WO2006090915A1 - Pyridyl acetic acid compounds - Google Patents
Pyridyl acetic acid compounds Download PDFInfo
- Publication number
- WO2006090915A1 WO2006090915A1 PCT/JP2006/304177 JP2006304177W WO2006090915A1 WO 2006090915 A1 WO2006090915 A1 WO 2006090915A1 JP 2006304177 W JP2006304177 W JP 2006304177W WO 2006090915 A1 WO2006090915 A1 WO 2006090915A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- group
- methyl
- optionally substituted
- methylphenyl
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/72—Nitrogen atoms
- C07D213/75—Amino or imino radicals, acylated by carboxylic or carbonic acids, or by sulfur or nitrogen analogues thereof, e.g. carbamates
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/48—Drugs for disorders of the endocrine system of the pancreatic hormones
- A61P5/50—Drugs for disorders of the endocrine system of the pancreatic hormones for increasing or potentiating the activity of insulin
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/24—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D213/54—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/55—Acids; Esters
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/24—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D213/54—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/56—Amides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D453/00—Heterocyclic compounds containing quinuclidine or iso-quinuclidine ring systems, e.g. quinine alkaloids
- C07D453/02—Heterocyclic compounds containing quinuclidine or iso-quinuclidine ring systems, e.g. quinine alkaloids containing not further condensed quinuclidine ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/10—Spiro-condensed systems
Definitions
- DPP-IV Dipeptidyl dipeptidase-IV
- DPP-IV is serine protease that specifically binds with a peptide containing proline (or alanine) at the 2nd from the N-terminal and cleaves the C-terminal side of the proline (or alanine) to produce dipeptide.
- DPP-IV has been shown to be the same molecule as CD26, and reported to be also involved in the immune system.
- DPP-IV While the role of DPP-IV in mammals has not been entirely clarified, it is- considered to play an important role in the metabolism of neuropeptides, activation of T cells, adhesion of cancer cells to endothelial cells, invasion of HIV into cells and the like. Particularly, from the aspect of glycometabolism, DPP-IV is involved in the inactivation of GLP-I (glucagon-like peptide-1) and GIP (Gastric inhibitory peptide/Glucose-dependent insulinotropic peptide) , which are incretins.
- GLP-I glucagon-like peptide-1
- GIP Gastric inhibitory peptide/Glucose-dependent insulinotropic peptide
- X is N or CR 5 (wherein R 5 is hydrogen or lower alkyl) ;
- R 1 and R 2 are independently hydrogen or lower alkyl;
- R 3 is heterocyclic group or aryl, each optionally substituted by lower alkyl and the like;
- R 4 is lower alkyl and the like, or a salt thereof, has been reported (see WO03/068757) .
- R 1 is a Q L - 6 alkyl group optionally substituted by a C3- 1 0 cycloalkyl group
- R 2 is a C2-6 alkyl group
- R 3 is a hydrogen atom, a Ci- 6 alkyl group or a halogen atom
- X is -OR 6 or -NR 4 R 5 wherein R 4 and R 6 are each independently a hydrogen atom, an optionally substituted hydrocarbon group or an optionally substituted heterocyclic group, R 5 is an optionally substituted hydrocarbon group, an optionally substituted heterocyclic group or an optionally substituted hydroxy group, or R 4 and R 5 optionally form, together with the adjacent nitrogen atom, an optionally substituted nitrogen-containing heterocycle, or a salt thereof
- the pharmaceutical agent of the aforementioned 7) which is an agent for the prophylaxis or treatment of diabetes, diabetic complications, impaired glucose tolerance or obesity; 9) a peptidase inhibitor comprising compound (I) or a prodrug thereof;
- compound (I) or a prodrug thereof for the production of an agent for the prophylaxis or treatment of diabetes, diabetic complications, impaired glucose tolerance or obesity;
- R 1 , R 2 and R 3 are each as defined above, or a salt thereof, to hydrolysis and deprotection; and the like.
- the compound of the present invention has a superior peptidase inhibitory action and is useful as an agent for the prophylaxis or treatment of diabetes and the like.
- Ci-6 alkyl group of the "C ⁇ -6 alkyl group optionally substituted by a C3-10 cycloalkyl group” for R 1 for example, methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec- butyl, tert-butyl, pentyl, isopentyl, neopentyl, 1-ethylpropyl, hexyl, isohexyl, 1, 1-dimethylbutyl, 2, 2-dimethylbutyl, 3,3- dimethylbutyl, 2-ethylbutyl and the like can be mentioned.
- C3- 1 0 cycloalkyl group for example, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, bicyclo [2.2.l]heptyl, bicyclo[2.2.2]octyl, bicyclo [3.2. l]octyl, bicyclo [3.2.2]nonyl, bicyclo[3.3.1]nonyl, bicyclo [4.2. ljnonyl, bicyclo[4.3.1] decyl, adamantyl and the like can be mentioned.
- R 1 is preferably a C 3 _ 6 alkyl group, more preferably a branched C 3 _ 6 alkyl group, particularly preferably isobutyl or neopentyl .
- R 2 for example, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl, tert-butyl, pentyl, isopentyl, neopentyl, 1-ethylpropyl, hexyl, isohexyl, 1,1- dimethylbutyl, 2, 2-dimethylbutyl, 3, 3-dimethylbutyl, 2- ethylbutyl and the like can be mentioned.
- R 2 is preferably ethyl or isobutyl.
- C 2 - 10 alkynyl group for example, ethynyl, 1- propynyl, 2-propynyl, 1-butynyl, 2-butynyl, 3-butynyl, 1- pentynyl, 2-pentynyl, 3-pentynyl, 4-pentynyl, 1-hexynyl, 2- hexynyl, 3-hexynyl, 4-hexynyl, 5-hexynyl, 1-heptynyl, 1-octynyl and the like can be mentioned.
- C 3 - 10 cycloalkyl group for example, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, bicyclo[2.2.1]heptyl, bicyclo [2.2.2]octyl, bicyclo [3.2.l]octyl, bicyclo [3.2.2 ]nonyl, bicyclo [3.3.1] nonyl, bicyclo [4.2. l]nonyl, bicyclo [4.3.1]decyl, adamantyl and the like can be mentioned.
- C 4 -10 cycloalkadienyl group for example, 2,4- cyclopentadien-1-yl, 2, 4-cyclohexadien-l-yl, 2, 5-cyclohexadien- 1-yl and the like can be mentioned.
- C3- 1 0 cycloalkyl group, C3-10 cycloalkenyl group and C 4 - 10 cycloalkadienyl group are each optionally condensed with a benzene ring, and, for example, indanyl, dihydronaphthyl, tetrahydronaphthyl, fluorenyl and the like can be mentioned.
- C ⁇ - 1 4 aryl group for example, phenyl, naphthyl, anthryl, phenanthryl, acenaphthylenyl, biphenylyl and the like can be mentioned. Of these, phenyl, 1-naphthyl, 2-naphthyl and the like are preferable.
- C7- 13 aralkyi group for example, benzyl, phenethyl, naphthylmethy1, biphenylylmethyl and the like can be mentioned.
- C 8 -i3 arylalkenyl group for example, styryl and the like can be mentioned.
- C3-10 cycloalkyl-Ci_ 5 alkyl group for example, cyclohexylmethyl and the like can be mentioned.
- Ci_io alkyl group, C2- 1 0 alkenyl group and C2- 1 0 alkynyl group optionally have 1 to 3 substituents at substitutable positions.
- substituents for example,
- an aromatic heterocyclic group e.g., thienyl, furyl, pyridyl, oxazolyl, thiazolyl, tetrazolyl, oxadiazolyl, pyrazinyl, quinolyl, indolyl
- an aromatic heterocyclic group e.g., thienyl, furyl, pyridyl, oxazolyl, thiazolyl, tetrazolyl, oxadiazolyl, pyrazinyl, quinolyl, indolyl
- 1 to 3 substituents selected from a carboxyl group, a carbamoyl group, a thiocarbamoyl group and a Ci- 6 alkoxy-carbonyl group (e.g., methoxycarbonyl, ethoxycarbonyl, propoxycarbonyl, tert- butoxycarbonyl) ;
- substituent (s) selected from a C ⁇ _6 alkyl group (e.g., methyl, ethyl), a Ci- 6 alkyl-carbonyl group (e.g., acetyl, isobutanoyl, isopentanoyl) and a Ci- 6 alkoxy-carbonyl group (e.g., methoxycarbonyl, ethoxycarbonyl
- Ci-6 alkylsulfonylamino group e.g., methylsulfonylamino
- Ci_6 alkoxy-carbonyl group e.g., methoxycarbonyl, ethoxycarbonyl, propoxycarbonyl, tert-butoxycarbonyl
- Ci_6 alkylsulfonyl group e.g., methylsulfonyl, ethylsulfonyl
- a carbamoyl group optionally mono- or di-substituted by Ci-6 alkyl group (s) (e.g., methyl, ethyl) optionally substituted by 1 to 3 halogen atoms (e.g., fluorine, chlorine, bromine, iodine)
- a thiocarbamoyl group optionally mono- or di-substituted by Ci-6 alkyl group (s) (e.g., methyl, ethyl) optionally substituted by 1 to 3 halogen atoms (e.g., fluorine, chlorine, bromine, iodine)
- a thiocarbamoyl group optionally mono- or di-substituted by Ci-6 alkyl group (s) (e.g., methyl, e
- Ci_ 6 alkyl group e.g., methyl, ethyl
- halogen atoms e.g., fluorine, chlorine, bromine, iodine
- a C2-6 alkenyloxy group e.g., ethenyloxy
- 1 to 3 halogen atoms e.g., fluorine, chlorine, bromine, iodine
- a C 6 - I4 aryloxy group e.g., phenyloxy, naphthyloxy
- Ci-6 alkyl-carbonyloxy group e.g., acetyloxy, tert- butylcarbonyloxy
- (22) a thiol group; (23) a Ci-6 alkylthio group (e.g., methylthio, ethylthio) optionally substituted by 1 to 3 halogen atoms (e.g., fluorine, chlorine, bromine, iodine) ;
- a nitroso group (31) a halogen atom (e.g., fluorine, chlorine, bromine, iodine); (32) a Ci-6 alkylsulfinyl group (e.g., methylsulfinyl) ; and the like can be mentioned.
