PHARMACEUTICAL COMPOSIΗONS CONTAINING VANILLOID AGONISTS IN COMBINAΗON WITH VANILLOID ANTAGONISTS
TECHNICAL FIELD OF THE INVENTION
5 The present invention relates to the use of vanilloid agonists and antagonists for the therapeutic desensitization of vanilloid-responsive neurons, and to pharmaceutical compositions which contain a combination of the vanilloid agonists and antagonists.
10 BACKGROUND OF THE INVENTION
Capsaicin stimulates and then desensitizes sensory afferent C-fiber neurons, Aδ fiber neurons, and other sensory neurons involved in pain perception and other perceptions. Capsaicin and analogs of capsaicin are known in the art and described by Reid et al. in U.S. Patent 5,403,868, hereby incorporated by reference
15 in its entirety.
Resiniferatoxin (RTX), tinyatoxin (TTX), and related pharmacologically active compounds are potent stimulants of capsaicin-sensitive neurons. RTX, TTX, and analogues thereof are known in the art and described in U.S. Patent 4,939,149 (Blumberg) and in U.S. Patent 5,021,450 (Blumberg), both hereby
20 incorporated in their entirety by reference. RTX, TTX, and analogues thereof described above and below, stimulate and then desensitize vanilloid-sensitive neurons, in a manner similar to that of capsaicin. Therefore RTX, TTX, and their analogues have medical and veterinary utility in the treatment of arthritis, asthma, bladder hyperreflexia, allergic responses including rhinitis, fever, pain (including
25 pain associated with cancer, peripheral neuropathies, and postherpetic neuralgia), and in biological processes mediated by tachykinins, including substance P and other neuropeptides depleted by capsaicin.
The effects of RTX are, in general, similar to capsaicin, but are much more potent, up to 20,000 times more potent. Both capsaicin and RTX are known to
30 affect a wide spectrum of mammals including humans, monkeys (and other primates), horses, dogs, mice and rats. Sensitivity to RTX is easily demonstrated in these mammals. For example, humans exhibit "gustatory sweating" when they consume hot peppers (which contain capsaicin) and are effectively treated with Zostrix™ (capsaicin) for a variety of indications. Likewise, horses are
35 therapeutically treated for arthritic-like indications by capsaicin creams and dogs are known to be adversely affected by pepper sprays.
A wide- variety of laboratory tests have also been used to demonstrate sensitivity to and potency of capsaicin-like molecules in the laboratory. These tests include the tail-flick test, yeast-induced inflammation test, the ethanol- induced gastric lesion test (Reid, supra). More specific tests employing 3H- resinferatoxin binding to membrane preparations or cultured dorsal root ganglions (G. Acs et al. Molecular Brain Research, 35, 173-182 (1996)), as well as ion flux studies and electrophysiological studies as described by Bevan et al. (Bevan et al., British Journal of Pharmacology, 107, 544-552 (1992), hereby incorporated by reference) are also known. Animals which exhibit a physiological or biochemical sensitivity to capsaicin or RTX are referred to as capsaicin-sensitive. Additionally neurons and other cells that bind or respond to capsaicin, or RTX, are referred to as capsaicin- sensitive neurons. However, capsaicin-sensitive neurons can be defined more specifically as those neurons that not only respond to capsaicin, but also become insensitive (desensitized) to capsaicin or RTX upon repeated capsaicin challenge (Buck et al., Pharmacol. Rev.. 38, 179-226 (1986) and Holzer, Pharmacol. Rev.. 43, 144-201 (1991)). It is this latter, more specific definition which is preferred herein.
It is generally understood in the art that the homovanillic acid moiety shared by capsaicin and RTX is central to the similarity of the pharmacological activity of these neuroactive compounds. Accordingly, the site of capsaicin activity is commonly referred to as a vanilloid receptor.
Capsaicin-like antagonists of capsaicin are also known. The most familiar example of these antagonists is capsazepine. Capsazepine and molecules similar to capsazepine are described in Walpole et al., J. Med. Chem., 37, 1942-1954
(1994), hereby incorporated by reference in its entirety. Capsazepine and related compounds are competitive antagonists of both capsaicin and capsaicin-like agonists and RTX and RTX-like agonists.
