Fused pyrazole derivatives and processes for their preparation
The invention relates to 4-amino-1 H-pyrazolo[3,4-d]pyrimidine derivatives and processes for their preparation, pharmaceutical formulations comprising such derivatives, and the use of these derivatives as pharmaceuticals.
The invention relates to 4-amino-1 H-pyrazolo[3,4-d]pyrimidine derivatives of the formula I
in which m, n and v are each, independently of one another, 0 or 1 ,
R is hydrogen or methyl,
RT is chlorine or methyl which is in each case located in position 3 of the phenyl radical,
X is the group NH(CH-R7), in which t is 0, 1 or 2 and R is hydrogen, or the group
(C[R3]-R4)q, in which q is 0, and
R2 is nitro, cyano, amino, dimethylamino-lower-alkyleneamino, 4-pyridylcarbonylamino,
2-methylpropanoylamino, tert-butyloxycarbonylamino, or methyl which is substituted by amino, C C5-alkanoylamino, tert-butyloxycarbonylamino or benzoylamino, and salts, solvates or tautomers of such compounds with the exception of 3-(3-aminobenzylamino)-
4-(3-chlorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine and its salts, solvates and tautomers.
When m and n are 0, the relevant phenyl rings have no substituents RT and R2. It is preferred for m and n each independently of one another to be 0 or 1. When m and/or n are 1 , the phenyl substituent Ri or R2 is located mainly in position 4, i.e. in the para position, or preferably in position 3, i.e. in the meta position.
When v is 0, the (Rι)m-phenyl radical is bonded directly to the nitrogen atom in position 4 of the 1 H-pyrazolo[3,4-d]pyrimidine derivatives.
The group NH(CH-R7)t represented by the symbol X is, when t is 0, the bivalent group NH. When t is 1-2 and R7 is hydrogen, the group NH(CH-R7), is the bivalent radical NHCH2 or NH-CH2-CH2, each of which is bonded via the nitrogen atom to the pyrazole ring and via the terminal carbon atom to the phenyl ring. X is preferably NH, NH-CH2 or NH-CH(CH3).
When the symbol X is the group (C[R3]-R4)q in which q is 0, the phenyl radical is bonded directly to the pyrazole ring.
Dimethylamino-lower-alkyleneamino R2 is a radical of the formula
-N=C(R8)-N(methyl)2 in which R8 is hydrogen or lower alkyl, and is, in particular, dimethylaminomethyleneamino of the formula (CH3)2N-CH=N-.
Salts of the compounds of the formula I are, since the latter have basic properties, acid addition salts with organic or inorganic acids, in particular the pharmaceutically acceptable non-toxic salts. Examples of suitable inorganic acids are carbonic acid (preferably in the form of carbonates or bicarbonates); hydrohalic acids such as hydrochloric acid; sulfuric acid; or phosphoric acid. Examples of suitable organic acids are carboxylic, phosphonic, sulfonic or sulfamic acids, for example acetic acid, propionic acid, octanoic acid, decanoic acid, dodecanoic acid, glycolic acid, lactic acid, 2-hydroxybutyric acid, gluconic acid, glucosemonocarboxylic acid, fumaric acid, succinic acid, adipic acid, pimelic acid, suberic acid, azelaic acid, malic acid, tartaric acid, citric acid, glucaric acid, galactaric acid, amino acids such as glutaric acid, aspartic acid, N-methylglycine, acetylaminoacetic acid, N-acetylasparagine or N-acetyicysteine, pyruvic acid, acetoacetic acid, phosphoserine, 2- or 3-glycerophosphoric acid, glucose-6-phosphoric acid, glucose-1 -phosphoric acid, fructose- 1 ,6-bisphosphoric acid, maleic acid, hydroxymaleic acid, methylmaleic acid, cyclohexane- carboxylic acid, adamantanecarboxylic acid, benzoic acid, salicylic acid, 1- or 3-hydroxynaphthalene-2-carboxylic acid, 3,4,5-trimethoxybenzoic acid, 2-phenoxybenzoic acid, 2-acetoxybenzoic acid, 4-aminosalicylic acid, phthalic acid, phenylacetic acid, mandelic acid, cinnamic acid, nicotinic acid, isonicotinic acid, glucuronic acid, galacturonic acid, methane- or ethanesulfonic acid, 2-hydroxyethanesulfonic acid, ethane-1 ,2-disulfonic acid, benzenesulfonic acid, 1-naphthalenesulfonic acid, 1 ,5-naphthalenedisulfonic acid, 2-, 3- or 4-methylbenzenesulfonic acid, methylsulfuric acid, ethylsulfuric acid, dodecylsulfuric
acid, N-cyclohexylsulfamic acid, N-methyl-, N-ethyl- or N-propylsulfamic acid or other organic protic acids such as ascorbic acid.
It is also possible to use pharmaceutically unsuitable salts such as picrates or perchlorates for isolation or purification. Only the pharmaceutically acceptable (in appropriate doses), non-toxic salts are used therapeutically and are therefore preferred.
The compounds of the formula I and intermediates which comprise a pyrazole residue for preparing them may, under certain conditions, for example when they are dissolved in particular solvents, be partly in a tautomeric form in which the hydrogen atom which is normally located on nitrogen N-1 has shifted to another suitable nitrogen atom, for example N-2, N-5 or N-7. The invention also relates to these tautomers.
The compounds of the formula I have valuable pharmacological properties. In particular, they show specific inhibitory effects which are of pharmacological interest. They act primarily as protein tyrosine kinase inhibitors and/or (additionally) as protein servine/theonine kinase inhibitors. For example, they show a potent inhibition of the tyrosine kinase activity of the receptor for epidermal growth factor (EGF) and of c-erbB2 kinase. These receptor-specific enzyme activities play a key role in signal transmission in a large number of mammalian cells, including human cells, in particular of epithelial cells, cells of the immune system and cells of the central and peripheral nervous system. For example, the EGF-induced activation of the receptor-associated protein tyrosine kinase (EGF-R-PTK) is in various types of cells a prerequisite for cell division and thus proliferation of the cell population. Multiplication of EGF receptor-specific tyrosine kinase inhibitors thus inhibits multiplication of the cells. Analogous statements apply to the other protein kinases mentioned hereinbefore and hereinafter.
The inhibition of the EGF receptor-specific protein tyrosine kinase (EGF-R-PTK) can be detected by known methods, for example using the recombinant intracellular domain of the EGF receptor (EGF-R ICD; see, for example, E. McGlynn et al., Europ. J. Biochem. 207, 265-275 (1992)). The compounds of the formula I inhibit the enzyme activity compared with the control without inhibitor by 50% (IC50) for example in a concentration of 0.0005 to 5, in particular of 0.001 to 0.1 mM.
The compounds of the formula I, likewise the micromolar range also show for example an inhibition of cell growth in EGF-dependent cell lines, for example the epidermoid BALB/c
mouse keratinocyte cell line (see Weissmann, B.A., and Aaronson, S.A., Cell 32, 599 (1983)) or the A431 cell line, which are acknowledged to be useful standard sources of EGF-dependent epithelial cells (see Carpenter, G., and Zendegni, J. Anal. Biochem. 153, 279-282 (1985)). The inhibitory effect of the compounds of the formula I is measured in a known test method (see Meyer et al., Int. J. Cancer 43. 851 (1989)) briefly as follows: BALB/cMK cells (10,000/microtiter plate well) are transferred into 96-well microtitre plates. The test compounds (dissolved in DMSO) are added in a series of concentrations (dilution series) so that the final concentration of DMSO is not greater than 1% (v/v). After the addition, the plates are incubated for three days, during which the control cultures without test substance are able to pass through at least three cell division cycles. The growth of the MK cells is measured by means of methylene blue staining: After the incubation, the cells are fixed with glutaraldehyde, washed with water and stained with 0.05% methylene blue. After a washing step, the dye is eluted with 3% HCI, and the optical density of each well of the microtitre plate is measured using a Titretek multiscan at 665 nm. IC50-values are determined by a computer-assisted system using the formula:
IC50 = [(ODxβst - ODStert)/(ODCOntrol " ODS,art)] X 100.
The IC50 in these experiments is reported as the concentration of the particular test compound which results in a cell count which is 50% less than the control without inhibitor. The compounds of the formula I show inhibitory effects in the micromolar range, for example an IC50 of about 0.1 to 10 mM, in particular from 0.4 to 4 mM.
The compounds of the formula I also show in vivo an inhibition of the growth of tumour cells, for example as shown by the test described hereinafter: The test is based on the inhibition of the growth of the human epidermoid carcinoma A431 (ATCC No. CRL 1555; American Type Culture Collection, Rockville, Maryland, USA; see Santon, J.B., et al., Cancer Research 46, 4701-4705 (1986) and Ozawa, S., et al., Int. J. Cancer 40, 706-710 (1987)), which is transplanted into female BALB/c nude mice (Bomholtgard, Denmark). This carcinoma shows a growth which correlates with the extent of EGF receptor expression. In the experiment, tumours which have been grown in vivo and have a volume of about 1 cm3 are removed surgically from experimental animals under sterile conditions. These tumours are comminuted and suspended in 10 volumes (w/v) of phosphate-buffered saline. The suspension is injected s.c. (0.2 ml/mouse in phosphate-buffered saline) into the left flank of the animals. Alternatively, 1 x 106 cells from an in-vitro culture in 0.2 ml of phosphate- buffered saline can be injected. Treatment with the test compound of the formula I is started
5 or 7 days after the transplantation when the tumours have reached a diameter of 4-5 mm. The particular active substance is administered (in various doses in different animal groups) once a day for 15 consecutive days. The tumour growth is determined by measuring the diameters of the tumours along three mutually perpendicular axes. The tumour volumes are calculated using the known formula p x L x D2/6 (see Evans, B.D., et al., Brit. J. Cancer 45, 466-8 (1982)). The results are reported as treated/control percentages (T/C x 100 = T/C%). With a dose of 3 to 50 mg/kg of the active substance, marked inhibition of tumour growth is found, for example T/C% values of less than 10, which denotes strong inhibition of tumour growth.
The compounds of the formula I also inhibit, besides or instead of the EGF-receptor protein tyrosine kinase, other protein tyrosine kinases involved in signal transmission mediated by trophic factors, for example abl kinase, kinases from the family of src kinases such as, in particular, c-src kinase (IC50 for example between 1 and 10 mM) and c-erbB2 kinase (HER- 2), and serine/threonine kinases, for example protein kinase C, all of which are involved in growth regulation and transformation of mammalian cells, including human cells.
The inhibition of c-erbB2 tyrosine kinase (HER-2) can be determined, for example, in analogy to a method used for EGF-R-PTK (see C. House et al., Europ. J. Biochem. 140. 363-367 (1984)). The c-erbB2 kinase can be isolated, and its activity determined, by protocols known per se, for example as disclosed by T. Akiyama et al., Science 232. 1644 (1986)).
The compounds of the formula I which inhibit the tyrosine kinase activity of the receptor for epidermal growth factor (EGF) or furthermore of the other protein tyrosine kinases mentioned are therefore useful, for example, for the treatment of benign or malignant tumors. They are able to bring about tumor regression and to prevent tumor metastasis and the growth of micrometastases. They can be used in particular for epidermal hyper- proliferation (psoriasis), for the treatment of neoplasms of an epithelial nature, for example carcinomas of the breast, and for leukaemias. The compounds of the formula I (especially the novel compounds) can furthermore be used to treat disorders of the immune system in which several or, preferably, single protein tyrosine and/or (furthermore) protein serine/ threonine kinases are involved; these compounds of the formula I can also be used for treating disorders of the central or peripheral nervous system in which signal transmission by several or, preferably, one protein tyrosine and/or (furthermore) protein serine/threonine kinase is involved.
The present invention also relates in general to the use of the compounds of the formula I for inhibiting said protein kinases.
The compounds according to the invention can be used either alone or in combination with other pharmacologically effective substances, for example together with inhibitors of the enzymes of polyamine synthesis, inhibitors of protein kinase C, inhibitors of other tyrosine kinases, cytokines, negative growth regulators, for example TGF-β or IFN-β, aromatase inhibitors, antioestrogens and/or cytostatics.
It is possible in the preferred subject-matters of the invention which are mentioned hereinafter to use in place of general definitions, where logical and expedient, the more specific definitions mentioned at the outset.
Preferred compounds of the formula I are those in which m, n and v are each, independently of one another, 0 or 1 ,
R is hydrogen or methyl,
Ri is chlorine or methyl, in each case located in position 3 of the phenyl radical,
X is the group NH(CH-R7), in which t is 0 or 1 and R is hydrogen, or the group (C[R3]-R4)q, in which q is 0, and
R2 is nitro, cyano, amino, dimethylamino-lower-alkyleneamino, 4-pyridyl-carbonylamino,
2-methylpropanoylamino, tert-butyloxycarbonylamino, or methyl which is substituted by amino, CrCs-alkanoylamino, tert-butyloxycarbonylamino or benzoylamino, and salts, solvates or tautomers of such compounds with the exception of 3-(3-amino-benzylamino)-4-
(3-chlorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine or a pharmaceutically acceptable salt, solvate or tautomer thereof.
The compounds of the formula I mentioned in Examples 71 and 73-93. and the pharmaceutically acceptable salts, solvates or tautomers thereof, are very particularly preferred.
The claimed compounds of the formula I show, in particular by comparison with other 4-amino-1 H-pyrazolo[3,4-d]pyrimidine derivatives, besides high activity in the inhibition of the EGF receptor-specific protein tyrosine kinase, one or more of the following advantageous properties: improved pharmacokinetic properties such as increased cell permeability and elevated blood levels, increased selectivity with respect to other kinases,
considerably lower or virtually no effect on abl kinases such as, in particular v-abl kinase, cdc2 kinase and c-src kinase and, presumably associated with this, fewer side effects.
The compounds of the formula I and their salts can be prepared in a manner known per se. The preparation process according to the invention comprises a) treating a compound of the formula II
in which R5 is hydrogen or methyl, R6 is alkoxy with 1-3 C atoms or nitro, r is an integer from 0 to 2, and the other substituents and symbols have the abovementioned meanings, with a suitable Lewis acid, or
b) reacting a compound of the formula XV
in which the symbols have the abovementioned meanings, with an amine of the formula XVI
in which v is 1 and the other symbols have the abovementioned meanings, or with a salt thereof in the presence of formic acid, or
c) reacting a compound of the formula XV
NH,
in which the symbols have the abovementioned meanings, with a formamide derivative of the formula XVII
in which v is 1 and the other symbols have the abovementioned meanings, or
d) reacting a compound of the formula XVIII
(XVIII)
in which the symbols have the abovementioned meanings, with an amine of the formula XVI
in which v is 0 or 1 , and the other symbols have the abovementioned meanings, or with a salt thereof, or
e) subjecting a compound of the formula XIX
(XIX)
in which v is 0 and the other symbols have the abovementioned meanings, to the conditions of a Dimroth rearrangement, and, if required, converting a compound of the formula I obtained by one of processes a) to e) into its salt, or converting a resulting salt of a compound of the formula I into the free compound.
The procedure for these process variants and the preperation of the starting materials are described in detail below:
General: If necessary, interfering functional groups in the starting materials are protected before the reaction in a manner known per se by protective groups which can easily be eliminated and which are then eliminated again after the reaction has taken place.
Process a): When R5 is hydrogen, a suitable and preferred Lewis acid is aluminium chloride. The reaction is carried out in an inert organic solvent, for example a hydrocarbon such as, preferably, an aromatic hydrocarbon such as, in particular, benzene or toluene, at a temperature between room temperature (about 20°C) and +200 °C, if necessary under protective gas, such as argon, and/or elevated pressure, preferably at the boiling point of the solvent used, that is to say under reflux. When R5 is methyl, boiling with polyphosphoric acid is preferably carried out.
Process b): The starting material of the formula XVI can be used as salt, for example as acetate. In addition to formic acid, it is possible to add other acids such as glacial acetic acid. The reaction is carried out at elevated temperature, preferably 100-250°C, such as, in particular, at 200°C.
Acid addition salts of compounds of the formula I are obtained in a manner known per se, for example by treatment with an acid or a suitable anion exchange reagent.
Acid addition salts can be converted in a conventional way into the free compounds, for example by treatment with a suitable basic agent.
The starting material of the formula II is obtained in the following way: first a compound of the formula III
in which X, R2 and n have the abovementioned meanings, is reacted with a hydrazine derivative of the formula IV
in which R5 is hydrogen or methyl, R6 is alkoxy with 1-3 C atoms or nitro, and r is an integer from 0 to 2, or with a salt thereof to give a pyrazole derivative of the formula V
in which the substituents have the abovementioned meanings. The process starts, for example, from a methanolic solution of a hydrazine derivative of the formula IV in the form of the dihydrochloride, to which firstly a methanolic sodium methoxide solution is added with cooling, for example with ice, and then a solution of a compound of the formula III in a suitable anhydrous alcohol, such as absolute ethanol, is added at room temperature. This is followed by heating to reflux for some hours.
The resulting compound of the formula V is reacted with formic acid to assemble the pyrimidine ring and give a compound of the formula VI
in which the substituents have the abovementioned meanings. A compound of the formula V is preferably heated to reflux in 85% strength formic acid for some hours.
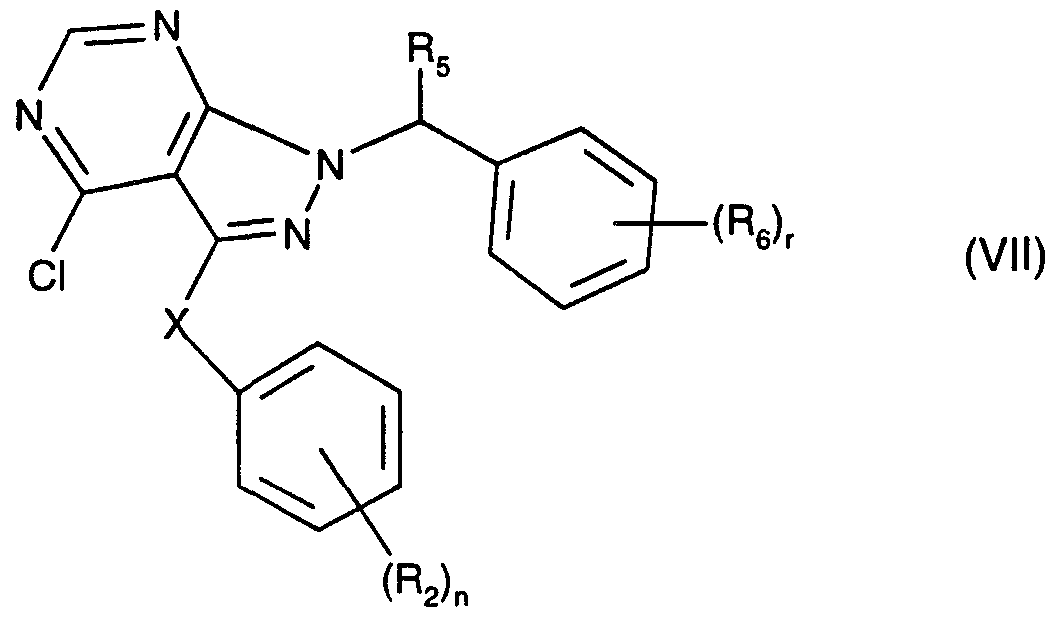
Replacement of the hydroxyl group in a compound of the formula VI by chlorine using phosphoryl chloride (phosphorus oxychloride, POCI3) or phosphorus trichloride (PCI3) results in a compound of the formula VII
in which the substituents have the abovementioned meanings. A compound of the formula VI is preferably heated to reflux in phosphoryl chloride under a protective gas such as argon for some hours.
The compound of the formula VII is then reacted with an aniline derivative of the formula VIII
(R, 1)'m
in which the symbols have the abovementioned meanings, preferably in a suitable solvent such as a suitable alcohol, for example ethanol, under a protective gas such as nitrogen, at elevated temperature, for example under reflux, to give the required starting material of the formula II.
The starting material of the formula III in which X is the group NH(CH2)t in which t has the abovementioned meaning is obtained, for example, by reacting 3,3-bismethylthio-2- cyanoacrylonitrile of the formula IX,
with an amine of the formula X
in which the symbols have the abovementioned meanings. The abovementioned 3,3-bis- methylthio-2-cyanoacrylonitrile of the formula IX is described, under the name "2,2-bis- methylmercapto-1-cyanoacryionitrile" by R. Gompper and W. Tόpel, Chem. Ber. 95, 2861 - 2870, in particular in the middle of page 2868, and can be prepared by addition of malononitrile of the formula CH2(CN)2 onto carbon disulfide in the presence of sodium methoxide in methanol, at a temperature between 5 and 20°C, followed by methylation of the resulting intermediate with dimethyl sulfate.