- a halogen atom e.g., fluorine, chlorine, bromine, iodine
- a Ci-6 alkylsulfinyl group e.g., methylsulfinyl
- a Ci- 6 alkyl group e.g., methyl, ethyl
- substituents selected from a halogen atom (e.g., fluorine, chlorine, bromine, iodine), a carboxyl group, a Q -6 alkoxy-carbonyl group (e.g., methoxycarbonyl, ethoxycarbonyl) , a carbamoyl group and a Ci-s alkoxy group (e.g., methoxy)
- a C 2 - 6 alkenyl group e.g., ethenyl, 1-propenyl
- substituents for example, those exemplarily recited for the substituents for the aforementioned Ci-io alkyl group and the like
- a halogen atom e.g., fluorine, chlorine, bromine, iodine
- a carboxyl group e.g., methyl, ethyl
- substituents
- pyrrolyl e.g., 1-pyrrolyl, 2-pyrrolyl, 3- pyrrolyl
- imidazolyl e.g., 1-imidazolyl, 2-imidazolyl, 4- imidazolyl, 5-imidazolyl
- pyrazolyl e.g., 1-pyrazolyl, 3- pyrazolyl, 4-pyrazolyl
- thiazolyl e.g., 2-thiazolyl, 4- thiazolyl, 5-thiazolyl
- isothiazolyl e.g., 2-thiazolyl, 4- thiazolyl, 5-thiazolyl
- isoxazolyl e.g., 3- isoxazolyl, 4-isoxazolyl, 5-isoxazolyl)
- oxadiazolyl e
- tetrazolyl e.g., tetrazol-1-yl, tetrazol- 5-yl
- fused aromatic heterocyclic groups such as quinolyl (e.g., 2- quinolyl, 3-quinolyl, 4-quinolyl) , quinazolyl (e.g., 2- quinazolyl, 4-quinazolyl) , quinoxalyl (e.g., 2-quinoxalyl) , benzofuryl (e.g., 2-benzofuryl, 3-benzofuryl) , benzothienyl (e.g., 2-benzothienyl, 3-benzothienyl) , benzoxazolyl (e.g., 2- benzoxazolyl) , benzothiazolyl (e.g., 2-benzothienyl, 3-benzothienyl) , benzoxazolyl (e.g., 2- benzoxazolyl) , benzothiazolyl (
- nitrogen-containing heterocycle of the "optionally substituted nitrogen-containing heterocycle” formed by R 4 and R 5 together with the adjacent nitrogen atom
- a 5- to 7-membered nitrogen-containing heterocycle containing, as a ring-constituting atom besides carbon atoms, at least one nitrogen atom and optionally further containing 1 or 2 heteroatoms selected from an oxygen atom, a sulfur atom and a nitrogen atom can be mentioned.
- the "nitrogen-containing heterocycle” pyrrolidine, imidazolidine, pyrazolidine, piperidine, piperazine, morpholine, thiomorpholine, oxopiperazine, homopiperidine, homopiperazine, thiazolidine, dihydroindole (e.g., 2, 3-dihydroindole) , dihydroisoindole (e.g., 1,3-dihydroisoindole) , tetrahydroquinoline (e.g., 1,2,3,4- tetrahydroquinoline) , triazaspirodecanedione (e.g., 1,3,8- triazaspiro [4.5] decane-2, 4-dione) , hexahydropyrazinooxazinone (e.g., hexahydropyrazino[2, 1-c] [1, 4] oxazin-4 (3H) -one)
- the nitrogen-containing heterocycle optionally has 1 to 3 (preferably 1 or 2) substituents at substitutable positions.
- substituents for example, those exemplarily recited for the substituents for the C3- 1 0 cycloalkyl group, which is exemplarily recited for the aforementioned "hydrocarbon group" of the "optionally substituted hydrocarbon group” for R 4 or R 5 , can be mentioned.
- hydrocarbon group of the "optionally substituted hydrocarbon group” for R 4 or R 5
- R 5 for example, a hydroxy group optionally substituted by a hydrocarbon group can be mentioned.
- hydrocarbon group here, those exemplarily recited for the aforementioned "hydrocarbon group” of the “optionally substituted hydrocarbon group” for R 4 , R 5 or R 6 can be mentioned.
- the "optionally substituted hydroxy group” is preferably a hydroxy group, a Ci_ 6 alkoxy group (e.g., methoxy, ethoxy) and the like.
- Ci-io alkyl group preferably methyl, ethyl
- a Ci-io alkyl group optionally substituted by 1 to 3 substituents selected from a Ci-6 alkoxy- carbonyl group, a Ci-s alkoxy group and a heterocyclic group (e.g., 2-thienyl) ;
- Ci-6 alkoxy group optionally substituted by 1 to 3 halogen atoms (e.g., fluorine, chlorine, bromine, iodine);
- a C 7 - I3 aralkyl group (preferably benzyl) optionally substituted by 1 to 3 Ci_ 5 alkylsulfonyl groups; (6) a heterocyclic group (e.g., pyridyl, isoxazolyl, pyrazolyl, thiadiazolyl, benzothiadiazolyl, oxodihydropyrazolyl, azabicyclooctyl, pyrrolidinyl) optionally substituted by 1 to 3 substituents selected from a C ⁇ _6 alkyl group, a Cs-i4 aryl group, a C7-13 aralkyl group and a Ci-6 alkoxy-carbonyl group; and (7) a Ci- 6 alkoxy group (only for R 5 ) ; are preferable.
- a C 7 - I3 aralkyl group preferably benzyl
- a heterocyclic group e.g., pyridyl, is
- nitrogen-containing heterocycle of the "optionally substituted nitrogen-containing heterocycle” formed by R 4 and R 5 together with the adjacent nitrogen atom
- nitrogen-containing heterocycle for example, pyrrolidine, piperidine, piperazine, morpholine, homopiperidine, homopiperazine, thiazolidine, dihydroindole, dihydroisoindole, tetrahydroquinoline, triazaspirodecanedione, hexahydropyrazinooxazinone and the like are preferable.
- substituents for the nitrogen-containing heterocycle for example, pyrrolidine, piperidine, piperazine, morpholine, homopiperidine, homopiperazine, thiazolidine, dihydroindole, dihydroisoindole, tetrahydroquinoline, triazaspirodecanedione, hexahydropyrazinooxazinone and the like are preferable.
- Ci-6 alkyl group optionally substituted by 1 to 3 substituents selected from a C ⁇ -6 alkoxy group and a C ⁇ -6 alkoxy- carbonyl group;
- a non-aromatic heterocyclic group e.g., pyrrolidinyl
- R 6 is preferably a hydrogen atom or an optionally substituted Ci-io alkyl group.
- a non-aromatic heterocyclic group e.g., oxodioxolyl
- R 6 is particularly preferably a hydrogen atom.
- R 1 is a C3-6 alkyl group (preferably isobutyl, neopentyl) ;
- R 2 is a C 2 ⁇ 6 alkyl group (preferably ethyl, isobutyl) ;
- R 3 is a Ci- 6 alkyl group (preferably methyl) ;
- X is -OR 6 or -NR 4 R 5 ,
- R 6 is a hydrogen atom, or a Ci-io alkyl group optionally substituted by a non-aromatic heterocyclic group (e.g., oxodioxolyl) optionally substituted by a Ci- 6 alkyl group;
- R 4 and R 5 are the same or different and each is
- Ci-io alkyl group preferably methyl, ethyl
- a Ci-io alkyl group optionally substituted by 1 to 3 substituents selected from a Ci-e alkoxy- carbonyl group, a Ci- ⁇ alkoxy group and a heterocyclic group (preferably 2-thienyl) ;
- Ci-6 alkoxy group optionally substituted by 1 to 3 halogen atoms (e.g., fluorine, chlorine, bromine, iodine);
- a C7-13 aralkyl group (preferably benzyl) optionally substituted by 1 to 3 Ci-e alkylsulfonyl groups; (6) a heterocyclic group (e.g., pyridyl, isoxazolyl, pyrazolyl, thiadiazolyl, benzothiadiazolyl, oxodihydropyrazolyl, azabicyclooctyl, pyrrolidinyl) optionally substituted by 1 to 3 substituents selected from a Ci-6 alkyl group, a C 6 -i4 aryl group, a C7-13 aralkyl group and a Ci-s alkoxy-carbonyl group; or (7) a Ci-6 alkoxy group (only for R 5 ) ; or
- Ci-6 alkyl group optionally substituted by 1 to 3 substituents selected from a Ci-6 alkoxy group and a C ⁇ alkoxy- carbonyl group;
- a halogen atom e.g., fluorine, chlorine, bromine, iodine.
- a pharmacologically acceptable salt is preferable.
- a salt with inorganic base a salt with organic base, a salt with inorganic acid, a salt with organic acid, a salt with basic or a,cidic amino acid and the like.
- the salt with inorganic base include alkali metal salts such as sodium salt, potassium salt and the like; alkaline earth metal salts such as calcium salt, magnesium salt and the like; aluminum salt; ammonium salt and the like.
