WO2007122411A1 - Diazepan-1-yl-sulfonyl derivatives for the treatment of metabolic syndrome - Google Patents
Diazepan-1-yl-sulfonyl derivatives for the treatment of metabolic syndrome Download PDFInfo
- Publication number
- WO2007122411A1 WO2007122411A1 PCT/GB2007/001507 GB2007001507W WO2007122411A1 WO 2007122411 A1 WO2007122411 A1 WO 2007122411A1 GB 2007001507 W GB2007001507 W GB 2007001507W WO 2007122411 A1 WO2007122411 A1 WO 2007122411A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- sulfonyl
- diazepane
- dichloropyridin
- compound
- phenyl
- Prior art date
Links
- YACXLISQUJIFCS-BYIUYBJJSA-N O=S(/C1=C/C=C\C=C/C/C=C\C=C1)(N1CCNCCC1)=O Chemical compound O=S(/C1=C/C=C\C=C/C/C=C\C=C1)(N1CCNCCC1)=O YACXLISQUJIFCS-BYIUYBJJSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D243/00—Heterocyclic compounds containing seven-membered rings having two nitrogen atoms as the only ring hetero atoms
- C07D243/06—Heterocyclic compounds containing seven-membered rings having two nitrogen atoms as the only ring hetero atoms having the nitrogen atoms in positions 1 and 4
- C07D243/08—Heterocyclic compounds containing seven-membered rings having two nitrogen atoms as the only ring hetero atoms having the nitrogen atoms in positions 1 and 4 not condensed with other rings
Definitions
- This invention relates to chemical compounds, or pharmaceutically acceptable salts thereof. These compounds possess human 11- ⁇ -hydroxysteroid dehydrogenase type 1 enzyme (1 l ⁇ HSDl) inhibitory activity and accordingly have value in the treatment of disease states including metabolic syndrome and are useful in methods of treatment of a warm-blooded animal, such as man.
- the invention also relates to processes for the manufacture of said compounds, to pharmaceutical compositions containing them and to their use in the manufacture of medicaments to inhibit ll ⁇ HSDlin a warm-blooded animal, such as man.
- Glucocorticoids Cortisol in man, corticosterone in rodents
- Glucocorticoids are also important in the differentiation of pre-adipocytes into mature adipocytes which are able to store triglycerides (Bujalska IJ et al. 1999; Endocrinology 140, 3188-3196).
- 1 l ⁇ HSDl knock-out mice show attenuated glucocorticoid-induced activation of gluconeogenic enzymes in response to fasting and lower plasma glucose levels in response to stress or obesity (Kotelevtsev Y et al. 1997; Proc. Natl. Acad. Sci USA 94, 14924-14929) indicating the utility of inhibition of 1 l ⁇ HSDl in lowering of plasma glucose and hepatic glucose output in type 2 diabetes. Furthermore, these mice express an anti-atherogenic lipoprotein profile, having low triglycerides, increased HDL cholesterol and increased apo-lipoprotein AI levels. (Morton NM et al. 2001 ; J. Biol. Chem.
- This phenotype is due to an increased hepatic expression of enzymes of fat catabolism and PP ARa. Again this indicates the utility of 1 l ⁇ HSDl inhibition in treatment of the dyslipidaemia of the metabolic syndrome.
- 1 l ⁇ HSDl transgenic mice When expressed under the control of an adipose specific promoter, 1 l ⁇ HSDl transgenic mice have high adipose levels of corticosterone, central obesity, insulin resistant diabetes, hyperlipidaemia and hyperphagia. Most importantly, the increased levels of 1 l ⁇ HSDl activity in the fat of these mice are similar to those seen in obese subjects. Hepatic 1 l ⁇ HSDl activity and plasma corticosterone levels were normal, however, hepatic portal vein levels of corticosterone were increased 3 fold and it is thought that this is the cause of the metabolic effects in liver.
- Glucocorticoids also decrease insulin secretion and this could exacerbate the effects of glucocorticoid induced insulin resistance.
- Pancreatic islets express ll ⁇ HSDl and carbenoxolone can inhibit the effects of 11-dehydocorticosterone on insulin release (Davani B et al. 2000; J. Biol. Chem. 275, 34841-34844).
- 11 ⁇ HSDl inhibitors may not only act at the tissue level on insulin resistance but also increase insulin secretion itself.
- Skeletal development and bone function is also regulated by glucocorticoid action.
- 11 ⁇ HSDl is present in human bone osteoclasts and osteoblasts and treatment of healthy volunteers with carbenoxolone showed a decrease in bone resorption markers with no change in bone formation markers (Cooper MS et al 2000; Bone 27, 375-381). Inhibition of 11 ⁇ HSDl activity in bone could be used as a protective mechanism in treatment of osteoporosis.
- Glucocorticoids may also be involved in diseases of the eye such as glaucoma.
- 11 ⁇ HSD 1 has been shown to affect intraocular pressure in man and inhibition of 11 ⁇ HSD 1 may be expected to alleviate the increased intraocular pressure associated with glaucoma (Rauz S et al. 2001; Investigative Opthalmology & Visual Science 42, 2037-2042).
- the WHO consultation has recommended the following definition which does not imply causal relationships and is suggested as a working definition to be improved upon in due course: s >
- the patient has at least one of the following conditions: glucose intolerance, impaired glucose tolerance (IGT) or diabetes mellitus and/or insulin resistance; together with two or more of the following:
- Ring A is phenyl or naphthyl, optionally substituted by 1, 2 or 3 substituents independently selected from R 1 ;
- R 1 is selected from hydroxy, halo, trifluoromethyl, C(OH)(CF 3 )-, Ci. 4 alkoxy, cyano, Ci. 4 alkylcarbonyl and Ring B is phenyl or pyridyl, optionally substituted in the 2-, T-, 3- or 3'- positions (relative to the point of attachment) (where available) by 1 or 2 substituents independently selected from R 2 , and/or substituted in the 4- position (relative to the point of attachment) (where available) with a substituent selected from R 3 ;
- R 2 is selected from halo, cyano, trifluoromethyl, Ci -4 alkyl and Ci_ 4 alkoxy;
- R 3 is selected from halo, cyano, Ci -3 alkylsulfonyl and -CONR 4 R 5 ;
- R 4 and R 5 are independently selected from hydrogen and Ci -4 alkyl; or
- R 4 and R 5 together with the nitrogen to which they are attached form an azetidinyl or pyrrolidinyl ring; provided that the compound of formula (1) is not:
- alkyl includes both straight and branched chain alkyl groups
- “Ci. 4 alkyl” includes propyl, isopropyl and t-buty ⁇ .
- references to individual alkyl groups such as 'propyl' are specific for the straight chained version only and references to individual branched chain alkyl groups such as 'isopropyl' are specific for the branched chain version only.
- references to other radicals therefore
- Ci- 4 alkylcarbonyl would include prop-1-ylcarbonyl and but-3-ylcarbonyl.
- halo refers to fiuoro, chloro, bromo and iodo.
- Examples of "Ci -4 alkoxy” include methoxy, ethoxy and propoxy. Examples of include formamido, acetamido and propionylamino. Examples of “Ci-salkylsulphonyl” include mesyl and ethylsulphonyl. include propionyl and acetyl. Examples of “Ci -3 alkyl” include methyl, ethyl, propyl and isopropyl. include the examples of "C 1-3 alkyl” as well as butyl, isobutyl, sec- butyl and tert-butyl.
- hydroxyCi_ 4 alkyl examples include hydroxymethyl, hydroxyethyl, l-hydroxyprop-2-yl and l-hydroxyprop-3-yl.
- C ⁇ alkylcarbonyl examples include methylcarbonyl, ethylcarbonyl, propylcarbonyl, isopropylcarbonyl and tert-butylcarbonyl.
- the present invention relates to the compounds of formula (1) as hereinbefore defined as well as to the salts thereof.
- Salts for use in pharmaceutical compositions will be pharmaceutically acceptable salts, but other salts may be useful in the production of the compounds of formula (1) and their pharmaceutically acceptable salts.
- a suitable pharmaceutically acceptable salt of a compound of the invention is, for example, an acid-addition salt of a compound of the invention which is sufficiently basic, for example, an acid-addition salt with, for example, an inorganic or organic acid, for example hydrochloric, hydrobromic, sulphuric, phosphoric, citric or maleic acid.
- a suitable pharmaceutically acceptable salt of a compound of the invention which is sufficiently acidic is an alkali metal salt, for example a sodium or potassium salt, an alkaline earth metal salt, for example a calcium or magnesium salt, an ammonium salt or a salt with an organic base which affords a physiologically-acceptable cation, for example a salt with methylamine, dimethylamine, trimethylamine, piperidine, morpholine or tris-(2-hydroxyethyi)amine.
- an alkali metal salt for example a sodium or potassium salt
- an alkaline earth metal salt for example a calcium or magnesium salt
- an ammonium salt or a salt with an organic base which affords a physiologically-acceptable cation
- a salt with methylamine, dimethylamine, trimethylamine, piperidine, morpholine or tris-(2-hydroxyethyi)amine for example a salt with methylamine, dimethylamine, trimethylamine, piperidine, morpholine or tris-(2-hydroxye
- Some compounds of the formula (1) may have chiral centres and/or geometric isomeric centres (E- and Z- isomers), and it is to be understood that the invention encompasses all such optical, diastereoisomers and geometric isomers that possess 1 l ⁇ HSDl inhibitory activity.
- the invention relates to any and all tautomeric forms of the compounds of the formula (1) that possess 1 l ⁇ HSDl inhibitoiy activity.
- Ring A is phenyl, optionally substituted by 1, 2 or 3 substituents independently selected from R 1 ;
- Ring A is naphthyl, optionally substituted by 1, 2 or 3 substituents independently selected from R 1 ;
- Ring A is phenyl, substituted by 1 or 2 substituents independently selected from R 1
- Ring A is naphthyl 5)
- R 1 is selected from hydroxy, fluoro, chloro, trifluoromethyl, Ci -4 alkyl, hydroxy C 1-4 alkyl, MeC(OH)(CF 3 )-, methoxy, cyano, methylcarbonyl and methylcarbonylamino
- R 1 is selected from hydroxy, fluoro, chloro, trifluoromethyl, Ci -4 alkyl, hydroxy C 1-4 alkyl, methoxy, cyano, methylcarbonyl and methylcarbonylamino
- R 1 is MeC(OH)(CF 3 )- 8) Ring B is phenyl
- Ring B is pyridyl
- Ring B is 2-pyridyl or 4-pyridyl
- Ring B is substituted in the 2- and/or T- positions (relative to the point of attachment) by one or two, as appropriate, substituents independently selected from R 2 , particularly wherein R 2 is selected from chloro, fluoro and trifluoromethyl
- Ring B is substituted on the 4- position (relative to the point of attachment) (where available) with a substituent selected from R 3
- R 2 is selected from fluoro, chloro, cyano, trifluoromethyl, Ci_ 4 alkyl and C 1-4 alkoxy
- R 2 is selected from fluoro, chloro, cyano, trifluoromethyl, Ci -4 alkyl and methoxy 15) R 2 is selected from chloro, fluoro and trifluoromethyl
- R 3 is selected from fluoro, chloro, cyano, Ci -3 alkylsulfonyl, -CONHMe and -CONMe 2 17) R 3 is -CONR 4 R 5 wherein R 4 and R 5 together with the nitrogen to which they are attached form an azetidinyl or pyrrolidinyl ring.
- suitable compounds of the invention are any one or more of the Examples or a salt thereof.
- suitable compounds of the invention are any one or more of the following or a salt thereof: l-(3,5-dichloropyridin-4-yl)-4-(4-fluorophenyl)sulfonyl-l,4-diazepane; l-(2,4-dichlorophenyl)sulfonyl-4-(3,5-dichloropyridin-4-yl)-l,4-diazepane; l-(3,5-dichloropyridin-4-yl)-4-(3-methylphenyl)sulfonyl-l,4-diazepane; 1 -(3 ,5 -dichloropyridin-4-yl)-4-(4-methylphenyl)sulfonyl- 1 ,4-diazepane;
- Another aspect of the present invention provides a process for preparing a compound of formula (1) or a pharmaceutically acceptable salt thereof which process (wherein variable groups are, unless otherwise specified, as defined in formula (I)) comprises any one of processes a) to c): a) reaction of a compound of Formula (2) with a compound of Formula (3):
- Examples of conversions of a compound of Formula (1) into another compound of Formula (1) include functional group interconversions such as hydrolysis, hydrogenation, hydrogenolysis, oxidation or reduction, and/or further functionalisation by standard reactions such as amide or metal-catalysed coupling, or nucleophilic displacement reactions.
- Suitable conditions for the above processes a) to c) are as follows.
- a) Process a) may be carried out in a suitable solvent such as DCM, DMF, pyridine or water, typically with the addition of a base such as triethylamine, DIPEA, pyridine, or aqueous sodium hydroxide.
- Process b) may be carried out without solvent, or in a suitable solvent such as DMA, DMF or xylene; typically the reaction is carried out without solvent at elevated temperature, using Microwave or conventional heating.
- Compounds of formula (4) may be made by reaction of a compound of Formula (10) with a compound of Formula (11):
- X 4 is a leaving group and R is hydrogen or a suitable protecting group, followed by removal of said protecting group if appropriate;
- Process c) is typically carried out in an anhydrous aprotic solvent such as THF or diethyl ether;
- suitable organometallic reagents for process c) are alkyl or aryl magnesium halides (Grignard reagent), alkyl or aryl lithium, or trimethyl (trifluoromethyl) silane (Ruppert's reagent).
- Suitable examples of leaving groups for processes a) to c) are: fluoro, chloro, bromo, iodo, mesylate, tosylate or triflate.
- Suitable examples of protecting groupsfor processes a) to c) are: tert-butyl oxycarbonyl (Boc), benzyl oxycarbonyl (Z), acetyl or trifluoracetyl.
- Boc tert-butyl oxycarbonyl
- Z benzyl oxycarbonyl
- acetyl or trifluoracetyl tert-butyl oxycarbonyl
- the reactions described above may be performed under standard conditions known to the person skilled in the art.
- the intermediates described above are commercially available, are known in the art or may be prepared by known procedures and/or by the procedures shown above.
- aromatic substitution reactions include the introduction of a nitro group using concentrated nitric acid, the introduction of an acyl group using, for example, an acyl halide and Lewis acid (such as aluminium trichloride) under Friedel Crafts conditions; the introduction of an alkyl group using an alkyl halide and Lewis acid (such as aluminium trichloride) under Friedel Crafts conditions; and the introduction of a halogeno group.
- modifications include the reduction of a nitro group to an amino group by for example, catalytic hydrogenation with a nickel catalyst or treatment with iron in the presence of hydrochloric acid with heating; oxidation of alkylthio to alkylsulphinyl or alkylsulphonyl.
- a suitable protecting group for an amino or alkylamino group is, for example, an acyl group, for example an alkanoyl group such as acetyl, an alkoxycarbonyl group, for example a methoxycarbonyl, ethoxycarbonyl or t-butoxycarbonyl group, an arylmethoxycarbonyl group, for example benzyloxycarbonyl, or an aroyl group, for example benzoyl.
- the deprotection conditions for the above protecting groups necessarily vary with the choice of protecting group.
