WO1999007732A1 - SELECTIVE FACTOR Xa INHIBITORS - Google Patents
SELECTIVE FACTOR Xa INHIBITORS Download PDFInfo
- Publication number
- WO1999007732A1 WO1999007732A1 PCT/US1998/016720 US9816720W WO9907732A1 WO 1999007732 A1 WO1999007732 A1 WO 1999007732A1 US 9816720 W US9816720 W US 9816720W WO 9907732 A1 WO9907732 A1 WO 9907732A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- alkyl
- compound
- aryl
- alkylaryl
- Prior art date
Links
- 0 CC(C1)*CC1(*)C1CCCCCC1 Chemical compound CC(C1)*CC1(*)C1CCCCCC1 0.000 description 2
- WEBZMGHVGASLFJ-UHFFFAOYSA-N CCc(cccc1)c1SC Chemical compound CCc(cccc1)c1SC WEBZMGHVGASLFJ-UHFFFAOYSA-N 0.000 description 1
- JMRIRFQZDMHJLV-SFHVURJKSA-N COC(CN(CC1=C(C[C@@H]2NS(Cc3ccccc3)(=O)=O)C=CCC1)C2=O)=O Chemical compound COC(CN(CC1=C(C[C@@H]2NS(Cc3ccccc3)(=O)=O)C=CCC1)C2=O)=O JMRIRFQZDMHJLV-SFHVURJKSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06139—Dipeptides with the first amino acid being heterocyclic
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
Definitions
- This invention relates to a novel class of bicyclic aryl azepinone compounds which are potent and highly selective inhibitors of factor Xa or factor Xa when assembled in the prothrombinase complex. These compounds show selectivity for factor Xa versus other proteases of the coagulation (e.g. thrombin, rVIla, fEXa) or the fibrinolytic cascades (e.g. plasminogen activators, plasmin).
- other proteases of the coagulation e.g. thrombin, rVIla, fEXa
- fibrinolytic cascades e.g. plasminogen activators, plasmin
- Blood coagulation protects mammalian species when the integrity of the blood vessel wall is damaged and uncontrolled loss of blood threatens survival. Coagulation, resulting in the clotting of blood, is an important component of hemostasis. Under normal hemostatic circumstances, there is maintained an acute balance of clot formation and clot removal (fibrinolysis).
- the blood coagulation cascade involves the conversion of a variety of inactive enzymes (zymogens) into active enzymes, which ultimately convert the soluble plasma protein fibrinogen into an insoluble matrix of highly cross-linked fibrin. (See
- thrombin A key enzyme in the coagulation cascade, as well as in hemostasis, is thrombin.
- Thrombin is intimately involved in the process of thrombus formation, but under normal circumstances can also play an anticoagulant role in hemostasis through its ability to convert protein C into activated protein C in a thrombomodulin-dependent manner.
- Thrombin plays a central role in thrombosis through its ability to catalyze the penultimate conversion of fibrinogen into fibrin and through its potent platelet activation activity.
- thrombin i.e. heparins, low- molecular weight heparins and coumarins. Thrombin is generated at the convergence of the intrinsic and extrinsic coagulation pathways by the prothrombinase complex.
- the prothrombinase complex is formed when activated Factor X (factor Xa) and its non- enzymatic cofactor, factor Va assemble on phospholipid surfaces in a Ca +2 -dependent fashion as reviewed by Mann, et al, "Surface-Dependent Reactions of the Vitamin K- Dependent Enzymes", Blood 76: 1-16 (1990).
- Factor Xa Factor X
- factor Va non-enzymatic cofactor
- the prothrombinase complex converts the zymogen prothrombin into the active procoagulant thrombin.
- prothrombinase complex The location of the prothrombinase complex at the convergence of the intrinsic and extrinsic coagulation pathways, and the significant amplification of thrombin generation (393,000-fold over uncomplexed factor Xa) mediated by the complex at a limited number of targeted catalytic units present at vascular lesion sites, suggests that inhibition of thrombin generation is an ideal method to block uncontrolled procoagulant activity.
- factor Xa appears to have a single physiologic substrate, namely prothrombin.
- Plasma contains an endogenous inhibitor of both the factor Vila-tissue factor (TF) complex and factor Xa called tissue factor pathway inhibitor (TFPI).
- TFPI is a Kunitz- type protease inhibitor with three tandem Kunitz domains. TFPI inhibits the TF/fVIIa complex in a two-step mechanism which includes the initial interaction of the second
- the second step involves the inhibition of the TF/fVIIa complex by formation of a quaternary complex TF/fVIIa/TFPI/fXa as described by Girard, et al, "Functional Significance of the Kunitz-type Inhibitory Domains of Lipoprotein-associated Coagulation Inhibitor", Nature 338:518-520 (1989).
- Polypeptides derived from hematophagous organisms have been reported which are highly potent and specific inhibitors of factor Xa.
- Tick Anticoagulant Peptide TRIP
- Ghilanten Anti-coagulant-antimetastatic Principle of the South American Leech, Haementeria ghilianii” , Biochem. Biophys. Res. Commun. 166: 1384-1389 (1990); Brankamp, et al, "Ghilantens: Anticoagulants, Antimetastatic Proteins from the South American Leech Haementeria ghilianii", J. Lab. Clin. Med. 115:89-97 (1990); Jacobs, et al, "Isolation and Characterization of a Coagulation Factor Xa Inhibitor from Black Fly
- Factor Xa inhibitory compounds which are not large polypeptide-type inhibitors have also been reported including: Tidwell, et al, "Strategies for Anticoagulation With Synthetic Protease Inhibitors. Xa Inhibitors Versus Thrombin Inhibitors", Thromb. Res.
- Bovine Factor Xa and Thrombin Comparison of Their Anticoagulant Efficiency", Thromb. Res. 54:245-252 (1989); Kam, et al, “Mechanism Based Isocoumarin Inhibitors for Trypsin and Blood Coagulation Serine Proteases: New Anticoagulants", Biochemistry 27:2547-2557 (1988); Hauptmann, et al, “Comparison of the Anticoagulant and Antithrombotic Effects of Synthetic Thrombin and Factor Xa Inhibitors", Thromb.
- Fractor Xa Inhibitors discloses pentapeptide X1-Y-I-R-X2 derivatives as factor Xa inhibitors. Said compounds are useful for inhibiting blood clotting in the treatment of thrombosis, stroke, and myocardial infarction.
- the present invention relates to novel peptide mimetic analogs, their pharmaceutically acceptable isomers, salts, hydrates, solvates and prodrug derivatives.
- the present invention includes pharmaceutical compositions comprising a pharmaceutically effective amount of the compounds of this invention and a pharmaceutically acceptable carrier. These compositions are useful as potent and specific inhibitors of blood coagulation in mammals.
- the invention relates to methods of using these inhibitors as therapeutic agents for disease states in mammals which have disorders of coagulation such as in the treatment or prevention of unstable angina, refractory angina, myocardial infarction, transient ischemic attacks, thrombotic stroke, embolic stroke, disseminated intravascular coagulation including the treatment of septic shock, deep venous thrombosis in the prevention of pulmonary embolism or the treatment of reocclusion or restenosis of reperfused coronary arteries.
- These compositions may optionally include anticoagulants, antiplatelet agents, and thrombolytic agents.