- the salt with organic base includes a salt with trimethylamine, triethylamine, pyridine, picoline, ethanolamine, diethanolamine, triethanolamine, tromethamine [tris (hydroxymethyl)methylamine] , tert-butylamine, cyclohexylamine, benzylamine, dicyclohexylamine, N,N- dibenzylethylenediamine and the like .
- the salt with inorganic acid include a salt with hydrochloric acid, hydrobromic acid, nitric acid, sulfuric acid, phosphoric acid and the like.
- the salt with organic acid include a salt with formic acid, acetic acid, trifluoroacetic acid, phthalic acid, fumaric acid, oxalic acid, tartaric acid, maleic acid, citric acid, succinic acid, malic acid, methanesulfonic acid, benzenesulfonic acid, p-toluenesulfonic acid and the like.
- Preferable examples of the salt with basic amino acid include a salt with arginine, lysin, ornithine and the like.
- the salt with acidic amino acid include a salt with aspartic acid, glutamic acid and the like.
- the salt with inorganic acid and the salt with organic acid are preferable, hydrochloride, trifluoroacetate and the like are more preferable.
- a prodrug of compound (I) is a compound that converts to compound (I) due to the reaction by enzyme, gastric acid and the like under the physiological conditions in the body; that is, a compound that converts to compound (I) by enzymatic oxidation, reduction, hydrolysis and the like, and a compound that converts to compound (I) by hydrolysis and the like by gastric acid and the like.
- Examples of a prodrug of compound (I) include a compound wherein an amino group of compound (I) is acylated, alkylated or phosphorylated (e.g., a compound where amino group of compound (I) is eicosanoylated, alanylated, pentylaminocarbonylatedy (5-methyl-2-oxo-l, 3-dioxolen-4- yl) methoxycarbonylated, tetrahydrofuranylated, pyrrolidylmethylated, pivaloyloxymethylated or tert-butylated) ; a compound wherein a hydroxy group of compound (I) is acylated, alkylated, phosphorylated or borated (e.g., a compound where a hydroxy group of compound (I) is acetylated, palmitoylated, propanoylated, pivaloylated, succinylated, fumarylated, alany
- a prodrug of compound (I) may be a compound that converts to compound (I) under physiological conditions as described in Development of Pharmaceutical Products, vol. 7, Molecule Design, 163-198, Hirokawa Shoten (1990) .
- the compound (I) may be labeled with an isotope (e.g., 3 H, n Cf 3S Sf 12S 1 and the like) and the like>
- an isotope e.g., 3 H, n Cf 3S Sf 12S 1 and the like
- the compound (I) may be an anhydride or a hydrate.
- the compound (I) and a prodrug thereof show low toxicity and can be used as an agent for the prophylaxis or treatment of various diseases to be mentioned later for mammals (e.g., human, mouse, rat, rabbit, dog, cat, cattle, horse, swine, simian) as they are or by admixing with a pharmacologically acceptable carrier and the like to give a pharmaceutical composition.
- mammals e.g., human, mouse, rat, rabbit, dog, cat, cattle, horse, swine, simian
- organic or inorganic carriers conventionally used as materials for pharmaceutical preparations are used as a pharmacologically acceptable carrier, which are added as an excipient, a lubricant, a binder, a disintegrant and the like for solid preparations; and a solvent, a dissolution aid, a suspending agent, an isotonicity agent, a buffer, a soothing agent and the like for liquid preparations.
- an additive for pharmaceutical preparations such as a preservative, an antioxidant, a coloring agent, a sweetening agent and the like can be used.
- excipient examples include lactose, sucrose, D-mannitol, D-sorbitol, starch, pregelatinized starch, dextrin, crystalline cellulose, low-substituted hydroxypropyl cellulose, sodium carboxymethylcellulose, powdered acacia, pullulan, light silicic anhydride, synthetic aluminum silicate, magnesium aluminate metasilicate and the like.
- Preferable examples of the lubricant include magnesium stearate, calcium stearate, talc, colloidal silica and the like.
- Preferable examples of the binder include pregelatinized starch, saccharose, gelatin, powdered acacia, methylcellulose, carboxymethylcellulose, sodium carboxymethylcellulose, crystalline cellulose, sucrose, D-mannitol, trehalose, dextrin, pullulan, hydroxypropyl cellulose, hydroxypropyl methylcellulose, polyvinylpyrrolidone and the like.
- disintegrant examples include lactose, sucrose, starch, carboxymethylcellulose, calcium carboxymethylcellulose, sodium croscarmellose, sodium carboxymethyl starch, light silicic anhydride, low-substituted hydroxypropyl cellulose and the like.
- solvent include water for injection, physiological brine, Ringer's solution, alcohol, propylene glycol, polyethylene glycol, sesame oil, corn oil, olive oil, cottonseed oil and the like.
- dissolution aid examples include polyethylene glycol, propylene glycol, D-mannitol, trehalose, benzyl benzoate, ethanol, trisaminomethane, cholesterol, triethanolamine, sodium carbonate, sodium citrate, sodium salicylate, sodium acetate and the like.
- the suspending agent include surfactants such as stearyltriethanolamine, sodium lauryl sulfate, lauryl aminopropionate, lecithin, benzalkonium chloride, benzethonium chloride, glycerol monostearate and the like; hydrophilic polymers such as polyvinyl alcohol, polyvinylpyrrolidone, sodium carboxymethylcellulose, methylcellulose, hydroxymethylcellulose, hydroxyethylcellulose, hydroxypropyl cellulose and the like; polysorbates, polyoxyethylene hydrogenated castor oil, and the like.
- surfactants such as stearyltriethanolamine, sodium lauryl sulfate, lauryl aminopropionate, lecithin, benzalkonium chloride, benzethonium chloride, glycerol monostearate and the like
- hydrophilic polymers such as polyvinyl alcohol, polyvinylpyrrolidone, sodium carboxymethylcellulose, methylcellulose, hydroxymethylcellulose, hydroxy
- the isotonicity agent include sodium chloride, glycerol, D-mannitol, D-sorbitol, glucose and the like.
- the buffer include phosphate buffer, acetate buffer, carbonate' buffer, citrate buffer and the like.
- the soothing agent include benzyl alcohol and the like.
- preservative examples include p- oxybenzoates, chlorobutanol, benzyl alcohol, phenethyl alcohol, dehydroacetic acid, sorbic acid and the like.
- antioxidant examples include sulfite, ascorbate and the like.
- the coloring agent include water- soluble edible tar pigments (e.g., foodcolors such as Food Color Red Nos. 2 and 3, Food Color Yellow Nos. 4 and 5, Food Color Blue Nos. 1 and 2 and the like), water insoluble lake pigments (e.g., aluminum salt of the aforementioned water-soluble edible tar pigment), natural pigments (e.g., beta carotene, chlorophil, red iron oxide) and the like.
- water- soluble edible tar pigments e.g., foodcolors such as Food Color Red Nos. 2 and 3, Food Color Yellow Nos. 4 and 5, Food Color Blue Nos. 1 and 2 and the like
- water insoluble lake pigments e.g., aluminum salt of the aforementioned water-soluble edible tar pigment
- natural pigments e.g., beta carotene, chlorophil, red iron oxide
- the dosage form, of the aforementioned pharmaceutical composition is, for example, an oral agent such as tablets (inclusive of sublingual tablets and orally disintegrable tablets) , capsules (inclusive of soft capsules and microcapsules) , granules, powders, troches, syrups, emulsions, suspensions and the like; or a parenteral agent such as injections (e.g., subcutaneous injections, intravenous injections, intramuscular injections, intraperitoneal injections, drip infusions), external agents (e.g., transdermal preparations, ointments), suppositories (e.g., rectal suppositories, vaginal suppositories) , pellets, nasal preparations, pulmonary preparations (inhalations), ophthalmic preparations and the like. These may be administered safely via an oral or parenteral route. These agents may be controlled-release preparations such as rapid-release preparations and sustained-release preparations (e.g., sustained-release micro
- the pharmaceutical 'composition can be produced according to a method conventionally used in the field of pharmaceutical preparation, such as the method described in Japan Pharmacopoeia and the like.
- the content of the compound of the present invention in the pharmaceutical composition varies depending on the dosage form, dose of the compound of the present invention and the like, it is, for example, about 0.1-100 wt%.
- the aforementioned oral agents may be coated with a coating base for the purpose of masking taste, enteric property or sustained release.
- the coating base examples include a sugar-coating base, a water-soluble film coating base, an enteric film coating base, a sustained-release film coating base and the like.
- sucrose may be used, if necessary, along with one or more species selected from talc, precipitated calcium carbonate, gelatin, powdered acacia, pullulan, carnauba wax and the like.
- water-soluble film coating base for example, cellulose polymers such as hydroxypropyl cellulose, hydroxypropyl methylcellulose, hydroxyethylcellulose, methylhydroxyethylcellulose and the like; synthetic polymers such as polyvinyl acetal diethylaminoacetate, aminoalkyl methacrylate copolymer E [Eudragit E, trade name, Roehm Pharma] , polyvinylpyrrolidone and the like; polysaccharides such as pullulan and the like; and the like are used.
- cellulose polymers such as hydroxypropyl cellulose, hydroxypropyl methylcellulose, hydroxyethylcellulose, methylhydroxyethylcellulose and the like
- synthetic polymers such as polyvinyl acetal diethylaminoacetate, aminoalkyl methacrylate copolymer E [Eudragit E, trade name, Roehm Pharma] , polyvinylpyrrolidone and the like
- polysaccharides
- enteric film coating base for example, cellulose polymers such as hydroxypropyl methylcellulose phthalate, hydroxypropyl methylcellulose acetate succinate, carboxymethylethylcellulose, cellulose acetate phthalate and the like; acrylic acid polymers such as methacrylic acid copolymer L [Eudragit L, trade name, Roehm Pharma] , methacrylic acid copolymer LD [Eudragit L-30D55, trade name, Roehm Pharma] , methacrylic acid copolymer S [Eudragit S, trade name, Roehm Pharma] and the like; natural products such as shellac and the like; and the like are used.