Despite the known properties of capsaicin and RTX, and the known properties of capsaicin-like antagonists, there has been no motivation heretofore to use a therapeutic combination of RTX-like capsaicin agonists and capsaicin-like antagonists to desensitize a vanilloid-responsive cell. In fact, heretofore it was believed that the acute response to capsaicin agonists was causally linked to the desensitization that follows. Prior to the present disclosure it would have been believed that administration of antagonists in sufficient quantities to attenuate the toxic response to capsaicin agonists would have attenuated the subsequent desensitization to an equal extent.
Accordingly it is a principle object of the present invention to provide a pharmaceutical composition which includes a therapeutically effective combination of RTX-like capsaicin agonists and capsaicin-like antagonists that is capable of desensitizing a vanilloid responsive cell with a reduced toxic response normally attendant the administration of the agonist alone.
It is another object of the present invention to provide a method for desensitizing a vanilloid-responsive cell which includes the administration of a therapeutically effective combination of a RTX-like capsaicin agonist and a capsaicin-like antagonist. It is yet another object of the invention to provide a pharmaceutical composition as described which has attenuated side effects. These and other objects and advantages of the present invention will be apparent from the following detailed description of the invention.
SUMMARY OF THE INVENTION
The present invention is predicated on the discovery that vanilloid- responsive cells can be desensitized by a therapeutically effective combination of a capsaicin agonist and a capsaicin antagonist. Advantageously, the therapeutic index of the combination therapy exceeds the therapeutic index of administering the agonist alone. The present invention thus provides a method of desensitizing a vanilloid-responsive cell comprising administering to a capsaicin-sensitive animal a therapeutically effective combination of capsaicin agonist and a capsaicin antagonist. In accordance with the invention, the capsaicin agonist can be of the formula
Formula I
wherein RQ is hydroxy or methoxy, R10 and Rπ, which may be the same or different, are independently selected from the group consisting of hydrogen, hydroxyl, or a C,.10 alkyl or aryl ester, R12 , R] , and R15, which may be the same or different, are independently selected from the group consisting of hydrogen, hydroxy, methoxy, sulfhydryl, nitro, amino, ethoxy, halo, OCOCH3, and O(CH2)2NH2, R13 is selected from the group consisting of hydroxy, OCOCH3 and O(CH2)2NH2.
The capsaicin agonist can also be of the formula
Formula I I
wherein R2 is hydroxy or methoxy, R3 is a loweralkylaryl or aryl group having 1 to 3 rings, R4 to R7, which may be the same or different, are independently selected from the group consisting of hydrogen, hydroxy, methoxy, sulfhydryl, nitro, amino, ethoxy, halo, and OCOCH3.
In accordance with the invention, the antagonist of capsaicin is of the formula
A— B-D „ Formul , a τ IτI τI
B = CH
2— X—
and wherein J, is independently selected from the group consisting of C,_
3 alkoxy, hydroxy, a halo, and a divalent alkyl which together with a nitrogen of X forms a heterocyclic ring of from 5 to 12 carbon atoms and wherein, when n is greater than one, each J, may be the same or different; n is 1-5; J
2 and J
3 are independently selected from hydrogen, and C,_
5 alkyl; D is selected from the group consisting of C
4.
20 alkyl, C
4.
20 alkenyl, C
4.
20 alkynyl, aryl of 1-3 rings substituted with C,.
g alkyl, a C
4.
20 alkyl, alkenyl or alkynyl substituted with an aryl of 1 to 3 rings, aryl, haloaryl, haloarylalkenyl, such that the vanilloid-responsive cell becomes less sensitive to an initial administration of capsaicin and such that the acute response to a capsaicin agonist is attenuated.
The present invention also provides a pharmaceutical composition comprising the an agonist of Formula I or Formula II, an antagonist of Formula III, and a pharmaceutically acceptable carrier. It will be appreciated that the capsaicin agonist and the antagonist can be used concurrently, e.g., in the same pharmaceutical composition or administered at the same time, or the agonist and antagonist may be used sequentially to reduce the toxic effects of the agonist and/or increase the therapeutic index.
BRIEF DESCRIPTION OF THE DRAWINGS
Figure 1 depicts the data demonstrating the enhanced therapeutic index of resiniferatoxin when capsazepine is co-administered.