The starting material of the formula III in which X is the group (C[R
3]-R )
q, in which q is 0, and the other symbols have the abovementioned meanings, that is to say a compound of the formula Ilia
in which R2 and n have the abovementioned meanings, is obtained, for example, by reacting a compound of the formula XI
in which R2 and n have the abovementioned meanings, with a tetracyano epoxide of the formula XII
The compounds of the formula XI are obtained, for example, from an aldehyde of the formula XIII
in which R
2 and n have the abovementioned meanings, which is first converted with sulfur and morpholine into a compound of the formula XIV
in which R2 and n have the abovementioned meanings, which is then converted with methyl iodide in acetone, followed by hydrogen sulfide in pyridine, into a compound of the formula XI.
Process b): The starting material of the formula XVI can be used as salt, for exmple as acetate. In addition to formic acid, it is possible to add other acids such as glacial acetic acid. The reaction is carried out at elevated temperature, preferably 100-250°C, such as, in particular, at 200°C.
The starting material of the formula XV is obtained from a compound of the formula III by reaction with hydrazine in a suitable solvent such as a suitable alkanol such as, preferably, methanol, for example at the reflux temperature.
Process c): The reaction is carried out at elevated temperature, preferably 100-250°C, such as, in particular, at 200°C, in the presence or, where possible, absence of a solvent, i.e. the formamide derivative of the formula XVII can itself act as solvent.
Process d): The reaction is carried out at elevated temperature, preferably 50-180°C, such as, in particular, at 120°C, in the presence, or, where possible, absence of a solvent, i.e. the amine derivative of the formula XVI can itself act as solvent. When v is 0, the amine derivative of the formula XVI is preferably used as salt, for example as hydrochloride. When v is 1 , the amine derivative of the formula XVI is preferably used as free amine.
The starting material of the formula XVIII is obtained from a compound of the formula XV by reaction with a suitable dimethylformamide acetal such as N,N-dimethylformamide diethyl acetal, in a suitable solvent such as a suitable aromatic hydrocarbon such as, in particular, toluene at elevated temperature, preferably 50-180°C, such as, in particular, under reflux.
Process e): The Dimroth rearrangement is carried out at elevated temperature, for example 70-200°C, preferably 80-150°C, for example under reflux, in a suitable water-containing solvent mixture, for example a mixture of water and a suitable ether such as a cyclic ether, for example dioxane, for example a dioxane/water mixture in the ratio of 1 :1 by volume.
The imine of the formula XIX is obtained, for example, from a compound of the formula XV in two stages as follows:
In the first stage, a compound of the formula XV is reacted with triethyl orthoformate of the formula HC(OC2H5)3 to give an ethoxymethyleneamino compound of the formula XX
in which the symbols have the abovementioned meanings.
The reaction is carried out at elevated temperature, preferably 50-180°C, such as, in particular, at 120°C, with the triethyl orthoformate itself acting as solvent. The ethanol formed in the reaction is continuously distilled out of the reaction mixture.
In the second stage, the resulting compound of the formula XX is reacted with an amine of the formula XVI in which v is 0 or 1 , and the other symbols have the abovementioned meanings, to give the required imine of the formula XIX. This reaction is carried out in a suitable solvent such as a suitable alcohol, for example an alkanol, such as, preferably, ethanol, at elevated temperature, preferably 50-180°C, such as, in particular, at 70-120°C, for example at the reflux temperature.
Alternatively, the imine of the formula XIX is obtained directly from a compound of the formula XVIII by reaction with an amine of the formula XVI [similar to Process d)] mixed with
a final product of the formula I. This reaction is carried out in a suitable solvent such as a suitable alcohol, for example an alkanol, such as, preferably, ethanol, at elevated temperature, preferably 50-180°C, such as, in particular, at 70-120°C, for example at the reflux temperature.
Acid addition salts of compounds of the formula I are obtained in a manner known per se, for example by treatment with an acid or a suitable anion exchange reagent.
Acid addition salts can be converted in a conventional way into the free compounds, for example by treatment with a suitable basic agent.
Mixtures of isomers can be fractionated in a manner known per se, for example by fractional crystallization, chromatography etc., into the individual isomers.
The processes described above, including the processes for eliminating protective groups and the additional process measures, are, unless indicated otherwise, carried out in a manner known per se, for example in the presence or absence of, preferably inert, solvents and diluents, if necessary in the presence of condensing agents or catalysts, at reduced or elevated temperature, for example in a temperature range from about -20°C to about 150°C, in particular from about 0°C to about +70°C, preferably from about +10°C to about +50°C, mainly at room temperature, in a suitable vessel, and, if necessary, in an inert gas, for example nitrogen, atmosphere.
Moreover, taking account of all the substituents present in the molecule, if necessary, for example in the presence of easily hydrolysable radicals, particularly mild reaction conditions should be used, such as short reaction times, use of mild acidic or basic agents in low concentration, stoichiometric ratios of amounts, choice of suitable catalysts, solvents, temperature and/or pressure conditions.
The invention also relates to those embodiments of the process which start from a compound which can be obtained as intermediate at any stage of the process, and the missing process steps are carried out or the process is stopped at any stage, or a starting material forms under the reaction conditions or is used in the form of a reactive derivative or salt. Moreover the starting materials preferably used are those which in the process result in the compounds described above as particularly valuable.
The present invention likewise relates to novel starting materials and/or intermediates, and to processes for their preparation. The starting materials used, and the reaction conditions chosen, are preferably such that the compounds mentioned as particularly preferred in this application are obtained.
The intermediates of the formulae V, VI, VII, XV, XVIII, XIX and XX are novel and the present invention likewise relates to them. Preferred intermediates are those which result in the preferred final products of the formula I.
The invention also relates to a method for the treatment of warm-blooded animals suffering from an oncosis, wherein an effective tumour-inhibiting amount of a compound of the formula I or of a pharmaceutically acceptable salt or solvate thereof is administered to warm-blooded animals requiring such a treatment. The invention additionally relates to the use of a compound of the formula I or of a pharmaceutically acceptable salt thereof for inhibiting EGF receptor-specific protein tyrosine kinase C in warm-blooded animals or for manufacturing pharmaceutical products for use for the therapeutic treatment of the human or animal body. This entails administering to a warm-blooded animal with a bodyweight of about 70 kg effective doses depending on the species, age, individual condition, mode of administration and the particular pathology, for example daily doses of about 5-5000 mg, in particular 200-2000 mg.
The invention also relates to pharmaceutical products which comprise an effective amount, in particular an amount effective for the prophylaxis or therapy of one of the abovementioned diseases, of the active substance together with pharmaceutically acceptable carriers which are suitable for topical, enteral, for example oral or rectal, or parenteral administration, and may be inorganic or organic, solid or liquid. Used for oral administration are, in particular, tablets or gelatin capsules which comprise the active ingredient together with diluents, for example lactose, dextrose, sucrose, mannitol, sorbitol, cellulose and/or glycerol, and/or lubricants, for example diatomaceous earth, talc, stearic acid or salts thereof, such as magnesium or calcium stearate, and/or polyethylene glycol. Tablets may likewise comprise binders, for example magnesium aluminium silicate, starches, such as maize, wheat or rice starch, gelatin, methyicellulose, sodium carboxymethylcellulose and/or polyvinylpyrrolidone, and, if required, disintegrants, for example starches, agar, alginic acid or a salt thereof, such as sodium alginate, and/or effervescent mixtures, or adsorbents, colorants, flavourings and sweeteners. The pharmacologically active compounds of the present invention can furthermore be used in the form of products which can be
administered parenterally or of infusion solutions. Solutions of this type are preferably isotonic aqueous solutions or suspensions, it being possible for the latter, for example in the case of lyophilized products which comprise the active substance alone or together with a carrier, for example manitol, to be prepared before use. The pharmaceutical products may be sterilized and/or comprise adjuncts, for example preservatives, stabilizers, wetting agents and/or emulsifiers, solubilizers, salts to regulate the osmotic pressure and/or buffers. The present pharmaceutical products which, if required, may comprise other pharmacologically active substances such as antibiotics, are manufactured in a manner known per se, for example using conventional mixing, granulating, coating, dissolving or lyophilizing processes, and comprise about 1% to 100%, in particular about 5% to about 90%, of the active substance(s).
The following Examples 71 and 73-93 illustrate the invention, without restricting it in any way. The other examples in some cases describe the preparation of starting materials and are provisionally left in this text for this reason. The ratio of the solvents or eluents in the solvent or eluent mixtures used is indicated in proportions by volume (v/v), and temperatures are stated in degrees Celsius.
Abbreviations: abs.: absolute
API-MS: atmospheric pressure ionisation mass spectroscopy
Boc: tert-butyioxycarbonyl
DIPE: diisopropyl ether
DMEU: 1 ,3-dimethyl-2-imidazolidinone
DMF: dimethylformamide
EI-MS: electron impact ionization mass spectroscopy
ESI-MS: electrospray ionisation mass spectroscopy
FAB-MS: fast atom bombardment mass spectroscopy sat.: saturated h: hour(s)
HV: high vacuum cone: concentrated min: minute(s)
RF: reflux
RT: room temperature
RE: rotary evaporator
Brine: saturated sodium chloride solution THF: tetrahydrofuran
HPLC Gradients:
Grad2o-100 20% → 1 00°o a) in ) over 20 min-
Grad5_4o 5% → 40% a) in b) over 20 min.
Eluent a): acetonitrile + 0.05% TFA; eluent b): water + 0.05% TFA. Column (250 x 4.6 mm) packed with C18 Nucleosil reversed phase material (silica gel covalently derivatized with octadecylsilanes and of average particle size 5 μm, Macherey & Nagel, Dϋren, FRG).
Detection by UV absorption at 254 nm. The retention times (tp,et) are indicated in minutes.
Flow rate: 1 ml/min.
Example 1 : With exclusion of air and moisture, a solution of 100 mg of 1 -benzyl-3,4- diphenylaminopyrazolo[3,4-d]pyrimidine in 2.5 ml of absolute benzene is added to a suspension of 203 mg of AICI3 in 2.5 ml of absolute benzene. The reaction mixture is stirred at 50°C for 1.5-2 h until thin-layer chromatography shows no starting material left, and is then stirred into about 30 ml of water. The precipitate is filtered off and dissolved in ethyl acetate. The ethyl acetate phase is washed several times with 5% aqueous sodium bicarbonate solution and then with saturated brine, dried and evaporated to dryness. The residue is crystallized from ethyl acetate/hexane, resulting in 3,4-diphenylamino-1 H- pyrazolo[3,4-d]pyrimidine as colourless crystals; melting point 263-264°C, FAB-MS: (M+H)+ = 303 (C17H14N6).
The starting material is obtained in the following way:
Stage 1.1 : 19.3 ml of a 5.4 normal sodium methoxide solution in 20 ml of methanol (extra pure) are added to 9.9 g of benzylhydrazine dihydrochloride in 20 ml of methanol (extra pure) while cooling in ice, stirred at room temperature for about 15 minutes and then added to a solution of 4.65 g of 2-cyano-3-methylthio-3-phenylaminoacrylonitrile in 150 ml of absolute ethanol. The mixture is heated to reflux for about 17 hours, cooled to room temperature and filtered with suction to remove insolubles. The filtrate is evaporated in a rotary evaporator, and the brown oily residue is chromatographed on 190 g of silica gel using methylene chloride/ethyl acetate mixtures as eluent. 5-Amino-1-benzyl-4-cyano-3- phenylaminopyrazole is obtained; melting point 139-140°C, FAB-MS: (M+H)+ = 290 (Cι7H15N5).
Stage 1.2: 1 g of 5-amino-1 -benzyl-4-cyano-3-phenylaminopyrazole and 6 ml of 85% strength aqueous formic acid are heated to reflux for 12 hours and then cooled to room temperature. The suspension is stirred with 20 ml of ethanol, and the crude product is filtered off with suction. This is suspended in water and again filtered off with suction. Recrystallization from tetrahydrofuran/cyclohexane affords crystalline 1 -benzyl-4-hydroxy-3- phenylaminopyrazolo[3,4-d]pyrimidine; melting point 246-247°C, FAB-MS: (M+H)+ = 318 (C18H15N50).
Staαe 1.3: 200 mg of 1-benzyl-4-hydroxy-3-phenylaminopyrazolo[3,4-d]pyrimidine are heated to reflux with 2 ml of POCI3 under argon for 5 hours, during which the suspension slowly becomes a solution. The pale brown solution is cooled to room temperature, evaporated to dryness and stirred with ice-water. The crude product is filtered off with suction and recrystallized from ethanol/water. Fine needles of 1 -benzyl-4-chloro-3- phenylaminopyrazolo[3,4-d]pyrimidine are obtained; melting point 135°C, FAB-MS: (M+H)+ = 336 (C1ΘH14CIN5).
Staαe 1.4: 1 g of 1-benzyl-4-chloro-3-phenylaminopyrazolo[3,4-d]pyrimidine is suspended in 8 ml of ethanol, 27 ml of aniline are added, and the mixture is heated to reflux under nitrogen for 2.5 hours until a thin-layer chromatogram shows that all the starting material has disappeared. The reaction mixture is evaporated to dryness, the residue is suspended in water, and the pH is adjusted to 8.5-9 with 0.1 N NaOH. The mixture is then extracted with ethyl acetate, and the ethyl acetate phase is dried and evaporated. The crude product is chromatographed on silica gel using toluene/ethyl acetate mixtures as eluent. Product- containing fractions from the column are stirred with cyclohexane/hexane, resulting in colourless crystals of 1-benzyl-3,4-diphenylaminopyrazolo[3,4-d]pyrimidine; FAB-MS: (M+H)+ = 393 (C24H20N6).
Example 2: Elimination of the benzyl protective group from 1 -benzyl-3-(3- chlorophenylamino)-4-phenylaminopyrazolo[3,4-d]pyrimidine in AlCls/benzene in a manner analogous to that described in Example 1 results in 3-(3-chlorophenylamino)-4- phenylamino-1 H-pyrazolo[3,4-d]pyrimidine; melting point 230°C, FAB-MS: (M+H)+ = 337 (C17H13CIN6).
The starting material is obtained in the following way:
Stage 2.1 : In analogy to stage 1.1 , 3-(3-chlorophenylamino)-2-cyano-3- methylthioacrylonitrile and benzylhydrazine dihydrochloride result in 5-amino-1-benzyl-3-(3- chlorophenylamino)-4-cyanopyrazole; melting point 146-148°C; FAB-MS: (M+H)+ = 324 (C17H14CIN5).
Staαe 2.2: In analogy to stage 1.2, boiling 5-amino-1-benzyl-3-(3-chlorophenylamino)-4- cyanopyrazole with formic acid results in 1 -benzyl-3-(3-chlorophenylamino)-4- hydroxypyrazolo[3,4-d]pyrimidine; melting point 234-236°C, FAB-MS: (M+H)+ = 352 (C18H14CIN50).
Staαe 2.3: In analogy to stage 1.3, boiling 1-benzyl-3-(3-chlorophenylamino)-4- hydroxypyrazolo[3,4-d]pyrimidine in POCI3 for 7 hours and crystallizing from ethanol result in 1-benzyl-4-chloro-3-(3-chlorophenylamino)pyrazolo[3,4-d]pyrimidine; melting point 148- 150°C, FAB-MS: (M+H)+ = 370 (C18H13CI2N5).
Staαe 2.4: In analogy to stage 1.4, boiling 1 -benzyl-4-chloro-3-(3-chlorophenyl- amino)pyrazolo[3,4-d]pyrimidine and aniline in ethanol results in 1 -benzyl-3-(3- chlorophenylamino)-4-phenylaminopyrazolo[3,4-d]pyrimidine; melting point 64-65°C; FAB- MS: (M+H)+ = 427 (C24H19CIN6).
Example 3: In analogy to Example 1 , elimination of the benzyl protective group from 1-benzyl-3,4-di(3-chlorophenylamino)pyrazolo[3,4-d]pyrimidine in AlCls/benzene results in 3,4-di(3-chlorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine; melting point 175°C, FAB-MS: (M+Hf = 371 (C17H12CI2N6).
The starting material is obtained in the following way:
Staαe 3.1 : In analogy to stage 1.4, boiling 1-benzyl-4-chloro-3-(3-chlorophenylamino)- pyrazolo[3,4-d]pyrimidine (see stage 2.3) and 3-chloroaniline in ethanol results in 1-benzyl- 3,4-di(3-chlorophenylamino)pyrazolo[3,4-d]pyrimidine; melting point 131-133°C, FAB-MS: (M+H)+ = 461 (C24H18CI2N6).
Example 4: In analogy to Example 1 , elimination of the benzyl protective group from 1-benzyl-4-(3-bromophenylamino)-3-(3-chlorophenylamino)pyrazolo[3,4-d]pyrimidine in AlCla/benzene results in 4-(3-bromophenylamino)-3-(3-chlorophenyiamino)-1 H- pyrazolo[3,4-d]pyrimidine; melting point 179-181 °C, FAB-MS: (M+H)+ = 415 (C17H12BrCIN6).
The starting material is obtained in the following way:
Staαe 4.1 : In analogy to stage 1.4, boiling 1-benzyl-4-chloro-3-(3-chlorophenylamino)- pyrazolo[3,4-d]pyrimidine (see stage 2.3) and 3-bromoaniline in ethanol results in 1-benzyl- 4-(3-bromophenylamino)-3-(3-chlorophenylamino)pyrazolo[3,4-d]pyrimidine; FAB-MS: (M+H)+ = 506 (C24H18CIBrN6).
Example 5: In analogy to Example 1 , elimination of the benzyl protective group from 1-benzyl-3-(3-chlorophenylamino)-4-(3-methylphenylamino)pyrazolo[3,4-d]pyrimidine in AlCls/benzene results in 3-(3-chlorophenylamino)-4-(3-methylphenylamino)-1 H- pyrazolo[3,4-d]pyrimidine; melting point 194-195°C, FAB-MS: (M+H)+ = 351 (C18H15CIN6).
The starting material is obtained in the following way:
Staαe 5.1 : In analogy to stage 1.4, boiling 1-benzyl-4-chloro-3-(3-chlorophenylamino)- pyrazolo[3,4-d]pyrimidine (see stage 2.3) and 3-methylaniline in ethanol results in 1-benzyl- 3-(3-chlorophenylamino)-4-(3-methylphenylamino)pyrazolo[3,4-d]pyrimidine; FAB-MS: (M+H)+ = 441 (C25H2ιCIN6).
Example 6: In analogy to Example 1 , elimination of the benzyl protective group from 1-benzyl-4-(3-[2-cyanoethyl]phenylamino)-3-phenylaminopyrazolo[3,4-d]pyrimidine in AlCls/benzene results in 4-(3-[2-cyanoethyl]phenylamino)-3-phenylamino-1 H- pyrazolo[3,4-d]pyrimidine; melting point 202°C, FAB-MS: (M+H)+ = 356 (C20Hι7N7).
The starting material is obtained in the following way:
Stage 6.1 : In analogy to stage 1.4, boiling 1-benzyl-4-chloro-3-phenylaminopyrazolo- [3,4-d]pyrimidine (see stage 1.3) and 3-[2-cyanoethyl]aniline in ethanol results in 1-benzyl-4- (3-[2-cyanoethyl]phenylamino)-3-phenylaminopyrazolo[3,4-d]pyrimidine; FAB-MS: (M+H)+ = 446 (C27H23N7).
Example 7: In analogy to Example 1 , elimination of the benzyl protective group from 1-benzyl-4-(4-[2-cyanoethyl]phenylamino)-3-phenylaminopyrazolo[3,4-d]pyrimidine in AlCls/benzene results in 4-(4-[2-cyanoethyl]phenylamino)-3-phenylamino-1 H-pyrazolo[3.4- djpyrimidine; melting point 268°C; FAB-MS: (M+H)+ = 356 (C20H17N7).
The starting material is obtained in the following way:
Staαe 7.1 : In analogy to stage 1.4, boiling 1-benzyl-4-chloro-3-phenylaminopyrazolo- [3,4-d]pyrimidine (see stage 1.3) and 4-[2-cyanoethyl]aniline in ethanol results in 1-benzyl-4- (4-[2-cyanoethyl]phenylamino)-3-phenylaminopyrazolo[3,4-]pyrimidine; FAB-MS: (M+H)+ = 446 (C27H23N7).
Example 8: In analogy to Example 1 , elimination of the benzyl protective group from 1-benzyl-4-(3-cyanomethylphenylamino)-3-phenylaminopyrazolo[3,4-d]pyrimidine in AlCls benzene results in 4-(3-cyanomethylphenylamino)-3-phenylamino-1 H- pyrazolo[3,4-d]pyrimidine.
The starting material is obtained in the following way:
Staαe 8.1 : In analogy to stage 1.4, boiling 1-benzyl-4-chloro-3-phenylaminopyrazolo- [3,4-d]pyrimidine (see stage 1.3) and 3-cyanomethylaniline in ethanol results in 1-benzyl-4- (3-cyanomethylphenylamino)-3-phenylaminopyrazolo[3,4-d]pyrimidine; melting point 80°C, FAB-MS: (M+H)+ = 432 (C26H21N7).
