JP6983771B2 - 幹細胞からt細胞を作製する方法および該t細胞を用いた免疫療法的方法 - Google Patents
幹細胞からt細胞を作製する方法および該t細胞を用いた免疫療法的方法 Download PDFInfo
- Publication number
- JP6983771B2 JP6983771B2 JP2018522655A JP2018522655A JP6983771B2 JP 6983771 B2 JP6983771 B2 JP 6983771B2 JP 2018522655 A JP2018522655 A JP 2018522655A JP 2018522655 A JP2018522655 A JP 2018522655A JP 6983771 B2 JP6983771 B2 JP 6983771B2
- Authority
- JP
- Japan
- Prior art keywords
- cells
- cell
- tcr
- ato
- antigen
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 210000001744 T-lymphocyte Anatomy 0.000 title claims description 485
- 238000000034 method Methods 0.000 title claims description 229
- 210000000130 stem cell Anatomy 0.000 title claims description 195
- 238000004519 manufacturing process Methods 0.000 title description 21
- 230000001024 immunotherapeutic effect Effects 0.000 title description 2
- 210000004027 cell Anatomy 0.000 claims description 490
- 239000000427 antigen Substances 0.000 claims description 193
- 102000036639 antigens Human genes 0.000 claims description 186
- 108091007433 antigens Proteins 0.000 claims description 186
- 210000002536 stromal cell Anatomy 0.000 claims description 104
- 239000000203 mixture Substances 0.000 claims description 98
- 108010019670 Chimeric Antigen Receptors Proteins 0.000 claims description 77
- 210000004700 fetal blood Anatomy 0.000 claims description 75
- 239000003446 ligand Substances 0.000 claims description 69
- 206010028980 Neoplasm Diseases 0.000 claims description 52
- 238000000338 in vitro Methods 0.000 claims description 46
- 230000014509 gene expression Effects 0.000 claims description 45
- 108091008874 T cell receptors Proteins 0.000 claims description 38
- 102000016266 T-Cell Antigen Receptors Human genes 0.000 claims description 37
- 210000003958 hematopoietic stem cell Anatomy 0.000 claims description 37
- 201000011510 cancer Diseases 0.000 claims description 34
- 210000001185 bone marrow Anatomy 0.000 claims description 30
- 210000005024 intraepithelial lymphocyte Anatomy 0.000 claims description 29
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 claims description 28
- 230000004069 differentiation Effects 0.000 claims description 27
- -1 IL-7 Chemical compound 0.000 claims description 26
- 108700018351 Major Histocompatibility Complex Proteins 0.000 claims description 24
- 230000020382 suppression by virus of host antigen processing and presentation of peptide antigen via MHC class I Effects 0.000 claims description 24
- 210000000581 natural killer T-cell Anatomy 0.000 claims description 23
- 210000004263 induced pluripotent stem cell Anatomy 0.000 claims description 22
- 210000005259 peripheral blood Anatomy 0.000 claims description 22
- 239000011886 peripheral blood Substances 0.000 claims description 22
- 239000003550 marker Substances 0.000 claims description 21
- 102100024222 B-lymphocyte antigen CD19 Human genes 0.000 claims description 19
- 102000004127 Cytokines Human genes 0.000 claims description 19
- 108090000695 Cytokines Proteins 0.000 claims description 19
- 101000980825 Homo sapiens B-lymphocyte antigen CD19 Proteins 0.000 claims description 19
- 238000012258 culturing Methods 0.000 claims description 19
- 210000001671 embryonic stem cell Anatomy 0.000 claims description 19
- 108010002586 Interleukin-7 Proteins 0.000 claims description 16
- 230000000735 allogeneic effect Effects 0.000 claims description 15
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 claims description 14
- 102100025570 Cancer/testis antigen 1 Human genes 0.000 claims description 14
- 101000856237 Homo sapiens Cancer/testis antigen 1 Proteins 0.000 claims description 14
- 102000004877 Insulin Human genes 0.000 claims description 14
- 108090001061 Insulin Proteins 0.000 claims description 14
- 229940125396 insulin Drugs 0.000 claims description 14
- 230000007720 allelic exclusion Effects 0.000 claims description 13
- 210000001778 pluripotent stem cell Anatomy 0.000 claims description 10
- 208000009329 Graft vs Host Disease Diseases 0.000 claims description 9
- 238000012239 gene modification Methods 0.000 claims description 9
- 208000024908 graft versus host disease Diseases 0.000 claims description 9
- 208000023275 Autoimmune disease Diseases 0.000 claims description 8
- 102100032702 Protein jagged-1 Human genes 0.000 claims description 8
- 239000011668 ascorbic acid Substances 0.000 claims description 8
- 229960005070 ascorbic acid Drugs 0.000 claims description 8
- 210000004369 blood Anatomy 0.000 claims description 8
- 239000008280 blood Substances 0.000 claims description 8
- 230000005017 genetic modification Effects 0.000 claims description 8
- 235000013617 genetically modified food Nutrition 0.000 claims description 8
- 235000010323 ascorbic acid Nutrition 0.000 claims description 7
- 210000004907 gland Anatomy 0.000 claims description 7
- 239000002773 nucleotide Substances 0.000 claims description 7
- 125000003729 nucleotide group Chemical group 0.000 claims description 7
- 238000000926 separation method Methods 0.000 claims description 7
- 210000000115 thoracic cavity Anatomy 0.000 claims description 7
- 102100032733 Protein jagged-2 Human genes 0.000 claims description 6
- 206010052779 Transplant rejections Diseases 0.000 claims description 5
- 230000036961 partial effect Effects 0.000 claims description 5
- 210000001704 mesoblast Anatomy 0.000 claims description 2
- 239000008194 pharmaceutical composition Substances 0.000 claims 3
- 206010010356 Congenital anomaly Diseases 0.000 claims 1
- 102100036462 Delta-like protein 1 Human genes 0.000 claims 1
- 102100033553 Delta-like protein 4 Human genes 0.000 claims 1
- 102100020715 Fms-related tyrosine kinase 3 ligand protein Human genes 0.000 claims 1
- 101710162577 Fms-related tyrosine kinase 3 ligand protein Proteins 0.000 claims 1
- 101000928537 Homo sapiens Delta-like protein 1 Proteins 0.000 claims 1
- 101000872077 Homo sapiens Delta-like protein 4 Proteins 0.000 claims 1
- 101000994437 Homo sapiens Protein jagged-1 Proteins 0.000 claims 1
- 101000994434 Homo sapiens Protein jagged-2 Proteins 0.000 claims 1
- 210000004475 gamma-delta t lymphocyte Anatomy 0.000 claims 1
- 239000004017 serum-free culture medium Substances 0.000 claims 1
- 102000017420 CD3 protein, epsilon/gamma/delta subunit Human genes 0.000 description 198
- 108050005493 CD3 protein, epsilon/gamma/delta subunit Proteins 0.000 description 198
- 102100034922 T-cell surface glycoprotein CD8 alpha chain Human genes 0.000 description 129
- 108090000623 proteins and genes Proteins 0.000 description 103
- 102100031573 Hematopoietic progenitor cell antigen CD34 Human genes 0.000 description 101
- 101000777663 Homo sapiens Hematopoietic progenitor cell antigen CD34 Proteins 0.000 description 101
- 102100025244 T-cell surface glycoprotein CD5 Human genes 0.000 description 69
- 239000002609 medium Substances 0.000 description 62
- 102000004169 proteins and genes Human genes 0.000 description 62
- 235000018102 proteins Nutrition 0.000 description 61
- 102000014736 Notch Human genes 0.000 description 58
- 108010070047 Notch Receptors Proteins 0.000 description 58
- 230000024245 cell differentiation Effects 0.000 description 41
- 102100036301 C-C chemokine receptor type 7 Human genes 0.000 description 35
- 101000716065 Homo sapiens C-C chemokine receptor type 7 Proteins 0.000 description 35
- 108090000765 processed proteins & peptides Proteins 0.000 description 33
- 102100027207 CD27 antigen Human genes 0.000 description 32
- 101000914511 Homo sapiens CD27 antigen Proteins 0.000 description 32
- 102000005962 receptors Human genes 0.000 description 32
- 108020003175 receptors Proteins 0.000 description 32
- 210000001519 tissue Anatomy 0.000 description 31
- 210000000612 antigen-presenting cell Anatomy 0.000 description 28
- 239000011159 matrix material Substances 0.000 description 28
- 102000010834 Extracellular Matrix Proteins Human genes 0.000 description 26
- 108010037362 Extracellular Matrix Proteins Proteins 0.000 description 26
- 239000013598 vector Substances 0.000 description 25
- 230000010076 replication Effects 0.000 description 24
- 239000003795 chemical substances by application Substances 0.000 description 23
- 239000013589 supplement Substances 0.000 description 23
- 101000738771 Homo sapiens Receptor-type tyrosine-protein phosphatase C Proteins 0.000 description 21
- 102100037422 Receptor-type tyrosine-protein phosphatase C Human genes 0.000 description 21
- 150000007523 nucleic acids Chemical class 0.000 description 21
- 108010002350 Interleukin-2 Proteins 0.000 description 20
- 230000002992 thymic effect Effects 0.000 description 20
- 210000001541 thymus gland Anatomy 0.000 description 20
- 102000004196 processed proteins & peptides Human genes 0.000 description 19
- 230000004044 response Effects 0.000 description 19
- 102000039446 nucleic acids Human genes 0.000 description 18
- 108020004707 nucleic acids Proteins 0.000 description 18
- 239000002243 precursor Substances 0.000 description 18
- 210000002744 extracellular matrix Anatomy 0.000 description 17
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 16
- 108010075704 HLA-A Antigens Proteins 0.000 description 16
- 102000011786 HLA-A Antigens Human genes 0.000 description 16
- 101710163270 Nuclease Proteins 0.000 description 16
- 210000000173 T-lymphoid precursor cell Anatomy 0.000 description 16
- 238000004458 analytical method Methods 0.000 description 16
- 230000035755 proliferation Effects 0.000 description 16
- 101000884271 Homo sapiens Signal transducer CD24 Proteins 0.000 description 15
- 101000914514 Homo sapiens T-cell-specific surface glycoprotein CD28 Proteins 0.000 description 15
- 102100021592 Interleukin-7 Human genes 0.000 description 15
- 241000699666 Mus <mouse, genus> Species 0.000 description 15
- 102100038081 Signal transducer CD24 Human genes 0.000 description 15
- 102100027213 T-cell-specific surface glycoprotein CD28 Human genes 0.000 description 15
- 229940100994 interleukin-7 Drugs 0.000 description 15
- 210000003289 regulatory T cell Anatomy 0.000 description 15
- 238000010361 transduction Methods 0.000 description 15
- 102100027581 Forkhead box protein P3 Human genes 0.000 description 14
- 101000861452 Homo sapiens Forkhead box protein P3 Proteins 0.000 description 14
- 108700014844 flt3 ligand Proteins 0.000 description 14
- 239000012679 serum free medium Substances 0.000 description 14
- 239000007787 solid Substances 0.000 description 14
- 230000026683 transduction Effects 0.000 description 14
- 102100031585 ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 Human genes 0.000 description 13
- 101000777636 Homo sapiens ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 Proteins 0.000 description 13
- 241000700605 Viruses Species 0.000 description 13
- 229940024606 amino acid Drugs 0.000 description 13
- 235000001014 amino acid Nutrition 0.000 description 13
- 150000001413 amino acids Chemical class 0.000 description 13
- 238000004113 cell culture Methods 0.000 description 13
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 12
- 101000917858 Homo sapiens Low affinity immunoglobulin gamma Fc region receptor III-A Proteins 0.000 description 12
- 101000917839 Homo sapiens Low affinity immunoglobulin gamma Fc region receptor III-B Proteins 0.000 description 12
- 101000581981 Homo sapiens Neural cell adhesion molecule 1 Proteins 0.000 description 12
- 102100029185 Low affinity immunoglobulin gamma Fc region receptor III-B Human genes 0.000 description 12
- 102100027347 Neural cell adhesion molecule 1 Human genes 0.000 description 12
- 108700042076 T-Cell Receptor alpha Genes Proteins 0.000 description 12
- 108700042077 T-Cell Receptor beta Genes Proteins 0.000 description 12
- 108700042074 T-Cell Receptor delta Genes Proteins 0.000 description 12
- 108700042082 T-Cell Receptor gamma Genes Proteins 0.000 description 12
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 12
- 239000012634 fragment Substances 0.000 description 12
- 230000003394 haemopoietic effect Effects 0.000 description 12
- 230000011664 signaling Effects 0.000 description 12
- 239000002356 single layer Substances 0.000 description 12
- 239000011701 zinc Substances 0.000 description 12
- 229910052725 zinc Inorganic materials 0.000 description 12
- 101001057504 Homo sapiens Interferon-stimulated gene 20 kDa protein Proteins 0.000 description 11
- 101001055144 Homo sapiens Interleukin-2 receptor subunit alpha Proteins 0.000 description 11
- 108010074328 Interferon-gamma Proteins 0.000 description 11
- 102000008070 Interferon-gamma Human genes 0.000 description 11
- 102100027268 Interferon-stimulated gene 20 kDa protein Human genes 0.000 description 11
- 230000004663 cell proliferation Effects 0.000 description 11
- 238000000684 flow cytometry Methods 0.000 description 11
- 229960003130 interferon gamma Drugs 0.000 description 11
- 230000001105 regulatory effect Effects 0.000 description 11
- 229940088594 vitamin Drugs 0.000 description 11
- 229930003231 vitamin Natural products 0.000 description 11
- 235000013343 vitamin Nutrition 0.000 description 11
- 239000011782 vitamin Substances 0.000 description 11
- WQZGKKKJIJFFOK-SVZMEOIVSA-N (+)-Galactose Chemical compound OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@H]1O WQZGKKKJIJFFOK-SVZMEOIVSA-N 0.000 description 10
- 241000894006 Bacteria Species 0.000 description 10
- 108020004414 DNA Proteins 0.000 description 10
- 102100037362 Fibronectin Human genes 0.000 description 10
- 108010067306 Fibronectins Proteins 0.000 description 10
- 101000946889 Homo sapiens Monocyte differentiation antigen CD14 Proteins 0.000 description 10
- 108010085895 Laminin Proteins 0.000 description 10
- 102000007547 Laminin Human genes 0.000 description 10
- 241001465754 Metazoa Species 0.000 description 10
- 102100035877 Monocyte differentiation antigen CD14 Human genes 0.000 description 10
- RJKFOVLPORLFTN-LEKSSAKUSA-N Progesterone Chemical compound C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H](C(=O)C)[C@@]1(C)CC2 RJKFOVLPORLFTN-LEKSSAKUSA-N 0.000 description 10
- 230000008901 benefit Effects 0.000 description 10
- 230000011712 cell development Effects 0.000 description 10
- 230000000694 effects Effects 0.000 description 10
- 210000002220 organoid Anatomy 0.000 description 10
- 102000040430 polynucleotide Human genes 0.000 description 10
- 108091033319 polynucleotide Proteins 0.000 description 10
- 239000002157 polynucleotide Substances 0.000 description 10
- 150000003839 salts Chemical class 0.000 description 10
- 210000002966 serum Anatomy 0.000 description 10
- 230000003612 virological effect Effects 0.000 description 10
- FPIPGXGPPPQFEQ-UHFFFAOYSA-N 13-cis retinol Natural products OCC=C(C)C=CC=C(C)C=CC1=C(C)CCCC1(C)C FPIPGXGPPPQFEQ-UHFFFAOYSA-N 0.000 description 9
- FPIPGXGPPPQFEQ-BOOMUCAASA-N Vitamin A Natural products OC/C=C(/C)\C=C\C=C(\C)/C=C/C1=C(C)CCCC1(C)C FPIPGXGPPPQFEQ-BOOMUCAASA-N 0.000 description 9
- 108010017070 Zinc Finger Nucleases Proteins 0.000 description 9
- 230000004913 activation Effects 0.000 description 9
- FPIPGXGPPPQFEQ-OVSJKPMPSA-N all-trans-retinol Chemical compound OC\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C FPIPGXGPPPQFEQ-OVSJKPMPSA-N 0.000 description 9
- 230000001580 bacterial effect Effects 0.000 description 9
- 230000006870 function Effects 0.000 description 9
- 238000010362 genome editing Methods 0.000 description 9
- 238000001727 in vivo Methods 0.000 description 9
- 229920001184 polypeptide Polymers 0.000 description 9
- 235000019155 vitamin A Nutrition 0.000 description 9
- 239000011719 vitamin A Substances 0.000 description 9
- 229940045997 vitamin a Drugs 0.000 description 9
- GHOKWGTUZJEAQD-ZETCQYMHSA-N (D)-(+)-Pantothenic acid Chemical compound OCC(C)(C)[C@@H](O)C(=O)NCCC(O)=O GHOKWGTUZJEAQD-ZETCQYMHSA-N 0.000 description 8
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 8
- 102000009027 Albumins Human genes 0.000 description 8
- 108010088751 Albumins Proteins 0.000 description 8
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 8
- ZAKOWWREFLAJOT-CEFNRUSXSA-N D-alpha-tocopherylacetate Chemical compound CC(=O)OC1=C(C)C(C)=C2O[C@@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1C ZAKOWWREFLAJOT-CEFNRUSXSA-N 0.000 description 8
- 102000004190 Enzymes Human genes 0.000 description 8
- 108090000790 Enzymes Proteins 0.000 description 8
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 8
- DFPAKSUCGFBDDF-UHFFFAOYSA-N Nicotinamide Chemical compound NC(=O)C1=CC=CN=C1 DFPAKSUCGFBDDF-UHFFFAOYSA-N 0.000 description 8
- AUNGANRZJHBGPY-SCRDCRAPSA-N Riboflavin Chemical compound OC[C@@H](O)[C@@H](O)[C@@H](O)CN1C=2C=C(C)C(C)=CC=2N=C2C1=NC(=O)NC2=O AUNGANRZJHBGPY-SCRDCRAPSA-N 0.000 description 8
- 230000027455 binding Effects 0.000 description 8
- 229960002685 biotin Drugs 0.000 description 8
- 239000011616 biotin Substances 0.000 description 8
- 235000020958 biotin Nutrition 0.000 description 8
- 229940098773 bovine serum albumin Drugs 0.000 description 8
- 229940088598 enzyme Drugs 0.000 description 8
- 238000002474 experimental method Methods 0.000 description 8
- OVBPIULPVIDEAO-LBPRGKRZSA-N folic acid Chemical compound C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-LBPRGKRZSA-N 0.000 description 8
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 description 8
- 230000004048 modification Effects 0.000 description 8
- 238000012986 modification Methods 0.000 description 8
- 210000000822 natural killer cell Anatomy 0.000 description 8
- 230000009257 reactivity Effects 0.000 description 8
- 229960001295 tocopherol Drugs 0.000 description 8
- 239000011732 tocopherol Substances 0.000 description 8
- 229940042585 tocopherol acetate Drugs 0.000 description 8
- 210000004881 tumor cell Anatomy 0.000 description 8
- 108010035532 Collagen Proteins 0.000 description 7
- 102000008186 Collagen Human genes 0.000 description 7
- 101000578784 Homo sapiens Melanoma antigen recognized by T-cells 1 Proteins 0.000 description 7
- 108700003486 Jagged-1 Proteins 0.000 description 7
- 102100028389 Melanoma antigen recognized by T-cells 1 Human genes 0.000 description 7
- 108091028043 Nucleic acid sequence Proteins 0.000 description 7
- 108700037966 Protein jagged-1 Proteins 0.000 description 7
- 210000003719 b-lymphocyte Anatomy 0.000 description 7
- 229920001436 collagen Polymers 0.000 description 7
- 108700041286 delta Proteins 0.000 description 7
- 239000005090 green fluorescent protein Substances 0.000 description 7
- 229910001410 inorganic ion Inorganic materials 0.000 description 7
- 108091033409 CRISPR Proteins 0.000 description 6
- 108091007741 Chimeric antigen receptor T cells Proteins 0.000 description 6
- 108010047041 Complementarity Determining Regions Proteins 0.000 description 6
- OMFXVFTZEKFJBZ-UHFFFAOYSA-N Corticosterone Natural products O=C1CCC2(C)C3C(O)CC(C)(C(CC4)C(=O)CO)C4C3CCC2=C1 OMFXVFTZEKFJBZ-UHFFFAOYSA-N 0.000 description 6
- 101001094545 Homo sapiens Retrotransposon-like protein 1 Proteins 0.000 description 6
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 6
- RJURFGZVJUQBHK-UHFFFAOYSA-N actinomycin D Natural products CC1OC(=O)C(C(C)C)N(C)C(=O)CN(C)C(=O)C2CCCN2C(=O)C(C(C)C)NC(=O)C1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)NC4C(=O)NC(C(N5CCCC5C(=O)N(C)CC(=O)N(C)C(C(C)C)C(=O)OC4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-UHFFFAOYSA-N 0.000 description 6
- DTOSIQBPPRVQHS-PDBXOOCHSA-N alpha-linolenic acid Chemical compound CC\C=C/C\C=C/C\C=C/CCCCCCCC(O)=O DTOSIQBPPRVQHS-PDBXOOCHSA-N 0.000 description 6
- 235000020661 alpha-linolenic acid Nutrition 0.000 description 6
- 239000003963 antioxidant agent Substances 0.000 description 6
- 235000006708 antioxidants Nutrition 0.000 description 6
- 210000004970 cd4 cell Anatomy 0.000 description 6
- 238000002659 cell therapy Methods 0.000 description 6
- OMFXVFTZEKFJBZ-HJTSIMOOSA-N corticosterone Chemical compound O=C1CC[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@H](CC4)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 OMFXVFTZEKFJBZ-HJTSIMOOSA-N 0.000 description 6
- 231100000433 cytotoxic Toxicity 0.000 description 6
- 230000001472 cytotoxic effect Effects 0.000 description 6
- 238000001943 fluorescence-activated cell sorting Methods 0.000 description 6
- 108091006047 fluorescent proteins Proteins 0.000 description 6
- 102000034287 fluorescent proteins Human genes 0.000 description 6
- 230000004927 fusion Effects 0.000 description 6
- 230000001965 increasing effect Effects 0.000 description 6
- PGHMRUGBZOYCAA-UHFFFAOYSA-N ionomycin Natural products O1C(CC(O)C(C)C(O)C(C)C=CCC(C)CC(C)C(O)=CC(=O)C(C)CC(C)CC(CCC(O)=O)C)CCC1(C)C1OC(C)(C(C)O)CC1 PGHMRUGBZOYCAA-UHFFFAOYSA-N 0.000 description 6
- PGHMRUGBZOYCAA-ADZNBVRBSA-N ionomycin Chemical compound O1[C@H](C[C@H](O)[C@H](C)[C@H](O)[C@H](C)/C=C/C[C@@H](C)C[C@@H](C)C(/O)=C/C(=O)[C@@H](C)C[C@@H](C)C[C@@H](CCC(O)=O)C)CC[C@@]1(C)[C@@H]1O[C@](C)([C@@H](C)O)CC1 PGHMRUGBZOYCAA-ADZNBVRBSA-N 0.000 description 6
- KQQKGWQCNNTQJW-UHFFFAOYSA-N linolenic acid Natural products CC=CCCC=CCC=CCCCCCCCC(O)=O KQQKGWQCNNTQJW-UHFFFAOYSA-N 0.000 description 6
- 229960004488 linolenic acid Drugs 0.000 description 6
- 108010082117 matrigel Proteins 0.000 description 6
- 230000035800 maturation Effects 0.000 description 6
- KIDHWZJUCRJVML-UHFFFAOYSA-N putrescine Chemical compound NCCCCN KIDHWZJUCRJVML-UHFFFAOYSA-N 0.000 description 6
- 238000001890 transfection Methods 0.000 description 6
- 230000003827 upregulation Effects 0.000 description 6
- 102100035248 Alpha-(1,3)-fucosyltransferase 4 Human genes 0.000 description 5
- 102100038080 B-cell receptor CD22 Human genes 0.000 description 5
- 102100022005 B-lymphocyte antigen CD20 Human genes 0.000 description 5
- 101001022185 Homo sapiens Alpha-(1,3)-fucosyltransferase 4 Proteins 0.000 description 5
- 101000884305 Homo sapiens B-cell receptor CD22 Proteins 0.000 description 5
- 101000897405 Homo sapiens B-lymphocyte antigen CD20 Proteins 0.000 description 5
- 101001046686 Homo sapiens Integrin alpha-M Proteins 0.000 description 5
- 101001015004 Homo sapiens Integrin beta-3 Proteins 0.000 description 5
- 101001043564 Homo sapiens Prolow-density lipoprotein receptor-related protein 1 Proteins 0.000 description 5
- 101000851376 Homo sapiens Tumor necrosis factor receptor superfamily member 8 Proteins 0.000 description 5
- 102100022338 Integrin alpha-M Human genes 0.000 description 5
- 102100032999 Integrin beta-3 Human genes 0.000 description 5
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 5
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 5
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 5
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 5
- 239000004472 Lysine Substances 0.000 description 5
- 241000699670 Mus sp. Species 0.000 description 5
- 102000003729 Neprilysin Human genes 0.000 description 5
- 108090000028 Neprilysin Proteins 0.000 description 5
- 102100021923 Prolow-density lipoprotein receptor-related protein 1 Human genes 0.000 description 5
- 101710170213 Protein jagged-2 Proteins 0.000 description 5