DETAILED DESCRIPTION OF THE INVENTION As observed for capsaicin, treatment of vanilloid sensitive cells with resiniferatoxin, and related compounds, causes an initial excitation and a subsequent desensitization to repeated applications of the compound (Szallasi et al, Neuroscience, 30, 515-520 (1989), Blumberg et al., Resiniferatoxin, An Ultrapotent Capsaicin Analog, in Capsaicin in the Study of Pain. J.N. Wood, ed., pages 45-62, Academic Press Publishers, London (1993)). The initial excitation is frequently manifested as pain in the patient or subject and can be quantified by
directly or indirectly measuring the calcium ion flux induced in vanilloid sensitive cells. Desensitization is a blunting or abrogation of response to capsaicin agonists measured at a time point a few hours after than the initial application of the agonist (e.g., 2-8 hours later, six hours later is a common time point for conducting this measurement). For example, desensitization can be effected in some cells with 250 pM RTX. Subsequent dosages of capsaicin agonists nearly 1000 times more potent (e.g., 200 nM RTX) will then fail to induce a strong response in vanilloid sensitive cells. This is a typical illustration of desensitization, and is believed to be the mechanism by which the commercially available analgesic Zostrix™ (capsaicin) functions.
It has been postulated for decades that the pungency of capsaicin analogs is proportional to the desensitization that follows (Jansco, Desensitization With Capsaicin and Related Acylamides as a Tool for Studying the Function of Pain Receptors, In Pharmacology of Pain, 9, 33-55, Pergamon Publishers, Oxford, U.K., (1968)). However, quite unexpectedly and surprisingly it has been found that the pungency of resiniferatoxin, tinyatoxin, related daphnanes, and C-20 homovanillyl phorbolesters is not proportional to the desensitization that follows.
It has now been found that resiniferatoxin, tinyatoxin, and related daphnanes and C-20 homovanillyl phorbol esters induce desensitization to further administration of capsaicin agonists, or other painful stimuli by a process which is distinct from the acute response (i.e., the initial excitation, sometimes referred to as the toxic response) initially induced by these compounds. Further, it has now been found that RTX, and the like, are capable of inducing desensitization even in the presence of competing concentrations of capsazepine and other antagonists, which are capable of blocking or significantly attenuating the acute phase response to the drug. Accordingly, in accordance with the invention it is possible to formulate pharmaceutical compositions with an enhanced therapeutic index for clinical and veterinary use in the desensitization of vanilloid sensitive cells by controlling the proportion of an RTX-like agonist with a capsaicin-like antagonist. As used herein, the therapeutic index of a pharmaceutical composition is a number representing the magnitude of desensitization produced by the administration of the pharmaceutical composition divided by the magnitude of the acute (toxic) response induced by the administration of the pharmaceutical composition. It is often useful to substitute the level of calcium ion influx in capsaicin responsive neurons for the direct measurement of response to resiniferatoxin (or the like) during the initial application of such capsaicin agonists, because calcium ion influx is more easily quantified than pain and other
indications of acute phase response. Similarly, desensitization can be quantified by observing the degree of inhibition of calcium ion flux upon a subsequent challenging application of capsaicin or the like.
The pharmaceutical compositions can be prepared as one discrete formulation or can be formulated from two or more discrete preparations. If the pharmaceutical composition is composed of multiple components, these components can be mixed before administration to an animal or may be co- administered to achieve the desired therapeutic benefit. Regardless of the initial form of the pharmaceutical composition, the pharmaceutical composition is formulated and administered such that the animal receives a capsaicin agonist and a capsaicin antagonist dissolved, suspended, or dispersed in a pharmaceutically acceptable carrier.
The capsaicin agonist which mediates the therapeutic desensitization is of the formula
Formula I
wherein RR =
wherein Re, is hydroxy or methoxy, R]0 and R, ,, which may be the same or different, are independently selected from the group consisting of hydrogen, hydroxyl, or a C^n alkyl or aryl ester (such as decanoate, benzoate, or phenylacetate), R12 , R14, and R15, which may be the same or different, and are independently selected from the group consisting of hydrogen, hydroxy, methoxy, sulfhydryl, nitro, amino, ethoxy, halo (i.e., fluoro, chloro, bromo, iodo), OCOCH3, and O(CH2)2NH2, R13 is selected from the group consisting of hydroxy, OCOCH3, and O(CH2)2NH2, or is of the formula
Formula I I
wherein R2 is hydroxy or methoxy, R3 is a loweralkylaryl or aryl group having from 1 to 3 rings, preferably an aryl group with 1 ring having 5-8 ring members, yet more preferably phenyl or C,_4 alkylphenyl, R4 to R7, may be the same or different, and are independently selected from the group consisting of hydrogen, hydroxy, methoxy, sulfhydryl, nitro, amino, ethoxy, halo (i.e., chloro, fluoro, bromo, iodo), OCOCH3. By loweralkyl is meant a alkyl moiety of 1 to 8 carbons, preferably 1 to 4 carbons.
