WO2008121064A1 - New imidazo[4,5-b]pyridine-6-halo-7-aryl/heteroaryl compounds 705 - Google Patents
New imidazo[4,5-b]pyridine-6-halo-7-aryl/heteroaryl compounds 705 Download PDFInfo
- Publication number
- WO2008121064A1 WO2008121064A1 PCT/SE2008/050357 SE2008050357W WO2008121064A1 WO 2008121064 A1 WO2008121064 A1 WO 2008121064A1 SE 2008050357 W SE2008050357 W SE 2008050357W WO 2008121064 A1 WO2008121064 A1 WO 2008121064A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- imidazo
- alkyl
- pyridin
- hydrogen
- optionally substituted
- Prior art date
Links
- 0 CCNc(c(*)c(*C)c(N)c1NCC)c1NCC Chemical compound CCNc(c(*)c(*C)c(N)c1NCC)c1NCC 0.000 description 3
- YZZAAQKAWUKIQJ-UHFFFAOYSA-N COC(c(cc1)ccc1-c([nH]1)nc2c1ncc(Cl)c2I)=O Chemical compound COC(c(cc1)ccc1-c([nH]1)nc2c1ncc(Cl)c2I)=O YZZAAQKAWUKIQJ-UHFFFAOYSA-N 0.000 description 1
- KGOHNBDUDKZRLZ-UHFFFAOYSA-N COC(c(cc1)ccc1-c([nH]1)nc2c1ncc(F)c2I)=O Chemical compound COC(c(cc1)ccc1-c([nH]1)nc2c1ncc(F)c2I)=O KGOHNBDUDKZRLZ-UHFFFAOYSA-N 0.000 description 1
- SPEZMRDIZLDMBO-UHFFFAOYSA-N OC(c(cc1)ccc1-c([nH]1)nc2c1ncc(Cl)c2I)=O Chemical compound OC(c(cc1)ccc1-c([nH]1)nc2c1ncc(Cl)c2I)=O SPEZMRDIZLDMBO-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
Definitions
- the present invention relates to new compounds of formula I, as a free base or a pharmaceutically acceptable salt, solvate or solvate of salt thereof, to pharmaceutical formulations containing said compounds and to the use of said compounds in therapy.
- the present invention further relates to a process for the preparation of compounds of formula I and to new intermediates used therein.
- Glycogen synthase kinase 3 is a serine / threonine protein kinase composed of two isoforms ( ⁇ and ⁇ ), which are encoded by distinct genes but are highly homologous within the catalytic domain. GSK3 is highly expressed in the central and peripheral nervous system. GSK3 phosphorylates several substrates including tau, ⁇ -catenin, glycogen synthase, pyruvate dehydrogenase and elongation initiation factor 2b (eIF2b). Insulin and growth factors activate protein kinase B, which phosphorylates GSK3 on serine 9 residue and inactivates it.
- eIF2b elongation initiation factor 2b
- AD dementias dementias, and taupathies.
- AD Alzheimer's disease
- Glycogen synthase kinase 3 ⁇ GSK3 ⁇
- Tau phosphorylating kinase selectively phosphorylates the microtubule associated protein Tau in neurons at sites that are hyperphosphorylated in AD brains.
- Hyperphosphorylated tau has lower affinity for microtubules and accumulates as paired helical filaments, which are the main components that constitute neurofibrillary tangles and neuropil threads in AD brains.
- Neurofibrillary tangles are consistently found in diseases such as AD, amyotrophic lateral sclerosis, parkinsonism- dementia of Gaum, corticobasal degeneration, dementia pugilistica and head trauma, Down's syndrome, postencephalatic parkinsonism, progressive supranuclear palsy, Niemann-Pick's Disease and Pick's Disease.
- GSK3 ⁇ preferentially labels neurofibrillary tangles and has been shown to be active in pre -tangle neurons in AD brains. GSK3 protein levels are also increased by 50% in brain tissue from AD patients.
- GSK3 ⁇ phosphorylates pyruvate dehydrogenase, a key enzyme in the glycolytic pathway and prevents the conversion of pyruvate to acetyl-Co-A (Hoshi et al, PNAS 1996, 93: 2719-2723).
- Acetyl-Co-A is critical for the synthesis of acetylcholine, a neurotransmitter with cognitive functions.
- Accumulation of amyloid- ⁇ is an early event in AD.
- GSK Tg mice show increased levels of amyloid- ⁇ in brain.
- PDAPP mice fed with Lithium show decreased amyloid- ⁇ levels in hippocampus and decreased amyloid plaque area (Su et al., Biochemistry 2004, 43: 6899-6908).
- GSK3 ⁇ inhibition may have beneficial effects in progression as well as the cognitive deficits associated with Alzheimer's disease and other above-referred to diseases.
- GSK3 ⁇ activity is increased in cellular and animal models of neurodegeneration such as cerebral ischemia or after growth factor deprivation.
- the active site phosphorylation was increased in neurons vulnerable to apoptosis, a type of cell death commonly thought to occur in chronic and acute degenerative diseases such as cognitive disorders, Alzheimer's Disease, Parkinson's Disease, amyotrophic lateral sclerosis, Huntington's Disease and HIV dementia and traumatic brain injury; and as in ischemic stroke.
- Lithium was neuroprotective in inhibiting apoptosis in cells and in the brain at doses that resulted in the inhibition of GSK3 ⁇ .
- GSK3 ⁇ inhibitors could be useful in attenuating the course of neurodegenerative diseases.
- Bipolar Disorders (BD) BD
- Bipolar Disorders are characterised by manic episodes and depressive episodes. Lithium has been used to treat BD based on its mood stabilising effects. The disadvantage of lithium is the narrow therapeutic window and the danger of overdosing that can lead to lithium intoxication. The discovery that lithium inhibits GSK3 at therapeutic concentrations has raised the possibility that this enzyme represents a key target of lithium's action in the brain (Stambolic et al, Curr. Biol. 1996, 68(12): 1664-1668, 1996; Klein and Melton; PNAS 1996, 93:8455-8459; Gould et al., Neuropsychopharmacology, 2005, 30:1223-1237).
- GSK3 inhibitor has been shown to reduce immobilisation time in forced swim test, a model to assess on depressive behavior (O'Brien et al., J Neurosci 2004, 24(30): 6791-6798).
- GSK3 has been associated with a polymorphism found in bipolar II disorder (Szczepankiewicz et al., Neuropsychobiology. 2006, 53: 51-56). Inhibition of GSK3 ⁇ may therefore be of therapeutic relevance in the treatment of BD as well as in AD patients that have affective disorders.
- GSK3 is involved in signal transduction cascades of multiple cellular processes, particularly during neural development.
- GSK3 ⁇ levels were 41 % lower in the schizophrenic patients than in comparison subjects.
- This study indicates that schizophrenia involves neurodevelopmental pathology and that abnormal GSK3 regulation could play a role in schizophrenia.
- reduced ⁇ -catenin levels have been reported in patients exhibiting schizophrenia (Cotter et al., Neuroreport 1998, 9(7): 1379-1383).
- Atypical antipsychotics such as olanzapine, clozapine, quetiapine, and ziprasidone, inhibits GSK3 by increasing ser9 phosphorylation suggesting that antipsychotics may exert their beneficial effects via GSK3 inhibition (Li X. et al., Int. J.of Neuropsychopharmacol, 2007, 10: 7-19, Epubl. 2006, May 4).
- Insulin stimulates glycogen synthesis in skeletal muscles via the dephosphorylation and thus activation of glycogen synthase.
- GSK3 phosphorylates and inactivates glycogen synthase via dephosphorylation.
- GSK3 is also over-expressed in muscles from Type II diabetic patients (Nikoulina et al., Diabetes 2000 Feb; 49(2): 263- 71). Inhibition of GSK3 increases the activity of glycogen synthase thereby decreasing glucose levels by its conversion to glycogen.
- GSK3 inhibitors lowered plasma glucose levels up to 50 % (Cline et al., Diabetes, 2002, 51:
- GSK3 inhibition may therefore be of therapeutic relevance in the treatment of Type I and Type II diabetes and diabetic neuropathy.
- GSK3 phosphorylates and degrades ⁇ -catenin.
- ⁇ -catenin is an effector of the pathway for keratonin synthesis, ⁇ -catenin stabilisation may be lead to increase hair development.
- Mice expressing a stabilised ⁇ -catenin by mutation of sites phosphorylated by GSK3 undergo a process resembling de novo hair morphogenesis (Gat et al., Cell, 1998, 95(5): 605-14)).
- the new follicles formed sebaceous glands and dermal papilla, normally established only in embryogenesis.
- GSK3 inhibition may offer treatment for baldness.
- GSK3 inhibitors provide anti-inflammatory effects.
- Inflammation is a common feature of a broad range of conditions including Alzheimer's Disease and mood disorders.
- GSK3 is overexpressed in ovarian, breast and prostate cancer cells and recent data suggests that GSK3b may have a role in contributing to cell proliferation and survival pathways in several solid tumor types.
- GSK3 plays an important role in several signal transduction systems which influence cell proliferation and survival such as WNT, PI3 Kinase and NFkB.
- GSK3b deficient MEFs indicate a crucial role in cell survival mediated NFkB pathway (Ougolkov AV and Billadeau DD. Future Oncol. 2006 Feb;2(l):91-100.).
- GSK3 inhibitors may inhibit growth and survival of solid tumors, including pancreatic, colon and prostate cancer.
- Bone-related disorders and conditions GSK3 inhibitors could be used for treatment of bone-related disorders or other conditions, which involves a need for new and increased bone formation. Remodeling of the skeleton is a continuous process, controlled by systemic hormones such as parathyroid hormone (PTH), local factors (e.g. prostaglandin E 2 ), cytokines and other biologically active substances.
- PTH parathyroid hormone
- local factors e.g. prostaglandin E 2
- cytokines cytokines
- Two cell types are of key importance: osteoblasts (responsible for bone formation) and osteoclasts (responsible for bone resorption).
- Osteoporosis is a skeletal disorder in which low bone mass and deterioration of bone microarchitecture lead to increased bone fragility and fracture risk.
- the two main strategies are to either inhibit bone resorption or to stimulate bone formation.
- the majority of drugs currently on the market for the treatment of osteoporosis act to increase bone mass by inhibiting osteoclastic bone resorption. It is recognized that a drug with the capacity to increase bone formation would be of great value in the treatment of osteoporosis as well as having the potential to enhance fracture healing in patients.
- the object of the present invention is to provide compounds having a selective inhibiting effect at GSK3. Accordingly, the present invention provides a compound of the formula I:
- Q is selected from halogen
- R 1 is C(O)NR b R c ;
- R 2 and R 4 are independently selected from hydrogen, halogen, CN, NO 2 , Ci- 3 alkyl, Ci- ⁇ haloalkyl;
- R 3 and R 5 are independently selected from hydrogen, halogen, Ci- 3 alkyl and Ci- 3 haloalkyl;
- A is aryl or heteroaryl, optionally substituted with one or more CN, CO 2 H, Ci- 6 alkyl, C 1 - ehaloalkyl, halo,C(O)R a , OR k , C(O)NR b R c or S(O) n R m , wherein said Ci -6 alkyl or Ci- ⁇ haloalkyl is optionally substituted by at least one CN, OR a or NR b R c ;
- R a is selected from hydrogen, and wherein said or Ci- ⁇ haloalkyl is optionally substituted with one or more
- R b and R c may, together with the atom to which they are attached, form a 4-, 5- or 6- membered heterocyclic ring containing one or more heteroatoms selected from N, O or S, wherein said heterocyclic ring is optionally substituted with one or more halo, OR a , NR d R e , is optionally further substituted with one or more
- R d and R e are independently selected from hydrogen, Ci_ 6 alkyl or Ci- ⁇ haloalkyl, wherein said Ci_ 6 alkyl or Ci- ⁇ haloalkyl is optionally substituted with one or more OR a ; or R k is Ci_ 6 alkyl or Ci- ⁇ haloalkyl, wherein said Ci_ 6 alkyl or Ci- ⁇ haloalkyl is optionally substituted with at least one CN, OR a , NR b R c , C(O)NR b R c or NR b C(O)R c ; R m is C 1-3 alkyl, optionally substituted with at least one halo, CN, OR a , NR b R c or C(O)NR b R c ; n is 0 to 2; as a base or a pharmaceutically acceptable salt, solvate or solvate of a salt thereof.
- compounds of formula I wherein A is aryl optionally substituted with one or more OR k .
- said A is phenyl, substituted with one OR k , wherein R k is Ci- 6 alkyl.
- said A is phenyl, substituted with one OR k , wherein OR k represents methoxy.
- R 2 , R 3 , R 4 and R 5 represent hydrogen.
- R 1 is C(O)NR b R c ;
- R 2 and R 4 are hydrogen
- R 3 and R 5 are hydrogen
- A is aryl optionally substituted with one OR k ;
- R b and R c together with the atom to which they are attached, form a 6-membered heterocyclic ring containing one or two heteroatoms selected from N, O or S, wherein said heterocyclic ring is optionally substituted with one and
- R k is Ci -6 alkyl.
- R 1 is C(O)NR b R c ;
- R 2 and R 4 are hydrogen; R 3 and R 5 are hydrogen;
- A is aryl optionally substituted with one OR k ;
- R b and R c together with the atom to which they are attached, form a 6-membered heterocyclic ring containing one N heteroatom, wherein said heterocyclic ring is substituted with one and R k is Ci -6 alkyl.
- R 1 is C(O)NR b R c ;
- R 2 and R 4 are hydrogen
- R 3 and R 5 are hydrogen
- A is aryl optionally substituted with one OR k ;
- R b and R c together with the atom to which they are attached, form a piperazine, wherein said piperazine is substituted with one and
- R k is Ci -6 alkyl.
- R 1 is C(O)NR b R c ;
- R 2 and R 4 are hydrogen;
- R 3 and R 5 are hydrogen;
- A is phenyl optionally substituted with one OR k ;
- R b and R c together with the atom to which they are attached, form a piperazine, wherein said piperazine is substituted with one and R k is Ci -6 alkyl.
- compounds of formula I selected from: 6-Bromo-7-(4-methoxyphenyl)-2- ⁇ 4-[(4-methylpiperazin- 1 -yl)carbonyl]phenyl ⁇ -3H- imidazo[4,5- ⁇ ]pyridine hydrochloride;
- alkyl includes both straight and branched chains as well as cyclic alkyl groups.
- d- 6 alkyl having 1 to 6 carbon atoms may be, but is not limited to, methyl, ethyl, n-propyl, /-propyl, n-butyl, /-butyl, 5-butyl, t-butyl, n-pentyl, /-pentyl, t-pentyl, neo-pentyl, n-hexyl, /-hexyl or cyclohexyl.
- Ci-3alkoxy includes both straight and branched chains .
- the term “Ci-3alkoxy” having 1 to 3 carbon atoms may be, but is not limited to, methoxy, ethoxy, n-propoxy or i- propoxy.
- halo or halogen refers to fluorine, chlorine, bromine and iodine.
- haloalkyl refers to an alkyl group, defined as above, in which one or several of the hydrogen substituents have been replaced by halogen substituents, in which the term halogen is defined as above.
- aryl refers to an optionally substituted monocyclic or bicyclic hydrocarbon ring system containing at least one unsaturated aromatic ring.
- the "aryl” may be fused with a C 5 - 7 Cycloalkyl ring to form a bicyclic hydrocarbon ring system.
- heteroaryl refers to an aromatic heterocycle having at least one heteroatom ring member such as sulfur, oxygen or nitrogen.
- Heteroaryl groups include monocyclic and polycyclic (e.g., having 2, 3 or 4 fused rings) systems. Examples of heteroaryl groups include without limitation, pyridyl (i.e., pyridinyl), pyrimidinyl, pyrazinyl, pyridazinyl, triazinyl, furyl (i.e.
- furanyl quinolyl, isoquinolyl, thienyl, imidazolyl, thiazolyl, indolyl, pyrryl, oxazolyl, benzofuryl, benzothienyl, benzthiazolyl, isoxazolyl, pyrazolyl, triazolyl, tetrazolyl, indazolyl, 1,2,4-thiadiazolyl, isothiazolyl, benzothienyl, purinyl, carbazolyl, fluorenonyl, benzimidazolyl, indolinyl, and the like.
- the heteroaryl group has from 1 to about 20 carbon atoms, and in further embodiments from about 3 to about 20 carbon atoms. In some embodiments, the heteroaryl group contains 3 to about 14, 4 to about 14, 3 to about 7 or 5 to 6 ring-forming atoms. In some embodiments, the heteroaryl or heteroaromatic group has 1 to about 4, 1 to about 3 or 1 to 2 heteroatoms. In some embodiments, the heteroaryl or heteroaromatic group has 1 heteroatom.
- heterocyclic ring containing one or more heteroatoms independently selected from N, O or S refers to a mono- or bicyclic- heterocyclic ring which may be saturated or partly saturated and which may optionally contain a carbonyl function and which may be, but is not limited to, azetidinyl, imidazolidinyl, imidazolinyl, morpholinyl, piperazinyl, piperidinyl, piperidonyl, pyrazolidinyl, pyrazolinyl, pyrrolidinyl, pyrrolinyl, 1 -methyl- 1 ,4-diazepane, tetrahydropyranyl or thiomorpholinyl.
- the heterocyclic ring contains a heteroatom selected from S or N, these atoms may optionally be in an oxidised form.
- hydrochloride includes monohydrochloride, dihydrochloride, trihydrochloride and tetrahydrochloride salts.
- a suitable pharmaceutically acceptable salt of the compound of the invention is, for example, an acid-addition salt, for example an inorganic or organic acid.
- a suitable pharmaceutically acceptable salt of the compounds of the invention is an alkali metal salt, an alkaline earth metal salt or a salt with an organic base that affords a physiologically-acceptable cation.
- Some compounds of formula I may have sterogenic centres and/or geometric isomeric centres (E- and Z-isomers), and it is to be understood that the invention encompasses all such optical, diastereoisomers and geometric isomers.
- the present invention relates to the use of compounds of formula I as hereinbefore defined as well as to the salts thereof.
- Salts for use in pharmaceutical compositions will be pharmaceutically acceptable salts, but other salts may be useful in the production of the compounds of formula I.
- An object of the invention is to provide compounds of formula I for therapeutic use, especially compounds that are useful for the prevention and/or treatment of conditions associated with glycogen synthase kinase-3 (GSK3) in mammals including man. Particularly, compounds of formula I exhibiting a selective affinity for GSK-3.
- GSK3 glycogen synthase kinase-3
- Another object of the invention is wherein a compound of formula (1) or a pharmaceutically acceptable salt, solvate or in vivo hydrolysable ester thereof, or a pharmaceutical composition or formulation comprising a compound of formula (1) is administered concurrently, simultaneously, sequentially or separately with another pharmaceutically active compound or compounds selected from the following:
- antidepressants such as agomelatine, amitriptyline, amoxapine, bupropion, citalopram, clomipramine, desipramine, doxepin duloxetine, elzasonan, escitalopram, fiuvoxamine, fluoxetine, gepirone, imipramine, ipsapirone, maprotiline, nortriptyline, nefazodone, paroxetine, phenelzine, pro trip ty line, ramelteon, reboxetine, robalzotan, sertraline, sibutramine, thionisoxetine, tranylcypromaine, trazodone, trimipramine, venlafaxine and equivalents and pharmaceutically active isomer(s) and metabolite(s) thereof.
- antidepressants such as agomelatine, amitriptyline, amoxapine, bupropion, citalopram,
- atypical antipsychotics including for example quetiapine and pharmaceutically active isomer(s) and metabolite(s) thereof.
- antipsychotics including for example amisulpride, aripiprazole, asenapine, benzisoxidil, bifeprunox, carbamazepine, clozapine, chlorpromazine, debenzapine, divalproex, duloxetine, eszopiclone, haloperidol, iloperidone, lamotrigine, loxapine, mesoridazine, olanzapine, paliperidone, perlapine, perphenazine, phenothiazine, phenylbutylpiperidine, pimozide, prochlorperazine, risperidone, sertindole, sulpiride, suproclone, suriclone, thioridazine, trifluoperazine, trimetozine, valproate, valproic acid, zopiclone, zotepine, ziprasidone
- anxiolytics including for example alnespirone, azapirones,benzodiazepines, barbiturates such as adinazolam, alprazolam, balezepam, bentazepam, bromazepam, brotizolam, buspirone, clonazepam, clorazepate, chlordiazepoxide, cyprazepam, diazepam, diphenhydramine, estazolam, fenobam, flunitrazepam, flurazepam, fosazepam, lorazepam, lormetazepam, meprobamate, midazolam, nitrazepam, oxazepam, prazepam, quazepam, reclazepam, tracazolate, trepipam, temazepam, triazolam, uldazepam, zolazepam and equivalents and pharmaceutically active
