WO2008083070A1 - Crf1 receptor ligands comprising fused bicyclic heteroaryl moieties - Google Patents
Crf1 receptor ligands comprising fused bicyclic heteroaryl moieties Download PDFInfo
- Publication number
- WO2008083070A1 WO2008083070A1 PCT/US2007/088534 US2007088534W WO2008083070A1 WO 2008083070 A1 WO2008083070 A1 WO 2008083070A1 US 2007088534 W US2007088534 W US 2007088534W WO 2008083070 A1 WO2008083070 A1 WO 2008083070A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- alkyl
- salt
- hydrogen
- crf
- Prior art date
Links
- 0 CCC(CC)[n](cc1*)c2c1nc(-c1c(C)cc(CO)[s]1)c(C)n2 Chemical compound CCC(CC)[n](cc1*)c2c1nc(-c1c(C)cc(CO)[s]1)c(C)n2 0.000 description 6
- LTFWYIGEZRNMNN-UHFFFAOYSA-N CCC(CC)[n](cc1C)c2c1nc(-c1c(C)nc(C=O)[s]1)c(C)n2 Chemical compound CCC(CC)[n](cc1C)c2c1nc(-c1c(C)nc(C=O)[s]1)c(C)n2 LTFWYIGEZRNMNN-UHFFFAOYSA-N 0.000 description 1
- LMJWYFDQEADRJH-UHFFFAOYSA-N CCC(COC)NCC(OCC)=O Chemical compound CCC(COC)NCC(OCC)=O LMJWYFDQEADRJH-UHFFFAOYSA-N 0.000 description 1
- PLOVSNNIAQSKIH-UHFFFAOYSA-N CCOC(CN(CC(C)C#N)C(COC)CI)=O Chemical compound CCOC(CN(CC(C)C#N)C(COC)CI)=O PLOVSNNIAQSKIH-UHFFFAOYSA-N 0.000 description 1
- NLMCPDODBJIHEG-PKPIPKONSA-N C[C@@H](C1)OC(C)CN1c1nc(Cl)c[s]1 Chemical compound C[C@@H](C1)OC(C)CN1c1nc(Cl)c[s]1 NLMCPDODBJIHEG-PKPIPKONSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
Definitions
- the present invention relates to novel substituted purines and heteroaryi fused pyridine, pyrazine, pyrimidine, and related compounds that bind with high selectivity and / or high affinity to CRF (Corticotropin Releasing Factor), preferably CRFl, receptor. It also relates to pharmaceutical compositions comprising such compounds and to their use in the treatment of psychiatric disorders and neurological diseases, including major depression, anxiety-related disorders, post-traumatic stress disorder, supranuclear palsy, substance abuse disorders and feeding disorders, as well as treatment of immunological, cardiovascular or heart-related diseases and digestive disorders including colonic hypersensitivity associated with psychopathological disturbance and stress, including Irritable Bowel Syndrome (IBS). Additionally this invention relates to the use such compounds as probes for the localization of CRF receptors in cells and tissues.
- CRF Corticotropin Releasing Factor
- Corticotropin releasing factor is a peptide that acts by binding to and modulating the signal transduction activities of specific cell surface receptors, including CRFl receptors and CRF2 receptors. CRF receptors are believed to play a significant role in integrating responses to stress.
- CRF has been shown to be involved in psychiatric disorders and neurological diseases including depression, anxiety-related disorders, gastrointestinal disorders, substance abuse disorders and feeding disorders.
- the invention provides novel compounds of Formulas A, B, C, D, E, F, G, H, i, J, K, L, M, N. O, P, Q, R, S, and T (below), and pharmaceutical compositions comprising such compounds and at least one pharmaceutically acceptable carrier or excipient.
- Such compounds bind to cell surface receptors, preferably G-coupled protein receptors, especially CRF receptors (including CRFl and CRF2 receptors) and most preferably CRFl receptors.
- Preferred compounds exhibit drug-like affinity (i.e., IC 50 or K, values ⁇ 1 micromolar in the assay for CRF receptor functional activity of Example 6, below) for CRF receptors, preferably CRFl receptors.
- preferred compounds also exhibit high specificity for CRF receptors (i.e., they exhibit high selectivity compared to their binding to non-CRF receptors). Preferably they exhibit high specificity for CRFl receptors.
- the invention provides compounds and pharmaceutically acceptable salts thereof that are compounds of a Formula seiected from the group consisting of:
- Z at each occurrence is independently S or O;
- R 2i is hydrogen, halogen, amino, (C1-C4)alkyi, C 2 -C 4 aikenyi, C 2 -C 4 alkyny], (C i -C4)alkoxy, hydroxy, halo(C1-C4)alkyl, ha!o(C1-C4)alkoxy, cyano, mono- or di- (C1-C4)alkylamino, (C ⁇ - C4)alkanoy], aminocarbonyl, or -S(O)n-(C1-C4)alkyh
- R 22 is hydrogen, halogen, (C1-C4)alkyl, C 2 -C 4 alkenyl, C 2 -C 4 alkynyl, (C1-C4)alkoxy, hydroxy, ImIo(C1 -C4)alkyl, halo(C!-C4)alkoxy, cyano, mono- or di- (C 1 -C4)alky!amino, (C1- C4)alkanoyl, aminocarbonyl, or -S(0)n-(C1-C4)alkyl;
- R 23 is (C1-C7)alkyl, (C2-C7)alkenyi, (C2-C7)alkynyl, (C6-C12)aryl, 4 to 12 membered heterocycloalkyl, or 5 to 12 membered heteroaryl, each of which is unsubstituted or substituted with one or more substituents independently selected at each occurrence from (C1-C7)alkyi, (C1- C7)alk
- R 24 is hydrogen, halogen, cyano, (C1-C3)alkyl, (C1-C3)alkoxy, hafo(C1-C7)alkyl. mono- or di- (C1-C7)alkylamino, -(CH2)n-R 10 , CONH 2 , or mono- or di-(C1-C4)alkyl-arninocarbonyl-;
- R 25 is hydrogen, halogen, cyano, (C1-C7)alkyl, (C1-C3)alkoxy, or haloalkyl, mono- or di- (C1- C7)alkylamino, -(CH2)n-R 10 , CONH 2 , or mono- or di-(C1-C4)alkyl-aminocarbonyl-;
- R 26 is hydrogen or (C1-C4)aikyl
- R 27 is (C1-C7)alkyl, (C2-C7)alkenyi, (C2-C7)alkynyi, (C6-C12)aryl, mono- or di- (C1- C7)alkylamino, 4 to 12 membered heterocycloalkyl, 5 to 12 membered heteroaryl, -(CH2)n-R 10 , - (CH2)n-CONH 2 , or mono- or di-(C1-C4)alkyl-aminocarbonyl-(CH2)n-, each of which is unsubstituted or substituted with one or more substituents independently selected at each occurrence from (C1-C7)alkyl, (C1-C7)alkoxy, halogen, amino, hydroxyl, 4 to 12 membered heterocycloalkyl, and 5 to 12 membered heteroaryl;
- R 2S is (C1-C7)alkyl, (C6-C12)aryl, 4 to 12 membered heterocycloalkyl, or 5 to 12 membered heteroaryl, each of which is unsubstituted or substituted with one or more substituents independently selected at each occurrence from alkyl, alkoxy, halogen, amino, hydroxy!, 4 to 12 membered heterocycloalkyl, and 5 to 12 membered heteroaryl;
- R 10 is N-linked and at each occurrence is independently acetidine, pyrrolidine, piperidine. hexahydroazepine, or octahydroazepine;
- R 2 is selected from the group consisting of:
- R 6 at each occurrence is independently hydrogen, halogen, cyano, (C1-C4)aikyl, halo(C1- C4)alkyl, (C1-C4)alkoxy, halo(C1-C4)alkoxy, or phenyl;
- R 7 is hydrogen, halogen, cyano, (C1-C4)alkyi, ha!o(CJ -C4)alkyl, (C1-C4)alkoxy, haio(C1- C4)alkoxy, mono- or di- (C1-C4)alkylamino, (C 1 -C4)alkylcarbonyl, -(CH2)n-R 10 , -(CH2)n- CONH 2 , mono- or di-(CI-C4)alkyl-aminocarbonyl-(CH2)n-, -S(O)n-(C1-C4)alkyl, phenyl,
- R 8 is hydrogen, halogen, haloalkyl, haioalkoxy, cyano, (C1-C4)alky], R a R b N-, carbamyl, -(C1- C2)alkoxy(C1-C2)alkyl, R ⁇ -C(O)-, Rl l-(CH2)n-,
- R a is hydrogen, (C1-C5)alkyl, (C3-C5)cycloalkyl, methoxy(C2-C4)alkyl, acetyl, (C1- C2)alkylsulfonyl. (C3)aikenyl, R 15 -(CH2)n-, or (C1-C2)alkyl, each of which is unsubstituted or substituted with cyano, fo ⁇ nyl, vinyl, or ethynyl; R b is hydrogen or (C1-C3)alkyl;
- RI 1 is hydrogen. (C1-C7)alkyi, (C3-C7)cycioalkyl, -O(C1-C4)alkyI, (C2-C7)alkenyl, ethynyl, mono- or di- (C1-C4)alkylamino, aryl, 4 to 12 membered heterocycloalkyl, or 5 to 12 membered heteroaryl, R15-(CH2)n-;
- R 15 is cyclopropyl, phenyl, O
- R 31 represents one or two substituents, at each occurrence independently selected from hydrogen, halogen, (C1-C4)alkyi, (C1-C4)alkoxy, hydroxy, hydroxyI(C1-C4)a!kyl, haloalkyl, haloalkoxy, cyano, amino, mono- or di- (C1-C4)a!kylami ⁇ o, (C1-C4)afkyl-CO-, CONH2, and -S(O)n-(C1- C4)alkyl;
- R 33 is (C1-C7)aikyL (C2-C7)alkenyl. (C2-C7)alkynyl. -(CH2)n-R 10 , -(CH2)n-CONH 2 , mono- or di-(C1-C4)alkyl-aminocarbonyl-(CH2)n-, aryl, 4 to 12 membered heterocycloalkyl, or 5 to 12 membered heteroaryl, each of which is unsubstituted or substituted one or more substituents independently selected at each occurrence from (C1-C7)alkyi, (C 1 -C7)alkoxy, halogen, amino, hydroxy!, 4 to 12 membered heterocycloalkyl, and 5 to 12 membered heteroaryl;
- R 35 at each occurrence is independently hydrogen, halogen, cyano, (C1-C3)alkyl, (C1-C7)alkyl, (C1-C3)a!koxy, halo(C1-C7)a!kyl, -(CH2)n-R 10 , -(CH2)n-CO mono- or di- (C1-C7)alkylamino, mono- or di- (C1-C7)alkylamino, or -CO mono- or di- (C1-C7)alkylamino;
- R 36 is hydrogen or (C1-C4)alkyl
- R 37 is hydrogen, halogen, (C1-C4)alkyl, or halo(C1-C4)a!kyi; n is 0, I, or 2; and
- X is CH2. CO, O, S, or SO2; or
- R " is selected from the group consisting of:
- R 1 is hydrogen, halogen, cyano, (C1-C3)alkyl, or methoxy;
- R 3 is hydrogen or (C1-C3)alkyl
- R 4 is hydrogen, halogen, hydroxyl, formyl, (C1-C3)alkoxy, (C2-C3)alkenyl,
- R c R d N-, or R 16 -C(O)-; or R 4 is (C3-C5)cycloalkyl or (C1-C7)alkyl, each substituted with zero, one, or two that are the same or different of hydroxy, (C1 -C2)alkoxy, or cyclopropyl;
- R 5 is hydrogen, methyl, or trifluoromethyl:
- R 16 is hydrogen, (C1-C3)alkyl, cyclopropyl. methoxy, or R c R d N-;
- R c and R d are independently hydrogen. (C1-C4)alkyl, or methoxy.
- the invention further provides methods of treating patients suffering from certain disorders with a therapeutically effective amount of at least one compound provided herein.
- disorders include CMS disorders, particularly affective disorders, anxiety disorders, stress-related disorders, eating disorders and substance abuse.
- the patient suffering from these disorders may be a human or other animal (preferably a mammal), such as a domesticated companion animal (pet) or a livestock animal.
- Preferred compounds for such therapeutic purposes are those that antagonize the binding of CRF to CRF receptors (preferably CRFl , or less preferably CRF2 receptors).
- CRF receptors preferably CRFl , or less preferably CRF2 receptors.
- the ability of compounds to act as antagonists can be measured as an IC 50 value as described below.
- the present invention provides pharmaceutical compositions comprising at least one compound or salt of Formulas A, B, C, D, E, F, G, H, i, J, K, L, M, N, O, P, Q, R, S, or T (preferably at least one compound or salt of Table I or Table 11 ) or the pharmaceutically acceptable salts (as defined below) thereof, which compositions are useful for the treatment of the above- recited disorders.
- the invention further provides methods of treating patients suffering from any of the above-recited disorders with an effective amount of a compound or composition provided herein.
- this invention relates to the use of the compounds provided (particularly labeled compounds) as probes for the localization of receptors in cells and tissues and as standards and reagents for use in determining the receptor-binding characteristics of test compounds.
- Preferred compounds exhibit a half-maximal inhibitory concentration (IC 50 ) of less than 5 micromoiar in the standard in vitro CRFl receptor binding assay provided in Example 7 below.
- Particularly preferred compounds exhibit an IC 50 of about 1 micromoiar or less, still more preferably an IC 50 of about 100 nanomolar or less even more preferably an IC 5 0 of about 10 nanomolar or less.
- Certain particularly preferred compounds exhibit an IC 50 of 1 nanomolar or less in such standard binding assay.
- HFLC instrumentation Analyses are performed using a Waters 600 series pump (Waters Corp.), a Waters 996 Diode Array detector and a Gilson 215 auto-sampler (Gilson Inc.). Data are acquired using MassLynx 4.0 software, with OpenLynx processing.
- MS instrumentation LC-MS experiments are performed using a Waters ZMD II Mass Spectrometer. MS conditions: EIectrospray positive ionization; capillary voltage 3.5kV; cone voltage 30V; desoivation and source temperature 250 °C and 100 °C respectively; mass range 120-800 with a scan time of 0.5 seconds and an inter scan delay of 0.1 min. Method 2.
- a Perkin Elmer HPLC system (tow Series 200 micro LC pumps, pump A and pump B, with a Series 200 autosampler) is used to perform flow injection.
- Mobile phase is a combination of 85% methanol (pump B) with 15% of water (pump A).
- the flow rate is 1.0 mL/min; and the injection volume is 3 ⁇ l.
- MS instrumentation LC-MS experiments are performed using a Sciex 150MA Mass Spectrometer.
- the Nebulizer gas is 10, and the Curtain gas is 12.
- the declustering potential is 30 V.
- the Focusing potential is 200 V, and the entrance potential is -10 V.
- Nebulizer current is -2.0 mA, and the temperature is 350 °C.
- the Nebulizer gas is 10, and the Curtain gas is 12.
- the declustering potential is -30 V.
- the Focusing potential is -200 V, and the entrance potential is 10 V.
- HPLC Instrumentation HPl 100 PUMP, HPl 100 UV detector with 220 nm, HTS/PAL autosampler from Leap Technology, data acquired by Micromass Ma
- HPLC conditions Sy ⁇ ergi 2U HYDRO-RP 20 x 4.0mm column, flow rate 1.0 mL/min, injection volume 5 ⁇ L.
- Compounds described herein and salts thereof are useful in treating a variety of conditions including affective disorders, anxiety disorders, stress disorders, eating disorders, inflammatory or chronic pain disorders, and substance abuse disorders. Hence they each may be used in the manufacture of a medicament for the treatment of each such condition.
- Affective disorders include all types of depression, bipolar disorder, cyclothymia, and dysthymia.
- Anxiety disorders include generalized anxiety disorder, panic, phobias and obsessive-compulsive disorder.
- Compounds described herein and salts thereof are useful in treating symptoms of affective disorders and anxiety disorders. These syptoms include increased awakenings, increased REM density, and related sleep disorders (e.g., insomnia) and de ⁇ natologic and related immunogenic inflammatory conditions including atopic dermatitis, urticaria, rheumatoid arthritis, and psoriasis.
- Stress-related disorders include post-traumatic stress disorder, hemorrhagic stress, stress-induced psychotic episodes, psychosocial dwarfism, stress headaches, stress-induced immune systems disorders such as stress-induced fever, and stress-related sleep disorders.
- Substance abuse disorders include alcohol abuse and drug addictions.
- Compounds described herein and salts thereof are particularly useful for the therapeutic reduction of the anxiogenic effects of drug or alcohol withdrawal in drug or alcohol habituated patients. They are also useful for the prevention of relapse of abusive alcohol or drug consumption following withdrawal.
- Eating disorders include anorexia nervosa, bulimia nervosa, and obesity.
- Compounds described herein and salts thereof are also useful in the treatment (e.g., symptomatic treatment) of a variety of neurological disorders including supranuclear palsy, AIDS related dementias, multlinfarct dementia, neurodegenerative disorders such as Alzheimer's disease, Parkinson's disease, and Huntington's disease, head trauma, spinal cord trauma, ischemic neuronal damage, epilepsy, amyotrophic lateral sclerosis, and disorders of pain perception such as fibromyalgia.
- neurological disorders including supranuclear palsy, AIDS related dementias, multlinfarct dementia, neurodegenerative disorders such as Alzheimer's disease, Parkinson's disease, and Huntington's disease, head trauma, spinal cord trauma, ischemic neuronal damage, epilepsy, amyotrophic lateral sclerosis, and disorders of pain perception such as fibromyalgia.
- compounds described herein and salts thereof are useful in the treatment (e.g.. symptomatic treatment) of a number of gastrointestinal, cardiovascular, hormonal, autoimmune and inflammatory conditions.
- Such conditions include irritable bowel syndrome (IBS), ulcers, Crohn's disease, spastic colon, diarrhea, post operative ilius. and colonic hypersensitivity associated with psychopathoiogical disturbances or stress; hypertension, tachycardia, congestive heart failure, infertility, and euthyroid sick syndrome; and inflammatory conditions effected by or associated with rheumatoid arthritis, osteoarthritis, pain, asthma, psoriasis and allergies.
- IBS irritable bowel syndrome
- ulcers Crohn's disease
- spastic colon diarrhea
- colonic hypersensitivity associated with psychopathoiogical disturbances or stress hypertension
- tachycardia congestive heart failure
- infertility infertility
- Compounds described herein and salts thereof are also useful as modulators of the CRFl receptor in the treatment of animal disorders associated with aberrant CRF levels.
- Such conditions include porcine stress syndrome, bovine shipping fever, equine paroxysmal fibrillation, and dysfunctions induced by confinement in chickens, sheering stress in sheep or human-animal interaction related stress in dogs, psychosocial dwarfism and hypoglycemia.
- Typical subjects to which compounds described herein and salts thereof may be administered include mammals, preferably primates, most preferably humans. For veterinary applications, a wide variety of subjects are suitable, e.g.
- livestock such as cattle, sheep, goats, cows, swine and the like; poultry such as chickens, ducks, geese, turkeys, and the like; and other domesticated animals particularly pets such as dogs and cats.
- rodents e.g. mice, rats, hamsters
- rabbits primates, and swine
- inbred pigs and the like e.g., cow, sheep, goats, cows, swine and the like
- body fluids e.g., blood, plasma, serum, CSF, lymph, cellular interstitial fluid, aqueous humor, saliva, synovial fluid, feces, or urine
- cell and tissue samples e.g., cell and tissue samples of the above subjects are suitable for use.
- test compounds e.g., a potential pharmaceutical
- Labeled compounds provided herein and salts thereof are also useful as tracers for positron emission tomography (PET) imaging or for single photon emission computerized tomography (SPECT).
- PET positron emission tomography
- SPECT single photon emission computerized tomography
- More particularly such compounds and salts are useful for demonstrating the presence of CRF receptors in cell or tissue samples. This may be done by preparing a plurality of matched cell or tissue samples, at least one of which is prepared as an experiment sample and at least one of which is prepared as a control sample.
- the experimental sample is prepared by contacting (under conditions that permit binding of CRF to CRF receptors within cell and tissue samples) at least one of the matched cell or tissue samples that has not previously been contacted with any compound or salt provided herein with an experimental solution comprising the detectably-labeled preparation of the selected compound or salt at a first measured molar concentration.
- control sample is prepared by in the same manner as the experimental sample and is incubated in a solution that contains the same ingredients as the experimental solution but that also contains an unlabelled preparation of the same compound or salt provided herein at a molar concentration that is greater than the first measured molar concentration.
- the experimental and control samples are then washed to remove unbound detectably-labeled compound.
- the amount of detectably-iabeled compound remaining bound to each sample is then measured and the amount of detectably-labeled compound in the experimental and control samples is compared.
- a comparison that indicates the detection of a greater amount of detectable label in the at least one washed experimental sample than is detected in any of the at least one washed control samples demonstrates the presence of CRF receptors in that experimental sample.
- the detectably-labeled compound used in this procedure may be labeled with any detectable label, such as a radioactive label, a biological tag such as biotin (which can be detected by binding to detectably-labeled avidin), an enzyme (e.g., alkaline phosphatase, beta galactosidase, or a like enzyme that can be detected its activity in a colorimetric assay) or a directly or indirectly luminescent label.
- a detectable label such as a radioactive label, a biological tag such as biotin (which can be detected by binding to detectably-labeled avidin), an enzyme (e.g., alkaline phosphatase, beta galactosidase, or a like enzyme that can be detected its activity in a colorimetric assay) or a directly or indirectly luminescent label.
- the amount of detectable label in an experimental or control sample may be measured by viewing the autoradiogram s and comparing the exposure density of the autoradiograms.
- the present invention also pertains to methods of inhibiting the binding of CRF to CRF receptors
- CFRl receptors which methods involve contacting a solution containing a compound provided herein with cells expressing CRF receptors, wherein the compound is present in the solution at a concentration sufficient to inhibit CRF binding to CRF receptors in vitro.
- This method includes inhibiting the binding of CRF to CRF receptors in vivo, e.g., in a patient given an amount of a compound or salt disclosed herein that would be sufficient to inhibit the binding of CRF to CRF receptors in vitro.
- such methods are useful in treating physiological disorders associated with excess concentrations of CRF.
- the amount of a compound that would be sufficient to inhibit the binding of CRF to the CRF receptor in vitro may be readily determined via a CRF receptor binding assay (see, e.g., Example 7), or from the IC 50 determined using a CRF receptor functional assay, such as the standard assay of CRF receptor activity of Example 6.
- the CRF receptors used to determine in vitro binding may be obtained from a variety of sources, e.g., from cells that naturally express CRF receptors, e.g. IMR32 cells, or from cells expressing cloned human CRF receptors.
- the present invention also pertains to methods for altering the activity of CRF receptors, said method comprising exposing cells expressing such receptors to an effective amount of a compound provided herein, wherein the compound is present in the solution at a concentration sufficient to specifically alter the signal transduction activity in response to CRF in cells expressing CRF receptors in vitro.
- Preferred cells for this purpose are those that express high levels of CRF receptors (i.e., equal to or greater than the number of CRFl receptors per cell found in differentiated ⁇ MR-32 human neuroblastoma ceJJs), with IMR-32 ceils being particularly preferred for testing the concentration of a compound required to alter the activity of CRFl receptors.
- This method includes altering the signal transduction activity of CRF receptors in vivo, e.g., in a patient given an amount of a compound or salt disclosed herein that would be sufficient to alter the signal transduction activity in response to CRF in cells expressing CRF receptors in vitro.
- the amount of a compound that would be sufficient to alter the signal transduction activity of CRF receptors in response to CRF in vitro may also be determined via an assay of CRF receptor mediated signal transduction, such as an assay wherein the binding of CRF to a cell surface
- CRF receptor effects a changes in reporter gene expression.
- the present invention also pertains to packaged pharmaceutical compositions for treating disorders responsive to CRF receptor modulation, e.g.. eating disorders, depression or stress.
- the packaged pharmaceutical compositions include a container holding dosage units comprising a therapeutically effective amount of at least one compound or salt disclosed herein and instructions for using the treating disorder responsive to CRFl receptor modulation in the patient.
- a “pharmaceutically acceptable salt” of a compound is an acid or base salt that is suitable for use in contact with the tissues of human beings or animals.
- Such salts include mineral and organic acid salts of basic residues such as amines, as well as alkali or organic salts of acidic residues such as carboxylic acids.
- Specific pharmaceutically acceptable salts include, but are not limited to, salts of acids such as hydrochloric, phosphoric, hydrobromic, malic, glycolic, fumaric, sulfuric, sulfamic, sulfanilic, formic, toluenesulfonic, methanesulfon ⁇ c.
- pharmaceutically acceptable salts of bases include, but are not limited to sodium, potassium, calcium, aluminum, lithium and ammonium salts.
- Such salts can be prepared by any conventional chemical method, such as reacting the free acid or base forms of these compounds with a stoichiometric amount of the appropriate base or acid in water or in an organic solvent, or in a mixture of the two; generally, the use of nonaqueous media, such as ether, ethyl acetate, ethanoi, isopropanol or acetonitrile, is preferred.
- nonaqueous media such as ether, ethyl acetate, ethanoi, isopropanol or acetonitrile
- prodrug' 1 is a compound that may not fully satisfy the structural requirements of the compounds provided herein, but is modified in vivo, following administration to a patient, to produce a compound a formula provided herein.
- a prodrug may be an acylated derivative of a compound as provided herein.
- Prodrugs include compounds wherein hydroxy, amine or sulfhydryl groups are bonded to any group that, when administered to a mammalian subject, cleaves to form a free hydroxy, amino, or sulfhydryl group, respectively.
- Examples of prodrugs include, but are not limited to, acetate, formate, benzoate and peptide derivatives of alcohol and amine functional groups within the compounds provided herein.
- Aikoxy is meant an alky! group as described above attached via an oxygen bridge.
- Aikoxy groups include Ci-C n alkoxy groups, for example methoxy, ethoxy, propoxy, isopropoxy, n-butoxy, sec-butoxy, tert-butoxy, n-pentoxy, 2-pentoxy, 3-pentoxy, isopentoxy, neopentoxy, hexoxy, 2- hexoxy, 3 -hexoxy, or 3-methyIpentoxy groups.
- Alkylsulfonyl indicates groups of the formula -(SO 2 )-alkyl, in which the sulfur atom is the point of attachment.
- Alkylsulfonyl groups include C r C n alky!sulfonyl groups, which have from I to n carbon atoms, such as methyl sulfonyl or pentylsuifonyl.
- C]-C n haloalkylsulfonyl is an aikylsulfonyl group that has from 1 to n carbon atoms and is substituted with at least one halogen (e.g., trifluoroinethylsulfonyl).
- a “cycioalkyl” is a group that comprises one or more saturated and/or partially saturated rings in which all ring members are carbon, such as cyclopropyl, cyclobutyl, cyclopentyl, and partially saturated variants of the foregoing, such as cyclopentenyl. Cycloalkyi groups do not comprise an aromatic ring or a heterocyclic ring. Certain cycioalkyl groups are C 3 -C 5 cyc ⁇ oalkyl, in which the cycioalkyl group contains a single ring having from 3 to 5 ring members, all of which are carbon.
- Aikoxy is meant an alkyl group as described above attached via an oxygen bridge.
- Aikoxy groups include C]-C 6 alkoxy and CpC 4 alkoxy groups, which have from 1 to 6 or from I to 4 carbon atoms, respectively.
- Methoxy, ethoxy, propoxy, isopropoxy, n-butoxy, sec-butoxy, tert ⁇ butoxy, n-pentoxy, 2-pentoxy, 3-pentoxy, isopentoxy, neopentoxy, hexoxy, 2-hexoxy, 3-hexoxy, and 3- methylpentoxy are representative aikoxy groups.
- Alkylsulfonyl indicates groups of the formula -(SC ⁇ -alkyl, in which the sulfur atom is the point of attachment.
- Alkylsulfonyl groups include C r C 6 alkylsulfonyI and C r C 4 alkyisulfonyl groups, which have from I to 6 or from 1 to 4 carbon atoms, respectively.
- Methylsuifonyl is one representative alkylsulfonyl group.
- halogen indicates fluorine, chlorine, bromine or iodine.
- a "substituent,” as used herein, indicates a molecular moiety that is covalently bonded to an atom within a molecule of interest.
- a ring substituent may be a moiety such as a halogen, alkyl group, haloaiky! group or other group that is covalently bonded to an atom (preferably a carbon or nitrogen atom) that is a ring member.
- Substituents of aromatic groups are generally covalently bonded to a ring carbon atom.
- substitution refers to replacing a hydrogen atom in a molecular structure with a substituent, such that the valence on the designated atom is not exceeded, and such that a chemically stable compound (i.e., a compound that can be isolated, characterized, and tested for biological activity) results from the substitution.
- a dash (“-" or "-") in a formula that is not between two letters or symbols is used to indicate a point of attachment for a substituent. For example, -CONH 2 is attached through the carbon atom.
- Table II that is effective, when administered to a human or non-human patient, to provide a therapeutic benefit such as an amelioration of symptoms, e.g., an amount effective to antagonize the effects of pathogenic levels of CRF or to treat the symptoms of any of the disorders listed above under the subheading "Methods of Treament * '. It will be apparent that the therapeutic benefit may be apparent after administration of a single dose, or may become apparent following repeated administration of the therapeutically effective dose according to a predetermined regimen, depending upon the indication for which the compound is administered.
- compositions described herein, and salts thereof may be administered orally, topically, transdermally, parenterally, by inhalation or spray or rectally or vaginally in dosage unit formulations containing conventional non-toxic pharmaceutically acceptable carriers, adjuvants and vehicles.
- parenteral as used herein includes subcutaneous, intravenous, intramuscular, intrathecal and like types of injection or infusion techniques.
- a pharmaceutical formulation comprising a compound provided herein and a pharmaceutically acceptable carrier.
- One or more compounds disclosed herein may be present in association with one or more non-toxic pharmaceutically acceptable carriers and/or diluents and/or adjuvants and if desired other active ingredients.
- compositions containing compounds disclosed herein may be in a form suitable for oral use. for example, as tablets, troches, lozenges, aqueous or oily suspensions, dispersible powders or granules, emulsion, hard or soft capsules, or syrups or elixirs.
- Compositions intended for oral use may be prepared according to any method known to the art for the manufacture of pharmaceutical compositions and such compositions may contain one or more agents selected from the group consisting of sweetening agents, flavoring agents, coloring agents and preserving agents in order to provide pharmaceutically elegant and palatable preparations. Tablets contain the active ingredient in admixture with non-toxic pharmaceutically acceptable excipients that are suitable for the manufacture of tablets.
- excipients may be for example, inert diluents, such as calcium carbonate, sodium carbonate, lactose, calcium phosphate or sodium phosphate; granulating and disintegrating agents, for example, corn starch, or alginic acid; binding agents, e.g., starch, gelatin or acacia, and lubricating agents, e.g., magnesium slearate, stearic acid or talc.
- the tablets may be uncoated or they may be coated by known techniques to delay disintegration and absorption in the gastrointestinal tract and thereby provide a sustained action over a longer period.
- a time delay material such as glyceryl monosterate or glyceryl distearate may be employed.
- Formulations for oral use may also be presented as hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent, for example, calcium carbonate, calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredient is mixed with water or an oil medium, e.g., peanut oil, liquid paraffin or olive oil.
- an inert solid diluent for example, calcium carbonate, calcium phosphate or kaolin
- water or an oil medium e.g., peanut oil, liquid paraffin or olive oil.
- Aqueous suspensions contain the active materials in admixture with excipients suitable for the manufacture of aqueous suspensions.
- excipients are suspending agents, e.g., sodium carboxymethylcellulose, methylcellulose, hydropropylmethylcellulose, sodium alginate, polyvinylpyrrolidone, gum tragacarsth and gum acacia; dispersing or wetting agents may be a naturally- occurring phosphatide, for example, lecithin, or condensation products of an alkyiene oxide with fatty acids, e.g., polyoxyethylene stearate, or condensation products of ethylene oxide with long chain aliphatic alcohols, e.g., heptadecaethyleneoxycetanol, or condensation products of ethylene oxide with partial esters derived from fatty acids and a hexitol such as polyoxyethylene sorbitol monooleate, or condensation products of ethylene oxide with partial esters derived from fatty acids and hexitol anhydrides,
- the aqueous suspensions may also contain one or more preservatives, e.g., ethyl, or n- ⁇ ro ⁇ yi p-hydroxybenzoate, one or more coloring agents, one or more flavoring agents, and one or more sweetening agents, such as sucrose or saccharin.
- preservatives e.g., ethyl, or n- ⁇ ro ⁇ yi p-hydroxybenzoate
- coloring agents e.g., ethyl, or n- ⁇ ro ⁇ yi p-hydroxybenzoate
- flavoring agents e.g., n- ⁇ ro ⁇ yi p-hydroxybenzoate
- sweetening agents such as sucrose or saccharin.
- Oily suspensions may be formulated by suspending the active ingredients in a vegetable oil, e.g., arachis oil, olive oil, sesame oil or coconut oil, or in a mineral oil such as liquid paraffin.
- the oily suspensions may contain a thickening agent, e.g., beeswax, hard paraffin or cetyl alcohol.
- Sweetening agents such as those set forth above, and flavoring agents may be added to provide palatable oral preparations. These compositions may be preserved by the addition of an anti-oxidant such as ascorbic acid.
- Dispersible powders and granules suitable for preparation of an aqueous suspension by the addition of water provide the active ingredient in admixture with a dispersing or wetting agent, suspending agent and one or more preservatives.
- a dispersing or wetting agent e.g., sodium EDTA
- suspending agent e.g., sodium EDTA
- preservatives e.g., sodium EDTA, sodium sulfate, sodium bicarbonate
- compositions provided herein may also be in the form of oil-in-water emulsions.
- the oily phase may be a vegetable oil, e.g., olive oil or arachis oil or a mineral oil e.g., liquid paraffin or mixtures of these.
- Suitable emulsifying agents may be naturally-occurring gums, e.g., gum acacia or gum tragacanth, naturally-occurring phosphatides, e.g..
- the emulsions may also contain sweetening and flavoring agents. Syrups and elixirs may be formulated with sweetening agents, e.g., glycerol, propylene glycol, sorbitol or sucrose. Such formulations may also contain a demulcent, a preservative and flavoring and coloring agents.
- the pharmaceutical compositions may be in the form of a sterile injectable aqueous or oleaginous suspension.
- This suspension may be formulated according to the known art using those suitable dispersing or wetting agents and suspending agents that have been mentioned above.
- the sterile injectable preparation may also be sterile injectable solution or suspension in a non-toxic parentally acceptable dilutent or solvent, e.g., as a solution in 1,3-butanediol.
- the acceptable vehicles and solvents that may be employed are water, Ringer's solution and isotonic sodium chloride solution.
- sterile, fixed oils are conventionally employed as a solvent or suspending medium.
- any bland fixed oil may be employed including synthetic mono- or diglycerides.
- fatty acids such as oleic acid find use in the preparation of injectables.
- compositions disclosed herein may also be administered in the form of suppositories, e.g., for rectal administration of the drug.
- suppositories e.g., for rectal administration of the drug.
- suitable non-irritating excipient that is solid at ordinary temperatures but liquid at body temperature and will therefore melt in the body to release the drug.
- Such materials include cocoa butter and polyethylene glycols.
- Compounds disclosed herein may be administered parenterally in a sterile medium.
- the drug depending on the vehicle and concentration used, can either be suspended or dissolved in the vehicle.
- one or more adjuvants such as preservatives, buffering agents, or local anesthetics can also be present in the vehicle.
- Dosage levels of the order of from about 0.05 mg to about 100 mg per kilogram of body weight per day are useful in the treatment of the above-indicated conditions, preferred dosages range from about 0.1 to about 30 mg per kg and more preferably from about 0.5 to about 5 mg per kg per subject per day.
- the amount of active ingredient that may be combined with the carrier materials to produce a single dosage form will vary depending upon the host treated and the particular mode of administration. Dosage unit forms will generally contain between from about 0.1 mg to about 750 mg of an active ingredient.
- Frequency of dosage may also vary depending on the compound used and the particular disease treated. However, for treatment of most CNS and gastrointestinal disorders, a dosage regimen of four times daily, preferably three times daily, more preferably two times daily and most preferably once daily is contemplated. For the u'eatment of stress and depression a dosage regimen of 1 or 2 times daily is particularly preferred. It will be understood, however, that the specific dose level for any particular patient will depend upon a variety of factors including the activity of the specific compound employed, the age, body weight, general health, sex, diet, time of administration, route of administration, and rate of excretion, drug combination (i.e. other drugs being used to treat the patient) and the severity of the particular disease undergoing therapy.
- Preferred compounds disclosed herein will have certain pharmacological properties. Such properties include, but are not limited to oral bioavailability, such that the preferred oral dosage forms discussed above can provide therapeutically effective levels of the compound in vivo. Penetration of the blood brain barrier is necessary for most compounds used to treat CNS disorders, while low brain levels of compounds used to treat periphereal disorders are generally preferred.
- Assays may be used to predict these desirable pharmacological properties. Assays used to predict bioavailability include transport across human intestinal cell monolayers, including Caco-2 ceil monolayers. Toxicity to cultured hepatocyctes may be used to predict compound toxicity, with non-toxic compounds being preferred. Penetration of the blood brain barrier of a compound in humans may be predicted from the brain levels of the compound in laboratosy animals given the compound, e.g., intravenously.
- Percentage of serum protein binding may be predicted from albumin binding assays. Examples of such assays are described in a review by Oravcova, et al. (Journal of Chromatography B (1996) volume
- Preferred compounds exhibit reversible serum protein binding. Preferably this binding is less than 99%, more preferably less than 95%, even more preferably less than 90%, and most preferably less than 80%.
- Frequency of administration is generally inversely proportional to the in vivo half-life of a compound.
- In vivo half-lives of compounds may be predicted from in vitro assays of microsomal half-life as described by Kuhnz and Gieschen (Drug Metabolism and Disposition, (1998) volume 26, pages 1120- 1 127). Preferred half lives are those allowing for a preferred frequency of administration.
- preferred compounds disclosed herein exhibit activity in the standard in vitro CRF receptor binding assay specified in Example 7 below.
- References herein to '"standard in vitro receptor binding assay” are intended to refer to this standard assay protocol.
- preferred compounds disclosed herein have an IC 50 (half-maximal inhibitory concentration) of about 1 micromolar or less, still more preferably and IC 50 of about 100 nanomolar or less even more preferably an IC 50 of about 10 nanomolar or less or even 1 nanomolar or less in such a defined standard in vitro CRF receptor binding assay.
- a suitable transition metal catalyst such as, but not limited to, palladium(I ⁇ ) acetate or tris(dibenzylideneacetone)dipaIladium(0)
- a ligand such as, but not limited to, l,l'-bis(diphenylphosphine)ferrocene, 2,2'-bis(diphenylphosphine)-l : l'-binaphthyl, dicyclohexyl(2-bi ⁇ henyl)phosphine, tricyclohexylphosphine, or tri-tert-butylphosphine
- a base such as sodium or potassium tert-butoxide in inert solvents such as, but not limited to, toluene, ethyleneglycol dimethyl ether, diglyme, DMF, or N-methylpyrrolidinone at temperatures ranging from ambient to 100 °C
- Resulting monochloropyrazi ⁇ c (b) can be further converted to compound (c) by displacing the halogen atom with a variety of nucleophiles (R 2 -[M]), in the presence or absence of a transition metal catalyst.
- the aforementioned nucleophiles may include sodium or potassium (thio)alkoxide.
- alkylamine, and organometallic reagent such as, but not limited to, alkyl Grignard reagents, alkylboronic acids, esters of alkylboronic acids, or alkylstannanes, or alkylaiuminum reagents.
- the aforementioned transition metal catalyst may be, but is not limited to, Ni(DPPP)C12, or palladium(Il) acetate or tris(dibenzylideneacetone)dipalladium(0), a ligand such as, but not limited to, 1,1'- bis(diphenylphosphine)ferrocene, 2,2'-bis(diphenylphosphine)-1,1 '-binaphthyl, dicyclohexyl(2- biphenyl)phosphine, tricyclohexylphosphine, or tri-tert-butyiphosphine, and a base such as sodium or potassium ter/-butoxide in inert solvents such as, but not limited to, toluene, ethyleneglycol dimethyl ether, diglyme, DMF, or N-methylpyrrolidinone at temperatures ranging from ambient to 100 °C.
- a ligand such as, but not limited to, 1,1'
- Halogenation of compound (c) may be accomplished by a variety of methods known in the art, including treatment with N-chlorosuccinimide, bromine, N-bromosuccinimide, pyridinium tribromide, triphenylphosphine dibromide, iodine, and N-iodosuccinimide in solvents such as. but not limited to, dichloromethane, acetic acid, or methyl sulfoxide.
- the secondary amine (d) may react with ally! halides or substituted allyl halides in a variety of solvents such as, but not limited to DMF or MeCN in the presence of base such as, but not limited, NaH or NaOH at room temperature or higher temperature.
- the al ⁇ ylated product (e) can undergo a transition metal catalyzed cyciization reaction (Heck reaction review: Chem. Rev. 2000, 100, 3009) in solvents such as, but not limited to DMF or MeCN at temperatures at room temperature or above in the presence of bases such as, but not limited to NaOH, potassium carbonate, triethylamine.
- Catalysts include, but are not limited to Pd(OAc)2 or Pd(PPh3)4.
- bromo derivative (Ia) may be converted to boronic acid derivative (2a) via a metallation reaction with alkyllitium reagents or metals such as lithium or magnesium in ethereal solvents such as, but not limited to THF or diethylether at temperatures below zero degrees celcius, followed by addition of trialkyl borate and hydrolysis to the boronic acid.
- the borate ester (3a) may be prepared from diborane reagents in the presence of palladium catalysts in solvents such as, but not limited to dioxane.
- dibromopyridine can undergo monosubstitution with nucleophilic nitrogen compounds such as amines to give the monobromopyridine.
- this reaction can be achieved in solvents such as toluene with catalytic palladium acetate and DPPF in the presence of potassium tert- butoxide at temperatures near or at the boiling point of the solvent.
- the subsequent reaction steps follow a procedure similar to the descriptions given in scheme 1.
- the bromine in the bromochloro bicycle shown in Scheme 3 can be selectively functionalized by metalation with a reagent such as alkyllithium and reacted with an electrophile.
- a reagent such as alkyllithium
- the bromine in the bromochloro bicycle shown in Scheme 4 can be selectively functionalized by metalation with a reagent such as alkyllithium and reacted with an electrophile.
- a reagent such as alkyllithium
- compound 6 can be prepared from 5 and ammonia in the presence of a suitable transition metal catalyst such as but not limited to copper (0), palladium(II) acetate or tris(dibenzylideneacetone)dipalladium(O), a ligand such as but not limited to 1,1'- bis(diphenylphosphine)ferrocene, 2,2 r -bis(diphenylphosphine)-l , 1 '-binaphthyl, dicyclohexyl(2- biphenyl)phosphine, tricyclohexylphosphine, or tri-tert-butylphosphine, in inert solvents such as, but not limited to, ethanol, methanol, toluene, ethyleneglycol dimethyl ether, diglyme, DMF, or
- Cyclization of the diaminopyrazine 6 to compound Ie can be accomplished by treatment with a number of reagents such as dimethoxymethylacetate or trialkylorthoesters such as triethylorthoformate or trimethylortoacetate in the presence or not of a suitable acidic catalyst such as, but not limited to, p-toluenesulfonic acid or sulfuric acid, in a solvent such as toluene, xylene, DMF, NMP 3 or methyl sulfoxide at temperatures ranging from O °C to 100 °C.
- a suitable acidic catalyst such as, but not limited to, p-toluenesulfonic acid or sulfuric acid
- a solvent such as toluene, xylene, DMF, NMP 3 or methyl sulfoxide at temperatures ranging from O °C to 100 °C.
- Preparation of the boronic acid or boronale 2e would follow the general
- 2,6-dichioro-3-nitropyrimidine can undergo monosubstitution with allylamines in solvents such as, but not limited to DMF in the presence of base such as potassium carbonate.
- the nitro group can be reduced selectively with reagents such as stannous(ll)ch!oride in acidic media.
- the amine can be converted to a halogen such as bromine by diazotization with a reagent such as sodium nitrite in an acid such as HBr.
- the general procedure for transition metal catalyzed cyclization followed by conversion of the chloride to the boronic acid or borate 2i is described in Scheme I .
- R2-U is selected from the group consisting of:
- R2-Hu is selected from:
- Part B 4-(4-Chloro-l,3-thiazol-2-yl)morpholine
- a solution of fresh sodium ethoxide in ethanol is made by dissolving sodium (339 rng, 14,75 mmol) in 100 mL of absolute ethanol.
- Ethyl N-[(lZ)-2-cyanoprop-l-en-l-yl]-N-[l-(methoxymethyl) ⁇ ropyl]- glycinate (7.5 g, 29.5 mmol) is then dissolved in this solution, and the resulting yellow reaction mixture is stirred at RT for 19 h.
- the basic solution is neutralized with an excess of Amberlite IRC-50 (H+) resin and evaporated to dryness. The residue is taken up in 100 mL Of CH 2 C1 2 and stirred for 5-10 min.
- Trimethylal ⁇ minum (5.9 mL of a 2.0M solution in hexancs, 1 1.79 mmol) is added dropwise to a stirred solution of methylamine (9.8 mL of a 2.0M solution in THF, 19.65 mmol) in 15 mL of DCE at -5 °C (ice/salt bath).
- the clear solution is stirred in a sealed tube for 1 h.
- Ethyl 3-amino-l-[l-(methoxy- methyl)propyl]-4-methyl-lH-p ⁇ rrole-2-carboxylate (3.0 g, 3.93 mmol) is then added, the cold bath is removed, and the reaction is heated to 90 °C for 16 h in the sealed tube.
- Part G 4-Methyl-2-morphoIin-4-yl-1.3-thiazole-5-carboxyIic acid
- Phosphorous oxychloride (1.0 mL) is added to N- ⁇ l-[l -(methoxymethyl)propyl]-4-methyl-2- (methyicarbamoyi> ⁇ H- ⁇ yrrol-3-yl ⁇ -4-methyl-2-mo ⁇ holiri-4-yl-l,3-thiazole-5-carboxamide (100 mg of crude material, ⁇ 0.21 mmol). and the reaction mixture is heated to 1 10 °C for 1.5 h. After cooling to RT, the excess POCI 3 is removed in vacuo, and ice water (10 mL) is added to the residue. The mixture is then extracted with CH 2 C1 2 (IO mL), and the organic extract is washed with sat.
- chemiluminescent ELISA system cAMP-Screen ® , Applied Biosystems, Bedford, MA
- cAMP-Screen ® Applied Biosystems, Bedford, MA
- IMR32 cells ⁇ TCC CCL 127) are grown to confluence in one or more T- 175 flasks, each flask is split evenly into the wells of two pre-coated 96-weil cAMP-Screen ® assay piates prior to the test treatment and ELlSA, each plate is then incubated for 16 hours and assayed.
- the following assay is referred to herein and in the claims as a standard in vitro CRF receptor binding assay.
- the CRF receptor binding is performed using a modified version of the assay described by Grigoriadis and De Souza ⁇ Methods in Neurosciences, Vol. 5, J 991).
- IMR-32 human neuroblastoma cells a cell-line that naturally expresses the CRFI receptor, are grown in IMR-32 Medium (see preceeding Example). The cells are grown to confluence and split three times (all splits and harvest are carried out using NO-ZYME — JRH Biosciences. Cat# 59226).
- the cells are first split 1 :2, incubated for 3 days and split 1 :3, and finally incubated for 4 days and split 1 :5.
- the cells are then incubated for an additional 4 days before being differentiated by treatment with 5-bromo-2'deoxyuridine (BrdU, Sigma, Cat# B9285).
- the medium is replaced every 3-4 days with IMR-32 medium w/2.5uM BrdU and the cells are harvested after 10 days of BrdU treatment and washed with calcium and magnesium-free PBS. .
- To prepare receptor containing membranes cells are homogenized in wash buffer (50 mM Tris
- Membrane pellets (containing CRF receptors) are re-suspended in 50 mM Tris buffer pH 7.7 containing 10 mM MgC1 2 and 2 mM EDTA and centrifuged for 10 minutes at 48,00Og, Membranes are washed again and brought to a final concentration of 1500 ug/ml in binding buffer (Tris buffer above with 0.1 % BSA, 15 mM bacitracin and 0.01 mg/ml aprotinin.).
- binding assay 100 ⁇ L of the membrane preparation are added to 96 well microtube plates containing 100 ⁇ L of 125 ⁇ -sauvagine (SA 2200 Ci/mmol, final concentration of 100 pM) and 50 ⁇ L of test compound. Binding is carried out at room temperature for 2 hours. Plates are then harvested on a BRANDEL 96 well cell harvester and filters are counted for gamma emissions on a Wallac 1205 BETAPLATE liquid scintillation counter. Nonspecific binding is defined by 1 mM cold CRF. IC 50 values are calculated with the non-linear curve fitting program RS/ 1 (BBN Software Products Corp., Cambridge, MA).
- the binding affinity (expressed as IC 50 or K, value) for those compounds and salts of Table I or Table II that have been tested generally ranges from about 0.5 nanomolar to about 10 micromolar.
- Compound or salt at 1, 2.5, 5, 10, 25, and less preferably 50 ⁇ M are preincubated with pooled human liver microsomes 1) in the presence and 2) in the absence of NADPH (final cone. 1 mM) in a shaking water bath at 37°C for 30 minutes. After 10-fold dilution of the preincubation mixture with 0.1M phosphate buffer, pH 7.4, containing a selective CYP probe substrate and NADPH (final cone. 1 mM), residual enzyme activity is measured and the extent of the inhibition shift between the two treatments is evaluated. A concentration-dependent and statistically significant decrease in each particular CYP activity in the presence vs. absence of NADPH indicates mechanism-based inhibition of that CYP isozyme.
- CYP1A2 furafylline
- CYP2C8 phenelzine
- CYP2C9 tienilic acid
- CYP2C19 ticlopidine
- CYP2D6 paroxetine
- CYP3A4 ribefradil
- LC- MS extrorphan
- LC-MS/MS 4'-hydroxytolbutamide, desethylamodiaquine,4- acetamidophenol, 4- hydroxy-S-mephenytoin
- HPLC HPLC (6 ⁇ -hydroxytestosterone).
- a mechanism based inhibitor ot the isozyme will show concentration dependence and a statistically significant decrease (student's t-test: p ⁇ 0.05) in activity after 30 minutes preincubation with NADPH as compared to preincubation in the absence of NADPH.
- Preferred compounds or salts disclosed herein do not cause nausea or vomiting when administered to a patient in a therapeutically effective amount.
- a convenient measure of this property is absence of emesis in more than I out of six dogs within one, preferably 3, or less preferably 6 hours of administration of a dose selected from 1. 30, 100, 300, and 1000mg/kg of body weight.
- Such absence of emesis is determined as follows: Doses of compound or salt are administered by oral intubation using a flexible tube fitted onto a syringe and utilizing a flush of approximately 10 mL of distilled water. Dogs are fed Certified Canine Diet, No. 5007 (PMl Nutrition International, St. Louis, MO) ad libitum in 400 gram amounts for approximately 4 hours daily.
- Radiolabeled probe compounds The compounds and salts disclosed herein are prepared as radiolabeled probes by carrying out their synthesis using precursors comprising at least one atom that is a radioisotope.
- the radioisotope is preferably selected from of at least one of carbon (preferably 14 C). hydrogen (preferably 3 H), sulfur (preferably 3:? S), or iodine (preferably 123 I).
- radiolabeled probes are conveniently synthesized by a radioisotope supplier specializing in custom synthesis of radiolabeled probe compounds.
- a radioisotope supplier specializing in custom synthesis of radiolabeled probe compounds.
- suppliers include Amersham Corporation, Arlington Heights, IL; Cambridge Isotope Laboratories, Inc. Andover, MA; SRJ Internationa!, Menlo Park, CA; Wizard Laboratories, West Sacramento, CA; CheraSyn Laboratories, Lexena, KS; American Radiolabeled Chemicals, Inc., St. Louis, MO; and Moravek Biochemicals Inc., Brea, CA.
- Tritium labeled probe compounds are also conveniently prepared catalytically via platinum- catalyzed exchange in tritiated acetic acid, acid-cata ⁇ yzed exchange in tritiated trifiuoroacetic acid, or heterogeneous-catalyzed exchange with tritium gas. Such preparations are also conveniently carried out as a custom radiolabeling by any of the suppliers listed in the preceding paragraph using the unlabeled compound as substrate. In addition, certain precursors may be subjected to tritium-halogen exchange with tritium gas, tritium gas reduction of unsaturated bonds, or reduction using sodium borotritide, as appropriate.
- Example 11 Receptor autoradiography
- Receptor autoradiography (receptor mapping) is carried out in vitro as described by Kuhar in sections 8.1.1 to 8.1.9 of Current Protocols in Pharmacology (1998) John Wiley & Sons, New York, using radiolabeled compounds provided herein prepared as described in the preceeding schemes and the preceding Example.
- Example 12 Microsomal in vitro half-life
- Compound half-life values may be determined via the following standard liver microsomal half-life assay. Pooled Human liver microsomes are obtained from XenoTech LLC, 3800 Cambridge St. Kansas's City, Kansas, 66103 (catalog # H0610). Such liver microsomes may also be obtained from In Vitro Technologies, 1450 South Rolling Road, Baltamore, MD 21227, or from Tissue Transformation Technologies, Edison Corporate Center, 175 May Street, Suite 600. Edison, NJ 08837.
- Reactions are preformed as follows: Reagents: Phosphate buffer: 19 mL 0.1 M NaH 2 PO 4 , 81 mL 0.1 Na 2 HPO 4 , adjusted to pH 7.4 with H 3 PO 4 .
- CoFactor Mixture 16.2 nig NADP, 45.4 mg Glucose-6-phosphate in 4 mL 100 mM MgCl 2 .
- Glucose-6-phosphate dehydrogenase 214.3 ⁇ L glucose-6-phosphate dehydrogenase suspension (Boehringer-Manheim catalog no. 0737224, distributed by Roche Molecular Biochemicals, 9115 Hague Road, P.O.
- test reactions are prepared, each containing 25 ⁇ L microsomes, 5 ⁇ L of a 100 uM solution of test compound, and 399 ⁇ L 0.1 M phosphate buffer.
- a seventh reaction is prepared as a positive control containing 25 ⁇ L microsomes, 399 ⁇ L 0.1 M phosphate buffer, and 5 ⁇ L of a 100 uM solution of a compound with known metabolic properties (e.g. diazepam or clozepine). Reactions are preincubated at 39°C for 10 minutes.
- 71 ⁇ L Starting Reaction Mixture is added to 5 of the 6 test reactions and to the positive control, 71 ⁇ L 100 mM MgCl 2 is added to the sixth test reaction, which is used as a negative control.
- Preferred compounds exhibit in vitro t] /2 values of greater than 10 minutes and less than 4 hours. Most preferred compounds exhibit in vitro t )/2 values of between 30 minutes and 1 hour in human liver microsomes.
- Example 15 Preparation of: 5-( " l -Ethvlpropvi)-3,7-dimethvl-2-r3-methyl-5-fmethvlsulfonvl)-2-thienvl]-5H-pvrrolor2.3-blpyrazine
- Example 16 Preparation of: l- ⁇ 5-[5-(l -Ethylpropyl)-3.7-dimethyl-5H-pyrrolo[2,3-blpyrazin-2-vlH-methyl-2-thienvl ⁇ ethanone
- Example 17 Preparation of: l- ⁇ 5-[5-(l -Ethylpropy])-3J-dimethyi-5H-pyrrolo[2,3-&lpyrazin-2-yl1-4-rnethyl-2-thienvUethanol
- the crude product obtained from previous step is taken into ethanol (3 mL). p-Toluenesulfonic acid (2 mg) is added followed by the addition of triethyl orthoformate (2 mLl). The resulting mixture is heated at 80 °C for 18 hours. The reaction mixture is concentrated and the residue is partitioned between water and ethyl acetate. The organic layer is dried with magnesium sulfate. The crude product is purified by silica gel column chromatography with hexanes/ethyl acetate to afford the title compound.
- Example 24 Preparation of: 2-(5-r5-( ⁇ R)-l-methoxybutan-2-yl)-3.7-dimethyl-5H-pyrrolor3.2-blpyrazin-2-ylV4-methylthiophen-2- yl)propan-2-ol l-(5- ⁇ 5-[(lR)-1-(Methoxymethyl)propyl]-3 ! 7-dimethy]-5H-pyrroIo[2,3-blpyrazin-2-yl ⁇ -4-methyl-2- thienyl)ethanone (28 mg) is taken into anhydrous THF (2 mL).
- a solution of zinc chloride (2.0M in THF, 96.5 mL, 48.2 mmol, 5.0 equiv.) is treated dropwise with methylmagnesium bromide (3.0 M in Et 2 O, 32.2 mL, 96.5 mmoi, 10.0 equiv.) at 0 °C.
- the mixture is warmed to room temp and stirred for 45 min.
- a solution of 2-amino-6-chloro pyrazine (1.25 g. 9.65 mmol, 1.0 equiv) and NiC1 2 (dppp) (157 mg, 0.29 mmol, 0.03 equiv) in THF (15 mL) is added to the freshly prepared dimethylzinc reagent.
- the compound is prepared following a previously reported procedure (WO 2005/023806) with slight modifications.
- To a solution of 2,6-dichloropyrazine (100 g. 0.671 mol, 1 ,0 equiv.) hi THF (500 mL) is added 175 mL (2.5 equiv.) of 40% aqueous methylamine solution followed by 117 ml (0.671 mo!, 1.0 equiv.) of N,N-diisopropylethyl amine.
- the resulting mixture is heated overnight in a sealed tube at 80 °C. After this period, the reaction mixture is allowed to cool to room temperature and then concentrated under reduced pressure to remove solvent.
- Part B 3.5-Dibromo-6-chloro-N-methyIpyrazin-2-amine
- the compound is prepared following a previously reported procedure (WO 2005/023806) with slight modifications.
- a cooled (0 "C) solution of crude 6-ehloro-N-methylpyrazin-2-amine (0.671 rnol, 1.0 equiv) in CH 2 C1 2 (600 mL) is added N-bromosuccinamide (299 g, 1.68 mol, 2.5 equiv.) in small portions over a period of 30 min.
- the resultant mixture is warmed to rt and allowed to stir. After 1 hr, the reaction mixture is diluted with water (500 mL) and the layers separated. The organic layer is washed with water (2 X 500 mL), brine (500 mL) and dried over Na 2 SO 4 .
- Part C S.S-Dibromo- ⁇ -chloro-N-fS-ethylpent- ⁇ -envD-N-methylpyrazin ⁇ -amine
- the compound is prepared following a previously reported procedure (WO 2005/023806) with slight modifications.
- the resultant suspension is allowed to stir for 1 h at rt and then treated with TBAI (245 mg, 0.664 mmol, 0.1 equiv) and 18-crown-6 (176 mg, 0.664 mmol, 0.1 equiv).
- Example 35 Preparation of: l-(5-r2.5-DimcthvI-7-rpentan-3-v ⁇ -5H-pvrrolor2,3-&1pvrazin-3-v ⁇ -4methylthiophe ⁇ -2-yl)ethanol
- the resultant anion is treated with a solution of carbon tetrabromide (338 mg, 1.02 mmoi, 1.5 equiv) in THF (1.0 mL) and stirred at -78 °C for Ih, before being quenched with water and allowed to warm to it.
- the product is extracted with EtOAc and the combined organic extracts are washed with brine, dried over Na 2 S ⁇ 4 and concentrated under reduced pressure.
- the resulting residue is purified by silica gei column chromatography using 10% EtOAc in hexanes to give the desired compound as a brown oil.
- a Teflon screw-cap viai is charged with 150 mg (0.38 mmoi, 1.0 equiv) of 3-(5-bromo-3-methylthiophen- 2-yl)- 2,5-dimethyl-7-(pentan-3-yl)-5H-pyrro!o[2,3-i]pyrazine and 2 mL of N ; N-dimethyIethanolamine.
- To the solution is added 12.5 mg of Cu powder, 12.5 mg of copper(l) iodide and 161 mg (0.76 mmol, 2.0 equiv) of morpholine.
- the reaction vial is evacuated and purged with nitrogen three times before being tightly capped and heated overnight at 120 0 C.
- administering does not result in prolongation of heart QT intervals (i.e., as determined by electrocardiography, e.g., in guinea pigs, minipigs or dogs).
- such doses of such preferred compounds When administered daily for 5 or preferably ten days, such doses of such preferred compounds also do not cause liver enlargement resulting in an increase of liver to body weight ratio of more than 100%, preferably not more than 75% and more preferably not more than 50% over matched controls in laboratory rodents (e.g., mice or rats). In another aspect such doses of such preferred compounds also preferably do not cause liver enlargement resulting in an increase of liver to body weight ratio of more than 50%, more preferably preferably not more than 25%, and most preferably not more than 10% over matched untreated controls in dogs or other non-rodent mammals.
- such doses of such preferred compounds also preferably do not promote the release of liver enzymes (e.g., ALT, LDH, or AST) from hepatocytes in vivo.
- liver enzymes e.g., ALT, LDH, or AST
- such doses do not elevate serum levels of such enzymes by more than 100%, more preferably not by more than 75% and most preferably not by more than 50% over matched untreated controls in laboratory rodents.
- concentrations (in culture media or other such solutions that are contacted and incubated with cells in vitro) equivalent to two, fold, preferably five-fold, and most preferably ten-fold the minimum in vivo therapeutic concentration do not cause release of any of such liver enzymes from hepatocytes into culture medium in vitro above baseline levels seen in media from untreated cells.
- preferred compounds exert their receptor-modulatory effects with high selectivity. This means that they do not bind to certain other receptors (other than CRF, preferably CRFl, receptors) with high affinity, but rather only bind to, activate, or inhibit the activity of such other receptors with affinity constants (note: greater affinity constants indicate weaker binding) of greater than 100 nanomolar, preferably greater than 1 micromolar, more preferably greater than 10 micromolar and most preferably greater than 300 micromolar.
- Such receptors preferably are selected from the group consisting of a) ion channel receptors, (preferably sodium ion channel receptors), b) neurotransmitter receptors (preferably selected from alpha- and beta-adrenergic receptors, muscarinic receptors - most preferably m 1 , m2, or m3 receptors, dopamine receptors, GABA A receptors and metabotropic glutamate receptors), c) histamine receptors, d) cytokine receptors (preferably selected from interleukin receptors, most preferabiy IL-8 receptors), e) bioactive peptide receptors (preferably selected from NPY and VIP receptors), f) neurokinin receptors g) bradykinin receptors (preferably selected from BKl receptors and BK2 receptors), and h) hormone receptors (preferably selected from thyrotropin releasing hormone receptors and melanocyte- concentrating hormone receptors).
- ion channel receptors preferably sodium ion channel receptors
Abstract
Substituted heteroaryl fused pyridine, pyrazine, and related heterobicyclic compounds that act as selective modulators of CRF1 receptors are provided. These compounds are useful in the treatment of a number of CNS and periphereal disorders, particularly stress, anxiety, depression, cardiovascular disorders, gastrointestinal disorders, and eating disorders. Methods of treatment of such disorders as well as packaged pharmaceutical compositions are also provided. Compounds provided herein are also useful as probes for the localization of CRF receptors and as standards in assays for CRF receptor binding. Methods of using the compounds in receptor localization studies are given.
Description
CRFl RECEPTOR LIGANDS COMPRISING FUSED BICYCLIC HETEROAR YL MOIETIES
This application claims the priority benefit of US Provisional Application Serial No. 60/882.696 filed December 29, 2006, any teachngs of which that are omitted herefrom being incorporated herein by reference.
FIELD QF THE INVENTION
The present invention relates to novel substituted purines and heteroaryi fused pyridine, pyrazine, pyrimidine, and related compounds that bind with high selectivity and / or high affinity to CRF (Corticotropin Releasing Factor), preferably CRFl, receptor. It also relates to pharmaceutical compositions comprising such compounds and to their use in the treatment of psychiatric disorders and neurological diseases, including major depression, anxiety-related disorders, post-traumatic stress disorder, supranuclear palsy, substance abuse disorders and feeding disorders, as well as treatment of immunological, cardiovascular or heart-related diseases and digestive disorders including colonic hypersensitivity associated with psychopathological disturbance and stress, including Irritable Bowel Syndrome (IBS). Additionally this invention relates to the use such compounds as probes for the localization of CRF receptors in cells and tissues.
BACKGROUND OF THE INVENTION
Corticotropin releasing factor (CRF), is a peptide that acts by binding to and modulating the signal transduction activities of specific cell surface receptors, including CRFl receptors and CRF2 receptors. CRF receptors are believed to play a significant role in integrating responses to stress.
CRF has been shown to be involved in psychiatric disorders and neurological diseases including depression, anxiety-related disorders, gastrointestinal disorders, substance abuse disorders and feeding disorders.
SUMMARY OF THE INVENTION
The invention provides novel compounds of Formulas A, B, C, D, E, F, G, H, i, J, K, L, M, N. O, P, Q, R, S, and T (below), and pharmaceutical compositions comprising such compounds and at least one pharmaceutically acceptable carrier or excipient. Such compounds bind to cell surface receptors, preferably G-coupled protein receptors, especially CRF receptors (including CRFl and CRF2 receptors) and most preferably CRFl receptors. Preferred compounds exhibit drug-like affinity (i.e., IC50 or K, values < 1 micromolar in the assay for CRF receptor functional activity of Example 6, below) for CRF receptors, preferably CRFl receptors. Additionally, preferred compounds also exhibit high specificity for
CRF receptors (i.e., they exhibit high selectivity compared to their binding to non-CRF receptors). Preferably they exhibit high specificity for CRFl receptors.
Thus, in certain aspects, the invention provides compounds and pharmaceutically acceptable salts thereof that are compounds of a Formula seiected from the group consisting of:

