WO2009100441A2 - Depot formulations - Google Patents
Depot formulations Download PDFInfo
- Publication number
- WO2009100441A2 WO2009100441A2 PCT/US2009/033574 US2009033574W WO2009100441A2 WO 2009100441 A2 WO2009100441 A2 WO 2009100441A2 US 2009033574 W US2009033574 W US 2009033574W WO 2009100441 A2 WO2009100441 A2 WO 2009100441A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- clonidine
- formulation
- depot formulation
- acid
- days
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
- A61K9/0024—Solid, semi-solid or solidifying implants, which are implanted or injected in body tissue
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1629—Organic macromolecular compounds
- A61K9/1641—Organic macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyethylene glycol, poloxamers
- A61K9/1647—Polyesters, e.g. poly(lactide-co-glycolide)
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/12—Carboxylic acids; Salts or anhydrides thereof
Definitions
- the present invention is directed to pharmaceutical depot formulations of basic active agents (e.g. clonidine) or pharmaceutically acceptable salts or prodrugs thereof, which release therapeutic levels of the active agent for at least 24 hours, preferably at least 3 days to about 5 days, more preferably from about 7 days to about 28 days, and most preferably for at least 3 months.
- basic active agents e.g. clonidine
- pharmaceutically acceptable salts or prodrugs thereof which release therapeutic levels of the active agent for at least 24 hours, preferably at least 3 days to about 5 days, more preferably from about 7 days to about 28 days, and most preferably for at least 3 months.
- Clonidine is an imidazoline derivative that exhibits alpha- 2 -adrenergic agonist activity and is used primarily for the treatment of systemic hypertension. Clonidine lowers blood pressure by activation of alpha- 2 -receptors in the cardiovascular control centers of the CNS which suppresses the outflow of sympathetic nervous system activity from the brain. Clonidine also exhibits analgesic effects which are mediated by pre- and possibly postsynaptic alpha- 2 -adrenergic receptors that block nociceptive transmission. Other known uses for clonidine include therapy for opioid detoxification, nicotine detoxification, sleep hyperhydrosis, insomnia, menopausal symptoms, ADHD, and Tourette's syndrome.
- clonidine is highly soluble and penetrates the blood brain barrier and the placenta. It is believed that that binding of clonidine to receptors is highest in the rostral venterolateral medulla in the brain stem (the final common pathway for sympathetic outflow) where it activates inhibitory neurons. The overall effect is to decrease sympathetic activity, to enhance parasympathetic tone, and to reduce circulating catecholamines.
- Clonidine is commercially available as tablets in 0.1 -mg, 0.2-mg, and 0.3-mg strengths under the tradename Catapres® by Boehringer Ingelheim. Clonidine is also available as a transdermal patch in 0.1-mg, 0.2-mg, and 0.3-mg under the tradename Catapres-TTS® by Boehringer Ingelheim. When administered orally, clonidine is almost completely absorbed from the gastrointestinal tract, but it is subject to the first pass effect. The biological half-life ranges from about four to six hours after oral administration, with wide interpatient variability.
- clonidine dizziness associated with orthostatic hypotension.
- Other adverse effects of clonidine can include dry mouth, constipation, nausea, daytime sleepiness, weakness, and lethargy, eye dryness and loss of libido.
- These side effects may be potentiated by the currently available immediate release tablets which typically exhibit a large peak to trough ratio given the typical TID (three times daily) or QID (four times daily) therapy. Further, given the short half-life of the drug, plasma levels could fall below the therapeutic range quickly in the event that a patient is late with the administration of a dose.
- the transdermal patch of clonidine would be expected to address some of the problems related to oral clonidine therapy.
- the transdermal patch has its own issues with respect to adverse effects and patient compliance.
- Clonidine transdermal patches exhibit contact dermatitis in many patients. Clonidine patches can also exhibit problems with adherence to the skin under certain conditions such as elevated humidity, sweating, showering, swimming, etc. This can also result in plasma levels falling below therapeutic levels in the event that a patient is unable to immediately reapply a new patch, resulting in a shortened duration of efficacy.
- the art does not contain or suggest a dosage form that can effectively provide therapeutic levels of an alpha-2-adrenergic agonist (e ⁇ g., clonidine) for an extended period of time, which can address the issues (ej*., compliance issues, plasma fluctuations) commonly associated with the commercially available immediate release tablets or patches.
- an alpha-2-adrenergic agonist e ⁇ g., clonidine
- Such a dosage form would need to take into account the high solubility of clonidine to assure that the dosage form does not "dose dump" and provide plasma levels which could potentially be toxic.
- an alpha- 2 -adrenergic agonist such as clonidine
- an alpha- 2 -adrenergic agonist such as clonidine
- an alpha- 2 -adrenergic agonist such as clonidine
- an alpha- 2 -adrenergic agonist such as clonidine
- the present invention which, in certain embodiments, is directed to a pharmaceutical depot formulation comprising clonidine or a pharmaceutically acceptable salt or prodrug thereof; the depot formulation providing a mean release rate of clonidine of less than about 5 mg over 24 hours and providing a release rate of at least about 0.1 mg of clonidine per 24 hour period for at least 24 hours, at least 3 to 5 days, at least 7 to 28 days or at least 3 months.
- the invention is directed to a pharmaceutical depot formulation comprising clonidine or a pharmaceutically acceptable salt or prodrug thereof and a dissolution modifying excipient; the depot formulation providing a mean release rate of clonidine of less than about 5 mg over 24 hours and providing a release rate of at least about 0.1 mg of clonidine per 24 hour period for at least 24 hours, at least 3 to 5 days, at least 7 to 28 days or at least 3 months.
- the invention is directed to a pharmaceutical depot formulation comprising a therapeutically effective amount of clonidine or a pharmaceutically acceptable salt or prodrug thereof, a dissolution modifying excipient and a pharmaceutically acceptable vehicle; the depot formulation after injection or implantation into a human patient providing a mean release rate of clonidine of less than about 5 mg over 24 hours and providing a release rate of at least about 0.1 mg of clonidine per 24 hour period for at least 24 hours, at least 3 to 5 days, at least 7 to 28 days or at least 3 months.
- the depot formulation of the present invention provides a release rate of at least about 0.1 mg of clonidine per 24 hour period for at least 24 hours, at least 3 days, at least 5 days, at least 7 days; at least 14 days; at least 1 month; or at least 3 months.
- the invention is directed to a pharmaceutical depot formulation comprising an active pharmaceutical ingredient in base form which is at least sparingly soluble in water; and a biocompatible polymer, wherein the depot formulation releases an amount of active pharmaceutical ingredient during the first twenty-four hour period that is approximately equal to, but no more than about three times the amount of active pharmaceutical ingredient released during the seventh twenty- four hour period.
- the invention is directed to a pharmaceutical depot formulation comprising clonidine base, salt or prodrug thereof; a biocompatible polymer, and an optional hydrophobic acid, wherein the depot formulation exhibits an in-vitro clonidine release profile comprising a first amount of clonidine released during a first twenty- four hour period of the profile, and a second amount of clonidine released during a second twenty-four hour period of the profile, wherein the second twenty-four hour period occurs at least six days after the first twenty-four hour period.
- the invention is directed to a pharmaceutical depot formulation comprising clonidine base, salt or prodrug thereof; a biocompatible polymer, and an optional carboxylic acid, preferably acetic acid, wherein the depot formulation exhibits an in-vitro clonidine release profile comprising a first amount of clonidine released during a first twenty-four hour period of the profile, and a second amount of clonidine released during a second twenty-four hour period of the profile, wherein the second twenty-four hour period occurs at least six days after the first twenty-four hour period.
- the present invention is directed to a method of preparing a pharmaceutical depot formulation as disclosed herein by a contacting an alpha- 2 - adrenergic agonist (ej*., clonidine or a pharmaceutically acceptable salt or prodrug thereof) with an excipient to retard the release of the clonidine, and incorporating the resultant mixture into a pharmaceutically acceptable vehicle suitable for injection.
- an alpha- 2 - adrenergic agonist ej*., clonidine or a pharmaceutically acceptable salt or prodrug thereof
- the present invention is directed to a method of preparing a pharmaceutical depot formulation as disclosed herein by incorporating an insoluble salt of an alpha- 2 -adrenergic agonist (e ⁇ g., clonidine), a fatty acid salt thereof, or a prodrug thereof, into a pharmaceutically acceptable vehicle suitable for injection.
- an alpha- 2 -adrenergic agonist e ⁇ g., clonidine
- clonidine means clonidine free base as well as any salt, hydrate, solvate, polymorph, or derivative thereof.
- alpha- 2 -adrenergic agonist means an agent that is capable of eliciting a therapeutic response by agonizing an alpha- 2 -adrenergic receptor.
- prodrug means a compound which is converted to a therapeutically active compound after administration. A prodrug is typically inactive or less active than the therapeutically active compound to which it is converted.
- salts means salts prepared from pharmaceutically acceptable non-toxic bases or acids including inorganic or organic bases, inorganic or organic acids and fatty acids. Salts derived from inorganic bases include aluminum, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, manganic salts, manganous, potassium, sodium, zinc, and the like.
- Salts derived from pharmaceutically acceptable organic non-toxic bases include salts of primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines, and basic ion exchange resins, such as arginine, betaine, caffeine, choline, N,N'-dibenzylethylenediamine, diethylamine, 2-diethylaminoethanol, 2- dimethylaminoethanol, ethanolamine, ethylenediamine, N-ethyl-morpholine, N- ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethyl amine, tripropylamine, tromethamine, and the like.
- basic ion exchange resins such
- salts may be prepared from pharmaceutically acceptable non-toxic acids, including inorganic and organic acids.
- acids include acetic, benzenesulfonic, benzoic, camphorsulfonic, citric, ethanesulfonic, formic, fumaric, gluconic, glutamic, hydrobromic, hydrochloric, isethionic, lactic, maleic, malic, mandelic, methanesulfonic, malonic, mucic, nitric, pamoic, pantothenic, phosphoric, propionic, succinic, sulfuric, tartaric, p-toluenesulfonic acid, trifluoroacetic acid, and the like.
- Fatty acid salts may also be used, e ⁇ g., fatty acid salts having greater than 2 carbons, greater than 8 carbons or greater than 16 carbons, such as butyric, caprioc, caprylic, capric, lauric, mystiric, palmitic, stearic, arachidic and the like.
- spot means a formulation that is suitable to be administered to a patient parenterally or by implantation that can release the alpha- 2 -adrenergic agonist (e.g., clonidine) over an extended period of time to provide a therapeutic effect.
- alpha- 2 -adrenergic agonist e.g., clonidine
- spot does not include a transdermal patch or intrathecal administration (e ⁇ g., epidural).
- biodegradable means that the excipient will degrade or erode in vivo to form smaller chemical species. Degradation can result, for example, by enzymatic, chemical, and/or physical processes.
- therapeutic plasma level means that plasma levels are achieved that are within the therapeutic window, i ⁇ e,, above the minimum therapeutic level and below the toxic level.
- sustained release means that the active agent is released from the depot formulation of the present invention over an extended period of time (e.g., at least 24 hours, at least 3 days, at least 5 days, at least 7 days; at least 14 days; at least 1 month or at least 3 months) as compared to an immediate release formulation.
- substantially zero order release means that the release rate of active agent from the depot formulation at one time is not substantially different as compared to the release rate of active agent from the depot formulation at another time. For purposes of this definition, a selected time would not include a lag time of active agent release that may be present upon initiation of therapy.
- effective average particle size means that 50% of the particle volume distribution falls below the designated measurement when measured by, for example, sieves, sedimentation field flow fractionation, photon correlation spectroscopy, light scattering, disk centrifugation, and other techniques known to those of skill in the art.
- the particle sizes distributions and values reported herein are deduced from sieves, a Malvern instrument, and visual microscopy (converted to volume) depending on, and made clear within, the context.
- Figure 1 depicts a graph of the particle size distribution for clonidine in Example 2.
- Figure 2 depicts the micrographs of lyophilized microspheres acquired by light microscope in Example 2.
- Figure 3 depicts a graph of the mean size distribution of clonidine encapsulated microspheres in Example 2.
- Figure 4 depicts the substantially zero order clonidine release profile from copolymer formulations comprising PLGA 50:50 and PLGA 75:25 as fabricated utilizing a double emulsion evaporation method.
- Figure 5 depicts the release rate profile for two formulations made according to
- Example 3 differing only by the molecular weight of the biodegradable polymer utilized in the formulation.
- Figure 6 depicts a graph of the release profiles from PLGA microspheres made from
- Figure 7 compares drug loading as a function of reagent drug-to-PLGA-polymer ratio for formulations made according to Example 4.
- Figure 8 depicts how the percent of clonidine released from microspheres of Example 4 is affected by the drug-to-polymer-ratio during the formulation process.
- Figure 9 depicts a graph of the dissolution profiles of clonidine release from PLGA microspheres with different properties from Example 4.
- Figure 10 depicts a graph of the clonidine release profiles from PLGA microspheres fabricated by the single emulsion co-solvent method of Example 5 and the effect of solvent / co-solvent composition thereon.
- Figures 11 compares the cumulative dissolution rate of the microsphere formulations A and B from Example 7 with an Alzet pump.
- Figure 12 compares release rates of formulations A and B from Example 7 with an Alzet pump on a non-cumulative basis.
- Figure 13 depicts the in vitro dissolution profile of Example 9.
- Figure 14 depicts the in vitro dissolution profile of samples from Example 10 and demonstrates the affect of particle size on release rate.
- Figure 15 depicts a graph of the in vitro dissolution profile batches from Example 11.
- Figure 16 depicts the rate of drug release from formulations prepared according to
- Example 3 with differing ratios of inner water phase to inner organic phase.
- Figure 17 depicts the rate of drug release from formulations prepared according to
- Example 3 with differing terminal groups (ester or carboxylic acid).
- Figure 18 depicts the dissolution profile for clonidine microspheres prepared according to Example 2 and containing clonidine having differing particle size distributions.
- Figure 19 depicts the rate of clonidine release based on microsphere homogenization time (and otherwise prepared according to Example 3).
- Figure 20 depicts the rate of clonidine release based on microsphere vortex time (and otherwise prepared according to Example 3).
- the physical and chemical properties of the active agent must be fully considered and accounted for.
- one of the more important features of the active ingredient is its solubility. If the solubility of the active ingredient is relatively high, then the formulation can have a tendency to dose dump either as a direct result of the solubility, or due to an unusually high "apparent" solubility, wherein the release rate (and hence metabolism) may be affected by external factors. In those cases where the therapeutically effective range is low relative to the actual or apparent solubility, the effect of a dose dump may not only cause causing untimely or premature release, but may also lead to toxic concentrations of the drug in the recipient.
- Clonidine is highly soluble (clonidine base has a solubility of about 1.8 mg/mL in water under nominal experimental conditions), and as such is susceptible to the dose dump phenomenon. Therefore, it is advantageous to provide a therapeutic dose of clonidine that is both low enough and consistently low enough to reduce or avoid the dose dumping phenomenon.
- the therapeutic levels of clonidine desired to promote a therapeutic effect can be large enough that the efficient loading of an injectable formulation (e.g. a depot carrier such as microspheres) remains a compelling objective.
- an injectable formulation e.g. a depot carrier such as microspheres
- the inventors of the present invention have overcome these obstacles to formulate the long acting depot formulations described herein.
- clonidine selectively stimulates receptors in the brain that monitor catecholamine levels (epinephrine and norepinephrine) in the blood to lower catecholamine production. The result is a lowered heart rate and blood pressure.
- catecholamine levels epinephrine and norepinephrine
- the result is a lowered heart rate and blood pressure.
- medical professionals have begun to expand the use of clonidine to treat disorders other than hypertension, e.g., in the treatment of attention-deficit hyperactivity disorder (ADHD).
- ADHD patients clonidine is often used either alone or in combination with other medications, such as stimulants, to moderate ADHD-associated impulsive and oppositional behavior, or to counteract the side effects of stimulants.
- clonidine formulation that is long acting.
- One such long acting formulation is an injectable depot formulation.
- the inventors have identified a myriad of factors that can impact the success of the depot formulation including, but not limited to, factors that affect the drug release rate, drug loading of the microspheres, and overall formulation stability.
- the rate of drug release can be tuned on the basis of the mole fraction of carboxylic acid terminal groups relative to ester terminal groups in the polymer.
- Zero order release or at least quasi zero order release, is desirable because it demonstrates that the active agent is released at a constant, or approximately constant, rate.
- the present invention is directed to a pharmaceutical depot formulation comprising clonidine or a pharmaceutically acceptable salt or prodrug thereof; the depot formulation after injection or implantation into a human patient providing a mean release rate of clonidine of less than about 5 mg over 24 hours and providing a release rate of at least about 0.1 mg of clonidine per 24 hour period for at least 24 hours, at least 3 to 5 days, at least 7 to 28 days or at least 3 months.
- the present invention is directed to clonidine or a pharmaceutically acceptable salt or prodrug thereof; wherein from about 10% to about 60% clonidine is released after 4 days, from about 20% to about 80% clonidine is released after 8 days and from about 30% to about 95% clonidine is released after 12 days and less than 100% clonidine is released after 16 days when tested in a pH 7.4 buffered solution at 37 0 C and lOOrpm.
- the present invention is directed to clonidine or a pharmaceutically acceptable salt or prodrug thereof; wherein from about 10% to about 60% clonidine is released after 4 days, from about 20% to about 80% clonidine is released after 8 days and from about 30% to about 95% clonidine is released after 12 days and less than 100% clonidine is released after 16 days when tested in a neutral pH buffered solution at 37°C and lOOrpm.
- This invention also overcomes the difficulties in formulating clonidine in controlled release formulations due to the high solubility and potency of the compound.
- the high solubility of clonidine can result in failure of a controlled release system. This can lead to "dose dumping" wherein a portion of, or an entire dose that is intended to be released over an extended period of time is released at a time shorter than intended, e ⁇ g., a faster release or an immediate release. This can result in a plasma level above the therapeutic level and result in increased incidence of adverse events. This is especially an issue with a potent drug such as clonidine where even a minimal "dose dump" can lead to toxic levels of the drug accumulating in the patient.
- clonidine hydrochloride in order to reduce the solubility of the clonidine to assist in obtaining a controlled release depot effect, clonidine is utilized as the free base or utilized in a salt which has relatively lower solubility.
- the present invention can utilize an insoluble salt such as a fatty acid salt.
- Representative fatty acid salts include salts of oleic acid or linoleic acid.
- fatty acid salts with between 8 to 20 carbons are used to produce salts with an aqueous solubility of less than about 2.0 mg/ml, and preferably less than about 1.0 mg/ml, such as clonidine palmeate and clonidine stearate. Most preferably, fatty acid salts with between 12 to 18 carbons are used. Other embodiments can utilize a lipid soluble salt of clonidine.
