WO1996000574A1 - Vitronectin receptor antagonists - Google Patents
Vitronectin receptor antagonists Download PDFInfo
- Publication number
- WO1996000574A1 WO1996000574A1 PCT/US1995/008146 US9508146W WO9600574A1 WO 1996000574 A1 WO1996000574 A1 WO 1996000574A1 US 9508146 W US9508146 W US 9508146W WO 9600574 A1 WO9600574 A1 WO 9600574A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- methyl
- oxo
- tetrahydro
- amino
- acetic acid
- Prior art date
Links
- 0 COC(C(*)Nc(ccc([N+]([O-])=O)c1)c1C#N)=O Chemical compound COC(C(*)Nc(ccc([N+]([O-])=O)c1)c1C#N)=O 0.000 description 4
- PAMIQIKDUOTOBW-UHFFFAOYSA-N CN1CCCCC1 Chemical compound CN1CCCCC1 PAMIQIKDUOTOBW-UHFFFAOYSA-N 0.000 description 1
- RXYPXQSKLGGKOL-UHFFFAOYSA-N CN1CCN(C)CC1 Chemical compound CN1CCN(C)CC1 RXYPXQSKLGGKOL-UHFFFAOYSA-N 0.000 description 1
- YLACBMHBZVYOAP-UHFFFAOYSA-N N#Cc1cc([N+]([O-])=O)ccc1F Chemical compound N#Cc1cc([N+]([O-])=O)ccc1F YLACBMHBZVYOAP-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D223/00—Heterocyclic compounds containing seven-membered rings having one nitrogen atom as the only ring hetero atom
- C07D223/14—Heterocyclic compounds containing seven-membered rings having one nitrogen atom as the only ring hetero atom condensed with carbocyclic rings or ring systems
- C07D223/16—Benzazepines; Hydrogenated benzazepines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/02—Nutrients, e.g. vitamins, minerals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D235/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings
- C07D235/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings condensed with carbocyclic rings or ring systems
- C07D235/04—Benzimidazoles; Hydrogenated benzimidazoles
- C07D235/06—Benzimidazoles; Hydrogenated benzimidazoles with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached in position 2
- C07D235/14—Radicals substituted by nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D243/00—Heterocyclic compounds containing seven-membered rings having two nitrogen atoms as the only ring hetero atoms
- C07D243/06—Heterocyclic compounds containing seven-membered rings having two nitrogen atoms as the only ring hetero atoms having the nitrogen atoms in positions 1 and 4
- C07D243/10—Heterocyclic compounds containing seven-membered rings having two nitrogen atoms as the only ring hetero atoms having the nitrogen atoms in positions 1 and 4 condensed with carbocyclic rings or ring systems
- C07D243/14—1,4-Benzodiazepines; Hydrogenated 1,4-benzodiazepines
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
Definitions
- This invention relates to pharmaceutically active compounds which inhibit the vitronectin receptor and are useful for the treatment of osteoporosis. '
- Bone formation is the result of the deposition of mineralized bone by osteoblast cells
- bone resorption is the result of the dissolution of bone matrix by osteoclast cells.
- Many bone diseases are brought about by an imbalance of bone formation relative to bone resorption. For instance, diseases such as osteoporosis are characterized by a net loss of bone matrix. Thus, agents which inhibit bone resorption are useful for the treatment of such diseases.
- An activated osteoclast resorbs bone by attaching to the bone matrix, and secreting proteolytic enzymes, organic acids and protons into the sealed compartment formed between its cell membrane and the bone matrix.
- the acidic environment and proteolytic enzymes effect the dissolution of bone in the sealed compartment to create pits, or lacuna, in the bone surface, which are apparent when the osteoclast detaches from the bone.
- osteoclasts are mediated through cell surface adhesion receptors called integrins.
- integrins cell surface adhesion receptors
- the vitronectin receptor, or the ⁇ 3 integrin is known to bind to bone matrix proteins, such as osteopontin, bone sialoprotein and thrombospondin, which contain the tri-peptide Arg-Gly-Asp (or RGD) motif.
- RGD tri-peptide Arg-Gly-Asp
- Endocrinology 1993, 132, 1411 has further shown that echistatin inhibits bone resorption in vivo in the rat.
- EP 528 587 and 528 586 report substituted phenyl derivatives which inhibit osteoclast mediated bone resorption.
- Bondinell, et al, in WO 93/00095 (PCT/US92/05463) and PCT US93/12436 disclose that certain compounds which have a substituted 6-7 bicyclic ring system are useful for inhibiting the fibrinogen receptor, which is an integrin ( ⁇ i b ⁇ 3 ) protein founds on platelets.
- Other 6-7 bicyclic ring systems which inhibit the fibrinogen receptor are disclosed by Blackburn et al. in WO 93/08174 (PCT/US92/08788).
- This invention comprises compounds of the formula (I) as described hereinafter, which have pharmacological activity for the inhibition of the vitronection receptor and are useful in the treatment of osteoporosis.
- This invention is also a pharmaceutical composition
- a pharmaceutical composition comprising a compound according to formula (I) and a pharmaceutically carrier.
- This invention is also a method of treating diseases which are mediated by ligands which bind to the vitronectin receptor.
- the compounds of this invention are useful for treating osteoporosis, atherosclerosis, restenosis and cancer.
- This invention comprises compounds of formula (I):
- R 1 is H, C ⁇ _6 alkyl or benzyl
- R2 is (CH 2 )nCO 2 H
- R 3 is H, C ⁇ _6alkyl, Ar-Co- ⁇ -alkyl, Het-Co_6alkyl, or C3_6cycloalkyl-Co_6alkyl;
- R 4 is W-U, Y-(CHR 5 )m-U or Z-C(O);
- R 5 and R 6 are independently chosen from H, C ⁇ _6alkyl, Ar-Co-6alkyl, Het-
- Co- ⁇ -alkyl and C3_6cycloalkyl-Co_6alkyl; m is 1 or 2; n is 1 or 2; U is NR'QO), C ⁇ NR 1 , CH CH, C ⁇ C, CH 2 -CH 2 , O-CH 2 , CH 2 -O or
- Rd is H, N(R! 2 , C ⁇ _4alkyl, CONtR ⁇ , OH, OR 1 , or Ar-C 0 -4alkyl
- R e is H, Ci-4alkyl, Het-Co-4alkyl or Ar-Co ⁇ alkyl; and pharmaceutically acceptable salts thereof, provided that R 3 is not phenylethyl when R 4 is (3-amidino)phenylaminocarbonyl and X-X' is NH-CH.
- the compounds of formula (I) inhibit the binding of vitronectin and other RGD -containing peptides to the vitronectin ( ⁇ 3) receptor.
- Inhibition of the vitronectin receptor on osteoclasts inhibits osteoclastic bone resorption and is useful in the treatment of diseases wherein bone resorption is associated with pathology, such as osteoporosis.
- X-X' is NH-CH or CH 2 -CH.
- U is NR i CO, CONR l or C ⁇ OCONR 1 . More preferably U is NR i CO.
- R a is hydroxy or amino.
- R a is amino.
- W is Preferably, W is H 2 N or (3-amidino)phenyl.
- Y is , OH or NHR 6 .
- R 5 is H, phenyl or C ⁇ _ 6 alkyl.
- R 6 is benzyl, 2-pyridinyl or H.
- Z is 1-piperazinyl or 1 -piperidinyl.
- R 4 is Y-(CH 2 ) m NCH CO.
- R e is H or substituted or unsubstituted phenyl, benzyl, 2- or 3- pyridinyl, 2- or 4-pyrimidinyl or 1-, 2- or 3-piperidinyl.
- Z is 1-piperazinyl
- R e is preferably 2- or 3- pyridinyl or 2- or 4-pyrimidinyl.
- Z is piperidinyl
- R e is preferably 1 -piperidinyl.
- Y is NH 2 or pyridinyl.
- n is 1.
- R 1 is H, methyl or phenylethyl.
- novel compounds of this invention are the following:
- Ci- 4 _ ⁇ lkyl as applied herein means an optionally substituted alkyl group of 1 to 4 carbon atoms, and includes methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl and t-butyl.
- Ci-6alkyl additionally includes pentyl, n-pentyl, isopentyl, neopentyl and hexyl and the simple aliphatic isomers thereof.
- Co-4alkyl and Co_6alkyl additionally indicate that no alkyl group need be present (e.g., that a covalent bond is present).
- a substituent on a C ⁇ _ 6 alkyl group may be on any carbon atom which results in a stable structure, and is available by conventional synthetic techniques. Suitable substituents are C ⁇ _ 4 alkyl, OR 1 , SR 1 , C ⁇ _ 4 alkyl, Ci- 4 alkylsulfonyl, C ⁇ _ 4 alkylsulfoxyl, -CN, N(R i ) 2 ⁇ CH 2 N(R 1 ) 2 , -NO 2 , -CF 3 , -CO 2 R' 3 -CONCR 1 ⁇ , -COR 1 . -NR i C ⁇ R 1 , OH, F, Cl, Br, I, or CF 3 S(O) r ,wherein r is 0 to 2.
- Ar, or aryl as applied herein, means phenyl or naphthyl, or phenyl or naphthyl substituted by one to three substituents, such as those defined above for alkyl, especially C ⁇ _ 4 alkyl, C ⁇ _4alkoxy, Ci ⁇ alkthio, trifluoroalkyl, OH, F, Cl, Br, I or a 1,2-methylene dioxy group.
- Het, or heterocycle indicates an optionally substituted five or six membered monocyclic ring, or a nine or ten-membered bicyclic ring containing one to three heteroatoms chosen from the group of nitrogen, oxygen and sulfur, which are stable and available by conventional chemical synthesis.