- cellulose polymers such as hydroxypropyl methylcellulose phthalate, hydroxypropyl methylcellulose acetate succinate, carboxymethylethylcellulose, cellulose acetate phthalate and the like
- acrylic acid polymers such as methacrylic acid copolymer L [Eudragit L, trade
- sustained-release film coating base for example, cellulose polymers such as ethylcellulose and the like; acrylic acid polymers such as aminoalkyl methacrylate copolymer RS [Eudragit RS, trade name, Roehm Pharma] , ethyl acrylate-methyl methacrylate copolymer suspension [Eudragit NE, trade name, Roehm Pharma] and the like; and the like are used.
- cellulose polymers such as ethylcellulose and the like
- acrylic acid polymers such as aminoalkyl methacrylate copolymer RS [Eudragit RS, trade name, Roehm Pharma] , ethyl acrylate-methyl methacrylate copolymer suspension [Eudragit NE, trade name, Roehm Pharma] and the like; and the like are used.
- Two or more kinds of the above-mentioned coating bases may be mixed in an appropriate ratio for use.
- a light shielding agent such as titanium oxide, ferric oxide and the like may be used during coating.
- the compound of the present invention shows low toxicity (e.g., acute toxicity, chronic toxicity, genetic toxicity, reproductive toxicity, vascular toxicity, carcinogenic) , causes fewer side effects and can be used as an agent for the prophylaxis or treatment or diagnosis of various diseases for mammals (e.g., human, cattle, horse, dog, cat, simian, mouse, rat, especially human) .
- neurotransmitters examples include neuropeptide Y and the like.
- peptidases examples include EC 3.4.11.1 (Leucyl aminopeptidase) , EC 3.4.11.2 (Membrane alanine aminopeptidase) , EC 3.4.11.3 (Cystinyl aminopeptidase), EC 3.4.11.4 (Tripeptide aminopeptidase), EC 3.4.11.5 (Prolyl aminopeptidase), EC 3.4.11.6 (Aminopeptidase B) , EC 3.4.11.7 (Glutamyl aminopeptidase), EC 3.4.11.9 (Xaa-Pro aminopeptidase), EC 3.4.11.10 (Bacterial leucyl aminopeptidase), EC 3.4.11.13 (Clostridial aminopeptidase), EC 3.4.11.14 (Cytosol alanyl aminopeptidase), EC 3.4.11.15 (Lysyl aminopeptidase), EC 3.4.11.1 (Leucyl aminopeptidase), EC 3.4.11.2 (Me
- aminopeptidase Ey EC 3.4.11.21 (Aspartyl aminopeptidase), EC 3.4.11.22 (Aminopeptidase I), EC 3.4.13.3 (Xaa-His dipeptidase) , EC 3.4.13.4 (Xaa-Arg dipeptidase), EC 3.4.13.5 (Xaa-methyl-His dipeptidase), EC 3.4.13.7 (Glu-Glu dipeptidase), EC 3.4.13.9 (Xaa-Pro dipeptidase), EC 3.4.13.12 (Met-Xaa dipeptidase), EC
- 3.4.14.11 are preferable. Especially preferred is EC 3.4.14.5 • (Dipeptidyl-peptidase IV) .
- IFG Impaired Fasting Glucose
- IFG a condition showing a fasting blood glucose level (glucose concentration of intravenous plasma) of not less than 110 mg/dl and less than 126 mg/dl
- IFG a condition showing a fasting blood glucose level (glucose concentration of intravenous plasma) of not less than 110 mg/dl and less than 126 mg/dl
- IFG a condition showing a 75g oral glucose tolerance test 2 h level (glucose concentration of intravenous plasma) of less than 140 mg/dl.
- the compound of the present invention can be also used for secondary prophylaxis and prevention of progression of the above-mentioned various diseases (e.g., cardiovascular event such as myocardial infarction and the like) .
- cardiovascular event such as myocardial infarction and the like
- sulfonylurea compound a compound having a sulfonylurea skeleton or a derivative thereof, such as • tolbutamide, glibenclamide, gliclazide, chlorpropamide, tolazamide, acetohexamide, glyclopyramide, glimepiride, glipizide, glybuzole and the like can be mentioned.
- Examples of the therapeutic agent for incontinentia or pollakiuria include flavoxate hydrochloride, oxybutynin hydrochloride, propiverine hydrochloride and the like.
- the combination drug is preferably an insulin preparation, an insulin sensitizer, an ⁇ glucosidase inhibitor, a biguanide, an insulin secretagogue (preferably sulfonylurea) and the like.
- an insulin secretagogue preferably sulfonylurea
- a biguanide preferably a biguanide
- an ⁇ glucosidase inhibitor preferably sulfonylurea
- a sustained treatment effect can be designed by selecting a combination drug having different action and mechanism from the compound of the present invention
- the compound of the present invention can be produced according to a method known per se, such as a method to be described in detail in the following, or an analogous method thereto.
- Compounds 1 to 14 in the following formulas may form a salt, and as such a salt, for example, salts similar to the salt of compound (I) can be mentioned.
- amino-protecting group for example, formyl group, Ci_ 6 alkyl-carbonyl group, Ci- 6 alkoxy-carbonyl group, benzoyl group, C 7 -I 0 aralkyl-carbonyl group (e.g., benzylcarbonyl) , C 7 - I4 aralkyloxy-carbonyl group (e.g., benzyloxycarbonyl, 9-fluorenylmethoxycarbonyl) , trityl group, phthaloyl group, N,N-dimethylaminomethylene group, substituted silyl group (e.g., trimethylsilyl, triethylsilyl, dimethylphenylsilyl, tert-butyldimethylsilyl, tert- butyldiethylsilyl) , C2-6 alkenyl group (e.g., 1-allyl) and the like can be mentioned. These groups are optionally
- hydroxy-protecting group for example, C ⁇ - 6 alkyl group, phenyl group, trityl group, C 7 -I 0 aralkyl group (e.g., benzyl) , formyl group, Ci-s alkyl-carbonyl group, benzoyl group, C 7 - I o aralkyl-carbonyl group (e.g., benzylcarbonyl), 2- tetrahydropyranyl group, 2-tetrahydrofuranyl group, substituted silyl group (e.g., trimethylsilyl, triethylsilyl, dimethylphenylsilyl, tert-butyldimethylsilyl, tert- butyldiethylsilyl), C2-6 alkenyl group (e.g., 1-allyl) and the like can be mentioned. These groups are optionally substituted by 1 to 3 substituents selected from halogen atom, Ci- S alkyl, Ci
- the amino-protecting group for P is preferably a Ci-6 alkoxy-carbonyl group (preferably Boc (tert-butoxycarbonyl) group) ) , a C 7 -i 4 aralkyloxy-carbonyl group (preferably Cbz (benxyloxycarbonyl) group, Etaoc (9-fluorenylmethoxycarbonyl group) ) and the like.
- the hydrolysis can be generally carried out in the presence of an acid or base.
- the acid for example, mineral acids (e.g., hydrochloric acid, hydrobromide acid, sulfuric acid, phosphoric acid), carboxylic acids (e.g., formic acid, acetic acid, propionic acid) and the like can be mentioned.
- mineral acids e.g., hydrochloric acid, hydrobromide acid, sulfuric acid, phosphoric acid
- carboxylic acids e.g., formic acid, acetic acid, propionic acid
- hydrochloric acid, sulfuric acid and the like are preferable.
- alkali metal salts such as lithium hydroxide, potassium hydroxide, sodium hydroxide, potassium carbonate, sodium carbonate, potassium hydrogencarbonate, sodium hydrogencarbonate and the like
- alkaline earth metal salts such as calcium hydroxide, barium hydroxide and the like
- amines such as trimethylamine, triethylamine, N, N-diisopropylethylamine, N-methylmorpholine and the like; and the like can be mentioned.
- potassium hydroxide, sodium hydroxide and the like are preferable.
- the amount of the acid or base to be used is generally 0.01 to 100 mol, preferably 0.1 to 50 mol, per 1 mol of compound JL.
- the reaction temperature is generally 0 0 C to 150°C, preferably 10 0 C to 100 0 C.
- the amino-protecting group can be eliminated according to a method known per se.
- Conversion to a leaving group can be carried out according to a conventional method, for example, by reacting with methanesulfonyl chloride in the presence of a suitable base, or by reacting with thionyl chloride in the presence of a suitable base, and the like.
- a suitable base used for conversion to a leaving group for example, N,N-diisopropylethylamine (DIEA) , triethylamine (TEA), pyridine, N, N-dimethylaniline and the like can be mentioned.
- Compound IJO can be produced, for example, by protecting the amino group of compound 9_. Protection of the amino group can be carried out according to a method known per se.
- Compound 9 can be produced, for example, by subjecting compound 8 to a reduction- reaction, thereby converting the 5- position substituent (i.e., cyano group) and the 3-position substituent (i.e., substituted oxycarbonyl group) to an aminomethyl group and a hydroxymethyl group, respectively.
- the reduction reaction of the cyano group and that of the substituted oxycarbonyl group can be carried out sequentially or simultaneously.
- either of the reduction reactions may be carried out first and, where necessary, the intermediate obtained upon completion of one reduction reaction may be isolated and purified and then the intermediate may be subjected to the other reduction reaction.
- Such a reduction reaction is carried out according to a conventional method in the presence of a reducing agent in a solvent that does not adversely affect the reaction.
