WO2006104030A1 - Thiazole compound - Google Patents
Thiazole compound Download PDFInfo
- Publication number
- WO2006104030A1 WO2006104030A1 PCT/JP2006/305940 JP2006305940W WO2006104030A1 WO 2006104030 A1 WO2006104030 A1 WO 2006104030A1 JP 2006305940 W JP2006305940 W JP 2006305940W WO 2006104030 A1 WO2006104030 A1 WO 2006104030A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- amino
- oxy
- indeno
- substituted
- Prior art date
Links
- -1 Thiazole compound Chemical class 0.000 title claims description 392
- 150000001875 compounds Chemical class 0.000 claims abstract description 376
- 150000003839 salts Chemical class 0.000 claims abstract description 91
- 125000003277 amino group Chemical group 0.000 claims abstract description 70
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims abstract description 51
- 125000005843 halogen group Chemical group 0.000 claims abstract description 49
- 125000002252 acyl group Chemical group 0.000 claims abstract description 42
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims abstract description 41
- 125000001931 aliphatic group Chemical group 0.000 claims abstract description 40
- 206010012601 diabetes mellitus Diseases 0.000 claims abstract description 26
- 125000002947 alkylene group Chemical group 0.000 claims abstract description 20
- 239000003795 chemical substances by application Substances 0.000 claims abstract description 15
- 230000000069 prophylactic effect Effects 0.000 claims abstract description 7
- 230000001225 therapeutic effect Effects 0.000 claims abstract description 5
- YACKEPLHDIMKIO-UHFFFAOYSA-N methylphosphonic acid Chemical compound CP(O)(O)=O YACKEPLHDIMKIO-UHFFFAOYSA-N 0.000 claims description 226
- 238000000034 method Methods 0.000 claims description 214
- 125000004495 thiazol-4-yl group Chemical group S1C=NC(=C1)* 0.000 claims description 143
- 125000001424 substituent group Chemical group 0.000 claims description 102
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 87
- 125000000623 heterocyclic group Chemical group 0.000 claims description 62
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 61
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 58
- 229910052757 nitrogen Inorganic materials 0.000 claims description 55
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 44
- 229910052760 oxygen Inorganic materials 0.000 claims description 44
- 239000001301 oxygen Substances 0.000 claims description 44
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 44
- 229910052717 sulfur Inorganic materials 0.000 claims description 43
- 125000005842 heteroatom Chemical group 0.000 claims description 42
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 claims description 40
- 125000000217 alkyl group Chemical group 0.000 claims description 40
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 40
- 239000011593 sulfur Substances 0.000 claims description 40
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 34
- 125000003545 alkoxy group Chemical group 0.000 claims description 28
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 claims description 27
- 125000004890 (C1-C6) alkylamino group Chemical group 0.000 claims description 27
- 125000004663 dialkyl amino group Chemical group 0.000 claims description 26
- 125000003282 alkyl amino group Chemical group 0.000 claims description 25
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 claims description 24
- 125000004103 aminoalkyl group Chemical group 0.000 claims description 20
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 18
- 125000002768 hydroxyalkyl group Chemical group 0.000 claims description 18
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 claims description 17
- 125000000041 C6-C10 aryl group Chemical group 0.000 claims description 15
- 239000003814 drug Substances 0.000 claims description 15
- 125000006376 (C3-C10) cycloalkyl group Chemical group 0.000 claims description 13
- 125000003118 aryl group Chemical group 0.000 claims description 13
- 125000000676 alkoxyimino group Chemical group 0.000 claims description 12
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 12
- 208000002705 Glucose Intolerance Diseases 0.000 claims description 11
- 125000004414 alkyl thio group Chemical group 0.000 claims description 11
- 125000006577 C1-C6 hydroxyalkyl group Chemical group 0.000 claims description 10
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 claims description 10
- 125000001188 haloalkyl group Chemical group 0.000 claims description 10
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 claims description 10
- 125000006619 (C1-C6) dialkylamino group Chemical group 0.000 claims description 9
- 125000004429 atom Chemical group 0.000 claims description 9
- 125000004043 oxo group Chemical group O=* 0.000 claims description 9
- 239000000126 substance Substances 0.000 claims description 9
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 8
- 201000010099 disease Diseases 0.000 claims description 8
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 8
- 125000000250 methylamino group Chemical group [H]N(*)C([H])([H])[H] 0.000 claims description 8
- 125000006583 (C1-C3) haloalkyl group Chemical group 0.000 claims description 7
- 125000000033 alkoxyamino group Chemical group 0.000 claims description 7
- 208000004104 gestational diabetes Diseases 0.000 claims description 7
- 230000003449 preventive effect Effects 0.000 claims description 7
- 125000000171 (C1-C6) haloalkyl group Chemical group 0.000 claims description 6
- 206010003210 Arteriosclerosis Diseases 0.000 claims description 6
- 208000014311 Cushing syndrome Diseases 0.000 claims description 6
- 208000031226 Hyperlipidaemia Diseases 0.000 claims description 6
- 208000008589 Obesity Diseases 0.000 claims description 6
- 208000011775 arteriosclerosis disease Diseases 0.000 claims description 6
- 201000001421 hyperglycemia Diseases 0.000 claims description 6
- 239000003112 inhibitor Substances 0.000 claims description 6
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 6
- 235000020824 obesity Nutrition 0.000 claims description 6
- 201000009104 prediabetes syndrome Diseases 0.000 claims description 6
- 201000009395 primary hyperaldosteronism Diseases 0.000 claims description 6
- 150000003431 steroids Chemical class 0.000 claims description 6
- 125000000979 2-amino-2-oxoethyl group Chemical group [H]C([*])([H])C(=O)N([H])[H] 0.000 claims description 5
- 208000002249 Diabetes Complications Diseases 0.000 claims description 5
- 206010018429 Glucose tolerance impaired Diseases 0.000 claims description 5
- 102000004877 Insulin Human genes 0.000 claims description 5
- 108090001061 Insulin Proteins 0.000 claims description 5
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 5
- 229940125396 insulin Drugs 0.000 claims description 5
- 238000004519 manufacturing process Methods 0.000 claims description 5
- 125000004442 acylamino group Chemical group 0.000 claims description 4
- 125000004453 alkoxycarbonyl group Chemical group 0.000 claims description 4
- 125000003368 amide group Chemical group 0.000 claims description 4
- 125000001951 carbamoylamino group Chemical group C(N)(=O)N* 0.000 claims description 4
- 208000007345 glycogen storage disease Diseases 0.000 claims description 4
- 238000011282 treatment Methods 0.000 claims description 4
- 208000004930 Fatty Liver Diseases 0.000 claims description 3
- 206010019708 Hepatic steatosis Diseases 0.000 claims description 3
- 206010020772 Hypertension Diseases 0.000 claims description 3
- 241001465754 Metazoa Species 0.000 claims description 3
- 125000006310 cycloalkyl amino group Chemical group 0.000 claims description 3
- 208000010706 fatty liver disease Diseases 0.000 claims description 3
- 229910052736 halogen Inorganic materials 0.000 claims description 3
- 150000002367 halogens Chemical class 0.000 claims description 3
- 231100000240 steatosis hepatitis Toxicity 0.000 claims description 3
- 125000004354 sulfur functional group Chemical group 0.000 claims description 3
- 125000006700 (C1-C6) alkylthio group Chemical group 0.000 claims description 2
- 206010061218 Inflammation Diseases 0.000 claims description 2
- 230000004054 inflammatory process Effects 0.000 claims description 2
- 210000003127 knee Anatomy 0.000 claims description 2
- 241001553014 Myrsine salicina Species 0.000 claims 2
- 101710099475 3'-phosphoadenosine 5'-phosphate phosphatase Proteins 0.000 claims 1
- 125000001963 4 membered heterocyclic group Chemical group 0.000 claims 1
- 101710196411 Fructose-1,6-bisphosphatase Proteins 0.000 claims 1
- 101710186733 Fructose-1,6-bisphosphatase, chloroplastic Proteins 0.000 claims 1
- 101710109119 Fructose-1,6-bisphosphatase, cytosolic Proteins 0.000 claims 1
- 101710198902 Fructose-1,6-bisphosphate aldolase/phosphatase Proteins 0.000 claims 1
- 230000000694 effects Effects 0.000 abstract description 8
- 125000001476 phosphono group Chemical group [H]OP(*)(=O)O[H] 0.000 abstract 1
- 125000001820 oxy group Chemical group [*:1]O[*:2] 0.000 description 266
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 231
- 238000005160 1H NMR spectroscopy Methods 0.000 description 190
- 238000004992 fast atom bombardment mass spectroscopy Methods 0.000 description 149
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 135
- VLKZOEOYAKHREP-UHFFFAOYSA-N methyl pentane Natural products CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 97
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 96
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 96
- 239000002904 solvent Substances 0.000 description 94
- 238000006243 chemical reaction Methods 0.000 description 89
- 230000002829 reductive effect Effects 0.000 description 80
- 239000000243 solution Substances 0.000 description 77
- 239000000203 mixture Substances 0.000 description 76
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 67
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 57
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 50
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 50
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 50
- 235000019441 ethanol Nutrition 0.000 description 48
- 239000012044 organic layer Substances 0.000 description 47
- 238000010898 silica gel chromatography Methods 0.000 description 47
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical class O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 47
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 46
- 239000007788 liquid Substances 0.000 description 42
- 239000010408 film Substances 0.000 description 41
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 38
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 38
- ZMXDDKWLCZADIW-UHFFFAOYSA-N dimethylformamide Substances CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 38
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 34
- 239000002253 acid Substances 0.000 description 34
- 229920006395 saturated elastomer Polymers 0.000 description 30
- 230000008569 process Effects 0.000 description 29
- YROXIXLRRCOBKF-UHFFFAOYSA-N sulfonylurea Chemical class OC(=N)N=S(=O)=O YROXIXLRRCOBKF-UHFFFAOYSA-N 0.000 description 27
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 26
- 125000006239 protecting group Chemical group 0.000 description 25
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 25
- 235000017557 sodium bicarbonate Nutrition 0.000 description 25
- 238000001816 cooling Methods 0.000 description 24
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 24
- 229910052739 hydrogen Inorganic materials 0.000 description 24
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 23
- 239000011541 reaction mixture Substances 0.000 description 23
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 22
- RGSFGYAAUTVSQA-UHFFFAOYSA-N Cyclopentane Chemical compound C1CCCC1 RGSFGYAAUTVSQA-UHFFFAOYSA-N 0.000 description 22
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 22
- 125000005605 benzo group Chemical group 0.000 description 20
- 150000002170 ethers Chemical class 0.000 description 20
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 20
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 19
- 239000002585 base Substances 0.000 description 19
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 18
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 18
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 18
- NCEMUOYFYLEORV-UHFFFAOYSA-N difluoromethylphosphonic acid Chemical compound OP(O)(=O)C(F)F NCEMUOYFYLEORV-UHFFFAOYSA-N 0.000 description 18
- 239000012299 nitrogen atmosphere Substances 0.000 description 18
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 17
- 239000003153 chemical reaction reagent Substances 0.000 description 17
- 230000035484 reaction time Effects 0.000 description 17
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 16
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 15
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 15
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 15
- 239000003054 catalyst Substances 0.000 description 15
- ITMCEJHCFYSIIV-UHFFFAOYSA-N triflic acid Chemical compound OS(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-N 0.000 description 15
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 14
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 14
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 14
- 125000004432 carbon atom Chemical group C* 0.000 description 14
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 14
- 239000007858 starting material Substances 0.000 description 14
- OISVCGZHLKNMSJ-UHFFFAOYSA-N 2,6-dimethylpyridine Chemical compound CC1=CC=CC(C)=N1 OISVCGZHLKNMSJ-UHFFFAOYSA-N 0.000 description 12
- GOJUJUVQIVIZAV-UHFFFAOYSA-N 2-amino-4,6-dichloropyrimidine-5-carbaldehyde Chemical group NC1=NC(Cl)=C(C=O)C(Cl)=N1 GOJUJUVQIVIZAV-UHFFFAOYSA-N 0.000 description 12
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 12
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 12
- 102000012195 Fructose-1,6-bisphosphatases Human genes 0.000 description 12
- 108010017464 Fructose-Bisphosphatase Proteins 0.000 description 12
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 12
- 229910052783 alkali metal Inorganic materials 0.000 description 12
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 12
- 238000007796 conventional method Methods 0.000 description 12
- ABLZXFCXXLZCGV-UHFFFAOYSA-N phosphonic acid group Chemical group P(O)(O)=O ABLZXFCXXLZCGV-UHFFFAOYSA-N 0.000 description 12
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 11
- 229910019142 PO4 Inorganic materials 0.000 description 11
- 150000001408 amides Chemical class 0.000 description 11
- 235000019270 ammonium chloride Nutrition 0.000 description 11
- NBIIXXVUZAFLBC-UHFFFAOYSA-M dihydrogenphosphate Chemical compound OP(O)([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-M 0.000 description 11
- DMEGYFMYUHOHGS-UHFFFAOYSA-N heptamethylene Natural products C1CCCCCC1 DMEGYFMYUHOHGS-UHFFFAOYSA-N 0.000 description 11
- 235000021317 phosphate Nutrition 0.000 description 11
- 238000003786 synthesis reaction Methods 0.000 description 11
- 150000007979 thiazole derivatives Chemical class 0.000 description 11
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 10
- 229920002472 Starch Polymers 0.000 description 10
- 150000001298 alcohols Chemical class 0.000 description 10
- 150000008282 halocarbons Chemical class 0.000 description 10
- NWVVVBRKAWDGAB-UHFFFAOYSA-N p-methoxyphenol Chemical compound COC1=CC=C(O)C=C1 NWVVVBRKAWDGAB-UHFFFAOYSA-N 0.000 description 10
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 10
- 239000010452 phosphate Substances 0.000 description 10
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 10
- 239000000843 powder Substances 0.000 description 10
- 238000006722 reduction reaction Methods 0.000 description 10
- 239000008107 starch Substances 0.000 description 10
- 235000019698 starch Nutrition 0.000 description 10
- 238000003756 stirring Methods 0.000 description 10
- 239000003826 tablet Substances 0.000 description 10
- 102000004190 Enzymes Human genes 0.000 description 9
- 108090000790 Enzymes Proteins 0.000 description 9
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 9
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 9
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 9
- 230000002218 hypoglycaemic effect Effects 0.000 description 9
- 229910052751 metal Inorganic materials 0.000 description 9
- 239000002184 metal Substances 0.000 description 9
- OELFLUMRDSZNSF-BRWVUGGUSA-N nateglinide Chemical compound C1C[C@@H](C(C)C)CC[C@@H]1C(=O)N[C@@H](C(O)=O)CC1=CC=CC=C1 OELFLUMRDSZNSF-BRWVUGGUSA-N 0.000 description 9
- UEZVMMHDMIWARA-UHFFFAOYSA-M phosphonate Chemical compound [O-]P(=O)=O UEZVMMHDMIWARA-UHFFFAOYSA-M 0.000 description 9
- 238000010992 reflux Methods 0.000 description 9
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Substances C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 9
- ASHGTJPOSUFTGB-UHFFFAOYSA-N 3-methoxyphenol Chemical compound COC1=CC=CC(O)=C1 ASHGTJPOSUFTGB-UHFFFAOYSA-N 0.000 description 8
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 8
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 8
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 8
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 8
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 8
- 239000013078 crystal Substances 0.000 description 8
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 7
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 7
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 7
- 150000001412 amines Chemical class 0.000 description 7
- PQNFLJBBNBOBRQ-UHFFFAOYSA-N benzocyclopentane Natural products C1=CC=C2CCCC2=C1 PQNFLJBBNBOBRQ-UHFFFAOYSA-N 0.000 description 7
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 7
- 125000006317 cyclopropyl amino group Chemical group 0.000 description 7
- 239000008103 glucose Substances 0.000 description 7
- 238000007254 oxidation reaction Methods 0.000 description 7
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N phenol group Chemical group C1(=CC=CC=C1)O ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 7
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 7
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 7
- LVEYOSJUKRVCCF-UHFFFAOYSA-N 1,3-bis(diphenylphosphino)propane Chemical compound C=1C=CC=CC=1P(C=1C=CC=CC=1)CCCP(C=1C=CC=CC=1)C1=CC=CC=C1 LVEYOSJUKRVCCF-UHFFFAOYSA-N 0.000 description 6
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 6
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 6
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 6
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 6
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 6
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 6
- UORVGPXVDQYIDP-UHFFFAOYSA-N borane Chemical compound B UORVGPXVDQYIDP-UHFFFAOYSA-N 0.000 description 6
- 239000000969 carrier Substances 0.000 description 6
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 6
- 229940117389 dichlorobenzene Drugs 0.000 description 6
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 description 6
- 238000001914 filtration Methods 0.000 description 6
- 230000002140 halogenating effect Effects 0.000 description 6
- GNOIPBMMFNIUFM-UHFFFAOYSA-N hexamethylphosphoric triamide Chemical compound CN(C)P(=O)(N(C)C)N(C)C GNOIPBMMFNIUFM-UHFFFAOYSA-N 0.000 description 6
- 230000002401 inhibitory effect Effects 0.000 description 6
- 150000002576 ketones Chemical class 0.000 description 6
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 6
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 6
- 230000009467 reduction Effects 0.000 description 6
- 239000012279 sodium borohydride Substances 0.000 description 6
- 229910000033 sodium borohydride Inorganic materials 0.000 description 6
- 238000012360 testing method Methods 0.000 description 6
- 229940124597 therapeutic agent Drugs 0.000 description 6
- IMFACGCPASFAPR-UHFFFAOYSA-N tributylamine Chemical compound CCCCN(CCCC)CCCC IMFACGCPASFAPR-UHFFFAOYSA-N 0.000 description 6
- OCJBOOLMMGQPQU-UHFFFAOYSA-N 1,4-dichlorobenzene Chemical compound ClC1=CC=C(Cl)C=C1 OCJBOOLMMGQPQU-UHFFFAOYSA-N 0.000 description 5
- UGEJOEBBMPOJMT-UHFFFAOYSA-N 3-(trifluoromethyl)phenol Chemical compound OC1=CC=CC(C(F)(F)F)=C1 UGEJOEBBMPOJMT-UHFFFAOYSA-N 0.000 description 5
- UGFAIRIUMAVXCW-UHFFFAOYSA-N Carbon monoxide Chemical compound [O+]#[C-] UGFAIRIUMAVXCW-UHFFFAOYSA-N 0.000 description 5
- 206010022489 Insulin Resistance Diseases 0.000 description 5
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 5
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 5
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 5
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 5
- WCUXLLCKKVVCTQ-UHFFFAOYSA-M Potassium chloride Chemical compound [Cl-].[K+] WCUXLLCKKVVCTQ-UHFFFAOYSA-M 0.000 description 5
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 5
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 5
- 230000002159 abnormal effect Effects 0.000 description 5
- 238000005575 aldol reaction Methods 0.000 description 5
- XHLHPRDBBAGVEG-UHFFFAOYSA-N alpha-Tetralone Natural products C1=CC=C2C(=O)CCCC2=C1 XHLHPRDBBAGVEG-UHFFFAOYSA-N 0.000 description 5
- 229910002091 carbon monoxide Inorganic materials 0.000 description 5
- 238000006880 cross-coupling reaction Methods 0.000 description 5
- 239000012043 crude product Substances 0.000 description 5
- 125000002883 imidazolyl group Chemical group 0.000 description 5
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 5
- 229910000027 potassium carbonate Inorganic materials 0.000 description 5
- 235000011181 potassium carbonates Nutrition 0.000 description 5
- 125000000714 pyrimidinyl group Chemical group 0.000 description 5
- 239000001632 sodium acetate Substances 0.000 description 5
- 235000017281 sodium acetate Nutrition 0.000 description 5
- 235000000346 sugar Nutrition 0.000 description 5
- 125000004044 trifluoroacetyl group Chemical group FC(C(=O)*)(F)F 0.000 description 5
- 239000008096 xylene Substances 0.000 description 5
- QVCUKHQDEZNNOC-UHFFFAOYSA-N 1,2-diazabicyclo[2.2.2]octane Chemical compound C1CC2CCN1NC2 QVCUKHQDEZNNOC-UHFFFAOYSA-N 0.000 description 4
- WNXJIVFYUVYPPR-UHFFFAOYSA-N 1,3-dioxolane Chemical compound C1COCO1 WNXJIVFYUVYPPR-UHFFFAOYSA-N 0.000 description 4
- SGUVLZREKBPKCE-UHFFFAOYSA-N 1,5-diazabicyclo[4.3.0]-non-5-ene Chemical compound C1CCN=C2CCCN21 SGUVLZREKBPKCE-UHFFFAOYSA-N 0.000 description 4
- BMIBJCFFZPYJHF-UHFFFAOYSA-N 2-methoxy-5-methyl-3-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridine Chemical compound COC1=NC=C(C)C=C1B1OC(C)(C)C(C)(C)O1 BMIBJCFFZPYJHF-UHFFFAOYSA-N 0.000 description 4
- XWKFPIODWVPXLX-UHFFFAOYSA-N 2-methyl-5-methylpyridine Natural products CC1=CC=C(C)N=C1 XWKFPIODWVPXLX-UHFFFAOYSA-N 0.000 description 4
- 125000004180 3-fluorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C(F)=C1[H] 0.000 description 4
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 4
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 4
- 208000012219 Autonomic Nervous System disease Diseases 0.000 description 4
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 4
- 206010012655 Diabetic complications Diseases 0.000 description 4
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 4
- QIGBRXMKCJKVMJ-UHFFFAOYSA-N Hydroquinone Chemical compound OC1=CC=C(O)C=C1 QIGBRXMKCJKVMJ-UHFFFAOYSA-N 0.000 description 4
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 4
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 4
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 4
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 4
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 4
- AYVGBNGTBQLJBG-UHFFFAOYSA-N [3-(hydroxymethyl)cyclopentyl]methanol Chemical compound OCC1CCC(CO)C1 AYVGBNGTBQLJBG-UHFFFAOYSA-N 0.000 description 4
- 125000000738 acetamido group Chemical group [H]C([H])([H])C(=O)N([H])[*] 0.000 description 4
- 230000009471 action Effects 0.000 description 4
- 229910000288 alkali metal carbonate Inorganic materials 0.000 description 4
- 150000001340 alkali metals Chemical class 0.000 description 4
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 4
- 229910021529 ammonia Inorganic materials 0.000 description 4
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 4
- 239000012298 atmosphere Substances 0.000 description 4
- 239000008280 blood Substances 0.000 description 4
- 210000004369 blood Anatomy 0.000 description 4
- 239000003638 chemical reducing agent Substances 0.000 description 4
- QTMDXZNDVAMKGV-UHFFFAOYSA-L copper(ii) bromide Chemical compound [Cu+2].[Br-].[Br-] QTMDXZNDVAMKGV-UHFFFAOYSA-L 0.000 description 4
- ZCXIFQYOEZLHSP-UHFFFAOYSA-N dicyanophosphorylformonitrile Chemical class N#CP(=O)(C#N)C#N ZCXIFQYOEZLHSP-UHFFFAOYSA-N 0.000 description 4
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 description 4
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 4
- 239000008187 granular material Substances 0.000 description 4
- 239000012442 inert solvent Substances 0.000 description 4
- 230000005764 inhibitory process Effects 0.000 description 4
- 239000008101 lactose Substances 0.000 description 4
- 229910052744 lithium Inorganic materials 0.000 description 4
- XGZVUEUWXADBQD-UHFFFAOYSA-L lithium carbonate Chemical compound [Li+].[Li+].[O-]C([O-])=O XGZVUEUWXADBQD-UHFFFAOYSA-L 0.000 description 4
- 229910052808 lithium carbonate Inorganic materials 0.000 description 4
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 4
- HQRPHMAXFVUBJX-UHFFFAOYSA-M lithium;hydrogen carbonate Chemical compound [Li+].OC([O-])=O HQRPHMAXFVUBJX-UHFFFAOYSA-M 0.000 description 4
- 201000007270 liver cancer Diseases 0.000 description 4
- 208000014018 liver neoplasm Diseases 0.000 description 4
- NUJOXMJBOLGQSY-UHFFFAOYSA-N manganese dioxide Chemical compound O=[Mn]=O NUJOXMJBOLGQSY-UHFFFAOYSA-N 0.000 description 4
- XFDWRONFBJOKHC-UHFFFAOYSA-N methylphosphonic acid;hydrochloride Chemical compound Cl.CP(O)(O)=O XFDWRONFBJOKHC-UHFFFAOYSA-N 0.000 description 4
- 239000012046 mixed solvent Substances 0.000 description 4
- DXASQZJWWGZNSF-UHFFFAOYSA-N n,n-dimethylmethanamine;sulfur trioxide Chemical group CN(C)C.O=S(=O)=O DXASQZJWWGZNSF-UHFFFAOYSA-N 0.000 description 4
- 230000007170 pathology Effects 0.000 description 4
- 239000003208 petroleum Substances 0.000 description 4
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 4
- 238000005954 phosphonylation reaction Methods 0.000 description 4
- 239000011591 potassium Substances 0.000 description 4
- 229910052700 potassium Inorganic materials 0.000 description 4
- 229960003975 potassium Drugs 0.000 description 4
- 235000007686 potassium Nutrition 0.000 description 4
- 229910000028 potassium bicarbonate Inorganic materials 0.000 description 4
- 235000015497 potassium bicarbonate Nutrition 0.000 description 4
- 239000011736 potassium bicarbonate Substances 0.000 description 4
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 4
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 4
- JWHOQZUREKYPBY-UHFFFAOYSA-N rubonic acid Natural products CC1(C)CCC2(CCC3(C)C(=CCC4C5(C)CCC(=O)C(C)(C)C5CC(=O)C34C)C2C1)C(=O)O JWHOQZUREKYPBY-UHFFFAOYSA-N 0.000 description 4
- JPJALAQPGMAKDF-UHFFFAOYSA-N selenium dioxide Chemical compound O=[Se]=O JPJALAQPGMAKDF-UHFFFAOYSA-N 0.000 description 4
- 229910000029 sodium carbonate Inorganic materials 0.000 description 4
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 4
- 235000019345 sodium thiosulphate Nutrition 0.000 description 4
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 4
- 150000003573 thiols Chemical class 0.000 description 4
- UMGDCJDMYOKAJW-UHFFFAOYSA-N thiourea Chemical compound NC(N)=S UMGDCJDMYOKAJW-UHFFFAOYSA-N 0.000 description 4
- WJKHJLXJJJATHN-UHFFFAOYSA-N triflic anhydride Chemical compound FC(F)(F)S(=O)(=O)OS(=O)(=O)C(F)(F)F WJKHJLXJJJATHN-UHFFFAOYSA-N 0.000 description 4
- 208000001072 type 2 diabetes mellitus Diseases 0.000 description 4
- 125000004066 1-hydroxyethyl group Chemical group [H]OC([H])([*])C([H])([H])[H] 0.000 description 3
- WNWHHMBRJJOGFJ-UHFFFAOYSA-N 16-methylheptadecan-1-ol Chemical class CC(C)CCCCCCCCCCCCCCCO WNWHHMBRJJOGFJ-UHFFFAOYSA-N 0.000 description 3
- MGDCBOKBTJIJBT-UHFFFAOYSA-N 2,2-difluoro-1,3-dimethylimidazolidine Chemical compound CN1CCN(C)C1(F)F MGDCBOKBTJIJBT-UHFFFAOYSA-N 0.000 description 3
- PAQZWJGSJMLPMG-UHFFFAOYSA-N 2,4,6-tripropyl-1,3,5,2$l^{5},4$l^{5},6$l^{5}-trioxatriphosphinane 2,4,6-trioxide Chemical compound CCCP1(=O)OP(=O)(CCC)OP(=O)(CCC)O1 PAQZWJGSJMLPMG-UHFFFAOYSA-N 0.000 description 3
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 3
- 125000004200 2-methoxyethyl group Chemical group [H]C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 3
- RHMPLDJJXGPMEX-UHFFFAOYSA-N 4-fluorophenol Chemical compound OC1=CC=C(F)C=C1 RHMPLDJJXGPMEX-UHFFFAOYSA-N 0.000 description 3
- 125000001255 4-fluorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1F 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- 239000005995 Aluminium silicate Substances 0.000 description 3
- 206010002383 Angina Pectoris Diseases 0.000 description 3
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 3
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 3
- 208000002177 Cataract Diseases 0.000 description 3
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical compound OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 3
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 3
- 108010010803 Gelatin Proteins 0.000 description 3
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 3
- 208000001089 Multiple system atrophy Diseases 0.000 description 3
- 206010031127 Orthostatic hypotension Diseases 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 3
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 3
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 3
- 240000008042 Zea mays Species 0.000 description 3
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 description 3
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 3
- 239000004480 active ingredient Substances 0.000 description 3
- 239000000654 additive Substances 0.000 description 3
- 150000001338 aliphatic hydrocarbons Chemical class 0.000 description 3
- 150000008041 alkali metal carbonates Chemical class 0.000 description 3
- 150000008044 alkali metal hydroxides Chemical class 0.000 description 3
- 235000012211 aluminium silicate Nutrition 0.000 description 3
- 235000001014 amino acid Nutrition 0.000 description 3
- 229940024606 amino acid Drugs 0.000 description 3
- 150000001413 amino acids Chemical class 0.000 description 3
- 125000004104 aryloxy group Chemical group 0.000 description 3
- 239000011230 binding agent Substances 0.000 description 3
- 230000036772 blood pressure Effects 0.000 description 3
- 238000009835 boiling Methods 0.000 description 3
- 229910000085 borane Inorganic materials 0.000 description 3
- 239000000872 buffer Substances 0.000 description 3
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- 239000002775 capsule Substances 0.000 description 3
- 208000026106 cerebrovascular disease Diseases 0.000 description 3
- 238000004440 column chromatography Methods 0.000 description 3
- 235000005822 corn Nutrition 0.000 description 3
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 3
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 3
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 3
- MTHSVFCYNBDYFN-UHFFFAOYSA-N diethylene glycol Chemical compound OCCOCCO MTHSVFCYNBDYFN-UHFFFAOYSA-N 0.000 description 3
- 150000002148 esters Chemical class 0.000 description 3
- 125000000031 ethylamino group Chemical group [H]C([H])([H])C([H])([H])N([H])[*] 0.000 description 3
- 238000000605 extraction Methods 0.000 description 3
- 239000000706 filtrate Substances 0.000 description 3
- 238000003682 fluorination reaction Methods 0.000 description 3
- 125000001153 fluoro group Chemical group F* 0.000 description 3
- 239000008273 gelatin Substances 0.000 description 3
- 229920000159 gelatin Polymers 0.000 description 3
- 235000019322 gelatine Nutrition 0.000 description 3
- 235000011852 gelatine desserts Nutrition 0.000 description 3
- 238000005658 halogenation reaction Methods 0.000 description 3
- 239000001257 hydrogen Substances 0.000 description 3
- 201000001881 impotence Diseases 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 3
- 208000017169 kidney disease Diseases 0.000 description 3
- 229910000103 lithium hydride Inorganic materials 0.000 description 3
- JILPJDVXYVTZDQ-UHFFFAOYSA-N lithium methoxide Chemical compound [Li+].[O-]C JILPJDVXYVTZDQ-UHFFFAOYSA-N 0.000 description 3
- 208000010125 myocardial infarction Diseases 0.000 description 3
- RLKHFSNWQCZBDC-UHFFFAOYSA-N n-(benzenesulfonyl)-n-fluorobenzenesulfonamide Chemical compound C=1C=CC=CC=1S(=O)(=O)N(F)S(=O)(=O)C1=CC=CC=C1 RLKHFSNWQCZBDC-UHFFFAOYSA-N 0.000 description 3
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 3
- 150000007530 organic bases Chemical class 0.000 description 3
- 125000004430 oxygen atom Chemical group O* 0.000 description 3
- 229910052763 palladium Inorganic materials 0.000 description 3
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 3
- 239000000546 pharmaceutical excipient Substances 0.000 description 3
- 239000000825 pharmaceutical preparation Substances 0.000 description 3
- 125000005936 piperidyl group Chemical group 0.000 description 3
- 125000003226 pyrazolyl group Chemical group 0.000 description 3
- 125000004076 pyridyl group Chemical group 0.000 description 3
- GHMLBKRAJCXXBS-UHFFFAOYSA-N resorcinol Chemical compound OC1=CC=CC(O)=C1 GHMLBKRAJCXXBS-UHFFFAOYSA-N 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 3
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 3
- 239000012312 sodium hydride Substances 0.000 description 3
- 229910000104 sodium hydride Inorganic materials 0.000 description 3
- 125000003003 spiro group Chemical group 0.000 description 3
- 238000006467 substitution reaction Methods 0.000 description 3
- 150000003462 sulfoxides Chemical class 0.000 description 3
- 125000004434 sulfur atom Chemical group 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- 230000035900 sweating Effects 0.000 description 3
- 239000010409 thin film Substances 0.000 description 3
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 3
- BDZBKCUKTQZUTL-UHFFFAOYSA-N triethyl phosphite Chemical compound CCOP(OCC)OCC BDZBKCUKTQZUTL-UHFFFAOYSA-N 0.000 description 3
- IMNIMPAHZVJRPE-UHFFFAOYSA-N triethylenediamine Chemical compound C1CN2CCN1CC2 IMNIMPAHZVJRPE-UHFFFAOYSA-N 0.000 description 3
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 3
- MPDDTAJMJCESGV-CTUHWIOQSA-M (3r,5r)-7-[2-(4-fluorophenyl)-5-[methyl-[(1r)-1-phenylethyl]carbamoyl]-4-propan-2-ylpyrazol-3-yl]-3,5-dihydroxyheptanoate Chemical compound C1([C@@H](C)N(C)C(=O)C2=NN(C(CC[C@@H](O)C[C@@H](O)CC([O-])=O)=C2C(C)C)C=2C=CC(F)=CC=2)=CC=CC=C1 MPDDTAJMJCESGV-CTUHWIOQSA-M 0.000 description 2
- 125000004502 1,2,3-oxadiazolyl group Chemical group 0.000 description 2
- PAMIQIKDUOTOBW-UHFFFAOYSA-N 1-methylpiperidine Chemical compound CN1CCCCC1 PAMIQIKDUOTOBW-UHFFFAOYSA-N 0.000 description 2
- ZVCPCZRXWGOICC-UHFFFAOYSA-N 1h-benzo[7]annulene Chemical compound C1=CC=CC=C2CC=CC=C21 ZVCPCZRXWGOICC-UHFFFAOYSA-N 0.000 description 2
- MEKOFIRRDATTAG-UHFFFAOYSA-N 2,2,5,8-tetramethyl-3,4-dihydrochromen-6-ol Chemical compound C1CC(C)(C)OC2=C1C(C)=C(O)C=C2C MEKOFIRRDATTAG-UHFFFAOYSA-N 0.000 description 2
- HVHZEKKZMFRULH-UHFFFAOYSA-N 2,6-ditert-butyl-4-methylpyridine Chemical compound CC1=CC(C(C)(C)C)=NC(C(C)(C)C)=C1 HVHZEKKZMFRULH-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 2
- 125000000022 2-aminoethyl group Chemical group [H]C([*])([H])C([H])([H])N([H])[H] 0.000 description 2
- IQHSSYROJYPFDV-UHFFFAOYSA-N 2-bromo-1,3-dichloro-5-(trifluoromethyl)benzene Chemical group FC(F)(F)C1=CC(Cl)=C(Br)C(Cl)=C1 IQHSSYROJYPFDV-UHFFFAOYSA-N 0.000 description 2
- UIIAXUWGKAAXNZ-UHFFFAOYSA-N 2-trimethylsilylethanol Chemical compound C[Si](C)(C)CCO.C[Si](C)(C)CCO UIIAXUWGKAAXNZ-UHFFFAOYSA-N 0.000 description 2
- SJTBRFHBXDZMPS-UHFFFAOYSA-N 3-fluorophenol Chemical compound OC1=CC=CC(F)=C1 SJTBRFHBXDZMPS-UHFFFAOYSA-N 0.000 description 2
- QOXOZONBQWIKDA-UHFFFAOYSA-N 3-hydroxypropyl Chemical group [CH2]CCO QOXOZONBQWIKDA-UHFFFAOYSA-N 0.000 description 2
- 125000004207 3-methoxyphenyl group Chemical group [H]C1=C([H])C(*)=C([H])C(OC([H])([H])[H])=C1[H] 0.000 description 2
- PLIKAWJENQZMHA-UHFFFAOYSA-N 4-aminophenol Chemical compound NC1=CC=C(O)C=C1 PLIKAWJENQZMHA-UHFFFAOYSA-N 0.000 description 2
- ZTWBCFVYMDBPPD-UHFFFAOYSA-N 4-bromo-7-hydroxy-2,3-dihydroinden-1-one Chemical compound OC1=CC=C(Br)C2=C1C(=O)CC2 ZTWBCFVYMDBPPD-UHFFFAOYSA-N 0.000 description 2
- RGUKYNXWOWSRET-UHFFFAOYSA-N 4-pyrrolidin-1-ylpyridine Chemical compound C1CCCN1C1=CC=NC=C1 RGUKYNXWOWSRET-UHFFFAOYSA-N 0.000 description 2
- 102100031126 6-phosphogluconolactonase Human genes 0.000 description 2
- 108010029731 6-phosphogluconolactonase Proteins 0.000 description 2
- HFMZPBSZKCDKOR-UHFFFAOYSA-N 7-hydroxy-2,3-dihydroinden-1-one Chemical compound OC1=CC=CC2=C1C(=O)CC2 HFMZPBSZKCDKOR-UHFFFAOYSA-N 0.000 description 2
- FJNCXZZQNBKEJT-UHFFFAOYSA-N 8beta-hydroxymarrubiin Natural products O1C(=O)C2(C)CCCC3(C)C2C1CC(C)(O)C3(O)CCC=1C=COC=1 FJNCXZZQNBKEJT-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 229940123208 Biguanide Drugs 0.000 description 2
- XNCOSPRUTUOJCJ-UHFFFAOYSA-N Biguanide Chemical compound NC(N)=NC(N)=N XNCOSPRUTUOJCJ-UHFFFAOYSA-N 0.000 description 2
- NCRDKKMWYBVLFZ-UHFFFAOYSA-N C(C)OC(CCC)[Sn](CCCC)(CCCC)CCCC Chemical compound C(C)OC(CCC)[Sn](CCCC)(CCCC)CCCC NCRDKKMWYBVLFZ-UHFFFAOYSA-N 0.000 description 2
- LIPBJDWXQMXEEJ-UHFFFAOYSA-N C1CC2=C(C=C(C=C2)OP(=O)(C(F)F)O)C(=O)C1 Chemical compound C1CC2=C(C=C(C=C2)OP(=O)(C(F)F)O)C(=O)C1 LIPBJDWXQMXEEJ-UHFFFAOYSA-N 0.000 description 2
- BJRYIMQWIVHEHB-UHFFFAOYSA-N COP(OOC(C=CC(Br)=C1CC2)=C1C2=O)=O Chemical compound COP(OOC(C=CC(Br)=C1CC2)=C1C2=O)=O BJRYIMQWIVHEHB-UHFFFAOYSA-N 0.000 description 2
- GAWIXWVDTYZWAW-UHFFFAOYSA-N C[CH]O Chemical group C[CH]O GAWIXWVDTYZWAW-UHFFFAOYSA-N 0.000 description 2
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 2
- 229910021590 Copper(II) bromide Inorganic materials 0.000 description 2
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical compound C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 2
- LCGLNKUTAGEVQW-UHFFFAOYSA-N Dimethyl ether Chemical compound COC LCGLNKUTAGEVQW-UHFFFAOYSA-N 0.000 description 2
- KRHYYFGTRYWZRS-UHFFFAOYSA-N Fluorane Chemical compound F KRHYYFGTRYWZRS-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 108010018962 Glucosephosphate Dehydrogenase Proteins 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 2
- 229920001543 Laminarin Polymers 0.000 description 2
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 2
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 2
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 2
- JRNVZBWKYDBUCA-UHFFFAOYSA-N N-chlorosuccinimide Chemical compound ClN1C(=O)CCC1=O JRNVZBWKYDBUCA-UHFFFAOYSA-N 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- 229920001214 Polysorbate 60 Polymers 0.000 description 2
- 208000017442 Retinal disease Diseases 0.000 description 2
- 206010038923 Retinopathy Diseases 0.000 description 2
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 2
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- 229940100389 Sulfonylurea Drugs 0.000 description 2
- 244000299461 Theobroma cacao Species 0.000 description 2
- 235000005764 Theobroma cacao ssp. cacao Nutrition 0.000 description 2
- 235000005767 Theobroma cacao ssp. sphaerocarpum Nutrition 0.000 description 2
- HVPHRLJLJVQMFF-UHFFFAOYSA-N [8-[tert-butyl(dimethyl)silyl]oxy-5,6,7,8-tetrahydronaphthalen-2-yl]methanol Chemical compound CC(C)(C)[Si](C)(C)OC1CCCC2=C1C=C(C=C2)CO HVPHRLJLJVQMFF-UHFFFAOYSA-N 0.000 description 2
- XJLXINKUBYWONI-DQQFMEOOSA-N [[(2r,3r,4r,5r)-5-(6-aminopurin-9-yl)-3-hydroxy-4-phosphonooxyoxolan-2-yl]methoxy-hydroxyphosphoryl] [(2s,3r,4s,5s)-5-(3-carbamoylpyridin-1-ium-1-yl)-3,4-dihydroxyoxolan-2-yl]methyl phosphate Chemical compound NC(=O)C1=CC=C[N+]([C@@H]2[C@H]([C@@H](O)[C@H](COP([O-])(=O)OP(O)(=O)OC[C@@H]3[C@H]([C@@H](OP(O)(O)=O)[C@@H](O3)N3C4=NC=NC(N)=C4N=C3)O)O2)O)=C1 XJLXINKUBYWONI-DQQFMEOOSA-N 0.000 description 2
- 238000002835 absorbance Methods 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 230000004913 activation Effects 0.000 description 2
- 230000000996 additive effect Effects 0.000 description 2
- 239000003463 adsorbent Substances 0.000 description 2
- 125000003158 alcohol group Chemical group 0.000 description 2
- 150000001299 aldehydes Chemical class 0.000 description 2
- 229910000102 alkali metal hydride Inorganic materials 0.000 description 2
- 150000008046 alkali metal hydrides Chemical class 0.000 description 2
- 150000004703 alkoxides Chemical class 0.000 description 2
- 125000004448 alkyl carbonyl group Chemical group 0.000 description 2
- 125000005233 alkylalcohol group Chemical group 0.000 description 2
- XXROGKLTLUQVRX-UHFFFAOYSA-N allyl alcohol Chemical compound OCC=C XXROGKLTLUQVRX-UHFFFAOYSA-N 0.000 description 2
- 238000005576 amination reaction Methods 0.000 description 2
- 125000004202 aminomethyl group Chemical group [H]N([H])C([H])([H])* 0.000 description 2
- 229940127003 anti-diabetic drug Drugs 0.000 description 2
- 230000003110 anti-inflammatory effect Effects 0.000 description 2
- 239000003472 antidiabetic agent Substances 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- XTKDAFGWCDAMPY-UHFFFAOYSA-N azaperone Chemical compound C1=CC(F)=CC=C1C(=O)CCCN1CCN(C=2N=CC=CC=2)CC1 XTKDAFGWCDAMPY-UHFFFAOYSA-N 0.000 description 2
- 150000001540 azides Chemical class 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- RNBGYGVWRKECFJ-ARQDHWQXSA-N beta-D-fructofuranose 1,6-bisphosphate Chemical compound O[C@H]1[C@H](O)[C@@](O)(COP(O)(O)=O)O[C@@H]1COP(O)(O)=O RNBGYGVWRKECFJ-ARQDHWQXSA-N 0.000 description 2
- UWTDFICHZKXYAC-UHFFFAOYSA-N boron;oxolane Chemical compound [B].C1CCOC1 UWTDFICHZKXYAC-UHFFFAOYSA-N 0.000 description 2
- 125000001246 bromo group Chemical group Br* 0.000 description 2
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 2
- 235000014121 butter Nutrition 0.000 description 2
- 235000001046 cacaotero Nutrition 0.000 description 2
- 229910000019 calcium carbonate Inorganic materials 0.000 description 2
- OSGAYBCDTDRGGQ-UHFFFAOYSA-L calcium sulfate Chemical compound [Ca+2].[O-]S([O-])(=O)=O OSGAYBCDTDRGGQ-UHFFFAOYSA-L 0.000 description 2
- 125000002837 carbocyclic group Chemical group 0.000 description 2
- 125000006297 carbonyl amino group Chemical group [H]N([*:2])C([*:1])=O 0.000 description 2
- MVPPADPHJFYWMZ-UHFFFAOYSA-N chlorobenzene Chemical compound ClC1=CC=CC=C1 MVPPADPHJFYWMZ-UHFFFAOYSA-N 0.000 description 2
- 229910052802 copper Inorganic materials 0.000 description 2
- 239000010949 copper Substances 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- PWAPCRSSMCLZHG-UHFFFAOYSA-N cyclopentylidene Chemical group [C]1CCCC1 PWAPCRSSMCLZHG-UHFFFAOYSA-N 0.000 description 2
- SCIGVHCNNXTQDB-UHFFFAOYSA-N decyl dihydrogen phosphate Chemical compound CCCCCCCCCCOP(O)(O)=O SCIGVHCNNXTQDB-UHFFFAOYSA-N 0.000 description 2
- AVGAIPMGSIOHFZ-UHFFFAOYSA-N dichloromethane;2-propan-2-yloxypropane Chemical compound ClCCl.CC(C)OC(C)C AVGAIPMGSIOHFZ-UHFFFAOYSA-N 0.000 description 2
- 235000014113 dietary fatty acids Nutrition 0.000 description 2
- CSJLBAMHHLJAAS-UHFFFAOYSA-N diethylaminosulfur trifluoride Chemical compound CCN(CC)S(F)(F)F CSJLBAMHHLJAAS-UHFFFAOYSA-N 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 description 2
- JMQGGPRJQOQKRT-UHFFFAOYSA-N diphenyl hydrogen phosphate;azide Chemical compound [N-]=[N+]=[N-].C=1C=CC=CC=1OP(=O)(O)OC1=CC=CC=C1 JMQGGPRJQOQKRT-UHFFFAOYSA-N 0.000 description 2
- 239000007884 disintegrant Substances 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 238000006911 enzymatic reaction Methods 0.000 description 2
- UFZOPKFMKMAWLU-UHFFFAOYSA-N ethoxy(methyl)phosphinic acid Chemical compound CCOP(C)(O)=O UFZOPKFMKMAWLU-UHFFFAOYSA-N 0.000 description 2
- DEFVIWRASFVYLL-UHFFFAOYSA-N ethylene glycol bis(2-aminoethyl)tetraacetic acid Chemical compound OC(=O)CN(CC(O)=O)CCOCCOCCN(CC(O)=O)CC(O)=O DEFVIWRASFVYLL-UHFFFAOYSA-N 0.000 description 2
- 238000001704 evaporation Methods 0.000 description 2
- 239000000194 fatty acid Substances 0.000 description 2
- 229930195729 fatty acid Natural products 0.000 description 2
- 239000000796 flavoring agent Substances 0.000 description 2
- 239000012025 fluorinating agent Substances 0.000 description 2
- 229910052731 fluorine Inorganic materials 0.000 description 2
- 235000013355 food flavoring agent Nutrition 0.000 description 2
- 238000009472 formulation Methods 0.000 description 2
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 2
- 239000003205 fragrance Substances 0.000 description 2
- 235000011187 glycerol Nutrition 0.000 description 2
- 150000004795 grignard reagents Chemical class 0.000 description 2
- 230000026030 halogenation Effects 0.000 description 2
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine hydrate Chemical compound O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 2
- 150000004678 hydrides Chemical class 0.000 description 2
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 2
- 230000003914 insulin secretion Effects 0.000 description 2
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 2
- 125000004491 isohexyl group Chemical group C(CCC(C)C)* 0.000 description 2
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 2
- HJOVHMDZYOCNQW-UHFFFAOYSA-N isophorone Chemical compound CC1=CC(=O)CC(C)(C)C1 HJOVHMDZYOCNQW-UHFFFAOYSA-N 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 125000001786 isothiazolyl group Chemical group 0.000 description 2
- 125000000842 isoxazolyl group Chemical group 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- DBTMGCOVALSLOR-VPNXCSTESA-N laminarin Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)OC1O[C@@H]1[C@@H](O)C(O[C@H]2[C@@H]([C@@H](CO)OC(O)[C@@H]2O)O)O[C@H](CO)[C@H]1O DBTMGCOVALSLOR-VPNXCSTESA-N 0.000 description 2
- 229920005610 lignin Polymers 0.000 description 2
- 150000002632 lipids Chemical class 0.000 description 2
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 2
- 229910000032 lithium hydrogen carbonate Inorganic materials 0.000 description 2
- 210000004185 liver Anatomy 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 2
- 235000019341 magnesium sulphate Nutrition 0.000 description 2
- 230000010534 mechanism of action Effects 0.000 description 2
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 2
- CYEBJEDOHLIWNP-UHFFFAOYSA-N methanethioamide Chemical compound NC=S CYEBJEDOHLIWNP-UHFFFAOYSA-N 0.000 description 2
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 2
- CQDGTJPVBWZJAZ-UHFFFAOYSA-N monoethyl carbonate Chemical compound CCOC(O)=O CQDGTJPVBWZJAZ-UHFFFAOYSA-N 0.000 description 2
- 125000002757 morpholinyl group Chemical group 0.000 description 2
- 238000000465 moulding Methods 0.000 description 2
- 208000031225 myocardial ischemia Diseases 0.000 description 2
- YKYONYBAUNKHLG-UHFFFAOYSA-N n-Propyl acetate Natural products CCCOC(C)=O YKYONYBAUNKHLG-UHFFFAOYSA-N 0.000 description 2
- 229910052759 nickel Inorganic materials 0.000 description 2
- 125000004433 nitrogen atom Chemical group N* 0.000 description 2
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- 125000002971 oxazolyl group Chemical group 0.000 description 2
- KJIFKLIQANRMOU-UHFFFAOYSA-N oxidanium;4-methylbenzenesulfonate Chemical compound O.CC1=CC=C(S(O)(=O)=O)C=C1 KJIFKLIQANRMOU-UHFFFAOYSA-N 0.000 description 2
- 239000007800 oxidant agent Substances 0.000 description 2
- 150000002923 oximes Chemical class 0.000 description 2
- 229960002085 oxycodone Drugs 0.000 description 2
- 208000033808 peripheral neuropathy Diseases 0.000 description 2
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 2
- 239000006187 pill Substances 0.000 description 2
- 125000004193 piperazinyl group Chemical group 0.000 description 2
- 125000003386 piperidinyl group Chemical group 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 2
- NTTOTNSKUYCDAV-UHFFFAOYSA-N potassium hydride Chemical compound [KH] NTTOTNSKUYCDAV-UHFFFAOYSA-N 0.000 description 2
- 229910000105 potassium hydride Inorganic materials 0.000 description 2
- 229940086066 potassium hydrogencarbonate Drugs 0.000 description 2
- 239000012286 potassium permanganate Substances 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- 229940090181 propyl acetate Drugs 0.000 description 2
- UMLDUMMLRZFROX-UHFFFAOYSA-N pyridin-2-ylboronic acid Chemical compound OB(O)C1=CC=CC=N1 UMLDUMMLRZFROX-UHFFFAOYSA-N 0.000 description 2
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 2
- 125000001422 pyrrolinyl group Chemical group 0.000 description 2
- 125000000168 pyrrolyl group Chemical group 0.000 description 2
- 239000000741 silica gel Substances 0.000 description 2
- 229910002027 silica gel Inorganic materials 0.000 description 2
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- HJUGFYREWKUQJT-UHFFFAOYSA-N tetrabromomethane Chemical compound BrC(Br)(Br)Br HJUGFYREWKUQJT-UHFFFAOYSA-N 0.000 description 2
- 125000003831 tetrazolyl group Chemical group 0.000 description 2
- 125000001113 thiadiazolyl group Chemical group 0.000 description 2
- 125000000335 thiazolyl group Chemical group 0.000 description 2
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 230000009466 transformation Effects 0.000 description 2
- 229910052723 transition metal Inorganic materials 0.000 description 2
- 150000003624 transition metals Chemical class 0.000 description 2
- 125000001425 triazolyl group Chemical group 0.000 description 2
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 2
- YFTHZRPMJXBUME-UHFFFAOYSA-N tripropylamine Chemical compound CCCN(CCC)CCC YFTHZRPMJXBUME-UHFFFAOYSA-N 0.000 description 2
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical compound CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 2
- 125000003774 valeryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- GTLDTDOJJJZVBW-UHFFFAOYSA-N zinc cyanide Chemical compound [Zn+2].N#[C-].N#[C-] GTLDTDOJJJZVBW-UHFFFAOYSA-N 0.000 description 2
- HHXBZEIOUIMZET-UHFFFAOYSA-N (2,3-dichlorophenyl)-phenylphosphane Chemical compound ClC1=CC=CC(PC=2C=CC=CC=2)=C1Cl HHXBZEIOUIMZET-UHFFFAOYSA-N 0.000 description 1
- QCSLIRFWJPOENV-UHFFFAOYSA-N (2-fluorophenyl)boronic acid Chemical compound OB(O)C1=CC=CC=C1F QCSLIRFWJPOENV-UHFFFAOYSA-N 0.000 description 1
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- MLIWQXBKMZNZNF-KUHOPJCQSA-N (2e)-2,6-bis[(4-azidophenyl)methylidene]-4-methylcyclohexan-1-one Chemical compound O=C1\C(=C\C=2C=CC(=CC=2)N=[N+]=[N-])CC(C)CC1=CC1=CC=C(N=[N+]=[N-])C=C1 MLIWQXBKMZNZNF-KUHOPJCQSA-N 0.000 description 1
- FIAINKIUSZGVGX-HSHFZTNMSA-N (2r)-2-aminopropanamide;hydrochloride Chemical compound Cl.C[C@@H](N)C(N)=O FIAINKIUSZGVGX-HSHFZTNMSA-N 0.000 description 1
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 1
- YPPPOKFOFWBRLT-JEDNCBNOSA-N (2s)-2,6-diaminohexanoic acid;pyridine Chemical compound C1=CC=NC=C1.NCCCC[C@H](N)C(O)=O YPPPOKFOFWBRLT-JEDNCBNOSA-N 0.000 description 1
- VURWDDZIWBGXCK-DKWTVANSSA-N (2s)-2-amino-3-hydroxypropanamide;hydrochloride Chemical group Cl.OC[C@H](N)C(N)=O VURWDDZIWBGXCK-DKWTVANSSA-N 0.000 description 1
- KNXQDJCZSVHEIW-UHFFFAOYSA-N (3-fluorophenyl)boronic acid Chemical compound OB(O)C1=CC=CC(F)=C1 KNXQDJCZSVHEIW-UHFFFAOYSA-N 0.000 description 1
- NLLGFYPSWCMUIV-UHFFFAOYSA-N (3-methoxyphenyl)boronic acid Chemical compound COC1=CC=CC(B(O)O)=C1 NLLGFYPSWCMUIV-UHFFFAOYSA-N 0.000 description 1
- COIQUVGFTILYGA-UHFFFAOYSA-N (4-hydroxyphenyl)boronic acid Chemical compound OB(O)C1=CC=C(O)C=C1 COIQUVGFTILYGA-UHFFFAOYSA-N 0.000 description 1
- VOAAEKKFGLPLLU-UHFFFAOYSA-N (4-methoxyphenyl)boronic acid Chemical compound COC1=CC=C(B(O)O)C=C1 VOAAEKKFGLPLLU-UHFFFAOYSA-N 0.000 description 1
- QGKMIGUHVLGJBR-UHFFFAOYSA-M (4z)-1-(3-methylbutyl)-4-[[1-(3-methylbutyl)quinolin-1-ium-4-yl]methylidene]quinoline;iodide Chemical class [I-].C12=CC=CC=C2N(CCC(C)C)C=CC1=CC1=CC=[N+](CCC(C)C)C2=CC=CC=C12 QGKMIGUHVLGJBR-UHFFFAOYSA-M 0.000 description 1
- DBASWUOOEDIDQL-UHFFFAOYSA-N (5-propan-2-yl-4H-indeno[1,2-d][1,3]thiazol-8-yl)oxymethylphosphonic acid Chemical compound CC(C)C1=C2CC3=C(C2=C(C=C1)OCP(=O)(O)O)N=CS3 DBASWUOOEDIDQL-UHFFFAOYSA-N 0.000 description 1
- 125000004844 (C1-C6) alkoxyimino group Chemical group 0.000 description 1
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 1
- ONRNRVLJHFFBJG-UHFFFAOYSA-N 1,2-di(imidazol-1-yl)ethane-1,2-dione Chemical compound C1=CN=CN1C(=O)C(=O)N1C=CN=C1 ONRNRVLJHFFBJG-UHFFFAOYSA-N 0.000 description 1
- CYSGHNMQYZDMIA-UHFFFAOYSA-N 1,3-Dimethyl-2-imidazolidinon Chemical compound CN1CCN(C)C1=O CYSGHNMQYZDMIA-UHFFFAOYSA-N 0.000 description 1
- LDVVTQMJQSCDMK-UHFFFAOYSA-N 1,3-dihydroxypropan-2-yl formate Chemical compound OCC(CO)OC=O LDVVTQMJQSCDMK-UHFFFAOYSA-N 0.000 description 1
- OGFAWKRXZLGJSK-UHFFFAOYSA-N 1-(2,4-dihydroxyphenyl)-2-(4-nitrophenyl)ethanone Chemical compound OC1=CC(O)=CC=C1C(=O)CC1=CC=C([N+]([O-])=O)C=C1 OGFAWKRXZLGJSK-UHFFFAOYSA-N 0.000 description 1
- VVFGLBKYBBUTRO-UHFFFAOYSA-N 1-[difluoromethyl(ethoxy)phosphoryl]oxyethane Chemical compound CCOP(=O)(C(F)F)OCC VVFGLBKYBBUTRO-UHFFFAOYSA-N 0.000 description 1
- ZQXCQTAELHSNAT-UHFFFAOYSA-N 1-chloro-3-nitro-5-(trifluoromethyl)benzene Chemical compound [O-][N+](=O)C1=CC(Cl)=CC(C(F)(F)F)=C1 ZQXCQTAELHSNAT-UHFFFAOYSA-N 0.000 description 1
- ZTIJJEYYNVBJBB-UHFFFAOYSA-N 1-chloropyrrolidine-2,5-dione;methylsulfinylmethane Chemical compound CS(C)=O.ClN1C(=O)CCC1=O ZTIJJEYYNVBJBB-UHFFFAOYSA-N 0.000 description 1
- ZBEQYOACPCLOIV-UHFFFAOYSA-N 1-chloropyrrolidine-2,5-dione;triphenylphosphane Chemical compound ClN1C(=O)CCC1=O.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 ZBEQYOACPCLOIV-UHFFFAOYSA-N 0.000 description 1
- 125000004899 1-ethylpropylamino group Chemical group C(C)C(CC)N* 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- UCNGGGYMLHAMJG-UHFFFAOYSA-N 1-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazole Chemical compound C1=NN(C)C=C1B1OC(C)(C)C(C)(C)O1 UCNGGGYMLHAMJG-UHFFFAOYSA-N 0.000 description 1
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 1
- 125000004214 1-pyrrolidinyl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- ZLTWIJREHQCJJL-UHFFFAOYSA-N 1-trimethylsilylethanol Chemical compound CC(O)[Si](C)(C)C ZLTWIJREHQCJJL-UHFFFAOYSA-N 0.000 description 1
- KPWDGTGXUYRARH-UHFFFAOYSA-N 2,2,2-trichloroethanol Chemical compound OCC(Cl)(Cl)Cl KPWDGTGXUYRARH-UHFFFAOYSA-N 0.000 description 1
- UTQNKKSJPHTPBS-UHFFFAOYSA-N 2,2,2-trichloroethanone Chemical group ClC(Cl)(Cl)[C]=O UTQNKKSJPHTPBS-UHFFFAOYSA-N 0.000 description 1
- 125000004206 2,2,2-trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 1
- QQGRFMIMXPWKPM-UHFFFAOYSA-N 2,3,4-tributylphenol Chemical compound CCCCC1=CC=C(O)C(CCCC)=C1CCCC QQGRFMIMXPWKPM-UHFFFAOYSA-N 0.000 description 1
- ZFFMLCVRJBZUDZ-UHFFFAOYSA-N 2,3-dimethylbutane Chemical group CC(C)C(C)C ZFFMLCVRJBZUDZ-UHFFFAOYSA-N 0.000 description 1
- IJZGBGLFLOKQIU-UHFFFAOYSA-N 2-(2H-benzotriazol-4-yloxy)-2-oxoacetic acid Chemical compound C1=CC2=NNN=C2C(=C1)OC(=O)C(=O)O IJZGBGLFLOKQIU-UHFFFAOYSA-N 0.000 description 1
- VLROJECCXBBKPZ-UHFFFAOYSA-N 2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenol Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC=CC=C1O VLROJECCXBBKPZ-UHFFFAOYSA-N 0.000 description 1
- AFABGHUZZDYHJO-UHFFFAOYSA-N 2-Methylpentane Chemical compound CCCC(C)C AFABGHUZZDYHJO-UHFFFAOYSA-N 0.000 description 1
- QQZOPKMRPOGIEB-UHFFFAOYSA-N 2-Oxohexane Chemical compound CCCCC(C)=O QQZOPKMRPOGIEB-UHFFFAOYSA-N 0.000 description 1
- MWPNPNLDBXDQCC-UHFFFAOYSA-N 2-amino-3-(diethylamino)propanoic acid Chemical compound CCN(CC)CC(N)C(O)=O MWPNPNLDBXDQCC-UHFFFAOYSA-N 0.000 description 1
- WKNMKGVLOWGGOU-UHFFFAOYSA-N 2-aminoacetamide;hydron;chloride Chemical compound Cl.NCC(N)=O WKNMKGVLOWGGOU-UHFFFAOYSA-N 0.000 description 1
- KJJPLEZQSCZCKE-UHFFFAOYSA-N 2-aminopropane-1,3-diol Chemical compound OCC(N)CO KJJPLEZQSCZCKE-UHFFFAOYSA-N 0.000 description 1
- 125000006176 2-ethylbutyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(C([H])([H])*)C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004913 2-ethylbutylamino group Chemical group C(C)C(CN*)CC 0.000 description 1
- QQMWIWDZVJNDAQ-UHFFFAOYSA-N 2-hydroxy-4-methyl-2,3-dihydroinden-1-one Chemical compound OC1C(C2=CC=CC(=C2C1)C)=O QQMWIWDZVJNDAQ-UHFFFAOYSA-N 0.000 description 1
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 description 1
- 125000004493 2-methylbut-1-yl group Chemical group CC(C*)CC 0.000 description 1
- VJROPLWGFCORRM-UHFFFAOYSA-N 2-methylbutan-1-amine Chemical compound CCC(C)CN VJROPLWGFCORRM-UHFFFAOYSA-N 0.000 description 1
- 125000004896 2-methylbutylamino group Chemical group CC(CN*)CC 0.000 description 1
- 125000003229 2-methylhexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- WNDXRJBYZOSNQO-UHFFFAOYSA-N 2-methylpentan-1-amine Chemical compound CCCC(C)CN WNDXRJBYZOSNQO-UHFFFAOYSA-N 0.000 description 1
- 125000005916 2-methylpentyl group Chemical group 0.000 description 1
- BSKHPKMHTQYZBB-UHFFFAOYSA-N 2-methylpyridine Chemical compound CC1=CC=CC=N1 BSKHPKMHTQYZBB-UHFFFAOYSA-N 0.000 description 1
- MNTLSHJBDKQYOF-UHFFFAOYSA-N 2-oxo-2-[[6-(trifluoromethyl)-2H-benzotriazol-4-yl]oxy]acetic acid Chemical class C1=C(C=C(C2=NNN=C21)OC(=O)C(=O)O)C(F)(F)F MNTLSHJBDKQYOF-UHFFFAOYSA-N 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- KKHIFLYUSFQMDJ-UHFFFAOYSA-N 3,4-dihydronaphthalen-1-ol Chemical compound C1=CC=C2C(O)=CCCC2=C1 KKHIFLYUSFQMDJ-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- 125000004208 3-hydroxyphenyl group Chemical group [H]OC1=C([H])C([H])=C([H])C(*)=C1[H] 0.000 description 1
- FAXDZWQIWUSWJH-UHFFFAOYSA-N 3-methoxypropan-1-amine Chemical compound COCCCN FAXDZWQIWUSWJH-UHFFFAOYSA-N 0.000 description 1
- 125000003469 3-methylhexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000005917 3-methylpentyl group Chemical group 0.000 description 1
- 125000004904 3-methylpentylamino group Chemical group CC(CCN*)CC 0.000 description 1
- NHDVZOXPHYFANX-UHFFFAOYSA-N 4,4-dimethyl-2,3-dihydronaphthalen-1-one Chemical compound C1=CC=C2C(C)(C)CCC(=O)C2=C1 NHDVZOXPHYFANX-UHFFFAOYSA-N 0.000 description 1
- YMIIQKHQXYRXGP-UHFFFAOYSA-N 4,5-dimethyl-2,3-dihydroinden-1-one Chemical compound CC1=CC=C2C(=O)CCC2=C1C YMIIQKHQXYRXGP-UHFFFAOYSA-N 0.000 description 1
- ZMEOTPGFFBPSCJ-UHFFFAOYSA-N 4,6-difluoro-7-hydroxy-2,3-dihydroinden-1-one Chemical compound C1CC(=O)C2=C1C(=CC(=C2O)F)F ZMEOTPGFFBPSCJ-UHFFFAOYSA-N 0.000 description 1
- UZFMOKQJFYMBGY-UHFFFAOYSA-N 4-hydroxy-TEMPO Chemical compound CC1(C)CC(O)CC(C)(C)N1[O] UZFMOKQJFYMBGY-UHFFFAOYSA-N 0.000 description 1
- 125000004172 4-methoxyphenyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C([H])C([H])=C1* 0.000 description 1
- QVIAMKXOQGCYCV-UHFFFAOYSA-N 4-methylpentan-1-amine Chemical compound CC(C)CCCN QVIAMKXOQGCYCV-UHFFFAOYSA-N 0.000 description 1
- 125000000439 4-methylpentoxy group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])C([H])([H])O* 0.000 description 1
- QOPRWBRNMPANKN-UHFFFAOYSA-N 5-methoxy-2,3-dihydroinden-1-one Chemical compound COC1=CC=C2C(=O)CCC2=C1 QOPRWBRNMPANKN-UHFFFAOYSA-N 0.000 description 1
- KWHUHTFXMNQHAA-UHFFFAOYSA-N 6,7,8,9-tetrahydrobenzo[7]annulen-5-one Chemical compound O=C1CCCCC2=CC=CC=C12 KWHUHTFXMNQHAA-UHFFFAOYSA-N 0.000 description 1
- GZADZXCHFHIRHN-UHFFFAOYSA-N 6,7-difluoro-2,3-dihydro-1H-inden-4-ol Chemical compound FC1=CC(O)=C2C(=C1F)CCC2 GZADZXCHFHIRHN-UHFFFAOYSA-N 0.000 description 1
- RMRKDYNVZWKAFP-UHFFFAOYSA-N 6-methoxy-3,4-dihydro-1h-naphthalen-2-one Chemical compound C1C(=O)CCC2=CC(OC)=CC=C21 RMRKDYNVZWKAFP-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- KLSJWNVTNUYHDU-UHFFFAOYSA-N Amitrole Chemical compound NC1=NC=NN1 KLSJWNVTNUYHDU-UHFFFAOYSA-N 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- CYOIVPQGTFLYFH-UHFFFAOYSA-N CC(C(Br)=C1CC2)=CC(OOP(OC)=O)=C1C2=O Chemical compound CC(C(Br)=C1CC2)=CC(OOP(OC)=O)=C1C2=O CYOIVPQGTFLYFH-UHFFFAOYSA-N 0.000 description 1
- IWWLOHRMSKEVDD-UHFFFAOYSA-N CC(C=C1)=C(CCC2=O)C2=C1OOP(OC)=O Chemical compound CC(C=C1)=C(CCC2=O)C2=C1OOP(OC)=O IWWLOHRMSKEVDD-UHFFFAOYSA-N 0.000 description 1
- PFSCWQJTCFYTKW-UHFFFAOYSA-N CC=C=C=C(C)C=C Chemical group CC=C=C=C(C)C=C PFSCWQJTCFYTKW-UHFFFAOYSA-N 0.000 description 1
- DXOOTIWGEWJMPO-UHFFFAOYSA-N CN(C1=CC=NC=C1)C.CC1=NC=CC=C1 Chemical compound CN(C1=CC=NC=C1)C.CC1=NC=CC=C1 DXOOTIWGEWJMPO-UHFFFAOYSA-N 0.000 description 1
- KEIPQLQSLOTHIR-UHFFFAOYSA-N COP(OOC(C=CC(F)=C1CC2)=C1C2=O)=O Chemical compound COP(OOC(C=CC(F)=C1CC2)=C1C2=O)=O KEIPQLQSLOTHIR-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 208000019736 Cranial nerve disease Diseases 0.000 description 1
- HTJDQJBWANPRPF-UHFFFAOYSA-N Cyclopropylamine Chemical compound NC1CC1 HTJDQJBWANPRPF-UHFFFAOYSA-N 0.000 description 1
- GSXOAOHZAIYLCY-UHFFFAOYSA-N D-F6P Natural products OCC(=O)C(O)C(O)C(O)COP(O)(O)=O GSXOAOHZAIYLCY-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 description 1
- 239000004129 EU approved improving agent Substances 0.000 description 1
- 241000588724 Escherichia coli Species 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical group C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- 239000005977 Ethylene Chemical group 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- DCXXMTOCNZCJGO-UHFFFAOYSA-N Glycerol trioctadecanoate Natural products CCCCCCCCCCCCCCCCCC(=O)OCC(OC(=O)CCCCCCCCCCCCCCCCC)COC(=O)CCCCCCCCCCCCCCCCC DCXXMTOCNZCJGO-UHFFFAOYSA-N 0.000 description 1
- 229920002527 Glycogen Polymers 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 101001028852 Homo sapiens Fructose-1,6-bisphosphatase 1 Proteins 0.000 description 1
- 101000635799 Homo sapiens Run domain Beclin-1-interacting and cysteine-rich domain-containing protein Proteins 0.000 description 1
- 229920002153 Hydroxypropyl cellulose Polymers 0.000 description 1
- 208000013016 Hypoglycemia Diseases 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- 239000002841 Lewis acid Substances 0.000 description 1
- NTIZESTWPVYFNL-UHFFFAOYSA-N Methyl isobutyl ketone Chemical compound CC(C)CC(C)=O NTIZESTWPVYFNL-UHFFFAOYSA-N 0.000 description 1
- UIHCLUNTQKBZGK-UHFFFAOYSA-N Methyl isobutyl ketone Natural products CCC(C)C(C)=O UIHCLUNTQKBZGK-UHFFFAOYSA-N 0.000 description 1
- 238000005654 Michaelis-Arbuzov synthesis reaction Methods 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- PHSPJQZRQAJPPF-UHFFFAOYSA-N N-alpha-Methylhistamine Chemical compound CNCCC1=CN=CN1 PHSPJQZRQAJPPF-UHFFFAOYSA-N 0.000 description 1
- LFTLOKWAGJYHHR-UHFFFAOYSA-N N-methylmorpholine N-oxide Chemical compound CN1(=O)CCOCC1 LFTLOKWAGJYHHR-UHFFFAOYSA-N 0.000 description 1
- UQFQONCQIQEYPJ-UHFFFAOYSA-N N-methylpyrazole Chemical compound CN1C=CC=N1 UQFQONCQIQEYPJ-UHFFFAOYSA-N 0.000 description 1
- 229910021586 Nickel(II) chloride Inorganic materials 0.000 description 1
- UHJFMAJYDZKDLJ-UHFFFAOYSA-N OBO.OC1=CC=CC=C1 Chemical compound OBO.OC1=CC=CC=C1 UHJFMAJYDZKDLJ-UHFFFAOYSA-N 0.000 description 1
- BNXLOMAJKIKRIO-UHFFFAOYSA-N OP(OC(F)F)=O Chemical compound OP(OC(F)F)=O BNXLOMAJKIKRIO-UHFFFAOYSA-N 0.000 description 1
- ZNEBRQOBFGOSLN-UHFFFAOYSA-N PCCC.C1(=CC=CC=C1)O.C1(=CC=CC=C1)O Chemical compound PCCC.C1(=CC=CC=C1)O.C1(=CC=CC=C1)O ZNEBRQOBFGOSLN-UHFFFAOYSA-N 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- GHUUBYQTCDQWRA-UHFFFAOYSA-N Pioglitazone hydrochloride Chemical compound Cl.N1=CC(CC)=CC=C1CCOC(C=C1)=CC=C1CC1C(=O)NC(=O)S1 GHUUBYQTCDQWRA-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- UEJYSALTSUZXFV-SRVKXCTJSA-N Rigin Chemical compound NCC(=O)N[C@@H](CCC(N)=O)C(=O)N1CCC[C@H]1C(=O)N[C@@H](CCCN=C(N)N)C(O)=O UEJYSALTSUZXFV-SRVKXCTJSA-N 0.000 description 1
- 102100030852 Run domain Beclin-1-interacting and cysteine-rich domain-containing protein Human genes 0.000 description 1
- BUGBHKTXTAQXES-UHFFFAOYSA-N Selenium Chemical compound [Se] BUGBHKTXTAQXES-UHFFFAOYSA-N 0.000 description 1
- GIIZNNXWQWCKIB-UHFFFAOYSA-N Serevent Chemical compound C1=C(O)C(CO)=CC(C(O)CNCCCCCCOCCCCC=2C=CC=CC=2)=C1 GIIZNNXWQWCKIB-UHFFFAOYSA-N 0.000 description 1
- 229920001800 Shellac Polymers 0.000 description 1
- 206010058339 Splenitis Diseases 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- FZNCGRZWXLXZSZ-CIQUZCHMSA-N Voglibose Chemical compound OCC(CO)N[C@H]1C[C@](O)(CO)[C@@H](O)[C@H](O)[C@H]1O FZNCGRZWXLXZSZ-CIQUZCHMSA-N 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- FIAINKIUSZGVGX-DKWTVANSSA-N [(2s)-1-amino-1-oxopropan-2-yl]azanium;chloride Chemical compound Cl.C[C@H](N)C(N)=O FIAINKIUSZGVGX-DKWTVANSSA-N 0.000 description 1
- JNSBEPKGFVENFS-UHFFFAOYSA-N [2-(trifluoromethyl)phenyl]boronic acid Chemical compound OB(O)C1=CC=CC=C1C(F)(F)F JNSBEPKGFVENFS-UHFFFAOYSA-N 0.000 description 1
- MKLAUAFKHQGDKZ-UHFFFAOYSA-N [5-(2-acetamidoethylcarbamoyl)-4H-indeno[1,2-d][1,3]thiazol-8-yl]oxymethylphosphonic acid Chemical compound CC(=O)NCCNC(=O)C1=C2CC3=C(C2=C(C=C1)OCP(=O)(O)O)N=CS3 MKLAUAFKHQGDKZ-UHFFFAOYSA-N 0.000 description 1
- LPCLHYYCZIHUSA-UHFFFAOYSA-N [5-(2-methoxyethylcarbamoyl)-4H-indeno[1,2-d][1,3]thiazol-8-yl]oxymethylphosphonic acid Chemical compound COCCNC(=O)C1=C2CC3=C(C2=C(C=C1)OCP(=O)(O)O)N=CS3 LPCLHYYCZIHUSA-UHFFFAOYSA-N 0.000 description 1
- AJJRNCOUKUBFRS-UHFFFAOYSA-N [7-(bromomethyl)-1,2,3,4-tetrahydronaphthalen-1-yl]oxy-tert-butyl-dimethylsilane Chemical compound CC(C)(C)[Si](C)(C)OC1CCCC2=C1C=C(C=C2)CBr AJJRNCOUKUBFRS-UHFFFAOYSA-N 0.000 description 1
- KKLWSPPIRBIEOV-UHFFFAOYSA-N [n'-(n'-butylcarbamimidoyl)carbamimidoyl]azanium;chloride Chemical compound Cl.CCCCN=C(N)N=C(N)N KKLWSPPIRBIEOV-UHFFFAOYSA-N 0.000 description 1
- WLLIXJBWWFGEHT-UHFFFAOYSA-N [tert-butyl(dimethyl)silyl] trifluoromethanesulfonate Chemical compound CC(C)(C)[Si](C)(C)OS(=O)(=O)C(F)(F)F WLLIXJBWWFGEHT-UHFFFAOYSA-N 0.000 description 1
- 230000005856 abnormality Effects 0.000 description 1
- 229940124532 absorption promoter Drugs 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- CDXSJGDDABYYJV-UHFFFAOYSA-N acetic acid;ethanol Chemical compound CCO.CC(O)=O CDXSJGDDABYYJV-UHFFFAOYSA-N 0.000 description 1
- LXNAVEXFUKBNMK-UHFFFAOYSA-N acetic acid;palladium Chemical compound [Pd].CC(O)=O.CC(O)=O LXNAVEXFUKBNMK-UHFFFAOYSA-N 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 125000002015 acyclic group Chemical group 0.000 description 1
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 235000010419 agar Nutrition 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 229910052936 alkali metal sulfate Inorganic materials 0.000 description 1
- 125000004183 alkoxy alkyl group Chemical group 0.000 description 1
- 125000004666 alkoxyiminoalkyl group Chemical group 0.000 description 1
- 150000001350 alkyl halides Chemical class 0.000 description 1
- 150000004791 alkyl magnesium halides Chemical class 0.000 description 1
- 150000001343 alkyl silanes Chemical class 0.000 description 1
- 238000005804 alkylation reaction Methods 0.000 description 1
- 150000004808 allyl alcohols Chemical class 0.000 description 1
- BHELZAPQIKSEDF-UHFFFAOYSA-N allyl bromide Chemical compound BrCC=C BHELZAPQIKSEDF-UHFFFAOYSA-N 0.000 description 1
- 125000000746 allylic group Chemical class 0.000 description 1
- AZDRQVAHHNSJOQ-UHFFFAOYSA-N alumane Chemical class [AlH3] AZDRQVAHHNSJOQ-UHFFFAOYSA-N 0.000 description 1
- 230000001548 androgenic effect Effects 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 1
- 125000002029 aromatic hydrocarbon group Chemical group 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 125000002785 azepinyl group Chemical group 0.000 description 1
- RQPZNWPYLFFXCP-UHFFFAOYSA-L barium dihydroxide Chemical compound [OH-].[OH-].[Ba+2] RQPZNWPYLFFXCP-UHFFFAOYSA-L 0.000 description 1
- 229910001863 barium hydroxide Inorganic materials 0.000 description 1
- 235000013405 beer Nutrition 0.000 description 1
- 239000000440 bentonite Substances 0.000 description 1
- 229910000278 bentonite Inorganic materials 0.000 description 1
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 235000019445 benzyl alcohol Nutrition 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 125000000051 benzyloxy group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])O* 0.000 description 1
- BGWGXPAPYGQALX-ARQDHWQXSA-N beta-D-fructofuranose 6-phosphate Chemical compound OC[C@@]1(O)O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O BGWGXPAPYGQALX-ARQDHWQXSA-N 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- OMAHFYGHUQSIEF-UHFFFAOYSA-N bis(2,5-dioxopyrrolidin-1-yl) oxalate Chemical compound O=C1CCC(=O)N1OC(=O)C(=O)ON1C(=O)CCC1=O OMAHFYGHUQSIEF-UHFFFAOYSA-N 0.000 description 1
- UXXXZMDJQLPQPH-UHFFFAOYSA-N bis(2-methylpropyl) carbonate Chemical compound CC(C)COC(=O)OCC(C)C UXXXZMDJQLPQPH-UHFFFAOYSA-N 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- KGBXLFKZBHKPEV-UHFFFAOYSA-N boric acid Chemical compound OB(O)O KGBXLFKZBHKPEV-UHFFFAOYSA-N 0.000 description 1
- 239000004327 boric acid Substances 0.000 description 1
- OPPWASLOVKWHCT-UHFFFAOYSA-N boric acid;phenol Chemical compound OB(O)O.OC1=CC=CC=C1 OPPWASLOVKWHCT-UHFFFAOYSA-N 0.000 description 1
- MCQRPQCQMGVWIQ-UHFFFAOYSA-N boron;methylsulfanylmethane Chemical compound [B].CSC MCQRPQCQMGVWIQ-UHFFFAOYSA-N 0.000 description 1
- 230000031709 bromination Effects 0.000 description 1
- 238000005893 bromination reaction Methods 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- IYYIVELXUANFED-UHFFFAOYSA-N bromo(trimethyl)silane Chemical compound C[Si](C)(C)Br IYYIVELXUANFED-UHFFFAOYSA-N 0.000 description 1
- 229960004111 buformin Drugs 0.000 description 1
- 125000004063 butyryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- VSGNNIFQASZAOI-UHFFFAOYSA-L calcium acetate Chemical compound [Ca+2].CC([O-])=O.CC([O-])=O VSGNNIFQASZAOI-UHFFFAOYSA-L 0.000 description 1
- 239000001639 calcium acetate Substances 0.000 description 1
- 235000011092 calcium acetate Nutrition 0.000 description 1
- 229960005147 calcium acetate Drugs 0.000 description 1
- 239000001110 calcium chloride Substances 0.000 description 1
- 229910001628 calcium chloride Inorganic materials 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 125000003739 carbamimidoyl group Chemical group C(N)(=N)* 0.000 description 1
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 1
- JJNHBFYGCSOONU-UHFFFAOYSA-M carbanide;cyclopenta-1,3-diene;dimethylaluminum;titanium(4+);chloride Chemical compound [CH3-].[Ti+3]Cl.C[Al]C.C=1C=C[CH-]C=1.C=1C=C[CH-]C=1 JJNHBFYGCSOONU-UHFFFAOYSA-M 0.000 description 1
- 235000014633 carbohydrates Nutrition 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical compound OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 230000006315 carbonylation Effects 0.000 description 1
- 238000005810 carbonylation reaction Methods 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 150000001733 carboxylic acid esters Chemical class 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 238000006555 catalytic reaction Methods 0.000 description 1
- 235000010980 cellulose Nutrition 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- KXZJHVJKXJLBKO-UHFFFAOYSA-N chembl1408157 Chemical compound N=1C2=CC=CC=C2C(C(=O)O)=CC=1C1=CC=C(O)C=C1 KXZJHVJKXJLBKO-UHFFFAOYSA-N 0.000 description 1
- 238000005660 chlorination reaction Methods 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 150000001805 chlorine compounds Chemical class 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 125000002668 chloroacetyl group Chemical group ClCC(=O)* 0.000 description 1
- QSHQKIURKJITMZ-BRPMRXRMSA-N cholane Chemical compound C1CC2CCCC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCC)[C@@]1(C)CC2 QSHQKIURKJITMZ-BRPMRXRMSA-N 0.000 description 1
- ZCDOYSPFYFSLEW-UHFFFAOYSA-N chromate(2-) Chemical compound [O-][Cr]([O-])(=O)=O ZCDOYSPFYFSLEW-UHFFFAOYSA-N 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 229910001179 chromel Inorganic materials 0.000 description 1
- 229910052804 chromium Inorganic materials 0.000 description 1
- 239000011651 chromium Substances 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- DOBRDRYODQBAMW-UHFFFAOYSA-N copper(i) cyanide Chemical compound [Cu+].N#[C-] DOBRDRYODQBAMW-UHFFFAOYSA-N 0.000 description 1
- GBRBMTNGQBKBQE-UHFFFAOYSA-L copper;diiodide Chemical compound I[Cu]I GBRBMTNGQBKBQE-UHFFFAOYSA-L 0.000 description 1
- 239000000287 crude extract Substances 0.000 description 1
- 238000012258 culturing Methods 0.000 description 1
- VXVVUHQULXCUPF-UHFFFAOYSA-N cycloheptanamine Chemical compound NC1CCCCCC1 VXVVUHQULXCUPF-UHFFFAOYSA-N 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- UKJLNMAFNRKWGR-UHFFFAOYSA-N cyclohexatrienamine Chemical group NC1=CC=C=C[CH]1 UKJLNMAFNRKWGR-UHFFFAOYSA-N 0.000 description 1
- PAFZNILMFXTMIY-UHFFFAOYSA-N cyclohexylamine Chemical compound NC1CCCCC1 PAFZNILMFXTMIY-UHFFFAOYSA-N 0.000 description 1
- NISGSNTVMOOSJQ-UHFFFAOYSA-N cyclopentanamine Chemical compound NC1CCCC1 NISGSNTVMOOSJQ-UHFFFAOYSA-N 0.000 description 1
- 125000006312 cyclopentyl amino group Chemical group [H]N(*)C1([H])C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000002704 decyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 230000001934 delay Effects 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- NKLCNNUWBJBICK-UHFFFAOYSA-N dess–martin periodinane Chemical compound C1=CC=C2I(OC(=O)C)(OC(C)=O)(OC(C)=O)OC(=O)C2=C1 NKLCNNUWBJBICK-UHFFFAOYSA-N 0.000 description 1
- WDPNDMPWBDGXDB-UHFFFAOYSA-N dichloro-hydroxy-imino-$l^{5}-phosphane Chemical compound NP(Cl)(Cl)=O WDPNDMPWBDGXDB-UHFFFAOYSA-N 0.000 description 1
- 125000004772 dichloromethyl group Chemical group [H]C(Cl)(Cl)* 0.000 description 1
- YNHIGQDRGKUECZ-UHFFFAOYSA-N dichloropalladium;triphenylphosphanium Chemical compound Cl[Pd]Cl.C1=CC=CC=C1[PH+](C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1[PH+](C=1C=CC=CC=1)C1=CC=CC=C1 YNHIGQDRGKUECZ-UHFFFAOYSA-N 0.000 description 1
- 230000029087 digestion Effects 0.000 description 1
- 150000004683 dihydrates Chemical class 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- BADXJIPKFRBFOT-UHFFFAOYSA-N dimedone Chemical compound CC1(C)CC(=O)CC(=O)C1 BADXJIPKFRBFOT-UHFFFAOYSA-N 0.000 description 1
- VAWRKITUPUFMHV-UHFFFAOYSA-N dimethoxyborane Chemical compound COBOC VAWRKITUPUFMHV-UHFFFAOYSA-N 0.000 description 1
- FPAFDBFIGPHWGO-UHFFFAOYSA-N dioxosilane;oxomagnesium;hydrate Chemical compound O.[Mg]=O.[Mg]=O.[Mg]=O.O=[Si]=O.O=[Si]=O.O=[Si]=O.O=[Si]=O FPAFDBFIGPHWGO-UHFFFAOYSA-N 0.000 description 1
- MKRTXPORKIRPDG-UHFFFAOYSA-N diphenylphosphoryl azide Chemical compound C=1C=CC=CC=1P(=O)(N=[N+]=[N-])C1=CC=CC=C1 MKRTXPORKIRPDG-UHFFFAOYSA-N 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 239000008298 dragée Substances 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- 239000002662 enteric coated tablet Substances 0.000 description 1
- XWBDWHCCBGMXKG-UHFFFAOYSA-N ethanamine;hydron;chloride Chemical compound Cl.CCN XWBDWHCCBGMXKG-UHFFFAOYSA-N 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- 150000002168 ethanoic acid esters Chemical class 0.000 description 1
- 125000005448 ethoxyethyl group Chemical group [H]C([H])([H])C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 1
- 125000005745 ethoxymethyl group Chemical group [H]C([H])([H])C([H])([H])OC([H])([H])* 0.000 description 1
- RIFGWPKJUGCATF-UHFFFAOYSA-N ethyl chloroformate Chemical compound CCOC(Cl)=O RIFGWPKJUGCATF-UHFFFAOYSA-N 0.000 description 1
- 239000007941 film coated tablet Substances 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- WBJINCZRORDGAQ-UHFFFAOYSA-N formic acid ethyl ester Natural products CCOC=O WBJINCZRORDGAQ-UHFFFAOYSA-N 0.000 description 1
- FATAVLOOLIRUNA-UHFFFAOYSA-N formylmethyl Chemical group [CH2]C=O FATAVLOOLIRUNA-UHFFFAOYSA-N 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- WIGIZIANZCJQQY-RUCARUNLSA-N glimepiride Chemical compound O=C1C(CC)=C(C)CN1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)N[C@@H]2CC[C@@H](C)CC2)C=C1 WIGIZIANZCJQQY-RUCARUNLSA-N 0.000 description 1
- 229960004346 glimepiride Drugs 0.000 description 1
- 230000004110 gluconeogenesis Effects 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- 125000005456 glyceride group Chemical group 0.000 description 1
- 229940096919 glycogen Drugs 0.000 description 1
- 229960000789 guanidine hydrochloride Drugs 0.000 description 1
- PJJJBBJSCAKJQF-UHFFFAOYSA-N guanidinium chloride Chemical compound [Cl-].NC(N)=[NH2+] PJJJBBJSCAKJQF-UHFFFAOYSA-N 0.000 description 1
- 125000002795 guanidino group Chemical group C(N)(=N)N* 0.000 description 1
- 208000019622 heart disease Diseases 0.000 description 1
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000003906 humectant Substances 0.000 description 1
- 150000007857 hydrazones Chemical class 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002430 hydrocarbons Chemical class 0.000 description 1
- 150000003840 hydrochlorides Chemical class 0.000 description 1
- JUINSXZKUKVTMD-UHFFFAOYSA-N hydrogen azide Chemical compound N=[N+]=[N-] JUINSXZKUKVTMD-UHFFFAOYSA-N 0.000 description 1
- 229910000042 hydrogen bromide Inorganic materials 0.000 description 1
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 1
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 239000008172 hydrogenated vegetable oil Substances 0.000 description 1
- 229940071870 hydroiodic acid Drugs 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- XNXVOSBNFZWHBV-UHFFFAOYSA-N hydron;o-methylhydroxylamine;chloride Chemical compound Cl.CON XNXVOSBNFZWHBV-UHFFFAOYSA-N 0.000 description 1
- 239000001863 hydroxypropyl cellulose Substances 0.000 description 1
- 235000010977 hydroxypropyl cellulose Nutrition 0.000 description 1
- 125000001841 imino group Chemical group [H]N=* 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 1
- 238000004255 ion exchange chromatography Methods 0.000 description 1
- 125000006316 iso-butyl amino group Chemical group [H]N(*)C([H])([H])C([H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000005929 isobutyloxycarbonyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])OC(*)=O 0.000 description 1
- NIZHERJWXFHGGU-UHFFFAOYSA-N isocyanato(trimethyl)silane Chemical compound C[Si](C)(C)N=C=O NIZHERJWXFHGGU-UHFFFAOYSA-N 0.000 description 1
- 125000004594 isoindolinyl group Chemical group C1(NCC2=CC=CC=C12)* 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 125000005932 isopentyloxycarbonyl group Chemical group 0.000 description 1
- 125000005928 isopropyloxycarbonyl group Chemical group [H]C([H])([H])C([H])(OC(*)=O)C([H])([H])[H] 0.000 description 1
- 125000005956 isoquinolyl group Chemical group 0.000 description 1
- 239000000644 isotonic solution Substances 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 150000007517 lewis acids Chemical class 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- XIXADJRWDQXREU-UHFFFAOYSA-M lithium acetate Chemical compound [Li+].CC([O-])=O XIXADJRWDQXREU-UHFFFAOYSA-M 0.000 description 1
- FUJCRWPEOMXPAD-UHFFFAOYSA-N lithium oxide Chemical compound [Li+].[Li+].[O-2] FUJCRWPEOMXPAD-UHFFFAOYSA-N 0.000 description 1
- 229910001947 lithium oxide Inorganic materials 0.000 description 1
- INHCSSUBVCNVSK-UHFFFAOYSA-L lithium sulfate Inorganic materials [Li+].[Li+].[O-]S([O-])(=O)=O INHCSSUBVCNVSK-UHFFFAOYSA-L 0.000 description 1
- 231100000053 low toxicity Toxicity 0.000 description 1
- 125000001288 lysyl group Chemical group 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- UEGPKNKPLBYCNK-UHFFFAOYSA-L magnesium acetate Chemical compound [Mg+2].CC([O-])=O.CC([O-])=O UEGPKNKPLBYCNK-UHFFFAOYSA-L 0.000 description 1
- 239000011654 magnesium acetate Substances 0.000 description 1
- 235000011285 magnesium acetate Nutrition 0.000 description 1
- 229940069446 magnesium acetate Drugs 0.000 description 1
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L magnesium chloride Substances [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- LWLPYZUDBNFNAH-UHFFFAOYSA-M magnesium;butane;bromide Chemical compound [Mg+2].[Br-].CCC[CH2-] LWLPYZUDBNFNAH-UHFFFAOYSA-M 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- OETHQSJEHLVLGH-UHFFFAOYSA-N metformin hydrochloride Chemical compound Cl.CN(C)C(=N)N=C(N)N OETHQSJEHLVLGH-UHFFFAOYSA-N 0.000 description 1
- 229960004329 metformin hydrochloride Drugs 0.000 description 1
- XZWYZXLIPXDOLR-UHFFFAOYSA-N metformin hydrochloride Natural products CN(C)C(=N)NC(N)=N XZWYZXLIPXDOLR-UHFFFAOYSA-N 0.000 description 1
- UKVIEHSSVKSQBA-UHFFFAOYSA-N methane;palladium Chemical compound C.[Pd] UKVIEHSSVKSQBA-UHFFFAOYSA-N 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- RHMQNXNXUZLEIY-UHFFFAOYSA-N methanol;2-propan-2-yloxypropane Chemical compound OC.CC(C)OC(C)C RHMQNXNXUZLEIY-UHFFFAOYSA-N 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- CAAULPUQFIIOTL-UHFFFAOYSA-L methyl phosphate(2-) Chemical compound COP([O-])([O-])=O CAAULPUQFIIOTL-UHFFFAOYSA-L 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- RIKFFJJHWCSHKF-UHFFFAOYSA-N methylphosphonic acid;dihydrochloride Chemical compound Cl.Cl.CP(O)(O)=O RIKFFJJHWCSHKF-UHFFFAOYSA-N 0.000 description 1
- 125000002816 methylsulfanyl group Chemical group [H]C([H])([H])S[*] 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- 229910052750 molybdenum Inorganic materials 0.000 description 1
- 239000011733 molybdenum Substances 0.000 description 1
- DAKZISABEDGGSV-UHFFFAOYSA-N n-(2-aminoethyl)acetamide Chemical compound CC(=O)NCCN DAKZISABEDGGSV-UHFFFAOYSA-N 0.000 description 1
- RWIVICVCHVMHMU-UHFFFAOYSA-N n-aminoethylmorpholine Chemical compound NCCN1CCOCC1 RWIVICVCHVMHMU-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- GSJRUEBQWPLHSN-UHFFFAOYSA-N n-methylmethanamine;oxolane Chemical compound CNC.C1CCOC1 GSJRUEBQWPLHSN-UHFFFAOYSA-N 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- 229960000698 nateglinide Drugs 0.000 description 1
- 125000005484 neopentoxy group Chemical group 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000005933 neopentyloxycarbonyl group Chemical group 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- QMMRZOWCJAIUJA-UHFFFAOYSA-L nickel dichloride Chemical compound Cl[Ni]Cl QMMRZOWCJAIUJA-UHFFFAOYSA-L 0.000 description 1
- 150000002823 nitrates Chemical class 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 125000002868 norbornyl group Chemical group C12(CCC(CC1)C2)* 0.000 description 1
- 239000012038 nucleophile Substances 0.000 description 1
- DCIKEGMDLASJHI-RXMQYKEDSA-N o-[[(4r)-2,2-dimethyl-1,3-dioxolan-4-yl]methyl]hydroxylamine Chemical compound CC1(C)OC[C@H](CON)O1 DCIKEGMDLASJHI-RXMQYKEDSA-N 0.000 description 1
- TVMXDCGIABBOFY-UHFFFAOYSA-N octane Chemical compound CCCCCCCC TVMXDCGIABBOFY-UHFFFAOYSA-N 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 235000019198 oils Nutrition 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 125000001979 organolithium group Chemical group 0.000 description 1
- 125000002524 organometallic group Chemical group 0.000 description 1
- 229910052762 osmium Inorganic materials 0.000 description 1
- SYQBFIAQOQZEGI-UHFFFAOYSA-N osmium atom Chemical compound [Os] SYQBFIAQOQZEGI-UHFFFAOYSA-N 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- VMWYXJBLVOBZTQ-UHFFFAOYSA-N oxolane;tetrabutylazanium Chemical compound C1CCOC1.CCCC[N+](CCCC)(CCCC)CCCC VMWYXJBLVOBZTQ-UHFFFAOYSA-N 0.000 description 1
- KRHGGZJSXWNNKS-UHFFFAOYSA-M oxolane;tetrabutylazanium;chloride Chemical class [Cl-].C1CCOC1.CCCC[N+](CCCC)(CCCC)CCCC KRHGGZJSXWNNKS-UHFFFAOYSA-M 0.000 description 1
- ZOUWOGOTHLRRLS-UHFFFAOYSA-N palladium;phosphane Chemical compound P.[Pd] ZOUWOGOTHLRRLS-UHFFFAOYSA-N 0.000 description 1
- 125000003538 pentan-3-yl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical class OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 150000002978 peroxides Chemical class 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- HXITXNWTGFUOAU-UHFFFAOYSA-N phenylboronic acid Chemical compound OB(O)C1=CC=CC=C1 HXITXNWTGFUOAU-UHFFFAOYSA-N 0.000 description 1
- RPGWZZNNEUHDAQ-UHFFFAOYSA-N phenylphosphine Chemical compound PC1=CC=CC=C1 RPGWZZNNEUHDAQ-UHFFFAOYSA-N 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- 150000003003 phosphines Chemical class 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- UHZYTMXLRWXGPK-UHFFFAOYSA-N phosphorus pentachloride Chemical compound ClP(Cl)(Cl)(Cl)Cl UHZYTMXLRWXGPK-UHFFFAOYSA-N 0.000 description 1
- CYQAYERJWZKYML-UHFFFAOYSA-N phosphorus pentasulfide Chemical compound S1P(S2)(=S)SP3(=S)SP1(=S)SP2(=S)S3 CYQAYERJWZKYML-UHFFFAOYSA-N 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 229960002827 pioglitazone hydrochloride Drugs 0.000 description 1
- 229960005235 piperonyl butoxide Drugs 0.000 description 1
- 239000013612 plasmid Substances 0.000 description 1
- 229920000128 polypyrrole Polymers 0.000 description 1
- 230000000291 postprandial effect Effects 0.000 description 1
- 235000011056 potassium acetate Nutrition 0.000 description 1
- CHKVPAROMQMJNQ-UHFFFAOYSA-M potassium bisulfate Chemical compound [K+].OS([O-])(=O)=O CHKVPAROMQMJNQ-UHFFFAOYSA-M 0.000 description 1
- 229910000343 potassium bisulfate Inorganic materials 0.000 description 1
- 239000001103 potassium chloride Substances 0.000 description 1
- 235000011164 potassium chloride Nutrition 0.000 description 1
- NNFCIKHAZHQZJG-UHFFFAOYSA-N potassium cyanide Chemical compound [K+].N#[C-] NNFCIKHAZHQZJG-UHFFFAOYSA-N 0.000 description 1
- 229910000160 potassium phosphate Inorganic materials 0.000 description 1
- 235000011009 potassium phosphates Nutrition 0.000 description 1
- OTYBMLCTZGSZBG-UHFFFAOYSA-L potassium sulfate Chemical compound [K+].[K+].[O-]S([O-])(=O)=O OTYBMLCTZGSZBG-UHFFFAOYSA-L 0.000 description 1
- 229910052939 potassium sulfate Inorganic materials 0.000 description 1
- 235000011151 potassium sulphates Nutrition 0.000 description 1
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- OVARTBFNCCXQKS-UHFFFAOYSA-N propan-2-one;hydrate Chemical compound O.CC(C)=O OVARTBFNCCXQKS-UHFFFAOYSA-N 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 125000001501 propionyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 1
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 1
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 239000012264 purified product Substances 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 125000005495 pyridazyl group Chemical group 0.000 description 1
- ABMYEXAYWZJVOV-UHFFFAOYSA-N pyridin-3-ylboronic acid Chemical compound OB(O)C1=CC=CN=C1 ABMYEXAYWZJVOV-UHFFFAOYSA-N 0.000 description 1
- TXQWFIVRZNOPCK-UHFFFAOYSA-N pyridin-4-ylmethanamine Chemical compound NCC1=CC=NC=C1 TXQWFIVRZNOPCK-UHFFFAOYSA-N 0.000 description 1
- ZDYVRSLAEXCVBX-UHFFFAOYSA-N pyridinium p-toluenesulfonate Chemical compound C1=CC=[NH+]C=C1.CC1=CC=C(S([O-])(=O)=O)C=C1 ZDYVRSLAEXCVBX-UHFFFAOYSA-N 0.000 description 1
- 125000005344 pyridylmethyl group Chemical group [H]C1=C([H])C([H])=C([H])C(=N1)C([H])([H])* 0.000 description 1
- HNJBEVLQSNELDL-UHFFFAOYSA-N pyrrolidin-2-one Chemical compound O=C1CCCN1 HNJBEVLQSNELDL-UHFFFAOYSA-N 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 229910052705 radium Inorganic materials 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 238000006268 reductive amination reaction Methods 0.000 description 1
- 238000001226 reprecipitation Methods 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 210000001525 retina Anatomy 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- QBERHIJABFXGRZ-UHFFFAOYSA-M rhodium;triphenylphosphane;chloride Chemical compound [Cl-].[Rh].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 QBERHIJABFXGRZ-UHFFFAOYSA-M 0.000 description 1
- 108010091078 rigin Proteins 0.000 description 1
- 102220094506 rs876659390 Human genes 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 229910052711 selenium Inorganic materials 0.000 description 1
- 239000011669 selenium Substances 0.000 description 1
- 150000007659 semicarbazones Chemical class 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- ZLGIYFNHBLSMPS-ATJNOEHPSA-N shellac Chemical compound OCCCCCC(O)C(O)CCCCCCCC(O)=O.C1C23[C@H](C(O)=O)CCC2[C@](C)(CO)[C@@H]1C(C(O)=O)=C[C@@H]3O ZLGIYFNHBLSMPS-ATJNOEHPSA-N 0.000 description 1
- 239000004208 shellac Substances 0.000 description 1
- 235000013874 shellac Nutrition 0.000 description 1
- 229940113147 shellac Drugs 0.000 description 1
- 125000003808 silyl group Chemical group [H][Si]([H])([H])[*] 0.000 description 1
- 235000020374 simple syrup Nutrition 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- WRIKHQLVHPKCJU-UHFFFAOYSA-N sodium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([Na])[Si](C)(C)C WRIKHQLVHPKCJU-UHFFFAOYSA-N 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 238000001179 sorption measurement Methods 0.000 description 1
- 210000000952 spleen Anatomy 0.000 description 1
- 239000007940 sugar coated tablet Substances 0.000 description 1
- HXJUTPCZVOIRIF-UHFFFAOYSA-N sulfolane Chemical compound O=S1(=O)CCCC1 HXJUTPCZVOIRIF-UHFFFAOYSA-N 0.000 description 1
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 1
- 150000003457 sulfones Chemical class 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 125000004213 tert-butoxy group Chemical group [H]C([H])([H])C(O*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- RBTVSNLYYIMMKS-UHFFFAOYSA-N tert-butyl 3-aminoazetidine-1-carboxylate;hydrochloride Chemical compound Cl.CC(C)(C)OC(=O)N1CC(N)C1 RBTVSNLYYIMMKS-UHFFFAOYSA-N 0.000 description 1
- 125000006318 tert-butyl amino group Chemical group [H]N(*)C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- AOCSUUGBCMTKJH-UHFFFAOYSA-N tert-butyl n-(2-aminoethyl)carbamate Chemical compound CC(C)(C)OC(=O)NCCN AOCSUUGBCMTKJH-UHFFFAOYSA-N 0.000 description 1
- YBRBMKDOPFTVDT-UHFFFAOYSA-N tert-butylamine Chemical compound CC(C)(C)N YBRBMKDOPFTVDT-UHFFFAOYSA-N 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- NZBUCABTIWJWAN-UHFFFAOYSA-N tetrabromomethane;triphenylphosphane Chemical compound BrC(Br)(Br)Br.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NZBUCABTIWJWAN-UHFFFAOYSA-N 0.000 description 1
- 229940126585 therapeutic drug Drugs 0.000 description 1
- 150000003557 thiazoles Chemical class 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 125000003944 tolyl group Chemical group 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- 238000013519 translation Methods 0.000 description 1
- 125000004665 trialkylsilyl group Chemical group 0.000 description 1
- 125000003866 trichloromethyl group Chemical group ClC(Cl)(Cl)* 0.000 description 1
- XPDWGBQVDMORPB-UHFFFAOYSA-N trifluoromethane acid Natural products FC(F)F XPDWGBQVDMORPB-UHFFFAOYSA-N 0.000 description 1
- PQDJYEQOELDLCP-UHFFFAOYSA-N trimethylsilane Chemical group C[SiH](C)C PQDJYEQOELDLCP-UHFFFAOYSA-N 0.000 description 1
- FTVLMFQEYACZNP-UHFFFAOYSA-N trimethylsilyl trifluoromethanesulfonate Chemical compound C[Si](C)(C)OS(=O)(=O)C(F)(F)F FTVLMFQEYACZNP-UHFFFAOYSA-N 0.000 description 1
- 125000004417 unsaturated alkyl group Chemical group 0.000 description 1
- 229910052720 vanadium Inorganic materials 0.000 description 1
- 229960001729 voglibose Drugs 0.000 description 1
- 238000010792 warming Methods 0.000 description 1
- 238000005303 weighing Methods 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6536—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having nitrogen and sulfur atoms with or without oxygen atoms, as the only ring hetero atoms
- C07F9/6539—Five-membered rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Definitions
- the present invention relates to a novel thiazole derivative and a pharmacologically acceptable salt thereof.
- the present invention also relates to a thiazole derivative having an excellent fructose 1,6-bisphosphatase (FBPase) inhibitory action, hypoglycemic action, lipid lowering action, anti-inflammatory action and the like, and a pharmacologically acceptable salt thereof.
- FBPase fructose 1,6-bisphosphatase
- the present invention relates to diabetes, hyperglycemia, glucose intolerance, obesity, hyperlipidemia, diabetic complications (for example, retina) containing a thiazole derivative or a pharmacologically acceptable salt thereof as an active ingredient.
- ischemic heart disease e.g, cerebrovascular disorder, angina pectoris, myocardial infarction, abnormal blood pressure, etc.
- arteriosclerosis e.g, cerebrovascular disorder, angina pectoris, myocardial infarction, abnormal blood pressure, etc.
- gestational diabetes Cushing syndrome, steroid diabetes, Glycogenosis, autonomic nervous system disorders (eg impotence, sweating abnormalities, orthostatic hypotension, etc.), surgical diabetes-like pathology, liver cancer and other therapeutic agents and Z or prophylactic agents (preferably diabetes therapeutic agents and Z or a preventive drug.
- the present invention provides a preventive or therapeutic agent for the above-mentioned diseases containing the above-mentioned compound as an active ingredient, a composition for preventing or treating the above-mentioned disease containing the above-mentioned compound as an active ingredient, the prevention or the above-mentioned disease.
- the present invention relates to the use of the above-mentioned compound for producing a medicament for treatment, or a method for preventing or treating the above-mentioned disease, wherein a pharmacologically effective amount of the above-mentioned compound is administered to a warm-blooded animal (preferably a human).
- Sulfonylurea (SU), (3) Fast-acting ⁇ Stimulates insulin secretion in a short time to lower postprandial blood glucose level, such as star cis (nateglinide), (4) A-Dalcosidase inhibitors such as Basin (Voglibose), Dalcoby (Valboose), which inhibits the action of saccharide-degrading enzymes (hyaldarcosidase), suppresses sugar digestion and delays absorption, and suppresses postprandial hyperglycemia (5) Gibetose B (buformin hydrochloride), which has a hypoglycemic effect by suppressing sugar production in the liver, inhibiting sugar absorption in the gastrointestinal tract, and improving insulin sensitivity in peripheral tissues.
- A-Dalcosidase inhibitors such as Basin (Voglibose), Dalcoby (Valboose), which inhibits the action of saccharide-degrading enzymes (hyaldarcosidase
- BG agents such as cholan (metformin hydrochloride), and (6) insulin resistance improving agents such as lactos (pioglitazone hydrochloride) which improve insulin resistance and exhibit hypoglycemic action have been used.
- cholan metalformin hydrochloride
- insulin resistance improving agents such as lactos (pioglitazone hydrochloride) which improve insulin resistance and exhibit hypoglycemic action have been used.
- lactos lactos
- FBPase is an enzyme deeply involved in gluconeogenesis in the liver, and inhibition of this enzyme has been reported to suppress sugar production and lower blood glucose levels. Therefore, FBPase inhibitors are expected to be antidiabetic drugs having a novel mechanism of action, and some FBPase inhibitors are disclosed as compounds that have a hypoglycemic action as FBPase inhibitors (see Patent Documents 1 and 2). See). However, the disclosed compounds do not have a tricyclic skeleton as a basic skeleton, and are completely different in structure from the compounds of the present invention.
- Patent Document 1 International Publication No. 01/47935 Pamphlet
- Patent Document 2 Pamphlet of International Publication No. 00/14095
- the present inventors have conducted intensive research for the purpose of developing therapeutic agents such as diabetes and Z or prophylactic agents having excellent activity and safety, and the novel thiazole derivatives have excellent FBPase inhibitory action, insulin resistance. It has antibacterial, hypoglycemic, and lipid lowering effects, hyperlipidemia, hyperglycemia, obesity, impaired glucose tolerance, non-insulin-resistant glucose intolerance, insulin resistance, diabetic complications, imaginary
- the present invention was completed by finding that blood heart disease, arteriosclerosis, gestational diabetes, Cushing syndrome, steroid diabetes, glycogenosis, autonomic nervous system disorder, surgical diabetes-like pathology, liver cancer and the like.
- the present invention relates to hyperlipidemia, hyperglycemia, obesity, impaired glucose tolerance (IGT) state, insulin resistant non-IGT: NGT ) Condition, hypertension, splenitis, fatty liver, diabetic complications (e.g.
- retinopathy cataract, nephropathy, end (Eg, cranial nerve disorders), arteriosclerosis (eg, cerebrovascular disorders, angina pectoris' myocardial infarction, abnormal blood pressure, etc.), gestational diabetes mellitus (GDM), Cushing syndrome, steroid diabetes, glucose Thiazole derivatives and their pharmacologically acceptable drugs useful as therapeutic or prophylactic agents for diseases, autonomic nervous system disorders (for example, impotence, abnormal sweating, orthostatic hypotension, etc.), surgical diabetes-like pathology, liver cancer, etc. Provide salt.
- arteriosclerosis eg, cerebrovascular disorders, angina pectoris' myocardial infarction, abnormal blood pressure, etc.
- GDM gestational diabetes mellitus
- Cushing syndrome steroid diabetes
- glucose Thiazole derivatives useful as therapeutic or prophylactic agents for diseases, autonomic nervous system disorders (for example, impotence, abnormal sweating, orthostatic hypotension, etc.), surgical diabetes-like pathology, liver
- the present invention provides:
- R lx and R 1Y are different from each other in hydrogen atom, halogen atom, C1-C3 alkyl group, P (0) (OH), OP (0) (OH), CH P (0 ) (OH), CF P (0) (OH) or OCH P (0) (OH)
- R 2 and R 3 are the same or different and each represents a hydrogen atom, a halogen atom, a -C6 alkyl group, a C1-C6 haloalkyl group, a C1-C6 aminoalkyl group (the aminoalkyl)
- the amino group of the group may be substituted with one or two groups selected from the substituent group ⁇ force), C1-C6 hydroxyalkyl group, CI-C6 alkylthio group, CI-C6 alkoxy group, CI-C6
- An alkoxyximino C1-C3 alkyl group (the alkoxyimino C1-C3 alkyl group may be substituted with a C1-C3 alkyl group), a hydroxyl group, a C1-C6 aliphatic acyl group (the acyl group is a substituted group)
- amino group CI-C6 alkyl
- Nitrogen, oxygen, and sulfur power 4 to 10-membered heterocyclic group containing the same or different 1 to 3 hetero atoms selected (the heterocyclic group is a substituent group ⁇ force) 1 to 5 selected groups may be substituted), nitrogen, oxygen and sulfur powers the same or different selected 1 -3 4- to 10-membered heterocyclic C1-C6 alkyl group containing 3 heteroatoms (the heterocyclic C1-C6 alkyl group is substituted with 1 to 5 groups selected from the substituent group ⁇ force) 4) 10-membered heterocyclic CI-C6 alkylamino group containing the same or different 1 to 3 heteroatoms selected (nitrogen, oxygen and sulfur power) (the heterocyclic CI-C6 alkylamino group) May be substituted with 1 to 5 groups selected from the substituent group a), represents a ureido group, a cyano group or a -tro group, R 4 represents a hydrogen atom, a C1-C3 alkyl group, A C
- R 1Y represents a hydrogen atom or a halogen atom.
- R 1Y is P ( ⁇ ) ( ⁇ H), OP (0) (OH), CH P (0) (OH), CF P
- R lx is a hydrogen atom, a halogen atom or C1
- a -C6 alkyl group is shown.
- R is P (0) (OH), OP (0) (OH), CH P (0) (OH), CF P (0) (OH) or OCH P
- R 2 and R 3 are the same or different and each represents a hydrogen atom, halogen
- CI-C6 alkyl group, CI-C6 haloalkyl group, CI-C6 aminoalkyl group (the amino group of the aminoamino group is substituted with one or two substituents selected from the substituent group a) CI-C6 hydroxyalkyl group, CI-C6 alkylthio group, CI-C6 alkoxy group, CI-C6 alkoxyimino CI-C3 alkyl group (the alkoxyimino CI-C3 alkyl group is C1-C3 alkyl)
- a hydroxyl group, a C1-C6 aliphatic acyl group (this acyl group may be substituted with a group selected from the substituent group ⁇ ), an amino group, C1 -C6 alkylamino group, CI-C6 dialkylamino group, CI-C6 aliphatic asilamino group, C6-C10 aryl group (the aryl group is substituted with
- nitrogen, oxygen and sulfur force 4-10 membered heterocyclic CI-C6 alkylamino group containing 1 to 3 heteroatoms selected from the same or different Heterocyclic CI-C6 alkylamino group is a substituent group a 1 to 5 substituents may be substituted with a selected group), a ureido group, a cyano group, or a -toto group, and R 4 represents a hydrogen atom, a C1-C3 alkyl group, a C1-C3 haloalkyl group, X represents a C1-C3 alkylene group (the alkylene group is substituted with one or two of an oxo group, a hydroxyl group, a halogen atom, a C1-C3 alkyl group, or a C3-6 cycloalkylidene group) May be).
- R lx is OCH P (0) (OH)
- R 2 and R 3 are the same or different, and hydrogen atom, halogen atom, CI-C6 alkyl group, CI-C6 alkoxy group, CI-C6 dialkylamino
- R 2 is a hydrogen atom or a C1-C6 alkyl group, or a pharmacologically acceptable salt thereof,
- R 3 is a halogen atom, a C1-C6 dialkylamino group, a C1-C6 aliphatic acyl group (the acyl group is substituted with a group selected from the substituent group a) Or a pharmacologically acceptable salt thereof, which is a C1-C6 alkyl group or a C1-C6 alkoxy group,
- R 1 is P (0) (OH), OP (0) (OH), CH P (0) (OH), CF P (0) (OH) or OCH P (
- R 2 and R 3 are the same or different and each represents a hydrogen atom or a halogen atom.
- CI-C6 alkyl group, CI-C6 haloalkyl group, CI-C6 aminoalkyl group (the amino group of the aminoalkyl group may be substituted with one or two substituents selected from group a force) ), CI-C6 hydroxyalkyl group, CI-C6 alkylthio group, CI-C6 alkoxy group, CI-C6 alkoxyimino CI-C3 alkyl group (the alkoxyimino CI-C3 alkyl group is a C1-C3 alkyl group) Optionally substituted), hydroxyl group, C1-C6 aliphatic acyl group (the acyl group may be substituted with a group selected by the substituent group ⁇ force), amino group, C1-C6 alkylamino Group, CI-C6 dialkylamino group, CI-C6 aliphatic acylamino group, C6-C10 aryl group (this aryl group
- Halogen atom, CI-C6 alkyl group, CI-C6 haloalkyl group, CI-C6 aminoalkyl Group (the amino group of the aminoalkyl group may be substituted with one or two C1-C6 alkyl groups), CI-C6 alkoxy group, CI-C6 alkylthio group, carboxy group, CI-C6 alkoxycarbo group Group, hydroxyl group, C1-C6 aliphatic asil group, amino group, C1-C6 alkylamino group, CI-C6 dialkylamino group, CI-C6 aliphatic asilamino group, cyano group, nitro group, C3-C10 cycloalkyl group , C6-C10 aryl group, same or different selected from nitrogen, oxygen and sulfur 4- to 10-membered heterocyclic group containing 1-3 heteroatoms, nitrogen, oxygen and sulfur power Or a compound represented by a 4- to 10-membered heterocyclic C1
- a medicament comprising the compound according to (1) to (20) above or a pharmacologically acceptable salt thereof,
- a therapeutic or prophylactic agent for diabetes comprising the compound according to (1) to (20) above or a pharmacologically acceptable salt thereof,
- a method for the prophylaxis or treatment of diabetes comprising administering to a warm-blooded animal a pharmacologically effective amount of the compound according to (1) to (20) above or a pharmacologically acceptable salt thereof.
- the “Cl-3 alkyl group” is a linear or branched alkyl group having 1 to 3 carbon atoms, and examples thereof include a methyl, ethyl or n-propyl group. Can do.
- R 1x and R 1Y , R 2 and R 3 C 1-6 alkoxyimino C 1-3 alkyl group substituents, R 4 and X C 1-3 alkylene substituents are preferably methyl groups is there
- the “Cl-6 alkyl group” is a linear or branched alkyl group having 1 to 6 carbon atoms.
- the “C1_3 alkyl group” Or methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, s-butyl, tert-butyl, n-pentyl, isopentyl, 2-methylbutyl, neopentyl, 1-ethylpropyl N-hexyl, isohexyl, 4-methylpentyl, 3-methylpentyl, 2-methylpentyl, 1-methylpentyl, 3,3-dimethylbutyl, 2,2-dimethylbutyl, 1,1-dimethylbutyl 1,2-dimethylbutyl, 1,3-dimethylbutyl, 2,3-dimethylbutyl, 2-ethylbutyl, heptyl, 1-methylhexyl, 2-methylhexyl, 3-methylhexyl, 4-methyl Xyl, 5-methylhexyl, 1-propyl, 1-prop
- R 2 and R 3 are preferably an alkyl group having 1 to 3 carbon atoms, and most preferably an ethyl group or a methyl group.
- the substituents of the substituent group a and the C1-C6 aminoalkyl group of the substituent group a are preferably an alkyl group having 1 to 3 carbon atoms, and most preferably a methyl group. .
- the “Cl-3 alkylene group” is an alkylene group having 1 to 3 carbon atoms, and examples thereof include methylene, methylmethylene, dimethylmethylene, ethylene, and propylene. Can do.
- X is preferably methylene or ethylene, and more preferably methylene.
- “/ C3-10 cycloalkyl group” means a condensed ring! /, Which may be a 3- to 10-membered saturated cyclic hydrocarbon group. Mention may be made of propyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, norbornyl, adamantyl groups.
- Substituent group a and the substituent of C1-C6 alkylamino group of substituent group a are preferably 3- to 7-membered saturated cyclic hydrocarbon groups, and more preferably cyclopropyl, cyclobutyl. Til, cyclopentyl, cyclohexyl.
- the “C3-6 cycloalkylidene group” is a divalent 3- to 6-membered saturated cyclic hydrocarbon group such as cyclopropylidene, cyclobutylidene, cyclopentylidene, Shi Mention may be made of the chlorohexylene group.
- X is preferably cyclopentylidene.
- the “Cl-6 haloalkyl group” is a group in which a halogen atom is substituted on the “Cl-6 alkyl group”, and examples thereof include trifluoromethyl, trichloromethyl, Difluoromethyl, dichloromethyl, dibromomethyl, fluoromethyl, 2,2,2-trifluoroethyl, 2,2,2-trichloroethinole, 2-bromoethinole, 2-chloroethinole, 2-funoleoloetinore, 2 -Rhodochinole, 3-chloropropinole, 4-funoleolobutinole, 6-hydrohexenole, 2,2-dibucolate, R 2 , R 3 , R 4 and substituents
- preferred is a trifluoromethyl group or a difluoromethyl group.
- the “C1-C6 hydroxyalkyl group” is a group in which a hydroxyl group is substituted on the “Cl-6 alkyl group”, and examples thereof include hydroxymethyl, hydroxyethyl, 1- And hydroxyethyl, hydroxypropyl, hydroxybutyl, hydroxypentyl, hydroxyhexyl groups, R 2 ,
- the substituent of the C1-C6 hydroxyalkyl group of the substituent group a, the substituent of the rubamoylalkylamino group of the substituent group a, and the substituent of the C1-C6 alkylamino group of the substituent group a Is preferably a hydroxymethyl group or a 1-hydroxyethyl group.
- the "C 1-6 aminoalkyl group” is a group obtained by substituting an amino group for the above-mentioned "Cl-6 alkyl group”.
- aminomethyl, aminoethyl, aminopropyl An aminobutyl group can be mentioned, and in R 2 , R 3 and the substituent group, aminomethyl is preferred.
- the “Cl-6 alkoxy group” is a group in which the “Cl-6 alkyl group” is bonded to an oxygen atom.
- substituent group a a methoxy group is preferred.
- R 2 , CI- of substituent group a and substituent group a The substituent for the C6 alkylamino group is preferably a methoxy or ethoxy group.
- the “Cl-6 alkoxy C1-6 alkyl group” is a group in which the “Cl-6 alkyl group” is bonded to the “Cl-6 alkoxy group”, for example, methoxymethyl, Methoxyethyl, ethoxymethyl, ethoxyethyl, n-propoxymethyl, n-propoxychetyl, isopropoxymethyl, n-butoxymethyl, isobutoxymethyl, s-butoxymethyl, tert-butoxymethyl, n-pentoxymethyl, Isopentoxymethyl, 2-methylbutoxymethyl, neopentoxymethyl, n-hexyloxymethyl, 4-methylpentoxymethyl, 3-methylpentoxymethyl, 2-methylpentoxymethyl, 3,3- Dimethylbutoxymethyl, 2,2-dimethylbutoxymethyl, 1,1-dimethylbutoxymethyl, 1,2-dimethylbutoxymethyl, 1,3-dimethylbutoxymethyl, 2,3 -A straight-chain or branched alkoxy group
- the “Cl-6 alkylthio group” is a group in which the “Cl-6 alkyl group” is bonded to a sulfur atom.
- a methylthio, ethylthio, or t-butylthio group In R 2 , R 3 and substituent group ⁇ , a methylthio group is preferred.
- the “ ⁇ 6 aliphatic acyl group” is a group in which an aliphatic hydrocarbon group having 1 to 6 carbon atoms is bonded to a carbonyl group.
- an aliphatic hydrocarbon group having 1 to 6 carbon atoms is bonded to a carbonyl group.
- R 2 , R 3 and substituent group ⁇ a formyl group,
- the “C 1-6 alkylamino group” is a group in which the “Cl-6 alkyl group” is bonded to an amino group, such as methylamino, ethylamino-containing isopropylamino, n-Butylamino-containing isobutylamino-containing S-Butylamino, tert-Butylamino-containing n-Pentylami-containing Isopentylami-containing 2-Methylbutylamino-containing Neopentylamino, 1-ethylpropylamino-containing n-Hexylami-containing Isohexylami-containing 4-methylpentylami containing 3 -Methylpentylamino, 2-methylpentylamino, 1-methylpentylamino, 3,3-dimethylbutylamino, 2,2-dimethylbutylamino, 1,1-dimethylbutylamino, 1,2-di
- the "C3-10 cycloalkylamino group” is a group in which the "C3-10 cycloalkyl” is bonded to an amino group, such as cyclopropylamino-containing cyclobutyl.
- an amino group such as cyclopropylamino-containing cyclobutyl.
- Examples include amide-containing cyclopentyl amide-containing cyclohexyl amide-containing cycloheptyl amide-containing norbornylamino groups and adamantylamino groups.
- a 3- to 7-membered saturated cyclic hydrocarbon amino group is preferable, and a cyclopropylamino-containing cyclopropylamino or cyclopentylamino-containing cyclohexylamino group is more preferable.
- the “Cl-6 alkoxyamino group” is a group in which the “Cl-6 alkoxy group” is bonded to an amino group, such as methoxyamino-containing ethoxyamino, n-propoxyamino, isopropoxy, and the like.
- the "Cl-6 dialkylamino group” is a group in which the same or different "Cl-6 alkyl group” force is substituted on an amino group, such as ⁇ , ⁇ -dimethylamino.
- the “ ⁇ 6 aliphatic acylamine group” is an aliphatic hydrocarbon group having 1 to 6 carbon atoms or a group in which a hydrogen atom is bonded to a carbonylamino group.
- formylamino Alkyl power such as acetylamino-containing propionylamino-containing butyrylamino, isobutyrylami-containing penta-nylamino, bivaloylami-containing valeryl-amido-isovalerylamino group
- a halogenated alkyl carbolumino group a lower alkoxyalkyl carboamino group such as methoxyacetylamino, an unsaturated alkyl carbolumino group such as (E) -2-methyl-2-butenoylamino, and the like.
- Amino groups R 3 and Substituent group a preferably a Asechiruamino group.
- the “C 1-6 alkoxyimino group” is a group in which the “Cl-6 alkoxy group” is bonded to an imino group, and includes, for example, methoxyimid ethoxyimine-containing n-propoxyimid-isolated groups.
- Examples include straight-chain or branched alkoxyimino groups having 1 to 6 carbon atoms such as 2,3-dimethylbutoxyimino, 2,3-dimethylbutoxyimino. That.
- the “Cl-6 alkoxyimino C1-3 alkyl group” is a group in which the “Cl-6 alkoxyximino group” is bonded to the C1-3 alkyl group, and includes, for example, methoxyimino Methyl, methoxyiminoethyl, methoxyimino n-butyl, ethoxyiminomethyl, ethoxyiminoethyl, n-propoxyiminomethyl, isopropoxyiminomethyl, n-butoxyiminomethyl, isobutoxyiminomethyl, s-butoxyiminomethyl, tert- Butoxyiminomethyl, n-vent Xyiminomethyl, isopentoxyiminomethyl, 2-methylbutoxyiminomethyl, neovent xyiminomethyl, n-hexyloxyiminomethyl, 4-methylpentoxyiminomethyl, 3-methylpentoxyiminomethyl, 2- Methoxyimino
- the “Cl-6 alkoxycarbo group” is a group in which the “Cl-6 alkoxy group” is bonded to a carbo group, such as methoxy carbo yl, ethoxy carbo ter.
- methoxy is preferably used.
- a carbo carbonyl, an ethoxy is preferably used
- the “C6-10 aryl group” is an aromatic hydrocarbon group having 6 to 10 carbon atoms, and examples thereof include a phenyl, indul, and naphthyl group.
- R 3 and substituent group a a phenyl group is preferred.
- a 4- to 10-membered heterocyclic group containing 1 to 3 heteroatoms which are the same or different and the nitrogen, oxygen and sulfur powers are also selected means a sulfur atom, an oxygen atom or a nitrogen atom.
- Aromatic heterocyclic groups such as lysyl, pyridazinyl, pyrimidinyl, birazinyl and morpholyl, thiomorpholinyl, pyrrolidyl, pyrrolinyl, imidazolidyl, imidazolyl, virazolyl, virazolyl, piperidyl, pipepe Mention may be made of partially or fully reduced groups corresponding to these groups such as rajur, tetrahydrobiral.
- the above “4- to 10-membered heterocyclic group” may be condensed with other cyclic groups, for example, isobenzazolyl, chromel, indolizyl, isoindolyl, indolyl, indazolyl, pre- Examples include nyl, quinolizinyl, isoquinolyl, quinolyl, phthalajuryl, naphthyridinyl, quinoxalyl, quinazolyl, and isoindolinyl groups.
- R 2 , R 3 and the substituent group a are preferably a 4- to 10-membered heterocyclic group containing at least one nitrogen atom and optionally containing an oxygen atom or a sulfur atom, such as pyrrolyl, azepinyl, pyrazolyl.
- a 4- to 10-membered heterocyclic C1-C6 alkyl group containing 1-3 heteroatoms which are the same or different and also have nitrogen, oxygen and sulfur powers selected means the above-mentioned "C1- C6 alkyl group ”is a group in which the above“ 4-10 membered heterocyclic ring containing 1-3 heteroatoms which are the same or different selected from nitrogen, oxygen and sulfur power ”is substituted, and R 2 and R 3 Is preferably a 4-, 10-membered heterocyclic methyl group containing the same or different 1-3 heteroatoms selected from nitrogen, oxygen and sulfur power, and more preferably pyridylmethyl, pyrrolidylmethyl. , Piperidylmethyl and morpholylmethyl groups.
- a 4- to 10-membered heterocyclic C1-C6 alkylamino group containing 1-3 heteroatoms which are the same or different and also have nitrogen, oxygen and sulfur powers selected means "C1 -C6 alkylamino group "is the same or different 1- ⁇ 4-10 membered heterocyclic ring containing 3 heteroatoms '' is a substituted group, and in R 2 , R 3 and the group of substituents, nitrogen, oxygen and sulfur forces are also preferably selected.
- a 4- to 10-membered heterocyclic ethylamino group containing 1 to 3 heteroatoms more preferably pyridylmethylamino, pyridylethylamino, pyrrolidylethylamino-piperidylethylamino-containing morpholyl. It is a tyramino group.
- the “halogen atom” is a fluorine atom, a chlorine atom, a bromine atom or an iodine atom, and R 1X , R 1Y , R 2 , Substituent group
- the substituents of ⁇ and Cl-3 alkylene groups are preferably fluorine atoms.
- the “C 1-6 force rubamoylalkylamino group” is a group obtained by substituting a force rubamoyl group for the “C1-C6 alkylamino group”.
- the thiazole derivative having the general formula (1), ( ⁇ ) or (II) of the present invention has a basic group, it can be converted into an acid addition salt according to a conventional method.
- Such salts include, for example, hydrohalic acid salts such as hydrofluoric acid, hydrochloric acid, hydrobromic acid, hydroiodic acid; nitrates, perchlorates, sulfates, phosphates, etc.
- Mineral acid salts Salts of lower alkane sulfonic acids such as methane sulfonic acid, trifluoro methane sulfonic acid and ethane sulfonic acid; Salts of allylic sulfonic acid such as benzene sulfonic acid and p-toluene sulfonic acid; Glutamic acid and aspartic acid
- a salt of an amino acid such as acetic acid, fumaric acid, tartaric acid, succinic acid, maleic acid, phosphoric acid, succinic acid, benzoic acid, mandelic acid, ascorbic acid, lactic acid, darconic acid, and carboxylic acid.
- Preferred is a salt of hydrohalic acid, and most preferred is hydrochloride.
- the thiazole derivative having the general formula (1), ( ⁇ ) or (II) when it has a carboxyl group, it can be converted into a metal salt according to a conventional method.
- such salts include alkali metal salts such as lithium, sodium, and potassium; alkaline earth metal salts such as calcium, norum, and magnesium; and aluminum salts. Alkali metal salts are preferred.
- the thiazole derivative having the general formula (1), ( ⁇ ) or (II) has various isomers. There are things to do. In the above general formulas 0), ( ⁇ ) or ( ⁇ ⁇ ⁇ ), these isomers and equivalent and unequal mixtures of these isomers are all represented by a single formula. Accordingly, the present invention includes all of these isomers and mixtures of these isomers in various proportions.
- the present invention relates to a case where the thiazole derivative having the general formula (1), ( ⁇ ) or (II) or a salt thereof forms a solvate (for example, a hydrate). Included.
- R 1X is preferably P (0) (OH), ⁇ ( ⁇ ), or
- OCH P (O) (OH) is OCH P (O) (OH), and more preferably OCH P (O) (OH).
- R 1Y is preferably a hydrogen atom, a fluorine atom, or OCH P ( ⁇ ) ( ⁇ H), more preferably
- R 1 is preferably P (0) (0H), 0P (0) (0H), or OCH P (0) (0H), and more preferably
- R 2 is preferably a C1-6 alkyl group, C1-6 alkoxy group, C 1-6 dialkylamino group, Ha androgenic atom, a hydrogen atom, more preferably a C1-6 alkyl group or hydrogen An atom, particularly preferably a methyl group or a hydrogen atom.
- R 3 is preferably a C1-6 alkyl group, a C1-6 alkoxy group, a dialkylamino group, an acyl group, a halogen atom, or a hydrogen atom, more preferably a hydrogen atom, a C1-6 alkyl group, A C1-6 alkoxy group, a dialkylamino group, and an acyl group, particularly preferably an ethyl group, a methyl group, or a methoxy group.
- R 4 is preferably a hydrogen atom.
- X is preferably methylene.
- a 6-alkyl group and a C3-C10 cycloalkyl group more preferably a methyl group, an ethyl group, a propyl group, and a cyclopropyl group.
- a 6-alkyl group and a C3-C10 cycloalkyl group more preferably a methyl group, an ethyl group, a propyl group, and a cyclopropyl group.
- Substituent group a is preferably a C1-6 alkoxy group, a hydroxyl group, an amino group, a monoalkylamino group, or a dialkylamino group when the Cl-6 aliphatic acyl group of R 2 is substituted. .
- substituent group a when replacing the C 1-6 aliphatic Ashiru group R 3, preferably, C1-6 alkyl Le group, C1-6 alkoxy group, a hydroxyl group, an amino group, a monoalkylamino Group, a dialkylamino group.
- Substituent group a is preferably a C1-6 alkyl group, a C1-6 neuroalkyl group, a C1-6 alkoxy group, a hydroxyl group, an amino group, when the C6-10 aryl group of R 2 is substituted.
- Dialkylamino group is preferably a C1-6 alkyl group, a C1-6 neuroalkyl group, a C1-6 alkoxy group, a hydroxyl group, an amino group, when the C6-10 aryl group of R 2 is substituted.
- a acyl group and a halogen atom A acyl group and a halogen atom.
- Substituent group a is preferably a C1-6 alkyl group, a C1-6 neuroalkyl group, a C1-6 alkoxy group, a hydroxyl group, an amino group, when the C6-10 aryl group of R 3 is substituted.
- Dialkylamino group is preferably a C1-6 alkyl group, a C1-6 neuroalkyl group, a C1-6 alkoxy group, a hydroxyl group, an amino group, when the C6-10 aryl group of R 3 is substituted.
- a acyl group and a halogen atom A acyl group and a halogen atom.
- substituent group a when replacing the 4-10 membered heterocyclic group containing nitrogen R 2, the same or different 1-3 heteroatom selected from oxygen and sulfur force, preferably , C1-6 alkyl group, C1-6 neuroalkyl group, C1-6 alkoxy group, hydroxyl group, amino group, dialkylamino group
- a acyl group and a halogen atom A acyl group and a halogen atom.
- substituent group a when replacing the 4-10 membered heterocyclic group containing nitrogen R 3, the same or different 1-3 heteroatom selected from oxygen and sulfur force, preferably , C1-6 alkyl group, C1-6 neuroalkyl group, C1-6 alkoxy group, hydroxyl group, amino group, dialkylamino group
- a acyl group and a halogen atom A acyl group and a halogen atom.
- Substituent group a is a nitrogen, oxygen and sulfur force of R 2
- a 4- to 10-membered heterocyclic C1-C6 alkyl group containing 1-3 identical or different hetero atoms is selected
- Substituent group a is a nitrogen, oxygen and sulfur force of R 3
- a 4- to 10-membered heterocyclic C1-C6 alkyl group containing 1-3 identical or different hetero atoms is selected
- Substituent group a is the same or different selected from nitrogen, oxygen and sulfur power of R 2 1-3
- a 4- to 10-membered heterocyclic C1-C6 alkylamino group containing a hetero atom is substituted, a C1-6 alkyl group, a C1-6 haloalkyl group, a C1-6 alkoxy group, a hydroxyl group, an amino group is preferable.
- a dialkylamino group, an acyl group, and a halogen atom is preferable.
- Substituent group a substitutes for a nitrogen, oxygen and sulfur power of R 3 and a 4-10 membered heterocyclic C1-C6 alkylamino group containing 1-3 identical or different heteroatoms.
- a C1-6 alkyl group, a C1-6 haloalkyl group, a C1-6 alkoxy group, a hydroxyl group, an amino group, a dialkylamino group, an acyl group, and a halogen atom are preferable.
- thiazole derivative having the general formula 0), ( ⁇ ) or (II) or a pharmacologically acceptable salt thereof according to the present invention include the compounds exemplified below. However, the present invention is not limited to the following exemplary compounds.
- R 1 A is OCH P (0) (OH)
- B is CF P (0) (OH)
- C CH P (0) (OH)
- D is OP (0) (OH)
- E P (O)
- the preferred ones are 1-1, 1-6, 1-7, 1-8, 1-10, 1-11, 1-12, 1-13 , 1-1 4, 1-15, 1-23, 1-24, 1-32, 1-34, 1-35, 1-37, 1-38, 1-40, 1-41, 1-49, 1-50, 1-5 1, 1-52, 1-53, 1-54, 1-55, 1-56, 1-57, 1-58, 1-59, 1-60, 1-61, 1 -64, 1-65, 1-7 0, 1-71, 1-76, 1-77, 1-78, 1-80, 1-81, 1-82, 1-92, 1-93, 1- 94, 1-95, 1-96, 1-9 7, 1-98, 1-99, 1-100, 1-101, 1-105, 1-149, 1-160, 1-164, 1-182 1-198, 1-199, 1-200, 1-201, 1-202, 1-203, 1-204, 1-205, 1-206, 1-207, 1-209, 1-231, 1 -243, 1-244, 1-245, 1-246, 1-252,
- the compound having the following general formula (II) of the present invention can be produced, for example, by the following method using a known compound as a starting material.
- the compound having the following general formula (I) or ( ⁇ ) can be produced according to the production method of the compound having the general formula (II).
- R lx , R 1Y , R 4 and X have the same meaning as described above.
- R 1A , R 1Ba and R 1Bb are the same or different and each represents a phosphoric acid group or a phosphonic acid group, wherein R 1 may be protected by a protective group
- R 2A, R 2Ba ⁇ beauty R 2Bb are the same or different, when shown a R 2 where (R 2 has an Amino group or a hydroxyl group, ⁇ A Amino group or hydroxyl group may be protected by a protecting group)
- R 3Ba and R 3Bb are the same or different, when shown a R 3 which (R 3 has an Amino group or a hydroxyl group, the Amino group or R 4Ba and R 4Bb are the same or different and each represents R 4 (when R 4 has an amino group or a hydroxyl group, the amino group or hydroxyl group is L 1 represents a hydrogen atom, a leaving group or R 3Bb , and Y, L 2a and L 2b may be
- the "protecting group" of a phosphate group in the definition of R 1A , R 1Ba and R 1Bb is a protecting group of a phosphate group used in the field of synthetic organic chemistry.
- the method is not particularly limited as long as it is performed.
- the method described in Green's Watts, "Protective groups in organic synthesis (3fe)" (Wiley-Interscience, USA) be able to.
- Preferable examples include a C1-6 alkyl group such as a methyl group and an ethyl group, and a C2-6 alkyl group such as an aryl group, and more preferable is a C1-3 alkyl group. Particularly preferred is an ethyl group.
- R 2A The hydroxyl “protecting group” in the definitions of R 3Ba , R 3Bb , R 4Ba and R 4Bb is not particularly limited as long as it is a hydroxyl protecting group used in the field of synthetic organic chemistry.
- Protective groups in organic synthesis (JR) preferably a methyl group such as a methyl group. Examples thereof include a C1-6 alkyl group, a benzyl group, an aryl group, a silyl group and an acyl group.
- R 2A, R 2Ba, R 2Bb, R 3Ba, R 3Bb, the term "protecting group" of Amino groups in the definition of R 4Ba and R 4Bb, the field of organic synthetic chemistry For example, Green's Watts, Protective Groups in Organic Synthesis 3
- the "leaving group” is not particularly limited as long as it is a leaving group used in the field of organic synthetic chemistry.
- L 2a and L 2b are preferably a halogen atom such as a bromine atom
- Y is preferably a lower aliphatic sulfo-oxy group, an aromatic sulfo-oxy group or a halogen atom, and more preferably Is an aromatic sulfo-loxy group.
- the step of producing the compound (ii) of the present invention comprises the following two steps.
- Step A is a step of constructing a phosphate group moiety of compound (II).
- Step B is a step for producing a compound (II) of the present invention by constructing a thiazole ring in the right part of the intermediate (2) obtained in Step A, and depending on the desired compound, Method a B method can be selected.
- R 1A group is added to compound (1) which can be prepared according to a known method (for example, Tetrahedron Asymmetry 14, 2003, p. 481, J. Chem. Res. Miniprint 11, 1995, p. 2544). Is a process for producing the compound (2).
- the ketone of compound (II) can be protected and deprotected as necessary, and the alcohol moiety can be converted to a halogenated group or a leaving group as necessary.
- a protecting group for ketones a protecting group generally known in the art of this field, for example, “Protective groups in organic synthesis", Green, by Watts, Protective groups in organic synthesis, Wiley—Interscience) [Can be used by this method, and preferably a protecting group such as acyclic or cyclic ketal, dithioketal, monothioketal, 0-substituted cyanohydrin, hydrazone, semicarbazone, oxime. Can be mentioned.
- the halogenating agent is not particularly limited as long as it is a reagent usually used in a halogenated reaction. J, Comprehensive Organic Transformation Larlock, V and H)
- bromination hydrogen bromide, phosphorus tribromide, carbon tetrabromide-triphenylphosphine, N-prosuccinimide-trif is preferred.
- phenylphosphine and chlorination preferred examples include hydrogen chloride, tetrasalt-carbon-triphenylphosphine, and N-chlorosuccinimide-triphenylphosphine.
- R 1 is OCH P (0) (OH) or ⁇ ( ⁇ )
- Reagent YR 1A may be generated in the reaction system.
- the solvent to be used is not particularly limited as long as it does not inhibit the reaction and dissolves the starting material, but is preferably a solvent such as jetyl ether, diisopropyl ether, tetrahydrofuran, dioxane or dimethoxyethane.
- Ethers formamide, ⁇ , ⁇ -dimethylformamide, ⁇ , ⁇ -dimethylacetamide, ⁇ ⁇ ⁇ -methyl-2-pyrrolidone, ⁇ -methylpyrrolidinone, amides such as hexamethylphosphorotriamide, dichloromethane, Halogenated hydrocarbons such as black mouth form, carbon tetrachloride, dichloroethane, black mouth benzene and dichlorobenzene, more preferably halogenated hydrocarbons, ethers or amides, most preferably Tetrahydrofuran, dimethoxyethane or ⁇ , ⁇ -dimethylformamide.
- the reaction is preferably carried out in the presence of a base, and the base to be used is not particularly limited as long as it is a base used in a usual alkylation reaction.
- a base for example, lithium carbonate, sodium carbonate, carbonate Alkali metal carbonates such as potassium; alkali metal bicarbonates such as lithium hydrogen carbonate, sodium hydrogen carbonate, potassium hydrogen carbonate; alkali metal hydrides such as lithium hydride, sodium hydride, potassium hydride; water Alkali metal hydroxides such as lithium oxide, sodium hydroxide, potassium hydroxide; alkaline metal alkoxides such as lithium methoxide, sodium methoxide, sodium ethoxide, potassium t-butoxide Or triethylamine, tributylamine, diisopropylethylamine, N-methylmorpholine, pyridine Lysine, 4- (N, N-dimethylamino) pyridine, ⁇ , ⁇ -dimethylaline, ⁇ , ⁇ -
- the reaction time is 0.5 to 24 hours, preferably 0.5 to 12 hours.
- R 1 is CH P (0) (OH)
- the hydroxyl group at the R 1 site of compound (1) is converted to the leaving group by a conventional method.
- the leaving group can be introduced according to a conventional method without particular limitation as long as it is a leaving group used in the field of synthetic organic chemistry.
- Preferred is a lower aliphatic sulfonyloxy group, an aromatic sulfo-loxy group or a halogen atom, and more preferred is a halogen atom.
- the phosphonylation is preferably performed in the presence of a solvent, and the solvent used is not particularly limited as long as it does not inhibit the reaction and dissolves the starting material.
- Ethers such as diisopropyl ether, tetrahydrofuran, dioxane, dimethoxyethane, formamide, ⁇ , ⁇ -dimethylformamide, ⁇ , ⁇ -dimethylacetamide, ⁇ -methyl-2-pyrrolidone, ⁇ -methylpyrrolidinone, hexane
- Amides such as methyl phosphorotriamide, sulfoxides such as dimethyl sulfoxide, and more preferably tetrahydrofuran, dimethyl sulfoxide or ⁇ , ⁇ -dimethylformamide.
- this reaction can be carried out without using a solvent.
- the reagent used is trialkyl phosphite, preferably triethyl phosphite.
- the reaction temperature is 0 ° C to 200 ° C, preferably 50 ° C to 180 ° C.
- the reaction time is 0.5 to 24 hours, preferably 0.5 to 12 hours.
- R 1 is P (0) (OH)
- the compound (1) is converted to a hydroxyl group at the R 1 site by a conventional method.
- the leaving group can be introduced according to a conventional method without particular limitation as long as it is a leaving group used in the field of organic synthetic chemistry.
- it is a C 1-3 alkylsulfo-oxy group, a C1-3 haloalkyl sulfo-oxy group, an aromatic sulfo-oxy group or a halogen atom, more preferably a trifluoromethane sulfo-oxy group, a halogen atom.
- the solvent used in the phosphonylation does not inhibit the reaction and does not dissolve the starting material.
- ethers such as jetyl ether, diisopropyl ether, tetrahydrofuran, dioxane, dimethoxyethane, formamide, ⁇ , ⁇ -dimethylformamide, ⁇ , ⁇ -dimethylacetate.
- Amides ⁇ -methyl-2-pyrrolidone, ⁇ -methylpyrrolidinone, amides such as hexamethyl phosphorotriamide, and sulfoxides such as dimethyl sulfoxide, more preferably tetrahydrofuran, dimethyl sulfoxide Sid or ⁇ , ⁇ -dimethylformamide.
- the reaction is preferably carried out in the presence of a catalyst, and the catalyst used is not particularly limited as long as it is a catalyst used in a normal phosphorylation reaction, but palladium acetate, bistriphenyl are not limited.
- a transition metal catalyst such as phosphine palladium dichloride or tetrakistriphenylphosphine palladium, preferably tetrakistriphenylphosphine palladium.
- the reaction is preferably carried out in the presence of a base, and the base to be used is not particularly limited as long as it is a base used for ordinary catalytic reaction, but for example, lithium carbonate, sodium carbonate, etc.
- Alkali metal carbonates such as potassium carbonate; alkali metal bicarbonates such as lithium hydrogen carbonate, sodium hydrogen carbonate, potassium hydrogen carbonate; alkali metals such as lithium hydride, sodium hydride, potassium hydride Hydrides; alkali metal hydroxides such as lithium hydroxide, sodium hydroxide, potassium hydroxide; alkali metals such as lithium methoxide, sodium methoxide, sodium ethoxide, potassium t-butoxide Alkoxides; or triethylamine, tributylamine, diisopropylethylamine, N-methylmorpholine, pyridine , 4- (N, N-dimethylamino) pyridine, ⁇ , ⁇ -dimethylaline, ⁇ , ⁇ -
- the reaction temperature is 0 ° C to 200 ° C, preferably 50 ° C to 150 ° C.
- the reaction time is 0.5 to 24 hours, preferably 0.5 to 12 hours.
- the solvent used is not particularly limited as long as it does not inhibit the reaction and dissolves the starting material.
- ethers such as jetyl ether, diisopropyl ether, tetrahydrofuran, dioxane and dimethoxyethane are preferred, and tetrahydrofuran is more preferred.
- the fluorinating agent is not particularly limited as long as it is usually used in an electrophilic fluorination reaction.
- aromatic sulfonimides such as N-fluorobenzenesulfonimide, N-fluoropyrididine, etc. -Um salt may be mentioned, and N-fluorobenzenesulfonimide is preferred.
- the reaction is preferably carried out in the presence of a base, and the base used is not particularly limited as long as it is a base used in a usual electrophilic fluorination reaction.
- a base for example, lithium isopropyl pyramide is used.
- Metal dialkylamides such as (LDA) and metal bis (trialkylsilyl) amides such as lithium bis (trimethylsilyl) amide.
- the reaction temperature is -78 ° C to 50 ° C, preferably -78 ° C to room temperature.
- the reaction time is 0.5 to 12 hours, preferably 0.5 to 6 hours.
- This step is a step for producing the compound (3), and is achieved by subjecting the compound (2a) produced in the step A to a carbolation reaction in the presence of a catalyst and an alcohol.
- the solvent used is not particularly limited as long as it does not inhibit the reaction and dissolves the starting material, but is preferably jetyl ether, diisopropyl ether, tetrahydrofuran, dioxane, dimethoxyethane, diethylene glycol dimethyl ether.
- Ethers such as formamide, ⁇ , ⁇ -dimethylformamide, ⁇ , ⁇ -dimethylacetamide, ⁇ ⁇ ⁇ -methyl-2-pyrrolidone, ⁇ -methylpyrrolidinone, and amides such as hexamethylphosphorotriamide I can make it. More preferred is ⁇ , ⁇ -dimethylformamide.
- the carbonylation reagent to be used is not particularly limited as long as it is used as a reagent in a normal carbo-Louis reaction, and examples thereof include carbon monoxide.
- the alcohol used is not particularly limited as long as it is used in a normal carbo-Louis reaction.
- C1-6 alcohol such as methanol and ethanol
- Aromatic alcohols such as benzyl alcohol
- C1-3 alcohols such as allylic alcohol
- halogenated alkyl alcohols such as 1, 1, 1 trichloroethanol
- trialkyl such as trimethylsilylethanol
- silylalkyl alcohols preferably C1-6 alcohol, aromatic alkyl alcohol, and trialkylsilylalkyl alcohol, and more preferably trialkylsilylalkyl alcohol.
- the catalyst to be used is not particularly limited as long as it is used as a catalyst in a normal carbolation reaction, but tetrakistriphenylphosphine palladium, palladium chloride, palladium acetate, bistriphenylphosphine. Transition metal catalysts such as palladium dichloride, and optionally triphenylphosphine, 1,3 bis (diphenylphosphino) propane (dppp), 1,1'-bis (diphenylphosphino) phenocene (dppf ), And the like. More preferably, the palladium acetate, the ligand is 1,3 bis (diphenol phosphino) propane (dppp).
- the base to be used is not particularly limited as long as it is used as a base in a usual carbolation reaction.
- an alkali such as cesium carbonate, sodium carbonate, potassium carbonate, lithium carbonate, Alkali metal bicarbonates such as metal carbonates, sodium bicarbonate, potassium bicarbonate, lithium bicarbonate; alkali metal hydrides such as lithium hydride, sodium hydride, potassium hydride;
- Inorganic bases such as sodium hydroxide, lithium hydroxide, barium hydroxide, alkali metal hydroxides such as lithium hydroxide, N-methylmorpholine, triethylamine, tripropylamine, tributylamine, diisopropyl Ethylamine, dicyclohexylamine, N-methylpiperidine, pyridine, 4-pyrrolidinopyridine, picoline 4- (N, N-dimethylamino) pyridine, 2,6-di (t-butyl) -4-methylpyridine, quino
- the reaction temperature is 0 ° C to 150 ° C, preferably 50 ° C to 100 ° C.
- the reaction time is 0.5 to 48 hours, preferably 2 to 24 hours.
- the P 1 moiety can be converted into various protecting groups as necessary.
- This step is a step for producing compound (4), and is achieved by halogenating compound (3) in an inert solvent in the presence or absence of a base.
- the solvent to be used is not particularly limited as long as it does not inhibit the reaction and dissolves the starting material, but alcohols such as methanol and ethanol, esters such as ethyl acetate, or dichloromethane, Halogenated hydrocarbons such as form can be mentioned, and preferred is chloroform, dichloromethane, ethyl acetate or ethanol.
- the halogenating agent to be used is not particularly limited, and examples thereof include halogenating agents described in "Comprehensive 0 rganic Transformation” (Larlock, VCH). Are bromine, odorous copper (11), and phenyltrimethylammotribromide.
- the base to be used is not particularly limited as long as it does not affect the portion other than the halogen atom substitution position in the compound (3), and examples thereof include lithium carbonate, sodium carbonate and potassium carbonate.
- the reaction temperature is 0 ° C to 100 ° C, preferably 0 ° C to 50 ° C.
- the reaction time is 0.5 hour to 48 hours, preferably 0.5 hour to 12 hours.
- This step is a step of producing compound (6), and compound (4) is added to an additive in an inert solvent. It is achieved by reacting with compound (5a) in the presence or absence of.
- the inert solvent used is not particularly limited as long as it is inert to this reaction.
- aliphatic hydrocarbons such as hexane, heptane, lignin, petroleum ether; benzene Aromatic hydrocarbons such as toluene, xylene; halogenated hydrocarbons such as dichloromethane, chloroform, carbon tetrachloride, dichloroethane, black benzene, dichlorobenzene; ethyl formate, ethyl acetate, propyl acetate, acetic acid Esters such as butyl and ethyl carbonate; ethers such as jetyl ether, diisopropyl ether, tetrahydrofuran, dioxane, dimethoxyethane, and diethylene glycol dimethyl ether; acetone, methyl ethyl ketone, methyl isobutyl ketone, isophor
- the additive used in the reaction is not particularly limited as long as it is a salt that accelerates the progress of the reaction.
- alkali metal acetates such as lithium acetate, sodium acetate, and potassium acetate
- lithium chloride Alkaline metal hydrochlorides such as sodium chloride and potassium chloride
- Alkali metal sulfates such as lithium sulfate, sodium sulfate and potassium sulfate
- Alkaline earth metal acetates such as magnesium acetate and calcium acetate
- Alkaline earth metal hydrochlorides such as magnesium and calcium chloride
- alkaline earth metal sulfates such as magnesium sulfate and calcium sulfate or triethylamine, tributylamine, diisopropylethylamine, ⁇ -methylmorpholine, pyridine 4- ( ⁇ , ⁇ -dimethylamino) pyridine,, ⁇ -dimethylamino, ⁇ , ⁇ - Jetylaline, 1,5
- the reaction temperature is 0 ° C to 100 ° C, preferably 50 ° C to 100 ° C.
- the reaction time is 1 hour to 48 hours, preferably 1 hour to 12 hours.
- This step is a step for producing the compound (7) and can be achieved by converting the carboxylic acid ester of the compound (6) into strong rubonic acid.
- a allyl group When a allyl group is used as a protecting group for carboxylic acid, it is removed by adding a nucleophile such as pyrrolidine in the presence of a palladium catalyst such as tetrakistriphenylphosphine palladium. be able to.
- a nucleophile such as pyrrolidine
- a palladium catalyst such as tetrakistriphenylphosphine palladium.
- This step is a step for producing the compound (8a), and is achieved by converting the carboxylic acid of the compound (7) to R 3Ba .
- the method power of Ba-5-l to Ba-5-4 can be selected.
- R 3Ba is an amino group, a CI-C6 alkylamino group, a CI-C6 dialkylamino group, a CI-C6 aliphatic asilamino group, nitrogen, oxygen and sulfur power.
- a 4- to 10-membered heterocyclic C1-C6 alkylamino group or an acyl group substituted with a C1-6 strength rubermoylalkylamino group by condensing the compound (7) and the amine corresponding to HR 3Ba Can also be manufactured. That is, the reaction is carried out by reacting a carboxylic acid with an active agent in an inert solvent and reacting with the amine compound after activation or simultaneously with the activation. In addition, you may add a base as needed.
- the solvent to be used is not particularly limited as long as it does not inhibit the reaction and dissolves the starting material, but is preferably dichloromethane, chloroform, carbon tetrachloride, dichloroethane, chloroform, Halogenated hydrocarbons such as chlorobenzene, ethers such as jetyl ether, diisopropyl ether, tetrahydrofuran, dioxane, dimethoxyethane, diethylene glycol dimethyl ether; formamide, ⁇ , ⁇ -dimethylformamide, Examples include amides such as ⁇ , ⁇ -dimethylacetamide, ⁇ -methyl-2-pyrrolidone, ⁇ -methylpyrrolidinone, and hexamethylphosphorotriamide. More preferred is ⁇ , ⁇ -dimethylolenolemamide, tetrahydrofuran or dichloromethane.
- Examples of the activator used include diimidazole compounds such as 1,1'-oxalyldiimidazole and ⁇ , ⁇ '-carbodiimidazole; ⁇ , ⁇ '-disuccine Succinic compounds such as imidyl carbonate; ⁇ , ⁇ '-bis (2-oxo-3-oxazolidyl) phosphoramidyl chloride compounds such as phosphoramidic chloride; ⁇ , ⁇ '- Disuccinimidyl oxalate (DSO), ⁇ , ⁇ '-Diphtal imidooxalate (DPO), ⁇ , ⁇ '-bis (norbornene-russuccinimidyl) oxalate ( ⁇ ), 1,1'-bis (benzo Triazolyl) oxalate ( ⁇ ), 1,1'-bis (6-chlorobenzobenzotriazolinole) oxalate (BCTO), 1,1'-bis (6-trifluor
- the base used is not particularly limited as long as it is used as a base in a normal reaction.
- the reaction temperature is from -10 ° C to 60 ° C, preferably room temperature.
- the reaction time is 30 minutes to 24 hours, preferably 2 hours to 20 hours.
- R 3Ba contains an optionally substituted amino group
- an ordinary method such as an alkyl group or an acyl group.
- an alcohol can be isolated in the presence of alcohol, and introduction of an amino group can be achieved by deprotection.
- the solvent used in the reaction for introducing the amino group is not particularly limited as long as it does not inhibit the reaction and dissolves the starting material, but preferably hexane, heptane, lignin, petroleum Aliphatic hydrocarbons such as ethers, aromatic hydrocarbons such as benzene, toluene, xylene, halogenated hydrocarbons such as dichloromethane, chloroform, carbon tetrachloride, dichloroethane, benzene, dichlorobenzene Ethers such as dimethyl ether, tetrahydrofuran, dioxane, dimethoxyethane, diethylene glycol dimethyl ether; formamide, dimethylformamide, dimethylacetamide, methyl-2-pyrrolidone, N-methylpyrrolidinone, hexane Amides such as methyl phosphorotriamide There can be mentioned. More preferably, it is toluene, benzene, dimethylol
- the alcohol used in the reaction for introducing an amino group is not particularly limited.
- methanol, ethanol, propanol, C1-6 alcohol such as t-butyl alcohol, and fragrance such as benzyl alcohol.
- Alkyl alcohols, allylic alcohols, 1, 1, 1 C1-6 alcohol, aromatic alkyl alcohol, aryl alcohol, and trialkylsilylalkyl alcohol are preferable, and tbutyl alcohol and aryl alcohol are more preferable.
- the reagent used in the reaction for introducing an amino group is diphenyl phosphate azide (DPPA).
- Metal azides such as sodium azide, hydrogen azide, and the like. Diphenyl phosphate azide is preferred.
- the reaction temperature is 0 ° C to 150 ° C, preferably 0 ° C to 100 ° C.
- the reaction time is 30 minutes to 48 hours, preferably 30 minutes to 24 hours.
- R 3Ba is a group containing an optionally substituted acyl group
- the carboxylic acid is converted to an alcohol by a conventional method, further acidified to an aldehyde, and then the corresponding substituent is introduced by an aldol reaction to form an alcohol. And can be produced by acidifying the alcohol by a conventional method.
- the reducing agent used in the reduction of carboxylic acid is not particularly limited as long as it is usually used for the reduction reaction of carboxylic acid.
- borane such as borane-tetrahydrofuran and borane-dimethylsulfide.
- Complex, borane, diborane and the like, and preferred is a borane complex.
- the solvent used in the reduction of the carboxylic acid is not particularly limited as long as it does not inhibit the reaction and dissolves the starting material to some extent.
- jetyl ether, diisopropyl ether, tetrahydrofuran, dioxane , Ethers such as dimethoxyethane and diethylene glycol dimethyl ether are preferred, and dimethoxyethane or tetrahydrofuran is preferred.
- the reaction temperature is 0 ° C to 100 ° C, preferably 0 ° C to room temperature.
- the reaction time is 0.5 to 48 hours, preferably 0.5 to 24 hours.
- the solvent used in the oxidation reaction is not particularly limited as long as it does not inhibit the reaction and dissolves the starting material to a certain extent.
- hexane, heptane, rig in Aliphatic hydrocarbons such as petroleum ether, aromatic hydrocarbons such as benzene, toluene and xylene, ethers such as jetyl ether, diisopropyl ether, tetrahydrofuran, dioxane, dimethoxyethane, and diethylene glycol dimethyl ether
- Halogenated hydrocarbons such as dichloromethane, chloroform, carbon tetrachloride, dichloroethane, black benzene, and dichlorobenzene are exemplified, and dichloromethane or chloroform is preferred.
- the oxidizing agent used in the oxidation reaction is not particularly limited as long as it is usually used for the acid-oxidation reaction to alcoholic ketones.
- manganese dioxide, Dess Martin reagent, dimethylsulfoxide examples include oxalyl chloride, dimethylsulfoxide N-chlorosuccinimide (NCS), and pyridi-mukuroguchi chromate (PCC).
- the reaction temperature is 0 ° C to 100 ° C, preferably room temperature.
- the reaction time is 0.5 to 48 hours, preferably 0.5 to 24 hours.
- the solvent used in the aldol reaction is not particularly limited as long as it does not inhibit the reaction and dissolves the starting material to some extent.
- hexane, heptane, rigin, petroleum ether Aliphatic hydrocarbons such as benzene, toluene, aromatic hydrocarbons such as xylene, jetyl ether, diisopropyl ether, tetrahydrofuran, dioxane, dimethoxyethane, ethers such as diethylene glycol dimethyl ether, dichloromethane , Halogenated hydrocarbons such as black mouth form, carbon tetrachloride, dichloroethane, black mouth benzene, and dichlorobenzene, and preferred are jetyl ether, dimethoxyethane, and tetrahydrofuran.
- Reagents used in the aldol reaction include organic lithium reagents such as alkyllithium, Grignard reagents such as alkylmagnesium halides, organic zinc reagents such as alkylzinc, organotin reagents such as alkyltin, and alkylsilanes.
- organic lithium reagents and the like can be mentioned.
- Organolithium reagents, Grignard reagents, and organic cage reagents are preferred, and Lewis acids, metal salts, and the like may coexist if necessary.
- the reaction temperature is -100 ° C to 100 ° C, preferably -78 ° C to room temperature.
- the reaction time is 0.5 to 48 hours, preferably 0.5 to 24 hours.
- R 3Ba is substituted and is a group containing an aminoalkyl group
- the aldehyde obtained by oxidation after the reduction step in Step Ba-5-3-3 is converted to the corresponding amino acid in the presence of a reducing agent. It can also be produced by carrying out a reductive amination reaction.
- the reducing agent used is not particularly limited as long as it is usually used for the reduction reaction.
- borohydride such as sodium borohydride, sodium triacetoxyborohydride, sodium cyanoborohydride, etc.
- hydride reagents such as alkali metals, preferably sodium triacetoxyborohydride and sodium cyanoborohydride.
- the solvent to be used is not particularly limited as long as it does not inhibit the reaction and dissolves the starting material to some extent.
- dichloromethane, chloroform, carbon tetrachloride, dichloroethane examples include halogenated hydrocarbons such as benzene and dichlorobenzene, alcohols such as methanol, ethanol and isopropanol, and ethers such as dioxane, jetyl ether and tetrahydrofuran, preferably dichloromethane, methanol or tetrahydrofuran. It is.
- the reaction temperature is from 0 ° C to the boiling point of the solvent used, and preferably from 0 ° C to room temperature.
- the reaction time is 0.5 to 48 hours, preferably 0.5 to 24 hours.
- This step is a step for producing the target compound (II).
- the hydroxyl group, amino group, carboxyl group and Z or phosphate group of the compound (8a) is protected, it should be deprotected according to a conventional method. It is achieved from Yuko. When there is no protecting group, this step is unnecessary.
- the target compound (II) can also be produced by this method. [0171] (Step Bb-1)
- This step is a step for producing compound (9), and is achieved by treating compound (2b) produced in step A under the same conditions as in Step Ba-2.
- This step is a step for producing compound (10), and is achieved by treating compound (9) under the same conditions as in Step Ba-3.
- This step is a step of producing compound (8b), and is achieved by converting L 1 of compound (10) to R.
- a cross-coupling reaction is performed in the presence of a catalyst, and then, if necessary, a conventional hydrolysis reaction, a hydrolyzate reduction reaction, an oxime reaction, a halogenation and a subsequent amination. This is achieved by performing a reaction or the like.
- the solvent used in the cross-coupling reaction is not particularly limited as long as it does not participate in this reaction, but alcohols such as methanol, ethanol and isopropanol, jetyl ether, tetrahydrofuran and dioxane.
- Ethers such as ethyl acetate, esters such as propyl acetate, formamide, dimethylformamide, dimethylacetamide, amides such as N-methyl-2-pyrrolidone, hexamethylphosphorotriamide, water, or these
- These mixed solvents are preferable, and alcohols, ethers, water, amides, or mixed solvents thereof are more preferable.
- the catalyst to be used is not particularly limited as long as it is usually used in a cross-coupling reaction, and preferably tetrakistriphenylphosphine palladium, dichlorodiphenylphosphine palladium, palladium acetate.
- Palladium catalysts such as nickel catalyst, nickel catalysts such as tetrakistriphenylphosphine nickel and nickel chloride, and copper catalysts such as copper iodide.
- Reagents for the cross-coupling reaction include arylboron acids such as phenylboric acid, heteroarylboronic acids such as pyridylboric acid, arylboronates such as phenolboric acid pinacol ester, and tributylphenol tin.
- arylboron acids such as phenylboric acid
- heteroarylboronic acids such as pyridylboric acid
- arylboronates such as phenolboric acid pinacol ester
- tributylphenol tin Trikylalkenyl such as trialkylaryl tin, tributyl beer tin, tributyl (1 ethoxy butyl) tin
- cyanine compounds such as niltin, sodium cyanide, potassium cyanide, zinc cyanide and copper cyanide.
- the reaction temperature is 0 ° C to 150 ° C, preferably 50 ° C to 100 ° C.
- the reaction time is 0.5 to 48 hours, preferably 0.5 to 24 hours.
- This step is a step for producing the target compound (II) and can be achieved by treating the compound (8b) under the same conditions as in Step Ba-6.
- X is an oxo group, a hydroxyl group, or an alkylene group substituted by 1 or 2 with a halogen atom
- the synthesis of the target compound (II) can be achieved by introducing a carbocyclic group with an acid of the methylene moiety and subsequently reducing the introduced carbonyl group or halogenating the carbocyclic group. it can.
- the oxidizing agent to be used is not particularly limited as long as it is usually used for the acid-acid reaction, and examples thereof include acid-chromium, potassium permanganate, selenium dioxide, oxygen monometallic catalyst, and the like. , Selenium diacid.
- the solvent to be used is not particularly limited as long as it does not inhibit the reaction and dissolves the starting material to some extent.
- halogenated hydrocarbons such as dichloromethane, chloroform, carbon tetrachloride, etc.
- ethers such as jetyl ether, diisopropyl ether, tetrahydrofuran, dioxane and dimethoxyethane, or mixed solvents thereof with acetic acid, etc., and preferably a mixed solvent of ethers and acetic acid. is there.
- the reaction temperature is 0 ° C to 150 ° C, preferably 0 ° C to 100 ° C.
- the reaction time is 30 minutes to 48 hours, preferably 1 hour to 12 hours.
- the reducing agent to be used is not particularly limited as long as it is usually used for the reduction of ketone power alcohol, but for example, sodium borohydride, sodium triacetoxyborohydride, sodium cyanoborohydride and the like. Hydride such as alkali metal borohydride A reagent.
- the solvent to be used is not particularly limited as long as it does not inhibit the reaction.
- halogens such as dichloromethane, chloroform, carbon tetrachloride, dichloroethane, chloroform, and dichloromethane are used.
- Hydrocarbons, alcohols such as methanol and ethanol, ethers such as dioxane, ether and tetrahydrofuran, and dichloromethane, methanol and tetrahydrofuran are preferred.
- the reaction temperature is from 0 ° C to the boiling point of the solvent used, and preferably from 0 ° C to room temperature.
- the reaction time is 0.5 to 48 hours, preferably 0.5 to 12 hours.
- the halogenating agent used is not particularly limited as long as it is used for the halogenation of ketones.
- DAST jetylaminosulfur trifluoride
- DFI 2,2-difluorodimethylimidazolidine
- salt Examples include thiol, phosphorus pentachloride and phosphorus pentabromide.
- the solvent to be used is not particularly limited as long as it does not inhibit the reaction, but aromatic hydrocarbons such as benzene, toluene, xylene, dichloromethane, chloroform, carbon tetrachloride, dichloroethane. And halogenated hydrocarbons such as: jetyl ether, tetrahydrofuran, dioxane, ethers such as dimethoxyethane, and -tolyls such as acetonitrile.
- aromatic hydrocarbons such as benzene, toluene, xylene, dichloromethane, chloroform, carbon tetrachloride, dichloroethane.
- halogenated hydrocarbons such as: jetyl ether, tetrahydrofuran, dioxane, ethers such as dimethoxyethane, and -tolyls such as acetonitrile.
- the reaction temperature is from 0 ° C to the boiling point of the solvent used, preferably from room temperature to 100 ° C.
- the reaction time is 0.5 to 48 hours, preferably 0.5 to 24 hours.
- the target compound 0) can be converted to an acid addition salt when it has a basic group according to a conventional method, and is preferably a hydrochloride.
- the target compound is collected from the reaction mixture according to a conventional method. For example, neutralize the reaction mixture as appropriate, and if insolubles exist, remove by filtration, add water and an immiscible organic solvent such as ethyl acetate, wash with water, etc. The organic layer containing is separated, dried over anhydrous magnesium sulfate and the like, and then distilled off to remove the solvent.
- the obtained target product can be obtained by a conventional method such as recrystallization, reprecipitation, or a method usually used for separation and purification of organic compounds, such as adsorption column chromatography, distribution.
- Elution with an appropriate eluent by combining a method using a synthetic adsorbent such as column chromatography, a method using ion exchange chromatography, or a normal phase or reverse phase column chromatography using silica gel or alkyl silica gel. By doing so, it can be separated and purified.
- a synthetic adsorbent such as column chromatography, a method using ion exchange chromatography, or a normal phase or reverse phase column chromatography using silica gel or alkyl silica gel.
- the thiazole derivative having the general formula (1), ( ⁇ ) or (II) of the present invention, a pharmacologically acceptable salt thereof and an ester thereof are administered in various forms.
- the dosage form is not particularly limited, and is determined according to the patient's age, gender and other conditions, the degree of disease, etc.
- they are administered orally.
- it is administered intravenously alone or mixed with a normal fluid such as glucose or amino acid, and further administered alone intramuscularly, intradermally, subcutaneously or intraperitoneally as necessary.
- suppositories it is administered rectally. Oral administration is preferred.
- carriers When forming into a tablet form, conventionally known carriers can be widely used as carriers.
- carriers for example, lactose, sucrose, sodium chloride salt, glucose, urea, starch, calcium carbonate, kaolin, crystalline cellulose Excipients such as carboxylic acid, water, ethanol, propanol, simple syrup, glucose solution, starch solution, gelatin solution, binders such as carboxymethylcellulose, shellac, methylcellulose, potassium phosphate, polypyrrole pyrrolidone, Dry starch, sodium alginate, agar powder, laminaran powder, sodium bicarbonate, calcium carbonate, polyoxyethylene sorbitan fatty acid esters, sodium lauryl sulfate, monoglyceride stearate, starch, lactose, etc., sucrose, stearin , Cacao butter, decay additives such as hydrogenated oil, 4th Absorption promoters such as ammonia base, sodium lauryl sulfate, humec
- conventionally known carriers can be widely used as carriers, for example, excipients such as glucose, lactose, starch, cocoa butter, hydrogenated vegetable oil, kaolin, talc, Examples thereof include binders such as gum arabic powder, tragacanth powder, gelatin and ethanol, and disintegrants such as laminaran strength.
- excipients such as glucose, lactose, starch, cocoa butter, hydrogenated vegetable oil, kaolin, talc
- examples thereof include binders such as gum arabic powder, tragacanth powder, gelatin and ethanol, and disintegrants such as laminaran strength.
- conventionally known carriers can be widely used as carriers, such as polyethylene glycol, cacao butter, higher alcohols, higher alcohol esters, gelatin, semi-synthetic glycerides, etc. Can be mentioned.
- liquids and suspensions are preferably sterilized and isotonic with blood, and when formed into these liquids, emulsions and suspensions, Any of those commonly used in this field can be used as a diluent, such as water, ethyl alcohol, propylene glycol, ethoxylated isostearyl alcohol, polyoxylated isostearyl alcohol, polyoxyethylene sorbitan fatty acid esters and the like. Can do.
- a sufficient amount of sodium chloride, glucose, or glycerin may be included in the pharmaceutical preparation to prepare an isotonic solution, and normal solubilizing agents, buffers, soothing agents, etc. may be added. It may be added.
- a colorant if necessary, a colorant, a preservative, a fragrance, a flavoring agent, a sweetening agent and the like and other pharmaceuticals may be contained.
- the amount of the active ingredient-compound contained in the above-mentioned pharmaceutical preparation is not particularly limited and is appropriately selected over a wide range. Usually 1 to 70% by weight, preferably 1 to 30% by weight in the total composition Appropriate amounts are included.
- the dosage varies depending on symptoms, age, body weight, administration method, dosage form, etc., but is usually 0.001 mg / kg (preferably 0.01 mg / kg, more preferably 1 day) for adults per day. Is 0.1 mg / kg), and 200 mg / kg (preferably 20 mg / kg, more preferably 10 mg / kg) as the upper limit can be administered once or several times.
- the novel thiazole derivative and pharmacologically acceptable salt thereof which are the compounds of the present invention, have excellent FBPase inhibitory action, hypoglycemic action, lipid-lowering action, anti-inflammatory action, etc. Glycemia, glucose intolerance, obesity, hyperlipidemia, diabetic complications (e.g. retinopathy, cataract, nephropathy, peripheral nerve disorders, etc.), ischemic heart disease, arteriosclerosis (e.g.
- diabetes-like pathology liver cancer and the like and Z or a preventive agent (preferably a therapeutic agent for Zuria and Z or a preventive agent).
- Example 1 [(2 amino-7bromo-8H-indeno [1,2-d] [l, 3] thiazol-4-yl) oxy] methyl phosphonate (49.5 mg, 0.114 mmo) 1) was dissolved in dichloromethane (1.1 mL), bromotrimethylsilane (151 L, 1.14 mmol) was added thereto, and the mixture was stirred at room temperature for 4 hours under a nitrogen atmosphere.
- Example 2 [(3-Oxo 2,3 dihydro-1H-indene-4-yl) oxy] methylphosphonate jetyl (259 mg, 0.868 mmol) prepared in (2a) was dissolved in dichloromethane (8.0 mL). Then, trimethylammonium tribromide (392 mg, 1.04 mmol) was added thereto, and the mixture was stirred at room temperature for 4 hours under a nitrogen atmosphere. [0218] A saturated aqueous sodium hydrogen carbonate solution was added to the reaction mixture, and the mixture was extracted with dichloromethane.
- Example 2 [(2Amino-8H-indeno [1,2-d] [l, 3] thiazol-4-yl) oxy] methylphosphonate jetyl (59.3 mg, 0.167 mmol) prepared in Example 2 (2b) was used to give the title object compound (44.3 mg, yield 89%) according to the method described in Example 1 (lc).
- Example 2 [(3-Oxo 2,3 dihydro-1H-indene-4-yl) oxy] methyl phosphonate (28.3 g, 94.9 mmol) prepared in (2a) was dissolved in ethanol (500 mL). Thereto was added cupric bromide (44.3 g, 190 mmol), and the mixture was stirred at 50 ° C. for 2.5 hours.
- reaction solution was poured into a saturated aqueous sodium hydrogen carbonate solution, and the resulting solid was removed by Celite filtration. The filtrate was distilled off under reduced pressure and then extracted with ethyl acetate. The obtained organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was removed under reduced pressure.
- Formamide (40.5 g, 899 mmol) was dissolved in tetrahydrofuran (300 mL), to which was added diphosphorus pentasulfide (40. Og, 180 mmol) at 0 ° C, and the mixture was stirred at room temperature for 17 hours, filtered, and thioformamide A tetrahydrofuran solution was prepared.
- Example 3 Using (8H-indeno [1,2-d] [l, 3] thiazole-4-yloxy) methylphosphonate jetyl (19.8 g, 58.3 mmol) prepared in Example 3 (3b) The title object compound (16.2 g, yield 98%) was obtained according to the method described in (lc).
- Example 1 According to the method described in Example 1 (la) using 7-hydroxy-4-phenolindanone (338 mg, 1.51 mmol) prepared in Example 4 (4a), the title target compound (165 mg yield) 29%).
- Example 4 Example 2 was carried out using [(3 oxo 7-phenol-2,3-dihydro 1H indene-4-yl) oxy] methylphosphonate (157 mg, 0.419 mmol) prepared in (4b). According to the method described in (2b), the title object compound (76.5 mg, yield 42%) Got.
- Example 4 [(2 amino-7-Fel-8H-indeno [1,2d] [1,3] thiazol-4-yl) oxy] methylphosphonate (72.9 mg, 0. 169 mmol) was used to give the title object compound (50.2 mg, 79% yield) according to the method described in Example 1 (lc).
- Example 5 [(3-Oxo 7- (3 fluorophenyl) 2,3 dihydrone 1H-indene-4-yl) oxy] methyl phosphonate jetyl (208 mg, 0.530 mmol) prepared in Example 5 (5a) was used to give the title object compound (79.9 mg, 34% yield) according to the method described in Example 2 (2b).
- Example 5 ⁇ [2 amino 7- (3 fluorophenol) 8H-indeno [1,2-d] [l, 3] thiazol-4-yl] oxy ⁇ methylphosphonate prepared in (5b) (60.9 mg, 0.136 mmol) was used to give the title object compound (53.4 mg, 100% yield) according to the method described in Example 1 (lc).
- Example 6 ⁇ [2 amino 7- (4 fluorophenol) 8H-indeno [1,2-d] [l, 3] thiazol-4-yl] oxy ⁇ methylphosphonate jetyl prepared in (6b) (56.3 mg, 0.126 mmol) and the title compound (47.6 mg, 96% yield) was obtained according to the method described in Example 1 (lc).
- Example 7 ⁇ [2Amino-7- (3 hydroxyphenol) 8H-indeno [1,2-d] [l, 3] thiazole-4-yl] oxy ⁇ methylphosphonate prepared in Example 7 (7b) (118 mg, 0.264 mmol) was used to give the title object compound (45.7 mg, 44% yield) according to the method described in Example 1 (lc).
- Example 8 [2Amino-7- (4 hydroxyphenol) 8H-indeno [1,2-d] [l, 3] thiazol-4-yl] oxy ⁇ methylphosphonate jetyl prepared in Example 8 (8b) (88 3 mg, 0.198 mmol) was used to give the title compound (58.5 mg, 76% yield) according to the method described in Example 1 (lc).
- Example 9 In accordance with the method described in Example 1 (la) using 7 hydroxy 1 4- (3 methoxyphenol) indan 1-one (237 mg, 0.932 mmol) prepared in Example 9 (9a), The target compound (251 mg, yield 67%) was obtained.
- Example 9 ⁇ [2 amino-l 7- (3-methoxyphenyl) 8H-indeno [1,2-d] [l, 3] thiazol-4-yl] oxy ⁇ methylphosphonate (16) prepared in (9c) .6mg , 0.036 mmol) and the title compound (10.6 mg, 73% yield) was obtained according to the method described in Example 1 (lc).
- Example 10 Using the method described in Example 1 (la) using 7 hydroxy 1 4- (4 methoxyphenol) indan 1-one (107 mg, 0.421 mmol) prepared in Example 10 (10a) The target compound (47. Omg, yield 28%) was obtained.
- Example 10 ⁇ [2 amino 7- (4-methoxyphenol) 8H-indeno [1,2-d] [l, 3] thiazole-4-yl] oxy ⁇ methylphosphone prepared in (10c) The title compound (17.6 mg, yield 91%) was obtained according to the method described in Example 1 (lc) using decyl acid (22. lmg, 0.048 mmol).
- Example 11 (11a) was used in Example 1 (la) using 7-hydroxy-4-([(t-butoxycarbol) amino] -phenolindanone (409 mg, 1.20 mmol).
- the title object compound (334 mg, yield 57%) was obtained according to the method described.
- Example 11 [(2 amino 7- ⁇ [4— (t-butoxycarbol) amino] phenol ⁇ -811—indeno [1, 2-d] [l, 3 ] Thiazol-4-yl) oxy] methyl phosphonate (96. lmg, 0.176 mmol) and according to the method described in Example l (lc), the title compound (52.2 mg, yield 76) %).
- Example 12 (12b) ( ⁇ 7- [3 (trifluoromethyl) phenol]-3 oxo-2,3 dihydro-1H-indene-4-yl ⁇ oxy) methylphosphonate (28) 4 mg, 0.642 mmol) were used to give the title object compound (116 mg, 36% yield) according to the method described in Example 2 (2b).
- Example 12 (12c) ( ⁇ 2 amino 7- [3 (trifluoromethyl) phenol] —8H-indeno [1, 2-d] [l, 3] thiazol-4-yl ⁇ oxy) methylphosphone
- the title object compound (86.8 mg, yield 88%) was obtained according to the method described in Example 1 (lc) by using acid jetyl (ll lm g , 0.222 mmol).
- Example 2 (2b) was used using the decyl phosphate (8-oxo-5,6,7,8-tetrahydronaphthalene-2-yl) (402 mg, 1.35 mmol) prepared in Example 14 (14a). ) To give the title object compound (183 mg, yield 40%).
- Example 14 Performed using the phosphoric acid (2 amino-4,5 dihydronaphtho [1,2-d] [l, 3] thiazol-8 yl) jetyl (147 mg, 0.415 mmol) prepared in Example 14 (14b) The title compound (86.5 mg, yield 70%) was obtained according to the method described in Example 1 (lc).
- Example 15 Dissolve trifluoromethanesulfonic acid (8-oxo5,6,7,8-tetrahydronaphthalene-2-yl) (10. lg, 34.2 mmol) prepared in Example 15 (15a) in toluene (137 mL) Then, add ethylene glycol (38. OmL, 684 mmol) and p-toluenesulfonic acid monohydrate (1.18 g, 6.20 mmol), and attach to a Dean 'Stark apparatus and heat to reflux for 12 hours. did.
- Trifluoromethanesulfonic acid prepared in Example 15 (15b) (3, 4, 1, dihydro 2, H-spiro [1, 3 dioxolane 1, 2, 1, naphthalene] -7, 1 yl) (6. 25 g, 18.5 mmol) was dissolved in tetrahydrofuran (62 mL), to which a complex of [1,1,1bis (diphenylphosphino) pheocene] dichloropalladium and dichloromethane (1: 1) (755 mg, 0.925 mmol) and butylmagnesium bromide (0.97 M tetrahydrofuran solution) (28.6 mL, 27.8 mmol) were added, and the mixture was stirred under a nitrogen atmosphere for 18 hours.
- Example 15 3, 15, 1, dihydro-1, 7, hydroxymethyl 1, 2, H sp, prepared in Example 15 (15c) Dissolve [1,3 dioxolane-1,2,1'-naphthalene] (2.29 g, 10.4 mmol) in dichloromethane (52 mL). Under C cooling, triphenylphosphine (2.87 g, 10.9 mmol) and carbon tetrabromide (3.62 g, 10.9 mmol) were added thereto, and the mixture was stirred at 0 ° C. for 1 hour in a nitrogen atmosphere.
- Triethyl phosphite (7.33 mL, 42.7 mmol) was added thereto, and the mixture was stirred at 150 ° C for 4 hours.
- Example 15 (3,4,1-dihydro-1,2, H-spiro [1,3 dixolan-2,1'-naphthalene] -7'-yl) methylphosphonate jetyl (1.45 g) prepared in Example 15 (15d) 4.26 mmol) was dissolved in tetrahydrofuran (43 mL), and sodium bis (trimethylsilyl) amide (1. OM tetrahydrolafuran solution) (12.8 mL, 12.8 mmol) was added thereto under cooling at 78 ° C. The mixture was stirred at 78 ° C for 1 hour in a nitrogen atmosphere.
- N-fluorobenzenesulfonimide (4.70 g, 14.9 mmol) was added to the reaction solution, and the mixture was stirred for 3 hours while warming to room temperature.
- To the reaction solution was added 0.1N hydrochloric acid, and the mixture was extracted with dichloromethane. The obtained organic layer was washed with water and saturated brine, dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure.
- Example 15 Using (8-oxo-5,6,7,8-tetrahydronaphthalene-2-yl) difluoromethylphosphonate jetyl (873 mg, 2.63 mmol) prepared in Example 15 (15e), Example 2 According to the method described in (2b), the title object compound (726 mg, yield 71%) was obtained.
- Phosphoric acid (8-oxo-5,5 dimethyl-1,5,6,7,8-tetrahydronaphthalene-2-yl) prepared in Example 16 (16a) is used. Jetyl (1.07 g, 3.28 mmol) is used. Then, according to the method described in Example 2 (2b), the title object compound (495 mg, yield 39%) was obtained.
- the title compound (1.84 g, 97% yield) was obtained according to the method described in Example 15 (15c) using 7,1 g (3.71 g, 10. lmmol).
- Example 17 (4,4,1, dimethyl-3,4, -dihydro-2, H-spiro [1,3 dioxolane-1,2,1'-naphthalene] -7'-yl) methylphosphonic acid prepared in Example 17 (17d)
- the title compound (1.81 g, 93% yield) was obtained according to the method described in Example 15 (15e) using jetyl (2.08 g, 5.38 mmol).
- Example 17 (17e) (5,5 dimethyl-1-oxo-5,6,7,8-tetrahydronaphthalene-2-yl) difluoromethylphosphonate jetyl (1.52 g, 4.22 mmol) To give the title object compound (1.02 g, yield 58%) according to the method described in Example 2 (2b).
- 6-Methoxy-2-tetralone (8.44 g, 47.9 mmol) is dissolved in N, N dimethylformamide (200 mL), and sodium hydrogen carbonate (5.23 g, 120 mmol) is added to the solution, and the mixture is stirred at room temperature for 0.5 hour. After stirring, 1,4-Jodobutane (15.6 g, 50.3 mmol) was added and stirred for another 3 hours.
- magnesium sulfate (7.62 g, 63.3 mmol) and potassium permanganate (20. Og, 127 mmol) were added, and the mixture was stirred at room temperature for 21 hours.
- reaction solution was filtered, 50% (W / V) aqueous sodium thiosulfate solution was added to the filtrate, filtered again, and extracted with ethyl acetate. The obtained organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Animal Behavior & Ethology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Public Health (AREA)
- Engineering & Computer Science (AREA)
- Diabetes (AREA)
- Emergency Medicine (AREA)
- Endocrinology (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Gastroenterology & Hepatology (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Disclosed is a therapeutic and/or prophylactic agent for diabetes or the like which is excellent in activity and safety. A compound represented by the general formula (I) or a pharmacologically acceptable salt thereof: wherein R1X and R1Y independently represent a hydrogen atom, a halogen atom, a C1-C6 alkyl group, P(O)(OH)2, OP(O)(OH)2, CH2P(O)(OH)2, CF2P(O)(OH)2 or OCH2P(O)(OH)2; R2 and R3 independently represent a hydrogen atom, a C1-C6 aliphatic acyl group which may be substituted or the like; R4 represents a hydrogen atom, an amino group or the like; and X represents a C1-C3 alkylene group which may be substituted.
Description
明 細 書 Specification
チアゾール化合物 Thiazole compounds
技術分野 Technical field
[0001] 本発明は、新規なチアゾール誘導体及びその薬理上許容される塩に関する。 [0001] The present invention relates to a novel thiazole derivative and a pharmacologically acceptable salt thereof.
[0002] また、本発明は、優れたフルクトース 1,6-ビスホスファターゼ(FBPase)阻害作用、 血糖降下作用、脂質低下作用、抗炎症作用等を有するチアゾール誘導体及びその 薬理上許容される塩に関する。 [0002] The present invention also relates to a thiazole derivative having an excellent fructose 1,6-bisphosphatase (FBPase) inhibitory action, hypoglycemic action, lipid lowering action, anti-inflammatory action and the like, and a pharmacologically acceptable salt thereof.
[0003] 更に本発明は、チアゾール誘導体又はその薬理上許容される塩を有効成分として 含有する糖尿病、高血糖症、耐糖能不全、肥満症、高脂血症、糖尿病合併症 (例え ば、網膜症、白内障、腎症、末梢神経の障害等)、虚血性心疾患、動脈硬化 (例えば 、脳血管障害、狭心症,心筋梗塞、血圧の異常等)、妊娠糖尿病、クッシング症候群 、ステロイド糖尿病、糖原病、自律神経系の障害 (例えば、インポテンス、発汗異常、 起立性低血圧等)、外科的糖尿病様病態、肝臓癌等の治療薬及び Z又は予防薬( 好適には糖尿病の治療薬及び Z又は予防薬である。 )に関する。 [0003] Further, the present invention relates to diabetes, hyperglycemia, glucose intolerance, obesity, hyperlipidemia, diabetic complications (for example, retina) containing a thiazole derivative or a pharmacologically acceptable salt thereof as an active ingredient. , Cataract, nephropathy, peripheral nerve disorder, etc.), ischemic heart disease, arteriosclerosis (eg, cerebrovascular disorder, angina pectoris, myocardial infarction, abnormal blood pressure, etc.), gestational diabetes, Cushing syndrome, steroid diabetes, Glycogenosis, autonomic nervous system disorders (eg impotence, sweating abnormalities, orthostatic hypotension, etc.), surgical diabetes-like pathology, liver cancer and other therapeutic agents and Z or prophylactic agents (preferably diabetes therapeutic agents and Z or a preventive drug.
[0004] 更に、本発明は上記化合物を有効成分として含有する上記疾病の予防薬若しくは 治療薬、上記化合物を有効成分として含有する上記疾病の予防若しくは治療のため の組成物、上記疾病の予防若しくは治療のための医薬を製造するための上記化合 物の使用、又は上記化合物の薬理的な有効量を温血動物 (好適には人間である)に 投与する上記疾病の予防若しくは治療方法に関する。 [0004] Further, the present invention provides a preventive or therapeutic agent for the above-mentioned diseases containing the above-mentioned compound as an active ingredient, a composition for preventing or treating the above-mentioned disease containing the above-mentioned compound as an active ingredient, the prevention or the above-mentioned disease. The present invention relates to the use of the above-mentioned compound for producing a medicament for treatment, or a method for preventing or treating the above-mentioned disease, wherein a pharmacologically effective amount of the above-mentioned compound is administered to a warm-blooded animal (preferably a human).
背景技術 Background art
[0005] 従来、糖尿病の治療においては、(1)インスリン注射、(2)脾臓からのインスリン分泌 を促進して血糖低下作用を示すオイダルコン (ダリベンクラミド)、ダリミクロン (ダリクラ ジド)、アマリール (グリメピリド)などのスルホニル尿素(SU)剤、(3)速効 ·短時間にィ ンスリン分泌を促進させて、食後血糖値を低下させるスターシス (ナテグリニド)などの フエ二ルァラニン誘導体、(4)小腸粘膜に存在する二糖類分解酵素(ひ—ダルコシダ ーゼ)の作用を阻害し、糖の消化を抑制し吸収を遅らせ、食後の高血糖を抑制する ベイスン(ボグリボース)、ダルコバイ(ァカルボース)等の a—ダルコシダーゼ阻害剤
、(5)肝臓での糖の産生の抑制、消化管力 の糖の吸収の抑制、末梢組織でのインス リン感受性の改善などにより、血糖降下作用を示すジべトス B (塩酸ブホルミン)、ダリ コラン (塩酸メトホルミン)等のビグアナイド (BG)剤、(6)インスリン抵抗性を改善して、 血糖降下作用を示すァクトス (塩酸ピオグリタゾン)等のインスリン抵抗性改善剤等が 使用されてきた。し力しながらこれらは急激な低血糖を招くなどと 、つた副作用の問 題等があり、糖尿病治療薬として解決すべき課題が多い。 [0005] Conventionally, in the treatment of diabetes, (1) insulin injection, (2) Eudalcon (Daribenclamide), Dalimicron (Dariclazide), Amaril (Glimepiride), etc., which promotes insulin secretion from the spleen and exhibits a hypoglycemic effect, etc. Sulfonylurea (SU), (3) Fast-acting ・ Stimulates insulin secretion in a short time to lower postprandial blood glucose level, such as star cis (nateglinide), (4) A-Dalcosidase inhibitors such as Basin (Voglibose), Dalcoby (Valboose), which inhibits the action of saccharide-degrading enzymes (hyaldarcosidase), suppresses sugar digestion and delays absorption, and suppresses postprandial hyperglycemia (5) Gibetose B (buformin hydrochloride), which has a hypoglycemic effect by suppressing sugar production in the liver, inhibiting sugar absorption in the gastrointestinal tract, and improving insulin sensitivity in peripheral tissues. Biguanide (BG) agents such as cholan (metformin hydrochloride), and (6) insulin resistance improving agents such as lactos (pioglitazone hydrochloride) which improve insulin resistance and exhibit hypoglycemic action have been used. However, these have problems such as side effects such as rapid hypoglycemia, and there are many problems to be solved as antidiabetic drugs.
[0006] このような背景から、毒性が低ぐ新しい作用機序による優れた血糖低下作用を有 する化合物を見出すことが試みられて 、る。 [0006] Against this background, an attempt has been made to find a compound having an excellent hypoglycemic effect by a new mechanism of action with low toxicity.
FBPaseは肝臓内での糖新生に深く関わっている酵素であり、この酵素を阻害すること により、糖産生を抑制し血糖値を低下させることが報告されている。よって、 FBPase阻 害剤は、新規な作用機序を有する糖尿病治療薬として期待され、力かる FBPase阻害 剤で血糖降下作用を有する化合物で開示されているものがある(特許文献 1、 2を参 照)。しかし、開示されている化合物は、基本骨格として 3環性の骨格を有さず、本発 明の化合物とは構造がまったく異なる。 FBPase is an enzyme deeply involved in gluconeogenesis in the liver, and inhibition of this enzyme has been reported to suppress sugar production and lower blood glucose levels. Therefore, FBPase inhibitors are expected to be antidiabetic drugs having a novel mechanism of action, and some FBPase inhibitors are disclosed as compounds that have a hypoglycemic action as FBPase inhibitors (see Patent Documents 1 and 2). See). However, the disclosed compounds do not have a tricyclic skeleton as a basic skeleton, and are completely different in structure from the compounds of the present invention.
特許文献 1:国際公開第 01/47935号パンフレット Patent Document 1: International Publication No. 01/47935 Pamphlet
特許文献 2:国際公開第 00/14095号パンフレット Patent Document 2: Pamphlet of International Publication No. 00/14095
発明の開示 Disclosure of the invention
発明が解決しょうとする課題 Problems to be solved by the invention
[0007] 本発明者らは、優れた活性及び安全性を有する糖尿病等の治療薬及び Z又は予 防薬の開発を目的として鋭意研究を行い、新規チアゾール誘導体が優れた FBPase 阻害作用、インスリン抵抗性改善作用、血糖降下作用、脂質低下作用を有し、高脂 血症、高血糖症、肥満症、耐糖能不全状態、インスリン抵抗性非耐糖能不全状態、 インスリン抵抗性、糖尿病合併症、虚血性心疾患、動脈硬化症、妊娠糖尿病、クッシ ング症候群、ステロイド糖尿病、糖原病、自律神経系の障害、外科的糖尿病様病態 、肝臓癌等を改善することを見出し、本発明を完成した。 [0007] The present inventors have conducted intensive research for the purpose of developing therapeutic agents such as diabetes and Z or prophylactic agents having excellent activity and safety, and the novel thiazole derivatives have excellent FBPase inhibitory action, insulin resistance. It has antibacterial, hypoglycemic, and lipid lowering effects, hyperlipidemia, hyperglycemia, obesity, impaired glucose tolerance, non-insulin-resistant glucose intolerance, insulin resistance, diabetic complications, imaginary The present invention was completed by finding that blood heart disease, arteriosclerosis, gestational diabetes, Cushing syndrome, steroid diabetes, glycogenosis, autonomic nervous system disorder, surgical diabetes-like pathology, liver cancer and the like.
[0008] 即ち、本発明は、高脂血症、高血糖症、肥満症、耐糖能不全 (impaired glucose tol erance : IGT)状態、インスリン抵抗性非而糖能不全 (insulin resistant non- IGT:NGT) 状態、高血圧症、脾炎、脂肪肝、糖尿病合併症 (例えば、網膜症、白内障、腎症、末
梢神経の障害等)、動脈硬化症 (例えば、脳血管障害、狭心症'心筋梗塞、血圧の異 常等)、妊娠糖尿病(gestational diabetes mellitus:GDM)、クッシング症候群、ステロ イド糖尿病、糖原病、自律神経系の障害 (例えば、インポテンス、発汗異常、起立性 低血圧等)、外科的糖尿病様病態、肝臓癌等の治療薬または予防薬として有用な、 チアゾール誘導体及びその薬理上許容される塩を提供する。 That is, the present invention relates to hyperlipidemia, hyperglycemia, obesity, impaired glucose tolerance (IGT) state, insulin resistant non-IGT: NGT ) Condition, hypertension, splenitis, fatty liver, diabetic complications (e.g. retinopathy, cataract, nephropathy, end (Eg, cranial nerve disorders), arteriosclerosis (eg, cerebrovascular disorders, angina pectoris' myocardial infarction, abnormal blood pressure, etc.), gestational diabetes mellitus (GDM), Cushing syndrome, steroid diabetes, glucose Thiazole derivatives and their pharmacologically acceptable drugs useful as therapeutic or prophylactic agents for diseases, autonomic nervous system disorders (for example, impotence, abnormal sweating, orthostatic hypotension, etc.), surgical diabetes-like pathology, liver cancer, etc. Provide salt.
課題を解決するための手段 Means for solving the problem
[0009] 本発明は、 [0009] The present invention provides:
(1)下記一般式 (I) (1) The following general formula (I)
[0010] [化 1] [0010] [Chemical 1]