R2i is hydrogen, halogen, amino, (C1-C4)alkyi, C2-C4aikenyi, C2-C4alkyny], (C i -C4)alkoxy, hydroxy, halo(C1-C4)alkyl, ha!o(C1-C4)alkoxy, cyano, mono- or di- (C1-C4)alkylamino, (Cϊ- C4)alkanoy], aminocarbonyl, or -S(O)n-(C1-C4)alkyh
R22 is hydrogen, halogen, (C1-C4)alkyl, C2-C4alkenyl, C2-C4alkynyl, (C1-C4)alkoxy, hydroxy, ImIo(C1 -C4)alkyl, halo(C!-C4)alkoxy, cyano, mono- or di- (C 1 -C4)alky!amino, (C1- C4)alkanoyl, aminocarbonyl, or -S(0)n-(C1-C4)alkyl;
R23 is (C1-C7)alkyl, (C2-C7)alkenyi, (C2-C7)alkynyl, (C6-C12)aryl, 4 to 12 membered heterocycloalkyl, or 5 to 12 membered heteroaryl, each of which is unsubstituted or substituted with one or more substituents independently selected at each occurrence from (C1-C7)alkyi, (C1- C7)alkoxy, halogen, amino, hydroxyl, 4 to 12 membered heterocycloalkyl, and 5 to 12 membered heteroaryl;
R24 is hydrogen, halogen, cyano, (C1-C3)alkyl, (C1-C3)alkoxy, hafo(C1-C7)alkyl. mono- or di- (C1-C7)alkylamino, -(CH2)n-R10, CONH2, or mono- or di-(C1-C4)alkyl-arninocarbonyl-;
R25 is hydrogen, halogen, cyano, (C1-C7)alkyl, (C1-C3)alkoxy, or haloalkyl, mono- or di- (C1- C7)alkylamino, -(CH2)n-R10, CONH2, or mono- or di-(C1-C4)alkyl-aminocarbonyl-;
R26 is hydrogen or (C1-C4)aikyl;
R27 is (C1-C7)alkyl, (C2-C7)alkenyi, (C2-C7)alkynyi, (C6-C12)aryl, mono- or di- (C1- C7)alkylamino, 4 to 12 membered heterocycloalkyl, 5 to 12 membered heteroaryl, -(CH2)n-R10, - (CH2)n-CONH2, or mono- or di-(C1-C4)alkyl-aminocarbonyl-(CH2)n-, each of which is unsubstituted or substituted with one or more substituents independently selected at each occurrence from (C1-C7)alkyl, (C1-C7)alkoxy, halogen, amino, hydroxyl, 4 to 12 membered heterocycloalkyl, and 5 to 12 membered heteroaryl;
R2S is (C1-C7)alkyl, (C6-C12)aryl, 4 to 12 membered heterocycloalkyl, or 5 to 12 membered heteroaryl, each of which is unsubstituted or substituted with one or more substituents independently selected at each occurrence from alkyl, alkoxy, halogen, amino, hydroxy!, 4 to 12 membered heterocycloalkyl, and 5 to 12 membered heteroaryl;
R10 is N-linked and at each occurrence is independently acetidine, pyrrolidine, piperidine. hexahydroazepine, or octahydroazepine; and
R2 is selected from the group consisting of:

R7 is hydrogen, halogen, cyano, (C1-C4)alkyi, ha!o(CJ -C4)alkyl, (C1-C4)alkoxy, haio(C1- C4)alkoxy, mono- or di- (C1-C4)alkylamino, (C 1 -C4)alkylcarbonyl, -(CH2)n-R10 , -(CH2)n- CONH2, mono- or di-(CI-C4)alkyl-aminocarbonyl-(CH2)n-, -S(O)n-(C1-C4)alkyl, phenyl,

R8 is hydrogen, halogen, haloalkyl, haioalkoxy, cyano, (C1-C4)alky], RaRbN-, carbamyl, -(C1- C2)alkoxy(C1-C2)alkyl, Rπ-C(O)-, Rl l-(CH2)n-,

Ra is hydrogen, (C1-C5)alkyl, (C3-C5)cycloalkyl, methoxy(C2-C4)alkyl, acetyl, (C1- C2)alkylsulfonyl. (C3)aikenyl, R15-(CH2)n-, or (C1-C2)alkyl, each of which is unsubstituted or substituted with cyano, foπnyl, vinyl, or ethynyl;
Rb is hydrogen or (C1-C3)alkyl;
RI 1 is hydrogen. (C1-C7)alkyi, (C3-C7)cycioalkyl, -O(C1-C4)alkyI, (C2-C7)alkenyl, ethynyl, mono- or di- (C1-C4)alkylamino, aryl, 4 to 12 membered heterocycloalkyl, or 5 to 12 membered heteroaryl, R15-(CH2)n-;
R15 is cyclopropyl, phenyl,
 O
O
R31 represents one or two substituents, at each occurrence independently selected from hydrogen, halogen, (C1-C4)alkyi, (C1-C4)alkoxy, hydroxy, hydroxyI(C1-C4)a!kyl, haloalkyl, haloalkoxy, cyano, amino, mono- or di- (C1-C4)a!kylamiπo, (C1-C4)afkyl-CO-, CONH2, and -S(O)n-(C1- C4)alkyl;
R33 is (C1-C7)aikyL (C2-C7)alkenyl. (C2-C7)alkynyl. -(CH2)n-R10 , -(CH2)n-CONH2, mono- or di-(C1-C4)alkyl-aminocarbonyl-(CH2)n-, aryl, 4 to 12 membered heterocycloalkyl, or 5 to 12 membered heteroaryl, each of which is unsubstituted or substituted one or more substituents independently selected at each occurrence from (C1-C7)alkyi, (C 1 -C7)alkoxy, halogen, amino, hydroxy!, 4 to 12 membered heterocycloalkyl, and 5 to 12 membered heteroaryl;
R35 at each occurrence is independently hydrogen, halogen, cyano, (C1-C3)alkyl, (C1-C7)alkyl, (C1-C3)a!koxy, halo(C1-C7)a!kyl, -(CH2)n-R10 , -(CH2)n-CO mono- or di- (C1-C7)alkylamino, mono- or di- (C1-C7)alkylamino, or -CO mono- or di- (C1-C7)alkylamino;
R36 is hydrogen or (C1-C4)alkyl;
R37 is hydrogen, halogen, (C1-C4)alkyl, or halo(C1-C4)a!kyi; n is 0, I, or 2; and
X is CH2. CO, O, S, or SO2; or
R" is selected from the group consisting of:

- 3 -

R1 is hydrogen, halogen, cyano, (C1-C3)alkyl, or methoxy;
R3 is hydrogen or (C1-C3)alkyl;
R4 is hydrogen, halogen, hydroxyl, formyl, (C1-C3)alkoxy, (C2-C3)alkenyl,
RcRdN-, or R16-C(O)-; or
R4 is (C3-C5)cycloalkyl or (C1-C7)alkyl, each substituted with zero, one, or two that are the same or different of hydroxy, (C1 -C2)alkoxy, or cyclopropyl;
R5 is hydrogen, methyl, or trifluoromethyl: R16 is hydrogen, (C1-C3)alkyl, cyclopropyl. methoxy, or RcRdN-; and
Rc and Rd are independently hydrogen. (C1-C4)alkyl, or methoxy.
The invention further provides methods of treating patients suffering from certain disorders with a therapeutically effective amount of at least one compound provided herein. These disorders include CMS disorders, particularly affective disorders, anxiety disorders, stress-related disorders, eating disorders and substance abuse. The patient suffering from these disorders may be a human or other animal (preferably a mammal), such as a domesticated companion animal (pet) or a livestock animal.
Preferred compounds for such therapeutic purposes are those that antagonize the binding of CRF to CRF receptors (preferably CRFl , or less preferably CRF2 receptors). The ability of compounds to act as antagonists can be measured as an IC50 value as described below.
According to yet another aspect, the present invention provides pharmaceutical compositions comprising at least one compound or salt of Formulas A, B, C, D, E, F, G, H, i, J, K, L, M, N, O, P, Q, R, S, or T (preferably at least one compound or salt of Table I or Table 11 ) or the pharmaceutically acceptable salts (as defined below) thereof, which compositions are useful for the treatment of the above- recited disorders. The invention further provides methods of treating patients suffering from any of the above-recited disorders with an effective amount of a compound or composition provided herein.
Additionally this invention relates to the use of the compounds provided (particularly labeled compounds) as probes for the localization of receptors in cells and tissues and as standards and reagents for use in determining the receptor-binding characteristics of test compounds. Preferred compounds exhibit a half-maximal inhibitory concentration (IC50) of less than 5 micromoiar in the standard in vitro CRFl receptor binding assay provided in Example 7 below. Particularly preferred compounds exhibit an IC50 of about 1 micromoiar or less, still more preferably an IC50 of about 100 nanomolar or less even more preferably an IC50 of about 10 nanomolar or less. Certain particularly preferred compounds exhibit an IC50 of 1 nanomolar or less in such standard binding assay.
DETAILED DESCRIPTION OF THE INVENTION
All compounds listed herein may be characterized by 1H-NMR and the following LCMS methods: Method I.
HFLC instrumentation: Analyses are performed using a Waters 600 series pump (Waters Corp.), a Waters 996 Diode Array detector and a Gilson 215 auto-sampler (Gilson Inc.). Data are acquired using MassLynx 4.0 software, with OpenLynx processing.
HPLC conditions: 4.6x50mm, XTerra MS C18, 5 mm column (Waters Corp.); UV 10 spectra/sec, 220, 254nm; flow rate 4.0 mL/min; injection volume I -10 μl; Gradient conditions - Mobile phase A 95% Water, 5% Methanol with 0.05% Formic acid; Mobile phase B 95% methanol, 5% Water with 0.025% Formic acid; Gradient: Time (min) %B
0 5
0.01 5
1.0 100
2.0 100 2.1 5
MS instrumentation: LC-MS experiments are performed using a Waters ZMD II Mass Spectrometer. MS conditions: EIectrospray positive ionization; capillary voltage 3.5kV; cone voltage 30V; desoivation and source temperature 250 °C and 100 °C respectively; mass range 120-800 with a scan time of 0.5 seconds and an inter scan delay of 0.1 min. Method 2.
Flow Injection Condition:
A Perkin Elmer HPLC system (tow Series 200 micro LC pumps, pump A and pump B, with a Series 200 autosampler) is used to perform flow injection. Mobile phase is a combination of 85% methanol (pump B) with 15% of water (pump A). The flow rate is 1.0 mL/min; and the injection volume is 3 μl. MS instrumentation: LC-MS experiments are performed using a Sciex 150MA Mass Spectrometer. MS conditions: Ion source is Heated Nebulizer (atmosphere pressure chemical ionization). The mass range is 100-1000 amu. Both positive and negative modes are in place. For positive ion mode, Nebulizer current is 2.0 mA, and the temperature is 350 °C. The Nebulizer gas is 10, and the Curtain gas is 12. The declustering potential is 30 V. The Focusing potential is 200 V, and the entrance potential is -10 V. For negative ion mode, Nebulizer current is -2.0 mA, and the temperature is 350 °C. The Nebulizer gas is 10, and the Curtain gas is 12. The declustering potential is -30 V. The Focusing potential is -200 V, and the entrance potential is 10 V. Method 3.
HPLC Instrumentation: HPl 100 PUMP, HPl 100 UV detector with 220 nm, HTS/PAL autosampler from Leap Technology, data acquired by Micromass Ma
HPLC conditions: Syπergi 2U HYDRO-RP 20 x 4.0mm column, flow rate 1.0 mL/min, injection volume 5 μL.
Gradient conditions: 0.3% foπnic acid in aqueous acetonitrile, 10-90% acetonitrile over 3 min, then 100% acetonitrile, end at 5 min. MS instrumentation: Micromass LCT-TOF MS
MS conditions: Scan in/z 100-1200, capillary voltage 3000V. cone voltage 25V, desolvation 200 °C and source temperature 100 °C. Methods of treatment
Compounds described herein and salts thereof are useful in treating a variety of conditions including affective disorders, anxiety disorders, stress disorders, eating disorders, inflammatory or chronic pain disorders, and substance abuse disorders. Hence they each may be used in the manufacture of a medicament for the treatment of each such condition.
Affective disorders include all types of depression, bipolar disorder, cyclothymia, and dysthymia. Anxiety disorders include generalized anxiety disorder, panic, phobias and obsessive-compulsive disorder. Compounds described herein and salts thereof are useful in treating symptoms of affective disorders and anxiety disorders. These syptoms include increased awakenings, increased REM density, and related sleep disorders (e.g., insomnia) and deπnatologic and related immunogenic inflammatory conditions including atopic dermatitis, urticaria, rheumatoid arthritis, and psoriasis.
Stress-related disorders include post-traumatic stress disorder, hemorrhagic stress, stress-induced psychotic episodes, psychosocial dwarfism, stress headaches, stress-induced immune systems disorders such as stress-induced fever, and stress-related sleep disorders.
Substance abuse disorders include alcohol abuse and drug addictions. Compounds described herein and salts thereof are particularly useful for the therapeutic reduction of the anxiogenic effects of drug or alcohol withdrawal in drug or alcohol habituated patients. They are also useful for the prevention of relapse of abusive alcohol or drug consumption following withdrawal.
Eating disorders include anorexia nervosa, bulimia nervosa, and obesity.
Compounds described herein and salts thereof are also useful in the treatment (e.g., symptomatic treatment) of a variety of neurological disorders including supranuclear palsy, AIDS related dementias, multlinfarct dementia, neurodegenerative disorders such as Alzheimer's disease, Parkinson's disease, and Huntington's disease, head trauma, spinal cord trauma, ischemic neuronal damage, epilepsy, amyotrophic lateral sclerosis, and disorders of pain perception such as fibromyalgia.
Additionally compounds described herein and salts thereof are useful in the treatment (e.g.. symptomatic treatment) of a number of gastrointestinal, cardiovascular, hormonal, autoimmune and inflammatory conditions. Such conditions include irritable bowel syndrome (IBS), ulcers, Crohn's disease, spastic colon, diarrhea, post operative ilius. and colonic hypersensitivity associated with psychopathoiogical disturbances or stress; hypertension, tachycardia, congestive heart failure, infertility, and euthyroid sick syndrome; and inflammatory conditions effected by or associated with rheumatoid arthritis, osteoarthritis, pain, asthma, psoriasis and allergies.
Compounds described herein and salts thereof are also useful as modulators of the CRFl receptor in the treatment of animal disorders associated with aberrant CRF levels. Such conditions include porcine stress syndrome, bovine shipping fever, equine paroxysmal fibrillation, and dysfunctions induced
by confinement in chickens, sheering stress in sheep or human-animal interaction related stress in dogs, psychosocial dwarfism and hypoglycemia. Typical subjects to which compounds described herein and salts thereof may be administered include mammals, preferably primates, most preferably humans. For veterinary applications, a wide variety of subjects are suitable, e.g. livestock such as cattle, sheep, goats, cows, swine and the like; poultry such as chickens, ducks, geese, turkeys, and the like; and other domesticated animals particularly pets such as dogs and cats. For diagnostic or research applications, a wide variety of mammals are suitable subjects including rodents (e.g. mice, rats, hamsters), rabbits, primates, and swine such as inbred pigs and the like. Additionally, for in vitro applications, such as in vitro diagnostic and research applications, body fluids (e.g., blood, plasma, serum, CSF, lymph, cellular interstitial fluid, aqueous humor, saliva, synovial fluid, feces, or urine) and cell and tissue samples of the above subjects are suitable for use.
Compounds described herein and salts thereof are also useful as standards and reagents in determining the ability of test compounds (e.g., a potential pharmaceutical) to bind to a CRF receptor.
Labeled compounds provided herein and salts thereof are also useful as tracers for positron emission tomography (PET) imaging or for single photon emission computerized tomography (SPECT).
More particularly such compounds and salts are useful for demonstrating the presence of CRF receptors in cell or tissue samples. This may be done by preparing a plurality of matched cell or tissue samples, at least one of which is prepared as an experiment sample and at least one of which is prepared as a control sample. The experimental sample is prepared by contacting (under conditions that permit binding of CRF to CRF receptors within cell and tissue samples) at least one of the matched cell or tissue samples that has not previously been contacted with any compound or salt provided herein with an experimental solution comprising the detectably-labeled preparation of the selected compound or salt at a first measured molar concentration. The control sample is prepared by in the same manner as the experimental sample and is incubated in a solution that contains the same ingredients as the experimental solution but that also contains an unlabelled preparation of the same compound or salt provided herein at a molar concentration that is greater than the first measured molar concentration.
The experimental and control samples are then washed to remove unbound detectably-labeled compound. The amount of detectably-iabeled compound remaining bound to each sample is then measured and the amount of detectably-labeled compound in the experimental and control samples is compared. A comparison that indicates the detection of a greater amount of detectable label in the at least one washed experimental sample than is detected in any of the at least one washed control samples demonstrates the presence of CRF receptors in that experimental sample.
The detectably-labeled compound used in this procedure may be labeled with any detectable label, such as a radioactive label, a biological tag such as biotin (which can be detected by binding to detectably-labeled avidin), an enzyme (e.g., alkaline phosphatase, beta galactosidase, or a like enzyme that can be detected its activity in a colorimetric assay) or a directly or indirectly luminescent label. When
tissue sections are used in this procedure and the detectably-labeled compound is radiolabeled, the bound, labeled compound may be detected autoradiographically to generate an autoradiogram. When autoradiography is used, the amount of detectable label in an experimental or control sample may be measured by viewing the autoradiogram s and comparing the exposure density of the autoradiograms. The present invention also pertains to methods of inhibiting the binding of CRF to CRF receptors
(preferably CFRl receptors) which methods involve contacting a solution containing a compound provided herein with cells expressing CRF receptors, wherein the compound is present in the solution at a concentration sufficient to inhibit CRF binding to CRF receptors in vitro. This method includes inhibiting the binding of CRF to CRF receptors in vivo, e.g., in a patient given an amount of a compound or salt disclosed herein that would be sufficient to inhibit the binding of CRF to CRF receptors in vitro. In one embodiment, such methods are useful in treating physiological disorders associated with excess concentrations of CRF. The amount of a compound that would be sufficient to inhibit the binding of CRF to the CRF receptor in vitro may be readily determined via a CRF receptor binding assay (see, e.g., Example 7), or from the IC50 determined using a CRF receptor functional assay, such as the standard assay of CRF receptor activity of Example 6. The CRF receptors used to determine in vitro binding may be obtained from a variety of sources, e.g., from cells that naturally express CRF receptors, e.g. IMR32 cells, or from cells expressing cloned human CRF receptors.
The present invention also pertains to methods for altering the activity of CRF receptors, said method comprising exposing cells expressing such receptors to an effective amount of a compound provided herein, wherein the compound is present in the solution at a concentration sufficient to specifically alter the signal transduction activity in response to CRF in cells expressing CRF receptors in vitro. Preferred cells for this purpose are those that express high levels of CRF receptors (i.e., equal to or greater than the number of CRFl receptors per cell found in differentiated ΪMR-32 human neuroblastoma ceJJs), with IMR-32 ceils being particularly preferred for testing the concentration of a compound required to alter the activity of CRFl receptors. This method includes altering the signal transduction activity of CRF receptors in vivo, e.g., in a patient given an amount of a compound or salt disclosed herein that would be sufficient to alter the signal transduction activity in response to CRF in cells expressing CRF receptors in vitro. The amount of a compound that would be sufficient to alter the signal transduction activity of CRF receptors in response to CRF in vitro may also be determined via an assay of CRF receptor mediated signal transduction, such as an assay wherein the binding of CRF to a cell surface
CRF receptor effects a changes in reporter gene expression.
The present invention also pertains to packaged pharmaceutical compositions for treating disorders responsive to CRF receptor modulation, e.g.. eating disorders, depression or stress. The packaged pharmaceutical compositions include a container holding dosage units comprising a therapeutically effective amount of at least one compound or salt disclosed herein and instructions for using the treating disorder responsive to CRFl receptor modulation in the patient.
Chemical description and terminology
Compounds are generally described herein using standard nomenclature. Compounds having asymmetric centers and compounds with carbon-carbon double bonds may occur in + or -and/or Z- or E- forms. Each isolated isomeric form of the compounds and all mixtures thereof are included in the present invention unless otherwise specified. All tautomeric forms that may exist for any compound are included. Where a general formula includes variables (e.g., R1, A, X), unless otherwise specified, each variable within such a formula is defined independently of any other variable, and any variable that occurs more than one time in a formula is defined independently at each occurrence,.
A "pharmaceutically acceptable salt" of a compound is an acid or base salt that is suitable for use in contact with the tissues of human beings or animals. Such salts include mineral and organic acid salts of basic residues such as amines, as well as alkali or organic salts of acidic residues such as carboxylic acids. Specific pharmaceutically acceptable salts include, but are not limited to, salts of acids such as hydrochloric, phosphoric, hydrobromic, malic, glycolic, fumaric, sulfuric, sulfamic, sulfanilic, formic, toluenesulfonic, methanesulfonϊc. benzene sulfonic, camphorsuifonic, ethane disulfonic, 2- hydroxyethylsulfonic, nitric, benzoic, 2-acetoxybenzoic, citric, tartaric, lactic, stearic, salicylic, glutamic, ascorbic, pamoic, succinic, fumaric, maleic, propionic, hydroxymaleic, hydroiodic, phenylacetic, alkaiioic such as acetic, HOOC-(CH2)n-COOH where n is 0-4, and the like. Similarly, pharmaceutically acceptable salts of bases include, but are not limited to sodium, potassium, calcium, aluminum, lithium and ammonium salts. Further pharmaceutically acceptable salts for the compounds provided herein include those listed in Remington's Pharmaceutical Sciences, 18th ed., Mack Publishing Company, Eastern, PA, p. 1418 (1990) and in particular, the discussion and Table II appearing under the heading "Salt Formation" spanning pages 1444-45 thereof, or the equivalent disclosure in Remington: The Science and Practice of Pharmacy, 21st ed., Lippincott Williams & WUkins, Philadelphia, PA (2005) , each of which is incorporated herein by reference for its disclosures regarding pharmaceutically acceptable salts. Such salts can be prepared by any conventional chemical method, such as reacting the free acid or base forms of these compounds with a stoichiometric amount of the appropriate base or acid in water or in an organic solvent, or in a mixture of the two; generally, the use of nonaqueous media, such as ether, ethyl acetate, ethanoi, isopropanol or acetonitrile, is preferred.
Each compound provided herein may, but need not, be formulated as a solvate (e.g., hydrate) or non-covalent complex. Tthe various crystal forms and polymorphs are also encompassed. Also provided herein are prodrugs of the compounds of the recited Formulas. A "prodrug'1 is a compound that may not fully satisfy the structural requirements of the compounds provided herein, but is modified in vivo, following administration to a patient, to produce a compound a formula provided herein. For example, a prodrug may be an acylated derivative of a compound as provided herein. Prodrugs include compounds wherein hydroxy, amine or sulfhydryl groups are bonded to any group that, when administered to a mammalian subject, cleaves to form a free hydroxy, amino, or sulfhydryl group, respectively. Examples
of prodrugs include, but are not limited to, acetate, formate, benzoate and peptide derivatives of alcohol and amine functional groups within the compounds provided herein.
Acyclic (aliphatic) carbon groups herein include the following straight or branched carbon chains: "alkyl" - C]-C0 (having one to n carbons) fuily saturated aliphatic groups, for example methyl, ethyl, propyl, isopropyl, n-butyl, sec-butyl, tert-buty], pentyl, 2-ρentyl, isopentyl, neopentyl, hexyl, 2- hexyl, 3-hexyl or 3-methylpentyl; and "alkenyl" -C2-Cn aliphatic groups containing at least one carbon- carbon double bond with no carbon-carbon triple bonds, such as ethenyl (HC=C-), I,3-butadienyl (HC=C-CH=CH-). or 2-methyl-l,3-butadienyl
By "aikoxy," as used herein, is meant an alky! group as described above attached via an oxygen bridge. Aikoxy groups include Ci-Cnalkoxy groups, for example methoxy, ethoxy, propoxy, isopropoxy, n-butoxy, sec-butoxy, tert-butoxy, n-pentoxy, 2-pentoxy, 3-pentoxy, isopentoxy, neopentoxy, hexoxy, 2- hexoxy, 3 -hexoxy, or 3-methyIpentoxy groups.
"Alkylsulfonyl" indicates groups of the formula -(SO2)-alkyl, in which the sulfur atom is the point of attachment. Alkylsulfonyl groups include CrCnalky!sulfonyl groups, which have from I to n carbon atoms, such as methyl sulfonyl or pentylsuifonyl. "C]-Cnhaloalkylsulfonyl" is an aikylsulfonyl group that has from 1 to n carbon atoms and is substituted with at least one halogen (e.g., trifluoroinethylsulfonyl).
A "cycioalkyl" is a group that comprises one or more saturated and/or partially saturated rings in which all ring members are carbon, such as cyclopropyl, cyclobutyl, cyclopentyl, and partially saturated variants of the foregoing, such as cyclopentenyl. Cycloalkyi groups do not comprise an aromatic ring or a heterocyclic ring. Certain cycioalkyl groups are C3-C5cycϊoalkyl, in which the cycioalkyl group contains a single ring having from 3 to 5 ring members, all of which are carbon.
By "aikoxy," as used herein, is meant an alkyl group as described above attached via an oxygen bridge. Aikoxy groups include C]-C6alkoxy and CpC4alkoxy groups, which have from 1 to 6 or from I to 4 carbon atoms, respectively. Methoxy, ethoxy, propoxy, isopropoxy, n-butoxy, sec-butoxy, tert~ butoxy, n-pentoxy, 2-pentoxy, 3-pentoxy, isopentoxy, neopentoxy, hexoxy, 2-hexoxy, 3-hexoxy, and 3- methylpentoxy are representative aikoxy groups.
"Alkylsulfonyl" indicates groups of the formula -(SC^-alkyl, in which the sulfur atom is the point of attachment. Alkylsulfonyl groups include CrC6alkylsulfonyI and CrC4alkyisulfonyl groups, which have from I to 6 or from 1 to 4 carbon atoms, respectively. Methylsuifonyl is one representative alkylsulfonyl group.
The term "halogen" indicates fluorine, chlorine, bromine or iodine.
A "substituent," as used herein, indicates a molecular moiety that is covalently bonded to an atom within a molecule of interest. For example, a ring substituent may be a moiety such as a halogen, alkyl group, haloaiky! group or other group that is covalently bonded to an atom (preferably a carbon or nitrogen atom) that is a ring member. Substituents of aromatic groups are generally covalently bonded to
a ring carbon atom. The term "substitution" refers to replacing a hydrogen atom in a molecular structure with a substituent, such that the valence on the designated atom is not exceeded, and such that a chemically stable compound (i.e., a compound that can be isolated, characterized, and tested for biological activity) results from the substitution. A dash ("-" or "-") in a formula that is not between two letters or symbols is used to indicate a point of attachment for a substituent. For example, -CONH2 is attached through the carbon atom.
Similarly, in structural drawings of substituents. such as (H-, ϊπ~, 0~\ and ^T; where ^ represents any drawn structural feature(s); * , ~ ", ~^ , and T each indicates the point of attachment for the substituent. Note: in structural drawings of substituents. +, "V, and 4r do NOT represent tert-butyl. The term '"therapeutically effective amount" means an amount of a compound or salt of Table I or
Table II that is effective, when administered to a human or non-human patient, to provide a therapeutic benefit such as an amelioration of symptoms, e.g., an amount effective to antagonize the effects of pathogenic levels of CRF or to treat the symptoms of any of the disorders listed above under the subheading "Methods of Treament*'. It will be apparent that the therapeutic benefit may be apparent after administration of a single dose, or may become apparent following repeated administration of the therapeutically effective dose according to a predetermined regimen, depending upon the indication for which the compound is administered. Pharmaceutical Preparations
Compounds described herein, and salts thereof may be administered orally, topically, transdermally, parenterally, by inhalation or spray or rectally or vaginally in dosage unit formulations containing conventional non-toxic pharmaceutically acceptable carriers, adjuvants and vehicles. The term parenteral as used herein includes subcutaneous, intravenous, intramuscular, intrathecal and like types of injection or infusion techniques. In addition, there is provided a pharmaceutical formulation comprising a compound provided herein and a pharmaceutically acceptable carrier. One or more compounds disclosed herein may be present in association with one or more non-toxic pharmaceutically acceptable carriers and/or diluents and/or adjuvants and if desired other active ingredients. The pharmaceutical compositions containing compounds disclosed herein may be in a form suitable for oral use. for example, as tablets, troches, lozenges, aqueous or oily suspensions, dispersible powders or granules, emulsion, hard or soft capsules, or syrups or elixirs. Compositions intended for oral use may be prepared according to any method known to the art for the manufacture of pharmaceutical compositions and such compositions may contain one or more agents selected from the group consisting of sweetening agents, flavoring agents, coloring agents and preserving agents in order to provide pharmaceutically elegant and palatable preparations. Tablets contain the active ingredient in admixture with non-toxic pharmaceutically acceptable excipients that are suitable for the manufacture of tablets. These excipients may be for example, inert diluents, such as
calcium carbonate, sodium carbonate, lactose, calcium phosphate or sodium phosphate; granulating and disintegrating agents, for example, corn starch, or alginic acid; binding agents, e.g., starch, gelatin or acacia, and lubricating agents, e.g., magnesium slearate, stearic acid or talc. The tablets may be uncoated or they may be coated by known techniques to delay disintegration and absorption in the gastrointestinal tract and thereby provide a sustained action over a longer period. For example, a time delay material such as glyceryl monosterate or glyceryl distearate may be employed.
Formulations for oral use may also be presented as hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent, for example, calcium carbonate, calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredient is mixed with water or an oil medium, e.g., peanut oil, liquid paraffin or olive oil.
Aqueous suspensions contain the active materials in admixture with excipients suitable for the manufacture of aqueous suspensions. Such excipients are suspending agents, e.g., sodium carboxymethylcellulose, methylcellulose, hydropropylmethylcellulose, sodium alginate, polyvinylpyrrolidone, gum tragacarsth and gum acacia; dispersing or wetting agents may be a naturally- occurring phosphatide, for example, lecithin, or condensation products of an alkyiene oxide with fatty acids, e.g., polyoxyethylene stearate, or condensation products of ethylene oxide with long chain aliphatic alcohols, e.g., heptadecaethyleneoxycetanol, or condensation products of ethylene oxide with partial esters derived from fatty acids and a hexitol such as polyoxyethylene sorbitol monooleate, or condensation products of ethylene oxide with partial esters derived from fatty acids and hexitol anhydrides, e.g., polyethylene sorbitan monooleate. The aqueous suspensions may also contain one or more preservatives, e.g., ethyl, or n-ρroρyi p-hydroxybenzoate, one or more coloring agents, one or more flavoring agents, and one or more sweetening agents, such as sucrose or saccharin.
Oily suspensions may be formulated by suspending the active ingredients in a vegetable oil, e.g., arachis oil, olive oil, sesame oil or coconut oil, or in a mineral oil such as liquid paraffin. The oily suspensions may contain a thickening agent, e.g., beeswax, hard paraffin or cetyl alcohol. Sweetening agents such as those set forth above, and flavoring agents may be added to provide palatable oral preparations. These compositions may be preserved by the addition of an anti-oxidant such as ascorbic acid.
Dispersible powders and granules suitable for preparation of an aqueous suspension by the addition of water provide the active ingredient in admixture with a dispersing or wetting agent, suspending agent and one or more preservatives. Suitable dispersing or wetting agents and suspending agents are exemplified by those already mentioned above. Additional excipients, e.g., sweetening, flavoring and coloring agents, may also be present.
Pharmaceutical compositions provided herein may also be in the form of oil-in-water emulsions. The oily phase may be a vegetable oil, e.g., olive oil or arachis oil or a mineral oil e.g., liquid paraffin or mixtures of these. Suitable emulsifying agents may be naturally-occurring gums, e.g., gum acacia or gum
tragacanth, naturally-occurring phosphatides, e.g.. soy bean, lecithin, and esters or partial esters derived from fatty acids and hexitol, anhydrides, e.g., sorbitan monoieate, and condensation products of the said partial esters with ethylene oxide, e.g., polyoxyethylene sorbitan monoieate. The emulsions may also contain sweetening and flavoring agents. Syrups and elixirs may be formulated with sweetening agents, e.g., glycerol, propylene glycol, sorbitol or sucrose. Such formulations may also contain a demulcent, a preservative and flavoring and coloring agents. The pharmaceutical compositions may be in the form of a sterile injectable aqueous or oleaginous suspension. This suspension may be formulated according to the known art using those suitable dispersing or wetting agents and suspending agents that have been mentioned above. The sterile injectable preparation may also be sterile injectable solution or suspension in a non-toxic parentally acceptable dilutent or solvent, e.g., as a solution in 1,3-butanediol. Among the acceptable vehicles and solvents that may be employed are water, Ringer's solution and isotonic sodium chloride solution. In addition, sterile, fixed oils are conventionally employed as a solvent or suspending medium. For this purpose any bland fixed oil may be employed including synthetic mono- or diglycerides. In addition, fatty acids such as oleic acid find use in the preparation of injectables.
Compounds disclosed herein may also be administered in the form of suppositories, e.g., for rectal administration of the drug. These compositions can be prepared by mixing the drug with a suitable non-irritating excipient that is solid at ordinary temperatures but liquid at body temperature and will therefore melt in the body to release the drug. Such materials include cocoa butter and polyethylene glycols.
Compounds disclosed herein may be administered parenterally in a sterile medium. The drug, depending on the vehicle and concentration used, can either be suspended or dissolved in the vehicle. Advantageously, one or more adjuvants such as preservatives, buffering agents, or local anesthetics can also be present in the vehicle. Dosage levels of the order of from about 0.05 mg to about 100 mg per kilogram of body weight per day are useful in the treatment of the above-indicated conditions, preferred dosages range from about 0.1 to about 30 mg per kg and more preferably from about 0.5 to about 5 mg per kg per subject per day. The amount of active ingredient that may be combined with the carrier materials to produce a single dosage form will vary depending upon the host treated and the particular mode of administration. Dosage unit forms will generally contain between from about 0.1 mg to about 750 mg of an active ingredient.
Frequency of dosage may also vary depending on the compound used and the particular disease treated. However, for treatment of most CNS and gastrointestinal disorders, a dosage regimen of four times daily, preferably three times daily, more preferably two times daily and most preferably once daily is contemplated. For the u'eatment of stress and depression a dosage regimen of 1 or 2 times daily is particularly preferred.
It will be understood, however, that the specific dose level for any particular patient will depend upon a variety of factors including the activity of the specific compound employed, the age, body weight, general health, sex, diet, time of administration, route of administration, and rate of excretion, drug combination (i.e. other drugs being used to treat the patient) and the severity of the particular disease undergoing therapy.
Preferred compounds disclosed herein will have certain pharmacological properties. Such properties include, but are not limited to oral bioavailability, such that the preferred oral dosage forms discussed above can provide therapeutically effective levels of the compound in vivo. Penetration of the blood brain barrier is necessary for most compounds used to treat CNS disorders, while low brain levels of compounds used to treat periphereal disorders are generally preferred.
Assays may be used to predict these desirable pharmacological properties. Assays used to predict bioavailability include transport across human intestinal cell monolayers, including Caco-2 ceil monolayers. Toxicity to cultured hepatocyctes may be used to predict compound toxicity, with non-toxic compounds being preferred. Penetration of the blood brain barrier of a compound in humans may be predicted from the brain levels of the compound in laboratosy animals given the compound, e.g., intravenously.
Percentage of serum protein binding may be predicted from albumin binding assays. Examples of such assays are described in a review by Oravcova, et al. (Journal of Chromatography B (1996) volume
677, pages 1-27). Preferred compounds exhibit reversible serum protein binding. Preferably this binding is less than 99%, more preferably less than 95%, even more preferably less than 90%, and most preferably less than 80%.
Frequency of administration is generally inversely proportional to the in vivo half-life of a compound. In vivo half-lives of compounds may be predicted from in vitro assays of microsomal half-life as described by Kuhnz and Gieschen (Drug Metabolism and Disposition, (1998) volume 26, pages 1120- 1 127). Preferred half lives are those allowing for a preferred frequency of administration.
As discussed above, preferred compounds disclosed herein exhibit activity in the standard in vitro CRF receptor binding assay specified in Example 7 below. References herein to '"standard in vitro receptor binding assay" are intended to refer to this standard assay protocol. Generally preferred compounds disclosed herein have an IC50 (half-maximal inhibitory concentration) of about 1 micromolar or less, still more preferably and IC50 of about 100 nanomolar or less even more preferably an IC50 of about 10 nanomolar or less or even 1 nanomolar or less in such a defined standard in vitro CRF receptor binding assay.
Preparation of Compounds
The compounds described herein, and salts thereof, may be synthesized using the methods described below, together with synthetic methods known in the art of synthetic organic chemistry, or variations thereon as appreciated by those skilled in the art. Preferred methods include, but are not limited to, those
described in the schemes set forth below. Those skilled in the art will recognize that the starting materials may be varied and additional steps employed in order to obtain particular compounds and salts.

The preparation of dibromopyrazine has been previously disclosed in WO 20051 15399. Scheme 1 outlines the synthesis of intermediates Ia, 2a and 3a utilized to prepare the compounds disclosed herein. Commercially available 2,6-dichloropyrazine (a) can undergo monosubstitution with nucleophilϊc nitrogen compounds to give pyrazine (b). Thus, 2,6-dichloropyraziπe (a) may react with an amine in solvents such as, but not limited to, dichloromethane, acetonitrile, THF, DMF, N-methylρyrrolidinone, methyl sulfoxide, methanol, ethanol, and isopropano! at temperatures ranging from 0 °C to the boiling point of the solvent, with or without the presence of a suitable transition metal catalyst such as, but not limited to, palladium(IΪ) acetate or tris(dibenzylideneacetone)dipaIladium(0), a ligand such as, but not limited to, l,l'-bis(diphenylphosphine)ferrocene, 2,2'-bis(diphenylphosphine)-l:l'-binaphthyl, dicyclohexyl(2-biρhenyl)phosphine, tricyclohexylphosphine, or tri-tert-butylphosphine, and a base such as sodium or potassium tert-butoxide in inert solvents such as, but not limited to, toluene, ethyleneglycol dimethyl ether, diglyme, DMF, or N-methylpyrrolidinone at temperatures ranging from ambient to 100 °C. Resulting monochloropyraziπc (b) can be further converted to compound (c) by displacing the halogen atom with a variety of nucleophiles (R2-[M]), in the presence or absence of a transition metal catalyst. The aforementioned nucleophiles may include sodium or potassium (thio)alkoxide. alkylamine, and organometallic reagent such as, but not limited to, alkyl Grignard reagents, alkylboronic acids, esters of alkylboronic acids, or alkylstannanes, or alkylaiuminum reagents. The aforementioned transition metal catalyst may be, but is not limited to, Ni(DPPP)C12, or palladium(Il) acetate or tris(dibenzylideneacetone)dipalladium(0), a ligand such as, but not limited to, 1,1'- bis(diphenylphosphine)ferrocene, 2,2'-bis(diphenylphosphine)-1,1 '-binaphthyl, dicyclohexyl(2- biphenyl)phosphine, tricyclohexylphosphine, or tri-tert-butyiphosphine, and a base such as sodium or
potassium ter/-butoxide in inert solvents such as, but not limited to, toluene, ethyleneglycol dimethyl ether, diglyme, DMF, or N-methylpyrrolidinone at temperatures ranging from ambient to 100 °C. Halogenation of compound (c) may be accomplished by a variety of methods known in the art, including treatment with N-chlorosuccinimide, bromine, N-bromosuccinimide, pyridinium tribromide, triphenylphosphine dibromide, iodine, and N-iodosuccinimide in solvents such as. but not limited to, dichloromethane, acetic acid, or methyl sulfoxide.
The secondary amine (d) may react with ally! halides or substituted allyl halides in a variety of solvents such as, but not limited to DMF or MeCN in the presence of base such as, but not limited, NaH or NaOH at room temperature or higher temperature. The alϊylated product (e) can undergo a transition metal catalyzed cyciization reaction (Heck reaction review: Chem. Rev. 2000, 100, 3009) in solvents such as, but not limited to DMF or MeCN at temperatures at room temperature or above in the presence of bases such as, but not limited to NaOH, potassium carbonate, triethylamine. Catalysts include, but are not limited to Pd(OAc)2 or Pd(PPh3)4. Other additives (see Jeffery, T., et.al Tet. Lett., 1998, 39, 5751) such as phase transfer catalysts, such as tetrabutylammonium salts and sodium formate may be added to give bromo derivative (Ia). The bromo derivative (Ia) may be converted to boronic acid derivative (2a) via a metallation reaction with alkyllitium reagents or metals such as lithium or magnesium in ethereal solvents such as, but not limited to THF or diethylether at temperatures below zero degrees celcius, followed by addition of trialkyl borate and hydrolysis to the boronic acid. Alternatively, the borate ester (3a) may be prepared from diborane reagents in the presence of palladium catalysts in solvents such as, but not limited to dioxane.

Commercially available dibromopyridine can undergo monosubstitution with nucleophilic nitrogen compounds such as amines to give the monobromopyridine. Thus, this reaction can be achieved in solvents such as toluene with catalytic palladium acetate and DPPF in the presence of potassium tert- butoxide at temperatures near or at the boiling point of the solvent. The subsequent reaction steps follow a procedure similar to the descriptions given in scheme 1.

The bromine in the bromochloro bicycle shown in Scheme 3 can be selectively functionalized by metalation with a reagent such as alkyllithium and reacted with an electrophile. The general procedure for the other steps in the scheme above is similar to the description given in Scheme I .

The bromine in the bromochloro bicycle shown in Scheme 4 can be selectively functionalized by metalation with a reagent such as alkyllithium and reacted with an electrophile. The general procedure for the other steps in the scheme above is similar to the description given in Scheme 1.

The halogen atom on position ortho to the amino group in the pyrazine nucleus of compound 5 can be selectively displaced by an amine. Thus, compound 6 can be prepared from 5 and ammonia in the presence of a suitable transition metal catalyst such as but not limited to copper (0), palladium(II) acetate or tris(dibenzylideneacetone)dipalladium(O), a ligand such as but not limited to 1,1'- bis(diphenylphosphine)ferrocene, 2,2r-bis(diphenylphosphine)-l , 1 '-binaphthyl, dicyclohexyl(2- biphenyl)phosphine, tricyclohexylphosphine, or tri-tert-butylphosphine, in inert solvents such as, but not limited to, ethanol, methanol, toluene, ethyleneglycol dimethyl ether, diglyme, DMF, or N- methylpyrrolidinone at temperatures ranging from ambient to 100 0C. Cyclization of the diaminopyrazine 6 to compound Ie can be accomplished by treatment with a number of reagents such as dimethoxymethylacetate or trialkylorthoesters such as triethylorthoformate or trimethylortoacetate in the
presence or not of a suitable acidic catalyst such as, but not limited to, p-toluenesulfonic acid or sulfuric acid, in a solvent such as toluene, xylene, DMF, NMP3 or methyl sulfoxide at temperatures ranging from O °C to 100 °C. Preparation of the boronic acid or boronale 2e would follow the general procedure described in Scheme 1.
Scheme 6
1f M) OR11J2
 2f
2f
From commercially available starting material, where R2=methyl, the general procedure for the scheme 6 is similar to the description given in Scheme 5.
Scheme 7
B ][ I ^ ϊ . NH31 CU1 EtOH-


The reaction steps from aminodibromopyrimidine to intermediates Ih and 2h are similar to the general procedure described in Scheme 1.
Scheme 9

Commercially available 2,6-dichioro-3-nitropyrimidine can undergo monosubstitution with allylamines in solvents such as, but not limited to DMF in the presence of base such as potassium carbonate. The nitro group can be reduced selectively with reagents such as stannous(ll)ch!oride in acidic media. The amine can be converted to a halogen such as bromine by diazotization with a reagent such as sodium nitrite in an acid such as HBr. The general procedure for transition metal catalyzed cyclization followed by conversion of the chloride to the boronic acid or borate 2i is described in Scheme I .
Scheme 10

1j 2J
An analogous procedure for the above Scheme 10 has been described in WO2005028480.
Scheme ] 1
Synthesis of compounds of Formulas A, B, C, D, E, F, G5 H, i, J, K, L, M5 N, O, P, Q3 R, S, or T by transition metal catalysis






Where R2-Hu is selected from:
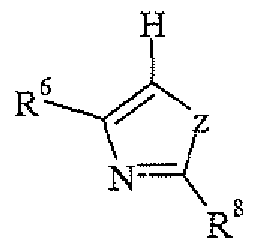
R , R , R , R , U, and z are defined supra.
Scheme 12

Scheme 13

Scheme 14 base / solvent

Table I











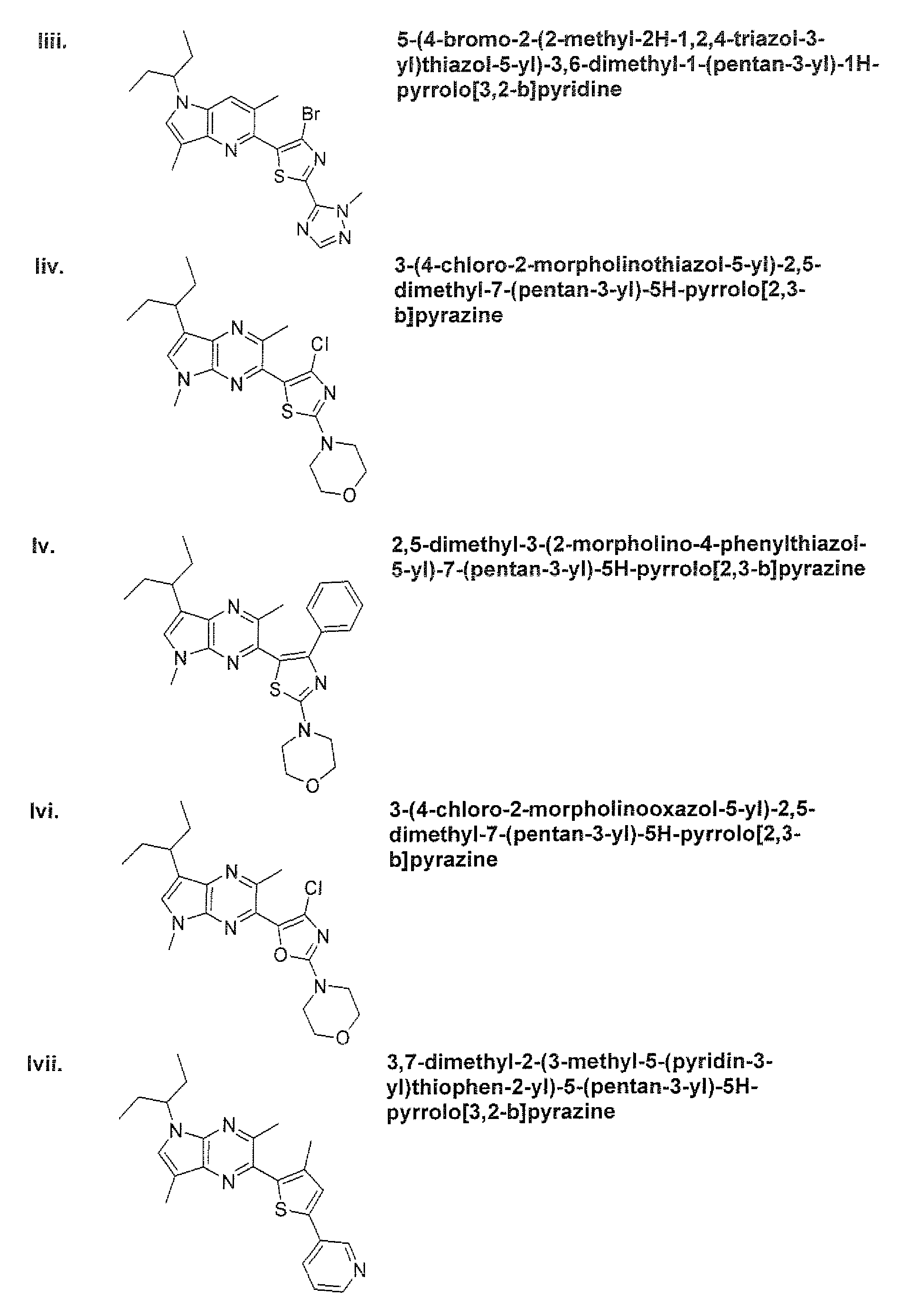










CXV.
CXVI.

Table II
Structure Name Mass K1 <
Compound M+H 50OnM

2-[2-{4-chϊoro-2- morpholfn-4-yl-1 ,3- thιazol-5-yl)-3,7- dιmethyl-5H- b) pyrrolo[2,3-b]pyraziπ-5- 422 02 yl]butan-1 -ol
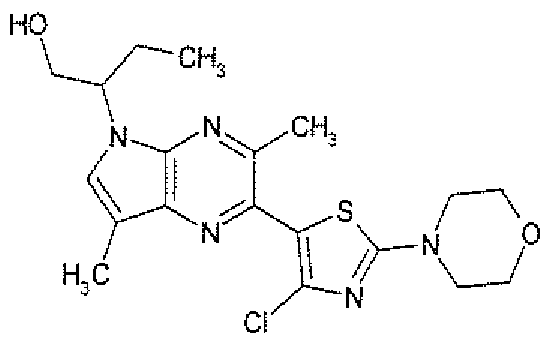
 2-(5-bromo-3-methyl-2- thienyl)-5-(1- ethylpropyl)-3,7- dimethyl-5H- pyrrolo[2,3-b]pyra2Jne 391.99
2-(5-bromo-3-methyl-2- thienyl)-5-(1- ethylpropyl)-3,7- dimethyl-5H- pyrrolo[2,3-b]pyra2Jne 391.99
1 -{5-[5-(1 -ethylpropyl)-
3,7-dimethyl-5H- pyrrolo[2,3-b]pyrazin-2- yi]-4-methy!-2-
J) thienyl}ethanone 356.06
5-[5-(1 -ethyipropyl)-
3,7-dimethyl-5H- pyrrolo[2,3-b]pyrazin-2- yl]-4-methyithioρhene- k) 2-carboxylic acid 358.04
5-(1-ethyipropyl)-3,7- dimethyl-2-[3-methyl-5-
(methylsulfonyl)-2- thienyi]-5H-pyrroio[2,3- 392.04 b]pyrazine
5-(1-ethylproρyl)-3,7- dimethyl-2-[3-methyi-5-
(morpholin-4- yicarbonyl)-2-thienyl]- m) 5H-pyrrolα[2,3- 427.10 bjpyrazine
1 -{5-[5-(1 -ethylpropyl)-
3,7-dimethy!-5H- pyrro[o[2,3-b]pyrazin-2- yi]-4-methyl-2- n) thienyl}ethanol 358.05