- the present invention provides compounds of general formula I:
- R and R ⁇ are independently selected from the group consisting of H, C ⁇ _ alkyl, C 3-8 cycloalkyl, C ⁇ .. 3 alkylaryl, C ⁇ _ 3 alkyl-C -8 cycloalkyl and aryl;
- R " is H, C ⁇ - 6 alkyl, or R ⁇ and R are taken together to form a carbocyclic ring; q is an integer from 0-2; r is an integer from 0-4; s is an integer from 0-1 ; t is an integer from 0-4;
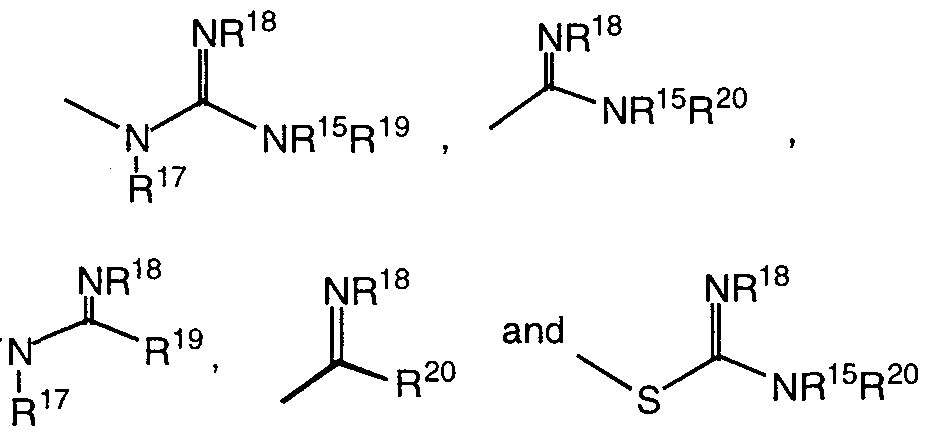
- A is selected from the group consisting of R 8 , -NR 8 R 9 ,
- R 8 , R 9 , R 10 and R 1 ' are independently selected from the group consisting of H,
- R 12 is selected from the group consisting of H, C ⁇ _ 6 alkyl, aryl and C ⁇ - alkylaryl, or can be taken together with R 10 or R 11 to form a 5-6 membered ring
- R 13 is selected from the group consisting of H, C 1-6 alkyl, aryl and C ⁇ _ 4 alkylaryl, or can be taken together with R u to form a 5-6 membered ring;
- D is selected from the group consisting of a direct link, C 3 . 8 cycloalkyl, C ⁇ -6 alkenyl, . 6 alkenylaryl, aryl and a five to ten membered heterocyclic ring system containing 1-4 heteroatoms selected from the group consisting of N, O and S;
- E is selected from the group consisting of a direct link, -CO-, -SO?-, -O-CO-, -NR l4 -SO 2 - and -NR 14 -CO-, where R 14 is selected from the group consisting of H, -OH, C ⁇ - 6 alkyl, aryl and C ⁇ - alkylaryl; G is selected from the group consisting of a direct link, C _ 8 cycloalkyl, aryl, and a five to ten membered heterocyclic ring system containing 1 -4 heteroatoms selected from the group consisting of N, O and S;
- J is selected from the group consisting of R 15 , -NR 15 R 16 ,
- R 15 , R 16 , R 17 and R 18 are independently selected from the group consisting of H,
- R 19 is selected from the group consisting of H, C ⁇ -6 alkyl, aryl and C ⁇ -4 alkylaryl, or can be taken together with R 17 or R 18 to form a 5-6 membered ring; and R is selected from the group consisting of H, C ⁇ -6 alkyl, aryl and C ⁇ - alkylaryl, or can be taken together with R 18 to form a 5-6 membered ring; with the proviso that when J is R 15 , then G must contain at least one N atom;
- K', K", K'" and K"" are independently selected from the group consisting of -CH-, -CR 4 -, -CR 5 - and -N-; with the proviso that no more than one of K', K", K'" and K"" are -CR 4 - and no more than one of K ⁇ K", K'" and K"" are -CR 5 -;
- R 4 and R 5 are independently selected from the group consisting of C ⁇ -6 alkyl, aryl, C ⁇ . 6 alkylaryl, C 1-4 alkyloxy, halogen, -NO 2 , -NR 6 R 7 , -NR 6 COR 7 , -OR 6 , -OCOR 6 , -COOR 6 ,
- R 6 and R 7 are independently selected from the group consisting of H, C ⁇ profession 3 alkylaryl and aryl; Q is selected from the group consisting of H,
- R > 2 ⁇ 1 and R ⁇ are independently selected from the group consisting of H, C ⁇ _ 3 alkyl and aryl; and T is selected from the group consisting of H, -COOR 23 , -CONR 23 R 24 , -CF , -CF 2 CF 3 and a group having the formula:
- R ⁇ and R" are independently selected from the group consisting of H, C ⁇ _ alkyl, aryl and C ⁇ -4 alkylaryl;
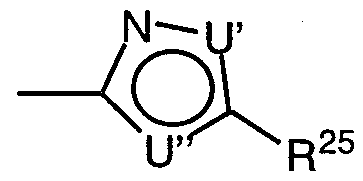
- U' and U" are independently selected from the group consisting of - O-, -S-, -N- and -NH-; with the proviso that at least one of U' or U" is -N- or -NH-;
- R 25 is selected from the group consisting of H, Ci ⁇ alkyl, C 2 - 6 alkenyl, Co- 6 alkylaryl, C 2 _ alkenylaryl, Co- 6 alkylheterocyclo, C 2 _ 6 alkenylheterocyclo, -CF and -CF 2 CF 3 ;
- V is selected from the group consisting of -S-, -SO-, -SO 2 -, -O- and -NR 26 -, where R 2 is selected from the group consisting of H, C ⁇ _ 6 alkyl and benzyl; and
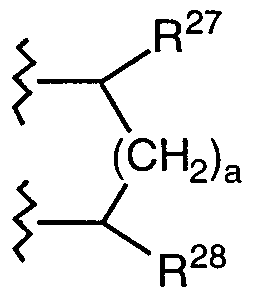
- a C 6 _ ⁇ o heterocyclic ring system substituted by R 29 and R 30 and containing 1-4 heteroatoms selected from N, S and O; where: a is an integer from 0-2; R 27 and R 28 are independently selected from the group consisting of H, C ⁇ _ 6 alkyl, aryl, C ⁇ _ 6 alkylaryl, -COOR 31 -CONR 31 R 32 , -CN and -CF 3 ; and R 29 and R 30 are independently selected from the group consisting of H, C ⁇ -6 alkyl, aryl, C ⁇ _ alkylaryl, C ⁇ _ 4 alkyloxy, halogen, -NO 2 , -NR R , -NR 31 COR 32 , -OR 31 , -OCOR 31 , -COOR 31 , -CONR 31 R 32 , -CN, -CF 3 , -SO 2 NR 31 R 32 and
- alkyl refers to saturated aliphatic groups including straight-chain, branched-chain, cyclic groups, and combinations thereof, having the number of carbon atoms specified, or if no number is specified, having up to 12 carbon atoms.
- cycloalkyl refers to a mono-, bi-, or tricyclic aliphatic ring having 3 to 12 carbon atoms, preferably 3 to 7 carbon atoms.
- alkenyl refers to unsaturated aliphatic groups including straight-chain, branched-chain, cyclic groups, and combinations thereof, having at least one double bond and having the number of carbon atoms specified.
- aryl refers to an unsubstituted or substituted aromatic ring(s), substituted with one, two or three substituents such as, by way of example and not limitation, C, .6 alkoxy, C 1 6 alkyl, C j _ 6 alkylamino, hydroxy, halogen, cyano (-CN), mercapto, nitro (-NO 2 ), thioalkoxy, carboxaldehyde, carboxyl, carboalkoxy, carboxamide, -NR ⁇ .”, -NR'COR", -OR, -OCOR, -COOR, -CONRR", -CF 3 , -SO ⁇ NRTT and C . . 6 alkyl-OR; aryl,
- C ⁇ _ 6 alkylaryl (where the R groups can be H, C ⁇ _ 6 alkyl, C ⁇ - 3 alkylaryl and aryl), including but not limited to carbocyclic aryl, heterocyclic aryl, biaryl and triaryl groups and the like, all of which may be optionally substituted.
- Preferred aryl groups include phenyl, halophenyl, C,_ 6 alkylphenyl, naphthyl, biphenyl, phenanthrenyl, naphthacenyl, and aromatic heterocyclics or heteroaryls, the latter of which is an aryl group containing one to four heteroatoms selected from the group consisting of nitrogen, oxygen and sulfur.
- Aryl groups preferably have 5-14 carbon atoms making up the ring(s) structure, while heteroaryls preferably have 1-4 heteroatoms, with the remaining 4-10 atoms being carbon atoms.
- heterocyclo and “heterocyclic ring system” as used herein refer to any saturated or unsaturated mono- or bicyclic ring system, containing from one to four heteroatoms, selected from the group consisting of nitrogen, oxygen and sulfur.
- a typical heterocyclic ring system will have five to ten members, 1-4 of which are heteroatoms.
- Typical examples of monocyclic ring systems include piperidinyl, pyrrolidinyl, pyridinyl, piperidonyl, pyrrolidonyl and thiazolyl, while examples of bicyclic ring systems include benzimidazolyl, benzothiazolyl and benzoxazolyl, all of which may be substituted.
- carrier ring refers to any saturated or unsaturated ring containing from three to six carbon atoms.
- alkylaryl and alkenylaryl refer to an alkyl group or alkenyl group, respectively, having the number of carbon atoms designated, appended to one, two, or three aryl groups.
- benzyl refers to -CH 2 -C 6 H 5 .
- alkyloxy refers to an alkyl group linked to an oxygen atom, such as methoxy, ethoxy, and so forth.
- halogen refers to Cl, Br, F or I substituents.
- direct link refers to a bond directly linking the substituents on each side of the direct link. When two adjacent substituents are defined as each being a “direct link”, it is considered to be a single bond.
- Two substituents are "taken together to form a 5-6 membered ring” means that an ethylene or a propylene bridge, respectively, is formed between the two substituents.
- pharmaceutically acceptable salts includes salts of compounds derived from the combination of a compound and an organic or inorganic acid. These compounds are useful in both free base and salt form. In practice, the use of the salt form amounts to use of the base form; both acid and base addition salts are within the scope of the present invention.