[0011] [式中、 Rlx及び R1Yは、それぞれ異なって水素原子、ハロゲン原子、 C1-C3アルキ ル基、 P(0)(OH)、 OP(0)(OH)、 CH P(0)(OH)、 CF P(0)(OH)又は OCH P(0)(OH) [In the formula, R lx and R 1Y are different from each other in hydrogen atom, halogen atom, C1-C3 alkyl group, P (0) (OH), OP (0) (OH), CH P (0 ) (OH), CF P (0) (OH) or OCH P (0) (OH)
2 2 2 2 2 2 2 2 を示し、 R2及び R3は、それぞれ同一若しくは異なって、水素原子、ハロゲン原子、 - C6アルキル基、 C1-C6ハロアルキル基、 C1-C6アミノアルキル基(該ァミノアルキル基 のァミノ基は、置換基群 α力 選択される基で 1又は 2個置換されていても良い)、 C1 -C6ヒドロキシアルキル基、 CI- C6アルキルチオ基、 CI- C6アルコキシ基、 CI- C6ァ ルコキシィミノ C1-C3アルキル基(該アルコキシィミノ C1-C3アルキル基は、 C1-C3ァ ルキル基で置換されていても良い)、水酸基、 C1-C6脂肪族ァシル基 (該ァシル基は 、置換基群 α力 選択される基で置換されていても良い)、アミノ基、 CI- C6アルキル アミノ基、 CI- C6ジアルキルァミノ基、 CI- C6脂肪族ァシルァミノ基、 C6- C10ァリール 基 (該ァリール基は、置換基群 α力 選択される基で 1乃至 5個置換されていても良 い)、窒素、酸素及び硫黄力 選択される同一若しくは異なった 1-3個のへテロ原子 を含む 4-10員複素環基 (該複素環基は、置換基群 α力 選択される基で 1乃至 5個 置換されていても良い)、窒素、酸素及び硫黄力 選択される同一若しくは異なった 1
-3個のへテロ原子を含む 4-10員複素環 C1-C6アルキル基 (該複素環 C1-C6アルキ ル基は、置換基群 α力 選択される基で 1乃至 5個置換されていても良い)、窒素、酸 素及び硫黄力 選択される同一若しくは異なった 1-3個のへテロ原子を含む 4-10員 複素環 CI- C6アルキルアミノ基 (該複素環 CI- C6アルキルアミノ基は、置換基群 aか ら選択される基で 1乃至 5個置換されていても良い)、ウレイド基、シァノ基又は-トロ 基を示し、 R4は、水素原子、 C1-C3アルキル基、 C1-C3ハロアルキル基、ハロゲン原 子又はアミノ基を示し、 Xは、 C1-C3アルキレン基 (該アルキレン基は、ォキソ基、水酸 基、ハロゲン原子、 C1-C3アルキル基又は C3-6シクロアルキリデン基で 1又は 2個置 換されていてもよい)、を示す。ただし、 R1Xが P(0)(OH)、 OP(OXOH)、 CH P(0)(OH) 2 2 2 2 2 2 2 2 and R 2 and R 3 are the same or different and each represents a hydrogen atom, a halogen atom, a -C6 alkyl group, a C1-C6 haloalkyl group, a C1-C6 aminoalkyl group (the aminoalkyl) The amino group of the group may be substituted with one or two groups selected from the substituent group α force), C1-C6 hydroxyalkyl group, CI-C6 alkylthio group, CI-C6 alkoxy group, CI-C6 An alkoxyximino C1-C3 alkyl group (the alkoxyimino C1-C3 alkyl group may be substituted with a C1-C3 alkyl group), a hydroxyl group, a C1-C6 aliphatic acyl group (the acyl group is a substituted group) Group group α force optionally substituted with a selected group), amino group, CI-C6 alkylamino group, CI-C6 dialkylamino group, CI-C6 aliphatic asilamino group, C6-C10 aryl group The aryl group is substituted with 1 to 5 substituents selected from the substituent group α force. Nitrogen, oxygen, and sulfur power 4 to 10-membered heterocyclic group containing the same or different 1 to 3 hetero atoms selected (the heterocyclic group is a substituent group α force) 1 to 5 selected groups may be substituted), nitrogen, oxygen and sulfur powers the same or different selected 1 -3 4- to 10-membered heterocyclic C1-C6 alkyl group containing 3 heteroatoms (the heterocyclic C1-C6 alkyl group is substituted with 1 to 5 groups selected from the substituent group α force) 4) 10-membered heterocyclic CI-C6 alkylamino group containing the same or different 1 to 3 heteroatoms selected (nitrogen, oxygen and sulfur power) (the heterocyclic CI-C6 alkylamino group) May be substituted with 1 to 5 groups selected from the substituent group a), represents a ureido group, a cyano group or a -tro group, R 4 represents a hydrogen atom, a C1-C3 alkyl group, A C1-C3 haloalkyl group, a halogen atom or an amino group, X is a C1-C3 alkylene group (the alkylene group is an oxo group, a hydroxyl group, a halogen atom, a C1-C3 alkyl group or a C3-6 cycloalkylidene group). 1 or 2 may be substituted with a group). However, R 1X is P (0) (OH), OP (OXOH), CH P (0) (OH)
2 2 2 2 2 2
、 CF P(0)(OH)又は OCH P(0)(OH)を示す場合は、 R1Yは水素原子、ハロゲン原子, CF P (0) (OH) or OCH P (0) (OH), R 1Y represents a hydrogen atom or a halogen atom.
2 2 2 2 2 2 2 2 2 2
又は CI- C6アルキル基を示し、 R1Yが P(〇)(〇H)、 OP(0)(OH)、 CH P(0)(OH)、 CF P Or CI-C6 alkyl group, R 1Y is P (〇) (〇H), OP (0) (OH), CH P (0) (OH), CF P
2 2 2 2 2 2 2 2 2 2
(0)(OH)又は OCH P(0)(OH)を示す場合は、 Rlxは水素原子、ハロゲン原子又は C1When (0) (OH) or OCH P (0) (OH) is indicated, R lx is a hydrogen atom, a halogen atom or C1
2 2 2 2 2 2
-C6アルキル基を示す。 A -C6 alkyl group is shown.
(置換基群 α ) (Substituent group α)
ハロゲン原子、 CI- C6アルキル基、 CI- C6ハロアルキル基、 CI- C6ヒドロキシアルキ ル基 (該ヒドロキシアルキル基のアルキル基は、 C1-C6ヒドロキシアルキル基で 1個置 換されていても良い)、 C1-C6アルコキシ C1-C6アルキル基、 C1-C6アミノアルキル基 (該ァミノアルキル基のアミノ基は、 C1-C6アルキル基で 1又は 2個置換されていても 良い)、 CI- C6アルコキシ基、 CI- C6アルキルチオ基、カルボキシ基、 CI- C6アルコ キシカルボ-ル基、水酸基、 C1-C6脂肪族ァシル基、アミノ基、 C1-C6アルキルアミノ 基 (該アルキルアミノ基のアルキル基は、水酸基、 C1-C6ヒドロキシアルキル基、 C3- C10シクロアルキル基、 CI- C6アルコキシ基、アミノ基、 CI- C6アルキルアミノ基、 C1- C6ジアルキルァミノ基又はアミド基で 1若しくは 2個置換されていても良い)、 C3-C10 シクロアルキルアミノ基、 C1-C6ジァルキルアミノ基、 C1-C6アルコキシァミノ基 (該ァ ルコキシァミノ基は、水酸基で 1又は 2個置換されていてもよい)、 C1-C6脂肪族ァシ ルァミノ基、シァノ基、ニトロ基、グァ -ジノ基、 C3- C10シクロアルキル基、 C6- C10ァ リール基、窒素、酸素及び硫黄力 選択される同一若しくは異なった 1-3個のへテロ 原子を含む 4-10員複素環基、窒素、酸素及び硫黄力 選択される同一若しくは異な
つた 1-3個のへテロ原子を含む 4-10員複素環 C1-C6アルキルアミノ基、 C1-6力ルバ モイルアルキルアミノ基(該カルバモイルアルキルアミノ基のアルキル基は、ヒドロキシ アルキル基で 1個置換されて 、てもよ 、) ]で表される化合物又はその薬理上許容さ れる塩、 Halogen atom, CI-C6 alkyl group, CI-C6 haloalkyl group, CI-C6 hydroxyalkyl group (the alkyl group of the hydroxyalkyl group may be replaced by one C1-C6 hydroxyalkyl group), C1-C6 alkoxy C1-C6 alkyl group, C1-C6 aminoalkyl group (the amino group of the aminoamino group may be substituted with one or two C1-C6 alkyl groups), CI-C6 alkoxy group, CI -C6 alkylthio group, carboxy group, CI-C6 alkoxycarbonyl group, hydroxyl group, C1-C6 aliphatic acyl group, amino group, C1-C6 alkylamino group (the alkyl group of the alkylamino group is a hydroxyl group, C1- 1 or 2 may be substituted with C6 hydroxyalkyl group, C3-C10 cycloalkyl group, CI-C6 alkoxy group, amino group, CI-C6 alkylamino group, C1-C6 dialkylamino group or amide group) , C3-C10 Chloroalkylamino group, C1-C6 dialkylamino group, C1-C6 alkoxyamino group (the alkyloxyamino group may be substituted with one or two hydroxyl groups), C1-C6 aliphatic acylamino group, cyano Group, nitro group, guazino group, C3-C10 cycloalkyl group, C6-C10 aryl group, nitrogen, oxygen and sulfur powers containing 1-3 identical or different heteroatoms selected 4-10 Member heterocyclic group, nitrogen, oxygen and sulfur powers selected same or different 4- to 10-membered heterocyclic ring containing 1 to 3 heteroatoms C1-C6 alkylamino group, C1-6 rubamoylalkylamino group (the alkyl group of the carbamoylalkylamino group is one hydroxyalkyl group) Or a pharmacologically acceptable salt thereof, which may be substituted, or
(2)下記一般式 (Γ ) (2) The following general formula (Γ)
[0013] [化 2] [0013] [Chemical 2]

[0014] [式中、 R は、 P(0)(OH)、 OP(0)(OH)、 CH P(0)(OH)、 CF P(0)(OH)又は OCH P [Wherein R is P (0) (OH), OP (0) (OH), CH P (0) (OH), CF P (0) (OH) or OCH P
2 2 2 2 2 2 2 2 2 2 2 2 2 2
(0)(OH)を示し、 R2及び R3は、それぞれ同一若しくは異なって、水素原子、ハロゲン(0) represents (OH), R 2 and R 3 are the same or different and each represents a hydrogen atom, halogen
2 2
原子、 CI- C6アルキル基、 CI- C6ハロアルキル基、 CI- C6アミノアルキル基(該ァミノ アルキル基のアミノ基は、置換基群 aカゝら選択される基で 1又は 2個置換されていても 良い)、 CI- C6ヒドロキシアルキル基、 CI- C6アルキルチオ基、 CI- C6アルコキシ基、 CI- C6アルコキシィミノ CI- C3アルキル基(該アルコキシィミノ CI- C3アルキル基は、 C1-C3アルキル基で置換されていても良い)、水酸基、 C1-C6脂肪族ァシル基 (該ァ シル基は、置換基群 αカゝら選択される基で置換されていても良い)、アミノ基、 C1-C6 アルキルアミノ基、 CI- C6ジアルキルァミノ基、 CI- C6脂肪族ァシルァミノ基、 C6- C10 ァリール基 (該ァリール基は、置換基群 a力 選択される基で 1乃至 5個置換されてい ても良い)、窒素、酸素及び硫黄力 選択される同一若しくは異なった 1-3個のへテロ 原子を含む 4-10員複素環基 (該複素環基は、置換基群 α力 選択される基で 1乃至 5個置換されていても良い)、窒素、酸素及び硫黄力 選択される同一若しくは異なつ た 1-3個のへテロ原子を含む 4-10員複素環 C1-C6アルキル基 (該複素環 C1-C6アル キル基は、置換基群 α力も選択される基で 1乃至 5個置換されていても良い)、窒素、 酸素及び硫黄力 選択される同一若しくは異なった 1-3個のへテロ原子を含む 4-10 員複素環 CI- C6アルキルアミノ基 (該複素環 CI- C6アルキルアミノ基は、置換基群 a
力も選択される基で 1乃至 5個置換されていても良い)、ウレイド基、シァノ基又は-ト 口基を示し、 R4は、水素原子、 C1-C3アルキル基、 C1-C3ハロアルキル基、ハロゲン 原子又はアミノ基を示し、 Xは、 C1-C3アルキレン基 (該アルキレン基は、ォキソ基、水 酸基、ハロゲン原子、 C1-C3アルキル基又は C3-6シクロアルキリデン基で 1又は 2個 置換されていてもよい)、を示す。 Atom, CI-C6 alkyl group, CI-C6 haloalkyl group, CI-C6 aminoalkyl group (the amino group of the aminoamino group is substituted with one or two substituents selected from the substituent group a) CI-C6 hydroxyalkyl group, CI-C6 alkylthio group, CI-C6 alkoxy group, CI-C6 alkoxyimino CI-C3 alkyl group (the alkoxyimino CI-C3 alkyl group is C1-C3 alkyl) A hydroxyl group, a C1-C6 aliphatic acyl group (this acyl group may be substituted with a group selected from the substituent group α), an amino group, C1 -C6 alkylamino group, CI-C6 dialkylamino group, CI-C6 aliphatic asilamino group, C6-C10 aryl group (the aryl group is substituted with 1 to 5 substituents selected from the group a force) ), Nitrogen, oxygen and sulfur power 1- or the same selected 4- to 10-membered heterocyclic group containing 3 heteroatoms (the heterocyclic group may be substituted with 1 to 5 substituents selected from the group α group), nitrogen, oxygen and sulfur groups 4- to 10-membered heterocyclic C1-C6 alkyl group containing 1 to 3 heteroatoms that are the same or different selected (the heterocyclic C1-C6 alkyl group is a group in which the substituent group α force is also selected. Optionally substituted with 1 to 5), nitrogen, oxygen and sulfur force 4-10 membered heterocyclic CI-C6 alkylamino group containing 1 to 3 heteroatoms selected from the same or different Heterocyclic CI-C6 alkylamino group is a substituent group a 1 to 5 substituents may be substituted with a selected group), a ureido group, a cyano group, or a -toto group, and R 4 represents a hydrogen atom, a C1-C3 alkyl group, a C1-C3 haloalkyl group, X represents a C1-C3 alkylene group (the alkylene group is substituted with one or two of an oxo group, a hydroxyl group, a halogen atom, a C1-C3 alkyl group, or a C3-6 cycloalkylidene group) May be).
(置換基群 α ) (Substituent group α)
ハロゲン原子、 CI- C6アルキル基、 CI- C6ハロアルキル基、 CI- C6ヒドロキシアルキ ル基 (該ヒドロキシアルキル基のアルキル基は、 C1-C6ヒドロキシアルキル基で 1個置 換されていても良い)、 C1-C6アルコキシ C1-C6アルキル基、 C1-C6アミノアルキル基 (該 C1-C6アミノアルキル基のアミノ基は、 C1-6アルキル基で 1若しくは 2個置換され ていても良い)、 C1-C6アルコキシ基、 C1-C6アルキルチオ基、カルボキシ基、 C1-C 6アルコキシカルボ-ル基、水酸基、 C1-C6脂肪族ァシル基、アミノ基、 C1-C6アルキ ルァミノ基 (該アルキルアミノ基のアルキル基は、水酸基、 C1-C6ヒドロキシアルキル 基、 C3- C10シクロアルキル基、 CI- C6アルコキシ基、アミノ基、 CI- C6アルキルアミノ 基、 C1-C6ジアルキルァミノ基又はアミド基で 1若しくは 2個置換されていても良い)、 C3- C10シクロアルキルアミノ基、 CI- C6ジアルキルァミノ基、 CI- C6アルコキシァミノ 基 (該アルコキシァミノ基は、水酸基で 1又は 2個置換されていてもよい)、 - C6脂 肪族ァシルァミノ基、シァノ基、ニトロ基、グァ -ジノ基、 C3-C10シクロアルキル基、 C 6-C10ァリール基、窒素、酸素及び硫黄力 選択される同一若しくは異なった 1-3個 のへテロ原子を含む 4-10員複素環基、窒素、酸素及び硫黄力 選択される同一若し くは異なった 1-3個のへテロ原子を含む 4-10員複素環 C1-C6アルキルアミノ基、 C1- 6力ルバモイルアルキルアミノ基(該カルバモイルアルキルアミノ基のアルキル基は、ヒ ドロキシアルキル基で 1個置換されて 、てもよ 、) ]で表される化合物又はその薬理上 許容される塩、 Halogen atom, CI-C6 alkyl group, CI-C6 haloalkyl group, CI-C6 hydroxyalkyl group (the alkyl group of the hydroxyalkyl group may be replaced by one C1-C6 hydroxyalkyl group), C1-C6 alkoxy C1-C6 alkyl group, C1-C6 aminoalkyl group (the amino group of the C1-C6 aminoalkyl group may be substituted with one or two C1-6 alkyl groups), C1-C6 An alkoxy group, a C1-C6 alkylthio group, a carboxy group, a C1-C6 alkoxycarbo group, a hydroxyl group, a C1-C6 aliphatic acyl group, an amino group, a C1-C6 alkylamino group (the alkyl group of the alkylamino group is 1 or 2 substituted with hydroxyl group, C1-C6 hydroxyalkyl group, C3-C10 cycloalkyl group, CI-C6 alkoxy group, amino group, CI-C6 alkylamino group, C1-C6 dialkylamino group or amide group You may) C3-C10 cycloalkylamino group, CI-C6 dialkylamino group, CI-C6 alkoxyamino group (the alkoxyamino group may be substituted with one or two hydroxyl groups), -C6 aliphatic Asylamino group, cyano group, nitro group, guazino group, C3-C10 cycloalkyl group, C6-C10 aryl group, nitrogen, oxygen and sulfur forces selected 1-3 identical or different heteroatoms Containing 4-10 membered heterocyclic group, nitrogen, oxygen and sulfur force Selected 4-10 membered heterocyclic C1-C6 alkylamino group containing the same or different 1-3 heteroatoms, C1- Or a pharmacologically acceptable salt thereof, wherein 6 is a rubamoylalkylamino group (the alkyl group of the carbamoylalkylamino group may be substituted with one hydroxyalkyl group)]
(3)上記(1)又は(2)において Rlxが、 OCH P(0)(OH)である化合物又はその薬理上 (3) In the above (1) or (2), R lx is OCH P (0) (OH)
2 2 twenty two
許容される塩、 Acceptable salts,
(4)上記(1)乃至(3)において、 R2及び R3がそれぞれ同一若しくは異なって、水素原 子、ハロゲン原子、 CI- C6アルキル基、 CI- C6アルコキシ基、 CI- C6ジアルキルァミノ
基又は - C6脂肪族ァシル基 (該ァシル基は、置換基群 aから選択される基で置換 されて 、ても良 、)である化合物又はその薬理上許容される塩、 (4) In the above (1) to (3), R 2 and R 3 are the same or different, and hydrogen atom, halogen atom, CI-C6 alkyl group, CI-C6 alkoxy group, CI-C6 dialkylamino A compound or a pharmacologically acceptable salt thereof, which is a group or a -C6 aliphatic isyl group (the acyl group may be substituted with a group selected from the substituent group a),
(5)上記(1)乃至 (4)において、 R2が水素原子又は C1-C6アルキル基である化合物 又はその薬理上許容される塩、 (5) In the above (1) to (4), R 2 is a hydrogen atom or a C1-C6 alkyl group, or a pharmacologically acceptable salt thereof,
(6)上記(1)乃至 (4)にお 、て R2が水素原子又はメチル基である化合物又はその薬 理上許容される塩、 (6) In the above (1) to (4), a compound wherein R 2 is a hydrogen atom or a methyl group or a pharmaceutically acceptable salt thereof,
(7)上記(1)乃至(6)において、 R3がハロゲン原子、 C1-C6ジァルキルアミノ基、 C1- C6脂肪族ァシル基 (該ァシル基は、置換基群 aから選択される基で置換されて!ヽて も良い)、 C1-C6アルキル基又は C1-C6アルコキシ基である化合物又はその薬理上 許容される塩、 (7) In the above (1) to (6), R 3 is a halogen atom, a C1-C6 dialkylamino group, a C1-C6 aliphatic acyl group (the acyl group is substituted with a group selected from the substituent group a) Or a pharmacologically acceptable salt thereof, which is a C1-C6 alkyl group or a C1-C6 alkoxy group,
(8)上記(1)乃至(6)において、 R3が - C6脂肪族ァシル基、 - C6アルキル基又 は C1-C6アルコキシ基である化合物又はその薬理上許容される塩、 (8) In the above (1) to (6), a compound or a pharmacologically acceptable salt thereof in which R 3 is a -C6 aliphatic acyl group, -C6 alkyl group or C1-C6 alkoxy group,
(9)上記(1)乃至(6)にお!/、て R3が C1-C6アルキル基又は C1-C6アルコキシ基であ る化合物又はその薬理上許容される塩、 (9) In the above (1) to (6), a compound in which R 3 is a C1-C6 alkyl group or a C1-C6 alkoxy group, or a pharmacologically acceptable salt thereof,
(10)上記(1)乃至(6)において R3カ チル基、ェチル基又はメトキシ基である化合物 又はその薬理上許容される塩、 (10) A compound or a pharmacologically acceptable salt thereof which is an R 3 cate group, an ethyl group or a methoxy group in the above (1) to (6),
(11)上記(1)乃至(10)にお 、て、 R4が水素原子又はアミノ基である化合物又はそ の薬理上許容される塩、 (11) In the above (1) to (10), a compound wherein R 4 is a hydrogen atom or an amino group, or a pharmacologically acceptable salt thereof,
(12)上記(1)乃至(10)において、 R4が水素原子である化合物又はその薬理上許容 される塩、 (12) In the above (1) to (10), a compound wherein R 4 is a hydrogen atom or a pharmacologically acceptable salt thereof,
(13)上記( 1)乃至( 12)にお!/、て、 Xが C 1-C3アルキレン基である化合物又はその 薬理上許容される塩、 (13) In the above (1) to (12), a compound in which X is a C 1 -C3 alkylene group or a pharmacologically acceptable salt thereof,
(14)上記(1)乃至(12)において、 Xがメチレン基である化合物又はその薬理上許 容される塩、 (14) In the above (1) to (12), a compound wherein X is a methylene group or a pharmacologically acceptable salt thereof,
(15)下記一般式 (II) (15) The following general formula (II)
[化 3]
 [Chemical 3]
[Chemical 3]
(Π) (Π)
[0017] [式中、 R1は、 P(0)(OH)、 OP(0)(OH)、 CH P(0)(OH)、 CF P(0)(OH)又は OCH P( [In the formula, R 1 is P (0) (OH), OP (0) (OH), CH P (0) (OH), CF P (0) (OH) or OCH P (
2 2 2 2 2 2 2 2 2 2 2 2 2 2
0)(OH)を示し、 R2及び R3は、それぞれ同一若しくは異なって、水素原子、ハロゲン原0) (OH), R 2 and R 3 are the same or different and each represents a hydrogen atom or a halogen atom.
2 2
子、 CI- C6アルキル基、 CI- C6ハロアルキル基、 CI- C6アミノアルキル基(該アミノア ルキル基のアミノ基は、置換基群 a力 選択される基で 1又は 2個置換されていても 良い)、 CI- C6ヒドロキシアルキル基、 CI- C6アルキルチオ基、 CI- C6アルコキシ基、 CI- C6アルコキシィミノ CI- C3アルキル基(該アルコキシィミノ CI- C3アルキル基は、 C1-C3アルキル基で置換されていても良い)、水酸基、 C1-C6脂肪族ァシル基 (該ァ シル基は、置換基群 α力 選択される基で置換されていても良い)、アミノ基、 C1-C6 アルキルアミノ基、 CI- C6ジアルキルァミノ基、 CI- C6脂肪族ァシルァミノ基、 C6- C10 ァリール基 (該ァリール基は、置換基群 a力 選択される基で 1乃至 5個置換されてい ても良い)、窒素、酸素及び硫黄力 選択される同一若しくは異なった 1-3個のへテロ 原子を含む 4-10員複素環基 (該複素環基は、置換基群 α力 選択される基で 1乃至 5個置換されていても良い)、窒素、酸素及び硫黄力 選択される同一若しくは異なつ た 1-3個のへテロ原子を含む 4-10員複素環 C1-C6アルキル基 (該複素環 C1-C6アル キル基は、置換基群 α力も選択される基で 1乃至 5個置換されていても良い)、窒素、 酸素及び硫黄力 選択される同一若しくは異なった 1-3個のへテロ原子を含む 4-10 員複素環 CI- C6アルキルアミノ基 (該複素環 CI- C6アルキルアミノ基は、置換基群 a カゝら選択される基で 1乃至 5個置換されていても良い)、シァノ基及び-トロ基を示し、 R4は、水素原子、 C1-C3アルキル基、 C1-C3ハロアルキル基、ハロゲン原子又はアミ ノ基を示し、 Xは、 C1-C3アルキレン基 (該アルキレン基は、 C1-C3アルキル基又は C 3-6シクロアルキリデン基で置換されていてもよい)、を示す。 , CI-C6 alkyl group, CI-C6 haloalkyl group, CI-C6 aminoalkyl group (the amino group of the aminoalkyl group may be substituted with one or two substituents selected from group a force) ), CI-C6 hydroxyalkyl group, CI-C6 alkylthio group, CI-C6 alkoxy group, CI-C6 alkoxyimino CI-C3 alkyl group (the alkoxyimino CI-C3 alkyl group is a C1-C3 alkyl group) Optionally substituted), hydroxyl group, C1-C6 aliphatic acyl group (the acyl group may be substituted with a group selected by the substituent group α force), amino group, C1-C6 alkylamino Group, CI-C6 dialkylamino group, CI-C6 aliphatic acylamino group, C6-C10 aryl group (this aryl group may be substituted with 1 to 5 groups selected from the substituent group a) , Nitrogen, oxygen and sulfur power (B) A 4-10 membered heterocyclic group containing an atom (the heterocyclic group may be substituted with 1 to 5 groups selected from the substituent group α force), nitrogen, oxygen and sulfur force Or a 4- to 10-membered heterocyclic C1-C6 alkyl group containing 1 to 3 heteroatoms (the heterocyclic C1-C6 alkyl group is a group in which the substituent group α force is also selected from 1 to 5 Optionally substituted), nitrogen, oxygen and sulfur power 4-10 membered heterocyclic CI- C6 alkylamino group containing 1-3 identical or different heteroatoms (the heterocyclic CI- The C6 alkylamino group may be substituted with 1 to 5 groups selected from the substituent group a), a cyano group and a -tro group, R 4 represents a hydrogen atom, a C1-C3 alkyl group Group, a C1-C3 haloalkyl group, a halogen atom or an amino group, X is a C1-C3 alkylene group (the alkylene group is a C1-C3 alkyl group or C 3-6 Optionally substituted with a cycloalkylidene group).
[0018] (置換基群 α ) [0018] (Substituent group α)
ハロゲン原子、 CI- C6アルキル基、 CI- C6ハロアルキル基、 CI- C6アミノアルキル
基 (該ァミノアルキル基のアミノ基は、 C1-C6アルキル基で 1又は 2個置換されていて も良い)、 CI- C6アルコキシ基、 CI- C6アルキルチオ基、カルボキシ基、 CI- C6アルコ キシカルボ-ル基、水酸基、 C1-C6脂肪族ァシル基、アミノ基、 C1-C6アルキルアミノ 基、 CI- C6ジアルキルァミノ基、 CI- C6脂肪族ァシルァミノ基、シァノ基、ニトロ基、 C3 -C10シクロアルキル基、 C6-C10ァリール基、窒素、酸素及び硫黄から選択される同 一若しくは異なった 1-3個のへテロ原子を含む 4-10員複素環基、窒素、酸素及び硫 黄力も選択される同一若しくは異なった 1-3個のへテロ原子を含む 4-10員複素環 C1 -C6アルキルアミノ基、 C1-6力ルバモイルアルキルアミノ基]で表される化合物又はそ の薬理上許容される塩、 Halogen atom, CI-C6 alkyl group, CI-C6 haloalkyl group, CI-C6 aminoalkyl Group (the amino group of the aminoalkyl group may be substituted with one or two C1-C6 alkyl groups), CI-C6 alkoxy group, CI-C6 alkylthio group, carboxy group, CI-C6 alkoxycarbo group Group, hydroxyl group, C1-C6 aliphatic asil group, amino group, C1-C6 alkylamino group, CI-C6 dialkylamino group, CI-C6 aliphatic asilamino group, cyano group, nitro group, C3-C10 cycloalkyl group , C6-C10 aryl group, same or different selected from nitrogen, oxygen and sulfur 4- to 10-membered heterocyclic group containing 1-3 heteroatoms, nitrogen, oxygen and sulfur power Or a compound represented by a 4- to 10-membered heterocyclic C1-C6 alkylamino group or a C1-6-powered rubamoylalkylamino group containing 1 to 3 different heteroatoms, or a pharmaceutically acceptable salt thereof. ,
(16) {[7- (アミノカルボ-ル) -8H-インデノ [1 ,2-d][l ,3]チアゾール -4-ィル]ォキシ }メチ ルホスホン酸、 {[7- (シクロプロピルカルボ-ル) -8H-インデノ [l,2-d][l,3]チアゾール- 4-ィル]ォキシ }メチルホスホン酸又はその薬理上許容される塩、 (16) {[7- (Aminocarbol) -8H-indeno [1,2-d] [l, 3] thiazol-4-yl] oxy} methylphosphonic acid, {[7- (cyclopropylcarbo- -8H-indeno [l, 2-d] [l, 3] thiazol-4-yl] oxy} methylphosphonic acid or a pharmacologically acceptable salt thereof,
( 17) [(7-フルォ口- 8H-インデノ [1 ,2-d][l ,3]チアゾール -4-ィル)ォキシ]メチルホスホ ン酸、 [(7-ァセチル -8H-インデノ [1 ,2-d][l ,3]チアゾール -4-ィル)ォキシ]メチルホスホ ン酸、 {[7- (ジメチルァミノ) -8H-インデノ [1 ,2-d][l ,3]チアゾール -4-ィル]ォキシ }メチル ホスホン酸、({7- [(メチルァミノ)カルボ-ル] -8H-インデノ [l,2-d][l,3]チアゾール -4-ィ ル}ォキシ)メチルホスホン酸又はその薬理上許容される塩、 (17) [(7-Fluoro-8H-indeno [1,2, d] [l, 3] thiazol-4-yl) oxy] methylphosphonic acid, [(7-acetyl-8H-indeno [1, 2-d] [l, 3] thiazol-4-yl) oxy] methylphosphonic acid, {[7- (dimethylamino) -8H-indeno [1,2, d] [l, 3] thiazole-4- Ru] oxy} methyl phosphonic acid, ({7-[(methylamino) carbol] -8H-indeno [l, 2-d] [l, 3] thiazole-4-yl} oxy) methylphosphonic acid or pharmacology thereof Top acceptable salt,
(18) [(7- {[(2-ァミノ- 2-ォキソェチル)ァミノ]カルボ-ル}- 8H-インデノ [1,2- d][l,3]チ ァゾール -4-ィル)ォキシ]メチルホスホン酸、 {[7-({[(lS)-2-ァミノ- 1-メチル - 2-ォキソ ェチル]アミノ}カルボ-ル)- 8H-インデノ [1 ,2- d][l ,3]チアゾール - 4-ィル]ォキシ }メチ ルホスホン酸、 {[7-({[(lR)- 2-ァミノ- 1-メチル - 2-ォキソェチル]アミノ}カルボ-ル) -8H -インデノ [l,2-d][l,3]チアゾール -4-ィル]ォキシ }メチルホスホン酸、 {[7-({[(lS)-l- (ァ ミノカルボ-ル) -3-メチルブチル]アミノ}カルボ-ル) -8H-インデノ [1 ,2-d][l ,3]チアゾ ール- 4-ィル]ォキシ }メチルホスホン酸、 {[7- ({[(1S)- 2-ァミノ- 1- (ヒドロキシメチル) -2- ォキソェチル]アミノ}カルボ-ル) -8H-インデノ [l,2-d][l,3]チアゾール -4-ィル]ォキシ } メチルホスホン酸又はその薬理上許容される塩、 (18) [(7- {[(2-Amino-2-oxoethyl) amino] carbol} -8H-indeno [1,2-d] [l, 3] thiazol-4-yl) oxy] Methylphosphonic acid, {[7-({[(lS) -2-amino-1-methyl-2-oxoethyl] amino} carbol) -8H-indeno [1,2-d] [l, 3] thiazole -4-yl] oxy} methylphosphonic acid, {[7-({[(lR)-2-amino-1-methyl-2-oxoethyl] amino} carbol) -8H -indeno [l, 2- d] [l, 3] thiazol-4-yl] oxy} methylphosphonic acid, {[7-({[(lS) -l- (aminocarbole) -3-methylbutyl] amino} carbole)- 8H-indeno [1,2-d] [l, 3] thiazol-4-yl] oxy} methylphosphonic acid, {[7- ({[(1S) -2-amino-1- (hydroxymethyl) -2-oxoethyl] amino} carbol) -8H-indeno [l, 2-d] [l, 3] thiazol-4-yl] oxy} methylphosphonic acid or a pharmacologically acceptable salt thereof,
( 19) [(7-メトキシ -8H-インデノ [1 ,2-d][l ,3]チアゾール -4-ィル)ォキシ]メチルホスホン 酸、 [(7-ェチル -8H-インデノ [1 ,2-d][l ,3]チアゾール -4-ィル)ォキシ]メチルホスホン
酸、 [(6,7-ジメチル-81"[-ィンデノ[1,2-(1][1,3]チァゾール-4-ィル)ォキシ]メチルホスホ ン酸又はその薬理上許容される塩、 (19) [(7-Methoxy-8H-indeno [1,2, d] [l, 3] thiazol-4-yl) oxy] methylphosphonic acid, [(7-Ethyl-8H-indeno [1,2,2- d] [l, 3] thiazole-4-yl) oxy] methylphosphone Acid, [(6,7-dimethyl-81 "[-indeno [1,2- (1] [1,3] thiazol-4-yl) oxy] methylphosphonic acid or a pharmacologically acceptable salt thereof,
(20) [(7-ブロモ-6-メチル-81"[-ィンデノ[1,2-(1][1,3]チァゾール-4-ィル)ォキシ]メチ ルホスホン酸、 [(6-メトキシ -8H-インデノ [l,2-d][l,3]チアゾール -4-ィル)ォキシ]メチ ルホスホン酸、 [(7-ブロモ -6-メトキシ -8H-インデノ [l,2-d][l,3]チアゾール -4-ィル)ォ キシ]メチルホスホン酸又はその薬理上許容される塩、 (20) [(7-Bromo-6-methyl-81 "[-indeno [1,2- (1] [1,3] thiazol-4-yl) oxy] methylphosphonic acid, [(6-methoxy- 8H-indeno [l, 2-d] [l, 3] thiazol-4-yl) oxy] methylphosphonic acid, [(7-bromo-6-methoxy-8H-indeno [l, 2-d] [l , 3] thiazole-4-yl) oxy] methylphosphonic acid or a pharmacologically acceptable salt thereof,
(21)上記(1)乃至(20)に記載の化合物又はその薬理上許容される塩を含有する 医薬、 (21) A medicament comprising the compound according to (1) to (20) above or a pharmacologically acceptable salt thereof,
(22)上記(1)乃至(20)に記載の化合物又はその薬理上許容される塩を含有する、 糖尿病の治療薬又は予防薬、 (22) A therapeutic or prophylactic agent for diabetes, comprising the compound according to (1) to (20) above or a pharmacologically acceptable salt thereof,
(23)上記(1)乃至(20)に記載の化合物又はその薬理上許容される塩を含有する、 高血糖症、肥満症、耐糖能不全状態、インスリン抵抗性非耐糖能不全状態、糖尿病 合併症、妊娠糖尿病、ステロイド糖尿病若しくは外科的糖尿病様病態の治療薬又は 予防薬、 (23) Hyperglycemia, obesity, impaired glucose tolerance, insulin-resistant non-glucose intolerance, diabetes, comprising the compound according to (1) to (20) or a pharmacologically acceptable salt thereof Remedies or preventives for diabetes, gestational diabetes, steroid diabetes or surgical diabetes-like conditions,
(24)上記(1)乃至(20)に記載の化合物又はその薬理上許容される塩を含有する、 高脂血症、高血圧症、膝炎、脂肪肝、動脈硬化症、クッシング症候群若しくは糖原病 の治療薬又は予防薬、 (24) Hyperlipidemia, hypertension, knee inflammation, fatty liver, arteriosclerosis, Cushing's syndrome or glycogen, containing the compound according to (1) to (20) above or a pharmacologically acceptable salt thereof Remedies or preventives for diseases,
(25)上記(1)乃至(20)に記載の化合物又はその薬理上許容される塩を含有する、 FBPase阻害剤、 (25) an FBPase inhibitor comprising the compound according to (1) to (20) above or a pharmacologically acceptable salt thereof,
(26)糖尿病の治療又は予防のための医薬を製造するための、上記(1)乃至(20)に 記載の化合物又はその薬理上許容される塩の使用、 (26) Use of the compound according to (1) to (20) above or a pharmacologically acceptable salt thereof for the manufacture of a medicament for treating or preventing diabetes,
(27)上記(1)乃至(20)に記載の化合物又はその薬理上許容される塩の薬理的な 有効量を温血動物に投与する糖尿病の予防若しくは治療方法である。 本発明において、「Cl-3アルキル基」とは、炭素原子を 1個乃至 3個有する直鎖状 又は分枝鎖状のアルキル基であり、例えば、メチル、ェチル若しくは n-プロピル基を 挙げることができる。 Rlx及び R1Y、 R2及び R3の C 1-6アルコキシィミノ C 1-3アルキル基の 置換基、 R4及び Xの C1-3アルキレンの置換基においては、好適にはメチル基である
[0020] 本発明にお 、て、「Cl-6アルキル基」とは、炭素原子を 1個乃至 6個有する直鎖状 又は分枝鎖状のアルキル基であり、例えば、前記「C1_3アルキル基」の例として挙げ た基又は、メチル、ェチル、 n-プロピル、イソプロピル、 n-ブチル、イソブチル、 s-ブチ ル、 tert-ブチル、 n-ペンチル、イソペンチル、 2-メチルブチル、ネオペンチル、 1-ェ チルプロピル、 n-へキシル、イソへキシル、 4-メチルペンチル、 3-メチルペンチル、 2- メチルペンチル、 1-メチルペンチル、 3,3-ジメチルブチル、 2,2-ジメチルブチル、 1,1- ジメチルブチル、 1,2-ジメチルブチル、 1,3-ジメチルブチル、 2,3-ジメチルブチル、 2- ェチルブチル、ヘプチル、 1-メチルへキシル、 2-メチルへキシル、 3-メチルへキシル 、 4-メチルへキシル、 5-メチルへキシル、 1-プロピルブチル、 4,4-ジメチルペンチル、 ォクチル、 1-メチルヘプチル、 2-メチルヘプチル、 3-メチルヘプチル、 4-メチルヘプ チル、 5-メチルヘプチル、 6-メチルヘプチル、 1-プロピルペンチル、 2-ェチルへキシ ル若しくは 5,5-ジメチルへキシル基を挙げることができる。 R2及び R3においては、好適 には炭素数 1乃至 3個のアルキル基であり、最も好適にはェチル基又はメチル基であ る。置換基群 a及び置換基群 aの C1-C6アミノアルキル基の置換基にお!、ては、好 適には炭素数 1乃至 3個のアルキル基であり、最も好適にはメチル基である。 (27) A method for the prophylaxis or treatment of diabetes, comprising administering to a warm-blooded animal a pharmacologically effective amount of the compound according to (1) to (20) above or a pharmacologically acceptable salt thereof. In the present invention, the “Cl-3 alkyl group” is a linear or branched alkyl group having 1 to 3 carbon atoms, and examples thereof include a methyl, ethyl or n-propyl group. Can do. R 1x and R 1Y , R 2 and R 3 C 1-6 alkoxyimino C 1-3 alkyl group substituents, R 4 and X C 1-3 alkylene substituents are preferably methyl groups is there In the present invention, the “Cl-6 alkyl group” is a linear or branched alkyl group having 1 to 6 carbon atoms. For example, the “C1_3 alkyl group” , Or methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, s-butyl, tert-butyl, n-pentyl, isopentyl, 2-methylbutyl, neopentyl, 1-ethylpropyl N-hexyl, isohexyl, 4-methylpentyl, 3-methylpentyl, 2-methylpentyl, 1-methylpentyl, 3,3-dimethylbutyl, 2,2-dimethylbutyl, 1,1-dimethylbutyl 1,2-dimethylbutyl, 1,3-dimethylbutyl, 2,3-dimethylbutyl, 2-ethylbutyl, heptyl, 1-methylhexyl, 2-methylhexyl, 3-methylhexyl, 4-methyl Xyl, 5-methylhexyl, 1-propyl To butyl, 4,4-dimethylpentyl, octyl, 1-methylheptyl, 2-methylheptyl, 3-methylheptyl, 4-methylheptyl, 5-methylheptyl, 6-methylheptyl, 1-propylpentyl, 2-ethyl Mention may be made of xyl or 5,5-dimethylhexyl groups. R 2 and R 3 are preferably an alkyl group having 1 to 3 carbon atoms, and most preferably an ethyl group or a methyl group. The substituents of the substituent group a and the C1-C6 aminoalkyl group of the substituent group a are preferably an alkyl group having 1 to 3 carbon atoms, and most preferably a methyl group. .
[0021] 本発明にお 、て、「Cl-3アルキレン基」とは、炭素原子を 1個乃至 3個のアルキレン 基であり、例えば、メチレン、メチルメチレン、ジメチルメチレン、エチレン、プロピレン を挙げることができる。 Xにおいては、好適にはメチレン又はエチレンであり、更に好 適にはメチレンである。 In the present invention, the “Cl-3 alkylene group” is an alkylene group having 1 to 3 carbon atoms, and examples thereof include methylene, methylmethylene, dimethylmethylene, ethylene, and propylene. Can do. X is preferably methylene or ethylene, and more preferably methylene.
[0022] 本発明にお!/、て、「C3-10シクロアルキル基」とは、縮環して!/、てもよ 、3乃至 10員飽 和環状炭化水素基であり、例えば、シクロプロピル、シクロブチル、シクロペンチル、 シクロへキシル、シクロへプチル、ノルボルニル、ァダマンチル基を挙げることができ る。置換基群 a及び置換基群 aの C1-C6アルキルアミノ基の置換基にお!、ては、好 適には 3乃至 7員飽和環状炭化水素基であり、更に好適にはシクロプロピル、シクロブ チル、シクロペンチル、シクロへキシルである。 In the present invention, “/ C3-10 cycloalkyl group” means a condensed ring! /, Which may be a 3- to 10-membered saturated cyclic hydrocarbon group. Mention may be made of propyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, norbornyl, adamantyl groups. Substituent group a and the substituent of C1-C6 alkylamino group of substituent group a are preferably 3- to 7-membered saturated cyclic hydrocarbon groups, and more preferably cyclopropyl, cyclobutyl. Til, cyclopentyl, cyclohexyl.
[0023] 本発明にお 、て、「C3-6シクロアルキリデン基」とは、 2価の 3乃至 6員飽和環状炭化 水素基であり、例えば、シクロプロピリデン、シクロブチリデン、シクロペンチリデン、シ
クロへキシリデン基を挙げることができる。 Xにおいては好適にはシクロペンチリデン である。 In the present invention, the “C3-6 cycloalkylidene group” is a divalent 3- to 6-membered saturated cyclic hydrocarbon group such as cyclopropylidene, cyclobutylidene, cyclopentylidene, Shi Mention may be made of the chlorohexylene group. X is preferably cyclopentylidene.
[0024] 本発明にお!/、て、「Cl-6ハロアルキル基」とは、前記「Cl-6アルキル基」にハロゲン 原子が置換した基であり、例えば、トリフルォロメチル、トリクロロメチル、ジフルォロメ チル、ジクロロメチル、ジブロモメチル、フルォロメチル、 2,2,2-トリフルォロェチル、 2, 2,2-トリクロロェチノレ、 2-ブロモェチノレ、 2-クロロェチノレ、 2-フノレオロェチノレ、 2-ョード ェチノレ、 3-クロ口プロピノレ、 4-フノレオロブチノレ、 6-ョードへキシノレ、 2,2-ジブ口モェチ ル基を挙げることができ、 R2、 R3、 R4及び置換基群ひにおいては、好適にはトリフルォ ロメチル又はジフルォロメチル基である。 In the present invention, the “Cl-6 haloalkyl group” is a group in which a halogen atom is substituted on the “Cl-6 alkyl group”, and examples thereof include trifluoromethyl, trichloromethyl, Difluoromethyl, dichloromethyl, dibromomethyl, fluoromethyl, 2,2,2-trifluoroethyl, 2,2,2-trichloroethinole, 2-bromoethinole, 2-chloroethinole, 2-funoleoloetinore, 2 -Rhodochinole, 3-chloropropinole, 4-funoleolobutinole, 6-hydrohexenole, 2,2-dibucolate, R 2 , R 3 , R 4 and substituents In the group, preferred is a trifluoromethyl group or a difluoromethyl group.
[0025] 本発明にお 、て、「C1-C6ヒドロキシアルキル基」とは前記「Cl-6アルキル基」に水 酸基が置換した基であり、例えば、ヒドロキシメチル、ヒドロキシェチル、 1-ヒドロキシェ チル、ヒドロキシプロピル、ヒドロキシブチル、ヒドロキシペンチル、ヒドロキシへキシル 基を挙げることができ、 R2、
 置換基群 a、置換基群 aの C1-C6ヒドロキシアルキル 基の置換基、置換基群 aの力ルバモイルアルキルアミノ基の置換基及び置換基群 aの C1-C6アルキルアミノ基の置換基においては、好適にはヒドロキシメチル基又は 1-ヒドロキシェチル基である。 In the present invention, the “C1-C6 hydroxyalkyl group” is a group in which a hydroxyl group is substituted on the “Cl-6 alkyl group”, and examples thereof include hydroxymethyl, hydroxyethyl, 1- And hydroxyethyl, hydroxypropyl, hydroxybutyl, hydroxypentyl, hydroxyhexyl groups, R 2 , In the substituent group a, the substituent of the C1-C6 hydroxyalkyl group of the substituent group a, the substituent of the rubamoylalkylamino group of the substituent group a, and the substituent of the C1-C6 alkylamino group of the substituent group a Is preferably a hydroxymethyl group or a 1-hydroxyethyl group.
置換基群 a、置換基群 aの C1-C6ヒドロキシアルキル 基の置換基、置換基群 aの力ルバモイルアルキルアミノ基の置換基及び置換基群 aの C1-C6アルキルアミノ基の置換基においては、好適にはヒドロキシメチル基又は 1-ヒドロキシェチル基である。 In the present invention, the “C1-C6 hydroxyalkyl group” is a group in which a hydroxyl group is substituted on the “Cl-6 alkyl group”, and examples thereof include hydroxymethyl, hydroxyethyl, 1- And hydroxyethyl, hydroxypropyl, hydroxybutyl, hydroxypentyl, hydroxyhexyl groups, R 2 , In the substituent group a, the substituent of the C1-C6 hydroxyalkyl group of the substituent group a, the substituent of the rubamoylalkylamino group of the substituent group a, and the substituent of the C1-C6 alkylamino group of the substituent group a Is preferably a hydroxymethyl group or a 1-hydroxyethyl group.
[0026] 本発明にお!/、て、「C 1-6アミノアルキル基」とは前記「Cl-6アルキル基」にァミノ基 が置換した基であり、例えば、アミノメチル、アミノエチル、ァミノプロピル、アミノブチ ル基を挙げることができ、 R2、 R3及び置換基群ひにおいては好適にはアミノメチルで ある。 [0026] In the present invention, the "C 1-6 aminoalkyl group" is a group obtained by substituting an amino group for the above-mentioned "Cl-6 alkyl group". For example, aminomethyl, aminoethyl, aminopropyl An aminobutyl group can be mentioned, and in R 2 , R 3 and the substituent group, aminomethyl is preferred.
[0027] 本発明において、「Cl-6アルコキシ基」とは、前記「Cl-6アルキル基」が酸素原子に 結合した基であり、例えば、メトキシ、エトキシ、 n-プロポキシ、イソプロポキシ、 n-ブト キシ、イソブトキシ、 s-ブトキシ、 tert-ブトキシ、 n-ペントキシ、イソペントキシ、 2-メチノレ ブトキシ、ネオペントキシ、 n-へキシルォキシ、 4-メチルペントキシ、 3-メチルペントキ シ、 2-メチルペントキシ、 3,3-ジメチルブトキシ、 2,2-ジメチルブトキシ、 1,1-ジメチル ブトキシ、 1,2-ジメチルブトキシ、 1,3-ジメチルブトキシ、 2,3-ジメチルブトキシのような 炭素数 1乃至 6個の直鎖又は分枝鎖アルコキシ基を挙げることができる。置換基群 a においては、好適にはメトキシ基である。 R2、 置換基群 a及び置換基群 aの CI-
C6アルキルアミノ基の置換基においては、好適にはメトキシ又はエトキシ基である。 In the present invention, the “Cl-6 alkoxy group” is a group in which the “Cl-6 alkyl group” is bonded to an oxygen atom. For example, methoxy, ethoxy, n-propoxy, isopropoxy, n- Butoxy, isobutoxy, s-butoxy, tert-butoxy, n-pentoxy, isopentoxy, 2-methylenobutoxy, neopentoxy, n-hexyloxy, 4-methylpentoxy, 3-methylpentoxy, 2-methylpentoxy, 3, 1 to 6 carbon atoms such as 3-dimethylbutoxy, 2,2-dimethylbutoxy, 1,1-dimethylbutoxy, 1,2-dimethylbutoxy, 1,3-dimethylbutoxy, 2,3-dimethylbutoxy Mention may be made of chain or branched alkoxy groups. In the substituent group a, a methoxy group is preferred. R 2 , CI- of substituent group a and substituent group a The substituent for the C6 alkylamino group is preferably a methoxy or ethoxy group.
[0028] 本発明において、「Cl-6アルコキシ C1-6アルキル基」とは、前記「Cl-6アルキル基 」が前記「Cl-6アルコキシ基」に結合した基であり、例えば、メトキシメチル、メトキシェ チル、エトキシメチル、エトキシェチル、 n-プロポキシメチル、 n-プロポキシェチル、ィ ソプロボキシメチル、 n-ブトキシメチル、イソブトキシメチル、 s-ブトキシメチル、 tert-ブ トキシメチル、 n-ペントキシメチル、イソペントキシメチル、 2-メチルブトキシメチル、ネ ォペントキシメチル、 n-へキシルォキシメチル、 4-メチルペントキシメチル、 3-メチル ペントキシメチル、 2-メチルペントキシメチル、 3,3-ジメチルブトキシメチル、 2,2-ジメチ ルブトキシメチル、 1,1-ジメチルブトキシメチル、 1,2-ジメチルブトキシメチル、 1,3-ジメ チルブトキシメチル、 2,3-ジメチルブトキシメチルのような炭素数 1乃至 6個の直鎖又 は分枝鎖アルコキシ基を挙げることができる。置換基群 αにおいては、好適にはメト キシメチル基である。 In the present invention, the “Cl-6 alkoxy C1-6 alkyl group” is a group in which the “Cl-6 alkyl group” is bonded to the “Cl-6 alkoxy group”, for example, methoxymethyl, Methoxyethyl, ethoxymethyl, ethoxyethyl, n-propoxymethyl, n-propoxychetyl, isopropoxymethyl, n-butoxymethyl, isobutoxymethyl, s-butoxymethyl, tert-butoxymethyl, n-pentoxymethyl, Isopentoxymethyl, 2-methylbutoxymethyl, neopentoxymethyl, n-hexyloxymethyl, 4-methylpentoxymethyl, 3-methylpentoxymethyl, 2-methylpentoxymethyl, 3,3- Dimethylbutoxymethyl, 2,2-dimethylbutoxymethyl, 1,1-dimethylbutoxymethyl, 1,2-dimethylbutoxymethyl, 1,3-dimethylbutoxymethyl, 2,3 -A straight-chain or branched alkoxy group having 1 to 6 carbon atoms such as dimethylbutoxymethyl. In the substituent group α, a methoxymethyl group is preferred.
[0029] 本発明にお 、て、「Cl-6アルキルチオ基」とは、前記「Cl-6アルキル基」が硫黄原 子に結合した基であり、例えば、メチルチオ、ェチルチオ、 t-プチルチオ基を挙げる ことができ、 R2、 R3及び置換基群 αにおいては、好適にはメチルチオ基である。 In the present invention, the “Cl-6 alkylthio group” is a group in which the “Cl-6 alkyl group” is bonded to a sulfur atom. For example, a methylthio, ethylthio, or t-butylthio group In R 2 , R 3 and substituent group α, a methylthio group is preferred.
[0030] 本発明において、「 - 6脂肪族ァシル基」とは、炭素数 1乃至 6個の脂肪族炭化水 素基がカルボニル基に結合した基であり、例えば、ホルミル、ァセチル、プロピオニル 、ブチリル、イソブチリル、ペンタノィル、ピバロィル、バレリル、イソバレリル基のような アルキルカルボ-ル基、クロロアセチル、ジクロロアセチル、トリクロロアセチル、トリフ ルォロアセチルのようなハロゲン化アルキルカルボ-ル基、メトキシァセチルのような 低級アルコキシアルキルカルボ-ル基、(Ε)-2-メチル -2-ブテノィルのような不飽和ァ ルキルカルボニル基等が挙げられる。 R2、 R3及び置換基群 αにおいて、好適にはホ ルミル基、ァセチル基、トリフルォロアセチル基である。 In the present invention, the “−6 aliphatic acyl group” is a group in which an aliphatic hydrocarbon group having 1 to 6 carbon atoms is bonded to a carbonyl group. For example, formyl, acetyl, propionyl, butyryl , Alkylbutyl groups such as isobutyryl, pentanoyl, pivalol, valeryl, isovaleryl groups, halogenated alkylcarbo groups such as chloroacetyl, dichloroacetyl, trichloroacetyl, trifluoroacetyl, lower alkoxy such as methoxyacetyl Examples thereof include an alkyl carbonyl group and an unsaturated alkyl carbonyl group such as (Ε) -2-methyl-2-butenoyl. In R 2 , R 3 and substituent group α, a formyl group, a acetyl group and a trifluoroacetyl group are preferred.
[0031] 本発明において、「C 1-6アルキルアミノ基」とは、前記「Cl-6アルキル基」がァミノ基 に結合した基であり、例えば、メチルァミノ、ェチルアミ入 n-プロピルアミ入イソプロ ピルァミノ、 n-ブチルアミ入イソブチルアミ入 S-ブチルァミノ、 tert-ブチルアミ入 n- ペンチルアミ入イソペンチルアミ入 2-メチルブチルアミ入ネオペンチルァミノ、 1-ェ チルプロピルアミ入 n-へキシルアミ入イソへキシルアミ入 4-メチルペンチルアミ入 3
-メチルペンチルァミノ、 2-メチルペンチルァミノ、 1-メチルペンチルァミノ、 3, 3-ジメチ ルブチルァミノ、 2,2-ジメチルブチルァミノ、 1,1-ジメチルブチルァミノ、 1,2-ジメチル ブチルアミ入 1,3-ジメチルブチルアミ入 2,3-ジメチルブチルアミ入若しくは 2-ェチ ルブチルァミノ基を挙げることができ、 R2、
 置換基群 α及び置換基群 αの C1-C6 アルキルアミノ基の置換基においては、好適にはメチルァミノ基、ェチルァミノ基、ィ ソプロピルアミノ基である。 In the present invention, the “C 1-6 alkylamino group” is a group in which the “Cl-6 alkyl group” is bonded to an amino group, such as methylamino, ethylamino-containing isopropylamino, n-Butylamino-containing isobutylamino-containing S-Butylamino, tert-Butylamino-containing n-Pentylami-containing Isopentylami-containing 2-Methylbutylamino-containing Neopentylamino, 1-ethylpropylamino-containing n-Hexylami-containing Isohexylami-containing 4-methylpentylami containing 3 -Methylpentylamino, 2-methylpentylamino, 1-methylpentylamino, 3,3-dimethylbutylamino, 2,2-dimethylbutylamino, 1,1-dimethylbutylamino, 1,2-dimethyl Butylamino-containing 1,3-dimethylbutylamido-containing 2,3-dimethylbutylamido-containing or 2-ethylbutylamino group, R 2 , The substituent of the substituent group α and the C1-C6 alkylamino group of the substituent group α is preferably a methylamino group, an ethylamino group, or an isopropylamino group.
置換基群 α及び置換基群 αの C1-C6 アルキルアミノ基の置換基においては、好適にはメチルァミノ基、ェチルァミノ基、ィ ソプロピルアミノ基である。 In the present invention, the “C 1-6 alkylamino group” is a group in which the “Cl-6 alkyl group” is bonded to an amino group, such as methylamino, ethylamino-containing isopropylamino, n-Butylamino-containing isobutylamino-containing S-Butylamino, tert-Butylamino-containing n-Pentylami-containing Isopentylami-containing 2-Methylbutylamino-containing Neopentylamino, 1-ethylpropylamino-containing n-Hexylami-containing Isohexylami-containing 4-methylpentylami containing 3 -Methylpentylamino, 2-methylpentylamino, 1-methylpentylamino, 3,3-dimethylbutylamino, 2,2-dimethylbutylamino, 1,1-dimethylbutylamino, 1,2-dimethyl Butylamino-containing 1,3-dimethylbutylamido-containing 2,3-dimethylbutylamido-containing or 2-ethylbutylamino group, R 2 , The substituent of the substituent group α and the C1-C6 alkylamino group of the substituent group α is preferably a methylamino group, an ethylamino group, or an isopropylamino group.
[0032] 本発明にお!/、て、「C3-10シクロアルキルアミノ基」とは、前記「C3-10シクロアルキル 」がァミノ基に結合した基であり、例えば、シクロプロピルアミ入シクロブチルアミ入シ クロペンチルアミ入シクロへキシルアミ入シクロへプチルアミ入ノルボルニルァミノ、 ァダマンチルァミノ基を挙げることができる。置換基群 αにおいては好適には 3乃至 7 員飽和環状炭化水素アミノ基であり、更に好適にはシクロプロピルアミ入シクロプチ ルァミノ、シクロペンチルアミ入シクロへキシルァミノ基である。 [0032] In the present invention, the "C3-10 cycloalkylamino group" is a group in which the "C3-10 cycloalkyl" is bonded to an amino group, such as cyclopropylamino-containing cyclobutyl. Examples include amide-containing cyclopentyl amide-containing cyclohexyl amide-containing cycloheptyl amide-containing norbornylamino groups and adamantylamino groups. In the substituent group α, a 3- to 7-membered saturated cyclic hydrocarbon amino group is preferable, and a cyclopropylamino-containing cyclopropylamino or cyclopentylamino-containing cyclohexylamino group is more preferable.
[0033] 本発明において、「Cl-6アルコキシァミノ基」とは、前記「Cl-6アルコキシ基」がアミ ノ基に結合した基であり、例えば、メトキシアミ入エトキシァミノ、 n-プロボキシァミノ、 イソプロポキシァミノ、 n-ブトキシアミ入イソブトキシアミ入 S-ブトキシァミノ、 tert-ブト キシァミノ、 n-ペントキシアミ入イソペントキシァミノ、 2-メチルブトキシアミ入ネオペン トキシアミ入 n-へキシルォキシアミ入 4-メチルペントキシアミ入 3-メチルペントキシ アミ入 2-メチルペントキシアミ入 3,3-ジメチルブトキシアミ入 2,2-ジメチルブトキシァ ミ入 1,1-ジメチルブトキシアミ入 1,2-ジメチルブトキシアミ入 1,3-ジメチルブトキシァ ミ入 2,3-ジメチルブトキシァミノのような炭素数 1乃至 6個の直鎖又は分枝鎖アルコキ シァミノ基を挙げることができる。置換基群 αにおいては、好適にはメトキシァミノ基、 エトキシァミノ基、 η -プロボキシァミノ基である。 In the present invention, the “Cl-6 alkoxyamino group” is a group in which the “Cl-6 alkoxy group” is bonded to an amino group, such as methoxyamino-containing ethoxyamino, n-propoxyamino, isopropoxy, and the like. Amino, n-butoxyamido-isobutoxyamido S-butoxyamino, tert-butoxyamino, n-pentoxyamido-isopentoxyamino, 2-methylbutoxyamido-neopentoxyami n-hexyloxyamime-containing 4-methylpentoxyamido With 3-methylpentoxyami with 2-methylpentoxyami with 3,3-dimethylbutoxyami with 2,2-dimethylbutoxy with 1,1-dimethylbutoxyami with 1,2-dimethylbutoxyami 1, Examples include straight or branched alkoxy groups having 1 to 6 carbon atoms such as 2,3-dimethylbutoxyamino containing 3-dimethylbutoxyamino.In the substituent group α, a methoxyamino group, an ethoxyamino group, and an η-propoxyamino group are preferable.
[0034] 本発明において、「Cl-6ジアルキルアミノ基」とは、ァミノ基に同一又は異なった前 記「Cl-6アルキル基」力 ^個置換した基であり、例えば、 Ν,Ν-ジメチルアミ入 Ν,Ν-ジ ェチルアミ入 Ν,Ν-ジ η-プロピルアミ入 Ν,Ν-ジイソプロピルアミ入 Ν,Ν-ジ η-ブチル アミ入 Ν,Ν-ジイソブチルアミ入 Ν,Ν-ジ S-ブチルアミ入 Ν,Ν-ジ tert-ブチルアミ入 N, N-ジ n-ペンチルアミ入 Ν,Ν-ジイソペンチルアミ入 Ν,Ν-ジ 2-メチルブチルアミ入 Ν, Ν-ジネオペンチルアミ入 Ν,Ν-ジ 1-ェチルプロピルアミ入 Ν,Ν-ジ η-へキシルァミノ、
Ν,Ν-ジイソへキシルアミ入 Ν,Ν-ジ 4-メチルペンチルアミ入 Ν,Ν-ジ 3-メチルペンチ ルァミノ、 Ν,Ν-ジ 2-メチルペンチルアミ入 Ν,Ν-ジ 1-メチルペンチルアミ入 Ν,Ν-ェチ ルメチルァミノ、 Ν,Ν-イソプロピルメチルアミノ基を挙げることができ、 R2、
 置換基 群 α及び置換基群 aの C1-C6アルキルアミノ基の置換基においては、好適にはジメ チルァミノ又はジェチルァミノ基である。 [0034] In the present invention, the "Cl-6 dialkylamino group" is a group in which the same or different "Cl-6 alkyl group" force is substituted on an amino group, such as Ν, Ν-dimethylamino. Ν, Ν-diethyl amide input Ν, ジ -di η-propyl amide input Ν, Ν-diisopropyl amide input Ν, Ν-di η-butyl amide input Ν, Ν-diisobutyl amide input Ν, Ν-diisobutyl amide input Ν, ジ -di S-butyl amide input Ν, Ν-Di tert-Butylamide N, N-Di n-Pentylami Ν, Ν-Diisopentylami Ν, Ν-Di 2-Methylbutylamide Ν, Ν-Dineopentylami Ν, Ν -Di 1-ethylpropylamino 入, Ν-di η-hexylamino, Ν, Ν-Diisohexylamide Ν, Ν-Di 4-methylpentylamide 入, Ν-Di 3-methylpentylamino, Ν, Ν-Di 2-methylpentylamide Ν, Ν-Di 1-methylpentylamide入, Ν-ethylmethylamino, Ν, Ν-isopropylmethylamino group, R 2 , The substituent of the C1-C6 alkylamino group in the substituent group α and the substituent group a is preferably a dimethylamino group or a jetylamino group.
置換基 群 α及び置換基群 aの C1-C6アルキルアミノ基の置換基においては、好適にはジメ チルァミノ又はジェチルァミノ基である。 [0034] In the present invention, the "Cl-6 dialkylamino group" is a group in which the same or different "Cl-6 alkyl group" force is substituted on an amino group, such as Ν, Ν-dimethylamino. Ν, Ν-diethyl amide input Ν, ジ -di η-propyl amide input Ν, Ν-diisopropyl amide input Ν, Ν-di η-butyl amide input Ν, Ν-diisobutyl amide input Ν, Ν-diisobutyl amide input Ν, ジ -di S-butyl amide input Ν, Ν-Di tert-Butylamide N, N-Di n-Pentylami Ν, Ν-Diisopentylami Ν, Ν-Di 2-Methylbutylamide Ν, Ν-Dineopentylami Ν, Ν -Di 1-ethylpropylamino 入, Ν-di η-hexylamino, Ν, Ν-Diisohexylamide Ν, Ν-Di 4-methylpentylamide 入, Ν-Di 3-methylpentylamino, Ν, Ν-Di 2-methylpentylamide Ν, Ν-Di 1-methylpentylamide入, Ν-ethylmethylamino, Ν, Ν-isopropylmethylamino group, R 2 , The substituent of the C1-C6 alkylamino group in the substituent group α and the substituent group a is preferably a dimethylamino group or a jetylamino group.
[0035] 本発明において、「 - 6脂肪族ァシルァミノ基」とは、炭素数 1乃至 6個の脂肪族炭 化水素基又は水素原子がカルボニルァミノ基に結合した基であり、例えば、ホルミル アミ入ァセチルアミ入プロピオニルアミ入ブチリルァミノ、イソブチリルアミ入ペンタ ノィルァミノ、ビバロイルアミ入バレリルアミ入イソバレリルアミノ基のようなアルキル力 ルボニルァミノ基、クロロアセチルアミ入ジクロロアセチルアミ入トリクロロアセチルァ ミ入トリフルォロアセチルァミノのようなハロゲン化アルキルカルボ-ルァミノ基、メトキ シァセチルァミノのような低級アルコキシアルキルカルボ-ルァミノ基、(E)-2-メチル- 2-ブテノィルァミノのような不飽和アルキルカルボ-ルァミノ基等が挙げられる。
 R3 及び置換基群 aのァミノ基の置換基において、好適にはァセチルァミノ基である。 In the present invention, the “−6 aliphatic acylamine group” is an aliphatic hydrocarbon group having 1 to 6 carbon atoms or a group in which a hydrogen atom is bonded to a carbonylamino group. For example, formylamino Alkyl power such as acetylamino-containing propionylamino-containing butyrylamino, isobutyrylami-containing penta-nylamino, bivaloylami-containing valeryl-amido-isovalerylamino group And a halogenated alkyl carbolumino group, a lower alkoxyalkyl carboamino group such as methoxyacetylamino, an unsaturated alkyl carbolumino group such as (E) -2-methyl-2-butenoylamino, and the like. In the substituents of Amino groups R 3 and Substituent group a, preferably a Asechiruamino group.
R3 及び置換基群 aのァミノ基の置換基において、好適にはァセチルァミノ基である。 In the present invention, the “−6 aliphatic acylamine group” is an aliphatic hydrocarbon group having 1 to 6 carbon atoms or a group in which a hydrogen atom is bonded to a carbonylamino group. For example, formylamino Alkyl power such as acetylamino-containing propionylamino-containing butyrylamino, isobutyrylami-containing penta-nylamino, bivaloylami-containing valeryl-amido-isovalerylamino group And a halogenated alkyl carbolumino group, a lower alkoxyalkyl carboamino group such as methoxyacetylamino, an unsaturated alkyl carbolumino group such as (E) -2-methyl-2-butenoylamino, and the like. In the substituents of Amino groups R 3 and Substituent group a, preferably a Asechiruamino group.
[0036] 本発明において、「C 1-6アルコキシィミノ基」とは、前記「Cl-6アルコキシ基」がィミノ 基に結合した基であり、例えば、メトキシイミ入エトキシイミ入 n-プロポキシイミ入イソ プロポキシィミノ、 n-ブトキシィミノ、イソブトキシィミノ、 S-ブトキシィミノ、 tert-ブトキシ イミ入 n-ペントキシイミ入イソペントキシイミ入 2-メチルブトキシイミ入ネオペントキ シィミノ、 n-へキシルォキシイミ入 4-メチルペントキシイミ入 3-メチルペントキシィミノ 、 2-メチルペントキシイミ入 3, 3-ジメチルブトキシイミ入 2, 2-ジメチルブトキシイミ入 1 , 1-ジメチルブトキシイミ入 1 ,2-ジメチルブトキシイミ入 1 ,3-ジメチルブトキシイミ入 2,3 -ジメチルブトキシィミノのような炭素数 1乃至 6個の直鎖又は分枝鎖アルコキシィミノ 基を挙げることができる。 In the present invention, the “C 1-6 alkoxyimino group” is a group in which the “Cl-6 alkoxy group” is bonded to an imino group, and includes, for example, methoxyimid ethoxyimine-containing n-propoxyimid-isolated groups. Propoxymino, n-Butoxyimino, Isobutoxymino, S-Butoxyimino, tert-Butoxyimi, n-Pentoxyimi, Isopentoxyimi, 2-Methylbutoxyimi, Neopentoxymino, n-Hexyloxyimi, 4-Methylpentoxy With 3-methylpentoxyimino, with 2-methylpentoxyimi, with 3,3-dimethylbutoxyimi, with 2,2-dimethylbutoxyimi, with 1,1-dimethylbutoxyimi and with 1,2-dimethylbutoxyimi 1 Examples include straight-chain or branched alkoxyimino groups having 1 to 6 carbon atoms such as 2,3-dimethylbutoxyimino, 2,3-dimethylbutoxyimino. That.
[0037] 本発明において、「Cl-6アルコキシィミノ C1-3アルキル基」とは、前記「Cl-6アルコ キシィミノ基」が上記 C1-3アルキル基に結合した基であり、例えば、メトキシイミノメチ ル、メトキシイミノエチル、メトキシィミノ n-ブチル、エトキシイミノメチル、エトキシィミノ ェチル、 n-プロポキシイミノメチル、イソプロポキシイミノメチル、 n-ブトキシィミノメチル 、イソブトキシイミノメチル、 s-ブトキシイミノメチル、 tert-ブトキシイミノメチル、 n-ベント
キシイミノメチル、イソペントキシイミノメチル、 2-メチルブトキシイミノメチル、ネオベント キシイミノメチル、 n-へキシルォキシイミノメチル、 4-メチルペントキシイミノメチル、 3-メ チルペントキシイミノメチル、 2-メチルペントキシイミノメチル、 3,3-ジメチルブトキシイミ ノメチル、 2,2-ジメチルブトキシイミノメチル、 1 , 1-ジメチルブトキシイミノメチル、 1 ,2-ジ メチルブトキシイミノメチル、 1 ,3-ジメチルブトキシイミノメチル、 2,3-ジメチルブトキシィ ミノメチルのような炭素数 1乃至 6個の直鎖又は分枝鎖アルコキシィミノアルキル基を 挙げることができ、 R2及び R3においては、好適にはエトキシィミノメチル又はメトキシィ ミノメチル基であり、更に好適にはメトキシィミノメチル基である。 In the present invention, the “Cl-6 alkoxyimino C1-3 alkyl group” is a group in which the “Cl-6 alkoxyximino group” is bonded to the C1-3 alkyl group, and includes, for example, methoxyimino Methyl, methoxyiminoethyl, methoxyimino n-butyl, ethoxyiminomethyl, ethoxyiminoethyl, n-propoxyiminomethyl, isopropoxyiminomethyl, n-butoxyiminomethyl, isobutoxyiminomethyl, s-butoxyiminomethyl, tert- Butoxyiminomethyl, n-vent Xyiminomethyl, isopentoxyiminomethyl, 2-methylbutoxyiminomethyl, neovent xyiminomethyl, n-hexyloxyiminomethyl, 4-methylpentoxyiminomethyl, 3-methylpentoxyiminomethyl, 2- Methylpentoxyiminomethyl, 3,3-dimethylbutoxyiminomethyl, 2,2-dimethylbutoxyiminomethyl, 1,1-dimethylbutoxyiminomethyl, 1,2-dimethylbutoxyiminomethyl, 1,3-dimethylbutoxyimino Examples thereof include straight chain or branched alkoxyiminoalkyl groups having 1 to 6 carbon atoms such as methyl and 2,3-dimethylbutoxyminomethyl. R 2 and R 3 are preferably ethoxylates. It is a minomethyl or methoxyminominomethyl group, more preferably a methoxyiminomethyl group.
[0038] 本発明において、「Cl-6アルコキシカルボ-ル基」とは、前記「Cl-6アルコキシ基」 がカルボ-ル基に結合した基であり、例えば、メトキシカルボ-ル、エトキシカルボ- ル、 n-プロポキシカルボニル、イソプロポキシカルボニル、 n-ブトキシカルボニル、イソ ブトキシカルボニル、 s-ブトキシカルボニル、 tert-ブトキシカルボニル、 n-ペントキシ カルボニル、イソペントキシカルボニル、 2-メチルブトキシカルボニル、ネオペントキシ カルボニル、 n-へキシルォキシカルボニル、 4-メチルペントキシカルボニル、 3-メチ ルペントキシカルボニル、 2-メチルペントキシカルボニル、 3, 3-ジメチルブトキシカル ボニル、 2, 2-ジメチルブトキシカルボニル、 1 , 1-ジメチルブトキシカルボニル、 1 ,2-ジメ チルブトキシカルボニル、 1 ,3-ジメチルブトキシカルボニル、 2,3-ジメチルブトキシカ ルポニルのような炭素数 1乃至 6個の直鎖又は分枝鎖アルコキシカルボ-ル基を挙げ ることができ、置換基群 αにおいては、好適にはメトキシカルボ-ル、エトキシカルボ -ル、 tert-ブトキシカルボ-ル基である。 In the present invention, the “Cl-6 alkoxycarbo group” is a group in which the “Cl-6 alkoxy group” is bonded to a carbo group, such as methoxy carbo yl, ethoxy carbo ter. N-propoxycarbonyl, isopropoxycarbonyl, n-butoxycarbonyl, isobutoxycarbonyl, s-butoxycarbonyl, tert-butoxycarbonyl, n-pentoxycarbonyl, isopentoxycarbonyl, 2-methylbutoxycarbonyl, neopentoxycarbonyl, n -Hexyloxycarbonyl, 4-methylpentoxycarbonyl, 3-methylpentoxycarbonyl, 2-methylpentoxycarbonyl, 3,3-dimethylbutoxycarbonyl, 2,2-dimethylbutoxycarbonyl, 1, 1-dimethyl Butoxycarbonyl, 1,2-dimethylbutoxycarbonyl, 1,3-dimethylbutane Examples thereof include straight-chain or branched alkoxycarbonyl groups having 1 to 6 carbon atoms such as tooxycarbonyl and 2,3-dimethylbutoxycarbonyl. In the substituent group α, methoxy is preferably used. A carbo carbonyl, an ethoxy carbo yl, and a tert-butoxy carbo yl group.
[0039] 本発明にお 、て、「C6-10ァリール基」とは、炭素数 6乃至 10個の芳香族炭化水素 基であり、例えば、フエ-ル、インデュル、ナフチル基を挙げることができ、
 R3及び 置換基群 aにおいては好適にはフエ-ル基である。 In the present invention, the “C6-10 aryl group” is an aromatic hydrocarbon group having 6 to 10 carbon atoms, and examples thereof include a phenyl, indul, and naphthyl group. , In R 3 and substituent group a, a phenyl group is preferred.
R3及び 置換基群 aにおいては好適にはフエ-ル基である。 In the present invention, the “C6-10 aryl group” is an aromatic hydrocarbon group having 6 to 10 carbon atoms, and examples thereof include a phenyl, indul, and naphthyl group. , In R 3 and substituent group a, a phenyl group is preferred.
[0040] 本発明において、「窒素、酸素及び硫黄力も選択される同一若しくは異なった 1-3 個のへテロ原子を含む 4-10員複素環基」とは、硫黄原子、酸素原子又は窒素原子を 1乃至 3個含む 4乃至 10員複素環基であり、例えばフリル、チェ-ル、ピロリル、ァゼピ -ル、ピラゾリル、イミダゾリル、ォキサゾリル、イソキサゾリル、チアゾリル、イソチアゾリ ル、 1 ,2,3-ォキサジァゾリル、トリァゾリル、テトラゾリル、チアジアゾリル、ビラニル、 ピ
リジル、ピリダジニル、ピリミジニル、ビラジニルのような芳香族複素環基及びモルホリ -ル、チオモルホリニル、ピロリジ -ル、ピロリニル、イミダゾリジ -ル、イミダゾリ-ル、 ビラゾリジ-ル、ビラゾリ-ル、ピベリジ-ル、ピぺラジュル、テトラヒドロビラ-ルのよう なこれらの基に対応する、部分若しくは完全還元型の基を挙げることができる。尚、 上記「4乃至 10員複素環基」は、他の環式基と縮環していてもよぐ例えば、イソベン ゾフラ -ル、クロメ-ル、インドリジ -ル、イソインドリル、インドリル、インダゾリル、プリ ニル、キノリジニル、イソキノリル、キノリル、フタラジュル、ナフチリジニル、キノキサリ -ル、キナゾリ-ル、イソインドリニル基を挙げることができる。 R2、 R3及び置換基群 a において好適には、窒素原子を少なくとも 1個含み、酸素原子又は硫黄原子を含ん でいてもよい 4乃至 10員複素環基であり、例えばピロリル、ァゼピニル、ピラゾリル、ィ ミダゾリル、ォキサゾリル、イソキサゾリル、チアゾリル、イソチアゾリル、 1,2,3-ォキサジ ァゾリル、トリァゾリル、テトラゾリル、チアジアゾリル、ピリジル、ピリダジ -ル、ピリミジ -ル、ピラジュルのような芳香族複素環基及びモルホリニル、チオモルホリニル、ピロ リジ -ル、ピロリニル、イミダゾリジ -ル、イミダゾリ-ル、ビラゾリジ-ル、ビラゾリ-ル、 ピベリジ-ル、ピペラジニル、テトラヒドロビラ-ルのようなこれらの基に対応する、部 分若しくは完全還元型の基を挙げることができ、さらに好適には、ピリジル、ピリミジル 、ピラゾリル、ピロリジニル、ピベリジニル、ピペラジニル、モルホリニル、テトラヒドロピ ラ-ル基である。 [0040] In the present invention, "a 4- to 10-membered heterocyclic group containing 1 to 3 heteroatoms which are the same or different and the nitrogen, oxygen and sulfur powers are also selected" means a sulfur atom, an oxygen atom or a nitrogen atom. A 4- to 10-membered heterocyclic group containing 1 to 3, such as furyl, chael, pyrrolyl, azepyr, pyrazolyl, imidazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, 1,2,3-oxadiazolyl, Triazolyl, tetrazolyl, thiadiazolyl, biranyl, pi Aromatic heterocyclic groups such as lysyl, pyridazinyl, pyrimidinyl, birazinyl and morpholyl, thiomorpholinyl, pyrrolidyl, pyrrolinyl, imidazolidyl, imidazolyl, virazolyl, virazolyl, piperidyl, pipepe Mention may be made of partially or fully reduced groups corresponding to these groups such as rajur, tetrahydrobiral. The above “4- to 10-membered heterocyclic group” may be condensed with other cyclic groups, for example, isobenzazolyl, chromel, indolizyl, isoindolyl, indolyl, indazolyl, pre- Examples include nyl, quinolizinyl, isoquinolyl, quinolyl, phthalajuryl, naphthyridinyl, quinoxalyl, quinazolyl, and isoindolinyl groups. R 2 , R 3 and the substituent group a are preferably a 4- to 10-membered heterocyclic group containing at least one nitrogen atom and optionally containing an oxygen atom or a sulfur atom, such as pyrrolyl, azepinyl, pyrazolyl. , Imidazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, 1,2,3-oxadiazolyl, triazolyl, tetrazolyl, thiadiazolyl, pyridyl, pyridazyl, pyrimidyl, pyrazol, and morpholinyl, thiomorpholinyl , Partially or fully reduced, corresponding to these groups such as pyrrolidyl, pyrrolinyl, imidazolyl, imidazolyl, virazolidyl, virazolyl, piperidyl, piperazinyl, tetrahydroviral And more preferably, pyridyl, pyrimidyl, Zoriru, pyrrolidinyl, piperidinyl, piperazinyl, morpholinyl, Tetorahidoropi La - a le group.
[0041] 本発明において、「窒素、酸素及び硫黄力も選択される同一若しくは異なった 1-3 個のへテロ原子を含む 4-10員複素環 C1-C6アルキル基」とは、前記「C1-C6アルキ ル基」に上記「窒素、酸素及び硫黄力 選択される同一若しくは異なった 1-3個のへ テロ原子を含む 4-10員複素環」が置換した基であり、 R2及び R3においては、好適には 窒素、酸素及び硫黄力 選択される同一若しくは異なった 1-3個のへテロ原子を含む 4-10員複素環メチル基であり、更に好適にはピリジルメチル、ピロリジルメチル、ピぺ リジルメチル、モルホリルメチル基である。 [0041] In the present invention, "a 4- to 10-membered heterocyclic C1-C6 alkyl group containing 1-3 heteroatoms which are the same or different and also have nitrogen, oxygen and sulfur powers selected" means the above-mentioned "C1- C6 alkyl group ”is a group in which the above“ 4-10 membered heterocyclic ring containing 1-3 heteroatoms which are the same or different selected from nitrogen, oxygen and sulfur power ”is substituted, and R 2 and R 3 Is preferably a 4-, 10-membered heterocyclic methyl group containing the same or different 1-3 heteroatoms selected from nitrogen, oxygen and sulfur power, and more preferably pyridylmethyl, pyrrolidylmethyl. , Piperidylmethyl and morpholylmethyl groups.
[0042] 本発明において、「窒素、酸素及び硫黄力も選択される同一若しくは異なった 1-3 個のへテロ原子を含む 4-10員複素環 C1-C6アルキルアミノ基」とは、前記「C1-C6ァ ルキルアミノ基」に上記「窒素、酸素及び硫黄力 選択される同一若しくは異なった 1-
3個のへテロ原子を含む 4-10員複素環」が置換した基であり、 R2、 R3及び置換基群ひ においては、好適には窒素、酸素及び硫黄力も選択される同一若しくは異なった 1-3 個のへテロ原子を含む 4-10員複素環ェチルァミノ基であり、更に好適にはピリジルメ チルァミノ、ピリジルェチルァミノ、ピロリジルェチルアミ入ピペリジルェチルアミ入モ ルホリルェチルァミノ基である。 [0042] In the present invention, "a 4- to 10-membered heterocyclic C1-C6 alkylamino group containing 1-3 heteroatoms which are the same or different and also have nitrogen, oxygen and sulfur powers selected" means "C1 -C6 alkylamino group "is the same or different 1- `` 4-10 membered heterocyclic ring containing 3 heteroatoms '' is a substituted group, and in R 2 , R 3 and the group of substituents, nitrogen, oxygen and sulfur forces are also preferably selected. And a 4- to 10-membered heterocyclic ethylamino group containing 1 to 3 heteroatoms, more preferably pyridylmethylamino, pyridylethylamino, pyrrolidylethylamino-piperidylethylamino-containing morpholyl. It is a tyramino group.
[0043] 本発明にお 、て、「ハロゲン原子」とは、弗素原子、塩素原子、臭素原子又は沃素 原子であり、 R1X、 R1Y、 R2、
 置換基群 α及び Cl-3アルキレン基の置換基にお いては、好適には、弗素原子である。 In the present invention, the “halogen atom” is a fluorine atom, a chlorine atom, a bromine atom or an iodine atom, and R 1X , R 1Y , R 2 , Substituent group The substituents of α and Cl-3 alkylene groups are preferably fluorine atoms.
置換基群 α及び Cl-3アルキレン基の置換基にお いては、好適には、弗素原子である。 In the present invention, the “halogen atom” is a fluorine atom, a chlorine atom, a bromine atom or an iodine atom, and R 1X , R 1Y , R 2 , Substituent group The substituents of α and Cl-3 alkylene groups are preferably fluorine atoms.
[0044] 本発明にお 、て、 「C 1-6力ルバモイルアルキルアミノ基」とは、前記「C1-C6アルキ ルァミノ基」に力ルバモイル基が置換した基であり、置換基群 aにおいて、好適にはIn the present invention, the “C 1-6 force rubamoylalkylamino group” is a group obtained by substituting a force rubamoyl group for the “C1-C6 alkylamino group”. Preferably
、力ルバモイルメチルァミノ基である。 , Force ruberamoylmethylamino group.
[0045] [0045]
本発明の前記一般式 (1)、(Γ)又は (II)を有するチアゾール誘導体は、塩基性基を有 するため、常法に従って酸付加塩にすることができる。そのような塩としては、例えば フッ化水素酸、塩酸、臭化水素酸、沃化水素酸のようなハロゲン化水素酸の塩;硝酸 塩、過塩素酸塩、硫酸塩、燐酸塩のような無機酸塩;メタンスルホン酸、トリフルォロメ タンスルホン酸、エタンスルホン酸のような低級アルカンスルホン酸の塩;ベンゼンス ルホン酸、 p-トルエンスルホン酸のようなァリールスルホン酸の塩;グルタミン酸、ァス パラギン酸のようなアミノ酸の塩;酢酸、フマール酸、酒石酸、蓚酸、マレイン酸、リン ゴ酸、コハク酸、安息香酸、マンデル酸、ァスコルビン酸、乳酸、ダルコン酸、クェン 酸のようなカルボン酸の塩を挙げることができる。好適にはハロゲン化水素酸の塩で あり、最も好適には塩酸塩である。 Since the thiazole derivative having the general formula (1), (Γ) or (II) of the present invention has a basic group, it can be converted into an acid addition salt according to a conventional method. Such salts include, for example, hydrohalic acid salts such as hydrofluoric acid, hydrochloric acid, hydrobromic acid, hydroiodic acid; nitrates, perchlorates, sulfates, phosphates, etc. Mineral acid salts; Salts of lower alkane sulfonic acids such as methane sulfonic acid, trifluoro methane sulfonic acid and ethane sulfonic acid; Salts of allylic sulfonic acid such as benzene sulfonic acid and p-toluene sulfonic acid; Glutamic acid and aspartic acid A salt of an amino acid such as acetic acid, fumaric acid, tartaric acid, succinic acid, maleic acid, phosphoric acid, succinic acid, benzoic acid, mandelic acid, ascorbic acid, lactic acid, darconic acid, and carboxylic acid. Can be mentioned. Preferred is a salt of hydrohalic acid, and most preferred is hydrochloride.
[0046] 更に、前記一般式 (1)、(Γ)又は (II)を有するチアゾール誘導体は、カルボキシル基を 有する場合、常法に従って金属塩にすることができる。そのような塩としては、例えば リチウム、ナトリウム、カリウムのようなアルカリ金属塩;カルシウム、ノ リウム、マグネシ ゥムのようなアルカリ土類金属塩;アルミニウム塩をあげることができる。好適にはアル カリ金属塩である。 Furthermore, when the thiazole derivative having the general formula (1), (Γ) or (II) has a carboxyl group, it can be converted into a metal salt according to a conventional method. Examples of such salts include alkali metal salts such as lithium, sodium, and potassium; alkaline earth metal salts such as calcium, norum, and magnesium; and aluminum salts. Alkali metal salts are preferred.
[0047] なお、前記一般式 (1)、(Γ)又は (II)を有するチアゾール誘導体は、種々の異性体を有
することがある。前記一般式 0)、(Γ)又は (Π)においては、これら異性体及びこれら異性 体の等量及び非等量混合物がすべて単一の式で示されている。従って、本発明は、 これらの異性体及びこれら異性体の種々の割合での混合物をすベて含むものである [0047] The thiazole derivative having the general formula (1), (Γ) or (II) has various isomers. There are things to do. In the above general formulas 0), (Γ) or (こ れ ら), these isomers and equivalent and unequal mixtures of these isomers are all represented by a single formula. Accordingly, the present invention includes all of these isomers and mixtures of these isomers in various proportions.
[0048] 更に本発明は、前記一般式 (1)、(Γ)又は (II)を有するチアゾール誘導体又はその塩 が溶媒和物(例えば水和物)を形成する場合には、これらもすベて含むものである。 [0048] Furthermore, the present invention relates to a case where the thiazole derivative having the general formula (1), (Γ) or (II) or a salt thereof forms a solvate (for example, a hydrate). Included.
[0049] [0049]
上記一般式 (1)、(Γ)又は (II)において、 R1Xは、好適には P(0)(OH)、 ΟΡ(ΟΧΟΗ)、又 In the general formula (1), (Γ) or (II), R 1X is preferably P (0) (OH), ΟΡ (ΟΧΟΗ), or
2 2 は〇CH P(〇)(〇H)であり、更に好適には OCH P(〇)(〇H)である。 2 2 is OCH P (O) (OH), and more preferably OCH P (O) (OH).
2 2 2 2 2 2 2 2
[0050] R1Yは、好適には水素原子、フッ素原子、又は OCH P(〇)(〇H)であり、更に好適に [0050] R 1Y is preferably a hydrogen atom, a fluorine atom, or OCH P (◯) (〇H), more preferably
2 2 twenty two
は水素原子である。 Is a hydrogen atom.
[0051] R1は、好適には P(0)(0H)、 0P(0)(0H)、又は OCH P(0)(0H)であり、更に好適に [0051] R 1 is preferably P (0) (0H), 0P (0) (0H), or OCH P (0) (0H), and more preferably
2 2 2 2 2 2 2 2
は OCH P(0)(0H)である。 Is OCH P (0) (0H).
2 2 twenty two
[0052] R2は、好適には C1-6アルキル基、 C1-6アルコキシ基、 C 1-6ジアルキルアミノ基、ハ ロゲン原子、水素原子であり、更に好適には C1-6アルキル基又は水素原子であり、 特に好適には、メチル基又は水素原子である。 [0052] R 2 is preferably a C1-6 alkyl group, C1-6 alkoxy group, C 1-6 dialkylamino group, Ha androgenic atom, a hydrogen atom, more preferably a C1-6 alkyl group or hydrogen An atom, particularly preferably a methyl group or a hydrogen atom.
[0053] R3は、好適には C1-6アルキル基、 C1-6アルコキシ基、ジアルキルアミノ基、ァシル 基、ハロゲン原子、水素原子であり、更に好適には水素原子、 C1-6アルキル基、 C1- 6アルコキシ基、ジアルキルアミノ基、ァシル基であり、特に好適にはェチル基、メチ ル基又はメトキシ基である。 [0053] R 3 is preferably a C1-6 alkyl group, a C1-6 alkoxy group, a dialkylamino group, an acyl group, a halogen atom, or a hydrogen atom, more preferably a hydrogen atom, a C1-6 alkyl group, A C1-6 alkoxy group, a dialkylamino group, and an acyl group, particularly preferably an ethyl group, a methyl group, or a methoxy group.
[0054] R4は、好適には水素原子である。 [0054] R 4 is preferably a hydrogen atom.
[0055] Xは、好適にはメチレンである。 [0055] X is preferably methylene.
[0056] 置換基群 aは、 R2の該ァミノアルキル基のアミノ基を置換する場合、好適には、 C1-[0056] substituent group a, when replacing the amino group of the Aminoarukiru group R 2, preferably, C1-
6アルキル基、 C3-C10シクロアルキル基であり、更に好適には、メチル基、ェチル基、 プロピル基、シクロプロピル基である。 A 6-alkyl group and a C3-C10 cycloalkyl group, more preferably a methyl group, an ethyl group, a propyl group, and a cyclopropyl group.
[0057] 置換基群 aは、 R3の該ァミノアルキル基のアミノ基を置換する場合、好適には、 C1-[0057] substituent group a, when replacing the amino group of the Aminoarukiru groups R 3, preferably, C1-
6アルキル基、 C3-C10シクロアルキル基であり、更に好適には、メチル基、ェチル基、 プロピル基、シクロプロピル基である。
[0058] 置換基群 aは、 R2の Cl-6脂肪族ァシル基を置換する場合、好適には、 C1-6アルコ キシ基、水酸基、アミノ基、モノアルキルアミノ基、ジアルキルアミノ基である。 A 6-alkyl group and a C3-C10 cycloalkyl group, more preferably a methyl group, an ethyl group, a propyl group, and a cyclopropyl group. [0058] Substituent group a is preferably a C1-6 alkoxy group, a hydroxyl group, an amino group, a monoalkylamino group, or a dialkylamino group when the Cl-6 aliphatic acyl group of R 2 is substituted. .
[0059] 置換基群 aは、 R3の C 1-6脂肪族ァシル基を置換する場合、好適には、 C1-6アルキ ル基、 C1-6アルコキシ基、水酸基、アミノ基、モノアルキルアミノ基、ジアルキルアミノ 基である。 [0059] substituent group a, when replacing the C 1-6 aliphatic Ashiru group R 3, preferably, C1-6 alkyl Le group, C1-6 alkoxy group, a hydroxyl group, an amino group, a monoalkylamino Group, a dialkylamino group.
[0060] 置換基群 aは、 R2の C6-10ァリール基を置換する場合、好適には、 C1-6アルキル 基、 C1-6ノヽロアルキル基、 C1-6アルコキシ基、水酸基、アミノ基、ジアルキルアミノ基[0060] Substituent group a is preferably a C1-6 alkyl group, a C1-6 neuroalkyl group, a C1-6 alkoxy group, a hydroxyl group, an amino group, when the C6-10 aryl group of R 2 is substituted. Dialkylamino group
、ァシル基、ハロゲン原子である。 A acyl group and a halogen atom.
[0061] 置換基群 aは、 R3の C6-10ァリール基を置換する場合、好適には、 C1-6アルキル 基、 C1-6ノヽロアルキル基、 C1-6アルコキシ基、水酸基、アミノ基、ジアルキルアミノ基[0061] Substituent group a is preferably a C1-6 alkyl group, a C1-6 neuroalkyl group, a C1-6 alkoxy group, a hydroxyl group, an amino group, when the C6-10 aryl group of R 3 is substituted. Dialkylamino group
、ァシル基、ハロゲン原子である。 A acyl group and a halogen atom.
[0062] 置換基群 aは、 R2の窒素、酸素及び硫黄力 選択される同一若しくは異なった 1-3 個のへテロ原子を含む 4-10員複素環基を置換する場合、好適には、 C1-6アルキル 基、 C1-6ノヽロアルキル基、 C1-6アルコキシ基、水酸基、アミノ基、ジアルキルアミノ基[0062] substituent group a, when replacing the 4-10 membered heterocyclic group containing nitrogen R 2, the same or different 1-3 heteroatom selected from oxygen and sulfur force, preferably , C1-6 alkyl group, C1-6 neuroalkyl group, C1-6 alkoxy group, hydroxyl group, amino group, dialkylamino group
、ァシル基、ハロゲン原子である。 A acyl group and a halogen atom.
[0063] 置換基群 aは、 R3の窒素、酸素及び硫黄力 選択される同一若しくは異なった 1-3 個のへテロ原子を含む 4-10員複素環基を置換する場合、好適には、 C1-6アルキル 基、 C1-6ノヽロアルキル基、 C1-6アルコキシ基、水酸基、アミノ基、ジアルキルアミノ基[0063] substituent group a, when replacing the 4-10 membered heterocyclic group containing nitrogen R 3, the same or different 1-3 heteroatom selected from oxygen and sulfur force, preferably , C1-6 alkyl group, C1-6 neuroalkyl group, C1-6 alkoxy group, hydroxyl group, amino group, dialkylamino group
、ァシル基、ハロゲン原子である。 A acyl group and a halogen atom.
[0064] 置換基群 aは、 R2の窒素、酸素及び硫黄力 選択される同一若しくは異なった 1-3 個のへテロ原子を含む 4-10員複素環 C1-C6アルキル基を置換する場合、好適には[0064] Substituent group a is a nitrogen, oxygen and sulfur force of R 2 When a 4- to 10-membered heterocyclic C1-C6 alkyl group containing 1-3 identical or different hetero atoms is selected Preferably
、 C1-6アルキル基、 C1-6ハロアルキル基、 C1-6アルコキシ基、水酸基、アミノ基、ジ アルキルアミノ基、ァシル基、ハロゲン原子である。 A C1-6 alkyl group, a C1-6 haloalkyl group, a C1-6 alkoxy group, a hydroxyl group, an amino group, a dialkylamino group, an acyl group, and a halogen atom.
[0065] 置換基群 aは、 R3の窒素、酸素及び硫黄力 選択される同一若しくは異なった 1-3 個のへテロ原子を含む 4-10員複素環 C1-C6アルキル基を置換する場合、好適には[0065] Substituent group a is a nitrogen, oxygen and sulfur force of R 3 When a 4- to 10-membered heterocyclic C1-C6 alkyl group containing 1-3 identical or different hetero atoms is selected Preferably
、 C1-6アルキル基、 C1-6ハロアルキル基、 C1-6アルコキシ基、水酸基、アミノ基、ジ アルキルアミノ基、ァシル基、ハロゲン原子である。 A C1-6 alkyl group, a C1-6 haloalkyl group, a C1-6 alkoxy group, a hydroxyl group, an amino group, a dialkylamino group, an acyl group, and a halogen atom.
[0066] 置換基群 aは、 R2の窒素、酸素及び硫黄力 選択される同一若しくは異なった 1-3
個のへテロ原子を含む 4-10員複素環 C1-C6アルキルアミノ基を置換する場合、好適 には、 C1-6アルキル基、 C1-6ハロアルキル基、 C1-6アルコキシ基、水酸基、アミノ基 、ジアルキルアミノ基、ァシル基、ハロゲン原子である。 [0066] Substituent group a is the same or different selected from nitrogen, oxygen and sulfur power of R 2 1-3 When a 4- to 10-membered heterocyclic C1-C6 alkylamino group containing a hetero atom is substituted, a C1-6 alkyl group, a C1-6 haloalkyl group, a C1-6 alkoxy group, a hydroxyl group, an amino group is preferable. A dialkylamino group, an acyl group, and a halogen atom.
[0067] 置換基群 aは、 R3の窒素、酸素及び硫黄力 選択される同一若しくは異なった 1-3 個のへテロ原子を含む 4-10員複素環 C1-C6アルキルアミノ基を置換する場合、好適 には、 C1-6アルキル基、 C1-6ハロアルキル基、 C1-6アルコキシ基、水酸基、アミノ基 、ジアルキルアミノ基、ァシル基、ハロゲン原子である。 [0067] Substituent group a substitutes for a nitrogen, oxygen and sulfur power of R 3 and a 4-10 membered heterocyclic C1-C6 alkylamino group containing 1-3 identical or different heteroatoms. In this case, a C1-6 alkyl group, a C1-6 haloalkyl group, a C1-6 alkoxy group, a hydroxyl group, an amino group, a dialkylamino group, an acyl group, and a halogen atom are preferable.
[0068] [0068]
本発明の、前記一般式 0)、(Γ)又は (II)を有するチアゾール誘導体又はその薬理上 許容される塩の具体例としては、次に例示する化合物を挙げることができる。但し、本 発明は下記の例示化合物に限定されるものではない。 Specific examples of the thiazole derivative having the general formula 0), (Γ) or (II) or a pharmacologically acceptable salt thereof according to the present invention include the compounds exemplified below. However, the present invention is not limited to the following exemplary compounds.
[0069] なお、以下の表 1及び表 2において「Me」はメチル基を、「Et」はェチル基を、「nPr」 はノルマルプロピル基を、「'Pr」はイソプロピル基を、「ePr」はシクロプロピル基を、「 u 」はイソブチル基を、「 u」はターシャリーブチル基を、「¾ex」はシクロへキシル基を、 「ePn」はシクロペンチル基を、「MeO」はメトキシ基を、「EtO」はエトキシ基を、「Ph」は フエ-ル基を、「Pyr」はピリジル基を、「Piperidinyl」はピベリジ-ル基を、「Pirazolyl」は ピラゾリル基を、「Piperadinyl」はピペラジ-ル基を、「Pyrrolidinyl」はピロリジ-ル基を 、「Pyrimidyl」はピリミジル基を、「Mor」はモルホリ -ル基を、「guanidino」はグァ -ジノ 基を、「cyclopentylidene」はシクロペンチリデン基をそれぞれ示す。 R1において、 Aは OCH P(0)(OH)、 Bは CF P(0)(OH)、 Cは CH P(0)(OH)、 Dは OP(0)(OH)、 Eは P(O) [0069] In Tables 1 and 2 below, "Me" represents a methyl group, "Et" represents an ethyl group, " n Pr" represents a normal propyl group, "'Pr" represents an isopropyl group, " e Pr "is a cyclopropyl group, a" u "isobutyl group," u "is a tertiary butyl group, a" ¾ex "is a cyclohexyl group, a" e Pn 'is cyclopentyl, "MeO" a methoxy `` EtO '' is an ethoxy group, `` Ph '' is a phenol group, `` Pyr '' is a pyridyl group, `` Piperidinyl '' is a piperidyl group, `` Pirazolyl '' is a pyrazolyl group, `` Piperadinyl '' Is a piperazil group, “Pyrrolidinyl” is a pyrrolidyl group, “Pyrimidyl” is a pyrimidyl group, “Mor” is a morpholyl group, “guanidino” is a guazino group, and “cyclopentylidene” is a cyclopentylidene group. Each pentylidene group is shown. In R 1, A is OCH P (0) (OH) , B is CF P (0) (OH) , C is CH P (0) (OH) , D is OP (0) (OH), E is P (O)
2 2 2 2 2 2 2 2 2 2 2 2 2 2
(OH)を示す。 (OH) is shown.
2 2
[0070] [化 4] [0070] [Chemical 4]