One particular example of a compound of Formula II is tinyatoxin. Another particular example of a compound of Formula II is resiniferatoxin. Use of resiniferatoxin is advantageous in that it is among the capsaicin analogs most widely utilized in current biomedical research. This research has revealed that resiniferatoxin is up to 20,000 times more potent than capsaicin. Surprisingly, it has now been found that competitive antagonists of resiniferatoxin and capsaicin (e.g., capsazepine) can inhibit the toxic effects of resiniferatoxin administration at least about ten-fold more effectively than they inhibit the desensitizing effects of resiniferatoxin. Therefore, the combination of resiniferatoxin, or the like, with capsazepine or the like, allows a greater therapeutic benefit to be derived from the administration of resiniferatoxin, because it is the toxic effects of capsaicin and resiniferatoxin that function to limit the amount of capsaicin or resiniferatoxin which can be administered to a patient and these toxic effects are suitably reduced
when capsazepine-like antagonists are co-administered with resiniferatoxin or the like.
The capsaicin antagonists useful accordance with the present invention are of the formula
A— B-D „ Formul . a τ I τI τI
B = CH, X-
and wherein J, is independently selected from the group consisting of C^ alkoxy, hydroxy, a halo, and a divalent alkyl which together with a nitrogen of X forms a heterocyclic ring of from 5 to 12 carbon atoms and wherein when n is greater than one, each J, may be the same or different; n is 1-5; J2 and J3 are independently selected from hydrogen, and C,.5 alkyl; D is selected from the group consisting of C4.20 alkyl, C4.20 alkenyl, C4.20 alkynyl, aryl of 1-3 rings substituted with C,.g alkyl, a C4.20 alkyl, alkenyl or alkynyl substituted with an aryl of 1 to 3 rings, aryl, haloaryl, haloarylalkenyl, such that the vanilloid-responsive cell becomes less sensitive to an initial administration of capsaicin and such that the acute response to a capsaicin agonist is attenuated. Compounds illustrative of Formula III include the hydroxy-halo benzene containing compounds, which include compounds of Formula III wherein n is 2 or 3, preferably 2, and a first occurrence of J, is hydroxy, and a second occurrence of
J, is halo, preferably chloro. Commonly, the hydroxy moiety is 4-hydroxy, and the halogen, e.g., is 3-chloro.
Another group of compounds illustrative of Formula III include methoxybenzene containing compounds of Formula III, wherein n is 1 or 2, preferably 1, and at least one occurrence of J, is methoxy, preferably 3-methoxy.
Another group of compounds illustrative of Formula III include the divalent-alkylbenzene containing compounds of Formula III. A well known example of this group is capsazepine. This group of compounds are illustrated by compounds wherein a first occurrence of J, is 2, N-propyl and additional occurrences of Jj include 3 -hydroxy and 4-hydroxy. A particularly preferred characteristic of the divalent-alkylbenzene containing compounds of Formula III are those in which the aryl ring of A is perpendicular the plane of the amide or thiourea of B. By way of illustration, the A ring is usually perpendicular to the plane of B when the ring formed in part by the divalent alkyl moiety has seven ring atoms and is attached at position 2 of the A ring.
Other compounds of Formula III of particular interest are those in which J2 is hydrogen.
Moreover, while the hydrophobic moiety represented by D in Formula III can vary, those D moieties of particular interest a C6.10 alkyl, alkenyl, or alkynyl, especially about Cg alkyl, and substituted or unsubstituted aryl alkenyl moieties such as iodophenylpropenamide.
Particular embodiments of Formula III that are useful in the context of the present invention include N-octyl N'(p-hydroxy m-chlorobenzyl) thiourea, N-octyl
N ' (p-hydroxy m-chlorophenyl)acetamide, N-(o-methoxybenzyl)-N ' -octy 1 thiourea, and 3-methoxybenzyl p-iodophenylpropenamide. The best known antagonist suitable for use in the present invention is capsazepine. To better illustrate the compounds of Formula III the following depictions are provided.
