WO2010064597A1 - Piperidine derivative - Google Patents
Piperidine derivative Download PDFInfo
- Publication number
- WO2010064597A1 WO2010064597A1 PCT/JP2009/070098 JP2009070098W WO2010064597A1 WO 2010064597 A1 WO2010064597 A1 WO 2010064597A1 JP 2009070098 W JP2009070098 W JP 2009070098W WO 2010064597 A1 WO2010064597 A1 WO 2010064597A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- ring
- reference example
- mmol
- nmr
- Prior art date
Links
- XDDPFYLZBCCPBX-UHFFFAOYSA-N C(CNCC1)C1c1cc(-c2ccccc2)ccn1 Chemical compound C(CNCC1)C1c1cc(-c2ccccc2)ccn1 XDDPFYLZBCCPBX-UHFFFAOYSA-N 0.000 description 1
- GMMIEOYPIKYMEE-UHFFFAOYSA-N Clc1nc(-c2c[s]cc2)ccn1 Chemical compound Clc1nc(-c2c[s]cc2)ccn1 GMMIEOYPIKYMEE-UHFFFAOYSA-N 0.000 description 1
- QRFYQWBMBLKLAU-UHFFFAOYSA-N Fc(cccc1-c2ccnc(C3CCNCC3)c2)c1F Chemical compound Fc(cccc1-c2ccnc(C3CCNCC3)c2)c1F QRFYQWBMBLKLAU-UHFFFAOYSA-N 0.000 description 1
- RKLKUKLBXGQPTL-UHFFFAOYSA-N Fc(cccc1-c2nccc(C3CCNCC3)n2)c1F Chemical compound Fc(cccc1-c2nccc(C3CCNCC3)n2)c1F RKLKUKLBXGQPTL-UHFFFAOYSA-N 0.000 description 1
- VYZKNRBBDSRALM-UHFFFAOYSA-N Fc(cccc1-c2ncnc(C3CCNCC3)c2)c1F Chemical compound Fc(cccc1-c2ncnc(C3CCNCC3)c2)c1F VYZKNRBBDSRALM-UHFFFAOYSA-N 0.000 description 1
- RJGVXFIVMMTHCU-UHFFFAOYSA-N Fc(cnc(C1CCNCC1)c1)c1-c1ccccc1 Chemical compound Fc(cnc(C1CCNCC1)c1)c1-c1ccccc1 RJGVXFIVMMTHCU-UHFFFAOYSA-N 0.000 description 1
- MLJCPPUJYVYODD-UHFFFAOYSA-N Fc1ccc(-c2cc(Cl)ncc2)c(F)c1 Chemical compound Fc1ccc(-c2cc(Cl)ncc2)c(F)c1 MLJCPPUJYVYODD-UHFFFAOYSA-N 0.000 description 1
- SLAHUTRJKUQFFS-UHFFFAOYSA-N O=C(Nc1ccc[n-]1)N(CC1)CCC1c1nc(-c(cc2)ccc2F)ccn1 Chemical compound O=C(Nc1ccc[n-]1)N(CC1)CCC1c1nc(-c(cc2)ccc2F)ccn1 SLAHUTRJKUQFFS-UHFFFAOYSA-N 0.000 description 1
- GKKGHBPSYCMVPJ-UHFFFAOYSA-N O=C(Nc1cccnn1)N(CC1)CCC1c1nc(-c2c[o]cc2)ccn1 Chemical compound O=C(Nc1cccnn1)N(CC1)CCC1c1nc(-c2c[o]cc2)ccn1 GKKGHBPSYCMVPJ-UHFFFAOYSA-N 0.000 description 1
- VOLPYROTYITAQJ-UHFFFAOYSA-N O=C(Nc1cccnn1)N(CC1)CCC1c1nc(-c2c[s]cc2)ccn1 Chemical compound O=C(Nc1cccnn1)N(CC1)CCC1c1nc(-c2c[s]cc2)ccn1 VOLPYROTYITAQJ-UHFFFAOYSA-N 0.000 description 1
- KWQDCPHXAMDQMM-UHFFFAOYSA-N O=C(Nc1cnccc1)N(CC1)CCC1c1nc(-c2cccc(F)c2)ccn1 Chemical compound O=C(Nc1cnccc1)N(CC1)CCC1c1nc(-c2cccc(F)c2)ccn1 KWQDCPHXAMDQMM-UHFFFAOYSA-N 0.000 description 1
- NQQXHEXFYVYRBJ-UHFFFAOYSA-N O=C(Nc1cnccc1)N(CC1)CCC1c1nc(-c2ccccc2)ccn1 Chemical compound O=C(Nc1cnccc1)N(CC1)CCC1c1nc(-c2ccccc2)ccn1 NQQXHEXFYVYRBJ-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/08—Drugs for disorders of the alimentary tract or the digestive system for nausea, cinetosis or vertigo; Antiemetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/14—Prodigestives, e.g. acids, enzymes, appetite stimulants, antidyspeptics, tonics, antiflatulents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/10—Drugs for disorders of the urinary system of the bladder
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/16—Masculine contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/18—Feminine contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/04—Drugs for disorders of the muscular or neuromuscular system for myasthenia gravis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
- A61P29/02—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID] without antiinflammatory effect
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/04—Immunostimulants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
Definitions
- the present invention relates to a novel piperidine derivative having an FAAH inhibitory action.
- Pain is a disease that can be serious for patients, reduce QOL, and make social life difficult. Pain is classified according to its cause into inflammatory pain, neuropathic pain, nociceptive pain, psychogenic pain and the like. Inflammatory pain is pain caused by inflammation caused by noxious mechanical stimulation, thermal stimulation, chemical stimulation, etc. applied from outside the living body. It is known that inflammatory cytokines and cyclooxygenase play an important role not only in the local area of inflammation but also in the spinal cord for the expression of inflammatory pain. Neuropathic pain is pathological pain due to abnormal function of the peripheral or central nervous system itself. Nociceptive pain is pain caused by the addition of a noxious stimulus that injures normal tissue or has the risk of it, and is divided into somatic pain and visceral pain.
- cyclooxygenase (COX) inhibitors such as indomethacin, cyclooxygenase II (COX-II) inhibitors such as celecoxib, central analgesics such as tramadol, and antipyretic analgesics such as acetaminophen are used. It has been. However, when a cyclooxygenase inhibitor is used for a long time, gastrointestinal disorders may occur as a side effect, which is problematic. In addition, cyclooxygenase II inhibitors have also been reported to cause gastric ulcers, and recently, cardiovascular side effects such as myocardial infarction and cerebral infarction have become a problem.
- opioid analgesics such as morphine
- anticonvulsants such as gabapentin and pregabalin are used.
- opioid analgesics such as morphine
- anticonvulsants such as gabapentin and pregabalin
- a cannabinoid receptor has been identified since around 1990 as a receptor for ⁇ 9-tetrahydrocannabinol ( ⁇ 9-THC), which is an active ingredient of cannabis.
- the CB1 receptor see Non-Patent Document 1
- its splice variant CB1a see Non-Patent Document 2
- the CB2 receptor see Non-Patent Document 3
- N-arachidonoylethanolamine was discovered from pig brain as an endogenous ligand of the CB1 receptor (see Non-Patent Document 4).
- Anandamide belongs to N-acylated ethanolamine as well as N-palmitoylethanolamine and N-oleoylethanolamine.
- Fatty acid amides containing these N-acylated ethanolamines have physiological functions such as pain (see Non-Patent Documents 5 and 6), adjustment of feeding (see Non-Patent Document 7), and promotion of sleep (see Non-Patent Document 8). It has been shown that it exerts its action.
- the biosynthesis or degradation pathway of fatty acid amides has been investigated since about 1980.
- transacylase produces N-acylphosphatidylethanolamine anandamide in a calcium-dependent manner (see Non-Patent Document 9), and then fatty acid amide is released by phospholipase D (see Non-Patent Document 10).
- the presence of enzyme activity that hydrolyzes fatty acid amides to the corresponding fatty acids and eliminates their physiological activity has been suggested, but the substance was not clear until the late 1990s.
- An active component that hydrolyzes oleamide was purified from rats, and cDNA was cloned (see Non-Patent Document 11).
- the enzyme produced by gene recombination hydrolyzes various fatty acid amides including oleamide and anandamide, and is named as fatty acid amide hydrolase (hereinafter abbreviated as “FAAH” in this specification). It was. Enzymes that biosynthesize fatty acid amides have not been fully elucidated. However, that fatty acid amides are produced from nerve cells in a calcium-dependent manner, that is, in a nerve activity-dependent manner (see Non-Patent Document 12), is extremely significant in therapeutic drug development. FAAH knockout mice have been created, FAAH inhibitors have been discovered, and the physiological significance of FAAH inhibition is being elucidated.
- FAAH knockout mice the content of fatty acid amide in the brain including anandamide is increased 10 to 15 times, but the exercise capacity, body weight, and body temperature are normal. However, a decrease in pain responsiveness was observed, and this correlated with the brain fatty acid amide content (see Non-Patent Document 13).
- Examples of FAAH inhibitors include trifluoromethyl ketone derivatives (see Non-Patent Document 14), ⁇ -ketoheterocyclic derivatives (see Non-Patent Document 15), sulfonyl fluoride derivatives (see Non-Patent Document 16), fluorophosphonate derivatives ( Non-patent document 17) Allyl carbamate derivatives (see non-patent document 18) and the like are known.
- FAAH and anandamide are involved in various diseases.
- FAAH inhibitors have brain / nerve cell protective effects and are useful as therapeutic agents for cerebrovascular disorders.
- FAAH there is much FAAH in the brain of an Alzheimer patient (refer nonpatent literature 19). It has been clarified by a test using rats that anti-Parkinson action is exhibited by increasing anandamide (see Non-Patent Document 20). It has been reported that FAAH decreases in miscarried women (see Non-Patent Document 21). Anandamide has been reported to suppress the growth of rectal cancer (see Non-Patent Document 22). FAAH knockout mice have been reported to be less susceptible to colitis and colitis (see Non-Patent Document 23).
- FAAH inhibitors exhibit an anxiolytic action (see Non-Patent Document 24).
- FAAH has been reported to be a hydrolase of oleylethanolamide, a satiety factor present in the small intestine (see Non-Patent Document 25).
- FAAH is a stearoylethanolamide hydrolase, and it has been reported that when stearoylethanolamide is administered to mice, feeding is suppressed (see Non-Patent Document 26). Since anandamide is an agonist of the nociceptor vanilloid receptor, the FAAH inhibitor acts like a vanilloid receptor agonist (eg, frequent urinary urinary incontinence preventive treatment interstitial cystitis) Is also expected (see Patent Document 1).
- FAAH is also an enzyme that hydrolyzes oleamide, which is an endogenous sleep substance. For this reason, FAAH inhibitors induce sleep by suppressing the degradation of oleamide (Patent Document 2).
- Patent Document 2 Japanese Patent Laid-Open No. 2002-202204 US2003 / 0092734A International Publication 2007/020888 Nature 1990, 346, 561 J. et al. Biol. Chem. 1995, 270, 3726. Eur. J. et al. Biochem. 1995, 232, 54 Science, 1992, 258, 1946. Nature, 1998, 394, 277 Pain, 1998, 76, 189 Nature, 2001, 414, 209. Science, 1995, 268, 1506. J. et al. Neurochem. 1983, 41, 1303 J.
- An object of the present invention is to provide a safe and excellent pain prevention / treatment agent.
- Ring Ar 1 represents an optionally substituted aromatic heterocycle
- a 1 represents CR 1 or N
- a 2 represents CR 2 or N
- a 3 represents CR 3 or N
- a 4 represents CR 4 or N
- At least one of A 4 represents N
- Ring Ar 2 represents an aromatic hydrocarbon ring that may be substituted with one or more halogen atoms, or an aromatic heterocycle that may be substituted with one or more halogen atoms
- R 1 , R 2 , R 3 , and R 4 each independently represent a hydrogen atom, a halogen atom, or an optionally substituted C 1-6 alkyl group.
- the compound represented by the formula (I) of the present invention or a salt thereof has an excellent FAAH inhibitory activity and is useful as a safe and excellent preventive / therapeutic agent for pain, depression or anxiety.
- halogen (atom) include fluorine (atom), chlorine (atom), bromine (atom), and iodine (atom).
- C 1-6 alkyl group include methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl, tert-butyl, pentyl, isopentyl, neopentyl, hexyl and the like.
- the ring Ar 1 represents an optionally substituted aromatic heterocycle.
- aromatic heterocycle represented by the ring Ar 1 include, for example, 5 to 14 members containing 1 or 2 heteroatoms selected from a nitrogen atom, a sulfur atom and an oxygen atom in addition to a carbon atom.
- it is a 5- to 10-membered, more preferably 5- or 6-membered aromatic heterocycle.
- thienyl eg, 2-thienyl, 3-thienyl
- furyl eg, 2-furyl, 3-furyl
- pyridyl eg, 2-pyridyl, 3-pyridyl, 4-pyridyl
- Thiazolyl eg, 2-thiazolyl, 4-thiazolyl, 5-thiazolyl
- oxazolyl eg, 2-oxazolyl, 4-oxazolyl
- pyrazinyl pyrimidinyl (eg, 2-pyrimidinyl, 4-pyrimidinyl)
- pyrrolyl eg, 1 -Pyrrolyl, 2-pyrrolyl, 3-pyrrolyl
- imidazolyl eg, 1-imidazolyl, 2-imidazolyl, 4-imidazolyl
- pyrazolyl eg, 1-pyrazolyl, 3-pyrazolyl, 4-pyrazolyl
- pyridazinyl
- the aromatic heterocycle is preferably a 5- or 6-membered aromatic heterocycle, more preferably pyridyl, pyridazinyl, or isoxazolyl, and still more preferably pyridazyl.
- the aromatic heterocycle may have 1 to 5, preferably 1 or 2, substituents at substitutable positions.
- An unsubstituted aromatic heterocyclic ring is also preferable.
- the substituent include a halogen atom (for example, fluorine, chlorine, bromine, iodine, etc.), a lower alkyl group (for example, methyl, ethyl, propyl, isopropyl, etc.) which may be halogenated, hydroxylated, or oxoated.
- C 1-6 alkyl groups such as butyl, isobutyl, sec-butyl, tert-butyl, pentyl, hexyl, etc .; halogenated such as fluoromethyl, chloromethyl, difluoromethyl, dichloromethyl, trifluoromethyl, trichloromethyl, etc.
- C 1-6 alkyl group also a C 1-6 alkyl group; hydroxymethyl, hydroxylated which may be C 1-6 alkyl groups such as hydroxyethyl, 2-oxopropyl, 2-oxobutyl may be oxo of such groups C 1 -6 alkyl group), cycloalkyl group (for example, cyclopropyl Le, cyclobutyl, cyclopentyl, and the like C 3-6 cycloalkyl groups such as cyclohexyl), a lower alkynyl group (e.g., ethynyl, 1-propynyl, etc.
- a lower alkynyl group e.g., ethynyl, 1-propynyl, etc.
- C 2-6 alkynyl group propargyl, etc. lower alkenyl groups (e.g., vinyl C 2-6 alkenyl groups such as allyl, isopropenyl, butenyl, isobutenyl, etc.), aralkyl groups (eg, C 7-11 aralkyl groups such as benzyl, ⁇ -methylbenzyl, phenethyl, etc.), aryl groups (eg, phenyl, C 6-10 aryl group such as naphthyl, etc., preferably phenyl group), aryloxy group (eg C 6-10 aryloxy group etc.), lower alkanoyl group (eg formyl; acetyl, propionyl, butyryl, isobutyryl etc.) the C 1-6 alkyl - carbonyl group), Ariruka Boniru (e.g., benzoyl group, C 6-10 aryl such as naphthoyl -
- the ring Ar 2 represents an aromatic hydrocarbon ring that may be substituted with one or more halogen atoms, or an aromatic heterocyclic ring that may be substituted with one or more halogen atoms.
- the “aromatic hydrocarbon ring” represented by the ring Ar 2 include C 6-14 aryl groups such as phenyl, 2-biphenylyl, 3-biphenylyl, 4-biphenylyl, and cyclooctatetraenyl.
- the aromatic hydrocarbon ring is preferably phenyl.
- the aromatic hydrocarbon ring may have 1 to 5, preferably 1 or 2, halogen atoms, preferably fluorine atoms, at substitutable positions.
- An unsubstituted aromatic hydrocarbon ring is also preferred.
- the “aromatic heterocycle” represented by the ring Ar 2 include the same “aromatic heterocycle” as described in the ring Ar 1 .
- the aromatic heterocycle is preferably furyl or thienyl.
- the aromatic heterocyclic ring may have 1 to 5, preferably 1 or 2, halogen atoms, preferably fluorine atoms, at substitutable positions.
- An unsubstituted aromatic heterocyclic ring is also preferable.
- a 1 represents CR 1 or N
- a 2 represents CR 2 or N
- a 3 represents CR 3 or N
- a 4 represents CR 4 or N
- a 1 , A 2 , A 3 , and A 4 represent N.
- 1 to 2 of A 1 , A 2 , A 3 , and A 4 are N, more preferably 2 are N.
- R 1 , R 2 , R 3 , and R 4 each independently represent a hydrogen atom, a halogen atom, or an optionally substituted C 1-6 alkyl group.
- Such optionally substituted C 1-6 alkyl groups include optionally halogenated or oxoated C 1-6 alkyl (eg, methyl, chloromethyl, difluoromethyl, trichloromethyl, trifluoromethyl, ethyl).
- R 1 , R 2 , R 3 and R 4 are preferably a hydrogen atom or a halogen atom (particularly a fluorine atom), more preferably a hydrogen atom.
- the partial structure of formula (I) May be substituted by a halogen atom (preferably a fluorine atom) And more preferably And more preferably It is.
- a halogen atom preferably a fluorine atom
- the compound represented by formula (I) is: Ring Ar 1 is a pyridine ring, a pyridazine ring, or an isoxazole ring substituted with a C 1-6 alkyl group, A ring Ar 2 may be substituted with one or two halogen atoms, a benzene ring, a furan ring, or a thiophene ring; Is a compound.
- the compound of formula (I) is of formula (Ia) [Where Ring Ar 3 represents a pyridazine ring optionally substituted with a halogen atom; Ring Ar 4 represents a benzene ring which may be substituted with a halogen atom. ] It is a compound represented by these.
- the ring Ar 3 may have 1 to 3 halogen atoms at substitutable positions.
- ring Ar 3 is unsubstituted.
- “unsubstituted” means having no substituent at a substitutable position on the ring Ar 3 in formula (Ia).
- the ring Ar 4 may have 1 to 5, preferably 1 or 2, halogen atoms at substitutable positions.
- An unsubstituted aromatic heterocyclic ring is also preferable.
- “unsubstituted” means having no substituent at a substitutable position on the ring Ar 4 in formula (Ia).
- the halogen atom include fluorine, chlorine, bromine, and iodine.
- the halogen atom is fluorine.
- the compound represented by the formula (Ia) is a compound in which the ring Ar 3 is unsubstituted and the ring Ar 4 is a benzene ring optionally having a halogen atom.
- the compound of formula (I) is: 4- (4-phenylpyrimidin-2-yl) -N-pyridazin-3-ylpiperidin-1-carboxamide; 4- [4- (2,3-difluorophenyl) pyrimidin-2-yl] -N-pyridazin-3-ylpiperidin-1-carboxamide; 4- [4- (3-Fluorophenyl) pyrimidin-2-yl] -N-pyridazin-3-ylpiperidin-1-carboxamide; or 4- [4- (4-fluorophenyl) pyrimidin-2-yl]- N-pyridazin-3-ylpiperidine-1-carboxamide.
- the salt of the compound represented by the formula (I) is preferably a pharmacologically acceptable salt, for example, a salt with an inorganic base, a salt with an organic base, a salt with an inorganic acid, a salt with an organic acid. And salts with basic or acidic amino acids.
- the salt with an inorganic base include alkali metal salts such as sodium salt and potassium salt; alkaline earth metal salts such as calcium salt and magnesium salt; aluminum salt and ammonium salt.
- the salt with an organic base include trimethylamine, triethylamine, pyridine, picoline, ethanolamine, diethanolamine, triethanolamine, tromethamine [tris (hydroxymethyl) methylamine], tert-butylamine, cyclohexylamine, benzylamine, And salts with dicyclohexylamine, N, N-dibenzylethylenediamine and the like.
- salt with inorganic acid examples include salts with hydrochloric acid, hydrobromic acid, nitric acid, sulfuric acid, phosphoric acid and the like.
- salts with organic acids include formic acid, acetic acid, trifluoroacetic acid, phthalic acid, fumaric acid, oxalic acid, tartaric acid, maleic acid, citric acid, succinic acid, malic acid, methanesulfonic acid, and benzenesulfonic acid And salts with p-toluenesulfonic acid and the like.
- Particularly preferred is a salt with trifluoroacetic acid.
- Preferable examples of the salt with basic amino acid include salts with arginine, lysine, ornithine and the like.
- Preferable examples of the salt with acidic amino acid include salts with aspartic acid, glutamic acid and the like.
- a prodrug of compound (I) is a compound that is converted to compound (I) by a reaction with an enzyme, gastric acid, or the like under physiological conditions in vivo, that is, compound (I) that is enzymatically oxidized, reduced, hydrolyzed, etc.
- Compound (I) prodrugs include compounds in which the amino group of compound (I) is acylated, alkylated or phosphorylated (eg, the amino group of compound (I) is eicosanoylated, alanylated, pentylaminocarbonylated) , (5-methyl-2-oxo-1,3-dioxolen-4-yl) methoxycarbonylation, tetrahydrofuranylation, tetrahydropyranylation, pyrrolidylmethylation, pivaloyloxymethylation or tert-butylation Compound); a compound wherein the hydroxy group of compound (I) is acylated, alkylated, phosphorylated or borated (eg, hydroxy group of compound (I) is acetylated, palmitoylated, propanoylated, pivaloylated, succinyl , Fumarylation, alanylation, dimethylaminomethylcarbonylation or
- prodrugs of Compound (I) can be obtained under the physiological conditions as described in Hirokawa Shoten 1990, “Development of Pharmaceuticals”, Volume 7, Molecular Design, pages 163 to 198. It may change to. Hereafter, the manufacturing method of this invention compound is demonstrated. In addition, you may use the compound used as a raw material compound as a salt, respectively. As such a salt, those exemplified as the salt of the aforementioned compound (I) can be used.
- Compound (I) of the present invention can be synthesized, for example, by the following scheme: [Wherein L 1 represents a leaving group, and other symbols are as defined above. ] It can manufacture by the manufacturing method 1 represented by this, or the method according to this.
- compound (II) and compound (III) are first subjected to a coupling reaction to produce compound (IV).
- the leaving group L 1 include halides such as chloride, bromide and iodide, and alkylsulfonyloxy groups such as methanesulfonyloxy group and trifluoromethanesulfonyloxy group.
- the coupling reaction is performed in the presence of a base and a catalyst with a solvent that does not affect the reaction.
- the amount of compound (III) to be used is about 0.5 to about 10 mol, preferably about 0.9 to about 3 mol, per 1 mol of compound (II).
- bases include basic salts such as sodium carbonate, potassium carbonate, cesium carbonate, sodium hydrogen carbonate, tripotassium phosphate, aromatic amines such as pyridine and lutidine, triethylamine, tripropylamine, tributylamine, cyclohexyldimethylamine, Tertiary amines such as 4-dimethylaminopyridine, N, N-dimethylaniline, N-methylpiperidine, N-methylpyrrolidine, N-methylmorpholine, sodium methoxide, sodium ethoxide, sodium tert-butoxide, potassium Metal alkoxides such as tributoxide are used.
- basic salts such as sodium carbonate, potassium carbonate, cesium carbonate, sodium hydrogen carbonate, tripotassium phosphate
- aromatic amines such as pyridine and lutidine, triethylamine, tripropylamine, tributylamine, cyclohexyldimethylamine
- the amount of the base to be used is about 0.5 to about 10 mol, preferably about 1 to about 5 mol equivalent, relative to compound (II), respectively.
- the catalyst used in this reaction include palladium catalysts such as palladium acetate, palladium chloride, tetrakis (triphenylphosphine) palladium, bis (dibenzylideneacetone) palladium, tris (dibenzylideneacetone) dipalladium, It can also be performed by adding a ligand.
- phosphine is preferable, and trialkylphosphine (for example, tributylphosphine, tricyclohexylphosphine and the like), triarylphosphine (for example, triphenylphosphine and the like), trialkoxyphosphine and the like can be mentioned.
- the amount of the palladium catalyst to be used is generally about 0.001 to about 5 mol, preferably about 0.01 to about 0.5 mol, relative to compound (II).
- the amount of “phosphine” to be used is generally about 0.001 to about 10 mol, preferably about 0.01 to about 1 mol, relative to compound (II).
- Examples of the solvent that does not affect the reaction include ethers such as tetrahydrofuran and 1,2-dimethoxyethane, alcohols such as methanol, ethanol and propanol, halogenated hydrocarbons such as chloroform, and aromatics such as benzene and toluene. Group hydrocarbons, nitriles such as acetonitrile and propionitrile, amides such as N, N-dimethylformamide, sulfoxides such as dimethyl sulfoxide, and water. These solvents may be used by mixing two or more kinds at an appropriate ratio. The amount of these solvents used is, for example, 1 to 100 times the volume of the compound (II).
- the reaction temperature is usually about ⁇ 50 ° C.
- the reaction time is usually about 0.5 to about 36 hours. In this reaction, the reaction time can be shortened by using a microwave reaction apparatus or the like.
- the compound (IV) thus obtained can be isolated and purified by known separation and purification means such as concentration, concentration under reduced pressure, solvent extraction, crystallization, recrystallization, phase transfer, chromatography, and the like. In addition, compound (IV) may be used for the next reaction without isolation.
- compound (IV) is reduced to produce compound (V).
- the reduction of compound (IV) can be performed using a metal catalyst in a hydrogen atmosphere.
- an appropriate acid catalyst may be added. Raney nickel, platinum oxide, metallic palladium, palladium carbon, etc. are used as the metal catalyst.
- the amount of the metal catalyst to be used is generally about 1 to about 1000 wt%, preferably about 5 to about 20 wt%, relative to compound (IV), respectively.
- organic acids such as formic acid, acetic acid, trifluoroacetic acid and p-toluenesulfonic acid, and mineral acids such as sulfuric acid, hydrochloric acid and hydrobromic acid are used.
- the amount of the acid catalyst to be used is about 0.1 to excess amount per 1 mol of compound (IV).
- This reaction is advantageously performed using a solvent inert to the reaction.
- a solvent is not particularly limited as long as the reaction proceeds.
- alcohols such as methanol, ethanol and propanol
- ethers such as diethyl ether, tetrahydrofuran, dioxane and 1,2-dimethoxyethane
- benzene, toluene Hydrocarbons such as cyclohexane and hexane
- amides such as N, N-dimethylformamide and N, N-dimethylacetamide
- organic acids such as acetic acid, water and the like are used.
- the hydrogen pressure is usually about 1 to about 100 atmospheres, preferably about 1 to about 5 atmospheres.
- the reaction time is usually about 30 minutes to about 48 hours, preferably about 1 to 24 hours.
- the reaction temperature is usually about 0 to about 120 ° C, preferably about 20 to about 80 ° C.
- the product (V) can be isolated from the reaction mixture according to a conventional method after removing the catalyst, and can be easily purified by a usual separation means (eg, recrystallization, distillation, chromatography, etc.). In addition, compound (V) may be used for the next reaction without isolation.
- the tert-butoxycarbonyl group of compound (V) is removed to produce compound (VI).
- This reaction is carried out by reacting an acid according to a conventional method in a solvent that does not adversely influence the reaction.
- the acid include hydrogen chloride, hydrogen bromide, sulfuric acid, trifluoroacetic acid, trifluoromethanesulfonic acid and the like.
- the amount of the acid used is preferably about 1 to about 100 molar equivalents relative to compound (V), respectively.
- solvents that do not affect the reaction include hydrocarbons such as hexane and cyclohexane, alcohols such as methanol, ethanol, and propanol, ethers such as diethyl ether, tetrahydrofuran, dioxane, and 1,2-dimethoxyethane, and acetic acid.
- Esters such as ethyl and methyl acetate, halogenated hydrocarbons such as chloroform and dichloromethane; aromatic hydrocarbons such as benzene and toluene; amides such as N, N-dimethylformamide; sulfoxides such as dimethyl sulfoxide Can be mentioned.
- the amount of these solvents used is, for example, 1 to 100 times the volume of the compound (V).
- the reaction temperature is usually about ⁇ 50 ° C. to about 250 ° C., preferably 0 ° C. to 120 ° C.
- the reaction time is usually about 0.5 to about 24 hours.
- the compound (VI) thus obtained can be isolated and purified by known separation and purification means, for example, concentration, concentration under reduced pressure, solvent extraction, crystallization, recrystallization, phase transfer, chromatography, etc. Compound (VI) may be used for the next reaction without isolation.
- compound (VI) is uread to produce compound (I). Ureaization is performed on the compound (VI) with respect to the isocyanate (VII), 2,2,2-trichloroethoxycarbamate (VIII), bis (2,2,2-trichloroethoxycarbamate) (IX), or , Aryl ester (X) can be reacted.
- the production of compound (I) by the reaction of compound (VI) and isocyanate (VII) is carried out in the presence of a base and isocyanate (VII) with a solvent that does not affect the reaction.
- Examples of the base include triethylamine, tributylamine, diisopropylethylamine, potassium carbonate, sodium carbonate, sodium hydride, potassium hydride and the like.
- the amount of base and isocyanate (VII) to be used is preferably about 1 to about 5 molar equivalents relative to compound (VI), respectively.
- Examples of the solvent that does not affect the reaction include ethers such as diethyl ether, tetrahydrofuran, dioxane, and 1,2-dimethoxyethane, halogenated hydrocarbons such as chloroform and dichloromethane, and aromatic hydrocarbons such as benzene and toluene.
- Amides such as N, N-dimethylformamide, and sulfoxides such as dimethyl sulfoxide. Two or more of these solvents may be mixed and used at an appropriate ratio. The amount of these solvents to be used is, for example, 1 to 100 times the volume of the compound (VI).
- the reaction temperature is usually about ⁇ 50 ° C. to about 250 ° C., preferably 0 ° C. to 120 ° C.
- the reaction time is usually about 0.5 to about 24 hours.
- the compound (I) thus obtained can be isolated and purified by known separation and purification means such as concentration, concentration under reduced pressure, solvent extraction, crystallization, recrystallization, phase transfer, chromatography, and the like.
- a base in a solvent that does not affect the reaction.
- the base include pyridine, triethylamine, diisopropylethylamine, potassium carbonate, sodium carbonate, sodium hydride, potassium hydride and the like.
- the amount of base, 2,2,2-trichloroethoxycarbamate (VIII), bis (2,2,2-trichloroethoxycarbamate) (IX), and aryl ester (X) used is preferably relative to compound (VI).
- solvents such as diethyl ether, tetrahydrofuran, dioxane and 1,2-dimethoxyethane, halogenated hydrocarbons such as dichloromethane and chloroform, and esters such as ethyl acetate and methyl acetate.
- Aromatic hydrocarbons such as benzene and toluene, amides such as N, N-dimethylformamide, and sulfoxides such as dimethyl sulfoxide. Two or more of these solvents may be mixed and used at an appropriate ratio.
- the amount of these solvents to be used is, for example, 1 to 100 times the volume of the compound (V).
- the reaction temperature is usually about ⁇ 50 ° C. to 200 ° C., preferably 0 ° C. to 120 ° C.
- the reaction time is usually about 0.5 to about 36 hours.
- the compound (I) thus obtained can be isolated and purified by known separation and purification means such as concentration, concentration under reduced pressure, solvent extraction, crystallization, recrystallization, phase transfer, chromatography, etc.
- Examples of the leaving group L 2 include halides such as chloride, bromide, iodide, and alkylsulfonyloxy groups such as methanesulfonyloxy group and trifluoromethanesulfonyloxy group.
- compound (II) and compound (XI) are first subjected to a coupling reaction to produce compound (XII).
- Compound (XII) can be synthesized by a method similar to the production of compound (IV) shown in Production Method 1.
- the compound (XII) thus obtained can be isolated and purified by known separation and purification means, for example, concentration, concentration under reduced pressure, solvent extraction, crystallization, recrystallization, phase transfer, chromatography, etc.
- compound (XII) may be used for the next reaction without isolation.
- compound (IV) is produced by a coupling reaction between compound (XII) and boronic acids or boronic esters.
- the coupling reaction is performed in the presence of a base and a catalyst with a solvent that does not affect the reaction.
- the amount of the boronic acid or boronic acid ester to be used is about 0.5 to about 10 mol, preferably about 0.9 to about 3 mol, per 1 mol of compound (XII).
- Examples of the base include basic salts such as sodium carbonate, potassium carbonate, cesium carbonate, sodium hydrogen carbonate, tripotassium phosphate, aromatic amines such as pyridine and lutidine, triethylamine, tripropylamine, tributylamine, cyclohexyldimethylamine, Tertiary amines such as 4-dimethylaminopyridine, N, N-dimethylaniline, N-methylpiperidine, N-methylpyrrolidine, N-methylmorpholine, sodium methoxide, sodium ethoxide, sodium tert-butoxide, potassium Metal alkoxides such as tributoxide are used.
- basic salts such as sodium carbonate, potassium carbonate, cesium carbonate, sodium hydrogen carbonate, tripotassium phosphate
- aromatic amines such as pyridine and lutidine, triethylamine, tripropylamine, tributylamine, cyclohexyldimethylamine
- the amount of the base to be used is about 0.5 to about 10 mol, preferably about 1 to about 5 molar equivalent, relative to compound (XII), respectively.
- the catalyst used in this reaction include palladium catalysts such as palladium acetate, palladium chloride, tetrakis (triphenylphosphine) palladium, bis (dibenzylideneacetone) palladium, tris (dibenzylideneacetone) dipalladium, It can also be performed by adding a ligand.
- phosphine is preferable, and trialkylphosphine (for example, tributylphosphine, tricyclohexylphosphine and the like), triarylphosphine (for example, triphenylphosphine and the like), trialkoxyphosphine and the like can be mentioned.
- the amount of the palladium catalyst to be used is generally about 0.001 to about 5 mol, preferably about 0.01 to about 0.5 mol, relative to compound (XII).
- the amount of the “phosphine” to be used is generally about 0.001 to about 10 mol, preferably about 0.01 to about 1 mol, relative to compound (XII).
- solvents such as tetrahydrofuran and 1,2-dimethoxyethane
- alcohols such as methanol, ethanol and propanol
- halogenated hydrocarbons such as chloroform
- aromatics such as benzene and toluene.
- These solvents may be used by mixing two or more kinds at an appropriate ratio. The amount of these solvents to be used is, for example, 1 to 100 times the volume of the compound (XII).
- the reaction temperature is usually about ⁇ 50 ° C. to about 250 ° C., preferably 0 ° C. to 120 ° C.
- the reaction time is usually about 0.5 to about 36 hours. In this reaction, the reaction time can be shortened by using a microwave reaction apparatus or the like.
- the compound (IV) thus obtained can be isolated and purified by known separation and purification means such as concentration, concentration under reduced pressure, solvent extraction, crystallization, recrystallization, phase transfer, chromatography, and the like. In addition, compound (IV) may be used for the next reaction without isolation.
- compound (I) has an isomer such as an optical isomer, a stereoisomer, a positional isomer, a rotational isomer, etc.
- any one of the isomers and a mixture are encompassed in compound (I).
- compound (I) has an optical isomer
- the optical isomer resolved from the racemate is also encompassed in compound (I).
- Each of these isomers can be obtained as a single product by a known synthesis method or separation method (concentration, solvent extraction, column chromatography, recrystallization, etc.).
- Compound (I) may be a crystal, and it is included in compound (I) regardless of whether the crystal form is a single crystal form or a mixture of crystal forms.
- the crystal can be produced by crystallization by applying a crystallization method known per se.
- Compound (I) may be a solvate (such as a hydrate) or a non-solvate, and both are encompassed in compound (I).
- Compounds labeled with isotopes eg, 3 H, 14 C, 35 S, 125 I, etc.
- a deuterium converter obtained by converting 1 H into 2 H (D) is also encompassed in compound (I).
- Compound (I) or a prodrug thereof (hereinafter sometimes collectively referred to as the compound of the present invention) is a mammal (eg, mouse, rat, hamster, rabbit, cat, dog, cow, sheep, monkey, human) ), It has an excellent FAAH inhibitory activity, and is therefore useful as a prophylactic / therapeutic agent for a disease state or disease involving FAAH.
- the compound of the present invention prevents pain, for example, inflammatory pain associated with osteoarthritis and rheumatoid arthritis, and pain associated with diabetic painful neuropathy, postherpetic pain, trigeminal neuralgia and other neuropathic pain. And useful for treatment.
- cerebrovascular disorders caused by brain / nerve cell disorders brain / nerve cell protective effects upon head injury or spinal cord injury, cardiac arrest Brain damage during post-resuscitation, brain function decline before and after brain surgery, hypoxia, hypoglycemia, brain or spinal cord trauma, drug poisoning, gas poisoning, diabetes, administration of antitumor agents, alcohol damage, etc.
- Huntington's disease Huntington's disease, prion disease, amyotrophic lateral sclerosis, spinocerebellar degeneration, eating disorder, obesity, frequent urination, urinary incontinence, rheumatism, osteoarthritis, interstitial cystitis, Crohn's disease, colitis , Colitis, colon cancer, colon cancer, contraception or AIDS, but is not limited thereto.
- the compound of the present invention further comprises nausea, nausea or vomiting due to anticancer agents; anorexia or anorexia of cachexia in cancer or infectious diseases (eg, AIDS); convulsions and pain due to multiple sclerosis , Tremor, nystagmus or nocturnal urine; chronic pain; Huntington's chorea; Tourette syndrome; levotoba-induced movement disorder; Depression; anxiety; psychiatric disease; Crohn's disease; Alzheimer's disease; interstitial cystitis; AIDS; colitis; colitis; colon cancer; rectal cancer; hypertriglyceridemia; It is also useful as a preventive / therapeutic agent or contraceptive for Parkinson's disease.
- the compounds of the present invention are useful for sleep disorders such as endogenous sleep disorders (eg, psychophysiological insomnia), extrinsic sleep disorders, and circadian rhythm disorders (eg, time zone change syndrome (time difference blur)), Sleep abnormalities such as shift work sleep disorder, irregular sleep awakening pattern, sleep phase regression syndrome, sleep phase advance syndrome, non-24-hour sleep awakening); sleep-related complications; internal or psychiatric disorders (eg, chronic obstructive lungs) It is useful as a preventive or therapeutic agent for sleep disorders associated with diseases, Alzheimer's disease, Parkinson's disease, cerebrovascular dementia, schizophrenia, depression, anxiety).
- the subject compounds are further useful in methods for the present invention, methods for prevention, treatment, modulation, amelioration or reduction of the risks of diseases, disorders and conditions described herein.
- the dosage of active ingredient in the compositions of the invention can vary, however, it is necessary that the amount of active ingredient is such that a suitable dosage form is obtained.
- the active ingredient can be administered to patients (animals and humans) in need of such treatment at dosages that provide optimal pharmaceutical efficacy.
- the selected dose depends on the desired therapeutic effect, the route of administration and the duration of treatment. Dosages will vary from patient to patient depending on the nature and severity of the disease, the patient's weight, the special diet performed later by the patient, the medications performed simultaneously, and other factors recognized by those skilled in the art.
- daily dose levels between 0.0001 mg / kg body weight and 10 mg / kg body weight are administered to patients, such as humans and the elderly, in order to obtain an effective inhibitory effect of FAAH.
- the range of doses that can be administered in a single dose or multiple doses is generally about 0.5 mg to 1.0 g per patient per day.
- the dosage range is about 0.5 mg to 500 mg per patient per day, more preferably about 0.5 mg to 200 mg per patient per day, even more preferably about 5 mg to 50 mg per patient per day.
- the pharmaceutical composition of the present invention may be provided in a solid dosage form preferably containing from about 0.5 mg to 500 mg of the active ingredient, more preferably from about 1 mg to 250 mg of the active ingredient.
- the pharmaceutical composition is preferably provided in a solid dosage form comprising about 1 mg, 5 mg, 10 mg, 25 mg, 50 mg, 100 mg, 200 mg or 250 mg of the active ingredient.
- the composition is preferably 1.0 to 1000 mg of active ingredient, especially active ingredient 1, 5, 10, 15, 20, 25, for symptomatic adjustment of the dose to the patient to be treated.
- the compounds may be administered on a regimen of 1 to 4 times per day, preferably once or twice per day.
- Compound (I) or a prodrug thereof has low toxicity (eg, acute toxicity, chronic toxicity, genotoxicity, reproductive toxicity, cardiotoxicity, drug interaction, carcinogenicity), and can be used as it is or pharmacologically.
- toxicity eg, acute toxicity, chronic toxicity, genotoxicity, reproductive toxicity, cardiotoxicity, drug interaction, carcinogenicity
- a carrier or the like By mixing with a carrier or the like to obtain a pharmaceutical composition, it is possible to prevent or prevent various diseases described below for mammals (eg, humans, mice, rats, rabbits, dogs, cats, cows, horses, pigs, monkeys). It can be used as a therapeutic agent.
- Examples of the dosage form of the pharmaceutical composition include tablets (including sugar-coated tablets, film-coated tablets, sublingual tablets, orally disintegrating tablets), capsules (including soft capsules and microcapsules), granules, powders, and lozenges.
- Oral preparations such as syrup, emulsion, suspension, film (eg, orally disintegrating film); and injection (eg, subcutaneous injection, intravenous injection, intramuscular injection, intraperitoneal injection, Intravenous preparations, external preparations (eg, transdermal preparations, ointments), suppositories (eg, rectal suppositories, vaginal suppositories), pellets, nasal preparations, pulmonary preparations (inhalants), eye drops, etc.
- Oral preparations are mentioned. These can be safely administered orally or parenterally (eg, topical, rectal, intravenous administration). These preparations may be controlled-release preparations such as immediate-release preparations or sustained-release preparations (eg, sustained-release microcapsules).
- the pharmaceutical composition can be produced by a method commonly used in the field of pharmaceutical technology, for example, a method described in the Japanese Pharmacopoeia.
- the content of the compound of the present invention in the pharmaceutical composition varies depending on the dosage form, the dose of the compound of the present invention, etc., but is, for example, about 0.1 to 100% by weight.
- the pharmacologically acceptable carrier various organic or inorganic carrier substances commonly used as pharmaceutical materials are used, and excipients, lubricants, binders, disintegrants in solid preparations; solvents in liquid preparations , Solubilizing agents, suspending agents, isotonic agents, buffers, soothing agents and the like. If necessary, preparation additives such as preservatives, antioxidants, colorants, sweeteners and the like can also be used.
- excipient examples include lactose, sucrose, D-mannitol, D-sorbitol, starch, pregelatinized starch, dextrin, crystalline cellulose, low-substituted hydroxypropylcellulose, sodium carboxymethylcellulose, gum arabic, pullulan, light Anhydrous silicic acid, synthetic aluminum silicate, magnesium magnesium metasilicate, etc. are mentioned.
- Preferable examples of the lubricant include magnesium stearate, calcium stearate, talc, colloidal silica and the like.
- Preferred examples of the binder include pregelatinized starch, sucrose, gelatin, gum arabic, methyl cellulose, carboxymethyl cellulose, sodium carboxymethyl cellulose, crystalline cellulose, sucrose, D-mannitol, trehalose, dextrin, pullulan, hydroxypropyl cellulose, hydroxy Examples thereof include propylmethylcellulose and polyvinylpyrrolidone.
- disintegrant examples include lactose, sucrose, starch, carboxymethyl cellulose, carboxymethyl cellulose calcium, croscarmellose sodium, carboxymethyl starch sodium, light anhydrous silicic acid, low substituted hydroxypropyl cellulose and the like.
- the solvent include water for injection, physiological saline, Ringer's solution, alcohol, propylene glycol, polyethylene glycol, sesame oil, corn oil, olive oil, cottonseed oil and the like.
- the solubilizer include polyethylene glycol, propylene glycol, D-mannitol, trehalose, benzyl benzoate, ethanol, trisaminomethane, cholesterol, triethanolamine, sodium carbonate, sodium citrate, sodium salicylate, sodium acetate. Etc.
- suspending agent examples include surfactants such as stearyltriethanolamine, sodium lauryl sulfate, laurylaminopropionic acid, lecithin, benzalkonium chloride, benzethonium chloride, glyceryl monostearate; for example, polyvinyl alcohol, Examples include hydrophilic polymers such as polyvinylpyrrolidone, sodium carboxymethylcellulose, methylcellulose, hydroxymethylcellulose, hydroxyethylcellulose, hydroxypropylcellulose; polysorbates, polyoxyethylene hydrogenated castor oil, and the like.
- surfactants such as stearyltriethanolamine, sodium lauryl sulfate, laurylaminopropionic acid, lecithin, benzalkonium chloride, benzethonium chloride, glyceryl monostearate
- polyvinyl alcohol examples include hydrophilic polymers such as polyvinylpyrrolidone, sodium carboxymethylcellulose, methylcellulose, hydroxymethylcellulose,
- Preferable examples of the isotonizing agent include sodium chloride, glycerin, D-mannitol, D-sorbitol, glucose and the like.
- Preferable examples of the buffer include buffer solutions such as phosphate, acetate, carbonate, citrate and the like.
- Preferable examples of the soothing agent include benzyl alcohol.
- Preferable examples of the preservative include p-hydroxybenzoates, chlorobutanol, benzyl alcohol, phenethyl alcohol, dehydroacetic acid, sorbic acid and the like.
- Preferable examples of the antioxidant include sulfite and ascorbate.
- Suitable examples of the colorant include water-soluble edible tar dyes (eg, edible dyes such as edible red Nos. 2 and 3, edible yellow Nos. 4 and 5, edible blue Nos. 1 and 2), water-insoluble lake dyes (Eg, the aluminum salt of the water-soluble edible tar dye), natural dyes (eg, ⁇ -carotene, chlorophyll, bengara) and the like.
- Preferable examples of the sweetening agent include saccharin sodium, dipotassium glycyrrhizinate, aspartame, stevia and the like.
- the compounds of the present invention may be combined with one or more other drugs in the treatment, prevention, amelioration, or reduction of the risk of a disease or condition for which the compounds of the present invention or other drugs may have utility In which case the drug combinations are safer or more effective than each other drug alone.
- Such other drugs may be administered simultaneously or sequentially with the compounds and compounds of the present invention, in the routes and amounts commonly used for such drugs.
- a pharmaceutical composition in unit dosage form containing such other drugs and the compound of the present invention is preferred.
- combination therapy may also include therapies in which the compound of the invention and one or more other drugs are administered on different overlapping schedules.
- the compounds of the present invention and the other active ingredients can each be used in a smaller dose than when used alone.
- the pharmaceutical compositions of the present invention include those that contain one or more other active ingredients, in addition to a compound of the present invention.
- the above combinations include combinations of a compound of the present invention not only with one other active compound, but also with two or more other active compounds.
- the compounds of the present invention are used in combination with other drugs used in the prevention, treatment, modulation, amelioration, or reduction of the risk of diseases or conditions for which compounds of the present invention are useful.
- Such other drugs may be administered concurrently or sequentially with the compounds and compounds of the present invention by the routes and amounts generally used for such drugs.
- a pharmaceutical composition containing such other drugs in addition to the compound of the present invention is preferred.
- the pharmaceutical compositions of the present invention include those that also contain one or more other active ingredients, in addition to a compound of the present invention.
- the weight ratio of the compound of the present invention to the second active ingredient may be varied and will depend upon the effective dose of each ingredient. In general, each effective dose is used. Thus, for example, when a compound of the present invention is combined with another drug, the weight ratio of the compound of the present invention to the other drug is generally about 1000: 1 to about 1: 1000, preferably about 200: 1 to The range is about 1: 200. Combinations of a compound of the present invention and other active ingredients will generally also be within the aforementioned range, but in each case, an effective dose of each active ingredient should be used. In such combinations, the compounds of the present invention and other active drugs can be administered separately or together. Furthermore, the administration of one element can be before, simultaneously with, or after administration of the other drug.
- Examples of a drug that can be used in combination with the compound of the present invention include, for example, nonsteroidal anti-inflammatory drugs (eg, meloxicam, teoxicam, indomethacin, ibuprofen, celecoxib, rofecoxib, aspirin, indomethacin, etc.
- nonsteroidal anti-inflammatory drugs eg, meloxicam, teoxicam, indomethacin, ibuprofen, celecoxib, rofecoxib, aspirin, indomethacin, etc.
- DMARDs Disease-modifying anti-rheumatic drugs
- antipyretic analgesics acetanilide, acetaminophen, phenacetin, etc.
- steroidal anti-inflammatory drugs hydrocortisone, prednisolone, methylprednisolone, betamethasone, dexamethasone, etc.
- narcotic analgesics Morphine, fentanyl, codeine phosphate, pethidine, oxycodone, etc., non-narcotic analgesics (eg, tramadol), local anesthetics (eg, lidocaine), anticonvulsants (gabapentin, bupivacaine, carbamazepine, phenytoin) ), Antiarrhythmic drugs (procaine, etc.), anti-cytokine drugs (TNF inhibitor, MAP kinase inhibitor, etc.), aldose reductase inhibitors (Kinedakku etc
- the concomitant drugs include anti-dementia therapeutic agents such as acetylcholinesterase inhibitors (eg, donepezil, rivastigmine, galantamine, zanapezil, etc.), ⁇ amyloid protein production, secretion, accumulation, aggregation and / or deposition inhibitors [ ⁇ secretase Inhibitors (eg, compounds described in WO98 / 38156, compounds described in WO02 / 2505, WO02 / 2506, WO02 / 2512, OM99-2 (WO01 / 00663)), ⁇ -secretase inhibitors (such as LY-450139), ⁇ -secretase Modulators (E2012 etc.), ⁇ amyloid protein aggregation inhibitors (eg, PTI-00703, ALZHEMED (NC-531), PPI-368 (Special Tables Hei 11-514333), PPI-558 (Special Tables Hei 2001-500852), SKF-74652
- ⁇ -amyloid antibody eg., ⁇ -amyloid vaccine, ⁇ -amyloid degrading enzyme, etc.
- brain function stimulating agents eg, aniracetam, nicergoline, etc.
- neurogenesis / Regenerative promoter eg, Akt / PKB activator, GSK-3 ⁇ inhibitor, etc.
- Parkinson's disease therapeutic agent eg, dopamine receptor agonist (L-dopa, bromocriptene, pergolide, taripexole, pripepexol, cabergoline) , Adamantadine, etc.), monoamine oxidase (MAO) inhibitors (deprenyl, sergiline (selegiline), remacemide, riluzole, etc.), anticholinergic agents (eg, trihexyphenidyl, biperiden, etc.)) , COMT inhibitors (eg, entacapone, etc.)
- COMT inhibitors
- the compound of the present invention By combining the compound of the present invention and a concomitant drug, (1) The dose can be reduced compared to the case where the compound of the present invention or the concomitant drug is administered alone. (2) By selecting a concomitant drug having a different mechanism of action from the compound of the present invention, the treatment period can be set longer. (3) By selecting a concomitant drug having a mechanism of action different from that of the compound of the present invention, the therapeutic effect can be sustained. (4) By using the compound of the present invention and the concomitant drug in combination, excellent effects such as a synergistic effect can be obtained.
- the administration timing of the compound of the present invention and the concomitant drug is not limited, and the compound of the present invention and the concomitant drug may be administered simultaneously to the administration subject. Alternatively, administration may be performed with a time difference.
- the dose of the concomitant drug may be determined according to the dose used clinically, and can be appropriately selected depending on the administration subject, administration route, disease, combination and the like.
- the administration form of the compound of the present invention and the concomitant drug for example, (1) administration of a single preparation obtained by simultaneously formulating the compound of the present invention and the concomitant drug, and (2) separate administration of the compound of the present invention and the concomitant drug Simultaneous administration of the two preparations obtained by formulation into the same administration route, (3) Time difference in the same administration route of the two preparations obtained by separately formulating the compound of the present invention and the concomitant drug.
- the present invention is further explained in detail by the following examples, formulation examples and experimental examples, which are merely examples and do not limit the present invention and do not depart from the scope of the present invention. It may be changed with.
- the starting compound and reaction product may form a salt that does not interfere with the reaction.
- Reference example 2 4-Phenyl-2-piperidin-4-ylpyrimidine dihydrochloride Tert-butyl 4- (4-phenylpyrimidin-2-yl) -3,6-dihydropyridine-1 (2H) -carboxylate (8.00 g, 24.0 mmol) obtained in Reference Example 1 in methanol (300 ml) To the solution, 10% palladium carbon (800 mg) was added under a nitrogen atmosphere, and the reaction solution was stirred overnight at room temperature under a hydrogen atmosphere of 3 atm. The reaction solution was filtered and the filtrate was concentrated.
- Reference example 4 4- (2,3-Difluorophenyl) -2-piperidin-4-ylpyrimidine dihydrochloride
- Reference Example 8 4- (2,3-Difluorophenyl) -6-piperidin-4-ylpyrimidine dihydrochloride In the same manner as in Reference Example 2, from the tert-butyl 4- [6- (2,3-difluorophenyl) pyrimidin-4-yl] -3,6-dihydropyridine-1 (2H) -carboxylate obtained in Reference Example 7. The title compound (5.70 g, 86%) was obtained as a solid.
- reaction mixture was diluted with ethyl acetate, washed with saturated brine, dried over sodium sulfate, and the solvent was evaporated under reduced pressure.
- Reference Example 18 4-Phenyl-2-piperidin-4-ylpyridine dihydrochloride To a solution of tert-butyl 4- (4-phenylpyridin-2-yl) piperidine-1-carboxylate (4.15 g, 12.3 mmol) obtained in Reference Example 17 in methanol (20 ml), 4.0 M hydrogen chloride. / Methanol solution (12.3 ml) was added and stirred at room temperature for 24 hours. The resulting solid was collected by filtration, washed with ethyl acetate and dried to give the title compound (3.77 g, 88%) as a solid.
- Reference Example 21 4- (2-Fluorophenyl) -2-piperidin-4-ylpyridine dihydrochloride Title compound (4.50 g, 67%) from tert-butyl 4- [4- (2-fluorophenyl) pyridin-2-yl] piperidine-1-carboxylate obtained in Reference Example 20 in the same manner as Reference Example 18 was obtained as a solid.
- Reference Example 24 4- (3-Fluorophenyl) -2-piperidin-4-ylpyridine dihydrochloride In the same manner as in Reference Example 18, from the tert-butyl 4- [4- (3-fluorophenyl) pyridin-2-yl] piperidine-1-carboxylate obtained in Reference Example 23, the title compound (3.50 g, 71% ) was obtained as a solid.
- Reference Example 26 4- (4-Fluorophenyl) -2-piperidin-4-ylpyridine dihydrochloride
- tert-butyl 4- (4-fluorophenyl) -3 ′, 6′-dihydro-2,4′-bipyridine-1 ′ (2′H) -carboxy obtained in Reference Example 25
- the tert-butyl 4- [4- (4-fluorophenyl) pyridin-2-yl] piperidine-1-carboxylate (4.80 g, quant.) was obtained as an oil from the late.
- Reference Example 29 4- (2,3-Difluorophenyl) -2-piperidin-4-ylpyridine dihydrochloride
- tert-butyl 4- (2,3-difluorophenyl) -3 ′, 6′-dihydro-2,4′-bipyridine-1 ′ (2′H) obtained in Reference Example 28
- the carboxylate gave tert-butyl 4- [4- (2,3-difluorophenyl) pyridin-2-yl] piperidine-1-carboxylate (6.50 g, 99%) as an oil.
- Reference Example 32 4- (2,4-Difluorophenyl) -2-piperidin-4-ylpyridine dihydrochloride
- tert-butyl 4- (2,4-difluorophenyl) -3 ′, 6′-dihydro-2,4′-bipyridine-1 ′ (2′H) obtained in Reference Example 31
- the carboxylate gave tert-butyl 4- [4- (2,4-difluorophenyl) pyridin-2-yl] piperidine-1-carboxylate (4.07 g, 98%) as an oil.
- Reference Example 36 4-furan-3-yl-2-piperidin-4-ylpyrimidine dihydrochloride To a solution of tert-butyl 4- (4-furan-3-ylpyrimidin-2-yl) piperidine-1-carboxylate (540 mg, 1.64 mmol) obtained in Reference Example 35 in ethyl acetate (5.4 ml) was added 4N A hydrogen chloride-ethyl acetate solution (5.4 ml) was added, and the mixture was stirred at room temperature overnight. The reaction solution was diluted with ethyl acetate, and the resulting crystals were collected by filtration to give the title compound (407 mg, 77%) as a solid.
- Reference Example 40 4-thiophen-3-yl-2-piperidin-4-ylpyrimidine dihydrochloride In the same manner as in Reference Example 36, the title compound (415 mg, 95%) was obtained as a solid from tert-butyl 4- (4-thiophen-3-ylpyrimidin-2-yl) piperidine-1-carboxylate obtained in Reference Example 39. Got as.
- Reference Example 44 4- (3-Fluorophenyl) -2-piperidin-4-ylpyrimidine dihydrochloride In the same manner as in Reference Example 36, the title compound (836 mg, 96%) was obtained from tert-butyl 4- [4- (3-fluorophenyl) pyrimidin-2-yl] piperidine-1-carboxylate obtained in Reference Example 43. Obtained as a solid.
- Reference Example 48 4- (4-Fluorophenyl) -2-piperidin-4-ylpyrimidine dihydrochloride To a solution of tert-butyl 4- [4- (4-fluorophenyl) pyrimidin-2-yl] piperidine-1-carboxylate (780 mg, 2.18 mmol) obtained in Reference Example 47 in ethyl acetate (7.8 ml). 4N hydrogen chloride-ethyl acetate solution (7.8 ml) was added, and the mixture was stirred at room temperature overnight. The reaction solution was diluted with ethyl acetate, and the resulting crystals were collected by filtration to give the title compound (686 mg, 95%) as a solid.
- Example 1 4- (4-Phenylpyrimidin-2-yl) -N-pyridin-3-ylpiperidin-1-carboxamide Triethylamine was added to a solution of 4-phenyl-2-piperidin-4-ylpyrimidine dihydrochloride (250 mg, 1.00 mmol) and 3-pyridine isocyanate (127 mg, 1.0 mmol) obtained in Reference Example 2 in tetrahydrofuran (4 ml). (210 mg, 2.1 mmol) was added and stirred at room temperature overnight. The reaction solution was diluted with ethyl acetate, washed with saturated brine, dried over sodium sulfate, and the solvent was evaporated under reduced pressure.
- Example 2 N- (3,4-dimethylisoxazol-5-yl) -4- (4-phenylpyrimidin-2-yl) piperidine-1-carboxamide 4-Phenyl-2-piperidin-4-ylpyrimidine dihydrochloride (250 mg, 1.00 mmol), (3,4-dimethylisoxazol-5-yl) carbamic acid 2,2,2 obtained in Reference Example 2 A mixture of trichloroethyl (295 mg, 1.00 mmol), diisopropylethylamine (268 mg, 2.10 mmol) in dimethyl sulfoxide (2.0 ml) was stirred at 80 ° C. overnight.
- reaction solution was diluted with ethyl acetate, washed with saturated brine, dried over sodium sulfate, and the solvent was evaporated under reduced pressure.
- Example 3 4- (4-Phenylpyrimidin-2-yl) -N-pyridazin-3-ylpiperidin-1-carboxamide 4-Phenyl-2-piperidin-4-ylpyrimidine dihydrochloride obtained in Reference Example 2 (250 mg, 1.0 mmol), pyridazin-3-ylcarbamate 2,2,2-trichloroethyl (281 mg, 1.0 Mmol), diisopropylethylamine (268 mg, 2.10 mmol) in dimethyl sulfoxide (2.0 ml) was stirred at 80 ° C. overnight.
- reaction solution was diluted with ethyl acetate, washed with saturated brine, dried over sodium sulfate, and the solvent was evaporated under reduced pressure.
- Example 4 4- [4- (2,3-Difluorophenyl) pyrimidin-2-yl] -N-pyridin-3-ylpiperidin-1-carboxamide trifluoroacetate 4- (2,3-difluorophenyl) -2-piperidin-4-ylpyrimidine dihydrochloride (250 mg, 0.900 mmol), 3-pyridine isocyanate (111 mg, 0.900 mmol) obtained in Reference Example 4 Triethylamine (184 mg, 1.80 mmol) was added to a tetrahydrofuran (4 ml) solution, and the mixture was stirred at room temperature overnight.
- reaction solution was diluted with ethyl acetate, washed with saturated brine, dried over sodium sulfate, and the solvent was evaporated under reduced pressure.
- the residue was recrystallized from hexane and methylene chloride and purified by high performance liquid chromatography (Fuji C18 HPLC column A solution 0.1% trifluoroacetic acid acetonitrile solution, solution B 0.1% trifluoroacetic acid aqueous solution) to give the title compound (300 mg, 83%).
- Example 5 4- [4- (2,3-Difluorophenyl) pyrimidin-2-yl] -N-pyridin-3-ylpiperidine-1-carboxamide 4- (2,3-Difluorophenyl) -2-piperidin-4-ylpyrimidine dihydrochloride (1.00 g, 2.87 mmol) obtained in Reference Example 4, 2,3-pyridin-3-ylcarbamic acid 2,2, Triethylamine (1.60 ml, 11.5 mmol) was added dropwise at room temperature to a suspension of 2-trichloroethyl (851 mg, 3.16 mmol) in acetone (5 ml), and the reaction solution was stirred at 65 ° C. overnight.
- Example 6 4- [4- (2,3-Difluorophenyl) pyrimidin-2-yl] -N- (3,4-dimethylisoxazol-5-yl) piperidine-1-carboxamide
- the title compound (222 mg, 59%) was obtained as a solid from 2,2,2-trichloroethyl carbamate.
- Example 7 4- [4- (2,3-Difluorophenyl) pyrimidin-2-yl] -N-pyridazin-3-ylpiperidin-1-carboxamide
- the title compound (229 mg, 63%) was obtained as a solid from trichloroethyl.
- Example 8 4- (6-Phenylpyrimidin-4-yl) -N-pyridin-3-ylpiperidin-1-carboxamide trifluoroacetate In the same manner as in Example 4, the title compound (123 mg, 54%) was obtained as a solid from 4-phenyl-6-piperidin-4-ylpyrimidine dihydrochloride obtained in Reference Example 6 and 3-pyridine isocyanate.
- Example 9 N- (3,4-Dimethylisoxazol-5-yl) -4- (6-phenylpyrimidin-4-yl) piperidine-1-carboxamide
- 4-phenyl-6-piperidin-4-ylpyrimidine dihydrochloride obtained in Reference Example 6 and (3,4-dimethylisoxazol-5-yl) carbamic acid 2,2,2
- the title compound (149 mg, 47%) was obtained as a solid from trichloroethyl.
- Example 10 4- (6-Phenylpyrimidin-4-yl) -N-pyridazin-3-ylpiperidin-1-carboxamide
- the title compound (104 mg) was obtained from 4-phenyl-6-piperidin-4-ylpyrimidine dihydrochloride obtained in Reference Example 6 and 2,2,2-trichloroethyl pyridazin-3-ylcarbamate. 34%) as a solid.
- Example 11 4- [6- (2,3-Difluorophenyl) pyrimidin-4-yl] -N-pyridin-3-ylpiperidine-1-carboxamide 4- (2,3-difluorophenyl) -6-piperidin-4-ylpyrimidine dihydrochloride (300 mg, 1.10 mmol), 3-pyridine isocyanate (135 mg, 1.1 mmol) obtained in Reference Example 8 Triethylamine (223 mg, 2.2 mmol) was added to a tetrahydrofuran (3 ml) solution, and the mixture was stirred overnight at room temperature.
- reaction solution was diluted with ethyl acetate, washed with saturated brine, dried over sodium sulfate, and the solvent was evaporated under reduced pressure. The residue was recrystallized from hexane and ethyl acetate to give the title compound (99.0 mg, 23%) as a solid.
- Example 12 4- [6- (2,3-Difluorophenyl) pyrimidin-4-yl] -N- (3,4-dimethylisoxazol-5-yl) piperidine-1-carboxamide
- the title compound (114 mg, 25%) was obtained as a solid from 2,2,2-trichloroethyl carbamate.
- Example 13 4- [6- (2,3-Difluorophenyl) pyrimidin-4-yl] -N-pyridazin-3-ylpiperidin-1-carboxamide trifluoroacetate 4- (2,3-difluorophenyl) -6-piperidin-4-ylpyrimidine dihydrochloride (300 mg, 1.10 mmol), pyridazin-3-ylcarbamic acid 2,2,2-trichloro obtained in Reference Example 8 A mixture of ethyl (295 mg, 1.10 mmol), diisopropylethylamine (281 mg, 2.20 mmol) in dimethyl sulfoxide (2.0 ml) was stirred at 80 ° C. overnight.
- reaction solution was diluted with ethyl acetate, washed with saturated brine, dried over sodium sulfate, and the solvent was evaporated under reduced pressure.
- the residue was purified by silica gel column chromatography (methylene chloride, 0.5% triethylamine added), and high performance liquid chromatography (Fuji C18 HPLC column A solution 0.1% trifluoroacetic acid acetonitrile solution, solution B 0.1% trifluoroacetic acid aqueous solution. To give the title compound (85.0 mg, 20%).
- Example 14 4- (2-Phenylpyrimidin-4-yl) -N-pyridin-3-ylpiperidine-1-carboxamide
- the title compound (334 mg, 89%) was obtained as a solid from 2-phenyl-4-piperidin-4-ylpyrimidine dihydrochloride obtained in Reference Example 11 and 3-pyridine isocyanate.
- Example 15 N- (3,4-dimethylisoxazol-5-yl) -4- (2-phenylpyrimidin-4-yl) piperidine-1-carboxamide
- 2-phenyl-4-piperidin-4-ylpyrimidine dihydrochloride obtained in Reference Example 11 and (3,4-dimethylisoxazol-5-yl) carbamic acid 2,2,2
- the title compound (200 mg, 51%) was obtained as a solid from trichloroethyl.
- Example 16 4- (2-Phenylpyrimidin-4-yl) -N-pyridazin-3-ylpiperidin-1-carboxamide
- the title compound (253 mg) was obtained from 2-phenyl-4-piperidin-4-ylpyrimidine dihydrochloride obtained in Reference Example 11 and 2,2,2-trichloroethyl pyridazin-3-ylcarbamate. 67%) as a solid.
- Example 17 4- [2- (2,3-Difluorophenyl) pyrimidin-4-yl] -N-pyridin-3-ylpiperidine-1-carboxamide
- the title compound (309 mg, 86%) was obtained from 2- (2,3-difluorophenyl) -4-piperidin-4-ylpyrimidine dihydrochloride obtained in Reference Example 15 and 3-pyridine isocyanate.
- 3-pyridine isocyanate was obtained as a solid.
- Example 18 4- [2- (2,3-Difluorophenyl) pyrimidin-4-yl] -N- (3,4-dimethylisoxazol-5-yl) piperidine-1-carboxamide trifluoroacetate
- 2- (2,3-difluorophenyl) -4-piperidin-4-ylpyrimidine dihydrochloride obtained in Reference Example 15 (250 mg, 0.90 mmol)
- reaction solution was diluted with ethyl acetate, washed with saturated brine, dried over sodium sulfate, and the solvent was evaporated under reduced pressure.
- the residue was purified by silica gel column chromatography (methylene chloride, 0.5% triethylamine added), and high performance liquid chromatography (Fuji C18 HPLC column A solution 0.1% trifluoroacetic acid acetonitrile solution, solution B 0.1% trifluoroacetic acid aqueous solution. To give the title compound (43 mg, 11%) as a solid.
- Example 19 4- [2- (2,3-Difluorophenyl) pyrimidin-4-yl] -N-pyridazin-3-ylpiperidin-1-carboxamide
- the title compound (63 mg, 17%) was obtained as an oil from trichloroethyl.
- Example 20 N- (3,4-dimethylisoxazol-5-yl) -4- (4-phenylpyridin-2-yl) piperidine-1-carboxamide Diisopropylethylamine (0.588 ml, 3.37 mmol) was added to a solution of 4-phenyl-2-piperidin-4-ylpyridine dihydrochloride (300 mg, 0.964 mmol) obtained in Reference Example 18 in dimethyl sulfoxide (3.2 ml). After stirring the reaction solution for 30 minutes, 2,3,4-trichloroethyl (3,4-dimethylisoxazol-5-yl) carbamate (333 mg, 1.16 mmol) was added at room temperature, Stir at 45 ° C.
- Example 21 N- (3,4-dimethylisoxazol-5-yl) -4- [4- (2-fluorophenyl) pyridin-2-yl] piperidine-1-carboxamide
- 4- (2-fluorophenyl) -2-piperidin-4-ylpyridine dihydrochloride obtained in Reference Example 21 and (3,4-dimethylisoxazol-5-yl) carbamic acid 2 , 2,2-Trichloroethyl gave the title compound (210 mg, 58%) as a solid.
- Example 22 N- (3,4-dimethylisoxazol-5-yl) -4- [4- (3-fluorophenyl) pyridin-2-yl] piperidine-1-carboxamide
- 4- (3-fluorophenyl) -2-piperidin-4-ylpyridine dihydrochloride obtained in Reference Example 24 and (3,4-dimethylisoxazol-5-yl) carbamic acid 2 , 2,2-Trichloroethyl gave the title compound (160 mg, 45%) as a solid.
- Example 23 N- (3,4-dimethylisoxazol-5-yl) -4- [4- (4-fluorophenyl) pyridin-2-yl] piperidine-1-carboxamide
- 4- (4-fluorophenyl) -2-piperidin-4-ylpyridine dihydrochloride obtained in Reference Example 26 and (3,4-dimethylisoxazol-5-yl) carbamic acid 2 , 2,2-Trichloroethyl gave the title compound (160 mg, 45%) as a solid.
- Example 24 4- [4- (2,3-Difluorophenyl) pyridin-2-yl] -N- (3,4-dimethylisoxazol-5-yl) piperidine-1-carboxamide
- 4- (2,3-difluorophenyl) -2-piperidin-4-ylpyridine dihydrochloride and (3,4-dimethylisoxazol-5-yl) carbamine obtained in Reference Example 29
- the title compound (110 mg, 31%) was obtained as a solid from the acid 2,2,2-trichloroethyl.
- Example 25 4- [4- (2,4-Difluorophenyl) pyridin-2-yl] -N- (3,4-dimethylisoxazol-5-yl) piperidine-1-carboxamide
- 4- (2,4-difluorophenyl) -2-piperidin-4-ylpyridine dihydrochloride and (3,4-dimethylisoxazol-5-yl) carbamine obtained in Reference Example 32
- the title compound (203 mg, 57%) was obtained as a solid from the acid 2,2,2-trichloroethyl.
- Example 26 4- (4-Furan-3-ylpyrimidin-2-yl) -N-pyridin-3-ylpiperidin-1-carboxamide 4-furan-3-yl-2-piperidin-4-ylpyrimidine dihydrochloride (120 mg, 0.397 mmol) obtained in Reference Example 36, 2,2,2-trichloroethyl pyridin-3-ylcarbamate ( Triethylamine (0.221 ml, 1.59 mmol) was added dropwise to a suspension of 126 mg, 0.437 mmol) in acetone (1 ml) at room temperature, and the reaction solution was stirred at 65 ° C. overnight.
- 2,2,2-trichloroethyl pyridin-3-ylcarbamate Triethylamine (0.221 ml, 1.59 mmol) was added dropwise to a suspension of 126 mg, 0.437 mmol) in acetone (1 ml) at room temperature, and the reaction solution was stirred at
- Example 28 4- (4-Furan-3-ylpyrimidin-2-yl) -N-pyridazin-3-ylpiperidin-1-carboxamide 4-furan-3-yl-2-piperidin-4-ylpyrimidine dihydrochloride (120 mg, 0.397 mmol), phenyl pyridazin-3-ylcarbamate (94.0 g, 0.437) obtained in Reference Example 36. To a suspension of (mmol) of acetone (1 ml), triethylamine (0.221 ml, 1.59 mmol) was added dropwise at room temperature, and the reaction solution was stirred at 45 ° C. for 3 hours.
- Example 29 N-pyridin-3-yl-4- (4-thiophen-3-ylpyrimidin-2-yl) piperidine-1-carboxamide
- 4-thiophen-3-yl-2-piperidin-4-ylpyrimidine dihydrochloride obtained in Reference Example 40 and 2,2,2-trichloroethyl pyridin-3-ylcarbamate The title compound (39.5 mg, 27%) was obtained as a solid.
- Example 30 N- (3,4-Dimethylisoxazol-5-yl) -4- (4-thiophen-3-ylpyrimidin-2-yl) piperidine-1-carboxamide
- 4-thiophen-3-yl-2-piperidin-4-ylpyrimidine dihydrochloride obtained in Reference Example 40 and (3,4-dimethylisoxazol-5-yl) carbamic acid 2 , 2,2-Trichloroethyl gave the title compound (108 mg, 69%) as a solid.
- Example 31 N-pyridazin-3-yl-4- (4-thiophen-3-ylpyrimidin-2-yl) piperidine-1-carboxamide
- the title compound (68.0 mg, 4-thiophen-3-yl-2-piperidin-4-ylpyrimidine dihydrochloride obtained in Reference Example 40 and phenyl pyridazin-3-ylcarbamate was obtained. 45%) was obtained as a solid.
- Example 32 4- [4- (3-Fluorophenyl) pyrimidin-2-yl] -N-pyridin-3-ylpiperidin-1-carboxamide
- Example 33 N- (3,4-dimethylisoxazol-5-yl) -4- [4- (3-fluorophenyl) pyrimidin-2-yl] piperidine-1-carboxamide
- the title compound (198 mg, 83%) was obtained as a solid from 4- (3-fluorophenyl) -2-piperidin-4-ylpyrimidine dihydrochloride obtained in Reference Example 44. Melting point 181-182 ° C.
- Example 34 4- [4- (3-Fluorophenyl) pyrimidin-2-yl] -N-pyridazin-3-ylpiperidin-1-carboxamide In the same manner as in Example 28, the title compound (149 mg, 65 %) As a solid. Melting point 163-164 ° C.
- Example 35 4- [4- (4-Fluorophenyl) pyrimidin-2-yl] -N-pyridin-3-ylpiperidin-1-carboxamide 4- (4-Fluorophenyl) -2-piperidin-4-ylpyrimidine dihydrochloride (200 mg, 0.606 mmol) obtained in Reference Example 48, 2,2,2-trichloroethyl pyridin-3-ylcarbamate Triethylamine (0.338 ml, 2.42 mmol) was added dropwise to a suspension of (180 mg, 0.666 mmol) in acetone (1 ml) at room temperature, and the reaction solution was stirred at 65 ° C. overnight.
- Example 36 N- (3,4-dimethylisoxazol-5-yl) -4- [4- (4-fluorophenyl) pyrimidin-2-yl] piperidine-1-carboxamide 4- (4-Fluorophenyl) -2-piperidin-4-ylpyrimidine dihydrochloride (200 mg, 0.606 mmol), (3,4-dimethylisoxazol-5-yl) carbamic acid obtained in Reference Example 48 Triethylamine (0.338 ml, 2.42 mmol) was added dropwise at room temperature to a suspension of 2,2,2-trichloroethyl (192 mg, 0.666 mmol) in acetone (1 ml), and the reaction solution was added at 45 ° C.
- Example 37 4- [4- (4-Fluorophenyl) pyrimidin-2-yl] -N-pyridazin-3-ylpiperidin-1-carboxamide 4- (4-Fluorophenyl) -2-piperidin-4-ylpyrimidine dihydrochloride (200 mg, 0.606 mmol), phenylpyridazin-3-ylcarbamate (143 mg, 0.666 mmol) obtained in Reference Example 48 ) In acetone (1 ml) was added dropwise triethylamine (0.338 ml, 2.42 mmol) at room temperature, and the reaction solution was stirred at 45 ° C. for 3 hours.
- Example 38 4- (5-Fluoro-4-phenylpyrimidin-2-yl) -N-pyridazin-3-ylpiperidin-1-carboxamide 5-fluoro-4-phenyl-2-piperidin-4-ylpyrimidine dihydrochloride (0.16 g, 0.48 mmol), phenyl pyridazin-3-ylcarbamate (0.13 g,. Triethylamine (0.20 g, 2.0 mmol) was added dropwise to a suspension of 60 mmol) in acetone (2 ml) at room temperature, and the reaction solution was stirred at 40 ° C. for 2 hours.
- Example 39 4- (5-Fluoro-4-phenylpyrimidin-2-yl) -N-pyridin-3-ylpiperidin-1-carboxamide 5-Fluoro-4-phenyl-2-piperidin-4-ylpyrimidine dihydrochloride (0.16 g, 0.48 mmol) obtained in Reference Example 51, 2,2,2-trichloroethyl pyridin-3-ylcarbamate Triethylamine (0.20 g, 2.0 mmol) was added dropwise at room temperature to a suspension of acetone (2 ml) in (0.24 g, 0.90 mmol), and the reaction solution was stirred at 40 ° C. for 2 hours.
- Example 1 (1) 10 mg of the compound of Example 1 (2) Lactose 60mg (3) Corn starch 35mg (4) Hydroxypropyl methylcellulose 3mg (5) Magnesium stearate 2mg A mixture of 10 mg of the compound obtained in Example 1, 60 mg of lactose and 35 mg of corn starch was granulated with 0.03 mL of 10 wt% aqueous hydroxypropylmethylcellulose solution (3 mg as hydroxypropylmethylcellulose), and then dried at 40 ° C. Sift through. The obtained granules are mixed with 2 mg of magnesium stearate and compressed. The resulting uncoated tablets are coated with a sugar coating with an aqueous suspension of sucrose, titanium dioxide, talc and gum arabic. The coated tablet is polished with beeswax to obtain a coated tablet.
- the cells were washed with PBS, suspended in a buffer (all of which had a final concentration of 10 mM Tris-HCl, 1 mM EDTA, 10 mM MgCl 2 ), and the cells were disrupted using a polytron homogenizer. After centrifugation at 2,000 rpm, the supernatant was collected and further centrifuged at 40,000 rpm, and the pellet was suspended in the above buffer to prepare an enzyme fraction.
- the fluorescence intensity of the plate was measured with an ARVO SX 1420 MULTILABEL COUNTER (manufactured by WALLAC) at excitation of 355 nm and emission of 460 nm.
- the inhibition rate of the test compound was calculated with the reaction containing no enzyme as the inhibition rate of 100%.
- mice Analgesic effect in mouse acetic acid rising test
- a suspension of test compound (10 mg / kg) was orally administered to mice, and 60 minutes after administration, a 0.6% acetic acid aqueous solution was intraperitoneally administered at 10 ml / kg body weight.
- Mice were housed in dedicated cages and the number of rising reactions was counted using a counter.
- the control group was tested for difference in mean value between the two groups (Student's t-test) to evaluate the analgesic effect.
- the compound represented by the formula (I) of the present invention or a salt thereof has an excellent FAAH inhibitory activity and is useful as a safe and excellent preventive / therapeutic agent for pain, depression or anxiety.
Landscapes
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- General Chemical & Material Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Immunology (AREA)
- Diabetes (AREA)
- Pain & Pain Management (AREA)
- Endocrinology (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Psychiatry (AREA)
- Rheumatology (AREA)
- Pulmonology (AREA)
- Hospice & Palliative Care (AREA)
- Physical Education & Sports Medicine (AREA)
- Oncology (AREA)
- Reproductive Health (AREA)
- Communicable Diseases (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Virology (AREA)
- Ophthalmology & Optometry (AREA)
- Psychology (AREA)
- Urology & Nephrology (AREA)
- Emergency Medicine (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
Abstract
A compound represented by formula (I) or a salt thereof. In formula (I), the symbols are as defined in the description.
Description
本発明は、FAAH阻害作用を有する新規なピペリジン誘導体に関する。
The present invention relates to a novel piperidine derivative having an FAAH inhibitory action.
痛みは患者にとって深刻であり、QOLを低下させ、社会生活を困難にすることもある疾患である。痛みはその原因によって炎症性疼痛、神経因性疼痛、侵害受容性疼痛、心因性疼痛などに分類される。炎症性疼痛は、生体外から加わる侵害性機械刺激・熱刺激・化学刺激等によって引き起こされる炎症に伴って引き起こされる疼痛である。炎症性疼痛発現には炎症局所のみならず、脊髄における炎症性サイトカインやシクロオキシゲナーゼが重要な役割を果たしていることが知られている。神経因性疼痛は末梢あるいは中枢神経系そのものの機能異常による病的な痛みである。侵害受容性疼痛は、正常な組織を傷害するか、その危険性を持つ侵害刺激が加わったために生じる痛みであり、体性痛や内臓痛に分けられる。
痛 み Pain is a disease that can be serious for patients, reduce QOL, and make social life difficult. Pain is classified according to its cause into inflammatory pain, neuropathic pain, nociceptive pain, psychogenic pain and the like. Inflammatory pain is pain caused by inflammation caused by noxious mechanical stimulation, thermal stimulation, chemical stimulation, etc. applied from outside the living body. It is known that inflammatory cytokines and cyclooxygenase play an important role not only in the local area of inflammation but also in the spinal cord for the expression of inflammatory pain. Neuropathic pain is pathological pain due to abnormal function of the peripheral or central nervous system itself. Nociceptive pain is pain caused by the addition of a noxious stimulus that injures normal tissue or has the risk of it, and is divided into somatic pain and visceral pain.
炎症性疼痛治療薬としては、インドメタシンなどのシクロオキシゲナーゼ(COX)阻害薬、セレコキシブなどのシクロオキシゲナーゼII(COX-II)阻害薬、トラマドールなどの中枢性鎮痛薬およびアセトアミノフェンなどの解熱鎮痛薬などが用いられている。しかし、シクロオキシゲナーゼ阻害薬を長期間使用すると、副作用として消化管障害が生じることがあり、問題となっている。また、シクロオキシゲナーゼII阻害薬についても胃潰瘍を起こす報告もあり、また最近になって心筋梗塞や脳梗塞など心循環器系の副作用も問題点になっている。
As therapeutic agents for inflammatory pain, cyclooxygenase (COX) inhibitors such as indomethacin, cyclooxygenase II (COX-II) inhibitors such as celecoxib, central analgesics such as tramadol, and antipyretic analgesics such as acetaminophen are used. It has been. However, when a cyclooxygenase inhibitor is used for a long time, gastrointestinal disorders may occur as a side effect, which is problematic. In addition, cyclooxygenase II inhibitors have also been reported to cause gastric ulcers, and recently, cardiovascular side effects such as myocardial infarction and cerebral infarction have become a problem.
神経因性疼痛治療薬としては、モルヒネなどのオピオイド性鎮痛薬やガバペンチンやプレガバリンなどの抗痙攣薬が使用されているが、長期使用に伴って増量を必要とする場合があることや、鎮静などの副作用が生じることが知られており、副作用がなく安全で使用できる薬剤がないのが現状である。
As neuropathic pain treatment drugs, opioid analgesics such as morphine and anticonvulsants such as gabapentin and pregabalin are used. However, there are no drugs that can be used safely and without side effects.
ところで、大麻活性成分であるΔ9-テトラヒドロカンナビノール(Δ9-THC)の受容体としてカンナビノイドレセプターが1990年頃から同定されている。CB1レセプター(非特許文献1参照)、そのスプライス変異体CB1a(非特許文献2参照)、並びにCB2レセプター(非特許文献3参照)が現在まで知られている。ほぼ同じ頃に、CB1受容体の内因性リガンドとして、N-アラキドノイルエタノールアミン(アナンダミド)が豚脳より発見された(非特許文献4参照)。アナンダミドは、N-パルミトイルエタノールアミン、N-オレオイルエタノールアミンと同様にN-アシル化エタノールアミンに属する。これらのN-アシル化エタノールアミンを含む脂肪酸アミドは、疼痛(非特許文献5および6参照)、摂食の調整(非特許文献7参照)、睡眠の促進(非特許文献8参照)等の生理作用を発揮することが明らかにされている。脂肪酸アミドの生合成、或いは分解経路は1980年頃から調べられている。先ず、カルシウム依存的にトランスアシラーゼがN-アシルフォスファチジルエタノールアミンアナンダミドを生成し(非特許文献9参照)、次いでフォスフォリパーゼDにより、脂肪酸アミドが遊離される(非特許文献10参照)。脂肪酸アミドを対応する脂肪酸に加水分解し、その生理活性を消失させる酵素活性の存在は示唆されていたが、その実体は1990年代後半まで明らかでなかった。ラットよりオレアミドを加水分解する活性成分が精製され、cDNAがクローニングされた(非特許文献11参照)。遺伝子組換えで作製された酵素はオレアミド、アナンダミドを含む種々の脂肪酸アミドを加水分解し、脂肪酸アミドヒドロラーゼ(Fatty acid amide hydrolase;以下、本明細書において「FAAH」と略すことがある。)と名づけられた。脂肪酸アミドを生合成する酵素は充分には明らかにされていない。しかし、脂肪酸アミドが、カルシウム依存的、即ち、神経活動依存的に神経細胞より産生されること(非特許文献12参照)は治療薬開発において極めて意義深い。FAAHノックアウトマウスが作出され、また、FAAH阻害薬が見出され、FAAH阻害の生理的意義が明らかにされつつある。FAAHノックアウトマウスでは、アナンダミドを始め脳内脂肪酸アミドの含量が10から15倍上昇しているが、運動能、体重、体温は正常である。しかし、疼痛反応性の低下が見られ、これは脳内脂肪酸アミド含量と相関していた(非特許文献13参照)。FAAH阻害剤としては、トリフルオロメチルケトン誘導体(非特許文献14参照)、α-ケトへテロ環誘導体(非特許文献15参照)スルフォニルフルオロライド誘導体(非特許文献16参照)フルオロフォスフォネート誘導体(非特許文献17参照)アリルカルバメート誘導体(非特許文献18参照)などが知られている。
Incidentally, a cannabinoid receptor has been identified since around 1990 as a receptor for Δ9-tetrahydrocannabinol (Δ9-THC), which is an active ingredient of cannabis. The CB1 receptor (see Non-Patent Document 1), its splice variant CB1a (see Non-Patent Document 2), and the CB2 receptor (see Non-Patent Document 3) are known to date. About the same time, N-arachidonoylethanolamine (anandamide) was discovered from pig brain as an endogenous ligand of the CB1 receptor (see Non-Patent Document 4). Anandamide belongs to N-acylated ethanolamine as well as N-palmitoylethanolamine and N-oleoylethanolamine. Fatty acid amides containing these N-acylated ethanolamines have physiological functions such as pain (see Non-Patent Documents 5 and 6), adjustment of feeding (see Non-Patent Document 7), and promotion of sleep (see Non-Patent Document 8). It has been shown that it exerts its action. The biosynthesis or degradation pathway of fatty acid amides has been investigated since about 1980. First, transacylase produces N-acylphosphatidylethanolamine anandamide in a calcium-dependent manner (see Non-Patent Document 9), and then fatty acid amide is released by phospholipase D (see Non-Patent Document 10). The presence of enzyme activity that hydrolyzes fatty acid amides to the corresponding fatty acids and eliminates their physiological activity has been suggested, but the substance was not clear until the late 1990s. An active component that hydrolyzes oleamide was purified from rats, and cDNA was cloned (see Non-Patent Document 11). The enzyme produced by gene recombination hydrolyzes various fatty acid amides including oleamide and anandamide, and is named as fatty acid amide hydrolase (hereinafter abbreviated as “FAAH” in this specification). It was. Enzymes that biosynthesize fatty acid amides have not been fully elucidated. However, that fatty acid amides are produced from nerve cells in a calcium-dependent manner, that is, in a nerve activity-dependent manner (see Non-Patent Document 12), is extremely significant in therapeutic drug development. FAAH knockout mice have been created, FAAH inhibitors have been discovered, and the physiological significance of FAAH inhibition is being elucidated. In FAAH knockout mice, the content of fatty acid amide in the brain including anandamide is increased 10 to 15 times, but the exercise capacity, body weight, and body temperature are normal. However, a decrease in pain responsiveness was observed, and this correlated with the brain fatty acid amide content (see Non-Patent Document 13). Examples of FAAH inhibitors include trifluoromethyl ketone derivatives (see Non-Patent Document 14), α-ketoheterocyclic derivatives (see Non-Patent Document 15), sulfonyl fluoride derivatives (see Non-Patent Document 16), fluorophosphonate derivatives ( Non-patent document 17) Allyl carbamate derivatives (see non-patent document 18) and the like are known.
他にもFAAHやアナンダミドがさまざまな疾患に関与していることが報告されている。我々はFAAH阻害剤が脳・神経細胞保護効果を有し、脳血管障害治療薬として有用であることを見出している。また、アルツハイマー患者の脳にFAAHが多いことが報告されている(非特許文献19参照)。アナンダミドを増やすことにより、抗パーキンソン作用を示すことがラットを用いた試験により明らかにされている(非特許文献20参照)。流産する女性でFAAHが減少することが報告されている(非特許文献21参照)。アナンダミドは、直腸ガンの増殖を抑えることが報告されている(非特許文献22参照)。FAAHノックアウトマウスは、大腸炎、結腸炎になりにくいことが報告されている(非特許文献23参照)。FAAH阻害薬が抗不安作用を示すことが報告されている(非特許文献24参照)。FAAHは、小腸に存在する満腹因子であるオレイルエタノールアミドの加水分解酵素であることが報告されている(非特許文献25参照)。FAAHは、ステアロイルエタノールアミドの加水分解酵素であり、ステアロイルエタノールアミドをネズミに投与すると摂食が抑制されることが報告されている(非特許文献26参照)。アナンダミドは、侵害受容体であるバニロイド受容体の作動薬であることから、FAAH阻害薬は、バニロイド受容体作動薬のような作用(例えば、頻尿・尿失禁予防治療作用間質性膀胱炎)も期待される(特許文献1参照)。
In addition, it has been reported that FAAH and anandamide are involved in various diseases. We have found that FAAH inhibitors have brain / nerve cell protective effects and are useful as therapeutic agents for cerebrovascular disorders. Moreover, it has been reported that there is much FAAH in the brain of an Alzheimer patient (refer nonpatent literature 19). It has been clarified by a test using rats that anti-Parkinson action is exhibited by increasing anandamide (see Non-Patent Document 20). It has been reported that FAAH decreases in miscarried women (see Non-Patent Document 21). Anandamide has been reported to suppress the growth of rectal cancer (see Non-Patent Document 22). FAAH knockout mice have been reported to be less susceptible to colitis and colitis (see Non-Patent Document 23). It has been reported that FAAH inhibitors exhibit an anxiolytic action (see Non-Patent Document 24). FAAH has been reported to be a hydrolase of oleylethanolamide, a satiety factor present in the small intestine (see Non-Patent Document 25). FAAH is a stearoylethanolamide hydrolase, and it has been reported that when stearoylethanolamide is administered to mice, feeding is suppressed (see Non-Patent Document 26). Since anandamide is an agonist of the nociceptor vanilloid receptor, the FAAH inhibitor acts like a vanilloid receptor agonist (eg, frequent urinary urinary incontinence preventive treatment interstitial cystitis) Is also expected (see Patent Document 1).
FAAHは内因性の睡眠物質であるオレアマイドを加水分解する酵素でもあり、このためFAAH阻害薬はオレアマイドの分解を抑制することにより睡眠を誘導する(特許文献2)。
特開平2002-202204号公報
US2003/0092734A
国際公開2007/020888
Nature 1990年、346巻、561頁
J.Biol.Chem.、1995年、270巻、3726頁
Eur.J.Biochem.、1995年、232巻、54頁
Science、1992年、258巻、1946頁
Nature、1998年、394巻、277頁
Pain、1998年、76巻、189頁
Nature、2001年、414巻、209頁
Science、1995年、268巻、1506頁
J.Neurochem.、1983年、41巻、1303頁
J.Neurochem.、1984年、42巻、1613頁
Nature、1996年、384巻、83頁
Nature、1994年、372巻、686頁
Proc.Natl.Acad.Sci.USA、2001年、98巻、9371頁
J.Am.Chem.Soc.、1996年、118巻、5938頁
Proc.Natl.Acad.Sci.USA、2000年、97巻、5044頁
Biochem.Biophys.Res.Commun.1997年、231巻、217頁
Biochem.Pharmacol.、1997年、53巻、255頁
Nat.Med.、2003年、9巻、76頁
The Journal of Neuroscience.、2003年、23巻、1136頁
Neuropsychophamacology.、2004年、29巻、1134頁
J Clin Endocrinol Metab.、2004年、89巻、5168頁
Gastroenterology.、2003年、125巻、677頁
J Clin Invest.、2004年、113巻、1202頁
Nature Medicine.、2003年、9巻、76頁
Nature.、2001年、414巻、209頁
FASEB Journal.、2004年、18巻、1580頁
FAAH is also an enzyme that hydrolyzes oleamide, which is an endogenous sleep substance. For this reason, FAAH inhibitors induce sleep by suppressing the degradation of oleamide (Patent Document 2).
Japanese Patent Laid-Open No. 2002-202204 US2003 / 0092734A International Publication 2007/020888 Nature 1990, 346, 561 J. et al. Biol. Chem. 1995, 270, 3726. Eur. J. et al. Biochem. 1995, 232, 54 Science, 1992, 258, 1946. Nature, 1998, 394, 277 Pain, 1998, 76, 189 Nature, 2001, 414, 209. Science, 1995, 268, 1506. J. et al. Neurochem. 1983, 41, 1303 J. et al. Neurochem. 1984, 42, 1613 Nature, 1996, 384, 83. Nature, 1994, 372, 686. Proc. Natl. Acad. Sci. USA, 2001, 98, 9371. J. et al. Am. Chem. Soc. 1996, 118, 5938. Proc. Natl. Acad. Sci. USA, 2000, 97, 5044 Biochem. Biophys. Res. Commun. 1997, Vol. 231, p. 217 Biochem. Pharmacol. 1997, 53, 255 Nat. Med. 2003, Volume 9, p. 76 The Journal of Neuroscience. 2003, 23, 1136 Neuropsychopharmacology. 2004, 29, 1134 J Clin Endocrinol Metab. 2004, 89, 5168. Gastroenterology. 2003, 125, 677 J Clin Invest. 2004, 113, 1202. Nature Medicine. 2003, Volume 9, p. 76 Nature. 2001, 414, 209 FASEB Journal. 2004, 18: 1580
現在、炎症性疼痛や神経因性疼痛として、非ステロイド抗炎症性薬(NSAID)や麻薬性鎮痛薬などが使用されているが、これらに比べて副作用のないより安全な疼痛治療薬の開発が切望されている。本発明の目的は、安全で優れた疼痛の予防・治療剤を提供することである。
Currently, non-steroidal anti-inflammatory drugs (NSAIDs) and narcotic analgesics are being used as inflammatory pain and neuropathic pain. Longed for. An object of the present invention is to provide a safe and excellent pain prevention / treatment agent.
本発明者らは、上記の目的を達成するために、下記の式(I)で表される化合物またはその塩(以下、化合物(I)と称する場合がある。)が、FAAH阻害活性を有し、種々の疼痛モデルにおいて優れた鎮痛作用を有することを見出し、本発明を完成するに至った。
すなわち、本発明は、
[1]
式(I)