- the clonidine free base or pharmaceutically acceptable salt thereof can be combined with a complexing agent, e.g., calcium chloride or magnesium chloride, in order to modify the release characteristics of the active agent.
- a complexing agent e.g., calcium chloride or magnesium chloride
- the clonidine free base or pharmaceutically acceptable salt thereof can be combined with a dissolution modifying excipient.
- the clonidine can be combined with the dissolution modifying excipient, ejj., by being dispersed in the excipient, being microencapsulated by the excipient, by being embedded in the excipient (e ⁇ g., in between layers), in intimate association with the excipient, or bonded to the excipient (e ⁇ g., by a covalent bond or by Vander Waals forces).
- a preferred dissolution modifying excipient is a biocompatible polymer.
- biocompatible polymers can be selected from the group consisting of a biodegradable polymer, a non-biodegradable polymer, or a combination thereof.
- the biodegradable polymer can be selected from, ej*., the group consisting of poly(lactide)s, poly(glycolide)s, poly(lactide-co-glycolide)s, poly(lactic acid)s, poly(glycolic acid)s, poly(lactic acid-co-glycolic acid)s, polyanhydrides, polyortho esters, polyetheresters, polycaprolactone, polyesteramides, polyphosphazenes, polysaccharides, proteinaceous polymers, soluble derivatives of polysaccharides, soluble derivatives of proteinaceous polymers, polypeptides, polyesters, and combinations thereof.
- the biodegradable polymer can be selected, e ⁇ g., from the group consisting of polymers having blocked carboxyl end groups, polymers having free carboxyl end groups, or combinations thereof.
- the blocking group can be, ⁇ jg., an alkyl radical.
- the biodegradable polymer has a molecular weight of from about 2,000 Daltons to about 2,000,000 Daltons; from about 5,000 Daltons to about 100,000 Daltons; or from about 20,000 Daltons to about 75,000 Daltons.
- the biodegradable polymer is a poly(lactide-co- glycolide)(PLGA).
- the poly(lactide-co-glycolide) has a lactide:glycolide ratio of about 10: 1 to about 1 : 10; about 5:1 to about 1:5 about 3: 1 to about 1:3; or about 1 : 1.
- the biodegradable polymer is poly (D-, L-, or D, L-lactic acid)(PLA).
- the biodegradable polymer is a proteinaceous polymer
- the polymer can be selected, ejj,, from the group consisting of gelatin, elastin, alkylated collagen, and alkylated elastin.
- the non-biodegradable polymers utilized in the present invention can be, e.g., selected from the group consisting of polyacrylates, polymers of ethylene-vinyl acetates and other acyl substituted cellulose acetates, non-degradable polyurethanes, polystyrenes, polyvinyl chloride, polyvinyl fluoride, poly(vinyl imidazole), chlorosulphonate polyolefms, polyethylene oxide, and combinations thereof.
- the clonidine or pharmaceutically acceptable salt or prodrug thereof preferably in particulate form, is contained in a pharmaceutically acceptable vehicle, with or without a dissolution modifying excipient.
- the vehicle is selected from the group consisting of a lipid, an oil, a wax, and a combination thereof.
- Suitable pharmaceutically acceptable vehicles which can be utilized in the present invention include cottonseed oil, soybean oil, safflower oil, hydrated peanut oil, olive oil, castor oil, tryglyceride mixtures, monoglyceride, silicone oil, isopropylmyristate, ethyloleate, paraffins, glycerol, propylene glycol, polyethylene glycol, medium chain fatty acids, long chain fatty acids or a combination thereof.
- the pharmaceutically acceptable vehicle is selected from the group consisting of water, ethanol, glycerin, polyethylene glycol, propylene glycol and a combination thereof.
- a viscosity increasing agent can be utilized.
- the viscosity increasing agent can also affect the dissolution of the clonidine or pharmaceutically acceptable salt thereof.
- the viscosity increasing agent can be selected, e ⁇ g., from the group consisting of a cellulose derivative, polyvinylpyrrolidone, alginates, chitosan, a dextran, gelatin, polyethylene glycols, polyoxyethylene ethers, polyoxypropylene ethers, polylactides, polyglycolides, polycaprolactones, polyanhydrides, polyamines, polyur ethanes, polyesteramides, polyortho esters, polydioxanones, polyacetals, polycarbonates, polyorthocarbonates, polyphosphazenes, succinates, polycarbonates, poly(maleic acid), poly(amino acids), polyhydroxycellulose, chitin, copolymers or terpolymers of the group consisting of a cellulose derivative
- a gelling agent can also be utilized in the pharmaceutical depot formulations of the present invention.
- the gelling agent can be utilized in formulations utilizing a vehicle, wherein the gelling agent also provides increased viscosity to the formulation.
- the gelling agent can be utilized, e.g., in a dry powder depot formulation such as when the formulation is administered by needleless injection, In such embodiments, the gelling agent will form a gel upon exposure to bodily fluids.
- the particles of clonidine become embedded, encapsulated or dispersed in the gel which controls or further controls the release of the active agent from the depot formulation.
- the gelling agent is selected, e ⁇ g., from the group consisting of polyethylene oxide; hydroxypropyl methylcellulose; polyvinyl alcohol; methyl cellulose; ethylcellulose; hydroxyethyl cellulose; sodium carboxymethyl cellulose; dextran; gelatin; pectin; sodium poly(acrylic acid); carboxymethyl cellulose and combinations thereof.
- the formulation can include dissolution modifying excipient which associates with the drug particles and reduces cohesive forces and reduces agglomeration of the particles.
- the dissolution modifying excipient may promote release, as agglomeration and cohesiveness of the particles can result in a reduction of release to a level that therapeutic plasma levels are not achieved by the pharmaceutical depot formulation.
- a dissolution modifying excipient balances the reduction of the solubility of the clonidine to impede release while at the same time reducing agglomeration and cohesiveness to promote release.
- Such dissolution modifying excipients can be selected, e ⁇ g., from the group consisting of an anionic compound, a cationic compound, a zwitterionic compound, a non-ionic compound, and an ionic compound.
- the dissolution modifying excipient is selected, e.g., from the group consisting of cetyl pyridiniuni chloride, albumin, gelatin, casein, phosphatides, dextran, glycerol, gum acacia, cholesterol, tragacanth, stearic acid, benzalkonium chloride, calcium stearate, glycerol monostearate, cetostearyl alcohol, cetomacrogol emulsifying wax, sorbitan esters, polyoxyethylene alkyl ethers, polyoxyethylene castor oil derivatives, polyoxyethylene sorbitan fatty acid esters, polyethylene glycols, dodecyl trimethyl ammonium bromide, polyoxyethylene stea
- a glucocorticoid can also be included in the pharmaceutical depot formulation to minimize this effect.
- the glucocorticoid can be selected, e.g., from the group consisting of alclometasone, desonide, flumethasone, hydrocortisone and its esters such as hydrocortisone butyrate or hydrocortisone acetate, clobetasone, triamcinolone acetonide, betamethasone, budenoside, desoximethasone, diflorosane, fluocinolone, fluoccinonide acetonide, fluocortolone, fluticasone, methylprednisolone aceponate, mometasone, rofleponide and combinations thereof.
- the pharmaceutical depot formulations of the present invention provide a therapeutic plasma level of clonidine for at least 24 hours, at least 3 days, at least 5 days, at least 7 days, at least 14 days; at least 1 month or at least 3 months.
- the clonidine or pharmaceutically acceptable salt or prodrug thereof is present in the depot formulation in micro- or nano-sized particles, e.g., in the form of particles having an effective average particle size of less than or equal to about 200 ⁇ m. In other preferred embodiments, the effective average particle size is less than or equal to about 2000 nm. In still other preferred embodiments, the effective average particle size is less than or equal to about 50 ⁇ m.
- micro- or nano- sizing of particles is utilized in order to increase the dissolution and bioavailability of poorly soluble drugs. See, Kesisoglou et al., Nanosizing - Oral formulation development and biopharmaceutical evaluation, Advanced Drug Delivery Reviews, 59(2007), 631- 644.
- the present invention provides a depot formulation that minimizes or prevents "dose dumping" given the further increase in the dissolution of the clonidine particles.
- the clonidine or pharmaceutically acceptable salt or prodrug thereof is in the form of particles having an effective average particle size selected from the group consisting of less than or equal to about 100 ⁇ m, less than or equal to about 50 ⁇ m, less than or equal to about 25 ⁇ m. In preferred embodiments, the clonidine or pharmaceutically acceptable salt or prodrug thereof is in the form of particles having an mean effective particle size of from about 5 ⁇ m to about 40 ⁇ m.
- the micro- or nano-sized clonidine of the present invention can be obtained by techniques such as milling or by spray drying. The milling process can be a dry process, e ⁇ g., utilizing a dry roller milling process.
- the milling process can be a wet process wherein the clonidine is dispersed in a liquid such as water, safflower oil, aqueous salt solutions, ethanol, n-butanol, hexane, glycol and the like.
- the dispersing and/or wetting agents can be selected from known organic and inorganic pharmaceutical excipients and can be present in an amount, e ⁇ g., from about 1% to about 80% by weight of the total mixing slurry.
- the grinding media utilized in the micro- or nano-sizing of the particles can be, e.g., rigid media that are preferably spherical or particulate in shape.
- grinding media in the form of other non-spherical shapes can be utilized in micro- or nano-sizing clonidine in the present invention.
- Grinding media utilized in the present invention include but is not limited to stainless steel, titania, agate, glass, alumina, zirconium silicate, and zirconium oxide (optionally stabilized with magnesia or yttrium).
- Polymeric materials such as polymeric resins can also be utilized as grinding materials.
- contamination from the media can be metabolized in vivo into biologically acceptable products which can be eliminated from the body.
- the grinding can take place in any suitable grinding mill such as an airjet mill, a roller mill, a ball mill, an attritor mill, a vibratory mill, a planetary mill, a sand mill and a bead mill.
- a high energy media mill is preferred when small particles are desired.
- the mill can contain a rotating shaft.
- the grinding media is separated from the milled clonidine particulate product (in either a dry or liquid dispersion form) using techniques such as by filtration, sieving through a mesh screen, and the like.
- a solution of clonidine in a solvent can be sprayed into an empty chamber under conditions that allow for a substantial amount of the solvent to be removed from the solution, such that nanometer-sized clonidine particles are formed.
- a clonidine solution is sprayed onto a fluidized bed of carrier particles.
- the bed is maintained at a temperature, e ⁇ g., from about 20° C to about 80° C or from about 25° C to about 50° C.
- This embodiment results in micro- or nano- particles of clonidine formed in a mixture with carrier particles.
- a carrier is mixed with the clonidine solution to be spray dried. This mixture is sprayed into a heated chamber and the nanoparticles formed in this process contain a mixture of clonidine and carrier.
- the carrier particles include, but are not limited to, saccharides, such as sugars and sugar alcohols (e ⁇ g., lactose, sucrose, mannitol, or sorbitol), starches, flour, cellulose preparations and/or salts such as carbonates, bicarbonates and phosphates (e.g., tricalcium phosphate or calcium hydrogen phosphate).
- saccharides such as sugars and sugar alcohols (e ⁇ g., lactose, sucrose, mannitol, or sorbitol)
- starches e.g., lactose, sucrose, mannitol, or sorbitol
- flour e.g., cellulose preparations and/or salts
- carbonates e.g., bicarbonates and phosphates (e.g., tricalcium phosphate or calcium hydrogen phosphate).
- the pharmaceutical depot formulation of the present invention can be suitable for administration via implantation or injection (e ⁇ g., intravenously, intramuscularly or subcutaneously) to the patient.
- Injection of the pharmaceutical depot formulation may be through a syringe with any size and shaped needle capable of administering the formulation without resulting in clogging of the formulation.
- the pharmaceutical depot formulation may be incorporated into a suitable liquid or gel composition for administration or may be in the form of microspheres, nanospheres, etc. Examples of apparatus and asceptic procedures useful for the formation of sterile microspheres are described, e.g., in U.S. Patent Nos. 5,945,126; 6,270,802; and 6,361,798, the disclosures of which are hereby incorporated by reference.
- a needleless injector may be used to inject the depot formulation. Needless injections use gas pressure to deliver the pharmaceutical depot formulation, which is typically in powder form.
- Needleless injections including apparatus and types of formulations useful therefore, are described, e.g., in U.S. Pat. Nos. 6,053,889; 6,013,050; 6,010,478; 6,004,286; and 5,899,880, the disclosures of which are hereby incorporated by reference.
- the embodiments of the invention utilizing needleless injection may result in improved compliance as non-compliance in many patients is due to anxiety associated with conventional needle injection. This may be especially true in pediatric patients (e.g., pediatric patients with ADHD).
- the present invention is directed to a clonidine prodrug of the formula:
- R represents an R moiety bound to any suitable location on the clonidine structure, ejj., on any nitrogen or carbon within the ring structures or on the nitrogen linking the two ring structures.
- the R moiety is R is selected from the group consisting of a formate radical of the formula COO ' ; a formate radical of the formula R 1 COO " , wherein R 1 is an alkylene having from 1-30 carbons, 5-25 carbons 10-20 carbons or 16-18 carbons; a moiety selected from the group consisting of polyethylene glycol fatty acid esters, fatty alkylol amide condensates, alkylarylsulfonates, fatty alcohol sulfates, dialkyl esters of sodium sulfosuccinate, fatty acid esters of sodium isothionate, polyoxyethylene ethers and thioethers, and long-chain quaternary ammonium chloride compounds; and a moiety selected from the group consisting of an amino acid, a dipeptide and a trip ep tide.
- the amino acid can be natural or synthetic.
- Amino acid moieties suitable for preparing the prodrugs of the present invention include valine, leucine, isoleucine, methionine, phenylalanine, asparagine, glutamic acid, glutamine, histidine, lysine, arginine, aspartic acid, glycine, alanine, serine, threonine, tyrosine, tryptophan, cysteine and proline.
- the amino acids can be D or L-amino acids; and can be alpha, beta, or gamma amino acids.
- amino acids can be gamma-carboxyglutamic acid, selenocysteine, desmosine, 6-N-methyllysine, epsilon-N,N,N-trimethyllysine, 3-methylhistidine, O- phosphoserine, 5-hydroxylysine, epsilon-N-acetyllysine, omega.-N-methylarginine, N- acetylserine, gamma-aminobutyric acid, citrulline, ornithine, azaserine, homocysteine, beta-cyanoalanine and S-adenosylmethionine.
- Synthetic amino acids include phenyl glycine, meta-tyrosine, para-amino phenylalanine, 3-(3-pyridyl)-L-alanine, and 4-(trifluoromethyl)-D-phenylalanine.
- R is selected from the group consisting of an aldehyde (- CHO), a ketone (-COR 2 ), a carboxylic acid (-COOH), an ester (-COOR 2 ), an amide (- CONR 2 R 3 ), an enone (-C(O)C(R 2 )CR 3 R 4 ), an acyl chloride (-C0C1) or an acid anhydride (-COOCOR ).
- R , R and R are independently selected from the group consisting of alkyl, alkenyl, aryl, heteroaryl, alkoxy, aryloxy and heteroaryloxy moieties wherein the alkyl moiety is selected from the group consisting of unsubstituted or substituted, straight- chain, branched-chain and cyclic alkyl moieties having 1-20 carbon atoms; wherein the alkenyl moiety is selected from the group consisting of unsubstituted and substituted, straight-chain, branched-chain and cyclic alkenyl moieties having 2-20 carbon atoms; wherein the aryl moiety is selected from the group consisting of unsubstituted and substituted phenyl, and phenalkyl moieties wherein the alkyl moiety contains 1-3 carbon atoms and the phenyl moiety is unsubstituted or substituted; and the heteroaryl moiety is a substituted or unsubstituted aromatic 5-
- Suitable straight-chain alkyl moieties include methyl, ethyl, propyl, butyl, hexyl, heptyl, octyl, dodecyl and palnityl moieties.
- branched-chain alkyl moieties examples include isopropyl, sec-butyl, t-butyl, 2-methylbutyl, 2-pentyl and 3-pentyl moieties.
- Suitable cyclic alkyl moieties include cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl moieties.
- alkenyl moieties examples include vinyl (ethenyl), 1-propenyl, i- butenyl, pentenyl, hexenyl, n-decenyl and c-pentenyl.
- the moieties may be substituted, generally with 1 or 2 substituents, wherein the substituents are independently selected from halo, hydroxy, alkoxy(alkoxy)x, hydroxyalkoxy(alkoxy)x, amino, mono- and dialkylamino, nitro, carboxyl, alkoxycarbonyl, and cyano moieties, wherein x is an integer from 0 to 3 and the alkoxy moieties contain from 1 to 5 carbon atoms.
- phenalkyl moieties wherein the alkyl moiety contains 1-3 carbon atoms includes benzyl, phenethyl and phenylpropyl moieties wherein the phenyl moiety may be substituted. When substituted, the phenyl moiety of the phenalkyl group may contain 1-3 substituents independently selected from alkyl, hydroxy, alkoxy, halo, amino, mono- and dialkylamino, nitro, carboxyl, alkoxycarbonyl and cyano moieties.
- heteroaryl moieties examples include pyridinyl, thienyl and imidazolyl.
- halo includes F, Cl, Br, and I.
- prodrugs of clonidine disclosed herein can be utilized in the depot formulations of the present invention or can be included in other pharmaceutical formulations, e ⁇ g., oral, intranasal, pulmonary, and transdermal formulations.
- the present invention is directed to a fatty acid salt of clonidine, e ⁇ g., fatty acid salts having greater than 2 carbons, greater than 8 carbons or greater than 16 carbons, such as butyric, caprioc, caprylic, capric, lauric, mystiric, palmitic, stearic, arachidic and the like.
- Representative fatty acid salts of the present invention include salts of oleic acid or linoleic acid.
- fatty acid salts with between 8 to 20 carbons are used to produce salts with an aqueous solubility of less than about 2.0 mg/ml, and preferably less than about 1.0 mg/ml, such as clonidine palmeate and clonidine stearate.
- fatty acid salts with between 12 to 18 carbons are used.
- Other embodiments can utilize a lipid soluble salt of clonidine.
- fatty acid and lipid salts of clonidine disclosed herein can be utilized in the depot formulations of the present invention or can be included in other pharmaceutical formulations, ej*., oral, intranasal, pulmonary, and transdermal formulations. While many of the embodiments disclosed herein reference clonidine, other alpha- 2 -adrenergic agonists can be utilized therein. Such agents include tizanidine, guanfacine, guanabenz, guanoxabenz, guanethidine and pharmaceutically acceptable salts thereof.
- the actual weight (mg) per dose depends on the actual loading of clonidine in microspheres. The weight per dose was therefore adjusted accordingly.