- 108700008625 Reporter Genes Proteins 0.000 description 5
- 102100036857 Tumor necrosis factor receptor superfamily member 8 Human genes 0.000 description 5
- 239000011324 bead Substances 0.000 description 5
- 238000005119 centrifugation Methods 0.000 description 5
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 5
- 238000010790 dilution Methods 0.000 description 5
- 239000012895 dilution Substances 0.000 description 5
- 238000001476 gene delivery Methods 0.000 description 5
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 5
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 5
- 208000015181 infectious disease Diseases 0.000 description 5
- 230000000977 initiatory effect Effects 0.000 description 5
- 230000003834 intracellular effect Effects 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 210000001616 monocyte Anatomy 0.000 description 5
- 239000000186 progesterone Substances 0.000 description 5
- 229960003387 progesterone Drugs 0.000 description 5
- LXNHXLLTXMVWPM-UHFFFAOYSA-N pyridoxine Chemical compound CC1=NC=C(CO)C(CO)=C1O LXNHXLLTXMVWPM-UHFFFAOYSA-N 0.000 description 5
- 241000894007 species Species 0.000 description 5
- 239000000126 substance Substances 0.000 description 5
- 150000003722 vitamin derivatives Chemical class 0.000 description 5
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 5
- MZOFCQQQCNRIBI-VMXHOPILSA-N (3s)-4-[[(2s)-1-[[(2s)-1-[[(1s)-1-carboxy-2-hydroxyethyl]amino]-4-methyl-1-oxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-[[2-[[(2s)-2,6-diaminohexanoyl]amino]acetyl]amino]-4-oxobutanoic acid Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN MZOFCQQQCNRIBI-VMXHOPILSA-N 0.000 description 4
- OYHQOLUKZRVURQ-NTGFUMLPSA-N (9Z,12Z)-9,10,12,13-tetratritiooctadeca-9,12-dienoic acid Chemical compound C(CCCCCCC\C(=C(/C\C(=C(/CCCCC)\[3H])\[3H])\[3H])\[3H])(=O)O OYHQOLUKZRVURQ-NTGFUMLPSA-N 0.000 description 4
- PHIQHXFUZVPYII-ZCFIWIBFSA-N (R)-carnitine Chemical compound C[N+](C)(C)C[C@H](O)CC([O-])=O PHIQHXFUZVPYII-ZCFIWIBFSA-N 0.000 description 4
- 239000001763 2-hydroxyethyl(trimethyl)azanium Substances 0.000 description 4
- 239000004475 Arginine Substances 0.000 description 4
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 4
- 102100035882 Catalase Human genes 0.000 description 4
- 108010053835 Catalase Proteins 0.000 description 4
- GHOKWGTUZJEAQD-UHFFFAOYSA-N Chick antidermatitis factor Natural products OCC(C)(C)C(O)C(=O)NCCC(O)=O GHOKWGTUZJEAQD-UHFFFAOYSA-N 0.000 description 4
- 235000019743 Choline chloride Nutrition 0.000 description 4
- 108091026890 Coding region Proteins 0.000 description 4
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 4
- AUNGANRZJHBGPY-UHFFFAOYSA-N D-Lyxoflavin Natural products OCC(O)C(O)C(O)CN1C=2C=C(C)C(C)=CC=2N=C2C1=NC(=O)NC2=O AUNGANRZJHBGPY-UHFFFAOYSA-N 0.000 description 4
- 108010024636 Glutathione Proteins 0.000 description 4
- SQUHHTBVTRBESD-UHFFFAOYSA-N Hexa-Ac-myo-Inositol Natural products CC(=O)OC1C(OC(C)=O)C(OC(C)=O)C(OC(C)=O)C(OC(C)=O)C1OC(C)=O SQUHHTBVTRBESD-UHFFFAOYSA-N 0.000 description 4
- 101001023379 Homo sapiens Lysosome-associated membrane glycoprotein 1 Proteins 0.000 description 4
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 4
- 102000003812 Interleukin-15 Human genes 0.000 description 4
- 108090000172 Interleukin-15 Proteins 0.000 description 4
- 108090001005 Interleukin-6 Proteins 0.000 description 4
- 102000004889 Interleukin-6 Human genes 0.000 description 4
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 4
- LEVWYRKDKASIDU-IMJSIDKUSA-N L-cystine Chemical compound [O-]C(=O)[C@@H]([NH3+])CSSC[C@H]([NH3+])C([O-])=O LEVWYRKDKASIDU-IMJSIDKUSA-N 0.000 description 4
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 description 4
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 4
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 4
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 4
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 4
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 4
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 4
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 4
- 102100035133 Lysosome-associated membrane glycoprotein 1 Human genes 0.000 description 4
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 4
- ZOKXTWBITQBERF-UHFFFAOYSA-N Molybdenum Chemical compound [Mo] ZOKXTWBITQBERF-UHFFFAOYSA-N 0.000 description 4
- OVBPIULPVIDEAO-UHFFFAOYSA-N N-Pteroyl-L-glutaminsaeure Natural products C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)NC(CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-UHFFFAOYSA-N 0.000 description 4
- 206010061535 Ovarian neoplasm Diseases 0.000 description 4
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 4
- 102100024616 Platelet endothelial cell adhesion molecule Human genes 0.000 description 4
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 4
- BUGBHKTXTAQXES-UHFFFAOYSA-N Selenium Chemical compound [Se] BUGBHKTXTAQXES-UHFFFAOYSA-N 0.000 description 4
- 102000019197 Superoxide Dismutase Human genes 0.000 description 4
- 108010012715 Superoxide dismutase Proteins 0.000 description 4
- JZRWCGZRTZMZEH-UHFFFAOYSA-N Thiamine Natural products CC1=C(CCO)SC=[N+]1CC1=CN=C(C)N=C1N JZRWCGZRTZMZEH-UHFFFAOYSA-N 0.000 description 4
- 102000004338 Transferrin Human genes 0.000 description 4
- 108090000901 Transferrin Proteins 0.000 description 4
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 4
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 4
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 4
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 4
- 229930003779 Vitamin B12 Natural products 0.000 description 4
- 108010031318 Vitronectin Proteins 0.000 description 4
- 102100035140 Vitronectin Human genes 0.000 description 4
- 239000000556 agonist Substances 0.000 description 4
- 230000000890 antigenic effect Effects 0.000 description 4
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 4
- 238000003556 assay Methods 0.000 description 4
- 239000011575 calcium Substances 0.000 description 4
- 229910052791 calcium Inorganic materials 0.000 description 4
- FAPWYRCQGJNNSJ-UBKPKTQASA-L calcium D-pantothenic acid Chemical compound [Ca+2].OCC(C)(C)[C@@H](O)C(=O)NCCC([O-])=O.OCC(C)(C)[C@@H](O)C(=O)NCCC([O-])=O FAPWYRCQGJNNSJ-UBKPKTQASA-L 0.000 description 4
- 229960002079 calcium pantothenate Drugs 0.000 description 4
- 230000021164 cell adhesion Effects 0.000 description 4
- 230000003915 cell function Effects 0.000 description 4
- 210000000170 cell membrane Anatomy 0.000 description 4
- 230000001413 cellular effect Effects 0.000 description 4
- 239000003153 chemical reaction reagent Substances 0.000 description 4
- SGMZJAMFUVOLNK-UHFFFAOYSA-M choline chloride Chemical compound [Cl-].C[N+](C)(C)CCO SGMZJAMFUVOLNK-UHFFFAOYSA-M 0.000 description 4
- 229960003178 choline chloride Drugs 0.000 description 4
- 238000003501 co-culture Methods 0.000 description 4
- AGVAZMGAQJOSFJ-WZHZPDAFSA-M cobalt(2+);[(2r,3s,4r,5s)-5-(5,6-dimethylbenzimidazol-1-yl)-4-hydroxy-2-(hydroxymethyl)oxolan-3-yl] [(2r)-1-[3-[(1r,2r,3r,4z,7s,9z,12s,13s,14z,17s,18s,19r)-2,13,18-tris(2-amino-2-oxoethyl)-7,12,17-tris(3-amino-3-oxopropyl)-3,5,8,8,13,15,18,19-octamethyl-2 Chemical compound [Co+2].N#[C-].[N-]([C@@H]1[C@H](CC(N)=O)[C@@]2(C)CCC(=O)NC[C@@H](C)OP(O)(=O)O[C@H]3[C@H]([C@H](O[C@@H]3CO)N3C4=CC(C)=C(C)C=C4N=C3)O)\C2=C(C)/C([C@H](C\2(C)C)CCC(N)=O)=N/C/2=C\C([C@H]([C@@]/2(CC(N)=O)C)CCC(N)=O)=N\C\2=C(C)/C2=N[C@]1(C)[C@@](C)(CC(N)=O)[C@@H]2CCC(N)=O AGVAZMGAQJOSFJ-WZHZPDAFSA-M 0.000 description 4
- 229910052802 copper Inorganic materials 0.000 description 4
- 239000010949 copper Substances 0.000 description 4
- 238000012136 culture method Methods 0.000 description 4
- 229960003067 cystine Drugs 0.000 description 4
- 230000016396 cytokine production Effects 0.000 description 4
- 230000018109 developmental process Effects 0.000 description 4
- 201000010099 disease Diseases 0.000 description 4
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 4
- 235000021186 dishes Nutrition 0.000 description 4
- BVTBRVFYZUCAKH-UHFFFAOYSA-L disodium selenite Chemical compound [Na+].[Na+].[O-][Se]([O-])=O BVTBRVFYZUCAKH-UHFFFAOYSA-L 0.000 description 4
- 229960000304 folic acid Drugs 0.000 description 4
- 235000019152 folic acid Nutrition 0.000 description 4
- 239000011724 folic acid Substances 0.000 description 4
- 230000002068 genetic effect Effects 0.000 description 4
- 229960003180 glutathione Drugs 0.000 description 4
- 238000009169 immunotherapy Methods 0.000 description 4
- CDAISMWEOUEBRE-GPIVLXJGSA-N inositol Chemical compound O[C@H]1[C@H](O)[C@@H](O)[C@H](O)[C@H](O)[C@@H]1O CDAISMWEOUEBRE-GPIVLXJGSA-N 0.000 description 4
- 229960000367 inositol Drugs 0.000 description 4
- 238000003780 insertion Methods 0.000 description 4
- 230000037431 insertion Effects 0.000 description 4
- 229910052742 iron Inorganic materials 0.000 description 4
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 description 4
- 229960000310 isoleucine Drugs 0.000 description 4
- 239000011777 magnesium Substances 0.000 description 4
- 229910052749 magnesium Inorganic materials 0.000 description 4
- 238000012423 maintenance Methods 0.000 description 4
- 230000001404 mediated effect Effects 0.000 description 4
- 239000012528 membrane Substances 0.000 description 4
- 210000004379 membrane Anatomy 0.000 description 4
- 210000002901 mesenchymal stem cell Anatomy 0.000 description 4
- 229930182817 methionine Natural products 0.000 description 4
- 230000000813 microbial effect Effects 0.000 description 4
- 229910052750 molybdenum Inorganic materials 0.000 description 4
- 239000011733 molybdenum Substances 0.000 description 4
- 238000004264 monolayer culture Methods 0.000 description 4
- 150000002772 monosaccharides Chemical class 0.000 description 4
- 229960003966 nicotinamide Drugs 0.000 description 4
- 235000005152 nicotinamide Nutrition 0.000 description 4
- 239000011570 nicotinamide Substances 0.000 description 4
- 229910052757 nitrogen Inorganic materials 0.000 description 4
- 229940055726 pantothenic acid Drugs 0.000 description 4
- 235000019161 pantothenic acid Nutrition 0.000 description 4
- 239000011713 pantothenic acid Substances 0.000 description 4
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 4
- 229910052698 phosphorus Inorganic materials 0.000 description 4
- 239000011574 phosphorus Substances 0.000 description 4
- 239000011591 potassium Substances 0.000 description 4
- 229910052700 potassium Inorganic materials 0.000 description 4
- RXWNCPJZOCPEPQ-NVWDDTSBSA-N puromycin Chemical compound C1=CC(OC)=CC=C1C[C@H](N)C(=O)N[C@H]1[C@@H](O)[C@H](N2C3=NC=NC(=C3N=C2)N(C)C)O[C@@H]1CO RXWNCPJZOCPEPQ-NVWDDTSBSA-N 0.000 description 4
- 230000002829 reductive effect Effects 0.000 description 4
- 238000011160 research Methods 0.000 description 4
- 229960002477 riboflavin Drugs 0.000 description 4
- 235000019192 riboflavin Nutrition 0.000 description 4
- 239000002151 riboflavin Substances 0.000 description 4
- CDAISMWEOUEBRE-UHFFFAOYSA-N scyllo-inosotol Natural products OC1C(O)C(O)C(O)C(O)C1O CDAISMWEOUEBRE-UHFFFAOYSA-N 0.000 description 4
- 229910052711 selenium Inorganic materials 0.000 description 4
- 239000011669 selenium Substances 0.000 description 4
- 229940091258 selenium supplement Drugs 0.000 description 4
- 239000011734 sodium Substances 0.000 description 4
- 229910052708 sodium Inorganic materials 0.000 description 4
- 229960001471 sodium selenite Drugs 0.000 description 4
- 235000015921 sodium selenite Nutrition 0.000 description 4
- 239000011781 sodium selenite Substances 0.000 description 4
- 230000000638 stimulation Effects 0.000 description 4
- 229960003495 thiamine Drugs 0.000 description 4
- 235000019157 thiamine Nutrition 0.000 description 4
- KYMBYSLLVAOCFI-UHFFFAOYSA-N thiamine Chemical compound CC1=C(CCO)SCN1CC1=CN=C(C)N=C1N KYMBYSLLVAOCFI-UHFFFAOYSA-N 0.000 description 4
- 239000011721 thiamine Substances 0.000 description 4
- 238000012546 transfer Methods 0.000 description 4
- 239000012581 transferrin Substances 0.000 description 4
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 4
- 239000004474 valine Substances 0.000 description 4
- 229910052720 vanadium Inorganic materials 0.000 description 4
- GPPXJZIENCGNKB-UHFFFAOYSA-N vanadium Chemical compound [V]#[V] GPPXJZIENCGNKB-UHFFFAOYSA-N 0.000 description 4
- 235000019163 vitamin B12 Nutrition 0.000 description 4
- 239000011715 vitamin B12 Substances 0.000 description 4
- 229940011671 vitamin b6 Drugs 0.000 description 4
- 108090000672 Annexin A5 Proteins 0.000 description 3
- 102000004121 Annexin A5 Human genes 0.000 description 3
- 238000010354 CRISPR gene editing Methods 0.000 description 3
- 108010092160 Dactinomycin Proteins 0.000 description 3
- 238000012413 Fluorescence activated cell sorting analysis Methods 0.000 description 3
- 108010010803 Gelatin Proteins 0.000 description 3
- 108010017080 Granulocyte Colony-Stimulating Factor Proteins 0.000 description 3
- 102000004269 Granulocyte Colony-Stimulating Factor Human genes 0.000 description 3
- 102000006354 HLA-DR Antigens Human genes 0.000 description 3
- 108010058597 HLA-DR Antigens Proteins 0.000 description 3
- 101001018097 Homo sapiens L-selectin Proteins 0.000 description 3
- 101000914484 Homo sapiens T-lymphocyte activation antigen CD80 Proteins 0.000 description 3
- 101000845170 Homo sapiens Thymic stromal lymphopoietin Proteins 0.000 description 3
- 108090000978 Interleukin-4 Proteins 0.000 description 3
- 102100033467 L-selectin Human genes 0.000 description 3
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 3
- 241000713666 Lentivirus Species 0.000 description 3
- 108010052285 Membrane Proteins Proteins 0.000 description 3
- 102000018697 Membrane Proteins Human genes 0.000 description 3
- 102100030335 Midkine Human genes 0.000 description 3
- 108010092801 Midkine Proteins 0.000 description 3
- 208000034578 Multiple myelomas Diseases 0.000 description 3
- 206010033128 Ovarian cancer Diseases 0.000 description 3
- 102100036467 Protein delta homolog 1 Human genes 0.000 description 3
- 239000005700 Putrescine Substances 0.000 description 3
- 102100027222 T-lymphocyte activation antigen CD80 Human genes 0.000 description 3
- 238000010459 TALEN Methods 0.000 description 3
- 108700012920 TNF Proteins 0.000 description 3
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 3
- 239000004473 Threonine Substances 0.000 description 3
- 102100031294 Thymic stromal lymphopoietin Human genes 0.000 description 3
- 108010043645 Transcription Activator-Like Effector Nucleases Proteins 0.000 description 3
- 102000004887 Transforming Growth Factor beta Human genes 0.000 description 3
- 108090001012 Transforming Growth Factor beta Proteins 0.000 description 3
- RJURFGZVJUQBHK-IIXSONLDSA-N actinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-IIXSONLDSA-N 0.000 description 3
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 3
- 230000003115 biocidal effect Effects 0.000 description 3
- 210000002798 bone marrow cell Anatomy 0.000 description 3
- 229940105657 catalase Drugs 0.000 description 3
- 239000002771 cell marker Substances 0.000 description 3
- 238000010367 cloning Methods 0.000 description 3
- 230000004186 co-expression Effects 0.000 description 3
- 230000000295 complement effect Effects 0.000 description 3
- 239000002299 complementary DNA Substances 0.000 description 3
- 229960000640 dactinomycin Drugs 0.000 description 3
- 210000004443 dendritic cell Anatomy 0.000 description 3
- 238000011161 development Methods 0.000 description 3
- 239000003814 drug Substances 0.000 description 3
- 239000012636 effector Substances 0.000 description 3
- 210000003386 epithelial cell of thymus gland Anatomy 0.000 description 3
- 210000003743 erythrocyte Anatomy 0.000 description 3
- 239000012091 fetal bovine serum Substances 0.000 description 3
- 230000008014 freezing Effects 0.000 description 3
- 238000007710 freezing Methods 0.000 description 3
- 229920000159 gelatin Polymers 0.000 description 3
- 239000008273 gelatin Substances 0.000 description 3
- 235000019322 gelatine Nutrition 0.000 description 3
- 235000011852 gelatine desserts Nutrition 0.000 description 3
- 238000002744 homologous recombination Methods 0.000 description 3
- 230000006801 homologous recombination Effects 0.000 description 3
- 210000005260 human cell Anatomy 0.000 description 3
- 230000003993 interaction Effects 0.000 description 3
- 230000014828 interferon-gamma production Effects 0.000 description 3
- 238000010212 intracellular staining Methods 0.000 description 3
- 230000000670 limiting effect Effects 0.000 description 3
- 150000002632 lipids Chemical class 0.000 description 3
- WPBNNNQJVZRUHP-UHFFFAOYSA-L manganese(2+);methyl n-[[2-(methoxycarbonylcarbamothioylamino)phenyl]carbamothioyl]carbamate;n-[2-(sulfidocarbothioylamino)ethyl]carbamodithioate Chemical compound [Mn+2].[S-]C(=S)NCCNC([S-])=S.COC(=O)NC(=S)NC1=CC=CC=C1NC(=S)NC(=O)OC WPBNNNQJVZRUHP-UHFFFAOYSA-L 0.000 description 3
- 244000005700 microbiome Species 0.000 description 3
- 229960004857 mitomycin Drugs 0.000 description 3
- 210000002501 natural regulatory T cell Anatomy 0.000 description 3
- 210000000653 nervous system Anatomy 0.000 description 3
- 108010008217 nidogen Proteins 0.000 description 3
- 239000002245 particle Substances 0.000 description 3
- 238000002360 preparation method Methods 0.000 description 3
- 239000000047 product Substances 0.000 description 3
- 230000006798 recombination Effects 0.000 description 3
- 238000005215 recombination Methods 0.000 description 3
- 230000007115 recruitment Effects 0.000 description 3
- 238000012216 screening Methods 0.000 description 3
- 238000012163 sequencing technique Methods 0.000 description 3
- 230000019491 signal transduction Effects 0.000 description 3
- 239000000758 substrate Substances 0.000 description 3
- ZRKFYGHZFMAOKI-QMGMOQQFSA-N tgfbeta Chemical compound C([C@H](NC(=O)[C@H](C(C)C)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CC(C)C)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CCSC)C(C)C)[C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O)C1=CC=C(O)C=C1 ZRKFYGHZFMAOKI-QMGMOQQFSA-N 0.000 description 3
- 230000001988 toxicity Effects 0.000 description 3
- 231100000419 toxicity Toxicity 0.000 description 3
- 230000005030 transcription termination Effects 0.000 description 3
- 239000013638 trimer Substances 0.000 description 3
- 239000003981 vehicle Substances 0.000 description 3
- 238000005406 washing Methods 0.000 description 3
- OZFAFGSSMRRTDW-UHFFFAOYSA-N (2,4-dichlorophenyl) benzenesulfonate Chemical compound ClC1=CC(Cl)=CC=C1OS(=O)(=O)C1=CC=CC=C1 OZFAFGSSMRRTDW-UHFFFAOYSA-N 0.000 description 2
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 2
- UZOVYGYOLBIAJR-UHFFFAOYSA-N 4-isocyanato-4'-methyldiphenylmethane Chemical compound C1=CC(C)=CC=C1CC1=CC=C(N=C=O)C=C1 UZOVYGYOLBIAJR-UHFFFAOYSA-N 0.000 description 2
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 2
- 108010006654 Bleomycin Proteins 0.000 description 2
- 208000003174 Brain Neoplasms Diseases 0.000 description 2
- 206010006187 Breast cancer Diseases 0.000 description 2
- 208000026310 Breast neoplasm Diseases 0.000 description 2
- 102100039521 C-type lectin domain family 9 member A Human genes 0.000 description 2
- 102100031170 CCN family member 3 Human genes 0.000 description 2
- 244000025254 Cannabis sativa Species 0.000 description 2
- 235000012766 Cannabis sativa ssp. sativa var. sativa Nutrition 0.000 description 2
- 235000012765 Cannabis sativa ssp. sativa var. spontanea Nutrition 0.000 description 2
- 206010008342 Cervix carcinoma Diseases 0.000 description 2
- 108010035563 Chloramphenicol O-acetyltransferase Proteins 0.000 description 2
- 206010009944 Colon cancer Diseases 0.000 description 2
- KDXKERNSBIXSRK-RXMQYKEDSA-N D-lysine Chemical compound NCCCC[C@@H](N)C(O)=O KDXKERNSBIXSRK-RXMQYKEDSA-N 0.000 description 2
- 101150011813 DLL1 gene Proteins 0.000 description 2
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 2
- 101100347633 Drosophila melanogaster Mhc gene Proteins 0.000 description 2
- 239000012591 Dulbecco’s Phosphate Buffered Saline Substances 0.000 description 2
- 108010042407 Endonucleases Proteins 0.000 description 2
- 102000004533 Endonucleases Human genes 0.000 description 2
- 208000000461 Esophageal Neoplasms Diseases 0.000 description 2
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 2
- 102100021186 Granulysin Human genes 0.000 description 2
- 101000888548 Homo sapiens C-type lectin domain family 9 member A Proteins 0.000 description 2
- 101100499372 Homo sapiens DLL1 gene Proteins 0.000 description 2
- 101001027128 Homo sapiens Fibronectin Proteins 0.000 description 2
- 101001040751 Homo sapiens Granulysin Proteins 0.000 description 2
- 101000928541 Homo sapiens Protein delta homolog 2 Proteins 0.000 description 2
- 101001061851 Homo sapiens V(D)J recombination-activating protein 2 Proteins 0.000 description 2
- 206010061598 Immunodeficiency Diseases 0.000 description 2
- 208000029462 Immunodeficiency disease Diseases 0.000 description 2
- 108060003951 Immunoglobulin Proteins 0.000 description 2
- 102100030703 Interleukin-22 Human genes 0.000 description 2
- 102100020880 Kit ligand Human genes 0.000 description 2
- GQYIWUVLTXOXAJ-UHFFFAOYSA-N Lomustine Chemical compound ClCCN(N=O)C(=O)NC1CCCCC1 GQYIWUVLTXOXAJ-UHFFFAOYSA-N 0.000 description 2
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 2
- 206010025323 Lymphomas Diseases 0.000 description 2
- 102100036203 Microfibrillar-associated protein 5 Human genes 0.000 description 2
- 102100037369 Nidogen-1 Human genes 0.000 description 2
- 206010030155 Oesophageal carcinoma Diseases 0.000 description 2
- 102000007079 Peptide Fragments Human genes 0.000 description 2
- 108010033276 Peptide Fragments Proteins 0.000 description 2
- 102100037765 Periostin Human genes 0.000 description 2
- 101710199268 Periostin Proteins 0.000 description 2
- 206010035226 Plasma cell myeloma Diseases 0.000 description 2
- 206010060862 Prostate cancer Diseases 0.000 description 2
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 2
- 102100036463 Protein delta homolog 2 Human genes 0.000 description 2
- 102000001183 RAG-1 Human genes 0.000 description 2
- 108060006897 RAG1 Proteins 0.000 description 2
- 239000012980 RPMI-1640 medium Substances 0.000 description 2
- 208000015634 Rectal Neoplasms Diseases 0.000 description 2
- 208000000453 Skin Neoplasms Diseases 0.000 description 2
- 208000021712 Soft tissue sarcoma Diseases 0.000 description 2
- 108010039445 Stem Cell Factor Proteins 0.000 description 2
- 208000005718 Stomach Neoplasms Diseases 0.000 description 2
- 230000006052 T cell proliferation Effects 0.000 description 2
- 102100025237 T-cell surface antigen CD2 Human genes 0.000 description 2
- 208000024313 Testicular Neoplasms Diseases 0.000 description 2
- 206010057644 Testis cancer Diseases 0.000 description 2
- 102000036693 Thrombopoietin Human genes 0.000 description 2