[式中、
環Ar1は、置換されていてもよい芳香族複素環を示す;
A1はCR1又はNを示し、A2はCR2又はNを示し、A3はCR3又はNを示し、A4はCR4又はNを示し、かつA1、A2、A3、及びA4のうち少なくとも1つはNを示す;
環Ar2は、1個以上のハロゲン原子で置換されていてもよい芳香族炭化水素環、又は1個以上のハロゲン原子で置換されていてもよい芳香族複素環を示す;
R1、R2、R3、及びR4はそれぞれ独立して、水素原子、ハロゲン原子、又は置換されていても良いC1-6アルキル基を示す。]
で示される化合物、又はその塩;
[2]
式(I)の部分構造

が、

である上記[1]記載の化合物;
[3]
式(I)の部分構造

が、

である上記[1]記載の化合物;
[4]
環Ar1が、ピリジン環、ピリダジン環、又はC1-6アルキル基で置換されたイソオキサゾール環である、上記[1]~[3]のいずれかに記載の化合物;
[5]
環Ar2が、1個または2個のハロゲン原子で置換されていてもよいベンゼン環、フラン環、又はチオフェン環である、上記[1]~[3]のいずれかに記載の化合物;
[6]
環Ar1が、ピリジン環、ピリダジン環、又はC1-6アルキル基で置換されたイソオキサゾール環であり、
環Ar2が、1個または2個のハロゲン原子で置換されていてもよいベンゼン環、フラン環、又はチオフェン環、
である、上記[1]~[3]のいずれかに記載の化合物;
[7]
式(I)が、式(Ia)

[式中、
環Ar3は、ハロゲン原子で置換されていてもよいピリダジン環を示す;
環Ar4は、ハロゲン原子で置換されていてもよいベンゼン環を示す。]
である、上記[1]記載の化合物;
[8]
4-(4-フェニルピリミジン-2-イル)-N-ピリダジン-3-イルピペリジン-1-カルボキサミドまたはその塩;
[9]
4-[4-(2,3-ジフルオロフェニル)ピリミジン-2-イル]-N-ピリダジン-3-イルピペリジン-1-カルボキサミドまたはその塩;
[10]
4-[4-(3-フルオロフェニル)ピリミジン-2-イル]-N-ピリダジン-3-イルピペリジン-1-カルボキサミドまたはその塩;
[11]
4-[4-(4-フルオロフェニル)ピリミジン-2-イル]-N-ピリダジン-3-イルピペリジン-1-カルボキサミドまたはその塩;
[12]
上記[1]記載の化合物のプロドラッグ;
[13]
上記[1]記載の化合物、又はそのプロドラッグを含有する医薬;
[14]
上記[1]記載の化合物、又はそのプロドラッグを含有するFAAH阻害剤;
[15]
上記[1]記載の化合物若しくはそのプロドラッグを含有する、痛み、うつ若しくは不安の予防剤、又は治療剤;
[16]
上記[1]記載の化合物、又はそのプロドラッグの有効量を哺乳動物に投与することを特徴とするFAAH阻害方法;
[17]
上記[1]記載の化合物若しくはそのプロドラッグの有効量を哺乳動物に投与することを特徴とする、痛み、うつ若しくは不安の予防方法、又は治療方法;
[18]
FAAH阻害剤を製造するための、上記[1]記載の化合物、又はそのプロドラッグの使用;
[19]
痛み、うつ若しくは不安の予防剤、又は治療剤を製造するための、上記[1]記載の化合物若しくはそのプロドラッグの使用;
を提供するものである。 In order to achieve the above object, the present inventors have found that a compound represented by the following formula (I) or a salt thereof (hereinafter sometimes referred to as compound (I)) has FAAH inhibitory activity. As a result, they have found that they have excellent analgesic action in various pain models, and have completed the present invention.
That is, the present invention
[1]
Formula (I)
[Where:
Ring Ar 1 represents an optionally substituted aromatic heterocycle;
A 1 represents CR 1 or N, A 2 represents CR 2 or N, A 3 represents CR 3 or N, A 4 represents CR 4 or N, and A 1 , A 2 , A 3 , And at least one of A 4 represents N;
Ring Ar 2 represents an aromatic hydrocarbon ring that may be substituted with one or more halogen atoms, or an aromatic heterocycle that may be substituted with one or more halogen atoms;
R 1 , R 2 , R 3 , and R 4 each independently represent a hydrogen atom, a halogen atom, or an optionally substituted C 1-6 alkyl group. ]
Or a salt thereof;
[2]
Partial structure of formula (I)
But,
The compound of the above-mentioned [1], which is
[3]
Partial structure of formula (I)
But,
The compound of the above-mentioned [1], which is
[4]
The compound according to any one of the above [1] to [3], wherein the ring Ar 1 is a pyridine ring, a pyridazine ring, or an isoxazole ring substituted with a C 1-6 alkyl group;
[5]
The compound according to any one of the above [1] to [3], wherein the ring Ar 2 is a benzene ring, a furan ring, or a thiophene ring optionally substituted with one or two halogen atoms;
[6]
Ring Ar 1 is a pyridine ring, a pyridazine ring, or an isoxazole ring substituted with a C 1-6 alkyl group,
A ring Ar 2 may be substituted with one or two halogen atoms, a benzene ring, a furan ring, or a thiophene ring;
The compound according to any one of the above [1] to [3],
[7]
Formula (I) is converted to Formula (Ia)
[Where
Ring Ar 3 represents a pyridazine ring optionally substituted with a halogen atom;
Ring Ar 4 represents a benzene ring which may be substituted with a halogen atom. ]
The compound of the above-mentioned [1], which is
[8]
4- (4-phenylpyrimidin-2-yl) -N-pyridazin-3-ylpiperidin-1-carboxamide or a salt thereof;
[9]
4- [4- (2,3-difluorophenyl) pyrimidin-2-yl] -N-pyridazin-3-ylpiperidin-1-carboxamide or a salt thereof;
[10]
4- [4- (3-fluorophenyl) pyrimidin-2-yl] -N-pyridazin-3-ylpiperidin-1-carboxamide or a salt thereof;
[11]
4- [4- (4-fluorophenyl) pyrimidin-2-yl] -N-pyridazin-3-ylpiperidin-1-carboxamide or a salt thereof;
[12]
A prodrug of the compound of the above-mentioned [1];
[13]
A medicament containing the compound of the above-mentioned [1] or a prodrug thereof;
[14]
A FAAH inhibitor comprising the compound according to [1] above or a prodrug thereof;
[15]
A prophylactic or therapeutic agent for pain, depression or anxiety, comprising the compound according to [1] above or a prodrug thereof;
[16]
A method for inhibiting FAAH, comprising administering an effective amount of the compound according to [1] above or a prodrug thereof to a mammal;
[17]
A method for preventing or treating pain, depression or anxiety, which comprises administering an effective amount of the compound or prodrug thereof according to [1] to a mammal;
[18]
Use of the compound of the above-mentioned [1] or a prodrug thereof for producing a FAAH inhibitor;
[19]
Use of the compound of the above-mentioned [1] or a prodrug thereof for producing a prophylactic or therapeutic agent for pain, depression or anxiety;
Is to provide.
すなわち、本発明は、
[1]
式(I)
環Ar1は、置換されていてもよい芳香族複素環を示す;
A1はCR1又はNを示し、A2はCR2又はNを示し、A3はCR3又はNを示し、A4はCR4又はNを示し、かつA1、A2、A3、及びA4のうち少なくとも1つはNを示す;
環Ar2は、1個以上のハロゲン原子で置換されていてもよい芳香族炭化水素環、又は1個以上のハロゲン原子で置換されていてもよい芳香族複素環を示す;
R1、R2、R3、及びR4はそれぞれ独立して、水素原子、ハロゲン原子、又は置換されていても良いC1-6アルキル基を示す。]
で示される化合物、又はその塩;
[2]
式(I)の部分構造
[3]
式(I)の部分構造
[4]
環Ar1が、ピリジン環、ピリダジン環、又はC1-6アルキル基で置換されたイソオキサゾール環である、上記[1]~[3]のいずれかに記載の化合物;
[5]
環Ar2が、1個または2個のハロゲン原子で置換されていてもよいベンゼン環、フラン環、又はチオフェン環である、上記[1]~[3]のいずれかに記載の化合物;
[6]
環Ar1が、ピリジン環、ピリダジン環、又はC1-6アルキル基で置換されたイソオキサゾール環であり、
環Ar2が、1個または2個のハロゲン原子で置換されていてもよいベンゼン環、フラン環、又はチオフェン環、
である、上記[1]~[3]のいずれかに記載の化合物;
[7]
式(I)が、式(Ia)
環Ar3は、ハロゲン原子で置換されていてもよいピリダジン環を示す;
環Ar4は、ハロゲン原子で置換されていてもよいベンゼン環を示す。]
である、上記[1]記載の化合物;
[8]
4-(4-フェニルピリミジン-2-イル)-N-ピリダジン-3-イルピペリジン-1-カルボキサミドまたはその塩;
[9]
4-[4-(2,3-ジフルオロフェニル)ピリミジン-2-イル]-N-ピリダジン-3-イルピペリジン-1-カルボキサミドまたはその塩;
[10]
4-[4-(3-フルオロフェニル)ピリミジン-2-イル]-N-ピリダジン-3-イルピペリジン-1-カルボキサミドまたはその塩;
[11]
4-[4-(4-フルオロフェニル)ピリミジン-2-イル]-N-ピリダジン-3-イルピペリジン-1-カルボキサミドまたはその塩;
[12]
上記[1]記載の化合物のプロドラッグ;
[13]
上記[1]記載の化合物、又はそのプロドラッグを含有する医薬;
[14]
上記[1]記載の化合物、又はそのプロドラッグを含有するFAAH阻害剤;
[15]
上記[1]記載の化合物若しくはそのプロドラッグを含有する、痛み、うつ若しくは不安の予防剤、又は治療剤;
[16]
上記[1]記載の化合物、又はそのプロドラッグの有効量を哺乳動物に投与することを特徴とするFAAH阻害方法;
[17]
上記[1]記載の化合物若しくはそのプロドラッグの有効量を哺乳動物に投与することを特徴とする、痛み、うつ若しくは不安の予防方法、又は治療方法;
[18]
FAAH阻害剤を製造するための、上記[1]記載の化合物、又はそのプロドラッグの使用;
[19]
痛み、うつ若しくは不安の予防剤、又は治療剤を製造するための、上記[1]記載の化合物若しくはそのプロドラッグの使用;
を提供するものである。 In order to achieve the above object, the present inventors have found that a compound represented by the following formula (I) or a salt thereof (hereinafter sometimes referred to as compound (I)) has FAAH inhibitory activity. As a result, they have found that they have excellent analgesic action in various pain models, and have completed the present invention.
That is, the present invention
[1]
Formula (I)
Ring Ar 1 represents an optionally substituted aromatic heterocycle;
A 1 represents CR 1 or N, A 2 represents CR 2 or N, A 3 represents CR 3 or N, A 4 represents CR 4 or N, and A 1 , A 2 , A 3 , And at least one of A 4 represents N;
Ring Ar 2 represents an aromatic hydrocarbon ring that may be substituted with one or more halogen atoms, or an aromatic heterocycle that may be substituted with one or more halogen atoms;
R 1 , R 2 , R 3 , and R 4 each independently represent a hydrogen atom, a halogen atom, or an optionally substituted C 1-6 alkyl group. ]
Or a salt thereof;
[2]
Partial structure of formula (I)
[3]
Partial structure of formula (I)
[4]
The compound according to any one of the above [1] to [3], wherein the ring Ar 1 is a pyridine ring, a pyridazine ring, or an isoxazole ring substituted with a C 1-6 alkyl group;
[5]
The compound according to any one of the above [1] to [3], wherein the ring Ar 2 is a benzene ring, a furan ring, or a thiophene ring optionally substituted with one or two halogen atoms;
[6]
Ring Ar 1 is a pyridine ring, a pyridazine ring, or an isoxazole ring substituted with a C 1-6 alkyl group,
A ring Ar 2 may be substituted with one or two halogen atoms, a benzene ring, a furan ring, or a thiophene ring;
The compound according to any one of the above [1] to [3],
[7]
Formula (I) is converted to Formula (Ia)
Ring Ar 3 represents a pyridazine ring optionally substituted with a halogen atom;
Ring Ar 4 represents a benzene ring which may be substituted with a halogen atom. ]
The compound of the above-mentioned [1], which is
[8]
4- (4-phenylpyrimidin-2-yl) -N-pyridazin-3-ylpiperidin-1-carboxamide or a salt thereof;
[9]
4- [4- (2,3-difluorophenyl) pyrimidin-2-yl] -N-pyridazin-3-ylpiperidin-1-carboxamide or a salt thereof;
[10]
4- [4- (3-fluorophenyl) pyrimidin-2-yl] -N-pyridazin-3-ylpiperidin-1-carboxamide or a salt thereof;
[11]
4- [4- (4-fluorophenyl) pyrimidin-2-yl] -N-pyridazin-3-ylpiperidin-1-carboxamide or a salt thereof;
[12]
A prodrug of the compound of the above-mentioned [1];
[13]
A medicament containing the compound of the above-mentioned [1] or a prodrug thereof;
[14]
A FAAH inhibitor comprising the compound according to [1] above or a prodrug thereof;
[15]
A prophylactic or therapeutic agent for pain, depression or anxiety, comprising the compound according to [1] above or a prodrug thereof;
[16]
A method for inhibiting FAAH, comprising administering an effective amount of the compound according to [1] above or a prodrug thereof to a mammal;
[17]
A method for preventing or treating pain, depression or anxiety, which comprises administering an effective amount of the compound or prodrug thereof according to [1] to a mammal;
[18]
Use of the compound of the above-mentioned [1] or a prodrug thereof for producing a FAAH inhibitor;
[19]
Use of the compound of the above-mentioned [1] or a prodrug thereof for producing a prophylactic or therapeutic agent for pain, depression or anxiety;
Is to provide.
本発明の式(I)で示される化合物でまたはその塩は、優れたFAAH阻害活性を有しており、安全で優れた痛み、うつまたは不安などの予防・治療剤として有用である。
The compound represented by the formula (I) of the present invention or a salt thereof has an excellent FAAH inhibitory activity and is useful as a safe and excellent preventive / therapeutic agent for pain, depression or anxiety.
本明細書において、「FAAH阻害活性を有する」とは、「脂肪酸アミドヒドロラーゼの活性を直接的または間接的に低下させる活性を有する」ことを意味する。
本明細書において、「ハロゲン(原子)」としては、例えば、フッ素(原子)、塩素(原子)、臭素(原子)、ヨウ素(原子)が挙げられる。
本明細書において、「C1-6アルキル基」としては、例えば、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、sec-ブチル、tert-ブチル、ペンチル、イソペンチル、ネオペンチル、ヘキシル等が挙げられる。 As used herein, “having FAAH inhibitory activity” means “having an activity that directly or indirectly decreases the activity of fatty acid amide hydrolase”.
In the present specification, examples of the “halogen (atom)” include fluorine (atom), chlorine (atom), bromine (atom), and iodine (atom).
In the present specification, examples of the “C 1-6 alkyl group” include methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl, tert-butyl, pentyl, isopentyl, neopentyl, hexyl and the like.
本明細書において、「ハロゲン(原子)」としては、例えば、フッ素(原子)、塩素(原子)、臭素(原子)、ヨウ素(原子)が挙げられる。
本明細書において、「C1-6アルキル基」としては、例えば、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、sec-ブチル、tert-ブチル、ペンチル、イソペンチル、ネオペンチル、ヘキシル等が挙げられる。 As used herein, “having FAAH inhibitory activity” means “having an activity that directly or indirectly decreases the activity of fatty acid amide hydrolase”.
In the present specification, examples of the “halogen (atom)” include fluorine (atom), chlorine (atom), bromine (atom), and iodine (atom).
In the present specification, examples of the “C 1-6 alkyl group” include methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl, tert-butyl, pentyl, isopentyl, neopentyl, hexyl and the like.
上記式(I)において、環Ar1は、置換されていてもよい芳香族複素環を示す。
環Ar1で示される「芳香族複素環」としては、例えば、炭素原子以外に窒素原子、硫黄原子および酸素原子から選ばれる1または2種、1~4個のヘテロ原子を含む5~14員、好ましくは5~10員、より好ましくは5または6員の芳香族複素環が挙げられる。具体的には、例えば、チエニル(例、2-チエニル、3-チエニル)、フリル(例、2-フリル、3-フリル)、ピリジル(例、2-ピリジル、3-ピリジル、4-ピリジル)、チアゾリル(例、2-チアゾリル、4-チアゾリル、5-チアゾリル)、オキサゾリル(例、2-オキサゾリル、4-オキサゾリル)、ピラジニル、ピリミジニル(例、2-ピリミジニル、4-ピリミジニル)、ピロリル(例、1-ピロリル、2-ピロリル、3-ピロリル)、イミダゾリル(例、1-イミダゾリル、2-イミダゾリル、4-イミダゾリル)、ピラゾリル(例、1-ピラゾリル、3-ピラゾリル、4-ピラゾリル)、ピリダジニル(例、3-ピリダジニル、4-ピリダジニル)、イソチアゾリル(例、3-イソチアゾリル、4-イソチアゾリル、5-イソチアゾリル)、およびイソオキサゾリル(例、3-イソオキサゾリル、4-イソオキサゾリル、5-イソオキサゾリル)が挙げられる。
当該芳香族複素環は、好ましくは、5または6員の芳香族複素環であり、より好ましくは、ピリジル、ピリダジニル、またはイソオキサゾリルであり、さらに好ましくは、ピリダジルである。
当該芳香族複素環は、置換可能な位置に、1~5個、好ましくは1または2個の置換基を有していてもよい。また、無置換の芳香族複素環も好ましい。
かかる置換基としては、例えばハロゲン原子(例えば、フッ素、塩素、臭素、ヨウ素等)、ハロゲン化、ヒドロキシ化、またはオキソ化されていてもよい低級アルキル基(例えば、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、sec-ブチル、tert-ブチル、ペンチル、ヘキシル等のC1-6アルキル基等;フルオロメチル、クロロメチル、ジフルオロメチル、ジクロロメチル、トリフルオロメチル、トリクロロメチル等のハロゲン化されていてもよいC1-6アルキル基;ヒドロキシメチル、ヒドロキシエチル等のヒドロキシ化されていてもよいC1-6アルキル基、2-オキソプロピル、2-オキソブチル基などのオキソ化されていてもよいC1-6アルキル基)、シクロアルキル基(例えば、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル等のC3-6シクロアルキル基等)、低級アルキニル基(例えば、エチニル、1-プロピニル、プロパルギル等のC2-6アルキニル基等)、低級アルケニル基(例えば、ビニル、アリル、イソプロペニル、ブテニル、イソブテニル等のC2-6アルケニル基等)、アラルキル基(例えばベンジル、α-メチルベンジル、フェネチル等のC7-11アラルキル基等)、アリール基(例えば、フェニル、ナフチル等のC6-10アリール基等、好ましくはフェニル基等)、アリールオキシ基(例えば、C6-10アリールオキシ基等)、低級アルカノイル基(例えば、ホルミル;アセチル、プロピオニル、ブチリル、イソブチリル等のC1-6アルキル-カルボニル基等)、アリールカルボニル(例えば、ベンゾイル基、ナフトイル基等のC6-10アリール-カルボニル基等)、低級アルカノイルオキシ基(例えば、ホルミルオキシ;アセチルオキシ、プロピオニルオキシ、ブチリルオキシ、イソブチリルオキシ等のC1-6アルキル-カルボニルオキシ基等)、アリールカルボニルオキシ基(例えば、ベンゾイルオキシ、ナフトイルオキシ等のC6-10アリール-カルボニルオキシ基等)、低級アルコキシカルボニル基(例えば、メトキシカルボニル、エトキシカルボニル、プロポキシカルボニル、イソプロポキシカルボニル、ブトキシカルボニル、イソブトキシカルボニル、tert-ブトキシカルボニル等のC1-6アルコキシ-カルボニル基等)、アラルキルオキシカルボニル(例えば、ベンジルオキシカルボニル等のC7-11アラルキルオキシカルボニル基等)、カルバモイル基、モノ-、ジ-またはトリ-ハロゲノ-低級アルキル基(例えば、クロロメチル、ジクロロメチル、トリフルオロメチル、2,2,2-トリフルオロエチル等のモノ-、ジ-またはトリ-ハロゲノ-C1-4アルキル基等)、オキソ基、アミジノ基、イミノ基、アミノ基、モノ-低級アルキルアミノ基(例えば、メチルアミノ、エチルアミノ、プロピルアミノ、イソプロピルアミノ、ブチルアミノ等のモノ-C1-4アルキルアミノ基等)、ジ-低級アルキルアミノ基(例えば、ジメチルアミノ、ジエチルアミノ、ジプロピルアミノ、ジイソプロピルアミノ、ジブチルアミノ、メチルエチルアミノ等のジ-C1-4アルキルアミノ基等)、低級アルキル-低級アルキルカルボニルアミノ基(例えば、N-メチルアセチル基等)、ハロゲン化されていてもよい低級アルキルカルボニルアミノ基(例えば、アセチルアミノ基、トリフルオロアセチルアミノ基等)、炭素原子と1個の窒素原子以外に酸素原子、硫黄原子および窒素原子から選ばれたヘテロ原子を1ないし3個含んでいてもよい3ないし6員の環状アミノ基(例えば、アジリジニル、アゼチジニル、ピロリジニル、ピロリニル、ピロリル、イミダゾリル、ピラゾリル、イミダゾリジニル、ピペリジル、モルホリニル、ジヒドロピリジル、ピリジル、N-メチルピペラジニル、N-エチルピペラジニル等の3ないし6員の環状アミノ基等)、アルキレンジオキシ基(例えば、メチレンジオキシ、エチレンジオキシ等のC1-3アルキレンジオキシ基等)、ヒドロキシ基、ニトロ基、シアノ基、メルカプト基、スルホ基、スルフィノ基、ホスホノ基、スルファモイル基、モノアルキルスルファモイル基(例えば、N-メチルスルファモイル、N-エチルスルファモイル、N-プロピルスルファモイル、N-イソプロピルスルファモイル、N-ブチルスルファモイル等のモノ-C1-6アルキルスルファモイル基等)、ジアルキルスルファモイル基(例えば、N,N-ジメチルスルファモイル、N,N-ジエチルスルファモイル、N,N-ジプロピルスルファモイル、N,N-ジブチルスルファモイル等のジ-C1-6アルキルスルファモイル基等)、アルキルチオ基(例えば、メチルチオ、エチルチオ、プロピルチオ、イソプロピルチオ、ブチルチオ、sec-ブチルチオ、tert-ブチルチオ等のC1-6アルキルチオ基等)、アリールチオ基(例えば、フェニルチオ、ナフチルチオ等のC6-10アリールチオ基等)、低級アルキルスルフィニル基(例えば、メチルスルフィニル、エチルスルフィニル、プロピルスルフィニル、ブチルスルフィニル等のC1-6アルキルスルフィニル基等)、アリールスルフィニル基(例えば、フェニルスルフィニル、ナフチルスルフィニル等のC6-10アリールスルフィニル基等)、低級アルキルスルホニル基(例えば、メチルスルホニル、エチルスルホニル、プロピルスルホニル、ブチルスルホニル等のC1-6アルキルスルホニル基等)、アリールスルホニル基(例えば、フェニルスルホニル、ナフチルスルホニル等のC6-10アリールスルホニル基等)等が挙げられる。
当該置換基は、好ましくはC1-6アルキル基であり、さらに好ましくはメチルである。 In the above formula (I), the ring Ar 1 represents an optionally substituted aromatic heterocycle.
Examples of the “aromatic heterocycle” represented by the ring Ar 1 include, for example, 5 to 14 members containing 1 or 2 heteroatoms selected from a nitrogen atom, a sulfur atom and an oxygen atom in addition to a carbon atom. Preferably, it is a 5- to 10-membered, more preferably 5- or 6-membered aromatic heterocycle. Specifically, for example, thienyl (eg, 2-thienyl, 3-thienyl), furyl (eg, 2-furyl, 3-furyl), pyridyl (eg, 2-pyridyl, 3-pyridyl, 4-pyridyl), Thiazolyl (eg, 2-thiazolyl, 4-thiazolyl, 5-thiazolyl), oxazolyl (eg, 2-oxazolyl, 4-oxazolyl), pyrazinyl, pyrimidinyl (eg, 2-pyrimidinyl, 4-pyrimidinyl), pyrrolyl (eg, 1 -Pyrrolyl, 2-pyrrolyl, 3-pyrrolyl), imidazolyl (eg, 1-imidazolyl, 2-imidazolyl, 4-imidazolyl), pyrazolyl (eg, 1-pyrazolyl, 3-pyrazolyl, 4-pyrazolyl), pyridazinyl (eg, 3-pyridazinyl, 4-pyridazinyl), isothiazolyl (eg, 3-isothiazolyl, 4-isothiazolyl, 5 Isothiazolyl), and isoxazolyl (e.g., 3-isoxazolyl, 4-isoxazolyl, 5-isoxazolyl) can be mentioned.
The aromatic heterocycle is preferably a 5- or 6-membered aromatic heterocycle, more preferably pyridyl, pyridazinyl, or isoxazolyl, and still more preferably pyridazyl.
The aromatic heterocycle may have 1 to 5, preferably 1 or 2, substituents at substitutable positions. An unsubstituted aromatic heterocyclic ring is also preferable.
Examples of the substituent include a halogen atom (for example, fluorine, chlorine, bromine, iodine, etc.), a lower alkyl group (for example, methyl, ethyl, propyl, isopropyl, etc.) which may be halogenated, hydroxylated, or oxoated. C 1-6 alkyl groups such as butyl, isobutyl, sec-butyl, tert-butyl, pentyl, hexyl, etc .; halogenated such as fluoromethyl, chloromethyl, difluoromethyl, dichloromethyl, trifluoromethyl, trichloromethyl, etc. also a C 1-6 alkyl group; hydroxymethyl, hydroxylated which may be C 1-6 alkyl groups such as hydroxyethyl, 2-oxopropyl, 2-oxobutyl may be oxo of such groups C 1 -6 alkyl group), cycloalkyl group (for example, cyclopropyl Le, cyclobutyl, cyclopentyl, and the like C 3-6 cycloalkyl groups such as cyclohexyl), a lower alkynyl group (e.g., ethynyl, 1-propynyl, etc. C 2-6 alkynyl group propargyl, etc.), lower alkenyl groups (e.g., vinyl C 2-6 alkenyl groups such as allyl, isopropenyl, butenyl, isobutenyl, etc.), aralkyl groups (eg, C 7-11 aralkyl groups such as benzyl, α-methylbenzyl, phenethyl, etc.), aryl groups (eg, phenyl, C 6-10 aryl group such as naphthyl, etc., preferably phenyl group), aryloxy group (eg C 6-10 aryloxy group etc.), lower alkanoyl group (eg formyl; acetyl, propionyl, butyryl, isobutyryl etc.) the C 1-6 alkyl - carbonyl group), Ariruka Boniru (e.g., benzoyl group, C 6-10 aryl such as naphthoyl - such carbonyl group), a lower alkanoyloxy group (e.g., formyloxy; acetyloxy, propionyloxy, butyryloxy, etc. isobutyryloxy C 1-6 Alkyl-carbonyloxy group etc.), arylcarbonyloxy group (eg C 6-10 aryl-carbonyloxy group such as benzoyloxy, naphthoyloxy etc.), lower alkoxycarbonyl group (eg methoxycarbonyl, ethoxycarbonyl, propoxycarbonyl etc.) , isopropoxycarbonyl, butoxycarbonyl, isobutoxycarbonyl, C 1-6 alkoxy, such as tert- butoxycarbonyl - such carbonyl group), an aralkyloxycarbonyl (e.g., benzyloxy carbonitrile C 7-11 aralkyloxycarbonyl group, etc.), a carbamoyl group such as Le, mono -, di- - or tri - halogeno - lower alkyl group (e.g., chloromethyl, dichloromethyl, trifluoromethyl, 2,2,2- Mono-, di- or tri-halogeno-C 1-4 alkyl groups such as fluoroethyl), oxo groups, amidino groups, imino groups, amino groups, mono-lower alkylamino groups (eg, methylamino, ethylamino, Mono-C 1-4 alkylamino groups such as propylamino, isopropylamino, butylamino, etc.), di-lower alkylamino groups (eg, dimethylamino, diethylamino, dipropylamino, diisopropylamino, dibutylamino, methylethylamino, etc.) di -C 1-4 alkylamino group, etc.), lower alkyl - lower alkyl Other than carbonylamino group (for example, N-methylacetyl group), optionally halogenated lower alkylcarbonylamino group (for example, acetylamino group, trifluoroacetylamino group, etc.), carbon atom and one nitrogen atom 3 to 6-membered cyclic amino group which may contain 1 to 3 heteroatoms selected from oxygen atom, sulfur atom and nitrogen atom (for example, aziridinyl, azetidinyl, pyrrolidinyl, pyrrolinyl, pyrrolyl, imidazolyl, pyrazolyl, Imidazolidinyl, piperidyl, morpholinyl, dihydropyridyl, pyridyl, 3- to 6-membered cyclic amino groups such as N-methylpiperazinyl and N-ethylpiperazinyl), alkylenedioxy groups (eg methylenedioxy, ethylenedi) C 1-3 alkylenedioxy groups oxy such like , Hydroxy group, nitro group, cyano group, mercapto group, sulfo group, sulfino group, phosphono group, sulfamoyl group, monoalkylsulfamoyl group (for example, N-methylsulfamoyl, N-ethylsulfamoyl, N- Mono-C 1-6 alkylsulfamoyl groups such as propylsulfamoyl, N-isopropylsulfamoyl, N-butylsulfamoyl, etc.), dialkylsulfamoyl groups (eg, N, N-dimethylsulfamoyl) N, N-diethylsulfamoyl, N, N-dipropylsulfamoyl, di-C 1-6 alkylsulfamoyl groups such as N, N-dibutylsulfamoyl), alkylthio groups (for example, methylthio, Ethylthio, propylthio, isopropylthio, butylthio, sec-butylthio, tert-butyl C 1-6 alkylthio groups such as thio), arylthio groups (eg, C 6-10 arylthio groups such as phenylthio, naphthylthio, etc.), lower alkylsulfinyl groups (eg, methylsulfinyl, ethylsulfinyl, propylsulfinyl, butylsulfinyl, etc.) C 1-6 alkylsulfinyl group, etc.), arylsulfinyl groups (eg, C 6-10 arylsulfinyl groups such as phenylsulfinyl, naphthylsulfinyl, etc.), lower alkylsulfonyl groups (eg, methylsulfonyl, ethylsulfonyl, propylsulfonyl, etc.) C 1-6 alkylsulfonyl groups such as butylsulfonyl), arylsulfonyl groups (for example, C 6-10 arylsulfonyl groups such as phenylsulfonyl, naphthylsulfonyl, etc.) and the like.
The substituent is preferably a C 1-6 alkyl group, and more preferably methyl.
環Ar1で示される「芳香族複素環」としては、例えば、炭素原子以外に窒素原子、硫黄原子および酸素原子から選ばれる1または2種、1~4個のヘテロ原子を含む5~14員、好ましくは5~10員、より好ましくは5または6員の芳香族複素環が挙げられる。具体的には、例えば、チエニル(例、2-チエニル、3-チエニル)、フリル(例、2-フリル、3-フリル)、ピリジル(例、2-ピリジル、3-ピリジル、4-ピリジル)、チアゾリル(例、2-チアゾリル、4-チアゾリル、5-チアゾリル)、オキサゾリル(例、2-オキサゾリル、4-オキサゾリル)、ピラジニル、ピリミジニル(例、2-ピリミジニル、4-ピリミジニル)、ピロリル(例、1-ピロリル、2-ピロリル、3-ピロリル)、イミダゾリル(例、1-イミダゾリル、2-イミダゾリル、4-イミダゾリル)、ピラゾリル(例、1-ピラゾリル、3-ピラゾリル、4-ピラゾリル)、ピリダジニル(例、3-ピリダジニル、4-ピリダジニル)、イソチアゾリル(例、3-イソチアゾリル、4-イソチアゾリル、5-イソチアゾリル)、およびイソオキサゾリル(例、3-イソオキサゾリル、4-イソオキサゾリル、5-イソオキサゾリル)が挙げられる。
当該芳香族複素環は、好ましくは、5または6員の芳香族複素環であり、より好ましくは、ピリジル、ピリダジニル、またはイソオキサゾリルであり、さらに好ましくは、ピリダジルである。
当該芳香族複素環は、置換可能な位置に、1~5個、好ましくは1または2個の置換基を有していてもよい。また、無置換の芳香族複素環も好ましい。
かかる置換基としては、例えばハロゲン原子(例えば、フッ素、塩素、臭素、ヨウ素等)、ハロゲン化、ヒドロキシ化、またはオキソ化されていてもよい低級アルキル基(例えば、メチル、エチル、プロピル、イソプロピル、ブチル、イソブチル、sec-ブチル、tert-ブチル、ペンチル、ヘキシル等のC1-6アルキル基等;フルオロメチル、クロロメチル、ジフルオロメチル、ジクロロメチル、トリフルオロメチル、トリクロロメチル等のハロゲン化されていてもよいC1-6アルキル基;ヒドロキシメチル、ヒドロキシエチル等のヒドロキシ化されていてもよいC1-6アルキル基、2-オキソプロピル、2-オキソブチル基などのオキソ化されていてもよいC1-6アルキル基)、シクロアルキル基(例えば、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル等のC3-6シクロアルキル基等)、低級アルキニル基(例えば、エチニル、1-プロピニル、プロパルギル等のC2-6アルキニル基等)、低級アルケニル基(例えば、ビニル、アリル、イソプロペニル、ブテニル、イソブテニル等のC2-6アルケニル基等)、アラルキル基(例えばベンジル、α-メチルベンジル、フェネチル等のC7-11アラルキル基等)、アリール基(例えば、フェニル、ナフチル等のC6-10アリール基等、好ましくはフェニル基等)、アリールオキシ基(例えば、C6-10アリールオキシ基等)、低級アルカノイル基(例えば、ホルミル;アセチル、プロピオニル、ブチリル、イソブチリル等のC1-6アルキル-カルボニル基等)、アリールカルボニル(例えば、ベンゾイル基、ナフトイル基等のC6-10アリール-カルボニル基等)、低級アルカノイルオキシ基(例えば、ホルミルオキシ;アセチルオキシ、プロピオニルオキシ、ブチリルオキシ、イソブチリルオキシ等のC1-6アルキル-カルボニルオキシ基等)、アリールカルボニルオキシ基(例えば、ベンゾイルオキシ、ナフトイルオキシ等のC6-10アリール-カルボニルオキシ基等)、低級アルコキシカルボニル基(例えば、メトキシカルボニル、エトキシカルボニル、プロポキシカルボニル、イソプロポキシカルボニル、ブトキシカルボニル、イソブトキシカルボニル、tert-ブトキシカルボニル等のC1-6アルコキシ-カルボニル基等)、アラルキルオキシカルボニル(例えば、ベンジルオキシカルボニル等のC7-11アラルキルオキシカルボニル基等)、カルバモイル基、モノ-、ジ-またはトリ-ハロゲノ-低級アルキル基(例えば、クロロメチル、ジクロロメチル、トリフルオロメチル、2,2,2-トリフルオロエチル等のモノ-、ジ-またはトリ-ハロゲノ-C1-4アルキル基等)、オキソ基、アミジノ基、イミノ基、アミノ基、モノ-低級アルキルアミノ基(例えば、メチルアミノ、エチルアミノ、プロピルアミノ、イソプロピルアミノ、ブチルアミノ等のモノ-C1-4アルキルアミノ基等)、ジ-低級アルキルアミノ基(例えば、ジメチルアミノ、ジエチルアミノ、ジプロピルアミノ、ジイソプロピルアミノ、ジブチルアミノ、メチルエチルアミノ等のジ-C1-4アルキルアミノ基等)、低級アルキル-低級アルキルカルボニルアミノ基(例えば、N-メチルアセチル基等)、ハロゲン化されていてもよい低級アルキルカルボニルアミノ基(例えば、アセチルアミノ基、トリフルオロアセチルアミノ基等)、炭素原子と1個の窒素原子以外に酸素原子、硫黄原子および窒素原子から選ばれたヘテロ原子を1ないし3個含んでいてもよい3ないし6員の環状アミノ基(例えば、アジリジニル、アゼチジニル、ピロリジニル、ピロリニル、ピロリル、イミダゾリル、ピラゾリル、イミダゾリジニル、ピペリジル、モルホリニル、ジヒドロピリジル、ピリジル、N-メチルピペラジニル、N-エチルピペラジニル等の3ないし6員の環状アミノ基等)、アルキレンジオキシ基(例えば、メチレンジオキシ、エチレンジオキシ等のC1-3アルキレンジオキシ基等)、ヒドロキシ基、ニトロ基、シアノ基、メルカプト基、スルホ基、スルフィノ基、ホスホノ基、スルファモイル基、モノアルキルスルファモイル基(例えば、N-メチルスルファモイル、N-エチルスルファモイル、N-プロピルスルファモイル、N-イソプロピルスルファモイル、N-ブチルスルファモイル等のモノ-C1-6アルキルスルファモイル基等)、ジアルキルスルファモイル基(例えば、N,N-ジメチルスルファモイル、N,N-ジエチルスルファモイル、N,N-ジプロピルスルファモイル、N,N-ジブチルスルファモイル等のジ-C1-6アルキルスルファモイル基等)、アルキルチオ基(例えば、メチルチオ、エチルチオ、プロピルチオ、イソプロピルチオ、ブチルチオ、sec-ブチルチオ、tert-ブチルチオ等のC1-6アルキルチオ基等)、アリールチオ基(例えば、フェニルチオ、ナフチルチオ等のC6-10アリールチオ基等)、低級アルキルスルフィニル基(例えば、メチルスルフィニル、エチルスルフィニル、プロピルスルフィニル、ブチルスルフィニル等のC1-6アルキルスルフィニル基等)、アリールスルフィニル基(例えば、フェニルスルフィニル、ナフチルスルフィニル等のC6-10アリールスルフィニル基等)、低級アルキルスルホニル基(例えば、メチルスルホニル、エチルスルホニル、プロピルスルホニル、ブチルスルホニル等のC1-6アルキルスルホニル基等)、アリールスルホニル基(例えば、フェニルスルホニル、ナフチルスルホニル等のC6-10アリールスルホニル基等)等が挙げられる。
当該置換基は、好ましくはC1-6アルキル基であり、さらに好ましくはメチルである。 In the above formula (I), the ring Ar 1 represents an optionally substituted aromatic heterocycle.
Examples of the “aromatic heterocycle” represented by the ring Ar 1 include, for example, 5 to 14 members containing 1 or 2 heteroatoms selected from a nitrogen atom, a sulfur atom and an oxygen atom in addition to a carbon atom. Preferably, it is a 5- to 10-membered, more preferably 5- or 6-membered aromatic heterocycle. Specifically, for example, thienyl (eg, 2-thienyl, 3-thienyl), furyl (eg, 2-furyl, 3-furyl), pyridyl (eg, 2-pyridyl, 3-pyridyl, 4-pyridyl), Thiazolyl (eg, 2-thiazolyl, 4-thiazolyl, 5-thiazolyl), oxazolyl (eg, 2-oxazolyl, 4-oxazolyl), pyrazinyl, pyrimidinyl (eg, 2-pyrimidinyl, 4-pyrimidinyl), pyrrolyl (eg, 1 -Pyrrolyl, 2-pyrrolyl, 3-pyrrolyl), imidazolyl (eg, 1-imidazolyl, 2-imidazolyl, 4-imidazolyl), pyrazolyl (eg, 1-pyrazolyl, 3-pyrazolyl, 4-pyrazolyl), pyridazinyl (eg, 3-pyridazinyl, 4-pyridazinyl), isothiazolyl (eg, 3-isothiazolyl, 4-isothiazolyl, 5 Isothiazolyl), and isoxazolyl (e.g., 3-isoxazolyl, 4-isoxazolyl, 5-isoxazolyl) can be mentioned.
The aromatic heterocycle is preferably a 5- or 6-membered aromatic heterocycle, more preferably pyridyl, pyridazinyl, or isoxazolyl, and still more preferably pyridazyl.
The aromatic heterocycle may have 1 to 5, preferably 1 or 2, substituents at substitutable positions. An unsubstituted aromatic heterocyclic ring is also preferable.
Examples of the substituent include a halogen atom (for example, fluorine, chlorine, bromine, iodine, etc.), a lower alkyl group (for example, methyl, ethyl, propyl, isopropyl, etc.) which may be halogenated, hydroxylated, or oxoated. C 1-6 alkyl groups such as butyl, isobutyl, sec-butyl, tert-butyl, pentyl, hexyl, etc .; halogenated such as fluoromethyl, chloromethyl, difluoromethyl, dichloromethyl, trifluoromethyl, trichloromethyl, etc. also a C 1-6 alkyl group; hydroxymethyl, hydroxylated which may be C 1-6 alkyl groups such as hydroxyethyl, 2-oxopropyl, 2-oxobutyl may be oxo of such groups C 1 -6 alkyl group), cycloalkyl group (for example, cyclopropyl Le, cyclobutyl, cyclopentyl, and the like C 3-6 cycloalkyl groups such as cyclohexyl), a lower alkynyl group (e.g., ethynyl, 1-propynyl, etc. C 2-6 alkynyl group propargyl, etc.), lower alkenyl groups (e.g., vinyl C 2-6 alkenyl groups such as allyl, isopropenyl, butenyl, isobutenyl, etc.), aralkyl groups (eg, C 7-11 aralkyl groups such as benzyl, α-methylbenzyl, phenethyl, etc.), aryl groups (eg, phenyl, C 6-10 aryl group such as naphthyl, etc., preferably phenyl group), aryloxy group (eg C 6-10 aryloxy group etc.), lower alkanoyl group (eg formyl; acetyl, propionyl, butyryl, isobutyryl etc.) the C 1-6 alkyl - carbonyl group), Ariruka Boniru (e.g., benzoyl group, C 6-10 aryl such as naphthoyl - such carbonyl group), a lower alkanoyloxy group (e.g., formyloxy; acetyloxy, propionyloxy, butyryloxy, etc. isobutyryloxy C 1-6 Alkyl-carbonyloxy group etc.), arylcarbonyloxy group (eg C 6-10 aryl-carbonyloxy group such as benzoyloxy, naphthoyloxy etc.), lower alkoxycarbonyl group (eg methoxycarbonyl, ethoxycarbonyl, propoxycarbonyl etc.) , isopropoxycarbonyl, butoxycarbonyl, isobutoxycarbonyl, C 1-6 alkoxy, such as tert- butoxycarbonyl - such carbonyl group), an aralkyloxycarbonyl (e.g., benzyloxy carbonitrile C 7-11 aralkyloxycarbonyl group, etc.), a carbamoyl group such as Le, mono -, di- - or tri - halogeno - lower alkyl group (e.g., chloromethyl, dichloromethyl, trifluoromethyl, 2,2,2- Mono-, di- or tri-halogeno-C 1-4 alkyl groups such as fluoroethyl), oxo groups, amidino groups, imino groups, amino groups, mono-lower alkylamino groups (eg, methylamino, ethylamino, Mono-C 1-4 alkylamino groups such as propylamino, isopropylamino, butylamino, etc.), di-lower alkylamino groups (eg, dimethylamino, diethylamino, dipropylamino, diisopropylamino, dibutylamino, methylethylamino, etc.) di -C 1-4 alkylamino group, etc.), lower alkyl - lower alkyl Other than carbonylamino group (for example, N-methylacetyl group), optionally halogenated lower alkylcarbonylamino group (for example, acetylamino group, trifluoroacetylamino group, etc.), carbon atom and one nitrogen atom 3 to 6-membered cyclic amino group which may contain 1 to 3 heteroatoms selected from oxygen atom, sulfur atom and nitrogen atom (for example, aziridinyl, azetidinyl, pyrrolidinyl, pyrrolinyl, pyrrolyl, imidazolyl, pyrazolyl, Imidazolidinyl, piperidyl, morpholinyl, dihydropyridyl, pyridyl, 3- to 6-membered cyclic amino groups such as N-methylpiperazinyl and N-ethylpiperazinyl), alkylenedioxy groups (eg methylenedioxy, ethylenedi) C 1-3 alkylenedioxy groups oxy such like , Hydroxy group, nitro group, cyano group, mercapto group, sulfo group, sulfino group, phosphono group, sulfamoyl group, monoalkylsulfamoyl group (for example, N-methylsulfamoyl, N-ethylsulfamoyl, N- Mono-C 1-6 alkylsulfamoyl groups such as propylsulfamoyl, N-isopropylsulfamoyl, N-butylsulfamoyl, etc.), dialkylsulfamoyl groups (eg, N, N-dimethylsulfamoyl) N, N-diethylsulfamoyl, N, N-dipropylsulfamoyl, di-C 1-6 alkylsulfamoyl groups such as N, N-dibutylsulfamoyl), alkylthio groups (for example, methylthio, Ethylthio, propylthio, isopropylthio, butylthio, sec-butylthio, tert-butyl C 1-6 alkylthio groups such as thio), arylthio groups (eg, C 6-10 arylthio groups such as phenylthio, naphthylthio, etc.), lower alkylsulfinyl groups (eg, methylsulfinyl, ethylsulfinyl, propylsulfinyl, butylsulfinyl, etc.) C 1-6 alkylsulfinyl group, etc.), arylsulfinyl groups (eg, C 6-10 arylsulfinyl groups such as phenylsulfinyl, naphthylsulfinyl, etc.), lower alkylsulfonyl groups (eg, methylsulfonyl, ethylsulfonyl, propylsulfonyl, etc.) C 1-6 alkylsulfonyl groups such as butylsulfonyl), arylsulfonyl groups (for example, C 6-10 arylsulfonyl groups such as phenylsulfonyl, naphthylsulfonyl, etc.) and the like.
The substituent is preferably a C 1-6 alkyl group, and more preferably methyl.
上記式(I)において、環Ar2は、1個以上のハロゲン原子で置換されていてもよい芳香族炭化水素環、または1個以上のハロゲン原子で置換されていてもよい芳香族複素環を示す。
環Ar2で示される「芳香族炭化水素環」としては、例えば、フェニル、2-ビフェニリル、3-ビフェニリル、4-ビフェニリル、およびシクロオクタテトラエニルなどのC6-14アリール基が挙げられる。
当該芳香族炭化水素環は、好ましくはフェニルである。
当該芳香族炭化水素環は、置換可能な位置に、1~5個、好ましくは1または2個のハロゲン原子、好ましくはフッ素原子を有していてもよい。また、無置換の芳香族炭化水素環も好ましい。
環Ar2で示される「芳香族複素環」としては、上記環Ar1に記載の「芳香族複素環」と同様のものが挙げられる。
当該芳香族複素環は、好ましくは、フリルまたはチエニルである。
当該芳香族複素環は、置換可能な位置に、1~5個、好ましくは1または2個のハロゲン原子、好ましくはフッ素原子を有していてもよい。また、無置換の芳香族複素環も好ましい。 In the above formula (I), the ring Ar 2 represents an aromatic hydrocarbon ring that may be substituted with one or more halogen atoms, or an aromatic heterocyclic ring that may be substituted with one or more halogen atoms. Show.
Examples of the “aromatic hydrocarbon ring” represented by the ring Ar 2 include C 6-14 aryl groups such as phenyl, 2-biphenylyl, 3-biphenylyl, 4-biphenylyl, and cyclooctatetraenyl.
The aromatic hydrocarbon ring is preferably phenyl.
The aromatic hydrocarbon ring may have 1 to 5, preferably 1 or 2, halogen atoms, preferably fluorine atoms, at substitutable positions. An unsubstituted aromatic hydrocarbon ring is also preferred.
Examples of the “aromatic heterocycle” represented by the ring Ar 2 include the same “aromatic heterocycle” as described in the ring Ar 1 .
The aromatic heterocycle is preferably furyl or thienyl.
The aromatic heterocyclic ring may have 1 to 5, preferably 1 or 2, halogen atoms, preferably fluorine atoms, at substitutable positions. An unsubstituted aromatic heterocyclic ring is also preferable.
環Ar2で示される「芳香族炭化水素環」としては、例えば、フェニル、2-ビフェニリル、3-ビフェニリル、4-ビフェニリル、およびシクロオクタテトラエニルなどのC6-14アリール基が挙げられる。
当該芳香族炭化水素環は、好ましくはフェニルである。
当該芳香族炭化水素環は、置換可能な位置に、1~5個、好ましくは1または2個のハロゲン原子、好ましくはフッ素原子を有していてもよい。また、無置換の芳香族炭化水素環も好ましい。
環Ar2で示される「芳香族複素環」としては、上記環Ar1に記載の「芳香族複素環」と同様のものが挙げられる。
当該芳香族複素環は、好ましくは、フリルまたはチエニルである。
当該芳香族複素環は、置換可能な位置に、1~5個、好ましくは1または2個のハロゲン原子、好ましくはフッ素原子を有していてもよい。また、無置換の芳香族複素環も好ましい。 In the above formula (I), the ring Ar 2 represents an aromatic hydrocarbon ring that may be substituted with one or more halogen atoms, or an aromatic heterocyclic ring that may be substituted with one or more halogen atoms. Show.
Examples of the “aromatic hydrocarbon ring” represented by the ring Ar 2 include C 6-14 aryl groups such as phenyl, 2-biphenylyl, 3-biphenylyl, 4-biphenylyl, and cyclooctatetraenyl.
The aromatic hydrocarbon ring is preferably phenyl.
The aromatic hydrocarbon ring may have 1 to 5, preferably 1 or 2, halogen atoms, preferably fluorine atoms, at substitutable positions. An unsubstituted aromatic hydrocarbon ring is also preferred.
Examples of the “aromatic heterocycle” represented by the ring Ar 2 include the same “aromatic heterocycle” as described in the ring Ar 1 .
The aromatic heterocycle is preferably furyl or thienyl.
The aromatic heterocyclic ring may have 1 to 5, preferably 1 or 2, halogen atoms, preferably fluorine atoms, at substitutable positions. An unsubstituted aromatic heterocyclic ring is also preferable.
上記式(I)において、A1はCR1またはNを示し、A2はCR2またはNを示し、A3はCR3またはNを示し、A4はCR4またはNを示し、かつA1、A2、A3、およびA4のうち少なくとも1つはNを示す。
好ましくは、A1、A2、A3、およびA4のうち1~2個がNであり、より好ましくは2個がNである。
R1、R2、R3、およびR4はそれぞれ独立して、水素原子、ハロゲン原子、または置換されていてもよいC1-6アルキル基を示す。
かかる置換されていてもよいC1-6アルキル基としては、ハロゲン化またはオキソ化されていてもよいC1-6アルキル(例えば、メチル、クロロメチル、ジフルオロメチル、トリクロロメチル、トリフルオロメチル、エチル、2-ブロモエチル、2,2,2-トリフルオロエチル、ペンタフルオロエチル、プロピル、3,3,3-トリフルオロプロピル、イソプロピル、ブチル、イソブチル、sec-ブチル、tert-ブチル、4,4,4-トリフルオロブチル、ペンチル、イソペンチル、ネオペンチル、5,5,5-トリフルオロペンチル、ヘキシル、6,6,6-トリフルオロヘキシル、2-オキソプロピル、2-オキソブチル等)が挙げられる。
当該R1、R2、R3、およびR4は、好ましくは、水素原子またはハロゲン原子(特にフッ素原子)、さらに好ましくは水素原子である。 In the above formula (I), A 1 represents CR 1 or N, A 2 represents CR 2 or N, A 3 represents CR 3 or N, A 4 represents CR 4 or N, and A 1 , A 2 , A 3 , and A 4 represent N.
Preferably, 1 to 2 of A 1 , A 2 , A 3 , and A 4 are N, more preferably 2 are N.
R 1 , R 2 , R 3 , and R 4 each independently represent a hydrogen atom, a halogen atom, or an optionally substituted C 1-6 alkyl group.
Such optionally substituted C 1-6 alkyl groups include optionally halogenated or oxoated C 1-6 alkyl (eg, methyl, chloromethyl, difluoromethyl, trichloromethyl, trifluoromethyl, ethyl). 2-bromoethyl, 2,2,2-trifluoroethyl, pentafluoroethyl, propyl, 3,3,3-trifluoropropyl, isopropyl, butyl, isobutyl, sec-butyl, tert-butyl, 4,4,4 -Trifluorobutyl, pentyl, isopentyl, neopentyl, 5,5,5-trifluoropentyl, hexyl, 6,6,6-trifluorohexyl, 2-oxopropyl, 2-oxobutyl and the like.
R 1 , R 2 , R 3 and R 4 are preferably a hydrogen atom or a halogen atom (particularly a fluorine atom), more preferably a hydrogen atom.
好ましくは、A1、A2、A3、およびA4のうち1~2個がNであり、より好ましくは2個がNである。
R1、R2、R3、およびR4はそれぞれ独立して、水素原子、ハロゲン原子、または置換されていてもよいC1-6アルキル基を示す。
かかる置換されていてもよいC1-6アルキル基としては、ハロゲン化またはオキソ化されていてもよいC1-6アルキル(例えば、メチル、クロロメチル、ジフルオロメチル、トリクロロメチル、トリフルオロメチル、エチル、2-ブロモエチル、2,2,2-トリフルオロエチル、ペンタフルオロエチル、プロピル、3,3,3-トリフルオロプロピル、イソプロピル、ブチル、イソブチル、sec-ブチル、tert-ブチル、4,4,4-トリフルオロブチル、ペンチル、イソペンチル、ネオペンチル、5,5,5-トリフルオロペンチル、ヘキシル、6,6,6-トリフルオロヘキシル、2-オキソプロピル、2-オキソブチル等)が挙げられる。
当該R1、R2、R3、およびR4は、好ましくは、水素原子またはハロゲン原子(特にフッ素原子)、さらに好ましくは水素原子である。 In the above formula (I), A 1 represents CR 1 or N, A 2 represents CR 2 or N, A 3 represents CR 3 or N, A 4 represents CR 4 or N, and A 1 , A 2 , A 3 , and A 4 represent N.
Preferably, 1 to 2 of A 1 , A 2 , A 3 , and A 4 are N, more preferably 2 are N.
R 1 , R 2 , R 3 , and R 4 each independently represent a hydrogen atom, a halogen atom, or an optionally substituted C 1-6 alkyl group.
Such optionally substituted C 1-6 alkyl groups include optionally halogenated or oxoated C 1-6 alkyl (eg, methyl, chloromethyl, difluoromethyl, trichloromethyl, trifluoromethyl, ethyl). 2-bromoethyl, 2,2,2-trifluoroethyl, pentafluoroethyl, propyl, 3,3,3-trifluoropropyl, isopropyl, butyl, isobutyl, sec-butyl, tert-butyl, 4,4,4 -Trifluorobutyl, pentyl, isopentyl, neopentyl, 5,5,5-trifluoropentyl, hexyl, 6,6,6-trifluorohexyl, 2-oxopropyl, 2-oxobutyl and the like.
R 1 , R 2 , R 3 and R 4 are preferably a hydrogen atom or a halogen atom (particularly a fluorine atom), more preferably a hydrogen atom.
すなわち、好ましくは、式(I)の部分構造