- Microspheres can be fabricated using the following procedures:
- step #5 Wash the microspheres from step #5 and collect by sieving the particles through sieves mesh #40 (424 ⁇ m), #60 (250 ⁇ m), #80 (180 ⁇ m), #100 (150 ⁇ m), #140 (107 ⁇ m), #270 (53 ⁇ m), and #400 (38 ⁇ m). The remainder of the collected filtrate will be centrifuged to retrieve microspheres of diameter less than 38 ⁇ m.
- microspheres Weigh microspheres and, according to the loading of the clonidine in the microspheres, extract sample sufficient for a weekly dose, then re-suspend in normal saline solution or phosphate buffer pH 7.4 for injection.
- microspheres are suspended in 0.1% aqueous Tween 80 solution and sonicated for 30 seconds and stirred for 2 minutes before analysis.
- Size distribution was analyzed by Malvern or dynamic light scattering particle size analyses. The particle size distributions are reported on a volume basis.
- microspheres are spread on the microscope slides and sizes of the microspheres are measured. The population or number distribution is converted to a volume distribution. Determination of drug loading in PLGA microspheres
- Encapsulation efficiency (%) mass of drug in microspheres x 100 / initial mass of drug used
- a 50 mg sample in this case the microsphere formulation, was weighed and transferred to a 15 mL centrifuge tube containing 10 niL 50 mM phosphate buffer pH 7.4 with addition of 0.1%v/v Tween80. Samples were incubated in Brunswick Scientific Water Bath Shaker at 100 rpm at 37 °C for a specified time. At each specified time point, the microspheres were swirled in the medium and centrifuged as necessary. 2 mL of sample was pipetted for HPLC analyses. 2 mL of fresh 50 mM phosphate buffer pH 7.4 was added to the centrifuge tubes and shaken gently to re-suspend the microspheres. The tubes were placed back in the 37°C water bath for further incubation.
- the particle size of clonidine base was reduced by probe sonication to the average mean size distribution of 3 to 10 ⁇ m utilizing Branson Ultrasonics Sonifier Cell Disrupters 400W at 50% power intensity with 1.5 cm probe for 30 minutes.
- aqueous containing l%w/w Tween 80 400mg of the micronized clonidine base was suspended in 600 mg of aqueous containing l%w/w Tween 80 (or without Tween 80) and the mixture was vortexed to form clonidine suspensions.
- the clonidine suspensions were homogenized with the PLGA solution for 3 minutes at 15,000 RPM to form the first wj/o emulsion.
- Microspheres were collected by sieving through sieves mesh #100 (150 ⁇ m) and #400 (38 ⁇ m). Microspheres size less than 38 ⁇ m were collected by centrifugation at -1200 RPM for 5 minutes. All microspheres were washed with deionized water. After washing, the microspheres were freeze dried.
- Figure 2 shows the micrographs of dried microspheres acquired by light microscope. Micrograph (A) was acquired with objective 20 x for microspheres ranged from 150 to 38 ⁇ m and micrographs (B) was acquired with objective 40x for microspheres less than 38 ⁇ m.
- the actual loading of the microspheres is about 7%w/w.
- FIG. 4 shows the dissolution profiles of clonidine released from copolymer PLGA 50:50 and PLGA 75:25 that were fabricated by the double emulsion evaporation method.
- the actual loading of clonidine in the microspheres made of PLGA 50:50 is 8%w/w
- PLGA 75:25 size range 150 to 38 ⁇ m
- PLGA75:25 average size 22 ⁇ m
- the encapsulation of clonidine base was prepared as described in Example 2 using different ratio of PLGA copolymer. Table 3 below shows the ratio of PLA to PGA and their respective inherent viscosity.
- Ester Figure 6 shows the release profiles of clonidine from PLGA 50:50, 65:35, and 75:25 microspheres.
- the processing parameters of all three batches were kept constant for three different ratio of PLA to PGA copolymers. All of these batches were homogenized at 13000 RPM for 1 minute to form the first emulsion and vortexed at 3000 RPM for 30 seconds to form second emulsions.
- the higher the mole fraction of glycolic acid the faster the degradation of the microspheres and therefore faster release of clonidine from the PLGA 50:50 microspheres compared to 65:25 and 75:25 microspheres.
- Figure 16 compares the release of clonidine from two formulations of PLGA 50:50 (0.82 dL/g in HFIP) microspheres prepared according to the double emulsion process described in examples 2 and 3 above.
- the ratio of the inner water phase to inner organic phase is changed from a 1:7 to a 1:10 and the effect is to increase the drug release rate.
- Figure 18 compares the dissolution rate of clonidine microspheres prepared according to the double emulsion process described in examples 2 and 3 above where the particle size of the clonidine active agent is different.
- the large-particle drug sample leads to faster dissolution than the small-particle drug formulation.
- Different batches of clonidine microspheres were manufactured having different microsphere particle size.
- the batch with average particle size equal to about 250 microns showed drug loading of about 13% w/w.
- the batch with particle sizes ranging from 38 to 160 microns showed drug loading of about eight percent, and the batch with particle sizes less than 38 microns showed drug loading of about three percent.
- microspheres were prepared with different of clonidine to PLGA ratio.
- the ratio of clonidine to polymer tested were 1: 1, 1 :2, 2,5:400, 3:4, 4:4, and 4:5.
- PLGA used was PLGA 75:25 with ester terminals and the inherent viscosity was 0.55- 0.75 dL/g.
- Drug and copolymer were dissolved in methylene chloride and homogenized at 13000 rpm in 5%w/w PVA (MW ⁇ 25K) solution for 1 to 2 minutes followed by vortexing for 1 minute.
- the emulsion was transferred into a 0.01%w/w of Tween 80 and 5%w/w of NaCl in 2% of PVA solution stirring at ⁇ 600 RPM for 5 to 12 hours.
- Sodium chloride helps to prevent mutual aggregation of microspheres and osmotic stress during solvent evaporation process.
- the microspheres were collected by vacuum filtration and centrifugation processes.
- Figure 7 shows the effect of drug loading with different reagent levels of clonidine and PLGA and thus different clonidine to PLGA copolymer ratios.
- There is a significant drug loading effect that ranged from 6% to 31% w/w with different drug-to- polymer ratios and for microspheres having size range from 149 to 38 ⁇ m.
- Microspheres smaller than 38 ⁇ m loading remained in the range of 3-6% w/w loading.
- Figure 8 shows that the dissolution release of clonidine from PLGA 75:25 (ester terminated groups) microspheres are affected by the drug to polymer ratio of the reagents during the formulation process.
- the burst release is reduced with the O/W system where the drug to polymer ratio is lower.
- co-solvent in the system increases the solubility of drug and polymer in organic phase.
- the co-solvent enhances the homogeneity of drug molecules dispersion in the microspheres.
- the microspheres were prepared following the procedure outlined in Example 4 and the co-solvent was added to the methylene chloride. For example, add 2%v/v of methanol into 98% v/v of methylene chloride and then dissolve the PLGA as described in Example 4. Results of clonidine release are shown in Figure 10.
- IPA is a non-aqueous solution to extract acetonitrile from polymer solution and to wash mineral oil from the surface of microsphere. Hexane can be used as alternative non-aqueous washing solution.
- the microspheres are harvested by rinsing with IPA using vacuum filtration unit and dried under vacuum for an hour. O/O emulsion method could produce more porous microspheres in a short period of time.
- a water/oil/water double emulsion (Formula A) and a water/oil and co-solvent single emulsion (Formula B) were prepared in accordance with Examples 3 and 4, respectively and their dissolutions were compared.
- the specific formulations are provided in the Tables below.
- Figure 11 compares the cumulative dissolution rate of the microsphere formulations A and B with an Alzet pump
- Figure 12 compares their respective release rates on a non-cumulative basis.
- the figures show that at least one of the formulations achieves a substantially zero order release rate as reflected by the Alzet pump data.
- the representations also demonstrate that the formulations are capable of releasing a therapeutically effective amount of active agent over an extended period of time.
- the samples were removed from Brunswick Scientific Water Bath Shaker and centrifuged for 5 minutes at 3000rpm. 2ml of sample was pipetted out of centrifuge tube and collect in a scintillation vial. 2ml of fresh Phosphate Buffer pH7.4 was then pipetted into the centrifuge tube containing the sample. The centrifuge tube samples were returned to the Brunswick Scientific Water Bath Shaker for further sampling. Samples remaining in scintillation vials were filtered through a glass acrodisc filter, and the aliquot was collected for HPLC analysis,
- Preparations of the salts involved dissolving a 1 : 1 molar equivalent of clonidine and the desired fatty acid into an organic solvent.
- Ig of clonidine was dissolved with 1.2g linoleic acid, l.lg palmitic acid, and 1.2g stearic acid in their respective containers into isopropanol.
- Other organic solvents were used such as methanol, ethanol, and chloroform and yielded similar if not the same results.
- Isopropanol was used for the rest of the study taking into consideration future manufacturing costs and the lower cost of isopropanol compared to the other solvents.
- the mixture was stirred until the clonidine and fatty acids were completely dissolved. The mixture was then allowed to air dry to evaporate the solvent and the solid waxy residue that remains was collected, mixed/crushed for uniformity, and allowed to air dry.
- the samples were then analyzed via DSC to observe a change in melting point and HPLC to record the solubility.
- Micronized clonidine base was suspended in double distilled water and the mixture was vortexed at 3000 rpm for 10 sec to disperse the drug.
- Polymer solution was prepared by dissolving 600 mg PLGA 50/50 (i.v. 0.26 - 0.54 dl/g, end capped) in 1 mL of methylene chloride. Polymer solution was added into above drug suspension and homogenized at 13,000 RPM for 1 minute to form the first emulsion. Then 1 mL of 2% (w/w) polyvinyl alcohol (PVA, 25 kD) solution was added.
- PVA polyvinyl alcohol
- the emulsion was added into 100 ml of 0.5% (w/w) PVA (25 kD) solution containing 2% (w/w) NaCl under stirring at ⁇ 600 rpm, and hardened for 4.5 hr at room temperature.
- the hardened microspheres were sieved through sieves of 106, 53 and 25 um sizes and washed by double-distilled water three times before further collection by vacuum filtration.
- the collected microspheres were then placed into a freeze-drier (Vertis Genesis 25 LE) and dried for 2 days after freezing the microspheres overnight.
- the size range of the microspheres collected was 53-106 um, and the encapsulation efficiency was measured to be 74%,
- the in vitro dissolution profile is shown in Figure 13 and demonstrates that formulations of the present invention can release therapeutically effective amounts of clonidine for extended periods of time.
- Clonidine base was dissolved in methylene chloride, and 250 mg of PLGA 50/50 (i.v. 0.26 - 0.54 dl/g, end-capped) was added and the mixture was vortexed until polymer dissolved completely. 300 mg of PLGA 85/15 (i.v. 0.55 - 0.75 dl/g, end-capped) was dissolved separately in 1 ml of methylene chloride. Polymer solution of PLGA 50/50 containing drug was then added into PLGA 85/15 solution and homogenized at 19,000 rpm for 30 sec. Then 1.4 niL of 2% (w/w) polyvinyl alcohol (PVA, 25 kD) solution was added.
- PVA polyvinyl alcohol
- the emulsion was added into 140 ml of 0.5% (w/w) PVA (25 kD) solution containing 2% (w/w) NaCl under stirring at ⁇ 700 rpm, and hardened for 4.5 hr at room temperature.
- the hardened microspheres were sieved through sieves of 106, 53 and 25 Dm sizes, and microspheres of 53-106 Dm and 25-53 Dm were collected and washed by double-distilled water three times before further collection under vacuum filtration.
- the collected microspheres were then placed into a freeze-drier (Vertis Genesis 25 LE) and dried for 2 days after freezing the microspheres overnight.
- the encapsulation efficiency was measured to be 57% for microspheres of 53- 106 Dm and 51% for microspheres of 25-53 Dm.
- the in-vitro dissolution profile is shown in Figure 14. The results indicate that as microsphere particle size increases, the release rate of the active agent decreases.
- Figure 15 presents the in-vitro release for specific batches of Example 11 and shows the substantially zero order release profile and extended release profile of depot formulations according to the present invention
Landscapes
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Public Health (AREA)
- Life Sciences & Earth Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Dermatology (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
In certain embodiments the present invention is directed to a pharmaceutical depot formulation comprising an active agent, such as an alpha-2-adrenergic agonist, that can release an effective amount of the active agent over an extended period of time.
Description
DEPOT FORMULATIONS
PRIORITY
This application claims priority to U.S. Provisional Patent Application No. 61/027,333, filed February 8, 2008, which is hereby incorporated by reference in its entirety.
FIELD OF THE INVENTION
The present invention is directed to pharmaceutical depot formulations of basic active agents (e.g. clonidine) or pharmaceutically acceptable salts or prodrugs thereof, which release therapeutic levels of the active agent for at least 24 hours, preferably at least 3 days to about 5 days, more preferably from about 7 days to about 28 days, and most preferably for at least 3 months.
BACKGROUND
Clonidine is an imidazoline derivative that exhibits alpha-2-adrenergic agonist activity and is used primarily for the treatment of systemic hypertension. Clonidine lowers blood pressure by activation of alpha-2 -receptors in the cardiovascular control centers of the CNS which suppresses the outflow of sympathetic nervous system activity from the brain. Clonidine also exhibits analgesic effects which are mediated by pre- and possibly postsynaptic alpha-2 -adrenergic receptors that block nociceptive transmission. Other known uses for clonidine include therapy for opioid detoxification, nicotine detoxification, sleep hyperhydrosis, insomnia, menopausal symptoms, ADHD, and Tourette's syndrome.
Among other physical and chemical properties, clonidine is highly soluble and penetrates the blood brain barrier and the placenta. It is believed that that binding of clonidine to receptors is highest in the rostral venterolateral medulla in the brain stem (the final common pathway for sympathetic outflow) where it activates inhibitory neurons.
The overall effect is to decrease sympathetic activity, to enhance parasympathetic tone, and to reduce circulating catecholamines.
Clonidine is commercially available as tablets in 0.1 -mg, 0.2-mg, and 0.3-mg strengths under the tradename Catapres® by Boehringer Ingelheim. Clonidine is also available as a transdermal patch in 0.1-mg, 0.2-mg, and 0.3-mg under the tradename Catapres-TTS® by Boehringer Ingelheim. When administered orally, clonidine is almost completely absorbed from the gastrointestinal tract, but it is subject to the first pass effect. The biological half-life ranges from about four to six hours after oral administration, with wide interpatient variability.
The most common side effect associated with clonidine is dizziness associated with orthostatic hypotension. Other adverse effects of clonidine can include dry mouth, constipation, nausea, daytime sleepiness, weakness, and lethargy, eye dryness and loss of libido. These side effects may be potentiated by the currently available immediate release tablets which typically exhibit a large peak to trough ratio given the typical TID (three times daily) or QID (four times daily) therapy. Further, given the short half-life of the drug, plasma levels could fall below the therapeutic range quickly in the event that a patient is late with the administration of a dose.
The transdermal patch of clonidine would be expected to address some of the problems related to oral clonidine therapy. However, the transdermal patch has its own issues with respect to adverse effects and patient compliance.
Clonidine transdermal patches exhibit contact dermatitis in many patients. Clonidine patches can also exhibit problems with adherence to the skin under certain conditions such as elevated humidity, sweating, showering, swimming, etc. This can also result in plasma levels falling below therapeutic levels in the event that a patient is unable to immediately reapply a new patch, resulting in a shortened duration of efficacy.
Prior to the invention disclosed herein, the art does not contain or suggest a dosage form that can effectively provide therapeutic levels of an alpha-2-adrenergic agonist (e^g., clonidine) for an extended period of time, which can address the issues (ej*., compliance issues, plasma fluctuations) commonly associated with the commercially available immediate release tablets or patches. Such a dosage form would
need to take into account the high solubility of clonidine to assure that the dosage form does not "dose dump" and provide plasma levels which could potentially be toxic.
OBJECTS A]NfD SUMMARY OF THE INVENTION
It is an object of the present invention to provide a pharmaceutical depot formulation containing a basic active agent that is suitable for injection or implantation into a human patient.
It is an object of the present invention to provide a pharmaceutical depot formulation containing an alpha-2-adrenergic agonist that is suitable for injection or implantation into a human patient.
It is an object of certain embodiments of the present invention to provide a pharmaceutical depot formulation containing an alpha-2-adrenergic agonist, such as clonidine, that can release an effective amount of the alpha-2-adrenergic agonist for at least 24 hours, at least 3 to 5 days, at least 7 to 28 days or at least 3 months.
It is an object of certain embodiments of the present invention to provide a pharmaceutical depot formulation containing an alpha-2-adrenergic agonist, such as clonidine, that can provide a substantially zero order release profile for at least about one day, or at least about five days, or at least about seven days, or at least about fourteen days, or at least about 28 days, or at least about 90 days.
It is an object of certain embodiments of the present invention to provide a pharmaceutical depot formulation containing an alpha-2-adrenergic agonist, such as clonidine, that can release an initial burst of the alpha-2 -adrenergic agonist and a substantially uniform release thereafter.
It is an object of certain embodiments of the present invention to provide a pharmaceutical depot formulation containing an alpha-2 -adrenergic agonist, such as clonidine, that can provide therapeutic levels of the alpha-2-adrenergic agonist over extended periods of time.
It is an object of the present invention to provide novel salts of clonidine which exhibit modified absorption characteristics as compared to clonidine base.
It is an object of the present invention to provide novel prodrugs of clonidine which exhibit modified absorption characteristics as compared to clonidine base.
The above objects and others can be achieved by the present invention which, in certain embodiments, is directed to a pharmaceutical depot formulation comprising clonidine or a pharmaceutically acceptable salt or prodrug thereof; the depot formulation providing a mean release rate of clonidine of less than about 5 mg over 24 hours and providing a release rate of at least about 0.1 mg of clonidine per 24 hour period for at least 24 hours, at least 3 to 5 days, at least 7 to 28 days or at least 3 months.
In certain embodiments, the invention is directed to a pharmaceutical depot formulation comprising clonidine or a pharmaceutically acceptable salt or prodrug thereof and a dissolution modifying excipient; the depot formulation providing a mean release rate of clonidine of less than about 5 mg over 24 hours and providing a release rate of at least about 0.1 mg of clonidine per 24 hour period for at least 24 hours, at least 3 to 5 days, at least 7 to 28 days or at least 3 months.
In certain embodiments, the invention is directed to a pharmaceutical depot formulation comprising a therapeutically effective amount of clonidine or a pharmaceutically acceptable salt or prodrug thereof, a dissolution modifying excipient and a pharmaceutically acceptable vehicle; the depot formulation after injection or implantation into a human patient providing a mean release rate of clonidine of less than about 5 mg over 24 hours and providing a release rate of at least about 0.1 mg of clonidine per 24 hour period for at least 24 hours, at least 3 to 5 days, at least 7 to 28 days or at least 3 months.