- anticonvulsants including for example carbamazepine, valproate, lamotrogine, gabapentin and equivalents and pharmaceutically active isomer(s) and metabolite(s) thereof.
- Alzheimer's therapies including for example donepezil, memantine, tacrine and equivalents and pharmaceutically active isomer(s) and metabolite(s) thereof.
- Parkinson's therapies including for example deprenyl, L-dopa, Requip, Mirapex, MAOB inhibitors such as selegine and rasagiline, comP inhibitors such as Tasmar, A-2 inhibitors, dopamine reuptake inhibitors, NMDA antagonists, Nicotine agonists, Dopamine agonists and inhibitors of neuronal nitric oxide synthase and equivalents and pharmaceutically active isomer(s) and metabolite(s) thereof.
- MAOB inhibitors such as selegine and rasagiline
- comP inhibitors such as Tasmar, A-2 inhibitors, dopamine reuptake inhibitors, NMDA antagonists, Nicotine agonists, Dopamine agonists and inhibitors of neuronal nitric oxide synthase and equivalents and pharmaceutically active isomer(s) and metabolite(s) thereof.
- migraine therapies including for example almotriptan, amantadine, bromocriptine, butalbital, cabergoline, dichloralphenazone, eletriptan, frovatriptan, lisuride, naratriptan, pergolide, pramipexole, rizatriptan, ropinirole, sumatriptan, zolmitriptan, zomitriptan, and equivalents and pharmaceutically active isomer(s) and metabolite(s) thereof.
- (ix) stroke therapies including for example abciximab, activase, NXY-059, citicoline, crobenetine, desmoteplase,repinotan, traxoprodil and equivalents and pharmaceutically active isomer(s) and metabolite(s) thereof.
- urinary incontinence therapies including for example darafenacin, falvoxate, oxybutynin, propiverine, robalzotan, solifenacin, tolterodine and and equivalents and pharmaceutically active isomer(s) and metabolite(s) thereof.
- neuropathic pain therapies including for example gabapentin, lidoderm, pregablin and equivalents and pharmaceutically active isomer(s) and metabolite(s) thereof.
- nociceptive pain therapies such as celecoxib, etoricoxib, lumiracoxib, rofecoxib, valdecoxib, diclofenac, loxoprofen, naproxen, paracetamol and equivalents and pharmaceutically active isomer(s) and metabolite(s) thereof.
- insomnia therapies including for example agomelatine, allobarbital, alonimid, amobarbital, benzoctamine, butabarbital, capuride, chloral, cloperidone, clorethate, dexclamol, ethchlorvynol, etomidate, glutethimide, halazepam, hydroxyzine, mecloqualone, melatonin, mephobarbital, methaqualone, midaflur, nisobamate, pentobarbital, phenobarbital, propofol, ramelteon, roletamide, triclofos,secobarbital, zaleplon, Zolpidem and equivalents and pharmaceutically active isomer(s) and metabolite(s) thereof.
- mood stabilizers including for example carbamazepine, divalproex, gabapentin, lamotrigine, lithium, olanzapine, quetiapine, valproate, valproic acid, verapamil, and equivalents and pharmaceutically active isomer(s) and metabolite(s) thereof.
- Such combination products employ the compounds of this invention within the dosage range described herein and the other pharmaceutically active compound or compounds within approved dosage ranges and/or the dosage described in the publication reference.
- Another aspect of the present invention provides a process for preparing a compound of formula I as a free base or a pharmaceutically acceptable salt thereof.
- suitable protecting groups will be added to, and subsequently removed from, the various reactants and intermediates in a manner that will be readily understood by one skilled in the art of organic synthesis.
- Conventional procedures for using such protecting groups as well as examples of suitable protecting groups are described, for example, in "Protective Groups in Organic Synthesis", T.W. Greene, P.G.M. Wuts, Wiley-Interscience, New York, 1999.
- aromatic substitution reactions include the introduction of a nitro group using concentrated nitric acid, the introduction of an acyl group using, for example, an acyl halide and Lewis acid (such as aluminium trichloride) under Friedel Crafts conditions; the introduction of an alkyl group using an alkyl halide and Lewis acid (such as aluminium trichloride) under Friedel Crafts conditions; and the introduction of a halogeno group.
- modifications include the reduction of a nitro group to an amino group by for example, catalytic hydrogenation with a nickel catalyst or treatment with iron in the presence of hydrochloric acid with heating; oxidation of alkylthio to alkylsulphinyl or alkylsulphonyl.
- a suitable catalyst e.g. o-benzotriazol-1-yl- ⁇ /, ⁇ /, ⁇ r, ⁇ r-tetramethyluroniumhexafluorophosphate or O-(7-azabenzotriazol-l-yl)- N,7V,7V ',7V '-tetramethyluronium hexafluorophosphate, in a solvent such as acetonitrile, dimethyl formamide, or a mixture thereof.
- a suitable base such as N, N- diisopropylethylamine may be used in the reaction, which can be performed at a temperature in the range of 0 0 C to +20 0 C.
- Conversion of a compound of type IV into a chloride of type V can be achieved by (a) first, reacting the compound of type IV with an appropriate oxidant, e.g. m- chloroperbenzoic acid, in a suitable solvent, e.g. acetic acid, at a temperature in the range of +20 0 C to +30 0 C; (b) second, reaction of the formed intermediate with neat phosphorus oxychloride at a temperature in the range of +100 0 C to +150 0 C using an oil bath or a microwave oven.
- an appropriate oxidant e.g. m- chloroperbenzoic acid
- a suitable solvent e.g. acetic acid
- Formation of an amide of type VIII from the corresponding acid VI and an amine VII can be performed by reacting VI and VII in the presence of a suitable catalyst, e.g. o-benzotriazol-l-yl- ⁇ /, ⁇ /,N',N'- tetramethyluroniumhexafluorophosphate or O-(7-azabenzotriazol- 1 -yl)-N,N,N',N'- tetramethyluronium hexafluorophosphate in a solvent such as acetonitrile, dimethyl formamide, or a mixture thereof.
- a suitable catalyst e.g. o-benzotriazol-l-yl- ⁇ /, ⁇ /,N',N'- tetramethyluroniumhexafluorophosphate or O-(7-azabenzotriazol- 1 -yl)-N,N,N',N'- tetramethyluronium hexafluorophosphat
- a suitable base such as 7V,7V-diisopropylethylamine may be used in the reaction, which can be performed at a temperature in the range of 0 0 C to +20 0 C.
- a solution of VI in a solvent such as dimethyl acetamide can be first reacted with an activating agent such as 1,1 '-carbonylbis( ⁇ H- imidazole) at a temperature in the range of +80 0 C to +120 0 C, and then reacted with the amine VII at a temperature in the range of +100 0 C to +150 0 C, using an oil bath or a microwave oven.
- a compound of type V can be transformed into the corresponding iodide IX by (a) first, treatment with HCl in a suitable solvent such as diethyl ether to give the hydrochloride salt, and (b) second, reaction of the salt with NaI in a suitable solvent, e.g acetonitrile, at a temperature in the range of +150 0 C to +175 0 C using an oil bath or a microwave oven.
- a suitable solvent such as diethyl ether
- the reaction may be carried out using a suitable palladium catalyst such as Pd(PPh 3 ) 4 , Pd(dppf)Cl 2 or Pd(OAc) 2 or Pd 2 (dba) 3 together with a suitable ligand such as P(ter£-butyl) 3 , 2-(dicyclohexylphosphino)- biphenyl or 2-(2',6'-dimethoxybiphenyl)-dicyclohexylphosphine or a nickel catalyst such as nickel on charcoal or Ni(dppe)Cl 2 together with zinc and sodium triphenylphosphinetrimetasulfonate.
- a suitable base such as an alkyl amine, e.g.
- trie thy lamine, or potassium carbonate, sodium carbonate, cesium carbonate, sodium hydroxide or cesium fluoride may be used in the reaction, which can be performed in the temperature range of +20 0 C to +160 0 C, using an oil bath or a microwave oven, in a suitable solvent or solvent mixture such as toluene, tetrahydrofuran, dimethoxyethane/water, 7V, ⁇ /-dimethylformamide or dioxane.
- the boronic acid or boronic ester may be formed in situ, by reaction of the corresponding aryl halide (e.g., the aryl bromide) with an alkyllithium reagent such as butyllithium to form an intermediate aryl lithium species, which then is reacted with a suitable boron compound, e.g., trimethyl borate, tributyl borate or triisopropyl borate.
- a suitable boron compound e.g., trimethyl borate, tributyl borate or triisopropyl borate.
- Another objective of the invention are processes for the preparation of a compound of general formula I, wherein, R 1 , R 2 , R 3 , R 4 , R 5 and A are, unless specified otherwise, defined as in formula I and Q is halo, comprising of:
- An ester of type XI may be transformed into a compound of type Ia (I, wherein A is as defined above and wherein R b and R c are as defined as in formula I and wherein R 1 are CO 2 R and wherein R is alkyl, for example methyl or ethyl) by (a) first, heating neat with an amine VII at a temperature in the range of +180 0 C to +220 0 C using an oil bath or a microwave oven, and (b) second, after cooling, adding a suitable catalyst such as o- benzotriazol- 1 -yl- ⁇ /, ⁇ / > N',N'-tetramethyluroniumhexafiuorophosphate or O-(7- azabenzotriazol-l-yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate and continuing the reaction at a temperature in the range of 0 0 C to +20 0 C.
- a suitable catalyst
- Formation of an amide of type Ia can also be performed by reacting a carboxylic acid of type XII (wherein R 1 is CO 2 H) with an amine of type VII (R b and R c are as defined as in formula I), as described for the preparation of VIII from VI and VII.
- a process for preparing a compound of formula I comprising of: (i) Condensation of a diamine II and a carboxylic acid of type III by first reacting the components in the presence of a suitable catalyst, optionally with an added base, and then heating the resulting intermediate in a suitable organic acid.
- the acid XII can be first reacted with an activating agent, and then reacted with the amine.
- the hydrochloric salt of a compound of formula I may be obtained from a compound of formula I by treatment with hydrochloric acid at a temperature in the range of 0 0 C to +25 0 C, in a suitable solvent such as dichloromethane, tetrahydrofuran or a dichloromethane/methanol mixture .
- spectra were recorded at 400 MHz for proton and 100 MHz for carbon-13.
- the following reference signals were used: the middle line OfDMSO-J 6 ⁇ 2.50 ( 1 H), ⁇ 39.51 ( 13 C); the middle line of CD 3 OD ⁇ 3.31 ( 1 H) or ⁇ 49.15 ( 13 C), CDCl 3 ⁇ 7.26 ( 1 H) and the middle line Of CDCl 3 ⁇ 77.16 ( 13 C) (unless otherwise indicated).
- Mass spectra were recorded on a Waters LCMS consisting of an Alliance 2795 (LC), Waters PDA 2996 and a ZQ single quadrupole mass spectrometer.
- the mass spectrometer was equipped with an electrospray ion source (ESI) operated in a positive or negative ion mode.
- the capillary voltage was 3 kV and cone voltage was 30 V.
- the mass spectrometer was scanned between m/z 100-700 with a scan time of 0.3s.
- mass spectra were recorded on a Waters LC-MS system (Sample Manager 2777C, 1525 ⁇ binary pump, 1500 Column Oven, ZQ, PDA2996 and ELS detector, Sedex 85). Separation was performed using a Zorbax column (C8, 3.0 x 50 mm, 3 ⁇ m). A four minutes linear gradient was used starting at 100 % A (A: 95:5 10 mM NH 4 OAc:MeOH ) and ending at 100% B (MeOH). The ZQ was equipped with a combined APPI/APCI ion source and scanned in the positive mode between m/z 120-800 with a scan time of 0.3 s.
- the APPI repeller and the APCI corona were set to 0.86 kV and 0.80 ⁇ A, respectively.
- the desolvation temperature (300 0 C), desolvation gas (400 L/Hr) and cone gas (5 L/Hr) were constant for both APCI and APPI mode.
- Microwave heating was performed in a Creator or Smith Synthesizer Single-mode microwave cavity producing continuous irradiation at 2450 MHz.
- a typical workup procedure after a reaction consisted of extraction of the product with a solvent such as ethyl acetate, washing with water followed by drying of the organic phase over MgSO 4 or Na 2 SO 4 , filtration and concentration of the solution in vacuo.
- TLC Thin layer chromatography
- Merck TLC-plates Silica gel 60 F 2 S 4
- Flash chromatography was preformed on a Combi Flash ® CompanionTM using RediSepTM normal-phase flash columns. Typical solvents used for flash chromatography was mixtures of heptane/ethyl acetate.
- SCX ion exchange columns were performed on Isolute ® columns. Chromatography through ion exchange columns were typically performed in solvents or solvent mixtures such a methanol and 10% ammonia in methanol.
- Preparative chromatography was run on a Waters autopurification HPLC with a diode array detector.
- Narrow gradients with MeCN/(95:5 0.1M NH 4 OAc :MeCN) were used at a flow rate of 20 ml/min.
- purification was achieved on a semi preparative Shimadzu LC-8A HPLC with a Shimadzu SPD-IOA UV-vis.
- -detector equipped with a Waters Symmetry ® column (C18, 5 ⁇ m, 100 mm x 19 mm).
- Narrow gradients with MeCN/0.1% trifluoroacetic acid in MiIIiQ Water were used at a flow rate of 10 ml/min.
- hydrochloride salts of the final products were typically performed by dissolution in solvents or solvent mixtures such as diethyl ether, tetrahydrofuran, dichloromethane/methanol, followed by addition of IM HCl in diethyl ether.
- Pd(PPh 3 ) 4 tris(tri-phenylphosphine)palladium
- Ni(dppe)Cl 2 (1 ,2-bis(diphenylphosphino)ethane)nickel(II) chloride;
- DIPEA (21.9 mL, 126 mmol) was added to a suspension of 5-chloro pyridine -2, 3-diamine (6.0 g, 42.0 mmol), terephtalic acid monomethyl ester (9.06 g 50.3 mmol) and ⁇ BTU (19.1 g 50.3mmol) in MeCN (100 mL) and the reaction mixture was stirred at r.t. for 1 h. A precipitate that formed was collected and washed with MeCN. The solid was distributed into microwave vials with HO Ac (4 mL) and heated to +200 0 C for 10 minutes. The product precipitated at r.t.
- 6-Bromo-7-iodo-2- ⁇ 4-[(4-methylpiperazin- 1 -yl)carbonyl]phenyl ⁇ -3H-imidazo[4,5- ⁇ ]pyridine (described in Example 9) (150 mg, 0.29 mmol), 4-methoxyphenyl boronic acid (43 mg, 0.29 mmol), sodium carbonate (123 mg, 1.16 mmol) and PdCl 2 (dppf)*DCM (12 mg, 0.014 mmol) were mixed in T ⁇ F: water 9:1 (3 ml). The mixture was heated in a microwave reactor at 130° for 17 minutes. The mixture was filtered through diatomeous earth and the filtrate was concentrated.
- Triethylamine (2.412 mL, 17.31 mmol) was added to a suspension of 5-fluoropyridine-2,3- diamine (2.2 g, 17.31 mmol), 4-(methoxycarbonyl)benzoic acid (3.12 g, 17.31 mmol) and O-benzotriazol-1-yl-tetramethyluronium hexafluorophosphate (6.56 g, 17.31 mmol) in acetonitrile (15 mL) and the reaction mixture was stirred at r.t. for 1 h.The precipitate that formed was collected and washed with MeCN. The solid was distributed into microwave vials with HOAc (4 mL) and heated to +200 0 C for 5 minutes.
- Methyl 4-(7-chloro-6-fluoro-3H-imidazo[4,5- ⁇ ]pyridin-2-yl)benzoate (1.20 g, 3.93 mmol) was suspended in T ⁇ F. Hydrochloric acid (1 M in diethyl ether, 4 ml) was added and the solvents were evaporated. Sodium iodide (8.83 g, 58.9 mmol) and acetonitrile (40 ml) was added and the mixture was heated to 160° for 30 min in a microwave reactor. The mixture was poured onto NaHCO ⁇ (aq) containing Na2S2C>3. The solid was collected by filtration and washed with water.
- 6-Fluoro-7-iodo-2- ⁇ 4-[(4-methylpiperazin- 1 -yl)carbonyl]phenyl ⁇ -3H-imidazo[4,5- ⁇ ]pyridine 74 mg, 0.16 mmol
- 4-methoxyphenylboronic acid 36 mg, 0.24 mmol
- Na 2 CO 3 67 mg, 0.64 mmol
- PdCl 2 ⁇ pPf)-CH 2 Cl 2 adduct 13 mg, 0.02 mmol
- the compounds of formula (I) defined in the present invention are well suited for inhibiting glycogen synthase kinase-3 (GSK3). Accordingly, said compound of the present invention is expected to be useful in the prevention and/or treatment of conditions associated with glycogen synthase kinase-3 activity, i.e. the compounds may be used to produce an inhibitory effect of GSK3 in mammals, including human, in need of such prevention and/or treatment. GSK3 is highly expressed in the central and peripheral nervous system and in other tissues. Thus, it is expected that compound of the invention is well suited for the prevention and/or treatment of conditions associated with glycogen synthase kinase-3 in the central and peripheral nervous system.
- the compound of the invention is expected to be suitable for prevention and/or treatment of conditions associated with cognitive disorders and predemented states, especially dementia, Alzheimer's Disease (AD), Cognitive Deficit in Schizophrenia (CDS), Mild Cognitive Impairment (MCI), Age-Associated Memory Impairment (AAMI), Age-Related Cognitive Decline (ARCD) and Cognitive Impairement No Dementia (CIND), diseases associated with neurofibrillar tangle pathologies, especially dementia, Alzheimer's Disease (AD), Cognitive Deficit in Schizophrenia (CDS), Mild Cognitive Impairment (MCI), Age-Associated Memory Impairment (AAMI), Age-Related Cognitive Decline (ARCD) and Cognitive Impairement No Dementia (CIND), diseases associated with neurofibrillar tangle pathologies,
- AD Alzheimer's Disease
- CDS Cognitive Deficit in Schizophrenia
- MCI Mild Cognitive Impairment
- AAMI Age-Associated Memory Impairment
- ARCD Age-Rel
- Frontotemporal dementia Frontotemporal dementia Parkinson's Type (FTDP), progressive supranuclear palsy (PSP), Pick's Disease, Niemann-Pick's Disease, corticobasal degeneration (CBD), traumatic brain injury (TBI) and dementia pugilistica.
- FTD Frontotemporal dementia
- FTDP Frontotemporal dementia Parkinson's Type
- PSP progressive supranuclear palsy
- Pick's Disease Pick's Disease
- Niemann-Pick's Disease corticobasal degeneration
- TBI traumatic brain injury
- dementia pugilistica dementia pugilistica
- One embodiment of the invention relates to the prevention and/or treatment of Alzheimer's Disease, especially the use in the delay of the disease progression of Alzheimer's Disease.
- PD Parkinson's Disease
- ALS amyotrophic lateral sclerosis
- MND motor neuron diseases
- ADD attention deficit disorder
- ADHD attention deficit hyperactivity disorder
- affective disorders are Bipolar Disorder including acute mania, bipolar depression, bipolar maintenance, major depressive disorders (MDD) including depression, major depression, mood stabilization, schizoaffective disorders including schizophrenia, and dysthymia.
- ADD attention deficit disorder
- ADHD attention deficit hyperactivity disorder
- MDD major depressive disorders
- schizoaffective disorders including schizophrenia, and dysthymia.
- Type I diabetes Type II diabetes
- diabetic neuropathy alopecia
- inflammatory diseases and cancer.
- One embodiment of the invention relates to the use of a compound of the formula (I) , as a free base or a pharmaceutically acceptable salt thereof, in the prevention and/or treatment of bone-related disorders or conditions in mammals.
- One aspect of the invention is directed to the use of a compound of the formula (I) , as a free base or a pharmaceutically acceptable salt thereof, to treat osteoporosis.
- One aspect of the invention is directed to the use of a compound of the formula (I), as a free base or a pharmaceutically acceptable salt thereof, to increase and promote bone formation in mammals.
- One aspect of the invention is directed to the use of a compound of the formula (I), as a free base or a pharmaceutically acceptable salt thereof, to increase bone mineral density in mammals.