Example 9: 1.3 g of 3-(3-chlorophenylamino)-4-(3-methylphenylamino)-1 H- pyrazolo[3,4-d]pyrimidine (see Example 5) are dissolved in 70 ml of ethanol, and 2 ml of 4 normal ethanolic hydrochloric acid solution are added at room temperature. The HCI salt starts to crystallize out after stirring for about 10 minutes. The solution is cooled to 0°C and the salt is completely crystallized by adding ethyl ether. Colourless crystals of 3-(3- chlorophenylamino)-4-(3-methylphenylamino)-1 H-pyrazolo[3,4-d]pyrimidine hydrochloride are obtained; melting point 260-263°C.
Example 10: 100 mg of 4-(3-[2-cyanoethyl]phenylamino)-3-phenylamino-1 H- pyrazolo[3,4-d]pyrimidine (see Example 6) are dissolved in 2.5 ml of absolute tetrahydrofuran and added dropwise over the course of 10 minutes to a suspension of 135 mg of aluminium trichloride in 2.5 ml of tetrahydrofuran and 46 mg of lithium aluminium hydride. The reaction is slightly exothermic. The mixture is heated to reflux for 2.5 hours until a thin-layer chromatogram shows no starting material left. The solution is cooled to 0°C, 5 ml of water are added, and the mixture is stirred at room temperature for 2 hours. It is then adjusted to pH 9 with 1 normal sodium hydroxide solution, insolubles are filtered off,
and the filtrate is evaporated. The residue is digested with tetrahydrofuran. Insolubles are again filtered off. The tetrahydrofuran filtrate is concentrated to about 3 ml, and about 15 ml of methylene chloride are added. After addition of 5 ml of hexane, crystals of 4-(3-[3- aminopropyl]phenylamino)-3-phenylamino-1 H-pyrazolo[3,4-d]pyrimidine separate out at 0°C.
Example 11 : Reduction of 4-(4-[2-cyanoethyl]phenylamino)-3-phenylamino-1 H- pyrazolo[3,4-d]pyrimidine (see Example 7) with Raney nickel in methanolic ammonia solution results in 4-(4-[3-aminopropyl]phenylamino)-3-phenylamino-1 H- pyrazolo[3,4-d]pyrimidine hydrochloride.
Example 12: Reduction of 4-(3-cyanomethylphenylamino)-3-phenylamino-1 H- pyrazolo[3,4-d]pyrimidine (see Example 8) with Raney nickel in methanolic ammonia solution results in 4-(3-[2-aminoethyl)phenylamino)-3-phenylamino-1 H- pyrazolo[3,4-d]pyrimidine.
Example 13: The following compounds of the formula I are obtained in analogy to the processes described in this text, for example in analogy to Examples 1 -3: a) 3-(4-chlorophenylamino)-4-(3-chlorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine, b) 4-(3-chlorophenylamino)-3-(3-fluorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine, c) 4-(3-chlorophenylamino)-3-(4-fluorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine, d) 4-(3-chlorophenylamino)-3-(4-methoxyphenylamino)-1 H-pyrazolo[3,4-d]pyrimidine, e) 4-(3-chlorophenylamino)-3-(4-hydroxyphenylamino)-1 H-pyrazolo[3,4-d]pyrimidine and f) 4-(3-chlorophenylamino)-3-(4-iodophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine, for example starting from the following compounds of the formula V:
aa) In analogy to stage 1.1 , 2-cyano-3-methylthio-3-(4-chlorophenyiamino)acrylonitrile and benzylhydrazine dihydrochloride result in 5-amino-1-benzyI-3-(4-chlorophenylamino)-4- cyanopyrazole; melting point 163-164 °C, FAB-MS (M+H)+: 324 (C17H14CIN5).
ba) In analogy to stage 1.1 , 2-cyano-3-methylthio-3-(3-fluorophenylamino)acrylonitrile and benzylhydrazine dihydrochloride result in 5-amino-1-benzyl-3-(3-fluorophenylamino)-4- cyanopyrazole; melting point 151 -152 °C, FAB-MS (M+H)+: 308 (C17H14FN5).
ca) In analogy to stage 1.1 , 2-cyano-3-methylthio-3-(4-fluorophenylamino)acrylonitrile and benzylhydrazine dihydrochloride result in 5-amino-1 -benzyl-3-(4-fluorophenylamino)-4- cyanopyrazole; melting point 167-168 °C, FAB-MS (M+H)+: 308 (Cι7H14FN5).
ea) In analogy to stage 1.1 , 2-cyano-3-methylthio-3-(4-methoxyphenylamino)acrylonitrile and benzylhydrazine dihydrochloride result in 5-amino-1-benzyl-3-(4- methoxyphenylamino)-4-cyanopyrazole.
fa) In analogy to stage 1.1 , 2-cyano-3-methylthio-3-(4-iodophenylamino)acrylonitrile and benzylhydrazine dihydrochloride result in 5-amino-1-benzyl-3-(4-iodophenylamino)-4- cyanopyrazole; FAB-MS (M+H)+: 416 (Cι7H14IN5).
Example 14: With exclusion of moisture, 79.2 g (295mmol) of N'-(3-benzylamino-4-cyano- 1 H-pyrazol-5-yl)-N,N-dimethylformamidine are suspended in 700 ml of methanol, 60.6 g (369 mmol) of 3-chloroaniline hydrochloride are added, and the mixture is boiled under reflux for 22 h. The resulting yellow reaction solution is cooled to = 50°C and poured into 2 litres of ice-water, 200 ml of sat. NaHC03 solution and 1 I of ethyl acetate. The aqueous phase is separated off and extracted twice with ethyl acetate. The organic phases are washed twice with water, sat. NaHC03 solution, water and brine, dried (Na2S04) and evaporated to a residual volume of = 1.5 litre. Seeding and dilution with 300 ml of diethyl ether affords crystalline 3-benzylamino-4-(3-chlorophenylamino)-1 H- pyrazolo[3,4-d]pyrimidine; melting point 214-217°C; TLC Rf = 0.29 (ethyl acetate:hexane = 1 :1).
The starting material is prepared as follows:
Staαe 14.1 : 43.6 ml (400 mmol) of benzylamine are added to a suspension of 68.4 g (400 mmol) of 3,3-bis(methylthio)-2-cyanoacryionitrile (Maybridge) in 400 ml of ethyl acetate. The clear solution is slowly heated to 70°C (→ MeSH evolution), stirred at this temperature for 1.5 h, cooled to RT and evaporated, resulting in crystalline 3-benzylamino- 3-methylthio-2-cyanoacrylonitrile; 1 H-NMR: (CD3OD) 7.36 (m, 5H), 4.77 (s, 2H), 2.59 (s, 3H).
Staαe 14.2: 24 ml (0.48 mol) of hydrazine hydrate are added dropwise to a solution of 92 g (0.4 mol) of 3-benzylamino-3-methylthio-2-cyanoacrylonitrile in 400 ml of methanol. The temperature rises to 40°C during this. It is slowly heated to boiling (→ MeSH evolution),
boiled for 2 h, cooled to RT and evaporated to a residual volume of = 200 ml. Dilution with diethyl ether, filtration and washing with diethyl ether afford
5-amino-3-benzylamino-1 H-pyrazole-4-carbonitrile [Spectrochimica Acta, 47A. 1635 (1991)]; melting point 150-152 °C; TLC: Rf = 0.41 (ethyl acetate).
Stage 14.3: A suspension of 74.3 g (348 mmol) of 5-amino-3-benzylamino-1 H-pyrazole-4- carbonitrile in 1.0 litre of toluene is boiled under reflux with 70,1 ml (95% pure; 409 mmol) of N,N-dimethylformamide diethyl acetal under an N2 atmosphere for 2 h. Cooling to RT, filtration with suction and washing with diethyl ether afford N'-(3-benzylamino-4-cyano-1 H- pyrazol-5-yl)-N,N-dimethylformamidine; melting point 197-200°C; TLC: Rf: 0.50 (ethyl acetate).
Stage 14.4: 60 g (0.47 mol) of 3-chloraniline are dissolved in 155 ml (0.56 mol) of 2.2 N methanolic HCI. Concentration and stirring the residue in diethyl ether, followed by filtration and drying, afford 3-chloraniline hydrochloride.
Example 15: 788 mg (4.8 mmol) of methyl 4-formylbenzoate and 300 mg of 5% Pt/C are added to a solution of 1.04 g (4.0 mmol) of 3-amino-4-(3-chlorophenylamino)-1 H- pyrazolo[3,4-d]pyrimidine in 40 ml of DMEU and 961 mg (16 mmol) of acetic acid, and hydrogenation is immediately carried out (reduction with NaCNBH3 is also possible in place of the hydrogenation). A further 788 mg of methyl 4-formylbenzoate are added after 2 and after 4 days. After 7 days, the catalyst is removed from the reaction mixture by filtration through Celite, and the filtrate is decolorized by treatment with active charcoal and is evaporated to a residue of = 4 g at 70°C under HV. Crystallization with diisopropyl ether/toluene affords a product of = 80% purity. Heating in a mixture of 30 ml of ethanol and = 5 ml of acetone, filtration hot, evaporation to half the volume and cooling afford 4-(3- chlorophenylamino)-3-(4-methoxycarbonylbenzylamino)-1 H-pyrazolo[3,4-d]pyrimidine; melting point 228°C; FAB-MS: (M+H)+ = 409.
The starting material is obtained in the following way:
Staαe 15.1 : Some of the solvent is distilled out of a suspension of 75.8 g (216 mmol) of 3-benzylamino-4-(3-chlorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine in 1.5 litres of benzene to remove residual water. This supension is then added, with exclusion of moisture, to 84 g of aluminium chloride (Fluka, Buchs/Switzerland) in 500 ml of benzene and heated at 80°C for 2.5 h. The reaction mixture is cooled to RT, the supernatant
benzene phase is poured into 2 kg of ice-water (a green oily residue remains behind), and the solid which separates out is filtered off with suction and thoroughly washed with water (_> K,). The benzene is evaporated off from the filtrate in a rotary evaporator, and the remaining aqueous phase is added together with 1 kg of ice to the green oily residue and hydrolysed at 40CC for 2 h. The crystalline product is filtered off with suction and washed with water (→ K2). K^ and K2 are taken up in 1 litre of methanol, acidified with 4 N aqueous HCI and partially evaporated. Water is added, and the methanol is completely evaporated off. The crystals are filtered off and washed with water. The same purification procedure is then repeated with half-saturated Na2C03 solution/methanol and water/methanol. Stirring in methanol at 50°C, precipitation with diethyl ether, filtration and drying afford 3-amino-4-(3- chlorophenyiamino)-1 H-pyrazolo[3,4-d]pyrimidine; melting point 232-234°C; TLC: Rf = 0.50 (ethyl acetate).
Example 16: 16.5 mg (0.39 mmol) of LiOH x H20 are added to a mixture of 98 mg (0.24 mmol) of 3-(4-methoxycarbonylbenzylamino)-4-(3-chlorophenylamino)- 1 H-pyrazolo[3,4-d]pyrimidine in 8 ml of methanol and 4 ml of water, and the mixture is stirred at 45°C for 3 days. The reaction mixture is evaporated, and the residue is taken up in ethanol and, after addition of active carbon, filtered to clarify and again evaporated. Precipitation from a solution in DMF using diethyl ether yields the lithium salt of 3-(4- carboxybenzylamino)-4-(3-chlorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine; HPLC: TRet(Grad.2o-100) = 9-7! FAB-MS: (M+H, acid)+ = 395, (M+Li)+ = 401.
Example 17: A solution of 261 mg (1.0 mmol) of 3-amino-4-(3-chlorophenylamino)-1H- pyrazolo[3,4-d]pyrimidine (see stage 15.1) and 245 mg (1.5 mmol) of 3-(methyiaminocarbonyl)benzaldehyde in 26 ml of methanol, 26 ml of DMEU and 180 mg (3.0 mmol) of acetic acid is stirred at RT for 1 h. Then 440 mg (7.0 mmol) of NaCNBH3 are added, and the mixture is stirred at RT for 9 days. The methanol is evaporated off from the reaction solution in an RE, and the residue is poured into 0.6 litre of water and stirred overnight. The product separates out during this. Filtration with suction, washing with water, stirring in 10 ml of boiling ethanol twice, cooling and filtration afford 4-(3- chlorophenylamino)-3-[3-(methylaminocarbonyl)benzylamino]-1 H-pyrazolo[3,4-d]pyrimidine; melting point 252-254°C; HPLC: TRet(Grad5.4o) = 16-9; FAB-MS: (M+H)+ = 408.
The starting material is prepared as follows:
Staαe 17.1 : A solution of 3 g of methyl 3-formylbenzoate and 20 ml of methylamine (8.03 M in ethanol) is stirred at RT for 3 days. Evaporation and crystallization from DIPE result in N-methyl-3-methyliminomethylbenzamide; melting point 87°C.
Staαe 17.2: A 2-phase mixture of 2.91 g of N-methyl-3-methyliminomethylbenzamide, 50 ml of methylene chloride and 30 ml of 1 N HCI is stirred at RT for 1.5 h. Removal of the organic phase, washing with 1 N HCI and brine, drying with Na2S04, evaporation and stirring with DIPE/hexane afford 3-(methylaminocarbonyl)benzaldehyde; melting point 101- 102°C.
Example 18: 4-(3-Chlorophenvlamino)-3-f4-(methvlaminocarbonyl)benzvlamino)-1 H- pyrazoio[3,4-d]pyrimidine is prepared in analogy to Example 17 from 261 mg (1.0 mmol) of 3-amino-4-(3-chiorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine (see stage 15.1) and 245 mg (1.5 mmol) of 4-methylaminocarbonyl)benzaldehyde; HPLC: TRet(Grad5_4o) = 16.3; 1 H-NMR: (DMSO-d6) 12.38 (s, HN), 8.93 (s, HN), 8.39 (m, 1H), 8.27 (s, 1 H), 7.94 (t, J=2, 1H), 7.81 (d, J=8, 2H), 7.68 (db, J=8, 1 H), 7.52 (d, J=8, 2H), 7.41 (t, J=8, 1 H), 7.17 (db, J=8, 1 H), 6.99 (tb, J=5, HN), 4.57 (d, J=5, 2H), 2.79 (d, J=4, 3H); FAB-MS: (M+H)+ = 408.
The starting material is prepared in analogy to stages 17.1 and 17.2:
Staαe 18.1 : N-Methyl-4-methyliminomethylbenzamide is obtained from 3 g of methyl 4-formylbenzoate and 20 ml of methylamine (8.03 M in ethanol); melting point 140-141 °C.
Staαe 18.2: 705 mg of N-methyl-4-methyliminomethylbenzamide are hydrolysed to 3-(methylaminocarbonyl)benzaldehyde; 1 H-NMR: (CDCI3) 10.06 (s, 1 H), 7.93 (s, 4H), 6.4 (sb, HN), 3.04 (d, J=5, 3H).
Example 19: 99.7 mg (0.60 mmol) of 3,5-dimethoxybenzaldehyde are added to a solution of 130 mg (0.50 mmol) of 3-amino-4-(3-chlorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine in 26 ml of methanol and 120 mg (2.0 mmol) of acetic acid. On stirring, a solid separates out and can be redissolved by adding 52 ml of DMEU. 220 mg (3.5 mmol) of NaCNBH3 are added, and the mixture is stirred at RT for 5 h. Since not all the 3-amino-4-(3- chlorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine has been converted (HPLC), two further 220 mg portions of NaCNBH3 are added, followed by stirring for 5 h each time. The reaction solution is then poured into 1 litre of water, stirred vigorously for 1 h and filtered.
Crystallization of the filtered residue from acetone affords 4-(3-chlorophenylamino)-3-(3,5- dimethoxybenzylamino)-1 H-pyrazolo[3,4-d]pyrimidine; melting point 208-209°C; HPLC: TRet(Grad2o-lθθ) = 11 -7; FAB-MS: (M+H)+ = 411.
Example 20: 228 mg (1.50 mmol) of 4-hydroxy-3-methoxybenzaldehyde are added (N2 atmosphere) to a solution of 261 mg (1.00 mmol) of 3-amino-4-(3-chlorophenylamino)-1 H- pyrazolo[3,4-d]pyrimidine in 26 ml of methanol, 26 ml of DMEU and 120 mg (2.0 mmol) of acetic acid. After 1 h, 440 mg (7.0 mmol) of NaCNBH3 are added to the clear solution, and the mixture is stirred at RT for 7 days. The reaction solution is poured into 0.8 I of water and stirred overnight, during which the product separates out. Filtration with suction, washing with water, stirring in hot ethyl acetate, cooling and filtration afford 4-(3-chlorophenylamino)- 3-(3-methoxy-4-hydroxybenzylamino)-1 H-pyrazolo[3,4-d]pyrimidine; melting point 223- 225°C; HPLC: TRet(Grad5_4o) = 19.8; FAB-MS: (M+H)+ = 397.
Example 21 : 225 mg (1.50 mmol) of 3-formylbenzoic acid are added under an N2 atmosphere to a solution of 261 mg (1.00 mmol) of 3-amino-4-(3-chlorophenylamino)-1H- pyrazolo[3,4-d]pyrimidine in 26 ml of methanol, 13 ml of DMEU and 120 mg (2.0 mmol) of acetic acid. The mixture is stirred for 1 h, during which a solid separates out, and then 440 mg (7.0 mmol) of NaCNBH3 are added. The suspension is stirred for 5 days, during which it is converted into a clear solution. The methanol is evaporated off in an RE. The residue is poured into 0.6 litre of water and stirred for 3 h. Filtration with suction, washing with water and diethyl ether, stirring in hot ethanol, cooling and filtration afford 3-(4- carboxybenzylamino)-4-(3-chlorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine (3-{[4-(3- chlorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidin-3-ylamino]methyl}benzoic acid); melting point 294-296°C; HPLC: TRet(Grad5.40) = 20.1 ; FAB-MS: (M+H)+ = 395.
Example 22: In analogy to Example 21 , firstly 1.00 mmol of 3-amino-4-(3- chlorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine in 26 ml of methanol, 13 ml of DMEU and 3.0 mmol of acetic acid is reacted with methyl 3-formylbenzoate, and then reduction is carried out with 7.00 mmol of NaCNBH3 (5-7 days). 4-(3-Chlorophenylamino)-3-(3- methoxycarbonylbenzyiamino)-1 H-pyrazolo[3,4-d]pyrimidine is obtained.
Example 23: In analogy to Example 21 , firstly 1.00 mmol of 3-amino-4-(3- chlorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine in 26 ml of methanol, 13 ml of DMEU and 3.0 mmol of acetic acid is reacted with 3,4,5-trimethoxybenzaldehyde, and then reduction is carried out with 7.00 mmol of NaCNBH3 (5-7 days). 4-(3-Chlorophenylamino)-3-(3,4,5-
trimethoxybenzylamino)-1 H-pyrazolo[3,4-d]pyrimidine is obtained; HPLC: T et(Grad5_4o) = 19.1.
Example 24: In analogy to Example 21 , firstly 1.00 mmol of 3-amino-4-(3- chiorophenyiamino)-1 H-pyrazolo[3,4-d]pyrimidine in 26 ml of methanol, 13 ml of DMEU and 3.0 mmol of acetic acid is reacted with 3,4-dimethoxybenzaldehyde, and then reduction is carried out with 7.00 mmol of NaCNBH3 (5-7 days). 4-(3-Chlorophenylamino)-3-(3,4- dimethoxybenzyiamino)-1 H-pyrazolo[3,4-d]pyrimidine is obtained; melting point 228-232°C; HPLC: TRet(Grad5.4o) = 19.0.
Example 25: In analogy to Example 21 , firstly 1.00 mmol of 3-amino-4-(3- chlorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine in 26 ml of methanol, 13 ml of DMEU and 3.0 mmol of acetic acid is reacted with 2,3,4-trimethoxybenzaldehyde, and then reduction is carried out with 7.00 mmol of NaCNBH3 (5-7 days). 4-(3-Chlorophenylamino)-3-(2,3,4- trimethoxybenzylamino)-1 H-pyrazolo[3,4-d]pyrimidine is obtained; melting point 168-169CC; HPLC: TRet(Grad5_4o) = 20.2.
Example 26: In analogy to Example 21 , firstly 1.00 mmol of 3-amino-4-(3- chlorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine in 26 ml of methanol, 13 ml of DMEU and 3.0 mmol of acetic acid is reacted with 3-hydroxy-4-methoxybenzaldehyde, and then reduction is carried out with 7.00 mmol of NaCNBH3 (5-7 days). 4-(3-Chlorophenylamino)-3- (3-hydroxy-4-methoxybenzylamino)-1 H-pyrazolo[3,4-d]pyrimidine is obtained; melting point 225-227°C; HPLC: TRet(Grad5.40) = 17.6.
Example 27: In analogy to Example 21 , firstly 1.00 mmol of 3-amino-4-(3- chlorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine in 26 ml of methanol, 13 ml of DMEU and 3.0 mmol of acetic acid is reacted with 4-hydroxy-3,5-dimethoxybenzaldehyde, and then reduction is carried out with 7.00 mmol of NaCNBH3 (5-7 days). 4-(3-Chlorophenylamino)-3- (4-hydroxy-3,5-dimethoxybenzylamino)-1 H-pyrazolo[3,4-d]pyrimidine is obtained; HPLC: TRet(Grad5_4o) = 17.0.