In certain embodiments, the depot formulation of the present invention provides a release rate of at least about 0.1 mg of clonidine per 24 hour period for at least 24 hours, at least 3 days, at least 5 days, at least 7 days; at least 14 days; at least 1 month; or at least 3 months.
In certain embodiments, the invention is directed to a pharmaceutical depot formulation comprising an active pharmaceutical ingredient in base form which is at least sparingly soluble in water; and a biocompatible polymer, wherein the depot formulation releases an amount of active pharmaceutical ingredient during the first twenty-four hour
period that is approximately equal to, but no more than about three times the amount of active pharmaceutical ingredient released during the seventh twenty- four hour period.
In certain embodiment, the invention is directed to a pharmaceutical depot formulation comprising clonidine base, salt or prodrug thereof; a biocompatible polymer, and an optional hydrophobic acid, wherein the depot formulation exhibits an in-vitro clonidine release profile comprising a first amount of clonidine released during a first twenty- four hour period of the profile, and a second amount of clonidine released during a second twenty-four hour period of the profile, wherein the second twenty-four hour period occurs at least six days after the first twenty-four hour period.
In certain embodiment, the invention is directed to a pharmaceutical depot formulation comprising clonidine base, salt or prodrug thereof; a biocompatible polymer, and an optional carboxylic acid, preferably acetic acid, wherein the depot formulation exhibits an in-vitro clonidine release profile comprising a first amount of clonidine released during a first twenty-four hour period of the profile, and a second amount of clonidine released during a second twenty-four hour period of the profile, wherein the second twenty-four hour period occurs at least six days after the first twenty-four hour period.
In certain embodiments, the present invention is directed to a method of preparing a pharmaceutical depot formulation as disclosed herein by a contacting an alpha-2- adrenergic agonist (ej*., clonidine or a pharmaceutically acceptable salt or prodrug thereof) with an excipient to retard the release of the clonidine, and incorporating the resultant mixture into a pharmaceutically acceptable vehicle suitable for injection.
In certain embodiments, the present invention is directed to a method of preparing a pharmaceutical depot formulation as disclosed herein by incorporating an insoluble salt of an alpha-2-adrenergic agonist (e^g., clonidine), a fatty acid salt thereof, or a prodrug thereof, into a pharmaceutically acceptable vehicle suitable for injection.
For purposes of the present invention, the term "clonidine" means clonidine free base as well as any salt, hydrate, solvate, polymorph, or derivative thereof.
The term "alpha-2-adrenergic agonist" means an agent that is capable of eliciting a therapeutic response by agonizing an alpha-2-adrenergic receptor.
The term "prodrug" means a compound which is converted to a therapeutically active compound after administration. A prodrug is typically inactive or less active than the therapeutically active compound to which it is converted.
The term "pharmaceutically acceptable salt" means salts prepared from pharmaceutically acceptable non-toxic bases or acids including inorganic or organic bases, inorganic or organic acids and fatty acids. Salts derived from inorganic bases include aluminum, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, manganic salts, manganous, potassium, sodium, zinc, and the like. Salts derived from pharmaceutically acceptable organic non-toxic bases include salts of primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines, and basic ion exchange resins, such as arginine, betaine, caffeine, choline, N,N'-dibenzylethylenediamine, diethylamine, 2-diethylaminoethanol, 2- dimethylaminoethanol, ethanolamine, ethylenediamine, N-ethyl-morpholine, N- ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethyl amine, tripropylamine, tromethamine, and the like. When the compound of the present invention is basic, salts may be prepared from pharmaceutically acceptable non-toxic acids, including inorganic and organic acids. Such acids include acetic, benzenesulfonic, benzoic, camphorsulfonic, citric, ethanesulfonic, formic, fumaric, gluconic, glutamic, hydrobromic, hydrochloric, isethionic, lactic, maleic, malic, mandelic, methanesulfonic, malonic, mucic, nitric, pamoic, pantothenic, phosphoric, propionic, succinic, sulfuric, tartaric, p-toluenesulfonic acid, trifluoroacetic acid, and the like. Fatty acid salts may also be used, e^g., fatty acid salts having greater than 2 carbons, greater than 8 carbons or greater than 16 carbons, such as butyric, caprioc, caprylic, capric, lauric, mystiric, palmitic, stearic, arachidic and the like.
The term "depot" means a formulation that is suitable to be administered to a patient parenterally or by implantation that can release the alpha-2 -adrenergic agonist (e.g., clonidine) over an extended period of time to provide a therapeutic effect. The term "depot" does not include a transdermal patch or intrathecal administration (e^g., epidural).
The term "biodegradable" means that the excipient will degrade or erode in vivo to form smaller chemical species. Degradation can result, for example, by enzymatic, chemical, and/or physical processes.
The term "therapeutic plasma level" means that plasma levels are achieved that are within the therapeutic window, i^e,, above the minimum therapeutic level and below the toxic level.
The term "sustained release" means that the active agent is released from the depot formulation of the present invention over an extended period of time (e.g., at least 24 hours, at least 3 days, at least 5 days, at least 7 days; at least 14 days; at least 1 month or at least 3 months) as compared to an immediate release formulation.
The term "substantially zero order release" means that the release rate of active agent from the depot formulation at one time is not substantially different as compared to the release rate of active agent from the depot formulation at another time. For purposes of this definition, a selected time would not include a lag time of active agent release that may be present upon initiation of therapy.
The term "effective average particle size" means that 50% of the particle volume distribution falls below the designated measurement when measured by, for example, sieves, sedimentation field flow fractionation, photon correlation spectroscopy, light scattering, disk centrifugation, and other techniques known to those of skill in the art. The particle sizes distributions and values reported herein are deduced from sieves, a Malvern instrument, and visual microscopy (converted to volume) depending on, and made clear within, the context.
BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1 depicts a graph of the particle size distribution for clonidine in Example 2. Figure 2 depicts the micrographs of lyophilized microspheres acquired by light microscope in Example 2.
Figure 3 depicts a graph of the mean size distribution of clonidine encapsulated microspheres in Example 2.
Figure 4 depicts the substantially zero order clonidine release profile from copolymer formulations comprising PLGA 50:50 and PLGA 75:25 as fabricated utilizing a double emulsion evaporation method.
Figure 5 depicts the release rate profile for two formulations made according to
Example 3 and differing only by the molecular weight of the biodegradable polymer utilized in the formulation.
Figure 6 depicts a graph of the release profiles from PLGA microspheres made from
PLA % mole ration 50:50; 65:35 and 75:25 with inherent viscosity 0.55-0.75 dL/g from
Example 3.
Figure 7 compares drug loading as a function of reagent drug-to-PLGA-polymer ratio for formulations made according to Example 4.
Figure 8 depicts how the percent of clonidine released from microspheres of Example 4 is affected by the drug-to-polymer-ratio during the formulation process.
Figure 9 depicts a graph of the dissolution profiles of clonidine release from PLGA microspheres with different properties from Example 4.
Figure 10 depicts a graph of the clonidine release profiles from PLGA microspheres fabricated by the single emulsion co-solvent method of Example 5 and the effect of solvent / co-solvent composition thereon.
Figures 11 compares the cumulative dissolution rate of the microsphere formulations A and B from Example 7 with an Alzet pump.
Figure 12 compares release rates of formulations A and B from Example 7 with an Alzet pump on a non-cumulative basis.
Figure 13 depicts the in vitro dissolution profile of Example 9.
Figure 14 depicts the in vitro dissolution profile of samples from Example 10 and demonstrates the affect of particle size on release rate.
Figure 15 depicts a graph of the in vitro dissolution profile batches from Example 11.
Figure 16 depicts the rate of drug release from formulations prepared according to
Example 3 with differing ratios of inner water phase to inner organic phase.
Figure 17 depicts the rate of drug release from formulations prepared according to
Example 3 with differing terminal groups (ester or carboxylic acid).
Figure 18 depicts the dissolution profile for clonidine microspheres prepared according to Example 2 and containing clonidine having differing particle size distributions. Figure 19 depicts the rate of clonidine release based on microsphere homogenization time (and otherwise prepared according to Example 3).
Figure 20 depicts the rate of clonidine release based on microsphere vortex time (and otherwise prepared according to Example 3).
DETAILED DESCRIPTION
When contemplating the viability of depot formulations, the physical and chemical properties of the active agent must be fully considered and accounted for. In many cases, one of the more important features of the active ingredient is its solubility. If the solubility of the active ingredient is relatively high, then the formulation can have a tendency to dose dump either as a direct result of the solubility, or due to an unusually high "apparent" solubility, wherein the release rate (and hence metabolism) may be affected by external factors. In those cases where the therapeutically effective range is low relative to the actual or apparent solubility, the effect of a dose dump may not only cause causing untimely or premature release, but may also lead to toxic concentrations of the drug in the recipient.
On the other hand, and somewhat in contrast, a non-trivial subset of the more soluble active ingredients exhibit therapeutic dose ranges that are not only sufficiently low to raise toxicity concerns, but sufficiently high that efficient and substantial drug loading in the carrier is important to remediate other safety, comfort and compliance concerns.
One example of such an active pharmaceutical agent is clonidine. Clonidine is highly soluble (clonidine base has a solubility of about 1.8 mg/mL in water under nominal experimental conditions), and as such is susceptible to the dose dump phenomenon. Therefore, it is advantageous to provide a therapeutic dose of clonidine that is both low enough and consistently low enough to reduce or avoid the dose dumping phenomenon. The therapeutic levels of clonidine desired to promote a therapeutic effect,
however, can be large enough that the efficient loading of an injectable formulation (e.g. a depot carrier such as microspheres) remains a compelling objective.
The inventors of the present invention have overcome these obstacles to formulate the long acting depot formulations described herein.
As an example, clonidine selectively stimulates receptors in the brain that monitor catecholamine levels (epinephrine and norepinephrine) in the blood to lower catecholamine production. The result is a lowered heart rate and blood pressure. Because of the mechanism of action of clonidine, medical professionals have begun to expand the use of clonidine to treat disorders other than hypertension, e.g., in the treatment of attention-deficit hyperactivity disorder (ADHD). In ADHD patients, clonidine is often used either alone or in combination with other medications, such as stimulants, to moderate ADHD-associated impulsive and oppositional behavior, or to counteract the side effects of stimulants.
In order to reduce patient compliance issues, particularly with ADHD patients, it would be beneficial to have a clonidine formulation that is long acting. One such long acting formulation is an injectable depot formulation.
In developing the injectable depot formulations of the present invention, which utilizes microsphere technology, the inventors have identified a myriad of factors that can impact the success of the depot formulation including, but not limited to, factors that affect the drug release rate, drug loading of the microspheres, and overall formulation stability.
For example, it has been determined herein that there can be a direct and proportional relationship between the rate of drug release from formulated microspheres and the hydrophylicity of the polymer. The rate of drug release will also increase with the porosity of the formulated microspheres and an increase in the ratio of the inner water phase to the inner organic phase (Figure 16). Moreover, in those embodiments where the microspheres comprise PLGA as the biocompatible polymer, the rate of drug release increases with an increase in the mole fraction of "G" (glycolic acid) therein (Figure 6). At an even more microscopic level, the rate of drug release can be tuned on the basis of the mole fraction of carboxylic acid terminal groups relative to ester terminal groups in the polymer. The greater the mole fraction of carboxylic acid terminal groups, the higher
the release rate of the active agent (Figure 17). Moreover the release rate can increase with an increase in the drug-to-polymer mass ratio at the reagent/manufacturing level as well as in the final formulated product (Figures 7 and 8). Other factors that can increase the release rate of the active agent are an increase in the active agent's particle size (Figure 18), lengthening the homogenization time for the carrier microspheres (Figure 19), and lengthening the vortex time of the microspheres during manufacture (Figure 20).
Among others, there is an inverse relationship between the drug release rate and the microsphere particle size (Figure 14), and between the drug release rate and the polymer molecular weight (Figure 5). Therefore an increase in either the microsphere particle size or the molecular weight of the biocompatible polymer utilized to formulate the compositions can reduce the rate of drug release.
Additionally, there is an inverse relationship between (substantially) zero order release properties and particle size. As the microsphere particles increase in volume, the release rate increasingly deviates from zero order (Figure 18). Zero order release, or at least quasi zero order release, is desirable because it demonstrates that the active agent is released at a constant, or approximately constant, rate.
With respect to drug loading, there can be an increase in drug loading with an increase in particle size, but there can also be an increase in the drug loading when a hydrophobic acid (e^g., palmitic acid) is introduced to the formulation, particularly those formulations comprising PLGA polymers (Table 9).
Further, it was found that many factors can contribute to an increase in stability of the final depot formulation (Table 9). For example, when an acid is introduced to the formulation, the stability of the formulation increases. The stability of the final formulation, however, can be inversely related to the mole fraction of "G" (glycolic acid) in PLGA formulations, and inversely related to the extent of drug loading. In other words, increasing the mole fraction of glycolic acid in PLGA and/or increasing the percent drug loading tend to decrease the stability of the final formulation.
As disclosed above, in certain embodiments the present invention is directed to a pharmaceutical depot formulation comprising clonidine or a pharmaceutically acceptable salt or prodrug thereof; the depot formulation after injection or implantation into a human patient providing a mean release rate of clonidine of less than about 5 mg over 24 hours
and providing a release rate of at least about 0.1 mg of clonidine per 24 hour period for at least 24 hours, at least 3 to 5 days, at least 7 to 28 days or at least 3 months.
In other embodiments, the present invention is directed to clonidine or a pharmaceutically acceptable salt or prodrug thereof; wherein from about 10% to about 60% clonidine is released after 4 days, from about 20% to about 80% clonidine is released after 8 days and from about 30% to about 95% clonidine is released after 12 days and less than 100% clonidine is released after 16 days when tested in a pH 7.4 buffered solution at 370C and lOOrpm.
In other embodiments, the present invention is directed to clonidine or a pharmaceutically acceptable salt or prodrug thereof; wherein from about 10% to about 60% clonidine is released after 4 days, from about 20% to about 80% clonidine is released after 8 days and from about 30% to about 95% clonidine is released after 12 days and less than 100% clonidine is released after 16 days when tested in a neutral pH buffered solution at 37°C and lOOrpm.
This invention also overcomes the difficulties in formulating clonidine in controlled release formulations due to the high solubility and potency of the compound. The high solubility of clonidine can result in failure of a controlled release system. This can lead to "dose dumping" wherein a portion of, or an entire dose that is intended to be released over an extended period of time is released at a time shorter than intended, e^g., a faster release or an immediate release. This can result in a plasma level above the therapeutic level and result in increased incidence of adverse events. This is especially an issue with a potent drug such as clonidine where even a minimal "dose dump" can lead to toxic levels of the drug accumulating in the patient.
The commercially available oral clonidine formulations utilize clonidine hydrochloride. In certain embodiments of the invention, in order to reduce the solubility of the clonidine to assist in obtaining a controlled release depot effect, clonidine is utilized as the free base or utilized in a salt which has relatively lower solubility. For example, the present invention can utilize an insoluble salt such as a fatty acid salt. Representative fatty acid salts include salts of oleic acid or linoleic acid. In preferred embodiments fatty acid salts with between 8 to 20 carbons are used to produce salts with an aqueous solubility of less than about 2.0 mg/ml, and preferably less than about 1.0
mg/ml, such as clonidine palmeate and clonidine stearate. Most preferably, fatty acid salts with between 12 to 18 carbons are used. Other embodiments can utilize a lipid soluble salt of clonidine.
In other embodiments, the clonidine free base or pharmaceutically acceptable salt thereof (regardless of the choice of salt form) can be combined with a complexing agent, e.g., calcium chloride or magnesium chloride, in order to modify the release characteristics of the active agent.
In other embodiments, the clonidine free base or pharmaceutically acceptable salt thereof (regardless of the choice of salt form) can be combined with a dissolution modifying excipient. The clonidine can be combined with the dissolution modifying excipient, ejj., by being dispersed in the excipient, being microencapsulated by the excipient, by being embedded in the excipient (e^g., in between layers), in intimate association with the excipient, or bonded to the excipient (e^g., by a covalent bond or by Vander Waals forces).
A preferred dissolution modifying excipient is a biocompatible polymer. Such biocompatible polymers can be selected from the group consisting of a biodegradable polymer, a non-biodegradable polymer, or a combination thereof.
The biodegradable polymer can be selected from, ej*., the group consisting of poly(lactide)s, poly(glycolide)s, poly(lactide-co-glycolide)s, poly(lactic acid)s, poly(glycolic acid)s, poly(lactic acid-co-glycolic acid)s, polyanhydrides, polyortho esters, polyetheresters, polycaprolactone, polyesteramides, polyphosphazenes, polysaccharides, proteinaceous polymers, soluble derivatives of polysaccharides, soluble derivatives of proteinaceous polymers, polypeptides, polyesters, and combinations thereof.
The biodegradable polymer can be selected, e^g., from the group consisting of polymers having blocked carboxyl end groups, polymers having free carboxyl end groups, or combinations thereof. In the blocked embodiments, the blocking group can be, βjg., an alkyl radical.
Preferably, the biodegradable polymer has a molecular weight of from about 2,000 Daltons to about 2,000,000 Daltons; from about 5,000 Daltons to about 100,000 Daltons; or from about 20,000 Daltons to about 75,000 Daltons.
In preferred embodiments, the biodegradable polymer is a poly(lactide-co- glycolide)(PLGA). In other preferred embodiments, the poly(lactide-co-glycolide) has a lactide:glycolide ratio of about 10: 1 to about 1 : 10; about 5:1 to about 1:5 about 3: 1 to about 1:3; or about 1 : 1. In other preferred embodiments, the biodegradable polymer is poly (D-, L-, or D, L-lactic acid)(PLA).
When the biodegradable polymer is a proteinaceous polymer, the polymer can be selected, ejj,, from the group consisting of gelatin, elastin, alkylated collagen, and alkylated elastin.
The non-biodegradable polymers utilized in the present invention can be, e.g., selected from the group consisting of polyacrylates, polymers of ethylene-vinyl acetates and other acyl substituted cellulose acetates, non-degradable polyurethanes, polystyrenes, polyvinyl chloride, polyvinyl fluoride, poly(vinyl imidazole), chlorosulphonate polyolefms, polyethylene oxide, and combinations thereof.
In certain embodiments, the clonidine or pharmaceutically acceptable salt or prodrug thereof, preferably in particulate form, is contained in a pharmaceutically acceptable vehicle, with or without a dissolution modifying excipient. In certain embodiments, the vehicle is selected from the group consisting of a lipid, an oil, a wax, and a combination thereof. Suitable pharmaceutically acceptable vehicles which can be utilized in the present invention include cottonseed oil, soybean oil, safflower oil, hydrated peanut oil, olive oil, castor oil, tryglyceride mixtures, monoglyceride, silicone oil, isopropylmyristate, ethyloleate, paraffins, glycerol, propylene glycol, polyethylene glycol, medium chain fatty acids, long chain fatty acids or a combination thereof.
