(1-H-INDAZO -3-Y ) -AMIDE DERIVATIVES AS GSK-3 INHIBITORS
This invention relates to novel compounds, in particular to novel indazole derivatives, to processes for the preparation of such compounds, to pharmaceutical compositions containing such compounds and to the use of such compounds in medicine. GSK-3 is a serine/threonine protein kinase composed of two isoforms (α and β) which are encoded by distinct genes. GSK-3 is one of several protein kinases which phosphorylates glycogen synthase (GS) (Embi et al, Eur. J. Biochem., (107), 519-527, (1980)). The α and β isoforms have a monomeric structure and are both found in mammalian cells. Both isoforms phospliorylate muscle glycogen synthase (Cross et al, BiochemicalJournal, (303), 21-26, (1994)) and these two isoforms show good homology between species (e.g. human and rabbit GSK-3α are 96% identical).
Type II diabetes (or Non-Insulin Dependent Diabetes Mellitus, NIDDM) is a multifactorial disease. Hyperglycaemia is due to insulin resistance in the liver, muscle and other tissues coupled with inadequate or defective secretion of insulin from pancreatic islets. Skeletal muscle is the major site for insulin-stimulated glucose uptake and in this tissue, glucose removed from the circulation is either metabolised through glycolysis and the TCA cycle, or stored as glycogen. Muscle glycogen deposition plays the more important role in glucose homeostasis and Type II diabetic subjects have defective muscle glycogen storage.
The stimulation of glycogen synthesis by insulin in skeletal muscle results from the dephosphorylation and activation of glycogen synthase (Villar-Palasi C. and Lamer J., Biochim. Biophys. Ada., (39), 171-173, (1960), Parker PJ. et al, Eur. J. Biochem., (130), 227-234, (1983) and Cohen P., Biochem. Soc. Trans., (21), 555-567, (1993)). The phosphorylation and dephosphorylation of GS are mediated by specific kinases and phosphatases. GSK-3 is responsible for phosphorylation and deactivation of GS, while glycogen bound protein phosphatase 1 (PP1G) dephosphorylates and activates GS. Insulin both inactivates GSK-3 and activates PP1G (Srivastava A.K. and Pandey S.K., Mol and Cellular Biochem., (182), 135-141, (1998)). Chen et al. (Diabetes, (43), 1234-1241, (1994)) found that there was no difference in the mRNA abundance of PP1G between patients with Type II diabetes and control patients, suggesting that an increase in GSK-3 activity might be important in Type II
diabetes. It has also recently been demonstrated that GSK-3 is overexpressed in Type II diabetic muscle and that an inverse correlation exists between skeletal muscle GSK-3 activity and insulin action (Nikoulina et al, Diabetes, (49), 263-271, (2000)). Overexpression of GSK-3β and constirutively active GSK-3β(S9A, S9E) mutants in 5 HEK-293 cells resulted in suppression of glycogen synthase activity (Eldar-Finkelman et al, PNAS, (93), 10228-10233, (1996)) and overexpression of GSK-3β in CHO cells, expressing both insulin receptor and insulin receptor substrate 1 (IRS-1), resulted in an impairment of insulin action (Eldar-Finkelman and Krebs, PNAS, (94), 9660-9664, (1997)). Recent evidence for the involvement of elevated GSK-3 activity and the 10 development of insulin resistance and type II diabetes in adipose tissue has emerged from studies undertaken in diabetes and obesity prone C57BL/6J mice (Eldar-Finkelman et al, Diabetes, (48), 1662-1666, (1999)).
GSK-3 has been shown to phospliorylate other proteins in vitro including the eukaryotic initiation factor eIF-2B at Serine540 (Welsh et al, FEBS Letts., (421), 125-
15 130, (1998)). This phosphorylation results in an inhibition of eIF-2B activity and leads to a reduction in this key regulatory step of translation. In disease states, such as diabetes, where there is elevated GSK-3 activity this could result in a reduction of translation and potentially contribute to the pathology of the disease.
Several aspects of GSK-3 functions and regulation in addition to modulation of 0 glycogen synthase activity indicate that inhibitors of this enzyme may be effective in treatment of disorders of the central nervous system. GSK-3 activity is subject to inhibitory phosphorylation by PI 3 kinase-mediated or Wnt-1 class-mediated signals that can be mimicked by treatment with Uthium, a low mM inhibitor of GSK-3 (Stambolic V., Ruel L. and Woodgett J.R., Curr. Biol, (6), 1664-8, (1996)). 5 GSK-3 inhibitors may be of value as neuroprotectants in treatment of acute stroke and other neurotraumatic injuries. Roles for PI 3-kinase signalling through PKB/akt to promote neuronal cell survival are well established, and GSK-3 is one of a number of PKB/akt substrates to be identified that can contribute to the inhibition of apoptosis via this pathway (Pap and Cooper, J. Biol. Chem., (273), 19929-19932, ((1998)). Evidence 0 suggests that astrocytic glycogen can provide an alternative energy source to facilitate neuronal survival under conditions of glucose deprivation (for example, see Ransom B.R. and Fern R., Glia, (21), 134-141, (1997) and references therein). Lithium is known to
protect cerebellar granule neurons from death (D'Mello et al, Exp. Cell Res., (211), 332- 338, (1994) and Nolonte et al, Neurosci. Letts., (172), 6-10, (1994)) and chronic lithium treatment has demonstrable efficacy in the middle cerebral artery occlusion model of stroke in rodents (Νonaka and Chuang, Neuroreport, (9), 2081-2084, (1998)). Wnt- induced axonal spreading and branching in neuronal culture models has been shown to correlate with GSK-3 inhibition (Lucas and Salinas, Dev. Biol, (192), 31-44, (1997)) suggesting additional value of GSK-3 inhibitors in promoting neuronal regeneration following neurotraumatic insult.
Tau and β-catenin, two known in vivo substrates of GSK-3, are of direct relevance in consideration of further aspects of the value of GSK-3 inhibitors in relation to treatment of chronic neurodegenerative conditions. Tau hyperphosphorylation is an early event in neurodegenerative conditions such as Alzheimer's disease (AD), and is postulated to promote microtubule disassembly. Lithium has been reported to reduce the phosphorylation of tau, enhance the binding of tau to microtubules, and promote microtubule assembly through direct and reversible inhibition of glycogen synthase kinase-3 (Hong M., Chen D.C., Klein P.S. and Lee V.M., J. Biol. Chem., (272), 25326- 32, (1997). β-catenin is phosphorylated by GSK-3 as part of a tripartite complex with axin, resulting in β-catenin being targetted for degradation (D eda et al, J. EMBO., (17), 1371-1384, (1998)). Inhibition of GSK-3 activity is a key mechanism by which cytosolic levels of catenin are stabilised and hence promote β-catenin-LEF-1/TCF transcnptional activity (Eastman, Grosschedl, Curr. Opin. Cell. Biol, (11), 233, (1999)). Rapid onset AD mutations in presenilin-1 (PS-1) have been shown to decrease the cytosolic β-catenin pool in transgenic mice. Further evidence suggests that such a reduction in available β- catenin may increase neuronal sensitivity to amyloid mediated death through inhibition of β-catenin-LEF-1/TCF transcriptional regulation of neuroprotective genes (Zhang et al, Nature, (395), 698-702, (1998)). A likely mechanism is suggested by the finding that mutant PS-1 protein confers decreased inactivation of GSK-3 compared with normal PS- 1 (Weihl C.C., Ghadge G.D., Kennedy S.G., Hay Ν, Miller RJ. and Roos R.P., J. Neurosci., (19), 5360-5369, (1999)). International Patent Application Publication Number WO 97/41854 (University of
Pennsylvania) discloses that an effective drug for the treatment of manic depression is lithium, but that there are serious drawbacks associated with this treatment. Whilst the
precise mechanism of action of this drug for treatment of manic depression remains to be fully defined, current models suggest that inhibition of GSK-3 is a relevant target that contributes to the modulation of AP-1 DNA binding activity observed with this compound (see Manji et al, J. Clin. Psychiatry, (60) (suppl 2), 27-39, (1999) for review). GSK-3 inhibitors may also be of value in treatment of schizophrenia. Reduced levels of β-catenin have been reported in schizophremc patients (Cotter D., Kerwin R., al- Sarraji S., Brion J.P., Chadwich A., Lovestone S., Anderton B., and Everall I., Neuroreport, (9), 1379-1383, (1998)) and defects inpre-pulse inhibition to startle response have been observed in schizophremc patients (Swerdlow et al, Arch. Gen. Psychiat., (51), 139-154, (1994)). Mice lacking the adaptor protein dishevelled- 1, an essential mediator of Wnt-induced inhibition of GSK-3, exhibit both a behavioural disorder and defects in pre-pulse inhibition to startle response (Lijam N., Paylor R., McDonald M.P., Crawley J.N., Deng C.X., Herrup K, Stevens K.E., Maccaferri G., McBain C.J., SussmanDJ., and Wynshaw-Boris A., Cell, (90), 895-905, (1997)). Together, these findings implicate deregulation of GSK-3 activity as contributing to schizophrenia. Hence, small molecule inhibitors of GSK-3 catalytic activity maybe effective in treatment of this mood disorder.