- heterocycles are benzofuryl, benzimidazole, benzopyran, benzothiophene, furan, imidazole, indoline, morpholine, piperidine, piperazine, pyrrole, pyrrolidine, tetrahydropyridine, pyridine, thiazole, thiophene, quinoline, isoquinoline, and tetra- and perhydro- quinoline and isoquinoline. Any accessible combination of up to three substituents on the Het ring, such as those defined above for alkyl that are available by chemical synthesis and are stable are within the scope of this invention.
- C3-7cycloalkyl refers to an optionally substituted carbocyclic system of three to seven carbon atoms, which may contain up to two unsaturated carbon- carbon bonds.
- Typical of C 3 _7cycloalkyl are cyclopropyl, cyclobutyl, cyclopentyl, cyclopentenyl, cyclohexyl, cyclohexenyl and cycloheptyl. Any combination of up to three substituents, such as those defined above for alkyl, on the cycloalkyl ring that is available by conventional chemical synthesis and is stable, is within the scope of this invention.
- Certain radical groups are abbreviated herein.
- t-Bu refers to the tertiary butyl radical
- Boc refers to the t-butyloxycarbonyl radical
- Fmoc refers to the fluorenylmethoxycarbonyl radical
- Ph refers to the phenyl radical
- Cbz refers to the benzyloxycarbonyl radical
- BrZ refers to the o-bromobenzyloxycarbonyl radical
- C1Z refers to the o-chlorobenzyloxycarbonyl radical
- Bn refers to the benzyl radical
- 4-MBzl refers to the 4-methyl benzyl radical
- Me refers to methyl
- Et refers to ethyl
- Ac refers to acetyl
- Alk refers to C1.4a.kyl
- Nph refers to 1- or 2-naphthyl
- cHex refers to cyclohexyl.
- MeArg is N ⁇ -methyl argin
- DCC refers to dicyclohexylcarbodiimide
- DMAP refers to dimethylaminopyridine
- DIEA refers to diisopropylethylamine
- EDC refers to N-ethyl-N'(dimethylaminopropyl)- carbodiimide.
- HOBt refers to 1 -hydroxybenzotriazole
- THF refers to tetrahydrofuran
- DMF refers to dimethyl formamide
- NBS refers to N-bromo- succinimide
- Pd/C refers to a palladium on carbon catalyst
- DPPA diphenylphosphoryl azide
- BOP refers to benzotriazol-1-yloxy- tris(dimethylamino)phosphonium hexafluorophosphate
- HF refers to hydrofluoric acid
- TEA refers to triethylamine
- TFA refers to trifluoroacetic acid
- PCC refers to pyridinium chlorochromate.
- EDC and HOBT or SOCl a carboxylic acid
- SOCl HOBT or SOCl
- Many additional methods for converting a carboxylic acid to an amide are known, and can be found in standard reference books, such as "Compendium of Organic Synthetic Methods", Vol. I - VI (published by Wiley-Interscience).
- the methyl ester of 1-2 is hydrolyzed using aqueous base, for example, aqueous LiOH in THF or aqueous NaOH in methanol, and the intermediate carboxylate salt is acidified with a suitable acid, for instance TFA or HC1, to afford the carboxylic acid 1-3.
- aqueous base for example, aqueous LiOH in THF or aqueous NaOH in methanol
- the intermediate carboxylate salt is acidified with a suitable acid, for instance TFA or HC1, to afford the carboxylic acid 1-3.
- a suitable acid for instance TFA or HC1
- the intermediate carboxylate salt can be isolated, if desired.
- the protecting group can be removed either prior or subsequently to the ester hydrolysis step, using methods suitable for selective deprotection of the specific protecting group employed. Such methods are described in Green, "Protective Groups in Organic Synthesis"
- Piperidine-containing compounds such as HI- 3 can be prepared either from a suitably N-protected piperidine derivative, according to the methods described in Schemes I and ⁇ , or from a pyridine precursor, such as HI-l.
- a pyridine precursor such as HI-l.
- the pyridine subunit of HI-l can be reduced to the corresponding piperidine group by hydrogenation over a suitable catalyst, preferably PtO 2 , in the presence of acid, such as HC1.
- the resulting piperidinium salt IH-2 is then converted to compound ⁇ i-3 by the methods described in Scheme I.
- the core 6-7 fused ring system is prepared from compounds of the general formula (II):
- Schemes IV - VI Representative methods for preparing the benzodiazepine nucleus are given by Schemes IV - VI.
- a representative method for preparing a benzazepine nucleus is given by Scheme VII.
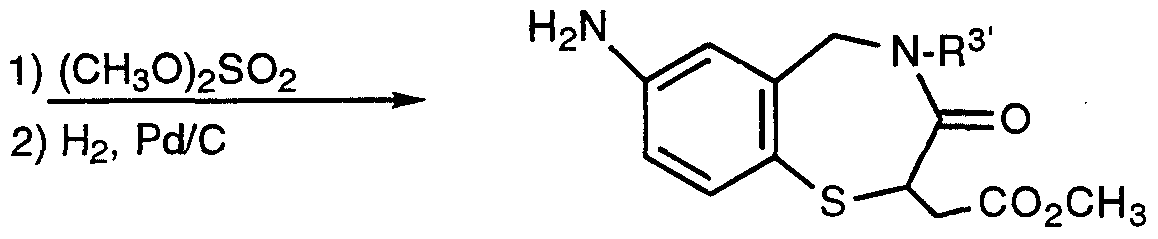
- a representative method for preparing a benzothiazepine is given by Scheme VIII.
- An benzoxazepine nucleus may be prepared in the same manner as Scheme VIII, except substituting a benzyl alcohol for a benzyl thiol.
- NBS N-bromosuccinimide
- Coupling reagents as used herein denote reagents which may be used to form peptide bonds. Typical coupling methods employ carbodiimides, activated anhydrides and esters and acyl halides. Reagents such as EDC, DCC, DPPA, BOP 10. reagent, HOBt, N-hydroxysuccinimide and oxalyl chloride are typical.
- Coupling methods to form peptide bonds are generally well known to the art.
- the methods of peptide synthesis generally set forth by Bodansky et al, THE PRACTICE OF PEPTIDE SYNTHESIS, Springer-Verlag, Berlin, 1984, Ali et al. in J. Med. Chem., 29, 984 (1986) and J. Med. Chem., 30, 2291 (1987) are generally 15 illustrative of the technique and are incorporated herein by reference.
- the amine or aniline is coupled via its free amino group to an appropriate carboxylic acid substrate using a suitable carbodiimide coupling agent, such as ⁇ , ⁇ ' dicyclohexyl carbodiimide (DCC), optionally in the presence of catalysts such as 1-hydroxybenzotriazole (HOBt) and dimethylamino pyridine 20 (DMAP).
- a suitable carbodiimide coupling agent such as ⁇ , ⁇ ' dicyclohexyl carbodiimide (DCC)
- catalysts such as 1-hydroxybenzotriazole (HOBt) and dimethylamino pyridine 20 (DMAP).
- HABt 1-hydroxybenzotriazole
- DMAP dimethylamino pyridine 20
- Other methods such as the formation of activated esters, anhydrides or acid halides, of the free carboxyl of a suitably protected acid substrate, and subsequent reaction with the free amine of a suitably protected amine, optionally in the presence of a base, are also suitable
- a protected Boc-amino acid or Cbz-amidino benzoic acid is treated in an anhydrous solvent, such as methylene chloride or tetrahydrofuran(THF), in the presence of a base, such as N-methyl morpholine, DMAP or a trialkylamine, with isobutyl chloroformate to form the "activated anhydride", which is subsequently reacted with the free amine of a second protected amino acid or aniline.
- anhydrous solvent such as methylene chloride or tetrahydrofuran(THF)
- a base such as N-methyl morpholine, DMAP or a trialkylamine
- Acid addition salts of the compounds are prepared in a standard manner in a suitable solvent from the parent compound and an excess of an acid, such as hydrochloric, hydrobromic, hydrofluoric, sulfuric, phosphoric, acetic, trifluoroacetic, maleic, succinic or methanesulfonic. Certain of the compounds form inner salts or zwitterions which may be acceptable.
- Cationic salts are prepared by treating the parent compound with an excess of an alkaline reagent, such as a hydroxide, carbonate or alkoxide, containing the appropriate cation; or with an appropriate organic amine. Cations such as Li + , Na + , K + , Ca ++ , Mg ++ and NH4" 1 " are specific examples of cations present in pharmaceutically acceptable salts.
- compositions of the compounds of formula (I) may be used in the manufacture of a medicament.
- Pharmaceutical compositions of the compounds of formula (I) prepared as hereinbefore described may be formulated as solutions or lyophilized powders for parenteral administration. Powders may be reconstituted by addition of a suitable diluent or other pharmaceutically acceptable carrier prior to use.
- the liquid formulation may be a buffered, isotonic, aqueous solution. Examples of suitable diluents are normal isotonic saline solution, standard 5% dextrose in water or buffered sodium or ammonium acetate solution.
- Such formulation is especially suitable for parenteral administration, but may also be used for oral administration or contained in a metered dose inhaler or nebulizer for insufflation. It may be desirable to add excipients such as polyvinylpyrrolidone, gelatin, hydroxy cellulose, acacia, polyethylene glycol, mannitol, sodium chloride or sodium citrate. Alternately, these compounds may be encapsulated, tableted or prepared in a emulsion or syrup for oral administration. Pharmaceutically acceptable solid or liquid carriers may be added to enhance or stabilize the composition, or to facilitate preparation of the composition.
- Solid carriers include starch, lactose, calcium sulfate dihydrate, terra alba, magnesium stearate or stearic acid, talc, pectin, acacia, agar or gelatin.
- Liquid carriers include syrup, peanut oil, olive oil, saline and water.
- the carrier may also include a sustained release material such as glyceryl monostearate or glyceryl distearate, alone or with a wax.
- the amount of solid carrier varies but, preferably, will be between about 20 mg to about 1 g per dosage unit.