(^) 〔〕0071l
-35 CH2 A H H C(=0)NH2 H-36 (CH2)2 A H H C(=0)NH2 H-37 CH2 A H H G(=0)NHMe H-38 CH2 A H H C(=0)NHEt H-39 CH2 A H H C(=0)NH"Pr H-40 CH2 A H H C(=0)NHPr H-41 CH2 A H H C(=0)NH°Pr H-42 CH2 A H H 0(=Ο)ΝΗ(Βυ H-43 CH2 A H H C(=0)NH°Pn H-44 CH2 A H H C(=0)NH°Hex H-45 (CH2)2 A H H C(=0)NHMe H-46 (CH2)3 A H H C(=0)NHMe H-47 cyclopentylidene-CH2 A H H C(=0)NH e H-48 CMe2CH2 A H H C(=0)NHMe H-49 CH2 A H H C(=0)NHOMe H-50 CH2 A H H C(=0)NHCH2 cPr H-51 CH2 A H H C(=0)NHCH2 4Bu H-52 CH2 A H H C(=0)NH(CH2)2OH H-53 CH2 A H H C(=0)NH(CH2)2O e H-54 CH2 A H H C(=0)NH(CH2)3OH H-55 CH2 A H H C(=0)NH(CHa)3OMe H-56 CH2 A H H C(=0)NHCH( e)CONH2 H-57 CH2 A H H C(=0)NH(CH2)2NH2 H-58 CH2 A H H C(=0)NH(CH2)2NMe2 H-59 CH2 A H H C(=0)CH2NMe2 H-60 CH2 A H H C(=0)CF3 H-61 CH2 A H H C(=0)CH2(1 -Pyrrolidinyl) H-62 CH2 A H H C(=0)CH2(1 -Piperadinyl) H-63 CH2 A H H C(=0)CH2(1 -Mor) H-64 CH2 A H H Br H-65 CH2 A H H F H-66 (CH2)2 A H H F H-67 (CH2)3 A H H F H-68 cyclopentylidene—Ch^ A H H F H-69 CMe2CH2 A H H F H
s/v:o£90sfcl>d ^ OSさ Ϊ O. (^) [] 0071l -35 CH 2 AHHC (= 0) NH 2 H-36 (CH 2 ) 2 AHHC (= 0) NH 2 H-37 CH 2 AHHG (= 0) NHMe H-38 CH 2 AHHC (= 0) NHEt H- 39 CH 2 AHHC (= 0) NH "Pr H-40 CH 2 AHHC (= 0) NHPr H-41 CH 2 AHHC (= 0) NH ° Pr H-42 CH 2 AHH 0 (= Ο) ΝΗ ( Βυ H -43 CH 2 AHHC (= 0) NH ° Pn H-44 CH 2 AHHC (= 0) NH ° Hex H-45 (CH 2 ) 2 AHHC (= 0) NHMe H-46 (CH 2 ) 3 AHHC (= 0) NHMe H-47 cyclopentylidene-CH 2 AHHC (= 0) NH e H-48 CMe 2 CH 2 AHHC (= 0) NHMe H-49 CH 2 AHHC (= 0) NHOMe H-50 CH 2 AHHC (= 0 ) NHCH 2 c Pr H-51 CH 2 AHHC (= 0) NHCH 2 4 Bu H-52 CH 2 AHHC (= 0) NH (CH 2 ) 2 OH H-53 CH 2 AHHC (= 0) NH (CH 2 ) 2 O e H-54 CH 2 AHHC (= 0) NH (CH 2 ) 3 OH H-55 CH 2 AHHC (= 0) NH (CH a ) 3 OMe H-56 CH 2 AHHC (= 0) NHCH ( e) CONH 2 H-57 CH 2 AHHC (= 0) NH (CH 2 ) 2 NH 2 H-58 CH 2 AHHC (= 0) NH (CH 2 ) 2 NMe 2 H-59 CH 2 AHHC (= 0) CH 2 NMe 2 H-60 CH 2 AHHC (= 0) CF 3 H-61 CH 2 AHHC (= 0) CH 2 (1 -Pyrrolidinyl) H-62 CH 2 AHHC (= 0) CH 2 (1 -Piperadinyl) H-63 CH 2 AHHC (= 0) CH 2 (1 -Mor) H-64 CH 2 AHH Br H-65 CH 2 AHHF H-66 (CH 2 ) 2 AHHF H-67 (CH 2 ) 3 AHHF H- 68 cyclopentylidene—Ch ^ AHHF H-69 CMe 2 CH 2 AHHFH s / v: o £ 90sfcl> d ^ OS Ϊ O.


Η H H V 2H0 ZLY-l Η HHV 2 H0 ZLY-l
Η X3H。 H H V JH0 ZLl-l Η X3H. HHV J H0 ZLl-l
Η ud。 H H V Z IZ.I-L Η u d. HHV Z IZ.IL
Η H H V ¾0 OLl-l Η H H V ¾0 OLl-l
Η H H V 5H0 691-1 HV HHV 5 H0 691-1
Η H H V ¾0 891-L Η H H V ¾0 891-L
Η H H V ZH0 LSl-l Η HHV Z H0 LSl-l
Η H H V ZH0 991-1 HV HHV Z H0 991-1
Η Jd。 H H V !H0 991-1 Η Jd. HHV ! H0 991-1
Η H H V 3H0 Wl-l Η HHV 3 H0 Wl-l
Η H H V ZH0 91-1 HV HHV Z H0 91-1
Η H H V HO) 391-1 Η H H V HO) 391-1
Η 3ΙΛ| H H V HO) 19ト L Η 3 ΙΛ | HHV HO) 19 to L
Η H H V ZH0 091- L Η HHV Z H0 091- L
2ΗΝ H H H 3 Z 0 691- L 2 ΗΝ HHH 3 Z 0 691- L
Η mo H H 3 ZH0 8SI-L Η mo HH 3 Z H0 8SI-L
Η H H 3 !H0 L91-1 Η HH 3 ! H0 L91-1
Η ή H H 3 ZH0 931- 1 Η ή HH 3 Z H0 931- 1
Η 8WHN(0=)0 H H 3 ZH0 SSI- 1 WH 8WHN (0 =) 0 HH 3 Z H0 SSI- 1
Η zHN(0=)0 H H 3 ZH0 Η z HN (0 =) 0 HH 3 Z H0
Η 」d。(0=)0 H H 3 ZH0 eg i- 1 」D. (0 =) 0 HH 3 Z H0 eg i- 1
Η »W(0=)0 H H 3 JH0 391-1. Η »W (0 =) 0 HH 3 J H0 391-1.
Η (·"八 d - HO H H Ξ Z Η (· "8 d-HO HH Ξ Z
Η H H H 3 HO 0 1- L Η H H H 3 HO 0 1- L
ΖΗΗ H H H a ZH0 6frl-l Ζ ΗΗ HHH a Z H0 6frl- l
ΖΗΝ H H H a - 1 Ζ ΗΝ HHH a - 1
ΖΗΝ H H H a zl-)Q_euapj|A^uado|oAo Lfl-l Ζ ΗΝ HHH a z l-) Q_euapj | A ^ uado | oAo Lfl-l
ΖΗΝ H H H a HO) 9PI-1 Ζ ΗΝ HHH a HO) 9PI- 1
'ΗΝ H H H a で HO) 'ΗΝ H H H a HO)
Η o H H a JH0 m-i Η o HH a J H0 mi
Η H H a SH0 Η HH a S H0
Η H H a ZH0 -i Η HH a Z H0 -i
Η H H H a ZH0 I l-i Η HHH a Z H0 I li
!ΗΝ H H H 0 'HO Ot-l-l ! ΗΝ HHH 0 'HO Ot-ll
6S0C/900idf/X3d 93 0ε01"0ΐ/900Ζ OAV
6S0C / 900idf / X3d 93 0ε01 "0ΐ / 900Ζ OAV


l76S0C/900Z<If/X3d 83 0£0 0l/900Z OAV
 l76S0C / 900Z <If / X3d 83 0 £ 0 0l / 900Z OAV
l76S0C / 900Z <If / X3d 83 0 £ 0 0l / 900Z OAV

o. X R1X R1Y R2 R3 R4-1 (CH2)2 H A H H H-2 (CH2)2 H A H H NH2-3 (CH2)3 H A H H NH2-4 cyclopentylidene— CH2 H A H H NH2-5 (CH2)2 H B H H NH2-6 (CH2)3 H B H H NH2-7 CMe2CH2 H B H H NH2-8 cyclopentylidene - CH2- H B H H NH2-9 (CH2)2 H C H H H-10 (CH2)2 H C H H NH2-11 (CH2)3 H C H H NH2-12 cyclopentylidene-CH2- H C H H NH2-13 (CH2)2 H D H H H-14 (CH2)3 H D H H H-15 (CH2)2 H D H H NH2-16 (CH2)2 H D Me H NH2 - 17 (CH2)2 H D Ph H NH2-18 (CH2)2 H D F H NH2 - 19 (CH2)2 H D H O e NH2-20 (CH2)2 H D H Br NH2-21 (CH2)2 H D H F NH2-22 (CH2)2 H D H NMe2 NH2-23 (CH2)2 H D H CH2(4-Pyr) NH2-24 (CH2)2 H D H C(=0)Me NH2-25 (CH2)2 H D H C(=0)°Pr NH2-26 (CH2)2 H D H C(=0)NH2 NH2-27 (CH2)3 H D H H NH2-28 cyclopentylidene-CH2- H D H H NH2-29 C e2CH2 H D H H NH2-30 (CH2)2 H D H H Me-31 (CH2)2 H D H H F-32 (CH2)2 H D H H CF3-33 (CH2)3 H E H H H
2-34 (CH2)2 H E H H NH2 o. XR 1X R 1Y R 2 R 3 R 4 -1 (CH 2 ) 2 HAHH H-2 (CH 2 ) 2 HAHH NH 2 -3 (CH 2 ) 3 HAHH NH 2 -4 cyclopentylidene— CH 2 HAHH NH 2 -5 (CH 2 ) 2 HBHH NH 2 -6 (CH 2 ) 3 HBHH NH 2 -7 CMe 2 CH 2 HBHH NH 2 -8 cyclopentylidene-CH 2 -HBHH NH 2 -9 (CH 2 ) 2 HCHH H-10 (CH 2 ) 2 HCHH NH 2 -11 (CH 2 ) 3 HCHH NH 2 -12 cyclopentylidene-CH 2 -HCHH NH 2 -13 (CH 2 ) 2 HDHH H-14 (CH 2 ) 3 HDHH H-15 (CH 2) 2 HDHH NH 2 -16 ( CH 2) 2 HD Me H NH 2 - 17 (CH 2) 2 HD Ph H NH 2 -18 (CH 2) 2 HDFH NH 2 - 19 (CH 2) 2 HDHO e NH 2 -20 (CH 2 ) 2 HDH Br NH 2 -21 (CH 2 ) 2 HDHF NH 2 -22 (CH 2 ) 2 HDH NMe 2 NH 2 -23 (CH 2 ) 2 HDH CH 2 (4-Pyr) NH 2 -24 (CH 2 ) 2 HDHC (= 0) Me NH 2 -25 (CH 2 ) 2 HDHC (= 0) ° Pr NH 2 -26 (CH 2 ) 2 HDHC (= 0) NH 2 NH 2 -27 (CH 2 ) 3 HDHH NH 2 -28 cyclopentylidene-CH 2 -HDHH NH 2 -29 Ce 2 CH 2 HDHH NH 2 -30 (CH 2 ) 2 HDHH Me-31 (CH 2 ) 2 HDHH F-32 (CH 2 ) 2 HDHH CF 3 -33 (CH 2 ) 3 HEHHH 2-34 (CH 2 ) 2 HEHH NH 2
2-35 (CH2)3 H E H H NH2 2-35 (CH 2 ) 3 HEHH NH 2
2-36 cyclopentylidene— CH2 - H E H H NH2 2-36 cyclopentylidene— CH 2 -HEHH NH 2
2-37 (CH2)2 H B H H H 上記表中、好適なものとしては、 1-1、 1-6、 1-7、 1-8、 1-10、 1-11, 1-12, 1-13, 1-1 4、 1-15、 1-23、 1-24、 1-32、 1-34、 1-35、 1-37、 1-38、 1-40、 1-41、 1-49、 1-50、 1-5 1、 1-52、 1-53、 1-54、 1-55、 1-56、 1-57、 1-58、 1-59、 1-60、 1-61、 1-64、 1-65、 1-7 0、 1-71、 1-76、 1-77、 1-78、 1-80、 1-81、 1-82、 1-92、 1-93、 1-94、 1-95、 1-96、 1-9 7、 1-98、 1-99、 1-100、 1-101、 1-105、 1-149、 1-160、 1-164、 1-182、 1-198、 1-199 、 1-200、 1-201、 1-202、 1-203、 1-204、 1-205、 1-206、 1-207、 1-209、 1-231、 1-243 、 1-244、 1-245、 1-246、 1-252、 1-253、 1-258、 1-263、 2-5、 2-7、 2-8、 2-13、 2-14、 2-15、 2-27、 2-28、 2-29、 2-33、 2-35又は 2-37が挙げられ、更に好適なものとしては 、 1-8、 1-10、 1-12、 1-15、 1-24、 1-35、 1-37、 1-65、 1-71、 1-82、 1-209、 1-231又 は 2-15が挙げられ、特に好適なものとしては、 {[7- (アミノカルボ-ル) -8H-インデノ [1, 2-d][l ,3]チアゾール -4-ィル]ォキシ }メチルホスホン酸、 {[7- (シクロプロピルカルボ- ル) -8H-インデノ [1 ,2-d][l ,3]チアゾール -4-ィル]ォキシ }メチルホスホン酸、 [(7-フル ォ口- 8H-インデノ [l,2-d][l,3]チアゾール -4-ィル)ォキシ]メチルホスホン酸、 [(7-メト キシ -8H-インデノ [1 ,2-d][l ,3]チアゾール -4-ィル)ォキシ]メチルホスホン酸、 [(7-ァセ チル -8H-インデノ [1 ,2-d][l ,3]チアゾール -4-ィル)ォキシ]メチルホスホン酸、 {[7- (ジメ チルァミノ) -8H-インデノ [1 ,2-d][l ,3]チアゾール -4-ィル]ォキシ }メチルホスホン酸、({ 7- [(メチルァミノ)カルボ-ル] -8H-インデノ [l,2-d][l,3]チアゾール -4-ィル }ォキシ)メ チルホスホン酸、 [(7-{[(2-ァミノ- 2-ォキソェチル)ァミノ]カルボ-ル}-8!"[-インデノ [1 ,2 -d][l,3]チアゾール -4-ィル)ォキシ]メチルホスホン酸、 {[7-({[(lS)-2-ァミノ- 1-メチル- 2-ォキソェチル]アミノ}カルボ-ル) -8H-インデノ [1 ,2-d][l ,3]チアゾール -4-ィル]ォキ シ}メチルホスホン酸、 {[7-({[(lR)- 2-ァミノ- 1-メチル - 2-ォキソェチル]アミノ}カルボ- ル) -8H-インデノ [1 ,2-d][l ,3]チアゾール -4-ィル]ォキシ }メチルホスホン酸、 {[7-({[(lS) -1- (ァミノカルボ-ル) -3-メチルブチル]アミノ}カルボ-ル) -8H-インデノ [l,2-d][l,3]
チアゾール -4-ィル]ォキシ }メチルホスホン酸、 {[7-({[(lS)-2-ァミノ- 1- (ヒドロキシメチ ル)- 2-ォキソェチル]アミノ}カルボ-ル)- 8H-インデノ [1,2- d][l, 3]チアゾール -4-ィル] ォキシ }メチルホスホン酸、 [(7-ェチル -8H-インデノ [1 ,2-d][l ,3]チアゾール -4-ィル)ォ キシ]メチルホスホン酸、 [(6,7-ジメチル -8H-インデノ [l,2-d][l,3]チアゾール -4-ィル) ォキシ]メチルホスホン酸、 [(7-ブロモ -6-メチル -8H-インデノ [l,2-d][l,3]チアゾール- 4-ィル)ォキシ]メチルホスホン酸、 [(6-メトキシ -8H-インデノ [l,2-d][l,3]チアゾール -4 -ィル)ォキシ]メチルホスホン酸、 [(7-ブロモ -6-メトキシ -8H-インデノ [l,2-d][l,3]チア ゾール -4-ィル)ォキシ]メチルホスホン酸又はその薬理上許容される塩が挙げられ、 最も好適なものとしては、 [(7-メトキシ- 8H-インデノ [1,2- d][l, 3]チアゾール -4-ィル)ォ キシ]メチルホスホン酸、 [(7-ェチル -8H-インデノ [1 ,2-d][l ,3]チアゾール -4-ィル)ォキ シ]メチルホスホン酸、 [(6,7-ジメチル -8H-インデノ [1 ,2-d][l ,3]チアゾール -4-ィル)ォ キシ]メチルホスホン酸又はその薬理上許容される塩が挙げられる。 2-37 (CH 2 ) 2 HBHHH In the above table, the preferred ones are 1-1, 1-6, 1-7, 1-8, 1-10, 1-11, 1-12, 1-13 , 1-1 4, 1-15, 1-23, 1-24, 1-32, 1-34, 1-35, 1-37, 1-38, 1-40, 1-41, 1-49, 1-50, 1-5 1, 1-52, 1-53, 1-54, 1-55, 1-56, 1-57, 1-58, 1-59, 1-60, 1-61, 1 -64, 1-65, 1-7 0, 1-71, 1-76, 1-77, 1-78, 1-80, 1-81, 1-82, 1-92, 1-93, 1- 94, 1-95, 1-96, 1-9 7, 1-98, 1-99, 1-100, 1-101, 1-105, 1-149, 1-160, 1-164, 1-182 1-198, 1-199, 1-200, 1-201, 1-202, 1-203, 1-204, 1-205, 1-206, 1-207, 1-209, 1-231, 1 -243, 1-244, 1-245, 1-246, 1-252, 1-253, 1-258, 1-263, 2-5, 2-7, 2-8, 2-13, 2-14 2-15, 2-27, 2-28, 2-29, 2-33, 2-35 or 2-37, and more preferable are 1-8, 1-10, 1-12 1-15, 1-24, 1-35, 1-37, 1-65, 1-71, 1-82, 1-209, 1-231 or 2-15, particularly suitable {[7- (aminocarbol) -8H-indeno [1,2-d] [l, 3] thiazol-4-yl] oxy} methylphosphonic acid, {[7- (cyclopropylcarbo- ) -8H-indeno [1,2-d] [l, 3] thiazol-4-yl] oxy} methylphosphonic acid, [(7-fluoro-8H-indeno [l, 2-d] [l , 3] thiazol-4-yl) oxy] methylphosphonic acid, [(7-methoxy-8H-indeno [1,2-d] [l, 3] thiazol-4-yl) oxy] methylphosphonic acid, [ (7-acetyl-8H-indeno [1,2-d] [l, 3] thiazol-4-yl) oxy] methylphosphonic acid, {[7- (dimethylamino) -8H-indeno [1,2 -d] [l, 3] thiazole-4-yl] oxy} methylphosphonic acid, ({7- [(methylamino) carbol] -8H-indeno [l, 2-d] [l, 3] thiazole- 4-yl} oxy) methylphosphonic acid, [(7-{[(2-amino-2-oxoethyl) amino] carbol} -8! "[-Indeno [1,2-d] [l, 3 ] Thiazole-4-i L) oxy] methylphosphonic acid, {[7-({[(lS) -2-amino-1-methyl-2-oxoethyl] amino} carbol) -8H-indeno [1,2-d] [l, 3] thiazole-4-yl] oxy} methylphosphonic acid, {[7-({[(lR) -2-amino-1-methyl-2-oxoethyl] amino} carbole) -8H-indeno [ 1, 2-d] [l, 3] thiazol-4-yl] oxy} methylphosphonic acid, {[7-({[(lS) -1- (aminocarbole) -3-methylbutyl] amino} carbo- -8H-indeno [l, 2-d] [l, 3] Thiazole-4-yl] oxy} methylphosphonic acid, {[7-({[(lS) -2-amino-1- (hydroxymethyl) -2-oxoethyl] amino} carbol) -8H-indeno [ 1,2-d] [l, 3] thiazol-4-yl] oxy} methylphosphonic acid, [(7-ethyl-8H-indeno [1,2-d] [l, 3] thiazol-4-yl ) Oxy] methylphosphonic acid, [(6,7-dimethyl-8H-indeno [l, 2-d] [l, 3] thiazol-4-yl) oxy] methylphosphonic acid, [(7-bromo-6- Methyl-8H-indeno [l, 2-d] [l, 3] thiazol-4-yl) oxy] methylphosphonic acid, [(6-methoxy-8H-indeno [l, 2-d] [l, 3] Thiazole-4-yl) oxy] methylphosphonic acid, [(7-bromo-6-methoxy-8H-indeno [l, 2-d] [l, 3] thiazol-4-yl) oxy] methylphosphonic acid or Pharmacologically acceptable salts thereof, and the most preferable one is [(7-methoxy-8H-indeno [1,2-d] [l, 3] thiazole -4-yl) oxy] methylphosphonic acid, [(7-ethyl-8H-indeno [1,2-d] [l, 3] thiazol-4-yl) oxy] methylphosphonic acid, [(6 , 7-dimethyl-8H-indeno [1,2-d] [l, 3] thiazol-4-yl) oxy] methylphosphonic acid or a pharmacologically acceptable salt thereof.
[0072] [0072]
本発明の、下記一般式(II)を有する化合物は、例えば、以下の方法により公知化合 物を出発原料として用いて、製造することができる。また、下記一般式 (I)又は (Γ)を有 する化合物は、一般式 (II)を有する化合物の製法に準じて製造することができる。 The compound having the following general formula (II) of the present invention can be produced, for example, by the following method using a known compound as a starting material. In addition, the compound having the following general formula (I) or (Γ) can be produced according to the production method of the compound having the general formula (II).
[0073] [化 5] [0073] [Chemical 5]

(り (Π) (Ri (Π)
[0074] 上記式中及び以下の記載にお!、て、 Rlx、 R1Y、
 R4及び Xは、前述したも のと同意義を示す。 [0074] In the above formula and the following description !, R lx , R 1Y , R 4 and X have the same meaning as described above.
R4及び Xは、前述したも のと同意義を示す。 [0074] In the above formula and the following description !, R lx , R 1Y , R 4 and X have the same meaning as described above.
A工程 Process A
[0075] [化 6]
[0075] [Chemical 6]

[0076] B工程 [0076] Process B
Ba工程 Ba process
[0077] [化 7] [0077] [Chemical 7]

(2a) (3) (4) (2a) (3) (4)
(Π) (Π)