- “Pharmaceutically acceptable acid addition salt” refers to those salts which retain the biological effectiveness and properties of the free bases and which are not biologically or otherwise undesirable, formed with inorganic acids such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid and the like, and organic acids such as acetic acid, propionic acid, glycolic acid, pyruvic acid, oxalic acid, maleic acid, malonic acid, succinic acid, fumaric acid, tartaric acid, citric acid, benzoic acid, cinnamic acid, mandelic acid, methanesulfonic acid, ethanesulfonic acid, p- toluenesulfonic acid, salicylic acid and the like.
- inorganic acids such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid and the like
- organic acids such as acetic acid, propionic acid, glycolic acid, pyruvic acid
- “Pharmaceutically acceptable base addition salts” include those derived from inorganic bases such as sodium, potassium, lithium, ammonium, calcium, magnesium, iron, zinc, copper, manganese, and aluminum bases, and the like. Particularly preferred are the ammonium, potassium, sodium, calcium and magnesium salts.
- Salts derived from pharmaceutically acceptable organic nontoxic bases include salts of primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines and basic ion exchange resins, such as isopropylamine, trimethylamine, diethylamine, triethylamine, tripropylamine, ethanolamine, 2-diethylaminoethanol, trimethamine, dicyclohexylamine, lysine, arginine, histidine, caffeine, procaine, hydrabamine, choline, betaine, ethylenediamine, glucosamine, methylglucamine, theobromine, purines, piperizine, piperidine, N-ethylpiperidine, polyamine resins and the like.
- Particularly preferred organic nontoxic bases are isopropylamine, diethylamine, ethanolamine, trimethamine, dicyclohexylamine, choline, and caffeine.
- Bio property for the purposes herein means an in vivo effector or antigenic function or activity that is directly or indirectly performed by a compound of this invention. Effector functions include receptor or ligand binding, any enzyme activity or enzyme modulatory activity, any carrier binding activity, any hormonal activity, any activity in promoting or inhibiting adhesion of cells to an extracellular matrix or cell surface molecules, or any structural role. Antigenic functions include possession of an epitope or antigenic site that is capable of reacting with antibodies raised against it. The biological properties of the compounds of the present invention can be readily characterized by the methods described in Examples 13 and 14 and by such other methods as are well known in the art.
- BOP refers to benzotriazol-l-yloxy-tris-(dimethylamino) phosphonium hexafluorophosphate.
- Bu refers to butyl.
- CBZ refers to carbobenzyloxy.
- DCM refers to dichloromethane.
- DIEA diisopropylethylamine.
- DMF N,N-dimethylformamide.
- EDC l-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride
- Et refers to ethyl.
- Et 2 O refers to diethyl ether.
- EtOAc refers to ethyl acetate.
- EtOH refers to ethanol.
- HF hydrogen fluoride.
- Me refers to methyl.
- MeOH refers to methanol.
- NHS refers to N-hydroxysuccinimide.
- Ph refers to phenyl
- p-TsOH refers to p-toluenesulfonic acid monohydrate.
- TFA refers to trifluoroacetic acid.
- THF refers to tetrahydrofuran.
- Tos refers to p-toluenesulfonyl.
- the compounds of this invention carbon atoms bonded to four non-identical substituents are asymmetric. Accordingly, the compounds may exist as diastereoisomers, enantiomers or mixtures thereof.
- the syntheses described herein may employ racemates, enantiomers or diastereomers as starting materials or intermediates. Diastereomeric products resulting from such syntheses may be separated by chromatographic or crystallization methods, or by other methods known in the art. Likewise, enantiomeric product mixtures may be separated using the same techniques or by other methods known in the art.
- Each of the asymmetric carbon atoms when present in the compounds of this invention, may be in one of two configurations (R or S) and both are within the scope of the present invention.
- the final products may, in some cases, contain a small amount of diastereomeric or enantiomeric products; however, these products do not affect their therapeutic or diagnostic application.
- This invention relates to a new class of bicyclic aryl azepinone compounds selected from those of general formula I which are potent and specific inhibitors of Xa, their pharmaceutically acceptable compositions thereof, and the methods of using them as therapeutic agents for disease states in mammals characterized by abnormal thrombosis:
- R 1 and R 2 are independently selected from the group consisting of H, C ⁇ _ 6 alkyl, C -8 cycloalkyl, C ⁇ - alkylaryl, C ⁇ . 3 alkyl-C -8 cycloalkyl and aryl;
- R is H, C ⁇ _ 6 alkyl, or R ⁇ and R are taken together to form a carbocyclic ring; q is an integer from 0-2; r is an integer from 0-4; s is an integer from 0- 1 ; t is an integer from 0-4;
- A is selected from the group consisting of R 8 , -NR 8 R 9 ,
- R 12 is selected from the group consisting of H, C ⁇ _ 6 alkyl, aryl and C ⁇ _ alkylaryl, or can be taken together with R 10 or R 11 to form a 5-6 membered ring
- R 13 is selected from the group consisting of H, C 1-6 alkyl, aryl and C ⁇ _ 4 alkylaryl, or can be taken together with R 1 ' to form a 5-6 membered ring
- D is selected from the group consisting of a direct link, C 3-8 cycloalkyl, C ⁇ -6 alkenyl, .
- E is selected from the group consisting of a direct link, -CO-, -SO 2 -, -O-CO-, -NR 14 -SO 2 - and -NR 14 -CO-, where R 14 is selected from the group consisting of H, -OH, C 1- alkyl, aryl and C ⁇ _ 4 alkylaryl;
- G is selected from the group consisting of a direct link, C 3 _ 8 cycloalkyl, aryl, and a five to ten membered heterocyclic ring system containing 1-4 heteroatoms selected from the group consisting of N, O and S;
- R 15 is selected from the group consisting of R 15 , -NR 15 R 16 , where R , 1 1 5 3 , R , 1'6°, R 17 and R 18 are independently selected from the group consisting of H,
- R , 19 is selected from the group consisting of H, Ci- ⁇ alkyl, aryl and C ⁇ _ 4 alkylaryl, or can be taken together with R 17 or R 18 to form a 5-6 membered ring; and R is selected from the group consisting of H, C ⁇ . _ 6 alkyl, aryl and Cj. 4 alkylaryl, or can be taken together with R to form a 5-6 membered ring; with the proviso that when J is R 15 , then G must contain at least one N atom;
- K', K", K'" and K"" are independently selected from the group consisting of -CH-, -CR 4 -, -CR 5 - and -N-; with the proviso that no more than one of K', K", K'" and K"" are -CR 4 - and no more than one of K ⁇ K", K'" and K"" are -CR 5 -;
- R 4 and R 5 are independently selected from the group consisting of C ⁇ -6 alkyl, aryl, C ⁇ -6 alkylaryl, C,. 4 alkyloxy, halogen, -NO 2 , -NR 6 R 7 , -NR 6 COR 7 , -OR 6 , -OCOR 6 , -COOR 6 , -CONR 6 R 7 , -CN, -CF 3 , -SO 2 NR 6 R 7 and C,. 6 alkyl-OR 6 ; where R 6 and R 7 are independently selected from the group consisting of H, C ⁇ - 6 alkyl, C ⁇ _ 3 alkylaryl and aryl;
- Q is selected from the group consisting of H,
- R >2 ⁇ 1 and R , 22 are independently selected from the group consisting of H, ⁇ alkyl and aryl; and T is selected from the group consisting of H, -COOR , -CONR " R , -CF 3 , -CF 2 CF 3 and a group having the formula:
- R 23 and R 24 are independently selected from the group consisting of H, C ⁇ . 6 alkyl, aryl and C ⁇ _ 4 alkylaryl;
- U' and U" are independently selected from the group consisting of - O-, -S-, -N- and -NH-; with the proviso that at least one of U' or U" is -N- or -NH-;
- R 25 is selected from the group consisting of H, C ⁇ _ 6 alkyl, C _ 6 alkenyl, C 0 - 6 alkylaryl, C 2- alkenylaryl, Co- 6 alkylheterocyclo, C .
- V is selected from the group consisting of -S-, -SO-, -SO 2 -, -O- and -NR 26 -, where R 26 is selected from the group consisting of H, C ⁇ . 6 alkyl and benzyl; and W is selected from the group consisting of:
- R 29 and R 30 a C ⁇ -io heterocyclic ring system substituted by R 29 and R 30 and containing 1-4 heteroatoms selected from N, S and O; where: a is an integer from 0-2; R 27 and R 28 are independently selected from the group consisting of H, C ⁇ _ 6 alkyl, aryl, C ⁇ - 6 alkylaryl, -COOR 31 , -CONR 31 R 32 , -CN and -CF 3 ; and R 29 and R 30 are independently selected from the group consisting of H, C 1-6 alkyl, aryl, C 1-6 alkylaryl, C 1- alkyloxy, halogen, -NO 2 , -NR 3, R 32 , -NR 31 COR 32 , -OR 31 , -OCOR 31 , -COOR 31 , -CONR 31 R 32 , -CN, -CF 3 , -SO 2 NR 31 R 32 and
- R and R substituents are H and C ⁇ _ 6 alkyl; more preferably H and methyl; most preferably H.