- the pharmaceutical preparations are made following the conventional techniques of pharmacy involving milling, mixing, granulating, and compressing, when necessary, for tablet forms; or milling, mixing and filling for hard gelatin capsule forms.
- a liquid carrier When a liquid carrier is used, the preparation will be in the form of a syrup, elixir, emulsion or an aqueous or non-aqueous suspension.
- Such a liquid formulation may be administered directly p.o. or filled into a soft gelatin capsule.
- the compounds of this invention may also be combined with excipients such as cocoa butter, glycerin, gelatin or polyethylene glycols and molded into a suppository.
- the compounds described herein are antagonists of the vitronectin receptor, and are useful for treating diseases wherein the underlying pathology is attributable to ligand or cell which interacts with the vitronectin receptor. For instance, these compounds are useful for the treatment of diseases wherein loss of the bone matrix creates pathology.
- the instant compounds are useful for the treatment of ostoeporosis, hyperparathyroidism, Paget's disease, hypercalcemia of malignancy, osteolytic lesions produced by bone metastasis, bone loss due to immobilization or sex hormone deficiency.
- the compounds of this invention are also believed to have utility as antitumor, ant ⁇ nflammatory, anti-angiogenic and anti-metastatic agents, and be useful in the treatment of cancer, atherosclerosis and restenosis.
- the peptide is administered either orally or parenterally to the patient, in a manner such that the concentration of drug is sufficient to inhibit bone resorption, or other such indication.
- the pharmaceutical composition containing the peptide is administered at an oral dose of between about 0.1 to about 50 mg/kg in a manner consistent with the condition of the patient. Preferably the oral dose would be about 0.5 to about 20 mg/kg.
- parenteral administration is preferred.
- An intravenous infusion of the peptide in 5% dextrose in water or normal saline, or a similar formulation with suitable excipients, is most effective, although an intramuscular bolus injection is also useful.
- the parenteral dose will be about 0.01 to about 100 mg/kg; preferably between 0.1 and 20 mg/kg.
- the compounds are administered one to four times daily at a level to achieve a total daily dose of about 0.4 to about 400 mg/kg/day.
- the precise level and method by which the compounds are administered is readily determined by one routinely skilled in the art by comparing the blood level of the agent to the concentration required to have a therapeutic effect.
- the compounds may be tested in one of several biological assays to determine the concentration of compound which is required to have a given pharmacological effect.
- Solid-Phase [ 3 H]-SK&F-107260 Binding to cc v ⁇ 3 Human placenta or human platelet ⁇ v ⁇ 3 (0.1-0.3 mg/mL) in buffer T (containing 2 mM CaCl 2 and 1% octylglucoside) was diluted with buffer T containing 1 mM CaCl 2 , 1 mM MnCl 2 , 1 mM MgCl 2 (buffer A) and 0.05% NaN 3 , and then immediately added to 96-well ELISA plates (Corning, New York, NY) at 0.1 mL per well. 0.1 - 0.2 ⁇ g of ⁇ v ⁇ 3 was added per well.
- the plates were incubated overnight at 4°C. At the time of the experiment, the wells were washed once with buffer A and were incubated with 0.1 mL of 3.5% bovine serum albumin in the same buffer for 1 hr at room temperature. Following incubation the wells were aspirated completely and washed twice with 0.2 mL buffer A.
- the IC 50 concentration of the antagonist to inhibit 50% binding of [ 3 H]-SK&F- 107260
- the Kj dissociation constant of the antagonist
- Ki IC 50 /(1 + IJK d )
- L and K d were the concentration and the dissociation constant of [ 3 H]-SK&F- 107260, respectively.
- Compounds of the present invention inhibit vitronectin binding to SK&F 107260 in the concentration range of 0.1 to 25 micromolar.
- Preferred compounds inhibit vitronectin binding at a concentration of less than 1 micromolar.
- Each experimental group consists of 5-6 male Sprague-Dawley rats.
- the rats are parathyroidectomized (by the vendor, Taconic Farms) 7 days prior to use. Twenty four hours prior to use, circulating ionized calcium levels are measured in whole blood immediately after it has been withdrawn by tail venipuncture into heparinized tubes. Rats are included if ionized Ca level (measured with a Ciba-Corning model 634 calcium pH analyzer) is ⁇ 1.2 mM/L. The rats are then put on a diet of calcium-free chow and deionized water. At the start of the experiment the rats weigh approximately lOOg.
- Baseline Ca levels are measured and the rats are administered control vehicle (saline) or compound (dissolved in saline) as a single intravenous (tail vein) bolus injection followed immediately by a single subcutaneous injection of either human parathyroid hormone 1-34 peptide (hPTHl-34, dose 0.2mg/kg in saline/0.1% bovine serum albumen, Bachem, Ca) or the PTH vehicle.
- hPTHl-34 human parathyroid hormone 1-34 peptide
- the calcemic response to PTH is measured 2h after compound/PTH administration.
- Each experimental group consists of 8-10 male Sprague-Dawley or Wistar rats of approximately 30-40g body weight at the start of the experiment.
- the agent being tested is administered by an appropriate route as single or multiple daily doses for a period of seven days.
- the rats Prior to administration of the first dose, the rats are given a single dose of a fluorescent marker (tetracycline 25mg/kg, or calcein lOmg/kg) that labels the position of bone forming surfaces at that point in time.
- a fluorescent marker tetracycline 25mg/kg, or calcein lOmg/kg
- the rats are killed and both forelimbs are removed at the elbow, the foot is removed at the ankle and the skin removed.
- the sample is frozen and mounted vertically on a microtome chuck.
- the rate of bone resorption is measured morphometrically in the medial-dorsal portion of the cortical bone. The measurement is done as follows: the amount of bone resorbed at the periosteal surface is equal to the distance by which the periosteal surface has advanced towards the fluorescent label which had been incorporated at the endosteal bone formation surface on day zero; this distance is calculated by subtracting the width of bone between the label and the periosteal surface on day 7 from the width on day zero; the resorption rate in microns per day is calculated by dividing the result by 7.
- the cells are washed x2 with cold RPMI-1640 by centrifugation (lOOOrpm, 5 mins at 4°C) and the cells are transferred to a sterile 15 ml centrifuge tube. The number of mononuclear cells are enumerated in an improved Neubauer counting chamber.
- Sufficient magnetic beads (5 / mononuclear cell), coated with goat anti-mouse IgG, are removed from their stock bottle and placed into 5 ml of fresh medium (this washes away the toxic azide preservative). The medium is removed by immobilizing the beads on a magnet and is replaced with fresh medium.
- the beads are mixed with the cells and the suspension is incubated for 30 mins on ice. The suspension is mixed frequently.
- the bead-coated cells are immobilized on a magnet and the remaining cells (osteoclast-rich fraction) are decanted into a sterile 50 ml centrifuge tube.
- Fresh medium is added to the bead-coated cells to dislodge any trapped osteoclasts. This wash process is repeated xlO. The bead-coated cells are discarded.
- the osteoclasts are enumerated in a counting chamber, using a large-bore disposable plastic pasteur to charge the chamber with the sample. • The cells are pelleted by centrifugation and the density of osteoclasts adjusted to 1.5xl0 4 /ml in EMEM medium, supplemented with 10% fetal calf serum and 1.7g/litre of sodium bicarbonate.
- the slices are washed in phosphate buffered saline and fixed in 2% gluteraldehyde (in 0.2M sodium cacodylate) for 5 mins.
- the column was washed with 50 mL cold buffer A.
- the lectin-retained GPIIb-IIIa was eluted with buffer A containing 10% dextrose. All procedures were performed at 4°C.
- the GPIIb-IIIa obtained was >95% pure as shown by SDS polyacrylamide gel electrophoresis.
- a mixture of phosphatidylserine (70%) and phosphatidylcholine (30%) (Avanti Polar Lipids) were dried to the walls of a glass tube under a stream of nitrogen.
- Purified GP ⁇ b- ⁇ la was diluted to a final concentration of 0.5 mg/mL and mixed with the phospholipids in a protein:phospholipid ratio of 1:3 (w:w). The mixture was resuspended and sonicated in a bath sonicator for 5 min.
- the mixture was then dialyzed overnight using 12,000-14,000 molecular weight cutoff dialysis tubing against a 1000-fold excess of 50 mM Tris-HCl, pH 7.4, 100 mM NaCl, 2 mM CaC12 (with 2 changes).
- the GPIIb-IIIa-containing liposomes wee centrifuged at 12,000g for 15 min and resuspended in the dialysis buffer at a final protein concentration of approximately 1 mg/mL. The liposomes were stored at -70C until needed.
- GPIIb-IIIa The binding to the fibrinogen receptor (GPIIb-IIIa) was assayed by an indirect competitive binding method using [ 3 H]-SK&F- 107260 as an RGD-type ligand.
- the binding assay was performed in a 96-well filtration plate assembly (Millipore Corporation, Bedford, MA) using 0.22 um hydrophilic durapore membranes.
- the wells were precoated with 0.2 mL of 10 ⁇ g/mL polylysine (Sigma Chemical Co., St. Louis, MO.) at room temperature for 1 h to block nonspecific binding.
- Various concentrations of unlabeled benzadiazapines were added to the wells in quadruplicate.
- [ 3 H]-SK&F- 107260 was applied to each well at a final concentration of 4.5 nM, followed by the addition of 1 ⁇ g of the purified platelet GPHb- ⁇ ia-containing liposomes. The mixtures were incubated for 1 h at room temperature. The GPIIb-IIIa-bound [3HJ-SK&F- 107260 was seperated from the unbound by filtration using a Millipore filtration manifold, followed by washing with ice-cold buffer (2 times, each 0.2 mL).
- IC50 concentration of the antagonist which inhibits specific binding of [ 3 H]-SK&F-107260 by 50% at equilibrium.