は、ハロゲン原子(好ましくは、フッ素原子)により置換されていてもよい

であり、より好ましくは

であり、さらに好ましくは

である。
That is, preferably the partial structure of formula (I)
May be substituted by a halogen atom (preferably a fluorine atom)
And more preferably
And more preferably
It is.
好ましくは、式(I)で表される化合物は、
環Ar1が、ピリジン環、ピリダジン環、又はC1-6アルキル基で置換されたイソオキサゾール環であり、
環Ar2が、1個または2個のハロゲン原子で置換されていてもよいベンゼン環、フラン環、又はチオフェン環、
である化合物である。 Preferably, the compound represented by formula (I) is:
Ring Ar 1 is a pyridine ring, a pyridazine ring, or an isoxazole ring substituted with a C 1-6 alkyl group,
A ring Ar 2 may be substituted with one or two halogen atoms, a benzene ring, a furan ring, or a thiophene ring;
Is a compound.
環Ar1が、ピリジン環、ピリダジン環、又はC1-6アルキル基で置換されたイソオキサゾール環であり、
環Ar2が、1個または2個のハロゲン原子で置換されていてもよいベンゼン環、フラン環、又はチオフェン環、
である化合物である。 Preferably, the compound represented by formula (I) is:
Ring Ar 1 is a pyridine ring, a pyridazine ring, or an isoxazole ring substituted with a C 1-6 alkyl group,
A ring Ar 2 may be substituted with one or two halogen atoms, a benzene ring, a furan ring, or a thiophene ring;
Is a compound.
より好ましくは、式(I)で表される化合物は、式(Ia)

[式中、
環Ar3は、ハロゲン原子で置換されていてもよいピリダジン環を示す;
環Ar4は、ハロゲン原子で置換されていてもよいベンゼン環を示す。]
で表される化合物である。 More preferably, the compound of formula (I) is of formula (Ia)
[Where
Ring Ar 3 represents a pyridazine ring optionally substituted with a halogen atom;
Ring Ar 4 represents a benzene ring which may be substituted with a halogen atom. ]
It is a compound represented by these.
環Ar3は、ハロゲン原子で置換されていてもよいピリダジン環を示す;
環Ar4は、ハロゲン原子で置換されていてもよいベンゼン環を示す。]
で表される化合物である。 More preferably, the compound of formula (I) is of formula (Ia)
Ring Ar 3 represents a pyridazine ring optionally substituted with a halogen atom;
Ring Ar 4 represents a benzene ring which may be substituted with a halogen atom. ]
It is a compound represented by these.
上記式(Ia)において、環Ar3は、置換可能な位置に、1~3個のハロゲン原子を有していてもよい。好ましくは、環Ar3は、無置換である。ここで、「無置換」とは、式(Ia)中の環Ar3上の置換可能な位置に置換基を有さないことを意味する。
上記式(Ia)において、環Ar4は、置換可能な位置に、1~5個、好ましくは1または2個のハロゲン原子を有していてもよい。また、無置換の芳香族複素環も好ましい。ここで「無置換」とは、式(Ia)中の環Ar4上の置換可能な位置に置換基を有さないことを意味する。
当該ハロゲン原子としては、例えば、フッ素、塩素、臭素、またはヨウ素が挙げられ、好ましくは、当該ハロゲン原子はフッ素である。
好ましくは、式(Ia)で表される化合物は、環Ar3が無置換であり、環Ar4がハロゲン原子を有していてもよいベンゼン環である化合物である。 In the above formula (Ia), the ring Ar 3 may have 1 to 3 halogen atoms at substitutable positions. Preferably, ring Ar 3 is unsubstituted. Here, “unsubstituted” means having no substituent at a substitutable position on the ring Ar 3 in formula (Ia).
In the above formula (Ia), the ring Ar 4 may have 1 to 5, preferably 1 or 2, halogen atoms at substitutable positions. An unsubstituted aromatic heterocyclic ring is also preferable. Here, “unsubstituted” means having no substituent at a substitutable position on the ring Ar 4 in formula (Ia).
Examples of the halogen atom include fluorine, chlorine, bromine, and iodine. Preferably, the halogen atom is fluorine.
Preferably, the compound represented by the formula (Ia) is a compound in which the ring Ar 3 is unsubstituted and the ring Ar 4 is a benzene ring optionally having a halogen atom.
上記式(Ia)において、環Ar4は、置換可能な位置に、1~5個、好ましくは1または2個のハロゲン原子を有していてもよい。また、無置換の芳香族複素環も好ましい。ここで「無置換」とは、式(Ia)中の環Ar4上の置換可能な位置に置換基を有さないことを意味する。
当該ハロゲン原子としては、例えば、フッ素、塩素、臭素、またはヨウ素が挙げられ、好ましくは、当該ハロゲン原子はフッ素である。
好ましくは、式(Ia)で表される化合物は、環Ar3が無置換であり、環Ar4がハロゲン原子を有していてもよいベンゼン環である化合物である。 In the above formula (Ia), the ring Ar 3 may have 1 to 3 halogen atoms at substitutable positions. Preferably, ring Ar 3 is unsubstituted. Here, “unsubstituted” means having no substituent at a substitutable position on the ring Ar 3 in formula (Ia).
In the above formula (Ia), the ring Ar 4 may have 1 to 5, preferably 1 or 2, halogen atoms at substitutable positions. An unsubstituted aromatic heterocyclic ring is also preferable. Here, “unsubstituted” means having no substituent at a substitutable position on the ring Ar 4 in formula (Ia).
Examples of the halogen atom include fluorine, chlorine, bromine, and iodine. Preferably, the halogen atom is fluorine.
Preferably, the compound represented by the formula (Ia) is a compound in which the ring Ar 3 is unsubstituted and the ring Ar 4 is a benzene ring optionally having a halogen atom.
さらに好ましくは、式(I)で表される化合物は:
4-(4-フェニルピリミジン-2-イル)-N-ピリダジン-3-イルピペリジン-1-カルボキサミド;
4-[4-(2,3-ジフルオロフェニル)ピリミジン-2-イル]-N-ピリダジン-3-イルピペリジン-1-カルボキサミド;
4-[4-(3-フルオロフェニル)ピリミジン-2-イル]-N-ピリダジン-3-イルピペリジン-1-カルボキサミド;または
4-[4-(4-フルオロフェニル)ピリミジン-2-イル]-N-ピリダジン-3-イルピペリジン-1-カルボキサミド
である。 More preferably, the compound of formula (I) is:
4- (4-phenylpyrimidin-2-yl) -N-pyridazin-3-ylpiperidin-1-carboxamide;
4- [4- (2,3-difluorophenyl) pyrimidin-2-yl] -N-pyridazin-3-ylpiperidin-1-carboxamide;
4- [4- (3-Fluorophenyl) pyrimidin-2-yl] -N-pyridazin-3-ylpiperidin-1-carboxamide; or 4- [4- (4-fluorophenyl) pyrimidin-2-yl]- N-pyridazin-3-ylpiperidine-1-carboxamide.
4-(4-フェニルピリミジン-2-イル)-N-ピリダジン-3-イルピペリジン-1-カルボキサミド;
4-[4-(2,3-ジフルオロフェニル)ピリミジン-2-イル]-N-ピリダジン-3-イルピペリジン-1-カルボキサミド;
4-[4-(3-フルオロフェニル)ピリミジン-2-イル]-N-ピリダジン-3-イルピペリジン-1-カルボキサミド;または
4-[4-(4-フルオロフェニル)ピリミジン-2-イル]-N-ピリダジン-3-イルピペリジン-1-カルボキサミド
である。 More preferably, the compound of formula (I) is:
4- (4-phenylpyrimidin-2-yl) -N-pyridazin-3-ylpiperidin-1-carboxamide;
4- [4- (2,3-difluorophenyl) pyrimidin-2-yl] -N-pyridazin-3-ylpiperidin-1-carboxamide;
4- [4- (3-Fluorophenyl) pyrimidin-2-yl] -N-pyridazin-3-ylpiperidin-1-carboxamide; or 4- [4- (4-fluorophenyl) pyrimidin-2-yl]- N-pyridazin-3-ylpiperidine-1-carboxamide.
式(I)で表される化合物の塩としては、薬理学的に許容される塩が好ましく、例えば、無機塩基との塩、有機塩基との塩、無機酸との塩、有機酸との塩、塩基性または酸性アミノ酸との塩等が挙げられる。
The salt of the compound represented by the formula (I) is preferably a pharmacologically acceptable salt, for example, a salt with an inorganic base, a salt with an organic base, a salt with an inorganic acid, a salt with an organic acid. And salts with basic or acidic amino acids.
無機塩基との塩の好適な例としては、ナトリウム塩、カリウム塩等のアルカリ金属塩;カルシウム塩、マグネシウム塩等のアルカリ土類金属塩;アルミニウム塩、アンモニウム塩等が挙げられる。
Preferable examples of the salt with an inorganic base include alkali metal salts such as sodium salt and potassium salt; alkaline earth metal salts such as calcium salt and magnesium salt; aluminum salt and ammonium salt.
有機塩基との塩の好適な例としては、トリメチルアミン、トリエチルアミン、ピリジン、ピコリン、エタノールアミン、ジエタノールアミン、トリエタノールアミン、トロメタミン[トリス(ヒドロキシメチル)メチルアミン]、tert-ブチルアミン、シクロヘキシルアミン、ベンジルアミン、ジシクロヘキシルアミン、N,N-ジベンジルエチレンジアミン等との塩が挙げられる。
Preferable examples of the salt with an organic base include trimethylamine, triethylamine, pyridine, picoline, ethanolamine, diethanolamine, triethanolamine, tromethamine [tris (hydroxymethyl) methylamine], tert-butylamine, cyclohexylamine, benzylamine, And salts with dicyclohexylamine, N, N-dibenzylethylenediamine and the like.
無機酸との塩の好適な例としては、塩酸、臭化水素酸、硝酸、硫酸、リン酸等との塩が挙げられる。
Preferable examples of the salt with inorganic acid include salts with hydrochloric acid, hydrobromic acid, nitric acid, sulfuric acid, phosphoric acid and the like.
有機酸との塩の好適な例としては、ギ酸、酢酸、トリフルオロ酢酸、フタル酸、フマル酸、シュウ酸、酒石酸、マレイン酸、クエン酸、コハク酸、リンゴ酸、メタンスルホン酸、ベンゼンスルホン酸、p-トルエンスルホン酸等との塩が挙げられる。特にトリフルオロ酢酸との塩が好ましい。
Preferable examples of salts with organic acids include formic acid, acetic acid, trifluoroacetic acid, phthalic acid, fumaric acid, oxalic acid, tartaric acid, maleic acid, citric acid, succinic acid, malic acid, methanesulfonic acid, and benzenesulfonic acid And salts with p-toluenesulfonic acid and the like. Particularly preferred is a salt with trifluoroacetic acid.
塩基性アミノ酸との塩の好適な例としては、アルギニン、リジン、オルニチン等との塩が挙げられる。
酸性アミノ酸との塩の好適な例としては、アスパラギン酸、グルタミン酸等との塩が挙げられる。 Preferable examples of the salt with basic amino acid include salts with arginine, lysine, ornithine and the like.
Preferable examples of the salt with acidic amino acid include salts with aspartic acid, glutamic acid and the like.
酸性アミノ酸との塩の好適な例としては、アスパラギン酸、グルタミン酸等との塩が挙げられる。 Preferable examples of the salt with basic amino acid include salts with arginine, lysine, ornithine and the like.
Preferable examples of the salt with acidic amino acid include salts with aspartic acid, glutamic acid and the like.
化合物(I)のプロドラッグは、生体内における生理条件下で酵素や胃酸等による反応により化合物(I)に変換する化合物、すなわち酵素的に酸化、還元、加水分解等を起こして化合物(I)に変化する化合物、胃酸等により加水分解等を起こして化合物(I)に変化する化合物をいう。化合物(I)のプロドラッグとしては、化合物(I)のアミノ基がアシル化、アルキル化またはリン酸化された化合物(例、化合物(I)のアミノ基がエイコサノイル化、アラニル化、ペンチルアミノカルボニル化、(5-メチル-2-オキソ-1,3-ジオキソレン-4-イル)メトキシカルボニル化、テトラヒドロフラニル化、テトラヒドロピラニル化、ピロリジルメチル化、ピバロイルオキシメチル化またはtert-ブチル化された化合物);化合物(I)のヒドロキシ基がアシル化、アルキル化、リン酸化またはほう酸化された化合物(例、化合物(I)のヒドロキシ基がアセチル化、パルミトイル化、プロパノイル化、ピバロイル化、サクシニル化、フマリル化、アラニル化、ジメチルアミノメチルカルボニル化またはテトラヒドロピラニル化された化合物);化合物(I)のカルボキシ基がエステル化またはアミド化された化合物(例、化合物(I)のカルボキシ基がエチルエステル化、フェニルエステル化、カルボキシメチルエステル化、ジメチルアミノメチルエステル化、ピバロイルオキシメチルエステル化、エトキシカルボニルオキシエチルエステル化、フタリジルエステル化、(5-メチル-2-オキソ-1,3-ジオキソレン-4-イル)メチルエステル化、シクロヘキシルオキシカルボニルエチルエステル化またはメチルアミド化された化合物);等が挙げられる。これらの化合物は自体公知の方法によって化合物(I)から製造することができる。
A prodrug of compound (I) is a compound that is converted to compound (I) by a reaction with an enzyme, gastric acid, or the like under physiological conditions in vivo, that is, compound (I) that is enzymatically oxidized, reduced, hydrolyzed, etc. A compound that changes to compound (I) by hydrolysis or the like due to gastric acid or the like. Compound (I) prodrugs include compounds in which the amino group of compound (I) is acylated, alkylated or phosphorylated (eg, the amino group of compound (I) is eicosanoylated, alanylated, pentylaminocarbonylated) , (5-methyl-2-oxo-1,3-dioxolen-4-yl) methoxycarbonylation, tetrahydrofuranylation, tetrahydropyranylation, pyrrolidylmethylation, pivaloyloxymethylation or tert-butylation Compound); a compound wherein the hydroxy group of compound (I) is acylated, alkylated, phosphorylated or borated (eg, hydroxy group of compound (I) is acetylated, palmitoylated, propanoylated, pivaloylated, succinyl , Fumarylation, alanylation, dimethylaminomethylcarbonylation or tetrahydation Compound obtained by esterifying or amidating the carboxy group of compound (I) (eg, carboxy group of compound (I) is ethyl esterified, phenyl esterified, carboxymethyl esterified, dimethylaminomethyl) Esterification, pivaloyloxymethyl esterification, ethoxycarbonyloxyethyl esterification, phthalidyl esterification, (5-methyl-2-oxo-1,3-dioxolen-4-yl) methyl esterification, cyclohexyloxycarbonylethyl Esterified or methylamidated compounds) and the like. These compounds can be produced from compound (I) by a method known per se.
また、化合物(I)のプロドラッグは、広川書店1990年刊、「医薬品の開発」、第7巻、分子設計、163頁から198頁に記載されているような、生理的条件で化合物(I)に変化するものであってもよい。
以下、本発明化合物の製造法について説明する。なお、原料化合物として用いられる化合物は、それぞれ塩として用いてもよい。このような塩としては、前記した化合物(I)の塩として例示したものなどが用いられる。 In addition, prodrugs of Compound (I) can be obtained under the physiological conditions as described in Hirokawa Shoten 1990, “Development of Pharmaceuticals”, Volume 7, Molecular Design, pages 163 to 198. It may change to.
Hereafter, the manufacturing method of this invention compound is demonstrated. In addition, you may use the compound used as a raw material compound as a salt, respectively. As such a salt, those exemplified as the salt of the aforementioned compound (I) can be used.
以下、本発明化合物の製造法について説明する。なお、原料化合物として用いられる化合物は、それぞれ塩として用いてもよい。このような塩としては、前記した化合物(I)の塩として例示したものなどが用いられる。 In addition, prodrugs of Compound (I) can be obtained under the physiological conditions as described in Hirokawa Shoten 1990, “Development of Pharmaceuticals”, Volume 7, Molecular Design, pages 163 to 198. It may change to.
Hereafter, the manufacturing method of this invention compound is demonstrated. In addition, you may use the compound used as a raw material compound as a salt, respectively. As such a salt, those exemplified as the salt of the aforementioned compound (I) can be used.
[製造方法1]
本発明の化合物(I)は、例えば、次のスキーム:

〔式中、L1は脱離基を表し、その他の記号は上記と同意義である。〕
で表される製造方法1、または、これに準ずる方法によって製造することができる。 [Production Method 1]
Compound (I) of the present invention can be synthesized, for example, by the following scheme:
[Wherein L 1 represents a leaving group, and other symbols are as defined above. ]
It can manufacture by the manufacturing method 1 represented by this, or the method according to this.
本発明の化合物(I)は、例えば、次のスキーム:
で表される製造方法1、または、これに準ずる方法によって製造することができる。 [Production Method 1]
Compound (I) of the present invention can be synthesized, for example, by the following scheme:
It can manufacture by the manufacturing method 1 represented by this, or the method according to this.
製造方法1に従って、まず、化合物(II)と化合物(III)とをカップリング反応に付し、化合物(IV)を製造する。
脱離基L1としてはハライド、例えばクロライド、ブロマイド、アイオダイド、またアルキルスルホニルオキシ基、例えばメタンスルホニルオキシ基、トリフルオロメタンスルホニルオキシ基などが挙げられる。
カップリング反応は、塩基および触媒の存在下、反応に影響を及ぼさない溶媒で行われる。化合物(III)の使用量は、化合物(II)1モルに対し約0.5~約10モル、好ましくは約0.9~約3モルである。
塩基としては、炭酸ナトリウム、炭酸カリウム、炭酸セシウム、炭酸水素ナトリウム、りん酸三カリウム等の塩基性塩類、ピリジン、ルチジン等の芳香族アミン類、トリエチルアミン、トリプロピルアミン、トリブチルアミン、シクロヘキシルジメチルアミン、4-ジメチルアミノピリジン、N,N-ジメチルアニリン、N-メチルピペリジン、N-メチルピロリジン、N-メチルモルホリン等の第3級アミン類、ナトリウムメトキシド、ナトリウムエトキシド、ナトリウム第三ブトキシド、カリウム第三ブトキシド等の金属アルコキシド類などが用いられる。
塩基の使用量は、それぞれ化合物(II)に対して約0.5~約10モル、好ましくは、約1~約5モル当量である。
本反応で用いられるは触媒としては、酢酸パラジウム、塩化パラジウム、テトラキス(トリフェニルホスフィン)パラジウム、ビス(ジベンジリデンアセトン)パラジウム、トリス(ジベンジリデンアセトン)ジパラジウム等のパラジウム触媒が挙げられ、また、配位子を添加して行うこともできる。配位子としてはホスフィンが好ましく、トリアルキルホスフィン(例えば、トリブチルホスフィン、トリシクロヘキシルホスフィンなど)、トリアリールホスフィン(例えば、トリフェニルホスフィンなど)、トリアルコキシホスフィン等が挙げられる。
パラジウム触媒の使用量は、化合物(II)に対し、通常約0.001ないし約5モル、好ましくは約0.01ないし約0.5モルである。「ホスフィン」の使用量は、化合物(II)に対し、通常約0.001ないし約10モル、好ましくは約0.01ないし約1モルである。
反応に影響を及ぼさない溶媒としては、例えば、テトラヒドロフラン、1,2-ジメトキシエタンなどのエーテル類、メタノール、エタノール、プロパノールなどのアルコール類、クロロホルムなどのハロゲン化炭化水素類、ベンゼン、トルエンなどの芳香族炭化水素類、アセトニトリル、プロピオニトリルなどのニトリル類、N,N-ジメチルホルムアミドなどのアミド類、ジメチルスルホキシドなどのスルホキシド類、水などが挙げられる。これらの溶媒は、2種以上を適宜の割合で混合して用いてもよい。これらの溶媒の使用量は、例えば、化合物(II)に対し、1~100容量倍である。
反応温度は、通常約-50℃~約250℃、好ましくは0℃~120℃である。
反応時間は、通常、約0.5~約36時間である。
本反応は、マイクロウェーブ反応装置などを用いることにより反応時間の短縮を図ることができる。
こうして得られる化合物(IV)は、公知の分離精製手段、例えば濃縮、減圧濃縮、溶媒抽出、晶出、再結晶、転溶、クラマトグラフィーなどにより、単離精製することができる。また、化合物(IV)は、単離せずに次の反応に用いてもよい。 According to production method 1, compound (II) and compound (III) are first subjected to a coupling reaction to produce compound (IV).
Examples of the leaving group L 1 include halides such as chloride, bromide and iodide, and alkylsulfonyloxy groups such as methanesulfonyloxy group and trifluoromethanesulfonyloxy group.
The coupling reaction is performed in the presence of a base and a catalyst with a solvent that does not affect the reaction. The amount of compound (III) to be used is about 0.5 to about 10 mol, preferably about 0.9 to about 3 mol, per 1 mol of compound (II).
Examples of bases include basic salts such as sodium carbonate, potassium carbonate, cesium carbonate, sodium hydrogen carbonate, tripotassium phosphate, aromatic amines such as pyridine and lutidine, triethylamine, tripropylamine, tributylamine, cyclohexyldimethylamine, Tertiary amines such as 4-dimethylaminopyridine, N, N-dimethylaniline, N-methylpiperidine, N-methylpyrrolidine, N-methylmorpholine, sodium methoxide, sodium ethoxide, sodium tert-butoxide, potassium Metal alkoxides such as tributoxide are used.
The amount of the base to be used is about 0.5 to about 10 mol, preferably about 1 to about 5 mol equivalent, relative to compound (II), respectively.
Examples of the catalyst used in this reaction include palladium catalysts such as palladium acetate, palladium chloride, tetrakis (triphenylphosphine) palladium, bis (dibenzylideneacetone) palladium, tris (dibenzylideneacetone) dipalladium, It can also be performed by adding a ligand. As the ligand, phosphine is preferable, and trialkylphosphine (for example, tributylphosphine, tricyclohexylphosphine and the like), triarylphosphine (for example, triphenylphosphine and the like), trialkoxyphosphine and the like can be mentioned.
The amount of the palladium catalyst to be used is generally about 0.001 to about 5 mol, preferably about 0.01 to about 0.5 mol, relative to compound (II). The amount of “phosphine” to be used is generally about 0.001 to about 10 mol, preferably about 0.01 to about 1 mol, relative to compound (II).
Examples of the solvent that does not affect the reaction include ethers such as tetrahydrofuran and 1,2-dimethoxyethane, alcohols such as methanol, ethanol and propanol, halogenated hydrocarbons such as chloroform, and aromatics such as benzene and toluene. Group hydrocarbons, nitriles such as acetonitrile and propionitrile, amides such as N, N-dimethylformamide, sulfoxides such as dimethyl sulfoxide, and water. These solvents may be used by mixing two or more kinds at an appropriate ratio. The amount of these solvents used is, for example, 1 to 100 times the volume of the compound (II).
The reaction temperature is usually about −50 ° C. to about 250 ° C., preferably 0 ° C. to 120 ° C.
The reaction time is usually about 0.5 to about 36 hours.
In this reaction, the reaction time can be shortened by using a microwave reaction apparatus or the like.
The compound (IV) thus obtained can be isolated and purified by known separation and purification means such as concentration, concentration under reduced pressure, solvent extraction, crystallization, recrystallization, phase transfer, chromatography, and the like. In addition, compound (IV) may be used for the next reaction without isolation.
脱離基L1としてはハライド、例えばクロライド、ブロマイド、アイオダイド、またアルキルスルホニルオキシ基、例えばメタンスルホニルオキシ基、トリフルオロメタンスルホニルオキシ基などが挙げられる。
カップリング反応は、塩基および触媒の存在下、反応に影響を及ぼさない溶媒で行われる。化合物(III)の使用量は、化合物(II)1モルに対し約0.5~約10モル、好ましくは約0.9~約3モルである。
塩基としては、炭酸ナトリウム、炭酸カリウム、炭酸セシウム、炭酸水素ナトリウム、りん酸三カリウム等の塩基性塩類、ピリジン、ルチジン等の芳香族アミン類、トリエチルアミン、トリプロピルアミン、トリブチルアミン、シクロヘキシルジメチルアミン、4-ジメチルアミノピリジン、N,N-ジメチルアニリン、N-メチルピペリジン、N-メチルピロリジン、N-メチルモルホリン等の第3級アミン類、ナトリウムメトキシド、ナトリウムエトキシド、ナトリウム第三ブトキシド、カリウム第三ブトキシド等の金属アルコキシド類などが用いられる。
塩基の使用量は、それぞれ化合物(II)に対して約0.5~約10モル、好ましくは、約1~約5モル当量である。
本反応で用いられるは触媒としては、酢酸パラジウム、塩化パラジウム、テトラキス(トリフェニルホスフィン)パラジウム、ビス(ジベンジリデンアセトン)パラジウム、トリス(ジベンジリデンアセトン)ジパラジウム等のパラジウム触媒が挙げられ、また、配位子を添加して行うこともできる。配位子としてはホスフィンが好ましく、トリアルキルホスフィン(例えば、トリブチルホスフィン、トリシクロヘキシルホスフィンなど)、トリアリールホスフィン(例えば、トリフェニルホスフィンなど)、トリアルコキシホスフィン等が挙げられる。
パラジウム触媒の使用量は、化合物(II)に対し、通常約0.001ないし約5モル、好ましくは約0.01ないし約0.5モルである。「ホスフィン」の使用量は、化合物(II)に対し、通常約0.001ないし約10モル、好ましくは約0.01ないし約1モルである。
反応に影響を及ぼさない溶媒としては、例えば、テトラヒドロフラン、1,2-ジメトキシエタンなどのエーテル類、メタノール、エタノール、プロパノールなどのアルコール類、クロロホルムなどのハロゲン化炭化水素類、ベンゼン、トルエンなどの芳香族炭化水素類、アセトニトリル、プロピオニトリルなどのニトリル類、N,N-ジメチルホルムアミドなどのアミド類、ジメチルスルホキシドなどのスルホキシド類、水などが挙げられる。これらの溶媒は、2種以上を適宜の割合で混合して用いてもよい。これらの溶媒の使用量は、例えば、化合物(II)に対し、1~100容量倍である。
反応温度は、通常約-50℃~約250℃、好ましくは0℃~120℃である。
反応時間は、通常、約0.5~約36時間である。
本反応は、マイクロウェーブ反応装置などを用いることにより反応時間の短縮を図ることができる。
こうして得られる化合物(IV)は、公知の分離精製手段、例えば濃縮、減圧濃縮、溶媒抽出、晶出、再結晶、転溶、クラマトグラフィーなどにより、単離精製することができる。また、化合物(IV)は、単離せずに次の反応に用いてもよい。 According to production method 1, compound (II) and compound (III) are first subjected to a coupling reaction to produce compound (IV).
Examples of the leaving group L 1 include halides such as chloride, bromide and iodide, and alkylsulfonyloxy groups such as methanesulfonyloxy group and trifluoromethanesulfonyloxy group.
The coupling reaction is performed in the presence of a base and a catalyst with a solvent that does not affect the reaction. The amount of compound (III) to be used is about 0.5 to about 10 mol, preferably about 0.9 to about 3 mol, per 1 mol of compound (II).
Examples of bases include basic salts such as sodium carbonate, potassium carbonate, cesium carbonate, sodium hydrogen carbonate, tripotassium phosphate, aromatic amines such as pyridine and lutidine, triethylamine, tripropylamine, tributylamine, cyclohexyldimethylamine, Tertiary amines such as 4-dimethylaminopyridine, N, N-dimethylaniline, N-methylpiperidine, N-methylpyrrolidine, N-methylmorpholine, sodium methoxide, sodium ethoxide, sodium tert-butoxide, potassium Metal alkoxides such as tributoxide are used.
The amount of the base to be used is about 0.5 to about 10 mol, preferably about 1 to about 5 mol equivalent, relative to compound (II), respectively.
Examples of the catalyst used in this reaction include palladium catalysts such as palladium acetate, palladium chloride, tetrakis (triphenylphosphine) palladium, bis (dibenzylideneacetone) palladium, tris (dibenzylideneacetone) dipalladium, It can also be performed by adding a ligand. As the ligand, phosphine is preferable, and trialkylphosphine (for example, tributylphosphine, tricyclohexylphosphine and the like), triarylphosphine (for example, triphenylphosphine and the like), trialkoxyphosphine and the like can be mentioned.
The amount of the palladium catalyst to be used is generally about 0.001 to about 5 mol, preferably about 0.01 to about 0.5 mol, relative to compound (II). The amount of “phosphine” to be used is generally about 0.001 to about 10 mol, preferably about 0.01 to about 1 mol, relative to compound (II).
Examples of the solvent that does not affect the reaction include ethers such as tetrahydrofuran and 1,2-dimethoxyethane, alcohols such as methanol, ethanol and propanol, halogenated hydrocarbons such as chloroform, and aromatics such as benzene and toluene. Group hydrocarbons, nitriles such as acetonitrile and propionitrile, amides such as N, N-dimethylformamide, sulfoxides such as dimethyl sulfoxide, and water. These solvents may be used by mixing two or more kinds at an appropriate ratio. The amount of these solvents used is, for example, 1 to 100 times the volume of the compound (II).
The reaction temperature is usually about −50 ° C. to about 250 ° C., preferably 0 ° C. to 120 ° C.
The reaction time is usually about 0.5 to about 36 hours.
In this reaction, the reaction time can be shortened by using a microwave reaction apparatus or the like.
The compound (IV) thus obtained can be isolated and purified by known separation and purification means such as concentration, concentration under reduced pressure, solvent extraction, crystallization, recrystallization, phase transfer, chromatography, and the like. In addition, compound (IV) may be used for the next reaction without isolation.
ついで、化合物(IV)を還元して、化合物(V)を製造する。
化合物(IV)の還元は、水素雰囲気下、金属触媒を用いて行うことができる。所望により適当な酸触媒を加えても良いよい。
金属触媒としては、ラネーニッケル、酸化白金、金属パラジウム、パラジウム炭素などが用いられる。金属触媒の使用量は、それぞれ化合物(IV)に対して、通常約1ないし約1000重量%、好ましくは約5ないし約20重量%である。
酸触媒としては、ギ酸、酢酸、トリフルオロ酢酸、p-トルエンスルホン酸等の有機酸、硫酸、塩酸、臭化水素酸等の鉱酸などが用いられる。酸触媒の使用量は、それぞれ化合物(IV)1モルに対し、約0.1ないし過剰量である。
本反応は、反応に不活性な溶媒を用いて行うのが有利である。このような溶媒としては、反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノールなどのアルコール類、ジエチルエーテル、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタンなどのエーテル類、ベンゼン、トルエン、シクロヘキサン、ヘキサンなどの炭化水素類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミドなどのアミド類、酢酸等の有機酸類、水等などが用いられる。また、これらの溶媒は2種以上を適宜の割合で混合して用いてもよい。水素圧は通常約1ないし約100気圧、好ましくは約1ないし約5気圧である。反応時間は通常約30分ないし約48時間、好ましくは約1ないし24時間である。反応温度は通常約0ないし約120℃、好ましくは約20ないし約80℃である。
生成物(V)は触媒を除いた後、常法に従って反応混合物から単離することもでき、通常の分離手段(例、再結晶、蒸留、クロマトグラフィー等)により容易に精製することができる。また、化合物(V)は単離せずに次の反応に用いてもよい。 Subsequently, compound (IV) is reduced to produce compound (V).
The reduction of compound (IV) can be performed using a metal catalyst in a hydrogen atmosphere. If desired, an appropriate acid catalyst may be added.
Raney nickel, platinum oxide, metallic palladium, palladium carbon, etc. are used as the metal catalyst. The amount of the metal catalyst to be used is generally about 1 to about 1000 wt%, preferably about 5 to about 20 wt%, relative to compound (IV), respectively.
As the acid catalyst, organic acids such as formic acid, acetic acid, trifluoroacetic acid and p-toluenesulfonic acid, and mineral acids such as sulfuric acid, hydrochloric acid and hydrobromic acid are used. The amount of the acid catalyst to be used is about 0.1 to excess amount per 1 mol of compound (IV).
This reaction is advantageously performed using a solvent inert to the reaction. Such a solvent is not particularly limited as long as the reaction proceeds. For example, alcohols such as methanol, ethanol and propanol, ethers such as diethyl ether, tetrahydrofuran, dioxane and 1,2-dimethoxyethane, benzene, toluene, Hydrocarbons such as cyclohexane and hexane, amides such as N, N-dimethylformamide and N, N-dimethylacetamide, organic acids such as acetic acid, water and the like are used. These solvents may be used by mixing two or more kinds at an appropriate ratio. The hydrogen pressure is usually about 1 to about 100 atmospheres, preferably about 1 to about 5 atmospheres. The reaction time is usually about 30 minutes to about 48 hours, preferably about 1 to 24 hours. The reaction temperature is usually about 0 to about 120 ° C, preferably about 20 to about 80 ° C.
The product (V) can be isolated from the reaction mixture according to a conventional method after removing the catalyst, and can be easily purified by a usual separation means (eg, recrystallization, distillation, chromatography, etc.). In addition, compound (V) may be used for the next reaction without isolation.
化合物(IV)の還元は、水素雰囲気下、金属触媒を用いて行うことができる。所望により適当な酸触媒を加えても良いよい。
金属触媒としては、ラネーニッケル、酸化白金、金属パラジウム、パラジウム炭素などが用いられる。金属触媒の使用量は、それぞれ化合物(IV)に対して、通常約1ないし約1000重量%、好ましくは約5ないし約20重量%である。
酸触媒としては、ギ酸、酢酸、トリフルオロ酢酸、p-トルエンスルホン酸等の有機酸、硫酸、塩酸、臭化水素酸等の鉱酸などが用いられる。酸触媒の使用量は、それぞれ化合物(IV)1モルに対し、約0.1ないし過剰量である。
本反応は、反応に不活性な溶媒を用いて行うのが有利である。このような溶媒としては、反応が進行する限り特に限定されないが、例えばメタノール、エタノール、プロパノールなどのアルコール類、ジエチルエーテル、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタンなどのエーテル類、ベンゼン、トルエン、シクロヘキサン、ヘキサンなどの炭化水素類、N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミドなどのアミド類、酢酸等の有機酸類、水等などが用いられる。また、これらの溶媒は2種以上を適宜の割合で混合して用いてもよい。水素圧は通常約1ないし約100気圧、好ましくは約1ないし約5気圧である。反応時間は通常約30分ないし約48時間、好ましくは約1ないし24時間である。反応温度は通常約0ないし約120℃、好ましくは約20ないし約80℃である。
生成物(V)は触媒を除いた後、常法に従って反応混合物から単離することもでき、通常の分離手段(例、再結晶、蒸留、クロマトグラフィー等)により容易に精製することができる。また、化合物(V)は単離せずに次の反応に用いてもよい。 Subsequently, compound (IV) is reduced to produce compound (V).
The reduction of compound (IV) can be performed using a metal catalyst in a hydrogen atmosphere. If desired, an appropriate acid catalyst may be added.
Raney nickel, platinum oxide, metallic palladium, palladium carbon, etc. are used as the metal catalyst. The amount of the metal catalyst to be used is generally about 1 to about 1000 wt%, preferably about 5 to about 20 wt%, relative to compound (IV), respectively.
As the acid catalyst, organic acids such as formic acid, acetic acid, trifluoroacetic acid and p-toluenesulfonic acid, and mineral acids such as sulfuric acid, hydrochloric acid and hydrobromic acid are used. The amount of the acid catalyst to be used is about 0.1 to excess amount per 1 mol of compound (IV).
This reaction is advantageously performed using a solvent inert to the reaction. Such a solvent is not particularly limited as long as the reaction proceeds. For example, alcohols such as methanol, ethanol and propanol, ethers such as diethyl ether, tetrahydrofuran, dioxane and 1,2-dimethoxyethane, benzene, toluene, Hydrocarbons such as cyclohexane and hexane, amides such as N, N-dimethylformamide and N, N-dimethylacetamide, organic acids such as acetic acid, water and the like are used. These solvents may be used by mixing two or more kinds at an appropriate ratio. The hydrogen pressure is usually about 1 to about 100 atmospheres, preferably about 1 to about 5 atmospheres. The reaction time is usually about 30 minutes to about 48 hours, preferably about 1 to 24 hours. The reaction temperature is usually about 0 to about 120 ° C, preferably about 20 to about 80 ° C.
The product (V) can be isolated from the reaction mixture according to a conventional method after removing the catalyst, and can be easily purified by a usual separation means (eg, recrystallization, distillation, chromatography, etc.). In addition, compound (V) may be used for the next reaction without isolation.
ついで、化合物(V)のtert-ブトキシカルボニル基を除去して、化合物(VI)を製造する。
この反応は、常法に従い、反応に悪影響をおよぼさない溶媒中で、酸を作用させることにより行われる。
酸としては、塩化水素、臭化水素、硫酸、トリフルオロ酢酸、トリフルオロメタンスルホン酸などがあげられる。酸の使用量は、それぞれ化合物(V)に対して好ましくは、約1~約100モル当量である。
反応に影響を及ぼさない溶媒としては、例えば、ヘキサン、シクロヘキサンなどの炭化水素類、メタノール、エタノール、プロパノールなどのアルコール類、ジエチルエーテル、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタンなどのエーテル類、酢酸エチル、酢酸メチルなどのエステル類、クロロホルム、ジクロロメタンなどのハロゲン化炭化水素類;ベンゼン、トルエンなどの芳香族炭化水素類;N,N-ジメチルホルムアミドなどのアミド類;ジメチルスルホキシドなどのスルホキシド類などが挙げられる。これらの溶媒は2種以上を適宜の割合で混合して用いてもよい。これらの溶媒の使用量は、例えば化合物(V)に対し、1~100容量倍である。
反応温度は、通常約-50℃~約250℃、好ましくは0℃~120℃である。
反応時間は、通常、約0.5~約24時間である。
こうして得られる化合物(VI)は、公知の分離精製手段、例えば濃縮、減圧濃縮、溶媒抽出、晶出、再結晶、転溶、クラマトグラフィーなどにより、単離精製することができる。また、化合物(VI)は、単離せずにつぎの反応に用いてもよい。 Next, the tert-butoxycarbonyl group of compound (V) is removed to produce compound (VI).
This reaction is carried out by reacting an acid according to a conventional method in a solvent that does not adversely influence the reaction.
Examples of the acid include hydrogen chloride, hydrogen bromide, sulfuric acid, trifluoroacetic acid, trifluoromethanesulfonic acid and the like. The amount of the acid used is preferably about 1 to about 100 molar equivalents relative to compound (V), respectively.
Examples of solvents that do not affect the reaction include hydrocarbons such as hexane and cyclohexane, alcohols such as methanol, ethanol, and propanol, ethers such as diethyl ether, tetrahydrofuran, dioxane, and 1,2-dimethoxyethane, and acetic acid. Esters such as ethyl and methyl acetate, halogenated hydrocarbons such as chloroform and dichloromethane; aromatic hydrocarbons such as benzene and toluene; amides such as N, N-dimethylformamide; sulfoxides such as dimethyl sulfoxide Can be mentioned. Two or more of these solvents may be mixed and used at an appropriate ratio. The amount of these solvents used is, for example, 1 to 100 times the volume of the compound (V).
The reaction temperature is usually about −50 ° C. to about 250 ° C., preferably 0 ° C. to 120 ° C.
The reaction time is usually about 0.5 to about 24 hours.
The compound (VI) thus obtained can be isolated and purified by known separation and purification means, for example, concentration, concentration under reduced pressure, solvent extraction, crystallization, recrystallization, phase transfer, chromatography, etc. Compound (VI) may be used for the next reaction without isolation.
この反応は、常法に従い、反応に悪影響をおよぼさない溶媒中で、酸を作用させることにより行われる。
酸としては、塩化水素、臭化水素、硫酸、トリフルオロ酢酸、トリフルオロメタンスルホン酸などがあげられる。酸の使用量は、それぞれ化合物(V)に対して好ましくは、約1~約100モル当量である。
反応に影響を及ぼさない溶媒としては、例えば、ヘキサン、シクロヘキサンなどの炭化水素類、メタノール、エタノール、プロパノールなどのアルコール類、ジエチルエーテル、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタンなどのエーテル類、酢酸エチル、酢酸メチルなどのエステル類、クロロホルム、ジクロロメタンなどのハロゲン化炭化水素類;ベンゼン、トルエンなどの芳香族炭化水素類;N,N-ジメチルホルムアミドなどのアミド類;ジメチルスルホキシドなどのスルホキシド類などが挙げられる。これらの溶媒は2種以上を適宜の割合で混合して用いてもよい。これらの溶媒の使用量は、例えば化合物(V)に対し、1~100容量倍である。
反応温度は、通常約-50℃~約250℃、好ましくは0℃~120℃である。
反応時間は、通常、約0.5~約24時間である。
こうして得られる化合物(VI)は、公知の分離精製手段、例えば濃縮、減圧濃縮、溶媒抽出、晶出、再結晶、転溶、クラマトグラフィーなどにより、単離精製することができる。また、化合物(VI)は、単離せずにつぎの反応に用いてもよい。 Next, the tert-butoxycarbonyl group of compound (V) is removed to produce compound (VI).
This reaction is carried out by reacting an acid according to a conventional method in a solvent that does not adversely influence the reaction.
Examples of the acid include hydrogen chloride, hydrogen bromide, sulfuric acid, trifluoroacetic acid, trifluoromethanesulfonic acid and the like. The amount of the acid used is preferably about 1 to about 100 molar equivalents relative to compound (V), respectively.
Examples of solvents that do not affect the reaction include hydrocarbons such as hexane and cyclohexane, alcohols such as methanol, ethanol, and propanol, ethers such as diethyl ether, tetrahydrofuran, dioxane, and 1,2-dimethoxyethane, and acetic acid. Esters such as ethyl and methyl acetate, halogenated hydrocarbons such as chloroform and dichloromethane; aromatic hydrocarbons such as benzene and toluene; amides such as N, N-dimethylformamide; sulfoxides such as dimethyl sulfoxide Can be mentioned. Two or more of these solvents may be mixed and used at an appropriate ratio. The amount of these solvents used is, for example, 1 to 100 times the volume of the compound (V).
The reaction temperature is usually about −50 ° C. to about 250 ° C., preferably 0 ° C. to 120 ° C.
The reaction time is usually about 0.5 to about 24 hours.
The compound (VI) thus obtained can be isolated and purified by known separation and purification means, for example, concentration, concentration under reduced pressure, solvent extraction, crystallization, recrystallization, phase transfer, chromatography, etc. Compound (VI) may be used for the next reaction without isolation.
ついで、化合物(VI)をウレア化して、化合物(I)を製造する。
ウレア化は化合物(VI)に対して、イソシアネート(VII)、あるいは、2,2,2-トリクロロエトキシカルバメート(VIII)、あるいは、ビス(2,2,2-トリクロロエトキシカルバメート)(IX)、あるいは、アリールエステル(X)を反応させることによって行うことができる。
化合物(VI)とイソシアネート(VII)との反応による化合物(I)の製造では、塩基とイソシアネート(VII)の存在下、反応に影響を及ぼさない溶媒で行われる。塩基としては、トリエチルアミン、トリブチルアミン、ジイソプロピルエチルアミン、炭酸カリウム、炭酸ナトリウム、水素化ナトリウム、水素化カリウムなどが挙げられる。
塩基とイソシアネート(VII)の使用量は、それぞれ化合物(VI)に対して好ましくは、約1ないし約5モル当量である。
反応に影響を及ぼさない溶媒としては、例えば、ジエチルエーテル、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタンなどのエーテル類、クロロホルム、ジクロロメタンなどのハロゲン化炭化水素類、ベンゼン、トルエンなどの芳香族炭化水素類、N,N-ジメチルホルムアミドなどのアミド類、ジメチルスルホキシドなどのスルホキシド類などが挙げられる。これらの溶媒は2種以上を適宜の割合で混合して用いてもよい。これらの溶媒の使用量は、例えば、化合物(VI)に対し、1ないし100容量倍である。
反応温度は、通常約-50℃ないし約250℃、好ましくは0℃ないし120℃である。
反応時間は、通常、約0.5ないし約24時間である。こうして得られる化合物(I)は、公知の分離精製手段、例えば濃縮、減圧濃縮、溶媒抽出、晶出、再結晶、転溶、クラマトグラフィーなどにより、単離精製することができる。 Next, compound (VI) is uread to produce compound (I).
Ureaization is performed on the compound (VI) with respect to the isocyanate (VII), 2,2,2-trichloroethoxycarbamate (VIII), bis (2,2,2-trichloroethoxycarbamate) (IX), or , Aryl ester (X) can be reacted.
The production of compound (I) by the reaction of compound (VI) and isocyanate (VII) is carried out in the presence of a base and isocyanate (VII) with a solvent that does not affect the reaction. Examples of the base include triethylamine, tributylamine, diisopropylethylamine, potassium carbonate, sodium carbonate, sodium hydride, potassium hydride and the like.
The amount of base and isocyanate (VII) to be used is preferably about 1 to about 5 molar equivalents relative to compound (VI), respectively.
Examples of the solvent that does not affect the reaction include ethers such as diethyl ether, tetrahydrofuran, dioxane, and 1,2-dimethoxyethane, halogenated hydrocarbons such as chloroform and dichloromethane, and aromatic hydrocarbons such as benzene and toluene. Amides such as N, N-dimethylformamide, and sulfoxides such as dimethyl sulfoxide. Two or more of these solvents may be mixed and used at an appropriate ratio. The amount of these solvents to be used is, for example, 1 to 100 times the volume of the compound (VI).
The reaction temperature is usually about −50 ° C. to about 250 ° C., preferably 0 ° C. to 120 ° C.
The reaction time is usually about 0.5 to about 24 hours. The compound (I) thus obtained can be isolated and purified by known separation and purification means such as concentration, concentration under reduced pressure, solvent extraction, crystallization, recrystallization, phase transfer, chromatography, and the like.
ウレア化は化合物(VI)に対して、イソシアネート(VII)、あるいは、2,2,2-トリクロロエトキシカルバメート(VIII)、あるいは、ビス(2,2,2-トリクロロエトキシカルバメート)(IX)、あるいは、アリールエステル(X)を反応させることによって行うことができる。
化合物(VI)とイソシアネート(VII)との反応による化合物(I)の製造では、塩基とイソシアネート(VII)の存在下、反応に影響を及ぼさない溶媒で行われる。塩基としては、トリエチルアミン、トリブチルアミン、ジイソプロピルエチルアミン、炭酸カリウム、炭酸ナトリウム、水素化ナトリウム、水素化カリウムなどが挙げられる。
塩基とイソシアネート(VII)の使用量は、それぞれ化合物(VI)に対して好ましくは、約1ないし約5モル当量である。
反応に影響を及ぼさない溶媒としては、例えば、ジエチルエーテル、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタンなどのエーテル類、クロロホルム、ジクロロメタンなどのハロゲン化炭化水素類、ベンゼン、トルエンなどの芳香族炭化水素類、N,N-ジメチルホルムアミドなどのアミド類、ジメチルスルホキシドなどのスルホキシド類などが挙げられる。これらの溶媒は2種以上を適宜の割合で混合して用いてもよい。これらの溶媒の使用量は、例えば、化合物(VI)に対し、1ないし100容量倍である。
反応温度は、通常約-50℃ないし約250℃、好ましくは0℃ないし120℃である。
反応時間は、通常、約0.5ないし約24時間である。こうして得られる化合物(I)は、公知の分離精製手段、例えば濃縮、減圧濃縮、溶媒抽出、晶出、再結晶、転溶、クラマトグラフィーなどにより、単離精製することができる。 Next, compound (VI) is uread to produce compound (I).
Ureaization is performed on the compound (VI) with respect to the isocyanate (VII), 2,2,2-trichloroethoxycarbamate (VIII), bis (2,2,2-trichloroethoxycarbamate) (IX), or , Aryl ester (X) can be reacted.
The production of compound (I) by the reaction of compound (VI) and isocyanate (VII) is carried out in the presence of a base and isocyanate (VII) with a solvent that does not affect the reaction. Examples of the base include triethylamine, tributylamine, diisopropylethylamine, potassium carbonate, sodium carbonate, sodium hydride, potassium hydride and the like.
The amount of base and isocyanate (VII) to be used is preferably about 1 to about 5 molar equivalents relative to compound (VI), respectively.
Examples of the solvent that does not affect the reaction include ethers such as diethyl ether, tetrahydrofuran, dioxane, and 1,2-dimethoxyethane, halogenated hydrocarbons such as chloroform and dichloromethane, and aromatic hydrocarbons such as benzene and toluene. Amides such as N, N-dimethylformamide, and sulfoxides such as dimethyl sulfoxide. Two or more of these solvents may be mixed and used at an appropriate ratio. The amount of these solvents to be used is, for example, 1 to 100 times the volume of the compound (VI).
The reaction temperature is usually about −50 ° C. to about 250 ° C., preferably 0 ° C. to 120 ° C.
The reaction time is usually about 0.5 to about 24 hours. The compound (I) thus obtained can be isolated and purified by known separation and purification means such as concentration, concentration under reduced pressure, solvent extraction, crystallization, recrystallization, phase transfer, chromatography, and the like.
化合物(VI)と2,2,2-トリクロロエトキシカルバメート(VIII)、あるいは、ビス(2,2,2-トリクロロエトキシカルバメート)(VIV)、あるいは、アリールエステル(X)との反応による化合物(I)の製造では、塩基の存在下、反応に影響を及ぼさない溶媒で行われる。塩基としては、ピリジン、トリエチルアミン、ジイソプロピルエチルアミン、炭酸カリウム、炭酸ナトリウム、水素化ナトリウム、水素化カリウムなどが挙げられる。
塩基と2,2,2-トリクロロエトキシカルバメート(VIII)、ビス(2,2,2-トリクロロエトキシカルバメート)(IX)、アリールエステル(X)の使用量は、それぞれ化合物(VI)に対して好ましくは、約1ないし約5モル当量である。
反応に影響を及ぼさない溶媒としては、例えば、ジエチルエーテル、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタンなどのエーテル類、ジクロロメタン、クロロホルムなどのハロゲン化炭化水素類、酢酸エチル、酢酸メチルなどのエステル類、ベンゼン、トルエンなどの芳香族炭化水素類、N,N-ジメチルホルムアミドなどのアミド類、ジメチルスルホキシドなどのスルホキシド類などが挙げられる。これらの溶媒は2種以上を適宜の割合で混合して用いてもよい。これらの溶媒の使用量は、例えば、化合物(V)に対し、1~100容量倍である。
反応温度は、通常約-50℃~200℃、好ましくは0℃~120℃である。
反応時間は、通常、約0.5~約36時間である。
こうして得られる化合物(I)は、公知の分離精製手段、例えば、濃縮、減圧濃縮、溶媒抽出、晶出、再結晶、転溶、クラマトグラフィーなどにより、単離精製することができる。 Compound (I) by reaction of Compound (VI) with 2,2,2-trichloroethoxycarbamate (VIII), bis (2,2,2-trichloroethoxycarbamate) (VII), or aryl ester (X) In the presence of a base in a solvent that does not affect the reaction. Examples of the base include pyridine, triethylamine, diisopropylethylamine, potassium carbonate, sodium carbonate, sodium hydride, potassium hydride and the like.
The amount of base, 2,2,2-trichloroethoxycarbamate (VIII), bis (2,2,2-trichloroethoxycarbamate) (IX), and aryl ester (X) used is preferably relative to compound (VI). Is from about 1 to about 5 molar equivalents.
Examples of the solvent that does not affect the reaction include ethers such as diethyl ether, tetrahydrofuran, dioxane and 1,2-dimethoxyethane, halogenated hydrocarbons such as dichloromethane and chloroform, and esters such as ethyl acetate and methyl acetate. , Aromatic hydrocarbons such as benzene and toluene, amides such as N, N-dimethylformamide, and sulfoxides such as dimethyl sulfoxide. Two or more of these solvents may be mixed and used at an appropriate ratio. The amount of these solvents to be used is, for example, 1 to 100 times the volume of the compound (V).
The reaction temperature is usually about −50 ° C. to 200 ° C., preferably 0 ° C. to 120 ° C.
The reaction time is usually about 0.5 to about 36 hours.
The compound (I) thus obtained can be isolated and purified by known separation and purification means such as concentration, concentration under reduced pressure, solvent extraction, crystallization, recrystallization, phase transfer, chromatography, etc.
塩基と2,2,2-トリクロロエトキシカルバメート(VIII)、ビス(2,2,2-トリクロロエトキシカルバメート)(IX)、アリールエステル(X)の使用量は、それぞれ化合物(VI)に対して好ましくは、約1ないし約5モル当量である。
反応に影響を及ぼさない溶媒としては、例えば、ジエチルエーテル、テトラヒドロフラン、ジオキサン、1,2-ジメトキシエタンなどのエーテル類、ジクロロメタン、クロロホルムなどのハロゲン化炭化水素類、酢酸エチル、酢酸メチルなどのエステル類、ベンゼン、トルエンなどの芳香族炭化水素類、N,N-ジメチルホルムアミドなどのアミド類、ジメチルスルホキシドなどのスルホキシド類などが挙げられる。これらの溶媒は2種以上を適宜の割合で混合して用いてもよい。これらの溶媒の使用量は、例えば、化合物(V)に対し、1~100容量倍である。
反応温度は、通常約-50℃~200℃、好ましくは0℃~120℃である。
反応時間は、通常、約0.5~約36時間である。
こうして得られる化合物(I)は、公知の分離精製手段、例えば、濃縮、減圧濃縮、溶媒抽出、晶出、再結晶、転溶、クラマトグラフィーなどにより、単離精製することができる。 Compound (I) by reaction of Compound (VI) with 2,2,2-trichloroethoxycarbamate (VIII), bis (2,2,2-trichloroethoxycarbamate) (VII), or aryl ester (X) In the presence of a base in a solvent that does not affect the reaction. Examples of the base include pyridine, triethylamine, diisopropylethylamine, potassium carbonate, sodium carbonate, sodium hydride, potassium hydride and the like.
The amount of base, 2,2,2-trichloroethoxycarbamate (VIII), bis (2,2,2-trichloroethoxycarbamate) (IX), and aryl ester (X) used is preferably relative to compound (VI). Is from about 1 to about 5 molar equivalents.
Examples of the solvent that does not affect the reaction include ethers such as diethyl ether, tetrahydrofuran, dioxane and 1,2-dimethoxyethane, halogenated hydrocarbons such as dichloromethane and chloroform, and esters such as ethyl acetate and methyl acetate. , Aromatic hydrocarbons such as benzene and toluene, amides such as N, N-dimethylformamide, and sulfoxides such as dimethyl sulfoxide. Two or more of these solvents may be mixed and used at an appropriate ratio. The amount of these solvents to be used is, for example, 1 to 100 times the volume of the compound (V).
The reaction temperature is usually about −50 ° C. to 200 ° C., preferably 0 ° C. to 120 ° C.
The reaction time is usually about 0.5 to about 36 hours.
The compound (I) thus obtained can be isolated and purified by known separation and purification means such as concentration, concentration under reduced pressure, solvent extraction, crystallization, recrystallization, phase transfer, chromatography, etc.
[製造方法2]
製造方法1に示した化合物(IV)は