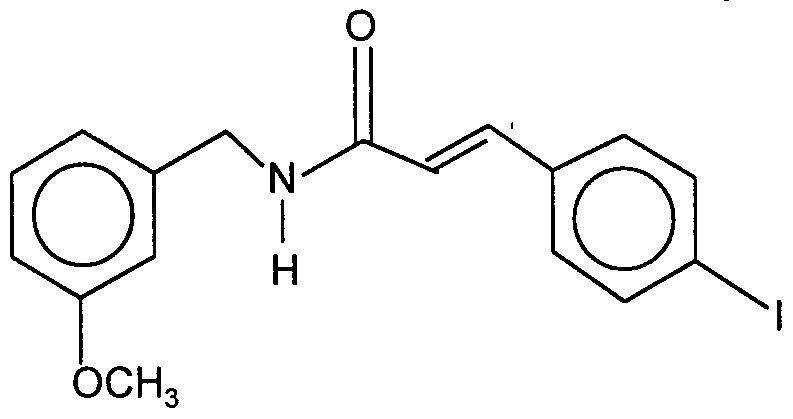
3-methoxybenzyl p-iodophenylpropenamide
N- (o-methoxybenzyl) , N'-octyl thiourea
N-octyl (p-hydroxy m-chlorophenyl) acetamide
N-octyl, N' - (p-hydroxy m-chlorobenzyl) thiourea
Capsazepine
Advantageously, any one or more compounds of Formula I or Formula II can be used in combination to practice the claimed method of desensitizing an vanilloid-responsive cell and, any one or more compounds of Formula I or Formula II can be combined with any one or more compound of Formula III in a pharmaceutically acceptable carrier.
The compounds useful in the context of the present inventive method can be administered to a capsaicin-sensitive animal, especially humans, non-human primates, horses, dogs, rats, and mice by any suitable means. Such administration means or routes include for example, orally, inhalation, parenterally, vaginally, rectally, intranasal, intravesicular, intraperitoneally, and, especially, topically. When applied topically the agonist should be present in a concentration sufficient to provide a therapeutic benefit (i.e., desensitization) in the presence of the antagonist. In one particular embodiment, this amount is from a 0.0001% to a 1% cream, preferably a 0.025% to 0.1% cream. To effect a suitable attenuation of the initial excitement without abrogating the desensitization, the relative strength of the antagonist (e.g., capsazepine) must be balanced against the relative strength of the antagonist. For example, if a relatively very weak agonist is selected for use with a strong antagonist, then the ratio of antagonist to agonist will be smaller than if a relatively strong agonist is used with the same antagonist. Accordingly, ratios of antagonist to agonist varying in the range of 1 : 1 up to 108 : 1 are contemplated. Advantageously, standard potencies can be derived by comparison to capsaicin or resiniferatoxin for agonists and by comparison to capsazepine for antagonists. After making such a routine determination, calculating the appropriate ratios for use in the context of the present invention would be straightforward. Specific ratios for using resiniferatoxin in combination with capsazepine and the rationale for selecting these ratios are disclosed in detail below.
When administered internally, the quantity of agonist and antagonist will vary according to the distribution properties associated with the pharmaceutically acceptable carrier or carriers and the distribution kinetics associated with that carrier when administered by a particular route in the patient or subject. The ratio of agonist to antagonist, however, should remain essentially constant. If the internally applied composition will be dispersed in the blood stream (e.g., if applied in liposomes administered intravenously) then one skilled in the art will appreciate that 6-8% of the weight of the patient will be derived from the blood volume. Accordingly, an agonist with a particular potency should be administered such that it is equivalent in its agonism to about 25 pM to about 25 nM resiniferatoxin and an antagonist with a particular potency should be administered such that it is equivalent in its antagonism to between 1 μM to 1 mM capsazepine. In one particular embodiment, this value will be approximately 10 to 35 times the dose required for maximal inhibition of acute phase response to capsaicin. Of course, the amount administered will vary according to such parameters as the severity of the indication treated, the age, weight, and general health of the patient
or subject, the carrier employed, and the route of administration. Further, the quantity employed will be in keeping with the entire therapeutic regime for an animal under the care of a clinician or veterinarian.