- 108010041111 Thrombopoietin Proteins 0.000 description 2
- 101800001703 Thymopentin Proteins 0.000 description 2
- 102400000160 Thymopentin Human genes 0.000 description 2
- 102000040945 Transcription factor Human genes 0.000 description 2
- 108091023040 Transcription factor Proteins 0.000 description 2
- 102000004357 Transferases Human genes 0.000 description 2
- 108090000992 Transferases Proteins 0.000 description 2
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 description 2
- 102100029591 V(D)J recombination-activating protein 2 Human genes 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- 238000011467 adoptive cell therapy Methods 0.000 description 2
- 230000004520 agglutination Effects 0.000 description 2
- 230000002776 aggregation Effects 0.000 description 2
- 238000004220 aggregation Methods 0.000 description 2
- 210000004102 animal cell Anatomy 0.000 description 2
- 229940045799 anthracyclines and related substance Drugs 0.000 description 2
- 239000003242 anti bacterial agent Substances 0.000 description 2
- 230000001745 anti-biotin effect Effects 0.000 description 2
- 230000000259 anti-tumor effect Effects 0.000 description 2
- 230000000840 anti-viral effect Effects 0.000 description 2
- 238000013459 approach Methods 0.000 description 2
- 239000007640 basal medium Substances 0.000 description 2
- 230000029918 bioluminescence Effects 0.000 description 2
- 238000005415 bioluminescence Methods 0.000 description 2
- 229960001561 bleomycin Drugs 0.000 description 2
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 2
- 210000001772 blood platelet Anatomy 0.000 description 2
- 206010006007 bone sarcoma Diseases 0.000 description 2
- 235000009120 camo Nutrition 0.000 description 2
- 229960004562 carboplatin Drugs 0.000 description 2
- 230000030833 cell death Effects 0.000 description 2
- 239000002458 cell surface marker Substances 0.000 description 2
- 201000010881 cervical cancer Diseases 0.000 description 2
- 235000005607 chanvre indien Nutrition 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- 239000013611 chromosomal DNA Substances 0.000 description 2
- 229960004316 cisplatin Drugs 0.000 description 2
- 238000003776 cleavage reaction Methods 0.000 description 2
- 208000029742 colonic neoplasm Diseases 0.000 description 2
- 239000000470 constituent Substances 0.000 description 2
- 238000010276 construction Methods 0.000 description 2
- 230000000139 costimulatory effect Effects 0.000 description 2
- 210000005220 cytoplasmic tail Anatomy 0.000 description 2
- 229940127089 cytotoxic agent Drugs 0.000 description 2
- 238000002784 cytotoxicity assay Methods 0.000 description 2
- 231100000263 cytotoxicity test Toxicity 0.000 description 2
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 2
- 238000002716 delivery method Methods 0.000 description 2
- 229940127276 delta-like ligand 3 Drugs 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- 235000014113 dietary fatty acids Nutrition 0.000 description 2
- 210000002249 digestive system Anatomy 0.000 description 2
- 238000007865 diluting Methods 0.000 description 2
- 230000005782 double-strand break Effects 0.000 description 2
- 230000003828 downregulation Effects 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 201000004101 esophageal cancer Diseases 0.000 description 2
- 239000013604 expression vector Substances 0.000 description 2
- 229930195729 fatty acid Natural products 0.000 description 2
- 239000000194 fatty acid Substances 0.000 description 2
- 150000004665 fatty acids Chemical class 0.000 description 2
- 230000001605 fetal effect Effects 0.000 description 2
- 239000012530 fluid Substances 0.000 description 2
- 229960002949 fluorouracil Drugs 0.000 description 2
- 206010017758 gastric cancer Diseases 0.000 description 2
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 2
- 229960005277 gemcitabine Drugs 0.000 description 2
- 230000009368 gene silencing by RNA Effects 0.000 description 2
- 238000010353 genetic engineering Methods 0.000 description 2
- 210000004392 genitalia Anatomy 0.000 description 2
- 210000001654 germ layer Anatomy 0.000 description 2
- 230000012010 growth Effects 0.000 description 2
- 239000003102 growth factor Substances 0.000 description 2
- 239000001963 growth medium Substances 0.000 description 2
- 201000010536 head and neck cancer Diseases 0.000 description 2
- 208000014829 head and neck neoplasm Diseases 0.000 description 2
- 239000000833 heterodimer Substances 0.000 description 2
- 239000000710 homodimer Substances 0.000 description 2
- 238000003384 imaging method Methods 0.000 description 2
- 230000001900 immune effect Effects 0.000 description 2
- 230000007813 immunodeficiency Effects 0.000 description 2
- 102000018358 immunoglobulin Human genes 0.000 description 2
- 239000003112 inhibitor Substances 0.000 description 2
- 108010074108 interleukin-21 Proteins 0.000 description 2
- 238000002955 isolation Methods 0.000 description 2
- 208000032839 leukemia Diseases 0.000 description 2
- 210000000265 leukocyte Anatomy 0.000 description 2
- 201000007270 liver cancer Diseases 0.000 description 2
- 208000014018 liver neoplasm Diseases 0.000 description 2
- 201000005202 lung cancer Diseases 0.000 description 2
- 208000020816 lung neoplasm Diseases 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 239000012092 media component Substances 0.000 description 2
- 201000001441 melanoma Diseases 0.000 description 2
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 2
- 210000003716 mesoderm Anatomy 0.000 description 2
- 230000003278 mimic effect Effects 0.000 description 2
- 238000002703 mutagenesis Methods 0.000 description 2
- 231100000350 mutagenesis Toxicity 0.000 description 2
- 230000035772 mutation Effects 0.000 description 2
- 230000006780 non-homologous end joining Effects 0.000 description 2
- 210000004940 nucleus Anatomy 0.000 description 2
- 201000008017 ovarian lymphoma Diseases 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- 229920000729 poly(L-lysine) polymer Polymers 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- AQHHHDLHHXJYJD-UHFFFAOYSA-N propranolol Chemical compound C1=CC=C2C(OCC(O)CNC(C)C)=CC=CC2=C1 AQHHHDLHHXJYJD-UHFFFAOYSA-N 0.000 description 2
- 229950010131 puromycin Drugs 0.000 description 2
- 230000008707 rearrangement Effects 0.000 description 2
- 206010038038 rectal cancer Diseases 0.000 description 2
- 201000001275 rectum cancer Diseases 0.000 description 2
- 230000000284 resting effect Effects 0.000 description 2
- 108091008146 restriction endonucleases Proteins 0.000 description 2
- 108010056030 retronectin Proteins 0.000 description 2
- 238000003757 reverse transcription PCR Methods 0.000 description 2
- 230000007017 scission Effects 0.000 description 2
- 210000003491 skin Anatomy 0.000 description 2
- 201000000849 skin cancer Diseases 0.000 description 2
- 210000004872 soft tissue Anatomy 0.000 description 2
- 238000010186 staining Methods 0.000 description 2
- 230000004936 stimulating effect Effects 0.000 description 2
- 201000011549 stomach cancer Diseases 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 230000008685 targeting Effects 0.000 description 2
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 2
- 201000003120 testicular cancer Diseases 0.000 description 2
- PSWFFKRAVBDQEG-YGQNSOCVSA-N thymopentin Chemical compound NC(N)=NCCC[C@H](N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@H](C(O)=O)CC1=CC=C(O)C=C1 PSWFFKRAVBDQEG-YGQNSOCVSA-N 0.000 description 2
- 229960004517 thymopentin Drugs 0.000 description 2
- WYWHKKSPHMUBEB-UHFFFAOYSA-N tioguanine Chemical compound N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 description 2
- 206010044285 tracheal cancer Diseases 0.000 description 2
- 230000035897 transcription Effects 0.000 description 2
- 238000013518 transcription Methods 0.000 description 2
- 230000002103 transcriptional effect Effects 0.000 description 2
- 238000013519 translation Methods 0.000 description 2
- VBEQCZHXXJYVRD-GACYYNSASA-N uroanthelone Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CS)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C(C)C)[C@@H](C)O)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCSC)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)CNC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CS)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CS)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC(N)=O)C(C)C)[C@@H](C)CC)C1=CC=C(O)C=C1 VBEQCZHXXJYVRD-GACYYNSASA-N 0.000 description 2
- 230000002861 ventricular Effects 0.000 description 2
- 229960004528 vincristine Drugs 0.000 description 2
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 2
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 2
- 230000003442 weekly effect Effects 0.000 description 2
- MWWSFMDVAYGXBV-MYPASOLCSA-N (7r,9s)-7-[(2r,4s,5s,6s)-4-amino-5-hydroxy-6-methyloxan-2-yl]oxy-6,9,11-trihydroxy-9-(2-hydroxyacetyl)-4-methoxy-8,10-dihydro-7h-tetracene-5,12-dione;hydrochloride Chemical compound Cl.O([C@@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 MWWSFMDVAYGXBV-MYPASOLCSA-N 0.000 description 1
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 1
- IAKHMKGGTNLKSZ-INIZCTEOSA-N (S)-colchicine Chemical compound C1([C@@H](NC(C)=O)CC2)=CC(=O)C(OC)=CC=C1C1=C2C=C(OC)C(OC)=C1OC IAKHMKGGTNLKSZ-INIZCTEOSA-N 0.000 description 1
- VVJYUAYZJAKGRQ-BGZDPUMWSA-N 1-[(2r,4r,5s,6r)-4,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]-5-methylpyrimidine-2,4-dione Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)C1 VVJYUAYZJAKGRQ-BGZDPUMWSA-N 0.000 description 1
- VSNHCAURESNICA-NJFSPNSNSA-N 1-oxidanylurea Chemical compound N[14C](=O)NO VSNHCAURESNICA-NJFSPNSNSA-N 0.000 description 1
- PMUNIMVZCACZBB-UHFFFAOYSA-N 2-hydroxyethylazanium;chloride Chemical compound Cl.NCCO PMUNIMVZCACZBB-UHFFFAOYSA-N 0.000 description 1
- 238000012604 3D cell culture Methods 0.000 description 1
- FWBHETKCLVMNFS-UHFFFAOYSA-N 4',6-Diamino-2-phenylindol Chemical compound C1=CC(C(=N)N)=CC=C1C1=CC2=CC=C(C(N)=N)C=C2N1 FWBHETKCLVMNFS-UHFFFAOYSA-N 0.000 description 1
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 description 1
- TVZGACDUOSZQKY-LBPRGKRZSA-N 4-aminofolic acid Chemical compound C1=NC2=NC(N)=NC(N)=C2N=C1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 TVZGACDUOSZQKY-LBPRGKRZSA-N 0.000 description 1
- 208000030507 AIDS Diseases 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 241000242759 Actiniaria Species 0.000 description 1
- 102100034540 Adenomatous polyposis coli protein Human genes 0.000 description 1
- 241000242764 Aequorea victoria Species 0.000 description 1
- HJCMDXDYPOUFDY-WHFBIAKZSA-N Ala-Gln Chemical compound C[C@H](N)C(=O)N[C@H](C(O)=O)CCC(N)=O HJCMDXDYPOUFDY-WHFBIAKZSA-N 0.000 description 1
- 241000242757 Anthozoa Species 0.000 description 1
- 102000006306 Antigen Receptors Human genes 0.000 description 1
- 108010083359 Antigen Receptors Proteins 0.000 description 1
- 108010024976 Asparaginase Proteins 0.000 description 1
- 102000015790 Asparaginase Human genes 0.000 description 1
- 239000012583 B-27 Supplement Substances 0.000 description 1
- 102100027314 Beta-2-microglobulin Human genes 0.000 description 1
- 102100026189 Beta-galactosidase Human genes 0.000 description 1
- 108700031361 Brachyury Proteins 0.000 description 1
- 102100026008 Breakpoint cluster region protein Human genes 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- 101150036984 CCN3 gene Proteins 0.000 description 1
- 101100004286 Caenorhabditis elegans best-5 gene Proteins 0.000 description 1
- 101100495352 Candida albicans CDR4 gene Proteins 0.000 description 1
- 102100025470 Carcinoembryonic antigen-related cell adhesion molecule 8 Human genes 0.000 description 1
- 235000014653 Carica parviflora Nutrition 0.000 description 1
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 1
- 108091006146 Channels Proteins 0.000 description 1
- 101710098119 Chaperonin GroEL 2 Proteins 0.000 description 1
- 208000030275 Chondronectin Diseases 0.000 description 1
- 241000243321 Cnidaria Species 0.000 description 1
- 108020004705 Codon Proteins 0.000 description 1
- 102000012422 Collagen Type I Human genes 0.000 description 1
- 108010022452 Collagen Type I Proteins 0.000 description 1
- 102100024203 Collagen alpha-1(XIV) chain Human genes 0.000 description 1
- 101710106877 Collagen alpha-1(XIV) chain Proteins 0.000 description 1
- 208000035473 Communicable disease Diseases 0.000 description 1
- 108010087195 Contactin 1 Proteins 0.000 description 1
- 102100024326 Contactin-1 Human genes 0.000 description 1
- 102100040500 Contactin-6 Human genes 0.000 description 1
- 101710107702 Contactin-6 Proteins 0.000 description 1
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 1
- 102000053602 DNA Human genes 0.000 description 1
- 230000004568 DNA-binding Effects 0.000 description 1
- WEAHRLBPCANXCN-UHFFFAOYSA-N Daunomycin Natural products CCC1(O)CC(OC2CC(N)C(O)C(C)O2)c3cc4C(=O)c5c(OC)cccc5C(=O)c4c(O)c3C1 WEAHRLBPCANXCN-UHFFFAOYSA-N 0.000 description 1
- 102100026662 Delta and Notch-like epidermal growth factor-related receptor Human genes 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- 102100024746 Dihydrofolate reductase Human genes 0.000 description 1
- 241001512730 Discosoma striata Species 0.000 description 1
- 108700019745 Disks Large Homolog 4 Proteins 0.000 description 1
- 102000047174 Disks Large Homolog 4 Human genes 0.000 description 1
- 206010059866 Drug resistance Diseases 0.000 description 1
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 1
- 108010014258 Elastin Proteins 0.000 description 1
- 102000016942 Elastin Human genes 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 101800003838 Epidermal growth factor Proteins 0.000 description 1
- YQYJSBFKSSDGFO-UHFFFAOYSA-N Epihygromycin Natural products OC1C(O)C(C(=O)C)OC1OC(C(=C1)O)=CC=C1C=C(C)C(=O)NC1C(O)C(O)C2OCOC2C1O YQYJSBFKSSDGFO-UHFFFAOYSA-N 0.000 description 1
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 description 1
- 108091092566 Extrachromosomal DNA Proteins 0.000 description 1
- 108010087819 Fc receptors Proteins 0.000 description 1
- 102000009109 Fc receptors Human genes 0.000 description 1
- 108010043685 GPI-Linked Proteins Proteins 0.000 description 1
- 102000002702 GPI-Linked Proteins Human genes 0.000 description 1
- 108700028146 Genetic Enhancer Elements Proteins 0.000 description 1
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 1
- 102100039620 Granulocyte-macrophage colony-stimulating factor Human genes 0.000 description 1
- 108010043121 Green Fluorescent Proteins Proteins 0.000 description 1
- 102000004144 Green Fluorescent Proteins Human genes 0.000 description 1
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000924577 Homo sapiens Adenomatous polyposis coli protein Proteins 0.000 description 1
- 101100273713 Homo sapiens CD2 gene Proteins 0.000 description 1
- 101000914320 Homo sapiens Carcinoembryonic antigen-related cell adhesion molecule 8 Proteins 0.000 description 1
- 101001054266 Homo sapiens Delta and Notch-like epidermal growth factor-related receptor Proteins 0.000 description 1
- 101000932480 Homo sapiens Fms-related tyrosine kinase 3 ligand Proteins 0.000 description 1
- 101001043807 Homo sapiens Interleukin-7 Proteins 0.000 description 1
- 101000927946 Homo sapiens LisH domain-containing protein ARMC9 Proteins 0.000 description 1
- 101000575378 Homo sapiens Microfibrillar-associated protein 2 Proteins 0.000 description 1
- 101000947695 Homo sapiens Microfibrillar-associated protein 5 Proteins 0.000 description 1
- 101000928535 Homo sapiens Protein delta homolog 1 Proteins 0.000 description 1
- 101000800116 Homo sapiens Thy-1 membrane glycoprotein Proteins 0.000 description 1
- 108091006905 Human Serum Albumin Proteins 0.000 description 1
- 102000008100 Human Serum Albumin Human genes 0.000 description 1
- 241000725303 Human immunodeficiency virus Species 0.000 description 1
- 108010028750 Integrin-Binding Sialoprotein Proteins 0.000 description 1
- 102000016921 Integrin-Binding Sialoprotein Human genes 0.000 description 1
- 102000014150 Interferons Human genes 0.000 description 1
- 108010050904 Interferons Proteins 0.000 description 1
- 102000000588 Interleukin-2 Human genes 0.000 description 1
- 108091092195 Intron Proteins 0.000 description 1
- 108091006975 Iron transporters Proteins 0.000 description 1
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 1
- NNJVILVZKWQKPM-UHFFFAOYSA-N Lidocaine Chemical compound CCN(CC)CC(=O)NC1=C(C)C=CC=C1C NNJVILVZKWQKPM-UHFFFAOYSA-N 0.000 description 1
- 102100036882 LisH domain-containing protein ARMC9 Human genes 0.000 description 1
- 108060001084 Luciferase Proteins 0.000 description 1
- 239000005089 Luciferase Substances 0.000 description 1
- 206010025327 Lymphopenia Diseases 0.000 description 1
- PWHULOQIROXLJO-UHFFFAOYSA-N Manganese Chemical compound [Mn] PWHULOQIROXLJO-UHFFFAOYSA-N 0.000 description 1
- 101150076359 Mhc gene Proteins 0.000 description 1
- 102100025599 Microfibrillar-associated protein 2 Human genes 0.000 description 1
- 101710147471 Microfibrillar-associated protein 5 Proteins 0.000 description 1
- VFKZTMPDYBFSTM-KVTDHHQDSA-N Mitobronitol Chemical compound BrC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CBr VFKZTMPDYBFSTM-KVTDHHQDSA-N 0.000 description 1
- ZRKWMRDKSOPRRS-UHFFFAOYSA-N N-Methyl-N-nitrosourea Chemical compound O=NN(C)C(N)=O ZRKWMRDKSOPRRS-UHFFFAOYSA-N 0.000 description 1
- 108010077854 Natural Killer Cell Receptors Proteins 0.000 description 1
- 102000010648 Natural Killer Cell Receptors Human genes 0.000 description 1
- 241001045988 Neogene Species 0.000 description 1
- 229930193140 Neomycin Natural products 0.000 description 1
- 108700024729 Nephroblastoma Overexpressed Proteins 0.000 description 1
- 102000009065 Netrin-1 Human genes 0.000 description 1
- 108010074223 Netrin-1 Proteins 0.000 description 1
- 206010029260 Neuroblastoma Diseases 0.000 description 1
- 239000004677 Nylon Substances 0.000 description 1
- 240000007594 Oryza sativa Species 0.000 description 1
- 235000007164 Oryza sativa Nutrition 0.000 description 1
- 102000004067 Osteocalcin Human genes 0.000 description 1
- 108090000573 Osteocalcin Proteins 0.000 description 1
- 102000004264 Osteopontin Human genes 0.000 description 1
- 108010081689 Osteopontin Proteins 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 1
- 102100033237 Pro-epidermal growth factor Human genes 0.000 description 1
- 108010009736 Protein Hydrolysates Proteins 0.000 description 1
- 108010076504 Protein Sorting Signals Proteins 0.000 description 1
- 101710119301 Protein delta homolog 1 Proteins 0.000 description 1
- 101000762949 Pseudomonas aeruginosa (strain ATCC 15692 / DSM 22644 / CIP 104116 / JCM 14847 / LMG 12228 / 1C / PRS 101 / PAO1) Exotoxin A Proteins 0.000 description 1
- LCTONWCANYUPML-UHFFFAOYSA-M Pyruvate Chemical compound CC(=O)C([O-])=O LCTONWCANYUPML-UHFFFAOYSA-M 0.000 description 1
- 238000012228 RNA interference-mediated gene silencing Methods 0.000 description 1
- 108091030071 RNAI Proteins 0.000 description 1
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 description 1
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 description 1
- 239000006146 Roswell Park Memorial Institute medium Substances 0.000 description 1
- 101710102802 Runt-related transcription factor 2 Proteins 0.000 description 1
- 206010039491 Sarcoma Diseases 0.000 description 1
- 241000242583 Scyphozoa Species 0.000 description 1
- 108091058545 Secretory proteins Proteins 0.000 description 1
- 102000040739 Secretory proteins Human genes 0.000 description 1
- 108091081021 Sense strand Proteins 0.000 description 1
- 229920002684 Sepharose Polymers 0.000 description 1
- 241000700584 Simplexvirus Species 0.000 description 1
- 108020004459 Small interfering RNA Proteins 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 108091081024 Start codon Proteins 0.000 description 1
- 230000006044 T cell activation Effects 0.000 description 1
- 230000024806 T cell lineage commitment Effects 0.000 description 1
- 230000005867 T cell response Effects 0.000 description 1
- 210000000662 T-lymphocyte subset Anatomy 0.000 description 1
- 101000588258 Taenia solium Paramyosin Proteins 0.000 description 1
- 108010009978 Tec protein-tyrosine kinase Proteins 0.000 description 1
- 108010008125 Tenascin Proteins 0.000 description 1
- 102000007000 Tenascin Human genes 0.000 description 1
- 102100029524 Thrombospondin-3 Human genes 0.000 description 1
- 102100029219 Thrombospondin-4 Human genes 0.000 description 1
- 102100033523 Thy-1 membrane glycoprotein Human genes 0.000 description 1
- 102000006601 Thymidine Kinase Human genes 0.000 description 1
- 108020004440 Thymidine kinase Proteins 0.000 description 1
- 102100040247 Tumor necrosis factor Human genes 0.000 description 1
- 108010084455 Zeocin Proteins 0.000 description 1
- JXXCENBLGFBQJM-FYZOBXCZSA-N [(2r)-3-carboxy-2-hydroxypropyl]-trimethylazanium;chloride Chemical compound [Cl-].C[N+](C)(C)C[C@H](O)CC(O)=O JXXCENBLGFBQJM-FYZOBXCZSA-N 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- 230000001070 adhesive effect Effects 0.000 description 1
- 229930013930 alkaloid Natural products 0.000 description 1
- 229960003896 aminopterin Drugs 0.000 description 1
- MWPLVEDNUUSJAV-UHFFFAOYSA-N anthracene Chemical compound C1=CC=CC2=CC3=CC=CC=C3C=C21 MWPLVEDNUUSJAV-UHFFFAOYSA-N 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 230000001640 apoptogenic effect Effects 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 229960003272 asparaginase Drugs 0.000 description 1
- DCXYFEDJOCDNAF-UHFFFAOYSA-M asparaginate Chemical compound [O-]C(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-M 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 230000005784 autoimmunity Effects 0.000 description 1
- 238000011130 autologous cell therapy Methods 0.000 description 1
- 210000002469 basement membrane Anatomy 0.000 description 1
- 210000003651 basophil Anatomy 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 108010005774 beta-Galactosidase Proteins 0.000 description 1
- 230000000975 bioactive effect Effects 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 210000000601 blood cell Anatomy 0.000 description 1
- RSIHSRDYCUFFLA-DYKIIFRCSA-N boldenone Chemical compound O=C1C=C[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 RSIHSRDYCUFFLA-DYKIIFRCSA-N 0.000 description 1
- 210000000988 bone and bone Anatomy 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 239000007975 buffered saline Substances 0.000 description 1
- 229960002092 busulfan Drugs 0.000 description 1
- 210000004899 c-terminal region Anatomy 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 238000007675 cardiac surgery Methods 0.000 description 1
- 229930188550 carminomycin Natural products 0.000 description 1
- XREUEWVEMYWFFA-CSKJXFQVSA-N carminomycin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=C(O)C=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XREUEWVEMYWFFA-CSKJXFQVSA-N 0.000 description 1