- metal hydride compounds such as sodium bis (2-methoxyethoxy) aluminum hydride, diisobutylaluminum hydride (DIBALH) and the like; metal hydride complex compounds such as sodium borohydride, sodium cyanoborohydride, lithium aluminum hydride, sodium aluminum hydride and the like; and the like can be mentioned.
- the amount of the reduction agent to be used is generally 0.1 to 20 r ⁇ ol per 1 mol of compound 8.
- alcohols such as methanol, ethanol, propanol, isopropanol, butanol, isobutanol, tert-butanol and the like; aromatic hydrocarbons such as benzene, toluene, xylene and the like; aliphatic hydrocarbons such as hexane, heptane and the like; ethers such as diethyl ether, diisopropyl ether, tert- butyl methyl ether, tetrahydrofuran, dioxane, dimethoxyethane and the like; esters such as methyl acetate, ethyl acetate, n- butyl acetate, tert-butyl acetate and the like; amides such as N,N-dimethylformamide, N, N-dimethylacetamide, N- methylpyrrolidone and the like are used.
- the reaction time is generally 0.1 to 100 hrs, preferably 0.1 to 40 hrs.
- the amount of the metal catalyst to be used is generally 0.001 to 1000 mol, preferably 0.01 to 100 mol, per 1 mol of compound 8_.
- hydrogen source for example, hydrogen gas, formic acid, amine salt of formic acid, phosphinate, hydrazine and the like can be mentioned.
- solvent that does not adversely affect the reaction for example, methanol, tetrahydrofuran, N,N-dimethylacetamide and the like can be mentioned.
- This reaction may be carried out, where necessary, in the presence of ammonia (e.g., aqueous ammonia, ammonia-methanol) . Reaction in the presence of ammonia suppresses side reactions and compound 9_ can be produced in a high yield.
- ammonia e.g., aqueous ammonia, ammonia-methanol
- Compound 8_ can be produced, for example, by oxidation of compound 1_.
- the oxidation reaction is carried out according to a conventional method in the presence of an oxidant (e.g., dilute nitric acid, cerium ammonium nitrate (CAN) ) in a solvent that does not adversely affect the reaction (e.g., dioxane, acetone) .
- an oxidant e.g., dilute nitric acid, cerium ammonium nitrate (CAN)
- a solvent that does not adversely affect the reaction e.g., dioxane, acetone
- Compound 1_ can be produced, for example, from compound 4_ and compound _6, according to a method known per se, such as a Hantzch/ s pyridine synthetic method described in " "Shin Jikken Kagaku Kouza (The Chemical Society of Japan ed.), Vol. 14, Synthesis and Reaction of Organic Compound IV, Maruzen (1978), page 2057, or a method analogous thereto.
- Compound _4 can be produced by a method known per se, for example, by subjecting compound 2_ and compound 3 to a known Knoevenagel condensation.
- Compound _6 can be produced by a reaction of compound .5 with ammonia or ammonium salt, according to a method known per se, such as methods described in Synthesis, (1999), vol. 11, p. 1951-1960; Journal of Chemical Society Perkin Transactions 1, (2002), p. 1663-1671 and the like or methods analogous thereto.
- the aforementioned compound 2_, compound 3 and compound 5 ⁇ can be produced by a method known per se.
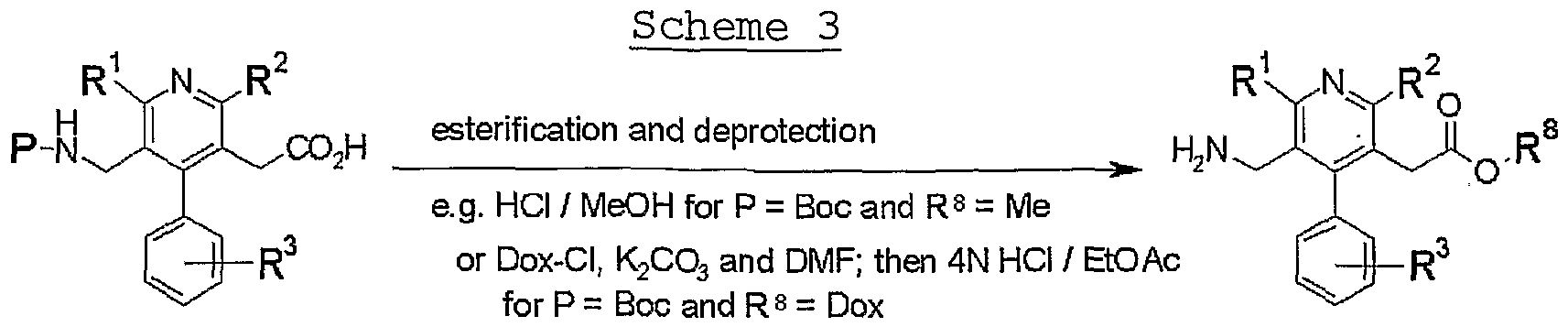
- Compound (I-b) which is a compound of the formula (I) wherein X is -OR 8 [R 8 is an optionally substituted hydrocarbon group or an optionally substituted heterocyclic group] , can be produced according to the following Scheme 3 or a method analogous thereto.
- Dox is (5-methyl-2-oxo-l, 3-dioxol-4-yl)methyl group, and other symbols are as defined above.
- compound V2_ is esterified and, where necessary, the amino-protecting group is eliminated simultaneously or subsequently to give compound (I-b) .
- esterification a method known per se, such as esterification with an alcohol (R 8 -0H) , esterification with an 0- alkylating agent (R 8 -L) and the like can be mentioned.
- acids generally used as an acid catalyst in condensation such as hydrochloric acid, sulfuric acid, p-toluenesulfonic acid, boron fluoride etherate and the like, can be mentioned.
- the amount of the acid catalyst to be used is preferably about 0.05 to- about 50 mol per 1 mol of compound 12.
- a reagent that activates compound _12_ e.g., dicyclohexylcarbodiimide (DCC), trifluoroacetic anhydride
- a reagent that activates alcohols e.g., combination of an organophosphorus compound (e.g., triphenylphosphine) and an electrophilic agent (e.g., diethyl azodicarboxylate)
- the amount of the dehydrating agent to be used is preferably about 1 to about 50 mol per 1 itiol of compound 12.
- ethers such as diethyl ether, tetrahydrofuran, dioxane and the like; halogenated hydrocarbons such as chloroform, dichloromethane and the like; aromatic hydrocarbons such as benzene, toluene, xylene and the like; amides such as N,N-dimethylformamide (DMF) and the like; sulfoxides such as dimethyl sulfoxide and the like, and the like can be mentioned.
- ethers such as diethyl ether, tetrahydrofuran, dioxane and the like
- halogenated hydrocarbons such as chloroform, dichloromethane and the like
- aromatic hydrocarbons such as benzene, toluene, xylene and the like
- amides such as N,N-dimethylformamide (DMF) and the like
- sulfoxides such as dimethyl sulfoxide and the like, and the like
- the reaction time is generally 0.5 to 20 hrs .
- bases generally used for O-alkylation of a carboxyl group such as amines (e.g., triethylamine, N- methylmorpholine, N,N-dimethylaniline) ; alkali metal salts (e.g., sodium hydrogencarbonate, sodium carbonate, potassium carbonate) ; and the like can be mentioned.
- amines e.g., triethylamine, N- methylmorpholine, N,N-dimethylaniline
- alkali metal salts e.g., sodium hydrogencarbonate, sodium carbonate, potassium carbonate
- the amount of each of the O-alkylating agent and base to be used is preferably about 1 to about 50 mol per 1 mol of compound 12.
- the solvent that does not • adversely affect the reaction for example, ethers such as tetrahydrofuran, dioxane and the like; halogenated hydrocarbons such as chloroform, dichloromethane and the like; aromatic hydrocarbons such as benzene, toluene, xylene and the like; amides such as N, N- dimethylformamide and the like; sulfoxides such as dimethyl sulfoxide and the like; and the like can be mentioned. These solvents may be used in a mixture at an appropriate ratio.
- the reaction time is generally 0.5 to 20 hrs.
- the amino-protecting group can be eliminated according to a method known per se.
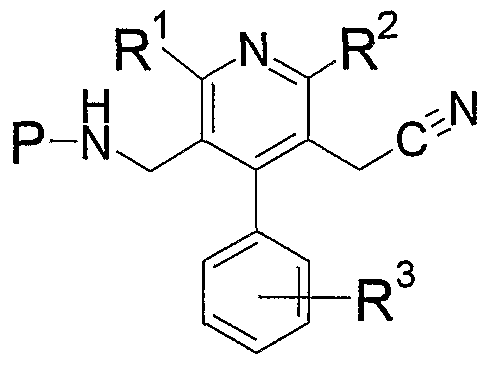
- compound Y ⁇ _ is condensed with compound 13, and then the amino-protecting group is eliminated to give compound (I-c) .
- the condensation is carried out according to a conventional method, for example, conventional peptide coupling method.
- a conventional method for example, conventional peptide coupling method.
- direct condensation of compound Y ⁇ ⁇ with compound 13 using a condensing agent reaction of a reactive derivative of compound Y ⁇ _ with compound Yh_ and the like can be mentioned.
- carbodiimide condensing reagents such as dicyclohexylcarbodiimide (DCC) , diisopropylcarbodiimide (DIPC), l-ethyl-3- (3- dimethylaminopropyl) carbodiimide (EDC), hydrochlorides thereof and the like; phosphoric acid condensing reagents such as diethyl cyanophosphate, diphenylphosphoryl azide and the like; carbonyldiimidazole, 2-chloro-l, 3-dimethylimidazolium tetrafluoroborate, 0- (7-azabenzotriazol-l-yl) -1, 1,3,3- tetramethyluronium hexafluorophosphate (HATU) and the like can be mentioned.