In other embodiments, the pharmaceutically acceptable vehicle is selected from the group consisting of water, ethanol, glycerin, polyethylene glycol, propylene glycol and a combination thereof.
In embodiments utilizing a vehicle, a viscosity increasing agent can be utilized. In certain embodiments, the viscosity increasing agent can also affect the dissolution of the clonidine or pharmaceutically acceptable salt thereof. The viscosity increasing agent can be selected, e^g., from the group consisting of a cellulose derivative, polyvinylpyrrolidone, alginates, chitosan, a dextran, gelatin, polyethylene glycols, polyoxyethylene ethers, polyoxypropylene ethers, polylactides, polyglycolides,
polycaprolactones, polyanhydrides, polyamines, polyur ethanes, polyesteramides, polyortho esters, polydioxanones, polyacetals, polycarbonates, polyorthocarbonates, polyphosphazenes, succinates, polycarbonates, poly(maleic acid), poly(amino acids), polyhydroxycellulose, chitin, copolymers or terpolymers of the foregoing, sucrose acetate isobutyrate, PLGA, PLA, stearic acid/NMP, and a combination thereof.
Irrespective of the use of a vehicle, a gelling agent can also be utilized in the pharmaceutical depot formulations of the present invention. The gelling agent can be utilized in formulations utilizing a vehicle, wherein the gelling agent also provides increased viscosity to the formulation. Alternatively, the gelling agent can be utilized, e.g., in a dry powder depot formulation such as when the formulation is administered by needleless injection, In such embodiments, the gelling agent will form a gel upon exposure to bodily fluids. In such an embodiment, the particles of clonidine become embedded, encapsulated or dispersed in the gel which controls or further controls the release of the active agent from the depot formulation. In certain embodiments, the gelling agent is selected, e^g., from the group consisting of polyethylene oxide; hydroxypropyl methylcellulose; polyvinyl alcohol; methyl cellulose; ethylcellulose; hydroxyethyl cellulose; sodium carboxymethyl cellulose; dextran; gelatin; pectin; sodium poly(acrylic acid); carboxymethyl cellulose and combinations thereof.
In certain embodiments when the clonidine is present as nanoparticles, the formulation can include dissolution modifying excipient which associates with the drug particles and reduces cohesive forces and reduces agglomeration of the particles. In this context, the dissolution modifying excipient may promote release, as agglomeration and cohesiveness of the particles can result in a reduction of release to a level that therapeutic plasma levels are not achieved by the pharmaceutical depot formulation. Thus, in certain embodiments, a dissolution modifying excipient balances the reduction of the solubility of the clonidine to impede release while at the same time reducing agglomeration and cohesiveness to promote release.
Such dissolution modifying excipients can be selected, e^g., from the group consisting of an anionic compound, a cationic compound, a zwitterionic compound, a non-ionic compound, and an ionic compound.
In other such embodiments, the dissolution modifying excipient is selected, e.g., from the group consisting of cetyl pyridiniuni chloride, albumin, gelatin, casein, phosphatides, dextran, glycerol, gum acacia, cholesterol, tragacanth, stearic acid, benzalkonium chloride, calcium stearate, glycerol monostearate, cetostearyl alcohol, cetomacrogol emulsifying wax, sorbitan esters, polyoxyethylene alkyl ethers, polyoxyethylene castor oil derivatives, polyoxyethylene sorbitan fatty acid esters, polyethylene glycols, dodecyl trimethyl ammonium bromide, polyoxyethylene stearates, colloidal silicon dioxide, phosphates, sodium dodecyl sulfate, carboxymethylcellulose calcium, hydroxypropyl celluloses, hypromellose, carboxymethylcellulose sodium, methylcellulose, hydroxyethylcellulose, hypromellose phthalate, noncrystalline cellulose, magnesium aluminum silicate, triethanolamine, polyvinyl alcohol, polyvinylpyrrolidone, 4-(l,l,3,3-tetramethylbutyl)-phenol polymer with ethylene oxide and formaldehyde, poloxamers; poloxamines, a charged phospholipid, dioctylsulfosuccinate, dialkylesters of sodium sulfosuccinic acid, sodium lauryl sulfate, alkyl aryl polyether sulfonates, mixtures of sucrose stearate and sucrose distearate, p-isononylphenoxypoly-(glycidol), decanoyl- N-methylglucamide; n-decyl β -D-glucopyranoside; n-decyl β -D-maltopyranoside; n- dodecyl /ϊ-D-glucopyranoside; n-dodecyl β -D-maltoside; heptanoyl-N-methylglucamide; n-heptyl- β -D-glucopyranoside; n-heptyl β -D-thioglucoside; n-hexyl β -D- glucopyranoside; nonanoyl-N-methylglucamide; n-noyl β -D-glucopyranoside; octanoyl- N-methylgluc amide; n-octyl- β -D-glucopyranoside; octyl β -D-thioglucopyranoside; lysozyme, PEG-phospholipid, PEG-cholesterol, PEG-cholesterol derivative, PEG- vitamin A, PEG-vitamin E, random copolymers of vinyl acetate and vinyl pyrrolidone, a cationic polymer, a cationic biopolymer, a cationic polysaccharide, a cationic cellulosic, a cationic alginate, a cationic nonpolymeric compound, a cationic phospholipids, cationic lipids, polymethylmethacrylate trimethylammonium bromide, sulfonium compounds, polyvinylpyrrolidone-2-dimethylaminoethyl methacrylate dimethyl sulfate, hexadecyltrimethyl ammonium bromide, phosphonium compounds, quarternary ammonium compounds, benzyl-di(2-chloroethyl)ethylammonium bromide, coconut trimethyl ammonium chloride, coconut trimethyl ammonium bromide, coconut methyl dihydroxyethyl ammonium chloride, coconut methyl dihydroxyethyl ammonium bromide, decyl triethyl ammonium chloride, decyl dimethyl hydroxyethyl ammonium
chloride, decyl dimethyl hydroxyethyl ammonium chloride bromide, Cπ-isdimethyl hydroxyethyl ammonium chloride, Ci2.i5dimethyl hydroxyethyl ammonium chloride bromide, coconut dimethyl hydroxyethyl ammonium chloride, coconut dimethyl hydroxyethyl ammonium bromide, myristyl trimethyl ammonium methyl sulphate, lauryl dimethyl benzyl ammonium chloride, lauryi dimethyl benzyl ammonium bromide, lauryl dimethyl (ethenoxy)4 ammonium chloride, lauryl dimethyl (ethenoxy)^ ammonium bromide, N-alkyl (Ci2-i8)dimethylbenzyl ammonium chloride, N-alkyl (Ci4-i8)dimethyl- benzyl ammonium chloride, N-tetradecylidmethylbenzyl ammonium chloride monohydrate, dimethyl di decyl ammonium chloride, N-alkyl and (C 12- 14) dimethyl 1- napthylmethyl ammonium chloride, trimethylammonium halide, alkyl- trimethylammonium salts, dialkyl-dimethylammonium salts, lauryl trimethyl ammonium chloride, ethoxylated alkyamidoalkyldialkylammonium salt, an ethoxylated trialkyl ammonium salt, dialkylbenzene dialkylammonium chloride, N-didecyldimethyl ammonium chloride, N-tetradecyldimethylbenzyl ammonium, chloride monohydrate, N- alkyl(Ci2-i4) dimethyl 1-naphthylmethyl ammonium chloride, dodecyldimethylbenzyl ammonium chloride, dialkyl benzenealkyl ammonium chloride, lauryl trimethyl ammonium chloride, alkylbenzyl methyl ammonium chloride, alkyl benzyl dimethyl ammonium bromide, C12 trimethyl ammonium bromides, C15 trimethyl ammonium bromides, C 17 trimethyl ammonium bromides, dodecylbenzyl triethyl ammonium chloride, poly-diallyldimethylammonium chloride (DADMAC), dimethyl ammonium chlorides, alkyl dimethylammonium halogenides, tricetyl methyl ammonium chloride, decyltrimethylammonium bromide, dodecyltriethylamrnonium bromide, tetradecyltrimethylammonium bromide, methyl trioctylammonium chloride, tetrabutylammonium bromide, benzyl trimethylammonium bromide, choline esters, benzalkonium chloride, stearalkonium chloride compounds, cetyl pyridinium bromide, cetyl pyridinium chloride, halide salts of quaternized polyoxyethylalkylamines, alkyl pyridinium salts; amines, amine salts, amine oxides, imide azolinium salts, protonated quaternary acrylamides, methylated quaternary polymers, cationic guar and a combination thereof.
As the administration of depot formulations may cause some irritation at the site of injection or implantation, a glucocorticoid can also be included in the pharmaceutical
depot formulation to minimize this effect. The glucocorticoid can be selected, e.g., from the group consisting of alclometasone, desonide, flumethasone, hydrocortisone and its esters such as hydrocortisone butyrate or hydrocortisone acetate, clobetasone, triamcinolone acetonide, betamethasone, budenoside, desoximethasone, diflorosane, fluocinolone, fluoccinonide acetonide, fluocortolone, fluticasone, methylprednisolone aceponate, mometasone, rofleponide and combinations thereof.
With one intent being to increase patient compliance, the pharmaceutical depot formulations of the present invention provide a therapeutic plasma level of clonidine for at least 24 hours, at least 3 days, at least 5 days, at least 7 days, at least 14 days; at least 1 month or at least 3 months.
In preferred embodiments, the clonidine or pharmaceutically acceptable salt or prodrug thereof is present in the depot formulation in micro- or nano-sized particles, e.g., in the form of particles having an effective average particle size of less than or equal to about 200 μm. In other preferred embodiments, the effective average particle size is less than or equal to about 2000 nm. In still other preferred embodiments, the effective average particle size is less than or equal to about 50 μm. Typically, micro- or nano- sizing of particles is utilized in order to increase the dissolution and bioavailability of poorly soluble drugs. See, Kesisoglou et al., Nanosizing - Oral formulation development and biopharmaceutical evaluation, Advanced Drug Delivery Reviews, 59(2007), 631- 644. As clonidine already has a high solubility, in embodiments utilizing nanosized particles of clonidine, the present invention provides a depot formulation that minimizes or prevents "dose dumping" given the further increase in the dissolution of the clonidine particles.
In certain embodiments, the clonidine or pharmaceutically acceptable salt or prodrug thereof is in the form of particles having an effective average particle size selected from the group consisting of less than or equal to about 100 μm, less than or equal to about 50 μm, less than or equal to about 25 μm. In preferred embodiments, the clonidine or pharmaceutically acceptable salt or prodrug thereof is in the form of particles having an mean effective particle size of from about 5 μm to about 40 μm. The micro- or nano-sized clonidine of the present invention can be obtained by techniques such as milling or by spray drying.
The milling process can be a dry process, e^g., utilizing a dry roller milling process. In other embodiments, the milling process can be a wet process wherein the clonidine is dispersed in a liquid such as water, safflower oil, aqueous salt solutions, ethanol, n-butanol, hexane, glycol and the like. The dispersing and/or wetting agents can be selected from known organic and inorganic pharmaceutical excipients and can be present in an amount, e^g., from about 1% to about 80% by weight of the total mixing slurry.
The grinding media utilized in the micro- or nano-sizing of the particles can be, e.g., rigid media that are preferably spherical or particulate in shape. However, grinding media in the form of other non-spherical shapes can be utilized in micro- or nano-sizing clonidine in the present invention. Grinding media utilized in the present invention include but is not limited to stainless steel, titania, agate, glass, alumina, zirconium silicate, and zirconium oxide (optionally stabilized with magnesia or yttrium).
Polymeric materials such as polymeric resins can also be utilized as grinding materials. In the case of biodegradable polymers, contamination from the media can be metabolized in vivo into biologically acceptable products which can be eliminated from the body.
The grinding can take place in any suitable grinding mill such as an airjet mill, a roller mill, a ball mill, an attritor mill, a vibratory mill, a planetary mill, a sand mill and a bead mill. A high energy media mill is preferred when small particles are desired. The mill can contain a rotating shaft.
After grinding is completed, the grinding media is separated from the milled clonidine particulate product (in either a dry or liquid dispersion form) using techniques such as by filtration, sieving through a mesh screen, and the like.
When spray drying is utilized, a solution of clonidine in a solvent can be sprayed into an empty chamber under conditions that allow for a substantial amount of the solvent to be removed from the solution, such that nanometer-sized clonidine particles are formed.
In another embodiment, a clonidine solution is sprayed onto a fluidized bed of carrier particles. The bed is maintained at a temperature, e^g., from about 20° C to about
80° C or from about 25° C to about 50° C. This embodiment results in micro- or nano- particles of clonidine formed in a mixture with carrier particles.
In a further embodiment, a carrier is mixed with the clonidine solution to be spray dried. This mixture is sprayed into a heated chamber and the nanoparticles formed in this process contain a mixture of clonidine and carrier.
In the spray drying embodiments disclosed herein, the carrier particles include, but are not limited to, saccharides, such as sugars and sugar alcohols (e^g., lactose, sucrose, mannitol, or sorbitol), starches, flour, cellulose preparations and/or salts such as carbonates, bicarbonates and phosphates (e.g., tricalcium phosphate or calcium hydrogen phosphate).
The pharmaceutical depot formulation of the present invention can be suitable for administration via implantation or injection (e^g., intravenously, intramuscularly or subcutaneously) to the patient.
Injection of the pharmaceutical depot formulation may be through a syringe with any size and shaped needle capable of administering the formulation without resulting in clogging of the formulation. The pharmaceutical depot formulation may be incorporated into a suitable liquid or gel composition for administration or may be in the form of microspheres, nanospheres, etc. Examples of apparatus and asceptic procedures useful for the formation of sterile microspheres are described, e.g., in U.S. Patent Nos. 5,945,126; 6,270,802; and 6,361,798, the disclosures of which are hereby incorporated by reference. Additionally, a needleless injector may be used to inject the depot formulation. Needless injections use gas pressure to deliver the pharmaceutical depot formulation, which is typically in powder form. Examples of Needleless injections, including apparatus and types of formulations useful therefore, are described, e.g., in U.S. Pat. Nos. 6,053,889; 6,013,050; 6,010,478; 6,004,286; and 5,899,880, the disclosures of which are hereby incorporated by reference.
The embodiments of the invention utilizing needleless injection may result in improved compliance as non-compliance in many patients is due to anxiety associated with conventional needle injection. This may be especially true in pediatric patients (e.g., pediatric patients with ADHD).
Clonidine Prodrugs
In certain embodiments, the present invention is directed to a clonidine prodrug of the formula:
A-R
wherein A is a radical of clonidine (formula I)

(I)
and R represents an R moiety bound to any suitable location on the clonidine structure, ejj., on any nitrogen or carbon within the ring structures or on the nitrogen linking the two ring structures.
In certain embodiments, the R moiety is R is selected from the group consisting of a formate radical of the formula COO'; a formate radical of the formula R1COO", wherein R1 is an alkylene having from 1-30 carbons, 5-25 carbons 10-20 carbons or 16-18 carbons; a moiety selected from the group consisting of polyethylene glycol fatty acid esters, fatty alkylol amide condensates, alkylarylsulfonates, fatty alcohol sulfates, dialkyl esters of sodium sulfosuccinate, fatty acid esters of sodium isothionate, polyoxyethylene ethers and thioethers, and long-chain quaternary ammonium chloride compounds; and a moiety selected from the group consisting of an amino acid, a dipeptide and a trip ep tide.
The amino acid can be natural or synthetic. Amino acid moieties suitable for preparing the prodrugs of the present invention include valine, leucine, isoleucine, methionine, phenylalanine, asparagine, glutamic acid, glutamine, histidine, lysine, arginine, aspartic acid, glycine, alanine, serine, threonine, tyrosine, tryptophan, cysteine
and proline. The amino acids can be D or L-amino acids; and can be alpha, beta, or gamma amino acids.
Other amino acids can be gamma-carboxyglutamic acid, selenocysteine, desmosine, 6-N-methyllysine, epsilon-N,N,N-trimethyllysine, 3-methylhistidine, O- phosphoserine, 5-hydroxylysine, epsilon-N-acetyllysine, omega.-N-methylarginine, N- acetylserine, gamma-aminobutyric acid, citrulline, ornithine, azaserine, homocysteine, beta-cyanoalanine and S-adenosylmethionine.
Synthetic amino acids include phenyl glycine, meta-tyrosine, para-amino phenylalanine, 3-(3-pyridyl)-L-alanine, and 4-(trifluoromethyl)-D-phenylalanine.
In other embodiments, R is selected from the group consisting of an aldehyde (- CHO), a ketone (-COR2), a carboxylic acid (-COOH), an ester (-COOR2), an amide (- CONR2R3), an enone (-C(O)C(R2)CR3R4), an acyl chloride (-C0C1) or an acid anhydride (-COOCOR ). In such embodiments, R , R and R are independently selected from the group consisting of alkyl, alkenyl, aryl, heteroaryl, alkoxy, aryloxy and heteroaryloxy moieties wherein the alkyl moiety is selected from the group consisting of unsubstituted or substituted, straight- chain, branched-chain and cyclic alkyl moieties having 1-20 carbon atoms; wherein the alkenyl moiety is selected from the group consisting of unsubstituted and substituted, straight-chain, branched-chain and cyclic alkenyl moieties having 2-20 carbon atoms; wherein the aryl moiety is selected from the group consisting of unsubstituted and substituted phenyl, and phenalkyl moieties wherein the alkyl moiety contains 1-3 carbon atoms and the phenyl moiety is unsubstituted or substituted; and the heteroaryl moiety is a substituted or unsubstituted aromatic 5-or 6-membered heterocyclic ring containing one or two heteroatoms selected from the group consisting of nitrogen, oxygen, and sulfur; and pharmaceutically acceptable salts thereof.
Examples of suitable straight-chain alkyl moieties include methyl, ethyl, propyl, butyl, hexyl, heptyl, octyl, dodecyl and palnityl moieties.
Examples of suitable branched-chain alkyl moieties include isopropyl, sec-butyl, t-butyl, 2-methylbutyl, 2-pentyl and 3-pentyl moieties.
Examples of suitable cyclic alkyl moieties include cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl moieties.
Examples of suitable "alkenyl" moieties include vinyl (ethenyl), 1-propenyl, i-
butenyl, pentenyl, hexenyl, n-decenyl and c-pentenyl.
The moieties may be substituted, generally with 1 or 2 substituents, wherein the substituents are independently selected from halo, hydroxy, alkoxy(alkoxy)x, hydroxyalkoxy(alkoxy)x, amino, mono- and dialkylamino, nitro, carboxyl, alkoxycarbonyl, and cyano moieties, wherein x is an integer from 0 to 3 and the alkoxy moieties contain from 1 to 5 carbon atoms.
