JP4601166B2 - 抗原特異的t細胞の直接的選択方法 - Google Patents
抗原特異的t細胞の直接的選択方法 Download PDFInfo
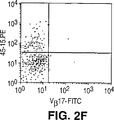
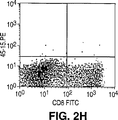
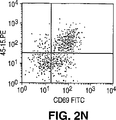
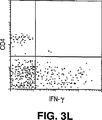
- Publication number
- JP4601166B2 JP4601166B2 JP2000548729A JP2000548729A JP4601166B2 JP 4601166 B2 JP4601166 B2 JP 4601166B2 JP 2000548729 A JP2000548729 A JP 2000548729A JP 2000548729 A JP2000548729 A JP 2000548729A JP 4601166 B2 JP4601166 B2 JP 4601166B2
- Authority
- JP
- Japan
- Prior art keywords
- cells
- cell
- antigen
- product
- specific
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 210000001744 T-lymphocyte Anatomy 0.000 title claims abstract description 196
- 239000000427 antigen Substances 0.000 title claims abstract description 188
- 108091007433 antigens Proteins 0.000 title claims abstract description 184
- 102000036639 antigens Human genes 0.000 title claims abstract description 184
- 238000000034 method Methods 0.000 title claims abstract description 141
- 210000004027 cell Anatomy 0.000 claims abstract description 574
- 238000002372 labelling Methods 0.000 claims abstract description 44
- 230000000638 stimulation Effects 0.000 claims abstract description 26
- 230000004044 response Effects 0.000 claims abstract description 22
- 239000006249 magnetic particle Substances 0.000 claims abstract description 9
- 102100034922 T-cell surface glycoprotein CD8 alpha chain Human genes 0.000 claims description 52
- 230000003248 secreting effect Effects 0.000 claims description 40
- 230000027455 binding Effects 0.000 claims description 36
- 102100036011 T-cell surface glycoprotein CD4 Human genes 0.000 claims description 30
- 230000014509 gene expression Effects 0.000 claims description 28
- 102000016266 T-Cell Antigen Receptors Human genes 0.000 claims description 21
- 230000009870 specific binding Effects 0.000 claims description 13
- 239000012634 fragment Substances 0.000 claims description 12
- 239000000126 substance Substances 0.000 claims description 12
- 101000914514 Homo sapiens T-cell-specific surface glycoprotein CD28 Proteins 0.000 claims description 10
- 102100027213 T-cell-specific surface glycoprotein CD28 Human genes 0.000 claims description 10
- 230000007503 antigenic stimulation Effects 0.000 claims description 10
- 102100027207 CD27 antigen Human genes 0.000 claims description 5
- -1 CD31 Proteins 0.000 claims description 5
- 101000914511 Homo sapiens CD27 antigen Proteins 0.000 claims description 5
- 108010092262 T-Cell Antigen Receptors Proteins 0.000 claims description 5
- 238000012258 culturing Methods 0.000 claims description 5
- 125000005647 linker group Chemical group 0.000 claims description 5
- 102000008949 Histocompatibility Antigens Class I Human genes 0.000 claims description 4
- 102100025012 Dipeptidyl peptidase 4 Human genes 0.000 claims description 3
- 101000908391 Homo sapiens Dipeptidyl peptidase 4 Proteins 0.000 claims description 3
- 101000851376 Homo sapiens Tumor necrosis factor receptor superfamily member 8 Proteins 0.000 claims description 3
- 102100036857 Tumor necrosis factor receptor superfamily member 8 Human genes 0.000 claims description 3
- 239000011230 binding agent Substances 0.000 claims description 3
- 150000002632 lipids Chemical class 0.000 claims description 3
- 108010029697 CD40 Ligand Proteins 0.000 claims description 2
- 102100032937 CD40 ligand Human genes 0.000 claims description 2
- 108010088652 Histocompatibility Antigens Class I Proteins 0.000 claims description 2
- 101001046686 Homo sapiens Integrin alpha-M Proteins 0.000 claims description 2