- DCC dicyclohexylcarbodiimide
- DIPC diisopropylcarbodiimide
- EDC l-ethyl-3- (3
- amides such as N, N- dimethylformamide, N, N-dimethylacetamide, N-methylpyrrolidone and the like
- sulfoxides such as dimethyl sulfoxide and the like
- halogenated hydrocarbons such as chloroform, dichloromethane and the like
- aromatic hydrocarbons such as benzene, toluene and the like
- ethers such as tetrahydrofuran, dioxane, diethyl ether, dimethoxyethane and the like
- esters such as methyl acetate, ethyl acetate and the like
- nitriles such as acetonitrile, propionitrile and the like
- water and the like can be mentioned.
- solvents may be used in a mixture at an appropriate ratio .
- the amount of compound 3 ⁇ 3 to be used is generally 1 to 10 mol, preferably 1 to 3 mol, per 1 mol of compound 12.
- the amount of the condensing agent to be used is generally 0.1 to 10 mol, preferably 0.3 to 3 mol, per 1 mol of compound 12.
- the amount of each of the above-mentioned condensation promoter and organic amine base to be used is generally 0.1 to 10 mol, preferably 0.3 to 3 mol, per 1 mol of compound 12.
- the reaction temperature is generally -30 0 C to 120 0 C, preferably -10 0 C to 100 0 C.
- the reaction time is generally 0.5 to 60 hrs.
- an acid anhydride for example, an acid anhydride, an acid halide (e.g., an acid chloride, an acid bromide), an imidazolide, a mixed acid anhydride (e.g., an anhydride with methyl carbonate, ethyl carbonate, isobutyl carbonate) , and the like can be mentioned.
- an acid anhydride e.g., an acid chloride, an acid bromide
- an imidazolide e.g., a mixed acid anhydride with methyl carbonate, ethyl carbonate, isobutyl carbonate
- the reaction is generally carried out in the presence of a base in a solvent that does not adversely affect the reaction.
- amines such as triethylamine, pyridine, N-methylmorpholine, N,N-dimethylaniline, A- dimethylaminopyridine and the like
- alkali metal salts such as lithium hydroxide, sodium hydroxide, potassium hydroxide, sodium hydrogencarbonate, sodium carbonate, potassium carbonate and the like, and the like can be mentioned.
- amides such as N,N-dimethylformamide, N,N- dimethylacetamide, N-methylpyrrolidone and the like; sulfoxides such as dimethyl sulfoxide and the like; halogenated hydrocarbons such as chloroform, dichloromethane and the like; aromatic hydrocarbons such as benzene, toluene and the like; ethers such as tetrahydrofuran, dioxane, diethyl ether, dimethoxyethane and the like; esters such as methyl acetate, ethyl acetate and the like; nitriles such as acetonitrile, propionitrile and the like; water; and the like can be mentioned. These solvents may be used in a mixture at an appropriate ratio. When the above-mentioned amides are used as a solvent that does not adversely affect the reaction, the reaction can also be carried out in the absence of
- the amount of compound 1_3 to be used is generally 1 to 10 mol, preferably 1 to 5 mol, per 1 r ⁇ ol of compound 12.
- the amount of the base to be used is generally 1 to 10 mol, preferably 1 to 5 mol, per 1 mol of compound 12.
- the reaction temperature is generally -30 0 C to 100 0 C, preferably -10 0 C to 100 0 C.
- the reaction time is generally 0.5 to 30 hrs.
- compound Y ⁇ _ is reacted with a chlorocarbonate (e.g., methyl chlorocarbonate, ethyl chlorocarbonate, isobutyl chlorocarbonate) in the presence of a base and the resulting compound is reacted with compound 13.
- a chlorocarbonate e.g., methyl chlorocarbonate, ethyl chlorocarbonate, isobutyl chlorocarbonate
- the amount of compound 1_3 to be used is generally 1 to 10 mol, preferably 1 to 5 mol, per 1 mol of compound 12.
- the amount of the base to be used is generally 1 to 10 mol, preferably 1 to 3 mol, per 1 mol of compound 12.
- the reaction temperature is generally -30 0 C to 120 0 C, preferably -10 0 C to 100 0 C.
- the reaction time is generally 0.5 to 20 hrs.
- an imidazolide is used, a corresponding imidazolide is obtained from compound _12_ and, for example, N,N'- carbonyldiimidazole (CDI), which is then reacted with compound
- the amount of compound 13 to be used is generally 1 to 10 mol, preferably 1 to 5 mol, per 1 mol of compound 12.
- the reaction temperature is generally -30 0 C to 120 0 C, preferably -10 0 C to 100 0 C.
- the reaction time is generally 0.5 to 20 hrs.
- the amino-protecting group can be eliminated according to a method known per se.
- the compound (I) thus obtained can be isolated and purified by a known separation and purification means, such as concentration, concentration under reduced pressure, solvent extraction, crystallization, recrystallization, phase transfer, chromatography and the like.
- compound (I) When compound (I) is obtained as a free compound, it can be converted to the object salt according to a method known per se or a method analogous thereto, and when it is obtained as a salt, it can be converted to a free compound or the object salt according to a method known per se or a method analogous thereto .
- compound (I) contains an optical isomer, a stereoisomer, a positional isomer or a rotational isomer, these are also encompassed in compound (I) , and can be obtained as a single product according to a synthetic method and separation method known per se.
- compound (I) has an optical isomer, an optical isomer resolved from this compound is also encompassed in compound (I) .
- the optical isomer can be produced according to a method known per se. To be specific, an optically active synthetic intermediate is used, or the final racemate product is subjected to optical resolution according to a conventional method to give an optical isomer.
- the method of optical resolution may be a method known per se, such as a fractional recrystallization method, a chiral column method, a dlastereomer method and the like.
- a salt of a racemate with an optically active compound e.g., (+) -mandelic acid, (-)-mandelic acid, (+) -tartaric acid, (-) -tartaric acid, (+) -1-phenethylamine, (-)- 1-phenethylamine, cinchonine, (-) -cinchonidine, brucine
- an optically active compound e.g., (+) -mandelic acid, (-)-mandelic acid, (+) -tartaric acid, (-) -tartaric acid, (+) -1-phenethylamine, (-)- 1-phenethylamine, cinchonine, (-) -cinchonidine, brucine
- an optically active compound e.g., (+) -mandelic acid, (-)-mandelic acid, (+) -tartaric acid, (-) -tartaric acid, (+) -1-phenethylamine,
- a racemate or a salt thereof is applied to a column for separation of an optical isomer (chiral column) to allow separation.
- a mixture of optical isomers is applied to a chiral column such as ENANTIO-OVM (manufactured by Tosoh Corporation) , CHIRAL series (manufactured by Daicel Chemical Industries, Ltd.) and the like, and developed with water, various buffers (e.g., phosphate buffer) and organic solvents (e.g., ethanol, methanol, isopropanol, acetonitrile, trifluoroacetic acid, diethylamine) solely or in admixture to separate the optical isomer.
- buffers e.g., phosphate buffer
- organic solvents e.g., ethanol, methanol, isopropanol, acetonitrile, trifluoroacetic acid, diethylamine
- a racemic mixture is prepared into a diastereomeric mixture by chemical reaction with an optically active reagent, which is prepared into a single substance by a typical separation means (e.g., fractional recrystallization, chromatography method) and the like, and subjected to a chemical treatment such as hydrolysis and the like to separate the optically active reagent moiety, whereby the optical isomer is obtained.
- a typical separation means e.g., fractional recrystallization, chromatography method
- compound (I) when compound (I) contains a hydroxy group or a primary or secondary amino group in a molecule, the compound and an optically active organic acid (e.g., MTPA [ ⁇ - methoxy- ⁇ ,- (trifluoromethyl)phenylacetic acid], (-)- menthoxyacetic acid) and the like are subjected to condensation to give an ester form diastereomer thereof or an amide form diastereomer thereof, respectively.
- compound (I) has a carboxyl group
- this compound and an optically active amine or an optically alcohol are subjected to condensation to give an amide form diastereomer thereof or an ester form diastereomer thereof, respectively.
- the separated diastereomer is converted to the optical isomer of the original compound by acidic hydrolysis or basic hydrolysis.
- Step D 8- ⁇ [5-(aminomethyl) -6-isobutyl-2-methyl-4- (4- methylphenyl) pyridin-3-yl] acetyl ⁇ hexahydropyrazino [2, 1- c][l, 4] oxazin-4 (3H) -one dihydrochloride tert-Butyl ( ⁇ 2-isobutyl- ⁇ -methyl-4- (4-methylphenyl) -5- [2- oxo-2- (4-oxohexahydropyrazino [2, 1-c] [1, 4] oxazin-8 (IH)- yl) ethyl] pyridin-3-yl ⁇ methyl) carbamate (0.42 g, 0.74 mmol) was dissolved in ethyl acetate (2 mL) , 4N hydrogen chloride ethyl acetate solution (3 mL) was added, and the mixture was stirred at room temperature for 3 hrs .
- Methyl 3-aminopent-2-enoate was obtained as a crude product (11.5 g) from methyl 3-oxopentanoate (12.0 g, 92 mmol) by a method similar to Step A of Reference Example 4.
- a mixture of 5-methyl-3-oxohexanenitrile (11.4 g, 91 mmol), p-tolualdehyde (11.0 g, 91 mmol), piperidine (0.90 mL, 9.1 mmol) , acetic acid (1.05 mL, 18 mmol) and toluene (200 mL) was heated under reflux for 12 hrs using a Dean-stark trap.