The term "phenalkyl moieties wherein the alkyl moiety contains 1-3 carbon atoms" includes benzyl, phenethyl and phenylpropyl moieties wherein the phenyl moiety may be substituted. When substituted, the phenyl moiety of the phenalkyl group may contain 1-3 substituents independently selected from alkyl, hydroxy, alkoxy, halo, amino, mono- and dialkylamino, nitro, carboxyl, alkoxycarbonyl and cyano moieties.
Examples of suitable "heteroaryl" moieties are pyridinyl, thienyl and imidazolyl.
The term "halo" includes F, Cl, Br, and I.
The prodrugs of clonidine disclosed herein can be utilized in the depot formulations of the present invention or can be included in other pharmaceutical formulations, e^g., oral, intranasal, pulmonary, and transdermal formulations.
Fatty Acid Salts of Clonidine
In certain embodiments, the present invention is directed to a fatty acid salt of clonidine, e^g., fatty acid salts having greater than 2 carbons, greater than 8 carbons or greater than 16 carbons, such as butyric, caprioc, caprylic, capric, lauric, mystiric, palmitic, stearic, arachidic and the like.
Representative fatty acid salts of the present invention include salts of oleic acid or linoleic acid. In preferred embodiments fatty acid salts with between 8 to 20 carbons are used to produce salts with an aqueous solubility of less than about 2.0 mg/ml, and preferably less than about 1.0 mg/ml, such as clonidine palmeate and clonidine stearate. Most preferably, fatty acid salts with between 12 to 18 carbons are used. Other embodiments can utilize a lipid soluble salt of clonidine.
The fatty acid and lipid salts of clonidine disclosed herein can be utilized in the depot formulations of the present invention or can be included in other pharmaceutical formulations, ej*., oral, intranasal, pulmonary, and transdermal formulations.
While many of the embodiments disclosed herein reference clonidine, other alpha-2 -adrenergic agonists can be utilized therein. Such agents include tizanidine, guanfacine, guanabenz, guanoxabenz, guanethidine and pharmaceutically acceptable salts thereof.
The following examples are set forth to assist in understanding the invention and should not, of course, be construed as specifically limiting the invention described and claimed herein. Such variations of the invention, including the substitution of all equivalents now known or later developed, which would be within the purview of those skilled in the art, and changes in formulation or minor changes in experimental design, are to be considered to fall within the scope of the invention incorporated herein.
EXAMPLES
EXAMPLE 1 Clonidine Weekly Depot Formulation Experimental Procedures
The following (PLGA) Polylactide-co-glycolide compositions were tested for incorporation into clonidine depot formulations:
(1:1 lactide:glycolide, MW 29,800; Inherent viscosity 800 0.38 dL/g); (1 :1 lactide:glycolide, MW 51,400; Inherent viscosity 0.60 dL/g); (1:1 lactide:glycolide, MW 81,600; Inherent viscosity 0.82 dL/g); and (1 : 1 lactide:glycolide, MW 122,000; Inherent viscosity 1.12 dL/g).
PLGA (1: 1 lactide:glycolide, MW 51,400; Inherent viscosity 0.60 dL/g) and PLGA (1:1 lactide:glycolide, MW 81,600; Inherent viscosity 0.82 dL/g) were then chosen to incorporate into two clonidine depot formulations in accordance to Tables 1 and 2 below:
Table 1

** Evaporated during process
The actual weight (mg) per dose depends on the actual loading of clonidine in microspheres. The weight per dose was therefore adjusted accordingly.
Fabrication of Microspheres
Microspheres can be fabricated using the following procedures:
1. Weigh 3 g (range: depends on batch size, typical 0.5-10 g) of PLGA into 10 g of methylene chloride (CH2CL2) or ethyl acetate.
2. Weigh 200 mg (range: 50mg-200mg) and dissolve into 0.8 niL of methylene chloride and 0.6 mL of deionized water (the solvent could be a mixture of methanol/water or water or phosphate buffer, range of volume: 50 μL to 5 mL).
3. Homogenize the mixture at 22,000 RPM (range: 10,000 to 30,000 RPM) for 2 min (range: 1 to 2 min).
4. Add 30 mL (range: 10-100 mL) of 5% w/w polyvinyl alcohol (PVA) (MW25,000) aqueous solution into mixture step #3 and homogenize at 1 1,000 RPM for 15 sec (range: 5-20 sec) to form water-in-oil-in water emulsion. Alternatively, after adding the PVA, the mixture can be vortex for 30 sec (range: 30 sec to 1 min). PVA molecular weight can be varied from 5,000 to 75,000 and the %w/w of PVA can be ranged from 0.1-5.0%w/w.
5. Transfer the emulsion into a 1.0 L (range: 100 mL to 100OmL) of (0.5% w/w) PVA and stir with magnetic or mechanical stirrer at ~ 600 RPM to evaporate CH2CL2 for overnight (range: 3-20 h).
6. Wash the microspheres from step #5 and collect by sieving the particles through sieves mesh #40 (424 μm), #60 (250μm), #80 (180 μm), #100 (150μm), #140 (107μm), #270 (53μm), and #400 (38 μm). The remainder of the collected filtrate will be centrifuged to retrieve microspheres of diameter less than 38 μm.
7. Dry microspheres by oven or freeze-dryer or vacuum oven.
8. Weigh microspheres and, according to the loading of the clonidine in the microspheres, extract sample sufficient for a weekly dose, then re-suspend in normal saline solution or phosphate buffer pH 7.4 for injection.
Microsphere Characterization Particle size analyses
Malvern or Dynamic Light Scattering analyses
• The microspheres are suspended in 0.1% aqueous Tween 80 solution and sonicated for 30 seconds and stirred for 2 minutes before analysis.
• Size distribution was analyzed by Malvern or dynamic light scattering particle size analyses. The particle size distributions are reported on a volume basis.
Microscopy analyses
• The microspheres are spread on the microscope slides and sizes of the microspheres are measured. The population or number distribution is converted to a volume distribution.
Determination of drug loading in PLGA microspheres
In triplicate, lOmg of microspheres were weighed and transferred to 10ml volumetric flasks. To each flask, 4ml of acetonitrile was added. After sonication for ten to 15 minutes, the solution was diluted to volume with SGF pH1.2 (prepared by dissolving 2.Og of sodium chloride in 60OmL of water and adding 7.OmL of concentrated hydrochloric acid and dilute with water to 1000ml). The solution was continued to sonicate for 1 h. The solution was filtered using a 1.0 μm glass acrodisc filter and an aliquot was collected for HPLC analysis. Drug loading was determined by the following equations:
Actual drug loading (%w/w) = mass of drug in microspheres x 100 / mass of drug loaded microspheres
Encapsulation efficiency (%) = mass of drug in microspheres x 100 / initial mass of drug used
In vitro Dissolution of Microspheres
About a 50 mg sample, in this case the microsphere formulation, was weighed and transferred to a 15 mL centrifuge tube containing 10 niL 50 mM phosphate buffer pH 7.4 with addition of 0.1%v/v Tween80. Samples were incubated in Brunswick Scientific Water Bath Shaker at 100 rpm at 37 °C for a specified time. At each specified time point, the microspheres were swirled in the medium and centrifuged as necessary. 2 mL of sample was pipetted for HPLC analyses. 2 mL of fresh 50 mM phosphate buffer pH 7.4 was added to the centrifuge tubes and shaken gently to re-suspend the microspheres. The tubes were placed back in the 37°C water bath for further incubation.
EXAMPLE 2 Preparation of microencapsulated clonidine by double emulsion
The particle size of clonidine base was reduced by probe sonication to the average mean size distribution of 3 to 10 μm utilizing Branson Ultrasonics Sonifier Cell Disrupters 400W at 50% power intensity with 1.5 cm probe for 30 minutes. The particle
size distribution was determined by Melvern Mastersize 2000 analysis. See Figure 1 which depicts the mean size distribution of clonidine base particles after probe sonification (dlo% = 0.79 μm , d50%= 9.67 μm, d90%= 23.87 μm). After achieving the desired particle size, the clonidine base was freeze dried (or oven dried).
In a 2-mL eppendorf tube, 400mg of the micronized clonidine base was suspended in 600 mg of aqueous containing l%w/w Tween 80 (or without Tween 80) and the mixture was vortexed to form clonidine suspensions. In a 40-mL glass vial, 2g of lactic acid/glycolic acid copolymer was dispensed (e.g., lactic acid/glycolic acid (PLGA) = 75/25 (mole%) with inherent viscosity 0.55-0.75dL/g) and dissolved in 5 mL of methylene chloride to form PLGA solution. Then, the clonidine suspensions were homogenized with the PLGA solution for 3 minutes at 15,000 RPM to form the first wj/o emulsion.
15-niL of 5%w/w polyvinyl alcohol (PVA) solution was added into the first emulsion and homogenized for 1 minute at 13000 RPM to form the second emulsion wi/o/ w2. (Note that the solution containing 5% PVA could be made of PBS pH9, 5% NaCl ensure higher osmotic pressure at the outer phase to enhance encapsulation efficiency). Then, the emulsion was transferred into a 1 liter of 0.5%w/w PVA (5%w/w NaCl may be added to increase osmority of the outer phase) solution continuously stirred at -600RPM to evaporate methylene chloride and harden the microspheres overnight.
Microspheres were collected by sieving through sieves mesh #100 (150 μm) and #400 (38 μm). Microspheres size less than 38 μm were collected by centrifugation at -1200 RPM for 5 minutes. All microspheres were washed with deionized water. After washing, the microspheres were freeze dried. Figure 2 shows the micrographs of dried microspheres acquired by light microscope. Micrograph (A) was acquired with objective 20x for microspheres ranged from 150 to 38 μm and micrographs (B) was acquired with objective 40x for microspheres less than 38 μm. Figure 3 shows the particle size distributions of microspheres that passed through sieve#400 (with opening 38 μm) acquired by Melvern Mastersizer 2000 particle size analyzer (dio% = 8.57 μm , dso%= 22.10 μm, d9o%= 39.85 μm). The actual loading of the microspheres is about 7%w/w.
Similar method and processing parameters can be applied to different types of PLGA copolymers (including different copolymer average molecular weight different,
PLA:PGA ratio, viscosity, conjugated with other polymer such as PEG). The size of the microspheres could be controlled by the speed and time of homogenization. For a smaller batch, the second emulsions could be formed by vortexing at 3000 RPM for 30 seconds to 2 minutes depending on the polymer viscosity. Figure 4 shows the dissolution profiles of clonidine released from copolymer PLGA 50:50 and PLGA 75:25 that were fabricated by the double emulsion evaporation method. The actual loading of clonidine in the microspheres made of PLGA 50:50 (size range 150 to 38 μm) is 8%w/w, PLGA 75:25 (size range 150 to 38 μm) is 10%w/w, and PLGA75:25 (average size 22 μm) is 7%w/w.
The data of Figure 4 demonstrate that substantially zero order release rates for greater than ten-day periods are achievable utilizing the depot formulations of the present invention.
EXAMPLE 3
Preparation of microencapsulated clonidine by double emulsion with different weight average molecular weight of lactic acid/glycolic acid copolymer 50/50 (%mole) with ester or carboxylate end group
The encapsulation of clonidine base was prepared as described in Example 2 using different ratio of PLGA copolymer. Table 3 below shows the ratio of PLA to PGA and their respective inherent viscosity.
Table 3
Poly (DL-Lactic-co Inherent Viscosity Weight avg. End Group glycolide) polymer Range Molecular dL/g Weight
50:50 0.15-0.25 -6700 Carboxylate
50:50 0.55-0.75 -51400 Ester
50:50 0.55-0.75 -32000 Carboxylate
50:50 0.76-0.94 -81600 Ester
Figure 6 shows the release profiles of clonidine from PLGA 50:50, 65:35, and 75:25 microspheres. The processing parameters of all three batches were kept constant for three different ratio of PLA to PGA copolymers. All of these batches were homogenized at 13000 RPM for 1 minute to form the first emulsion and vortexed at 3000 RPM for 30 seconds to form second emulsions.
As shown from the Figure 6, the higher the mole fraction of glycolic acid, the faster the degradation of the microspheres and therefore faster release of clonidine from the PLGA 50:50 microspheres compared to 65:25 and 75:25 microspheres.
Figure 16 compares the release of clonidine from two formulations of PLGA 50:50 (0.82 dL/g in HFIP) microspheres prepared according to the double emulsion process described in examples 2 and 3 above. Here, the ratio of the inner water phase to inner organic phase is changed from a 1:7 to a 1:10 and the effect is to increase the drug release rate.
Figure 18 compares the dissolution rate of clonidine microspheres prepared according to the double emulsion process described in examples 2 and 3 above where the particle size of the clonidine active agent is different. The large-particle drug sample leads to faster dissolution than the small-particle drug formulation.
Different batches of clonidine microspheres were manufactured having different microsphere particle size. The batch with average particle size equal to about 250 microns showed drug loading of about 13% w/w. The batch with particle sizes ranging from 38 to 160 microns showed drug loading of about eight percent, and the batch with particle sizes less than 38 microns showed drug loading of about three percent.
EXAMPLE 4 Single Emulsion System or Oil in Water Emulsion System
In this system, microspheres were prepared with different of clonidine to PLGA ratio. The ratio of clonidine to polymer tested were 1: 1, 1 :2, 2,5:400, 3:4, 4:4, and 4:5. PLGA used was PLGA 75:25 with ester terminals and the inherent viscosity was 0.55- 0.75 dL/g. Drug and copolymer were dissolved in methylene chloride and homogenized at 13000 rpm in 5%w/w PVA (MW ~25K) solution for 1 to 2 minutes followed by vortexing for 1 minute. The emulsion was transferred into a 0.01%w/w of Tween 80 and
5%w/w of NaCl in 2% of PVA solution stirring at ~ 600 RPM for 5 to 12 hours. Sodium chloride helps to prevent mutual aggregation of microspheres and osmotic stress during solvent evaporation process. The microspheres were collected by vacuum filtration and centrifugation processes.
Figure 7 shows the effect of drug loading with different reagent levels of clonidine and PLGA and thus different clonidine to PLGA copolymer ratios. There is a significant drug loading effect that ranged from 6% to 31% w/w with different drug-to- polymer ratios and for microspheres having size range from 149 to 38 μm. Microspheres smaller than 38 μm loading remained in the range of 3-6% w/w loading.
Figure 8 shows that the dissolution release of clonidine from PLGA 75:25 (ester terminated groups) microspheres are affected by the drug to polymer ratio of the reagents during the formulation process. The burst release is reduced with the O/W system where the drug to polymer ratio is lower.
The O/W emulsions procedures can be applied to different of PLGA copolymers ratio <le. 50:50 and 75:25), different molecular weight of copolymers with either ester or carboxylate end-groups as listed in Table 3 in Example 3. Figure 9 illustrates that the release profile of e.g. clonidine from the microspheres can be achieved by choosing appropriate combinations of polymer properties, such as acid or ester end groups or viscosity of the copolymer.
EXAMPLE 5 Co-solvent Single Emulsion System
The addition of co-solvent in the system increases the solubility of drug and polymer in organic phase. The co-solvent enhances the homogeneity of drug molecules dispersion in the microspheres. The microspheres were prepared following the procedure outlined in Example 4 and the co-solvent was added to the methylene chloride. For example, add 2%v/v of methanol into 98% v/v of methylene chloride and then dissolve the PLGA as described in Example 4. Results of clonidine release are shown in Figure 10.
EXAMPLE 6
Oil in Oil Emulsion System
1000 mg of clonidine base and 2000 mg of PLGA (75:25 (%mol), ester terminated, 52k) were dissolved in 10 ml of acetonitrile. The solution was then added drop wise to a beaker containing 50ml of mineral oil stirred continuously with a magnetic stirrer. 2% of Span 85 was added into mineral oil to prevent adhering of microspheres. (The microspheres are not stable in mineral oil.) Once the microspheres formed, the resulting emulsion was transferred to a beaker containing 300 ml of isopropyl alcohol (IPA) stirred continuously with a magnetic stir bar. IPA is a non-aqueous solution to extract acetonitrile from polymer solution and to wash mineral oil from the surface of microsphere. Hexane can be used as alternative non-aqueous washing solution. The microspheres are harvested by rinsing with IPA using vacuum filtration unit and dried under vacuum for an hour. O/O emulsion method could produce more porous microspheres in a short period of time.
EXAMPLE 7
A water/oil/water double emulsion (Formula A) and a water/oil and co-solvent single emulsion (Formula B) were prepared in accordance with Examples 3 and 4, respectively and their dissolutions were compared. The specific formulations are provided in the Tables below.
Table 4


Figure 11 compares the cumulative dissolution rate of the microsphere formulations A and B with an Alzet pump, and Figure 12 compares their respective release rates on a non-cumulative basis. The figures show that at least one of the formulations achieves a substantially zero order release rate as reflected by the Alzet pump data. The
representations also demonstrate that the formulations are capable of releasing a therapeutically effective amount of active agent over an extended period of time.
The following procedure was used to measure dissolution for Examples 2-7:
About 25mg of the sample was weighed and transferred into a 25ml volumetric flask. 2.5ml of Methylene Chloride was then added to dissolve the sample, and the solution was then diluted to volume with SGF and mixed well. The aqueous phase was filtered through a glass acrodisc filter and the aliquot was collected for HPLC analysis.
To prepare the sample for dissolution, 20 to 50mg of the sample was weighed and transferred into a centrifuge tube. 10ml of Phosphate Buffer pH 7.4 was added. The samples were incubated in a Brunswick Scientific Water Bath Shaker at 370C and lOOrpm. Samples were collected at appropriate time points until 100% release was released.
To collect the samples, the samples were removed from Brunswick Scientific Water Bath Shaker and centrifuged for 5 minutes at 3000rpm. 2ml of sample was pipetted out of centrifuge tube and collect in a scintillation vial. 2ml of fresh Phosphate Buffer pH7.4 was then pipetted into the centrifuge tube containing the sample. The centrifuge tube samples were returned to the Brunswick Scientific Water Bath Shaker for further sampling. Samples remaining in scintillation vials were filtered through a glass acrodisc filter, and the aliquot was collected for HPLC analysis,
EXAMPLE 8 Clonidine Fatty Acid Salt Preparation
Preparations of the salts involved dissolving a 1 : 1 molar equivalent of clonidine and the desired fatty acid into an organic solvent. In this example, Ig of clonidine was dissolved with 1.2g linoleic acid, l.lg palmitic acid, and 1.2g stearic acid in their respective containers into isopropanol. Other organic solvents were used such as methanol, ethanol, and chloroform and yielded similar if not the same results. Isopropanol was used for the rest of the study taking into consideration future manufacturing costs and the lower cost of isopropanol compared to the other solvents. The mixture was stirred until the
clonidine and fatty acids were completely dissolved. The mixture was then allowed to air dry to evaporate the solvent and the solid waxy residue that remains was collected, mixed/crushed for uniformity, and allowed to air dry.
The samples were then analyzed via DSC to observe a change in melting point and HPLC to record the solubility.