Example 28: In analogy to Example 21 , firstly 1.00 mmol of 3-amino-4-(3- chlorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine in 26 ml of methanol, 13 ml of DMEU and 3.0 mmol of acetic acid is reacted with 3,4-methylenedioxybenzaldehyde, and then reduction is carried out with 7.00 mmol of NaCNBH3 (5-7 days). 4-(3-Chlorophenylamino)-3-
(3,4-methylenedioxybenzyiamino)-1 H-pyrazolo[3,4-d]pyrimidine is obtained; melting point 220-222°C; HPLC: TRet(Grad5.40) = 22.5°C.
Example 29: In analogy to Example 21 , firstly 1.00 mmol of 3-amino-4-(3- chlorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine in 26 ml of methanol, 13 ml of DMEU and 3.0 mmol of acetic acid is reacted with 2,3-methylenedioxybenzaldehyde, and then reduction is carried out with 7.00 mmol of NaCNBH3 (5-7 days). 4-(3-Chlorophenylamino)-3- (2,3-methylenedioxybenzylamino)-1 H-pyrazolo[3,4-d]pyrimidine is obtained; melting point 214-215°C; HPLC: TRet(Grad5.40) = 24.1.
Example 30: In analogy to Example 21 , firstly 1.00 mmol of 3-amino-4-(3- chlorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine in 26 ml of methanol, 13 ml of DMEU and 3.0 mmol of acetic acid is reacted with 3-chlorobenzaldehyde, and then reduction is carried out with 7.00 mmol of NaCNBH3 (5-7 days).
3-(3-chlorobenzylamino)-4-(3-chlorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine is obtained; melting point 158-163°C; HPLC: T et(Grad o-i oo) = 12-4-
Example 31 : In analogy to Example 21 , firstly 1.00 mmol of 3-amino-4-(3- chlorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine in 26 ml of methanol, 13 ml of DMEU and 3.0 mmol of acetic acid is reacted with 3-chloro-4-hydroxybenzaldehyde, and then reduction is carried out with 7.00 mmol of NaCNBH3 (5-7 days). 3-(3-chloro-4-hydroxybenzylamino)-4- (3-chlorophenylamino)-1H-pyrazolo[3,4-d]pyrimidine is obtained; melting point 220°C; HPLC: TRet(Grad5_4o) = 18.7.
Example 32: In analogy to Example 21 , firstly 1.00 mmol of 3-amino-4-(3- chlorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine in 26 ml of methanol, 13 ml of DMEU and 3.0 mmol of acetic acid is reacted with 3-chloro-4-methoxybenzaldehyde, and then reduction is carried out with 7.00 mmol of NaCNBH3 (5-7 days). 3-(3-chloro-4- methoxybenzylamino)-4-(3-chlorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine is obtained; melting point 205-208°C; HPLC: T et(Grad2o-lθθ) = 12-2-
Example 33: Processes analogous to those described in this text result in a) 4-(3-chlorophenylamino)-3-[1 -(3-chlorophenyl)ethylamino]-1 H-pyrazolo[3,4-d]pyrimidine, b) 4-(3-chlorophenylamino)-3-(1-phenylethylamino)-1 H-pyrazolo[3,4-d]pyrimidine and c) 4-(3-chlorophenylamino)-3-[3-(dimethylaminocarbonyl)benzyiamino]-1H- pyrazolo[3,4-d]pyrimidine.
Example 34: 1.31 g (7.98 mmol) of 3-chloroaniline hydrochloride (see stage 14.4) are added to a suspension of 2.05 g (7.6 mmol) of N'-[3-(4-methoxyphenyl)-4-cyano-1 H-pyrazol-5-yl]- N,N-dimethylformamidine in 24 ml of methanol, and the mixture is boiled under reflux. After 13 h, a further 561 mg (3.42 mmol) of 3-chloroaniline hydrochloride are added. After boiling for a total of 18 h, the suspension is cooled. 4-(3-Chlorophenylamino)-3-(4-methoxyphenyl)- 1 H-pyrazolo[3,4-d]pyrimidine is filtered off, washed with hexane and dried: melting point 268-269°C; HPLC: TRet(Grad20-ioo) = 13-8! FAB-MS: (M+H)+ = 352.
The starting material is prepared as follows:
Staαe 34.1 : Under an N2 atmosphere, a suspension of 1.87 g (8.16 mmol) of 5-amino-3-(4- methoxyphenyl)-1 H-pyrazole-4-carbonitrile (for preparation, see: J. Heterocyclic Chem. 2∑, 647 (1990)) in 33 ml of toluene is boiled under reflux with 1.64 ml (95% pure; 9.1 mmol) of N,N-dimethylformamide diethyl acetal for 3.5 h. Crystallization by cooling to 0°C, filtration with suction and washing with hexane afford N'-[3-(4-methoxyphenyl)-4-cyano-1 H-pyrazol- 5-yl]-N,N-dimethylformamidine; melting point 169-171 °C; TLC: R, = 0.18 (ethyl acetate-.hexane = 1:1); MS: (M)+ = 269.
Example 35: With exclusion of moisture, 62 mg (0.38 mmol) of 3-chloroaniline hydrochloride (see stage 14.4) are added to 92 mg (0.36 mmol) of N'-[3-(4-aminophenyl)-4-cyano-1 H- pyrazol-5-yl]-N,N-dimethylformamidine in 1 ml of methanol, and the mixture is boiled under reflux for 28 h. The pale yellow reaction solution is cooled, evaporated and chromatographed (Si02, methylene chloride-ethanol [10:1]). Crystallization from isopropanol affords 3-(4-aminophenyl)-4-(3-chlorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine; TLC: Rf = 0.33 (methylene chloride:methanol = 10:1); FAB-MS: (M)+ = 336; HPLC: TRet(Grad20-i oo) = 9.2, TRet(Grad5_4o) = 18.8.
3-(4-Aminophenyl)-4-(3-chlorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine can also be prepared, as alternative to the above process, from 3-(4-fe/?-butoxycarbonylaminophenyl)-4- (3-chlorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine (Example 36) by stirring in 4 N HCI solution in dioxane for 3 hours and filtering off the product as hydrochloride.
The starting material is prepared as follows:
Staαe 35.1 : Under an N2 atmosphere, 0.92 g (6.38 mmol) of tetracyanoethylene oxide (Fluka, Buchs/Switzerland) are added to 618.5 g (2.9 mmol) of methyl 4-nitrodithiobenzoate (for preparation, see J. prakt. Chem. 331, 243 (1989)) in 5 ml of toluene and then heated to boiling for 4 h. The reaction mixture is cooled, 10 g of silica gel are added, and the mixture is evaporated in an RE. Loading of the residue onto a silica gel column and elution with hexane/ethyl acetate (2:1) result in 3-(4-nitrophenyl)-3-methylthio-2-cyanoacrylonitrile; TLC: R, = 0.46 (ethyl acetate:hexane = 1 :2); MS: (M)+ = 245.
Staαe 35.2: 0.05 ml (1.00 mmol) of hydrazine hydrate is added dropwise to 245.3 mg (1.00 mmol) of 3-(4-nitrophenyl)-3-methylthio-2-cyanoacrylonitrile in 1.3 ml of methanol, and the mixture is heated to boiling for 8 h. The reaction mixture is evaporated, and the residue is stirred with ethyl acetate and filtered off. 5-Amino-3-(4-nitrophenyl)-1 H-pyrazole-4- carbonitrile is obtained: TLC: Rf = 0.14 (ethyl acetate: hexane = 1 :1 ).
Staαe 35.3: 57.3 mg (0.25 mmol) of 5-amino-3-(4-nitrophenyl)-1 H-pyrazole-4-carbonitrile and 79.3 μl of N,N-dimethylformamide dibenzyl acetal are heated to boiling in 1 ml of toluene overnight. Filtration of the suspension and washing with hexane afford N'-[3-(4- nitrophenyl)-4-cyano-1 H-pyrazol-5-yl]-N,N-dimethylformamidine; TLC: Rf = 0.15 (ethyl acetate:hexane = 2:1); TRet(Grad u-ioo) = 8-8-
Staαe 35.4: 142 mg (0.50 mmol) of N'-[3-(4-nitrophenyl)-4-cyano-1 H-pyrazol-5-yl]-N,N- dimethylformamidine are hydrogenated in the presence of 30 mg of Pd/C (5%) in 20 ml of THF. Filtration, evaporation and crystallization from ethyl acetate/diethyl ether/hexane afford N'-[3-(4-aminophenyl)-4-cyano-1 H-pyrazol-5-yl]-N,N-dimethylformamidine; TLC: Rf = 0.07 (ethyl acetate:hexane = 3:1); FAB-MS: (M+H)+ = 255.
Example 36: With exclusion of air, 172 mg (1.05 mmol) of 3-chloroaniiine hydrochloride (see stage 14.4) are added to 248 mg (0.70 mmol) of N'-[3-(4-ferf-butoxycarbonylaminophenyl)- 4-cyano-1 H-pyrazol-5-yl]-N,N-dimethylformamidine in 2 ml of methanol, and the mixture is heated to boiling for 19 h. The reaction mixture is evaporated and chromatographed (Si02) methylene chlohde/ethanol [20:1]). Stirring with diethyl ether/hexane affords 3-(4-fert- butoxycarbonylaminophenyl)-4-(3-chlorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine; melting point 165-168 °C (decomposition); FAB-MS: (M)+ = 436; HPLC: TRet(Grad20.100) = 15.4.
The starting material is prepared as follows:
Staαe 36.1 : 254 mg (1.0 mmol) of N'-[3-(4-aminophenyl)-4-cyano-1 H-pyrazol-5-yl]-N,N- dimethylformamidine (stage 35.4) are heated in 4 ml of dioxane and 436.5 mg (2.0 mmol) of di-tert-butyl dicarbonate at 80°C under an argon atmosphere for 7 h. Cooling, evaporation to a remaining volume of = 1 ml, addition of = 2 ml of hexane and filtration yield N'-[3-(4-fe/t- butoxycarbonylaminophenyl)-4-cyano-1 H-pyrazol-5-yl]-N,N-dimethylformamidine; melting point 230-233°C (decomposition); TLC: Rf = 0.47 (methylene chloride:methanol = 10:1 ); MS: (M)+ = 354.
Example 37: 172.2 mg (1.05 mmol) of 3-chloroaniline hydrochloride (see stage 14.4) are added with exclusion of air to 199 mg (0.70 mmol) of N'-[3-(3-nitrophenyl)-4-cyano-1 H- pyrazol-5-yl]-N,N-dimethylformamidine in 2 ml of methanol, and the mixture is boiled under reflux for 19 h. Cooling, filtration and washing with isopropanol and hexane afford 4-(3- chlorophenylamino)-3-(3-nitrophenyl)-1 H-pyrazolo[3,4-d]pyrimidine; TLC: Rf = 0.55 (methylene chloride:methanol = 10:1 ); HPLC: T et(Grad20-i oo) = 14-2: 1 H-NMR: (DMSO- d6) 9.05 (s, HN), 8.54 (s, 2H), 8.32 (dd, J=8, 2, 1 H), 8.22 (d, J=8, 1 H), 7.8 (m, 2H), 7.49 (d, J=8, 1 H), 7.33 (t, J=8, 1 H), 7.10 (dd, J=8, 2, 1H).
The starting material is prepared as follows:
Staαe 37.1 : 13.47 g (0.42 mmol) of sulfur and 60.72 g of triethylamine are mixed in 65 ml of DMF under an N2 atmosphere. At 0-5°C, a solution of 30 g (175 mmol) of 3-nitrobenzyl chloride in 35 ml of DMF is added dropwise, and the mixture is stirred at 0-5°C for 1.5 h, at RT for 3 h and finally at 40°C (→ exothermic) for 4 h. The orange reaction mixture is then cooled to 0°C, and 13 ml (208 mmol) of methyl iodide are added. The red suspension is stirred at 0-5°C overnight, poured into ice-water and stirred for 1 h, ethyl acetate is added, and the sulfur is filtered off. The aqueous phase is removed and extracted once with ethyl acetate; the organic phases are washed 3 times with water and brine, dried with Na2S0 and evaporated. Column chromatography (Si02, ethyl acetate/hexane [1 :4]) and crystallization from a solution in diethyl ether by adding hexane and cooling to -70°C result in methyl 3-nitrodithiobenzoate; melting point 38°C.
Staαe 37.2: 550 mg (3.8 mmol) of tetracyanoethylene oxide (Fluka, Buchs/Switzerland) are added under an N2 atmosphere to 500 mg (2.34 mmol) of methyl 3-nitrodithiobenzoate in 4 ml of toluene, and the mixture is then heated at 50°C for 10 h. 5 g of silica gel are added to the reaction mixture, which is evaporated. Loading of the residue onto a silica gel
column, elution with hexane/ethyl acetate [1 :2], treatment with active carbon, evaporation and crystallization from diethyl ether (-70°C) afford
3-(3-nitrophenyl)-3-methylthio-2-cyanoacrylonitrile; MS: (M)+ = 245, (M-SMe)+ = 198, (M- N02-Me)+ = 184; IR: (KBr) 2222s, 1531 s, 1514s, 1353s.
Staαe 37.3: 0.24 ml (4.8 mmol) of hydrazine hydrate is added dropwise to 1.00 g (4.08 mmol) of 3-(3-nitrophenyl)-3-methylthio-2-cyanoacrylonitrile in 5.3 ml of methanol and the mixture is heated to boiling for 1.5 h. The reaction mixture is cooled in ice-water, and the precipitate is filtered off and washed with diethyl ether/isopropanol (2:1 ). 5-Amino-3-(3- nitrophenyl)-1 H-pyrazole-4-carbonitrile is obtained; TLC: Rf = 0.4 (methylene chloride:methanol = 10:1); TRet(Grad20-ioo) =10-8: S: (M)+ = 229- (M-N02)+ = 183.
Staαe 37.4: 229 mg (1.00 mmol) of 5-amino-3-(3-nitrophenyl)-1 H-pyrazole-4-carbonitrile and 317 μl of N,N-dimethylformamide dibenzyl acetal are heated to boiling in 8 ml of toluene overnight. Cooling, filtration of the suspension and washing with hexane afford N'-[3-(3- nitrophenyl)-4-cyano-1 H-pyrazol-5-yl]-N,N-dimethylformamidine; melting point 221-225°C; τRet(Grad20-10θ) = 8-7-
Example 38: 110 mg (0.30 mmol) of 3-(3-nitrophenyl)-4-(3-chlorophenylamino)-1 H- pyrazolo[3,4-d]pyrimidine are hydrogenated in the presence of 30 mg of Raney nickel in 3 ml of methanol and 3 ml of THF. After the catalyst has been filtered off, 15 g of silica gel are added to the filtrate, which is evaporated. Loading of the powder onto a silica gel column and elution with methylene chloride/ethanol (15:1 ) and then stirring of the crude product with diethyl ether/hexane afford 3-(3-aminophenyl)-4-(3-chlorophenylamino)-1 H- pyrazolo[3,4-d]pyrimidine); melting point: 264-266 °C; TLC: Rf = 0.45 (methylene chloride/methanol 10:1 ); HPLC: TRet(Grad2n-ioo) = 9-7-
Example 39: The following compounds are obtained by processes analogous to those described in this text: a) 4-(3-chlorophenylamino)-3-(4-[3-methylbutanoylamino]phenyl)-1 H-pyrazolo[3,4-d]- pyrimidine, b) 4-(3-chlorophenylamino)-3-(3-[3-methylbutanoylamino]phenyl)-1 H-pyrazolo[3,4-d]- pyrimidine, c) 4-(3-chlorophenylamino)-3-(4-propanoylaminophenyl)-1 H-pyrazolo[3,4-d]pyrimidine, d) 4-(3-chlorophenylamino)-3-(3-propanoylaminophenyl)-1 H-pyrazolo[3,4-d]pyrimidine, e) 4-(3-chlorophenylamino)-3-(4-pivaloylaminophenyl)-1 H-pyrazolo[3,4-d]pyrimidine,
f) 4-(3-chlorophenylamino)-3-(3-pivaloylaminophenyl)-1 H-pyrazolo[3,4-d]pyrimidine, g) 3-(4-acetylaminophenyl)-4-(3-chlorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine, h) 3-(3-acetylaminophenyl)-4-(3-chlorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine, i) 4-(3-chlorophenylamino)-3-(4-[{N,N-dimethylamino}methyleneamino]phenyl)-1 H- pyrazolo[3,4-d]pyrimidine, j) 4-(3-chlorophenylamino)-3-(3-[{N,N-dimethylamino}methyleneamino]phenyl)-1 H- pyrazolo[3,4-d]pyrimidine, k) 4-(3-chlorophenylamino)-3-(4-[2-thienylcarbonylamino]phenyl)-1H-pyrazolo[3,4-d]- pyrimidine,
I) 4-(3-chlorophenylamino)-3-(3-[2-thienylcarbonylamino]phenyl)-1 H-pyrazolo[3,4-d]- pyrimidine, m) 4-(3-chlorophenylamino)-3-(4-[2-furylcarbonylamino]phenyl)-1 H-pyrazolo[3,4-d]- pyrimidine, n) 4-(3-chlorophenylamino)-3-(3-[2-furylcarbonylamino]phenyl)-1 H-pyrazolo[3,4- djpyrimidine, o) 4-(3-chlorophenylamino)-3-(4-[2-pyridylcarbonylamino]phenyl)-1 H-pyrazolo[3,4-d]- pyrimidine, p) 4-(3-chlorophenyiamino)-3-(3-[2-pyridylcarbonylamino]phenyl)-1 H-pyrazolo[3,4-d]- pyrimidine, q) 4-(3-chlorophenylamino)-3-(4-methylsulfonylaminophenyl)-1 H-pyrazolo[3,4-d]pyrimidine, r) 4-(3-chlorophenylamino)-3-(3-methylsulfonylaminophenyl)-1 H-pyrazolo[3,4-d]pyrimidine, s) 4-(3-chlorophenylamino)-3-(4-[4-methylbenzenesulfonylamino]phenyl)-1 H- pyrazolo[3,4-d]pyrimidine, t) 4-(3-chlorophenylamino)-3-(3-[4-methylbenzenesulfonylamino]phenyl)-1 H-pyrazolo[3,4-d]- pyrimidine, u) 3-(4-tert-butoxycarbonylaminophenyl)-4-(3-chlorophenylamino)-1 H-pyrazolo[3,4-d]- pyrimidine, v) 3-(3-tert-butoxycarbonylaminophenyl)-4-(3-chlorophenylamino)-1 H-pyrazolo[3,4-d]- pyrimidine, w) 3-(4-benzyloxycarbonylaminophenyl)-4-(3-chlorophenylamino)-1 H-pyrazolo[3,4-d]- pyrimidine and x) 3-(3-benzyloxycarbonylaminophenyl)-4-(3-chlorophenylamino)-1 H-pyrazolo[3,4-d]- pyrimidin.
Example 40: 4-Chlorophenylarnino)-3-(4-methoxyphenylamino)-1 H- pyrazolof3.4-d1pyrimidine
A suspension of 1 g (3.52 mmol) of 4-cyano-5-(dimethylaminomethyleneamino)-3-(4- methoxyphenylamino)pyrazole and 0.9 g (5.48 mmol) of 3-chloroaniline hydrochloride [for preparation, see: Justus Liebigs Ann. Chem. 176, 45 (1875)] in 7 ml of methanol is heated under reflux for 17 h. It is then cooled to room temperature, the reaction mixture is made alkaline (pH 10) by adding 1 N sodium hydroxide solution and is filtered, and the residue on the filter is washed with a methanol/water mixture (1:1). Recrystallization of the crude product from methanol/water results in the title compound with a water content of 1.69% H20; melting point 223-224°C (decomposition).
The starting material is prepared as follows:
Staαe 40.1 : 4-Cvano-5-(dimethylaminomethyleneamino)-3-(4- methoxyphenylamino)pyrazole
A suspension of 5.41 g (23.6 mmol) of 5-amino-4-cyano-3-(4-methoxyphenylamino)pyrazole
[for preparation see: J. Heterocyclic Chem. 27, 775 (1990)] in 4.8 ml (27.2 mmol) of N,N- dimethylformamide diethyl acetal (97%) and 100 ml of toluene is heated under reflux for
5 h. It is then cooled to room temperature and filtered, and the residue on the filter is washed with toluene. Recrystallization of the crude product from methanol/water results in the title compound; melting point 232-234°C (decomposition).
Example 41 : 3-(4-Acetylaminophenylamino)-4-(3-chlorophenylamino)-1 H-pyrazolor3.4-d1- pyrimidine
A suspension of 6.32 g (20.3 mmol) of 3-(4-acetylaminophenylamino)-4-cyano-5- (dimethylaminomethyleneamino)pyrazole and 4 g (24.4 mmol) of 3-chioroaniline hydrochloride in 50 ml of methanol is heated under reflux for 96 h and then cooled to room temperature. The mixture is filtered, and the residue on the filter is digested with 50 ml of 1 N sodium hydroxide solution for 1/2 h. Filtration and washing with water result in the title compound; melting point 290-291 °C (decomposition).