(2b) 0) (10) (2b) 0) (10)
Bb-3工程 Bb- 4ェ甲呈 (Π)
 Bb-3 process Bb-4
Bb-3 process Bb-4
[0080] 上記工程中及び以下の記載において、 R1A、 R1Ba及び R1Bbは、同一又は異なって、リ ン酸基又はホスホン酸基が保護基により保護されていてもよい R1を示し、 R2A、 R2Ba及 び R2Bbは、同一又は異なって、 R2を示し (R2がァミノ基又は水酸基を有する場合、該ァ
ミノ基又は水酸基は、保護基により保護されていてもよい)、 R3Ba及び R3Bbは、同一又 は異なって、 R3を示し (R3がァミノ基又は水酸基を有する場合、該ァミノ基又は水酸基 は、保護基により保護されていてもよい)、 R4Ba及び R4Bbは、同一又は異なって、 R4を示 し (R4がァミノ基又は水酸基を有する場合、該ァミノ基又は水酸基は、保護基により保 護されていてもよい)、 L1は、水素原子、脱離基又は R3Bbを示し、 Y、 L2a及び L2bは、同 一又は異なって、水素原子又は脱離基を示し、 P1は、カルボン酸の保護基を示し、 t は、 0又は 1の整数を示す。 [0080] In the above steps and in the following description, R 1A , R 1Ba and R 1Bb are the same or different and each represents a phosphoric acid group or a phosphonic acid group, wherein R 1 may be protected by a protective group, R 2A, R 2Ba及beauty R 2Bb are the same or different, when shown a R 2 where (R 2 has an Amino group or a hydroxyl group,該A Amino group or hydroxyl group may be protected by a protecting group), R 3Ba and R 3Bb are the same or different, when shown a R 3 which (R 3 has an Amino group or a hydroxyl group, the Amino group or R 4Ba and R 4Bb are the same or different and each represents R 4 (when R 4 has an amino group or a hydroxyl group, the amino group or hydroxyl group is L 1 represents a hydrogen atom, a leaving group or R 3Bb , and Y, L 2a and L 2b may be the same or different and each represents a hydrogen atom or a leaving group. P 1 represents a protecting group for carboxylic acid, and t represents an integer of 0 or 1.
[0081] 上記工程中及び以下の記載において、 R1A、 R1Ba及び R1Bbの定義におけるリン酸基 の「保護基」とは、有機合成化学の分野で使用されるリン酸基の保護基であれば特に 限定はされないが、例えば、グリーン'ワッツ著、「プロテクティブ グループス イン ォ一!ニック シンセシス第 3fe(Protective groups in organic synthesis)」 (米国、 Wile y- Interscience社)に記載の方法により行うことができる。好適には、メチル基、ェチル 基のような C1-6アルキル基、ァリル基のような C2-6ァルケ-ル基などを挙げることが でき、更に好適には、 C1-3アルキル基であり、特に好適には、ェチル基である。 [0081] In the above process and the following description, the "protecting group" of a phosphate group in the definition of R 1A , R 1Ba and R 1Bb is a protecting group of a phosphate group used in the field of synthetic organic chemistry. The method is not particularly limited as long as it is performed. For example, the method described in Green's Watts, "Protective groups in organic synthesis (3fe)" (Wiley-Interscience, USA) be able to. Preferable examples include a C1-6 alkyl group such as a methyl group and an ethyl group, and a C2-6 alkyl group such as an aryl group, and more preferable is a C1-3 alkyl group. Particularly preferred is an ethyl group.
[0082] 上記工程中及び以下の記載において、 R2A、
 R3Ba、 R3Bb、 R4Ba及び R4Bbの 定義における水酸基の「保護基」とは、有機合成化学の分野で使用される水酸基の 保護基であれば特に限定はされないが、例えば、グリーン 'ワッツ著、「プロテクティブ クノレープス イン 才ー ニック シンセシス第 3|iR(Protective groups in organic syn thesis )J (米国、 Wiley-Interscience社)に記載の方法により行うことができる。好適に は、メチル基のような C1-6アルキル基、ベンジル基、ァリル基、シリル基、ァシル基を 挙げることがでできる。 [0082] In the above process and in the following description, R 2A , The hydroxyl “protecting group” in the definitions of R 3Ba , R 3Bb , R 4Ba and R 4Bb is not particularly limited as long as it is a hydroxyl protecting group used in the field of synthetic organic chemistry. Protective groups in organic synthesis (JR) (Wiley-Interscience, USA), preferably a methyl group such as a methyl group. Examples thereof include a C1-6 alkyl group, a benzyl group, an aryl group, a silyl group and an acyl group.
R3Ba、 R3Bb、 R4Ba及び R4Bbの 定義における水酸基の「保護基」とは、有機合成化学の分野で使用される水酸基の 保護基であれば特に限定はされないが、例えば、グリーン 'ワッツ著、「プロテクティブ クノレープス イン 才ー ニック シンセシス第 3|iR(Protective groups in organic syn thesis )J (米国、 Wiley-Interscience社)に記載の方法により行うことができる。好適に は、メチル基のような C1-6アルキル基、ベンジル基、ァリル基、シリル基、ァシル基を 挙げることがでできる。 [0082] In the above process and in the following description, R 2A , The hydroxyl “protecting group” in the definitions of R 3Ba , R 3Bb , R 4Ba and R 4Bb is not particularly limited as long as it is a hydroxyl protecting group used in the field of synthetic organic chemistry. Protective groups in organic synthesis (JR) (Wiley-Interscience, USA), preferably a methyl group such as a methyl group. Examples thereof include a C1-6 alkyl group, a benzyl group, an aryl group, a silyl group and an acyl group.
[0083] 上記工程中及び以下の記載において、 R2A、 R2Ba、 R2Bb、 R3Ba、 R3Bb、 R4Ba及び R4Bbの 定義におけるァミノ基の「保護基」とは、有機合成化学の分野で使用されるァミノ基の 保護基であれば特に限定はされないが、例えば、グリーン 'ワッツ著、「プロテクティブ クノレープス イン 才ー ニック シンセシス第 3|iR(Protective groups in organic syn thesis )J (米国、 Wiley-Interscience社)に記載の方法により行うことができる。好適に は、 t-ブトキシカルボ-ル基のような C1-6アルコキシカルボ-ル基、ベンジルォキシ カルボ-ル基、ァリルォキシカルボ-ル基を挙げることができ、さらに好適には、 t-ブ
トキシカルボ-ル基又はァリルォキシカルボ-ル基である。 [0083] In the step in and the following description, R 2A, R 2Ba, R 2Bb, R 3Ba, R 3Bb, the term "protecting group" of Amino groups in the definition of R 4Ba and R 4Bb, the field of organic synthetic chemistry For example, Green's Watts, Protective Groups in Organic Synthesis 3 | iR (Protective groups in organic syn thesis) J (USA, Wiley -Interscience), preferably a C1-6 alkoxy carbo ol group such as a t-butoxy carbo ol group, a benzyloxy carbo ol group, an aryloxy carbo ol group. And more preferably t-butyl A toxoxycarbonyl group or an aryloxycarboxyl group.
[0084] 上記工程中及び以下の記載において、「脱離基」とは、有機合成化学の分野で使 用される脱離基であれば特に限定はされないが、
 L2a及び L2bにおいて好適には、 臭素原子のようなハロゲン原子であり、 Yにおいて好適には、低級脂肪族スルホ-ル ォキシ基、芳香族スルホ -ル基ォキシ又はハロゲン原子であり、更に好適には、芳香 族スルホ-ルォキシ基である。 [0084] In the above process and the following description, the "leaving group" is not particularly limited as long as it is a leaving group used in the field of organic synthetic chemistry. L 2a and L 2b are preferably a halogen atom such as a bromine atom, and Y is preferably a lower aliphatic sulfo-oxy group, an aromatic sulfo-oxy group or a halogen atom, and more preferably Is an aromatic sulfo-loxy group.
L2a及び L2bにおいて好適には、 臭素原子のようなハロゲン原子であり、 Yにおいて好適には、低級脂肪族スルホ-ル ォキシ基、芳香族スルホ -ル基ォキシ又はハロゲン原子であり、更に好適には、芳香 族スルホ-ルォキシ基である。 [0084] In the above process and the following description, the "leaving group" is not particularly limited as long as it is a leaving group used in the field of organic synthetic chemistry. L 2a and L 2b are preferably a halogen atom such as a bromine atom, and Y is preferably a lower aliphatic sulfo-oxy group, an aromatic sulfo-oxy group or a halogen atom, and more preferably Is an aromatic sulfo-loxy group.
[0085] 本発明の化合物 (Π)を製造する工程は、以下の 2工程からなる。 [0085] The step of producing the compound (ii) of the present invention comprises the following two steps.
すなわち、 That is,
(1) A工程は、化合物 (II)のリン酸基部分を構築する工程である。 (1) Step A is a step of constructing a phosphate group moiety of compound (II).
(2) B工程は、 A工程で得られた中間体 (2)の右側部分にチアゾール環を構築し、本 発明の化合物 (II)を製造する工程であり、所望する化合物に応じて a法、 b法を選択で きる。 (2) Step B is a step for producing a compound (II) of the present invention by constructing a thiazole ring in the right part of the intermediate (2) obtained in Step A, and depending on the desired compound, Method a B method can be selected.
[0086] 以下、各工程につき、説明する。 [0086] Hereinafter, each step will be described.
工程) Process)
(第 A- 1工程) (Process A-1)
本工程は、公知の方法(例えば、 Tetrahedron Asymmetry 14, 2003, p.481, J. Che m. Res. Miniprint 11, 1995, p.2544など)に準じて調製できる化合物 (1)に R1A基を導 入し、化合物 (2)を製造する工程である。 In this step, R 1A group is added to compound (1) which can be prepared according to a known method (for example, Tetrahedron Asymmetry 14, 2003, p. 481, J. Chem. Res. Miniprint 11, 1995, p. 2544). Is a process for producing the compound (2).
[0087] 尚、化合物 (II)のケトンは必要に応じて、保護'脱保護することができ、アルコール部 分は必要に応じて、ハロゲンィ匕又は脱離基への変換を行うこともできる。 [0087] The ketone of compound (II) can be protected and deprotected as necessary, and the alcohol moiety can be converted to a halogenated group or a leaving group as necessary.
[0088] ケトンの保護基としては、一般にこの分野の技術において周知の保護基を、例えば 、グリーン 'ワッツ著、「プロテクティブ グループス イン オーガニック シンセシス第 3te(Protective groups in organic synthesisノ」、未国、 Wiley— Interscience社)【こ 载 の方法により用いることができ、好適には、非環状又は環状のケタール、ジチオケタ ール、モノチオケタール、 0-置換シァノヒドリン、ヒドラゾン、セミカルバゾン、ォキシム のような保護基を挙げることができる。 [0088] As a protecting group for ketones, a protecting group generally known in the art of this field, for example, "Protective groups in organic synthesis", Green, by Watts, Protective groups in organic synthesis, Wiley—Interscience) [Can be used by this method, and preferably a protecting group such as acyclic or cyclic ketal, dithioketal, monothioketal, 0-substituted cyanohydrin, hydrazone, semicarbazone, oxime. Can be mentioned.
[0089] ハロゲン化剤としては、通常、ハロゲンィ匕反応に用いられる試薬であれば特に限定 ίまな ヽカ、 [列? J 、 Comprehensive Organic Transformation Larlock、 Vし H)【こ g己
載されているようなハロゲン化剤を挙げることができ、臭素化の場合、好適には、臭化 水素、三臭化リン、四臭化炭素-トリフエ-ルホスフィン、 N -プロモコハク酸イミド-トリフ ェニルホスフィン、塩素化の場合、好適には、塩化水素、四塩ィ匕炭素-トリフエニルホ スフイン、 N-クロロコハク酸イミド-トリフエ-ルホスフィンをあげることができる。 [0089] The halogenating agent is not particularly limited as long as it is a reagent usually used in a halogenated reaction. J, Comprehensive Organic Transformation Larlock, V and H) In the case of bromination, hydrogen bromide, phosphorus tribromide, carbon tetrabromide-triphenylphosphine, N-prosuccinimide-trif is preferred. In the case of phenylphosphine and chlorination, preferred examples include hydrogen chloride, tetrasalt-carbon-triphenylphosphine, and N-chlorosuccinimide-triphenylphosphine.
[0090] (A- 1- 1) [0090] (A- 1- 1)
R1が OCH P(0)(OH)又は ΟΡ(ΟΧΟΗ)の場合、化合物 (1)を、試薬 Y- R1Aと処理する If R 1 is OCH P (0) (OH) or ΟΡ (ΟΧΟΗ), treat compound (1) with reagent Y-R 1A
2 2 2 2 2 2
こと〖こより達成される。試薬 Y-R1Aは反応系中で発生させても良い。 It is achieved from Kotoko. Reagent YR 1A may be generated in the reaction system.
[0091] 使用される溶媒は反応を阻害せず、出発物質を溶解するものであれば特に限定は ないが、好適には、ジェチルエーテル、ジイソプロピルエーテル、テトラヒドロフラン、 ジォキサン、ジメトキシェタンのようなエーテル類、ホルムアミド、 Ν,Ν-ジメチルホルム アミド、 Ν,Ν-ジメチルァセトアミド、 Ν-メチル -2-ピロリドン、 Ν-メチルピロリジノン、へキ サメチルホスホロトリアミドのようなアミド類、ジクロロメタン、クロ口ホルム、四塩化炭素 、ジクロロエタン、クロ口ベンゼン、ジクロロベンゼンのようなハロゲン化炭化水素類で あり、さらに好適にはハロゲンィ匕炭化水素類、エーテル類又はアミド類であり、最も好 適にはテトラヒドロフラン、ジメトキシェタン又は Ν,Ν-ジメチルホルムアミドである。 [0091] The solvent to be used is not particularly limited as long as it does not inhibit the reaction and dissolves the starting material, but is preferably a solvent such as jetyl ether, diisopropyl ether, tetrahydrofuran, dioxane or dimethoxyethane. Ethers, formamide, Ν, Ν-dimethylformamide, Ν, Ν-dimethylacetamide, ア ミ ド -methyl-2-pyrrolidone, Ν-methylpyrrolidinone, amides such as hexamethylphosphorotriamide, dichloromethane, Halogenated hydrocarbons such as black mouth form, carbon tetrachloride, dichloroethane, black mouth benzene and dichlorobenzene, more preferably halogenated hydrocarbons, ethers or amides, most preferably Tetrahydrofuran, dimethoxyethane or Ν, Ν-dimethylformamide.
[0092] 反応は、好適には塩基存在下行なわれ、使用される塩基としては、通常のアルキル 化反応に使用される塩基であれば特に限定はないが、例えば、炭酸リチウム、炭酸 ナトリウム、炭酸カリウムのようなアルカリ金属炭酸塩類;炭酸水素リチウム、炭酸水素 ナトリウム、炭酸水素カリウムのようなアルカリ金属重炭酸塩類;水素化リチウム、水素 化ナトリウム、水素化カリウムのようなアルカリ金属水素化物類;水酸化リチウム、水酸 化ナトリウム、水酸ィ匕カリウムのようなアルカリ金属水酸ィ匕物類;リチウムメトキシド、ナ トリウムメトキシド、ナトリウムエトキシド、カリウム t-ブトキシドのようなアルカリ金属アル コキシド類;又はトリェチルァミン、トリブチルァミン、ジイソプロピルェチルァミン、 N-メ チルモルホリン、ピリジン、 4-(N,N-ジメチルァミノ)ピリジン、 Ν,Ν-ジメチルァ-リン、 Ν, Ν-ジェチルァ-リン、 1,5-ジァザビシクロ [4.3.0]ノナ- 5-ェン、 1,4-ジァザビシクロ [2.2. 2]オクタン (DABCO)、 1,8-ジァザビシクロ [5.4.0]- 7-ゥンデセン (DBU)のような有機アミ ン類;であり、好適には有機アミン類 (最も好適にはトリエチルァミン)又はアルカリ金 属炭酸塩類 (最も好適には炭酸カリウム)である。
[0093] 反応温度は、 0°C乃至 150°Cであり、好適には室温乃至 120°Cである。 [0092] The reaction is preferably carried out in the presence of a base, and the base to be used is not particularly limited as long as it is a base used in a usual alkylation reaction. For example, lithium carbonate, sodium carbonate, carbonate Alkali metal carbonates such as potassium; alkali metal bicarbonates such as lithium hydrogen carbonate, sodium hydrogen carbonate, potassium hydrogen carbonate; alkali metal hydrides such as lithium hydride, sodium hydride, potassium hydride; water Alkali metal hydroxides such as lithium oxide, sodium hydroxide, potassium hydroxide; alkaline metal alkoxides such as lithium methoxide, sodium methoxide, sodium ethoxide, potassium t-butoxide Or triethylamine, tributylamine, diisopropylethylamine, N-methylmorpholine, pyridine Lysine, 4- (N, N-dimethylamino) pyridine, Ν, Ν-dimethylaline, Ν, Ν-jetylaline, 1,5-diazabicyclo [4.3.0] non-5-ene, 1,4- Organic amines such as diazabicyclo [2.2.2] octane (DABCO), 1,8-diazabicyclo [5.4.0] -7-undecene (DBU); preferably organic amines (most preferably Triethylamine) or alkali metal carbonates (most preferably potassium carbonate). [0093] The reaction temperature is 0 ° C to 150 ° C, preferably room temperature to 120 ° C.
[0094] 反応時間は、 0.5時間乃至 24時間であり、好適には 0.5時間乃至 12時間である。 [0094] The reaction time is 0.5 to 24 hours, preferably 0.5 to 12 hours.
[0095] (A-1-2) [0095] (A-1-2)
R1が CH P(0)(OH)の場合、化合物 (1)の R1の部位の水酸基に常法により脱離基にWhen R 1 is CH P (0) (OH), the hydroxyl group at the R 1 site of compound (1) is converted to the leaving group by a conventional method.
2 2 twenty two
変換した後に、アルブゾフ反応を用いてホスホニルイ匕することによつても製造できる。 After conversion, it can also be prepared by phosphonylation using the Arbuzov reaction.
[0096] 脱離基は、有機合成化学の分野で使用される脱離基であれば特に限定はなぐ常 法に従って導入が可能である。好適には、低級脂肪族スルホニルォキシ基、芳香族 スルホ -ルォキシ基又はハロゲン原子であり、更に好適には、ハロゲン原子である。 [0096] The leaving group can be introduced according to a conventional method without particular limitation as long as it is a leaving group used in the field of synthetic organic chemistry. Preferred is a lower aliphatic sulfonyloxy group, an aromatic sulfo-loxy group or a halogen atom, and more preferred is a halogen atom.
[0097] ホスホニル化は、好適には、溶媒存在下行なわれ、使用される溶媒は反応を阻害 せず、出発物質を溶解するものであれば特に限定はないが、好適には、ジェチルェ 一テル、ジイソプロピルエーテル、テトラヒドロフラン、ジォキサン、ジメトキシェタンの ようなエーテル類、ホルムアミド、 Ν,Ν-ジメチルホルムアミド、 Ν,Ν-ジメチルァセトアミド 、 Ν-メチル -2-ピロリドン、 Ν-メチルピロリジノン、へキサメチルホスホロトリアミドのよう なアミド類、ジメチルスルホキシドのようなスルホキシド類であり、さらに好適には、テト ラヒドロフラン、ジメチルスルホキシド又は Ν,Ν-ジメチルホルムアミドである。また、本 反応は、溶媒を使用しなくても行なうことができる。 [0097] The phosphonylation is preferably performed in the presence of a solvent, and the solvent used is not particularly limited as long as it does not inhibit the reaction and dissolves the starting material. , Ethers such as diisopropyl ether, tetrahydrofuran, dioxane, dimethoxyethane, formamide, Ν, Ν-dimethylformamide, Ν, Ν-dimethylacetamide, Ν-methyl-2-pyrrolidone, Ν-methylpyrrolidinone, hexane Amides such as methyl phosphorotriamide, sulfoxides such as dimethyl sulfoxide, and more preferably tetrahydrofuran, dimethyl sulfoxide or Ν, Ν-dimethylformamide. In addition, this reaction can be carried out without using a solvent.
[0098] 使用される試薬としては、亜リン酸トリアルキルであり、好適には亜リン酸トリェチル である。 [0098] The reagent used is trialkyl phosphite, preferably triethyl phosphite.
[0099] 反応温度は、 0°C乃至 200°Cであり、好適には 50°C乃至 180°Cである。 [0099] The reaction temperature is 0 ° C to 200 ° C, preferably 50 ° C to 180 ° C.
[0100] 反応時間は、 0.5時間乃至 24時間であり、好適には 0.5時間乃至 12時間である。 [0100] The reaction time is 0.5 to 24 hours, preferably 0.5 to 12 hours.
[0101] (A-1-3) [0101] (A-1-3)
R1が P(0)(OH)の場合、化合物 (1) R1の部位の水酸基に常法により脱離基に変換し When R 1 is P (0) (OH), the compound (1) is converted to a hydroxyl group at the R 1 site by a conventional method.
2 2
た後に、クロスカップリング反応を用いてホスホニルイ匕することによって製造できる。 Thereafter, it can be prepared by phosphonylation using a cross-coupling reaction.
[0102] 脱離基は、有機合成化学の分野で使用される脱離基であれば特に限定はなぐ常 法に従って導入が可能である。好適には、 C 1-3アルキルスルホ-ルォキシ基、 C1-3 ハロアルキルスルホ-ルォキシ基、芳香族スルホ -ル基ォキシ又はハロゲン原子で あり、更に好適には、トリフルォロメタンスルホ-ルォキシ基、ハロゲン原子である。 [0102] The leaving group can be introduced according to a conventional method without particular limitation as long as it is a leaving group used in the field of organic synthetic chemistry. Preferably, it is a C 1-3 alkylsulfo-oxy group, a C1-3 haloalkyl sulfo-oxy group, an aromatic sulfo-oxy group or a halogen atom, more preferably a trifluoromethane sulfo-oxy group, a halogen atom. Is an atom.
[0103] ホスホニル化において使用される溶媒は反応を阻害せず、出発物質を溶解するも
のであれば特に限定はないが、好適には、ジェチルエーテル、ジイソプロピルエーテ ル、テトラヒドロフラン、ジォキサン、ジメトキシェタンのようなエーテル類、ホルムアミド 、 Ν,Ν-ジメチルホルムアミド、 Ν,Ν-ジメチルァセトアミド、 Ν-メチル -2-ピロリドン、 Ν-メ チルピロリジノン、へキサメチルホスホロトリアミドのようなアミド類、ジメチルスルホキシ ドのようなスルホキシド類であり、さらに好適には、テトラヒドロフラン、ジメチルスルホキ シド又は Ν,Ν-ジメチルホルムアミドである。 [0103] The solvent used in the phosphonylation does not inhibit the reaction and does not dissolve the starting material. Is not particularly limited, and preferably ethers such as jetyl ether, diisopropyl ether, tetrahydrofuran, dioxane, dimethoxyethane, formamide, Ν, Ν-dimethylformamide, Ν, Ν-dimethylacetate. Amides, Ν-methyl-2-pyrrolidone, Ν-methylpyrrolidinone, amides such as hexamethyl phosphorotriamide, and sulfoxides such as dimethyl sulfoxide, more preferably tetrahydrofuran, dimethyl sulfoxide Sid or Ν, Ν-dimethylformamide.
[0104] 反応は、好適には触媒存在下行なわれ、使用される触媒としては、通常のホスホ- ル化反応に使用される触媒であれば特に限定はないが、酢酸パラジウム、ビストリフ ェ-ルホスフィンパラジウムジクロリド、テトラキストリフエ-ルホスフィンパラジウムのよう な遷移金属触媒であり、好適にはテトラキストリフエニルホスフィンパラジウムである。 [0104] The reaction is preferably carried out in the presence of a catalyst, and the catalyst used is not particularly limited as long as it is a catalyst used in a normal phosphorylation reaction, but palladium acetate, bistriphenyl are not limited. A transition metal catalyst such as phosphine palladium dichloride or tetrakistriphenylphosphine palladium, preferably tetrakistriphenylphosphine palladium.
[0105] 反応は、好適には塩基存在下行なわれ、使用される塩基としては、通常の触媒反 応に使用される塩基であれば特に限定はないが、例えば、炭酸リチウム、炭酸ナトリ ゥム、炭酸カリウムのようなアルカリ金属炭酸塩類;炭酸水素リチウム、炭酸水素ナトリ ゥム、炭酸水素カリウムのようなアルカリ金属重炭酸塩類;水素化リチウム、水素化ナ トリウム、水素化カリウムのようなアルカリ金属水素化物類;水酸化リチウム、水酸化ナ トリウム、水酸ィ匕カリウムのようなアルカリ金属水酸ィ匕物類;リチウムメトキシド、ナトリウ ムメトキシド、ナトリウムエトキシド、カリウム t-ブトキシドのようなアルカリ金属アルコキシ ド類;又はトリェチルァミン、トリブチルァミン、ジイソプロピルェチルァミン、 N-メチル モルホリン、ピリジン、 4-(N,N-ジメチルァミノ)ピリジン、 Ν,Ν-ジメチルァ-リン、 Ν,Ν-ジ ェチルァ-リン、 1,5-ジァザビシクロ [4.3.0]ノナ- 5-ェン、 1,4-ジァザビシクロ [2.2.2]ォ クタン (DABCO)、 1,8-ジァザビシクロ [5.4.0]- 7-ゥンデセン (DBU)のような有機アミン類 であり、好適には、トリェチルァミンである。 [0105] The reaction is preferably carried out in the presence of a base, and the base to be used is not particularly limited as long as it is a base used for ordinary catalytic reaction, but for example, lithium carbonate, sodium carbonate, etc. Alkali metal carbonates such as potassium carbonate; alkali metal bicarbonates such as lithium hydrogen carbonate, sodium hydrogen carbonate, potassium hydrogen carbonate; alkali metals such as lithium hydride, sodium hydride, potassium hydride Hydrides; alkali metal hydroxides such as lithium hydroxide, sodium hydroxide, potassium hydroxide; alkali metals such as lithium methoxide, sodium methoxide, sodium ethoxide, potassium t-butoxide Alkoxides; or triethylamine, tributylamine, diisopropylethylamine, N-methylmorpholine, pyridine , 4- (N, N-dimethylamino) pyridine, Ν, Ν-dimethylaline, Ν, Ν-diethylaline, 1,5-diazabicyclo [4.3.0] non-5-ene, 1,4- Organic amines such as diazabicyclo [2.2.2] octane (DABCO) and 1,8-diazabicyclo [5.4.0] -7-undecene (DBU), preferably triethylamine.
[0106] 反応温度は、 0°C乃至 200°Cであり、好適には 50°C乃至 150°Cである。 [0106] The reaction temperature is 0 ° C to 200 ° C, preferably 50 ° C to 150 ° C.
[0107] 反応時間は、 0.5時間乃至 24時間であり、好適には 0.5時間乃至 12時間である。 [0107] The reaction time is 0.5 to 24 hours, preferably 0.5 to 12 hours.
[0108] (A-1-4) [0108] (A-1-4)
R1が CF P(0)(OH)の場合、 R1が CH Ρ(0)(ΟΗ)である化合物 (2)のケトンを保護したWhen R 1 is CF P (0) (OH), protected the ketone of compound (2) where R 1 is CH Ρ (0) (ΟΗ)
2 2 2 2 2 2 2 2
後に弗素化剤により弗素化することによつても製造できる。 It can also be produced by subsequent fluorination with a fluorinating agent.
[0109] 使用される溶媒は反応を阻害せず、出発物質を溶解するものであれば特に限定は
ないが、好適には、ジェチルエーテル、ジイソプロピルエーテル、テトラヒドロフラン、 ジォキサン、ジメトキシェタンのようなエーテル類であり、さらに好適にはテトラヒドロフ ランである。 [0109] The solvent used is not particularly limited as long as it does not inhibit the reaction and dissolves the starting material. Although not preferred, ethers such as jetyl ether, diisopropyl ether, tetrahydrofuran, dioxane and dimethoxyethane are preferred, and tetrahydrofuran is more preferred.
[0110] 弗素化剤としては、通常、求電子的弗素化反応に用いるものであれば特に限定は ないが、 N-フルォロベンゼンスルホンイミドのような芳香族スルホンイミド類、 N-フル ォロピリジ-ゥム塩を挙げることができ、好適には、 N-フルォロベンゼンスルホンイミド である。 [0110] The fluorinating agent is not particularly limited as long as it is usually used in an electrophilic fluorination reaction. However, aromatic sulfonimides such as N-fluorobenzenesulfonimide, N-fluoropyrididine, etc. -Um salt may be mentioned, and N-fluorobenzenesulfonimide is preferred.
[0111] 反応は、好適には塩基存在下行なわれ、使用される塩基としては、通常の求電子 的弗素化反応に使用される塩基であれば特に限定はないが、例えば、リチウムイソプ 口ピルアミド(LDA)のような金属ジアルキルアミド類、リチウム ビス(トリメチルシリル) アミドのような金属ビス(トリアルキルシリル)アミド類である。 [0111] The reaction is preferably carried out in the presence of a base, and the base used is not particularly limited as long as it is a base used in a usual electrophilic fluorination reaction. For example, lithium isopropyl pyramide is used. Metal dialkylamides such as (LDA) and metal bis (trialkylsilyl) amides such as lithium bis (trimethylsilyl) amide.
[0112] 反応温度は、 -78°C乃至 50°Cであり、好適には- 78°C乃至室温である。 [0112] The reaction temperature is -78 ° C to 50 ° C, preferably -78 ° C to room temperature.
[0113] 反応時間は、 0.5時間乃至 12時間であり、好適には 0.5時間乃至 6時間である。 [0113] The reaction time is 0.5 to 12 hours, preferably 0.5 to 6 hours.
[0114] (B工程) [0114] (Process B)
(Ba法) (Ba method)
(第 Ba- 1工程) (Process Ba-1)
本工程は、化合物 (3)を製造する工程であり、 A工程で製造したィ匕合物 (2a)に触媒、 及びアルコール存在下、カルボ-ル化反応を行うことにより達成される。 This step is a step for producing the compound (3), and is achieved by subjecting the compound (2a) produced in the step A to a carbolation reaction in the presence of a catalyst and an alcohol.
[0115] 使用される溶媒は反応を阻害せず、出発物質を溶解するものであれば特に限定は ないが、好適にはジェチルエーテル、ジイソプロピルエーテル、テトラヒドロフラン、ジ ォキサン、ジメトキシェタン、ジエチレングリコールジメチルエーテルのようなエーテル 類;ホルムアミド、 Ν,Ν-ジメチルホルムアミド、 Ν,Ν-ジメチルァセトアミド、 Ν-メチル -2- ピロリドン、 Ν-メチルピロリジノン、へキサメチルホスホロトリアミドのようなアミド類を挙 げることができる。さらに好適には Ν,Ν-ジメチルホルムアミドである。 [0115] The solvent used is not particularly limited as long as it does not inhibit the reaction and dissolves the starting material, but is preferably jetyl ether, diisopropyl ether, tetrahydrofuran, dioxane, dimethoxyethane, diethylene glycol dimethyl ether. Ethers such as formamide, Ν, Ν-dimethylformamide, Ν, Ν-dimethylacetamide, ア ミ ド -methyl-2-pyrrolidone, Ν-methylpyrrolidinone, and amides such as hexamethylphosphorotriamide I can make it. More preferred is Ν, Ν-dimethylformamide.
[0116] 使用されるカルボニル化試薬としては、通常のカルボ-ルイ匕反応において試薬とし て使用されるものであれば、特に限定はないが、、一酸ィ匕炭素を挙げることができる。 [0116] The carbonylation reagent to be used is not particularly limited as long as it is used as a reagent in a normal carbo-Louis reaction, and examples thereof include carbon monoxide.
[0117] 使用されるアルコールは通常のカルボ-ルイ匕反応において使用されるものであれ ば、特に限定はないが、例えば、メタノール、エタノールのような C1-6アルコール、ベ
ンジルアルコールのような芳香族アルキルアルコール、ァリルアルコールのような C1- 3ァルケ-ルアルコール、 1, 1, 1 トリクロ口エタノールノのようなハロゲン化アルキル アルコール、トリメチルシリルエタノールのような、トリアルキルシリルアルキルアルコー ルが挙げられ、好適には、 C1-6アルコール、芳香族アルキルアルコール、トリアルキ ルシリルアルキルアルコールであり、更に好適にはトリアルキルシリルアルキルアルコ ールである。 [0117] The alcohol used is not particularly limited as long as it is used in a normal carbo-Louis reaction. For example, C1-6 alcohol such as methanol and ethanol, Aromatic alcohols such as benzyl alcohol, C1-3 alcohols such as allylic alcohol, halogenated alkyl alcohols such as 1, 1, 1 trichloroethanol, trialkyl such as trimethylsilylethanol Examples thereof include silylalkyl alcohols, preferably C1-6 alcohol, aromatic alkyl alcohol, and trialkylsilylalkyl alcohol, and more preferably trialkylsilylalkyl alcohol.
[0118] 使用される触媒は通常のカルボ-ル化反応において触媒として使用されるもので あれば、特に限定はないが、テトラキストリフエニルホスフィンパラジウム、塩化パラジ ゥム、酢酸パラジウム、ビストリフエ-ルホスフィンパラジウムジクロリドのような遷移金 属触媒、及び必要に応じてトリフエ-ルホスフィン、 1, 3 ビス(ジフエ-ルホスフイノ) プロパン(dppp)、 1, 1 '—ビス(ジフエ-ルホスフイノ)フエ口セン(dppf)、のような配位 子が好適である。更に好適には、酢酸パラジウム、配位子は 1, 3 ビス(ジフエ-ル ホスフイノ)プロパン(dppp)である。 [0118] The catalyst to be used is not particularly limited as long as it is used as a catalyst in a normal carbolation reaction, but tetrakistriphenylphosphine palladium, palladium chloride, palladium acetate, bistriphenylphosphine. Transition metal catalysts such as palladium dichloride, and optionally triphenylphosphine, 1,3 bis (diphenylphosphino) propane (dppp), 1,1'-bis (diphenylphosphino) phenocene (dppf ), And the like. More preferably, the palladium acetate, the ligand is 1,3 bis (diphenol phosphino) propane (dppp).
[0119] 使用される塩基は通常のカルボ-ル化反応において塩基として使用されるもので あれば、特に限定はないが、例えば、炭酸セシウム、炭酸ナトリウム、炭酸カリウム、炭 酸リチウムのようなアルカリ金属炭酸塩類、炭酸水素ナトリウム、炭酸水素カリウム、炭 酸水素リチウムのようなアルカリ金属炭酸水素塩類;水素化リチウム、水素化ナトリウ ム、水素化カリウムのようなアルカリ金属水素化物類;水酸ィ匕ナトリウム、水酸化力リウ ム、水酸化バリウム、水酸化リチウムのようなアルカリ金属水酸化物類等の無機塩基 類、 N-メチルモルホリン、トリエチルァミン、トリプロピルァミン、トリブチルァミン、ジイソ プロピルェチルァミン、ジシクロへキシルァミン、 N-メチルピペリジン、ピリジン、 4-ピロ リジノピリジン、ピコリン、 4-(N,N-ジメチルァミノ)ピリジン、 2,6-ジ (t-ブチル )-4-メチル ピリジン、キノリン、 Ν,Ν-ジメチルァニリン、 Ν,Ν-ジェチルァニリン、 1,5-ジァザビシクロ [4.3.0]ノナ- 5-ェン (DBN)、 1,4-ジァザビシクロ [2.2.2]ォクタン(0 8じ0)、 1,8-ジァザ ビシクロ [5.4.0]ゥンデ力- 7-ェン (DBU)のような有機塩基類が好適である。更に好適に は、有機塩基類であり、特に好適にはトリエチルァミンである。 [0119] The base to be used is not particularly limited as long as it is used as a base in a usual carbolation reaction. For example, an alkali such as cesium carbonate, sodium carbonate, potassium carbonate, lithium carbonate, Alkali metal bicarbonates such as metal carbonates, sodium bicarbonate, potassium bicarbonate, lithium bicarbonate; alkali metal hydrides such as lithium hydride, sodium hydride, potassium hydride; Inorganic bases such as sodium hydroxide, lithium hydroxide, barium hydroxide, alkali metal hydroxides such as lithium hydroxide, N-methylmorpholine, triethylamine, tripropylamine, tributylamine, diisopropyl Ethylamine, dicyclohexylamine, N-methylpiperidine, pyridine, 4-pyrrolidinopyridine, picoline 4- (N, N-dimethylamino) pyridine, 2,6-di (t-butyl) -4-methylpyridine, quinoline, Ν, Ν-dimethylaniline, Ν, Ν-jetylaniline, 1,5-diazabicyclo [4.3 .0] Nona 5-Yen (DBN), 1,4-Diazabicyclo [2.2.2] Octane (0 8 0 0), 1,8-Zazabicyclo [5.4.0] Unde Force-7-Yen ( Organic bases such as DBU) are preferred. More preferred are organic bases, and particularly preferred is triethylamine.
[0120] 反応温度は、 0°C乃至 150°Cであり、好適には 50°C乃至 100°Cである。 [0120] The reaction temperature is 0 ° C to 150 ° C, preferably 50 ° C to 100 ° C.
[0121] 反応時間は、 0.5時間乃至 48時間であり、好適には 2時間乃至 24時間である。
[0122] 本工程にて、必要に応じて P1部分を各種保護基に変換することも可能である。 [0121] The reaction time is 0.5 to 48 hours, preferably 2 to 24 hours. [0122] In this step, the P 1 moiety can be converted into various protecting groups as necessary.
[0123] (第 Ba—2工程) [0123] (Process Ba-2)
本工程は、化合物 (4)を製造する工程であり、化合物 (3)を不活性溶媒中、塩基存在 下又は非存在下、ハロゲンィ匕することにより達成される。 This step is a step for producing compound (4), and is achieved by halogenating compound (3) in an inert solvent in the presence or absence of a base.
[0124] 使用される溶媒は反応を阻害せず、出発物質を溶解するものであれば特に限定は ないが、メタノール、エタノールのようなアルコール類、酢酸ェチルのようなエステル 類又はジクロロメタン、クロ口ホルムのようなハロゲン化炭化水素類を挙げることができ 、好適にはクロ口ホルム、ジクロロメタン、酢酸ェチル又はエタノールである。 [0124] The solvent to be used is not particularly limited as long as it does not inhibit the reaction and dissolves the starting material, but alcohols such as methanol and ethanol, esters such as ethyl acetate, or dichloromethane, Halogenated hydrocarbons such as form can be mentioned, and preferred is chloroform, dichloromethane, ethyl acetate or ethanol.
[0125] 使用されるハロゲン化剤としては、特に限定はないが、例えば、 "Comprehensive 0 rganic Transformation" (Larlock、 VCH)に記載されているようなハロゲン化剤を挙げ ることができ、好適には、臭素、臭ィ匕銅 (11)、フエ-ルトリメチルアンモ-ゥムトリブロミド である。 [0125] The halogenating agent to be used is not particularly limited, and examples thereof include halogenating agents described in "Comprehensive 0 rganic Transformation" (Larlock, VCH). Are bromine, odorous copper (11), and phenyltrimethylammotribromide.
[0126] 使用される塩基としては、化合物 (3)におけるハロゲン原子の置換位置以外の部分 に影響を与えないものであれば特に限定されず、例えば、炭酸リチウム、炭酸ナトリウ ム、炭酸カリウムのようなアルカリ金属炭酸塩類;炭酸水素リチウム、炭酸水素ナトリウ ム、炭酸水素カリウムのようなアルカリ金属重炭酸塩類;リチウムメトキシド、ナトリウムメ トキシド、ナトリウムエトキシド、カリウム- 1-ブトキシドのような金属アルコキシド類;トリエ チルァミン、トリブチルァミン、ジイソプロピルェチルァミン、 N-メチルモルホリン、ピリ ジン、 2,6-ルチジン、 4-(N,N-ジメチルァミノ)ピリジン、 Ν,Ν-ジメチルァ-リン、 Ν,Ν-ジ ェチルァ-リン、 1,5-ジァザビシクロ [4.3.0]ノナ- 5-ェン、 1,4-ジァザビシクロ [2.2.2]ォ クタン(DABCO)、 1,8-ジァザビシクロ [5.4.0]-7-ゥンデセン(DBU)のような有機アミン 類;ブチルリチウム、リチウム ジイソプロピルアミド(LDA)、リチウム ビス(トリメチルシ リル)アミドのような有機金属塩基類または上記塩基の組み合わせを挙げることができ る。好適には、有機アミン類である。 [0126] The base to be used is not particularly limited as long as it does not affect the portion other than the halogen atom substitution position in the compound (3), and examples thereof include lithium carbonate, sodium carbonate and potassium carbonate. Alkali metal carbonates; alkali metal bicarbonates such as lithium bicarbonate, sodium bicarbonate, potassium bicarbonate; metal alkoxides such as lithium methoxide, sodium methoxide, sodium ethoxide, potassium 1-butoxide Triethylamine, tributylamine, diisopropylethylamine, N-methylmorpholine, pyridine, 2,6-lutidine, 4- (N, N-dimethylamino) pyridine, Ν, Ν-dimethylamine, Ν, Ν -Jetylaline, 1,5-diazabicyclo [4.3.0] non-5-ene, 1,4-diazabicyclo [2.2.2] octane (DABCO), 1,8-diaza Organic amines such as cyclo [5.4.0] -7-undecene (DBU); organometallic bases such as butyllithium, lithium diisopropylamide (LDA), lithium bis (trimethylsilyl) amide, or combinations of the above bases Can be mentioned. Preferred are organic amines.
[0127] 反応温度は、 0°C乃至 100°Cであり、好適には 0°C乃至 50°Cである。 [0127] The reaction temperature is 0 ° C to 100 ° C, preferably 0 ° C to 50 ° C.
[0128] 反応時間は、 0.5時間乃至 48時間であり、好適には 0.5時間乃至 12時間である。 [0128] The reaction time is 0.5 hour to 48 hours, preferably 0.5 hour to 12 hours.
[0129] (第 Ba- 3工程) [0129] (Process Ba-3)
本工程は、化合物 (6)を製造する工程であり、化合物 (4)を、不活性溶媒中、添加物
の存在下又は非存在下、化合物 (5a)と反応させることにより達成される。 This step is a step of producing compound (6), and compound (4) is added to an additive in an inert solvent. It is achieved by reacting with compound (5a) in the presence or absence of.
[0130] 使用される不活性溶媒としては、本反応に不活性なものであれば特に限定はない 力 例えば、へキサン、ヘプタン、リグ口イン、石油エーテルのような脂肪族炭化水素 類;ベンゼン、トルエン、キシレンのような芳香族炭化水素類;ジクロロメタン、クロロホ ルム、四塩化炭素、ジクロロエタン、クロ口ベンゼン、ジクロロベンゼンのようなハロゲ ン化炭化水素類;ギ酸ェチル、酢酸ェチル、酢酸プロピル、酢酸ブチル、炭酸ジェチ ルのようなエステル類;ジェチルエーテル、ジイソプロピルエーテル、テトラヒドロフラ ン、ジォキサン、ジメトキシェタン、ジエチレングリコールジメチルエーテルのようなェ テル類;アセトン、メチルェチルケトン、メチルイソブチルケトン、イソホロン、シクロ へキサノンのようなケトン類;ァセトニトリル、イソブチル二トリルのような-トリル類;ホル ムアミド、 Ν,Ν-ジメチルホルムアミド、 Ν,Ν-ジメチルァセトアミド、へキサメチルリン酸ト リアミドのようなアミド類;ジメチルスルホキシドようなスルホキシド類;又はスルホランの ようなスルホン類;メタノール、エタノール、プロパノールなどのアルコール類であり、 好適には、エーテル類、アルコール類であり、最も好適には、テトラヒドロフラン、エタ ノールである。 [0130] The inert solvent used is not particularly limited as long as it is inert to this reaction. For example, aliphatic hydrocarbons such as hexane, heptane, lignin, petroleum ether; benzene Aromatic hydrocarbons such as toluene, xylene; halogenated hydrocarbons such as dichloromethane, chloroform, carbon tetrachloride, dichloroethane, black benzene, dichlorobenzene; ethyl formate, ethyl acetate, propyl acetate, acetic acid Esters such as butyl and ethyl carbonate; ethers such as jetyl ether, diisopropyl ether, tetrahydrofuran, dioxane, dimethoxyethane, and diethylene glycol dimethyl ether; acetone, methyl ethyl ketone, methyl isobutyl ketone, isophorone, cyclohexane Ketones such as hexanone; -Tolyls such as tonitrile, isobutylnitryl; amides such as formamide, Ν, Ν-dimethylformamide, Ν, Ν-dimethylacetamide, hexamethylphosphoric triamide; sulfoxides such as dimethyl sulfoxide; or Sulfones such as sulfolane; alcohols such as methanol, ethanol and propanol; preferred are ethers and alcohols; and most preferred are tetrahydrofuran and ethanol.
[0131] 反応に使用される添加物としては、反応の進行を加速させる塩であれば特に限定 はないが、例えば、酢酸リチウム、酢酸ナトリウム、酢酸カリウムのようなアルカリ金属 酢酸塩類;塩化リチウム、塩ィ匕ナトリウム、塩化カリウムのようなアルカリ金属塩酸塩類 ;硫酸リチウム、硫酸ナトリウム、硫酸カリウムのようなアルカリ金属硫酸塩類;酢酸マ グネシゥム、酢酸カルシウムのようなアルカリ土類金属酢酸塩類;塩ィ匕マグネシウム、 塩化カルシウムのようなアルカリ土類金属塩酸塩類;硫酸マグネシウム、硫酸カルシ ゥムのようなアルカリ土類金属硫酸塩類又はトリェチルァミン、トリブチルァミン、ジイソ プロピルェチルァミン、 Ν-メチルモルホリン、ピリジン、 4-(Ν,Ν-ジメチルァミノ)ピリジン 、 Ν,Ν-ジメチルァ-リン、 Ν,Ν-ジェチルァ-リン、 1,5-ジァザビシクロ [4.3.0]ノナ- 5-ェ ン、 1,4-ジァザビシクロ [2.2.2]オクタン (DABCO)、 1,8-ジァザビシクロ [5.4.0]- 7-ゥン デセン (DBU)のような有機アミン類の酢酸;塩酸;硫酸塩類などであり、好適にはアル カリ金属酢酸塩類であり、最も好適には酢酸ナトリウムである。 [0131] The additive used in the reaction is not particularly limited as long as it is a salt that accelerates the progress of the reaction. For example, alkali metal acetates such as lithium acetate, sodium acetate, and potassium acetate; lithium chloride, Alkaline metal hydrochlorides such as sodium chloride and potassium chloride; Alkali metal sulfates such as lithium sulfate, sodium sulfate and potassium sulfate; Alkaline earth metal acetates such as magnesium acetate and calcium acetate; Alkaline earth metal hydrochlorides such as magnesium and calcium chloride; alkaline earth metal sulfates such as magnesium sulfate and calcium sulfate or triethylamine, tributylamine, diisopropylethylamine, Ν-methylmorpholine, pyridine 4- (Ν, Ν-dimethylamino) pyridine,, Ν-dimethylamino, ァ, Ν- Jetylaline, 1,5-diazabicyclo [4.3.0] non-5-ene, 1,4-diazabicyclo [2.2.2] octane (DABCO), 1,8-diazabicyclo [5.4.0] -7-u Acetic acid of organic amines such as ndecene (DBU); hydrochloric acid; sulfates, etc., preferably alkali metal acetates, and most preferably sodium acetate.
[0132] 反応温度は、 0°C乃至 100°Cであり、好適には 50°C乃至 100°Cである。
[0133] 反応時間は 1時間乃至 48時間であり、好適には 1時間乃至 12時間である。 [0132] The reaction temperature is 0 ° C to 100 ° C, preferably 50 ° C to 100 ° C. [0133] The reaction time is 1 hour to 48 hours, preferably 1 hour to 12 hours.
[0134] (第 Ba- 4工程) [0134] (Process Ba-4)
本工程は、化合物 (7)を製造する工程であり、化合物 (6)のカルボン酸エステルを力 ルボン酸にすることにより達成される。 This step is a step for producing the compound (7) and can be achieved by converting the carboxylic acid ester of the compound (6) into strong rubonic acid.
[0135] カルボン酸の保護基の除去はその種類によって異なる力 一般にこの分野の技術 において周知の方法、例えば、グリーン'ワッツ著、「プロテクティブ グループス イン オーカニック シンセシス第 3版 (Protective groups in organic synthesis )」 (米国、 Wi ley-Interscience社)に記載の方法により行うことができる。 [0135] Removal of protecting groups on carboxylic acids depends on their type Generally, methods well known in the art, such as Green's Watts, "Protective groups in organic synthesis, 3rd edition (Protective groups in organic synthesis) "(Wiley-Interscience, USA).
[0136] カルボン酸の保護基として、ァリル基を使用した場合には、テトラキストリフエ-ルホ スフインパラジウムなどのパラジウム触媒存在下、ピロリジンなどの求核剤を加えて除 去する方法で実施することができる。 [0136] When a allyl group is used as a protecting group for carboxylic acid, it is removed by adding a nucleophile such as pyrrolidine in the presence of a palladium catalyst such as tetrakistriphenylphosphine palladium. be able to.
[0137] (第 Ba- 5工程) [0137] (Process Ba-5)
本工程は、化合物 (8a)を製造する工程であり、化合物 (7)のカルボン酸を R3Baに変換 することにより達成される。所望する R3Baの種類に応じて、 Ba-5-l乃至 Ba-5-4の方法 力ら選択することができる。 This step is a step for producing the compound (8a), and is achieved by converting the carboxylic acid of the compound (7) to R 3Ba . Depending on the type of R 3Ba desired, the method power of Ba-5-l to Ba-5-4 can be selected.
[0138] (第 Ba— 5—1工程) [0138] (Step Ba— 5—1 step)
R3Baが、アミノ基、 CI- C6アルキルアミノ基、 CI- C6ジァルキルアミノ基、 CI- C6脂肪 族ァシルァミノ基、窒素、酸素及び硫黄力 選択される同一若しくは異なった 1-3個 のへテロ原子を含む 4-10員複素環 C1-C6アルキルアミノ基又は C1-6力ルバモイルァ ルキルアミノ基に置換されているァシル基の場合、化合物 (7)と H-R3Baに相当するアミ ンを縮合することによつても製造できる。即ち、不活性溶媒中、カルボン酸を活性ィ匕 剤と反応させ、活性化した後、又は活性ィ匕すると同時にアミンィ匕合物と反応させること によって行われる。なお、必要に応じて塩基を加えて行っても良い。 R 3Ba is an amino group, a CI-C6 alkylamino group, a CI-C6 dialkylamino group, a CI-C6 aliphatic asilamino group, nitrogen, oxygen and sulfur power. In the case of a 4- to 10-membered heterocyclic C1-C6 alkylamino group or an acyl group substituted with a C1-6 strength rubermoylalkylamino group, by condensing the compound (7) and the amine corresponding to HR 3Ba Can also be manufactured. That is, the reaction is carried out by reacting a carboxylic acid with an active agent in an inert solvent and reacting with the amine compound after activation or simultaneously with the activation. In addition, you may add a base as needed.
[0139] 使用される溶媒は反応を阻害せず、出発物質を溶解するものであれば特に限定は ないが、好適にはジクロロメタン、クロ口ホルム、四塩化炭素、ジクロロエタン、クロ口べ ンゼン、ジクロロベンゼンのようなハロゲン化炭化水素類、ジェチルエーテル、ジイソ プロピルエーテル、テトラヒドロフラン、ジォキサン、ジメトキシェタン、ジエチレングリコ ールジメチルエーテルのようなエーテル類;ホルムアミド、 Ν,Ν-ジメチルホルムアミド、
Ν,Ν-ジメチルァセトアミド、 Ν-メチル -2-ピロリドン、 Ν-メチルピロリジノン、へキサメチ ルホスホロトリアミドのようなアミド類を挙げることができる。さらに好適には Ν,Ν-ジメチ ノレホノレムアミド、テトラヒドロフラン又はジクロロメタンである。 [0139] The solvent to be used is not particularly limited as long as it does not inhibit the reaction and dissolves the starting material, but is preferably dichloromethane, chloroform, carbon tetrachloride, dichloroethane, chloroform, Halogenated hydrocarbons such as chlorobenzene, ethers such as jetyl ether, diisopropyl ether, tetrahydrofuran, dioxane, dimethoxyethane, diethylene glycol dimethyl ether; formamide, Ν, Ν-dimethylformamide, Examples include amides such as Ν, Ν-dimethylacetamide, Ν-methyl-2-pyrrolidone, Ν-methylpyrrolidinone, and hexamethylphosphorotriamide. More preferred is Ν, Ν-dimethylolenolemamide, tetrahydrofuran or dichloromethane.
[0140] 使用される活性化剤としては、例えば、 1,1'-ォキザリルジイミダゾール、 Ν,Ν'-カル ボ -ルジイミダゾールのようなジイミダゾール化合物類; Ν,Ν'-ジサクシンィミジルカー ボネートのようなコハク酸化合物類; Ν,Ν'-ビス (2-ォキソ -3-ォキサゾリジ-ル)ホスホロ アミジッククロライドのようなホスホロアミジッククロライド化合物類; Ν,Ν'-ジサクシンイミ ジルォキザレート (DSO) 、 Ν,Ν'-ジフタールイミドォキザレート (DPO) 、 Ν,Ν'-ビス(ノ ールボルネ -ルサクシンィミジル)ォキザレート (ΒΝΟ) 、 1,1'-ビス(ベンゾトリァゾリル )ォキザレート (ΒΒΤΟ)、 1,1'-ビス (6-クロ口べンゾトリアゾリノレ)ォキザレート (BCTO)、 1, 1'-ビス (6-トリフルォロメチルベンゾトリアゾリル)ォキザレート (BTBO)のようなォキザレ ート化合物類; N-ェチル -5-フエ-ルイソォキサゾリゥム- 3しスルホナートのような N- 低級アルキル- 5-ァリールイソォキサゾリゥム- 3'-スルホナート類;ジェチルホスホリル サイアナイド (DEPC)のようなホスホリルサイアナイド類、 1-ェチル -3- (3-ジメチルァミノ プロピル)カルボジイミド (WSC)-N-ヒドロキシサクシイミド、 1-ェチル -3-(3-ジメチルァ ミノプロピル)カルボジイミド (WSC)-N-ヒドロキシベンゾトリァゾール、 1,3-ジシクロへキ シルカルボジイミド (DCC)カルボジイミド- N-ヒドロキシサクシイミド、 1 ,3-ジシクロへキ シルカルボジイミド (DCC)カルボジイミド- N-ヒドロキシベンゾトリアゾールのようなカル ボジイミド- N-ヒドロキシサクシイミド類;ジ- 2-ピリジルジセレニド一トリフエニルホスフィ ンのようなジヘテロァリールジセレニド―トリアリールホスフィン類;ジピリジルジスルフ イド一トリフエ-ルホスフィンのようなジスルフイド一トリアリールホスフィン類; P-二トロべ ンゼンスルホ -ルトリアゾリドのようなァリールスルホ -ルトリアゾリド類: 2-クロル- 1-メ チルピリジ-ゥム ョーダイドのような 2-ハロ- 1-低級アルキルピリジ-ゥム ハライド;ジ フエ-ルホスホリルアジド (DPPA)のようなジァリールホスホリルアジド類;クロ口炭酸ェ チル、クロ口炭酸イソブチルのような炭酸低級アルキルハライド; 1-プロパンホスホン 酸環状無水物(T3P)のようなホスホン酸環状無水物である。好適には、ジェチルホス ホリルサイアナイド (DEPC)のようなホスホリルサイアナイド類である。 [0140] Examples of the activator used include diimidazole compounds such as 1,1'-oxalyldiimidazole and Ν, Ν'-carbodiimidazole; Ν, Ν'-disuccine Succinic compounds such as imidyl carbonate; Ν, Ν'-bis (2-oxo-3-oxazolidyl) phosphoramidyl chloride compounds such as phosphoramidic chloride; Ν, Ν'- Disuccinimidyl oxalate (DSO), Ν, Ν'-Diphtal imidooxalate (DPO), Ν, Ν'-bis (norbornene-russuccinimidyl) oxalate (ΒΝΟ), 1,1'-bis (benzo Triazolyl) oxalate (ΒΒΤΟ), 1,1'-bis (6-chlorobenzobenzotriazolinole) oxalate (BCTO), 1,1'-bis (6-trifluoromethylbenzotriazolyl) Oxalates such as oxalate (BTBO); N-ethyl-5-fluoro N-lower alkyl-5-arylisoxazolium-3'-sulfonates such as -ruisoxazolium-3 and sulfonate; phosphoryl cyanides such as jetyl phosphoryl cyanide (DEPC) 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide (WSC) -N-hydroxysuccinimide, 1-ethyl-3- (3-dimethylaminopropyl) carbodiimide (WSC) -N-hydroxybenzotriazole, 1 1,3-dicyclocarbodiimide (DCC) carbodiimide-N-hydroxysuccinimide, 1,3-dicyclocyclohexylcarbodiimide (DCC) carbodiimide-N-hydroxybenzotriazole and other carbodiimide-N-hydroxysuccinimides Diheteroaryl diselenide-triarylphosphines such as di-2-pyridyl diselenide-triphenylphosphine; Disulfide monotriaryl phosphines such as lysyl disulfide monotriphosphine; P-ditrobenzene sulfo-tritriazolides such as arylsulfol triazolides: Halo-1-lower alkylpyridyl halides; diarylphosphoryl azides such as diphenylphosphoryl azide (DPPA); carbonic acid lower alkyl halides such as ethyl carbonate and isobutyl carbonate; 1- A phosphonic acid cyclic anhydride such as propanephosphonic acid cyclic anhydride (T3P). Preferred are phosphoryl cyanides such as jetyl phosphoryl cyanide (DEPC).
[0141] 使用される塩基は通常の反応において塩基として使用されるものであれば、特に限
定はないが、例えば、 N-メチルモルホリン、トリエチルァミン、トリプロピルァミン、トリブ チルァミン、ジイソプロピルェチルァミン、ジシクロへキシルァミン、 N-メチルビベリジ ン、ピリジン、 4-ピロリジノピリジン、ピコリン、 4-(N,N-ジメチルァミノ)ピリジン、 2,6-ジ (t -ブチル)- 4-メチルピリジン、キノリン、 Ν,Ν-ジメチルァニリン、 Ν,Ν-ジェチルァニリン、 1,5-ジァザビシクロ [4.3.0]ノナ- 5-ェン (DBN)、 1,4-ジァザビシクロ [2.2.2]オクタン (DA BCO)、 1,8-ジァザビシクロ [5.4.0]ゥンデ力- 7-ェン (DBU)のような有機塩基類が好適 である。更に好適には、ジイソプロピルェチルァミン、トリェチルァミンである。 [0141] The base used is not particularly limited as long as it is used as a base in a normal reaction. For example, N-methylmorpholine, triethylamine, tripropylamine, tributylamine, diisopropylethylamine, dicyclohexylamine, N-methylbiveridine, pyridine, 4-pyrrolidinopyridine, picoline, 4 -(N, N-dimethylamino) pyridine, 2,6-di (t-butyl) -4-methylpyridine, quinoline, Ν, Ν-dimethylaniline, Ν, Ν-jetylaniline, 1,5-diazabicyclo [4.3. 0] Nonah 5-Yen (DBN), 1,4-Dazabicyclo [2.2.2] Octane (DA BCO), 1,8-Zazabicyclo [5.4.0] Unde force-7-Yen (DBU) Organic bases are preferred. More preferred are diisopropylethylamine and triethylamine.
[0142] 反応温度は、 - 10°C乃至 60°Cであり、好適には室温である。 [0142] The reaction temperature is from -10 ° C to 60 ° C, preferably room temperature.
[0143] 反応時間は 30分間乃至 24時間であり、好適には 2時間乃至 20時間である。 [0143] The reaction time is 30 minutes to 24 hours, preferably 2 hours to 20 hours.
[0144] (第 Ba- 5- 2工程) [0144] (Process Ba-5- 2)
R3Baが、置換していても良いアミノ基を含む場合、化合物 (7)にアミノ基を導入後、ァ ルキルイ匕又はァシルイ匕等の常法により R3Baに相当する置換基に変換することによって も製造することができる。本ァミノ化工程にて、アルコールを共存させることによりカー ノメート体として単離し、脱保護によりァミノ基の導入を達成することもできる。 When R 3Ba contains an optionally substituted amino group, after introducing the amino group into the compound (7), it is converted into a substituent corresponding to R 3Ba by an ordinary method such as an alkyl group or an acyl group. Can also be manufactured. In this amination step, an alcohol can be isolated in the presence of alcohol, and introduction of an amino group can be achieved by deprotection.
[0145] アミノ基を導入する反応において使用される溶媒は、反応を阻害せず、出発物質を 溶解するものであれば特に限定はないが、好適にはへキサン、ヘプタン、リグ口イン、 石油エーテルのような脂肪族炭化水素類、ベンゼン、トルエン、キシレンのような芳香 族炭化水素類、ジクロロメタン、クロ口ホルム、四塩化炭素、ジクロロェタン、クロ口ベン ゼン、ジクロロベンゼンのようなハロゲン化炭化水素類、ジェチルエーテル、ジィソプ 口ピルエーテル、テトラヒドロフラン、ジォキサン、ジメトキシェタン、ジエチレングリコー ルジメチルエーテルのようなエーテル類;ホルムアミド、ジメチルホルムアミド、ジメチ ルァセトアミド、メチル -2-ピロリドン、 N-メチルピロリジノン、へキサメチルホスホロトリア ミドのようなアミド類を挙げることができる。さらに好適にはトルエン、ベンゼン、ジメチ ノレホノレムアミド、テトラヒドロフラン又はジクロロメタンである。 [0145] The solvent used in the reaction for introducing the amino group is not particularly limited as long as it does not inhibit the reaction and dissolves the starting material, but preferably hexane, heptane, lignin, petroleum Aliphatic hydrocarbons such as ethers, aromatic hydrocarbons such as benzene, toluene, xylene, halogenated hydrocarbons such as dichloromethane, chloroform, carbon tetrachloride, dichloroethane, benzene, dichlorobenzene Ethers such as dimethyl ether, tetrahydrofuran, dioxane, dimethoxyethane, diethylene glycol dimethyl ether; formamide, dimethylformamide, dimethylacetamide, methyl-2-pyrrolidone, N-methylpyrrolidinone, hexane Amides such as methyl phosphorotriamide There can be mentioned. More preferably, it is toluene, benzene, dimethylolenolemamide, tetrahydrofuran or dichloromethane.
[0146] アミノ基を導入する反応において使用されるアルコールは、通特に限定はないが、 例えば、メタノール、エタノール、プロパノール、 t—ブチルアルコールのような C1-6ァ ルコール、ベンジルアルコールのような芳香族アルキルアルコール、ァリルアルコー ル、 1, 1, 1 トリクロ口エタノール、トリメチルシリルエタノールのようなトリアルキルシリ
ルアルキルアルコールが挙げられ、好適には、 C1-6アルコール、芳香族アルキルァ ルコール、ァリルアルコール、トリアルキルシリルアルキルアルコールであり、更に好 適には t ブチルアルコール、ァリルアルコールである。 [0146] The alcohol used in the reaction for introducing an amino group is not particularly limited. For example, methanol, ethanol, propanol, C1-6 alcohol such as t-butyl alcohol, and fragrance such as benzyl alcohol. Alkyl alcohols, allylic alcohols, 1, 1, 1 C1-6 alcohol, aromatic alkyl alcohol, aryl alcohol, and trialkylsilylalkyl alcohol are preferable, and tbutyl alcohol and aryl alcohol are more preferable.
[0147] アミノ基を導入する反応において使用される試薬は、ジフエニルリン酸アジド (DPPA[0147] The reagent used in the reaction for introducing an amino group is diphenyl phosphate azide (DPPA).
)のようなジァリールリン酸アジド類、アジィ匕ナトリウムのような金属アジド、アジ化水素 などが挙げられ、好適には、ジフエ-ルリン酸アジドである。 ), Metal azides such as sodium azide, hydrogen azide, and the like. Diphenyl phosphate azide is preferred.
[0148] 反応温度は、 0°C乃至 150°Cであり、好適には 0°C乃至 100°Cである。 [0148] The reaction temperature is 0 ° C to 150 ° C, preferably 0 ° C to 100 ° C.
[0149] 反応時間は、 30分乃至 48時間であり、好適には 30分乃至 24時間である。 [0149] The reaction time is 30 minutes to 48 hours, preferably 30 minutes to 24 hours.
[0150] (第 Ba- 5- 3工程) [0150] (Process Ba-5-3)
R3Baが、置換されていても良いァシル基を含む基の場合、カルボン酸を常法により アルコールとし、さらに酸ィ匕してアルデヒドとした後、アルドール反応で相当する置換 基を導入してアルコールを得、そのアルコールを常法により酸ィ匕することにより製造 することができる。 In the case where R 3Ba is a group containing an optionally substituted acyl group, the carboxylic acid is converted to an alcohol by a conventional method, further acidified to an aldehyde, and then the corresponding substituent is introduced by an aldol reaction to form an alcohol. And can be produced by acidifying the alcohol by a conventional method.
[0151] カルボン酸の還元において使用される還元剤は、通常、カルボン酸の還元反応に 用いるものであれば特に限定はないが、例えば、ボラン一テトラヒドロフラン、ボラン一 ジメチルスルフイドのようなボランの錯体、ボラン、ジボランなどが挙げられ、好適には 、ボラン錯体である。 [0151] The reducing agent used in the reduction of carboxylic acid is not particularly limited as long as it is usually used for the reduction reaction of carboxylic acid. For example, borane such as borane-tetrahydrofuran and borane-dimethylsulfide. Complex, borane, diborane and the like, and preferred is a borane complex.
[0152] カルボン酸の還元において使用される溶媒としては、反応を阻害せず、出発物質 をある程度溶解するものであれば特に限定はないが、例えば、ジェチルエーテル、ジ イソプロピルエーテル、テトラヒドロフラン、ジォキサン、ジメトキシェタン、ジエチレング リコールジメチルエーテルのようなエーテル類が挙げられ、好適には、ジメトキシエタ ン又はテトラヒドロフランである。 [0152] The solvent used in the reduction of the carboxylic acid is not particularly limited as long as it does not inhibit the reaction and dissolves the starting material to some extent. For example, jetyl ether, diisopropyl ether, tetrahydrofuran, dioxane , Ethers such as dimethoxyethane and diethylene glycol dimethyl ether are preferred, and dimethoxyethane or tetrahydrofuran is preferred.
[0153] カルボン酸の還元において反応温度は、 0°C乃至 100°Cであり、好適には 0°C乃至 室温である。 [0153] In the reduction of carboxylic acid, the reaction temperature is 0 ° C to 100 ° C, preferably 0 ° C to room temperature.
[0154] カルボン酸の還元において反応時間は、 0.5時間乃至 48時間であり、好適には 0.5 時間乃至 24時間である。 [0154] In the reduction of the carboxylic acid, the reaction time is 0.5 to 48 hours, preferably 0.5 to 24 hours.
[0155] 酸化反応において使用される溶媒としては、反応を阻害せず、出発物質をある程 度溶解するものであれば特に限定はないが、例えば、へキサン、ヘプタン、リグ口イン
、石油エーテルのような脂肪族炭化水素類、ベンゼン、トルエン、キシレンのような芳 香族炭化水素類、ジェチルエーテル、ジイソプロピルエーテル、テトラヒドロフラン、ジ ォキサン、ジメトキシェタン、ジエチレングリコールジメチルエーテルのようなエーテル 類ジクロロメタン、クロ口ホルム、四塩化炭素、ジクロロエタン、クロ口ベンゼン、ジクロロ ベンゼンのようなハロゲン化炭化水素類が挙げられ、好適には、ジクロロメタン又はク ロロホルムである。 [0155] The solvent used in the oxidation reaction is not particularly limited as long as it does not inhibit the reaction and dissolves the starting material to a certain extent. For example, hexane, heptane, rig in , Aliphatic hydrocarbons such as petroleum ether, aromatic hydrocarbons such as benzene, toluene and xylene, ethers such as jetyl ether, diisopropyl ether, tetrahydrofuran, dioxane, dimethoxyethane, and diethylene glycol dimethyl ether Halogenated hydrocarbons such as dichloromethane, chloroform, carbon tetrachloride, dichloroethane, black benzene, and dichlorobenzene are exemplified, and dichloromethane or chloroform is preferred.
[0156] 酸化反応において使用される酸化剤は、通常、アルコール力 ケトンへの酸ィ匕反応 に用いるものであれば特に限定はないが、例えば、二酸化マンガン、デス'マーチン 試薬、ジメチルスルホキシドーォキザリルクロリド、ジメチルスルホキシドー N—クロロス クシンイミド (NCS)、ピリジ-ゥムクロ口クロメート(PCC)などが挙げられる。 [0156] The oxidizing agent used in the oxidation reaction is not particularly limited as long as it is usually used for the acid-oxidation reaction to alcoholic ketones. For example, manganese dioxide, Dess Martin reagent, dimethylsulfoxide Examples include oxalyl chloride, dimethylsulfoxide N-chlorosuccinimide (NCS), and pyridi-mukuroguchi chromate (PCC).
[0157] 酸化反応において反応温度は、 0°C乃至 100°Cであり、好適には室温である。 [0157] In the oxidation reaction, the reaction temperature is 0 ° C to 100 ° C, preferably room temperature.
[0158] 酸化反応において反応時間は、 0.5時間乃至 48時間であり、好適には 0.5時間乃至 24時間である。 [0158] In the oxidation reaction, the reaction time is 0.5 to 48 hours, preferably 0.5 to 24 hours.
[0159] アルドール反応において使用される溶媒としては、反応を阻害せず、出発物質をあ る程度溶解するものであれば特に限定はないが、例えば、へキサン、ヘプタン、リグ口 イン、石油エーテルのような脂肪族炭化水素類、ベンゼン、トルエン、キシレンのよう な芳香族炭化水素類、ジェチルエーテル、ジイソプロピルエーテル、テトラヒドロフラ ン、ジォキサン、ジメトキシェタン、ジエチレングリコールジメチルエーテルのようなェ 一テル類、ジクロロメタン、クロ口ホルム、四塩化炭素、ジクロロェタン、クロ口ベンゼン 、ジクロロベンゼンのようなハロゲン化炭化水素類が挙げられ、好適には、ジェチル エーテル、ジメトキシェタン又はテトラヒドロフランである。 [0159] The solvent used in the aldol reaction is not particularly limited as long as it does not inhibit the reaction and dissolves the starting material to some extent. For example, hexane, heptane, rigin, petroleum ether Aliphatic hydrocarbons such as benzene, toluene, aromatic hydrocarbons such as xylene, jetyl ether, diisopropyl ether, tetrahydrofuran, dioxane, dimethoxyethane, ethers such as diethylene glycol dimethyl ether, dichloromethane , Halogenated hydrocarbons such as black mouth form, carbon tetrachloride, dichloroethane, black mouth benzene, and dichlorobenzene, and preferred are jetyl ether, dimethoxyethane, and tetrahydrofuran.
[0160] アルドール反応において使用される試薬としては、アルキルリチウム等の有機リチウ ム試薬、アルキルマグネシウムハライド等のグリニャール試薬、アルキル亜鉛等の有 機亜鉛試薬、アルキルスズ等の有機スズ試薬、アルキルシラン等の有機ケィ素試薬 等が挙げられ、好適には有機リチウム試薬、グリニャール試薬、有機ケィ素試薬であ り必要に応じて、ルイス酸、金属塩等を共存させても良い。 [0160] Reagents used in the aldol reaction include organic lithium reagents such as alkyllithium, Grignard reagents such as alkylmagnesium halides, organic zinc reagents such as alkylzinc, organotin reagents such as alkyltin, and alkylsilanes. Organic lithium reagents and the like can be mentioned. Organolithium reagents, Grignard reagents, and organic cage reagents are preferred, and Lewis acids, metal salts, and the like may coexist if necessary.
[0161] アルドール反応において反応温度は、 -100°C乃至 100°Cであり、好適には- 78°C乃 至室温である。
[0162] アルドール反応において反応時間は、 0.5時間乃至 48時間であり、好適には 0.5時 間乃至 24時間である。 [0161] In the aldol reaction, the reaction temperature is -100 ° C to 100 ° C, preferably -78 ° C to room temperature. [0162] In the aldol reaction, the reaction time is 0.5 to 48 hours, preferably 0.5 to 24 hours.
[0163] (第 Ba— 5— 4工程) [0163] (No. Ba-5-5 process)
R3Baが置換されて 、ても良!、ァミノアルキル基を含む基である場合、第 Ba- 5- 3工程 の還元工程後、酸化することにより得られたアルデヒドを還元剤存在下、相当するアミ ンと還元的ァミノ化反応を行うことによつても製造することができる。 In the case where R 3Ba is substituted and is a group containing an aminoalkyl group, the aldehyde obtained by oxidation after the reduction step in Step Ba-5-3-3 is converted to the corresponding amino acid in the presence of a reducing agent. It can also be produced by carrying out a reductive amination reaction.
[0164] 使用される還元剤は、通常、還元反応に用いるものであれば特に限定はないが、 例えば、水素化ホウ素ナトリウム、トリァセトキシ水素化ホウ素ナトリウム、シァノ水素化 ホウ素ナトリウムのような水素化ホウ素アルカリ金属などのヒドリド試薬が挙げられ、好 適には、トリァセトキシ水素化ホウ素ナトリウム、シァノ水素化ホウ素ナトリウムである。 [0164] The reducing agent used is not particularly limited as long as it is usually used for the reduction reaction. For example, borohydride such as sodium borohydride, sodium triacetoxyborohydride, sodium cyanoborohydride, etc. Examples thereof include hydride reagents such as alkali metals, preferably sodium triacetoxyborohydride and sodium cyanoborohydride.
[0165] 使用される溶媒としては、反応を阻害せず、出発物質をある程度溶解するものであ れば特に限定はないが、例えば、ジクロロメタン、クロ口ホルム、四塩化炭素、ジクロロ ェタン、クロ口ベンゼン、ジクロロベンゼンのようなハロゲン化炭化水素類、メタノール 、エタノール、イソプロパノールのようなアルコール類、ジォキサン、ジェチルエーテ ル、テトラヒドロフランのようなエーテル類が挙げられ、好適には、ジクロロメタン、メタノ ール又はテトラヒドロフランである。 [0165] The solvent to be used is not particularly limited as long as it does not inhibit the reaction and dissolves the starting material to some extent. For example, dichloromethane, chloroform, carbon tetrachloride, dichloroethane, Examples include halogenated hydrocarbons such as benzene and dichlorobenzene, alcohols such as methanol, ethanol and isopropanol, and ethers such as dioxane, jetyl ether and tetrahydrofuran, preferably dichloromethane, methanol or tetrahydrofuran. It is.
[0166] 反応温度は、 0°C乃至使用溶媒の沸点であり、好適には 0°C乃至室温である。 [0166] The reaction temperature is from 0 ° C to the boiling point of the solvent used, and preferably from 0 ° C to room temperature.
[0167] 反応時間は、 0.5時間乃至 48時間であり、好適には 0.5時間乃至 24時間である。 [0167] The reaction time is 0.5 to 48 hours, preferably 0.5 to 24 hours.
[0168] (第 Ba—6工程) [0168] (Process Ba-6)
本工程は、 目的化合物 (II)を製造する工程であり、化合物 (8a)の水酸基、アミノ基、 カルボキシル基及び Z又はリン酸基が保護されて ヽる場合、常法に従って脱保護す ること〖こより達成される。保護基を有しない場合、本工程は、不要である。 This step is a step for producing the target compound (II). When the hydroxyl group, amino group, carboxyl group and Z or phosphate group of the compound (8a) is protected, it should be deprotected according to a conventional method. It is achieved from Yuko. When there is no protecting group, this step is unnecessary.
[0169] 保護基の除去はその種類によって異なるが、一般に有機合成化学の技術において 周知の方法、例えば、グリーン'ワッツ著、「プロテクティブ グループス イン オーガ ニック ンンセンス第 3版 (Protective groups in organic synthesis )」 (未国、 Wiley- Inte rscience社)に記載の方法により行うことができる。 [0169] The removal of protecting groups varies depending on the type, but generally methods well known in the art of organic synthetic chemistry, such as Green's Watts, "Protective groups in organic synthesis, 3rd edition (Protective groups in organic synthesis). (Uncounted, Wiley-Inscience).
[0170] (Bb法) [0170] (Bb method)
本法によっても、 目的化合物 (II)が製造できる。
[0171] (第 Bb- 1工程) The target compound (II) can also be produced by this method. [0171] (Step Bb-1)
本工程は、化合物 (9)を製造する工程であり、工程 Aで製造される化合物 (2b)を第 Ba -2工程と同様な条件で処理することにより達成される。 This step is a step for producing compound (9), and is achieved by treating compound (2b) produced in step A under the same conditions as in Step Ba-2.
[0172] (第 Bb-2工程) [0172] (Process Bb-2)
本工程は、化合物 (10)を製造する工程であり、化合物 (9)を第 Ba-3工程と同様な条 件で処理することにより達成される。 This step is a step for producing compound (10), and is achieved by treating compound (9) under the same conditions as in Step Ba-3.
[0173] (第 Bb-3工程) [0173] (Process Bb-3)
本工程は、化合物 (8b)を製造する工程であり、化合物 (10)の L1を R に変換すること により達成される。 L1から R の変換は、触媒存在下、クロスカップリング反応を行い、 その後、必要に応じて常法の加水分解反応、加水分解物の還元反応、ォキシムィ匕 反応、ハロゲン化とそれに続くアミノ化反応等を行うことによって達成される。 This step is a step of producing compound (8b), and is achieved by converting L 1 of compound (10) to R. For the conversion of L 1 to R, a cross-coupling reaction is performed in the presence of a catalyst, and then, if necessary, a conventional hydrolysis reaction, a hydrolyzate reduction reaction, an oxime reaction, a halogenation and a subsequent amination. This is achieved by performing a reaction or the like.
[0174] ただし、 L1が R3Bbを示す場合は、この工程を省略することができる。 However, when L 1 represents R 3Bb , this step can be omitted.
[0175] クロスカップリング反応において使用される溶媒としては、本反応に関与しないもの であれば特に限定はないが、メタノール、エタノール、イソプロパノールのようなアルコ ール類、ジェチルエーテル、テトラヒドロフラン、ジォキサンのようなエーテル類、酢酸 ェチル、酢酸プロピルのようなエステル類、ホルムアミド、ジメチルホルムアミド、ジメチ ルァセトアミド、 N—メチル—2—ピロリドン、へキサメチルホスホロトリアミドのようなアミ ド類、水、又はこれらの混合溶媒が好適であり、更に好適には、アルコール類、エー テル類、水、アミド類、又はこれらの混合溶媒である。 [0175] The solvent used in the cross-coupling reaction is not particularly limited as long as it does not participate in this reaction, but alcohols such as methanol, ethanol and isopropanol, jetyl ether, tetrahydrofuran and dioxane. Ethers such as ethyl acetate, esters such as propyl acetate, formamide, dimethylformamide, dimethylacetamide, amides such as N-methyl-2-pyrrolidone, hexamethylphosphorotriamide, water, or these These mixed solvents are preferable, and alcohols, ethers, water, amides, or mixed solvents thereof are more preferable.
[0176] 使用される触媒としては、通常、クロスカップリング反応に使用されるものであれば、 特に限定はないが、好適には、テトラキストリフエニルホスフィンパラジウム、ジクロロジ フエ-ルホスフィンパラジウム、酢酸パラジウムなどのパラジウム触媒、テトラキストリフ ェ-ルホスフィンニッケル、塩化ニッケルなどのニッケル触媒、ヨウ化銅のような銅触 媒などが挙げられる。 [0176] The catalyst to be used is not particularly limited as long as it is usually used in a cross-coupling reaction, and preferably tetrakistriphenylphosphine palladium, dichlorodiphenylphosphine palladium, palladium acetate. Palladium catalysts such as nickel catalyst, nickel catalysts such as tetrakistriphenylphosphine nickel and nickel chloride, and copper catalysts such as copper iodide.
[0177] クロスカップリング反応の試薬としては、フエ-ルホウ酸などのァリールボロン酸、ピ リジルホウ酸などのへテロアリールボロン酸、フエ-ルホウ酸ピナコールエステルなど のァリールボロン酸エステル、トリブチルフエ-ルスズなどのトリアルキルァリールスズ 、トリブチルビ-ルスズ、トリブチル(1 エトキシビュル)スズなどのトリアルキルアルケ
ニルスズ、シアン化ナトリウム、シアン化カリウム、シアン化亜鉛、シアン化銅などのシ アンィ匕化合物などが挙げられる。 [0177] Reagents for the cross-coupling reaction include arylboron acids such as phenylboric acid, heteroarylboronic acids such as pyridylboric acid, arylboronates such as phenolboric acid pinacol ester, and tributylphenol tin. Trialkylalkenyl such as trialkylaryl tin, tributyl beer tin, tributyl (1 ethoxy butyl) tin Examples include cyanine compounds such as niltin, sodium cyanide, potassium cyanide, zinc cyanide and copper cyanide.
[0178] 反応温度は、 0°C乃至 150°Cであり、好適には 50°C乃至 100°Cである。 [0178] The reaction temperature is 0 ° C to 150 ° C, preferably 50 ° C to 100 ° C.
[0179] 反応時間は、 0.5時間乃至 48時間であり、好適には 0.5時間乃至 24時間である。 [0179] The reaction time is 0.5 to 48 hours, preferably 0.5 to 24 hours.
[0180] (第 Bb- 4工程) [0180] (Process Bb-4)
本工程は、 目的化合物 (II)を製造する工程であり、化合物 (8b)を第 Ba-6工程と同様 な条件で処理することにより達成される。 This step is a step for producing the target compound (II) and can be achieved by treating the compound (8b) under the same conditions as in Step Ba-6.
[0181] [0181]
なお、 Xがォキソ基、水酸基、ハロゲン原子で 1又は 2個置換されているアルキレン 基である場合、第 Bb-3〜6工程及び第 Bb-2〜4工程のいずれかの工程の間に、メチ レン部分の酸ィ匕によるカルボ-ル基の導入と、それに引き続き導入されたカルボニル 基の還元又はカルボ-ル基のハロゲンィ匕を行うことにより、 目的化合物 (II)の合成を 達成することができる。 In the case where X is an oxo group, a hydroxyl group, or an alkylene group substituted by 1 or 2 with a halogen atom, during any of the steps Bb-3 to 6 and Bb-2 to 4 The synthesis of the target compound (II) can be achieved by introducing a carbocyclic group with an acid of the methylene moiety and subsequently reducing the introduced carbonyl group or halogenating the carbocyclic group. it can.
[0182] (メチレン部分の酸化反応) [0182] (Oxidation reaction of methylene moiety)
使用される酸化剤は、通常、酸ィ匕反応に用いるものであれば特に限定はないが、 例えば酸ィ匕クロム、過マンガン酸カリウム、二酸化セレン、酸素一金属触媒などであり 、好適には、二酸ィ匕セレンである。 The oxidizing agent to be used is not particularly limited as long as it is usually used for the acid-acid reaction, and examples thereof include acid-chromium, potassium permanganate, selenium dioxide, oxygen monometallic catalyst, and the like. , Selenium diacid.
[0183] 使用される溶媒は、反応を阻害せず、出発物質をある程度溶解するものであれば 特に限定はないが、例えば、ジクロロメタン、クロ口ホルム、四塩化炭素のようなハロゲ ン化炭化水素類や、ジェチルエーテル、ジイソプロピルエーテル、テトラヒドロフラン、 ジォキサン、ジメトキシェタンのようなエーテル類、または、それらと酢酸等の混合溶 媒が挙げられ、好適には、エーテル類と酢酸との混合溶媒である。 [0183] The solvent to be used is not particularly limited as long as it does not inhibit the reaction and dissolves the starting material to some extent. For example, halogenated hydrocarbons such as dichloromethane, chloroform, carbon tetrachloride, etc. And ethers such as jetyl ether, diisopropyl ether, tetrahydrofuran, dioxane and dimethoxyethane, or mixed solvents thereof with acetic acid, etc., and preferably a mixed solvent of ethers and acetic acid. is there.
[0184] 反応温度は、 0°C乃至 150°Cであり、好適には 0°C乃至 100°Cである。 [0184] The reaction temperature is 0 ° C to 150 ° C, preferably 0 ° C to 100 ° C.
[0185] 反応時間は、 30分乃至 48時間であり、好適には 1時間乃至 12時間である。 [0185] The reaction time is 30 minutes to 48 hours, preferably 1 hour to 12 hours.
[0186] (カルボニル基の還元反応) [0186] (Reduction reaction of carbonyl group)
使用される還元剤は、通常、ケトン力 アルコールの還元に用いるものであれば特 に限定はないが、例えば、水素化ホウ素ナトリウム、トリァセトキシ水素化ホウ素ナトリ ゥム、シァノ水素化ホウ素ナトリウムのような水素化ホウ素アルカリ金属などのヒドリド
試薬が挙げられる。 The reducing agent to be used is not particularly limited as long as it is usually used for the reduction of ketone power alcohol, but for example, sodium borohydride, sodium triacetoxyborohydride, sodium cyanoborohydride and the like. Hydride such as alkali metal borohydride A reagent.
[0187] 使用される溶媒としては、反応を阻害しないものであれば特に限定はないが、例え ば、ジクロロメタン、クロ口ホルム、四塩化炭素、ジクロロェタン、クロ口ベンゼン、ジクロ 口ベンゼンのようなハロゲン化炭化水素類、メタノール、エタノールのようなアルコー ル類、ジォキサン、エーテル、テトラヒドロフランのようなエーテル類が挙げられ、好適 には、ジクロロメタン、メタノール又はテトラヒドロフランである。 [0187] The solvent to be used is not particularly limited as long as it does not inhibit the reaction. For example, halogens such as dichloromethane, chloroform, carbon tetrachloride, dichloroethane, chloroform, and dichloromethane are used. Hydrocarbons, alcohols such as methanol and ethanol, ethers such as dioxane, ether and tetrahydrofuran, and dichloromethane, methanol and tetrahydrofuran are preferred.
[0188] 反応温度は、 0°C乃至使用溶媒の沸点であり、好適には 0°C乃至室温である。 [0188] The reaction temperature is from 0 ° C to the boiling point of the solvent used, and preferably from 0 ° C to room temperature.
[0189] 反応時間は、 0.5時間乃至 48時間であり、好適には 0.5時間乃至 12時間である。 [0189] The reaction time is 0.5 to 48 hours, preferably 0.5 to 12 hours.
[0190] (カルボニル基のハロゲン化反応) [0190] (Halogenation reaction of carbonyl group)
使用されるハロゲン化剤は、ケトンのハロゲンィ匕に用いるものであれば特に限定は ないが、例えば、ジェチルアミノサルファートリフルオライド(DAST)、 2,2-ジフルォロ ジメチルイミダゾリジン (DFI)、塩ィ匕チォ -ル、五塩化りん、五臭化りんなどが挙げら れる。 The halogenating agent used is not particularly limited as long as it is used for the halogenation of ketones. For example, jetylaminosulfur trifluoride (DAST), 2,2-difluorodimethylimidazolidine (DFI), salt Examples include thiol, phosphorus pentachloride and phosphorus pentabromide.
[0191] 使用される溶媒としては、反応を阻害しないものであれば特に限定はないが、ベン ゼン、トルエン、キシレンのような芳香族炭化水素類、ジクロロメタン、クロ口ホルム、四 塩化炭素、ジクロロェタンようなハロゲンィ匕炭化水素類、ジェチルエーテル、テトラヒド 口フラン、ジォキサン、ジメトキシェタンのようなェ一テル類、ァセトニトリルのような-ト リル類などが挙げられる。 [0191] The solvent to be used is not particularly limited as long as it does not inhibit the reaction, but aromatic hydrocarbons such as benzene, toluene, xylene, dichloromethane, chloroform, carbon tetrachloride, dichloroethane. And halogenated hydrocarbons such as: jetyl ether, tetrahydrofuran, dioxane, ethers such as dimethoxyethane, and -tolyls such as acetonitrile.
[0192] 反応温度は、 0°C乃至使用溶媒の沸点であり、好適には室温乃至 100°Cである。 [0192] The reaction temperature is from 0 ° C to the boiling point of the solvent used, preferably from room temperature to 100 ° C.
[0193] 反応時間は、 0.5時間乃至 48時間であり、好適には 0.5時間乃至 24時間である。 [0193] The reaction time is 0.5 to 48 hours, preferably 0.5 to 24 hours.
[0194] [0194]
また、 目的化合物 0)は、常法に従って塩基性基を有する場合は酸付加塩にするこ とができ、好適には塩酸塩である。 The target compound 0) can be converted to an acid addition salt when it has a basic group according to a conventional method, and is preferably a hydrochloride.
[0195] 上記各工程の反応終了後、 目的化合物は常法に従って、反応混合物から採取さ れる。例えば、反応混合物を適宜中和し、又、不溶物が存在する場合には濾過によ り除去した後、水と酢酸ェチルのような混和しない有機溶媒を加え、水等で洗浄後、 目的化合物を含む有機層を分離し、無水硫酸マグネシウム等で乾燥後、溶剤を留去 すること〖こよって得られる。
[0196] 得られた目的物は必要ならば常法、例えば再結晶、再沈殿、又は、通常、有機化 合物の分離精製に慣用されている方法、例えば、吸着カラムクロマトグラフィー法、分 配カラムクロマトグラフィー法等の合成吸着剤を使用する方法、イオン交換クロマトグ ラフィーを使用する方法、又は、シリカゲル若しくはアルキルィ匕シリカゲルによる順相 •逆相カラムクロマトグラフィー法を適宜組み合わせ、適切な溶離剤で溶出することに よって分離、精製することができる。 [0195] After completion of the reaction in each of the above steps, the target compound is collected from the reaction mixture according to a conventional method. For example, neutralize the reaction mixture as appropriate, and if insolubles exist, remove by filtration, add water and an immiscible organic solvent such as ethyl acetate, wash with water, etc. The organic layer containing is separated, dried over anhydrous magnesium sulfate and the like, and then distilled off to remove the solvent. [0196] If necessary, the obtained target product can be obtained by a conventional method such as recrystallization, reprecipitation, or a method usually used for separation and purification of organic compounds, such as adsorption column chromatography, distribution. Elution with an appropriate eluent by combining a method using a synthetic adsorbent such as column chromatography, a method using ion exchange chromatography, or a normal phase or reverse phase column chromatography using silica gel or alkyl silica gel. By doing so, it can be separated and purified.
[0197] 本発明の前記一般式 (1)、(Γ)又は (II)を有するチアゾール誘導体、その薬理上許容 される塩及びそのエステルは、種々の形態で投与される。その投与形態としては特に 限定はなぐ各種製剤形態、患者の年齢、性別その他の条件、疾患の程度等に応じ て決定される。例えば錠剤、丸剤、散剤、顆粒剤、シロップ剤、液剤、懸濁剤、乳剤、 顆粒剤およびカプセル剤の場合には経口投与される。また注射剤の場合には単独 であるいはぶどう糖、アミノ酸等の通常の補液と混合して静脈内投与され、更には必 要に応じて単独で筋肉内、皮内、皮下もしくは腹腔内投与される。坐剤の場合には 直腸内投与される。好適には経口投与である。 [0197] The thiazole derivative having the general formula (1), (Γ) or (II) of the present invention, a pharmacologically acceptable salt thereof and an ester thereof are administered in various forms. The dosage form is not particularly limited, and is determined according to the patient's age, gender and other conditions, the degree of disease, etc. For example, in the case of tablets, pills, powders, granules, syrups, solutions, suspensions, emulsions, granules and capsules, they are administered orally. In the case of an injection, it is administered intravenously alone or mixed with a normal fluid such as glucose or amino acid, and further administered alone intramuscularly, intradermally, subcutaneously or intraperitoneally as necessary. In the case of suppositories, it is administered rectally. Oral administration is preferred.
[0198] これらの各種製剤は、常法に従って主薬に賦形剤、結合剤、崩壊剤、潤沢剤、溶 解剤、矯味矯臭、コーティング剤等既知の医薬製剤分野において通常使用しうる既 知の補助剤を用いて製剤化することができる。 [0198] These various preparations are known in general that can be commonly used in the field of known pharmaceutical preparations such as excipients, binders, disintegrants, lubricants, solubilizers, flavoring agents, and coating agents. It can be formulated using adjuvants.
[0199] 錠剤の形態に成形するに際しては、担体としてこの分野で従来公知のものを広く使 用でき、例えば乳糖、白糖、塩ィ匕ナトリウム、ぶどう糖、尿素、澱粉、炭酸カルシウム、 カオリン、結晶セルロース、ケィ酸等の賦形剤、水、エタノール、プロパノール、単シロ ップ、ぶどう糖液、澱粉液、ゼラチン溶液、カルボキシメチルセルロース、セラック、メ チルセルロース、リン酸カリウム、ポリビュルピロリドン等の結合剤、乾燥澱粉、アルギ ン酸ナトリウム、カンテン末、ラミナラン末、炭酸水素ナトリウム、炭酸カルシウム、ポリ ォキシエチレンソルビタン脂肪酸エステル類、ラウリル硫酸ナトリウム、ステアリン酸モ ノグリセリド、澱粉、乳糖等の崩壊剤、白糖、ステアリン、カカオバター、水素添加油等 の崩壊抑制剤、第 4級アンモ-ゥム塩基、ラウリル硫酸ナトリウム等の吸収促進剤、グ リセリン、澱粉等の保湿剤、澱粉、乳糖、カオリン、ベントナイト、コロイド状ケィ酸等の 吸着剤、精製タルク、ステアリン酸塩、硼酸末、ポリエチレングリコール等の滑沢剤等
が例示できる。更に錠剤は必要に応じ通常の剤皮を施した錠剤、例えば糖衣錠、ゼ ラチン被包錠、腸溶被錠、フィルムコーティング錠あるいは二重錠、多層錠とすること ができる。 [0199] When forming into a tablet form, conventionally known carriers can be widely used as carriers. For example, lactose, sucrose, sodium chloride salt, glucose, urea, starch, calcium carbonate, kaolin, crystalline cellulose Excipients such as carboxylic acid, water, ethanol, propanol, simple syrup, glucose solution, starch solution, gelatin solution, binders such as carboxymethylcellulose, shellac, methylcellulose, potassium phosphate, polypyrrole pyrrolidone, Dry starch, sodium alginate, agar powder, laminaran powder, sodium bicarbonate, calcium carbonate, polyoxyethylene sorbitan fatty acid esters, sodium lauryl sulfate, monoglyceride stearate, starch, lactose, etc., sucrose, stearin , Cacao butter, decay additives such as hydrogenated oil, 4th Absorption promoters such as ammonia base, sodium lauryl sulfate, humectants such as glycerin and starch, adsorbents such as starch, lactose, kaolin, bentonite and colloidal key acid, purified talc, stearate, boric acid powder , Lubricants such as polyethylene glycol, etc. Can be illustrated. Furthermore, the tablets can be made into tablets with ordinary coatings as necessary, for example, sugar-coated tablets, gelatin-encapsulated tablets, enteric-coated tablets, film-coated tablets, double tablets, and multilayer tablets.
[0200] 丸剤の形態に成形するに際しては、担体としてこの分野で従来公知のものを広く使 用でき、例えばぶどう糖、乳糖、澱粉、カカオ脂、硬化植物油、カオリン、タルク等の 賦形剤、アラビアゴム末、トラガント末、ゼラチン、エタノール等の結合剤、ラミナラン力 ンテン等の崩壊剤等が例示できる。 [0200] In molding into a pill form, conventionally known carriers can be widely used as carriers, for example, excipients such as glucose, lactose, starch, cocoa butter, hydrogenated vegetable oil, kaolin, talc, Examples thereof include binders such as gum arabic powder, tragacanth powder, gelatin and ethanol, and disintegrants such as laminaran strength.
[0201] 坐剤の形態に成形するに際しては、担体としてこの分野で従来公知のものを広く使 用でき、例えばポリエチレングリコール、カカオ脂、高級アルコール、高級アルコール のエステル類、ゼラチン、半合成グリセライド等を挙げることができる。 [0201] In molding into a suppository, conventionally known carriers can be widely used as carriers, such as polyethylene glycol, cacao butter, higher alcohols, higher alcohol esters, gelatin, semi-synthetic glycerides, etc. Can be mentioned.
[0202] 注射剤として調製される場合には、液剤および懸濁剤は殺菌され、且つ血液と等 張であるのが好ましぐこれら液剤、乳剤および懸濁剤の形態に成形するに際しては 、希釈剤としてこの分野において慣用されているものを全て使用でき、例えば水、ェ チルアルコール、プロピレングリコール、エトキシ化イソステアリルアルコール、ポリオ キシ化イソステアリルアルコール、ポリオキシエチレンソルビタン脂肪酸エステル類等 を挙げることができる。なお、この場合、等張性の溶液を調製するに十分な量の食塩 、ぶどう糖、あるいはグリセリンを医薬製剤中に含有せしめてもよぐまた通常の溶解 補助剤、緩衝剤、無痛化剤等を添加してもよい。 [0202] When prepared as injections, liquids and suspensions are preferably sterilized and isotonic with blood, and when formed into these liquids, emulsions and suspensions, Any of those commonly used in this field can be used as a diluent, such as water, ethyl alcohol, propylene glycol, ethoxylated isostearyl alcohol, polyoxylated isostearyl alcohol, polyoxyethylene sorbitan fatty acid esters and the like. Can do. In this case, a sufficient amount of sodium chloride, glucose, or glycerin may be included in the pharmaceutical preparation to prepare an isotonic solution, and normal solubilizing agents, buffers, soothing agents, etc. may be added. It may be added.
[0203] 更に必要に応じて着色剤、保存剤、香料、風味剤、甘味剤等や他の医薬品を含有 せしめてもよい。 [0203] Further, if necessary, a colorant, a preservative, a fragrance, a flavoring agent, a sweetening agent and the like and other pharmaceuticals may be contained.
[0204] 上記医薬製剤中に含まれる有効成分ィ匕合物の量は、特に限定されず広範囲に適 宜選択される力 通常全組成物中 1〜70重量%、好ましくは 1〜30重量%含まれる量 とするのが適当である。 [0204] The amount of the active ingredient-compound contained in the above-mentioned pharmaceutical preparation is not particularly limited and is appropriately selected over a wide range. Usually 1 to 70% by weight, preferably 1 to 30% by weight in the total composition Appropriate amounts are included.
[0205] その投与量は、症状、年令、体重、投与方法および剤型等によって異なるが、通常 は成人に対して 1日、下限として 0.001mg/kg (好ましくは 0.01mg/kg、更に好ましくは 0 .lmg/kg)であり、上限として 200mg/kg (好ましくは 20mg/kg、更に好ましくは 10mg/kg) を 1回な 、し数回投与することができる。 [0205] The dosage varies depending on symptoms, age, body weight, administration method, dosage form, etc., but is usually 0.001 mg / kg (preferably 0.01 mg / kg, more preferably 1 day) for adults per day. Is 0.1 mg / kg), and 200 mg / kg (preferably 20 mg / kg, more preferably 10 mg / kg) as the upper limit can be administered once or several times.
発明の効果
[0206] 本発明の化合物である、新規チアゾール誘導体及びその薬理上許容される塩は、 優れた FBPase阻害作用、血糖降下作用、脂質低下作用、抗炎症作用等を有し、糖 尿病、高血糖症、耐糖能不全、肥満症、高脂血症、糖尿病合併症 (例えば、網膜症 、白内障、腎症、末梢神経の障害等)、虚血性心疾患、動脈硬化 (例えば、脳血管障 害、狭心症'心筋梗塞、血圧の異常等)、妊娠糖尿病、クッシング症候群、ステロイド 糖尿病、糖原病、自律神経系の障害 (例えば、インポテンス、発汗異常、起立性低血 圧等)、外科的糖尿病様病態、肝臓癌等の治療薬及び Z又は予防薬 (好適には糖 尿病の治療薬及び Z又は予防薬である。 )として有用である。 The invention's effect [0206] The novel thiazole derivative and pharmacologically acceptable salt thereof, which are the compounds of the present invention, have excellent FBPase inhibitory action, hypoglycemic action, lipid-lowering action, anti-inflammatory action, etc. Glycemia, glucose intolerance, obesity, hyperlipidemia, diabetic complications (e.g. retinopathy, cataract, nephropathy, peripheral nerve disorders, etc.), ischemic heart disease, arteriosclerosis (e.g. cerebrovascular disorder) , Angina pectoris, myocardial infarction, abnormal blood pressure, etc.), gestational diabetes, Cushing syndrome, steroid diabetes, glycogenosis, autonomic nervous system disorders (eg, impotence, abnormal sweating, orthostatic hypotension), surgical It is useful as a therapeutic agent for diabetes-like pathology, liver cancer and the like and Z or a preventive agent (preferably a therapeutic agent for Zuria and Z or a preventive agent).
発明を実施するための最良の形態 BEST MODE FOR CARRYING OUT THE INVENTION
[0207] 次に実施例、試験例および製剤例をあげて本発明を更に詳細に説明するが、本発 明はこれらに限定されるものではない。 [0207] Next, the present invention will be described in more detail with reference to Examples, Test Examples and Formulation Examples, but the present invention is not limited thereto.
実施例 Example
[0208] <実施例 1 > <Example 1>
[ (2 アミノー 7 ブロモー 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォ キシ]メチルホスホン酸 (例示化合物番号 1-105) [(2 Amino-7 bromo-8H-indeno [1, 2-d] [l, 3] thiazole-4-yl) oxy] methylphosphonic acid (Exemplified Compound No. 1-105)
(la) [ (7—ブロモ 3—ォキソ 2, 3 ジヒドロ一 1H—インデン一 4—ィル)ォキシ] メチルホスホン酸ジェチル (la) [(7-Bromo-3-oxo2,3 dihydro-1H-indene-4-yl) oxy] Jetyl methylphosphonate
4 ブロモ 7 ヒドロキシインダン一 1—オン(201mg、 0. 885mmol)を N, N— ジメチルホルムアミド(5. 5mL)に溶解し、そこへ炭酸カリウム(135mg、 0. 977mm ol)及びジェトキシホスホリルメチル p トルエンスルホナート(319mg、 0. 977mm ol)を加え、窒素雰囲気下、 80°Cで 20時間攪拌した。 4 Bromo 7-hydroxyindan 1-one (201 mg, 0.885 mmol) was dissolved in N, N-dimethylformamide (5.5 mL), and potassium carbonate (135 mg, 0.977 mmol) and ketoxyphosphorylmethyl p Toluenesulfonate (319 mg, 0.977 mmol) was added, and the mixture was stirred at 80 ° C. for 20 hours under a nitrogen atmosphere.
[0209] 冷却後、反応液に水を加え、ジクロロメタンで抽出した。得られた有機層を飽和食 塩水で洗浄し、無水硫酸ナトリウムで乾燥し、減圧下溶媒を留去した。得られた残渣 をシリカゲルカラムクロマトグラフィー (へキサン:酢酸ェチル、 1 : 1— 0 : 1、 vZv)を用 いて精製し、標記目的化合物 (174mg、収率 52%)を得た。 [0209] After cooling, water was added to the reaction mixture, and the mixture was extracted with dichloromethane. The obtained organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, and the solvent was distilled off under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane: ethyl acetate, 1: 1—0: 1, vZv) to obtain the title compound (174 mg, yield 52%).
[0210] IR(KBr) v 1010, 1029, 1255, 1288, 1473, 1588, 1710 cm"1; [0210] IR (KBr) v 1010, 1029, 1255, 1288, 1473, 1588, 1710 cm "1;
max max
1H NMR(CDC1 , 500MHz): δ 1.37 (6H, t, J=7.3 Hz), 2.66-2.72 (2H, m), 2.99—3.0 1H NMR (CDC1, 500MHz): δ 1.37 (6H, t, J = 7.3 Hz), 2.66-2.72 (2H, m), 2.99—3.0
3 Three
5 (2H, m), 4.33 (4H, dq, J=7.3 Hz, 7.3 Hz), 4.42 (2H, d, J=8.8 Hz), 6.81 (1H, d, J=
8.8 Hz), 7.66 (1H, d, J=8.8 Hz); 5 (2H, m), 4.33 (4H, dq, J = 7.3 Hz, 7.3 Hz), 4.42 (2H, d, J = 8.8 Hz), 6.81 (1H, d, J = 8.8 Hz), 7.66 (1H, d, J = 8.8 Hz);
MS(FAB) m/z: 377 (M+H)+. MS (FAB) m / z: 377 (M + H) + .
(lb) [ (2 アミノー 7 ブロモー 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4ーィ ル)ォキシ]メチルホスホン酸ジェチル (lb) [(2 amino-7 bromo-8H-indeno [1, 2-d] [l, 3] thiazol-4-yl) oxy] methyl phosphonate
実施例 1 ( la)で製造した [ (7 ブロモ 3 ォキソ 2, 3 -ジヒドロ 1H インデ ンー4 ィル)ォキシ]メチルホスホン酸ジェチル(174mg、 0. 461mmol)を酢酸ェ チルークロロホルム(1 : 1、 VZV、 lmL)に溶解し、そこへ臭化第二銅(206mg、 0. 922mmol)をカ卩え、 2時間加熱還流した。 [(7 Bromo-3oxo2,3-dihydro-1H indene-4-yl) oxy] methyl phosphonate (174 mg, 0.461 mmol) prepared in Example 1 (la) was mixed with ethyl chloroformate (1: 1, VZV). , 1 mL), and cupric bromide (206 mg, 0.922 mmol) was added thereto, and the mixture was heated to reflux for 2 hours.
[0211] 冷却後、反応液をろ過し、減圧下溶媒を留去した。得られた残渣をシリカゲルカラ ムクロマトグラフィー (酢酸ェチル)を用いて精製し、 [ (2, 7 ジブ口モー 3—ォキソ一 2, 3 ジヒドロ 1H—インデン— 4—ィル)ォキシ]メチルホスホン酸ジェチルの粗生 成物を得た。 [0211] After cooling, the reaction solution was filtered, and the solvent was distilled off under reduced pressure. The resulting residue was purified using silica gel column chromatography (ethyl acetate) and [(2,7 dib-methyl 3-oxo-1,3-dihydro 1H-indene-4-yl) oxy] methylphosphonate A crude product was obtained.
[0212] これをエタノール(3. 5mL)に溶解し、そこへチォ尿素(41. 5mg、 0. 545mmol) 及び酢酸ナトリウム(58. 7mg、0. 716mmol)をカ卩え、 2時間加熱還流した。 [0212] This was dissolved in ethanol (3.5 mL), and thiourea (41.5 mg, 0.545 mmol) and sodium acetate (58.7 mg, 0.716 mmol) were added thereto and heated to reflux for 2 hours. .
[0213] 冷却後、減圧下溶媒を留去し、得られた残渣に水を加え、クロ口ホルムで抽出した 。得られた有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、減圧下溶媒 を留去した。得られた残渣をシリカゲルカラムクロマトグラフィー (ジクロロメタン:メタノ ール、 50 : 1— 30 : 1、 VZV)を用いて精製し、標記目的化合物(52. 3mg、収率 26 %)を得た。 [0213] After cooling, the solvent was distilled off under reduced pressure, and water was added to the resulting residue, followed by extraction with black mouth form. The obtained organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure. The resulting residue was purified using silica gel column chromatography (dichloromethane: methanol, 50: 1-30: 1, VZV) to obtain the title object compound (52.3 mg, yield 26%).
[0214] IR(KBr) v 789, 977, 1025, 1243, 1537, 3130 cm"1; [0214] IR (KBr) v 789, 977, 1025, 1243, 1537, 3130 cm "1;
max max
1H NMR(CD OD, 500MHz): δ 1.32 (6H, t, J=7.1 Hz), 3.65 (2H, s), 4.27 (4H, dq, 1H NMR (CD OD, 500MHz): δ 1.32 (6H, t, J = 7.1 Hz), 3.65 (2H, s), 4.27 (4H, dq,
3 Three
J=7.1 Hz, 7.1 Hz), 4.67 (2H, d, J=7.8 Hz), 7.03 (1H, d, J=8.8 Hz), 7.26 (1H, d, J=8 .8 Hz); J = 7.1 Hz, 7.1 Hz), 4.67 (2H, d, J = 7.8 Hz), 7.03 (1H, d, J = 8.8 Hz), 7.26 (1H, d, J = 8.8 Hz);
MS(FAB) m/z: 433 (M+H)+. MS (FAB) m / z: 433 (M + H) + .
(lc) [ (2 アミノー 7 ブロモー 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4ーィ ル)ォキシ]メチルホスホン酸 (lc) [(2 amino-7 bromo-8H-indeno [1, 2-d] [l, 3] thiazol-4-yl) oxy] methylphosphonic acid
実施例 1 (lb)で製造した [ (2 アミノー 7 ブロモ— 8H—インデノ [1, 2-d] [l, 3 ]チアゾールー 4 ィル)ォキシ]メチルホスホン酸ジェチル(49. 5mg、 0. 114mmo
1)をジクロロメタン(1. lmL)に溶解し、そこへブロモトリメチルシラン(151 L、 1. 14 mmol)を加え、窒素雰囲気下、室温で 4時間撹拌した。 Example 1 (lb) [(2 amino-7bromo-8H-indeno [1,2-d] [l, 3] thiazol-4-yl) oxy] methyl phosphonate (49.5 mg, 0.114 mmo) 1) was dissolved in dichloromethane (1.1 mL), bromotrimethylsilane (151 L, 1.14 mmol) was added thereto, and the mixture was stirred at room temperature for 4 hours under a nitrogen atmosphere.
[0215] 減圧下溶媒を留去し、得られた残渣に水を加えた。析出した粗結晶を水、メタノー ル及びジェチルエーテルで洗浄して、標記目的化合物(40. 4mg、収率 94%)を得 た。 [0215] The solvent was distilled off under reduced pressure, and water was added to the resulting residue. The precipitated crude crystals were washed with water, methanol and jetyl ether to obtain the title object compound (40.4 mg, yield 94%).
[0216] IR(KBr) v 930, 1081, 1259, 1468, 1583, 2909 cm"1; [0216] IR (KBr) v 930, 1081, 1259, 1468, 1583, 2909 cm "1;
max max
1H NMR(DMSO-d , 500MHz): δ 3.64 (2H, s), 4.29 (2H, d, J=5.9 Hz), 7.08 (IH, d 1H NMR (DMSO-d, 500MHz): δ 3.64 (2H, s), 4.29 (2H, d, J = 5.9 Hz), 7.08 (IH, d
6 6
, J=8.8 Hz), 7.28 (IH, d, J=8.8 Hz), 7.55-7.73 (2H, br); , J = 8.8 Hz), 7.28 (IH, d, J = 8.8 Hz), 7.55-7.73 (2H, br);
MS(FAB) m/z: 375 (M— H)— . MS (FAB) m / z: 375 (M— H) —.
<実施例 2> <Example 2>
[ (2 アミノー 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキシ]メチル ホスホン酸 (例示化合物番号 1-92) [(2 Amino-8H-indeno [1,2-d] [l, 3] thiazol-4-yl) oxy] methylphosphonic acid (Exemplified Compound No. 1-92)
(2a) [ (3—ォキソ 2, 3 ジヒドロ一 1H—インデン一 4—ィル)ォキシ]メチルホスホ ン酸ジェチノレ (2a) [(3-Oxo 2, 3 dihydro-1H-indene-4-yl) oxy] methylphosphonic acid jetinore
7 ヒドロキシインダン一 1—オン(200mg、 1. 35mmol)を使用して、実施例 l (la )に記載した方法に従い、標記目的化合物 (282mg、収率 70%)を得た。 The title object compound (282 mg, yield 70%) was obtained according to the method described in Example 1 (la) using 7-hydroxyindan-1-one (200 mg, 1.35 mmol).
[0217] IR(KBr) v 786, 834, 1032, 1256, 1476, 1601, 1708 cm"1; [0217] IR (KBr) v 786, 834, 1032, 1256, 1476, 1601, 1708 cm "1;
max max
1H NMR(CDC1 , 400MHz) δ 1.37 (6H, t, J=7.1 Hz), 2.63—2.68 (2H, m), 3.07—3.1 1H NMR (CDC1, 400MHz) δ 1.37 (6H, t, J = 7.1 Hz), 2.63-2.68 (2H, m), 3.07-3.1
3 Three
2 (2H, m), 4.35 (4H, dq, J=7.1 Hz, 7.1 Hz), 4.43 (2H, d, J=9.5 Hz), 6.84 (IH, d, J= 8.1 Hz), 7.08 (IH, d, J=8.1 Hz), 7.52 (IH, dd, J=8.1 Hz, 8.1 Hz); 2 (2H, m), 4.35 (4H, dq, J = 7.1 Hz, 7.1 Hz), 4.43 (2H, d, J = 9.5 Hz), 6.84 (IH, d, J = 8.1 Hz), 7.08 (IH, d, J = 8.1 Hz), 7.52 (IH, dd, J = 8.1 Hz, 8.1 Hz);
MS(FAB) m/z: 299 (M+H)+. MS (FAB) m / z: 299 (M + H) + .
(2b) [ (2 ァミノ— 8H—インデノ [1, 2-d] [l, 3]チアゾール—4—ィル)ォキシ]メ チノレホスホン酸ジェチノレ (2b) [(2 amino-8H-indeno [1,2-d] [l, 3] thiazol-4-yl) oxy] methinorephosphonic acid jetinore
実施例 2 (2a)で製造した [ (3—ォキソ 2, 3 ジヒドロ一 1H—インデン一 4—ィル )ォキシ]メチルホスホン酸ジェチル(259mg、 0. 868mmol)をジクロロメタン(8. 0 mL)に溶解し、そこへ三臭化フエ-ルトリメチルアンモ -ゥム(392mg、 1. 04mmol) を加え、窒素雰囲気下、室温で 4時間撹拌した。
[0218] 反応液に飽和炭酸水素ナトリウム水溶液を加え、ジクロロメタンで抽出した。得られ た有機層を水及び飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、減圧下溶媒 を留去して [ (2 ブロモ 3 ォキソ 2, 3 ジヒドロ 1H インデン 4 ィノレ)ォ キシ]メチルホスホン酸ジェチルの粗生成物を得た。 Example 2 [(3-Oxo 2,3 dihydro-1H-indene-4-yl) oxy] methylphosphonate jetyl (259 mg, 0.868 mmol) prepared in (2a) was dissolved in dichloromethane (8.0 mL). Then, trimethylammonium tribromide (392 mg, 1.04 mmol) was added thereto, and the mixture was stirred at room temperature for 4 hours under a nitrogen atmosphere. [0218] A saturated aqueous sodium hydrogen carbonate solution was added to the reaction mixture, and the mixture was extracted with dichloromethane. The obtained organic layer was washed with water and saturated brine, dried over anhydrous sodium sulfate, and the solvent was distilled off under reduced pressure to remove [(2 bromo 3 oxo 2, 3 dihydro 1H indene 4 ynole) oxy] methylphosphonic acid. A crude product of jetyl was obtained.
[0219] これをエタノール(3. 5mL)に溶解し、そこへチォ尿素(76. 9mg、 1. Olmmol)及 び酢酸ナトリウム(107mg、 1. 30mmol)を加え、 2時間加熱還流した。 [0219] This was dissolved in ethanol (3.5 mL), thiourea (76.9 mg, 1. Olmmol) and sodium acetate (107 mg, 1.30 mmol) were added thereto, and the mixture was heated to reflux for 2 hours.
[0220] 冷却後、減圧下溶媒を留去し、残渣に水を加え、クロ口ホルムで抽出した。得られ た有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、減圧下溶媒を留去 した。得られた残渣をシリカゲルカラムクロマトグラフィー (ジクロロメタン:メタノール、 5 0 : 1 - 30 : 1, VZV)を用いて精製し、標記目的化合物(73. 8mg、収率 25%)を得 た。 [0220] After cooling, the solvent was evaporated under reduced pressure, water was added to the residue, and the mixture was extracted with black mouth form. The obtained organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure. The obtained residue was purified by silica gel column chromatography (dichloromethane: methanol, 50: 1-30: 1, VZV) to obtain the title object compound (73.8 mg, yield 25%).
[0221] IR(KBr) v 1019, 1247, 1539, 3142, 3269 cm"1; [0221] IR (KBr) v 1019, 1247, 1539, 3142, 3269 cm "1;
max max
1H NMR(CD OD, 400MHz): δ 1.32 (6H, t, J=7.1 Hz), 3.68 (2H, s), 4.26 (4H, dq, 1H NMR (CD OD, 400MHz): δ 1.32 (6H, t, J = 7.1 Hz), 3.68 (2H, s), 4.26 (4H, dq,
3 Three
J=7.1 Hz, 7.1 Hz), 4.67 (2H, d, J=7.3 Hz), 7.04 (1H, d, J=8.1 Hz), 7.09—7.18 (2H, m); J = 7.1 Hz, 7.1 Hz), 4.67 (2H, d, J = 7.3 Hz), 7.04 (1H, d, J = 8.1 Hz), 7.09—7.18 (2H, m);
MS(FAB) m/z: 355 (M+H)+. MS (FAB) m / z: 355 (M + H) + .
(2c) [ (2 アミノー 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキシ]メ チノレホスホン酸 (2c) [(2 Amino-8H-indeno [1, 2-d] [l, 3] thiazol-4-yl) oxy] methinorephosphonic acid
実施例 2 (2b)で製造した [ (2 アミノー 8H—インデノ [1, 2-d] [l, 3]チアゾール —4—ィル)ォキシ]メチルホスホン酸ジェチル(59. 3mg、0. 167mmol)を使用し て、実施例 1 (lc)に記載した方法に従い、標記目的化合物 (44. 3mg、収率 89%)を 得た。 Example 2 [(2Amino-8H-indeno [1,2-d] [l, 3] thiazol-4-yl) oxy] methylphosphonate jetyl (59.3 mg, 0.167 mmol) prepared in Example 2 (2b) Was used to give the title object compound (44.3 mg, yield 89%) according to the method described in Example 1 (lc).
[0222] IR(KBr) v 635, 933, 1071, 1154, 1258, 1471, 1585, 1636, 2919 cm"1; [0222] IR (KBr) v 635, 933, 1071, 1154, 1258, 1471, 1585, 1636, 2919 cm "1;
max max
1H NMR(DMSO-d , 400MHz): δ 3.73 (2H, s), 4.20 (2H, d, J=8.8 Hz), 7.03-7.10 ( 1H NMR (DMSO-d, 400MHz): δ 3.73 (2H, s), 4.20 (2H, d, J = 8.8 Hz), 7.03-7.10 (
6 6
1H, m), 7.12-7.19 (2H, m), 8.30-8.50 (2H, br); 1H, m), 7.12-7.19 (2H, m), 8.30-8.50 (2H, br);
MS(FAB) m/z: 299 (M+H)+. MS (FAB) m / z: 299 (M + H) + .
<実施例 3 >
(8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィルォキシ)メチルホスホン酸 (3a) [ (2 ブロモ 3—ォキソ 2, 3 ジヒドロ一 1H—インデン一 4—ィル)ォキシ] メチルホスホン酸ジェチル(例示化合物番号 1-1) <Example 3> (8H—Indeno [1,2-d] [l, 3] thiazole-4-yloxy) methylphosphonic acid (3a) [(2 Bromo-3-oxo2,3 dihydro-1H-indene-1-yl) oxy] methylphosphonic acid Jetyl (Exemplified Compound No. 1-1)
実施例 2 (2a)で製造した [ (3—ォキソ 2, 3 ジヒドロ一 1H—インデン一 4—ィル )ォキシ]メチルホスホン酸ジェチル(28. 3g、 94. 9mmol)をエタノール(500mL) に溶解し、そこへ臭化第二銅 (44. 3g、 190mmol)を加え、 50°Cで 2. 5時間撹拌し た。 Example 2 [(3-Oxo 2,3 dihydro-1H-indene-4-yl) oxy] methyl phosphonate (28.3 g, 94.9 mmol) prepared in (2a) was dissolved in ethanol (500 mL). Thereto was added cupric bromide (44.3 g, 190 mmol), and the mixture was stirred at 50 ° C. for 2.5 hours.
[0223] 氷冷下、反応液を飽和炭酸水素ナトリウム水溶液に注ぎ、生じた固体をセライトろ 過により除去した。ろ液を減圧下、溶媒を留去した後、酢酸ェチルで抽出した。得ら れた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥し、減圧下溶媒を [0223] Under ice-cooling, the reaction solution was poured into a saturated aqueous sodium hydrogen carbonate solution, and the resulting solid was removed by Celite filtration. The filtrate was distilled off under reduced pressure and then extracted with ethyl acetate. The obtained organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was removed under reduced pressure.
*y した。 * y.
[0224] 得られた残渣をシリカゲルカラムクロマトグラフィー (酢酸ェチル:メタノール、 10 : 1、 VZV)を用いて精製し、さらにジクロロメタンジイソプロピルエーテルより再結晶し、標 記目的化合物(33. 5g、収率 94%)を得た。 [0224] The obtained residue was purified using silica gel column chromatography (ethyl acetate: methanol, 10: 1, VZV) and recrystallized from dichloromethane diisopropyl ether to give the target compound (33.5 g, yield). 94%).
[0225] IR(KBr) v 1036, 1255, 1433, 1475, 1596, 1717, 2912, 2944, 2954, 2984 cm"1; [0225] IR (KBr) v 1036, 1255, 1433, 1475, 1596, 1717, 2912, 2944, 2954, 2984 cm "1;
max max
1H NMR(CDC1 , 500MHz): δ 1.38 (6H, t, J=6.8 Hz), 3.37 (IH, dd, J=3.4 Hz, 18. 1H NMR (CDC1, 500 MHz): δ 1.38 (6H, t, J = 6.8 Hz), 3.37 (IH, dd, J = 3.4 Hz, 18.
3 Three
1 Hz), 3.77 (IH, dd, J=7.3 Hz, 18.1 Hz), 4.31—4.39 (4H, m), 4.44 (2H, dd, J=2.9 Hz , 9.8 Hz), 4.60 (IH, dd, J=3.4 Hz, 7.3 Hz), 6.90 (IH, d, J=7.8 Hz), 7.04 (IH, d, J=7 .8 Hz), 7.60 (IH, dd, J=7.8 Hz, 7.8 Hz); 1 Hz), 3.77 (IH, dd, J = 7.3 Hz, 18.1 Hz), 4.31—4.39 (4H, m), 4.44 (2H, dd, J = 2.9 Hz, 9.8 Hz), 4.60 (IH, dd, J = 3.4 Hz, 7.3 Hz), 6.90 (IH, d, J = 7.8 Hz), 7.04 (IH, d, J = 7.8 Hz), 7.60 (IH, dd, J = 7.8 Hz, 7.8 Hz);
MS(FAB) m/z: 377, 379 (M+H)+. MS (FAB) m / z: 377, 379 (M + H) + .
(3b) (8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィルォキシ)メチルホスホン 酸ジェチル (3b) (8H—Indeno [1,2-d] [l, 3] thiazole-4-yloxy) methylphosphonic acid jetyl
ホルムアミド(40. 5g、 899mmol)をテトラヒドロフラン(300mL)に溶解し、そこへ 0 °Cで五硫化二リン (40. Og、 180mmol)を加え、室温で 17時間撹拌した後、ろ過し、 チォホルムアミドのテトラヒドロフラン溶液を調製した。 Formamide (40.5 g, 899 mmol) was dissolved in tetrahydrofuran (300 mL), to which was added diphosphorus pentasulfide (40. Og, 180 mmol) at 0 ° C, and the mixture was stirred at room temperature for 17 hours, filtered, and thioformamide A tetrahydrofuran solution was prepared.
[0226] 実施例 3 (3a)で製造した [ (2 ブロモー 3 ォキソ—2, 3 ジヒドロー 1H—インデ ンー4 ィル)ォキシ]メチルホスホン酸ジェチル(33. 5g、 88. 8mmol)をテトラヒド 口フラン(500mL)に溶解し、そこへチォホルムアミドのテトラヒドロフラン溶液をカロえ、
5.5時間加熱還流した。 [0226] [(2 Bromo-3oxo-2,3 dihydro-1H-indene-4-yl) oxy] methyl phosphonate (33.5 g, 88.8 mmol) prepared in Example 3 (3a) was added to tetrahydrofuran ( 500 mL), and then add a solution of thioformamide in tetrahydrofuran. Heated to reflux for 5.5 hours.
[0227] 室温まで冷却後、減圧下溶媒を留去した後、ジクロロメタンで希釈した。得られた有 機層を飽和炭酸水素ナトリウム水溶液及び飽和食塩水で洗浄し、無水硫酸マグネシ ゥムで乾燥し、減圧下溶媒を留去した。 [0227] After cooling to room temperature, the solvent was distilled off under reduced pressure and then diluted with dichloromethane. The obtained organic layer was washed with a saturated aqueous sodium hydrogen carbonate solution and saturated brine, dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure.
[0228] 得られた残渣をシリカゲルカラムクロマトグラフィー (酢酸ェチル:メタノール、 20 : 1、[0228] The obtained residue was subjected to silica gel column chromatography (ethyl acetate: methanol, 20: 1,
VZV)を用いて精製し、さらにジクロロメタンジイソプロピルエーテルより再結晶し、標 記目的化合物(19. 8g、収率 66%)を得た。 VZV) and further recrystallized from dichloromethane diisopropyl ether to obtain the target compound (19.8 g, yield 66%).
[0229] IR(KBr) v 975, 1025, 1248, 1394, 1493, 1581, 2909, 2930, 2984, 3465 cm"1; [0229] IR (KBr) v 975, 1025, 1248, 1394, 1493, 1581, 2909, 2930, 2984, 3465 cm "1;
max max
1H NMR(CDC1 , 500MHz) δ 1.36 (6H, t, J=6.8 Hz), 3.87 (2H, s), 4.33 (4H, dq, J 1H NMR (CDC1, 500MHz) δ 1.36 (6H, t, J = 6.8 Hz), 3.87 (2H, s), 4.33 (4H, dq, J
3 Three
=6.8 Hz, 6.8 Hz), 4.65 (2H, d, J=9.1 Hz), 7.06 (1H, dd, J=1.5 Hz, 7.3 Hz), 7.20-7.2 5 (2H, m), 8.84 (1H, s); = 6.8 Hz, 6.8 Hz), 4.65 (2H, d, J = 9.1 Hz), 7.06 (1H, dd, J = 1.5 Hz, 7.3 Hz), 7.20-7.2 5 (2H, m), 8.84 (1H, s );
MS(FAB) m/z: 339 (M+H)+. MS (FAB) m / z: 339 (M + H) + .
(3c) (8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィルォキシ)メチルホスホン 酸 (3c) (8H-indeno [1,2-d] [l, 3] thiazole-4-yloxy) methylphosphonic acid
実施例 3 (3b)で製造した(8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィルォ キシ)メチルホスホン酸ジェチル(19. 8g、 58. 3mmol)を使用して、実施例 l (lc)に 記載した方法に従い、標記目的化合物(16. 2g、収率 98%)を得た。 Example 3 Using (8H-indeno [1,2-d] [l, 3] thiazole-4-yloxy) methylphosphonate jetyl (19.8 g, 58.3 mmol) prepared in Example 3 (3b) The title object compound (16.2 g, yield 98%) was obtained according to the method described in (lc).
[0230] IR(KBr) v 1073, 1258, 1475, 1580, 2903, 3086, 3418 cm"1; [0230] IR (KBr) v 1073, 1258, 1475, 1580, 2903, 3086, 3418 cm "1;
max max
1H NMR(CD OD, 500MHz): δ 3.94 (2H, s), 4.37 (2H, d, J=10.7 Hz), 7.10 (1H, d, 1H NMR (CD OD, 500 MHz): δ 3.94 (2H, s), 4.37 (2H, d, J = 10.7 Hz), 7.10 (1H, d,
3 Three
J=7.8 Hz), 7.25 (1H, dd, J=1.0 Hz, 7.8 Hz), 7.30 (1H, dd, J=7.8 Hz, 7.8 Hz), 9.20 ( 1H, s); J = 7.8 Hz), 7.25 (1H, dd, J = 1.0 Hz, 7.8 Hz), 7.30 (1H, dd, J = 7.8 Hz, 7.8 Hz), 9.20 (1H, s);
MS(FAB) m/z: 282 (M— H)— . <実施例 4> MS (FAB) m / z: 282 (M—H) —. <Example 4>
[ (2 アミノー 7 フエ-ルー 8H—インデノ [1, 2— d] [l, 3]チアゾール—4—ィル) ォキシ]メチルホスホン酸 (例示化合物番号 1-94) [(2 Amino-7 Ferrue 8H—Indeno [1, 2—d] [l, 3] Thiazole-4-yl) oxy] methylphosphonic acid (Exemplified Compound No. 1-94)
(4a) 7 -ヒドロキシ 4 フエ-ルインダン 1 オン (4a) 7-Hydroxy 4 feltindan 1 on
4 ブロモ 7 ヒドロキシインダン一 1—オン(400mg、 1. 76mmol)を 1, 2 ジメ
トキシェタン(8. 8mL)に溶解し、そこへテトラキス(トリフエ-ルホスフィン)パラジウム (102mg、 0. 088mmol)、フエ-ルホウ酸(322mg、 2. 64mmol)及び 2M炭酸カリ ゥム水溶液(1. 76mL、 3. 52mmol)をカ卩え、 4時間加熱還流した。 4 Bromo 7-hydroxyindan 1-one (400 mg, 1.76 mmol) in 1, 2 dimethyl Dissolve in toxetane (8.8 mL) and add tetrakis (triphenylphosphine) palladium (102 mg, 0.088 mmol), phenol boric acid (322 mg, 2.64 mmol) and 2M aqueous potassium carbonate solution (1.76 mL). 3.52 mmol) and heated to reflux for 4 hours.
[0231] 冷却後、減圧下溶媒を留去し、残渣に水を加え、酢酸ェチルで抽出した。得られた 有機層を飽和塩ィ匕アンモ-ゥム水溶液及び飽和食塩水で洗浄し、無水硫酸ナトリウ ムで乾燥し、減圧下溶媒を留去した。得られた残渣をシリカゲルカラムクロマトグラフ ィー (へキサン:酢酸ェチル、 VZV)を用いて精製し、標記目的化合物(347 mg、収率 88%)を得た。 [0231] After cooling, the solvent was distilled off under reduced pressure, water was added to the residue, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with a saturated aqueous solution of sodium chloride and saturated brine, dried over anhydrous sodium sulfate, and the solvent was distilled off under reduced pressure. The obtained residue was purified using silica gel column chromatography (hexane: ethyl acetate, VZV) to obtain the title object compound (347 mg, yield 88%).
[0232] IR(KBr) v 1163, 1478, 1599, 1694, 3352 cm"1; [0232] IR (KBr) v 1163, 1478, 1599, 1694, 3352 cm "1;
max max
1H NMR(CDC1 , 500MHz): δ 2.73 (2H, dd, J=5.6 Hz, 5.6 Hz), 3.18 (2H, dd, J=5. 1H NMR (CDC1, 500 MHz): δ 2.73 (2H, dd, J = 5.6 Hz, 5.6 Hz), 3.18 (2H, dd, J = 5.
3 Three
6 Hz, 5.6 Hz), 6.87 (IH, d, J=8.3 Hz), 7.33—7.48 (5H, m), 7.52 (IH, d, J=8.3 Hz), 9 .31 (IH, s); 6 Hz, 5.6 Hz), 6.87 (IH, d, J = 8.3 Hz), 7.33—7.48 (5H, m), 7.52 (IH, d, J = 8.3 Hz), 9.31 (IH, s);
MS(EI) m/z: 224 M+. MS (EI) m / z: 224 M + .
(4b) [ (3—ォキソ 7 フエ-ル一 2, 3 ジヒドロ一 IH—インデン一 4—ィル)ォキ シ]メチルホスホン酸ジェチル (4b) [(3-Oxo 7-phenol 2, 3 Dihydro-IH-indene 4-yl) oxy] Jetyl methylphosphonate
実施例 4 (4a)で製造した 7 ヒドロキシー4 フエ-ルインダン— 1 オン(338mg 、 1. 51mmol)を使用して、実施例 1 (la)に記載した方法に従い、標記目的化合物( 165mgゝ収率 29%)を得た。 According to the method described in Example 1 (la) using 7-hydroxy-4-phenolindanone (338 mg, 1.51 mmol) prepared in Example 4 (4a), the title target compound (165 mg yield) 29%).
[0233] IR(KBr) v 1022, 1236, 1480, 1715, 2983 cm"1; [0233] IR (KBr) v 1022, 1236, 1480, 1715, 2983 cm "1;
max max
1H NMR(CDC1 , 400MHz) δ 1.38 (6H, t, J=7.1 Hz), 2.60—2.66 (2H, m), 3.05—3.1 1H NMR (CDC1, 400MHz) δ 1.38 (6H, t, J = 7.1 Hz), 2.60—2.66 (2H, m), 3.05—3.1
3 Three
0 (2H, m), 4.30-4.39 (4H, m), 4.46 (2H, d, J=9.4 Hz), 6.94 (IH, d, J=8.2 Hz), 7.32- 7.46 (5H, m), 7.49 (IH, d, J=8.2 Hz); 0 (2H, m), 4.30-4.39 (4H, m), 4.46 (2H, d, J = 9.4 Hz), 6.94 (IH, d, J = 8.2 Hz), 7.32-7.46 (5H, m), 7.49 (IH, d, J = 8.2 Hz);
MS(FAB) m/z: 375 (M+H)+. MS (FAB) m / z: 375 (M + H) + .
(4c) [ (2 アミノー 7 フヱ-ルー 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキシ]メチルホスホン酸ジェチル (4c) [(2 Amino-7Fu-Lu 8H—Indeno [1,2-d] [l, 3] thiazole-4-yl) oxy] methylphosphonate
実施例 4 (4b)で製造した [ (3 ォキソ 7 フエ-ル - 2, 3-ジヒドロ 1H イン デンー4 ィル)ォキシ]メチルホスホン酸ジェチル(157mg、 0. 419mmol)を使用 して、実施例 2 (2b)に記載した方法に従い、標記目的化合物 (76. 5mg、収率 42%)
を得た。 Example 4 Example 2 was carried out using [(3 oxo 7-phenol-2,3-dihydro 1H indene-4-yl) oxy] methylphosphonate (157 mg, 0.419 mmol) prepared in (4b). According to the method described in (2b), the title object compound (76.5 mg, yield 42%) Got.
[0234] IR(KBr) v 970, 1023, 1253, 1535, 3123, 3331 cm"1; [0234] IR (KBr) v 970, 1023, 1253, 1535, 3123, 3331 cm "1;
max max
1H NMR(CD OD, 400MHz): δ 1.33 (6H, t, J=7.0 Hz), 3.70 (2H, s), 4.23—4.34 (4 1H NMR (CD OD, 400MHz): δ 1.33 (6H, t, J = 7.0 Hz), 3.70 (2H, s), 4.23–4.34 (4
3 Three
H, m), 4.69 (2H, d, J=7.4 Hz), 7.07-7.15 (2H, m), 7.28-7.49 (5H, m); H, m), 4.69 (2H, d, J = 7.4 Hz), 7.07-7.15 (2H, m), 7.28-7.49 (5H, m);
MS(FAB) m/z: 431 (M+H)+. MS (FAB) m / z: 431 (M + H) + .
(4d) [ (2 アミノー 7 フヱ-ルー 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキシ]メチルホスホン酸 (4d) [(2 Amino-7Fu-Lu 8H—Indeno [1, 2-d] [l, 3] thiazole-4-yl) oxy] methylphosphonic acid
実施例 4 (4c)で製造した [ ( 2 ァミノ一 7 フエ-ル -8H-インデノ [ 1 , 2 d] [ 1 , 3]チアゾールー 4 ィル)ォキシ]メチルホスホン酸ジェチル(72. 9mg、0. 169m mol)を使用して、実施例 1 (lc)に記載した方法に従い、標記目的化合物 (50. 2mg 、収率 79%)を得た。 Example 4 [(2 amino-7-Fel-8H-indeno [1,2d] [1,3] thiazol-4-yl) oxy] methylphosphonate (72.9 mg, 0. 169 mmol) was used to give the title object compound (50.2 mg, 79% yield) according to the method described in Example 1 (lc).
[0235] IR(KBr) v 925, 1072, 1157, 1267, 1475, 1627, 3056 cm"1; [0235] IR (KBr) v 925, 1072, 1157, 1267, 1475, 1627, 3056 cm "1;
max max
1H NMR(DMSO-d , 500MHz) 1H NMR (DMSO-d, 500MHz)
6 : δ 3.80 (2H, s), 4.27 (2H, d, J=8.3 Hz), 7.17 (IH, d 6: δ 3.80 (2H, s), 4.27 (2H, d, J = 8.3 Hz), 7.17 (IH, d
, J=8.3 Hz), 7.22 (IH, d, J=8.3 Hz), 7.37 (IH, dd, J=7.3 Hz, 7.3 Hz), 7.46 (2H, dd, J=7.3 Hz, 7.3 Hz), 7.57 (2H, d, J=7.3 Hz), 8.10—8.50 (2H, br); , J = 8.3 Hz), 7.22 (IH, d, J = 8.3 Hz), 7.37 (IH, dd, J = 7.3 Hz, 7.3 Hz), 7.46 (2H, dd, J = 7.3 Hz, 7.3 Hz), 7.57 (2H, d, J = 7.3 Hz), 8.10-8.50 (2H, br);
MS(FAB) m/z: 373 (M- H)— . MS (FAB) m / z: 373 (M- H) —.
<実施例 5 > <Example 5>
{ [2 アミノー 7—(3 フルオロフェ-ル) 8H—インデノ [1, 2-d] [l, 3]チアゾ 一ルー 4 ィル]ォキシ }メチルホスホン酸(例示化合物番号 1-96) {[2 Amino-7- (3 Fluorophenol) 8H-Indeno [1, 2-d] [l, 3] thiazo 4-l] oxy} methylphosphonic acid (Exemplary Compound No. 1-96)
(5a) [ (3—ォキソ 7— (3 フルオロフェ-ル)一2, 3 ジヒドロ一 1H—インデン一 4 ィル)ォキシ]メチルホスホン酸ジェチル (5a) [(3-Oxo 7- (3 Fluorophenyl) -1,2,3 dihydro-1 1H-indene-4-yl) oxy] methyl phosphonate
実施例 1 ( la)で製造した [ (7 ブロモ 3 ォキソ 2, 3 -ジヒドロ 1H インデ ン— 4—ィル)ォキシ]メチルホスホン酸ジェチル(200mg、 0. 530mmol)を 1, 2— ジメトキシェタン(10. 6mL)に溶解し、そこへテトラキス(トリフエ-ルホスフィン)パラ ジゥム(61. 3mg、 0. 053mmol)、 3 フルォロベンゼンボロン酸(11 lmg、 0. 795 mmol)及び 2M炭酸カリウム水溶液(530 /z L、 1. 06mmol)を加え、 5時間加熱還 流した。
[0236] 冷却後、減圧下溶媒を留去し、残渣に水を加え、酢酸ェチルで抽出した。得られた 有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、減圧下溶媒を留去し た。得られた残渣をシリカゲルカラムクロマトグラフィー (へキサン:酢酸ェチル、 1 : 1 - 0 : 1、 VZV)を用いて精製し、標記目的化合物(208mg、収率 100%)を得た。 [(7 Bromo 3oxo2,3-dihydro 1H indene-4-yl) oxy] methylphosphonate (200 mg, 0.530 mmol) prepared in Example 1 (la) was replaced with 1,2-dimethoxyethane (200 mg, 0.530 mmol). 10. 6 mL), tetrakis (triphenylphosphine) paradium (61.3 mg, 0.053 mmol), 3 fluorobenzeneboronic acid (11 lmg, 0.795 mmol) and 2M aqueous potassium carbonate solution (530 / zL, 1.06 mmol) was added and heated to reflux for 5 hours. [0236] After cooling, the solvent was distilled off under reduced pressure, water was added to the residue, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane: ethyl acetate, 1: 1-0: 1, VZV) to obtain the title object compound (208 mg, yield 100%).
[0237] IR(Liquid film) v 975, 1030, 1255, 1478, 1713 cm"1; [0237] IR (Liquid film) v 975, 1030, 1255, 1478, 1713 cm "1;
max max
1H NMR(CDC1 , 400MHz) δ 1.38 (6H, t, J=7.2 Hz), 2.62—2.68 (2H, m), 3.04—3.1 1H NMR (CDC1, 400MHz) δ 1.38 (6H, t, J = 7.2 Hz), 2.62−2.68 (2H, m), 3.04−3.1
3 Three
0 (2H, m), 4.27-4.40 (4H, m), 4.46 (2H, d, J=9.8 Hz), 6.90-7.70 (6H, m); 0 (2H, m), 4.27-4.40 (4H, m), 4.46 (2H, d, J = 9.8 Hz), 6.90-7.70 (6H, m);
MS(FAB) m/z: 393 (M+H)+. MS (FAB) m / z: 393 (M + H) + .
(5b) { [2 アミノー 7—(3 フルオロフヱ-ル) 8H インデノ [1, 2— d] [l, 3]チ ァゾール 4 ィル]ォキシ }メチルホスホン酸ジェチル (5b) {[2 Amino-7- (3 fluorophenyl) 8H Indeno [1, 2— d] [l, 3] Thiazole 4-yl] oxy} methyl phosphonate
実施例 5 (5a)で製造した [(3—ォキソ 7— (3 フルオロフェ -ル) 2, 3 ジヒド 口 1H—インデン— 4—ィル)ォキシ]メチルホスホン酸ジェチル(208mg、 0. 530 mmol)を使用して、実施例 2 (2b)に記載した方法に従 、、標記目的化合物 (79. 9 mg、収率 34%)を得た。 Example 5 [(3-Oxo 7- (3 fluorophenyl) 2,3 dihydrone 1H-indene-4-yl) oxy] methyl phosphonate jetyl (208 mg, 0.530 mmol) prepared in Example 5 (5a) Was used to give the title object compound (79.9 mg, 34% yield) according to the method described in Example 2 (2b).
[0238] IR(KBr) v 788, 1023, 1245, 1524, 3135 cm"1; [0238] IR (KBr) v 788, 1023, 1245, 1524, 3135 cm "1;
max max
1H NMR(DMSO-d , 500MHz) δ 1.26 (6H, t, J=7.1 Hz), 3.80 (2H, s), 4.19-4.27 ( 1H NMR (DMSO-d, 500MHz) δ 1.26 (6H, t, J = 7.1 Hz), 3.80 (2H, s), 4.19-4.27 (
6 6
4H, m), 4.63 (2H, d, J=8.3 Hz), 7.00-7.54 (6H, m); 4H, m), 4.63 (2H, d, J = 8.3 Hz), 7.00-7.54 (6H, m);
MS(FAB) m/z: 449 (M+H)+. MS (FAB) m / z: 449 (M + H) + .
(5c) { [2 アミノー 7— (3 フルオロフヱ-ル) 8H—インデノ [1, 2— d] [l, 3]チ ァゾール 4—ィル]ォキシ }メチルホスホン酸 (5c) {[2 Amino-7- (3 fluorophenyl) 8H-Indeno [1, 2— d] [l, 3] Thiol 4-yl] oxy} methylphosphonic acid
実施例 5 (5b)で製造した { [2 ァミノ一 7— (3 フルオロフェ -ル) 8H—インデ ノ [1, 2— d] [l, 3]チアゾール—4—ィル]ォキシ }メチルホスホン酸ジェチル(60. 9 mg、 0. 136mmol)を使用して、実施例 1 (lc)に記載した方法に従い、標記目的化 合物 (53. 4mg、収率 100%)を得た。 Example 5 {[2 amino 7- (3 fluorophenol) 8H-indeno [1,2-d] [l, 3] thiazol-4-yl] oxy} methylphosphonate prepared in (5b) (60.9 mg, 0.136 mmol) was used to give the title object compound (53.4 mg, 100% yield) according to the method described in Example 1 (lc).
[0239] IR(KBr) v 1070, 1264, 1473, 1579, 2925 cm"1; [0239] IR (KBr) v 1070, 1264, 1473, 1579, 2925 cm "1;
max max
1H NMR(DMSO-d , 400MHz): δ 3.83 (2H, s), 4.28 (2H, d, J=8.2 Hz), 7.14-7.51 ( 1H NMR (DMSO-d, 400MHz): δ 3.83 (2H, s), 4.28 (2H, d, J = 8.2 Hz), 7.14-7.51 (
6 6
6H, m), 8.06-8.28 (2H, br); 6H, m), 8.06-8.28 (2H, br);
MS(FAB) m/z: 391 (M- H)— .
<実施例 6 > MS (FAB) m / z: 391 (M- H) —. <Example 6>
{ [2 アミノー 7—(4 フルオロフェ-ル) 8H—インデノ [1, 2-d] [l, 3]チアゾ 一ルー 4 ィル]ォキシ }メチルホスホン酸(例示化合物番号 1-95) {[2 Amino-7- (4 Fluorophenol) 8H-Indeno [1, 2-d] [l, 3] thiazo-l-l 4-yl] oxy} methylphosphonic acid (Exemplified Compound No. 1-95)
(6a) [ (3-ォキソ 7— (4 フルオロフェ -ル) 2, 3 ジヒドロ 1H インデン 4 ィル)ォキシ]メチルホスホン酸ジェチル (6a) [(3-oxo 7- (4 fluorophenol) 2,3 dihydro 1H indene 4yl) oxy] methyl phosphonate
4 フルォロベンゼンボロン酸(148mg、 1. 06mmol)を使用して、実施例 5 (5a) に記載した方法に従い、標記目的化合物 (203mg、収率 98%)を得た。 4 Using the fluorobenzeneboronic acid (148 mg, 1.06 mmol) and according to the method described in Example 5 (5a), the title object compound (203 mg, 98% yield) was obtained.
[0240] IR(KBr) v 807, 1033, 1257, 1482, 1697 cm"1; [0240] IR (KBr) v 807, 1033, 1257, 1482, 1697 cm "1;
max max
1H NMR(CDC1 , 400MHz): δ 1.38 (6H, t, J=7.1 Hz), 2.61-2.67 (2H, m), 3.01—3.0 1H NMR (CDC1, 400 MHz): δ 1.38 (6H, t, J = 7.1 Hz), 2.61-2.67 (2H, m), 3.01—3.0
3 Three
7 (2H, m), 4.35 (4H, dq, J=7.1 Hz, 7.1 Hz), 4.46 (2H, d, J=9.4 Hz), 6.93 (IH, d, J= 8.2 Hz), 7.08-7.48 (5H, m); 7 (2H, m), 4.35 (4H, dq, J = 7.1 Hz, 7.1 Hz), 4.46 (2H, d, J = 9.4 Hz), 6.93 (IH, d, J = 8.2 Hz), 7.08-7.48 ( 5H, m);
MS(FAB) m/z: 393 (M+H)+. MS (FAB) m / z: 393 (M + H) + .
(6b) { [2 アミノー 7—(4 フルオロフヱ-ル) 8H インデノ [1, 2— d] [l, 3]チ ァゾール 4 ィル]ォキシ }メチルホスホン酸ジェチル (6b) {[2 Amino 7- (4 fluorophenyl) 8H Indeno [1, 2— d] [l, 3] Thiol 4 yl] oxy} Jetyl methylphosphonate
実施例 6 (6a)で製造した [(3—ォキソ 7— (4 フルオロフェ -ル) 2, 3 ジヒド 口 1H—インデン— 4—ィル)ォキシ]メチルホスホン酸ジェチル(194mg、 0. 494 mmol)を使用して、実施例 2 (2b)に記載した方法に従 、、標記目的化合物 (58. 2 mg、収率 26%)を得た。 EXAMPLE 6 [(3-Oxo 7- (4 fluorophenyl) 2,3 dihydrone 1H-indene-4-yl) oxy] methyl phosphonate (194 mg, 0.494 mmol) prepared in Example 6 (6a) Was used to give the title object compound (58.2 mg, 26% yield) according to the method described in Example 2 (2b).
[0241] IR(KBr) v 1023, 1052, 1214, 1244, 1507, 1542, 3116 cm"1; [0241] IR (KBr) v 1023, 1052, 1214, 1244, 1507, 1542, 3116 cm "1;
max max
1H NMR(DMSO-d , 400MHz): δ 1.26 (6H, t, J=7.1 Hz), 3.73 (2H, s), 4.21 (4H, d 1H NMR (DMSO-d, 400MHz): δ 1.26 (6H, t, J = 7.1 Hz), 3.73 (2H, s), 4.21 (4H, d
6 6
q, J=7.1 Hz, 7.1 Hz), 4.61 (2H, d, J=8.2 Hz), 6.99—7.05 (2H, br), 7.08 (IH, d, J=8.6 Hz), 7.13 (IH, d, J=8.6 Hz), 7.22-7.28 (2H, m), 7.55-7.62 (2H, m); q, J = 7.1 Hz, 7.1 Hz), 4.61 (2H, d, J = 8.2 Hz), 6.99—7.05 (2H, br), 7.08 (IH, d, J = 8.6 Hz), 7.13 (IH, d, J = 8.6 Hz), 7.22-7.28 (2H, m), 7.55-7.62 (2H, m);
MS(FAB) m/z: 449 (M+H)+. MS (FAB) m / z: 449 (M + H) + .
(6c) { [2 アミノー 7— (4 フルオロフヱ-ル) 8H—インデノ [1, 2— d] [l, 3]チ ァゾール 4—ィル]ォキシ }メチルホスホン酸 (6c) {[2 Amino-7- (4 fluorophenyl) 8H-Indeno [1, 2- d] [l, 3] Thiol 4-yl] oxy} methylphosphonic acid
実施例 6 (6b)で製造した { [2 ァミノ一 7— (4 フルオロフェ -ル) 8H—インデ ノ [1, 2— d] [l, 3]チアゾール—4—ィル]ォキシ }メチルホスホン酸ジェチル(56. 3
mg、 0. 126mmol)を使用して、実施例 1 (lc)に記載した方法に従い、標記目的化 合物 (47. 6mg、収率 96%)を得た。 Example 6 {[2 amino 7- (4 fluorophenol) 8H-indeno [1,2-d] [l, 3] thiazol-4-yl] oxy} methylphosphonate jetyl prepared in (6b) (56.3 mg, 0.126 mmol) and the title compound (47.6 mg, 96% yield) was obtained according to the method described in Example 1 (lc).
[0242] IR(KBr) v 1072, 1159, 1225, 1480, 1594, 1630, 2926 cm"1; [0242] IR (KBr) v 1072, 1159, 1225, 1480, 1594, 1630, 2926 cm "1;
max max
1H NMR(DMSO-d , 400MHz): δ 3.78 (2H, s), 4.26 (2H, d, J=8.2 Hz), 7.13 (1H, d 1H NMR (DMSO-d, 400MHz): δ 3.78 (2H, s), 4.26 (2H, d, J = 8.2 Hz), 7.13 (1H, d
6 6
, J=8.6 Hz), 7.19 (1H, d, J=8.6 Hz), 7.22-7.30 (2H, m), 7.56-7.63 (2H, m), 8.10-8. 45 (2H, br); , J = 8.6 Hz), 7.19 (1H, d, J = 8.6 Hz), 7.22-7.30 (2H, m), 7.56-7.63 (2H, m), 8.10-8. 45 (2H, br);
MS(FAB) m/z: 393 (M+H)+ MS (FAB) m / z: 393 (M + H) +
<実施例 7> <Example 7>
{ [2 アミノー 7— (3 ヒドロキシフエ-ル)一 8H—インデノ [1, 2— d] [l, 3]チアゾ 一ルー 4 ィル]ォキシ }メチルホスホン酸(例示化合物番号 1-97) {[2 amino-7- (3 hydroxyphenol) 1 8H-indeno [1, 2- d] [l, 3] thiazo 1-ru 4 yl] oxy} methylphosphonic acid (Exemplified Compound No. 1-97)
(7a) { [3—ォキソ 7— (3 ヒドロキシフエ-ル) 2, 3 ジヒドロ一 1H—インデン 4 ィル]ォキシ }メチルホスホン酸ジェチル (7a) {[3-oxo 7- (3 hydroxyphenol) 2, 3 dihydro-1H-indene 4yl] oxy} methyl phosphonate jetyl
3 ヒドロキシベンゼンボロン酸ピナコールエステル(467mg、 2. 12mmol)を使用 して、実施例 5 (5a)に記載した方法に従い、標記目的化合物 (414mg、収率 100%) を得た。 3 Using the hydroxybenzeneboronic acid pinacol ester (467 mg, 2.12 mmol) according to the method described in Example 5 (5a), the title object compound (414 mg, 100% yield) was obtained.
[0243] IR(KBr) v 1031, 1246, 1481, 1579, 1711, 3235 cm"1; [0243] IR (KBr) v 1031, 1246, 1481, 1579, 1711, 3235 cm "1;
max max
1H NMR(CDC1 , 400MHz): δ 1.37 (6H, t, J=7.0 Hz), 2.41—2.48 (2H, m), 3.03—3.0 1H NMR (CDC1, 400 MHz): δ 1.37 (6H, t, J = 7.0 Hz), 2.41—2.48 (2H, m), 3.03—3.0
3 Three
9 (2H, m), 4.31-4.41 (4H, m), 4.40 (2H, d, J=9.8 Hz), 6.78-6.87 (3H, m), 7.04-7.07 (1H, m), 7.22-7.28 (1H, m), 7.33 (1H, s), 7.43 (1H, d, J=8.2 Hz); 9 (2H, m), 4.31-4.41 (4H, m), 4.40 (2H, d, J = 9.8 Hz), 6.78-6.87 (3H, m), 7.04-7.07 (1H, m), 7.22-7.28 ( 1H, m), 7.33 (1H, s), 7.43 (1H, d, J = 8.2 Hz);
MS(FAB) m/z: 391 (M+H)+. MS (FAB) m / z: 391 (M + H) + .
(7b) { [2 アミノー 7— (3 ヒドロキシフヱ-ル) 8H—インデノ [1, 2— d] [l, 3]チ ァゾール 4 ィル]ォキシ }メチルホスホン酸ジェチル (7b) {[2 Amino-7- (3 Hydroxyl) 8H-Indeno [1, 2- d] [l, 3] Thiazole 4-yl] oxy} methyl phosphonate
実施例 7 (7a)で製造した [ (3—ォキソ 7— (3 ヒドロキシフエ-ル) 2, 3 ジヒ ドロー 1H—インデンー4 ィル)ォキシ]メチルホスホン酸ジェチル(378mg、 0. 96 8mmol)を使用して、実施例 2 (2b)に記載した方法に従い、標記目的化合物 (131 mg、収率 30%)を得た。 EXAMPLE 7 Using [(3-oxo7- (3 hydroxyphenol) 2,3 dihydro 1H-indene-4-yl) oxy] methylphosphonate (378 mg, 0.96 8 mmol) prepared in (7a) Then, according to the method described in Example 2 (2b), the title object compound (131 mg, yield 30%) was obtained.
[0244] IR(Liquid film) v 1025, 1213, 1522, 1608, 3186, 3301 cm"1;
H NMR(DMSO-d , 400MHz): δ 1.26 (6H, t, J=7.0 Hz), 3.71 (2H, s), 4.21 (4H, d [0244] IR (Liquid film) v 1025, 1213, 1522, 1608, 3186, 3301 cm "1; H NMR (DMSO-d, 400MHz): δ 1.26 (6H, t, J = 7.0 Hz), 3.71 (2H, s), 4.21 (4H, d
6 6
q, J=7.0 Hz, 7.0 Hz), 4.61 (2H, d, J=8.6 Hz), 6.70-7.50 (8H, m), 9.44 (IH, s); q, J = 7.0 Hz, 7.0 Hz), 4.61 (2H, d, J = 8.6 Hz), 6.70-7.50 (8H, m), 9.44 (IH, s);
MS(FAB) m/z: 447 (M+H)+. MS (FAB) m / z: 447 (M + H) + .
(7c) { [2 アミノー 7— (3 ヒドロキシフヱ-ル) 8H—インデノ [1, 2— d] [l, 3]チ ァゾール 4—ィル]ォキシ }メチルホスホン酸 (7c) {[2 Amino-7- (3 hydroxyphenyl) 8H-Indeno [1, 2- d] [l, 3] Thiazole 4-yl] oxy} methylphosphonic acid
実施例 7 (7b)で製造した { [2 アミノー 7— (3 ヒドロキシフエ-ル) 8H—インデ ノ [1, 2— d] [l, 3]チアゾール—4—ィル]ォキシ }メチルホスホン酸ジェチル(118 mg、 0. 264mmol)を使用して、実施例 1 (lc)に記載した方法に従い、標記目的化 合物 (45. 7mg、収率 44%)を得た。 Example 7 {[2Amino-7- (3 hydroxyphenol) 8H-indeno [1,2-d] [l, 3] thiazole-4-yl] oxy} methylphosphonate prepared in Example 7 (7b) (118 mg, 0.264 mmol) was used to give the title object compound (45.7 mg, 44% yield) according to the method described in Example 1 (lc).
[0245] IR(KBr) v 1068, 1159, 1213, 1585, 1624, 3105 cm"1; [0245] IR (KBr) v 1068, 1159, 1213, 1585, 1624, 3105 cm "1;
max max
1H NMR(DMSO-d , 400MHz): δ 3.77 (2H, s), 4.25 (2H, d, J=8.2 Hz), 6.71—7.50 ( 1H NMR (DMSO-d, 400MHz): δ 3.77 (2H, s), 4.25 (2H, d, J = 8.2 Hz), 6.71-7.50 (
6 6
6H, m), 8.20-8.50 (2H, br); 6H, m), 8.20-8.50 (2H, br);
MS(FAB) m/z: 389 (M- H)— . MS (FAB) m / z: 389 (M- H) —.
<実施例 8 > <Example 8>
{ [2 アミノー 7— (4 ヒドロキシフエ-ル)一 8H—インデノ [1, 2— d] [l, 3]チアゾ 一ルー 4 ィル]ォキシ }メチルホスホン酸(例示化合物番号 1-93) {[2 Amino-7- (4 hydroxyphenol) 1 8H-indeno [1, 2- d] [l, 3] thiazo 1-ru 4 yl] oxy} methylphosphonic acid (Exemplary Compound No. 1-93)
(8a) { [3—ォキソ 7— (4 ヒドロキシフエ-ル) 2, 3 ジヒドロ一 1H—インデン 4 ィル]ォキシ }メチルホスホン酸ジェチル (8a) {[3-oxo 7- (4 hydroxyphenol) 2, 3 dihydro-1H-indene 4yl] oxy} methyl phosphonate jetyl
4ーヒドロキシベンゼンボロン酸(292mg、 2. 12mmol)を使用して、実施例 5 (5a) に記載した方法に従い、標記目的化合物 (321mg、収率 78%)を得た。 The title object compound (321 mg, 78% yield) was obtained according to the method described in Example 5 (5a) using 4-hydroxybenzeneboronic acid (292 mg, 2.12 mmol).
[0246] IR(KBr) v 1029, 1221, 1483, 1713, 3196 cm"1; [0246] IR (KBr) v 1029, 1221, 1483, 1713, 3196 cm "1;
max max
1H NMR(CDC1 , 400MHz) δ 1.40 (6H, t, J=7.0 Hz), 2.57—2.63 (2H, m), 2.98—3.0 1H NMR (CDC1, 400MHz) δ 1.40 (6H, t, J = 7.0 Hz), 2.57-2.63 (2H, m), 2.98-3.0
3 Three
4 (2H, m), 4.29-4.40 (4H, m), 4.50 (2H, d, J=9.0 Hz), 6.76 (IH, s), 6.83 (2H, d, J=8 .4 Hz), 6.91 (IH, d, J=8.4 Hz), 7.11 (2H, d, J=8.4 Hz), 7.37 (IH, d, J=8.4 Hz); MS(FAB) m/z: 391 (M+H)+. 4 (2H, m), 4.29-4.40 (4H, m), 4.50 (2H, d, J = 9.0 Hz), 6.76 (IH, s), 6.83 (2H, d, J = 8.4 Hz), 6.91 (IH, d, J = 8.4 Hz), 7.11 (2H, d, J = 8.4 Hz), 7.37 (IH, d, J = 8.4 Hz); MS (FAB) m / z: 391 (M + H) + .
(8b) { [2 アミノー 7— (4 ヒドロキシフヱ-ル) 8H—インデノ [1, 2— d] [l, 3]チ ァゾール 4 ィル]ォキシ }メチルホスホン酸ジェチル
実施例 8 (8a)で製造した { [3—ォキソ 7— (4—ヒドロキシフエ-ル) 2, 3 ジヒ ドロ 1H—インデン— 4—ィル]ォキシ }メチルホスホン酸ジェチル(287mg、 0. 73 5mmol)を使用して、実施例 2 (2b)に記載した方法に従 、、標記目的化合物 (95. 7 mg、収率 29%)を得た。 (8b) {[2 Amino 7- (4 Hydroxyl) 8H-Indeno [1, 2- d] [l, 3] Thiazole 4-yl] oxy} Jetyl methylphosphonate Example 8 {[3-Oxo 7- (4-hydroxyphenol) 2,3 dihydro 1H-indene-4-yl] oxy} prepared in Example 8 (8a) Jetyl methylphosphonate (287 mg, 0.73 5 mmol ) To give the title object compound (95.7 mg, 29% yield) according to the method described in Example 2 (2b).
[0247] IR(KBr) v 1024, 1232, 1509, 1610, 3176, 3285 cm"1; [0247] IR (KBr) v 1024, 1232, 1509, 1610, 3176, 3285 cm "1;
max max
1H NMR(DMSO-d , 400MHz): δ 1.25 (6H, t, J=7.1 Hz), 3.71 (2H, s), 4.21 (4H, d 1H NMR (DMSO-d, 400MHz): δ 1.25 (6H, t, J = 7.1 Hz), 3.71 (2H, s), 4.21 (4H, d
6 6
q, J=7.1 Hz, 7.1 Hz), 4.59 (2H, d, J=8.2 Hz), 6.81 (2H, d, J=8.4 Hz), 6.92-7.20 (4H , m), 7.35 (2H, d, J=8.4 Hz), 9.46 (1H, s); q, J = 7.1 Hz, 7.1 Hz), 4.59 (2H, d, J = 8.2 Hz), 6.81 (2H, d, J = 8.4 Hz), 6.92-7.20 (4H, m), 7.35 (2H, d, J = 8.4 Hz), 9.46 (1H, s);
MS(FAB) m/z: 447 (M+H)+. MS (FAB) m / z: 447 (M + H) + .
(8c) { [2 アミノー 7— (4 ヒドロキシフヱ-ル) 8H—インデノ [1, 2— d] [l, 3]チ ァゾール 4—ィル]ォキシ }メチルホスホン酸 (8c) {[2 Amino-7- (4 Hydroxyphenol) 8H-Indeno [1, 2— d] [l, 3] Thiazole 4-yl] oxy} methylphosphonic acid
実施例 8 (8b)で製造した [2 アミノー 7— (4 ヒドロキシフエ-ル) 8H—インデノ [1, 2— d] [l, 3]チアゾール—4—ィル]ォキシ }メチルホスホン酸ジェチル(88. 3m g、 0. 198mmol)を使用して、実施例 1 (lc)に記載した方法に従い、標記目的化合 物 (58. 5mg、収率 76%)を得た。 Example 8 [2Amino-7- (4 hydroxyphenol) 8H-indeno [1,2-d] [l, 3] thiazol-4-yl] oxy} methylphosphonate jetyl prepared in Example 8 (8b) (88 3 mg, 0.198 mmol) was used to give the title compound (58.5 mg, 76% yield) according to the method described in Example 1 (lc).
[0248] IR(KBr) v 1061, 1173, 1264, 1478, 1612, 3131 cm"1; [0248] IR (KBr) v 1061, 1173, 1264, 1478, 1612, 3131 cm "1;
max max
1H NMR(DMSO-d , 400MHz): δ 3.77 (2H, s), 4.22 (2H, d, J=8.6 Hz), 6.81 (2H, d 1H NMR (DMSO-d, 400MHz): δ 3.77 (2H, s), 4.22 (2H, d, J = 8.6 Hz), 6.81 (2H, d
6 6
, J=8.6 Hz), 7.09 (1H, d, J=8.6 Hz), 7.15 (1H, d, J=8.6 Hz), 7.36 (2H, d, J=8.6 Hz), 8.35-8.60 (2H, br); , J = 8.6 Hz), 7.09 (1H, d, J = 8.6 Hz), 7.15 (1H, d, J = 8.6 Hz), 7.36 (2H, d, J = 8.6 Hz), 8.35-8.60 (2H, br );
MS(FAB) m/z: 389 (M- H)— . MS (FAB) m / z: 389 (M- H) —.
<実施例 9 > <Example 9>
{ [2 アミノー 7— (3—メトキシフエ-ル)一 8H—インデノ [1, 2— d] [l, 3]チアゾー ルー 4 ィル]ォキシ }メチルホスホン酸(例示化合物番号 1-98) {[2 amino-7- (3-methoxyphenol) 1 8H-indeno [1, 2- d] [l, 3] thiazol 4-yl] oxy} methylphosphonic acid (Exemplary Compound No. 1-98)
( 9a) 7 ヒドロキシ一 4— ( 3—メトキシフエ-ル)インダン 1 オン (9a) 7-hydroxy 4- (3-methoxyphenol) indan 1-on
3—メトキシベンゼンボロン酸(501mg、 3. 30mmol)を使用して、実施例 4 (4a)に 記載した方法に従い、標記目的化合物 (242mg、収率 43%)を得た。 The title object compound (242 mg, 43% yield) was obtained according to the method described in Example 4 (4a) using 3-methoxybenzeneboronic acid (501 mg, 3.30 mmol).
[0249] IR(KBr) v 1042, 1222, 1476, 1614, 1676, 3301 cm"1;
H NMR(CDC1 , 500MHz): δ 2.70-2.75 (2H, m), 3.16—3.21 (2H, m), 3.85 (3H, s), [0249] IR (KBr) v 1042, 1222, 1476, 1614, 1676, 3301 cm "1; H NMR (CDC1, 500 MHz): δ 2.70-2.75 (2H, m), 3.16—3.21 (2H, m), 3.85 (3H, s),
3 Three
6.86 (IH, d, J=8.3 Hz), 6.88—7.02 (3H, m), 7.36 (IH, dd, J=8.3 Hz, 8.3 Hz), 7.52 ( IH, d, J=8.3 Hz), 9.31 (IH, s); 6.86 (IH, d, J = 8.3 Hz), 6.88—7.02 (3H, m), 7.36 (IH, dd, J = 8.3 Hz, 8.3 Hz), 7.52 (IH, d, J = 8.3 Hz), 9.31 ( IH, s);
MS(EI) m/z: 254 M+. MS (EI) m / z: 254 M + .
(9b) { [7- (3—メトキシフエ-ル) 3—ォキソ 2, 3 ジヒドロ一 1H—インデン一 4 ィル]ォキシ }メチルホスホン酸ジェチル (9b) {[7- (3-Methoxyphenol) 3-oxo2, 3 dihydro-1H-indene-4-yl] oxy} methyl phosphonate
実施例 9 (9a)で製造した 7 ヒドロキシ一 4— (3 メトキシフエ-ル)インダン一 1 オン(237mg、 0. 932mmol)を使用して、実施例 1 (la)に記載した方法に従い、標 記目的化合物 (251mg、収率 67%)を得た。 In accordance with the method described in Example 1 (la) using 7 hydroxy 1 4- (3 methoxyphenol) indan 1-one (237 mg, 0.932 mmol) prepared in Example 9 (9a), The target compound (251 mg, yield 67%) was obtained.
[0250] IR(Liquid film) v 1029, 1231, 1480, 1589, 1711, 2983 cm"1; [0250] IR (Liquid film) v 1029, 1231, 1480, 1589, 1711, 2983 cm "1;
max max
1H NMR(CDC1 , 400MHz) δ 1.38 (6H, t, J=7.1 Hz), 2.60—2.66 (2H, m), 3.06—3.1 1H NMR (CDC1, 400MHz) δ 1.38 (6H, t, J = 7.1 Hz), 2.60—2.66 (2H, m), 3.06—3.1
3 Three
1 (2H, m), 3.84 (3H, s), 4.35 (4H, dq, J=7.1 Hz, 7.1 Hz), 4.46 (2H, d, J=9.4 Hz), 6. 86-6.97 (4H, m), 7.31-7.37 (IH, m), 7.50 (IH, d, J=8.2 Hz); 1 (2H, m), 3.84 (3H, s), 4.35 (4H, dq, J = 7.1 Hz, 7.1 Hz), 4.46 (2H, d, J = 9.4 Hz), 6. 86-6.97 (4H, m ), 7.31-7.37 (IH, m), 7.50 (IH, d, J = 8.2 Hz);
MS(FAB) m/z: 405 (M+H)+. MS (FAB) m / z: 405 (M + H) + .
(9c) { [2 ァミノ一 7— (3—メトキシフエ-ル)一 8H—インデノ [1, 2— d] [l, 3]チア ゾール 4 ィル]ォキシ }メチルホスホン酸ジェチル (9c) {[2 amino 1- 7- (3-methoxyphenol) 1 8H-indeno [1, 2- d] [l, 3] thiazole 4-yl] oxy} methyl phosphonate
実施例 9 (9b)で製造した { [7— (3—メトキシフエ-ル) 3—ォキソ 2, 3 ジヒド 口 1H—インデン— 4—ィル]ォキシ }メチルホスホン酸ジェチル(237mg、 0. 586 mmol)を使用して、実施例 2 (2b)に記載した方法に従い、標記目的化合物 (16. 6 mg、収率 6%)を得た。 EXAMPLE 9 {[7- (3-Methoxyphenol) 3-oxo 2,3 dihydric 1H-indene-4-yl] oxy} prepared in Example 9 (9b) Jetyl methylphosphonate (237 mg, 0.586 mmol) To give the title object compound (16.6 mg, yield 6%) according to the method described in Example 2 (2b).
[0251] 1H NMR(CD OD— CDC1 , 400MHz): δ 1.36 (6Η, t, J=7.1 Hz), 3.71 (2H, s), 3.83 ( [0251] 1H NMR (CD OD—CDC1, 400MHz): δ 1.36 (6Η, t, J = 7.1 Hz), 3.71 (2H, s), 3.83 (
3 3 3 3
3H, s), 4.27 (4H, dq, J=7.1 Hz, 7.1 Hz), 4.67 (2H, d, J=7.4 Hz), 6.85—7.13 (5H, m), 7.32 (IH, dd, J=7.8 Hz, 7.8 Hz). 3H, s), 4.27 (4H, dq, J = 7.1 Hz, 7.1 Hz), 4.67 (2H, d, J = 7.4 Hz), 6.85—7.13 (5H, m), 7.32 (IH, dd, J = 7.8 Hz, 7.8 Hz).
MS(EI) m/z: 461 (M+H)+. MS (EI) m / z: 461 (M + H) + .
(9d){[2 ァミノ一 7— (3—メトキシフヱ-ル)一 8H—インデノ [1, 2— d] [l, 3]チア ゾール— 4—ィル]ォキシ }メチルホスホン酸 (9d) {[2 amino 1- 7- (3-methoxyphenyl) 1 8H-indeno [1, 2-d] [l, 3] thiazole-4-yl] oxy} methylphosphonic acid
実施例 9 (9c)で製造した {[2 ァミノ一 7— (3—メトキシフヱ-ル) 8H—インデノ [ 1, 2-d] [l, 3]チアゾールー 4 ィル]ォキシ }メチルホスホン酸ジェチル(16. 6mg
、 0. 036mmol)を使用して、実施例 1 (lc)に記載した方法に従い、標記目的化合 物 (10. 6mg、収率 73%)を得た。 Example 9 {[2 amino-l 7- (3-methoxyphenyl) 8H-indeno [1,2-d] [l, 3] thiazol-4-yl] oxy} methylphosphonate (16) prepared in (9c) .6mg , 0.036 mmol) and the title compound (10.6 mg, 73% yield) was obtained according to the method described in Example 1 (lc).
[0252] IR(KBr) v 932, 1072, 1219, 1476, 1596, 2934 cm"1; [0252] IR (KBr) v 932, 1072, 1219, 1476, 1596, 2934 cm "1;
max max
1H NMR(DMSO-d , 400MHz): δ 3.77-3.82 (5H, m), 4.26 (2H, d, J=8.2 Hz), 6.88 1H NMR (DMSO-d, 400MHz): δ 3.77-3.82 (5H, m), 4.26 (2H, d, J = 8.2 Hz), 6.88
6 6
-6.94 (IH, m), 7.08 (IH, s), 7.10 (IH, d, J=7.8 Hz), 7.14 (IH, d, J=8.6 Hz), 7.18 (1 H, d, J=8.6 Hz), 7.34 (IH, dd, J=7.8 Hz, 7.8 Hz); -6.94 (IH, m), 7.08 (IH, s), 7.10 (IH, d, J = 7.8 Hz), 7.14 (IH, d, J = 8.6 Hz), 7.18 (1 H, d, J = 8.6 Hz) ), 7.34 (IH, dd, J = 7.8 Hz, 7.8 Hz);
MS(FAB) m/z: 403 (M- H)— . MS (FAB) m / z: 403 (M- H) —.
<実施例 10 > <Example 10>
{[2 アミノー 7— (4—メトキシフエ-ル)一 8H—インデノ [1, 2— d] [l, 3]チアゾー ルー 4 ィル]ォキシ }メチルホスホン酸(例示化合物番号 1-100) {[2 amino-7- (4-methoxyphenol) 1 8H-indeno [1, 2- d] [l, 3] thiazol 4-yl] oxy} methylphosphonic acid (Exemplified Compound No. 1-100)
( 1 Oa) 7 ヒドロキシ一 4— (4—メトキシフエ-ル)インダン 1 オン (1 Oa) 7 Hydroxy 4- (4-methoxyphenol) indan 1 on
4ーメトキシベンゼンボロン酸(501mg、 3. 30mmol)を使用して、実施例 4 (4a)に 記載した方法に従い、標記目的化合物 (113mg、収率 20%)を得た。 The title object compound (113 mg, yield 20%) was obtained according to the method described in Example 4 (4a) using 4-methoxybenzeneboronic acid (501 mg, 3.30 mmol).
[0253] IR(KBr) v 1028, 1166, 1260, 1482, 1696, 3350 cm"1; [0253] IR (KBr) v 1028, 1166, 1260, 1482, 1696, 3350 cm "1;
max max
1H NMR(CDC1 , 500MHz): δ 2.69—2.75 (2H, m), 3.13—3.19 (2H, m), 3.86 (3H, s), 1H NMR (CDC1, 500 MHz): δ 2.69—2.75 (2H, m), 3.13—3.19 (2H, m), 3.86 (3H, s),
3 Three
6.85 (IH, d, J=8.3 Hz), 6.96—7.01 (2H, m), 7.31—7.36 (2H, m), 7.48 (IH, d, J=8.3 Hz), 9.27 (IH, s). 6.85 (IH, d, J = 8.3 Hz), 6.96—7.01 (2H, m), 7.31—7.36 (2H, m), 7.48 (IH, d, J = 8.3 Hz), 9.27 (IH, s).
MS(EI) m/z: 254 M+. MS (EI) m / z: 254 M + .
(10b) { [7— (4—メトキシフエ-ル) 3—ォキソ 2, 3 ジヒドロ一 1H—インデン一 4 ィル]ォキシ }メチルホスホン酸ジェチル (10b) {[7- (4-Methoxyphenol) 3-oxo2, 3 dihydro-1H-indene-4-yl] oxy} methyl phosphonate jetyl
実施例 10 (10a)で製造した 7 ヒドロキシ一 4— (4 メトキシフエ-ル)インダン一 1 —オン(107mg、 0. 421mmol)を使用して、実施例 1 (la)に記載した方法に従い、 標記目的化合物 (47. Omg、収率 28%)を得た。 Using the method described in Example 1 (la) using 7 hydroxy 1 4- (4 methoxyphenol) indan 1-one (107 mg, 0.421 mmol) prepared in Example 10 (10a) The target compound (47. Omg, yield 28%) was obtained.
[0254] IR(KBr) v 819, 1035, 1254, 1702, 2923 cm"1; [0254] IR (KBr) v 819, 1035, 1254, 1702, 2923 cm "1;
max max
1H NMR(CDC1 , 500MHz): δ 1.39 (6H, t, J=7.2 Hz), 2.61—2.67 (2H, m), 3.05—3.1 1H NMR (CDC1, 500 MHz): δ 1.39 (6H, t, J = 7.2 Hz), 2.61—2.67 (2H, m), 3.05—3.1
3 Three
0 (2H, m), 3.86 (3H, s), 4.36 (4H, dq, J=7.2 Hz, 7.2 Hz), 4.47 (2H, d, J=9.8 Hz), 6. 94 (IH, d, J=8.3 Hz), 6.96—7.00 (2H, m), 7.30-7.34 (2H, m), 7.48 (IH, d, J=8.3 Hz)
MS(FAB) m/z: 405 (M+H)+. 0 (2H, m), 3.86 (3H, s), 4.36 (4H, dq, J = 7.2 Hz, 7.2 Hz), 4.47 (2H, d, J = 9.8 Hz), 6. 94 (IH, d, J = 8.3 Hz), 6.96—7.00 (2H, m), 7.30-7.34 (2H, m), 7.48 (IH, d, J = 8.3 Hz) MS (FAB) m / z: 405 (M + H) + .
(10c) { [2 ァミノ一 7— (4—メトキシフエ-ル)一 8H—インデノ [1, 2— d] [l, 3]チ ァゾール 4 ィル]ォキシ }メチルホスホン酸ジェチル (10c) {[2 Amino 7- (4-Methoxyphenol) 1 8H-Indeno [1, 2- d] [l, 3] Thiazole 4-yl] oxy} Methylphosphonate
実施例 10 (10b)で製造した { [7— (4—メトキシフエ-ル) 3—ォキソ 2, 3 ジヒ ドロー 1H—インデンー4 ィル]ォキシ }メチルホスホン酸ジェチル(46. 5mg、0. 1 15mmol)を使用して、実施例 2 (2b)に記載した方法に従い、標記目的化合物 (22. lmg、収率 42%)を得た。 EXAMPLE 10 {[7- (4-Methoxyphenol) 3-oxo2,3 dihydro 1H-indene-4-yl] oxy} methyl phosphonate (46.5 mg, 0.1 15 mmol) prepared in Example 10 (10b) To give the title object compound (22. lmg, yield 42%) according to the method described in Example 2 (2b).
[0255] IR(KBr) v 1026, 1242, 1508, 2928, 3110, 3332 cm"1; [0255] IR (KBr) v 1026, 1242, 1508, 2928, 3110, 3332 cm "1;
max max
1H NMR(CD OD, 400MHz): δ 1.34 (6H, t, J=7.2 Hz), 3.69 (2H, s), 3.83 (3H, s), 1H NMR (CD OD, 400MHz): δ 1.34 (6H, t, J = 7.2 Hz), 3.69 (2H, s), 3.83 (3H, s),
3 Three
4.27 (4H, dq, J=7.2 Hz, 7.2 Hz), 4.68 (2H, d, J=7.8 Hz), 6.97 (2H, d, J=8.6 Hz), 7. 07 (IH, d, J=8.6 Hz), 7.11 (IH, d, J=8.6 Hz), 7.39 (2H, d, J=8.6 Hz); 4.27 (4H, dq, J = 7.2 Hz, 7.2 Hz), 4.68 (2H, d, J = 7.8 Hz), 6.97 (2H, d, J = 8.6 Hz), 7. 07 (IH, d, J = 8.6 Hz), 7.11 (IH, d, J = 8.6 Hz), 7.39 (2H, d, J = 8.6 Hz);
MS(FAB) m/z: 461 (M+H)+. MS (FAB) m / z: 461 (M + H) + .
(lOd) { [2 アミノー 7— (4—メトキシフヱ-ル) 8H—インデノ [1, 2 d] [l, 3]チ ァゾール 4—ィル]ォキシ }メチルホスホン酸 (lOd) {[2 amino-7- (4-methoxyphenyl) 8H-indeno [1, 2 d] [l, 3] thiazole 4-yl] oxy} methylphosphonic acid
実施例 10 (10c)で製造した { [2 ァミノ一 7— (4—メトキシフエ-ル) 8H—イン デノ [1, 2— d] [l, 3]チアゾール—4—ィル]ォキシ }メチルホスホン酸ジェチル(22 . lmg、 0. 048mmol)を使用して、実施例 1 (lc)に記載した方法に従い、標記目的 化合物 (17. 6mg、収率 91%)を得た。 Example 10 {[2 amino 7- (4-methoxyphenol) 8H-indeno [1,2-d] [l, 3] thiazole-4-yl] oxy} methylphosphone prepared in (10c) The title compound (17.6 mg, yield 91%) was obtained according to the method described in Example 1 (lc) using decyl acid (22. lmg, 0.048 mmol).
[0256] IR(KBr) v 1060, 1178, 1250, 1480, 1610, 2927 cm"1; [0256] IR (KBr) v 1060, 1178, 1250, 1480, 1610, 2927 cm "1;
max max
1H NMR(DMSO-d , 400MHz): δ 3.76 (2H, s), 3.79 (3H, s), 4.24 (2H, d, J=8.2 Hz 1H NMR (DMSO-d, 400MHz): δ 3.76 (2H, s), 3.79 (3H, s), 4.24 (2H, d, J = 8.2 Hz
6 6
), 6.98 (2H, d, J=8.6 Hz), 7.09 (IH, d, J=8.4 Hz), 7.16 (IH, d, J=8.4 Hz), 7.47 (2H, d, J=8.6 Hz), 7.90-8.25 (2H, br); ), 6.98 (2H, d, J = 8.6 Hz), 7.09 (IH, d, J = 8.4 Hz), 7.16 (IH, d, J = 8.4 Hz), 7.47 (2H, d, J = 8.6 Hz), 7.90-8.25 (2H, br);
MS(FAB) m/z: 403 (M- H)— . MS (FAB) m / z: 403 (M- H) —.
<実施例 11 > <Example 11>
{ [2 アミノー 7—(4 アミノフヱ-ル) 8H—インデノ [1, 2-d] [l, 3]チアゾール 4 ィル]ォキシ }メチルホスホン酸(例示化合物番号 1-99)
( 11 a) 7 ヒドロキシ— 4— [ (t ブトキシカルボ-ル)ァミノ]フエ-ルインダン 1— オン {[2 amino-7- (4 aminophenyl) 8H-indeno [1, 2-d] [l, 3] thiazole 4-yl] oxy} methylphosphonic acid (Exemplified Compound No. 1-99) (11a) 7Hydroxy—4 — [(t-Butoxycarbol) amino] Phenol Indan 1—On
4— [ (t—ブトキシカルボ-ル)ァミノ]フエ-ルボロン酸(626mg、 2. 64mmol)を使 用して、実施例 4 (4a)に記載した方法に従い、標記目的化合物 (421mg、収率 71% )を得た。 4-[(t-Butoxycarbol) amino] phenol boronic acid (626 mg, 2.64 mmol) was used according to the method described in Example 4 (4a) to give the title compound (421 mg, yield). 71%).
[0257] IR(KBr) v 1157, 1233, 1287, 1528, 1594, 1662, 1726, 3366 cm"1; [0257] IR (KBr) v 1157, 1233, 1287, 1528, 1594, 1662, 1726, 3366 cm "1;
max max
1H NMR(CDC1 , 500MHz) δ 1.54 (9H, s), 2.68-2.77 (2H, m), 3.10—3.19 (2H, m), 1H NMR (CDC1, 500MHz) δ 1.54 (9H, s), 2.68-2.77 (2H, m), 3.10-3.19 (2H, m),
3 Three
6.50-6.58 (IH, br), 6.85 (IH, d, J=8.3 Hz), 7.31-7.37 (2H, m), 7.40-7.46 (2H, m), 7.48 (IH, d, J=8.3 Hz), 9.29 (IH, s); 6.50-6.58 (IH, br), 6.85 (IH, d, J = 8.3 Hz), 7.31-7.37 (2H, m), 7.40-7.46 (2H, m), 7.48 (IH, d, J = 8.3 Hz) , 9.29 (IH, s);
MS(FAB) m/z: 340 (M+H)+. MS (FAB) m / z: 340 (M + H) + .
( 1 lb) [ (7— { [4— (t ブトキシカルボ-ル)ァミノ]フエ-ル} 3 ォキソ—2, 3— ジヒドロ 1H—インデン 4 ィル)ォキシ]メチルホスホン酸ジェチル (1 lb) [(7— {[4— (t-Butoxycarbol) amino] phenol} 3 oxo-2,3-dihydro 1H-indene 4 yl) oxy] methylphosphonate
実施例 11 ( 11 a)で製造した 7 ヒドロキシ— 4— [ (t ブトキシカルボ-ル)ァミノ]フ ェ-ルインダンー1 オン (409mg、 1. 20mmol)を使用して、実施例 1 (la)に記載 した方法に従い、標記目的化合物 (334mg、収率 57%)を得た。 Example 11 (11a) was used in Example 1 (la) using 7-hydroxy-4-([(t-butoxycarbol) amino] -phenolindanone (409 mg, 1.20 mmol). The title object compound (334 mg, yield 57%) was obtained according to the method described.
[0258] IR(KBr) v 1031, 1163, 1238, 1715, 2980, 3266 cm"1; [0258] IR (KBr) v 1031, 1163, 1238, 1715, 2980, 3266 cm "1;
max max
1H NMR(CDC1 , 500MHz): δ 1.39 (6H, t, J=7.2 Hz), 1.59 (9H, s), 2.61—2.67 (2H, 1H NMR (CDC1, 500MHz): δ 1.39 (6H, t, J = 7.2 Hz), 1.59 (9H, s), 2.61—2.67 (2H,
3 Three
m), 3.04-3.10 (2H, m), 4.36 (4H, dq, J=7.2 Hz, 7.2 Hz), 4.47 (2H, d, J=9.3 Hz), 6. 54-6.58 (IH, br), 6.94 (IH, d, J=8.3 Hz), 7.30-7.34 (2H, m), 7.42-7.46 (2H, m), 7. 48 (IH, d, J=8.3 Hz); m), 3.04-3.10 (2H, m), 4.36 (4H, dq, J = 7.2 Hz, 7.2 Hz), 4.47 (2H, d, J = 9.3 Hz), 6. 54-6.58 (IH, br), 6.94 (IH, d, J = 8.3 Hz), 7.30-7.34 (2H, m), 7.42-7.46 (2H, m), 7. 48 (IH, d, J = 8.3 Hz);
MS(FAB) m/z: 490 (M+H)+. MS (FAB) m / z: 490 (M + H) + .
(11c) [ (2—ァミノ— 7— { [4— (t—ブトキシカルボ-ル)ァミノ]フエ-ル}— 8H—ィ ンデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキシ]メチルホスホン酸ジェチル 実施例 11 ( 1 lb)で製造した [ (7— { [4一(t—ブトキシカルボ-ル)ァミノ]フ -ル} — 3 ォキソ 2, 3 ジヒドロ一 1 H—インデン 4 ィル)ォキシ]メチルホスホン酸 ジェチル(330mg、 0. 674mmol)を使用して、実施例 2 (2b)に記載した方法に従 い、標記目的化合物 (96. 8mg、収率 26%)を得た。 (11c) [(2—Amino— 7— {[4— (t-Butoxycarbol) amino] phenol} — 8H—Indeno [1, 2-d] [l, 3] thiazole- 4 yl ) Oxy] methyl phosphonate Jetyl prepared in Example 11 (1 lb) [(7— {[4 (t-Butoxycarbole) amino] fur} — 3 oxo 2, 3 dihydro 1 H-indene 4- (yl) oxy] methylphosphonate Jetyl (330 mg, 0.674 mmol) was used to give the title object compound (96.8 mg, 26% yield) according to the method described in Example 2 (2b). It was.
[0259] 1H NMR(CD OD, 400MHz): δ 1.33 (6Η, t, J=7.0 Hz), 1.53 (9H, s), 3.69 (2H, s),
4.22-4.32 (4H, m), 4.65-4.70 (2H, m), 6.74-7.48 (6H, m). [0259] 1H NMR (CD OD, 400MHz): δ 1.33 (6Η, t, J = 7.0 Hz), 1.53 (9H, s), 3.69 (2H, s), 4.22-4.32 (4H, m), 4.65-4.70 (2H, m), 6.74-7.48 (6H, m).
MS(EI) m/z: 546 (M+H)+. MS (EI) m / z: 546 (M + H) + .
(l id) { [2 ァミノ一 7— (4—アミノフヱ-ル)一 8H—インデノ [1, 2 d] [l, 3]チア ゾール 4—ィル]ォキシ }メチルホスホン酸 (l id) {[2 amino 1- 7- (4-aminophenol) 1 8H-indeno [1, 2 d] [l, 3] thiazole 4-yl] oxy} methylphosphonic acid
実施例 11 ( 1 lc)で製造した [ (2 ァミノ 7— { [4— (t—ブトキシカルボ-ル)アミ ノ]フエ-ル}ー811—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキシ]メチル ホスホン酸ジェチル(96. lmg、 0. 176mmol)を使用して、実施例 l (lc)に記載し た方法に従い、標記目的化合物 (52. 2mg、収率 76%)を得た。 Example 11 [(2 amino 7- {[4— (t-butoxycarbol) amino] phenol} -811—indeno [1, 2-d] [l, 3 ] Thiazol-4-yl) oxy] methyl phosphonate (96. lmg, 0.176 mmol) and according to the method described in Example l (lc), the title compound (52.2 mg, yield 76) %).
[0260] IR(KBr) v 922, 1061, 1154, 1266, 1480, 1624, 2924 cm"1; [0260] IR (KBr) v 922, 1061, 1154, 1266, 1480, 1624, 2924 cm "1;
max max
1H NMR(DMSO-d , 500MHz): δ 3.78 (2H, s), 4.25 (2H, d, J=8.3 Hz), 6.70 (2H, d 1H NMR (DMSO-d, 500MHz): δ 3.78 (2H, s), 4.25 (2H, d, J = 8.3 Hz), 6.70 (2H, d
6 6
, J=8.3 Hz), 7.09 (IH, d, J=8.3 Hz), 7.15 (IH, d, J=8.3 Hz), 7.27 (2H, d, J=8.3 Hz); MS(FAB) m/z: 389 (M- H)— . , J = 8.3 Hz), 7.09 (IH, d, J = 8.3 Hz), 7.15 (IH, d, J = 8.3 Hz), 7.27 (2H, d, J = 8.3 Hz); MS (FAB) m / z : 389 (M- H) —.
<実施例 12 > <Example 12>
( {2 アミノー 7— [3 (トリフルォロメチル)フ -ル ] 8H—インデノ [1, 2-d] [l , 3]チアゾールー 4ーィル }ォキシ)メチルホスホン酸(例示化合物番号 1-101) ( 12a) 7 ヒドロキシ一 4— [ 3 (トリフルォロメチル)フエ-ル]インダン 1 オン ({2 amino-7- [3 (trifluoromethyl) fur] 8H-indeno [1,2-d] [l, 3] thiazol-4-yl} oxy) methylphosphonic acid (Exemplary Compound No. 1-101) ( 12a) 7-Hydroxy mono 4-— [3 (trifluoromethyl) phenol] indan 1-one
3 トリフルォロメチルベンゼンボロン酸(627mg、 3. 30mmol)を使用して、実施 例 4 (4a)に記載した方法に従い、標記目的化合物 (344mg、収率 54%)を得た。 3 The title object compound (344 mg, 54% yield) was obtained using trifluoromethylbenzeneboronic acid (627 mg, 3.30 mmol) according to the method described in Example 4 (4a).
[0261] IR(KBr) v 1122, 1179, 1301, 1332, 1664 cm"1; [0261] IR (KBr) v 1122, 1179, 1301, 1332, 1664 cm "1;
max max
1H NMR(CDC1 , 500MHz): δ 2.73-2.78 (2H, m), 3.14—3.18 (2H, m), 6.90 (IH, d, 1H NMR (CDC1, 500 MHz): δ 2.73-2.78 (2H, m), 3.14—3.18 (2H, m), 6.90 (IH, d,
3 Three
J=8.3 Hz), 7.52 (IH, d, J=8.3 Hz), 7.54-7.69 (4H, m), 9.35 (IH, s); J = 8.3 Hz), 7.52 (IH, d, J = 8.3 Hz), 7.54-7.69 (4H, m), 9.35 (IH, s);
MS(EI) m/z: 292 M+. MS (EI) m / z: 292 M + .
(12b) ({7— [3— (トリフルォロメチル)フエ-ル]— 3—ォキソ 2, 3 ジヒドロ一 1H —インデン— 4—ィル }ォキシ)メチルホスホン酸ジェチル (12b) ({7— [3— (Trifluoromethyl) phenol] — 3 —oxo 2, 3 dihydro-1H —indene-4-yl} oxy) methyl phosphonate
実施例 12 (12a)で製造した 7 ヒドロキシ一 4— [3— (トリフルォロメチル)フエ-ル ]インダン— 1—オン(341mg、 1. 17mmol)を使用して、実施例 1 (la)に記載した 方法に従い、標記目的化合物 (303mg、収率 59%)を得た。
[0262] IR(Liquid film) v 1031, 1126, 1256, 1289, 1339, 1713, 2986 cm ; EXAMPLE 12 (la) 7 Hydroxy 4- [3- (trifluoromethyl) phenol] indan-1-one (341 mg, 1.17 mmol) prepared in Example 12 (12a) was used to prepare Example 1 (la) The title object compound (303 mg, yield 59%) was obtained according to the method described in 1). [0262] IR (Liquid film) v 1031, 1126, 1256, 1289, 1339, 1713, 2986 cm;
max max
1H NMR(CDC1 , 400MHz) δ 1.39 (6H, t, J=7.1 Hz), 2.63—2.68 (2H, m), 3.03—3.0 1H NMR (CDC1, 400MHz) δ 1.39 (6H, t, J = 7.1 Hz), 2.63-2.68 (2H, m), 3.03-3.0
3 Three
9 (2H, m), 4.35 (4H, dq, J=7.1 Hz, 7.1 Hz), 4.47 (2H, d, J=9.4 Hz), 6.97 (IH, d, J= 8.2 Hz), 7.50 (IH, d, J=8.2 Hz), 7.54-7.65 (4H, m); 9 (2H, m), 4.35 (4H, dq, J = 7.1 Hz, 7.1 Hz), 4.47 (2H, d, J = 9.4 Hz), 6.97 (IH, d, J = 8.2 Hz), 7.50 (IH, d, J = 8.2 Hz), 7.54-7.65 (4H, m);
MS(FAB) m/z: 443 (M+H)+. MS (FAB) m / z: 443 (M + H) + .
(12c) ( {2 アミノー 7— [3 (トリフルォロメチル)フエ-ル ] 8H—インデノ [1, 2 d] [1, 3]チアゾールー 4ーィル }ォキシ)メチルホスホン酸ジェチル (12c) ({2 amino-7- [3 (trifluoromethyl) phenol] 8H-indeno [1, 2 d] [1, 3] thiazole-4-yl} oxy) methylphosphonic acid jetyl
実施例 12 ( 12b)で製造した( { 7— [3 (トリフルォロメチル)フエ-ル] - 3 ォキソ - 2, 3 ジヒドロ一 1H—インデン一 4—ィル }ォキシ)メチルホスホン酸ジェチル(28 4mg、 0. 642mmol)を使用して、実施例 2 (2b)に記載した方法に従 、、標記目的 化合物 (116mg、収率 36%)を得た。 Example 12 (12b) ({7- [3 (trifluoromethyl) phenol]-3 oxo-2,3 dihydro-1H-indene-4-yl} oxy) methylphosphonate (28) 4 mg, 0.642 mmol) were used to give the title object compound (116 mg, 36% yield) according to the method described in Example 2 (2b).
[0263] IR(KBr) v 1025, 1124, 1246, 1335, 1539, 3127 cm"1; [0263] IR (KBr) v 1025, 1124, 1246, 1335, 1539, 3127 cm "1;
max max
1H NMR(CD OD-CDC1 , 400MHz): δ 1.35 (6H, t, J=7.1 Hz), 3.71 (2H, s), 4.28 ( 1H NMR (CD OD-CDC1, 400MHz): δ 1.35 (6H, t, J = 7.1 Hz), 3.71 (2H, s), 4.28 (
3 3 3 3
4H, dq, J=7.1 Hz, 7.1 Hz), 4.70 (2H, d, J=7.8 Hz), 7.13 (IH, d, J=8.4 Hz), 7.17 (IH , d, J=8.4 Hz), 7.58-7.65 (2H, m), 7.71-7.76 (2H, m); 4H, dq, J = 7.1 Hz, 7.1 Hz), 4.70 (2H, d, J = 7.8 Hz), 7.13 (IH, d, J = 8.4 Hz), 7.17 (IH, d, J = 8.4 Hz), 7.58 -7.65 (2H, m), 7.71-7.76 (2H, m);
MS(FAB) m/z: 499 (M+H)+. MS (FAB) m / z: 499 (M + H) + .
(12d) ( {2 アミノー 7— [3 (トリフルォロメチル)フエ-ル ] 8H—インデノ [1, 2 d] [1, 3]チアゾールー 4ーィル }ォキシ)メチルホスホン酸 (12d) ({2 amino-7- [3 (trifluoromethyl) phenol] 8H-indeno [1, 2 d] [1, 3] thiazol-4-yl} oxy) methylphosphonic acid
実施例 12 ( 12c)で製造した( { 2 ァミノ 7— [3 (トリフルォロメチル)フエ-ル] —8H—インデノ [1, 2-d] [l, 3]チアゾールー 4ーィル }ォキシ)メチルホスホン酸 ジェチル(l l lmg、 0. 222mmol)を使用して、実施例 1 (lc)に記載した方法に従 い、標記目的化合物 (86. 8mg、収率 88%)を得た。 Example 12 (12c) ({2 amino 7- [3 (trifluoromethyl) phenol] —8H-indeno [1, 2-d] [l, 3] thiazol-4-yl} oxy) methylphosphone The title object compound (86.8 mg, yield 88%) was obtained according to the method described in Example 1 (lc) by using acid jetyl (ll lm g , 0.222 mmol).
[0264] IR(KBr) v 1070, 1126, 1167, 1266, 1336, 1631, 3077 cm"1; [0264] IR (KBr) v 1070, 1126, 1167, 1266, 1336, 1631, 3077 cm "1;
max max
1H NMR(DMSO-d , 500MHz): δ 3.81 (2H, s), 4.29 (2H, d, J=8.3 Hz), 7.21 (IH, d 1H NMR (DMSO-d, 500MHz): δ 3.81 (2H, s), 4.29 (2H, d, J = 8.3 Hz), 7.21 (IH, d
6 6
, J=8.3 Hz), 7.25 (IH, d, J=8.3 Hz), 7.65-7.75 (2H, m), 7.85—7.93 (2H, m); , J = 8.3 Hz), 7.25 (IH, d, J = 8.3 Hz), 7.65-7.75 (2H, m), 7.85—7.93 (2H, m);
MS(FAB) m/z: 441 (M— H)— . MS (FAB) m / z: 441 (M— H) —.
<実施例 13 >
•^[ε 'i][p-s ' I ] ^ -us - ^ . -z) ^ C^W ^ (qs I ) ε I m 一 /— 、 ^ [ε 'I][P—S 'ι],^ベ —H8—,^ —s) 峯氺ニ 邈べ ίί(3ει)<Example 13> ^ [Ε 'i] [ps' I] ^ -us-^. -Z) ^ C ^ W ^ (qs I) ε I m i / —, ^ [ε 'I] [P—S' ι] , ^ Be —H8 —, ^ —s) 峯 氺 ニ 邈 べ ίί ( 3 ει)
(w ) ι ε: ζ/ω (9VH)S (w) ι ε: ζ / ω (9VH) S
•(ZH Z'L=[ 'P 'Ηΐ) \Z'L '(ZH S"8=f 'P 'Ηΐ) IZ'L '(ZH S"8 'ZH S"Z=f 'PP 'Ηΐ) 3ΓΖ '( ω 'Η ) 6ε· — 8 '(s Ή2) '(ω 'Η9) 92Ί-6ΖΊ 9 '· (zH )0 'ac dつ) WN Hx • (ZH Z'L = ['P' Ηΐ) \ Z'L '(ZH S "8 = f' P 'Ηΐ) IZ'L' ( Z HS" 8 ' Z HS "Z = f' PP 'Ηΐ ) 3ΓΖ '(ω' Η) 6ε · — 8 '(s Ή2)' (ω 'Η9) 92Ί-6ΖΊ 9' · ( z H) 0 'ac d) WN H x
_ω。 6Πε 'f 9l '8SST ' ΖΙ '^ΖΖΙ 'ΖΖΟΙ Λ ( 1)ΗΙ [Ζ920]_ω. 6Πε 'f 9l' 8SST 'ΖΙ' ^ ΖΖΙ 'ΖΖΟΙ Λ (1) ΗΙ [Ζ920]
° %ε ¾τ
 °% ε ¾τ
°% ε ¾τ
Μ^\ {^zrnm^ ^a¾r¾¾a°^ss ·χ §ui9^S) ( — —べ^べ —
 (Βε ι ) ε i P}¾? Μ ^ \ {^ zrnm ^ ^ a¾r¾¾a ° ^ ss · χ §ui 9 ^ S ) (— —Be ^ — ( Β ε ι) ε i P} ¾?
(Βε ι ) ε i P}¾? Μ ^ \ {^ zrnm ^ ^ a¾r¾¾a ° ^ ss · χ §ui 9 ^ S ) (— —Be ^ — ( Β ε ι) ε i P} ¾?
( / 一 /— 、 ^ [ε 'i][p-s 'ΐ],^ベ Η8—,^ s) ^fi(qsx) (/ 1 / —, ^ [ε 'i] [p-s' ΐ], ^ be Η8 —, ^ s) ^ fi (qsx)
•+(H+ ) 382: ζ/ω (9VH)S • + (H +) 382: ζ / ω (9VH) S
•(ZH VI • (ZH VI
'ZH Z"Z=f 'PP 'Ηΐ) WL '(ω 'ΗΖ) ZZ'L-ZZ'L '(ω 'Η ) 82^-82^ '(ΖΗ 2"9 'ΖΗ 2"9=f ' PP 'ΗΖ) ZVZ '(ω 'ΗΖ) U — 99 '(ω 'Η9) ΐ ·ΐ— 9ε·ΐ 9 : (zH )0 ' DaD)H N Ηχ 'ZH Z "Z = f' PP 'Ηΐ) WL' (ω 'ΗΖ) ZZ'L-ZZ'L' (ω 'Η) 82 ^ -82 ^' ( Ζ Η 2" 9 ' Ζ Η 2 "9 = f 'PP' ΗΖ) ZVZ '(ω' ΗΖ) U — 99 '(ω' Η9) ΐ · ΐ— 9ε · ΐ 9: ( z H) 0 'DaD) HN Η χ
·τ_ωο 9862 'ΖΐΖΐ 'Ζ09ΐ '^ΐ '^ΖΙ 'ΖΖΟΙ '898 Λ ( 1)ΗΙ [9920]· Τ _ωο 9862 'ΖΐΖΐ' Ζ09ΐ '^ ΐ' ^ ΖΙ 'ΖΖΟΙ' 898 Λ (1) ΗΙ [9920]
(%οοι*¾ί、3π^8ε)呦^; ]Κ¾目 ¾遨
 χ:ο-χ:χ / ェ邈 :ベ ^ )— ム マ / f 、 ί ^ 。 つ辛爵 (% οοι * ¾ί 、 3π ^ 8ε ) 呦 ^ ;] Κ¾ 目 ¾ 遨 χ: ο-χ: χ / 邈 : Be ^) — Muma / f, ί ^. Spicy baron
χ:ο-χ:χ / ェ邈 :ベ ^ )— ム マ / f 、 ί ^ 。 つ辛爵 (% οοι * ¾ί 、 3π ^ 8ε ) 呦 ^ ;] Κ¾ 目 ¾ 遨 χ: ο-χ: χ / 邈 : Be ^) — Muma / f, ί ^. Spicy baron
¾瀚缀止 s教
 ¾ 瀚 缀 定 s
¾ 瀚 缀 定 s
。 つ 翻 S
 aouiuio ·Ζ lrf8 ε) ^エ; ^邈べ ri亜、 ¾ iOUIUIoz 'ζ ベ ^ ^エ (Η^-¾· ^ W -\
aouiuio ·Ζ lrf8 ε) ^エ; ^邈べ ri亜、 ¾ iOUIUIoz 'ζ ベ ^ ^エ (Η^-¾· ^ W -\
 ( / — 一 /— : ^ [ε 'I][P—S
( / — 一 /— : ^ [ε 'I][P—S
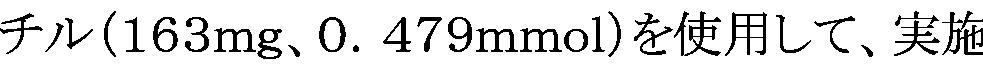 . Tsu S aouiuio · Ζ l rf 8 ε) ^ e; ^ 邈 be ri, ¾ i OUIUI oz 'ζ be ^^ e (Η ^ -¾ · ^ W-\ (/ — One / —: ^ [ε 'I] [P—S
. Tsu S aouiuio · Ζ l rf 8 ε) ^ e; ^ 邈 be ri, ¾ i OUIUI oz 'ζ be ^^ e (Η ^ -¾ · ^ W-\ (/ — One / —: ^ [ε 'I] [P—S
0l76S0C/900Zdf/X3d εζ 0C0l70l/900Z OAV
に記載の方法に従い、標記目的化合物(96. 6mg、収率 71%)を得た。 0l76S0C / 900Zdf / X3d εζ 0C0l70l / 900Z OAV To give the title compound (96.6 mg, yield 71%).
[0268] IR(KBr) v 931, 1016, 1080, 1245, 1469, 1593, 2988 cm"1; [0268] IR (KBr) v 931, 1016, 1080, 1245, 1469, 1593, 2988 cm "1;
max max
1H NMR(DMSO-d , 400MHz): δ 3.75 (2H, s), 7.04 (IH, d, J=8.1 Hz), 7.15 (IH, d 1H NMR (DMSO-d, 400MHz): δ 3.75 (2H, s), 7.04 (IH, d, J = 8.1 Hz), 7.15 (IH, d
6 6
d, J=7.3 Hz, 8.1 Hz), 7.27 (IH, d, J=7.3 Hz), 7.80—8.05 (br, 2H); d, J = 7.3 Hz, 8.1 Hz), 7.27 (IH, d, J = 7.3 Hz), 7.80—8.05 (br, 2H);
MS(ESI) m/z: 283 (M— H)— . MS (ESI) m / z: 283 (M— H) —.
<実施例 14 > <Example 14>
リン酸 二水素 (2 アミノー 4, 5 ジヒドロナフト[1, 2— d] [l, 3]チアゾールー 8 ィル)(例示化合物番号 2-15) Dihydrogen phosphate (2 amino-4,5 dihydronaphtho [1,2-d] [l, 3] thiazole-8 yl) (Exemplified Compound No. 2-15)
(14a)リン酸 ジェチル(8—ォキソ 5, 6, 7, 8—テトラヒドロナフタレン一 2—ィル) 7 ヒドロキシ一 3, 4 ジヒドロナフタレン一 1 (2H)—オン(1. OOg、 6. 17mmol) を使用して、実施例 13 (13a)に記載の方法に従い、標記目的化合物(1. 65g、収率 90%)を得た。 (14a) Jetyl phosphate (8-oxo-5,6,7,8-tetrahydronaphthalene-2-yl) 7 hydroxy-1,3,4 dihydronaphthalene-1- (2H) -one (1. OOg, 6.17 mmol) To give the title object compound (1.65 g, yield 90%) according to the method described in Example 13 (13a).
[0269] IR(Liquid film) v 968, 1031, 1224, 1270, 1491, 1687, 2986 cm"1; [0269] IR (Liquid film) v 968, 1031, 1224, 1270, 1491, 1687, 2986 cm "1;
max max
1H NMR(CDC1 , 400MHz): δ 1.34—1.39 (6H, m), 2.09-2.17 (2H, m), 2.65 (2H, dd 1H NMR (CDC1, 400 MHz): δ 1.34—1.39 (6H, m), 2.09-2.17 (2H, m), 2.65 (2H, dd
3 Three
, J=6.6 Hz, 6.6 Hz), 2.94 (2H, dd, J=6.2 Hz, 6.2 Hz), 4.17-4.28 (4H, m), 7.24 (IH, d, J=8.1 Hz), 7.35-7.40 (IH, m), 7.78-7.82 (IH, m); , J = 6.6 Hz, 6.6 Hz), 2.94 (2H, dd, J = 6.2 Hz, 6.2 Hz), 4.17-4.28 (4H, m), 7.24 (IH, d, J = 8.1 Hz), 7.35-7.40 ( IH, m), 7.78-7.82 (IH, m);
MS(EI) m/z: 298 M+. MS (EI) m / z: 298 M + .
(14b)リン酸(2 アミノー 4, 5 ジヒドロナフト[1, 2-d] [l, 3]チアゾール 8—ィ ル)ジェチル (14b) Phosphate (2 amino-4,5 dihydronaphtho [1,2-d] [l, 3] thiazole 8-yl) jetyl
実施例 14 (14a)で製造したリン酸 ジェチル (8—ォキソ 5, 6, 7, 8—テトラヒド ロナフタレン一 2—ィル)(402mg、 1. 35mmol)を使用して、実施例 2 (2b)に記載し た方法に従い、標記目的化合物 (183mg、収率 40%)を得た。 Example 2 (2b) was used using the decyl phosphate (8-oxo-5,6,7,8-tetrahydronaphthalene-2-yl) (402 mg, 1.35 mmol) prepared in Example 14 (14a). ) To give the title object compound (183 mg, yield 40%).
[0270] IR(KBr) v 988, 1038, 1262, 1537, 1650, 3127, 3363 cm"1; [0270] IR (KBr) v 988, 1038, 1262, 1537, 1650, 3127, 3363 cm "1;
max max
1H NMR(CD OD, 400MHz): δ 1.36 (6H, t, J=7.3 Hz), 2.77-2.84 (2H, m), 2.94—3. 1H NMR (CD OD, 400 MHz): δ 1.36 (6H, t, J = 7.3 Hz), 2.77-2.84 (2H, m), 2.94—3.
3 Three
01 (2H, m), 4.24 (4H, dq, J=7.3 Hz, 7.3 Hz), 6.95—7.00 (IH, m), 7.19 (IH, d, J=8.8 Hz), 7.40-7.44 (IH, m); 01 (2H, m), 4.24 (4H, dq, J = 7.3 Hz, 7.3 Hz), 6.95—7.00 (IH, m), 7.19 (IH, d, J = 8.8 Hz), 7.40-7.44 (IH, m );
MS(FAB) m/z: 355 (M+H)+.
(14c)リン酸 二水素 (2 アミノー 4, 5 ジヒドロナフト[1, 2— d] [l, 3]チアゾー ルー 8—ィノレ) MS (FAB) m / z: 355 (M + H) + . (14c) Dihydrogen phosphate (2 amino-4,5 dihydronaphtho [1, 2—d] [l, 3] thiazol 8—inore)
実施例 14 (14b)で製造したリン酸(2 アミノー 4, 5 ジヒドロナフト[1, 2-d] [l, 3]チアゾールー 8 ィル)ジェチル(147mg、 0. 415mmol)を使用して、実施例 1 ( lc)に記載した方法に従い、標記目的化合物 (86. 5mg、収率 70%)を得た。 Example 14 Performed using the phosphoric acid (2 amino-4,5 dihydronaphtho [1,2-d] [l, 3] thiazol-8 yl) jetyl (147 mg, 0.415 mmol) prepared in Example 14 (14b) The title compound (86.5 mg, yield 70%) was obtained according to the method described in Example 1 (lc).
[0271] IR(KBr) v 940, 1078, 1204, 1507, 1601, 1634, 3065 cm"1; [0271] IR (KBr) v 940, 1078, 1204, 1507, 1601, 1634, 3065 cm "1;
max max
1H NMR(DMSO-d , 500MHz): δ 2.73 (2H, dd, J=7.8 Hz, 7.8 Hz), 2.88 (2H, dd, J 1H NMR (DMSO-d, 500MHz): δ 2.73 (2H, dd, J = 7.8 Hz, 7.8 Hz), 2.88 (2H, dd, J
6 6
=7.8 Hz, 7.8 Hz), 6.83—6.87 (IH, m), 7.13 (IH, d, J=7.8 Hz), 7.22-7.30 (br, 2H), 7. 42-7.44 (IH, m); = 7.8 Hz, 7.8 Hz), 6.83—6.87 (IH, m), 7.13 (IH, d, J = 7.8 Hz), 7.22-7.30 (br, 2H), 7. 42-7.44 (IH, m);
MS(FAB) m/z: 297 (M- H)— . MS (FAB) m / z: 297 (M- H) —.
<実施例 15 > <Example 15>
(2 アミノー 4, 5 ジヒドロナフト[1, 2-d] [l, 3]チアゾールー 8 ィル)ジフルォ ロメチルホスホン酸 (例示化合物番号 2-5) (2 amino-4,5 dihydronaphtho [1,2-d] [l, 3] thiazole-8yl) difluoromethylphosphonic acid (Exemplified Compound No. 2-5)
(15a)トリフルォロメタンスルホン酸(8—ォキソ 5, 6, 7, 8—テトラヒドロナフタレン 2—ィル) (15a) trifluoromethanesulfonic acid (8-oxo 5, 6, 7, 8-tetrahydronaphthalene 2-yl)
7 ヒドロキシ一 3, 4 ジヒドロナフタレン一 1 (2H)—オン(5. 54g、 34. 2mmol) をジクロロメタン(114mL)に溶解し、 30°C冷却下、そこへ 2, 6—ルチジン(4. 78 mL、 41. Ommol)、 4 ジメチルァミノピリジン(835mg、 6. 83mmol)及びトリフル ォロメタンスルホン酸無水物(6. 90mL、 41. Ommol)を加え、窒素雰囲気下、室温 まで昇温しながら 3時間撹拌した。 7 Hydroxy mono 3,4 Dihydronaphthalene mono 1 (2H) -one (5.54 g, 34.2 mmol) was dissolved in dichloromethane (114 mL), and cooled to 30 ° C, 2, 6-lutidine (4.78) was added thereto. mL, 41. Ommol), 4 dimethylaminopyridine (835 mg, 6.83 mmol) and trifluoromethanesulfonic anhydride (6.90 mL, 41. Ommol) were added, and the temperature was raised to room temperature under a nitrogen atmosphere. Stir for hours.
[0272] 反応液に水を加え、ジクロロメタンで抽出した。得られた有機層を飽和炭酸水素ナト リウム水溶液及び飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、減圧下溶媒を 留去した。得られた残渣をシリカゲルカラムクロマトグラフィー (へキサン:酢酸ェチル、 7 : 1—4 : 1、 VZV)により精製し、標記目的化合物(10. lg、収率 100%)を得た。 [0272] Water was added to the reaction mixture, and the mixture was extracted with dichloromethane. The obtained organic layer was washed with a saturated aqueous sodium hydrogen carbonate solution and saturated brine, dried over anhydrous sodium sulfate, and the solvent was distilled off under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane: ethyl acetate, 7: 1-4: 1, VZV) to obtain the title object compound (10. lg, yield 100%).
[0273] IR(Liquid film) v 613, 837, 898, 927, 1142, 1217, 1427, 1695 cm"1; [0273] IR (Liquid film) v 613, 837, 898, 927, 1142, 1217, 1427, 1695 cm "1;
max max
1H NMR(CDC1 , 500MHz): δ 2.14-2.21 (2H, m), 2.69 (2H, dd, J=6.8 Hz, 6.8 Hz) 1H NMR (CDC1, 500MHz): δ 2.14-2.21 (2H, m), 2.69 (2H, dd, J = 6.8 Hz, 6.8 Hz)
3 Three
, 3.00 (2H, dd, J=5.9 Hz, 5.9 Hz), 7.36—7.39 (2H, m), 7.91 (IH, s);
MS(EI) m/z: 294 M+. , 3.00 (2H, dd, J = 5.9 Hz, 5.9 Hz), 7.36—7.39 (2H, m), 7.91 (IH, s); MS (EI) m / z: 294 M + .
(15b)トリフルォロメタンスルホン酸 (3,, 4,一ジヒドロ一 2, H—スピロ [1, 3 ジォ キソラン一 2, 1 '—ナフタレン]— 7'—ィル) (15b) trifluoromethanesulfonic acid (3, 4, 1, dihydro-1, 2, H-spiro [1, 3 dixolane 1, 2, 1'-naphthalene] -7'-yl)
実施例 15 (15a)で製造したトリフルォロメタンスルホン酸(8—ォキソ 5, 6, 7, 8 —テトラヒドロナフタレン一 2—ィル)(10. lg、 34. 2mmol)をトルエン(137mL)に 溶解し、そこへエチレングリコール(38. OmL、 684mmol)及び p トルエンスルホン 酸一水和物(1. 18g、 6. 20mmol)をカ卩え、ディーン 'スターク装置を付して, 12時 間加熱還流した。 Example 15 Dissolve trifluoromethanesulfonic acid (8-oxo5,6,7,8-tetrahydronaphthalene-2-yl) (10. lg, 34.2 mmol) prepared in Example 15 (15a) in toluene (137 mL) Then, add ethylene glycol (38. OmL, 684 mmol) and p-toluenesulfonic acid monohydrate (1.18 g, 6.20 mmol), and attach to a Dean 'Stark apparatus and heat to reflux for 12 hours. did.
[0274] 反応液に飽和炭酸水素ナトリウム水溶液を加え、酢酸ェチルで抽出した。得られた 有機層を水及び飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、減圧下溶媒を 留去した。得られた残渣をシリカゲルカラムクロマトグラフィー (へキサン:酢酸ェチル、 10 : 1— 5 : 1、 VZV)により精製し、標記目的化合物(8. 83g、収率 76%)を得た。 [0274] A saturated aqueous sodium hydrogen carbonate solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with water and saturated brine, dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane: ethyl acetate, 10: 1-5: 1, VZV) to obtain the title object compound (8.83 g, yield 76%).
[0275] IR(Liquid film) v 605, 852, 925, 1061, 1143, 1212, 1423, 2952 cm"1; [0275] IR (Liquid film) v 605, 852, 925, 1061, 1143, 1212, 1423, 2952 cm "1;
max max
1H NMR(CDC1 , 500MHz) : δ 1.94—2.02 (4H, m), 2.78-2.84 (2H, m), 4.07-4.24 (4 1H NMR (CDC1, 500 MHz): δ 1.94—2.02 (4H, m), 2.78-2.84 (2H, m), 4.07-4.24 (4
3 Three
H, m), 7.13 (1H, dd, J=2.0 Hz, 7.8 Hz), 7.17 (1H, d, J=7.8 Hz), 7.37 (1H, d, J=2.0 Hz); H, m), 7.13 (1H, dd, J = 2.0 Hz, 7.8 Hz), 7.17 (1H, d, J = 7.8 Hz), 7.37 (1H, d, J = 2.0 Hz);
MS(FAB) m/z: 339 (M+H)+. MS (FAB) m / z: 339 (M + H) + .
(15c) 3,, 4,一ジヒドロ一 7,一ヒドロキシメチル一 2,H—スピロ [1, 3 ジォキソラン - 2, 1 '—ナフタレン] (15c) 3, 4, 1, dihydro-1, 7, hydroxymethyl 1, 2, H-spiro [1, 3 dioxolane-2, 1'-naphthalene]
実施例 15 ( 15b)で製造したトリフルォロメタンスルホン酸 (3,, 4,一ジヒドロ 2, H—スピロ [1, 3 ジォキソラン一 2, 1,一ナフタレン]— 7,一ィル)(6. 25g、 18. 5 mmol)をテトラヒドロフラン(62mL)に溶解し、そこへ [1,1,一ビス(ジフエ-ルホスフ イノ)フエ口セン]ジクロロパラジウムとジクロロメタンの複合体(1 : 1) (755mg、 0. 925 mmol)及び臭化ビュルマグネシウム(0. 97M テトラヒドロフラン溶液)(28. 6mL、 27. 8mmol)を加え、窒素雰囲気下、 18時間攪拌した。 Trifluoromethanesulfonic acid prepared in Example 15 (15b) (3, 4, 1, dihydro 2, H-spiro [1, 3 dioxolane 1, 2, 1, naphthalene] -7, 1 yl) (6. 25 g, 18.5 mmol) was dissolved in tetrahydrofuran (62 mL), to which a complex of [1,1,1bis (diphenylphosphino) pheocene] dichloropalladium and dichloromethane (1: 1) (755 mg, 0.925 mmol) and butylmagnesium bromide (0.97 M tetrahydrofuran solution) (28.6 mL, 27.8 mmol) were added, and the mixture was stirred under a nitrogen atmosphere for 18 hours.
[0276] 反応液に飽和塩化アンモ-ゥム水溶液を加え、減圧下テトラヒドロフランを留去し、 酢酸ェチルで抽出した。得られた有機層を水及び飽和食塩水で洗浄し、無水硫酸 ナトリウムで乾燥し、減圧下溶媒を留去した。得られた残渣をシリカゲルカラムクロマト
グラフィー (へキサン:酢酸ェチル、 9 : 1、 V/V)により精製し、 7,—ビュル— 3,, 4, —ジヒドロ一 2,H—スピロ [1, 3 ジォキソラン一 2, 1,一ナフタレン]の粗精製物を 得た。 [0276] Saturated aqueous ammonium chloride solution was added to the reaction mixture, tetrahydrofuran was distilled off under reduced pressure, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with water and saturated brine, dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure. The obtained residue was subjected to silica gel column chromatography. Purified by chromatography (Hexane: Ethyl acetate, 9: 1, V / V), 7, -Bur-3,4, -Dihydro-1,2, H-spiro [1,3 Dioxolan-1,2,1,1-naphthalene A crudely purified product was obtained.
[0277] これを t ブチルアルコール(86mL)、水(86mL)及びテトラヒドロフラン(34mL) に溶解し、そこへ 4—メチルモルホリン N—ォキシド(50% 水溶液)(6. 03g、 25. 7mmol)及びオスミウム酸カリウム(IV)二水和物(317mg、 0. 860mmol)を加え、 8 時間攪拌した。 [0277] This was dissolved in t-butyl alcohol (86 mL), water (86 mL) and tetrahydrofuran (34 mL), and then 4-methylmorpholine N-oxide (50% aqueous solution) (6.03 g, 25.7 mmol) and osmium. Potassium (IV) acid dihydrate (317 mg, 0.860 mmol) was added and stirred for 8 hours.
[0278] 反応液に炭酸水素ナトリウム(17. 3g、 206mmol)、過ヨウ素酸カリウム(12. 7g、 5 5. Ommol)及び水(150mL)をカ卩え、 14時間攪拌した。 [0278] Sodium hydrogen carbonate (17.3 g, 206 mmol), potassium periodate (12.7 g, 5 5. Ommol) and water (150 mL) were added to the reaction solution, and the mixture was stirred for 14 hours.
[0279] 反応液に飽和チォ硫酸ナトリウム水溶液を加え、酢酸ェチルで抽出した。得られた 有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、減圧下溶媒を留去し、 3' , 4'—ジヒドロ一 7'—ホルミル一 2' H—スピロ [1, 3 ジォキソラン一 2, 1 '—ナフ タレン]の粗精製物を得た。 [0279] A saturated aqueous sodium thiosulfate solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, the solvent was distilled off under reduced pressure, and 3 ', 4'-dihydro-1,7'-formyl-1,2' H-spiro [1, 3 A crude product of dioxolan-1,2,1'-naphthalene] was obtained.
[0280] これをメタノール(77mL)に溶解し、そこへ水素化ホウ素ナトリウム(1. 17g、 30. 8 mmol)をカ卩え、 0.5時間攪拌した。 [0280] This was dissolved in methanol (77 mL), sodium borohydride (1.17 g, 30.8 mmol) was added thereto, and the mixture was stirred for 0.5 hr.
[0281] 反応液に飽和塩化アンモ-ゥム水溶液を加え、減圧下メタノールを留去し、ジクロロ メタンで抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾 燥し、減圧下溶媒を留去した。得られた残渣をシリカゲルカラムクロマトグラフィー (へ キサン:酢酸ェチル、 4 : 1— 0 : 1、 VZV)により精製し、標記目的化合物(2. 32g、収 率 57%)を得た。 [0281] A saturated aqueous ammonium chloride solution was added to the reaction mixture, methanol was distilled off under reduced pressure, and the mixture was extracted with dichloromethane. The obtained organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane: ethyl acetate, 4: 1—0: 1, VZV) to obtain the title object compound (2.32 g, yield 57%).
[0282] IR(Liquid film) v 937, 1062, 1132, 2943, 3415 cm"1; [0282] IR (Liquid film) v 937, 1062, 1132, 2943, 3415 cm "1;
max max
1H NMR(CDC1 , 500MHz) : δ 1.92—1.99 (4H, m), 2.76-2.83 (2H, m), 4.07-4.16 (2 1H NMR (CDC1, 500MHz): δ 1.92—1.99 (4H, m), 2.76-2.83 (2H, m), 4.07-4.16 (2
3 Three
H, m), 4.18-4.26 (2H, m), 4.65 (2H, d, J=4.9 Hz), 7.10 (1H, d, J=7.8 Hz), 7.22-7.2 7 (1H, m), 7.47-7.50 (1H, m); H, m), 4.18-4.26 (2H, m), 4.65 (2H, d, J = 4.9 Hz), 7.10 (1H, d, J = 7.8 Hz), 7.22-7.2 7 (1H, m), 7.47- 7.50 (1H, m);
MS(EI) m/z: 220 M+. MS (EI) m / z: 220 M + .
(15d) (3,, 4,一ジヒドロ一 2,H—スピロ [1, 3 ジォキソラン一 2, 1,一ナフタレン] - 7' ィル)メチルホスホン酸ジェチル (15d) (3, 4, 1, dihydro-1, 2, H-spiro [1, 3 dioxolane 1, 2, 1, naphthalene] -7'yl) methylphosphonate
実施例 15 ( 15c)で製造した 3,, 4,一ジヒドロ一 7,一ヒドロキシメチル一 2, H スピ
口 [1, 3 ジォキソラン一 2, 1 '—ナフタレン] (2. 29g、 10. 4mmol)をジクロロメタン (52mL)に溶解し、 0。C冷却下、そこへトリフエ-ルホスフィン(2. 87g、 10. 9mmol) 及び四臭化炭素(3. 62g、 10. 9mmol)を加え、窒素雰囲気下、 0°Cで 1時間攪拌し た。 3, 15, 1, dihydro-1, 7, hydroxymethyl 1, 2, H sp, prepared in Example 15 (15c) Dissolve [1,3 dioxolane-1,2,1'-naphthalene] (2.29 g, 10.4 mmol) in dichloromethane (52 mL). Under C cooling, triphenylphosphine (2.87 g, 10.9 mmol) and carbon tetrabromide (3.62 g, 10.9 mmol) were added thereto, and the mixture was stirred at 0 ° C. for 1 hour in a nitrogen atmosphere.
[0283] 反応液に飽和炭酸水素ナトリウム水溶液を加え、ジクロロメタンで抽出した。得られ た有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、減圧下溶媒を留去 した。得られた残渣をシリカゲルカラムクロマトグラフィー (へキサン:酢酸ェチル、 6 : 1 、 V/V)により精製し、 7,—ブロモメチル—3,, 4,—ジヒドロ 2,H—スピロ [1, 3— ジォキソラン一 2, 1,一ナフタレン]の粗精製物を得た。 [0283] Saturated aqueous sodium hydrogen carbonate solution was added to the reaction mixture, and the mixture was extracted with dichloromethane. The obtained organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane: ethyl acetate, 6: 1, V / V), and 7, -bromomethyl-3,4, -dihydro-2, H-spiro [1, 3- A crude product of dioxolane-1,2,1, naphthalene] was obtained.
[0284] これを亜リン酸トリェチル(7. 33mL、 42. 7mmol)を加え、 150°Cで 4時間攪拌し た。 [0284] Triethyl phosphite (7.33 mL, 42.7 mmol) was added thereto, and the mixture was stirred at 150 ° C for 4 hours.
[0285] 反応液力 減圧下亜リン酸トリェチルを留去し、得られた残渣をシリカゲルカラムク 口マトグラフィー (へキサン:酢酸ェチル、 1 : 2— 0 : 1、 VZV)により精製し、標記目的 化合物(1. 48g、収率 42%)を得た。 [0285] Reaction solution power Triethyl phosphite was distilled off under reduced pressure, and the resulting residue was purified by silica gel column chromatography (hexane: ethyl acetate, 1: 2—0: 1, VZV) to give the title The objective compound (1.48 g, yield 42%) was obtained.
[0286] IR(Liquid film) v 965, 1028, 1061, 1248, 2942, 3469 cm"1; [0286] IR (Liquid film) v 965, 1028, 1061, 1248, 2942, 3469 cm "1;
max max
1H NMR(CDC1 , 500MHz) δ 1.25 (6H, t, J=6.8 Hz), 1.93—1.97 (4H, m), 2.74-2.8 1H NMR (CDC1, 500MHz) δ 1.25 (6H, t, J = 6.8 Hz), 1.93-1.97 (4H, m), 2.74-2.8
3 Three
0 (2H, m), 3.13 (2H, d, J=21.5 Hz), 3.96—4.05 (4H, m), 4.07-4.14 (2H, m), 4.16-4.2 4 (2H, m), 7.04 (1H, d, J=7.8 Hz), 7.17 (1H, d, J=7.8 Hz), 7.40 (1H, s); 0 (2H, m), 3.13 (2H, d, J = 21.5 Hz), 3.96—4.05 (4H, m), 4.07-4.14 (2H, m), 4.16-4.2 4 (2H, m), 7.04 (1H , d, J = 7.8 Hz), 7.17 (1H, d, J = 7.8 Hz), 7.40 (1H, s);
MS(FAB) m/z: 341 (M+H)+. MS (FAB) m / z: 341 (M + H) + .
(15e) (8 ォキソ 5, 6, 7, 8—テトラヒドロナフタレンー2 ィル)ジフルォロメチル ホスホン酸ジェチル (15e) (8oxo5,6,7,8-tetrahydronaphthalene-2-yl) difluoromethyl phosphonate jetyl
実施例 15 (15d)で製造した(3,, 4,一ジヒドロ一 2, H—スピロ [1, 3 ジォキソラン - 2, 1 '—ナフタレン]— 7'—ィル)メチルホスホン酸ジェチル(1. 45g、 4. 26mmol )をテトラヒドロフラン (43mL)に溶解し、 78°C冷却下、そこへナトリウムビス(トリメチ ルシリル)アミド(1. OM テトラヒドロラフラン溶液)(12. 8mL、 12. 8mmol)を加え、 窒素雰囲気下、 78°Cで 1時間攪拌した。 Example 15 (3,4,1-dihydro-1,2, H-spiro [1,3 dixolan-2,1'-naphthalene] -7'-yl) methylphosphonate jetyl (1.45 g) prepared in Example 15 (15d) 4.26 mmol) was dissolved in tetrahydrofuran (43 mL), and sodium bis (trimethylsilyl) amide (1. OM tetrahydrolafuran solution) (12.8 mL, 12.8 mmol) was added thereto under cooling at 78 ° C. The mixture was stirred at 78 ° C for 1 hour in a nitrogen atmosphere.
[0287] 反応液に N—フルォロベンゼンスルホンイミド(4. 70g、 14. 9mmol)をカ卩え、室温 まで昇温しながら 3時間攪拌した。
[0288] 反応液に 0. 1規定塩酸を加え、ジクロロメタンで抽出した。得られた有機層を水及 び飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、減圧下溶媒を留去した。得ら れた残渣をシリカゲルカラムクロマトグラフィー (へキサン:酢酸ェチル、 2: 1— 1: 1、 V /V)により精製し、 (3' , 4,一ジヒドロ一 2,H—スピロ [1, 3 ジォキソラン一 2, 1,一 ナフタレン ]一 7,一ィル)ジフルォロメチルホスホン酸ジェチルの粗精製物を得た。 [0287] N-fluorobenzenesulfonimide (4.70 g, 14.9 mmol) was added to the reaction solution, and the mixture was stirred for 3 hours while warming to room temperature. [0288] To the reaction solution was added 0.1N hydrochloric acid, and the mixture was extracted with dichloromethane. The obtained organic layer was washed with water and saturated brine, dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure. The resulting residue was purified by silica gel column chromatography (hexane: ethyl acetate, 2: 1—1: 1, V / V), and (3 ′, 4, 1 dihydro-1, 2, H-spiro [1, 3 Dioxolan-1,2,1, naphthalene] 1,7,1yl) A crude product of diethyldifluoromethylphosphonate was obtained.
[0289] これをアセトン(21mL)及び水(21mL)に溶解し、 p—トルエンスルホン酸ピリジ- ゥム(107mg、 0. 426mmol)を加え、 2時間加熱還流した。 [0289] This was dissolved in acetone (21 mL) and water (21 mL), p-toluenesulfonic acid pyridinium (107 mg, 0.426 mmol) was added, and the mixture was heated to reflux for 2 hours.
[0290] 反応液に飽和炭酸水素ナトリウム水溶液を加え、減圧下アセトンを留去し、酢酸ェ チルで抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥 し、減圧下溶媒を留去した。得られた残渣をシリカゲルカラムクロマトグラフィー (へキ サン:酢酸ェチル、 2 : 1— 1 : 1、 VZV)により精製し、標記目的化合物(998mg、収 率 71%)を得た。 [0290] A saturated aqueous sodium hydrogen carbonate solution was added to the reaction mixture, and acetone was distilled off under reduced pressure, followed by extraction with ethyl acetate. The obtained organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane: ethyl acetate, 2: 1—1: 1, VZV) to obtain the title object compound (998 mg, yield 71%).
[0291] 1H NMR(CDC1 , 500MHz): δ 1.34 (6Η, t, J=6.8 Hz), 2.14-2.21 (2H, m), 2.69 (2H [0291] 1H NMR (CDC1, 500MHz): δ 1.34 (6Η, t, J = 6.8 Hz), 2.14-2.21 (2H, m), 2.69 (2H
3 Three
, dd, J=6.8 Hz, 6.8 Hz), 3.01 (2H, dd, J=5.9 Hz, 5.9 Hz), 4.16—4.29 (4H, m), 7.37 ( 1H, d, J=7.8 Hz), 7.73 (1H, d, J=7.8 Hz), 8.26 (1H, s). , dd, J = 6.8 Hz, 6.8 Hz), 3.01 (2H, dd, J = 5.9 Hz, 5.9 Hz), 4.16—4.29 (4H, m), 7.37 (1H, d, J = 7.8 Hz), 7.73 ( 1H, d, J = 7.8 Hz), 8.26 (1H, s).
MS(EI) m/z: 333 (M+H)+. MS (EI) m / z: 333 (M + H) + .
(15f) (2 アミノー 4, 5 ジヒドロナフト[1, 2-d] [l, 3]チアゾールー 8 ィル)ジ フルォロメチルホスホン酸ジェチル (15f) (2 amino-4,5 dihydronaphtho [1,2-d] [l, 3] thiazole-8yl) difluoromethylphosphonate jetyl
実施例 15 (15e)で製造した (8—ォキソ 5, 6, 7, 8—テトラヒドロナフタレン一 2— ィル)ジフルォロメチルホスホン酸ジェチル(873mg、 2. 63mmol)を使用して、実施 例 2 (2b)に記載した方法に従い、標記目的化合物 (726mg、収率 71%)を得た。 Example 15 Using (8-oxo-5,6,7,8-tetrahydronaphthalene-2-yl) difluoromethylphosphonate jetyl (873 mg, 2.63 mmol) prepared in Example 15 (15e), Example 2 According to the method described in (2b), the title object compound (726 mg, yield 71%) was obtained.
[0292] IR(KBr) v 1016, 1047, 1255, 1538, 3148, 3341 cm"1; [0292] IR (KBr) v 1016, 1047, 1255, 1538, 3148, 3341 cm "1;
max max
1H NMR(CD OD, 500MHz): δ 1.31 (6H, t, J=6.8 Hz), 2.84 (2H, dd, J=7.8 Hz, 7. 1H NMR (CD OD, 500 MHz): δ 1.31 (6H, t, J = 6.8 Hz), 2.84 (2H, dd, J = 7.8 Hz, 7.
3 Three
8 Hz), 3.06 (2H, dd, J=7.8 Hz, 7.8 Hz), 4.13—4.26 (4H, m), 7.32 (1H, d, J=7.8 Hz), 7.35 (1H, d, J=7.8 Hz), 7.82 (1H, s); 8 Hz), 3.06 (2H, dd, J = 7.8 Hz, 7.8 Hz), 4.13—4.26 (4H, m), 7.32 (1H, d, J = 7.8 Hz), 7.35 (1H, d, J = 7.8 Hz ), 7.82 (1H, s);
MS(FAB) m/z: 389 (M+H)+. MS (FAB) m / z: 389 (M + H) + .
(15g) (2 アミノー 4, 5 ジヒドロナフト[1, 2-d] [l, 3]チアゾールー 8 ィル)ジ フルォロメチルホスホン酸
実施例 15 (15f)で製造した(2 アミノー 4, 5 ジヒドロナフト[1, 2-d] [l, 3]チ ァゾールー 8 ィル)ジフルォロメチルホスホン酸ジェチル(726mg、 1. 88mmol)を 使用して、実施例 1 (lc)に記載した方法に従い、標記目的化合物 (506mg、収率 81 %)を得た。 (15g) (2 amino-4,5 dihydronaphtho [1,2-d] [l, 3] thiazole-8yl) difluoromethylphosphonic acid Example 15 Using (2 amino-4,5 dihydronaphtho [1,2-d] [l, 3] thiazol-8yl) difluoromethylphosphonate jetyl (726 mg, 1.88 mmol) prepared in Example 15 (15f) Then, according to the method described in Example 1 (lc), the title object compound (506 mg, yield 81%) was obtained.
[0293] IR(KBr) v 570, 926, 1081, 1140, 1238, 1619, 2959 cm"1; [0293] IR (KBr) v 570, 926, 1081, 1140, 1238, 1619, 2959 cm "1;
max max
1H NMR(DMSO-d , 500MHz): δ 2.62-2.72 (2H, m), 2.83—2.91 (2H, m), 7.19 (1H 1H NMR (DMSO-d, 500MHz): δ 2.62-2.72 (2H, m), 2.83—2.91 (2H, m), 7.19 (1H
6 6
, d, J=7.8 Hz), 7.34 (1H, d, J=7.8 Hz), 7.88 (1H, s); , d, J = 7.8 Hz), 7.34 (1H, d, J = 7.8 Hz), 7.88 (1H, s);
MS(FAB) m/z: 331 (M- H)— . MS (FAB) m / z: 331 (M- H) —.
<実施例 16 > <Example 16>
リン酸 二水素 (2 アミノー 5, 5 ジメチルー 4, 5 ジヒドロナフト[1, 2— d] [l, 3 ]チアゾールー 8 ィル)(例示化合物番号 2-29) Dihydrogen phosphate (2 amino-5,5 dimethyl-4,5 dihydronaphtho [1,2-d] [l, 3] thiazole-8 yl) (Exemplified Compound No. 2-29)
(16a)リン酸(8—ォキソ 5, 5 ジメチル一 5, 6, 7, 8—テトラヒドロナフタレン一 2 ィル) ジェチル (16a) Phosphoric acid (8-oxo-5,5 dimethyl-1,5,6,7,8-tetrahydronaphthalene-2-yl) Jetyl
7 ヒドロキシ一 4, 4 ジメチノレ一 3, 4 ジヒドロナフタレン一 1 (2H)—オン(711 mg、 3. 74mmol)を使用して、実施例 13 (13a)に記載の方法に従い、標記目的化 合物(1. 13g、収率 93%)を得た。 7 Hydroxy 1,4 Dimethinole 3,4 Dihydronaphthalene 1 (2H) -one (711 mg, 3.74 mmol) was used according to the method described in Example 13 (13a) to give the title compound. (1.13 g, 93% yield) was obtained.
[0294] IR(Liquid film) v 966, 1031, 1272, 1487, 1687, 2964 cm"1; [0294] IR (Liquid film) v 966, 1031, 1272, 1487, 1687, 2964 cm "1;
max max
1H NMR(CDC1 , 500MHz): δ 1.34—1.40 (12H, m), 2.01 (2H, dd, J=6.8 Hz, 6.8 Hz 1H NMR (CDC1, 500MHz): δ 1.34—1.40 (12H, m), 2.01 (2H, dd, J = 6.8 Hz, 6.8 Hz
3 Three
), 2.73 (2H, dd, J=6.8 Hz, 6.8 Hz), 4.18—4.28 (4H, m), 7.38-7.44 (2H, m), 7.77 (1H , s); ), 2.73 (2H, dd, J = 6.8 Hz, 6.8 Hz), 4.18—4.28 (4H, m), 7.38-7.44 (2H, m), 7.77 (1H, s);
MS(FAB) m/z: 327 (M+H)+. MS (FAB) m / z: 327 (M + H) + .
(16b)リン酸(2 アミノー 5, 5 ジメチノレ一 4, 5 ジヒドロナフト[1, 2— d] [l, 3]チ ァゾール— 8—ィル)ジェチル (16b) Phosphoric acid (2 amino-5,5 dimethylolene 4,5 dihydronaphtho [1,2-d] [l, 3] thiazol-8-yl) jetyl
実施例 16 (16a)で製造したリン酸 (8—ォキソ 5, 5 ジメチル一 5, 6, 7, 8—テト ラヒドロナフタレン一 2—ィル) ジェチル(1. 07g、 3. 28mmol)を使用して、実施例 2 (2b)に記載した方法に従い、標記目的化合物 (495mg、収率 39%)を得た。 Phosphoric acid (8-oxo-5,5 dimethyl-1,5,6,7,8-tetrahydronaphthalene-2-yl) prepared in Example 16 (16a) is used. Jetyl (1.07 g, 3.28 mmol) is used. Then, according to the method described in Example 2 (2b), the title object compound (495 mg, yield 39%) was obtained.
[0295] IR(KBr) v 988, 1038, 1266, 1541, 1650, 3144, 3348 cm"1; [0295] IR (KBr) v 988, 1038, 1266, 1541, 1650, 3144, 3348 cm "1;
max
H NMR(CD OD, 500MHz): δ 1.32 (6H, s), 1.35 (6H, t, J=7.3 Hz), 2.70 (2H, s), 4max H NMR (CD OD, 500 MHz): δ 1.32 (6H, s), 1.35 (6H, t, J = 7.3 Hz), 2.70 (2H, s), 4
3 Three
.23 (4H, dq, J=7.3 Hz, 7.3 Hz), 7.00—7.05 (IH, m), 7.34 (IH, d, J=7.8 Hz), 7.43-7. 46 (IH, m); .23 (4H, dq, J = 7.3 Hz, 7.3 Hz), 7.00—7.05 (IH, m), 7.34 (IH, d, J = 7.8 Hz), 7.43-7. 46 (IH, m);
MS(FAB) m/z: 383 (M+H)+. MS (FAB) m / z: 383 (M + H) + .
(16c)リン酸 二水素 (2 アミノー 5, 5 ジメチルー 4, 5 ジヒドロナフト[1, 2 d ] [1, 3]チアゾールー 8 ィル) (16c) Dihydrogen phosphate (2 amino-5,5 dimethyl-4,5 dihydronaphtho [1, 2 d] [1, 3] thiazole-8 yl)
実施例 16 (16b)で製造したリン酸(2 ァミノ一 5, 5 ジメチルー 4, 5 ジヒドロナ フト [1, 2— d] [l, 3]チアゾール 8—ィル)ジェチル(101mg、 0. 264mmol)を使 用して、実施例 1 (lc)に記載した方法に従い、標記目的化合物 (81. 4mg、収率 95 %)を得た。 Phosphoric acid prepared in Example 16 (16b) (2-amino-1,5 dimethyl-4,5 dihydronaphtho [1,2-d] [l, 3] thiazole 8-yl) jetyl (101 mg, 0.264 mmol) To give the title object compound (81.4 mg, yield 95%) according to the method described in Example 1 (lc).
[0296] IR(KBr) v 983, 1108, 1164, 1507, 1632, 2955 cm"1; [0296] IR (KBr) v 983, 1108, 1164, 1507, 1632, 2955 cm "1;
max max
1H NMR(DMSO-d , 500MHz): δ 1.26 (6H, s), 2.66 (2H, s), 6.91—6.96 (IH, m), 7. 1H NMR (DMSO-d, 500 MHz): δ 1.26 (6H, s), 2.66 (2H, s), 6.91-6.96 (IH, m), 7.
6 6
13-7.25 (2H, br), 7.27 (IH, d, J=7.8 Hz), 7.46-7.49 (IH, m); 13-7.25 (2H, br), 7.27 (IH, d, J = 7.8 Hz), 7.46-7.49 (IH, m);
MS(FAB) m/z: 325 (M- H)— . MS (FAB) m / z: 325 (M- H) —.
<実施例 17 > <Example 17>
(2 アミノー 5, 5 ジメチルー 4, 5 ジヒドロナフト[1, 2-d] [l, 3]チアゾール 8 ィル)ジフルォロメチルホスホン酸(例示化合物番号 2-7) (2 amino-5,5 dimethyl-4,5 dihydronaphtho [1,2-d] [l, 3] thiazole 8yl) difluoromethylphosphonic acid (Exemplified Compound No. 2-7)
(17a)トリフルォロメタンスルホ-ル(8—ォキソ—5, 5 ジメチル— 5, 6, 7, 8—テト ラヒドロナフタレン 2—ィノレ) (17a) trifluoromethanesulfurol (8-oxo-5,5 dimethyl-5,6,7,8-tetrahydronaphthalene 2-inole)
7 ヒドロキシ一 4, 4 ジメチノレ一 3, 4 ジヒドロナフタレン一 1 (2H)—オン(2. 77 g、 14. 6mmol)を使用して、実施例 15 (15a)に記載した方法に従い、標記目的化 合物 (4. 69g、収率 100%)を得た。 7 Hydroxy 1,4 Dimethinole 3,4 Dihydronaphthalene 1 (2H) -one (2.77 g, 14.6 mmol) was used to prepare the title compound according to the method described in Example 15 (15a). The compound (4.69 g, yield 100%) was obtained.
[0297] IR(Liquid film) v 925, 1142, 1212, 1427, 1695, 2968 cm"1; [0297] IR (Liquid film) v 925, 1142, 1212, 1427, 1695, 2968 cm "1;
max max
1H NMR(CDC1 , 500MHz): δ 1.41 (6H, s), 2.05 (2H, dd, J=6.8 Hz, 6.8 Hz), 2.77 1H NMR (CDC1, 500 MHz): δ 1.41 (6H, s), 2.05 (2H, dd, J = 6.8 Hz, 6.8 Hz), 2.77
3 Three
(2H, dd, J=6.8 Hz, 6.8 Hz), 7.27 (IH, d, J=2.9 Hz), 7.41 (IH, dd, J=2.9 Hz, 8.8 Hz) , 7.52 (IH, d, J=8.8 Hz); (2H, dd, J = 6.8 Hz, 6.8 Hz), 7.27 (IH, d, J = 2.9 Hz), 7.41 (IH, dd, J = 2.9 Hz, 8.8 Hz), 7.52 (IH, d, J = 8.8 Hz);
MS(FAB) m/z: 323 (M+H)+.
(17b)トリフルォロメタンスルホン酸 {4,, 4,一ジメチルー 3,, 4,ージヒドロー 2,H— スピロ [1, 3 ジォキソラン一 2, 1,一ナフタレン]— 7,一ィル } MS (FAB) m / z: 323 (M + H) + . (17b) trifluoromethanesulfonic acid {4, 4, 4, dimethyl-3, 4, 4-dihydro-2, H-spiro [1, 3 dioxolane-1, 2, 1, naphthalene] -7, 1il}
実施例 17 ( 17a)で製造したトリフルォロメタンスルホニル (8 -ォキソ 5, 5 ジメ チル— 5, 6, 7, 8—テトラヒドロナフタレン— 2—ィル)(4. 63g、 14. 4mmol)を使用 して、実施例 15 (15b)に記載した方法に従い、標記目的化合物 (3. 82g、収率 73% )を得た。 Using trifluoromethanesulfonyl (8-oxo-5,5 dimethyl-5,6,7,8-tetrahydronaphthalene-2-yl) (4.63 g, 14.4 mmol) prepared in Example 17 (17a) Then, according to the method described in Example 15 (15b), the title object compound (3.82 g, yield 73%) was obtained.
[0298] IR(Liquid film) v 928, 1060, 1143, 1213, 1424, 2964 cm"1; [0298] IR (Liquid film) v 928, 1060, 1143, 1213, 1424, 2964 cm "1;
max max
1H NMR(CDC1 , 500MHz) δ 1.30 (6H, s), 1.84—1.89 (2H, m), 1.98—2.02 (2H, m), 1H NMR (CDC1, 500MHz) δ 1.30 (6H, s), 1.84—1.89 (2H, m), 1.98—2.02 (2H, m),
3 Three
4.08-4.15 (2H, m), 4.16—4.23 (2H, m), 7.17 (1H, dd, J=2.9 Hz, 8.8 Hz), 7.34 (1H, d, J=2.9 Hz), 7.37 (1H, d, J=8.8 Hz); 4.08-4.15 (2H, m), 4.16—4.23 (2H, m), 7.17 (1H, dd, J = 2.9 Hz, 8.8 Hz), 7.34 (1H, d, J = 2.9 Hz), 7.37 (1H, d , J = 8.8 Hz);
MS(FAB) m/z: 367 (M+H)+. MS (FAB) m / z: 367 (M + H) + .
(17c) 4,, 4,一ジメチルー 3,, 4,一ジヒドロ一 7,一ヒドロキシメチル一 2,H—スピロ [ 1, 3 ジ才キソラン 2, 1 ' ナフタレン] (17c) 4, 4, 1, dimethyl-3, 4, 1, dihydro-1, 7, hydroxymethyl 1, 2, H-spiro [1, 3 di-aged xolan 2, 1 'naphthalene]
実施例 17 (17b)で製造したトリフルォロメタンスルホン酸 {4' , 4'—ジメチル— 3' , 4,一ジヒドロ一 2,H—スピロ [1, 3 ジォキソラン一 2, 1,一ナフタレン]— 7,一ィル (3. 71g、 10. lmmol)を使用して、実施例 15 (15c)に記載した方法に従い、標記 目的化合物 (1. 84g、収率 97%)を得た。 Trifluoromethanesulfonic acid {4 ', 4'-dimethyl-3', 4, didihydro-1,2, H-spiro [1,3 dioxolane-1,2,1, mononaphthalene]-prepared in Example 17 (17b)- The title compound (1.84 g, 97% yield) was obtained according to the method described in Example 15 (15c) using 7,1 g (3.71 g, 10. lmmol).
[0299] IR(Liquid film) v 939, 1060, 1089, 1126, 1171, 2958, 3411 cm"1; [0299] IR (Liquid film) v 939, 1060, 1089, 1126, 1171, 2958, 3411 cm "1;
max max
1H NMR(CDC1 , 500MHz) δ 1.30 (6H, s), 1.82—1.87 (2H, m), 1.97—2.03 (2H, m), 1H NMR (CDC1, 500MHz) δ 1.30 (6H, s), 1.82—1.87 (2H, m), 1.97—2.03 (2H, m),
3 Three
4.08-4.16 (2H, m), 4.18-4.27 (2H, m), 4.65 (2H, d, J=5.9 Hz), 7.29-7.32 (2H, m), 7.47 (1H, s); 4.08-4.16 (2H, m), 4.18-4.27 (2H, m), 4.65 (2H, d, J = 5.9 Hz), 7.29-7.32 (2H, m), 7.47 (1H, s);
MS(EI) m/z: 248 M+. MS (EI) m / z: 248 M + .
(17d) (4,, 4,一ジメチルー 3,, 4,一ジヒドロ一 2,H—スピロ [1, 3 ジォキソラン一 2, 1 '—ナフタレン ]—7' ィル)メチルホスホン酸ジェチル (17d) (4, 4, 4, 1-dimethyl-3, 4, 1-dihydro-1, 2, H-spiro [1, 3 dioxolane 1, 2, 1'-naphthalene] -7'yl) methylphosphonate
実施例 17 ( 17c)で製造した 4 ' , 4 ' ジメチル 3 ' , 4 ' ジヒドロ 7 ' ヒドロキシ メチル 2' H—スピロ [1, 3 ジォキソラン一 2, 1 '—ナフタレン] (1. 79g、 7. 21m mol)を使用して、実施例 15 (15d)に記載した方法に従い、標記目的化合物 (2. 13 g、収率 80%)を得た。
[0300] IR(Liquid film) v 966, 1028, 1060, 1251, 2960, 3457 cm ; 4 ', 4' dimethyl 3 ', 4' dihydro 7 'hydroxymethyl 2' H-spiro [1,3 dioxolane-1,2,1'-naphthalene] (1.79 g, 7. The title compound (2.13 g, yield 80%) was obtained according to the method described in Example 15 (15d) using 21 mmol). [0300] IR (Liquid film) v 966, 1028, 1060, 1251, 2960, 3457 cm;
max max
1H NMR(CDC1 , 500MHz) : δ 1.24 (6H, t, J=7.3 Hz), 1.28 (6H, s), 1.81—1.85 (2H, 1H NMR (CDC1, 500MHz): δ 1.24 (6H, t, J = 7.3 Hz), 1.28 (6H, s), 1.81-1.85 (2H,
3 Three
m), 1.96-2.01 (2H, m), 3.12 (2H, d, J=21.5 Hz), 3.96—4.05 (4H, m), 4.07-4.14 (2H, m), 4.16-4.23 (2H, m), 7.22-7.25 (2H, m), 7.38 (1H, s); m), 1.96-2.01 (2H, m), 3.12 (2H, d, J = 21.5 Hz), 3.96—4.05 (4H, m), 4.07-4.14 (2H, m), 4.16-4.23 (2H, m) , 7.22-7.25 (2H, m), 7.38 (1H, s);
MS(EI) m/z: 368 M+. MS (EI) m / z: 368 M + .
(17e) (5, 5 ジメチル一 8—ォキソ 5, 6, 7, 8—テトラヒドロナフタレン一 2—ィル )ジフルォロメチルホスホン酸ジェチル (17e) (5,5 Dimethyl 8-oxo 5, 6, 7, 8-tetrahydronaphthalene 2-yl) difluoromethylphosphonic acid jetyl
実施例 17 (17d)で製造した (4,, 4,一ジメチルー 3,, 4,ージヒドロー 2,H—スピロ [1, 3 ジォキソラン一 2, 1 '—ナフタレン]— 7'—ィル)メチルホスホン酸ジェチル( 2. 08g、 5. 38mmol)を使用して、実施例 15 (15e)に記載した方法に従い、標記目 的化合物 (1. 81g、収率 93%)を得た。 (4,4,1, dimethyl-3,4, -dihydro-2, H-spiro [1,3 dioxolane-1,2,1'-naphthalene] -7'-yl) methylphosphonic acid prepared in Example 17 (17d) The title compound (1.81 g, 93% yield) was obtained according to the method described in Example 15 (15e) using jetyl (2.08 g, 5.38 mmol).
[0301] IR(Liquid film) v 1021, 1269, 1691, 2968 cm"1; [0301] IR (Liquid film) v 1021, 1269, 1691, 2968 cm "1;
max max
1H NMR(CDC1 , 500MHz) δ 1.34 (6H, t, J=6.8 Hz), 1.41 (6H, s), 2.04 (2H, dd, J 1H NMR (CDC1, 500MHz) δ 1.34 (6H, t, J = 6.8 Hz), 1.41 (6H, s), 2.04 (2H, dd, J
3 Three
=6.8 Hz, 6.8 Hz), 2.75 (2H, dd, J=6.8 Hz, 6.8 Hz), 4.17—4.30 (4H, m), 7.53 (1H, d, J=7.8 Hz), 7.79 (1H, d, J=7.8 Hz), 8.24 (1H, s). = 6.8 Hz, 6.8 Hz), 2.75 (2H, dd, J = 6.8 Hz, 6.8 Hz), 4.17—4.30 (4H, m), 7.53 (1H, d, J = 7.8 Hz), 7.79 (1H, d, J = 7.8 Hz), 8.24 (1H, s).
MS(EI) m/z: 360 M+. MS (EI) m / z: 360 M + .
(17f) (2 アミノー 5, 5 ジメチノレー 4, 5 ジヒドロナフト[1, 2 d] [l, 3]チアゾー ルー 8—ィル)ジフルォロメチルホスホン酸ジェチル (17f) (2 amino-5,5 dimethylenoyl 4,5 dihydronaphtho [1,2d] [l, 3] thiazolu 8-yl) difluoromethylphosphonate jetyl
実施例 17 (17e)で製造した(5, 5 ジメチル一 8—ォキソ 5, 6, 7, 8—テトラヒド ロナフタレン一 2—ィル)ジフルォロメチルホスホン酸ジェチル(1. 52g、 4. 22mmol )を使用して、実施例 2 (2b)に記載した方法に従い、標記目的化合物 (1. 02g、収率 58%)を得た。 Example 17 (17e) (5,5 dimethyl-1-oxo-5,6,7,8-tetrahydronaphthalene-2-yl) difluoromethylphosphonate jetyl (1.52 g, 4.22 mmol) To give the title object compound (1.02 g, yield 58%) according to the method described in Example 2 (2b).
[0302] IR(KBr) v 1018, 1047, 1258, 1542, 1653, 3123, 3356 cm"1; [0302] IR (KBr) v 1018, 1047, 1258, 1542, 1653, 3123, 3356 cm "1;
max max
1H NMR(CD OD, 500MHz) : δ 1.31 (6H, t, J=7.3 Hz), 1.36 (6H, s), 2.75 (2H, s), 1H NMR (CD OD, 500 MHz): δ 1.31 (6H, t, J = 7.3 Hz), 1.36 (6H, s), 2.75 (2H, s),
3 Three
4.14-4.26 (4H, m), 7.41 (1H, d, J=7.8 Hz), 7.49 (1H, d, J=7.8 Hz), 7.87 (1H, s); MS(FAB) m/z: 417 (M+H)+. 4.14-4.26 (4H, m), 7.41 (1H, d, J = 7.8 Hz), 7.49 (1H, d, J = 7.8 Hz), 7.87 (1H, s); MS (FAB) m / z: 417 ( M + H) + .
(17g) (2 アミノー 5, 5 ジメチノレー 4, 5 ジヒドロナフト[1, 2— d] [l, 3]チアゾ 一ルー 8—ィル)ジフルォロメチルホスホン酸
実施例 17 (17f)で製造した(2 ァミノ 5, 5-ジメチル -4, 5 ジヒドロナフト[1 , 2-d] [l, 3]チアゾールー 8 ィル)ジフルォロメチルホスホン酸ジェチル(995mg 、 2. 39mmol)を使用して、実施例 1 (lc)に記載した方法に従い、標記目的化合物( 650mgゝ収率 76%)を得た。 (17g) (2 Amino-5, 5 Dimethinolere 4, 5 Dihydronaphtho [1, 2—d] [l, 3] thiazo-Lu 8-yl) Difluoromethylphosphonic acid Example 17 (17f) (2-amino-5,5-dimethyl-4,5 dihydronaphtho [1,2-d] [l, 3] thiazol-8 yl) difluoromethylphosphonic acid jetyl (995 mg, 2 39 mmol) was used to give the title compound (650 mg yield 76%) according to the method described in Example 1 (lc).
[0303] IR(KBr) v 927, 1072, 1243, 1604, 1638, 2964 cm"1; [0303] IR (KBr) v 927, 1072, 1243, 1604, 1638, 2964 cm "1;
max max
1H NMR(DMSO-d , 500MHz): δ 1.23 (6H, s), 2.52 (2H, s), 7.37 (1H, d, J=7.8 Hz 1H NMR (DMSO-d, 500MHz): δ 1.23 (6H, s), 2.52 (2H, s), 7.37 (1H, d, J = 7.8 Hz
6 6
), 7.42 (1H, d, J=7.8 Hz), 8.00 (1H, s); ), 7.42 (1H, d, J = 7.8 Hz), 8.00 (1H, s);
MS(ESI) m/z: 359 (M— H)— . MS (ESI) m / z: 359 (M— H) —.
<実施例 18 > <Example 18>
リン酸 二水素 (2,一アミノー 4,H—スピロ [シクロペンタン 1, 5,一ナフト[1, 2— d] [l, 3]チアゾール] 8' ィル)(例示化合物番号 2-28) Dihydrogen phosphate (2,1-amino-4, H-spiro [cyclopentane 1,5, one-naphtho [1,2-d] [l, 3] thiazole] 8'-yl) (Exemplified compound number 2-28)
(18a) 6,ーメトキシー3,, 4,ージヒドロー 2,H—スピロ [シクロペンタン 1, 1,ーナ フタレン] 2'—才ン (18a) 6, -Methoxy-3,4, -dihydro-2, H-spiro [cyclopentane 1,1, -naphthalene] 2 '-
6—メトキシ一 2—テトラロン(8. 44g、47. 9mmol)を N, N ジメチルホルムアミド( 200mL)に溶解し、そこへ水素ィ匕ナトリウム(5. 23g、 120mmol)をカロえ、室温で 0.5 時間撹拌した後に、 1, 4ージョードブタン(15. 6g、 50. 3mmol)を加え、さらに 3時 間撹拌した。 6-Methoxy-2-tetralone (8.44 g, 47.9 mmol) is dissolved in N, N dimethylformamide (200 mL), and sodium hydrogen carbonate (5.23 g, 120 mmol) is added to the solution, and the mixture is stirred at room temperature for 0.5 hour. After stirring, 1,4-Jodobutane (15.6 g, 50.3 mmol) was added and stirred for another 3 hours.
[0304] 反応液に水を加え、酢酸ェチルで抽出した。得られた有機層を飽和食塩水で洗浄 し、無水硫酸マグネシウムで乾燥し、減圧下溶媒を留去した。 [0304] Water was added to the reaction solution, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure.
[0305] 得られた残渣をシリカゲルカラムクロマトグラフィー (へキサン:酢酸ェチル、 10 : 1、[0305] The obtained residue was subjected to silica gel column chromatography (hexane: ethyl acetate, 10: 1,
VZV)を用いて精製し、標記目的化合物 (8. 13g、収率 74%)を得た。 VZV) to give the title compound (8.13 g, yield 74%).
[0306] IR(Liquid film) v 1040, 1262, 1500, 1579, 1610, 1708, 2836, 2868, 2952 cm"1; [0306] IR (Liquid film) v 1040, 1262, 1500, 1579, 1610, 1708, 2836, 2868, 2952 cm "1;
max max
1H NMR(CDC1 , 400MHz): δ 1.76—1.85 (6H, m), 2.26-2.37 (2H, m), 2.67 (2H, dd 1H NMR (CDC1, 400MHz): δ 1.76—1.85 (6H, m), 2.26-2.37 (2H, m), 2.67 (2H, dd
3 Three
, J=6.6 Hz, 6.6 Hz), 3.05 (2H, dd, J=6.6 Hz, 6.6 Hz), 3.80 (3H, s), 6.70 (1H, d, J=2 .9 Hz), 6.80 (1H, dd, J=2.9 Hz, 8.8 Hz), 7.18 (1H, d, J=8.8 Hz); , J = 6.6 Hz, 6.6 Hz), 3.05 (2H, dd, J = 6.6 Hz, 6.6 Hz), 3.80 (3H, s), 6.70 (1H, d, J = 2.9 Hz), 6.80 (1H, dd, J = 2.9 Hz, 8.8 Hz), 7.18 (1H, d, J = 8.8 Hz);
MS(EI) m/z: 230 M+. MS (EI) m / z: 230 M + .
(18b) 6'—メトキシー 3' , 4'—ジヒドロー 2' H—スピロ [シクロペンタン 1, 1 'ーナ
フタレン] (18b) 6'-Methoxy-3 ', 4'-dihydro-2' H-spiro [cyclopentane 1,1'-na Phthalene]
実施例 18 ( 18a)で製造した 6,一メトキシ一 3,, 4,一ジヒドロ一 2, H スピロ [シクロ ペンタン 1, 1,一ナフタレン ] 2,一オン(5. 45g、 23. 7mmol)をジエチレングリ コール(250mL)〖こ溶解し、ヒドラジン一水和物(25. OmL、 515mmol)及び水酸化 カリウム(45. 0g、 802mmol)を加え、ディーン 'スターク装置を付して、 180°Cで 1. 5時間撹拌した。 6, 18-methoxy-1, 3, 4, 1-dihydro-1, 2, H spiro [cyclopentane 1, 1, 1-naphthalene] 2, 1-one (5.45 g, 23.7 mmol) prepared in Example 18 (18a). Dissolve diethylene glycol (250 mL), add hydrazine monohydrate (25. OmL, 515 mmol) and potassium hydroxide (45.0 g, 802 mmol), add Dean 'Stark apparatus at 180 ° C. 1. Stir for 5 hours.
[0307] 室温まで冷却後、反応液に水を加え、塩酸により中和した後、酢酸ェチルで抽出し た。得られた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥し、減圧 下溶媒を留去した。 [0307] After cooling to room temperature, water was added to the reaction solution, neutralized with hydrochloric acid, and extracted with ethyl acetate. The obtained organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure.
[0308] 得られた残渣をシリカゲルカラムクロマトグラフィー (へキサン:酢酸ェチル、 10 : 1、 [0308] The obtained residue was subjected to silica gel column chromatography (hexane: ethyl acetate, 10: 1,
VZV)を用いて精製し、標記目的化合物 (4. 16g、収率 81%)を得た。 VZV) to give the title object compound (4.16 g, yield 81%).
[0309] IR(Liquid film) v 1046, 1126, 1245, 1450, 1500, 1573, 1610, 2867, 2938 cm"1; [0309] IR (Liquid film) v 1046, 1126, 1245, 1450, 1500, 1573, 1610, 2867, 2938 cm "1;
max max
1H NMR(CDC1 , 400MHz) : δ 1.62-1.67 (2H, m), 1.69-1.88 (10H, m), 2.74 (IH, d 1H NMR (CDC1, 400MHz): δ 1.62-1.67 (2H, m), 1.69-1.88 (10H, m), 2.74 (IH, d
3 Three
d, J=5.9 Hz, 6.6 Hz), 3.77 (3H, s), 6.56 (IH, d, J=2.9 Hz), 6.73 (IH, dd, J=2.9 Hz, 8.8 Hz), 7.19 (IH, d, J=8.8 Hz); d, J = 5.9 Hz, 6.6 Hz), 3.77 (3H, s), 6.56 (IH, d, J = 2.9 Hz), 6.73 (IH, dd, J = 2.9 Hz, 8.8 Hz), 7.19 (IH, d , J = 8.8 Hz);
MS(EI) m/z: 216 M+. MS (EI) m / z: 216 M + .
(18c) 6,ーメトキシ 2,, 3,ージヒドロー 4,H—スピロ [シクロペンタン 1, 1,ーナ フタレン] 4'一才ン (18c) 6, -methoxy 2,3, -dihydro-4, H-spiro [cyclopentane 1,1, -naphthalene] 4 '
実施例 18 ( 18b)で製造した 6,一メトキシ 3,, 4,一ジヒドロ一 2, H スピロ [シクロ ペンタン 1, 1,一ナフタレン] (5. 26g、 23. Ommol)をアセトン一水(3 : 1、 VZV、 6,18-methoxy 3,4,1-dihydro-1,2, H spiro [cyclopentane 1,1,1-naphthalene] (5.26 g, 23. Ommol) prepared in Example 18 (18b) was added to acetone monohydrate (3 : 1, VZV,
150mL)に溶解し、硫酸マグネシウム(7. 62g、 63. 3mmol)及び過マンガン酸カリ ゥム(20. Og、 127mmol)をカ卩え、室温で 21時間撹拌した。 150 mL), magnesium sulfate (7.62 g, 63.3 mmol) and potassium permanganate (20. Og, 127 mmol) were added, and the mixture was stirred at room temperature for 21 hours.
[0310] 反応液をろ過し、ろ液に 50% (W/V)チォ硫酸ナトリウム水溶液をカ卩え、再びろ過 し、酢酸ェチルで抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸マグ ネシゥムで乾燥し、減圧下溶媒を留去した。 [0310] The reaction solution was filtered, 50% (W / V) aqueous sodium thiosulfate solution was added to the filtrate, filtered again, and extracted with ethyl acetate. The obtained organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure.
[0311] 得られた残渣をシリカゲルカラムクロマトグラフィー (へキサン:酢酸ェチル、 10 : 1、[0311] The obtained residue was subjected to silica gel column chromatography (hexane: ethyl acetate, 10: 1,
VZV)を用いて精製し、標記目的化合物(5. 03g、収率 90%)を得た。 VZV) to give the title compound (5.03 g, yield 90%).
[0312] IR(Liquid film) v 1040, 1234, 1285, 1493, 1609, 1685, 2870, 2951 cm"1;
•ΐ '(ΖΗ o"z=f '; Ήε) ζε·ΐ '(ζΗ o"z=f '; Ήε) ιζε'ΐ 9 : (ΖΗ )Ο ' ιつ αつ) WN HX [0312] IR (Liquid film) v 1040, 1234, 1285, 1493, 1609, 1685, 2870, 2951 cm "1; • ΐ '(ΖΗ o "z = f'; Ήε) ζε · ΐ '( ζ Η o" z = f'; Ήε) ιζε'ΐ 9: ( Ζ Η) Ο 'ια α) WN H X
·τ_ωο es62 '9162 'ZLSZ '8891 '909ΐ '8ΐ^ΐ '6ΐ2ΐ 'ISO! Λ (ω¾ pmbn)HI [STSO] · Τ _ωο es62 '9162' ZLSZ '8891' 909ΐ '8ΐ ^ ΐ' 6ΐ2ΐ 'ISO! Λ (ω¾ pmbn) HI [STSO]
(%06 ¾τ ^ιΐ£) ^ 目 ¾遨 w^ ^-xm^ (^SD z m 第、ェっ ¾ί¾¾(ΐου υ6"[ ·"[、3u¾SS)ベ —^— [ベ ^ 'ΐ Ί- ^ -^(% 06 ¾τ ^ ιΐ £) ^ Eye ¾ 遨 w ^ ^ -xm ^ (^ SD zm No. ¾ί¾¾ (ΐ ου υ 6 "[·" [, 3u ¾SS) Be — ^ — [Be ^ 'ΐ Ί-^-^
( - ,9-_ Λ^^ - Λ 'τ ,、
 9ΐζ: ζ/ω (m)S (-, 9-_ Λ ^^-Λ 'τ, 9ΐζ: ζ / ω (m) S
9ΐζ: ζ/ω (m)S (-, 9-_ Λ ^^-Λ 'τ, 9ΐζ: ζ / ω (m) S
•(ZH 6'Ζ=ί 'Ρ 'Ηΐ) 83"Ζ '(ΖΗ 8·8=ί" 'Ρ 'Ηΐ) 92" Ζ '(ΖΗ 8·8 'ΖΗ 6'Ζ=ί 'ΡΡ 'Ηΐ) 80· ^ 'Ηΐ) 66'S '(ΖΗ 9·9 'ΖΗ 9"9=f 'PP 'ΗΖ) IL'Z ' • (ZH 6'Ζ = ί 'Ρ' Ηΐ) 83 "Ζ '(ΖΗ 8 · 8 = ί"' Ρ 'Ηΐ) 92 "Ζ' (ΖΗ 8 · 8 ' Ζ Η 6'Ζ = ί' ΡΡ ' Ηΐ) 80 · ^ 'Ηΐ) 66'S' (ΖΗ 9 · 9 ' Ζ Η 9 "9 = f' PP 'ΗΖ) IL'Z'
(ΖΗ 9·9 'ΖΗ 9"9=f 'PP 'ΗΖ) WZ '(ω 'Η8) 86·ΐ— 9Ζ·ΐ 9 '· (ζΗ )0 つ αつ) Ηχ (ΖΗ 9 · 9 ' Ζ Η 9 "9 = f' PP 'ΗΖ) WZ' (ω 'Η8) 86 · ΐ— 9Ζ · ΐ 9' · ( ζ )) 0 α) Η χ
ί ΙΙΙΟ ΐ- ί ΙΙΙΟ ΐ-
8εεε 'f 6z 'em ' 9sz '\ 'ΖΟΘΪ 'm '9 'θΐετ H 'mi Λ ( i) [mo 8εεε 'f 6z' em '9sz' \ 'ΖΟΘΪ' m '9' θΐετ H 'mi Λ (i) (mo
99