- XREUEWVEMYWFFA-UHFFFAOYSA-N carminomycin I Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=C(O)C=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XREUEWVEMYWFFA-UHFFFAOYSA-N 0.000 description 1
- 229960005243 carmustine Drugs 0.000 description 1
- 229950001725 carubicin Drugs 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 230000020411 cell activation Effects 0.000 description 1
- 230000004956 cell adhesive effect Effects 0.000 description 1
- 230000022131 cell cycle Effects 0.000 description 1
- 230000032823 cell division Effects 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 230000022534 cell killing Effects 0.000 description 1
- 230000011748 cell maturation Effects 0.000 description 1
- 239000006285 cell suspension Substances 0.000 description 1
- 230000003833 cell viability Effects 0.000 description 1
- 238000006243 chemical reaction Methods 0.000 description 1
- 230000002759 chromosomal effect Effects 0.000 description 1
- 231100000313 clinical toxicology Toxicity 0.000 description 1
- 230000006690 co-activation Effects 0.000 description 1
- 230000001149 cognitive effect Effects 0.000 description 1
- 238000004737 colorimetric analysis Methods 0.000 description 1
- 239000000356 contaminant Substances 0.000 description 1
- 238000011109 contamination Methods 0.000 description 1
- 239000002872 contrast media Substances 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- 230000000875 corresponding effect Effects 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 229960004397 cyclophosphamide Drugs 0.000 description 1
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 description 1
- 210000000805 cytoplasm Anatomy 0.000 description 1
- 230000001086 cytosolic effect Effects 0.000 description 1
- 231100000599 cytotoxic agent Toxicity 0.000 description 1
- 239000002254 cytotoxic agent Substances 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- 229960003901 dacarbazine Drugs 0.000 description 1
- 229960000975 daunorubicin Drugs 0.000 description 1
- 238000012350 deep sequencing Methods 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 230000003112 degranulating effect Effects 0.000 description 1
- RSIHSRDYCUFFLA-UHFFFAOYSA-N dehydrotestosterone Natural products O=C1C=CC2(C)C3CCC(C)(C(CC4)O)C4C3CCC2=C1 RSIHSRDYCUFFLA-UHFFFAOYSA-N 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 108020001096 dihydrofolate reductase Proteins 0.000 description 1
- 238000006471 dimerization reaction Methods 0.000 description 1
- XEYBRNLFEZDVAW-ARSRFYASSA-N dinoprostone Chemical compound CCCCC[C@H](O)\C=C\[C@H]1[C@H](O)CC(=O)[C@@H]1C\C=C/CCCC(O)=O XEYBRNLFEZDVAW-ARSRFYASSA-N 0.000 description 1
- 229960002986 dinoprostone Drugs 0.000 description 1
- 125000002228 disulfide group Chemical group 0.000 description 1
- 210000003317 double-positive, alpha-beta immature T lymphocyte Anatomy 0.000 description 1
- 229960004679 doxorubicin Drugs 0.000 description 1
- 238000009510 drug design Methods 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 210000003981 ectoderm Anatomy 0.000 description 1
- 230000002900 effect on cell Effects 0.000 description 1
- 229920002549 elastin Polymers 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 210000001900 endoderm Anatomy 0.000 description 1
- 230000008519 endogenous mechanism Effects 0.000 description 1
- 210000002472 endoplasmic reticulum Anatomy 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 210000003979 eosinophil Anatomy 0.000 description 1
- 210000001339 epidermal cell Anatomy 0.000 description 1
- 229940116977 epidermal growth factor Drugs 0.000 description 1
- 210000002514 epidermal stem cell Anatomy 0.000 description 1
- 108010015749 epinectin Proteins 0.000 description 1
- 229960001904 epirubicin Drugs 0.000 description 1
- 239000003797 essential amino acid Substances 0.000 description 1
- 235000020776 essential amino acid Nutrition 0.000 description 1
- ZMMJGEGLRURXTF-UHFFFAOYSA-N ethidium bromide Chemical compound [Br-].C12=CC(N)=CC=C2C2=CC=C(N)C=C2[N+](CC)=C1C1=CC=CC=C1 ZMMJGEGLRURXTF-UHFFFAOYSA-N 0.000 description 1
- 229960005542 ethidium bromide Drugs 0.000 description 1
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 1
- 229960005420 etoposide Drugs 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 230000007717 exclusion Effects 0.000 description 1
- 210000003754 fetus Anatomy 0.000 description 1
- 108060002895 fibrillin Proteins 0.000 description 1
- 102000013370 fibrillin Human genes 0.000 description 1
- 210000002950 fibroblast Anatomy 0.000 description 1
- 238000002189 fluorescence spectrum Methods 0.000 description 1
- 239000007850 fluorescent dye Substances 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 238000010230 functional analysis Methods 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 230000030279 gene silencing Effects 0.000 description 1
- 238000012252 genetic analysis Methods 0.000 description 1
- 239000003862 glucocorticoid Substances 0.000 description 1
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 1
- ZWCXYZRRTRDGQE-SORVKSEFSA-N gramicidina Chemical compound C1=CC=C2C(C[C@H](NC(=O)[C@@H](CC(C)C)NC(=O)[C@H](CC=3C4=CC=CC=C4NC=3)NC(=O)[C@@H](CC(C)C)NC(=O)[C@H](CC=3C4=CC=CC=C4NC=3)NC(=O)[C@@H](CC(C)C)NC(=O)[C@H](CC=3C4=CC=CC=C4NC=3)NC(=O)[C@H](C(C)C)NC(=O)[C@H](C(C)C)NC(=O)[C@@H](C(C)C)NC(=O)[C@H](C)NC(=O)[C@H](NC(=O)[C@H](C)NC(=O)CNC(=O)[C@@H](NC=O)C(C)C)CC(C)C)C(=O)NCCO)=CNC2=C1 ZWCXYZRRTRDGQE-SORVKSEFSA-N 0.000 description 1
- 210000003714 granulocyte Anatomy 0.000 description 1
- 210000003780 hair follicle Anatomy 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 238000007490 hematoxylin and eosin (H&E) staining Methods 0.000 description 1
- 230000011132 hemopoiesis Effects 0.000 description 1
- 229960002897 heparin Drugs 0.000 description 1
- 229920000669 heparin Polymers 0.000 description 1
- 108010038082 heparin proteoglycan Proteins 0.000 description 1
- 210000003897 hepatic stem cell Anatomy 0.000 description 1
- 238000013537 high throughput screening Methods 0.000 description 1
- 230000007236 host immunity Effects 0.000 description 1
- 102000052622 human IL7 Human genes 0.000 description 1
- 108700020610 human chondronectin Proteins 0.000 description 1
- 102000043667 human chondronectin Human genes 0.000 description 1
- 210000004408 hybridoma Anatomy 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 239000012642 immune effector Substances 0.000 description 1
- 102000027596 immune receptors Human genes 0.000 description 1
- 108091008915 immune receptors Proteins 0.000 description 1
- 210000000987 immune system Anatomy 0.000 description 1
- 208000026278 immune system disease Diseases 0.000 description 1
- 230000036039 immunity Effects 0.000 description 1
- 238000003125 immunofluorescent labeling Methods 0.000 description 1
- 230000002163 immunogen Effects 0.000 description 1
- 229940121354 immunomodulator Drugs 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 230000001976 improved effect Effects 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000011503 in vivo imaging Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 229910017053 inorganic salt Inorganic materials 0.000 description 1
- 230000008611 intercellular interaction Effects 0.000 description 1
- 229940079322 interferon Drugs 0.000 description 1
- 230000017307 interleukin-4 production Effects 0.000 description 1
- 210000005061 intracellular organelle Anatomy 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- 238000011813 knockout mouse model Methods 0.000 description 1
- 229960004194 lidocaine Drugs 0.000 description 1
- 108700041430 link Proteins 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 230000004807 localization Effects 0.000 description 1
- 229960002247 lomustine Drugs 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 210000004324 lymphatic system Anatomy 0.000 description 1
- 210000004698 lymphocyte Anatomy 0.000 description 1
- 210000003738 lymphoid progenitor cell Anatomy 0.000 description 1
- 231100001023 lymphopenia Toxicity 0.000 description 1
- 229920002521 macromolecule Polymers 0.000 description 1
- 210000002540 macrophage Anatomy 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 description 1
- 210000001161 mammalian embryo Anatomy 0.000 description 1
- 229910052748 manganese Inorganic materials 0.000 description 1
- 239000011572 manganese Substances 0.000 description 1
- 210000003593 megakaryocyte Anatomy 0.000 description 1
- 229960001924 melphalan Drugs 0.000 description 1
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 1
- 229960001428 mercaptopurine Drugs 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 229960000485 methotrexate Drugs 0.000 description 1
- 239000011325 microbead Substances 0.000 description 1
- CFCUWKMKBJTWLW-BKHRDMLASA-N mithramycin Chemical compound O([C@@H]1C[C@@H](O[C@H](C)[C@H]1O)OC=1C=C2C=C3C[C@H]([C@@H](C(=O)C3=C(O)C2=C(O)C=1C)O[C@@H]1O[C@H](C)[C@@H](O)[C@H](O[C@@H]2O[C@H](C)[C@H](O)[C@H](O[C@@H]3O[C@H](C)[C@@H](O)[C@@](C)(O)C3)C2)C1)[C@H](OC)C(=O)[C@@H](O)[C@@H](C)O)[C@H]1C[C@@H](O)[C@H](O)[C@@H](C)O1 CFCUWKMKBJTWLW-BKHRDMLASA-N 0.000 description 1
- 229960005485 mitobronitol Drugs 0.000 description 1
- 239000003226 mitogen Substances 0.000 description 1
- 230000011278 mitosis Effects 0.000 description 1
- 229960001156 mitoxantrone Drugs 0.000 description 1
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 1
- 239000002362 mulch Substances 0.000 description 1
- 210000002894 multi-fate stem cell Anatomy 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 210000000663 muscle cell Anatomy 0.000 description 1
- 210000003914 myeloid leukocyte Anatomy 0.000 description 1
- 101150091879 neo gene Proteins 0.000 description 1
- 229960004927 neomycin Drugs 0.000 description 1
- 210000005036 nerve Anatomy 0.000 description 1
- 210000001178 neural stem cell Anatomy 0.000 description 1
- 230000007472 neurodevelopment Effects 0.000 description 1
- 210000000440 neutrophil Anatomy 0.000 description 1
- 238000010899 nucleation Methods 0.000 description 1
- 230000030648 nucleus localization Effects 0.000 description 1
- 229920001778 nylon Polymers 0.000 description 1
- QYSGYZVSCZSLHT-UHFFFAOYSA-N octafluoropropane Chemical compound FC(F)(F)C(F)(F)C(F)(F)F QYSGYZVSCZSLHT-UHFFFAOYSA-N 0.000 description 1
- 238000010397 one-hybrid screening Methods 0.000 description 1
- 230000005868 ontogenesis Effects 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 229940045681 other alkylating agent in atc Drugs 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- 210000000496 pancreas Anatomy 0.000 description 1
- 201000002528 pancreatic cancer Diseases 0.000 description 1
- 208000008443 pancreatic carcinoma Diseases 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 210000004976 peripheral blood cell Anatomy 0.000 description 1
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 1
- CWCMIVBLVUHDHK-ZSNHEYEWSA-N phleomycin D1 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC[C@@H](N=1)C=1SC=C(N=1)C(=O)NCCCCNC(N)=N)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C CWCMIVBLVUHDHK-ZSNHEYEWSA-N 0.000 description 1
- 239000013612 plasmid Substances 0.000 description 1
- 229960003171 plicamycin Drugs 0.000 description 1
- 230000008488 polyadenylation Effects 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 201000000742 primary sclerosing cholangitis Diseases 0.000 description 1
- 210000001978 pro-t lymphocyte Anatomy 0.000 description 1
- CPTBDICYNRMXFX-UHFFFAOYSA-N procarbazine Chemical compound CNNCC1=CC=C(C(=O)NC(C)C)C=C1 CPTBDICYNRMXFX-UHFFFAOYSA-N 0.000 description 1
- 229960000624 procarbazine Drugs 0.000 description 1
- 230000000750 progressive effect Effects 0.000 description 1
- 230000000770 proinflammatory effect Effects 0.000 description 1
- 229960003712 propranolol Drugs 0.000 description 1
- XEYBRNLFEZDVAW-UHFFFAOYSA-N prostaglandin E2 Natural products CCCCCC(O)C=CC1C(O)CC(=O)C1CC=CCCCC(O)=O XEYBRNLFEZDVAW-UHFFFAOYSA-N 0.000 description 1
- 239000003531 protein hydrolysate Substances 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 235000008160 pyridoxine Nutrition 0.000 description 1
- 239000011677 pyridoxine Substances 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 230000007420 reactivation Effects 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 230000001172 regenerating effect Effects 0.000 description 1
- 230000008929 regeneration Effects 0.000 description 1
- 238000011069 regeneration method Methods 0.000 description 1
- 230000008439 repair process Effects 0.000 description 1
- 230000008672 reprogramming Effects 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 230000002207 retinal effect Effects 0.000 description 1
- 235000009566 rice Nutrition 0.000 description 1
- 231100000279 safety data Toxicity 0.000 description 1
- 239000000523 sample Substances 0.000 description 1
- 108010038379 sargramostim Proteins 0.000 description 1
- HVAKSLUOHARFLM-UHFFFAOYSA-N selenium;sodium Chemical compound [Se][Na] HVAKSLUOHARFLM-UHFFFAOYSA-N 0.000 description 1
- 238000002741 site-directed mutagenesis Methods 0.000 description 1
- 210000001626 skin fibroblast Anatomy 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 239000000243 solution Substances 0.000 description 1
- 210000001988 somatic stem cell Anatomy 0.000 description 1
- 125000006850 spacer group Chemical group 0.000 description 1
- 230000009870 specific binding Effects 0.000 description 1
- 230000003595 spectral effect Effects 0.000 description 1
- 230000002269 spontaneous effect Effects 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 231100000617 superantigen Toxicity 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 230000002195 synergetic effect Effects 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 description 1
- 229960001278 teniposide Drugs 0.000 description 1
- 210000001550 testis Anatomy 0.000 description 1
- 238000010257 thawing Methods 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 108010060803 thrombospondin 3 Proteins 0.000 description 1
- 108010060815 thrombospondin 4 Proteins 0.000 description 1
- 229960003087 tioguanine Drugs 0.000 description 1
- 230000000451 tissue damage Effects 0.000 description 1
- 231100000827 tissue damage Toxicity 0.000 description 1
- 239000011573 trace mineral Substances 0.000 description 1
- 235000013619 trace mineral Nutrition 0.000 description 1
- 230000009261 transgenic effect Effects 0.000 description 1
- 238000002054 transplantation Methods 0.000 description 1
- 230000004614 tumor growth Effects 0.000 description 1
- 210000003171 tumor-infiltrating lymphocyte Anatomy 0.000 description 1
- 241001430294 unidentified retrovirus Species 0.000 description 1
- 238000010200 validation analysis Methods 0.000 description 1
- JXLYSJRDGCGARV-CFWMRBGOSA-N vinblastine Chemical compound C([C@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-CFWMRBGOSA-N 0.000 description 1
- 229960003048 vinblastine Drugs 0.000 description 1
- 239000013603 viral vector Substances 0.000 description 1
- 238000001429 visible spectrum Methods 0.000 description 1
- 239000002676 xenobiotic agent Substances 0.000 description 1
Images


























































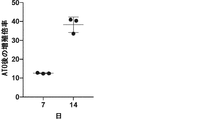




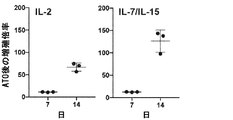






















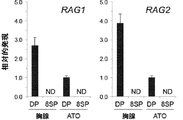









































































Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/12—Materials from mammals; Compositions comprising non-specified tissues or cells; Compositions comprising non-embryonic stem cells; Genetically modified cells
- A61K35/14—Blood; Artificial blood
- A61K35/17—Lymphocytes; B-cells; T-cells; Natural killer cells; Interferon-activated or cytokine-activated lymphocytes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/461—Cellular immunotherapy characterised by the cell type used
- A61K39/4611—T-cells, e.g. tumor infiltrating lymphocytes [TIL], lymphokine-activated killer cells [LAK] or regulatory T cells [Treg]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/463—Cellular immunotherapy characterised by recombinant expression
- A61K39/4631—Chimeric Antigen Receptors [CAR]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/463—Cellular immunotherapy characterised by recombinant expression
- A61K39/4632—T-cell receptors [TCR]; antibody T-cell receptor constructs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/464—Cellular immunotherapy characterised by the antigen targeted or presented
- A61K39/4643—Vertebrate antigens
- A61K39/4644—Cancer antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/705—Receptors; Cell surface antigens; Cell surface determinants
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/705—Receptors; Cell surface antigens; Cell surface determinants
- C07K14/70503—Immunoglobulin superfamily
- C07K14/7051—T-cell receptor (TcR)-CD3 complex
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/0018—Culture media for cell or tissue culture
- C12N5/0037—Serum-free medium, which may still contain naturally-sourced components
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/0062—General methods for three-dimensional culture
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0603—Embryonic cells ; Embryoid bodies
- C12N5/0606—Pluripotent embryonic cells, e.g. embryonic stem cells [ES]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0634—Cells from the blood or the immune system
- C12N5/0636—T lymphocytes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0652—Cells of skeletal and connective tissues; Mesenchyme
- C12N5/0662—Stem cells
- C12N5/0663—Bone marrow mesenchymal stem cells (BM-MSC)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0652—Cells of skeletal and connective tissues; Mesenchyme
- C12N5/0669—Bone marrow stromal cells; Whole bone marrow
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0696—Artificially induced pluripotent stem cells, e.g. iPS
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/60—Immunoglobulins specific features characterized by non-natural combinations of immunoglobulin fragments
- C07K2317/62—Immunoglobulins specific features characterized by non-natural combinations of immunoglobulin fragments comprising only variable region components
- C07K2317/622—Single chain antibody (scFv)
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/01—Fusion polypeptide containing a localisation/targetting motif
- C07K2319/03—Fusion polypeptide containing a localisation/targetting motif containing a transmembrane segment
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2500/00—Specific components of cell culture medium
- C12N2500/05—Inorganic components
- C12N2500/10—Metals; Metal chelators
- C12N2500/20—Transition metals
- C12N2500/22—Zinc; Zn chelators
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2500/00—Specific components of cell culture medium
- C12N2500/05—Inorganic components
- C12N2500/10—Metals; Metal chelators
- C12N2500/20—Transition metals
- C12N2500/24—Iron; Fe chelators; Transferrin
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2500/00—Specific components of cell culture medium
- C12N2500/05—Inorganic components
- C12N2500/10—Metals; Metal chelators
- C12N2500/20—Transition metals
- C12N2500/24—Iron; Fe chelators; Transferrin
- C12N2500/25—Insulin-transferrin; Insulin-transferrin-selenium
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2500/00—Specific components of cell culture medium
- C12N2500/30—Organic components
- C12N2500/32—Amino acids
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2500/00—Specific components of cell culture medium
- C12N2500/30—Organic components
- C12N2500/38—Vitamins
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2500/00—Specific components of cell culture medium
- C12N2500/90—Serum-free medium, which may still contain naturally-sourced components
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2500/00—Specific components of cell culture medium
- C12N2500/99—Serum-free medium
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/125—Stem cell factor [SCF], c-kit ligand [KL]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/145—Thrombopoietin [TPO]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/10—Growth factors
- C12N2501/15—Transforming growth factor beta (TGF-β)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/20—Cytokines; Chemokines
- C12N2501/23—Interleukins [IL]
- C12N2501/2302—Interleukin-2 (IL-2)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/20—Cytokines; Chemokines
- C12N2501/23—Interleukins [IL]
- C12N2501/2304—Interleukin-4 (IL-4)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/20—Cytokines; Chemokines
- C12N2501/23—Interleukins [IL]
- C12N2501/2306—Interleukin-6 (IL-6)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/20—Cytokines; Chemokines
- C12N2501/23—Interleukins [IL]
- C12N2501/2307—Interleukin-7 (IL-7)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/20—Cytokines; Chemokines
- C12N2501/23—Interleukins [IL]
- C12N2501/2315—Interleukin-15 (IL-15)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/20—Cytokines; Chemokines
- C12N2501/23—Interleukins [IL]
- C12N2501/2321—Interleukin-21 (IL-21)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2501/00—Active agents used in cell culture processes, e.g. differentation
- C12N2501/20—Cytokines; Chemokines
- C12N2501/25—Tumour necrosing factors [TNF]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2513/00—3D culture
Description
本出願は、2015年10月30日に提出された米国仮特許出願第62/248,931号、2015年12月9日に提出された米国仮特許出願第62/265,204号、および2016年7月7日に提出された米国仮特許出願第62/359,456号の優先権の恩典を主張するものである。上記の各開示の全内容が、免責条項なしに参照により本明細書に具体的に組み入れられる。
本発明は、米国国立衛生研究所によって授与された助成金番号HL066992の下で政府支援によりなされた。政府は、本発明において一定の権利を有する。
本発明は、概して、細胞培養および発生の分野に関する。より特には、幹細胞または前駆細胞からのT細胞の産生に関する。
現在の改変T細胞療法(TCR改変およびCAR-T手法を含む)は、自家末梢血T細胞を遺伝子改変することに依存する(すなわち、改変のためのT細胞は、それらを受容するであろう同じ患者から単離される)。同種異系(非自己)レシピエントに移植すると、ドナーT細胞の同種反応性により、移植片対宿主疾患(GVHD)として知られるレシピエントにおける組織損傷の症候群をもたらし得るため、自家手法が要される。しかしながら、現在後期臨床試験中である自家細胞療法の商品化への急速な後押しにもかかわらず、患者特異的な自家改変T細胞療法の使用は、極めて労働および費用集約的であり、かつ拡張性が不確定である。さらに、自家改変T細胞療法の使用は、そこから正常なT細胞が十分に回収され得ない患者(例えば、リンパ球減少患者)またはT細胞が機能的に損なわれている患者(例えば、HIV/AIDS患者;加齢に伴う免疫機能不全の結果として高齢患者)においては、除外されるか、効力が小さい。これら多くの懸案事項を考慮すると、同種反応性でない、すぐに使える改変T細胞療法を生み出す方法は、養子細胞療法の分野における満たされていない大きな商業的ニーズである。

幹細胞または前駆細胞からT細胞の組成物を調製するための方法であって、該方法は、
a)Notchリガンドを発現する間質細胞の選択集団;
b)幹細胞または前駆細胞の選択集団
を含む三次元(3D)細胞凝集体を培養する工程を含み、該3D細胞凝集体は、T細胞への幹細胞または前駆細胞のインビトロでの分化に充分な期間、インスリン、ビオチン、トランスフェリン、およびアルブミンを含む血清不含培地中で培養される、方法。
[本発明1002]
幹細胞または前駆細胞からT細胞の組成物を調製するための方法であって、該方法は、
a)Notchリガンドを発現する間質細胞の選択集団;
b)幹細胞または前駆細胞の選択集団
を含む三次元(3D)細胞凝集体を培養する工程を含み、該3D細胞凝集体は、T細胞への幹細胞または前駆細胞のインビトロでの分化に充分な期間、インスリンを含む血清不含培地中で培養される、方法。
[本発明1003]
Notchリガンドが外因性Notchリガンドである、本発明1002の方法。
[本発明1004]
3D細胞凝集体を形成させるための幹細胞または前駆細胞および間質細胞の遠心分離をさらに含む、本発明1002または1003の方法。
[本発明1005]
培地が、外部から添加されたアスコルビン酸をさらに含む、本発明1002〜1004のいずれかの方法。
[本発明1006]
培地が、外部から添加されたFLT3リガンド(FLT3L)、インターロイキン7(IL-7)、幹細胞因子(SCF)、トロンボポエチン(TPO)、幹細胞因子(SCF)、トロンボポエチン(TPO)、IL-2、IL-4、IL-6、IL-15、IL-21、TNF-アルファ、TGF-ベータ、インターフェロン-ガンマ、インターフェロン-ラムダ、TSLP、チモペンチン、プレオトロフィン(pleotrophin)、ミッドカイン、またはそれらの組み合わせをさらに含む、本発明1002〜1005のいずれかの方法。