The following table contains the summary of the results:
Table 6

EXAMPLE 9
Micronized clonidine base was suspended in double distilled water and the mixture was vortexed at 3000 rpm for 10 sec to disperse the drug. Polymer solution was prepared by dissolving 600 mg PLGA 50/50 (i.v. 0.26 - 0.54 dl/g, end capped) in 1 mL of methylene chloride. Polymer solution was added into above drug suspension and homogenized at 13,000 RPM for 1 minute to form the first emulsion. Then 1 mL of 2% (w/w) polyvinyl alcohol (PVA, 25 kD) solution was added. After vortexing at 3000 rpm for 30 sec, the emulsion was added into 100 ml of 0.5% (w/w) PVA (25 kD) solution containing 2% (w/w) NaCl under stirring at ~ 600 rpm, and hardened for 4.5 hr at room temperature. The hardened microspheres were sieved through sieves of 106, 53 and 25 um sizes and washed by double-distilled water three times before further collection by vacuum filtration. The collected microspheres were then placed into a freeze-drier (Vertis
Genesis 25 LE) and dried for 2 days after freezing the microspheres overnight. The size range of the microspheres collected was 53-106 um, and the encapsulation efficiency was measured to be 74%, The in vitro dissolution profile is shown in Figure 13 and demonstrates that formulations of the present invention can release therapeutically effective amounts of clonidine for extended periods of time.
EXAMPLE 10
Clonidine base was dissolved in methylene chloride, and 250 mg of PLGA 50/50 (i.v. 0.26 - 0.54 dl/g, end-capped) was added and the mixture was vortexed until polymer dissolved completely. 300 mg of PLGA 85/15 (i.v. 0.55 - 0.75 dl/g, end-capped) was dissolved separately in 1 ml of methylene chloride. Polymer solution of PLGA 50/50 containing drug was then added into PLGA 85/15 solution and homogenized at 19,000 rpm for 30 sec. Then 1.4 niL of 2% (w/w) polyvinyl alcohol (PVA, 25 kD) solution was added. After vortexing at 3000 rpm for 1 min, the emulsion was added into 140 ml of 0.5% (w/w) PVA (25 kD) solution containing 2% (w/w) NaCl under stirring at ~ 700 rpm, and hardened for 4.5 hr at room temperature. The hardened microspheres were sieved through sieves of 106, 53 and 25 Dm sizes, and microspheres of 53-106 Dm and 25-53 Dm were collected and washed by double-distilled water three times before further collection under vacuum filtration. The collected microspheres were then placed into a freeze-drier (Vertis Genesis 25 LE) and dried for 2 days after freezing the microspheres overnight. The encapsulation efficiency was measured to be 57% for microspheres of 53- 106 Dm and 51% for microspheres of 25-53 Dm. The in-vitro dissolution profile is shown in Figure 14. The results indicate that as microsphere particle size increases, the release rate of the active agent decreases.
EXAMPLE 11 Microsphere Stability Testing
Microspheres were manufactured using varying amounts of PLGA and PLA to determine drug load, as set forth below in Tables 7 and 8:
Table 7: PLGA Batches

Table 8: PLA Batches
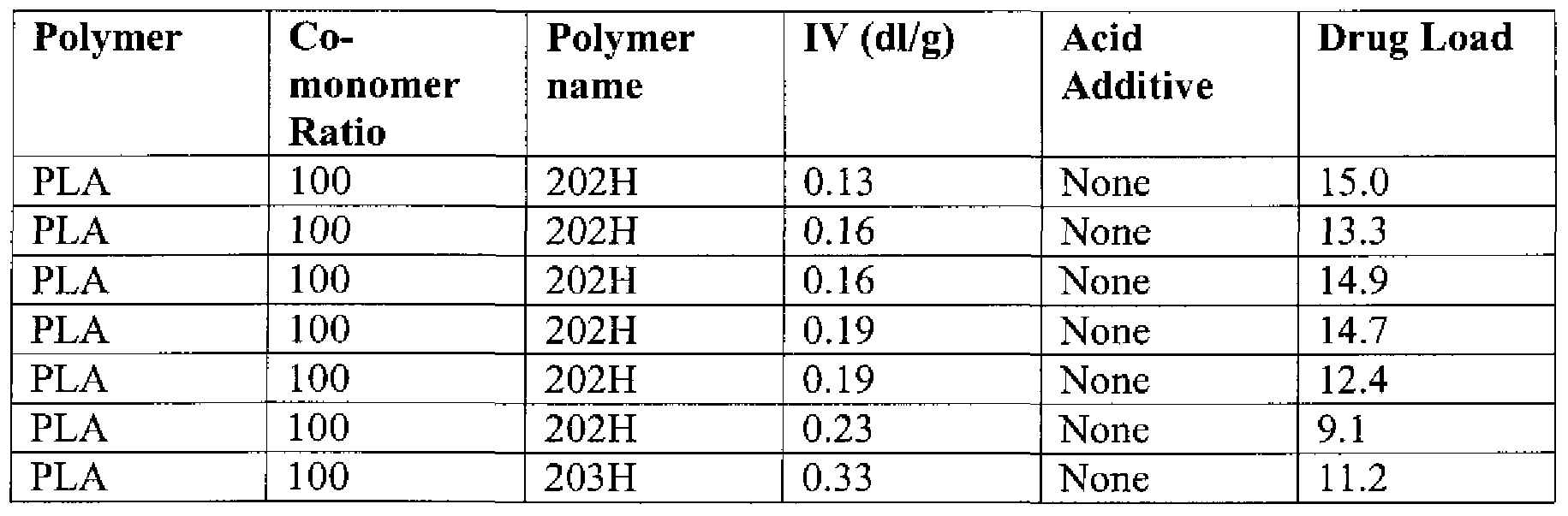
The effect of molecular weight on stability was then tested for specific formulations, as set forth in Table 9 below.
Table 9

Claims
1. A pharmaceutical depot formulation comprising: clonidine or a pharmaceutically acceptable salt or prodrug thereof; the depot formulation providing a mean in vitro release rate of clonidine of less than or equal to about 5 mg per day and releasing at least about 0.1 mg of clonidine per day for at least one day.
2. The depot formulation of claim 1, further comprising a dissolution modifying excipient.
3. The depot formulation of claim 1, comprising clonidine base.
4. The depot formulation of claim 1, comprising clonidine hydrochloride.
5. The depot formulation of claim 1 , comprising an insoluble salt of clonidine.
6. The depot formulation of claim 5, wherein the insoluble salt is a fatty acid salt.
7. The depot formulation of claim 1, comprising a therapeutically effective amount of a prodrug of clonidine represented by Formula I:
A-R
wherein A is a radical of clonidine; and
R is selected from the group consisting of a formate radical of the formula COO'; a formate radical of the formula R1COO", wherein R1 is an alkylene having from 1-30 carbons; a moiety selected from the group consisting of polyethylene glycol fatty acid esters, fatty alkylol amide condensates, alkylarylsulfonates, fatty alcohol sulfates, dialkyl esters of sodium sulfo succinate, fatty acid esters of sodium isothionate, polyoxyethylene ethers and thioethers, and long-chain quaternary ammonium chloride compounds; and a moiety selected from the group consisting of an amino acid, a dipeptide and a tripeptide.
8. The depot formulation of claim 1, comprising a therapeutically effective amount of a prodrug of clonidine represented by Formula I:
A-B
wherein A is a radical of clonidine; and
R is selected from the group consisting of an aldehyde (-CHO), a ketone (-COR ), a carboxylic acid (-COOH), an ester (-COOR2), an amide (-CONR2R3), an enone (- C(O)C(R2)CR3R4), an acyl chloride (-COC1) or an acid anhydride (-COOCOR2). In such embodiments, R2, R3 and R4 are independently selected from the group consisting of alky], alkenyl, aryl, heteroaryl, alkoxy, aryloxy and heteroaryloxy moieties wherein the alkyl moiety is selected from the group consisting of unsubstituted or substituted, straight-chain, branched-chain and cyclic alkyl moieties having 1-20 carbon atoms; wherein the alkenyl moiety is selected from the group consisting of unsubstituted and substituted, straight-chain, branched-chain and cyclic alkenyl moieties having 2-20 carbon atoms; wherein the aryl moiety is selected from the group consisting of unsubstituted and substituted phenyl, and phenalkyl moieties wherein the alkyl moiety contains 1-3 carbon atoms and the phenyl moiety is unsubstituted or substituted; and the heteroaryl moiety is a substituted or unsubstituted aromatic 5-or 6-membered heterocyclic ring containing one or two heteroatoms selected from the group consisting of nitrogen, oxygen, and sulfur; and pharmaceutically acceptable salts thereof.
9. The depot formulation of claim 1, wherein the clonidine or pharmaceutically effective salt or prodrug thereof is in the form of particles having an effective average particle size of less than about 50 um.
10. The depot formulation of claim 2, wherein the dissolution modifying excipient is a biocompatible polymer.
11. The depot formulation of claim 10, wherein the biocompatible polymer is selected from the group consisting of a biodegradable polymer, a non-biodegradable polymer, or a combination thereof.
12. The depot formulation of claim 11, wherein the biodegradable polymer is selected from the group consisting of poly(lactide)s, poly(glycolide)s, poly(lactide-co-glycolide)s, poly(lactic acid)s, poly(glycolic acid)s, poly(lactic acid-co-glycolic acid)s, polyanhydrides, polyorthoesters, polyetheresters, polycaprolactone, polyesteramides, polyphosphazenes, polysaccharides, proteinaceous polymers, soluble derivatives of polysaccharides, soluble derivatives of proteinaceous polymers, polypeptides, polyesters, and combinations thereof.
13. The depot formulation of claim 10, wherein the biodegradable polymer has a molecular weight of about 2,000 Daltons to about 2,000,000 Daltons when formulation is complete.
14. The depot formulation of claim 13, wherein the biodegradable polymer is a poly(lactide-co-glycolide) with a molecular weight of about 5,000 Daltons to about 100,000 Daltons.
15. The depot formulation of claim 13, wherein the poly(lactide-co-glycolide) has a molecular weight of about 20,000 Daltons to about 75,000 Daltons.
16. The depot formulation of claim 13, wherein the poly (lactide-co-glycolide) has a lactide: glycolide ratio of about 10:1 to about 1:10; about 5:1 to about 1:5 about 3:1 to about 1 :3 ; or about 1: 1.
17. The depot formulation of claim 1, providing a release rate of clonidine (as measured in a phosphate buffer at pH 7.4, 0.1% Tween 80, 37 degrees Celsius, and water bath shaker speed of 100 RPM) of at least about 0.1 mg per day for at least 3 days.
18. The depot formulation of claim 1, providing a release rate of clonidine (as measured in a phosphate buffer at pH 7.4, 0.1% Tween 80, 37 degrees Celsius, and water bath shaker speed of 100 RPM) of at least about 0.1 mg per day for at least 5 days.
19. The depot formulation of claim 1, providing a release rate of clonidine (as measured in a phosphate buffer at pH 7.4, 0.1% Tween 80, 37 degrees Celsius, and water bath shaker speed of 100 RPM) of at least about 0.1 mg per day for at least 7 days.
20. The depot formulation of claim 1, providing a release rate of clonidine (as measured in a phosphate buffer at pH 7.4, 0.1% Tween 80, 37 degrees Celsius, and water bath shaker speed of 100 RPM) of at least about 0.1 mg per day for at least 14 days.
21. The depot formulation of claim 1, providing a release rate of clonidine (as measured in a phosphate buffer at pH 7.4, 0.1% Tween 80, 37 degrees Celsius, and water bath shaker speed of 100 RPM) of at least about 0.1 mg per day for at least 1 month,
22. The depot formulation of claim 1, wherein from about 10% to about 60% clonidine is released after 4 days, from about 20% to about 80% clonidine is released after 8 days and from about 30% to about 95% clonidine is released after 12 days and less than 100% clonidine is released after 16 days when tested in a pH 7.4 buffered solution at 37°C and lOOrpm.
23. The depot formulation of claim 1, wherein from about 10% to about 60% clonidine is released after 4 days, from about 20% to about 80% clonidine is released after 8 days and from about 30% to about 95% clonidine is released after 12 days and less than 100% clonidine is released after 16 days when tested in a neutral pH buffered solution at 37°C and lOOrpm.
24. A pharmaceutical depot formulation comprising: a therapeutically effective amount of clonidine or a pharmaceutically acceptable salt or prodrug thereof; wherein from about 10% to about 60% clonidine is released after 4 days, from about 20% to about 80% clonidine is released after 8 days and from about 30% to about 95% clonidine is released after 12 days and less than 100% clonidine is released after 16 days when tested in a pH 7.4 buffered solution at 37°C and lOOrpm.
25. The depot formulation of claim 24, further comprising a dissolution modifying excipient.
26. A method of preparing a pharmaceutical depot formulation comprising preparing a mixture by contacting clonidine or a pharmaceutically acceptable salt thereof with an excipient to decrease the solubility of the clonidine, and incorporating the mixture into a pharmaceutically acceptable vehicle suitable for injection.
27. A method of preparing a pharmaceutical depot formulation comprising incorporating an insoluble salt of clonidine into a pharmaceutically acceptable vehicle suitable for injection.
28. A prodrug of clonidine represented by Formula I:
A-R
wherein A is a radical of clonidine; and
R is selected from the group consisting of a formate radical of the formula COO"; a formate radical of the formula R1COO", wherein R1 is an alkylene having from 1-30 carbons; a moiety selected from the group consisting of polyethylene glycol fatty acid esters, fatty alkylol amide condensates, alkylarylsulfonates, fatty alcohol sulfates, dialkyl esters of sodium sulfosuccinate, fatty acid esters of sodium isothionate, polyoxyethylene ethers and thioethers, and long-chain quaternary ammonium chloride compounds; and a moiety selected from the group consisting of an amino acid, a dipeptide and a tripeptide.
29. The prodrug of clonidine of claim 28, wherein the amino acid is natural or synthetic.
30. The prodrug of clonidine of claim 28, wherein the amino acid is an L-amino acid.
31. The prodrug of clonidine of claim 28, wherein the amino acid is a D-amino acid.
32. The prodrug of clonidine of claim 28, wherein the amino acid is an alpha, beta, or gamma amino acid.
33. The prodrug of clonidine of claim 28, wherein the amino acid selected from the group consisting of valine, leucine, isoleucine, methionine, phenylalanine, asparagine, glutamic acid, glutamine, histidine, lysine, arginine, aspartic acid, glycine, alanine, serine, threonine, tyrosine, tryptophan, cysteine and proline.
34. The prodrug of clonidine of claim 28, wherein the amino acid selected from the group consisting of gamma- carboxyglutamic acid, selenocysteine, desmosine, 6-N- methyllysine, epsilon-N,N,N-trimethyllysine, 3-methylhistidine, O-phosphoserine, 5- hydroxylysine, epsilon-N-acetyllysine, omega.-N-methylarginine, N-acetylserine, gamma-aminobutyric acid, citrulline, ornithine, azaserine, homocysteine, beta- cyanoalanine and S-adenosylmethionine.
35. The prodrug of clonidine of claim 28, wherein the amino acid selected from the group consisting of phenyl glycine, meta-tyrosine, para-amino phenylalanine, 3-(3- pyridyl)-L-alanine, and 4-(trifluoromethyl)-D-phenylalanine.
36. A pharmaceutical depot formulation comprising: a. an active pharmaceutical ingredient in hase form, said active pharmaceutical ingredient being at least sparingly soluble in water; and b. a biocompatible polymer, wherein said depot formulation releases an amount of active pharmaceutical ingredient during the first twenty- four hour period equal to the amount of active pharmaceutical ingredient released during the seventh twenty-four hour period.
37. The formulation of claim 36, wherein said active pharmaceutical ingredient has a water solubility of about 0.1 mg/mL or greater.
38. The formulation of claim 36, wherein said active pharmaceutical ingredient has a water solubility of about 1.0 mg/mL or greater.
39. The formulation of claim 36, wherein said active pharmaceutical ingredient has a water solubility of from about 1.0 mg/mL to about 2.0 mg/mL.
40. The formulation of claim 36, wherein said active pharmaceutical ingredient is an amine.
41. The formulation of claim 40, wherein said active pharmaceutical ingredient is clonidine.
42. The formulation of claim 36, wherein said biocompatible polymer is selected from the group consisting of PLA, PLGA and PGA.
43. The formulation of claim 42, wherein the biocompatible polymer is PLGA.
44. The formulation of claim 42, wherein the biocompatible polymer is PLA.
45. The formulation of claim 43, further comprising an organic acid.
46. The formulation of claim 45, wherein said organic acid is a hydrophobic acid.
47. The formulation of claim 46, wherein said hydrophobic acid is selected from the group consisting of stearic acid, linoleic acid, and palmitic acid.
48. The formulation of claim 47, wherein said hydrophobic acid is palmitic acid.
49. The formulation of claim 41, comprising from about 0.1 mg to about 10 mg of clonidine.
50. The formulation of claim 41, comprising from about 0.1 mg to about 1.0 mg of clonidine.
51. The formulation of claim 36, wherein said amount of active pharmaceutical agent released is measured in a pH 7.4 Phosphate buffered solution at 37°C and lOOrpm.
52. The formulation of claim 51, wherein the release of active pharmaceutical ingredient during the fourteenth twenty- four hour period is equal to the amount of active pharmaceutical ingredient released during the first twenty-four hour period.
53. The formulation of claim 52, wherein the release of active pharmaceutical ingredient during the thirtieth twenty-four hour period is equal to the amount of active pharmaceutical ingredient released during the first twenty- four hour period.
54. The formulation of claim 53, wherein the release of active pharmaceutical ingredient during the ninetieth twenty-four hour period is equal to the amount of active pharmaceutical ingredient released during the first twenty-four hour period.
55. A pharmaceutical depot formulation comprising:
(a) clonidine base, salt or prodrug thereof;
(b) a biocompatible polymer, and
(c) optionally a hydrophobic acid, wherein said depot formulation exhibits an in vitro clonidine release profile comprising
(i) a first amount of clonidine released during a first twenty- four hour period of said profile, and
(ii) a second amount of said clonidine released during a second twenty-four hour period of said profile, wherein said second twenty-four hour period occurs at least six days after said first twenty-four hour period.
56. The depot formulation of claim 55, wherein said biocompatible polymer is selected from the group consisting of PLA and PLGA.
57. The depot formulation of claim 56, wherein said hydrophobic acid is selected from the group consisting of stearic acid, linoleic acid, and palmitic acid.
58. The depot formulation of claim 55, wherein the in vitro clonidine release profile further comprises a third amount of clonidine released during a third twenty-four hour period of said profile, said third twenty-four hour period occurring at least fourteen days following said first twenty-four hour period.