〔式中、L2は脱離基を表し、その他の記号は上記と同意義である。〕
で表される製造方法2、または、これに準ずる方法によっても製造することができる。 [Production Method 2]
Compound (IV) shown in Production Method 1 is
[Wherein L 2 represents a leaving group, and other symbols are as defined above. ]
It can manufacture also by the manufacturing method 2 represented by this, or the method according to this.
製造方法1に示した化合物(IV)は
で表される製造方法2、または、これに準ずる方法によっても製造することができる。 [Production Method 2]
Compound (IV) shown in Production Method 1 is
It can manufacture also by the manufacturing method 2 represented by this, or the method according to this.
脱離基L2としてはハライド、例えばクロライド、ブロマイド、アイオダイド、またアルキルスルホニルオキシ基、例えばメタンスルホニルオキシ基、トリフルオロメタンスルホニルオキシ基などが挙げられる。
製造方法2に従って、まず、化合物(II)と化合物(XI)とをカップリング反応に付し、化合物(XII)を製造する。化合物(XII)は製造方法1に示した化合物(IV)の製造と同様の方法によって合成することができる。
こうして得られる化合物(XII)は、公知の分離精製手段、例えば濃縮、減圧濃縮、溶媒抽出、晶出、再結晶、転溶、クラマトグラフィーなどにより、単離精製することができる。また、化合物(XII)は、単離せずに次の反応に用いてもよい。 Examples of the leaving group L 2 include halides such as chloride, bromide, iodide, and alkylsulfonyloxy groups such as methanesulfonyloxy group and trifluoromethanesulfonyloxy group.
According to production method 2, compound (II) and compound (XI) are first subjected to a coupling reaction to produce compound (XII). Compound (XII) can be synthesized by a method similar to the production of compound (IV) shown in Production Method 1.
The compound (XII) thus obtained can be isolated and purified by known separation and purification means, for example, concentration, concentration under reduced pressure, solvent extraction, crystallization, recrystallization, phase transfer, chromatography, etc. In addition, compound (XII) may be used for the next reaction without isolation.
製造方法2に従って、まず、化合物(II)と化合物(XI)とをカップリング反応に付し、化合物(XII)を製造する。化合物(XII)は製造方法1に示した化合物(IV)の製造と同様の方法によって合成することができる。
こうして得られる化合物(XII)は、公知の分離精製手段、例えば濃縮、減圧濃縮、溶媒抽出、晶出、再結晶、転溶、クラマトグラフィーなどにより、単離精製することができる。また、化合物(XII)は、単離せずに次の反応に用いてもよい。 Examples of the leaving group L 2 include halides such as chloride, bromide, iodide, and alkylsulfonyloxy groups such as methanesulfonyloxy group and trifluoromethanesulfonyloxy group.
According to production method 2, compound (II) and compound (XI) are first subjected to a coupling reaction to produce compound (XII). Compound (XII) can be synthesized by a method similar to the production of compound (IV) shown in Production Method 1.
The compound (XII) thus obtained can be isolated and purified by known separation and purification means, for example, concentration, concentration under reduced pressure, solvent extraction, crystallization, recrystallization, phase transfer, chromatography, etc. In addition, compound (XII) may be used for the next reaction without isolation.
ついで、化合物(XII)とボロン酸類、あるいはボロン酸エステル類とのカップリング反応により、化合物(IV)を製造する。
カップリング反応は、塩基および触媒の存在下、反応に影響を及ぼさない溶媒で行われる。
ボロン酸類、あるいはボロン酸エステル類の使用量は、化合物(XII)1モルに対し約0.5~約10モル、好ましくは約0.9~約3モルである。
塩基としては、炭酸ナトリウム、炭酸カリウム、炭酸セシウム、炭酸水素ナトリウム、リン酸三カリウム等の塩基性塩類、ピリジン、ルチジン等の芳香族アミン類、トリエチルアミン、トリプロピルアミン、トリブチルアミン、シクロヘキシルジメチルアミン、4-ジメチルアミノピリジン、N,N-ジメチルアニリン、N-メチルピペリジン、N-メチルピロリジン、N-メチルモルホリン等の第3級アミン類、ナトリウムメトキシド、ナトリウムエトキシド、ナトリウム第三ブトキシド、カリウム第三ブトキシド等の金属アルコキシド類などが用いられる。
塩基の使用量は、それぞれ化合物(XII)に対して約0.5~約10モル、好ましくは、約1~約5モル当量である。
本反応で用いられるは触媒としては、酢酸パラジウム、塩化パラジウム、テトラキス(トリフェニルホスフィン)パラジウム、ビス(ジベンジリデンアセトン)パラジウム、トリス(ジベンジリデンアセトン)ジパラジウム等のパラジウム触媒が挙げられ、また、配位子を添加して行うこともできる。配位子としてはホスフィンが好ましく、トリアルキルホスフィン(例えば、トリブチルホスフィン、トリシクロヘキシルホスフィンなど)、トリアリールホスフィン(例えば、トリフェニルホスフィンなど)、トリアルコキシホスフィン等が挙げられる。
パラジウム触媒の使用量は、化合物(XII)に対し、通常約0.001ないし約5モル、好ましくは約0.01ないし約0.5モルである。「ホスフィン」の使用量は、化合物(XII)に対し、通常約0.001ないし約10モル、好ましくは約0.01ないし約1モルである。
反応に影響を及ぼさない溶媒としては、例えば、テトラヒドロフラン、1,2-ジメトキシエタンなどのエーテル類、メタノール、エタノール、プロパノールなどのアルコール類、クロロホルムなどのハロゲン化炭化水素類、ベンゼン、トルエンなどの芳香族炭化水素類、アセトニトリル、プロピオニトリルなどのニトリル類、N,N-ジメチルホルムアミドなどのアミド類、ジメチルスルホキシドなどのスルホキシド類、水などが挙げられる。これらの溶媒は、2種以上を適宜の割合で混合して用いてもよい。これらの溶媒の使用量は、例えば、化合物(XII)に対し、1~100容量倍である。
反応温度は、通常約-50℃~約250℃、好ましくは0℃~120℃である。
反応時間は、通常、約0.5~約36時間である。
本反応は、マイクロウェーブ反応装置などを用いることにより反応時間の短縮を図ることができる。
こうして得られる化合物(IV)は、公知の分離精製手段、例えば濃縮、減圧濃縮、溶媒抽出、晶出、再結晶、転溶、クラマトグラフィーなどにより、単離精製することができる。また、化合物(IV)は、単離せずに次の反応に用いてもよい。 Next, compound (IV) is produced by a coupling reaction between compound (XII) and boronic acids or boronic esters.
The coupling reaction is performed in the presence of a base and a catalyst with a solvent that does not affect the reaction.
The amount of the boronic acid or boronic acid ester to be used is about 0.5 to about 10 mol, preferably about 0.9 to about 3 mol, per 1 mol of compound (XII).
Examples of the base include basic salts such as sodium carbonate, potassium carbonate, cesium carbonate, sodium hydrogen carbonate, tripotassium phosphate, aromatic amines such as pyridine and lutidine, triethylamine, tripropylamine, tributylamine, cyclohexyldimethylamine, Tertiary amines such as 4-dimethylaminopyridine, N, N-dimethylaniline, N-methylpiperidine, N-methylpyrrolidine, N-methylmorpholine, sodium methoxide, sodium ethoxide, sodium tert-butoxide, potassium Metal alkoxides such as tributoxide are used.
The amount of the base to be used is about 0.5 to about 10 mol, preferably about 1 to about 5 molar equivalent, relative to compound (XII), respectively.
Examples of the catalyst used in this reaction include palladium catalysts such as palladium acetate, palladium chloride, tetrakis (triphenylphosphine) palladium, bis (dibenzylideneacetone) palladium, tris (dibenzylideneacetone) dipalladium, It can also be performed by adding a ligand. As the ligand, phosphine is preferable, and trialkylphosphine (for example, tributylphosphine, tricyclohexylphosphine and the like), triarylphosphine (for example, triphenylphosphine and the like), trialkoxyphosphine and the like can be mentioned.
The amount of the palladium catalyst to be used is generally about 0.001 to about 5 mol, preferably about 0.01 to about 0.5 mol, relative to compound (XII). The amount of the “phosphine” to be used is generally about 0.001 to about 10 mol, preferably about 0.01 to about 1 mol, relative to compound (XII).
Examples of the solvent that does not affect the reaction include ethers such as tetrahydrofuran and 1,2-dimethoxyethane, alcohols such as methanol, ethanol and propanol, halogenated hydrocarbons such as chloroform, and aromatics such as benzene and toluene. Group hydrocarbons, nitriles such as acetonitrile and propionitrile, amides such as N, N-dimethylformamide, sulfoxides such as dimethyl sulfoxide, and water. These solvents may be used by mixing two or more kinds at an appropriate ratio. The amount of these solvents to be used is, for example, 1 to 100 times the volume of the compound (XII).
The reaction temperature is usually about −50 ° C. to about 250 ° C., preferably 0 ° C. to 120 ° C.
The reaction time is usually about 0.5 to about 36 hours.
In this reaction, the reaction time can be shortened by using a microwave reaction apparatus or the like.
The compound (IV) thus obtained can be isolated and purified by known separation and purification means such as concentration, concentration under reduced pressure, solvent extraction, crystallization, recrystallization, phase transfer, chromatography, and the like. In addition, compound (IV) may be used for the next reaction without isolation.
カップリング反応は、塩基および触媒の存在下、反応に影響を及ぼさない溶媒で行われる。
ボロン酸類、あるいはボロン酸エステル類の使用量は、化合物(XII)1モルに対し約0.5~約10モル、好ましくは約0.9~約3モルである。
塩基としては、炭酸ナトリウム、炭酸カリウム、炭酸セシウム、炭酸水素ナトリウム、リン酸三カリウム等の塩基性塩類、ピリジン、ルチジン等の芳香族アミン類、トリエチルアミン、トリプロピルアミン、トリブチルアミン、シクロヘキシルジメチルアミン、4-ジメチルアミノピリジン、N,N-ジメチルアニリン、N-メチルピペリジン、N-メチルピロリジン、N-メチルモルホリン等の第3級アミン類、ナトリウムメトキシド、ナトリウムエトキシド、ナトリウム第三ブトキシド、カリウム第三ブトキシド等の金属アルコキシド類などが用いられる。
塩基の使用量は、それぞれ化合物(XII)に対して約0.5~約10モル、好ましくは、約1~約5モル当量である。
本反応で用いられるは触媒としては、酢酸パラジウム、塩化パラジウム、テトラキス(トリフェニルホスフィン)パラジウム、ビス(ジベンジリデンアセトン)パラジウム、トリス(ジベンジリデンアセトン)ジパラジウム等のパラジウム触媒が挙げられ、また、配位子を添加して行うこともできる。配位子としてはホスフィンが好ましく、トリアルキルホスフィン(例えば、トリブチルホスフィン、トリシクロヘキシルホスフィンなど)、トリアリールホスフィン(例えば、トリフェニルホスフィンなど)、トリアルコキシホスフィン等が挙げられる。
パラジウム触媒の使用量は、化合物(XII)に対し、通常約0.001ないし約5モル、好ましくは約0.01ないし約0.5モルである。「ホスフィン」の使用量は、化合物(XII)に対し、通常約0.001ないし約10モル、好ましくは約0.01ないし約1モルである。
反応に影響を及ぼさない溶媒としては、例えば、テトラヒドロフラン、1,2-ジメトキシエタンなどのエーテル類、メタノール、エタノール、プロパノールなどのアルコール類、クロロホルムなどのハロゲン化炭化水素類、ベンゼン、トルエンなどの芳香族炭化水素類、アセトニトリル、プロピオニトリルなどのニトリル類、N,N-ジメチルホルムアミドなどのアミド類、ジメチルスルホキシドなどのスルホキシド類、水などが挙げられる。これらの溶媒は、2種以上を適宜の割合で混合して用いてもよい。これらの溶媒の使用量は、例えば、化合物(XII)に対し、1~100容量倍である。
反応温度は、通常約-50℃~約250℃、好ましくは0℃~120℃である。
反応時間は、通常、約0.5~約36時間である。
本反応は、マイクロウェーブ反応装置などを用いることにより反応時間の短縮を図ることができる。
こうして得られる化合物(IV)は、公知の分離精製手段、例えば濃縮、減圧濃縮、溶媒抽出、晶出、再結晶、転溶、クラマトグラフィーなどにより、単離精製することができる。また、化合物(IV)は、単離せずに次の反応に用いてもよい。 Next, compound (IV) is produced by a coupling reaction between compound (XII) and boronic acids or boronic esters.
The coupling reaction is performed in the presence of a base and a catalyst with a solvent that does not affect the reaction.
The amount of the boronic acid or boronic acid ester to be used is about 0.5 to about 10 mol, preferably about 0.9 to about 3 mol, per 1 mol of compound (XII).
Examples of the base include basic salts such as sodium carbonate, potassium carbonate, cesium carbonate, sodium hydrogen carbonate, tripotassium phosphate, aromatic amines such as pyridine and lutidine, triethylamine, tripropylamine, tributylamine, cyclohexyldimethylamine, Tertiary amines such as 4-dimethylaminopyridine, N, N-dimethylaniline, N-methylpiperidine, N-methylpyrrolidine, N-methylmorpholine, sodium methoxide, sodium ethoxide, sodium tert-butoxide, potassium Metal alkoxides such as tributoxide are used.
The amount of the base to be used is about 0.5 to about 10 mol, preferably about 1 to about 5 molar equivalent, relative to compound (XII), respectively.
Examples of the catalyst used in this reaction include palladium catalysts such as palladium acetate, palladium chloride, tetrakis (triphenylphosphine) palladium, bis (dibenzylideneacetone) palladium, tris (dibenzylideneacetone) dipalladium, It can also be performed by adding a ligand. As the ligand, phosphine is preferable, and trialkylphosphine (for example, tributylphosphine, tricyclohexylphosphine and the like), triarylphosphine (for example, triphenylphosphine and the like), trialkoxyphosphine and the like can be mentioned.
The amount of the palladium catalyst to be used is generally about 0.001 to about 5 mol, preferably about 0.01 to about 0.5 mol, relative to compound (XII). The amount of the “phosphine” to be used is generally about 0.001 to about 10 mol, preferably about 0.01 to about 1 mol, relative to compound (XII).
Examples of the solvent that does not affect the reaction include ethers such as tetrahydrofuran and 1,2-dimethoxyethane, alcohols such as methanol, ethanol and propanol, halogenated hydrocarbons such as chloroform, and aromatics such as benzene and toluene. Group hydrocarbons, nitriles such as acetonitrile and propionitrile, amides such as N, N-dimethylformamide, sulfoxides such as dimethyl sulfoxide, and water. These solvents may be used by mixing two or more kinds at an appropriate ratio. The amount of these solvents to be used is, for example, 1 to 100 times the volume of the compound (XII).
The reaction temperature is usually about −50 ° C. to about 250 ° C., preferably 0 ° C. to 120 ° C.
The reaction time is usually about 0.5 to about 36 hours.
In this reaction, the reaction time can be shortened by using a microwave reaction apparatus or the like.
The compound (IV) thus obtained can be isolated and purified by known separation and purification means such as concentration, concentration under reduced pressure, solvent extraction, crystallization, recrystallization, phase transfer, chromatography, and the like. In addition, compound (IV) may be used for the next reaction without isolation.
化合物(I)が、光学異性体、立体異性体、位置異性体、回転異性体等の異性体を有する場合には、いずれか一方の異性体も混合物も化合物(I)に包含される。例えば、化合物(I)に光学異性体が存在する場合には、ラセミ体から分割された光学異性体も化合物(I)に包含される。これらの異性体は、自体公知の合成手法、分離手法(濃縮、溶媒抽出、カラムクロマトグラフィー、再結晶など)によりそれぞれを単品として得ることができる。
化合物(I)は、結晶であってもよく、結晶形が単一であっても結晶形混合物であっても化合物(I)に包含される。結晶は、自体公知の結晶化法を適用して、結晶化することによって製造することができる。
化合物(I)は、溶媒和物(例えば、水和物等)であっても、無溶媒和物であってもよく、いずれも化合物(I)に包含される。
同位元素(例、3H、14C、35S、125Iなど)などで標識された化合物も、化合物(I)に包含される。さらに、1Hを2H(D)に変換した重水素変換体も、化合物(I)に包含される。 When compound (I) has an isomer such as an optical isomer, a stereoisomer, a positional isomer, a rotational isomer, etc., any one of the isomers and a mixture are encompassed in compound (I). For example, when compound (I) has an optical isomer, the optical isomer resolved from the racemate is also encompassed in compound (I). Each of these isomers can be obtained as a single product by a known synthesis method or separation method (concentration, solvent extraction, column chromatography, recrystallization, etc.).
Compound (I) may be a crystal, and it is included in compound (I) regardless of whether the crystal form is a single crystal form or a mixture of crystal forms. The crystal can be produced by crystallization by applying a crystallization method known per se.
Compound (I) may be a solvate (such as a hydrate) or a non-solvate, and both are encompassed in compound (I).
Compounds labeled with isotopes (eg, 3 H, 14 C, 35 S, 125 I, etc.) are also encompassed in compound (I). Further, a deuterium converter obtained by converting 1 H into 2 H (D) is also encompassed in compound (I).
化合物(I)は、結晶であってもよく、結晶形が単一であっても結晶形混合物であっても化合物(I)に包含される。結晶は、自体公知の結晶化法を適用して、結晶化することによって製造することができる。
化合物(I)は、溶媒和物(例えば、水和物等)であっても、無溶媒和物であってもよく、いずれも化合物(I)に包含される。
同位元素(例、3H、14C、35S、125Iなど)などで標識された化合物も、化合物(I)に包含される。さらに、1Hを2H(D)に変換した重水素変換体も、化合物(I)に包含される。 When compound (I) has an isomer such as an optical isomer, a stereoisomer, a positional isomer, a rotational isomer, etc., any one of the isomers and a mixture are encompassed in compound (I). For example, when compound (I) has an optical isomer, the optical isomer resolved from the racemate is also encompassed in compound (I). Each of these isomers can be obtained as a single product by a known synthesis method or separation method (concentration, solvent extraction, column chromatography, recrystallization, etc.).
Compound (I) may be a crystal, and it is included in compound (I) regardless of whether the crystal form is a single crystal form or a mixture of crystal forms. The crystal can be produced by crystallization by applying a crystallization method known per se.
Compound (I) may be a solvate (such as a hydrate) or a non-solvate, and both are encompassed in compound (I).
Compounds labeled with isotopes (eg, 3 H, 14 C, 35 S, 125 I, etc.) are also encompassed in compound (I). Further, a deuterium converter obtained by converting 1 H into 2 H (D) is also encompassed in compound (I).
化合物(I)またはそのプロドラッグ(以下、これらをまとめて本発明化合物と称する場合がある)は、哺乳動物(例えば、マウス、ラット、ハムスター、ウサギ、ネコ、イヌ、ウシ、ヒツジ、サル、ヒト)に対して、優れたFAAH阻害活性を有しているので、FAAHが関与する病態または疾患の予防・治療剤として有用である。
Compound (I) or a prodrug thereof (hereinafter sometimes collectively referred to as the compound of the present invention) is a mammal (eg, mouse, rat, hamster, rabbit, cat, dog, cow, sheep, monkey, human) ), It has an excellent FAAH inhibitory activity, and is therefore useful as a prophylactic / therapeutic agent for a disease state or disease involving FAAH.
下記の実施例において示される通り、FAAH阻害活性を有する本発明化合物の投与により疼痛モデルにおける閾値が低下する。したがって、本発明化合物は疼痛、例えば、変形性関節症および関節リウマチ炎に伴う炎症性疼痛ならびに糖尿病性有痛性神経障害に伴う疼痛、帯状疱疹後疼痛、三叉神経痛などの神経因性疼痛の予防および治療に有用である。
さらに、本発明化合物がその予防、治療に有用であると考えられる疾患として、脳・神経細胞の障害に起因する脳血管障害、頭部外傷または脊髄損傷に際しての脳・神経細胞保護効果、心停止後蘇生時の脳障害、脳手術前および後の脳機能低下、低酸素症、低血糖、脳または脊髄の外傷、薬物中毒、ガス中毒、糖尿病、抗腫瘍剤投与、アルコール等による神経系の障害、ハンチントン舞踏病、プリオン病、筋萎縮性側索硬化症、脊髄小脳変性症、摂食障害、肥満、頻尿、尿失禁、リウマチ、変形性関節炎、間質性膀胱炎、クローン病、大腸炎、結腸炎、結腸癌、大腸癌、避妊あるいはAIDSが挙げられるが、これらに限定されない。
本発明化合物は、上記の公知の知見から、更に、抗癌剤による悪心、吐き気もしくは嘔吐;癌もしくは感染症(例、AIDSなど)における拒食もしくは悪液質の食欲不振;多発性硬化症による痙攣、疼痛、振戦、眼振もしくは夜尿;慢性疼痛;ハンチントン舞踏病;ツレット症候群;レボトバ惹起運動障害;運動機能障害;喘息;緑内障;アレルギー;炎症;癲癇;自己免疫疾患;下痢;肥満;睡眠障害;うつ;不安;精神疾患;クローン病;アルツハイマー病;間質性膀胱炎;AIDS;大腸炎;結腸炎;結腸癌;直腸癌;高トリグセリド血症;高脂血症;糖尿病;動脈硬化症;およびパーキンソン病の予防・治療剤あるいは避妊薬としても有用である。 As shown in the Examples below, administration of a compound of the present invention having FAAH inhibitory activity lowers the threshold in a pain model. Accordingly, the compound of the present invention prevents pain, for example, inflammatory pain associated with osteoarthritis and rheumatoid arthritis, and pain associated with diabetic painful neuropathy, postherpetic pain, trigeminal neuralgia and other neuropathic pain. And useful for treatment.
Further, as the diseases that the compounds of the present invention are considered to be useful for their prevention and treatment, cerebrovascular disorders caused by brain / nerve cell disorders, brain / nerve cell protective effects upon head injury or spinal cord injury, cardiac arrest Brain damage during post-resuscitation, brain function decline before and after brain surgery, hypoxia, hypoglycemia, brain or spinal cord trauma, drug poisoning, gas poisoning, diabetes, administration of antitumor agents, alcohol damage, etc. , Huntington's disease, prion disease, amyotrophic lateral sclerosis, spinocerebellar degeneration, eating disorder, obesity, frequent urination, urinary incontinence, rheumatism, osteoarthritis, interstitial cystitis, Crohn's disease, colitis , Colitis, colon cancer, colon cancer, contraception or AIDS, but is not limited thereto.
Based on the above-mentioned known findings, the compound of the present invention further comprises nausea, nausea or vomiting due to anticancer agents; anorexia or anorexia of cachexia in cancer or infectious diseases (eg, AIDS); convulsions and pain due to multiple sclerosis , Tremor, nystagmus or nocturnal urine; chronic pain; Huntington's chorea; Tourette syndrome; levotoba-induced movement disorder; Depression; anxiety; psychiatric disease; Crohn's disease; Alzheimer's disease; interstitial cystitis; AIDS; colitis; colitis; colon cancer; rectal cancer; hypertriglyceridemia; It is also useful as a preventive / therapeutic agent or contraceptive for Parkinson's disease.
さらに、本発明化合物がその予防、治療に有用であると考えられる疾患として、脳・神経細胞の障害に起因する脳血管障害、頭部外傷または脊髄損傷に際しての脳・神経細胞保護効果、心停止後蘇生時の脳障害、脳手術前および後の脳機能低下、低酸素症、低血糖、脳または脊髄の外傷、薬物中毒、ガス中毒、糖尿病、抗腫瘍剤投与、アルコール等による神経系の障害、ハンチントン舞踏病、プリオン病、筋萎縮性側索硬化症、脊髄小脳変性症、摂食障害、肥満、頻尿、尿失禁、リウマチ、変形性関節炎、間質性膀胱炎、クローン病、大腸炎、結腸炎、結腸癌、大腸癌、避妊あるいはAIDSが挙げられるが、これらに限定されない。
本発明化合物は、上記の公知の知見から、更に、抗癌剤による悪心、吐き気もしくは嘔吐;癌もしくは感染症(例、AIDSなど)における拒食もしくは悪液質の食欲不振;多発性硬化症による痙攣、疼痛、振戦、眼振もしくは夜尿;慢性疼痛;ハンチントン舞踏病;ツレット症候群;レボトバ惹起運動障害;運動機能障害;喘息;緑内障;アレルギー;炎症;癲癇;自己免疫疾患;下痢;肥満;睡眠障害;うつ;不安;精神疾患;クローン病;アルツハイマー病;間質性膀胱炎;AIDS;大腸炎;結腸炎;結腸癌;直腸癌;高トリグセリド血症;高脂血症;糖尿病;動脈硬化症;およびパーキンソン病の予防・治療剤あるいは避妊薬としても有用である。 As shown in the Examples below, administration of a compound of the present invention having FAAH inhibitory activity lowers the threshold in a pain model. Accordingly, the compound of the present invention prevents pain, for example, inflammatory pain associated with osteoarthritis and rheumatoid arthritis, and pain associated with diabetic painful neuropathy, postherpetic pain, trigeminal neuralgia and other neuropathic pain. And useful for treatment.
Further, as the diseases that the compounds of the present invention are considered to be useful for their prevention and treatment, cerebrovascular disorders caused by brain / nerve cell disorders, brain / nerve cell protective effects upon head injury or spinal cord injury, cardiac arrest Brain damage during post-resuscitation, brain function decline before and after brain surgery, hypoxia, hypoglycemia, brain or spinal cord trauma, drug poisoning, gas poisoning, diabetes, administration of antitumor agents, alcohol damage, etc. , Huntington's disease, prion disease, amyotrophic lateral sclerosis, spinocerebellar degeneration, eating disorder, obesity, frequent urination, urinary incontinence, rheumatism, osteoarthritis, interstitial cystitis, Crohn's disease, colitis , Colitis, colon cancer, colon cancer, contraception or AIDS, but is not limited thereto.
Based on the above-mentioned known findings, the compound of the present invention further comprises nausea, nausea or vomiting due to anticancer agents; anorexia or anorexia of cachexia in cancer or infectious diseases (eg, AIDS); convulsions and pain due to multiple sclerosis , Tremor, nystagmus or nocturnal urine; chronic pain; Huntington's chorea; Tourette syndrome; levotoba-induced movement disorder; Depression; anxiety; psychiatric disease; Crohn's disease; Alzheimer's disease; interstitial cystitis; AIDS; colitis; colitis; colon cancer; rectal cancer; hypertriglyceridemia; It is also useful as a preventive / therapeutic agent or contraceptive for Parkinson's disease.
また、FAAHは、内因性の睡眠物質であるオレアマイドを加水分解する酵素であり、このためFAAH阻害薬は、オレアマイドの分解を抑制することにより睡眠を誘導する。したがって、本発明化合物は、睡眠障害、例えば、内在因性睡眠障害(例、精神生理性不眠)、外在因性睡眠障害、および概日リズム障害(例、時間帯域変化症候群(時差ボケ)、交代勤務睡眠障害、不規則型睡眠覚醒パターン、睡眠相後退症候群、睡眠相前進症候群、非24時間睡眠覚醒)等の睡眠異常;睡眠時随伴症;内科または精神科障害(例、慢性閉塞性肺疾患、アルツハイマー病、パーキンソン病、脳血管性痴呆、統合失調症、うつ病、不安神経症)に伴う睡眠障害の予防または治療剤として有用である。
In addition, FAAH is an enzyme that hydrolyzes oleamide, which is an endogenous sleep substance. Therefore, FAAH inhibitors induce sleep by suppressing the degradation of oleamide. Accordingly, the compounds of the present invention are useful for sleep disorders such as endogenous sleep disorders (eg, psychophysiological insomnia), extrinsic sleep disorders, and circadian rhythm disorders (eg, time zone change syndrome (time difference blur)), Sleep abnormalities such as shift work sleep disorder, irregular sleep awakening pattern, sleep phase regression syndrome, sleep phase advance syndrome, non-24-hour sleep awakening); sleep-related complications; internal or psychiatric disorders (eg, chronic obstructive lungs) It is useful as a preventive or therapeutic agent for sleep disorders associated with diseases, Alzheimer's disease, Parkinson's disease, cerebrovascular dementia, schizophrenia, depression, anxiety).
対象の化合物はさらに、本発明のための方法、本明細書に記載される疾病、障害及び状態の危険性の予防、治療、調節、寛解又は低下のための方法において有用である。本発明の組成物中の活性成分の用量は変動し得るが、しかしながら、前記活性成分の量が、適切な剤形を得るようになっていることが必要である。前記活性成分は、最適な薬学的効能を提供する用量でこのような治療を要する患者(動物及びヒト)へ投与され得る。選択された用量は、望まれる治療効果、投与の経路、治療の期間に依存する。投与量は、疾病の性質及び重度、患者の体重、患者によって後で遂行される特別食、同時に実施する投薬、及び当業者が認識する他の因子に依存して、患者によって変動する。一般的に、FAAHの効果的な阻害作用を得るために、毎日、0.0001mg/体重kgと10mg/体重kgとの間の用量レベルが、患者、例えばヒト及び高齢者へ投与される。単回投与又は複数投与で投与され得る用量の範囲は一般的に、患者あたり1日あたり約0.5mgないし1.0gである。好ましくは、投与量範囲は、患者あたり1日あたり約0.5mgないし500mg、より好ましくは患者あたり1日あたり約0.5mgないし200mg、さらにより好ましくは患者あたり1日あたり約5mgないし50mgである。本発明の医薬組成物は、好ましくは約0.5mgないし500mgの活性成分を含む、より好ましくは約1mgないし250mgの活性成分を含む固体剤形中で提供され得る。前記医薬組成物は、好ましくは約1mg、5mg、10mg、25mg、50mg、100mg、200mg又は250mgの活性成分を含む固体剤形中に提供される。経口投与の場合、前記組成物は好ましくは、活性成分1.0ないし1000mg、特に、治療すべき患者への用量の対症調整のために、活性成分1、5、10、15、20、25、50、75、100、150、200、250、300、400、500、600、750、800、900及び1000mgを含有する錠剤の形態で提供される。前記化合物は、1日あたり1ないし4回の、好ましくは1日あたり1回又は2回の投与計画で投与され得る。
The subject compounds are further useful in methods for the present invention, methods for prevention, treatment, modulation, amelioration or reduction of the risks of diseases, disorders and conditions described herein. The dosage of active ingredient in the compositions of the invention can vary, however, it is necessary that the amount of active ingredient is such that a suitable dosage form is obtained. The active ingredient can be administered to patients (animals and humans) in need of such treatment at dosages that provide optimal pharmaceutical efficacy. The selected dose depends on the desired therapeutic effect, the route of administration and the duration of treatment. Dosages will vary from patient to patient depending on the nature and severity of the disease, the patient's weight, the special diet performed later by the patient, the medications performed simultaneously, and other factors recognized by those skilled in the art. Generally, daily dose levels between 0.0001 mg / kg body weight and 10 mg / kg body weight are administered to patients, such as humans and the elderly, in order to obtain an effective inhibitory effect of FAAH. The range of doses that can be administered in a single dose or multiple doses is generally about 0.5 mg to 1.0 g per patient per day. Preferably, the dosage range is about 0.5 mg to 500 mg per patient per day, more preferably about 0.5 mg to 200 mg per patient per day, even more preferably about 5 mg to 50 mg per patient per day. . The pharmaceutical composition of the present invention may be provided in a solid dosage form preferably containing from about 0.5 mg to 500 mg of the active ingredient, more preferably from about 1 mg to 250 mg of the active ingredient. The pharmaceutical composition is preferably provided in a solid dosage form comprising about 1 mg, 5 mg, 10 mg, 25 mg, 50 mg, 100 mg, 200 mg or 250 mg of the active ingredient. For oral administration, the composition is preferably 1.0 to 1000 mg of active ingredient, especially active ingredient 1, 5, 10, 15, 20, 25, for symptomatic adjustment of the dose to the patient to be treated. Provided in the form of tablets containing 50, 75, 100, 150, 200, 250, 300, 400, 500, 600, 750, 800, 900 and 1000 mg. The compounds may be administered on a regimen of 1 to 4 times per day, preferably once or twice per day.
化合物(I)またはそのプロドラッグは、毒性(例、急性毒性、慢性毒性、遺伝毒性、生殖毒性、心毒性、薬物相互作用、癌原性)が低く、そのまま、または薬理学的に許容し得る担体等と混合して医薬組成物とすることにより、哺乳動物(例、ヒト、マウス、ラット、ウサギ、イヌ、ネコ、ウシ、ウマ、ブタ、サル)に対して、後述する各種疾患の予防・治療剤として用いることができる。
Compound (I) or a prodrug thereof has low toxicity (eg, acute toxicity, chronic toxicity, genotoxicity, reproductive toxicity, cardiotoxicity, drug interaction, carcinogenicity), and can be used as it is or pharmacologically. By mixing with a carrier or the like to obtain a pharmaceutical composition, it is possible to prevent or prevent various diseases described below for mammals (eg, humans, mice, rats, rabbits, dogs, cats, cows, horses, pigs, monkeys). It can be used as a therapeutic agent.
前記医薬組成物の剤形としては、例えば、錠剤(糖衣錠、フィルムコーティング錠、舌下錠、口腔内崩壊錠を含む)、カプセル剤(ソフトカプセル、マイクロカプセルを含む)、顆粒剤、散剤、トローチ剤、シロップ剤、乳剤、懸濁剤、フィルム剤(例、口腔内崩壊フィルム)等の経口剤;および注射剤(例、皮下注射剤、静脈内注射剤、筋肉内注射剤、腹腔内注射剤、点滴剤)、外用剤(例、経皮製剤、軟膏剤)、坐剤(例、直腸坐剤、膣坐剤)、ペレット、経鼻剤、経肺剤(吸入剤)、点眼剤等の非経口剤が挙げられる。これらはそれぞれ経口的あるいは非経口的(例、局所、直腸、静脈投与)に安全に投与できる。
これらの製剤は、速放性製剤または徐放性製剤等の放出制御製剤(例、徐放性マイクロカプセル)であってもよい。 Examples of the dosage form of the pharmaceutical composition include tablets (including sugar-coated tablets, film-coated tablets, sublingual tablets, orally disintegrating tablets), capsules (including soft capsules and microcapsules), granules, powders, and lozenges. Oral preparations such as syrup, emulsion, suspension, film (eg, orally disintegrating film); and injection (eg, subcutaneous injection, intravenous injection, intramuscular injection, intraperitoneal injection, Intravenous preparations, external preparations (eg, transdermal preparations, ointments), suppositories (eg, rectal suppositories, vaginal suppositories), pellets, nasal preparations, pulmonary preparations (inhalants), eye drops, etc. Oral preparations are mentioned. These can be safely administered orally or parenterally (eg, topical, rectal, intravenous administration).
These preparations may be controlled-release preparations such as immediate-release preparations or sustained-release preparations (eg, sustained-release microcapsules).
これらの製剤は、速放性製剤または徐放性製剤等の放出制御製剤(例、徐放性マイクロカプセル)であってもよい。 Examples of the dosage form of the pharmaceutical composition include tablets (including sugar-coated tablets, film-coated tablets, sublingual tablets, orally disintegrating tablets), capsules (including soft capsules and microcapsules), granules, powders, and lozenges. Oral preparations such as syrup, emulsion, suspension, film (eg, orally disintegrating film); and injection (eg, subcutaneous injection, intravenous injection, intramuscular injection, intraperitoneal injection, Intravenous preparations, external preparations (eg, transdermal preparations, ointments), suppositories (eg, rectal suppositories, vaginal suppositories), pellets, nasal preparations, pulmonary preparations (inhalants), eye drops, etc. Oral preparations are mentioned. These can be safely administered orally or parenterally (eg, topical, rectal, intravenous administration).
These preparations may be controlled-release preparations such as immediate-release preparations or sustained-release preparations (eg, sustained-release microcapsules).
医薬組成物は、製剤技術分野において慣用の方法、例えば、日本薬局方に記載の方法等により製造することができる。
なお、医薬組成物中の本発明化合物の含量は、剤形、本発明化合物の投与量等により異なるが、例えば、約0.1~100重量%である。 The pharmaceutical composition can be produced by a method commonly used in the field of pharmaceutical technology, for example, a method described in the Japanese Pharmacopoeia.
The content of the compound of the present invention in the pharmaceutical composition varies depending on the dosage form, the dose of the compound of the present invention, etc., but is, for example, about 0.1 to 100% by weight.
なお、医薬組成物中の本発明化合物の含量は、剤形、本発明化合物の投与量等により異なるが、例えば、約0.1~100重量%である。 The pharmaceutical composition can be produced by a method commonly used in the field of pharmaceutical technology, for example, a method described in the Japanese Pharmacopoeia.
The content of the compound of the present invention in the pharmaceutical composition varies depending on the dosage form, the dose of the compound of the present invention, etc., but is, for example, about 0.1 to 100% by weight.
ここにおいて、薬理学的に許容し得る担体としては、製剤素材として慣用の各種有機あるいは無機担体物質が用いられ、固形製剤における賦形剤、滑沢剤、結合剤、崩壊剤;液状製剤における溶剤、溶解補助剤、懸濁化剤、等張化剤、緩衝剤、無痛化剤等として配合される。また必要に応じて、防腐剤、抗酸化剤、着色剤、甘味剤等の製剤添加物を用いることもできる。
Here, as the pharmacologically acceptable carrier, various organic or inorganic carrier substances commonly used as pharmaceutical materials are used, and excipients, lubricants, binders, disintegrants in solid preparations; solvents in liquid preparations , Solubilizing agents, suspending agents, isotonic agents, buffers, soothing agents and the like. If necessary, preparation additives such as preservatives, antioxidants, colorants, sweeteners and the like can also be used.
賦形剤の好適な例としては、乳糖、白糖、D-マンニトール、D-ソルビトール、デンプン、α化デンプン、デキストリン、結晶セルロース、低置換度ヒドロキシプロピルセルロース、カルボキシメチルセルロースナトリウム、アラビアゴム、プルラン、軽質無水ケイ酸、合成ケイ酸アルミニウム、メタケイ酸アルミン酸マグネシウム等が挙げられる。
Preferable examples of the excipient include lactose, sucrose, D-mannitol, D-sorbitol, starch, pregelatinized starch, dextrin, crystalline cellulose, low-substituted hydroxypropylcellulose, sodium carboxymethylcellulose, gum arabic, pullulan, light Anhydrous silicic acid, synthetic aluminum silicate, magnesium magnesium metasilicate, etc. are mentioned.
滑沢剤の好適な例としては、ステアリン酸マグネシウム、ステアリン酸カルシウム、タルク、コロイドシリカ等が挙げられる。
結合剤の好適な例としては、α化デンプン、ショ糖、ゼラチン、アラビアゴム、メチルセルロース、カルボキシメチルセルロース、カルボキシメチルセルロースナトリウム、結晶セルロース、白糖、D-マンニトール、トレハロース、デキストリン、プルラン、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、ポリビニルピロリドン等が挙げられる。 Preferable examples of the lubricant include magnesium stearate, calcium stearate, talc, colloidal silica and the like.
Preferred examples of the binder include pregelatinized starch, sucrose, gelatin, gum arabic, methyl cellulose, carboxymethyl cellulose, sodium carboxymethyl cellulose, crystalline cellulose, sucrose, D-mannitol, trehalose, dextrin, pullulan, hydroxypropyl cellulose, hydroxy Examples thereof include propylmethylcellulose and polyvinylpyrrolidone.
結合剤の好適な例としては、α化デンプン、ショ糖、ゼラチン、アラビアゴム、メチルセルロース、カルボキシメチルセルロース、カルボキシメチルセルロースナトリウム、結晶セルロース、白糖、D-マンニトール、トレハロース、デキストリン、プルラン、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、ポリビニルピロリドン等が挙げられる。 Preferable examples of the lubricant include magnesium stearate, calcium stearate, talc, colloidal silica and the like.
Preferred examples of the binder include pregelatinized starch, sucrose, gelatin, gum arabic, methyl cellulose, carboxymethyl cellulose, sodium carboxymethyl cellulose, crystalline cellulose, sucrose, D-mannitol, trehalose, dextrin, pullulan, hydroxypropyl cellulose, hydroxy Examples thereof include propylmethylcellulose and polyvinylpyrrolidone.
崩壊剤の好適な例としては、乳糖、白糖、デンプン、カルボキシメチルセルロース、カルボキシメチルセルロースカルシウム、クロスカルメロースナトリウム、カルボキシメチルスターチナトリウム、軽質無水ケイ酸、低置換度ヒドロキシプロピルセルロース等が挙げられる。
Preferable examples of the disintegrant include lactose, sucrose, starch, carboxymethyl cellulose, carboxymethyl cellulose calcium, croscarmellose sodium, carboxymethyl starch sodium, light anhydrous silicic acid, low substituted hydroxypropyl cellulose and the like.
溶剤の好適な例としては、注射用水、生理的食塩水、リンゲル液、アルコール、プロピレングリコール、ポリエチレングリコール、ゴマ油、トウモロコシ油、オリーブ油、綿実油等が挙げられる。
溶解補助剤の好適な例としては、ポリエチレングリコール、プロピレングリコール、D-マンニトール、トレハロース、安息香酸ベンジル、エタノール、トリスアミノメタン、コレステロール、トリエタノールアミン、炭酸ナトリウム、クエン酸ナトリウム、サリチル酸ナトリウム、酢酸ナトリウム等が挙げられる。 Preferable examples of the solvent include water for injection, physiological saline, Ringer's solution, alcohol, propylene glycol, polyethylene glycol, sesame oil, corn oil, olive oil, cottonseed oil and the like.
Preferable examples of the solubilizer include polyethylene glycol, propylene glycol, D-mannitol, trehalose, benzyl benzoate, ethanol, trisaminomethane, cholesterol, triethanolamine, sodium carbonate, sodium citrate, sodium salicylate, sodium acetate. Etc.
溶解補助剤の好適な例としては、ポリエチレングリコール、プロピレングリコール、D-マンニトール、トレハロース、安息香酸ベンジル、エタノール、トリスアミノメタン、コレステロール、トリエタノールアミン、炭酸ナトリウム、クエン酸ナトリウム、サリチル酸ナトリウム、酢酸ナトリウム等が挙げられる。 Preferable examples of the solvent include water for injection, physiological saline, Ringer's solution, alcohol, propylene glycol, polyethylene glycol, sesame oil, corn oil, olive oil, cottonseed oil and the like.
Preferable examples of the solubilizer include polyethylene glycol, propylene glycol, D-mannitol, trehalose, benzyl benzoate, ethanol, trisaminomethane, cholesterol, triethanolamine, sodium carbonate, sodium citrate, sodium salicylate, sodium acetate. Etc.
懸濁化剤の好適な例としては、ステアリルトリエタノールアミン、ラウリル硫酸ナトリウム、ラウリルアミノプロピオン酸、レシチン、塩化ベンザルコニウム、塩化ベンゼトニウム、モノステアリン酸グリセリン等の界面活性剤;例えば、ポリビニルアルコール、ポリビニルピロリドン、カルボキシメチルセルロースナトリウム、メチルセルロース、ヒドロキシメチルセルロース、ヒドロキシエチルセルロース、ヒドロキシプロピルセルロース等の親水性高分子;ポリソルベート類、ポリオキシエチレン硬化ヒマシ油等が挙げられる。
Suitable examples of the suspending agent include surfactants such as stearyltriethanolamine, sodium lauryl sulfate, laurylaminopropionic acid, lecithin, benzalkonium chloride, benzethonium chloride, glyceryl monostearate; for example, polyvinyl alcohol, Examples include hydrophilic polymers such as polyvinylpyrrolidone, sodium carboxymethylcellulose, methylcellulose, hydroxymethylcellulose, hydroxyethylcellulose, hydroxypropylcellulose; polysorbates, polyoxyethylene hydrogenated castor oil, and the like.
等張化剤の好適な例としては、塩化ナトリウム、グリセリン、D-マンニトール、D-ソルビトール、ブドウ糖等が挙げられる。
緩衝剤の好適な例としては、リン酸塩、酢酸塩、炭酸塩、クエン酸塩等の緩衝液等が挙げられる。
無痛化剤の好適な例としては、ベンジルアルコール等が挙げられる。 Preferable examples of the isotonizing agent include sodium chloride, glycerin, D-mannitol, D-sorbitol, glucose and the like.
Preferable examples of the buffer include buffer solutions such as phosphate, acetate, carbonate, citrate and the like.
Preferable examples of the soothing agent include benzyl alcohol.
緩衝剤の好適な例としては、リン酸塩、酢酸塩、炭酸塩、クエン酸塩等の緩衝液等が挙げられる。
無痛化剤の好適な例としては、ベンジルアルコール等が挙げられる。 Preferable examples of the isotonizing agent include sodium chloride, glycerin, D-mannitol, D-sorbitol, glucose and the like.
Preferable examples of the buffer include buffer solutions such as phosphate, acetate, carbonate, citrate and the like.
Preferable examples of the soothing agent include benzyl alcohol.
防腐剤の好適な例としては、パラオキシ安息香酸エステル類、クロロブタノール、ベンジルアルコール、フェネチルアルコール、デヒドロ酢酸、ソルビン酸等が挙げられる。
抗酸化剤の好適な例としては、亜硫酸塩、アスコルビン酸塩等が挙げられる。
着色剤の好適な例としては、水溶性食用タール色素(例、食用赤色2号および3号、食用黄色4号および5号、食用青色1号および2号等の食用色素)、水不溶性レーキ色素(例、前記水溶性食用タール色素のアルミニウム塩)、天然色素(例、β-カロチン、クロロフィル、ベンガラ)等が挙げられる。
甘味剤の好適な例としては、サッカリンナトリウム、グリチルリチン酸二カリウム、アスパルテーム、ステビア等が挙げられる。 Preferable examples of the preservative include p-hydroxybenzoates, chlorobutanol, benzyl alcohol, phenethyl alcohol, dehydroacetic acid, sorbic acid and the like.
Preferable examples of the antioxidant include sulfite and ascorbate.
Suitable examples of the colorant include water-soluble edible tar dyes (eg, edible dyes such as edible red Nos. 2 and 3, edible yellow Nos. 4 and 5, edible blue Nos. 1 and 2), water-insoluble lake dyes (Eg, the aluminum salt of the water-soluble edible tar dye), natural dyes (eg, β-carotene, chlorophyll, bengara) and the like.
Preferable examples of the sweetening agent include saccharin sodium, dipotassium glycyrrhizinate, aspartame, stevia and the like.
抗酸化剤の好適な例としては、亜硫酸塩、アスコルビン酸塩等が挙げられる。
着色剤の好適な例としては、水溶性食用タール色素(例、食用赤色2号および3号、食用黄色4号および5号、食用青色1号および2号等の食用色素)、水不溶性レーキ色素(例、前記水溶性食用タール色素のアルミニウム塩)、天然色素(例、β-カロチン、クロロフィル、ベンガラ)等が挙げられる。
甘味剤の好適な例としては、サッカリンナトリウム、グリチルリチン酸二カリウム、アスパルテーム、ステビア等が挙げられる。 Preferable examples of the preservative include p-hydroxybenzoates, chlorobutanol, benzyl alcohol, phenethyl alcohol, dehydroacetic acid, sorbic acid and the like.
Preferable examples of the antioxidant include sulfite and ascorbate.
Suitable examples of the colorant include water-soluble edible tar dyes (eg, edible dyes such as edible red Nos. 2 and 3, edible yellow Nos. 4 and 5, edible blue Nos. 1 and 2), water-insoluble lake dyes (Eg, the aluminum salt of the water-soluble edible tar dye), natural dyes (eg, β-carotene, chlorophyll, bengara) and the like.
Preferable examples of the sweetening agent include saccharin sodium, dipotassium glycyrrhizinate, aspartame, stevia and the like.
本発明の化合物は、本発明の化合物又は他の薬物が有用性を有し得る疾病又は状態の危険性の治療、予防、寛解、又は低下において、1つ又はそれ以上の他の薬物との組み合わせで使用され得、その場合、薬物の組み合わせは互いに、いずれの薬物単独よりも安全であるかまたはより効果的である。このような他の薬物は、当該薬物に関して一般的に使用される経路及び量で、本発明の化合物と同時に又は連続して投与され得る。本発明の化合物が1つ又はそれ以上の他の薬物と同時に使用される場合には、このような他の薬物及び本発明の化合物を含有する単位剤形中の医薬組成物が好ましい。しかしながら、組み合わせ療法には、本発明の化合物及び1つ以上の他の薬物が異別の重複スケジュールで投与される療法も含まれ得る。1つ又はそれ以上の他の活性成分との組み合わせで使用される場合には、本発明の化合物及び前記他の活性成分が、各々単独で使用される場合よりも少量の用量で使用され得ることも想定される。従って、本発明の医薬組成物には、本発明の化合物に加え、1つ又はそれ以上の他の活性成分を含有するものが含まれる。上述の組み合わせには、本発明の化合物と、1つの他の活性化合物だけでなく、2つ又はそれ以上の他の活性化合物との組み合わせが含まれる。
The compounds of the present invention may be combined with one or more other drugs in the treatment, prevention, amelioration, or reduction of the risk of a disease or condition for which the compounds of the present invention or other drugs may have utility In which case the drug combinations are safer or more effective than each other drug alone. Such other drugs may be administered simultaneously or sequentially with the compounds and compounds of the present invention, in the routes and amounts commonly used for such drugs. When a compound of the present invention is used contemporaneously with one or more other drugs, a pharmaceutical composition in unit dosage form containing such other drugs and the compound of the present invention is preferred. However, combination therapy may also include therapies in which the compound of the invention and one or more other drugs are administered on different overlapping schedules. When used in combination with one or more other active ingredients, the compounds of the present invention and the other active ingredients can each be used in a smaller dose than when used alone. Is also envisaged. Accordingly, the pharmaceutical compositions of the present invention include those that contain one or more other active ingredients, in addition to a compound of the present invention. The above combinations include combinations of a compound of the present invention not only with one other active compound, but also with two or more other active compounds.
同様に、本発明の化合物は、本発明の化合物が有用である疾病又は状態の危険性の予防、治療、調節、寛解、又は低下において使用される他の薬物との組み合わせにおいて使用される。このような他の薬物は、当該薬物に対して一般的に使用される経路及び量で、本発明の化合物と同時に又は連続して投与され得る。本発明の化合物が1つ又はそれ以上の他の薬物と同時に使用される場合には、本発明の化合物に加えて、このような他の薬物を含有する医薬組成物が好ましい。従って、本発明の医薬組成物には、本発明の化合物に加え、1つ又はそれ以上の他の活性成分も含有するものが含まれる。
Similarly, the compounds of the present invention are used in combination with other drugs used in the prevention, treatment, modulation, amelioration, or reduction of the risk of diseases or conditions for which compounds of the present invention are useful. Such other drugs may be administered concurrently or sequentially with the compounds and compounds of the present invention by the routes and amounts generally used for such drugs. When a compound of the present invention is used contemporaneously with one or more other drugs, a pharmaceutical composition containing such other drugs in addition to the compound of the present invention is preferred. Accordingly, the pharmaceutical compositions of the present invention include those that also contain one or more other active ingredients, in addition to a compound of the present invention.
第二の活性成分に対する本発明の化合物の重量比は変動し得、各成分の効果的な用量に依存する。一般的に、各々の効果的な用量が使用される。従って、例えば、本発明の化合物が別の薬物と組み合わされるとき、他の薬物に対する本発明の化合物の重量比は一般的に、約1000:1ないし約1:1000、好ましくは約200:1ないし約1:200の範囲である。本発明の化合物と他の活性成分との組み合わせも一般的に、上述の範囲内であるが、各場合において、各活性成分の効果的な用量が使用されるべきである。このような組み合わせにおいて、本発明の化合物及び他の活性薬物は、個別に又は共に投与され得る。さらに、1つの要素の投与は、他の薬物の投与前、投与と同時、投与後であり得る。
The weight ratio of the compound of the present invention to the second active ingredient may be varied and will depend upon the effective dose of each ingredient. In general, each effective dose is used. Thus, for example, when a compound of the present invention is combined with another drug, the weight ratio of the compound of the present invention to the other drug is generally about 1000: 1 to about 1: 1000, preferably about 200: 1 to The range is about 1: 200. Combinations of a compound of the present invention and other active ingredients will generally also be within the aforementioned range, but in each case, an effective dose of each active ingredient should be used. In such combinations, the compounds of the present invention and other active drugs can be administered separately or together. Furthermore, the administration of one element can be before, simultaneously with, or after administration of the other drug.
本発明化合物と併用し得る薬物(以下、併用薬物と略記する場合がある)としては、例えば、非ステロイド性抗炎症薬(例、メロキシカム、テオキシカム、インドメタシン、イブプロフェン、セレコキシブ、ロフェコキシブ、アスピリン、インドメタシン等)、疾患修飾性抗リウマチ薬(DMARDs)、解熱鎮痛剤(アセトアニリド、アセトアミノフェン、フェナセチン等)、ステロイド性抗炎症薬(ヒドロコルチゾン、プレドニゾロン、メチルプレドニゾロン、ベタメサゾン、デキサメサゾンなど)、麻薬性鎮痛薬(モルヒネ、フェンタニール、リン酸コデイン、ペチジン、オキシコドン等)、非麻薬性鎮痛薬(トラマドール等)、局所麻酔薬(リドカイン等)、抗痙攣薬(ガバペンチン、ブピバカイン、カルバマゼピン、フェニトイン等)、抗不整脈薬(プロカイン等)、抗サイトカイン薬(TNF阻害薬、MAPキナーゼ阻害薬等)、アルドース還元酵素阻害薬(キネダック等)、カンナビノイド作動薬(テトラヒドロカンナビノール等)など挙げられる。さらに併用薬物としては、抗認知症治療薬、例えば、アセチルコリンエステラーゼ阻害剤(例、ドネペジル、リバスチグミン、ガランタミン、ザナペジル等)、βアミロイド蛋白産生、分泌、蓄積、凝集および/または沈着抑制剤[βセクレターゼ阻害剤(例、WO98/38156記載の化合物、WO02/2505、WO02/2506、WO02/2512記載の化合物、OM99-2(WO01/00663))、γセクレターゼ阻害剤(LY-450139等)、γセクレターゼモジュレーター(E2012等)、βアミロイド蛋白凝集阻害剤(例、PTI-00703、ALZHEMED(NC-531)、PPI-368(特表平11-514333)、PPI-558(特表平2001-500852)、SKF-74652(Biochem. J.(1999),340(1),283-289))、βアミロイド抗体、βアミロイドワクチン、βアミロイド分解酵素等]、脳機能賦活薬(例、アニラセタム、ニセルゴリン等)、神経新生・再生促進薬(例、Akt/PKB活性化剤、GSK-3β阻害剤など)、パーキンソン病治療薬[(例、ドーパミン受容体作動薬(L-ドーパ、ブロモクリプテン、パーゴライド、タリペキソール、プラシペキソール、カベルゴリン、アダマンタジン等)、モノアミン酸化酵素(MAO)阻害薬(デプレニル、セルジリン(セレギリン)、レマセミド(remacemide),リルゾール(riluzole)等)、抗コリン剤(例、トリヘキシフェニジル、ビペリデン等)、)、COMT阻害剤(例、エンタカポン等)]、筋萎縮性側索硬化症治療薬(例、リルゾール等、神経栄養因子等)、痴呆の進行に伴う異常行動、徘徊等の治療薬(例、鎮静剤、抗不安剤等)、アポトーシス阻害薬(例、CPI-1189、IDN-6556、CEP-1347等)、神経分化・再生促進剤(レテプリニム(Leteprinim)、キサリプローデン(Xaliproden;SR-57746-A)、 SB-216763等)、抗うつ剤(例、MAO阻害薬、3環性抗うつ薬、選択的セロトニン取り込み阻害薬SSRI、選択的セロトニン・ノルアドレナリン取り込み阻害薬SNRI、トリプル取り込み阻害薬、CRF拮抗薬等)、抗不安薬(例、ベンゾジアゼピン系薬剤等)、睡眠薬(例、ベンゾジアゼピン系、非ベンゾジアゼピン系薬剤、メラトニン作動薬等)、血栓溶解剤(例、ティシュープラスミノーゲンアクチベーター、ウロキナーゼ等)、抗凝固剤(例、アルガトロバン、ワーファリン等)、第10因子阻害剤、トロンボキサン合成酵素阻害剤(例、オザグレル等)、抗酸化剤(例、エダラボン等)、抗浮腫剤(例、グリセロール、マンニトール等)、コレステロール低下薬等の高脂血症治療薬[スタチン系(例、プラバスタチンナトリウム、アトロバスタチン、シンバスタチン、ロスバスタチン等)、フィブラート(例、クロフィブラート等)、スクアレン合成酵阻害剤]、降圧剤(ACE阻害薬、アンジオテンシンII拮抗薬、レニン阻害薬等)、抗糖尿病薬(例、インスリン製剤、スルホニルウレア剤などのインスリン分泌促進剤、PPAR作動薬、PPAR部分作動薬などのインスリン抵抗性改善薬、DPPIV阻害薬等)、抗がん剤(例、白金製剤、5-FU、アントラサイクリン系、分子標的薬、ホルモン療法剤等)、頻尿、尿失禁治療薬(例、ハルンケア、ソリフェナシン等)、過活動膀胱治療薬(例、トルテロジン等)、骨粗鬆症治療薬(例、ビタミンD製剤、PTH、カルシウム受容体拮抗薬、カルシトニン、リセドロネート、アレンドロネート等)等が挙げられる。
Examples of a drug that can be used in combination with the compound of the present invention (hereinafter sometimes abbreviated as a concomitant drug) include, for example, nonsteroidal anti-inflammatory drugs (eg, meloxicam, teoxicam, indomethacin, ibuprofen, celecoxib, rofecoxib, aspirin, indomethacin, etc. ), Disease-modifying anti-rheumatic drugs (DMARDs), antipyretic analgesics (acetanilide, acetaminophen, phenacetin, etc.), steroidal anti-inflammatory drugs (hydrocortisone, prednisolone, methylprednisolone, betamethasone, dexamethasone, etc.), narcotic analgesics ( Morphine, fentanyl, codeine phosphate, pethidine, oxycodone, etc., non-narcotic analgesics (eg, tramadol), local anesthetics (eg, lidocaine), anticonvulsants (gabapentin, bupivacaine, carbamazepine, phenytoin) ), Antiarrhythmic drugs (procaine, etc.), anti-cytokine drugs (TNF inhibitor, MAP kinase inhibitor, etc.), aldose reductase inhibitors (Kinedakku etc.), cannabinoid agonists (tetrahydrocannabinol, etc.) and the like. Further, the concomitant drugs include anti-dementia therapeutic agents such as acetylcholinesterase inhibitors (eg, donepezil, rivastigmine, galantamine, zanapezil, etc.), β amyloid protein production, secretion, accumulation, aggregation and / or deposition inhibitors [β secretase Inhibitors (eg, compounds described in WO98 / 38156, compounds described in WO02 / 2505, WO02 / 2506, WO02 / 2512, OM99-2 (WO01 / 00663)), γ-secretase inhibitors (such as LY-450139), γ-secretase Modulators (E2012 etc.), β amyloid protein aggregation inhibitors (eg, PTI-00703, ALZHEMED (NC-531), PPI-368 (Special Tables Hei 11-514333), PPI-558 (Special Tables Hei 2001-500852), SKF-74652 ( Biochem. J. (1999), 340 (1), 283-289)), β-amyloid antibody, β-amyloid vaccine, β-amyloid degrading enzyme, etc.], brain function stimulating agents (eg, aniracetam, nicergoline, etc.), neurogenesis / Regenerative promoter (eg, Akt / PKB activator, GSK-3β inhibitor, etc.), Parkinson's disease therapeutic agent [(eg, dopamine receptor agonist (L-dopa, bromocriptene, pergolide, taripexole, pripepexol, cabergoline) , Adamantadine, etc.), monoamine oxidase (MAO) inhibitors (deprenyl, sergiline (selegiline), remacemide, riluzole, etc.), anticholinergic agents (eg, trihexyphenidyl, biperiden, etc.)) , COMT inhibitors (eg, entacapone, etc.)], amyotrophic lateral sclerosis drugs (eg, riluzole, etc.), progression of dementia Abnormal behavior associated with cancer, therapeutic agents such as epilepsy (eg, sedatives, anxiolytics, etc.), apoptosis inhibitors (eg, CPI-1189, IDN-6556, CEP-1347, etc.), neuronal differentiation / regeneration promoters (leteprinim) (Leteprinim), xaliproden (SR-57746-A), SB-216763, etc.), antidepressants (eg, MAO inhibitors, tricyclic antidepressants, selective serotonin uptake inhibitors SSRI, selective serotonin Noradrenaline uptake inhibitor SNRI, triple uptake inhibitor, CRF antagonist, etc.), anxiolytics (eg, benzodiazepines, etc.), hypnotics (eg, benzodiazepines, non-benzodiazepines, melatonin agonists, etc.), thrombolytic agents (Eg, tissue plasminogen activator, urokinase, etc.), anticoagulants (eg, argatroban, warfarin, etc.), factor 10 inhibitor, thromboxane Antilipidemic agents (eg, ozagrel, etc.), antioxidants (eg, edaravone, etc.), anti-edema agents (eg, glycerol, mannitol, etc.), antihyperlipidemic drugs such as cholesterol-lowering drugs [statins (eg, Pravastatin sodium, atorvastatin, simvastatin, rosuvastatin, etc.), fibrates (eg, clofibrate, etc.), squalene synthesis fermentation inhibitors], antihypertensives (ACE inhibitors, angiotensin II antagonists, renin inhibitors, etc.), antidiabetics ( Examples, insulin preparations, insulin secretagogues such as sulfonylureas, PPAR agonists, insulin resistance improvers such as PPAR partial agonists, DPPIV inhibitors, etc.), anticancer agents (eg, platinum preparations, 5-FU, Anthracyclines, molecular targeted drugs, hormone therapy drugs, etc., frequent urination, urinary incontinence drugs (eg, Harncare, Sorif) Nashin etc.), overactive bladder therapeutic agent (e.g., tolterodine, etc.), therapeutic agents for osteoporosis (e.g., vitamin D preparation, PTH, calcium receptor antagonist, calcitonin, risedronate, alendronate, etc.) and the like.
本発明化合物と併用薬物とを組み合わせることにより、
(1)本発明化合物または併用薬物を単独で投与する場合に比べて、その投与量を軽減することができる、
(2)本発明化合物と作用機序が異なる併用薬物を選択することにより、治療期間を長く設定することができる、
(3)本発明化合物と作用機序が異なる併用薬物を選択することにより、治療効果の持続を図ることができる、
(4)本発明化合物と併用薬物とを併用することにより、相乗効果が得られる、などの優れた効果を得ることができる。 By combining the compound of the present invention and a concomitant drug,
(1) The dose can be reduced compared to the case where the compound of the present invention or the concomitant drug is administered alone.
(2) By selecting a concomitant drug having a different mechanism of action from the compound of the present invention, the treatment period can be set longer.
(3) By selecting a concomitant drug having a mechanism of action different from that of the compound of the present invention, the therapeutic effect can be sustained.
(4) By using the compound of the present invention and the concomitant drug in combination, excellent effects such as a synergistic effect can be obtained.
(1)本発明化合物または併用薬物を単独で投与する場合に比べて、その投与量を軽減することができる、
(2)本発明化合物と作用機序が異なる併用薬物を選択することにより、治療期間を長く設定することができる、
(3)本発明化合物と作用機序が異なる併用薬物を選択することにより、治療効果の持続を図ることができる、
(4)本発明化合物と併用薬物とを併用することにより、相乗効果が得られる、などの優れた効果を得ることができる。 By combining the compound of the present invention and a concomitant drug,
(1) The dose can be reduced compared to the case where the compound of the present invention or the concomitant drug is administered alone.
(2) By selecting a concomitant drug having a different mechanism of action from the compound of the present invention, the treatment period can be set longer.
(3) By selecting a concomitant drug having a mechanism of action different from that of the compound of the present invention, the therapeutic effect can be sustained.
(4) By using the compound of the present invention and the concomitant drug in combination, excellent effects such as a synergistic effect can be obtained.
以下、本発明化合物と併用薬物とを組み合わせて用いる場合、本発明化合物と併用薬物の投与時期は限定されず、本発明化合物と併用薬物とを、投与対象に対し、同時に投与してもよいし、時間差をおいて投与してもよい。併用薬物の投与量は、臨床上用いられている投与量に準ずればよく、投与対象、投与ルート、疾患、組み合わせなどにより適宜選択することができる。
Hereinafter, when the compound of the present invention and a concomitant drug are used in combination, the administration timing of the compound of the present invention and the concomitant drug is not limited, and the compound of the present invention and the concomitant drug may be administered simultaneously to the administration subject. Alternatively, administration may be performed with a time difference. The dose of the concomitant drug may be determined according to the dose used clinically, and can be appropriately selected depending on the administration subject, administration route, disease, combination and the like.
本発明化合物と併用薬物の投与形態としては、例えば、(1)本発明化合物と併用薬物とを同時に製剤化して得られる単一の製剤の投与、(2)本発明化合物と併用薬物とを別々に製剤化して得られる2種の製剤の同一投与経路での同時投与、(3)本発明化合物と併用薬物とを別々に製剤化して得られる2種の製剤の同一投与経路での時間差をおいての投与、(4)本発明化合物と併用薬物とを別々に製剤化して得られる2種の製剤の異なる投与経路での同時投与、(5)本発明化合物と併用薬物とを別々に製剤化して得られる2種の製剤の異なる投与経路での時間差をおいての投与(例えば、本発明化合物;併用薬物の順序での投与、あるいは逆の順序での投与)などが挙げられる。
As the administration form of the compound of the present invention and the concomitant drug, for example, (1) administration of a single preparation obtained by simultaneously formulating the compound of the present invention and the concomitant drug, and (2) separate administration of the compound of the present invention and the concomitant drug Simultaneous administration of the two preparations obtained by formulation into the same administration route, (3) Time difference in the same administration route of the two preparations obtained by separately formulating the compound of the present invention and the concomitant drug. (4) Simultaneous administration of two preparations obtained by separately formulating the compound of the present invention and a concomitant drug by different administration routes, (5) Formulating the compound of the present invention and the concomitant drug separately Administration of the two types of preparations obtained at different times by different administration routes (for example, the compound of the present invention; administration in the order of concomitant drugs, or administration in the reverse order).
本発明は、更に以下の実施例、製剤例及び実験例によって詳しく説明されるが、これらの例は単なる実施であって、本発明を限定するものではなく、また本発明の範囲を逸脱しない範囲で変化させてもよい。なお、以下の製造法の説明において、原料となる化合物および反応生成物は反応に支障とならない塩を形成していてもよい。
The present invention is further explained in detail by the following examples, formulation examples and experimental examples, which are merely examples and do not limit the present invention and do not depart from the scope of the present invention. It may be changed with. In the following description of the production method, the starting compound and reaction product may form a salt that does not interfere with the reaction.
以下の参考例、実施例中の「室温」は、通常、約10℃~約35℃を示す。
その他の本文中で用いられている略号は下記の意味を示す。
s: シングレット(singlet)
d: ダブレット(doublet)
t: トリプレット(triplet)
q: クァルテット(quartet)
m: マルチプレット(multiplet)
br: ブロード(broad)
J: カップリング定数(coupling constant)
Hz: ヘルツ(Hertz)
CDCl3 : 重クロロホルム
DMSO-d6: 重ジメチルスルホキシド
1H NMR: プロトン核磁気共鳴
(R,R)-Me-BPE: (+)-1,2-ビス((2R,5R)-2,5-ジメチルホスホラノ)エタン
(S,S)-Et-FerroTANE: (-)-1,1’-ビス((2S,4S)-2,4-ジエチルホスホタノ)フェロセン “Room temperature” in the following Reference Examples and Examples usually indicates about 10 ° C. to about 35 ° C.
Other abbreviations used in the text have the following meanings.
s: singlet
d: Doublet
t: triplet
q: quartet
m: multiplet
br: broad
J: coupling constant
Hz: Hertz
CDCl 3 : deuterated chloroform DMSO-d 6 : deuterated dimethyl sulfoxide
1 H NMR: proton nuclear magnetic resonance (R, R) -Me-BPE: (+)-1,2-bis ((2R, 5R) -2,5-dimethylphosphorano) ethane (S, S) -Et -FerroTANE: (-)-1,1'-bis ((2S, 4S) -2,4-diethylphosphotano) ferrocene
その他の本文中で用いられている略号は下記の意味を示す。
s: シングレット(singlet)
d: ダブレット(doublet)
t: トリプレット(triplet)
q: クァルテット(quartet)
m: マルチプレット(multiplet)
br: ブロード(broad)
J: カップリング定数(coupling constant)
Hz: ヘルツ(Hertz)
CDCl3 : 重クロロホルム
DMSO-d6: 重ジメチルスルホキシド
1H NMR: プロトン核磁気共鳴
(R,R)-Me-BPE: (+)-1,2-ビス((2R,5R)-2,5-ジメチルホスホラノ)エタン
(S,S)-Et-FerroTANE: (-)-1,1’-ビス((2S,4S)-2,4-ジエチルホスホタノ)フェロセン “Room temperature” in the following Reference Examples and Examples usually indicates about 10 ° C. to about 35 ° C.
Other abbreviations used in the text have the following meanings.
s: singlet
d: Doublet
t: triplet
q: quartet
m: multiplet
br: broad
J: coupling constant
Hz: Hertz
CDCl 3 : deuterated chloroform DMSO-d 6 : deuterated dimethyl sulfoxide
1 H NMR: proton nuclear magnetic resonance (R, R) -Me-BPE: (+)-1,2-bis ((2R, 5R) -2,5-dimethylphosphorano) ethane (S, S) -Et -FerroTANE: (-)-1,1'-bis ((2S, 4S) -2,4-diethylphosphotano) ferrocene
参考例1
tert-ブチル 4-(4-フェニルピリミジン-2-イル)-3,6-ジヒドロピリジン-1(2H)-カルボキシラート

2-クロロ-4-フェニルピリミジン(6.00g、31.5ミリモル)、tert-ブチル 4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-3,6-ジヒドロピリジン-1(2H)-カルボキシラート(10.7g、34.7ミリモル)、炭酸ナトリウム(6.68g、63.0ミリモル)の1,4-ジオキサン(100ml)/水(25ml)溶液に、テトラキストリフェニルホスフィンパラジウム(1.82g、1.60ミリモル)を室温で加え、80℃で終夜攪拌した。反応液を酢酸エチルで希釈し、飽和食塩水で洗浄し、硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(石油エーテル:酢酸エチル=25:1、0.5%トリエチルアミン添加)で精製し、表題化合物(8.05g、76%)を固体として得た。
1H NMR(CDCl3)δ:1.43(9H,s),2.76(2H,s),3.60(2H,t,J=5.4Hz),4.14(2H,d,J=2.4Hz),7.20-7.30(1H,brs),7.40-7.50(4H,m),8.02-8.11(2H,m),8.65(1H,d,J=5.2Hz)。 Reference example 1
tert-Butyl 4- (4-phenylpyrimidin-2-yl) -3,6-dihydropyridine-1 (2H) -carboxylate
2-Chloro-4-phenylpyrimidine (6.00 g, 31.5 mmol), tert-butyl 4- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) -3 , 6-Dihydropyridine-1 (2H) -carboxylate (10.7 g, 34.7 mmol), sodium carbonate (6.68 g, 63.0 mmol) in 1,4-dioxane (100 ml) / water (25 ml) Was added tetrakistriphenylphosphine palladium (1.82 g, 1.60 mmol) at room temperature and stirred at 80 ° C. overnight. The reaction mixture was diluted with ethyl acetate, washed with saturated brine, dried over sodium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (petroleum ether: ethyl acetate = 25: 1, 0.5% triethylamine was added) to obtain the title compound (8.05 g, 76%) as a solid.
1 H NMR (CDCl 3 ) δ: 1.43 (9H, s), 2.76 (2H, s), 3.60 (2H, t, J = 5.4 Hz), 4.14 (2H, d, J = 2.4 Hz), 7.20-7.30 (1H, brs), 7.40-7.50 (4H, m), 8.02-8.11 (2H, m), 8.65 ( 1H, d, J = 5.2 Hz).
tert-ブチル 4-(4-フェニルピリミジン-2-イル)-3,6-ジヒドロピリジン-1(2H)-カルボキシラート
1H NMR(CDCl3)δ:1.43(9H,s),2.76(2H,s),3.60(2H,t,J=5.4Hz),4.14(2H,d,J=2.4Hz),7.20-7.30(1H,brs),7.40-7.50(4H,m),8.02-8.11(2H,m),8.65(1H,d,J=5.2Hz)。 Reference example 1
tert-Butyl 4- (4-phenylpyrimidin-2-yl) -3,6-dihydropyridine-1 (2H) -carboxylate
1 H NMR (CDCl 3 ) δ: 1.43 (9H, s), 2.76 (2H, s), 3.60 (2H, t, J = 5.4 Hz), 4.14 (2H, d, J = 2.4 Hz), 7.20-7.30 (1H, brs), 7.40-7.50 (4H, m), 8.02-8.11 (2H, m), 8.65 ( 1H, d, J = 5.2 Hz).
参考例2
4-フェニル-2-ピペリジン-4-イルピリミジン二塩酸塩

参考例1で得たtert-ブチル 4-(4-フェニルピリミジン-2-イル)-3,6-ジヒドロピリジン-1(2H)-カルボキシラート(8.00g、24.0ミリモル)のメタノール(300ml)溶液に、窒素雰囲気下、10%パラジウム炭素(800mg)を加え、反応溶液を3気圧の水素雰囲気下、室温で終夜攪拌した。反応溶液をろ過し、ろ液を濃縮した。残渣を酢酸エチル(50ml)に溶解させ、飽和塩化水素/酢酸エチル溶液(25ml)を加えて室温で2時間撹拌した。生じた固体をろ取し、酢酸エチルで洗浄し、乾燥して表題化合物(7.91g、quant.)を固体として得た。
1H NMR(DMSO-d6)δ:1.99-2.12(2H,m),2.14-2.22(2H,m),3.00(2H,q,J=11.7Hz),3.17-3.35(3H,m),7.48-7.57(3H,m),8.00(1H,d,J=5.6Hz),8.21(2H,dd,J=7.6,4.4Hz),8.84(1H,d,J=9.2Hz),9.17(1H,brs),9.41(1H,brs)。 Reference example 2
4-Phenyl-2-piperidin-4-ylpyrimidine dihydrochloride
Tert-butyl 4- (4-phenylpyrimidin-2-yl) -3,6-dihydropyridine-1 (2H) -carboxylate (8.00 g, 24.0 mmol) obtained in Reference Example 1 in methanol (300 ml) To the solution, 10% palladium carbon (800 mg) was added under a nitrogen atmosphere, and the reaction solution was stirred overnight at room temperature under a hydrogen atmosphere of 3 atm. The reaction solution was filtered and the filtrate was concentrated. The residue was dissolved in ethyl acetate (50 ml), saturated hydrogen chloride / ethyl acetate solution (25 ml) was added, and the mixture was stirred at room temperature for 2 hr. The resulting solid was collected by filtration, washed with ethyl acetate, and dried to give the title compound (7.91 g, quant.) As a solid.
1 H NMR (DMSO-d 6 ) δ: 1.99-2.12 (2H, m), 2.14-2.22 (2H, m), 3.00 (2H, q, J = 11.7 Hz) ), 3.17-3.35 (3H, m), 7.48-7.57 (3H, m), 8.00 (1H, d, J = 5.6 Hz), 8.21 (2H, dd) , J = 7.6, 4.4 Hz), 8.84 (1H, d, J = 9.2 Hz), 9.17 (1H, brs), 9.41 (1H, brs).
4-フェニル-2-ピペリジン-4-イルピリミジン二塩酸塩
1H NMR(DMSO-d6)δ:1.99-2.12(2H,m),2.14-2.22(2H,m),3.00(2H,q,J=11.7Hz),3.17-3.35(3H,m),7.48-7.57(3H,m),8.00(1H,d,J=5.6Hz),8.21(2H,dd,J=7.6,4.4Hz),8.84(1H,d,J=9.2Hz),9.17(1H,brs),9.41(1H,brs)。 Reference example 2
4-Phenyl-2-piperidin-4-ylpyrimidine dihydrochloride
1 H NMR (DMSO-d 6 ) δ: 1.99-2.12 (2H, m), 2.14-2.22 (2H, m), 3.00 (2H, q, J = 11.7 Hz) ), 3.17-3.35 (3H, m), 7.48-7.57 (3H, m), 8.00 (1H, d, J = 5.6 Hz), 8.21 (2H, dd) , J = 7.6, 4.4 Hz), 8.84 (1H, d, J = 9.2 Hz), 9.17 (1H, brs), 9.41 (1H, brs).
参考例3
tert-ブチル 4-[4-(2,3-ジフルオロフェニル)ピリミジン-2-イル]-3,6-ジヒドロピリジン-1(2H)-カルボキシラート

参考例1と同様にして2-クロロ-4-(2,3-ジフルオロフェニル)ピリミジンから表題化合物(6.11g、62%)を固体として得た。
1H NMR(CDCl3)δ:1.48(9H,s),2.78(2H,d,J=0.8Hz),3.65(2H,s),4.19(2H,s),7.17-7.36(3H,m),7.62(1H,dd,J=5.2,2.0Hz),7.96(1H,t,J=9.0Hz),8.75(1H,d,J=5.2Hz)。 Reference example 3
tert-Butyl 4- [4- (2,3-difluorophenyl) pyrimidin-2-yl] -3,6-dihydropyridine-1 (2H) -carboxylate
In the same manner as in Reference Example 1, the title compound (6.11 g, 62%) was obtained as a solid from 2-chloro-4- (2,3-difluorophenyl) pyrimidine.
1 H NMR (CDCl 3 ) δ: 1.48 (9H, s), 2.78 (2H, d, J = 0.8 Hz), 3.65 (2H, s), 4.19 (2H, s) 7.17-7.36 (3H, m), 7.62 (1H, dd, J = 5.2, 2.0 Hz), 7.96 (1H, t, J = 9.0 Hz), 8. 75 (1H, d, J = 5.2 Hz).
tert-ブチル 4-[4-(2,3-ジフルオロフェニル)ピリミジン-2-イル]-3,6-ジヒドロピリジン-1(2H)-カルボキシラート
1H NMR(CDCl3)δ:1.48(9H,s),2.78(2H,d,J=0.8Hz),3.65(2H,s),4.19(2H,s),7.17-7.36(3H,m),7.62(1H,dd,J=5.2,2.0Hz),7.96(1H,t,J=9.0Hz),8.75(1H,d,J=5.2Hz)。 Reference example 3
tert-Butyl 4- [4- (2,3-difluorophenyl) pyrimidin-2-yl] -3,6-dihydropyridine-1 (2H) -carboxylate
1 H NMR (CDCl 3 ) δ: 1.48 (9H, s), 2.78 (2H, d, J = 0.8 Hz), 3.65 (2H, s), 4.19 (2H, s) 7.17-7.36 (3H, m), 7.62 (1H, dd, J = 5.2, 2.0 Hz), 7.96 (1H, t, J = 9.0 Hz), 8. 75 (1H, d, J = 5.2 Hz).
参考例4
4-(2,3-ジフルオロフェニル)-2-ピペリジン-4-イルピリミジン二塩酸塩

参考例2と同様にして、参考例3で得たtert-ブチル 4-[4-(2,3-ジフルオロフェニル)ピリミジン-2-イル]-3,6-ジヒドロピリジン-1(2H)-カルボキシラートから表題化合物(4.65g、82%)を固体として得た。
1H NMR(DMSO-d6)δ:1.95-2.09(2H,m),2.10-2.20(2H,m),3.01(2H,q,J=11.6Hz),3.15-3.25(1H,m),3.25-3.45(2H,m),7.34-7.45(1H,m),7.56-7.68(1H,m),7.77(1H,dd,J=5.2,2.0Hz),7.84(1H,t,J=7.2Hz),8.89(1H,d,J=5.2Hz),9.04(1H,brs),9.33(1H,brs)。 Reference example 4
4- (2,3-Difluorophenyl) -2-piperidin-4-ylpyrimidine dihydrochloride
In the same manner as in Reference Example 2, tert-butyl 4- [4- (2,3-difluorophenyl) pyrimidin-2-yl] -3,6-dihydropyridine-1 (2H) -carboxylate obtained in Reference Example 3 Gave the title compound (4.65 g, 82%) as a solid.
1 H NMR (DMSO-d 6 ) δ: 1.95-2.09 (2H, m), 2.10-2.20 (2H, m), 3.01 (2H, q, J = 11.6 Hz) ), 3.15-3.25 (1H, m), 3.25-3.45 (2H, m), 7.34-7.45 (1H, m), 7.56-7.68 (1H) M), 7.77 (1H, dd, J = 5.2, 2.0 Hz), 7.84 (1H, t, J = 7.2 Hz), 8.89 (1H, d, J = 5. 2 Hz), 9.04 (1H, brs), 9.33 (1H, brs).
4-(2,3-ジフルオロフェニル)-2-ピペリジン-4-イルピリミジン二塩酸塩
1H NMR(DMSO-d6)δ:1.95-2.09(2H,m),2.10-2.20(2H,m),3.01(2H,q,J=11.6Hz),3.15-3.25(1H,m),3.25-3.45(2H,m),7.34-7.45(1H,m),7.56-7.68(1H,m),7.77(1H,dd,J=5.2,2.0Hz),7.84(1H,t,J=7.2Hz),8.89(1H,d,J=5.2Hz),9.04(1H,brs),9.33(1H,brs)。 Reference example 4
4- (2,3-Difluorophenyl) -2-piperidin-4-ylpyrimidine dihydrochloride
1 H NMR (DMSO-d 6 ) δ: 1.95-2.09 (2H, m), 2.10-2.20 (2H, m), 3.01 (2H, q, J = 11.6 Hz) ), 3.15-3.25 (1H, m), 3.25-3.45 (2H, m), 7.34-7.45 (1H, m), 7.56-7.68 (1H) M), 7.77 (1H, dd, J = 5.2, 2.0 Hz), 7.84 (1H, t, J = 7.2 Hz), 8.89 (1H, d, J = 5. 2 Hz), 9.04 (1H, brs), 9.33 (1H, brs).
参考例5
tert-ブチル 4-(6-フェニルピリミジン-4-イル)-3,6-ジヒドロピリジン-1(2H)-カルボキシラート

参考例1と同様にして4-クロロ-6-フェニルピリミジンから表題化合物(1.64g、47%)を固体として得た。
1H NMR(CDCl3)δ:1.50(9H,s),2.62-2.75(2H,m),3.69(2H,t,J=5.4Hz),4.20(2H,d,J=2.8Hz),6.99(1H,s),7.47-7.54(3H,m),7.71(1H,s),8.03-8.12(2H,m),9.19(1H,s)。 Reference Example 5
tert-Butyl 4- (6-phenylpyrimidin-4-yl) -3,6-dihydropyridine-1 (2H) -carboxylate
The title compound (1.64 g, 47%) was obtained as a solid from 4-chloro-6-phenylpyrimidine in the same manner as in Reference Example 1.
1 H NMR (CDCl 3 ) δ: 1.50 (9H, s), 2.62-2.75 (2H, m), 3.69 (2H, t, J = 5.4 Hz), 4.20 ( 2H, d, J = 2.8 Hz), 6.99 (1H, s), 7.47-7.54 (3H, m), 7.71 (1H, s), 8.03-8.12 ( 2H, m), 9.19 (1H, s).
tert-ブチル 4-(6-フェニルピリミジン-4-イル)-3,6-ジヒドロピリジン-1(2H)-カルボキシラート
1H NMR(CDCl3)δ:1.50(9H,s),2.62-2.75(2H,m),3.69(2H,t,J=5.4Hz),4.20(2H,d,J=2.8Hz),6.99(1H,s),7.47-7.54(3H,m),7.71(1H,s),8.03-8.12(2H,m),9.19(1H,s)。 Reference Example 5
tert-Butyl 4- (6-phenylpyrimidin-4-yl) -3,6-dihydropyridine-1 (2H) -carboxylate
1 H NMR (CDCl 3 ) δ: 1.50 (9H, s), 2.62-2.75 (2H, m), 3.69 (2H, t, J = 5.4 Hz), 4.20 ( 2H, d, J = 2.8 Hz), 6.99 (1H, s), 7.47-7.54 (3H, m), 7.71 (1H, s), 8.03-8.12 ( 2H, m), 9.19 (1H, s).
参考例6
4-フェニル-6-ピペリジン-4-イルピリミジン二塩酸塩