 {5-[5-(1-ethylpropyl}-
{5-[5-(1-ethylpropyl}-
3,7-dimethyl-5H- pyrroio[2,3-b]pyrazin-2- yl]-4-methyl-2-
V) thienyl}methano! 344.04
5-(1-ethylpropyl)-2-[5-
(methoxymethyl)-3- methyi-2-thienyl]-3,7- w) dimethyl-5H- 358.05 pyrrolo[2,3-b]pyrazine


385.05
2-
371.03


CC) ethylpropyl)-2,5- 420.08 dimethyi-5H- pyrrofoJ2,3-b]pyrazine
1 -{5-[7-(1-ethyϊpropyf)-
2,5-dimethyl-SH- pyrrolo[2,3-b]pyrazin-3- dd) y!j-4-methyf~2- 356.03 thienyl}ethanone
1 -{5-[7-( 1 -ethylpropy!)- 2.5-dimethyi-5H- pyrrolo[2 , 3-b]py razi n-3- ee) yl]-4-methyl-2- 358.05 thienyl}ethanol
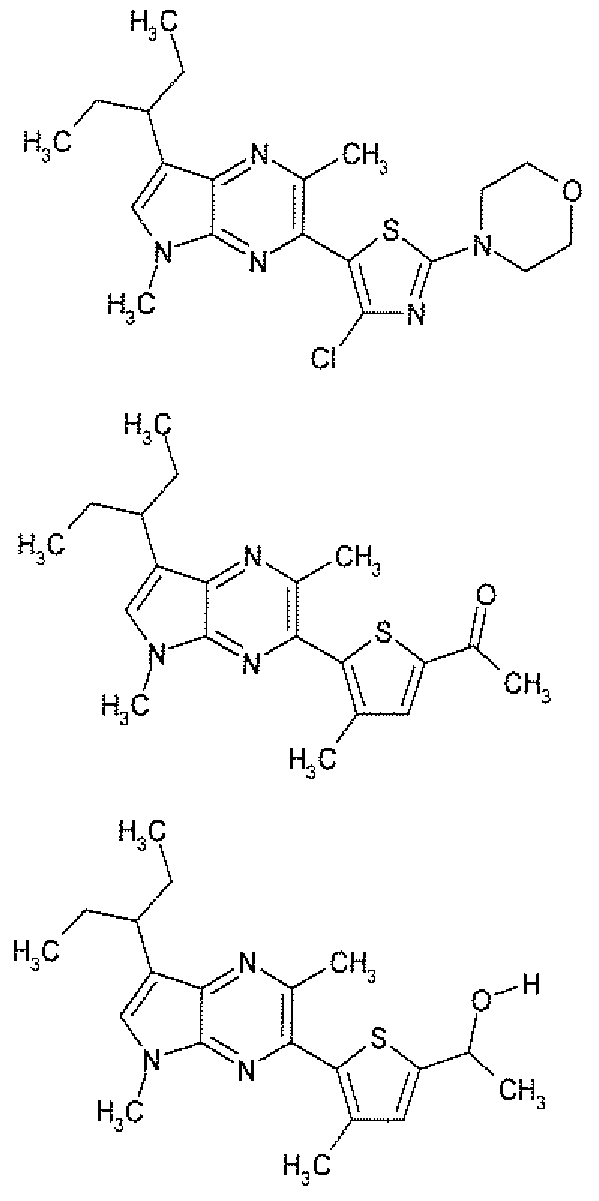
7-(1-ethylpropyl)-2,5- dimethyl-3-[3-methyl-5-
(methylsuifonyl)-2- thienyl]-5H-pyrrolo[2,3- b]pyrazine 392.01


4-{5-|7-(1-ethylpropyl)- 2,5-d'imethyl-5H- pyrrolo[2,3-b]pyrazin-3- yl]-4-methyl-2- hh) thieny[}tetrahydro-2H- 414.08 pyran-4-of
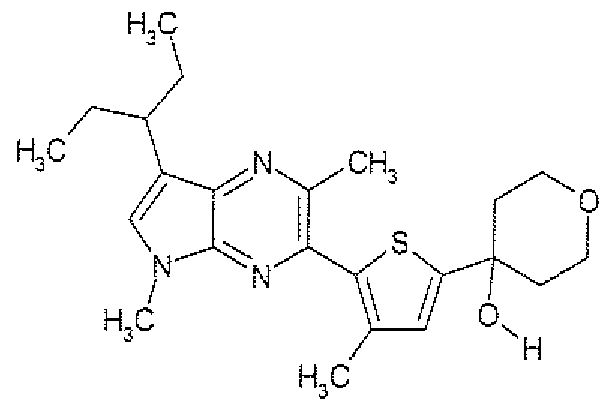










EXAMPLES
The following examples serve Io further illustrate methods for preparing compounds and salts disclosed herein. Additional synthetic methods relating to the synthesis of compounds and salts disclosed herein have been published in copending United States Patent Application publication 200501 13379, US Serial No. 10/933,834, filed September 3, 2004, entitled "Heteroaryl fused pyridines, pyrazines and pyrimidines as CRFl receptor ligands" in the disclosure of the Examples (through Example 49) in numbered paragraphs 0284-0863, pages 17-90, which disclosure is hereby incorporated by reference for its teachings of such synthetic methods.
10 Example 1: Preparation of:
3.7-dimethyl-2-(3-methyl-5-('2-methyl-2I-I-1.2.4-triazol-3-vi)thiophen-2-v])-5-(pentan-3-yπ-5H- pyiτolor3,2-blpyrazine

Example 2: Preparation of:
2-(4-Chloro-2-mθφholinothiazol-5-yl)-3.7-dimethyl-5-fpentan-3-yl)-5H-pyrrolof3.2-blpyrazine
Part A: 2,4-Dichloro-l,3-thiazole
CI'~~XN CI
Reflux a mixture of 2,4-thiazolidinedione (12.5 g, 107 mmol), phosphorousoxychloride (70 mL). and pyridine (8.5 mL) for 3 hrs and then cool to room temperature. Remove volatiles under vacuo, pour the residue into ice/water (30 mL) slowly, extract the resulting mixture with dichloromethane (2 x 50 mL) and dry combined organic extracts with MgSO4 Filter the mixture and concentrate in vacuo. Purify the crude product by silica gel column chromatography to afford desired product as colorless crystals. 1H NMR (400 MHz, CDCl3) 7.00 (1 H, s).
Part B: 4-(4-Chloro-l,3-thiazol-2-yl)morpholine

In a sealed tube dissolve 2,4-dichloro-l,3-thiazole (1,00 g, 6.49 mmol), morpholine (1.13 g, 32.99 mmol) in dry THF (4 mL) and then add Cs2CO3 (3.17 g, 9.74 mmol). Heat the resultant mixture at 1 10 °C for 3 hrs. Concentrate the reaction mixture in vacuo and treat the residue with dichloromethane (30 mL). Filter the insoluble material and evaporate the filtrate in vacuo to afford off-white solid. !H NMR (400 MHz, CDC13) 6.33 (1 H, s), 3.80 (4 H, t, J = 9 Hz), 3.45 (4 H1 1, J = 9 Hz).
Part C: Title compound:

(R)-2-[2-(4-chϊoro-2-moφho]in-4»yl-I,3-thiazo!-5-yl)-3,7-dimethyl-5H-pyrro]o[2,3-b]pyraziπ-5-yl]butan- l-ol

Example 4: Preparation of: 2,5-Dimethyl-3-(3-methyl-5-(methyl5ulfonyl)lhiophen-2-yl)-7-(pentan-3-vl)-5H-pyrro1or2.3-51pyi'azine

A Teflon screw-cap vial is charged with 60 mg (0.15 mmol, 1.0 equiv) of 3-(5-bromo-3-melhylthiophen-
2-yl)- 2,5-dimethyl-7-(pentan-3-yl)-5H-pyrroIo[2,3-A]pyrazine and 0.5 mL of DMSO. To the solution is added 4.2 mg (90% purity, 0.0075 mmol, 0.05 equiv) of (CuOTf)rbenzene complex, 22 mg ( 85% purity, 0.18 mmol, 1 .2 equiv) of sodium methanesulfmate and 1.7 mg (0.015 mmol, 0.1 equiv) of trans- 1,2- diaminocyclohexane. The reaction via! is evacuated and purged with nitrogen three times before being tightly capped and heated overnight at 1 10 °C. The reaction mixture is cooled to rt, diluted with water and extracted with EtOAc (3 X 10 mL). The combined organic extracts are washed with brine, dried over Na2SO4 and concentrated under reduced pressure. The resulting residue is purified by silica gel column chromatography using initially 10% EtOAc in hexanes and then 30% EtOAc in hexanes to give the desired compound as a yellow oil. MS = 392.01 (M +1). !Η NMR (400 MHz, CDC13) δ 7.58 (IH, s), 7.26 (IH, s), 3.85 (3H, s) 3.24 (3H, s), 2.96 (IH, m), 2.58 (3H, s), 2.19 (3H, s), 1.80-1.84 (4H, m), 0.88 (6H, t, J= 7.6 Hz).
Example 5: Preparation of:
5-ri-fMethoxymethyi)propyl]-3J-diirιethyl-2-f4-methyl-2-morpholin-4-yi-l ,3-thiazol-5-vπ-3.S-dihydro-
4H-pyrrolor312-d"jpyrimidin-4-one
Part A: Ethyl N-[T-(methoxymethyl)propyllglycinate

Diisopropyiethylamine (33.8 mL, 193.8 mmol) and ethyl bromoacetate (10.7 mL, 96.9 mmol) are added sequentially to a solution of 2-amino-I-methoxybutane (10 g, 96.9 mmol) in 100 mL Of CH3CN at RT. The reaction mixture is stirred for 4 h, and then concentrated in vacuo. The residue is partitioned between CH2CI2 and water (60 mL each), and the layers are separated. The aqueous layer is extracted twice more with 50 mL of CH2C12. The combined organic extracts are dried (Na2SO4), filtered, and evaporated in vacuo to give the title compound as a yellow oil, which needed no further purification. Mass spec. (190.13, M+H). Part B: 2-Methyl-3-oxopropanenitrile

A solution of propionitrile (17.1 mL, 240 mmol), ethyl formate (39 mL, 480 mmol), and absolute ethanol (2.0 mL) in 30 mL of anhydrous ether is added dropwise over about 30 mln. to a well-stirred suspension of sodium hydride (9.6 g, 240 mmol, 60% in mineral oil) in 100 mL of ether at RT. After the addition, the milky-white suspension is stirred for 16 h. The ether and excess ethyl formate are removed in vacuo (bath temp. < 30 °C), and 50 ml of water are cautiously added to the resulting solid. The mixture is then neutralized to pH= 4-5 with acetic acid and extracted with CH3Cl2 (3 x). The combined CH2C12 extracts are dried (Na2SO-O, filtered, and evaporated m vacuo (<30 °C) to give the title compound as a yellow/brown oil. This material is used without any further purification. Part C: Ethyl N-f(lZ)-2-cyanoptOp-l-en-l-y]]-N-ri-(methoxymethyl)propyl]glycinate

To a solution of 2-methyl-3-oxoρropanenitrile (14 g, 169 mmol) in 70 mL of methanol is added ethyl N-
[l-(methoxymethyl)propyl]glycinate (16 g, 84.5 mmol) and trifluoroacetic acid (1.0 mL), followed by enough powdered NaHCOj to neutralize the mixture. After stirring for 22 h at RT, the solution is evaporated to dryness, and the residue is partitioned between CH2CI2 and water (70 mL each). The organic layer is washed with water (40 mL) and brine (40 mL), dried (Na2SO4). filtered, and evaporated
in vacuo to give a golden oil. Purification by silica gel column chromatography (gradient from CH2C12 to 1 % MeOH/CH2C12) affords the title compound as yellow oil. Mass spec. (255.14, M+H).


Part F: Ethyl 4-methyi-2-morpholin-4-yl-1.3-thiazole-5-carboxylate

A mixture of ethyl 2-bromo-4-methyl-ls3-thiazole-5-carboxylate (950 mg, 3.80 mmol), morpholine (0.5 mL, 5.70 mmol), and potassium carbonate (1.1 g, 7.60 mmol) in 4.0 ml of NMP is heated to 130 °C for
1.5 h. The reaction is cooled to RT, and the NMP is removed in vacuo. The residue is partitioned between CH1CI2 and water (20 ml each), and the layers are separated. The aqueous layer is extracted once more with 20 ml of CH2CI2, and the combined organic extracts are dried (Na2SQ4), filtered, and evaporated in vacuo to give the title compound as an off-white solid. Mass spec. (257.06, M+H).
Part G: 4-Methyl-2-morphoIin-4-yl-1.3-thiazole-5-carboxyIic acid

A mixture of ethyl 4-methyl-2-moφholin-4-yl-l,3-thiazole-5-carboxylate (980 mg, 3.82 mmol) and 5 mL of ION NaOH in 10 mL of ethanol is stirred at 50 °C for 1 h. After cooling, the ethanol is removed in vacuo, and the remaining aqueous residue is diluted with water, cooled to 0 °C, and acidified to pH=5-6 with cone. HC1. The resulting solid is collected by vacuum filtration and dried in vacuo to give the title compound as a while soϋd. Mass spec. (229.01, M+H).
Part H: N-{ I-ri-(Methoxyniethyl)ρroρyl]-4-methyl-2-(methylcarbamoyl)-lH-pyrrol-3-yl}-4-methyl-2- moφholin-4-yl- 1 ,3-thiazole-5-carboxamide


Phosphorous oxychloride (1.0 mL) is added to N-{ l-[l -(methoxymethyl)propyl]-4-methyl-2- (methyicarbamoyi>ΪH-ρyrrol-3-yl}-4-methyl-2-moφholiri-4-yl-l,3-thiazole-5-carboxamide (100 mg of crude material, ~0.21 mmol). and the reaction mixture is heated to 1 10 °C for 1.5 h. After cooling to RT, the excess POCI3 is removed in vacuo, and ice water (10 mL) is added to the residue. The mixture is then
extracted with CH2C12 (IO mL), and the organic extract is washed with sat. NaHCO3 (IO mL) and brine (10 mL), dried (Na2SO4), filtered, and evaporated in vacuo to give a brown oily residue. Purification by silica gel column chromatography (gradient from 80% EtOAc/Hex to EtOAc) affords the title compound as a giass-like, brown solid. 1H NMR (300 MHz, DMSO-d6) 7.40 (IH, s), 5.3 (IH, bs), 3.70 (5H, m), 3.55 (IH, m), 3.39 (7H, m), 3.18 (3H, s), 2.10 (3H, s), 2.07 (3 H, s), 1.77 (2H, m), 0.69 (3H5 t, J 7.2). Mass spec. (432.10, M+H). Example 6: Assay for CRF receptor functional activity
As discussed above, the following assay is referred to herein and in the claims as a standard in vitro CRF receptor functional assay. A chemiluminescent ELISA system (cAMP-Screen®, Applied Biosystems, Bedford, MA) is used according to the manufacturers instructions to quantify the levels of
3',5'-cycIic AMP (cAMP) in extracts prepared from the human neuroblastoma cell line 1MR32, which endogenously expresses the human CRFl receptor. IMR32 cells (ΛTCC CCL 127) are grown to confluence in one or more T- 175 flasks, each flask is split evenly into the wells of two pre-coated 96-weil cAMP-Screen® assay piates prior to the test treatment and ELlSA, each plate is then incubated for 16 hours and assayed. Culture is in ΪMR-32 Medium: EMEM w/Earle's BSS (JRH Biosciences, Cat# 5141 1 ) pius, as supplements, 2mM L-GIutamine, 10% Fetal Bovine Serum, 25mM HEPES (pH 7.2), ImM
Sodium Pyruvate and Non-Essential Amino Acids (JRH Biosciences, Cat# 58572) for 16-20 hours. For antagonist mode characterization, compounds are used to dose dependently antagonize the cAMP production response of CRF. An EC6O response of CRF is used to stimulate 1MR32 cells to produce cAMP. A co-incubation strategy is used to determine the potency of compounds to reverse the CRF stimulated cAMP response. IC50 and Kt values are calculated using non-linear regression analysis. For agonist mode characterization, compounds are used alone to treat the cells.
Example 7: Assay for CRF Receptor Binding Activity
As discussed above, the following assay is referred to herein and in the claims as a standard in vitro CRF receptor binding assay. The CRF receptor binding is performed using a modified version of the assay described by Grigoriadis and De Souza {Methods in Neurosciences, Vol. 5, J 991). IMR-32 human neuroblastoma cells, a cell-line that naturally expresses the CRFI receptor, are grown in IMR-32 Medium (see preceeding Example). The cells are grown to confluence and split three times (all splits and harvest are carried out using NO-ZYME — JRH Biosciences. Cat# 59226). The cells are first split 1 :2, incubated for 3 days and split 1 :3, and finally incubated for 4 days and split 1 :5. The cells are then incubated for an additional 4 days before being differentiated by treatment with 5-bromo-2'deoxyuridine (BrdU, Sigma, Cat# B9285). The medium is replaced every 3-4 days with IMR-32 medium w/2.5uM BrdU and the cells are harvested after 10 days of BrdU treatment and washed with calcium and magnesium-free PBS. . To prepare receptor containing membranes, cells are homogenized in wash buffer (50 mM Tris
HC1, 10 mM MgC12, 2 mM EGTA, pH 7.4) and centrifuged at 48,000 x g for 10 minutes at 4°C. The
pellet is re-suspended in wash buffer and the homogenization and centrifugation steps are performed two additional times.
Membrane pellets (containing CRF receptors) are re-suspended in 50 mM Tris buffer pH 7.7 containing 10 mM MgC12 and 2 mM EDTA and centrifuged for 10 minutes at 48,00Og, Membranes are washed again and brought to a final concentration of 1500 ug/ml in binding buffer (Tris buffer above with 0.1 % BSA, 15 mM bacitracin and 0.01 mg/ml aprotinin.). For the binding assay, 100 μL of the membrane preparation are added to 96 well microtube plates containing 100 μL of 125ϊ-sauvagine (SA 2200 Ci/mmol, final concentration of 100 pM) and 50 μL of test compound. Binding is carried out at room temperature for 2 hours. Plates are then harvested on a BRANDEL 96 well cell harvester and filters are counted for gamma emissions on a Wallac 1205 BETAPLATE liquid scintillation counter. Nonspecific binding is defined by 1 mM cold CRF. IC50 values are calculated with the non-linear curve fitting program RS/ 1 (BBN Software Products Corp., Cambridge, MA). The binding affinity (expressed as IC50 or K, value) for those compounds and salts of Table I or Table II that have been tested generally ranges from about 0.5 nanomolar to about 10 micromolar. Preferred compounds or salts of Formulas A, B, C, D, E, F, G, H, I J, K, L, M, N, O, P, Q, R, S, or T exhibit IC50 values of less than or equal to 1.5 micromolar, more preferred compounds or salts exhibit IC50 or Kj values of less than 500 nanomolar, still more preferred compounds or salts exhibit IC50 or K1 values of less than 100 nanomolar, and most preferred compounds or salts are those othat exhibit IC50 or K1 values of less than 10 nanomolar. Preferred of these compounds or salts are those of Table I or Table II . Example 8: Evaluation of mechanism-based inhibition of Cytochrome P45Θ (CYP) 1A2, 2C8, 2C9, 2C19, 2D6 and 3A4 in human liver microsomes
Compound or salt at 1, 2.5, 5, 10, 25, and less preferably 50 μM are preincubated with pooled human liver microsomes 1) in the presence and 2) in the absence of NADPH (final cone. 1 mM) in a shaking water bath at 37°C for 30 minutes. After 10-fold dilution of the preincubation mixture with 0.1M phosphate buffer, pH 7.4, containing a selective CYP probe substrate and NADPH (final cone. 1 mM), residual enzyme activity is measured and the extent of the inhibition shift between the two treatments is evaluated. A concentration-dependent and statistically significant decrease in each particular CYP activity in the presence vs. absence of NADPH indicates mechanism-based inhibition of that CYP isozyme. Vehicle and known selective mechanism-based inhibitors of each CYP isozyme are also separartely incubated in parallel with the compound or salt to serve as negative and positive controls. Positive controls include furafylline (CYP1A2), gemfibrozil glucuronide, or, less preferably, phenelzine (CYP2C8), tienilic acid (CYP2C9), ticlopidine (CYP2C19), paroxetine (CYP2D6), and ribefradil (CYP3A4). For all CYPs evaluated, the activity tests are conducted under linear conditions as follows:
Microsome Cone. Microsome Cone, sn „ ■. .« .. - - I 1- .-
- „ . . ^. * x- .. . ^ L- O t- X . Probe Substrate Incubation
Enzyme in Preincubation Activity Assay Probe Substrate _ , ... _. , . .
3 , ,, , , i, Cone. (uM) Time mm
(mg/ml rng/m
CYP 1A2 1 0 1 Phenacetin 35 30
CYP2C8 0 1 0 01 Amodiaquine 2 5 10
CYP2C9 5 0 5 Tolbutamide 400 10
CYP2C19 2 0 2 S-Mephenytoiπ 105 40
CYP2D6 1 0.1 Dextromethorphan 15 5
CYP3A4 2 0 2 Testosterone 250 10
For each assay, the formation of the CYP specific metabolites is monitored and quantified by LC- MS (dextrorphan), LC-MS/MS (4'-hydroxytolbutamide, desethylamodiaquine,4- acetamidophenol, 4- hydroxy-S-mephenytoin), or HPLC (6β-hydroxytestosterone). For each isozyme tested, a mechanism based inhibitor ot the isozyme will show concentration dependence and a statistically significant decrease (student's t-test: p<0.05) in activity after 30 minutes preincubation with NADPH as compared to preincubation in the absence of NADPH. Absence of mechanism-based inhibition is demonstrated for each isozyme when no statistically significant changes in one or more (preferably all) of CYPI A2, CYP2C8, CYP2C9, CYP2C19, CYP2D6 and CYP3A4 activities are observed when human liver microsomes are preincubated with a preferred compound or salt disclosed herein at concentrations of 1. 2.5, 5, 10, 25, and 50 μM with vs. without NADPH.
Example 9: Assay for emesis in dogs
Preferred compounds or salts disclosed herein do not cause nausea or vomiting when administered to a patient in a therapeutically effective amount. A convenient measure of this property is absence of emesis in more than I out of six dogs within one, preferably 3, or less preferably 6 hours of administration of a dose selected from 1. 30, 100, 300, and 1000mg/kg of body weight. Such absence of emesis is determined as follows: Doses of compound or salt are administered by oral intubation using a flexible tube fitted onto a syringe and utilizing a flush of approximately 10 mL of distilled water. Dogs are fed Certified Canine Diet, No. 5007 (PMl Nutrition International, St. Louis, MO) ad libitum in 400 gram amounts for approximately 4 hours daily. Food is presented at approximately the same time each day. Food is provided 1-2 h prior to dosing. Water is provided ad libitum, via automated watering system. Observations are made 4 times after compound or salt administration at 30 minutes. 1, 3 and 6 hours post dosing. Observed emesis events are recorded during the observation period. Example 10: Preparation of radiolabeled probe compounds The compounds and salts disclosed herein are prepared as radiolabeled probes by carrying out their synthesis using precursors comprising at least one atom that is a radioisotope. The radioisotope is preferably selected from of at least one of carbon (preferably 14C). hydrogen (preferably 3H), sulfur (preferably 3:?S), or iodine (preferably 123I). Such radiolabeled probes are conveniently synthesized by a radioisotope supplier specializing in custom synthesis of radiolabeled probe compounds. Such suppliers
include Amersham Corporation, Arlington Heights, IL; Cambridge Isotope Laboratories, Inc. Andover, MA; SRJ Internationa!, Menlo Park, CA; Wizard Laboratories, West Sacramento, CA; CheraSyn Laboratories, Lexena, KS; American Radiolabeled Chemicals, Inc., St. Louis, MO; and Moravek Biochemicals Inc., Brea, CA. Tritium labeled probe compounds are also conveniently prepared catalytically via platinum- catalyzed exchange in tritiated acetic acid, acid-cataϊyzed exchange in tritiated trifiuoroacetic acid, or heterogeneous-catalyzed exchange with tritium gas. Such preparations are also conveniently carried out as a custom radiolabeling by any of the suppliers listed in the preceding paragraph using the unlabeled compound as substrate. In addition, certain precursors may be subjected to tritium-halogen exchange with tritium gas, tritium gas reduction of unsaturated bonds, or reduction using sodium borotritide, as appropriate. Example 11: Receptor autoradiography
Receptor autoradiography (receptor mapping) is carried out in vitro as described by Kuhar in sections 8.1.1 to 8.1.9 of Current Protocols in Pharmacology (1998) John Wiley & Sons, New York, using radiolabeled compounds provided herein prepared as described in the preceeding schemes and the preceding Example. Example 12: Microsomal in vitro half-life
Compound half-life values (t] <2 values) may be determined via the following standard liver microsomal half-life assay. Pooled Human liver microsomes are obtained from XenoTech LLC, 3800 Cambridge St. Kansas's City, Kansas, 66103 (catalog # H0610). Such liver microsomes may also be obtained from In Vitro Technologies, 1450 South Rolling Road, Baltamore, MD 21227, or from Tissue Transformation Technologies, Edison Corporate Center, 175 May Street, Suite 600. Edison, NJ 08837. Reactions are preformed as follows: Reagents: Phosphate buffer: 19 mL 0.1 M NaH2PO4, 81 mL 0.1 Na2HPO4, adjusted to pH 7.4 with H3PO4. CoFactor Mixture: 16.2 nig NADP, 45.4 mg Glucose-6-phosphate in 4 mL 100 mM MgCl2. Glucose-6-phosphate dehydrogenase: 214.3 μL glucose-6-phosphate dehydrogenase suspension (Boehringer-Manheim catalog no. 0737224, distributed by Roche Molecular Biochemicals, 9115 Hague Road, P.O. Box 50414, Indianapolis, IN 46250) is diluted into 1285.7 μL distilled water. Starting Reaction Mixture: 3 mL CoFactor Mixture, 1.2 mL Glucose-6-phosphate dehydrogenase. Reaction:
6 test reactions are prepared, each containing 25 μL microsomes, 5 μL of a 100 uM solution of test compound, and 399 μL 0.1 M phosphate buffer. A seventh reaction is prepared as a positive control containing 25 μL microsomes, 399 μL 0.1 M phosphate buffer, and 5 μL of a 100 uM solution of a compound with known metabolic properties (e.g. diazepam or clozepine). Reactions are preincubated at 39°C for 10 minutes. 71 μL Starting Reaction Mixture is added to 5 of the 6 test reactions and to the
positive control, 71 μL 100 mM MgCl2 is added to the sixth test reaction, which is used as a negative control. At each time point (0, 1 , 3, 5, and 10 minutes) 75 μL of each reaction mix is pipetted into a well of a 96-we!l deep-well plate containing 75 μL ice-cold acetonitrile. Samples are vortexed and centrifuged 10 minutes at 3500 rpm (SORVAL T 6000D centrifuge, HlOOOB rotor). 75 μL of supernatant from each reaction is transferred to a well of a 96-weIl plate containing 150 μL of a 0.5 uM solution of a compound with a known LCMS profile (internal standard) per well. LCMS analysis of each sample is carried out and the amount of unmetabolized test compound is measured as AUC7 compound concentration vs time is plotted, and the ti« value of the test compound is extrapolated.
Preferred compounds exhibit in vitro t]/2 values of greater than 10 minutes and less than 4 hours. Most preferred compounds exhibit in vitro t)/2 values of between 30 minutes and 1 hour in human liver microsomes.
Example 13: Preparation of:
5-π-Ethylpropyπ-3.7-dimethyl-2-f3-methvl-5-morpholin-4-yl-2-thienyl)-5H-pyrrolor2.3-b1pvrazine
Part A: N-Alivi-3.5-dibromo-N-(T -ethvlpropyl)-6-methvlpvrazin-2 -amine