[本発明1007]
培地がビタミンをさらに含む、本発明1002〜1006のいずれかの方法。
[本発明1008]
ビタミンが、ビオチン、酢酸DLアルファトコフェロール、DLアルファ-トコフェロール、ビタミンA、塩化コリン、パントテン酸カルシウム、パントテン酸、葉酸ニコチンアミド、ピリドキシン、リボフラビン、チアミン、イノシトール、ビタミンB12、またはそれらの組み合わせもしくはそれらの塩を含む、本発明1007の方法。
[本発明1009]
ビタミンが、ビオチン、酢酸DLアルファトコフェロール、DLアルファ-トコフェロール、ビタミンA、またはそれらの組み合わせもしくは塩を含む、本発明1008の方法。
[本発明1010]
培地がタンパク質をさらに含む、本発明1002〜1009のいずれかの方法。
[本発明1011]
タンパク質が、アルブミンもしくはウシ血清アルブミン、BSAの一画分、カタラーゼ、インスリン、トランスフェリン、スーパーオキシドジスムターゼ、またはそれらの組み合わせを含む、本発明1010の方法。
[本発明1012]
培地が、コルチコステロン、D-ガラクトース、エタノールアミン、グルタチオン、L-カルニチン、リノール酸、リノレン酸、プロゲステロン、プトレシン、亜セレン酸ナトリウム、もしくはトリヨード(triodo)-I-サイロニン、またはそれらの組み合わせをさらに含む、本発明1002〜1011のいずれかの方法。
[本発明1013]
培地が、B-27(登録商標)サプリメント、ゼノフリーB-27(登録商標)サプリメント、GS21(商標)サプリメント、またはそれらの組み合わせを含む、本発明1002〜1012のいずれかの方法。
[本発明1014]
培地が、アミノ酸、単糖、無機イオンを含むまたはさらに含む、本発明1002〜1013のいずれかの方法。
[本発明1015]
アミノ酸が、アルギニン、シスチン、イソロイシン、ロイシン、リジン、メチオニン、グルタミン、フェニルアラニン、トレオニン、トリプトファン、ヒスチジン、チロシン、もしくはバリン、またはそれらの組み合わせを含む、本発明1014の方法。
[本発明1016]
無機イオンが、ナトリウム、カリウム、カルシウム、マグネシウム、窒素、もしくはリン、またはそれらの組み合わせもしくは塩を含む、本発明1014の方法。
[本発明1017]
培地が、モリブデン、バナジウム、鉄、亜鉛、セレン、銅、もしくはマンガン、またはそれらの組み合わせをさらに含む、本発明1002〜1016のいずれかの方法。
[本発明1018]
3D細胞凝集体が、規定された外因性細胞外マトリックスを含む、本発明1002〜1017のいずれかの方法。
[本発明1019]
3D細胞凝集体が、外因性マトリックスまたは足場を含まない、本発明1002〜1017のいずれかの方法。
[本発明1020]
間質細胞が、Notchリガンドをコードする外因性ヌクレオチド配列を有する、本発明1002〜1019のいずれかの方法。
[本発明1021]
Notchリガンドが、完全な、部分的な、または改変されたDLL4、DLL1、JAG1、JAG2、またはそれらの組み合わせである、本発明1002〜1020のいずれかの方法。
[本発明1022]
幹細胞または前駆細胞が、胚性幹細胞(ESC)、誘導多能性幹細胞(iPSC)、ヒト胚性中胚葉前駆細胞、造血幹細胞もしくは前駆細胞、骨髄から単離された細胞、臍帯血から単離された細胞、末梢血から単離された細胞、胸腺から単離された細胞、またはインビトロでESCもしくはiPSCから分化させた細胞より選択される、本発明1002〜1021のいずれかの方法。
[本発明1023]
間質細胞:幹細胞または前駆細胞の比率が約1:5〜1:20である、本発明1002〜1022のいずれかの方法。
[本発明1024]
間質細胞が、マウス間質細胞株、ヒト間質細胞株、初代間質細胞の選択集団、インビトロで多能性幹細胞から分化した間質細胞の選択集団、またはそれらの組み合わせである、本発明1002〜1023のいずれかの方法。
[本発明1025]
間質細胞が、インビトロで造血幹細胞または前駆細胞から分化した間質細胞の選択集団である、本発明1002〜1023のいずれかの方法。
[本発明1026]
間質細胞が、外因性ヒト主要組織適合複合体(MHC)を発現する、本発明1002〜1025のいずれかの方法。
[本発明1027]
間質細胞が、外因性抗原特異的共刺激分子またはサイトカインを発現する、本発明1002〜1026のいずれかの方法。
[本発明1028]
間質細胞が外因性抗原を発現する、本発明1002〜1027のいずれかの方法。
[本発明1029]
幹細胞または前駆細胞が以前に凍結されている、本発明1002〜1028のいずれかの方法。
[本発明1030]
幹細胞または前駆細胞が凍結されたことがない、本発明1002〜1028のいずれかの方法。
[本発明1031]
幹細胞または前駆細胞が、外因性T細胞受容体(TCR)もしくはキメラ抗原受容体(CAR)、またはその両方を発現する、本発明1002〜1030のいずれかの方法。
[本発明1032]
幹細胞または前駆細胞が、内因性T細胞受容体(TCR)もしくはキメラ抗原受容体(CAR)、またはその両方を発現する、本発明1002〜1030のいずれかの方法。
[本発明1033]
幹細胞または前駆細胞が、外因性インバリアントナチュラルキラーT細胞(iNKT)関連TCRを発現する、本発明1002〜1032のいずれかの方法。
[本発明1034]
幹細胞または前駆細胞が、外因性抗原特異的TCR、選択もしくはスクリーニングマーカー、インビボ追跡マーカーもしくはインビボイメージングマーカーを発現する、またはHLA遺伝子座、ナチュラルキラー細胞受容体もしくはリガンドの外因性遺伝子改変を有する、本発明1002〜1033のいずれかの方法。
[本発明1035]
幹細胞または前駆細胞の数が1,000〜200,000個である、本発明1002〜1034のいずれかの方法。
[本発明1036]
培養が約2〜10週間の期間である、本発明1002〜1035のいずれかの方法。
[本発明1037]
培養が細胞継代を伴わない、本発明1002〜1036のいずれかの方法。
[本発明1038]
四量体もしくは抗TCR抗体を用いて、CD4 + CD8 - T細胞、CD4 - CD8 + T細胞、CD34 + CD7 + CD1a + 細胞、CD3+ TCRab+細胞、CD3+ TCRgd+細胞、CD3+ TCRab+ CD4+ CD8-細胞、CD3+ TCRab+ CD8+ CD4-細胞、CD3+ TCRab+ CD4+ CD8- CD45RO- CD45RA+細胞、CD3+ TCRab+ CD8+ CD4- CD45RO- CD45RA+細胞、CD3+ TCRab+ CD4+ CD8- CD45RO- CD45RA+ CCR7+細胞、CD3+ TCRab+ CD8+ CD4- CD45RO- CD45RA+ CCR7+細胞、CD3+ TCRab+ CD4+ CD8- CD45RO- CD45RA+ CD27+細胞、CD3+ TCRab+ CD8+ CD4- CD45RO- CD45RA+ CD27+細胞、CD34 + CD7 + CD1a + 細胞、CD34+CD5+CD7+細胞、CD34+CD5+CD7-細胞、ナチュラルキラーT細胞、調節性T細胞、上皮内リンパ球(IEL)、抗原特異的T細胞の数を、改変された抗原を用いてCAR T細胞の数を、蛍光マーカーを用いて形質導入されたT細胞の数を、またはそれらの組み合わせを検出する工程をさらに含む、本発明1002〜1037のいずれかの方法。
[本発明1039]
四量体もしくは抗TCR抗体を用いて、CD4 + CD8 - T細胞、CD4 - CD8 + T細胞、CD34 + CD7 + CD1a + 細胞、CD3+ TCRab+細胞、CD3+ TCRgd+細胞、CD3+ TCRab+ CD4+ CD8-細胞、CD3+ TCRab+ CD8+ CD4-細胞、CD3+ TCRab+ CD4+ CD8- CD45RO- CD45RA+細胞、CD3+ TCRab+ CD8+ CD4- CD45RO- CD45RA+細胞、CD3+ TCRab+ CD4+ CD8- CD45RO- CD45RA+ CCR7+細胞、CD3+ TCRab+ CD8+ CD4- CD45RO- CD45RA+ CCR7+細胞、CD3+ TCRab+ CD4+ CD8- CD45RO- CD45RA+ CD27+細胞、CD3+ TCRab+ CD8+ CD4- CD45RO- CD45RA+ CD27+細胞、CD34 + CD7 + CD1a + 細胞、CD34+CD5+CD7+細胞、CD34+CD5+CD7-細胞、ナチュラルキラーT細胞、調節性T細胞、上皮内リンパ球(IEL)、抗原特異的T細胞を、改変された抗原を用いてCAR T細胞を、蛍光マーカーを用いて形質導入されたT細胞を、またはそれらの組み合わせを選択する工程をさらに含む、本発明1002〜1038のいずれかの方法。
[本発明1040]
四量体もしくは抗TCR抗体を用いて、CD4 + CD8 - T細胞、CD4 - CD8 + T細胞、CD34 + CD7 + CD1a + 細胞、CD3+ TCRab+細胞、CD3+ TCRgd+細胞、CD3+ TCRab+ CD4+ CD8-細胞、CD3+ TCRab+ CD8+ CD4-細胞、CD3+ TCRab+ CD4+ CD8- CD45RO- CD45RA+細胞、CD3+ TCRab+ CD8+ CD4- CD45RO- CD45RA+細胞、CD3+ TCRab+ CD4+ CD8- CD45RO- CD45RA+ CCR7+細胞、CD3+ TCRab+ CD8+ CD4- CD45RO- CD45RA+ CCR7+細胞、CD3+ TCRab+ CD4+ CD8- CD45RO- CD45RA+ CD27+細胞、CD3+ TCRab+ CD8+ CD4- CD45RO- CD45RA+ CD27+細胞、CD34 + CD7 + CD1a + 細胞、CD34+CD5+CD7+細胞、CD34+CD5+CD7-細胞、ナチュラルキラーT細胞、調節性T細胞、上皮内リンパ球(IEL)、抗原特異的T細胞の数を、改変された抗原を用いてCAR T細胞の数を、蛍光マーカーを用いて形質導入されたT細胞の数を、またはそれらの組み合わせを増加させる工程をさらに含む、本発明1002〜1039のいずれかの方法。
[本発明1041]
3D細胞凝集体由来のT細胞を、それを必要としている対象に投与する工程をさらに含む、本発明1002〜1040のいずれかの方法。
[本発明1042]
3D細胞凝集体由来のT細胞を分化させる工程をさらに含む、本発明1002〜1040のいずれかの方法。
[本発明1043]
3D細胞凝集体由来のT細胞が、対立遺伝子排除により内因性TCRを発現しない、本発明1002〜1042のいずれかの方法。
[本発明1044]
3D細胞凝集体由来のT細胞が、外因性TCRまたはCARを発現する、本発明1002〜1043のいずれかの方法。
[本発明1045]
前記期間が2〜16週間である、本発明1002〜1044のいずれかの方法。
[本発明1046]
T細胞が陽性選択を受ける、本発明1002〜1045のいずれかの方法。
[本発明1047]
細胞凝集体が腫瘍細胞または腫瘍抗原をさらに含む、本発明1002〜1046のいずれかの方法。
[本発明1048]
T細胞から内因的に発現したTCRを単離する工程をさらに含む、本発明1002〜1047のいずれかの方法。
[本発明1049]
T細胞を刺激する工程をさらに含む、本発明1048の方法。
[本発明1050]
T細胞を抗原提示細胞で刺激する、本発明1049の方法。
[本発明1051]
抗原提示細胞が腫瘍抗原を提示する、本発明1050の方法。
[本発明1052]
a)i)Notchリガンドを発現する間質細胞の選択集団;
ii)幹細胞または前駆細胞の選択集団
を含む三次元(3D)細胞凝集体と、
b)インスリンを含む血清不含培地と
を含む、細胞培養組成物。
[本発明1053]
Notchリガンドが外因性Notchリガンドである、本発明1052の組成物。
[本発明1054]
培地が、外部から添加されたアスコルビン酸をさらに含む、本発明1052または1053の組成物。
[本発明1055]
培地が、外部から添加されたFLT3リガンド(FLT3L)、インターロイキン7(IL-7)、幹細胞因子(SCF)、トロンボポエチン(TPO)、IL-2、IL-4、IL-6、IL-15、IL-21、TNF-アルファ、TGF-ベータ、インターフェロン-ガンマ、インターフェロン-ラムダ、TSLP、チモペンチン、プレオトロフィン、ミッドカイン、またはそれらの組み合わせをさらに含む、本発明1052〜1054のいずれかの組成物。
[本発明1056]
培地がビタミンをさらに含む、本発明1052〜1055のいずれかの組成物。
[本発明1057]
ビタミンが、ビオチン、酢酸DLアルファトコフェロール、DLアルファ-トコフェロール、ビタミンA、塩化コリン、パントテン酸カルシウム、パントテン酸、葉酸ニコチンアミド、ピリドキシン、リボフラビン、チアミン、イノシトール、ビタミンB12、またはそれらの組み合わせもしくはそれらの塩を含む、本発明1056の組成物。
[本発明1058]
ビタミンが、ビオチン、酢酸DLアルファトコフェロール、DLアルファ-トコフェロール、ビタミンA、またはそれらの組み合わせもしくは塩を含む、本発明1056の組成物。
[本発明1059]
培地がタンパク質をさらに含む、本発明1052〜1058のいずれかの組成物。
[本発明1060]
タンパク質が、アルブミンもしくはウシ血清アルブミン、BSAの一画分、カタラーゼ、インスリン、トランスフェリン、スーパーオキシドジスムターゼ、またはそれらの組み合わせを含む、本発明1059の組成物。
[本発明1061]
培地が、コルチコステロン、D-ガラクトース、エタノールアミン、グルタチオン、L-カルニチン、リノール酸、リノレン酸、プロゲステロン、プトレシン、亜セレン酸ナトリウム、もしくはトリヨード-I-サイロニン、またはそれらの組み合わせをさらに含む、本発明1052〜1060のいずれかの組成物。
[本発明1062]
培地が、B-27(登録商標)サプリメント、ゼノフリーB-27(登録商標)サプリメント、GS21(商標)サプリメント、またはそれらの組み合わせを含む、本発明1052〜1061のいずれかの組成物。
[本発明1063]
培地が、アミノ酸、単糖、もしくは無機イオン、またはそれらの組み合わせを含むまたはさらに含む、本発明1052〜1062のいずれかの組成物。
[本発明1064]
アミノ酸が、アルギニン、シスチン、イソロイシン、ロイシン、リジン、メチオニン、グルタミン、フェニルアラニン、トレオニン、トリプトファン、ヒスチジン、チロシン、もしくはバリン、またはそれらの組み合わせを含む、本発明1063の組成物。
[本発明1065]
無機イオンが、ナトリウム、カリウム、カルシウム、マグネシウム、窒素、もしくはリン、またはそれらの組み合わせもしくは塩を含む、本発明1063の組成物。
[本発明1066]
培地が、モリブデン、バナジウム、鉄、亜鉛、セレン、銅、もしくはマンガン、またはそれらの組み合わせをさらに含む、本発明1052〜1065のいずれかの組成物。
[本発明1067]
3D細胞凝集体が、規定された外因性細胞外マトリックスを含む、本発明1052〜1066のいずれかの組成物。
[本発明1068]
3D細胞凝集体が、外因性マトリックスまたは足場を含まない、本発明1052〜1067のいずれかの組成物。
[本発明1069]
間質細胞が、Notchリガンドをコードする外因性ヌクレオチド配列を有する、本発明1052〜1068のいずれかの組成物。
[本発明1070]
Notchリガンドが、完全な、部分的な、または改変されたDLL4、DLL1、JAG1、JAG2、またはそれらの組み合わせである、本発明1052〜1069のいずれかの組成物。
[本発明1071]
幹細胞または前駆細胞が、ESC、iPSC、ヒト胚性中胚葉前駆細胞、造血幹細胞もしくは前駆細胞、骨髄から単離された細胞、臍帯血から単離された細胞、末梢血から単離された細胞、胸腺から単離された細胞、またはインビトロでESCもしくはiPSCから分化させた細胞より選択される、本発明1052〜1070のいずれかの組成物。
[本発明1072]
間質細胞:幹細胞または前駆細胞の比率が約1:5〜1:20である、本発明1052〜1071のいずれかの組成物。
[本発明1073]
間質細胞が、マウス間質細胞株、ヒト間質細胞株、初代間質細胞の選択集団、インビトロで多能性幹細胞から分化した間質細胞の選択集団、またはそれらの組み合わせである、本発明1052〜1072のいずれかの組成物。
[本発明1074]
間質細胞が、インビトロで造血幹細胞または前駆細胞から分化した間質細胞の選択集団である、本発明1052〜1073のいずれかの組成物。
[本発明1075]
幹細胞または前駆細胞が以前に凍結されている、本発明1052〜1074のいずれかの組成物。
[本発明1076]
幹細胞または前駆細胞が凍結されたことがない、本発明1052〜1074のいずれかの組成物。59.間質細胞が、外因性ヒト主要組織適合複合体(MHC)を発現する、本発明1052〜1076のいずれかの組成物。
[本発明1077]
間質細胞が、外因性抗原特異的共刺激分子、サイトカイン、抗原、もしくは細胞外マトリックスタンパク質、またはT細胞の分化、増殖、もしくは機能を調節する他の任意の生理活性分子もしくは遺伝子を発現する、本発明1052〜1076のいずれかの組成物。
[本発明1078]
幹細胞または前駆細胞が、外因性T細胞受容体(TCR)もしくはキメラ抗原受容体(CAR)、またはその両方を発現する、本発明1052〜1077のいずれかの組成物。
[本発明1079]
幹細胞または前駆細胞が、外因性インバリアントナチュラルキラーT細胞(iNKT)関連TCRを発現する、本発明1052〜1078のいずれかの組成物。
[本発明1080]
幹細胞または前駆細胞が、外因性抗原特異的TCRを発現する、またはT細胞の分化、増殖、もしくは機能を調節する遺伝子の外因性遺伝子改変を有する、本発明1052〜1079のいずれかの組成物。
[本発明1081]
幹細胞または前駆細胞の数が1〜200,000個である、本発明1052〜1080のいずれかの組成物。
[本発明1082]
本発明1052〜1081のいずれかの細胞培養組成物を培養し、それによってT細胞を産生する工程を含む、T細胞を産生するための方法。
[本発明1083]
前駆細胞が外因性抗原特異的TCRまたはCARを発現する、抗原特異的T細胞を産生するための方法としてさらに規定される、本発明1082の方法。
[本発明1084]
四量体もしくは抗TCR抗体を用いて、CD4 + CD8 - T細胞、CD4 - CD8 + T細胞、CD34 + CD7 + CD1a + 細胞、CD3+ TCRab+細胞、CD3+ TCRgd+細胞、CD3+ TCRab+ CD4+ CD8-細胞、CD3+ TCRab+ CD8+ CD4-細胞、CD3+ TCRab+ CD4+ CD8- CD45RO- CD45RA+細胞、CD3+ TCRab+ CD8+ CD4- CD45RO- CD45RA+細胞、CD3+ TCRab+ CD4+ CD8- CD45RO- CD45RA+ CCR7+細胞、CD3+ TCRab+ CD8+ CD4- CD45RO- CD45RA+ CCR7+細胞、CD3+ TCRab+ CD4+ CD8- CD45RO- CD45RA+ CD27+細胞、CD3+ TCRab+ CD8+ CD4- CD45RO- CD45RA+ CD27+細胞、CD34 + CD7 + CD1a + 細胞、CD34+CD5+CD7+細胞、CD34+CD5+CD7-細胞、ナチュラルキラーT細胞、調節性T細胞、上皮内リンパ球(IEL)、抗原特異的T細胞の数を、改変された抗原を用いてCAR T細胞の数を、蛍光マーカーを用いて形質導入されたT細胞の数を、またはそれらの組み合わせを検出する工程をさらに含む、本発明1082または1083の方法。
[本発明1085]
四量体もしくは抗TCR抗体を用いて、CD4 + CD8 - T細胞、CD4 - CD8 + T細胞、CD34 + CD7 + CD1a + 細胞、CD34 + CD7 + CD1a + 細胞、CD3+ TCRab+細胞、CD3+ TCRgd+細胞、CD3+ TCRab+ CD4+ CD8-、CD3+ TCRab+ CD8+ CD4-細胞、CD3+ TCRab+ CD4+ CD8- CD45RO- CD45RA+細胞、CD3+ TCRab+ CD8+ CD4- CD45RO- CD45RA+細胞、CD3+ TCRab+ CD4+ CD8- CD45RO- CD45RA+ CCR7+細胞、CD3+ TCRab+ CD8+ CD4- CD45RO- CD45RA+ CCR7+細胞、CD3+ TCRab+ CD4+ CD8- CD45RO- CD45RA+ CD27+細胞、CD3+ TCRab+ CD8+ CD4- CD45RO- CD45RA+ CD27+細胞、CD34 + CD7 + CD1a + 細胞、CD34+CD5+CD7+細胞、CD34+CD5+CD7-細胞、ナチュラルキラーT細胞、調節性T細胞、上皮内リンパ球(IEL)、抗原特異的T細胞の数を、改変された抗原を用いてCAR T細胞の数を、蛍光マーカーを用いて形質導入されたT細胞の数を、またはそれらの組み合わせを増加させる工程をさらに含む、本発明1082〜1084のいずれかの方法。
[本発明1086]
キメラ抗原受容体(CAR)を含む単離されたT細胞またはT細胞の集団であって、該T細胞は上皮内リンパ球表現型を有する、単離されたT細胞またはT細胞の集団。
[本発明1087]
T細胞がTCR-である、本発明1087のT細胞。
[本発明1088]
CD4- CD8+、CD4+ CD8-、CD4+ CD8+、CD4- CD8-、TCRab+、TCRgd+、CD5+CD7+、CD5+CD7+CD3-CD4-CD8-、CD5+CD7+CD3-CD4-CD8aa、またはそれらの組み合わせである、本発明1086のT細胞。
[本発明1089]
CD5+CD7+CD3-CD4-CD8-である、本発明1088の単離されたT細胞。
[本発明1090]
CD5+CD7+CD3-CD4-CD8aaである、本発明1089の単離されたT細胞。
[本発明1091]
CARがCD19 CARを含む、本発明1086〜1090のいずれかの単離されたT細胞。
[本発明1092]
外因性TCRをさらに含む、本発明1086〜1091のいずれかの単離されたT細胞。
[本発明1093]
CD3+である、本発明1092の単離されたT細胞。
[本発明1094]
CD4 + CD8 - T細胞、CD4 - CD8 + T細胞、CD34 + CD7 + CD1a + 細胞、CD3+ TCRab+、CD3+ TCRgd+細胞、CD3+ TCRab+ CD4+ CD8-細胞、CD3+ TCRab+ CD8+ CD4-細胞、CD3+ TCRab+ CD4+ CD8- CD45RO- CD45RA+細胞、CD3+ TCRab+ CD8+ CD4- CD45RO- CD45RA+細胞、CD3+ TCRab+ CD4+ CD8- CD45RO- CD45RA+ CCR7+細胞、CD3+ TCRab+ CD8+ CD4- CD45RO- CD45RA+ CCR7+細胞、CD3+ TCRab+ CD4+ CD8- CD45RO- CD45RA+ CD27+細胞、CD3+ TCRab+ CD8+ CD4- CD45RO- CD45RA+ CD27+、CD34 + CD7 + CD1a + 細胞、CD34+CD5+CD7+細胞、CD34+CD5+CD7-細胞、ナチュラルキラーT細胞、調節性T細胞、抗原特異的T細胞、上皮内リンパ球T細胞、あるいはCD45+、CD11b+、CD11b-、CD15+、CD15-、CD24+、CD24-、CD114+、CD114-、CD182+、CD182-、CD4+、CD4-、CD14+、CD14-、CD11a+、CD11a-、CD91+、CD91-、CD16+、CD16-、CD3+、CD3-、CD25+、CD25-、Foxp3+、Fox3p-、CD8+、CD8-、CD19+、CD19-、CD20+、CD20-、CD24+、CD24、CD38+、CD38-、CD22+、CD22-、CD61+、CD61-、CD16+、CD16-、CD56+、CD56-、CD31+、CD31-、CD30+、CD30-、CD38+、もしくはCD38-、またはそれらの組み合わせである細胞である、本発明1086〜1093のいずれかの単離されたT細胞。
[本発明1095]
外因性TCRまたはCARを発現し、かつCD4 + CD8 - T細胞、CD4 - CD8 + T細胞、CD34 + CD7 + CD1a + 細胞、CD3+ TCRab+、CD3+ TCRgd+、CD3+ TCRab+ CD4+ CD8-、CD3+ TCRab+ CD8+ CD4-、CD3+ TCRab+ CD4+ CD8- CD45RO- CD45RA+、CD3+ TCRab+ CD8+ CD4- CD45RO- CD45RA+、CD3+ TCRab+ CD4+ CD8- CD45RO- CD45RA+ CCR7+、CD3+ TCRab+ CD8+ CD4- CD45RO- CD45RA+ CCR7+、CD3+ TCRab+ CD4+ CD8- CD45RO- CD45RA+ CD27+、CD3+ TCRab+ CD8+ CD4- CD45RO- CD45RA+ CD27+、CD34 + CD7 + CD1a + 細胞、CD34+CD5+CD7+、CD34+CD5+CD7-、ナチュラルキラーT細胞、調節性T細胞、抗原特異的T細胞、上皮内リンパ球T細胞、CD45+、CD11b+、CD11b-、CD15+、CD15-、CD24+、CD24-、CD114+、CD114-、CD182+、CD182-、CD4+、CD4-、CD14+、CD14-、CD11a+、CD11a-、CD91+、CD91-、CD16+、CD16-、CD3+、CD3-、CD25+、CD25-、Foxp3+、Fox3p-、CD8+、CD8-、CD19+、CD19-、CD20+、CD20-、CD24+、CD24、CD38+、CD38-、CD22+、CD22-、CD61+、CD61-、CD16+、CD16-、CD56+、CD56-、CD31+、CD31-、CD30+、CD30-、CD38+、もしくはCD38-細胞、またはそれらの組み合わせを有する細胞である、単離されたT細胞またはT細胞の集団。
[本発明1096]
T細胞がT細胞の集団であり、かつ該T細胞の集団は、成熟ナイーブCD8またはCD4シングルポジティブ細胞である細胞を少なくとも50%含む、本発明1086〜1096のいずれかのT細胞。
[本発明1097]
外因性インバリアントナチュラルキラーT細胞(iNKT)関連TCRを発現する、本発明1086〜1096のいずれかのT細胞。
[本発明1098]
幹細胞または前駆細胞からインビトロで分化させた、本発明1086〜1097のいずれかのT細胞。
[本発明1099]
幹細胞または前駆細胞が、ESC、iPSC、ヒト胚性中胚葉前駆細胞、造血幹細胞もしくは前駆細胞、骨髄から単離された細胞、臍帯血から単離された細胞、末梢血から単離された細胞、胸腺から単離された細胞、またはインビトロでESCもしくはiPSCから分化させた細胞より選択される、本発明1098のT細胞。
[本発明1100]
内因性TCRが対立遺伝子排除により抑制されている、本発明1086〜1099のいずれかのT細胞。
[本発明1101]
外因性TCRを含むインビトロで分化させたT細胞またはT細胞先駆体を患者に投与する工程を含む、患者を治療するための方法。
[本発明1102]
T細胞が、外因性TCRおよび付加的な抗原またはリガンド認識受容体を含む、本発明1101の方法。
[本発明1103]
外因性TCRがNKT由来のTCRを含む、本発明1101または1102の方法。
[本発明1104]
外因性TCRが、TCR-アルファおよびTCR-ベータ遺伝子から発現されたタンパク質を含む、本発明1101〜1103のいずれかの方法。
[本発明1105]
外因性TCRが、TCR-ガンマおよびTCR-デルタ遺伝子から発現されたタンパク質を含む、本発明1101〜1103のいずれかの方法。
[本発明1106]
外因性TCRが、TCR-アルファおよびTCR-ベータ遺伝子から発現されたタンパク質を含み、かつ抗原認識受容体が、TCR-ガンマおよびTCR-デルタ遺伝子から発現されたタンパク質を含む、本発明1101〜1103のいずれかの方法。
[本発明1107]
外因性TCRが、TCR-ガンマおよびTCR-デルタ遺伝子から発現されたタンパク質を含み、かつ抗原認識受容体が、TCR-アルファおよびTCR-ベータ遺伝子から発現されたタンパク質を含む、本発明1101〜1103のいずれかの方法。
[本発明1108]
付加的な抗原認識受容体がTCR分子ではない、本発明1102の方法。
[本発明1109]
外因性TCRが、TCRシグナル伝達を模倣する改変された分子である、本発明1101〜1108のいずれかの方法。
[本発明1110]
付加的な抗原認識受容体がキメラ抗原受容体(CAR)である、本発明1102または1108の方法。
[本発明1111]
CARが腫瘍抗原特異的CARである、本発明1110の方法。
[本発明1112]
外因性TCRが第一の抗原に特異的であり、かつ付加的な抗原認識受容体が第二の抗原に特異的である、本発明1102〜1111のいずれかの方法。
[本発明1113]
第一および第二の抗原が、患者のがん細胞によって発現されるがん細胞抗原である、本発明1112の方法。
[本発明1114]
外因性TCRが、ウイルス特異的TCR、外来物特異的TCR、がん細胞特異的TCR、細菌特異的TCR、または癌-精巣抗原特異的TCRである、本発明1101〜1113のいずれかの方法。
[本発明1115]
T細胞が、レシピエントに対して同種異系である、本発明1101〜1114のいずれかの方法。
[本発明1116]
患者ががんを有する、本発明1101〜1115のいずれかの方法。
[本発明1117]
対象におけるがんを治療するためのものである、本発明1116の方法。
[本発明1118]
がんが、肺癌、前立腺癌、卵巣癌、精巣癌、脳がん、皮膚癌、黒色腫、結腸癌、直腸癌、胃癌、食道癌、気管癌、頭頸部癌、膵臓癌、肝臓癌、乳癌、卵巣癌、リンパ腫および多発性骨髄腫を含めたリンパ系がん、白血病、骨または軟部組織の肉腫、子宮頸癌、ならびに外陰癌より選択される、本発明1116または1117の方法。
[本発明1119]
外因性TCRによって認識される抗原の投与をさらに含む、本発明1101〜1118のいずれかの方法。
[本発明1120]
抗原が、精製される、他の分子にコンジュゲートされる、または細胞もしくは細胞様ビヒクルによって提示される、本発明1119の方法。
[本発明1121]
外因性TCRが同種反応性でない、本発明1101〜1120のいずれかの方法。
[本発明1122]
外因性TCRが不活性である、本発明1101〜1121のいずれかの方法。
[本発明1123]
一部の態様において、CARは、ウイルス抗原特異的CARまたは細菌抗原特異的CARである。
[本発明1124]
患者が、微生物感染症を有するまたは有する危険性がある、本発明1101〜1115または1119〜1123のいずれかの方法。
[本発明1125]
第一および第二の抗原が、同じウイルスタイプによって、または該ウイルスタイプに感染した細胞によって発現されるウイルス抗原である、本発明1124の方法。
[本発明1126]
第一および第二の抗原が、同じ細菌によって、または該細菌に感染した細胞において発現される細菌細胞抗原である、本発明1124の方法。
[本発明1127]
外因性TCRがNY-ESO-1特異的TCRである、本発明1101〜1123のいずれかの方法。
[本発明1128]
患者への抗原提示細胞の投与をさらに含む、本発明1101〜1127のいずれかの方法。
[本発明1129]
抗原提示細胞が樹状細胞である、本発明1128の方法。
[本発明1130]
抗原提示細胞が人工抗原提示細胞である、本発明1128の方法。
[本発明1131]
抗原提示細胞に、外因性TCRまたはCARによって認識される抗原がロードされている、本発明1128〜1130のいずれかの方法。
[本発明1132]
T細胞がFOXP3の発現をさらに含む、本発明1101〜1131のいずれかの方法。
[本発明1133]
T細胞が、FOXP3を発現するように操作されるまたは選択される、本発明1132の方法。
[本発明1134]
FOXP3発現が構成的である、本発明1133の方法。
[本発明1135]
対象が、自己免疫疾患、移植片対宿主疾患(GVHD)、または移植片拒絶を有するまたは有する危険性がある、本発明1132〜1134のいずれかの方法。
[本発明1136]
自己免疫疾患、GVHD、または移植片拒絶を治療するためのものである、本発明1135の方法。
[本発明1137]
外因性TCRを含む、インビトロで分化させたT細胞またはT細胞先駆体。
[本発明1138]
TCRが、ウイルス特異的TCR、外来物特異的TCR、がん細胞特異的TCR、細菌特異的TCR、または癌-精巣抗原特異的TCRである、本発明1137のT細胞。
[本発明1139]
付加的な抗原認識受容体をさらに含む、本発明1137または1138のT細胞。
[本発明1140]
外因性TCRが、TCR-アルファおよびTCR-ベータ遺伝子から発現されたタンパク質を含む、本発明1138または1139のT細胞。
[本発明1141]
外因性TCRが、TCR-ガンマおよびTCR-デルタ遺伝子から発現されたタンパク質を含む、本発明1138または1139のT細胞。
[本発明1142]
外因性TCRが、TCR-アルファおよびTCR-ベータ遺伝子から発現されたタンパク質を含み、かつ抗原認識受容体が、TCR-ガンマおよびTCR-デルタ遺伝子から発現されたタンパク質を含む、本発明1139のT細胞。
[本発明1143]
外因性TCRが、TCR-ガンマおよびTCR-デルタ遺伝子から発現されたタンパク質を含み、かつ抗原認識受容体が、TCR-アルファおよびTCR-ベータ遺伝子から発現されたタンパク質を含む、本発明1139のT細胞。
[本発明1144]
付加的な抗原認識受容体がTCR分子ではない、本発明1139のT細胞。
[本発明1145]
付加的な抗原認識受容体がキメラ抗原受容体(CAR)である、本発明1139または1144のT細胞。
[本発明1146]
CARが腫瘍抗原特異的CARである、本発明1145のT細胞。
[本発明1147]
CARがウイルス抗原特異的CARである、本発明1145のT細胞。
[本発明1148]
外因性TCRが第一の抗原に特異的であり、かつ付加的な抗原認識受容体が第二の抗原に特異的である、本発明1138〜1144のいずれかのT細胞。
[本発明1149]
第一および第二の抗原が、がんの細胞によって発現されるがん細胞抗原である、本発明1148のT細胞。
[本発明1150]
第一および第二の抗原が、ウイルスまたは該ウイルスに感染した細胞によって発現されるウイルス抗原である、本発明1148のT細胞。
[本発明1151]
第一および第二の抗原が、細菌の細胞または該細菌に感染した細胞によって発現される細菌抗原である、本発明1148のT細胞。
[本発明1152]
外因性TCRがNY-ESO-1特異的TCRである、本発明1137〜1149のいずれかのT細胞。
[本発明1153]
FOXP3の発現をさらに含む、本発明1137〜1152のいずれかのT細胞。
[本発明1154]
FOXP3を発現するように操作されるまたは選択される、本発明1153のT細胞。
[本発明1155]
FOXP3発現が構成的である、本発明1154のT細胞。
[本発明1156]
抗原にコンジュゲートした作用物質を患者に投与する工程を含む、外因性TCR発現T細胞を有する患者における外因性TCR発現T細胞への作用物質の送達のための方法であって、該抗原は該外因性TCRによって認識される、方法。
[本発明1157]
外因性TCRが不活性である、本発明1156の方法。
[本発明1158]
作用物質が除去剤である、本発明1156または1157の方法。
[本発明1159]
T細胞を含む組成物と、外因性TCRに特異的に結合する作用物質とを接触させて、作用物質-TCR発現細胞複合体を作製する工程、および
組成物から該作用物質-TCR発現細胞複合体を精製する工程
を含む、本発明1137〜1155のいずれかのT細胞のインビトロ選択および単離のための方法。
[本発明1160]
作用物質が抗体である、本発明1159の方法。
[本発明1161]
作用物質がペプチド-MHC多量体である、本発明1159の方法。
[本発明1162]
作用物質を固体支持体に付着させる、本発明1159〜1161のいずれかの方法。
[本発明1163]
作用物質に特異的に結合する二次分子の投与をさらに含む、本発明1159〜1161のいずれかの方法。
[本発明1164]
二次分子を固体支持体に付着させる、本発明1163の方法。
[本発明1165]
組成物からの固体支持体および結合分子の分離をさらに含む、本発明1162〜1164のいずれかの方法。
[本発明1166]
固体支持体および結び付いた分子を1回または複数回洗浄する工程をさらに含む、本発明1165の方法。
[本発明1167]
作用物質または二次分子を蛍光分子とコンジュゲートさせる、本発明1159〜1165のいずれかの方法。
[本発明1168]
作用物質-TCR発現細胞複合体を精製する工程が、フローサイトメトリー、蛍光活性化細胞選別(FACS)、ビーズ、および/またはカラムを含む、本発明1159〜1167のいずれかの方法。
[本発明1169]
作用物質-TCR発現細胞複合体を精製する工程が、他のT細胞マーカーに基づく精製を含む、本発明1159〜1168のいずれかの方法。
[本発明1170]
他のT細胞マーカーが、CD4、CD8、CD45RA、CD45RO、CCR7/CD197、CD62L、CD27、CD28、およびCD1aのうちの1つまたは複数である、本発明1169の方法。
[本発明1171]
作用物質-TCR発現細胞複合体から作用物質を解離させる工程をさらに含む、本発明1159〜1170のいずれかの方法。
[本発明1172]
精製されたTCR発現細胞を培養する工程をさらに含む、本発明1159〜1166のいずれかの方法。
[本発明1173]
精製されたTCR発現細胞を凍結する工程をさらに含む、本発明1159〜1172のいずれかの方法。
[本発明1174]
a)外因性TCRまたはTCR誘導体を幹細胞または前駆細胞に移入する工程;および
b)該幹細胞または前駆細胞をT細胞またはT細胞先駆体に分化させる工程
を含む、本発明1137〜1155のいずれかのT細胞を作製する方法。
[本発明1175]
幹細胞または前駆細胞を、分化剤との接触の前に、同時に、および/または後に、同族MHCおよび/またはペプチド分子と接触させる、本発明1174の方法。
[本発明1176]
幹細胞または前駆細胞をT細胞に分化させる工程が、幹細胞または前駆細胞を、Notchリガンドを発現する間質細胞と共培養することを含む、本発明1174または1175の方法。
[本発明1177]
間質細胞がOP9細胞、MS5、またはHS27細胞である、本発明1176の方法。
[本発明1178]
Notchリガンドがデルタ様1(Dll1)である、本発明1176または1177の方法。
[本発明1179]
共培養した幹細胞または前駆細胞および間質細胞を、Flt-3リガンドおよび/またはIL-7と接触させる工程をさらに含む、本発明1176〜1178のいずれかの方法。
[本発明1180]
幹細胞または免疫前駆細胞をT細胞に分化させる工程が、本発明1002〜1052のいずれかの方法を含む、本発明1174または1175の方法。
[本発明1181]
対象におけるT細胞の数を増加させるための、または対象における疾患もしくは病状を治療するための方法であって、有効量の、本発明1002〜1052のいずれかに従って調製されたT細胞もしくは抗原特異的T細胞、または本発明1137〜1155もしくは1086〜1100のいずれかのT細胞を対象に投与する工程を含む、方法。
[本発明1182]
対象が、自己免疫疾患、がん、感染症、免疫不全、またはそれらの組み合わせを有するまたはその危険性があると判定されている、本発明1181の方法。
[本発明1183]
T細胞が調節性T細胞である、本発明1181または1182の方法。
[本発明1184]
対象が、自己免疫疾患を有するまたはその危険性があると判定されている、本発明1183の方法。
[本発明1185]
三次元(3D)細胞凝集体からT細胞を産生するのに有効な濃度の血清不含培地を用いて3D細胞凝集体を培養する工程を含む、自己抗原と反応しないT細胞を産生するための方法であって、該3D細胞凝集体は、a)Notchリガンドを発現する間質細胞の選択集団、およびb)幹細胞または前駆細胞の選択集団を含み、a)またはb)の1種または複数種の細胞は、外因性自己抗原を発現し;それによって該3D細胞凝集体は、自己抗原と反応しないT細胞を産生する、方法。
[本発明1186]
a)またはb)の細胞のうちの1種または複数種が外因性自己MHCを発現する、本発明1185の方法。
本発明の他の目的、特質、および利点は、以下の詳細な説明から明白になるであろう。しかしながら、この詳細な説明から、本発明の精神および範囲の内にある様々な変化および改変が当業者に明白になるであろうため、本発明の好ましい態様が示されているものの、詳細な説明および具体的な例は、例証としてのみ与えられることが理解されるべきである。
I.序論
レシピエント患者に対して同種異系であり得る非同種反応性T細胞を産生するための組成物および方法が記載される。これは、より費用効果が高くより労働集約度が低い療法、および自家T細胞療法が可能でない個体における免疫療法を提供する、当技術分野における重大な改善である。さらに、改変T細胞の生成のための新規な組成物および方法が提供される。これらの組成物および方法は、一部には、高度に標準化された血清不含構成要素および間質細胞株を用いて、ヒトHSPCからの堅牢かつ再現性の高いT細胞分化を促す、人工胸腺オルガノイド(ATO)3D培養物を含む細胞培養組成物の発見に基づいて提供される。ある特定の態様において、ATOにおけるT細胞分化は、内因性の胸腺リンパ球形成を厳密に模倣し、かつ単層共培養とは対照的に、多様なTCRレパートリーおよび抗原ナイーブ表現型を有する機能的CD3+ TCRαβ+ CD8+およびCD4+ T細胞の効率的な陽性選択を支持することが発見された。
・ATOは、最も重要なことには、成熟ナイーブT細胞(CD4SPおよびCD8SPの両方)を含む、造血幹細胞・前駆細胞(HSPC)からのT細胞分化のすべてのステージを生成する。
・ATOにおいてHSPCから成熟T細胞を生成し得る能力は、HSPCにおいて遺伝子(例えば、TCRまたはCAR)を発現させることによって、ATOシステムが改変T細胞を生成するように改変されることを可能にする。
・ATOシステムは、間質区画(MS5)において遺伝子を発現させることによっても改変され得る。
・ATO培養物は血清不含であり得、ゆえにウシ胎仔血清(FCS)のロット間変動の制約、またはFCS含有培地を用いた臨床的橋渡し(translation)の問題を有しない。
「外因性TCR」という用語は、インビトロで細胞内に移入される(すなわち、遺伝子移入/形質導入/トランスフェクション技法によって)、TCR遺伝子またはTCR遺伝子誘導体を指す。外因性TCR遺伝子は、レシピエント細胞のゲノム内に挿入される。一部の態様において、挿入はランダム挿入である。TCR遺伝子のランダム挿入は、当技術分野において公知の方法によって容易に達成される。一部の態様において、TCR遺伝子は、内因性遺伝子座(内因性TCR遺伝子の遺伝子座など)内に挿入される。一部の態様において、細胞は、内因性遺伝子座ではない遺伝子座に挿入される1つまたは複数のTCR遺伝子を含む。一部の態様において、細胞は、マーカーまたは耐性遺伝子などの異種配列をさらに含む。
T細胞受容体またはTCRとは、抗原のフラグメントを主要組織適合複合体(MHC)分子に結合したペプチドとして認識することに関与する、Tリンパ球(T細胞)の表面に見出される分子である。TCRは、2つの異なるタンパク質鎖から構成される(つまり、それはヘテロ二量体である)。ヒトにおけるT細胞の95%において、TCRは、アルファ(α;本明細書において「a」とも称される)およびベータ(β-本明細書において「b」とも称される)鎖からなり、一方でT細胞の5%において、TCRは、ガンマおよびデルタ(γ/δ)鎖からなる。この比率は、個体発生の間および罹患した状態において、ならびに種々の種において変化する。
人工胸腺オルガノイド(ATO)などの3D培養組成物は、幹細胞からのヒトT細胞のインビトロ生成のための、最適化された非常に効率的でかつ高度に再現性のあるすぐに使える解決策である。T細胞分化の既存の実験モデルとは対照的に、3D培養組成物のある特定の局面は、血清不含条件を用い、ヒト胸腺組織または専売的足場材料を回避し、かつ幹細胞からの十分に機能的な成熟ヒトT細胞の陽性選択および堅牢な生成を促す。インビトロT細胞発生のための潜在的に商業的なプラットフォームとして、3D培養組成物は、競合技術と比較して、効率、再現性、拡張性、ならびに費用および労働の低下を提供する。非限定的な商業的適用には、ヒトT細胞発生のインビトロ実験モデル化、および色々な幹細胞供給源からの改変T細胞免疫療法のインビトロ産生が含まれ得る。
本明細書において記載される3D細胞凝集体の培養のための、ならびにT細胞の産生および/またはその陽性/陰性選択のための、細胞培養条件が提供され得る。ある特定の局面において、選択集団の開始細胞は、少なくとももしくは約104、105、106、107、108、109、1010、1011、1012、1013個の細胞、またはその中に導き出せる任意の値域を含み得る。開始細胞集団は、少なくとももしくは約10、101、102、103、104、105、106、107、108細胞/ml、またはその中に導き出せる任意の値域の播種密度を有し得る。
3D細胞凝集体またはその子孫細胞を培養するために用いられる培養容器には、それがその中で幹細胞を培養し得る限り、フラスコ、組織培養用フラスコ、ディッシュ、ペトリディッシュ、組織培養用ディッシュ、マルチディッシュ、マイクロプレート、マイクロウェルプレート、マルチプレート、マルチウェルプレート、マイクロスライド、チャンバースライド、チューブ、トレー、CellSTACK(登録商標)チャンバー、培養バッグ、およびローラーボトルが含まれ得るが、それらに特に限定されるわけではない。幹細胞は、培養の必要性に応じて、少なくとももしくは約0.2、0.5、1、2、5、10、20、30、40、50ml、100ml、150ml、200ml、250ml、300ml、350ml、400ml、450ml、500ml、550ml、600ml、800ml、1000ml、1500ml、またはその中に導き出せる任意の値域の容量で培養され得る。ある特定の態様において、培養容器は、生物学的に活性な環境を支持する任意の装置またはシステムを指し得るバイオリアクターであり得る。バイオリアクターは、少なくとももしくは約2、4、5、6、8、10、15、20、25、50、75、100、150、200、500リットル、1、2、4、6、8、10、15立方メートル、またはその中に導き出せる任意の値域の容量を有し得る。
様々な規定されたマトリックス構成要素が、培養法または組成物において用いられ得る。例えば、参照によりその全体として組み入れられるLudwig et al.(2006a;2006b)に記載されるように、多能性細胞成長のための固体支持体を提供する手段として、組み合わせでの組換えコラーゲンIV、フィブロネクチン、ラミニン、およびビトロネクチンを用いて、培養表面をコーティングし得る。
ある特定の態様において、外因性核酸を含有する細胞は、発現ベクターまたは外因性核酸内にマーカーを含ませることによって、インビトロまたはインビボで同定され得る。そのようなマーカーは、細胞に同定可能な変化を付与し得、発現ベクターを含有する細胞の容易な同定を可能にする。一般的に、選択マーカーは、選択を可能にする特性を付与するものであり得る。陽性選択マーカーは、該マーカーの存在がその選択を可能にするものであり得、一方で陰性選択マーカーは、その存在がその選択を阻止するものである。陽性選択マーカーの例は、薬物耐性マーカーである。
ある特定の態様において、改変されたヌクレアーゼを用いて、本明細書において用いられる任意の細胞、特に培養法または組成物における間質細胞または幹細胞もしくは前駆細胞などの開始細胞、の遺伝子改変のための外因性核酸配列を導入し得る。
ある特定の態様において、ベクターは、本明細書において用いられる任意の細胞、特に培養法または組成物における間質細胞または幹細胞もしくは前駆細胞などの開始細胞、の遺伝子改変のための外因性核酸配列を含むように構築され得る。これらのベクターの構成要素および送達法についての詳細は、下記で開示される。
当業者であれば、標準的組換え技法によりベクターを構築する十分な能力を備えているであろう(例えば、両方とも参照により本明細書に組み入れられる、Maniatis et al., 1988およびAusubel et al., 1994を参照されたい)。
ベクターに内包された真核生物発現カセットは、(5'から3'方向に)タンパク質コード配列に機能的に連結された真核生物転写プロモーター、介在配列を含むスプライシングシグナル、および転写終結/ポリアデニル化配列を特に含有する。
「プロモーター」とは、転写の開始および割合が制御される核酸配列の領域である制御配列である。それは、そこでRNAポリメラーゼおよび他の転写因子などの調節タンパク質および分子が結合して、核酸配列の特異的転写を開始し得る、遺伝子エレメントを含有し得る。「機能的に配置された」、「機能的に連結された」、「制御下にある」、および「転写制御下にある」という語句は、プロモーターが、その配列の転写開始および/または発現を制御する核酸配列との関連で、正しい機能的位置および/または配向にあることを意味する。
適切なプロテアーゼ切断部位および自己切断ペプチドは、当業者に公知である(例えば、Ryan et al., 1997;Scymczak et al., 2004を参照されたい)。プロテアーゼ切断部位の例は、ポティウイルスNIaプロテアーゼ(例えば、タバコエッチウイルス(tobacco etch virus)プロテアーゼ)、ポティウイルスHCプロテアーゼ、ポティウイルスP1(P35)プロテアーゼ、ビモウイルス(byovirus)Nlaプロテアーゼ、ビモウイルスRNA-2コードプロテアーゼ、アフトウイルス(aphthovirus)Lプロテアーゼ、エンテロウイルス(enterovirus)2Aプロテアーゼ、ライノウイルス2Aプロテアーゼ、ピコルナ3Cプロテアーゼ、コモウイルス24Kプロテアーゼ、ネポウイルス(nepovirus)24Kプロテアーゼ、RTSV(イネツングロ(rice tungro)球形ウイルス)3C様プロテアーゼ、PY/IF(パースニップ黄色斑点ウイルス)3C様プロテアーゼ、トロンビン、Xa因子、およびエンテロキナーゼの切断部位である。その高い切断ストリンジェンシーにより、TEV(タバコエッチウイルス)プロテアーゼ切断部位が用いられ得る。
ベクターは、そのうちのいずれかを標準的組換え技術と併せて用いてベクターを消化し得る、多数の制限酵素部位を含有する核酸領域であるマルチクローニングサイト(MCS)を含み得る(例えば、参照により本明細書に組み入れられる、Carbonelli et al., 1999、Levenson et al., 1998、およびCocea, 1997を参照されたい)。「制限酵素消化」とは、核酸分子内の特異的位置でのみ機能する酵素による核酸分子の触媒的切断を指す。これらの制限酵素の多くは市販されている。そのような酵素の使用は、当業者によって広く理解されている。しばしば、ベクターを、MCS内で切り込む制限酵素を用いて直線化またはフラグメント化して、外因性配列がベクターにライゲーションされることを可能にする。「ライゲーション」とは、互いに連続していても連続していなくてもよい2つの核酸フラグメント間でホスホジエステル結合を形成する過程を指す。制限酵素およびライゲーション反応を伴う技法は、組換え技術の技術分野における当業者に周知である。