- the integer "r” is preferably 3.
- the integer "s” is preferably 0.
- the integer "t” is preferably from 0-1.
- R 8 , R 9 , R 10 and R 1 1 are independently selected from the group consisting of H and C ⁇ _ 6 alkyl; and are more preferably independently selected from the group consisting of H and methyl.
- R 12 is H, C ⁇ . 6 alkyl or taken together with R 10 or R 1 ' to form a 5-6 membered ring; and is more preferably H or methyl.
- R 13 is H, C ⁇ . alkyl or taken together with R 10 to form a 5-6 membered ring; and is more preferably H or methyl.
- D is preferably selected from the group consisting of a direct link, C 3 . 8 cycloalkyl, aryl and a five to ten membered heterocyclic ring system containing 1-4 heteroatoms selected from the group consisting of N, O and S.
- E is preferably a direct link, -CO- or -SO 2 -.
- G is preferably a direct link.
- R 15 , R 16 , R 17 , R 18 , R 19 and R 20 are independently selected from the group consisting of H and C ⁇ -6 alkyl, more preferably H and methyl.
- K', K", K'" and K"" are -CH-; more preferably K', K", K'" and K"" are all -CH-.
- R 4 or R 5 is preferably halogen.
- Q is preferably: ° where R 21 is preferably H and R 22 is preferably H.
- T is preferably H, -COOR 23 , -CONR 23 R 24 or a group having the formula:
- R ⁇ is preferably H.
- R is preferably C ⁇ -4 alkylaryl.
- V is preferably -S-, -O- or -NR -, where R" is preferably H or methyl, more preferably H.
- W is preferably selected from the group consisting of:
- W is more preferably
- R “9 and R” are preferably independently selected from the group consisting of H, -O-R 31 , -COOR 31 , -CONR 31 R 32 or -CF 3 ; more preferably H.
- R 27 is preferably H and R 28 is preferably H.
- U' is preferably O
- U" is preferably N
- R 25 is preferably -CF 3 or -CF CF 3 .
- s is 0; R 2 and R 3 are H; K'. K". K'" and K"" are -CH-; and Q is -C(O)-T.
- R 2 and R 3 are H; K'. K". K'" and K"" are -CH-; and Q is -C(O)-T.
- R 15 , R 17 , R 18 and R 9 are all H; and Q is -C(O)-T. This is also illustrated as a preferred group of compounds defined by the general structural formula HI as:
- This invention also encompasses all pharmaceutically acceptable isomers, salts, hydrates and solvates of the compounds of formulas I, II and m.
- the compounds of formulas I, II and IE can exist in various isomeric and tautomeric forms, and all such forms are meant to be included in the invention, along with pharmaceutically acceptable salts, hydrates and solvates of such isomers and tautomers.
- the compounds of this invention may be isolated as the free acid or base or converted to salts of various inorganic and organic acids and bases. Such salts are within the scope of this invention. Non-toxic and physiologically compatible salts are particularly useful although other less desirable salts may have use in the processes of isolation and purification.
- the free acid or free base form of a compound of one of the formulas above can be reacted with one or more molar equivalents of the desired acid or base in a solvent or solvent mixture in which the salt is insoluble, or in a solvent like water after which the solvent is removed by evaporation, distillation or freeze drying.
- the free acid or base form of the product may be passed over an ion exchange resin to form the desired salt or one salt form of the product may be converted to another using the same general process.
- prodrug refers to a pharmacologically inactive derivative of a parent drug molecule that requires biotransformation, either spontaneous or enzymatic, within the organism to release the active drug.
- Prodrugs are variations or derivatives of the compounds of this invention which have groups cleavable under metabolic conditions. Prodrugs become the compounds of the invention which are pharmaceutically active in vivo, when they undergo solvolysis under physiological conditions or undergo enzymatic degradation. Prodrug compounds of this invention may be called single, double, triple etc., depending on the number of biotransformation steps required to release the active drug within the organism, and indicating the number of functionalities present in a precursor-type form.
- Prodrug forms often offer advantages of solubility, tissue compatibility, or delayed release in the mammalian organism (see, Bundgard, Design of Prodrugs, pp. 7-9, 21-24, Elsevier, Amsterdam 1985 and Silverman, The Organic Chemistry of Drug Design and Drug Action, pp. 352-401, Academic Press, San Diego, CA, 1992).
- Prodrugs commonly known in the art include acid derivatives well known to practitioners of the art, such as, for example, esters prepared by reaction of the parent acids with a suitable alcohol, or amides prepared by reaction of the parent acid compound with an amine, or basic groups reacted to form an acylated base derivative.
- the prodrug derivatives of this invention may be combined with other features herein taught to enhance bioavailability.
- the invention encompasses compounds of general structural formula TV, where R , R and R 3 are H; q is 1; r is 3; s is 0; G is a direct link; J is -NH-C(NH)NH 2 ; K ⁇ K", K'" and
- the invention encompasses compounds of general structural formula V, where R , R and R 3 are H; q is 2; t is 1; A is H; D is phenyl; E is -SO 2 -; K', K", K'" and K"" are -CH-;
- the invention encompasses compounds of general structural formula VI, where q is 1 ; r is 3; s is 0; t is 1; A is H; D is phenyl; E is -SO?-; G is a direct link; J is -NH-
- the invention encompasses compounds of general structural formula VII, where R , R " and R 3 are H; q is 0; r is 3; s is 0; t is 1; A is H; D is phenyl; E is -SO?-; G is a direct link; J is -NH-C(NH)NH?; and K', K", K'" and K"" are -CH-:
- the invention encompasses compounds of general structural formula VIE where R 1 , R 2 and R 3 are H; q and t are 1 ; r is 3; s is 0; A is H; D is phenyl; E is -SO 2 -; G is a direct
- J is -NH-C(NH)NH 2 ; and Q is
- the compounds of this invention find utility as therapeutic agents for disease states in mammals which have disorders of coagulation such as in the treatment or prevention of unstable angina, refractory angina, myocardial infarction, transient ischemic attacks, thrombotic stroke, embolic stroke, disseminated intravascular coagulation including the treatment of septic shock, deep venous thrombosis in the prevention of pulmonary embolism or the treatment of reocclusion or restenosis of reperfused coronary arteries. Further, these compounds are useful for the treatment or prophylaxis of those diseases which involve the production and/or action of factor
- Xa/prothrombinase complex This includes a number of thrombotic and prothrombotic states in which the coagulation cascade is activated which include but are not limited to, deep venous thrombosis, pulmonary embolism, myocardial infarction, stroke, thromboembolic complications of surgery and peripheral arterial occlusion.
- a method for preventing or treating a condition in a mammal characterized by undesired thrombosis comprises administering to the mammal a therapeutically effective amount of a compound of this invention.
- diseases treatable or preventable by the administration of compounds of this invention include, without limitation, occlusive coronary thrombus formation resulting from either thrombolytic therapy or percutaneous transluminal coronary angioplasty, thrombus formation in the venous vasculature, disseminated intravascular coagulopathy, a condition wherein there is rapid consumption of coagulation factors and systemic coagulation which results in the formation of life-threatening thrombi occurring throughout the microvasculature leading to widespread organ failure, hemorrhagic stroke, renal dialysis, blood oxygenation, and cardiac catheterization.
- the compounds of the invention also find utility in a method for inhibiting the coagulation biological samples, which comprises the administration of a compound of the invention.
- the compounds of the present invention may also be used in combination with other therapeutic or diagnostic agents.
- the compounds of this invention may be coadministered along with other compounds typically prescribed for these conditions according to generally accepted medical practice such as anticoagulant agents, thrombolytic agents, or other antithrombotics, including platelet aggregation inhibitors, tissue plasminogen activators, urokinase, prourokinase, strep tokinase, heparin, aspirin, or warfarin.
- the compounds of the present invention may act in a synergistic fashion to prevent reocclusion following a successful thrombolytic therapy and/or reduce the time to reperfusion. These compounds may also allow for reduced doses of the thrombolytic agents to be used and therefore minimize potential hemorrhagic side-effects.
- the compounds of this invention can be utilized in vivo, ordinarily in mammals such as primates, (e.g. humans), sheep, horses, cattle, pigs, dogs, cats, rats and mice, or in vitro.
- the biological properties of the compounds of the present invention can be readily characterized by methods that are well known in the art, for example by the in vitro protease activity assays and in vivo studies to evaluate antithrombotic efficacy, and effects on hemostasis and hematological parameters, such as are illustrated in the examples. Diagnostic applications of the compounds of this invention will typically utilize formulations in the form of solutions or suspensions. In the management of thrombotic disorders the compounds of this invention may be utilized in compositions such as tablets, capsules or elixirs for oral administration, suppositories, sterile solutions or suspensions or injectable administration, and the like, or incorporated into shaped articles.