The finding that transient β-catenin stabilisation may play a role in hair development (Gat et al, Cell, (95), 605-614, (1998)) suggests that GSK-3 inhibitors could be used in the treatment of baldness.
Studies on fibroblasts from the GSK-3β knockout mouse (Hoeflich K.P. et al, Nature, (406), 86-90, (2000)) support a role for this kinase in positively regulating the activity of NFkB. This transcription factor mediates cellular responses to a number of inflammatory stimuli. Therefore, pharmacologic inhibition of GSK-3 maybe of use in treating inflammatory disorders through the negative regulation of NFkB activity.
The compounds of the present invention are indazole derivatives. Other indazole derivatives have been described previously for use in alternative medicinal applications. For example, International Application, Publication Number WO 93/23404 describes a series of bicyclic compounds, including substituted indazoles, which are stated to modulate endothelin activity and may accordingly be of use in the treatment of conditions such as asthma, hypertension, renal failure and endotoxin shock. International Application, Publication Number WO 94/14780 describes a series of heterocyclic
compounds, including indazoles, which are stated to have biological activity as neuronal nitric oxide synthase inhibitors, and as such may be useful in the treatment of, for example, cerebral ischaemia.
We have now discovered that a series of indazoles are potent and selective inhibitors of GSK-3. These compounds are indicated to be useful for the treatment and/or prophylaxis of conditions associated with a need for inhibition of GSK-3, such as diabetes, conditions associated with diabetes, chronic neurodegenerative conditions including dementias such as Alzheimer's disease, Parkinson's disease, progressive supranuclear palsy, subacute sclerosing panencephalitic parkinsonism, postencephalitic parkinsonism, pugilistic encephalitis, guam parkinsonism-dementia complex, Pick's disease, corticobasal degeneration, frontotemporal dementia, Huntingdon's disease, ADDS associated dementia, amyotrophic lateral sclerosis, multiple sclerosis and neurotraumatic diseases such as acute stroke, mood disorders such as schizophrenia and bipolar disorders, promotion of functional recovery post stroke, cerebral bleeding (for example, due to solitary cerebral amyloid angiopathy), hair loss, obesity, atherosclerotic cardiovascular disease, hypertension, polycystic ovary syndrome, syndrome X, ischaemia, traumatic brain injury, cancer, leukopenia, Down's syndrome, Lewy body disease, inflammation, and immunodeficiency.
Accordingly, in a first aspect, the present invention provides a compound of formula ®,
or a salt thereof, or a solvate thereof, wherein,
Rl is unsubstituted or substituted alkyl, unsubstituted or substituted cycloalkyl, unsubstituted or substituted alkenyl, unsubstituted or substituted cycloalkenyl, unsubstituted or substituted aryl, aralkyl wherein the aryl and the alkyl moieties may each independently be unsubstituted or substituted, aralkenyl wherein the aryl and alkenyl
moieties may each independently be unsubstituted or substituted, unsubstituted or substituted heterocyclyl, or heterocyclylalkyl wherein the heterocyclyl and the alkyl moieties may each independently be unsubstituted or substituted, unsubstituted or substituted heteroaryl, heteroarylalkyl wherem the heteroaryl and the alkyl moieties may each independently be unsubstituted or substituted;
R2 is H;
R3 is H, halo, alkyl, unsubstituted or substituted aryl or unsubstituted or substituted heteroaryl;
R4 is H, unsubstituted or substituted aryl; unsubstituted or substituted heteroaryl, unsubstituted or substituted heterocyclyl; or -X-aryl;
R5 is H;
X is O, NH, S or S(O)n and n is 1 or 2; with the proviso that when R1, R2 and R^ are as defined in relation to formula (I), and R^ is H or halo, then R4 is unsubstituted or substituted aryl; unsubstituted or substituted heteroaryl, unsubstituted or substituted heterocyclyl; or -X-aryl.
Suitably, R* is alkyl, alkenyl, cycloC3_g alkyl, cycloC3_g alkenyl, di- alkylaminoalkyl, arylalkyl, arylalkenyl, heterocyclyl wherein the heterocyclyl group may be optionally substituted by one or more groups selected from alkyl, arylalkyl and alkoxyalkyl; heterocyclylalkyl wherein the heterocyclyl group may be optionally substituted by one or more groups selected from alkoxyalkyl, aryloxyalkyl, arylalkyl and alkyl; heteroarylalkyl wherein the heteroaryl may be optionally substituted by one or more groups selected from alkyl; heteroaryl wherein the heteroaryl may be optionally substituted by one or more groups selected from aryl and heteroaryl; aryl wherein the aryl group may be optionally substituted by heterocyclylalkyl and di-alkylaminoalkyl; or alkoxyalkyl wherein the alkoxy group may be optionally substituted by alkoxy. More suitably, R is alkyl, cycloC3_g alkyl or heterocyclyl wherein the heterocyclyl group is substituted by arylalkyl. Preferably, Rl is n-propyl, cyclopropyl, cyclopentyl and N- benzylpyrrolidin-3-yl. Suitably, R^ is H, halo or aryl. Preferably R^ is H, bromo or phenyl.
Suitably, R4 is H, aryl wherein the aryl group may be optionally substituted by one or more substituents selected from halo, -SO2NH2, -OH, -NHSO2Me and -Sθ2Me; heteroaryl wherein the heteroaryl group may be optionally substituted by one or more substituents selected from halo, alkyl and oxo; heterocyclyl wherein the heterocyclyl group may be optionally substituted by benzyloxycarbonyl; or -X-aryl. Preferably, R4 is H, phenyl, 2-fluorophenyl, 3-fluorophenyl, 4-fluorophenyl, 2,3-difluorophenyl, 2,5- difluorophenyl, 3,5-difluoroρhenyl, 3-H2NSO2-Ph, 4-H2NSO2-PI1, 3-MeSO2NH-Ph, 4-
MeSO2NH-Ph, 4-methanesulfonylphenyl, 4-hydroxyphenyl, 2-furyl, 3-furyl, 2-pyrrolyl,
2-thienyl, 3-thienyl, 4-methylthien-2-yl, 5-bromotbien-2-yl, indol-5-yl, pyrid-2-on-5-yl, - NHPh, -OPh, -SPh, -SO2Ph, piperazin-1-yl or 4-benzyloxycarbonylpiperazin-l-yl. In a preferred aspect of the present invention there is provided a subset of compounds of formula (I), of formula (IA),
(IA)
or a salt thereof, or a solvate thereof, wherein,
Rl is alkyl, cyck>C3_g alkyl or heterocyclyl wherein the heterocyclyl group is substituted by arylalkyl;
R2 is H; R3 is H, halo or aryl; and
R4 is H, aryl wherein the aryl group may be optionally substituted by one or more substituents selected from halo, -SO2NH2, -OH, -NHSO2Me and -SO2Me; heteroaryl wherein the heteroaryl group may be optionally substituted by one or more substituents selected from halo, alkyl and oxo; heterocyclyl wherein the heterocyclyl group may be optionally substituted by aryloxycarbonyl; or -X-aryl;
with the proviso that when R , R2 and R^ are as defined in relation to formula (IA), and
R3 is H or halo, then R4 is aryl wherein the aryl group may be optionally substituted by one or more substituents selected from halo, -SO2NH2, -OH, -NHSO2Me and -SO2Me; heteroaryl wherein the heteroaryl group may be optionally substituted by one or more substituents selected from halo, alkyl and oxo; heterocyclyl wherein the heterocyclyl group may be optionally substituted by aryloxycarbonyl; or -X-aryl.
In a further preferred aspect of the present invention there is provided a subset of compounds of formula (I), of formula (IB),
(IB)
or a salt thereof, or a solvate thereof, wherein,
Rl is n-propyl, cyclopropyl, cyclopentyl or N-benzylρyrrolidin-3-yl;
R2 is H; R3 is H, bromo or phenyl;
R4 is H, phenyl, 2-fluorophenyl, 3 -fluorophenyl, 4-fluorophenyl, 2,3-difluorophenyl, 2,5- difluorophenyl, 3,5-difluorophenyl, 3-H2NSO2-Ph, 4-H2NSO2-Ph, 3-MeSO2NH-Ph, 4-
MeSO2 H-Ph, 4-methanesulfonylphenyl, 4-hydroxyphenyl, 2-furyl, 3-furyl, 2-pyrrolyl,
2-thienyl, 3-thienyl, 4-methylthien-2-yl, 5-bromothien-2-yl, indol-5-yl, pyrid-2-on-5-yl, - NHPh, -OPh, -SPh, -SO2PI1, piperazin-1-yl or 4-benzyloxycarbonylpiperazin-l-yl; and
R5 is H; with the proviso that when R , R2 and R^ are as defined in relation to formula (IB), and
R3 is H or bromo, then R4 is phenyl, 2-fluorophenyl, 3-fluorophenyl, 4-fluorophenyl, 2,3-difluorophenyl, 2,5-difluorophenyl, 3,5-difluorophenyl, 3-H2NSO2-Ph, 4-H2NSO2- Ph, 3-MeSO2NH-Ph, 4-MeSθ2NH-Ph, 4-methanesulfonylphenyl, 4-hydroxyphenyl, 2-
furyl, 3-furyl, 2-pyrrolyl, 2-thienyl, 3-thienyl, 4-methylthien-2-yl, 5-bromothien-2-yl, indol-5-yl, pyrid-2-on-5-yl, -NHPh, -OPh, -SPh, -SO2PI1, piperazin-1-yl or 4- benzyloxycarbonylpiperazin-1-yl.