- Step B methyl 5-cyano-2-ethyl-6-isobutyl-4 ⁇ (4- methylphenyl) nicotinate
- Step C methyl 5- (aminomethyl) -2-ethyl-6-isobutyl-4- (4- methylpheny1) nicotinate
- Step E tert-butyl ⁇ [5- (cyanomethyl) -6-ethyl-2-isobutyl-4- (4- methylpheny1) pyridin-3-yl]methyl ⁇ carbamate
- Step F [5- (aminomethyl)-2-ethyl-6-isobutyl-4- (4- methylphenyl) pyridin-3-yl] acetic acid tert-Butyl ⁇ [5- (cyanomethyl) -6-ethyl-2-isobutyl-4- (4- methylphenyl)pyridin-3-yl]methyl ⁇ carbamate (15.6 g, 37 mmol) was suspended in 6N hydrochloric acid (60 mL) , and the suspension was stirred at 90 0 C for 24 hrs. The reaction mixture was concentrated under reduced pressure. The residue was dissolved in water (150 mL) , and the solution was washed with ethyl acetate.
- Example 3 [5- (aminomethyl) -2-ethyl-4- (4-methylphenyl) -6- neopentylpyridin-3-yl] acetic acid dihydrochloride [5- ⁇ [ (tert-Butoxycarbonyl) amino]methyl ⁇ -2-ethyl-4- (4- methylphenyl) -6-neopentylpyridin-3-yl] acetic acid (0.14 g, 0.31 mmol) prepared in Reference Example 4 was suspended in 6N hydrochloric acid (5 mL) , and the suspension was stirred at room temperature for 3 hr. The reaction mixture was concentrated under reduced pressure, and the residue was triturated from diisopropyl ether to give the title compound (0.13 g, yield 99%) as a white powder.
- Step A tert-butyl ⁇ [5- ⁇ 2- [ (2S) -2- (aminocarbonyl)pyrrolidin-l- yl] -2-oxoethyl ⁇ 6-ethyl-4- (4-methylphenyl) -2-neopentylpyridin-3- yl]methyl ⁇ carbamate
- Step B l- ⁇ [5- (aminomethyl) -2-ethyl-4- (4-methylphenyl) -6- neopentylpyridin-3-yl] acetyl ⁇ -L-prolinamide
- tert-butyl ⁇ [5- ⁇ 2-[ (2S) -2- (aminocarbonyl) pyrrolidin-1-yl] -2-oxoethyl ⁇ -6-ethyl-4- (4- methylphenyl) -2-neopentylpyridin-3-yl]methyl ⁇ carbamate (0.14 g, 0.26 mmol) in ethyl acetate (2 mL) was added 4N hydrogen chloride ethyl acetate solution (3 mL) , and the mixture was stirred at room temperature for 3 hr.
- Step B 2- [5- (aminomethyl)-2-ethyl-4- (4-methylphenyl) -6- neopentylpyridin-3-yl] -N- [3- (methylsulfonyl) phenyl] acetamide dihydrochloride
- Step A (5-methyl-2-oxo-l,3-dioxol-4-yl)methyl [5- ⁇ [ (tert- butoxycarbonyl) amino] methyl ⁇ -2-ethyl-4- (4-methylphenyl) -6- neopentylpyridin-3-yl] acetate
- the reaction mixture was diluted with dichloromethane (2 mL) , and washed successively with saturated aqueous sodium hydrogencarbonate and water. The organic layer was separated, trifluoroacetic acid (1 mL) was added and the mixture was stirred at room temperature for 1 hr. The reaction mixture was concentrated under reduced pressure, and the residue was purified by HPLC to give the title compound (0.035 g, yield 81%) MS 500 (M+l) .
- Examples 54-94 were prepared by a method similar to Example 10 from [5- ⁇ [(tert- butoxycarbonyl) amino]methyl ⁇ -2-ethyl-4- (4-methylphenyl) -6- neopentylpyridin-3-yl] acetic acid and an amine corresponding to Table 4 or a free amine prepared from a salt of the amine.
- the reaction was carried out according to the method of Raymond et al. (Diabetes, vol. 47, pp. 1253-1258, 1998) using a 96 well flat-bottomed plate at 30 0 C.
- a dimethylformamide solution (1 ⁇ L) containing the test compound was added to a mixture of water (69 ⁇ L) , 1 M Tris-hydrochloride buffer- (10 ⁇ L, pH 7.5) and 1 mM aqueous Gly-Pro-p-NA solution (100 ⁇ L) to prepare a mixed solution.
- Plasma (20 ⁇ L) prepared from blood of SD rat according to a conventional method was added to the above-mentioned mixed solution and the enzyme reaction was started at 30 0 C. The absorbance after 0 hr.
- the dipeptidyl peptidase IV inhibitory activity of the test compound group is expressed in IC50 value (nM) and shown in Table 5.
- Table 5 The dipeptidyl peptidase IV inhibitory activity of the test compound group is expressed in IC50 value (nM) and shown in Table 5.
- the dipeptidyl peptidase IV inhibitory activity of the test compound group is expressed in IC 5 0 value (nM) and shown in i ° Table 6.
- the 'compound of the present invention has a superior dipeptidyl peptidase IV inhibitory activity, and is 15 useful as an agent for the prophylaxis or treatment of diabetes and the like.
- Formulation Example 1 production of capsules
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Diabetes (AREA)
- Public Health (AREA)
- Endocrinology (AREA)
- Veterinary Medicine (AREA)
- General Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Obesity (AREA)
- Emergency Medicine (AREA)
- Hematology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Pyridine Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description





















Claims


Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2006217677A AU2006217677A1 (en) | 2005-02-25 | 2006-02-24 | Pyridyl acetic acid compounds |
| BRPI0607433-2A BRPI0607433A2 (en) | 2005-02-25 | 2006-02-24 | compound or a salt thereof, prodrug, pharmaceutical agent, peptidase inhibitor, use of a compound or prodrug thereof, and methods for preventing or treating disease, inhibiting peptidase in a mammal, and production of a compound |
| CA002598934A CA2598934A1 (en) | 2005-02-25 | 2006-02-24 | Pyridyl acetic acid compounds |
| US11/817,088 US20090088419A1 (en) | 2005-02-25 | 2006-02-24 | Pyridyl acetic acid compounds |
| EP06715236A EP1851202A1 (en) | 2005-02-25 | 2006-02-24 | Pyridyl acetic acid compounds |
| IL185399A IL185399A0 (en) | 2005-02-25 | 2007-08-20 | Pyridyl acetic acid compounds |
| NO20074668A NO20074668L (en) | 2005-02-25 | 2007-09-13 | Pyridal acetic acid compounds |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2005052018 | 2005-02-25 | ||
| JP2005-052018 | 2005-02-25 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2006090915A1 true WO2006090915A1 (en) | 2006-08-31 |
Family
ID=36370977
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2006/304177 WO2006090915A1 (en) | 2005-02-25 | 2006-02-24 | Pyridyl acetic acid compounds |
Country Status (17)
| Country | Link |
|---|---|
| US (1) | US20090088419A1 (en) |
| EP (1) | EP1851202A1 (en) |
| KR (1) | KR20070106794A (en) |
| CN (1) | CN101166725A (en) |
| AR (1) | AR055563A1 (en) |
| AU (1) | AU2006217677A1 (en) |
| BR (1) | BRPI0607433A2 (en) |
| CA (1) | CA2598934A1 (en) |
| CR (1) | CR9369A (en) |
| IL (1) | IL185399A0 (en) |
| MA (1) | MA29323B1 (en) |
| NO (1) | NO20074668L (en) |
| PE (1) | PE20061099A1 (en) |
| RU (1) | RU2007135339A (en) |
| TW (1) | TW200640862A (en) |
| WO (1) | WO2006090915A1 (en) |
| ZA (1) | ZA200708144B (en) |
Cited By (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008017381A1 (en) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, processes for preparing them, medicaments comprising these compounds, and their use |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| GB2463788A (en) * | 2008-09-29 | 2010-03-31 | Amira Pharmaceuticals Inc | Pyridinyl antagonists of prostaglandin D2 receptors |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| EP2308847A1 (en) | 2009-10-09 | 2011-04-13 | EMC microcollections GmbH | Substituted pyridines as inhibitors of dipeptidyl peptidase IV and their application for the treatment of diabetes and related diseases |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| US8648073B2 (en) | 2009-12-30 | 2014-02-11 | Fochon Pharma, Inc. | Certain dipeptidyl peptidase inhibitors |
| WO2014064215A1 (en) | 2012-10-24 | 2014-05-01 | INSERM (Institut National de la Santé et de la Recherche Médicale) | TPL2 KINASE INHIBITORS FOR PREVENTING OR TREATING DIABETES AND FOR PROMOTING β-CELL SURVIVAL |
| WO2016151018A1 (en) | 2015-03-24 | 2016-09-29 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Method and pharmaceutical composition for use in the treatment of diabetes |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CL2007001873A1 (en) | 2006-06-27 | 2008-01-04 | Takeda Pharmaceutical | ((3s)) - 6 - ((¨2,6-dimethyl-4- (3- (methylsulfonyl) -propoxy) biphenyl-3-yl) methoxy-2,3-dihydro-1-benzofuran-3-yl acid ) acetic or salt thereof |
| CN109305957B (en) * | 2017-07-26 | 2021-08-03 | 上海医药工业研究院 | Phenylpyridine compound and application thereof in DPP-4 enzyme inhibitor |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003068757A1 (en) * | 2002-02-13 | 2003-08-21 | F. Hoffmann-La Roche Ag | Novel pyridin- and pyrimidin-derivatives |
| WO2005042488A1 (en) * | 2003-10-31 | 2005-05-12 | Takeda Pharmaceutical Company Limited | Pyridine compounds as inhibitors of dipeptidyl peptidase iv |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0648212B1 (en) * | 1992-07-03 | 2001-10-24 | Smithkline Beecham Plc | Benzoxazole and benzothiazole derivatives as pharmaceutical |
| US5827865A (en) * | 1995-03-09 | 1998-10-27 | Smithkline Beecham P.L.C. | Heterocyclic compounds as pharmaceutical |
| US6153632A (en) * | 1997-02-24 | 2000-11-28 | Rieveley; Robert B. | Method and composition for the treatment of diabetes |