- 101000935043 Homo sapiens Integrin beta-1 Proteins 0.000 claims description 2
- 102100022338 Integrin alpha-M Human genes 0.000 claims description 2
- 102100025304 Integrin beta-1 Human genes 0.000 claims description 2
- 102100024616 Platelet endothelial cell adhesion molecule Human genes 0.000 claims description 2
- 102100031585 ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 Human genes 0.000 claims 1
- 102000017420 CD3 protein, epsilon/gamma/delta subunit Human genes 0.000 claims 1
- 108050005493 CD3 protein, epsilon/gamma/delta subunit Proteins 0.000 claims 1
- 101000777636 Homo sapiens ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 Proteins 0.000 claims 1
- 101001137987 Homo sapiens Lymphocyte activation gene 3 protein Proteins 0.000 claims 1
- 101000934346 Homo sapiens T-cell surface antigen CD2 Proteins 0.000 claims 1
- 101000716102 Homo sapiens T-cell surface glycoprotein CD4 Proteins 0.000 claims 1
- 101000934341 Homo sapiens T-cell surface glycoprotein CD5 Proteins 0.000 claims 1
- 101000946843 Homo sapiens T-cell surface glycoprotein CD8 alpha chain Proteins 0.000 claims 1
- 102000017578 LAG3 Human genes 0.000 claims 1
- 102100025237 T-cell surface antigen CD2 Human genes 0.000 claims 1
- 102100025244 T-cell surface glycoprotein CD5 Human genes 0.000 claims 1
- 238000000926 separation method Methods 0.000 abstract description 44
- 230000005291 magnetic effect Effects 0.000 abstract description 37
- 238000004458 analytical method Methods 0.000 abstract description 34
- 238000000684 flow cytometry Methods 0.000 abstract description 13
- 239000000463 material Substances 0.000 abstract description 5
- 238000005119 centrifugation Methods 0.000 abstract description 4
- 230000002285 radioactive effect Effects 0.000 abstract 1
- 239000000047 product Substances 0.000 description 135
- 108090000765 processed proteins & peptides Proteins 0.000 description 78
- 108010074328 Interferon-gamma Proteins 0.000 description 65
- 102100037850 Interferon gamma Human genes 0.000 description 62
- 239000012636 effector Substances 0.000 description 40
- 102000004127 Cytokines Human genes 0.000 description 34
- 108090000695 Cytokines Proteins 0.000 description 34
- 108700018351 Major Histocompatibility Complex Proteins 0.000 description 33
- 230000020382 suppression by virus of host antigen processing and presentation of peptide antigen via MHC class I Effects 0.000 description 33
- 206010028980 Neoplasm Diseases 0.000 description 30
- 239000011159 matrix material Substances 0.000 description 29
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 28
- 210000000612 antigen-presenting cell Anatomy 0.000 description 23
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 23
- 230000028327 secretion Effects 0.000 description 23
- 238000011282 treatment Methods 0.000 description 23
- 239000003607 modifier Substances 0.000 description 21
- 208000023275 Autoimmune disease Diseases 0.000 description 20
- 201000010099 disease Diseases 0.000 description 20
- 102000004196 processed proteins & peptides Human genes 0.000 description 19
- 230000004913 activation Effects 0.000 description 17
- 239000002609 medium Substances 0.000 description 17
- 108010004729 Phycoerythrin Proteins 0.000 description 16
- 108091008874 T cell receptors Proteins 0.000 description 16
- 201000011510 cancer Diseases 0.000 description 16
- 108090000623 proteins and genes Proteins 0.000 description 16
- 210000001151 cytotoxic T lymphocyte Anatomy 0.000 description 15
- 206010020751 Hypersensitivity Diseases 0.000 description 14
- 108090000978 Interleukin-4 Proteins 0.000 description 14
- 102000004388 Interleukin-4 Human genes 0.000 description 14