。: . :
3^S^iD 翻 s Ί ^ t^( ^i -gg、¾ •D^r^-^m 、つ ^nra 3 ^ S ^ iD translation s Ί ^ t ^ (^ i -gg, ¾ • D ^ r ^-^ m, ^ n ra
\) ^H^{\ommz 'ζζ §εχ,s)ベ 一^— ([ベ ^ 'ΐ 'τ一べ\) ^ H ^ {\ omm z 'ζζ § εχ, s) be ^^ ([be ^' ΐ 'τ
J - ー [ベ ^ J--[Be ^
-{I ' I - ^ -^ ^'] 0^-u ( - - ί ε '.S-^ci^- {9(Ρ8Ι)- { I 'I-^-^ ^'] 0 ^ -u ( --ί ε ' .S- ^ ci ^- { 9 (Ρ8Ι)
•(ΖΗ 6 =1" 'Ρ 'Ηΐ) 6VL '(ΖΗ 8·8=ί" 'Ρ 'Ηΐ) 82 •Ζ '(ΖΗ 8·8 'ΖΗ 6'Ζ=ί 'ΡΡ 'Ηΐ) ΟΓ Ήε) fS'£ '(ΖΗ 9·9 'ΖΗ 9·9=ί" 'ΡΡ 'ΗΖ) OL'Z ' (ΖΗ 9·9 'ΖΗ 9·9=ί" 'ΡΡ 'ΗΖ) WZ '(ω 'Η8) 86"T-ZZ"T 9 '· (ζΗ )0 ' つ αつ) Ητ • ( Ζ Η 6 = 1 ”'Ρ' Ηΐ) 6VL '( Ζ Η 8 · 8 = ί”' Ρ 'Ηΐ) 82 • Ζ' (ΖΗ 8 · 8 ' Ζ Η 6'Ζ = ί' ΡΡ 'Ηΐ ) ΟΓ Ήε) fS '£' ( Ζ Η 9 · 9 ' Ζ Η 9 · 9 = ί "' ΡΡ 'ΗΖ) OL'Z' (ΖΗ 9 · 9 ' Ζ Η 9 · 9 = ί"' ΡΡ 'ΗΖ ) WZ '(ω' Η8) 86 "T-ZZ" T 9 '· ( ζ Η) 0' α ') Η τ
0l76S0C/900Zdf/X3d 98 0C0l70l/900Z OAV
(H+n) see: ζ/ω (evd)S 0l76S0C / 900Zdf / X3d 98 0C0l70l / 900Z OAV (H + n) see: ζ / ω (evd) S
•(ZH οτ=ί 'p Ή ΐ) WL '(Jq ΉΖ) fZ'L '(ΖΗ ε·8=ί" 'P 'Ηΐ) 6ΓΖ '(ΖΗ ε·8 'ΖΗ 0"2=f 'PP 'Ηΐ) 06·9 '(s ' HZ) 69 '(ω 'ΗΖ) S8"T-SZ"T '(ω 'Η9) SZ'I— ε9·ΐ 9 '· (zH OOS '9P-0S a)H N Hx • (ZH οτ = ί 'p Ή ΐ) WL' (Jq ΉΖ) fZ'L '( Ζ Η ε · 8 = ί "' P 'Ηΐ) 6ΓΖ' (ΖΗ ε · 8 ' Ζ Η 0" 2 = f 'PP' Ηΐ) 06 · 9 '( s ' HZ) 69 '(ω' ΗΖ) S8 "T-SZ" T '(ω' Η9) SZ'I— ε9 · ΐ 9 '· ( z H OOS' 9P -0S a) HNH x
ί IIIO i- ί IIIO i-
TS^S ' 90S 'ZLSZ '9ZLZ 'SS9T '0091 'ZOST '39Π 'ΖΟΠ Λ ( 1)ΗΙ [ TSO] ο^ ¾(%0 *¾ί
 αοπ υ TS ^ S '90S' ZLSZ '9ZLZ' SS9T '0091' ZOST '39 Π 'ΖΟΠ Λ (1) ΗΙ [TSO] ο ^ ¾ (% 0 * ¾ί αοπ υ
αοπ υ TS ^ S '90S' ZLSZ '9ZLZ' SS9T '0091' ZOST '39 Π 'ΖΟΠ Λ (1) ΗΙ [TSO] ο ^ ¾ (% 0 * ¾ί αοπ υ
S90 Ό
 / <8 [ /— 、 ^ [ε Ί][Ρ-2 ー 'S ' "[—ベ ベ 一 Η^— 一 ) (^WMi^ (J81 ) 81 P}¾? S90 Ό / <8 [/ —, ^ [ε Ί] [Ρ-2 ー 'S'"[—Bebe one Η ^ — one) (^ WMi ^ (J81) 81 P} ¾?
/ <8 [ /— 、 ^ [ε Ί][Ρ-2 ー 'S ' "[—ベ ベ 一 Η^— 一 ) (^WMi^ (J81 ) 81 P}¾? S90 Ό / <8 [/ —, ^ [ε Ί] [Ρ-2 ー 'S'"[—Bebe one Η ^ — one) (^ WMi ^ (J81) 81 P} ¾?
( / <8 [ /— 、 ^ [ε Ί][Ρ-2 Ί (/ <8 [/ —, ^ [ε Ί] [Ρ-2 Ί
]4 一 <g
 ] 4 <g
] 4 <g
( ) 60f : ζ/ω (9VH)S () 60f: ζ / ω (9VH) S
•(ΖΗ 6'Ζ=ί 'Ρ 'Ηΐ) SS"Z '(ΖΗ 8·8=ί" 'Ρ 'Ηΐ) LVL '(ΖΗ 8· 8 'ΖΗ 6'Ζ=ί 'ΡΡ 'Ηΐ) 66·9 ^ 'ΗΖ) S8"S '(ω Ή^) LZ' - Y '(s 'ΗΖ) 9L'Z '(ω 'ΗΖ) S • (ΖΗ 6'Ζ = ί 'Ρ' Ηΐ) SS "Z '(ΖΗ 8 · 8 = ί"' Ρ 'Ηΐ) LVL' (ΖΗ 8 · 8 ' Ζ Η 6'Ζ = ί' ΡΡ 'Ηΐ) 66 · 9 ^ 'ΗΖ) S8 "S' (ω Ή ^) LZ '-Y' ( s ' ΗΖ) 9L'Z '(ω' ΗΖ) S
6·ΐ— 88·ΐ '(ω 'Η9) 08"T-SZ"T '(ΖΗ S"Z=f '; 'Η9) 9£Ί 9 '· (ΖΗ 003 ^ Ιつ) Ηχ 6 · ΐ— 88 · ΐ '(ω' Η9) 08 "T-SZ" T '(ΖΗ S "Z = f';'Η9) 9 £ Ί 9' · ( Ζ Η 003 ^ Ι つ) Η χ
·τ_ωο εεε 'θετε ' s62 's ss '039ΐ '^ΟΘΪ Ό½Ϊ ' Θ^Ϊ 'οεοτ ( i) [9ΐεο]· Τ _ωο εεε 'θετε' s62 's ss'039ΐ' ^ ΟΘΪ Ό½Ϊ 'Θ ^ Ϊ' οεοτ (i) [9ΐεο]
。 (%6ε ? φ ra0 )呦^; 目 ¾遨
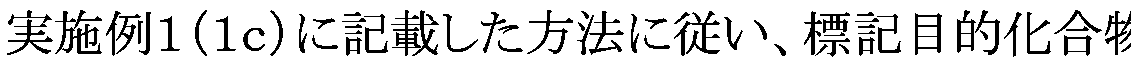 . (% 6ε? Φ ra 0 ) 呦 ^; Eye ¾ 遨
. (% 6ε? Φ ra 0 ) 呦 ^; Eye ¾ 遨
rarazo Ί ^ll£) ( / 一' 9— [べ 一'!; ' I - ^ ^ ^^ a^y:-U ( rarazo Ί ^ ll £) (/ 1 '9— [Be 1'!;'I-^ ^ ^^ a ^ y: -U (
(98i)8i m( 9 8i) 8i m

_V-Z 'S 'l- ^ ^ ^^ ^-U^-Z^J.- ,Ζ) 邈べ fi(J8I) _V-Z 'S' l- ^ ^ ^^ ^ -U ^ -Z ^ J.-, Ζ) fibe fi (J8I)
(w ) see: ζ/ω (evd)S (w) see: ζ / ω (evd) S
•(ZH Z 'ΖΗ ΖΊ=ί 'PP 'HI) OL'L '(z • (ZH Z ' Ζ Η ΖΊ = ί' PP 'HI) OL'L' ( z
H 9·8 'ΖΗ UZ 'ΖΗ Ζ'1=ί 'PPP 'Ηΐ) WL '(ΖΗ 9"8=f 'P 'Ηΐ) WL '(ω 'Hf) 62^-81^ H 9/8 ' Ζ Η UZ' Ζ Η Ζ'1 = ί 'PPP' Ηΐ) WL '( Ζ Η 9 "8 = f' P 'Ηΐ) WL' (ω 'Hf) 62 ^ -81 ^
'(ΖΗ Ζ·9 'ΖΗ Z"9=f 'PP ΉΖ) OL'Z '(ΖΗ Ζ·9 'ΖΗ Z"9=f 'PP 'ΗΖ) 9'Z '(ω 'Η8) 86"ΐ-6Ζ '(ΖΗ Ζ · 9' Ζ Η Z "9 = f 'PP ΉΖ) OL'Z' ( Ζ Η Ζ · 9 ' Ζ Η Z" 9 = f' PP 'ΗΖ) 9'Z' (ω 'Η8) 86 "ΐ-6Ζ
0l76S0C/900Zdf/X3d Z8 0C0l70l/900Z OAV
<実施例 19 > 0l76S0C / 900Zdf / X3d Z8 0C0l70l / 900Z OAV <Example 19>
(2,一アミノー 4,H—スピロ [シクロペンタン一 1, 5,一ナフト [1, 2-d] [l, 3]チアゾ ール] 8,一ィル)ジフルォロメチルホスホン酸(例示化合物番号 2-8) (2,1-amino-4, H-spiro [cyclopentane-1,5,1-naphtho [1,2-d] [l, 3] thiazol] 8,1-yl) difluoromethylphosphonic acid (exemplary compound (Number 2-8)
(19a)トリフルォロメタンスルホン酸 (4,一ォキソ 3,, 4,一ジヒドロ一 2,H—スピロ [シクロペンタン一 1, 1 '—ナフタレン]— 6'—ィノレ) (19a) trifluoromethanesulfonic acid (4, 1-oxo-3, 4, 1-dihydro-1, 2, H-spiro [cyclopentane-1,1,1'-naphthalene] -6'-inore)
実施例 18 (18d)で製造した 6,一ヒドロキシ一 2,, 3,一ジヒドロ一 4, H—スピロ [シク 口ペンタン一 1, 1,一ナフタレン]— 4,一オン(4. 23g、 19. 6mmol)をジクロロメタン (lOOmL)に溶解し、 30。Cで 2, 6—ルチジン(2. 8mL、 24. 0mmol)、4 ジメチ ルァミノピリジン(480mg、 3. 93mmol)及び無水トリフルォロメタンスルホン酸(4. 0 mL、 23. 8mmol)を加え、徐々に室温まで昇温しながら、 4時間撹拌した。 6,18-hydroxy-1,2,3,1-dihydro-1,4, H-spiro [cyclopentane-1,1,1, naphthalene] -4,1one (4.23 g, 19) prepared in Example 18 (18d) 6 mmol) is dissolved in dichloromethane (lOOmL) 30. Add 2,6-lutidine (2.8 mL, 24.0 mmol), 4 dimethylaminopyridine (480 mg, 3.93 mmol) and trifluoromethanesulfonic anhydride (4.0 mL, 23.8 mmol) to C, and gradually add to room temperature. The mixture was stirred for 4 hours while raising the temperature to 4 ° C.
[0318] 反応液に水を加え、酢酸ェチルで抽出した。得られた有機層を希塩酸及び飽和食 塩水で洗浄し、無水硫酸マグネシウムで乾燥し、減圧下溶媒を留去した。 [0318] Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with dilute hydrochloric acid and saturated brine, dried over anhydrous magnesium sulfate, and the solvent was distilled off under reduced pressure.
[0319] 得られた残渣をシリカゲルカラムクロマトグラフィー (へキサン:酢酸ェチル、 10 : 1、 VZV)を用いて精製し、標記目的化合物(6. 79g、収率 99%)を得た。 [0319] The obtained residue was purified by silica gel column chromatography (hexane: ethyl acetate, 10: 1, VZV) to obtain the title object compound (6.79 g, yield 99%).
[0320] IR(Liquid film) v 1141, 1212, 1246, 1427, 1480, 1695, 2875, 2956, 3075 cm"1; [0320] IR (Liquid film) v 1141, 1212, 1246, 1427, 1480, 1695, 2875, 2956, 3075 cm "1;
max max
1H NMR(CDC1 , 400MHz) : δ 1.82—1.99 (8H, m), 2.08 (2H, dd, J=5.9 Hz, 6.8 Hz) 1H NMR (CDC1, 400MHz): δ 1.82—1.99 (8H, m), 2.08 (2H, dd, J = 5.9 Hz, 6.8 Hz)
3 Three
, 2.74 (2H, dd, J=5.9 Hz, 6.8 Hz), 7.40 (1H, dd, J=2.9 Hz, 8.8 Hz), 7.46 (1H, d, J= 8.8 Hz), 7.88 (1H, d, J=2.9 Hz); , 2.74 (2H, dd, J = 5.9 Hz, 6.8 Hz), 7.40 (1H, dd, J = 2.9 Hz, 8.8 Hz), 7.46 (1H, d, J = 8.8 Hz), 7.88 (1H, d, J = 2.9 Hz);
MS(FAB) m/z: 349 (M+H)+. MS (FAB) m / z: 349 (M + H) + .
(19b)トリフルォロメタンスルホン酸 (4,一ヒドロキシ一 3,, 4,一ジヒドロ一 2,H—ス ピロ [シクロペンタン 1, 1 '—ナフタレン ]—6'—ィノレ) (19b) trifluoromethanesulfonic acid (4,1hydroxy-1,3,4,1dihydro-1,2, H-spiro [cyclopentane 1,1'-naphthalene] -6'-inole)
実施例 19 (19a)で製造したトリフルォロメタンスルホン酸 (4,一ォキソ一 3' , 4'—ジ ヒドロ一 2, H—スピロ [シクロペンタン一 1, 1,一ナフタレン]— 6,一ィル)(1. 64g、 4 . 71mmol)をメタノール(25mL)に溶解し、 0°Cで水素化ホウ素ナトリウム (270mg、 7. 14mmol)をカ卩え、 0.5時間撹拌した。 Trifluoromethanesulfonic acid prepared in Example 19 (19a) (4,1-oxo-3 ', 4'-dihydro-1, H-spiro [cyclopentane-1,1,1, naphthalene] -6, (1.64 g, 4.71 mmol) was dissolved in methanol (25 mL), sodium borohydride (270 mg, 7.14 mmol) was added at 0 ° C., and the mixture was stirred for 0.5 hr.
[0321] 反応液にアセトンを加えた後、減圧下溶媒を留去した。得られた残渣を酢酸ェチル で希釈し、得られた有機層を飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥し、 減圧下溶媒を留去した。
[0322] 得られた残渣をシリカゲルカラムクロマトグラフィー(へキサン:酢酸ェチル、 5 : 1, V[0321] After adding acetone to the reaction solution, the solvent was distilled off under reduced pressure. The obtained residue was diluted with ethyl acetate, and the obtained organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. [0322] The obtained residue was subjected to silica gel column chromatography (hexane: ethyl acetate, 5: 1, V
/V)を用いて精製し、標記目的化合物(1. 65g、収率 100%)を得た。 / V) to give the title object compound (1.65 g, yield 100%).
[0323] IR(Liquid film) v 1142, 1213, 1247, 1424, 1486, 1607, 2871, 2949, 3325, 3580 max [0323] IR (Liquid film) v 1142, 1213, 1247, 1424, 1486, 1607, 2871, 2949, 3325, 3580 max
-1 -1
cm ; cm ;
1H NMR(CDC1 , 400MHz) : δ 1.59—1.67 (1H, m), 1.70—1.95 (11H, m), 2.06—2.16 ( 1H NMR (CDC1, 400 MHz): δ 1.59—1.67 (1H, m), 1.70—1.95 (11H, m), 2.06—2.16 (
3 Three
1H, m), 4.68-4.76 (1H, m), 7.13 (1H, dd, J=2.7 Hz, 8.8 Hz), 7.33 (1H, d, J=8.6 Hz ), 7.38 (1H, d, J=2.7 Hz); 1H, m), 4.68-4.76 (1H, m), 7.13 (1H, dd, J = 2.7 Hz, 8.8 Hz), 7.33 (1H, d, J = 8.6 Hz), 7.38 (1H, d, J = 2.7 Hz);
MS(FAB) m/z: 350 M+. MS (FAB) m / z: 350 M + .
(19c)トリフルォロメタンスルホン酸 [4,一(t一ブチルジメチルシリルォキシ)一 3,, 4'ージヒドロー 2' H—スピロ [シクロペンタン 1, 1 '—ナフタレン ]—6'—ィノレ] 実施例 19 (19b)で製造したトリフルォロメタンスルホン酸 (4,一ヒドロキシ一 3,, 4, —ジヒドロ一 2,H—スピロ [シクロペンタン一 1, 1,一ナフタレン]— 6,一ィル)(1. 65 g、 4. 71mmol)をジクロロメタン(25mL)に溶解し、 0°Cで 2, 6—ルチジン(1. 6mL 、 13. 7mmol)及びトリフルォロメタンスルホン酸 tーブチルジメチルシリル(1. 3mL、 5. 66mmol)を加え、 1時間撹拌した。 (19c) Trifluoromethanesulfonic acid [4,1- (t-butyldimethylsilyloxy) -1,3,4, -dihydro-2 'H-spiro [cyclopentane 1,1'-naphthalene] -6'-inore] Implementation Trifluoromethanesulfonic acid prepared in Example 19 (19b) (4,1-hydroxy-1,3,4, —dihydro-1,2, H-spiro [cyclopentane-1,1,1, naphthalene] —6,1 yl) (1.65 g, 4.71 mmol) was dissolved in dichloromethane (25 mL), and 2,6-lutidine (1.6 mL, 13.7 mmol) and t-butyldimethylsilyl trifluoromethanesulfonate (1 3 mL, 5.66 mmol) was added and stirred for 1 hour.
[0324] 反応液に水を加え、酢酸ェチルで抽出した。得られた有機層を飽和炭酸水素ナトリ ゥム水溶液及び飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥し、減圧下溶媒 を留去した。 [0324] Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with saturated aqueous sodium hydrogen carbonate solution and saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure.
[0325] 得られた残渣をシリカゲルカラムクロマトグラフィー (へキサン:酢酸ェチル、 20 : 1、 [0325] The obtained residue was subjected to silica gel column chromatography (hexane: ethyl acetate, 20: 1,
VZV)を用いて精製し、標記目的化合物(2. 10g、収率 96%)を得た。 VZV) to give the title object compound (2.10 g, yield 96%).
[0326] IR(Liquid film) v 1089, 1124, 1144, 1212, 1248, 1362, 1426, 1485, 2861, 2954 max [0326] IR (Liquid film) v 1089, 1124, 1144, 1212, 1248, 1362, 1426, 1485, 2861, 2954 max
-1 -1
cm ; cm ;
1H NMR(CDC1 , 400MHz) δ 0.16 (3H, s), 0.18 (3H, s), 0.96 (9H, s), 1.55-2.05 ( 1H NMR (CDC1, 400MHz) δ 0.16 (3H, s), 0.18 (3H, s), 0.96 (9H, s), 1.55-2.05 (
3 Three
12H, m), 4.70 (1H, dd, J=5.1 Hz, 9.4 Hz), 7.08 (1H, dd, J=2.7 Hz, 9.0 Hz), 7.29 (1 H, d, J=9.0 Hz), 7.30 (1H, d, J=2.7 Hz); 12H, m), 4.70 (1H, dd, J = 5.1 Hz, 9.4 Hz), 7.08 (1H, dd, J = 2.7 Hz, 9.0 Hz), 7.29 (1 H, d, J = 9.0 Hz), 7.30 ( 1H, d, J = 2.7 Hz);
MS(FAB) m/z: 463 (M-H)+. MS (FAB) m / z: 463 (MH) + .
( 19d) 4,一(t一ブチルジメチルシリルォキシ)一 3,, 4,一ジヒドロ一 2, H—スピロ [シ クロペンタン一 1, 1,一ナフタレン]— 6'—力ノレボン酸メチノレ
実施例 19 (19c)で製造したトリフルォロメタンスルホン酸 [4'一(tーブチルジメチル シリルォキシ)一 3,, 4,一ジヒドロ一 2,H—スピロ [シクロペンタン一 1, 1,一ナフタレ ン]— 6,—ィル] (2. 10g、 4. 52mmol)をメタノール(20mL)に溶解し、トリエチルァ ミン(2. OmL、 14. 3mmol)、 1, 3 ビス(ジフエ-ルホスフイノ)プロパン(560mg、 1. 36mmol)及び酢酸パラジウム(304mg、 1. 35mmol)をカ卩え、一酸化炭素置換 をし、一酸化炭素雰囲気下、 60°Cで 21時間撹拌した。 (19d) 4, 1, (t-Butyldimethylsilyloxy) 1, 3, 4, 1, Dihydro 1, 2, H-spiro [cyclopentane 1, 1, 1, naphthalene] — 6'— Example 19 Trifluoromethanesulfonic acid [4 ′-(tert-butyldimethylsilyloxy) -1,3,4,1-dihydro-1,2, H-spiro [cyclopentane-1,1,1, naphthalene] —prepared in 19c 6, -yl] (2. 10 g, 4.52 mmol) was dissolved in methanol (20 mL) and triethylamine (2. OmL, 14.3 mmol), 1,3 bis (diphenylphosphino) propane (560 mg, 1 36 mmol) and palladium acetate (304 mg, 1.35 mmol) were added, carbon monoxide substitution was performed, and the mixture was stirred at 60 ° C. for 21 hours in a carbon monoxide atmosphere.
[0327] 室温まで冷却後、減圧下溶媒を留去した。得られた残渣を酢酸ェチルで希釈し、 得られた有機層を飽和炭酸水素ナトリウム水溶液及び飽和食塩水で洗浄し、無水硫 酸マグネシウムで乾燥し、減圧下溶媒を留去した。 [0327] After cooling to room temperature, the solvent was distilled off under reduced pressure. The obtained residue was diluted with ethyl acetate, and the obtained organic layer was washed with saturated aqueous sodium hydrogen carbonate solution and saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure.
[0328] 得られた残渣をシリカゲルカラムクロマトグラフィー (へキサン:酢酸ェチル、 10 : 1、 VZV)を用いて精製し、標記目的化合物(1. 69g、収率 100%)を得た。 The obtained residue was purified by silica gel column chromatography (hexane: ethyl acetate, 10: 1, VZV) to obtain the title object compound (1.69 g, yield 100%).
[0329] IR(Liquid film) v 1088, 1111, 1251, 1292, 1436, 1472, 1614, 1725, 2859, 2952 max [0329] IR (Liquid film) v 1088, 1111, 1251, 1292, 1436, 1472, 1614, 1725, 2859, 2952 max
1 1
cm ; cm ;
1H NMR(CDC1 , 400MHz) : δ 0.17 (3Η, s), 0.21 (3Η, s), 0.97 (9H, s), 1.56—1.65 ( 1H NMR (CDC1, 400MHz): δ 0.17 (3Η, s), 0.21 (3Η, s), 0.97 (9H, s), 1.56—1.65 (
3 Three
1H, m), 1.65-2.04 (11H, m), 3.89 (3H, s), 4.76 (1H, dd, J=4.7 Hz, 8.6 Hz), 7.30 (1 1H, m), 1.65-2.04 (11H, m), 3.89 (3H, s), 4.76 (1H, dd, J = 4.7 Hz, 8.6 Hz), 7.30 (1
H, d, J=8.6 Hz), 7.85 (1H, dd, J=2.0 Hz, 8.6 Hz), 8.11 (1H, d, J=2.0 Hz); H, d, J = 8.6 Hz), 7.85 (1H, dd, J = 2.0 Hz, 8.6 Hz), 8.11 (1H, d, J = 2.0 Hz);
MS(FAB) m/z: 375 (M+H)+. MS (FAB) m / z: 375 (M + H) + .
(19e) 4,一(tーブチルジメチルシリルォキシ)ー6,ーヒドロキシメチルー 3,, 4,ージ ヒドロ一 2, H—スピロ [シクロペンタン一 1, 1,一ナフタレン] (19e) 4,1 (t-Butyldimethylsilyloxy) -6, -Hydroxymethyl-3,4, -Dihydro-1, H-spiro [Cyclopentane-1,1,1, Naphthalene]
実施例 19 ( 19d)で製造した 4 ' (t プチルジメチルシリルォキシ) 3 ' , 4 ' ジヒ ドロ一 2 ' H スピロ [シクロペンタン一 1, 1 '—ナフタレン] 6 ' カルボン酸メチル( 4 '(t-Ptyldimethylsilyloxy) 3', 4 'Dihydrol 2' H Spiro [cyclopentane-1,1,1'-naphthalene] 6 'Methyl carboxylate prepared in Example 19 (19d)
I. 69g、 4. 51mmol)をテトラヒドロフラン(30mL)に溶解し、 0°Cで水素化アルミ- ゥムリチウム(257mg、 6. 77mmol)をカ卩え、 0.5時間撹拌した。 I. 69 g, 4.51 mmol) was dissolved in tetrahydrofuran (30 mL), and aluminum lithium hydride (257 mg, 6.77 mmol) was added at 0 ° C. and stirred for 0.5 hour.
[0330] 反応液に 1N水酸ィ匕ナトリウム溶液及びセライトを加えた後、ろ過し、減圧下溶媒を [0330] A 1N sodium hydroxide solution and celite were added to the reaction solution, followed by filtration. The solvent was removed under reduced pressure.
*y した。 * y.
[0331] 得られた残渣をシリカゲルカラムクロマトグラフィー(へキサン:酢酸ェチル、 3 : 1、 V [0331] The obtained residue was subjected to silica gel column chromatography (hexane: ethyl acetate, 3: 1, V
ZV)を用いて精製し、標記目的化合物(1. 51g、収率 97%)を得た。 ZV) to give the title object compound (1.51 g, yield 97%).
[0332] IR(Liquid film) v 1037, 1085, 1253, 1361, 1462, 1472, 1496, 2859, 2952, 3362
-1 [0332] IR (Liquid film) v 1037, 1085, 1253, 1361, 1462, 1472, 1496, 2859, 2952, 3362 -1
cm ; cm ;
1H NMR(CDC1 , 500MHz) δ 0.16 (3H, s), 0.18 (3H, s), 0.95 (9H, s), 1.50 (IH, t, 1H NMR (CDC1, 500MHz) δ 0.16 (3H, s), 0.18 (3H, s), 0.95 (9H, s), 1.50 (IH, t,
3 Three
J=5.9 Hz), 1.55-1.62 (IH, m), 1.66-1.92 (9H, m), 1.92-2.02 (2H, m), 4.64 (2H, d, J=8.6 Hz), 4.75 (IH, dd, J=4.9 Hz, 8.8 Hz), 7.23 (IH, dd, J=2.0 Hz, 8.3 Hz), 7.26 ( IH, d, J=8.3 Hz), 7.38 (IH, d, J=2.0 Hz); J = 5.9 Hz), 1.55-1.62 (IH, m), 1.66-1.92 (9H, m), 1.92-2.02 (2H, m), 4.64 (2H, d, J = 8.6 Hz), 4.75 (IH, dd , J = 4.9 Hz, 8.8 Hz), 7.23 (IH, dd, J = 2.0 Hz, 8.3 Hz), 7.26 (IH, d, J = 8.3 Hz), 7.38 (IH, d, J = 2.0 Hz);
MS(FAB) m/z: 345 (M— H)+. MS (FAB) m / z: 345 (M— H) +.
(19f) 6,ーブロモメチルー 4 tーブチルジメチルシリルォキシ)ー3 4 ジヒド 2 H—スピロ [シクロペンタン 1, 1,一ナフタレン] (19f) 6, -Bromomethyl-4 t-butyldimethylsilyloxy) -3 4 Dihydr 2 H-spiro [cyclopentane 1, 1, mononaphthalene]
実施例 19 ( 19e)で製造した 4,一(t ブチルジメチルシルォキシ) 6 ヒドロキシ メチ 3 4 ジヒドロ一 2 H—スピロ [シクロペンタン一 1, 1 ナフタレン] (1. 64g 4. 71mmol)を使用して、実施例 15 (15d)に記載した方法に従い、標記目的 化合物(1. 68g、収率 94%)を得た。 Using 4,1- (t-butyldimethylsiloxy) 6-hydroxymethyl 34-dihydro-1 2 H-spiro [cyclopentane-1,1, naphthalene] (1.64 g 4.71 mmol) prepared in Example 19 (19e) Then, according to the method described in Example 15 (15d), the title object compound (1.68 g, yield 94%) was obtained.
[0333] IR(Liquid film) v 1087, 1252, 1333, 1361, 1445, 1462, 1472, 1494, 2858, 2953 max [0333] IR (Liquid film) v 1087, 1252, 1333, 1361, 1445, 1462, 1472, 1494, 2858, 2953 max
-1 -1
cm ; cm ;
1H NMR(CDC1 , 400MHz) δ 0.16 (3H, s), 0.19 (3H, s), 0.96 (9H, s), 1.52-1.62 ( 1H NMR (CDC1, 400MHz) δ 0.16 (3H, s), 0.19 (3H, s), 0.96 (9H, s), 1.52-1.62 (
3 Three
IH, m), 1.64-1.89 (9H, m), 1.89-2.02 (2H, m), 4.47 (IH, d, J=10.2 Hz), 4.49 (IH, d , J=10.2 Hz), 4.73 (IH, dd, J=4.7 Hz, 8.6 Hz), 7.21-7.27 (2H, m), 7.41 (IH, s); MS(FAB) m/z: 407, 409 (M— H)+. IH, m), 1.64-1.89 (9H, m), 1.89-2.02 (2H, m), 4.47 (IH, d, J = 10.2 Hz), 4.49 (IH, d, J = 10.2 Hz), 4.73 (IH , dd, J = 4.7 Hz, 8.6 Hz), 7.21-7.27 (2H, m), 7.41 (IH, s); MS (FAB) m / z: 407, 409 (M- H) +.
(19g) [4 tーブチルジメチルシリルォキシ) 3 4,ージヒドロー 2 H—スピロ [ シクロペンタン 1, 1 ナフタレン ]—6' ィル]メチルホスホン酸ジェチル (19g) [4 t-Butyldimethylsilyloxy) 3 4, -dihydro-2 H-spiro [cyclopentane 1,1 naphthalene] -6'yl] methyl phosphonate
実施例 19 ( 19f )で製造した 6 ' -ブロモメチル 4 —(t ブチルジメチルシリルォ キシ)ー3 4,ージヒドロー 2 H—スピロ [シクロペンタン 1, 1,一ナフタレン] (1. 6 8g 4. lOmmol)を使用して、実施例 15 (15d)に記載した方法に従い、標記目的化 合物(1. 86g、収率 97%)を得た。 6′-Bromomethyl 4 — (t butyldimethylsilyloxy) -3 4,4-dihydro-2 H-spiro [cyclopentane 1,1, mononaphthalene] (1.6 6 g 4. lOmmol) prepared in Example 19 (19f) ) To give the title object compound (1.86 g, 97% yield) according to the method described in Example 15 (15d).
[0334] IR(Liquid film) v 1031, 1057, 1254, 1362, 1391, 1467, 1473, 1497, 2859, 2952 max [0334] IR (Liquid film) v 1031, 1057, 1254, 1362, 1391, 1467, 1473, 1497, 2859, 2952 max
-1 -1
cm ; cm ;
XH NMR(CDC1 500MHz): δ 0.15 (3Η s), 0.17 (3Η s), 0.94 (9Η s), 1.25 (3Η t X H NMR (CDC1 500MHz): δ 0.15 (3Η s), 0.17 (3Η s), 0.94 (9Η s), 1.25 (3Η t
3 Three
J=7.0 Hz), 1.26 (3H t J=7.0 Hz), 1.52-1.60 (1H m), 1.62-2.00 (11H, m), 3.10 (2
H, d, J=21.1 Hz), 3.97-4.08 (4H, m), 4.72 (1H, dd, J=4.7 Hz, 8.2 Hz), 7.14-7.25 (3 H, m); J = 7.0 Hz), 1.26 (3H t J = 7.0 Hz), 1.52-1.60 (1H m), 1.62-2.00 (11H, m), 3.10 (2 H, d, J = 21.1 Hz), 3.97-4.08 (4H, m), 4.72 (1H, dd, J = 4.7 Hz, 8.2 Hz), 7.14-7.25 (3 H, m);
MS(FAB) m/z: 465 (M-H)+. MS (FAB) m / z: 465 (MH) + .
( 19h) [4,一(t一ブチルジメチルシリルォキシ)一 3,, 4,一ジヒドロ一 2, H—スピロ [ シクロペンタン 1, 1 '—ナフタレン ]—6' ィル]ジフルォロメチルホスホン酸ジェチ ル (19h) [4,1 (t-Butyldimethylsilyloxy) -1,3,4,1-dihydro-1,2, H-spiro [cyclopentane 1,1'-naphthalene] -6'yl] difluoromethylphosphone Acid jet
実施例 19 ( 19g)で製造した [4,—(t プチルジメチルシリルォキシ) 3,, 4, ジ ヒドロ一 2,H—スピロ [シクロペンタン一 1, 1,一ナフタレン]— 6,一ィル]メチルホスホ ン酸ジェチル(1. 86g、 3. 99mmol)を使用して、実施例 15 (15e)に記載した方法 に従い、標記目的化合物(1. 69g、収率 84%)を得た。 [4,-(t-butyldimethylsilyloxy) 3,4, dihydro-1, H-spiro [cyclopentane-1,1,1, naphthalene] -6,1 The title object compound (1.69 g, yield 84%) was obtained by following the method described in Example 15 (15e) using [ethyl] l] methylphosphonate (1.86 g, 3.99 mmol).
[0335] IR(Liquid film) v 1023, 1048, 1124, 1272, 1362, 1392, 1473, 2860, 2953 cm"1; [0335] IR (Liquid film) v 1023, 1048, 1124, 1272, 1362, 1392, 1473, 2860, 2953 cm "1;
max max
1H NMR(CDC1 , 400MHz) δ 0.16 (3H, s), 0.18 (3H, s), 0.95 (9H, s), 1.30 (3H, t, 1H NMR (CDC1, 400MHz) δ 0.16 (3H, s), 0.18 (3H, s), 0.95 (9H, s), 1.30 (3H, t,
3 Three
J=7.0 Hz), 1.33 (3H, t, J=7.0 Hz), 1.55—1.63 (1H, m), 1.66—2.02 (11H, m), 4.08-4. 30 (4H, m), 4.75 (1H, dd, J=4.7 Hz, 8.2 Hz), 7.32 (1H, d, J=8.6 Hz), 7.45 (1H, d, J =8.6 Hz), 7.62 (1H, d, J=8.6 Hz); J = 7.0 Hz), 1.33 (3H, t, J = 7.0 Hz), 1.55—1.63 (1H, m), 1.66—2.02 (11H, m), 4.08-4.30 (4H, m), 4.75 (1H , dd, J = 4.7 Hz, 8.2 Hz), 7.32 (1H, d, J = 8.6 Hz), 7.45 (1H, d, J = 8.6 Hz), 7.62 (1H, d, J = 8.6 Hz);
MS(FAB) m/z: 501 (M-H)+. MS (FAB) m / z: 501 (MH) + .
(19i) (4,一ヒドロキシ一 3,, 4,一ジヒドロ一 2,H—スピロ [シクロペンタン一 1, 1,一 ナフタレン] 6'—ィル)ジフルォロメチルホスホン酸ジェチル (19i) (4,1-hydroxy-1,3,4,1-dihydro-1,2, H-spiro [cyclopentane-1,1,1,1-naphthalene] 6'-yl) difluoromethylphosphonate jetyl
実施例 19 ( 19h)で製造した [4,一(t ブチルジメチルシロキシ) 3,, 4,一ジヒド 口一 2' H—スピロ [シクロペンタン一 1, 1,一ナフタレン]— 6'—ィル]ジフルォロメチ ルホスホン酸ジェチル(1. 69g、 3. 36mmol)をテトラヒドロフラン(10mL)に溶解し 、 1. OMフッ化テトラブチルアンモ-ゥム一テトラヒドロフラン溶液(4. 0mL、 4. OOm mol)を加え、室温で 6.5時間撹拌した。 [19] (4, 1 (t-butyldimethylsiloxy) 3, 4, 1 dihydride 1 'H-spiro [cyclopentane-1,1,1, naphthalene] -6'-yl prepared in Example 19 (19h) ] Dissolve difluoromethyl phosphonate (1.69 g, 3.36 mmol) in tetrahydrofuran (10 mL), 1. Add OM tetrabutylammonium monotetrahydrofuran solution (4.0 mL, 4. OOm mol), Stir at room temperature for 6.5 hours.
[0336] 反応液を減圧下溶媒を留去し、得られた残渣を酢酸ェチルで希釈後、得られた有 機層を水及び飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥し、減圧下溶媒を[0336] The solvent was removed from the reaction solution under reduced pressure, and the resulting residue was diluted with ethyl acetate. The resulting organic layer was washed with water and saturated brine, dried over anhydrous magnesium sulfate, and dried under reduced pressure. Solvent
*y した。 * y.
[0337] 得られた残渣をシリカゲルカラムクロマトグラフィー(へキサン:酢酸ェチル、 2 : 1, V ZV)を用いて精製し、標記目的化合物(1. 05g、収率 81%)を得た。
[0338] IR(KBr) v 1015, 1042, 1060, 1257, 1392, 1412, 1448, 1478, 2869, 2942, 3420 max The obtained residue was purified by silica gel column chromatography (hexane: ethyl acetate, 2: 1, V ZV) to obtain the title object compound (1.05 g, yield 81%). [0338] IR (KBr) v 1015, 1042, 1060, 1257, 1392, 1412, 1448, 1478, 2869, 2942, 3420 max
-1 -1
cm ; cm ;
1H NMR(CDC1 , 500MHz) : δ 1.335 (3H, t, J=7.0 Hz), 1.340 (3H, t, J=7.0 Hz), 1. 1H NMR (CDC1, 500 MHz): δ 1.335 (3H, t, J = 7.0 Hz), 1.340 (3H, t, J = 7.0 Hz), 1.
3 Three
60-1.68 (IH, m), 1.74-1.95 (11H, m), 2.03-2.11 (IH, m), 4.14-4.30 (4H, m), 4.74- 4.79 (IH, m), 7.36 (IH, d, J=8.3 Hz), 7.48 (IH, d, J=8.3 Hz), 7.67 (IH, s); 60-1.68 (IH, m), 1.74-1.95 (11H, m), 2.03-2.11 (IH, m), 4.14-4.30 (4H, m), 4.74- 4.79 (IH, m), 7.36 (IH, d , J = 8.3 Hz), 7.48 (IH, d, J = 8.3 Hz), 7.67 (IH, s);
MS(EI) m/z: 388 M+. MS (EI) m / z: 388 M + .
(19j) (4' ォキソ 3' , 4'—ジヒドロー 2' H—スピロ [シクロペンタン 1, 1 ' ナフ タレン ]—6 '—ィル)ジフルォロメチルホスホン酸ジェチル (19j) (4'oxo3 ', 4'-dihydro-2'H-spiro [cyclopentane 1,1'naphthalene] -6'-yl) difluoromethylphosphonate jetyl
実施例 19 ( 19i)で製造した(4, -ヒドロキシ 3,, 4,一ジヒドロ 2, H スピロ [シ クロペンタン 1, 1 '—ナフタレン ]—6' ィル)ジフルォロメチルホスホン酸ジェチル (1. 05g、 2. 70mmol)をジクロロメタン(20mL)に溶解し、二酸化マンガン(10. 5g 、 121mmol)を加え、室温で 2.5時間撹拌した。 Example 19 (19i) (4, -hydroxy 3,4, monodihydro 2, H spiro [cyclopentane 1,1'-naphthalene] -6'yl) difluoromethylphosphonate jetyl (1. 05 g, 2.70 mmol) was dissolved in dichloromethane (20 mL), manganese dioxide (10.5 g, 121 mmol) was added, and the mixture was stirred at room temperature for 2.5 hours.
[0339] 反応液をろ過し、減圧下溶媒を留去した。 [0339] The reaction solution was filtered, and the solvent was distilled off under reduced pressure.
[0340] 得られた残渣をシリカゲルカラムクロマトグラフィー(へキサン:酢酸ェチル、 1 : 1, V [0340] The obtained residue was subjected to silica gel column chromatography (hexane: ethyl acetate, 1: 1, V
/V)を用いて精製し、標記目的化合物(959mg、収率 92%)を得た。 / V) to give the title object compound (959 mg, yield 92%).
[0341] IR(Liquid film) v 1021, 1043, 1274, 1612, 1691, 2873, 2954 cm"1; [0341] IR (Liquid film) v 1021, 1043, 1274, 1612, 1691, 2873, 2954 cm "1;
max max
1H NMR(CDC1 , 500MHz) δ 1.34 (6H, t, J=7.0 Hz), 1.80—2.02 (8H, m),2.08 (2H, 1H NMR (CDC1, 500MHz) δ 1.34 (6H, t, J = 7.0 Hz), 1.80—2.02 (8H, m), 2.08 (2H,
3 Three
dd, J=6.8 Hz, 6.8 Hz), 2.73 (2H, dd, J=6.8 Hz, 6.8 Hz), 4.17-4.30 (4H, m), 7.46 (1 H, d, J=8.3 Hz), 7.77 (IH, d, J=8.3 Hz), 8.23 (IH, s); dd, J = 6.8 Hz, 6.8 Hz), 2.73 (2H, dd, J = 6.8 Hz, 6.8 Hz), 4.17-4.30 (4H, m), 7.46 (1 H, d, J = 8.3 Hz), 7.77 ( IH, d, J = 8.3 Hz), 8.23 (IH, s);
MS(EI) m/z: 386 M+. MS (EI) m / z: 386 M + .
(19k) (3,一ブロモー 4,一ォキソ一 3,, 4,一ジヒドロ一 2, H—スピロ [シクロペンタン — 1, 1 ' ナフタレン ]—6' ィル)ジフルォロメチルホスホン酸ジェチル (19k) (3,1-bromo-4,1-oxo-3,4,1-dihydro-1,2, H-spiro [cyclopentane — 1, 1 'naphthalene] -6'yl) difluoromethylphosphonate jetyl
実施例 19 ( 1 ¾ )で製造した (4, -ォキソ 3,, 4,一ジヒドロ一 2, H スピロ [シクロ ペンタン 1, 1 '—ナフタレン ]—6' ィル)ジフルォロメチルホスホン酸ジェチル(4 57mg、 1. 18mmol)を使用して、実施例 3 (3a)に記載した方法に従い、標記目的 化合物(528mg、収率 96%)を得た。 Example 19 (1, ¾) (4, -oxo 3,4,1 dihydro-1,2, H spiro [cyclopentane 1,1'-naphthalene] -6 'yl) difluoromethylphosphonate jetyl ( 4 57 mg, 1.18 mmol) was used to give the title object compound (528 mg, 96% yield) according to the method described in Example 3 (3a).
[0342] IR(KBr) v 1026, 1071, 1133, 1174, 1202, 1268, 1613, 1695, 2874, 2965 cm"1; [0342] IR (KBr) v 1026, 1071, 1133, 1174, 1202, 1268, 1613, 1695, 2874, 2965 cm "1;
max max
1H NMR(CDC1 , 400MHz) δ 1.35 (3H, t, J=7.0 Hz), 1.36 (3H, t, J=7.0 Hz), 1.68
-1.78 (IH, m), 1.78-2.01 (5H, m), 2.08—2.18 (IH, m), 2.24-2.34 (IH, m), 2.55-2.65 (2H, m), 4.16-4.34 (4H, m), 5.04 (IH, dd, J=7.4 Hz, 11.3 Hz), 7.46 (IH, d, J=8.21H NMR (CDC1, 400MHz) δ 1.35 (3H, t, J = 7.0 Hz), 1.36 (3H, t, J = 7.0 Hz), 1.68 -1.78 (IH, m), 1.78-2.01 (5H, m), 2.08—2.18 (IH, m), 2.24-2.34 (IH, m), 2.55-2.65 (2H, m), 4.16-4.34 (4H, m), 5.04 (IH, dd, J = 7.4 Hz, 11.3 Hz), 7.46 (IH, d, J = 8.2
Hz), 7.82 (IH, d, J=8.2 Hz), 8.26 (IH, s); Hz), 7.82 (IH, d, J = 8.2 Hz), 8.26 (IH, s);
MS(FAB) m/z: 465, 467 (M+H)+. MS (FAB) m / z: 465, 467 (M + H) + .
(191) (2,一アミノー 4,H—スピロ [シクロペンタン一 1, 5,一ナフト[1, 2 d] [l, 3] チアゾール] 8'—ィル)ジフルォロメチルホスホン酸ジェチル (191) (2,1-Amino-4, H-spiro [cyclopentane-1,5,1-naphtho [1,2d] [l, 3] thiazole] 8'-yl) difluoromethylphosphonate jetyl
実施例 19 (19k)で製造した(3,ーブロモー 4,一ォキソー3,, 4,ージヒドロー 2,H —スピロ [シクロペンタン一 1, 1,一ナフタレン]— 6'—ィル)ジフルォロメチルホスホ ン酸ジェチル(103mg、 0. 221mmol)を使用して、実施例 1 (lb)に記載した方法 に従 、、標記目的化合物(52mg、収率 53%)を得た。 (3, -Bromo-4,1oxo3,4, -dihydro-2, H-spiro [cyclopentane-1,1,1, naphthalene] -6'-yl) difluoromethyl prepared in Example 19 (19k) The title object compound (52 mg, 53% yield) was obtained by using the method described in Example 1 (lb) using jetyl phosphonate (103 mg, 0.221 mmol).
[0343] IR(KBr) v 1020, 1126, 1162, 1239, 1259, 1358, 1652, 2874, 2958, 3133, 3346 max [0343] IR (KBr) v 1020, 1126, 1162, 1239, 1259, 1358, 1652, 2874, 2958, 3133, 3346 max
-1 -1
cm ; cm ;
1H NMR(CDC1 , 500MHz): δ 1.32 (6Η, t, J=7.0 Hz), 1.73—1.85 (6H, m), 1.90—2.0 1H NMR (CDC1, 500MHz): δ 1.32 (6Η, t, J = 7.0 Hz), 1.73—1.85 (6H, m), 1.90—2.0
3 Three
0 (2H, m), 2.79 (2H, s), 4.11—4.28 (4H, m), 5.74—5.81 (2H, m), 7.31 (IH, d, J=8.3 Hz), 7.43 (IH, d, J=8.3 Hz), 7.93 (IH, s); 0 (2H, m), 2.79 (2H, s), 4.11—4.28 (4H, m), 5.74—5.81 (2H, m), 7.31 (IH, d, J = 8.3 Hz), 7.43 (IH, d, J = 8.3 Hz), 7.93 (IH, s);
MS(FAB) m/z: 443 (M+H)+. MS (FAB) m / z: 443 (M + H) + .
(19m) (2,一アミノー 4,H—スピロ [シクロペンタン一 1, 5,一ナフト[1, 2 d] [l, 3 ]チアゾール] 8,一ィル)ジフルォロメチルホスホン酸 (19m) (2,1amino-4, H-spiro [cyclopentane-1,5,1naphtho [1,2d] [l, 3] thiazole] 8,1yl) difluoromethylphosphonic acid
実施例 19 ( 191)で製造した(2,一ァミノ一 4, H スピロ [シクロペンタン 1, 5,一 ナフト[1, 2-d] [l, 3]チアゾール] 8,一ィル)ジフルォロメチルホスホン酸ジェチ ル(28mg、 0. O63mmol)を使用して、実施例 1 (lc)に記載した方法に従い、標記 目的化合物(17mg、収率 70%)を得た。 (2,1, amino-1,4 H spiro [cyclopentane 1,5,1 naphtho [1,2-d] [l, 3] thiazole] 8,1 yl) difluro prepared in Example 19 (191) The title compound (17 mg, yield 70%) was obtained according to the method described in Example 1 (lc) using ethyl lomethylphosphonate (28 mg, 0.063 mmol).
[0344] IR(KBr) v 1078, 1178, 1240, 1595, 1633, 2744, 2876, 2952, 3067, 3290, 3481 max [0344] IR (KBr) v 1078, 1178, 1240, 1595, 1633, 2744, 2876, 2952, 3067, 3290, 3481 max
-1 -1
cm ; cm ;
XH NMR(DMSO-d , 500MHz): δ 1.59-1.80 (8H, m), 2.57 (2Η, br s), 7.27 (1H, d, X H NMR (DMSO-d, 500MHz): δ 1.59-1.80 (8H, m), 2.57 (2Η, br s), 7.27 (1H, d,
6 6
J=7.8 Hz), 7.40 (1H, d, J=7.8 Hz), 7.97 (1H, s); J = 7.8 Hz), 7.40 (1H, d, J = 7.8 Hz), 7.97 (1H, s);
MS(FAB) m/z: 385 (M— H)+.
<実施例 20> MS (FAB) m / z: 385 (M— H) +. <Example 20>
リン酸 二水素 (4, 5 ジヒドロナフト[1, 2— d] [l, 3]チアゾールー 8 ィル)(例 示化合物番号 2-13) Dihydrogen phosphate (4,5 dihydronaphtho [1,2-d] [l, 3] thiazole-8 yl) (example compound number 2-13)
(20a)リン酸 ジェチル (4, 5 ジヒドロナフト[1, 2— d] [l, 3]チアゾールー 8—ィ ル) (20a) Jetyl phosphate (4,5 dihydronaphtho [1,2-d] [l, 3] thiazole-8-yl)
実施例 14 (14a)で製造したリン酸 ジェチル (8—ォキソ 5, 6, 7, 8—テトラヒド ロナフタレン— 2—ィル)(262mg、 0. 878mmol)を使用して、実施例 3 (3a— 3b)に 記載した方法に従い、標記目的化合物(94. 4mg、収率 32%)を得た。 Example 3 (3a) was used using the decyl phosphate (8-oxo-5,6,7,8-tetrahydronaphthalene-2-yl) (262 mg, 0.887 mmol) prepared in Example 14 (14a). — The title object compound (94.4 mg, yield 32%) was obtained according to the method described in 3b).
[0345] IR(Liquid film) v 922, 985, 1031, 1273, 1496, 1610, 2985, 3480 cm"1; [0345] IR (Liquid film) v 922, 985, 1031, 1273, 1496, 1610, 2985, 3480 cm "1;
max max
1H NMR(CDC1 , 400MHz) δ 1.37 (6H, t, J=7.0 Hz), 3.04 (2H x2, s x2), 4.19—4.2 1H NMR (CDC1, 400MHz) δ 1.37 (6H, t, J = 7.0 Hz), 3.04 (2H x2, s x2), 4.19—4.2
3 Three
6 (4H, m), 7.09-7.11 (IH, m), 7.17 (IH, d, J=8.2 Hz), 7.73-7.74 (IH, m), 8.65 (IH, s); 6 (4H, m), 7.09-7.11 (IH, m), 7.17 (IH, d, J = 8.2 Hz), 7.73-7.74 (IH, m), 8.65 (IH, s);
MS(FAB) m/z: 340 (M+H)+. MS (FAB) m / z: 340 (M + H) + .
(20b)リン酸 二水素 (4, 5 ジヒドロナフト[1, 2— d] [l, 3]チアゾールー 8—ィル ) (20b) Dihydrogen phosphate (4, 5 dihydronaphtho [1, 2—d] [l, 3] thiazole-8-yl)
実施例 20 (20a)で製造したリン酸 ジェチル (4, 5 ジヒドロナフト[1, 2— d] [l , 3]チアゾール—8—ィル)(88. 9mg、 0. 262mmol)を使用して、実施例 l (lc)に 記載した方法に従 、、標記目的化合物 (44. lmg、収率 59%)を得た。 Using Jetyl Phosphate (4,5 Dihydronaphtho [1,2-d] [l, 3] thiazol-8-yl) (88.9 mg, 0.262 mmol) prepared in Example 20 (20a) According to the method described in Example l (lc), the title object compound (44. lmg, yield 59%) was obtained.
[0346] IR(KBr) v 984, 1204, 1497, 2839, 2896, 2937, 3089, 3422 cm"1; [0346] IR (KBr) v 984, 1204, 1497, 2839, 2896, 2937, 3089, 3422 cm "1;
max max
1H NMR(DMSO-d , 400MHz): δ 2.95—3.06 (4H, m), 6.97 (IH, ddd, J=1.2 Hz, 2.7 1H NMR (DMSO-d, 400 MHz): δ 2.95—3.06 (4H, m), 6.97 (IH, ddd, J = 1.2 Hz, 2.7
6 6
Hz, 8.2 Hz), 7.24 (IH, d, J=8.2 Hz), 7.64 (IH, dd, J=1.2 Hz, 2.3 Hz), 8.99 (IH, s); MS(FAB) m/z: 282 (M— H)— . Hz, 8.2 Hz), 7.24 (IH, d, J = 8.2 Hz), 7.64 (IH, dd, J = 1.2 Hz, 2.3 Hz), 8.99 (IH, s); MS (FAB) m / z: 282 ( M— H) —.
<実施例 21 > <Example 21>
リン酸 二水素 (2 アミノー 5, 6 ジヒドロー 4H べンゾ [6, 7]シクロへプタ [1, 2 -d] [l, 3]チアゾールー 9 ィル)(例示化合物番号 2-27) Dihydrogen phosphate (2 amino-5, 6 dihydro-4H benzo [6, 7] cyclohepta [1, 2 -d] [l, 3] thiazole-9 yl) (exemplary compound number 2-27)
(21a)リン酸 ジェチル (9 ォキソ 6, 7, 8, 9ーテトラヒドロー 5H べンゾ [7]ァ ヌレン一 2—ィノレ)
3 ヒドロキシ一 6, 7, 8, 9—テトラヒドロー 5H ベンゾ [7]ァヌレン一 5—オン(19 9mg、 1. 13mmol)を使用して、実施例 13 (13a)に記載の方法に従い、標記目的 化合物(321mg、 91%)を得た。 (21a) Jetyl phosphate (9 oxo 6, 7, 8, 9-tetrahydro-5H benzo [7] nulene 2-ynole) 3 Hydroxy-1,6,8,9-tetrahydro-5H Benzo [7] anulene-1-5-one (19 9 mg, 1.13 mmol) was used according to the method described in Example 13 (13a) to give the title object compound (321 mg, 91%) was obtained.
[0347] IR(Liquid film) v 960, 982, 1032, 1267, 1488, 1679, 2941 cm"1; [0347] IR (Liquid film) v 960, 982, 1032, 1267, 1488, 1679, 2941 cm "1;
max max
1H NMR(CDC1 , 500MHz): δ 1.37 (6H, t, J=7.3 Hz), 1.78—1.90 (4H, m), 2.71-2.7 1H NMR (CDC1, 500MHz): δ 1.37 (6H, t, J = 7.3 Hz), 1.78—1.90 (4H, m), 2.71-2.7
3 Three
5 (2H, m), 2.91 (2H, dd, J=5.9 Hz, 5.9 Hz), 4.18-4.27 (4H, m), 7.18 (IH, d, J=8.8 Hz), 7.29-7.33 (IH, m), 7.51-7.54 (IH, m); 5 (2H, m), 2.91 (2H, dd, J = 5.9 Hz, 5.9 Hz), 4.18-4.27 (4H, m), 7.18 (IH, d, J = 8.8 Hz), 7.29-7.33 (IH, m ), 7.51-7.54 (IH, m);
MS(FAB) m/z: 313 (M+H)+. MS (FAB) m / z: 313 (M + H) + .
(21b)リン酸 (2 アミノー 5, 6 ジヒドロ一 4H ベンゾ [6, 7]シクロへプタ [1, 2— d] [l, 3]チアゾールー 9 ィル) ジェチル (21b) Phosphate (2 amino-5, 6 dihydro-l 4H benzo [6,7] cyclohepta [1,2-d] [l, 3] thiazole-9yl) Jetyl
実施例 21 (21a)で製造したリン酸 ジェチル (9 ォキソ 6, 7, 8, 9—テトラヒド 口一 5H ベンゾ [7]ァヌレン一 2—ィル)(282mg、 0. 903mmol)を使用して、実施 例 2 (2b)に記載した方法に従い、標記目的化合物 (106mg、収率 32%)を得た。 Using Jetyl Phosphate (9 oxo 6, 7, 8, 9-tetrahydr 1H benzo [7] annulene 2-yl) (282 mg, 0.903 mmol) prepared in Example 21 (21a) The title object compound (106 mg, yield 32%) was obtained according to the method described in Example 2 (2b).
[0348] IR(KBr) v 987, 1035, 1266, 1558, 1654, 3119, 3290 cm"1; [0348] IR (KBr) v 987, 1035, 1266, 1558, 1654, 3119, 3290 cm "1;
max max
1H NMR(CD OD, 500MHz): δ 1.35 (6H, t, J=7.3 Hz), 2.05—2.14 (2H, m), 2.77-2. 1H NMR (CD OD, 500 MHz): δ 1.35 (6H, t, J = 7.3 Hz), 2.05—2.14 (2H, m), 2.77-2.
3 Three
82 (4H, m), 4.23 (4H, dq, J=7.3 Hz, 7.3 Hz), 6.99—7.03 (IH, m), 7.17 (IH, d, J=7.8 Hz), 7.63-7.67 (IH, m); 82 (4H, m), 4.23 (4H, dq, J = 7.3 Hz, 7.3 Hz), 6.99—7.03 (IH, m), 7.17 (IH, d, J = 7.8 Hz), 7.63-7.67 (IH, m );
MS(FAB) m/z: 369 (M+H)+. MS (FAB) m / z: 369 (M + H) + .
(21c)リン酸 二水素 (2 アミノー 5, 6 ジヒドロー 4H べンゾ [6, 7]シクロヘプ タ [1, 2-d] [l, 3]チアゾールー 9 ィル) (21c) Dihydrogen phosphate (2 amino-5, 6 dihydro-4H benzo [6, 7] cyclohepta [1, 2-d] [l, 3] thiazole-9 yl)
実施例 21 (21b)で製造したリン酸 (2 アミノー 5, 6 ジヒドロ一 4H ベンゾ [6, 7]シクロへプタ [1, 2-d] [l, 3]チアゾールー 9 ィル) ジェチル(65. 4mg、 0. 1 78mmol)を使用して、実施例 1 (lc)に記載した方法に従い、標記目的化合物 (47. 5mg、収率 85%)を得た。 Phosphoric acid prepared in Example 21 (21b) (2 amino-5,6 dihydro-l 4H benzo [6,7] cyclohepta [1,2-d] [l, 3] thiazol-9 yl) jetyl (65. 4 mg, 0.1 78 mmol) was used to give the title object compound (47.5 mg, 85% yield) according to the method described in Example 1 (lc).
[0349] IR(KBr) v 949, 1052, 1225, 1506, 1635, 2937 cm"1; [0349] IR (KBr) v 949, 1052, 1225, 1506, 1635, 2937 cm "1;
max max
1H NMR(DMSO-d , 500MHz): δ 1.90—1.98 (2H, m), 2.69-2.74 (2H, m), 2.78 (2H 1H NMR (DMSO-d, 500MHz): δ 1.90—1.98 (2H, m), 2.69-2.74 (2H, m), 2.78 (2H
6 6
, dd, J=6.8 Hz, 6.8 Hz), 6.87—6.92 (IH, m), 6.95—7.04 (2H, br), 7.10 (IH, d, J=7.8 Hz), 7.66-7.71 (IH, m);
MS(FAB) m/z: 311 (M— H)— . , dd, J = 6.8 Hz, 6.8 Hz), 6.87—6.92 (IH, m), 6.95—7.04 (2H, br), 7.10 (IH, d, J = 7.8 Hz), 7.66-7.71 (IH, m) ; MS (FAB) m / z: 311 (M— H) —.
<実施例 22 > <Example 22>
リン酸 二水素 (5, 6 ジヒドロー 4H べンゾ [6, 7]シクロへプタ [1, 2— d] [l, 3] チアゾールー 9 ィル)(例示化合物番号 2-14) Dihydrogen phosphate (5, 6 dihydro-4H benzo [6, 7] cyclohepta [1, 2-d] [l, 3] thiazole-9 yl) (Exemplified Compound No. 2-14)
(22a)リン酸 ジェチル (5, 6 ジヒドロー 4H—べンゾ [6, 7]シクロへプタ [1, 2— d] [l, 3]チアゾールー 9 ィル) (22a) Jetyl phosphate (5, 6 dihydro-4H-benzo [6, 7] cyclohepta [1, 2—d] [l, 3] thiazole-9 yl)
実施例 21 (21a)で製造したリン酸 ジェチル (9 ォキソ 6, 7, 8, 9—テトラヒド 口一 5H ベンゾ [7]ァヌレン一 2—ィル)(321mg、 1. 03mmol)を使用して、実施 例 3 (3a— 3b)に記載した方法に従い、標記目的化合物(19. 3mg、収率 5%)を得 た。 Using Jetyl phosphate prepared in Example 21 (21a) (9 oxo 6, 7, 8, 9-tetrahydr 1H benzo [7] annulene 2-yl) (321 mg, 1.03 mmol) The title object compound (19.3 mg, yield 5%) was obtained according to the method described in Example 3 (3a-3b).
[0350] 1H NMR(CDC1 , 400MHz) δ 1.36 (6Η, dt, J=7.0 Hz, 7.0 Hz), 2.16-2.22 (2H, m), [0350] 1H NMR (CDC1, 400MHz) δ 1.36 (6Η, dt, J = 7.0 Hz, 7.0 Hz), 2.16-2.22 (2H, m),
3 Three
2.75-2.78 (2H, m), 3.01 (2H, t, J=7.0 Hz), 4.18—4.26 (4H, m), 7.12-7.13 (2H, m), 7.85 (IH, s), 8.65 (IH, s); 2.75-2.78 (2H, m), 3.01 (2H, t, J = 7.0 Hz), 4.18—4.26 (4H, m), 7.12-7.13 (2H, m), 7.85 (IH, s), 8.65 (IH, s);
MS(FAB) m/z: 354 (M+H)+. MS (FAB) m / z: 354 (M + H) + .
(22b)リン酸 二水素 (5, 6 ジヒドロー 4H べンゾ [6, 7]シクロへプタ [1, 2— d] [1, 3]チアゾールー 9 ィル) (22b) Dihydrogen phosphate (5, 6 dihydro-4H benzo [6, 7] cyclohepta [1, 2-d] [1, 3] thiazole-9 yl)
実施例 22 (22a)で製造したリン酸 ジェチル (5, 6 ジヒドロー 4H べンゾ [6, 7 ]シクロへプタ [1, 2-d] [l, 3]チアゾール 9—ィル)(18. 7mg、 0. 053mmol)を 使用して、実施例 l (lc)に記載した方法に従い、標記目的化合物(11. 3mg、収率 72%)を得た。 Jetyl phosphate (5, 6 dihydro-4H benzo [6, 7] cyclohepta [1, 2-d] [l, 3] thiazole 9-yl) prepared in Example 22 (22a) (18. The title object compound (11.3 mg, yield 72%) was obtained according to the method described in Example 1 (lc) using 7 mg, 0.053 mmol).
[0351] IR(KBr) v 963, 1200, 1497, 1609, 2860, 2933, 3088 cm"1; [0351] IR (KBr) v 963, 1200, 1497, 1609, 2860, 2933, 3088 cm "1;
max max
1H NMR(DMSO-d , 400MHz): δ 2.04—2.11 (2H, m), 2.72-2.75 (2H, m), 3.02—3.0 1H NMR (DMSO-d, 400 MHz): δ 2.04—2.11 (2H, m), 2.72-2.75 (2H, m), 3.02—3.0
6 6
5 (2H, m), 7.00 (IH, ddd, J=1.2 Hz, 2.7 Hz, 8.2 Hz), 7.20 (IH, d, J=8.2 Hz), 7.81— 7.82 (IH, m), 8.98 (IH, s); 5 (2H, m), 7.00 (IH, ddd, J = 1.2 Hz, 2.7 Hz, 8.2 Hz), 7.20 (IH, d, J = 8.2 Hz), 7.81—7.82 (IH, m), 8.98 (IH, s);
MS(FAB) m/z: 296 (M— H)— . MS (FAB) m / z: 296 (M— H) —.
<実施例 23 >
(2 アミノー 5, 6 ジヒドロー 4H べンゾ [6, 7]シクロへプタ [1, 2— d] [l, 3]チア ゾールー 9 ィル)ホスホン酸(例示化合物番号 2-35) <Example 23> (2 amino-5,6 dihydro-4H benzo [6,7] cyclohepta [1,2-d] [l, 3] thiazol-9yl) phosphonic acid (Exemplary Compound No. 2-35)
(23a)トリフルォロメタンスルホン酸 (9—ォキソ 6, 7, 8, 9—テトラヒドロ一 5H— ベンゾ [7]ァヌレンー2 ィル) (23a) trifluoromethanesulfonic acid (9-oxo 6, 7, 8, 9-tetrahydro-5H-benzo [7] annulene-2-yl)
3 ヒドロキシ一 6, 7, 8, 9—テトラヒドロー 5H ベンゾ [7]ァヌレン一 5—オン(27 lmg、 1. 54mmol)を使用して、実施例 15 (15a)に記載の方法に従い、標記目的 化合物 (422mg、収率 89%)を得た。 3 Hydroxy 1, 6, 7, 8, 9-tetrahydro-5H Benzo [7] annulen-5-one (27 lmg, 1.54 mmol) was used according to the method described in Example 15 (15a) to give the title object compound (422 mg, 89% yield) was obtained.
[0352] IR(Liquid film) v 607, 891, 1141, 1216, 1427, 1686, 2948 cm"1; [0352] IR (Liquid film) v 607, 891, 1141, 1216, 1427, 1686, 2948 cm "1;
max max
1H NMR(CDC1 , 500MHz): δ 1.81—1.95 (4H, m), 2.75-2.80 (2H, m), 2.95—3.00 (2 1H NMR (CDC1, 500MHz): δ 1.81—1.95 (4H, m), 2.75-2.80 (2H, m), 2.95—3.00 (2
3 Three
H, m), 7.28-7.35 (2H, m), 7.62-7.65 (1H, m); H, m), 7.28-7.35 (2H, m), 7.62-7.65 (1H, m);
MS(EI) m/z: 308 M+. MS (EI) m / z: 308 M + .
(23b) (9—ォキソ 6, 7, 8, 9—テトラヒドロ一 5H ベンゾ [7]ァヌレン一 2—ィル) ホスホン酸 ジェチノレ (23b) (9-oxo 6, 7, 8, 9-tetrahydro-5H benzo [7] annulene-2-yl) phosphonic acid jetinore
実施例 23 (23a)で製造したトリフルォロメタンスルホン酸 (9 ォキソ—6, 7, 8, 9 —テトラヒドロ一 5H ベンゾ [7]ァヌレン一 2—ィル)(377mg、 1. 22mmol)を N, N —ジメチルホルムアミド(6. lmL)に溶解し、そこへテトラキス(トリフエ-ルホスフィン) ノ《ラジウム(70. 7mg、 0. 061mmol)、卜リエチルァミン(1. 70mL、 12. 2mmol)及 び亜リン酸ジェチル(315 レ 2. 45mmol)をカ卩え、窒素雰囲気下、 100°Cで 3時 間撹拌した。 Trifluoromethanesulfonic acid (9oxo-6,7,8,9-tetrahydro-5H benzo [7] annulene-2-yl) (377 mg, 1.22 mmol) prepared in Example 23 (23a) Dissolve in N-dimethylformamide (6. lmL), then add tetrakis (triphenylphosphine) -no-radium (70.7 mg, 0.061 mmol), triethylamine (1.70 mL, 12.2 mmol) and phosphorus Jetyl acid (315 x 2.45 mmol) was added and stirred at 100 ° C for 3 hours under nitrogen atmosphere.
[0353] 反応液に飽和塩化アンモ-ゥム水溶液を加え、クロ口ホルムで抽出した。得られた 有機層を水及び飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、減圧下溶媒を 留去した。得られた残渣をシリカゲルカラムクロマトグラフィー (へキサン:酢酸ェチル、 1: 2、 VZV)により精製し、標記目的化合物(361mg、収率 100%)を得た。 [0353] A saturated aqueous ammonium chloride solution was added to the reaction mixture, and the mixture was extracted with black mouth form. The obtained organic layer was washed with water and saturated brine, dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure. The obtained residue was purified by silica gel column chromatography (hexane: ethyl acetate, 1: 2, VZV) to obtain the title object compound (361 mg, yield 100%).
[0354] IR(Liquid film) v 971, 1023, 1248, 1681, 2941 cm"1; [0354] IR (Liquid film) v 971, 1023, 1248, 1681, 2941 cm "1;
max max
1H NMR(CDC1 , 500MHz): δ 1.30—1.40 (6H, m), 1.79—1.87 (2H, m), 1.87—1.95 (2 1H NMR (CDC1, 500MHz): δ 1.30—1.40 (6H, m), 1.79—1.87 (2H, m), 1.87—1.95 (2
3 Three
H, m), 2.72-2.79 (2H, m), 2.95—3.02 (2H, m), 4.05-4.20 (4H, m), 7.30-7.35 (1H, m ), 7.85-7.92 (1H, m), 8.07—8.14 (1H, m); H, m), 2.72-2.79 (2H, m), 2.95—3.02 (2H, m), 4.05-4.20 (4H, m), 7.30-7.35 (1H, m), 7.85-7.92 (1H, m), 8.07—8.14 (1H, m);
MS(EI) m/z: 296 M+.
(23c) (2 アミノー 5, 6 ジヒドロー 4H べンゾ [6, 7]シクロへプタ [1, 2— d] [l, 3]チアゾーノレ 9ーィノレ)ホスホン酸 ジェチノレ MS (EI) m / z: 296 M + . (23c) (2 amino-5,6 dihydro-4H benzo [6,7] cyclohepta [1,2-d] [l, 3] thiazonole 9-inore) phosphonic acid jetinore
実施例 23 (23b)で製造した(9—ォキソ 6, 7, 8, 9—テトラヒドロ一 5H ベンゾ[ 7]ァヌレン一 2—ィル)ホスホン酸 ジェチル(361mg、 1. 22mmol)を使用して、実 施例 2 (2b)に記載した方法に従い、標記目的化合物 (133mg、収率 32%)を得た。 Using (9-oxo 6, 7, 8, 9-tetrahydro-5H benzo [7] annulene-2-yl) phosphonic acid jetyl prepared in Example 23 (23b) (361 mg, 1.22 mmol) The title target compound (133 mg, yield 32%) was obtained according to the method described in Example 2 (2b).
[0355] IR(KBr) v 977, 1025, 1228, 1556, 1655, 3151, 3300 cm"1; [0355] IR (KBr) v 977, 1025, 1228, 1556, 1655, 3151, 3300 cm "1;
max max
1H NMR(CD OD, 500MHz): δ 1.33 (6H, t, J=7.3 Hz), 2.07—2.15 (2H, m), 2.80—2. 1H NMR (CD OD, 500 MHz): δ 1.33 (6H, t, J = 7.3 Hz), 2.07—2.15 (2H, m), 2.80—2.
3 Three
90 (4H, m), 4.05-4.15 (4H, m), 7.35 (IH, dd, J=4.9 Hz, 7.8 Hz), 7.56 (IH, dd, J=7. 8 Hz, 12.7 Hz), 8.25 (IH, d, J=13.7 Hz); 90 (4H, m), 4.05-4.15 (4H, m), 7.35 (IH, dd, J = 4.9 Hz, 7.8 Hz), 7.56 (IH, dd, J = 7.8 Hz, 12.7 Hz), 8.25 ( IH, d, J = 13.7 Hz);
MS(EI) m/z: 352 M+. MS (EI) m / z: 352 M + .
(23d) (2 アミノー 5, 6 ジヒドロー 4H べンゾ [6, 7]シクロへプタ [1, 2— d] [l, 3]チアゾーノレ - 9—ィノレ)ホスホン酸 (23d) (2 amino-5, 6 dihydro-4H benzo [6, 7] cyclohepta [1,2-d] [l, 3] thiazolole-9-inole) phosphonic acid
実施例 23 (23c)で製造した(2 アミノー 5, 6 ジヒドロー 4H べンゾ [6, 7]シク 口へプタ [1, 2-d] [l, 3]チアゾーノレ一 9—ィノレ)ホスホン酸 ジェチノレ(82. 3mg、 0. 234mmol)を使用して、実施例 1 (lc)に記載した方法に従い、標記目的化合物 ( 53. 9mg、収率 78%)を得た。 Example 23 (23c) (2 amino-5,6 dihydro-4H benzo [6,7] sicester hepta [1,2-d] [l, 3] thiazonole-9-inole) phosphonic acid jetinore (82.3 mg, 0.234 mmol) was used to give the title object compound (53.9 mg, 78% yield) according to the method described in Example 1 (lc).
[0356] IR(KBr) v 924, 1066, 1129, 1632, 2934 cm"1; [0356] IR (KBr) v 924, 1066, 1129, 1632, 2934 cm "1;
max max
1H NMR(DMSO-d , 500MHz): δ 1.92—2.00 (2H, m), 2.79-2.87 (4H, m), 6.87—6.9 1H NMR (DMSO-d, 500 MHz): δ 1.92—2.00 (2H, m), 2.79-2.87 (4H, m), 6.87—6.9
6 6
4 (2H, br), 7.22 (IH, dd, J=3.9 Hz, 7.8 Hz), 7.42 (IH, ddd, J=2.0 Hz, 7.8 Hz, 12.7 Hz), 8.35 (IH, dd, J=2.0 Hz, 14.7 Hz); 4 (2H, br), 7.22 (IH, dd, J = 3.9 Hz, 7.8 Hz), 7.42 (IH, ddd, J = 2.0 Hz, 7.8 Hz, 12.7 Hz), 8.35 (IH, dd, J = 2.0 Hz) , 14.7 Hz);
MS(FAB) m/z: 295 (M— H)— . MS (FAB) m / z: 295 (M— H) —.
<実施例 24 > <Example 24>
(5, 6 ジヒドロー 4H べンゾ [6, 7]シクロへプタ [1, 2-d] [l, 3]チアゾーノレー9 ィル)ホスホン酸 (例示化合物番号 2-33) (5,6 dihydro-4H benzo [6,7] cyclohepta [1,2-d] [l, 3] thiazonole 9yl) phosphonic acid (Exemplified Compound No. 2-33)
(24a) (5, 6 ジヒドロー 4H べンゾ [6, 7]シクロへプタ [1, 2— d] [l, 3]チアゾー ルー 9 ィル)ホスホン酸ジェチル (24a) (5, 6 dihydro-4H benzo [6, 7] cyclohepta [1, 2—d] [l, 3] thiazol 9 yl) jetyl phosphonate
実施例 23 (23b)で製造した(9—ォキソ 6, 7, 8, 9—テトラヒドロ一 5H—ベンゾ[
7]ァヌレン一 2—ィル)ホスホン酸 ジェチル(318mg、 1. 07mmol)を使用して、実 施例 3 (3a— 3b)に記載した方法に従い、標記目的化合物(146mg、収率 40%)を 得た。 Example 9 (9-oxo 6, 7, 8, 9-tetrahydro-5H-benzo [ 7] Annulen-2-yl) Phosphonic acid Jetyl (318 mg, 1.07 mmol) was used according to the method described in Example 3 (3a-3b) and the title compound (146 mg, 40% yield) Got.
[0357] IR(Liquid film) v 966, 1024, 1052, 1243, 1443, 2936, 2982, 3466 cm"1; [0357] IR (Liquid film) v 966, 1024, 1052, 1243, 1443, 2936, 2982, 3466 cm "1;
max max
1H NMR(CDC1 , 400MHz) : δ 1.33 (6H, t, J=7.1 Hz), 2.21-2.27 (2H, m), 2.81—2.8 1H NMR (CDC1, 400MHz): δ 1.33 (6H, t, J = 7.1 Hz), 2.21-2.27 (2H, m), 2.81—2.8
3 Three
4 (2H, m), 2.92 (2H, t, J=7.1 Hz), 4.06-4.18 (4H, m), 7.29-7.32 (IH, m), 7.70 (IH, ddd, J=1.6 Hz, 6.7 Hz, 12.9 Hz), 8.44 (IH, dd, J=1.6 Hz, 14.1 Hz) 8.68 (IH, s); MS(FAB) m/z: 338 (M+H)+. 4 (2H, m), 2.92 (2H, t, J = 7.1 Hz), 4.06-4.18 (4H, m), 7.29-7.32 (IH, m), 7.70 (IH, ddd, J = 1.6 Hz, 6.7 Hz , 12.9 Hz), 8.44 (IH, dd, J = 1.6 Hz, 14.1 Hz) 8.68 (IH, s); MS (FAB) m / z: 338 (M + H) + .
(24b) (5, 6 ジヒドロー 4H べンゾ [6, 7]シクロへプタ [1, 2— d] [l, 3]チアゾー ル一 9 ィル)ホスホン酸 (24b) (5, 6 dihydro-4H benzo [6, 7] cyclohepta [1,2-d] [l, 3] thiazol-9 yl) phosphonic acid
実施例 24 (24a)で製造した(5, 6 ジヒドロー 4H べンゾ [6, 7]シクロへプタ [1, 2-d] [l, 3]チアゾールー 9 ィル)ホスホン酸ジェチル(143mg、 0. 424mmol) を使用して、実施例 1 (lc)に記載した方法に従い、標記目的化合物(53. 5mg、収 率 45%)を得た。 Example 24 (24, a) (5,6 dihydro-4H benzo [6,7] cyclohepta [1,2-d] [l, 3] thiazol-9yl) jetyl phosphonate (143 mg, 0 424 mmol) was used to give the title object compound (53.5 mg, yield 45%) according to the method described in Example 1 (lc).
[0358] IR(KBr) v 931, 991, 1136, 2326, 2862, 2933, 3078 cm"1; [0358] IR (KBr) v 931, 991, 1136, 2326, 2862, 2933, 3078 cm "1;
max max
1H NMR(DMSO-d , 400MHz) : δ 2.06—2.12 (2H, m), 2.82-2.79 (2H, m), 3.06 (2H 1H NMR (DMSO-d, 400MHz): δ 2.06—2.12 (2H, m), 2.82-2.79 (2H, m), 3.06 (2H
6 6
, t, J=7.0 Hz), 7.28-7.31 (IH, m), 7.48-7.53 (IH, m), 8.37 (IH, d, J=14.1 Hz), 9.01 (IH, s); , t, J = 7.0 Hz), 7.28-7.31 (IH, m), 7.48-7.53 (IH, m), 8.37 (IH, d, J = 14.1 Hz), 9.01 (IH, s);
MS(FAB) m/z: 280 (M— H)— . <実施例 25 > MS (FAB) m / z: 280 (M— H) —. <Example 25>
[ (7 フルオロー 8H インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキシ]メチ ルホスホン酸(例示化合物番号 1-65) [(7 Fluoro-8H indeno [1,2-d] [l, 3] thiazole-4-yl) oxy] methylphosphonic acid (Exemplified Compound No. 1-65)
(25a) [ (7 フルォロ一 3—ォキソ 2, 3 ジヒドロ一 1H—インデン一 4—ィル)ォキ シ]メチルホスホン酸ジェチル (25a) [(7 Fluoro-3-oxo2, 3 dihydro-1H-indene-4-yl) oxy] methyl phosphonate
4 フルォロ一 7 ヒドロキシインダン一 1—オン(7. 88g、 47. 4mmol)を使用して 、実施例 1 (la)に記載した方法に従い、標記目的化合物(12. 0g、収率 80%)を得
[0359] IR(KBr) v 979, 1009, 1042, 1096, 1229, 1238, 1255, 1291, 1336, 1399, 1441, 1 max 4 Using the fluoro 7-hydroxyindan 1-one (7.88 g, 47.4 mmol) according to the method described in Example 1 (la), the title compound (12.0 g, 80% yield) was obtained. Gain [0359] IR (KBr) v 979, 1009, 1042, 1096, 1229, 1238, 1255, 1291, 1336, 1399, 1441, 1 max
493, 1614, 1706, 2909, 2942, 2985, 3044, 3074 cm"1; 493, 1614, 1706, 2909, 2942, 2985, 3044, 3074 "1;
1H NMR(CDC1 , 400MHz) δ 1.37 (6H, t, J=7.0 Hz), 2.67-2.72 (2H, m), 3.07—3.1 1H NMR (CDC1, 400MHz) δ 1.37 (6H, t, J = 7.0 Hz), 2.67-2.72 (2H, m), 3.07-3.1
3 Three
2 (2H, m), 4.31 (2H, q, J=7.0 Hz), 4.33 (2H, q, J=7.0 Hz), 4.41 (2H, d, J=9.4 Hz), 6 .85 (IH, dd, J=3.3 Hz, 8.6 Hz), 7.21 (IH, dd, J=7.8 Hz, 8.6 Hz); 2 (2H, m), 4.31 (2H, q, J = 7.0 Hz), 4.33 (2H, q, J = 7.0 Hz), 4.41 (2H, d, J = 9.4 Hz), 6.85 (IH, dd , J = 3.3 Hz, 8.6 Hz), 7.21 (IH, dd, J = 7.8 Hz, 8.6 Hz);
MS(FAB) m/z: 317 (M+H)+. MS (FAB) m / z: 317 (M + H) + .
(25b) [ (7 フルオロー 8H インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキ シ]メチルホスホン酸ジェチル (25b) [(7 Fluoro-8H indeno [1, 2-d] [l, 3] thiazole-4-yl) oxy] methyl phosphonate
実施例 25 (25a)で製造した [ (7 フルォロ 3 ォキソ 2, 3 -ジヒドロ 1H ィ ンデン—4—ィル)ォキシ]メチルホスホン酸ジェチル(12. Og、 37. 9mmol)を使用 して、実施例 3 (3a— 3b)に記載した方法に従い、標記目的化合物(8. 02g、収率 5 9%)を得た。 Example 25 Using [(7 Fluoro-3oxo2,3-dihydro-1Hindene-4-yl) oxy] methylphosphonate (12. Og, 37.9 mmol) prepared in Example 25 (25a) 3 The title object compound (8.02 g, yield 59%) was obtained according to the method described in 3 (3a-3b).
[0360] IR(KBr) v 979, 1018, 1049, 1085, 1237, 1252, 1274, 1387, 1432, 1509, 1593, max [0360] IR (KBr) v 979, 1018, 1049, 1085, 1237, 1252, 1274, 1387, 1432, 1509, 1593, max
2900, 2983 cm"1; 2900, 2983 cm "1;
1H NMR(CDC1 , 400MHz) δ 1.36 (6H, t, J=7.0 Hz), 3.90 (2H, s), 4.30 (2H, q, J= 1H NMR (CDC1, 400MHz) δ 1.36 (6H, t, J = 7.0 Hz), 3.90 (2H, s), 4.30 (2H, q, J =
3 Three
7.0 Hz), 4.32 (2H, q, J=7.0 Hz), 4.63 (2H, d, J=8.6 Hz), 6.92 (IH, dd, J=8.6 Hz, 8. 6 Hz), 7.05 (IH, dd, J=3.5 Hz, 8.6 Hz), 8.86 (IH, s); 7.0 Hz), 4.32 (2H, q, J = 7.0 Hz), 4.63 (2H, d, J = 8.6 Hz), 6.92 (IH, dd, J = 8.6 Hz, 8.6 Hz), 7.05 (IH, dd , J = 3.5 Hz, 8.6 Hz), 8.86 (IH, s);
MS(FAB) m/z: 358 (M+H)+. MS (FAB) m / z: 358 (M + H) + .
(25c) [ (7 フルオロー 8H インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキ シ]メチノレホスホン酸 (25c) [(7 Fluoro-8H indeno [1, 2-d] [l, 3] thiazole-4-yl) oxy] methinorephosphonic acid
実施例 25 (25b)で製造した [ (7 フルォロ— 8H—インデノ [1, 2— d] [l, 3]チア ゾールー 4 ィル)ォキシ]メチルホスホン酸ジェチル(82mg、 0. 229mmol)を使用 して、実施例 1 (lc)に記載した方法に従い、標記目的化合物(64mg、収率 93%)を 得た。 EXAMPLE 25 [(7 Fluoro-8H-indeno [1,2-d] [l, 3] thiazol-4-yl) oxy] methylphosphonate (82 mg, 0.229 mmol) prepared in Example 25 (25b) was used. Then, according to the method described in Example 1 (lc), the title object compound (64 mg, 93% yield) was obtained.
[0361] IR(KBr) v 950, 1008, 1076, 1123, 1244, 1387, 1432, 1485, 1509, 1598, 1629, 2 max [0361] IR (KBr) v 950, 1008, 1076, 1123, 1244, 1387, 1432, 1485, 1509, 1598, 1629, 2 max
323, 2841, 2903, 3087 cm"1; 323, 2841, 2903, 3087 cm "1;
1H NMR(DMSO-d , 500MHz) : δ 2.07 (2H, s), 4.02 (2H, s), 4.33 (2H, d, J=8.8 Hz 1H NMR (DMSO-d, 500MHz): δ 2.07 (2H, s), 4.02 (2H, s), 4.33 (2H, d, J = 8.8 Hz
6 6
), 7.06 (IH, dd, J=8.8 Hz, 8,8 Hz), 7.16 (IH, dd, J=3.9 Hz, 9.3 Hz), 9.21 (IH, s);
MS(FAB) m/z: 300 (M— H)— . ), 7.06 (IH, dd, J = 8.8 Hz, 8,8 Hz), 7.16 (IH, dd, J = 3.9 Hz, 9.3 Hz), 9.21 (IH, s); MS (FAB) m / z: 300 (M— H) —.
<実施例 26 > <Example 26>
[ (7 クロロー 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキシ]メチル ホスホン酸 (例示化合物番号 1-70) [(7 Chloro-8H—indeno [1,2-d] [l, 3] thiazol-4-yl) oxy] methylphosphonic acid (Exemplified Compound No. 1-70)
(26a) [ (7 クロ口一 3—ォキソ 2, 3 ジヒドロ一 1H—インデン一 4—ィル)ォキシ] メチルホスホン酸ジェチル (26a) [(7 Black-and-one 3-oxo 2, 3 Dihydro- 1H-Indene 4-yl) oxy] Jetyl methylphosphonate
4 クロ口一 7 ヒドロキシインダン一 1—オン(6. OOg、 32. 9mmol)を使用して、 実施例 1 (la)に記載した方法に従い、標記目的化合物(7. 79g、収率 71%)を得た 4 The title compound (7. 79 g, 71% yield) was prepared according to the method described in Example 1 (la) using 7-hydroxyindan 1-one (6. OOg, 32.9 mmol). Got
[0362] IR(KBr) v 980, 1010, 1030, 1256, 1289, 1474, 1591, 1709, 2940, 2982, 3397 c max [0362] IR (KBr) v 980, 1010, 1030, 1256, 1289, 1474, 1591, 1709, 2940, 2982, 3397 c max
-1 -1
m ; m;
1H NMR(CDC1 , 400MHz) δ 1.37 (6H, t, J=7.0 Hz), 2.65-2.72 (2H, m), 3.04—3.1 1H NMR (CDC1, 400MHz) δ 1.37 (6H, t, J = 7.0 Hz), 2.65-2.72 (2H, m), 3.04—3.1
3 Three
0 (2H, m), 4.32 (2H, q, J=7.0 Hz), 4.33 (2H, q, J=7.0 Hz), 4.42 (2H, d, J=9.8 Hz), 6 .86 (1H, d, J=8.6 Hz), 7.50 (1H, d, J=8.6 Hz); 0 (2H, m), 4.32 (2H, q, J = 7.0 Hz), 4.33 (2H, q, J = 7.0 Hz), 4.42 (2H, d, J = 9.8 Hz), 6.86 (1H, d , J = 8.6 Hz), 7.50 (1H, d, J = 8.6 Hz);
MS(FAB) m/z: 333 (M+H)+. MS (FAB) m / z: 333 (M + H) + .
(26b) [ (7 クロロー 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキシ] メチルホスホン酸ジェチル (26b) [(7 Chloro-8H-indeno [1, 2-d] [l, 3] thiazol-4-yl) oxy] methyl phosphonate
実施例 26 (26a)で製造した [ (7 クロ口一 3—ォキソ 2, 3 ジヒドロ一 1H—イン デン 4 ィル)ォキシ]メチルホスホン酸ジェチル(7. 75g、 24. Ommol)を使用し て、実施例 3 (3a— 3b)に記載した方法に従い、標記目的化合物(5. 07g、収率 56 %)を得た。 Using [(7-Chrono-3-oxo-2,3-dihydro-1H-indene-4-yl) oxy] methylphosphonate jetyl (7.75 g, 24. Ommol) prepared in Example 26 (26a) The title compound (5.07 g, yield 56%) was obtained according to the method described in Example 3 (3a-3b).
[0363] IR(KBr) v 980, 1023, 1049, 1083, 1233, 1256, 1271, 1389, 1427, 1490, 2980, max [0363] IR (KBr) v 980, 1023, 1049, 1083, 1233, 1256, 1271, 1389, 1427, 1490, 2980, max
3091 cm"1; 3091 cm "1;
1H NMR(CDC1 , 400MHz) δ 1.35 (6H, t, J=7.0 Hz), 3.86 (2H, s), 4.29 (2H, q, J= 1H NMR (CDC1, 400MHz) δ 1.35 (6H, t, J = 7.0 Hz), 3.86 (2H, s), 4.29 (2H, q, J =
3 Three
7.0 Hz), 4.31 (2H, q, J=7.0 Hz), 4.62 (2H, d, J=8.6 Hz), 7.03 (1H, d, J=8.6 Hz), 7.1 8 (1H, d, J=8.6 Hz), 8.83 (1H, s); 7.0 Hz), 4.31 (2H, q, J = 7.0 Hz), 4.62 (2H, d, J = 8.6 Hz), 7.03 (1H, d, J = 8.6 Hz), 7.1 8 (1H, d, J = 8.6 Hz), 8.83 (1H, s);
MS(FAB) m/z: 374 (M+H)+.
( / — 一 /—、 ^ [ε 'i][p-s ] (qzs)MS (FAB) m / z: 374 (M + H) + . (/ — One / —, ^ [ε 'i] [ps] (qzs)