参考例2と同様にして、参考例5で得たtert-ブチル 4-(6-フェニルピリミジン-4-イル)-3,6-ジヒドロピリジン-1(2H)-カルボキシラートから表題化合物(3.30g、74%)を固体として得た。
1H NMR(DMSO-d6)δ:1.90-2.09(4H,m),2.92-3.11(3H,m),3.30-3.40(2H,m),7.46-7.60(3H,m),7.97(1H,d,J=1.2Hz),8.13-8.23(2H,m),9.01(1H,brs),9.15(1H,d,J=1.2Hz),9.30(1H,brs)。 Reference Example 6
4-Phenyl-6-piperidin-4-ylpyrimidine dihydrochloride
In the same manner as in Reference Example 2, the title compound (3.30 g) was obtained from tert-butyl 4- (6-phenylpyrimidin-4-yl) -3,6-dihydropyridine-1 (2H) -carboxylate obtained in Reference Example 5. 74%) as a solid.
1 H NMR (DMSO-d 6 ) δ: 1.90-2.09 (4H, m), 2.92-3.11 (3H, m), 3.30-3.40 (2H, m), 7.46-7.60 (3H, m), 7.97 (1H, d, J = 1.2 Hz), 8.13-8.23 (2H, m), 9.01 (1H, brs), 9.15 (1H, d, J = 1.2 Hz), 9.30 (1H, brs).
4-フェニル-6-ピペリジン-4-イルピリミジン二塩酸塩
1H NMR(DMSO-d6)δ:1.90-2.09(4H,m),2.92-3.11(3H,m),3.30-3.40(2H,m),7.46-7.60(3H,m),7.97(1H,d,J=1.2Hz),8.13-8.23(2H,m),9.01(1H,brs),9.15(1H,d,J=1.2Hz),9.30(1H,brs)。 Reference Example 6
4-Phenyl-6-piperidin-4-ylpyrimidine dihydrochloride
1 H NMR (DMSO-d 6 ) δ: 1.90-2.09 (4H, m), 2.92-3.11 (3H, m), 3.30-3.40 (2H, m), 7.46-7.60 (3H, m), 7.97 (1H, d, J = 1.2 Hz), 8.13-8.23 (2H, m), 9.01 (1H, brs), 9.15 (1H, d, J = 1.2 Hz), 9.30 (1H, brs).
参考例7
tert-ブチル 4-[6-(2,3-ジフルオロフェニル)ピリミジン-4-イル]-3,6-ジヒドロピリジン-1(2H)-カルボキシラート

参考例1と同様にして4-クロロ-6-(2,3-ジフルオロフェニル)ピリミジンから表題化合物(7.10g、68%)を固体として得た。
1H NMR(CDCl3)δ:1.48(9H,s),2.65(2H,d,J=2.8Hz),3.66(2H,t,J=5.4Hz),4.18(2H,d,J=2.8Hz),6.98(1H,s),7.18-7.35(2H,m),7.80(1H,s),7.87(1H,t,J=7.2Hz),9.21(1H,s)。 Reference Example 7
tert-Butyl 4- [6- (2,3-difluorophenyl) pyrimidin-4-yl] -3,6-dihydropyridine-1 (2H) -carboxylate
The title compound (7.10 g, 68%) was obtained as a solid from 4-chloro-6- (2,3-difluorophenyl) pyrimidine in the same manner as in Reference Example 1.
1 H NMR (CDCl 3 ) δ: 1.48 (9H, s), 2.65 (2H, d, J = 2.8 Hz), 3.66 (2H, t, J = 5.4 Hz), 4. 18 (2H, d, J = 2.8 Hz), 6.98 (1H, s), 7.18-7.35 (2H, m), 7.80 (1H, s), 7.87 (1H, t, J = 7.2 Hz), 9.21 (1H, s).
tert-ブチル 4-[6-(2,3-ジフルオロフェニル)ピリミジン-4-イル]-3,6-ジヒドロピリジン-1(2H)-カルボキシラート
1H NMR(CDCl3)δ:1.48(9H,s),2.65(2H,d,J=2.8Hz),3.66(2H,t,J=5.4Hz),4.18(2H,d,J=2.8Hz),6.98(1H,s),7.18-7.35(2H,m),7.80(1H,s),7.87(1H,t,J=7.2Hz),9.21(1H,s)。 Reference Example 7
tert-Butyl 4- [6- (2,3-difluorophenyl) pyrimidin-4-yl] -3,6-dihydropyridine-1 (2H) -carboxylate
1 H NMR (CDCl 3 ) δ: 1.48 (9H, s), 2.65 (2H, d, J = 2.8 Hz), 3.66 (2H, t, J = 5.4 Hz), 4. 18 (2H, d, J = 2.8 Hz), 6.98 (1H, s), 7.18-7.35 (2H, m), 7.80 (1H, s), 7.87 (1H, t, J = 7.2 Hz), 9.21 (1H, s).
参考例8
4-(2,3-ジフルオロフェニル)-6-ピペリジン-4-イルピリミジン二塩酸塩

参考例2と同様にして参考例7で得たtert-ブチル 4-[6-(2,3-ジフルオロフェニル)ピリミジン-4-イル]-3,6-ジヒドロピリジン-1(2H)-カルボキシラートから表題化合物(5.70g、86%)を固体として得た。
1H NMR(DMSO-d6)δ:1.90-2.12(4H,m),2.99(2H,q,J=11.2Hz),3.06-3.21(1H,m),3.30-3.40(2H,m),7.33-7.47(1H,m),7.63(1H,d,J=8.4Hz),7.75-7.88(2H,m),9.15(1H,brs),9.26(1H,s),9.35(1H,brs)。 Reference Example 8
4- (2,3-Difluorophenyl) -6-piperidin-4-ylpyrimidine dihydrochloride
In the same manner as in Reference Example 2, from the tert-butyl 4- [6- (2,3-difluorophenyl) pyrimidin-4-yl] -3,6-dihydropyridine-1 (2H) -carboxylate obtained in Reference Example 7. The title compound (5.70 g, 86%) was obtained as a solid.
1 H NMR (DMSO-d 6 ) δ: 1.90-2.12 (4H, m), 2.99 (2H, q, J = 11.2 Hz), 3.06-3.21 (1H, m ), 3.30-3.40 (2H, m), 7.33-7.47 (1H, m), 7.63 (1H, d, J = 8.4 Hz), 7.75-7.88. (2H, m), 9.15 (1H, brs), 9.26 (1H, s), 9.35 (1H, brs).
4-(2,3-ジフルオロフェニル)-6-ピペリジン-4-イルピリミジン二塩酸塩
1H NMR(DMSO-d6)δ:1.90-2.12(4H,m),2.99(2H,q,J=11.2Hz),3.06-3.21(1H,m),3.30-3.40(2H,m),7.33-7.47(1H,m),7.63(1H,d,J=8.4Hz),7.75-7.88(2H,m),9.15(1H,brs),9.26(1H,s),9.35(1H,brs)。 Reference Example 8
4- (2,3-Difluorophenyl) -6-piperidin-4-ylpyrimidine dihydrochloride
1 H NMR (DMSO-d 6 ) δ: 1.90-2.12 (4H, m), 2.99 (2H, q, J = 11.2 Hz), 3.06-3.21 (1H, m ), 3.30-3.40 (2H, m), 7.33-7.47 (1H, m), 7.63 (1H, d, J = 8.4 Hz), 7.75-7.88. (2H, m), 9.15 (1H, brs), 9.26 (1H, s), 9.35 (1H, brs).
参考例9
4-クロロ-2-フェニルピリミジン

2-フェニル-4-メトキシピリミジン(9.00g、48.6ミリモル)のフェニルホスホン酸ジクロリド(36ml)溶液を140℃で終夜撹拌した。反応溶液を氷水(200ml)で希釈し、炭酸ナトリウムを用いてpH=8とし、塩化メチレンで抽出した。抽出液を飽和食塩水で洗浄し、硫酸ナトリウムで乾燥し、溶媒を減圧下留去して表題化合物(8.75g、94%)を固体として得た。
1H NMR(CDCl3)δ:7.21(1H,d,J=5.2Hz),7.38-7.52(3H,m),8.42(2H,d,J=7.4,2.2Hz),8.65(1H,d,J=5.2Hz)。 Reference Example 9
4-chloro-2-phenylpyrimidine
A solution of 2-phenyl-4-methoxypyrimidine (9.00 g, 48.6 mmol) in phenylphosphonic dichloride (36 ml) was stirred at 140 ° C. overnight. The reaction solution was diluted with ice water (200 ml), adjusted to pH = 8 using sodium carbonate, and extracted with methylene chloride. The extract was washed with saturated brine, dried over sodium sulfate, and the solvent was evaporated under reduced pressure to give the title compound (8.75 g, 94%) as a solid.
1 H NMR (CDCl 3 ) δ: 7.21 (1H, d, J = 5.2 Hz), 7.38-7.52 (3H, m), 8.42 (2H, d, J = 7.4) , 2.2 Hz), 8.65 (1H, d, J = 5.2 Hz).
4-クロロ-2-フェニルピリミジン
1H NMR(CDCl3)δ:7.21(1H,d,J=5.2Hz),7.38-7.52(3H,m),8.42(2H,d,J=7.4,2.2Hz),8.65(1H,d,J=5.2Hz)。 Reference Example 9
4-chloro-2-phenylpyrimidine
1 H NMR (CDCl 3 ) δ: 7.21 (1H, d, J = 5.2 Hz), 7.38-7.52 (3H, m), 8.42 (2H, d, J = 7.4) , 2.2 Hz), 8.65 (1H, d, J = 5.2 Hz).
参考例10
tert-ブチル 4-(2-フェニルピリミジン-4-イル)-3,6-ジヒドロピリジン-1(2H)-カルボキシラート

参考例1と同様にして、参考例9で得た4-クロロ-2-フェニルピリミジンから表題化合物(7.08g、67%)を固体として得た。
1H NMR(CDCl3)δ:1.51(9H,s),2.73(2H,d,J=5.6Hz),3.69(2H,t,J=5.6Hz),4.21(2H,d,J=1.6Hz),7.03(1H,s),7.21(1H,d,J=5.6Hz),7.45-7.56(3H,m),8.45-8.55(2H,m),8.75(1H,d,J=5.2Hz)。 Reference Example 10
tert-Butyl 4- (2-phenylpyrimidin-4-yl) -3,6-dihydropyridine-1 (2H) -carboxylate
In the same manner as in Reference Example 1, the title compound (7.08 g, 67%) was obtained as a solid from 4-chloro-2-phenylpyrimidine obtained in Reference Example 9.
1 H NMR (CDCl 3 ) δ: 1.51 (9H, s), 2.73 (2H, d, J = 5.6 Hz), 3.69 (2H, t, J = 5.6 Hz), 4. 21 (2H, d, J = 1.6 Hz), 7.03 (1H, s), 7.21 (1H, d, J = 5.6 Hz), 7.45-7.56 (3H, m), 8.45-8.55 (2H, m), 8.75 (1H, d, J = 5.2 Hz).
tert-ブチル 4-(2-フェニルピリミジン-4-イル)-3,6-ジヒドロピリジン-1(2H)-カルボキシラート
1H NMR(CDCl3)δ:1.51(9H,s),2.73(2H,d,J=5.6Hz),3.69(2H,t,J=5.6Hz),4.21(2H,d,J=1.6Hz),7.03(1H,s),7.21(1H,d,J=5.6Hz),7.45-7.56(3H,m),8.45-8.55(2H,m),8.75(1H,d,J=5.2Hz)。 Reference Example 10
tert-Butyl 4- (2-phenylpyrimidin-4-yl) -3,6-dihydropyridine-1 (2H) -carboxylate
1 H NMR (CDCl 3 ) δ: 1.51 (9H, s), 2.73 (2H, d, J = 5.6 Hz), 3.69 (2H, t, J = 5.6 Hz), 4. 21 (2H, d, J = 1.6 Hz), 7.03 (1H, s), 7.21 (1H, d, J = 5.6 Hz), 7.45-7.56 (3H, m), 8.45-8.55 (2H, m), 8.75 (1H, d, J = 5.2 Hz).
参考例11
2-フェニル-4-ピペリジン-4-イルピリミジン二塩酸塩

参考例2と同様にして、参考例10で得たtert-ブチル 4-(2-フェニルピリミジン-4-イル)-3,6-ジヒドロピリジン-1(2H)-カルボキシラートから表題化合物(6.38g、97%)を固体として得た。
1H NMR(DMSO-d6)δ:1.95-2.18(4H,m),2.91-3.15(3H,m),3.30-3.40(2H,m),7.36(1H,d,J=5.2Hz),7.46-7.55(3H,m),8.34-8.47(2H,m),8.83(1H,d,J=5.2Hz),9.12(1H,brs),9.43(1H,brs)。 Reference Example 11
2-Phenyl-4-piperidin-4-ylpyrimidine dihydrochloride
In the same manner as in Reference Example 2, the title compound (6.38 g) was obtained from tert-butyl 4- (2-phenylpyrimidin-4-yl) -3,6-dihydropyridine-1 (2H) -carboxylate obtained in Reference Example 10. 97%) as a solid.
1 H NMR (DMSO-d 6 ) δ: 1.95-2.18 (4H, m), 2.91-3.15 (3H, m), 3.30-3.40 (2H, m), 7.36 (1H, d, J = 5.2 Hz), 7.46-7.55 (3H, m), 8.34-8.47 (2H, m), 8.83 (1H, d, J = 5.2 Hz), 9.12 (1H, brs), 9.43 (1H, brs).
2-フェニル-4-ピペリジン-4-イルピリミジン二塩酸塩
1H NMR(DMSO-d6)δ:1.95-2.18(4H,m),2.91-3.15(3H,m),3.30-3.40(2H,m),7.36(1H,d,J=5.2Hz),7.46-7.55(3H,m),8.34-8.47(2H,m),8.83(1H,d,J=5.2Hz),9.12(1H,brs),9.43(1H,brs)。 Reference Example 11
2-Phenyl-4-piperidin-4-ylpyrimidine dihydrochloride
1 H NMR (DMSO-d 6 ) δ: 1.95-2.18 (4H, m), 2.91-3.15 (3H, m), 3.30-3.40 (2H, m), 7.36 (1H, d, J = 5.2 Hz), 7.46-7.55 (3H, m), 8.34-8.47 (2H, m), 8.83 (1H, d, J = 5.2 Hz), 9.12 (1H, brs), 9.43 (1H, brs).
参考例12
2-(2,3-ジフルオロフェニル)-4-メトキシピリミジン

2-クロロ-4-メトキシピリミジン(30.0g、210ミリモル)、2,3-ジフルオロフェニルボロン酸(50.5g、320ミリモル)、炭酸ナトリウム(44.5g、420ミリモル)の1,4-ジオキサン(240ml)/水(60ml)溶液に、テトラキストリフェニルホスフィンパラジウム(11.6g、10.0ミリモル)を室温で加え、80℃で終夜攪拌した。反応液を酢酸エチルで希釈し、飽和食塩水で洗浄し、硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(石油エーテル:酢酸エチル=50:1、0.5%トリエチルアミン添加)で精製し、ヘキサンと塩化メチレンから再結晶して表題化合物(25.6g、55%)を固体として得た。
1H NMR(DMSO-d6)δ:3.95(3H,s),6.85-6.96(1H,m),7.22-7.34(1H,m),7.46-7.60(1H,m),8.86(1H,t,J=6.4Hz),8.62(1H,d,J=8.8Hz)。 Reference Example 12
2- (2,3-Difluorophenyl) -4-methoxypyrimidine
2-chloro-4-methoxypyrimidine (30.0 g, 210 mmol), 2,3-difluorophenylboronic acid (50.5 g, 320 mmol), sodium carbonate (44.5 g, 420 mmol) of 1,4-dioxane Tetrakistriphenylphosphine palladium (11.6 g, 10.0 mmol) was added to a (240 ml) / water (60 ml) solution at room temperature and stirred at 80 ° C. overnight. The reaction mixture was diluted with ethyl acetate, washed with saturated brine, dried over sodium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (petroleum ether: ethyl acetate = 50: 1, 0.5% triethylamine added) and recrystallized from hexane and methylene chloride to give the title compound (25.6 g, 55%) as a solid Obtained.
1 H NMR (DMSO-d 6 ) δ: 3.95 (3H, s), 6.85-6.96 (1H, m), 7.22-7.34 (1H, m), 7.46- 7.60 (1H, m), 8.86 (1H, t, J = 6.4 Hz), 8.62 (1H, d, J = 8.8 Hz).
2-(2,3-ジフルオロフェニル)-4-メトキシピリミジン
1H NMR(DMSO-d6)δ:3.95(3H,s),6.85-6.96(1H,m),7.22-7.34(1H,m),7.46-7.60(1H,m),8.86(1H,t,J=6.4Hz),8.62(1H,d,J=8.8Hz)。 Reference Example 12
2- (2,3-Difluorophenyl) -4-methoxypyrimidine
1 H NMR (DMSO-d 6 ) δ: 3.95 (3H, s), 6.85-6.96 (1H, m), 7.22-7.34 (1H, m), 7.46- 7.60 (1H, m), 8.86 (1H, t, J = 6.4 Hz), 8.62 (1H, d, J = 8.8 Hz).
参考例13
4-クロロ-2-(2,3-ジフルオロフェニル)ピリミジン

参考例9と同様にして、参考例12で得た2-(2,3-ジフルオロフェニル)-4-メトキシピリミジンから表題化合物(9.00g、88%)を固体として得た。
1H NMR(DMSO-d6)δ:7.30-7.40(1H,m),7.63(1H,q,J=4.0Hz),7.75(1H,d,J=5.2Hz),7.85(1H,t,J=7.4Hz),8.94(1H,d,J=5.2Hz)。 Reference Example 13
4-Chloro-2- (2,3-difluorophenyl) pyrimidine
In the same manner as in Reference Example 9, the title compound (9.00 g, 88%) was obtained as a solid from 2- (2,3-difluorophenyl) -4-methoxypyrimidine obtained in Reference Example 12.
1 H NMR (DMSO-d 6 ) δ: 7.30-7.40 (1H, m), 7.63 (1H, q, J = 4.0 Hz), 7.75 (1H, d, J = 5) .2 Hz), 7.85 (1 H, t, J = 7.4 Hz), 8.94 (1 H, d, J = 5.2 Hz).
4-クロロ-2-(2,3-ジフルオロフェニル)ピリミジン
1H NMR(DMSO-d6)δ:7.30-7.40(1H,m),7.63(1H,q,J=4.0Hz),7.75(1H,d,J=5.2Hz),7.85(1H,t,J=7.4Hz),8.94(1H,d,J=5.2Hz)。 Reference Example 13
4-Chloro-2- (2,3-difluorophenyl) pyrimidine
1 H NMR (DMSO-d 6 ) δ: 7.30-7.40 (1H, m), 7.63 (1H, q, J = 4.0 Hz), 7.75 (1H, d, J = 5) .2 Hz), 7.85 (1 H, t, J = 7.4 Hz), 8.94 (1 H, d, J = 5.2 Hz).
参考例14
tert-ブチル 4-[2-(2,3-ジフルオロフェニル)ピリミジン-4-イル]-3,6-ジヒドロピリジン-1(2H)-カルボキシラート

参考例1と同様にして、参考例13で得た4-クロロ-2-(2,3-ジフルオロフェニル)ピリミジンから表題化合物(5.50g、56%)を固体として得た。
1H NMR(CDCl3)δ:1.50(9H,s),2.68(2H,d,J=1.6Hz),3.58-3.74(2H,m),4.21(2H,s),7.05(1H,s),7.13-7.22(1H,m),7.23-7.37(2H,m),7.88(1H,t,J=7.2Hz),8.81(1H,d,J=5.2Hz)。 Reference Example 14
tert-Butyl 4- [2- (2,3-difluorophenyl) pyrimidin-4-yl] -3,6-dihydropyridine-1 (2H) -carboxylate
In the same manner as in Reference Example 1, the title compound (5.50 g, 56%) was obtained as a solid from 4-chloro-2- (2,3-difluorophenyl) pyrimidine obtained in Reference Example 13.
1 H NMR (CDCl 3 ) δ: 1.50 (9H, s), 2.68 (2H, d, J = 1.6 Hz), 3.58-3.74 (2H, m), 4.21 ( 2H, s), 7.05 (1H, s), 7.13-7.22 (1H, m), 7.23-7.37 (2H, m), 7.88 (1H, t, J = 7.2 Hz), 8.81 (1H, d, J = 5.2 Hz).
tert-ブチル 4-[2-(2,3-ジフルオロフェニル)ピリミジン-4-イル]-3,6-ジヒドロピリジン-1(2H)-カルボキシラート
1H NMR(CDCl3)δ:1.50(9H,s),2.68(2H,d,J=1.6Hz),3.58-3.74(2H,m),4.21(2H,s),7.05(1H,s),7.13-7.22(1H,m),7.23-7.37(2H,m),7.88(1H,t,J=7.2Hz),8.81(1H,d,J=5.2Hz)。 Reference Example 14
tert-Butyl 4- [2- (2,3-difluorophenyl) pyrimidin-4-yl] -3,6-dihydropyridine-1 (2H) -carboxylate
1 H NMR (CDCl 3 ) δ: 1.50 (9H, s), 2.68 (2H, d, J = 1.6 Hz), 3.58-3.74 (2H, m), 4.21 ( 2H, s), 7.05 (1H, s), 7.13-7.22 (1H, m), 7.23-7.37 (2H, m), 7.88 (1H, t, J = 7.2 Hz), 8.81 (1H, d, J = 5.2 Hz).
参考例15
2-(2,3-ジフルオロフェニル)-4-ピペリジン-4-イルピリミジン二塩酸塩

参考例2と同様にして、参考例14で得たtert-ブチル 4-[2-(2,3-ジフルオロフェニル)ピリミジン-4-イル]-3,6-ジヒドロピリジン-1(2H)-カルボキシラートから表題化合物(1.13g、90%)を固体として得た。
1H NMR(DMSO-d6)δ:1.88-2.02(2H,m),2.03-2.15(2H,m),3.30-3.40(2H,m),3.05-3.16(1H,m),3.34(2H,d,J=12.0Hz),7.34(1H,dd,J=13.4,7.8Hz),7.44(1H,d,J=5.2Hz),7.58(1H,dd,J=7.8,6.4Hz),7.83(1H,t,J=7.2Hz),8.70-9.09(2H,m),9.13-9.51(1H,brs)。 Reference Example 15
2- (2,3-Difluorophenyl) -4-piperidin-4-ylpyrimidine dihydrochloride
In the same manner as in Reference Example 2, tert-butyl 4- [2- (2,3-difluorophenyl) pyrimidin-4-yl] -3,6-dihydropyridine-1 (2H) -carboxylate obtained in Reference Example 14 Gave the title compound (1.13 g, 90%) as a solid.
1 H NMR (DMSO-d 6 ) δ: 1.88-2.02 (2H, m), 2.03-2.15 (2H, m), 3.30-3.40 (2H, m), 3.05-3.16 (1H, m), 3.34 (2H, d, J = 12.0 Hz), 7.34 (1H, dd, J = 13.4, 7.8 Hz), 7.44 (1H, d, J = 5.2 Hz), 7.58 (1H, dd, J = 7.8, 6.4 Hz), 7.83 (1H, t, J = 7.2 Hz), 8.70− 9.09 (2H, m), 9.13-9.51 (1H, brs).
2-(2,3-ジフルオロフェニル)-4-ピペリジン-4-イルピリミジン二塩酸塩
1H NMR(DMSO-d6)δ:1.88-2.02(2H,m),2.03-2.15(2H,m),3.30-3.40(2H,m),3.05-3.16(1H,m),3.34(2H,d,J=12.0Hz),7.34(1H,dd,J=13.4,7.8Hz),7.44(1H,d,J=5.2Hz),7.58(1H,dd,J=7.8,6.4Hz),7.83(1H,t,J=7.2Hz),8.70-9.09(2H,m),9.13-9.51(1H,brs)。 Reference Example 15
2- (2,3-Difluorophenyl) -4-piperidin-4-ylpyrimidine dihydrochloride
1 H NMR (DMSO-d 6 ) δ: 1.88-2.02 (2H, m), 2.03-2.15 (2H, m), 3.30-3.40 (2H, m), 3.05-3.16 (1H, m), 3.34 (2H, d, J = 12.0 Hz), 7.34 (1H, dd, J = 13.4, 7.8 Hz), 7.44 (1H, d, J = 5.2 Hz), 7.58 (1H, dd, J = 7.8, 6.4 Hz), 7.83 (1H, t, J = 7.2 Hz), 8.70− 9.09 (2H, m), 9.13-9.51 (1H, brs).
参考例16
tert-ブチル 4-フェニル-3’,6’-ジヒドロ-2,4’-ビピリジン-1’(2’H)-カルボキシラート

2-クロロ-4-フェニルピリジン(3.51g、18.5ミリモル)、tert-ブチル 4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-3,6-ジヒドロピリジン-1(2H)-カルボキシラート(6.20g、19.4ミリモル)、炭酸カリウム(7.67g、55.5ミリモル)の1,2-ジメトキシエタン(40ml)/水(20ml)溶液に、テトラキストリフェニルホスフィンパラジウム(1.07g、0.925ミリモル)を室温で加え、90℃で17時間攪拌した。反応液に水を注ぎ、塩化メチレンで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:8)で精製して表題化合物(4.10g、66%)を固体として得た。
1H NMR(CDCl3)δ:1.49(9H,s),2.70-2.73(2H,m),3.68(2H,brs),4.16(2H,d,J=2.8Hz),6.66-6.68(1H,m),7.38(1H,dd,J=4.8,1.6Hz),7.43-7.52(3H,m),7.52(1H,brs),7.62-7.65(2H,m),8.61(1H,dd,J=4.8,0.8Hz)。 Reference Example 16
tert-Butyl 4-phenyl-3 ′, 6′-dihydro-2,4′-bipyridine-1 ′ (2′H) -carboxylate
2-Chloro-4-phenylpyridine (3.51 g, 18.5 mmol), tert-butyl 4- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) -3 , 6-Dihydropyridine-1 (2H) -carboxylate (6.20 g, 19.4 mmol), potassium carbonate (7.67 g, 55.5 mmol) in 1,2-dimethoxyethane (40 ml) / water (20 ml) Tetrakistriphenylphosphine palladium (1.07 g, 0.925 mmol) was added to the solution at room temperature and stirred at 90 ° C. for 17 hours. Water was poured into the reaction solution and extracted with methylene chloride. The extract was dried over anhydrous magnesium sulfate and the solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography (ethyl acetate: hexane = 1: 8) to give the title compound (4.10 g, 66%) as a solid.
1 H NMR (CDCl 3 ) δ: 1.49 (9H, s), 2.70-2.73 (2H, m), 3.68 (2H, brs), 4.16 (2H, d, J = 2.8 Hz), 6.66-6.68 (1 H, m), 7.38 (1 H, dd, J = 4.8, 1.6 Hz), 7.43-7.52 (3 H, m), 7.52 (1H, brs), 7.62-7.65 (2H, m), 8.61 (1H, dd, J = 4.8, 0.8 Hz).
tert-ブチル 4-フェニル-3’,6’-ジヒドロ-2,4’-ビピリジン-1’(2’H)-カルボキシラート
1H NMR(CDCl3)δ:1.49(9H,s),2.70-2.73(2H,m),3.68(2H,brs),4.16(2H,d,J=2.8Hz),6.66-6.68(1H,m),7.38(1H,dd,J=4.8,1.6Hz),7.43-7.52(3H,m),7.52(1H,brs),7.62-7.65(2H,m),8.61(1H,dd,J=4.8,0.8Hz)。 Reference Example 16
tert-Butyl 4-phenyl-3 ′, 6′-dihydro-2,4′-bipyridine-1 ′ (2′H) -carboxylate
1 H NMR (CDCl 3 ) δ: 1.49 (9H, s), 2.70-2.73 (2H, m), 3.68 (2H, brs), 4.16 (2H, d, J = 2.8 Hz), 6.66-6.68 (1 H, m), 7.38 (1 H, dd, J = 4.8, 1.6 Hz), 7.43-7.52 (3 H, m), 7.52 (1H, brs), 7.62-7.65 (2H, m), 8.61 (1H, dd, J = 4.8, 0.8 Hz).
参考例17
tert-ブチル 4-(4-フェニルピリジン-2-イル)ピペリジン-1-カルボキシラート

参考例16で得たtert-ブチル 4-フェニル-3’,6’-ジヒドロ-2,4’-ビピリジン-1’(2’H)-カルボキシラート(4.10g、12.2ミリモル)のエタノール(40ml)懸濁液に、窒素雰囲気下、10%パラジウム炭素(50.0mg)を加え、反応溶液を水素雰囲気下、室温で24時間攪拌した。反応溶液をろ過し、ろ液を濃縮して表題化合物(4.15g、quant.)を油状物として得た。
1H NMR(CDCl3)δ:1.47(9H,s),1.73-1.84(2H,m),1.97(2H,d,J=13.2Hz),2.86-2.96(3H,m),4.29(2H,brs),7.35-7.37(2H,m),7.44-7.51(3H,m),7.62-7.64(2H,m),8.58(1H,dd,J=4.8,1.2Hz)。 Reference Example 17
tert-Butyl 4- (4-phenylpyridin-2-yl) piperidine-1-carboxylate
Tert-Butyl 4-phenyl-3 ′, 6′-dihydro-2,4′-bipyridine-1 ′ (2′H) -carboxylate (4.10 g, 12.2 mmol) ethanol obtained in Reference Example 16 To the (40 ml) suspension, 10% palladium carbon (50.0 mg) was added under a nitrogen atmosphere, and the reaction solution was stirred at room temperature under a hydrogen atmosphere for 24 hours. The reaction solution was filtered, and the filtrate was concentrated to give the title compound (4.15 g, quant.) As an oil.
1 H NMR (CDCl 3 ) δ: 1.47 (9H, s), 1.73-1.84 (2H, m), 1.97 (2H, d, J = 13.2 Hz), 2.86- 2.96 (3H, m), 4.29 (2H, brs), 7.35-7.37 (2H, m), 7.44-7.51 (3H, m), 7.62-7. 64 (2H, m), 8.58 (1H, dd, J = 4.8, 1.2 Hz).
tert-ブチル 4-(4-フェニルピリジン-2-イル)ピペリジン-1-カルボキシラート
1H NMR(CDCl3)δ:1.47(9H,s),1.73-1.84(2H,m),1.97(2H,d,J=13.2Hz),2.86-2.96(3H,m),4.29(2H,brs),7.35-7.37(2H,m),7.44-7.51(3H,m),7.62-7.64(2H,m),8.58(1H,dd,J=4.8,1.2Hz)。 Reference Example 17
tert-Butyl 4- (4-phenylpyridin-2-yl) piperidine-1-carboxylate
1 H NMR (CDCl 3 ) δ: 1.47 (9H, s), 1.73-1.84 (2H, m), 1.97 (2H, d, J = 13.2 Hz), 2.86- 2.96 (3H, m), 4.29 (2H, brs), 7.35-7.37 (2H, m), 7.44-7.51 (3H, m), 7.62-7. 64 (2H, m), 8.58 (1H, dd, J = 4.8, 1.2 Hz).
参考例18
4-フェニル-2-ピペリジン-4-イルピリジン二塩酸塩

参考例17で得たtert-ブチル 4-(4-フェニルピリジン-2-イル)ピペリジン-1-カルボキシラート(4.15g、12.3ミリモル)のメタノール(20ml)溶液に4.0Mの塩化水素/メタノール溶液(12.3ml)を加え、室温で24時間撹拌した。生じた固体をろ取し、酢酸エチルで洗浄し、乾燥して表題化合物(3.77g、88%)を固体として得た。
1H NMR(DMSO-d6)δ:2.10-2.20(4H,m),2.97-3.06(2H,m),3.35-3.42(3H,m),7.57-7.63(3H,m),7.96-7.98(2H,m),8.01(1H,brs),8.08(1H,brs),8.77(1H,d,J=6.0Hz),9.08(1H,brs),9.24(1H,brs)。 Reference Example 18
4-Phenyl-2-piperidin-4-ylpyridine dihydrochloride
To a solution of tert-butyl 4- (4-phenylpyridin-2-yl) piperidine-1-carboxylate (4.15 g, 12.3 mmol) obtained in Reference Example 17 in methanol (20 ml), 4.0 M hydrogen chloride. / Methanol solution (12.3 ml) was added and stirred at room temperature for 24 hours. The resulting solid was collected by filtration, washed with ethyl acetate and dried to give the title compound (3.77 g, 88%) as a solid.
1 H NMR (DMSO-d 6 ) δ: 2.10-2.20 (4H, m), 2.97-3.06 (2H, m), 3.35-3.42 (3H, m), 7.57-7.63 (3H, m), 7.96-7.98 (2H, m), 8.01 (1H, brs), 8.08 (1H, brs), 8.77 (1H, d, J = 6.0 Hz), 9.08 (1H, brs), 9.24 (1H, brs).
4-フェニル-2-ピペリジン-4-イルピリジン二塩酸塩
1H NMR(DMSO-d6)δ:2.10-2.20(4H,m),2.97-3.06(2H,m),3.35-3.42(3H,m),7.57-7.63(3H,m),7.96-7.98(2H,m),8.01(1H,brs),8.08(1H,brs),8.77(1H,d,J=6.0Hz),9.08(1H,brs),9.24(1H,brs)。 Reference Example 18
4-Phenyl-2-piperidin-4-ylpyridine dihydrochloride
1 H NMR (DMSO-d 6 ) δ: 2.10-2.20 (4H, m), 2.97-3.06 (2H, m), 3.35-3.42 (3H, m), 7.57-7.63 (3H, m), 7.96-7.98 (2H, m), 8.01 (1H, brs), 8.08 (1H, brs), 8.77 (1H, d, J = 6.0 Hz), 9.08 (1H, brs), 9.24 (1H, brs).
参考例19
tert-ブチル 4-(2-フルオロフェニル)-3’,6’-ジヒドロ-2,4’-ビピリジン-1’(2’H)-カルボキシラート

参考例16と同様にして2-クロロ-4-(2-フルオロフェニル)ピリジンから表題化合物(6.47g、89%)を固体として得た。
1H NMR(CDCl3)δ:1.49(9H,s),2.68-2.71(2H,m),3.66(2H,brs),4.15-4.16(2H,m),6.66-6.67(1H,m),7.17-7.29(2H,m),7.34-7.36(1H,m),7.39-7.44(1H,m),7.48(1H,td,J=7.6,1.6Hz),7.55(1H,brs),8.63(1H,dd,J=5.4,0.6Hz)。 Reference Example 19
tert-Butyl 4- (2-fluorophenyl) -3 ′, 6′-dihydro-2,4′-bipyridine-1 ′ (2′H) -carboxylate
The title compound (6.47 g, 89%) was obtained as a solid from 2-chloro-4- (2-fluorophenyl) pyridine in the same manner as in Reference Example 16.
1 H NMR (CDCl 3 ) δ: 1.49 (9H, s), 2.68-2.71 (2H, m), 3.66 (2H, brs), 4.15-4.16 (2H, m), 6.66-6.67 (1H, m), 7.17-7.29 (2H, m), 7.34-7.36 (1H, m), 7.39-7.44 ( 1H, m), 7.48 (1H, td, J = 7.6, 1.6 Hz), 7.55 (1H, brs), 8.63 (1H, dd, J = 5.4, 0.6 Hz) ).
tert-ブチル 4-(2-フルオロフェニル)-3’,6’-ジヒドロ-2,4’-ビピリジン-1’(2’H)-カルボキシラート
1H NMR(CDCl3)δ:1.49(9H,s),2.68-2.71(2H,m),3.66(2H,brs),4.15-4.16(2H,m),6.66-6.67(1H,m),7.17-7.29(2H,m),7.34-7.36(1H,m),7.39-7.44(1H,m),7.48(1H,td,J=7.6,1.6Hz),7.55(1H,brs),8.63(1H,dd,J=5.4,0.6Hz)。 Reference Example 19
tert-Butyl 4- (2-fluorophenyl) -3 ′, 6′-dihydro-2,4′-bipyridine-1 ′ (2′H) -carboxylate
1 H NMR (CDCl 3 ) δ: 1.49 (9H, s), 2.68-2.71 (2H, m), 3.66 (2H, brs), 4.15-4.16 (2H, m), 6.66-6.67 (1H, m), 7.17-7.29 (2H, m), 7.34-7.36 (1H, m), 7.39-7.44 ( 1H, m), 7.48 (1H, td, J = 7.6, 1.6 Hz), 7.55 (1H, brs), 8.63 (1H, dd, J = 5.4, 0.6 Hz) ).
参考例20
tert-ブチル 4-[4-(2-フルオロフェニル)ピリジン-2-イル]ピペリジン-1-カルボキシラート

参考例17と同様にして参考例19で得たtert-ブチル 4-(2-フルオロフェニル)-3’,6’-ジヒドロ-2,4’-ビピリジン-1’(2’H)-カルボキシラートから表題化合物(6.51g、quant.)を油状物として得た。
1H NMR(CDCl3)δ:1.49(9H,s),1.72-1.83(2H,m),1.97(2H,d,J=13.6Hz),2.86-2.95(3H,m),4.28(2H,brs),7.17-7.28(2H,m),7.32-7.34(2H,m),7.38-7.43(1H,m),7.46(1H,td,J=7.7,2.0Hz),8.60(1H,dd,J=5.2,0.8Hz)。 Reference Example 20
tert-Butyl 4- [4- (2-fluorophenyl) pyridin-2-yl] piperidine-1-carboxylate
Tert-Butyl 4- (2-fluorophenyl) -3 ′, 6′-dihydro-2,4′-bipyridine-1 ′ (2′H) -carboxylate obtained in Reference Example 19 in the same manner as Reference Example 17 Gave the title compound (6.51 g, quant.) As an oil.
1 H NMR (CDCl 3 ) δ: 1.49 (9H, s), 1.72-1.83 (2H, m), 1.97 (2H, d, J = 13.6 Hz), 2.86- 2.95 (3H, m), 4.28 (2H, brs), 7.17-7.28 (2H, m), 7.32-7.34 (2H, m), 7.38-7. 43 (1H, m), 7.46 (1H, td, J = 7.7, 2.0 Hz), 8.60 (1H, dd, J = 5.2, 0.8 Hz).
tert-ブチル 4-[4-(2-フルオロフェニル)ピリジン-2-イル]ピペリジン-1-カルボキシラート
1H NMR(CDCl3)δ:1.49(9H,s),1.72-1.83(2H,m),1.97(2H,d,J=13.6Hz),2.86-2.95(3H,m),4.28(2H,brs),7.17-7.28(2H,m),7.32-7.34(2H,m),7.38-7.43(1H,m),7.46(1H,td,J=7.7,2.0Hz),8.60(1H,dd,J=5.2,0.8Hz)。 Reference Example 20
tert-Butyl 4- [4- (2-fluorophenyl) pyridin-2-yl] piperidine-1-carboxylate
1 H NMR (CDCl 3 ) δ: 1.49 (9H, s), 1.72-1.83 (2H, m), 1.97 (2H, d, J = 13.6 Hz), 2.86- 2.95 (3H, m), 4.28 (2H, brs), 7.17-7.28 (2H, m), 7.32-7.34 (2H, m), 7.38-7. 43 (1H, m), 7.46 (1H, td, J = 7.7, 2.0 Hz), 8.60 (1H, dd, J = 5.2, 0.8 Hz).
参考例21
4-(2-フルオロフェニル)-2-ピペリジン-4-イルピリジン二塩酸塩

参考例18と同様にして参考例20で得たtert-ブチル 4-[4-(2-フルオロフェニル)ピリジン-2-イル]ピペリジン-1-カルボキシラートから表題化合物(4.50g、67%)を固体として得た。
1H NMR(DMSO-d6)δ:2.09-2.21(4H,m),3.01-3.04(2H,m),3.41(3H,d,J=11.6Hz),7.45(2H,q,J=7.2Hz),7.64(1H,q,J=7.2Hz),7.81(1H,t,J=7.2Hz),7.91(1H,brs),7.95(1H,d,J=4.0Hz),8.82(1H,d,J=5.2Hz),9.29(1H,brs),9.44(1H,brs)。 Reference Example 21
4- (2-Fluorophenyl) -2-piperidin-4-ylpyridine dihydrochloride
Title compound (4.50 g, 67%) from tert-butyl 4- [4- (2-fluorophenyl) pyridin-2-yl] piperidine-1-carboxylate obtained in Reference Example 20 in the same manner as Reference Example 18 Was obtained as a solid.
1 H NMR (DMSO-d 6 ) δ: 2.09-2.21 (4H, m), 3.01-3.04 (2H, m), 3.41 (3H, d, J = 11.6 Hz) ), 7.45 (2H, q, J = 7.2 Hz), 7.64 (1H, q, J = 7.2 Hz), 7.81 (1H, t, J = 7.2 Hz), 7.91 (1H, brs), 7.95 (1H, d, J = 4.0 Hz), 8.82 (1H, d, J = 5.2 Hz), 9.29 (1H, brs), 9.44 (1H , Brs).
4-(2-フルオロフェニル)-2-ピペリジン-4-イルピリジン二塩酸塩
1H NMR(DMSO-d6)δ:2.09-2.21(4H,m),3.01-3.04(2H,m),3.41(3H,d,J=11.6Hz),7.45(2H,q,J=7.2Hz),7.64(1H,q,J=7.2Hz),7.81(1H,t,J=7.2Hz),7.91(1H,brs),7.95(1H,d,J=4.0Hz),8.82(1H,d,J=5.2Hz),9.29(1H,brs),9.44(1H,brs)。 Reference Example 21
4- (2-Fluorophenyl) -2-piperidin-4-ylpyridine dihydrochloride
1 H NMR (DMSO-d 6 ) δ: 2.09-2.21 (4H, m), 3.01-3.04 (2H, m), 3.41 (3H, d, J = 11.6 Hz) ), 7.45 (2H, q, J = 7.2 Hz), 7.64 (1H, q, J = 7.2 Hz), 7.81 (1H, t, J = 7.2 Hz), 7.91 (1H, brs), 7.95 (1H, d, J = 4.0 Hz), 8.82 (1H, d, J = 5.2 Hz), 9.29 (1H, brs), 9.44 (1H , Brs).
参考例22
tert-ブチル 4-(3-フルオロフェニル)-3’,6’-ジヒドロ-2,4’-ビピリジン-1’(2’H)-カルボキシラート

参考例16と同様にして、2-クロロ-4-(3-フルオロフェニル)ピリジンから表題化合物(4.77g、93%)を固体として得た。
1H NMR(CDCl3)δ:1.49(9H,s),2.69-2.72(2H,m),3.68(2H,brs),4.16(2H,d,J=2.0Hz),6.67-6.70(1H,m),7.12-7.17(1H,m),7.31-7.36(2H,m),7.41-7.49(2H,m),7.54(1H,brs),8.63(1H,dd,J=5.2,0.8Hz)。 Reference Example 22
tert-Butyl 4- (3-fluorophenyl) -3 ′, 6′-dihydro-2,4′-bipyridine-1 ′ (2′H) -carboxylate
In the same manner as in Reference Example 16, the title compound (4.77 g, 93%) was obtained as a solid from 2-chloro-4- (3-fluorophenyl) pyridine.
1 H NMR (CDCl 3 ) δ: 1.49 (9H, s), 2.69-2.72 (2H, m), 3.68 (2H, brs), 4.16 (2H, d, J = 2.0 Hz), 6.67-6.70 (1 H, m), 7.12-7.17 (1 H, m), 7.31-7.36 (2 H, m), 7.41-7. 49 (2H, m), 7.54 (1H, brs), 8.63 (1H, dd, J = 5.2, 0.8 Hz).
tert-ブチル 4-(3-フルオロフェニル)-3’,6’-ジヒドロ-2,4’-ビピリジン-1’(2’H)-カルボキシラート
1H NMR(CDCl3)δ:1.49(9H,s),2.69-2.72(2H,m),3.68(2H,brs),4.16(2H,d,J=2.0Hz),6.67-6.70(1H,m),7.12-7.17(1H,m),7.31-7.36(2H,m),7.41-7.49(2H,m),7.54(1H,brs),8.63(1H,dd,J=5.2,0.8Hz)。 Reference Example 22
tert-Butyl 4- (3-fluorophenyl) -3 ′, 6′-dihydro-2,4′-bipyridine-1 ′ (2′H) -carboxylate
1 H NMR (CDCl 3 ) δ: 1.49 (9H, s), 2.69-2.72 (2H, m), 3.68 (2H, brs), 4.16 (2H, d, J = 2.0 Hz), 6.67-6.70 (1 H, m), 7.12-7.17 (1 H, m), 7.31-7.36 (2 H, m), 7.41-7. 49 (2H, m), 7.54 (1H, brs), 8.63 (1H, dd, J = 5.2, 0.8 Hz).
参考例23
tert-ブチル 4-[4-(3-フルオロフェニル)ピリジン-2-イル]ピペリジン-1-カルボキシラート

参考例17と同様にして、参考例22で得たtert-ブチル 4-(3-フルオロフェニル)-3’,6’-ジヒドロ-2,4’-ビピリジン-1’(2’H)-カルボキシラートから表題化合物(4.80g、quant.)を油状物として得た。
1H NMR(CDCl3)δ:1.49(9H,s),1.73-1.84(2H,m),1.97(2H,d,J=13.2Hz),2.86-2.96(3H,m),4.29(2H,brs),7.11-7.16(1H,m),7.30-7.34(3H,m),7.39-7.49(2H,m),8.60(1H,dd,J=4.8,1.2Hz)。 Reference Example 23
tert-Butyl 4- [4- (3-fluorophenyl) pyridin-2-yl] piperidine-1-carboxylate
In the same manner as in Reference Example 17, tert-butyl 4- (3-fluorophenyl) -3 ′, 6′-dihydro-2,4′-bipyridine-1 ′ (2′H) -carboxy obtained in Reference Example 22 The title compound (4.80 g, quant.) Was obtained from oil as an oil.
1 H NMR (CDCl 3 ) δ: 1.49 (9H, s), 1.73-1.84 (2H, m), 1.97 (2H, d, J = 13.2 Hz), 2.86- 2.96 (3H, m), 4.29 (2H, brs), 7.11-7.16 (1H, m), 7.30-7.34 (3H, m), 7.39-7. 49 (2H, m), 8.60 (1H, dd, J = 4.8, 1.2 Hz).
tert-ブチル 4-[4-(3-フルオロフェニル)ピリジン-2-イル]ピペリジン-1-カルボキシラート
1H NMR(CDCl3)δ:1.49(9H,s),1.73-1.84(2H,m),1.97(2H,d,J=13.2Hz),2.86-2.96(3H,m),4.29(2H,brs),7.11-7.16(1H,m),7.30-7.34(3H,m),7.39-7.49(2H,m),8.60(1H,dd,J=4.8,1.2Hz)。 Reference Example 23
tert-Butyl 4- [4- (3-fluorophenyl) pyridin-2-yl] piperidine-1-carboxylate
1 H NMR (CDCl 3 ) δ: 1.49 (9H, s), 1.73-1.84 (2H, m), 1.97 (2H, d, J = 13.2 Hz), 2.86- 2.96 (3H, m), 4.29 (2H, brs), 7.11-7.16 (1H, m), 7.30-7.34 (3H, m), 7.39-7. 49 (2H, m), 8.60 (1H, dd, J = 4.8, 1.2 Hz).
参考例24
4-(3-フルオロフェニル)-2-ピペリジン-4-イルピリジン二塩酸塩

参考例18と同様にして、参考例23で得たtert-ブチル 4-[4-(3-フルオロフェニル)ピリジン-2-イル]ピペリジン-1-カルボキシラートから表題化合物(3.50g、71%)を固体として得た。
1H NMR(DMSO-d6)δ:2.10-2.23(4H,m),2.99-3.08(2H,m),3.40-3.43(3H,m),7.46(1H,td,J=8.4,2.4Hz),7.63-7.69(1H,m),7.86(1H,d,J=8.0Hz),7.93(1H,d,J=10.0Hz),8.07(1H,s),8.12(1H,d,J=5.2Hz),8.81(1H,d,J=5.6Hz),9.23(1H,brs),9.39(1H,brs)。 Reference Example 24
4- (3-Fluorophenyl) -2-piperidin-4-ylpyridine dihydrochloride
In the same manner as in Reference Example 18, from the tert-butyl 4- [4- (3-fluorophenyl) pyridin-2-yl] piperidine-1-carboxylate obtained in Reference Example 23, the title compound (3.50 g, 71% ) Was obtained as a solid.
1 H NMR (DMSO-d 6 ) δ: 2.10-2.23 (4H, m), 2.99-3.08 (2H, m), 3.40-3.43 (3H, m), 7.46 (1H, td, J = 8.4, 2.4 Hz), 7.63-7.69 (1H, m), 7.86 (1H, d, J = 8.0 Hz), 7.93 (1H, d, J = 10.0 Hz), 8.07 (1 H, s), 8.12 (1 H, d, J = 5.2 Hz), 8.81 (1 H, d, J = 5.6 Hz) , 9.23 (1H, brs), 9.39 (1H, brs).
4-(3-フルオロフェニル)-2-ピペリジン-4-イルピリジン二塩酸塩
1H NMR(DMSO-d6)δ:2.10-2.23(4H,m),2.99-3.08(2H,m),3.40-3.43(3H,m),7.46(1H,td,J=8.4,2.4Hz),7.63-7.69(1H,m),7.86(1H,d,J=8.0Hz),7.93(1H,d,J=10.0Hz),8.07(1H,s),8.12(1H,d,J=5.2Hz),8.81(1H,d,J=5.6Hz),9.23(1H,brs),9.39(1H,brs)。 Reference Example 24
4- (3-Fluorophenyl) -2-piperidin-4-ylpyridine dihydrochloride
1 H NMR (DMSO-d 6 ) δ: 2.10-2.23 (4H, m), 2.99-3.08 (2H, m), 3.40-3.43 (3H, m), 7.46 (1H, td, J = 8.4, 2.4 Hz), 7.63-7.69 (1H, m), 7.86 (1H, d, J = 8.0 Hz), 7.93 (1H, d, J = 10.0 Hz), 8.07 (1 H, s), 8.12 (1 H, d, J = 5.2 Hz), 8.81 (1 H, d, J = 5.6 Hz) , 9.23 (1H, brs), 9.39 (1H, brs).
参考例25
tert-ブチル 4-(4-フルオロフェニル)-3’,6’-ジヒドロ-2,4’-ビピリジン-1’(2’H)-カルボキシラート

参考例16と同様にして2-クロロ-4-(4-フルオロフェニル)ピリジンから表題化合物(4.77g、93%)を固体として得た。
1H NMR(CDCl3)δ:1.50(9H,s),2.69-2.72(2H,m),3.68(2H,brs),4.16(2H,d,J=2.4Hz),6.67-6.68(1H,m),7.16-7.21(2H,m),7.33(1H,dd,J=5.2,1.6Hz),7.52(1H,brs),7.60-7.63(2H,m),8.61(1H,dd,J=5.2,0.8Hz)。 Reference Example 25
tert-Butyl 4- (4-fluorophenyl) -3 ′, 6′-dihydro-2,4′-bipyridine-1 ′ (2′H) -carboxylate
In the same manner as in Reference Example 16, the title compound (4.77 g, 93%) was obtained as a solid from 2-chloro-4- (4-fluorophenyl) pyridine.
1 H NMR (CDCl 3 ) δ: 1.50 (9H, s), 2.69-2.72 (2H, m), 3.68 (2H, brs), 4.16 (2H, d, J = 2.4 Hz), 6.67-6.68 (1 H, m), 7.16-7.21 (2 H, m), 7.33 (1 H, dd, J = 5.2, 1.6 Hz), 7.52 (1H, brs), 7.60-7.63 (2H, m), 8.61 (1H, dd, J = 5.2, 0.8 Hz).
tert-ブチル 4-(4-フルオロフェニル)-3’,6’-ジヒドロ-2,4’-ビピリジン-1’(2’H)-カルボキシラート
1H NMR(CDCl3)δ:1.50(9H,s),2.69-2.72(2H,m),3.68(2H,brs),4.16(2H,d,J=2.4Hz),6.67-6.68(1H,m),7.16-7.21(2H,m),7.33(1H,dd,J=5.2,1.6Hz),7.52(1H,brs),7.60-7.63(2H,m),8.61(1H,dd,J=5.2,0.8Hz)。 Reference Example 25
tert-Butyl 4- (4-fluorophenyl) -3 ′, 6′-dihydro-2,4′-bipyridine-1 ′ (2′H) -carboxylate
1 H NMR (CDCl 3 ) δ: 1.50 (9H, s), 2.69-2.72 (2H, m), 3.68 (2H, brs), 4.16 (2H, d, J = 2.4 Hz), 6.67-6.68 (1 H, m), 7.16-7.21 (2 H, m), 7.33 (1 H, dd, J = 5.2, 1.6 Hz), 7.52 (1H, brs), 7.60-7.63 (2H, m), 8.61 (1H, dd, J = 5.2, 0.8 Hz).
参考例26
4-(4-フルオロフェニル)-2-ピペリジン-4-イルピリジン二塩酸塩

参考例17と同様にして、参考例25で得たtert-ブチル 4-(4-フルオロフェニル)-3’,6’-ジヒドロ-2,4’-ビピリジン-1’(2’H)-カルボキシラートからtert-ブチル 4-[4-(4-フルオロフェニル)ピリジン-2-イル]ピペリジン-1-カルボキシラート(4.80g、quant.)を油状物として得た。この化合物(4.80g、13.5ミリモル)をメタノール(27ml)に溶解させ、4.0Mの塩化水素/メタノール溶液(13.5ml、53.9ミリモル)を加え、室温で24時間撹拌した。生じた固体をろ取し、酢酸エチルで洗浄し、乾燥して表題化合物(3.80g、86%)を固体として得た。
1H NMR(DMSO-d6)δ:2.07-2.19(4H,m),2.99-3.02(2H,m),3.37-3.40(3H,m),7.44(2H,t,J=8.6Hz),7.99-8.05(4H,m),8.75(1H,d,J=5.6Hz),9.12(1H,brs),9.29(1H,brs)。 Reference Example 26
4- (4-Fluorophenyl) -2-piperidin-4-ylpyridine dihydrochloride
In the same manner as in Reference Example 17, tert-butyl 4- (4-fluorophenyl) -3 ′, 6′-dihydro-2,4′-bipyridine-1 ′ (2′H) -carboxy obtained in Reference Example 25 The tert-butyl 4- [4- (4-fluorophenyl) pyridin-2-yl] piperidine-1-carboxylate (4.80 g, quant.) Was obtained as an oil from the late. This compound (4.80 g, 13.5 mmol) was dissolved in methanol (27 ml), 4.0 M hydrogen chloride / methanol solution (13.5 ml, 53.9 mmol) was added, and the mixture was stirred at room temperature for 24 hours. The resulting solid was collected by filtration, washed with ethyl acetate and dried to give the title compound (3.80 g, 86%) as a solid.
1 H NMR (DMSO-d 6 ) δ: 2.07-2.19 (4H, m), 2.99-3.02 (2H, m), 3.37-3.40 (3H, m), 7.44 (2H, t, J = 8.6 Hz), 7.99-8.05 (4H, m), 8.75 (1H, d, J = 5.6 Hz), 9.12 (1H, brs) ), 9.29 (1H, brs).
4-(4-フルオロフェニル)-2-ピペリジン-4-イルピリジン二塩酸塩
1H NMR(DMSO-d6)δ:2.07-2.19(4H,m),2.99-3.02(2H,m),3.37-3.40(3H,m),7.44(2H,t,J=8.6Hz),7.99-8.05(4H,m),8.75(1H,d,J=5.6Hz),9.12(1H,brs),9.29(1H,brs)。 Reference Example 26
4- (4-Fluorophenyl) -2-piperidin-4-ylpyridine dihydrochloride
1 H NMR (DMSO-d 6 ) δ: 2.07-2.19 (4H, m), 2.99-3.02 (2H, m), 3.37-3.40 (3H, m), 7.44 (2H, t, J = 8.6 Hz), 7.99-8.05 (4H, m), 8.75 (1H, d, J = 5.6 Hz), 9.12 (1H, brs) ), 9.29 (1H, brs).
参考例27
2-クロロ-4-(2,3-ジフルオロフェニル)ピリジン

4-ブロモ-2-クロロピリジン(5.00g、26.0ミリモル)、2,3-ジフルオロフェニルボロン酸(4.10g、26.0ミリモル)、炭酸カリウム(7.18g、52.0ミリモル)のメタノール(90ml)溶液に、テトラキストリフェニルホスフィンパラジウム(1.50g、1.30ミリモル)を室温で加え、50℃で17時間攪拌した。反応液に水を注ぎ、塩化メチレンで抽出した。抽出液を無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:8)で精製して表題化合物(4.30g、73%)を固体として得た。
1H NMR(CDCl3)δ:7.18-7.31(3H,m),7.41-7.43(1H,m),7.52-7.53(1H,m),8.48(1H,dd,J=5.0,1.6Hz)。 Reference Example 27
2-Chloro-4- (2,3-difluorophenyl) pyridine
4-Bromo-2-chloropyridine (5.00 g, 26.0 mmol), 2,3-difluorophenylboronic acid (4.10 g, 26.0 mmol), potassium carbonate (7.18 g, 52.0 mmol) Tetrakistriphenylphosphine palladium (1.50 g, 1.30 mmol) was added to a methanol (90 ml) solution at room temperature, and the mixture was stirred at 50 ° C. for 17 hours. Water was poured into the reaction solution and extracted with methylene chloride. The extract was dried over anhydrous magnesium sulfate and the solvent was removed under reduced pressure. The residue was purified by silica gel column chromatography (ethyl acetate: hexane = 1: 8) to give the title compound (4.30 g, 73%) as a solid.
1 H NMR (CDCl 3 ) δ: 7.18-7.31 (3H, m), 7.41-7.43 (1H, m), 7.52-7.53 (1H, m), 8. 48 (1H, dd, J = 5.0, 1.6 Hz).
2-クロロ-4-(2,3-ジフルオロフェニル)ピリジン
1H NMR(CDCl3)δ:7.18-7.31(3H,m),7.41-7.43(1H,m),7.52-7.53(1H,m),8.48(1H,dd,J=5.0,1.6Hz)。 Reference Example 27
2-Chloro-4- (2,3-difluorophenyl) pyridine
1 H NMR (CDCl 3 ) δ: 7.18-7.31 (3H, m), 7.41-7.43 (1H, m), 7.52-7.53 (1H, m), 8. 48 (1H, dd, J = 5.0, 1.6 Hz).
参考例28
tert-ブチル 4-(2,3-ジフルオロフェニル)-3’,6’-ジヒドロ-2,4’-ビピリジン-1’(2’H)-カルボキシラート

参考例16と同様にして、参考例27で得た2-クロロ-4-(2,3-ジフルオロフェニル)ピリジンから表題化合物(6.50g、98%)を固体として得た。
1H NMR(CDCl3)δ:1.49(9H,s),2.68-2.71(2H,m),3.67(2H,t,J=5.4Hz),4.15-4.16(2H,m),6.66-6.69(1H,m),7.18-7.28(3H,m),7.32-7.34(1H,m),7.53(1H,brs),8.65(1H,dd,J=5.2,0.8Hz)。 Reference Example 28
tert-Butyl 4- (2,3-difluorophenyl) -3 ′, 6′-dihydro-2,4′-bipyridine-1 ′ (2′H) -carboxylate
In the same manner as in Reference Example 16, the title compound (6.50 g, 98%) was obtained as a solid from 2-chloro-4- (2,3-difluorophenyl) pyridine obtained in Reference Example 27.
1 H NMR (CDCl 3 ) δ: 1.49 (9H, s), 2.68-2.71 (2H, m), 3.67 (2H, t, J = 5.4 Hz), 4.15- 4.16 (2H, m), 6.66-6.69 (1H, m), 7.18-7.28 (3H, m), 7.32-7.34 (1H, m), 7. 53 (1H, brs), 8.65 (1H, dd, J = 5.2, 0.8 Hz).
tert-ブチル 4-(2,3-ジフルオロフェニル)-3’,6’-ジヒドロ-2,4’-ビピリジン-1’(2’H)-カルボキシラート
1H NMR(CDCl3)δ:1.49(9H,s),2.68-2.71(2H,m),3.67(2H,t,J=5.4Hz),4.15-4.16(2H,m),6.66-6.69(1H,m),7.18-7.28(3H,m),7.32-7.34(1H,m),7.53(1H,brs),8.65(1H,dd,J=5.2,0.8Hz)。 Reference Example 28
tert-Butyl 4- (2,3-difluorophenyl) -3 ′, 6′-dihydro-2,4′-bipyridine-1 ′ (2′H) -carboxylate
1 H NMR (CDCl 3 ) δ: 1.49 (9H, s), 2.68-2.71 (2H, m), 3.67 (2H, t, J = 5.4 Hz), 4.15- 4.16 (2H, m), 6.66-6.69 (1H, m), 7.18-7.28 (3H, m), 7.32-7.34 (1H, m), 7. 53 (1H, brs), 8.65 (1H, dd, J = 5.2, 0.8 Hz).
参考例29
4-(2,3-ジフルオロフェニル)-2-ピペリジン-4-イルピリジン二塩酸塩

参考例17と同様にして、参考例28で得たtert-ブチル 4-(2,3-ジフルオロフェニル)-3’,6’-ジヒドロ-2,4’-ビピリジン-1’(2’H)-カルボキシラートからtert-ブチル 4-[4-(2,3-ジフルオロフェニル)ピリジン-2-イル]ピペリジン-1-カルボキシラート(6.50g、99%)を油状物として得た。この化合物(6.50g、17.4ミリモル)をメタノール(35ml)に溶解させ、4.0Mの塩化水素/メタノール溶液(17.4ml、69.4ミリモル)を加え、室温で24時間撹拌した。生じた固体をろ取し、酢酸エチルで洗浄し、乾燥して表題化合物(4.88g、81%)を固体として得た。
1H NMR(DMSO-d6)δ:2.04-2.17(4H,m),3.01-3.06(2H,m),3.32-3.41(3H,m),7.42(1H,q,J=4.8Hz),7.56(1H,t,J=6.8Hz),7.64(1H,q,J=9.2Hz),7.80(2H,brs),8.79(1H,d,J=5.2Hz),9.11(1H,brs),9.31(1H,brs)。 Reference Example 29
4- (2,3-Difluorophenyl) -2-piperidin-4-ylpyridine dihydrochloride
In the same manner as in Reference Example 17, tert-butyl 4- (2,3-difluorophenyl) -3 ′, 6′-dihydro-2,4′-bipyridine-1 ′ (2′H) obtained in Reference Example 28 The carboxylate gave tert-butyl 4- [4- (2,3-difluorophenyl) pyridin-2-yl] piperidine-1-carboxylate (6.50 g, 99%) as an oil. This compound (6.50 g, 17.4 mmol) was dissolved in methanol (35 ml), 4.0 M hydrogen chloride / methanol solution (17.4 ml, 69.4 mmol) was added, and the mixture was stirred at room temperature for 24 hours. The resulting solid was collected by filtration, washed with ethyl acetate, and dried to give the title compound (4.88 g, 81%) as a solid.
1 H NMR (DMSO-d6) δ: 2.04-2.17 (4H, m), 3.01-3.06 (2H, m), 3.32-3.41 (3H, m), 7 .42 (1H, q, J = 4.8 Hz), 7.56 (1H, t, J = 6.8 Hz), 7.64 (1H, q, J = 9.2 Hz), 7.80 (2H, brs), 8.79 (1H, d, J = 5.2 Hz), 9.11 (1H, brs), 9.31 (1H, brs).
4-(2,3-ジフルオロフェニル)-2-ピペリジン-4-イルピリジン二塩酸塩
1H NMR(DMSO-d6)δ:2.04-2.17(4H,m),3.01-3.06(2H,m),3.32-3.41(3H,m),7.42(1H,q,J=4.8Hz),7.56(1H,t,J=6.8Hz),7.64(1H,q,J=9.2Hz),7.80(2H,brs),8.79(1H,d,J=5.2Hz),9.11(1H,brs),9.31(1H,brs)。 Reference Example 29
4- (2,3-Difluorophenyl) -2-piperidin-4-ylpyridine dihydrochloride
1 H NMR (DMSO-d6) δ: 2.04-2.17 (4H, m), 3.01-3.06 (2H, m), 3.32-3.41 (3H, m), 7 .42 (1H, q, J = 4.8 Hz), 7.56 (1H, t, J = 6.8 Hz), 7.64 (1H, q, J = 9.2 Hz), 7.80 (2H, brs), 8.79 (1H, d, J = 5.2 Hz), 9.11 (1H, brs), 9.31 (1H, brs).
参考例30
2-クロロ-4-(2,4-ジフルオロフェニル)ピリジン

参考例27と同様にして4-ブロモ-2-クロロピリジンから表題化合物(3.70g、79%)を固体として得た。
1H NMR(CDCl3)δ:6.95-7.05(2H,s),7.37-7.39(1H,m),7.43-7.47(1H,m),7.49(1H,brs),8.47(1H,d,J=13.2Hz)。 Reference Example 30
2-Chloro-4- (2,4-difluorophenyl) pyridine
In the same manner as in Reference Example 27, the title compound (3.70 g, 79%) was obtained as a solid from 4-bromo-2-chloropyridine.
1 H NMR (CDCl 3 ) δ: 6.95-7.05 (2H, s), 7.37-7.39 (1H, m), 7.43-7.47 (1H, m), 7. 49 (1H, brs), 8.47 (1H, d, J = 13.2 Hz).
2-クロロ-4-(2,4-ジフルオロフェニル)ピリジン
1H NMR(CDCl3)δ:6.95-7.05(2H,s),7.37-7.39(1H,m),7.43-7.47(1H,m),7.49(1H,brs),8.47(1H,d,J=13.2Hz)。 Reference Example 30
2-Chloro-4- (2,4-difluorophenyl) pyridine
1 H NMR (CDCl 3 ) δ: 6.95-7.05 (2H, s), 7.37-7.39 (1H, m), 7.43-7.47 (1H, m), 7. 49 (1H, brs), 8.47 (1H, d, J = 13.2 Hz).
参考例31
tert-ブチル 4-(2,4-ジフルオロフェニル)-3’,6’-ジヒドロ-2,4’-ビピリジン-1’(2’H)-カルボキシラート
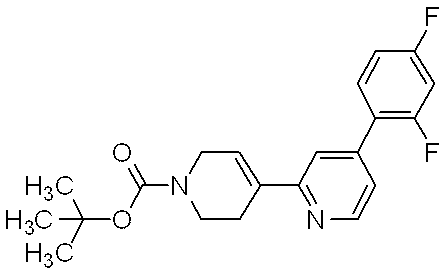
参考例16と同様にして、参考例30で得た2-クロロ-4-(2,4-ジフルオロフェニル)ピリジンから表題化合物(4.15g、68%)を固体として得た。
1H NMR(CDCl3)δ:1.50(9H,s),2.67-2.71(2H,m),3.67(2H,t,J=5.6Hz),4.15-4.16(2H,m),6.65-6.67(1H,m),6.93-7.04(2H,m),7.29-7.31(1H,m),7.43-7.49(1H,m),7.50(1H,brs),8.62(1H,dd,J=5.2,0.8Hz)。 Reference Example 31
tert-Butyl 4- (2,4-difluorophenyl) -3 ′, 6′-dihydro-2,4′-bipyridine-1 ′ (2′H) -carboxylate
In the same manner as in Reference Example 16, the title compound (4.15 g, 68%) was obtained as a solid from 2-chloro-4- (2,4-difluorophenyl) pyridine obtained in Reference Example 30.
1 H NMR (CDCl 3 ) δ: 1.50 (9H, s), 2.67-2.71 (2H, m), 3.67 (2H, t, J = 5.6 Hz), 4.15- 4.16 (2H, m), 6.65-6.67 (1H, m), 6.93-7.04 (2H, m), 7.29-7.31 (1H, m), 7. 43-7.49 (1H, m), 7.50 (1H, brs), 8.62 (1H, dd, J = 5.2, 0.8 Hz).
tert-ブチル 4-(2,4-ジフルオロフェニル)-3’,6’-ジヒドロ-2,4’-ビピリジン-1’(2’H)-カルボキシラート
1H NMR(CDCl3)δ:1.50(9H,s),2.67-2.71(2H,m),3.67(2H,t,J=5.6Hz),4.15-4.16(2H,m),6.65-6.67(1H,m),6.93-7.04(2H,m),7.29-7.31(1H,m),7.43-7.49(1H,m),7.50(1H,brs),8.62(1H,dd,J=5.2,0.8Hz)。 Reference Example 31
tert-Butyl 4- (2,4-difluorophenyl) -3 ′, 6′-dihydro-2,4′-bipyridine-1 ′ (2′H) -carboxylate
1 H NMR (CDCl 3 ) δ: 1.50 (9H, s), 2.67-2.71 (2H, m), 3.67 (2H, t, J = 5.6 Hz), 4.15- 4.16 (2H, m), 6.65-6.67 (1H, m), 6.93-7.04 (2H, m), 7.29-7.31 (1H, m), 7. 43-7.49 (1H, m), 7.50 (1H, brs), 8.62 (1H, dd, J = 5.2, 0.8 Hz).
参考例32
4-(2,4-ジフルオロフェニル)-2-ピペリジン-4-イルピリジン二塩酸塩

参考例17と同様にして、参考例31で得たtert-ブチル 4-(2,4-ジフルオロフェニル)-3’,6’-ジヒドロ-2,4’-ビピリジン-1’(2’H)-カルボキシラートからtert-ブチル 4-[4-(2,4-ジフルオロフェニル)ピリジン-2-イル]ピペリジン-1-カルボキシラート(4.07g、98%)を油状物として得た。この化合物(4.07g、10.9ミリモル)をメタノール(36ml)に溶解させ、4.0Mの塩化水素/メタノール溶液(10.1ml、43.5ミリモル)を加え、室温で24時間撹拌した。生じた固体をろ取し、酢酸エチルで洗浄し、乾燥して表題化合物(3.40g、82%)を固体として得た。
1H NMR(DMSO-d6)δ:2.01-2.15(4H,m),2.98-3.07(2H,m),3.28(1H,t,J=11.4Hz),3.39(2H,d,J=12.4Hz),7.33(1H,td,J=8.4,2.0Hz),7.49-7.55(1H,m),7.74-7.87(3H,m),8.75(1H,d,J=5.2Hz),9.01(1H,brs),9.21(1H,brs)。 Reference Example 32
4- (2,4-Difluorophenyl) -2-piperidin-4-ylpyridine dihydrochloride
In the same manner as in Reference Example 17, tert-butyl 4- (2,4-difluorophenyl) -3 ′, 6′-dihydro-2,4′-bipyridine-1 ′ (2′H) obtained in Reference Example 31 The carboxylate gave tert-butyl 4- [4- (2,4-difluorophenyl) pyridin-2-yl] piperidine-1-carboxylate (4.07 g, 98%) as an oil. This compound (4.07 g, 10.9 mmol) was dissolved in methanol (36 ml), 4.0 M hydrogen chloride / methanol solution (10.1 ml, 43.5 mmol) was added, and the mixture was stirred at room temperature for 24 hours. The resulting solid was collected by filtration, washed with ethyl acetate and dried to give the title compound (3.40 g, 82%) as a solid.
1 H NMR (DMSO-d 6 ) δ: 2.01-2.15 (4H, m), 2.98-3.07 (2H, m), 3.28 (1H, t, J = 11.4 Hz ), 3.39 (2H, d, J = 12.4 Hz), 7.33 (1H, td, J = 8.4, 2.0 Hz), 7.49-7.55 (1H, m), 7 .74-7.87 (3H, m), 8.75 (1H, d, J = 5.2 Hz), 9.01 (1H, brs), 9.21 (1H, brs).
4-(2,4-ジフルオロフェニル)-2-ピペリジン-4-イルピリジン二塩酸塩
1H NMR(DMSO-d6)δ:2.01-2.15(4H,m),2.98-3.07(2H,m),3.28(1H,t,J=11.4Hz),3.39(2H,d,J=12.4Hz),7.33(1H,td,J=8.4,2.0Hz),7.49-7.55(1H,m),7.74-7.87(3H,m),8.75(1H,d,J=5.2Hz),9.01(1H,brs),9.21(1H,brs)。 Reference Example 32
4- (2,4-Difluorophenyl) -2-piperidin-4-ylpyridine dihydrochloride
1 H NMR (DMSO-d 6 ) δ: 2.01-2.15 (4H, m), 2.98-3.07 (2H, m), 3.28 (1H, t, J = 11.4 Hz ), 3.39 (2H, d, J = 12.4 Hz), 7.33 (1H, td, J = 8.4, 2.0 Hz), 7.49-7.55 (1H, m), 7 .74-7.87 (3H, m), 8.75 (1H, d, J = 5.2 Hz), 9.01 (1H, brs), 9.21 (1H, brs).
参考例33
2-クロロ-4-フラン-3-イルピリミジン