ほとんどの転写される真核生物RNA分子は、RNAスプライシングを受けて、一次転写産物からイントロンを除去する。真核生物ゲノム配列を含有するベクターは、タンパク質発現のための転写産物の適正なプロセシングを確実にするために、ドナーおよび/またはアクセプターのスプライシング部位を要し得る(例えば、参照により本明細書に組み入れられる、Chandler et al., 1997を参照されたい)。
ベクターまたは構築物は、少なくとも1つの終結シグナルを含み得る。「終結シグナル」または「ターミネーター」は、RNAポリメラーゼによるRNA転写産物の特異的終結に関与するDNA配列から構成される。ゆえに、ある特定の態様において、RNA転写産物の産生を終わらせる終結シグナルが企図される。ターミネーターは、望ましいメッセージレベルを達成するためにインビボで必要であり得る。
発現、特に真核生物発現では、典型的に、転写産物の適正なポリアデニル化をもたらすポリアデニル化シグナルが含まれる。ポリアデニル化シグナルの性質は、実践の成功に極めて重要ではないと考えられ、そして任意のそのような配列が採用され得る。例示的な態様は、好都合でありかつ様々な標的細胞において十分に機能することが知られる、SV40ポリアデニル化シグナルまたはウシ成長ホルモンポリアデニル化シグナルを含む。ポリアデニル化は、転写産物の安定性を増加させ得る、または細胞質輸送を促し得る。
宿主細胞内でベクターを繁殖させるために、それは、複製部位の1つまたは複数の起点(「ori」と称されることが多い)、例えば上記のEBVのoriP、または分化プログラミングにおいて類似のもしくは上昇した機能を有する遺伝子操作されたoriPに相当する核酸配列を含有し得、それは、そこで複製が開始される特異的核酸配列である。あるいは、上記の他の染色体外で複製するウイルスの複製起点、または自律複製配列(ARS)が採用され得る。
培養組成物または方法の開始細胞内への遺伝子改変または外因性核酸の導入は、本明細書において記載されるまたは当業者に公知であろう、細胞の形質転換のための核酸送達のための任意の適切な方法を用い得る。そのような方法には、エクスビボトランスフェクション(Wilson et al., 1989、Nabel et al, 1989)による、マイクロインジェクション(Harland and Weintraub, 1985;参照により本明細書に組み入れられる、米国特許第5,789,215号)を含めたインジェクション(それぞれが参照により本明細書に組み入れられる、米国特許第5,994,624号、第5,981,274号、第5,945,100号、第5,780,448号、第5,736,524号、第5,702,932号、第5,656,610号、第5,589,466号、および第5,580,859号)による;エレクトロポレーション(参照により本明細書に組み入れられる、米国特許第5,384,253号;Tur-Kaspa et al., 1986;Potter et al., 1984)による;リン酸カルシウム沈殿(Graham and Van Der Eb, 1973;Chen and Okayama, 1987;Rippe et al., 1990)による;ポリエチレングリコールが後に続くDEAE-デキストラン(Gopal, 1985)を用いることによる;直接超音波負荷(Fechheimer et al., 1987)による;リポソーム仲介性トランスフェクション(Nicolau and Sene, 1982;Fraley et al., 1979;Nicolau et al., 1987;Wong et al., 1980;Kaneda et al., 1989;Kato et al., 1991)および受容体仲介性トランスフェクション(Wu and Wu, 1987;Wu and Wu, 1988)による;微粒子銃(PCT出願第WO 94/09699号および第95/06128号;米国特許第5,610,042号;第5,322,783号、第5,563,055号、第5,550,318号、第5,538,877号、および第5,538,880号、そしてそれぞれが参照により本明細書に組み入れられる)による;炭化ケイ素繊維との撹拌(Kaeppler et al., 1990;それぞれが参照により本明細書に組み入れられる、米国特許第5,302,523号および第5,464,765号)による;アグロバクテリウム(Agrobacterium)仲介性形質転換(それぞれが参照により本明細書に組み入れられる、米国特許第5,591,616号および第5,563,055号)による;プロトプラストのPEG仲介性形質転換(Omirulleh et al., 1993;それぞれが参照により本明細書に組み入れられる、米国特許第4,684,611号および第4,952,500号)による;乾燥/阻害仲介性DNA取り込み(Potrykus et al., 1985)による;ならびに、そのような方法の任意の組み合わせなどのDNAの直接送達が含まれるが、それらに限定されるわけではない。これらなどの技法の適用により、オルガネラ、細胞、組織、または生物は、安定にまたは一時的に形質転換され得る。
ある特定の態様において、核酸は、例えばリポソームなどの脂質複合体に封入され得る。リポソームは、リン脂質二重層膜および内側の水性媒体によって特徴付けされる小胞構造である。多重膜リポソームは、水性媒体によって分離された多数の脂質層を有する。リン脂質が過剰な水溶液中に懸濁された場合、それらは自然発生的に形成される。脂質構成要素は、閉じた構造の形成前に自己再編成を受け、かつ脂質二重層の間に水および溶解した溶質を封入する(Ghosh and Bachhawat, 1991)。Lipofectamine(Gibco BRL)またはSuperfect(Qiagen)と複合した核酸も企図される。用いられるリポソームの量は、リポソームならびに用いられる細胞の性質によって変動し得、例えば1〜10百万個の細胞あたり約5〜約20μgのベクターDNAが企図され得る。
ある特定の態様において、核酸は、エレクトロポレーションを介して細胞内に導入される。エレクトロポレーションは、細胞とDNAとの懸濁液の高電圧放電への曝露を伴う。レシピエント細胞は、機械的創傷により、形質転換に対してより感受性を高くされ得る。また、用いられるベクターの量は、用いられる細胞の性質によって変動し得、例えば1〜10百万個の細胞あたり約5〜約20μgのベクターDNAが企図され得る。
他の態様において、核酸は、リン酸カルシウム沈殿を用いて細胞に導入される。この技法を用いて、ヒトKB細胞にアデノウイルス5のDNAがトランスフェクトされている(Graham and Van Der Eb, 1973)。またこの様式で、マウスL(A9)、マウスC127、CHO、CV-1、BHK、NIH3T3、およびHeLa細胞に、ネオマイシンマーカー遺伝子がトランスフェクトされ(Chen and Okayama, 1987)、そしてラット肝細胞に色々なマーカー遺伝子がトランスフェクトされた(Rippe et al., 1990)。
別の態様において、核酸は、ポリエチレングリコールが後に続くDEAE-デキストランを用いて細胞内に送達される。この様式で、レポータープラスミドは、マウス骨髄腫および赤白血球病細胞内に導入された(Gopal, 1985)。
本明細書において記載される培養組成物を用いて、下記で例示される商業的または臨床的な適用を有するT細胞を産生し得る。
多能性幹細胞または造血幹細胞もしくは前駆細胞などの開始細胞は、選択されたT細胞系統に沿った分化のためのある特定の組成物または方法において用いられ得る。間質細胞を用いて、幹細胞または前駆細胞と共培養し得る。
間質細胞は、任意の器官の、例えば骨髄、胸腺、子宮粘膜(子宮内膜)、前立腺、および卵巣における、結合組織細胞である。それらは、その器官の実質細胞の機能を支持する細胞である。線維芽細胞(間葉系間質細胞/MSCとしても知られる)および周皮細胞は、中でも最もよく見られるタイプの間質細胞である。
造血幹細胞および前駆細胞の重大な医学的潜在性が理由で、胚性幹細胞からの造血前駆細胞の分化のための方法を改善しようと試みる実質的な仕事が行われている。ヒト成体において、主として骨髄に存在する造血幹細胞は、血液系のすべての細胞に分化する造血(CD34+)前駆細胞の異質集団を産生する。成体ヒトにおいて、造血前駆体は増殖分化し、毎日何千億という成熟血液細胞の生成をもたらす。造血前駆細胞は、臍帯血にも存在している。インビトロにおいて、ヒト胚性幹細胞は、造血前駆細胞に分化し得る。造血前駆細胞は、下記のように、末梢血のサンプルからも増殖または富化され得る。造血細胞は、ヒト起源、マウス起源、または他の任意の哺乳類種のものであり得る。
造血幹細胞(HSC)は、通常骨髄に存在するが、血中に押し込まれ得、末梢血中の多数のHSCを収集するために臨床的に用いられる動員と称される過程である。一般的に好まれる1種の動員剤は、顆粒球コロニー刺激因子(G-CSF)である。
本明細書において記載される組成物および方法に適した細胞は、造血幹細胞および前駆細胞であり得、インビトロで多能性幹細胞の分化からも調製され得る。一部の態様において、本明細書において記載される方法において用いられる細胞は、ATO内に直接播種される多能性幹細胞(胚性幹細胞または誘導多能性幹細胞)である。さらなる態様において、本明細書において記載される方法および組成物において用いられる細胞は、中胚葉前駆体、血管内皮前駆体、または造血前駆体などであるがそれらに限定されない、PSCの誘導体または子孫である。
ある特定の態様における細胞は、分化の前または後のいずれに、細胞の遺伝子操作によって1つまたは複数の遺伝子変更を含有するように作製され得る(US 2002/0168766)。外因性核酸もしくはポリヌクレオチドが、人工的操作の任意の適切な手段によって細胞内に移入されている場合、または細胞が、ポリヌクレオチドを受け継いでいる元の変更された細胞の子孫である場合、細胞は「遺伝子変更された」、「遺伝子改変された」、または「トランスジェニック」であると言われる。例えば、細胞は、それらが、制限された発生系統細胞にまたは最終分化した細胞に進行する前または後のいずれかに、テロメラーゼ逆転写酵素を発現するように細胞を遺伝子変更することによって、それらの複製潜在力を増加させるように加工され得る(US 2003/0022367)。
そのような障害に対して検査で陽性と出ている個体、障害の1つもしくは複数の症状を有する個体、またはそのような病状もしくは関連する病状を発症する危険性があると思われる個体に対して、方法が採用され得る。一部の態様において、本明細書において記載される組成物および方法を用いて、本明細書において列挙されるおよび/または当技術分野において公知の障害の炎症性または自己免疫性構成要素を治療する。
自己免疫性溶血、ベック病、クリオグロブリン血症、デュピュイトラン拘縮、水晶体過敏性眼内炎(endophthalmia phacoanaphylactica)、アレルギー性腸炎、癩性結節性紅斑、特発性顔面麻痺、慢性疲労症候群、リウマチ性熱病、ハンマン・リッチ病、感音性(sensoneural)聴力喪失、発作性ヘモグロビン尿症(haemoglobinuria paroxysmatica)、性腺機能低下症、限局性回腸炎(ileitis regionalis)、白血球減少症、伝染性単核球症(mononucleosis infectiosa)、横断性(traverse)脊髄炎、原発性特発性粘液水腫、ネフローゼ、交感性眼炎(ophthalmia symphatica)、肉芽腫性精巣炎、膵炎、急性多発性神経根炎、壊疽性膿皮症、ケルヴァン甲状腺炎(thyreoiditis)、後天性脾臓萎縮症、非悪性胸腺腫、白斑、毒素性ショック症候群、食中毒、T細胞の浸潤に伴う病状、白血球粘着不全症、サイトカインおよびTリンパ球によって仲介される急性および遅延型過敏症と関連した免疫応答、白血球漏出を伴う疾患、多臓器傷害症候群、抗原-抗体複合体仲介性疾患、抗糸球体基底膜疾患、アレルギー性神経炎、自己免疫性多腺性内分泌障害、卵巣炎、原発性粘液水腫、自己免疫性萎縮性胃炎、交感性眼炎、リウマチ性疾患、混合性結合組織疾患、ネフローゼ症候群、膵島炎、多内分泌腺不全、自己免疫性多腺性症候群I型、成人発症型特発性副甲状腺機能低下症(AOIH)、拡張型心筋症などの心筋症、後天性表皮(epidermolisis)水疱症(EBA)、ヘモクロマトーシス、心筋炎、ネフローゼ症候群、原発性硬化性胆管炎、化膿性または非化膿性副鼻腔炎、急性または慢性副鼻腔炎、篩骨洞炎、前頭洞炎、上顎洞炎、または蝶形骨洞炎、好酸球増多症、肺浸潤性好酸球増多症、好酸球増多筋痛症候群、レフラー(Loffler's)症候群、慢性好酸球性肺炎(pneumonia)、熱帯性肺好酸球増多症、気管支肺アスペルギルス症、アスペルギルス腫、または好酸球含有肉芽腫などの好酸球関連障害、アナフィラキシー、血清反応陰性脊椎関節炎(spondyloarthritides)、多内分泌腺自己免疫疾患、硬化性胆管炎、強膜、上強膜、慢性粘膜皮膚カンジダ症、ブルトン症候群、一過性乳児低ガンマグロブリン血症、ウィスコット・アルドリッチ症候群、毛細血管拡張性運動失調症候群、血管拡張症、膠原病と関連した自己免疫障害、リウマチ、神経学的疾患、リンパ節炎、血圧応答の低下、血管機能不全、組織傷害、心血管虚血、痛覚過敏、腎虚血、脳虚血、および血管新生を伴う疾患、アレルギー性過敏性障害、糸球体腎炎、再灌流傷害、虚血再灌流障害、心筋または他の組織の再灌流傷害、リンパ腫様気管気管支炎、炎症性皮膚症、急性炎症性構成要素による皮膚症、多臓器不全、水疱性疾患、腎皮質壊死、急性化膿性髄膜炎または他の中枢神経系炎症性障害、眼および眼窩の炎症性障害、顆粒球輸血関連症候群、サイトカイン誘導性毒性、ナルコレプシー、急性重症炎症、慢性難治性炎症、腎盂炎、動脈内過形成、消化性潰瘍、弁膜炎、移植片対宿主疾患、接触過敏症、喘息性気道過反応、ならびに子宮内膜症も企図される。
以下の実施例は、本発明の好ましい態様を実証するために含まれる。後に続く実施例において開示される技法は、本発明の実践において十分に機能することを本発明者によって発見された技法であり、ゆえにその実践のための好ましい形式をなすと考えられ得ることは、当業者によって解されるべきである。しかしながら、当業者であれば、本開示に照らして、本発明の精神および範囲から逸脱することなく、多くの変化が、開示される具体的な態様においてもたらされ得、かつなおも同様または類似の結果を達成し得ることを解するべきである。
ヒトT細胞発生のためのすぐに使える3Dオルガノイドシステムの開発−目標は、OP9-DL1システムによって例示される、Notchリガンドを形質導入された間質細胞株の可能性を高めて、T細胞の分化および選択における3D相互作用の役割を調べるための標準化された人工胸腺オルガノイドシステムを開発することであった。しかしながら、OP9-DL1細胞は、長期培養下で安定ではなく、T支持潜在力を維持しかつ脂質生成形質転換を阻止するために数日ごとに細胞変化を要する。3D培養物は数週間安定であることを要されるため、長期にわたるインタクトな培養物を可能にするであろう培養および細胞構成要素を同定することが求められた。さらに、OP9-DL1システムの効力は、ウシ胎仔血清(FCS)ロットの生物学的変動に対して非常に感受性が高く、ゆえに本発明者らは、T細胞分化を再現可能に支持し得る血清不含培養条件も同定しようとした。
改変T細胞療法は、がんおよび慢性ウイルス感染症の治療のための前例のない機会を提供する。造血幹細胞・前駆細胞(HSPC)から改変T細胞を直接生成し得る能力は、同種反応性を含めた、末梢血T細胞の使用に伴う主な治療的制約を克服する潜在力を有する。臍帯血、骨髄、および末梢血HSPCからの、天然のおよびTCR改変されたヒトT細胞の非常に効率的なインビトロでの分化および陽性選択を支持する、臨床的に関連する人工胸腺オルガノイド(ATO)システムが本明細書において記載される。ATO由来T細胞は、ナイーブ表現型、多様なTCRレパートリー、ならびにTCR依存的活性化および増殖を呈した。ATO由来TCR改変T細胞もナイーブ表現型に成熟し、さらに対立遺伝子排除の誘導と一致した内因性TCR発現の完全に近い欠如を示した。ゆえに、ATOは、養子細胞療法のためのナイーブの非同種反応性の改変T細胞の生成のための簡便でかつ直接的な方法を提示する。
1.インビトロヒトT細胞分化のための最適化された人工胸腺オルガノイドシステムの開発
1つの目標は、HSPCからのヒトT細胞のインビトロ陽性選択および成熟を支持し得る臨床的に橋渡し可能なオルガノイドシステムを開発することであった。初代胸腺組織の使用を回避するために、DLL1を形質導入された間質細胞株を、3Dオルガノイド培養においてヒトT細胞発生を支持し得る能力について検査した。本発明者らおよび他の研究者らは、OP9-DL1システムにおけるT細胞分化が、ウシ胎仔血清の特異的ロットおよび間質細胞の頻繁な変化に依存して非常に可変的であることを観察したため、本発明者らは、オルガノイド培養においてT細胞分化を一貫して支持し得る血清不含条件を同定することも求めた。専売的足場材料の使用を回避するために、本発明者らは、間質細胞が遠心分離によってHSPCと凝集しかつ空気-流体界面で細胞培養インサート上に配備される、胸腺組織に基づくオルガノイドにおいて有効であることが示された圧縮再凝集技法を用いた(図3A)。これらの3D培養において、本発明者らは、T細胞枯渇したCD34+臍帯血(CB)HSPCからのヒトT細胞分化を強く支持するものとして、ヒトDLL1を形質導入されたMS-5マウス骨髄間質細胞株(以後、MS5-hDLL1)を同定した。さらに、本発明者らは、MS5-hDLL1オルガノイド培養において堅牢なヒトT細胞分化を一貫して支持する新規な血清不含培地として、B27、神経幹および胚性幹細胞培養において用いられる多構成要素添加物、ならびにFLT3L、IL-7、ならびにアスコルビン酸を補充されたRPMI(以後、「RB27」)を同定した。
次に、長期ATOにおけるT細胞分化を、出生後ヒト胸腺におけるものと比較した。12週目のCB ATOは、胸腺と同程度の頻度のT系統コミットした(CD5+CD7+)およびCD34+のT細胞前駆体を示し(図4A)、一方でDPおよびSP頻度は、胸腺においてよりも、12週目のATOにおいてより進歩したT細胞成熟を示唆した(図4A)。胸腺に見られるように、ATOにおけるCD3+細胞の大多数はTCRαβ+であり、より少なくはあるが一致したTCRγδ+集団を有した(図4A)。ATO由来CD3+TCRαβ+細胞の中で、成熟CD8SPおよびCD4SP T細胞の生成は、6〜12週目の間に増加した(図4Bおよび図12A)。胸腺とは対照的に、ATOは、CD8SP T細胞に対して比例的により少ないCD4SP T細胞を呈し、おそらくCD4+ T細胞発生のより遅い動態を反映し;CD4+細胞は、分析された最も遠い時点の12週目まで頻度が増加し続けた(図4Bおよび図12A)。
すべての臨床的に関連するHSPC供給源、すなわち成体骨髄(BM)、G-CSF動員末梢血(MPB)、および非動員末梢血(PB)からの、先駆体およびCD3+TCRαβ+ T細胞の同程度の頻度を有する、効率的なT細胞分化が見られた(図5A〜Bおよび図13A、B)。全細胞増殖も、HSPC供給源にわたって匹敵した(図13C)。CB、BM、またはMPB由来の高度に富化された造血幹細胞(HSC)画分(Lin-CD34+CD38)は、同程度に堅牢なT細胞分化を実証し(図5C〜Dおよび図13D〜E)、これらの供給源からのT細胞潜在性が、既存のリンパ系コミットした前駆体とは無関係であることを示唆した。
次に、本発明者らは、ATOにおいて生成された成熟T細胞のTCR多様性および機能を特徴付けした。共通TCR Vβセグメントに対するATO由来CD3+TCRαβ+CD8SP T細胞についてのフローサイトメトリーは、ヒト胸腺由来の対応するCD8SP T細胞のものに著しく類似した多様性を明らかにした(図6A)。重要なことには、偏ったVβ使用法もクローン選択も観察されず、それぞれ、非従来的なT細胞サブセットまたはクローン増殖した成熟T細胞の、ATOにおける優勢に反論した。
次に、本発明者らは、HSPCからのTCR改変T細胞のインビトロ生成にATOを適応させた。CB CD34+CD3- HSPCに、NY-ESO-1157〜165に特異的なHLA-A*02:01拘束TCRのαおよびβ鎖をコードするレンチウイルスベクターを形質導入した。6週目の時点で、TCR形質導入ATOは、モック形質導入対照と同程度の頻度のCD5+CD7+ Tコミットした細胞を示したが、CD3+TCRαβ+ T細胞を際立って増加させ、その大多数は、四量体、または形質導入されたVβ13.1鎖に対する抗体での染色によって見られるように、形質導入されたTCRを発現した(図7A)。CD8SP細胞の頻度は、モック形質導入対照由来の四量体+細胞とCD3+TCRαβ+細胞との間で同程度であったが、四量体+CD8SP細胞は、成熟ナイーブ表現型(すなわち、CD45RA+CD45RO-CD27+CCR7+CD1a低)への加速した成熟を呈示した(図7A)。非形質導入ATOと同様に、エフェクター/メモリー表現型への分化は観察されなかった(図7A)。
胸腺における陽性選択は、T細胞先駆体上のTCRと胸腺間質細胞および造血細胞上の自己MHCとの間の相互作用によって仲介される。ゆえに、ATOにおける「自己」MHCの造血系または間質系発現が、TCR改変T細胞の陽性選択を増強し得るかどうかが検討された。HLA-A*02:01陽性または陰性ドナーを用いて、ATO内での陽性選択に対する自己MHCの造血系発現の効果を検査し、そして間質細胞MHC発現を、HLA-A*02:01を形質導入されたMS5-hDLL1細胞を用いてATOを生成することによって検査した(図8A)。すべての場合において、HSPCにHLA-A*02:01拘束NY-ESO-1特異的TCRを形質導入し、そして四量体+CD3+CD8SP T細胞の頻度を、陽性選択の読み出しとして用いた。HLA-A*02:01の造血系発現は、四量体+CD8SP T細胞の陽性選択に対して穏やかな効果のみを発揮した(図8B)。対照的に、ATO間質細胞におけるHLA-A*02:01の発現は、四量体+CD8SP T細胞の陽性選択を際立って増強し、かつドナー造血系HLA-02:01発現と相乗的であった(図8B)。MHC改変ATOにおけるTCR改変T細胞は、CCR7の上方調節を含めた、成熟ナイーブ表現型へのより大きな成熟も呈し(図8C)、増強した陽性選択と一致した。要約すると、TCR形質導入HSPCおよびMHC形質導入間質細胞を含むATOは、インビトロでのヒトT細胞の陽性選択をモデル化するための多目的システム、ならびに養子細胞療法のためのインビトロ由来TCR改変T細胞の陽性選択および成熟を増強するための簡便な方法である。
インビトロにおいて胸腺リンパ球形成を誠実に再現し得る能力は、抗原ナイーブ状態および内因性TCR発現の欠如を含めた、望ましい治療的形質を有する改変T細胞の産生のための固有の機会を与える。本明細書で実証されるように、標準化されたすぐに使える構成要素を用いて、ATOシステムは、HSPCからの胸腺リンパ球形成を誠実に再現し、胸腺または末梢血由来のナイーブT細胞に厳密に似ている成熟CD3+TCRαβ+CD8SPおよびCD4SP T細胞の産生に至る。
1.ヒトCD34+CD3- HSPCの単離
新生児臍帯血を、UCLAにおいて正常分娩からの捨てられた胎盤から得た。骨髄(BM)を、UCLAにおいて同種異系BMドナー収集物からの捨てられた材料を通じて健常成体ドナーから得た、またはAllCells Inc.(Alameda, CA)から購入した。G-CSF動員末梢血を、UCLAにおいて同種異系幹細胞移植供与のためにアフェレーシスを受けた同意している健常成体ドナーから得た。非動員末梢血を、UCLA CFARウイルス学中核部(Virology Core)を通じて健常成体ドナーから得た。すべての組織サンプルを、UCLA IRBに承認されたプロトコールまたは適用除外の下で得た。すべてのサンプルを、Ficoll-Paque(GE Healthcare Life Sciences, Pittsburgh, PA)勾配遠心分離、それに続くCD34 MicroBead Kit UltraPure(Miltenyi, Auburn CA)を用いた磁気細胞選別(MACS)によるCD34+細胞の陽性選択によって、単核細胞について富化した。別様に付記されていない限り、CD34+細胞富化画分を、MACS後に冷凍保存した。使用前に、細胞を融解し、かつ残余のT細胞を、CD34+CD3-細胞を選別することによってFACSにより排除し、それを直ちにATOに播種または下記のように形質導入した。一部の実験では、ATOに播種する前に、HSCをLin-CD34+CD38-細胞についてFACS選別によって富化した。HSPCのHLA型付けは、高分解能配列特異的オリゴヌクレオチド(SSO)ビーズを用いて、UCLA免疫遺伝学センター(Immunogenetics Center)によって実施された。
CD34+ HSPCを、上記のように新鮮なBM吸引物から富化し、かつ、以下のマーカー:全HSPC(CD34+)、HSC(CD34+CD38-CD45RA)、LMPP(CD34+CD38+CD45RA+CD10-CD62L高)、CD24- CLP(CD34+CD38+CD45RA+CD10+CD24-)、およびCD24+ CLP(CD34+CD38+CD45RA+CD10 CD24+)と組み合わせた、CD45の陽性発現および系統マーカーの発現不在(CD3、CD14、CD19、CD56、およびCD235a;「Lin-」)に基づき、幹/前駆体集団についてFACSによって直ちに選別した。
出生後ヒト胸腺を、ロサンゼルス子ども病院(Children's Hospital Los Angeles)(CHLA)において、心臓手術を受けた患者からの捨てられた廃棄物として、IRB適用除外の下で得た。胸腺フラグメントをRPMI中で細かく刻み、かつピペッティングによって分断して懸濁液中に胸腺細胞を放出し、その後に70μmナイロン製ストレーナーの通過が続いた。細胞を、新鮮な同日または翌日に分析した。胸腺およびATO由来のT細胞前駆体についてのフローサイトメトリー分析は、以下の表面表現型:初期胸腺前駆体(ETP;CD34+CD7-CD1a-)、CD1a- プロ-T(CD34+CD7+CD1a-)、およびCD1a+ プロ-T(CD34+CD7+CD1a+);または、CD5- プロ-T(プロ-T1;CD34+CD7+CD5-)およびCD5+ プロ-T(プロ-T2;CD34+CD7+CD5+)を用いた。胸腺およびATO由来のT細胞および先駆体は、以下の表現型:全T系統細胞(CD7+CD5+)、ダブルネガティブ(DN;CD4-CD8-)、CD4未成熟シングルポジティブ(CD4ISP;CD5+CD4+CD3)、ダブルポジティブ(DP;CD4+CD8+)、CD8SP(CD3+TCRαβ+CD8+CD4-)、CD4SP(CD3+TCRαβ+CD8-CD4+)、未成熟ナイーブ(CD8SPまたはCD4SPであるCD45RA-CD45RO+)、成熟ナイーブ(CD8SPまたはCD4SPであるCD45RA+CD45RO-)と組み合わせた、CD14-CD56-として規定された。未成熟のおよび成熟したナイーブ表現型を、CD1a、CD27、CD28、およびCCR7に対して共染色することによって確認した。
MS-5マウス間質細胞株を贈与として得た。MS5-hDLL1を生成するために、MS-5細胞に、ヒトDLL1およびGFPをコードするレンチウイルスベクターを形質導入した。最も高い5%GFP発現細胞をFACSによって選別し、かつDMEM/10% FCS中で継代した。安定な発現を、培養の数週間後にGFP発現に対するフローサイトメトリーによって確認した。MS5-hDLL1細胞にヒトHLA-A*02:01レンチウイルスベクター(Dr. David Baltimore、Caltechからの贈与)を形質導入し、それに続くヒトHLA-A2を認識する抗体(BB7.2)(Biolegend, San Diego, CA)を用いた形質導入された細胞のFACS選別によって、MS5-hDLL1-A2.1細胞を作製した。OP9-DL1細胞株は、Dr. Juan Carlos Zuniga-Pflucker(トロント大学)からの贈与であり、そして0.1%ゼラチンコーティングされたフラスコ中にてMEMα(ThermoFisher Scientific, Grand Island, NY)/20% FBS中で継代した。K562細胞株はATCCから得た。K562-CD80/HLA-A*02:01/NY-ESO-1 aAPC細胞株は、Dr. Antoni Ribas(UCLA)からの贈与であった。
MS5-hDLL1(またはMS-5もしくはOP9-DL1、付記されるとおり)細胞をトリプシン処理によって収集し、かつ、RPMI 1640(Corning, Manassas, VA)、4% B27サプリメント(ThermoFisher Scientific, Grand Island, NY)、PBS中で再構成された30mM L-アスコルビン酸2-リン酸セスキマグネシウム塩水和物(Sigma-Aldrich, St. Louis, MO)、1%ペニシリン/ストレプトマイシン(Gemini Bio-Products, West Sacramento, CA)、1% Glutamax(ThermoFisher Scientific, Grand Island, NY)、5ng/ml rhFLT3L、および5ng/ml rhIL-7(Peprotech, Rocky Hill, NJ)から構成される血清不含ATO培養培地(「RB27」)中に再懸濁した。RB27は毎週新鮮にされた。表示される実験において、4%ゼノフリーB27がB27の代わりに置換された。実験に応じて、1.5〜6×105個のMS5-hDLL1細胞を、1.5mlエッペンドルフチューブ中にてATOあたり5×102〜1×105個の精製CD34+CD3-細胞(または他のHSPC集団、表示されるとおり)と組み合わせ、かつスイングバケット遠心分離機にて4℃で5分間300gで遠心分離した。上清を慎重に除去し、かつ細胞ペレットを短時間のボルテックスによって再懸濁した。各ATOに対して、0.4μm Millicellトランスウェルインサート(EMD Millipore, Billerica, MA;Cat. PICM0RG50)を、1ウェルあたり1mlのRB27を含有する6ウェルプレートに置いた。ATOをプレーティングするために、インサートを取り出してプレートの端に休ませ、過剰な培地を流出させた。細胞スラリーをATOあたり5〜8μlに調整し、20μlピペットチップ内に吸い上げ、ピペットチップの端部に液滴を形成させて細胞インサート上に穏やかに堆積させることによってプレーティングした。細胞インサートを、1mLのRB27を含有するウェルに置き戻した。細胞インサート周辺からの吸引、それに続く1mlの新鮮なRB27/サイトカインでの置き換えによって、培地を3〜4日間ごとに完全に交換した。ATOをこの形式で最高12週間培養した。表示される時点で、各ウェルにFACSバッファー(PBS/0.5%ウシ血清アルブミン(album)/2mM EDTA)を添加し、かつ1ml「P1000」ピペットでピペッティングすることによってATOを短時間分散させ、それに続く70μmナイロン製ストレーナーの通過によって、ATO細胞を収集した。一部の実験では、MS5-hDLL1細胞の単一細胞懸濁液を、ATOにおける使用前に表示される線量でγ照射した。
OP9-DL1単層培養を、当技術分野において以前に記載されたように準備した。簡潔には、OP9-DL1を、使用の1〜2日前に、0.1%ゼラチンコーティングされた12ウェルプレートに播種して、70〜80%コンフルエンスに到達させた。培地を単層から吸引し、かつ1×104〜1.5×104個の精製CD34+CD3- HSPCを、MEMα、20% FBS、30mM L-アスコルビン酸、5ng/ml rhFLT3L、および5ng/ml rhIL-7から構成される2mlの培地中、間質単層上にプレーティングした。一部の実験では、MS-5またはMS5-hDLL1がOP9-DL1の代わりに置換され、かつRB27が培養培地として置換された。細胞を収集し、70μmナイロン製ストレーナーを通して濾過し、かつ新鮮な培地中に再プレーティングすることによって、細胞を4〜5日間ごとに新たな間質細胞単層に移した。コンフルエントの場合、細胞を、新鮮な間質層を含有する複数のウェルに分けた。培養を最高10週間維持した。
ヒトDLL1の全長コード配列を、ヒトuniversal reference RNAセット(Agilent Technologies, Santa Clara, CA)からのRT-PCRによって、第三世代レンチウイルスベクターpCCL-c-MNDU3-X-IRES-eGFP(Dr. Donald Kohn、UCLAからの贈与)内にクローニングした。HLA-A*02:01レンチウイルスベクターpHAGE6-HGHSS-HLAA2.1-IRES-ZsGreenは、Dr. David Baltimore(Caltech)からの贈与であった。HLA-A*02:01/NY-ESO-1157〜165に特異的なコドン最適化TCRのαおよびβ鎖をコードする第三世代レンチウイルスベクターは以前に記載されており、そしてDr. Antoni Ribas(UCLA)からの贈与であった。レンチウイルス粒子のパッケージングおよび濃度は、以前に記載されたように実施された。簡潔には、293T細胞(ATCC)に、TransIT 293T(Mirus Bio, Madison, WI)を用いて、レンチウイルスベクタープラスミド、pCMV-ΔR8.9、およびpCAGGS-VSVGを17時間共トランスフェクトし、その後に20mM酪酸ナトリウムによる8時間の処理が続き、その後に血清不含UltraCulture中での48時間の細胞上清の生成が続いた。上清を、メーカーのプロトコールに従い、Amicon Ultra-15 100Kフィルター(EMD Millipore, Billerica, MA)を用いたタンジェンシャルフロー濾過によって濃縮し、かつ-80Cで保管した。HSPC形質導入に関しては、1×105〜1×106個のFACS選別されたCD34+CD3- HSPCを、50ng/mlの組換えヒトSCF、FLT3L、およびTPO、ならびに10ng/ml IL-3(Peprotech, Rocky Hill, NJ)を補充された1ml X-VIVO-15(Lonza, Basel, Switzerland)中、20μg/mlレトロネクチン(Clontech, Mountain View, CA)でコーティングされた6ウェル非処理プレートに12〜18時間プレーティングし、その後、濃縮されたレンチウイルス上清を100の感染多重度(MOI)で添加した。モック形質導入細胞を、同一の条件で、しかしベクターの添加なしで培養した。細胞を形質導入の24時間後に収集し、洗浄し、かつATOに播種した。示されていない限り、TCR形質導入HSPCは、HLA-A*02:01+ CBユニット由来であった。
ヘマトキシリンおよびエオシン(H&E)画像に関しては、ATOをHistogel(ThermoFisher Scientific, Grand Island, NY)に包埋し、かつ10%中性緩衝ホルマリン(ThermoFisher Scientific, Grand Island, NY)中で一晩固定した。5μm切片およびH&E染色は、UCLAトランスレーショナル病理学中核研究室(Translational Pathology Core Laboratory)(TPCL)によって実施された。免疫蛍光イメージングに関しては、各ATO周辺の培養インサートをメスで切り込み、それに続いて膜およびATOをTissue-Tek OCT(VWR Radnor, PA)に包埋し、ドライアイス上で凍結させることによって、ATOを単離した。5μmの凍結切片を10%中性緩衝ホルマリン中で固定し、1:50希釈の抗CD3(クローンUCHT1;Biolegend, San Diego, CA)で4℃にて一晩染色し、続いてその後、室温でAlexaFluor 594コンジュゲート抗マウスIgG (H+L)(Jackson ImmunoResearch, West Grove, PA)とインキュベーションした。H&Eおよび免疫蛍光画像を、AxioCam MRMおよびAxioVisionソフトウェアを備えたZeiss AzioImager M2(Zeiss, Jena, Germany)で取得した。
ATO由来のCD8SP、CD4SP、またはDP細胞を、抗CD8および抗CD4抗体を用いたFACSによって選別し、かつ5%熱不活性化ヒトAB血清(Gemini Bio-Products, West Sacramento, CA)を有する200μl AIM V(ThermoFisher Scientific, Grand Island, NY)中、96ウェルU底プレートにプレーティングした。PMA/イオノマイシン/タンパク質輸送阻害剤カクテルまたは対照タンパク質輸送阻害剤カクテル(両方とも、eBioscience, San Diego, CA製)を各ウェルに添加し、かつ一晩インキュベートした。細胞を、固定、ならびに細胞内染色バッファーキット(eBioscience, San Diego, CA)による透過化、ならびにIFNγおよびIL-4に対する抗体(Biolegend, San Diego, CA)によるサイトカイン染色の前に、CD3、CD4、およびCD8(Biolegend, San Diego, CA)、ならびにUV455固定可能な生存率色素(eBioscience, San Diego, CA)で染色した。
CFSE増殖アッセイに関しては、ATO由来CD8SP T細胞をFACSによって選別し、かつメーカーのプロトコールに従い、5μM CFSE(Biolegend, San Diego, CA)で標識した。標識された細胞を、5% AB血清および20ng/ml rhIL-2(Peprotech, Rocky Hill, NJ)またはIL-2のみを補充されたAIM V中、抗CD3/CD28ビーズ(ThermoFisher Scientific, Grand Island, NY)とともにインキュベートし、CD25(Biolegend, San Diego, CA)に対して共染色し、かつ5日目にフローサイトメトリーによって分析した。インビトロ細胞増殖アッセイに関しては、1×104個のFACS選別されたATO由来CD8SP T細胞を、20ng/ml rhIL-2および1×抗CD3/CD28四量体抗体複合体(Stem Cell Technologies, Vancouver, BC, Canada)を補充された200μl Immunocult XF培地(Stem Cell Technologies, Vancouver, BC, Canada)中、96ウェルU底プレートにプレーティングした。必要に応じてより大きなウェルに再プレーティングするとともに、新鮮な培地およびサイトカインを2〜3日間ごとに添加した。新鮮な抗CD3/CD28を7および14日目に添加した。細胞を血球計数器で毎週カウントした。
5×104個の全ATO由来CD8SP T細胞を、6週目のTCR形質導入ATOから選別し、200μl AIM V/5%ヒトAB血清中、96ウェルU底プレートにて、CD80を発現するK562に基づくaAPC、およびHLA-A*02:01/B2M/NY-ESO-1157〜165一本鎖三量体(Dr. Antoni Ribas、UCLAからの贈与)、または親K562細胞とともに、5:1のT細胞:K562比で一晩共培養した。CD170a-APC抗体(Biolegend, San Diego, CA)を、培養の最後の4時間、タンパク質輸送阻害剤カクテル(eBioscience, San Diego, CA)と一緒に1:50の最終希釈でウェルに添加し、その後に、上記の固定、透過化、および細胞内サイトカイン染色が続いた。
7週目のATOまたは出生後胸腺由来の全細胞を、IOTest Beta Mark TCR Vキット(Beckman Coulter, Indianapolis, IN)と併せて、CD3、CD4、CD8、およびTCRγδに対して染色した。CD3+TCRgd-CD8+CD4-細胞を、Vβ分析のためにゲート制御した。TCR形質導入ATOに関しては、6週目の全ATO細胞を、APCコンジュゲートHLA-A*02:01/NY-ESO-1157〜165四量体(MBL International, Woburn, MA)でも標識し、かつVβ分析のためにCD3+TCRγδ-四量体+CD8+CD4-に対してゲート制御した。
すべてのフローサイトメトリー染色は、PBS/0.5% BSA/2mM EDTA中、氷上で30分間実施された。抗体染色の前に、FcX(Biolegend, San Diego, CA)をすべてのサンプルに5分間添加した。四量体共染色に関しては、氷上での付加的な20分間の抗体の添加前に、PEまたはAPCコンジュゲート四量体(MBL International, Woburn, MA)を、室温で10分間、1:50の最終希釈で細胞に添加した。分析前に、DAPIをすべてのサンプルに添加した。分析は、UCLA広域幹細胞研究センターフローサイトメトリー中核部(Broad Stem Cell Research Center Flow Cytometry Core)にて、LSRII Fortessa、およびARIAまたはARIA-H機器でのFACS(BD Biosciences, San Jose, CA)で実施された。すべての分析に関して、DAPI+細胞をゲートをかけて外し、かつ単一細胞を、FSC-H対FSC-WおよびSSC-H対SSC-Wプロファイルに基づいてゲート制御した。表面および細胞内染色に用いられた抗体クローンは、Biolegend(San Diego, CA):CD1a(HI149)、CD3(UCHT1)、CD4(RPA-T4)、CD5(UCHT2)、CD8(RPA-T8)、CD10(6H6)、CD14(M5E2)、CD19(HIB19)、CD24(ML5)、CD25(BC96)、CD27(O323)、CD28(CD28.2)、CD31(WM59)、CD34(581)、CD38(HIT2)、CD45(HI30)、CD45RA(HI100)、CD45RO(UCHL1)、CD56(HCD56)、CD107a(H4A3)、CD127(A019D5)、CD235a(HI264)、CCR7(G043H7)、HLA-A2(BB7.2)、インターフェロンγ(4S.B3)、IL-4(MP4-25D2)、TCRαβ(IP26)、TCRγδ(B1)、Vβ13.1(H131)、ヒト系統カクテル(CD3、CD14、CD19、CD20、CD56);または、BD Biosciences(San Jose, CA):CD7(M-T701)、およびCD62L(DREG-56)、から得た。
改変T細胞療法は、がんおよび慢性ウイルス感染症の治療のための前例のない機会を提供する。造血幹細胞・前駆細胞(HSPC)から改変T細胞を直接生成し得る能力は、同種反応性を含めた、末梢血T細胞の使用に伴う主な治療的制約を克服する潜在力を有する。本実施例は、臍帯血、骨髄、および末梢血HSPCからの、天然のおよびTCR改変されたヒトT細胞の非常に効率的なインビトロ分化および陽性選択を支持する、臨床的に関連する人工胸腺オルガノイド(ATO)システムを記載する。ATO由来T細胞は、ナイーブ表現型、多様なTCRレパートリー、ならびにTCR依存的活性化および増殖を呈した。ATO由来TCR改変T細胞もナイーブ表現型に成熟し、さらにインビトロおよびインビボでの抗原特異的腫瘍殺傷、ならびに対立遺伝子排除の誘導と一致した内因性TCR Vβ発現の完全に近い欠如を示した。ゆえに、ATOは、養子細胞療法のためのナイーブでかつ潜在的に非同種反応性の改変T細胞の生成のための効率的な方法を提示する。
A.インビトロヒトT細胞分化のための最適化された人工胸腺オルガノイドシステムの開発
目標は、HSPCからのヒトT細胞のインビトロ陽性選択および成熟を支持し得る臨床的に橋渡し可能なオルガノイドシステムを開発することであった。初代胸腺組織の使用を回避するために、本発明者らは、DLL1を形質導入された間質細胞株を、3Dオルガノイド培養においてヒトT細胞発生を支持し得る能力について検査した。OP9-DL1システムにおけるT細胞分化は、ウシ胎仔血清の特異的ロットに依存して非常に可変的であることが観察され、そして本発明者らは、オルガノイド培養においてT細胞分化を一貫して支持し得る血清不含条件を同定することを求めた。専売的足場材料の使用を回避するために、間質細胞が遠心分離によってHSPCと凝集しかつ空気-流体界面で細胞培養インサート上に配備される、胸腺組織に基づくオルガノイドにおいて有効であることが示された圧縮再凝集技法が用いられた(図3A)。これらの3D培養において、本発明者らは、T細胞枯渇したCD34+臍帯血(CB)HSPCからのヒトT細胞分化を強く支持するものとして、ヒトDLL1を形質導入されたMS-5マウス骨髄間質細胞株(以後、MS5-hDLL1)を同定した。さらに、本発明者らは、MS5-hDLL1オルガノイド培養において堅牢なヒトT細胞分化を一貫して支持する新規な血清不含培地として、B27、神経幹および胚性幹細胞培養において用いられる多構成要素添加物、ならびにFLT3L、IL-7、ならびにアスコルビン酸を補充されたRPMI(以後、「RB27」)を同定した。
次に、ATOにおけるT細胞分化を、出生後ヒト胸腺におけるものと比較した。12週目のCB ATOは、胸腺と同程度の頻度のT系統コミットした(CD5+CD7+)およびCD34+の前駆体を示した(図4A)。胸腺に見られるように、ATOにおけるCD3+ T細胞の大多数はTCRαβ+であったが、容易に検出可能なTCRγδ+集団も一貫して見られた(図4A)。ATO由来CD3+TCRαβ+細胞の中で、成熟CD8SPおよびCD4SP T細胞の生成は、6〜12週目の間に増加した(図4Bおよび図12A〜C)。胸腺とは対照的に、ATOは、CD8SP T細胞に対して比例的により少ないCD4SP T細胞を呈した。
すべての臨床的に関連するHSPC供給源、すなわち成体骨髄(BM)、G-CSF動員末梢血(MPB)、および非動員末梢血(PB)からの、先駆体およびCD3+TCRαβ+ T細胞の同程度の頻度を有する、ATOにおける効率的なT細胞分化が見られた(図3A〜B、15A〜B、13A〜B)。これらの供給源由来およびまた胸腺由来のHSPCは、T細胞分化の種々の動態を呈示したが(図15A)、しかしながらT細胞産出量は、HSPC供給源にわたって匹敵した(図3d)。CB、BM、またはMPB由来の高度に富化された造血幹細胞(HSC)画分(lin-CD34+CD38-)は、同程度に堅牢なT細胞分化を実証した(図5C〜D、図13D〜E)。
胸腺と同様に、RAG1およびRAG2は、ATO由来DPにおいて発現した(図25A)。ATO由来のCD3+TCRαβ+CD8SP(図16A)およびCD3+TCRαβ+CD4SP(図25B)T細胞におけるTCR Vβファミリー使用法についてのフローサイトメトリー分析は、ヒト胸腺由来の対応するT細胞のものに著しく類似した多様性を明らかにした。加えて、胸腺CD8SP細胞およびPBナイーブCD8+ T細胞におけるものに匹敵する、CD3+TCRαβ+CD8SPにおける非常に多様なTCRレパートリーが、TCR VαおよびVβ CDR3領域のディープシーケンシングによって見られた(図16B〜C)。重要なことには、偏ったVαまたはVβ使用法は観察されず、非従来的なT細胞サブセットまたはクローン増殖させた成熟T細胞の、ATOにおける優勢に反論した。
次に、本発明者らは、HSPCからのTCR改変T細胞のインビトロ生成にATOを適応させた。CB CD34+CD3- HSPCに、NY-ESO-1157〜165ペプチドに特異的なHLA-A*02:01拘束TCRのコドン最適化αおよびβ鎖をコードするレンチウイルスベクターを形質導入した。7週目の時点で、TCR形質導入ATOは、モック形質導入対照と同程度の頻度のCD5+CD7+ T系統細胞を示したが、CD3+TCRαβ+ T細胞を際立って増加させ、その大多数は、四量体、または形質導入されたVβ13.1鎖に対する抗体での染色によって見られるように、形質導入されたTCRを発現した(図7A)。CD8SP細胞の頻度は、モック形質導入対照由来のCD3+TCRαβ+四量体+細胞とCD3+TCRαβ+細胞との間で同程度であったが、四量体+CD8SP細胞は、成熟ナイーブ表現型(すなわち、CD45RA+CD45RO-CD27+CCR7+CD1a低)への加速した成熟を呈示した(図7A)。留意すべきことに、四量体+CD8SP細胞は、NK細胞および上皮内リンパ球と関連したマーカーであるCD16またはCD56の発現なく、従来のCD8αβ T細胞表現型を呈示した(図17A)。
胸腺における陽性選択は、T細胞先駆体上のTCRと胸腺間質細胞および造血細胞上の自己MHCとの間の相互作用によって仲介される。ゆえに、ATOにおける「自己」MHCの間質系発現の増加が、TCR改変T細胞の陽性選択を増強し得るかどうかが検討された。HLA-A*02:01陽性HPSCに、HLA-A*02:01拘束NY-ESO-1特異的TCRを形質導入し、かつ対照MS5-hDLL1間質、またはHLA-A*02:01を形質導入されたMS5-hDLL1と組み合わせた(図27A)。ATO間質細胞におけるHLA-A*02:01の発現は、四量体+CD3+CD8SP T細胞の陽性を増強し(図27A)、一方で成熟ナイーブ表現型への正常な分化を維持した(図27B)。
次に、本発明者らは、ATO由来TCR改変T細胞の抗原特異的細胞傷害性を検査した。NY-ESO-1特異的TCR形質導入ATOから単離されかつインビトロで36時間活性化された精製CD8SP T細胞は、NY-ESO-1発現細胞株(HLA-A*02:01/NY-ESO-1 pMHC複合体を形質導入したK562細胞;および、NY-ESO-1抗原が内因的に発現しているHLA-A*02:01+ U266多発性骨髄腫細胞株)においてアポトーシスを強力に誘導したが、親K562細胞、または非関連のHLA-A*02:01/MART-1 pMHCを発現するK562細胞に対してはほとんど活性を示さなかった(図18A〜B)。さらに、抗原特異的細胞傷害性は、長期にわたる(14日間)インビトロ増殖後に保たれ、従来のT細胞表現型の保持と一致し(図18C);細胞傷害性は、同じ期間増殖させたTCR形質導入PB CD8+ T細胞のものと同程度であった(図18C)。これらの結果と一致して、増殖させたATO由来TCR改変T細胞は、同族(K562-ESO)pMHC抗原を発現するK562腫瘍を皮下生着させたNSGマウスにおいて疾患負荷を有意に制御し得たが、非関連の(K562-MART1)pMHC抗原ではそうではなかった(図18D〜F)。
インビトロにおいて胸腺リンパ球形成を忠実に再現し得る能力は、抗原ナイーブ状態および内因性TCR発現の欠如を含めた、望ましい治療的形質を有する改変T細胞の産生のための固有の機会を与える。本明細書で実証されるように、標準化されたすぐに使える構成要素を用いて、ATOシステムは、HSPCからのT細胞のコミットメントおよび分化の正常なステージを効率的に開始、維持し、胸腺および末梢血由来のナイーブの従来のT細胞に近似する成熟CD3+TCRαβ+CD8SPおよびCD3+TCRαβ+CD4SP T細胞の産生に至る。
A.ヒトCD34+CD3- HSPCの単離