• (s • ( s
'Ηΐ) S8'8 '(zH8"8=f 'P 'Ηΐ) SS- '(ΖΗ8·8=1" 'P 'Ηΐ) Ζ6·9 '(zH0"6=f 'P 'ΗΖ) 09·, '(ω 'Ηΐ) S8'8' ( z H8 "8 = f 'P' Ηΐ) SS- '(ΖΗ8 · 8 = 1"' P 'Ηΐ) Ζ6 · 9' ( z H0 "6 = f 'P' ΗΖ) 09, '(ω
'Η ) 9£'f-LZ'f '(s Ή2) ZS'£ '(zH0"Z=f '; 'Η9) SS"T 9 '· (zH )0 つ dつ) WN Hx 'Η) 9 £'f-LZ'f'(s Ή2) ZS' £ '(zH0 "Z = f';'Η9)SS" T 9' · ( z H) 0 d) WN H x
S60S '8Z6 S60S '8Z6
Z 'TZST '88 1 'S6ST '0Ζ2ΐ 'SSST 'SSST 'ΐ80ΐ '6雨 'ΖΖΟΙ '6Z6 'S08 Λ ( 1)ΗΙ [S9S0]Z 'TZST '88 1' S6ST '0Ζ2ΐ' SSST 'SSST' ΐ80ΐ '6 Rain' ΖΖΟΙ '6Z6' S08 Λ (1) ΗΙ [S9S0]
。 .
(%89 ¾τ ¾ ·υ)呦^: ]Κ¾目 ¾遨 (qs-^s)spM^ 、ェっ ¾ί (ΐου υ8 ·6ε §ο · 91 )
 →- ^ -ui - α^ ^ - ε 'Ζ- ^-£ - -ζ)] ^niiif (βΐ)χ P}¾?(% 89 ¾τ ¾ · υ) 呦 ^:] Κ¾ 目 ¾ 遨 (qs- ^ s) spM ^, ί ( ΐου υ 8 · 6ε § ο · 91) →-^ -ui-α ^ ^-ε 'Ζ- ^-£--ζ)] ^ niiif ( β ΐ) χ P} ¾?
→- ^ -ui - α^ ^ - ε 'Ζ- ^-£ - -ζ)] ^niiif (βΐ)χ P}¾?(% 89 ¾τ ¾ · υ) 呦 ^:] Κ¾ 目 ¾ 遨 (qs- ^ s) spM ^, ί ( ΐου υ 8 · 6ε § ο · 91) →-^ -ui-α ^ ^-ε 'Ζ- ^-£--ζ)] ^ niiif ( β ΐ) χ P} ¾?
^エ 邈べ ^ [ ^ D
( / — 一 /—、 ^ [ε Ί][Ρ-2
 (/ — One / —, ^ [ε Ί] [Ρ-2
(/ — One / —, ^ [ε Ί] [Ρ-2
09- ΐ各粱呦^ ^ )邈べ 09- ΐEach 粱 呦 ^ ^)
< 爾第 > <爾 No.>
•_(H- ) 9TS: ζ/ω (9VH)S • _ (H-) 9TS: ζ / ω (9VH) S
•(s 'Ηΐ) IZ'6 '(ΖΗ ε·6=ί" 'P 'Ηΐ) 6Z'L '(ΖΗ 0·6 =f ' Ρ 'Ηΐ) ZZ'L '(ΖΗ 0"6=f 'Ρ 'ΗΖ) 9£'f '(s 'ΗΖ) Ζ6Τ 9 '· (zH )0 <9p-OS a)H N Ηχ • ( s 'Ηΐ) IZ'6' ( Ζ Η ε · 6 = ί "'P' Ηΐ) 6Z'L '( Ζ Η 0 · 6 = f' Ρ 'Ηΐ) ZZ'L' ( Ζ Η 0" 6 = f 'Ρ' ΗΖ) 9 £ 'f' ( s 'ΗΖ) Ζ6Τ 9' · ( z H) 0 <9 p-OS a) HN Η χ
'S80S Ί06Ζ 'ΖΖΖΖ '083ΐ '26^ΐ 'S ^T 'εβεΐ '892ΐ 'εΖΟΐ '236 Λ ( i) 9S0] 'S80S Ί06Ζ' ΖΖΖΖ '083ΐ '26 ^ ΐ' S ^ T 'εβεΐ' 892ΐ 'εΖΟΐ' 236 Λ (i) 9S0]
(%06 ¾τ
 (% 06 ¾τ
(% 06 ¾τ
¾f¾¾ (TOUIUIXO^ Ό、3raog"[) ^エ 、邈べ ^ [/^^ ( / — 一 /— ¾f¾¾ (TOUIUIXO ^ Ό, 3raog "[) ^ d, ^ ^ // ^^ (/ — One / —
、
 ,
,
邈べ ^ ^^
0l76S0C/900Zdf/X3d εοΐ· 0C0l70l/900Z OAV
]メチノレホスホン酸 0l76S0C / 900Zdf / X3d εοΐ0C0l70l / 900Z OAV ] Metinorephosphonic acid
実施例 27 (27a)で製造した [ (7 ブロモ— 8H—インデノ [1, 2 d] [l, 3]チアゾ 一ルー 4—ィル)ォキシ]メチルホスホン酸ジェチル(300mg、 0. 717mmol)を使用 して、実施例 1 (lc)に記載した方法に従い、標記目的化合物(260mg、収率 100% )を得た。 Example 27 [(7 Bromo-8H-indeno [1,2d] [l, 3] thiazo 4-l) oxy] methylphosphonate (300 mg, 0.717 mmol) prepared in Example 27 (27a) was used. Then, according to the method described in Example 1 (lc), the title object compound (260 mg, yield 100%) was obtained.
[0366] IR(KBr) v 936, 1061, 1177, 1264, 1401, 1429, 1490, 1573, 1639, 2322, 3126 c max [0366] IR (KBr) v 936, 1061, 1177, 1264, 1401, 1429, 1490, 1573, 1639, 2322, 3126 c max
-1 -1
m ; m;
1H NMR(DMSO-d , 400MHz): δ 3.90 (2H, s), 4.12 (2H, d, J=9.8Hz), 7.12 (1H, d, 1H NMR (DMSO-d, 400MHz): δ 3.90 (2H, s), 4.12 (2H, d, J = 9.8Hz), 7.12 (1H, d,
6 6
J=8.6Hz), 7.42 (1H, d, J=8.6Hz), 9.13 (1H, s); J = 8.6Hz), 7.42 (1H, d, J = 8.6Hz), 9.13 (1H, s);
MS(FAB) m/z: 360 (M— H)— . MS (FAB) m / z: 360 (M— H) —.
<実施例 28 > <Example 28>
[ (7 ピリジンー3—ィルー 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォ キシ]メチルホスホン酸 (例示化合物番号 1-6) [(7 Pyridine-3-yl 8H-indeno [1, 2-d] [l, 3] thiazole-4-yl) oxy] methylphosphonic acid (Exemplified Compound No. 1-6)
(28a) [ (7 ピリジンー3—ィルー 8H—インデノ [1, 2 d] [l, 3]チアゾールー 4 ィル)ォキシ]メチルホスホン酸ジェチル (28a) [(7 Pyridine-3-yl 8H-indeno [1, 2 d] [l, 3] thiazol-4-yl) oxy] methyl phosphonate
実施例 27 (27a)で製造した [ (7 ブロモ— 8H—インデノ [1, 2 d] [l, 3]チアゾ 一ルー 4 ィル)ォキシ]メチルホスホン酸ジェチル(200mg、 0. 478mmol)をェタノ ール(9. 6mL)に溶解し、そこへテトラキス(トリフエ-ルホスフィン)パラジウム(27. 6 mg、 0. 239mmol)、 3 ピリジルボロン酸ピナコールエステル(196mg、 0. 956m mol)及び 2M炭酸カリウム水溶液(0. 717mL、 1. 43mmol)を加え、 9時間加熱還 流した。 Example 27 [(7 Bromo-8H-indeno [1,2d] [l, 3] thiazo 4-yl) oxy] methylphosphonate (200 mg, 0.478 mmol) prepared in Example 27 (27a) (99.6 mL), tetrakis (triphenylphosphine) palladium (27.6 mg, 0.239 mmol), 3 pyridylboronic acid pinacol ester (196 mg, 0.956 mmol) and 2M aqueous potassium carbonate solution (0.771 mL, 1.43 mmol) was added, and the mixture was refluxed for 9 hours.
[0367] 冷却後、減圧下溶媒を留去し、残渣を塩基性シリカゲルカラムクロマトグラフィー( へキサン:酢酸ェチル、 50 : 50— 0 : 100、 V/V)を用 、て精製し、標記目的化合物 (99. 3mg、収率 50%)を得た。 [0367] After cooling, the solvent was distilled off under reduced pressure, and the residue was purified using basic silica gel column chromatography (hexane: ethyl acetate, 50: 50—0: 100, V / V) for the title purpose. The compound (99.3 mg, yield 50%) was obtained.
[0368] IR(KBr) v 977, 1034, 1252, 1405, 2980 cm"1; [0368] IR (KBr) v 977, 1034, 1252, 1405, 2980 cm "1;
max max
1H NMR(CDC1 , 400MHz) δ 1.37 (6H, t, J=7.1 Hz), 3.88 (2H, s), 4.34 (4H, dq, J 1H NMR (CDC1, 400MHz) δ 1.37 (6H, t, J = 7.1 Hz), 3.88 (2H, s), 4.34 (4H, dq, J
3 Three
=7.1 Hz, 7.1 Hz), 4.67 (2H, d, J=8.6 Hz), 7.18 (1H, d, J=8.6 Hz), 7.21 (1H, d, J=8.
6 Hz), 7.38 (1H, ddd, J=0.8 Hz, 4.7 Hz, 7.8 Hz), 7.78 (1H, ddd, J=1.6 Hz, 2.4 Hz, 7.8 Hz), 8.61 (1H, dd, J=1.6 Hz, 4.7 Hz), 8.75 (1H, dd, J=0.8 Hz, 2.4 Hz), 8.84 (1 H, s); = 7.1 Hz, 7.1 Hz), 4.67 (2H, d, J = 8.6 Hz), 7.18 (1H, d, J = 8.6 Hz), 7.21 (1H, d, J = 8. 6 Hz), 7.38 (1H, ddd, J = 0.8 Hz, 4.7 Hz, 7.8 Hz), 7.78 (1H, ddd, J = 1.6 Hz, 2.4 Hz, 7.8 Hz), 8.61 (1H, dd, J = 1.6 Hz) , 4.7 Hz), 8.75 (1H, dd, J = 0.8 Hz, 2.4 Hz), 8.84 (1 H, s);
MS(FAB) m/z: 417 (M+H)+. MS (FAB) m / z: 417 (M + H) + .
(28b) [ (7 ピリジンー3—ィルー 8H—インデノ [1, 2— d] [l, 3]チアゾールー 4 ィル)ォキシ]メチルホスホン酸 (28b) [(7 Pyridine-3-yl 8H-indeno [1, 2- d] [l, 3] thiazole-4-yl) oxy] methylphosphonic acid
実施例 28 (28a)で製造した [ (7 ピリジン 3—ィルー 8H—インデノ [ 1 , 2 d] [ 1 , 3]チアゾールー 4 ィル)ォキシ]メチルホスホン酸ジェチル(69. 3mg、0. 166m mol)を使用して、実施例 1 (lc)に記載した方法に従い、標記目的化合物 (46. Img 、収率 77%)を得た。 Example 28 [(7 Pyridine 3-yl 8H-indeno [1,2d] [1,3] thiazol-4-yl) oxy] methylphosphonate jetyl (69.3 mg, 0.166 mmol) prepared in Example 28 (28a) To give the title object compound (46. Img, yield 77%) according to the method described in Example 1 (lc).
[0369] IR(KBr) v 936, 1065, 1261, 1508, 3069 cm"1; [0369] IR (KBr) v 936, 1065, 1261, 1508, 3069 cm "1;
max max
1H NMR(D O+NaHCO , 400MHz): δ 3.60 (2H, s), 4.12 (2H, d, J=9.8 Hz), 7.11-7 1H NMR (D 2 O + NaHCO 3, 400 MHz): δ 3.60 (2H, s), 4.12 (2H, d, J = 9.8 Hz), 7.11-7
2 3 twenty three
.21 (2H, m), 7.47 (1H, dd, J=5.3 Hz, 7.6 Hz), 7.81 (1H, d, J=7.4 Hz), 8.42-8.47 (2 H, m), 8.98 (1H, s); .21 (2H, m), 7.47 (1H, dd, J = 5.3 Hz, 7.6 Hz), 7.81 (1H, d, J = 7.4 Hz), 8.42-8.47 (2 H, m), 8.98 (1H, s );
MS(ESI) m/z: 359 (M— H)— . MS (ESI) m / z: 359 (M— H) —.
<実施例 29 > <Example 29>
[ (7 ピリジンー4ーィルー 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォ キシ]メチルホスホン酸 (例示化合物番号 1-7) [(7 Pyridine-4-yl 8H-indeno [1, 2-d] [l, 3] thiazole-4-yl) oxy] methylphosphonic acid (Exemplified Compound No. 1-7)
(29a) [ (7 ピリジンー4ーィルー 8H—インデノ [1, 2 d] [l, 3]チアゾールー 4 ィル)ォキシ]メチルホスホン酸ジェチル (29a) [(7 Pyridine-4-yl 8H-indeno [1, 2 d] [l, 3] thiazol-4-yl) oxy] methyl phosphonate
4 ピリジルボロン酸ピナコールエステル(196mg、 0. 956mmol)を使用して、実 施例 28 (28a)に記載した方法に従い、標記目的化合物(75. 3mg、収率 38%)を得 た。 4 Using the pyridylboronic acid pinacol ester (196 mg, 0.956 mmol) and according to the method described in Example 28 (28a), the title object compound (75.3 mg, 38% yield) was obtained.
[0370] IR(KBr) v 974, 1025, 1049, 1257, 1599, 2982 cm"1; [0370] IR (KBr) v 974, 1025, 1049, 1257, 1599, 2982 cm "1;
max max
1H NMR(CDC1 , 500MHz): δ 1.37 (6H, t, J=7.1 Hz), 3.93 (2H, s), 4.31—4.39 (4H, 1H NMR (CDC1, 500MHz): δ 1.37 (6H, t, J = 7.1 Hz), 3.93 (2H, s), 4.31–4.39 (4H,
3 Three
m), 4.68 (2H, d, J=8.8 Hz), 7.20 (1H, d, J=8.3 Hz), 7.27 (1H, d, J=8.3 Hz), 7.42-7. 44 (2H, m), 8.69-8.71 (2H, m), 8.87 (1H, s);
 m), 4.68 (2H, d, J = 8.8 Hz), 7.20 (1H, d, J = 8.3 Hz), 7.27 (1H, d, J = 8.3 Hz), 7.42-7. 44 (2H, m), 8.69-8.71 (2H, m), 8.87 (1H, s);
m), 4.68 (2H, d, J = 8.8 Hz), 7.20 (1H, d, J = 8.3 Hz), 7.27 (1H, d, J = 8.3 Hz), 7.42-7. 44 (2H, m), 8.69-8.71 (2H, m), 8.87 (1H, s);
邈べ ^ [/^^ ( /y- 一 /— 、 ^ [ε Ί][Ρ-2
 ](qOS)^^ [/ ^^ (/ y- ichi / —, ^ [ε Ί] [Ρ-2 ] (qOS)
](qOS)^^ [/ ^^ (/ y- ichi / —, ^ [ε Ί] [Ρ-2 ] (qOS)
(um) sif : ζ/ω (isa)S (um) sif: ζ / ω (isa) S
•(s < • ( s <
HI) Z'6 '(s 'ΗΖ) ΐ6·8 '(s 'Ηΐ) 68·8 '(ω 'ΗΖ) ^τΐ-ΖΖ'Ι '(ΖΗ 8·8=ί" 'Ρ 'ΗΖ) 69· '(ω HI) Z'6 '( s ' ΗΖ) ΐ6 ・ 8' ( s ' Ηΐ) 68 ・ 8 '(ω' ΗΖ) ^ τΐ-ΖΖ'Ι '( Ζ Η 8 ・ 8 = ί "' Ρ 'ΗΖ) 69 · '(ω
'Η ) 6ε·— ιε· '(s 'Η ΐ6·ε '(ζΗ ΓΖ=Γ '; Ή9) 8ε·ΐ 9: (ZH OOS 'ει αつ) WN HX 'Η) 6ε · — ιε ·' (s 'Η ΐ6 · ε' ( ζ Η ΓΖ = Γ '; Ή9) 8ε · ΐ 9: (ZH OOS' ε ι α) WN H X
1110 ζιη 'ζιζι ' ζι 'εκπ 'ζτοτ Ί 6 Λ ( i) βζεο] (%ι *¾ί ^ΐ2) ί¾?^ ^目 ¾遨 ^エ 邈べ ^ [/^^ ( /y- 一 /— 、 ^ [ε Ί][Ρ-2
 1 110 ζιη 'ζιζι' ζι 'εκπ' ζτοτ Ί 6 Λ (i) βζεο] (% ι * ¾ί ^ ΐ2) ί¾? ^ ^ 目 ¾ 遨 ^ エ 邈 べ ^ [/ ^^ (/ y- , ^ [Ε Ί] [Ρ-2
1 110 ζιη 'ζιζι' ζι 'εκπ' ζτοτ Ί 6 Λ (i) βζεο] (% ι * ¾ί ^ ΐ2) ί¾? ^ ^ 目 ¾ 遨 ^ エ 邈 べ ^ [/ ^^ (/ y- , ^ [Ε Ί] [Ρ-2
(8- ΐ各粱呦^ ^ ^)邈べ [ ^ (8- ΐEach 粱 呦 ^ ^ ^)
( / — 一 /— 、 ^ [ε Ί][Ρ-2
 ] (/ — One / —, ^ [ε Ί] [Ρ-2 ]
] (/ — One / —, ^ [ε Ί] [Ρ-2 ]
<οε圏第 > <οε range>
•_(H- ) 6SS: z/ui (IS3)S • _ (H-) 6SS: z / ui (IS3) S
•(s 'Ηΐ) Z • ( s ' Ηΐ) Z
0·6 '(ω 'Η 8 ·8—„·8 '(ω 'Η ^l-Wl '(ΖΗ 9·8=ί" 'Ρ 'Ηΐ) WL '(ΖΗ 9·8=ί" 'Ρ Ή ΐ) ΐΓΖ '(ΖΗ ·6=ί" 'Ρ 'ΗΖ) OVf '(s Ή2) ΐ9·ε 9 '· (zH )0 'SODH¾N+0¾H N ΗΧ 0 · 6 '(ω' Η 8 · 8— „· 8 '(ω' Η ^ l-Wl '( Ζ Η 9 · 8 = ί"' Ρ 'Ηΐ) WL' ( Ζ Η 9 · 8 = ί "'Ρ Ή ΐ) ΐΓΖ' ( ΖΗ · 6 = ί "'Ρ' ΗΖ) OVf '(s Ή2) ΐ9 · ε 9' · (z H) 0 'S ODH¾N + 0¾H N Η Χ
·τ_ωο O OS '^92ΐ '0901 '9S6 Λ ( 1)ΗΙ [TZSO] · Τ _ωο O OS '^ 92ΐ' 0901 '9S6 Λ (1) ΗΙ [TZSO]
0^ ¾(%99*¾ί 0 ^ ¾ (% 99 * ¾ί
·βε) 目 ¾遨 w^ ^-xm^ (°D m ^n¾r¾¾ cioni Βε) eyes ¾ 遨 w ^ ^ -xm ^ (° D m ^ n¾r¾¾ cioni
^91 Ό §^9 ·89) ^エ; ^邈べ ^ [ ^^ ( / — 一 /— 、 ^[ε ' ΐ][Ρ— Ζ 'ΐ], ^ベ — Η8— / — —ベ f — Ζ)] -^ΨΜ^ ^6Z)6Z M ^ 91 § § ^ 9 · 89) ^ d; ^ 邈 be ^ [^^ (/ — one / —, ^ [ε 'ΐ] [Ρ— Ζ' ΐ], ^ be — Η8— / — —be f — Ζ)]-^ ΨΜ ^ ^ 6Z) 6Z M
邈べ ^ [/^^ ^ ^ [/ ^^
— — /— 、 ^ [ε Ί][Ρ-2
 ](q6S) — — / —, ^ [Ε Ί] [Ρ-2 ] (q6S)
](q6S) — — / —, ^ [Ε Ί] [Ρ-2 ] (q6S)
•+(H+ ) ΐ : z/ui (eVH)S • + (H +) ΐ: z / ui (eVH) S
0l76S0C/900Zdf/X3d 901· 0C0l70l/900Z OAV
1, 3]チアゾールー 4 ィル)ォキシ]メチルホスホン酸ジェチル(103mg、 0. 247m mol)を使用して、実施例 1 (lc)に記載した方法に従い、標記目的化合物(78. 3mg 、収率 88%)を得た。 0l76S0C / 900Zdf / X3d 9010C0l70l / 900Z OAV 1,3] thiazol-4-yl) oxy] methylphosphonate (103 mg, 0.247 mmol) according to the method described in Example 1 (lc) and the title compound (78.3 mg, yield 88). %).
[0373] IR(KBr) v 944, 1074, 1265, 1415, 3060 cm"1; [0373] IR (KBr) v 944, 1074, 1265, 1415, 3060 cm "1;
max max
1H NMR(DMSO-d , 500MHz) δ 4.16 (2H, s), 4.42 (2H, d, J=9.3 Hz), 7.35 (1H, d 1H NMR (DMSO-d, 500MHz) δ 4.16 (2H, s), 4.42 (2H, d, J = 9.3 Hz), 7.35 (1H, d
6 6
, J=8.8 Hz), 7.40 (1H, d, J=8.8 Hz), 9.11 (2H, s), 9.21 (1H, s), 9.22 (1H, s); , J = 8.8 Hz), 7.40 (1H, d, J = 8.8 Hz), 9.11 (2H, s), 9.21 (1H, s), 9.22 (1H, s);
MS(ESI) m/z: 360 (M— H)— . MS (ESI) m / z: 360 (M— H) —.
<実施例 31 > <Example 31>
[ (7 ァセチルー 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキシ]メ チルホスホン酸(例示化合物番号 1-15) [(7-acetyl-8H-indeno [1,2-d] [l, 3] thiazole-4-yl) oxy] methylphosphonic acid (Exemplified Compound No. 1-15)
(31a) [ (7 ァセチルー 8H—インデノ [1, 2 d] [l, 3]チアゾールー 4 ィル)ォキ シ]メチルホスホン酸ジェチル (31a) [(7 Acetyl-8H-indeno [1, 2 d] [l, 3] thiazole-4-yl) oxy] demethyl methylphosphonate
実施例 27 (27a)で製造した [ (7 ブロモ 8H—インデノ [1, 2— d] [l, 3]チアゾ 一ルー 4—ィル)ォキシ]メチルホスホン酸ジェチル(16. 7g、 40. Ommol)をジメチ ルホルムアミド(170mL)に溶解し、ビス(トリフエ-ルホスフィン)パラジウムジクロリド( 5. 62g、 8. Ommol)、トリブチル(1—エトキシビュル)スズ(28. 9g、 80. Omol)をカロ え、 80°Cで 3時間攪拌した。室温まで冷却後、減圧下溶媒を留去し、酢酸ェチルを 加えた。得られた有機層を硫酸水素カリウム水溶液、飽和炭酸水素ナトリウム水溶液 、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥し、減圧下溶媒を留去した。 Example 27 [(7 Bromo 8H-indeno [1,2-d] [l, 3] thiazo-l-l-yl) oxy] methylphosphonate (16.7 g, 40. Ommol) prepared in (27a) Was dissolved in dimethylformamide (170 mL), and bis (triphenylphosphine) palladium dichloride (5.62 g, 8. Ommol) and tributyl (1-ethoxybutyl) tin (28.9 g, 80. Omol) were calorieated. The mixture was stirred at 80 ° C for 3 hours. After cooling to room temperature, the solvent was distilled off under reduced pressure, and ethyl acetate was added. The obtained organic layer was washed successively with aqueous potassium hydrogen sulfate solution, saturated aqueous sodium hydrogen carbonate solution and saturated brine, dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure.
[0374] 得られた残渣をエタノールに溶解後、 3M塩酸をカ卩ぇ pHを 1〜2に調製して室温で 2 時間攪拌した。反応液に酢酸ェチルを加え、飽和炭酸水素ナトリウム水溶液、飽和 食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥後、減圧下溶媒を留去した。得られ た残渣をシリカゲルクロマトグラフィー(酢酸ェチル:メタノール、 100 : 0— 80 : 20、 V ZV)を用いて精製し、標記目的化合物(9. 13g、収率 60%)を得た。 [0374] After the obtained residue was dissolved in ethanol, 3M hydrochloric acid was added to adjust the pH to 1-2, and the mixture was stirred at room temperature for 2 hours. Ethyl acetate was added to the reaction solution, washed successively with saturated aqueous sodium hydrogen carbonate solution and saturated brine, dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure. The obtained residue was purified by silica gel chromatography (ethyl acetate: methanol, 100: 0-80: 20, V ZV) to obtain the title object compound (9.13 g, yield 60%).
[0375] IR(KBr) v 838, 982, 1027, 1239, 1258, 1575, 1671, 2990, 3057 cm"1; [0375] IR (KBr) v 838, 982, 1027, 1239, 1258, 1575, 1671, 2990, 3057 cm "1;
max max
1H NMR(CDC1 , 400MHz) δ 1.35 (6H, t, J=7.0 Hz), 2.64 (3H, s), 4.25 (2H, s), 4 1H NMR (CDC1, 400MHz) δ 1.35 (6H, t, J = 7.0 Hz), 2.64 (3H, s), 4.25 (2H, s), 4
3 Three
.31 (2H, q, J=7.0Hz), 4.33 (2H, q, J=7.0Hz), 4.66 (2H, d, J=8.6Hz), 7.12 (1H, d, J= 8.6Hz), 7.82 (1H, d, J=8.6Hz), 8.83 (1H, s);
MS(FAB) m/z: 382 (M+H)+. .31 (2H, q, J = 7.0Hz), 4.33 (2H, q, J = 7.0Hz), 4.66 (2H, d, J = 8.6Hz), 7.12 (1H, d, J = 8.6Hz), 7.82 (1H, d, J = 8.6Hz), 8.83 (1H, s); MS (FAB) m / z: 382 (M + H) + .
(31b) [ (7 ァセチルー 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキ シ]メチノレホスホン酸 (31b) [(7 Acetyl-8H-indeno [1, 2-d] [l, 3] thiazol-4-yl) oxy] methinorephosphonic acid
実施例 31 (31a)で製造した [ (7 ァセチルー 8H—インデノ [1, 2— d] [l, 3]チア ゾールー 4 ィル)ォキシ]メチルホスホン酸ジェチル(140mg、 0. 367mmol)を使 用して、実施例 l (lc)に記載した方法 (この場合はトリエチルァミン(37mg、 0. 367 mmol)を共存させる)に従 、、標記目的化合物(107mg、収率 90%)を得た。 EXAMPLE 31 [(7-acetyl-8H-indeno [1,2-d] [l, 3] thiazol-4-yl) oxy] methylphosphonate (140 mg, 0.367 mmol) prepared in Example 31 (31a) was used. According to the method described in Example l (lc) (in this case, triethylamine (37 mg, 0.367 mmol) was allowed to coexist), the title object compound (107 mg, yield 90%) was obtained.
[0376] IR(KBr) v 937, 998, 1072, 1261, 1360, 1387, 1571, 1669, 3086, 3413 cm"1; [0376] IR (KBr) v 937, 998, 1072, 1261, 1360, 1387, 1571, 1669, 3086, 3413 cm "1;
max max
1H NMR(DMSO-d , 400MHz): δ 2.62 (3H, s), 4.45 (1H, d, J=9.0Hz), 7.32 (1H, d, 1H NMR (DMSO-d, 400MHz): δ 2.62 (3H, s), 4.45 (1H, d, J = 9.0Hz), 7.32 (1H, d,
6 6
J=8.8Hz), 7.96 (1H, d, J=8.8Hz), 9.18 (1H, s); J = 8.8Hz), 7.96 (1H, d, J = 8.8Hz), 9.18 (1H, s);
MS(FAB) m/z: 324 (M- H)— . MS (FAB) m / z: 324 (M- H) —.
<実施例 32 > <Example 32>
{ [7—(1ーヒドロキシェチル)ー811—ィンデノ[1, 2-d] [l, 3]チアゾールー 4ーィ ル]ォキシ }メチルホスホン酸 (例示化合物番号 1-76) {[7- (1-Hydroxyethyl) -811-indeno [1, 2-d] [l, 3] thiazol-4-yl] oxy} methylphosphonic acid (Exemplified Compound No. 1-76)
(32a) { [7—(1ーヒドロキシェチル) 8H インデノ [1, 2— d] [l, 3]チアゾールー 4 ィル]ォキシ }メチルホスホン酸ジェチル (32a) {[7- (1-Hydroxyethyl) 8H Indeno [1, 2— d] [l, 3] thiazole-4-yl] oxy} Jetyl methylphosphonate
実施例 31 (31a)で製造した [ (7 ァセチル— 8H—インデノ [1, 2 d] [l, 3]チア ゾールー 4 ィル)ォキシ]メチルホスホン酸ジェチル(190mg、 0. 498mmol)をメタ ノール(4mL)に溶解し、水素化ホウ素ナトリウム(57mg、 1. 49mmol)をカ卩え、室温 で 0.5時間攪拌した。反応液に酢酸ェチルを加え、有機層を希塩酸、飽和飽和炭酸 水素ナトリウム水溶液、飽和食塩水で順次洗浄し、無水硫酸ナトリウムで乾燥し、減 圧下溶媒を留去した。得られた残渣をシリカゲルクロマトグラフィー(酢酸ェチル:メタ ノール、 100 : 0— 85 : 15、 VZV)を用いて精製し、標記目的化合物(180mg、収率 94%)を得た。 Example 31 [(7-acetyl-8H-indeno [1,2d] [l, 3] thiazol-4-yl) oxy] methylphosphonate (190 mg, 0.498 mmol) prepared in Example 31 (31a) was converted to methanol ( 4 mL), sodium borohydride (57 mg, 1.49 mmol) was added, and the mixture was stirred at room temperature for 0.5 hour. Ethyl acetate was added to the reaction solution, and the organic layer was washed successively with dilute hydrochloric acid, saturated saturated aqueous sodium hydrogen carbonate solution and saturated brine, dried over anhydrous sodium sulfate, and the solvent was distilled off under reduced pressure. The obtained residue was purified by silica gel chromatography (ethyl acetate: methanol, 100: 0-85: 15, VZV) to obtain the title compound (180 mg, yield 94%).
[0377] IR(KBr) v 798, 838, 978, 1018, 1040, 1088, 1228, 1264, 1431, 1508, 2984, 33 max [0377] IR (KBr) v 798, 838, 978, 1018, 1040, 1088, 1228, 1264, 1431, 1508, 2984, 33 max
91 cm"1; 91 cm "1;
1H NMR(CDC1 , 400MHz) δ 1.35 (6H, t, J=7.0 Hz), 1.56 (3H, d, J=6.3Hz), 2.07 1H NMR (CDC1, 400MHz) δ 1.35 (6H, t, J = 7.0 Hz), 1.56 (3H, d, J = 6.3Hz), 2.07
3 Three
(1H, br s), 3.86 (1H, d, J=21.9Hz), 3.93 (1H, d, J=21.9Hz), 4.30 (2H, q, J=7.0Hz),
m辛爵¾瀚缀止
 (1H, br s), 3.86 (1H, d, J = 21.9Hz), 3.93 (1H, d, J = 21.9Hz), 4.30 (2H, q, J = 7.0Hz), m
(1H, br s), 3.86 (1H, d, J = 21.9Hz), 3.93 (1H, d, J = 21.9Hz), 4.30 (2H, q, J = 7.0Hz), m
m ^ ^ ^ ^ っ ^エ邈 4§ ^ ^ ^ ΨΜ^ [6 εο] m ^ ^ ^ ^ っ ^ d 邈 4§ ^ ^ ^ ΨΜ ^ [6 εο]
^/^ L^(\omm£S Ί マ fH 邈 ^、つ ¾¾^nra ) 一 ^ ^ [ε 'i][p-s
 ^ / ^ L ^ (\ omm £ S Ί ma fH 邈 ^, ¾¾ ^ n ra ) One ^ ^ [ε 'i] [ps
^ / ^ L ^ (\ omm £ S Ί ma fH 邈 ^, ¾¾ ^ n ra ) One ^ ^ [ε 'i] [ps
^エ;^邈べ ^ { [ / 一,— /— 、
 ^ E; ^ 邈 ^ ^ [[/ One, — / —,
^ E; ^ 邈 ^ ^ [[/ One, — / —,
(08- ΐ各粱呦 ^)邈べ ^ { ^ [ / 一 一 /— 、 ^ [ε Ί][Ρ-2 Ί] ^^-Η8- -ζ]} (08- ΐeach 粱 呦 ^) 邈 ^ {^ [/ Ichiichi / —, ^ [ε Ί] [Ρ-2 Ί] ^^-Η8- -ζ]}
<εε圏第 > <εε range>
·_(Η- ) 92S: ζ/ω (9VH)S · _ (Η-) 92S : ζ / ω (9VH) S
• (s 'Ηΐ) 9Γ6 '(zH9"8=f 'Ρ 'Ηΐ) SS^ '(ΖΗ9·8=1" 'Ρ 'Ηΐ) ΐΓΖ ' • ( s 'Ηΐ) 9Γ6' ( z H9 "8 = f 'Ρ' Ηΐ) SS ^ '(ΖΗ9 · 8 = 1"' Ρ 'Ηΐ) ΐΓΖ'
'Ηΐ) 36^ '(ζΗε·9=ί" 'Ηΐ) Z6'f '(zH9"8=f 'Ρ 'ΗΖ) WZZ=i 'Ρ 'Ηΐ) 66·ε'Ηΐ) 36 ^' (ζΗε · 9 = ί "'Ηΐ) Z6'f' ( z H9" 8 = f 'Ρ' ΗΖ) WZZ = i 'Ρ' Ηΐ) 66 · ε
Z'ZZ=[ 'Ρ 'Ηΐ) WZ '(zHZ"9=f 'Ρ Ήε) 6ε·ΐ 9 '· (zH )0 <9p-0S a)H N Ηχ Z'ZZ = ['Ρ' Ηΐ) WZ '(zHZ "9 = f' Ρ Ήε) 6ε · ΐ 9 '· ( z H) 0 <9 p-0S a) HN Η χ
'26οε '8εεε 'ssei 'soei ' \ 'ιζη 'εβετ 'T ST 'εζοτ Os6 Λ ( i) [8ζεο]'26 οε '8εεε' ssei 'soei' \ 'ιζη' εβετ 'T ST' εζοτ Os6 Λ (i) [8ζεο]
。 (%86 ¾ί. (% 86 ¾ί
、31!¾£1)呦^;] (¾|目¾遨
 ·0、3raoz
·0、3raoz
 Ί][Ρ 邈べ ^ { ^: [ / 一 一 /— 、 ^ [ε 'i][p-s Ί]
Ί][Ρ 邈べ ^ { ^: [ / 一 一 /— 、 ^ [ε 'i][p-s Ί]
 , 31! ¾ £ 1) 呦 ^ ;] (¾ | 目 ¾ 遨 · 0, 3raoz Ί] [Ρ 邈 ^ {^: [/ 1/1, ^ [ε 'i] [ps Ί]
, 31! ¾ £ 1) 呦 ^ ;] (¾ | 目 ¾ 遨 · 0, 3raoz Ί] [Ρ 邈 ^ {^: [/ 1/1, ^ [ε 'i] [ps Ί]
• (s 'Ηΐ) ΐ8·8 '(zH9"8=f 'Ρ 'Ηΐ) SZ'L '(ΖΗ• ( s ' Ηΐ) ΐ8 · 8 '( z H9 "8 = f' Ρ 'Ηΐ) SZ'L' ( Ζ Η
0l76S0C/900Zdf/X3d 601· 0C0l70l/900Z OAV
[0380] 得られた残渣をシリカゲルクロマトグラフィー(へキサン:酢酸ェチル、 1 : 8— 1 : 10、0l76S0C / 900Zdf / X3d 601 0C0l70l / 900Z OAV [0380] The obtained residue was subjected to silica gel chromatography (hexane: ethyl acetate, 1: 8—1: 10,
V/V)を用いて精製し、標記目的化合物(92. 8mg、収率 86%)を得た。 V / V) to give the title object compound (92.8 mg, yield 86%).
[0381] IR(KBr) v 1248, 1274, 1384, 1509, 1602, 2912, 2932, 2976, 3056 cm"1; [0381] IR (KBr) v 1248, 1274, 1384, 1509, 1602, 2912, 2932, 2976, 3056 cm "1;
max max
1H NMR(CDC1 , 400MHz): δ 1.35 (6H, t, J=7.0 Hz), 2.20 (0.45H, s), 2.28 (2.55H 1H NMR (CDC1, 400MHz): δ 1.35 (6H, t, J = 7.0 Hz), 2.20 (0.45H, s), 2.28 (2.55H
3 Three
, s), 3.79 (0.3H, s), 3.83 (0.45H, s), 4.02 (2.55H, s), 4.13 (1.7H, s), 4.31 (2H, q, J= 7.0 Hz), 4.33 (2H, q, J=7.0 Hz), 4.62 (2H, d, J=9.0 Hz), 7.05 (0.15H, d, J=8.6 Hz), 7.07 (1H, d, J=8.6 Hz), 7.34 (0.85H, d, J=8.6 Hz), 8.81 (1H, s); , s), 3.79 (0.3H, s), 3.83 (0.45H, s), 4.02 (2.55H, s), 4.13 (1.7H, s), 4.31 (2H, q, J = 7.0 Hz), 4.33 ( 2H, q, J = 7.0 Hz), 4.62 (2H, d, J = 9.0 Hz), 7.05 (0.15H, d, J = 8.6 Hz), 7.07 (1H, d, J = 8.6 Hz), 7.34 (0.85 H, d, J = 8.6 Hz), 8.81 (1H, s);
MS(FAB) m/z: 411 (M+H)+. MS (FAB) m / z: 411 (M + H) + .
(33b) { [7— (N—メトキシェタンイミドイル)— 8H—インデノ [1, 2-d] [l, 3]チアゾ 一ルー 4 ィル]ォキシ }メチルホスホン酸 (33b) {[7- (N-Methoxyethanimidyl)-8H-indeno [1, 2-d] [l, 3] thiazo 4-lu] oxy} methylphosphonic acid
実施例 33 (33a)で製造した { [7— (N—メトキシェタンイミドイル) 8H—インデノ [ 1, 2-d] [l, 3]チアゾール—4—ィル]ォキシ }メチルホスホン酸ジェチル(90. 3m g、 0. 220mmol)を使用して、実施例 1 (lc)に記載した方法に従い、標記目的化合 物(65. 3mg、収率 84%)を得た。 Example 33 {[7- (N-methoxyethaneimidoyl) 8H-indeno [1,2-d] [l, 3] thiazol-4-yl] oxy} prepared in Example 33 (33a) Jetyl methylphosphonate ( 90.3 mg, 0.220 mmol) was used to obtain the title compound (65.3 mg, 84% yield) according to the method described in Example 1 (lc).
[0382] IR(KBr) v 1272, 1384, 1492, 1510, 1565, 1603, 2818, 2902, 2936, 3099 cm"1; [0382] IR (KBr) v 1272, 1384, 1492, 1510, 1565, 1603, 2818, 2902, 2936, 3099 cm "1;
max max
1H NMR(DMSO-d , 400MHz) δ 2.14 (0.3H, s), 2.24 (2.7H, s), 3.72 (0.3H, s), 3. 1H NMR (DMSO-d, 400MHz) δ 2.14 (0.3H, s), 2.24 (2.7H, s), 3.72 (0.3H, s), 3.
6 6
83 (0.2H, s), 3.95 (2.7H, s), 4.13 (1.8H, s), 4.36 (2H, d, J=8.7 Hz), 7.11 (O.IH, d, J =8.6 Hz), 7.17 (O.IH, d, J=8.6 Hz), 7.22 (0.9H, d, J=8.6 Hz), 7.43 (0.9H, d, J=8.6 Hz), 9.15 (1H, s); 83 (0.2H, s), 3.95 (2.7H, s), 4.13 (1.8H, s), 4.36 (2H, d, J = 8.7 Hz), 7.11 (O.IH, d, J = 8.6 Hz), 7.17 (O.IH, d, J = 8.6 Hz), 7.22 (0.9H, d, J = 8.6 Hz), 7.43 (0.9H, d, J = 8.6 Hz), 9.15 (1H, s);
MS(FAB) m/z: 353 (M- H)— . MS (FAB) m / z: 353 (M- H) —.
<実施例 34 > <Example 34>
{ [7- (N, N ジメチルダリシル)— 8H—インデノ [1, 2-d] [l, 3]チアゾール—4 ィル]ォキシ }メチルホスホン酸塩酸塩 (例示化合物番号 1-59) {[7- (N, N dimethyldalysyl) — 8H—indeno [1, 2-d] [l, 3] thiazole-4-yl] oxy} methylphosphonate hydrochloride (Exemplary Compound No. 1-59)
(34a) { [7—(ブ口モアセチル) 8H—インデノ [1, 2— d] [l, 3]チアゾールー 4 ィル]ォキシ }メチルホスホン酸ジェチル (34a) {[7- (Bucchimoacetyl) 8H-indeno [1, 2- d] [l, 3] thiazole-4-yl] oxy} methyl phosphonate
実施例 31 (31a)で製造した [ (7 ァセチル— 8H—インデノ [1, 2 d] [l, 3]チア ゾールー 4 ィル)ォキシ]メチルホスホン酸ジェチル(1. 5g、 3. 93mmol)をジクロ ロメタン(40mL)に溶解し、そこへジイソプロピルェチルァミン(1. 37mL、 7. 86mm
ol)、トリメチルシリルトリフルォロメタンスルホナート(1. 07mL、 5. 90mmol)をカロえ 室温で 0.5時間攪拌した。さらに、 N ブロモスクシンイミド(1. 05g、 5. 90mmol)を 加え 0. 5時間攪拌した。 EXAMPLE 31 [(7-acetyl-8H-indeno [1,2d] [l, 3] thiazol-4-yl) oxy] methylphosphonate jetyl (1.5 g, 3.93 mmol) prepared in (31a) was dissolved in dichloromethane. Dissolve in methane (40mL) and diisopropyl ethyramine (1.37mL, 7.86mm) ol) and trimethylsilyl trifluoromethanesulfonate (1.07 mL, 5.90 mmol) were prepared and stirred at room temperature for 0.5 hour. Further, N bromosuccinimide (1.05 g, 5.90 mmol) was added and stirred for 0.5 hour.
[0383] 反応液に中性リン酸緩衝液をカ卩え、さらに希塩酸をカ卩えて酢酸ェチルで抽出した。 [0383] A neutral phosphate buffer was added to the reaction solution, and further diluted hydrochloric acid was added and extracted with ethyl acetate.
得られた有機層を飽和炭酸水素ナトリウム水溶液及び飽和食塩水で洗浄し、無水硫 酸ナトリウムで乾燥し、減圧下溶媒を留去した。 The obtained organic layer was washed with a saturated aqueous sodium hydrogen carbonate solution and saturated brine, dried over anhydrous sodium sulfate, and the solvent was distilled off under reduced pressure.
[0384] 得られた残渣をシリカゲルクロマトグラフィー (酢酸ェチル:メタノール、 50 : 1、 V/V[0384] The obtained residue was subjected to silica gel chromatography (ethyl acetate: methanol, 50: 1, V / V
)を用いて精製し、標記目的化合物 (741mg、収率 41%)を得た。 To give the title object compound (741 mg, yield 41%).
[0385] IR(KBr) v 1230, 1242, 1267, 1384, 1569, 1604, 1670, 2900, 2929, 2976 cm"1; [0385] IR (KBr) v 1230, 1242, 1267, 1384, 1569, 1604, 1670, 2900, 2929, 2976 cm "1;
max max
1H NMR(CDC1 , 400MHz) δ 1.36 (6H, t, J=7.0 Hz), 4.28 (2H, s), 4.30—4.38 (4H, 1H NMR (CDC1, 400MHz) δ 1.36 (6H, t, J = 7.0 Hz), 4.28 (2H, s), 4.30-4.38 (4H,
3 Three
m), 4.50 (2H, s), 4.69 (2H, d, J=9.0 Hz), 7.16 (1H, d, J=9.0 Hz), 7.86 (1H, d, J=9.0 Hz), 8.87 (1H, s); m), 4.50 (2H, s), 4.69 (2H, d, J = 9.0 Hz), 7.16 (1H, d, J = 9.0 Hz), 7.86 (1H, d, J = 9.0 Hz), 8.87 (1H, s);
MS(FAB) m/z: 460 (M+H)+. MS (FAB) m / z: 460 (M + H) + .
(34b) [7- (N, N ジメチルダリシル) 8H—インデノ [1, 2— d] [l, 3]チアゾー ルー 4 ィル]ォキシ }メチルホスホン酸ジェチル (34b) [7- (N, N dimethyldaricyl) 8H-indeno [1, 2-d] [l, 3] thiazoluro 4-yl] oxy} methyl phosphonate
実施例 34 (34a)で製造した { [7 (プロモアセチル) 8H—インデノ [1, 2 d] [1 , 3]チアゾールー 4 ィル]ォキシ }メチルホスホン酸ジェチル(100mg、 0. 217mm ol)をジクロロメタン(7mL)に溶解し、そこへ 2. OM ジメチルァミン テトラヒドロフラ ン溶液 (0. 22mL、 0. 435mmol)をカ卩ぇ窒素雰囲気下、 2時間攪拌した。反応液に 水を加え、酢酸ェチルで抽出した。得られた有機層を飽和炭酸水素ナトリウム水溶液 及び飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、減圧下溶媒を留去した。得 られた残渣をシリカゲルクロマトグラフィー(ジクロロメタン:メタノール、 20 : 1— 8 : 1、 V /V)を用いて精製し、標記目的化合物(39. 4mg、収率 43%)を得た。 EXAMPLE 34 {[7 (Promoacetyl) 8H-indeno [1,2d] [1,3] thiazol-4-yl] oxy} methylphosphonate (100 mg, 0.217 mmol) prepared in Example 34 (34a) was dissolved in dichloromethane. (OM) was dissolved in (7 mL), and 2. OM dimethylamine tetrahydrofuran solution (0.22 mL, 0.435 mmol) was stirred in a nitrogen atmosphere for 2 hours. Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with saturated aqueous sodium hydrogen carbonate solution and saturated brine, dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure. The obtained residue was purified by silica gel chromatography (dichloromethane: methanol, 20: 1-8: 1, V / V) to obtain the title object compound (39.4 mg, yield 43%).
[0386] IR(KBr) v 1246, 1386, 1572, 1679, 2909, 2931, 2980, 3425 cm"1; [0386] IR (KBr) v 1246, 1386, 1572, 1679, 2909, 2931, 2980, 3425 cm "1;
max max
1H NMR(CDC1 , 400MHz) δ 1.35 (6H, t, J=7.0 Hz), 2.46 (6H, s), 3.85 (2H, s), 4 1H NMR (CDC1, 400MHz) δ 1.35 (6H, t, J = 7.0 Hz), 2.46 (6H, s), 3.85 (2H, s), 4
3 Three
.26 (2H, s), 4.32 (2H, q, J=7.0 Hz), 4.34 (2H, q, J=7.0 Hz), 4.66 (2H, d, J=9.0 Hz), 7.10 (1H, d, J=8.6 Hz), 7.90 (1H, d, J=8.6 Hz), 8.83 (1H, s); .26 (2H, s), 4.32 (2H, q, J = 7.0 Hz), 4.34 (2H, q, J = 7.0 Hz), 4.66 (2H, d, J = 9.0 Hz), 7.10 (1H, d, J = 8.6 Hz), 7.90 (1H, d, J = 8.6 Hz), 8.83 (1H, s);
MS(FAB) m/z: 425 (M+H)+.
(34c) { [7- (N, N ジメチルダリシル)— 8H—インデノ [1, 2— d] [l, 3]チアゾー ルー 4—ィル]ォキシ }メチルホスホン酸塩酸塩 MS (FAB) m / z: 425 (M + H) + . (34c) {[7- (N, N dimethyldalysyl) — 8H—indeno [1, 2— d] [l, 3] thiazole 4-yl] oxy} methylphosphonate hydrochloride
実施例 34 (34b)で製造した [7— (N, N ジメチルダリシル)— 8H—インデノ [1, 2 -d] [l, 3]チアゾール—4—ィル]ォキシ }メチルホスホン酸ジェチル(38. 4mg、0 . O90mmol)を濃塩酸に溶解し、 100°Cで 8時間攪拌した。 Example 34 [7- (N, N dimethyldaricyl) -8H-indeno [1,2-d] [l, 3] thiazol-4-yl] oxy} prepared in Example 34 (34b) Jetyl methylphosphonate (38 (4 mg, 0.090 mmol) was dissolved in concentrated hydrochloric acid and stirred at 100 ° C. for 8 hours.
冷却後、反応液にァセトニトリルを加え、析出した結晶をろ取した。得られた結晶をァ セトニトリルで洗浄し、標記目的化合物(27. 9mg、収率 76%)を得た。 After cooling, acetonitrile was added to the reaction solution, and the precipitated crystals were collected by filtration. The obtained crystals were washed with acetonitrile to give the title object compound (27.9 mg, yield 76%).
[0387] IR(KBr) v 1242, 1573, 1683, 2928, 2963, 3046, 3394 cm"1; [0387] IR (KBr) v 1242, 1573, 1683, 2928, 2963, 3046, 3394 cm "1;
max max
1H NMR(D O, 500MHz): δ 3.03 (6H, s), 3.77 (2H, s), 4.15—4.24 (2H, m), 4.77 (2 1H NMR (D 2 O, 500 MHz): δ 3.03 (6H, s), 3.77 (2H, s), 4.15—4.24 (2H, m), 4.77 (2
2 2
H, s), 6.96-6.99 (1H, m), 7.58-7.64 (1H, m), 9.11 (1H, s); H, s), 6.96-6.99 (1H, m), 7.58-7.64 (1H, m), 9.11 (1H, s);
MS(FAB) m/z: 367 (M— H)— (free form). MS (FAB) m / z: 367 (M— H) — (free form).
<実施例 35 > <Example 35>
{ [7 (ピロリジン 1 ィルァセチル)—8H—インデノ [1, 2-d] [l, 3]チアゾール 4 ィル]ォキシ }メチルホスホン酸塩酸塩 (例示化合物番号 1-61 ) {[7 (Pyrrolidine 1-Ilacetyl) —8H-Indeno [1, 2-d] [l, 3] thiazole 4-yl] oxy} Methylphosphonate hydrochloride (Exemplary Compound No. 1-61)
(35a) { [7 (ピロリジン 1 ィルァセチル)—8H—インデノ [1, 2— d] [l, 3]チア ゾール 4 ィル]ォキシ }メチルホスホン酸ジェチル (35a) {[7 (Pyrrolidine 1-ilacetyl) -8H-indeno [1,2-d] [l, 3] thiazol-4-yl] oxy} methyl phosphonate
実施例 34 (34a)で製造した { [7 (プロモアセチル) 8H—インデノ [1, 2 d] [1 , 3]チアゾールー 4 ィル]ォキシ }メチルホスホン酸ジェチル(138mg、 0. 300mm ol)をジクロロメタン(5mL)に溶解し、そこへピロリジン(0. 05mL、 0. 600mmol)を 加え窒素雰囲気下、 2時間攪拌した。 EXAMPLE 34 {[7 (Promoacetyl) 8H-indeno [1,2d] [1,3] thiazol-4-yl] oxy} prepared in Example 34 (34a) methyl phosphonate (138 mg, 0.300 mmol) was dissolved in dichloromethane. (5 mL) was dissolved, pyrrolidine (0.05 mL, 0.600 mmol) was added thereto, and the mixture was stirred under a nitrogen atmosphere for 2 hours.
[0388] 反応液に水を加え、酢酸ェチルで抽出した。得られた有機層を飽和炭酸水素ナトリ ゥム水溶液及び飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、減圧下溶媒を[0388] Water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with saturated aqueous sodium hydrogen carbonate solution and saturated brine, dried over anhydrous sodium sulfate, and the solvent was removed under reduced pressure.
*y した。 * y.
[0389] 得られた残渣をシリカゲルクロマトグラフィー(ジクロロメタン:メタノール、 10: 1、 VZ [0389] The obtained residue was subjected to silica gel chromatography (dichloromethane: methanol, 10: 1, VZ
V)を用いて精製し、標記目的化合物(60. lmg、収率 45%)を得た。 V) was used to obtain the title object compound (60. lmg, yield 45%).
[0390] IR(KBr) v 1243, 1268, 1569, 1667, 2792, 2822, 2934, 2965 cm"1; [0390] IR (KBr) v 1243, 1268, 1569, 1667, 2792, 2822, 2934, 2965 cm "1;
max max
1H NMR(CDC1 , 400MHz) δ 1.35 (6H, t, J=7.0 Hz), 1.83—1.87 (4H, m), 2.68-2.7 1H NMR (CDC1, 400MHz) δ 1.35 (6H, t, J = 7.0 Hz), 1.83—1.87 (4H, m), 2.68-2.7
3 Three
2 (4H, m), 3.99 (2H, s), 4.27 (2H, s), 4.32 (2H, q, J=7.0 Hz), 4.33 (2H, q, J=7.0 Hz)
, 4.66 (2H, d, J=8.6 Hz), 7.09 (IH, d, J=8.6 Hz), 7.90 (IH, d, J=8.6 Hz), 8.82 (IH, s); 2 (4H, m), 3.99 (2H, s), 4.27 (2H, s), 4.32 (2H, q, J = 7.0 Hz), 4.33 (2H, q, J = 7.0 Hz) , 4.66 (2H, d, J = 8.6 Hz), 7.09 (IH, d, J = 8.6 Hz), 7.90 (IH, d, J = 8.6 Hz), 8.82 (IH, s);
MS(FAB) m/z: 451 (M+H)+. MS (FAB) m / z: 451 (M + H) + .
(35b) { [7— (ピロリジン— 1—ィルァセチル)—8H—インデノ [1, 2— d] [l, 3]チア ゾール 4 ィル]ォキシ }メチルホスホン酸塩酸塩 (35b) {[7- (Pyrrolidine-1-ylacetyl) -8H-indeno [1,2-d] [l, 3] thiazole 4-yl] oxy} methylphosphonate hydrochloride
実施例 35 (35a)で製造した { [7— (ピロリジン— 1—ィルァセチル) 8H—インデノ Prepared in Example 35 (35a) {[7- (pyrrolidine-1-ylacetyl) 8H-indeno
[1, 2-d] [l, 3]チアゾール—4—ィル]ォキシ }メチルホスホン酸ジェチル(59. 3m g、 0. 132mmol)を濃塩酸に溶解し、 100°Cで 8時間攪拌した。 [1,2-d] [l, 3] thiazole-4-yl] oxy} Jetyl methylphosphonate (59.3 mg, 0.132 mmol) was dissolved in concentrated hydrochloric acid and stirred at 100 ° C. for 8 hours.
[0391] 冷却後、反応液にァセトニトリルを加え、析出した結晶をろ取した。得られた結晶を ァセトニトリルで洗浄し、標記目的化合物(56. Omg、収率 99%)を得た。 [0391] After cooling, acetonitrile was added to the reaction solution, and the precipitated crystals were collected by filtration. The obtained crystals were washed with acetonitrile to give the title object compound (56. Omg, yield 99%).
[0392] IR(KBr) v 1238, 1574, 1684, 2691, 2833, 1958, 3405 cm"1; [0392] IR (KBr) v 1238, 1574, 1684, 2691, 2833, 1958, 3405 cm "1;
max max
1H NMR(D O, 400MHz): δ 2.06—2.20 (4H, m), 3.16—3.23 (2H, m), 3.82—3.89 (4H 1H NMR (D 2 O, 400 MHz): δ 2.06—2.20 (4H, m), 3.16—3.23 (2H, m), 3.82—3.89 (4H
2 2
, m), 4.22 (2H, d, J=9.4 Hz), 4.86 (2H, s), 7.02 (IH, d, J=8.2 Hz), 7.62 (IH, d, J=9. 0 Hz), 9.15 (IH, s); , m), 4.22 (2H, d, J = 9.4 Hz), 4.86 (2H, s), 7.02 (IH, d, J = 8.2 Hz), 7.62 (IH, d, J = 9.0 Hz), 9.15 (IH, s);
MS(FAB) m/z: 395 (M+H)+ (free form). MS (FAB) m / z: 395 (M + H) + (free form).
<実施例 36 > <Example 36>
{ [7—(ァミノカルボ-ル) 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル] ォキシ }メチルホスホン酸 (例示化合物番号 1-35) {[7- (Aminocarbol) 8H-indeno [1, 2-d] [l, 3] thiazol-4-yl] oxy} methylphosphonic acid (Exemplified Compound No. 1-35)
(36a) 7- [ (ジエトキシホスホリル)メトキシ] 1一才キソインダンー4一力ルボン酸ァ リル (36a) 7- [(Diethoxyphosphoryl) methoxy] 1 year old xoindane 4
実施例 1 ( la)で製造した [ (7 ブロモ 3 ォキソ 2, 3 -ジヒドロ 1H インデ ン— 4—ィル)ォキシ]メチルホスホン酸ジェチル(40. 0g、 106mmol)を、 N, N—ジ メチルホルムアミド(150mL)に溶解し、そこへ 2 トリメチルシリルエタノール(75mL 、 523mmol)、トリェチルァミン(50. OmL、 359mmol)、 1, 3 ビス(ジフエ-ルホス フイノ)プロノ ン(13. 2g、 32. Ommol)及び酢酸パラジウム(7. 80g、 31. 9mmol) を加え、一酸化炭素置換をし、一酸化炭素雰囲気下、 70°Cで 30時間撹拌した。 [(7 Bromo-3oxo2,3-dihydro-1H indene-4-yl) oxy] methylphosphonate jetyl (40.0 g, 106 mmol) prepared in Example 1 (la) was converted to N, N-dimethylformamide. (150 mL), 2 trimethylsilylethanol (75 mL, 523 mmol), triethylamine (50. OmL, 359 mmol), 1,3 bis (diphenylphosphino) pronone (13.2 g, 32. Ommol) and acetic acid Palladium (7.80 g, 31.9 mmol) was added to perform carbon monoxide substitution, and the mixture was stirred at 70 ° C. for 30 hours in a carbon monoxide atmosphere.
[0393] 室温まで冷却後、セライトろ過をした。得られたろ液にトリェチルァミン(30. OmL、 215mmol)及び臭化ァリル(18. OmL、 208mmol)を加え、 50°Cで 5時間撹拌した
[0394] 室温まで冷却後、反応液に水を加え、酢酸ェチルで抽出した。得られた有機層を 飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥し、減圧下溶媒を留去した。 [0393] After cooling to room temperature, the mixture was filtered through Celite. To the obtained filtrate was added triethylamine (30. OmL, 215 mmol) and allyl bromide (18. OmL, 208 mmol), and the mixture was stirred at 50 ° C for 5 hours. [0394] After cooling to room temperature, water was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure.
[0395] 得られた残渣をテトラヒドロフラン (400mL)に溶解し、そこへ 1. OMフッ化テトラブ チルアンモ-ゥムーテトラヒドロフラン溶液(65. OmL、 65. Ommol)を加え、室温で 4 時間撹拌した。 [0395] The obtained residue was dissolved in tetrahydrofuran (400 mL), and 1. OM fluorinated tetrabutylammonium chloride tetrahydrofuran solution (65. OmL, 65. Ommol) was added thereto, followed by stirring at room temperature for 4 hours.
[0396] 反応液にトリエチノレアミン(30. OmL、 215mmol)及び臭ィ匕ァリノレ(13. OmL、 150 mmol)を加え、室温で 2時間撹拌した。 [0396] Triethynoleamine (30. OmL, 215 mmol) and odorinoleol (13. OmL, 150 mmol) were added to the reaction solution, and the mixture was stirred at room temperature for 2 hours.
[0397] 反応液を減圧下溶媒留去し、酢酸ェチルで希釈し、得られた有機層を飽和食塩水 で洗浄し、無水硫酸マグネシウムで乾燥し、減圧下溶媒を留去した。 [0397] The reaction mixture was evaporated under reduced pressure, diluted with ethyl acetate, and the resulting organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure.
[0398] 得られた残渣をシリカゲルカラムクロマトグラフィー (酢酸ェチル)を用いて精製し、標 記目的化合物(28. 7g、収率 71%)を得た。 [0398] The obtained residue was purified using silica gel column chromatography (ethyl acetate) to obtain the target compound (28.7 g, yield 71%).
[0399] IR(KBr) v 983, 1032, 1161, 1251, 1265, 1491, 1588, 1597, 1707, 1714, 2989 c max [0399] IR (KBr) v 983, 1032, 1161, 1251, 1265, 1491, 1588, 1597, 1707, 1714, 2989 c max
-1 -1
m ; m;
1H NMR(CDC1 , 400MHz) δ 1.38 (6H, t, J=7.0 Hz), 2.64-2.70 (2H, m), 3.43—3.4 1H NMR (CDC1, 400MHz) δ 1.38 (6H, t, J = 7.0 Hz), 2.64-2.70 (2H, m), 3.43—3.4
3 Three
8 (2H, m), 4.34 (2H, q, J=7.0 Hz), 4.36 (2H, q, J=7.0 Hz), 4.48 (2H, d, J=9.0 Hz), 4 .83 (2H, dt, J=1.2 Hz, J=5.5 Hz), 5.31 (IH, dd, J=1.2 Hz, J=10.6 Hz), 5.41 (IH, dd, J=1.2 Hz, J=17.2 Hz), 5.99—6.11 (IH, m), 6.93 (IH, d, J=8.6 Hz), 8.28 (IH, d, J=8. 6 Hz); 8 (2H, m), 4.34 (2H, q, J = 7.0 Hz), 4.36 (2H, q, J = 7.0 Hz), 4.48 (2H, d, J = 9.0 Hz), 4.83 (2H, dt , J = 1.2 Hz, J = 5.5 Hz), 5.31 (IH, dd, J = 1.2 Hz, J = 10.6 Hz), 5.41 (IH, dd, J = 1.2 Hz, J = 17.2 Hz), 5.99-6.11 ( IH, m), 6.93 (IH, d, J = 8.6 Hz), 8.28 (IH, d, J = 8.6 Hz);
MS(FAB) m/z: 383 (M+H)+. MS (FAB) m / z: 383 (M + H) + .
(36b) 4 [ (ジエトキシホスホリル)メトキシ]— 8H—インデノ [1, 2-d] [l, 3]チアゾ ール 7—カルボン酸ァリル (36b) 4 [(Diethoxyphosphoryl) methoxy] -8H-indeno [1,2-d] [l, 3] thiazol 7-Carboxylate
実施例 36 (36a)で製造した 7— [ (ジエトキシホスホリル)メトキシ]― 1 ォキソイン ダンー4一力ルボン酸ァリル(28. 7g、 75. lmmol) )を使用して、実施例 3 (3a— 3b )に記載した方法に従 、、標記目的化合物(18. 6g、収率 71%)を得た。 Example 36 7-[(Diethoxyphosphoryl) methoxy] -1-oxoindan-4 gallic arborate (28.7 g, 75. lmmol)) prepared in Example 36 (36a) was used to produce Example 3 (3a— According to the method described in 3b), the title object compound (18.6 g, yield 71%) was obtained.
[0400] IR(KBr) v 980, 1026, 1083, 1109, 1146, 1262, 1390, 1580, 1711, 2984 cm"1; [0400] IR (KBr) v 980, 1026, 1083, 1109, 1146, 1262, 1390, 1580, 1711, 2984 cm "1;
max max
1H NMR(CDC1 , 400MHz) δ 1.35 (6H, t, J=6.8 Hz), 4.25 (2H, s), 4.33 (2H, q, J= 1H NMR (CDC1, 400MHz) δ 1.35 (6H, t, J = 6.8 Hz), 4.25 (2H, s), 4.33 (2H, q, J =
3 Three
6.8 Hz), 4.35 (2H, q, J=6.8 Hz), 4.67 (2H, d, J=8.8 Hz), 4.86 (2H, dt, J=1.5 Hz, J=5
·ΐ)、 ^Κ^^ ( ΰ :,^ ^ — ε)— S— ^エ—" [邈 ^、(Iorara6 ·8 6.8 Hz), 4.35 (2H, q, J = 6.8 Hz), 4.67 (2H, d, J = 8.8 Hz), 4.86 (2H, dt, J = 1.5 Hz, J = 5 · Ϊ́), ^ Κ ^^ (ΰ:, ^ ^ — ε) — S— ^ E— "[邈 ^, (I orara 6 · 8
I
 、 ^ マ ^I , ^ Ma ^
、 ^ マ ^I , ^ Ma ^
-Ν 'Ν ¾ (louiuig ·ε、 ·ΐ)邈ベ^ / 一 Ζ— /— 、 ^ [ε Ί][Ρ-2 Ί -Ν 'Ν ¾ (louiuig · ε, · ΐ) 邈 Be ^ / Ζ Ζ— / —, ^ [ε Ί] [Ρ-2 Ί
], ^ベ — Η8— [ ^^ ( ίί 、^!ェ Ο ]— ·η¾ί§ (39ε) 9εί^¾ϊ第], ^ Be — Η8— [^^ (ίί, ^! Ο]) · · η¾ί§ ( 3 9ε) 9εί ^ ¾ϊ 第
^エ 邈べ ^ { ^: [ / ^ E 邈 ^^ ^ [
— 一 /— 、 ^ [ε Ί][Ρ-2 'ΐ],^ベ —Η8—( ^ / ^ )—ζ]}(Ρ9ε) — One / —, ^ [ε Ί] [Ρ-2 'ΐ], ^ Be —Η8— (^ / ^) —ζ]} (Ρ9ε)
•+(H+ ) 8S: ζ/ω (9VH)S • + (H +) 8S: ζ / ω (9VH) S
•(s 'Ηΐ) 9Γ6 '(ΖΗ 8"8=f 'P 'Ηΐ) S8"Z • ( s 'Ηΐ) 9Γ6' ( Ζ Η 8 "8 = f 'P' Ηΐ) S8" Z
'(ZH 8"8=f 'P 'Ηΐ) SZ'L '(ZH S"6=f 'P 'ΗΖ) OL'f '(ZH 0"Z=f 'b 'ΗΖ) 9Z'f '(ZH 0"Z=f ' b ΉΖ) fZ'f '(s Ή2) ε '(ΖΗ 0"Z=f 'Η9) S2"T 9: (ZH )0 <9p-OS a)H N Ηχ '(ZH 8 "8 = f' P 'Ηΐ) SZ'L' ( Z HS" 6 = f 'P' ΗΖ) OL'f '( Z H 0 "Z = f' b 'ΗΖ) 9Z'f' ( Z H 0 "Z = f ' b ΉΖ) fZ'f' (s Ή2) ε '( Ζ Η 0" Z = f' Η9) S2 "T 9: (ZH) 0 <9 p-OS a) HN Η χ
·τ_ωο s ε 'TOTS '^862 '8262 'LUZ 'L19Z '9Ζ Ζ 'ΟΟΖΐ Τ _ωο s ε 'TOTS' ^ 862 '8262' LUZ 'L19Z' 9Ζ Ζ 'ΟΟΖΐ
'Ζ09ΐ 'ΐ83ΐ '603ΐ '88ST '6ΖΖΙ '39Π 'ΐδΐΐ '280ΐ ' ^Οΐ '^Οΐ '6Z6 Λ ( 1)ΗΙ [SOW)] 'Ζ09ΐ' ΐ83ΐ '603ΐ' 88ST '6ΖΖΙ '39 Π' ΐδΐΐ '280ΐ' ^ Οΐ '^ Οΐ' 6Z6 Λ (1) ΗΙ (SOW)]
(%6 *¾ί §69 ·9) 目 ¾遨
 #¾ ¾ "¥Di 缀氺マ (H 峯氺邈 ¾Ή靱 つ辛爵 瀚缀止 S教 ' M^^ [10^0](% 6 * ¾ί §69 9) Eye ¾ 遨 # ¾ ¾ "¥ Di 缀 氺 マ (H 峯 氺 邈 ¾ Ή
#¾ ¾ "¥Di 缀氺マ (H 峯氺邈 ¾Ή靱 つ辛爵 瀚缀止 S教 ' M^^ [10^0](% 6 * ¾ί §69 9) Eye ¾ 遨 # ¾ ¾ "¥ Di 缀 氺 マ (H 峯 氺 邈 ¾ Ή
。 つ 翻 S っ。 OS、"¾^ij¾(Ioura¾8 'I §01 ·2 . Tsu S OS, "¾ ^ ij¾ (I oura ¾8 'I §01 2
)マ
 )
)
/ -ェ fH、(IOUIUI8 ·εε
 / -FH , (I OUIUI 8 · εε
/ -FH , (I OUIUI 8 · εε
^ ¾ (louiui^ ·8Ι §6 ·Ζ) / (! 邈べ^ 一 Ζ— /— 、 ^ [ε Ί][Ρ-2 Ί ^ ¾ (louiui ^ · 8Ι §6 · Ζ) / (! 邈 ベ ^ 一 Ζ— / —, ^ [ε Ί] [Ρ-2 Ί
], ^ベ — Η8— [ 、^ ^^ェ 、) ]— ^niiii (q9s) 9εί^¾?第 ], ^ Be — Η8— [, ^ ^^ e,]] — ^ niiii (q9s) 9εί ^ ¾?
邈べ^ / 一 Ζ— /—
 z : ζ/ω (9VH)S ^ Be ^ / One Ζ— z: ζ / ω (9VH) S
z : ζ/ω (9VH)S ^ Be ^ / One Ζ— z: ζ / ω (9VH) S
•(s • ( s
'Ηΐ) S8'8 '(ΖΗ 8·8=ί" 'Ρ 'Ηΐ) ΐ0·8 '(ΖΗ 8·8=ί" 'Ρ 'Ηΐ) ΐΓΖ '(ω 'Ηΐ) εΐ·9- εθ·9 '(ΖΗ ΐ •Ζΐ=ί" 'ΖΗ 6'Ζ 'ΖΗ ΖΊ=ί 'ΡΡΡ 'Ηΐ) W '(ZH S T=f 'ΖΗ 3"ΐ=Γ 'ΡΡ 'Ηΐ) 2S"S '(ΖΗ 6· 'Ηΐ) S8'8' ( Ζ Η 8 · 8 = ί "'Ρ' Ηΐ) ΐ0 · 8 '( Ζ Η 8 · 8 = ί"' Ρ 'Ηΐ) ΐΓΖ' (ω 'Ηΐ) εΐ · 9- εθ · 9 '( Ζ Η ΐ • Ζΐ = ί "' ΖΗ 6'Ζ ' Ζ Η ί = ί' ΡΡΡ 'Ηΐ) W' (ZH ST = f ' Ζ Η 3" ΐ = Γ' ΡΡ 'Ηΐ) 2S "S '(ΖΗ 6 ·
0l76S0C/900Zdf/X3d S 0C0l70l/900Z OAV
[0403] 反応液に水を加え、クロ口ホルムで抽出した。得られた有機層を飽和食塩水で洗浄 し、無水硫酸マグネシウムで乾燥し、減圧下溶媒を留去した。 0l76S0C / 900Zdf / X3d S 0C0l70l / 900Z OAV [0403] Water was added to the reaction solution, and the mixture was extracted with black mouth form. The obtained organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure.
[0404] 得られた粗製の目的化合物を、メタノールージイソプロピルエーテルより再結晶し、 標記目的化合物(1. 26g、収率 87%)を得た。 [0404] The obtained crude target compound was recrystallized from methanol-diisopropyl ether to obtain the title target compound (1.26 g, yield 87%).
[0405] IR(KBr) v 1034, 1089, 1162, 1237, 1314, 1343, 1389, 1426, 1440, 1510, 1583, max [0405] IR (KBr) v 1034, 1089, 1162, 1237, 1314, 1343, 1389, 1426, 1440, 1510, 1583, max
1626, 1678, 2900, 2927, 2983, 3176, 3369 cm"1; 1626, 1678, 2900, 2927, 2983, 3176, 3369 cm "1;
1H NMR(CD OD, 400MHz): δ 1.33 (6H, t, J=7.0 Hz), 4.22 (2H, s), 4.28-4.37 (4 1H NMR (CD OD, 400MHz): δ 1.33 (6H, t, J = 7.0 Hz), 4.22 (2H, s), 4.28-4.37 (4
3 Three
H, m), 4.74 (2H, d, J=8.6 Hz), 7.22 (IH, d, J=8.6 Hz), 7.67 (IH, d, J=8.6 Hz), 9.02 (IH, s); H, m), 4.74 (2H, d, J = 8.6 Hz), 7.22 (IH, d, J = 8.6 Hz), 7.67 (IH, d, J = 8.6 Hz), 9.02 (IH, s);
MS(FAB) m/z: 383 (M+H)+. MS (FAB) m / z: 383 (M + H) + .
(366) { [7—(ァミノカルボニル)ー811—ィンデノ[1, 2-d] [l, 3]チアゾールー 4 ィル]ォキシ }メチルホスホン酸 (366) {[7- (Aminocarbonyl) -811-indeno [1, 2-d] [l, 3] thiazole-4-yl] oxy} methylphosphonic acid
実施例 36 (36d)で製造した { [7— (ァミノカルボ-ル)— 8H—インデノ [1, 2— d] [ Prepared in Example 36 (36d) {[7— (Aminocarbol) — 8H—Indeno [1, 2— d] [
I, 3]チアゾールー 4 ィル]ォキシ }メチルホスホン酸ジェチル(1. 34mg、 3. 50m ol)をァセトニトリル(20mL)に溶解し、そこへョードトリメチルシラン(2. 5mL、 17. 6 mmol)を加え、窒素雰囲気下、室温で 2時間撹拌した。 I, 3] thiazole-4-yl] oxy} Jetyl methylphosphonate (1.34 mg, 3.50 mol) is dissolved in acetonitrile (20 mL), where it is trimethylsilane (2.5 mL, 17.6 mmol). And stirred at room temperature for 2 hours under a nitrogen atmosphere.
[0406] 減圧下溶媒を留去し、得られた残渣に水を加えた。析出した紫色粗結晶をチォ硫 酸ナトリウム水溶液で洗浄し脱色した後、メタノールで洗浄して、標記目的化合物(9 14mg、収率 80%)を得た。 [0406] The solvent was distilled off under reduced pressure, and water was added to the resulting residue. The precipitated purple crude crystals were washed with an aqueous sodium thiosulfate solution and decolorized, and then washed with methanol to obtain the title object compound (914 mg, yield 80%).
[0407] IR(KBr) v 1073, 1157, 1220, 1277, 1390, 1491, 1580, 1664, 2338, 2767, 2854, max [0407] IR (KBr) v 1073, 1157, 1220, 1277, 1390, 1491, 1580, 1664, 2338, 2767, 2854, max
2924, 3095, 3188, 3343 cm"1; 2924, 3095, 3188, 3343 cm "1;
1H NMR(DMSO-d , 500MHz): δ 4.21 (2H, s), 4.39 (2H, d, J=8.8 Hz), 7.21 (IH, d 1H NMR (DMSO-d, 500MHz): δ 4.21 (2H, s), 4.39 (2H, d, J = 8.8 Hz), 7.21 (IH, d
6 6
, J=8.8 Hz), 7.28 (IH, br s), 7.66 (IH, d, J=8.8 Hz), 7.83 (IH, br s), 9.16 (IH, s); , J = 8.8 Hz), 7.28 (IH, br s), 7.66 (IH, d, J = 8.8 Hz), 7.83 (IH, br s), 9.16 (IH, s);
MS(FAB) m/z: 325 (M- H)— . MS (FAB) m / z: 325 (M- H) —.
<実施例 37 > <Example 37>
({7 [ (メチルァミノ)カルボ-ル] 8H インデノ [1, 2— d] [l, 3]チアゾールー 4 ーィル }ォキシ)メチルホスホン酸 (例示化合物番号 1-37) ({7 [(Methylamino) carbol] 8H Indeno [1, 2— d] [l, 3] thiazole-4-yl} oxy) methylphosphonic acid (Exemplified Compound No. 1-37)
(37a) ({7 [ (メチルァミノ)カルボ-ル ] 8H—インデノ [1, 2— d] [l, 3]チアゾー
ルー 4ーィル }ォキシ)メチルホスホン酸ジェチル (37a) ({7 [(Methylamino) carbol] 8H-indeno [1, 2- d] [l, 3] thiazole Roux 4-yl} oxy) demethyl methylphosphonate
実施例 36 (36c)で製造した 4 [ (ジエトキシホスホリル)メトキシ]— 8H インデノ [ 1, 2-d] [l, 3]チアゾール—7—カルボン酸(1. 35g、 3. 52mmol)及び塩酸メチ ルァミン (484mg、 7. 74mmol)を使用して、実施例 36 (36d)に記載した方法に従 い、標記目的化合物 (1. 35g、収率 93%)を得た。 Example 36 4 [(Diethoxyphosphoryl) methoxy] -8H indeno [1,2-d] [l, 3] thiazole-7-carboxylic acid (1.35 g, 3.52 mmol) and hydrochloric acid prepared in (36c) The title object compound (1.35 g, 93% yield) was obtained using methylamine (484 mg, 7.74 mmol) according to the method described in Example 36 (36d).
[0408] IR(KBr) v 1026, 1048, 1162, 1248, 1269, 1390, 1430, 1504, 1545, 1582, 1650, max [0408] IR (KBr) v 1026, 1048, 1162, 1248, 1269, 1390, 1430, 1504, 1545, 1582, 1650, max
2906, 2932, 2981, 3071, 3307 cm"1; 2906, 2932, 2981, 3071, 3307 cm "1;
1H NMR(CDC1 , 500MHz) δ 1.35 (6H, t, J=6.8 Hz), 3.03 (2H, d, J=4.9 Hz), 4.20 1H NMR (CDC1, 500MHz) δ 1.35 (6H, t, J = 6.8 Hz), 3.03 (2H, d, J = 4.9 Hz), 4.20
3 Three
(2H, s), 4.29-4.37 (4H, m), 4.62 (2H, d, J=8.8 Hz), 6.24 (IH, br s), 7.04 (IH, d, J= 8.3 Hz), 7.41 (IH, d, J=8.3 Hz), 8.83 (IH, s); (2H, s), 4.29-4.37 (4H, m), 4.62 (2H, d, J = 8.8 Hz), 6.24 (IH, br s), 7.04 (IH, d, J = 8.3 Hz), 7.41 (IH , d, J = 8.3 Hz), 8.83 (IH, s);
MS(FAB) m/z: 397 (M+H)+. MS (FAB) m / z: 397 (M + H) + .
(37b) ({7 [ (メチルァミノ)カルボ-ル ] 8H—インデノ [1, 2-d] [l, 3]チアゾー ルー 4ーィル }ォキシ)メチルホスホン酸 (37b) ({7 [(Methylamino) carbol] 8H-indeno [1, 2-d] [l, 3] thiazol 4-yl} oxy) methylphosphonic acid
実施例 37 (37a)で製造した({ 7 [ (メチルァミノ)カルボ-ル]— 8H インデノ [ 1 , 2-d] [l, 3]チアゾールー 4ーィル }ォキシ)メチルホスホン酸ジェチル(1. 02g、 3 . 50mol)を使用して、実施例 1 (lc)に記載した方法に従い、標記目的化合物 (845 mg、収率 97%)を得た。 Example 37 ({7 [(Methylamino) carbol] —8H indeno [1,2-d] [l, 3] thiazol-4-yl} oxy) methylphosphonate (1.02 g, 3 The title object compound (845 mg, yield 97%) was obtained according to the method described in Example 1 (lc) using 50 mol).
[0409] IR(KBr) v 1075, 1154, 1266, 1277, 1383, 1412, 1430, 1486, 1504, 1548, 1581, max [0409] IR (KBr) v 1075, 1154, 1266, 1277, 1383, 1412, 1430, 1486, 1504, 1548, 1581, max
1660, 2329, 2906, 2938, 3084, 3389 cm"1; 1660, 2329, 2906, 2938, 3084, 3389 cm "1;
1H NMR(DMSO-d , 500MHz) 1H NMR (DMSO-d, 500MHz)
6 : δ 2.78 (3H, d, J=4.4 Hz), 4.19 (2H, s), 4.39 (2H, d 6: δ 2.78 (3H, d, J = 4.4 Hz), 4.19 (2H, s), 4.39 (2H, d
, J=8.8 Hz), 7.23 (IH, d, J=8.8 Hz), 7.58 (IH, d, J=8.8 Hz), 8.25—8.31 (IH, m), 9.1 6 (IH, s); , J = 8.8 Hz), 7.23 (IH, d, J = 8.8 Hz), 7.58 (IH, d, J = 8.8 Hz), 8.25—8.31 (IH, m), 9.1 6 (IH, s);
MS(FAB) m/z: 339 (M- H)— . MS (FAB) m / z: 339 (M- H) —.
<実施例 38 > <Example 38>
( {7 [ (メトキシァミノ)カルボ-ル ] 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4ーィル }ォキシ)メチルホスホン酸(例示化合物番号 1-49) ({7 [(Methoxyamino) carbol] 8H-indeno [1, 2-d] [l, 3] thiazol-4-yl} oxy) methylphosphonic acid (Exemplary Compound No. 1-49)
(38a) ({7— [ (メトキシァミノ)カルボ-ル]— 8H—インデノ [1, 2 d] [l, 3]チアゾ 一ルー 4ーィル }ォキシ)メチルホスホン酸ジェチル
実施例 36 (36c)で製造した 4 [ (ジエトキシホスホリル)メトキシ]— 8H インデノ [ 1, 2-d] [l, 3]チアゾール 7—カルボン酸(150mg、 0. 391mmol)及びメトキシ ルァミン塩酸塩(39mg、 0. 470mmol)、卜リエチルァミン(48mg、 0. 470mmol)を 使用して、実施例 36 (36d)に記載した方法に従い、標記目的化合物(161mg、収 率 99%)を得た。 (38a) ({7— [(Methoxyamino) carbol] — 8H—indeno [1, 2 d] [l, 3] thiazo 1-l 4-yl} oxy) methylphosphonate Example 36 4 [(Diethoxyphosphoryl) methoxy] -8H Indeno [1,2-d] [l, 3] thiazole 7-carboxylic acid (150 mg, 0.391 mmol) and methoxylamine hydrochloride prepared in (36c) (39 mg, 0.470 mmol) and triethylamine (48 mg, 0.470 mmol) were used to give the title object compound (161 mg, 99% yield) according to the method described in Example 36 (36d).
[0410] IR(KBr) v 812, 983, 1028, 1054, 1082, 1222, 1267, 1584、 1672, 2979, 3183 cm max [0410] IR (KBr) v 812, 983, 1028, 1054, 1082, 1222, 1267, 1584, 1672, 2979, 3183 cm max
-1 -1
1H NMR(CDC1 , 400MHz) δ 1.35 (6H, t, J=7.0 Hz), 3.92 (3H, s), 3.97 (2H, s), 4 1H NMR (CDC1, 400MHz) δ 1.35 (6H, t, J = 7.0 Hz), 3.92 (3H, s), 3.97 (2H, s), 4
3 Three
.28-4.40 (4H, m), 4.44 (2H, d, J=9.4 Hz), 6.85 (1H, d, J=8.6 Hz), 7.42 (1H, d, J=8. 6 Hz), 8.78 (1H, s), 9.62 (1H, s); .28-4.40 (4H, m), 4.44 (2H, d, J = 9.4 Hz), 6.85 (1H, d, J = 8.6 Hz), 7.42 (1H, d, J = 8.6 Hz), 8.78 ( 1H, s), 9.62 (1H, s);
MS(FAB) m/z: 413 (M+H)+. MS (FAB) m / z: 413 (M + H) + .
(38b) ({7— [ (メトキシァミノ)カルボ-ル]— 8H—インデノ [1, 2— d] [l, 3]チアゾ 一ルー 4ーィル }ォキシ)メチルホスホン酸 (38b) ({7— [(Methoxyamino) carbol] — 8H—indeno [1, 2— d] [l, 3] thiazo 4-Lu} oxy) methylphosphonic acid
実施例 38 (38a)で製造した({ 7 [ (メトキシァミノ)カルボ二ル]— 8H インデノ [ 1 , 2 d] [l, 3]チアゾールー 4ーィル }ォキシ)メチルホスホン酸ジェチル(150mg、 0. 364mmol)を使用して、実施例 36 (36e)に記載した方法に従い、標記目的化合 物(106mg、収率 82%)を得た。 Example 38 ({7 [(Methoxyamino) carbonyl] -8H indeno [1,2d] [l, 3] thiazol-4-yl} oxy) methylphosphonate (150 mg, 0.364 mmol) prepared in Example 38 (38a) To give the title object compound (106 mg, 82% yield) according to the method described in Example 36 (36e).
[0411] IR(KBr) v 948, 1072, 1268, 1382, 1430, 1483, 1582, 1667, 2938, 3098, 3185 c max [0411] IR (KBr) v 948, 1072, 1268, 1382, 1430, 1483, 1582, 1667, 2938, 3098, 3185 c max
1 1
m ; m;
1H NMR(DMSO-d , 400MHz): δ 3.72 (3Η, s), 4.18 (2Η, s), 4.39 (2H, d, J=8.6Hz) 1H NMR (DMSO-d, 400MHz): δ 3.72 (3Η, s), 4.18 (2Η, s), 4.39 (2H, d, J = 8.6Hz)
6 6
, 7.22 (1H, d, J=8.6Hz), 7.48 (1H, d, J=8.6Hz), 9.15 (1H, s), 11.53 (1H, s); , 7.22 (1H, d, J = 8.6Hz), 7.48 (1H, d, J = 8.6Hz), 9.15 (1H, s), 11.53 (1H, s);
MS(FAB) m/z: 355 (M- H)— . MS (FAB) m / z: 355 (M- H) —.
<実施例 39 > <Example 39>
[ (7— { [ (2—メトキシェチル)ァミノ]カルボ-ル}— 8H—インデノ [1, 2-d] [l, 3] チアゾール 4 ィル)ォキシ]メチルホスホン酸(例示化合物番号 1-53) [(7— {[(2-Methoxyethyl) amino] carbol} — 8H-indeno [1,2-d] [l, 3] thiazole-4-yl) oxy] methylphosphonic acid (Exemplary Compound No. 1-53)
(39a) [ (7- { [ (2—メトキシェチル)ァミノ]カルボ二ル} 8H—インデノ [1, 2 d] [ 1, 3]チアゾール 4 ィル)ォキシ]メチルホスホン酸ジェチル (39a) [(7- {[(2-Methoxyethyl) amino] carbonyl} 8H-indeno [1, 2 d] [1, 3] thiazole 4 yl) oxy] methyl phosphonate
実施例 36 (36c)で製造した 4 [ (ジエトキシホスホリル)メトキシ]— 8H インデノ [
1, 2-d] [l, 3]チアゾール 7—カルボン酸(150mg、 0. 391mmol)及び 2—メト キシェチルァミン(35mg、 0. 470mmol)を使用して、実施例 36 (36d)に記載した 方法に従い、標記目的化合物(160mg、収率 93%)を得た。 Example 36 4 [(Diethoxyphosphoryl) methoxy] -8H indeno prepared in (36c) [ 1,2-d] [l, 3] thiazole The method described in Example 36 (36d) using 7-carboxylic acid (150 mg, 0.391 mmol) and 2-methoxetylamine (35 mg, 0.470 mmol). To give the title compound (160 mg, 93% yield).
[0412] IR(KBr) v 1233, 1270, 1506, 1541, 1647, 2899, 2936, 2980, 3287 cm"1; [0412] IR (KBr) v 1233, 1270, 1506, 1541, 1647, 2899, 2936, 2980, 3287 cm "1;
max max
1H NMR(CDC1 , 500MHz): δ 1.35 (6H, t, J=7.3 Hz), 3.41 (3H, s), 3.59 (2H, t, J= 1H NMR (CDC1, 500MHz): δ 1.35 (6H, t, J = 7.3 Hz), 3.41 (3H, s), 3.59 (2H, t, J =
3 Three
4.9 Hz), 3.67 (2H, q, J=4.9 Hz), 4.24 (2H, s), 4.32 (2H, q, J=7.3 Hz), 4.35 (2H, q, J =7.3 Hz), 4.66 (2H, d, J=8.8 Hz), 6.43-6.50 (IH, m), 7.09 (IH, d, J=8.8 Hz), 7.46 ( IH, d, J=8.8 Hz), 8.85 (IH, s); 4.9 Hz), 3.67 (2H, q, J = 4.9 Hz), 4.24 (2H, s), 4.32 (2H, q, J = 7.3 Hz), 4.35 (2H, q, J = 7.3 Hz), 4.66 (2H , d, J = 8.8 Hz), 6.43-6.50 (IH, m), 7.09 (IH, d, J = 8.8 Hz), 7.46 (IH, d, J = 8.8 Hz), 8.85 (IH, s);
MS(FAB) m/z: 441 (M+H)+. MS (FAB) m / z: 441 (M + H) + .
(39b) [ (7— { [ (2—メトキシェチル)ァミノ]カルボ二ル} 8H—インデノ [1, 2-d] [ 1, 3]チアゾール 4 ィル)ォキシ]メチルホスホン酸 (39b) [(7— {[(2-Methoxyethyl) amino] carbonyl} 8H-indeno [1, 2-d] [1, 3] thiazole 4-yl) oxy] methylphosphonic acid
実施例 39 (39a)で製造した [ (7— { [ (2—メトキシェチル)ァミノ]カルボ-ル} 8H インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキシ]メチルホスホン酸ジェチ ル(158mg、 0. 359mmol)を使用して、実施例 1 (lc)に記載した方法に従い、標記 目的化合物(95. 7mg、収率 69%)を得た。 Example 39 [(7 — {[(2-Methoxyethyl) amino] carbol} 8H indeno [1,2-d] [1,2-d] [l, 3] thiazol-4-yl) oxy] methylphosphonic acid prepared in (39a) The title compound (95.7 mg, 69% yield) was obtained according to the method described in Example 1 (lc) using 1 mg (158 mg, 0.359 mmol).
[0413] IR(KBr) v 1272, 1486, 1548, 1580, 1644, 2835, 2929, 2977, 3079, 3298 cm"1; [0413] IR (KBr) v 1272, 1486, 1548, 1580, 1644, 2835, 2929, 2977, 3079, 3298 cm "1;
max max
1H NMR(DMSO-d , 400MHz): δ 3.28 (3H, s), 3.43 (2H, t, J=5.1 Hz), 3.47 (2H, d 1H NMR (DMSO-d, 400MHz): δ 3.28 (3H, s), 3.43 (2H, t, J = 5.1 Hz), 3.47 (2H, d
6 6
, J=5.1 Hz), 4.19 (2H, s), 4.39 (2H, d, J=9.0 Hz), 7.22 (IH, d, J=8.6 Hz), 7.6 (IH, d , J=8.6 Hz), 8.36-8.39 (IH, m), 9.15 (IH, s); , J = 5.1 Hz), 4.19 (2H, s), 4.39 (2H, d, J = 9.0 Hz), 7.22 (IH, d, J = 8.6 Hz), 7.6 (IH, d, J = 8.6 Hz), 8.36-8.39 (IH, m), 9.15 (IH, s);
MS(FAB) m/z: 383 (M— H)— . MS (FAB) m / z: 383 (M— H) —.
<実施例 40 > <Example 40>
[ (7— { [ (3 ヒドロキシプロピル)ァミノ]カルボ-ル} - 8H インデノ [ 1 , 2— d] [ 1 , 3]チアゾールー 4 ィル)ォキシ]メチルホスホン酸(例示化合物番号 1-54) [(7— {[(3 Hydroxypropyl) amino] carbol}-8H Indeno [1, 2 — d] [1, 3] thiazole-4-yl) oxy] methylphosphonic acid (Exemplary Compound No. 1-54)
(40a) [ (7— { [ (3 ヒドロキシプロピル)ァミノ]カルボ-ル}—811—インデノ [1, 2— d] [1, 3]チアゾールー 4 ィル)ォキシ]メチルホスホン酸ジェチル (40a) [(7— {[(3 Hydroxypropyl) amino] carbol} —811—Indeno [1, 2— d] [1, 3] thiazole-4-yl) oxy] methyl phosphonate
実施例 36 (36c)で製造した 4 [ (ジエトキシホスホリル)メトキシ]— 8H インデノ [ 1, 2— d] [l, 3]チアゾール 7—カルボン酸(100mg、 0. 261mmol)及び 3 アミ ノー 1 プロパノール(28mg、 0. 37mmol)を使用して、実施例 36 (36d)に記載し
·τ_ωο πεε '9^9ΐ 'τεεΐ 'εβοτ 'εεοτ ( i) [9iw)] Example 36 4 [(Diethoxyphosphoryl) methoxy] -8H indeno [1,2-d] [l, 3] thiazole 7-carboxylic acid (100 mg, 0.261 mmol) and 3 amino 1 prepared in 36c Propanol (28 mg, 0.37 mmol) was used as described in Example 36 (36d). · Τ _ωο πεε '9 ^ 9ΐ' τεεΐ 'εβοτ' εεοτ (i) [9iw)]
(%^ *¾ί §^9 ·Ζ9)呦^ ]Κ¾目 ¾遨 (% ^ * ¾ί § ^ 9 · Ζ9) 呦 ^] Κ¾ 目 ¾ 遨
(P9S) 9εί^¾ϊ第、ェっ ¾Τ¾¾(ΐοππυειε ·ο、3ΐΐιε ·ΖΖ) ^Λ^^ ^(P9S) 9εί ^ ¾ϊ 第, ¾Τ¾¾ (ΐοππυ ε ι ε · ο, 3ΐΐι ε · ΖΖ) ^ Λ ^^ ^
], ^ベ — Η8— [ ^^ ( ίί 、^!ェ Ο ] ·η¾ί§ (39ε) 9εί^¾ϊ第], ^ Be — Η8— [^^ (ίί, ^! Ο] · η¾ί§ ( 3 9ε) 9εί ^ ¾ϊ 第
^エ 邈べ ^ [/^^ ( / 一 /— : ^ [ε 'χ] [
 ^ E 邈 be ^ [/ ^^ (/ 1 / —: ^ [ε 'χ] [
^ E 邈 be ^ [/ ^^ (/ 1 / —: ^ [ε 'χ] [
(os- ΐ各粱呦^: ^ ^)邈べ ^ i^J^ ( / →- /— 、 ^ [ (os- ΐeach 粱 呦 ^: ^^) ^ ^ i ^ J ^ (/ →-/ —, ^ [
S Ί][Ρ-2
 ]}— Ζ)] S Ί] [Ρ-2 ]} — Ζ)]
]}— Ζ)] S Ί] [Ρ-2 ]} — Ζ)]
•_(H- ) S8S: ζ/ω (9VH)S • _ (H-) S8S: ζ / ω (9VH) S
•(s 'Ηΐ) ΖΓ6 '(ω 'Ηΐ) ΐε·8- 6S'8 '(ΖΗ ε·8=ί" 'Ρ 'Ηΐ) 09· '(ΖΗ ε·8=ί" 'Ρ 'Ηΐ) fZ'L '(ΖΗ ε·6=ί" 'Ρ 'ΗΖ) Off '(s 'ΗΖ) 6Vf '(ω • ( s ' Ηΐ) ΖΓ6 '(ω' Ηΐ) ΐε · 8-6S'8 '( Ζ Η ε · 8 = ί "' Ρ 'Ηΐ) 09 ·' (ΖΗ ε · 8 = ί"'Ρ' Ηΐ ) fZ'L '( Ζ Η ε · 6 = ί "' Ρ 'ΗΖ) Off' ( s 'ΗΖ) 6Vf' (ω
Ήζ) 0 -8Υ£ '(ω 'Η ε·ε— ιε·ε '(ω Ήε) 9"ΐ 9: (ZH OOS ,9p-os a)H N HX Ήζ) 0 -8Υ £ '(ω' Η ε · ε— ιε · ε '(ω Ήε) 9 "ΐ 9: (ZH OOS , 9 p-os a) HNH X
·τ xsee '6 οε 'is6s ' ss Os9i 'ssei 'ε½ΐ 'soei 'ssw ' 9si Λ ( i) [siw)]· Τ xsee '6 οε' is6s 'ss Os9i' ssei 'ε½ΐ' soei 'ssw' 9si Λ (i) [siw)]
。 ra9 -oD ^ 目 ¾遨ヽ . ra 9 -oD ^ eye ¾ 遨 ヽ
^ _η¾¾^(99ε)9είϋ¾?第 ^a¾r¾¾a°^ ss Ό ·66) ^ ェ; ^邈べ ^ [ 、^ ( / — 一 /— 、 ^[ε Ί][Ρ-2 'ΐ], べ —Η8 ^ _η¾¾ ^ (99ε) 9είϋ¾? 第 ^ a¾r¾¾a ° ^ ss Ό · 66) ^ ee; ^ Η8
- { -^ ^ [ ^ {Λ^ ^^ ^ -ε)]}-ζ)] -^ΨΜ^ i^o ) O -{-^ ^ [^ (Λ ^ ^^ ^ -ε)]}-ζ)]-^ ΨΜ ^ i ^ o) O
邈べ ^ [ ^: ( / 一 /—/: ^ [ε Ί] [ρ ^ ^ [^: (/ 一 / — /: ^ [ε Ί] [ρ
—ζ ' I ] ^ -us - { /-^ /^ [ ^ . {Λ^Ά ^^ ^ - ε) ] } - ) ] (qo^) —Ζ 'I] ^ -us-{/-^ / ^ [^. (Λ ^ Ά ^^ ^-ε)]}-)] (qo ^)
(w ) iff : z/ui (evd)S (w) iff: z / ui (evd) S
•(S 'HI) εε·8 '(ΖΗ • ( S 'HI) εε8' ( Ζ Η
S"8=f 'Ρ 'Ηΐ) WL '(ΖΗ S"8=f 'Ρ 'Ηΐ) Ϊ0"Ζ '(ω 'Ηΐ) Ζ8·9— 6Ζ·9 '(ΖΗ S"6=f 'Ρ 'ΗΖ) 8 S "8 = f 'Ρ' Ηΐ) WL '(ΖΗ S" 8 = f' Ρ 'Ηΐ) Ϊ0 "Ζ' (ω 'Ηΐ) Ζ8 · 9— 6Ζ · 9' ( Ζ Η S" 6 = f ' Ρ 'ΗΖ) 8
'(ω Ή^) L£'f-6Z'f '(s Ή2) fVf '(ΖΗ 6"S=f 'Η2) Τ '(ΖΗ 6"S=f 'Η2) 99Τ '(ω Ή ^) L £' f-6Z'f '(s Ή2) fVf' (ΖΗ 6 "S = f 'Η2) Τ' (ΖΗ 6" S = f 'Η2) 99Τ
'(ΖΗ 6"S=f '; u!nb Ή £S'l '(ΖΗ S"Z=f '; 'Η9) SS"T 9: (ZH OOS つ dつ) WN Hx '(ΖΗ 6 "S = f'; u! Nb Ή £ S'l '( Ζ Η S" Z = f';'Η9) SS "T 9: (ZH OOS and d) WN H x
^ ura 83^ε '^εε Ό62ε 'osez 'es6S Os9i 'esei ' si 'eoei Λ ( i) [ww)]^ ura 83 ^ ε '^ εε Ό62ε' osez 'es6S Os9i' esei 'si' eoei Λ (i) [ww)]
(%06 ¾ί
 (% 06 ¾ί
(% 06 ¾ί
0l76S0C/900Zdf/X3d 031- 0C0l70l/900Z OAV
εθ·9 '(ΖΗ 9·8=ί" 'Ρ 'ΗΖ) 99· '(ΖΗ ΓΖ '^Η ΓΖ=Γ '¾Ρ 'Hf) ZZ' '(s 'Η2) ZZ' '(ΖΗ ε·9 0l76S0C / 900Zdf / X3d 031- 0C0l70l / 900Z OAV εθ · 9 '(Ζ Η 9 · 8 = ί "' Ρ 'ΗΖ) 99 ·' (Ζ Η ΓΖ '^ Η ΓΖ = Γ' ¾Ρ 'Hf) ZZ''(s' Η2) ZZ '' (Ζ Η ε9
=f 'Ρ 'ΗΖ) 6Ζ'£ '(ΖΗ ΓΖ=Γ 'Η9) SS"T '(s Ή6) ΐΟ·ΐ 9 '· (ζΗ )0 ^ Ιつ) Ηχ = f 'Ρ' ΗΖ) 6Ζ '£' ( Ζ Η ΓΖ = Γ 'Η9) SS "T' (s Ή6) ΐΟ · ΐ 9 '· ( ζ Η) 0 ^ Ι) Η χ
:ト ω。 goes ' S62 '6^9ΐ 'sssi 'δεοΐ ( i) [8iw)] (%ο *¾ί
 : G ω. goes 'S62' 6 ^ 9ΐ 'sssi' δεοΐ (i) [8iw)] (% ο * ¾ί
: G ω. goes 'S62' 6 ^ 9ΐ 'sssi' δεοΐ (i) [8iw)] (% ο * ¾ί
(Ρ9ε)
 ' )べ^ ^(Ρ9ε) ') Be ^ ^
' )べ^ ^(Ρ9ε) ') Be ^ ^
^ ^ S. (1OUIUI60S Ό、3υ¾8)邈ベ^ / 一 Ζ— /— 、 ^ [ε Ί][Ρ-2 Ί ^ ^ S. (1 OUIUI 60S Ό , 3υ ¾8) 邈 Be ^ / Ζ Ζ — / —, ^ [ε Ί] [Ρ-2 Ί
], ^ベ — Η8— [ ^^ ( ίί 、^!ェ Ο ] ·η¾ί§ (39ε) 9εί^¾ϊ第], ^ Be — Η8— [^^ (ίί, ^! Ο] · η¾ί§ ( 3 9ε) 9εί ^ ¾ϊ 第
^エ 邈べ ^ { / 一 /— 、 ^ ^ D ^ ^ {/ One / —, ^
[ε 'i][p-s ' I ] - [ /—^ /^ { ^Λ^ ·^^) ] - } ) (^S^) [ε 'i] [p-s' I]-[/ — ^ / ^ {^ Λ ^ · ^^)]-}) (^ S ^)
( ΐ S- ΐ各粱呦^: ^ ^)邈べ ^ {^J {^y→- /— 、
 ]—Ζ}) (ΐ S- ΐ each 粱 呦 ^: ^ ^) ^ ^ {^ J {^ y →-/ —, ] —Ζ})
]—Ζ}) (ΐ S- ΐ each 粱 呦 ^: ^ ^) ^ ^ {^ J {^ y →-/ —, ] —Ζ})
•_(H- ) 6ZS: z/ui (IS3)S • _ (H-) 6ZS: z / ui (IS3) S
•(s 'Ηΐ) ΖΓ6 '(ΖΗ 9"S=f '; 'Ηΐ) ε ·8 '(ΖΗ 8·8=ί" 'Ρ 'Ηΐ) 29"Ζ '(ΖΗ 8·8=ί" 'Ρ 'Ηΐ) fZ •Ζ '(ΖΗ 8·8=ί" 'Ρ 'ΗΖ) 6S' '(s 'ΗΖ) OZ'f '(ΖΗ ΐ·9 'ΖΗ Γ9=Γ 'ΡΡ 'ΗΖ) 9ΐ·ε '(ω 'Ηΐ) 6 • ( s 'Ηΐ) ΖΓ6' ( Ζ Η 9 "S = f ';' Ηΐ) ε · 8 '( Ζ Η 8 · 8 = ί"' Ρ 'Ηΐ) 29 "Ζ' (ΖΗ 8 · 8 = ί "'Ρ' Ηΐ) fZ • Ζ '(ΖΗ 8 · 8 = ί"' Ρ 'ΗΖ) 6S''( s ' ΗΖ) OZ'f '( Ζ Η ΐ · 9' Ζ Η Γ9 = Γ 'ΡΡ' ΗΖ) 9ΐ · ε '(ω' Ηΐ) 6
0·ΐ— SO'I '(ω 'ΗΖ) LV0-ZV0 '(ω Ή2) 9 '· (zH 00S <9p-OS a)H N Ηχ 0 · ΐ— SO'I '(ω' ΗΖ) LV0-ZV0 '(ω Ή2) 9' · ( z H 00S <9 p-OS a) HN Η χ
·τ_ωο 6SS 'ZS9T 'ΐ½ΐ '292ΐ '036 Λ ( 1)ΗΙ [ΖΐΜ)] 0^ ¾(%98*¾ί u¾ ·9 )呦 目 2暴 m
 、Su¾ ·ΐ9) ^ ェ; ^邈べ ^ [ 、^ ( / — 一 /— 、 ^[ε Ί][Ρ-2 'ΐ], べ —Η8 邈べ ^ [/^^ ( / 一 /—/: ^ [ε 'χ] [· Τ _ωο 6SS 'ZS9T' ΐ½ΐ '292ΐ' 036 Λ (1) ΗΙ [ΖΐΜ)] 0 ^ ¾ (% 98 * ¾ί u¾ · 9) , Su ¾ · ΐ9) ^ é; ^ 邈 be ^ [, ^ (/ — one / —, ^ [ε Ί] [Ρ-2 'ΐ], be —Η8 邈 be ^ [/ ^^ (/ one / — /: ^ [Ε 'χ] [
、Su¾ ·ΐ9) ^ ェ; ^邈べ ^ [ 、^ ( / — 一 /— 、 ^[ε Ί][Ρ-2 'ΐ], べ —Η8 邈べ ^ [/^^ ( / 一 /—/: ^ [ε 'χ] [· Τ _ωο 6SS 'ZS9T' ΐ½ΐ '292ΐ' 036 Λ (1) ΗΙ [ΖΐΜ)] 0 ^ ¾ (% 98 * ¾ί u¾ · 9) , Su ¾ · ΐ9) ^ é; ^ 邈 be ^ [, ^ (/ — one / —, ^ [ε Ί] [Ρ-2 'ΐ], be —Η8 邈 be ^ [/ ^^ (/ one / — /: ^ [Ε 'χ] [
V-Z { /-^ /^[ ^ {Λ^-^Λ^Ά ^ ^) ] } - ζ) ] (qx^)V-Z {/-^ / ^ [^ (Λ ^-^ Λ ^ Ά ^ ^)]}-ζ)] (qx ^)
(w ) L£ : z/ui (evd)S (w) L £: z / ui (evd) S
•(s 'HI) S8"8 '(ZH 9"8=f 'P 'HI) 9VL '(ΖΗ 9"8=f 'P 'Ηΐ) 60· '(ω 'Ηΐ) 6Γ9— W9 '(ΖΗ 9"8=f 'Ρ 'ΗΖ) 9'f '(ΖΗ ΓΖ '^Η ΓΖ=Γ '¾Ρ 'H • (s 'HI) S8 "8' ( Z H 9" 8 = f 'P' HI) 9VL '( Ζ Η 9 "8 = f' P 'Ηΐ) 60 ·' (ω 'Ηΐ) 6Γ9— W9' ( Ζ Η 9 "8 = f 'Ρ' ΗΖ) 9'f '( Ζ Η ΓΖ' ^ Η ΓΖ = Γ '¾Ρ' H
) εε· '(s Ή2) '(ζΗ ο" 'ΖΗ re=f 'ρρ Ήε) εε·ε '(ζΗ ΓΖ=Γ Ή9) ss'i '(ω Ή ΐ) εΐ·ΐ— εθ·ΐ '(ω Ή2) Ϊ9 -33 '(ω Ή2) Z2O-LZO 9 '· (ζΗ )0 ^ Ιつ) Ητ ) εε · '(s Ή2)' ( ζ Η ο "'ΖΗ re = f' ρρ Ήε) εε · ε '( ζ Ζ ΓΖ = Γ Ή9) ss'i' (ω Ή ΐ) εΐ · ΐ— εθ · ΐ '(ω Ή2) Ϊ9 -33' (ω Ή2) Z2O-LZO 9 '· ( ζ Η) 0 ^ Ι) Η τ
0l76S0C/900Zdf/X3d 1-31. 0C0l70l/900Z OAV
-6.11 (IH, m), 7.11 (IH, d, J=8.6 Hz), 7.43 (IH, d, J=8.6 Hz), 8.85 (IH, s); 0l76S0C / 900Zdf / X3d 1-31. 0C0l70l / 900Z OAV -6.11 (IH, m), 7.11 (IH, d, J = 8.6 Hz), 7.43 (IH, d, J = 8.6 Hz), 8.85 (IH, s);
MS(FAB) m/z: 453 (M+H)+. MS (FAB) m / z: 453 (M + H) + .
(42b) ({7 [ (ネオペンチルァミノ)カルボ-ル ]—8H—インデノ [1, 2-d] [l, 3] チアゾール 4—ィル }ォキシ)メチルホスホン酸 (42b) ({7 [(Neopentylamino) carbol] -8H-indeno [1,2-d] [l, 3] thiazole 4-yl} oxy) methylphosphonic acid
実施例 42 (42a)で製造した({ 7 [ (ネオペンチルァミノ)カルボ-ル ] 8H—イン デノ [1, 2— d] [l, 3]チアゾールー 4ーィル }ォキシ)メチルホスホン酸ジェチル(61 . 3mg、 0. 135mmol)を使用して、実施例 1 (lc)に記載した方法に従い、標記目的 化合物(23. 9mg、収率 45%)を得た。 Example 42 ({7 [(Neopentylamino) carbol] 8H-indeno [1,2-d] [l, 3] thiazol-4-yl} oxy) methylphosphonic acid jetty prepared in Example 42 (42a) 61.3 mg, 0.135 mmol) was used to give the title object compound (23.9 mg, 45% yield) according to the method described in Example 1 (lc).
[0419] IR(KBr) v 954, 1261, 1543, 1638, 2957 cm"1; [0419] IR (KBr) v 954, 1261, 1543, 1638, 2957 cm "1;
max max
1H NMR(DMSO-d , 500MHz): δ 0.93 (9H, s), 3.12 (2H, d, J=6.3 Hz), 4.17 (2H, s 1H NMR (DMSO-d, 500MHz): δ 0.93 (9H, s), 3.12 (2H, d, J = 6.3 Hz), 4.17 (2H, s
6 6
), 4.39 (2H, d, J=9.3 Hz), 7.24 (IH, d, J=8.5 Hz), 7.59 (IH, d, J=8.5 Hz), 8.26 (IH, t, J=6.3 Hz), 9.17 (IH, s); ), 4.39 (2H, d, J = 9.3 Hz), 7.24 (IH, d, J = 8.5 Hz), 7.59 (IH, d, J = 8.5 Hz), 8.26 (IH, t, J = 6.3 Hz), 9.17 (IH, s);
MS(ESI) m/z: 395 (M— H)— . MS (ESI) m / z: 395 (M— H) —.
<実施例 43 > <Example 43>
{ [7- ({[2- (ジメチルァミノ)ェチル]アミノ}カルボ-ル) 8H—インデノ [1, 2— d] [1, 3]チアゾール 4 ィル]ォキシ }メチルホスホン酸(例示化合物番号 1-58) 実施例 36 (36c)で製造した 4 [ (ジエトキシホスホリル)メトキシ]— 8H インデノ [ 1, 2-d] [l, 3]チアゾール 7—カルボン酸(150mg、 0. 391mmol)及び N, N— ジメチルエチレンジァミン(0. 085mL、 0. 587mmol)を使用して、実施例 36 (36d) に記載した方法に従い、 { [7- ({[2- (ジメチルァミノ)ェチル]アミノ}カルボ-ル) 8H—インデノ [1, 2— d] [l, 3]チアゾール—4—ィル]ォキシ }メチルホスホン酸ジェ チル(121mg、収率 68%)を得、更に実施例 l (lc)に記載した方法に従い、標記目 的化合物(78mg、収率 61%)を得た。 {[7- ({[2- (Dimethylamino) ethyl] amino} carbol) 8H-indeno [1,2-d] [1,3] thiazole-4-yl] oxy} methylphosphonic acid (Exemplary Compound No. 1- 58) 4 [(Diethoxyphosphoryl) methoxy] -8H indeno [1,2-d] [l, 3] thiazole 7-carboxylic acid (150 mg, 0.391 mmol) prepared in Example 36 (36c) and N, According to the method described in Example 36 (36d) using N-dimethylethylenediamine (0.085 mL, 0.587 mmol), {[7- ({[2- (dimethylamino) ethyl] amino} carbohydrate -H) 8H-indeno [1,2-d] [l, 3] thiazole-4-yl] oxy} methyl phosphonate (121 mg, yield 68%) was obtained, and Example l (lc) The title compound (78 mg, 61% yield) was obtained according to the method described.
[0420] IR(KCl) V 3287, 3036, 2962, 2691, 1650, 1581, 1542, 1504, 1486, 1430, 1383, max [0420] IR (KCl) V 3287, 3036, 2962, 2691, 1650, 1581, 1542, 1504, 1486, 1430, 1383, max
1263, 1165, 1070, 946 cm"1; 1263, 1165, 1070, 946 cm "1;
1H NMR(DMSO-d , 400MHz) : δ 2.87 (6Η, s), 3.30 (2Η, dd, J=5.8 Hz, 5.8Hz), 1H NMR (DMSO-d, 400MHz): δ 2.87 (6Η, s), 3.30 (2Η, dd, J = 5.8 Hz, 5.8Hz),
6 6
3.62 (2H, dd, J=5.8 Hz, 11.6Hz), 4.18 (2H, s), 4.38 (2H, d, J=9.0 Hz), 7.26 (IH, d, J=8.7Hz), 7.67 (IH, d, J=8.7Hz), 9.20 (IH, s).
MS(FAB) m/z: 396 (M- H)— . 3.62 (2H, dd, J = 5.8 Hz, 11.6Hz), 4.18 (2H, s), 4.38 (2H, d, J = 9.0 Hz), 7.26 (IH, d, J = 8.7Hz), 7.67 (IH, d, J = 8.7Hz), 9.20 (IH, s). MS (FAB) m / z: 396 (M- H) —.
<実施例 44 > <Example 44>
[ (7— { [ (ピリジン一 4—ィルメチル)ァミノ]カルボ-ル } 8H—インデノ [ 1 , 2— d] [ 1 , 3]チアゾールー 4 ィル)ォキシ]メチルホスホン酸(例示化合物番号 1-10) 実施例 36 (36c)で製造した 4 [ (ジエトキシホスホリル)メトキシ]— 8H インデノ [ 1, 2-d] [l, 3]チアゾール 7—カルボン酸(150mg、 0. 587mmol)及び 4 ピコ リルアミン(0. 060mL、 0. 587mmol)を使用して、実施例 36 (36d)に記載した方法 に従 、、 [ (7 { [(ピリジン 4 ィルメチル)ァミノ]カルボ-ル } 8H—インデノ [1 , 2— d] [l, 3]チアゾールー 4 ィル)ォキシ]メチルホスホン酸ジェチル(107mg、収 率 58%)を得、更に実施例 1 (lc)に記載した方法に従い、標記目的化合物(110m g、収率 99%)を得た。 [(7— {[(Pyridine mono-4-ylmethyl) amino] carbol} 8H-indeno [1,2-d] [1,3] thiazol-4-yl) oxy] methylphosphonic acid (Exemplified Compound No. 1-10 ) 4 [(Diethoxyphosphoryl) methoxy] -8H indeno [1,2-d] [l, 3] thiazole 7-carboxylic acid (150 mg, 0.587 mmol) and 4 picolylamine prepared in Example 36 (36c) (0.060 mL, 0.587 mmol) and according to the method described in Example 36 (36d), [(7 {[(Pyridine-4-ylmethyl) amino] carbol} 8H-indeno [1, 2-d] [l, 3] thiazol-4-yl) oxy] methyl phosphonate (107 mg, 58% yield) was obtained, and the title target compound (110 mg, 110 mg, 58%) was further obtained according to the method described in Example 1 (lc). Yield 99%).
[0421] IR(KCl) V 3265, 3075, 1641, 1581, 1543, 1272, 1075, 1014, 955 cm"1; [0421] IR (KCl) V 3265, 3075, 1641, 1581, 1543, 1272, 1075, 1014, 955 cm "1;
max max
1H NMR(DMSO-d , 400MHz): δ 4.20 (2Η, s), 4.43 (2Η, d, J=9.0 Hz), 4.76 (2H, 1H NMR (DMSO-d, 400MHz): δ 4.20 (2Η, s), 4.43 (2Η, d, J = 9.0 Hz), 4.76 (2H,
6 6
d, J=5.9 Hz), 7.32 (IH, d, J=8.8 Hz), 7.79 (IH, d, J=8.8 Hz), 8.04 (2H, d, J=6.7 Hz) , 8.68 (2H, d, J=6.7 Hz), 9.18 (IH, s). d, J = 5.9 Hz), 7.32 (IH, d, J = 8.8 Hz), 7.79 (IH, d, J = 8.8 Hz), 8.04 (2H, d, J = 6.7 Hz), 8.68 (2H, d, J = 6.7 Hz), 9.18 (IH, s).
MS (FAB) m/z :416 (M— H)— . MS (FAB) m / z: 416 (M— H) —.
<実施例 45 > <Example 45>
{ [7—(シクロプロピルカルボ-ル) 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル]ォキシ }メチルホスホン酸 (例示化合物番号 1-24) {[7- (Cyclopropylcarbol) 8H-indeno [1, 2-d] [l, 3] thiazol-4-yl] oxy} methylphosphonic acid (Exemplified Compound No. 1-24)
(45a) { [7—(ヒドロキシメチル) 8H—インデノ [1, 2— d] [l, 3]チアゾールー 4 ィル]ォキシ }メチルホスホン酸ジェチル (45a) {[7- (Hydroxymethyl) 8H-indeno [1,2-d] [l, 3] thiazole-4-yl] oxy} methyl phosphonate
実施例 36 (36c)で製造した 4 [ (ジエトキシホスホリル)メトキシ]— 8H インデノ [ 1, 2-d] [l, 3]チアゾール—7—カルボン酸(11. 7g、 30. 5mmol)をテトラヒドロフ ラン(305mL)に溶解し、 0°C冷却下、そこへボラン一テトラヒドロフランコンプレックス (1. OM テトラヒドロフラン溶液)(70. 2mL、 70. 2mmol)を加え、窒素雰囲気下、 室温まで昇温しながら 5時間撹拌した。 Example 36 4 [(Diethoxyphosphoryl) methoxy] -8H indeno [1,2-d] [l, 3] thiazole-7-carboxylic acid (11.7 g, 30.5 mmol) prepared in (36c) was added to tetrahydro Dissolve in furan (305 mL), add borane-tetrahydrofuran complex (1. OM tetrahydrofuran solution) (70.2 mL, 70.2 mmol) to 0 ° C under cooling, and raise the temperature to room temperature in a nitrogen atmosphere. Stir for 5 hours.
[0422] 反応液に飽和塩化アンモ-ゥム水溶液を加え、クロ口ホルムで抽出した。得られた 有機層を水及び飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、減圧下溶媒を
留去し、標記目的化合物(10. lg、収率 89%)を得た。 [0422] A saturated aqueous ammonium chloride solution was added to the reaction mixture, and the mixture was extracted with black mouth form. The obtained organic layer was washed with water and saturated brine, dried over anhydrous sodium sulfate, and the solvent was removed under reduced pressure. Distilled off to give the title object compound (10. lg, 89% yield).
[0423] IR(KBr) v 980, 1023, 1251, 1509, 3381 cm"1; [0423] IR (KBr) v 980, 1023, 1251, 1509, 3381 cm "1;
max max
1H NMR(CDC1 , 400MHz) : δ 1.35 (6H, t, J=7.0 Hz), 3.87 (2H, s), 4.31 (4H, dq, J 1H NMR (CDC1, 400MHz): δ 1.35 (6H, t, J = 7.0 Hz), 3.87 (2H, s), 4.31 (4H, dq, J
3 Three
=7.0 Hz, 7.0 Hz), 4.58 (2H, d, J=8.6 Hz), 4.77 (2H, s), 7.00 (1H, d, J=8.4 Hz), 7.21 (1H, d, J=8.4 Hz), 8.81 (1H, s); = 7.0 Hz, 7.0 Hz), 4.58 (2H, d, J = 8.6 Hz), 4.77 (2H, s), 7.00 (1H, d, J = 8.4 Hz), 7.21 (1H, d, J = 8.4 Hz) , 8.81 (1H, s);
MS(FAB) m/z: 370 (M+H)+. MS (FAB) m / z: 370 (M + H) + .
(45b) [ (7 ホルミル—8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキ シ]メチルホスホン酸ジェチル (45b) [(7 formyl-8H-indeno [1,2-d] [l, 3] thiazole-4-yl) oxy] demethyl methylphosphonate
実施例 45 (45a)で製造した { [7— (ヒドロキシメチル) 8H—インデノ [1, 2 d] [1, 3]チアゾール—4—ィル]ォキシ }メチルホスホン酸ジェチル(10. lg、 27. 3mmol) をジクロロメタン(273mL)に溶解し、 0°C冷却下、そこへデス マーチンペルョージ ナン(11. 6g、 27. 3mmol)をカ卩え、窒素雰囲気下、 0°Cで 1時間攪拌した。 {45-) {[7- (Hydroxymethyl) 8H-indeno [1,2d] [1,3] thiazol-4-yl] oxy} methyl phosphonate jetyl (10. lg, 27. 3mmol) was dissolved in dichloromethane (273mL), and desmartin peroxide (11.6g, 27.3mmol) was added to it under cooling at 0 ° C, and at 0 ° C for 1 hour under nitrogen atmosphere. Stir.
[0424] 反応液に飽和チォ硫酸ナトリウム水溶液をカ卩え、ジクロロメタンで抽出した。得られ た有機層を水及び飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、減圧下溶媒 を留去した。得られた残渣をシリカゲルクロマトグラフィー (酢酸ェチル:メタノール、 1 : 0— 30 : 1、 VZV)により精製し、標記目的化合物(7. 07g、収率 71%)を得た。 [0424] A saturated aqueous sodium thiosulfate solution was added to the reaction mixture, and the mixture was extracted with dichloromethane. The obtained organic layer was washed with water and saturated brine, dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure. The obtained residue was purified by silica gel chromatography (ethyl acetate: methanol, 1: 0-30: 1, VZV) to obtain the title object compound (7.07 g, yield 71%).
[0425] IR(KBr) v 1028, 1253, 1579, 1691, 3058 cm"1; [0425] IR (KBr) v 1028, 1253, 1579, 1691, 3058 cm "1;
max max
1H NMR(CDC1 , 400MHz) δ 1.37 (6H, t, J=7.0 Hz), 4.27 (2H, s), 4.30—4.40 (4H, 1H NMR (CDC1, 400MHz) δ 1.37 (6H, t, J = 7.0 Hz), 4.27 (2H, s), 4.30—4.40 (4H,
3 Three
m), 4.70 (2H, d, J=9.0 Hz), 7.24 (1H, d, J=8.2 Hz), 7.75 (1H, d, J=8.2 Hz), 8.89 (1 H, s), 10.11 (1H, s); m), 4.70 (2H, d, J = 9.0 Hz), 7.24 (1H, d, J = 8.2 Hz), 7.75 (1H, d, J = 8.2 Hz), 8.89 (1 H, s), 10.11 (1H , s);
MS(FAB) m/z: 368 (M+H)+. MS (FAB) m / z: 368 (M + H) + .
(45c) ({7 [シクロプロピル(ヒドロキシ)メチル ] 8H インデノ [1, 2-d] [l, 3] チアゾール 4 ィル }ォキシ)メチルホスホン酸ジェチル (45c) ({7 [cyclopropyl (hydroxy) methyl] 8H indeno [1, 2-d] [l, 3] thiazole 4 yl} oxy) methyl phosphonate jetyl
実施例 45 (45b)で製造した [ (7 ホルミル— 8H—インデノ [1, 2— d] [l, 3]チア ゾールー 4 ィル)ォキシ]メチルホスホン酸ジェチル(123mg、 335 μ mol)をテトラ ヒドロフラン(34mL)に溶解し、 0°C冷却下、そこヘシクロプロピルマグネシウムブロミ ド(0. 5M テトラヒドロフラン溶液) (2. 34mL、 1. 17mmol)を加え、窒素雰囲気下 、室温まで昇温しながら 1. 5時間撹拌した。
[0426] 反応液に飽和塩化アンモ -ゥム水溶液を加え、酢酸ェチルで抽出した。得られた 有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、減圧下溶媒を留去し た。得られた残渣をシリカゲルクロマトグラフィー (ジクロロメタン:メタノール、 100 : 1— 30 : 1, VZV)により精製し、標記目的化合物(35. 6mg、収率 26%)を得た。 EXAMPLE 45 [(7-Formyl-8H-indeno [1,2-d] [l, 3] thiazol-4-yl) oxy] methylphosphonate jetyl (123 mg, 335 μmol) prepared in Example 45 (45b) was dissolved in tetrahydrofuran. (34 mL), and at 0 ° C cooling, add hecyclopropylmagnesium bromide (0.5 M tetrahydrofuran solution) (2.34 mL, 1.17 mmol), and raise the temperature to room temperature in a nitrogen atmosphere. 1. Stir for 5 hours. [0426] To the reaction solution was added a saturated aqueous ammonium chloride solution, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure. The obtained residue was purified by silica gel chromatography (dichloromethane: methanol, 100: 1-30: 1, VZV) to obtain the title object compound (35.6 mg, yield 26%).
[0427] IR(Thin film) v 978, 1027, 1240, 1508, 3399 cm"1; [0427] IR (Thin film) v 978, 1027, 1240, 1508, 3399 cm "1;
max max
1H NMR(CDC1 , 400MHz): δ 0.33—0.72 (4H, m), 1.33—1.42 (1H, m), 1.35 (6H, t, 1H NMR (CDC1, 400 MHz): δ 0.33—0.72 (4H, m), 1.33—1.42 (1H, m), 1.35 (6H, t,
3 Three
J=7.0 Hz), 3.93-3.96 (2H, m), 4.25—4.36 (5H, m), 4.60 (2H, d, J=9.0 Hz), 7.04 (1H, d, J=8.4 Hz), 7.33 (1H, d, J=8.4 Hz), 8.81 (1H, s); J = 7.0 Hz), 3.93-3.96 (2H, m), 4.25—4.36 (5H, m), 4.60 (2H, d, J = 9.0 Hz), 7.04 (1H, d, J = 8.4 Hz), 7.33 ( 1H, d, J = 8.4 Hz), 8.81 (1H, s);
MS(FAB) m/z: 410 (M+H)+. MS (FAB) m / z: 410 (M + H) + .
(45d) { [7- (シクロプロピルカルボ-ル)—8H—インデノ [1, 2— d] [1, 3]チアゾ ール 4 ィル]ォキシ }メチルホスホン酸ジェチル (45d) {[7- (Cyclopropylcarbol) —8H-indeno [1, 2— d] [1, 3] thiazol 4-yl] oxy} methyl phosphonate
実施例 45 (45c)で製造した({7— [シクロプロピル (ヒドロキシ)メチル ]—8H—イン デノ [1, 2— d] [l, 3]チアゾールー 4ーィル }ォキシ)メチルホスホン酸ジェチル(34 . lmg、 83. 3 /z mol)を使用して、実施例 45 (45b)に記載した方法に従い、標記目 的化合物(27. 6mg、収率 81%)を得た。 Example 45 ({7- [cyclopropyl (hydroxy) methyl] -8H-indeno [1,2-d] [l, 3] thiazol-4-yl} oxy) methylphosphonate (34) prepared in Example 45 (45c) The title compound (27.6 mg, 81% yield) was obtained according to the method described in Example 45 (45b) using 1 mg, 83.3 / z mol).
[0428] IR(Thin film) v 1026, 1058, 1233, 1386, 1573, 1657 cm"1; [0428] IR (Thin film) v 1026, 1058, 1233, 1386, 1573, 1657 cm "1;
max max
1H NMR(CDC1 , 400MHz): δ 1.01—1.07 (2H, m), 1.22-1.27 (2H, m), 1.35 (6H, t, 1H NMR (CDC1, 400 MHz): δ 1.01—1.07 (2H, m), 1.22-1.27 (2H, m), 1.35 (6H, t,
3 Three
J=7.0 Hz), 2.63-2.71 (1H, m), 4.23 (2H, s), 4.28-4.37 (4H, m), 4.67 (2H, d, J=8.6 Hz), 7.14 (1H, d, J=8.8 Hz), 8.01 (1H, d, J=8.8 Hz), 8.83 (1H, s); J = 7.0 Hz), 2.63-2.71 (1H, m), 4.23 (2H, s), 4.28-4.37 (4H, m), 4.67 (2H, d, J = 8.6 Hz), 7.14 (1H, d, J = 8.8 Hz), 8.01 (1H, d, J = 8.8 Hz), 8.83 (1H, s);
MS(FAB) m/z: 408 (M+H)+. MS (FAB) m / z: 408 (M + H) + .
(45e) { [7- (シクロプロピルカルボ-ル)—8H—インデノ [1, 2 d] [1, 3]チアゾ 一ルー 4 ィル]ォキシ }メチルホスホン酸 (45e) {[7- (Cyclopropylcarbol) —8H-indeno [1, 2 d] [1, 3] thiazo 1-ru 4 yl] oxy} methylphosphonic acid
実施例 45 (45d)で製造した { [7 (シクロプロピルカルボ-ル)—8H—インデノ [1 , 2— d] [l, 3]チアゾール—4—ィル]ォキシ }メチルホスホン酸ジェチル(25. 6mg 、 0. 628mmol)を使用して、実施例 1 (lc)に記載した方法に従い、標記目的化合 物(16. 5mg、収率 75%)を得た。 {45 (45d) {[7 (Cyclopropylcarbol) -8H-indeno [1,2, -d] [l, 3] thiazole-4-yl] oxy} jethyl phosphonate (25. The title compound (16.5 mg, yield 75%) was obtained according to the method described in Example 1 (lc) using 6 mg, 0.628 mmol).
[0429] IR(KBr) v 949, 1057, 1230, 1387, 1573, 1658, 3087 cm"1; [0429] IR (KBr) v 949, 1057, 1230, 1387, 1573, 1658, 3087 cm "1;
max max
1H NMR(DMSO-d , 400MHz): δ 0.99—1.06 (4H, m), 2.86—2.94 (1H, m), 4.21 (2H
/v: O£900ifcl£ OSさAV 1H NMR (DMSO-d, 400MHz): δ 0.99—1.06 (4H, m), 2.86—2.94 (1H, m), 4.21 (2H / v: O £ 900ifcl £ OS AV
)慫/ aェムムK ½,T0:oτ¾ λAar:::-;uヽsou, I . ·
 ) 慫 / aemmu K ½, T0: oτ¾ λAar :::-; u ヽ sou, I.
) 慫 / aemmu K ½, T0: oτ¾ λAar :::-; u ヽ sou, I.
0¾¾ (\驊皿 3 J5οτ 060 OCHへ 4ΛΛΛ>.ίヽ : :.〜 1- , , , . , , 0¾¾ (¥ 驊 3 J5οτ 060 To OCH 4ΛΛΛ> .ί ヽ::. ~ 1-,,,.,,
(/οτειπ。 。 (/ οτειπ.
)()() (舊? G∞ε oεεΐ 5εΐ應寸8ΓHfΗ 3Η1ΗτΗ Ν=ZΖΖ - - - - *.·. ) () () (舊? G∞ε oεεΐ 5εΐDimension 8ΓHfΗ 3Η1ΗτΗ Ν = ZΖΖ----* ...
(()()(pp寸寸∞ε寸∞ε寸02寸6ΐss6Ϊ2ΐHΗ sΗ1ΗssΗZΖ ι 1 * * - - - - - -....... (() () (pp dimension∞ε dimension∞ε dimension 02 dimension 6 寸 ss6Ϊ2ΐHΗsΗ1ΗssΗZΖι 1 * *------.......
99. 99.
)ΐ∞Η -
MS(FAB) m/z: 438 (M+H)+. ) ΐ∞Η- MS (FAB) m / z: 438 (M + H) + .
(46b) { [7 (トリフルォロアセチル) 8H インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル]ォキシ }メチルホスホン酸ジェチル (46b) {[7 (Trifluoroacetyl) 8H indeno [1, 2-d] [l, 3] thiazol-4-yl] oxy} methyl phosphonate
実施例 46 (46a)で製造した { [7— (2, 2, 2 トリフルォロ一 1—ヒドロキシェチル) — 8H—インデノ [1, 2-d] [l, 3]チアゾール—4—ィル]ォキシ }メチルホスホン酸 ジェチル(155mg、 0. 355mmol)を使用して、実施例 45 (45b)に記載した方法に 従い、標記目的化合物(59mg、収率 37%)を得た。 Prepared in Example 46 (46a) {[7- (2, 2, 2 trifluoro 1-hydroxyethyl) — 8H-indeno [1, 2-d] [l, 3] thiazole-4-yl] The title object compound (59 mg, 37% yield) was obtained by following the method described in Example 45 (45b) using oxy} methyl phosphonate (155 mg, 0.355 mmol).
[0432] IR(KBr) v 912, 982, 1038, 1145, 1192, 1248, 1283, 1569, 1694, 2984, 3086 cm max [0432] IR (KBr) v 912, 982, 1038, 1145, 1192, 1248, 1283, 1569, 1694, 2984, 3086 cm max
-1 -1
1H NMR(CDC1 , 400MHz) δ 1.36 (6H, t, J= 7.0Hz) 4.27 (2H, s), 4.25—4.38 (4H, 1H NMR (CDC1, 400MHz) δ 1.36 (6H, t, J = 7.0Hz) 4.27 (2H, s), 4.25—4.38 (4H,
3 Three
m), 4.69 (2H, d, J= 9.0Hz), 7.18 (IH, d, J= 9.0Hz), 7.96 (IH, d, J= 9.0Hz) 8.86 (IH , s) ; m), 4.69 (2H, d, J = 9.0Hz), 7.18 (IH, d, J = 9.0Hz), 7.96 (IH, d, J = 9.0Hz) 8.86 (IH, s);
MS(FAB) m/z: 436 (M+H)+. MS (FAB) m / z: 436 (M + H) + .
(46c) { [7 (トリフルォロアセチル) 8H インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル]ォキシ }メチルホスホン酸 (46c) {[7 (Trifluoroacetyl) 8H Indeno [1, 2-d] [l, 3] thiazole-4-yl] oxy} methylphosphonic acid
実施例 46 (46b)で製造した { [7— (トリフルォロアセチル) 8H—インデノ [1, 2— d] [l, 3]チアゾール—4—ィル]ォキシ }メチルホスホン酸ジェチル(52mg、 0. 119 mmol)を使用して、実施例 1 (lc)に記載した方法 (この場合はトリエチルァミン(12m g、 0. 119mmol)を共存させる)に従い、標記目的化合物(24mg、収率 53%)を得 た。 {[7- (Trifluoroacetyl) 8H-indeno [1,2-d] [l, 3] thiazol-4-yl] oxy} prepared in Example 46 (46b)} methylphosphonate (52 mg, 0 119 mmol) according to the method described in Example 1 (lc) (in this case co-existing triethylamine (12 mg, 0.119 mmol)) (24 mg, 53% yield) )
[0433] IR(KBr) v 919, 1035, 1074, 1140, 1201, 1285, 1570, 1697, 3085, 3369 cm"1; [0433] IR (KBr) v 919, 1035, 1074, 1140, 1201, 1285, 1570, 1697, 3085, 3369 cm "1;
max max
1H NMR(DMSO-d , 400MHz): δ 4.29 (2H, s), 4.51 (2H, d, J= 9.0Hz), 7.44 (IH, d 1H NMR (DMSO-d, 400MHz): δ 4.29 (2H, s), 4.51 (2H, d, J = 9.0Hz), 7.44 (IH, d
6 6
, J= 9.0Hz), 7.87-7.91 (IH, m), 9.17 (IH, s); , J = 9.0Hz), 7.87-7.91 (IH, m), 9.17 (IH, s);
MS(FAB) m/z: 378 (M- H)— . MS (FAB) m / z: 378 (M- H) —.
<実施例 47 > <Example 47>
{ [7 (ジメチルァミノ) 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル]ォキ シ}メチルホスホン酸 (例示化合物番号 1-82) {[7 (Dimethylamino) 8H-indeno [1, 2-d] [l, 3] thiazole-4-yl] oxy} methylphosphonic acid (Exemplified Compound No. 1-82)
(47a) [ (7— { [ (ァリルォキシ)カルボニル]アミノ}— 8H—インデノ [1, 2— d] [l, 3]
チアゾール 4 ィル)ォキシ]メチルホスホン酸ジェチル (47a) [(7— {[(Aryloxy) carbonyl] amino} — 8H—indeno [1, 2— d] [l, 3] Thiazole 4-yl) oxy] methyl phosphonate
実施例 36 (36c)で製造した 4 [ (ジエトキシホスホリル)メトキシ]— 8H インデノ [ 1, 2-d] [l, 3]チアゾール—7—カルボン酸(10. 3g、 26. 9mmol)を N, N ジメ チルホルムアミド(134mL)に溶解し、 0°C冷却下、そこへトリェチルァミン(4. l lmL 、 29. 6mmol)及びジフエ-ルホスホン酸アジド(6. 96mL、 32. 3mmol)を加え、 窒素雰囲気下、室温まで昇温しながら 4時間撹拌した。 Example 36 4 [(Diethoxyphosphoryl) methoxy] -8H indeno [1,2-d] [l, 3] thiazole-7-carboxylic acid (10.3 g, 26.9 mmol) prepared in (36c) was replaced with N , N Dimethylformamide (134 mL), and at 0 ° C cooling, to this was added triethylamine (4. l lmL, 29.6 mmol) and diphenylphosphonic acid azide (6.96 mL, 32.3 mmol), nitrogen. The mixture was stirred for 4 hours while raising the temperature to room temperature.
[0434] 反応液にァリルアルコール(18. 4mL、 269mmol)を加え、 90°Cで 1. 5時間撹拌 した。 [0434] To the reaction solution was added aryl alcohol (18.4 mL, 269 mmol), and the mixture was stirred at 90 ° C for 1.5 hours.
[0435] 反応液に水を加え、減圧下 N, N ジメチルホルムアミドを留去し、クロ口ホルムで 抽出した。得られた有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、減 圧下溶媒を留去した。得られた残渣をシリカゲルクロマトグラフィー (酢酸ェチル)によ り精製し、標記目的化合物(9. 23g、収率 78%)を得た。 [0435] Water was added to the reaction solution, and N, N dimethylformamide was distilled off under reduced pressure, followed by extraction with chloroform. The obtained organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, and the solvent was distilled off under reduced pressure. The obtained residue was purified by silica gel chromatography (ethyl acetate) to obtain the title object compound (9.23 g, yield 78%).
[0436] IR(KBr) v 1027, 1235, 1535, 1722 cm"1; [0436] IR (KBr) v 1027, 1235, 1535, 1722 cm "1;
max max
1H NMR(CDC1 , 400MHz): δ 1.35 (6H, t, J=7.1 Hz), 3.79 (2H, s), 4.31 (4H, dq, J 1H NMR (CDC1, 400MHz): δ 1.35 (6H, t, J = 7.1 Hz), 3.79 (2H, s), 4.31 (4H, dq, J
3 Three
=7.1 Hz, 7.1 Hz), 4.60 (2H, d, J=8.6 Hz), 4.66—4.70 (2H, m), 5.27 (IH, d, J=10.6 H z), 5.38 (IH, d, J=17.2 Hz), 5.92—6.03 (IH, m), 6.50—6.60 (IH, br), 7.03 (IH, d, J= 9.0 Hz), 7.38-7.48 (IH, br), 8.81 (IH, s); = 7.1 Hz, 7.1 Hz), 4.60 (2H, d, J = 8.6 Hz), 4.66—4.70 (2H, m), 5.27 (IH, d, J = 10.6 H z), 5.38 (IH, d, J = 17.2 Hz), 5.92—6.03 (IH, m), 6.50—6.60 (IH, br), 7.03 (IH, d, J = 9.0 Hz), 7.38-7.48 (IH, br), 8.81 (IH, s);
MS(FAB) m/z: 439 (M+H)+. MS (FAB) m / z: 439 (M + H) + .
(47b) [ (7 アミノー 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキシ] メチルホスホン酸ジェチル (47b) [(7 Amino-8H-indeno [1, 2-d] [l, 3] thiazole-4-yl) oxy] Jetyl methylphosphonate
実施例 47 (47a)で製造した [ (7— { [ (ァリルォキシ)カルボ-ル]アミノ}—8H—ィ ンデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキシ]メチルホスホン酸ジェチル(9 . 23g、21. lmmol)をテトラヒドロフラン(21 lmL)に溶解し、そこへテトラキス(トリフ ェ-ルホスフィン)パラジウム(1. 22g、 1. 06mmol)及びジメドン(29. 6g、 211mm ol)を加え、窒素雰囲気下、室温で 6時間撹拌した。 Example 47 [(7 — {[(Aryloxy) carbol] amino} —8H-indeno [1,2-d] [l, 3] thiazol-4-yl) oxy] methylphosphonic acid prepared in (47a) Jetyl (9.23 g, 21. lmmol) was dissolved in tetrahydrofuran (21 lmL), and tetrakis (triphenylphosphine) palladium (1.22 g, 1.06 mmol) and dimedone (29.6 g, 211 mmol) And stirred at room temperature for 6 hours under a nitrogen atmosphere.
[0437] 減圧下テトラヒドロフランを留去し、ジクロロメタンを加えた。得られた有機層を飽和 炭酸水素ナトリウム水溶液及び飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、 減圧下溶媒を留去した。得られた残渣をシリカゲルクロマトグラフィー(酢酸ェチル:メ
タノール、 100 : 0— 97 : 3、 VZV)により精製し、標記目的化合物(4. 22g、収率 56[0437] Tetrahydrofuran was distilled off under reduced pressure, and dichloromethane was added. The obtained organic layer was washed with a saturated aqueous sodium hydrogen carbonate solution and saturated brine, dried over anhydrous sodium sulfate, and the solvent was distilled off under reduced pressure. The obtained residue was subjected to silica gel chromatography (ethyl acetate: The product was purified by the following method: Tanol, 100: 0—97: 3, VZV) to give the title compound (4.22 g, yield 56).
%)を得た。 %).
[0438] IR(Thin film) v 975, 1026, 1242, 1509 cm"1; [0438] IR (Thin film) v 975, 1026, 1242, 1509 cm "1;
max max
1H NMR(CDC1 , 400MHz) δ 1.35 (6H, t, J=7.0 Hz), 3.50—3.67 (2H, br), 3.64 (2 1H NMR (CDC1, 400MHz) δ 1.35 (6H, t, J = 7.0 Hz), 3.50-3.67 (2H, br), 3.64 (2
3 Three
H, s), 4.29 (4H, dq, J=7.0 Hz, 7.0 Hz), 4.59 (2H, d, J=8.2 Hz), 6.58 (1H, d, J=8.4 Hz), 6.95 (1H, d, J=8.4 Hz), 8.82 (1H, s); H, s), 4.29 (4H, dq, J = 7.0 Hz, 7.0 Hz), 4.59 (2H, d, J = 8.2 Hz), 6.58 (1H, d, J = 8.4 Hz), 6.95 (1H, d, J = 8.4 Hz), 8.82 (1H, s);
MS(FAB) m/z: 355 (M+H)+. MS (FAB) m / z: 355 (M + H) + .
(47c) { [7 (ジメチルァミノ) 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4ーィ ル]ォキシ }メチルホスホン酸ジェチル (47c) {[7 (Dimethylamino) 8H-indeno [1, 2-d] [l, 3] thiazol-4-yl] oxy} methyl phosphonate jetyl
実施例 47 (47b)で製造し [ (7 ァミノ 8H—インデノ [1, 2— d] [l, 3]チアゾー ルー 4—ィル)ォキシ]メチルホスホン酸ジェチル(4. 20g、 11. 9mmol)を N, N ジ メチルホルムアミド(119mL)に溶解し、炭酸水素ナトリウム(2. 49g、 29. 6mmol) 及びヨウ化メチル(1. 63mL、 26. 2mmol)をカ卩え、窒素雰囲気下、室温で 19時間 撹拌した。 Example 47 (47b) was prepared from [(7 amino 8H-indeno [1,2-d] [l, 3] thiazol 4-yl) oxy] methylphosphonate jetyl (4.20 g, 11.9 mmol). Dissolve in N, N dimethylformamide (119 mL), and add sodium bicarbonate (2.49 g, 29.6 mmol) and methyl iodide (1.63 mL, 26.2 mmol). Stir for hours.
[0439] 反応液に飽和炭酸水素ナトリウム水溶液を加え、酢酸ェチルで抽出した。得られた 有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、減圧下溶媒を留去し た。得られた残渣をシリカゲルクロマトグラフィー(へキサン:酢酸ェチル、 50 : 50— 0 : 100、 V/V)により精製し、標記目的化合物(2. 92g、収率 64%)を得た。 [0439] A saturated aqueous sodium hydrogen carbonate solution was added to the reaction mixture, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure. The obtained residue was purified by silica gel chromatography (hexane: ethyl acetate, 50: 50—0: 100, V / V) to obtain the title object compound (2.92 g, yield 64%).
[0440] IR(Liquid film) v 973, 1026, 1249, 1509, 2981 cm"1; [0440] IR (Liquid film) v 973, 1026, 1249, 1509, 2981 cm "1;
max max
1H NMR(CDC1 , 400MHz) δ 1.35 (6H, t, J=7.0 Hz), 2.82 (6H, s), 3.86 (2H, s), 4 1H NMR (CDC1, 400MHz) δ 1.35 (6H, t, J = 7.0 Hz), 2.82 (6H, s), 3.86 (2H, s), 4
3 Three
.31 (4H, dq, J=7.0 Hz, 7.0 Hz), 4.60 (2H, d, J=8.6 Hz), 6.88 (1H, d, J=8.6 Hz), 7.0 2 (1H, d, J=8.6 Hz), 8.80 (1H, s); .31 (4H, dq, J = 7.0 Hz, 7.0 Hz), 4.60 (2H, d, J = 8.6 Hz), 6.88 (1H, d, J = 8.6 Hz), 7.0 2 (1H, d, J = 8.6 Hz), 8.80 (1H, s);
MS(FAB) m/z: 383 (M+H)+. MS (FAB) m / z: 383 (M + H) + .
(47(1) { [7—(ジメチルァミノ)ー811—ィンデノ[1, 2-d] [l, 3]チアゾールー 4ーィ ル]ォキシ }メチノレホスホン酸 (47 (1) {[7- (Dimethylamino) -811-indeno [1, 2-d] [l, 3] thiazol-4-yl] oxy} methinorephosphonic acid
実施例 47 (47c)で製造した { [7 (ジメチルァミノ) 8H—インデノ [1, 2 d] [1, 3]チアゾールー 4 ィル]ォキシ }メチルホスホン酸ジェチル(147mg、 384 μ mol) を使用して、実施例 1 (lc)に記載した方法に従い、標記目的化合物(88. 8mg、収
率 71%)を得た。 Using {[7 (dimethylamino) 8H-indeno [1,2d] [1,3] thiazol-4-yl] oxy} methylphosphonate jetyl (147 mg, 384 μmol) prepared in Example 47 (47c) In accordance with the method described in Example 1 (lc), the title target compound (88.8 mg, Rate 71%).
[0441] IR(KBr) v 942, 1057, 1268, 1508, 3399 cm"1; [0441] IR (KBr) v 942, 1057, 1268, 1508, 3399 cm "1;
max max
1H NMR(DMSO-d , 500MHz): δ 2.78 (6H, s), 3.95 (2H, s), 4.28 (2H, d, J=8.3 Hz 1H NMR (DMSO-d, 500MHz): δ 2.78 (6H, s), 3.95 (2H, s), 4.28 (2H, d, J = 8.3 Hz
6 6
), 6.89 (1H, d, J=8.8 Hz), 7.09 (1H, d, J=8.8 Hz), 9.19 (1H, s); ), 6.89 (1H, d, J = 8.8 Hz), 7.09 (1H, d, J = 8.8 Hz), 9.19 (1H, s);
MS(FAB) m/z: 325 (M- H)— . MS (FAB) m / z: 325 (M- H) —.
<実施例 48 > <Example 48>
{ [7 (ァセチルァミノ) 8H インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル]ォ キシ }メチルホスホン酸 (例示化合物番号 1-81) {[7 (Acetylamino) 8H Indeno [1, 2-d] [l, 3] thiazole-4-yl] oxy} methylphosphonic acid (Exemplified Compound No. 1-81)
(48a) { [7 (ァセチルァミノ) 8H インデノ [1, 2 d] [l, 3]チアゾールー 4ーィ ル]ォキシ }メチルホスホン酸ジェチル (48a) {[7 (Acetylamino) 8H Indeno [1, 2 d] [l, 3] thiazol-4-yl] oxy} Methylphosphonate jetyl
実施例 47 (47b)で製造した [ (7 アミノー 8H—インデノ [1, 2— d] [l, 3]チアゾ 一ルー 4—ィル)ォキシ]メチルホスホン酸ジェチル(63. 6mg、0. 179mmol)をテト ラヒドロフラン(1. 8mL)に溶解し、そこへトリェチルァミン(0. 624mL、 0. 449mmo 1)及び無水酢酸 (0. 338mL、 0. 358mmol)を加え、窒素雰囲気下、室温で 4時間 撹拌した。 Example 47 [(7 Amino-8H-indeno [1,2-d] [l, 3] thiazo 4-luyl) oxy] methylphosphonate (63.6 mg, 0.179 mmol) prepared in Example 47 (47b) Was dissolved in tetrahydrofuran (1.8 mL), and to this was added triethylamine (0.624 mL, 0.449 mmo 1) and acetic anhydride (0.3338 mL, 0.358 mmol), and the mixture was stirred at room temperature for 4 hours under a nitrogen atmosphere. .
[0442] 反応液に水を加え、クロ口ホルムで抽出した。得られた有機層を飽和食塩水で洗浄 し、無水硫酸ナトリウムで乾燥し、減圧下溶媒を留去した。得られた粗結晶をジイソプ 口ピルエーテルで洗净して、標記目的化合物(62. 8mg、収率 89%)を得た。 [0442] Water was added to the reaction solution, and the mixture was extracted with black mouth form. The obtained organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure. The obtained crude crystals were washed with diisopropyl propyl ether to obtain the title object compound (62.8 mg, yield 89%).
[0443] IR(KBr) v 1021, 1271, 1512, 1680, 3247 cm"1; [0443] IR (KBr) v 1021, 1271, 1512, 1680, 3247 cm "1;
max max
1H NMR(CDC1 , 500MHz) δ 1.37 (6H, t, J=7.1 Hz), 2.24 (3H, s), 3.72 (2H, s), 4 1H NMR (CDC1, 500MHz) δ 1.37 (6H, t, J = 7.1 Hz), 2.24 (3H, s), 3.72 (2H, s), 4
3 Three
.30-4.40 (4H, m), 4.49 (2H, d, J=8.8 Hz), 6.85 (1H, d, J=8.8 Hz), 7.29 (1H, d, J=8. 8 Hz), 7.78-7.83 (1H, br), 8.75 (1H, s); .30-4.40 (4H, m), 4.49 (2H, d, J = 8.8 Hz), 6.85 (1H, d, J = 8.8 Hz), 7.29 (1H, d, J = 8.8 Hz), 7.78- 7.83 (1H, br), 8.75 (1H, s);
MS(ESI) m/z: 397 (M+H)+. MS (ESI) m / z: 397 (M + H) + .
(48b) { [7 (ァセチルァミノ) 8H インデノ [1, 2-d] [l, 3]チアゾールー 4ーィ ル]ォキシ }メチノレホスホン酸 (48b) {[7 (Acetylamino) 8H Indeno [1, 2-d] [l, 3] thiazol-4-yl] oxy} methinorephosphonic acid
実施例 48 (48a)で製造した { [7— (ァセチルァミノ) 8H—インデノ [1, 2 d] [1 , 3]チアゾールー 4 ィル]ォキシ }メチルホスホン酸ジェチル(57. 6mg、0. 146m mol)を使用して、実施例 1 (lc)に記載した方法に従い、標記目的化合物 (42. 4mg
、収率 85%)を得た。 Example 48 {[7- (Acetyllumino) 8H-indeno [1,2d] [1,3] thiazol-4-yl] oxy} methylphosphonate jetyl (57.6 mg, 0.146 mmol) prepared in Example 48 (48a) In accordance with the method described in Example 1 (lc), the title target compound (42.4 mg Yield 85%).
[0444] IR(KBr) v 949, 1264, 1510, 1665, 3254 cm"1; [0444] IR (KBr) v 949, 1264, 1510, 1665, 3254 cm "1;
max max
1H NMR(DMSO-d , 500MHz): δ 2.09 (3H, s), 3.89 (2H, s), 4.33 (2H, d, J=8.8 Hz 1H NMR (DMSO-d, 500MHz): δ 2.09 (3H, s), 3.89 (2H, s), 4.33 (2H, d, J = 8.8 Hz
6 6
), 7.12 (1H, d, J=8.8 Hz), 7.37 (1H, d, J=8.8 Hz), 9.19 (1H, s), 9.59 (1H, s); ), 7.12 (1H, d, J = 8.8 Hz), 7.37 (1H, d, J = 8.8 Hz), 9.19 (1H, s), 9.59 (1H, s);
MS(ESI) m/z: 339 (M— H)— . MS (ESI) m / z: 339 (M— H) —.
<実施例 49 > <Example 49>
({7 [ (ジメチルァミノ)メチル] 8H インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル }ォキシ)メチルホスホン酸塩酸塩 (例示化合物番号 1-77) ({7 [(Dimethylamino) methyl] 8H Indeno [1, 2-d] [l, 3] thiazole-4-yl} oxy) methylphosphonic acid hydrochloride (Exemplary Compound No. 1-77)
(49a) ({7 [ (ジメチルァミノ)メチル ] 8H—インデノ [1, 2— d] [l, 3]チアゾール 4ーィル }ォキシ)メチルホスホン酸ジェチル (49a) ({7 [(Dimethylamino) methyl] 8H-indeno [1,2-d] [l, 3] thiazole-4-yl} oxy) methyl phosphonate
実施例 45 (45b)で製造した [ (7 ホルミル— 8H—インデノ [1, 2— d] [l, 3]チア ゾールー 4 ィル)ォキシ]メチルホスホン酸ジェチル(200mg、 0. 544mmol)を 1, 2 ジクロロェタン(5. 4mL)に溶解し、そこへジメチルァミン(2. OM テトラヒドロフラ ン溶液)(0. 817mL、 1. 63mmol)及びトリァセトキシ水素化ホウ素ナトリウム(130 mg、 0. 612mol)をカ卩え、窒素雰囲気下、室温で 18時間撹拌した。 Example 45 [(7 formyl-8H-indeno [1,2-d] [l, 3] thiazol-4-yl) oxy] methylphosphonate (200 mg, 0.544 mmol) prepared in Example 45 (45b) 2 Dissolve in dichloroethane (5.4 mL), and add dimethylamine (2. OM tetrahydrofuran solution) (0.817 mL, 1.63 mmol) and sodium triacetoxyborohydride (130 mg, 0.612 mol). The mixture was stirred at room temperature for 18 hours under a nitrogen atmosphere.
[0445] 反応液に飽和塩化アンモ-ゥム水溶液を加え、クロ口ホルムで抽出した。得られた 有機層を飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、減圧下溶媒を留去し た。得られた残渣を塩基性シリカゲルクロマトグラフィー(へキサン:酢酸ェチル、 2 : 1 — 1 : 2、 V/V)により精製し、標記目的化合物(162mg、収率 75%)を得た。 [0445] To the reaction solution was added a saturated aqueous ammonium chloride solution, and the mixture was extracted with black mouth form. The obtained organic layer was washed with saturated brine, dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure. The obtained residue was purified by basic silica gel chromatography (hexane: ethyl acetate, 2: 1—1: 2, V / V) to obtain the title object compound (162 mg, yield 75%).
[0446] IR(KBr) v 981, 1022, 1071, 1245, 1505 cm"1; [0446] IR (KBr) v 981, 1022, 1071, 1245, 1505 cm "1;
max max
1H NMR(CDC1 , 400MHz) δ 1.35 (6H, t, J=7.1 Hz), 2.23 (6H, s), 3.46 (2H, s), 3 1H NMR (CDC1, 400MHz) δ 1.35 (6H, t, J = 7.1 Hz), 2.23 (6H, s), 3.46 (2H, s), 3
3 Three
.90 (2H, s), 4.32 (4H, dq, J=7.1 Hz, 7.1 Hz), 4.63 (2H, d, J=9.0 Hz), 7.00 (1H, d, J =8.6 Hz), 7.14 (1H, d, J=8.6 Hz), 8.80 (1H, s); .90 (2H, s), 4.32 (4H, dq, J = 7.1 Hz, 7.1 Hz), 4.63 (2H, d, J = 9.0 Hz), 7.00 (1H, d, J = 8.6 Hz), 7.14 (1H , d, J = 8.6 Hz), 8.80 (1H, s);
MS(FAB) m/z: 397 (M+H)+. MS (FAB) m / z: 397 (M + H) + .
(49b) ({7 [ (ジメチルァミノ)メチル ] 8H—インデノ [1, 2-d] [l, 3]チアゾール 4ーィル }ォキシ)メチルホスホン酸塩酸塩 (49b) ({7 [(Dimethylamino) methyl] 8H-indeno [1, 2-d] [l, 3] thiazole 4-yl} oxy) methylphosphonate hydrochloride
実施例 49 (49a)で製造した( { 7 [ (ジメチルァミノ)メチル] - 8H インデノ [1, 2 -d] [l, 3]チアゾールー 4ーィル }ォキシ)メチルホスホン酸ジェチル(108mg、 0.
272mmol)を使用して、実施例 34 (34c)に記載した方法に従い、標記目的化合物( 81. 4mg、収率 80%)を得た。 Example 49 ({7 [(Dimethylamino) methyl] -8H indeno [1,2-d] [l, 3] thiazole-4-yl} oxy) methylphosphonate (108 mg, 0. 272 mmol) was used to give the title object compound (81.4 mg, 80% yield) according to the method described in Example 34 (34c).
[0447] IR(KBr) v 945, 1016, 1201, 1276, 1491, 2677 cm"1; [0447] IR (KBr) v 945, 1016, 1201, 1276, 1491, 2677 cm "1;
max max
1H NMR(D O, 400MHz): δ 2.89 (6H, s), 3.92 (2H, s), 4.20-4.44 (4H, m), 7.08-7. 1H NMR (D 2 O, 400 MHz): δ 2.89 (6H, s), 3.92 (2H, s), 4.20-4.44 (4H, m), 7.08-7.
2 2
18 (1H, m), 7.31-7.41 (1H, m), 9.30 (1H, s); 18 (1H, m), 7.31-7.41 (1H, m), 9.30 (1H, s);
MS(ESI) m/z: 339 (M— H)— (free form). MS (ESI) m / z: 339 (M— H) — (free form).
<実施例 50 > <Example 50>
{ [7 (ピロリジン 1ーィルメチル)—8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル]ォキシ }メチルホスホン酸塩酸塩 (例示化合物番号 1-11 ) {[7 (Pyrrolidine 1-ylmethyl) -8H-indeno [1, 2-d] [l, 3] thiazole-4-yl] oxy} methylphosphonate hydrochloride (Exemplified Compound No. 1-11)
(50a) { [7 (ピロリジン 1ーィルメチル)—8H—インデノ [1, 2— d] [l, 3]チアゾ ール 4 ィル]ォキシ }メチルホスホン酸ジェチル (50a) {[7 (Pyrrolidine 1-ylmethyl) -8H-indeno [1,2-d] [l, 3] thiazol 4-yl] oxy} methyl phosphonate
実施例 45 (45b)で製造した [ (7 ホルミル— 8H—インデノ [1, 2— d] [l, 3]チア ゾールー 4 ィル)ォキシ]メチルホスホン酸ジェチル(150mg、 0. 410mmol)とピロ リジン(0. 409mL, 0. 490mmol)を使用して、実施 ί列 49 (49a)【こ記載した方法【こ 従 、、標記目的化合物(78. Omg、収率 45%)を得た。 Example 45 (45 form) [(7 formyl-8H-indeno [1,2-d] [l, 3] thiazol-4-yl) oxy] methyl phosphonate (150 mg, 0.410 mmol) and pyrrolidine (0.409 mL, 0.490 mmol) was used to obtain the title compound (78. Omg, 45% yield).
[0448] IR(KBr) v 981, 1024, 1255, 1271, 1509 cm"1; [0448] IR (KBr) v 981, 1024, 1255, 1271, 1509 cm "1;
max max
1H NMR(CDC1 , 400MHz): δ 1.35 (6H, t, J=7.1 Hz), 1.74—1.80 (4H, m), 2.46-2.5 1H NMR (CDC1, 400 MHz): δ 1.35 (6H, t, J = 7.1 Hz), 1.74—1.80 (4H, m), 2.46-2.5
3 Three
3 (4H, m), 3.67 (2H, s), 3.91 (2H, s), 4.32 (4H, dq, J=7.1 Hz, 7.1 Hz), 4.62 (2H, d, J=9.0 Hz), 6.99 (1H, d, J=8.2 Hz), 7.18 (1H, d, J=8.2 Hz), 8.80 (1H, s); 3 (4H, m), 3.67 (2H, s), 3.91 (2H, s), 4.32 (4H, dq, J = 7.1 Hz, 7.1 Hz), 4.62 (2H, d, J = 9.0 Hz), 6.99 ( 1H, d, J = 8.2 Hz), 7.18 (1H, d, J = 8.2 Hz), 8.80 (1H, s);
MS(FAB) m/z: 423 (M+H)+. MS (FAB) m / z: 423 (M + H) + .
(50b) { [7— (ピロリジン— 1—ィルメチル)—8H—インデノ [1, 2-d] [l, 3]チアゾ ール— 4—ィル]ォキシ }メチルホスホン酸塩酸塩 (50b) {[7- (Pyrrolidine-1-ylmethyl) -8H-indeno [1, 2-d] [l, 3] thiazol-4-yl] oxy} methylphosphonate hydrochloride
実施例 50 (50a)で製造した { [7— (ピロリジン— 1—ィルメチル) 8H—インデノ [1 , 2-d] [l, 3]チアゾール—4—ィル]ォキシ }メチルホスホン酸ジェチル(74. 2mg 、 0. 176mmol)を使用して、実施例 34 (34c)に記載した方法に従い、標記目的化 合物(61. 4mg、収率 87%)を得た。 EXAMPLE 50 {[7- (Pyrrolidine-1-ylmethyl) 8H-indeno [1,2-d] [l, 3] thiazol-4-yl] oxy} prepared in Example 50 (50a) Jetyl methylphosphonate (74. 2 mg, 0.176 mmol) was used to give the title object compound (61.4 mg, 87% yield) according to the method described in Example 34 (34c).
[0449] IR(KBr) v 1011, 1073, 1495, 2671 cm"1; [0449] IR (KBr) v 1011, 1073, 1495, 2671 cm "1;
max max
1H NMR(D O, 400MHz): δ 1.92-2.05 (2H, m), 2.12-2.33 (2H, m), 3.15—3.26 (2H
•(ULIOJ s g) (H- ) 6ZS: z/m (IS3)S 1H NMR (DO, 400MHz): δ 1.92-2.05 (2H, m), 2.12-2.33 (2H, m), 3.15—3.26 (2H • (ULIOJ sg) (H-) 6ZS: z / m (IS3) S
•(s 'Ηΐ) ΐε·6 '(• ( s 'Ηΐ) ΐε · 6' (
ZH Ζ'8=ί 'P 'Ηΐ) LZ'L '(ZH 2"8=f 'P 'Ηΐ) ZY L '(ω Ή^) ZZ' -^Z' '(s 'ΗΖ) Z6'£ '(ω ' HZ) £ -£-W£ '(ω 'ΗΖ) 80·ε— 96 '(ω 'Η9) S6'I- 8S'I 9 '· (ΖΗ兩 0 '。 Η匪 Hx ZH Ζ'8 = ί 'P' Ηΐ) LZ'L '(ZH 2 "8 = f' P 'Ηΐ) ZY L' (ω Ή ^) ZZ '-^ Z''( s ' ΗΖ) Z6' £ '(ω' HZ) £-£ -W £ '(ω' ΗΖ) 80 · ε— 96 '(ω' Η9) S6'I-8S'I 9 '· ( Ζ Η 兩 0'. Η 匪 H x
:ト 6S92 'L6fl 'Ζ60ΐ 'SZOT '2ΐ0ΐ Λ ( 1)ΗΙ [ISW)] 。 ¾¾(%S8 ¾ί ^ε ·08)呦^ 目 Ό : 6S92 'L6fl' Ζ60ΐ 'SZOT' 2ΐ0ΐ Λ (1) ΗΙ [ISW)]. ¾¾ (% S8 ¾ί ^ ε · 08) 呦 ^ Eye Ό
3ΐ«ΐ0ΐ)
 2 Ί 3ΐ «ΐ0ΐ) 2 Ί
2 Ί 3ΐ «ΐ0ΐ) 2 Ί
^邈^邈べ ^ { ^: [ / 一 /— 、
 -ζ]} (qi9) ^ 邈 ^ 邈 be ^ {^: [/ One / —, -ζ]} (qi9)
-ζ]} (qi9) ^ 邈 ^ 邈 be ^ {^: [/ One / —, -ζ]} (qi9)
·+(Η+ ) L£f : z/ui (eVH)S · + (Η +) L £ f: z / ui (eVH) S
•(s 'Ηΐ) S8'8 '(ΖΗ Ζ'8=ί 'P 'Ηΐ) 9ΓΖ '(ZH S'8=f 'P 'Ηΐ) 00· '(ZH 0'6=f 'Ρ 'ΗΖ) W '(ΖΗ ΓΖ 'ΖΗ ΓΖ=Γ '&Ρ 'Η SS^ '(s 'HS) ε6·ε '(s 'Η ΐ3·ε '(in Ή^) Ζ • ( s 'Ηΐ) S8'8' ( Ζ Η Ζ'8 = ί 'P' Ηΐ) 9ΓΖ '(ZH S'8 = f' P 'Ηΐ) 00' (ZH 0'6 = f 'Ρ'')W' ( Ζ Η ΓΖ 'ΖΗ ΓΖ = Γ'& Ρ 'Η SS ^' (s ' HS ) ε6 · ε ' (s ' Η · 3 · ε ' (in Ή ^) Ζ
ΥΖ-ΖΖ'Ζ '(ω 'Η9) 09·ΐ— 8ε·ΐ '(ΖΗ ΓΖ=Γ 'Η9) SS"T 9: (ΖΗ )0 ^ Ιつ) Ηχ ΥΖ-ΖΖ'Ζ '(ω' Η9) 09 · ΐ— 8ε · ΐ '( Ζ Η ΓΖ = Γ' Η9) SS "T 9: (ΖΗ) 0 ^ Ι つ) Η χ
·τ_ωο 9262 'ΐΖ2ΐ 'eSST '920ΐ '8Z6 Λ ( 1)ΗΙ [0SW)] · Τ _ωο 9262 'ΐΖ2ΐ' eSST '920ΐ' 8Z6 Λ (1) ΗΙ [0SW)]