- Another aspect of the invention is directed to the use of a compound of the formula (I), as a free base or a pharmaceutically acceptable salt thereof, to reduce the rate of fracture and/or increase the rate of fracture healing in mammals.
- Another aspect of the invention is directed to the use of a compound of the formula (I), as a free base or a pharmaceutically acceptable salt thereof, to increase cancellous bone formation and/or new bone formation in mammals.
- Another aspect of the invention is directed to a method of prevention and/or treatment of bone-related disorders comprising administering to a mammal in need of such prevention and/or treatment, a therapeutically effective amount of a compound of the formula (I) as a free base or a pharmaceutically acceptable salt thereof.
- Another aspect of the invention is directed to a method of prevention and/or treatment of osteoporosis comprising administering to a mammal in need of such prevention and/or treatment, a therapeutically effective amount of a compound of the formula (I) as a free base or a pharmaceutically acceptable salt thereof.
- Another aspect of the invention is directed to a method of increasing bone formation comprising administering to a mammal in need of such treatment, a therapeutically effective amount of a compound of the formula (I) as a free base or a pharmaceutically acceptable salt thereof.
- Another aspect of the invention is directed to a method of increasing bone mineral density comprising administering to a mammal in need of such treatment, a therapeutically effective amount of a compound of the formula (I) as a free base or a pharmaceutically acceptable salt thereof.
- Another aspect of the invention is directed to a method of reducing the incidence of fracture comprising administering to a mammal in need of such treatment, a therapeutically effective amount of a compound of the formula (I) as a free base or a pharmaceutically acceptable salt thereof.
- Another aspect of the invention is directed to a method of enhancing fracture healing comprising administering to a mammal in need of such treatment, a therapeutically effective amount of a compound of the formula (I) as a free base or a pharmaceutically acceptable salt thereof.
- Another aspect of the invention is directed to said methods and wherein said mammal is a human.
- Another aspect of the invention is directed to said methods and wherein said mammal is a vertibrate animal, preferably but not limited to bigger animals such as horses, camels, dromedars but not limited thereto.
- GSK3 inhibitors in primary and secondary ostopeorosis, where primary osteoporosis includes postmenopausal osteoporosis and senile osteoporosis in both men and women, and secondary osteoporosis includes cortison induced osteoporosis, as well as any other type of induced secondary osteoporosis, are included in the term osteoporosis.
- these GSK3 inhibitors may also be used in treatments of myeloma. These GSK3 inhibitors may be administered locally or systemically, in different formulation regimes, to treat these conditions.
- the promotion and increasing of bone formation makes the compounds of the formula (I) suitable to reducing the incidence of fracture, to reduce the rate of fracture and/or increase the rate of fracture healing, to increase cancellous bone formation and/or new bone formation in mammals.
- the use to promote and increase new bone formation may be in connection with surgery.
- This invention can be used during surgery, where the treating surgeon will place the invention locally in an appropriate formulation, near the deficient bone and/or in the body cavity.
- the bone may for instance have been broken, and utilizing the invention as described and claimed herein will then be placed in or near the fracture during open fracture repair.
- bone pieces may be missing (e.g. after tumour removal or severe casualties), and utilizing the invention as described and claimed herein will then be placed near the site of constructive bone surgery.
- the compounds of formula I are also useful as pharmacological tools in the development and standardisation of in vitro and in vivo test systems for the evaluation of the effects of inhibitors of GSK3 related activity in laboratory animals such as cats, dogs, rabbits, monkeys, rats and mice, as part of the search for new therapeutics agents.
- a pharmaceutical composition comprising a compound of formula I, as a free base or a pharmaceutically acceptable salt, solvate or solvate of salt thereof, for use in the prevention and/or treatment of conditions associated with glycogen synthase kinase-3.
- composition used in accordance with the present invention may be in a form suitable for oral administration, for example as a tablet, pill, syrup, powder, granule or capsule, for parenteral injection (including intravenous, subcutaneous, intramuscular, intravascular or infusion) as a sterile solution, suspension or emulsion, for topical administration as an ointment, patch or cream, for rectal administration as a suppository and for local administration in a body cavity or in a bone cavity.
- parenteral injection including intravenous, subcutaneous, intramuscular, intravascular or infusion
- a sterile solution sterile solution
- suspension or emulsion for topical administration as an ointment, patch or cream
- rectal administration as a suppository and for local administration in a body cavity or in a bone cavity.
- Suitable daily doses of the compounds of the formula (I) used in the treatment of a mammal, including human, are approximately from 0.01 to 250 mg/kg bodyweight at peroral administration and from about 0.001 to 250 mg/kg bodyweight at parenteral administration.
- the typical daily dose of the active ingredients varies within a wide range and will depend on various factors such as the relevant indication, the route of administration, the age, weight and sex of the patient and may be determined by a physician.
- the dosage form and the dose of the medicament may vary and will depend on various factors such as, for example the individual requirement of the animal treated.
- a suitable pharmaceutically acceptable salt of the compound of formula (I) useful in accordance to the invention is, for example, an acid-addition salt, which is sufficiently basic, for example an inorganic or organic acid.
- a suitable pharmaceutically acceptable salt of the compounds of the invention, which is sufficiently acidic is an alkali metal salt, an alkaline earth metal salt or a salt with an organic base, which affords a physiologically-acceptable cation.
- the dose required for the therapeutic or preventive treatment of a particular disease, disorder or a particular condition will necessarily be varied depending on the host treated, the route of administration and the severity of the illness or injury being treated.
- the term “therapy” also includes “prevention” unless there are specific indications to the contrary.
- the terms “therapeutic” and “therapeutically” should be construed accordingly.
- disorder also includes “condition” unless there are specific indications to the contrary.
- Typical K 1 values for the compounds of the present invention are in the range of about 0.001 to about 10,000 nM. Other values for K 1 are in the range of about 0.001 to about 1000 nM. Further values for K 1 are in the range of about 0.001 nM to about 300 nM.
Landscapes
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Diabetes (AREA)
- Physical Education & Sports Medicine (AREA)
- Emergency Medicine (AREA)
- Obesity (AREA)
- Endocrinology (AREA)
- Hospice & Palliative Care (AREA)
- Psychiatry (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Hematology (AREA)
- Rheumatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The present invention relates to new compounds of formula (I) as a free base or a pharmaceutically acceptable salt, solvate or solvate of salt thereof, a process for their preparation and new intermediates used therein, pharmaceutical formulations containing said therapeutically active compounds and to the use of said active compounds in therapy.
Description
NEW IMIDAZO[4,5-ό]PYRIDINE-6-HALO-7-ARYL/HETEROARYL COMPOUNDS
705
The present invention relates to new compounds of formula I, as a free base or a pharmaceutically acceptable salt, solvate or solvate of salt thereof, to pharmaceutical formulations containing said compounds and to the use of said compounds in therapy. The present invention further relates to a process for the preparation of compounds of formula I and to new intermediates used therein.
BACKGROUND OF THE INVENTION
Glycogen synthase kinase 3 (GSK3) is a serine / threonine protein kinase composed of two isoforms (α and β), which are encoded by distinct genes but are highly homologous within the catalytic domain. GSK3 is highly expressed in the central and peripheral nervous system. GSK3 phosphorylates several substrates including tau, β-catenin, glycogen synthase, pyruvate dehydrogenase and elongation initiation factor 2b (eIF2b). Insulin and growth factors activate protein kinase B, which phosphorylates GSK3 on serine 9 residue and inactivates it.
Alzheimer 's Disease (AD) dementias, and taupathies.
AD is characterized by cognitive decline, cholinergic dysfunction and neuronal death, neurofibrillary tangles and senile plaques consisting of amyloid-β deposits. The sequence of these events in AD is unclear, but is believed to be related. Glycogen synthase kinase 3β (GSK3β) or Tau phosphorylating kinase selectively phosphorylates the microtubule associated protein Tau in neurons at sites that are hyperphosphorylated in AD brains. Hyperphosphorylated tau has lower affinity for microtubules and accumulates as paired helical filaments, which are the main components that constitute neurofibrillary tangles and neuropil threads in AD brains. This results in depolymerization of microtubules, which leads to dying back of axons and neuritic dystrophy. Neurofibrillary tangles are consistently found in diseases such as AD, amyotrophic lateral sclerosis, parkinsonism- dementia of Gaum, corticobasal degeneration, dementia pugilistica and head trauma, Down's syndrome, postencephalatic parkinsonism, progressive supranuclear palsy,
Niemann-Pick's Disease and Pick's Disease. Addition of amyloid-β to primary hippocampal cultures results in hyperphosphorylation of tau and a paired helical filaments- like state via induction of GSK3β activity, followed by disruption of axonal transport and neuronal death (Imahori and Uchida, J. Biochem. 1997, 121:179-188). GSK3β preferentially labels neurofibrillary tangles and has been shown to be active in pre -tangle neurons in AD brains. GSK3 protein levels are also increased by 50% in brain tissue from AD patients. Furthermore, GSK3β phosphorylates pyruvate dehydrogenase, a key enzyme in the glycolytic pathway and prevents the conversion of pyruvate to acetyl-Co-A (Hoshi et al, PNAS 1996, 93: 2719-2723). Acetyl-Co-A is critical for the synthesis of acetylcholine, a neurotransmitter with cognitive functions. Accumulation of amyloid-β is an early event in AD. GSK Tg mice show increased levels of amyloid-β in brain. Also, PDAPP mice fed with Lithium show decreased amyloid-β levels in hippocampus and decreased amyloid plaque area (Su et al., Biochemistry 2004, 43: 6899-6908). Thus, GSK3β inhibition may have beneficial effects in progression as well as the cognitive deficits associated with Alzheimer's disease and other above-referred to diseases.
Chronic and Acute Neurodegenerative Diseases
Growth factor mediated activation of the PI3K /Akt pathway has been shown to play a key role in neuronal survival. The activation of this pathway results in GSK3β inhibition. Recent studies (Bhat et. al., PNAS 2000, 97: 11074-11079) indicate that GSK3β activity is increased in cellular and animal models of neurodegeneration such as cerebral ischemia or after growth factor deprivation. For example, the active site phosphorylation was increased in neurons vulnerable to apoptosis, a type of cell death commonly thought to occur in chronic and acute degenerative diseases such as cognitive disorders, Alzheimer's Disease, Parkinson's Disease, amyotrophic lateral sclerosis, Huntington's Disease and HIV dementia and traumatic brain injury; and as in ischemic stroke. Lithium was neuroprotective in inhibiting apoptosis in cells and in the brain at doses that resulted in the inhibition of GSK3β. Thus GSK3β inhibitors could be useful in attenuating the course of neurodegenerative diseases.
Bipolar Disorders (BD)
Bipolar Disorders are characterised by manic episodes and depressive episodes. Lithium has been used to treat BD based on its mood stabilising effects. The disadvantage of lithium is the narrow therapeutic window and the danger of overdosing that can lead to lithium intoxication. The discovery that lithium inhibits GSK3 at therapeutic concentrations has raised the possibility that this enzyme represents a key target of lithium's action in the brain (Stambolic et al, Curr. Biol. 1996, 68(12): 1664-1668, 1996; Klein and Melton; PNAS 1996, 93:8455-8459; Gould et al., Neuropsychopharmacology, 2005, 30:1223-1237). GSK3 inhibitor has been shown to reduce immobilisation time in forced swim test, a model to assess on depressive behavior (O'Brien et al., J Neurosci 2004, 24(30): 6791-6798). GSK3 has been associated with a polymorphism found in bipolar II disorder (Szczepankiewicz et al., Neuropsychobiology. 2006, 53: 51-56). Inhibition of GSK3β may therefore be of therapeutic relevance in the treatment of BD as well as in AD patients that have affective disorders.
Schizophrenia
Accumulating evidence implicates abnormal activity of GSK3 in mood disorders and schizophrenia. GSK3 is involved in signal transduction cascades of multiple cellular processes, particularly during neural development. (Kozlovsky et al., Am. J. Psychiatry, 2000, 157, 5: 831 -833) found that GSK3β levels were 41 % lower in the schizophrenic patients than in comparison subjects. This study indicates that schizophrenia involves neurodevelopmental pathology and that abnormal GSK3 regulation could play a role in schizophrenia. Furthermore, reduced β-catenin levels have been reported in patients exhibiting schizophrenia (Cotter et al., Neuroreport 1998, 9(7): 1379-1383). Atypical antipsychotics such as olanzapine, clozapine, quetiapine, and ziprasidone, inhibits GSK3 by increasing ser9 phosphorylation suggesting that antipsychotics may exert their beneficial effects via GSK3 inhibition (Li X. et al., Int. J.of Neuropsychopharmacol, 2007, 10: 7-19, Epubl. 2006, May 4).
Diabetes
Insulin stimulates glycogen synthesis in skeletal muscles via the dephosphorylation and thus activation of glycogen synthase. Under resting conditions, GSK3 phosphorylates and
inactivates glycogen synthase via dephosphorylation. GSK3 is also over-expressed in muscles from Type II diabetic patients (Nikoulina et al., Diabetes 2000 Feb; 49(2): 263- 71). Inhibition of GSK3 increases the activity of glycogen synthase thereby decreasing glucose levels by its conversion to glycogen. In animal models of diabetes, GSK3 inhibitors lowered plasma glucose levels up to 50 % (Cline et al., Diabetes, 2002, 51:
2903-2910; Ring et al., Diabetes 2003, 52: 588-595). GSK3 inhibition may therefore be of therapeutic relevance in the treatment of Type I and Type II diabetes and diabetic neuropathy.
Alopecia
GSK3 phosphorylates and degrades β-catenin. β-catenin is an effector of the pathway for keratonin synthesis, β-catenin stabilisation may be lead to increase hair development. Mice expressing a stabilised β-catenin by mutation of sites phosphorylated by GSK3 undergo a process resembling de novo hair morphogenesis (Gat et al., Cell, 1998, 95(5): 605-14)). The new follicles formed sebaceous glands and dermal papilla, normally established only in embryogenesis. Thus GSK3 inhibition may offer treatment for baldness.
Inflammatory disease
The discovery that GSK3 inhibitors provide anti-inflammatory effects has raised the possibility of using GSK3 inhibitors for therapeutic intervention in inflammatory diseases. (Martin et al., Nat. Immunol. 2005, 6(8): 777-784; Jope et al., Neurochem. Res. 2006, DOI 10.1007/sl 1064-006-9128-5)). Inflammation is a common feature of a broad range of conditions including Alzheimer's Disease and mood disorders.
Cancer
GSK3 is overexpressed in ovarian, breast and prostate cancer cells and recent data suggests that GSK3b may have a role in contributing to cell proliferation and survival pathways in several solid tumor types. GSK3 plays an important role in several signal transduction systems which influence cell proliferation and survival such as WNT, PI3 Kinase and NFkB. GSK3b deficient MEFs indicate a crucial role in cell survival mediated NFkB pathway (Ougolkov AV and Billadeau DD. Future Oncol. 2006 Feb;2(l):91-100.). Thus,
GSK3 inhibitors may inhibit growth and survival of solid tumors, including pancreatic, colon and prostate cancer.
Bone-related disorders and conditions GSK3 inhibitors could be used for treatment of bone-related disorders or other conditions, which involves a need for new and increased bone formation. Remodeling of the skeleton is a continuous process, controlled by systemic hormones such as parathyroid hormone (PTH), local factors (e.g. prostaglandin E2), cytokines and other biologically active substances. Two cell types are of key importance: osteoblasts (responsible for bone formation) and osteoclasts (responsible for bone resorption). Via the RANK, RANK ligand and osteoprotegerin regulatory system these two cell types interact to maintain normal bone turnover (Bell NH, Current Drug Targets - Immune, Endocrine & Metabolic Disorders, 2001, 1:93-102).
Osteoporosis is a skeletal disorder in which low bone mass and deterioration of bone microarchitecture lead to increased bone fragility and fracture risk. To treat osteoporosis, the two main strategies are to either inhibit bone resorption or to stimulate bone formation. The majority of drugs currently on the market for the treatment of osteoporosis act to increase bone mass by inhibiting osteoclastic bone resorption. It is recognized that a drug with the capacity to increase bone formation would be of great value in the treatment of osteoporosis as well as having the potential to enhance fracture healing in patients.
Recent in vitro studies suggest a role of GSK3β in osteoblast differentiation. First, it has been shown that glucocorticoids inhibit cell cycle progression during osteoblast differentiation in culture. The mechanism behind this is activation of GSK3β in osteoblasts, resulting in c-Myc down-regulation and impediment of the Gi/S cell cycle transition. The attenuated cell cycle and reduced c-Myc level are returned to normal when GSK3β is inhibited using lithium chloride (Smith et al, J. Biol. Chem, 2002, 277: 18191- 18197). Secondly, inhibition of GSK3β in the pluripotent mesenchymal cell line C3H10T1/2 leads to a significant increase in endogenous β-catenin signaling activity. This, in turn, induces expression of alkaline phosphatase mRNA and protein, a marker of early
osteoblast differentiation (Bain et al., Biochem. Biophys. Res. Commun., 2003, 301: 84- 91).
DISCLOSURE OF THE INVENTION The object of the present invention is to provide compounds having a selective inhibiting effect at GSK3. Accordingly, the present invention provides a compound of the formula I:

wherein
Q is selected from halogen; X is

R1 is C(O)NRbRc; R2 and R4 are independently selected from hydrogen, halogen, CN, NO2, Ci-3alkyl, Ci- βhaloalkyl;
R3 and R5 are independently selected from hydrogen, halogen, Ci-3alkyl and Ci-3haloalkyl;
A is aryl or heteroaryl, optionally substituted with one or more CN, CO2H, Ci-6alkyl, C1- ehaloalkyl, halo,C(O)Ra, ORk, C(O)NRbRc or S(O)nRm, wherein said Ci-6alkyl or Ci- βhaloalkyl is optionally substituted by at least one CN, ORa or NRbRc;
Ra is selected from hydrogen,
 and
and
 wherein said
wherein said
 or Ci- βhaloalkyl is optionally substituted with one or more
or Ci- βhaloalkyl is optionally substituted with one or more

Rb and Rc may, together with the atom to which they are attached, form a 4-, 5- or 6- membered heterocyclic ring containing one or more heteroatoms selected from N, O or S, wherein said heterocyclic ring is optionally substituted with one or more halo, ORa, NRdRe,
 is optionally further substituted with one or more
is optionally further substituted with one or more

Rd and Re are independently selected from hydrogen, Ci_6alkyl or Ci-όhaloalkyl, wherein said Ci_6alkyl or Ci-όhaloalkyl is optionally substituted with one or more ORa; or Rk is Ci_6alkyl or Ci-όhaloalkyl, wherein said Ci_6alkyl or Ci-όhaloalkyl is optionally substituted with at least one CN, ORa, NRbRc, C(O)NRbRc or NRbC(O)Rc; Rm is C1-3alkyl, optionally substituted with at least one halo, CN, ORa, NRbRc or C(O)NRbRc; n is 0 to 2; as a base or a pharmaceutically acceptable salt, solvate or solvate of a salt thereof.
In one aspect of the invention, there is provided compounds of formula I, wherein Q is halogen, said halogen is selected from chloro and bromo.
In another aspect of the invention, there is provided compounds of formula I, wherein A is aryl optionally substituted with one or more ORk. In one embodiment of this aspect, said A is phenyl, substituted with one ORk, wherein Rk is Ci-6alkyl. In one embodiment of this aspect, said A is phenyl, substituted with one ORk, wherein ORk represents methoxy.
In another aspect of the invention, there is provided compounds of formula I, wherein X is

and R2, R3, R4 and R5 represent hydrogen.
In another aspect of the invention, there is provided compounds of formula I, wherein R is C(O)NRbRc, wherein Rb and Rc, together with the atom to which they are attached, form a 6-membered heterocyclic ring containing one N heteroatom, wherein said heterocyclic ring is substituted with one Ci^alkyl.
In another aspect of the invention, there is provided compounds of formula I, wherein Q is halogen X is

R2 and R4 are hydrogen;
R3 and R5 are hydrogen;
A is aryl optionally substituted with one ORk;
Rb and Rc, together with the atom to which they are attached, form a 6-membered heterocyclic ring containing one or two heteroatoms selected from N, O or S, wherein said heterocyclic ring is optionally substituted with one
 and
and
Rk is Ci-6alkyl.
In another aspect of the invention, there is provided compounds of formula I, wherein Q is halogen, X is

R1 is C(O)NRbRc;
R2 and R4 are hydrogen; R3 and R5 are hydrogen;
A is aryl optionally substituted with one ORk;
Rb and Rc, together with the atom to which they are attached, form a 6-membered heterocyclic ring containing one N heteroatom, wherein said heterocyclic ring is substituted with one
 and Rk is Ci-6alkyl.
In another aspect of the invention, there is provided compounds of formula I, wherein Q is halogen X is
and Rk is Ci-6alkyl.
In another aspect of the invention, there is provided compounds of formula I, wherein Q is halogen X is

R2 and R4 are hydrogen;
R3 and R5 are hydrogen;
A is aryl optionally substituted with one ORk;
Rb and Rc, together with the atom to which they are attached, form a piperazine, wherein said piperazine is substituted with one
 and
and
Rk is Ci-6alkyl.
In another aspect of the invention, there is provided compounds of formula I, wherein Q is halogen X is

R1 is C(O)NRbRc; R2 and R4 are hydrogen; R3 and R5 are hydrogen; A is phenyl optionally substituted with one ORk;
Rb and Rc, together with the atom to which they are attached, form a piperazine, wherein said piperazine is substituted with one
 and Rk is Ci-6alkyl.
and Rk is Ci-6alkyl.
In another aspect of the invention, there is provided compounds of formula I, selected from:
6-Bromo-7-(4-methoxyphenyl)-2- {4-[(4-methylpiperazin- 1 -yl)carbonyl]phenyl} -3H- imidazo[4,5-ό]pyridine hydrochloride;
6-Chloro-7-(4-methoxyphenyl)-2- {4-[(4-methylpiperazin- 1 -yl)carbonyl]phenyl} -3H- imidazo[4,5-ό]pyridine hydrochloride; and 6-Fluoro-7-(4-methoxyphenyl)-2- {4-[(4-methylpiperazin- 1 -yl)carbonyl]phenyl} -3H- imidazo[4,5-ό]pyridine; as a free base or an alternative pharmaceutically acceptable salt, solvate or solvate of a salt thereof.
In another aspect of the invention, there is provided compounds selected from:
Methyl 4-(6-bromo-3H-imidazo[4,5-ό]pyridin-2-yl)benzoate;
Methyl 4-(6-chloro-3H-imidazo[4,5-ό]pyridin-2-yl)benzoate;
Methyl 4-(6-bromo-7-chloro-3H-imidazo[4,5-ό]pyridin-2-yl)benzoate;
Methyl 4-(6,7-dichloro-3H-imidazo[4,5-ό]pyridin-2-yl)benzoate; Methyl 4-(6-bromo-7-iodo-3H-imidazo[4,5-ό]pyridin-2-yl)benzoate;
Methyl 4-(6-chloro-7-iodo-3H-imidazo[4,5-b]pyridin-2-yl)benzoate;
4-(6-Bromo-7-iodo-3H-imidazo[4,5-ό]pyridin-2-yl)benzoic acid;
4-(6-Chloro-7-iodo-3H-imidazo[4,5-ό]pyridin-2-yl)benzoic acid;
6-Bromo-7-iodo-2- {4-[(4-methylpiperazin- 1 -yl)carbonyl]phenyl} -3H-imidazo[4,5- όjpyridine; and
6-Chloro-7-iodo-2- {4-[(4-methylpiperazin- 1 -yl)carbonyl]phenyl} -3H-imidazo[4,5- ό]pyridine.
These compounds are useful as intermediates in the process of preparing a compound according to formula I.
Listed below are definitions of various terms used in the specification and claims to describe the present invention.
In this specification the term "alkyl" includes both straight and branched chains as well as cyclic alkyl groups. The term Ci-3alkyl having 1 to 3 carbon atoms and may be, but is not limited to, methyl, ethyl, n-propyl, /-propyl or cyclopropyl. The term d-6alkyl having 1 to
6 carbon atoms and may be, but is not limited to, methyl, ethyl, n-propyl, /-propyl, n-butyl, /-butyl, 5-butyl, t-butyl, n-pentyl, /-pentyl, t-pentyl, neo-pentyl, n-hexyl, /-hexyl or cyclohexyl.
The term "Ci-3alkoxy" includes both straight and branched chains . The term "Ci-3alkoxy" having 1 to 3 carbon atoms may be, but is not limited to, methoxy, ethoxy, n-propoxy or i- propoxy.
The term "halo" or "halogen" refers to fluorine, chlorine, bromine and iodine.
The term "haloalkyl" refers to an alkyl group, defined as above, in which one or several of the hydrogen substituents have been replaced by halogen substituents, in which the term halogen is defined as above.
The term "aryl" refers to an optionally substituted monocyclic or bicyclic hydrocarbon ring system containing at least one unsaturated aromatic ring. The "aryl" may be fused with a C5-7Cycloalkyl ring to form a bicyclic hydrocarbon ring system. Examples and suitable values of the term "aryl", but not limiting,are phenyl, naphthyl, indanyl or tetralinyl.
As used herein, "heteroaryl" refers to an aromatic heterocycle having at least one heteroatom ring member such as sulfur, oxygen or nitrogen. Heteroaryl groups include monocyclic and polycyclic (e.g., having 2, 3 or 4 fused rings) systems. Examples of heteroaryl groups include without limitation, pyridyl (i.e., pyridinyl), pyrimidinyl, pyrazinyl, pyridazinyl, triazinyl, furyl (i.e. furanyl), quinolyl, isoquinolyl, thienyl, imidazolyl, thiazolyl, indolyl, pyrryl, oxazolyl, benzofuryl, benzothienyl, benzthiazolyl, isoxazolyl, pyrazolyl, triazolyl, tetrazolyl, indazolyl, 1,2,4-thiadiazolyl, isothiazolyl, benzothienyl, purinyl, carbazolyl, fluorenonyl, benzimidazolyl, indolinyl, and the like. In some embodiments, the heteroaryl group has from 1 to about 20 carbon atoms, and in further embodiments from about 3 to about 20 carbon atoms. In some embodiments, the heteroaryl group contains 3 to about 14, 4 to about 14, 3 to about 7 or 5 to 6 ring-forming atoms. In some embodiments, the heteroaryl or heteroaromatic group has 1 to about 4, 1 to
about 3 or 1 to 2 heteroatoms. In some embodiments, the heteroaryl or heteroaromatic group has 1 heteroatom.
The term "4-, 5- or 6-membered heterocyclic ring containing one or more heteroatoms independently selected from N, O or S" refers to a mono- or bicyclic- heterocyclic ring which may be saturated or partly saturated and which may optionally contain a carbonyl function and which may be, but is not limited to, azetidinyl, imidazolidinyl, imidazolinyl, morpholinyl, piperazinyl, piperidinyl, piperidonyl, pyrazolidinyl, pyrazolinyl, pyrrolidinyl, pyrrolinyl, 1 -methyl- 1 ,4-diazepane, tetrahydropyranyl or thiomorpholinyl. In the case where the heterocyclic ring contains a heteroatom selected from S or N, these atoms may optionally be in an oxidised form.
The term "hydrochloride" includes monohydrochloride, dihydrochloride, trihydrochloride and tetrahydrochloride salts.
A suitable pharmaceutically acceptable salt of the compound of the invention is, for example, an acid-addition salt, for example an inorganic or organic acid. In addition a suitable pharmaceutically acceptable salt of the compounds of the invention is an alkali metal salt, an alkaline earth metal salt or a salt with an organic base that affords a physiologically-acceptable cation.
Some compounds of formula I may have sterogenic centres and/or geometric isomeric centres (E- and Z-isomers), and it is to be understood that the invention encompasses all such optical, diastereoisomers and geometric isomers.
The present invention relates to the use of compounds of formula I as hereinbefore defined as well as to the salts thereof. Salts for use in pharmaceutical compositions will be pharmaceutically acceptable salts, but other salts may be useful in the production of the compounds of formula I.
It is to be understood that the present invention relates to any and all tautomeric forms of the compounds of formula I.
An object of the invention is to provide compounds of formula I for therapeutic use, especially compounds that are useful for the prevention and/or treatment of conditions associated with glycogen synthase kinase-3 (GSK3) in mammals including man. Particularly, compounds of formula I exhibiting a selective affinity for GSK-3.
Another object of the invention is wherein a compound of formula (1) or a pharmaceutically acceptable salt, solvate or in vivo hydrolysable ester thereof, or a pharmaceutical composition or formulation comprising a compound of formula (1) is administered concurrently, simultaneously, sequentially or separately with another pharmaceutically active compound or compounds selected from the following:
(i) antidepressants such as agomelatine, amitriptyline, amoxapine, bupropion, citalopram, clomipramine, desipramine, doxepin duloxetine, elzasonan, escitalopram, fiuvoxamine, fluoxetine, gepirone, imipramine, ipsapirone, maprotiline, nortriptyline, nefazodone, paroxetine, phenelzine, pro trip ty line, ramelteon, reboxetine, robalzotan, sertraline, sibutramine, thionisoxetine, tranylcypromaine, trazodone, trimipramine, venlafaxine and equivalents and pharmaceutically active isomer(s) and metabolite(s) thereof.
(ii) atypical antipsychotics including for example quetiapine and pharmaceutically active isomer(s) and metabolite(s) thereof.
(iii) antipsychotics including for example amisulpride, aripiprazole, asenapine, benzisoxidil, bifeprunox, carbamazepine, clozapine, chlorpromazine, debenzapine, divalproex, duloxetine, eszopiclone, haloperidol, iloperidone, lamotrigine, loxapine, mesoridazine, olanzapine, paliperidone, perlapine, perphenazine, phenothiazine, phenylbutylpiperidine, pimozide, prochlorperazine, risperidone, sertindole, sulpiride, suproclone, suriclone, thioridazine, trifluoperazine, trimetozine, valproate, valproic acid, zopiclone, zotepine, ziprasidone and equivalents and pharmaceutically active isomer(s) and metabolite(s) thereof.
(iv) anxiolytics including for example alnespirone, azapirones,benzodiazepines, barbiturates such as adinazolam, alprazolam, balezepam, bentazepam, bromazepam, brotizolam, buspirone, clonazepam, clorazepate, chlordiazepoxide, cyprazepam, diazepam, diphenhydramine, estazolam, fenobam, flunitrazepam, flurazepam, fosazepam, lorazepam, lormetazepam, meprobamate, midazolam, nitrazepam, oxazepam, prazepam, quazepam, reclazepam, tracazolate, trepipam, temazepam, triazolam, uldazepam, zolazepam and equivalents and pharmaceutically active isomer(s) and metabolite(s) thereof.
(v) anticonvulsants including for example carbamazepine, valproate, lamotrogine, gabapentin and equivalents and pharmaceutically active isomer(s) and metabolite(s) thereof.
(vi) Alzheimer's therapies including for example donepezil, memantine, tacrine and equivalents and pharmaceutically active isomer(s) and metabolite(s) thereof.
(vii) Parkinson's therapies including for example deprenyl, L-dopa, Requip, Mirapex, MAOB inhibitors such as selegine and rasagiline, comP inhibitors such as Tasmar, A-2 inhibitors, dopamine reuptake inhibitors, NMDA antagonists, Nicotine agonists, Dopamine agonists and inhibitors of neuronal nitric oxide synthase and equivalents and pharmaceutically active isomer(s) and metabolite(s) thereof.
(viii) migraine therapies including for example almotriptan, amantadine, bromocriptine, butalbital, cabergoline, dichloralphenazone, eletriptan, frovatriptan, lisuride, naratriptan, pergolide, pramipexole, rizatriptan, ropinirole, sumatriptan, zolmitriptan, zomitriptan, and equivalents and pharmaceutically active isomer(s) and metabolite(s) thereof.
(ix) stroke therapies including for example abciximab, activase, NXY-059, citicoline, crobenetine, desmoteplase,repinotan, traxoprodil and equivalents and pharmaceutically active isomer(s) and metabolite(s) thereof.
(x) urinary incontinence therapies including for example darafenacin, falvoxate, oxybutynin, propiverine, robalzotan, solifenacin, tolterodine and and equivalents and pharmaceutically active isomer(s) and metabolite(s) thereof.
(xi) neuropathic pain therapies including for example gabapentin, lidoderm, pregablin and equivalents and pharmaceutically active isomer(s) and metabolite(s) thereof.
(xii) nociceptive pain therapies such as celecoxib, etoricoxib, lumiracoxib, rofecoxib, valdecoxib, diclofenac, loxoprofen, naproxen, paracetamol and equivalents and pharmaceutically active isomer(s) and metabolite(s) thereof.
(xiii) insomnia therapies including for example agomelatine, allobarbital, alonimid, amobarbital, benzoctamine, butabarbital, capuride, chloral, cloperidone, clorethate, dexclamol, ethchlorvynol, etomidate, glutethimide, halazepam, hydroxyzine, mecloqualone, melatonin, mephobarbital, methaqualone, midaflur, nisobamate, pentobarbital, phenobarbital, propofol, ramelteon, roletamide, triclofos,secobarbital, zaleplon, Zolpidem and equivalents and pharmaceutically active isomer(s) and metabolite(s) thereof.
(xiv) mood stabilizers including for example carbamazepine, divalproex, gabapentin, lamotrigine, lithium, olanzapine, quetiapine, valproate, valproic acid, verapamil, and equivalents and pharmaceutically active isomer(s) and metabolite(s) thereof.
Such combination products employ the compounds of this invention within the dosage range described herein and the other pharmaceutically active compound or compounds within approved dosage ranges and/or the dosage described in the publication reference.
Methods of Preparation Another aspect of the present invention provides a process for preparing a compound of formula I as a free base or a pharmaceutically acceptable salt thereof. Throughout the following description of such processes it is understood that, where appropriate, suitable
protecting groups will be added to, and subsequently removed from, the various reactants and intermediates in a manner that will be readily understood by one skilled in the art of organic synthesis. Conventional procedures for using such protecting groups as well as examples of suitable protecting groups are described, for example, in "Protective Groups in Organic Synthesis", T.W. Greene, P.G.M. Wuts, Wiley-Interscience, New York, 1999. It will be appreciated that certain of the various ring substituents in the compounds of the present invention may be introduced by standard aromatic substitution reactions or generated by conventional functional group modifications either prior to or immediately following the processes mentioned above, and as such are included in the process aspect of the invention. Such reactions and modifications include, for example, introduction of a substituent by means of an aromatic substitution reaction, reduction of substituents, alkylation of substituents and oxidation of substituents. The reagents and reaction conditions for such procedures are well known in the chemical art. Particular examples of aromatic substitution reactions include the introduction of a nitro group using concentrated nitric acid, the introduction of an acyl group using, for example, an acyl halide and Lewis acid (such as aluminium trichloride) under Friedel Crafts conditions; the introduction of an alkyl group using an alkyl halide and Lewis acid (such as aluminium trichloride) under Friedel Crafts conditions; and the introduction of a halogeno group. Particular examples of modifications include the reduction of a nitro group to an amino group by for example, catalytic hydrogenation with a nickel catalyst or treatment with iron in the presence of hydrochloric acid with heating; oxidation of alkylthio to alkylsulphinyl or alkylsulphonyl.
Methods of Preparation of Intermediates
The processes for preparation of the intermediates, wherein R1, R2, R3, R4, R5 and A are, unless otherwise specified, as defined in formula I and Q is halo comprise of the following:

(a) First, reacting II and III in the presence of a suitable catalyst, e.g. o-benzotriazol-1-yl- Λ/,Λ/,Λr,Λr-tetramethyluroniumhexafluorophosphate or O-(7-azabenzotriazol-l-yl)- N,7V,7V ',7V '-tetramethyluronium hexafluorophosphate, in a solvent such as acetonitrile, dimethyl formamide, or a mixture thereof. A suitable base such as N, N- diisopropylethylamine may be used in the reaction, which can be performed at a temperature in the range of 0 0C to +20 0C.
(b) Second, heating the resulting intermediate in a suitable organic acid, e.g. acetic acid, at a temperature in the range of +150 0C to +200 0C using an oil bath or a microwave oven.

(ii) Conversion of a compound of type IV into a chloride of type V can be achieved by (a) first, reacting the compound of type IV with an appropriate oxidant, e.g. m- chloroperbenzoic acid, in a suitable solvent, e.g. acetic acid, at a temperature in the range of +20 0C to +30 0C; (b) second, reaction of the formed intermediate with neat phosphorus oxychloride at a temperature in the range of +100 0C to +150 0C using an oil bath or a microwave oven.

(iii) Hydrolysis of an ester of type Va (V, R1 is CO2R, wherein R is alkyl, for example, ethyl or methyl) to the corresponding acid VI might be effected by reaction with a suitable base, such as lithium, sodium or potassium hydroxide, or potassium carbonate, in mixtures of water and a suitable cosolvent, e.g. tetrahydrofuran or methanol, at a temperature in the range of +20 0C to +120 0C using an oil bath or a microwave oven.

(iv) Formation of an amide of type VIII from the corresponding acid VI and an amine VII (wherein Rb and Rc are as defined in formula I) can be performed by reacting VI and VII in the presence of a suitable catalyst, e.g. o-benzotriazol-l-yl-Λ/,Λ/,N',N'- tetramethyluroniumhexafluorophosphate or O-(7-azabenzotriazol- 1 -yl)-N,N,N',N'- tetramethyluronium hexafluorophosphate in a solvent such as acetonitrile, dimethyl formamide, or a mixture thereof. A suitable base such as 7V,7V-diisopropylethylamine may be used in the reaction, which can be performed at a temperature in the range of 0 0C to +20 0C. Alternatively, a solution of VI in a solvent such as dimethyl acetamide can be first reacted with an activating agent such as 1,1 '-carbonylbis( \H- imidazole) at a temperature in the range of +80 0C to +120 0C, and then reacted with the amine VII at a temperature in the range of +100 0C to +150 0C, using an oil bath or a microwave oven.
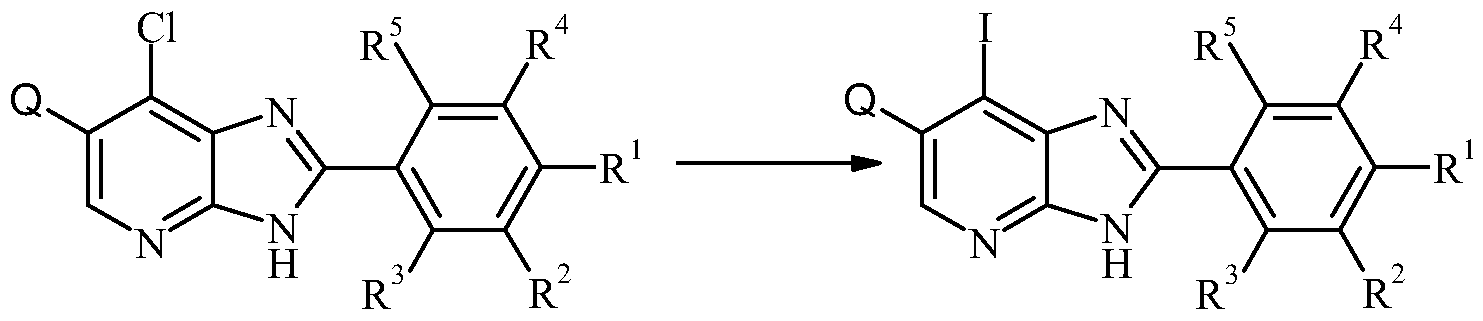
(V) (IX)
(vi) A compound of type V can be transformed into the corresponding iodide IX by (a) first, treatment with HCl in a suitable solvent such as diethyl ether to give the hydrochloride salt, and (b) second, reaction of the salt with NaI in a suitable solvent, e.g acetonitrile, at a temperature in the range of +150 0C to +175 0C using an oil bath or a microwave oven.

(vii) Cross-coupling of a compound of formula Va or IXa ( IX, wherein Q is as defined above and R1 is CO2R wherein R is alkyl, for example methyl or ethyl) with a suitable aryl species X to give a compound of formula XI may be carried out by reaction with an appropriate aryl boronic acid or an aryl boronic ester. The reaction may be carried out using a suitable palladium catalyst such as Pd(PPh3)4, Pd(dppf)Cl2 or Pd(OAc)2 or Pd2(dba)3 together with a suitable ligand such as P(ter£-butyl)3, 2-(dicyclohexylphosphino)- biphenyl or 2-(2',6'-dimethoxybiphenyl)-dicyclohexylphosphine or a nickel catalyst such as nickel on charcoal or Ni(dppe)Cl2 together with zinc and sodium triphenylphosphinetrimetasulfonate. A suitable base such as an alkyl amine, e.g. trie thy lamine, or potassium carbonate, sodium carbonate, cesium carbonate, sodium hydroxide or cesium fluoride may be used in the reaction, which can be performed in the temperature range of +20 0C to +160 0C, using an oil bath or a microwave oven, in a suitable solvent or solvent mixture such as toluene, tetrahydrofuran, dimethoxyethane/water, 7V,Λ/-dimethylformamide or dioxane. The boronic acid or boronic ester may be formed in situ, by reaction of the corresponding aryl halide (e.g., the aryl bromide) with an alkyllithium reagent such as butyllithium to form an intermediate aryl lithium species, which then is reacted with a suitable boron compound, e.g., trimethyl borate, tributyl borate or triisopropyl borate.


Methods of Preparation of End Products
Another objective of the invention are processes for the preparation of a compound of general formula I, wherein, R1, R2, R3, R4, R5 and A are, unless specified otherwise, defined as in formula I and Q is halo, comprising of:

(V): X = Cl (I) (IX): X = I
(i) Cross-coupling of a compound of formula V (Q=Cl) or IX (Q=I) with a suitable aryl species X to give a compound of formula I can be carried out as described above for the cross-coupling of Va or IXa ( IX, wherein Q is as defined above and R1 is CO2R wherein R is alkyl, for example methyl or ethyl) to give XI.

(ii) An ester of type XI may be transformed into a compound of type Ia (I, wherein A is as defined above and wherein Rb and Rc are as defined as in formula I and wherein R1 are CO2R and wherein R is alkyl, for example methyl or ethyl) by (a) first, heating neat with an amine VII at a temperature in the range of +180 0C to +220 0C using an oil bath or a
microwave oven, and (b) second, after cooling, adding a suitable catalyst such as o- benzotriazol- 1 -yl-Λ/,Λ/>N',N'-tetramethyluroniumhexafiuorophosphate or O-(7- azabenzotriazol-l-yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate and continuing the reaction at a temperature in the range of 0 0C to +20 0C.