Particularly preferred compounds of formula (I) which are of special interest as agents useful in the treatment and/or prophylaxis of conditions associated with a need for inhibition of GSK-3 are provided in Table 1 below.
Certain compounds of formula (I) may contain chiral atoms and/or multiple bonds, and hence may exist in one or more stereoisomeric forms. The present invention encompasses all of the isomeric forms of the compounds of formula (I) whether as individual isomers or as mixtures of isomers, including geometric isomers and racemic modifications.
As used herein the term "alkyl" as a group or part of a group refers to a straight or branched chain saturated aliphatic hydrocarbon radical containing 1 to 12 carbon atoms, suitably 1 to 6 carbon atoms. Such alkyl groups in particular include methyl ("Me"), ethyl ("Et"), n-propyl ("Prn"), ώo-propyl ("Pr1"), n-butyl ("Bun"), sec-butyl ("Bus"), tert- butyl ("Bu*"), pentyl and hexyl. Where appropriate, such alkyl groups may be substituted by one or more groups selected from halo (such as fluoro, chloro, bromo), -CN, -CF3, -
OH, -OCF3, C2-6 alkenyl, C^s alkynyl, C\. alkoxy, aryl and di-Cι_6 alkylamino.
As used herein the term "alkenyl" as a group or part of a group refers to a straight or branched chain mono- or poly-unsaturated aliphatic hydrocarbon radical containing 2 to 12 carbon atoms, suitably 2 to 6 carbon atoms. References to "alkenyl" groups include groups which may be in the E- or Z-form or mixtures thereof. Such alkenyl groups in particular include ethenyl, propenyl, butenyl, pentenyl and hexenyl. Where appropriate, such alkenyl groups may be substituted by one or more groups selected from halo (such as fluoro, chloro, bromo), -CN, -CF3, -OH, -OCF3, Cι _6 alkyl, C3_6 alkynyl, C . alkoxy, aryl and di-Cι_6 alkylamino.
As used herein the term "alkynyl" refers to hydrocarbon groups of either straight or branched configuration with one or more carbon-carbon triple bonds which may occur at any stable point in the chain, containing 3 to 12 carbon atoms, suitably 3 to 6 carbon atoms. Such alkynyl groups in particular include propynyl, butynyl and pentynyl. Where appropriate, such alkynyl groups may be substituted by one or more groups selected from
halo (such as fluoro, chloro, bromo), -CN, -CF3, -OH, -OCF3, C\^ alkyl, C2-6 alkenyl,
C\. alkoxy, aryl and di-Cι_6 alkylamino.
As used herein, the term "alkoxy" as a group or part of a group refers to an alkyl ether radical, wherein the term "alkyl" is defined above. Such alkoxy groups in particular include methoxy, ethoxy, n-propoxy, tso-propoxy, n-butoxy, zso-butoxy, sec-butoxy and tert-butoxy. Where appropriate, such alkoxy groups may be substituted by one or more groups selected from halo (such as fluoro, chloro, bromo), -CN, -CF3, -OH, -OCF3, Cj.g alkyl, C2_6 alkenyl, C2- alkynyl, aryl and di-Ci^g alkylamino.
As used herein, the term "aryl" as a group or part of a group refers to a carbocyclic aromatic radical. Suitably such aryl groups are 5-6 membered monocyclic groups or 8-10 membered fused bicyclic groups, especially phenyl ("Ph"), biphenyl and naphthyl, particularly phenyl. Such aryl groups may be optionally substituted with one or more substituents, which maybe the same or different, selected from halo (such as fluoro, chloro, bromo), -CN, -CF3, -OH, -OCF3, -NO2, C\- alkyl, C2-6 alkenyl, 0,^-6 alkynyl, C j _g alkoxy and di-C \ _g alkylamino.
As used herein, the term "heteroaryl" as a group or part of a group refers to stable heterocyclic aromatic single and fused rings containing one or more hetero atoms independently selected from nitrogen, oxygen and sulfur. A fused heteroaryl ring system may include carbocyclic rings and need include only one heteroaryl ring. Such heteroaryl groups include furyl, thienyl, pyridazinyl, pyridyl, quinolinyl, indolyl, thiazolyl, benzoxazolyl, and benzothiazolyl. Each ring may be optionally substituted with one or more substituents, which maybe the same or different, selected from halo (such as fluoro, chloro, bromo), -CN, -CF3, -OH, -NO2, -OCF3, Cχ_6 alkyl, C2-6 alkenyl, C3_6 alkynyl, Cι_6 alkoxy, aryl, heteroaryl, and di-C 1 -6 alkylamino. As used herein, the terms "heterocyclyl" and "heterocyclic" as a group or part of a group refer to stable heterocyclic non-aromatic single and fused rings containing one or more hetero atoms independently selected from nitrogen, oxygen and sulfur. A fused heterocyclyl ring system may include carbocyclic rings and need include only one heterocyclic ring. Such heterocyclyl groups include piperazinyl, piperidinyl and n orpholinyl. Each ring may be optionally substituted with one or more substituents, which may be the same or different, selected from halo (such as fluoro, chloro, bromo), -
CN, -CF3, -OH, -NO2, -OCF3, Cι _6 alkyl, C2_6 alkenyl, C3_6 alkynyl, C _6 alkoxy, aryl, heteroaryl, and di-Cι_6 alkylamino.
As used herein the terms "halo" include iodo, bromo, chloro or fluoro, suitably bromo, chloro and fluoro, especially bromo and chloro. Composite terms such as "alkoxyalkyl" and "arylalkyl" refer to substituents comprising two interlinked groups, with the group named latterly in the term being the linking group, so that "alkoxyalkyl" means -(alkyl)-(alkoxy) whilst "arylalkyl" means - (alkyl)-(aryl).
The compounds of formula (I) or their salts or solvates are preferably in pharmaceutically acceptable or substantially pure form. By pharmaceutically acceptable form is meant, inter alia, having a pharmaceutically acceptable level of purity excluding normal pharmaceutical additives such as diluents and carriers, and including no material considered toxic at normal dosage levels.
A substantially pure form will generally contain at least 50% (excluding normal pharmaceutical additives), preferably 75%, more preferably 90% and still more preferably 95% of the compound of formula (I) or its salt or solvate.
One preferred pharmaceutically acceptable form is the crystalline form, including such form in pharmaceutical composition. In the case of salts and solvates the additional ionic and solvent moieties must also be non-toxic. Suitable salts are pharmaceutically acceptable salts.
Suitable pharmaceutically acceptable salts include the acid addition salts with the conventional pharmaceutical acids, for example maleic, hydrochloric, hydrobromic, phosphoric, acetic, fumaric, salicylic, citric, lactic, mandelic, tartaric, succinic, benzoic, ascorbic and methanesulfonic. Suitable pharmaceutically acceptable salts include salts of acidic moieties of the compounds of formula (I) when they are present, for example salts of carboxy groups or phenolic hydroxy groups.
Suitable salts of acidic moieties include metal salts, such as for example aluminium, alkali metal salts such as lithium, sodium or potassium, alkaline earth metal salts such as calcium or magnesium and ammonium or substituted ammonium salts, for example those with lower alkylamines such as triethylamine, hydroxyalkylamines such as 2-hydroxyethylamine, bis-(2-hy<koxyethyl)-amine or tri-(2-hydroxyethyl)-amine,
cycloalkylamines such as bicyclohexylamine, or with procaine, dibenzylpiperidine, N-benzyl-β-phenethylamine, dehydroabietylamine, N,N'-bisdehydroabietylamine, glucamine, N-methylglucamine or bases of the pyridine type such as pyridine, collidine, quinine or quinoline. Suitable solvates are pharmaceutically acceptable solvates.
Suitable pharmaceutically acceptable solvates include hydrates. For the avoidance of doubt when used herein the term "diabetes" includes diabetes mellitus, especially Type 2 diabetes, and conditions associated with diabetes mellitus. The term "conditions associated with diabetes" includes those conditions associated with the pre-diabetic state, conditions associated with diabetes mellitus itself and complications associated with diabetes mellitus.