59. The depot formulation of claim 58, wherein the in vitro clonidine release profile further comprises a fourth amount of clonidine released during a fourth twenty- four hour period of said profile, said fourth twenty-four hour period occurring at least thirty days following said first twenty- four hour period.
60. The depot formulation of claim 59, wherein the in vitro clonidine release profile further comprises a fifth amount of clonidine released during a fifth twenty-four hour period of said profile, said fifth twenty-four hour period occurring at least ninety days following said first twenty- four hour period.
61. The depot formulation of claim 59, wherein said first amount released in said first twenty-four hour period comprises less than twenty percent of the total amount of clonidine in said formulation.
62. The depot formulation of claim 55, wherein said profile is a substantially zero order release profile.
63. The depot formulation of claim 55, wherein the polymer is in the form of microspheres.
64. The depot formulation of claim 63, wherein the average molecular weight of said polymer does not decrease by more than 40% when measured after thirty days of storage at 250C and 60% relative humidity.
65. The depot formulation of claim 63, wherein the weight average molecular weight of said polymer does not decrease by more than 20% when measured after thirty days of storage at 250C and 60% relative humidity.
66. The depot formulation of claim 63, wherein the weight average molecular weight of said polymer does not decrease by more than 40% when measured after thirty days of storage at 25°C and 60% relative humidity.
67. The depot formulation of claim 63, wherein the weight average molecular weight of said polymer does not decrease by more than 20% when measured after thirty days of storage at 25°C and 60% relative humidity.
68. The depot formulation of claim 55, wherein the mass of clonidine in said formulation is not less than about eight percent of the total mass of the formulation.
69. The depot formulation of claim 55, wherein the mass of clonidine in said formulation is not less than nine percent of the total mass of the formulation.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US2733308P | 2008-02-08 | 2008-02-08 | |
| US61/027,333 | 2008-02-08 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2009100441A2 true WO2009100441A2 (en) | 2009-08-13 |
| WO2009100441A3 WO2009100441A3 (en) | 2010-01-14 |
Family
ID=40952737
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2009/033574 WO2009100441A2 (en) | 2008-02-08 | 2009-02-09 | Depot formulations |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2009100441A2 (en) |
Cited By (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20090264489A1 (en) * | 2008-04-18 | 2009-10-22 | Warsaw Orthopedic, Inc. | Method for Treating Acute Pain with a Formulated Drug Depot in Combination with a Liquid Formulation |
| WO2009129439A2 (en) * | 2008-04-18 | 2009-10-22 | Medtronic, Inc. | Medical devices and methods including polymers having biologically active agents therein |
| WO2009129460A2 (en) * | 2008-04-18 | 2009-10-22 | Warsaw Orthopedic, Inc. | Clonidine formulations in a biodegradable polymer carrier |
| US20110182849A1 (en) * | 2010-01-28 | 2011-07-28 | Warsaw Orthopedic, Inc. | Compositions and methods for treating an intervertebral disc using bulking agents or sealing agents |
| WO2011133370A1 (en) * | 2010-04-23 | 2011-10-27 | Medtronic, Inc. | Shelf stable pharmaceutical depot |
| US20120142643A1 (en) * | 2010-12-03 | 2012-06-07 | Warsaw Orthopedic, Inc. | Clonidine and gaba compounds in a biodegradable polymer carrier |
| US8231891B2 (en) | 2009-07-31 | 2012-07-31 | Warsaw Orthopedic, Inc. | Implantable drug depot for weight control |
| US8470360B2 (en) | 2008-04-18 | 2013-06-25 | Warsaw Orthopedic, Inc. | Drug depots having different release profiles for reducing, preventing or treating pain and inflammation |
| US20130266631A1 (en) * | 2012-04-05 | 2013-10-10 | Warsaw Orthopedic, Inc. | Clonidine compounds in a biodegradable matrix |
| US8617583B2 (en) | 2009-07-17 | 2013-12-31 | Warsaw Orthopedic, Inc. | Alpha adrenergic receptor agonists for prevention or treatment of a hematoma, edema, and/or deep vein thrombosis |
| US8629172B2 (en) | 2008-04-18 | 2014-01-14 | Warsaw Orthopedic, Inc. | Methods and compositions for treating post-operative pain comprising clonidine |
| US8722079B2 (en) | 2008-04-18 | 2014-05-13 | Warsaw Orthopedic, Inc. | Methods for treating conditions such as dystonia and post-stroke spasticity with clonidine |
| US8822546B2 (en) | 2008-12-01 | 2014-09-02 | Medtronic, Inc. | Flowable pharmaceutical depot |
| US8889173B2 (en) | 2008-04-18 | 2014-11-18 | Warsaw Orthopedic, Inc. | Alpha adrenergic receptor agonists for treatment of pain and/or inflammation |
| US8956641B2 (en) | 2008-04-18 | 2015-02-17 | Warsaw Orthopedic, Inc. | Alpha adrenergic receptor agonists for treatment of inflammatory diseases |
| US9005634B2 (en) | 2011-04-13 | 2015-04-14 | Medtronic, Inc. | Shelf stable pharmaceutical depot |
| US9072727B2 (en) | 2008-04-18 | 2015-07-07 | Warsaw Orthopedic, Inc. | Alpha adrenergic receptor agonists for treatment of degenerative disc disease |
| US9132085B2 (en) | 2008-04-18 | 2015-09-15 | Warsaw Orthopedic, Inc. | Compositions and methods for treating post-operative pain using clonidine and bupivacaine |
| US9132119B2 (en) | 2008-04-18 | 2015-09-15 | Medtronic, Inc. | Clonidine formulation in a polyorthoester carrier |
| US9358223B2 (en) | 2009-10-26 | 2016-06-07 | Warsaw Orthopedic, Inc. | Formulation for preventing or reducing bleeding at a surgical site |
| WO2016110865A1 (en) * | 2015-01-06 | 2016-07-14 | Cellix Bio Private Limited | Compositions and methods for the treatment of inflammation and pain |
| US9492461B2 (en) | 2008-04-18 | 2016-11-15 | Warsaw Orthopedic, Inc. | Methods and compositions for treating intervertebral disc herniations |
| US9610243B2 (en) | 2008-04-18 | 2017-04-04 | Warsaw Orthopedic, Inc. | Clonidine compounds in a biodegradable polymer |
| US10653619B2 (en) | 2009-03-23 | 2020-05-19 | Medtronic, Inc. | Drug depots for treatment of pain and inflammation |
| USRE48948E1 (en) | 2008-04-18 | 2022-03-01 | Warsaw Orthopedic, Inc. | Clonidine compounds in a biodegradable polymer |
| US11357756B2 (en) * | 2017-01-20 | 2022-06-14 | Warsaw Orthopedic, Inc. | Anesthetic compositions and methods comprising imidazoline compounds |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20070196415A1 (en) * | 2002-11-14 | 2007-08-23 | Guohua Chen | Depot compositions with multiple drug release rate controls and uses thereof |
| US20070232529A1 (en) * | 2000-08-22 | 2007-10-04 | New River Pharmaceuticals Inc. | Active agent delivery systems and methods for protecting and administering active agents |
| US20070243228A1 (en) * | 2006-04-13 | 2007-10-18 | Mckay William F | Drug depot implant designs and methods of implantation |
| US20070251530A1 (en) * | 2006-04-28 | 2007-11-01 | Medtronic, Inc. | Screening candidates for implantable infusion devices |
-
2009
- 2009-02-09 WO PCT/US2009/033574 patent/WO2009100441A2/en active Application Filing
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20070232529A1 (en) * | 2000-08-22 | 2007-10-04 | New River Pharmaceuticals Inc. | Active agent delivery systems and methods for protecting and administering active agents |
| US20070196415A1 (en) * | 2002-11-14 | 2007-08-23 | Guohua Chen | Depot compositions with multiple drug release rate controls and uses thereof |
| US20070243228A1 (en) * | 2006-04-13 | 2007-10-18 | Mckay William F | Drug depot implant designs and methods of implantation |
| US20070251530A1 (en) * | 2006-04-28 | 2007-11-01 | Medtronic, Inc. | Screening candidates for implantable infusion devices |
Cited By (49)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8889173B2 (en) | 2008-04-18 | 2014-11-18 | Warsaw Orthopedic, Inc. | Alpha adrenergic receptor agonists for treatment of pain and/or inflammation |
| US9833548B2 (en) | 2008-04-18 | 2017-12-05 | Warsaw Orthopedic, Inc. | Methods and compositions for treating post-operative pain comprising clonidine |
| WO2009129460A2 (en) * | 2008-04-18 | 2009-10-22 | Warsaw Orthopedic, Inc. | Clonidine formulations in a biodegradable polymer carrier |
| US9387197B2 (en) | 2008-04-18 | 2016-07-12 | Warsaw Orthopedic, Inc. | Methods for treating conditions such as dystonia and post-stroke spasticity with clonidine |
| WO2009129460A3 (en) * | 2008-04-18 | 2010-02-25 | Warsaw Orthopedic, Inc. | Clonidine formulations in a biodegradable polymer carrier |
| USRE48948E1 (en) | 2008-04-18 | 2022-03-01 | Warsaw Orthopedic, Inc. | Clonidine compounds in a biodegradable polymer |
| US9492461B2 (en) | 2008-04-18 | 2016-11-15 | Warsaw Orthopedic, Inc. | Methods and compositions for treating intervertebral disc herniations |
| US8946277B2 (en) | 2008-04-18 | 2015-02-03 | Warsaw Orthopedic, Inc. | Clonidine formulations in a biodegradable polymer carrier |
| US9775800B2 (en) | 2008-04-18 | 2017-10-03 | Warsaw Orthopedic, Inc. | Compositions and methods for treating post-operative pain using clonidine and bupivacaine |
| US8470360B2 (en) | 2008-04-18 | 2013-06-25 | Warsaw Orthopedic, Inc. | Drug depots having different release profiles for reducing, preventing or treating pain and inflammation |
| US9770438B2 (en) | 2008-04-18 | 2017-09-26 | Warsaw Orthopedic, Inc. | Clonidine formulation in a polyorthoester carrier |
| US8557273B2 (en) | 2008-04-18 | 2013-10-15 | Medtronic, Inc. | Medical devices and methods including polymers having biologically active agents therein |
| US20090264489A1 (en) * | 2008-04-18 | 2009-10-22 | Warsaw Orthopedic, Inc. | Method for Treating Acute Pain with a Formulated Drug Depot in Combination with a Liquid Formulation |
| US8956641B2 (en) | 2008-04-18 | 2015-02-17 | Warsaw Orthopedic, Inc. | Alpha adrenergic receptor agonists for treatment of inflammatory diseases |
| US8722079B2 (en) | 2008-04-18 | 2014-05-13 | Warsaw Orthopedic, Inc. | Methods for treating conditions such as dystonia and post-stroke spasticity with clonidine |
| US9351959B2 (en) | 2008-04-18 | 2016-05-31 | Warsaw Orthopedic, Inc. | Alpha adreneric receptor agonists for treatment of degenerative disc disease |
| WO2009129439A3 (en) * | 2008-04-18 | 2010-01-28 | Medtronic, Inc. | Medical devices and methods including polymers having biologically active agents therein |
| WO2009129439A2 (en) * | 2008-04-18 | 2009-10-22 | Medtronic, Inc. | Medical devices and methods including polymers having biologically active agents therein |
| US8629172B2 (en) | 2008-04-18 | 2014-01-14 | Warsaw Orthopedic, Inc. | Methods and compositions for treating post-operative pain comprising clonidine |
| US8968767B2 (en) | 2008-04-18 | 2015-03-03 | Warsaw Orthopedic, Inc. | Drug depots having different release profiles for reducing, preventing or treating pain and inflammation |
| US8999368B2 (en) | 2008-04-18 | 2015-04-07 | Warsaw Orthopedic, Inc. | Medical devices and methods including polymers having biologically active agents therein |
| US9763917B2 (en) | 2008-04-18 | 2017-09-19 | Warsaw Orthopedic, Inc. | Clonidine formulations in a biodegradable polymer carrier |
| AU2009236115B2 (en) * | 2008-04-18 | 2015-05-21 | Medtronic, Inc. | Clonidine formulations in a biodegradable polymer carrier |
| US9610243B2 (en) | 2008-04-18 | 2017-04-04 | Warsaw Orthopedic, Inc. | Clonidine compounds in a biodegradable polymer |
| AU2009236094B2 (en) * | 2008-04-18 | 2015-06-18 | Medtronic, Inc. | Medical devices and methods including polymers having biologically active agents therein |
| US9072727B2 (en) | 2008-04-18 | 2015-07-07 | Warsaw Orthopedic, Inc. | Alpha adrenergic receptor agonists for treatment of degenerative disc disease |
| US9132085B2 (en) | 2008-04-18 | 2015-09-15 | Warsaw Orthopedic, Inc. | Compositions and methods for treating post-operative pain using clonidine and bupivacaine |
| US9132119B2 (en) | 2008-04-18 | 2015-09-15 | Medtronic, Inc. | Clonidine formulation in a polyorthoester carrier |
| US9211285B2 (en) | 2008-04-18 | 2015-12-15 | Warsaw Orthopedic, Inc. | Methods and compositions for treating post-operative pain comprising clonidine |
| US9265733B2 (en) | 2008-04-18 | 2016-02-23 | Warsaw Orthopedic, Inc. | Drug depots having different release profiles for reducing, preventing or treating pain and inflammation |
| US9585872B2 (en) | 2008-04-18 | 2017-03-07 | Warsaw Orthopedic, Inc. | Clonidine formulations in a biodegradable polymer carrier |
| US8822546B2 (en) | 2008-12-01 | 2014-09-02 | Medtronic, Inc. | Flowable pharmaceutical depot |
| US10653619B2 (en) | 2009-03-23 | 2020-05-19 | Medtronic, Inc. | Drug depots for treatment of pain and inflammation |
| US8617583B2 (en) | 2009-07-17 | 2013-12-31 | Warsaw Orthopedic, Inc. | Alpha adrenergic receptor agonists for prevention or treatment of a hematoma, edema, and/or deep vein thrombosis |
| US8231891B2 (en) | 2009-07-31 | 2012-07-31 | Warsaw Orthopedic, Inc. | Implantable drug depot for weight control |
| US9358223B2 (en) | 2009-10-26 | 2016-06-07 | Warsaw Orthopedic, Inc. | Formulation for preventing or reducing bleeding at a surgical site |
| US20110182849A1 (en) * | 2010-01-28 | 2011-07-28 | Warsaw Orthopedic, Inc. | Compositions and methods for treating an intervertebral disc using bulking agents or sealing agents |
| US9050274B2 (en) * | 2010-01-28 | 2015-06-09 | Warsaw Orthopedic, Inc. | Compositions and methods for treating an intervertebral disc using bulking agents or sealing agents |
| WO2011133370A1 (en) * | 2010-04-23 | 2011-10-27 | Medtronic, Inc. | Shelf stable pharmaceutical depot |
| US20120142643A1 (en) * | 2010-12-03 | 2012-06-07 | Warsaw Orthopedic, Inc. | Clonidine and gaba compounds in a biodegradable polymer carrier |
| US9301946B2 (en) * | 2010-12-03 | 2016-04-05 | Warsaw Orthopedic, Inc. | Clonidine and GABA compounds in a biodegradable polymer carrier |
| US9005634B2 (en) | 2011-04-13 | 2015-04-14 | Medtronic, Inc. | Shelf stable pharmaceutical depot |
| US20130266631A1 (en) * | 2012-04-05 | 2013-10-10 | Warsaw Orthopedic, Inc. | Clonidine compounds in a biodegradable matrix |
| US9511018B2 (en) * | 2012-04-05 | 2016-12-06 | Warsaw Orthopedic, Inc. | Clonidine compounds in a biodegradable matrix |
| CN107428697A (en) * | 2015-01-06 | 2017-12-01 | 塞尔利克斯生物私人有限公司 | For treating the composition and method of inflammation and pain |
| WO2016110865A1 (en) * | 2015-01-06 | 2016-07-14 | Cellix Bio Private Limited | Compositions and methods for the treatment of inflammation and pain |
| JP2018502915A (en) * | 2015-01-06 | 2018-02-01 | セリックス バイオ プライヴェート リミテッドCellix Bio Private Limited | Compositions and methods for the treatment of inflammation and pain |
| JP2020100623A (en) * | 2015-01-06 | 2020-07-02 | セリックス バイオ プライヴェート リミテッドCellix Bio Private Limited | Compositions and methods for treatment of inflammation and pain |
| US11357756B2 (en) * | 2017-01-20 | 2022-06-14 | Warsaw Orthopedic, Inc. | Anesthetic compositions and methods comprising imidazoline compounds |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2009100441A3 (en) | 2010-01-14 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2009100441A2 (en) | Depot formulations | |
| EP1429731B1 (en) | Nanoparticulate insulin formulations | |
| EP1658053B1 (en) | Novel compositions of sildenafil free base | |
| EP1895984B1 (en) | Nanoparticulate imatinib mesylate formulations | |
| CA2587710C (en) | Injectable nanoparticulate olanzapine formulations | |
| JP5707405B2 (en) | An oral solid dosage form containing nanoparticles and a method of formulating the dosage form using fish gelatin | |
| US20120121653A1 (en) | Novel mometasone compositions and methods of making and using the same | |
| US20060204588A1 (en) | Formulations of a nanoparticulate finasteride, dutasteride or tamsulosin hydrochloride, and mixtures thereof | |
| CA2492488A1 (en) | Liquid dosage compositions of stable nanoparticulate active agents | |
| KR20090015994A (en) | Nanoparticulate posaconazole formulations | |
| EP2279727A2 (en) | Nanoparticulate aripiprazole formulations | |
| KR20080017065A (en) | Nanoparticulate acetaminophen formulations | |
| EP1996161A2 (en) | Nanoparticulate carvedilol formulations | |
| EP1835889A1 (en) | Nanoparticulate tacrolimus formulations | |
| CA2601312A1 (en) | Injectable compositions of nanoparticulate immunosuppressive compounds | |
| JP2011121972A (en) | Fluticasone formulation | |
| FR2945950A1 (en) | ANTICANCER NANOPARTICLE COMPOSITIONS AND METHODS FOR PREPARING THE SAME | |
| KR20090023729A (en) | Compositions comprising nanoparticulate meloxicam and controlled release hydrocodone | |
| EP1898882B1 (en) | Nanoparticulate ebastine formulations | |
| JP2018199685A (en) | Nanoparticles of indirubin, derivatives thereof and methods for making and using the same | |
| US20090297596A1 (en) | Nanoparticulate and Controlled Release Compositions Comprising a Platelet Aggregation Inhibitor | |
| EP1855651A1 (en) | Nanoparticulate compositions of heterocyclic amide derivatives | |
| EP2043606A2 (en) | Nanoparticulate kinase inhibitor formulations |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09708208 Country of ref document: EP Kind code of ref document: A2 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 09708208 Country of ref document: EP Kind code of ref document: A2 |