The starting material is prepared as follows:
Staαe 41.1 : 3-(4-Acetylaminophenylamino)-2-cvano-3-methylthioacrylonitrile A mixture of 5 g (29.4 mmol) of 3,3-bismethylthio-2-cyanoacrylonitrile [for preparation, see: Qhem. Ber. 95, 2861 (1962)], 4.41 g (29.4 mmol) of 4-aminoacetanilide (Fluka) and 30 ml of toluene is heated under reflux for 20 h. Cooling to room temperature, filtration and washing
of the residue on the filter with toluene results in the title compound; melting point 258- 259°C (decomposition).
Staαe 41.2: 3-(4-Acetylaminophenylamino)-5-amino-4-cvanopyrazole A mixture of 7.46 g (27.4 mmol) of 3-(4-acety!aminophenylamino)-2-cyano-3- methylthioacrylonitrile, 1.63 ml (32.9 mmol) of hydrazine hydrate and 40 ml of methanol is heated under reflux for 5 h and then evaporated in vacuo. Recrystallization of the residue from methanol results in the title compound; melting point 245-246cC (decomposition).
Staαe 41.3: 3-(4-Acetylaminophenylamino)-4-cvano-5-(dimethylaminomethyleneamino pyrazole
A suspension of 5.3 g (20.7 mmol) of 3-(4-acetylaminophenylamino)-5-amino-4- cyanopyrazole in 4.4 ml (24.9 mmol) of N,N-dimethylformamide diethyl acetal (97%) and
100 ml of toluene is heated under reflux for 4 h. It is then cooled to room temperature and filtered, and the residue on the filter is washed with toluene, resulting in the title compound with a water content of 4.22% H20; melting point 275-276°C (decomposition).
Example 42: 4-(3-Chlorophenylamino)-3-(4-hvdroxyphenylamino)-1 H-pyrazolof3.4-d]- pyrimidine
A solution of 5 ml (51.9 mmol) of boron tribromide in 20 ml of methylene chloride is added dropwise over the course of 30 min to a suspension of 5 g (13.63 mmol) of 4-(3- chlorophenylamino)-3-(4-methoxyphenylamino)-1 H-pyrazolo[3,4-d]pyrimidine in 100 ml of methylene chloride which is cooled to 0°C under a nitrogen atmosphere. The reaction mixture is stirred at 20°C for 14 h and then filtered, and the material on the filter is digested with 85 ml of water at RT for 1/2 h. The mixture is filtered, and the residue on the filter is partitioned between 50 ml of sat. sodium bicarbonate solution, 50 ml of water and 135 ml of THF. Removal of the organic phase, extraction of the aqueous phase once more with 35 ml of THF, washing of the combined THF extracts with brine and drying of the organic phase over sodium sulfate are followed by evaporation in vacuo. The crystalline residue is digested with 65 ml of boiling ethyl acetate, cooled to RT and filtered, resulting in the crude title compound. Recrystallization of a sample from ethyl acetate affords the pure title compound; melting point > 260°C; TLC Rf = 0.54 (toluene/isopropanol/conc. ammonia [70:29:1]).
Example 43: 4-(3-Chlorophenylamino)-3-(4-dimethylaminophenylamino)-1 H-pyrazolof3.4-d]- pyrimidine
A mixture of 5 g (16.8 mmol) of 4-cyano-5-(dimethylaminomethyleneamino)-3-(4- dimethylaminophenylamino)pyrazole, 3.3 g (20.1 mmol) of 3-chloroaniline hydrochloride and 40 ml of DMF is heated at 130°C with stirring for 8 h. It is then cooled to RT, water is added dropwise to the reaction mixture and, after filtration, the residue on the filter is dissolved in about 20 ml of DMF. After precipitation with water and subsequent recrystallization from DMF/water, the resulting crystals are suspended in 10 ml of water. 6 ml of 1 N hydrochloric acid are added, the mixture is briefly heated to reflux and filtered, and 3 ml of 2 N sodium hydroxide solution are added to the filtrate. The crystalline precipitate is filtered off, washed with water and dried under high vacuum at 120CC, resulting in the title compound with a water content of 2.32%; melting point 250-255°C (decomposition).
The starting material is prepared as follows:
Staαe 43.1 : 4-Cvano-5-(dimethylaminomethyleneamino)-3-(4-dimethylaminophenylamino)- pyrazole
A suspension of 10 g (41.3 mmol) of 5-amino-4-cyano-3-(4-dimethylaminophenylamino)- pyrazole [for preparation, see: Arch. Pharm. (Weinheim) 326, 245 (1993)] in 8.75 ml
(49.5 mmol) of N,N-dimethylformamide diethyl acetal (97%) and 200 ml of toluene is heated under reflux for 1 h. It is then cooled to about 30°C and filtered, and the residue on the filter is washed with toluene, resulting in the title compound; melting point 281-282°C
(decomposition).
Example 44: 4-(3-Chlorophenylamino)-3-(4-methoxybenzylamino)-1 H-pyrazolor3.4-d1- pyrimidine
A mixture of 4.475 g (15 mmol) of 4-cyano-5-(dimethylaminomethyleneamino)-3-(4- methoxybenzylamino)pyrazole, 2.83 g (17.25 mmol) of 3-chloroaniline hydrochloride and
80 ml of ethanol is heated under reflux for 16 h. Cooling to about 5°C, filtration and washing of the residue on the filter with diethyl ether yield the title compound; melting point 222-
223°C.
The starting material is prepared as follows:
Stage 44.1 : 2-Cvano-3-(4-methoxybenzylamino)-3-methylthioacrylonitrile A mixture of 17.03 g (0.1 mol) of 3,3-bismethylthio-2-cyanoacrylonitrile, 12.98 ml (0.1 mol) of 4-methoxybenzylamine and 60 ml of ethanol is heated under reflux for 2 h. About 90 ml of hexane are then added dropwise to the hot solution, and it is allowed to cool to RT. The title compound separates out in the form of colourless crystals and is filtered off, washed with diethyl ether and dried under HV; melting point 100-101 °C.
Stage 44.2: 5-Amino-4-cvano-3-(4-methoxybenzylamino)pyrazole A mixture of 15 g (57.8 mmol) of 2-cyano-3-(4-methoxybenzylamino)-3- methylthioacrylonitrile, 3.03 ml (61.1 mmol) of hydrazine hydrate and 40 ml of methanol is stirred at RT for 1 h, heated under reflux for 2 h and then evaporated in vacuo. Recrystallization of the residue from ethanol/hexane results in the title compound; melting point 148-149°C.
Stage 44.3: 4-Cvano-5-(dimethylaminomethyleneamino)-3-(4-methoxybenzylamino)pyrazole A mixture of 6.08 g (25 mmol) of 5-amino-4-cyano-3-(4-methoxybenzylamino)pyrazole, 5.14 ml (30 mmol) of N,N-dimethylformamide diethyl acetal and 90 ml of toluene is heated under reflux for 3 h. It is then cooled to about 5°C and filtered, and the residue on the filter is washed with toluene, resulting in the title compound; melting point 147-148°C.
Example 45: 4-(3-Chlorophenylamino)-3-(3-methoxybenzylamino)-1 H-pyrazolo[3.4-d]- pyrimidine (title compound I) and 5-(3-chlorophenyl)-1 ,5-dihvdro-4-imino-3-(3- methoxybenzylamino)-4H-pyrazolo[3.4-dlpyrimidine hydrochloride (title compound II) A mixture of 5.97 g (20 mmol) of 4-cyano-5-(dimethylaminomethyleneamino)-3-(3- methoxybenzylamino)pyrazole, 3.77 g (23 mmol) of 3-chloroaniline hydrochloride and 100 ml of ethanol is heated under reflux for 36 h. Cooling to about 10°C, filtration and washing of the material on the filter with ethanol result in title compound II; melting point 251-253°C (decomposition).
The filtrate is evaporated in vacuo, the oily residue is partitioned between ethyl acetate and water, and the organic phase is washed with brine, dried over sodium sulfate and concentrated to a volume of about 25 ml in an RE, whereupon a crystalline precipitate separates out. Filtration and washing of the residue on the filter with a little ethyl acetate and diethyl ether yield the title compound I (see Example 52); melting point 194-195°C.
The starting material is prepared as follows:
Staαe 45.1 : 2-Cvano-3-(3-methoxybenzylamino)-3-methylthioacrylonitrile
A mixture of 17.03 g (0.1 mol) of 3,3-bismethylthio-2-cyanoacryionitrile, 12.78 ml (0.1 mol) of 3-methoxybenzylamine and 60 mi of ethyl acetate is heated under reflux for 2 h and then evaporated in vacuo, resulting in the oily title compound; TLC Rf = 0.28 (toluene/isopropanol
[9:1]).
Staαe 45.2: 5-Amino-4-cvano-3-(3-methoxybenzylamino)pyrazole A mixture of 25.57 g (98.6 mmol) of 2-cyano-3-(3-methoxybenzylamino)-3-methylthio- acrylonitrile, 5.16 ml (104 mmol) of hydrazine hydrate and 140 ml of methanol is stirred at RT for 1 h, heated under reflux for 2.5 h and then evaporated in vacuo. Recrystallization of the residue from ethanol/hexane results in the title compound; melting point 151 -153°C.
Stage 45.3: 4-Cvano-5-(dimethylaminomethyleneamino)-3-(3-methoxybenzylamino)pyrazole A mixture of 9.12 g (37.5 mmol) of 5-amino-4-cyano-3-(3-methoxybenzylamino)pyrazole, 7.71 ml (45 mmol) of N,N-dimethylformamide diethyl acetal and 130 ml of toluene is heated under reflux for 3 h and then about 40 ml of hexane are added dropwise. On cooling to 10°C, a crystalline precipitate forms. Filtration and washing of the residue on the filter with diethyl ether afford the title compound; melting point 136-137°C.
Example 46: 4-Benzylamino-3-(3-methylphenylamino)-1 H-pyrazolor3.4-d1pyrimidine A mixture of 1 g (3.74 mmol) of 4-cyano-5-(dimethylaminomethyleneamino)-3-(3- methylphenylamino)pyrazole and 10 ml of benzylamine is stirred at 120°C under a nitrogen atmosphere for 4 h and then evaporated under HV. The crystalline residue is digested with 10 ml of acetonitrile and filtered, and the material on the filter is recrystallized from 40 ml of acetonitrile, resulting in the title compound; melting point 200-201 °C.
The starting material is prepared as follows:
Stage 46.1 : 4-Cvano-5-(dimethylaminomethyleneamino)-3-(3-methylphenylamino)pyrazole A suspension of 10.71 g (50.22 mmol) of 5-amino-4-cyano-3-(3- methylphenylamino)pyrazole (for preparation, see: Arch. Pharm. (Weinheim) 326, 245 (1993)], 9.9 ml (57.8 mmol) of N,N-dimethylformamide diethyl acetal and 150 ml of toluene is heated under reflux for 4 h. It is then cooled to RT and filtered, and the residue on the filter is washed with toluene and diethyl ether, resulting in the title compound; melting point 260-261 °C.
Examole 47: (S)-3-(3-Methylphenylamino)-4-(1-phenylethylamino)-1 H-pyrazolor3.4-d]- pyrimidine hydrochloride
A mixture of 1 g (3.74 mmol) of 4-cyano-5-(dimethylaminomethyleneamino)-3-(3- methylphenylamino)pyrazole and 10 ml of (S)-l-phenylethylamine is stirred at 120°C under a nitrogen atmosphere for 24 h and then evaporated under HV. The oily residue is purified by flash chromatography on silica gel of particle size 0.04-0.06 mm using ethyl acetate/hexane (7:3). The product-containing fractions are evaporated and the resinous residue is dissolved in a mixture of 5 ml of ethanol and 0.8 ml of 4 N hydrochloric acid. Renewed evaporation and recrystallization of the residue from ethanol/diethyl ether affords the title compound; melting point 170-180°C (decomposition); [α]D 20 = +253.8 ± 1° (c = 1.015%, methanol)
Example 48: (R)-3-(3-Methylphenylamino)-4-(1 -phenylethylaminoH H-pyrazolor3,4-dl- pyrimidine hydrochloride
The title compound is obtained in analogy to Example 47 starting from 1.5 g (5.61 mmol) of 4-cyano-5-(dimethylaminomethyleneamino)-3-(3-methylphenylamino)pyrazole and 10 ml of (R)-l-phenylethylamine; melting point 170-180°C (decomposition); [α]D 20 = -249.4 ± 1° (c = 0.987%, methanol).
Example 49: 4-Benzylamino-3-(3-chlorophenylamino)-1 H-pyrazolo[3.4-dlpyrimidine A mixture of 1 g (3.46 mmol) of 3-(3-chlorophenylamino)-4-cyano-5-(dimethylamino- methyleneamino)pyrazole and 10 ml of benzylamine is stirred at 120°C under a nitrogen atmosphere for 3 h and then evaporated under HV. The crystalline residue is digested with 10 ml of acetonitrile, cooled to 0°C and filtered, and the material on the filter is crystallized from 35 ml of acetonitrile, resulting in the title compound; melting point 216-218CC.
The starting material is prepared as follows:
Stage 49.1 : 5-Amino-3-(3-chlorophenylamino)-4-cvanopyrazole
A mixture of 15 g (60.1 mmol) of 3-(3-chlorophenyiamino)-2-cyano-3-methylthioacrylonitrile
(for preparation, see: EP 0 010 396 A1], 3.13 ml (63.2 mmol) of hydrazine hydrate and
100 ml of methanol is heated under reflux for 3 h and then evaporated in vacuo.
Recrystallization of the residue from ethyl acetate/hexane results in the title compound; melting point 195-200°C.
Yield: 86.4%
Stage 49.2: 3-(3-Chlorophenylamino)-4-cvano-5-(dimethylaminomethyleneamino)pyrazole The title compound is obtained in analogy to Example E.3 starting from 11.7 g (50.07 mmol) of 5-amino-3-(3-chlorophenylamino)-4-cyanopyrazole, 9.43 ml (55.07 mmol) of N,N-dimethylformamide diethyl acetal and 140 ml of toluene after heating under reflux for 3.5 h; melting point > 260°C; TLC Rf = 0.87 (methylene chloride/methanol [9:1]).
Example 50: 4-(3-Chlorophenylamino)-3-(3-methoχyphenylamino -1 H-pyrazolor3.4-d]- pyrimidine
A mixture of 1.38 g (4.85 mmol) of 4-cyano-5-(dimethylaminomethyleneamino)-3-(3- methoxyphenylamino)pyrazole, 0.915 g (5.58 mmol) of 3-chloroaniline hydrochloride and
20 ml of methanol is heated under reflux for 16 h. Evaporation of the reaction mixture in vacuo and recrystallization of the residue from methanol/water yield the title compound; melting point 202-203°C.
The starting material is prepared as follows:
Stage 50.1 5-Amino-4-cvano-3-(3-methoxyphenylamino)pyrazole
The title compound is obtained in analogy to Example 49.1 starting from 10 g (40.77 mmol) of 2-cyano-3-(3-methoxyphenylamino)-3-methylthioacrylonitrile [for preparation, see: Bioorg. Med. Chem. Lett. 4, 615 (1994)], 2.12 ml (42.8 mmol) of hydrazine hydrate and 70 ml of methanol after heating under reflux for 3.5 h; melting point 177-178°C.
Stage 50.2: 4-Cvano-5-(dimethylaminomethyleneamino -3-(3-methoxyphenylamino)- pyrazole
The title compound is obtained in analogy to Example 44.3 starting from 1.25 g (5.45 mmol) of 5-amino-4-cyano-3-(3-methoxyphenylamino)pyrazole, 1.23 ml (7.18 mmol) of
N,N-dimethylformamide diethyl acetal and 25 ml of toluene after heating under reflux for
4.5 h; melting point 227-231 °C.
Example 51 : 4-(3-Chlorophenylamino)-3-(3-methoxyphenylamino)-1 H-pyrazolor3.4-d1- pyrimidine
A mixture of 0.9 g (2.45 mmol) of 5-(3-chlorophenyl)-1 ,5-dihydro-4-imino-3-(3- methoxyphenylamino)-4H-pyrazolo[3,4-d]pyrimidine, 18 ml of dioxane and 18 ml of water is heated under reflux for 10 h. Cooling to 0°C, filtration and recrystallization from methanol/water and from methanol afford the title compound; melting point 203-204°C.
The starting material is prepared as follows:
Staoe 51.1 : 4-Cyano-5-(ethoxymethyleneamino)-3-(3-methoxyphenylamino)pyrazole A mixture of 4 g (17.45 mmol) of 5-amino-4-cyano-3-(3-methoxyphenylamino)pyrazole (stage 50.1 ) and 50 ml of triethyl orthoformate is heated at 120°C with stirring for 1.5 h, taking care that the ethanol formed during the reaction distils out of the reaction mixture. The reaction mixture is cooled to 30°C and, after addition of about 100 ml of diethyl ether, cooled further to 0°C and filtered. Washing with diethyl ether affords the title compound; melting point 181-183°C.
Stage 51.2: 5-(3-ChlorophenvP-1 ,5-dihvdro-4-imino-3-(3-methoxyphenylamino)-4H- pyrazolo[3.4-dlpyrimidine
A mixture of 3.85 g (13.49 mmol) of 4-cyano-5-(ethoxymethyleneamino)-3-(3- methoxyphenylamino)pyrazole, 2.84 ml (27.02 mmol) of 3-chloroaniline and 75 ml of ethanol is heated under reflux for 2 h. It is then cooled to RT and filtered. Washing of the crystals with ethanol and diethyl ether affords the title compound; melting point 190-192°C.
Example 52: 4-(3-Chlorophenylamino)-3-(3-methoxybenzylamino)-1 H-pyrazolo 3.4-d]- pyrimidine
A mixture of 0.417 g (1 mmol) of 5-(3-chlorophenyl)-1 ,5-dihydro-4-imino-3-(3- methoxybenzylamino)-4H-pyrazolo[3,4-d]pyrimidine hydrochloride (Example 45, title compound II), 20 ml of dioxane, 19 ml of water and 1 ml of 1 N sodium hydroxide solution is heated under reflux for 10 h. Cooling to about 5°C, filtration, washing of the residue on the filter with water and drying under HV at 120°C yield the title compound (see Example 45); melting point 192-193°C.
Example 53: 3-(3-Hvdroxyphenyl)-4-(3-chlorophenylamino)-1 H-pyrazolo[3,4-dlpyrimidine A mixture of 44.2 mg (0.1 mmol) of 1-benzyl-4-(3-chlorophenylamino)-3-(3- methoxyphenyl)pyrazolo[3,4-d]pyrimidine, 80 mg (0.6 mmol) of anhydrous aluminium chloride and 2 ml of toluene is heated under reflux with exclusion of air and moisture for 2 h. The reaction mixture is then poured into ice-water and extracted with ethyl acetate, and the organic phase is washed with a saturated solution of sodium bicarbonate in water and with brine, dried over sodium sulfate and evaporated in vacuo. Purification of the residue by flash chromatography on silica gel (230-400 mesh) using methylene chloride and methylene chioride/methanol mixtures (19:1 and 17:3) results in the title compound; melting point > 220°C; FAB-MS: (M+H)+ = 338.
The starting material is prepared as follows:
Stage 53.1 : 2-Cvano-3-(3-methoxyphenyl)-3-methylthioacrylonitrile A mixture of 4.88 g (24.61 mmol) of methyl 3-methoxydithiobenzoate (for preparation, see: Chem. Ber. 120, 1757 (1987)], 4.26 g (29.56 mmol) of tetracyanoethylene oxide (Fluka) and 35 ml of toluene is stirred at -5°C for 1 h and at 20°C for 21 h. Then 2 ml of triethylamine are added to the reaction mixture, which is subsequently concentrated by passing a stream of nitrogen through the reaction mixture for about 15 h. The oily residue is purified by flash chromatography on silica gel (230-400 mesh) using hexane and hexane/ethyl acetate mixtures of increasing ethyl acetate content (5%, 7.5%, 10%, 12.5% and 20% ethyl acetate). The product-containing fractions are evaporated in vacuo, resulting in the title compound as oil; TLC Rf = 0.48 (toluene/acetone [9:1]); FAB-MS: (M+H)+ = 231 (C12H10N2OS).
Staαe 53.2: 5-Amino-1 -benzyl-4-cvano-3-(3-methoxyphenyl)pyrazole 11.94 ml of a 5.4 N sodium methoxide solution in methanol (64.47 mmol) are added to a suspension of 6 g (30.62 mmol) of benzylhydrazine dihydrochloride in 15 ml of ethanol at 5°C, and the mixture is stirred at 5-10°C for about 15 min and then added to a suspension of 2.95 g (12.81 mmol) of 2-cyano-3-(3-methoxyphenyl)-3-methylthioacrylonitrile in 95 ml of ethanol. The reaction mixture is heated under reflux for 1 h and then cooled to RT and filtered. The residue on the filter is washed with ethanol, and the filtrate is evaporated in vacuo. The oily residue is purified by flash chromatography on silica gel (230-400 mesh) using a toluene/acetone mixture (19:1). The combined product-containing fractions are evaporated, and the residue is digested with diisopropyl ether. Filtration results in the title compound; melting point 147-149°C; FAB-MS: (M+H)+ = 305.