The term "conditions associated with the pre-diabetic state" includes conditions such as insulin resistance, impaired glucose tolerance and hyperinsulinaemia. The term "conditions associated with diabetes mellitus itself includes hyperglycaemia, insulin resistance and obesity. Further conditions associated with diabetes mellitus itself include hypertension and cardiovascular disease, especially atherosclerosis and conditions associated with insulin resistance. Conditions associated with insulin resistance include polycystic ovarian syndrome and steroid induced insulin resistance. The term "complications associated with diabetes mellitus" includes renal disease, especially renal disease associated with Type II diabetes, neuropathy and retinopathy. Renal diseases associated with Type II diabetes include nephropathy, glomerulonephritis, glomerular sclerosis, nephrotic syndrome, hypertensive nephrosclerosis and end stage renal disease. The term "neurotraumatic diseases" includes both open or penetrating head trauma, such as caused by surgery, or a closed head trauma injury, such as caused by an injury to the head region, ischaemic stroke including acute stroke, particularly to the brain area, transient ischaemic attacks following coronary by-pass and cognitive decline following other transient ischaemic conditions. According to a further aspect of the present invention there is provided a process for the preparation of a compound of formula (I), or a salt and/or solvate thereof, which process comprises reacting a compound of formula (II),

wherein R
2, R3, R4 nd R^ are as defined in relation to formula (I) with a compound of formula (HI),
wherein R is as defined in relation to formula (I) and X is a suitable leaving group and thereafter, if required, carrying out one or more of the following optional steps: (i) converting a compound of formula (I) to a further compound of formula (I); (ii) removing any necessary protecting group;
(iii) preparing an appropriate derivative of the compound so formed.
Suitably X is chloro. It will be appreciated that compounds of formula (III) may also include related carboxylic acid anhydrides.
The reaction between the compounds of formulae (II) and (HI) is carried out in a suitable solvent, under conventional conditions, at a suitable temperature, providing a suitable rate of formation of the required product, over a suitable reaction time. A suitable solvent is pyridine. Suitable reaction temperatures include those in the range of 20°C to 220°C and, as appropriate, the reflux temperature of the solvent. Suitable reaction times are those in the range 12 to 72 hours. The reaction products are isolated using conventional methods. Conventional methods of heating and cooling may be employed, for example thermostatically controlled oil baths and ice/salt baths respectively. The reaction products are typically purified by conventional methods, such as crystallisation, chromatography and trituration. Crystalline product may be obtained by standard methods. In a preferred aspect, a compound of formula (III), such as n-butyric anhydride, is added to a solution of a compound of formula (II) in pyridine. The reaction mixture is
heated under reflux for 22 hours and allowed to cool to ambient temperature. The resulting solution is concentrated in vacuo and purified by column chromatography using one or more suitable solvents, such as 10% v/v methanol in chloroform, to afford the desired compound of formula (I). In a further preferred aspect, to a stirred solution of a compound of formula (II) in dry pyridine under argon is added a compound of formula (IH), such as cyclopropanecarbonyl chloride. The reaction mixture is stirred for 12 hours at room temperature. The resulting solution is concentrated in vacuo, and the crude oil is dissolved in ethyl acetate and washed with saturated sodium bicarbonate solution. The organic extract is dried over magnesium sulfate and further concentrated in vacuo. The resulting solid is triturated with a suitable solvent, such as dichloromethane, to afford the desired compound of formula (I).
It will be appreciated that treatment of a compound of formula (If) with a compound of formula (in), according to the above-mentioned process, may lead to the formation of a bw-acylated intermediate species of formula (IN),
wherein R2, R^, R4 and R^ are as defined in relation to formula (I). In a further aspect of the present invention, there is provided a process for the preparation of a compound of formula (I), or a salt and/or solvate thereof, which process comprises reacting a compound of formula (IV) with a nucleophile and thereafter, if required, carrying out one or more of the following optional steps: (i) converting a compound of formula (I) to a further compound of formula (I); (ii) removing any necessary protecting group;
(iii) preparing an appropriate derivative of the compound so formed.
The reaction between the compound of formula (TV) and a nucleophile is carried out optionally in a suitable solvent, under conventional conditions, at a suitable temperature, providing a suitable rate of formation of the required product, over a suitable reaction time. Suitably the reaction is performed using the nucleophile as a solvent. A suitable nucleophile is an amine, such as a primary or secondary amine. Suitable reaction temperatures include those in the range of 20°C to 100°C and, as appropriate, the reflux temperature of the solvent. Suitable reaction times are those in the range 1 to 48 hours. The reaction products are isolated using conventional methods. Conventional methods of heating and cooling may be employed, for example thermostatically controlled oil baths and ice/salt baths respectively. The reaction products are typically purified by conventional methods, such as crystallisation, chromatography and trituration. Crystalline product may be obtained by standard methods.
In a preferred aspect, a suitable nucleophile, such as piperidine, and water are added to a compound of formula (IV) under argon. The reaction mixture is stirred for 3 hours at ambient temperature. The resulting mixture is concentrated in vacuo and the crude product purified by preparative HPLC using one or more suitable solvents, such as 10-90% acetonitrile/0.1 % trifluoroacetic acid in water/0.1 % trifluoroacetic acid. The appropriate fractions are concentrated to dryness to afford the desired compound of formula (I). The above-mentioned conversions of a compound of formula (I) into another compound of formula (I) include any conversion, which maybe effected using conventional procedures, but in particular the said conversions include any combination of:
(i) converting one group R into another group R ; (ii) converting one group R^ into another group R^; and
(iii) converting one group R4 into another group R4.
The above-mentioned conversions (i), (ii) and (iii) may be performed using any appropriate method under conditions determined by the particular groups chosen.
Suitable conversions of one group R^ into another group R^, as in conversion (iii) above, include:
(a) converting a group R3 which represents halo, such as bromo, into another group R3 which represents aryl, such as phenyl. Such a conversion may be performed
using an appropriate arylation procedure, for example, by treating a compound of formula
(I) wherein R^ is halo, such as bromo, with an arylboronic acid or an arylboronate, such as phenylboronic acid.
Suitable conversions of one group R4 into another group R4, as in conversion (iv) above, include:
(b) converting a group R4 which represents -X-aryl, such as -SPh, into another group R4 which represents -X-aryl, such as -SO2PI1. Such a conversion may be performed using an appropriate oxidation procedure, for example, by treating a compound of formula (I) wherein R^ is -X-aryl, such as -SPh, with an appropriate oxidising agent such as wetβ-chloroperbenzoic acid.
It will be appreciated that the synthesis of compounds of formula (I) may involve the use of conventional protecting groups such as tert-butoxycarbonyl ("Boc"). Such protection/de-protection procedures may be performed using any appropriate method under conditions determined by the particular groups chosen. For example, a compound of formula (N),
wherein Rl, R
2, R^ and R^ are as defined in relation to formula (I), may be converted to a compound of formula (I) where R
4 is -ΝHAr by reaction with a suitable acid, such as hydrochloric acid in 1,4-dioxane. Compounds of formula (II) may be prepared by reaction of a compound of formula (VI),
wherein,
R2, R3, R4 and R^ are as defined in relation to formula (I), with hydrazine or a hydrate thereof. The reaction between the compound of formula (VI) and hydrazine or a hydrate thereof, is carried out in a suitable solvent at a suitable temperature, generally an elevated temperature, providing a suitable rate of formation of the required product, over a suitable reaction time. Suitable solvents include pyridine and ethanol. Suitable reaction temperatures include those in the range of 60 °C to 220 °C and, as appropriate, the reflux temperature of the solvent. Suitable reaction times are those in the range 1-72 hours. The reaction products are isolated using conventional methods. Typically, the reaction mixture is cooled, the product isolated by filtration, and dried. Conventional methods of heating and cooling may be employed, for example thermostatically controlled oil baths and ice/salt baths respectively. The reaction products may, if desired, be purified by conventional methods, such as crystallisation, chromatography and trituration.
In a preferred aspect, hydrazine hydrate is added to a stirred solution of the compound of formula (VI) in pyridine. The reaction mixture is stirred under reflux for 72 hours and cooled. The resulting mixture is concentrated in vacuo, and the residue dissolved in ethyl acetate and washed with aqueous sodium bicarbonate solution. The organic extract is dried over magnesium sulfate and concentrated in vacuo. The crude product is triturated with a suitable solvent, such as dichloromethane to afford the desired compound of formula (II).
Certain compounds of formula (fl) are believed to be novel and accordingly form a further aspect of the present invention. As mentioned hereinbefore, compounds of formula (IV) may be prepared by reaction of a compound of formula (II) with a compound of formula (in), in the presence of a suitable solvent such as pyridine.