(XII) (Ia)
(iv) Formation of an amide of type Ia can also be performed by reacting a carboxylic acid of type XII (wherein R1 is CO2H) with an amine of type VII (Rb and Rc are as defined as in formula I), as described for the preparation of VIII from VI and VII.
Consequently, in one aspect of the present invention, there is provided a process for preparing a compound of formula I, wherein R1, R2, R3, R4, R5 and A are, unless specified otherwise, defined as in formula I and Q is halo, comprising of: (i) Condensation of a diamine II and a carboxylic acid of type III by first reacting the components in the presence of a suitable catalyst, optionally with an added base, and then heating the resulting intermediate in a suitable organic acid.
(ii) Cross-coupling of a compound of formula V (X=Cl) or IX (X=I) with a suitable aryl species X by reaction in the presence of a suitable metal catalyst, optionally with an added organic or inorganic base. (iii) An ester of type XI may be coupled with an amine VII to give a compound of type Ia (I, R1=C(O)NRbRc, wherein Rb and Rc are as defined as in formula I) by first heating XI with the neat amine VII, and then adding a suitable catalyst and continuing the reaction, (iv) Formation of an amide of type Ia can also be performed by reacting a carboxylic acid of type XII with an amine of type VII, in the presence of a suitable catalyst, optionally with an added amine base. Alternatively, the acid XII can be first reacted with an activating agent, and then reacted with the amine.
The hydrochloric salt of a compound of formula I may be obtained from a compound of formula I by treatment with hydrochloric acid at a temperature in the range of 0 0C to +25 0C, in a suitable solvent such as dichloromethane, tetrahydrofuran or a dichloromethane/methanol mixture .
General Methods
All solvents used were analytical grade and commercially available anhydrous solvents were routinely used for reactions. Reactions were typically run under an inert atmosphere of nitrogen or argon.
1H and 13C NMR spectra were recorded at 400 MHz for proton, 376 MHz for fluorine- 19 and 100 MHz for carbon- 13, either on a Varian Unity+ 400 NMR Spectrometer equipped with a 5mm BBO probehead with Z-gradients, or a Bruker Avance 400 NMR spectrometer equipped with a 60 μl dual inverse flow probehead with Z-gradients, or a Bruker DPX400 NMR spectrometer equipped with a 4-nucleus probehead equipped with Z-gradients, or a Bruker Avance 600 NMR spectrometer equipped with a 5mm BBI probehead with Z- gradients. Unless specifically noted in the examples, spectra were recorded at 400 MHz for proton and 100 MHz for carbon-13. The following reference signals were used: the middle line OfDMSO-J6 δ 2.50 (1H), δ 39.51 (13C); the middle line of CD3OD δ 3.31 (1H) or δ 49.15 (13C), CDCl3 δ 7.26 (1H) and the middle line Of CDCl3 δ 77.16 (13C) (unless otherwise indicated).
Mass spectra were recorded on a Waters LCMS consisting of an Alliance 2795 (LC), Waters PDA 2996 and a ZQ single quadrupole mass spectrometer. The mass spectrometer was equipped with an electrospray ion source (ESI) operated in a positive or negative ion mode. The capillary voltage was 3 kV and cone voltage was 30 V. The mass spectrometer was scanned between m/z 100-700 with a scan time of 0.3s. Separations were performed on either Waters X-Terra MS C8 (3.5 μm, 50 or 100 mm x 2.1 mm i.d.) or an ACE 3 AQ (100 mm x 2.1 mm i.d.) obtained from ScantecLab. Flow rates were regulated to 1.0 or 0.3 mL/min, respectively. The column temperature was set to 40 0C. A linear gradient was applied using a neutral or acidic mobile phase system, starting at 100% A (A:95:5 0.1M NH4OAc:MeCN or 95:5 8 mM HCOOH:MeCN) ending at 100% B (MeCN).
Alternatively, mass spectra were recorded on a Waters LC-MS system (Sample Manager 2777C, 1525μ binary pump, 1500 Column Oven, ZQ, PDA2996 and ELS detector, Sedex 85). Separation was performed using a Zorbax column (C8, 3.0 x 50 mm, 3 μm). A four minutes linear gradient was used starting at 100 % A (A: 95:5 10 mM NH4OAc:MeOH ) and ending at 100% B (MeOH). The ZQ was equipped with a combined APPI/APCI ion source and scanned in the positive mode between m/z 120-800 with a scan time of 0.3 s. The APPI repeller and the APCI corona were set to 0.86 kV and 0.80 μA, respectively. In addition, the desolvation temperature (3000C), desolvation gas (400 L/Hr) and cone gas (5 L/Hr) were constant for both APCI and APPI mode.
Microwave heating was performed in a Creator or Smith Synthesizer Single-mode microwave cavity producing continuous irradiation at 2450 MHz.
HPLC analyses were performed on an Agilent HPlOOO system consisting of G1379A Micro Vacuum Degasser, G1312A Binary Pump, G1367A Well plate auto-sampler, G1316A Thermostatted Column Compartment and G1315B Diode Array Detector. Column: X-Terra MS, Waters, 3.0 x 100 mm, 3.5 μm. The column temperature was set to 40 0C and the flow rate to 1.0 ml/min. The Diode Array Detector was scanned from 210- 300 nm, step and peak width were set to 2 nm and 0.05 min, respectively. A linear gradient was applied, starting at 100 % A (95:5 10 mM NH4OAc:MeCN) and ending at 100% B (B: acetonitrile), in 4 min.
A typical workup procedure after a reaction consisted of extraction of the product with a solvent such as ethyl acetate, washing with water followed by drying of the organic phase over MgSO4 or Na2SO4, filtration and concentration of the solution in vacuo.
Thin layer chromatography (TLC) was performed on Merck TLC-plates (Silica gel 60 F2S4) and UV visualized the spots. Flash chromatography was preformed on a Combi Flash® Companion™ using RediSep™ normal-phase flash columns. Typical solvents used for flash chromatography was mixtures of heptane/ethyl acetate. SCX ion exchange columns were performed on Isolute® columns. Chromatography through ion exchange columns
were typically performed in solvents or solvent mixtures such a methanol and 10% ammonia in methanol.
Preparative chromatography was run on a Waters autopurification HPLC with a diode array detector. Column: XTerra MS C8, 19 x 300 mm, 10 μm. Narrow gradients with MeCN/(95:5 0.1M NH4OAc :MeCN) were used at a flow rate of 20 ml/min. Alternatively, purification was achieved on a semi preparative Shimadzu LC-8A HPLC with a Shimadzu SPD-IOA UV-vis. -detector equipped with a Waters Symmetry® column (C18, 5 μm, 100 mm x 19 mm). Narrow gradients with MeCN/0.1% trifluoroacetic acid in MiIIiQ Water were used at a flow rate of 10 ml/min.
Alternatively preparative chromatography was run on a Waters FractionLynx system with a Autosampler combined Automated Fraction Collector (Waters 2767), Gradient Pump (Waters 2525), Regeneration Pump (Waters 600), Make Up Pump (Waters 515), Waters Active Splitter, Column Switch (Waters CFO), PDA (Waters 2996) and Waters ZQ mass spectrometer. Column; XBridge™ Prep C8 5μm OBD™ 19 x 100mm, with guard column; XTerra ® Prep MS C8 lOμm 19 x 10mm Cartridge. A gradient from 100% A (95% 0.1M NH4OAc in MiIIiQ water and 5% MeCN) to 100% B (100% MeCN) was applied for LC-separation at flow rate 25ml/min. The PDA was scanned from 210-350nm. The ZQ mass spectrometer was run with ESI in positive mode. The Capillary Voltage was 3kV and the Cone Voltage was 30V. Mixed triggering, UV and MS signal, determined the fraction collection.
The formation of hydrochloride salts of the final products were typically performed by dissolution in solvents or solvent mixtures such as diethyl ether, tetrahydrofuran, dichloromethane/methanol, followed by addition of IM HCl in diethyl ether.
The following abbreviations have been used: aq. aqueous;
CDI carbonyl diimidazole; CH2Cl2 dichloromethane;
DIPEA AWV-diisopropylethylamine;
DMF AWV-dimethylformamide;
ether diethyl ether;
EtOAc ethyl acetate;
EtOH ethanol;
HBTU o-benzotriazol- 1 -yl-A^A^AT-tetramethyluronium hexafluorophosphate;
HCl hydrochloride;
HOAc acetic acid;
(1-Pr)2EiN N-N-diisopropylethylamine; m-CPBA 3-chloroperoxybenzoic acid;
MeCN acetonitrile;
MeOH methanol;
NaHCO3 sodium hydrogen carbonate;
Na2SO4 sodium sulphate;
NH3 ammonia;
NH4OAc ammonium acetate;
Pd(OAc)2 palladium diacetate;
PdCl2(dppf)*DCM (1,1 '-bis(diphenylphosphino)ferrocen)palladium(II) chloride dichloromethane adduct;
Pd2(dba)3 tris(dibenzylideneacetone)dipalladium;
Pd(dppf)Cl2 (1,1 '-bis(diphenylphosphino)ferrocen)palladium(II) chloride;
Pd(PPh3)4 tris(tri-phenylphosphine)palladium;
Ni(dppe)Cl2 (1 ,2-bis(diphenylphosphino)ethane)nickel(II) chloride;
POCl3 trichlorophosphorous oxide;
RT retention time (on HPLC or LCMS); r.t. room temperature;
THF tetrahydrofuran;
TSTU o-(N-succinimidyl)-N,N,N',N'-tetramethyluronium tetrafluoroborate.
Compounds have been named either using ACD/Name, version 8.08, software from Advanced Chemistry Development, Inc. (ACD/Labs), Toronto ON, Canada, www.acdlabs.com, 2004 or named according to IUPAC conventions.
WORKING EXAMPLES
Below follows a number of non-limiting examples of the compounds of the present invention.
Example 1
Methyl 4-(6-bromo-3H-imidazo [4,5-6] pyridin-2-yl)benzoate

DIPEA (16.6 mL, 95.7 mmol) was added to a suspension of 5-bromo pyridine-2, 3 -diamine (6.0 g, 31.9 mmol), terepthalic acid monomethyl ester (6.89 g, 38.3 mmol) and ΗBTU
(14.5 g, 38.3 mmol) in MeCN (100 mL) and the reaction mixture was stirred at r.t. for 1 h. A precipitate that formed was collected and washed with MeCN. The solid was distributed into microwave vials with HO Ac (4 mL) and heated to +200 0C for 5 minutes. The product precipitated at r.t. and was filtered, washed with HOAc and MeCN and dried to afford 8.58 g (81% yield) of the title compound.
1U NMR (CDCl3) δ ppm ); 8.15 (d, J=1.52 Hz, 1 H), 8.07 - 8.09 (m, 2 H), 7.97 (d, J=8.84 Hz, 2 H), 7.59 (d, J=I.52 Hz, 1 H), 3.75 (s, 3 H); MS (APPI) m/z (M+ 1) 332, 334.
Example 2 Methyl 4-(6-chloro-3H-imidazo [4,5-6] pyridin-2-yl)benzoate

DIPEA (21.9 mL, 126 mmol) was added to a suspension of 5-chloro pyridine -2, 3-diamine (6.0 g, 42.0 mmol), terephtalic acid monomethyl ester (9.06 g 50.3 mmol) and ΗBTU (19.1 g 50.3mmol) in MeCN (100 mL) and the reaction mixture was stirred at r.t. for 1 h. A precipitate that formed was collected and washed with MeCN. The solid was distributed into microwave vials with HO Ac (4 mL) and heated to +200 0C for 10 minutes. The product precipitated at r.t. and was filtered, washed with HOAc and MeCN and dried to afford 10.3g (85% yield) of the title compound.
1U NMR (CDCl3) δ ppm ); 7.92 - 7.98 (m, 3 H), 7.84 (d, J=8.84 Hz, 2 H), 7.38 (d, J=I.77 Hz, 1 H), 3.59 - 3.63 (m, 3 H); MS (APPI) m/z (M+ 1) 288, 290.
Example 3
Methyl 4-(6-bromo-7-chloro-3H-imidazo [4,5-6] pyridin-2-yl)benzoate

Methyl 4-(6-bromo-3H-imidazo[4,5-ό]pyridin-2-yl)benzoate (6.7 g, 20.2 mmol), which was obtained from Example 1 and m-CPBA (70%, 17.75 g, 60.3 mmol) in HO Ac was stirred at r.t. for 18 h. An additional 2 equivalents of m-CPBA (70%, 9.06 g, 40.6 mmol) was added to the reaction mixture, and stirring was continued for 6h. The solvent was evaporated in vacuo and the residue was crystallized from EtOH. The solid was mixed with POCl3 and heated in a microwave reactor at +120 0C for 5 minutes. After cooling to r.t., the mixture was poured into ice/water mixture and the precipitate that formed was collected, washed with water and dried, affording the title compound in 6.1 g (83%) yield. 1U NMR (400 MHz, DMSO-J6) □ ppm; 8.59 (s, 1 H), 8.40(d, J=8.53 Hz, 2 H), 8.15 (d, J=8.53 Hz, 2 H), 3.91 (s, 3 H); MS (APPI) m/z (M+ 1) 368.
Example 4 Methyl 4-(6,7-dichloro-3H-imidazo [4,5-6] pyridin-2-yl)benzoate

Methyl 4-(6-chloro-3H-imidazo[4,5-ό]pyridin-2-yl)benzoate (8.3 g, 28.8 mmol), which was obtained from Example 2 and m-CPBA (70%, 19.4 g, 86.5 mmol) in HO Ac was stirred at r.t. for 18 h. An additional 2 equvalents of m-CPBA (70%, 9.06 g, 40.6 mmol) was added to the reaction mixture, and stirring was continued for 6h. The solvent was evaporated in vacuo and the residue was crystallized from EtOH. The solid was mixed with POCl3 and heated in a microwave reactor at +120 0C for 5 minutes. After cooling to
r.t, the mixture was poured into ice/water mixture and the precipitate that formed was collected, washed with water and dried, affording the title compound in 9.3 g (81%) yield.
1R NMR (400 MHz, DMSO-J6) □ ppm; 8.53 (s, 1 H), 8.41 (d, J=8.53 Hz, 2 H), 8.15 (d, J=8.53 Hz, 2 H), 3.91 (s, 3 H); MS (APPI) m/z (M+ 1) 322, 324.
Example 5
Methyl 4-(6-br omo-7-iodo-3H-imidazo [4,5-6] pyridin-2-yl)benzoate
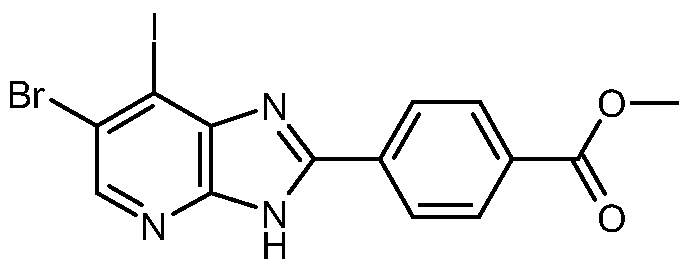
Example 6
Methyl 4-(6-chloro-7-iodo-3H-imidazo [4,5-6] pyridin-2-yl)benzoate

Methyl 4-(6,7-dichloro-3H-imidazo[4,5-ό]pyridin-2-yl)benzoate ( 2.0 g, 6.2 mmol), obtained from Example 4 was dissolved in DCM/MeOΗ (9: 1 , 20 mL), and HCl (IM in Et2O, 2 mL) was added followed by addition OfEt2O until precipitation formed. The solid was collected by filtration and dried. The hydrochloride was mixed with sodium iodide
(18.6 g, 124 mmol) and MeCN 10 (mL) and heated in a microwave reactor at +160 0C for 20 minutes. The mixture was added to Na2S2O3 (10%, aq.). The precipitate was filtered and washed with water and dried in vacuo to afford 1.9 g (76%) yield of the title compound. The mixture was used in the next step without further purification. 1R NMR (400 MHz, DMSO-J6) □ ppm; 8.40 (d, J=8.53 Hz, 1 H), 8.34 (s, 1 H), 8.13 (d, J=8.28 Hz, 2 H), 3.90 (s, 3 H); MS (APPI) m/z (M+ 1) 414, 416.
Example 7
4-(6-Br omo-7-iodo-3H-imidazo [4,5-6] pyridin-2-yl)benzoic acid

Methyl 4-(6-bromo-7-iodo-3H-imidazo[4,5-ό]pyridin-2-yl)benzoate (as described in Example 5) (4.5 g, 9.8 mmol) and lithiumhydroxide hydrate (4.2 g, 100 mmol) was dissolved in TΗF: water 9:1 (45 ml). The mixture was divided into three vials and was heated at 100° C by microwave irradiation for 10 minutes. The reaction mixture was made acidic by the addition of hydrochloric acid (2M). The solid formed was collected by filtration and washed with water and dried under vacuum to give the title compound (3.6 g, 82%) which was used without further purification. MS (ESI) m/z (M-I) 442; 444.
Example 8
4-(6-Chloro-7-iodo-3H-imidazo [4,5-6] pyridin-2-yl)benzoic acid

Methyl 4-(6-Chloro-7-iodo-3H-imidazo[4,5-ό]pyridin-2-yl)benzoate (as described in Example 6) (1.13 g, 2.74 mmol) and lithiumhydroxide hydrate (0.23 g, 5.47 mmol) was dissolved in TΗF: water 9:1 (45 ml). The mixture was heated at 100° C by microwave irradiation for 10 minutes. The reaction mixture was made acidic by the addition of
hydrochloric acid (2M). The solid formed was collected by filtration and washed with water and dried under vacuum to give the title compound (0.95 g, 86%) which was used without further purification. MS (APPI) m/z (M+ 1) 400, 402.
Example 9
6-Bromo-7-iodo-2- {4- [(4-methylpiperazin- l-yl)carbonyl] phenyl}-3H-imidazo [4,5- b] pyridine

Example 10
6-Chloro-7-iodo-2-{4-[(4-methylpiperazin-l-yl)carbonyl]phenyl}-3H-imidazo[4,5- b] pyridine

Et3N (0.21 mL, 1.50 mmol.) was added to a suspension of 4-(6-chloro-7-iodo-3H- imidazo[4,5-ό]pyridin-2-yl)benzoic acid (as described in Example 8) (0.20 g, 0.50 mmol) and TSTU (0.226 g, 0.75 mmol) in DMF (5mL) and the reaction mixture was stirred at room temperature for 30 minutes. N-Methylpiperazine (0.083 g, 0.75 mmol) was added and the reaction mixture was stirred for lh.The solvent was evaporated and the crude product mixture was used directly in the next step without further purification. 1R NMR (400 MHz, DMSO-J6) D ppm; 8.34 (s, 1 H), 8.31 (d, J=8.28 Hz, 2 H), 7.58 (d, J=8.28 Hz, 2 H), 2.21 (s, 3 H), 1.91 (s, 1 H); MS (ESI) m/z (M+ 1) 480, 482.
Example 11
6-Bromo-7-(4-methoxyphenyl)-2-{4-[(4-methylpiperazin-l-yl)carbonyl]phenyl}-3H- imidazo [4,5-6] pyridine

6-Bromo-7-iodo-2- {4-[(4-methylpiperazin- 1 -yl)carbonyl]phenyl} -3H-imidazo[4,5- ό]pyridine (described in Example 9) (150 mg, 0.29 mmol), 4-methoxyphenyl boronic acid (43 mg, 0.29 mmol), sodium carbonate (123 mg, 1.16 mmol) and PdCl2(dppf)*DCM (12 mg, 0.014 mmol) were mixed in TΗF: water 9:1 (3 ml). The mixture was heated in a microwave reactor at 130° for 17 minutes. The mixture was filtered through diatomeous earth and the filtrate was concentrated. The residue was purified by preparative ΗPLC which afforded the product as a base. The base was dissolved in dichloromethane and hydrochloric acid (0.1 ml, IM in diethyl ether) was added. The solvent was evaporated to give the hydrochloride salt of the title compound as a solid (30 mg, 19%).
1U NMR (400 MHz, DMSO-J6) D ppm 10.60 (br. s., 1 H) 8.57 (s, 1 H) 8.20 - 8.31 (m, 2 H) 7.57 - 7.64 (m, 4 H) 7.14 (d, 2 H) 5.76 (s, 3 H) 3.87 (s, 3 H) 3.10 (br. s., 2 H) 2.79 (s, 3 H); MS (ESI) m/z (M+l).
Example 12
6-Chloro-7-(4-methoxyphenyl)-2-{4-[(4-methylpiperazin-l-yl)carbonyl]phenyl}-3H- imidazo [4,5-6] pyridine