- 239000000872 buffer Substances 0.000 description 14
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 13
- 238000000338 in vitro Methods 0.000 description 13
- 238000011534 incubation Methods 0.000 description 13
- 239000002953 phosphate buffered saline Substances 0.000 description 13
- 239000004698 Polyethylene Substances 0.000 description 12
- 230000030741 antigen processing and presentation Effects 0.000 description 12
- 210000003719 b-lymphocyte Anatomy 0.000 description 12
- 239000011324 bead Substances 0.000 description 12
- 210000004698 lymphocyte Anatomy 0.000 description 12
- 102000004169 proteins and genes Human genes 0.000 description 12
- 238000010186 staining Methods 0.000 description 12
- 108010002350 Interleukin-2 Proteins 0.000 description 11
- 102000000588 Interleukin-2 Human genes 0.000 description 11
- 230000006870 function Effects 0.000 description 11
- 238000002955 isolation Methods 0.000 description 11
- 239000000203 mixture Substances 0.000 description 11
- 235000018102 proteins Nutrition 0.000 description 11
- 102100034540 Adenomatous polyposis coli protein Human genes 0.000 description 10
- 101000924577 Homo sapiens Adenomatous polyposis coli protein Proteins 0.000 description 10
- 239000003153 chemical reaction reagent Substances 0.000 description 10
- 238000001514 detection method Methods 0.000 description 10
- 230000000694 effects Effects 0.000 description 10
- 238000005516 engineering process Methods 0.000 description 10
- 238000007885 magnetic separation Methods 0.000 description 10
- 101000738771 Homo sapiens Receptor-type tyrosine-protein phosphatase C Proteins 0.000 description 9
- 102100037422 Receptor-type tyrosine-protein phosphatase C Human genes 0.000 description 9
- 208000026935 allergic disease Diseases 0.000 description 9
- 238000013459 approach Methods 0.000 description 9
- 241000700605 Viruses Species 0.000 description 8
- 230000007815 allergy Effects 0.000 description 8
- MHMNJMPURVTYEJ-UHFFFAOYSA-N fluorescein-5-isothiocyanate Chemical compound O1C(=O)C2=CC(N=C=S)=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 MHMNJMPURVTYEJ-UHFFFAOYSA-N 0.000 description 8
- 238000001727 in vivo Methods 0.000 description 8
- 238000002826 magnetic-activated cell sorting Methods 0.000 description 8
- 229960000814 tetanus toxoid Drugs 0.000 description 8
- 210000001519 tissue Anatomy 0.000 description 8
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 7
- 239000012980 RPMI-1640 medium Substances 0.000 description 7
- 239000013566 allergen Substances 0.000 description 7
- 238000003556 assay Methods 0.000 description 7
- 239000002775 capsule Substances 0.000 description 7
- 239000000499 gel Substances 0.000 description 7
- 230000002209 hydrophobic effect Effects 0.000 description 7
- 208000015181 infectious disease Diseases 0.000 description 7
- 206010022000 influenza Diseases 0.000 description 7
- 108091033319 polynucleotide Proteins 0.000 description 7
- 102000040430 polynucleotide Human genes 0.000 description 7
- 239000002157 polynucleotide Substances 0.000 description 7
- 229920001184 polypeptide Polymers 0.000 description 7
- 230000002829 reductive effect Effects 0.000 description 7
- 210000002955 secretory cell Anatomy 0.000 description 7
- 108020004414 DNA Proteins 0.000 description 6
- 241000712431 Influenza A virus Species 0.000 description 6
- 230000000735 allogeneic effect Effects 0.000 description 6
- 210000000170 cell membrane Anatomy 0.000 description 6
- 239000006285 cell suspension Substances 0.000 description 6
- 230000000139 costimulatory effect Effects 0.000 description 6
- 238000004519 manufacturing process Methods 0.000 description 6