| US6291495B1 (en) * | 1997-02-24 | 2001-09-18 | Robert B. Rieveley | Method and composition for the treatment of diabetes |
| US7115750B1 (en) * | 1999-09-20 | 2006-10-03 | Takeda Pharmaceutical Company Limited | Melanin concentrating hormone antagonist |
| EP1527049B8 (en) * | 2002-08-08 | 2008-10-29 | Takeda Pharmaceutical Company Limited | Fused heterocyclic compounds as peptidase inhibitors |
| WO2004081001A1 (en) * | 2003-02-13 | 2004-09-23 | Banyu Pharmaceutical Co., Ltd. | Novel 2-pyridinecarboxamide derivatives |
| BRPI0416238A (en) * | 2003-11-05 | 2007-01-02 | Hoffmann La Roche | compounds, process for their preparation, pharmaceutical composition comprising them, use of the compounds and methods for treating and / or preventing diseases that are modulated by ppar (delta) and / or ppar (alpha) agonists |
| NZ546444A (en) * | 2003-11-05 | 2009-09-25 | Hoffmann La Roche | Phenyl derivatives as PPAR agonists |
-
2006
- 2006-02-24 ZA ZA200708144A patent/ZA200708144B/en unknown
- 2006-02-24 CN CNA2006800139906A patent/CN101166725A/en active Pending
- 2006-02-24 TW TW095106239A patent/TW200640862A/en unknown
- 2006-02-24 PE PE2006000223A patent/PE20061099A1/en not_active Application Discontinuation
- 2006-02-24 CA CA002598934A patent/CA2598934A1/en not_active Abandoned
- 2006-02-24 EP EP06715236A patent/EP1851202A1/en not_active Withdrawn
- 2006-02-24 WO PCT/JP2006/304177 patent/WO2006090915A1/en active Application Filing
- 2006-02-24 US US11/817,088 patent/US20090088419A1/en not_active Abandoned
- 2006-02-24 BR BRPI0607433-2A patent/BRPI0607433A2/en not_active IP Right Cessation
- 2006-02-24 RU RU2007135339/04A patent/RU2007135339A/en not_active Application Discontinuation
- 2006-02-24 AU AU2006217677A patent/AU2006217677A1/en not_active Abandoned
- 2006-02-24 AR ARP060100686A patent/AR055563A1/en not_active Application Discontinuation
- 2006-02-24 KR KR1020077021956A patent/KR20070106794A/en not_active Application Discontinuation
-
2007
- 2007-08-20 IL IL185399A patent/IL185399A0/en unknown
- 2007-09-05 CR CR9369A patent/CR9369A/en not_active Application Discontinuation
- 2007-09-10 MA MA30205A patent/MA29323B1/en unknown
- 2007-09-13 NO NO20074668A patent/NO20074668L/en not_active Application Discontinuation
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003068757A1 (en) * | 2002-02-13 | 2003-08-21 | F. Hoffmann-La Roche Ag | Novel pyridin- and pyrimidin-derivatives |
| WO2005042488A1 (en) * | 2003-10-31 | 2005-05-12 | Takeda Pharmaceutical Company Limited | Pyridine compounds as inhibitors of dipeptidyl peptidase iv |
Cited By (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008017381A1 (en) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, processes for preparing them, medicaments comprising these compounds, and their use |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| US8049015B2 (en) | 2008-09-29 | 2011-11-01 | Panmira Pharmaceuticals, Llc | Heteroaryl antagonists of prostaglandin D2 receptors |
| GB2463788B (en) * | 2008-09-29 | 2010-12-15 | Amira Pharmaceuticals Inc | Heteroaryl antagonists of prostaglandin D2 receptors |
| GB2463788A (en) * | 2008-09-29 | 2010-03-31 | Amira Pharmaceuticals Inc | Pyridinyl antagonists of prostaglandin D2 receptors |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| EP2308847A1 (en) | 2009-10-09 | 2011-04-13 | EMC microcollections GmbH | Substituted pyridines as inhibitors of dipeptidyl peptidase IV and their application for the treatment of diabetes and related diseases |
| US8648073B2 (en) | 2009-12-30 | 2014-02-11 | Fochon Pharma, Inc. | Certain dipeptidyl peptidase inhibitors |
| US9340523B2 (en) | 2009-12-30 | 2016-05-17 | Fochon Pharma, Inc. | Certain dipeptidyl peptidase inhibitors |
| WO2011107494A1 (en) | 2010-03-03 | 2011-09-09 | Sanofi | Novel aromatic glycoside derivatives, medicaments containing said compounds, and the use thereof |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2011161030A1 (en) | 2010-06-21 | 2011-12-29 | Sanofi | Heterocyclic substituted methoxyphenyl derivatives having an oxo group, method for producing same, and use thereof as gpr40 receptor modulators |
| WO2012004270A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | Spirocyclically substituted 1,3-propane dioxide derivatives, methods for the production thereof and use of the same as medicament |
| WO2012010413A1 (en) | 2010-07-05 | 2012-01-26 | Sanofi | Aryloxy-alkylene substituted hydroxyphenyl hexynoic acids, methods for the production thereof and use of the same as medicament |
| WO2012004269A1 (en) | 2010-07-05 | 2012-01-12 | Sanofi | (2-aryloxy-acetylamino)-phenyl-propionic acid derivatives, method for producing same and use thereof as pharmaceuticals |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2013037390A1 (en) | 2011-09-12 | 2013-03-21 | Sanofi | 6-(4-hydroxy-phenyl)-3-styryl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2013045413A1 (en) | 2011-09-27 | 2013-04-04 | Sanofi | 6-(4-hydroxy-phenyl)-3-alkyl-1h-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2014064215A1 (en) | 2012-10-24 | 2014-05-01 | INSERM (Institut National de la Santé et de la Recherche Médicale) | TPL2 KINASE INHIBITORS FOR PREVENTING OR TREATING DIABETES AND FOR PROMOTING β-CELL SURVIVAL |
| WO2016151018A1 (en) | 2015-03-24 | 2016-09-29 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Method and pharmaceutical composition for use in the treatment of diabetes |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2598934A1 (en) | 2006-08-31 |
| AR055563A1 (en) | 2007-08-22 |
| US20090088419A1 (en) | 2009-04-02 |
| KR20070106794A (en) | 2007-11-05 |
| PE20061099A1 (en) | 2006-12-05 |
| BRPI0607433A2 (en) | 2009-09-08 |
| RU2007135339A (en) | 2009-03-27 |
| ZA200708144B (en) | 2008-11-26 |
| MA29323B1 (en) | 2008-03-03 |
| AU2006217677A1 (en) | 2006-08-31 |
| NO20074668L (en) | 2007-11-15 |
| CR9369A (en) | 2007-10-22 |
| EP1851202A1 (en) | 2007-11-07 |
| TW200640862A (en) | 2006-12-01 |
| IL185399A0 (en) | 2008-02-09 |
| CN101166725A (en) | 2008-04-23 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1851202A1 (en) | Pyridyl acetic acid compounds | |
| KR100858259B1 (en) | Pyridine compounds as inhibitors of dipeptidyl peptidase iv | |
| US20090286791A1 (en) | Amide Compounds | |
| US7547710B2 (en) | Fused heterocyclic compounds as peptidase inhibitors | |
| JPWO2007119833A1 (en) | Nitrogen-containing heterocyclic compounds | |
| EP1845081A1 (en) | Amide compound | |
| JP2006131559A (en) | Nitrogen-containing heterocyclic compound | |
| WO2008011130A2 (en) | Amide compounds | |
| JP4542757B2 (en) | Fused heterocyclic compounds | |
| SA05260027B1 (en) | Pyridine compound | |
| WO2009128481A1 (en) | Nitrogenated 5-membered heterocyclic compound | |
| JP2009196966A (en) | Pyrazolidinedione derivative | |
| WO2011055770A1 (en) | Fused heterocyclic compound | |
| JP4473698B2 (en) | Pyridine compounds | |
| JP2010265216A (en) | Heterocyclic compound | |
| JP4025345B2 (en) | Pyridyl acetic acid compound | |
| RU2353617C2 (en) | Pyridin derivatives as dipeptidyl peptidase iv inhibitors | |
| JPWO2013147026A1 (en) | Aromatic ring compounds | |
| JP2007224044A (en) | Pyridyl acetic acid compound |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200680013990.6 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 185399 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/a/2007/010218 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2598934 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11817088 Country of ref document: US Ref document number: 12007501829 Country of ref document: PH Ref document number: 2006715236 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: CR2007-009369 Country of ref document: CR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 3398/KOLNP/2007 Country of ref document: IN Ref document number: 2006217677 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 561603 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 07097893 Country of ref document: CO |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020077021956 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10287 Country of ref document: GE Ref document number: 1200701949 Country of ref document: VN Ref document number: 2007135339 Country of ref document: RU Ref document number: A20071156 Country of ref document: BY |
|
| ENP | Entry into the national phase |
Ref document number: 2006217677 Country of ref document: AU Date of ref document: 20060224 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2006217677 Country of ref document: AU |
|
| WWP | Wipo information: published in national office |
Ref document number: 2006715236 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: PI0607433 Country of ref document: BR Kind code of ref document: A2 |