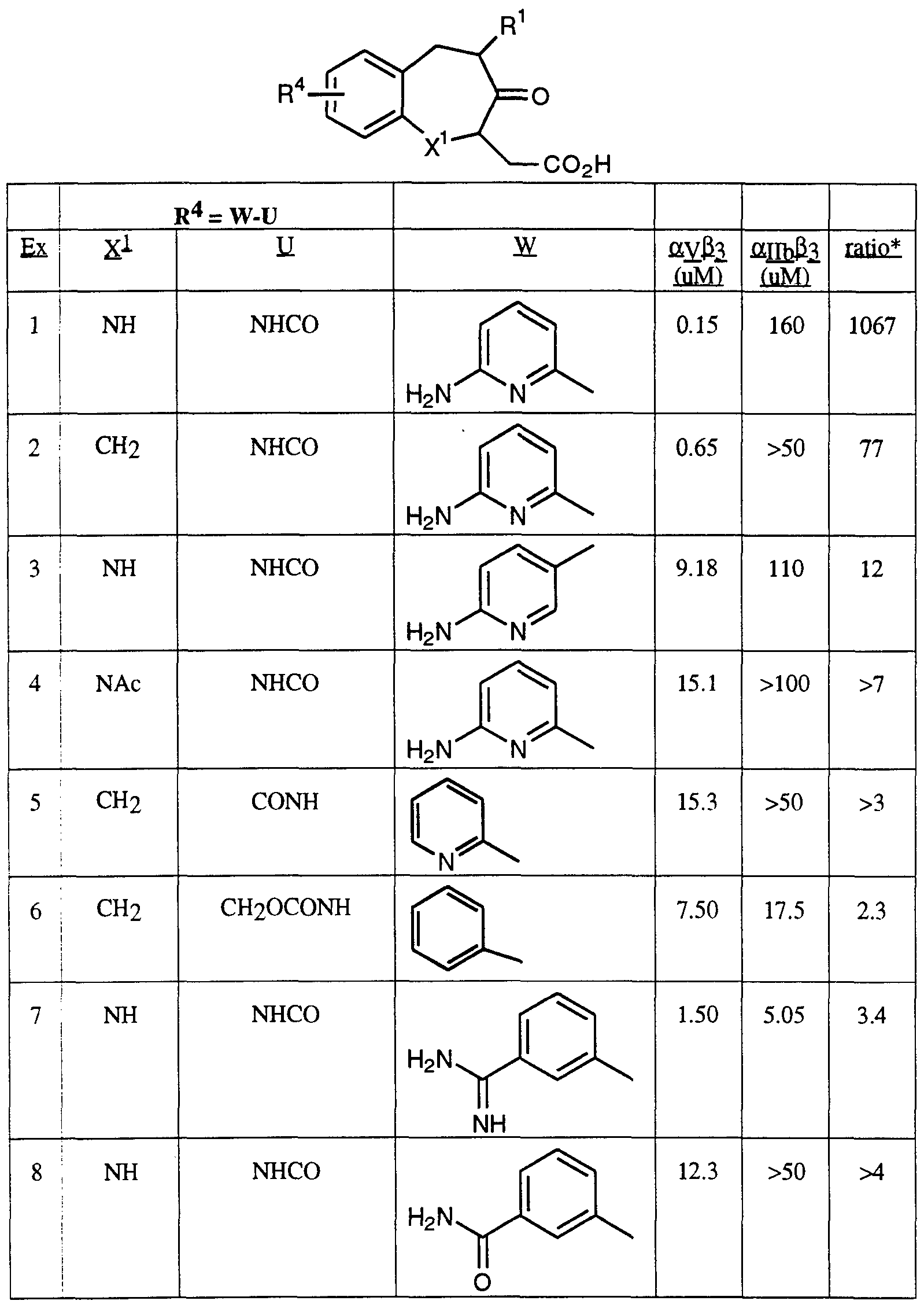
- Preferred compounds of this invention have an affinity for the vitronectin receptor relative to the fibrinogen receptor of greater than 3:1. More preferred compounds have a ratio of activity of greater than 10:1.
- Table 1 The comparative results of the enhanced binding of the compounds of this invention to the vitronecton receptor relative to the fibrinogen receptor are given in Table 1 below:
- the compounds of the instant invention were tested for their ability to inhibit the migration and proliferation of smooth muscle tissue in an artery or vein in order to assess their ability to prevent restenosis of an artery, such as that which typically occurs following angioplasty.
- Rat or human aortic smooth muscle cells were used. The cell migration was monitored in a Transwell cell culture chamber by using a polycarbonate membrane with pores of 8 urn (Costar). The lower surface of the filter was coated with vitronectin. Cells were suspended in DMEM supplemented with 0.2% bovine serum albumin at a concentration of 2.5 - 5.0 x 10 6 cells/mL, and were pretreated with test compound at various concentrations for 20 min at 20°C. The solvent alone was used as control. 0.2 mL of the cell suspension was placed in the upper compartment of the chamber. The lower compartment contained 0.6 mL of DMEM supplemented with 0.2% bovine serum albumin.
- Incubation was carried out at 37 °C in an atmosphere of 95% air/5% CO 2 for 24 hr. After incubation, the non- migrated cells on the upper surface of the filter were removed by gentle scraping. The filter was then fixed in methanol and stained with 10% Giemsa stain. Migration was measured either by a) counting the number of cells that had migrated to the lower surface of the filter or by b) extracting the stained cells with 10% acetic acid followed by determining the absorbance at 600 nM.
- Nuclear magnetic resonance spectra were recorded at either 250 or 400 MHz using, respectively, a Bruker AM 250 or Bruker AC 400 spectrometer.
- CDCI 3 is deuteriochloroform
- DMSO-d ⁇ is hexadeuteriodimethylsulfoxide
- CD 3 OD is tetradeuteriomethanol. Chemical shifts are reported in parts per million (5) downfield from the internal standard tetramethylsilane.
- ODS refers to an octadecylsilyl derivatized silica gel chromatographic support. 5 ⁇ Apex-ODS indicates an octadecylsilyl derivatized silica gel chromatographic support having a nominal particle size of 5 ⁇ , made by Jones Chromatography, Littleton, Colorado.
- YMC ODS-AQ® is an ODS chromatographic support and is a registered trademark of YMC Co. Ltd., Kyoto, Japan.
- PRP-1® is a polymeric (styrene-divinylbenzene) chromatographic support, and is a registered trademark of Hamilton Co., Reno, Nevada.
- Celite® is a filter aid composed of acid-washed diatomaceous silica, and is a registered trademark of Manville Corp., Denver, Colorado.
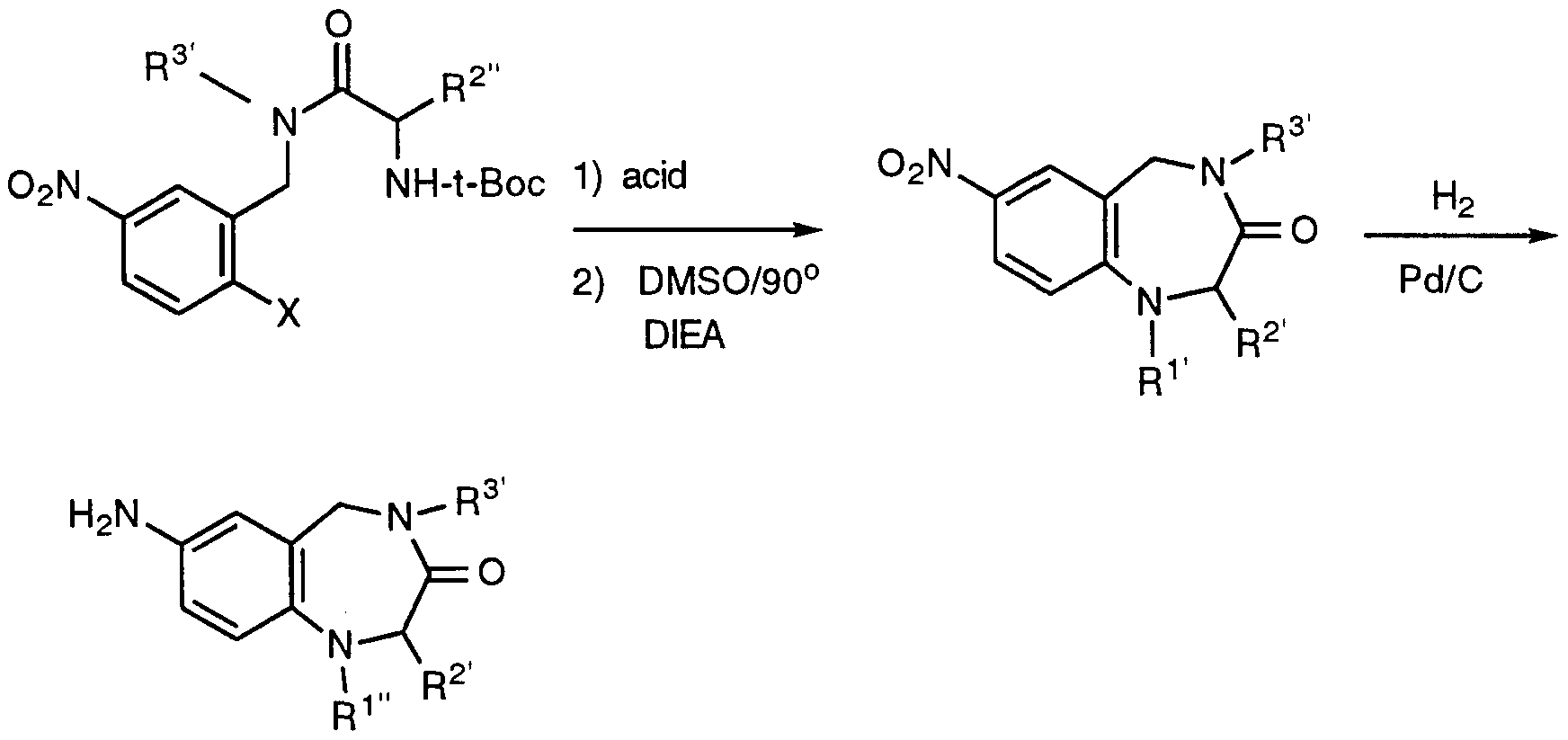
- Methyl ( ⁇ )-8-carboxy-2-methyl-3-oxo-2,3,4,5-tetrahaydro-lH-2- benzazepine-4-acetate was prepared by the method disclosed in Example 1 of WO 94/14776, except substituting 4-bromo-3-methylbenzoic acid for 3-bromo-4- methylbenzoic acid and substituting methylamine for phenethylamine therein.