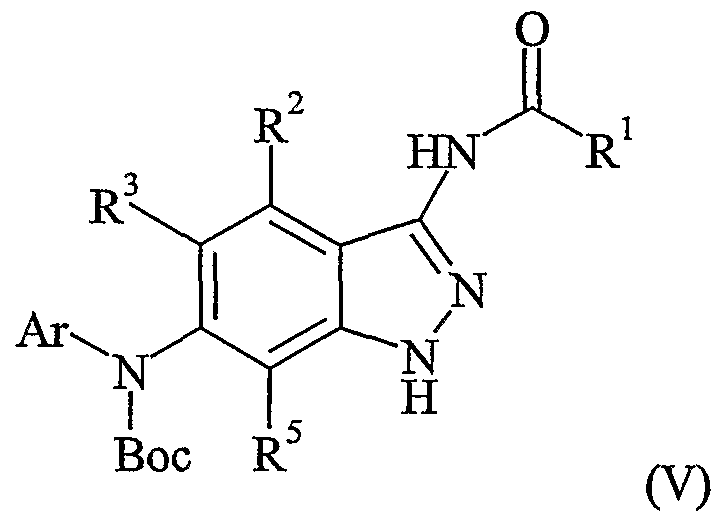
Compounds of formula (V) may be prepared by reaction of a compound of formula (VII),
wherein, R2, R3, R4 and R^ are as defined in relation to formula (I), with a compound of formula (HI). The synthesis of compounds of formula (V) by reaction of a compound of formula (NH) with a compound of formula (Hi) may be undertaken using analogous acylation conditions to those used for the preparation of compounds of formula (I) as described hereinbefore.
Compounds of formula (VI) may be prepared by reaction of a compound of formula (NTH),
wherein R2, R3 and R^ are as defined in relation to formula (I), with a compound of formula (LX),
wherein, 4 is as defined in relation to formula (I) and R^ is H or alkyl, in the presence of a suitable catalyst and a suitable base.
The reaction between the compound of formula (VTH) and a compound of formula (IX) is carried out in a suitable solvent under an inert atmosphere in the presence of a suitable catalyst and a suitable base, at a suitable temperature, generally an elevated temperature, providing a suitable rate of formation of the required product, over a suitable reaction time. Suitable solvents include a mixture of dimethylformamide, ethanol and water. Suitable catalysts are tetrakis(triphenylphosρhine)palladium(0) and PdCl2(dρpf).
A suitable base is KOAc. Suitable reaction temperatures include those in the range of 60 °C to 220 °C and, as appropriate, the reflux temperature of the solvent. Suitable reaction times are those in the range 1-72 hours. The reaction products are isolated using conventional methods. Typically, the reaction mixture is cooled, the product isolated by filtration, and dried. Conventional methods of heating and cooling may be employed, for example thermostatically controlled oil baths and ice/salt baths respectively. The reaction products may, if desired, be purified by conventional methods, such as crystallisation, chromatography and trituration. In a preferred aspect, to a stirred solution of a compound of formula (VIH), a compound of formula (IX), and potassium acetate in dimethylformamide, ethanol and water is added PdCl2(dppf). The resulting mixture is stirred at 100 °C for 4 hours, allowed to cool and is filtered through celite. The filtrate is added to ethyl acetate and washed with brine. The organic extract is separated, dried with magnesium sulfate and concentrated in vacuo. The crude solid is purified by chromatography using one or more suitable solvents, such as 10% diethyl ether/60-80 petrol, to afford the desired compound of formula (I).
Compounds of formula (NT) may also be prepared by reaction of a compound of formula (X),
wherein, R
2, R^ and R
$ are as defined in relation to formula (I), with a compound of formula (XI),
R— Y (XI)
wherein, R4 is as defined in relation to formula (I) and Y is halo, such as bromo, in the presence of a suitable catalyst.
The reaction between the compound of formula (X) and a compound of formula (XI) is carried out in a suitable solvent in the presence of a suitable catalyst and a suitable base, at a suitable temperature, generally an elevated temperature, providing a suitable rate of formation of the required product, over a suitable reaction time. Suitable solvents include a mixture of dimethylformamide, ethanol and water. A suitable catalyst is PdCl2(dppf). A suitable base is KOAc. Suitable reaction temperatures include those in the range of 60 °C to 220 °C and, as appropriate, the reflux temperature of the solvent. Suitable reaction times are those in the range 1-72 hours. The reaction products are isolated using conventional methods. Typically, the reaction mixture is cooled, the product isolated by filtration, and dried. Conventional methods of heating and cooling may be employed, for example thermostatically controlled oil baths and ice/salt baths respectively. The reaction products may, if desired, be purified by conventional methods, such as crystallisation, chromatography and trituration.
Compounds of formula (VI) wherein R4 is -X-aryl, where X is S, may be prepared by reaction of a compound of formula (XII),
wherein, R2, R3 and R^ are as defined in relation to formula (I), with a compound of formula (XTfl),
Ar— SH (XIH)
wherein Ar is aryl as defined in relation to formula (I), in the presence of a suitable base. The reaction between the compound of formula (XII) and a compound of formula (Xπi) is carried out in a suitable solvent in the presence of a suitable base, at a suitable temperature, providing a suitable rate of formation of the required product, over a suitable reaction time. A suitable solvent is dimethylformamide. A suitable base is sodium hydride. Suitable reaction temperatures include those in the range of 20 °C to 100 °C. Suitable reaction times are those in the range 1-48 hours. The reaction products are isolated using conventional methods. Typically, the reaction mixture is cooled, the product isolated by filtration, and dried. Conventional methods of heating and cooling may be employed, for example thermostatically controlled oil baths and ice/salt baths respectively. The reaction products may, if desired, be purified by conventional methods, such as crystallisation, chromatography and trituration.
In a preferred aspect, a compound of formula (XIH) is added to a compound of formula (XII) in the presence of sodium hydride in dimethylformamide. The reaction mixture is stirred at ambient temperature, under argon for 16 hours. The resulting solution is concentrated to dryness in vacuo and purified by preparative HPLC using one or more suitable solvents, such as 10-90% acetonitrile (containing 0.1% trifluoroacetic acid) in water (containing 0.1% trifluoroacetic acid). The appropriate fractions are concentrated to dryness to afford the desired compound of formula (VI).
Compounds of formula (VH) may be prepared by analogous methods to those mentioned hereinbefore for the preparation of compounds of formula (II).
Compounds of formula (X) may be prepared by reaction of a compound of formula (Vπi) with bis(pinacolato)diboron in the presence of a suitable catalyst, such as l,r-Bis(diphenylphosphino)ferrocenedichloropalladium(II), i.e. PdCl2(dppf).
Compounds of formula (HI), (VIH), (LX), (XI), (XH) and (XIH) are either commercially available or are prepared by analogy with known conventional procedures such as those in standard reference texts of synthetic methodology, for example, J. March, Advanced Organic Chemistry, 4th Edition, 1992, Wiley Interscience. Compounds of formula (IV), (V), (VII) and (X) are believed to be novel and accordingly form a further aspect of the present invention.
Compounds of formulae (I), (II), (V) and (VII) may exist as tautomers. The present invention encompasses all tautomeric forms of the compounds of (I), (II), (V) and (VII).
As stated above, the compounds of formula (I), or pharmaceutically acceptable salts or solvates thereof, are indicated to be useful as inhibitors of glycogen synthase kinase-3.
The invention therefore provides a compound of formula (I), or a pharmaceutically acceptable salt or solvate thereof, for use as an inhibitor of GSK-3. Accordingly, the present invention also provides a method for the treatment of conditions associated with a need for inhibition of GSK-3 such as diabetes, conditions associated with diabetes, chronic neurodegenerative conditions including dementias such as Alzheimer's disease, Parkinson's disease, progressive supranuclear palsy, subacute sclerosing panencephalitic parkinsonism, postencephalitic parkinsonism, pugilistic encephalitis, guam parkinsonism-dementia complex, Pick's disease, corticobasal degeneration, frontotemporal dementia, Huntingdon's disease, AIDS associated dementia, amyotrophic lateral sclerosis, multiple sclerosis and neurotraurnatic diseases such as acute stroke, mood disorders such as schizophrenia and bipolar disorders, promotion of functional recovery post stroke, cerebral bleeding (for example, due to solitary cerebral amyloid angiopathy), hair loss, obesity, atherosclerotic cardiovascular disease, hypertension, polycystic ovary syndrome, syndrome X, ischaemia, traumatic brain injury, cancer, leukopenia, Down's syndrome, Lewy body disease, inflammation, and immunodeficiency, which method comprises the administration of a pharmaceutically effective, non-toxic amount of a compound of formula (I) or a pharmaceutically acceptable salt or solvate thereof. The present invention further provides a compound of formula (I), or a pharmaceutically acceptable salt or solvate thereof, for use as an inhibitor of glycogen synthase kinase-3, and especially for use in the treatment of conditions associated with a need for the inhibition of GSK-3, such as diabetes, conditions associated with diabetes, chronic neurodegenerative conditions including dementias such as Alzheimer's disease, Parkinson's disease, progressive supranuclear palsy, subacute sclerosing panencephalitic parkinsonism, postencephalitic parkinsonism, pugilistic encephalitis, guam parkinsonism- dementia complex, Pick's disease, corticobasal degeneration, frontotemporal dementia,
Huntingdon's disease, ADDS associated dementia, amyotrophic lateral sclerosis, multiple sclerosis and neurotraurnatic diseases such as acute stroke, mood disorders such as schizophrenia and bipolar disorders, promotion of functional recovery post stroke, cerebral bleeding (for example, due to solitary cerebral amyloid angiopathy), hair loss, obesity, atherosclerotic cardiovascular disease, hypertension, polycystic ovary syndrome, syndrome X, ischaemia, traumatic brain injury, cancer, leukopenia, Down's syndrome, Lewy body disease, inflammation, and immunodeficiency.