Staαe 53.3: 1 -Benzyl-4-hvdroxy-3-(3-methoxyphenyl)pyrazolo[3.4-dlpyrimidine A mixture of 2.55 g (8.38 mmol) of 5-amino-1-benzyl-4-cyano-3-(3-methoxyphenyl)pyrazole and 20 ml of 85% strength aqueous formic acid is heated under reflux for 21 h and then cooled to RT and, while stirring, a little water is added. Filtration and washing of the material on the filter with water results in the title compound; melting point 183-185°C; FAB-MS: (M+H)+ = 333.
Staαe 53.4: 1 -Benzyl-4-chloro-3-(3-methoxyphenvπpyrazolof3,4-d1pyrimidine A mixture of 2.48 g (7.46 mmol) of 1-benzyl-4-hydroxy-3-(3-methoxyphenyl)pyrazolo[3,4-d]- pyrimidine and 30 ml of phosphorus oxychloride is heated under reflux for 10 h and then slowly poured into ice-water with stirring. The mixture is then stirred at 0-10°C for a further 2 h and filtered, and the material on the filter is washed with water. The crude product is dissolved in methylene chloride, and the organic phase is washed with brine, dried over sodium sulfate and evaporated in vacuo. Purification of the residue by flash chromatography on silica gel (230-400 mesh) using methylene chloride and methylene chloride/methanol mixtures (99:1 and 49:1 ) result in the title compound; melting point 99- 101 °C; FAB-MS: (M+H)+ = 351.
Stage 53.5: 1 -Benzyl-4-(3-chlorophenylamino)-3-(3-methoxyphenyl)pyrazolof3.4-dl- pyrimidine
A mixture of 70 mg (0.2 mmol) of 1 -benzyl-4-chloro-3-(3-methoxyphenyl)pyrazolo[3,4-d]- pyrimidine, 115.6 μl (1.1 mmol) of 3-chloroaniline and 7 ml of 1-butanol is heated under reflux for 20 h and then evaporated in vacuo. The residue is digested with hexane and filtered. Repeated digestion of the residue on the filter with diisopropyl ether and with ethanol results in the title compound; melting point 118-119°C.
Example 54: 4-(3-Chlorophenylamino)-3-(4-hvdroxyphenyl)-1 H-pyrazolor3.4-dlpyrimidine A mixture of 0.442 g (1.2 mmol of 4-(3-chlorophenylamino)-3-(4-methoxyphenyl)-1 H- pyrazolo[3,4-d]pyrimidine [see Example 34], 1.16 g (8.7 mmol) of anhydrous aluminium chloride and 20 ml of benzene is heated under reflux with exclusion of air and moisture for 45 min. The reaction mixture is then poured into ice-water and filtered, and the residue on the filter is washed with water and partitioned between a 5% strength solution of sodium bicarbonate in water and ethyl acetate. The organic phase is washed with brine, dried over sodium sulfate and concentrated in vacuo, and a little hexane is added, whereupon the required product precipitates. Washing with hexane and purification of the crude product by flash chromatography on silica gel (230-400 mesh) using an ethyl acetate/hexane mixture (7:3) result in the title compound; melting point > 220°C; MS (M)+ = 337.
Example 55: (R)-3-(3-Chlorophenylamino)-4-(1-phenylethylamino)-1 H-pyrazolo[3.4 d - pyrimidine hydrochloride
A mixture of 1.5 g (5.19 mmol) of 3-(3-chlorophenylamino)-4-cyano-5-(dimethylamino- methyleneamino)pyrazole (stage 49.2) and 10 ml of (R)-1 -phenylethylamine is stirred at 120°C under a nitrogen atmosphere for 40 h and then evaporated under HV. The oily
residue is purified by flash chromatography on silica gel of particle size 0.04-0.06 mm using ethyl acetate/hexane (7:3). The product-containing fractions are evaporated, and the resinous residue is dissolved in a mixture of 5 ml of ethanol and 1.4 ml of 4 N hydrochloric acid. Renewed evaporation and recrystallization of the residue from ethanol/diethyl ether and from ethanol afford the title compound; melting point 150-152°C; EI-MS: (M)+ = 364; [α]D 20 = -233.7 ± 2.1 ° (c = 0.486%, methanol).
Example 56: (S)-3-(3-Chlorophenylamino)-4-(1 -phenylethylamino)-1 H-pyrazolo[3.4-d- pyrimidine hydrochloride
0.4 g (1.1 mmol) of (S)-3-(3-chlorophenylamino)-1 ,5-dihydro-4-imino-5-(1-phenylethyl)-4H- pyrazolo[3,4-d]pyrimidine is stirred as melt at 220°C for 15 min. The crude product obtained after cooling to RT is purified by flash chromatography under silica gel of particle size 0.04-0.06 mm using ethyl acetate/hexane (7:3). The product-containing fractions are evaporated, and the resinous residue is dissolved in ethanol. Addition of 0.6 ml of 4 N hydrochloric acid, evaporation in vacuo and recrystallization of the residue from ethanol afford the title compound; melting point 153-155°C; EI-MS: (M)+ = 364; [αfo20 = +232.7 ± 2.2° (c = 0.465%, methanol).
The starting material is prepared as follows:
Stage 56.1 : 3-(3-Chlorophenylamino)-4-cvano-5-(ethoxymethyleneamino)pyrazole The title compound is obtained in analogy to stage 51.1 starting from 4.5 g (19.26 mmol) of 5-amino-3-(3-chiorophenylamino)-4-cyanopyrazole (stage 49.1 ) and 50 ml of triethyl orthoformate after stirring at 120°C for 2 h; melting point 197-199°C.
Stage 56.2: (S)-1 ,5-Dihvdro-3-(3-chlorophenylamino)-4-imino-5-(1-phenylethyl)-4H- pyrazolor3,4-dlpyrimidine
The title compound is obtained in analogy to stage 51.2 starting from 1.2 g (4.14 mmol) of
3-(3-chlorophenylamino)-4-cyano-5-(ethoxymethyleneamino)pyrazole, 0.632 ml (4.97 mmol) of (S)-l-phenylethylamine and 12 ml of ethanol after heating under reflux for 2 h; melting point 214-215°C; [α]D 20 = -279.6 ± 1° (c = 0.98%, methanol).
Example 57: 3-(4-Acetylaminobenzylamino)-4-(3-chlorophenylamino)-1 H-pyrazolo[3.4-d]- pyrimidine
A mixture of 0.34 g (1.04 mmol) of 3-(4-acetylaminobenzylamino)-4-cyano-5-
(dimethylaminomethyleneamino)pyrazole, 0.189 g (1.15 mmol) of 3-chloroaniline
hydrochloride and 4 ml of methanol is heated under reflux for 20 h. Cooling to about 20°C, filtration and washing of the residue on the filter with diethyl ether yield the title compound; melting point > 260°C; TLC R( = 0.34 (toluene/isopropanol/conc. ammonia [70:29:1]).
The starting material is prepared as follows:
Stage 57.1 : 3-(4-Aminobenzylamino)-2-cvano-3-methylthioacrylonitrile A mixture of 17.03 g (0.1 mol) of 3,3-bismethylthio-2-cyanoacrylonitrile, 11.34 ml (0.1 mol) of 4-aminobenzylamine (Aldrich) and 60 ml of ethanol is stirred at 50°C for 2 h and then evaporated in vacuo. Purification of the residue by flash chromatography on silica gel of particle size 0.04-0.06 mm using toluene/isopropanol mixtures (49:1 , 97:3, 24:1 and 9:1) result in the title compound; melting point 100-102°C.
Stage 57.2: 5-Amino-3-(4-aminobenzylamino)-4-cvanopyrazole A mixture of 15.71 g (64.3 mmol) of 3-(4-aminobenzylamino)-2-cyano-3-methylthio- acrylonitrile, 3.35 ml (67.5 mmol) of hydrazine hydrate and 90 ml of methanol is stirred at 20°C for 1 h, heated under reflux for 4 h and then evaporated in vacuo. Digestion of the crystalline residue with 70 ml of diisopropyl ether, filtration and digestion once again with 100 ml of isopropanol afford the title compound; melting point 168-170°C.
Stage 57.3: 3-(4-Acetylaminobenzylamino)-5-amino-4-cvanopyrazole A solution of 1.087 ml (11.5 mmol) of acetic anhydride in 20 ml of THF is added dropwise over 15 min to a stirred suspension of 2.5 g (10.95 mmol) of 5-amino-3-(4- aminobenzylamino)-4-cyanopyrazole in 50 ml of THF cooled to 0°C. The reaction mixture is stirred at RT for a further 2 h, during which there is initial formation of a solution, from which crystalline product gradually separates out. It is filtered and the material on the filter is washed with THF and diethyl ether. Drying under HV (8 h, 110°C) results in the title compound with a THF content of 21.06%; melting point 124-126°C (decomposition); FAB- MS: (M+H)+ = 271 (C13Hι4N60).
Stage 57.4: 3-(4-Acetylaminobenzylamino)-4-cvano-5-(dimethylaminomethyleneamino)- pyrazole
A mixture of 0.48 g (1.4 mmol) of 3-(4-acetylaminobenzylamino)-5-amino-4-cyanopyrazole with a THF content of 21.06%, 0.245 ml (1.43 mmol) of N,N-dimethylformamide diethyl acetal and 4 ml of toluene is heated under reflux for 4.5 h. It is then cooled to about 0°C
and filtered, and the residue on the filter is washed with toluene, resulting in the title compound; melting point 206-211°C.
Example 58: 3-(4-Aminophenylamino)-4-(3-chlorophenylamino)-1 H-pyrazolor3.4-d]- pyrimidine hydrochloride
A mixture of 0.25 g (0.553 mmol) of 3-[4-(N-BOC-amino)phenylamino]-4-(3- chlorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine (see Example 65) and 10 ml of 3 N methanolic hydrochloric acid is stirred at RT for 20 h. Then 10 ml of diethyl ether are added to the reaction mixture and, after filtration, the residue on the filter is digested with hot methanol. After cooling, filtration and washing of the residue on the filter with diethyl ether, the crystals are dried at 100°C under high vacuum for 15 h and then left to stand in the ambient atmosphere for 24 h, resulting in the title compound with a hydrogen chloride content of 10.97% and a water content of 4.34%; melting point 211-213°C; EI-MS: (M)+ = 351.
Example 59: 4-(3-Chlorophenylamino)-3-(3-hvdroxyphenylamino)-1 H-pyrazolof3.4-d1- pyrimidine
A mixture of 0.5 g (1.36 mmol) of 4-(3-chlorophenylamino)-3-(3-methoxyphenylamino)-1H- pyrazolo[3,4-d]pyrimidine (Example 50), 0.873 g (6.55 mmol) of anhydrous aluminium chloride and 15 ml of benzene is heated at 80°C with exclusion of air and moisture for 9 h. The benzene phase is then decanted off, the resinous residue is partitioned between ethyl acetate and water, and the organic phase is washed with water and with a saturated solution of sodium bicarbonate in water, dried over sodium sulfate and evaporated in vacuo. The residue is purified by flash chromatography on silica gel of particle size 0.04-0.06 mm using methylene chloride/methanol mixtures (50:1 and 20:1). The product-containing fractions are combined and concentrated to a volume of about 10 ml , whereupon the required product crystallizes out. Filtration and washing of the residue on the filter with diethyl ether afford the title compound; melting point 265-266°C; EI-MS: (M)+ = 352.
Example 60: 4-Benzylamino-3-(3-chlorophenylamino)-1 H-pyrazolo 3.4-dlpyrimidine A mixture of 1 g (4.28 mmol) of 5-amino-3-(3-chlorophenylamino)-4-cyanopyrazole (stage 49.1), 3.67 g (34.25 mmol) of benzylamine, 0.245 ml (4.28 mmol) of glacial acetic acid and 0.73 ml (19.35 mmol) of formic acid is heated at 200°C for 20 h. The reaction mixture is cooled to RT and 30 ml of ice-water and 5 ml of ethanol are added, the mixture is stirred for 20 min and filtered, and the material on the filter is washed with water. Recrystallization from isopropyl alcohol affords the title compound; melting point 215-218°C.
Example 61 : 4-Benzylamino-3-(3-chlorophenylamino -1 H-pyrazolo,3,4-dlpyrimidine A mixture of 1 g (4.28 mmol) of 5-amino-3-(3-chlorophenylamino)-4-cyanopyrazole (stage 49.1) and 1.75 g (12.95 mmol) of N-benzylformamide (Aldrich) is heated at 200°C for 20 h. The reaction mixture is purified by flash chromatography on silica gel of particle size 0.04-0.06 mm using ethyl acetate. Evaporation of the product-containing fractions and recrystallization of the residue from ethyl acetate/hexane result in the title compound; melting point 215-218°C; FAB-MS: (M+H)+ = 351.
Example 62: 3-(4-Aminobenzylamino)-4-(3-chlorophenylamino)-1 H-pyrazolo[3.4-d]- pyrimidine
A mixture of 0.11 g (0.27 mmol) of 3-(4-acetylaminobenzylamino)-4-(3-chlorophenyiamino)- 1 H-pyrazolo[3,4-d]pyrimidine (Example 57), 3 ml of water and 1.5 ml of 30% strength sodium hydroxide solution is heated at 100°C for 4 h. After cooling to RT, the reaction mixture is extracted with ethyl acetate, and the extract is washed with brine, dried over sodium sulfate and evaporated in vacuo. The residue is purified by flash chromatography on silica gel of particle size 0.04-0.06 mm using methylene chloride/methanol mixtures (50:1 and 20:1). The product-containing fractions are evaporated, and the residue is recrystallized from ethyl acetate/hexane, resulting in the title compound; melting point 170-171 °C; EI-MS: (M)+ = 365.
Example 63: 4-(3-Chlorophenylamino)-3-(4-ethoxyphenylamino)-1 H-pyrazolo[3.4-d]- pyrimidine
A mixture of 5.8 g (19.44 mmol) of 4-cyano-5-(dimethylaminomethyleneamino)-3-(4- ethoxyphenylamino)pyrazole, 3.51 g (21.4 mmol) of 3-chloroaniline hydrochloride and 40 ml of methanol is heated under reflux for 15 h. Cooling to 10°C, filtration and washing of the residue on the filter with methanol and diethyl ether yield the title compound; melting point
232-233°C; EI-MS: (M)+ = 380.
The starting material is prepared as follows:
Stage 63.1 : 2-Cvano-3-(4-ethoxyphenylamino)-3-methylthioacrylonitrile A mixture of 17.03 g (0.1 mol) of 3,3-bismethylthio-2-cyanoacrylonitrile, 12.94 ml (0.1 mol) of 4-ethoxyaniline and 60 ml of ethanol is heated under reflux for 2 h. Then 120 ml of diethyl ether are added dropwise to the stirred reaction mixture at about 30°C, and it is
cooled to 0°C. The title compound separates out in the form of colourless crystals and is filtered off, washed with diethyl ether and dried under HV; melting point 141-142°C.
Staαe 63.2: 5-Amino-4-cvano-3-(4-ethoxyphenylamino)pyrazole A mixture of 21.5 g (82.9 mmol) of 2-cyano-3-(4-ethoxyphenylamino)-3-methylthio- acrylonitrile, 4.31 ml (87 mmol) of hydrazine hydrate and 110 ml of methanol is heated under reflux for 7 h and then evaporated in vacuo. Recrystallization of the residue from ethyl acetate/hexane results in the title compound; melting point 166-167°C.
Staαe 63.3: 4-Cvano-5-(dimethylaminomethyleneamino)-3-(4-ethoxyphenylamino)pyrazole A suspension of 4.86 g (19.98 mmol) of 5-amino-4-cyano-3-(4-ethoxyphenylamino)pyrazole in 3.94 ml (23 mmol) of N,N-dimethylformamide diethyl acetal and 60 ml of toluene is heated under reflux for 2 h. It is then cooled to 20°C and filtered, and the residue from the filter is washed with toluene, resulting in the title compound; melting point 246-247°C (decomposition).
Example 64: The following compounds are obtained by processes analogous to those described in this text: a) 4-(3-chlorophenylamino)-3-(3,4-dimethoxyphenylamino)-1 H-pyrazolo[3,4-d]pyrimidine
b) 4-(3-chlorophenylamino)-3-(3,5-dimethoxyphenylamino)-1 H-pyrazolo[3,4-d]pyrimidine
c) 4-(3-chlorophenylamino)-3-(3-formylamino-4-methoxyphenylamino)-1 H-pyrazolo[3,4-d]- pyrimidine
d) 3-(3-acetylamino-4-methoxyphenylamino)-4-(3-chlorophenylamino)-1 H-pyrazolo[3,4-d]- pyrimidine
e) 3-(4-acetylamino-3-methoxyphenylamino)-4-(3-chlorophenylamino)-1 H-pyrazolo[3,4-d]- pyrimidine
f) 4-(3-chlorophenylamino)-3-(4-formylaminophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine
g) 4-(3-chlorophenylamino)-3-(4-formylaminobenzylamino)-1 H-pyrazolo[3,4-d]pyrimidine
h) 4-(3-chlorophenylamino)-3-(4-propionylaminobenzylamino)-1 H-pyrazolo[3,4-d]pyrimidine
i) 3-(4-aminomethylphenylamino)-4-(3-chlorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine hydrochloride
k) 3-(3-aminomethylphenylamino)-4-(3-chlorophenylamino)-1 H-pyrazolo[3,4-d]pyhmidine hydrochloride
l) 3-[4-(N-BOC-aminomethyl)phenylamino]-4-(3-chlorophenylamino)-1H-pyrazolo[3,4-d]- pyrimidine [precursor of i)],
m) 3-[3-(N-BOC-aminomethyl)phenylamino]-4-(3-chlorophenylamino)-1 H- pyrazolo[3,4-d]pyrimidine [precursor of k)]
The compounds a) to e) mentioned in Example 64 are preferably prepared in analogy to Example 63, compound f) is preferably prepared in analogy to Example 41 , compounds g) and h) are preferably obtained in analogy to Example 57, and compounds i) to m) in analogy to Example 58.
Example 65: 3-r4-(N-BOC-amino)phenylaminol-4-(3-chlorophenylamino)-1 H-pyrazolo[3.4-d]- pyrimidine
A mixture of 2 g (4.43 mmol) of 1 ,5-dihydro-3-[4-(N-BOC-amino)phenylamino]-4-imino-5-(3- chlorophenyl)-4H-pyrazolo[3,4-d]pyrimidine, 40 ml of dioxane and 40 ml of water is heated at 120°C with stirring for 22 h. Cooling to 20°C, filtration and washing of the residue on the filter with dioxane result in the title compound; melting point 256-258°C.
The starting material is prepared as follows:
Staαe 65.1 : 3-r4-(N-BOC-Amino)phenylamino]-2-cvano-3-methylthioacrylonitrile
A mixture of 13.89 g (81.58 mmol) of 3,3-bismethylthio-2-cyanoacryionitrile, 17 g
(81.63 mmol) of N-BOC-1 ,4-phenylenediamine (Fluka) and 200 ml of methanol is heated under reflux for 5 h. Cooling to room temperature, filtration and washing of the residue on the filter with methanol result in the title compound; melting point 190°C. Evaporation of the filtrate and recrystallization of the residue from 50 ml of methanol result in a further batch of the title compound; melting point 190-191 °C.
Stage 65.2: 5-Amino-3-r4-N-BOC-amino)phenylamino1-4-cvanopyrazole
The title compound is obtained in analogy to Example 49.1 starting from 23.1 g
(69.91 mmol) of 3-[4-(N-BOC-amino)phenylamino]-2-cyano-3-methylthioacrylonitrile, 4.15 ml
(83.75 mmol) of hydrazine hydrate and 200 ml of methanol after heating under reflux for
5 h; melting point 166-168°C.
Stage 65.3: 3-r4-(N-BOC-Amino)phenylamino1-4-cvano-5-(ethoxymethyleneamino yrazole A mixture of 13.8 g (43.9 mmol) of 5-amino-3-[4-(N-BOC-amino)phenylamino]-4- cyanopyrazole and 138 ml of triethyl orthoformate is heated at 120CC with stirring for 3 h, taking care that the ethanol formed during the reaction distils out of the reaction mixture. Cooling to RT, filtration and washing of the material on the filter with ethanol result in the title compound; melting point 180-182°C.
Stage 65.4: 1 ,5-Dihvdro-3-r4-(N-BOC-amino)phenylaminol-4-imino-5-(3-chlorophenyl)-4H- pyrazolof3.4-d yrimidine
A mixture of 7 g (18.9 mmol) of 3-[4-(N-BOC-amino)phenylamino]-4-cyano-5-
(ethoxymethyleneamino)pyrazole, 3.97 ml (37.78 mmol) of 3-chloroaniline and 150 ml of ethanol is heated under reflux for 9 h and stirred at RT for a further 15 h. Filtration and washing of the residue on the filter with cold ethanol afford the title compound; melting point
229-234°C (decomposition); FAB-MS: (M+H)+ = 452.