- 4-(2-Methylpyridyl) piperazine was prepared by the procedure of Cross and Dickinson, US 4,806,536 (Feb. 21, 1989).
- reaction mixture was partitioned between CH2CI2 (150 mL) and 5% NaHCO3 (70 mL). The organic layer was separated, washed sequentially with 5% NaHCO3 (50 mL) and H2O (50 mL), dried (Na2SO4), and concentrated to leave a yellowish solid (0.69 g).
- Methyl 2-(2-pyridinyl)aminoacetate hydrochloride (1.0 g, 5 mmol) was partitioned between EtOAc and 10% K 2 CO 3 . The organic layer was washed with brine, dried (MgSO 4 ), and concentrated. The residue was dissolved in CH 2 C1 2 (10 mL), and di-tert-butyl dicarbonate (1.2 g, 5 mmol) was added. The reaction was stirred at RT under argon overnight, then was concentrated.
- Methyl 2-(N-tert-butoxycarbonyl-N-2-pyridinyl)aminoacetate was saponified according to the procedure of Example 6(b) to give the title compound (74%) as a white solid.
- Methyl ( ⁇ )-8-[(2-[tert-butoxycarbonyl]aminoacetyl)amino]-2-methyl-3-oxo- 2,3,4,5-tetrahydro-lH-2-benzazepine-4-acetate was saponified according to the procedure of example 6(b). The resulting product was stirred in 1 : 1 CH2CI2/TFA for 2 h, then was concentrated.
Abstract
Description


























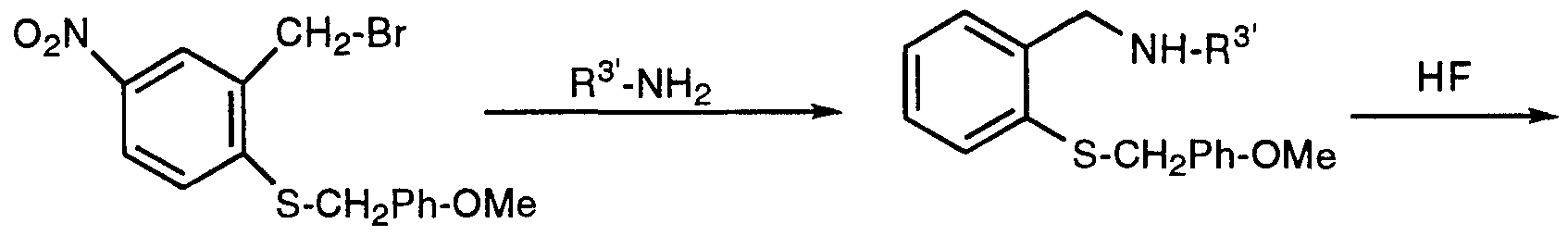




Claims





Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP95925353A EP0762882A4 (en) | 1994-06-29 | 1995-06-29 | Vitronectin receptor antagonists |
| JP8503418A JPH10504807A (en) | 1994-06-29 | 1995-06-29 | Vitronectin receptor antagonist |
| US09/679,982 US6458784B1 (en) | 1994-06-29 | 2000-10-05 | Vitronectin receptor antagonists |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US26769594A | 1994-06-29 | 1994-06-29 | |
| US08/267,695 | 1994-06-29 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US48264798A Continuation | 1994-06-29 | 1998-11-10 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1996000574A1 true WO1996000574A1 (en) | 1996-01-11 |
Family
ID=23019798
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US1995/008146 WO1996000574A1 (en) | 1994-06-29 | 1995-06-29 | Vitronectin receptor antagonists |
Country Status (4)
| Country | Link |
|---|---|
| EP (1) | EP0762882A4 (en) |
| JP (1) | JPH10504807A (en) |
| WO (1) | WO1996000574A1 (en) |
| ZA (1) | ZA955391B (en) |
Cited By (54)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0738150A1 (en) * | 1994-01-07 | 1996-10-23 | Smithkline Beecham Corporation | Bicyclic fibrinogen antagonists |
| WO1997008145A1 (en) * | 1995-08-30 | 1997-03-06 | G.D. Searle & Co. | Meta-guanidine, urea, thiourea or azacyclic amino benzoic acid derivatives as integrin antagonists |
| EP0777657A1 (en) * | 1994-08-22 | 1997-06-11 | Smithkline Beecham Corporation | Bicyclic compounds |
| EP0796855A1 (en) | 1996-03-20 | 1997-09-24 | Hoechst Aktiengesellschaft | Inhibitors of bone resorption and vitronectin receptor antagonists |
| WO1997036862A1 (en) * | 1996-03-29 | 1997-10-09 | G.D. Searle & Co. | META-SUBSTITUTED PHENYLENE DERIVATIVES AND THEIR USE AS ALPHAvBETA3 INTEGRIN ANTAGONISTS OR INHIBITORS |
| EP0820991A2 (en) * | 1996-07-24 | 1998-01-28 | Hoechst Aktiengesellschaft | Cycloalkyl derivatives as bone resorption inhibitors and vitronectin receptor antagonists |
| EP0820988A2 (en) * | 1996-07-24 | 1998-01-28 | Hoechst Aktiengesellschaft | Imino derivatives as bone resorption inhibitors and vitronectin receptor antagonists |
| WO1998014192A1 (en) * | 1996-10-02 | 1998-04-09 | Smithkline Beecham Corporation | Vitronectin receptor antagonists |
| WO1998015278A1 (en) * | 1996-10-07 | 1998-04-16 | Smithkline Beecham Corporation | Method for stimulating bone formation |
| EP0854145A2 (en) * | 1996-12-20 | 1998-07-22 | Hoechst Aktiengesellschaft | Vitronectin receptor antagonists, their production and their use |
| EP0854140A2 (en) | 1996-12-20 | 1998-07-22 | Hoechst Aktiengesellschaft | Vitronectin receptor antagonists, their production and their use |
| WO1998046220A1 (en) * | 1997-04-14 | 1998-10-22 | Merck & Co., Inc. | Combination therapy for the prevention and treatment of osteoporosis |
| EP0895475A1 (en) * | 1995-12-29 | 1999-02-10 | Smithkline Beecham Corporation | Vitronectin receptor antagonists |
| EP0906103A4 (en) * | 1995-12-29 | 1999-04-07 | ||
| EP0928194A1 (en) * | 1996-08-29 | 1999-07-14 | Merck & Co., Inc. | Compositions and methods for administering integrin receptor antagonists |
| US5939412A (en) * | 1992-06-26 | 1999-08-17 | Smithkline Beecham Corporation | Bicyclic fibrinogen antagonists |
| US5977101A (en) * | 1995-06-29 | 1999-11-02 | Smithkline Beecham Corporation | Benzimidazoles/Imidazoles Linked to a Fibrinogen Receptor Antagonist Template Having Vitronectin Receptor Antagonist Activity |
| EP1007051A1 (en) * | 1997-08-04 | 2000-06-14 | Smithkline Beecham Corporation | Integrin receptor antagonists |
| WO2000035488A2 (en) * | 1998-12-18 | 2000-06-22 | Du Pont Pharmaceuticals Company | Vitronectin receptor antagonist pharmaceuticals |
| WO2000035492A2 (en) * | 1998-12-18 | 2000-06-22 | Du Pont Pharmaceuticals Company | Vitronectin receptor antagonist pharmaceuticals |
| EP1017387A1 (en) * | 1997-09-24 | 2000-07-12 | Smithkline Beecham Corporation | Vitronectin receptor antagonist |
| US6100423A (en) * | 1995-08-30 | 2000-08-08 | G. D. Searle & Co. | Amino benzenepropanoic acid compounds and derivatives thereof |
| WO2000046215A1 (en) * | 1999-02-03 | 2000-08-10 | Merck & Co., Inc. | Benzazepine derivatives as alpha-v integrin receptor antagonists |
| WO2000035887A3 (en) * | 1998-12-18 | 2000-11-16 | Du Pont Pharmaceuticals Company | Vitronectin receptor antagonist pharmaceuticals |
| US6160099A (en) * | 1998-11-24 | 2000-12-12 | Jonak; Zdenka Ludmila | Anti-human αv β3 and αv β5 antibodies |
| US6218387B1 (en) | 1996-12-20 | 2001-04-17 | Hoechst Aktiengesellschaft | Vitronectin receptor anatagonists, their preparation and their use |
| US6313119B1 (en) | 1998-01-23 | 2001-11-06 | Adventis Pharma Deutschland Gmbh | Sulfonamide derivatives as inhibitors of bone resorption and as inhibitors of cell adhesion |
| US6322770B1 (en) | 1998-03-31 | 2001-11-27 | Dupont Pharmaceuticals Company | Indazole vitronectin receptor antagonist pharmaceuticals |
| WO2001097861A2 (en) * | 2000-06-21 | 2001-12-27 | Bristol-Myers Squibb Pharma Company | Vitronectin receptor antagonist pharmaceuticals |
| US6340679B1 (en) | 1999-02-13 | 2002-01-22 | Aventis Pharma Deutschland Gmbh | Guanidine derivatives as inhibitors of cell adhesion |
| WO2002018377A1 (en) * | 2000-08-29 | 2002-03-07 | Pharmacia Corporation | Compounds containing a bicyclic ring system useful as alpha v beta 3 antagonists |
| US6458784B1 (en) | 1994-06-29 | 2002-10-01 | Smithkline Beecham Corporation | Vitronectin receptor antagonists |
| US6482821B2 (en) | 1996-12-20 | 2002-11-19 | Hoechst Aktiengellschaft | Vitronectin receptor antagonists, their preparation and their use |
| US6511649B1 (en) | 1998-12-18 | 2003-01-28 | Thomas D. Harris | Vitronectin receptor antagonist pharmaceuticals |
| US6524553B2 (en) | 1998-03-31 | 2003-02-25 | Bristol-Myers Squibb Pharma Company | Quinolone vitronectin receptor antagonist pharmaceuticals |
| US6537520B1 (en) | 1998-03-31 | 2003-03-25 | Bristol-Myers Squibb Pharma Company | Pharmaceuticals for the imaging of angiogenic disorders |
| US6548663B1 (en) | 1998-03-31 | 2003-04-15 | Bristol-Myers Squibb Pharma Company | Benzodiazepine vitronectin receptor antagonist pharmaceuticals |
| US6569402B1 (en) | 1998-12-18 | 2003-05-27 | Bristol-Myers Squibb Pharma Company | Vitronectin receptor antagonist pharmaceuticals |