The present invention also provides the use of a compound of formula (I), or a pharmaceutically acceptable salt or solvate thereof, for the manufacture of a medicament for the treatment of conditions associated with a need for the inhibition of GSK-3, such as diabetes, conditions associated with diabetes, chronic neurodegenerative conditions including dementias such as Alzheimer's disease, Parkinson's disease, progressive supranuclear palsy, subacute sclerosing panencephalitic parkinsonism, postencephalitic parkinsonism, pugilistic encephalitis, guam parkinsonism-dementia complex, Pick's disease, corticobasal degeneration, frontotemporal dementia, Huntingdon's disease, ADDS associated dementia, amyotrophic lateral sclerosis, multiple sclerosis and neurotraurnatic diseases such as acute stroke, mood disorders such as schizophrenia and bipolar disorders, promotion of functional recovery post stroke, cerebral bleeding (for example, due to solitary cerebral amyloid angiopathy), hair loss, obesity, atherosclerotic cardiovascular disease, hypertension, polycystic ovary syndrome, syndrome X, ischaemia, traumatic brain injury, cancer, leukopenia, Down's syndrome, Lewy body disease, inflammation, and immunodeficiency.
In a further aspect of this invention, there is provided a compound of formula (I), or a pharmaceutically acceptable salt or solvate thereof, for use as an active therapeutic substance.
Preferably, the compounds of formula (I), or pharmaceutically acceptable salts or solvates thereof, are administered as pharmaceutically acceptable compositions.
Accordingly, the invention also provides a pharmaceutical composition which comprises a compound of formula (I), or a pharmaceutically acceptable salt or solvate thereof, and a pharmaceutically acceptable carrier.
The active compounds are usually administered as the sole medicament agent but they may be administered in combination with other medicament agents as dictated by the severity and type of disease being treated.
The said combination comprises co-administration of a compound of formula (I), or a pharmaceutically acceptable salt or solvate thereof, and an additional medicament agent or the sequential administration of a compound of formula (I), or a pharmaceutically acceptable derivative thereof, and the additional medicament agent.
Co-administration includes administration of a pharmaceutical composition which contains both a compound of formula (I), or a pharmaceutically acceptable salt or solvate thereof, and the additional medicament agent or the essentially simultaneous administration of separate pharmaceutical compositions of a compound of formula (I), or a pharmaceutically acceptable salt or solvate thereof, and the additional medicament agent.
The compositions of the invention are preferably adapted for oral administration. However, they may be adapted for other modes of administration. The compositions may be in the form of tablets, capsules, powders, granules, lozenges, suppositories, reconstitutable powders, or liquid preparations, such as oral or sterile parenteral solutions or suspensions. In order to obtain consistency of administration it is preferred that a composition of the invention is in the form of a unit dose. Preferably the composition are in unit dosage form. A unit dose will generally contain from 0.1 to 1000 mg of the active compound.
Generally an effective administered amount of a compound of the invention will depend on the relative efficacy of the compound chosen, the severity of the disorder being treated and the weight of the sufferer. However, active compounds will typically be administered once or more times a day for example 2, 3 or 4 times daily, with typical total daily doses in the range of from 0.1 to 800 mg/kg/day.
Suitable dose forms for oral administration may be tablets and capsules and may contain conventional excipients such as binding agents, for example syrup, acacia, gelatin, sorbitol, tragacanth, or polyvinylpyrrolidone; fillers, for example lactose, sugar, maize starch, calcium phosphate, sorbitol or glycine; tabletting lubricants, for example magnesium stearate; disintegrants, for example starch, polyvinylpyrrolidone, sodium
starch glycollate or microcrystalline cellulose; or pharmaceutically acceptable wetting agents such as sodium lauryl sulfate.
The solid oral compositions may be prepared by conventional methods of blending, filling or tabletting. Repeated blending operations may be used to distribute the active agent throughout those compositions employing large quantities of fillers. Such operations are of course conventional in the art. The tablets may be coated according to methods well known in normal pharmaceutical practice, in particular with an enteric coating.
Oral liquid preparations may be in the form of, for example, emulsions, syrups, or elixirs, or may be presented as a dry product for reconstitution with water or other suitable vehicle before use. Such liquid preparations may contain conventional additives such as suspending agents, for example sorbitol, syrup, methyl cellulose, gelatin, hydroxyethylcellulose, carboxymethylcellulose, aluminium stearate gel, hydrogenated edible fats; emulsifying agents, for example lecithin, sorbitan monooleate, or acacia; non-aqueous vehicles (which may include edible oils), for example almond oil, fractionated coconut oil, oily esters such as esters of glycerine, propylene glycol, or ethyl alcohol; preservatives, for example methyl or propyl p-hydroxybenzoate or sorbic acid; and if desired conventional flavouring or colouring agents.
For parenteral administration, fluid unit dosage forms are prepared utilizing the compound and a sterile vehicle, and, depending on the concentration used, can be either suspended or dissolved in the vehicle. In preparing solutions the compound can be dissolved in water for injection and filter sterilized before filling into a suitable vial or ampoule and sealing. Advantageously, adjuvants such as a local anaesthetic, a preservative and buffering agents can be dissolved in the vehicle. To enhance the stability, the composition can be frozen after filling into the vial and the water removed under vacuum. Parenteral suspensions are prepared in substantially the same manner, except that the compound is suspended in the vehicle instead of being dissolved, and sterilization cannot be accomplished by filtration. The compound can be sterilized by exposure to ethylene oxide before suspending in the sterile vehicle. Advantageously, a surfactant or wetting agent is included in the composition to facilitate uniform distribution of the compound.
The formulations mentioned herein are carried out using standard methods such as those described or referred to in reference texts such as the British and US Pharmacopoeias, Remington's Pharmaceutical Sciences (Mack Publishing Co.), Martindale The Extra Pharmacopoeia (London, The Pharmaceutical Press) or the above- mentioned publications.
Suitable methods for preparing and suitable unit dosages for the additional medicament agent, such as the antidiabetic agent mentioned herein include those methods and dosages described or referred to in the above-mentioned reference texts.
GSK-3 Assay
GSK-3 assays used to test the compounds of the invention include the following protocol which is based on the ability of the kinase to phosphorylate a biotinylated 26 mer peptide, Biot- KYRRAAVPPSPSLSRHSSPHQ(S)EDEEE, the sequence of which is derived from the phosphorylation site of glycogen synthase, where (S) is a pre- phosphorylated serine as in glycogen synthase in vivo and the three consensus sites for GSK-3 specific phosphorylation are underlined. The phosphorylated biotinylated peptide is then captured onto Streptavidin coated SPA beads (Amersham Technology), where the signal from the 3p [s amplified via the scintillant contained in the beads.
Using microtitre plates, GSK-3 was assayed in 50 mM MOPS buffer, pH 7.0, containing 5% glycerol, 0.01% Tween-20, 7.5 mM 2-mercaptoethanol, 10 mM magnesium acetate, 8 uM of the above peptide, and 10 uM [33p]_ATP. After incubation at room temperature, the reaction was stopped by addition of 50 mM EDTA solution containing the Streptavidin coated SPA beads to give a final 0.2 mgs. Following centrifugation, the microtitre plates are counted in a Trilux 1450 microbeta liquid scintillation counter (Wallac). IC50 values are generated for each compound by fitting to a four parameter model.
The most potent compounds of the present invention show IC50 values in the range of 1 to 500 nM.
No adverse toxicological effects are expected for the compounds of the invention, when administered in accordance with the invention.
The following Descriptions and Examples illustrate the invention, but do not limit it in any way.
Synthetic Method A
Example 1
N-(5-Bromo-lH-indazol-3-yl)butyramide n-Butyric anhydride (0.80 mL, 4.9 mmol) was added to a solution of 5-bromo-lH- indazol-3-ylamine (1.06 g, 5.0 mmol) in pyridine (5 mL). The reaction mixture was stirred at reflux for 22 hours, then allowed to cool. The solution was concentrated and purified by column chromatography (10% v/v methanol in chloroform) affording the title compound as a solid. MS (APCI+ve): [M+H]+ at m/z 282/284 (CπH12BrN3O requires [M+H]+ at m/z 282/284).
1HNMR 6 (DMSO-d6): 0.9 (3H, t), 1.6 (2H, appq), 2.4 (2H, t), 7.4 (2H, apps), 8.0 (1H, s), 10.4 (1H, s), 12.8 (1H, s).