Example 66: 3-(4-Aminomethylphenylamino)-4-(3-chlorophenylamino)-1 H-pyrazolor3.4-d1- pyrimidine (see Example 64i)
A mixture of 25 g (53.65 mmol) of 3-[4-(N-Boc-aminomethyl)phenylamino]-4-(3- chlorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine and 350 ml of 3 N methanolic hydrochloric acid is stirred at 50°C for 30 min. It is then allowed to cool to RT and is filtered, the residue on the filter is washed with methanol and diethyl ether, and the material on the filter is partitioned between saturated aqueous sodium carbonate solution and ethyl acetate. The organic phase is washed with water and brine and dried over sodium sulfate, filtered and evaporated in vacuo. Recrystallization of the residue from methanol/water results in the title compound in the form of the monohydrate; melting point 207-209°C; ESI-MS: (M+H)+ = 366.
Example 67: 4-(3-Chlorophenylamino)-3-(4-formylaminomethylphenylamino)-1 H-pyrazolo- r3,4-dlpyrimidine (see Example 64p)
A mixture of 250 mg (0.651 mmol) of 3-(4-aminomethylphenylamino)-4-(3- chiorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine hydrate and 10 ml of ethyl formate is
heated under reflux for 20 h. It is then cooled to RT and is filtered, and the residue on the filter is recrystallized from DMF/ethyl acetate, resulting in the title compound with a water content of 1.0%; melting point 249-251 °C; ESI-MS (M-H)' = 392.
Example 68: 3-(4-(Acetylaminomethylphenylamino)-4-(3-chlorophenylamino)-1 H-pyrazolo- [3,4-dlpyrimidine (See Example 64r)
A solution of 0.0517 ml (0.547 mmol) of acetic anhydride in 0.5 ml of THF is added dropwise to a stirred suspension of 200 mg (0.521 mmol) of 3-(4-aminomethylphenylamino)- 4-(3-chlorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine hydrate in 3 ml of THF cooled to 0°C. The reaction mixture is then stirred at RT for 30 min, resulting in a clear solution, out of which a crystalline precipitate separates after addition of 7 ml of diethyl ether. Filtration, washing of the residue on the filter with diethyl ether and recrystallization from methanol/diethyl ether afford the title compound with a water content of 0.66%; melting point 193-195°C; ESI-MS: (M-H)" = 406.
Example 69: 3-(3-Aminomethylphenylamino)-4-(3-chlorophenylamino)-1 H-pyrazolo- [3.4-dlpyrimidine (see Example 64k)
A mixture of 12 g (25.75 mmol) of 3-[3-(N-Boc-aminomethyl)phenylamino]-4-(3- chlorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine and 200 ml of 3 N methanolic hydrochloric acid is stirred at RT for 15 h. The viscous reaction mixture is diluted with methanol and filtered, and the material on the filter is washed with diethyl ether, resulting in the crude dihydrochloride of the title compound (melting point > 260°C). This is dissolved in 107 ml of water, and 80 ml of a saturated solution of sodium dicarbonate in water is added to this stirred solution at RT. The mixture is then stirred at 20°C for 1.5 h and filtered, and the residue on the filter is washed with water. Drying under high vacuum at 120°C results in the title compound with a water content of 1.79%; melting point 212-214°C.
The starting material is prepared as follows:
Stage 69.1 : 3-[3-(N-Boc-aminomethvπphenylamino1-2-cvano-3-methylthioacrylonitrile A mixture of 102.1 g (600 mmol) of 3,3-bismethylthio-2-cyanoacrylonitrile, 133.3 g (600 mmol) of 3-(N-Boc-aminomethyl)aniline (WO 93/00095) and 1100 ml of methanol is heated under reflux for 6.5 h and then evaporated in vacuo. Crystallization of the residue from ethyl acetate, filtration and washing of the material on the filter with ethyl acetate afford the title compound, melting point 150-151 °C.
Staαe 6g.2: 5-Amino-3-[3-(N-Boc-aminomethyl)phenylamino1-4-cvanopyrazole A mixture of 145.4 g (422 mmol) of 3-[3-(N-Boc-aminomethyl)phenylamino]-2-cyano-3- methylthioacrylonitrile, 21.g6 ml (443 mmol) of hydrazine hydrate and 1000 ml of methanol is heated under reflux for 2 h and then evaporated in vacuo, resulting in the crude title compound (content: go.7%) as resinous residue; TLC Rf = 0.28 (methylene chloride/methanol [9:1]).
Staαe 69.3: 3-r3-(N-Boc-Aminomethyl)phenylamino]-4-cyano-5-dimethylaminomethylene- aminopyrazole
A mixture of 152.7 g (422 mmol) of 5-amino-3-[3-(N-Boc-aminomethyl)phenylamino]-4- cyanopyrazole (90.7%), 78.g ml (460.4 mmol) of N,N-dimethylformamide diethyl acetal,
1350 ml of toluene and 150 ml of ethanol is stirred at 40°C for 2 h. Cooling to 15°C, filtration and washing of the crystals with diethyl ether result in the title compound; melting point 182-
184°C.
Staαe 6g.4: 3-r3-(N-Boc-Aminomethyl)phenylamino]-4-(3-chlorophenylamino)-1 H-pyrazolo- [3.4-dlpyrimidine
A mixture of 25 g (65.2 mmol) of 3-[3-(N-Boc-aminomethyl)phenylamino]-4-cyano-5- dimethylaminomethyleneaminopyrazole, 10.7 g (65.2 mmol) of 3-chloroaniline hydrochloride [for preparation, see: Justus Liebigs Ann. Chem. 176, 45 (1875)] and 250 ml of methanol is heated under reflux for 15 h and then evaporated in vacuo. The oily residue is purified by flash chromatography on silica gel of particle size 0.04-0.06 mm using ethyl acetate. The product-containing fractions are evaporated, and the residue is recrystallized from methanol, resulting in the title compound; melting point 147-148°C.
Example 70: 3-(3-Acetylaminomethylphenylamino)-4-(3-chlorophenylamino)-1 H-pyrazolo- r3.4-dlpyrimidine (see Example 64s)
A mixture of 300 mg (0.805 mmol) of 3-(3-aminomethylphenylamino)-4-(3- chlorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine (water content: 1.79%), 0.0814 ml (0.861 mmol) of acetic anhydride and 5 ml of THF is stirred at RT for 2 h. Addition of 2 ml of diethyl ether to the reaction mixture, filtration and washing of the residue on the filter with diethyl ether afford the title compound with a water content of 1.09%; melting point 210°C; ESI-MS: (M-H)" = 406.
Example 71 : 4-(3-Chlorophenylamino)-3-(3-nitrobenzylamino)-1 H-pyrazolor3,4-dlpyrimidine 0.907 g (6 mmol) of 3-nitrobenzaldehyde is added to a solution of 1.043 g (4 mmol) of 3-amino-4-(3-chlorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine [see stage 15.1] in 100 ml of methanol, 100 ml of DMEU and 0.48 g (8 mmol) of acetic acid, and the mixture is stirred at RT for 1 h. Then 2.07 g (28 mmol) of sodium cyanoborohydride (85%) are added to the reaction mixture, which is then stirred at 20°C for 7 days. After renewed addition of 2.07 g (28 mmol) of sodium cyanoborohydride (85%), the reaction mixture is stirred at RT for a further 7 days and then poured into 3.2 I of water. Stirring overnight, filtration and recrystallization of the residue on the filter from ethanol afford the title compound; melting point 195-196°C; ESI-MS: (M+H)+ = 396.
Example 72: 3-(3-Aminobenzylamino)-4-(3-chlorophenylamino)-1 H- pyrazolof3,4-dlpyrimidine
A mixture of 0.15 g (0.379 mmol) of 4-(3-chlorophenylamino)-3-(3-nitrobenzylamino)-1H- pyrazolo[3,4-d]pyrimidine, 4 ml of methanol, 4 ml of THF and 40 mg of Raney nickel is hydrogenated at RT under atmospheric pressure until hydrogen uptake ceases. The mixture is filtered, the filtrate is evaporated in vacuo, and the residue is recrystallized from ethyl acetate/hexane, resulting in the title compound with a water content of 1.22%; melting point 165-170°C; ESI-MS: (M+H)+ = 366.
An alternative synthesis for the title compound is illustrated in the following reaction scheme:
WO 93/19063
6 3-[3-(N-Boc-amino)-benzylamino]-4-(3-chlorophenylamino)-1 H- pyrazolo[3,4-d]pyrimidine
3-(3-aminobenzylamino)-4-(3-chlorop enylamino)-1H- pyrazolo[3,4-d]pyrimidine
Example 73: 3-r4-(N-Boc-Aminomethyl)phenylaminol-4-(3-chlorophenylamino)-1 H-pyrazolo- [3.4-dlPyrimidine
A mixture of 94 g (245.1 mmol) of 3-[4-(N-Boc-aminomethyl)phenylamino]-4-cyano-5- dimethylaminomethyleneaminopyrazole, 44.2 g (269.4 mmol) of 3-chloroaniline hydrochloride [for preparation, see: Justus Liebigs Ann. Chem. 176, 45 (1875)] and 1000 ml of ethanol is heated under reflux for 60 h and then evaporated in vacuo. The residue is dissolved in ethyl acetate, and the organic phase is washed with ice-cold 0.5 N hydrochloric acid, water and brine, dried over sodium sulfate and evaporated in vacuo. Recrystallization of the residue from ethyl acetate/hexane and from methanol/methylene chloride results in the title compound; melting point 196°C; ESI-MS: (M-H)" = 464.
The starting material is prepared as follows:
Staαe 73.1 : 3-f4-(N-Boc-aminomethyl)phenylamino1-2-cvano-3-methylthioacrylonitrile A mixture of 86.1 g (505.6 mmol) of 3,3-bismethylthio-2-cyanoacrylonitrile [for preparation, see: Chem. Ber. 95, 2861 (1962)], 112.4 g (505.6 mmol) of 4-(N-Boc-aminomethyI)aniline [for preparation, see: WO 93/00095] and 750 ml of methanol is heated under reflux for 5 h and then concentrated in vacuo until crystallization starts. The title compound obtained after filtration melts at 154-157°C; ESI-MS: (M-H)" = 343.
Staαe 73.2: 5-Amino-3-[4-(N-Boc-aminomethyl)phenylamino1-4-cyanopyrazole A mixture of 117.8 g (342 mmol) of 3-[4-(N-Boc-aminomethyl)phenylamino]-2-cyano-3- methylthioacrylonitrile, 20.54 ml (41.45 mmol) of hydrazine hydrate and 750 ml of methanol is heated under reflux for 3 h and then evaporated in vacuo at 50CC, resulting in the crude title compound (content: 95.4%) as amorphous residue; TLC Rf = 0.28 (methylene chloride/methanol [9:1]).
Staαe 73.3: 3-r4-(N-Boc-aminomethyl)phenylamino1-4-cvano-5-dimethylaminomethylene- aminopyrazole
A mixture of 117.7 g (341.9 mmol) of 5-amino-3-[4-(N-Boc-aminomethyl)phenylamino]-4- cyanopyrazole (content: 95.4%), 76.3 ml (445.2 mmol) of N,N-dimethylformamide diethyl acetal and 1200 ml of toluene is heated under reflux for 5 h. Cooling to 20°C, filtration and washing of the crystals with toluene and diethyl ether result in the title compound; melting point 227-228°C; ESI-MS: (M-H)" = 382.
Example 74: 4-(3-Chlorophenylamino)-3-(4-propionylaminomethylphenylamino)-1 H- pyrazolor3.4-dlPyrimidine
A solution of 0.0705 ml (0.547 mmol) of propionic anhydride in 0.5 ml of THF is added dropwise to a stirred suspension of 200 mg (0.521 mmol) of 3-(4-aminomethylphenylamino)- 4-(3-chlorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine hydrate in 3 ml of THF cooled to 0°C. The reaction mixture is then stirred at RT for 1 h, resulting in a clear solution, out of which a crystalline precipitate separates after addition of 7 ml of diethyl ether. Filtration and washing of the residue on the filter with diethyl ether afford the title compound with a water content of 1.05%; melting point 207-209°C; ESI-MS: (M-H)' = 420.
Example 75: 4-(3-Chlorophenylamino)-3-(4-valerylaminomethylphenylamino)-1 H-pyrazolo- r3.4-dlpyrimidine
A solution of 0.118 ml (O.δgβ mmol) of valeric anhydride in 0.5 ml of THF is added dropwise to a stirred suspension of 200 mg (0.521 mmol) of 3-(4-aminomethylphenylamino)-4-(3- chlorophenylamino)-1 H-pyrazolo[3,4-d]pyrimidine hydrate in 3 ml of THF cooled to 0°C. The reaction mixture is then stirred at RT for 2.5 h, resulting in a clear solution, out of which a crystalline precipitate separates after addition of 5 ml of diethyl ether and 4 ml of hexane. Filtration and washing of the residue on the filter with diethyl ether afford the title compound with a water content of 1.0%; melting point 202-204°C; ESI-MS: (M-H)" = 448.
Example 76: 3-(4-Benzoylaminomethylphenylamino)-4-(3-chlorophenylamino)-1 H-pyra- zolof3.4-d]pyrimidine
A solution of 136.2 mg (0.602 mmol) of benzoate anhydride in 1 ml of THF is added to a stirred suspension of 200 mg (0.521 mmol) of 3-(4-aminomethylphenyiamino)-4-(3-chloro- phenylamino)-1 H-pyrazolo[3,4-d]pyrimidine hydrate in 3 ml of THF cooled to 0°. The reaction mixture is stirred at RT for 2 h, resulting initially in a clear solution out of which a crystalline precipitate soon separates. Filtration and washing of the residue on the filter with diethyl ether result in the title compound with a water content of 4.74%; melting point 241- 243°C; ESI-MS: (M-H)' = 468.
Example 77: 4-(3-Chlorophenylamino)-3-(3-propionylaminomethylphenylamino)-1 H-pyra- zolo,3,4-dlpyrimidine
A mixture of 300 mg (0.805 mmol) of 3-(3-aminomethylphenylamino)-4-(3-chlorophenyl- amino)-1 H-pyrazolo[3,4-d]pyrimidine (water content: 1.7g%), 0.109 ml (0.846 mmol) of propionic anhydride and 5 ml of THF is stirred at RT for 2 h. Addition of 10 ml of diethyl ether to the reaction mixture, filtration and washing of the residue on the filter with diethyl
ether afford the title compound with a water content of 2.09%; melting point 210°C; ESI-MS: (M+H)+ = 422.
Example 78: 3-r3-(N-Boc-aminomethyl)benzylaminol-4-(3-chlorophenylamino)-1 H-pyra- zolo[3.4-d]pyrimidine
A mixture of 7.5 g (18.87 mmol) of 3-[3-(N-Boc-aminomethyl)benzylamino]-4-cyano-5-di- methylaminomethyleneaminopyrazole, 3.41 g (20.79 mmol) of 3-chloroaniline hydrochloride and 40 ml of methanol is heated under reflux for 24 h and then evaporated in vacuo. The residue is purified by flash chromatography on silica gel of particle size 0.04-0.06 mm using ethyl acetate/methanol (49:1 and 19:1). Evaporation of the product-containing fractions in vacuo and recrystallization of the residue from ethyl acetate/hexane afford the title compound; melting point 178-179°C (decomposition).
The starting material is prepared as follows:
Staαe 78.1 : 3- 3-(N-Boc-aminomethyl)benzylaminol-2-cvano-3-methylthioacrylonitrile A mixture of 11.71 g (68.78 mmol) of 3,3-bismethylthio-2-cyanoacrylonitrile, 16.25 g (68.76 mmol) of 3-(N-Boc-aminomethyl)benzylamine [for preparation, see: J. Med. Chem. 32, 391-396 (1989)] and 50 ml of ethyl acetate is heated under reflux for 2 h and then evaporated in vacuo. The residue is purified by flash chromatography on silica gel of particle size 0.04-0.06 mm using ethyl acetate/hexane (1 :2 and 1 :1). Evaporation of the product-containing fractions in vacuo affords the oily title compound; TLC Rf = 0.22 (ethyl acetate/ hexane [1 :1]); ESI-MS: (M-H)" = 357.
Staαe 78.2: 5-Amino-3-r3-(N-Boc-aminomethyl)benzylaminol-4-cvanopyrazole A mixture of 23.8 g (66.39 mmol) of 3-[3-(N-Boc-aminomethyl)benzylamino]-2-cyano- 3-methylthioacrylonitrile, 3.46 ml (69.81 mmol) of hydrazine hydrate and 100 ml of methanol is heated under reflux for 5 h and then evaporated in vacuo. The residue is purified by flash chromatography on silica gel of particle size 0.04-0.06 mm using ethyl acetate and ethyl acetate/methanol (19:1). Evaporation of the product-containing fractions in vacuo, recrystallization of the residue from ethyl acetate/diethyl ether and washing of the residue on the filter with diethyl ether afford the title compound; melting point 152-153°C.
Staαe 78.3: 3-r3-(N-Boc-aminomethyl)benzylamino1-4-cyano-5-dimethylaminomethylene- aminopyrazole
A mixture of 15 g (43.81 mmol) of 5-amino-3-[3-(N-Boc-aminomethyI)benzylamino]-4-cyano- pyrazole, 10.14 ml (59.16 mmol) of N,N-dimethylformamide diethyl acetal, 200 ml of toluene and 30 ml of ethanol is stirred at 100°C for 4 h and then evaporated in vacuo.
Crystallization of the residue from diethyl ether affords the title compound; melting point
123-125°C (decomposition).
Example 79: 3-(3-Aminomethylbenzylamino)-4-(3-chlorophenylamino)-1 H-pyrazolo- [3.4-dlpyrimidine dihydrochloride
A mixture of 4.22 g (8.79 mmol) of 3-[3-(N-Boc-aminomethyl)benzylamino]-4-(3-chloro- phenylamino)-1 H-pyrazolo[3,4-d]pyrimidine and 60 ml of 3 N methanolic hydrochloric acid is stirred at RT for 16 h and then 30 ml of methanol and 100 ml of diethyl ether are added. Filtration and washing of the residue on the filter with diethyl ether afford the title compound as monohydrate; melting point 210-220°C (decomposition).
Example 80: 4-(3-Chlorophenylamino)-3-f4-(dimethylaminomethyleneamino)benzylaminol- 1 H-pyrazolo 3,4-d1pyrimidine
A mixture of 366 mg (1 mmol) of 3-(4-aminobenzylamino)-4-(3-chlorophenylamino)- 1 H-pyrazolo[3,4-d]pyrimidine (cf. Example 62), 427 mg (2.9 mmol) of N,N-di- methylformamide diethyl acetal and 5 ml of toluene is stirred at 100°C for 2.5 h. Cooling to room temperature, filtration and washing of the residue on the filter with toluene results in the title compound with a water content of 0.85%; melting point 198-200°C.
Example 81 : 3-(4-Aminomethylbenzylamino)-4-(3-chlorophenylamino)-1 H-pyrazolo- [3.4-dlpyrimidine 2.5 hydrochloride
A mixture of 3 g (6.25 mmol) of 3-[4-(N-Boc-aminomethyl)benzylamino]-4-(3-chlorophenyl- amino)-1 H-pyrazolo[3,4-d]pyrimidine and 50 ml of 3 N methanolic hydrochloric acid is stirred at RT for 3.5 h and then filtered. Washing of the residue on the filter with methanol and diethyl ether results in the title compound with a water content of 2.24%; melting point > 260°C.
The starting material is prepared as follows:
Staαe 81.1 : 3-r4-(N-Boc-aminomethvπbenzylaminol-2-cvano-3-methylthioacrylonitrile A mixture of 10.39 g (61 mmol) of 3,3-bismethylthio-2-cyanoacrylonitrile, 14.41 g (61 mmol) of 4-(N-Boc-aminomethyl)benzylamine [for preparation, see: J. Med. Chem. 32, 391-396 (198g)] and 300 ml of ethyl acetate is heated under reflux for 2 h and then evaporated in vacuo. The residue is purified by flash chromatography on silica gel of particle size 0.04-0.06 mm using ethyl acetate/hexane (1 :2 and 1 :1). Evaporation of the product- containing fractions and recrystallization of the residue from ethyl acetate/hexane result in the title compound; melting point 93-95°C.
Staαe 81.2: 5-Amino-3-[4-(N-Boc-aminomethyl)benzylamino1-4-cvanopyrazole A mixture of 12.8 g (35.7 mmol) of 3-[4-(N-Boc-aminomethyl)benzylamino]-2-cyano- 3-methylthioacrylonitrile, 1.93 g (38.3 mmol) of hydrazine hydrate and 50 ml of methanol is heated under reflux for 5 h and then evaporated in vacuo. Recrystallization of the residue from ethyl acetate/diethyl ether and washing of the residue on the filter with diethyl ether afford the title compound; melting point 148-150°C.