| US6723727B1 (en) * | 1996-12-20 | 2004-04-20 | Hoechst Aktiengesellschaft | Substituted purine derivatives, processes for their preparation, their use, and compositions comprising them |
| FR2847254A1 (en) * | 2002-11-19 | 2004-05-21 | Aventis Pharma Sa | New 4-amino-6-piperidinyl-pyrimidine derivatives are vitronectin receptor antagonists useful e.g. for treating osteoporosis, cancer metastasis, restenosis or retinopathy |
| US6794518B1 (en) | 1998-12-18 | 2004-09-21 | Bristol-Myers Squibb Pharma Company | Vitronectin receptor antagonist pharmaceuticals |
| EP1466615A1 (en) | 1998-09-16 | 2004-10-13 | MERCK PATENT GmbH | Pharmaceutical composition |
| US6825188B2 (en) | 1996-10-02 | 2004-11-30 | Smithkline Beecham Corporation | Vitronectin receptor antagonists |
| US6833373B1 (en) | 1998-12-23 | 2004-12-21 | G.D. Searle & Co. | Method of using an integrin antagonist and one or more antineoplastic agents as a combination therapy in the treatment of neoplasia |
| US6858598B1 (en) | 1998-12-23 | 2005-02-22 | G. D. Searle & Co. | Method of using a matrix metalloproteinase inhibitor and one or more antineoplastic agents as a combination therapy in the treatment of neoplasia |
| WO2005120477A2 (en) | 2004-06-07 | 2005-12-22 | Merck & Co., Inc. | N- (2-benzyl) -2-phenylbutanamides as androgen receptor modulators |
| WO2007084670A2 (en) | 2006-01-18 | 2007-07-26 | Merck Patent Gmbh | Specific therapy using integrin ligands for treating cancer |
| WO2008087025A2 (en) | 2007-01-18 | 2008-07-24 | Merck Patent Gmbh | Specific therapy and medicament using integrin ligands for treating cancer |
| WO2009063990A1 (en) | 2007-11-16 | 2009-05-22 | Ube Industries, Ltd. | Benzazepinone compound |
| WO2010136168A2 (en) | 2009-05-25 | 2010-12-02 | Merck Patent Gmbh | Continuous administration of integrin ligands for treating cancer |
| US7846932B2 (en) | 2004-05-18 | 2010-12-07 | Galapagos Sas | Methods for the use of pyrimidine derivatives which are antagonists of vitronectin receptor |
| EP2292251A1 (en) | 2001-04-24 | 2011-03-09 | Merck Patent GmbH | Combination therapy using anti-angiogenic agents and TNF-alpha |
| WO2015181676A1 (en) | 2014-05-30 | 2015-12-03 | Pfizer Inc. | Carbonitrile derivatives as selective androgen receptor modulators |
| WO2023275715A1 (en) | 2021-06-30 | 2023-01-05 | Pfizer Inc. | Metabolites of selective androgen receptor modulators |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1993000095A2 (en) * | 1991-06-28 | 1993-01-07 | Smithkline Beecham Corporation | Bicyclic fibrinogen antagonists |
| WO1993008174A1 (en) * | 1991-10-18 | 1993-04-29 | Genentech, Inc. | NONPEPTIDYL INTEGRIN INHIBITORS HAVING SPECIFICITY FOR THE GPIIbIIIa RECEPTOR |
| WO1994014776A2 (en) * | 1992-12-21 | 1994-07-07 | Smithkline Beecham Corporation | Bicyclic fibrinogen antagonists |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1995004534A1 (en) * | 1993-08-06 | 1995-02-16 | Smithkline Beecham Corporation | Endothelin receptor antagonists |
| MA23420A1 (en) * | 1994-01-07 | 1995-10-01 | Smithkline Beecham Corp | BICYCLIC FIBRINOGEN ANTAGONISTS. |
-
1995
- 1995-06-29 JP JP8503418A patent/JPH10504807A/en not_active Ceased
- 1995-06-29 ZA ZA955391A patent/ZA955391B/en unknown
- 1995-06-29 WO PCT/US1995/008146 patent/WO1996000574A1/en not_active Application Discontinuation
- 1995-06-29 EP EP95925353A patent/EP0762882A4/en not_active Withdrawn
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1993000095A2 (en) * | 1991-06-28 | 1993-01-07 | Smithkline Beecham Corporation | Bicyclic fibrinogen antagonists |
| WO1993008174A1 (en) * | 1991-10-18 | 1993-04-29 | Genentech, Inc. | NONPEPTIDYL INTEGRIN INHIBITORS HAVING SPECIFICITY FOR THE GPIIbIIIa RECEPTOR |
| WO1994014776A2 (en) * | 1992-12-21 | 1994-07-07 | Smithkline Beecham Corporation | Bicyclic fibrinogen antagonists |
Non-Patent Citations (5)
| Title |
|---|
| BIOORGANIC AND MEDICINAL CHEMISTRY, Volume 2, Number 9, issued 1994, BONDINELL et al., "Design of a Potent and Orally Active Nonpeptide Platelet Fibrinogen Receptor (GPIIb/IIIa) Antagonist", pages 897-908. * |
| JOURNAL OF AMERICAN CHEMICAL SOCIETY, Volume 115, Number 19, issued September 1993, KU et al., "Direct Design of a Potent Non-Peptide Fibrinogen Receptor Antagonist Based on the Structure and Conformation of a Highly Constrained Cyclic RGD Peptide", pages 8861-8862. * |
| JOURNAL OF MEDICINAL CHEMISTRY, Volume 38, Number 1, issued January 1995, KU et al., "Potent Non-Peptide Fibrinogen Receptor Antagonists Which Present an Alternative Pharmacophore", pages 9-12. * |
| See also references of EP0762882A4 * |
| TETRAHEDRON LETTERS, Volume 36, Number 3, issued 1995, MILLER et al., "Synthesis of a 2-Benzazepine Analog of a Potent, Nonpeptide GPIIb/IIIa Antagonist", pages 373-376. * |
Cited By (115)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6127359A (en) * | 1991-06-28 | 2000-10-03 | Smithkline Beecham Corporation | Bicyclic fibrinogen antagonists |
| US5939412A (en) * | 1992-06-26 | 1999-08-17 | Smithkline Beecham Corporation | Bicyclic fibrinogen antagonists |
| EP0738150A4 (en) * | 1994-01-07 | 1997-03-19 | Smithkline Beecham Corp | Bicyclic fibrinogen antagonists |
| US6117866A (en) * | 1994-01-07 | 2000-09-12 | Smithkline Beecham Corporation | Bicyclic fibrinogen antagonists |
| EP0738150A1 (en) * | 1994-01-07 | 1996-10-23 | Smithkline Beecham Corporation | Bicyclic fibrinogen antagonists |
| US6458784B1 (en) | 1994-06-29 | 2002-10-01 | Smithkline Beecham Corporation | Vitronectin receptor antagonists |
| EP0777657A1 (en) * | 1994-08-22 | 1997-06-11 | Smithkline Beecham Corporation | Bicyclic compounds |
| EP0777657A4 (en) * | 1994-08-22 | 1997-07-16 | ||
| US5977101A (en) * | 1995-06-29 | 1999-11-02 | Smithkline Beecham Corporation | Benzimidazoles/Imidazoles Linked to a Fibrinogen Receptor Antagonist Template Having Vitronectin Receptor Antagonist Activity |
| US6831199B1 (en) | 1995-08-30 | 2004-12-14 | G. D. Searle & Co. | Pyrimidine compounds and derivatives thereof |
| US6100423A (en) * | 1995-08-30 | 2000-08-08 | G. D. Searle & Co. | Amino benzenepropanoic acid compounds and derivatives thereof |
| US6028223A (en) * | 1995-08-30 | 2000-02-22 | G. D. Searle & Co. | Meta-guanidine, urea, thiourea or azacyclic amino benzoic acid compounds and derivatives thereof |
| WO1997008145A1 (en) * | 1995-08-30 | 1997-03-06 | G.D. Searle & Co. | Meta-guanidine, urea, thiourea or azacyclic amino benzoic acid derivatives as integrin antagonists |
| EP0906103A4 (en) * | 1995-12-29 | 1999-04-07 | ||
| EP0895475A1 (en) * | 1995-12-29 | 1999-02-10 | Smithkline Beecham Corporation | Vitronectin receptor antagonists |
| US6159964A (en) * | 1995-12-29 | 2000-12-12 | Smithkline Beecham Corporation | Vitronectin receptor antagonists |
| EP0906103A1 (en) * | 1995-12-29 | 1999-04-07 | Smithkline Beecham Corporation | Vitronectin receptor antagonists |
| EP0895475A4 (en) * | 1995-12-29 | 2000-08-23 | Smithkline Beecham Corp | Vitronectin receptor antagonists |
| US6620820B2 (en) | 1996-03-20 | 2003-09-16 | Hoechst Aktiengesellschaft | Inhibitors of bone reabsorption and antagonists of vitronectin receptors |
| EP0796855A1 (en) | 1996-03-20 | 1997-09-24 | Hoechst Aktiengesellschaft | Inhibitors of bone resorption and vitronectin receptor antagonists |
| WO1997036862A1 (en) * | 1996-03-29 | 1997-10-09 | G.D. Searle & Co. | META-SUBSTITUTED PHENYLENE DERIVATIVES AND THEIR USE AS ALPHAvBETA3 INTEGRIN ANTAGONISTS OR INHIBITORS |
| US6399620B1 (en) | 1996-07-24 | 2002-06-04 | Aventis Pharma S.A. | Cycloalkyl derivatives as inhibitors of bone resporption and vitronectin receptor antagonists |