- Subjects in need of treatment can be administered dosages that will provide optimal efficacy.
- the dose and method of administration will vary from subject to subject and be dependent upon such factors as the type of mammal being treated, its sex, weight, diet, concurrent medication, overall clinical condition, the particular compounds employed, the specific use for which these compounds are employed, and other factors which those skilled in the medical arts will recognize.
- Formulations of the compounds of this invention are prepared for storage or administration by mixing the compound having a desired degree of purity with physiologically acceptable carriers, excipients, stabilizers etc., and may be provided in sustained release or timed release formulations.
- Acceptable carriers or diluents for therapeutic use are well known in the pharmaceutical field, and are described, for example, in Remington's Pharmaceutical Sciences, Mack Publishing Co., (A.R. Gennaro edit. 1985).
- Such materials are nontoxic to the recipients at the dosages and concentrations employed, and include buffers such as phosphate, citrate, acetate and other organic acid salts, antioxidants such as ascorbic acid, low molecular weight (less than about ten residues) peptides such as polyarginine, proteins, such as serum albumin, gelatin, or immunoglobulins, hydrophilic polymers such as polyvinylpyrrolidinone, amino acids such as glycine, glutamic acid, aspartic acid, or arginine, monosaccharides, disaccharides, and other carbohydrates including cellulose or its derivatives, glucose, mannose or dextrins, chelating agents such as EDTA, sugar alcohols such as mannitol or sorbitol, counterions such as sodium and/or nonionic surfactants such as Tween, Pluronics or polyethyleneglycol.
- buffers such as phosphate, citrate, acetate and other organic acid salts
- antioxidants such as as
- Dosage formulations of the compounds of this invention to be used for therapeutic administration must be sterile. Sterility is readily accomplished by filtration through sterile membranes such as 0.2 micron membranes, or by other conventional methods. Formulations typically will be stored in lyophilized form or as an aqueous solution.
- the pH of the preparations of this invention typically will be 3-11, more preferably 5-9 and most preferably 7-8. It will be understood that use of certain of the foregoing excipients, carriers, or stabilizers will result in the formation of cyclic polypeptide salts.
- While the preferred route of administration is by injection, other methods of administration are also anticipated such as orally, intravenously (bolus and/or infusion), subcutaneously, intramuscularly, colonically, rectally, nasally, transdermally or intraperitoneally, employing a variety of dosage forms such as suppositories, implanted pellets or small cylinders, aerosols, microencapsulation, oral dosage formulations and topical formulations such as ointments, drops and dermal patches.
- the compounds of this invention are desirably incorporated into shaped articles such as implants which may employ inert materials such as biodegradable polymers or synthetic silicones, for example, Silastic, silicone rubber or other polymers commercially available.
- the compounds of the invention may also be administered in the form of liposome delivery systems, such as small unilamellar vesicles, large unilamellar vesicles and multilamellar vesicles.
- Liposomes can be formed from a variety of lipids, such as cholesterol, stearylamine or phosphatidylcholines.
- the compounds of this invention may also be delivered by the use of antibodies, antibody fragments, growth factors, hormones, or other targeting moieties, to which the compound molecules are coupled.
- the compounds of this invention may also be coupled with suitable polymers as targetable drug carriers.
- Such polymers can include polyvinylpyrrolidinone, pyran copolymer, polyhydroxy-propyl-methacrylamide-phenol, polyhydroxyethyl-aspartamide-phenol, or polyethyleneoxide-polylysine substituted with palmitoyl residues.
- compounds of the invention may be coupled to a class of biodegradable polymers useful in achieving controlled release of a drug, for example polylactic acid, polyglycolic acid, copolymers of polylactic and polyglycolic acid, polyepsilon caprolactone, polyhydroxy butyric acid, polyorthoesters, polyacetals, polydihydropyrans, polycyanoacrylates and cross linked or amphipathic block copolymers of hydrogels.
- Polymers and semipermeable polymer matrices may be formed into shaped articles, such as valves, stents, tubing, prostheses and the like.
- Therapeutic compound liquid formulations generally are placed into a container having a sterile access port, for example, an intravenous solution bag or vial having a stopper pierceable by hypodermic injection needle.
- Therapeutically effective dosages may be determined by either in vitro or in vivo methods. For each particular compound of the present invention, individual determinations may be made to determine the optimal dosage required.
- the range of therapeutically effective dosages will be influenced by the route of administration, the therapeutic objectives and the condition of the patient. For injection by hypodermic needle, it may be assumed the dosage is delivered into the body's fluids. For other routes of administration, the absorption efficiency must be individually determined for each compound by methods well known in pharmacology. Accordingly, it may be necessary for the therapist to titer the dosage and modify the route of administration as required to obtain the optimal therapeutic effect.
- the determination of effective dosage levels that is, the dosage levels necessary to achieve the desired result, will be readily determined by one skilled in the art.
- the compounds of the invention can be administered orally or parenterally in an effective amount within the dosage range of about 0.1 to 100 mg/kg, preferably about 0.5 to 50 mg/kg and more preferably about 1 to 20 mg/kg on a regimen in a single or 2 to 4 divided daily doses and/or continuous infusion.
- a compound or mixture of compounds of this invention is compounded with a physiologically acceptable vehicle, carrier, excipient, binder, preservative, stabilizer, dye, flavor etc., as called for by accepted pharmaceutical practice.
- a physiologically acceptable vehicle carrier, excipient, binder, preservative, stabilizer, dye, flavor etc.
- the amount of active ingredient in these compositions is such that a suitable dosage in the range indicated is obtained.
- Typical adjuvants which may be incorporated into tablets, capsules and the like are binders such as acacia, corn starch or gelatin, and excipients such as microcrystalline cellulose, disintegrating agents like corn starch or alginic acid, lubricants such as magnesium stearate, sweetening agents such as sucrose or lactose, or flavoring agents.
- binders such as acacia, corn starch or gelatin
- excipients such as microcrystalline cellulose, disintegrating agents like corn starch or alginic acid, lubricants such as magnesium stearate, sweetening agents such as sucrose or lactose, or flavoring agents.
- lubricants such as magnesium stearate
- sweetening agents such as sucrose or lactose
- flavoring agents such as sucrose or lactose
- flavoring agents such as sucrose or lactose
- a dosage form is a capsule, in addition to the above materials it may also contain liquid carriers such as water,
- dissolution or suspension of the active compound in a vehicle such as an oil or a synthetic fatty vehicle like ethyl oleate, or into a liposome may be desired.
- Buffers, preservatives, antioxidants and the like can be incorporated according to accepted pharmaceutical practice.
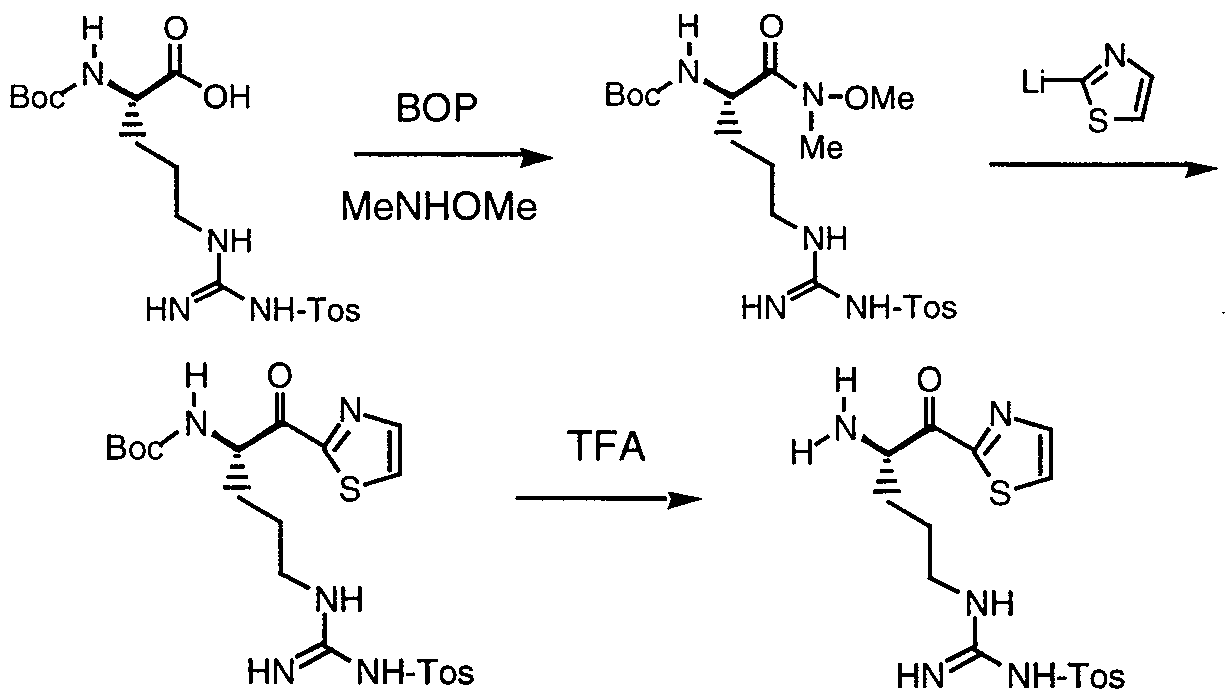
- Preparation of the Disclosed Compounds The compounds of the present invention may be synthesized by either solid or liquid phase methods described and referenced in standard textbooks, or by a combination of both methods. These methods are well known in the art. See, Bodanszky, "The
- the compounds of this invention may be preferably prepared by coupling the carboxylic acid of formula (a) to the amine of formula (b) by the standard amide bond formation strategies:
- the compounds of formula (b) wherein Q is -C(O)-T, where T is H may be prepared by the methods disclosed in WO 93/15756, supra; Vlasuk, et al, WO 94/17817; Abelman, et al, WO 94/21673; Webb, et al., WO 94/08941; Veber, et al., WO 94/25051;
- the compounds of formula (b) wherein Q is -C(O)-T, where T is -CF 3 or -CF 2 CF may be prepared by the methods disclosed in Schacht, et al, GB 2287027, the disclosure of which is inco ⁇ orated herein by reference.