- 238000002360 preparation method Methods 0.000 description 6
- 102000005962 receptors Human genes 0.000 description 6
- 108020003175 receptors Proteins 0.000 description 6
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 6
- 210000003171 tumor-infiltrating lymphocyte Anatomy 0.000 description 6
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 6
- 102000003814 Interleukin-10 Human genes 0.000 description 5
- 108090000174 Interleukin-10 Proteins 0.000 description 5
- 239000012298 atmosphere Substances 0.000 description 5
- 210000004369 blood Anatomy 0.000 description 5
- 239000008280 blood Substances 0.000 description 5
- 238000004132 cross linking Methods 0.000 description 5
- 230000009089 cytolysis Effects 0.000 description 5
- 230000001472 cytotoxic effect Effects 0.000 description 5
- 238000009792 diffusion process Methods 0.000 description 5
- 230000001404 mediated effect Effects 0.000 description 5
- 239000011325 microbead Substances 0.000 description 5
- 239000002245 particle Substances 0.000 description 5
- XJMOSONTPMZWPB-UHFFFAOYSA-M propidium iodide Chemical compound [I-].[I-].C12=CC(N)=CC=C2C2=CC=C(N)C=C2[N+](CCC[N+](C)(CC)CC)=C1C1=CC=CC=C1 XJMOSONTPMZWPB-UHFFFAOYSA-M 0.000 description 5
- 230000001629 suppression Effects 0.000 description 5
- 210000004881 tumor cell Anatomy 0.000 description 5
- 238000002255 vaccination Methods 0.000 description 5
- MZOFCQQQCNRIBI-VMXHOPILSA-N (3s)-4-[[(2s)-1-[[(2s)-1-[[(1s)-1-carboxy-2-hydroxyethyl]amino]-4-methyl-1-oxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-[[2-[[(2s)-2,6-diaminohexanoyl]amino]acetyl]amino]-4-oxobutanoic acid Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN MZOFCQQQCNRIBI-VMXHOPILSA-N 0.000 description 4
- 108090001008 Avidin Proteins 0.000 description 4
- 102100021260 Galactosylgalactosylxylosylprotein 3-beta-glucuronosyltransferase 1 Human genes 0.000 description 4
- 101000894906 Homo sapiens Galactosylgalactosylxylosylprotein 3-beta-glucuronosyltransferase 1 Proteins 0.000 description 4
- 101000914484 Homo sapiens T-lymphocyte activation antigen CD80 Proteins 0.000 description 4
- 108090001090 Lectins Proteins 0.000 description 4
- 102000004856 Lectins Human genes 0.000 description 4
- 230000005867 T cell response Effects 0.000 description 4
- 102100027222 T-lymphocyte activation antigen CD80 Human genes 0.000 description 4
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 4
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 4
- 206010053613 Type IV hypersensitivity reaction Diseases 0.000 description 4
- 239000000556 agonist Substances 0.000 description 4
- 239000005557 antagonist Substances 0.000 description 4
- 229960002685 biotin Drugs 0.000 description 4
- 235000020958 biotin Nutrition 0.000 description 4
- 239000011616 biotin Substances 0.000 description 4
- 239000006143 cell culture medium Substances 0.000 description 4
- 238000006243 chemical reaction Methods 0.000 description 4
- 239000003795 chemical substances by application Substances 0.000 description 4
- 230000021615 conjugation Effects 0.000 description 4
- 230000004069 differentiation Effects 0.000 description 4
- 239000003814 drug Substances 0.000 description 4
- 239000007850 fluorescent dye Substances 0.000 description 4
- 150000004676 glycans Chemical class 0.000 description 4
- 210000002443 helper t lymphocyte Anatomy 0.000 description 4
- 230000028993 immune response Effects 0.000 description 4
- 230000001506 immunosuppresive effect Effects 0.000 description 4
- 238000009169 immunotherapy Methods 0.000 description 4
- 230000001965 increasing effect Effects 0.000 description 4
- 230000003993 interaction Effects 0.000 description 4