| EP0820988A2 (en) * | 1996-07-24 | 1998-01-28 | Hoechst Aktiengesellschaft | Imino derivatives as bone resorption inhibitors and vitronectin receptor antagonists |
| US6005117A (en) * | 1996-07-24 | 1999-12-21 | Hoechst Aktiengesellschaft | Imino compounds, process for their preparation and their use as victronectin antagonists |
| US7348333B2 (en) | 1996-07-24 | 2008-03-25 | Aventis Pharma S.A. | Cycloalkyl derivatives as inhibitors of bone resorption and vitronectin receptor antagonists |
| EP0820988A3 (en) * | 1996-07-24 | 2000-05-17 | Hoechst Aktiengesellschaft | Imino derivatives as bone resorption inhibitors and vitronectin receptor antagonists |
| EP0820991A3 (en) * | 1996-07-24 | 2000-05-17 | Hoechst Aktiengesellschaft | Cycloalkyl derivatives as bone resorption inhibitors and vitronectin receptor antagonists |
| EP0820991A2 (en) * | 1996-07-24 | 1998-01-28 | Hoechst Aktiengesellschaft | Cycloalkyl derivatives as bone resorption inhibitors and vitronectin receptor antagonists |
| EP0928194A1 (en) * | 1996-08-29 | 1999-07-14 | Merck & Co., Inc. | Compositions and methods for administering integrin receptor antagonists |
| EP0928194A4 (en) * | 1996-08-29 | 2001-01-17 | Merck & Co Inc | Compositions and methods for administering integrin receptor antagonists |
| US6825188B2 (en) | 1996-10-02 | 2004-11-30 | Smithkline Beecham Corporation | Vitronectin receptor antagonists |
| AU733417B2 (en) * | 1996-10-02 | 2001-05-17 | Smithkline Beecham Corporation | Vitronectin receptor antagonists |
| KR100589578B1 (en) * | 1996-10-02 | 2006-06-15 | 스미스클라인 비참 코포레이션 | Vitronectin Receptor Antagonist |
| JP2001501936A (en) * | 1996-10-02 | 2001-02-13 | スミスクライン・ビーチャム・コーポレイション | Vitronectin receptor antagonist |
| CN1114403C (en) * | 1996-10-02 | 2003-07-16 | 史密丝克莱恩比彻姆公司 | Vitronectin receptor antagonists |
| CZ299076B6 (en) * | 1996-10-02 | 2008-04-16 | Smithkline Beecham Corporation | Vitronectin derivatives |
| WO1998014192A1 (en) * | 1996-10-02 | 1998-04-09 | Smithkline Beecham Corporation | Vitronectin receptor antagonists |
| JP2010006838A (en) * | 1996-10-02 | 2010-01-14 | Smithkline Beecham Corp | Vitronectin receptor antagonist |
| EA002419B1 (en) * | 1996-10-02 | 2002-04-25 | Смитклайн Бичам Корпорейшн | Vitronectin receptor antagonists |
| EP0946180A4 (en) * | 1996-10-07 | 2003-07-23 | Smithkline Beecham Corp | Method for stimulating bone formation |
| WO1998015278A1 (en) * | 1996-10-07 | 1998-04-16 | Smithkline Beecham Corporation | Method for stimulating bone formation |
| EP0946180A1 (en) * | 1996-10-07 | 1999-10-06 | Smithkline Beecham Corporation | Method for stimulating bone formation |
| US6218387B1 (en) | 1996-12-20 | 2001-04-17 | Hoechst Aktiengesellschaft | Vitronectin receptor anatagonists, their preparation and their use |
| EP0854145A3 (en) * | 1996-12-20 | 2000-03-22 | Hoechst Aktiengesellschaft | Vitronectin receptor antagonists, their production and their use |
| US6723727B1 (en) * | 1996-12-20 | 2004-04-20 | Hoechst Aktiengesellschaft | Substituted purine derivatives, processes for their preparation, their use, and compositions comprising them |
| EP0854145A2 (en) * | 1996-12-20 | 1998-07-22 | Hoechst Aktiengesellschaft | Vitronectin receptor antagonists, their production and their use |
| US6011045A (en) * | 1996-12-20 | 2000-01-04 | Hoechst Aktiengesellschaft | Vitronectin receptor antagonists, their preparation and their use |
| US5990145A (en) * | 1996-12-20 | 1999-11-23 | Hoechst Aktiengesellschaft | Vitronectin receptor antagonists, their preparation and their use |
| US6482821B2 (en) | 1996-12-20 | 2002-11-19 | Hoechst Aktiengellschaft | Vitronectin receptor antagonists, their preparation and their use |
| EP0854140A2 (en) | 1996-12-20 | 1998-07-22 | Hoechst Aktiengesellschaft | Vitronectin receptor antagonists, their production and their use |
| EP0854140A3 (en) * | 1996-12-20 | 2000-03-08 | Hoechst Aktiengesellschaft | Vitronectin receptor antagonists, their production and their use |
| WO1998046220A1 (en) * | 1997-04-14 | 1998-10-22 | Merck & Co., Inc. | Combination therapy for the prevention and treatment of osteoporosis |
| EP1007051A1 (en) * | 1997-08-04 | 2000-06-14 | Smithkline Beecham Corporation | Integrin receptor antagonists |
| EP1007051A4 (en) * | 1997-08-04 | 2001-08-29 | Smithkline Beecham Corp | Integrin receptor antagonists |
| EP1017387A4 (en) * | 1997-09-24 | 2004-08-18 | Smithkline Beecham Corp | Vitronectin receptor antagonist |
| EP1017387A1 (en) * | 1997-09-24 | 2000-07-12 | Smithkline Beecham Corporation | Vitronectin receptor antagonist |
| US6747148B2 (en) | 1998-01-23 | 2004-06-08 | Hoechst Marion Roussel Deutschland Gmbh | Sulfonamide derivatives as inhibitors of bone resorption and as inhibitors of cell adhesion |
| US6313119B1 (en) | 1998-01-23 | 2001-11-06 | Adventis Pharma Deutschland Gmbh | Sulfonamide derivatives as inhibitors of bone resorption and as inhibitors of cell adhesion |
| US7052673B2 (en) | 1998-03-31 | 2006-05-30 | Bristol-Myers Squibb Pharma Company | Pharmaceuticals for the imaging of angiogenic disorders |
| US6524553B2 (en) | 1998-03-31 | 2003-02-25 | Bristol-Myers Squibb Pharma Company | Quinolone vitronectin receptor antagonist pharmaceuticals |
| US6322770B1 (en) | 1998-03-31 | 2001-11-27 | Dupont Pharmaceuticals Company | Indazole vitronectin receptor antagonist pharmaceuticals |
| US6537520B1 (en) | 1998-03-31 | 2003-03-25 | Bristol-Myers Squibb Pharma Company | Pharmaceuticals for the imaging of angiogenic disorders |
| US6548663B1 (en) | 1998-03-31 | 2003-04-15 | Bristol-Myers Squibb Pharma Company | Benzodiazepine vitronectin receptor antagonist pharmaceuticals |
| EP1466615A1 (en) | 1998-09-16 | 2004-10-13 | MERCK PATENT GmbH | Pharmaceutical composition |
| US6160099A (en) * | 1998-11-24 | 2000-12-12 | Jonak; Zdenka Ludmila | Anti-human αv β3 and αv β5 antibodies |
| US6818201B2 (en) | 1998-12-18 | 2004-11-16 | Bristol-Myers Squibb Pharma Company | Vitronectin receptor antagonist pharmaceuticals |
| US7321045B2 (en) | 1998-12-18 | 2008-01-22 | Bristol-Myers Squibb Pharma Company | Vitronectin receptor antagonist pharmaceuticals |
| US6569402B1 (en) | 1998-12-18 | 2003-05-27 | Bristol-Myers Squibb Pharma Company | Vitronectin receptor antagonist pharmaceuticals |
| WO2000035492A2 (en) * | 1998-12-18 | 2000-06-22 | Du Pont Pharmaceuticals Company | Vitronectin receptor antagonist pharmaceuticals |
| US6683163B2 (en) | 1998-12-18 | 2004-01-27 | Bristol-Myers Squibb Pharma Company | Vitronectin receptor antagonist pharmaceuticals |
| US6689337B2 (en) | 1998-12-18 | 2004-02-10 | Bristol-Myers Squibb Pharma Company | Vitronectin receptor antagonist pharmaceuticals |
| US6558649B1 (en) | 1998-12-18 | 2003-05-06 | Bristol-Myers Squibb Pharma Company | Vitronectin receptor antagonist pharmaceuticals |
| WO2000035887A3 (en) * | 1998-12-18 | 2000-11-16 | Du Pont Pharmaceuticals Company | Vitronectin receptor antagonist pharmaceuticals |
| US6743412B2 (en) | 1998-12-18 | 2004-06-01 | Bristol-Myers Squibb Pharma Company | Vitronectin receptor antagonist pharmaceuticals |
| US7332149B1 (en) | 1998-12-18 | 2008-02-19 | Bristol-Myers Squibb Pharma Company | Vitronectin receptor antagonist pharmaceuticals |
| US7090828B2 (en) | 1998-12-18 | 2006-08-15 | Bristol-Myers Squibb Pharma Company | Vitronectin receptor antagonist pharmaceuticals |
| US6511648B2 (en) | 1998-12-18 | 2003-01-28 | Bristol-Myers Squibb Pharma Company | Vitronectin receptor antagonist pharmaceuticals |
| US6794518B1 (en) | 1998-12-18 | 2004-09-21 | Bristol-Myers Squibb Pharma Company | Vitronectin receptor antagonist pharmaceuticals |