A solution of 3,5-dibromo-N-(l-ethylpropyl)-6-methylpyrazin-2-arnine (25 g, 74.2 mmol; prepared according to patent WO 2004/018437.) in 100 mL of THF is treated with sodium hydride (3.56 g, 89.0 mmol, 60% in mineral oil) by portionwise addition. A mixture of tetrabutylammonium iodide (2.74 g, 7.42 mmoi) and 18-crown-6 (1.96 g, 7.42 mmol) is then added, followed by dropwise addition of allylbromide (12,6 mL, 148.4 mmol). The reaction mixture is stirred at RT for 48 h. Water (50-60 mL) is added, and the THF is removed in vacuo. The remaining aqueous residue is extracted with CH2CI2 (2 x 100 mL), and the combined CH2C12 extracts are washed with brine (100 mL), dried (Na2Sθ4), filtered, and evaporated in vacuo to give a dark brown oil. This oil is flushed through a plug of silica gel eluting with 3 %EtO Ac/Hex to give the title compound as brown oil. Mass spec. (377.91, M+H). Pai t B. 2-Brorno-3.7-uimethyl-5-(pentan-3 -yl r5H-ρyrroloP ,2-bipyrazine

Allyl-(3,5-dibromo-6-methyl-pyrazin-2-yl)-(l-ethyi-piOpyI)-amine (18 g, 47 mmol) is taken into anhydrous DMF (10OmL). Pd(OAc)2 (430 mg. 0.04 eq.), N(C2Hj)3 (16 mL, 2.4 eq.), tetrabutylammonium bromide (2.3 g, 0, 15 eq.), sodium foπnate (0.81 g, 0.25 eq.) are added . The resulting mixture is heated at 45 °C for 10 hours. Reaction mixture is diluted with 25% ethyl acetate/hexanes and washed with water, brine and dried with Na2SO4. The crude product is purified by silica gel column chromatograph to give
desired product. 'HNMR (CDC13, ppm): 7.19(s,lH); 4.4-4.6(ms IH); 2.72(s, 3H); 2.38(s, 3H); 2.7-2.97(m,
4H); 0.75(t, J=12Hz, 9.6H). LC/MS:[M+l]+ : 295.96.
Part C: 3.7-Dimetiivl-2-(3-i-nethvlthiophen-2-vl)-5-fpentan-3-vl)-5H-ρvrroIor312-blpvi-azine


3,7-Dimethyl-2-(3-methylthiophen-2-yI)-5-(pentan-3-yI)-5H-pyrrolo[3,2-b]pyra2ine (395 mg, 1.26 mmoi) is taken into anhydrous THF (5 mL) and cooled to -78 °C. n-Butyilithium (1.6 M hexanes, 0.83 mL) is added dropwise and stirred for Ihour. Carbon tetrabromide (626 mg, 1.89 mmoi) is added dropwise as a solution of THF (3 mL) and stirred at -78 °C for 1 hour. Reaction is quenched by carefully adding water.
The resulting mixture is extracted with ethyl acetate and dried with Na2SO4. Crude product is purified by silica gel column chromatography with 3% ethyl acetae/hexanes to give product as oil. 1H NMR (CDCl3): 7.26(s, IH); 6.90(s, IH); 4.5-4.7(m, IH); 2.53(s, 3H); 2.38(s, 3H); 2.08(s, 3H); 1.8-2.0(m, 4H); 0.77(t,
J-6.4Hz, 6H). LC/MS:[M+i;T: 391.99
Part E: Title compound

A mixture of 2-(5-bromo-3-methyl-2-thienyl)-5-( 1 -ethylpropyl)-3,7-dimethyl-5H-pyrrolo[2,3-b]pyrazine (130 mg), morpholine (40 mg), CuI/Cu (15mg/15mg), potassium phosphate (140 mg) in N,N- dimethylethanoi (2.5 mL) is heated at 120 °C overnight. Reaction mixture is diluted with ethyl acetate and washed with water and dried with MgSO4. Crude product is purified by column chromatograph with ethyl
acetate/hexanes to give the title compound. 1H NMR (CDC13): 7.20(s, I H); 6.0(s, IH); 4.55-4.62(m, IH); 3.84(t, J=4.8Hz, 4H); 3.14(t, J=.8Hz, 4H); 2.55(s, 3H); 2.38(s, 3H); 2.03(s, 3H); 1.78-2.0(m, 4H); 0.78(1, J=7.2Hz, 6H). LC/MS:[M+1]+: 399.08 Example 14: Preparation of: 2-(5-Cycloρroρyl-3-methyl-thiophen-2-yl)-5-π -ethyl-proρyl')-3,7-dimetliyl-5H-pyrrolo[2.3--?lpyrazine

A mixture of 2-(5-bromo-3-methyl-2-thienyl)-5-(I-ethylpropyl)-3,7-dimethy!-5Η-pyπOlo[2,3-b]pyrazine (72 mg), cyclopropylboronic acid (47 mg), potassium phosphate (134 mg), tricyclohexylphosphine (5 mg), palladium acetate(2 mg) in toluene (2 niL) and water (O.lmL) is heated at 100 °C for 3 hours. The reaction mixture is diluted with ethyl acetate and washed with brine and dried with magnesium sulfate. The crude product is purified by silica gel column chromatography with hexanes/ethyl acetate to afford the title compound. 1H NMR (CDC13): 7.19(s, IH); 6.60(s, IH); 4,5-4.7(m, IH); 2.53(s, 3H); 2.38(s, 3H); 2.04(s, 3H); 2.0-2.1 (m, IH); 1.8-2.0(m, 4H); 0.9-1.0(m, 2H); 0.7-0.82(m, 8H). LC/MS:[M+1]": 354.04. Example 15: Preparation of: 5-("l -Ethvlpropvi)-3,7-dimethvl-2-r3-methyl-5-fmethvlsulfonvl)-2-thienvl]-5H-pvrrolor2.3-blpyrazine

A mixture of 2-(5-bromo-3-methyI-2-thieny])-5-(I-ethylpropyl)-3,7-dimethyl-5H-pyrroIo[2,3-b]pyrazine (71 mg, 0.18 mmol), sodium methanes ulfinate (22 mg), (CuOTf)2PhH (5 mg), trans-1,2- diaminocyclohexane (2 mg) in DMSO (2 niL) is heated at 110 °C for 16 hours. To the reaction mixture water is added and extracted with ethyl acetate and dried with MgSO1). The crude product is purified by prep-TLC plate developed with hexanes/ethyl acetate to give desired product, 1H NMR (CDC13): 7.57(s, IH); 7.29(s, I H); 4.5-4.7(m, IH); 3.21(s, 3H); 2.53(s, 3H); 2.40(s, 3H); 2.15(s, 3H); 1 8-2.0(m, 4H); 0.79(t, J=7.6Hz, 6H). LC/MS:[M+I] ' : 392.04. Example 16: Preparation of: l-{5-[5-(l -Ethylpropyl)-3.7-dimethyl-5H-pyrrolo[2,3-blpyrazin-2-vlH-methyl-2-thienvl}ethanone


Reaction mixture is concentrated. The crude product is purified by prep-TLC plate developed by hexanes/ethyl acetate to give product as oil. 1H NMR (CDCl3): 7.22(s, IH); 6.83(s, lH);5.05-5.15(m, IH);
4.5-4.65(m, IH); 2.53(s, 3H); 2.38(s, 3H); 2.08(s, 3H); I .8-2.0(m, 4H); 1.62(d, J=6.4, 3H); 0.79(t,
J=7.6Hz, 6H). LC/MS:[M+1]+: 358.05
Example 18: Preparation of:
5-f1-Ethylpi-opyl)-3.7-dimethvi-2-["3-methyl-5-(moφholin-4-ylcarbonyl)-2-thienyl]-5H-pyrroio[2.3- b]pyrazine
Part A: 5-(3,7-Dimethyl-5-fpentan-3-yl)-5H-pyrrolor3.2-blρyrazin-2-yl)-4-methylthiophene-2-carboxylic acid


5-(l-Ethyl-propyiV3J-dimethyl-2-r3-methvi-5-f3-methyl-ri .2,41oxadiazoi-5-ylVt]iiophen-2-vl1-5H- pyrrolor2.3-Mρyrazine

[5-(l-Ethyi-piOpyl)-3,7-dimethyl-5H-pyrrolo[2,3-έ]pyrazin-2-yl]-4-methyl-thiophene-2-carboxylic acid (220 mg) is taken into anhydrous dichloromethane (5 mL). Oxalyl chloride (I mLi) is added at room temperature followed by the addition of 0.1 mL of DMF. The reaction is stirred at room temperature for 1 hour before concentrated. The residue is taken into anhydrous pyridine (2 mL). To this solution N- hydroxyacetamidine (600 mg) is added. The resulting reaction mixture is heated at 110 °C for 20 hours. The mixture is concentrated and the residue is partitioned between water and ethyl acetate. The extraction is dried with magnesium sulfate. The crude product is purified by silica gel column chromatography with hexanes/ethyl acetate to afford the title compound. !Η NMR (CDCl3): 7.72(s, IH); 7.26(s, IH); 4.5-4.7(m, IH); 2.56(s, 3H); 2.55(s, 3H); 2.45(s, 3H); 2.19(S1 3H); 1.8-2.0(m, 4H); 0.80(t, J=7.2Hz, 6H). LC/MS:[M+1]+: 396.03.
Example 20: Preparation of:
5-(l-Ethyl-propyn-3.7-dimethyl-2-(3-ir)ethyl-5-[ 1,3,4]oxadiazol-2-yl-thiophen-2-yl)-5H-ρyrrolor2,3- b]pyrazine
Part A: 5-[5-(1-Ethv1-propyl)-3,7-dimethyl-5H-pyrrolo[2.3-b]pyrazin-2-vl]-4-methyl-thiophene-2- carboxylic acid hydrazide

5-[5-(l-Ethyl-propyl)-3,7-dimethyl-5H-pyrrolo[2J3-6]pyrazin-2-yl]-4-methyl-thiophene-2-carboxylic acid (I SO mg) is taken into anhydrous dichloromethane (5 mL). Oxalyl chloride (I mL) is added at room temperature followed by the addition of 0.1 mL of DMF. The reaction is stirred at room temperature for 1 hour before concentrated. The residue is taken into anhydrous dichloromethane (10 mL). To this solution anhydrous hydrazine (0.5 mLl) is added. The resulting reaction mixture is stirred at room temperature for 16 hours. To the mixture water is added and separated and extracted with dichloromethane. The combined organic solution is washed with brine and dried with magnesium sulfate. The crude product is used directly for the next step without further purification. Part B: Title compound

The crude product obtained from previous step is taken into ethanol (3 mL). p-Toluenesulfonic acid (2 mg) is added followed by the addition of triethyl orthoformate (2 mLl). The resulting mixture is heated at 80 °C for 18 hours. The reaction mixture is concentrated and the residue is partitioned between water and ethyl acetate. The organic layer is dried with magnesium sulfate. The crude product is purified by silica gel column chromatography with hexanes/ethyl acetate to afford the title compound. 1H NMR (CDCl3): 8.40(s, IH); 7.66(s5 IH); 7.26(s, IH); 4.5-4.7(m, IH); 2.56(s, 3H); 2.40(s, 3H); 2.19(s, 3H); 1.8-2.0(m, 4H); 0.78(t, J=7.2Hz, 6H). LC/MS:[M+1]+: 382.01. Example 21: Preparation of:
4-f5-(5-((R)-l-Methoxybutan-2-yl)-3.7-dimethyi-5H-pyrrolor3,2-blρyrazin-2-vπ-4-methylthiophen-2-yl)- tetrahyd ro-2H-pyran-4 -ol

500 mL, 3-necked flask equipped with condenser, internal temperature probe, No inlet is charged with a solution of 2,6-dichloropyrazine (30 g, 0.2 mol), diisopropylethylamine (38.8 g, 0.3 mol) and (R)-2- amino-1-butanoi (23.2 g, 0.26 mol) in n-propanol (100 mL). The solution is heated at 100 °C for 36 hours. The reaction is concentrated and the residue is partitioned between ethyl acetate and water. Organic layer is washed with brine and dried with Na2SU4. Crude product is obtained after concentration. 1H NMR
(CDC13): 7.79(s, IH); 7.77(s, I H); 4.86(d, J=SHz, IH); 3.85-3.9(m, IH); 3.75-3.82(m;lH); 3.6-3.81(m, IH); 2.24(s, IH); 1.55-1.8(m, 2H); 0.99(1, J=7.2Hz, 3H). LC/MS:[M+1]+: 201.00 Part B: Preparation of f6-Chloro-pyraziiv2-yD-((R)-l-methoxymethyl-propyl)-arnine


(6-Chioro-pyrazin-2-yi)-((R)-l~methoxymethyl-propyl)-arnine crude product (-28 g) of previous step is taken into anhydrous THF (250 mL). NiC12(dppp) (2.7Ig) is added. Methylmagnesium bromide (3.0M ether. 84 mL) is added dropwise at room temperature. The resulting reaction mixture is stirred for 1 hour after addition. It is then cooied to 0 °C and quenched carefully by adding water (100 mL) and saturated NHjCl (200 mL). It is separated and extracted with ethyl acetate and dried with MgSCv Crude product is obtained after concentration. It is used for next step without further purification. Part D: (3,5-Dibromo-6-methyl-pyrazin-2-yi)-((R)- 1 -methoxymethyl-propyD-amine

((R)-l-Methoxymethyl-propyI)-(6-methyl-pyrazin-2-yl)-amine crude product (22 g) of previous step is taken into anhydrous dichloromethane (200 mL) and cooied to 0-5 0C N-Bromosuccinamide (40 g) is added in one portion and stirred for 30 minutes. Reaction is separated and washed with water, Na2S2O5 (5%) and brine. The crude is purified by running through a silica gel pad eluted with (15%ethyl acetate/hexanes) to give product. 1H NMR (CDC13): 5.23(d, J=8.4Hz, IH); 4.05-4.15(m, IH); 3.4- 3.55(m,2H); 3.38(s, 3H); 2.44(s, 3H): l .ό-1.8(m, 2H); 0.95(t, J=7.6Hz, 3H). Part E: Allyl-f 3.5-dibromo-6-rnethyl-pyrazin-2-yl)-((R>- 1 -methoxymethy]-propyl)-amine


2-Bromo-5-((R)-l-methoxymethyl-propyl)-3,7-dimethyl-5H-ρyiτolo[2,3-δ]py]'azine is prepared using a procedure similar to 2-bromo-5-(ϊ-ethyl-propy1)-3,7-diniethyI-5H-pyrrolo[2J3-i]pyrazine. 1H NMR
(CDCl3): 7.29(s, IH); 4.7-4.8(m, IH); 3.7-3.8(m, J H); 3.58-3.62(m, IH); 3.30(s, 3H); 2.71(s, 3H); 2.34(s, 3H); ] . 8-2.0(m, 2H); 0.78(t, J=7.2Hz, 3H). LC/MS:[M+1]+: 313.00 Part G: 5-r(lR)-l-(methoxymethyl)piOpyl]-3,7-dimethv]-2-(3-methvl-2-thienyl)-5H-pyrrolor2.3- bipyrazine

5-[(IR)-l-(methoxymethyl)propyl]-3,7-dimethyl-2-(3-methyl-2-thienyl)-5H-pyrrolo[2,3-b]pyrazine is prepared using a procedure similar to 3,7-dimethyl-2-(3-methylthiophen-2-yI)-5-(pentan-3-yl)-5H- pyrrolo[3,2-b]pyrazme. 1H NMR(CDC13): 7.33(s, IH); 7.29(d, J=4.8Hz, IH); 6.93(d, J=4.8Hz, IH); 4.8- 4.9(m, IH); 3.77-3.82(m, IH); 3.62-3.79(m, IH); 3.34(s, 3H); 2.51(s, 3H); 2.38(s, 3H); 2.12(s, 3H); 1.8- 2.0(m, 2H); 0.79(t, J-6.8Hz, 3H). LC/MS:[M+lf: 330.03. Part H: Title compound

1 -(5-f5-(fR)4-Methoxybutan-2-yl)-3.7-dimethyl-5H-pyrrolo|"3,2-blpyrazin-2-yl)-4-ιτtethv1thiophen-2- ypethanone

5-[( 1 R)- 1 -(Methoxyrnethy i)propy ij-3,7»dimethyi-2-(3-rnethyi-2-thienyl)-5H-pyrrolo[2,3-b]ρyrazinc (323 mg) is taken into anhydrous THF (5 mL) and cooled to -78 °C. H-BuLi (1.6M hexanes, 0.74 mL, 1.2 eq.) is added dropwise and stirred for 2 hours before it is quenched with N-methoxy-N-methylacetamide (1.5 eq.). The reaction is then wanned naturally to room temperature and stirred for 2 hours before IN HC1 (2 mL) is added. Na2CO3 (2M) is added to adjust pH to ~9 and extracted with ethyl acetate. The crude product is purified by silica gel column chromatography eluted with hexanes/ethyl acetate to give title compound. 1H NMR (CDCl3): 7.56(s, IH); 7.36(s, IH); 4.8-4.9(m, IH); 3.78-3.82(m, IH); 3.6-3.70(m, IH); 3.34(s, 3H); 2.56(s, 3H); 2.53(s, 3H); 2.38(s: 3H); 2.18-2.28(m, 2H); 2.08(s, 3H); 1.9-2.0(m, 2H); 0.84(t, J=7.2Hz, 3H). LC/MS:[M+1]H : 372.02 Example 23: Preparation of:
1-(5-(5-((R)-I -Methoxybutaii-2-yl)-3,7-dimethyi-5H-pyiTolor3.2-b1pyrazin-2-yl)-4-methylthiophen-2- vDethanol

Using a procedure analogous Io that of Example 17. above for l-{5-[5-(l-ethylpropyi)~3,7-dirnethyl-5H- pyiτolo[2,3-£]pyrazin-2-y]]-4-methyI-2-thieny!}ethanol, l-(5-{5-[(lR)-l-(methoxymethyl)propyl]-3,7- dimethyl-5H-pyrrolo[2,3-b]pyrazin-2-y!}-4-methyI-2-thienyl)ethanone produces the title compound. 1H
NMR (CDC13): 7.33(s, IH): 6.82(s, IH); 5.05-5.15(m, I H); 4.8-4.9(m, IH); 3.78-3.82(m. IH); 3.6-
3.70(m. IH); 3.34(s, 3H); 2.53(s, 3H); 2.38(s, 3H); 2.07(s. 3H); 1.9-2.0(m, 2H); 1.61(d, J=6.4Hz, 3H);
0.85(t. J=7.2Hz, 3H). LC/MS:[M+1]*: 374.05
Example 24: Preparation of: 2-(5-r5-(ΪR)-l-methoxybutan-2-yl)-3.7-dimethyl-5H-pyrrolor3.2-blpyrazin-2-ylV4-methylthiophen-2- yl)propan-2-ol
 l-(5-{5-[(lR)-1-(Methoxymethyl)propyl]-3!7-dimethy]-5H-pyrroIo[2,3-blpyrazin-2-yl}-4-methyl-2- thienyl)ethanone (28 mg) is taken into anhydrous THF (2 mL). To this solution methyimagnesium bromide (3.0M ether, 0.2 irtL) is added and stirred for 2 hours. Reaction is then quenched with methanol and concentrated. The residue Js purified by ρreρ-TLC plate developed by hexanes/ethy! acetate to give the title compound. 1H NMR (CDCl3): 7.32(s, IH); 6.79(s, IH); 4.8-4.9(m, IH); 3.78-3.82(m, IH); 3.6- 3.7O(m, IH); 3.48(d, J=4.4Hz, IH); 3.33(s, 3H); 2.53(s, 3H); 2.38(s, 3H): 2.07(s, 3H); 1.9-2.0(m, 2H); 1.68(s, 6H); 0.84(t, J=7.6Hz, 3H). LC/MS:[M+1]+: 388.06 Example 25: Preparation of: 2-(3-Chloro-2-thienvi)-5-rnR)-l-(methoxymethyl)propyl1-3,7-dimethyl-5H-pyrrolo["2J-blpyrazine
l-(5-{5-[(lR)-1-(Methoxymethyl)propyl]-3!7-dimethy]-5H-pyrroIo[2,3-blpyrazin-2-yl}-4-methyl-2- thienyl)ethanone (28 mg) is taken into anhydrous THF (2 mL). To this solution methyimagnesium bromide (3.0M ether, 0.2 irtL) is added and stirred for 2 hours. Reaction is then quenched with methanol and concentrated. The residue Js purified by ρreρ-TLC plate developed by hexanes/ethy! acetate to give the title compound. 1H NMR (CDCl3): 7.32(s, IH); 6.79(s, IH); 4.8-4.9(m, IH); 3.78-3.82(m, IH); 3.6- 3.7O(m, IH); 3.48(d, J=4.4Hz, IH); 3.33(s, 3H); 2.53(s, 3H); 2.38(s, 3H): 2.07(s, 3H); 1.9-2.0(m, 2H); 1.68(s, 6H); 0.84(t, J=7.6Hz, 3H). LC/MS:[M+1]+: 388.06 Example 25: Preparation of: 2-(3-Chloro-2-thienvi)-5-rnR)-l-(methoxymethyl)propyl1-3,7-dimethyl-5H-pyrrolo["2J-blpyrazine

2-Bromo-3-chlorothiophene (987 mg, 5 mmol; J. Med. Chem. 1981, 959-964.) is added to Rieke Zn suspension in THF(5 g/dL, 10 mL) and reflux for 3 hours. The solution is filtered to a mixture of 2- Bromo-5-((R)-l-methoxymethyI-proρyl)-3,7-dimethyl-5H-pyrrolo[2,3-ό]pyrazine (628 mg, 2 mmol) and PdC12(dppf)CΗ2C12 (82 mg). The resulting mixture is heated at 70 °C overnight. Reaction mixture is partitioned between ethyl acetate and water and separated. Organic layer is washed with water and brine and dried with MgSC^. Crude product is purified by silica gel column chromatography with ethyl acetate/hexanes to give tile compound. 1H NMR (CDC13): 7.35(s, I H); 7.39(d, J=5.6Hz, I H); 7.00(d, J=5.6 Hz, I H); 4.8-4.9(m, IH); 3.77-3.82(m, IH); 3.60-3.63(m, IH); 3.33(s, 3H); 2.56(s, 3H); 2.38(s, 3H); 1.84-2.0(m, 2H): 0.84 (I5 J
 =7.2Hz, 3H). 349.99 Example 26: Preparation of: l-(((5-(3,7-Dimethyl-5-(ρentan-3-vi)-5H-ρvnOlo|"3.2-blpyrazin-2-yl)-4-methylthiazol-2- yl)methylamino)methyl)cyclohexanol
=7.2Hz, 3H). 349.99 Example 26: Preparation of: l-(((5-(3,7-Dimethyl-5-(ρentan-3-vi)-5H-ρvnOlo|"3.2-blpyrazin-2-yl)-4-methylthiazol-2- yl)methylamino)methyl)cyclohexanol
Part A: 2-r2-π .3-Dioxolan-2-vn-4-methyl-1.3-thiazol-5-yl]-5-(l -ethylpropylV3.7-dimethyl-5H- ρyrro1or2.3-blpyrazine

Part B: 5-f5-π -Ethylpropyl)-3.7-dimethvI-5H-ρyrrolor2.3-blpyrazin-2-yl]-4-methyl-1.3-thiazoIe-2- carbaldehvde


5-[5-fl-EthylpropylV3,7-dimethyl-5H-pynOlo[2.3-blpyrazin-2-yll-N,N,4-trimethyl-l,3-thiazole-2- carboxamide
Part A: 5-(l -Ethylpropyl)-3.7-dimethyl-2-(4-methyl-L3-thiazoi-5-vi)-5H-pyrro]of2.3-blpyrazine

To 50 mL of dry and degassed DMF is added 2-bromo-5-(l-ethylpropyl)-3,7-dimethyl-5H-pyrroIo[2,3- bjpyrazine (5.4 g, 18.2 mmol), 4-methylthiazole (2.0 mL, 21.8 mmol), palladium acetate (82 mg, 0.364 mmol), triphenylphosphine (191 mg, 0.728 mmol), copper iodide (173 mg, 0.91 mmol), and cesium carbonate (11.9 g, 36.4 mmoi), and the reaction mixture is heated to 120 °C for 14 h. The DMF is removed in vacuo, and the residue is partitioned between CH2Ci2 and water (100 ml each). The CH2Cl2 layer is separated, dried (Na2SO4), filtered, and evaporated in vacuo to give a brown oily residue. Part B: 545-(l -EthylρiOpyi)-3 J-dimethyl-5H-pyiτolor2,3-b1pyra2in-2-yll-4-methyl-1.3-thiazole-2- carboxylic acid


To a stirred mixture of 5-[5-(l-ethylpropyI)-3,7-dimethyl-5H-ρyrrolo[2,3-b]pyrazin-2-yI]-4-methyl-l,3- thiazole-2-carboxylic acid (50 mg, 0.14 mmoi) and dimethylamine (0.14 mL, 0.28 mmol, 2.0M in THF) in 1.0 mL Of CH2Cl2 is added 2-chioro-l ,3-dimetbylimtdazolinium chloride (DMC, 36 mg, 0.21 mmol) by porrionwise addition. After IO minutes, the mixture is concentrated in vacuo and purified by preparative chromatography (90% EtOAc/Hex) to give the title compound as a white solid. 1H NMR (400 MHz, CDC13) 7.26 (IH, s), 4.60 (IH, m), 3.60 (3H, s), 3.16 (3H, s). 2.54 (3H5 s), 2.38 (3H, s), 2.37 (3H, s), 1.89 (4H, m), 0.78 (6H. t, J 6.8). Mass spec. (386.03, M+H). Example 28: Preparation of. (2RV2-r2-{4-Chloro-24f2R.6S)-2,6-dimethylmoφholin-4-vil-1.3-thiazoi-5-v]i-3.7-dimethvi-5H- pyrroio[2,3-b]pyrazin-5-yl)butaii-l -ol
Part A: (2R.6S)-4-C4-Chloro-l >thiazoi-2-vl)-2.6-dimethγlmoi-pholine


A mixture of 2R-(2-bromo-3,7-dimethyi-5H-pyrroio[2,3-b]pyrazin-5-yl)butan-l-oI (75 mg, 0.25 mmol), (2R,6S)-4-(4-chloro-l ,3-thiazol-2-yi)-2,6-dimethyimorphoiine (70 mg, 0.30 mmol), palladium acetate (] .1 mg, 0.005 mmol), tiϊphenylphosphine (2.6 mg, 0.01 mmol), copper (I) iodide (2.4 mg, 0.0125 mmol), and cesium carbonate (163 mg, 0.50 mmol) in 2.0 mL of dry and degassed DMF is heated to 120 °C for 15 h. The DMF is removed in vacuo, and the residue is purified by column chromatography (gradient from Hex to 50% EtOAc/Hex) to afford the title compound as light brown solid. 1H NMR (400 MHz,
DMSO-d6) 7.73 (IH5 s), 4.85 (IH, bs), 4.59 (IH, m), 3.71 (6H, m), 2.71 (2H, t, J 11.6), 2.53 (3H, s), 2.24 (3H5 s), 1.85 (2H, m), 1.15 (3H, s), 1.14 (3H5 s), 0.68 (3H, X, J 12). Mass spec. (451.07, M+H). Example 29: Preparation of:
2-(4-Ch]oro-2-moφholin-4-yl-1.24hiazol-5-yl)-5-(dicvcloproρyImethylV3,7-dimethyl-5H-pyiτolof2.3- ήipyrazine
Part A: 2-Amino-6-methy] pyrazine