2,4-ジクロロピリミジン(1.00g、6.71ミリモル)、フラン-3-イルボロン酸(826.1mg、7.38ミリモル)、2N炭酸ナトリウム水溶液(6.7ml)の1,2-ジメトキシエタン(10ml)溶液に、窒素雰囲気下、テトラキストリフェニルホスフィンパラジウム(931mg、0.805ミリモル)を室温で加え、マイクロウェーブリアクター(Initiator TM、Biotage社)によって、105℃で40分間攪拌した。反応液に水を注ぎ、酢酸エチルで抽出した。抽出液を水で洗浄し、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:4)で精製し、表題化合物(916mg、76%)を固体として得た。
1H NMR(CDCl3)δ:6.88-6.93(1H,m),7.29-7.35(1H,m),7.52-7.56(1H,m),8.21-8.25(1H,m),8.55(1H,d,J=4.9Hz)。 Reference Example 33
2-Chloro-4-furan-3-ylpyrimidine
1,2-Dimethoxyethane in 2,4-dichloropyrimidine (1.00 g, 6.71 mmol), furan-3-ylboronic acid (826.1 mg, 7.38 mmol), 2N aqueous sodium carbonate solution (6.7 ml) Tetrakistriphenylphosphine palladium (931 mg, 0.805 mmol) was added to the (10 ml) solution at room temperature under a nitrogen atmosphere, and the mixture was stirred at 105 ° C. for 40 minutes by a microwave reactor (Initiator ™, Biotage). Water was poured into the reaction mixture, and the mixture was extracted with ethyl acetate. The extract was washed with water and dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure. The residue was purified by silica gel column chromatography (ethyl acetate: hexane = 1: 4) to give the title compound (916 mg, 76%) as a solid.
1 H NMR (CDCl 3 ) δ: 6.88-6.93 (1H, m), 7.29-7.35 (1H, m), 7.52-7.56 (1H, m), 8. 21-8.25 (1H, m), 8.55 (1H, d, J = 4.9 Hz).
2-クロロ-4-フラン-3-イルピリミジン
1H NMR(CDCl3)δ:6.88-6.93(1H,m),7.29-7.35(1H,m),7.52-7.56(1H,m),8.21-8.25(1H,m),8.55(1H,d,J=4.9Hz)。 Reference Example 33
2-Chloro-4-furan-3-ylpyrimidine
1 H NMR (CDCl 3 ) δ: 6.88-6.93 (1H, m), 7.29-7.35 (1H, m), 7.52-7.56 (1H, m), 8. 21-8.25 (1H, m), 8.55 (1H, d, J = 4.9 Hz).
参考例34
tert-ブチル 4-(4-フラン-3-イルピリミジン-2-イル)-3,6-ジヒドロピリジン-1(2H)-カルボキシラート

参考例33で得た2-クロロ-4-フラン-3-イルピリミジン(500mg、2.77ミリモル)、tert-ブチル 4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-3,6-ジヒドロピリジン-1(2H)-カルボキシラート(942mg、3.05ミリモル)、2N炭酸ナトリウム水溶液(4.2ml)の1,2-ジメトキシエタン(7.5ml)溶液に、窒素雰囲気下、テトラキストリフェニルホスフィンパラジウム(384mg、0.332ミリモル)を室温で加え、マイクロウェーブリアクター(Initiator TM、Biotage社)によって、105℃で40分間攪拌した。反応液に水を注ぎ、酢酸エチルで抽出した。抽出液を水で洗浄し、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:4)で精製し、表題化合物(782mg、86%)を油状物として得た。
1H NMR(CDCl3)δ:1.50(9H,s),2.77(2H,brs),3.59-3.71(2H,m),4.12-4.27(2H,m),6.89-6.96(1H,m),7.20(1H,d,J=5.3Hz),7.24(1H,brs),7.53(1H,t,J=1.7Hz),8.17(1H,s),8.64(1H,d,J=5.3Hz)。 Reference Example 34
tert-Butyl 4- (4-furan-3-ylpyrimidin-2-yl) -3,6-dihydropyridine-1 (2H) -carboxylate
2-Chloro-4-furan-3-ylpyrimidine obtained in Reference Example 33 (500 mg, 2.77 mmol), tert-butyl 4- (4,4,5,5-tetramethyl-1,3,2- Dioxaborolan-2-yl) -3,6-dihydropyridine-1 (2H) -carboxylate (942 mg, 3.05 mmol), 1,2-dimethoxyethane (7.5 ml) in 2N aqueous sodium carbonate (4.2 ml) Tetrakistriphenylphosphine palladium (384 mg, 0.332 mmol) was added to the solution at room temperature under a nitrogen atmosphere, and the mixture was stirred at 105 ° C. for 40 minutes by a microwave reactor (Initiator ™, Biotage). Water was poured into the reaction mixture, and the mixture was extracted with ethyl acetate. The extract was washed with water and dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure. The residue was purified by silica gel column chromatography (ethyl acetate: hexane = 1: 4) to give the title compound (782 mg, 86%) as an oil.
1 H NMR (CDCl 3 ) δ: 1.50 (9H, s), 2.77 (2H, brs), 3.59-3.71 (2H, m), 4.12-4.27 (2H, m), 6.89-6.96 (1H, m), 7.20 (1H, d, J = 5.3 Hz), 7.24 (1H, brs), 7.53 (1H, t, J = 1.7 Hz), 8.17 (1 H, s), 8.64 (1 H, d, J = 5.3 Hz).
tert-ブチル 4-(4-フラン-3-イルピリミジン-2-イル)-3,6-ジヒドロピリジン-1(2H)-カルボキシラート
1H NMR(CDCl3)δ:1.50(9H,s),2.77(2H,brs),3.59-3.71(2H,m),4.12-4.27(2H,m),6.89-6.96(1H,m),7.20(1H,d,J=5.3Hz),7.24(1H,brs),7.53(1H,t,J=1.7Hz),8.17(1H,s),8.64(1H,d,J=5.3Hz)。 Reference Example 34
tert-Butyl 4- (4-furan-3-ylpyrimidin-2-yl) -3,6-dihydropyridine-1 (2H) -carboxylate
1 H NMR (CDCl 3 ) δ: 1.50 (9H, s), 2.77 (2H, brs), 3.59-3.71 (2H, m), 4.12-4.27 (2H, m), 6.89-6.96 (1H, m), 7.20 (1H, d, J = 5.3 Hz), 7.24 (1H, brs), 7.53 (1H, t, J = 1.7 Hz), 8.17 (1 H, s), 8.64 (1 H, d, J = 5.3 Hz).
参考例35
tert-ブチル 4-(4-フラン-3-イルピリミジン-2-イル)ピペリジン-1-カルボキシラート

参考例34で得たtert-ブチル 4-(4-フラン-3-イルピリミジン-2-イル)-3,6-ジヒドロピリジン-1(2H)-カルボキシラート(780mg、2.38ミリモル)のテトラヒドロフラン(12ml)溶液に、窒素雰囲気下、パラジウム炭素(156mg)を加え、反応溶液を水素雰囲気下、室温で6時間攪拌した。反応溶液をろ過し、ろ液を濃縮した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:4)で精製し、表題化合物(542mg、69%)を油状物として得た。
1H NMR(CDCl3)δ:1.48(9H,s),1.77-1.95(2H,m),1.96-2.08(2H,m),2.79-3.10(3H,m),4.09-4.35(2H,m),6.91(1H,d,J=1.9Hz),7.21(1H,d,J=4.9Hz),7.48-7.55(1H,m),8.16(1H,s),8.61(1H,d,J=4.9Hz)。 Reference Example 35
tert-Butyl 4- (4-furan-3-ylpyrimidin-2-yl) piperidine-1-carboxylate
Tert-butyl 4- (4-furan-3-ylpyrimidin-2-yl) -3,6-dihydropyridine-1 (2H) -carboxylate (780 mg, 2.38 mmol) obtained in Reference Example 34 in tetrahydrofuran ( 12 ml), palladium on carbon (156 mg) was added to the solution under a nitrogen atmosphere, and the reaction solution was stirred at room temperature for 6 hours under a hydrogen atmosphere. The reaction solution was filtered and the filtrate was concentrated. The residue was purified by silica gel column chromatography (ethyl acetate: hexane = 1: 4) to give the title compound (542 mg, 69%) as an oil.
1 H NMR (CDCl 3 ) δ: 1.48 (9H, s), 1.77-1.95 (2H, m), 1.96-2.08 (2H, m), 2.79-3. 10 (3H, m), 4.09-4.35 (2H, m), 6.91 (1H, d, J = 1.9 Hz), 7.21 (1H, d, J = 4.9 Hz), 7.48-7.55 (1 H, m), 8.16 (1 H, s), 8.61 (1 H, d, J = 4.9 Hz).
tert-ブチル 4-(4-フラン-3-イルピリミジン-2-イル)ピペリジン-1-カルボキシラート
1H NMR(CDCl3)δ:1.48(9H,s),1.77-1.95(2H,m),1.96-2.08(2H,m),2.79-3.10(3H,m),4.09-4.35(2H,m),6.91(1H,d,J=1.9Hz),7.21(1H,d,J=4.9Hz),7.48-7.55(1H,m),8.16(1H,s),8.61(1H,d,J=4.9Hz)。 Reference Example 35
tert-Butyl 4- (4-furan-3-ylpyrimidin-2-yl) piperidine-1-carboxylate
1 H NMR (CDCl 3 ) δ: 1.48 (9H, s), 1.77-1.95 (2H, m), 1.96-2.08 (2H, m), 2.79-3. 10 (3H, m), 4.09-4.35 (2H, m), 6.91 (1H, d, J = 1.9 Hz), 7.21 (1H, d, J = 4.9 Hz), 7.48-7.55 (1 H, m), 8.16 (1 H, s), 8.61 (1 H, d, J = 4.9 Hz).
参考例36
4-フラン-3-イル-2-ピペリジン-4-イルピリミジン二塩酸塩

参考例35で得たtert-ブチル 4-(4-フラン-3-イルピリミジン-2-イル)ピペリジン-1-カルボキシラート(540mg、1.64ミリモル)の酢酸エチル(5.4ml)溶液に4N塩化水素-酢酸エチル溶液(5.4ml)を加え、室温で終夜攪拌した。反応溶液を酢酸エチルで希釈し、生じた結晶をろ取して表題化合物(407mg、77%)を固体として得た。
1H NMR(DMSO-d6)δ:1.94-2.26(4H,m),2.93-3.13(2H,m),3.13-3.27(1H,m),3.27-3.41(2H,m),7.12-7.19(1H,m),7.75(1H,d,J=5.3Hz),7.85-7.92(1H,m),8.58-8.65(1H,m),8.80(1H,d,J=5.3Hz),9.09(1H,brs),9.32(1H,brs)。 Reference Example 36
4-furan-3-yl-2-piperidin-4-ylpyrimidine dihydrochloride
To a solution of tert-butyl 4- (4-furan-3-ylpyrimidin-2-yl) piperidine-1-carboxylate (540 mg, 1.64 mmol) obtained in Reference Example 35 in ethyl acetate (5.4 ml) was added 4N A hydrogen chloride-ethyl acetate solution (5.4 ml) was added, and the mixture was stirred at room temperature overnight. The reaction solution was diluted with ethyl acetate, and the resulting crystals were collected by filtration to give the title compound (407 mg, 77%) as a solid.
1 H NMR (DMSO-d 6 ) δ: 1.94-2.26 (4H, m), 2.93-3.13 (2H, m), 3.13-3.27 (1H, m), 3.27-3.41 (2H, m), 7.12-7.19 (1H, m), 7.75 (1H, d, J = 5.3 Hz), 7.85-7.92 (1H M), 8.58-8.65 (1H, m), 8.80 (1H, d, J = 5.3 Hz), 9.09 (1H, brs), 9.32 (1H, brs).
4-フラン-3-イル-2-ピペリジン-4-イルピリミジン二塩酸塩
1H NMR(DMSO-d6)δ:1.94-2.26(4H,m),2.93-3.13(2H,m),3.13-3.27(1H,m),3.27-3.41(2H,m),7.12-7.19(1H,m),7.75(1H,d,J=5.3Hz),7.85-7.92(1H,m),8.58-8.65(1H,m),8.80(1H,d,J=5.3Hz),9.09(1H,brs),9.32(1H,brs)。 Reference Example 36
4-furan-3-yl-2-piperidin-4-ylpyrimidine dihydrochloride
1 H NMR (DMSO-d 6 ) δ: 1.94-2.26 (4H, m), 2.93-3.13 (2H, m), 3.13-3.27 (1H, m), 3.27-3.41 (2H, m), 7.12-7.19 (1H, m), 7.75 (1H, d, J = 5.3 Hz), 7.85-7.92 (1H M), 8.58-8.65 (1H, m), 8.80 (1H, d, J = 5.3 Hz), 9.09 (1H, brs), 9.32 (1H, brs).
参考例37
2-クロロ-4-チオフェン-3-イルピリミジン

参考例33と同様にして2,4-ジクロロピリミジンとチオフェン-3-イルボロン酸から表題化合物(1.02g、77%)を固体として得た。
1H NMR(CDCl3)δ:7.42-7.50(2H,m),7.65-7.72(1H,m),8.18-8.25(1H,m),8.58(1H,d,J=5.3Hz)。 Reference Example 37
2-Chloro-4-thiophen-3-ylpyrimidine
The title compound (1.02 g, 77%) was obtained as a solid from 2,4-dichloropyrimidine and thiophen-3-ylboronic acid in the same manner as in Reference Example 33.
1 H NMR (CDCl 3 ) δ: 7.42-7.50 (2H, m), 7.65-7.72 (1H, m), 8.18-8.25 (1H, m), 8. 58 (1H, d, J = 5.3 Hz).
2-クロロ-4-チオフェン-3-イルピリミジン
1H NMR(CDCl3)δ:7.42-7.50(2H,m),7.65-7.72(1H,m),8.18-8.25(1H,m),8.58(1H,d,J=5.3Hz)。 Reference Example 37
2-Chloro-4-thiophen-3-ylpyrimidine
1 H NMR (CDCl 3 ) δ: 7.42-7.50 (2H, m), 7.65-7.72 (1H, m), 8.18-8.25 (1H, m), 8. 58 (1H, d, J = 5.3 Hz).
参考例38
tert-ブチル 4-(4-チオフェン-3-イルピリミジン-2-イル)-3,6-ジヒドロピリジン-1(2H)-カルボキシラート

参考例34と同様にして、参考例37で得た2-クロロ-4-チオフェン-3-イルピリミジンから表題化合物(793mg、90%)を油状物として得た。
1H NMR(CDCl3)δ:1.50(9H,s),2.80(2H,brs),3.60-3.71(2H,m),4.15-4.26(2H,m),7.19-7.32(1H,m),7.36(1H,d,J=5.3Hz),7.43(1H,dd,J=5.3,3.0Hz),7.68-7.76(1H,m),8.12-8.18(1H,m),8.67(1H,d,J=5.3Hz)。 Reference Example 38
tert-Butyl 4- (4-thiophen-3-ylpyrimidin-2-yl) -3,6-dihydropyridine-1 (2H) -carboxylate
In the same manner as in Reference Example 34, the title compound (793 mg, 90%) was obtained as an oil from 2-chloro-4-thiophen-3-ylpyrimidine obtained in Reference Example 37.
1 H NMR (CDCl 3 ) δ: 1.50 (9H, s), 2.80 (2H, brs), 3.60-3.71 (2H, m), 4.15-4.26 (2H, m), 7.19-7.32 (1H, m), 7.36 (1H, d, J = 5.3 Hz), 7.43 (1H, dd, J = 5.3, 3.0 Hz), 7.68-7.76 (1H, m), 8.12-8.18 (1H, m), 8.67 (1H, d, J = 5.3 Hz).
tert-ブチル 4-(4-チオフェン-3-イルピリミジン-2-イル)-3,6-ジヒドロピリジン-1(2H)-カルボキシラート
1H NMR(CDCl3)δ:1.50(9H,s),2.80(2H,brs),3.60-3.71(2H,m),4.15-4.26(2H,m),7.19-7.32(1H,m),7.36(1H,d,J=5.3Hz),7.43(1H,dd,J=5.3,3.0Hz),7.68-7.76(1H,m),8.12-8.18(1H,m),8.67(1H,d,J=5.3Hz)。 Reference Example 38
tert-Butyl 4- (4-thiophen-3-ylpyrimidin-2-yl) -3,6-dihydropyridine-1 (2H) -carboxylate
1 H NMR (CDCl 3 ) δ: 1.50 (9H, s), 2.80 (2H, brs), 3.60-3.71 (2H, m), 4.15-4.26 (2H, m), 7.19-7.32 (1H, m), 7.36 (1H, d, J = 5.3 Hz), 7.43 (1H, dd, J = 5.3, 3.0 Hz), 7.68-7.76 (1H, m), 8.12-8.18 (1H, m), 8.67 (1H, d, J = 5.3 Hz).
参考例39
tert-ブチル 4-(4-チオフェン-3-イルピリミジン-2-イル)ピペリジン-1-カルボキシラート

参考例38で得たtert-ブチル 4-(4-チオフェン-3-イルピリミジン-2-イル)-3,6-ジヒドロピリジン-1(2H)-カルボキシラート(790mg、2.30ミリモル)の酢酸エチル(12ml)溶液に、窒素雰囲気下、パラジウム炭素(158mg)を加え、反応溶液を3気圧の水素雰囲気下、室温で6時間攪拌した。反応溶液をろ過し、ろ液を濃縮した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:4)で精製し、表題化合物(477mg、60%)を油状物として得た。
1H NMR(CDCl3)δ:1.48(9H,s),1.80-1.97(2H,m),1.98-2.11(2H,m),2.82-2.98(2H,m),2.99-3.12(1H,m),4.12-4.34(2H,m),7.37(1H,d,J=5.3Hz),7.42(1H,dd,J=5.3,3.0Hz),7.67-7.74(1H,m),8.11-8.17(1H,m),8.65(1H,d,J=5.3Hz)。 Reference Example 39
tert-Butyl 4- (4-thiophen-3-ylpyrimidin-2-yl) piperidine-1-carboxylate
Tert-butyl 4- (4-thiophen-3-ylpyrimidin-2-yl) -3,6-dihydropyridine-1 (2H) -carboxylate (790 mg, 2.30 mmol) of ethyl acetate obtained in Reference Example 38 (12 ml) To the solution was added palladium carbon (158 mg) under a nitrogen atmosphere, and the reaction solution was stirred at room temperature for 6 hours under a hydrogen atmosphere of 3 atm. The reaction solution was filtered and the filtrate was concentrated. The residue was purified by silica gel column chromatography (ethyl acetate: hexane = 1: 4) to give the title compound (477 mg, 60%) as an oil.
1 H NMR (CDCl 3 ) δ: 1.48 (9H, s), 1.80-1.97 (2H, m), 1.98-2.11 (2H, m), 2.82-2. 98 (2H, m), 2.99-3.12 (1H, m), 4.12-4.34 (2H, m), 7.37 (1H, d, J = 5.3 Hz), 7. 42 (1H, dd, J = 5.3, 3.0 Hz), 7.67-7.74 (1H, m), 8.11-8.17 (1H, m), 8.65 (1H, d , J = 5.3 Hz).
tert-ブチル 4-(4-チオフェン-3-イルピリミジン-2-イル)ピペリジン-1-カルボキシラート
1H NMR(CDCl3)δ:1.48(9H,s),1.80-1.97(2H,m),1.98-2.11(2H,m),2.82-2.98(2H,m),2.99-3.12(1H,m),4.12-4.34(2H,m),7.37(1H,d,J=5.3Hz),7.42(1H,dd,J=5.3,3.0Hz),7.67-7.74(1H,m),8.11-8.17(1H,m),8.65(1H,d,J=5.3Hz)。 Reference Example 39
tert-Butyl 4- (4-thiophen-3-ylpyrimidin-2-yl) piperidine-1-carboxylate
1 H NMR (CDCl 3 ) δ: 1.48 (9H, s), 1.80-1.97 (2H, m), 1.98-2.11 (2H, m), 2.82-2. 98 (2H, m), 2.99-3.12 (1H, m), 4.12-4.34 (2H, m), 7.37 (1H, d, J = 5.3 Hz), 7. 42 (1H, dd, J = 5.3, 3.0 Hz), 7.67-7.74 (1H, m), 8.11-8.17 (1H, m), 8.65 (1H, d , J = 5.3 Hz).
参考例40
4-チオフェン-3-イル-2-ピペリジン-4-イルピリミジン二塩酸塩

参考例36と同様にして、参考例39で得たtert-ブチル 4-(4-チオフェン-3-イルピリミジン-2-イル)ピペリジン-1-カルボキシラートから表題化合物(415mg、95%)を固体として得た。
1H NMR(DMSO-d6)δ:1.96-2.14(2H,m),2.14-2.27(2H,m),2.97-3.13(2H,m),3.14-3.27(1H,m),3.28-3.42(2H,m),7.71-7.77(1H,m),7.81-7.90(2H,m),8.48-8.55(1H,m),8.80(1H,d,J=5.3Hz),8.92(1H,brs),9.17(1H,brs)。 Reference Example 40
4-thiophen-3-yl-2-piperidin-4-ylpyrimidine dihydrochloride
In the same manner as in Reference Example 36, the title compound (415 mg, 95%) was obtained as a solid from tert-butyl 4- (4-thiophen-3-ylpyrimidin-2-yl) piperidine-1-carboxylate obtained in Reference Example 39. Got as.
1 H NMR (DMSO-d 6 ) δ: 1.96-2.14 (2H, m), 2.14-2.27 (2H, m), 2.97-3.13 (2H, m), 3.14-3.27 (1H, m), 3.28-3.42 (2H, m), 7.71-7.77 (1H, m), 7.81-7.90 (2H, m ), 8.48-8.55 (1H, m), 8.80 (1H, d, J = 5.3 Hz), 8.92 (1H, brs), 9.17 (1H, brs).
4-チオフェン-3-イル-2-ピペリジン-4-イルピリミジン二塩酸塩
1H NMR(DMSO-d6)δ:1.96-2.14(2H,m),2.14-2.27(2H,m),2.97-3.13(2H,m),3.14-3.27(1H,m),3.28-3.42(2H,m),7.71-7.77(1H,m),7.81-7.90(2H,m),8.48-8.55(1H,m),8.80(1H,d,J=5.3Hz),8.92(1H,brs),9.17(1H,brs)。 Reference Example 40
4-thiophen-3-yl-2-piperidin-4-ylpyrimidine dihydrochloride
1 H NMR (DMSO-d 6 ) δ: 1.96-2.14 (2H, m), 2.14-2.27 (2H, m), 2.97-3.13 (2H, m), 3.14-3.27 (1H, m), 3.28-3.42 (2H, m), 7.71-7.77 (1H, m), 7.81-7.90 (2H, m ), 8.48-8.55 (1H, m), 8.80 (1H, d, J = 5.3 Hz), 8.92 (1H, brs), 9.17 (1H, brs).
参考例41
2-クロロ-4-(3-フルオロフェニル)ピリミジン

参考例33と同様にして2,4-ジクロロピリミジンと3-フルオロフェニルボロン酸から表題化合物(780mg、56%)を固体として得た。
1H NMR(CDCl3)δ:7.20-7.30(1H,m),7.44-7.55(1H,m),7.64(1H,d,J=4.9Hz),7.79-7.91(2H,m),8.68(1H,d,J=4.9Hz)。 Reference Example 41
2-Chloro-4- (3-fluorophenyl) pyrimidine
The title compound (780 mg, 56%) was obtained as a solid from 2,4-dichloropyrimidine and 3-fluorophenylboronic acid in the same manner as in Reference Example 33.
1 H NMR (CDCl 3 ) δ: 7.20-7.30 (1H, m), 7.44-7.55 (1H, m), 7.64 (1H, d, J = 4.9 Hz), 7.79-7.91 (2H, m), 8.68 (1 H, d, J = 4.9 Hz).
2-クロロ-4-(3-フルオロフェニル)ピリミジン
1H NMR(CDCl3)δ:7.20-7.30(1H,m),7.44-7.55(1H,m),7.64(1H,d,J=4.9Hz),7.79-7.91(2H,m),8.68(1H,d,J=4.9Hz)。 Reference Example 41
2-Chloro-4- (3-fluorophenyl) pyrimidine
1 H NMR (CDCl 3 ) δ: 7.20-7.30 (1H, m), 7.44-7.55 (1H, m), 7.64 (1H, d, J = 4.9 Hz), 7.79-7.91 (2H, m), 8.68 (1 H, d, J = 4.9 Hz).
参考例42
tert-ブチル 4-[4-(3-フルオロフェニル)ピリミジン-2-イル]-3,6-ジヒドロピリジン-1(2H)-カルボキシラート

参考例34と同様にして、参考例41で得た2-クロロ-4-(3-フルオロフェニル)ピリミジンから表題化合物(1.19g、90%)を油状物として得た。
1H NMR(CDCl3)δ:1.50(9H,s),2.82(2H,brs),3.63-3.72(2H,m),4.18-4.26(2H,m),7.16-7.25(1H,m),7.27-7.39(1H,m),7.42-7.54(2H,m),7.84-7.93(2H,m),8.75(1H,d,J=5.3Hz)。 Reference Example 42
tert-Butyl 4- [4- (3-fluorophenyl) pyrimidin-2-yl] -3,6-dihydropyridine-1 (2H) -carboxylate
In the same manner as in Reference Example 34, the title compound (1.19 g, 90%) was obtained as an oil from 2-chloro-4- (3-fluorophenyl) pyrimidine obtained in Reference Example 41.
1 H NMR (CDCl 3 ) δ: 1.50 (9H, s), 2.82 (2H, brs), 3.63-3.72 (2H, m), 4.18-4.26 (2H, m), 7.16-7.25 (1H, m), 7.27-7.39 (1H, m), 7.42-7.54 (2H, m), 7.84-7.93 ( 2H, m), 8.75 (1H, d, J = 5.3 Hz).
tert-ブチル 4-[4-(3-フルオロフェニル)ピリミジン-2-イル]-3,6-ジヒドロピリジン-1(2H)-カルボキシラート
1H NMR(CDCl3)δ:1.50(9H,s),2.82(2H,brs),3.63-3.72(2H,m),4.18-4.26(2H,m),7.16-7.25(1H,m),7.27-7.39(1H,m),7.42-7.54(2H,m),7.84-7.93(2H,m),8.75(1H,d,J=5.3Hz)。 Reference Example 42
tert-Butyl 4- [4- (3-fluorophenyl) pyrimidin-2-yl] -3,6-dihydropyridine-1 (2H) -carboxylate
1 H NMR (CDCl 3 ) δ: 1.50 (9H, s), 2.82 (2H, brs), 3.63-3.72 (2H, m), 4.18-4.26 (2H, m), 7.16-7.25 (1H, m), 7.27-7.39 (1H, m), 7.42-7.54 (2H, m), 7.84-7.93 ( 2H, m), 8.75 (1H, d, J = 5.3 Hz).
参考例43
tert-ブチル 4-[4-(3-フルオロフェニル)ピリミジン-2-イル]ピペリジン-1-カルボキシラート

参考例35と同様にして、参考例42で得たtert-ブチル 4-[4-(3-フルオロフェニル)ピリミジン-2-イル]-3,6-ジヒドロピリジン-1(2H)-カルボキシラートから表題化合物(943mg、79%)を油状物として得た。
1H NMR(CDCl3)δ:1.49(9H,s),1.82-2.00(2H,m),2.01-2.13(2H,m),2.83-3.01(2H,m),3.03-3.17(1H,m),4.09-4.40(2H,m),7.15-7.24(1H,m),7.41-7.57(2H,m),7.81-7.90(2H,m),8.73(1H,d,J=5.3Hz)。 Reference Example 43
tert-Butyl 4- [4- (3-fluorophenyl) pyrimidin-2-yl] piperidine-1-carboxylate
In the same manner as in Reference Example 35, the title was obtained from tert-butyl 4- [4- (3-fluorophenyl) pyrimidin-2-yl] -3,6-dihydropyridine-1 (2H) -carboxylate obtained in Reference Example 42. The compound (943 mg, 79%) was obtained as an oil.
1 H NMR (CDCl 3 ) δ: 1.49 (9H, s), 1.82-2.00 (2H, m), 2.01-2.13 (2H, m), 2.83-3. 01 (2H, m), 3.03-3.17 (1H, m), 4.09-4.40 (2H, m), 7.15-7.24 (1H, m), 7.41- 7.57 (2H, m), 7.81-7.90 (2H, m), 8.73 (1H, d, J = 5.3 Hz).
tert-ブチル 4-[4-(3-フルオロフェニル)ピリミジン-2-イル]ピペリジン-1-カルボキシラート
1H NMR(CDCl3)δ:1.49(9H,s),1.82-2.00(2H,m),2.01-2.13(2H,m),2.83-3.01(2H,m),3.03-3.17(1H,m),4.09-4.40(2H,m),7.15-7.24(1H,m),7.41-7.57(2H,m),7.81-7.90(2H,m),8.73(1H,d,J=5.3Hz)。 Reference Example 43
tert-Butyl 4- [4- (3-fluorophenyl) pyrimidin-2-yl] piperidine-1-carboxylate
1 H NMR (CDCl 3 ) δ: 1.49 (9H, s), 1.82-2.00 (2H, m), 2.01-2.13 (2H, m), 2.83-3. 01 (2H, m), 3.03-3.17 (1H, m), 4.09-4.40 (2H, m), 7.15-7.24 (1H, m), 7.41- 7.57 (2H, m), 7.81-7.90 (2H, m), 8.73 (1H, d, J = 5.3 Hz).
参考例44
4-(3-フルオロフェニル)-2-ピペリジン-4-イルピリミジン二塩酸塩
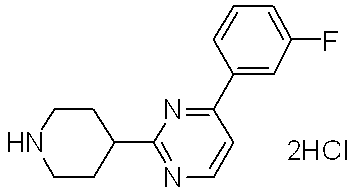
参考例36と同様にして、参考例43で得たtert-ブチル 4-[4-(3-フルオロフェニル)ピリミジン-2-イル]ピペリジン-1-カルボキシラートから表題化合物(836mg、96%)を固体として得た。
1H NMR(DMSO-d6)δ:1.98-2.16(2H,m),2.17-2.30(2H,m),2.97-3.15(2H,m),3.17-3.43(3H,m),7.36-7.48(1H,m),7.56-7.69(1H,m),7.98-8.15(3H,m),8.89(1H,d,J=5.3Hz),8.97(1H,brs),9.25(1H,brs)。 Reference Example 44
4- (3-Fluorophenyl) -2-piperidin-4-ylpyrimidine dihydrochloride
In the same manner as in Reference Example 36, the title compound (836 mg, 96%) was obtained from tert-butyl 4- [4- (3-fluorophenyl) pyrimidin-2-yl] piperidine-1-carboxylate obtained in Reference Example 43. Obtained as a solid.
1 H NMR (DMSO-d 6 ) δ: 1.98-2.16 (2H, m), 2.17-2.30 (2H, m), 2.97-3.15 (2H, m), 3.17-3.43 (3H, m), 7.36-7.48 (1H, m), 7.56-7.69 (1H, m), 7.98-8.15 (3H, m ), 8.89 (1H, d, J = 5.3 Hz), 8.97 (1H, brs), 9.25 (1H, brs).
4-(3-フルオロフェニル)-2-ピペリジン-4-イルピリミジン二塩酸塩
1H NMR(DMSO-d6)δ:1.98-2.16(2H,m),2.17-2.30(2H,m),2.97-3.15(2H,m),3.17-3.43(3H,m),7.36-7.48(1H,m),7.56-7.69(1H,m),7.98-8.15(3H,m),8.89(1H,d,J=5.3Hz),8.97(1H,brs),9.25(1H,brs)。 Reference Example 44
4- (3-Fluorophenyl) -2-piperidin-4-ylpyrimidine dihydrochloride
1 H NMR (DMSO-d 6 ) δ: 1.98-2.16 (2H, m), 2.17-2.30 (2H, m), 2.97-3.15 (2H, m), 3.17-3.43 (3H, m), 7.36-7.48 (1H, m), 7.56-7.69 (1H, m), 7.98-8.15 (3H, m ), 8.89 (1H, d, J = 5.3 Hz), 8.97 (1H, brs), 9.25 (1H, brs).
参考例45
2-クロロ-4-(4-フルオロフェニル)ピリミジン

2,4-ジクロロピリミジン(1.00g、6.71ミリモル)、4-フルオロフェニルボロン酸(1.03g、7.38ミリモル)、2N炭酸ナトリウム水溶液(6.7ml)の1,2-ジメトキシエタン(10ml)溶液に、窒素雰囲気下、テトラキストリフェニルホスフィンパラジウム(931mg、0.805ミリモル)を室温で加え、マイクロウェーブリアクター(Initiator TM、Biotage社)によって、105℃で40分間攪拌した。反応液に水を注ぎ、酢酸エチルで抽出した。抽出液を水で洗浄し、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:4)で精製し、表題化合物(930mg、66%)を固体として得た。
1H NMR(CDCl3)δ:7.15-7.25(2H,m),7.61(1H,d,J=5.3Hz),8.08-8.16(2H,m),8.64(1H,d,J=5.3Hz)。 Reference Example 45
2-Chloro-4- (4-fluorophenyl) pyrimidine
1,2-Dimethoxyethane in 2,4-dichloropyrimidine (1.00 g, 6.71 mmol), 4-fluorophenylboronic acid (1.03 g, 7.38 mmol), 2N aqueous sodium carbonate solution (6.7 ml) Tetrakistriphenylphosphine palladium (931 mg, 0.805 mmol) was added to the (10 ml) solution at room temperature under a nitrogen atmosphere, and the mixture was stirred at 105 ° C. for 40 minutes by a microwave reactor (Initiator ™, Biotage). Water was poured into the reaction mixture, and the mixture was extracted with ethyl acetate. The extract was washed with water and dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (ethyl acetate: hexane = 1: 4) to give the title compound (930 mg, 66%) as a solid.
1 H NMR (CDCl 3 ) δ: 7.15-7.25 (2H, m), 7.61 (1H, d, J = 5.3 Hz), 8.08-8.16 (2H, m), 8.64 (1H, d, J = 5.3 Hz).
2-クロロ-4-(4-フルオロフェニル)ピリミジン
1H NMR(CDCl3)δ:7.15-7.25(2H,m),7.61(1H,d,J=5.3Hz),8.08-8.16(2H,m),8.64(1H,d,J=5.3Hz)。 Reference Example 45
2-Chloro-4- (4-fluorophenyl) pyrimidine
1 H NMR (CDCl 3 ) δ: 7.15-7.25 (2H, m), 7.61 (1H, d, J = 5.3 Hz), 8.08-8.16 (2H, m), 8.64 (1H, d, J = 5.3 Hz).
参考例46
tert-ブチル 4-[4-(4-フルオロフェニル)ピリミジン-2-イル]-3,6-ジヒドロピリジン-1(2H)-カルボキシラート

参考例45で得た2-クロロ-4-(4-フルオロフェニル)ピリミジン(700mg、3.36ミリモル)、tert-ブチル 4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-3,6-ジヒドロピリジン-1(2H)-カルボキシラート(1.04g、3.36ミリモル)、2N炭酸ナトリウム水溶液(5.0ml)の1,2-ジメトキシエタン(10ml)溶液に、窒素雰囲気下、テトラキストリフェニルホスフィンパラジウム(465mg、0.403ミリモル)を室温で加え、マイクロウェーブリアクター(Initiator TM、Biotage社)によって、105℃で40分間攪拌した。反応液に水を注ぎ、酢酸エチルで抽出した。抽出液を水で洗浄し、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:4)で精製し、表題化合物(1.12g、94%)を油状物として得た。
1H NMR(CDCl3)δ:1.50(9H,s),2.82(2H,brs),3.67(2H,t,J=5.7Hz),4.17-4.25(2H,m),7.14-7.23(2H,m),7.31(1H,brs),7.48(1H,d,J=5.3Hz),8.10-8.19(2H,m),8.72(1H,d,J=5.3Hz)。 Reference Example 46
tert-Butyl 4- [4- (4-fluorophenyl) pyrimidin-2-yl] -3,6-dihydropyridine-1 (2H) -carboxylate
2-chloro-4- (4-fluorophenyl) pyrimidine (700 mg, 3.36 mmol) obtained in Reference Example 45, tert-butyl 4- (4,4,5,5-tetramethyl-1,3,2) -Dioxaborolan-2-yl) -3,6-dihydropyridine-1 (2H) -carboxylate (1.04 g, 3.36 mmol), 2N aqueous sodium carbonate (5.0 ml) in 1,2-dimethoxyethane (10 ml) ) Tetrakistriphenylphosphine palladium (465 mg, 0.403 mmol) was added to the solution at room temperature under a nitrogen atmosphere, and the mixture was stirred at 105 ° C. for 40 minutes by a microwave reactor (Initiator ™, Biotage). Water was poured into the reaction mixture, and the mixture was extracted with ethyl acetate. The extract was washed with water and dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (ethyl acetate: hexane = 1: 4) to give the title compound (1.12 g, 94%) as an oil.
1 H NMR (CDCl 3 ) δ: 1.50 (9H, s), 2.82 (2H, brs), 3.67 (2H, t, J = 5.7 Hz), 4.17-4.25 ( 2H, m), 7.14-7.23 (2H, m), 7.31 (1H, brs), 7.48 (1H, d, J = 5.3 Hz), 8.10-8.19 ( 2H, m), 8.72 (1H, d, J = 5.3 Hz).
tert-ブチル 4-[4-(4-フルオロフェニル)ピリミジン-2-イル]-3,6-ジヒドロピリジン-1(2H)-カルボキシラート
1H NMR(CDCl3)δ:1.50(9H,s),2.82(2H,brs),3.67(2H,t,J=5.7Hz),4.17-4.25(2H,m),7.14-7.23(2H,m),7.31(1H,brs),7.48(1H,d,J=5.3Hz),8.10-8.19(2H,m),8.72(1H,d,J=5.3Hz)。 Reference Example 46
tert-Butyl 4- [4- (4-fluorophenyl) pyrimidin-2-yl] -3,6-dihydropyridine-1 (2H) -carboxylate
1 H NMR (CDCl 3 ) δ: 1.50 (9H, s), 2.82 (2H, brs), 3.67 (2H, t, J = 5.7 Hz), 4.17-4.25 ( 2H, m), 7.14-7.23 (2H, m), 7.31 (1H, brs), 7.48 (1H, d, J = 5.3 Hz), 8.10-8.19 ( 2H, m), 8.72 (1H, d, J = 5.3 Hz).
参考例47
tert-ブチル 4-[4-(4-フルオロフェニル)ピリミジン-2-イル]ピペリジン-1-カルボキシラート

参考例46で得たtert-ブチル 4-[4-(4-フルオロフェニル)ピリミジン-2-イル]-3,6-ジヒドロピリジン-1(2H)-カルボキシラート(1.12g、3.15ミリモル)のエタノール(14ml)-テトラヒドロフラン(7ml)溶液に、窒素雰囲気下、パラジウム炭素(224mg)を加え、反応溶液を水素雰囲気下、室温で6時間攪拌した。反応溶液をろ過し、ろ液を濃縮した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:4)で精製し、表題化合物(782mg、70%)を油状物として得た。
1H NMR(CDCl3)δ:1.48(9H,s),1.81-1.99(2H,m),1.99-2.12(2H,m),2.81-3.00(2H,m),3.01-3.15(1H,m),4.11-4.38(2H,m),7.11-7.24(2H,m),7.49(1H,d,J=5.3Hz),8.06-8.19(2H,m),8.69(1H,d,J=5.3Hz)。 Reference Example 47
tert-Butyl 4- [4- (4-fluorophenyl) pyrimidin-2-yl] piperidine-1-carboxylate
Tert-Butyl 4- [4- (4-fluorophenyl) pyrimidin-2-yl] -3,6-dihydropyridine-1 (2H) -carboxylate obtained in Reference Example 46 (1.12 g, 3.15 mmol) To an ethanol (14 ml) -tetrahydrofuran (7 ml) solution was added palladium carbon (224 mg) under a nitrogen atmosphere, and the reaction solution was stirred at room temperature for 6 hours under a hydrogen atmosphere. The reaction solution was filtered and the filtrate was concentrated. The residue was purified by silica gel column chromatography (ethyl acetate: hexane = 1: 4) to give the title compound (782 mg, 70%) as an oil.
1 H NMR (CDCl 3 ) δ: 1.48 (9H, s), 1.81-1.99 (2H, m), 1.99-2.12 (2H, m), 2.81-3. 00 (2H, m), 3.01-3.15 (1H, m), 4.11-4.38 (2H, m), 7.11-7.24 (2H, m), 7.49 ( 1H, d, J = 5.3 Hz), 8.06-8.19 (2H, m), 8.69 (1H, d, J = 5.3 Hz).
tert-ブチル 4-[4-(4-フルオロフェニル)ピリミジン-2-イル]ピペリジン-1-カルボキシラート
1H NMR(CDCl3)δ:1.48(9H,s),1.81-1.99(2H,m),1.99-2.12(2H,m),2.81-3.00(2H,m),3.01-3.15(1H,m),4.11-4.38(2H,m),7.11-7.24(2H,m),7.49(1H,d,J=5.3Hz),8.06-8.19(2H,m),8.69(1H,d,J=5.3Hz)。 Reference Example 47
tert-Butyl 4- [4- (4-fluorophenyl) pyrimidin-2-yl] piperidine-1-carboxylate
1 H NMR (CDCl 3 ) δ: 1.48 (9H, s), 1.81-1.99 (2H, m), 1.99-2.12 (2H, m), 2.81-3. 00 (2H, m), 3.01-3.15 (1H, m), 4.11-4.38 (2H, m), 7.11-7.24 (2H, m), 7.49 ( 1H, d, J = 5.3 Hz), 8.06-8.19 (2H, m), 8.69 (1H, d, J = 5.3 Hz).
参考例48
4-(4-フルオロフェニル)-2-ピペリジン-4-イルピリミジン二塩酸塩

参考例47で得たtert-ブチル 4-[4-(4-フルオロフェニル)ピリミジン-2-イル]ピペリジン-1-カルボキシラート(780mg、2.18ミリモル)の酢酸エチル(7.8ml)溶液に4N塩化水素-酢酸エチル溶液(7.8ml)を加え、室温で終夜攪拌した。反応溶液を酢酸エチルで希釈し、生じた結晶をろ取して表題化合物(686mg、95%)を固体として得た。
1H NMR(DMSO-d6)δ:1.98-2.16(2H,m),2.17-2.30(2H,m),2.97-3.15(2H,m),3.17-3.42(3H,m),7.35-7.48(2H,m),8.00(1H,d,J=5.3Hz),8.25-8.38(2H,m),8.86(1H,d,J=5.3Hz),9.06(1H,brs),9.32(1H,brs)。 Reference Example 48
4- (4-Fluorophenyl) -2-piperidin-4-ylpyrimidine dihydrochloride
To a solution of tert-butyl 4- [4- (4-fluorophenyl) pyrimidin-2-yl] piperidine-1-carboxylate (780 mg, 2.18 mmol) obtained in Reference Example 47 in ethyl acetate (7.8 ml). 4N hydrogen chloride-ethyl acetate solution (7.8 ml) was added, and the mixture was stirred at room temperature overnight. The reaction solution was diluted with ethyl acetate, and the resulting crystals were collected by filtration to give the title compound (686 mg, 95%) as a solid.
1 H NMR (DMSO-d 6 ) δ: 1.98-2.16 (2H, m), 2.17-2.30 (2H, m), 2.97-3.15 (2H, m), 3.17-3.42 (3H, m), 7.35-7.48 (2H, m), 8.00 (1H, d, J = 5.3 Hz), 8.25-8.38 (2H M), 8.86 (1H, d, J = 5.3 Hz), 9.06 (1H, brs), 9.32 (1H, brs).
4-(4-フルオロフェニル)-2-ピペリジン-4-イルピリミジン二塩酸塩
1H NMR(DMSO-d6)δ:1.98-2.16(2H,m),2.17-2.30(2H,m),2.97-3.15(2H,m),3.17-3.42(3H,m),7.35-7.48(2H,m),8.00(1H,d,J=5.3Hz),8.25-8.38(2H,m),8.86(1H,d,J=5.3Hz),9.06(1H,brs),9.32(1H,brs)。 Reference Example 48
4- (4-Fluorophenyl) -2-piperidin-4-ylpyrimidine dihydrochloride
1 H NMR (DMSO-d 6 ) δ: 1.98-2.16 (2H, m), 2.17-2.30 (2H, m), 2.97-3.15 (2H, m), 3.17-3.42 (3H, m), 7.35-7.48 (2H, m), 8.00 (1H, d, J = 5.3 Hz), 8.25-8.38 (2H M), 8.86 (1H, d, J = 5.3 Hz), 9.06 (1H, brs), 9.32 (1H, brs).
参考例49
tert-ブチル 4-(5-フルオロ-4-フェニルピリミジン-2-イル)-3,6-ジヒドロピリジン-1(2H)-カルボキシラート

2,4-ジクロロ-5-フルオロピリミジン(5.29g、31.7ミリモル)、フェニルボロン酸(3.86g、31.7ミリモル)、2N炭酸ナトリウム水溶液(98ml)の1,2-ジメトキシエタン(200ml)溶液に、窒素雰囲気下、テトラキストリフェニルホスフィンパラジウム(7.33g、6.34ミリモル)を室温で加え、100℃で5時間攪拌した。反応液に水を注ぎ、酢酸エチルで抽出した。抽出液を水で洗浄し、無水硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=10:1)で精製して白色固体(4.75g)を得た。得られた固体(2.0g)、tert-ブチル 4-(4,4,5,5-テトラメチル-1,3,2-ジオキサボロラン-2-イル)-3,6-ジヒドロピリジン-1(2H)-カルボキシラート(0.98g、3.2ミリモル)、2N炭酸ナトリウム水溶液(10ml)の1,2-ジメトキシエタン(30ml)溶液に、窒素雰囲気下、テトラキストリフェニルホスフィンパラジウム(0.733g、0.634ミリモル)を室温で加え、100℃で5時間攪拌した。反応液に水を注ぎ、酢酸エチルで抽出した。抽出液を水で洗浄し、無水硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=10:1)で精製して表題化合物(1.0g、21%)を白色固体として得た。
1H NMR(CDCl3)δ:1.50(9H,s),2.75-2.85(2H,m),3.64-3.68(2H,m),4.17-4.23(2H,m),7.17-7.24(1H,m),7.51-7.55(3H,m),8.16-8.19(2H,m),8.57(1H,d,J=3.3Hz)。 Reference Example 49
tert-Butyl 4- (5-fluoro-4-phenylpyrimidin-2-yl) -3,6-dihydropyridine-1 (2H) -carboxylate
2,4-Dichloro-5-fluoropyrimidine (5.29 g, 31.7 mmol), phenylboronic acid (3.86 g, 31.7 mmol), 1,2-dimethoxyethane (98 ml) in 2N aqueous sodium carbonate (98 ml) 200 ml) under nitrogen atmosphere, tetrakistriphenylphosphine palladium (7.33 g, 6.34 mmol) was added at room temperature and stirred at 100 ° C. for 5 hours. Water was poured into the reaction mixture, and the mixture was extracted with ethyl acetate. The extract was washed with water and dried over anhydrous sodium sulfate, and the solvent was distilled off under reduced pressure. The residue was purified by silica gel column chromatography (ethyl acetate: hexane = 10: 1) to give a white solid (4.75 g). The resulting solid (2.0 g), tert-butyl 4- (4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) -3,6-dihydropyridine-1 (2H) -Carboxylate (0.98 g, 3.2 mmol), a solution of 2N aqueous sodium carbonate (10 ml) in 1,2-dimethoxyethane (30 ml) under a nitrogen atmosphere, tetrakistriphenylphosphine palladium (0.733 g,. 634 mmol) at room temperature and stirred at 100 ° C. for 5 hours. Water was poured into the reaction mixture, and the mixture was extracted with ethyl acetate. The extract was washed with water and dried over anhydrous sodium sulfate, and the solvent was distilled off under reduced pressure. The residue was purified by silica gel column chromatography (ethyl acetate: hexane = 10: 1) to give the title compound (1.0 g, 21%) as a white solid.
1 H NMR (CDCl 3 ) δ: 1.50 (9H, s), 2.75-2.85 (2H, m), 3.64-3.68 (2H, m), 4.17-4. 23 (2H, m), 7.17-7.24 (1H, m), 7.51-7.55 (3H, m), 8.16-8.19 (2H, m), 8.57 ( 1H, d, J = 3.3 Hz).
tert-ブチル 4-(5-フルオロ-4-フェニルピリミジン-2-イル)-3,6-ジヒドロピリジン-1(2H)-カルボキシラート
1H NMR(CDCl3)δ:1.50(9H,s),2.75-2.85(2H,m),3.64-3.68(2H,m),4.17-4.23(2H,m),7.17-7.24(1H,m),7.51-7.55(3H,m),8.16-8.19(2H,m),8.57(1H,d,J=3.3Hz)。 Reference Example 49
tert-Butyl 4- (5-fluoro-4-phenylpyrimidin-2-yl) -3,6-dihydropyridine-1 (2H) -carboxylate
1 H NMR (CDCl 3 ) δ: 1.50 (9H, s), 2.75-2.85 (2H, m), 3.64-3.68 (2H, m), 4.17-4. 23 (2H, m), 7.17-7.24 (1H, m), 7.51-7.55 (3H, m), 8.16-8.19 (2H, m), 8.57 ( 1H, d, J = 3.3 Hz).
参考例50
tert-ブチル 4-(5-フルオロ-4-フェニルピリミジン-2-イル)ピペリジン-1-カルボキシラート

参考例49で得たtert-ブチル 4-(5-フルオロ-4-フェニルピリミジン-2-イル)-3,6-ジヒドロピリジン-1(2H)-カルボキシラート(0.75g、2.11ミリモル)のエタノール(100ml)溶液に、窒素雰囲気下で、10%パラジウム炭素(50%含水、220mg)を加えた後、水素雰囲気下、室温で18時間攪拌した。反応液をセライトろ過し、ろ液を減圧濃縮し残留物を得た。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=5:1)で精製して表題化合物(620mg、82%)を油状物として得た。
1H NMR(CDCl3)δ:1.48(9H,s),1.81-2.10(4H,m),2.87-3.13(3H,m),4.17-4.30(2H,m),7.51-7.53(3H,m),8.14-8.16(2H,m),8.55(1H,d,J=3.9Hz)。 Reference Example 50
tert-Butyl 4- (5-fluoro-4-phenylpyrimidin-2-yl) piperidine-1-carboxylate
Of tert-butyl 4- (5-fluoro-4-phenylpyrimidin-2-yl) -3,6-dihydropyridine-1 (2H) -carboxylate (0.75 g, 2.11 mmol) obtained in Reference Example 49 After adding 10% palladium carbon (containing 50% water, 220 mg) to an ethanol (100 ml) solution under a nitrogen atmosphere, the mixture was stirred at room temperature for 18 hours under a hydrogen atmosphere. The reaction solution was filtered through Celite, and the filtrate was concentrated under reduced pressure to obtain a residue. The residue was purified by silica gel column chromatography (ethyl acetate: hexane = 5: 1) to give the title compound (620 mg, 82%) as an oil.
1 H NMR (CDCl 3 ) δ: 1.48 (9H, s), 1.81-2.10. (4H, m), 2.87-3.13 (3H, m), 4.17-4. 30 (2H, m), 7.51-7.53 (3H, m), 8.14-8.16 (2H, m), 8.55 (1H, d, J = 3.9 Hz).
tert-ブチル 4-(5-フルオロ-4-フェニルピリミジン-2-イル)ピペリジン-1-カルボキシラート
1H NMR(CDCl3)δ:1.48(9H,s),1.81-2.10(4H,m),2.87-3.13(3H,m),4.17-4.30(2H,m),7.51-7.53(3H,m),8.14-8.16(2H,m),8.55(1H,d,J=3.9Hz)。 Reference Example 50
tert-Butyl 4- (5-fluoro-4-phenylpyrimidin-2-yl) piperidine-1-carboxylate
1 H NMR (CDCl 3 ) δ: 1.48 (9H, s), 1.81-2.10. (4H, m), 2.87-3.13 (3H, m), 4.17-4. 30 (2H, m), 7.51-7.53 (3H, m), 8.14-8.16 (2H, m), 8.55 (1H, d, J = 3.9 Hz).
参考例51
5-フルオロ-4-フェニル-2-ピペリジン-4-イルピリミジン二塩酸塩

参考例50で得たtert-ブチル 4-(5-フルオロ-4-フェニルピリミジン-2-イル)ピペリジン-1-カルボキシラート(580mg、1.62ミリモル)のメタノール懸濁溶液に4N塩化水素-酢酸エチル溶液(10ml、40ミリモル)を加え、室温で4時間攪拌した。ジイソプロピルエーテル(20ml)を加えて、氷冷下、1時間攪拌した。析出物をろ取し、ジイソプロピルエーテルで洗浄して表題化合物(400mg、75%)を白色固体として得た。
1H NMR(DMSO-d6)δ:1.96-2.24(4H,m),3.00-3.12(2H,m),3.20-3.43(4H,m),7.59-7.63(3H,m),8.06-8.09(2H,m),8.82(1H,brs),8.92(1H,d,J=3.3Hz),9.10(1H,brs)。 Reference Example 51
5-Fluoro-4-phenyl-2-piperidin-4-ylpyrimidine dihydrochloride
To a suspension of tert-butyl 4- (5-fluoro-4-phenylpyrimidin-2-yl) piperidine-1-carboxylate (580 mg, 1.62 mmol) in methanol obtained in Reference Example 50 in 4N hydrogen chloride-acetic acid. Ethyl solution (10 ml, 40 mmol) was added and stirred at room temperature for 4 hours. Diisopropyl ether (20 ml) was added, and the mixture was stirred for 1 hour under ice cooling. The precipitate was collected by filtration and washed with diisopropyl ether to give the title compound (400 mg, 75%) as a white solid.
1 H NMR (DMSO-d 6 ) δ: 1.96-2.24 (4H, m), 3.00-3.12 (2H, m), 3.20-3.43 (4H, m), 7.59-7.63 (3H, m), 8.06-8.09 (2H, m), 8.82 (1H, brs), 8.92 (1H, d, J = 3.3 Hz), 9.10 (1H, brs).
5-フルオロ-4-フェニル-2-ピペリジン-4-イルピリミジン二塩酸塩
1H NMR(DMSO-d6)δ:1.96-2.24(4H,m),3.00-3.12(2H,m),3.20-3.43(4H,m),7.59-7.63(3H,m),8.06-8.09(2H,m),8.82(1H,brs),8.92(1H,d,J=3.3Hz),9.10(1H,brs)。 Reference Example 51
5-Fluoro-4-phenyl-2-piperidin-4-ylpyrimidine dihydrochloride
1 H NMR (DMSO-d 6 ) δ: 1.96-2.24 (4H, m), 3.00-3.12 (2H, m), 3.20-3.43 (4H, m), 7.59-7.63 (3H, m), 8.06-8.09 (2H, m), 8.82 (1H, brs), 8.92 (1H, d, J = 3.3 Hz), 9.10 (1H, brs).
実施例1
4-(4-フェニルピリミジン-2-イル)-N-ピリジン-3-イルピペリジン-1-カルボキサミド

参考例2で得た4-フェニル-2-ピペリジン-4-イルピリミジン二塩酸塩(250mg、1.00ミリモル)、3-ピリジンイソシアネート(127mg、1.0ミリモル)のテトラヒドロフラン(4ml)溶液にトリエチルアミン(210mg、2.1ミリモル)を加え、室温で終夜撹拌した。反応溶液を酢酸エチルで希釈し、飽和食塩水で洗浄し、硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。残渣をヘキサンと塩化メチレンから再結晶して表題化合物(345mg、92%)を固体として得た。
1H NMR(DMSO-d6)δ:1.68-1.85(2H,m),2.00-2.18(2H,m),3.01(2H,t,J=12.8Hz),3.07-3.18(1H,m),4.20-4.30(2H,m),7.23(1H,dd,J=8.4,4.8Hz),7.45-7.58(3H,m),7.82-7.94(2H,m),8.11(1H,dd,J=4.8,1.6Hz),8.15-8.23(2H,m),8.62(1H,s),8.70(1H,s),8.78(1H,d,J=5.2Hz)。 Example 1
4- (4-Phenylpyrimidin-2-yl) -N-pyridin-3-ylpiperidin-1-carboxamide
Triethylamine was added to a solution of 4-phenyl-2-piperidin-4-ylpyrimidine dihydrochloride (250 mg, 1.00 mmol) and 3-pyridine isocyanate (127 mg, 1.0 mmol) obtained in Reference Example 2 in tetrahydrofuran (4 ml). (210 mg, 2.1 mmol) was added and stirred at room temperature overnight. The reaction solution was diluted with ethyl acetate, washed with saturated brine, dried over sodium sulfate, and the solvent was evaporated under reduced pressure. The residue was recrystallized from hexane and methylene chloride to give the title compound (345 mg, 92%) as a solid.
1 H NMR (DMSO-d 6 ) δ: 1.68-1.85 (2H, m), 2.00-2.18 (2H, m), 3.01 (2H, t, J = 12.8 Hz) ), 3.07-3.18 (1H, m), 4.20-4.30 (2H, m), 7.23 (1H, dd, J = 8.4, 4.8 Hz), 7.45. -7.58 (3H, m), 7.82-7.94 (2H, m), 8.11 (1H, dd, J = 4.8, 1.6 Hz), 8.15-8.23 ( 2H, m), 8.62 (1H, s), 8.70 (1H, s), 8.78 (1H, d, J = 5.2 Hz).
4-(4-フェニルピリミジン-2-イル)-N-ピリジン-3-イルピペリジン-1-カルボキサミド
1H NMR(DMSO-d6)δ:1.68-1.85(2H,m),2.00-2.18(2H,m),3.01(2H,t,J=12.8Hz),3.07-3.18(1H,m),4.20-4.30(2H,m),7.23(1H,dd,J=8.4,4.8Hz),7.45-7.58(3H,m),7.82-7.94(2H,m),8.11(1H,dd,J=4.8,1.6Hz),8.15-8.23(2H,m),8.62(1H,s),8.70(1H,s),8.78(1H,d,J=5.2Hz)。 Example 1
4- (4-Phenylpyrimidin-2-yl) -N-pyridin-3-ylpiperidin-1-carboxamide
1 H NMR (DMSO-d 6 ) δ: 1.68-1.85 (2H, m), 2.00-2.18 (2H, m), 3.01 (2H, t, J = 12.8 Hz) ), 3.07-3.18 (1H, m), 4.20-4.30 (2H, m), 7.23 (1H, dd, J = 8.4, 4.8 Hz), 7.45. -7.58 (3H, m), 7.82-7.94 (2H, m), 8.11 (1H, dd, J = 4.8, 1.6 Hz), 8.15-8.23 ( 2H, m), 8.62 (1H, s), 8.70 (1H, s), 8.78 (1H, d, J = 5.2 Hz).
実施例2
N-(3,4-ジメチルイソオキサゾール-5-イル)-4-(4-フェニルピリミジン-2-イル)ピペリジン-1-カルボキサミド

参考例2で得た4-フェニル-2-ピペリジン-4-イルピリミジン二塩酸塩(250mg、1.00ミリモル)、(3,4-ジメチルイソオキサゾール-5-イル)カルバミン酸2,2,2-トリクロロエチル(295mg、1.00ミリモル)、ジイソプロピルエチルアミン(268mg、2.10ミリモル)のジメチルスルホキシド(2.0ml)の混合液を80℃で終夜攪拌した。反応溶液を酢酸エチルで希釈し、飽和食塩水で洗浄し、硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(塩化メチレン:メタノール=100:1、0.5%トリエチルアミン添加)で精製し、ヘキサンと塩化メチレンから再結晶して表題化合物(158mg、40%)を固体として得た。
1H NMR(DMSO-d6)δ:1.68-1.84(5H,m),2.01-2.10(2H,m),2.09(3H,s),3.00(2H,t,J=12.8Hz),3.06-3.17(1H,m),4.12-4.20(2H,m),7.44-7.58(3H,m),7.88(1H,d,J=5.2Hz),8.15-8.25(2H,m),8.78(1H,d,J=5.2Hz),9.12(1H,s)。 Example 2
N- (3,4-dimethylisoxazol-5-yl) -4- (4-phenylpyrimidin-2-yl) piperidine-1-carboxamide
4-Phenyl-2-piperidin-4-ylpyrimidine dihydrochloride (250 mg, 1.00 mmol), (3,4-dimethylisoxazol-5-yl) carbamic acid 2,2,2 obtained in Reference Example 2 A mixture of trichloroethyl (295 mg, 1.00 mmol), diisopropylethylamine (268 mg, 2.10 mmol) in dimethyl sulfoxide (2.0 ml) was stirred at 80 ° C. overnight. The reaction solution was diluted with ethyl acetate, washed with saturated brine, dried over sodium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (methylene chloride: methanol = 100: 1, 0.5% triethylamine added) and recrystallized from hexane and methylene chloride to obtain the title compound (158 mg, 40%) as a solid.
1 H NMR (DMSO-d 6 ) δ: 1.68-1.84 (5H, m), 2.01-2.10 (2H, m), 2.09 (3H, s), 3.00 ( 2H, t, J = 12.8 Hz), 3.06-3.17 (1H, m), 4.12-4.20 (2H, m), 7.44-7.58 (3H, m), 7.88 (1H, d, J = 5.2 Hz), 8.15-8.25 (2H, m), 8.78 (1H, d, J = 5.2 Hz), 9.12 (1H, s ).
N-(3,4-ジメチルイソオキサゾール-5-イル)-4-(4-フェニルピリミジン-2-イル)ピペリジン-1-カルボキサミド
1H NMR(DMSO-d6)δ:1.68-1.84(5H,m),2.01-2.10(2H,m),2.09(3H,s),3.00(2H,t,J=12.8Hz),3.06-3.17(1H,m),4.12-4.20(2H,m),7.44-7.58(3H,m),7.88(1H,d,J=5.2Hz),8.15-8.25(2H,m),8.78(1H,d,J=5.2Hz),9.12(1H,s)。 Example 2
N- (3,4-dimethylisoxazol-5-yl) -4- (4-phenylpyrimidin-2-yl) piperidine-1-carboxamide
1 H NMR (DMSO-d 6 ) δ: 1.68-1.84 (5H, m), 2.01-2.10 (2H, m), 2.09 (3H, s), 3.00 ( 2H, t, J = 12.8 Hz), 3.06-3.17 (1H, m), 4.12-4.20 (2H, m), 7.44-7.58 (3H, m), 7.88 (1H, d, J = 5.2 Hz), 8.15-8.25 (2H, m), 8.78 (1H, d, J = 5.2 Hz), 9.12 (1H, s ).
実施例3
4-(4-フェニルピリミジン-2-イル)-N-ピリダジン-3-イルピペリジン-1-カルボキサミド

参考例2で得た4-フェニル-2-ピペリジン-4-イルピリミジン二塩酸塩(250mg、1.0mmol)、ピリダジン-3-イルカルバミン酸2,2,2-トリクロロエチル(281mg、1.0ミリモル)、ジイソプロピルエチルアミン(268mg、2.10ミリモル)のジメチルスルホキシド(2.0ml)の混合液を80℃で終夜攪拌した。反応溶液を酢酸エチルで希釈し、飽和食塩水で洗浄し、硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(塩化メチレン:メタノール=100:1、0.5%トリエチルアミン添加)で精製し、ヘキサンと塩化メチレンから再結晶して表題化合物(277mg、74%)を固体として得た。
1H NMR(DMSO-d6)δ:1.71-1.87(2H,m),2.02-2.10(2H,m),3.03(2H,t,J=12.8Hz),3.09-3.17(1H,m),4.20-4.35(2H,m),7.45-7.60(4H,m),7.88(1H,d,J=5.6Hz),7.97(1H,dd,J=9.2,1.2Hz),8.12-8.26(2H,m),8.72-8.85(2H,m),9.83(1H,s)。 Example 3
4- (4-Phenylpyrimidin-2-yl) -N-pyridazin-3-ylpiperidin-1-carboxamide
4-Phenyl-2-piperidin-4-ylpyrimidine dihydrochloride obtained in Reference Example 2 (250 mg, 1.0 mmol), pyridazin-3-ylcarbamate 2,2,2-trichloroethyl (281 mg, 1.0 Mmol), diisopropylethylamine (268 mg, 2.10 mmol) in dimethyl sulfoxide (2.0 ml) was stirred at 80 ° C. overnight. The reaction solution was diluted with ethyl acetate, washed with saturated brine, dried over sodium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (methylene chloride: methanol = 100: 1, 0.5% triethylamine added) and recrystallized from hexane and methylene chloride to obtain the title compound (277 mg, 74%) as a solid.
1 H NMR (DMSO-d 6 ) δ: 1.71-1.87 (2H, m), 2.02-2.10 (2H, m), 3.03 (2H, t, J = 12.8 Hz) ), 3.09-3.17 (1H, m), 4.20-4.35 (2H, m), 7.45-7.60 (4H, m), 7.88 (1H, d, J = 5.6 Hz), 7.97 (1H, dd, J = 9.2, 1.2 Hz), 8.12-8.26 (2H, m), 8.72-8.85 (2H, m) , 9.83 (1H, s).
4-(4-フェニルピリミジン-2-イル)-N-ピリダジン-3-イルピペリジン-1-カルボキサミド
1H NMR(DMSO-d6)δ:1.71-1.87(2H,m),2.02-2.10(2H,m),3.03(2H,t,J=12.8Hz),3.09-3.17(1H,m),4.20-4.35(2H,m),7.45-7.60(4H,m),7.88(1H,d,J=5.6Hz),7.97(1H,dd,J=9.2,1.2Hz),8.12-8.26(2H,m),8.72-8.85(2H,m),9.83(1H,s)。 Example 3
4- (4-Phenylpyrimidin-2-yl) -N-pyridazin-3-ylpiperidin-1-carboxamide
1 H NMR (DMSO-d 6 ) δ: 1.71-1.87 (2H, m), 2.02-2.10 (2H, m), 3.03 (2H, t, J = 12.8 Hz) ), 3.09-3.17 (1H, m), 4.20-4.35 (2H, m), 7.45-7.60 (4H, m), 7.88 (1H, d, J = 5.6 Hz), 7.97 (1H, dd, J = 9.2, 1.2 Hz), 8.12-8.26 (2H, m), 8.72-8.85 (2H, m) , 9.83 (1H, s).
実施例4
4-[4-(2,3-ジフルオロフェニル)ピリミジン-2-イル]-N-ピリジン-3-イルピペリジン-1-カルボキサミドトリフルオロ酢酸塩

参考例4で得た4-(2,3-ジフルオロフェニル)-2-ピペリジン-4-イルピリミジン二塩酸塩(250mg、0.900ミリモル)、3-ピリジンイソシアネート(111mg、0.900ミリモル)のテトラヒドロフラン(4ml)溶液にトリエチルアミン(184mg、1.80ミリモル)を加え、室温で終夜撹拌した。反応溶液を酢酸エチルで希釈し、飽和食塩水で洗浄し、硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。残渣をヘキサンと塩化メチレンから再結晶し、高速液体クロマトグラフィー(FujiC18HPLCカラムA液0.1%トリフルオロ酢酸アセトニトリル溶液、B液0.1%トリフルオロ酢酸水溶液)により精製して表題化合物(300mg、83%)を得た。
1H NMR(DMSO-d6)δ:1.70-1.84(2H,m),2.00-2.10(2H,m),3.06(2H,t,J=12.4Hz),3.12-3.24(1H,m),4.20-4.30(2H,m),7.38(1H,dd,J=13.0,7.2Hz),7.55-7.68(1H,m),7.69-7.94(3H,m),8.22-8.43(2H,m),8.88(1H,d,J=5.2Hz),8.92-9.07(1H,m),9.22-9.42(1H,m). Example 4
4- [4- (2,3-Difluorophenyl) pyrimidin-2-yl] -N-pyridin-3-ylpiperidin-1-carboxamide trifluoroacetate
4- (2,3-difluorophenyl) -2-piperidin-4-ylpyrimidine dihydrochloride (250 mg, 0.900 mmol), 3-pyridine isocyanate (111 mg, 0.900 mmol) obtained in Reference Example 4 Triethylamine (184 mg, 1.80 mmol) was added to a tetrahydrofuran (4 ml) solution, and the mixture was stirred at room temperature overnight. The reaction solution was diluted with ethyl acetate, washed with saturated brine, dried over sodium sulfate, and the solvent was evaporated under reduced pressure. The residue was recrystallized from hexane and methylene chloride and purified by high performance liquid chromatography (Fuji C18 HPLC column A solution 0.1% trifluoroacetic acid acetonitrile solution, solution B 0.1% trifluoroacetic acid aqueous solution) to give the title compound (300 mg, 83%).
1 H NMR (DMSO-d 6 ) δ: 1.70-1.84 (2H, m), 2.00-2.10 (2H, m), 3.06 (2H, t, J = 12.4 Hz) ), 3.12-3.24 (1H, m), 4.20-4.30 (2H, m), 7.38 (1H, dd, J = 13.0, 7.2 Hz), 7.55 -7.68 (1H, m), 7.69-7.94 (3H, m), 8.22-8.43 (2H, m), 8.88 (1H, d, J = 5.2Hz) , 8.92-9.07 (1H, m), 9.22-9.42 (1H, m).
4-[4-(2,3-ジフルオロフェニル)ピリミジン-2-イル]-N-ピリジン-3-イルピペリジン-1-カルボキサミドトリフルオロ酢酸塩
1H NMR(DMSO-d6)δ:1.70-1.84(2H,m),2.00-2.10(2H,m),3.06(2H,t,J=12.4Hz),3.12-3.24(1H,m),4.20-4.30(2H,m),7.38(1H,dd,J=13.0,7.2Hz),7.55-7.68(1H,m),7.69-7.94(3H,m),8.22-8.43(2H,m),8.88(1H,d,J=5.2Hz),8.92-9.07(1H,m),9.22-9.42(1H,m). Example 4
4- [4- (2,3-Difluorophenyl) pyrimidin-2-yl] -N-pyridin-3-ylpiperidin-1-carboxamide trifluoroacetate
1 H NMR (DMSO-d 6 ) δ: 1.70-1.84 (2H, m), 2.00-2.10 (2H, m), 3.06 (2H, t, J = 12.4 Hz) ), 3.12-3.24 (1H, m), 4.20-4.30 (2H, m), 7.38 (1H, dd, J = 13.0, 7.2 Hz), 7.55 -7.68 (1H, m), 7.69-7.94 (3H, m), 8.22-8.43 (2H, m), 8.88 (1H, d, J = 5.2Hz) , 8.92-9.07 (1H, m), 9.22-9.42 (1H, m).
実施例5
4-[4-(2,3-ジフルオロフェニル)ピリミジン-2-イル]-N-ピリジン-3-イルピペリジン-1-カルボキサミド