新生児へその緒臍帯血を、UCLAにおいて分娩からの捨てられた臍帯および胎盤ユニットから得た。骨髄(BM)を、UCLAにおいて同種異系BMドナー収集物からの捨てられた材料を通じて健常成体ドナーから得た、またはAllCells Inc.(Alameda, CA)から購入した。G-CSF動員末梢血を、UCLAにおいて同種異系幹細胞移植供与のためにアフェレーシスを受けた同意している健常成体ドナーから得た。非動員末梢血を、UCLA CFARウイルス学中核部を通じて健常成体ドナーから得た。すべての組織サンプルを、UCLA IRBに承認されたプロトコールまたは適用除外の下で得た。すべてのサンプルを、Ficoll-Paque(GE Healthcare Life Sciences, Pittsburgh, PA)勾配遠心分離、それに続くCD34 MicroBead Kit UltraPure(Miltenyi, Auburn CA)を用いた磁気細胞選別(MACS)によるCD34+細胞の陽性選択によって、単核細胞について富化した。別様に付記されていない限り、CD34+細胞富化画分を、MACS後に冷凍保存した。使用前に、細胞を融解し、かつ残余のT細胞を、CD34+CD3-細胞を選別することによってFACSにより枯渇させ、それを直ちにATOに播種または下記のように形質導入した。一部の実験では、ATOに播種する前に、HSCをLin-CD34+CD38-細胞についてFACSによって富化した。TCR形質導入実験において用いられたHSPCは、HLA-A*02:01+ CBユニット由来のものであった。高分解能HLA-A2型付けは、配列特異的オリゴヌクレオチド(SSO)ビーズを用いて、UCLA免疫遺伝学センターによって実施された。
CD34+ HSPCを、上記のように新鮮なBM吸引物から富化し、かつ、以下のマーカー:全HSPC(CD34+)、HSC(CD34+CD38-CD45RA-)、LMPP(CD34+CD38+CD45RA+CD10-CD62L高)、CD24- CLP(CD34+CD38+CD45RA+CD10+CD24-)、およびCD24+ CLP(CD34+CD38+CD45RA+CD10CD24+)と組み合わせた、CD45の陽性発現および系統マーカーの発現不在(CD3、CD14、CD19、CD56、およびCD235a;「Lin-」)に基づき、幹/前駆体集団についてFACSによって直ちに選別した。
出生後ヒト胸腺を、ロサンゼルス子ども病院(CHLA)において、心臓手術を受けた患者からの捨てられた廃棄物として、IRB適用除外の下で得た。胸腺フラグメントをRPMI中で細かく刻み、かつピペッティングによって分断して懸濁液中に胸腺細胞を放出し、その後に70μmナイロン製ストレーナーの通過が続いた。細胞を、新鮮な同日または翌日に分析した。胸腺およびATO由来のT細胞前駆体についてのフローサイトメトリー分析は、以下の表面表現型:初期胸腺前駆体(ETP;CD34+CD7-CD1a-)、CD1a- プロ-T(CD34+CD7+CD1a-)、およびCD1a+ プロ-T(CD34+CD7+CD1a+);または、CD5- プロ-T(プロ-T1;CD34+CD7+CD5-)およびCD5+ プロ-T(プロ-T2;CD34+CD7+CD5+)を用いた。胸腺およびATO由来のT細胞および先駆体は、以下の表現型:全T系統細胞(CD7+CD5+)、ダブルネガティブ(DN;CD4-CD8-)、CD4未成熟シングルポジティブ(CD4 ISP;CD5+CD4+CD3-)、ダブルポジティブ(DP;CD4+CD8+)、CD8SP(CD3+TCRαβ+CD8+CD4-)、CD4SP(CD3+TCRαβ+CD8-CD4+)、未成熟ナイーブ(CD8SPまたはCD4SPであるCD45RA-CD45RO+)、成熟ナイーブ(CD8SPまたはCD4SPであるCD45RA+CD45RO-)と組み合わせた、CD14-CD56-として規定された。未成熟のおよび成熟したナイーブ表現型を、CD1a、CD27、CD28、およびCCR7に対して共染色することによって確認した。
胸腺T細胞を、上記のように胸腺細胞調製物から単離し、そして末梢血および臍帯血CD8+ T細胞を、上記のように単核細胞画分から単離した。すべての供給源からのCD8+ T細胞単離は、CD8+ T細胞単離キット(Miltenyi)を用いた、CD8SP T細胞についての磁気ビーズ富化によるものであった。一部の実験では、胸腺T細胞をFACSによってさらに精製して、CD4 ISPまたはDP先駆体およびPB T細胞を枯渇させて、ナイーブT細胞(CD45RO-CCR7+)を単離した。
MS-5マウス間質細胞株を贈与として得た。MS5-hDLL1を生成するために、MS-5細胞に、ヒトDLL1およびGFPをコードするレンチウイルスベクターを形質導入した。最も高い5%GFP発現細胞をFACSによって選別し、かつDMEM/10% FCS中で継代した。安定な発現を、培養の数週間後にGFP発現に対するフローサイトメトリーによって確認し、そしてDLL1発現を、qRT-PCRおよびDNAシーケンシングによって確認した。MS5-hDLL1細胞にヒトHLA-A*02:01レンチウイルスベクター(Dr. David Baltimore、Caltechからの贈与)を形質導入し、それに続くヒトHLA-A2を認識する抗体(BB7.2)(Biolegend, San Diego, CA)を用いた形質導入された細胞のFACS選別によって、MS5-hDLL1-A2.1細胞を作製した。OP9-DL1細胞株(マウスDll1を発現する)は、Dr. Juan Carlos Zuniga-Pflucker(トロント大学)からの贈与であり、そして0.1%ゼラチンコーティングされたフラスコ中にてMEMα(ThermoFisher Scientific, Grand Island, NY)/20% FBS中で継代した。K562細胞株はATCCから得、そしてRPMI/10% FCS中で維持した。K562 aAPCを、全長ヒトCD80およびHLA-A*02:01/B2M/NY-ESO-1157〜165またはMART-126〜35一本鎖三量体(SCT;Dr. David Baltimore、Caltechからの贈与)をコードするレンチウイルスベクターによるK562細胞の共形質導入によって生成した。K562標的細胞を、いずれかのSCT単独による形質導入によって作製した。K562インビボ標的細胞を、ホタルルシフェラーゼレンチウイルスベクター(Dr. Donald Kohn、UCLAからの贈与)、それに続くいずれかのSCTによる逐次的な形質導入によって作製した。K562形質導入体を、使用前にFACS選別した。U266多発性骨髄腫細胞株は、Dr. John Chute(UCLA)からの贈与であり、そしてRPMI/10% FCS中で維持した。
MS5-hDLL1(またはMS-5もしくはOP9-DL1、付記されるとおり)細胞をトリプシン処理によって収集し、かつ、RPMI 1640(Corning, Manassas, VA)、4% B27サプリメント(ThermoFisher Scientific, Grand Island, NY)、PBS中で再構成された30μM L-アスコルビン酸2-リン酸セスキマグネシウム塩水和物(Sigma-Aldrich, St. Louis, MO)、1%ペニシリン/ストレプトマイシン(Gemini Bio-Products, West Sacramento, CA)、1% Glutamax(ThermoFisher Scientific, Grand Island, NY)、5ng/ml rhFLT3L、および5ng/ml rhIL-7(Peprotech, Rocky Hill, NJ)から構成される血清不含ATO培養培地(「RB27」)中に再懸濁した。RB27は毎週新鮮にされた。表示される実験において、4%ゼノフリーB27がB27の代わりに置換された。実験に応じて、1.5〜6×105個のMS5-hDLL1細胞を、1.5mlエッペンドルフチューブ中にてATOあたり3×102〜1×105個の精製CD34+CD3-細胞(または他のHSPC集団、表示されるとおり)と組み合わせ、かつスイングバケット遠心分離機にて4℃で5分間300gで遠心分離した。上清を慎重に除去し、かつ細胞ペレットを短時間のボルテックスによって再懸濁した。各ATOに対して、0.4μm Millicellトランスウェルインサート(EMD Millipore, Billerica, MA;Cat. PICM0RG50)を、1ウェルあたり1mlのRB27を含有する6ウェルプレートに置いた。ATOをプレーティングするために、インサートを取り出してプレートの端に休ませ、過剰な培地を流出させた。細胞スラリーをATOあたり5〜8μlに調整し、20μlピペットチップ内に吸い上げ、ピペットチップの端部に液滴を形成させて細胞インサート上に穏やかに堆積させることによってプレーティングした。細胞インサートを、1mLのRB27を含有するウェルに置き戻した。細胞インサート周辺からの吸引、それに続く1mlの新鮮なRB27/サイトカインでの置き換えによって、培地を3〜4日間ごとに完全に交換した。ATOをこの形式で最高20週間培養した。表示される時点で、各ウェルにFACSバッファー(PBS/0.5%ウシ血清アルブミン/2mM EDTA)を添加し、かつ1ml「P1000」ピペットでピペッティングすることによってATOを短時間分散させ、それに続く70μmナイロン製ストレーナーの通過によって、ATO細胞を収集した。一部の実験では、MS5-hDLL1細胞の単一細胞懸濁液を、ATOにおける使用前に表示される線量でγ照射した。
OP9-DL1単層培養を、以前に記載されたように準備した。簡潔には、OP9-DL1細胞を、使用の1〜2日前に、0.1%ゼラチンコーティングされた12ウェルプレートに播種して、70〜80%コンフルエンスに到達させた。培地を単層から吸引し、かつ1×104〜1.5×104個の精製CD34+CD3- HSPCを、MEMα、20% FBS、30μM L-アスコルビン酸、5ng/ml rhFLT3L、および5ng/ml rhIL-7から構成される2mlの培地中、間質単層上にプレーティングした。一部の実験では、MS-5またはMS5-hDLL1がOP9-DL1の代わりに置換され、かつRB27が培養培地として置換された。細胞を収集し、70μmナイロン製ストレーナーを通して濾過し、かつ新鮮な培地中に再プレーティングすることによって、細胞を4〜5日間ごとに新たな間質細胞単層に移した。コンフルエントの場合、細胞を、新鮮な間質層を含有する複数のウェルに分けた。培養を最高10週間維持した。
ヒトDLL1の全長コード配列を、ヒトuniversal reference RNAセット(Agilent Technologies, Santa Clara, CA)からのRT-PCRによって、第三世代レンチウイルスベクターpCCL-c-MNDU3-X-IRES-eGFP(Dr. Donald Kohn、UCLAからの贈与)内にクローニングした。ヒトCD80を、pCCL-c-MNDU3内に同様にクローニングした。HLA-A*02:01/NY-ESO-1157〜165に特異的なTCRのコドン最適化αおよびβ(Vb13.1)鎖をコードする第三世代レンチウイルスベクター(1G4 TCRクローンに由来する)は以前に記載されており、そしてDr. Antoni Ribas(UCLA)からの贈与であった。コドン最適化HLA-A*02:01/MART-126〜35特異的TCR(F5 TCRクローンに由来する)は、Dr. Donald Kohn(UCLA)からの贈与であった。HLA-A*02:01およびHLA-A*02:01/B2M/NY-ESO-1157〜165またはHLA-A*02:01/B2M/MART-126〜35一本鎖三量体に対するコード配列は、Dr. David Baltimore(Caltech)からの贈与であり、そしてpCCL-c-MNDU3-X-IRES-mStrawberry内にサブクローニングされた。レンチウイルス粒子のパッケージングおよび濃度は、以前に記載されたように実施された。簡潔には、293T細胞(ATCC)に、TransIT 293T(Mirus Bio, Madison, WI)を用いて、レンチウイルスベクタープラスミド、pCMV-ΔR8.9、およびpCAGGS-VSVGを17時間共トランスフェクトし、その後に20mM酪酸ナトリウムによる8時間の処理が続き、その後に血清不含UltraCulture中での48時間の細胞上清の生成が続いた。上清を、4℃で40分間4000xgでAmicon Ultra-15 100Kフィルター(EMD Millipore, Billerica, MA)を用いたタンジェンシャルフロー濾過によって濃縮し、かつアリコートとして-80Cで保管した。HSPC形質導入に関しては、1×105〜1×106個のFACS選別されたCD34+CD3- HSPCを、50ng/mlの組換えヒトSCF、FLT3L、およびTPO、ならびに10ng/ml IL-3(Peprotech, Rocky Hill, NJ)を補充された1ml X-VIVO-15(Lonza, Basel, Switzerland)中、20μg/mlレトロネクチン(Clontech, Mountain View, CA)でコーティングされた6ウェル非処理プレートに12〜18時間プレーティングし、その後、濃縮されたレンチウイルス上清を100の感染多重度(MOI)で添加した。モック形質導入細胞を、ベクターの添加なしの同一の条件で培養した。細胞を形質導入の24時間後に収集し、洗浄し、かつATOに播種した。末梢血T細胞の形質導入に関しては、以前に記載されたように、健常ドナー由来のCD8+ T細胞を、CD8+ T細胞単離キット(Miltenyi)を用いた磁気陰性選択によって単離し、かつ形質導入の4日前に、抗CD3/CD28ビーズ(ThermoFisher Scientific)および20ng/ml IL-2を有するAIM V/5%ヒトAB中で活性化/増殖させた。形質導入されたT細胞を、その後、使用前にIL-2(20ng/ml)中で増殖させた。
ヘマトキシリンおよびエオシン(H&E)画像に関しては、ATOをHistogel(ThermoFisher Scientific, Grand Island, NY)に包埋し、かつ10%中性緩衝ホルマリン(ThermoFisher Scientific, Grand Island, NY)中で一晩固定した。5μm切片およびH&E染色は、UCLAトランスレーショナル病理学中核研究室(TPCL)によって実施された。免疫蛍光イメージングに関しては、各ATO周辺の培養インサートをメスで切り込み、それに続いて膜およびATOをTissue-Tek OCT(VWR Radnor, PA)に包埋し、ドライアイス上で凍結させることによって、ATOを単離した。5μmの凍結切片を10%中性緩衝ホルマリン中で固定し、1:50希釈の抗CD3(クローンUCHT1;Biolegend, San Diego, CA)で4℃にて一晩染色し、続いてその後、室温でAlexaFluor 594コンジュゲート抗マウスIgG (H+L)(Jackson ImmunoResearch, West Grove, PA)とインキュベーションした。H&Eおよび免疫蛍光画像を、AxioCam MRMおよびAxioVisionソフトウェアを備えたZeiss AzioImager M2(Zeiss, Jena, Germany)で取得した。
ATO由来の成熟したCD8SPまたはCD4SP細胞を、CD8+またはCD4+単離キット(Miltenyi)を用いた磁気陰性選択によって単離し、かつFACSによって選別して、CD45RO+細胞(未成熟ナイーブT細胞およびCD4ISP先駆体を含有する)をさらに枯渇させた。精製されたT細胞集団を、5%ヒトAB血清(Gemini Bio-Products, West Sacramento, CA)を有する200μl AIM V(ThermoFisher Scientific, Grand Island, NY)中、96ウェルU底プレートにプレーティングした。PMA/イオノマイシン/タンパク質輸送阻害剤カクテルまたは対照タンパク質輸送阻害剤カクテル(eBioscience, San Diego, CA)を各ウェルに添加し、かつ6時間インキュベートした。細胞を、固定、ならびに細胞内染色バッファーキット(eBioscience, San Diego, CA)による透過化、ならびにIFNγ、TNFα、IL-2、IL-4、またはIL-17Aに対する抗体(Biolegend, San Diego, CA)による細胞内染色の前に、CD3、CD4、およびCD8(Biolegend, San Diego, CA)、ならびにUV455固定可能な生存率色素(eBioscience, San Diego, CA)で染色した。
CFSE増殖アッセイに関しては、ATO由来CD8SPまたはCD4SP T細胞を、上記のように陰性選択MACSによって単離し(上記のCD4SP T細胞のさらなるFACS精製とともに)、かつメーカーのプロトコールに従い、5μM CFSE(Biolegend, San Diego, CA)で標識した。標識された細胞を、5% AB血清および20ng/ml rhIL-2(Peprotech, Rocky Hill, NJ)を補充されたAIM V中、抗CD3/CD28ビーズ(ThermoFisher Scientific, Grand Island, NY)とともにインキュベートし、かつCD25または4-1BB(Biolegend, San Diego, CA)に対して共染色し、かつ5日目にフローサイトメトリーによって分析した。一部の実験では、メーカーのプロトコールに従い、標識化に関して、CFSEがCellTrace Violet(CTV; ThermoFisher)の代わりに置換された。インビトロ細胞増殖アッセイに関しては、上記のように単離された5×103〜1×104個のATO由来CD8SPまたはCD4SP T細胞を、200μl中96ウェルU底プレートにプレーティングし、かつ抗CD3/28ビーズ、および20ng/mL IL-2または5ng/mL IL-7のいずれか、および5ng/mL IL-15(Peprotech)で活性化/増殖させた。ビーズを4日目に除去し、かつ必要に応じてより大きなウェルに再プレーティングするとともに、新鮮な培地およびサイトカインを2〜3日間ごとに添加した。細胞を血球計数器で毎週カウントした。一部の実験では、細胞を、新鮮な抗CD3/CD28ビーズで14日目に再刺激した。
1×105個の全ATO由来CD8SP T細胞を、上記のようにMACSによって6週目のTCR形質導入ATOから単離し、かつ200μl AIM V/5%ヒトAB血清中、96ウェルU底プレートにて、CD80を発現するK562由来aAPC、およびHLA-A*02:01/B2M/NY-ESO-1157〜165もしくはHLA-A*02:01/B2M/MART-126〜35のいずれかの一本鎖三量体、または親K562細胞とともに、4:1のT細胞:K562比で6時間共培養した。CD170a-APC抗体(Biolegend, San Diego, CA)を、培養の持続期間の間、タンパク質輸送阻害剤カクテル(eBioscience, San Diego, CA)と一緒に1:50の最終希釈でウェルに添加した。次いで、上記のように、細胞を表面マーカーに対して染色し、固定し、透過化し、かつサイトカインに対して細胞内染色した。
7週目のATOまたは出生後胸腺由来の全細胞を、IOTest Beta Mark TCR Vキット(Beckman Coulter, Indianapolis, IN)と併せて、CD3、CD4、CD8、およびTCRγδに対して染色した。CD3+TCRγδ-CD8+CD4-細胞を、Vβ分析のためにゲート制御し、かつVβファミリー使用法を、チューブごとの3種の異なるVβ抗体に当たる、FITC+、PE+、またはFITC+PE+細胞パーセントによって判定した。TCR形質導入ATOのVβ分析に関しては、6〜7週目のATO由来の全細胞を、表面抗体染色の前に、APCコンジュゲートHLA-A*02:01/NY-ESO-1157〜165四量体(MBL International, Woburn, MA)で10分間付加的に標識し、かつ細胞を、Vβ分析のためにCD3+TCRγδ-四量体+CD8+CD4-に対してゲート制御した。
全RNAを、メーカーの指示書に従いRNeasy Microキット(Qiagen)を用いて、40,000〜200,000個のATOもしくは胸腺CD8SP、またはPB CD45RO- CCR7+ナイーブCD8+ T 細胞からFACSによって精製した。RNAの濃度および質を、Agilent RNA 6000 Nanoチップを用いて判定した。再編成されたTCR可変遺伝子を含む標的cDNAライブラリーを、以下のような改変を有するSMARTer PCR cDNA合成キット(Clontech)を用いて、5'-RACEによって調製した。第一鎖cDNAを、メーカーのプロトコールを用いるがポリ-dTプライマー(5'-T30VN-3')を置換して、3.5〜500ngの全RNAから調製した。二本鎖TCRαおよびTCRβ cDNAライブラリーを、Advantage 2 PCRキット(Clontech)を用いて、セミネステッドPCRによって別個に調製した。TCRα cDNAの初回増幅は、メーカーの順方向Universal Primer Mixと、TRAC
CD8SP T細胞を、上記のとおり、機械的分断および磁気陰性選択によってATOから単離した。T細胞を、抗CD3/CD28ビーズ(ThermoFisher Scientific)および20ng/ml IL-2を有するAIM V/5%ヒトAB血清中、96ウェル丸底プレートで36時間活性化した。長期増殖のため、細胞をIL-2中で最高14日間さらに培養した。細胞傷害性アッセイに関しては、AIM V/5%ヒトAB血清中、1ウェルあたり1×105個の細胞から開始して、T細胞の2倍の連続希釈を、96ウェル丸底プレート中にウェルごとにプレーティングした。HLA-A*02:01/NY-ESO-1157〜165もしくはHLA-A*02:01/MART-126〜35一本鎖三量体を形質導入されたK562標的細胞、またはNY-ESO-1を内因的に発現するHLA-A*02:01+ U266を、1ウェルあたり5×104個の細胞でプレーティングした。標的細胞のアポトーシス細胞死を、9時間の時点でアネキシンV/DAPI染色によって定量化した。抗原特異的T細胞パーセントを、四量体染色によって判定し、かつそれを用いて、各ウェルのエフェクター:標的(E:T)比を遡及的に算出した。T細胞を受けたウェルから、T細胞を受けていないウェルにおけるアネキシンV+標的細胞パーセントを差し引くことによって、T細胞特異的細胞死を算出した。
すべての動物実験は、UCLA学長動物実験委員会(UCLA Chancellor's Animal Research Committee)によって承認されたプロトコールの下で行われた。4〜6週齢のNOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ(NSG)マウス(Jackson Laboratory, Bar Harbor, Maine)に、HLA-A*02:01/NY-ESO-1157〜165またはMART-126〜35一本鎖三量体およびホタルルシフェラーゼ(上記)を形質導入された2×105個のK562標的細胞を皮下に埋め込んだ。マウスを、ルシフェリンの腹腔内注射によって、3日目に腫瘍生物発光に対して撮像した。上記のように、ATO由来CD8SP T細胞を単離し、かつ14日間活性化/増殖させた。腫瘍の埋め込み後の3日目に、5.7×106個(注射の日の四量体染色によって判定される、4.5×106個の抗原特異的T細胞を含有する)を、眼窩後方静脈を通じて注射した。対照マウスへのPBSの注射も実施した。腫瘍生物発光を少なくとも21日間3〜4日間ごとに繰り返し、その後、疾患負荷基準に基づいてマウスを屠殺した。
すべてのフローサイトメトリー染色は、PBS/0.5% BSA/2mM EDTA中、氷上で30分間実施された。抗体染色の前に、FcX(Biolegend, San Diego, CA)をすべてのサンプルに5分間添加した。四量体共染色に関しては、氷上での付加的な20分間の抗体の添加前に、PEまたはAPCコンジュゲートHLA-A*02:01/NY-ESO-1157〜165またはHLA-A*02:01/MART-126〜35四量体(MBL International, Woburn, MA)を、室温で10分間、1:50の最終希釈で細胞に添加した。分析前に、DAPIをすべてのサンプルに添加した。分析は、UCLA広域幹細胞研究センターフローサイトメトリー中核部にて、LSRII Fortessa、およびARIAまたはARIA-H機器でのFACS(BD Biosciences, San Jose, CA)で実施された。すべての分析に関して、DAPI+細胞をゲートをかけて外し、かつ単一細胞を、FSC-H対FSC-WおよびSSC-H対SSC-Wに基づいてゲート制御した。表面および細胞内染色に用いられた抗体クローンは、Biolegend(San Diego, CA):CD1a(HI149)、CD3(UCHT1)、CD4(RPA-T4)、CD5(UCHT2)、CD8(SK1)、CD10(6H6)、CD14(M5E2)、CD19(HIB19)、CD24(ML5)、CD25(BC96)、CD27(O323)、CD28(CD28.2)、CD31(WM59)、CD34(581)、CD38(HIT2)、CD45(HI30)、CD45RA(HI100)、CD45RO(UCHL1)、CD56(HCD56)、CD107a(H4A3)、CD127(A019D5)、CD235a(HI264)、CCR7(G043H7)、HLA-A2(BB7.2)、インターフェロンγ(4S.B3)、IL-2(MQ1-17H12)、IL-4(MP4-25D2)、IL-17A(BL168)、TCRαβ(IP26)、TCRγδ(B1)、TNFα(Mab11)、Vβ13.1(H131)、ヒト系統カクテル(CD3、CD14、CD19、CD20、CD56);ならびに、BD Biosciences(San Jose, CA):CD7(M-T701)、およびCD62L(DREG-56)、から得た。
ATOシステムがより橋渡し的であるために、他の間質細胞株を、マウス起源由来であるMS5-hDLL1を置き換え得るそれらの能力について検査した。HS27a細胞はヒト骨髄細胞株であり、そしてそれは低レベルの成長因子を分泌するため有力な候補である。HS27a細胞にhDLL1を形質導入することによって、Notchリガンド(hDLL1)を発現するHS27a細胞株を生成した。高レベルのhDLL1を発現する細胞をFACSによって精製し、増殖させた。Notchシグナル伝達の非存在下で、ATOシステムにおけるHS27a細胞は、T細胞分化を支持し得なかった(図28A〜C)。MS5間質細胞に加えて、HS27aは、3つの異なる臍帯血サンプル調製物において、ATOシステムにおけるT細胞分化を支持し得ることが見出された、図29A〜C。図30に示されるように、HS27a-DLL1 ATOにおいて見られるCD8+細胞の堅牢な集団における細胞の大多数は、それらが(the)CD3およびTCRabを発現しないため、従来のCD8SP細胞ではない。成熟マーカーの使用は、HS27a-hDLL1間質細胞が、3つの異なる臍帯血調製物からのTCRab+CD3+ CD8SPおよびCD4SP細胞の生成を有して、成熟T細胞の分化を支持し得ることを示している(図31A〜C)。しかしながら、効率は、MS5-hDLL1 ATOにおいてよりも低い。
本明細書において記載されるATOシステムは、(種々の供給源由来の)ヒト造血幹細胞からインビトロで機能的ナイーブT細胞を生成する効率的でかつ堅牢なモデルである。このシステムは、ヒト胚性幹細胞(hESC)から成熟した機能的ナイーブヒトT細胞を生成するために適応している。本発明者らは、最初期の中胚葉コミットした前駆体に相当しかつすべての中胚葉系統(平滑筋、心筋細胞、造血系統、間葉系統)を生じさせ得る、胚性中胚葉前駆体(hEMP)集団を同定した。このhEMP集団は、Epcamマーカー(CD326)の発現の喪失およびCD56の発現の増大によって規定され、そしてFACSによって容易に単離され得る(例えば、Evseenko et al., Proc Natl Acad Sci U S A. 2010 Aug 3;107(31):13742-7を参照されたい)。簡潔には、MEF(マウス胎仔線維芽細胞)上で培養されたhESCは、80〜90%コンフルエンスでマトリゲルに移され得る。いったん移されると(0日目)、細胞は、アクチビン、BMP4、VEGF、bFGFを含む培地中で培養される。1〜2日後、BMP4、VEGF、およびbFGFを含む新たな培地が添加される。3〜4日目に、細胞は、EPCAMのそれらの喪失およびCD56の増大に基づいて選別され得る。hEMP集団を単離し、かつそれとMS5-hDLL1間質細胞とを凝集させることによって、ES由来ATOシステムを作製した。そのプロトコールは、RPMI-B27培地(SCF/IL7/Flt3Lの添加を有する)の使用によるT細胞分化の誘導前に、ATOにEGM2培地および造血系サイトカイン(SBおよびSFT3)を供給する、2週間の造血系誘導を要する。hEMPのステージにおける3D凝集は、T細胞の生成を支持することが見出された。OP9細胞(ATOではない)上でhEMPから分化した単離されたCD34+からのATOの生成は、T細胞分化を支持しなかった(データ示さず)。幹細胞の選択集団としてのhESCによるATOに由来するT細胞集団が図32に示されている。hESC由来のATOシステムにおけるT細胞の分化の動態、およびCD8ab SP細胞の生成が図33に示されている。図34〜35に示されるように、hESC由来のATOにおいて生成されたT細胞は、臍帯血細胞を用いて観察されるものと同様に、成熟ナイーブ表現型のマーカーを発現する。図36に示されるように、hESCの使用は、複数種の幹細胞供給源にわたって再現可能である。図36は、3種の異なるhESC株由来のATOシステム(4週目)からのT細胞分化を示している。ATOシステムは、iPSC株からの成熟T細胞の生成を可能にした(図37)。さらに、未分化hESC(単離されたhEMPの代わりに)の3D凝集、それに続く以前に記載された種々の分化培地の使用は、同様にT細胞の生成を可能にした(図38)。さらに、複数種のNotchリガンドがATOシステムにおいて用いられ得る。図39に示されるように、ATOシステムは、間質細胞株が、hDLL1の代わりにhJAG1を発現する場合、hESCからのT細胞成熟も支持し得る。hESC由来のATOシステムにおいて生成されたT細胞は、多様なTCR Vbレパートリーを示すことも見出された(図40)。図41に示されるように、hESC由来T細胞は、抗CD3/CD28およびIL2に応答して、増殖およびCD25上方調節を呈する。単離された細胞をCTV(セルトラッカーバイオレット)で染色し、かつCD3/CD28活性化ビーズとともに5日間インキュベートした。細胞は、CTVの希釈によって示される複数回の細胞分裂、およびCD25の発現によって示される活性化を受けた(図41A)。さらなる実験において、単離された細胞を、PMA/イオノマイシンで6時間処理し、そして細胞内染色は、刺激に応答した細胞傷害性サイトカイン(IFNg、IL2、TNFa)の産生を示した(図41B)。
B27サプリメントの構成要素を検査して、ATOシステムへのそれらの相対的な寄与を判定した。下記の表は、B27サプリメントの構成要素を示す。
本調査において、本発明者らは、ATOにおける正常なT細胞分化に対するCARシグナル伝達の効果を判定し、ATO由来CAT-T細胞のインビトロおよびインビボ機能ならびに抗腫瘍効力を判定し、かつCARがTCR対立遺伝子排除を仲介し得る能力を判定しようと追及した。この目標に向けて、CD19標的化CARを臍帯血CD34+CD3- HSPC内に形質導入した。次いで、これらの細胞をATOシステムに4〜6週間の期間供した。
ヒトES細胞からのATO由来CAR-T細胞は、PMA/イオノマイシンに応答してサイトカインを産生する。5週目のATOからのH1またはH1-CAR由来CD45+細胞を、PMA/イオノマイシンで6時間処理した。活性化は、インターフェロンガンマおよびIL-2に対する細胞内染色によって示される。
A.末梢血(PB)T細胞にTCRを形質導入する標準的な免疫療法手法を上回る、HSPC/ATOにおいてTCRを発現させることの一部の利点
PB T細胞は、それらが特異的な抗腫瘍TCRを発現するように形質導入された場合、既存の多様なTCRレパートリーを有する。ゆえに、内因性TCR鎖とトランスジェニックTCRとの誤対合が自家細胞に関して生じ得;これは、抗腫瘍特異性の減退、または新規な自己反応性の誤対合TCRの生成をもたらし得る。ATO由来TCR改変T細胞における内因性TCRβ再編成の対立遺伝子排除により、形質導入されたおよび内因性のTCR鎖間での誤対合の危険性は、大幅に低下するはずである。
CART細胞療法の通常の手法は、PBにおいてCARを発現させることである。ゆえに、そのように生成されたCART細胞は、内因性発現により多様なTCRも発現する。対照的に、HSPCのCAR形質導入は、上皮内リンパ球(IEL)と同様に、CD3-TCR-表現型を有するT細胞を産生する。TCR発現の非存在は、同種異系レシピエントにおいてGvHDを阻止するはずであり、普遍的な生成物の可能性を提示する。
ベルギーのBart Vandekerckhoveのグループは、TCR形質導入されたCD34+胸腺プロ-T細胞またはCD34+動員末梢血(MPB)HSPCのいずれかから未成熟T細胞先駆体を生成するために、OP9-DL1を用いている(Snauwaert, et. al, Leukemia, 2014)。しかしながら、このシステムにおいて、細胞の<1%が、14日目(胸腺プロ-T)または33日目(MPB)の時点で成熟CD8+ T細胞であった。TCR形質導入細胞の大多数は、DPステージで停止し、T細胞成熟を支持し得るOP9-DL1の乏しい能力と一致した。本方法は、より多くの成熟CD8+ T細胞を産生する。
OP9-DL1(またはDll4)は、以前は、hPSC(hESCまたはhiPSCのいずれか)からのT細胞分化のための唯一のシステムであった。OP9-DL1では、PSCからの細胞収量は非常に乏しく、T細胞分化はCB HSPCよりもさらに悪い。対照的に、hPSC(hESCまたはiPSCの両方)からのT細胞分化は、ATOシステムにおいて非常に効率的である。成熟ナイーブCD8およびCD4 SP細胞は、CB HSPCよりもPSCからさらにより迅速に産生される。hPSCから成熟T細胞を産生する能力は、本当にすぐに使える普遍的な生成物が、hPSCの遺伝子編集により容易に生成されて、免疫反応性遺伝子を除去し得るまたは置き換え得ることを意味する。単一のPSC由来T細胞生成物が、複数の潜在的患者に利用可能であると考えられ、患者特異的生成物を作製することにおいて存在する時間および費用の現在の制約を克服する。加えて、化学療法後のリンパ球減少症患者から充分な自家T細胞を収集するという問題は、回避されると考えられる。
前の実施例は、CD34+臍帯血細胞およびMS5-hDLL1間質を用いたATOシステムと、インビトロヒトT細胞分化のための技術プロトコールの現状:OP9-Dll1システムとの間の比較を示している。OP9-DLL1に対するATOシステムの優位性のさらなる証拠を提供するために、3つの異なる臍帯血ドナー(E37、E43、E68)からのATOを生成し、かつOP9-DLL1間質上での分化と比較した。図68は、4週目の時点での、両システムにおけるCD34+およびT細胞前駆体の維持を示している(図68)。両システムは、CD5およびCD7の発現によって示されるように、T細胞系統への細胞のコミットメントを可能にする。しかしながら、ATOシステムは、CD4CD8ダブルポジティブ細胞(DP)の生成において非常に優れている(図69)。すでに4週目の時点で、ATOシステムは、TCRab+CD3+細胞の堅牢な集団の生成を可能にし、かつそれらの一部はすでにCD8SPであるが、一方でOP9-hDLL1は、はるかにより非効率的であり、かつOP9-hDLL1システムから産生された生存細胞の量は、治療的および商業的な生存率に対して十分なT細胞の産生を可能にしない(図70)。図70におけるデータの数字表記は、図71に示されている。
以下の参考文献は、それらが、本明細書において明記されるものに補足的な、例示的な手続きのまたは他の詳細を提供する程度まで、参照により本明細書に具体的に組み入れられる。
Claims (17)
- 幹細胞または前駆細胞から成熟CD4 + CD8 - または成熟CD8ab + CD4 - T細胞の組成物を調製するための方法であって、該方法は、
a)外因性Notchリガンドを発現する間質細胞の選択細胞株;
b)造血幹細胞または前駆細胞の集団
を含む三次元(3D)細胞凝集体を培養する工程を含み、
該3D細胞凝集体が、T細胞への幹細胞または前駆細胞のインビトロでの分化に充分な期間、インスリン、FLT3L、IL-7およびアスコルビン酸を含む、血清不含培地中で培養され;
該造血幹細胞または前駆細胞が、(i)胚性幹細胞(ESC)、誘導多能性幹細胞(iPSC)もしくはヒト胚性中胚葉前駆細胞由来の培養物中で生成された、または(ii)骨髄、臍帯血、胸腺、末梢血もしくは動員血から単離された、
前記方法。 - 3D細胞凝集体を形成させるための幹細胞または前駆細胞および間質細胞の遠心分離をさらに含む、請求項1記載の方法。
- 間質細胞が、完全な、部分的な、または改変されたNotchリガンドをコードする外因性ヌクレオチド配列を有し、該Notchリガンドが、DLL4、DLL1、JAG1、JAG2、またはそれらの組み合わせである、請求項1記載の方法。
- 間質細胞が、マウス間質細胞株、ヒト間質細胞株、インビトロで多能性幹細胞から分化した間質細胞の選択集団、またはそれらの組み合わせである、請求項1記載の方法。
- 間質細胞が、外因性ヒト主要組織適合複合体(MHC)、外因性抗原特異的共刺激分子もしくはサイトカイン、外因性抗原、またはそれらの組み合わせを発現する、請求項1記載の方法。
- 幹細胞または前駆細胞が、外因性T細胞受容体(TCR)、キメラ抗原受容体(CAR)、およびHLA発現または機能の遺伝子改変の1つまたは複数を含む、請求項1記載の方法。
- 3D細胞凝集体由来のT細胞が、対立遺伝子排除または遺伝子改変により内因性TCRを発現しない、または該T細胞が内因性TCR遺伝子の遺伝子座に挿入された外因性TCR遺伝子を含む、請求項1記載の方法。
- 3D細胞凝集体由来のT細胞が、外因性TCRおよびCARの一方または両方を発現する、および該T細胞が先天性様T細胞に特徴的なマーカーを発現する、請求項1記載の方法。
- 請求項1記載の方法により産生されたT細胞の集団。
- T細胞が先天性様T細胞に特徴的なマーカーを発現する、請求項9記載のT細胞の集団。
- T細胞が、外因性TCRおよびCARの一方または両方を発現する、請求項9記載のT細胞の集団。
- CARがCD19 CARを含む、請求項11記載のT細胞の集団。
- 外因性TCRがNY-ESO-1特異的TCRである、請求項11記載のT細胞の集団。
- T細胞が、ナチュラルキラーT細胞、ガンマ-デルタT細胞、サプレッサーT細胞、抗原特異的T細胞、上皮内リンパ球T細胞、もしくはそれらの組み合わせである、請求項9記載のT細胞の集団。
- 請求項9記載のT細胞の集団を含む、患者を治療するための薬学的組成物。
- 患者におけるがん、自己免疫疾患、移植片対宿主疾患または移植片拒絶を治療するための請求項9記載のT細胞の集団を含む、薬学的組成物。
- T細胞が患者に対して同種異系である、請求項15または16記載の薬学的組成物。
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2021189828A JP7402211B2 (ja) | 2015-10-30 | 2021-11-24 | 幹細胞からt細胞を作製する方法および該t細胞を用いた免疫療法的方法 |
Applications Claiming Priority (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562248931P | 2015-10-30 | 2015-10-30 | |
| US62/248,931 | 2015-10-30 | ||
| US201562265204P | 2015-12-09 | 2015-12-09 | |
| US62/265,204 | 2015-12-09 | ||
| US201662359456P | 2016-07-07 | 2016-07-07 | |
| US62/359,456 | 2016-07-07 | ||
| PCT/US2016/059375 WO2017075389A1 (en) | 2015-10-30 | 2016-10-28 | Methods of generating t-cells from stem cells and immunotherapeutic methods using the t-cells |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2021189828A Division JP7402211B2 (ja) | 2015-10-30 | 2021-11-24 | 幹細胞からt細胞を作製する方法および該t細胞を用いた免疫療法的方法 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2018533364A JP2018533364A (ja) | 2018-11-15 |
| JP2018533364A5 JP2018533364A5 (ja) | 2019-12-05 |
| JP6983771B2 true JP6983771B2 (ja) | 2021-12-17 |
Family
ID=58631163
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2018522655A Active JP6983771B2 (ja) | 2015-10-30 | 2016-10-28 | 幹細胞からt細胞を作製する方法および該t細胞を用いた免疫療法的方法 |
| JP2021189828A Active JP7402211B2 (ja) | 2015-10-30 | 2021-11-24 | 幹細胞からt細胞を作製する方法および該t細胞を用いた免疫療法的方法 |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2021189828A Active JP7402211B2 (ja) | 2015-10-30 | 2021-11-24 | 幹細胞からt細胞を作製する方法および該t細胞を用いた免疫療法的方法 |
Country Status (14)
| Country | Link |
|---|---|
| US (2) | US11154573B2 (ja) |
| EP (1) | EP3368660A4 (ja) |
| JP (2) | JP6983771B2 (ja) |
| KR (1) | KR20180092951A (ja) |
| CN (1) | CN108463548B (ja) |
| AU (2) | AU2016343682A1 (ja) |
| BR (1) | BR112018008648A2 (ja) |
| CA (1) | CA3003145A1 (ja) |
| IL (1) | IL258899B2 (ja) |
| MX (1) | MX2018005274A (ja) |
| NZ (1) | NZ742915A (ja) |
| SG (1) | SG11201803419PA (ja) |
| TW (2) | TWI812584B (ja) |
| WO (1) | WO2017075389A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2022033812A (ja) * | 2015-10-30 | 2022-03-02 | ザ リージェンツ オブ ザ ユニバーシティ オブ カリフォルニア | 幹細胞からt細胞を作製する方法および該t細胞を用いた免疫療法的方法 |
Families Citing this family (57)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011140441A2 (en) | 2010-05-06 | 2011-11-10 | Children's Hospital Medical Center | Methods and systems for converting precursor cells into intestinal tissues through directed differentiation |
| AU2015267148B2 (en) | 2014-05-28 | 2021-07-29 | Children's Hospital Medical Center | Methods and systems for converting precursor cells into gastric tissues through directed differentiation |
| US10927160B2 (en) * | 2014-07-09 | 2021-02-23 | The Regents Of The University Of California | Engineered invariant natural killer T (iNKT) cells and methods of making and using thereof |
| EP3207123A1 (en) | 2014-10-17 | 2017-08-23 | Children's Hospital Center D/b/a Cincinnati Children's Hospital Medical Center | In vivo model of human small intestine using pluripotent stem cells and methods of making and using same |
| WO2017192997A1 (en) | 2016-05-05 | 2017-11-09 | Children's Hospital Medical Center | Methods for the in vitro manufacture of gastric fundus tissue and compositions related to same |
| NZ753873A (en) | 2016-12-05 | 2023-01-27 | Children’S Hospital Medical Center | Colonic organoids and methods of making and using same |
| WO2018111981A1 (en) * | 2016-12-13 | 2018-06-21 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Methods of preparing an isolated or purified population of thymic emigrant cells and methods of treatment using same |
| KR102426447B1 (ko) * | 2017-05-26 | 2022-07-29 | 카이트 파마 인코포레이티드 | 배아 중간엽 전구 세포를 제조하고 사용하는 방법 |
| WO2018232318A1 (en) * | 2017-06-16 | 2018-12-20 | Mayo Foundation For Medical Education And Research | Materials and methods for increasing immune responses |
| KR20200054178A (ko) | 2017-08-09 | 2020-05-19 | 주노 쎄러퓨티크스 인코퍼레이티드 | 유전자 조작된 세포 조성물 및 관련 조성물의 제조 방법 |
| WO2019060336A1 (en) | 2017-09-20 | 2019-03-28 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | IN VITRO GENERATION OF THYMIC ORGANOID FROM HUMAN PLURIPOTENT STEM CELLS |
| EP3717907A1 (en) * | 2017-11-30 | 2020-10-07 | Novartis AG | Bcma-targeting chimeric antigen receptor, and uses thereof |
| US20210128616A1 (en) * | 2017-12-08 | 2021-05-06 | Juno Therapeutics, Inc. | Phenotypic markers for cell therapy and related methods |
| CN111902532A (zh) | 2018-01-28 | 2020-11-06 | 日内瓦大学 | 用于癌症治疗的精氨酸酶抑制 |
| US20210040449A1 (en) | 2018-02-16 | 2021-02-11 | Kite Pharma, Inc. | Modified pluripotent stem cells and methods of making and use |
| IL278044B2 (en) * | 2018-04-19 | 2024-04-01 | Baylor College Medicine | Reprogramming of CD4 T cells to CD8 T cells through forced expression of CD8AB and type 1-restricted T cell receptors |
| CN110499291B (zh) * | 2018-05-16 | 2023-11-24 | 上海赛比曼生物科技有限公司 | 无血清培养制备嵌合抗原受体t细胞的方法 |
| JP2021526838A (ja) * | 2018-06-12 | 2021-10-11 | ザ リージェンツ オブ ザ ユニバーシティ オブ カリフォルニアThe Regents Of The University Of California | 幹細胞の操作によるinkt細胞に基づく既製の細胞療法 |
| EP3806962A1 (en) | 2018-06-13 | 2021-04-21 | Novartis AG | Bcma chimeric antigen receptors and uses thereof |
| AU2019302207A1 (en) * | 2018-07-13 | 2021-03-04 | Kyoto University | Method for producing γδ T cells |
| WO2020047099A1 (en) * | 2018-08-28 | 2020-03-05 | Fred Hutchinson Cancer Research Center | Methods and compositions for adoptive t cell therapy incorporating induced notch signaling |
| WO2020123015A1 (en) * | 2018-10-01 | 2020-06-18 | The Trustees Of Columbia University In The City Of New York | Three-dimensional co-culture system for high-throughput testing of therapeutics and diagnostics |
| US20210355190A1 (en) * | 2018-10-31 | 2021-11-18 | Mayo Foundation For Medical Education And Research | Methods and materials for treating cancer |