^ [ε 'i][p-s ' I ] - ) ]
 ^ [ε 'i] [ps' I]-)]
^ [ε 'i] [ps' I]-)]
Λ^-^ ^ ^Υ:^^ { -v- /— 、 ^[ε Ί][Ρ-2 Ί] ^^-Η8- -Ζ]} (^19) 一 /— 、 ^ [ε Ί][Ρ-2
 -Ζ]} Λ ^-^ ^ ^ Υ: ^^ {-v- / —, ^ [ε Ί] [Ρ-2 Ί] ^^-Η8- -Ζ]} (^ 19) One / —, ^ [ε Ί] [Ρ-2 -Ζ]}
-Ζ]} Λ ^-^ ^ ^ Υ: ^^ {-v- / —, ^ [ε Ί] [Ρ-2 Ί] ^^-Η8- -Ζ]} (^ 19) One / —, ^ [ε Ί] [Ρ-2 -Ζ]}
•(ULIOJ s g) (Η- ) S9S: ζ/ω (IS3)S • (ULIOJ sg) (Η-) S9S: ζ / ω (IS3) S
•(s 'Ηΐ) 2S"6 '(ω 'Ηΐ) 0VL- 2-L '(ω Ή • (s 'Ηΐ) 2S "6' (ω 'Ηΐ) 0VL- 2-L' (ω Ή
0l76S0C/900Zdf/X3d εεΐ· 0C0l70l/900Z OAV
<実施例 52 > 0l76S0C / 900Zdf / X3d εεΐ0C0l70l / 900Z OAV <Example 52>
{ [7 (モルホリン— 4ーィルメチル)—8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル]ォキシ }メチルホスホン酸塩酸塩 (例示化合物番号 1-13) {[7 (Morpholine-4-ylmethyl) -8H-indeno [1, 2-d] [l, 3] thiazole-4-yl] oxy} methylphosphonate hydrochloride (Exemplary Compound No. 1-13)
(52a) { [7 (モルホリン— 4ーィルメチル)—8H—インデノ [1, 2— d] [l, 3]チアゾ ール 4 ィル]ォキシ }メチルホスホン酸ジェチル (52a) {[7 (Morpholine-4-ylmethyl) -8H-indeno [1, 2-d] [l, 3] thiazol 4-yl] oxy} methyl phosphonate
実施例 45 (45b)で製造した [ (7 ホルミル— 8H—インデノ [1, 2— d] [l, 3]チア ゾールー 4 ィル)ォキシ]メチルホスホン酸ジェチル(150mg、 0. 0. 410mmol)と モルホリン(0. 043mL、 0. 490mmol)を使用して、実施例 49 (49a)に記載した方 法に従い、標記目的化合物(149mg、収率 83%)を得た。 Example 45 [(7-Formyl-8H-indeno [1,2-d] [l, 3] thiazol-4-yl) oxy] methylphosphonate (150 mg, 0.0.410 mmol) prepared in Example 45 (45b) The title object compound (149 mg, 83% yield) was obtained according to the method described in Example 49 (49a) using morpholine (0.043 mL, 0.490 mmol).
[0452] IR(KBr) v 978, 1020, 1113, 1253, 1272 cm"1; [0452] IR (KBr) v 978, 1020, 1113, 1253, 1272 cm "1;
max max
1H NMR(CDC1 , 400MHz) δ 1.35 (6H, t, J=7.1 Hz), 2.41-2.46 (4H, m), 3.55 (2H 1H NMR (CDC1, 400MHz) δ 1.35 (6H, t, J = 7.1 Hz), 2.41-2.46 (4H, m), 3.55 (2H
3 Three
, s), 3.65-3.69 (4H, m), 3.93 (2H, s), 4.32 (4H, dq, J=7.1 Hz, 7.1 Hz), 4.62 (2H, d, J=8.6 Hz), 6.99 (1H, d, J=8.2 Hz), 7.14 (1H, d, J=8.2 Hz), 8.81 (1H, s); , s), 3.65-3.69 (4H, m), 3.93 (2H, s), 4.32 (4H, dq, J = 7.1 Hz, 7.1 Hz), 4.62 (2H, d, J = 8.6 Hz), 6.99 (1H , d, J = 8.2 Hz), 7.14 (1H, d, J = 8.2 Hz), 8.81 (1H, s);
MS(FAB) m/z: 439 (M+H)+. MS (FAB) m / z: 439 (M + H) + .
(52b) { [7 (モルホリン— 4ーィルメチル)—8H—インデノ [1, 2— d] [l, 3]チアゾ ール— 4—ィル]ォキシ }メチルホスホン酸塩酸塩 (52b) {[7 (morpholine-4-ylmethyl) -8H-indeno [1, 2-d] [l, 3] thiazol-4-yl] oxy} methylphosphonate hydrochloride
実施例 52 (52a)で製造した { [7— (モルホリン一 4—ィルメチル) 8H—インデノ [ 1, 2-d] [l, 3]チアゾール—4—ィル]ォキシ }メチルホスホン酸ジェチル(96. 2m g、 0. 219mmol)を使用して、実施例 34 (34c)に記載した方法に従い、標記目的化 合物(82. 4mg、収率 90%)を得た。 Example 52 {[7- (Morpholine-4-ylmethyl) 8H-indeno [1,2-d] [l, 3] thiazol-4-yl] oxy} prepared in Example 52 (52a) Jetyl methylphosphonate (96. 2 mg, 0.219 mmol) was used to give the title compound (82.4 mg, 90% yield) according to the method described in Example 34 (34c).
[0453] IR(KBr) v 958, 1012, 1127, 1270, 1497, 2599 cm"1; [0453] IR (KBr) v 958, 1012, 1127, 1270, 1497, 2599 cm "1;
max max
1H NMR(D O, 400MHz): δ 3.26—3.36 (2H, m), 3.44—3.53 (2H, m), 3.73—3.84 (2H 1H NMR (D O, 400 MHz): δ 3.26—3.36 (2H, m), 3.44—3.53 (2H, m), 3.73—3.84 (2H
2 2
, m), 3.94 (2H, s), 4.04—4.13 (2H, m), 4.27 (2H, d, J=10.2 Hz), 4.39 (2H, s), 7.11 (1 H, d, J=8.6 Hz), 7.39 (1H, d, J=8.6 Hz), 9.28 (1H, s); , m), 3.94 (2H, s), 4.04—4.13 (2H, m), 4.27 (2H, d, J = 10.2 Hz), 4.39 (2H, s), 7.11 (1 H, d, J = 8.6 Hz ), 7.39 (1H, d, J = 8.6 Hz), 9.28 (1H, s);
MS(ESI) m/z: 381 (M— H)— (free form). MS (ESI) m / z: 381 (M— H) — (free form).
<実施例 53 > <Example 53>
[ (7—メトキシ 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキシ]メチ ルホスホン酸 (例示化合物番号 1-71)
Z '286 '0901 'S ZI 'fl£l '£S£l '££fl 'Π3ΐ 'S2SZ 'S062 '980S 'T8SS Λ (ID 1)HI [9SW)] [(7-methoxy 8H-indeno [1,2-d] [l, 3] thiazol-4-yl) oxy] methylphosphonic acid (Exemplified Compound No. 1-71) Z '286' 0901 'S ZI' fl £ l '£ S £ l' ££ fl 'Π3ΐ' S2SZ 'S062' 980S 'T8SS Λ (ID 1) HI [9SW)]
(%i8*¾i sra6ii)
 (°D ip}« っ (% i8 * ¾i sra6ii) (° D ip} «っ
(°D ip}« っ (% i8 * ¾i sra6ii) (° D ip} «っ
¾f¾¾ (Touiuigg^ ·〇 ^u^ZI) ^エ; ^邈べ ^ [ ^^ ( / — 一 /— 、 ^[ε 'i][p-s 'x] ^^-H8-/w- )]^a¾ (qs9)s9P«^ ¾f¾¾ (Touiuigg ^ · 〇 ^ u ^ ZI) ^ エ; ^ 邈 べ ^ [^^ (/ — One / —, ^ [ε 'i] [ps' x] ^^-H8- / w-)] ^ a¾ (qs9) s9P «^
邈べ Spear
^: ( / — 一 /—/: ^ [ε Ί][Ρ-2
 ^: (/ — One / — /: ^ [ε Ί] [Ρ-2
^: (/ — One / — /: ^ [ε Ί] [Ρ-2
(H+n) οζε: z/ui ' (evd) (H + n) οζε: z / ui '(evd)
•(s 'HI) 26"8 '( zH6"8=f 'P 'Ηΐ) 90"Z '(zH6"8=f 'P 'Ηΐ) SZ'9 '(zW8=f 'P 'HZ) Z9'f '(^ 'H ZVf-0 ε· '(s Ήε) 68·ε '(s Ήζ) β∑τ '(^ Ή9) ε·ΐ— 8ε·ΐ 9 兩 ο ' ι α 匪 • ( s 'HI) 26 "8' ( z H6" 8 = f 'P' Ηΐ) 90 "Z '(zH6" 8 = f' P 'Ηΐ) SZ'9' (zW8 = f 'P' HZ) Z9'f '(^' H ZVf-0 ε · '( s Ήε) 68 · ε' ( s Ήζ) β∑τ '(^ Ή9) ε · ΐ— 8ε · ΐ 9 兩 ο' ι α 匪
ト ura 88Ζ 'SZ6 'LZOl 'LfOl '0 ΖΙ '603ΐ '0091 'S6ZT Λ (Iつ [SS^O] *¾τ §^969) ^ 目 ¾遨 w^ ^-xm^ (qs— ) m っ Ura 88Ζ 'SZ6' LZOl 'LfOl' 0 ΖΙ '603ΐ' 0091 'S6ZT Λ (I [SS ^ O] * ¾τ § ^ 969) ^ ^ ¾ 遨 w ^ ^ -xm ^ (qs—) m
¾f¾¾ ( 'Ζ ^エ; ^邈べ ^ [ ^^ ( / 一,—ベ^べ ^エ 、邈べ ^ [/
 (qs9) ¾f¾¾ ('Ζ ^ エ; ^ 邈 べ ^ [^^ (/ 1 、 ― ベ ^ ベ ^ エ 、 邈 ^ ^ / (qs9)
(qs9) ¾f¾¾ ('Ζ ^ エ; ^ 邈 べ ^ [^^ (/ 1 、 ― ベ ^ ベ ^ エ 、 邈 ^ ^ / (qs9)
•+(1+ ) Z9S: ζ/ω ' (9VH) • + (1+) Z9S: ζ / ω '(9VH)
6"Z=f 'P 'Ηΐ) 96·9 '(ΖΗ 6 6 "Z = f 'P' Ηΐ) 96 9 '( Ζ Η 6
"Z=f 'P 'Ηΐ) f9'9 '(ΖΗ ε·6=ί" 'P 'ΗΖ) 6Vf '(ω Ή^) -^ '(s 'Ηε) ggT '(ω 'Η "Z = f 'P' Ηΐ) f9'9 '( Ζ Η ε · 6 = ί"' P 'ΗΖ) 6Vf' (ω Ή ^)-^ '(s' Ηε ) ggT '(ω' Η
96 — 00·ε '(ω Ή2) S9 — 99 '(ω 'Η9) SS'I— 6ε·ΐ 9 : (ΖΗ )0 ' つ αつ) Ηχ 96 — 00 · ε '(ω Ή2) S9 — 99' (ω 'Η9) SS'I— 6ε · ΐ 9: (ΖΗ) 0' and α) Η χ
τ OSOT '6WH '892ΐ ' ^ΐ ' 6fl '0091 'Z\L\ Λ (ID 1)HI τ OSOT '6WH' 892ΐ '^ ΐ' 6fl '0091' Z \ L \ Λ (ID 1) HI
° °
(%S8 ¾τ s^9 ) ^ 目 ¾遨 w^ ^-xm^ (bD m ^エ 、邈べ ^ [/
 (¾S9) (% S8 ¾τ s ^ 9) ^ Eye ¾ 遨 w ^ ^ -xm ^ ( b D m ^ D, ^ ^ ( ¾ S9)
(¾S9) (% S8 ¾τ s ^ 9) ^ Eye ¾ 遨 w ^ ^ -xm ^ ( b D m ^ D, ^ ^ ( ¾ S9)
0l76S0C/900Zdf/X3d 0C0l70l/900Z OAV
 : ζ/ω (9VH)S0l76S0C / 900Zdf / X3d 0C0l70l / 900Z OAV : Ζ / ω (9VH) S
: ζ/ω (9VH)S0l76S0C / 900Zdf / X3d 0C0l70l / 900Z OAV : Ζ / ω (9VH) S
•(s 'Ηΐ) 60·8 '(ΖΗ 8"Ζ 'ΖΗ 9·ΐ=ί" 'ΡΡ 'Ηΐ) 08"Ζ '(ΖΗ 8"Ζ=Γ 'Ρ 'Ηΐ) Ζ • (s 'Ηΐ) 60 · 8' ( Ζ Η 8 "Ζ 'ΖΗ 9 · ΐ = ί"' ΡΡ 'Ηΐ) 08 "Ζ' (ΖΗ 8" Ζ = Γ 'Ρ' Ηΐ) Ζ
VL '(ω 'Ηΐ) 08·,— 8Ζ· '(s Ήε) 68·ε '(ω 'ΗΖ) ^Z-ZVZ '(ω 'ΗΖ) SO - 66·ΐ '(ω 'ΗΖ VL '(ω' Ηΐ) 08 ·, — 8Ζ · '( s Ήε) 68 · ε' (ω 'ΗΖ) ^ Z-ZVZ' (ω 'ΗΖ) SO-66 · ΐ' (ω 'ΗΖ
) ΐ8·ΐ— ΐΓΐ '(s Ή6) 36 '(s Ήε) 6Γ0 '(s Ήε) Ζΐ 9 : (ΖΗ兩 0 ' ϋα )Η匪 Ηχ ) ΐ8 · ΐ— ΐΓΐ '(s Ή6) 36' ( s Ήε) 6Γ0 '( s Ήε) Ζΐ 9: ( Ζ Η 兩 0' ϋα) Η 匪 Η χ
·τ_ωο 0 6Ζ '6262 'ZS82 '02Ζΐ 'ΐ62ΐ '832ΐ 'SOU '2Ζ0ΐ Λ ( 1)ΗΙ [8SW)]· Τ _ωο 0 6Ζ '6262' ZS82 '02 Ζΐ 'ΐ62ΐ' 832ΐ 'SOU' 2Ζ0ΐ Λ (1) ΗΙ [8SW)]
。
 (p. (p
(p. (p
6ΐ)6ΐί^¾ϊ第 -α¾ί¾¾ (lorauig -Qz §ο ·οι)ベ ^ c^ ^i 一 'ε 's ' ^ 濯べ^ 一 s6ΐ) 6ΐί ^ ¾ϊ 第 -α¾ί¾¾ (lorauig -Qz §ο · οι) Be ^ c ^ ^ i One 'ε' s' ^ Rinse ^ One s
·+(Η- ) ΐ^ε '6εε: ζ/ω (HVH)S · + (Η-) ΐ ^ ε '6εε: ζ / ω (HVH) S
•(ΖΗ =1" 'Ρ 'Ηΐ) 03"Ζ '(ΖΗ 2"8 'ΖΗ =1" 'ΡΡ 'Ηΐ) ΖΊ '(ΖΗ Ζ· 8=1" 'Ρ 'Ηΐ) ε6·9 '(ω 'Ηΐ) S ^-O ^ '(ω 'Η2) 6 - 09 '(ω 'Η2) SO — ΐ6·ΐ '(ω 'H2 • (ΖΗ = 1 ”'Ρ' Ηΐ) 03” Ζ '(ΖΗ 2 ”8' Ζ Η = 1” 'ΡΡ' Ηΐ) ΖΊ '( Ζ Η Ζ · 8 = 1 ”' Ρ 'Ηΐ) ε6 · 9 '(ω' Ηΐ) S ^ -O ^ '(ω' Η2) 6-09 '(ω' Η2) SO — ΐ6 · ΐ '(ω' H2
) 6ΓΗ9·ΐ '(s 'Η6) 6·0 '(s Ήε) Ζΐ '(s Ήε) 3Γ0 9 : (ΖΗ兩 0 ' ϋα )Η匪 Ηχ ) 6ΓΗ9 · ΐ '( s ' Η6) 6 · 0' ( s Ήε) Ζΐ '( s Ήε) 3Γ0 9: ( Ζ Η 兩 0' ϋα) Η 匪 Η χ
ί ΙΙΙΟ ΐ- ί ΙΙΙΟ ΐ-
1 6Ζ ΊΖ6Ζ '9882 '8S82 '8 ^ΐ '90 ΐ 'T9ST 'L ZI 'ΖΟΠ 'S80T Λ ( Ρ Π)ΗΙ [L W1 6Ζ ΊΖ6Ζ '9882' 8S82 '8 ^ ΐ '90 ΐ' T9ST 'L ZI' ΖΟΠ 'S80T Λ (Ρ Π) ΗΙ (LW
。 (%οοι*¾ί ¾ 目 ¾遨 w^m^ . (% οοι * ¾ί ¾ eye ¾ 遨 w ^ m ^
-i^W ^ (^g I ) g I M 、ェっ隱 (ΐ〇画 ¾ 'Ζ£) {9ζζ '6 '^861 -i ^ W ^ (^ g I) g I M, 隱 隱 (ΐ〇 画 ¾ 'Ζ £) {9ζζ' 6 '^ 861
' -raoqo ' Ο ' ) Λ^^^ ^ ^→ 'ε 'Ζ ' I - Jpcl^^ - 1 - βτΰ Ι - L ^ ^- 'ε 'Z ' I - (^^ / ^ /^^-^ - ¾ - 1 - - L (^^9) '-raoqo' Ο ') Λ ^^^ ^ ^ →' ε 'Ζ' I-Jpcl ^^-1-βτΰ Ι-L ^ ^-'ε' Z 'I-(^^ / ^ / ^^- ^-¾-1--L (^^ 9)
< s圏第 > <S zone number>
'_(Η-η) ζΐ£·2/^ ' (9VH) sn'_ (Η-η) ζΐ £ · 2 / ^' (9VH) sn
•(s 'Ηΐ) 8Γ6 '(zH6"8=f 'P 'Ηΐ) ΐΓΖ '(ΖΗ6·8=1" 'Ρ 'Ηΐ) 88·9 '( ΖΗ8·8=1" 'Ρ 'ΗΖ) 6Z'f '(s Ήε) 8·ε '(s Ή S8T 9 :(ΖΗ兩 0 ' OSW。) 匪 Hx • ( s 'Ηΐ) 8Γ6' ( z H6 "8 = f 'P' Ηΐ) ΐΓΖ '(ΖΗ6 · 8 = 1"' Ρ 'Ηΐ) 88 · 9' (ΖΗ8 · 8 = 1 "'Ρ' ΗΖ) 6Z'f '(s Ήε) 8 · ε' (s Ή S8T 9 : ( Ζ Η 兩 0 'OSW.) 匪 H x
ト ω。 82 '06 G ω. 82 '06
0l76S0C/900Zdf/X3d 9ε ΐ· 0C0l70l/900Z OAV
(54c) 8—(tーブチルジメチルシリルォキシ) 2 ヒドロキシメチルー 5, 6, 7, 8—テ トラヒドロナフタレン 0l76S0C / 900Zdf / X3d 9ε 00C0l70l / 900Z OAV (54c) 8- (tert-Butyldimethylsilyloxy) 2 hydroxymethyl-5, 6, 7, 8-tetrahydronaphthalene
実施例 54 (54b)で製造した 8— (t—プチルジメチルシリルォキシ)一5, 6, 7, 8— テトラヒドロナフタレン一 2—カルボン酸メチル(88. 3mg、 0. 28mmol)を使用して、 実施例 19 (19e)に記載した方法に従い、標記目的化合物(73. 5mg、収率 91%)を 得た。 Using the methyl 8- (t-butyldimethylsilyloxy) -1,5,6,7,8-tetrahydronaphthalene-2-carboxylate (88.3 mg, 0.28 mmol) prepared in Example 54 (54b) The title compound (73.5 mg, yield 91%) was obtained according to the method described in Example 19 (19e).
[0459] IR(KBr) v 999, 1021, 1074, 1255, 2857, 2891, 2929, 3265 cm"1; [0459] IR (KBr) v 999, 1021, 1074, 1255, 2857, 2891, 2929, 3265 cm "1;
max max
1H NMR(CDC1 , 400MHz) : δ 0.16 (3H, s), 0.17 (3H, s), 0.94 (9H, s), 1.68—1.81 ( 1H NMR (CDC1, 400MHz): δ 0.16 (3H, s), 0.17 (3H, s), 0.94 (9H, s), 1.68—1.81 (
3 Three
2H, m), 1.94-2.03 (2H, m), 2.66—2.84 (2H, m), 4.63 (2H, m), 4.75-4.78 (IH, m), 7. 05 (IH, d, J=7.4 Hz), 7.15 (IH, dd, J=2.0 Hz, 7.4 Hz), 7.35 (IH, s); 2H, m), 1.94-2.03 (2H, m), 2.66—2.84 (2H, m), 4.63 (2H, m), 4.75-4.78 (IH, m), 7. 05 (IH, d, J = 7.4 Hz), 7.15 (IH, dd, J = 2.0 Hz, 7.4 Hz), 7.35 (IH, s);
MS(FAB) m/z: 291 (M- H)— . MS (FAB) m / z: 291 (M- H) —.
(54d) 7 ブロモメチルー 1一(tーブチルジメチルシリルォキシ) 1, 2, 3, 4ーテトラ ヒドロナフタレン (54d) 7 Bromomethyl-1 (t-butyldimethylsilyloxy) 1, 2, 3, 4-tetrahydronaphthalene
実施例 54 (54c)で製造した 8— (t—プチルジメチルシリルォキシ) 2 ヒドロキシ メチル—5, 6, 7, 8—テトラヒドロナフタレン(8. 37g、 28. 6mmol)を使用して、実施 例 15 (15d)に記載した方法に従い、標記目的化合物(10. 2g、収率 100%)を得た Example 54 Using 8- (t-butyldimethylsilyloxy) 2 hydroxymethyl-5,6,7,8-tetrahydronaphthalene (8.37 g, 28.6 mmol) prepared in Example 54 (54c) 15 According to the method described in (15d), the title object compound (10.2 g, yield 100%) was obtained.
[0460] IR(Liquid film) v 1079, 1100, 1254, 2858, 2886, 2931, 2950 cm"1; [0460] IR (Liquid film) v 1079, 1100, 1254, 2858, 2886, 2931, 2950 cm "1;
max max
1H NMR(CDC1 , 400MHz) : δ 0.16 (3H, s), 0.17 (3H, s), 0.94 (9H, s), 1.68—1.80 ( 1H NMR (CDC1, 400MHz): δ 0.16 (3H, s), 0.17 (3H, s), 0.94 (9H, s), 1.68—1.80 (
3 Three
2H, m), 1.93-2.02 (2H, m), 2.65-2.83 (2H, m), 4.47 (IH, d, J=12.9 Hz), 4.47 (IH, d , J=12.9 Hz), 4.73-4.76 (IH, m), 7.01 (IH, d, J=7.8 Hz), 7.16 (IH, dd, J=2.0 Hz, 7. 8 Hz), 7.38 (IH, d, J=1.6 Hz); 2H, m), 1.93-2.02 (2H, m), 2.65-2.83 (2H, m), 4.47 (IH, d, J = 12.9 Hz), 4.47 (IH, d, J = 12.9 Hz), 4.73-4.76 (IH, m), 7.01 (IH, d, J = 7.8 Hz), 7.16 (IH, dd, J = 2.0 Hz, 7.8 Hz), 7.38 (IH, d, J = 1.6 Hz);
MS(FAB) m/z: 355 (M+H)+. MS (FAB) m / z: 355 (M + H) + .
(54e) [8— (t—ブチルジメチルシロキシ) 5, 6, 7, 8—テトラヒドロナフタレン一 2— ィル]メチルホスホン酸ジェチル (54e) [8- (t-Butyldimethylsiloxy) 5, 6, 7, 8-tetrahydronaphthalene 2-yl] methylphosphonic acid jetyl
実施例 54 ( 54d)で製造した 7 -ブロモメチル 1— (t ブチルジメチルシリルォキ シ)一 1, 2, 3, 4—テトラヒドロナフタレン(10. 7g、 30. lmmol)を使用して、実施例 15 (15d)に記載した方法に従い、標記目的化合物(9. 89g、収率 80%)を得た。
[0461] IR(Liquid film) v 963, 1029, 1057, 1076, 1099, 1253, 2858, 2932 cm ; Example 54 Using 7-bromomethyl 1- (t-butyldimethylsilyloxy) -1,2,3,4-tetrahydronaphthalene (10.7 g, 30. lmmol) prepared in Example 54 (54d), According to the method described in 15 (15d), the title object compound (9. 89 g, yield 80%) was obtained. [0461] IR (Liquid film) v 963, 1029, 1057, 1076, 1099, 1253, 2858, 2932 cm;
max max
1H NMR(CDC1 , 400MHz): δ 0.15 (3H, s), 0.17 (3H, s), 0.93 (9H, s), 1.25 (3H, t, 1H NMR (CDC1, 400 MHz): δ 0.15 (3H, s), 0.17 (3H, s), 0.93 (9H, s), 1.25 (3H, t,
3 Three
J=7.0 Hz), 1.26 (3H, t, J=7.0 Hz), 1.70—1.80 (2H, m), 1.93—2.00 (2H, m), 2.64-2.8 2 (2H, m), 3.12 (2H, d, J=21.5 Hz), 3.98—4.07 (4H, m), 4.74-4.76 (IH, m), 7.00 (IH , d, J=7.8 Hz), 7.11 (IH, dt, J=2.0 Hz, 7.8 Hz), 7.24-7.26 (IH, m); J = 7.0 Hz), 1.26 (3H, t, J = 7.0 Hz), 1.70—1.80 (2H, m), 1.93—2.00 (2H, m), 2.64-2.8 2 (2H, m), 3.12 (2H, d, J = 21.5 Hz), 3.98—4.07 (4H, m), 4.74-4.76 (IH, m), 7.00 (IH, d, J = 7.8 Hz), 7.11 (IH, dt, J = 2.0 Hz, 7.8 Hz), 7.24-7.26 (IH, m);
MS(FAB) m/z: 411 (M- H)— . MS (FAB) m / z: 411 (M- H) —.
(54f) [8—(tーブチルジメチルシロキシ)一5, 6, 7, 8—テトラヒドロナフタレンー2— ィル]ジフルォロメチルホスホン酸ジェチル (54f) [8- (t-Butyldimethylsiloxy) -1,5,7,7-tetrahydronaphthalene-2-yl] difluoromethylphosphonate jetyl
実施例 54 (54e)で製造した [8— (t—ブチルジメチルシロキシ)—5, 6, 7, 8—テト ラヒドロナフタレン一 2—ィル]メチルホスホン酸ジェチル(105mg、 0. 254mmol)を 使用して、実施例 15 (15e)に記載した方法に従い、標記目的化合物(89. 3mg、収 率 78%)を得た。 Using [8- (t-butyldimethylsiloxy) -5,6,7,8-tetrahydronaphthalene-2-yl] methylphosphonate jetyl (105 mg, 0.254 mmol) prepared in Example 54 (54e) Then, according to the method described in Example 15 (15e), the title object compound (89.3 mg, yield 78%) was obtained.
[0462] IR(Liquid film) v 1024, 1057, 1080, 1271, 2859, 2934, 2952 cm"1; [0462] IR (Liquid film) v 1024, 1057, 1080, 1271, 2859, 2934, 2952 cm "1;
max max
1H NMR(CDC1 , 400MHz): δ 0.16 (3H, s), 0.17 (3H, s), 0.93 (9H, s), 1.30 (3H, d 1H NMR (CDC1, 400MHz): δ 0.16 (3H, s), 0.17 (3H, s), 0.93 (9H, s), 1.30 (3H, d
3 Three
t, J=0.8 Hz, 7.0 Hz), 1.33 (3H, dt, J=0.8 Hz, 7.0 Hz), 1.73—1.82 (2H, m), 1.97-2.03 (2H, m), 2.70-2.88 (2H, m), 4.09-4.27 (4H, m), 4.77-4.80 (IH, m), 7.14 (IH, d, J= t, J = 0.8 Hz, 7.0 Hz), 1.33 (3H, dt, J = 0.8 Hz, 7.0 Hz), 1.73—1.82 (2H, m), 1.97-2.03 (2H, m), 2.70-2.88 (2H, m), 4.09-4.27 (4H, m), 4.77-4.80 (IH, m), 7.14 (IH, d, J =
7.8 Hz), 7.40 (IH, d, J=7.8 Hz), 7.62 (IH, s); 7.8 Hz), 7.40 (IH, d, J = 7.8 Hz), 7.62 (IH, s);
MS(FAB) m/z: 447 (M— H)— . MS (FAB) m / z: 447 (M— H) —.
(54g) (8—ヒドロキシ一 5, 6, 7, 8—テトラヒドロナフタレン一 2—ィル)ジフルォロメ チノレホスホン酸ジェチノレ (54g) (8-hydroxy-1,5,6,7,8-tetrahydronaphthalene-1-yl) difluoromethinole
実施例 54 (54f)で製造した [8— (t—ブチルジメチルシロキシ)—5, 6, 7, 8—テト ラヒドロナフタレンー2—ィル]ジフルォロメチルホスホン酸ジェチル(7. 97g、 17. 8 mmol)を使用して、実施例 19 (19i)に記載した方法に従い、標記目的化合物 (4. 6 5g、収率 78%)を得た。 [8— (t-Butyldimethylsiloxy) -5,6,7,8-tetrahydronaphthalen-2-yl] difluoromethylphosphonate decyl ester (7.97 g, 17) prepared in Example 54 (54f) 8 mmol) and the title compound (4.65 g, 78% yield) was obtained according to the method described in Example 19 (19i).
[0463] IR(Liquid film) v 1023, 1045, 1122, 1262, 2869, 2940, 2987, 3429 cm"1; [0463] IR (Liquid film) v 1023, 1045, 1122, 1262, 2869, 2940, 2987, 3429 cm "1;
max max
1H NMR(CDC1 , 400MHz): δ 1.33 (6H, t, J=7.0 Hz), 1.74-2.06 (4H, m), 2.71-2.8 1H NMR (CDC1, 400MHz): δ 1.33 (6H, t, J = 7.0 Hz), 1.74-2.06 (4H, m), 2.71-2.8
3 Three
9 (2H, m), 4.11-4.27 (4H, m), 4.77-4.81 (IH, m), 7.16 (IH, d, J=7.8 Hz), 7.40 (IH, d, J=7.8 Hz), 7.67 (IH, s);
MS(EI) m/z: 334 M+. 9 (2H, m), 4.11-4.27 (4H, m), 4.77-4.81 (IH, m), 7.16 (IH, d, J = 7.8 Hz), 7.40 (IH, d, J = 7.8 Hz), 7.67 (IH, s); MS (EI) m / z: 334 M + .
(54h) (8 ォキソ 5, 6, 7, 8—テトラヒドロナフタレンー2 ィル)ジフルォロメチル ホスホン酸ジェチル (54h) (8oxo5,6,7,8-tetrahydronaphthalene-2-yl) difluoromethyl phosphonate jetyl
実施例 54 (54g)で製造した (8—ヒドロキシ一 5, 6, 7, 8—テトラヒドロナフタレン一 2 ィル)ジフルォロメチルホスホン酸ジェチル(4. 60g、 13. 8mmol)を使用して、 実施例 19 (19j)に記載した方法に従い、標記目的化合物(3. 92g、収率 86%)を得 た。 Performed using (8-hydroxy-1,5,6,7,8-tetrahydronaphthalene-2-yl) difluoromethylphosphonate decyl (4.60 g, 13.8 mmol) prepared in Example 54 (54 g). The title object compound (3.92 g, yield 86%) was obtained according to the method described in Example 19 (19j).
[0464] IR(Liquid film) v 1020, 1042, 1274, 1615, 1690, 2944, 2987 cm"1; [0464] IR (Liquid film) v 1020, 1042, 1274, 1615, 1690, 2944, 2987 cm "1;
max max
1H NMR(CDC1 , 400MHz) δ 1.34 (6H, t, J=7.0 Hz), 2.14-2.20 (2H, m), 2.69 (2H 1H NMR (CDC1, 400MHz) δ 1.34 (6H, t, J = 7.0 Hz), 2.14-2.20 (2H, m), 2.69 (2H
3 Three
, t, J=6.3 Hz), 3.02 (2H, t, J=5.9 Hz), 4.17-4.28 (4H, m), 7.37 (IH, d, J=8.2 Hz), 7. 73 (IH, d, J=8.2 Hz), 8.26 (IH, s); , t, J = 6.3 Hz), 3.02 (2H, t, J = 5.9 Hz), 4.17-4.28 (4H, m), 7.37 (IH, d, J = 8.2 Hz), 7. 73 (IH, d, J = 8.2 Hz), 8.26 (IH, s);
MS(EI) m/z: 332 M+. MS (EI) m / z: 332 M + .
(54i) (4, 5 ジヒドロナフト[1, 2-d] [l, 3]チアゾールー 8 ィル)ジフルォロメチ ルホスホン酸ジェチル (54i) (4,5 Dihydronaphtho [1,2-d] [l, 3] thiazole-8 yl) difluoromethyl jetyl phosphonate
実施例 54 (54h)で製造した(8—ォキソ 5, 6, 7, 8—テトラヒドロナフタレン一 2— ィル)ジフルォロメチルホスホン酸ジェチル(3. 78g、 11. 4mmol)を使用して、実施 例 3 (3a— 3b)に記載した方法に従 、、標記目的化合物(2. 09g、収率 49%)を得た Performed using (8-oxo-5,6,7,8-tetrahydronaphthalene-2-yl) difluoromethylphosphonate jetyl (3.78 g, 11.4 mmol) prepared in Example 54 (54h). According to the method described in Example 3 (3a-3b), the title target compound (2.09 g, yield 49%) was obtained.
[0465] IR(Liquid film) v 1020, 1043, 1124, 1231, 2913, 2940, 2986 cm"1; [0465] IR (Liquid film) v 1020, 1043, 1124, 1231, 2913, 2940, 2986 cm "1;
max max
1H NMR(CDC1 , 400MHz) δ 1.34 (6H, t, J=7.0 Hz), 3.10 (2H x2, s x2), 4.15—4.2 1H NMR (CDC1, 400MHz) δ 1.34 (6H, t, J = 7.0 Hz), 3.10 (2H x2, s x2), 4.15—4.2
3 Three
7 (4H, m), 7.30 (IH, d, J=7.8 Hz), 7.47 (IH, d, J=7.8 Hz), 8.17 (IH, s), 8.67 (IH, s) MS(EI) m/z: 373 M+. 7 (4H, m), 7.30 (IH, d, J = 7.8 Hz), 7.47 (IH, d, J = 7.8 Hz), 8.17 (IH, s), 8.67 (IH, s) MS (EI) m / z: 373 M + .
(54j) (4, 5 ジヒドロナフト[1, 2-d] [l, 3]チアゾールー 8 ィル)ジフルォロメチ ノレホスホン酸 (54j) (4, 5 Dihydronaphtho [1, 2-d] [l, 3] thiazole-8 yl) difluoromethinorephosphonic acid
実施例 54 (54i)で製造した (4, 5 ジヒドロナフト[1, 2-d] [l, 3]チアゾールー 8 ィル)ジフルォロメチルホスホン酸ジェチル(2. 07g、 5. 55mmol)を使用して、実 施例 1 (lc)に記載した方法に従い、標記目的化合物(1. 56g、収率 89%)を得た。
[0466] IR(KBr) v 1057, 1117, 1234, 1253, 1429, 1441, 3103, 3414 cm ; Using (4,5 dihydronaphtho [1,2-d] [l, 3] thiazol-8 yl) difluoromethylphosphonate jetyl (2.07 g, 5.55 mmol) prepared in Example 54 (54i) Then, according to the method described in Example 1 (lc), the title object compound (1.56 g, yield 89%) was obtained. [0466] IR (KBr) v 1057, 1117, 1234, 1253, 1429, 1441, 3103, 3414 cm;
max max
1H NMR(DMSO-d , 400MHz): δ 3.07 (2H x2, s x2), 7.35 (IH, d, J=7.8 Hz), 7.39 1H NMR (DMSO-d, 400 MHz): δ 3.07 (2H x2, s x2), 7.35 (IH, d, J = 7.8 Hz), 7.39
6 6
(IH, d, J=8.2 Hz), 7.98 (IH, s), 9.01 (IH, s); (IH, d, J = 8.2 Hz), 7.98 (IH, s), 9.01 (IH, s);
MS(FAB) m/z: 316 (M— H)— . MS (FAB) m / z: 316 (M— H) —.
<実施例 55 > <Example 55>
[ (7—メチルー 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキシ]メチル ホスホン酸 (例示化合物番号 1-160) [(7-Methyl-8H-indeno [1,2-d] [l, 3] thiazol-4-yl) oxy] methylphosphonic acid (Exemplified Compound No. 1-160)
(55a) [ (7—メチル 3—ォキソ 2, 3 ジヒドロ一 1H—インデン一 4—ィル)ォキシ ]メチルホスホン酸ジェチル (55a) [(7-Methyl-3-oxo2,3 dihydro-1H-indene-4-yl) oxy] methyl phosphonate
7 ヒドロキシ一 4—メチルインダン一 1—オン(1. 00g、6. 17mmol)を使用して、 実施例 1 (la)に記載した方法に従い、標記目的化合物(1. 72g、収率 89%)を得た 7 Using the title compound (1.72 g, 89% yield) according to the method described in Example 1 (la) using hydroxy 4-methylindan-1-one (1.00 g, 6.17 mmol) Got
[0467] IR(KBr) v 977, 1032, 1053, 1250, 1288, 1493, 1591, 1706, 2926, 2979 cm"1; [0467] IR (KBr) v 977, 1032, 1053, 1250, 1288, 1493, 1591, 1706, 2926, 2979 cm "1;
max max
1H NMR(CDC1 , 400MHz): δ 1.37 (6H, t, J=7.0 Hz), 2.27 (3H, s), 2.62—2.69 (2H, 1H NMR (CDC1, 400 MHz): δ 1.37 (6H, t, J = 7.0 Hz), 2.27 (3H, s), 2.62—2.69 (2H,
3 Three
m), 2.92-2.99 (2H, m), 4.33 (2H, q, J=7.0 Hz), 4.35 (2H, q, J=7.0 Hz), 4.41 (2H, d , J=9.8 Hz), 6.78 (IH, d, J=8.2 Hz), 7.32 (IH, d, J=8.2 Hz); m), 2.92-2.99 (2H, m), 4.33 (2H, q, J = 7.0 Hz), 4.35 (2H, q, J = 7.0 Hz), 4.41 (2H, d, J = 9.8 Hz), 6.78 ( IH, d, J = 8.2 Hz), 7.32 (IH, d, J = 8.2 Hz);
MS(FAB) m/z: 313 (M+H)+. MS (FAB) m / z: 313 (M + H) + .
(55b) [ (7—メチルー 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキシ ]メチルホスホン酸ジェチル (55b) [(7-Methyl-8H-indeno [1,2-d] [l, 3] thiazole-4-yl) oxy] methyl phosphonate
実施例 55 ( 55a)で製造した [ ( 7 メチル 3 ォキソ 2, 3 ジヒドロ一 1H—イン デン 4 ィル)ォキシ]メチルホスホン酸ジェチル(1. 72g、 5. 51mmol)を使用し て、実施例 3 (3a— 3b)に記載した方法に従い、標記目的化合物(1. 39g、収率 71 %)を得た。 Example 3 Using [(7-methyl-3-oxo-2,3-dihydro-1H-indene-4-yl) oxy] methylphosphonate (1.72 g, 5.51 mmol) prepared in Example 55 (55a), Example 3 According to the method described in (3a-3b), the title object compound (1.39 g, yield 71%) was obtained.
[0468] IR(KBr) v 979, 1051, 1058, 1084, 1255, 1275, 1388, 1430, 1509, 1593, 1614, max [0468] IR (KBr) v 979, 1051, 1058, 1084, 1255, 1275, 1388, 1430, 1509, 1593, 1614, max
2903, 2980, 3088 cm"1; 2903, 2980, 3088 cm "1;
1H NMR(CDC1 , 400MHz) δ 1.35 (6H, t, J=7.0 Hz), 2.35 (3H, s), 3.73 (2H, s), 4 1H NMR (CDC1, 400MHz) δ 1.35 (6H, t, J = 7.0 Hz), 2.35 (3H, s), 3.73 (2H, s), 4
3 Three
.32 (2H, q, J=7.0 Hz), 4.33 (2H, q, J=7.0 Hz), 4.63 (2H, d, J=8.8 Hz), 6.99 (IH, d, J=8.3 Hz), 7.04 (IH, d, J=8.3 Hz), 8.84 (IH, s);
MS(FAB) m/z: 354 (M+H)+. .32 (2H, q, J = 7.0 Hz), 4.33 (2H, q, J = 7.0 Hz), 4.63 (2H, d, J = 8.8 Hz), 6.99 (IH, d, J = 8.3 Hz), 7.04 (IH, d, J = 8.3 Hz), 8.84 (IH, s); MS (FAB) m / z: 354 (M + H) + .
(55c) [ (7—メチルー 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキシ ]メチノレホスホン酸 (55c) [(7-Methyl-8H-indeno [1,2-d] [l, 3] thiazole-4-yl) oxy] methinorephosphonic acid
実施例 55 (55b)で製造した [ (7—メチル 8H—インデノ [1, 2— d] [l, 3]チアゾ 一ルー 4 ィル)ォキシ]メチルホスホン酸ジェチル(56mg、 0. 158mmol)を使用し て、実施例 1 (lc)に記載した方法に従い、標記目的化合物 (43mg、収率 92%)を 得た。 Example 55 Using [(7-methyl 8H-indeno [1,2-d] [l, 3] thiazo-l4yl) oxy] methylphosphonate (56 mg, 0.158 mmol) prepared in Example 55 (55b) Then, according to the method described in Example 1 (lc), the title object compound (43 mg, yield 92%) was obtained.
[0469] IR(KBr) v 958, 1064, 1250, 1385, 1428, 1510, 1612, 2304, 2749, 2900, 2923, max [0469] IR (KBr) v 958, 1064, 1250, 1385, 1428, 1510, 1612, 2304, 2749, 2900, 2923, max
3088, 3371 cm"1; 3088, 3371 cm "1;
1H NMR(DMSO-d , 500MHz): δ 2.30 (3H, s), 3.85 (2H, s), 4.30 (2H, d, J=8.8 Hz 1H NMR (DMSO-d, 500MHz): δ 2.30 (3H, s), 3.85 (2H, s), 4.30 (2H, d, J = 8.8 Hz
6 6
), 7.03 (1H, d, J=8.3 Hz), 7.05 (1H, d, J=8.3 Hz), 9.18 (1H, s); ), 7.03 (1H, d, J = 8.3 Hz), 7.05 (1H, d, J = 8.3 Hz), 9.18 (1H, s);
MS(FAB) m/z: 296 (M— H)— . MS (FAB) m / z: 296 (M— H) —.
<実施例 56 > <Example 56>
[ (7-イソプロピル -8H—インデノ [ 1 , 2 - d]チアゾール 4 ィル)ォキシ]メチル ホスホン酸 (例示化合物番号 1-164) [(7-Isopropyl-8H-indeno [1,2-d] thiazole-4-yl) oxy] methylphosphonic acid (Exemplary Compound No. 1-164)
(56a) [ (7 イソプロべ-ルー 8H—インデノ [1, 2 d]チアゾールー 4 ィル)ォキシ ]メチルホスホン酸ジェチル (56a) [(7 Isoproberu 8H-indeno [1,2d] thiazole-4-yl) oxy] methyl phosphonate
実施例 31 (31a)で製造した [ (7 ァセチル— 8H—インデノ [1, 2 d] [l, 3]チア ゾール—4—ィル)ォキシ]メチルホスホン酸ジェチル(2. 54g、6. 66mmol)をテトラ ヒドロフラン(67mL)に溶解し、 0°C冷却下、そこへ Tebbe試薬(0. 5M トルエン溶 液)(16. OmL、 7. 99mmol)をカ卩え、窒素雰囲気下、室温で 2時間攪拌した。 Example 31 [(7 Acetyl-8H-indeno [1,2d] [l, 3] thiazol-4-yl) oxy] methylphosphonate jetyl (2.54 g, 6.66 mmol) prepared in (31a) Is dissolved in tetrahydrofuran (67 mL), cooled to 0 ° C, Tebbe reagent (0.5 M toluene solution) (16. OmL, 7.99 mmol) is added to it, and nitrogen atmosphere is used for 2 hours at room temperature. Stir.
[0470] 反応液に水および飽和炭酸水素ナトリウム水溶液を加え、塩化メチレンで抽出した 。得られた有機層を水及び飽和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、減圧 下溶媒を留去した。得られた残渣をシリカゲルクロマトグラフィー (酢酸ェチル:メタノ ール)を用いて精製し、標記目的化合物(1. 23g、収率 49%)を得た。 [0470] Water and a saturated aqueous sodium hydrogen carbonate solution were added to the reaction mixture, and the mixture was extracted with methylene chloride. The obtained organic layer was washed with water and saturated brine, dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure. The obtained residue was purified by silica gel chromatography (ethyl acetate: methanol) to obtain the title compound (1.23 g, yield 49%).
[0471] IR(Liquid film) v 975, 1026, 1248, 1508, 2982 cm"1; [0471] IR (Liquid film) v 975, 1026, 1248, 1508, 2982 cm "1;
max max
1H NMR(CDC1 , 400MHz) δ 1.35 (6H, t, J=7.0 Hz), 2.14—2.16 (3H, m), 3.90 (2H 1H NMR (CDC1, 400MHz) δ 1.35 (6H, t, J = 7.0 Hz), 2.14-2.16 (3H, m), 3.90 (2H
3 Three
, s), 4.27-4.36 (4H, m), 4.63 (2H, d, J=8.6 Hz), 5.11-5.13 (1H, m), 5.24-5.26 (1H,
m), 7.03 (IH, d, J=8.6 Hz), 7.13 (IH, d, J=8.6 Hz), 8.81 (IH, s); , s), 4.27-4.36 (4H, m), 4.63 (2H, d, J = 8.6 Hz), 5.11-5.13 (1H, m), 5.24-5.26 (1H, m), 7.03 (IH, d, J = 8.6 Hz), 7.13 (IH, d, J = 8.6 Hz), 8.81 (IH, s);
MS(FAB) m/z: 380 (M+H)+. MS (FAB) m / z: 380 (M + H) + .
(56b) [ (7 イソプロピル—8H—インデノ [1, 2— d]チアゾールー 4 ィル)ォキシ]メ チノレホスホン酸ジェチノレ (56b) [(7 Isopropyl-8H-indeno [1,2-d] thiazol-4-yl) oxy] methinorephosphonic acid jetinore
実施例 56 (56a)で製造した [ (7—イソプロべ-ル— 8H—インデノ [1, 2 d] [l, 3 ]チアゾールー 4 ィル)ォキシ]メチルホスホン酸ジェチル(1. 10g、 2. 90mmol)を ベンゼン(58mL)に溶解し、そこへクロロトリス(トリフエ-ルホスフィン)ロジウム(537 mg、 0. 580mmol)を加え、水素雰囲気下、 40°Cで 7時間攪拌した。 Example 56 [(7-Isopropyl-8H-indeno [1,2d] [l, 3] thiazol-4-yl) oxy] methylphosphonate (1.10 g, 2.90 mmol) prepared in Example 56 (56a) ) Was dissolved in benzene (58 mL), chlorotris (triphenylphosphine) rhodium (537 mg, 0.580 mmol) was added thereto, and the mixture was stirred at 40 ° C. for 7 hours in a hydrogen atmosphere.
[0472] 減圧下溶媒を留去し、得られた残渣をシリカゲルクロマトグラフィー (へキサン:酢酸 ェチル)を用いて精製し、標記目的化合物(887mg、収率 80%)を得た。 [0472] The solvent was distilled off under reduced pressure, and the obtained residue was purified by silica gel chromatography (hexane: ethyl acetate) to obtain the title object compound (887 mg, yield 80%).
[0473] 1H NMR(CDC1 , 400MHz): δ 1.29 (3Η, s), 1.31 (3H, s), 1.35 (6H, t, J=7.0 Hz), 3 [0473] 1H NMR (CDC1, 400MHz): δ 1.29 (3Η, s), 1.31 (3H, s), 1.35 (6H, t, J = 7.0 Hz), 3
3 Three
.05-3.15 (IH, m), 3.83 (2H, s), 4.33 (4H, dq, J=7.0 Hz, 7.0 Hz), 4.64 (2H, d, J=8.6 Hz), 7.06 (IH, d, J=8.6 Hz), 7.14 (IH, d, J=8.6 Hz), 8.83 (IH, s); .05-3.15 (IH, m), 3.83 (2H, s), 4.33 (4H, dq, J = 7.0 Hz, 7.0 Hz), 4.64 (2H, d, J = 8.6 Hz), 7.06 (IH, d, J = 8.6 Hz), 7.14 (IH, d, J = 8.6 Hz), 8.83 (IH, s);
MS(ES) m/z: 382 (M+H)+. MS (ES) m / z: 382 (M + H) + .
(56c) [ (7 イソプロピル—8H—インデノ [1, 2 d]チアゾールー 4 ィル)ォキシ]メ チノレホスホン酸 (56c) [(7 Isopropyl-8H-indeno [1,2d] thiazole-4-yl) oxy] methinorephosphonic acid
実施例 56 (56b)で製造した [ (7 イソプロピル—8H—インデノ [1, 2— d]チアゾー ルー 4—ィル)ォキシ]メチルホスホン酸ジェチル(77. 8mg、 0. 204mmol)を使用し て、実施例 1 (lc)に記載した方法に従い、標記目的化合物(52. lmg、収率 79%) を得た。 Using [(7 isopropyl-8H-indeno [1,2-d] thiazol 4-yl) oxy] methylphosphonate (77.8 mg, 0.204 mmol) prepared in Example 56 (56b) The title object compound (52. lmg, yield 79%) was obtained according to the method described in Example 1 (lc).
[0474] IR(KBr) v 951, 1273, 1490, 2960 cm"1; [0474] IR (KBr) v 951, 1273, 1490, 2960 cm "1;
max max
1H NMR(DMSO-d , 400MHz): δ 1.23 (3H, s), 1.26 (3H, s), 3.05—3.13 (IH, m), 3. 1H NMR (DMSO-d, 400MHz): δ 1.23 (3H, s), 1.26 (3H, s), 3.05—3.13 (IH, m), 3.
6 6
94 (2H, s), 4.30 (2H, d, J=9.0 Hz), 7.10 (IH, d, J=8.6 Hz), 7.13 (IH, d, J=8.6 Hz), 9.16 (IH, s); 94 (2H, s), 4.30 (2H, d, J = 9.0 Hz), 7.10 (IH, d, J = 8.6 Hz), 7.13 (IH, d, J = 8.6 Hz), 9.16 (IH, s);
MS(ESI) m/z: 324 (M— H)— . MS (ESI) m / z: 324 (M— H) —.
<実施例 57 > <Example 57>
( {7 [ (ェチルァミノ)カルボ-ル ] 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ーィル }ォキシ)メチルホスホン酸 (例示化合物番号 1-38)
実施例 36 (36c)で製造した 4— [ (ジエトキシホスホリル)メトキシ]— 8H—インデノ [ 1, 2-d] [l, 3]チアゾール— 7—カルボン酸(100mg、 0. 261mmol)を使用して、 実施例 36 (36d)及び実施例 1 (lc)に記載した方法に順次従い、標記目的化合物( 77. 3mg、収率 87%)を得た。 ({7 [(Ethylamino) carbol] 8H-indeno [1, 2-d] [l, 3] thiazole-4-yl} oxy) methylphosphonic acid (Exemplary Compound No. 1-38) Example 36 Using 4-[(diethoxyphosphoryl) methoxy] -8H-indeno [1,2-d] [l, 3] thiazole-7-carboxylic acid (100 mg, 0.261 mmol) prepared in (36c) Then, the title target compound (77.3 mg, yield 87%) was obtained by sequentially following the methods described in Example 36 (36d) and Example 1 (lc).
[0475] 但し、実施例 36 (36d)と同様の工程において、塩化アンモ-ゥムの代わりにェチル ァミン塩酸塩(31. 9mg、0. 391mmol)を用いた。 However, in the same process as in Example 36 (36d), ethylamine hydrochloride (31.9 mg, 0.391 mmol) was used instead of ammonium chloride.
[0476] IR(KBr) v 1075, 1266, 1543, 1650, 3302 cm"1; [0476] IR (KBr) v 1075, 1266, 1543, 1650, 3302 cm "1;
max max
1H NMR(DMSO-d , 400MHz) δ 1.14 (3H, t, J=7.0 Hz), 3.24-3.33 (2H, m), 4.19 1H NMR (DMSO-d, 400MHz) δ 1.14 (3H, t, J = 7.0 Hz), 3.24-3.33 (2H, m), 4.19
6 6
(2H, s), 4.38 (2H, d, J=9.0 Hz), 7.21 (IH, d, J=8.6 Hz), 7.57 (IH, d, J=8.6 Hz), 8.2 8-8.33 (IH, m), 9.14 (IH, s); (2H, s), 4.38 (2H, d, J = 9.0 Hz), 7.21 (IH, d, J = 8.6 Hz), 7.57 (IH, d, J = 8.6 Hz), 8.2 8-8.33 (IH, m ), 9.14 (IH, s);
MS(ESI) m/z: 353 (M— H)— . MS (ESI) m / z: 353 (M— H) —.
<実施例 58 > <Example 58>
({7— [ (イソプロピルァミノ)カルボ-ル]— 8H—インデノ [1, 2— d] [l, 3]チアゾー ルー 4ーィル }ォキシ)メチルホスホン酸(例示化合物番号 1-40) ({7- [(Isopropylamino) carbol] — 8H-indeno [1, 2— d] [l, 3] thiazol 4-yl} oxy) methylphosphonic acid (Exemplified Compound No. 1-40)
実施例 36 (36c)で製造した 4— [ (ジエトキシホスホリル)メトキシ]— 8H—インデノ [ 1, 2-d] [l, 3]チアゾール— 7—カルボン酸(100mg、 0. 261mmol)を使用して、 実施例 36 (36d)及び実施例 1 (lc)に記載した方法に順次従い、標記目的化合物( 75. 7mg、収率 81%)を得た。 Example 36 Using 4-[(diethoxyphosphoryl) methoxy] -8H-indeno [1,2-d] [l, 3] thiazole-7-carboxylic acid (100 mg, 0.261 mmol) prepared in (36c) Then, the title target compound (75.7 mg, yield 81%) was obtained by sequentially following the methods described in Example 36 (36d) and Example 1 (lc).
[0477] 但し、実施例 36 (36d)と同様の工程において、塩ィ匕アンモ-ゥムの代わりにイソプ 口ピノレアミン(33. 3 1^、0. 391mmol)を用! /、た。 [0477] However, in the same process as in Example 36 (36d), isopropanol pinoleamine (33.31 ^, 0.391 mmol) was used instead of salt ammonium.
[0478] IR(KBr) v 944, 1266, 1544, 1626, 3309 cm"1; [0478] IR (KBr) v 944, 1266, 1544, 1626, 3309 cm "1;
max max
1H NMR(DMSO-d , 400MHz): δ 1.18 (6H, d, J=6.7 Hz), 4.05—4.15 (IH, m), 4.17 1H NMR (DMSO-d, 400MHz): δ 1.18 (6H, d, J = 6.7 Hz), 4.05—4.15 (IH, m), 4.17
6 6
(2H, s), 4.37 (2H, d, J=9.0 Hz), 7.20 (IH, d, J=8.6 Hz), 7.57 (IH, d, J=8.6 Hz), 8.0 7-8.11 (IH, m), 9.14 (IH, s); (2H, s), 4.37 (2H, d, J = 9.0 Hz), 7.20 (IH, d, J = 8.6 Hz), 7.57 (IH, d, J = 8.6 Hz), 8.0 7-8.11 (IH, m ), 9.14 (IH, s);
MS(ESI) m/z: 367 (M— H)— . MS (ESI) m / z: 367 (M— H) —.
<実施例 59 > <Example 59>
({7— [ (シクロプロピルァミノ)カルボ-ル ]—8H—インデノ [1, 2-d] [l, 3]チアゾ 一ルー 4ーィル }ォキシ)メチルホスホン酸(例示化合物番号 1-41)
実施例 36 (36c)で製造した 4 [(ジエトキシホスホリル)メトキシ]— 8H インデノ [ 1, 2-d][l, 3]チアゾール—7—カルボン酸(80. Omg、0. 209mmol)を使用して 、実施例 36 (36d)及び実施例 1 (lc)に記載した方法に順次従い、標記目的化合物 (32. lmg、収率 48%)を得た。 ({7-[(Cyclopropylamino) carbol] -8H-indeno [1,2-d] [l, 3] thiazo 4-lu} oxy) methylphosphonic acid (Exemplary Compound No. 1-41) Example 36 Using 4 [(diethoxyphosphoryl) methoxy] -8H indeno [1,2-d] [l, 3] thiazole-7-carboxylic acid (80. Omg, 0.209 mmol) prepared in (36c) Then, the title target compound (32. lmg, yield 48%) was obtained by sequentially following the methods described in Example 36 (36d) and Example 1 (lc).
[0479] 但し、実施例 36 (36d)と同様の工程において、塩化アンモ-ゥムの代わりにシクロ プロピノレアミン(21. 7μレ 0. 313mmol)を用!/、た。 However, in the same process as in Example 36 (36d), cyclopropinoleamine (21.7 μl 0.313 mmol) was used instead of ammonium chloride.
[0480] IR(KBr) v 943, 1268, 1502, 1640, 3272 cm"1; [0480] IR (KBr) v 943, 1268, 1502, 1640, 3272 cm "1;
max max
1H NMR(DMSO-d , 400MHz): δ 0.55-0.62 (2H, m), 0.66—0.73 (2H, m), 2.82-2.9 1H NMR (DMSO-d, 400 MHz): δ 0.55-0.62 (2H, m), 0.66—0.73 (2H, m), 2.82-2.9
6 6
0 (IH, m), 4.19 (2H, s), 4.38 (2H, d, J=9.0 Hz), 7.22 (IH, d, J=8.6 Hz), 7.56 (IH, d , J=8.6 Hz), 8.30-8.35 (IH, m), 9.17 (IH, s); 0 (IH, m), 4.19 (2H, s), 4.38 (2H, d, J = 9.0 Hz), 7.22 (IH, d, J = 8.6 Hz), 7.56 (IH, d, J = 8.6 Hz), 8.30-8.35 (IH, m), 9.17 (IH, s);
MS(FAB) m/z: 365 (M- H)— . MS (FAB) m / z: 365 (M- H) —.
<実施例 60> <Example 60>
[(7— {[(3—メトキシプロピル)ァミノ]カルボ二ル}— 8H—インデノ [1, 2-d][l, 3] チアゾール 4 ィル)ォキシ]メチルホスホン酸(例示化合物番号 1-55) [(7— {[(3-Methoxypropyl) amino] carbonyl} — 8H-indeno [1,2-d] [l, 3] thiazole-4-yl) oxy] methylphosphonic acid (Exemplified Compound No. 1-55 )
実施例 36 (36c)で製造した 4 [(ジエトキシホスホリル)メトキシ]— 8H インデノ [ 1, 2-d][l, 3]チアゾール— 7—カルボン酸(100mg、 0. 261mmol)を使用して、 実施例 36 (36d)及び実施例 1 (lc)に記載した方法に順次従い、標記目的化合物( 55. Omg、収率 61%)を得た。 Example 4 Using 4 [(diethoxyphosphoryl) methoxy] -8H indeno [1,2-d] [l, 3] thiazole-7-carboxylic acid (100 mg, 0.261 mmol) prepared in (36c) The title target compound (55. Omg, 61% yield) was obtained by sequentially following the methods described in Example 36 (36d) and Example 1 (lc).
[0481] 但し、実施例 36 (36d)と同様の工程において、塩ィ匕アンモ-ゥムの代わりに 3—メト キシプロピルァミン(0. 038mL、0. 365mmol)を用いた。 [0481] However, in the same process as in Example 36 (36d), 3-methoxypropylamine (0.038 mL, 0.365 mmol) was used instead of salt ammonium.
[0482] IR(KBr) v 1267, 1486, 1503, 1543, 1583, 1651, 2873, 2926, 3081, 3285 cm"1; [0482] IR (KBr) v 1267, 1486, 1503, 1543, 1583, 1651, 2873, 2926, 3081, 3285 cm "1;
max max
1H NMR(DMSO-d , 500MHz): δ 1.78 (2H, m), 3.25 (3H, s), 3.31 (2H, m), 3.40 (2 1H NMR (DMSO-d, 500MHz): δ 1.78 (2H, m), 3.25 (3H, s), 3.31 (2H, m), 3.40 (2
6 6
H, t, J=6.4 Hz), 4.19 (2H, s), 4.40 (2H, d, J=8.8 Hz), 7.24 (IH, d, J=8.7 Hz), 7.60 ( IH, d, J=8.7 Hz), 8.32 (IH, t, J=5.4 Hz), 9.17 (IH, s); H, t, J = 6.4 Hz), 4.19 (2H, s), 4.40 (2H, d, J = 8.8 Hz), 7.24 (IH, d, J = 8.7 Hz), 7.60 (IH, d, J = 8.7 Hz), 8.32 (IH, t, J = 5.4 Hz), 9.17 (IH, s);
MS(FAB) m/z: 397 (M- H)— . MS (FAB) m / z: 397 (M- H) —.
<実施例 61> <Example 61>
[(7— {[(2 ヒドロキシェチル)ァミノ]カルボ-ル}— 8H—インデノ [1, 2— d][l, 3 ]チアゾール 4 ィル)ォキシ]メチルホスホン酸(例示化合物番号 1-52)
実施例 36 (36c)で製造した 4 [ (ジエトキシホスホリル)メトキシ]— 8H インデノ [ 1, 2-d] [l, 3]チアゾール 7—カルボン酸(150mg、 0. 391mmol)を使用して、 実施例 36 (36d)及び実施例 1 (lc)に記載した方法に順次従い、標記目的化合物( 12. 3mg、収率 8%)を得た。 [(7 — {[(2 Hydroxyethyl) amino] carbol} —8H-indeno [1,2-d] [l, 3] thiazole-4-yl) oxy] methylphosphonic acid (Exemplified Compound No. 1-52 ) Example 36 Using 4 [(diethoxyphosphoryl) methoxy] -8H indeno [1,2-d] [l, 3] thiazole 7-carboxylic acid (150 mg, 0.391 mmol) prepared in (36c) The title target compound (12.3 mg, yield 8%) was obtained by sequentially following the methods described in Example 36 (36d) and Example 1 (lc).
[0483] 但し、実施例 36 (36d)と同様の工程にお!、て、塩化アンモ-ゥムの代わりにェタノ ールァミン(0. 033mL、0. 548mmol)を用いた。 [0483] However, ethanolamine (0.033 mL, 0.548 mmol) was used in the same step as Example 36 (36d) instead of ammonium chloride.
[0484] IR(KBr) v 1068, 1269, 1544, 1580, 1644, 2929, 3082, 3310 cm"1; [0484] IR (KBr) v 1068, 1269, 1544, 1580, 1644, 2929, 3082, 3310 cm "1;
max max
1H NMR(DMSO-d , 500MHz): δ 3.32—3.36 (2H, m), 3.53 (2H, t, J=6.4 Hz), 4.20 1H NMR (DMSO-d, 500MHz): δ 3.32—3.36 (2H, m), 3.53 (2H, t, J = 6.4 Hz), 4.20
6 6
(2H, s), 4.40 (2H, d, J=8.8 Hz), 7.24 (IH, d, J=8.8 Hz), 7.63 (IH, d, J=8.8 Hz), 8.2 8-8.30 (IH, m), 9.17 (IH, s); (2H, s), 4.40 (2H, d, J = 8.8 Hz), 7.24 (IH, d, J = 8.8 Hz), 7.63 (IH, d, J = 8.8 Hz), 8.2 8-8.30 (IH, m ), 9.17 (IH, s);
MS(FAB) m/z: 369 (M— H)— . MS (FAB) m / z: 369 (M— H) —.
<実施例 62 > <Example 62>
{ [7- ({ [2 ヒロドキシ—1— (ヒドロキシメチル)ェチル]アミノ}カルボ-ル) 8H— インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル]ォキシ }メチルホスホン酸(例示化 合物番号 1-198) {[7- ({[2 Hydroxy-1- (hydroxymethyl) ethyl] amino} carbol) 8H— Indeno [1, 2-d] [l, 3] thiazol-4-yl] oxy} methylphosphonic acid (example (Compound number 1-198)
実施例 36 (36c)で製造した 4 [ (ジエトキシホスホリル)メトキシ]— 8H インデノ [ 1, 2-d] [l, 3]チアゾール 7—カルボン酸(150mg、 0. 391mmol)及び塩ィ匕ァ ンモ-ゥムの代わりに 2 アミノー 1, 3 プロパンジオール(49. 9mg、0. 548mmol )を使用して、実施例 36 (36d)に記載した方法に従い、 { [7— ({ [2 ヒロドキシ—1 —(ヒドロキシメチル)ェチル]アミノ}カルボ-ル)— 8H—インデノ [1, 2— d] [l, 3] チアゾール—4—ィル]ォキシ }メチルホスホン酸ジェチル(144mg、収率 80%)を得 た。 Example 36 4 [(Diethoxyphosphoryl) methoxy] -8H indeno [1,2-d] [l, 3] thiazole 7-carboxylic acid (150 mg, 0.391 mmol) prepared in (36c) and salt In accordance with the method described in Example 36 (36d) using 2 amino-1,3 propanediol (49.9 mg, 0.548 mmol) instead of ammonia, {[7— ({[2 hydroxy- 1 — (Hydroxymethyl) ethyl] amino} carbol) — 8H-indeno [1, 2-d] [l, 3] thiazole-4-yl] oxy} methyl phosphonate (144 mg, 80% yield) Got.
[0485] { [7- ({ [2 ヒロドキシ—1— (ヒドロキシメチル)ェチル]アミノ}カルボ-ル) 8H —インデノ [1, 2— d] [l, 3]チアゾール—4—ィル]ォキシ }メチルホスホン酸ジェチ ル(70. 6mg、0. 155mmol)を使用して、実施例 36 (36e)に記載した方法に従い、 標記目的化合物 (48. 7mg、収率 79%)を得た。 [0485] {[7- ({[2 Hydroxy-1- (hydroxymethyl) ethyl] amino} carbol) 8H —Indeno [1, 2— d] [l, 3] thiazole-4-yl] oxy } The title object compound (48.7 mg, 79% yield) was obtained according to the method described in Example 36 (36e) using methylphosphonate (70.6 mg, 0.155 mmol).
[0486] IR(KBr) v 1063, 1159, 1261, 1539, 1636, 3113, 3350 cm"1; [0486] IR (KBr) v 1063, 1159, 1261, 1539, 1636, 3113, 3350 cm "1;
max max
1H NMR(DMSO-d , 400MHz): δ 3.53 (4H, d, J=5.9 Hz), 3.93—4.01 (IH, m), 4.18
(2H, s), 4.39 (2H, d, J=9.0 Hz), 7.22 (IH, d, J=8.7 Hz), 7.62 (IH, d, J=8.7 Hz), 7.8 2 (IH, d, J=7.8 Hz), 9.15 (IH, s); 1H NMR (DMSO-d, 400MHz): δ 3.53 (4H, d, J = 5.9 Hz), 3.93—4.01 (IH, m), 4.18 (2H, s), 4.39 (2H, d, J = 9.0 Hz), 7.22 (IH, d, J = 8.7 Hz), 7.62 (IH, d, J = 8.7 Hz), 7.8 2 (IH, d, J = 7.8 Hz), 9.15 (IH, s);
MS(FAB) m/z: 399 (M— H)— . MS (FAB) m / z: 399 (M— H) —.
<実施例 63 > <Example 63>
[ (7— { [ (2 アミノエチル)ァミノ]カルボ-ル}— 8H—インデノ [1, 2— d] [l, 3]チ ァゾールー 4 ィル)ォキシ]メチルホスホン酸(例示化合物番号 1-57) [(7— {[(2 Aminoethyl) amino] carbol} — 8H—Indeno [1, 2— d] [l, 3] Thiazole-4-yl) oxy] methylphosphonic acid (Exemplary Compound No. 1-57 )
実施例 36 (36c)で製造した 4 [ (ジエトキシホスホリル)メトキシ]— 8H インデノ [ 1, 2-d] [l, 3]チアゾールー 7—力ルボン酸(150mg、 0. 39mmol)を使用して、 実施例 36 (36d)及び実施例 36 (36e)に記載した方法に順次従い、標記目的化合 物(119mg、収率 83%)を得た。 Example 36 Using 4 [(diethoxyphosphoryl) methoxy] -8H indeno [1,2-d] [l, 3] thiazole-7-strength rubonic acid (150 mg, 0.39 mmol) prepared in (36c) The title target compound (119 mg, 83% yield) was obtained by sequentially following the methods described in Example 36 (36d) and Example 36 (36e).
[0487] 但し、実施例 36 (36d)と同様の工程において、塩ィ匕アンモ-ゥムの代わりに t—ブ チル (2 アミノエチル)カーバメート(75mg、 0. 47mmol)を用いた。 However, in the same step as Example 36 (36d), t-butyl (2 aminoethyl) carbamate (75 mg, 0.47 mmol) was used in place of the salt ammonium.
[0488] IR(KBr) v 1060, 1228, 1253, 1506, 1542, 1583, 1639, 2123, 3068, 3138 cm"1; [0488] IR (KBr) v 1060, 1228, 1253, 1506, 1542, 1583, 1639, 2123, 3068, 3138 cm "1;
max max
1H NMR(DMSO-d +D O+NaHCO , 400MHz): δ 3.02 (2H, s), 3.38 (2H, t, J=5.1 1H NMR (DMSO-d + D 2 O + NaHCO 3, 400 MHz): δ 3.02 (2H, s), 3.38 (2H, t, J = 5.1
6 2 3 6 2 3
Hz), 3.75 (2H, t, J=5.1 Hz ), 4.01 (2H, d, J=9.8 Hz), 6.95 (IH, d, J=8.6 Hz), 7.60 (1 Hz), 3.75 (2H, t, J = 5.1 Hz), 4.01 (2H, d, J = 9.8 Hz), 6.95 (IH, d, J = 8.6 Hz), 7.60 (1
H, d, J=8.6 Hz), 9.00 (IH, s); H, d, J = 8.6 Hz), 9.00 (IH, s);
MS(ESI) m/z: 368 (M— H)— . MS (ESI) m / z: 368 (M— H) —.
<実施例 64 > <Example 64>
[ (7— { [ (2 ピペリジン 1 ィルェチル)ァミノ]カルボ-ル}—8H—インデノ [1, 2 d] [1, 3]チアゾールー 4 ィル)ォキシ]メチルホスホン酸(例示化合物番号 1-199 ) [(7— {[(2 Piperidine 1 ylethyl) amino] carbol} —8H-indeno [1, 2 d] [1, 3] thiazol-4-yl) oxy] methylphosphonic acid (Exemplary Compound No. 1-199)
実施例 36 (36c)で製造した 4 [ (ジエトキシホスホリル)メトキシ]— 8H インデノ [ Example 36 4 [(Diethoxyphosphoryl) methoxy] -8H indeno prepared in (36c) [
I, 2-d] [l, 3]チアゾール 7—カルボン酸(150mg、 0. 391mmol)を使用して、 実施例 36 (36d)及び実施例 1 (lc)に記載した方法に順次従い、標記目的化合物( 127mgゝ収率 60%)を得た。 I, 2-d] [l, 3] thiazole 7-carboxylic acid (150 mg, 0.391 mmol) was used in turn following the procedure described in Example 36 (36d) and Example 1 (lc) The target compound (127 mg yield 60%) was obtained.
[0489] 但し、実施例 36 (36d)と同様の工程において、塩ィ匕アンモ-ゥムの代わりに 1— (2 [0489] However, in the same process as in Example 36 (36d), 1— (2
—アミノエチル)ピぺリジン(85. OmL、0. 596mmol)を用いた。 —Aminoethyl) piperidine (85. OmL, 0.596 mmol) was used.
[0490] IR(KCl) V 3251, 2944, 2653, 1646, 1582, 1543, 1484, 1434, 1385, 1263, 1187,
1071, 1002, 950, 770, 677; [0490] IR (KCl) V 3251, 2944, 2653, 1646, 1582, 1543, 1484, 1434, 1385, 1263, 1187, 1071, 1002, 950, 770, 677;
1H NMR(DMSO-d , 400MHz): δ 1.32-1.48 (2H, m), 1.61-1.78 (2H, m), 1.81-1.9 1H NMR (DMSO-d, 400MHz): δ 1.32-1.48 (2H, m), 1.61-1.78 (2H, m), 1.81-1.9
6 6
0 (2H, m), 2.91-3.05 (2H, m), 3.21—3.29 (2H, m), 3.54—3.62 (2H, m), 3.62—3.71 (2 0 (2H, m), 2.91-3.05 (2H, m), 3.21—3.29 (2H, m), 3.54—3.62 (2H, m), 3.62—3.71 (2
H, m), 4.24 (2H, s), 4.41 (2H, d, J=9.0 Hz), 7.30 (IH, d, J=8.8 Hz), 7.69 (IH, d, J= 8.8 Hz), 8.63 (IH, t, J=5.7 Hz), 9.18 (IH, s); H, m), 4.24 (2H, s), 4.41 (2H, d, J = 9.0 Hz), 7.30 (IH, d, J = 8.8 Hz), 7.69 (IH, d, J = 8.8 Hz), 8.63 ( IH, t, J = 5.7 Hz), 9.18 (IH, s);
MS(FAB) m/z: 436 (M- H)— . MS (FAB) m / z: 436 (M- H) —.
<実施例 65 > <Example 65>
[ (7— { [ (2 モルホリン 4 ィルェチル)ァミノ]カルボ-ル} 8H—インデノ [1, 2 d] [1, 3]チアゾールー 4 ィル)ォキシ]メチルホスホン酸(例示化合物番号 1-200 ) [(7— {[(2 morpholine 4 ylethyl) amino] carbol} 8H—indeno [1, 2 d] [1, 3] thiazol-4-yl) oxy] methylphosphonic acid (Exemplary Compound No. 1-200)
実施例 36 (36c)で製造した 4 [ (ジエトキシホスホリル)メトキシ]— 8H インデノ [ Example 36 4 [(Diethoxyphosphoryl) methoxy] -8H indeno prepared in (36c) [
I, 2-d] [l, 3]チアゾール 7—カルボン酸(150mg、 0. 391mmol)及び塩ィ匕ァ ンモ-ゥムの代わりに 2 アミノエチルモルホリン(0. 072mL、0. 548mmol)を使用 して、実施例 36 (36d)に記載した方法に従い、 [ (7— { [ (2 モルホリン— 4—ィルェ チル)ァミノ]カルボ-ル}ー811—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル) ォキシ]メチルホスホン酸ジェチル(171mg、収率 88%)を得た。 I, 2-d] [l, 3] thiazole 7-carboxylic acid (150 mg, 0.391 mmol) and 2-aminoethylmorpholine (0.072 mL, 0.548 mmol) in place of salt-molybdenum Then, according to the method described in Example 36 (36d), [(7 — {[(2 morpholine—4-ylethyl) amino] carbol} -811—indeno [1, 2-d] [l, 3] thiazole-4-yl) oxy] methylphosphonate (171 mg, yield 88%) was obtained.
[0491] [ (7- { [ (2 モルホリン 4 ィルェチル)ァミノ]カルボ-ル}—8H—インデノ [1, 2 d] [l, 3]チアゾールー 4 ィル)ォキシ]メチルホスホン酸ジェチル(80. Omg、 0. 161mmol)を使用して、実施例 1 (lc)に記載した方法に従い、標記目的化合物( 53. Omg、収率 75%)を得た。 [0491] [(7- {[(2 morpholine 4 ylethyl) amino] carbol} —8H-indeno [1, 2 d] [l, 3] thiazol-4-yl) oxy] methyl phosphonate (80. Omg 0.161 mmol) was used to give the title object compound (53. Omg, 75% yield) according to the method described in Example 1 (lc).
[0492] IR(KBr) v 1056, 1268, 1505, 1542, 1584, 1645, 3399 cm"1; [0492] IR (KBr) v 1056, 1268, 1505, 1542, 1584, 1645, 3399 cm "1;
max max
1H NMR(DMSO-d , 400MHz): δ 2.80—3.61 (10H, m), 3.88—3.98 (4H, m), 4.09 (2 1H NMR (DMSO-d, 400MHz): δ 2.80—3.61 (10H, m), 3.88—3.98 (4H, m), 4.09 (2
6 6
H, d, J=9.8 Hz), 6.95 (IH, d, J=8.6 Hz), 7.52 (IH, d, J=8.6 Hz), 8.86-8.94 (IH, m), 9.10 (IH, s); H, d, J = 9.8 Hz), 6.95 (IH, d, J = 8.6 Hz), 7.52 (IH, d, J = 8.6 Hz), 8.86-8.94 (IH, m), 9.10 (IH, s);
MS(FAB) m/z: 438 (M - H)— . MS (FAB) m / z: 438 (M-H) —.
<実施例 66 > <Example 66>
{ [7— ({ [2— (ァセチルァミノ)ェチル]アミノ}カルボニル) 8H—インデノ [1, 2-d ] [1, 3]チアゾールー 4 ィル]ォキシ }メチルホスホン酸(例示化合物番号 1-201)
実施例 36 (36c)で製造した 4 [ (ジエトキシホスホリル)メトキシ]— 8H インデノ [ 1, 2-d] [l, 3]チアゾールー 7—力ルボン酸(150mg、 0. 39mmol)を使用して、 実施例 36 (36d)及び実施例 36 (36e) に記載された方法に順次従!ヽ、標記目的化 合物(133mg、収率 84%)を得た。 {[7- ({[2- (Acetylamino) ethyl] amino} carbonyl) 8H-indeno [1, 2-d] [1, 3] thiazole-4-yl] oxy} methylphosphonic acid (Exemplary Compound No. 1-201) Example 36 Using 4 [(diethoxyphosphoryl) methoxy] -8H indeno [1,2-d] [l, 3] thiazole-7-strength rubonic acid (150 mg, 0.39 mmol) prepared in (36c) The title title compound (133 mg, 84% yield) was obtained in order according to the methods described in Example 36 (36d) and Example 36 (36e).
[0493] 但し、実施例 36 (36d)と同様の工程において、塩化アンモ-ゥムの代わりに N ァ セチルエチレンジァミン(48mg、 0. 47mmol)を用いた。 However, in the same step as Example 36 (36d), N-acetylethylenediamine (48 mg, 0.47 mmol) was used instead of ammonium chloride.
[0494] IR(KBr) v 952, 991, 1070, 1269, 1432, 1547, 1581, 1638, 3086, 3277 cm"1; [0494] IR (KBr) v 952, 991, 1070, 1269, 1432, 1547, 1581, 1638, 3086, 3277 cm "1;
max max
1H NMR(DMSO-d , 400MHz) δ 1.82 (3H, s), 3.18-3.24 (2H, m), 3.27-3.34 (2H, 1H NMR (DMSO-d, 400MHz) δ 1.82 (3H, s), 3.18-3.24 (2H, m), 3.27-3.34 (2H,
6 6
m), 4.19 (2H, s), 4.39 (2H, d, J=8.6 Hz), 7.22 (IH, d, J=8.6 Hz), 7.59 (IH, d, J=8.6 Hz), 7.96 (IH, t, J=5.5 Hz), 8.34 (IH, t, J=5.7 Hz) 9.15 (IH, s); m), 4.19 (2H, s), 4.39 (2H, d, J = 8.6 Hz), 7.22 (IH, d, J = 8.6 Hz), 7.59 (IH, d, J = 8.6 Hz), 7.96 (IH, t, J = 5.5 Hz), 8.34 (IH, t, J = 5.7 Hz) 9.15 (IH, s);
MS(FAB) m/z: 410 (M— H)— . MS (FAB) m / z: 410 (M— H) —.
<実施例 67 > <Example 67>
[ (7— { [ (2 ァミノ 2—ォキソェチル)ァミノ]カルボ-ル}—8H—インデノ [1, 2- d] [1, 3]チアゾールー 4 ィル)ォキシ]メチルホスホン酸(例示化合物番号 1-34) 実施例 36 (36c)で製造した 4 [ (ジエトキシホスホリル)メトキシ]— 8H インデノ [ 1, 2-d] [l, 3]チアゾール 7—カルボン酸(150mg、 0. 361mmol)及び塩ィ匕ァ ンモ-ゥムの代わりにグリシンアミド塩酸塩(52. Omg、 0. 470mmol)を使用して、実 施例 36 (36d)に記載した方法に従い、 [ (7- { [ (2 アミノー 2—ォキソェチル)アミ ノ]カルボ-ル}ー811—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキシ]メ チルホスホン酸ジェチル(72. 3mg、収率 42%)を得た。 [(7— {[(2 amino-2-oxoethyl) amino] carbol} -8H-indeno [1,2-d] [1,3] thiazol-4-yl) oxy] methylphosphonic acid (Exemplary Compound No. 1- 34) 4 [(Diethoxyphosphoryl) methoxy] -8H indeno [1,2-d] [l, 3] thiazole 7-carboxylic acid (150 mg, 0.361 mmol) and salt prepared in Example 36 (36c) According to the method described in Example 36 (36d) using glycinamide hydrochloride (52. Omg, 0.470 mmol) instead of モ -moum, [(7- {[(2 amino-2 (Oxoethyl) amino] carbol} -811-indeno [1,2-d] [l, 3] thiazol-4-yl) oxy] methyl phosphonate (72.3 mg, 42% yield) was obtained. .
[0495] [ (7— { [ (2 アミノー 2—ォキソェチル)ァミノ]カルボ-ル} 8H—インデノ [1, 2 [0495] [(7— {[(2 Amino-2-oxoethyl) amino] carbol} 8H—Indeno [1, 2
-d] [l, 3]チアゾールー 4 ィル)ォキシ]メチルホスホン酸ジェチル(67. 5mg、0 . 154mmol)を使用して、実施例 1 (lc)に記載した方法に従い、標記目的化合物(2 6. Omg、収率 43%)を得た。 -d] [l, 3] thiazol-4-yl) oxy] methyl phosphonate (67.5 mg, 0.154 mmol) according to the method described in Example 1 (lc) and the title compound (2 6 Omg, 43% yield).
[0496] IR(KBr) v 1273, 1485, 1545, 1581, 1651, 3091, 3311 cm"1; [0496] IR (KBr) v 1273, 1485, 1545, 1581, 1651, 3091, 3311 cm "1;
max max
1H NMR(DMSO-d , 500MHz): δ 3.83 (2H, d, J=5.8 Hz), 4.21 (2H, s), 4.41 (2H, d 1H NMR (DMSO-d, 500MHz): δ 3.83 (2H, d, J = 5.8 Hz), 4.21 (2H, s), 4.41 (2H, d
6 6
, J=9.3 Hz), 7.04 (IH, brs), 7.26 (IH, d, J=8.8 Hz), 7.38 (IH, brs), 7.70 (IH, d, J=8 .8 Hz), 8.52 (IH, t, J=5.9 Hz), 9.17 (IH, s);
MS(FAB) m/z: 382 (M— H)— . , J = 9.3 Hz), 7.04 (IH, brs), 7.26 (IH, d, J = 8.8 Hz), 7.38 (IH, brs), 7.70 (IH, d, J = 8.8 Hz), 8.52 (IH , t, J = 5.9 Hz), 9.17 (IH, s); MS (FAB) m / z: 382 (M— H) —.
<実施例 68 > <Example 68>
{ [7— ({ [ (IS)— 2 アミノー 1—メチル 2 ォキソェチル]ァミノ }カルボ-ル) - 8 H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル]ォキシ }メチルホスホン酸(例示 化合物番号 1-56) {[7— ({[(IS) — 2 Amino-1-methyl-2-oxoethyl] amino} carbole) -8 H-indeno [1, 2-d] [1, 2-d] [l, 3] thiazole-4-yl] oxy} methylphosphones Acid (Ex. Compound No. 1-56)
実施例 36 (36c)で製造した 4 [ (ジエトキシホスホリル)メトキシ]— 8H インデノ [ 1, 2-d] [l, 3]チアゾール 7—カルボン酸(150mg、 0. 391mmol)を使用して、 実施例 36 (36d)及び実施例 1 (lc)に記載した方法に順次従い、標記目的化合物( 130mg、収率 84%)を得た。 Example 36 Using 4 [(diethoxyphosphoryl) methoxy] -8H indeno [1,2-d] [l, 3] thiazole 7-carboxylic acid (150 mg, 0.391 mmol) prepared in (36c) The title target compound (130 mg, yield 84%) was obtained by sequentially following the methods described in Example 36 (36d) and Example 1 (lc).
[0497] 但し、実施例 36 (36d)と同様の工程において、塩ィ匕アンモ-ゥムの代わりに L ァ ラニンアミド塩酸塩(58. 6mg、 0. 470mmol)を用いた。 However, in the same process as in Example 36 (36d), L-alaninamide hydrochloride (58.6 mg, 0.470 mmol) was used in place of the salt ammonium.
[0498] IR(KBr) v 1270, 1485, 1544, 1582, 1649, 3095, 3308 cm"1; [0498] IR (KBr) v 1270, 1485, 1544, 1582, 1649, 3095, 3308 cm "1;
max max
1H NMR(DMSO-d , 400MHz) δ 1.34 (3H, d, J=7.0 Hz), 4.19 (2H, s), 4.40 (2H, d 1H NMR (DMSO-d, 400MHz) δ 1.34 (3H, d, J = 7.0 Hz), 4.19 (2H, s), 4.40 (2H, d
6 6
, J=9.0 Hz), 4.39-4.43 (IH, m), 6.99 (IH, drs), 7.23 (IH, d, J=8.7 Hz), 7.37 (IH, br s), 7.70 (IH, d, J=8.7 Hz), 8.29 (IH, d, J=7.0 Hz), 9.16 (IH, s); , J = 9.0 Hz), 4.39-4.43 (IH, m), 6.99 (IH, drs), 7.23 (IH, d, J = 8.7 Hz), 7.37 (IH, br s), 7.70 (IH, d, J = 8.7 Hz), 8.29 (IH, d, J = 7.0 Hz), 9.16 (IH, s);
MS(FAB) m/z: 396 (M— H)— . MS (FAB) m / z: 396 (M— H) —.
<実施例 69 > <Example 69>
{ [7— ({ [ (1R)—2 ァミノ— 1—メチル—2—ォキソェチル]アミノ}カルボ-ル)—8 H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル]ォキシ }メチルホスホン酸(例示 化合物番号 1-56) {[7— ({[(1R) —2 Amino— 1-methyl-2-oxoethyl] amino} carbol) —8 H—Indeno [1, 2-d] [l, 3] thiazole- 4 yl] Oxy} methylphosphonic acid (Ex. Compound No. 1-56)
実施例 36 (36c)で製造した 4 [ (ジエトキシホスホリル)メトキシ]— 8H インデノ [ 1, 2-d] [l, 3]チアゾール 7—カルボン酸(152mg、 0. 396mmol)および塩ィ匕 アンモ-ゥムの代わりに D—ァラニンアミド塩酸塩(70mg、 0. 562mmol)を使用して 、実施例 36 (36d)に記載した方法に従い、 { [7— ({ [ (1R)— 2 ァミノ一 1—メチル —2—ォキソェチル]アミノ}カルボ-ル)— 8H—インデノ [1, 2— d] [l, 3]チアゾー ル— 4—ィル]ォキシ }メチルホスホン酸ジェチル(144mg、収率 80%)を得た。 Example 36 4 [(Diethoxyphosphoryl) methoxy] -8H indeno [1,2-d] [l, 3] thiazole 7-carboxylic acid (152 mg, 0.396 mmol) prepared in (36c) and salt According to the method described in Example 36 (36d) using D-alaninamide hydrochloride (70 mg, 0.562 mmol) instead of -um, {[7— ({[(1R) — 2 —Methyl —2-Oxoethyl] amino} carbol) — 8H-indeno [1, 2-d] [l, 3] thiazol-4-yl] oxy} Jetyl methylphosphonate (144 mg, 80% yield) Got.
[0499] { [7— ({ [ (1R)—2 ァミノ— 1—メチル—2—ォキソェチル]アミノ}カルボ-ル)― 8H—インデノ [1, 2— d] [l, 3]チアゾール—4—ィル]ォキシ }メチルホスホン酸ジェ
チル(110mg、 0. 243mmol)を使用して、実施例 1 (lc)に記載した方法に従い、標 記目的化合物(27mg、収率 28%)を得た。 [0499] {[7— ({[(1R) —2 Amino- 1-methyl-2-oxoethyl] amino} carbol) -8H-indeno [1, 2— d] [l, 3] thiazole-4 —Il] oxy} methylphosphonate The title compound (27 mg, 28% yield) was obtained according to the method described in Example 1 (lc) using chill (110 mg, 0.243 mmol).
[0500] IR(KBr) v 1059, 1184, 1255, 1431, 1502, 1529, 1583, 1643, 1677, 2934, 2982, max [0500] IR (KBr) v 1059, 1184, 1255, 1431, 1502, 1529, 1583, 1643, 1677, 2934, 2982, max
3080, 3396 cm"1; 3080, 3396 cm "1;
1H NMR(DMSO-d , 400MHz): δ 1.34 (3H, d, J=7.3 Hz), 4.10—4.21 (4H, m), 4.37 1H NMR (DMSO-d, 400MHz): δ 1.34 (3H, d, J = 7.3 Hz), 4.10—4.21 (4H, m), 4.37
6 6
-4.45 (IH, m), 6.99 (IH, s), 7.24 (IH , d, J=8.6 Hz), 7.40 (IH, s), 7.71 (IH, d, J=8. 6 Hz), 8.34 (IH, d, J=6.8 Hz), 9.12 (IH, s); -4.45 (IH, m), 6.99 (IH, s), 7.24 (IH, d, J = 8.6 Hz), 7.40 (IH, s), 7.71 (IH, d, J = 8.6 Hz), 8.34 ( IH, d, J = 6.8 Hz), 9.12 (IH, s);
MS(FAB) m/z: 396 (M- H)— . MS (FAB) m / z: 396 (M- H) —.
<実施例 70 > <Example 70>
{ [7- ({ [ (IS)— 1—(ァミノカルボ-ル) 3—メチルブチル]アミノ}カルボ-ル) {[7- ({[(IS) — 1- (Aminocarbole) 3-methylbutyl] amino} carbole)
8H—インデノ [1, 2— d] [l, 3]チアゾール—4—ィル]ォキシ }メチルホスホン酸(例 示化合物番号 1-202) 8H-indeno [1,2-d] [l, 3] thiazole-4-yl] oxy} methylphosphonic acid (example compound number 1-202)
実施例 36 (36c)で製造した 4 [ (ジエトキシホスホリル)メトキシ]— 8H インデノ [ Example 36 4 [(Diethoxyphosphoryl) methoxy] -8H indeno prepared in (36c) [
1, 2-d] [l, 3]チアゾールー 7—力ルボン酸(150mg、 0. 39mmol)を使用して、 実施例 36 (36d)及び実施例 36 (36e)に記載した方法に順次従い、標記目的化合 物(33mg、収率 19%)を得た。 1, 2-d] [l, 3] thiazole-7-strength rubonic acid (150 mg, 0.39 mmol) and sequentially followed the method described in Example 36 (36d) and Example 36 (36e) The title compound was obtained (33 mg, 19% yield).
[0501] 但し、実施例 36 (36d)と同様の工程において、塩化アンモ-ゥムの代わりに L-ロイ シナミド塩酸塩(78mg、 0. 47mmol)を用いた。 [0501] However, L-leucinamide hydrochloride (78 mg, 0.47 mmol) was used in place of ammonium chloride in the same step as Example 36 (36d).
[0502] IR(KBr) v 946, 993, 1071, 1260, 1501, 1534, 1582, 1647, 1672, 2956, 3307 cm max [0502] IR (KBr) v 946, 993, 1071, 1260, 1501, 1534, 1582, 1647, 1672, 2956, 3307 cm max
-1 -1
1H NMR(DMSO-d , 400MHz): δ 0.90 (3H, d, J=9.0 Hz), 0.92 (3H, d, J=9.0 Hz), 1H NMR (DMSO-d, 400MHz): δ 0.90 (3H, d, J = 9.0 Hz), 0.92 (3H, d, J = 9.0 Hz),
6 6
1.47-1.58 (IH, m), 1.62-1.76 (2H, m), 4.13 (IH, d, J=23.5 Hz), 4.20 (IH, d, J=23.5 Hz), 4.39 (2H, d, J=9.0 Hz), 4.38—4.49 (IH, m), 6.96 (IH, s), 7.23 (IH, d, J=8.6 H z), 7.38 (IH, s), 7.69 (IH, d, J=8.6 Hz), 8.25 (IH, d, J=7.8 Hz), 9.14 (IH, s); 1.47-1.58 (IH, m), 1.62-1.76 (2H, m), 4.13 (IH, d, J = 23.5 Hz), 4.20 (IH, d, J = 23.5 Hz), 4.39 (2H, d, J = 9.0 Hz), 4.38—4.49 (IH, m), 6.96 (IH, s), 7.23 (IH, d, J = 8.6 H z), 7.38 (IH, s), 7.69 (IH, d, J = 8.6 Hz) ), 8.25 (IH, d, J = 7.8 Hz), 9.14 (IH, s);
MS(FAB) m/z: 438 (M- H)— . MS (FAB) m / z: 438 (M- H) —.
<実施例 71 > <Example 71>
{ [7— ({ [ (IS)— 2 ァミノ一 1— (ヒドロキシメチル) - 2-ォキソェチル]ァミノ }カル ボ-ル)— 8H—インデノ [1, 2-d] [l, 3]チアゾール—4—ィル]ォキシ }メチルホス
ホン酸 (例示化合物番号 1-203) {[7— ({[(IS) — 2 amino 1- (hydroxymethyl) -2-oxoethyl] amino} carbole) -8H—indeno [1, 2-d] [l, 3] thiazole— 4-yl] oxy} methylphos Phosphonic acid (Exemplified compound number 1-203)
実施例 36 (36c)で製造した 4 [ (ジエトキシホスホリル)メトキシ]— 8H インデノ [ 1, 2-d] [l, 3]チアゾール 7—カルボン酸(150mg、 0. 391mmol)及び塩ィ匕ァ ンモ-ゥムの代わりに Lーセリンアミド塩酸塩(77. Omg、 0. 548mmol)を使用して、 実施例 36 (36d)に記載した方法に従い、 { [7- ({ [ (lS) 2 アミノー 1—(ヒドロキ シメチル)— 2—ォキソェチル]アミノ}カルボ-ル)— 8H—インデノ [1, 2— d] [l, 3] チアゾール—4—ィル]ォキシ }メチルホスホン酸ジェチル(175mg、収率 95%)を得 た。 Example 36 4 [(Diethoxyphosphoryl) methoxy] -8H indeno [1,2-d] [l, 3] thiazole 7-carboxylic acid (150 mg, 0.391 mmol) prepared in (36c) and salt Substituting L-serine amide hydrochloride (77. Omg, 0.548 mmol) in place of ammonia, following the procedure described in Example 36 (36d), {[7- ({[(lS) 2 amino 1 — (Hydroxymethyl) —2-oxoethyl] amino} carbol) —8H-indeno [1, 2-d] [l, 3] thiazole-4-yl] oxy} methyl phosphonate (175 mg, yield 95) %).
[0503] { [ 7— ( { [ ( 1 S)— 2 ァミノ一 1 (ヒドロキシメチル) 2 ォキソェチル]ァミノ)力 ルポ-ル)—8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル]ォキシ }メチルホ スホン酸ジェチル(81. 8mg、0. 172mmol)を使用して、実施例 36 (36e)に記載し た方法に従い、標記目的化合物(28. 3mg、収率 33%)を得た。 [0503] {[7— ({[(1 S) — 2 Amino 1 (hydroxymethyl) 2 oxoethyl] amino) Force)-8H—Indeno [1, 2-d] [l, 3] thiazole 4- [yl] oxy} methyl phosphonate (81.8 mg, 0.172 mmol) and the title compound (28.3 mg, 33% yield) according to the method described in Example 36 (36e). Got.
[0504] IR(KBr) v 1072, 1270, 1488, 1581, 1647, 1679, 3318 cm"1; [0504] IR (KBr) v 1072, 1270, 1488, 1581, 1647, 1679, 3318 cm "1;
max max
1H NMR(DMSO-d , 500MHz): δ 3.73 (2H, d, J=5.4 Hz), 4.20 (2H, s), 4.40 (2H, d 1H NMR (DMSO-d, 500MHz): δ 3.73 (2H, d, J = 5.4 Hz), 4.20 (2H, s), 4.40 (2H, d
6 6
, J=8.8 Hz), 4.42-4.46 (IH, m), 4.94 (IH, brs), 7.11 (IH, brs), 7.27 (IH, d, J=8.3 H z), 7.40 (IH, brs), 7.74 (IH, d, J=8.3 Hz), 8.07 (IH, d, J=7.8 Hz), 9.17 (IH, s); , J = 8.8 Hz), 4.42-4.46 (IH, m), 4.94 (IH, brs), 7.11 (IH, brs), 7.27 (IH, d, J = 8.3 H z), 7.40 (IH, brs), 7.74 (IH, d, J = 8.3 Hz), 8.07 (IH, d, J = 7.8 Hz), 9.17 (IH, s);
MS(FAB) m/z: 412 (M- H)— . MS (FAB) m / z: 412 (M- H) —.
<実施例 72 > <Example 72>
{ [7— ( { [ァミノ (ィミノ)メチル]アミノ}カルボ-ル)— 8H—インデノ [1, 2-d] [l, 3] チアゾールー 4 ィル]ォキシ }メチルホスホン酸(例示化合物番号 1-205) {[7— ({[Amino (imino) methyl] amino} carbol) — 8H—Indeno [1, 2-d] [l, 3] Thiazole-4-yl] oxy} methylphosphonic acid (Exemplary Compound No. 1- 205)
実施例 36 (36c)で製造した 4 [ (ジエトキシホスホリル)メトキシ]— 8H インデノ [ 1, 2-d] [l, 3]チアゾール 7—カルボン酸(230mg、 0. 600mmol)及び塩ィ匕ァ ンモ-ゥムの代わりにグァ-ジン塩酸塩(860mg、 9. OOmmol)を使用して、実施例 36 (36d)に記載した方法に従い、 { [7- ( { [ァミノ (ィミノ)メチル]アミノ}カルボニル) — 8H—インデノ [1, 2-d] [l, 3]チアゾール—4—ィル]ォキシ }メチルホスホン酸 ジェチル(132mg、収率 55%)を得た。 Example 36 4 [(Diethoxyphosphoryl) methoxy] -8H indeno [1,2-d] [l, 3] thiazole 7-carboxylic acid (230 mg, 0.600 mmol) and salt prepared in (36c) According to the method described in Example 36 (36d) using guanidine hydrochloride (860 mg, 9. OOmmol) instead of ammonia, {[7- ({[Amino (imino) methyl] amino } Carbonyl) —8H-indeno [1,2-d] [l, 3] thiazol-4-yl] oxy} methyl phosphonate jetyl (132 mg, yield 55%) was obtained.
[0505] { [7— ( { [ァミノ (ィミノ)メチル]アミノ}カルボ二ル)— 8H—インデノ [1, 2-d] [l, 3 ]チアゾールー 4 ィル]ォキシ }メチルホスホン酸ジェチル(61. 4mg、 0. 145mmo
1)を使用して、実施例 36 (36e)に記載した方法に従 、、標記目的化合物 (44.9mg 、収率 84%)を得た。 [0505] {[7— ({[Amino (imino) methyl] amino} carbonyl) — 8H-indeno [1, 2-d] [l, 3] thiazole-4-yl] oxy} Jetyl methylphosphonate (61 4mg, 0.145mmo Using 1), the title object compound (44.9 mg, yield 84%) was obtained according to the method described in Example 36 (36e).
[0506] IR(KBr) v 1156, 1271, 1567, 1602, 1697, 3172, 3333 cm"1; [0506] IR (KBr) v 1156, 1271, 1567, 1602, 1697, 3172, 3333 cm "1;
max max
1H NMR(D O+NaHCO , 500MHz): δ 3.18—3.29 (2H, m), 3.98 (2H, d, J=9.3 Hz), 1H NMR (D 2 O + NaHCO 3, 500 MHz): δ 3.18—3.29 (2H, m), 3.98 (2H, d, J = 9.3 Hz),
2 3 twenty three
6.90 (IH, d, J=8.8 Hz), 7.65 (IH, d, J=8.8 Hz), 8.85 (IH, s); 6.90 (IH, d, J = 8.8 Hz), 7.65 (IH, d, J = 8.8 Hz), 8.85 (IH, s);
MS(ESI) m/z: 367 (M— H)— . MS (ESI) m / z: 367 (M— H) —.
<実施例 73> <Example 73>
({7-[({[(2R) -2, 3 ジヒドロキシプロピル]ォキシ }ァミノ)カルボ-ル ] 8H— インデノ [1, 2— d][l, 3]チアゾールー 4ーィル }ォキシ)メチルホスホン酸(例示化 合物番号 1-204) ({7-[({[(2R) -2,3 dihydroxypropyl] oxy} amino) carbol] 8H-indeno [1,2-d] [l, 3] thiazol-4-yl} oxy) methylphosphonic acid ( (Example compound number 1-204)
実施例 36 (36c)で製造した 4 [(ジエトキシホスホリル)メトキシ]— 8H インデノ [ 1, 2-d][l, 3]チアゾール 7—カルボン酸(200mg、 0.522mmol)及び塩ィ匕ァ ンモ-ゥムの代わりに(4R)— 4— [(アミノォキシ)メチル ]—2, 2 ジメチル一 1, 3- ジォキソラン(123mg、 0.835mmol)を使用して、実施例 36 (36d)に記載した方法 に従い、 ({7-[({[(4R)-2, 2 ジメチル一 1, 3 ジォキソラン一 4—ィル]メトキシ }ァミノ)カルボ-ル ]—8H—インデノ [1, 2-d][l, 3]チアゾールー 4ーィル }ォキシ )メチルホスホン酸ジェチル(250mg、収率 94%)を得た。 Example 36 4 [(Diethoxyphosphoryl) methoxy] -8H Indeno [1,2-d] [l, 3] thiazole 7-carboxylic acid (200 mg, 0.522 mmol) and salt guanmo prepared in (36c) The method described in Example 36 (36d) using (4R) -4-[(aminoxy) methyl] -2,2 dimethyl-1,3-dioxolane (123 mg, 0.835 mmol) instead of -um. ({7-[({[(4R) -2, 2 dimethyl-1,1,3 dixolan-4-yl] methoxy} amino) carbol] —8H-indeno [1,2-d] [l , 3] thiazole-4-yl} oxy) methyl phosphonate (250 mg, 94% yield).
[0507] ({7-[({[(4R)-2, 2 ジメチル一 1, 3 ジォキソラン一 4—ィル]メトキシ}ァミノ )カルボ-ル] 8H インデノ [1, 2— d][l, 3]チアゾールー 4ーィル }ォキシ)メチ ルホスホン酸ジェチル(60. Omg、 0.117mmol)を使用して、実施例 l(lc)に記載 した方法に従い、標記目的化合物(20.2mg、収率 41%)を得た。 [0507] ({7-[({[(4R) -2, 2 dimethyl-1,1,3 dixolan-4-yl] methoxy} amino) carbol] 8H indeno [1, 2— d] [l, 3] The thiazole-4-yl} oxy) methyl phosphonate (60. Omg, 0.117mmol) was used to give the title compound (20.2mg, 41% yield) according to the method described in Example l (lc). Obtained.
[0508] IR(KBr) v 1272, 1485, 1584, 1673, 2935, 3101, 3365 cm"1; [0508] IR (KBr) v 1272, 1485, 1584, 1673, 2935, 3101, 3365 cm "1;
max max
1H NMR(DMSO-d , 500MHz): δ 3.40—3.45 (2H, m), 3.75—3.83 (2H, m), 3.97—4.0 1H NMR (DMSO-d, 500MHz): δ 3.40—3.45 (2H, m), 3.75—3.83 (2H, m), 3.97—4.0
6 6
0 (IH, m), 4.19 (2H, s), 4.40 (2H, d, J=8.8 Hz), 7.25 (IH, d, J=8.7 Hz), 7.52 (IH, d , J=8.7 Hz), 9.18 (IH, s), 11.65 (IH, brs); 0 (IH, m), 4.19 (2H, s), 4.40 (2H, d, J = 8.8 Hz), 7.25 (IH, d, J = 8.7 Hz), 7.52 (IH, d, J = 8.7 Hz), 9.18 (IH, s), 11.65 (IH, brs);
MS(FAB) m/z: 415 (M- H)— . MS (FAB) m / z: 415 (M- H) —.
<実施例 74> <Example 74>
( {7 [(シクロプロピルァミノ)メチル ] 8H—インデノ [1, 2-d][l, 3]チアゾール
4ーィル }ォキシ)メチルホスホン酸塩酸塩(例示化合物番号 1-182) 実施例 45 (45b)で製造した [ (7 ホルミル— 8H—インデノ [1, 2— d] [l, 3]チア ゾールー 4 ィル)ォキシ]メチルホスホン酸ジェチル(500mg、 1. 36mmol)及びジ メチルァミンの代わりにシクロプロピルアミン(867mg、 4. O9mmol)を使用して、実 施例 49 (49a)に記載した方法に従い、 ({7- [ (シクロプロピルァミノ)メチル ] 8H インデノ [1, 2-d] [l, 3]チアゾールー 4ーィル }ォキシ)メチルホスホン酸ジェチ ル (462mg、収率 83%)を得た。 ({7 [(Cyclopropylamino) methyl] 8H-indeno [1, 2-d] [l, 3] thiazole 4-oxyl) oxy) methylphosphonate hydrochloride (Exemplified Compound No. 1-182) Prepared in Example 45 (45b) [(7 formyl-8H-indeno [1, 2—d] [l, 3] thiazol-4 (L) oxy] methyl phosphonate (500 mg, 1.36 mmol) and cyclopropylamine (867 mg, 4. O9 mmol) in place of dimethylamine and following the procedure described in Example 49 (49a), ({ 7-[(Cyclopropylamino) methyl] 8H indeno [1,2-d] [l, 3] thiazol-4-yl} oxy) methylphosphonic acid ethyl ester (462 mg, 83% yield) was obtained.
({ 7 [ (シクロプロピルァミノ)メチル ] 8H—インデノ [1, 2-d] [l, 3]チアゾール — 4—ィル }ォキシ)メチルホスホン酸ジェチル(436mg、 1. 07mmol)を使用して、 実施例 34 (34c)に記載した方法に従い、標記目的化合物 (406mg、収率 98%)を 得た。 ({7 [(Cyclopropylamino) methyl] 8H-indeno [1,2-d] [l, 3] thiazole — 4-yl} oxy) using methyl phosphonate jetyl (436 mg, 1.07 mmol) The title object compound (406 mg, yield 98%) was obtained according to the method described in Example 34 (34c).
[0509] IR(KBr) v 1002, 1236, 1438, 1491, 2744, 2927, 3380 cm"1; [0509] IR (KBr) v 1002, 1236, 1438, 1491, 2744, 2927, 3380 cm "1;
max max
1H NMR(DMSO-d , 400MHz): δ 0.75-0.81 (2H, m), 0.90—0.94 (2H, m), 2.72-2.7 1H NMR (DMSO-d, 400MHz): δ 0.75-0.81 (2H, m), 0.90—0.94 (2H, m), 2.72-2.7
6 6
8 (IH, m), 4.17 (2H, s), 4.25-4.29 (2H, m), 4.36 (2H, d, J=9.0 Hz), 7.23 (IH, d, J=8 .6 Hz), 7.40 (IH, d, J=8.6 Hz), 9.18 (IH, s), 9.25-9.33 (2H, m); 8 (IH, m), 4.17 (2H, s), 4.25-4.29 (2H, m), 4.36 (2H, d, J = 9.0 Hz), 7.23 (IH, d, J = 8.6 Hz), 7.40 (IH, d, J = 8.6 Hz), 9.18 (IH, s), 9.25-9.33 (2H, m);
MS(FAB) m/z: 351 (M— H)— (free form). MS (FAB) m / z: 351 (M— H) — (free form).
<実施例 75 > <Example 75>
({7 [ (ジェチルァミノ)メチル] 8H インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル }ォキシ)メチルホスホン酸塩酸塩 (例示化合物番号 1-78) ({7 [(Jetylamino) methyl] 8H Indeno [1, 2-d] [l, 3] thiazole-4-yl} oxy) methylphosphonate hydrochloride (Exemplary Compound No. 1-78)
実施例 45 (45b)で製造した [ (7 ホルミル— 8H—インデノ [1, 2— d] [l, 3]チア ゾールー 4 ィル)ォキシ]メチルホスホン酸ジェチル(150mg、 0. 408mmol)及び ジメチルァミンの代わりにジェチルァミン(50. 7 /z L、 0. 490mmol)を使用して、実 施例 49 (49a)に記載した方法に従 、、( { 7— [ (ジェチルァミノ)メチル]— 8H—イン デノ [1, 2-d] [l, 3]チアゾールー 4ーィル }ォキシ)メチルホスホン酸ジェチル(85 . Omgゝ収率 49%)を得た。 Of [(7 formyl-8H-indeno [1,2-d] [l, 3] thiazol-4-yl) oxy] methylphosphonate (150 mg, 0.408 mmol) and dimethylamine prepared in Example 45 (45b) Instead of jetylamine (50.7 / z L, 0.490 mmol) and following the procedure described in Example 49 (49a), ({7— [(Jetylamino) methyl] — 8H-indene [1,2-d] [l, 3] thiazol-4-yl} oxy) methyl phosphonate jetyl (85.Omg yield 49%).
[0510] ({7 [ (ジェチルァミノ)メチル] 8H インデノ [1, 2-d] [l, 3]チアゾールー 4 ーィル }ォキシ)メチルホスホン酸ジェチル(29. 8mg、 0. 070mmol)を使用して、実 施例 34 (34c)に記載した方法に従い、標記目的化合物(21. 8mg、収率 77%)を得
た。 [0510] ({7 [(Jetylamino) methyl] 8H indeno [1, 2-d] [l, 3] thiazol-4-yl} oxy) methylphosphonate (29.8 mg, 0.070 mmol) The title compound (21.8 mg, yield 77%) was obtained according to the method described in Example 34 (34c). It was.
[0511] IR(KBr) v 1011, 1072, 1280, 1491, 2938 cm"1; [0511] IR (KBr) v 1011, 1072, 1280, 1491, 2938 cm "1;
max max
1H NMR(D O, 400MHz) δ 1.34 (6H, t, J=7.3 Hz), 3.26 (4H, q, J=7.3 Hz), 3.95 ( 1H NMR (D O, 400MHz) δ 1.34 (6H, t, J = 7.3 Hz), 3.26 (4H, q, J = 7.3 Hz), 3.95 (
2 2
2H, s), 4.29 (2H, d, J=10.2 Hz), 4.33 (2H, s), 7.14 (IH, d, J=8.6 Hz), 7.38 (IH, d, J =8.6 Hz), 9.35 (IH, s); 2H, s), 4.29 (2H, d, J = 10.2 Hz), 4.33 (2H, s), 7.14 (IH, d, J = 8.6 Hz), 7.38 (IH, d, J = 8.6 Hz), 9.35 ( IH, s);
MS(FAB) m/z: 367 (M— H)— (free form). MS (FAB) m / z: 367 (M— H) — (free form).
<実施例 76 > <Example 76>
({7— [ (4ーメチルビペラジン 1 ィル)メチル ]—8H—インデノ [1, 2-d] [l, 3] チアゾールー 4ーィル }ォキシ)メチルホスホン酸二塩酸塩(例示化合物番号 1-14) 実施例 45 (45b)で製造した [ (7 ホルミル— 8H—インデノ [1, 2— d] [l, 3]チア ゾールー 4 ィル)ォキシ]メチルホスホン酸ジェチル(150mg、 0. 408mmol)及び ジメチルァミンの代わりに N—メチルビペラジン(54. 4 レ0. 490mmol)を使用し て、実施例 49 (49a)に記載した方法に従い、({7—[ (4ーメチルピぺラジンー1ーィ ル)メチル] 8H インデノ [1, 2-d] [l, 3]チアゾールー 4ーィル }ォキシ)メチル ホスホン酸ジェチル(130mg、収率 71%)を得た。 ({7- [(4-Methylbiperazine 1-yl) methyl] -8H-indeno [1, 2-d] [l, 3] thiazole-4-yl} oxy) methylphosphonic acid dihydrochloride (Exemplary Compound No. 1 -14) Jetyl [(7 formyl-8H-indeno [1,2-d] [l, 3] thiazol-4-yl) oxy] methylphosphonate prepared in Example 45 (45b) (150 mg, 0.408 mmol) And according to the method described in Example 49 (49a) using N-methylbiperazine (54.4 les 0.490 mmol) instead of dimethylamine, ({7- [(4-methylpiperazine-1-yl) methyl 8H Indeno [1,2-d] [l, 3] thiazole-4-yl} oxy) methyl phosphonate (130 mg, 71% yield) was obtained.
[0512] ({7—[ (4ーメチルピぺラジンー1ーィル)メチル]ー811—ィンデノ[1, 2— d] [l, 3 ]チアゾールー 4ーィル }ォキシ)メチルホスホン酸ジェチル(73. 9mg、 0. 164mmo 1)を使用して、実施例 34 (34c)に記載した方法に従い、標記目的化合物(76. 7mg 、収率 100%)を得た。 [0512] ({7-[(4-Methylpiperazine-1-yl) methyl] -811-indeno [1,2-d] [l, 3] thiazol-4-yl} oxy) methyl phosphonate jetyl (73.9 mg, 0. The title compound (76.7 mg, yield 100%) was obtained according to the method described in Example 34 (34c) using 164 mmo 1).
[0513] IR(KBr) v 946, 1007, 1491, 2670 cm"1; [0513] IR (KBr) v 946, 1007, 1491, 2670 cm "1;
max max
1H NMR(D O, 400MHz): δ 3.03 (3H, s), 3.35—4.00 (8H, m), 3.89 (2H, s), 4.18 (2 1H NMR (D 2 O, 400 MHz): δ 3.03 (3H, s), 3.35—4.00 (8H, m), 3.89 (2H, s), 4.18 (2
2 2
H, d, J=10.2 Hz), 4.48 (2H, s), 7.02 (IH, d, J=8.6 Hz), 7.34 (IH, d, J=8.6 Hz), 9.14 (IH, s); H, d, J = 10.2 Hz), 4.48 (2H, s), 7.02 (IH, d, J = 8.6 Hz), 7.34 (IH, d, J = 8.6 Hz), 9.14 (IH, s);
MS(ESI) m/z: 394 (M— H)— (free form). MS (ESI) m / z: 394 (M— H) — (free form).
<実施例 77 > <Example 77>
[ (5, 7 ジフルオロー 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキシ ]メチルホスホン酸 (例示化合物番号 1-246) [(5, 7 Difluoro-8H-indeno [1, 2-d] [l, 3] thiazole-4-yl) oxy] methylphosphonic acid (Exemplified Compound No. 1-246)
4, 6 ジフルオロー 7 ヒドロキシインダン一 1—オン(4. 05g、 22. Ommol)を使
用して、実施例 1 (la)、実施例 3 (3a)及び実施例 3 (3b)に記載した方法に順次従い 、 [ (5, 7 ジフルオロー 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキ シ]メチルホスホン酸ジェチル(1. 22g、収率 15%)を得た。 4, 6 Difluoro-7 hydroxyindan 1-one (4.05 g, 22. Ommol) was used. Then, following the procedures described in Example 1 (la), Example 3 (3a) and Example 3 (3b), [(5, 7 Difluoro-8H-indeno [1, 2-d] [l , 3] thiazole-4-yl) oxy] methyl phosphonate (1.22 g, 15% yield).
[0514] [ (5, 7 ジフルオロー 8H—インデノ [1, 2— d] [l, 3]チアゾールー 4 ィル)ォキ シ]メチルホスホン酸ジェチル(46. Omg、0. 123mmol)を使用して、実施例 l (lc) に記載した方法に順従い、標記目的化合物(28. Omg、収率 71%)を得た。 [0514] Using ((5,7 difluoro-8H-indeno [1,2-d] [l, 3] thiazol-4-yl) oxy] methyl phosphonate (46. Omg, 0.123 mmol) The title target compound (28. Omg, yield 71%) was obtained by following the method described in Example l (lc).
[0515] IR(KBr) v 945, 1011, 1103, 1148, 1194, 1250, 1274, 1392, 1441, 1520, 1629, max [0515] IR (KBr) v 945, 1011, 1103, 1148, 1194, 1250, 1274, 1392, 1441, 1520, 1629, max
2293, 2923, 2950, 3084, 3099, 3433 cm"1; 2293, 2923, 2950, 3084, 3099, 3433 cm "1;
1H NMR(DMSO-d , 400MHz): δ 3.99 (2H, s), 4.44 (2H, d, J=8.8 Hz), 7.18 (1H, d 1H NMR (DMSO-d, 400MHz): δ 3.99 (2H, s), 4.44 (2H, d, J = 8.8 Hz), 7.18 (1H, d
6 6
d, J=8.8 Hz, 10.7 Hz), 9.27 (1H, s); d, J = 8.8 Hz, 10.7 Hz), 9.27 (1H, s);
MS(FAB) m/z: 318 (M— H)— . MS (FAB) m / z: 318 (M— H) —.
<実施例 78 > <Example 78>
[ (6, 7 ジフルオロー 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキシ [(6, 7 Difluoro-8H—indeno [1, 2-d] [l, 3] thiazole-4-yl) oxy
]メチルホスホン酸 (例示化合物番号 1-207) ] Methylphosphonic acid (Exemplified compound number 1-207)
4,5 ジフルオロー 7 ヒドロキシインダン(5. OOg、 27. 2mmol)を使用して、実施 例 1 (la)、実施例 3 (3a)及び実施例 3 (3b)に記載した方法に順次従い、 [ (6, 7— ジフルオロー 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキシ]メチルホ スホン酸ジェチル(2. 63g、収率 26%)を得た。 Using 4,5 difluoro-7-hydroxyindane (5. OOg, 27.2 mmol) and following the procedures described in Example 1 (la), Example 3 (3a) and Example 3 (3b) in turn, (6,7-Difluoro-8H-indeno [1,2-d] [l, 3] thiazol-4-yl) oxy] methylphosphoric acid jetyl (2.63 g, yield 26%) was obtained.
[0516] [ (6, 7 ジフルオロー 8H—インデノ [1, 2— d] [l, 3]チアゾールー 4 ィル)ォキ シ]メチルホスホン酸ジェチル(113mg、 0. 30mmol)を使用して、実施例 l (lc)に 記載した方法に従 、、標記目的化合物(74mg、収率 78%)を得た。 [0516] Examples using [(6,7 difluoro-8H-indeno [1,2-d] [l, 3] thiazol-4-yl) oxy] methyl phosphonate (113 mg, 0.30 mmol) According to the method described in l (lc), the title object compound (74 mg, yield 78%) was obtained.
[0517] 1H NMR(DMSO-d , 400MHz): δ 4.08 (2Η, s), 4.34 (2H, d, J=8.6 Hz), 7.32 (1H, [0517] 1H NMR (DMSO-d, 400MHz): δ 4.08 (2Η, s), 4.34 (2H, d, J = 8.6 Hz), 7.32 (1H,
6 6
dd, J=5.9 Hz, 7.0 Hz), 9.19 (1H, s). dd, J = 5.9 Hz, 7.0 Hz), 9.19 (1H, s).
<実施例 79 > <Example 79>
[ (7 ェチルー 8H インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキシ]メチ ルホスホン酸(例示化合物番号 1-209) [(7 ethyl-8H indeno [1,2-d] [l, 3] thiazole-4-yl) oxy] methylphosphonic acid (Exemplary Compound No. 1-209)
(79a) [ (7 ェチルー 8H—インデノ [1, 2— d] [l, 3]チアゾールー 4 ィル)ォキシ ]メチルホスホン酸ジェチル
4 ェチル 7 ヒドロキシインダン一 1—オン(7. 05g、 40. Ommol)を使用して、 実施例 1 (la)、実施例 3 (3a)及び実施例 3 (3b)に記載した方法に順次従い、標記 目的化合物 (4. 32g、収率 29%)を得た。 (79a) [(7 ethyl-8H-indeno [1,2-d] [l, 3] thiazol-4-yl) oxy] methyl phosphonate The procedure described in Example 1 (la), Example 3 (3a) and Example 3 (3b) was followed sequentially using 4 ethyl 7-hydroxyindan 1-one (7.05 g, 40. Ommol). The title compound (4.32 g, yield 29%) was obtained.
[0518] IR(KBr) v 978, 1016, 1256, 1511, 2979 cm"1; [0518] IR (KBr) v 978, 1016, 1256, 1511, 2979 cm "1;
max max
1H NMR(CDC1 , 400MHz) δ 1.28 (3H, t, J=7.6 Hz), 1.35 (6H, t, J=7.1 Hz), 2.70 1H NMR (CDC1, 400MHz) δ 1.28 (3H, t, J = 7.6 Hz), 1.35 (6H, t, J = 7.1 Hz), 2.70
3 Three
(2H, q, J=7.6 Hz), 3.78 (2H, s), 4.31 (4H, dq, J=7.1 Hz, 7.1 Hz), 4.62 (2H, d, J=8.6 Hz), 7.01 (1H, d, J=8.6 Hz), 7.05 (1H, d, J=8.6 Hz), 8.81 (1H, s); (2H, q, J = 7.6 Hz), 3.78 (2H, s), 4.31 (4H, dq, J = 7.1 Hz, 7.1 Hz), 4.62 (2H, d, J = 8.6 Hz), 7.01 (1H, d , J = 8.6 Hz), 7.05 (1H, d, J = 8.6 Hz), 8.81 (1H, s);
MS(FAB) m/z: 368 (M+H)+ . MS (FAB) m / z: 368 (M + H) + .
(79b) [ (7 ェチルー 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキシ ]メチノレホスホン酸 (79b) [(7 Ethyl-8H-indeno [1, 2-d] [l, 3] thiazol-4-yl) oxy] methinorephosphonic acid
実施例 79 (79a)で製造した [ (7 ェチル—8H—インデノ [1, 2— d] [l, 3]チアゾ 一ルー 4 ィル)ォキシ]メチルホスホン酸ジェチル(100mg、 0. 272mmol)を使用 して、実施例 1 (lc)に記載した方法に従い、標記目的化合物(82. 2mg、収率 97% )を得た。 EXAMPLE 79 Using [(7 ethyl-8H-indeno [1,2-d] [l, 3] thiazo 4-ruyl) oxy] methylphosphonate (100 mg, 0.272 mmol) prepared in Example 79 (79a) Then, according to the method described in Example 1 (lc), the title object compound (82.2 mg, yield 97%) was obtained.
[0519] IR(KBr) v 926, 1013, 1260, 1511, 2959 cm"1; [0519] IR (KBr) v 926, 1013, 1260, 1511, 2959 cm "1;
max max
1H NMR(DMSO-d , 400MHz) δ 1.23 (3H, t, J=7.4 Hz), 2.66 (2H, q, J=7.4 Hz), 3 1H NMR (DMSO-d, 400MHz) δ 1.23 (3H, t, J = 7.4 Hz), 2.66 (2H, q, J = 7.4 Hz), 3
6 6
.90 (2H, s), 4.30 (2H, d, J=9.0 Hz), 7.03-7.10 (2H, m), 9.16 (1H, s); .90 (2H, s), 4.30 (2H, d, J = 9.0 Hz), 7.03-7.10 (2H, m), 9.16 (1H, s);
MS(FAB) m/z: 310 (M— H)— . MS (FAB) m / z: 310 (M— H) —.
<実施例 80 > <Example 80>
[ (6, 7 ジメチルー 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキシ] メチルホスホン酸 (例示化合物番号 1-231) [(6, 7 Dimethyl-8H—indeno [1, 2-d] [l, 3] thiazole-4-yl) oxy] methylphosphonic acid (Exemplified Compound No. 1-231)
(80a) [ (6,7 ジメチルー 3—ォキソ 2, 3 ジヒドロ一 1H—インデン一 4—ィル)ォ キシ]メチルホスホン酸ジェチル (80a) [(6,7 Dimethyl-3-oxo-2,3 dihydro-1H-indene-4-yl) oxy] methyl phosphonate
7 ヒドロキシ一 4, 5 ジメチルインダン一 1—オン(5. OOg、 28. 4mmol)を使用 して、実施例 1 (la)に記載した方法に従い、標記目的化合物(7. 26g、収率 78%) を得た。 7 According to the method described in Example 1 (la) using hydroxy 1,4,5 dimethylindan 1-one (5. OOg, 28.4 mmol), the title compound (7.26 g, 78% yield) was obtained. )
[0520] IR(KBr) v 1029, 1244, 1705 cm"1; [0520] IR (KBr) v 1029, 1244, 1705 cm "1;
max max
1H NMR(CDC1 , 400MHz) δ 1.36 (6H, t, J=7.1 Hz), 2.16 (3H, s), 2.33 (3H, s), 2
( / — 一 /— 、 ^ [ε Ί][Ρ-2 Ί] ^^-Η8- -έ^-9-¾ΰ :- )] 1H NMR (CDC1, 400MHz) δ 1.36 (6H, t, J = 7.1 Hz), 2.16 (3H, s), 2.33 (3H, s), 2 (/ — One / —, ^ [ε Ί] [Ρ-2 Ί] ^^-Η8- -έ ^ -9-¾ΰ:-)]
<確爾第 > <Certificate No.>
• _(Η-η) οτε: ζ/ω (9VH)S • _ (Η-η) οτε: ζ / ω (9VH) S
•(s 'Ηΐ) εΐ·6 '(s 'Ηΐ) 36"9 '(ΖΗ 0"6=Γ 'Ρ Ή • ( s ' Ηΐ) εΐ · 6 '( s ' Ηΐ) 36 "9' ( Ζ Η 0" 6 = Γ 'Ρ Ή
Ζ) 6Z'f '(s 'ΗΖ) WZ '(s Ήε) SZ'Z '(s Ήε) 6VZ 9 : (zH )0 ,9p-os a)H N Ηχ Ζ) 6Z'f '( s ΗΖε) WZ' ( s Ήε) SZ'Z '( s Ήε) 6VZ 9 : ( z H) 0 , 9 p-os a) HN Η χ
9Ϊ62 'L6fl '½2ΐ 'Ζ60ΐ ' 96 Λ ( 1)ΗΙ [ SO] 9Ϊ62 'L6fl' ½2ΐ 'Ζ60ΐ' 96 Λ (1) ΗΙ [SO]
° (% 6 ° (% 6
¾τ ra ·6 )
 、ェっ ¾r ¾τ ra 6) , ¾r
、ェっ ¾r ¾τ ra 6) , ¾r
¾ (louiui22,2 ·ο、3rao{H) ^エ; ^邈べ ^ [ ^^ ( / — 一 /— 、
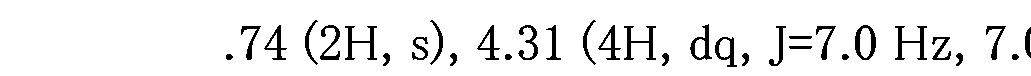 '9) (qos) os m ¾ (louiui22,2 · ο, 3rao {H) ^ e; ^ 邈 be ^ [^^ (/ — one / —, '9) (qos) os m
'9) (qos) os m ¾ (louiui22,2 · ο, 3rao {H) ^ e; ^ 邈 be ^ [^^ (/ — one / —, '9) (qos) os m
邈べ ^ [ 、^ ^ ^ [, ^
( / →- [S Ί][Ρ-2 ' I ] ^^^ - Η8 - - L '9)] (308) (/ →-[S Ί] [Ρ-2 'I] ^^^-Η8--L' 9)] ( 3 08)
• +(H+ ) 89S: ζ/ω (9VH)S • + (H +) 89S: ζ / ω (9VH) S
•(s 'Ηΐ) 6Ζ·• ( s ' Ηΐ) 6Ζ
8 '(s Ήΐ) 88·9 '(ΖΗ 9·8=ί" 'Ρ 'ΗΖ) £9'f '(ΖΗ 0"Ζ '^Η 0"Z=f '¾Ρ 'Η ) ΐε· '(s 'ΗΖ) fL' ε '(s Ήε) ε '(s Ήε) wz '(ΖΗ o"z=f Ή9) ss'i 9 '· (ΖΗ )Ο 'ει αつ) WN HX 8 '(s Ήΐ) 88 · 9' ( Ζ Η 9 · 8 = ί "'Ρ' ΗΖ) £ 9'f '( Ζ Η 0"Ζ' ^ Η 0 "Z = f '¾Ρ' Η) ΐε · '( s ' ΗΖ) fL 'ε' (s Ήε) ε '( s Ήε) wz' ( Ζ Η o "z = f Ή9) ss'i 9 '· ( Ζ Η) Ο' ε ι α) WN H X
·τ_ωο 0862 '9S2T '90Π ' ^Οΐ '^Οΐ Λ ( 1)ΗΙ [ΙΖ 0 w目 ¾遨
 · Τ _ωο 0862 '9S2T '90 Π' ^ Οΐ '^ Οΐ Λ (1) ΗΙ [ΙΖ 0 w eyes ¾ 遨
· Τ _ωο 0862 '9S2T '90 Π' ^ Οΐ '^ Οΐ Λ (1) ΗΙ [ΙΖ 0 w eyes ¾ 遨
¾ (louiui2 -zz §92 ·Ζ) ^エ; ^邈べ ^ [ 、^ ( / 一,—ベ^べ — ¾ (louiui2 -zz § 92 · Ζ) ^ e; ^ 邈 be ^ [, ^ (/ 1 、 —Be ^
ΗΙ-
 (B08) 08p}¾?ΗΙ- ( B 08) 08p} ¾?
(B08) 08p}¾?ΗΙ- ( B 08) 08p} ¾?
^エ 邈べ ^ ^ D
( / — 一 /— 、 ^ [ε Ί][Ρ-2
 '9)] ( 08) (/ — One / —, ^ [ε Ί] [Ρ-2 '9)] (08)
'9)] ( 08) (/ — One / —, ^ [ε Ί] [Ρ-2 '9)] (08)
• +(H+ ) LZZ: ζ/ω (9VH)S • + (H +) LZZ: ζ / ω (9VH) S
•(s 'Ηΐ) Z9"9 '(ΖΗ '6=f • ( s ' Ηΐ) Z9 "9 '( Ζ Η' 6 = f
'P ΉΖ) Off '(ΖΗ ΓΖ 'ZH VL=[ 'bp 'Hf) ZZ'f '(ω 'ΗΖ) Z6 — '(ω Ή2) 99 — S9' 'P ΉΖ) Off' ( Ζ Η ΓΖ 'ZH VL = [' bp 'Hf) ZZ'f' (ω 'ΗΖ) Z6 —' (ω Ή2) 99 — S9 '
0l76S0C/900Zdf/X3d Z9I- 0C0l70l/900Z OAV
を使用して、実施例 1 (la)に記載した方法に従い、 [ (7 プロモー 6—メチルー 3— ォキソ 2, 3 ジヒドロ 1H インデン 4 ィル)ォキシ]メチルホスホン酸ジェチ ル(9. 38g、収率 76%)を得た。 0l76S0C / 900Zdf / X3d Z9I- 0C0l70l / 900Z OAV In accordance with the method described in Example 1 (la), [(7 Promo 6-methyl-3-oxo 2,3 dihydro 1H indene 4 yl) oxy] methylphosphonic acid ethyl ester (9.38 g, yield) 76%).
[0523] [ (7 ブロモ 6—メチル 3—ォキソ 2, 3 ジヒドロ一 1H—インデン一 4—ィル )ォキシ]メチルホスホン酸ジェチル(600mg、 1. 53mmol)を使用して、実施例 3 (3 a)、実施例 3 (3b)及び実施例 1 (lc)に記載した方法に順次従い、標記目的化合物 (320mg、収率 55%)を得た。 [0523] [(7 Bromo 6-methyl 3-oxo 2,3 dihydro-1H-indene-4-yl) oxy] methyl phosphonate (600 mg, 1.53 mmol) was used in Example 3 (3 a ), Example 3 (3b) and Example 1 (lc) were sequentially followed to give the title object compound (320 mg, yield 55%).
[0524] IR(KBr) v 1101, 1189, 1211, 1304, 1423, 2917, 3083, 3419 cm"1; [0524] IR (KBr) v 1101, 1189, 1211, 1304, 1423, 2917, 3083, 3419 cm "1;
max max
1H NMR(DMSO-d , 400MHz): δ 2.41 (3H, s), 3.89 (2H, s), 4.34 (2H, d, J=9.0 Hz 1H NMR (DMSO-d, 400MHz): δ 2.41 (3H, s), 3.89 (2H, s), 4.34 (2H, d, J = 9.0 Hz
6 6
), 7.19 (1H, s), 9.15 (1H, s); ), 7.19 (1H, s), 9.15 (1H, s);
MS(FAB) m/z: 374 (M- H)— . MS (FAB) m / z: 374 (M- H) —.
<実施例 82 > <Example 82>
[ (6—メトキシ 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキシ]メチ ルホスホン酸(例示化合物番号 1-244) [(6-Methoxy8H-indeno [1,2-d] [l, 3] thiazole-4-yl) oxy] methylphosphonic acid (Exemplary Compound No. 1-244)
4 ブロモ 7 ヒドロキシ一 5—メトキシインダン一 1—オン(4. 19g、 16. 3mmol) を使用して、実施例 1 (la)に記載した方法に従い、 { [ (7 プロモー 6—メトキシー 3 -ォキソ 2, 3 ジヒドロ一 1H インデン 4 ィル)ォキシ]メチル }ホスホン酸ジ ェチル (4. 59g、収率 69%)を得た。 4 Bromo 7 Hydroxy 1 5-Methoxyindane 1-one (4.19 g, 16.3 mmol) was used according to the procedure described in Example 1 (la) {[((7 Promo 6-Methoxy-3-oxo) 2,3 dihydro-1H indene 4yl) oxy] methyl} phosphonate diethyl (4.59 g, yield 69%) was obtained.
{ [ (7—ブロモ 6—メトキシ一 3—ォキソ 2, 3 ジヒドロ一 1H—インデン一 4—ィ ル)ォキシ]メチル }ホスホン酸ジェチル(708mg、 1. 74mmol)をエタノール 酢酸( 1 : 1、 30mL)に溶解し、そこへパラジウム—炭素(70mg)および酢酸ナトリウム(143 mg、 1. 74mmol)を加え、水素雰囲気下、室温で 2時間 30分攪拌した。反応液をセ ライトろ過し、減圧下溶媒を留去した後、酢酸ェチルで希釈した。得られた有機層を 飽和食塩水で洗浄し、無水硫酸マグネシウムで乾燥し、減圧下溶媒を留去した。得 られた残渣をシリカゲルクロマトグラフィー (酢酸ェチル)を用いて精製し、 { [ (6—メトキ シ一 3 ォキソ 2, 3 ジヒドロ一 1H インデン 4 ィル)ォキシ]メチル }ホスホン 酸ジェチル (556mg、収率 97%)を得た。 {[(7-Bromo 6-methoxy-1-oxo-2,3 dihydro-l 1H-indene-4-yl) oxy] methyl} jetyl phosphonate (708 mg, 1.74 mmol) in ethanol acetic acid (1: 1, 30 mL ), Palladium-carbon (70 mg) and sodium acetate (143 mg, 1.74 mmol) were added thereto, and the mixture was stirred at room temperature for 2 hours 30 minutes in a hydrogen atmosphere. The reaction mixture was filtered through celite, the solvent was distilled off under reduced pressure, and the mixture was diluted with ethyl acetate. The obtained organic layer was washed with saturated brine, dried over anhydrous magnesium sulfate, and the solvent was evaporated under reduced pressure. The resulting residue was purified using silica gel chromatography (ethyl acetate) to obtain {[(6-Methoxy-3-oxo2,3-dihydro-1H indene-4-yl) oxy] methyl} jetyl phosphonate (556 mg, yield). Rate 97%).
{ [ (6—メトキシ一 3—ォキソ 2, 3 ジヒドロ一 1H—インデン一 4—ィル)ォキシ]メ
チル }ホスホン酸ジェチル(552mg、 1. 68mmol)を使用して、実施例 3 (3a)及び実 施例 3 (3b)に記載した方法に順次従い、 [ (6—メトキシー 8H インデノ [1, 2-d] [l , 3]チアゾールー 4 ィル)ォキシ]メチルホスホン酸ジェチル(86mg、収率 14%)を 得た。 {[(6-Methoxy-1-3-oxo-2, 3-dihydro-1H-indene-4-yl) oxy] The procedure described in Example 3 (3a) and Example 3 (3b) was followed in turn using til} jetyl phosphonate (552 mg, 1.68 mmol) and [(6-methoxy-8H indeno [1, 2 -d] [l, 3] thiazole-4-yl) oxy] methyl phosphonate (86 mg, 14% yield) was obtained.
[ (6—メトキシ 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキシ]メチ ルホスホン酸ジェチル(74mg、 0. 200mmol)を使用して、実施例 1 (lc)に記載した 方法に従い、標記目的化合物(58mg、収率 93%)を得た。 [(6-Methoxy8H-indeno [1,2-d] [l, 3] thiazol-4-yl) oxy] methyl phosphonate (74 mg, 0.200 mmol) was used in Example 1 (lc). The title object compound (58 mg, yield 93%) was obtained according to the method described.
[0525] IR(KBr) v 1070, 1108, 1164, 1194, 1299, 1317, 1396, 1426, 1488, 1533, 1592 max [0525] IR (KBr) v 1070, 1108, 1164, 1194, 1299, 1317, 1396, 1426, 1488, 1533, 1592 max
, 1617, 2841, 2905, 3075, 3403 cm"1; , 1617, 2841, 2905, 3075, 3403 cm "1;
1H NMR(DMSO-d , 400MHz): δ 3.80 (3Η, s), 3.90 (2Η, s), 4.34 (2H, d, J=8.8 Hz 1H NMR (DMSO-d, 400MHz): δ 3.80 (3Η, s), 3.90 (2Η, s), 4.34 (2H, d, J = 8.8 Hz
6 6
), 6.73 (1H, d, J=2.0 Hz), 6.88 (1H, d, J=2.0 Hz), 9.12 (1H, s); ), 6.73 (1H, d, J = 2.0 Hz), 6.88 (1H, d, J = 2.0 Hz), 9.12 (1H, s);
MS(FAB) m/z: 312 (M- H)— . MS (FAB) m / z: 312 (M- H) —.
<実施例 83 > <Example 83>
[ (7—ブロモー 6—メトキシ—8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル) ォキシ]メチルホスホン酸 (例示化合物番号 1-245) [(7-Bromo-6-methoxy-8H-indeno [1,2-d] [l, 3] thiazole-4-yl) oxy] methylphosphonic acid (Exemplary Compound No. 1-245)
4 ブロモ 7 ヒドロキシ一 5—メトキシインダン一 1—オン(1. OOg、 3. 89mmol) を使用して、実施例 1 (la)、実施例 3 (3a)及び実施例 3 (3b)に記載した方法に順次 従い、 [ (7 ブロモー 6—メトキシ 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキシ]メチルホスホン酸ジェチル(648mg、収率 37%)を得た。 4 Bromo 7 Hydroxy 1-Methoxyindane 1-one (1. OOg, 3.89 mmol) was used to describe in Example 1 (la), Example 3 (3a) and Example 3 (3b). By sequentially following the method, [(7 bromo-6-methoxy8H-indeno [1,2-d] [l, 3] thiazol-4-yl) oxy] methylphosphonate jetyl (648 mg, yield 37%) was obtained.
[0526] [ (7—ブロモー 6—メトキシ—8H—インデノ [1, 2— d] [l, 3]チアゾールー 4ーィル )ォキシ]メチルホスホン酸ジェチル(100mg、 0. 223mmol)を使用して、実施例 1 ( lc)に記載した方法に従い、標記目的化合物(80. Omg、収率 91%)を得た。 [0526] Examples using [(7-bromo-6-methoxy-8H-indeno [1,2-d] [l, 3] thiazol-4-yl) oxy] methylphosphonate (100 mg, 0.223 mmol) The title object compound (80. Omg, yield 91%) was obtained according to the method described in 1 (lc).
[0527] IR(KBr) v 1070, 1209, 1362, 1429, 1468, 1579, 3090 cm"1; [0527] IR (KBr) v 1070, 1209, 1362, 1429, 1468, 1579, 3090 cm "1;
max max
1H NMR(DMSO-d , 400MHz): δ 3.87 (2H, s), 3.90 (3H, s), 4.42 (2H, d, J=9.0 Hz 1H NMR (DMSO-d, 400MHz): δ 3.87 (2H, s), 3.90 (3H, s), 4.42 (2H, d, J = 9.0 Hz
6 6
), 6.95 (1H, s), 9.12 (1H, s); ), 6.95 (1H, s), 9.12 (1H, s);
MS(FAB) m/z: 390 (M— H)— . MS (FAB) m / z: 390 (M— H) —.
<実施例 84 > <Example 84>
[ (8 ォキソ 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキシ]メチル
ホスホン酸 (例示化合物番号 1-253) [(8 oxo 8H—indeno [1, 2-d] [l, 3] thiazol-4-yl) oxy] methyl Phosphonic acid (Exemplary compound number 1-253)
(84a) [ (8 ォキソ—8H—インデノ [1, 2 d] [l, 3]チアゾールー 4 ィル)ォキシ ]メチルホスホン酸ジェチル (84a) [(8-oxo-8H-indeno [1,2d] [l, 3] thiazol-4-yl) oxy] methyl phosphonate
実施例 3 (3b)で製造した(8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィルォ キシ)メチルホスホン酸ジェチル(150mg、 0. 442mmol)を 1, 4 ジォキサン(2. 2 mL)と酢酸(0. 2mL)に溶解し、そこへ二酸化セレン(98. lmg、 0. 884mmol)を 加え、 6時間加熱還流した。 Example 3 (8H-indeno [1,2-d] [l, 3] thiazole-4-yloxy) methylphosphonate jetyl (150 mg, 0.442 mmol) prepared in (3b) was prepared from 1,4 dioxane (2.2 mL). ) And acetic acid (0.2 mL), selenium dioxide (98. lmg, 0.884 mmol) was added thereto, and the mixture was heated to reflux for 6 hours.
[0528] 冷却後、ろ過した後減圧下溶媒を留去し、残渣に水を加え、酢酸ェチルで抽出し た。得られた有機層を飽和炭酸水素ナトリウム水溶液及び飽和食塩水で洗浄し、無 水硫酸ナトリウムで乾燥し、減圧下溶媒を留去した。得られた残渣をシリカゲルカラム クロマトグラフィー (酢酸ェチル:メタノール、 30 : 1、 VZV)を用いて精製し、標記目的 化合物(105mg、収率 67%)を得た。 [0528] After cooling and filtering, the solvent was evaporated under reduced pressure, water was added to the residue, and the mixture was extracted with ethyl acetate. The obtained organic layer was washed with a saturated aqueous sodium hydrogen carbonate solution and saturated brine, dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure. The obtained residue was purified by silica gel column chromatography (ethyl acetate: methanol, 30: 1, VZV) to obtain the title object compound (105 mg, yield 67%).
[0529] IR(KBr) v 985, 1019, 1248, 1485, 1712 cm"1; [0529] IR (KBr) v 985, 1019, 1248, 1485, 1712 cm "1;
max max
1H NMR(CDC1 , 400MHz) δ 1.36 (6H, t, J=7.1 Hz), 4.28 (4H, dq, J=7.1 Hz, 7.1 1H NMR (CDC1, 400MHz) δ 1.36 (6H, t, J = 7.1 Hz), 4.28 (4H, dq, J = 7.1 Hz, 7.1
3 Three
Hz), 4.59 (2H, d, J=9.0 Hz), 7.13 (1H, dd, J=2.0 Hz, 7.1 Hz), 7.20-7.27 (2H, m), 9. 07 (1H, s); Hz), 4.59 (2H, d, J = 9.0 Hz), 7.13 (1H, dd, J = 2.0 Hz, 7.1 Hz), 7.20-7.27 (2H, m), 9. 07 (1H, s);
MS(FAB) m/z: 354 (M+H)+ . MS (FAB) m / z: 354 (M + H) + .
(84b) [ (8 ォキソ—8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキシ ]メチノレホスホン酸 (84b) [(8-oxo-8H-indeno [1,2-d] [l, 3] thiazol-4-yl) oxy] methinorephosphonic acid
実施例 84 (84a)で製造した [ (8—ォキソ 8H—インデノ [1, 2 d] [l, 3]チアゾ 一ルー 4—ィル)ォキシ]メチルホスホン酸ジェチル(59. 3mg、0. 168mmol)を使 用して、実施例 1 (lc)に記載した方法に従い、標記目的化合物 (46. 7mg、収率 94 %)を得た。 Example 84 [(8-oxo 8H-indeno [1,2d] [l, 3] thiazo-l 4-yl) oxy] methylphosphonic acid jetyl (59.3 mg, 0.168 mmol) prepared in Example 84 (84a) Was used to give the title object compound (46.7 mg, yield 94%) according to the method described in Example 1 (lc).
[0530] IR(KBr) v 958, 1262, 1483, 1707, 3095 cm"1; [0530] IR (KBr) v 958, 1262, 1483, 1707, 3095 cm "1;
max max
1H NMR(DMSO-d , 400MHz): δ 4.38 (2H, d, J=9.0 Hz), 7.16 (1H, dd, J=0.8 Hz, 1H NMR (DMSO-d, 400 MHz): δ 4.38 (2H, d, J = 9.0 Hz), 7.16 (1H, dd, J = 0.8 Hz,
6 6
7.0 Hz), 7.29 (1H, dd, J=7.0 Hz, 8.6 Hz), 7.35 (1H, dd, J=0.8 Hz, 8.6 Hz), 9.56 (1 H, s); 7.0 Hz), 7.29 (1H, dd, J = 7.0 Hz, 8.6 Hz), 7.35 (1H, dd, J = 0.8 Hz, 8.6 Hz), 9.56 (1 H, s);
MS(FAB) m/z: 296 (M- H)— .
<実施例 85 > MS (FAB) m / z: 296 (M- H) —. <Example 85>
[ (8 ヒドロキシ 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキシ]メ チルホスホン酸(例示化合物番号 1-258) [(8Hydroxy 8H-indeno [1,2-d] [l, 3] thiazol-4-yl) oxy] methylphosphonic acid (Exemplary Compound No. 1-258)
実施例 84 (84a)で製造した [ (8—ォキソ 8H—インデノ [1, 2— d] [l, 3]チアゾ 一ルー 4 ィル)ォキシ]メチルホスホン酸ジェチル(350mg、 0. 991mmol)を使用 して、実施例 32 (32a)に記載した方法に従い、 [ (8 ヒドロキシ一 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキシ]メチルホスホン酸ジェチル(222mg、収 率 63%)を得た。 Example 84 Using [(8-oxo 8H-indeno [1,2-d] [l, 3] thiazo 4-l) oxy] methylphosphonate (350 mg, 0.999 mmol) prepared in Example 84 (84a) Then, according to the method described in Example 32 (32a), [(8-hydroxy-8H-indeno [1,2-d] [l, 3] thiazol-4-yl) oxy] methyl phosphonate (222 mg, yield) 63%) was obtained.
[0531] [ (8 ヒドロキシ 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキシ]メ チルホスホン酸ジェチル(107mg、 0. 301mmol)を使用して、実施例 l (lc)に記載 した方法に従い、標記目的化合物(77. 3mg、収率 86%)を得た。 [0531] [(8Hydroxy 8H-indeno [1,2-d] [l, 3] thiazol-4-yl) oxy] methyl phosphonate (107 mg, 0.301 mmol) was used in Example l (lc ) To give the title object compound (77.3 mg, yield 86%).
[0532] IR(KBr) v 1041, 1264, 1476, 1497, 1589, 3089, 3279 cm"1; [0532] IR (KBr) v 1041, 1264, 1476, 1497, 1589, 3089, 3279 cm "1;
max max
1H NMR(DMSO-d , 400MHz): δ 4.30—4.36 (2H, m), 5.57 (1H, s), 6.12 (1H, brs), 1H NMR (DMSO-d, 400MHz): δ 4.30—4.36 (2H, m), 5.57 (1H, s), 6.12 (1H, brs),
6 6
7.13-7.15 (1H, m), 7.20-7.26 (2H, m), 9.19 (1H, s); 7.13-7.15 (1H, m), 7.20-7.26 (2H, m), 9.19 (1H, s);
MS(FAB) m/z: 298 (M— H)— . MS (FAB) m / z: 298 (M— H) —.
<実施例 86 > <Example 86>
[ (8, 8 ジフルオロー 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキシ ]メチルホスホン酸 (例示化合物番号 1-263) [(8,8 difluoro-8H-indeno [1,2-d] [l, 3] thiazole-4-yl) oxy] methylphosphonic acid (Exemplified Compound No. 1-263)
(86a) [ (8, 8 ジフルオロー 8H—インデノ [1, 2— d] [l, 3]チアゾールー 4 ィル) ォキシ]メチルホスホン酸ジェチル (86a) [(8,8 Difluoro-8H-indeno [1,2-d] [l, 3] thiazol-4-yl) oxy] methyl phosphonate
実施例 84 (84a)で製造した [ (8—ォキソ 8H—インデノ [1, 2— d] [l, 3]チアゾ 一ルー 4 ィル)ォキシ]メチルホスホン酸ジェチル(315mg、 0. 89mmol)をトルェ ン(6mL)、 1, 3 ジメチルー 2—イミダゾリジノン(3mL)に溶解し、 2, 2 ジフルォロ - 1, 3 ジメチルイミダゾリジン(969mg、 7. 14mmol)を加え、 100。Cで 16時間カロ 熱した。減圧下溶媒を留去した後、残渣をエタノール (3mL)に溶解し、水素化ホウ 素ナトリウム(34mg、 0. 89mmol)をカ卩え、未反応の原料をアルコール体へと還元し た。減圧下溶媒を留去し、残渣に水を加え、酢酸ェチルで抽出した後、有機層を飽 和食塩水で洗浄し、無水硫酸ナトリウムで乾燥し、減圧下溶媒を留去した。得られた
残渣をシリカゲルカラムクロマトグラフィー (酢酸ェチル:メタノール、 100 : 0— 90 : 10 、 VZV)を用いて精製し、標記目的化合物(18. 6mg、収率 5. 5%)を得た。 Example 84 [(8-oxo8H-indeno [1,2-d] [l, 3] thiazo 4-l) oxy] methyl phosphonate (315 mg, 0.89 mmol) prepared in Example 84 (84a) (6 mL), 1,3 dimethyl-2-imidazolidinone (3 mL), add 2,2 difluoro-1,3 dimethylimidazolidine (969 mg, 7.14 mmol), 100. C heated for 16 hours at C. After evaporating the solvent under reduced pressure, the residue was dissolved in ethanol (3 mL), sodium borohydride (34 mg, 0.89 mmol) was added, and the unreacted raw material was reduced to the alcohol form. The solvent was distilled off under reduced pressure, water was added to the residue, and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated brine and dried over anhydrous sodium sulfate, and the solvent was distilled off under reduced pressure. Obtained The residue was purified by silica gel column chromatography (ethyl acetate: methanol, 100: 0-90: 10, VZV) to obtain the title object compound (18.6 mg, yield 5.5%).
[0533] 1H NMR(CDC1 , 400MHz): δ 1.35 (6Η, t, J=7.1 Hz), 4.25—4.35 (4H, m), 4.59 (2 [0533] 1H NMR (CDC1, 400MHz): δ 1.35 (6Η, t, J = 7.1 Hz), 4.25—4.35 (4H, m), 4.59 (2
3 Three
H, d, J=9.0 Hz), 7.10-7.30 (3H, m), 8.95 (IH, s); H, d, J = 9.0 Hz), 7.10-7.30 (3H, m), 8.95 (IH, s);
MS(EI) m/z: 376 (M+H)+. MS (EI) m / z: 376 (M + H) + .
(86b) [ (8, 8 ジフルオロー 8H—インデノ [1, 2— d] [l, 3]チアゾールー 4 ィル) 才キシ]メチノレホスホン酸 (86b) [(8, 8 Difluoro-8H—indeno [1, 2— d] [l, 3] thiazole-4 yl)
実施例 86 (86a)で製造した [ (8, 8 ジフルオロー 8H インデノ [1, 2— d] [l, 3] チアゾールー 4 ィル)ォキシ]メチルホスホン酸ジェチル(18. 6mg、 0. 05mmol) を使用して、実施例 1 (lc)に記載した方法に従い、標記目的化合物 (4. 6mg、収率 29%)を得た。 Example 86 Using [(8,8 difluoro-8H indeno [1,2-d] [l, 3] thiazol-4-yl) oxy] methylphosphonate (18.6 mg, 0.05 mmol) prepared in Example 86 (86a) Then, according to the method described in Example 1 (lc), the title object compound (4.6 mg, yield 29%) was obtained.
[0534] 1H NMR(DMSO-d , 400MHz): δ 4.35 (2Η, d, J=8.8 Hz), 7.26-7.42 (3H, m), 9.4 [0534] 1H NMR (DMSO-d, 400 MHz): δ 4.35 (2Η, d, J = 8.8 Hz), 7.26-7.42 (3H, m), 9.4
6 6
3 (IH, s); 3 (IH, s);
MS(FAB) m/z: 318 (M— H)— . MS (FAB) m / z: 318 (M— H) —.
<実施例 87 > <Example 87>
({7 [ (ァミノカルボ-ル)ァミノ] 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ーィル }ォキシ)メチルホスホン酸 (例示化合物番号 1-206) ({7 [(Aminocarbole) amino] 8H-indeno [1, 2-d] [l, 3] thiazole-4-yl} oxy) methylphosphonic acid (Exemplary Compound No. 1-206)
(87a) ({7— [ (ァミノカルボ-ル)ァミノ]— 8H—インデノ [1, 2— d] [l, 3]チアゾー ルー 4ーィル }ォキシ)メチルホスホン酸ジェチル (87a) ({7— [(Aminocarbole) amino] — 8H—Indeno [1, 2— d] [l, 3] thiazol 4-yl} oxy) Jetyl methylphosphonate
実施例 47 (47b)で製造した [ (7 アミノー 8H—インデノ [1, 2— d] [l, 3]チアゾ 一ルー 4—ィル)ォキシ]メチルホスホン酸ジェチル(51. 6mg、0. 146mmol)をテト ラヒドロフラン(1. 5mL)に溶解し、そこへ 85%トリメチルシリルイソシァネート(68. 3 μレ 0. 437mmol)を加え、 60°Cで 5時間撹拌した。 Example 47 [(7 Amino-8H-indeno [1,2-d] [l, 3] thiazo 4-l) oxy] methyl phosphonate (51.6 mg, 0.146 mmol) prepared in Example 47 (47b) Was dissolved in tetrahydrofuran (1.5 mL), 85% trimethylsilyl isocyanate (68.3 μl, 0.437 mmol) was added thereto, and the mixture was stirred at 60 ° C. for 5 hours.
[0535] 冷却後、減圧下溶媒を留去し、得られた結晶を酢酸ェチルで洗浄し、標記目的化 合物(29. 5mg、収率 51%)を得た。 [0535] After cooling, the solvent was distilled off under reduced pressure, and the obtained crystals were washed with ethyl acetate to give the title object compound (29.5 mg, yield 51%).
[0536] IR(KBr) v 1026, 1230, 1539, 1668, 3283 cm"1; [0536] IR (KBr) v 1026, 1230, 1539, 1668, 3283 cm "1;
max max
1H NMR(CDC1 , 400MHz) : δ 1.40 (6H, t, J=7.0 Hz), 3.48 (2H, s), 4.27-4.42 (4H, 1H NMR (CDC1, 400 MHz): δ 1.40 (6H, t, J = 7.0 Hz), 3.48 (2H, s), 4.27-4.42 (4H,
3 Three
m), 4.43 (2H, d, J=9.4 Hz), 5.05-5.19 (2H, br), 6.65 (IH, d, J=8.6 Hz), 7.15 (IH, d
, J=8.6 Hz), 7.34-7.42 (1H, br), 8.66 (1H, s); m), 4.43 (2H, d, J = 9.4 Hz), 5.05-5.19 (2H, br), 6.65 (IH, d, J = 8.6 Hz), 7.15 (IH, d , J = 8.6 Hz), 7.34-7.42 (1H, br), 8.66 (1H, s);
MS(FAB) m/z: 398 (M+H)+ . MS (FAB) m / z: 398 (M + H) + .
(87b) ({7— [(ァミノカルボ-ル)ァミノ]— 8H—インデノ [1, 2— d][l, 3]チアゾー ルー 4ーィル }ォキシ)メチルホスホン酸 (87b) ({7 — [(Aminocarbole) amino] — 8H—Indeno [1, 2—d] [l, 3] thiazol 4-yl} oxy) methylphosphonic acid
実施例 87 (87a)で製造した( { 7 [ (ァミノカルボ-ル)ァミノ] - 8H インデノ [ 1 , 2-d][l, 3]チアゾールー 4ーィル }ォキシ)メチルホスホン酸ジェチル(24. 7mg、 0. 062mmol)を使用して、実施例 1 (lc)に記載した方法に従い、標記目的化合物( 18. 5mg、収率 87%)を得た。 Example 87 (87a) ({7 [(Aminocarbole) amino]-8H indeno [1,2-d] [l, 3] thiazol-4-yl} oxy) methylphosphonic acid jetyl (24.7 mg, 0 062 mmol) was used to give the title object compound (18.5 mg, 87% yield) according to the method described in Example 1 (lc).
[0537] IR(KBr) v 948, 1264, 1519, 1664, 3346 cm"1; [0537] IR (KBr) v 948, 1264, 1519, 1664, 3346 cm "1;
max max
1H NMR(DMSO-d , 400MHz): δ 3.82 (2H, s), 4.28 (2H, d, J=9.0 Hz), 5.93—6.00 ( 1H NMR (DMSO-d, 400MHz): δ 3.82 (2H, s), 4.28 (2H, d, J = 9.0 Hz), 5.93—6.00 (
6 6
2H, br), 7.04 (1H, d, J=8.8 Hz), 7.59 (1H, d, J=8.8 Hz), 8.01—8.05 (1H, br), 9.17 (1 H, s); 2H, br), 7.04 (1H, d, J = 8.8 Hz), 7.59 (1H, d, J = 8.8 Hz), 8.01—8.05 (1H, br), 9.17 (1 H, s);
MS(ESI) m/z: 340 (M— H)— . MS (ESI) m / z: 340 (M— H) —.
<実施例 88> <Example 88>
[(7 シァノー 8H—インデノ [1, 2-d][l, 3]チアゾールー 4 ィル)ォキシ]メチル ホスホン酸 (例示化合物番号 1-32) [(7 cyano 8H—indeno [1, 2-d] [l, 3] thiazole-4-yl) oxy] methyl phosphonic acid (Exemplified Compound No. 1-32)
(88a) [(7 シァノー 8H—インデノ [1, 2— d][l, 3]チアゾールー 4 ィル)ォキシ] メチルホスホン酸ジェチル (88a) [(7 cyano 8H-indeno [1, 2- d] [l, 3] thiazol-4-yl) oxy] jetyl methylphosphonate
実施例 27 (27a)で製造した [(7 ブロモ— 8H—インデノ [1, 2 d][l, 3]チアゾ 一ルー 4—ィル)ォキシ]メチルホスホン酸ジェチル(332mg、 0. 80mmol)をジメチ ルホルムアミド(8mL)に溶解し、シアン化亜鉛(188mg、 1.60mmol)、塩化リチウム (68mg、 1.60mmol)、テトラキストリフエ-ルホスフィンパラジウム(444mg、 0. 40m mol)を加え、 120°Cで 0. 5時間加熱した。減圧下溶媒を留去した後、残渣に水を加 え酢酸ェチルで抽出した。有機層を飽和炭酸水素ナトリゥム水溶液及び飽和食塩水 で洗浄した後、無水硫酸ナトリウムで乾燥し減圧下溶媒を留去した。得られた残渣を シリカゲルカラムクロマトグラフィー (酢酸ェチル:メタノール、 100:0— 80 :20、 V/V )を用いて精製し、標記目的化合物(181mg、収率 62%)を得た。 EXAMPLE 27 [(7 Bromo-8H-indeno [1,2d] [l, 3] thiazol 4-yl) oxy] methylphosphonate jetyl (332 mg, 0.80 mmol) prepared in Example 27 (27a) Dissolve in formaldehyde (8 mL), add zinc cyanide (188 mg, 1.60 mmol), lithium chloride (68 mg, 1.60 mmol), tetrakistriphenylphosphine palladium (444 mg, 0.40 mmol) at 120 ° C. 0. Heated for 5 hours. After evaporating the solvent under reduced pressure, water was added to the residue and the mixture was extracted with ethyl acetate. The organic layer was washed with saturated aqueous sodium hydrogen carbonate solution and saturated brine, dried over anhydrous sodium sulfate, and the solvent was evaporated under reduced pressure. The obtained residue was purified by silica gel column chromatography (ethyl acetate: methanol, 100: 0-80: 20, V / V) to obtain the title compound (181 mg, yield 62%).
[0538] IR(KBr) v 983, 1023, 1062, 1245, 1278, 1427, 1501, 1578, 2226, 2991, 3079 c max
-1 [0538] IR (KBr) v 983, 1023, 1062, 1245, 1278, 1427, 1501, 1578, 2226, 2991, 3079 c max -1
m ; m;
1H NMR(CDC1 , 400MHz): δ 1.35 (6H, t, J=7.0 Hz), 4.06 (2H, s), 4.27-4.37 (4H 1H NMR (CDC1, 400MHz): δ 1.35 (6H, t, J = 7.0 Hz), 4.06 (2H, s), 4.27-4.37 (4H
3 Three
, m), 4.66 (2H, d, J=9.0 Hz), 7.15 (IH, d, J=8.6 Hz), 7.55 (IH, d, J=8.6 Hz), 8.89 (1 , m), 4.66 (2H, d, J = 9.0 Hz), 7.15 (IH, d, J = 8.6 Hz), 7.55 (IH, d, J = 8.6 Hz), 8.89 (1
H, s); H, s);
MS(FAB) m/z: 365 (M+H)+. MS (FAB) m / z: 365 (M + H) + .
(88b) [ (7 シァノー 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4 ィル)ォキシ] メチノレホスホン酸 (88b) [(7 Siano 8H-indeno [1, 2-d] [l, 3] thiazole-4-yl) oxy] methinorephosphonic acid
実施例 88 (88a)で製造した [ (7 シァノ 8H—インデノ [1, 2— d] [l, 3]チアゾ 一ルー 4—ィル)ォキシ]メチルホスホン酸ジェチル(160mg、 0. 44mmol)を使用し て実施例 1 (lc)に記載した方法に従い、標記目的化合物(88mg、収率 65%)を得 た。 Example 88 Using (7Siano 8H-indeno [1,2-d] [l, 3] thiazo-l-l-yl) oxy] methylphosphonate (160 mg, 0.44 mmol) prepared in Example 88 (88a) Then, according to the method described in Example 1 (lc), the title object compound (88 mg, yield 65%) was obtained.
[0539] IR(KBr) v 953, 1072, 1274, 1384, 1431, 1504, 1580, 1602, 2223, 3095, 3414 c max [0539] IR (KBr) v 953, 1072, 1274, 1384, 1431, 1504, 1580, 1602, 2223, 3095, 3414 c max
-1 -1
m ; m;
1H NMR(DMSO-d , 400MHz): δ 4.14 (2H, s), 4.39 (2H, d, J=8.6 Hz), 7.32 (IH, 1H NMR (DMSO-d, 400 MHz): δ 4.14 (2H, s), 4.39 (2H, d, J = 8.6 Hz), 7.32 (IH,
6 6
d, J=8.6Hz), 7.64 (IH, d, J=8.6Hz), 9.17 (IH, s); d, J = 8.6Hz), 7.64 (IH, d, J = 8.6Hz), 9.17 (IH, s);
MS(FAB) m/z: 307 (M- H)— . MS (FAB) m / z: 307 (M- H) —.
<実施例 89 > <Example 89>
{ [7—(1ーメチルー111ーピラゾールー4ーィル)ー811—ィンデノ[1, 2-d] [l, 3] チアゾールー 4 ィル]ォキシ }メチルホスホン酸(例示化合物番号 1-252) {[7- (1-Methyl-111-pyrazole-4-yl) -811-indeno [1, 2-d] [l, 3] thiazole-4-yl] oxy} methylphosphonic acid (Exemplary Compound No. 1-252)
実施例 27 (27a)で製造した [ (7 ブロモ— 8H—インデノ [1, 2 d] [l, 3]チアゾ 一ルー 4—ィル)ォキシ]メチルホスホン酸ジェチル(300mg、 0. 717mmol)及び 3 ピリジルボロン酸ピナコールエステルの代わりに 1 メチルピラゾール 4 ボロン 酸ピナコールエステル(273mg、 1. 43mmol)を使用して、実施例 28 (28a)に記載 した方法に従い、 { [7- (1—メチル 1H ピラゾール一 4—ィル) 8H—インデノ [ Example 27 [(7 Bromo-8H-indeno [1,2d] [l, 3] thiazo 4-luyl) oxy] methylphosphonate jetyl (300 mg, 0.717 mmol) and 3 prepared in Example 27 (27a) According to the method described in Example 28 (28a) using 1 methylpyrazole 4 boronic acid pinacol ester (273 mg, 1.43 mmol) instead of pyridylboronic acid pinacol ester, {[7- (1-methyl 1H pyrazole 1—4) 8H—Indeno [
I, 2-d] [l, 3]チアゾール—4—ィル]ォキシ }メチルホスホン酸ジェチル(254mg 、収率 84%)を得た。 I, 2-d] [l, 3] thiazole-4-yl] oxy} jethyl phosphonate (254 mg, 84% yield) was obtained.
[0540] { [7—(1ーメチルー111ーピラゾールー4ーィル)ー811—ィンデノ[1, 2— d] [l, 3 ]チアゾールー 4 ィル]ォキシ }メチルホスホン酸ジェチル(119mg、 0. 284mmol)
を使用して、実施例 1 (lc)に記載した方法に従い、標記目的化合物(91. 5mg、収 率 89%)を得た。 [0540] {[7- (1-Methyl-111-pyrazole-4-yl) -811-indeno [1, 2-d] [l, 3] thiazole-4-yl] oxy} Jetyl methylphosphonate (119 mg, 0.284 mmol) Was used to give the title object compound (91.5 mg, yield 89%) according to the method described in Example 1 (lc).
[0541] IR(KBr) v 928, 1071, 1271, 1562, 3412 cm"1; [0541] IR (KBr) v 928, 1071, 1271, 1562, 3412 cm "1;
max max
1H NMR(DMSO-d , 500MHz): δ 3.91 (3H, s), 4.09 (2H, s), 4.36 (2H, d, J=8.8 Hz 1H NMR (DMSO-d, 500MHz): δ 3.91 (3H, s), 4.09 (2H, s), 4.36 (2H, d, J = 8.8 Hz
6 6
), 7.20 (IH, d, J=8.8 Hz), 7.44 (IH, d, J=8.8 Hz), 7.91 (IH, s), 8.17 (IH, s), 9.21 (1 H, s); ), 7.20 (IH, d, J = 8.8 Hz), 7.44 (IH, d, J = 8.8 Hz), 7.91 (IH, s), 8.17 (IH, s), 9.21 (1 H, s);
MS(FAB) m/z: 362 (M— H)— MS (FAB) m / z: 362 (M— H) —
<実施例 90 > <Example 90>
{ [7—(メトキシカルボ-ル) 8H—インデノ [1, 2-d] [l, 3]チアゾールー 4ーィル ]ォキシ }メチルホスホン酸 (例示化合物番号 1-23) {[7- (methoxycarbol) 8H-indeno [1,2-d] [l, 3] thiazol-4-yl] oxy} methylphosphonic acid (Exemplified Compound No. 1-23)
実施例 1 ( la)で製造した [ (7 ブロモ 3 ォキソ 2, 3 -ジヒドロ 1H インデ ン 4 ィル)ォキシ]メチルホスホン酸ジェチル(1. 40g、 3. 71mmol)および 2 ト リメチルシリルエタノールの代わりにメタノール(20mL、 493mmol)を使用して、実施 例 36 (36a)に記載した方法に従 、、 7 [ (ジエトキシホスホリル)メトキシ] 1 ォキ ソインダン 4 カルボン酸メチル( 1. 08g、収率 81 %)を得た。 Instead of [(7 bromo 3 -oxo 2,3-dihydro 1H indene 4 yl) oxy] methyl phosphonate (1.40 g, 3.71 mmol) and 2 trimethylsilylethanol prepared in Example 1 (la) In accordance with the method described in Example 36 (36a), 7 [(diethoxyphosphoryl) methoxy] 1-oxoindane 4 methyl carboxylate (1.08 g, yield), using methanol (20 mL, 493 mmol). 81%).
[0542] 7 [ (ジエトキシホスホリル)メトキシ]— 1 ォキソインダンー4一力ルボン酸メチル( 529mg、 1. 48mmol)を使用して、実施例 3 (3a)及び実施例 3 (3b)に記載した方法 に順次従い、 { [7—(メトキシカルボ-ル) 8H—インデノ [1, 2-d] [l, 3]チアゾー ル— 4—ィル]ォキシ }メチルホスホン酸ジェチル(400mg、収率 69%)を得た。 [0542] 7 [(Diethoxyphosphoryl) methoxy] — 1 oxoindane-4 The method described in Example 3 (3a) and Example 3 (3b) using mono-methyl methyl boronate (529 mg, 1.48 mmol) {[7- (methoxycarbol) 8H-indeno [1,2-d] [l, 3] thiazol-4-yl] oxy} jetyl methylphosphonate (400 mg, 69% yield) Got.
[0543] { [7- (メトキシカルボ-ル)—8H—インデノ [1, 2 d] [1, 3]チアゾールー 4ーィ ル]ォキシ }メチルホスホン酸ジェチル(154mg、 0. 388mmol)を使用して、実施例 1 (lc)に記載した方法に従い、標記目的化合物(132mg、収率 100%)を得た。 [0543] {[7- (Methoxycarbol) -8H-indeno [1,2d] [1,3] thiazol-4-yl] oxy} using methyl phosphonate jetyl (154mg, 0.388mmol) The title object compound (132 mg, yield 100%) was obtained according to the method described in Example 1 (lc).
[0544] IR(KBr) v 957, 1001, 1071, 1112, 1146, 1268, 1362, 1385, 1431, 1488, 1507, max [0544] IR (KBr) v 957, 1001, 1071, 1112, 1146, 1268, 1362, 1385, 1431, 1488, 1507, max
1581, 1713, 2952, 3086, 3414 cm"1; 1581, 1713, 2952, 3086, 3414 cm "1;
1H NMR(DMSO-d , 400MHz): δ 3.87 (3H, s), 4.24 (2H, s), 4.43 (2H, d, J=8.8 Hz) 1H NMR (DMSO-d, 400MHz): δ 3.87 (3H, s), 4.24 (2H, s), 4.43 (2H, d, J = 8.8 Hz)
6 6
, 7.31 (IH, d, J=8.6 Hz), 7.86 (IH, d, J=8.6 Hz), 9.18 (IH, s); , 7.31 (IH, d, J = 8.6 Hz), 7.86 (IH, d, J = 8.6 Hz), 9.18 (IH, s);
MS(FAB) m/z: 340 (M- H)— . MS (FAB) m / z: 340 (M- H) —.
<試験例 1 >
FBPase阻害活性の測定 <Test Example 1> Measurement of FBPase inhibitory activity
国際公開第 00/14095号パンフレットの実施例に従って調製したヒト肝臓 FBPaseをコ ードするプラスミド pET- hlFBPaseを、 E.coli/BL21株に形質転換した。これを培養して 得た菌体をトリス塩酸緩衝液 (pH 7.4)中にて超音波粉砕した後、粗抽出液を遠心分 離(10,000rpm, 10分)し、上清を回収して酵素源として使用した。酵素活性の測定は 、 FBPaseの酵素反応によって生じたフルクトース -6-リン酸を、アツセィ系に共存させ たグルコース- 6-リン酸デヒドロゲナーゼ及びフォスフォグルコイソメラーゼの作用によ る NADPの還元反応により定量することにより行なった。 Plasmid pET-hlFBPase encoding human liver FBPase prepared according to the example of WO 00/14095 pamphlet was transformed into E. coli / BL21 strain. The cells obtained by culturing the cells are ultrasonically pulverized in Tris-HCl buffer (pH 7.4), and then the crude extract is centrifuged (10,000 rpm, 10 minutes). The supernatant is recovered and the enzyme is recovered. Used as a source. Enzyme activity was measured by reducing NADP by the action of glucose-6-phosphate dehydrogenase and phosphoglucoisomerase coexisting with fructose-6-phosphate produced by the enzyme reaction of FBPase. It was done by doing.
[0545] 具体的には、 0.1 M KCKSigma P— 3911)、 5mM EGTA(Sigma E— 4378)、 50mMトリス 塩酸緩衝液 (pH 7.4) (Sigma T- 4003)、 0.2mM NADP+(Sigma N- 0505)、 0.15mM FBP (フルクトースビスフォスフェート) (Sigma F-4757)、 ImM MgCl (Sigma M- 8266)、 200 [0545] Specifically, 0.1 M KCKSigma P-3911), 5 mM EGTA (Sigma E-4378), 50 mM Tris HCl buffer (pH 7.4) (Sigma T-4003), 0.2 mM NADP + (Sigma N-0505) 0.15mM FBP (fructose bisphosphate) (Sigma F-4757), ImM MgCl (Sigma M-8266), 200
2 2
μ g/ml BSA(Sigma A- 7906)、 2U/mlグルコース- 6-リン酸デヒドロゲナーゼ (Sigma G- 5 760)、 1.25U/mlフォスフォグルコイソメラーゼ (Sigma P- 5381)の混合液 (94 1、それぞ れの溶液の濃度は、最終調整濃度である)及び被験化合物液 (1 μ 1,最終調整濃度 10 μ g/mlから 1/3希釈系列又は 1/5希釈系列)を 96ゥエルプレートに分注し、そこに酵素 液 (5 μ 1)を添加し室温で酵素反応を開始させた(上記最終調整濃度は、全ての反応 溶液混合時の濃度を表す)。反応開始後 0分と 15分における 340應の吸光度をマイク 口プレートリーダー (ArvoSX, PerkinElmer Life Sciences社)にて測定し、その吸光度の 増加分を酵素活性とみなした。 μg / ml BSA (Sigma A-7906), 2U / ml glucose-6-phosphate dehydrogenase (Sigma G-5 760), 1.25U / ml phosphoglucoisomerase (Sigma P-5381) (94 1 The concentration of each solution is the final adjusted concentration) and the test compound solution (1 μ1, final adjusted concentration 10 μg / ml to 1/3 dilution series or 1/5 dilution series) 96 The enzyme solution (5 μ1) was added to the plate, and the enzyme reaction was started at room temperature (the above final adjusted concentration represents the concentration when all the reaction solutions were mixed). The absorbance at 340 ° at 0 minutes and 15 minutes after the start of the reaction was measured with a microphone plate reader (ArvoSX, PerkinElmer Life Sciences), and the increase in absorbance was regarded as the enzyme activity.
[0546] 各反応より得られた酵素活性の値から被験化合物の阻害曲線を作成した。その作 成した阻害曲線力 酵素活性の抑制率 50%に相当する被験化合物濃度を導き出し 、IC 値とした。 [0546] From the enzyme activity values obtained from each reaction, an inhibition curve of a test compound was prepared. The concentration of the test compound corresponding to the inhibition curve power enzyme inhibition rate of 50% was derived and used as the IC value.
50 50
[0547] 各被験化合物の IC 値を、以下の表 3に示す。 [0547] IC values of each test compound are shown in Table 3 below.
50 50
(表 3)
実施例 I Cso 実施例 icso (Table 3) Example I Cso Example ic so
番号 (π ) 番号 (πΗ) Number (π) Number (πΗ)
27 4 63 3 27 4 63 3
30 3 64 2 30 3 64 2
34 5 65 8 34 5 65 8
35 5 66 4 35 5 66 4
36 1 67 8 36 1 67 8
37 2 68 4 37 2 68 4
38 4 69 Π 38 4 69 Π
39 4 70 15 39 4 70 15
40 4 71 9 40 4 71 9
41 4 72 10 41 4 72 10
43 3 73 5 43 3 73 5
44 I 74 2 44 I 74 2
45 \ 75 9 . 45 \ 75 9.
51 5 78 9 51 5 78 9
53 5 79 Π 53 5 79 Π
57 3 80 10 57 3 80 10
58 3 81 Ί 7 58 3 81 Ί 7
59 5 82 8 59 5 82 8
60 6 83 14 60 6 83 14
62 6 87 12 62 6 87 12
表 3より、本発明の化合物は、優れた FBPase阻害活性を有しているため、糖尿病治 療薬として有用であることがわかる。 From Table 3, it can be seen that the compound of the present invention has an excellent FBPase inhibitory activity and is therefore useful as a therapeutic drug for diabetes.
<製剤例> <Formulation example>
(1)カプセル剤 (1) Capsule
実施例 36の化合物 50 mg Example 36 Compound 50 mg
ラタトース 110 mg Lattose 110 mg
コーン'スターチ 58 mg Corn 'starch 58 mg
2 mg 2 mg
合計 220 mg 220 mg total
上記で示される各成分の粉末を良く混合し、 60メッシュの篩 (メッシュの基準は Tyler 基準による)を通す。得られる粉末 220mgをはかり分け、ゼラチンカプセル (No.3)に充 填し、カプセル剤を調製する。
(2)錠剤 Mix well the powder of each component shown above and pass through a 60 mesh sieve (mesh standard is Tyler standard). Weigh out 220 mg of the resulting powder and fill into gelatin capsules (No. 3) to prepare capsules. (2) Tablet
実施例 37の化合物 50 mg Example 37 Compound 50 mg
ラタトース 85 mg Lattose 85 mg
コーン'スターチ 34 mg Corn 'starch 34 mg
微結晶セルロース 20 mg Microcrystalline cellulose 20 mg
'"ソ _ Ling '"Seo _ Ling
合計 190 mg 190 mg total
上記で示される各成分の粉末を良く混合し、各 190mg重量の錠剤に圧縮成型する 。必要ならば、これらの錠剤は糖またはフィルムで被覆してもよい。 The powder of each component shown above is mixed well and compressed into tablets each weighing 190 mg. If necessary, these tablets may be coated with sugar or film.
(3)顆粒剤 (3) Granules
実施例 45の化合物 50 mg Compound of Example 45 50 mg
ラタ卜ース 799 mg Ratose 799 mg
コーン'スターチ 150 mg Corn 'starch 150 mg
ヒドロキシプロピルセルロース 1 mg Hydroxypropylcellulose 1 mg
合計 1000 mg 1000 mg total
上記で示される各成分の粉末を良く混合し、純水で湿らし、バスケット式顆粒化機 で顆粒ィ匕し、乾燥して顆粒剤を得る。
The powders of the components shown above are mixed well, moistened with pure water, granulated with a basket type granulator, and dried to obtain granules.
Claims
請求の範囲 The scope of the claims
下記一般式 (I) The following general formula (I)