Staαe 81.3: 3-f4-(N-Boc-aminomethvhbenzylaminol-4-cvano-5-dimethylaminomethylene- aminopyrazole
A mixture of 6.61 g (19.3 mmol) of 5-amino-3-[4-(N-Boc-aminomethyl)benzylamino]-4- cyano-pyrazole, 3.7 ml (21.6 mmol) of N,N-dimethylformamide diethyl acetal and 100 ml of toluene is stirred at 100°C for 4 h. Cooling to RT, filtration and washing of the residue on the filter with toluene result in the title compound; melting point 178-181 °C.
Staαe 81.4: 3-f4-(N-Boc-aminomethy| benzylaminol-4-(3-chlorophenylamino)-1 H-pyra- zolof3,4-dlpyrimidine
A mixture of 7.5 g (18.87 mmol) of 3-[4-(N-Boc-aminomethyl)benzylamino]-4-cyano-5-di- methylaminomethyleneaminopyrazole, 3.41 g (20.79 mmol) of 3-chloroaniiine hydrochloride and 40 ml of methanol is heated under reflux for 18 h and then evaporated in vacuo. The residue is purified by flash chromatography on silica gel of particle size 0.04-0.06 mm using ethyl acetate and ethyl acetate/methanol (9g:1 and 20:1 ). Evaporation of the product- containing fractions in vacuo and recrystallization of the residue from ethyl acetate/diethyl ether afford the title compound; melting point 202-205°C.
Example 82: The following compounds are obtained by processes analogous to those described in this text:
a) 4-(3-chlorophenylamino)-3-[4-(dimethylaminomethyleneamino)phenylamino]-1 H-pyra- zolo[3,4-d]pyrimidine (water content 1.5%); melting point 226-228°C, ESI-MS: (M+H)+ = 407, b) 4-(3-chlorophenylamino)-3-[3-isonicotinoylaminophenyl]-1 H-pyrazolo[3,4-d]pyrimidine; melting point 318-322°C; ESI-MS: (M+H)+ = 442; TLC R = 0.22 (methylene chloride/ methanol [10:1]), c) 4-(3-chlorophenylamino)-3-[4-isonicotinoylaminophenyl]-1 H-pyrazolo[3,4-d]pyrimidine; melting point 309-311°C; ESI-MS: (M+H)+ = 442, d) 4-(3-chlorophenylamino)-3-(4-isobutyrylaminophenyl)-1 H-pyrazolo[3,4-d]pyrimidine; melting point 322-324°C; ESI-MS: (M+H)+ = 407, e) (S)-3-[4-(N-Boc-aminomethyl)phenylamino]-4-(1 -phenylethylamino)-1 H-pyrazolo[3,4-d]- pyrimidine, f) (S)-3-(4-aminomethylphenylamino)-4-(1 -phenylethylamino)-1 H-pyrazolo[3,4-d]pyrimidine, g) (R)-3-[4-(N-Boc-aminomethyl)phenylamino]-4-(1 -phenylethylamino)-1 H-pyrazolo[3,4-d]- pyrimidine, h) (R)-3-(4-aminomethylphenylamino)-4-(1 -phenylethylamino)-1 H-pyrazolo[3,4-d]pyrimidine, i) (S)-3-[3-(N-Boc-aminomethyl)phenylamino]-4-(1 -phenylethylamino)-1 H-pyrazolo[3,4-d]- pyrimidine, j) (S)-3-(3-aminomethylphenylamino)-4-(1 -phenylethylamino)-1 H-pyrazolo[3,4-d]pyrimidine, k) (R)-3-[3-(N-Boc-aminomethyl)phenylamino]-4-(1 -phenylethylamino)-1 H-pyrazolo[3,4-d]- pyrimidine,
I) (R)-3-(3-aminomethylphenylamino)-4-(1 -phenylethylamino)-1 H-pyrazolo[3,4-d]pyrimidine
(water content 1.83%); melting point 95-98°C; ESI-MS: (M+H)+ = 358;
[α]D 20 = -196.2 ± 2.0° (c = 0.49g%, methanol), m) 4-(3-chlorophenylamino)-3-(4-cyanobenzylamino)-1 H-pyrazolo[3,4-d]pyrimidine, n) 4-(3-chlorophenylamino)-3-(3-cyanobenzylamino)-1 H-pyrazolo[3,4-d]pyrimidine and o) 4-(3-chlorophenylamino)-3-(2-cyanobenzylamino)-1 H-pyrazolo[3,4-d]pyrimidine.
Example 83: 3-Benzylamino-4-(3-methylphenylamino)-1 H-pyrazolo[3.4-d]pyrimidine 800 mg (2.98 mmol) of N'-(3-benzylamino-4-cyano-1 H-pyrazol-5-yl)-N,N-dimethyl- formamidine (see stage 14.3) are suspended in 15 ml of methanol with exclusion of moisture, 728 mg (5.07 mmol) of m-toluidine hydrochloride are added, and the mixture is boiled under reflux for 15 h. A further 364 mg of m-toluidine hydrochloride are added and boiling is continued for 12 h. The reaction mixture is stirred into 200 ml of water, and the title compound is filtered off: analysis calculated for C19H18N6 (θ 0.07 H2O): C 68.81%,
H 5.51%, N 25.34%, H20 0.38%; found C 68.81%, H 5.65%, N 25.30%, H20 0.3g%; HPLC: TRet(Grad2o-l 00/20) = 1 -°; MS: (M)+ = 331 -
Example 84: 3-Benzylamino-4-[1 (ffl-phenylethylaminol-1 H-pyrazolor3.4-dlpyrimidine 687 mg (4.36 mmol) of 1 (f?)-phenylethylamine hydrochloride are added with exclusion of moisture to 779 mg (2.9 mmol) of N'-(3-benzylamino-4-cyano-1 H-pyrazol-5-yl)-N,N-dimethyl- formamidine (see stage 14.3) in 10 ml of methanol and boiled under reflux for 9 h. A further 458 mg of 1 (ft)-phenylethylamine hydrochloride are added and boiling is continued for 18 h. The reaction mixture is stirred into 160 ml of water, and the precipitate is filtered off. Column chromatography (Si02; methylene chloride/acetone 3:1 → 1:1) yields firstly 3-benzylamino- 4-methoxy-1 H-pyrazolo[3,4-d]pyrimidine (melting point 146-148°C), followed by the title compound, which is crystallized from ethyl acetate: melting point: 194-196°C; HPLC: TRet(Grad20-l 00/20) = 11 -6; FAB-MS: (M+H)+ = 345.
Example 85: 3-Benzylamino-4-[1 (S)-phenylethylaminol-1 H-pyrazolof3,4-dlpyrimidine In analogy to Example 84, 390 mg (1.45 mmol) of N'-(3-benzylamino-4-cyano-1 H-pyrazol- 5-yl)-N,N-dimethylformamidine (see stage 14.3) in 7 ml of methanol are reacted with 748 mg of 1 (S)-phenylethylamine hydrochloride to give the title compound; melting point 195-196°C.
Example 86: The following compounds are obtained by processes analogous to those described in this text: a) (R)-3-(3-nitrobenzylamino)-4-(1 -phenylethylamino)-1 H-pyrazolo[3,4-d]pyrimidine, b) (S)-3-(3-nitrobenzylamino)-4-(1 -phenylethylamino)-1 H-pyrazolo[3,4-d]pyrimidine, c) (R)-3-[3-(tert-butyloxycarbonylamino)benzylamino]-4-(1 -phenylethylamino)-1 H-pyrazolo- [3,4-d]pyrimidine (see Example 89), d) (S)-3-[3-(tert-butyloxycarbonylamino)benzylamino]-4-(1 -phenylethylamino)-1 H-pyrazolo- [3,4-d]pyrimidine (see Example 87), e) (R)-3-(3-aminobenzylamino)-4-(1 -phenylethylamino)-1 H-pyrazolo[3,4-d]pyrimidine dimesylate, f) (S)-3-(3-aminobenzylamino)-4-(1 -phenylethylamino)-1 H-pyrazolo[3,4-d]pyrimidine dimesylate (see Example 88), g) (R)-3-(4-nitrobenzylamino)-4-(1 -phenyiethylamino)-1 H-pyrazolo[3,4-d]pyrimidine, h) (S)-3-(4-nitrobenzylamino)-4-(1 -phenylethylamino)-1 H-pyrazolo[3,4-d]pyrimidine, i) (R)-3-[4-(tert-butyloxycarbonylamino)benzylamino]-4-(1 -phenylethylamino)-1 H-pyrazolo- [3,4-d]pyrimidine,
j) (S)-3-[4-(tert-butyloxycarbonylamino)benzylamino]-4-(1 -phenylethylamino)-1 H-pyrazolo-
[3,4-d]pyrimidine, k) (R)-3-(4-aminobenzylamino)-4-(1 -phenylethylamino)-1 H-pyrazolo[3,4-d]pyrimidine dimesylate,
I) (S)-3-(4-aminobenzylamino)-4-(1 -phenylethylamino)-1 H-pyrazolo[3,4-d]pyrimidine dimesylate, m) (R)-3-[2-(3-nitrophenyl)ethylamino]-4-(1 -phenylethylamino)-1 H-pyrazolo[3,4-d]pyrimidine, n) (S)-3-[2-(3-nitrophenyl)ethylamino]-4-(1 -phenylethylamino)-1 H-pyrazolo[3,4-d]pyrimidine, o) (R)-3-[2-(3-aminophenyl)ethylamino]-4-(1 -phenylethylamino)-1 H-pyrazolo[3,4- djpyrimidine dimesylate, p) (S)-3-[2-(3-aminophenyl)ethylamino]-4-(1 -phenyiethylamino)-1 H-pyrazolo[3,4-d]pyrimidine dimesylate, q) (R)-3-[2-(4-nitrophenyl)ethylamino]-4-(1 -phenylethylamino)-1 H-pyrazolo[3,4-d]pyrimidine, r) (S)-3-[2-(4-nitrophenyl)ethylamino]-4-(1 -phenylethylamino)-1 H-pyrazolo[3,4-d]pyrimidine, s) (R)-3-[2-(4-aminophenyl)ethylamino]-4-(1 -phenylethylamino)-1 H-pyrazolo[3,4-d]pyrimidine dimesylate and t) (S)-3-[2-(4-aminophenyl)ethylamino]-4-(1 -phenylethylamino)-1 H-pyrazolo[3,4-d]pyrimidine dimesylate.
Example 87: (S)-3-[3-(N-Boc-amino)benzylaminol-4-(1 -phenylethylamino)-1 H-pyrazolo- [3.4-dlpyrimidine (see Example 86d)
6.78 ml of ortho-phosphoric acid (85%) are added to a solution of 1.096 g (5 mmol) of S(-)-1-phenylethylamine (Fluka) in 100 ml of ethanol. The crystalline residue after evaporation in vacuo is dried in vacuo under about 100 mbar (2 h, 120°C) and then dissolved in 5 ml of DMF. 0.979 g (2.5 mmol) of 3-[3-(N-Boc-amino)benzylamino]-4-cyano- 5-dimethylaminomethyleneaminopyrazole (97.9%) is added to the solution, which is then stirred at 110°C for 6 h. The reaction mixture is then evaporated in vacuo, and the residue is partitioned between ethyl acetate and 10 ml of 20% strength aqueous potassium carbonate solution. The organic phase is dried over anhydrous sodium sulfate and evaporated, and the residue is purified by flash chromatography on silica gel of particle size 0.04-0.06 mm using ethyl acetate/hexane mixtures (2:1 and 5:1). Evaporation of the product-containing fractions in vacuo results in the title compound as resin; ESI-MS: (M+H)+ = 460; TLC Rf = 0.53 [methylene chloride:methanol:conc. ammonia (30%) = 350:50:1]
The starting material is prepared as follows:
Staαe 87.1 : 3-(N-Boc-amino)benzylamine
50 g of Raney nickel are added to a solution of 182.5 g (0.8362 mol) of 3-(N-Boc-amino)- benzonitrile (for preparation, see: WO 93/19063) in 1500 ml of ethanolic ammonia solution (about 8% NH3). Hydrogenation is carried out under a pressure of 4 bar for 10 h and, after filtration, the filtrate is evaporated in vacuo. The crystalline residue is recrystallized from a mixture of 300 ml of ethyl acetate and 400 ml of hexane. Filtration, washing of the material on the filter with hexane and drying in vacuo result in the title compound; melting point 133-134°C; ESI-MS: (M+H)+ = 223. Evaporation of the mother liquor and recrystallization of the residue from ethyl acetate/hexane afford a 2nd batch of the title compound; melting point 132-133°C.
Staαe 87.2: 3-r3-(N-Boc-amino)benzylaminol-2-cvano-3-methylthioacrylonitrile A mixture of 179 g (0.8053 mol) of 3-(N-Boc-amino)benzylamine and 137.1 g of 3,3-bis- methylthio-2-cyanoacrylonitrile [for preparation, see: Chem. Ber. 95, 2861 (1962)] and 1000 ml of ethyl acetate is heated under reflux for 2 h. 1000 ml of hexane are then added to the reaction mixture. The crystals obtained on cooling to 0°C are filtered off, and the residue on the filter is washed with hexane. Drying in vacuo (about 100 mbar, 4 h, 80°C) results in the title compound; melting point 145-146°C; ESI-MS: (M+H)+ = 345.
Staαe 87.3: 5-Amino-3-[3-(N-Boc-amino)benzylamino1-4-cvanopyrazole A mixture of 267.5 g (0.7766 mol) of 3-[3-(N-Boc-amino)benzylamino]-2-cyano-3-methyl- thioacrylonitrile, 40.41 ml (0.8153 mol) of hydrazine hydrate and 800 ml of methanol is heated under reflux for 4 h and then evaporated in vacuo. Crystallization of the residue from about 1400 ml of ethyl acetate/hexane (1 :1), filtration at 0°C, washing of the material on the filter with hexane and drying under HV (8 h, 90°C) results in the title compound with an ethyl acetate content of 8.2%; melting point (decomposition) 113-114°C; ESI-MS: (M+H)+ = 32g.
Staαe 87.4: 3-[3-(N-Boc-amino)benzylamino]-4-cvano-5-dimethylaminomethyleneamino- pyrazole
A mixture of 246.6 g (0.6893 mol) of 5-amino-3-[3-(N-Boc-amino)benzylamino]-4-cyano- pyrazole, 177 ml (1.0018 mol) of N,N-dimethylformamide diethyl acetal (97%), 1000 ml of toluene and 200 ml of ethanol is heated under reflux for 2 h. About 1 litre of hexane is then added to the hot reaction mixture, which is subsequently cooled in an ice bath. The crystalline precipitate which forms is filtered off and washed with 300 ml of hexane. Drying
in vacuo under about 100 mbar (14 h, 90°C) results in the title compound with a toluene content of 2.1%; melting point 112-115 °C; ESI-MS: (M+H)+ = 384.
Example 88: (S)-3-(3-Aminobenzylamino)-4-(1-phenylethylamino)-1 H-pyrazolor3.4-d]- pyrimidine (see Example 86f)
A mixture of 0.35 g (0.762 mmol) of (S)-3-[3-(N-Boc-amino)benzylamino]-4-(1-phenylethyl- amino)-1 H-pyrazolo[3,4-d]pyrimidine (see Example 87) and 5 ml of 3 N methanolic hydrochloric acid is stirred at RT for 15 h. The crystalline precipitate is filtered off, and the material on the filter is washed with diethyl ether and dried in vacuo under about 100 mbar (6 h, 80°C). The crystals [melting point (decomposition) 25g-261°C] are then dissolved in 5 ml of water. 0.6 ml of saturated aqueous sodium carbonate solution is added to the stirred solution, whereupon a suspension forms and is then stirred at RT for 20 min. Filtration, washing of the residue on the filter with water and drying under HV (8 h, 100°C) afford the title compound with a water content of 1.03%; melting point gβ-102°C; [CC]D20 = +230.1 ± 1.0° (c = 1.000%, methanol).
Example 8 : (R)-3-[3-(N-Boc-amino)benzylaminol-4-(1 -phenylethylamino)-1 H-pyrazolo- [3.4-d]pyrimidine (see Example 86c)
The title compound is obtained as a resin in analogy to Example 87, using 1.0g6 g (5 mmol) of R(+)-1 -phenylethylamine (Fluka); ESI-MS: (M+H)+ = 460; TLC-Rf = 0.52 [methylene chloride:methanol:conc. ammonia (30%) = 350:50:1].
Example θ: (R)-3-(3-Aminobenzylamino)-4-(1 -phenylethylaminoH H-pyrazolo[3.4-dl- pyrimidine (see Example 86e)
The crude title compound is obtained in analogy to Example 88 starting from 0.35 g (0.762 mmol) of (R)-3-[3-(N-Boc-amino)benzylamino]-4-(1-phenylethylamino)-1 H-pyrazolo- [3,4-d]pyrimidine (see Example 8 ) and 5 ml of 3 N methanolic hydrochloric acid. This is purified by partitioning between 15 ml of ethyl acetate and 5 ml of 20% strength aqueous potassium carbonate solution. The organic phase is dried over anhydrous sodium sulfate and then evaporated in vacuo, and the residue is recrystallized from ethyl acetate/hexane. Filtration and drying under HV (8 h, 80°C) afford the title compound; melting point 144-146°C; [α]D 20 = -23g.3 ± 1.0° (c = 0.g74%, methanol).
Examole gi : 4-(3-Chlorophenylamino)-3-(3-methylaminobenzylamino)-1 H-pyrazolor3,4-d1- pyrimidine
0.466 g (1 mmol) of 3-[3-(N-Boc-amino)benzylamino]-4-(3-chlorophenylamino)-1 H-pyrazolo- [3,4-d]pyrimidine (see Example 93) is added to a solution of 0.15 g (395 mmol) of lithium aluminium hydride in 12.5 ml of THF at 0°C. The reaction mixture is then heated under reflux under a nitrogen atmosphere for 24 h and subsequently, at 0°C, a mixture of 0.25 ml of water and 5 ml of THF and a mixture of 0.25 ml of 1 N sodium hydroxide solution and 5 ml of THF are added dropwise. The residue on the filter after filtration is washed with THF, and the filtrate is evaporated in vacuo. The residue is purified by flash chromatography on silica gel of particle size 0.04-0.06 mm using ethyl acetate and an ethyl acetate/methanol mixture (100:1 ). Evaporation of the product-containing fractions in vacuo and recrystallization of the residue from acetonitrile result in the title compound; melting point 192-193°C; ESI-MS: (M+H)+ = 380.
Example 92 (see also Example 82j): The following compound is obtained in analogy to the processes described in this text: (S)-3-(3-aminomethylphenylamino)-4-(1 -phenylethylamino)- 1 H-pyrazolo[3,4-d]pyrimidine»1.58 HCI (water content 5.83%); melting point > 200°C (decomposition); ESI-MS: (M+H)+ = 358; [α]D 20 = +160.0 ± 2.1° (c = 0.470%, methanol).
Example 93: 3-r3-(N-Boc-amino)benzylamino]-4-(3-chlorophenylamino)-1 H-pyrazolor3.4-d]- pyrimidine
A mixture of 242.4 g (0.6189 mol) of 3-[3-(N-Boc-amino)benzylamino]-4-cyano-5-dimethyl- aminomethyleneaminopyrazole (g7.g%; see stage 87.4), 120 g (0.7316 mol) of 3-chloro- aniline hydrochloride (for preparation, see: Justus Liebigs Ann. Chem. 176, 45 (1875)) and 1000 ml of methanol is heated under reflux for 38 h. The resulting suspension is cooled to 20°C and then 100 ml of water are slowly added and, after filtration, the residue on the filter is washed with 150 ml of methanol. Drying under about 100 mbar (4 h, 90°C) and under HV (8 h, 100°C) results in the title compound; melting point (decomposition) 210-211°C; ESI-MS: (M+H)+ = 466.
Example 94: Dry-filled capsules
5000 capsules, each of which contains as active ingredient 0.25 g of one of the compounds of the formula I mentioned in Examples 1 to 93, are produced in the following way:
Composition:
Active ingredient 1250 g
Talc 180 g
Wheat starch 120 g
Magnesium stearate 80 g
Lactose 20 g
Production process: The specified substances are powdered and forced through a screen with a mesh width of 0.6 mm. 0.33 g portions of the mixture are filled into gelatin capsules by means of a capsule-filling machine.
Example 95: soft-gelatin capsules
5000 soft-gelatin capsules, each of which contains 0.05 g of one of the compounds of the formula I mentioned in Examples 1 to 93 as active ingredient, are produced in the following way:
Composition:
Active ingredient 250 g
PEG 400 1 I
Tween 80 1 I
Production process: The powdered active ingredient is suspended in PEG 400 (polyethylene glycol with Mr between about 380 and about 420, Fluka, Switzerland) and ©Tween 80 (polyoxyethylene sorbitan monolaurate, Atlas Chem. Ind., Inc., USA, supplied by Fluka, Switzerland), and ground in a wet pulverizer to a particle size of about 1 to 3 mm. 0.43 g portions of the mixture are then filled into soft-gelatin capsules by means of a capsule-filling machine.