- 239000002523 lectin Substances 0.000 description 4
- 239000002502 liposome Substances 0.000 description 4
- 210000001165 lymph node Anatomy 0.000 description 4
- 239000012528 membrane Substances 0.000 description 4
- 210000000056 organ Anatomy 0.000 description 4
- 230000036961 partial effect Effects 0.000 description 4
- OXNIZHLAWKMVMX-UHFFFAOYSA-N picric acid Chemical compound OC1=C([N+]([O-])=O)C=C([N+]([O-])=O)C=C1[N+]([O-])=O OXNIZHLAWKMVMX-UHFFFAOYSA-N 0.000 description 4
- 229920000642 polymer Polymers 0.000 description 4
- 229920001282 polysaccharide Polymers 0.000 description 4
- 239000005017 polysaccharide Substances 0.000 description 4
- 230000001105 regulatory effect Effects 0.000 description 4
- 210000002966 serum Anatomy 0.000 description 4
- 230000004936 stimulating effect Effects 0.000 description 4
- 239000000758 substrate Substances 0.000 description 4
- 229950002929 trinitrophenol Drugs 0.000 description 4
- 230000005951 type IV hypersensitivity Effects 0.000 description 4
- 208000027930 type IV hypersensitivity disease Diseases 0.000 description 4
- 229920001817 Agar Polymers 0.000 description 3
- 241000894006 Bacteria Species 0.000 description 3
- 102100025221 CD70 antigen Human genes 0.000 description 3
- 102100025137 Early activation antigen CD69 Human genes 0.000 description 3
- 108060003393 Granulin Proteins 0.000 description 3
- 108010088729 HLA-A*02:01 antigen Proteins 0.000 description 3
- 101000934356 Homo sapiens CD70 antigen Proteins 0.000 description 3
- 101000934374 Homo sapiens Early activation antigen CD69 Proteins 0.000 description 3
- 101001057504 Homo sapiens Interferon-stimulated gene 20 kDa protein Proteins 0.000 description 3
- 101001055144 Homo sapiens Interleukin-2 receptor subunit alpha Proteins 0.000 description 3
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 3
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 3
- 102100037877 Intercellular adhesion molecule 1 Human genes 0.000 description 3
- 102000008070 Interferon-gamma Human genes 0.000 description 3
- 108010038453 Interleukin-2 Receptors Proteins 0.000 description 3
- 102000010789 Interleukin-2 Receptors Human genes 0.000 description 3
- 102100026878 Interleukin-2 receptor subunit alpha Human genes 0.000 description 3
- 108010002616 Interleukin-5 Proteins 0.000 description 3
- 102100039897 Interleukin-5 Human genes 0.000 description 3
- 241000124008 Mammalia Species 0.000 description 3
- 229930182555 Penicillin Natural products 0.000 description 3
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 3
- 108010008038 Synthetic Vaccines Proteins 0.000 description 3
- 108091023040 Transcription factor Proteins 0.000 description 3
- 102000040945 Transcription factor Human genes 0.000 description 3
- 208000036142 Viral infection Diseases 0.000 description 3
- 239000008272 agar Substances 0.000 description 3
- 230000000890 antigenic effect Effects 0.000 description 3
- 238000000149 argon plasma sintering Methods 0.000 description 3
- 230000006287 biotinylation Effects 0.000 description 3
- 238000007413 biotinylation Methods 0.000 description 3
- 210000002798 bone marrow cell Anatomy 0.000 description 3
- 239000002458 cell surface marker Substances 0.000 description 3
- 230000016396 cytokine production Effects 0.000 description 3
- 230000001461 cytolytic effect Effects 0.000 description 3
- 208000035475 disorder Diseases 0.000 description 3
- 229940079593 drug Drugs 0.000 description 3
- 239000002158 endotoxin Substances 0.000 description 3
- 238000003114 enzyme-linked immunosorbent spot assay Methods 0.000 description 3
- 239000000284 extract Substances 0.000 description 3
- 230000036039 immunity Effects 0.000 description 3