| US6511649B1 (en) | 1998-12-18 | 2003-01-28 | Thomas D. Harris | Vitronectin receptor antagonist pharmaceuticals |
| WO2000035492A3 (en) * | 1998-12-18 | 2001-01-18 | Du Pont Pharm Co | Vitronectin receptor antagonist pharmaceuticals |
| WO2000035488A2 (en) * | 1998-12-18 | 2000-06-22 | Du Pont Pharmaceuticals Company | Vitronectin receptor antagonist pharmaceuticals |
| WO2000035488A3 (en) * | 1998-12-18 | 2000-11-09 | Du Pont Pharm Co | Vitronectin receptor antagonist pharmaceuticals |
| US7018611B2 (en) | 1998-12-18 | 2006-03-28 | Bristol-Myers Squibb Pharma Company | Vitronectin receptor antagonist pharmaceuticals |
| US6833373B1 (en) | 1998-12-23 | 2004-12-21 | G.D. Searle & Co. | Method of using an integrin antagonist and one or more antineoplastic agents as a combination therapy in the treatment of neoplasia |
| US6858598B1 (en) | 1998-12-23 | 2005-02-22 | G. D. Searle & Co. | Method of using a matrix metalloproteinase inhibitor and one or more antineoplastic agents as a combination therapy in the treatment of neoplasia |
| US6232308B1 (en) | 1999-02-03 | 2001-05-15 | Merck & Co., Inc. | Bezazepine derivatives as αv integrin receptor antagonists |
| WO2000046215A1 (en) * | 1999-02-03 | 2000-08-10 | Merck & Co., Inc. | Benzazepine derivatives as alpha-v integrin receptor antagonists |
| US6340679B1 (en) | 1999-02-13 | 2002-01-22 | Aventis Pharma Deutschland Gmbh | Guanidine derivatives as inhibitors of cell adhesion |
| WO2001097861A2 (en) * | 2000-06-21 | 2001-12-27 | Bristol-Myers Squibb Pharma Company | Vitronectin receptor antagonist pharmaceuticals |
| WO2001097861A3 (en) * | 2000-06-21 | 2003-02-27 | Bristol Myers Squibb Pharma Co | Vitronectin receptor antagonist pharmaceuticals |
| WO2002018377A1 (en) * | 2000-08-29 | 2002-03-07 | Pharmacia Corporation | Compounds containing a bicyclic ring system useful as alpha v beta 3 antagonists |
| EP2292251A1 (en) | 2001-04-24 | 2011-03-09 | Merck Patent GmbH | Combination therapy using anti-angiogenic agents and TNF-alpha |
| FR2847254A1 (en) * | 2002-11-19 | 2004-05-21 | Aventis Pharma Sa | New 4-amino-6-piperidinyl-pyrimidine derivatives are vitronectin receptor antagonists useful e.g. for treating osteoporosis, cancer metastasis, restenosis or retinopathy |
| KR101140752B1 (en) | 2002-11-19 | 2012-05-03 | 갈라파고스 엔.브이. | Novel vitronectin receptor antagonist derivatives, method for preparing same, use thereof as medicines and pharmaceutical compositions containing same |
| EP2070914A1 (en) | 2002-11-19 | 2009-06-17 | Galapagos SAS | New antagonist derivatives of the vitronectin receiver, method for preparing same, application of same as medicine and pharmaceutical compositions containing them |
| US7763621B2 (en) | 2002-11-19 | 2010-07-27 | Galapagos Sas | Vitronectin receptor antagonist derivatives, method for preparing same, use thereof as medicines and pharmaceutical compositions containing same |
| WO2004048375A1 (en) * | 2002-11-19 | 2004-06-10 | Proskelia Sas | Novel vitronectin receptor antagonist derivatives, method for preparing same, use thereof as medicines and pharmaceutical compositions containing same |
| EP2368891A1 (en) | 2004-05-18 | 2011-09-28 | Galapagos SAS | Pyrimidines derivatives as antagonists of the vitronectine receptors |
| US8765766B2 (en) | 2004-05-18 | 2014-07-01 | Galapagos Nv | Pyrimidine derivatives which are antagonists of the vitronectin receptor |
| US8133896B2 (en) | 2004-05-18 | 2012-03-13 | Galapagos Nv | Pyrimidine derivatives which are antagonist of the vitronectin receptor |
| US7846932B2 (en) | 2004-05-18 | 2010-12-07 | Galapagos Sas | Methods for the use of pyrimidine derivatives which are antagonists of vitronectin receptor |
| WO2005120477A2 (en) | 2004-06-07 | 2005-12-22 | Merck & Co., Inc. | N- (2-benzyl) -2-phenylbutanamides as androgen receptor modulators |
| WO2007084670A2 (en) | 2006-01-18 | 2007-07-26 | Merck Patent Gmbh | Specific therapy using integrin ligands for treating cancer |
| EP2335733A1 (en) | 2006-01-18 | 2011-06-22 | Merck Patent GmbH | Specific therapy using integrin ligands for treating cancer |
| EP2338518A1 (en) | 2006-01-18 | 2011-06-29 | Merck Patent GmbH | Specific therapy using integrin ligands for treating cancer |
| EP2441464A1 (en) | 2007-01-18 | 2012-04-18 | Merck Patent GmbH | Specific therapy and medicament using integrin ligands for treating cancer |
| WO2008087025A2 (en) | 2007-01-18 | 2008-07-24 | Merck Patent Gmbh | Specific therapy and medicament using integrin ligands for treating cancer |
| EP2578225A1 (en) | 2007-07-18 | 2013-04-10 | Merck Patent GmbH | Specific Therapy and Medicament Using Integrin Ligands for Treating Cancer |
| KR20100106391A (en) | 2007-11-16 | 2010-10-01 | 우베 고산 가부시키가이샤 | Benzazepinone compound |
| WO2009063990A1 (en) | 2007-11-16 | 2009-05-22 | Ube Industries, Ltd. | Benzazepinone compound |
| US8377922B2 (en) | 2007-11-16 | 2013-02-19 | Ube Industries, Ltd. | Benzazepinone compound |
| WO2010136168A2 (en) | 2009-05-25 | 2010-12-02 | Merck Patent Gmbh | Continuous administration of integrin ligands for treating cancer |
| WO2015181676A1 (en) | 2014-05-30 | 2015-12-03 | Pfizer Inc. | Carbonitrile derivatives as selective androgen receptor modulators |
| US10328082B2 (en) | 2014-05-30 | 2019-06-25 | Pfizer Inc. | Methods of use and combinations |
| WO2023275715A1 (en) | 2021-06-30 | 2023-01-05 | Pfizer Inc. | Metabolites of selective androgen receptor modulators |
Also Published As
| Publication number | Publication date |
|---|---|
| JPH10504807A (en) | 1998-05-12 |
| ZA955391B (en) | 1996-02-09 |
| EP0762882A4 (en) | 2002-09-11 |
| EP0762882A1 (en) | 1997-03-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0762882A1 (en) | Vitronectin receptor antagonists | |
| US6239138B1 (en) | Vitronectin receptor antagonist | |
| CA2192764C (en) | Integrin receptor antagonists | |
| EP0674623B1 (en) | Bicyclic fibrinogen antagonists | |
| US6159964A (en) | Vitronectin receptor antagonists | |
| WO1996026190A1 (en) | Integrin receptor antagonists | |
| EP1007051A1 (en) | Integrin receptor antagonists | |
| AU1354097A (en) | Vitronectin receptor antagonists | |
| US6117910A (en) | Bicyclic fibrinogen antagonists | |
| EP0946180A1 (en) | Method for stimulating bone formation | |
| US5977101A (en) | Benzimidazoles/Imidazoles Linked to a Fibrinogen Receptor Antagonist Template Having Vitronectin Receptor Antagonist Activity | |
| EP1027337A1 (en) | Integrin receptor antagonists | |
| US6008213A (en) | Integrin receptor antagonists | |
| US6458784B1 (en) | Vitronectin receptor antagonists | |
| US6458814B1 (en) | Vitronectin receptor antagonists | |
| US6403578B1 (en) | Bicyclic fibrinogen antagonists | |
| US20010034445A1 (en) | Vitronectin receptor antagonists | |
| US20020032187A1 (en) | Method for stimulating bone formation | |
| KR100459621B1 (en) | Integrin Receptor Antagonists | |
| US20020055499A1 (en) | Integrin receptor antagonists | |
| PL191595B1 (en) | Antagonistic integrin receptors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): JP US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LU MC NL PT SE |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| ENP | Entry into the national phase |
Ref country code: US Ref document number: 1996 765753 Date of ref document: 19961220 Kind code of ref document: A Format of ref document f/p: F |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1995925353 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1995925353 Country of ref document: EP |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1995925353 Country of ref document: EP |