- an acyl group such as an alkanoyl or alkoxycarbonyl group or an aroyl group may be removed for example, by hydrolysis with a suitable base such as an alkali metal hydroxide, for example lithium or sodium hydroxide.
- a suitable base such as an alkali metal hydroxide, for example lithium or sodium hydroxide.
- an acyl group such as a /-butoxycarbonyl group may be removed, for example, by treatment with a suitable acid as hydrochloric, sulphuric or phosphoric acid or trifluoroacetic acid and an arylmethoxycarbonyl group such as a benzyloxycarbonyl group may be removed, for example, by hydrogenation over a catalyst such as palladium-on-carbon, or by treatment with a Lewis acid for example boron tris(trifluoroacetate).
- a suitable alternative protecting group for a primary amino group is, for example, a phthaloyl group which may be removed by treatment with an alkylamine, for example hydroxylamine, or with hydrazine.
- a suitable protecting group for a hydroxy group is, for example, an acyl group, for example an alkanoyl group such as acetyl, an aroyl group, for example benzoyl, or an arylmethyl group, for example benzyl.
- the deprotection conditions for the above protecting groups will necessarily vary with the choice of protecting group.
- an acyl group such as an alkanoyl or an aroyl group may be removed, for example, by hydrolysis with a suitable base such as an alkali metal hydroxide, for example lithium or sodium hydroxide.
- a suitable base such as an alkali metal hydroxide, for example lithium or sodium hydroxide.
- an arylmethyl group such as a benzyl group may be removed, for example, by hydrogenation over a catalyst such as palladium-on-carbon.
- a suitable protecting group for a carboxy group is, for example, an esterifying group, for example a methyl or an ethyl group which may be removed, for example, by hydrolysis with a base such as sodium hydroxide, or for example a /-butyl group which may be removed, for example, by treatment with an acid, for example an organic acid such as trifluoroacetic acid, or for example a benzyl group which may be removed, for example, by hydrogenation over a catalyst such as palladium-on-carbon.
- a base such as sodium hydroxide
- a /-butyl group which may be removed, for example, by treatment with an acid, for example an organic acid such as trifluoroacetic acid, or for example a benzyl group which may be removed, for example, by hydrogenation over a catalyst such as palladium-on-carbon.
- the protecting groups may be removed at any convenient stage in the synthesis using conventional techniques well known in the chemical art.
- cortisone to the active steroid Cortisol by 1 l ⁇ HSDl oxo-reductase activity
- a competitive immunoassay ⁇ 1 kit (Assay Designs, Inc: 800 Technology Drive, Ann Arbor, MI 48108, USA. Cortisol Enzyme Immunoassay kit: Cat No. 901-071).
- DMSO dimethyl sulphoxide
- the assay was carried out in a total volume of 200 ⁇ l consisting of cortisone (Sigma, Poole, Dorset, UK, l ⁇ M), glucose-6-phosphate (Roche Diagnostics, ImM), NADPH (Roche Diagnostics, lOO ⁇ M), glucose-6-phosphate dehydrogenase (Roche Diagnostics, 12.5 ⁇ g/ml), EDTA (Sigma, Poole, Dorset, UK, ImM), assay buffer (K 2 HPO 4 /KH 2 PO 4 , 10OmM) pH 7.5, recombinant 1 l ⁇ HSDl (5 ⁇ g/ml) plus test compound.
- the assay plates were incubated for 30 minutes at 37°C after which time the reaction was stopped by the addition of 20 ⁇ l of ImM glycerrhetinic acid.
- compositions of the invention which comprises a compound of formula (I), or a pharmaceutically acceptable salt thereof, as defined hereinbefore in association with a pharmaceutically-acceptable diluent or carrier.
- the compositions of the invention may be in a form suitable for oral use (for example as tablets, lozenges, hard or soft capsules, aqueous or oily suspensions, emulsions, dispersible powders or granules, syrups or elixirs), for topical use (for example as creams, ointments, gels, or aqueous or oily solutions or suspensions), for administration by inhalation (for example as a finely divided powder or a liquid aerosol), for administration by insufflation (for example as a finely divided powder) or for parenteral administration (for example as a sterile aqueous or oily solution for intravenous, subcutaneous, intramuscular or intramuscular dosing or as a suppository for rectal do
- compositions of the invention may be obtained by conventional procedures using conventional pharmaceutical excipients, well known in the art.
- compositions intended for oral use may contain, for example, one or more colouring, sweetening, flavouring and/or preservative agents.
- Suitable pharmaceutically acceptable excipients for a tablet formulation include, for example, inert diluents such as lactose, sodium carbonate, calcium phosphate or calcium carbonate, granulating and disintegrating agents such as corn starch or algenic acid; binding agents such as starch; lubricating agents such as magnesium stearate, stearic acid or talc; preservative agents such as ethyl or propyl p_-hydroxybenzoate, and anti-oxidants, such as ascorbic acid.
- Tablet formulations may be uncoated or coated either to modify their disintegration and the subsequent absorption of the active ingredient within the gastrointestinal tract, or to improve their stability and/or appearance, in either case, using conventional coating agents and procedures well known in the art.
- Compositions for oral use may be in the form of hard gelatin capsules in which the active ingredient is mixed with an inert solid diluent, for example, calcium carbonate, calcium phosphate or kaolin, or as soft gelatin capsules in which the active ingredient is mixed with water or an oil such as peanut oil, liquid paraffin, or olive oil.
- an inert solid diluent for example, calcium carbonate, calcium phosphate or kaolin
- water or an oil such as peanut oil, liquid paraffin, or olive oil.
- Aqueous suspensions generally contain the active ingredient in finely powdered form together with one or more suspending agents, such as sodium carboxymethylcellulose, methylcellulose, hydroxypropylmethylcellulose, sodium alginate, polyvinylpyrrolidone, gum tragacanth and gum acacia; dispersing or wetting agents such as lecithin or condensation products of an alkylene oxide with fatty acids (for example polyoxethylene stearate), or condensation products of ethylene oxide with long chain aliphatic alcohols, for example lieptadecaethyleneoxycetanol, or condensation products of ethylene oxide with partial esters derived from fatty acids and a hexitol such as polyoxyethylene sorbitol monooleate, or condensation products of ethylene oxide with long chain aliphatic alcohols, for example heptadecaethyleneoxycetanol, or condensation products of ethylene oxide with partial esters derived from fatty acids and a hexitol such as polyoxyethylene sorbitol mono
- the aqueous suspensions may also contain one or more preservatives (such as ethyl or propyl p-hydroxybenzoate, anti-oxidants (such as ascorbic acid), colouring agents, flavouring agents, and/or sweetening agents (such as sucrose, saccharine or aspartame).
- Oily suspensions may be formulated by suspending the active ingredient in a vegetable oil (such as arachis oil, olive oil, sesame oil or coconut oil) or in a mineral oil (such as liquid paraffin).
- the oily suspensions may also contain a thickening agent such as beeswax, hard paraffin or cetyl alcohol. Sweetening agents such as those set out above, and flavouring agents may be added to provide a palatable oral preparation. These compositions may be preserved by the addition of an anti-oxidant such as ascorbic acid.
- Dispersible powders and granules suitable for preparation of an aqueous suspension by the addition of water generally contain the active ingredient together with a dispersing or wetting agent, suspending agent and one or more preservatives. Suitable dispersing or wetting agents and suspending agents are exemplified by those already mentioned above. Additional excipients such as sweetening, flavouring and colouring agents, may also be present.
- the pharmaceutical compositions of the invention may also be in the form of oil-in-water emulsions.
- the oily phase may be a vegetable oil, such as olive oil or arachis oil, or a mineral oil, such as for example liquid paraffin or a mixture of any of these.
- Suitable emulsifying agents may be, for example, naturally-occurring gums such as gum acacia or gum tragacanth, naturally-occurring phosphatides such as soya bean, lecithin, an esters or partial esters derived from fatty acids and hexitol anhydrides (for example sorbitan monooleate) and condensation products of the said partial esters with ethylene oxide such as polyoxyethylene sorbitan monooleate.
- the emulsions may also contain sweetening, flavouring and preservative agents.
- Syrups and elixirs may be formulated with sweetening agents such as glycerol, propylene glycol, sorbitol, aspartame or sucrose, and may also contain a demulcent, preservative, flavouring and/or colouring agent.
- sweetening agents such as glycerol, propylene glycol, sorbitol, aspartame or sucrose, and may also contain a demulcent, preservative, flavouring and/or colouring agent.
- compositions may also be in the form of a sterile injectable aqueous or oily suspension, which may be formulated according to known procedures using one or more of the appropriate dispersing or wetting agents and suspending agents, which have been mentioned above.
- a sterile injectable preparation may also be a sterile injectable solution or suspension in a non-toxic parenterally-acceptable diluent or solvent, for example a solution in 1,3-butanediol.
- Compositions for administration by inhalation may be in the form of a conventional pressurised aerosol arranged to dispense the active ingredient either as an aerosol containing finely divided solid or liquid droplets.
- Conventional aerosol propellants such as volatile fluorinated hydrocarbons or hydrocarbons may be used and the aerosol device is conveniently arranged to dispense a metered quantity of active ingredient.
- the amount of active ingredient that is combined with one or more excipients to produce a single dosage form will necessarily vary depending upon the host treated and the particular route of administration.
- a formulation intended for oral administration to humans will generally contain, for example, from 0.5 mg to 2 g of active agent compounded with an appropriate and convenient amount of excipients which may vary from about 5 to about 98 percent by weight of the total composition.
- Dosage unit forms will generally contain about 1 mg to about 500 mg of an active ingredient.
- metabolic syndrome relates to metabolic syndrome as defined in 1) and/or 2) or any other recognised definition of this syndrome.
- Synonyms for "metabolic syndrome” used in the art include Reaven's Syndrome, Insulin Resistance Syndrome and Syndrome X. It is to be understood that where the term “metabolic syndrome” is used herein it also refers to Reaven's Syndrome, Insulin
- a compound of formula (1), or a pharmaceutically acceptable salt thereof, as defined hereinbefore for use as a medicament there is provided a compound of formula (1), or a pharmaceutically acceptable salt thereof, as defined hereinbefore for use as a medicament.
- production of or producing an 11 ⁇ HSDl inhibitory effect refers to the treatment of metabolic syndrome.
- production of an 11 ⁇ HSDl inhibitory effect is referred to this refers to the treatment of diabetes, obesity, hyperlipidaemia, hyperglycaemia, hyperinsulinemia or hypertension, particularly diabetes and obesity.
- production of an 11 ⁇ HSDl inhibitory effect is referred to this refers to the treatment of glaucoma, osteoporosis, tuberculosis, dementia, cognitive disorders or depression.
- an 11 ⁇ HSD 1 inhibitory effect refers to the treatment of cognitive disorders, such as improving the cognitive ability of an individual, for example by improvement of verbal fluency, verbal memory or logical memory, or for treatment of mild cognitive disorders. See for example WO03/086410 and references contained therein, and Proceedings of National Academy of Sciences (PNAS), 2001, 98(8), 4717-4721.
- production of an 1 l ⁇ HSDl inhibitory effect is referred to this refers to the treatment of, delaying the onset of and/or reducing the risk of atherosclerosis - see for example J. Experimental Medicine, 2005, 202(4), 517-527.
- ll ⁇ HSDl inhibitory effect refers to the treatment of Alzheimers and/or neurodegenerative disorders.
- a method for producing an 11 ⁇ HSDl inhibitory effect in a wa ⁇ n-blooded animal, such as man, in need of such treatment which comprises administering to said animal an effective amount of a compound of formula (1), or a pharmaceutically acceptable salt thereof.
- a compound of formula (1) or a pharmaceutically acceptable salt thereof.
- (1), or a pharmaceutically acceptable salt thereof are also useful as pharmacological tools in the development and standardisation of in vitro and in vivo test systems for the evaluation of the effects of inhibitors of ll ⁇ HSDl in laboratory animals such as cats, dogs, rabbits, monkeys, rats and mice, as part of the search for new therapeutic agents.
- the inhibition of 1 l ⁇ HSDl described herein may be applied as a sole therapy or may involve, in addition to the subject of the present invention, one or more other substances and/or treatments. Such conjoint treatment may be achieved by way of the simultaneous, sequential or separate administration of the individual components of the treatment. Simultaneous treatment may be in a single tablet or in separate tablets.
- agents than might be co-administered with 11 ⁇ HSDl inhibitors, particularly those of the present invention may include the following main categories of treatment:
- Insulin secretagogues including sulphonylureas (for example glibenclamide, glipizide), prandial glucose regulators (for example repaglinide, nateglinide), glucagon-like peptide 1 agonist (GLPl agonist) (for example exenatide, liraglutide) and dipeptidyl peptidase IV inhibitors (DPP-IV inhibitors);
- sulphonylureas for example glibenclamide, glipizide
- prandial glucose regulators for example repaglinide, nateglinide
- GLPl agonist glucagon-like peptide 1 agonist
- DPP-IV inhibitors dipeptidyl peptidase IV inhibitors
- Insulin sensitising agents including PPAR ⁇ agonists (for example pioglitazone and rosiglitazone);
- Agents that suppress hepatic glucose output for example metformin
- Agents designed to reduce the absorption of glucose from the intestine for example acarbose
- Agents designed to treat the complications of prolonged hyperglycaemia e.g. aldose reductase inhibitors
- anti-diabetic agents including phosotyrosine phosphatase inhibitors, glucose 6 - phosphatase inhibitors, glucagon receptor antagonists, glucokinase activators, glycogen phosphorylase inhibitors, fructose 1,6 bisphosphastase inhibitors, glutamine:fructose -6-phosphate amidotransferase inhibitors