A solution of zinc chloride (2.0M in THF, 96.5 mL, 48.2 mmol, 5.0 equiv.) is treated dropwise with methylmagnesium bromide (3.0 M in Et2O, 32.2 mL, 96.5 mmoi, 10.0 equiv.) at 0 °C. The mixture is wanned to room temp and stirred for 45 min. A solution of 2-amino-6-chloro pyrazine (1.25 g. 9.65 mmol, 1.0 equiv) and NiC12(dppp) (157 mg, 0.29 mmol, 0.03 equiv) in THF (15 mL) is added to the freshly prepared dimethylzinc reagent. The reaction mixture is stirred under for 3h and cooied to room temp. The reaction is quenched carefully with sat'd NH4CI solution and allowed to stir overnight. The layers are separated and the aqueous layer is extracted with EtOAc (2 X 30 mL). The combined organic extracts are washed with brine, dried over Na2Sθ4 and concentrated under reduced pressure to give a cream-colored solid that is used without further purification. MS = 110.08 (M +1). 1H NMR (400 MHz, CDCI3) δ 7.78 (IH, s), 7.72 (IH, s), 4.59 (2H, bs), 2.35 (3H, s). Part B: 2-Amino-3.5-dibromo-6-methyl pyrazine

A solution of 2-amino-6-m ethyl pyrazine (700 mg, 6.41 mmol, LO equiv.) in THF (15 ml) is cooled to - 10 0C and N-bromosuccinimide (2.34 g, 13.15 mmol, 2.05 equiv.) is added portion-wise over a period of 10 min. The reaction mixture Is warmed to 0 °C and stirred for 1 h. Hexanes (10 mL) is added and the mixture is stirred for an additional 30 min at 0 3C, following which it is directly transferred to a pad of silica gel. The crude product is purified by column chromatography using a solvent gradient of 10% EtOAc in hexanes to 30% EtOAc in hexanes. MS = 267.82 (M +1). 1H ΝMR (400 MHz, CDCl3) 4.95 (2H, bs) 2.47 (3H, s).
Part C: N-AllyI-3.5-dibiOmo-6-methylpyrazin-2 -amine

A solution of 2-amino-3,5-dibrorno-6-methyi pyrazine (550 mg, 2,06 mmoi, 1.0 equiv) in THF (10 mL) is treated with LHMDS (1.0 M in THF, 2.47 mL, 2.47 mrnoi, 1.2 equiv) and stirred for 1.5 h at room temp. To the resultant mixture is added allyl bromide (0.36 mL, 4.12 mmol, 2.0 equiv) and stirred overnight at
room temp. Sat'd NH4C1 solution is added and the iayers are separated and extracted with EtOAc. The combined organic extracts are washed with brine, dried over Na2SO4 and concentrated under reduced pressure. The resulting residue is purified by silica gel column chromatography using 10% EtOAc in hexanes to give the desired compound as a yelϊow solid.
Part D: 2-Bromo-3 J-trimethyl-5-H-pyriOior3.2-έ1pyraziiie

To a solution of N-alIyl-3,5-dibromo-6-methylpyrazin-2-amine (630 mg, 2.05 mmol, 1.0 equiv) in DMF (5 mL) and triethylamine (0.69 mL, 4.92 mmol, 2.4 equiv) is added sodium formate (35 mg, 0.51 mmol, 0.25 equiv), tetrabutylammonium bromide (99 mg, 0.31 mmol, 0.15 equiv) and lastly palladium acetate (46 mg, 0.21 mmol, 0.1 equiv). The reaction flask is evacuated and purged with nitrogen three times and the mixture stirred overnight at 50 "C. On completion, the reaction is cooled to rt and diluted with water (15 mL) and extracted with EtOAc (3 X 15 mL). The combined organic extracts are washed with brine, dried over Na2SO4 and concentrated under reduced pressure to give a brown solid, which is used without further purification. MS = 226.03 (M +1). ]H NMR (400 MHz, CDCl3) δ 8.43 (IH, bs), 7.28 (IH, s), 2.74 (3H, s), 2.38 (3H, s). Part E: 2-Bromo-5-(dicyc]opjOpylmethvi)-3.7-trimethyi-5-H-pyrro]o[3.2-£1pyrazine

A solution of diisopropylazodicarboxylate (3.2 g, 16.0 mmol, 1.2 equiv) in THF (15 mL) is cooled to -20 °C and treated with a solution of triphenylphosphine (4.2g, 16.0, 1.2 equiv.) in THF (15 mL) and stirred for 30 min at -20 "C. Next, a solution of 2-bromo-3,7-trimethyl-5-H-pyrroIo[3,2-£]ρyrazine (3.0 g, 13.3 mmol, 1.0 equiv.) in TΗF (20 mL) is added and the resulting mixture stirred for 30 min at -20 0C. After this period, dicyclopropyimethanol (2.7 g, 24.0 mmol, 1.8 mmol) is added dropwise to the reaction mixture at -20 0C. which is then warmed to room temp and stirred for 2 days. The solvent is removed under reduced pressure and the residue purified by column chromatography using a solvent gradient of 5% EtOAc in hexnes to 10% EtOAc in hexanes to give a yellow solid that contained the desired product along with inseparable impurities as indicated by TLC. MS = 319.98 (M +1). Part F: Title compound

A Teflon screw-cap vial is charged with crude 3-chloro-2,5-dimethyl-7-(pentan-3-yl)-5H-pyrrolo[2J3- b]pyrazine (100 mg, 0.31 mmoL 1.0 equiv) and 2 mL of DMF. To the solution is added 4-(4- chlorothiazoI-2-yl)morphoIine (77 mg, 0.37 mmol, 1.2 equiv) , triphenylphosphine (17 mg, 0.062 mmoi, 0.20 equiv), copper(l) iodide (15 mg, 0.078 mmoi, 0.25 equiv), cesium carbonate (203 mg, 0.62 mmol, 2.0 equiv), and palladium acetate (3.5 mg, 0.016 mmol, 0.05 equiv). The reaction vial is evacuated and purged with nitrogen three times before being tightly capped and heated overnight at 120 °C. The reaction mixture is cooled to rt, diluted with water and extracted with EtOAc (3 X 10 mL). The combined organic extracts are washed with brine, dried over NaiSC^ and concentrated under reduced pressure. The resulting residue is purified by preparative thin layer chromatography using 30% EtOAc in hexanes to give the desired compound as yellow foam. MS = 444.04 (M +] ). 1H NMR (400 MHz, OMSO-dg) δ 7.93 (IH, s), 3.72-3.74 (4H, m), 3.37-3.44 (5H, m), 2.52 (3H, s) 2.27 (3 H, s), 1.51-1.58 (2H, m), 0.61-0.68 (2H, m), 0.53-0.59 (2H, m), 0.30-0.36 (2H, m), 0.13-0.19 (2H, m). Example 30: Preparation of: 2-(4-Chloro-2-morρholin-4-yl-1.2-thiazol-5-yl)-5-(l-cycloρropylethvπ-3,7-dimethyl-5H-pyiτolo[2.3- j>1pyrazine
Part A: 2-(4-ChioiO-2-morpholinι-4-yl-1 ,2-thiazol-5-ylV3.7-dimethyl-SH-pviτolof2.3--?lpyrazine


To a solution of 2-(4-chloro-2-morpholin-4-yl-l,2-thiazol-5-yl)-3,7-dimethyϊ-5H-pytτolo[2,3-ό]pyrazine (35 mg, 0.1 mmol, 1.0 equiv.), triphenylphosphine (29 mg, 0.11 mmol, 1 .1 equiv) and 1- cyclopropylethanol (10.3 mg, 0.1 1 mmol, 1.1 equiv) in TΗF (2 mL) is added diisopropylazodicarboxyiate (22.2 mg. 0.12 equiv, 1.2 equiv). The reaction mixture is allowed to warm to it and stirred for 2 days. The solvent is removed under reduced pressure and the residue is purified by preparative TLC using 30% EtOAc in hexanes to give the desired product. MS = 418.01 (M +1). 1H NMR (400 MHz, DMSO-O δ 8.03 (IH, s), 4.00-4.06 (m, IH), 3.69-3.74 (4H, m), 3.40-3.46 (4H, m), 2.29 (3H, s) 2.11 (3H, s), 1.54 (3H, d, J = 6.4 Hz), 1.40-1.42 (IH, m), 1.51-1.58 (2H, m), 0.60-0.64 (IH, m), 0.45-0.49 (IH, m), 0.38- 0.42 (IH, m), 0.22-0.27 (I H, m). Example 31: Preparation of:
3-f4-Chloro-2-morphoHn-4-vJ-1.3-thiazoI-5-vI)-7-(l-ethvipropyiV2.5-dimcthyl-5H-pyrrolor2.3- b]pyrazine
Part A: 6-Chloro-7V-methvipyrazin-2-amine

The compound is prepared following a previously reported procedure (WO 2005/023806) with slight modifications. To a solution of 2,6-dichloropyrazine (100 g. 0.671 mol, 1 ,0 equiv.) hi THF (500 mL) is added 175 mL (2.5 equiv.) of 40% aqueous methylamine solution followed by 117 ml (0.671 mo!, 1.0 equiv.) of N,N-diisopropylethyl amine. The resulting mixture is heated overnight in a sealed tube at 80 °C. After this period, the reaction mixture is allowed to cool to room temperature and then concentrated under reduced pressure to remove solvent. The residue is taken up in water (500 mL) and extracted with EtOAc (2 X 500 mL). The combined organic extracts are washed with brine, dried over Na2SO4 and concentrated to give a solid that is used for the next step without further purification. Part B: 3.5-Dibromo-6-chloro-N-methyIpyrazin-2-amine

The compound is prepared following a previously reported procedure (WO 2005/023806) with slight modifications. To a cooled (0 "C) solution of crude 6-ehloro-N-methylpyrazin-2-amine (0.671 rnol, 1.0 equiv) in CH2C12 (600 mL) is added N-bromosuccinamide (299 g, 1.68 mol, 2.5 equiv.) in small portions over a period of 30 min. The resultant mixture is warmed to rt and allowed to stir. After 1 hr, the reaction
mixture is diluted with water (500 mL) and the layers separated. The organic layer is washed with water (2 X 500 mL), brine (500 mL) and dried over Na2SO4. Following filtration, the solvent is removed under reduced pressure and the residue is purified by silica gel column chromatography using 15% EtOAc in hexanes to give the desired compound as a yellow solid. 1H NMR (300 MHz, CDCl3) δ 5.34 (IH. bs), 3.04 (3H, d, J = 4.8 Hz).
Part C: S.S-Dibromo-ό-chloro-N-fS-ethylpent-Σ-envD-N-methylpyrazin^-amine

The compound is prepared following a previously reported procedure (WO 2005/023806) with slight modifications. To a solution of 3,5-dibronio-6-chloro-N-methylρyrazin-2-amine (2 g, 6.64 mmol, 1.0 equiv.) in THF (10 mL) at rt is added NaH (60 % suspension in mineral oil, 318 mg, 7.96 mmol, 1.2 equiv.) over a period of 20 min. The resultant suspension is allowed to stir for 1 h at rt and then treated with TBAI (245 mg, 0.664 mmol, 0.1 equiv) and 18-crown-6 (176 mg, 0.664 mmol, 0.1 equiv). Next, 1.2 g (7.96 mmol, 1.2 equiv.) of l-chloro-3-ethylpent-2-ene (prepared according to the procedure outlined in patent WO 2005/023806) is added via syringe over a period of 5 min and the resultant mixture is stirred overnight at ambient temperature. Following this period, the reaction is diluted with water (30 mL) and extracted with EtOAc (2 X 30 mL). The combined organic extracts are washed with brine, dried over N^SO4 and concentrated under reduced pressure. The resulting residue is purified by silica gel column chromatography using 10% EtOAc in hexanes to give the desired compound as brown oil. 1H NMR (300 MHz, CDCI3) δ 5.22 (IH, d, J = 6.6 Hz), 4.09 (2H. d, J = 6.6 Hz), 3.02 (3H, s), 2.05-2.17 (4H, m), 1.02 (6H, t, J= 7.5 Hz).
Part D: 2-Bromo-3-chloro-5-methyl-7-(pentan-3-yl)-5H-py-τolof2,3-blpyrazine

To a solution of 3,5-dibromo-6-chloro-N(3-ethyipent-2-enyl)-N-methylpyrazin-2-amine (1.93 g, 4.85 mmol, 1.0 equiv) in DMF (10 mL) and triethylamine (1.6 mL, 1 1.64 mmol, 2.4 equiv) is added sodium formate (83 mg, 1 .21 mmol, 0.25 equiv), tetrabutylammonium bromide (235 mg, 0.73 mmol, 0.15 equiv) and lastly palladium acetate (55 rng, 0.24 mmol, 0.05 equiv). The reaction flask is evacuated and purged with nitrogen three times and the mixture stirred overnight at 50 °C. On completion, the reaction is cooled to rt and diluted with water (40 mL) and extracted with EtOAc (3 X 30 mL). The combined organic extracts are washed with brine, dried over Na2SO4 and concentrated under reduced pressure. The resulting residue is purified by silica gel column chromatography using 15% EtOAc in hexanes to give the desired compound as brown oil. MS = 316.02 (M +1). !Η ΝMR (400 MHz, CDCI3) δ 5.22 (IH, d, J = 6.6 Hz), 4.09 (2H1 d, J= 6.6 Hz), 3.02 (3 H, s), 2.05-2.17 (4H, m), 1.02 (6H, q. J= 7.5 Hz). Part E: 3-Chloro-2.,5-dimethyl-7-(pentan-3-yl)-5H-py-τoloF2,3-blρyrazine

A solution of 2-bromo-3-ch!oro-5-methyl-7-(pentan-3-yJ)-5H-ρyrrolo[2s3-b]ρyrazine (1.28 g, 4.04 mmol, 1.0 equiv) in TΗF (50 mL) is cooled to -78 °C and treated with t-BuLi (1.7 M in pentane, 5.0 mL, 8.5 mmol. 2.1 equiv). After stirring at -78 °C for 10 min, the reaction mixture is treated with iodomethane (2.29g, 16.16 mmol. 4.0 equiv) and stirred for an additional 1 hr at -78 °C. The reaction is quenched by pouring into ice and saturated NaΗCO.i solution (15 mL). The aqueous layer is extracted with EtOAc (2 X 30 mL) and the combined organic extracts are washed with brine, dried over NaiSC^ and concentrated under reduced pressure. The resulting brown oil is used without further purification. Part F: Title compound

A Teflon screw-cap via) is charged with 100 mg (0.40 mmol, 1.0 equiv) of crude 3-chloro-2,5-dimethyI-
7-(pentan-3-yI)-5H-pyrrolo[2,3-b]ρyrazine and 2 mL of DMF. To the solution is added 98 mg (0.48 mmol, 1.2 equiv) of 4-(4-chlorothiazol-2-yI)morphoIine, 21 mg (0.08 mmol, 0.20 equiv) of tripbenylphosphine, 19 mg (0.1 mmol, 0.25 equiv) of copper(I) iodide, 261 mg (0.8 mmoi, 2.0 equiv) of cesium carbonate, and lastly 5 rag (0.02 mmol, 0.05 equiv) of palladium acetate. The reaction vial is evacuated and purged with nitrogen three times before being tightly capped and heated overnight at 1 10 °C, The reaction mixture is cooled to it, diluted with water and extracted with EtOAc (3 X 10 mL). The combined organic extracts are washed with brine, dried over NaiSO^ and concentrated under reduced pressure. The resulting residue is purified by preparative thin layer chromatography using 30% EtOAc in hexanes to give the desired compound as a yellow solid (26 mg, 15% yield). MS = 420.08 (M +1). 1H NMR (300 MHz, CDC13) δ 7.22 (IH, s), 3.82-3.85 (4H, m), 3.84 (3H, s), 3.44-3.56 (4H, m), 2.96 (IH, m) 2.70 (3 H, s), 1.73-1 .85 (4H, m), 0.86 (6H, q, J = 7.5 Hz). Example 32: Preparation of: 2.5-Dimethyi-3-(3-methylthioρhen-2-yl)-7-(pentan-3-ylV5H-ρyiτoIor2.3-b1pvrazine

A 10 mL round-bottom flask is charged with 250 mg (0.99 mmol, 1.0 equiv) of 3-chloro-2,5-dimethyl-7-
(pentan-3-yI)-5H-ρyrrolo[2,3-b]pyrazine and 41 mg (0.05 mmol, 0.05 equiv.) of PdC12(dppf). The solids are dissolved in 3 mL (0.5 M in TΗF, 1.5 mmol, 1.5 equiv) of 3-methyi-2-thienylzinc bromide and the
resultant mixture heated overnight at 70 °C. The reaction mixture is cooled to it, diluted with water and the product is extracted with EtOAc. The combined organic extracts are washed with brine, dried over Na2SC>4 and concentrated under reduced pressure. The resulting residue is purified by silica gel column chromatography using 10% EtOAc in hexanes to give the desired compound as yellow oil. MS = 314.05 (M +1). 1H NMR (400 MHz, CDC13) δ 7.33 (IH, d, J = 5.2 Hz), 7.18 (IH, s), 6.96 (IH, d, J = 5.2 Hz), 3.85 (3H1 s), 2.96 (IH, m), 2.58 (3H, s), 2.17 (3H, s). 1.77-1.85 (4H5 m), 0.88 (6H, t, J= 7.2 Hz). Example 33: Preparation of:
4-(5-(2,5-Dimethyl-7-(pentan-3-yi)-5H-pyrrolof2.3-έlpyrazin-3-yl)-4-methylthiophen-2-yl)-tetrahydro- 2H-pyran-4-ol

To a -78 0C solution of 2,5-dimethyi-3-(3-metliylthioρhen-2-yI)-7-(pentan-3-yl)-5Η-pyrroIo[2,3- bjpyrazine (135 mg, 0,43 mmol, 1.0 equiv) in 5 mL of THF is added 65 μL (0.43 mmol, 1.0 equiv) of TMEDA and 0.28 mL (1.7 M in pentane, 0.47 mmol, 1.1 equiv) of /-BuLi and the mixture is stirred at -78 "C for 1 h. The resultant solution is treated with 2.4 mL of tetrahydropyran-4-one (0.2 M in toluene, 0.47 mL, 1.1 equiv) and the mixture is allowed to warm to rt and stirred overnight. The reaction is quenched with water and the product is extracted with EtOAc. The combined organic extracts are washed with brine, dried over Na2SO4 and concentrated under reduced pressure. The resulting residue is purified by silica gel column chromatography using 30% EtOAc in hexanes to give the desired compound as a foam. MS = 414.08 (M +1). 1H NMR (400 MHz, CDC13) δ 7.18 (I H, s), 6.87 (IH, s), 3.84 (3H, s), 3.84-3.96 (4H, m) 2.96 (IH, m), 2.59 (3H, s), 2.20-2.27 (2H, m) 2.13 (3H, s), 1.75-1.95 (6H, m), 0.88 (6H, X, J= 7.6 Hz).
Example 34: Preparation of: l-f5-(2.5-Dimethyl-7-(pentan-3-yl)-5H-pyrrolor2.3-Z?lpyrazin-3-yl)-4-methylthiophen-2-yl)ethaiione


A solution of l-(5-(2,5-dimethyl-7-(pentan-3-yI)-5H-pyrrolo[2,3-έ]pyrazin-3-yi)-4-methylthiophen-2- yl)ethanone (20 mg, 0.056 mmol, 1.0 equiv) in MeOH (0.5 mL) is treated with solid sodium borohydride (3 mg, 0.079 mmoi, 1.4 equiv) and stirred at ambient temperature. After 30 min, the reaction is quenched with water and the product is extracted with EtOAc. The resulting residue is purified by preparative thin layer chromatography using 30% EtOAc in hexanes to give the desired compound as a white foam. MS = 358.05 (M +1). 1H NMR (400 MHz, CDCi3) δ 7.18 (IH, s), 6.85 (IH, s), 5.12 (IH, q, J = 6.4 Hz), 3.84 (3H, s), 2.97 (IH, m), 2.60 (3H, s), 2.12 (3H, s), 1.78-1.85 (4HS m), 1.64 (3H, d, J - 6.4 Hz), 0.88 (6H, t, J= 12 Hz). Example 36: Preparation of:
3-(5-Bromo-3-methy!thiophen-2-yl)- 2,5-dimethyl-7-(pentan-3-yD-5H-pyiτoJo[2,3-J>]pyrazine

To a -78 °C solution of 2,5-dimethyl-3-(3-methylthiophen-2-yl)-7-(pentan-3-yl)-5Η-pyrrolo[2,3- bjpyrazine (214 mg, 0.68 mmol, 1.0 equiv) in 5 mL of THF is added 0.47 mL of n-BuLi (1.6 M in hexanes, 0.75 mmol, 1.1 equiv) and the mixture is stirred for 15 min at -78 °C. The resultant anion is treated with a solution of carbon tetrabromide (338 mg, 1.02 mmoi, 1.5 equiv) in THF (1.0 mL) and stirred at -78 °C for Ih, before being quenched with water and allowed to warm to it. The product is extracted with EtOAc and the combined organic extracts are washed with brine, dried over Na2Sθ4 and concentrated under reduced pressure. The resulting residue is purified by silica gei column chromatography using 10% EtOAc in hexanes to give the desired compound as a brown oil.
Example 37: Preparation of: 2,5-Dimetliyl-3-('3-methyl-5-morpholinothiophen-2-vπ-7-fpentan-3-yl')-5H-pyrrolor2,3-61pyrazine

A Teflon screw-cap viai is charged with 150 mg (0.38 mmoi, 1.0 equiv) of 3-(5-bromo-3-methylthiophen- 2-yl)- 2,5-dimethyl-7-(pentan-3-yl)-5H-pyrro!o[2,3-i]pyrazine and 2 mL of N;N-dimethyIethanolamine. To the solution is added 12.5 mg of Cu powder, 12.5 mg of copper(l) iodide and 161 mg (0.76 mmol, 2.0 equiv) of morpholine. The reaction vial is evacuated and purged with nitrogen three times before being tightly capped and heated overnight at 120 0C. The reaction mixture is cooled to it, diluted with water and extracted with EtOAc (3 X 10 mL). The combined organic extracts arc washed with brine, dried over Na2SO4 and concentrated under reduced pressure. The resulting residue is purified by preparative thin layer chromatography using 20% EtOAc in hexanes to give the desired compound as yellow foam. MS = 399.08 (M +1). 1H ΝMR (400 MHz, CDCU) δ 7.15 (IH, s), 6.02 (IH, s), 3.85-3.87 (4H, m) 3.83 (3H, s), 3.15-3.18 (4H, m) 2.95 (I H, m), 2.07 (3H, s), 1.74-1.84 (4H, m), 0.87 (6H, t, J- 7.6 Hz). Additional Aspects of Preferred Compounds The most preferred compounds and salts of those herein are suitable for use in treating human patients. Accordingly, such preferred compounds are non-toxic. They do not exhibit single or multiple dose acute or long-term toxicity, mutagenicity (e.g., as determined in a bacterial reverse mutation assay such as an Ames test), teratogenicity, tumorogenicity, or the like, and rarely trigger adverse effects (side effects) when administered at therapeutically effective dosages. Preferably, administration of such preferred compounds at certain doses (i.e., doses yielding therapeutically effective in vivo concentrations or preferably doses of I, 30, 100, 300, or 1000 mg/kg - 300 being most preferred - administered parenterally or prefrerably orally) does not result in prolongation of heart QT intervals (i.e., as determined by electrocardiography, e.g., in guinea pigs, minipigs or dogs). When administered daily for 5 or preferably ten days, such doses of such preferred compounds also do not cause liver enlargement resulting in an increase of liver to body weight ratio of more than 100%, preferably not more than 75% and more preferably not more than 50% over matched controls in laboratory rodents (e.g., mice or rats). In another aspect such doses of such preferred compounds also preferably do not cause liver enlargement resulting in an increase of liver to body weight ratio of more than 50%, more preferably preferably not more than 25%, and most preferably not more than 10% over matched untreated controls in dogs or other non-rodent mammals.
In yet another aspect such doses of such preferred compounds also preferably do not promote the release of liver enzymes (e.g., ALT, LDH, or AST) from hepatocytes in vivo. Preferably such doses do not elevate serum levels of such enzymes by more than 100%, more preferably not by more than 75% and
most preferably not by more than 50% over matched untreated controls in laboratory rodents. Similarly, concentrations (in culture media or other such solutions that are contacted and incubated with cells in vitro) equivalent to two, fold, preferably five-fold, and most preferably ten-fold the minimum in vivo therapeutic concentration do not cause release of any of such liver enzymes from hepatocytes into culture medium in vitro above baseline levels seen in media from untreated cells.
Because side effects are often due to undesirable receptor activation or antagonism, preferred compounds exert their receptor-modulatory effects with high selectivity. This means that they do not bind to certain other receptors (other than CRF, preferably CRFl, receptors) with high affinity, but rather only bind to, activate, or inhibit the activity of such other receptors with affinity constants (note: greater affinity constants indicate weaker binding) of greater than 100 nanomolar, preferably greater than 1 micromolar, more preferably greater than 10 micromolar and most preferably greater than 300 micromolar. Such receptors preferably are selected from the group consisting of a) ion channel receptors, (preferably sodium ion channel receptors), b) neurotransmitter receptors (preferably selected from alpha- and beta-adrenergic receptors, muscarinic receptors - most preferably m 1 , m2, or m3 receptors, dopamine receptors, GABAA receptors and metabotropic glutamate receptors), c) histamine receptors, d) cytokine receptors (preferably selected from interleukin receptors, most preferabiy IL-8 receptors), e) bioactive peptide receptors (preferably selected from NPY and VIP receptors), f) neurokinin receptors g) bradykinin receptors (preferably selected from BKl receptors and BK2 receptors), and h) hormone receptors (preferably selected from thyrotropin releasing hormone receptors and melanocyte- concentrating hormone receptors).
Claims
WHAT IS CLAIMED IS:
] . compound of a formula selected from the group consisting of:





or a pharmaceutically acceptable salt thereof, wherein:
Z at each occurrence is independently S or O;
R2! is hydrogen, halogen, amino, (C1-C4)alkyL C2-C4alkenyl, C2-C4alkynyl, tC1-C4)alkoxy, hydroxy, halo(C1-C4)alkyl halo(C1-C4)alkoxy, cyano, mono- or di- (C1-C4)alkylamiπo, (CI- C4)alkanoyl, aminocarbonyl, or -S(O)n-(C1-C4)alkyI; R22 is hydrogen, halogen, (C1-C4)alkyl, C2-C4aikenyl, C2-C4alkynyl, (C 1 -C4)alkoxy, hydroxy, halo(C1-C4)alkyl, halo(C1 -C4)aϊkoxy, cyano, mono- or di- (CI-C4)alkyiamino, (C1- C4)alkanoyl, aminocarbonyl, or -S(O)n-(C1-C4)alkyI;
R23 is (C1-C7)alky!, (C2-C7)alkenyl, (C2-C7)alkynyL (C6-C12)ary!, 4 to 12 membered heterocycloalkyl, or 5 to 12 membered heteroaryl, each of which is unsubstituted or substituted with one or more substituents independently selected at each occurrence from (C1-C7)alkyL (C1- C7)alkoxy, halogen, amino, hydroxyl, 4 to 12 membered heterocycloalkyl, and 5 to 12 membered heteroaryl;
R24 is hydrogen, halogen, cyano, (C1-C3)a!kyl, (C1-C3)alkoxy, halo(C1-C7)alkyI, mono- or di- (C1-C7)alkylamino, -(CH2)n-R!0, CONH2, or mono- or di-(C1-C4)alkyl-aminocarbonyϊ-;
R25 is hydrogen, halogen, cyano5 (C1 -C7)alkyl, (C1-C3)alkoxy, or haloalkyi, mono- or di- (C1- C7)alkylamino, -(CH2)n-Ri0, CONH2, or mono- or di-(C1-C4)alky]-aminocarbony!-;
R26 is hydrogen or (C1-C4)alkyl;
R27 is (C1-C7)alkyl, (C2-C7)alkenyl, (C2-C7)alkynyl, (C6-C12)aryl, mono- or di- (CI-
C7)a!kylamino, 4 to 12 membered heterocycloalkyl, 5 to 12 membered heteroaryl, -(CH2)n-R10, - (CH2)n-CONH2, or mono- or di-(C1-C4)alkyl-amiπocarbonyl-(CH2)n-s each of which is unsubstituted or substituted with one or more substituents independently selected at each occurrence from (C1-C7)alkyl, (C1-C7)alkoxy, halogen, amino, hydroxyl, 4 to 12 membered heterocycloalkyl, and 5 to 12 membered heteroaryl;
R28 is (C1-C7)alkyl, (C6-C 12)aryl, 4 to 12 membered heterocycloalkyl, or 5 to 12 membered heteroaryl, each of which is unsubstituted or substituted with one or more substituents independently selected at each occurrence from alkyl, alkoxy, halogen, amino, hydroxyl. 4 to 12 membered heterocycloalkyl, and 5 to 12 membered heteroaryl; Ri0 is N-linked and at each occurrence is independently acetidiπe, pyrrolidine, piperidine, hexahydroazepine, or octahydroazepine; and
R2 is selected from the group consisting of:  wherein:
wherein:
R6 at each occurrence is independently hydrogen, halogen, cyano, (C1-C4)alkyI,halo(C1- C4)alkyl, (C1-C4)alkoxy, halo(C3-C4)alkoxy, or phenyl;
R7 is hydrogen, halogen, cyano, (C1-C4)alkyl, HaIo(C 1-C4)alkyi, (CI-C4)a!koxy, halo(C1- C4)alkoxy, mono- or di- (C1-C4)alkylamino, (C1-C4)alkylcarbonyl, -(CH2)n-R10 , -(CH2)n- CONH2, mono- or di-(CI-C4)a]kyl-aminocarbonyl-(CH2)n-, -S(0)n-(C1-C4)alkyl, phenyl,