6-Chloro-7-iodo-2- {4-[(4-methylpiperazin- 1 -yl)carbonyl]phenyl} -3H-imidazo[4,5- όjpyridine (as described in Example 10) (0.501 mmol), 4-methoxyphenyl boronic acid (152 mg, 1.0 mmol), sodium carbonate (319 mg, 3.0 mmol) and PdCl2(dppf)*DCM (20 mg, 0.025 mmol) were mixed in TΗF: water 9:1 (4 ml). The mixture was heated in a microwave reactor at 150° for 15 minutes. The mixture was filtered through diatomeous earth and the filtrate was concentrated. The residue was purified by preparative ΗPLC which afforded the product as a base. The base was dissolved in dichloromethane and hydrochloric acid (0.1 ml, IM in diethyl ether) was added. The product precipitated, was filtered and dried in vacou to afford the hydrochloride salt of the title compound as a solid (36 mg, 16%). NMR data from the free base.
1U NMR (400 MHz, DMSO-J6) □ ppm; 8.37 (s, 1 H), 8.25 (d, J=7.97 Hz, 2 H), 7.61 - 7.71 (m, 2 H), 7.50 (d, J=7.97 Hz, 2 H), 7.12 (d, J=8.58 Hz, 2 H), 3.86 (s, 3 H), 3.25 - 3.69 (m, 4 H), 2.54 (s, 3 H), 2.25 - 2.41 (m, 4 H); MS (ESI) m/z (M+l) 462, 464.
Example 13
Methyl 4-(6-fluoro-3H-imidazo [4,5-b] pyridin-2-yl)benzoate

Triethylamine (2.412 mL, 17.31 mmol) was added to a suspension of 5-fluoropyridine-2,3- diamine (2.2 g, 17.31 mmol), 4-(methoxycarbonyl)benzoic acid (3.12 g, 17.31 mmol) and O-benzotriazol-1-yl-tetramethyluronium hexafluorophosphate (6.56 g, 17.31 mmol) in acetonitrile (15 mL) and the reaction mixture was stirred at r.t. for 1 h.The precipitate that formed was collected and washed with MeCN. The solid was distributed into microwave vials with HOAc (4 mL) and heated to +200 0C for 5 minutes. The product precipitated at r.t. and was filtered, washed with HOAc and MeCN and dried to afford methyl 4-(6-fluoro- 3H-imidazo[4,5-b]pyridin-2-yl)benzoate (3.70 g, 79 %) MS ESI m/z 272 M+
Example 14
Methyl 4-(7-chlor o-6-fluoro-3H-imidazo [4,5-b] pyridin-2-yl)benzoate

Methyl 4-(6-fluoro-3H-imidazo[4,5-b]pyridin-2-yl)benzoate (2.7 g, 9.95 mmol) and 3- chloroperoxybenzoic acid (7.36 g, 29.86 mmol) were mixed with acetic acid (100 mL) and stirred at R.T for 18h. The solvent was evaporated. EtOH was added and the product mixture was allowed to stand at R.T o.n. The precipitate was filtered and dried. The intermediate (1.34 g, 4.66 mmol) was suspended in POCl3 (30 ml). The mixture was heated at 90° for 10 min in a microwave reactor. The mixture was poured onto ice and NaΗCOβ
(aq). The solid was isolated by filtration and washed with water. The solid was dried under vacuum at 50° to give a solid (1.2 g, 84%) that was used without further purification. A sample was purified by preparative ΗPLC for NMR analysis
1Η NMR (400 MHz, DMSO-J6) δ ppm 8.52 (d, 1 H) 8.39 (d, 2 H) 8.33 (d, 1 H) 8.14 (d, 2 H) 3.90 (s, 3 H)
MS (ESI) m/z 306; 308 (M+ 1).
Example 15
Methyl 4-(6-fluoro-7-iodo-3H-imidazo [4,5-6] pyridin-2-yl)benzoate
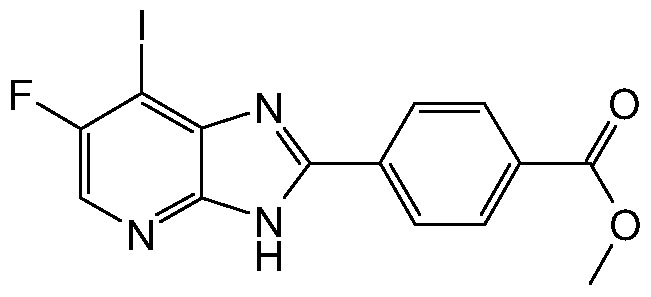
Methyl 4-(7-chloro-6-fluoro-3H-imidazo[4,5-ό]pyridin-2-yl)benzoate (1.20 g, 3.93 mmol) was suspended in TΗF. Hydrochloric acid (1 M in diethyl ether, 4 ml) was added and the solvents were evaporated. Sodium iodide (8.83 g, 58.9 mmol) and acetonitrile (40 ml) was added and the mixture was heated to 160° for 30 min in a microwave reactor. The mixture was poured onto NaHCOβ (aq) containing Na2S2C>3. The solid was collected by filtration and washed with water. The solid was dried under vacuum to give a solid (0.96 g, 62 %) that was used without further purification. A sample was purified by preparative HPLC for NMR analysis. IH NMR (400 MHz, DMSO-J6) δ ppm 8.38 (d, 2 H) 8.28 (s, 1 H) 8.14 (d, 2 H) 3.90 (s, 3 H)
MS (ESI) m/z 398 (M+ 1)
Example 16
4-(6-Fluoro-7-iodo-3H-imidazo [4,5-6] pyridin-2-yl)benzoic acid

Methyl 4-(6-fluoro-7-iodo-3H-imidazo[4,5-ό]pyridin-2-yl)benzoate (0.84 g, 2.12 mmol) and lithium hydroxide monohydrate (0.89 g, 21 mmol) were mixed in THF (18 mL) and water (2 mL). The mixture was heated to 100 0C for 15 min in a microwave reactor. The mixture was concentrated. Hyrochloric acid (aq, 2M) was added until acidic pH. The solid was isolated by filtration and was washed with water and dried under vacuum to give 0.759 g (94%) which was used without further purification.
MS (ESI) m/z 384 (M+ 1), m/z 382 (M-I).
Example 17
6-Fluoro-7-iodo-2- {4- [(4-methylpiperazin- l-yl)carbonyl] phenyl}-3H-imidazo [4,5- b] pyridine

4-(6-Fluoro-7-iodo-3H-imidazo[4,5-ό]pyridin-2-yl)benzoic acid (250 mg, 0.65 mmol) was dissolved in DMF (8 mL). O-(N-Succinimidyl)-N,N,N',N'-tetramethyl-uronium tetrafluoroborate (246 mg, 0.82 mmol) and triethylamine (0.273 mL, 1.96 mmol) were added. The mixture was stirred at ambient temperature for 15 minutes. 1-Methylpiperazine (0.109 mL, 0.98 mmol) was added and the mixture was stirred for 16 h. The mixture was diluted with NaΗCOβ (aq) and was extracted with dichloromethane (x3). The combined organic phases were dried (Na2SC>4) and the solvents were evaporated. The residue was purified by preparative ΗPLC.
The fractions containing product were pooled. NaΗCOβ (aq) was added and the mixture was extracted with dichloromethane (x3). The organic phase was dried (Na2SC>4) and evaporated to give a solid (74 mg, 24%). 1Η NMR (400 MHz, DMSO-J6) δ ppm 8.22 - 8.34 (m, 3 H) 7.56 - 7.61 (m, 2 H) 3.65 (s, 2 H) 2.24 - 2.43 (m, 4 H) 2.20 (s, 3 H) MS (ESI) m/z 466 (M+ 1).
Example 18
6-Fluoro-7-(4-methoxyphenyl)-2- {4- [(4-methylpiperazin- l-yl)carbonyl] phenyl}-3H- imidazo [4,5b] pyridine

6-Fluoro-7-iodo-2- {4-[(4-methylpiperazin- 1 -yl)carbonyl]phenyl} -3H-imidazo[4,5- ό]pyridine (74 mg, 0.16 mmol), 4-methoxyphenylboronic acid (36 mg, 0.24 mmol), Na2CO3 (67 mg, 0.64 mmol) and PdCl2^pPf)-CH2Cl2 adduct (13 mg, 0.02 mmol) were mixed in THF (1.8 mL) and water (0.2 mL). The mixture was heated to 130 0C in a microwave reactor for 20 min. The mixture was filtered through diatomeous earth and the filtrated was concentrated. The residue was purified by preparative HPLC. The pure fractions were combined, NaHCO3 (aq) was added and the mixture was extracted with dichloromethane (x3). The organic phase was dried (Na2SO^ and evaporated to give a solid. The solid was dissolved in dichloromethane (3 ml) and methanol (3 drops). Hydrochloric acid (IM in diethyl ether, 0.05 ml) was added and the solvent was evaporated. The solid was dried under vacuum at 40° to give the hydrochloride salt of the title compound (28 mg, 31%). IH NMR (400 MHz, DMSO-J6) δ ppm 10.89 (s, 1 H) 8.44 (d, 1 H) 8.32 (d, 2 H) 8.00 (d, 2 H) 7.66 (d, 2 H) 7.17 (d, 2 H) 4.15-4.65 (m, 8 H) 3.87 (s, 3 H) 2.78 (d, 3 H) MS (ESI) m/z 446 (M+ 1).
Medical use
Surprisingly, it has been found that the compounds of formula (I) defined in the present invention, are well suited for inhibiting glycogen synthase kinase-3 (GSK3). Accordingly, said compound of the present invention is expected to be useful in the prevention and/or treatment of conditions associated with glycogen synthase kinase-3 activity, i.e. the compounds may be used to produce an inhibitory effect of GSK3 in mammals, including human, in need of such prevention and/or treatment.
GSK3 is highly expressed in the central and peripheral nervous system and in other tissues. Thus, it is expected that compound of the invention is well suited for the prevention and/or treatment of conditions associated with glycogen synthase kinase-3 in the central and peripheral nervous system. In particular, the compound of the invention is expected to be suitable for prevention and/or treatment of conditions associated with cognitive disorders and predemented states, especially dementia, Alzheimer's Disease (AD), Cognitive Deficit in Schizophrenia (CDS), Mild Cognitive Impairment (MCI), Age-Associated Memory Impairment (AAMI), Age-Related Cognitive Decline (ARCD) and Cognitive Impairement No Dementia (CIND), diseases associated with neurofibrillar tangle pathologies,
Frontotemporal dementia (FTD), Frontotemporal dementia Parkinson's Type (FTDP), progressive supranuclear palsy (PSP), Pick's Disease, Niemann-Pick's Disease, corticobasal degeneration (CBD), traumatic brain injury (TBI) and dementia pugilistica.
One embodiment of the invention relates to the prevention and/or treatment of Alzheimer's Disease, especially the use in the delay of the disease progression of Alzheimer's Disease.
Other conditions are selected from the group consisting of Down's syndrome, vascular dementia, Parkinson's Disease (PD), postencephelatic parkinsonism, dementia with Lewy bodies, HIV dementia, Huntington's Disease, amyotrophic lateral sclerosis (ALS), motor neuron diseases (MND, Creuztfeld-Jacob's disease and prion diseases.
Other conditions are selected from the group consisting of attention deficit disorder (ADD), attention deficit hyperactivity disorder (ADHD) and affective disorders, wherein the affective disorders are Bipolar Disorder including acute mania, bipolar depression, bipolar maintenance, major depressive disorders (MDD) including depression, major depression, mood stabilization, schizoaffective disorders including schizophrenia, and dysthymia.
Other conditions are selected from the group consisting of Type I diabetes, Type II diabetes, diabetic neuropathy, alopecia, inflammatory diseases and cancer.
One embodiment of the invention relates to the use of a compound of the formula (I) , as a free base or a pharmaceutically acceptable salt thereof, in the prevention and/or treatment of bone-related disorders or conditions in mammals.
One aspect of the invention is directed to the use of a compound of the formula (I) , as a free base or a pharmaceutically acceptable salt thereof, to treat osteoporosis.
One aspect of the invention is directed to the use of a compound of the formula (I), as a free base or a pharmaceutically acceptable salt thereof, to increase and promote bone formation in mammals.
One aspect of the invention is directed to the use of a compound of the formula (I), as a free base or a pharmaceutically acceptable salt thereof, to increase bone mineral density in mammals.
Another aspect of the invention is directed to the use of a compound of the formula (I), as a free base or a pharmaceutically acceptable salt thereof, to reduce the rate of fracture and/or increase the rate of fracture healing in mammals.
Another aspect of the invention is directed to the use of a compound of the formula (I), as a free base or a pharmaceutically acceptable salt thereof, to increase cancellous bone formation and/or new bone formation in mammals.
Another aspect of the invention is directed to a method of prevention and/or treatment of bone-related disorders comprising administering to a mammal in need of such prevention and/or treatment, a therapeutically effective amount of a compound of the formula (I) as a free base or a pharmaceutically acceptable salt thereof.
Another aspect of the invention is directed to a method of prevention and/or treatment of osteoporosis comprising administering to a mammal in need of such prevention and/or treatment, a therapeutically effective amount of a compound of the formula (I) as a free base or a pharmaceutically acceptable salt thereof.
Another aspect of the invention is directed to a method of increasing bone formation comprising administering to a mammal in need of such treatment, a therapeutically effective amount of a compound of the formula (I) as a free base or a pharmaceutically acceptable salt thereof.
Another aspect of the invention is directed to a method of increasing bone mineral density comprising administering to a mammal in need of such treatment, a therapeutically effective amount of a compound of the formula (I) as a free base or a pharmaceutically acceptable salt thereof.
Another aspect of the invention is directed to a method of reducing the incidence of fracture comprising administering to a mammal in need of such treatment, a therapeutically effective amount of a compound of the formula (I) as a free base or a pharmaceutically acceptable salt thereof.
Another aspect of the invention is directed to a method of enhancing fracture healing comprising administering to a mammal in need of such treatment, a therapeutically effective amount of a compound of the formula (I) as a free base or a pharmaceutically acceptable salt thereof.
Another aspect of the invention is directed to said methods and wherein said mammal is a human.
Another aspect of the invention is directed to said methods and wherein said mammal is a vertibrate animal, preferably but not limited to bigger animals such as horses, camels, dromedars but not limited thereto.
The use of the GSK3 inhibitors, the compounds of formula (I), in primary and secondary ostopeorosis, where primary osteoporosis includes postmenopausal osteoporosis and senile osteoporosis in both men and women, and secondary osteoporosis includes cortison induced osteoporosis, as well as any other type of induced secondary osteoporosis, are
included in the term osteoporosis. In addition to this, these GSK3 inhibitors may also be used in treatments of myeloma. These GSK3 inhibitors may be administered locally or systemically, in different formulation regimes, to treat these conditions.
The promotion and increasing of bone formation makes the compounds of the formula (I) suitable to reducing the incidence of fracture, to reduce the rate of fracture and/or increase the rate of fracture healing, to increase cancellous bone formation and/or new bone formation in mammals.
The use to promote and increase new bone formation may be in connection with surgery.
This invention can be used during surgery, where the treating surgeon will place the invention locally in an appropriate formulation, near the deficient bone and/or in the body cavity. The bone may for instance have been broken, and utilizing the invention as described and claimed herein will then be placed in or near the fracture during open fracture repair. In some instances bone pieces may be missing (e.g. after tumour removal or severe casualties), and utilizing the invention as described and claimed herein will then be placed near the site of constructive bone surgery.
Non-medical use
In addition to their use in therapeutic medicine, the compounds of formula I as a free base or a pharmaceutically acceptable salt thereof, are also useful as pharmacological tools in the development and standardisation of in vitro and in vivo test systems for the evaluation of the effects of inhibitors of GSK3 related activity in laboratory animals such as cats, dogs, rabbits, monkeys, rats and mice, as part of the search for new therapeutics agents.
Pharmaceutical compositions
According to one aspect of the present invention there is provided a pharmaceutical composition comprising a compound of formula I, as a free base or a pharmaceutically acceptable salt, solvate or solvate of salt thereof, for use in the prevention and/or treatment of conditions associated with glycogen synthase kinase-3.
The composition used in accordance with the present invention may be in a form suitable for oral administration, for example as a tablet, pill, syrup, powder, granule or capsule, for parenteral injection (including intravenous, subcutaneous, intramuscular, intravascular or infusion) as a sterile solution, suspension or emulsion, for topical administration as an ointment, patch or cream, for rectal administration as a suppository and for local administration in a body cavity or in a bone cavity.
Suitable daily doses of the compounds of the formula (I) used in the treatment of a mammal, including human, are approximately from 0.01 to 250 mg/kg bodyweight at peroral administration and from about 0.001 to 250 mg/kg bodyweight at parenteral administration. The typical daily dose of the active ingredients varies within a wide range and will depend on various factors such as the relevant indication, the route of administration, the age, weight and sex of the patient and may be determined by a physician.
For veterinary use the amounts of different components, the dosage form and the dose of the medicament may vary and will depend on various factors such as, for example the individual requirement of the animal treated.
A suitable pharmaceutically acceptable salt of the compound of formula (I) useful in accordance to the invention is, for example, an acid-addition salt, which is sufficiently basic, for example an inorganic or organic acid. In addition a suitable pharmaceutically acceptable salt of the compounds of the invention, which is sufficiently acidic, is an alkali metal salt, an alkaline earth metal salt or a salt with an organic base, which affords a physiologically-acceptable cation.
The dose required for the therapeutic or preventive treatment of a particular disease, disorder or a particular condition will necessarily be varied depending on the host treated, the route of administration and the severity of the illness or injury being treated.
In the context of the present specification, the term "therapy" also includes "prevention" unless there are specific indications to the contrary. The terms "therapeutic" and "therapeutically" should be construed accordingly.
5 In the context of the present specification, the term "disorder" also includes "condition" unless there are specific indications to the contrary.
Pharmacology
Determination of ATP competition in Scintillation Proximity GSKS β Assay. i o GSK3β scintillation proximity assay.
The competition experiments were carried out in duplicate with 10 different concentrations of the inhibitors in clear-bottom microtiter plates (Wallac, Finland). A biotinylated peptide substrate, Biotin-Ala-Ala-Glu-Glu-Leu-Asp-Ser-Arg-Ala-Gly-Ser(PO3H2)-Pro-Gln-Leu (AstraZeneca, Lund), was added at a final concentration of 1 μM in an assay buffer is containing 1 mU recombinant human GSK3β (Dundee University, UK), 12 mM morpholinepropanesulfonic acid (MOPS), pH 7.0, 0.3 mM EDTA, 0.01% β- mercaptorethanol, 0.004 % Brij 35 (a natural detergent), 0.5 % glycerol and 0.5 μg BSA/25 μl. The reaction was initiated by the addition of 0.04 μCi [γ-33P]ATP (Amersham, UK) and unlabelled ATP at a final concentration of 1 μM and assay volume of 25 μl. After
20 incubation for 20 minutes at room temperature, each reaction was terminated by the addition of 25 μl stop solution containing 5 mM EDTA, 50 μM ATP, 0.1 % Triton X-100 and 0.25 mg streptavidin coated Scintillation Proximity Assay (SPA) beads (Amersham, UK). After 6 hours the radioactivity was determined in a liquid scintillation counter (1450 MicroBeta Trilux, Wallac). The inhibition curves were analysed by non-linear regression
25 using GraphPad Prism, USA. The Km value of ATP for GSK3β, used to calculate the inhibition constants (K1) of the various compounds, was 20 μM.
The following abbreviations have been used: MOPS Morpholinepropanesulfonic acid
30 EDTA Ethylenediaminetetraacetic acid
BSA Bovin Serum Albumin
ATP Adenosine Triphosphate
SPA Scintillation Proximity Assay
GSK3 Glycogen synthase kinase 3
Results
Typical K1 values for the compounds of the present invention are in the range of about 0.001 to about 10,000 nM. Other values for K1 are in the range of about 0.001 to about 1000 nM. Further values for K1 are in the range of about 0.001 nM to about 300 nM.
Table 1. Specimen results from assay.
Claims
1. A compound of formula I
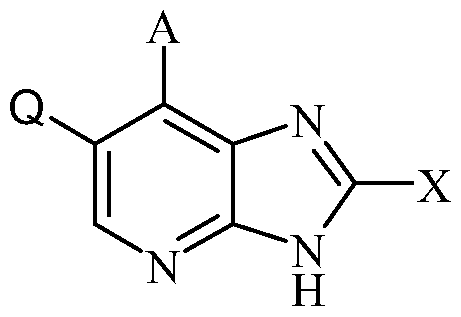
wherein
Q is selected from halogen;
X is