Synthetic Method B Example 2 N-(5-Phenyl-lH-indazol-3-yl)-butyramide
Tefrakis(triphenylphosphine)palladium(0) (20 mg, 0.02 mmol) was added to a stirred solution of phenylboronic acid (56 mg, 0.46 mmol), N-(5-bromo-lH-indazol-3- yl)butyramide (100 mg, 0.35 mmol) and sodium carbonate (0.5 mL of 2M aqueous solution) in 1,2-dimethoxyethane (1 mL) and ethanol (0.5 mL). The resulting suspension was stirred at reflux for 20 hours, concentrated in vacuo and water (25 mL) added. The aqueous layer was extracted with ethyl acetate (x3) and the combined organic extracts were washed with brine, dried and concentrated. Purification by column chromatography (2% v/v methanol in dichloromethane) afforded the title compound as a solid.
MS (APCI+ve): [M+H]+ at m/z 280 (C17H17N3O requires [M+H]+ at m/z 280).
1H NMR δ (DMSO-d6): 0.9 (3H, t), 1.6 (2H, appq), 2.4 (2H, t), 7.7-7.3 (7H, m), 8.0 (1H, s), 10.3 (1H, s), 12.6 (1H, s).
Synthetic Method C
Example 3
Cyclopentanecarboxylic acid (6-phenylamino-lH-indazoI-3-yl)-amide
[3 -[( 1 -Cycloρentyl-methanoyl)-amino] - 1 H-indazol-6-yl] -phenyl-carbamic acid 1,1- dimethylethyl ester (100 mg, 0.24 mmol) was dissolved in 4N hydrochloric acid in dioxane (5 mL) and the reaction mixture was stirred at room temperature for 2 hours. The reaction mixture was concentrated and the resulting crude oil purified by chromatography (2% methanol/dichloromethane) to afford a beige solid. The solid was washed with water and diethyl ether to afford the title compound as a solid. MS (ACPI+ve): [M+H]+ at m/z 321 (C19H20N4O requires [M+H]+ at m/z 321).
1H NMR δ (DMSO-d6): 1.58-1.90 (8H, m), 2.86-2.92 (1H, m), 7.75-7.79 (1H, dd), 7.84- 7.90 (1H, t), 7.97-7.98 (1H, d), 7.13-7.16 (2H, m), 7.24-7.30 (2H, m), 7.59-7.63 (1H, d), 8.29 (1H, s), 10.17 (1H, s), 12.09 (1H, s).
Description 1
2-Chloro-4-phenylamino-benzonitrile
To a stirred and degassed solution of 4-bromo-2-chlorobenzonitrile (2 g, 9.2 mmol), aniline (1 ml, 11.1 mmol), sodium tert-butoxide (1.3 g, 12.9 mmol) and BINAP (0.9 g,
1.4 mmol) in dry toluene was added Pd2(dba)3 (0.4 g, 0.5 mmol). The reaction mixture was stirred at 80°C for 18 hours, allowed to cool and filtered through celite. The filtrate was concentrated and the crude residue was purified by chromatography (20% ethyl acetate/60-80 petrol) to afford the title compound as a solid.
MS (ACPI+ve): [M+H]+ at m/z 229/231 (C13H9C1N2 requires [M+H]+ at m/z 229/231).
1H NMR (DMSO-d6): 6.97-7.01 (1H, dd), 7.07-7.10 (2H, m), 7.19-7.23 (2H, m), 7.35- 7.39 (2H, m), 7.65-7.68 (1H, d), 9.15 (1H, s).
Description 2
(3-Chloro-4-cyanophenyl)-phenyI-carbamic acid 1,1 -dimethylethyl ester
To a stirred solution of 2-chloro-4-phenylaminobenzonitrile (1.8 g, 7.9 mmol) in dry dimethylformamide (20 mL) was added at 0°C sodium hydride (0.5 g, 11.8 mmol) and the reaction mixture was stirred under argon for 1 hour. Di-tert-butyl dicarbonate (2.6 g, 11.8 mmol) was added and the reaction mixture was stirred at room temperature for 18
hours. The reaction mixture was quenched with water and concentrated. The crude oil was taken up in ethyl acetate (100 mL) and washed with brine (100 mL). The organic extract was dried (magnesium sulfate) and concentrated. The crude solid was purified by chromatography (20% ethyl acetate/60-80 petrol) to afford the title compound as a solid. MS (ACPI+ve): [M+H]+ at m z 329/331 (C18H17ClN2O2 requires [M+H]+ at m z 329/331).
1H NMR (DMSO-d6): 1.38 (9H, s), 7.20-7.22 (IH, dd), 7.27-7.29 (2H, m), 7.35-7.37 (IH, m), 7.43-7.45 (2H, m), 7.60-7.61 (IH, d), 7.88-7.90 (IH, d).
The general method of Description 1 was also employed to introduce the 4- benzyloxycarbonylpiperazin-1-yl substituent of Example 33 using 1- benzyloxycarbonylpiperazine and 4-bromo-2-chlorobenzonitrile
Synthetic Method D Example 4
Cyclopropanecarbox lie acid (6-thiophen-2-yl-lH-indazol-3-yl)-amide
To a stirred solution of 6-thiophen-2-yl-lH-indazol-3-ylamine (320 mg, 1.5 mmol) in dry pyridine (5 mL) under argon was added cyclopropanecarbonyl chloride (0.135 mL, 1.5 mmol) and the reaction was stirred at room temperature for 12 hours. The solution was concentrated. The crude oil was taken up in ethyl acetate (100 mL) and washed with saturated sodium bicarbonate solution in water (100 mL). The organic extract was dried (magnesium sulfate) and concentrated. The crude solid was triturated with dichloromethane to afford the title compound as a solid. MS (ACPI+ve): [M+H]+ at m/z 284 (C15H13N3OS requires [M+H]+ at m/z 284). 1H NMR (DMSO-d6): 0.82-0.90 (4H, m), 1.89-1.96 (IH, m), 7.14-7.18 (IH, m), 7.33- 7.40 (IH, dd), 7.55-7.61 (3H, m), 7.80-7.84 (IH, d), 10.7 (IH, s), 12.65 (IH, s).
Description 3 2-Chloro-4-thiophen-2-yl-benzonitrile To a stirred and degassed solution of 4-bromo-2-chlorobenzonitrile (1 g, 4.6 mmol), 2- thiophene boronic acid (0.7 g, 5.5 mmol) and potassium acetate (1.4 g, 13.9 mmol) in dimethylformamide (10 mL), ethanol (5 mL) and water (5 mL) was added PdCl2(dppf)
(113 mg, 0.1 mmol). The reaction mixture was stirred at 100°C for 4 hours, allowed to cool and filtered through celite. The filtrate was taken up in ethyl acetate (100 mL) and was washed with brine (3 x 100 mL). The organic extract was dried (magnesium sulfate) and concentrated. The crude solid was purified by chromatography (10% diethyl ether/60-80 petrol) to afford the title compound as a solid.
MS (ACPI+ve): [M+H]+ at m/z 220/222 (CnH6ClNS requires [M+H]+ at m/z 220/222). 1H NMR (DMSO-d6): 7.21-7.24 (IH, m), 7.75-7.84 (3H, m), 7.97-8.00 (IH, d), 8.06-8.07 (lH, d).
Description 4
6-Thiophen-2-yl-lH-indazol-3-ylamine
To a stirred solution of 2-chloro-4-thiophen-2-yl-benzonitrile (900 mg, 4.1 mmol) in pyridine (5 mL) was added hydrazine hydrate (2 mL, 41 mmol). The reaction mixture was stirred at reflux for 72 hours, allowed to cool and concentrated. The crude oil was taken up in ethyl acetate (100 mL) and washed with saturated sodium bicarbonate solution in water (100 mL). The organic extract was dried (magnesium sulfate) and concentrated. The crude solid was triturated with dichloromethane to afford the title compound as a solid. MS (ACPI+ve): [M+H]+ at m/z 216 (CnH9N3S requires [M+H]+ at m/z 216). 1H NMR (DMSO-d6): 5.37 (2H, s), 7.12-7.16 (IH, t), 7.21-7.25 (IH, dd), 7.43 (IH, s), 7.52-7.54 (2H, m), 7.69-7.72 (IH, d), 11.42 (IH, s).
An alternative method for the introduction of a substituent R^ is provided the method of Description 5 with a subsequent coupling step. In the latter process an appropriate aryl or heteroaryl halide is reacted with the compound of Description 5 by the general procedure of Description 3 employing a suitable palladium catalyst.