A therapeutically effective amount of the combined pharmacologically active ingredients of the present invention can be used in a pharmaceutical composition, which of course, will depend upon the particular means or route of administration, as well as other practical considerations. Accordingly, there is a wide variety of suitable formulations of the pharmaceutical compositions of the present invention. Formulations suitable for oral administration can comprise (a) liquid solutions, such as water or saline, (b) capsules, sachets or tablets, each containing a predetermined amount of the active ingredient, as solids or granules, (c) suspensions in an appropriate liquid, and (d) suitable emulsions. Tablet forms can include one or more of lactose, mannitol, corn starch, potato starch, microcrystalline cellulose, acacia, gelatin, colloidal silicon dioxide, croscarmellose sodium, talc, magnesium stearate, stearic acid, and other excipients, colorants, diluents, buffering agents, moistening agents, preservatives, flavoring agents, and pharmacologically compatible carriers. Lozenge forms can comprise the active ingredient in a flavor, usually sucrose and acacia or tragacanth, as well as pastilles comprising the active ingredient in an inert base, such as gelatin and glycerin or sucrose and acacia emulsions, gels, and the like containing, in addition to the active ingredient, such carriers as are known in the art.
One particular embodiment among the preferred embodiments of the present invention comprises a combination of a resiniferatoxin-like capsaicin agonist, in combination with a capsazepine- like capsaicin antagonist, in a cream, paste, emulsion, or the like for application to the skin of a human, domesticated horse, or a dog kept as a pet (as opposed to a research animal), especially to the skin of a human. The dosages of the pharmacologically active ingredients for such a pharmaceutical composition are disclosed above. All pharmaceutical compositions disclosed above can comprise formulations may include diluents, such as water and alcohols, for example, ethanol, benzyl alcohol, and the polyethylene alcohols, either with or without the addition of a pharmaceutically acceptable surfactant, suspending agent, or emulsifying agent. Capsule forms can be of the ordinary hard- or soft-shelled gelatin type containing, for example, surfactants, lubricants, and inert fillers, such as lactose, sucrose, calcium phosphate, and corn starch. Tablet forms can include one or more of lactose, sucrose, mannitol, corn starch, potato starch, alginic acid,
microcrystalline cellulose, acacia, gelatin, guar gum, colloidal silicon dioxide, croscarmellose sodium, talc, magnesium stearate, calcium stearate, zinc stearate, stearic acid, and other excipients, colorants, diluents, buffering agents, disintegrating agents, moistening agents, preservatives, flavoring agents, and pharmacologically compatible excipients. Lozenge forms can comprise the active ingredient in a flavor, usually sucrose and acacia or tragacanth, as well as pastilles comprising the active ingredient in an inert base, such as gelatin and glycerin, or sucrose and acacia, emulsions, gels, and the like containing, in addition to the active ingredient, such excipients as are known in the art. Regardless of the form of the combined capsaicin agonists and antagonists, the present invention provides a method of desensitizing a vanilloid-responsive cells that employs administering to a capsaicin-sensitive animal a therapeutically effective combination of capsaicin agonist and an antagonist of capsaicin such that the vanilloid-responsive cell becomes less sensitive to further or repeated administration of capsaicin and such that the acute response to the capsaicin agonist is attenuated.
Many but not all animals are sensitive to vanilloids. Humans, monkeys, horses, dogs, rats, and mice are all suitably sensitive to vanilloids. It is routine to determine whether any other animal is also sensitive to vanilloids such as capsaicin and resiniferatoxin. Ingestion of vanilloids in sensitive animals often causes gustatory sweating, while application to mucosa or skin is often accompanied by a burning sensation or behavior which would indicate the perception of a burning sensation (e.g., tail-flicking or eye-wiping).
Cells which are sensitive to vanilloids as meant herein exhibit a calcium ion influx upon a first exposure to capsaicin and the like upon a first administration and exhibit a reduced response to similar or even elevated concentrations of capsaicin a short period later (e.g., 2 to 8 hours later, often measured 6 hours later). The calcium uptake can be measured by any suitable means. Suitable measurements include behavioral measurements such as eye-wiping, electrophysiological measurement of calcium-dependent ion flows, or monitoring of the uptake of radioactive calcium.
Primates are frequently about ten- fold less sensitive to vanilloids than rats. Therefore, optimum concentrations of either the agonists or antagonists for primates, including humans will typically relate to optimum concentrations observed for rats by 0.1 to 1000 times, preferably 5 to 100 times, and most preferably 5 to 50 times the concentrations.