参考例4で得た4-(2,3-ジフルオロフェニル)-2-ピペリジン-4-イルピリミジン二塩酸塩(1.00g、2.87ミリモル)、ピリジン-3-イルカルバミン酸2,2,2-トリクロロエチル(851mg、3.16ミリモル)のアセトン(5ml)の懸濁液にトリエチルアミン(1.60ml、11.5ミリモル)を室温で滴下し、反応溶液を65℃で終夜攪拌した。反応液に水を注ぎ、酢酸エチルで抽出した。抽出液を水で洗浄し、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル)で精製し、ヘキサンとテトラヒドロフランから再結晶して表題化合物(773mg、68%)を固体として得た。
1H NMR(DMSO-d6)δ:1.70-1.89(2H,m),1.98-2.14(2H,m),2.93-3.10(2H,m),3.10-3.25(1H,m),4.16-4.32(2H,m),7.26(1H,dd,J=8.1,4.7Hz),7.34-7.47(1H,m),7.56-7.70(1H,m),7.77(1H,dd,J=5.1,2.3Hz),7.83-7.95(2H,m),8.14(1H,dd,J=4.7,1.5Hz),8.65(1H,d,J=2.3Hz),8.73(1H,s),8.91(1H,d,J=5.1Hz)。 Example 5
4- [4- (2,3-Difluorophenyl) pyrimidin-2-yl] -N-pyridin-3-ylpiperidine-1-carboxamide
4- (2,3-Difluorophenyl) -2-piperidin-4-ylpyrimidine dihydrochloride (1.00 g, 2.87 mmol) obtained in Reference Example 4, 2,3-pyridin-3-ylcarbamic acid 2,2, Triethylamine (1.60 ml, 11.5 mmol) was added dropwise at room temperature to a suspension of 2-trichloroethyl (851 mg, 3.16 mmol) in acetone (5 ml), and the reaction solution was stirred at 65 ° C. overnight. Water was poured into the reaction mixture, and the mixture was extracted with ethyl acetate. The extract was washed with water and dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (ethyl acetate) and recrystallized from hexane and tetrahydrofuran to give the title compound (773 mg, 68%) as a solid.
1 H NMR (DMSO-d 6 ) δ: 1.70-1.89 (2H, m), 1.98-2.14 (2H, m), 2.93-3.10 (2H, m), 3.10-3.25 (1H, m), 4.16-4.32 (2H, m), 7.26 (1H, dd, J = 8.1, 4.7 Hz), 7.34-7 .47 (1H, m), 7.56-7.70 (1H, m), 7.77 (1H, dd, J = 5.1, 2.3 Hz), 7.83-7.95 (2H, m), 8.14 (1H, dd, J = 4.7, 1.5 Hz), 8.65 (1H, d, J = 2.3 Hz), 8.73 (1H, s), 8.91 ( 1H, d, J = 5.1 Hz).
4-[4-(2,3-ジフルオロフェニル)ピリミジン-2-イル]-N-ピリジン-3-イルピペリジン-1-カルボキサミド
1H NMR(DMSO-d6)δ:1.70-1.89(2H,m),1.98-2.14(2H,m),2.93-3.10(2H,m),3.10-3.25(1H,m),4.16-4.32(2H,m),7.26(1H,dd,J=8.1,4.7Hz),7.34-7.47(1H,m),7.56-7.70(1H,m),7.77(1H,dd,J=5.1,2.3Hz),7.83-7.95(2H,m),8.14(1H,dd,J=4.7,1.5Hz),8.65(1H,d,J=2.3Hz),8.73(1H,s),8.91(1H,d,J=5.1Hz)。 Example 5
4- [4- (2,3-Difluorophenyl) pyrimidin-2-yl] -N-pyridin-3-ylpiperidine-1-carboxamide
1 H NMR (DMSO-d 6 ) δ: 1.70-1.89 (2H, m), 1.98-2.14 (2H, m), 2.93-3.10 (2H, m), 3.10-3.25 (1H, m), 4.16-4.32 (2H, m), 7.26 (1H, dd, J = 8.1, 4.7 Hz), 7.34-7 .47 (1H, m), 7.56-7.70 (1H, m), 7.77 (1H, dd, J = 5.1, 2.3 Hz), 7.83-7.95 (2H, m), 8.14 (1H, dd, J = 4.7, 1.5 Hz), 8.65 (1H, d, J = 2.3 Hz), 8.73 (1H, s), 8.91 ( 1H, d, J = 5.1 Hz).
実施例6
4-[4-(2,3-ジフルオロフェニル)ピリミジン-2-イル]-N-(3,4-ジメチルイソオキサゾール-5-イル)ピペリジン-1-カルボキサミド

実施例2と同様にして、参考例4で得た4-(2,3-ジフルオロフェニル)-2-ピペリジン-4-イルピリミジン二塩酸塩と(3,4-ジメチルイソオキサゾール-5-イル)カルバミン酸2,2,2-トリクロロエチルから表題化合物(222mg、59%)を固体として得た。
1H NMR(DMSO-d6)δ:1.67-1.83(5H,m),1.95-2.05(2H,m),2.09(3H,s),3.00(2H,t,J=12.2Hz),3.06-3.22(1H,m),4.00-4.15(2H,m),7.34-7.42(1H,m),7.55-7.68(1H,m),7.74(1H,dd,J=5.2,1.6Hz),7.86(1H,t,J=7.4Hz),8.88(1H,d,J=5.2Hz),9.14(1H,s)。 Example 6
4- [4- (2,3-Difluorophenyl) pyrimidin-2-yl] -N- (3,4-dimethylisoxazol-5-yl) piperidine-1-carboxamide
In the same manner as in Example 2, 4- (2,3-difluorophenyl) -2-piperidin-4-ylpyrimidine dihydrochloride obtained in Reference Example 4 and (3,4-dimethylisoxazol-5-yl) The title compound (222 mg, 59%) was obtained as a solid from 2,2,2-trichloroethyl carbamate.
1 H NMR (DMSO-d 6 ) δ: 1.67-1.83 (5H, m), 1.95-2.05 (2H, m), 2.09 (3H, s), 3.00 ( 2H, t, J = 12.2 Hz), 3.06-3.22 (1H, m), 4.00-4.15 (2H, m), 7.34-7.42 (1H, m), 7.55-7.68 (1H, m), 7.74 (1H, dd, J = 5.2, 1.6 Hz), 7.86 (1H, t, J = 7.4 Hz), 8.88 (1H, d, J = 5.2 Hz), 9.14 (1H, s).
4-[4-(2,3-ジフルオロフェニル)ピリミジン-2-イル]-N-(3,4-ジメチルイソオキサゾール-5-イル)ピペリジン-1-カルボキサミド
1H NMR(DMSO-d6)δ:1.67-1.83(5H,m),1.95-2.05(2H,m),2.09(3H,s),3.00(2H,t,J=12.2Hz),3.06-3.22(1H,m),4.00-4.15(2H,m),7.34-7.42(1H,m),7.55-7.68(1H,m),7.74(1H,dd,J=5.2,1.6Hz),7.86(1H,t,J=7.4Hz),8.88(1H,d,J=5.2Hz),9.14(1H,s)。 Example 6
4- [4- (2,3-Difluorophenyl) pyrimidin-2-yl] -N- (3,4-dimethylisoxazol-5-yl) piperidine-1-carboxamide
1 H NMR (DMSO-d 6 ) δ: 1.67-1.83 (5H, m), 1.95-2.05 (2H, m), 2.09 (3H, s), 3.00 ( 2H, t, J = 12.2 Hz), 3.06-3.22 (1H, m), 4.00-4.15 (2H, m), 7.34-7.42 (1H, m), 7.55-7.68 (1H, m), 7.74 (1H, dd, J = 5.2, 1.6 Hz), 7.86 (1H, t, J = 7.4 Hz), 8.88 (1H, d, J = 5.2 Hz), 9.14 (1H, s).
実施例7
4-[4-(2,3-ジフルオロフェニル)ピリミジン-2-イル]-N-ピリダジン-3-イルピペリジン-1-カルボキサミド

実施例3と同様にして、参考例4で得た4-(2,3-ジフルオロフェニル)-2-ピペリジン-4-イルピリミジン二塩酸塩とピリダジン-3-イルカルバミン酸2,2,2-トリクロロエチルから表題化合物(229mg、63%)を固体として得た。
1H NMR(DMSO-d6)δ:1.64-1.85(2H,m),1.92-2.05(2H,m),3.02(2H,t,J=12.0Hz),3.09-3.21(1H,m),4.20-4.30(2H,m),7.30-7.40(1H,m),7.54(1H,dd,J=9.2,4.8Hz),7.61(1H,q,J=8.8Hz),7.73(1H,dd,J=5.2,2.4Hz),7.84-7.88(1H,m),7.97(1H,dd,J=9.2,1.2Hz),8.80(1H,dd,J=4.4,1.2Hz),8.87(1H,d,J=5.2Hz),9.86(1H,s)。 Example 7
4- [4- (2,3-Difluorophenyl) pyrimidin-2-yl] -N-pyridazin-3-ylpiperidin-1-carboxamide
In the same manner as in Example 3, 4- (2,3-difluorophenyl) -2-piperidin-4-ylpyrimidine dihydrochloride obtained in Reference Example 4 and pyridazin-3-ylcarbamic acid 2,2,2- The title compound (229 mg, 63%) was obtained as a solid from trichloroethyl.
1 H NMR (DMSO-d 6 ) δ: 1.64-1.85 (2H, m), 1.92-2.05 (2H, m), 3.02 (2H, t, J = 12.0 Hz) ), 3.09-3.21 (1H, m), 4.20-4.30 (2H, m), 7.30-7.40 (1H, m), 7.54 (1H, dd, J) = 9.2, 4.8 Hz), 7.61 (1H, q, J = 8.8 Hz), 7.73 (1H, dd, J = 5.2, 2.4 Hz), 7.84-7. 88 (1H, m), 7.97 (1H, dd, J = 9.2, 1.2 Hz), 8.80 (1H, dd, J = 4.4, 1.2 Hz), 8.87 (1H , D, J = 5.2 Hz), 9.86 (1H, s).
4-[4-(2,3-ジフルオロフェニル)ピリミジン-2-イル]-N-ピリダジン-3-イルピペリジン-1-カルボキサミド
1H NMR(DMSO-d6)δ:1.64-1.85(2H,m),1.92-2.05(2H,m),3.02(2H,t,J=12.0Hz),3.09-3.21(1H,m),4.20-4.30(2H,m),7.30-7.40(1H,m),7.54(1H,dd,J=9.2,4.8Hz),7.61(1H,q,J=8.8Hz),7.73(1H,dd,J=5.2,2.4Hz),7.84-7.88(1H,m),7.97(1H,dd,J=9.2,1.2Hz),8.80(1H,dd,J=4.4,1.2Hz),8.87(1H,d,J=5.2Hz),9.86(1H,s)。 Example 7
4- [4- (2,3-Difluorophenyl) pyrimidin-2-yl] -N-pyridazin-3-ylpiperidin-1-carboxamide
1 H NMR (DMSO-d 6 ) δ: 1.64-1.85 (2H, m), 1.92-2.05 (2H, m), 3.02 (2H, t, J = 12.0 Hz) ), 3.09-3.21 (1H, m), 4.20-4.30 (2H, m), 7.30-7.40 (1H, m), 7.54 (1H, dd, J) = 9.2, 4.8 Hz), 7.61 (1H, q, J = 8.8 Hz), 7.73 (1H, dd, J = 5.2, 2.4 Hz), 7.84-7. 88 (1H, m), 7.97 (1H, dd, J = 9.2, 1.2 Hz), 8.80 (1H, dd, J = 4.4, 1.2 Hz), 8.87 (1H , D, J = 5.2 Hz), 9.86 (1H, s).
実施例8
4-(6-フェニルピリミジン-4-イル)-N-ピリジン-3-イルピペリジン-1-カルボキサミドトリフルオロ酢酸塩

実施例4と同様にして、参考例6で得た4-フェニル-6-ピペリジン-4-イルピリミジン二塩酸塩と3-ピリジンイソシアネートから表題化合物(123mg、54%)を固体として得た。
1H NMR(DMSO-d6)δ:1.62-1.87(2H,m),1.90-2.05(2H,m),2.91-3.16(3H,m),4.30-4.40(2H,m),7.50-7.60(3H,m),7.89-7.95(1H,m),8.02-8.05(1H,m),8.20-8.30(2H,m),8.40-8.50(2H,m),9.13(2H,d,J=16.8Hz),9.54(1H,s)。 Example 8
4- (6-Phenylpyrimidin-4-yl) -N-pyridin-3-ylpiperidin-1-carboxamide trifluoroacetate
In the same manner as in Example 4, the title compound (123 mg, 54%) was obtained as a solid from 4-phenyl-6-piperidin-4-ylpyrimidine dihydrochloride obtained in Reference Example 6 and 3-pyridine isocyanate.
1 H NMR (DMSO-d 6 ) δ: 1.62-1.87 (2H, m), 1.90-2.05 (2H, m), 2.91-3.16 (3H, m), 4.30-4.40 (2H, m), 7.50-7.60 (3H, m), 7.89-7.95 (1H, m), 8.02-8.05 (1H, m ), 8.20-8.30 (2H, m), 8.40-8.50 (2H, m), 9.13 (2H, d, J = 16.8 Hz), 9.54 (1 H, s) ).
4-(6-フェニルピリミジン-4-イル)-N-ピリジン-3-イルピペリジン-1-カルボキサミドトリフルオロ酢酸塩
1H NMR(DMSO-d6)δ:1.62-1.87(2H,m),1.90-2.05(2H,m),2.91-3.16(3H,m),4.30-4.40(2H,m),7.50-7.60(3H,m),7.89-7.95(1H,m),8.02-8.05(1H,m),8.20-8.30(2H,m),8.40-8.50(2H,m),9.13(2H,d,J=16.8Hz),9.54(1H,s)。 Example 8
4- (6-Phenylpyrimidin-4-yl) -N-pyridin-3-ylpiperidin-1-carboxamide trifluoroacetate
1 H NMR (DMSO-d 6 ) δ: 1.62-1.87 (2H, m), 1.90-2.05 (2H, m), 2.91-3.16 (3H, m), 4.30-4.40 (2H, m), 7.50-7.60 (3H, m), 7.89-7.95 (1H, m), 8.02-8.05 (1H, m ), 8.20-8.30 (2H, m), 8.40-8.50 (2H, m), 9.13 (2H, d, J = 16.8 Hz), 9.54 (1 H, s) ).
実施例9
N-(3,4-ジメチルイソオキサゾール-5-イル)-4-(6-フェニルピリミジン-4-イル)ピペリジン-1-カルボキサミド

実施例2と同様にして、参考例6で得た4-フェニル-6-ピペリジン-4-イルピリミジン二塩酸塩と(3,4-ジメチルイソオキサゾール-5-イル)カルバミン酸2,2,2-トリクロロエチルから表題化合物(149mg、47%)を固体として得た。
1H NMR(DMSO-d6)δ:1.70-1.80(5H,m),1.90-2.00(2H,m),2.12(3H,s),2.91-3.06(3H,m),4.15-4.25(2H,m),7.50-7.60(3H,m),8.03(1H,s),8.15-8.32(2H,m),9.15(2H,brs)。 Example 9
N- (3,4-Dimethylisoxazol-5-yl) -4- (6-phenylpyrimidin-4-yl) piperidine-1-carboxamide
In the same manner as in Example 2, 4-phenyl-6-piperidin-4-ylpyrimidine dihydrochloride obtained in Reference Example 6 and (3,4-dimethylisoxazol-5-yl) carbamic acid 2,2,2 The title compound (149 mg, 47%) was obtained as a solid from trichloroethyl.
1 H NMR (DMSO-d 6 ) δ: 1.70-1.80 (5H, m), 1.90-2.00 (2H, m), 2.12 (3H, s), 2.91- 3.06 (3H, m), 4.15-4.25 (2H, m), 7.50-7.60 (3H, m), 8.03 (1H, s), 8.15-8. 32 (2H, m), 9.15 (2H, brs).
N-(3,4-ジメチルイソオキサゾール-5-イル)-4-(6-フェニルピリミジン-4-イル)ピペリジン-1-カルボキサミド
1H NMR(DMSO-d6)δ:1.70-1.80(5H,m),1.90-2.00(2H,m),2.12(3H,s),2.91-3.06(3H,m),4.15-4.25(2H,m),7.50-7.60(3H,m),8.03(1H,s),8.15-8.32(2H,m),9.15(2H,brs)。 Example 9
N- (3,4-Dimethylisoxazol-5-yl) -4- (6-phenylpyrimidin-4-yl) piperidine-1-carboxamide
1 H NMR (DMSO-d 6 ) δ: 1.70-1.80 (5H, m), 1.90-2.00 (2H, m), 2.12 (3H, s), 2.91- 3.06 (3H, m), 4.15-4.25 (2H, m), 7.50-7.60 (3H, m), 8.03 (1H, s), 8.15-8. 32 (2H, m), 9.15 (2H, brs).
実施例10
4-(6-フェニルピリミジン-4-イル)-N-ピリダジン-3-イルピペリジン-1-カルボキサミド

実施例3と同様にして、参考例6で得た4-フェニル-6-ピペリジン-4-イルピリミジン二塩酸塩とピリダジン-3-イルカルバミン酸2,2,2-トリクロロエチルから表題化合物(104mg、34%)を固体として得た。
1H NMR(DMSO-d6)δ:1.64-1.85(2H,m),1.90-2.00(2H,m),2.86-3.08(3H,m),4.30-4.40(2H,m),7.45-7.60(4H,m),7.94-8.06(2H,m),8.22-8.35(2H,m),8.83(1H,d,J=3.6Hz),9.14(1H,s),9.87(1H,s)。 Example 10
4- (6-Phenylpyrimidin-4-yl) -N-pyridazin-3-ylpiperidin-1-carboxamide
In the same manner as in Example 3, the title compound (104 mg) was obtained from 4-phenyl-6-piperidin-4-ylpyrimidine dihydrochloride obtained in Reference Example 6 and 2,2,2-trichloroethyl pyridazin-3-ylcarbamate. 34%) as a solid.
1 H NMR (DMSO-d 6 ) δ: 1.64-1.85 (2H, m), 1.90-2.00 (2H, m), 2.86-3.08 (3H, m), 4.30-4.40 (2H, m), 7.45-7.60 (4H, m), 7.94-8.06 (2H, m), 8.22-8.35 (2H, m) ), 8.83 (1H, d, J = 3.6 Hz), 9.14 (1H, s), 9.87 (1H, s).
4-(6-フェニルピリミジン-4-イル)-N-ピリダジン-3-イルピペリジン-1-カルボキサミド
1H NMR(DMSO-d6)δ:1.64-1.85(2H,m),1.90-2.00(2H,m),2.86-3.08(3H,m),4.30-4.40(2H,m),7.45-7.60(4H,m),7.94-8.06(2H,m),8.22-8.35(2H,m),8.83(1H,d,J=3.6Hz),9.14(1H,s),9.87(1H,s)。 Example 10
4- (6-Phenylpyrimidin-4-yl) -N-pyridazin-3-ylpiperidin-1-carboxamide
1 H NMR (DMSO-d 6 ) δ: 1.64-1.85 (2H, m), 1.90-2.00 (2H, m), 2.86-3.08 (3H, m), 4.30-4.40 (2H, m), 7.45-7.60 (4H, m), 7.94-8.06 (2H, m), 8.22-8.35 (2H, m) ), 8.83 (1H, d, J = 3.6 Hz), 9.14 (1H, s), 9.87 (1H, s).
実施例11
4-[6-(2,3-ジフルオロフェニル)ピリミジン-4-イル]-N-ピリジン-3-イルピペリジン-1-カルボキサミド

参考例8で得た4-(2,3-ジフルオロフェニル)-6-ピペリジン-4-イルピリミジン二塩酸塩(300mg、1.10ミリモル)、3-ピリジンイソシアネート(135mg、1.1ミリモル)のテトラヒドロフラン(3ml)溶液にトリエチルアミン(223mg、2.2ミリモル)を加え、室温で終夜撹拌した。反応溶液を酢酸エチルで希釈し、飽和食塩水で洗浄し、硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。残渣をヘキサンと酢酸エチルから再結晶して表題化合物(99.0mg、23%)を固体として得た。
1H NMR(DMSO-d6)δ:1.67-1.83(2H,m),1.95-2.05(2H,m),2.93-3.12(3H,m),4.25-4.35(2H,m),7.28(1H,dd,J=8.4,4.4Hz),7.37-7.46(1H,m),7.65(1H,q,J=8.4Hz),7.80-7.95(3H,m),8.16(1H,dd,J=4.6,1.4Hz),8.67(1H,d,J=2.4Hz),8.75(1H,s),9.26(1H,s)。 Example 11
4- [6- (2,3-Difluorophenyl) pyrimidin-4-yl] -N-pyridin-3-ylpiperidine-1-carboxamide
4- (2,3-difluorophenyl) -6-piperidin-4-ylpyrimidine dihydrochloride (300 mg, 1.10 mmol), 3-pyridine isocyanate (135 mg, 1.1 mmol) obtained in Reference Example 8 Triethylamine (223 mg, 2.2 mmol) was added to a tetrahydrofuran (3 ml) solution, and the mixture was stirred overnight at room temperature. The reaction solution was diluted with ethyl acetate, washed with saturated brine, dried over sodium sulfate, and the solvent was evaporated under reduced pressure. The residue was recrystallized from hexane and ethyl acetate to give the title compound (99.0 mg, 23%) as a solid.
1 H NMR (DMSO-d 6 ) δ: 1.67-1.83 (2H, m), 1.95-2.05 (2H, m), 2.93-3.12 (3H, m), 4.25-4.35 (2H, m), 7.28 (1H, dd, J = 8.4, 4.4 Hz), 7.37-7.46 (1H, m), 7.65 (1H , Q, J = 8.4 Hz), 7.80-7.95 (3H, m), 8.16 (1H, dd, J = 4.6, 1.4 Hz), 8.67 (1H, d, J = 2.4 Hz), 8.75 (1H, s), 9.26 (1H, s).
4-[6-(2,3-ジフルオロフェニル)ピリミジン-4-イル]-N-ピリジン-3-イルピペリジン-1-カルボキサミド
1H NMR(DMSO-d6)δ:1.67-1.83(2H,m),1.95-2.05(2H,m),2.93-3.12(3H,m),4.25-4.35(2H,m),7.28(1H,dd,J=8.4,4.4Hz),7.37-7.46(1H,m),7.65(1H,q,J=8.4Hz),7.80-7.95(3H,m),8.16(1H,dd,J=4.6,1.4Hz),8.67(1H,d,J=2.4Hz),8.75(1H,s),9.26(1H,s)。 Example 11
4- [6- (2,3-Difluorophenyl) pyrimidin-4-yl] -N-pyridin-3-ylpiperidine-1-carboxamide
1 H NMR (DMSO-d 6 ) δ: 1.67-1.83 (2H, m), 1.95-2.05 (2H, m), 2.93-3.12 (3H, m), 4.25-4.35 (2H, m), 7.28 (1H, dd, J = 8.4, 4.4 Hz), 7.37-7.46 (1H, m), 7.65 (1H , Q, J = 8.4 Hz), 7.80-7.95 (3H, m), 8.16 (1H, dd, J = 4.6, 1.4 Hz), 8.67 (1H, d, J = 2.4 Hz), 8.75 (1H, s), 9.26 (1H, s).
実施例12
4-[6-(2,3-ジフルオロフェニル)ピリミジン-4-イル]-N-(3,4-ジメチルイソオキサゾール-5-イル)ピペリジン-1-カルボキサミド

参考例2と同様にして、参考例8で得た4-(2,3-ジフルオロフェニル)-6-ピペリジン-4-イルピリミジン二塩酸塩と(3,4-ジメチルイソオキサゾール-5-イル)カルバミン酸2,2,2-トリクロロエチルから表題化合物(114mg、25%)を固体として得た。
1H NMR(DMSO-d6)δ:1.61-1.76(5H,m),1.85-1.95(2H,m),2.09(3H,s),2.87-3.06(3H,m),4.10-4.20(2H,m),7.31-7.40(1H,m),7.60(1H,q,J=6.8Hz),7.74-7.85(2H,m),9.11(1H,s),9.21(1H,s)。 Example 12
4- [6- (2,3-Difluorophenyl) pyrimidin-4-yl] -N- (3,4-dimethylisoxazol-5-yl) piperidine-1-carboxamide
In the same manner as in Reference Example 2, 4- (2,3-difluorophenyl) -6-piperidin-4-ylpyrimidine dihydrochloride obtained in Reference Example 8 and (3,4-dimethylisoxazol-5-yl) The title compound (114 mg, 25%) was obtained as a solid from 2,2,2-trichloroethyl carbamate.
1 H NMR (DMSO-d 6 ) δ: 1.61-1.76 (5H, m), 1.85-1.95 (2H, m), 2.09 (3H, s), 2.87- 3.06 (3H, m), 4.10-4.20 (2H, m), 7.31-7.40 (1H, m), 7.60 (1H, q, J = 6.8 Hz), 7.74-7.85 (2H, m), 9.11 (1H, s), 9.21 (1H, s).
4-[6-(2,3-ジフルオロフェニル)ピリミジン-4-イル]-N-(3,4-ジメチルイソオキサゾール-5-イル)ピペリジン-1-カルボキサミド
1H NMR(DMSO-d6)δ:1.61-1.76(5H,m),1.85-1.95(2H,m),2.09(3H,s),2.87-3.06(3H,m),4.10-4.20(2H,m),7.31-7.40(1H,m),7.60(1H,q,J=6.8Hz),7.74-7.85(2H,m),9.11(1H,s),9.21(1H,s)。 Example 12
4- [6- (2,3-Difluorophenyl) pyrimidin-4-yl] -N- (3,4-dimethylisoxazol-5-yl) piperidine-1-carboxamide
1 H NMR (DMSO-d 6 ) δ: 1.61-1.76 (5H, m), 1.85-1.95 (2H, m), 2.09 (3H, s), 2.87- 3.06 (3H, m), 4.10-4.20 (2H, m), 7.31-7.40 (1H, m), 7.60 (1H, q, J = 6.8 Hz), 7.74-7.85 (2H, m), 9.11 (1H, s), 9.21 (1H, s).
実施例13
4-[6-(2,3-ジフルオロフェニル)ピリミジン-4-イル]-N-ピリダジン-3-イルピペリジン-1-カルボキサミドトリフルオロ酢酸塩

参考例8で得た4-(2,3-ジフルオロフェニル)-6-ピペリジン-4-イルピリミジン二塩酸塩(300mg、1.10mmol)、ピリダジン-3-イルカルバミン酸2,2,2-トリクロロエチル(295mg、1.10ミリモル)、ジイソプロピルエチルアミン(281mg、2.20ミリモル)のジメチルスルホキシド(2.0ml)の混合液を80℃で終夜攪拌した。反応溶液を酢酸エチルで希釈し、飽和食塩水で洗浄し、硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(塩化メチレン、0.5%トリエチルアミン添加)で精製し、高速液体クロマトグラフィー(FujiC18HPLCカラムA液0.1%トリフルオロ酢酸アセトニトリル溶液、B液0.1%トリフルオロ酢酸水溶液)により精製して表題化合物(85.0mg、20%)を得た。
1H NMR(DMSO-d6)δ:1.64-1.85(2H,m),1.90-2.00(2H,m),2.94-3.17(3H,m),4.30-4.40(2H,m),7.35-7.46(1H,m),7.58-7.70(2H,m),7.79-7.90(2H,m),8.07(1H,d,J=9.2Hz),8.88(1H,d,J=3.6Hz),9.25(1H,s)。 Example 13
4- [6- (2,3-Difluorophenyl) pyrimidin-4-yl] -N-pyridazin-3-ylpiperidin-1-carboxamide trifluoroacetate
4- (2,3-difluorophenyl) -6-piperidin-4-ylpyrimidine dihydrochloride (300 mg, 1.10 mmol), pyridazin-3-ylcarbamic acid 2,2,2-trichloro obtained in Reference Example 8 A mixture of ethyl (295 mg, 1.10 mmol), diisopropylethylamine (281 mg, 2.20 mmol) in dimethyl sulfoxide (2.0 ml) was stirred at 80 ° C. overnight. The reaction solution was diluted with ethyl acetate, washed with saturated brine, dried over sodium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (methylene chloride, 0.5% triethylamine added), and high performance liquid chromatography (Fuji C18 HPLC column A solution 0.1% trifluoroacetic acid acetonitrile solution, solution B 0.1% trifluoroacetic acid aqueous solution. To give the title compound (85.0 mg, 20%).
1 H NMR (DMSO-d 6 ) δ: 1.64-1.85 (2H, m), 1.90-2.00 (2H, m), 2.94-3.17 (3H, m), 4.30-4.40 (2H, m), 7.35-7.46 (1H, m), 7.58-7.70 (2H, m), 7.79-7.90 (2H, m) ), 8.07 (1H, d, J = 9.2 Hz), 8.88 (1H, d, J = 3.6 Hz), 9.25 (1H, s).
4-[6-(2,3-ジフルオロフェニル)ピリミジン-4-イル]-N-ピリダジン-3-イルピペリジン-1-カルボキサミドトリフルオロ酢酸塩
1H NMR(DMSO-d6)δ:1.64-1.85(2H,m),1.90-2.00(2H,m),2.94-3.17(3H,m),4.30-4.40(2H,m),7.35-7.46(1H,m),7.58-7.70(2H,m),7.79-7.90(2H,m),8.07(1H,d,J=9.2Hz),8.88(1H,d,J=3.6Hz),9.25(1H,s)。 Example 13
4- [6- (2,3-Difluorophenyl) pyrimidin-4-yl] -N-pyridazin-3-ylpiperidin-1-carboxamide trifluoroacetate
1 H NMR (DMSO-d 6 ) δ: 1.64-1.85 (2H, m), 1.90-2.00 (2H, m), 2.94-3.17 (3H, m), 4.30-4.40 (2H, m), 7.35-7.46 (1H, m), 7.58-7.70 (2H, m), 7.79-7.90 (2H, m) ), 8.07 (1H, d, J = 9.2 Hz), 8.88 (1H, d, J = 3.6 Hz), 9.25 (1H, s).
実施例14
4-(2-フェニルピリミジン-4-イル)-N-ピリジン-3-イルピペリジン-1-カルボキサミド

実施例1と同様にして、参考例11で得た2-フェニル-4-ピペリジン-4-イルピリミジン二塩酸塩と3-ピリジンイソシアネートから表題化合物(334mg、89%)を固体として得た。
1H NMR(DMSO-d6)δ:1.62-1.82(2H,m),1.90-2.10(2H,m),2.86-3.07(3H,m),4.25-4.35(2H,m),7.23(1H,dd,J=8.4,4.8Hz),7.36(1H,d,J=4.8Hz),7.40-7.50(3H,m),7.88(1H,d,J=8.4Hz),8.12(1H,d,J=4.4Hz),8.39(2H,t,J=3.0Hz),8.64(1H,s),8.72(1H,s),8.78(1H,d,J=4.8Hz)。 Example 14
4- (2-Phenylpyrimidin-4-yl) -N-pyridin-3-ylpiperidine-1-carboxamide
In the same manner as in Example 1, the title compound (334 mg, 89%) was obtained as a solid from 2-phenyl-4-piperidin-4-ylpyrimidine dihydrochloride obtained in Reference Example 11 and 3-pyridine isocyanate.
1 H NMR (DMSO-d 6 ) δ: 1.62-1.82 (2H, m), 1.90-2.10 (2H, m), 2.86-3.07 (3H, m), 4.25-4.35 (2H, m), 7.23 (1H, dd, J = 8.4, 4.8 Hz), 7.36 (1H, d, J = 4.8 Hz), 7.40 -7.50 (3H, m), 7.88 (1H, d, J = 8.4 Hz), 8.12 (1H, d, J = 4.4 Hz), 8.39 (2H, t, J = 3.0 Hz), 8.64 (1 H, s), 8.72 (1 H, s), 8.78 (1 H, d, J = 4.8 Hz).
4-(2-フェニルピリミジン-4-イル)-N-ピリジン-3-イルピペリジン-1-カルボキサミド
1H NMR(DMSO-d6)δ:1.62-1.82(2H,m),1.90-2.10(2H,m),2.86-3.07(3H,m),4.25-4.35(2H,m),7.23(1H,dd,J=8.4,4.8Hz),7.36(1H,d,J=4.8Hz),7.40-7.50(3H,m),7.88(1H,d,J=8.4Hz),8.12(1H,d,J=4.4Hz),8.39(2H,t,J=3.0Hz),8.64(1H,s),8.72(1H,s),8.78(1H,d,J=4.8Hz)。 Example 14
4- (2-Phenylpyrimidin-4-yl) -N-pyridin-3-ylpiperidine-1-carboxamide
1 H NMR (DMSO-d 6 ) δ: 1.62-1.82 (2H, m), 1.90-2.10 (2H, m), 2.86-3.07 (3H, m), 4.25-4.35 (2H, m), 7.23 (1H, dd, J = 8.4, 4.8 Hz), 7.36 (1H, d, J = 4.8 Hz), 7.40 -7.50 (3H, m), 7.88 (1H, d, J = 8.4 Hz), 8.12 (1H, d, J = 4.4 Hz), 8.39 (2H, t, J = 3.0 Hz), 8.64 (1 H, s), 8.72 (1 H, s), 8.78 (1 H, d, J = 4.8 Hz).
実施例15
N-(3,4-ジメチルイソオキサゾール-5-イル)-4-(2-フェニルピリミジン-4-イル)ピペリジン-1-カルボキサミド

実施例2と同様にして、参考例11で得た2-フェニル-4-ピペリジン-4-イルピリミジン二塩酸塩と(3,4-ジメチルイソオキサゾール-5-イル)カルバミン酸2,2,2-トリクロロエチルから表題化合物(200mg、51%)を固体として得た。
1H NMR(DMSO-d6)δ:1.64-1.82(5H,m),1.90-2.00(2H,m),2.12(3H,s),2.89-3.08(3H,m),4.10-4.20(2H,m),7.37(1H,d,J=4.8Hz),7.51(3H,t,J=3.2Hz),8.39(2H,dd,J=6.4,2.8Hz),8.79(1H,d,J=5.2Hz),9.15(1H,s)。 Example 15
N- (3,4-dimethylisoxazol-5-yl) -4- (2-phenylpyrimidin-4-yl) piperidine-1-carboxamide
In the same manner as in Example 2, 2-phenyl-4-piperidin-4-ylpyrimidine dihydrochloride obtained in Reference Example 11 and (3,4-dimethylisoxazol-5-yl) carbamic acid 2,2,2 The title compound (200 mg, 51%) was obtained as a solid from trichloroethyl.
1 H NMR (DMSO-d 6 ) δ: 1.64-1.82 (5H, m), 1.90-2.00 (2H, m), 2.12 (3H, s), 2.89- 3.08 (3H, m), 4.10-4.20 (2H, m), 7.37 (1H, d, J = 4.8 Hz), 7.51 (3H, t, J = 3.2 Hz) ), 8.39 (2H, dd, J = 6.4, 2.8 Hz), 8.79 (1H, d, J = 5.2 Hz), 9.15 (1H, s).
N-(3,4-ジメチルイソオキサゾール-5-イル)-4-(2-フェニルピリミジン-4-イル)ピペリジン-1-カルボキサミド
1H NMR(DMSO-d6)δ:1.64-1.82(5H,m),1.90-2.00(2H,m),2.12(3H,s),2.89-3.08(3H,m),4.10-4.20(2H,m),7.37(1H,d,J=4.8Hz),7.51(3H,t,J=3.2Hz),8.39(2H,dd,J=6.4,2.8Hz),8.79(1H,d,J=5.2Hz),9.15(1H,s)。 Example 15
N- (3,4-dimethylisoxazol-5-yl) -4- (2-phenylpyrimidin-4-yl) piperidine-1-carboxamide
1 H NMR (DMSO-d 6 ) δ: 1.64-1.82 (5H, m), 1.90-2.00 (2H, m), 2.12 (3H, s), 2.89- 3.08 (3H, m), 4.10-4.20 (2H, m), 7.37 (1H, d, J = 4.8 Hz), 7.51 (3H, t, J = 3.2 Hz) ), 8.39 (2H, dd, J = 6.4, 2.8 Hz), 8.79 (1H, d, J = 5.2 Hz), 9.15 (1H, s).
実施例16
4-(2-フェニルピリミジン-4-イル)-N-ピリダジン-3-イルピペリジン-1-カルボキサミド

実施例3と同様にして、参考例11で得た2-フェニル-4-ピペリジン-4-イルピリミジン二塩酸塩とピリダジン-3-イルカルバミン酸2,2,2-トリクロロエチルから表題化合物(253mg、67%)を固体として得た。
1H NMR(DMSO-d6)δ:1.63-1.85(2H,m),1.90-2.00(2H,m),2.91-3.06(3H,m),4.30-4.40(2H,m),7.36(1H,d,J=5.2Hz),7.50(3H,t,J=3.2Hz),7.55(1H,dd,J=8.8,4.4Hz),8.00(1H,dd,J=8.8,0.8Hz),8.34-8.43(2H,m),8.78(1H,d,J=5.2Hz),8.81(1H,d,J=4.4Hz),9.86(1H,s)。 Example 16
4- (2-Phenylpyrimidin-4-yl) -N-pyridazin-3-ylpiperidin-1-carboxamide
In the same manner as in Example 3, the title compound (253 mg) was obtained from 2-phenyl-4-piperidin-4-ylpyrimidine dihydrochloride obtained in Reference Example 11 and 2,2,2-trichloroethyl pyridazin-3-ylcarbamate. 67%) as a solid.
1 H NMR (DMSO-d 6 ) δ: 1.63-1.85 (2H, m), 1.90-2.00 (2H, m), 2.91-3.06 (3H, m), 4.30-4.40 (2H, m), 7.36 (1H, d, J = 5.2 Hz), 7.50 (3H, t, J = 3.2 Hz), 7.55 (1H, dd , J = 8.8, 4.4 Hz), 8.00 (1H, dd, J = 8.8, 0.8 Hz), 8.34-8.43 (2H, m), 8.78 (1H, d, J = 5.2 Hz), 8.81 (1H, d, J = 4.4 Hz), 9.86 (1H, s).
4-(2-フェニルピリミジン-4-イル)-N-ピリダジン-3-イルピペリジン-1-カルボキサミド
1H NMR(DMSO-d6)δ:1.63-1.85(2H,m),1.90-2.00(2H,m),2.91-3.06(3H,m),4.30-4.40(2H,m),7.36(1H,d,J=5.2Hz),7.50(3H,t,J=3.2Hz),7.55(1H,dd,J=8.8,4.4Hz),8.00(1H,dd,J=8.8,0.8Hz),8.34-8.43(2H,m),8.78(1H,d,J=5.2Hz),8.81(1H,d,J=4.4Hz),9.86(1H,s)。 Example 16
4- (2-Phenylpyrimidin-4-yl) -N-pyridazin-3-ylpiperidin-1-carboxamide
1 H NMR (DMSO-d 6 ) δ: 1.63-1.85 (2H, m), 1.90-2.00 (2H, m), 2.91-3.06 (3H, m), 4.30-4.40 (2H, m), 7.36 (1H, d, J = 5.2 Hz), 7.50 (3H, t, J = 3.2 Hz), 7.55 (1H, dd , J = 8.8, 4.4 Hz), 8.00 (1H, dd, J = 8.8, 0.8 Hz), 8.34-8.43 (2H, m), 8.78 (1H, d, J = 5.2 Hz), 8.81 (1H, d, J = 4.4 Hz), 9.86 (1H, s).
実施例17
4-[2-(2,3-ジフルオロフェニル)ピリミジン-4-イル]-N-ピリジン-3-イルピペリジン-1-カルボキサミド

実施例1と同様にして、参考例15で得た2-(2,3-ジフルオロフェニル)-4-ピペリジン-4-イルピリミジン二塩酸塩と3-ピリジンイソシアネートから表題化合物(309mg、86%)を固体として得た。
1H NMR(DMSO-d6)δ:1.62-1.76(2H,m),1.90-2.00(2H,m),2.91-3.08(3H,m),4.25-4.35(2H,m),7.25(1H,dd,J=8.4,4.8Hz),7.27-7.36(1H,m),7.49(1H,d,J=5.2Hz),7.52-7.62(1H,m),7.80-7.94(2H,m),8.13(1H,dd,J=4.4,1.2Hz),8.64(1H,d,J=2.4Hz),8.74(1H,s),8.87(1H,d,J=5.2Hz)。 Example 17
4- [2- (2,3-Difluorophenyl) pyrimidin-4-yl] -N-pyridin-3-ylpiperidine-1-carboxamide
In the same manner as in Example 1, the title compound (309 mg, 86%) was obtained from 2- (2,3-difluorophenyl) -4-piperidin-4-ylpyrimidine dihydrochloride obtained in Reference Example 15 and 3-pyridine isocyanate. Was obtained as a solid.
1 H NMR (DMSO-d 6 ) δ: 1.62-1.76 (2H, m), 1.90-2.00 (2H, m), 2.91-3.08 (3H, m), 4.25-4.35 (2H, m), 7.25 (1 H, dd, J = 8.4, 4.8 Hz), 7.27-7.36 (1 H, m), 7.49 (1 H , D, J = 5.2 Hz), 7.52-7.62 (1H, m), 7.80-7.94 (2H, m), 8.13 (1H, dd, J = 4.4). 1.2 Hz), 8.64 (1 H, d, J = 2.4 Hz), 8.74 (1 H, s), 8.87 (1 H, d, J = 5.2 Hz).
4-[2-(2,3-ジフルオロフェニル)ピリミジン-4-イル]-N-ピリジン-3-イルピペリジン-1-カルボキサミド
1H NMR(DMSO-d6)δ:1.62-1.76(2H,m),1.90-2.00(2H,m),2.91-3.08(3H,m),4.25-4.35(2H,m),7.25(1H,dd,J=8.4,4.8Hz),7.27-7.36(1H,m),7.49(1H,d,J=5.2Hz),7.52-7.62(1H,m),7.80-7.94(2H,m),8.13(1H,dd,J=4.4,1.2Hz),8.64(1H,d,J=2.4Hz),8.74(1H,s),8.87(1H,d,J=5.2Hz)。 Example 17
4- [2- (2,3-Difluorophenyl) pyrimidin-4-yl] -N-pyridin-3-ylpiperidine-1-carboxamide
1 H NMR (DMSO-d 6 ) δ: 1.62-1.76 (2H, m), 1.90-2.00 (2H, m), 2.91-3.08 (3H, m), 4.25-4.35 (2H, m), 7.25 (1 H, dd, J = 8.4, 4.8 Hz), 7.27-7.36 (1 H, m), 7.49 (1 H , D, J = 5.2 Hz), 7.52-7.62 (1H, m), 7.80-7.94 (2H, m), 8.13 (1H, dd, J = 4.4). 1.2 Hz), 8.64 (1 H, d, J = 2.4 Hz), 8.74 (1 H, s), 8.87 (1 H, d, J = 5.2 Hz).
実施例18
4-[2-(2,3-ジフルオロフェニル)ピリミジン-4-イル]-N-(3,4-ジメチルイソオキサゾール-5-イル)ピペリジン-1-カルボキサミドトリフルオロ酢酸塩

実施例2と同様にして、参考例15で得た2-(2,3-ジフルオロフェニル)-4-ピペリジン-4-イルピリミジン二塩酸塩(250mg、0.90ミリモル)、(3,4-ジメチルイソオキサゾール-5-イル)カルバミン酸2,2,2-トリクロロエチル(258mg、0.9ミリモル)、ジイソプロピルエチルアミン(235mg、1.80ミリモル)のジメチルスルホキシド(2.0ml)の混合液を80℃で終夜攪拌した。反応溶液を酢酸エチルで希釈し、飽和食塩水で洗浄し、硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(塩化メチレン、0.5%トリエチルアミン添加)で精製し、高速液体クロマトグラフィー(FujiC18HPLCカラムA液0.1%トリフルオロ酢酸アセトニトリル溶液、B液0.1%トリフルオロ酢酸水溶液)により精製して表題化合物(43mg、11%)を固体として得た。
1H NMR(DMSO-d6)δ:1.52-1.81(5H,m),1.90-2.00(2H,m),2.11(3H,s),2.88-3.11(3H,m),4.15-4.25(2H,m),7.28-7.42(1H,m),7.47(1H,d,J=5.2Hz),7.58(1H,q,J=8.4Hz),7.85(1H,t,J=6.6Hz),8.86(1H,d,J=4.8Hz),9.15(1H,s)。 Example 18
4- [2- (2,3-Difluorophenyl) pyrimidin-4-yl] -N- (3,4-dimethylisoxazol-5-yl) piperidine-1-carboxamide trifluoroacetate
In the same manner as in Example 2, 2- (2,3-difluorophenyl) -4-piperidin-4-ylpyrimidine dihydrochloride obtained in Reference Example 15 (250 mg, 0.90 mmol), (3,4- A mixture of 2,2,2-trichloroethyl dimethylisoxazol-5-yl) carbamate (258 mg, 0.9 mmol), diisopropylethylamine (235 mg, 1.80 mmol) and dimethyl sulfoxide (2.0 ml) Stir overnight at ° C. The reaction solution was diluted with ethyl acetate, washed with saturated brine, dried over sodium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (methylene chloride, 0.5% triethylamine added), and high performance liquid chromatography (Fuji C18 HPLC column A solution 0.1% trifluoroacetic acid acetonitrile solution, solution B 0.1% trifluoroacetic acid aqueous solution. To give the title compound (43 mg, 11%) as a solid.
1 H NMR (DMSO-d 6 ) δ: 1.52-1.81 (5H, m), 1.90-2.00 (2H, m), 2.11 (3H, s), 2.88- 3.11 (3H, m), 4.15-4.25 (2H, m), 7.28-7.42 (1H, m), 7.47 (1H, d, J = 5.2 Hz), 7.58 (1H, q, J = 8.4 Hz), 7.85 (1H, t, J = 6.6 Hz), 8.86 (1H, d, J = 4.8 Hz), 9.15 (1H , S).
4-[2-(2,3-ジフルオロフェニル)ピリミジン-4-イル]-N-(3,4-ジメチルイソオキサゾール-5-イル)ピペリジン-1-カルボキサミドトリフルオロ酢酸塩
1H NMR(DMSO-d6)δ:1.52-1.81(5H,m),1.90-2.00(2H,m),2.11(3H,s),2.88-3.11(3H,m),4.15-4.25(2H,m),7.28-7.42(1H,m),7.47(1H,d,J=5.2Hz),7.58(1H,q,J=8.4Hz),7.85(1H,t,J=6.6Hz),8.86(1H,d,J=4.8Hz),9.15(1H,s)。 Example 18
4- [2- (2,3-Difluorophenyl) pyrimidin-4-yl] -N- (3,4-dimethylisoxazol-5-yl) piperidine-1-carboxamide trifluoroacetate
1 H NMR (DMSO-d 6 ) δ: 1.52-1.81 (5H, m), 1.90-2.00 (2H, m), 2.11 (3H, s), 2.88- 3.11 (3H, m), 4.15-4.25 (2H, m), 7.28-7.42 (1H, m), 7.47 (1H, d, J = 5.2 Hz), 7.58 (1H, q, J = 8.4 Hz), 7.85 (1H, t, J = 6.6 Hz), 8.86 (1H, d, J = 4.8 Hz), 9.15 (1H , S).
実施例19
4-[2-(2,3-ジフルオロフェニル)ピリミジン-4-イル]-N-ピリダジン-3-イルピペリジン-1-カルボキサミド

実施例3と同様にして、参考例15で得た2-(2,3-ジフルオロフェニル)-4-ピペリジン-4-イルピリミジン二塩酸塩とピリダジン-3-イルカルバミン酸2,2,2-トリクロロエチルから表題化合物(63mg、17%)を油状物として得た。
1H NMR(DMSO-d6)δ:1.62-1.82(2H,m),1.90-2.00(2H,m),2.91-3.11(3H,m),4.30-4.40(2H,m),7.33(1H,q,J=6.4Hz),7.48(1H,d,J=5.2Hz),7.52-7.64(2H,m),7.85(1H,t,J=7.4Hz),7.99(1H,dd,J=9.0,1.0Hz),8.82(1H,dd,J=4.6,1.0Hz),8.86(1H,d,J=5.2Hz),9.87(1H,s)。 Example 19
4- [2- (2,3-Difluorophenyl) pyrimidin-4-yl] -N-pyridazin-3-ylpiperidin-1-carboxamide
In the same manner as in Example 3, 2- (2,3-difluorophenyl) -4-piperidin-4-ylpyrimidine dihydrochloride obtained in Reference Example 15 and pyridazin-3-ylcarbamic acid 2,2,2- The title compound (63 mg, 17%) was obtained as an oil from trichloroethyl.
1 H NMR (DMSO-d 6 ) δ: 1.62-1.82 (2H, m), 1.90-2.00 (2H, m), 2.91-3.11 (3H, m), 4.30-4.40 (2H, m), 7.33 (1H, q, J = 6.4 Hz), 7.48 (1H, d, J = 5.2 Hz), 7.52-7.64 (2H, m), 7.85 (1H, t, J = 7.4 Hz), 7.99 (1H, dd, J = 9.0, 1.0 Hz), 8.82 (1H, dd, J = 4.6, 1.0 Hz), 8.86 (1H, d, J = 5.2 Hz), 9.87 (1H, s).
4-[2-(2,3-ジフルオロフェニル)ピリミジン-4-イル]-N-ピリダジン-3-イルピペリジン-1-カルボキサミド
1H NMR(DMSO-d6)δ:1.62-1.82(2H,m),1.90-2.00(2H,m),2.91-3.11(3H,m),4.30-4.40(2H,m),7.33(1H,q,J=6.4Hz),7.48(1H,d,J=5.2Hz),7.52-7.64(2H,m),7.85(1H,t,J=7.4Hz),7.99(1H,dd,J=9.0,1.0Hz),8.82(1H,dd,J=4.6,1.0Hz),8.86(1H,d,J=5.2Hz),9.87(1H,s)。 Example 19
4- [2- (2,3-Difluorophenyl) pyrimidin-4-yl] -N-pyridazin-3-ylpiperidin-1-carboxamide
1 H NMR (DMSO-d 6 ) δ: 1.62-1.82 (2H, m), 1.90-2.00 (2H, m), 2.91-3.11 (3H, m), 4.30-4.40 (2H, m), 7.33 (1H, q, J = 6.4 Hz), 7.48 (1H, d, J = 5.2 Hz), 7.52-7.64 (2H, m), 7.85 (1H, t, J = 7.4 Hz), 7.99 (1H, dd, J = 9.0, 1.0 Hz), 8.82 (1H, dd, J = 4.6, 1.0 Hz), 8.86 (1H, d, J = 5.2 Hz), 9.87 (1H, s).
実施例20
N-(3,4-ジメチルイソオキサゾール-5-イル)-4-(4-フェニルピリジン-2-イル)ピペリジン-1-カルボキサミド

参考例18で得た4-フェニル-2-ピペリジン-4-イルピリジン二塩酸塩(300mg、0.964ミリモル)のジメチルスルホキシド(3.2ml)溶液にジイソプロピルエチルアミン(0.588ml、3.37ミリモル)を室温で加え、反応溶液を30分撹拌した後、(3,4-ジメチルイソオキサゾール-5-イル)カルバミン酸2,2,2-トリクロロエチル(333mg、1.16ミリモル)を室温で加え、45℃で17時間撹拌した。反応溶液に水を注ぎ、塩化メチレンで抽出した。抽出液を飽和食塩水、水で洗浄し、硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル:ヘキサン=1:1)で精製して表題化合物(210mg、58%)を固体として得た。
1H NMR(DMSO-d6)δ:1.87-1.98(2H,m),1.90(3H,s),2.05-2.21(2H,m),2.21(3H,s),2.98-3.13(3H,m),4.21(2H,d,J=13.6Hz),6.52(1H,brs),7.37-7.39(2H,m),7.43-7.52(3H,m),7.62-7.65(2H,m),8.60(1H,dd,J=3.6,2.4Hz)。 Example 20
N- (3,4-dimethylisoxazol-5-yl) -4- (4-phenylpyridin-2-yl) piperidine-1-carboxamide
Diisopropylethylamine (0.588 ml, 3.37 mmol) was added to a solution of 4-phenyl-2-piperidin-4-ylpyridine dihydrochloride (300 mg, 0.964 mmol) obtained in Reference Example 18 in dimethyl sulfoxide (3.2 ml). After stirring the reaction solution for 30 minutes, 2,3,4-trichloroethyl (3,4-dimethylisoxazol-5-yl) carbamate (333 mg, 1.16 mmol) was added at room temperature, Stir at 45 ° C. for 17 hours. Water was poured into the reaction solution and extracted with methylene chloride. The extract was washed with saturated brine and water, dried over sodium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (ethyl acetate: hexane = 1: 1) to give the title compound (210 mg, 58%) as a solid.
1 H NMR (DMSO-d 6 ) δ: 1.87-1.98 (2H, m), 1.90 (3H, s), 2.05-2.21 (2H, m), 2.21 ( 3H, s), 2.98-3.13 (3H, m), 4.21 (2H, d, J = 13.6 Hz), 6.52 (1H, brs), 7.37-7.39 ( 2H, m), 7.43-7.52 (3H, m), 7.62-7.65 (2H, m), 8.60 (1H, dd, J = 3.6, 2.4 Hz).
N-(3,4-ジメチルイソオキサゾール-5-イル)-4-(4-フェニルピリジン-2-イル)ピペリジン-1-カルボキサミド
1H NMR(DMSO-d6)δ:1.87-1.98(2H,m),1.90(3H,s),2.05-2.21(2H,m),2.21(3H,s),2.98-3.13(3H,m),4.21(2H,d,J=13.6Hz),6.52(1H,brs),7.37-7.39(2H,m),7.43-7.52(3H,m),7.62-7.65(2H,m),8.60(1H,dd,J=3.6,2.4Hz)。 Example 20
N- (3,4-dimethylisoxazol-5-yl) -4- (4-phenylpyridin-2-yl) piperidine-1-carboxamide
1 H NMR (DMSO-d 6 ) δ: 1.87-1.98 (2H, m), 1.90 (3H, s), 2.05-2.21 (2H, m), 2.21 ( 3H, s), 2.98-3.13 (3H, m), 4.21 (2H, d, J = 13.6 Hz), 6.52 (1H, brs), 7.37-7.39 ( 2H, m), 7.43-7.52 (3H, m), 7.62-7.65 (2H, m), 8.60 (1H, dd, J = 3.6, 2.4 Hz).
実施例21
N-(3,4-ジメチルイソオキサゾール-5-イル)-4-[4-(2-フルオロフェニル)ピリジン-2-イル]ピペリジン-1-カルボキサミド

実施例20と同様にして、参考例21で得た4-(2-フルオロフェニル)-2-ピペリジン-4-イルピリジン二塩酸塩と(3,4-ジメチルイソオキサゾール-5-イル)カルバミン酸2,2,2-トリクロロエチルから表題化合物(210mg、58%)を固体として得た。
1H NMR(DMSO-d6)δ:1.67-1.77(2H,m),1.76(3H,s),1.89-1.92(2H,m),2.13(3H,s),2.93-3.04(3H,m),4.19(2H,d,J=12.8Hz),7.34-7.40(2H,m),7.42-7.44(1H,m),7.49-7.55(2H,m),7.66(1H,td,J=7.9,1.6Hz),8.60(1H,d,J=4.8Hz),9.15(1H,brs)。 Example 21
N- (3,4-dimethylisoxazol-5-yl) -4- [4- (2-fluorophenyl) pyridin-2-yl] piperidine-1-carboxamide
In the same manner as in Example 20, 4- (2-fluorophenyl) -2-piperidin-4-ylpyridine dihydrochloride obtained in Reference Example 21 and (3,4-dimethylisoxazol-5-yl) carbamic acid 2 , 2,2-Trichloroethyl gave the title compound (210 mg, 58%) as a solid.
1 H NMR (DMSO-d 6 ) δ: 1.67-1.77 (2H, m), 1.76 (3H, s), 1.89-1.92 (2H, m), 2.13 ( 3H, s), 2.93-3.04 (3H, m), 4.19 (2H, d, J = 12.8 Hz), 7.34-7.40 (2H, m), 7.42- 7.44 (1H, m), 7.49-7.55 (2H, m), 7.66 (1H, td, J = 7.9, 1.6 Hz), 8.60 (1H, d, J = 4.8 Hz), 9.15 (1H, brs).
N-(3,4-ジメチルイソオキサゾール-5-イル)-4-[4-(2-フルオロフェニル)ピリジン-2-イル]ピペリジン-1-カルボキサミド
1H NMR(DMSO-d6)δ:1.67-1.77(2H,m),1.76(3H,s),1.89-1.92(2H,m),2.13(3H,s),2.93-3.04(3H,m),4.19(2H,d,J=12.8Hz),7.34-7.40(2H,m),7.42-7.44(1H,m),7.49-7.55(2H,m),7.66(1H,td,J=7.9,1.6Hz),8.60(1H,d,J=4.8Hz),9.15(1H,brs)。 Example 21
N- (3,4-dimethylisoxazol-5-yl) -4- [4- (2-fluorophenyl) pyridin-2-yl] piperidine-1-carboxamide
1 H NMR (DMSO-d 6 ) δ: 1.67-1.77 (2H, m), 1.76 (3H, s), 1.89-1.92 (2H, m), 2.13 ( 3H, s), 2.93-3.04 (3H, m), 4.19 (2H, d, J = 12.8 Hz), 7.34-7.40 (2H, m), 7.42- 7.44 (1H, m), 7.49-7.55 (2H, m), 7.66 (1H, td, J = 7.9, 1.6 Hz), 8.60 (1H, d, J = 4.8 Hz), 9.15 (1H, brs).
実施例22
N-(3,4-ジメチルイソオキサゾール-5-イル)-4-[4-(3-フルオロフェニル)ピリジン-2-イル]ピペリジン-1-カルボキサミド

実施例20と同様にして、参考例24で得た4-(3-フルオロフェニル)-2-ピペリジン-4-イルピリジン二塩酸塩と(3,4-ジメチルイソオキサゾール-5-イル)カルバミン酸2,2,2-トリクロロエチルから表題化合物(160mg、45%)を固体として得た。
1H NMR(DMSO-d6)δ:1.74-1.81(2H,m),1.77(3H,s),1.90-1.93(2H,m),2.13(3H,s),2.93-3.02(3H,m),4.21(2H,d,J=13.6Hz),7.29-7.34(1H,m),7.54-7.60(2H,m),7.68-7.74(3H,m),8.58(1H,d,J=5.2Hz),9.15(1H,brs)。 Example 22
N- (3,4-dimethylisoxazol-5-yl) -4- [4- (3-fluorophenyl) pyridin-2-yl] piperidine-1-carboxamide
In the same manner as in Example 20, 4- (3-fluorophenyl) -2-piperidin-4-ylpyridine dihydrochloride obtained in Reference Example 24 and (3,4-dimethylisoxazol-5-yl) carbamic acid 2 , 2,2-Trichloroethyl gave the title compound (160 mg, 45%) as a solid.
1 H NMR (DMSO-d 6 ) δ: 1.74-1.81 (2H, m), 1.77 (3H, s), 1.90-1.93 (2H, m), 2.13 ( 3H, s), 2.93-3.02 (3H, m), 4.21 (2H, d, J = 13.6 Hz), 7.29-7.34 (1H, m), 7.54- 7.60 (2H, m), 7.68-7.74 (3H, m), 8.58 (1H, d, J = 5.2 Hz), 9.15 (1H, brs).
N-(3,4-ジメチルイソオキサゾール-5-イル)-4-[4-(3-フルオロフェニル)ピリジン-2-イル]ピペリジン-1-カルボキサミド
1H NMR(DMSO-d6)δ:1.74-1.81(2H,m),1.77(3H,s),1.90-1.93(2H,m),2.13(3H,s),2.93-3.02(3H,m),4.21(2H,d,J=13.6Hz),7.29-7.34(1H,m),7.54-7.60(2H,m),7.68-7.74(3H,m),8.58(1H,d,J=5.2Hz),9.15(1H,brs)。 Example 22
N- (3,4-dimethylisoxazol-5-yl) -4- [4- (3-fluorophenyl) pyridin-2-yl] piperidine-1-carboxamide
1 H NMR (DMSO-d 6 ) δ: 1.74-1.81 (2H, m), 1.77 (3H, s), 1.90-1.93 (2H, m), 2.13 ( 3H, s), 2.93-3.02 (3H, m), 4.21 (2H, d, J = 13.6 Hz), 7.29-7.34 (1H, m), 7.54- 7.60 (2H, m), 7.68-7.74 (3H, m), 8.58 (1H, d, J = 5.2 Hz), 9.15 (1H, brs).
実施例23
N-(3,4-ジメチルイソオキサゾール-5-イル)-4-[4-(4-フルオロフェニル)ピリジン-2-イル]ピペリジン-1-カルボキサミド

実施例20と同様にして、参考例26で得た4-(4-フルオロフェニル)-2-ピペリジン-4-イルピリジン二塩酸塩と(3,4-ジメチルイソオキサゾール-5-イル)カルバミン酸2,2,2-トリクロロエチルから表題化合物(160mg、45%)を固体として得た。
1H NMR(DMSO-d6)δ:1.71-1.77(2H,m),1.79(3H,s),1.89(2H,d,J=10.8Hz),2.11(3H,s),2.91-3.01(3H,m),4.19(2H,d,J=13.2Hz),7.34(2H,t,J=8.8Hz),7.51-7.53(1H,m),7.60(1H,s),7.86-7.89(2H,m),8.54(1H,d,J=5.2Hz),9.12(1H,brs)。 Example 23
N- (3,4-dimethylisoxazol-5-yl) -4- [4- (4-fluorophenyl) pyridin-2-yl] piperidine-1-carboxamide
In the same manner as in Example 20, 4- (4-fluorophenyl) -2-piperidin-4-ylpyridine dihydrochloride obtained in Reference Example 26 and (3,4-dimethylisoxazol-5-yl) carbamic acid 2 , 2,2-Trichloroethyl gave the title compound (160 mg, 45%) as a solid.
1 H NMR (DMSO-d 6 ) δ: 1.71-1.77 (2H, m), 1.79 (3H, s), 1.89 (2H, d, J = 10.8 Hz), 2. 11 (3H, s), 2.91-3.01 (3H, m), 4.19 (2H, d, J = 13.2 Hz), 7.34 (2H, t, J = 8.8 Hz), 7.51-7.53 (1H, m), 7.60 (1H, s), 7.86-7.89 (2H, m), 8.54 (1H, d, J = 5.2 Hz), 9.12 (1H, brs).
N-(3,4-ジメチルイソオキサゾール-5-イル)-4-[4-(4-フルオロフェニル)ピリジン-2-イル]ピペリジン-1-カルボキサミド
1H NMR(DMSO-d6)δ:1.71-1.77(2H,m),1.79(3H,s),1.89(2H,d,J=10.8Hz),2.11(3H,s),2.91-3.01(3H,m),4.19(2H,d,J=13.2Hz),7.34(2H,t,J=8.8Hz),7.51-7.53(1H,m),7.60(1H,s),7.86-7.89(2H,m),8.54(1H,d,J=5.2Hz),9.12(1H,brs)。 Example 23
N- (3,4-dimethylisoxazol-5-yl) -4- [4- (4-fluorophenyl) pyridin-2-yl] piperidine-1-carboxamide
1 H NMR (DMSO-d 6 ) δ: 1.71-1.77 (2H, m), 1.79 (3H, s), 1.89 (2H, d, J = 10.8 Hz), 2. 11 (3H, s), 2.91-3.01 (3H, m), 4.19 (2H, d, J = 13.2 Hz), 7.34 (2H, t, J = 8.8 Hz), 7.51-7.53 (1H, m), 7.60 (1H, s), 7.86-7.89 (2H, m), 8.54 (1H, d, J = 5.2 Hz), 9.12 (1H, brs).
実施例24
4-[4-(2,3-ジフルオロフェニル)ピリジン-2-イル]-N-(3,4-ジメチルイソオキサゾール-5-イル)ピペリジン-1-カルボキサミド

実施例20と同様にして、参考例29で得た4-(2,3-ジフルオロフェニル)-2-ピペリジン-4-イルピリジン二塩酸塩と(3,4-ジメチルイソオキサゾール-5-イル)カルバミン酸2,2,2-トリクロロエチルから表題化合物(110mg、31%)を固体として得た。
1H NMR(DMSO-d6)δ:1.71-1.76(2H,m),1.76(3H,s),1.91(2H,d,J=12.0Hz),2.13(3H,s),2.93-3.02(3H,m),4.20(2H,d,J=12.8Hz),7.36-7.59(5H,m),8.63(1H,d,J=4.4Hz),9.15(1H,brs)。 Example 24
4- [4- (2,3-Difluorophenyl) pyridin-2-yl] -N- (3,4-dimethylisoxazol-5-yl) piperidine-1-carboxamide
In the same manner as in Example 20, 4- (2,3-difluorophenyl) -2-piperidin-4-ylpyridine dihydrochloride and (3,4-dimethylisoxazol-5-yl) carbamine obtained in Reference Example 29 The title compound (110 mg, 31%) was obtained as a solid from the acid 2,2,2-trichloroethyl.
1 H NMR (DMSO-d 6 ) δ: 1.71-1.76 (2H, m), 1.76 (3H, s), 1.91 (2H, d, J = 12.0 Hz), 2. 13 (3H, s), 2.93-3.02 (3H, m), 4.20 (2H, d, J = 12.8 Hz), 7.36-7.59 (5H, m), 8. 63 (1H, d, J = 4.4 Hz), 9.15 (1H, brs).
4-[4-(2,3-ジフルオロフェニル)ピリジン-2-イル]-N-(3,4-ジメチルイソオキサゾール-5-イル)ピペリジン-1-カルボキサミド
1H NMR(DMSO-d6)δ:1.71-1.76(2H,m),1.76(3H,s),1.91(2H,d,J=12.0Hz),2.13(3H,s),2.93-3.02(3H,m),4.20(2H,d,J=12.8Hz),7.36-7.59(5H,m),8.63(1H,d,J=4.4Hz),9.15(1H,brs)。 Example 24
4- [4- (2,3-Difluorophenyl) pyridin-2-yl] -N- (3,4-dimethylisoxazol-5-yl) piperidine-1-carboxamide
1 H NMR (DMSO-d 6 ) δ: 1.71-1.76 (2H, m), 1.76 (3H, s), 1.91 (2H, d, J = 12.0 Hz), 2. 13 (3H, s), 2.93-3.02 (3H, m), 4.20 (2H, d, J = 12.8 Hz), 7.36-7.59 (5H, m), 8. 63 (1H, d, J = 4.4 Hz), 9.15 (1H, brs).
実施例25
4-[4-(2,4-ジフルオロフェニル)ピリジン-2-イル]-N-(3,4-ジメチルイソオキサゾール-5-イル)ピペリジン-1-カルボキサミド

実施例20と同様にして、参考例32で得た4-(2,4-ジフルオロフェニル)-2-ピペリジン-4-イルピリジン二塩酸塩と(3,4-ジメチルイソオキサゾール-5-イル)カルバミン酸2,2,2-トリクロロエチルから表題化合物(203mg,57%)を固体として得た。
1H NMR(DMSO-d6)δ:1.66-1.75(2H,m),1.74(3H,s),1.90(2H,d,J=11.6Hz),2.13(3H,s),2.92-3.03(3H,m),4.19(2H,d,J=13.2Hz),7.24-7.29(1H,m),7.40-7.48(3H,m),7.71-7.77(1H,m),8.60(1H,d,J=5.2Hz),9.15(1H,brs)。 Example 25
4- [4- (2,4-Difluorophenyl) pyridin-2-yl] -N- (3,4-dimethylisoxazol-5-yl) piperidine-1-carboxamide
In the same manner as in Example 20, 4- (2,4-difluorophenyl) -2-piperidin-4-ylpyridine dihydrochloride and (3,4-dimethylisoxazol-5-yl) carbamine obtained in Reference Example 32 The title compound (203 mg, 57%) was obtained as a solid from the acid 2,2,2-trichloroethyl.
1 H NMR (DMSO-d 6 ) δ: 1.66-1.75 (2H, m), 1.74 (3H, s), 1.90 (2H, d, J = 11.6 Hz), 2. 13 (3H, s), 2.92-3.03 (3H, m), 4.19 (2H, d, J = 13.2 Hz), 7.24-7.29 (1 H, m), 7. 40-7.48 (3H, m), 7.71-7.77 (1H, m), 8.60 (1H, d, J = 5.2 Hz), 9.15 (1H, brs).
4-[4-(2,4-ジフルオロフェニル)ピリジン-2-イル]-N-(3,4-ジメチルイソオキサゾール-5-イル)ピペリジン-1-カルボキサミド
1H NMR(DMSO-d6)δ:1.66-1.75(2H,m),1.74(3H,s),1.90(2H,d,J=11.6Hz),2.13(3H,s),2.92-3.03(3H,m),4.19(2H,d,J=13.2Hz),7.24-7.29(1H,m),7.40-7.48(3H,m),7.71-7.77(1H,m),8.60(1H,d,J=5.2Hz),9.15(1H,brs)。 Example 25
4- [4- (2,4-Difluorophenyl) pyridin-2-yl] -N- (3,4-dimethylisoxazol-5-yl) piperidine-1-carboxamide
1 H NMR (DMSO-d 6 ) δ: 1.66-1.75 (2H, m), 1.74 (3H, s), 1.90 (2H, d, J = 11.6 Hz), 2. 13 (3H, s), 2.92-3.03 (3H, m), 4.19 (2H, d, J = 13.2 Hz), 7.24-7.29 (1 H, m), 7. 40-7.48 (3H, m), 7.71-7.77 (1H, m), 8.60 (1H, d, J = 5.2 Hz), 9.15 (1H, brs).
実施例26
4-(4-フラン-3-イルピリミジン-2-イル)-N-ピリジン-3-イルピペリジン-1-カルボキサミド

参考例36で得た4-フラン-3-イル-2-ピペリジン-4-イルピリミジン二塩酸塩(120mg、0.397ミリモル)、ピリジン-3-イルカルバミン酸2,2,2-トリクロロエチル(126mg、0.437ミリモル)のアセトン(1ml)の懸濁液にトリエチルアミン(0.221ml、1.59ミリモル)を室温で滴下し、反応溶液を65℃で終夜攪拌した。反応液に水を注ぎ、酢酸エチルで抽出した。抽出液を水で洗浄し、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル)で精製し、ヘキサンと酢酸エチルから再結晶して表題化合物(41.8mg、30%)を固体として得た。 融点145-146℃
1H NMR(DMSO-d6)δ:1.67-1.68(2H,m),1.95-2.09(2H,m),2.92-3.15(3H,m),4.16-4.30(2H,m),7.10-7.17(1H,m),7.26(1H,dd,J=8.1,4.3Hz),7.63(1H,d,J=5.3Hz),7.81-7.94(2H,m),8.10-8.18(1H,m),8.55(1H,s),8.63-8.68(1H,m),8.70-8.76(2H,m). Example 26
4- (4-Furan-3-ylpyrimidin-2-yl) -N-pyridin-3-ylpiperidin-1-carboxamide
4-furan-3-yl-2-piperidin-4-ylpyrimidine dihydrochloride (120 mg, 0.397 mmol) obtained in Reference Example 36, 2,2,2-trichloroethyl pyridin-3-ylcarbamate ( Triethylamine (0.221 ml, 1.59 mmol) was added dropwise to a suspension of 126 mg, 0.437 mmol) in acetone (1 ml) at room temperature, and the reaction solution was stirred at 65 ° C. overnight. Water was poured into the reaction mixture, and the mixture was extracted with ethyl acetate. The extract was washed with water and dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (ethyl acetate) and recrystallized from hexane and ethyl acetate to give the title compound (41.8 mg, 30%) as a solid. Melting point: 145-146 ° C
1 H NMR (DMSO-d 6 ) δ: 1.67-1.68 (2H, m), 1.95-2.09 (2H, m), 2.92-3.15 (3H, m), 4.16-4.30 (2H, m), 7.10-7.17 (1H, m), 7.26 (1H, dd, J = 8.1, 4.3 Hz), 7.63 (1H , D, J = 5.3 Hz), 7.81-7.94 (2H, m), 8.10-8.18 (1H, m), 8.55 (1H, s), 8.63-8 .68 (1H, m), 8.70-8.76 (2H, m).
4-(4-フラン-3-イルピリミジン-2-イル)-N-ピリジン-3-イルピペリジン-1-カルボキサミド
1H NMR(DMSO-d6)δ:1.67-1.68(2H,m),1.95-2.09(2H,m),2.92-3.15(3H,m),4.16-4.30(2H,m),7.10-7.17(1H,m),7.26(1H,dd,J=8.1,4.3Hz),7.63(1H,d,J=5.3Hz),7.81-7.94(2H,m),8.10-8.18(1H,m),8.55(1H,s),8.63-8.68(1H,m),8.70-8.76(2H,m). Example 26
4- (4-Furan-3-ylpyrimidin-2-yl) -N-pyridin-3-ylpiperidin-1-carboxamide
1 H NMR (DMSO-d 6 ) δ: 1.67-1.68 (2H, m), 1.95-2.09 (2H, m), 2.92-3.15 (3H, m), 4.16-4.30 (2H, m), 7.10-7.17 (1H, m), 7.26 (1H, dd, J = 8.1, 4.3 Hz), 7.63 (1H , D, J = 5.3 Hz), 7.81-7.94 (2H, m), 8.10-8.18 (1H, m), 8.55 (1H, s), 8.63-8 .68 (1H, m), 8.70-8.76 (2H, m).
実施例27
N-(3,4-ジメチルイソオキサゾール-5-イル)-4-(4-フラン-3-イルピリミジン-2-イル)ピペリジン-1-カルボキサミド