(I) (I)
[式中、 R1X及び R1Yは、それぞれ異なって水素原子、ハロゲン原子、 CI- C3アルキル 基、 P(0)(OH)、 OP(0)(OH)、 CH P(0)(0H)、 CF P(0)(0H)又は OCH P(0)(0H)を [Wherein R 1X and R 1Y are different from each other in hydrogen atom, halogen atom, CI-C3 alkyl group, P (0) (OH), OP (0) (OH), CH P (0) (0H) , CF P (0) (0H) or OCH P (0) (0H)
2 2 2 2 2 2 2 2 示し、 R2及び R3は、それぞれ同一若しくは異なって、水素原子、ハロゲン原子、 C1-C 6アルキル基、 C1-C6ハロアルキル基、 C1-C6アミノアルキル基(該ァミノアルキル基 のァミノ基は、置換基群 α力 選択される基で 1又は 2個置換されていても良い)、 C1 -C6ヒドロキシアルキル基、 CI- C6アルキルチオ基、 CI- C6アルコキシ基、 CI- C6ァ ルコキシィミノ C1-C3アルキル基(該アルコキシィミノ C1-C3アルキル基は、 C1-C3ァ ルキル基で置換されていても良い)、水酸基、 C1-C6脂肪族ァシル基 (該ァシル基は 、置換基群 α力 選択される基で置換されていても良い)、アミノ基、 CI- C6アルキル アミノ基、 CI- C6ジアルキルァミノ基、 CI- C6脂肪族ァシルァミノ基、 C6- C10ァリール 基 (該ァリール基は、置換基群 α力 選択される基で 1乃至 5個置換されていても良 い)、窒素、酸素及び硫黄力 選択される同一若しくは異なった 1-3個のへテロ原子 を含む 4-10員複素環基 (該複素環基は、置換基群 α力 選択される基で 1乃至 5個 置換されていても良い)、窒素、酸素及び硫黄力 選択される同一若しくは異なった 1 -3個のへテロ原子を含む 4-10員複素環 C1-C6アルキル基 (該複素環 C1-C6アルキ ル基は、置換基群 α力 選択される基で 1乃至 5個置換されていても良い)、窒素、酸 素及び硫黄力 選択される同一若しくは異なった 1-3個のへテロ原子を含む 4-10員 複素環 CI- C6アルキルアミノ基 (該複素環 CI- C6アルキルアミノ基は、置換基群 aか ら選択される基で 1乃至 5個置換されていても良い)、ウレイド基、シァノ基又は-トロ 基を示し、 R4は、水素原子、 C1-C3アルキル基、 C1-C3ハロアルキル基、ハロゲン原
子又はアミノ基を示し、 Xは、 C1-C3アルキレン基 (該アルキレン基は、ォキソ基、水酸 基、ハロゲン原子、 C1-C3アルキル基又は C3-6シクロアルキリデン基で 1又は 2個置 換されていてもよい)、を示す。ただし、 Rlxが P(0)(OH)、 OP(OXOH)、 CH P(0)(OH) 2 2 2 2 2 2 2 2 and R 2 and R 3 are the same or different and each represents a hydrogen atom, a halogen atom, a C1-C6 alkyl group, a C1-C6 haloalkyl group, a C1-C6 aminoalkyl group The amino group of the aminoamino group may be substituted with one or two groups selected from the substituent group α force), C1-C6 hydroxyalkyl group, CI-C6 alkylthio group, CI-C6 alkoxy group, CI- C6 alkyloximino C1-C3 alkyl group (the alkoxyimino C1-C3 alkyl group may be substituted with a C1-C3 alkyl group), a hydroxyl group, a C1-C6 aliphatic asil group (the acyl group is Substituent group α force (may be substituted with selected group), amino group, CI-C6 alkylamino group, CI-C6 dialkylamino group, CI-C6 aliphatic asilamino group, C6-C10 aryl group ( The aryl group is substituted with 1 to 5 groups selected from the substituent group α force. Nitrogen, oxygen, and sulfur power 4 to 10-membered heterocyclic group containing the same or different 1 to 3 hetero atoms selected (the heterocyclic group is a substituent group α force) 1 to 5 optionally substituted with selected groups), nitrogen, oxygen and sulfur forces 4-10 membered heterocycles containing the same or different 1 to 3 heteroatoms selected C1-C6 alkyl A group (the heterocyclic ring C1-C6 alkyl group may be substituted with 1 to 5 groups selected from the substituent group α force), nitrogen, oxygen and sulfur force selected the same or different 1 -A 4-10 membered heterocyclic CI-C6 alkylamino group containing 3 heteroatoms (the heterocyclic CI-C6 alkylamino group is substituted with 1 to 5 groups selected from the substituent group a) may be), a ureido group, Shiano group or - indicates Toro group, R 4 is a hydrogen atom, C1-C3 alkyl group, C1-C3 haloalkyl group, a halo NHara X represents a C1-C3 alkylene group (the alkylene group is substituted with one or two of an oxo group, a hydroxyl group, a halogen atom, a C1-C3 alkyl group, or a C3-6 cycloalkylidene group). May be). However, R lx is P (0) (OH), OP (OXOH), CH P (0) (OH)
2 2 2 2 2 2
、 CF P(0)(OH)又は OCH P(0)(OH)を示す場合は、 R1Yは水素原子、ハロゲン原子, CF P (0) (OH) or OCH P (0) (OH), R 1Y represents a hydrogen atom or a halogen atom.
2 2 2 2 2 2 2 2 2 2
又は CI- C6アルキル基を示し、 R1Yが P(〇)(〇H)、 OP(0)(OH)、 CH P(0)(OH)、 CF P Or CI-C6 alkyl group, R 1Y is P (〇) (〇H), OP (0) (OH), CH P (0) (OH), CF P
2 2 2 2 2 2 2 2 2 2
(0)(OH)又は OCH P(0)(OH)を示す場合は、 Rlxは水素原子、ハロゲン原子又は C1When (0) (OH) or OCH P (0) (OH) is indicated, R lx is a hydrogen atom, a halogen atom or C1
2 2 2 2 2 2
-C6アルキル基を示す。 A -C6 alkyl group is shown.
(置換基群ひ) (Substituent group)
ハロゲン原子、 CI- C6アルキル基、 CI- C6ハロアルキル基、 CI- C6ヒドロキシアルキ ル基 (該ヒドロキシアルキル基のアルキル基は、 C1-C6ヒドロキシアルキル基で 1個置 換されていても良い)、 C1-C6アルコキシ C1-C6アルキル基、 C1-C6アミノアルキル基 (該ァミノアルキル基のアミノ基は、 C1-C6アルキル基で 1又は 2個置換されていても 良い)、 CI- C6アルコキシ基、 CI- C6アルキルチオ基、カルボキシ基、 CI- C6アルコ キシカルボ-ル基、水酸基、 C1-C6脂肪族ァシル基、アミノ基、 C1-C6アルキルアミノ 基 (該アルキルアミノ基のアルキル基は、水酸基、 C1-C6ヒドロキシアルキル基、 C3- C10シクロアルキル基、 CI- C6アルコキシ基、アミノ基、 CI- C6アルキルアミノ基、 C1- C6ジアルキルァミノ基又はアミド基で 1若しくは 2個置換されていても良い)、 C3-C10 シクロアルキルアミノ基、 C1-C6ジァルキルアミノ基、 C1-C6アルコキシァミノ基 (該ァ ルコキシァミノ基は、水酸基で 1又は 2個置換されていてもよい)、 C1-C6脂肪族ァシ ルァミノ基、シァノ基、ニトロ基、グァ -ジノ基、 C3- C10シクロアルキル基、 C6- C10ァ リール基、窒素、酸素及び硫黄力 選択される同一若しくは異なった 1-3個のへテロ 原子を含む 4-10員複素環基、窒素、酸素及び硫黄力 選択される同一若しくは異な つた 1-3個のへテロ原子を含む 4-10員複素環 C1-C6アルキルアミノ基、 C1-6力ルバ モイルアルキルアミノ基(該カルバモイルアルキルアミノ基のアルキル基は、ヒドロキシ アルキル基で 1個置換されて 、てもよ 、) ]で表される化合物又はその薬理上許容さ れる塩。 Halogen atom, CI-C6 alkyl group, CI-C6 haloalkyl group, CI-C6 hydroxyalkyl group (the alkyl group of the hydroxyalkyl group may be replaced by one C1-C6 hydroxyalkyl group), C1-C6 alkoxy C1-C6 alkyl group, C1-C6 aminoalkyl group (the amino group of the aminoamino group may be substituted with one or two C1-C6 alkyl groups), CI-C6 alkoxy group, CI -C6 alkylthio group, carboxy group, CI-C6 alkoxycarbonyl group, hydroxyl group, C1-C6 aliphatic acyl group, amino group, C1-C6 alkylamino group (the alkyl group of the alkylamino group is a hydroxyl group, C1- 1 or 2 may be substituted with C6 hydroxyalkyl group, C3-C10 cycloalkyl group, CI-C6 alkoxy group, amino group, CI-C6 alkylamino group, C1-C6 dialkylamino group or amide group) , C3-C10 A chloroalkylamino group, a C1-C6 dialkylamino group, a C1-C6 alkoxyamino group (the alkyloxyamino group may be substituted with one or two hydroxyl groups), a C1-C6 aliphatic acylamino group, a cyano group. Group, nitro group, guazino group, C3-C10 cycloalkyl group, C6-C10 aryl group, nitrogen, oxygen and sulfur powers containing 1-3 identical or different heteroatoms selected 4-10 4-membered heterocyclic ring containing 1 to 3 heteroatoms selected from the same or different hetero atoms, nitrogen, oxygen and sulfur power C1-C6 alkylamino group, C1-6 power rubermoylalkylamino group (The alkyl group of the carbamoylalkylamino group may be substituted with one hydroxyalkyl group.)] Or a pharmacologically acceptable salt thereof.
下記一般式 (Γ ) The following general formula (Γ)
[化 2]
 [Chemical 2]
[Chemical 2]
( ) ()
[式中、 Rlxは、 P(0)(OH)、 OP(0)(OH)、 CH P(0)(OH)、 CF P(0)(OH)又は OCH P [ Where R lx is P (0) (OH), OP (0) (OH), CH P (0) (OH), CF P (0) (OH) or OCH P
2 2 2 2 2 2 2 2 2 2 2 2 2 2
(0)(OH)を示し、 R2及び R3は、それぞれ同一若しくは異なって、水素原子、ハロゲン(0) represents (OH), R 2 and R 3 are the same or different and each represents a hydrogen atom, halogen
2 2
原子、 CI- C6アルキル基、 CI- C6ハロアルキル基、 CI- C6アミノアルキル基(該ァミノ アルキル基のアミノ基は、置換基群 aカゝら選択される基で 1又は 2個置換されていても 良い)、 CI- C6ヒドロキシアルキル基、 CI- C6アルキルチオ基、 CI- C6アルコキシ基、 CI- C6アルコキシィミノ CI- C3アルキル基(該アルコキシィミノ CI- C3アルキル基は、 C1-C3アルキル基で置換されていても良い)、水酸基、 C1-C6脂肪族ァシル基 (該ァ シル基は、置換基群 αカゝら選択される基で置換されていても良い)、アミノ基、 C1-C6 アルキルアミノ基、 CI- C6ジアルキルァミノ基、 CI- C6脂肪族ァシルァミノ基、 C6- C10 ァリール基 (該ァリール基は、置換基群 a力 選択される基で 1乃至 5個置換されてい ても良い)、窒素、酸素及び硫黄力 選択される同一若しくは異なった 1-3個のへテロ 原子を含む 4-10員複素環基 (該複素環基は、置換基群 α力 選択される基で 1乃至 5個置換されていても良い)、窒素、酸素及び硫黄力 選択される同一若しくは異なつ た 1-3個のへテロ原子を含む 4-10員複素環 C1-C6アルキル基 (該複素環 C1-C6アル キル基は、置換基群 α力も選択される基で 1乃至 5個置換されていても良い)、窒素、 酸素及び硫黄力 選択される同一若しくは異なった 1-3個のへテロ原子を含む 4-10 員複素環 CI- C6アルキルアミノ基 (該複素環 CI- C6アルキルアミノ基は、置換基群 a から選択される基で 1乃至 5個置換されていても良い)、ウレイド基、シァノ基又は-ト 口基を示し、 R4は、水素原子、 C1-C3アルキル基、 C1-C3ハロアルキル基、ハロゲン 原子又はアミノ基を示し、 Xは、 C1-C3アルキレン基 (該アルキレン基は、ォキソ基、水 酸基、ハロゲン原子、 C1-C3アルキル基又は C3-6シクロアルキリデン基で 1又は 2個 置換されていてもよい)、を示す。 Atom, CI-C6 alkyl group, CI-C6 haloalkyl group, CI-C6 aminoalkyl group (the amino group of the aminoamino group is substituted with one or two substituents selected from the substituent group a) CI-C6 hydroxyalkyl group, CI-C6 alkylthio group, CI-C6 alkoxy group, CI-C6 alkoxyimino CI-C3 alkyl group (the alkoxyimino CI-C3 alkyl group is C1-C3 alkyl) A hydroxyl group, a C1-C6 aliphatic acyl group (this acyl group may be substituted with a group selected from the substituent group α), an amino group, C1 -C6 alkylamino group, CI-C6 dialkylamino group, CI-C6 aliphatic asilamino group, C6-C10 aryl group (the aryl group is substituted with 1 to 5 substituents selected from the group a force) ), Nitrogen, oxygen and sulfur power 1- or the same selected 4- to 10-membered heterocyclic group containing 3 heteroatoms (the heterocyclic group may be substituted with 1 to 5 substituents selected from the group α group), nitrogen, oxygen and sulfur groups 4- to 10-membered heterocyclic C1-C6 alkyl group containing 1 to 3 heteroatoms that are the same or different selected (the heterocyclic C1-C6 alkyl group is a group in which the substituent group α force is also selected. Optionally substituted with 1 to 5), nitrogen, oxygen and sulfur force 4-10 membered heterocyclic CI-C6 alkylamino group containing 1 to 3 heteroatoms selected from the same or different Heterocyclic CI-C6 alkylamino group may be substituted with 1 to 5 groups selected from the substituent group a), represents a ureido group, a cyano group or a -toco group, and R 4 represents a hydrogen atom. An atom, a C1-C3 alkyl group, a C1-C3 haloalkyl group, a halogen atom or an amino group, X is a C1-C3 alkylene group (the alkylene group is Seo group, water group, a halogen atom, a may) be substituted one or two an C1-C3 alkyl group or a C3-6 cycloalkylidene group.
(置換基群ひ)
ハロゲン原子、 CI- C6アルキル基、 CI- C6ハロアルキル基、 CI- C6ヒドロキシアルキ ル基 (該ヒドロキシアルキル基のアルキル基は、 C1-C6ヒドロキシアルキル基で 1個置 換されていても良い)、 C1-C6アルコキシ C1-C6アルキル基、 C1-C6アミノアルキル基 (該 C1-C6アミノアルキル基のアミノ基は、 C1-6アルキル基で 1若しくは 2個置換され ていても良い)、 C1-C6アルコキシ基、 C1-C6アルキルチオ基、カルボキシ基、 C1-C 6アルコキシカルボ-ル基、水酸基、 C1-C6脂肪族ァシル基、アミノ基、 C1-C6アルキ ルァミノ基 (該アルキルアミノ基のアルキル基は、水酸基、 C1-C6ヒドロキシアルキル 基、 C3- C10シクロアルキル基、 CI- C6アルコキシ基、アミノ基、 CI- C6アルキルアミノ 基、 C1-C6ジアルキルァミノ基又はアミド基で 1若しくは 2個置換されていても良い)、 C3- C10シクロアルキルアミノ基、 CI- C6ジアルキルァミノ基、 CI- C6アルコキシァミノ 基 (該アルコキシァミノ基は、水酸基で 1又は 2個置換されていてもよい)、 - C6脂 肪族ァシルァミノ基、シァノ基、ニトロ基、グァ -ジノ基、 C3-C10シクロアルキル基、 C 6-C10ァリール基、窒素、酸素及び硫黄力 選択される同一若しくは異なった 1-3個 のへテロ原子を含む 4-10員複素環基、窒素、酸素及び硫黄力 選択される同一若し くは異なった 1-3個のへテロ原子を含む 4-10員複素環 C1-C6アルキルアミノ基、 C1- 6力ルバモイルアルキルアミノ基(該カルバモイルアルキルアミノ基のアルキル基は、ヒ ドロキシアルキル基で 1個置換されて 、てもよ 、) ]で表される化合物又はその薬理上 許容される塩。 (Substituent group) Halogen atom, CI-C6 alkyl group, CI-C6 haloalkyl group, CI-C6 hydroxyalkyl group (the alkyl group of the hydroxyalkyl group may be replaced by one C1-C6 hydroxyalkyl group), C1-C6 alkoxy C1-C6 alkyl group, C1-C6 aminoalkyl group (the amino group of the C1-C6 aminoalkyl group may be substituted with one or two C1-6 alkyl groups), C1-C6 An alkoxy group, a C1-C6 alkylthio group, a carboxy group, a C1-C6 alkoxycarbo group, a hydroxyl group, a C1-C6 aliphatic acyl group, an amino group, a C1-C6 alkylamino group (the alkyl group of the alkylamino group is 1 or 2 substituted with hydroxyl group, C1-C6 hydroxyalkyl group, C3-C10 cycloalkyl group, CI-C6 alkoxy group, amino group, CI-C6 alkylamino group, C1-C6 dialkylamino group or amide group You may) C3-C10 cycloalkylamino group, CI-C6 dialkylamino group, CI-C6 alkoxyamino group (the alkoxyamino group may be substituted with one or two hydroxyl groups), -C6 aliphatic Asylamino group, cyano group, nitro group, guazino group, C3-C10 cycloalkyl group, C6-C10 aryl group, nitrogen, oxygen and sulfur forces selected 1-3 identical or different heteroatoms Containing 4-10 membered heterocyclic group, nitrogen, oxygen and sulfur force Selected 4-10 membered heterocyclic C1-C6 alkylamino group containing the same or different 1-3 heteroatoms, C1- Or a pharmacologically acceptable salt thereof. (6) A rubamoylalkylamino group (the alkyl group of the carbamoylalkylamino group may be substituted with one hydroxyalkyl group)].
[3] 請求項 1又は 2において Rlxが、 OCH P(0)(OH)である化合物又はその薬理上許容 [3] The compound according to claim 1 or 2, wherein R lx is OCH P (0) (OH), or a pharmacologically acceptable salt thereof.
2 2 twenty two
される塩。 Salt.
[4] 請求項 1乃至 3において、 R2及び R3がそれぞれ同一若しくは異なって、水素原子、 ハロゲン原子、 C1-C6アルキル基、 C1-C6アルコキシ基、 C1-C6ジァルキルアミノ基 又は - C6脂肪族ァシル基 (該ァシル基は、置換基群 a力も選択される基で置換さ れて 、ても良 、)である化合物又はその薬理上許容される塩。 [4] In Claims 1 to 3, each of R 2 and R 3 is the same or different and is a hydrogen atom, a halogen atom, a C1-C6 alkyl group, a C1-C6 alkoxy group, a C1-C6 dialkylamino group, or a -C6 aliphatic. A compound or a pharmacologically acceptable salt thereof which is an acyl group (the acyl group may be substituted with a group in which substituent group a force is also selected).
[5] 請求項 1乃至 4において、 R2が水素原子又は - C6アルキル基である化合物又は その薬理上許容される塩。 [5] The compound or pharmacologically acceptable salt thereof according to any one of claims 1 to 4, wherein R 2 is a hydrogen atom or a -C6 alkyl group.
[6] 請求項 1乃至 4にお 、て R2が水素原子又はメチル基である化合物又はその薬理上 許容される塩。
[6] The compound or pharmacologically acceptable salt thereof according to any one of claims 1 to 4, wherein R 2 is a hydrogen atom or a methyl group.
[7] 請求項 1乃至 6において、 R3がハロゲン原子、 C1-C6ジァルキルアミノ基、 CI- C6脂 肪族ァシル基 (該ァシル基は、置換基群 aから選択される基で置換されて!、ても良 い)、 C1-C6アルキル基又は C1-C6アルコキシ基である化合物又はその薬理上許容 される塩。 [7] In Claims 1 to 6, R 3 is a halogen atom, a C1-C6 dialkylamino group, a CI-C6 aliphatic acyl group (the acyl group is substituted with a group selected from the substituent group a! Or a pharmacologically acceptable salt thereof, which is a C1-C6 alkyl group or a C1-C6 alkoxy group.
[8] 請求項 1乃至 6において、 R3が C1-C6脂肪族ァシル基、 C1-C6アルキル基又は C1-[8] In Claims 1 to 6, R 3 is a C1-C6 aliphatic acyl group, a C1-C6 alkyl group or C1-
C6アルコキシ基である化合物又はその薬理上許容される塩。 A compound having a C6 alkoxy group or a pharmacologically acceptable salt thereof.
[9] 請求項 1乃至 6において R3が C1-C6アルキル基又は C1-C6アルコキシ基である化 合物又はその薬理上許容される塩。 [9] The compound or pharmacologically acceptable salt thereof according to any one of claims 1 to 6, wherein R 3 is a C1-C6 alkyl group or a C1-C6 alkoxy group.
[10] 請求項 1乃至 6において R3カ^チル基、ェチル基又はメトキシ基である化合物又は その薬理上許容される塩。 [10] The compound or pharmacologically acceptable salt thereof according to any one of [1] to [6], wherein the compound is an R 3 cate group, an ethyl group or a methoxy group.
[11] 請求項 1乃至 10において、 R4が水素原子又はアミノ基である化合物又はその薬理 上許容される塩。 [11] The compound or pharmacologically acceptable salt thereof according to any one of claims 1 to 10, wherein R 4 is a hydrogen atom or an amino group.
[12] 請求項 1乃至 10において、 R4が水素原子である化合物又はその薬理上許容される 塩。 [12] The compound or pharmacologically acceptable salt thereof according to any one of claims 1 to 10, wherein R 4 is a hydrogen atom.
[13] 請求項 1乃至 12において、 Xが C1-C3アルキレン基である化合物又はその薬理上 許容される塩。 [13] The compound or pharmacologically acceptable salt thereof according to any one of claims 1 to 12, wherein X is a C1-C3 alkylene group.
[14] 請求項 1乃至 12において、 Xカ チレン基である化合物又はその薬理上許容される 塩。 [14] The compound or the pharmacologically acceptable salt thereof according to any one of [1] to [12], wherein the compound is an X alkylene group.
[15] 下記一般式 (II) [15] The following general formula (II)
[化 3] [Chemical 3]

[式中、 R1は、 P(0)(OH)、 OP(0)(OH)、 CH P(0)(OH)、 CF P(0)(OH)又は OCH P( [Wherein R 1 is P (0) (OH), OP (0) (OH), CH P (0) (OH), CF P (0) (OH) or OCH P (
2 2 2 2 2 2 2 2 2 2 2 2 2 2
0)(OH)を示し、 R2及び R3は、それぞれ同一若しくは異なって、水素原子、ハロゲン原0) (OH), R 2 and R 3 are the same or different and each represents a hydrogen atom or a halogen atom.
2 2
子、 CI- C6アルキル基、 CI- C6ノヽロアルキル基、 CI- C6アミノアルキル基(該アミノア
ルキル基のアミノ基は、置換基群 a力 選択される基で 1又は 2個置換されていても 良い)、 CI- C6ヒドロキシアルキル基、 CI- C6アルキルチオ基、 CI- C6アルコキシ基、 CI- C6アルコキシィミノ CI- C3アルキル基(該アルコキシィミノ CI- C3アルキル基は、 C1-C3アルキル基で置換されていても良い)、水酸基、 C1-C6脂肪族ァシル基 (該ァ シル基は、置換基群 αカゝら選択される基で置換されていても良い)、アミノ基、 C1-C6 アルキルアミノ基、 CI- C6ジアルキルァミノ基、 CI- C6脂肪族ァシルァミノ基、 C6- C10 ァリール基 (該ァリール基は、置換基群 a力 選択される基で 1乃至 5個置換されてい ても良い)、窒素、酸素及び硫黄力 選択される同一若しくは異なった 1-3個のへテロ 原子を含む 4-10員複素環基 (該複素環基は、置換基群 α力 選択される基で 1乃至 5個置換されていても良い)、窒素、酸素及び硫黄力 選択される同一若しくは異なつ た 1-3個のへテロ原子を含む 4-10員複素環 C1-C6アルキル基 (該複素環 C1-C6アル キル基は、置換基群 α力も選択される基で 1乃至 5個置換されていても良い)、窒素、 酸素及び硫黄力 選択される同一若しくは異なった 1-3個のへテロ原子を含む 4-10 員複素環 CI- C6アルキルアミノ基 (該複素環 CI- C6アルキルアミノ基は、置換基群 a カゝら選択される基で 1乃至 5個置換されていても良い)、シァノ基及び-トロ基を示し、 R4は、水素原子、 C1-C3アルキル基、 C1-C3ハロアルキル基、ハロゲン原子又はアミ ノ基を示し、 Xは、 C1-C3アルキレン基 (該アルキレン基は、 C1-C3アルキル基又は C 3-6シクロアルキリデン基で置換されていてもよい)、を示す。 CI-C6 alkyl group, CI-C6 neuroalkyl group, CI-C6 aminoalkyl group (the amino group) The amino group of the alkyl group may be substituted with one or two groups selected from the substituent group a) CI-C6 hydroxyalkyl group, CI-C6 alkylthio group, CI-C6 alkoxy group, CI- C6 alkoxyimino CI-C3 alkyl group (the alkoxyimino CI-C3 alkyl group may be substituted with a C1-C3 alkyl group), a hydroxyl group, a C1-C6 aliphatic acyl group (the acyl group is The substituent group may be substituted with a group selected from α group, amino group, C1-C6 alkylamino group, CI-C6 dialkylamino group, CI-C6 aliphatic acylamino group, C6-C10 Aryl group (the aryl group may be substituted with 1 to 5 groups selected by the substituent group a force), nitrogen, oxygen and sulfur force selected 1-3 identical or different hetero 4- to 10-membered heterocyclic group containing an atom (the heterocyclic group is a group selected from the substituent group α force 1 4 to 10-membered heterocyclic C1-C6 alkyl group containing 1 to 3 heteroatoms, the same or different, selected from nitrogen, oxygen and sulfur power (optionally substituted up to 5) The C1-C6 alkyl group may be substituted with 1 to 5 substituents with a group that also selects the α group of the substituent group), nitrogen, oxygen, and sulfur groups. 4- to 10-membered heterocyclic CI-C6 alkylamino group containing an atom (the heterocyclic CI-C6 alkylamino group may be substituted with 1 to 5 groups selected from substituent group a) , Shiano group and - indicates Toro group, R 4 is a hydrogen atom, C1-C3 alkyl group, C1-C3 haloalkyl group, a halogen atom or an amino group, X is C1-C3 alkylene group (the alkylene group Represents a C1-C3 alkyl group or a C3-6 cycloalkylidene group).
(置換基群ひ) (Substituent group)
ハロゲン原子、 CI- C6アルキル基、 CI- C6ハロアルキル基、 CI- C6アミノアルキル 基 (該ァミノアルキル基のアミノ基は、 C1-C6アルキル基で 1又は 2個置換されていて も良い)、 CI- C6アルコキシ基、 CI- C6アルキルチオ基、カルボキシ基、 CI- C6アルコ キシカルボ-ル基、水酸基、 C1-C6脂肪族ァシル基、アミノ基、 C1-C6アルキルアミノ 基、 CI- C6ジァルキルアミノ基、 CI- C6脂肪族ァシルァミノ基、シァノ基、ニトロ基、 C3 - C10シクロアルキル基、 C6-C10ァリール基、窒素、酸素及び硫黄から選択される同 一若しくは異なった 1-3個のへテロ原子を含む 4-10員複素環基、窒素、酸素及び硫 黄力も選択される同一若しくは異なった 1-3個のへテロ原子を含む 4-10員複素環 C1 -C6アルキルアミノ基、 C1-6力ルバモイルアルキルアミノ基]で表される化合物又はそ
の薬理上許容される塩。 A halogen atom, a CI-C6 alkyl group, a CI-C6 haloalkyl group, a CI-C6 aminoalkyl group (the amino group of the aminoamino group may be substituted with one or two C1-C6 alkyl groups), CI- C6 alkoxy group, CI-C6 alkylthio group, carboxy group, CI-C6 alkoxycarbonyl group, hydroxyl group, C1-C6 aliphatic acyl group, amino group, C1-C6 alkylamino group, CI-C6 dialkylamino group, CI- Containing 1-3 identical or different heteroatoms selected from C6 aliphatic asylamino, cyano, nitro, C3-C10 cycloalkyl, C6-C10 aryl, nitrogen, oxygen and sulfur 4 -10-membered heterocyclic group, nitrogen, oxygen and sulfur power are also selected 4 to 10-membered heterocyclic ring containing 1 to 3 heteroatoms C1-C6 alkylamino group, C1-6 rubamoyl A compound represented by an alkylamino group] or The pharmacologically acceptable salt of
[16] {[7- (ァミノカルボ-ル) -8H-インデノ [1 ,2-d][l ,3]チアゾール -4-ィル]ォキシ }メチルホ スホン酸、 {[7- (シクロプロピルカルボ-ル) -8H-インデノ [l,2-d][l,3]チアゾール -4-ィ ル]ォキシ }メチルホスホン酸又はその薬理上許容される塩。 [16] {[7- (Aminocarbol) -8H-indeno [1,2-d] [l, 3] thiazol-4-yl] oxy} methylphosphonic acid, {[7- (cyclopropylcarbo- -8H-indeno [l, 2-d] [l, 3] thiazol-4-yl] oxy} methylphosphonic acid or a pharmacologically acceptable salt thereof.
[17] [(7-フルオロ- 8H-インデノ [l,2-d][l,3]チアゾール -4-ィル)ォキシ]メチルホスホン酸 、 [(7-ァセチル -8H-インデノ [1 ,2-d][l ,3]チアゾール -4-ィル)ォキシ]メチルホスホン酸 、 {[7- (ジメチルァミノ) -8H-インデノ [1 ,2-d][l ,3]チアゾール -4-ィル]ォキシ }メチルホス ホン酸、({7- [(メチルァミノ)カルボ-ル] -8H-インデノ [1 ,2-d][l ,3]チアゾール -4-ィル } ォキシ)メチルホスホン酸又はその薬理上許容される塩。 [17] [(7-Fluoro-8H-indeno [l, 2-d] [l, 3] thiazol-4-yl) oxy] methylphosphonic acid, [(7-acetyl-8H-indeno [1,2- d] [l, 3] thiazol-4-yl) oxy] methylphosphonic acid, {[7- (dimethylamino) -8H-indeno [1,2-d] [l, 3] thiazol-4-yl] oxy } Methylphosphonic acid, ({7-[(methylamino) carbol] -8H-indeno [1,2-d] [l, 3] thiazol-4-yl} oxy) methylphosphonic acid or its pharmacologically acceptable Salt.
[18] [(7- {[(2-ァミノ- 2-ォキソェチル)ァミノ]カルボ-ル}- 8H-インデノ [1,2- d][l,3]チアゾ ール -4-ィル)ォキシ]メチルホスホン酸、 {[7-({[(lS)-2-ァミノ- 1-メチル - 2-ォキソェチ ル]アミノ}カルボ-ル) -8H-インデノ [l,2-d][l,3]チアゾール -4-ィル]ォキシ }メチルホス ホン酸、 {[7-({[(lR)- 2-ァミノ- 1-メチル - 2-ォキソェチル]アミノ}カルボ-ル) -8H-インデ ノ [l,2-d][l,3]チアゾール -4-ィル]ォキシ }メチルホスホン酸、 {[7-({[(lS)-l- (アミノカル ボ-ル )-3-メチルブチル]アミノ}カルボ-ル) -8H-インデノ [l,2-d][l,3]チアゾール -4- ィル]ォキシ }メチルホスホン酸、 {[7-({[(lS)-2-ァミノ- 1- (ヒドロキシメチル) - 2-ォキソェ チル]アミノ}カルボ-ル) -8H-インデノ [l,2-d][l,3]チアゾール -4-ィル]ォキシ }メチルホ スホン酸又はその薬理上許容される塩。 [18] [(7- {[(2-Amino-2-oxoethyl) amino] carbol} -8H-indeno [1,2-d] [l, 3] thiazol-4-yl) oxy ] Methylphosphonic acid, {[7-({[(lS) -2-amino-1-methyl-2-oxo] amino} carbol) -8H-indeno [l, 2-d] [l, 3] Thiazole-4-yl] oxy} methylphosphonic acid, {[7-({[(lR) -2-amino-1-methyl-2-oxoethyl] amino} carbol) -8H-indeno [l, 2-d] [l, 3] thiazol-4-yl] oxy} methylphosphonic acid, {[7-({[(lS) -l- (aminocarbole) -3-methylbutyl] amino} carbole ) -8H-indeno [l, 2-d] [l, 3] thiazole-4-yl] oxy} methylphosphonic acid, {[7-({[(lS) -2-amino-1- (hydroxymethyl) -2-oxoethyl] amino} carbol) -8H-indeno [l, 2-d] [l, 3] thiazol-4-yl] oxy} methylphosphonic acid or a pharmacologically acceptable salt thereof.
[19] [(7-メトキシ -8H-インデノ [l,2-d][l,3]チアゾール -4-ィル)ォキシ]メチルホスホン酸、 [(7-ェチル -8H-インデノ [1 ,2-d][l ,3]チアゾール -4-ィル)ォキシ]メチルホスホン酸、 [( 6,7-ジメチル-81"[-ィンデノ[1,2-(1][1,3]チァゾール-4-ィル)ォキシ]メチルホスホン酸 又はその薬理上許容される塩。 [19] [(7-Methoxy-8H-indeno [l, 2-d] [l, 3] thiazol-4-yl) oxy] methylphosphonic acid, [(7-ethyl-8H-indeno [1, 2- d] [l, 3] thiazol-4-yl) oxy] methylphosphonic acid, [(6,7-dimethyl-81 "[-indeno [1,2- (1] [1,3] thiazol-4-yl L) oxy] methylphosphonic acid or a pharmacologically acceptable salt thereof.
[20] [(7-ブロモ -6-メチル -8H-インデノ [1 ,2-d][l ,3]チアゾール -4-ィル)ォキシ]メチルホ スホン酸、 [(6-メトキシ -8H-インデノ [l,2-d][l,3]チアゾール -4-ィル)ォキシ]メチルホ スホン酸、 [(7-ブロモ -6-メトキシ -8H-インデノ [l,2-d][l,3]チアゾール -4-ィル)ォキシ] メチルホスホン酸又はその薬理上許容される塩。 [20] [(7-Bromo-6-methyl-8H-indeno [1,2-d] [l, 3] thiazol-4-yl) oxy] methylphosphonic acid, [(6-methoxy-8H-indeno [l, 2-d] [l, 3] thiazol-4-yl) oxy] methylphosphonic acid, [(7-bromo-6-methoxy-8H-indeno [l, 2-d] [l, 3] Thiazole-4-yl) oxy] methylphosphonic acid or a pharmacologically acceptable salt thereof.
[21] 請求項 1乃至 20に記載の化合物又はその薬理上許容される塩を含有する医薬。 [21] A medicament comprising the compound according to any one of [1] to [20] or a pharmacologically acceptable salt thereof.
[22] 請求項 1乃至 20に記載の化合物又はその薬理上許容される塩を含有する、糖尿
病の治療薬又は予防薬。 [22] A diabetes comprising the compound according to any one of claims 1 to 20 or a pharmacologically acceptable salt thereof. A therapeutic or prophylactic agent for disease.
[23] 請求項 1乃至 20に記載の化合物又はその薬理上許容される塩を含有する、高血 糖症、肥満症、耐糖能不全状態、インスリン抵抗性非耐糖能不全状態、糖尿病合併 症、妊娠糖尿病、ステロイド糖尿病若しくは外科的糖尿病様病態の治療薬又は予防 薬。 [23] Hyperglycaemia, obesity, impaired glucose tolerance, insulin resistant non-glucose intolerance, diabetes complications, comprising the compound according to claim 1 or 20 or a pharmacologically acceptable salt thereof, Therapeutic or prophylactic agent for gestational diabetes, steroid diabetes or surgical diabetes-like condition.
[24] 請求項 1乃至 20に記載の化合物又はその薬理上許容される塩を含有する、高脂 血症、高血圧症、膝炎、脂肪肝、動脈硬化症、クッシング症候群若しくは糖原病の治 療薬又は予防薬。 [24] Treatment of hyperlipidemia, hypertension, knee inflammation, fatty liver, arteriosclerosis, Cushing's syndrome or glycogenosis, comprising the compound according to claim 1 or 20 or a pharmacologically acceptable salt thereof. Medicinal or preventive medicine.
[25] 請求項 1乃至 20に記載の化合物又はその薬理上許容される塩を含有する、 FBPas e阻害剤。 [25] An FBPase inhibitor comprising the compound according to any one of claims 1 to 20 or a pharmacologically acceptable salt thereof.
[26] 糖尿病の治療又は予防のための医薬を製造するための、請求項 1乃至 20に記載 の化合物又はその薬理上許容される塩の使用。 [26] Use of the compound according to claim 1 or a pharmacologically acceptable salt thereof for the manufacture of a medicament for treating or preventing diabetes.
[27] 請求項 1乃至 20に記載の化合物又はその薬理上許容される塩の薬理的な有効量 を温血動物に投与する、糖尿病の予防若しくは治療方法。
[27] A method for preventing or treating diabetes, comprising administering to a warm-blooded animal a pharmacologically effective amount of the compound according to claim 1 to 20 or a pharmacologically acceptable salt thereof.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2005089010 | 2005-03-25 | ||
| JP2005-089010 | 2005-03-25 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2006104030A1 true WO2006104030A1 (en) | 2006-10-05 |
Family
ID=37053287
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2006/305940 WO2006104030A1 (en) | 2005-03-25 | 2006-03-24 | Thiazole compound |
Country Status (2)
| Country | Link |
|---|---|
| TW (1) | TW200722432A (en) |
| WO (1) | WO2006104030A1 (en) |
Cited By (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008017381A1 (en) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, processes for preparing them, medicaments comprising these compounds, and their use |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| EP2058308A1 (en) | 2007-11-12 | 2009-05-13 | Merck Sante | Benzimidazoledihydrothiadiazinone derivatives used as fructose-1,6-biphosphatase inhibitors and pharmaceutical compositions containing same. |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1994018212A1 (en) * | 1993-02-15 | 1994-08-18 | Otsuka Pharmaceutical Factory, Inc. | Phosphonic diester derivative |
| JP2003503486A (en) * | 1999-06-30 | 2003-01-28 | シナプティック・ファーマスーティカル・コーポレーション | Selective NPY (Y5) antagonist |
| JP2004018489A (en) * | 2002-06-19 | 2004-01-22 | Otsuka Pharmaceut Factory Inc | Acat-1 inhibitor |
-
2006
- 2006-03-24 TW TW095110187A patent/TW200722432A/en unknown
- 2006-03-24 WO PCT/JP2006/305940 patent/WO2006104030A1/en active Application Filing
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1994018212A1 (en) * | 1993-02-15 | 1994-08-18 | Otsuka Pharmaceutical Factory, Inc. | Phosphonic diester derivative |
| JP2003503486A (en) * | 1999-06-30 | 2003-01-28 | シナプティック・ファーマスーティカル・コーポレーション | Selective NPY (Y5) antagonist |
| JP2004018489A (en) * | 2002-06-19 | 2004-01-22 | Otsuka Pharmaceut Factory Inc | Acat-1 inhibitor |
Cited By (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008017381A1 (en) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, processes for preparing them, medicaments comprising these compounds, and their use |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| EP2058308A1 (en) | 2007-11-12 | 2009-05-13 | Merck Sante | Benzimidazoledihydrothiadiazinone derivatives used as fructose-1,6-biphosphatase inhibitors and pharmaceutical compositions containing same. |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| TW200722432A (en) | 2007-06-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2006104030A1 (en) | Thiazole compound | |
| TWI774712B (en) | Modulator of cystic fibrosis transmembrane conductance regulator, pharmaceutical compositions, methods of treatment, and process for making the modulator | |
| TWI718207B (en) | Benzofuran derivatives, preparation method thereof and medicinal application thereof | |
| WO2021052499A1 (en) | Fused pyridone compound, and preparation method therefor and use thereof | |
| CN112805267B (en) | Carboxamide and sulfonamide derivatives as TEAD modulators | |
| TWI518085B (en) | Prodrug of substituted polycyclic carbamoyl pyridone derivative | |
| CN102459172B (en) | Tetracyclic compound | |
| CN113286794A (en) | KRAS mutein inhibitors | |
| EP3555076B1 (en) | Novel phenyl propionic acid derivatives and uses thereof | |
| CN108929263B (en) | Arylamide Kv2.1 inhibitor and preparation method, pharmaceutical composition and application thereof | |
| TWI700284B (en) | Nitrogen-containing tricyclic derivertives having hiv reprication inhibitory activity | |
| WO2022214102A1 (en) | Heterocyclic compound acting as kras g12d inhibitor | |
| WO2018171633A1 (en) | Macrocyclic derivative of pyrazol[3,4-d]pyrimidin-3-one, pharmaceutical composition and use thereof | |
| AU2020215380A1 (en) | Heterocyclic compound and use thereof | |
| CN103003233A (en) | Novel hydroxamic acid derivative | |
| WO2007020936A1 (en) | Bicyclo heterocyclic compound having antifungal action | |
| EP3884939B1 (en) | 3-phosphoglycerate dehydrogenase inhibitors and uses thereof | |
| CN105939997A (en) | Pyrazole derivatives and uses thereof as inhibitors of DLK | |
| WO2019057123A1 (en) | Polycyclic compound acting as ido inhibitor and/or ido-hdac dual inhibitor | |
| CA2868321A1 (en) | Kynurenine-3-monooxygenase inhibitors, pharmaceutical compositions, and methods of use thereof | |
| WO2022268230A1 (en) | Compound as kif18a inhibitor | |
| WO2017024180A1 (en) | Vinyl fluoride cyclopropyl fused thiazin-2-amine compounds as beta-secretase inhibitors and methods of use | |
| CN102015636A (en) | Azetidines and cyclobutanes as histamine H3 receptor antagonists | |
| TW202229300A (en) | Modulators of cystic fibrosis transmembrane conductance regulator | |
| CN101981031A (en) | Tricyclic compounds having corticotropin-releasing factor antagonistic activity and pharmaceutical compositions containing them |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| NENP | Non-entry into the national phase |
Ref country code: RU |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 06729888 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: JP |