Particular examples of vanilloid responsive cells in rats and humans are sensory afferent neurons, such as C-fiber sensory afferent neurons or a Aδ-fiber sensory afferent neuron. It is also known in the art that some neurons of the central nervous system contain vanilloid receptors and can be treated in accordance with the present invention.
In selecting a dosage of capsaicin antagonist, it is often useful to select a dosage that is 10-30 times the dose that is required to inhibit an acute response to capsaicin. In this regard, it is useful to determine either the dosage of antagonist at which less than 10% of the maximum calcium ion influx occurs in capsaicin sensitive cells and multiply that dosage by between 10 and 30 times or to determine a dosage 10-30 times the Kj with respect to an optimal dose of capsaicin. Since the antagonist capsazepine has been studied more extensively than any other capsaicin antagonist, its use as the antagonist in the present inventive composition and method is among the preferred embodiments.
Example 1 The following example demonstrates the relative affinity of various compounds useful in the context of the present invention for inhibiting the specific binding of 3H-resiniferatoxin to rat dorsal root ganglion membrane preparations and for stimulation of calcium (45Ca) uptake in cultured rat dorsal root ganglion neurons.
Female Sprague-Dawley rats (150-160 g body weight) were allowed access to food and water ad libitum. Anesthasized rats were killed by decapitation. The spinal columns were removed aseptically and dorsal root ganglia were dissected out and collected in DMEM containing 0.5% heat inactivated fetal bovine serum, 25 mM HEPES, 1 mM Na-pyruvate, and antibiotics. Ganglia were digested with 0.125% collagenase solution, washed twice in medium and triturated to form a single cell suspension. The cells were pelleted through a cushion of DMEM containing 15% fatty acid free BSA to remove myelin debris.
Resuspended cells were plated in laminin-coated 24-well plates at a density of 2 x 104 cells/well in 500 μl medium, treated with 10"4 M cytosine arabinoside for 24 hours, and cultured for 5 days in DMEM containing 10% fetal bovine serum, 10% horse serum, antibiotics, and 200 ng/ml mouse submaxillary gland 2.5 S nerve growth factor.
Alternatively, dispersed ganglia from six rats were placed in two 150 mm laminin coated dishes to achieve neuronal enrichment. After 18 hours, the non-
neuronal cells were firmly attached to the dish. Neuronal cells, which were weakly adherent, were selectively dislodged, washed in DMEM and re-plated at a density of 5 x 103 cells/well (96 well plate) in 100 μl serum-free medium and used for resiniferatoxin binding assays and calcium uptake assays immediately. Membrane preparations were then prepared as described by Szallasi et al.,
Resiniferatoxin In: P.M. Conn (Ed.), Methods in Neurosciences, 8, 368-380 Neurotoxins, Academic Press, Orlando, FL, USA, (1990). Tritiated resiniferatoxin binding assays were carried out as described, ibid. Six to 500 pM 3H- resiniferatoxin was used for binding analysis. Nonspecific binding was determined in the presence of 100 nM radioinert resiniferatoxin. Binding data used to calculate the data of Table 1 were expressed as fmol/mg protein.
Calcium uptake by these ganglia was determined by culturing the ganglia as above and treating with 1 μCi/ml 45Ca and increasing concentrations of the different compounds. Cells were washed six times by filtration. Calcium uptake was determined by scintillation counting.
Table 1. Effectiveness of various vanilloids in rat dorsal root ganglion cells.
Inhibition of 3H-RTX Stimulation of binding Calciu:m Uptake
Hill Hill
Ki (nM) Coefficient EC 50 Coefficient
RTX 0.0385 1.95 0.94 1.15
RTX-amide1 25.7 1.92 26.5 1.07
RTX-thiourea2 68.5 1.98 149 0.93
TTX 0.173 1.82 1.46 1.18
PDB-HVA3 7980 1.72 1410 1.11
Capsaicin 4930 1.81 340 1.08
Capsazepine 3890 1.74 -300 0.96
1 Resiniferonol 20-homovanillylamide 2 4-hydroxy-3-methoxyphenyl thiourea
3 Phorbol 12 , 13-dibenzoate 20-homovanillylamide
Example 2 The following example compares the sensitivity of rats and monkeys to resiniferatoxin mediated desensitization, resiniferatoxin mediated calcium uptake, and capsaicin mediated calcium uptake. This information allows one skilled in the art to extend the data derived from rats to the treatment of primates, especially monkeys and humans.