- V is -S-, -O-, -SO- or -SO 2 -
- V is -S-, -O-, -SO- or -SO 2 -
- the starting compound of formula (a) is either a known compound or can be produced by known methods (Heitsch, et al., Canadian Patent No. 2,071,744; Sugihara, et al., Canadian Patent No. 2,126,026; Baker, et al., EP 365,992; U.S. Pat. No. 4,251,438; Carr, et al., U.S. Pat. No. 4,341,698; Goldman, et al, U.S. Pat. No. 5,120,718; Biswanath, et al, U.S. Pat. No. 5,164,388; Duggan, et al, U.S. Pat. No. 5,281,585; Sugihara, et al, U.S.
- the reagent used in the eighth step of the Scheme can be synthesized as follows:
- reaction mixture was concentrated to dryness, and the residue was purified by flash column chromatography on silica gel (30% ethyl acetate in hexane, followed by 50% ethyl acetate in hexane) to give the desired product (182 mg, 67%) as a white solid.
- Example 13 Determination of ICsn
- the compounds of the present invention are first dissolved in a buffer to give solutions containing concentrations such that assay concentrations range from 0-100 ⁇ M.
- concentrations such that assay concentrations range from 0-100 ⁇ M.
- a synthetic chromogenic substrate would be added to a solution containing a test compound and the enzyme of interest and the residual catalytic activity of that enzyme would then be determined spectrophotometrically.
- the IC 50 of a compound is determined from the substrate turnover.
- the IC 50 is the concentration of test compound giving 50% inhibition of the substrate turnover.
- Preferred compounds of the invention desirably have an IC 50 of less than 500 nM in the factor Xa assay, preferably less than 200 nM, and more preferably less than 100 nM.
- Preferred compounds of the invention desirably have an IC 50 of less than 4.0 ⁇ M in the prothrombinase assay, preferably less than 200 nM, and more preferably less than 10 nM.
- Preferred compounds of the invention desirably have an IC 50 of greater than 1.0 ⁇ M in the thrombin assay, preferably greater than 10.0 ⁇ M, and more preferably greater than 100.0 ⁇ M.
- IC 50 of greater than 1.0 ⁇ M in the thrombin assay, preferably greater than 10.0 ⁇ M, and more preferably greater than 100.0 ⁇ M.
- Factor Xa and thrombin assays are performed at room temperature, in 0.02 M Tris HCl buffer, pH 7.5, containing 0.15 M NaCl.
- the rates of hydrolysis of the para- nitroanilide substrate S-2765 (Chromogenix) for factor Xa, and the substrate Chromozym TH (Boehringer Mannheim) for thrombin following preincubation of the enzyme with the test compound for 5 minutes at room temperature are determined using a Softmax 96-well plate reader (Molecular Devices), monitored at 405 nm to measure the time dependent appearance of p-nitroanilide.
- the prothrombinase inhibition assay is performed in a plasma free system with modifications to the method as described by Sinha, et al, Thromb. Res., 75:427-436 (1994).
- the activity of the prothrombinase complex is determined by measuring the time course of thrombin generation using the p-nitroanilide substrate Chromozym TH.
- the assay consists of a 5 minute preincubation of selected compounds to be tested as inhibitors with the complex formed from factor Xa (0.5 nM), factor Va (2 nM), phosphatidyl serine:phosphatidyl choline (25:75, 20 ⁇ M) in 20 mM Tris HCl buffer, pH 7.5, containing 0.15 M NaCl, 5 mM CaCl? and 0.1% bovine serum albumin. Aliquots from the complex-test compound mixture are added to prothrombin ( 1 nM) and Chromozym TH (0.1 mM). The rate of substrate cleavage is monitored at 405 nm for two minutes. Several concentrations of a given test compound are assayed in duplicate. A standard curve of thrombin generation by an equivalent amount of untreated complex is then used for determination of percent inhibition.
- the compounds of the invention exhibited inhibitory activity in the Factor Xa assay described above.
- the preferred compounds of the invention have IC 50 values less than 100 nM.
- the antithrombotic efficacy of the compounds of this invention can readily be evaluated using a series of studies in rabbits, as described below. These studies are also useful in evaluating a compounds effects on hemostasis and its the hematological parameters. Antithrombotic Efficacy in a Rabbit Model of Venous Thrombosis
- Rabbits are anesthetized with I.M. injections of Ketamine, Xylazine, and Acepromazine cocktail.
- a standardized protocol consists of insertion of a thrombogenic cotton thread and copper wire apparatus into the abdominal vena cava of the anesthetized rabbit.
- a non- occlusive thrombus is allowed to develop in the central venous circulation and inhibition of thrombus growth is then used as a measure of the antithrombotic activity of the compound being evaluated.
- Test agents or control saline are administered through a marginal ear vein catheter.
- a femoral vein catheter is used for blood sampling prior to and during steady state infusion of the compound being evaluated. Initiation of thrombus formation will begin immediately after advancement of the cotton thread apparatus into the central venous circulation.