| CN109762843A (zh) * | 2018-12-29 | 2019-05-17 | 武汉波睿达生物科技有限公司 | 一种利用脐血来源的cd3阳性t细胞制备通用car-t细胞的方法 |
| CN109652376B (zh) * | 2019-01-08 | 2021-10-15 | 创芯国际生物科技(广州)有限公司 | 一种用于卵巢癌组织3d培养的培养基 |
| BR112021016698A2 (pt) * | 2019-02-24 | 2021-10-13 | Gamida-Cell Ltd. | Método para realçar o potencial de retorno e/ou retenção de célula t gama delta, método para realçar a expressão de célula t gama delta cd62l, método para transplantar células em um sujeito, e, composição de célula terapêutica |
| CN113767167A (zh) * | 2019-04-26 | 2021-12-07 | 科罗拉多大学董事会,法人 | 从诱导多能干细胞体内产生功能性和患者特异性胸腺组织 |
| CN110106144B (zh) * | 2019-05-17 | 2021-10-08 | 苏州大学 | 一种诱导造血干细胞向前t系细胞分化的方法和试剂盒 |
| JP2022536693A (ja) * | 2019-06-12 | 2022-08-18 | ザ リージェンツ オブ ザ ユニバーシティ オブ カリフォルニア | 操作されたオフザシェルフ免疫細胞およびそれらの使用方法 |
| JPWO2021006316A1 (ja) * | 2019-07-10 | 2021-01-14 | ||
| GB201911958D0 (en) * | 2019-08-20 | 2019-10-02 | Adaptimmune Ltd | Methods of t cell production |
| CN114555788A (zh) * | 2019-09-06 | 2022-05-27 | 阿维塔斯有限公司 | 免疫细胞工程化用于体外细胞治疗 |
| CN110499288A (zh) * | 2019-09-17 | 2019-11-26 | 青岛华赛伯曼医学细胞生物有限公司 | 体外诱导造血干细胞向胸腺t细胞分化的试剂盒及其应用 |
| KR102081418B1 (ko) * | 2019-09-24 | 2020-05-26 | 주식회사 이뮤니스바이오 | 말초혈액단핵구 유래 조절 t 세포 배양용 조성물 및 이를 이용한 조절 t 세포 배양방법 |
| CN110872575A (zh) * | 2019-10-21 | 2020-03-10 | 中冠赛尔生物科技(北京)有限公司 | 一种iNKT细胞的体外扩增方法 |
| WO2021085576A1 (ja) | 2019-11-01 | 2021-05-06 | 国立大学法人京都大学 | T細胞の製造方法 |
| CA3160487A1 (en) * | 2019-11-05 | 2021-05-14 | City Of Hope | Generation of chimeric antigen receptor modified t cells from stem cells and therapeutic uses thereof |
| EP4164706A1 (en) | 2019-12-04 | 2023-04-19 | Centre Hospitalier Universitaire Vaudois (CHUV) | Device and process for tissue-engineering and regenerative medicine |
| GB202006903D0 (en) * | 2020-05-11 | 2020-06-24 | Adaptimmune Ltd | Modified iPSCs |
| WO2021252898A1 (en) * | 2020-06-12 | 2021-12-16 | Pact Pharma, Inc. | Compositions and methods of manufacturing autologous t cell therapies |
| CN111849913B (zh) * | 2020-07-31 | 2021-05-25 | 南京北恒生物科技有限公司 | 工程化免疫细胞及其用途 |
| AU2021325670A1 (en) * | 2020-08-10 | 2023-04-20 | Janssen Biotech, Inc. | Materials and methods for producing bioengineered virus specific lymphocytes |
| WO2022083667A1 (en) * | 2020-10-21 | 2022-04-28 | Nanjing Legend Biotech Co., Ltd. | Polypeptides comprising a vaccine specific tcr and a chimeric antigen receptor and uses thereof |
| WO2022147014A2 (en) * | 2020-12-28 | 2022-07-07 | The Regents Of The University Of California | Engineered gamma delta t cells and methods of making and using thereof |
| WO2022235482A1 (en) * | 2021-05-03 | 2022-11-10 | Rutgers, The State University Of New Jersey | Immunotherapy for inflammatory bowel disease and/or cancer |
| WO2022238890A1 (en) * | 2021-05-10 | 2022-11-17 | Bazaz Zohre | Method of producing antibodies from single plasma cells |
| CA3220136A1 (en) * | 2021-05-14 | 2022-11-17 | Appia Bio, Inc. | Production of engineered t cells from stem cells |
| CA3213581A1 (en) * | 2021-05-25 | 2022-12-01 | Kai Ling LIANG | Generating t cell precursors via agonizing tumor necrosis factor receptor 2 |
| CN117957313A (zh) * | 2021-07-19 | 2024-04-30 | 修复免疫股份有限公司 | 从多能干细胞生产免疫细胞群的方法 |
| WO2023019213A1 (en) * | 2021-08-13 | 2023-02-16 | Simcere Innovation, Inc. | Three-dimensional culturing of pluripotent stem cells to produce hematopoietic stem cells |
| AU2022349176A1 (en) * | 2021-09-27 | 2024-04-11 | Kyoto University | Method for producing t cell |
| AU2022366817A1 (en) * | 2021-10-14 | 2024-05-02 | Appia Bio, Inc. | Engineering stem cell t cells with multiple t cell receptors |
| WO2023149555A1 (ja) * | 2022-02-04 | 2023-08-10 | 国立大学法人京都大学 | T細胞の製造方法 |
| CN114807031A (zh) * | 2022-05-13 | 2022-07-29 | 山东赛恩福干细胞工程集团有限公司 | 一种人外周血免疫细胞库和干细胞库的构建方法 |
| CN115125203B (zh) * | 2022-07-08 | 2023-10-27 | 珠海贝索细胞科学技术有限公司 | 一种Th2细胞体外培养方法 |
| WO2024020365A1 (en) * | 2022-07-18 | 2024-01-25 | The Children's Medical Center Corporation | Methods of t cell differentiation and compositions thereof |
| CN115181730B (zh) * | 2022-07-29 | 2023-05-16 | 创芯国际生物科技(广州)有限公司 | 一种胃间质瘤类器官培养基及培养方法 |
Family Cites Families (103)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NL8200523A (nl) | 1982-02-11 | 1983-09-01 | Univ Leiden | Werkwijze voor het in vitro transformeren van planteprotoplasten met plasmide-dna. |
| US4714680B1 (en) | 1984-02-06 | 1995-06-27 | Univ Johns Hopkins | Human stem cells |
| US4683202A (en) | 1985-03-28 | 1987-07-28 | Cetus Corporation | Process for amplifying nucleic acid sequences |
| US4952500A (en) | 1988-02-01 | 1990-08-28 | University Of Georgia Research Foundation, Inc. | Cloning systems for Rhodococcus and related bacteria |
| US5703055A (en) | 1989-03-21 | 1997-12-30 | Wisconsin Alumni Research Foundation | Generation of antibodies through lipid mediated DNA delivery |
| US5302523A (en) | 1989-06-21 | 1994-04-12 | Zeneca Limited | Transformation of plant cells |
| US5550318A (en) | 1990-04-17 | 1996-08-27 | Dekalb Genetics Corporation | Methods and compositions for the production of stably transformed, fertile monocot plants and cells thereof |
| US7705215B1 (en) | 1990-04-17 | 2010-04-27 | Dekalb Genetics Corporation | Methods and compositions for the production of stably transformed, fertile monocot plants and cells thereof |
| US5322783A (en) | 1989-10-17 | 1994-06-21 | Pioneer Hi-Bred International, Inc. | Soybean transformation by microparticle bombardment |
| US5484956A (en) | 1990-01-22 | 1996-01-16 | Dekalb Genetics Corporation | Fertile transgenic Zea mays plant comprising heterologous DNA encoding Bacillus thuringiensis endotoxin |
| US5384253A (en) | 1990-12-28 | 1995-01-24 | Dekalb Genetics Corporation | Genetic transformation of maize cells by electroporation of cells pretreated with pectin degrading enzymes |
| US5389640A (en) | 1991-03-01 | 1995-02-14 | Minnesota Mining And Manufacturing Company | 1-substituted, 2-substituted 1H-imidazo[4,5-c]quinolin-4-amines |
| WO1993004169A1 (en) | 1991-08-20 | 1993-03-04 | Genpharm International, Inc. | Gene targeting in animal cells using isogenic dna constructs |
| WO1993005021A1 (en) | 1991-09-06 | 1993-03-18 | Yoshitomi Pharmaceutical Industries, Ltd. | 4-amino(alkyl)cyclohexane-1-carboxamide compound and use thereof |
| US5610042A (en) | 1991-10-07 | 1997-03-11 | Ciba-Geigy Corporation | Methods for stable transformation of wheat |
| DE69334225D1 (de) | 1992-07-07 | 2008-07-31 | Japan Tobacco Inc | Verfahren zur transformation einer monokotyledon pflanze |
| US5702932A (en) | 1992-07-20 | 1997-12-30 | University Of Florida | Microinjection methods to transform arthropods with exogenous DNA |
| BR9306802A (pt) | 1992-07-27 | 1998-12-08 | Pioneer Hi Bred Int | Processo independente de genótipos para produção de planta de soja transgénica e processo de regeneração de plantas de soja a partir de nodos cotiledonais |
| DE4228457A1 (de) | 1992-08-27 | 1994-04-28 | Beiersdorf Ag | Herstellung von heterodimerem PDGF-AB mit Hilfe eines bicistronischen Vektorsystems in Säugerzellen |
| GB9222888D0 (en) | 1992-10-30 | 1992-12-16 | British Tech Group | Tomography |
| US5656610A (en) | 1994-06-21 | 1997-08-12 | University Of Southern California | Producing a protein in a mammal by injection of a DNA-sequence into the tongue |
| FR2722208B1 (fr) | 1994-07-05 | 1996-10-04 | Inst Nat Sante Rech Med | Nouveau site interne d'entree des ribosomes, vecteur le contenant et utilisation therapeutique |
| US5736524A (en) | 1994-11-14 | 1998-04-07 | Merck & Co.,. Inc. | Polynucleotide tuberculosis vaccine |
| US5843780A (en) | 1995-01-20 | 1998-12-01 | Wisconsin Alumni Research Foundation | Primate embryonic stem cells |
| US5728581A (en) | 1995-06-07 | 1998-03-17 | Systemix, Inc. | Method of expanding hematopoietic stem cells, reagents and bioreactors for use therein |
| US5780448A (en) | 1995-11-07 | 1998-07-14 | Ottawa Civic Hospital Loeb Research | DNA-based vaccination of fish |
| US20030059913A1 (en) | 1995-11-20 | 2003-03-27 | Walter Weyler | Novel Microorganisms with ability to degrade indole and novel enzymes therefrom |
| US5928906A (en) | 1996-05-09 | 1999-07-27 | Sequenom, Inc. | Process for direct sequencing during template amplification |
| US5945100A (en) | 1996-07-31 | 1999-08-31 | Fbp Corporation | Tumor delivery vehicles |
| US5981274A (en) | 1996-09-18 | 1999-11-09 | Tyrrell; D. Lorne J. | Recombinant hepatitis virus vectors |
| JP2001508302A (ja) | 1997-01-10 | 2001-06-26 | ライフ テクノロジーズ,インコーポレイテッド | 胚性幹細胞血清置換 |
| US5994624A (en) | 1997-10-20 | 1999-11-30 | Cotton Incorporated | In planta method for the production of transgenic plants |
| AU729377B2 (en) | 1997-10-23 | 2001-02-01 | Asterias Biotherapeutics, Inc. | Methods and materials for the growth of primate-derived primordial stem cells in feeder-free culture |
| US20030044978A1 (en) | 1998-02-05 | 2003-03-06 | Novartis Corporation | Expanded and genetically modified populations of human hematopoietic stem cells |
| US6110929A (en) | 1998-07-28 | 2000-08-29 | 3M Innovative Properties Company | Oxazolo, thiazolo and selenazolo [4,5-c]-quinolin-4-amines and analogs thereof |
| US6667176B1 (en) | 2000-01-11 | 2003-12-23 | Geron Corporation | cDNA libraries reflecting gene expression during growth and differentiation of human pluripotent stem cells |
| US20020168766A1 (en) | 2000-01-11 | 2002-11-14 | Gold Joseph D. | Genetically altered human pluripotent stem cells |
| US6541485B1 (en) | 1999-06-10 | 2003-04-01 | 3M Innovative Properties Company | Urea substituted imidazoquinolines |
| US6331539B1 (en) | 1999-06-10 | 2001-12-18 | 3M Innovative Properties Company | Sulfonamide and sulfamide substituted imidazoquinolines |
| US6451810B1 (en) | 1999-06-10 | 2002-09-17 | 3M Innovative Properties Company | Amide substituted imidazoquinolines |
| CN1423523A (zh) * | 1999-11-17 | 2003-06-11 | 罗切斯特大学 | 来自人体内的免疫系统 |
| CA2394831A1 (en) * | 2000-01-11 | 2001-07-19 | Maxygen, Inc. | Monocyte-derived dendritic cell subsets |
| US6602711B1 (en) | 2000-02-21 | 2003-08-05 | Wisconsin Alumni Research Foundation | Method of making embryoid bodies from primate embryonic stem cells |
| WO2001088100A1 (fr) | 2000-05-16 | 2001-11-22 | Kyowa Hakko Kogyo Co., Ltd. | Nouvelle methode permettant de declencher une differentiation de cellules embryonnaires dans des cellules de l'ectoderme et leurs applications |
| JP5943533B2 (ja) | 2000-05-17 | 2016-07-06 | アステリアス バイオセラピューティクス インコーポレイテッド | 神経前駆細胞の集団 |
| CA2427996A1 (en) | 2000-11-22 | 2002-06-06 | Geron Corporation | Tolerizing allografts of pluripotent stem cells |
| JP2002184539A (ja) | 2000-12-14 | 2002-06-28 | Auto Network Gijutsu Kenkyusho:Kk | コネクタ |
| PT1370552E (pt) | 2001-03-23 | 2007-04-30 | Bayer Pharmaceuticals Corp | Inibidores de rho-quinase |
| DE60211317T2 (de) | 2001-03-23 | 2007-04-12 | Bayer Corp. | Rho-kinase inhibitoren |
| CA2351156A1 (en) | 2001-07-04 | 2003-01-04 | Peter W. Zandstra | A bioprocess for the generation of pluripotent cell derived cells and tissues |
| CA2453438C (en) | 2001-07-12 | 2016-04-05 | Geron Corporation | Cells of the cardiomyocyte lineage produced from human pluripotent stem cells |
| US20030211603A1 (en) | 2001-08-14 | 2003-11-13 | Earp David J. | Reprogramming cells for enhanced differentiation capacity using pluripotent stem cells |
| JP2003097552A (ja) | 2001-09-21 | 2003-04-03 | Nsk Ltd | 摩擦付加装置および直動案内装置 |
| US20030062225A1 (en) | 2001-10-03 | 2003-04-03 | Stucky Paul A. | Elevator load bearing assembly having a detectable element that is indicative of local strain |
| WO2003050251A2 (en) | 2001-12-07 | 2003-06-19 | Geron Corporation | Hematopoietic cells from human embryonic stem cells |
| US20040014755A1 (en) | 2002-01-10 | 2004-01-22 | Dhanapalan Nagarathnam | Rho-kinase inhibitors |
| WO2003062227A1 (en) | 2002-01-23 | 2003-07-31 | Bayer Pharmaceuticals Corporation | Rho-kinase inhibitors |
| CA2473510A1 (en) | 2002-01-23 | 2003-07-31 | Bayer Pharmaceuticals Corporation | Pyrimidine derivatives as rho-kinase inhibitors |
| WO2003062404A1 (en) | 2002-01-25 | 2003-07-31 | Gamida-Cell Ltd. | Methods of expanding stem and progenitor cells and expanded cell populations obtained thereby |
| AU2003304106C1 (en) | 2002-05-17 | 2010-10-28 | Mount Sinai School Of Medicine Of New York University | Mesoderm and definitive endoderm cell populations |
| US20040039796A1 (en) | 2002-08-08 | 2004-02-26 | Virtual Radio, Inc. | Personalized cyber disk jockey and Internet radio advertising |
| US7737153B2 (en) | 2002-10-28 | 2010-06-15 | Bayer Schering Pharma Aktiengesellschaft | Heteroaryloxy-substituted phenylaminopyrimidines as rho-kinase inhibitors |
| US7986994B2 (en) | 2002-12-04 | 2011-07-26 | Medtronic, Inc. | Method and apparatus for detecting change in intrathoracic electrical impedance |
| US7575925B2 (en) * | 2002-12-10 | 2009-08-18 | Sunnybrook Health Sciences Centre | Cell preparations comprising cells of the T cell lineage and methods of making and using them |
| US7485432B2 (en) | 2003-02-27 | 2009-02-03 | 3M Innovative Properties Company | Selective modulation of TLR-mediated biological activity |
| US20060205071A1 (en) | 2003-07-17 | 2006-09-14 | Gamida-Cell Ltd. | Methods for ex-vivo expanding stem/progenitor cells |
| US7795404B1 (en) | 2003-08-08 | 2010-09-14 | Five Prime Therapeutics, Inc. | Human soluble notch receptor ligands |
| WO2005021728A2 (en) | 2003-08-27 | 2005-03-10 | Stemcells California, Inc. | Enriched pancreatic stem cell and progenitor cell populations, and methods for identifying, isolating and enriching for these populations |
| WO2005070120A2 (en) * | 2004-01-09 | 2005-08-04 | Serologicals Investment Company, Inc. | Cell culture media |
| WO2005080554A1 (ja) | 2004-02-23 | 2005-09-01 | Kyoto University | 胚性幹細胞の分化調節方法 |
| US7452718B2 (en) | 2004-03-26 | 2008-11-18 | Geron Corporation | Direct differentiation method for making cardiomyocytes from human embryonic stem cells |
| WO2005123902A1 (ja) | 2004-06-18 | 2005-12-29 | Riken | 無血清浮遊培養による胚性幹細胞の神経分化誘導法 |
| US20080038820A1 (en) | 2004-06-22 | 2008-02-14 | Rudy-Reil Diane E | Induction of pluripotent stem cells into mesodermal lineages |
| NZ553235A (en) | 2004-09-08 | 2009-11-27 | Wisconsin Alumni Res Found | Culturing human pluripotent stem cells |
| US20080254003A1 (en) | 2004-12-22 | 2008-10-16 | Robert Passier | Differentiation of Human Embryonic Stem Cells and Cardiomyocytes and Cardiomyocyte Progenitors Derived Therefrom |
| WO2007027226A2 (en) | 2005-04-28 | 2007-03-08 | Board Of Regents, The University Of Texas System | Systems and methods for the production of differentiated cells |
| US8021867B2 (en) | 2005-10-18 | 2011-09-20 | Duke University | Rationally-designed meganucleases with altered sequence specificity and DNA-binding affinity |
| US20070116680A1 (en) | 2005-11-18 | 2007-05-24 | Rensselaer Polytechnic Institute | Stem cells within gel microenvironments |
| EP1815863A1 (en) | 2006-02-03 | 2007-08-08 | Nederlandse Organisatie voor toegepast-natuurwetenschappelijk Onderzoek TNO | Use of TLR3 agonists for the treatment of neurodegenerative disorders |
| EP1999249B8 (en) | 2006-03-30 | 2012-02-15 | The University Court Of The University of Edinburgh | Culture medium containing kinase inhibitors. and uses thereof |
| CN101595107A (zh) | 2006-06-30 | 2009-12-02 | 先灵公司 | 能提高p53活性的有取代哌啶及其用途 |
| WO2008006583A1 (en) | 2006-07-14 | 2008-01-17 | Novartis Ag | Pyrimidine derivatives as alk-5 inhibitors |
| JP5419279B2 (ja) | 2007-01-17 | 2014-02-19 | ウィスコンシン アラムニ リサーチ ファンデーション | 改良された幹細胞の培養 |
| US9175260B2 (en) | 2007-01-30 | 2015-11-03 | TheUniversity of Georgia Research Foundation, Inc. | Early mesoderm cells, a stable population of mesendoderm cells that has utility for generation of endoderm and mesoderm lineages and multipotent migratory cells (MMC) |
| US8377886B2 (en) | 2007-09-14 | 2013-02-19 | Albert Einstein College Of Medicine Of Yeshiva University | Use of gamma secretase inhibitors and notch pathway inhibitors for treatment and prevention of renal disease |
| US10350243B2 (en) * | 2008-01-30 | 2019-07-16 | Memorial Sloan-Kettering Cancer Center | Methods for off-the-shelf-tumor immunotherapy using allogeneic T-cell precursors |
| US10059923B2 (en) * | 2008-01-30 | 2018-08-28 | Memorial Sloan Kettering Cancer Center | Methods for off-the-shelf tumor immunotherapy using allogeneic T-cell precursors |
| CN102083979B (zh) * | 2008-05-09 | 2015-01-07 | 新加坡科技研究局 | Hbv表位反应性外源t细胞受体(tcr)及其用途 |
| DE102008040322A1 (de) | 2008-07-10 | 2010-09-16 | Dilo Trading Ag | Energiespeicher-Kreislaufsystem unter Verwendung von Lithium-Ionen-Zellen |
| JP2012508561A (ja) * | 2008-09-11 | 2012-04-12 | ユニバーシティ オブ フロリダ リサーチファウンデーション インコーポレイティッド | T細胞を産生するためのシステム及び方法 |
| PE20100362A1 (es) | 2008-10-30 | 2010-05-27 | Irm Llc | Derivados de purina que expanden las celulas madre hematopoyeticas |
| AU2009317161B2 (en) * | 2008-11-24 | 2014-09-11 | Helmholtz Zentrum Munchen Deutsches Forschungszentrum Fur Gesundheit Und Umwelt Gmbh | High affinity T cell receptor and use thereof |
| KR101720961B1 (ko) | 2009-02-27 | 2017-03-29 | 셀룰러 다이내믹스 인터내셔널, 인코포레이티드 | 다능성 세포의 분화 |
| EP2539441B1 (en) | 2010-02-22 | 2016-09-28 | Université Pierre et Marie Curie (Paris 6) | Cell culture medium for the growth and differentiation of cells of the hematopoietic lineage |
| GB201006768D0 (en) | 2010-04-22 | 2010-06-09 | Cancer Rec Tech Ltd | Method for obtaining dendritic cells |
| CA2802721C (en) | 2010-06-25 | 2015-08-18 | Idera Pharmaceuticals, Inc. | Novel agonists of toll-like receptor 3 and methods of their use |
| WO2014039044A1 (en) * | 2012-09-06 | 2014-03-13 | The United States Of America, As Represented By The Secretary, Department Of Health And Human Services | Methods of producing t memory stem cell populations |
| ES2824024T3 (es) | 2012-10-10 | 2021-05-11 | Sangamo Therapeutics Inc | Compuestos modificadores de células T y usos de los mismos |
| US20160017302A1 (en) * | 2013-03-05 | 2016-01-21 | Baylor College Of Medicine | Heparanase expression in human t lymphocytes |
| US9409760B1 (en) | 2013-12-05 | 2016-08-09 | Paul Lichtefeld, Sr. | Fluid dispenser |
| US9410501B2 (en) | 2014-04-25 | 2016-08-09 | Rohr, Inc. | Translating sleeve actuation system and apparatus |
| EP3298131B1 (en) | 2015-05-20 | 2023-04-26 | The Regents of The University of California | Method for generating human dendritic cells for immunotherapy |
| AU2016343682A1 (en) | 2015-10-30 | 2018-06-14 | The Regents Of The University Of California | Methods of generating T-cells from stem cells and immunotherapeutic methods using the T-cells |
-
2016
- 2016-10-28 AU AU2016343682A patent/AU2016343682A1/en not_active Abandoned
- 2016-10-28 BR BR112018008648A patent/BR112018008648A2/pt active Search and Examination
- 2016-10-28 IL IL258899A patent/IL258899B2/en unknown
- 2016-10-28 WO PCT/US2016/059375 patent/WO2017075389A1/en active Application Filing
- 2016-10-28 TW TW105135186A patent/TWI812584B/zh active
- 2016-10-28 JP JP2018522655A patent/JP6983771B2/ja active Active
- 2016-10-28 EP EP16860902.2A patent/EP3368660A4/en active Pending
- 2016-10-28 SG SG11201803419PA patent/SG11201803419PA/en unknown
- 2016-10-28 KR KR1020187015441A patent/KR20180092951A/ko unknown
- 2016-10-28 NZ NZ742915A patent/NZ742915A/en unknown
- 2016-10-28 CA CA3003145A patent/CA3003145A1/en active Pending
- 2016-10-28 MX MX2018005274A patent/MX2018005274A/es unknown
- 2016-10-28 TW TW112127839A patent/TW202344686A/zh unknown
- 2016-10-28 US US15/772,224 patent/US11154573B2/en active Active
- 2016-10-28 CN CN201680077607.7A patent/CN108463548B/zh active Active
-
2021
- 2021-09-17 US US17/478,875 patent/US20220096553A1/en active Pending
- 2021-11-24 JP JP2021189828A patent/JP7402211B2/ja active Active
-
2023
- 2023-04-11 AU AU2023202195A patent/AU2023202195A1/en active Pending
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2022033812A (ja) * | 2015-10-30 | 2022-03-02 | ザ リージェンツ オブ ザ ユニバーシティ オブ カリフォルニア | 幹細胞からt細胞を作製する方法および該t細胞を用いた免疫療法的方法 |
| JP7402211B2 (ja) | 2015-10-30 | 2023-12-20 | ザ リージェンツ オブ ザ ユニバーシティ オブ カリフォルニア | 幹細胞からt細胞を作製する方法および該t細胞を用いた免疫療法的方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| TW202344686A (zh) | 2023-11-16 |
| US11154573B2 (en) | 2021-10-26 |
| SG11201803419PA (en) | 2018-05-30 |
| IL258899B1 (en) | 2023-11-01 |
| WO2017075389A8 (en) | 2018-05-24 |
| TW201730334A (zh) | 2017-09-01 |
| AU2023202195A1 (en) | 2023-05-11 |
| JP2022033812A (ja) | 2022-03-02 |
| US20190231817A1 (en) | 2019-08-01 |
| AU2016343682A1 (en) | 2018-06-14 |
| CN108463548A (zh) | 2018-08-28 |
| TWI812584B (zh) | 2023-08-21 |
| IL258899B2 (en) | 2024-03-01 |
| JP7402211B2 (ja) | 2023-12-20 |
| BR112018008648A2 (pt) | 2018-11-27 |
| CA3003145A1 (en) | 2017-05-04 |
| WO2017075389A1 (en) | 2017-05-04 |
| IL258899A (en) | 2018-06-28 |
| EP3368660A1 (en) | 2018-09-05 |
| US20220096553A1 (en) | 2022-03-31 |
| JP2018533364A (ja) | 2018-11-15 |
| NZ742915A (en) | 2024-02-23 |
| MX2018005274A (es) | 2019-09-19 |
| CN108463548B (zh) | 2023-04-18 |
| EP3368660A4 (en) | 2019-04-24 |
| KR20180092951A (ko) | 2018-08-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6983771B2 (ja) | 幹細胞からt細胞を作製する方法および該t細胞を用いた免疫療法的方法 | |
| AU2019287483B2 (en) | Stem cell-engineered iNKT cell-based off-the-shelf cellular therapy | |
| EP3982979A1 (en) | Engineered off-the-shelf immune cells and methods of use thereof | |
| JP2022078215A (ja) | 多能性幹細胞からhlaホモ接合免疫細胞への指向分化方法 | |
| WO2020201230A1 (en) | Car for use in the treatment of hvg disease | |
| EP2150611A1 (en) | Novel thymic cellular populations and uses thereof | |
| US20230002727A1 (en) | Thymus organoids bioengineered from human pluripotent stem cells | |
| CN117480249A (zh) | 包含未重排的t细胞受体(tcr)基因座的干细胞及其使用方法 | |
| US20210002612A1 (en) | Novel method for obtaining t cells from pluripotent stem cells, and uses thereof | |
| CROOKS et al. | Patent 3003145 Summary | |
| O'Brien | Induced Human Pluripotent Stem Cell–derived NK Cells as an Alternative Source of Lymphocytes for Anti-Cancer Immunotherapy | |
| WO2024097800A1 (en) | Off-the-shelf therapeutic cells with multiplex genomic engineering for targeting kallikrein-2 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20191025 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20191025 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20201130 |
|
| RD04 | Notification of resignation of power of attorney |
Free format text: JAPANESE INTERMEDIATE CODE: A7424 Effective date: 20210127 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20210224 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20210609 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20210907 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20211006 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20211027 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20211124 |
|
| R150 | Certificate of patent or registration of utility model |
Ref document number: 6983771 Country of ref document: JP Free format text: JAPANESE INTERMEDIATE CODE: R150 |