Using methods similar to those described in Example 1, the data of Table 2 were collected. These data show that monkeys are also capsaicin-sensitive, but are approximately 3-6 fold less sensitive than rats. Therefore, the present inventive
pharmaceutical compositions should be formulated 3-6 fold more concentrated with respect to the active ingredients when used for monkeys, rather than rats.
Table 2. Relative sensitivity to vanilloids of rat and monkey.
Rat Monkey Ratio
RTX desensitization 0.081 nM 0.46 nM 5.7 RTX Ca uptake 1.24 nM 6.2 nM 5.0 Capsaicin Ca uptake 316 nM 880 nM 2.8
Example 3 This example sets forth the effect of pre-treating vanilloid sensitive cells with resiniferatoxin. Ganglia, prepared as in Example 1, were subjected to varying concentrations of resiniferatoxin either six hours preceding administration of 3 μM capsaicin or simultaneously with 3 μM capsaicin. No utilized concentration of resiniferatoxin had any effect on calcium uptake in response to 3 μM capsaicin when the compounds were co-administered. When cells were pretreated with concentrations of resiniferatoxin ranging from 0.1 to 0.5 nM, a dose dependent inhibition of calcium uptake was observed upon subsequent challenge with 3 μM capsaicin (six hours later). Treatments utilizing between 0.5 to 2.0 nM resiniferatoxin, were sufficient to completely block detectable calcium uptake upon challenge with capsaicin six hours later.
Example 4 This example sets forth that co-administration of a capsaicin antagonist (e.g., capsazepine) with a capsaicin agonist (e.g., resiniferatoxin) results in effective desensitization of vanilloid sensitive cells with a concomitant reduction in acute phase toxicity. That is, co-administration of antagonists with agonists results in an increase in the therapeutic index relative to the administration of agonists alone.
Control and capsazepine pretreated cells were challenged with different concentrations of resiniferatoxin to induce 45Ca uptake. The data points derived from these experiments are depicted in Figure 1 by squares. It is apparent that the co-administration of capsazepine (indicated by filled squares) increases the amount of resiniferatoxin required to induce a given level of calcium uptake by about tenfold relative to cells not administered capsazepine (indicated by open squares. The large shift to the right of the data from the cells treated with capsazepine indicates
that there is a reduction in the calcium influx mediated by moderate levels of resiniferatoxin in the acute (toxic) phase of administration. That is, e.g., a rat administered 3 nM resiniferatoxin would experience severe pain in the absence of 10 μM capsazepine, but nearly no pain in the presence of the capsazepine. The effect on desensitization of co-administration of capsazepine was also examined. In Figure 1, the circles indicate the reduction in the level of calcium uptake as a function of pretreatment with a given concentration of resiniferatoxin and when challenged six hours later with 3 μM capsaicin. The filled circles depict the results when the cells are co-administered 10 μM capsazepine and the indicated concentration of resiniferatoxin, and the open circles depict the results from when the cells are administered resiniferatoxin alone. The small degree of a shift to the right of the line formed by the open circles relative to the line formed by the filled circles indicates that the (therapeutic) desensitization mediated by pretreatment with 100 to 250 pM resiniferatoxin is not adversely affected by the co-administration of capsazepine.
All the data depicted in Figure 1 , therefore show that the presence of capsazepine increases the therapeutic index of resiniferatoxin by about 10-fold. These data also indicate that a ratio of 100 to 250 pM resiniferatoxin to about 10 μM capsazepine effectively increases the therapeutic index of resiniferatoxin. It should be appreciated that this ratio is central to the functionality of the instant embodiment of the present invention, rather than the particular quantity of resiniferatoxin or capsazepine. While not being bound by any particular theory, this appears to be true because of the competitive nature of capsazepine inhibition.
All of the references cited herein, including patents, patent applications, and publications, are hereby incorporated in their entireties by reference.
While this invention has been described with an emphasis upon preferred embodiments, it will be obvious to those of ordinary skill in the art that variations of the preferred embodiments may be used and that it is intended that the invention may be practiced otherwise than as specifically described herein. Accordingly, this invention includes all modifications encompassed within the spirit and scope of the invention as defined by the following claims.