R2 and R4 are independently selected from hydrogen, halogen, CN, NO2, Ci-3alkyl, C1-
3haloalkyl;
R3 and R5 are independently selected from hydrogen, halogen, Ci-3alkyl and Ci-3haloalkyl;
A is aryl or heteroaryl, optionally substituted with one or more CN, CO2H, Ci-6alkyl, C1- ghaloalkyl, halo,C(O)Ra, ORk, C(O)NRbRc or S(O)nRm, wherein said Ci-6alkyl or Ci- όhaloalkyl is optionally substituted by at least one CN, ORa or NRbRc;
Ra is selected from hydrogen, Ci-3alkyl and Ci-3haloalkyl, wherein said Ci-3alkyl or Ci-
3haloalkyl is optionally substituted with one or more Ci-3alkoxy;
Rb and Rc may, together with the atom to which they are attached, form a 4-, 5- o 6- membered heterocyclic ring containing one or more heteroatoms selected from N, O or S, wherein said heterocyclic ring is optionally substituted with one or more halo, ORa, NRdRe,
Ci-3alkyl or Ci-3haloalkyl, wherein said Ci-3alkyl or Ci-3haloalkyl is optionally further substituted with one or more Ci-3alkoxy;
Rd and Re are independently selected from hydrogen,  wherein said Ci_6alkyl or Ci-όhaloalkyl is optionally substituted with one or more ORa; or Rk is
wherein said Ci_6alkyl or Ci-όhaloalkyl is optionally substituted with one or more ORa; or Rk is  wherein said
wherein said  is optionally substituted with at least one CN, ORa, NRbRc, C(O)NRbRc or NRbC(O)Rc;
is optionally substituted with at least one CN, ORa, NRbRc, C(O)NRbRc or NRbC(O)Rc;
Rm is Ci-3alkyl, optionally substituted with at least one halo, CN, ORa, NRbRc or
C(O)NRbRc; n is 0 to 2; as a base or a pharmaceutically acceptable salt, solvate or solvate of a salt thereof.
2. A compound according to claim 1, wherein said halogen in Q is selected from chloro, bromo and fluoro.
3. A compound according to claim 2, wherein Rb and Rc in C(O)NRbRc, together with the atom to which they are attached, form a 6-membered heterocyclic ring containing one or more heteroatoms selected from N, O or S, wherein said heterocyclic ring optionally is substituted with one 
4. A compound according to any one of claims 1 to 3, wherein said A is phenyl, optionally substituted with one ORk .
5. A compound according to claim 4, wherein Rk is Ci-6alkyl .
6. A compound according to any one of claims 1 to 6, wherein X is

and wherein said R2, R3, R4 and R5 represent hydrogen.
7. A compound according to claim 1 , wherein
Q is halogen
X is 
R1 is C(O)NRbRc; R2 and R4 are hydrogen; R3 and R5 are hydrogen; A is aryl optionally substituted with one ORk;
Rb and Rc, together with the atom to which they are attached, form a 6-membered heterocyclic ring containing one or two heteroatoms selected from N, O or S, wherein said heterocyclic ring is optionally substituted with one C1-3alkyl; and Rk is Ci-βalkyl.
8. A compound according to claim 1, wherein Q is halogen X is

R2 and R4 are hydrogen;
R3 and R5 are hydrogen;
A is aryl optrionally substituted with one ORk;
Rb and Rc, together with the atom to which they are attached, form a 6-membered heterocyclic ring containing two heteroatoms selected from N, wherein said heterocyclic ring is substituted with one C1-3alkyl; and
Rk is Ci-βalkyl.
9. A compound according to claim 1, wherein Q is halogen X is 
R1 is C(O)NRbRc; R2 and R4 are hydrogen; R3 and R5 are hydrogen; A is aryl optionally substituted with one ORk;
Rb and Rc, together with the atom to which they are attached, form a piperazine, wherein said piperazine is substituted with one C1-3alkyl; and Rk is Ci-6alkyl.
10. A compound according to claim 1, wherein Q is halogen X is

R1 is C(O)NRbRc; R2 and R4 are hydrogen;
R3 and R5 are hydrogen;
A is phenyl optionally substituted with one ORk;
Rb and Rc, together with the atom to which they are attached, form a piperazine, wherein said piperazine is substituted with one C^alkyl; and Rk is Ci-6alkyl.
11. A compound according to claim 1 , selected from:
6-Bromo-7-(4-methoxyphenyl)-2- {4-[(4-methylpiperazin- 1 -yl)carbonyl]phenyl} -3H- imidazo[4,5-ό]pyridine hydrochloride, 6-Chloro-7-(4-methoxyphenyl)-2- {4-[(4-methylpiperazin- 1 -yl)carbonyl]phenyl} -3H- imidazo[4,5-ό]pyridine hydrochloride and 6-Fluoro-7-(4-methoxyphenyl)-2- {4-[(4-methylpiperazin- 1 -yl)carbonyl]phenyl} -3H- imidazo [4,5 -ό]pyridine as a base or an alternative pharmaceutically acceptable salt, solvate or solvate of a salt thereof.
12. A compound as defined in any one of claims 1 to 11 for use in therapy.
13. Use of a compound as defined in any one of claims 1 to 11 in the prevention and/or treatment of cognitive disorders, dementia, Alzheimer's Disease (AD), Cognitive Deficit in Schizophrenia (CDS), Mild Cognitive Impairment (MCI), Age-Associated Memory
Impairment (AAMI), Age-Related Cognitive Decline (ARCD), Cognitive Impairement No Dementia (CIND), Frontotemporal dementia (FTD), Frontotemporal dementia Parkinson's Type (FTDP), progressive supranuclear palsy (PSP), Pick's Disease, Niemann-Pick's Disease, corticobasal degeneration, traumatic brain injury (TBI), dementia pugilistica, Down's syndrome, vascular dementia, Parkinson's Disease (PD), postencephelatic parkinsonism, dementia with Lewy bodies, HIV dementia, Huntington's Disease, amyotrophic lateral sclerosis (ALS), motor neuron diseases (MND, Creuztfeld-Jacob's disease, prion diseases, attention deficit disorder (ADD), attention deficit hyperactivity disorder (ADHD), affective disorders, Bipolar Disorder including acute mania, bipolar depression, bipolar maintenance, major depressive disorders (MDD) including depression, major depression, mood stabilization, schizoaffective disorders including schizophrenia, or dysthymia, diabetes, alopecia, or bone-related disorders including osteoporosis and increased bone formation.
14. The use according to claim 13, wherein the disease is Alzheimer's Disease.
15. A compound selected from:
Methyl 4-(6-bromo-3H-imidazo[4,5-ό]pyridin-2-yl)benzoate; Methyl 4-(6-chloro-3H-imidazo[4,5-ό]pyridin-2-yl)benzoate; Methyl 4-(6-fluoro-3H-imidazo[4,5-b]pyridin-2-yl)benzoate; Methyl 4-(6-bromo-7-chloro-3H-imidazo[4,5-ό]pyridin-2-yl)benzoate; Methyl 4-(6,7-dichloro-3H-imidazo[4,5-ό]pyridin-2-yl)benzoate;
Methyl 4-(7-chloro-6-fluoro-3H-imidazo[4,5-b]pyridin-2-yl)benzoate
Methyl 4-(6-bromo-7-iodo-3H-imidazo[4,5-ό]pyridin-2-yl)benzoate;
Methyl 4-(6-chloro-7-iodo-3H-imidazo[4,5-b]pyridin-2-yl)benzoate; Methyl 4-(6-fluoro-7-iodo-3H-imidazo[4,5-ό]pyridin-2-yl)benzoate;
4-(6-Bromo-7-iodo-3H-imidazo[4,5-ό]pyridin-2-yl)benzoic acid;
4-(6-Chloro-7-iodo-3H-imidazo[4,5-ό]pyridin-2-yl)benzoic acid;
4-(6-Fluoro-7-iodo-3H-imidazo[4,5-ό]pyridin-2-yl)benzoic acid;
6-Bromo-7-iodo-2- {4-[(4-methylpiperazin- 1 -yl)carbonyl]phenyl} -3H-imidazo[4,5- ό]pyridine;
6-Chloro-7-iodo-2- {4-[(4-methylpiperazin- 1 -yl)carbonyl]phenyl} -3H-imidazo[4,5- ό]pyridine and
6-Fluoro-7-iodo-2- {4-[(4-methylpiperazin- 1 -yl)carbonyl]phenyl} -3H-imidazo[4,5- ό]pyridine.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US90903807P | 2007-03-30 | 2007-03-30 | |
| US60/909,038 | 2007-03-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2008121064A1 true WO2008121064A1 (en) | 2008-10-09 |
Family
ID=39808539
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/SE2008/050357 WO2008121064A1 (en) | 2007-03-30 | 2008-03-28 | New imidazo[4,5-b]pyridine-6-halo-7-aryl/heteroaryl compounds 705 |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2008121064A1 (en) |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2018011138A1 (en) | 2016-07-11 | 2018-01-18 | Kancera Ab | 2-phenylimidazo[4,5-b]pyridin-7-amine derivates useful as inhibitors of mammalian tyrosine kinase ror1 activity |
| US10550113B2 (en) | 2015-02-02 | 2020-02-04 | Kancera Ab | 2-phenyl-3H-imidazo[4,5-B]pyridine derivates useful as inhibitors of mammalian tyrosine kinase ROR1 activity |
| US11660303B2 (en) | 2016-07-11 | 2023-05-30 | Kancera Ab | 2-phenylimidazo[4,5-b]pyridin-7-amine derivates useful as inhibitors of mammalian tyrosine kinase ROR1 activity |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999007703A1 (en) * | 1997-08-05 | 1999-02-18 | Pfizer Products Inc. | 4-AMINOPYRROLE(3,2-d) PYRIMIDINES AS NEUROPEPTIDE Y RECEPTOR ANTAGONISTS |
| WO2004016611A1 (en) * | 2002-08-14 | 2004-02-26 | Astrazeneca Ab | Use of and some novel imidazopyridines |
| US20060189617A1 (en) * | 2005-02-18 | 2006-08-24 | Wyeth | Imidazo[4,5-b]pyridine antagonists of gonadotropin releasing hormone receptor |
| WO2007040438A2 (en) * | 2005-10-03 | 2007-04-12 | Astrazeneca Ab | Novel imidazo [4,5 -b] pyridine derivatives as inhibitors of glycogen synthase kinase 3 for use in the treatment of dementia and neurodegenerative disorders |
| WO2007072017A2 (en) * | 2005-12-22 | 2007-06-28 | The Institute Of Cancer Research | Enzyme inhibitors |
-
2008
- 2008-03-28 WO PCT/SE2008/050357 patent/WO2008121064A1/en active Application Filing
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999007703A1 (en) * | 1997-08-05 | 1999-02-18 | Pfizer Products Inc. | 4-AMINOPYRROLE(3,2-d) PYRIMIDINES AS NEUROPEPTIDE Y RECEPTOR ANTAGONISTS |
| WO2004016611A1 (en) * | 2002-08-14 | 2004-02-26 | Astrazeneca Ab | Use of and some novel imidazopyridines |
| US20060189617A1 (en) * | 2005-02-18 | 2006-08-24 | Wyeth | Imidazo[4,5-b]pyridine antagonists of gonadotropin releasing hormone receptor |
| WO2007040438A2 (en) * | 2005-10-03 | 2007-04-12 | Astrazeneca Ab | Novel imidazo [4,5 -b] pyridine derivatives as inhibitors of glycogen synthase kinase 3 for use in the treatment of dementia and neurodegenerative disorders |
| WO2007072017A2 (en) * | 2005-12-22 | 2007-06-28 | The Institute Of Cancer Research | Enzyme inhibitors |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US10550113B2 (en) | 2015-02-02 | 2020-02-04 | Kancera Ab | 2-phenyl-3H-imidazo[4,5-B]pyridine derivates useful as inhibitors of mammalian tyrosine kinase ROR1 activity |
| WO2018011138A1 (en) | 2016-07-11 | 2018-01-18 | Kancera Ab | 2-phenylimidazo[4,5-b]pyridin-7-amine derivates useful as inhibitors of mammalian tyrosine kinase ror1 activity |
| US11008318B2 (en) | 2016-07-11 | 2021-05-18 | Kancera Ab | 2-phenylimidazo[4,5-b]pyridin-7-amine derivates useful as inhibitors of mammalian tyrosine kinase ROR1 activity |
| US11660303B2 (en) | 2016-07-11 | 2023-05-30 | Kancera Ab | 2-phenylimidazo[4,5-b]pyridin-7-amine derivates useful as inhibitors of mammalian tyrosine kinase ROR1 activity |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20100234593A1 (en) | Imidazo[4,5-B]Pyridine-7-Carboxamides 704 | |
| WO2008121064A1 (en) | New imidazo[4,5-b]pyridine-6-halo-7-aryl/heteroaryl compounds 705 | |
| AU2006297948B2 (en) | Novel imidazo [4,5 -b] pyridine derivatives as inhibitors of glycogen synthase kinase 3 for use in the treatment of dementia and neurodegenerative disorders | |
| JP4465188B2 (en) | New compounds | |
| EP2456440B1 (en) | Quinolinone pde2 inhibitors | |
| EP2434885B1 (en) | Alkoxy tetrahydro-pyridopyrimidine pde10 inhibitors | |
| US20120101132A1 (en) | New Substituted Oxindole Derivative | |
| KR20090024817A (en) | Imidazol-pyrimidine derivatives for treatment of diseases related to glycogen synthase kinase (gsk3) | |
| AU2006232620A1 (en) | Substituted heterocycles and their use as CHK1, PDK1 and PAK inhibitors | |
| WO2011002409A1 (en) | 5h-pyrrolo[3,4-£>]pyrazin-7-amine derivatives inhibitors of beta-secretase | |
| WO2011053559A1 (en) | Aryl aminopyridine pde10 inhibitors | |
| US20080255106A1 (en) | Novel 2-Phenyl-Imidazo[4,5-B]Pyridine Derivatives as Inhibitors of Glycogen Synthase Kinase for the Treatment of Dementia and Neurodegenerative Disorders | |
| JP6283688B2 (en) | Novel pyrazole-substituted imidazopyrazine as casein kinase 1D / E inhibitor | |
| SK10252003A3 (en) | Pyrimidine compounds | |
| WO2016145614A1 (en) | Triazolyl pyrimidinone compounds as pde2 inhibitors | |
| JP2009510160A (en) | Use of pyrimidine derivatives in the manufacture of a medicament for preventing and / or treating Alzheimer's disease | |
| US20240158391A1 (en) | Derivatives of 4-(imidazo[1,2-a]pyridin-3-yl)-n-(pyridin-3-yl) pyrimidin-2- amine for treating proliferative diseases and conditions | |
| AU2014282977B2 (en) | 1-sulfonyl piperidine derivatives as modulators of prokineticin receptors | |
| US20110118275A1 (en) | OXAZOLO[4,5-c]PYRIDINE SUBSTITUTED PYRAZINE |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 08724303 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 08724303 Country of ref document: EP Kind code of ref document: A1 |