- Anti-obesity agents for example sibutramine and orlistat
- Anti- dyslipidaemia agents such as, HMG-CoA reductase inhibitors (statins, eg pravastatin); PP ARa agonists (f ⁇ brates, eg gemfibrozil); bile acid sequestrants (cholestyramine); cholesterol absorption inhibitors (plant stanols, synthetic inhibitors); ileal bile acid absorption inhibitors (IBATi), cholesterol ester transfer protein inhibitors and nicotinic acid and analogues (niacin and slow release formulations);
- Antihypertensive agents such as, ⁇ blockers (eg atenolol, inderal); ACE inhibitors (eg lisinopril); calcium antagonists (eg. nifedipine); angiotensin receptor antagonists (eg candesartan), ⁇ antagonists and diuretic agents (eg. furosemide, benzthiazide); l l)Haemostasis modulators such as, antithrombotics, activators of fibrinolysis and antiplatelet agents; thrombin antagonists; factor Xa inhibitors; factor Vila inhibitors); antiplatelet agents (eg.
- ⁇ blockers eg atenolol, inderal
- ACE inhibitors eg lisinopril
- calcium antagonists eg. nifedipine
- angiotensin receptor antagonists eg candesartan
- ⁇ antagonists and diuretic agents eg. furosemide, benzthiazide
- Anti-inflammatory agents such as non-steroidal anti-infammatory drugs (eg. aspirin) and steroidal anti-inflammatory agents (eg. cortisone).
- LCMS were recorded on a system comprising Waters 2790 LC equipped with a Waters 996 Photodiode array detector and Micromass ZMD MS (using a Phenomenex® Synergi 4 ⁇ MAX-RP 8OA 50x2 mm column), or a Waters 2795 LC equipped with a Waters 2996 Photodiode array detector and Micromass ZQ MS (using a Phenomenex® Gemini 5 ⁇ C18 1 lOAngstrom 50x2 mm column), and eluting with a flow rate of 1.1 ml/min with 5% (Water/ Acetonitrile (1:1) + 1% formic acid) and a gradient increasing from 0-95% of acetonitrile over the first 4 minutes, the balance (95-0%) being water; mass spectra (MS) (loop) were recorded on a Micromass LCT equipped with HP 1100 detector, or a Thermo LCQ; unless otherwise stated the mass ion quoted is (M+H +
- an Isolute SCX-2 column is referred to, this means an "ion exchange" extraction cartridge for adsorption of basic compounds, i.e. a polypropylene tube containing a benzenesulphonic acid based strong cation exchange sorbent, used according to the manufacturers instructions obtained from International Sorbent Technologies Limited, Dyffryn Business Park, Hengeod, Mid Glamorgan, UK, CF82 7RJ; viii) where an Isolute-NH2 column is referred to, this means an "ion exchange" extraction cartridge for adsorption of acidic compounds, i.e. a polypropylene tube containing a amino silane covalently bonded to a silica particle used according to the manufacturers instructions obtained from International Sorbent Technologies Limited, Dyffryn Business Park, Hengeod,
- Isco CombiFlash Optix-10 parallel flash chromatography system is referred to this means an automated chromatography workstation capable of cany ing out up to 10 purifications in parallel via flash chromatography using pre packed silica cartridges; x) where a "Biotage 9Og silica column” is referred to this means an automated chromatography workstation capable of carrying out up to 4 purifications in parallel via flash chromatography using pre packed silica cartridges, eg Si 12+M available from Biotage Inc.
- 1,4-Diazepane (5.49g, 54.8 mmol) and 3,4,5-trichloropyridine (2g, 3.0 mmol) were combined without solvent, and the mixture heated with stirring at 90 0 C for 2.5 hours.
- the reaction mixture was cooled to room temperature and the product purified by flash column chromatography (CombiFlash ® Companion TM , 4Og silica column, eluting with a 10:1 mixture of DCM and methanol) to give the title compound as a yellow oil (2.06 g, 76% );
- 1 H NMR 400 MHz, CDCl 3 ) ⁇ 1.88 - 1.94 (2H, m), 3.02 - 3.04 (2H, m), 3.09 (2H, t), 3.34 - 3.37 (4H, m), 8.39 (2H, s), m/z 246 (M+H) + [I].
- Example 1 1 -(3,5-dichloropyridin-4-yl)-4-(4-fluorophenyl)sulfonyl- 1 ,4-diazepane
- Example 2 1 -(2,4-dichlorophenyl)sulfonyl-4-(3 ,5-dichloropyridin-4-yl)- 1 ,4-diazepane
- Example 3 1 -(3 ,5 -dichloropyridin-4-yl)-4-(3-methylphenyl)sulfony 1-1 ,4-diazepane
- Example 4 l-(3,5-dichloropyridin-4-yl)-4-(4-methylphenyl)sulfonyl-l,4-diazepane
- Example 5 l-(3,5-dichloropyridin-4-yl)-4-(2-methoxyphenyl)sulfonyl-l,
- Example 16 1 -(3 , 5 -dichloropyridin-4-yl)-4- [3 -(trifiuoromethyl)phenyl] sulfonyl- 1 ,4- diazepane
- Example 17 2-[[4-(3,5-dichloropyridin-4-yl)-l,4-diazepan-l-yl]sulfonyl]benzonitrile
- Example 18 1 -(3 -chlorophenyl)sulfonyl-4-(3, 5 -dichloropyridin-4-yl)- 1,4-diazepane
- Example 19 1 -(3 ,5-dichloropyridin-4-yl)-4-(2-methylphenyl)sulfonyl- 1 ,4-diazepane
- 1,4-diazepane (1.3 g, 13 mmol) and 2-chloro-3-(trifluoromethyl) pyridine (473 mg, 2.6 mmol) were combined without solvent, and the mixture heated with stirring at 90 0 C for 3.5 hours.
- the reaction mixture was cooled to room temperature, dissolved in EtOAc, and the solution washed sequentially with water (10 ml) and brine (10 ml).
- Example 21 1 -(2-chlorophenyl)sulfonyl-4-(3 -chloropyridin-2-yl)- 1 ,4-diazepane
- Example 25 1 -(2-chlorophenyl)sulfonyl-4-(2,6-dichlorophenyl)- 1 ,4-diazepane
- Example 28 l-[4-[[4-[3-(trifluoromethyl)pyridin-2-yl]-l,4-diazepan-l- yl] sulfonyljphenyl] ethanone
- Example 33 1,1,1 -trifluoro-2-[4-[[4-[3-(lrifluoromethyl)pyridin-2-yl]-l ,4-diazepan- 1 - yl]sulfonyl]phenyl]propan-2-ol
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Diabetes (AREA)
- General Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Emergency Medicine (AREA)
- Endocrinology (AREA)
- Child & Adolescent Psychology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Pyridine Compounds (AREA)
Abstract
Compounds of formula (I): wherein variable groups are defined within; their use in the inhibition of 11βHSD1, processes for making them and pharmaceutical compositions comprising them are described.
Description
DIAZEPAN-1-YL-SULFONYL DERIVATIVES FOR THE TREATMENT OF METABOLIC SYNDROME
This invention relates to chemical compounds, or pharmaceutically acceptable salts thereof. These compounds possess human 11-β-hydroxysteroid dehydrogenase type 1 enzyme (1 lβHSDl) inhibitory activity and accordingly have value in the treatment of disease states including metabolic syndrome and are useful in methods of treatment of a warm-blooded animal, such as man. The invention also relates to processes for the manufacture of said compounds, to pharmaceutical compositions containing them and to their use in the manufacture of medicaments to inhibit llβHSDlin a warm-blooded animal, such as man. Glucocorticoids (Cortisol in man, corticosterone in rodents) are counter regulatory hormones i.e. they oppose the actions of insulin (Dallman MF, Strack AM, Akana SF et al. 1993; Front Neuroendocrinol 14, 303-347). They regulate the expression of hepatic enzymes involved in gluconeogenesis and increase substrate supply by releasing glycerol from adipose tissue (increased lipolysis) and amino acids from muscle (decreased protein synthesis and increased protein degradation). Glucocorticoids are also important in the differentiation of pre-adipocytes into mature adipocytes which are able to store triglycerides (Bujalska IJ et al. 1999; Endocrinology 140, 3188-3196). This may be critical in disease states where glucocorticoids induced by "stress" are associated with central obesity which itself is a strong risk factor for type 2 diabetes, hypertension and cardiovascular disease (Bjorntorp P & Rosmond R 2000; Int. J. Obesity 24, S80-S85)
It is now well established that glucocorticoid activity is controlled not simply by secretion of Cortisol but also at the tissue level by intracellular interconversion of active Cortisol and inactive cortisone by the 11-beta hydroxysteroid dehydrogenases, 1 lβHSDl (which activates cortisone) and 1 lβHSD2 (which inactivates Cortisol) (Sandeep TC & Walker BR 2001 Trends in Endocrinol & Metab. 12, 446-453). That this mechanism may be important in man was initially shown using carbenoxolone (an anti-ulcer drug which inhibits both 1 lβHSDl and 2) treatment which (Walker BR et al. 1995; J. Clin. Endocrinol. Metab. 80, 3155-3159) leads to increased insulin sensitivity indicating that 1 lβHSDl may well be regulating the effects of insulin by decreasing tissue levels of active glucocorticoids (Walker BR et al. 1995; J. Clin. Endocrinol. Metab. 80, 3155-3159).
Clinically, Cushing's syndrome is associated with Cortisol excess which in turn is associated with glucose intolerance, central obesity (caused by stimulation of pre-adipocyte differentiation in this depot), dyslipidaemia and hypertension. Cushing's syndrome shows a number of clear parallels with metabolic syndrome. Even though the metabolic syndrome is not generally associated with excess circulating Cortisol levels (Jessop DS et al. 2001; J. Clin. Endocrinol. Metab. 86, 4109-4114) abnormally high 1 lβHSDl activity within tissues would be expected to have the same effect. In obese men it was shown that despite having similar or lower plasma Cortisol levels than lean controls, 1 lβHSDl activity in subcutaneous fat was greatly enhanced (Rask E et al. 2001; J. Clin. Endocrinol. Metab. 1418-1421). Furthermore, the central fat, associated with the metabolic syndrome expresses much higher levels of 1 lβHSDl activity than subcutaneous fat (Bujalska IJ et al. 1997; Lancet 349, 1210-1213). Thus there appears to be a link between glucocorticoids, 1 lβHSDl and the metabolic syndrome.
1 lβHSDl knock-out mice show attenuated glucocorticoid-induced activation of gluconeogenic enzymes in response to fasting and lower plasma glucose levels in response to stress or obesity (Kotelevtsev Y et al. 1997; Proc. Natl. Acad. Sci USA 94, 14924-14929) indicating the utility of inhibition of 1 lβHSDl in lowering of plasma glucose and hepatic glucose output in type 2 diabetes. Furthermore, these mice express an anti-atherogenic lipoprotein profile, having low triglycerides, increased HDL cholesterol and increased apo-lipoprotein AI levels. (Morton NM et al. 2001 ; J. Biol. Chem. 276, 41293-41300). This phenotype is due to an increased hepatic expression of enzymes of fat catabolism and PP ARa. Again this indicates the utility of 1 lβHSDl inhibition in treatment of the dyslipidaemia of the metabolic syndrome.
The most convincing demonstration of a link between the metabolic syndrome and 11 βHSD 1 comes from recent studies of transgenic mice over-expressing 11 βHSD 1
(Masuzaki H et al. 2001; Science 294, 2166-2170). When expressed under the control of an adipose specific promoter, 1 lβHSDl transgenic mice have high adipose levels of corticosterone, central obesity, insulin resistant diabetes, hyperlipidaemia and hyperphagia. Most importantly, the increased levels of 1 lβHSDl activity in the fat of these mice are similar to those seen in obese subjects. Hepatic 1 lβHSDl activity and plasma corticosterone
levels were normal, however, hepatic portal vein levels of corticosterone were increased 3 fold and it is thought that this is the cause of the metabolic effects in liver.
Overall it is now clear that the complete metabolic syndrome can be mimicked in mice simply by overexpressing llβHSDl in fat alone at levels similar to those in obese man. 11 βHSDl tissue distribution is widespread and overlapping with that of the glucocorticoid receptor. Thus, 11 βHSDl inhibition could potentially oppose the effects of glucocorticoids in a number of physiological/pathological roles. llβHSDl is present in human skeletal muscle and glucocorticoid opposition to the anabolic effects of insulin on protein turnover and glucose metabolism are well documented (Whorwood CB et al. 2001; J. Clin. Endocrinol. Metab. 86, 2296-2308). Skeletal muscle must therefore be an important target for 11 βHSDl based therapy.
Glucocorticoids also decrease insulin secretion and this could exacerbate the effects of glucocorticoid induced insulin resistance. Pancreatic islets express llβHSDl and carbenoxolone can inhibit the effects of 11-dehydocorticosterone on insulin release (Davani B et al. 2000; J. Biol. Chem. 275, 34841-34844). Thus in treatment of diabetes 11 βHSDl inhibitors may not only act at the tissue level on insulin resistance but also increase insulin secretion itself.
Skeletal development and bone function is also regulated by glucocorticoid action. 11 βHSDl is present in human bone osteoclasts and osteoblasts and treatment of healthy volunteers with carbenoxolone showed a decrease in bone resorption markers with no change in bone formation markers (Cooper MS et al 2000; Bone 27, 375-381). Inhibition of 11 βHSDl activity in bone could be used as a protective mechanism in treatment of osteoporosis.
Glucocorticoids may also be involved in diseases of the eye such as glaucoma. 11 βHSD 1 has been shown to affect intraocular pressure in man and inhibition of 11 βHSD 1 may be expected to alleviate the increased intraocular pressure associated with glaucoma (Rauz S et al. 2001; Investigative Opthalmology & Visual Science 42, 2037-2042).
There appears to be a convincing link between 11 βHSDl and the metabolic syndrome both in rodents and in humans. Evidence suggests that a drug which specifically inhibits 11 βHSDl in type 2 obese diabetic patients will lower blood glucose by reducing hepatic gluconeogenesis, reduce central obesity, improve the atherogenic lipoprotein phenotype,
lower blood pressure and reduce insulin resistance. Insulin effects in muscle will be enhanced and insulin secretion from the beta cells of the islet may also be increased.
Currently there are two main recognised definitions of metabolic syndrome.
1) The Adult Treatment Panel (ATP III 2001 JMA) definition of metabolic syndrome 5 indicates that it is present if the patient has three or more of the following symptoms:
> Waist measuring at least 40 inches (102 cm) for men, 35 inches (88 cm) for women;
> Serum triglyceride levels of at least 150 mg/dl (1.69 mmol/1);
> HDL cholesterol levels of less than 40 mg/dl (1.04 mmol/1) in men, less than 50 mg/dl (1.29 mmol/1) in women; o > Blood pressure of at least 135/80 mm Hg; and / or
> Blood sugar (serum glucose) of at least 110 mg/dl (6.1 mmol/1).
2) The WHO consultation has recommended the following definition which does not imply causal relationships and is suggested as a working definition to be improved upon in due course: s > The patient has at least one of the following conditions: glucose intolerance, impaired glucose tolerance (IGT) or diabetes mellitus and/or insulin resistance; together with two or more of the following:
> Raised Arterial Pressure;
> Raised plasma triglycerides o > Central Obesity
> Microalbuminuria
We have found that the compounds defined in the present invention, or a pharmaceutically acceptable salt thereof, are effective l lβHSDl inhibitors, and accordingly have value in the treatment of disease states associated with metabolic syndrome. 5 Accordingly there is provided a compound of formula (1) or a salt thereof:

R1 is selected from hydroxy, halo, trifluoromethyl,
 C(OH)(CF3)-, Ci.4alkoxy, cyano, Ci.4alkylcarbonyl and
C(OH)(CF3)-, Ci.4alkoxy, cyano, Ci.4alkylcarbonyl and
 Ring B is phenyl or pyridyl, optionally substituted in the 2-, T-, 3- or 3'- positions (relative to the point of attachment) (where available) by 1 or 2 substituents independently selected from R2, and/or substituted in the 4- position (relative to the point of attachment) (where available) with a substituent selected from R3;
Ring B is phenyl or pyridyl, optionally substituted in the 2-, T-, 3- or 3'- positions (relative to the point of attachment) (where available) by 1 or 2 substituents independently selected from R2, and/or substituted in the 4- position (relative to the point of attachment) (where available) with a substituent selected from R3;
R2 is selected from halo, cyano, trifluoromethyl, Ci-4alkyl and Ci_4alkoxy;
R3 is selected from halo, cyano, Ci-3alkylsulfonyl and -CONR4R5;
R4 and R5 are independently selected from hydrogen and Ci-4alkyl; or
R4 and R5 together with the nitrogen to which they are attached form an azetidinyl or pyrrolidinyl ring; provided that the compound of formula (1) is not:
4-[[4-(3,5-dichloropyridin-4-yl)-l,4-diazepan-l-yl]sulfonyl]benzonitrile; l-(2-chlorophenyl)sulfonyl-4-(2-fluoropyridin-4-yl)-l,4-diazepane; l-(2-chlorophenyl)sulfonyl-4-(2-chloropyridin-4-yl)-l,4-diazepane; l-(5-chloro-3-methyl-pyridin-2-yl)-4-(2-chlorophenyl)sulfonyl-l,4-diazepane; 2-[4-(2-methylphenyl)sulfonyl-l,4-diazepan-l-yl]pyridine-3-carbonitrile;
2-[4-(2-methoxyphenyl)sulfonyl-l,4-diazepan-l-yl]pyridine-3-carbonitrile;
4-[4-(2-cyanophenyl)sulfonyl-l,4-diazepan-l-yl]-3-fluoro-N,N-dimethyl-benzamide;
3-fluoro-4-[4-(2-methoxyphenyl)sulfonyl-l,4-diazepan-l-yl]-N,N-dimethyl-benzamide;
1 -(4-methylphenyl)sulfonyl-4-(2-methoxyphenyl)- 1 ,4-diazepane; or l-(4-fluorophenyl)sulfonyl-4-(3-trifluoiOmethyl-4-cyanophenyl)-l,4-diazepane.
In this specification the term "alkyl" includes both straight and branched chain alkyl groups For example, "Ci.4alkyl" includes propyl, isopropyl and t-buty\. However, references to individual alkyl groups such as 'propyl' are specific for the straight chained version only and references to individual branched chain alkyl groups such as 'isopropyl' are specific for the branched chain version only. A similar convention applies to other radicals therefore
"Ci-4alkylcarbonyl" would include prop-1-ylcarbonyl and but-3-ylcarbonyl. The term "halo" refers to fiuoro, chloro, bromo and iodo.
Where optional substituents are chosen from "one or more" groups it is to be understood that this definition includes all substituents being chosen from one of the specified groups or the substituents being chosen from two or more of the specified groups.
Examples of "Ci-4alkoxy" include methoxy, ethoxy and propoxy. Examples of
 include formamido, acetamido and propionylamino. Examples of "Ci-salkylsulphonyl" include mesyl and ethylsulphonyl.
include formamido, acetamido and propionylamino. Examples of "Ci-salkylsulphonyl" include mesyl and ethylsulphonyl.
 include propionyl and acetyl. Examples of "Ci-3alkyl" include methyl, ethyl, propyl and isopropyl.
include propionyl and acetyl. Examples of "Ci-3alkyl" include methyl, ethyl, propyl and isopropyl.
 include the examples of "C1-3alkyl" as well as butyl, isobutyl, sec- butyl and tert-butyl. Examples of "hydroxyCi_4alkyl" include hydroxymethyl, hydroxyethyl, l-hydroxyprop-2-yl and l-hydroxyprop-3-yl. Examples of "C^alkylcarbonyl" include methylcarbonyl, ethylcarbonyl, propylcarbonyl, isopropylcarbonyl and tert-butylcarbonyl.
include the examples of "C1-3alkyl" as well as butyl, isobutyl, sec- butyl and tert-butyl. Examples of "hydroxyCi_4alkyl" include hydroxymethyl, hydroxyethyl, l-hydroxyprop-2-yl and l-hydroxyprop-3-yl. Examples of "C^alkylcarbonyl" include methylcarbonyl, ethylcarbonyl, propylcarbonyl, isopropylcarbonyl and tert-butylcarbonyl.
The present invention relates to the compounds of formula (1) as hereinbefore defined as well as to the salts thereof. Salts for use in pharmaceutical compositions will be pharmaceutically acceptable salts, but other salts may be useful in the production of the compounds of formula (1) and their pharmaceutically acceptable salts.
A suitable pharmaceutically acceptable salt of a compound of the invention is, for example, an acid-addition salt of a compound of the invention which is sufficiently basic, for example, an acid-addition salt with, for example, an inorganic or organic acid, for example hydrochloric, hydrobromic, sulphuric, phosphoric, citric or maleic acid. In addition a suitable pharmaceutically acceptable salt of a compound of the invention which is sufficiently acidic is an alkali metal salt, for example a sodium or potassium salt, an alkaline earth metal salt, for example a calcium or magnesium salt, an ammonium salt or a salt with an organic base which affords a physiologically-acceptable cation, for example a salt with methylamine, dimethylamine, trimethylamine, piperidine, morpholine or tris-(2-hydroxyethyi)amine. Some compounds of the formula (1) may have chiral centres and/or geometric isomeric centres (E- and Z- isomers), and it is to be understood that the invention encompasses all such optical, diastereoisomers and geometric isomers that possess 1 lβHSDl inhibitory activity.
The invention relates to any and all tautomeric forms of the compounds of the formula (1) that possess 1 lβHSDl inhibitoiy activity.
It is also to be understood that certain compounds of the formula (1) can exist in solvated as well as unsolvated forms such as, for example, hydrated forms. It is to be
understood that the invention encompasses all such solvated forms which possess 1 lβHSDl inhibitory activity.
In one embodiment of the invention are provided compounds of formula (1); in an alternative embodiment are provided pharmaceutically-acceptable salts of compounds of formula (1).
Particular values of variable groups are as follows. Such values may be used where appropriate with any of the definitions, claims or embodiments defined hereinbefore or hereinafter.
1) Ring A is phenyl, optionally substituted by 1, 2 or 3 substituents independently selected from R1;
2) Ring A is naphthyl, optionally substituted by 1, 2 or 3 substituents independently selected from R1;
3) Ring A is phenyl, substituted by 1 or 2 substituents independently selected from R1
4) Ring A is naphthyl 5) R1 is selected from hydroxy, fluoro, chloro, trifluoromethyl, Ci-4alkyl, hydroxy C1-4alkyl, MeC(OH)(CF3)-, methoxy, cyano, methylcarbonyl and methylcarbonylamino
6) R1 is selected from hydroxy, fluoro, chloro, trifluoromethyl, Ci-4alkyl, hydroxy C1-4alkyl, methoxy, cyano, methylcarbonyl and methylcarbonylamino
7) R1 is MeC(OH)(CF3)- 8) Ring B is phenyl
9) Ring B is pyridyl
10) Ring B is 2-pyridyl or 4-pyridyl
11) Ring B is substituted in the 2- and/or T- positions (relative to the point of attachment) by one or two, as appropriate, substituents independently selected from R2, particularly wherein R2 is selected from chloro, fluoro and trifluoromethyl
12) Ring B is substituted on the 4- position (relative to the point of attachment) (where available) with a substituent selected from R3
13) R2 is selected from fluoro, chloro, cyano, trifluoromethyl, Ci_4alkyl and C1-4alkoxy
14) R2 is selected from fluoro, chloro, cyano, trifluoromethyl, Ci-4alkyl and methoxy 15) R2 is selected from chloro, fluoro and trifluoromethyl
16) R3 is selected from fluoro, chloro, cyano, Ci-3alkylsulfonyl, -CONHMe and -CONMe2
17) R3 is -CONR4R5 wherein R4 and R5 together with the nitrogen to which they are attached form an azetidinyl or pyrrolidinyl ring.
In another aspect of the invention, suitable compounds of the invention are any one or more of the Examples or a salt thereof. In another aspect of the invention, suitable compounds of the invention are any one or more of the following or a salt thereof: l-(3,5-dichloropyridin-4-yl)-4-(4-fluorophenyl)sulfonyl-l,4-diazepane; l-(2,4-dichlorophenyl)sulfonyl-4-(3,5-dichloropyridin-4-yl)-l,4-diazepane; l-(3,5-dichloropyridin-4-yl)-4-(3-methylphenyl)sulfonyl-l,4-diazepane; 1 -(3 ,5 -dichloropyridin-4-yl)-4-(4-methylphenyl)sulfonyl- 1 ,4-diazepane;
1 -(3,5-dichloropyridin-4-yl)-4-(2-methoxyphenyl)sulfonyl- 1 ,4-diazepane; l-(3,5-dichloropyridin-4-yl)-4-(3-methoxyphenyl)sulfonyl-l,4-diazepane; l-(3,5-dichloropyridin-4-yl)-4-(4-methoxyphenyl)sulfonyl-l,4-diazepane; l-(3,5-dichloropyridin-4-yl)-4-[4-(trifluoromethyl)phenyl]sulfonyl-l,4-diazepane; l-[4-[[4-(3,5-dichloropyridin-4-yl)-l,4-diazepan-l-yl]sulfonyl]phenyl]ethanone;
1 -(3 ,5-dichloropyridin-4-yl)-4-naphthalen- 1 -ylsulfonyl- 1 ,4-diazepane; l-(3,5-dichloropyridin-4-yl)-4-(4-propan-2-ylphenyl)sulfonyl-l,4-diazepane;
1 -(3,5-dichloropyridin-4-yl)-4-naphthalen-2 -ylsulfonyl- 1 ,4-diazepane;
1 -(2-chlorophenyl)sulfonyl-4-(3 ,5-dichloropyridin-4-yl)- 1 ,4-diazepane; l-(3,5-dichloropyridin-4-yl)-4-[2-(trifluoromethyl)phenyl]sulfonyl-l,4-diazepane;
N-[4-[[4-(3,5-dichloropyridin-4-yl)-l,4-diazepan-l-yl]sulfonyl]phenyl]acetamide;
1 -(3 , 5 -dichloropyridin-4-yl)-4- [3 -(trifluoromethyl)phenyl] sulfonyl- 1 ,4-diazepane;
2-[[4-(3,5-dichloropyridin-4-yl)-l,4-diazepan-l-yl]sulfonyl]benzonitrile; l-(3-chlorophenyl)sulfonyl-4-(3,5-dichloropyridin-4-yl)-l,4-diazepane; l-(3,5-dichloropyridin-4-yl)-4-(2-methylphenyl)sulfonyl-l,4-diazepane; l-(2-chloiOphenyl)sulfonyl-4-[3-(trifluoromethyl)pyridin-2-yl]-l,4-diazepane;
1 -(2-chlorophenyl)sulfonyl-4-(3-chloropyridin-2-yl)- 1 ,4-diazepane; l-(2-chlorophenyl)sulfonyl-4-[2-(trifluoromethyl)phenyl]-l,4-diazepane;
1 -(2-chlorophenyl)-4-(2-chlorophenyl)sulfonyl- 1 ,4-diazepane; 1 -(2-chlorophenyl)sulfonyl-4-(4-fluorophenyl)- 1 ,4-diazepane;
1 -(2-chlorophenyl)sulfonyl-4-(2,6-dichlorophenyl)- 1 ,4-diazepane;
4-[4-(2-chlorophenyl)sulfonyl-l,4-diazepan-l-yl]-3-(trifluoromethyl)benzonitrile;
1 -(2-methylphenyl)sulfonyl-4-[3 -(trifluoromethyl)pyridin-2-yl] - 1 ,4-diazepane;
1 - [4- [ [4- [3 -(trifluoromethyl)pyridin-2-yl] - 1 ,4-diazepan- 1 -yl] sulfonyljphenyl] ethanone;
1 -(2-chlorophenyl)sulfonyl-4-(2-fluoro-4-methylsulfonyl-phenyl)- 1 ,4-diazepane;
4-[4-(2-chlorophenyl)sulfonyl-l,4-diazepan-l-yl]-3-fluoro-N,N-dimethyl-benzamide; 2- [4- [ [4-(3 ,5 -dichloropyridin-4-yl)- 1 ,4-diazepan- 1 -yl] sulfonyl]phenyl] -1,1,1 -trifluoro-prop an-
2-ol;
2-[4-[4-(I51 , 1 -trifluoro-2 -hydroxy ~propan-2-yl)phenyl] sulfonyl- 1 ,4-diazepan- 1 -yl]pyridine-3 - carbonitrile; and
1,1,1 -trifluoro-2-[4-[[4-[3-(trifluoromethyl)pyridin-2-yl]- 1 ,4-diazepan- 1 - yl]sulfonyl]phenyl]propan-2-ol.
Another aspect of the present invention provides a process for preparing a compound of formula (1) or a pharmaceutically acceptable salt thereof which process (wherein variable groups are, unless otherwise specified, as defined in formula (I)) comprises any one of processes a) to c): a) reaction of a compound of Formula (2) with a compound of Formula (3):