Description 5 2-Chloro-4-(4,4,5,5-tetramethyl-[l,3,2]dioxaborolan-2-yl)-benzonitrile l,r-Bis(diphenylphosphino)ferrocenedichloropalladium(π) (1.7 g, 2.1 mmol) was added to a stirred and degassed solution of 4-bromo-2-chlorobenzonitrile (15 g, 69.3 mmol), bis(pinacolato)diboron (21.1 g, 83.2 mmol) and potassium acetate (20.4 g, 207.9 mmol)
in dimethyl sulfoxide (200 mL). The reaction mixture was stirred at 100°C under argon for 18 hours, then allowed to cool. The solution was filtered through celite, then ethyl acetate (500 mL) was added and the organic phase was washed three times with brine (500 mL). The organic layer was dried (magnesium sulfate), concentrated and purified by chromatography (30% v/v ethyl acetate in petroleum ether) to afford a brown solid. Trituration with diethyl ether afforded the title compound as a solid. 1H NMR δ (DMSO-d6): 1.31 (12H, s), 7.73-7.76 (IH, dd), 7.82 (IH, app s), 7.96-8.00 (lH, d).
Examples containing a phenol substituent may be prepared by cleavage of an appropriate ether by the general procedure of Description 6. The product can then be acylated by a general method such as Synthetic Method D.
Description 6 4-(3-Amino-lH-mdazol-6-yl)-phenol
6-(4-Methoxy-phenyl)-lH-indazol-3-ylamine (150 mg, 0.63 mmol) was stirred at reflux for 4 hour in a 48% hydrobromic acid aqueous solution (5 mL). The reaction mixture was allowed to cool and was concentrated. The crude solid was triturated with water to afford the title compound as a solid. 1H NMR (DMSO-d6): 5.38 (2H, s), 6.83-6.86 (2H, d), 7.11-7.15 (IH, d), 7.31 (IH, s), 7.48-7.52 (2H, d), 7.71-7.77 (IH, d), 9.51 (IH, s), 11.35 (IH, s).
Where a ketone is present as an aryl substituent a hydrazone may be formed in the the cyclization step. Subsequent cleavage back to a carbonyl compound is exemplified in Description 7.
Description 7 l-[4-(3-Amino-lH-indazol-6-yl)-phenyl]-ethanone
6-[4-(l-Hydrazonoethyl)phenyl]-lH-indazol-3-ylamine (100 mg, 0.38 mmol) was stirred at reflux for 18 hours in a 4N hydrochloric solution in dioxane (10 mL) and water (1 mL). The reaction mixture was allowed to cool and was concentrated. The crude solid was taken up in ethyl acetate (50 mL) and washed with saturated sodium bicarbonate solution
in water (50 mL). The organic extract was dried (magnesium sulfate) and concentrated. The resulting solid was purified by chromatography (ethyl acetate) to afford the title compound as a solid.
MS (ACPI+ve): [M+H]+ at m/z 252 (C15H13N3O requires [M+H]+ at m/z 252). 1H NMR (DMSO-d6): 2.62 (3H, s), 5.41 (2H, s), 7.25-7.29 (IH, dd), 7.52 (IH, s), 7.78- 7.87 (3H, m), 8.03-8.06 (2H, d), 11.53 (IH, s).
Synthetic Method E Example 11 Cyclopropanecarboxylic acid (6-benzenesulfanyl-lH-indazol-3-yl)-amide
6-Phenylsulfanyl-lH-indazol-3-ylamine (303 mg, 1.26 mmol) and cyclopropylcarbonyl chloride (114 μL, 1.26 mmol) were dissolved in anhydrous pyridine (5 mL). The solution was stirred for 16 hours under argon at ambient temperature. A further amount of cyclopropyl carbonyl chloride (114 μL, 1.26 mmol) was added and the reaction stirred for a further 2 hours at the same temperature. Water (2 mL) was added and the solution concentrated to dryness. Piperidine (20 mL) and water (3 mL) were added and the solution stirred at ambient temperature under argon for 3 hours. The solvents were removed in vacuo and the product purified by preparative HPLC (10-90% acetonitrile (0.1% trifluoroacetic acid) in water (0.1% trifluoroacetic acid) over 10 minutes, C18 column). The appropriate fractions were concentrated to dryness to yield the title compound, as a solid.
MS (APCI+ve): [M+H]+ at m/z 310 (C17H15N3OS requires [M+H]+ at m/z 310.)
IH NMR δ (DMSO-d6) : 0.78-0.84 (4H, m), 1.91 (IH, br m), 6.97 (IH, dd), 7.26 (IH, s), 7.37 (5H, m), 7.77 (IH, d), 10.67 (IH, s), 12.58 (IH, s).
The starting material for Synthetic Method E is prepared as shown below in Descriptions 8 and 9.
Description 8 2-Chloro-4-phenylsulfanyl-benzonitrile
Thiophenol (3.63 mL, 35.4 mmol) was added to 2-chloro-4-fluoroberιzonitrile (5 g, 32.1 mmol), sodium hydride (2.56 g, 64.2 mmol) in dimethylformamide (20 mL). The
resulting solution was stirred at ambient temperature under argon for 16 hours. The solution was concentrated to dryness and purified by preparative HPLC (10-90% acetonitrile (0.1% trifluoroacetic acid) in water (0.1% trifluoroacetic acid) over 10 minutes, C18 column). The appropriate fractions were concentrated to dryness to yield the title compound as a solid.
MS (APCI-ve): [M-H]" at m z 168/170 (C13H8NC1S requires [M-H]" at m/z 244/246. Observed ion fits for loss of phenyl group).
IH NMR δ (DMSO-d6): 7.14 (IH, dd), 7.35 (IH, d), 7.56 (5H, m), 7.84 (IH, d).
Description 9
6-Phenylsulfanyl-lH-indazol-3-ylamine
Hydrazine hydrate (4 mL, 128 mmol) was added to 2-chloro-4-phenylsulfanyl- benzonitrile (1 g, 4.07 mmol) in pyridine (10 mL). The solution was heated at reflux for 96 hours, allowed to cool and concentrated to dryness. Purification was achieved by preparative HPLC (10-90% acetonitrile (0.1% trifluoroacetic acid) in water (0.1% trifluoroacetic acid) over 10 minutes, C18 column). The appropriate fractions were concentrated to dryness to yield the title compound as a solid.
MS (APCI+ve): [M+H]+ at m/z 242 (C13HπN3S requires [M+HJ+ at m/z 242.) IH NMR δ (DMSO-d6): 4.00-6.00 (2H, v. br s), 6.98 (IH, dd), 7.11 (IH, s), 7.42 (5H, m), 7.84 (IH, d). The pyrazole NH was not observed.
Synthetic Method F
Example 16
Cyclopentanecarboxylic acid (6-benzenesulfonyl-lH-indazol-3-yl)-amide Cyclopentanecarboxylic acid (6-benzenesulfanyl-lH-indazol-3-yl)-amide (100 mg, 0.324 mmol) and weto-chloroperbenzoic acid (168 mg, 0.972 mmol) were dissolved in dichloromethane (30 mL). The solution was stirred for 16 hours under argon at ambient temperature. A further 150 mL dichloromethane was added to dissolve the precipitate and the organic layer was washed with saturated sodium bicarbonate solution (3 x 150 mL). The organic layer was concentrated and purified by preparative HPLC (10-90% acetonitrile (0.1% trifluoroacetic acid) in water (0.1% trifluoroacetic acid) over 10
minutes, C18 column). The appropriate fractions were concentrated to dryness to yield the title compound as a solid.
MS (APCI+ve): [M+H]+ at m/z 370 (C19H19N3O3S requires [M+H]+ at m/z 370.) H NMR δ (DMSO-d6) : 1.54-1.89 (8H, m), 2.90 (IH, quint), 7.50 (IH, dd), 7.62 (2H, 5 t), 7.69 (IH, t), 7.95-7.99 (3H, m), 8.08 (IH, s), 10.48 (IH, s), 13.22 (IH, br s).
Synthetic Method G
Example 31
Cyclopropanecarboxy lie acid (6-piperazin-l-yl-lH-indazol-3-yl)-amide
10 10% Palladium on charcoal (16 mg) was added to a solution of 4-(3-[(l-cyclopropyl- methanoyl)-amino]-lH-indazol-6-yl)-piperazine-l-carboxylic acid benzyl ester (160 mg, 0.38 mmol) in ethanol (10 mL) and the reaction mixture was stirred at room temperature under 50 psi of hydrogen for 48 hours. The solution was filtered through celite and concentrated. The crude solid was purified by chromatography on silica gel (10%
15 methanol/dichloromethane then 10% (2N ammonia in methanol)/dichloromethane) to afford the title compound as a solid.
MS (ACPI+ve): [M+H]+ at m/z 286 (C15H19N5O requires [M+H]+ at m/z 286). 1H NMR δ (DMSO-d6): 0.78-0.83 (4H, m), 1.86-1.93 (IH, m), 2.77-2.78 (4H, m), 2.88- 2.92 (4H, m), 6.62 (IH, apps), 6.79-6.84 (IH, dd), 7.60-7.63 (IH, d), 10.50 (IH, s), 12.14 0 (lH, s).
The further Examples described herein were prepared by analogy with Synthetic Methods A-G described above.
5
0
Table 1