- the rabbits are euthanized and the thrombus excised by surgical dissection and characterized by weight and histology. Blood samples are then analyzed for changes in hematological and coagulation parameters.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Cardiology (AREA)
- Biophysics (AREA)
- Urology & Nephrology (AREA)
- Vascular Medicine (AREA)
- Hematology (AREA)
- Heart & Thoracic Surgery (AREA)
- Biochemistry (AREA)
- Diabetes (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Peptides Or Proteins (AREA)
Abstract
Description





















































Claims












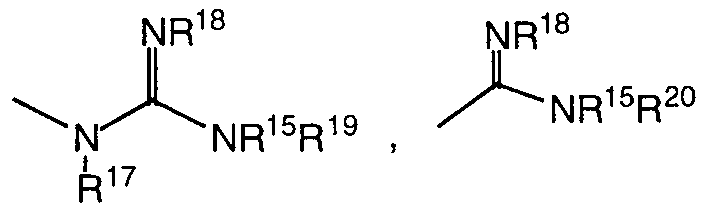






Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA002300478A CA2300478A1 (en) | 1997-08-11 | 1998-08-11 | Selective factor xa inhibitors |
| JP2000506234A JP2001512742A (en) | 1997-08-11 | 1998-08-11 | Selective factor Xa inhibitor |
| NZ502877A NZ502877A (en) | 1997-08-11 | 1998-08-11 | Bicyclic aryl azepinone selective factor Xa inhibitors for treating thrombosis related diseases |
| EP98938486A EP0994892A1 (en) | 1997-08-11 | 1998-08-11 | SELECTIVE FACTOR Xa INHIBITORS |
| AU87005/98A AU753842B2 (en) | 1997-08-11 | 1998-08-11 | Selective factor Xa inhibitors |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US90802997A | 1997-08-11 | 1997-08-11 | |
| US8418597P | 1997-08-11 | 1997-08-11 | |
| US08/908,029 | 1997-08-11 | ||
| US60/084,185 | 1997-08-11 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1999007732A1 true WO1999007732A1 (en) | 1999-02-18 |
Family
ID=26770692
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1998/016720 WO1999007732A1 (en) | 1997-08-11 | 1998-08-11 | SELECTIVE FACTOR Xa INHIBITORS |
Country Status (5)
| Country | Link |
|---|---|
| EP (1) | EP0994892A1 (en) |
| JP (1) | JP2001512742A (en) |
| CA (1) | CA2300478A1 (en) |
| NZ (1) | NZ502877A (en) |
| WO (1) | WO1999007732A1 (en) |
Cited By (35)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6133256A (en) * | 1997-04-14 | 2000-10-17 | Cor Therapeutics Inc | Selective factor Xa inhibitors |
| US6194435B1 (en) | 1996-10-11 | 2001-02-27 | Cor Therapeutics, Inc. | Lactams as selective factor Xa inhibitors |
| US6204268B1 (en) | 1997-04-14 | 2001-03-20 | Cor Therapeutics, Inc | Selective factor Xa inhibitors |
| US6211183B1 (en) | 1997-04-14 | 2001-04-03 | Cor Therapeutics, Inc. | Selective factor Xa inhibitors |
| US6218382B1 (en) | 1997-08-11 | 2001-04-17 | Cor Therapeutics, Inc | Selective factor Xa inhibitors |
| US6228854B1 (en) | 1997-08-11 | 2001-05-08 | Cor Therapeutics, Inc. | Selective factor Xa inhibitors |
| US6262047B1 (en) | 1996-10-11 | 2001-07-17 | Cor Therapeutics, Inc. | Selective factor Xa inhibitors |
| US6297233B1 (en) | 1999-02-09 | 2001-10-02 | Bristol-Myers Squibb Company | Lactam inhibitors of FXa and method |
| WO2001079261A1 (en) * | 2000-04-14 | 2001-10-25 | Corvas International, Inc. | Tetrahydro-azepinone derivatives as thrombin inhibitors |
| US6333321B1 (en) | 1997-08-11 | 2001-12-25 | Cor Therapeutics, Inc. | Selective factor Xa inhibitors |
| US6369063B1 (en) | 1997-04-14 | 2002-04-09 | Cor Therapeutics, Inc. | Selective factor Xa inhibitors |
| US6369080B2 (en) | 1996-10-11 | 2002-04-09 | Cor Therapeutics, Inc. | Selective factor Xa inhibitors |
| US6469029B1 (en) | 1999-09-13 | 2002-10-22 | 3-Dimensional Pharmaceuticals, Inc. | Azacycloalkanone serine protease inhibitors |
| US6511973B2 (en) | 2000-08-02 | 2003-01-28 | Bristol-Myers Squibb Co. | Lactam inhibitors of FXa and method |
| US6525076B1 (en) | 1996-10-11 | 2003-02-25 | Millennium Pharmaceuticals, Inc. | Selective factor Xa inhibitors |
| EP2982668A2 (en) | 2002-12-03 | 2016-02-10 | Pharmacyclics LLC | 2-(2-hydroxybiphenyl-3-yl)-1h-benzoimidazole-5-carboxamidine derivatives as factor viia inhibitors for the treatment of thromboembolic disorders |
| US9266892B2 (en) | 2012-12-19 | 2016-02-23 | Incyte Holdings Corporation | Fused pyrazoles as FGFR inhibitors |
| US9388185B2 (en) | 2012-08-10 | 2016-07-12 | Incyte Holdings Corporation | Substituted pyrrolo[2,3-b]pyrazines as FGFR inhibitors |
| US9533984B2 (en) | 2013-04-19 | 2017-01-03 | Incyte Holdings Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US9533954B2 (en) | 2010-12-22 | 2017-01-03 | Incyte Corporation | Substituted imidazopyridazines and benzimidazoles as inhibitors of FGFR3 |
| US9580423B2 (en) | 2015-02-20 | 2017-02-28 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US9611267B2 (en) | 2012-06-13 | 2017-04-04 | Incyte Holdings Corporation | Substituted tricyclic compounds as FGFR inhibitors |
| US9708318B2 (en) | 2015-02-20 | 2017-07-18 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US9890156B2 (en) | 2015-02-20 | 2018-02-13 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US10611762B2 (en) | 2017-05-26 | 2020-04-07 | Incyte Corporation | Crystalline forms of a FGFR inhibitor and processes for preparing the same |
| US10851105B2 (en) | 2014-10-22 | 2020-12-01 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US11174257B2 (en) | 2018-05-04 | 2021-11-16 | Incyte Corporation | Salts of an FGFR inhibitor |
| US11407750B2 (en) | 2019-12-04 | 2022-08-09 | Incyte Corporation | Derivatives of an FGFR inhibitor |
| US11466004B2 (en) | 2018-05-04 | 2022-10-11 | Incyte Corporation | Solid forms of an FGFR inhibitor and processes for preparing the same |
| US11566028B2 (en) | 2019-10-16 | 2023-01-31 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US11591329B2 (en) | 2019-07-09 | 2023-02-28 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US11607416B2 (en) | 2019-10-14 | 2023-03-21 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US11628162B2 (en) | 2019-03-08 | 2023-04-18 | Incyte Corporation | Methods of treating cancer with an FGFR inhibitor |
| US11897891B2 (en) | 2019-12-04 | 2024-02-13 | Incyte Corporation | Tricyclic heterocycles as FGFR inhibitors |
| US11939331B2 (en) | 2021-06-09 | 2024-03-26 | Incyte Corporation | Tricyclic heterocycles as FGFR inhibitors |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1995035311A1 (en) * | 1994-06-17 | 1995-12-28 | Corvas International, Inc. | 3-amino-2-oxo-1-piperidineacetic derivatives as enzyme inhibitors |
| WO1996019491A1 (en) * | 1994-12-22 | 1996-06-27 | Biochem Pharma Inc. | Heterocyclic keto arginine peptides as thrombin inhibitors |
| WO1998016523A2 (en) * | 1996-10-11 | 1998-04-23 | Cor Therapeutics, Inc. | Selective factor xa inhibitors |
-
1998
- 1998-08-11 CA CA002300478A patent/CA2300478A1/en not_active Abandoned
- 1998-08-11 EP EP98938486A patent/EP0994892A1/en not_active Withdrawn
- 1998-08-11 JP JP2000506234A patent/JP2001512742A/en active Pending
- 1998-08-11 WO PCT/US1998/016720 patent/WO1999007732A1/en not_active Application Discontinuation
- 1998-08-11 NZ NZ502877A patent/NZ502877A/en unknown
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1995035311A1 (en) * | 1994-06-17 | 1995-12-28 | Corvas International, Inc. | 3-amino-2-oxo-1-piperidineacetic derivatives as enzyme inhibitors |
| WO1996019491A1 (en) * | 1994-12-22 | 1996-06-27 | Biochem Pharma Inc. | Heterocyclic keto arginine peptides as thrombin inhibitors |
| WO1998016523A2 (en) * | 1996-10-11 | 1998-04-23 | Cor Therapeutics, Inc. | Selective factor xa inhibitors |
Non-Patent Citations (2)
| Title |
|---|
| J. E. SEMPLE: "Design, synthesis, and evolution of a novel, selective, and orally bioavailable class of thrombin inhibitors: P1-argininal derivatives incorporating P3-4 lactam sulfonamide moieties", JOURNAL OF MEDICINAL CHEMISTRY, vol. 39, no. 23, 1996, pages 4531 - 36, XP002081550 * |
| KUNITADA S ET AL: "FACTOR XA INHIBITORS", CURRENT PHARMACEUTICAL DESIGN, vol. 2, no. 5, October 1996 (1996-10-01), pages 531 - 542, XP002057653 * |
Cited By (56)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6194435B1 (en) | 1996-10-11 | 2001-02-27 | Cor Therapeutics, Inc. | Lactams as selective factor Xa inhibitors |
| US6262047B1 (en) | 1996-10-11 | 2001-07-17 | Cor Therapeutics, Inc. | Selective factor Xa inhibitors |
| US6525076B1 (en) | 1996-10-11 | 2003-02-25 | Millennium Pharmaceuticals, Inc. | Selective factor Xa inhibitors |
| US6369080B2 (en) | 1996-10-11 | 2002-04-09 | Cor Therapeutics, Inc. | Selective factor Xa inhibitors |
| US6133256A (en) * | 1997-04-14 | 2000-10-17 | Cor Therapeutics Inc | Selective factor Xa inhibitors |
| US6204268B1 (en) | 1997-04-14 | 2001-03-20 | Cor Therapeutics, Inc | Selective factor Xa inhibitors |
| US6211183B1 (en) | 1997-04-14 | 2001-04-03 | Cor Therapeutics, Inc. | Selective factor Xa inhibitors |