(2) (3)
wherein X1 is a leaving group; or b) reaction of a compound of Formula (4) with a compound of Formula (5):

(4) (5) wherein X2 is a leaving group; c) reaction of a compound of Formula (6) with an organometallic reagent to give a compound of Formula (7):

(6) (7) and thereafter if necessary or desirable: i) converting a compound of the formula (1) into another compound of the formula (1); ii) removing any protecting groups; iii) resolving enantiomers; iv) forming a salt thereof.
Examples of conversions of a compound of Formula (1) into another compound of Formula (1), well known to those skilled in the art, include functional group interconversions such as hydrolysis, hydrogenation, hydrogenolysis, oxidation or reduction, and/or further functionalisation by standard reactions such as amide or metal-catalysed coupling, or nucleophilic displacement reactions.
Suitable conditions for the above processes a) to c) are as follows. a) Process a) may be carried out in a suitable solvent such as DCM, DMF, pyridine or water, typically with the addition of a base such as triethylamine, DIPEA, pyridine, or aqueous sodium hydroxide.
Compounds of formula (3) may be made by reaction of a compound of Formula (8) with a compound of Formula (9):

(8) (9) wherein X3 is a leaving group and R is hydrogen or a suitable protecting group, followed by removal of said protecting group if appropriate; b) Process b) may be carried out without solvent, or in a suitable solvent such as DMA, DMF or xylene; typically the reaction is carried out without solvent at elevated temperature, using Microwave or conventional heating.
Compounds of formula (4) may be made by reaction of a compound of Formula (10) with a compound of Formula (11):

(10) (11)
wherein X4 is a leaving group and R is hydrogen or a suitable protecting group, followed by removal of said protecting group if appropriate; c) Process c) is typically carried out in an anhydrous aprotic solvent such as THF or diethyl ether; examples of suitable organometallic reagents for process c) are alkyl or aryl magnesium halides (Grignard reagent), alkyl or aryl lithium, or trimethyl (trifluoromethyl) silane (Ruppert's reagent). Suitable examples of leaving groups for processes a) to c) are: fluoro, chloro, bromo, iodo, mesylate, tosylate or triflate.
Suitable examples of protecting groupsfor processes a) to c) are: tert-butyl oxycarbonyl (Boc), benzyl oxycarbonyl (Z), acetyl or trifluoracetyl. The reactions described above may be performed under standard conditions known to the person skilled in the art. The intermediates described above are commercially available, are known in the art or may be prepared by known procedures and/or by the procedures shown above.
It will be appreciated that certain of the various ring substituents in the compounds of the present invention may be introduced by standard aromatic substitution reactions or generated by conventional functional group modifications either prior to or immediately following the processes mentioned above, and as such are included in the process aspect of the invention. Such reactions and modifications include, for example, introduction of a substituent by means of an aromatic substitution reaction, reduction of substituents, alkylation of substituents and oxidation of substituents. The reagents and reaction conditions for such procedures are well known in the chemical art. Particular examples of aromatic substitution reactions include the introduction of a nitro group using concentrated nitric acid, the introduction of an acyl group using, for example, an acyl halide and Lewis acid (such as aluminium trichloride) under Friedel Crafts conditions; the introduction of an alkyl group using an alkyl halide and Lewis acid (such as aluminium trichloride) under Friedel Crafts
conditions; and the introduction of a halogeno group. Particular examples of modifications include the reduction of a nitro group to an amino group by for example, catalytic hydrogenation with a nickel catalyst or treatment with iron in the presence of hydrochloric acid with heating; oxidation of alkylthio to alkylsulphinyl or alkylsulphonyl. It will also be appreciated that in some of the reactions mentioned herein it may be necessary/desirable to protect any sensitive groups in the compounds. The instances where protection is necessary or desirable and suitable methods for protection are known to those skilled in the art. Conventional protecting groups may be used in accordance with standard practice (for illustration see T.W. Green, Protective Groups in Organic Synthesis, John Wiley and Sons, 1991). Thus, if reactants include groups such as amino, carboxy or hydroxy it may be desirable to protect the group in some of the reactions mentioned herein.
A suitable protecting group for an amino or alkylamino group is, for example, an acyl group, for example an alkanoyl group such as acetyl, an alkoxycarbonyl group, for example a methoxycarbonyl, ethoxycarbonyl or t-butoxycarbonyl group, an arylmethoxycarbonyl group, for example benzyloxycarbonyl, or an aroyl group, for example benzoyl. The deprotection conditions for the above protecting groups necessarily vary with the choice of protecting group. Thus, for example, an acyl group such as an alkanoyl or alkoxycarbonyl group or an aroyl group may be removed for example, by hydrolysis with a suitable base such as an alkali metal hydroxide, for example lithium or sodium hydroxide. Alternatively an acyl group such as a /-butoxycarbonyl group may be removed, for example, by treatment with a suitable acid as hydrochloric, sulphuric or phosphoric acid or trifluoroacetic acid and an arylmethoxycarbonyl group such as a benzyloxycarbonyl group may be removed, for example, by hydrogenation over a catalyst such as palladium-on-carbon, or by treatment with a Lewis acid for example boron tris(trifluoroacetate). A suitable alternative protecting group for a primary amino group is, for example, a phthaloyl group which may be removed by treatment with an alkylamine, for example hydroxylamine, or with hydrazine.
A suitable protecting group for a hydroxy group is, for example, an acyl group, for example an alkanoyl group such as acetyl, an aroyl group, for example benzoyl, or an arylmethyl group, for example benzyl. The deprotection conditions for the above protecting groups will necessarily vary with the choice of protecting group. Thus, for example, an acyl group such as an alkanoyl or an aroyl group may be removed, for example, by hydrolysis with a suitable base such as an alkali metal hydroxide, for example lithium or sodium hydroxide.
Alternatively an arylmethyl group such as a benzyl group may be removed, for example, by hydrogenation over a catalyst such as palladium-on-carbon.
A suitable protecting group for a carboxy group is, for example, an esterifying group, for example a methyl or an ethyl group which may be removed, for example, by hydrolysis with a base such as sodium hydroxide, or for example a /-butyl group which may be removed, for example, by treatment with an acid, for example an organic acid such as trifluoroacetic acid, or for example a benzyl group which may be removed, for example, by hydrogenation over a catalyst such as palladium-on-carbon.
The protecting groups may be removed at any convenient stage in the synthesis using conventional techniques well known in the chemical art.
As stated hereinbefore the compounds defined in the present invention possess llβHSDl inhibitory activity. These properties may be assessed using the following assay. Assay
The conversion of cortisone to the active steroid Cortisol by 1 lβHSDl oxo-reductase activity, can be measured using a competitive immunoassay^ 1) kit (Assay Designs, Inc: 800 Technology Drive, Ann Arbor, MI 48108, USA. Cortisol Enzyme Immunoassay kit: Cat No. 901-071).
The evaluation of compounds described herein was carried out using a baculovirus expressed N terminal 6-His tagged full length human 1 lβHSDl enzyme(*2). The enzyme was purified from a detergent solublised cell lysate, using a copper chelate column. Inhibitors of 1 lβHSDl reduce the conversion of cortisone to Cortisol, which is identified by an increase in signal, in the above assay.
Compounds to be tested were dissolved in dimethyl sulphoxide (DMSO) to 1OmM and diluted further in assay buffer to 10 fold the final assay concentration and 10% DMSO. Diluted compounds were then plated into transparent 96 well plates (Matrix, Hudson NH, USA).
The assay was carried out in a total volume of 200μl consisting of cortisone (Sigma, Poole, Dorset, UK, lμM), glucose-6-phosphate (Roche Diagnostics, ImM), NADPH (Roche Diagnostics, lOOμM), glucose-6-phosphate dehydrogenase (Roche Diagnostics, 12.5μg/ml), EDTA (Sigma, Poole, Dorset, UK, ImM), assay buffer (K2HPO4/KH2PO4, 10OmM) pH 7.5, recombinant 1 lβHSDl (5μg/ml) plus test compound. The assay plates were incubated for 30
minutes at 37°C after which time the reaction was stopped by the addition of 20μl of ImM glycerrhetinic acid.
These samples were diluted 1:10 in EIA assay buffer (Assay Designs, Inc.) and Cortisol measured according to the manufactures instructions.
The amount of Cortisol produced in each reaction, was calculated from a Cortisol standard curve using Origin 6.0 (Microcal software, Northampton MA, USA) and expressed as a percentage of the maximum signal. Hence a % inhibition was calculated for each concentration of the tested compound. This data was then used to calculate IC50 values for each compound (Origin 6.0 Microcal software, Northampton MA, USA). *1 P. Tijssen, "Practise and theory of enzyme immunoassays"(1985), Elsevier, Amsterdam. *2 The Journal of Biological Chemistry, Vol. 26, No 25, ppl6653 - 16658 Compounds of the present invention typically show an IC5O of less than 30μM, and preferably less than 5 μM.
For example, the following results were obtained:

The compositions of the invention may be obtained by conventional procedures using conventional pharmaceutical excipients, well known in the art. Thus, compositions intended for oral use may contain, for example, one or more colouring, sweetening, flavouring and/or preservative agents.
Suitable pharmaceutically acceptable excipients for a tablet formulation include, for example, inert diluents such as lactose, sodium carbonate, calcium phosphate or calcium carbonate, granulating and disintegrating agents such as corn starch or algenic acid; binding agents such as starch; lubricating agents such as magnesium stearate, stearic acid or talc; preservative agents such as ethyl or propyl p_-hydroxybenzoate, and anti-oxidants, such as ascorbic acid. Tablet formulations may be uncoated or coated either to modify their disintegration and the subsequent absorption of the active ingredient within the gastrointestinal tract, or to improve their stability and/or appearance, in either case, using conventional coating agents and procedures well known in the art.
Compositions for oral use may be in the form of hard gelatin capsules in which the active ingredient is mixed with an inert solid diluent, for example, calcium carbonate, calcium phosphate or kaolin, or as soft gelatin capsules in which the active ingredient is mixed with water or an oil such as peanut oil, liquid paraffin, or olive oil.
Aqueous suspensions generally contain the active ingredient in finely powdered form together with one or more suspending agents, such as sodium carboxymethylcellulose,
methylcellulose, hydroxypropylmethylcellulose, sodium alginate, polyvinylpyrrolidone, gum tragacanth and gum acacia; dispersing or wetting agents such as lecithin or condensation products of an alkylene oxide with fatty acids (for example polyoxethylene stearate), or condensation products of ethylene oxide with long chain aliphatic alcohols, for example lieptadecaethyleneoxycetanol, or condensation products of ethylene oxide with partial esters derived from fatty acids and a hexitol such as polyoxyethylene sorbitol monooleate, or condensation products of ethylene oxide with long chain aliphatic alcohols, for example heptadecaethyleneoxycetanol, or condensation products of ethylene oxide with partial esters derived from fatty acids and a hexitol such as polyoxyethylene sorbitol monooleate, or condensation products of ethylene oxide with partial esters derived from fatty acids and hexitol anhydrides, for example polyethylene sorbitan monooleate. The aqueous suspensions may also contain one or more preservatives (such as ethyl or propyl p-hydroxybenzoate, anti-oxidants (such as ascorbic acid), colouring agents, flavouring agents, and/or sweetening agents (such as sucrose, saccharine or aspartame). Oily suspensions may be formulated by suspending the active ingredient in a vegetable oil (such as arachis oil, olive oil, sesame oil or coconut oil) or in a mineral oil (such as liquid paraffin). The oily suspensions may also contain a thickening agent such as beeswax, hard paraffin or cetyl alcohol. Sweetening agents such as those set out above, and flavouring agents may be added to provide a palatable oral preparation. These compositions may be preserved by the addition of an anti-oxidant such as ascorbic acid.
Dispersible powders and granules suitable for preparation of an aqueous suspension by the addition of water generally contain the active ingredient together with a dispersing or wetting agent, suspending agent and one or more preservatives. Suitable dispersing or wetting agents and suspending agents are exemplified by those already mentioned above. Additional excipients such as sweetening, flavouring and colouring agents, may also be present.
The pharmaceutical compositions of the invention may also be in the form of oil-in-water emulsions. The oily phase may be a vegetable oil, such as olive oil or arachis oil, or a mineral oil, such as for example liquid paraffin or a mixture of any of these. Suitable emulsifying agents may be, for example, naturally-occurring gums such as gum acacia or gum tragacanth, naturally-occurring phosphatides such as soya bean, lecithin, an esters or partial esters derived from fatty acids and hexitol anhydrides (for example sorbitan monooleate) and
condensation products of the said partial esters with ethylene oxide such as polyoxyethylene sorbitan monooleate. The emulsions may also contain sweetening, flavouring and preservative agents.
Syrups and elixirs may be formulated with sweetening agents such as glycerol, propylene glycol, sorbitol, aspartame or sucrose, and may also contain a demulcent, preservative, flavouring and/or colouring agent.
The pharmaceutical compositions may also be in the form of a sterile injectable aqueous or oily suspension, which may be formulated according to known procedures using one or more of the appropriate dispersing or wetting agents and suspending agents, which have been mentioned above. A sterile injectable preparation may also be a sterile injectable solution or suspension in a non-toxic parenterally-acceptable diluent or solvent, for example a solution in 1,3-butanediol.
Compositions for administration by inhalation may be in the form of a conventional pressurised aerosol arranged to dispense the active ingredient either as an aerosol containing finely divided solid or liquid droplets. Conventional aerosol propellants such as volatile fluorinated hydrocarbons or hydrocarbons may be used and the aerosol device is conveniently arranged to dispense a metered quantity of active ingredient.
For further information on formulation the reader is referred to Chapter 25.2 in Volume 5 of Comprehensive Medicinal Chemistry (Corwin Hansch; Chairman of Editorial Board), Pergamon Press 1990.
The amount of active ingredient that is combined with one or more excipients to produce a single dosage form will necessarily vary depending upon the host treated and the particular route of administration. For example, a formulation intended for oral administration to humans will generally contain, for example, from 0.5 mg to 2 g of active agent compounded with an appropriate and convenient amount of excipients which may vary from about 5 to about 98 percent by weight of the total composition. Dosage unit forms will generally contain about 1 mg to about 500 mg of an active ingredient. For further information on Routes of Administration and Dosage Regimes the reader is referred to Chapter 25.3 in Volume 5 of Comprehensive Medicinal Chemistry (Corwin Hansch; Chairman of Editorial Board), Pergamon Press 1990.
We have found that the compounds defined in the present invention, or a pharmaceutically acceptable salt thereof, are effective 1 lβHSDl inhibitors, and accordingly
have value in the treatment of disease states associated with metabolic syndrome.
It is to be understood that where the term "metabolic syndrome" is used herein, this relates to metabolic syndrome as defined in 1) and/or 2) or any other recognised definition of this syndrome. Synonyms for "metabolic syndrome" used in the art include Reaven's Syndrome, Insulin Resistance Syndrome and Syndrome X. It is to be understood that where the term "metabolic syndrome" is used herein it also refers to Reaven's Syndrome, Insulin
Resistance Syndrome and Syndrome X.
According to a further aspect of the present invention there is provided a compound of formula (1), or a pharmaceutically acceptable salt thereof, as defined hereinbefore for use in a method of prophylactic or therapeutic treatment of a warm-blooded animal, such as man. Thus according to this aspect of the invention there is provided a compound of formula (1), or a pharmaceutically acceptable salt thereof, as defined hereinbefore for use as a medicament.
According to another feature of the invention there is provided the use of a compound of formula (1), or a pharmaceutically acceptable salt thereof, as defined hereinbefore in the manufacture of a medicament for use in the production of an 1 lβHSDl inhibitory effect in a warm-blooded animal, such as man.
Where production of or producing an 11 βHSDl inhibitory effect is referred to suitably this refers to the treatment of metabolic syndrome. Alternatively, where production of an 11 βHSDl inhibitory effect is referred to this refers to the treatment of diabetes, obesity, hyperlipidaemia, hyperglycaemia, hyperinsulinemia or hypertension, particularly diabetes and obesity. Alternatively, where production of an 11 βHSDl inhibitory effect is referred to this refers to the treatment of glaucoma, osteoporosis, tuberculosis, dementia, cognitive disorders or depression. Alternatively, where production of an 11 βHSD 1 inhibitory effect is referred to this refers to the treatment of cognitive disorders, such as improving the cognitive ability of an individual, for example by improvement of verbal fluency, verbal memory or logical memory, or for treatment of mild cognitive disorders. See for example WO03/086410 and references contained therein, and Proceedings of National Academy of Sciences (PNAS), 2001, 98(8), 4717-4721.
Alternatively, where production of an 1 lβHSDl inhibitory effect is referred to this refers to the treatment of, delaying the onset of and/or reducing the risk of atherosclerosis - see for example J. Experimental Medicine, 2005, 202(4), 517-527.
Alternatively, where production of an llβHSDl inhibitory effect is referred to this refers to the treatment of Alzheimers and/or neurodegenerative disorders.
According to a further feature of this aspect of the invention there is provided a method for producing an 11 βHSDl inhibitory effect in a waπn-blooded animal, such as man, in need of such treatment which comprises administering to said animal an effective amount of a compound of formula (1), or a pharmaceutically acceptable salt thereof. In addition to their use in therapeutic medicine, the compounds of formula formula
(1), or a pharmaceutically acceptable salt thereof, are also useful as pharmacological tools in the development and standardisation of in vitro and in vivo test systems for the evaluation of the effects of inhibitors of llβHSDl in laboratory animals such as cats, dogs, rabbits, monkeys, rats and mice, as part of the search for new therapeutic agents. The inhibition of 1 lβHSDl described herein may be applied as a sole therapy or may involve, in addition to the subject of the present invention, one or more other substances and/or treatments. Such conjoint treatment may be achieved by way of the simultaneous, sequential or separate administration of the individual components of the treatment. Simultaneous treatment may be in a single tablet or in separate tablets. For example agents than might be co-administered with 11 βHSDl inhibitors, particularly those of the present invention, may include the following main categories of treatment:
1) Insulin and insulin analogues;
2) Insulin secretagogues including sulphonylureas (for example glibenclamide, glipizide), prandial glucose regulators (for example repaglinide, nateglinide), glucagon-like peptide 1 agonist (GLPl agonist) (for example exenatide, liraglutide) and dipeptidyl peptidase IV inhibitors (DPP-IV inhibitors);
3) Insulin sensitising agents including PPARγ agonists (for example pioglitazone and rosiglitazone);
4) Agents that suppress hepatic glucose output (for example metformin); 5) Agents designed to reduce the absorption of glucose from the intestine (for example acarbose);
6) Agents designed to treat the complications of prolonged hyperglycaemia; e.g. aldose reductase inhibitors
7) Other anti-diabetic agents including phosotyrosine phosphatase inhibitors, glucose 6 - phosphatase inhibitors, glucagon receptor antagonists, glucokinase activators, glycogen phosphorylase inhibitors, fructose 1,6 bisphosphastase inhibitors, glutamine:fructose -6-phosphate amidotransferase inhibitors
8) Anti-obesity agents (for example sibutramine and orlistat);
9) Anti- dyslipidaemia agents such as, HMG-CoA reductase inhibitors (statins, eg pravastatin); PP ARa agonists (fϊbrates, eg gemfibrozil); bile acid sequestrants (cholestyramine); cholesterol absorption inhibitors (plant stanols, synthetic inhibitors); ileal bile acid absorption inhibitors (IBATi), cholesterol ester transfer protein inhibitors and nicotinic acid and analogues (niacin and slow release formulations);
10) Antihypertensive agents such as, β blockers (eg atenolol, inderal); ACE inhibitors (eg lisinopril); calcium antagonists (eg. nifedipine); angiotensin receptor antagonists (eg candesartan), α antagonists and diuretic agents (eg. furosemide, benzthiazide); l l)Haemostasis modulators such as, antithrombotics, activators of fibrinolysis and antiplatelet agents; thrombin antagonists; factor Xa inhibitors; factor Vila inhibitors); antiplatelet agents (eg. aspirin, clopidogrel); anticoagulants (heparin and Low molecular weight analogues, hirudin) and warfarin; and 12) Anti-inflammatory agents, such as non-steroidal anti-infammatory drugs (eg. aspirin) and steroidal anti-inflammatory agents (eg. cortisone).
In the above other pharmaceutical composition, process, method, use and medicament manufacture features, the alternative and preferred embodiments of the compounds of the invention described herein also apply. Examples
The invention will now be illustrated in the following Examples, in which standard techniques known to the skilled chemist and techniques analogous to those described in these Examples may be used where appropriate, and in which, unless otherwise stated: (i) evaporations were carried out by rotary evaporation in vacuo and where applicable were carried out after removal of residual solids such as drying agents by filtration;
(ii) all reactions were carried out under an inert atmosphere at ambient temperature, typically in the range 18-25°C, with solvents of HPLC grade under anhydrous conditions, unless otherwise stated; (iii) yields are given for illustration only and are not necessarily the maximum attainable; (iv) the structures of the end products of the formula (1) were generally confirmed by proton nuclear magnetic resonance (NMR) and mass spectral techniques; magnetic resonance chemical shift values were measured in deuterated DMSO (unless otherwise stated) on the delta scale (ppm downfield from tetramethylsilane); proton data is quoted unless otherwise stated; spectra were recorded on a Bruker Avance 400MHz spectrometer; and peak multiplicities are shown as follows: s, singlet; d, doublet; dd, double doublet; t, triplet; tt, triple triplet; q, quartet; tq, triple quartet; m, multiplet; br, broad;
LCMS were recorded on a system comprising Waters 2790 LC equipped with a Waters 996 Photodiode array detector and Micromass ZMD MS (using a Phenomenex® Synergi 4μ MAX-RP 8OA 50x2 mm column), or a Waters 2795 LC equipped with a Waters 2996 Photodiode array detector and Micromass ZQ MS (using a Phenomenex® Gemini 5μ C18 1 lOAngstrom 50x2 mm column), and eluting with a flow rate of 1.1 ml/min with 5% (Water/ Acetonitrile (1:1) + 1% formic acid) and a gradient increasing from 0-95% of acetonitrile over the first 4 minutes, the balance (95-0%) being water; mass spectra (MS) (loop) were recorded on a Micromass LCT equipped with HP 1100 detector, or a Thermo LCQ; unless otherwise stated the mass ion quoted is (M+H+);
(v) intermediates were fully characterised unless otherwise stated and purity was assessed by
LCMS and NMR analysis;
(vi) where solutions were dried magnesium sulphate was the drying agent;
(vii) where an Isolute SCX-2 column is referred to, this means an "ion exchange" extraction cartridge for adsorption of basic compounds, i.e. a polypropylene tube containing a benzenesulphonic acid based strong cation exchange sorbent, used according to the manufacturers instructions obtained from International Sorbent Technologies Limited, Dyffryn Business Park, Hengeod, Mid Glamorgan, UK, CF82 7RJ; viii) where an Isolute-NH2 column is referred to, this means an "ion exchange" extraction cartridge for adsorption of acidic compounds, i.e. a polypropylene tube containing a amino silane covalently bonded to a silica particle used according to the manufacturers instructions
obtained from International Sorbent Technologies Limited, Dyffryn Business Park, Hengeod,
Mid Glamorgan, UK, CF82 7RJ; ix) where as Isco CombiFlash Optix-10 parallel flash chromatography system is referred to this means an automated chromatography workstation capable of cany ing out up to 10 purifications in parallel via flash chromatography using pre packed silica cartridges; x) where a "Biotage 9Og silica column" is referred to this means an automated chromatography workstation capable of carrying out up to 4 purifications in parallel via flash chromatography using pre packed silica cartridges, eg Si 12+M available from Biotage Inc. A
Dy ax Corp. Company; and xi) where a "Genevac HT4" is referred to, this means a centrifugal evaporator capable of the simultaneous evaporation of multiple samples supplied by Genevac Ltd, The Sovereign
Centre, Farthing Road, Ipswich, Suffolk IPl 5AP, UK.
(xii) the following abbreviations may be used hereinbefore or hereinafter: -
DCM Dichloromethane
DIPEA Di-isopropyl ethylamine
DMA Dimethyl acetamide
DMF Dimethyl formamide
EtOAc Ethyl acetate
Examples 1-19
General Method:

A solution of l-(3,5-dichloropyridin-4-yl)-l,4-diazepane (100 mg, 0.41 mmol) in DCM (1 ml) was added to the appropriate sulfonyl chloride (0.41 mmol) under a nitrogen atomosphere. DIPEA (0.22 ml, 1.22 mmol) was then added and the mixture stirred overnight at ambient temperature. The reaction mixtures were washed with water and the products isolated by
flash column chromatography (Combi-Flash Optix 10 ® on a 12g silica cartridge, eluting with a 1:1 mixture of EtOAc and isohexane) to give colourless solids.
The requisite l-(3,5-dichloropyridin-4~yl)-l,4-diazepane starting material was prepared as follows:
l-(3,5-dichloropyridin-4-yl)-l,4-diazepane

1,4-Diazepane (5.49g, 54.8 mmol) and 3,4,5-trichloropyridine (2g, 3.0 mmol) were combined without solvent, and the mixture heated with stirring at 90 0C for 2.5 hours. The reaction mixture was cooled to room temperature and the product purified by flash column chromatography (CombiFlash ® Companion ™ , 4Og silica column, eluting with a 10:1 mixture of DCM and methanol) to give the title compound as a yellow oil (2.06 g, 76% ); 1H NMR (400 MHz, CDCl3) δ 1.88 - 1.94 (2H, m), 3.02 - 3.04 (2H, m), 3.09 (2H, t), 3.34 - 3.37 (4H, m), 8.39 (2H, s), m/z 246 (M+H)+ [I].
[1] Note: The m/z value for the (M+H)+ molecular ion is based on the 35Cl isotope. When there are multiple chlorine atoms in the molecule, the m/z is based on the first peak of the isotope pattern.
The following compounds were synthesised as a multiparallel array:


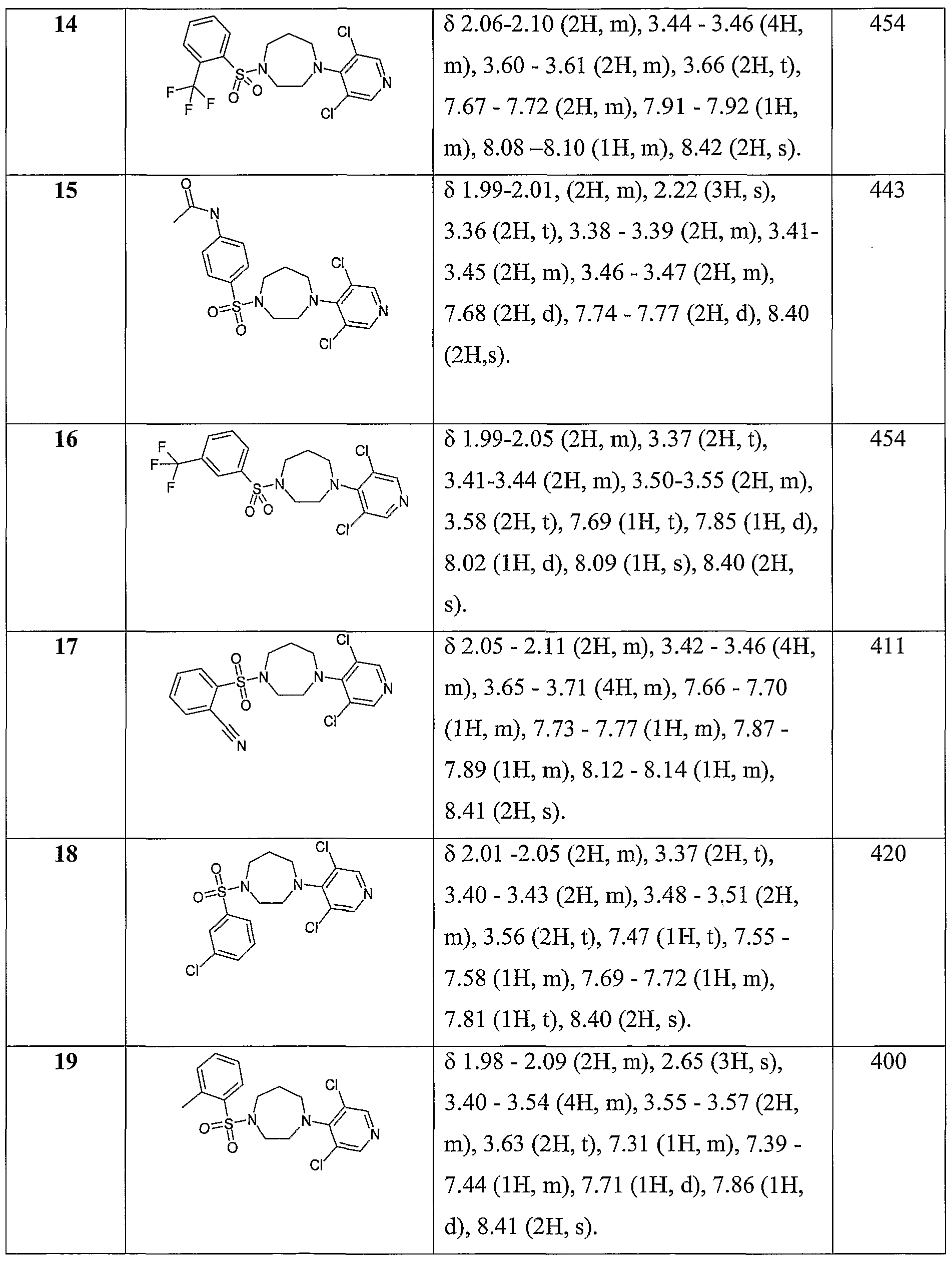
Example 1 : 1 -(3,5-dichloropyridin-4-yl)-4-(4-fluorophenyl)sulfonyl- 1 ,4-diazepane
Example 2: 1 -(2,4-dichlorophenyl)sulfonyl-4-(3 ,5-dichloropyridin-4-yl)- 1 ,4-diazepane Example 3 : 1 -(3 ,5 -dichloropyridin-4-yl)-4-(3-methylphenyl)sulfony 1-1 ,4-diazepane Example 4: l-(3,5-dichloropyridin-4-yl)-4-(4-methylphenyl)sulfonyl-l,4-diazepane Example 5: l-(3,5-dichloropyridin-4-yl)-4-(2-methoxyphenyl)sulfonyl-l,4-diazepane Example 6: l-(3,5-dichloropyridin-4-yl)-4-(3-methoxyplienyl)sulfonyl-l,4-diazepane Example 7 : 1 -(3 ,5-dichloropyridin-4-yl)-4-(4-methoxyphenyl)sulfonyl- 1 ,4-diazepane Example 8: l-(3,5-dichloropyridin-4-yl)-4-[4-(trifluoromethyl)phenyl]sulfonyl-l,4-diazepane Example 9 : 1 - [4- [ [4-(3 , 5 -dichloropyridin-4-yl)- 1 ,4-diazepan- 1 -yl] sulfonyljphenyl] ethanone Example 10: 1 -(3 , 5 -dichloropyridin-4-yl)-4-naphthalen- 1 -ylsulfonyl- 1 ,4-diazepane Example 11: l-(3,5-dichloropyridin-4-yl)-4-(4-propan-2-ylphenyl)sulfonyl-l,4-diazepane Example 12: 1 -(3 ,5-dichloropyridin-4-yl)-4-naphthalen-2 -ylsulfonyl- 1 ,4-diazepane Example 13: 1 -(2-chlorophenyl)sulfonyl-4-(3 ,5 -dichloropyridin-4-yl)- 1 ,4-diazepane Example 14: l-(3,5-dichloropyridin-4-yl)-4-[2-(trifluoromethyl)phenyl]sulfonyl-l,4- diazepane Example 15: N-[4-[[4-(3,5-dichloropyridin-4-yl)-l,4-diazepan-l- yl]sulfonyl]phenyl]acetamide
Example 16: 1 -(3 , 5 -dichloropyridin-4-yl)-4- [3 -(trifiuoromethyl)phenyl] sulfonyl- 1 ,4- diazepane Example 17: 2-[[4-(3,5-dichloropyridin-4-yl)-l,4-diazepan-l-yl]sulfonyl]benzonitrile Example 18: 1 -(3 -chlorophenyl)sulfonyl-4-(3, 5 -dichloropyridin-4-yl)- 1,4-diazepane Example 19: 1 -(3 ,5-dichloropyridin-4-yl)-4-(2-methylphenyl)sulfonyl- 1 ,4-diazepane
Example 20: l-(2-chlorophenyl)sulfonyl-4-f3-(trifluoromethyl)pyridin-2-yll-l,4- diazepane

A solution of l-[3-(trifluoromethyl)pyridin-2-yl]-l,4-diazepane (Intermediate 2, 322 mg, 1.31 mmol) in DCM (20 ml) was treated sequentially with triethylamine (0.22 ml, 1.56 mmol) and a solution of 2-chlorobenzenesulfonylchloride (305 mg, 1.44 mmol) in DCM (2 ml). The
reaction mixture was stirred for 1 hr at ambient temperature and then quenched with saturated sodium bicarbonate solution (10 ml); the 2-phase mixture was passed through a phase separation cartridge and the organic eluent evaporated to give a yellow oil (538 mg). The product was isolated by flash column chromatography (CombiFlash ® Companion ™ , 12g silica cartridge, eluting with isohexane containing EtOAc, 0 — 30% v/v gradient)) to give the title compound as a colourless gum, 1H NMR (400 MHz, DMSO-d6) δl .92 - 1.98 (2H, m), 3.42 (2H, t), 3.54 - 3.62 (6H, m), 7.01 - 7.04 (IH, m), 7.51 - 7.55 (IH, m), 7.62 - 7.68 (2H, m), 7.96 - 7.98 (2H, m), 8.40 - 8.41 (IH, m), m/z 420, (M+H)+ [I].
The requisite l-[3-(trifluoromethyl)pyridin-2-yl]-l,4-diazepane (Intermediate 2) starting material was prepared as follows:

1,4-diazepane (1.3 g, 13 mmol) and 2-chloro-3-(trifluoromethyl) pyridine (473 mg, 2.6 mmol) were combined without solvent, and the mixture heated with stirring at 90 0C for 3.5 hours. The reaction mixture was cooled to room temperature, dissolved in EtOAc, and the solution washed sequentially with water (10 ml) and brine (10 ml). The organic solution was dried (MgSO4) and evaporated to give the title compound as a yellow oil (593 mg, 93%) which was used without purification; 1H NMR (400 MHz, DMSO-d6) 1.76 - 1.82 (2H, m), 2.74 (2H, t), 2.88 - 2.91 (2H, m), 3.48 (2H, t), 3.53 - 3.58 (2H, m), 6.89 - 6.92 (IH, m), 7.92 - 7.94 (IH, m), 8.35 - 8.36 (IH, m), m/z 246 (M+H)+.
Analogous methods to those above were used to prepare intermediates for the Examples below (see table for Intermediates 3-10). It will be appreciated that variations of this methodology may be used to prepare individual sulfonamides, eg. solvent, base, temperature and duration of reaction, and method of purification may be varied. Similarly, it will be appreciated that the diazepane intermediates may be prepared under a variety of conditions, eg. with or without solvent, with conventional or microwave heating, at a temperature in the range 5O0C - 2000C, and with varying ratios of reactants.
Examples 21 - 30
The following Examples were prepared using a method essentially similar to that described above, using the appropriate commercially available sulfonyl chlorides, and the appropriate diazepane intermediates (described in the Intermediates Table, below).



Example 21: 1 -(2-chlorophenyl)sulfonyl-4-(3 -chloropyridin-2-yl)- 1 ,4-diazepane
Example 22: l-(2-chlorophenyl)sulfonyl-4-[2-(trifluoromethyl)phenyl]-l,4-diazepane
Example 23: l-(2-chlorophenyl)-4-(2-chlorophenyl)sulfonyl-l,4-diazepane
Example 24: 1 -(2-chlorophenyl)sulfonyl-4-(4-fluorophenyl)- 1 ,4-diazepane
Example 25: 1 -(2-chlorophenyl)sulfonyl-4-(2,6-dichlorophenyl)- 1 ,4-diazepane
Example 26: 4-[4-(2-chlorophenyl)sulfonyl-l,4-diazepan-l-yl]~3-
(trifluoromethyl)benzonitrile
Example 27: l-(2-methylphenyl)sulfonyl-4-[3-(trifluoromethyl)pyridin-2-yl]-l ,4-diazepane
Example 28: l-[4-[[4-[3-(trifluoromethyl)pyridin-2-yl]-l,4-diazepan-l- yl] sulfonyljphenyl] ethanone
Example 29: l-(2-chlorophenyl)sulfonyl-4-(2-fluoro-4-methylsulfonyl-phenyl)-l,4-diazepane
Example 30: 4- [4-(2-chlorophenyl)sulfonyl- 1 ,4-diazepan- 1 -yl] -3 -fluoro-N,N-dimethyl- benzamide
Intermediates


Example 31: 2-[4-f[4-(3.,5-dichloropyridin-4-yl)-l,4-diazepan-l-yl1sulfonvnphenyll-l,l,l- trifluoro-propan-2-ol

Examples 32 - 33
The following Examples were prepared by a method essentially similar to that described for Example 31 :
007/001507
34

[A] The requisite 2-[4-(4-acetylphenyl)sulfonyl-l,4-diazepan-l-yl]pyridine-3-carbonitrile intermediate was prepared by a method essentially similar to that described for Example 28 and used without characterisation.
[B] The requisite ketone starting material is Example 28.
Example 32: 2-[4-[4-(l,l,l-trifluoro-2-hydroxy-propan-2-yl)phenyl]sulfonyl-l,4-diazepan-l- yl]pyridine-3-carbonitrile
Example 33: 1,1,1 -trifluoro-2-[4-[[4-[3-(lrifluoromethyl)pyridin-2-yl]-l ,4-diazepan- 1 - yl]sulfonyl]phenyl]propan-2-ol
Claims
1. A compound of formula (1) or a salt thereof:

(1) wherein:
Ring A is phenyl or naphthyl, optionally substituted by 1, 2 or 3 substituents independently selected from R1 ;
R1 is selected from hydroxy, halo, trifluoromethyl, Ci-4alkyl, hydroxy Ci.4alkyl, Ci-3alkyl
C(OH)(CF3)-, C^alkoxy, cyano, C^alkylcarbonyl and Ci-4alkylcarbonylamino;
Ring B is phenyl or pyridyl, optionally substituted in the 2-, T-, 3- or 3'- positions (relative to the point of attachment) (where available) by 1 or 2 substituents independently selected from R2, and/or substituted in the 4- position (relative to the point of attachment) (where available) with a substituent selected from R3;
R2 is selected from halo, cyano, trifluoromethyl, Ci.4alkyl and Ci_4alkoxy;
R3 is selected from halo, cyano, Ci.3alkylsulfonyl and -CONR4R5;
R4 and R5 are independently selected from hydrogen and Ci-4alkyl; or R4 and R5 together with the nitrogen to which they are attached form an azetidinyl or pyrrolidinyl ring; provided that the compound of formula (1) is not:
4-[[4-(3,5-dichloropyridin-4-yl)-l,4-diazepan-l-yl]sulfonyl]benzonitrile;
1 -(2-chlorophenyl)sulfonyl-4-(2-fluoropyridin-4-yl)- 1 ,4-diazepane; 1 -(2-chlorophenyl)sulfonyl-4-(2-chloropyridin-4-yl)- 1 ,4-diazepane; l-(5-chloro-3-methyl-pyridin-2-yl)-4-(2-chlorophenyl)sulfonyl-l,4-diazepane;
2-[4-(2-methylphenyl)sulfonyl-l,4-diazepan-l-yl]pyridine-3-carbonitrile;
2-[4-(2-methoxyphenyl)sulfonyl-l,4-diazepan-l-yl]pyridine-3-carbonitrile;
4-[4-(2-cyanophenyl)sulfonyl-l,4-diazepan-l-yl]-3-fiuoro-N,N-dimethyl-benzamide; 3-fluoro-4-[4-(2-methoxyphenyl)sulfonyl-l,4-diazepan-l-yl]-N,N-dimethyl-benzamide; 01507
36
1 -(4-methylphenyl)sulfonyl-4-(2-methoxyphenyl)- 1 ,4-diazepane; or
1 -(4-fluorophenyl)sulfonyl-4-(3 -trifluoromethyl-4-cyanophenyl)- 1 ,4-diazepane.
2. A compound according to claim 1 wherein Ring A is phenyl, optionally substituted by 1, 2 or 3 substituents independently selected from R1.
3. A compound according to either claim 1 or claim 2 wherein Ring B is phenyl, optionally substituted in the 2-, T-, 3- or 3'- positions (relative to the point of attachment) (where available) by 1 or 2 substituents independently selected from R2, and/or substituted in the 4- position (relative to the point of attachment) (where available) with a substituent selected from R3.
4. A compound according to either claim 1 or claim 2 wherein Ring B is phenyl.
5. A compound according any one of claims 1 to 4 wherein R1 is selected from hydroxy, fluoro, chloro, trifluoromethyl, C^alkyl, hydroxy C1-4alkyl, methoxy, cyano, methylcarbonyl and methylcarbonylamino.
6. A compound according any one of claims 1 to 5 wherein R2 is selected from fluoro, chloro, cyano, trifluoromethyl, Ci-4alkyl and Ci^alkoxy.
7. A compound according any one of claims 1 to 6 wherein R3 is selected from fluoro, chloro, cyano, C1-3alkylsulfonyl, -CONHMe and -CONMe2 or R3 is -CONR4R5 wherein R4 and R5 together with the nitrogen to which they are attached form an azetidinyl or pyrrolidinyl ring.
8. A compound according to claim 1, which is one of the following or a salt thereof: l-(3,5-dichloropyridin-4-yl)-4-(4-fluoiOphenyl)sulfonyl-l,4-diazepane; l-(2,4-dichlorophenyl)sulfonyl-4-(3,5-dichloropyridin-4-yl)-l,4-diazepane; l-(3,5-dichloropyridin-4-yl)-4-(3-methylphenyl)sulfonyl-l,4-diazepane; l-(3,5-dichloropyridin-4-yl)-4-(4-methylphenyl)sulfonyl-l,4-diazepane; l-(3,5-dichloropyridin-4-yl)-4-(2-methoxyphenyl)sulfonyl-l,4-diazepane; l-(3,5-dichloropyridin-4-yl)-4-(3-methoxyphenyl)sulfonyl-l,4-diazepane; l-(3,5-dichloropyridin-4-yl)-4-(4-methoxyphenyl)sulfonyl-l,4-diazepane; l-(3,5-dichloropyridin-4-yl)-4-[4-(trifluoromethyl)phenyl]sulfonyl-l,4-diazepane; l-[4-[[4-(3,5 -dichloropyridin-4-y I)- 1 ,4-diazepan- 1 -yl] sulfonyl]phenyl] ethanone; 1 -(3 ,5 -dichloropyridin-4-yl)-4-naphthalen- 1 -ylsulfonyl- 1 ,4-diazepane; l-(3,5-dichloropyridin-4-yl)-4-(4-propan-2-ylphenyl)sulfonyl-l,4-diazepane; l-(3,5-dichloropyridin-4-yl)-4-naphthalen-2-ylsulfonyl-l,4-diazepane; l-(2-chlorophenyl)sulfonyl-4-(3,5-dichloropyridin-4-yl)-l,4-diazepane; l-(3,5-dichloropyridin-4-yl)-4-[2-(trifluoromethyl)phenyl]sulfonyl-l,4-diazepane; N-[4-[[4-(3,5-dichloropyridin-4-yl)-l,4-diazepan-l-yl]sulfonyl]phenyl]acetamide; l-(3,5-dichloiOpyridin-4-yl)-4-[3-(trifluoromethyl)phenyl]sulfonyl-l,4-diazepane;
2-[[4-(3,5-dichloropyridin-4-yl)-l,4-diazepan-l-yl]sulfonyl]benzonitrile; l-(3-chlorophenyl)sulfonyl-4-(3,5-dichloropyridin-4-yl)-l,4-diazepane;
1 -(3 ,5 -dichloropyridin-4-yl)-4-(2-methylphenyl)sulfonyl- 1 ,4-diazepane; l-(2-ciιlorophenyl)sulfonyl-4-[3-(trifluoromethyl)pyridin-2-yl]-l,4-diazepane;
1 -(2-chlorophenyl)sulfonyl-4-(3 -chloropyridin-2-yl)- 1 ,4-diazepane ; l-(2-chlorophenyl)sulfonyl-4-[2-(trifluoromethyl)phenyl]-l,4-diazepane;
1 -(2-chlorophenyl)-4-(2-chlorophenyl)sulfonyl- 1 ,4-diazepane;
1 -(2-chlorophenyl)sulfonyl-4-(4-fluorophenyl)- 1 ,4-diazepane; l-(2-chlorophenyl)sulfonyl-4-(2,6-dichlorophenyl)-l,4-diazepane;
4-[4-(2-chlorophenyl)sulfonyl-l,4-diazepan-l-yl]-3-(trifluoromethyl)benzonitrile; l-(2-methylphenyl)sulfonyl-4-[3-(trifluoromethyl)pyridin-2-yl]-l,4-diazepane;
1 - [4- [ [4- [3 -(trifluoromethyl)pyridin-2-yl]- 1 ,4-diazepan- 1 -yl] sulfonyl]phenyl] ethanone ; l-(2-chlorophenyl)sulfonyl-4-(2-fluoro-4-methylsulfonyl-phenyl)-l,4-diazepane; 4-[4-(2-chlorophenyl)sulfonyl-l,4-diazepan-l-yl]-3-fluoro-N,N-dimethyl-benzamide;
2-[4-[[4-(3 ,5 -dichloropyridin-4-yl)- 1 ,4-diazepan- 1 -yl]sulfonyl]phenyl]- 1 ,1,1 -trifluoro-propan-
2-ol;
2-[4-[4-(l , 1 , 1 ~trifluoro-2-hydroxy-propan-2-yl)phenyl]sulfonyl- 1 ,4-diazepan- l-yl]pyridine-3 - carbonitrile; and 1,1,1 -trifluoro-2- [4- [[4- [3 -(trifluoromethyl)pyridin-2-yl]-l ,4-diazepan- 1 - yl] sulfonyl]phenyl]propan-2-ol . 01507
38
9. A pharmaceutical composition which comprises a compound of formula (I), or a pharmaceutically acceptable salt thereof, as claimed in claiml in association with a pharmaceutically-acceptable diluent or carrier.
10. A compound of formula (1), or a pharmaceutically acceptable salt thereof, as claimed in claim 1, for use in a method of prophylactic or therapeutic treatment of a warm-blooded animal, such as man.
11. A compound of formula (1), or a pharmaceutically acceptable salt thereof, as claimed in claim 1 for use as a medicament.
12. The use of a compound of formula (1), or a pharmaceutically acceptable salt thereof, as claimed in claim 1 in the manufacture of a medicament for use in the production of an
1 lβHSDl inhibitory effect in a warm-blooded animal, such as man.
13. A process for preparing a compound according to claim 1 which process (wherein variable groups are, unless otherwise specified, as defined in formula (1) in claim 1) comprises any one of processes a) to c): a) reaction of a compound of Formula (2) with a compound of Formula (3):

(2) (3)
wherein X1 is a leaving group; or b) reaction of a compound of Formula (4) with a compound of Formula (5): 
(4) (5) wherein X2 is a leaving group; c) reaction of a compound of Formula (6) with an organometallic reagent to give a compound of Formula (7):

(6) (7) and thereafter if necessary or desirable: i) converting a compound of the formula (1) into another compound of the formula (1); ii) removing any protecting groups; iii) resolving enantiomers; iv) forming a salt thereof.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US79526006P | 2006-04-26 | 2006-04-26 | |
| US60/795,260 | 2006-04-26 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007122411A1 true WO2007122411A1 (en) | 2007-11-01 |
Family
ID=38227722
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB2007/001507 WO2007122411A1 (en) | 2006-04-26 | 2007-04-25 | Diazepan-1-yl-sulfonyl derivatives for the treatment of metabolic syndrome |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2007122411A1 (en) |
Cited By (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2015073774A1 (en) * | 2013-11-14 | 2015-05-21 | Novira Therapeutics, Inc. | Azepane derivatives and methods of treating hepatitis b infections |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0156433A2 (en) * | 1984-03-26 | 1985-10-02 | Janssen Pharmaceutica N.V. | Anti-virally active pyridazinamines |
| JP2001261657A (en) * | 2000-03-17 | 2001-09-26 | Yamanouchi Pharmaceut Co Ltd | Cyanophenyl derivative |
| WO2005115983A1 (en) * | 2004-04-07 | 2005-12-08 | Kalypsys, Inc. | Aryl sulfonamide and sulfonyl compounds as modulators of ppar and methods of treating metabolic disorders |
-
2007
- 2007-04-25 WO PCT/GB2007/001507 patent/WO2007122411A1/en active Application Filing
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0156433A2 (en) * | 1984-03-26 | 1985-10-02 | Janssen Pharmaceutica N.V. | Anti-virally active pyridazinamines |
| JP2001261657A (en) * | 2000-03-17 | 2001-09-26 | Yamanouchi Pharmaceut Co Ltd | Cyanophenyl derivative |
| WO2005115983A1 (en) * | 2004-04-07 | 2005-12-08 | Kalypsys, Inc. | Aryl sulfonamide and sulfonyl compounds as modulators of ppar and methods of treating metabolic disorders |
Non-Patent Citations (11)
| Title |
|---|
| BARF T ET AL: "Recent progress in 11-[beta]-hydroxysteroid dehydrogenase type 1 (11-[beta]-HSD1) inhibitor development", DRUGS OF THE FUTURE, BARCELONA, ES, vol. 31, no. 3, March 2006 (2006-03-01), pages 231 - 243, XP002424785, ISSN: 0377-8282 * |
| DATABASE REGISTRY CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; 2002, XP002442424, Database accession no. 439927-80-7 * |
| DATABASE REGISTRY CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; 2003, XP002442419, Database accession no. 478256-91-6 * |
| DATABASE REGISTRY CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; 2003, XP002442420, Database accession no. 478256-89-2 * |
| DATABASE REGISTRY CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; 2003, XP002442421, Database accession no. 478256-87-0 * |
| DATABASE REGISTRY CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; 2003, XP002442422, Database accession no. 478256-86-9 * |
| DATABASE REGISTRY CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; 2003, XP002442423, Database accession no. 478256-84-7 * |
| DATABASE REGISTRY CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; 2004, XP002442415, Database accession no. 693776-93-1 * |
| DATABASE REGISTRY CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; 2004, XP002442416, Database accession no. 693776-91-9 * |
| DATABASE REGISTRY CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; 2004, XP002442417, Database accession no. 685115-86-0 * |
| DATABASE REGISTRY CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; 2004, XP002442418, Database accession no. 651005-66-2 * |
Cited By (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2015073774A1 (en) * | 2013-11-14 | 2015-05-21 | Novira Therapeutics, Inc. | Azepane derivatives and methods of treating hepatitis b infections |
| US9115113B2 (en) | 2013-11-14 | 2015-08-25 | Novira Therapeutics, Inc. | Azepane derivatives and methods of treating hepatitis B infections |
| JP2016537368A (en) * | 2013-11-14 | 2016-12-01 | ノヴィラ・セラピューティクス・インコーポレイテッド | Azepan derivative and method for treating hepatitis B infection |
| US9758489B2 (en) | 2013-11-14 | 2017-09-12 | Novira Therapeutics, Inc. | Azepane derivatives and methods of treating hepatitis B infections |
| AU2014348518B2 (en) * | 2013-11-14 | 2019-01-03 | Novira Therapeutics, Inc. | Azepane derivatives and methods of treating hepatitis B infections |
| US10364228B2 (en) | 2013-11-14 | 2019-07-30 | Novira Therapeutics, Inc. | Azepane derivatives and methods of treating hepatitis B infections |
| CN106255684B (en) * | 2013-11-14 | 2019-08-23 | 诺维拉治疗公司 | The method of azepan derivatives and treatment hepatitis B infection |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2007122411A1 (en) | Diazepan-1-yl-sulfonyl derivatives for the treatment of metabolic syndrome | |
| WO2007135427A1 (en) | 1,4-disubstituted piperazine and 1,4-disubstituted azepane as 11 -beta-hydroxysteroid dehydrogenase 1 inhibitors | |
| US20100022589A1 (en) | Pyridine-3-carboxamide compounds and their use for inhibiting 11-beta-hydroxysteroid dehydrogenase | |
| AU2009211215B2 (en) | Novel crystalline forms of 4- [4- (2-adamantylcarbam0yl) -5-tert-butyl-pyrazol-1-yl] benzoic acid | |
| US20070112000A1 (en) | Chemical compounds | |
| US5238942A (en) | Substituted quinazolinones bearing acidic functional groups as angiotensin ii antagonists | |
| US20060058315A1 (en) | 2-Oxo-ethanesulfonamide derivates | |
| US8344016B2 (en) | Pyrazole derivatives as 11-beta-HSD1 inhibitors | |
| US20070219266A1 (en) | N-Acylated-3-(Benzoyl)-Pyrrolidines as 11-Beta-HSD1 Inhibitors Useful for the Treatment of Metabolic Disorders | |
| EP1820504A1 (en) | Imine compound | |
| AU2005315430A1 (en) | Oxadiazole derivatives as DGAT inhibitors | |
| JP2009539955A (en) | Oxazole derivatives and their use in the treatment of diabetes and obesity | |
| EP3813813A1 (en) | Compounds | |
| CA2072775A1 (en) | Substituted triazolinones | |
| AU2008326226B2 (en) | 4-[4-(2-adamantylcarbamoyl)-5-tert-butyl-pyrazol-1-yl]benzoic acid - 465 | |
| CN101668524B (en) | Pyrazole derivatives as 11-beta-hsd1 inhibitors | |
| BR112013013490B1 (en) | compounds and pharmaceutical composition | |
| WO2004076414A2 (en) | Novel compounds | |
| JP3688714B2 (en) | Aminophenol derivatives |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07732545 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 07732545 Country of ref document: EP Kind code of ref document: A1 |