- 238000010348 incorporation Methods 0.000 description 3
- 229960003130 interferon gamma Drugs 0.000 description 3
- 230000003834 intracellular effect Effects 0.000 description 3
- 239000003446 ligand Substances 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 210000002540 macrophage Anatomy 0.000 description 3
- 239000003550 marker Substances 0.000 description 3
- 210000001616 monocyte Anatomy 0.000 description 3
- 230000035772 mutation Effects 0.000 description 3
- 210000000822 natural killer cell Anatomy 0.000 description 3
- 244000052769 pathogen Species 0.000 description 3
- 230000008506 pathogenesis Effects 0.000 description 3
- 230000001575 pathological effect Effects 0.000 description 3
- 229940049954 penicillin Drugs 0.000 description 3
- PHEDXBVPIONUQT-RGYGYFBISA-N phorbol 13-acetate 12-myristate Chemical compound C([C@]1(O)C(=O)C(C)=C[C@H]1[C@@]1(O)[C@H](C)[C@H]2OC(=O)CCCCCCCCCCCCC)C(CO)=C[C@H]1[C@H]1[C@]2(OC(C)=O)C1(C)C PHEDXBVPIONUQT-RGYGYFBISA-N 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- 230000000717 retained effect Effects 0.000 description 3
- 230000019491 signal transduction Effects 0.000 description 3
- 239000007787 solid Substances 0.000 description 3
- 229960005322 streptomycin Drugs 0.000 description 3
- 208000024891 symptom Diseases 0.000 description 3
- 238000013518 transcription Methods 0.000 description 3
- 230000035897 transcription Effects 0.000 description 3
- 230000009385 viral infection Effects 0.000 description 3
- 230000003612 virological effect Effects 0.000 description 3
- 229920000936 Agarose Polymers 0.000 description 2
- 102100024222 B-lymphocyte antigen CD19 Human genes 0.000 description 2
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 2
- 108010084313 CD58 Antigens Proteins 0.000 description 2
- 102000000844 Cell Surface Receptors Human genes 0.000 description 2
- 108010001857 Cell Surface Receptors Proteins 0.000 description 2
- 229920001661 Chitosan Polymers 0.000 description 2
- 108010062580 Concanavalin A Proteins 0.000 description 2
- 229920002307 Dextran Polymers 0.000 description 2
- SHIBSTMRCDJXLN-UHFFFAOYSA-N Digoxigenin Natural products C1CC(C2C(C3(C)CCC(O)CC3CC2)CC2O)(O)C2(C)C1C1=CC(=O)OC1 SHIBSTMRCDJXLN-UHFFFAOYSA-N 0.000 description 2
- 238000011510 Elispot assay Methods 0.000 description 2
- 230000010190 G1 phase Effects 0.000 description 2
- 102000003886 Glycoproteins Human genes 0.000 description 2
- 108090000288 Glycoproteins Proteins 0.000 description 2
- 101000980825 Homo sapiens B-lymphocyte antigen CD19 Proteins 0.000 description 2
- 101000959820 Homo sapiens Interferon alpha-1/13 Proteins 0.000 description 2
- 241000725303 Human immunodeficiency virus Species 0.000 description 2
- 206010061598 Immunodeficiency Diseases 0.000 description 2
- 208000029462 Immunodeficiency disease Diseases 0.000 description 2
- 108060003951 Immunoglobulin Proteins 0.000 description 2
- 206010062016 Immunosuppression Diseases 0.000 description 2
- 206010061218 Inflammation Diseases 0.000 description 2
- 108010064593 Intercellular Adhesion Molecule-1 Proteins 0.000 description 2
- 102100040019 Interferon alpha-1/13 Human genes 0.000 description 2
- 102000003996 Interferon-beta Human genes 0.000 description 2
- 108090000467 Interferon-beta Proteins 0.000 description 2
- 239000000232 Lipid Bilayer Substances 0.000 description 2
- 108091054437 MHC class I family Proteins 0.000 description 2
- 102000043131 MHC class II family Human genes 0.000 description 2
- 108091054438 MHC class II family Proteins 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 2