Rs is hydrogen, halogen, haloalkyl, haloalkoxy, cyano, (C1-C4)alkyl, R3R1T^-, carbamyl, -(C1-
C2)aIkoxy(C1-C2)alkyl, Rn-C(0)-, Rl l-(CH2)n-,


Ra is hydrogen, (C1-C5)alkyi, (C3-C5)cycloalky], methoxy(C2-C4)aIkyl, acetyl, (C1- C2)alkylsu!fonyl, (C3)alkenyl, R15-(CH2)n-, or (C1-C2)alkyl, each of which is unsubstituted or substituted with cyano, formyl, vinyl, or ethynyl;
Rb is hydrogen or (C1-C3)alkyl;
RI 1 is hydrogen, (C1-C7)alkyl, (C3-C7)cycloalkyl, -O(C1-C4)aIkyl, (C2-C7)alkenyl, elhynyl, mono- or di- (C1-C4)alkylamino, aiyl. 4 to 12 membered heterocycloalkyl, or 5 to 12 membered heteroaryl, R15-(CH2)n-;
R is cyclopropyi, phenyl, 
R31 represents one or two substituents, at each occurrence independently selected from hydrogen, halogen, (C1-C4)alkyl, (CI-C4)aikoxy, hydroxy, hydroxy 1(C I -C4) alky L haloalkyl, haloalkoxy, cyano, amino, mono- or di- (C1-C4)alkylamino, (C1-C4)alkyI-CO-, CONH2, and -S(O)n-(C1 - C4)alkyl;
R33 is (C1-C7)alkyl, (C2-C7)alkenyi, (C2-C7)alkynyl, -(CH2)n-R10 , -(CH2)n -CONH2, mono- or di-(CI -C4)alkyl-aminocarbonyI-(CH2)n-, aryl, 4 to 12 membered heterocycloalkyl, or 5 to 32 membered heteroaryl, each of which is unsubstituted or substituted one or more substituents independently selected at each occurrence from (C1-C7)a!kyl, (C1-C7)alkoxy, halogen, amino, hydroxy!, 4 to 12 membered helerocycloalkyl, and 5 to 12 membered heteroaryl;
R33 at each occurrence is independently hydrogen, halogen, cyano, (C1-C3)alkyl, (C1-C7)alkyl, (C1-C3)aikoxy, halo(C1-C7)alkyl, -(CH2)n-R]0 , -(CH2)n-CO mono- or di- (C1-C7)alkylamino, mono- or di- (C1-C7)alkylamino, or -CO mono- or di- (C1-C7)alkylamino;
R36 is hydrogen or (C1 -C4)alkyl; R37 is hydrogen, halogen, (C1-C4)alkyl, or haio(C1-C4)aϊkyl; n is 0, 1, or 2; and X is CH2, CO, O, S, or SO2; or R2 is selected from the group consisting of:


R1 is hydrogen, halogen, cyano, (C1-C3)a!kyl, or methoxy;
R3 is hydrogen or (C1-C3)alkyl;
R4 is hydrogen, halogen, hydroxy!, formyi, (C]-C3)alkoxy, (C2-C3)alkenyl,
RcRdN-, or R16-C(O)s or
R4 is (C3-C5)cycloalkyl or (CI-C7)alkyi, each substituted with zero, one, or two that are the same or different of hydroxy, (C1-C2)alkoxy, or cyclopropyl;
R^ is hydrogen, methyl, or trifluoromethyl;
R16 is hydrogen, (CI-C3)a!kyI, cyclopropyl. methoxy, or RcRdN-; and
Rc and Rd are independently hydrogen, (C1-C4)a!kyi, or methoxy.
2. A compound or salt of claim 1 wherein the compound is capable of exhibiting an IC50 vaiue of less than or equal to 1 micromolar in a standard in vitro CRF receptor binding assay.
3. A compound or salt of claim 1 wherein, in a standard in vitro CRF receptor binding assay the compound exhibits an IC50 value of less than or equal to 100 nanomolar.
4. A compound or salt of claim 1 wherein, in a standard in vitro CRF receptor binding assay, the compound exhibits an lC^o value of less than or equal to 10 nanomolar,
5. A method for treating at least one syptom of an anxiety disorder, a stress-related disorder. or an eating disorder, comprising administering to a patient in need of such treatment a therapeutically effective amount of a compound or salt of claim 1.
6. A method for treating at least one syptom of an depression or bipolar disorder, comprising administering to a patient in need of such treatment a therapeutically effective amount of a compound or salt of claim I .
7. A method for treating anorexia nervosa, bulimia nervosa, or obesity, comprising administering to a patient in need of such treatment a therapeutically effective amount of a compound or salt of claim 1.
8. A method for treating Irritable Bowel Syndrome or Crohn's disease, comprising administering to a patient in need of such treatment a therapeutically effective amount of a compound or salt of claim 1.
9. A method for demonstrating the presence of CRF receptors in cell or tissue samples, said method comprising: preparing a plurality of matched cell or tissue samples; preparing at least one control sample by contacting (under conditions that permit binding of CRF to CRF receptors within cell and tissue samples) at least one of the matched eel! or tissue samples (that has not previously been contacted with any compound or salt of any of Claims 1-42) with a control solution comprising a detectably-labeled preparation of a selected compound or salt of claim 1 at a first measured molar concentration, said control solution further comprising an unlabeled preparation of the selected compound or salt at a second measured molar concentration, which second measured concentration is greater than said first measured concentration; preparing at least one experimental sample by contacting (under conditions that permit binding of CRF to CRF receptors within cell and tissue samples) at least one of the matched cell or tissue samples (that has not previously been contacted with any compound or salt of claim 1) with an experimental solution comprising the detectably-labeled preparation of the selected compound or salt at the first measured molar concentration, said experimental solution not further comprising an unlabelied preparation of any compound or salt of claim 1 at a concentration greater than or equal to said first measured concentration; washing the at least one control sample to remove unbound selected compound or salt to produce at least one washed control sample; washing the at least one experimental sample to remove unbound selected compound or salt Io produce at least one washed experimental sample; measuring the amount of detectable label of any remaining bound detectably-labeled selected compound or salt in the at least one washed control sample; measuring the amount detectable label of any remaining bound detectably-labeied selected compound or salt in the at least one washed experimental sample; and comparing the amount of detectable label measured in each of the at least one washed experimental sample to the amount of detectable label measured in each of the at least one washed control sample; wherein, a comparison that indicates the detection of a greater amount of detectable label in the at least one washed experimental sample than is detected in any of the at least one washed control samples demonstrates the presence of CRF receptors in that experimental sample.
10. A method of inhibiting the binding of CRF to a CRF 1 Receptor, which method comprises: contacting a solution comprising CRF and compound or salt of claim 1 with a cell expressing the CRF receptor, wherein the compound or salt is present in the solution at a concentration sufficient to inhibit in vitro CRF binding to IMR32 cells.
11. The method of C1aim J 0 wherein the eel! expressing the CRF receptor is a neuronal cell that is contacted in vivo in an animal, and wherein the solution is a body fluid of said animal.
12. The method of Claim 1 1 wherein the animal is a human patient.
13. A pharmaceutical composition comprising a pharmaceutically acceptable carrier and compound or salt of claim 1.
14. A package comprising a pharmaceutical composition of claim 13 in a container and further comprising indicia comprising at least one of: instructions for using the composition to treat a patient suffering from an anxiety disorder, or instructions for using the composition to treat a patient suffering from a stress-related disorder, or instructions for using the composition to treat a patient suffering from an eating disorder.
15. A package comprising a pharmaceutical composition of claim 13 in a container and further comprising indicia comprising at least one of: instructions for using the composition to treat a patient suffering from depression or instructions for using the composition to treat a patient suffering from a bipolar disorder,
16. A compound or salt of claim 1 which, when administered orally to dogs at a dose selected from 1, 30, 100, 300, and 1000mg/kg of body weight, does not produce emesis in more than I out of six dogs within three hours of administration.
17. A compound or salt of claim 1 which, at a concentration selected from 5, 10, 15, 20 and 25 micromolar in in vitro cytochrome P450 assays, does not exhibit mechanism -based inhibition of any of CYP 1 A2, CYP2C8, CYP2C9, CYP2C 19, CYP2D6 and CYP3 A4 activities.
18. The compound or salt of claim 17 wherein the concentration is 25 micromolar.
19. A compound or salt of claim 1 which, at a concentration selected from 5, 10, 15, 20 and 25 micromolar in an in vitro CYP2C8 assay does not produce irreversible inhibition of CYP2C8.
20. The method of Claim 5, wherein the symptom of a stress disorder is a sleep disorder or a dermatologic disorder.
21. A compound or salt or composition of any of claims 1-4, 13, and 16-19 wherein the compound is of a formula selected from Formula A. Formula E5 and Formula i.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US88269606P | 2006-12-29 | 2006-12-29 | |
| US60/882,696 | 2006-12-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2008083070A1 true WO2008083070A1 (en) | 2008-07-10 |
Family
ID=39588975
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2007/088534 WO2008083070A1 (en) | 2006-12-29 | 2007-12-21 | Crf1 receptor ligands comprising fused bicyclic heteroaryl moieties |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2008083070A1 (en) |
Cited By (34)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011092293A3 (en) * | 2010-02-01 | 2011-10-20 | Novartis Ag | Cyclohexyl amide derivatives as crf receptor antagonists |
| EP2532661A1 (en) | 2011-06-10 | 2012-12-12 | Syngenta Participations AG | Novel insecticides |
| EP2567959A1 (en) | 2011-09-12 | 2013-03-13 | Sanofi | 6-(4-Hydroxy-phenyl)-3-styryl-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US8476282B2 (en) | 2008-11-03 | 2013-07-02 | Intellikine Llc | Benzoxazole kinase inhibitors and methods of use |
| WO2013156433A1 (en) | 2012-04-17 | 2013-10-24 | Syngenta Participations Ag | Insecticidally active thiazole derivatives |
| WO2013178362A1 (en) * | 2012-05-31 | 2013-12-05 | Phenex Pharmaceuticals Ag | Carboxamide or sulfonamide substituted thiazoles and related derivatives as modulators for the orphan nuclear receptor ror[gamma] |
| US8637542B2 (en) | 2008-03-14 | 2014-01-28 | Intellikine, Inc. | Kinase inhibitors and methods of use |
| US8765746B2 (en) | 2010-10-13 | 2014-07-01 | Millennium Pharmaceuticals, Inc. | Heteroaryls and uses thereof |
| US20140206884A1 (en) * | 2010-03-16 | 2014-07-24 | Basf Se | Process Using Grignard Reagents |
| US8796271B2 (en) | 2010-08-11 | 2014-08-05 | Millennium Pharmaceuticals, Inc. | Heteroaryls and uses thereof |
| US8796314B2 (en) | 2009-01-30 | 2014-08-05 | Millennium Pharmaceuticals, Inc. | Heteroaryls and uses thereof |
| US8859768B2 (en) | 2010-08-11 | 2014-10-14 | Millennium Pharmaceuticals, Inc. | Heteroaryls and uses thereof |
| US8993580B2 (en) | 2008-03-14 | 2015-03-31 | Intellikine Llc | Benzothiazole kinase inhibitors and methods of use |
| US9029411B2 (en) | 2008-01-25 | 2015-05-12 | Millennium Pharmaceuticals, Inc. | Thiophenes and uses thereof |
| US9062038B2 (en) | 2010-08-11 | 2015-06-23 | Millennium Pharmaceuticals, Inc. | Heteroaryls and uses thereof |
| US9090601B2 (en) | 2009-01-30 | 2015-07-28 | Millennium Pharmaceuticals, Inc. | Thiazole derivatives |
| WO2015112642A1 (en) | 2014-01-21 | 2015-07-30 | Neurocrine Biosciences, Inc. | Crf1 receptor antagonists for the treatment of congenital adrenal hyperplasia |
| US9096611B2 (en) | 2008-07-08 | 2015-08-04 | Intellikine Llc | Kinase inhibitors and methods of use |
| US9139589B2 (en) | 2009-01-30 | 2015-09-22 | Millennium Pharmaceuticals, Inc. | Heteroaryls and uses thereof |
| JP2015227295A (en) * | 2014-05-30 | 2015-12-17 | 東ソー株式会社 | Production method of dithienobenzodithiophene |
| US9266886B2 (en) | 2014-02-03 | 2016-02-23 | Vitae Pharmaceuticals, Inc. | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| US9295673B2 (en) | 2011-02-23 | 2016-03-29 | Intellikine Llc | Combination of mTOR inhibitors and P13-kinase inhibitors, and uses thereof |
| US9359349B2 (en) | 2007-10-04 | 2016-06-07 | Intellikine Llc | Substituted quinazolines as kinase inhibitors |
| US9481674B1 (en) | 2016-06-10 | 2016-11-01 | Vitae Pharmaceuticals, Inc. | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| US9663515B2 (en) | 2014-11-05 | 2017-05-30 | Vitae Pharmaceuticals, Inc. | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| US9724354B2 (en) | 2013-03-22 | 2017-08-08 | Millennium Pharmaceuticals, Inc. | Combination of catalytic mTORC1/2 inhibitors and selective inhibitors of Aurora A kinase |
| US9796710B2 (en) | 2014-10-14 | 2017-10-24 | Vitae Pharmaceuticals, Inc. | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| US9845308B2 (en) | 2014-11-05 | 2017-12-19 | Vitae Pharmaceuticals, Inc. | Isoindoline inhibitors of ROR-gamma |
| US10301261B2 (en) | 2015-08-05 | 2019-05-28 | Vitae Pharmaceuticals, Llc | Substituted indoles as modulators of ROR-gamma |
| US10829481B2 (en) | 2016-01-29 | 2020-11-10 | Vitae Pharmaceuticals, Llc | Benzimidazole derivatives as modulators of ROR-gamma |
| US10913739B2 (en) | 2017-07-24 | 2021-02-09 | Vitae Pharmaceuticals, LLC (121374) | Inhibitors of RORγ |
| US11008340B2 (en) | 2015-11-20 | 2021-05-18 | Vitae Pharmaceuticals, Llc | Modulators of ROR-gamma |
| US11186573B2 (en) | 2017-07-24 | 2021-11-30 | Vitae Pharmaceuticals, Llc | Inhibitors of ROR gamma |
| US11547697B2 (en) | 2009-08-17 | 2023-01-10 | Millennium Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005023806A2 (en) * | 2003-09-05 | 2005-03-17 | Neurogen Corporation | Heteroaryl fused pyridines, pyrazines and pyrimidines as crf1 receptor ligands |
-
2007
- 2007-12-21 WO PCT/US2007/088534 patent/WO2008083070A1/en active Application Filing
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005023806A2 (en) * | 2003-09-05 | 2005-03-17 | Neurogen Corporation | Heteroaryl fused pyridines, pyrazines and pyrimidines as crf1 receptor ligands |
Cited By (58)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9359349B2 (en) | 2007-10-04 | 2016-06-07 | Intellikine Llc | Substituted quinazolines as kinase inhibitors |
| US9029411B2 (en) | 2008-01-25 | 2015-05-12 | Millennium Pharmaceuticals, Inc. | Thiophenes and uses thereof |
| US8637542B2 (en) | 2008-03-14 | 2014-01-28 | Intellikine, Inc. | Kinase inhibitors and methods of use |
| US9637492B2 (en) | 2008-03-14 | 2017-05-02 | Intellikine Llc | Benzothiazole kinase inhibitors and methods of use |
| US8993580B2 (en) | 2008-03-14 | 2015-03-31 | Intellikine Llc | Benzothiazole kinase inhibitors and methods of use |
| US9828378B2 (en) | 2008-07-08 | 2017-11-28 | Intellikine Llc | Kinase inhibitors and methods of use |
| US9096611B2 (en) | 2008-07-08 | 2015-08-04 | Intellikine Llc | Kinase inhibitors and methods of use |
| US9345706B2 (en) | 2008-11-03 | 2016-05-24 | Intellikine, Llc | Benzoxazole kinase inhibitors and methods of use |
| US8476431B2 (en) | 2008-11-03 | 2013-07-02 | Itellikine LLC | Benzoxazole kinase inhibitors and methods of use |
| US8476282B2 (en) | 2008-11-03 | 2013-07-02 | Intellikine Llc | Benzoxazole kinase inhibitors and methods of use |
| US9090601B2 (en) | 2009-01-30 | 2015-07-28 | Millennium Pharmaceuticals, Inc. | Thiazole derivatives |
| US8796314B2 (en) | 2009-01-30 | 2014-08-05 | Millennium Pharmaceuticals, Inc. | Heteroaryls and uses thereof |
| US9139589B2 (en) | 2009-01-30 | 2015-09-22 | Millennium Pharmaceuticals, Inc. | Heteroaryls and uses thereof |
| US11547697B2 (en) | 2009-08-17 | 2023-01-10 | Millennium Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| WO2011092293A3 (en) * | 2010-02-01 | 2011-10-20 | Novartis Ag | Cyclohexyl amide derivatives as crf receptor antagonists |
| US20140206884A1 (en) * | 2010-03-16 | 2014-07-24 | Basf Se | Process Using Grignard Reagents |
| US9167817B2 (en) * | 2010-03-16 | 2015-10-27 | Basf Se | Process using Grignard reagents |
| US8796268B2 (en) | 2010-08-11 | 2014-08-05 | Millennium Pharmaceuticals, Inc. | Heteroaryls and uses thereof |
| US9062038B2 (en) | 2010-08-11 | 2015-06-23 | Millennium Pharmaceuticals, Inc. | Heteroaryls and uses thereof |
| US8859768B2 (en) | 2010-08-11 | 2014-10-14 | Millennium Pharmaceuticals, Inc. | Heteroaryls and uses thereof |
| US8796271B2 (en) | 2010-08-11 | 2014-08-05 | Millennium Pharmaceuticals, Inc. | Heteroaryls and uses thereof |
| US8765746B2 (en) | 2010-10-13 | 2014-07-01 | Millennium Pharmaceuticals, Inc. | Heteroaryls and uses thereof |
| US9295673B2 (en) | 2011-02-23 | 2016-03-29 | Intellikine Llc | Combination of mTOR inhibitors and P13-kinase inhibitors, and uses thereof |
| EP2532661A1 (en) | 2011-06-10 | 2012-12-12 | Syngenta Participations AG | Novel insecticides |
| EP2567959A1 (en) | 2011-09-12 | 2013-03-13 | Sanofi | 6-(4-Hydroxy-phenyl)-3-styryl-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013156433A1 (en) | 2012-04-17 | 2013-10-24 | Syngenta Participations Ag | Insecticidally active thiazole derivatives |
| CN107007597A (en) * | 2012-05-31 | 2017-08-04 | 菲尼克斯药品股份公司 | It is used as the thiazole of orphan nuclear receptor ROR γ instrumentalities through formamide or sulfonamide substitutions and the pharmaceutical applications of related derivatives |
| TWI511960B (en) * | 2012-05-31 | 2015-12-11 | Phenex Pharmaceuticals Ag | Carboxamide or sulfonamide substituted thiazoles and related derivatives as modulators for the orphan nuclear receptor rorγ |
| AU2013270036B2 (en) * | 2012-05-31 | 2016-12-01 | Phenex Pharmaceuticals Ag | Carboxamide or sulfonamide substituted thiazoles and related derivatives as modulators for the orphan nuclear receptor ROR(gamma) |
| JP2015521193A (en) * | 2012-05-31 | 2015-07-27 | フェネックス ファーマシューティカルス アーゲー | Carboxamide or sulfonamide substituted thiazoles and related derivatives as modulators of the orphan nuclear receptor RORγ |
| EA028330B1 (en) * | 2012-05-31 | 2017-11-30 | Фенекс Фармасьютикалз Аг | CARBOXAMIDE OR SULFONAMIDE SUBSTITUTED THIAZOLES AND RELATED DERIVATIVES AS MODULATORS FOR THE ORPHAN NUCLEAR RECEPTOR RORγ |
| KR101742954B1 (en) | 2012-05-31 | 2017-06-02 | 페넥스 파마슈티컬스 아게 | Carboxamide or sulfonamide substituted thiazoles and related derivatives as modulators for the orphan nuclear receptor ror[gamma] |
| US10301272B2 (en) | 2012-05-31 | 2019-05-28 | Phenex Pharmaceuticals Ag | Carboxamide or sulfonamide substituted thiazoles and related derivatives as modulators for the orphan nuclear receptor ROR[γ] |
| WO2013178362A1 (en) * | 2012-05-31 | 2013-12-05 | Phenex Pharmaceuticals Ag | Carboxamide or sulfonamide substituted thiazoles and related derivatives as modulators for the orphan nuclear receptor ror[gamma] |
| US9724354B2 (en) | 2013-03-22 | 2017-08-08 | Millennium Pharmaceuticals, Inc. | Combination of catalytic mTORC1/2 inhibitors and selective inhibitors of Aurora A kinase |
| WO2015112642A1 (en) | 2014-01-21 | 2015-07-30 | Neurocrine Biosciences, Inc. | Crf1 receptor antagonists for the treatment of congenital adrenal hyperplasia |
| US11311544B2 (en) | 2014-01-21 | 2022-04-26 | Neurocrine Biosciences, Inc. | Treatment of congenital adrenal hyperplasia |
| US10905690B2 (en) | 2014-01-21 | 2021-02-02 | Neurocrine Biosciences, Inc. | Treatment of congenital adrenal hyperplasia |
| US11730739B2 (en) | 2014-01-21 | 2023-08-22 | Neurocrine Biosciences, Inc. | Treatment of congenital adrenal hyperplasia |
| US9624217B2 (en) | 2014-02-03 | 2017-04-18 | Vitae Pharmaceuticals, Inc. | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| US10047085B2 (en) | 2014-02-03 | 2018-08-14 | Vitae Pharmaceuticals, Inc. | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| US10399976B2 (en) | 2014-02-03 | 2019-09-03 | Vitae Pharmaceuticals, Llc | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| US10807980B2 (en) | 2014-02-03 | 2020-10-20 | Vitae Pharmaceuticals, Llc | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| US9266886B2 (en) | 2014-02-03 | 2016-02-23 | Vitae Pharmaceuticals, Inc. | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| US11535614B2 (en) | 2014-02-03 | 2022-12-27 | Vitae Pharmaceuticals, Llc | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| JP2015227295A (en) * | 2014-05-30 | 2015-12-17 | 東ソー株式会社 | Production method of dithienobenzodithiophene |
| US10087184B2 (en) | 2014-10-14 | 2018-10-02 | Vitae Pharmaceuticals, Inc. | Dihydropyrrolopyridine inhibitors of RORγ |
| US9796710B2 (en) | 2014-10-14 | 2017-10-24 | Vitae Pharmaceuticals, Inc. | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| US11001583B2 (en) | 2014-11-05 | 2021-05-11 | Vitae Pharmaceuticals, Llc | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| US9845308B2 (en) | 2014-11-05 | 2017-12-19 | Vitae Pharmaceuticals, Inc. | Isoindoline inhibitors of ROR-gamma |
| US9663515B2 (en) | 2014-11-05 | 2017-05-30 | Vitae Pharmaceuticals, Inc. | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| US10829448B2 (en) | 2015-08-05 | 2020-11-10 | Vitae Pharmaceuticals, Llc | Substituted benzoimidazoles as modulators of ROR-γ |
| US10301261B2 (en) | 2015-08-05 | 2019-05-28 | Vitae Pharmaceuticals, Llc | Substituted indoles as modulators of ROR-gamma |
| US11008340B2 (en) | 2015-11-20 | 2021-05-18 | Vitae Pharmaceuticals, Llc | Modulators of ROR-gamma |
| US10829481B2 (en) | 2016-01-29 | 2020-11-10 | Vitae Pharmaceuticals, Llc | Benzimidazole derivatives as modulators of ROR-gamma |
| US9481674B1 (en) | 2016-06-10 | 2016-11-01 | Vitae Pharmaceuticals, Inc. | Dihydropyrrolopyridine inhibitors of ROR-gamma |
| US11186573B2 (en) | 2017-07-24 | 2021-11-30 | Vitae Pharmaceuticals, Llc | Inhibitors of ROR gamma |
| US10913739B2 (en) | 2017-07-24 | 2021-02-09 | Vitae Pharmaceuticals, LLC (121374) | Inhibitors of RORγ |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2008083070A1 (en) | Crf1 receptor ligands comprising fused bicyclic heteroaryl moieties | |
| CN100369912C (en) | Heteroaryl substituted fused bicyclic heteroaryl compounds as GABAA receptor ligands | |
| WO2005115399A2 (en) | Imidazopyrazines, imidazopyridines, ans imidazopyrimidines as crf1 receptor ligands | |
| KR101803984B1 (en) | Heteroaromatic phenylimidazole derivatives as pde10a enzyme inhibitors | |
| US20060199823A1 (en) | Heteroaryl fused pyridines, pyrazines and pyrimidines as CRF1 receptor ligands | |
| US20050070542A1 (en) | 5-Aryl-pyrazolo[4,3-d]pyrimidines, pyridines, and pyrazines and related compounds | |
| KR20070120963A (en) | Imidazopyridazine compounds | |
| US20080004269A1 (en) | Pyrazolylmethy Heteroaryl Derivatives | |
| TWI487705B (en) | Heteroaromatic aryl triazole derivatives as pde10a enzyme inhibitors | |
| WO2007133756A2 (en) | Crf1 receptor ligands comprising heteroaryl fused bicycles | |
| JP2008007515A (en) | Specific alkylene diamine-substituted heterocycle | |
| TWI736578B (en) | Certain protein kinase inhibitors | |
| BRPI0809500A2 (en) | AND METHODS FOR MODULATING THE ACTIVITY OF A PROTEIN KINASE AND TREATING A DISEASE | |
| KR20100041711A (en) | Substituted pyrrolopyridines and pyrazolopyridines as kinase modulators | |
| KR20120123276A (en) | 2-arylimidazole derivatives as pde10a enzyme inhibitors | |
| Dwyer et al. | Discovery of pyrazolo [1, 5-a] pyrimidine-based Pim inhibitors: A template-based approach | |
| KR20140009192A (en) | Imidazole derivatives as pde10a enzyme inhibitors | |
| US6395766B1 (en) | Tetrahydroindolone derivatives as gabaaalpha5 ligands for enhancing cognition | |
| NO328580B1 (en) | 4- (2-Butylamino) -2,7-dimethyl-8- (2-methyl-6-methoxypyrid-3-yl) pyrazolo [1,5-A] -1,3,5-triazine, a pharmaceutically acceptable the drug thereof, the pharmaceutical composition, the use of the compound and the manufactured article comprising it | |
| US20080139580A1 (en) | Imidazo-pyrimidines and triazolo-pyrimidines: benzodiazepine receptor ligands | |
| Zhao et al. | Discovery of thieno [3, 2-c] pyridin-4-amines as novel Bruton’s tyrosine kinase (BTK) inhibitors | |
| BR112015002262A2 (en) | compounds, methods for the treatment of an inflammatory condition, rheumatoid arthritis, asthma, and immune disorder; pharmaceutical composition, use of the compound and invention | |
| KR20170005860A (en) | Fused triazole derivatives as phosphodiesterase 10a inhibitors | |
| JP2023552469A (en) | Aldehyde dehydrogenase inhibitors and their therapeutic uses | |
| EP1880998A1 (en) | Imidazo-pyrimidines and triazolo-pyrimidines: benzodiazepine receptor ligands |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07865948 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 07865948 Country of ref document: EP Kind code of ref document: A1 |