参考例36で得た4-フラン-3-イル-2-ピペリジン-4-イルピリミジン二塩酸塩(120mg、0.397ミリモル)、(3,4-ジメチルイソオキサゾール-5-イル)カルバミン酸2,2,2-トリクロロエチル(126mg、0.437ミリモル)のアセトン(1ml)の懸濁液にトリエチルアミン(0.221ml、1.59ミリモル)を室温で滴下し、反応溶液を45℃で3時間攪拌した。反応液に水を注ぎ、酢酸エチルで抽出した。抽出液を水で洗浄し、無水硫酸マグネシウムで乾燥し、少量のシリカゲルに通し、酢酸エチルで溶出し、溶媒を減圧下留去した。残渣をヘキサンとテトラヒドロフランから再結晶して表題化合物(107mg、73%)を固体として得た。 融点190-191℃
1H NMR(DMSO-d6)δ:1.65-1.84(5H,m),1.92-2.05(2H,m),2.13(3H,s),2.92-3.15(3H,m),4.07-4.22(2H,m),7.10-7.15(1H,m),7.63(1H,d,J=5.3Hz),7.85(1H,t,J=1.7Hz),8.52-8.58(1H,m),8.73(1H,d,J=5.3Hz),9.14(1H,s)。 Example 27
N- (3,4-Dimethylisoxazol-5-yl) -4- (4-furan-3-ylpyrimidin-2-yl) piperidine-1-carboxamide
4-furan-3-yl-2-piperidin-4-ylpyrimidine dihydrochloride (120 mg, 0.397 mmol), (3,4-dimethylisoxazol-5-yl) carbamic acid 2 obtained in Reference Example 36 , 2,2-Trichloroethyl (126 mg, 0.437 mmol) in acetone (1 ml) was added dropwise triethylamine (0.221 ml, 1.59 mmol) at room temperature, and the reaction solution was stirred at 45 ° C. for 3 hours. Stir. Water was poured into the reaction mixture, and the mixture was extracted with ethyl acetate. The extract was washed with water, dried over anhydrous magnesium sulfate, passed through a small amount of silica gel, eluted with ethyl acetate, and the solvent was evaporated under reduced pressure. The residue was recrystallized from hexane and tetrahydrofuran to give the title compound (107 mg, 73%) as a solid. Melting point 190-191 ° C
1 H NMR (DMSO-d 6 ) δ: 1.65 to 1.84 (5H, m), 1.92 to 2.05 (2H, m), 2.13 (3H, s), 2.92— 3.15 (3H, m), 4.07-4.22 (2H, m), 7.10-7.15 (1H, m), 7.63 (1H, d, J = 5.3 Hz), 7.85 (1H, t, J = 1.7 Hz), 8.52-8.58 (1H, m), 8.73 (1H, d, J = 5.3 Hz), 9.14 (1H, s ).
N-(3,4-ジメチルイソオキサゾール-5-イル)-4-(4-フラン-3-イルピリミジン-2-イル)ピペリジン-1-カルボキサミド
1H NMR(DMSO-d6)δ:1.65-1.84(5H,m),1.92-2.05(2H,m),2.13(3H,s),2.92-3.15(3H,m),4.07-4.22(2H,m),7.10-7.15(1H,m),7.63(1H,d,J=5.3Hz),7.85(1H,t,J=1.7Hz),8.52-8.58(1H,m),8.73(1H,d,J=5.3Hz),9.14(1H,s)。 Example 27
N- (3,4-Dimethylisoxazol-5-yl) -4- (4-furan-3-ylpyrimidin-2-yl) piperidine-1-carboxamide
1 H NMR (DMSO-d 6 ) δ: 1.65 to 1.84 (5H, m), 1.92 to 2.05 (2H, m), 2.13 (3H, s), 2.92— 3.15 (3H, m), 4.07-4.22 (2H, m), 7.10-7.15 (1H, m), 7.63 (1H, d, J = 5.3 Hz), 7.85 (1H, t, J = 1.7 Hz), 8.52-8.58 (1H, m), 8.73 (1H, d, J = 5.3 Hz), 9.14 (1H, s ).
実施例28
4-(4-フラン-3-イルピリミジン-2-イル)-N-ピリダジン-3-イルピペリジン-1-カルボキサミド

参考例36で得た4-フラン-3-イル-2-ピペリジン-4-イルピリミジン二塩酸塩(120mg、0.397ミリモル)、ピリダジン-3-イルカルバミン酸フェニル(94.0g、0.437ミリモル)のアセトン(1ml)の懸濁液にトリエチルアミン(0.221ml、1.59ミリモル)を室温で滴下し、反応溶液を45℃で3時間攪拌した。反応液に水を注ぎ、酢酸エチルで抽出した。抽出液を水で洗浄し、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル)で精製し、ヘキサンと酢酸エチルから再結晶して表題化合物(51.5mg、37%)を固体として得た。 融点146-147℃
1H NMR(DMSO-d6)δ:1.66-1.88(2H,m),1.92-2.11(2H,m),2.94-3.17(3H,m),4.17-4.42(2H,m),7.13(1H,s),7.50-7.69(2H,m),7.84(1H,s),8.01(1H,d,J=8.7Hz),8.55(1H,s),8.73(1H,d,J=5.3Hz),8.83(1H,d,J=3.8Hz),9.86(1H,s)。 Example 28
4- (4-Furan-3-ylpyrimidin-2-yl) -N-pyridazin-3-ylpiperidin-1-carboxamide
4-furan-3-yl-2-piperidin-4-ylpyrimidine dihydrochloride (120 mg, 0.397 mmol), phenyl pyridazin-3-ylcarbamate (94.0 g, 0.437) obtained in Reference Example 36. To a suspension of (mmol) of acetone (1 ml), triethylamine (0.221 ml, 1.59 mmol) was added dropwise at room temperature, and the reaction solution was stirred at 45 ° C. for 3 hours. Water was poured into the reaction mixture, and the mixture was extracted with ethyl acetate. The extract was washed with water and dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (ethyl acetate) and recrystallized from hexane and ethyl acetate to give the title compound (51.5 mg, 37%) as a solid. Melting point: 146-147 ° C
1 H NMR (DMSO-d 6 ) δ: 1.66-1.88 (2H, m), 1.92-2.11 (2H, m), 2.94-3.17 (3H, m), 4.17-4.42 (2H, m), 7.13 (1H, s), 7.50-7.69 (2H, m), 7.84 (1H, s), 8.01 (1H, d, J = 8.7 Hz), 8.55 (1H, s), 8.73 (1H, d, J = 5.3 Hz), 8.83 (1H, d, J = 3.8 Hz), 9. 86 (1H, s).
4-(4-フラン-3-イルピリミジン-2-イル)-N-ピリダジン-3-イルピペリジン-1-カルボキサミド
1H NMR(DMSO-d6)δ:1.66-1.88(2H,m),1.92-2.11(2H,m),2.94-3.17(3H,m),4.17-4.42(2H,m),7.13(1H,s),7.50-7.69(2H,m),7.84(1H,s),8.01(1H,d,J=8.7Hz),8.55(1H,s),8.73(1H,d,J=5.3Hz),8.83(1H,d,J=3.8Hz),9.86(1H,s)。 Example 28
4- (4-Furan-3-ylpyrimidin-2-yl) -N-pyridazin-3-ylpiperidin-1-carboxamide
1 H NMR (DMSO-d 6 ) δ: 1.66-1.88 (2H, m), 1.92-2.11 (2H, m), 2.94-3.17 (3H, m), 4.17-4.42 (2H, m), 7.13 (1H, s), 7.50-7.69 (2H, m), 7.84 (1H, s), 8.01 (1H, d, J = 8.7 Hz), 8.55 (1H, s), 8.73 (1H, d, J = 5.3 Hz), 8.83 (1H, d, J = 3.8 Hz), 9. 86 (1H, s).
実施例29
N-ピリジン-3-イル-4-(4-チオフェン-3-イルピリミジン-2-イル)ピペリジン-1-カルボキサミド
実施例26と同様にして、参考例40で得た4-チオフェン-3-イル-2-ピペリジン-4-イルピリミジン二塩酸塩とピリジン-3-イルカルバミン酸2,2,2-トリクロロエチルから表題化合物(39.5mg、27%)を固体として得た。 融点160-161℃(酢酸エチル-ヘキサン)
1H NMR(DMSO-d6)δ:1.69-1.88(2H,m),1.97-2.10(2H,m),2.94-3.18(3H,m),4.16-4.30(2H,m),7.26(1H,dd,J=8.3,5.3Hz),7.71(1H,dd,J=4.9,3.0Hz),7.78(1H,d,J=5.3Hz),7.82-7.87(1H,m),7.87-7.93(1H,m),8.14(1H,dd,J=4.9,1.5Hz),8.48(1H,dd,J=3.0,1.5Hz),8.66(1H,d,J=2.3Hz),8.73(1H,s),8.76(1H,d,J=5.3Hz)。 Example 29
N-pyridin-3-yl-4- (4-thiophen-3-ylpyrimidin-2-yl) piperidine-1-carboxamide
In the same manner as in Example 26, from 4-thiophen-3-yl-2-piperidin-4-ylpyrimidine dihydrochloride obtained in Reference Example 40 and 2,2,2-trichloroethyl pyridin-3-ylcarbamate The title compound (39.5 mg, 27%) was obtained as a solid. Melting point 160-161 ° C (ethyl acetate-hexane)
1 H NMR (DMSO-d 6 ) δ: 1.69-1.88 (2H, m), 1.97-2.10 (2H, m), 2.94-3.18 (3H, m), 4.16-4.30 (2H, m), 7.26 (1H, dd, J = 8.3, 5.3 Hz), 7.71 (1H, dd, J = 4.9, 3.0 Hz) , 7.78 (1H, d, J = 5.3 Hz), 7.82-7.87 (1H, m), 7.87-7.93 (1H, m), 8.14 (1H, dd, J = 4.9, 1.5 Hz), 8.48 (1H, dd, J = 3.0, 1.5 Hz), 8.66 (1H, d, J = 2.3 Hz), 8.73 (1H , S), 8.76 (1H, d, J = 5.3 Hz).
N-ピリジン-3-イル-4-(4-チオフェン-3-イルピリミジン-2-イル)ピペリジン-1-カルボキサミド
1H NMR(DMSO-d6)δ:1.69-1.88(2H,m),1.97-2.10(2H,m),2.94-3.18(3H,m),4.16-4.30(2H,m),7.26(1H,dd,J=8.3,5.3Hz),7.71(1H,dd,J=4.9,3.0Hz),7.78(1H,d,J=5.3Hz),7.82-7.87(1H,m),7.87-7.93(1H,m),8.14(1H,dd,J=4.9,1.5Hz),8.48(1H,dd,J=3.0,1.5Hz),8.66(1H,d,J=2.3Hz),8.73(1H,s),8.76(1H,d,J=5.3Hz)。 Example 29
N-pyridin-3-yl-4- (4-thiophen-3-ylpyrimidin-2-yl) piperidine-1-carboxamide
1 H NMR (DMSO-d 6 ) δ: 1.69-1.88 (2H, m), 1.97-2.10 (2H, m), 2.94-3.18 (3H, m), 4.16-4.30 (2H, m), 7.26 (1H, dd, J = 8.3, 5.3 Hz), 7.71 (1H, dd, J = 4.9, 3.0 Hz) , 7.78 (1H, d, J = 5.3 Hz), 7.82-7.87 (1H, m), 7.87-7.93 (1H, m), 8.14 (1H, dd, J = 4.9, 1.5 Hz), 8.48 (1H, dd, J = 3.0, 1.5 Hz), 8.66 (1H, d, J = 2.3 Hz), 8.73 (1H , S), 8.76 (1H, d, J = 5.3 Hz).
実施例30
N-(3,4-ジメチルイソオキサゾール-5-イル)-4-(4-チオフェン-3-イルピリミジン-2-イル)ピペリジン-1-カルボキサミド

実施例27と同様にして、参考例40で得た4-チオフェン-3-イル-2-ピペリジン-4-イルピリミジン二塩酸塩と(3,4-ジメチルイソオキサゾール-5-イル)カルバミン酸2,2,2-トリクロロエチルから表題化合物(108mg、69%)を固体として得た。融点180-181℃(酢酸エチル-ヘキサン)
1H NMR(DMSO-d6)δ:1.68-1.86(5H,m),1.94-2.07(2H,m),2.13(3H,s),2.94-3.17(3H,m),4.06-4.22(2H,m),7.72(1H,dd,J=5.3,3.0Hz),7.78(1H,d,J=5.3Hz),7.82-7.87(1H,m),8.48(1H,dd,J=3.0,1.5Hz),8.76(1H,d,J=5.3Hz),9.14(1H,s)。 Example 30
N- (3,4-Dimethylisoxazol-5-yl) -4- (4-thiophen-3-ylpyrimidin-2-yl) piperidine-1-carboxamide
In the same manner as in Example 27, 4-thiophen-3-yl-2-piperidin-4-ylpyrimidine dihydrochloride obtained in Reference Example 40 and (3,4-dimethylisoxazol-5-yl) carbamic acid 2 , 2,2-Trichloroethyl gave the title compound (108 mg, 69%) as a solid. Melting point 180-181 ° C (ethyl acetate-hexane)
1 H NMR (DMSO-d 6 ) δ: 1.68-1.86 (5H, m), 1.94-2.07 (2H, m), 2.13 (3H, s), 2.94- 3.17 (3H, m), 4.06-4.22 (2H, m), 7.72 (1H, dd, J = 5.3, 3.0 Hz), 7.78 (1H, d, J = 5.3 Hz), 7.82-7.87 (1 H, m), 8.48 (1 H, dd, J = 3.0, 1.5 Hz), 8.76 (1 H, d, J = 5. 3 Hz), 9.14 (1 H, s).
N-(3,4-ジメチルイソオキサゾール-5-イル)-4-(4-チオフェン-3-イルピリミジン-2-イル)ピペリジン-1-カルボキサミド
1H NMR(DMSO-d6)δ:1.68-1.86(5H,m),1.94-2.07(2H,m),2.13(3H,s),2.94-3.17(3H,m),4.06-4.22(2H,m),7.72(1H,dd,J=5.3,3.0Hz),7.78(1H,d,J=5.3Hz),7.82-7.87(1H,m),8.48(1H,dd,J=3.0,1.5Hz),8.76(1H,d,J=5.3Hz),9.14(1H,s)。 Example 30
N- (3,4-Dimethylisoxazol-5-yl) -4- (4-thiophen-3-ylpyrimidin-2-yl) piperidine-1-carboxamide
1 H NMR (DMSO-d 6 ) δ: 1.68-1.86 (5H, m), 1.94-2.07 (2H, m), 2.13 (3H, s), 2.94- 3.17 (3H, m), 4.06-4.22 (2H, m), 7.72 (1H, dd, J = 5.3, 3.0 Hz), 7.78 (1H, d, J = 5.3 Hz), 7.82-7.87 (1 H, m), 8.48 (1 H, dd, J = 3.0, 1.5 Hz), 8.76 (1 H, d, J = 5. 3 Hz), 9.14 (1 H, s).
実施例31
N-ピリダジン-3-イル-4-(4-チオフェン-3-イルピリミジン-2-イル)ピペリジン-1-カルボキサミド

実施例28と同様にして、参考例40で得た4-チオフェン-3-イル-2-ピペリジン-4-イルピリミジン二塩酸塩とピリダジン-3-イルカルバミン酸フェニルから表題化合物(68.0mg、45%)を固体として得た。融点132-133℃(酢酸エチル-ヘキサン)
1H NMR(DMSO-d6)δ:1.69-1.90(2H,m),1.93-2.11(2H,m),2.94-3.19(3H,m),4.18-4.39(2H,m),7.57(1H,dd,J=9.0,4.5Hz),7.71(1H,dd,J=4.5,3.0Hz),7.78(1H,d,J=5.3Hz),7.81-7.88(1H,m),7.96-8.07(1H,m),8.44-8.51(1H,m),8.76(1H,d,J=5.3Hz),8.83(1H,d,J=3.0Hz),9.87(1H,s)。 Example 31
N-pyridazin-3-yl-4- (4-thiophen-3-ylpyrimidin-2-yl) piperidine-1-carboxamide
In the same manner as in Example 28, the title compound (68.0 mg, 4-thiophen-3-yl-2-piperidin-4-ylpyrimidine dihydrochloride obtained in Reference Example 40 and phenyl pyridazin-3-ylcarbamate was obtained. 45%) was obtained as a solid. Melting point 132-133 ° C (ethyl acetate-hexane)
1 H NMR (DMSO-d 6 ) δ: 1.69-1.90 (2H, m), 1.93-2.11 (2H, m), 2.94-3.19 (3H, m), 4.18-4.39 (2H, m), 7.57 (1H, dd, J = 9.0, 4.5 Hz), 7.71 (1H, dd, J = 4.5, 3.0 Hz) 7.78 (1H, d, J = 5.3 Hz), 7.81-7.88 (1H, m), 7.96-8.07 (1H, m), 8.44-8.51 ( 1H, m), 8.76 (1H, d, J = 5.3 Hz), 8.83 (1H, d, J = 3.0 Hz), 9.87 (1H, s).
N-ピリダジン-3-イル-4-(4-チオフェン-3-イルピリミジン-2-イル)ピペリジン-1-カルボキサミド
1H NMR(DMSO-d6)δ:1.69-1.90(2H,m),1.93-2.11(2H,m),2.94-3.19(3H,m),4.18-4.39(2H,m),7.57(1H,dd,J=9.0,4.5Hz),7.71(1H,dd,J=4.5,3.0Hz),7.78(1H,d,J=5.3Hz),7.81-7.88(1H,m),7.96-8.07(1H,m),8.44-8.51(1H,m),8.76(1H,d,J=5.3Hz),8.83(1H,d,J=3.0Hz),9.87(1H,s)。 Example 31
N-pyridazin-3-yl-4- (4-thiophen-3-ylpyrimidin-2-yl) piperidine-1-carboxamide
1 H NMR (DMSO-d 6 ) δ: 1.69-1.90 (2H, m), 1.93-2.11 (2H, m), 2.94-3.19 (3H, m), 4.18-4.39 (2H, m), 7.57 (1H, dd, J = 9.0, 4.5 Hz), 7.71 (1H, dd, J = 4.5, 3.0 Hz) 7.78 (1H, d, J = 5.3 Hz), 7.81-7.88 (1H, m), 7.96-8.07 (1H, m), 8.44-8.51 ( 1H, m), 8.76 (1H, d, J = 5.3 Hz), 8.83 (1H, d, J = 3.0 Hz), 9.87 (1H, s).
実施例32
4-[4-(3-フルオロフェニル)ピリミジン-2-イル]-N-ピリジン-3-イルピペリジン-1-カルボキサミド

実施例26と同様にして、参考例44で得た4-(3-フルオロフェニル)-2-ピペリジン-4-イルピリミジン二塩酸塩とピリジン-3-イルカルバミン酸2,2,2-トリクロロエチルから表題化合物(84.9mg、37%)を固体として得た。 融点149-150℃(酢酸エチル-ヘキサン)
1H NMR(DMSO-d6)δ:1.70-1.93(2H,m),1.96-2.17(2H,m),2.94-3.25(3H,m),4.10-4.38(2H,m),7.19-7.34(1H,m),7.34-7.49(1H,m),7.53-7.71(1H,m),7.82-7.24(5H,m),8.66(1H,d,J=1.9Hz),8.74(1H,s),8.86(1H,d,J=5.3Hz)。 Example 32
4- [4- (3-Fluorophenyl) pyrimidin-2-yl] -N-pyridin-3-ylpiperidin-1-carboxamide
In the same manner as in Example 26, 4- (3-fluorophenyl) -2-piperidin-4-ylpyrimidine dihydrochloride obtained in Reference Example 44 and 2,2,2-trichloroethyl pyridin-3-ylcarbamate Gave the title compound (84.9 mg, 37%) as a solid. Melting point 149-150 ° C (ethyl acetate-hexane)
1 H NMR (DMSO-d 6 ) δ: 1.70-1.93 (2H, m), 1.96-2.17 (2H, m), 2.94-3.25 (3H, m), 4.10-4.38 (2H, m), 7.19-7.34 (1H, m), 7.34-7.49 (1H, m), 7.53-7.71 (1H, m) ), 7.82-7.24 (5H, m), 8.66 (1H, d, J = 1.9 Hz), 8.74 (1H, s), 8.86 (1H, d, J = 5) .3 Hz).
4-[4-(3-フルオロフェニル)ピリミジン-2-イル]-N-ピリジン-3-イルピペリジン-1-カルボキサミド
1H NMR(DMSO-d6)δ:1.70-1.93(2H,m),1.96-2.17(2H,m),2.94-3.25(3H,m),4.10-4.38(2H,m),7.19-7.34(1H,m),7.34-7.49(1H,m),7.53-7.71(1H,m),7.82-7.24(5H,m),8.66(1H,d,J=1.9Hz),8.74(1H,s),8.86(1H,d,J=5.3Hz)。 Example 32
4- [4- (3-Fluorophenyl) pyrimidin-2-yl] -N-pyridin-3-ylpiperidin-1-carboxamide
1 H NMR (DMSO-d 6 ) δ: 1.70-1.93 (2H, m), 1.96-2.17 (2H, m), 2.94-3.25 (3H, m), 4.10-4.38 (2H, m), 7.19-7.34 (1H, m), 7.34-7.49 (1H, m), 7.53-7.71 (1H, m) ), 7.82-7.24 (5H, m), 8.66 (1H, d, J = 1.9 Hz), 8.74 (1H, s), 8.86 (1H, d, J = 5) .3 Hz).
実施例33
N-(3,4-ジメチルイソオキサゾール-5-イル)-4-[4-(3-フルオロフェニル)ピリミジン-2-イル]ピペリジン-1-カルボキサミド

実施例27と同様にして、参考例44で得た4-(3-フルオロフェニル)-2-ピペリジン-4-イルピリミジン二塩酸塩とから表題化合物(198mg、83%)を固体として得た。融点181-182℃(酢酸エチル-ヘキサン)
1H NMR(DMSO-d6)δ:1.69-1.88(5H,m),1.96-2.10(2H,m),2.13(3H,s),2.97-3.22(3H,m),4.09-4.22(2H,m),7.36-7.47(1H,m),7.56-7.66(1H,m),7.98(1H,d,J=5.3Hz),8.01-8.13(2H,m),8.86(1H,d,J=5.3Hz),9.15(1H,s)。 Example 33
N- (3,4-dimethylisoxazol-5-yl) -4- [4- (3-fluorophenyl) pyrimidin-2-yl] piperidine-1-carboxamide
In the same manner as in Example 27, the title compound (198 mg, 83%) was obtained as a solid from 4- (3-fluorophenyl) -2-piperidin-4-ylpyrimidine dihydrochloride obtained in Reference Example 44. Melting point 181-182 ° C. (ethyl acetate-hexane)
1 H NMR (DMSO-d 6 ) δ: 1.69-1.88 (5H, m), 1.96-2.10 (2H, m), 2.13 (3H, s), 2.97- 3.22 (3H, m), 4.09-4.22 (2H, m), 7.36-7.47 (1H, m), 7.56-7.66 (1H, m), 7. 98 (1H, d, J = 5.3 Hz), 8.01-8.13 (2H, m), 8.86 (1H, d, J = 5.3 Hz), 9.15 (1H, s).
N-(3,4-ジメチルイソオキサゾール-5-イル)-4-[4-(3-フルオロフェニル)ピリミジン-2-イル]ピペリジン-1-カルボキサミド
1H NMR(DMSO-d6)δ:1.69-1.88(5H,m),1.96-2.10(2H,m),2.13(3H,s),2.97-3.22(3H,m),4.09-4.22(2H,m),7.36-7.47(1H,m),7.56-7.66(1H,m),7.98(1H,d,J=5.3Hz),8.01-8.13(2H,m),8.86(1H,d,J=5.3Hz),9.15(1H,s)。 Example 33
N- (3,4-dimethylisoxazol-5-yl) -4- [4- (3-fluorophenyl) pyrimidin-2-yl] piperidine-1-carboxamide
1 H NMR (DMSO-d 6 ) δ: 1.69-1.88 (5H, m), 1.96-2.10 (2H, m), 2.13 (3H, s), 2.97- 3.22 (3H, m), 4.09-4.22 (2H, m), 7.36-7.47 (1H, m), 7.56-7.66 (1H, m), 7. 98 (1H, d, J = 5.3 Hz), 8.01-8.13 (2H, m), 8.86 (1H, d, J = 5.3 Hz), 9.15 (1H, s).
実施例34
4-[4-(3-フルオロフェニル)ピリミジン-2-イル]-N-ピリダジン-3-イルピペリジン-1-カルボキサミド

実施例28と同様にして、参考例44で得た4-(3-フルオロフェニル)-2-ピペリジン-4-イルピリミジン二塩酸塩とピリダジン-3-イルカルバミン酸フェニルから表題化合物(149mg、65%)を固体として得た。融点163-164℃(酢酸エチル-ヘキサン)
1H NMR(DMSO-d6)δ:1.72-1.91(2H,m),1.99-2.14(2H,m),2.96-3.24(3H,m),4.23-4.37(2H,m),7.36-7.46(1H,m),7.52-7.66(2H,m),7.94-8.14(4H,m),8.79-8.90(2H,m),9.87(1H,s)。 Example 34
4- [4- (3-Fluorophenyl) pyrimidin-2-yl] -N-pyridazin-3-ylpiperidin-1-carboxamide
In the same manner as in Example 28, the title compound (149 mg, 65 %) As a solid. Melting point 163-164 ° C. (ethyl acetate-hexane)
1 H NMR (DMSO-d 6 ) δ: 1.72-1.91 (2H, m), 1.99-2.14 (2H, m), 2.96-3.24 (3H, m), 4.23-4.37 (2H, m), 7.36-7.46 (1H, m), 7.52-7.66 (2H, m), 7.94-8.14 (4H, m) ), 8.79-8.90 (2H, m), 9.87 (1H, s).
4-[4-(3-フルオロフェニル)ピリミジン-2-イル]-N-ピリダジン-3-イルピペリジン-1-カルボキサミド
1H NMR(DMSO-d6)δ:1.72-1.91(2H,m),1.99-2.14(2H,m),2.96-3.24(3H,m),4.23-4.37(2H,m),7.36-7.46(1H,m),7.52-7.66(2H,m),7.94-8.14(4H,m),8.79-8.90(2H,m),9.87(1H,s)。 Example 34
4- [4- (3-Fluorophenyl) pyrimidin-2-yl] -N-pyridazin-3-ylpiperidin-1-carboxamide
1 H NMR (DMSO-d 6 ) δ: 1.72-1.91 (2H, m), 1.99-2.14 (2H, m), 2.96-3.24 (3H, m), 4.23-4.37 (2H, m), 7.36-7.46 (1H, m), 7.52-7.66 (2H, m), 7.94-8.14 (4H, m) ), 8.79-8.90 (2H, m), 9.87 (1H, s).
実施例35
4-[4-(4-フルオロフェニル)ピリミジン-2-イル]-N-ピリジン-3-イルピペリジン-1-カルボキサミド

参考例48で得た4-(4-フルオロフェニル)-2-ピペリジン-4-イルピリミジン二塩酸塩(200mg、0.606ミリモル)、ピリジン-3-イルカルバミン酸2,2,2-トリクロロエチル(180mg、0.666ミリモル)のアセトン(1ml)の懸濁液にトリエチルアミン(0.338ml、2.42ミリモル)を室温で滴下し、反応溶液を65℃で終夜攪拌した。反応液に水を注ぎ、酢酸エチルで抽出した。抽出液を水で洗浄し、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル)で精製し、ヘキサンと酢酸エチルから再結晶して表題化合物(87.8mg、38%)を固体として得た。 融点156-157℃
1H NMR(DMSO-d6)δ:1.71-1.90(2H,m),1.99-2.14(2H,m),2.94-3.22(3H,m),4.16-4.33(2H,m),7.26(1H,dd,J=8.5,4.7Hz),7.33-7.45(2H,m),7.85-7.96(2H,m),8.09-8.19(1H,m),8.24-8.36(2H,m),8.67(1H,d,J=2.3Hz),8.74(1H,s),8.82(1H,d,J=4.7Hz)。 Example 35
4- [4- (4-Fluorophenyl) pyrimidin-2-yl] -N-pyridin-3-ylpiperidin-1-carboxamide
4- (4-Fluorophenyl) -2-piperidin-4-ylpyrimidine dihydrochloride (200 mg, 0.606 mmol) obtained in Reference Example 48, 2,2,2-trichloroethyl pyridin-3-ylcarbamate Triethylamine (0.338 ml, 2.42 mmol) was added dropwise to a suspension of (180 mg, 0.666 mmol) in acetone (1 ml) at room temperature, and the reaction solution was stirred at 65 ° C. overnight. Water was poured into the reaction mixture, and the mixture was extracted with ethyl acetate. The extract was washed with water and dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (ethyl acetate) and recrystallized from hexane and ethyl acetate to give the title compound (87.8 mg, 38%) as a solid. Melting point 156-157 ° C
1 H NMR (DMSO-d 6 ) δ: 1.71-1.90 (2H, m), 1.99-2.14 (2H, m), 2.94-3.22 (3H, m), 4.16-4.33 (2H, m), 7.26 (1H, dd, J = 8.5, 4.7 Hz), 7.33-7.45 (2H, m), 7.85-7 .96 (2H, m), 8.09-8.19 (1H, m), 8.24-8.36 (2H, m), 8.67 (1H, d, J = 2.3 Hz), 8 .74 (1H, s), 8.82 (1H, d, J = 4.7 Hz).
4-[4-(4-フルオロフェニル)ピリミジン-2-イル]-N-ピリジン-3-イルピペリジン-1-カルボキサミド
1H NMR(DMSO-d6)δ:1.71-1.90(2H,m),1.99-2.14(2H,m),2.94-3.22(3H,m),4.16-4.33(2H,m),7.26(1H,dd,J=8.5,4.7Hz),7.33-7.45(2H,m),7.85-7.96(2H,m),8.09-8.19(1H,m),8.24-8.36(2H,m),8.67(1H,d,J=2.3Hz),8.74(1H,s),8.82(1H,d,J=4.7Hz)。 Example 35
4- [4- (4-Fluorophenyl) pyrimidin-2-yl] -N-pyridin-3-ylpiperidin-1-carboxamide
1 H NMR (DMSO-d 6 ) δ: 1.71-1.90 (2H, m), 1.99-2.14 (2H, m), 2.94-3.22 (3H, m), 4.16-4.33 (2H, m), 7.26 (1H, dd, J = 8.5, 4.7 Hz), 7.33-7.45 (2H, m), 7.85-7 .96 (2H, m), 8.09-8.19 (1H, m), 8.24-8.36 (2H, m), 8.67 (1H, d, J = 2.3 Hz), 8 .74 (1H, s), 8.82 (1H, d, J = 4.7 Hz).
実施例36
N-(3,4-ジメチルイソオキサゾール-5-イル)-4-[4-(4-フルオロフェニル)ピリミジン-2-イル]ピペリジン-1-カルボキサミド

参考例48で得た4-(4-フルオロフェニル)-2-ピペリジン-4-イルピリミジン二塩酸塩(200mg、0.606ミリモル)、(3,4-ジメチルイソオキサゾール-5-イル)カルバミン酸2,2,2-トリクロロエチル(192mg、0.666ミリモル)のアセトン(1ml)の懸濁液にトリエチルアミン(0.338ml、2.42ミリモル)を室温で滴下し、反応溶液を45℃で3時間攪拌した。反応液に水を注ぎ、酢酸エチルで抽出した。抽出液を水で洗浄し、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をヘキサンと酢酸エチルから再結晶して表題化合物(171mg、72%)を固体として得た。融点145-146℃
1H NMR(DMSO-d6)δ:1.68-1.88(5H,m),1.96-2.10(2H,m),2.13(3H,s),2.95-3.21(3H,m),4.09-4.23(2H,m),7.33-7.46(2H,m),7.92(1H,d,J=5.3Hz),8.25-8.36(2H,m),8.82(1H,d,J=5.3Hz),9.14(1H,s)。 Example 36
N- (3,4-dimethylisoxazol-5-yl) -4- [4- (4-fluorophenyl) pyrimidin-2-yl] piperidine-1-carboxamide
4- (4-Fluorophenyl) -2-piperidin-4-ylpyrimidine dihydrochloride (200 mg, 0.606 mmol), (3,4-dimethylisoxazol-5-yl) carbamic acid obtained in Reference Example 48 Triethylamine (0.338 ml, 2.42 mmol) was added dropwise at room temperature to a suspension of 2,2,2-trichloroethyl (192 mg, 0.666 mmol) in acetone (1 ml), and the reaction solution was added at 45 ° C. for 3 hours. Stir for hours. Water was poured into the reaction mixture, and the mixture was extracted with ethyl acetate. The extract was washed with water and dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was recrystallized from hexane and ethyl acetate to give the title compound (171 mg, 72%) as a solid. Melting point: 145-146 ° C
1 H NMR (DMSO-d 6 ) δ: 1.68-1.88 (5H, m), 1.96-2.10 (2H, m), 2.13 (3H, s), 2.95- 3.21 (3H, m), 4.09-4.23 (2H, m), 7.33-7.46 (2H, m), 7.92 (1H, d, J = 5.3 Hz), 8.25-8.36 (2H, m), 8.82 (1 H, d, J = 5.3 Hz), 9.14 (1 H, s).
N-(3,4-ジメチルイソオキサゾール-5-イル)-4-[4-(4-フルオロフェニル)ピリミジン-2-イル]ピペリジン-1-カルボキサミド
1H NMR(DMSO-d6)δ:1.68-1.88(5H,m),1.96-2.10(2H,m),2.13(3H,s),2.95-3.21(3H,m),4.09-4.23(2H,m),7.33-7.46(2H,m),7.92(1H,d,J=5.3Hz),8.25-8.36(2H,m),8.82(1H,d,J=5.3Hz),9.14(1H,s)。 Example 36
N- (3,4-dimethylisoxazol-5-yl) -4- [4- (4-fluorophenyl) pyrimidin-2-yl] piperidine-1-carboxamide
1 H NMR (DMSO-d 6 ) δ: 1.68-1.88 (5H, m), 1.96-2.10 (2H, m), 2.13 (3H, s), 2.95- 3.21 (3H, m), 4.09-4.23 (2H, m), 7.33-7.46 (2H, m), 7.92 (1H, d, J = 5.3 Hz), 8.25-8.36 (2H, m), 8.82 (1 H, d, J = 5.3 Hz), 9.14 (1 H, s).
実施例37
4-[4-(4-フルオロフェニル)ピリミジン-2-イル]-N-ピリダジン-3-イルピペリジン-1-カルボキサミド

参考例48で得た4-(4-フルオロフェニル)-2-ピペリジン-4-イルピリミジン二塩酸塩(200mg、0.606ミリモル)、ピリダジン-3-イルカルバミン酸フェニル(143mg、0.666ミリモル)のアセトン(1ml)の懸濁液にトリエチルアミン(0.338ml、2.42ミリモル)を室温で滴下し、反応溶液を45℃で3時間攪拌した。反応液に水を注ぎ、酢酸エチルで抽出した。抽出液を水で洗浄し、無水硫酸マグネシウムで乾燥し、溶媒を減圧下留去した。残渣をシリカゲルカラムクロマトグラフィー(酢酸エチル)で精製し、ヘキサンと酢酸エチルから再結晶して表題化合物(130mg、57%)を固体として得た。融点115-116℃
1H NMR(DMSO-d6)δ:1.71-1.91(2H,m),1.96-2.14(2H,m),2.98-3.24(3H,m),4.21-4.39(2H,m),7.31-7.45(2H,m),7.57(1H,dd,J=9.1,4.5Hz),7.92(1H,d,J=5.3Hz),8.01(1H,d,J=9.1Hz),8.23-8.37(2H,m),8.75-8.91(2H,m),9.86(1H,s)。 Example 37
4- [4- (4-Fluorophenyl) pyrimidin-2-yl] -N-pyridazin-3-ylpiperidin-1-carboxamide
4- (4-Fluorophenyl) -2-piperidin-4-ylpyrimidine dihydrochloride (200 mg, 0.606 mmol), phenylpyridazin-3-ylcarbamate (143 mg, 0.666 mmol) obtained in Reference Example 48 ) In acetone (1 ml) was added dropwise triethylamine (0.338 ml, 2.42 mmol) at room temperature, and the reaction solution was stirred at 45 ° C. for 3 hours. Water was poured into the reaction mixture, and the mixture was extracted with ethyl acetate. The extract was washed with water and dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The residue was purified by silica gel column chromatography (ethyl acetate) and recrystallized from hexane and ethyl acetate to give the title compound (130 mg, 57%) as a solid. Melting point 115-116 ° C
1 H NMR (DMSO-d 6 ) δ: 1.71-1.91 (2H, m), 1.96-2.14 (2H, m), 2.98-3.24 (3H, m), 4.21-4.39 (2H, m), 7.31-7.45 (2H, m), 7.57 (1H, dd, J = 9.1, 4.5 Hz), 7.92 (1H , D, J = 5.3 Hz), 8.01 (1H, d, J = 9.1 Hz), 8.23-8.37 (2H, m), 8.75-8.91 (2H, m) , 9.86 (1H, s).
4-[4-(4-フルオロフェニル)ピリミジン-2-イル]-N-ピリダジン-3-イルピペリジン-1-カルボキサミド
1H NMR(DMSO-d6)δ:1.71-1.91(2H,m),1.96-2.14(2H,m),2.98-3.24(3H,m),4.21-4.39(2H,m),7.31-7.45(2H,m),7.57(1H,dd,J=9.1,4.5Hz),7.92(1H,d,J=5.3Hz),8.01(1H,d,J=9.1Hz),8.23-8.37(2H,m),8.75-8.91(2H,m),9.86(1H,s)。 Example 37
4- [4- (4-Fluorophenyl) pyrimidin-2-yl] -N-pyridazin-3-ylpiperidin-1-carboxamide
1 H NMR (DMSO-d 6 ) δ: 1.71-1.91 (2H, m), 1.96-2.14 (2H, m), 2.98-3.24 (3H, m), 4.21-4.39 (2H, m), 7.31-7.45 (2H, m), 7.57 (1H, dd, J = 9.1, 4.5 Hz), 7.92 (1H , D, J = 5.3 Hz), 8.01 (1H, d, J = 9.1 Hz), 8.23-8.37 (2H, m), 8.75-8.91 (2H, m) , 9.86 (1H, s).
実施例38
4-(5-フルオロ-4-フェニルピルイミジン-2-イル)-N-ピリダジン-3-イルピペリジン-1-カルボキサミド

参考例51で得た5-フルオロ-4-フェニル-2-ピペリジン-4-イルピルイミジン二塩酸塩(0.16g、0.48ミリモル)、ピリダジン-3-イルカルバミン酸フェニル(0.13g、0.60ミリモル)のアセトン(2ml)の懸濁液にトリエチルアミン(0.20g、2.0ミリモル)を室温で滴下し、反応溶液を40℃で2時間攪拌した。反応液に水(4ml)を滴下し、室温で1時間攪拌した。生じた結晶をろ過し、水で洗浄して粗結晶を固体として得た。得られた粗結晶を再結晶(酢酸エチル)して、表題化合物(140mg、77%)を白色結晶として得た。融点135-136℃
1H NMR(DMSO-d6)δ:1.72-1.86(2H,m),2.00-2.10(2H,m),3.02-3.24(3H,m),4.27-4.31(2H,m),7.54-7.62(4H,m),7.99-8.10(3H,m),8.83-8.84(1H,m),8.89-8.90(1H,m),9.87(1H,s)。 Example 38
4- (5-Fluoro-4-phenylpyrimidin-2-yl) -N-pyridazin-3-ylpiperidin-1-carboxamide
5-fluoro-4-phenyl-2-piperidin-4-ylpyrimidine dihydrochloride (0.16 g, 0.48 mmol), phenyl pyridazin-3-ylcarbamate (0.13 g,. Triethylamine (0.20 g, 2.0 mmol) was added dropwise to a suspension of 60 mmol) in acetone (2 ml) at room temperature, and the reaction solution was stirred at 40 ° C. for 2 hours. Water (4 ml) was added dropwise to the reaction solution, and the mixture was stirred at room temperature for 1 hour. The resulting crystals were filtered and washed with water to obtain crude crystals as a solid. The obtained crude crystals were recrystallized (ethyl acetate) to give the title compound (140 mg, 77%) as white crystals. Melting point 135-136 ° C
1 H NMR (DMSO-d 6 ) δ: 1.72-1.86 (2H, m), 2.00-2.10 (2H, m), 3.02-3.24 (3H, m), 4.27-4.31 (2H, m), 7.54-7.62 (4H, m), 7.9-8.10 (3H, m), 8.83-8.84 (1H, m ), 8.89-8.90 (1H, m), 9.87 (1H, s).
4-(5-フルオロ-4-フェニルピルイミジン-2-イル)-N-ピリダジン-3-イルピペリジン-1-カルボキサミド
1H NMR(DMSO-d6)δ:1.72-1.86(2H,m),2.00-2.10(2H,m),3.02-3.24(3H,m),4.27-4.31(2H,m),7.54-7.62(4H,m),7.99-8.10(3H,m),8.83-8.84(1H,m),8.89-8.90(1H,m),9.87(1H,s)。 Example 38
4- (5-Fluoro-4-phenylpyrimidin-2-yl) -N-pyridazin-3-ylpiperidin-1-carboxamide
1 H NMR (DMSO-d 6 ) δ: 1.72-1.86 (2H, m), 2.00-2.10 (2H, m), 3.02-3.24 (3H, m), 4.27-4.31 (2H, m), 7.54-7.62 (4H, m), 7.9-8.10 (3H, m), 8.83-8.84 (1H, m ), 8.89-8.90 (1H, m), 9.87 (1H, s).
実施例39
4-(5-フルオロ-4-フェニルピルイミジン-2-イル)-N-ピリジン-3-イルピペリジン-1-カルボキサミド

参考例51で得た5-フルオロ-4-フェニル-2-ピペリジン-4-イルピルイミジン二塩酸塩(0.16g、0.48ミリモル)、ピリジン-3-イルカルバミン酸2,2,2-トリクロロエチル(0.24g、0.90ミリモル)のアセトン(2ml)の懸濁液にトリエチルアミン(0.20g、2.0ミリモル)を室温で滴下し、反応溶液を40℃で2時間攪拌した。反応液に水(4ml)を滴下し、室温で1時間攪拌した。反応溶液を酢酸エチルで希釈し、水を加えて抽出した。抽出液を無水硫酸ナトリウムで乾燥し、溶媒を減圧下留去した。残渣を再結晶(ヘキサン-酢酸エチル)して、表題化合物(10mg、6%)を白色結晶として得た。 融点148-150℃
1H NMR(DMSO-d6)δ:1.71-1.85(2H,m),2.00-2.10(2H,m),2.98-3.25(3H,m),4.20-4.30(2H,m),7.24-7.28(1H,m),7.57-7.62(3H,m),7.88-7.91(1H,m),8.07-8.15(3H,m),8.64-8.66(1H,m),8.73(1H,s),8.89-8.90(1H,m). Example 39
4- (5-Fluoro-4-phenylpyrimidin-2-yl) -N-pyridin-3-ylpiperidin-1-carboxamide
5-Fluoro-4-phenyl-2-piperidin-4-ylpyrimidine dihydrochloride (0.16 g, 0.48 mmol) obtained in Reference Example 51, 2,2,2-trichloroethyl pyridin-3-ylcarbamate Triethylamine (0.20 g, 2.0 mmol) was added dropwise at room temperature to a suspension of acetone (2 ml) in (0.24 g, 0.90 mmol), and the reaction solution was stirred at 40 ° C. for 2 hours. Water (4 ml) was added dropwise to the reaction solution, and the mixture was stirred at room temperature for 1 hour. The reaction solution was diluted with ethyl acetate, and extracted by adding water. The extract was dried over anhydrous sodium sulfate, and the solvent was distilled off under reduced pressure. The residue was recrystallized (hexane-ethyl acetate) to give the title compound (10 mg, 6%) as white crystals. Melting point 148-150 ° C
1 H NMR (DMSO-d 6 ) δ: 1.71-1.85 (2H, m), 2.00-2.10 (2H, m), 2.98-3.25 (3H, m), 4.20-4.30 (2H, m), 7.24-7.28 (1H, m), 7.57-7.62 (3H, m), 7.88-7.91 (1H, m ), 8.07-8.15 (3H, m), 8.64-8.66 (1H, m), 8.73 (1H, s), 8.89-8.90 (1H, m).
4-(5-フルオロ-4-フェニルピルイミジン-2-イル)-N-ピリジン-3-イルピペリジン-1-カルボキサミド
1H NMR(DMSO-d6)δ:1.71-1.85(2H,m),2.00-2.10(2H,m),2.98-3.25(3H,m),4.20-4.30(2H,m),7.24-7.28(1H,m),7.57-7.62(3H,m),7.88-7.91(1H,m),8.07-8.15(3H,m),8.64-8.66(1H,m),8.73(1H,s),8.89-8.90(1H,m). Example 39
4- (5-Fluoro-4-phenylpyrimidin-2-yl) -N-pyridin-3-ylpiperidin-1-carboxamide
1 H NMR (DMSO-d 6 ) δ: 1.71-1.85 (2H, m), 2.00-2.10 (2H, m), 2.98-3.25 (3H, m), 4.20-4.30 (2H, m), 7.24-7.28 (1H, m), 7.57-7.62 (3H, m), 7.88-7.91 (1H, m ), 8.07-8.15 (3H, m), 8.64-8.66 (1H, m), 8.73 (1H, s), 8.89-8.90 (1H, m).
製剤例1
(1)実施例1の化合物 10mg
(2)乳糖 60mg
(3)コーンスターチ 35mg
(4)ヒドロキシプロピルメチルセルロース 3mg
(5)ステアリン酸マグネシウム 2mg
実施例1で得られた化合物10mgと乳糖60mgおよびコーンスターチ35mgとの混合物を、10重量%ヒドロキシプロピルメチルセルロース水溶液0.03mL(ヒドロキシプロピルメチルセルロースとして3mg)を用いて顆粒化した後、40℃で乾燥し篩過する。得られた顆粒をステアリン酸マグネシウム2mgと混合し、圧縮する。得られる素錠を、蔗糖、二酸化チタン、タルクおよびアラビアゴムの水懸濁液による糖衣でコーティングする。コーティングが施された錠剤をミツロウで艶出してコート錠を得る。 Formulation Example 1
(1) 10 mg of the compound of Example 1
(2) Lactose 60mg
(3) Corn starch 35mg
(4) Hydroxypropyl methylcellulose 3mg
(5) Magnesium stearate 2mg
A mixture of 10 mg of the compound obtained in Example 1, 60 mg of lactose and 35 mg of corn starch was granulated with 0.03 mL of 10 wt% aqueous hydroxypropylmethylcellulose solution (3 mg as hydroxypropylmethylcellulose), and then dried at 40 ° C. Sift through. The obtained granules are mixed with 2 mg of magnesium stearate and compressed. The resulting uncoated tablets are coated with a sugar coating with an aqueous suspension of sucrose, titanium dioxide, talc and gum arabic. The coated tablet is polished with beeswax to obtain a coated tablet.
(1)実施例1の化合物 10mg
(2)乳糖 60mg
(3)コーンスターチ 35mg
(4)ヒドロキシプロピルメチルセルロース 3mg
(5)ステアリン酸マグネシウム 2mg
実施例1で得られた化合物10mgと乳糖60mgおよびコーンスターチ35mgとの混合物を、10重量%ヒドロキシプロピルメチルセルロース水溶液0.03mL(ヒドロキシプロピルメチルセルロースとして3mg)を用いて顆粒化した後、40℃で乾燥し篩過する。得られた顆粒をステアリン酸マグネシウム2mgと混合し、圧縮する。得られる素錠を、蔗糖、二酸化チタン、タルクおよびアラビアゴムの水懸濁液による糖衣でコーティングする。コーティングが施された錠剤をミツロウで艶出してコート錠を得る。 Formulation Example 1
(1) 10 mg of the compound of Example 1
(2) Lactose 60mg
(3) Corn starch 35mg
(4) Hydroxypropyl methylcellulose 3mg
(5) Magnesium stearate 2mg
A mixture of 10 mg of the compound obtained in Example 1, 60 mg of lactose and 35 mg of corn starch was granulated with 0.03 mL of 10 wt% aqueous hydroxypropylmethylcellulose solution (3 mg as hydroxypropylmethylcellulose), and then dried at 40 ° C. Sift through. The obtained granules are mixed with 2 mg of magnesium stearate and compressed. The resulting uncoated tablets are coated with a sugar coating with an aqueous suspension of sucrose, titanium dioxide, talc and gum arabic. The coated tablet is polished with beeswax to obtain a coated tablet.
実験例1
FAAH阻害活性の測定
(1)細胞画分の調製
PCR法にて増幅したヒトFAAH遺伝子をpcDNA3.1ベクターに挿入し、発現プラスミドを構築した。K562細胞株に本プラスミドを導入し、自体公知の方法でヒトFAAHを安定発現させた細胞株K562/ヒトFAAHを構築した。K562/ヒトFAAHを培地(RPMI1640培地にFBS(ウシ胎児血清)を終濃度10%およびG418を終濃度300μg/mLになるよう添加したもの)を用いてCO2培養器で37℃下培養後、細胞を回収した。細胞をPBSで洗浄後、バッファー(いずれも終濃度10mMのTris-HCl、1mMのEDTA、10mMのMgCl2)に懸濁し、ポリトロンホモジナイザーを用いて細胞を破砕した。2,000rpmで遠心後、上清を回収しさらに40,000rpmで遠心分離し、ペレットを上記のバッファーに懸濁し、酵素画分を調製した。
(2)酵素反応
96ウエル・プレート(Costar社製)を用い、各濃度の被検化合物、酵素画分(終濃度37.5μg/mL)および基質AMCアラキドニルアミド(AMCAA:CAYMAN CHEMICAL社製:終濃度3μM)を反応バッファー(いずれも終濃度125mMのTris-HCl(pH9.0)、1mMのEDTA、0.4mMのHEPES、0.2%のグリセロール、0.02%のTriton X-100および0.3%のBSA)50μL中で37℃、90分間反応させた。反応後プレートをARVO SX 1420 MULTILABEL COUNTER(WALLAC 社製)にて励起355nm、発光460nmで蛍光強度を測定した。酵素を含まない反応を阻害率100%として、被検化合物の阻害率を算出した。 Experimental example 1
Measurement of FAAH inhibitory activity (1) Preparation of cell fraction The human FAAH gene amplified by the PCR method was inserted into a pcDNA3.1 vector to construct an expression plasmid. This plasmid was introduced into the K562 cell line, and a cell line K562 / human FAAH in which human FAAH was stably expressed by a method known per se was constructed. After culturing at 37 ° C. in a CO 2 incubator using K562 / human FAAH in a medium (RPMI1640 medium supplemented with FBS (fetal bovine serum) to a final concentration of 10% and G418 to a final concentration of 300 μg / mL), Cells were collected. The cells were washed with PBS, suspended in a buffer (all of which had a final concentration of 10 mM Tris-HCl, 1 mM EDTA, 10 mM MgCl 2 ), and the cells were disrupted using a polytron homogenizer. After centrifugation at 2,000 rpm, the supernatant was collected and further centrifuged at 40,000 rpm, and the pellet was suspended in the above buffer to prepare an enzyme fraction.
(2) Enzyme reaction Using a 96-well plate (manufactured by Costar), each concentration of test compound, enzyme fraction (final concentration 37.5 μg / mL), and substrate AMC arachidonil amide (AMCAA: CAYMAN CHEMICAL): Final concentration 3 μM) in reaction buffer (both 125 mM Tris-HCl pH 9.0), 1 mM EDTA, 0.4 mM HEPES, 0.2% glycerol, 0.02% Triton X-100 and The reaction was carried out in 50 μL of 0.3% BSA) at 37 ° C. for 90 minutes. After the reaction, the fluorescence intensity of the plate was measured with an ARVO SX 1420 MULTILABEL COUNTER (manufactured by WALLAC) at excitation of 355 nm and emission of 460 nm. The inhibition rate of the test compound was calculated with the reaction containing no enzyme as the inhibition rate of 100%.
FAAH阻害活性の測定
(1)細胞画分の調製
PCR法にて増幅したヒトFAAH遺伝子をpcDNA3.1ベクターに挿入し、発現プラスミドを構築した。K562細胞株に本プラスミドを導入し、自体公知の方法でヒトFAAHを安定発現させた細胞株K562/ヒトFAAHを構築した。K562/ヒトFAAHを培地(RPMI1640培地にFBS(ウシ胎児血清)を終濃度10%およびG418を終濃度300μg/mLになるよう添加したもの)を用いてCO2培養器で37℃下培養後、細胞を回収した。細胞をPBSで洗浄後、バッファー(いずれも終濃度10mMのTris-HCl、1mMのEDTA、10mMのMgCl2)に懸濁し、ポリトロンホモジナイザーを用いて細胞を破砕した。2,000rpmで遠心後、上清を回収しさらに40,000rpmで遠心分離し、ペレットを上記のバッファーに懸濁し、酵素画分を調製した。
(2)酵素反応
96ウエル・プレート(Costar社製)を用い、各濃度の被検化合物、酵素画分(終濃度37.5μg/mL)および基質AMCアラキドニルアミド(AMCAA:CAYMAN CHEMICAL社製:終濃度3μM)を反応バッファー(いずれも終濃度125mMのTris-HCl(pH9.0)、1mMのEDTA、0.4mMのHEPES、0.2%のグリセロール、0.02%のTriton X-100および0.3%のBSA)50μL中で37℃、90分間反応させた。反応後プレートをARVO SX 1420 MULTILABEL COUNTER(WALLAC 社製)にて励起355nm、発光460nmで蛍光強度を測定した。酵素を含まない反応を阻害率100%として、被検化合物の阻害率を算出した。 Experimental example 1
Measurement of FAAH inhibitory activity (1) Preparation of cell fraction The human FAAH gene amplified by the PCR method was inserted into a pcDNA3.1 vector to construct an expression plasmid. This plasmid was introduced into the K562 cell line, and a cell line K562 / human FAAH in which human FAAH was stably expressed by a method known per se was constructed. After culturing at 37 ° C. in a CO 2 incubator using K562 / human FAAH in a medium (RPMI1640 medium supplemented with FBS (fetal bovine serum) to a final concentration of 10% and G418 to a final concentration of 300 μg / mL), Cells were collected. The cells were washed with PBS, suspended in a buffer (all of which had a final concentration of 10 mM Tris-HCl, 1 mM EDTA, 10 mM MgCl 2 ), and the cells were disrupted using a polytron homogenizer. After centrifugation at 2,000 rpm, the supernatant was collected and further centrifuged at 40,000 rpm, and the pellet was suspended in the above buffer to prepare an enzyme fraction.
(2) Enzyme reaction Using a 96-well plate (manufactured by Costar), each concentration of test compound, enzyme fraction (final concentration 37.5 μg / mL), and substrate AMC arachidonil amide (AMCAA: CAYMAN CHEMICAL): Final concentration 3 μM) in reaction buffer (both 125 mM Tris-HCl pH 9.0), 1 mM EDTA, 0.4 mM HEPES, 0.2% glycerol, 0.02% Triton X-100 and The reaction was carried out in 50 μL of 0.3% BSA) at 37 ° C. for 90 minutes. After the reaction, the fluorescence intensity of the plate was measured with an ARVO SX 1420 MULTILABEL COUNTER (manufactured by WALLAC) at excitation of 355 nm and emission of 460 nm. The inhibition rate of the test compound was calculated with the reaction containing no enzyme as the inhibition rate of 100%.

実験例2
マウス酢酸ライジング試験における鎮痛効果
試験化合物の懸濁液(10mg/kg)をマウス経口投与し、投与60分後に0.6%酢酸水溶液を10ml/kg体重で腹腔内投与し、直後から20分後まで専用ケージにマウスを収容しカウンターにてライジング反応数を計測した。対照群に対して2群間で平均値の差の検定(Student’s t-test)を行い、鎮痛効果を評価した。 Experimental example 2
Analgesic effect in mouse acetic acid rising test A suspension of test compound (10 mg / kg) was orally administered to mice, and 60 minutes after administration, a 0.6% acetic acid aqueous solution was intraperitoneally administered at 10 ml / kg body weight. Mice were housed in dedicated cages and the number of rising reactions was counted using a counter. The control group was tested for difference in mean value between the two groups (Student's t-test) to evaluate the analgesic effect.
マウス酢酸ライジング試験における鎮痛効果
試験化合物の懸濁液(10mg/kg)をマウス経口投与し、投与60分後に0.6%酢酸水溶液を10ml/kg体重で腹腔内投与し、直後から20分後まで専用ケージにマウスを収容しカウンターにてライジング反応数を計測した。対照群に対して2群間で平均値の差の検定(Student’s t-test)を行い、鎮痛効果を評価した。 Experimental example 2
Analgesic effect in mouse acetic acid rising test A suspension of test compound (10 mg / kg) was orally administered to mice, and 60 minutes after administration, a 0.6% acetic acid aqueous solution was intraperitoneally administered at 10 ml / kg body weight. Mice were housed in dedicated cages and the number of rising reactions was counted using a counter. The control group was tested for difference in mean value between the two groups (Student's t-test) to evaluate the analgesic effect.

本発明の式(I)で示される化合物でまたはその塩は、優れたFAAH阻害活性を有しており、安全で優れた痛み、うつまたは不安などの予防・治療剤として有用である。
The compound represented by the formula (I) of the present invention or a salt thereof has an excellent FAAH inhibitory activity and is useful as a safe and excellent preventive / therapeutic agent for pain, depression or anxiety.
Claims (19)
- 式(I)

環Ar1は、置換されていてもよい芳香族複素環を示す;
A1はCR1又はNを示し、A2はCR2又はNを示し、A3はCR3又はNを示し、A4はCR4又はNを示し、かつA1、A2、A3、及びA4のうち少なくとも1つはNを示す;
環Ar2は、1個以上のハロゲン原子で置換されていてもよい芳香族炭化水素環、又は1個以上のハロゲン原子で置換されていてもよい芳香族複素環を示す;
R1、R2、R3、及びR4はそれぞれ独立して、水素原子、ハロゲン原子、又は置換されていても良いC1-6アルキル基を示す。]
で示される化合物、又はその塩。 Formula (I)
Ring Ar 1 represents an optionally substituted aromatic heterocycle;
A 1 represents CR 1 or N, A 2 represents CR 2 or N, A 3 represents CR 3 or N, A 4 represents CR 4 or N, and A 1 , A 2 , A 3 , And at least one of A 4 represents N;
Ring Ar 2 represents an aromatic hydrocarbon ring that may be substituted with one or more halogen atoms, or an aromatic heterocycle that may be substituted with one or more halogen atoms;
R 1 , R 2 , R 3 , and R 4 each independently represent a hydrogen atom, a halogen atom, or an optionally substituted C 1-6 alkyl group. ]
Or a salt thereof. - 式(I)の部分構造


- 式(I)の部分構造


- 環Ar1が、ピリジン環、ピリダジン環、又はC1-6アルキル基で置換されたイソオキサゾール環である、請求項1~3のいずれか一項に記載の化合物。 The compound according to any one of claims 1 to 3, wherein the ring Ar 1 is a pyridine ring, a pyridazine ring, or an isoxazole ring substituted with a C 1-6 alkyl group.
- 環Ar2が、1個または2個のハロゲン原子で置換されていてもよいベンゼン環、フラン環、又はチオフェン環である、請求項1~3のいずれか一項に記載の化合物。 The compound according to any one of claims 1 to 3, wherein the ring Ar 2 is a benzene ring, a furan ring, or a thiophene ring optionally substituted with one or two halogen atoms.
- 環Ar1が、ピリジン環、ピリダジン環、又はC1-6アルキル基で置換されたイソオキサゾール環であり、
環Ar2が、1個または2個のハロゲン原子で置換されていてもよいベンゼン環、フラン環、又はチオフェン環、
である、請求項1~3のいずれか一項に記載の化合物。 Ring Ar 1 is a pyridine ring, a pyridazine ring, or an isoxazole ring substituted with a C 1-6 alkyl group,
A ring Ar 2 may be substituted with one or two halogen atoms, a benzene ring, a furan ring, or a thiophene ring;
The compound according to any one of claims 1 to 3, wherein - 式(I)が、式(Ia)

環Ar3は、ハロゲン原子で置換されていてもよいピリダジン環を示す;
環Ar4は、ハロゲン原子で置換されていてもよいベンゼン環を示す。]
である、請求項1記載の化合物。 Formula (I) is converted to Formula (Ia)
Ring Ar 3 represents a pyridazine ring optionally substituted with a halogen atom;
Ring Ar 4 represents a benzene ring which may be substituted with a halogen atom. ]
The compound of claim 1, wherein - 4-(4-フェニルピリミジン-2-イル)-N-ピリダジン-3-イルピペリジン-1-カルボキサミドまたはその塩。 4- (4-phenylpyrimidin-2-yl) -N-pyridazin-3-ylpiperidin-1-carboxamide or a salt thereof.
- 4-[4-(2,3-ジフルオロフェニル)ピリミジン-2-イル]-N-ピリダジン-3-イルピペリジン-1-カルボキサミドまたはその塩。 4- [4- (2,3-difluorophenyl) pyrimidin-2-yl] -N-pyridazin-3-ylpiperidin-1-carboxamide or a salt thereof.
- 4-[4-(3-フルオロフェニル)ピリミジン-2-イル]-N-ピリダジン-3-イルピペリジン-1-カルボキサミドまたはその塩。 4- [4- (3-Fluorophenyl) pyrimidin-2-yl] -N-pyridazin-3-ylpiperidin-1-carboxamide or a salt thereof.
- 4-[4-(4-フルオロフェニル)ピリミジン-2-イル]-N-ピリダジン-3-イルピペリジン-1-カルボキサミドまたはその塩。 4- [4- (4-fluorophenyl) pyrimidin-2-yl] -N-pyridazin-3-ylpiperidin-1-carboxamide or a salt thereof.
- 請求項1記載の化合物のプロドラッグ。 A prodrug of the compound according to claim 1.
- 請求項1記載の化合物、又はそのプロドラッグを含有する医薬。 A pharmaceutical comprising the compound according to claim 1 or a prodrug thereof.
- 請求項1記載の化合物、又はそのプロドラッグを含有するFAAH阻害剤。 A FAAH inhibitor containing the compound according to claim 1 or a prodrug thereof.
- 請求項1記載の化合物若しくはそのプロドラッグを含有する、痛み、うつ若しくは不安の予防剤、又は治療剤。 A prophylactic or therapeutic agent for pain, depression or anxiety, comprising the compound according to claim 1 or a prodrug thereof.
- 請求項1記載の化合物、又はそのプロドラッグの有効量を哺乳動物に投与することを特徴とするFAAH阻害方法。 A method for inhibiting FAAH, comprising administering an effective amount of the compound according to claim 1 or a prodrug thereof to a mammal.
- 請求項1記載の化合物若しくはそのプロドラッグの有効量を哺乳動物に投与することを特徴とする、痛み、うつ若しくは不安の予防方法、又は治療方法。 A method for preventing or treating pain, depression or anxiety, which comprises administering an effective amount of the compound or prodrug thereof according to claim 1 to a mammal.
- FAAH阻害剤を製造するための、請求項1記載の化合物、又はそのプロドラッグの使用。 Use of the compound according to claim 1 or a prodrug thereof for producing a FAAH inhibitor.
- 痛み、うつ若しくは不安の予防剤、又は治療剤を製造するための、請求項1記載の化合物若しくはそのプロドラッグの使用。 Use of the compound according to claim 1 or a prodrug thereof for producing a prophylactic or therapeutic agent for pain, depression or anxiety.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008306366 | 2008-12-01 | ||
| JP2008-306366 | 2008-12-01 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2010064597A1 true WO2010064597A1 (en) | 2010-06-10 |
Family
ID=42233246
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2009/070098 WO2010064597A1 (en) | 2008-12-01 | 2009-11-30 | Piperidine derivative |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2010064597A1 (en) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011078369A1 (en) * | 2009-12-25 | 2011-06-30 | 持田製薬株式会社 | Novel aryl urea derivative |
| WO2011085216A2 (en) | 2010-01-08 | 2011-07-14 | Ironwood Pharmaceuticals, Inc. | Use of faah inhibitors for treating parkinson's disease and restless legs syndrome |
| WO2011123719A2 (en) | 2010-03-31 | 2011-10-06 | Ironwood Pharmaceuticals, Inc. | Use of faah inhibitors for treating abdominal, visceral and pelvic pain |
| JP2013518046A (en) * | 2010-01-25 | 2013-05-20 | シーエイチディーアイ ファウンデーション,インコーポレーテッド | Specific kynurenine-3-monooxygenase inhibitors and pharmaceutical compositions thereof and methods of use thereof |
| EP2709609A1 (en) * | 2011-05-17 | 2014-03-26 | Shionogi & Co., Ltd. | Heterocyclic compounds |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006054652A1 (en) * | 2004-11-18 | 2006-05-26 | Takeda Pharmaceutical Company Limited | Amide compound |
| JP2006528642A (en) * | 2003-07-24 | 2006-12-21 | ユーロ−セルティーク エス.エイ. | Useful therapeutics to treat pain |
| JP2007509915A (en) * | 2003-10-29 | 2007-04-19 | メルク シャープ エンド ドーム リミテッド | 4-Fluoro-4- (pyridin-2-yl) piperidine-1-carboxamide derivatives and related compounds that modulate the function of vanilloid 1 receptor (VR1) for the treatment of pain |
| JP2007522233A (en) * | 2004-02-11 | 2007-08-09 | アムジエン・インコーポレーテツド | Vanilloid receptor ligands and their use in therapy |
| WO2008023720A1 (en) * | 2006-08-23 | 2008-02-28 | Astellas Pharma Inc. | Urea compound or salt thereof |
| WO2008047229A2 (en) * | 2006-10-18 | 2008-04-24 | Pfizer Products Inc. | Biaryl ether urea compounds |
| WO2008153752A2 (en) * | 2007-05-25 | 2008-12-18 | Janssen Pharmaceutica N.V. | Heteroaryl-substituted urea modulators of fatty acid amide hydrolase |
| WO2009127948A1 (en) * | 2008-04-17 | 2009-10-22 | Pfizer Inc. | 4- [3- (aryloxy) benzylidene] -3-methyl piperidine 5-membered aryl carboxamide compounds useful as faah inhibitors |
| WO2009127943A1 (en) * | 2008-04-17 | 2009-10-22 | Pfizer Inc. | Ether benzylidene piperidine 5-membered aryl carboxamide compounds useful as faah inhibitors |
-
2009
- 2009-11-30 WO PCT/JP2009/070098 patent/WO2010064597A1/en active Application Filing
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2006528642A (en) * | 2003-07-24 | 2006-12-21 | ユーロ−セルティーク エス.エイ. | Useful therapeutics to treat pain |
| JP2007509915A (en) * | 2003-10-29 | 2007-04-19 | メルク シャープ エンド ドーム リミテッド | 4-Fluoro-4- (pyridin-2-yl) piperidine-1-carboxamide derivatives and related compounds that modulate the function of vanilloid 1 receptor (VR1) for the treatment of pain |
| JP2007522233A (en) * | 2004-02-11 | 2007-08-09 | アムジエン・インコーポレーテツド | Vanilloid receptor ligands and their use in therapy |
| WO2006054652A1 (en) * | 2004-11-18 | 2006-05-26 | Takeda Pharmaceutical Company Limited | Amide compound |
| WO2008023720A1 (en) * | 2006-08-23 | 2008-02-28 | Astellas Pharma Inc. | Urea compound or salt thereof |
| WO2008047229A2 (en) * | 2006-10-18 | 2008-04-24 | Pfizer Products Inc. | Biaryl ether urea compounds |
| WO2008153752A2 (en) * | 2007-05-25 | 2008-12-18 | Janssen Pharmaceutica N.V. | Heteroaryl-substituted urea modulators of fatty acid amide hydrolase |
| WO2009127948A1 (en) * | 2008-04-17 | 2009-10-22 | Pfizer Inc. | 4- [3- (aryloxy) benzylidene] -3-methyl piperidine 5-membered aryl carboxamide compounds useful as faah inhibitors |
| WO2009127943A1 (en) * | 2008-04-17 | 2009-10-22 | Pfizer Inc. | Ether benzylidene piperidine 5-membered aryl carboxamide compounds useful as faah inhibitors |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011078369A1 (en) * | 2009-12-25 | 2011-06-30 | 持田製薬株式会社 | Novel aryl urea derivative |
| WO2011085216A2 (en) | 2010-01-08 | 2011-07-14 | Ironwood Pharmaceuticals, Inc. | Use of faah inhibitors for treating parkinson's disease and restless legs syndrome |
| JP2013518046A (en) * | 2010-01-25 | 2013-05-20 | シーエイチディーアイ ファウンデーション,インコーポレーテッド | Specific kynurenine-3-monooxygenase inhibitors and pharmaceutical compositions thereof and methods of use thereof |
| WO2011123719A2 (en) | 2010-03-31 | 2011-10-06 | Ironwood Pharmaceuticals, Inc. | Use of faah inhibitors for treating abdominal, visceral and pelvic pain |
| EP2709609A1 (en) * | 2011-05-17 | 2014-03-26 | Shionogi & Co., Ltd. | Heterocyclic compounds |
| JP2014515349A (en) * | 2011-05-17 | 2014-06-30 | 塩野義製薬株式会社 | Heterocyclic compounds |
| EP2709609A4 (en) * | 2011-05-17 | 2015-08-26 | Shionogi & Co | Heterocyclic compounds |
| US9156830B2 (en) | 2011-05-17 | 2015-10-13 | Shionogi & Co., Ltd. | Heterocyclic compounds |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JPWO2009048101A1 (en) | Amide compounds | |
| US10323026B2 (en) | Heterocyclic compound | |
| JP6832342B2 (en) | Heterocyclic compound | |
| ES2641478T3 (en) | 3-Acetylamino-1- (phenyl-heteroaryl-aminocarbonyl or phenyl-heteroaryl-carbonylamino) benzene derivatives for the treatment of hyperproliferative disorders | |
| JP5143916B2 (en) | New bicyclic heterocyclic compounds | |
| ES2831832T3 (en) | Cyano-substituted indole compounds and uses thereof as LSD1 inhibitors | |
| EA028232B1 (en) | Heterocyclyl compounds as mek inhibitors | |
| JPWO2019065791A1 (en) | Heterocyclic compound | |
| ES2871673T3 (en) | KCNQ2-5 channel activator | |
| JP2017001991A (en) | Novel benzoxazolone compound | |
| JP2018199712A (en) | Arylamide kinase inhibitor | |
| US20220298112A1 (en) | N-(PHENYL)-Indole-3-Sulfonamide Derivatives And Related Compounds As GPR17 Modulators For Treating CNS Disorders Such As Multiple Sclerosis | |
| WO2010064597A1 (en) | Piperidine derivative | |
| AU2021219097A1 (en) | P2X3 modulators | |
| JP2021046428A (en) | Pyridopyrimidinones and their use as nmda receptor modulators | |
| JP5222737B2 (en) | Tricyclic compounds and their pharmaceutical uses | |
| KR101820541B1 (en) | 8-oxodihydropurine derivative | |
| WO2010117014A1 (en) | Triazine derivative | |
| JP6197971B1 (en) | KCNQ2-5 channel-related disease prevention and / or treatment agent | |
| JP2012031152A (en) | Medicine made of new bicyclic heterocyclic compound | |
| WO2006132438A1 (en) | 1,3-benzothiazinone derivative and use thereof | |
| RU2791533C2 (en) | Heterocyclic compound | |
| JP2023518299A (en) | Androgen receptor modulation by small molecule enantiomers |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09830363 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 09830363 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: JP |