| US6369063B1 (en) | 1997-04-14 | 2002-04-09 | Cor Therapeutics, Inc. | Selective factor Xa inhibitors |
| US6218382B1 (en) | 1997-08-11 | 2001-04-17 | Cor Therapeutics, Inc | Selective factor Xa inhibitors |
| US6228854B1 (en) | 1997-08-11 | 2001-05-08 | Cor Therapeutics, Inc. | Selective factor Xa inhibitors |
| US6333321B1 (en) | 1997-08-11 | 2001-12-25 | Cor Therapeutics, Inc. | Selective factor Xa inhibitors |
| US6297233B1 (en) | 1999-02-09 | 2001-10-02 | Bristol-Myers Squibb Company | Lactam inhibitors of FXa and method |
| US6469029B1 (en) | 1999-09-13 | 2002-10-22 | 3-Dimensional Pharmaceuticals, Inc. | Azacycloalkanone serine protease inhibitors |
| US6962942B2 (en) | 1999-09-13 | 2005-11-08 | 3-Dimensional Pharmaceuticals, Inc. | Azacycloalkanone serine protease inhibitors |
| WO2001079261A1 (en) * | 2000-04-14 | 2001-10-25 | Corvas International, Inc. | Tetrahydro-azepinone derivatives as thrombin inhibitors |
| US6511973B2 (en) | 2000-08-02 | 2003-01-28 | Bristol-Myers Squibb Co. | Lactam inhibitors of FXa and method |
| EP2982668A2 (en) | 2002-12-03 | 2016-02-10 | Pharmacyclics LLC | 2-(2-hydroxybiphenyl-3-yl)-1h-benzoimidazole-5-carboxamidine derivatives as factor viia inhibitors for the treatment of thromboembolic disorders |
| US9533954B2 (en) | 2010-12-22 | 2017-01-03 | Incyte Corporation | Substituted imidazopyridazines and benzimidazoles as inhibitors of FGFR3 |
| US10213427B2 (en) | 2010-12-22 | 2019-02-26 | Incyte Corporation | Substituted imidazopyridazines and benzimidazoles as inhibitors of FGFR3 |
| US10813930B2 (en) | 2010-12-22 | 2020-10-27 | Incyte Corporation | Substituted imidazopyridazines and benzimidazoles as inhibitors of FGFR3 |
| US11840534B2 (en) | 2012-06-13 | 2023-12-12 | Incyte Corporation | Substituted tricyclic compounds as FGFR inhibitors |
| US11053246B2 (en) | 2012-06-13 | 2021-07-06 | Incyte Corporation | Substituted tricyclic compounds as FGFR inhibitors |
| US9611267B2 (en) | 2012-06-13 | 2017-04-04 | Incyte Holdings Corporation | Substituted tricyclic compounds as FGFR inhibitors |
| US10131667B2 (en) | 2012-06-13 | 2018-11-20 | Incyte Corporation | Substituted tricyclic compounds as FGFR inhibitors |
| US9388185B2 (en) | 2012-08-10 | 2016-07-12 | Incyte Holdings Corporation | Substituted pyrrolo[2,3-b]pyrazines as FGFR inhibitors |
| US9745311B2 (en) | 2012-08-10 | 2017-08-29 | Incyte Corporation | Substituted pyrrolo[2,3-b]pyrazines as FGFR inhibitors |
| US9266892B2 (en) | 2012-12-19 | 2016-02-23 | Incyte Holdings Corporation | Fused pyrazoles as FGFR inhibitors |
| US10040790B2 (en) | 2013-04-19 | 2018-08-07 | Incyte Holdings Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US10450313B2 (en) | 2013-04-19 | 2019-10-22 | Incyte Holdings Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US9533984B2 (en) | 2013-04-19 | 2017-01-03 | Incyte Holdings Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US10947230B2 (en) | 2013-04-19 | 2021-03-16 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US11530214B2 (en) | 2013-04-19 | 2022-12-20 | Incyte Holdings Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US10851105B2 (en) | 2014-10-22 | 2020-12-01 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US10738048B2 (en) | 2015-02-20 | 2020-08-11 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US11014923B2 (en) | 2015-02-20 | 2021-05-25 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US10632126B2 (en) | 2015-02-20 | 2020-04-28 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US10251892B2 (en) | 2015-02-20 | 2019-04-09 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US9708318B2 (en) | 2015-02-20 | 2017-07-18 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US10214528B2 (en) | 2015-02-20 | 2019-02-26 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US9801889B2 (en) | 2015-02-20 | 2017-10-31 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US11667635B2 (en) | 2015-02-20 | 2023-06-06 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US9580423B2 (en) | 2015-02-20 | 2017-02-28 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US10016438B2 (en) | 2015-02-20 | 2018-07-10 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US11173162B2 (en) | 2015-02-20 | 2021-11-16 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US9890156B2 (en) | 2015-02-20 | 2018-02-13 | Incyte Corporation | Bicyclic heterocycles as FGFR4 inhibitors |
| US10611762B2 (en) | 2017-05-26 | 2020-04-07 | Incyte Corporation | Crystalline forms of a FGFR inhibitor and processes for preparing the same |
| US11472801B2 (en) | 2017-05-26 | 2022-10-18 | Incyte Corporation | Crystalline forms of a FGFR inhibitor and processes for preparing the same |
| US11174257B2 (en) | 2018-05-04 | 2021-11-16 | Incyte Corporation | Salts of an FGFR inhibitor |
| US11466004B2 (en) | 2018-05-04 | 2022-10-11 | Incyte Corporation | Solid forms of an FGFR inhibitor and processes for preparing the same |
| US11628162B2 (en) | 2019-03-08 | 2023-04-18 | Incyte Corporation | Methods of treating cancer with an FGFR inhibitor |
| US11591329B2 (en) | 2019-07-09 | 2023-02-28 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US11607416B2 (en) | 2019-10-14 | 2023-03-21 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US11566028B2 (en) | 2019-10-16 | 2023-01-31 | Incyte Corporation | Bicyclic heterocycles as FGFR inhibitors |
| US11407750B2 (en) | 2019-12-04 | 2022-08-09 | Incyte Corporation | Derivatives of an FGFR inhibitor |
| US11897891B2 (en) | 2019-12-04 | 2024-02-13 | Incyte Corporation | Tricyclic heterocycles as FGFR inhibitors |
| US11939331B2 (en) | 2021-06-09 | 2024-03-26 | Incyte Corporation | Tricyclic heterocycles as FGFR inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2001512742A (en) | 2001-08-28 |
| NZ502877A (en) | 2001-11-30 |
| EP0994892A1 (en) | 2000-04-26 |
| CA2300478A1 (en) | 1999-02-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0994892A1 (en) | SELECTIVE FACTOR Xa INHIBITORS | |
| AU4898897A (en) | Selective factor xa inhibitors | |
| WO1998016523A2 (en) | Selective factor xa inhibitors | |
| WO1998016523A9 (en) | Selective factor xa inhibitors | |
| WO1999007731A1 (en) | SELECTIVE FACTOR Xa INHIBITORS | |
| US6204268B1 (en) | Selective factor Xa inhibitors | |
| AU4904797A (en) | Selective factor xa inhibitors | |
| EP0994893B1 (en) | Selective factor xa inhibitors containing a fused azepinone structure | |
| US6262047B1 (en) | Selective factor Xa inhibitors | |
| AU741099B2 (en) | Selective factor Xa inhibitors | |
| US6369063B1 (en) | Selective factor Xa inhibitors | |
| EP0975625A1 (en) | SELECTIVE FACTOR Xa INHIBITORS | |
| US6525076B1 (en) | Selective factor Xa inhibitors | |
| US6333321B1 (en) | Selective factor Xa inhibitors | |
| US6369080B2 (en) | Selective factor Xa inhibitors | |
| US6194435B1 (en) | Lactams as selective factor Xa inhibitors | |
| US6218382B1 (en) | Selective factor Xa inhibitors | |
| WO1998016524A1 (en) | HETEROCYCLIC DERIVATIVES AS FACTOR Xa INHIBITORS | |
| AU720513C (en) | Selective factor Xa inhibitors | |
| AU753842B2 (en) | Selective factor Xa inhibitors | |
| AU746596B2 (en) | Selective factor Xa inhibitors | |
| EP0939758A1 (en) | HETEROCYCLIC DERIVATIVES AS FACTOR Xa INHIBITORS | |
| AU8826998A (en) | Selective factor xa inhibitors containing a fused azepinone structure | |
| MXPA00001443A (en) | SELECTIVE FACTOR Xa INHIBITORS |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GE GH GM HR HU ID IL IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG UZ VN YU ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW SD SZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2000/001443 Country of ref document: MX |
|
| NENP | Non-entry into the national phase |
Ref country code: KR |
|
| ENP | Entry into the national phase |
Ref country code: JP Ref document number: 2000 506234 Kind code of ref document: A Format of ref document f/p: F |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 502877 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1998938486 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2300478 Country of ref document: CA Ref country code: CA Ref document number: 2300478 Kind code of ref document: A Format of ref document f/p: F |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 87005/98 Country of ref document: AU |
|
| WWP | Wipo information: published in national office |
Ref document number: 1998938486 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWG | Wipo information: grant in national office |
Ref document number: 87005/98 Country of ref document: AU |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1998938486 Country of ref document: EP |