- 108010047620 Phytohemagglutinins Proteins 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- 108020004511 Recombinant DNA Proteins 0.000 description 2
- 108010090804 Streptavidin Proteins 0.000 description 2
- 230000017274 T cell anergy Effects 0.000 description 2
- 102000004887 Transforming Growth Factor beta Human genes 0.000 description 2
- 108090001012 Transforming Growth Factor beta Proteins 0.000 description 2
- 206010052779 Transplant rejections Diseases 0.000 description 2
- 102100031988 Tumor necrosis factor ligand superfamily member 6 Human genes 0.000 description 2
- 108050002568 Tumor necrosis factor ligand superfamily member 6 Proteins 0.000 description 2
- GBOGMAARMMDZGR-UHFFFAOYSA-N UNPD149280 Natural products N1C(=O)C23OC(=O)C=CC(O)CCCC(C)CC=CC3C(O)C(=C)C(C)C2C1CC1=CC=CC=C1 GBOGMAARMMDZGR-UHFFFAOYSA-N 0.000 description 2
- 208000027418 Wounds and injury Diseases 0.000 description 2
- 208000030961 allergic reaction Diseases 0.000 description 2
- 125000003277 amino group Chemical group 0.000 description 2
- 238000011398 antitumor immunotherapy Methods 0.000 description 2
- 230000001580 bacterial effect Effects 0.000 description 2
- 230000033228 biological regulation Effects 0.000 description 2
- 239000012472 biological sample Substances 0.000 description 2
- 210000004556 brain Anatomy 0.000 description 2
- 230000022131 cell cycle Effects 0.000 description 2
- 230000032823 cell division Effects 0.000 description 2
- 238000012412 chemical coupling Methods 0.000 description 2
- 238000007385 chemical modification Methods 0.000 description 2
- 238000002512 chemotherapy Methods 0.000 description 2
- 238000010367 cloning Methods 0.000 description 2
- 238000003501 co-culture Methods 0.000 description 2
- 230000000295 complement effect Effects 0.000 description 2
- 208000010247 contact dermatitis Diseases 0.000 description 2
- 238000010168 coupling process Methods 0.000 description 2
- 230000003229 cytophilic effect Effects 0.000 description 2
- 231100000433 cytotoxic Toxicity 0.000 description 2
- 230000003013 cytotoxicity Effects 0.000 description 2
- 231100000135 cytotoxicity Toxicity 0.000 description 2
- 210000004443 dendritic cell Anatomy 0.000 description 2
- 230000000779 depleting effect Effects 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 238000002405 diagnostic procedure Methods 0.000 description 2
- 235000014113 dietary fatty acids Nutrition 0.000 description 2
- QONQRTHLHBTMGP-UHFFFAOYSA-N digitoxigenin Natural products CC12CCC(C3(CCC(O)CC3CC3)C)C3C11OC1CC2C1=CC(=O)OC1 QONQRTHLHBTMGP-UHFFFAOYSA-N 0.000 description 2
- SHIBSTMRCDJXLN-KCZCNTNESA-N digoxigenin Chemical compound C1([C@@H]2[C@@]3([C@@](CC2)(O)[C@H]2[C@@H]([C@@]4(C)CC[C@H](O)C[C@H]4CC2)C[C@H]3O)C)=CC(=O)OC1 SHIBSTMRCDJXLN-KCZCNTNESA-N 0.000 description 2
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 2
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 2
- 210000003743 erythrocyte Anatomy 0.000 description 2
- 230000007717 exclusion Effects 0.000 description 2
- 239000000194 fatty acid Substances 0.000 description 2
- 229930195729 fatty acid Natural products 0.000 description 2
- 150000004665 fatty acids Chemical class 0.000 description 2
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 2
- 229930004094 glycosylphosphatidylinositol Natural products 0.000 description 2
- IPCSVZSSVZVIGE-UHFFFAOYSA-N hexadecanoic acid Chemical compound CCCCCCCCCCCCCCCC(O)=O IPCSVZSSVZVIGE-UHFFFAOYSA-N 0.000 description 2
- 238000003384 imaging method Methods 0.000 description 2
- 210000000987 immune system Anatomy 0.000 description 2
- 230000003053 immunization Effects 0.000 description 2
- 238000002649 immunization Methods 0.000 description 2
- 238000003018 immunoassay Methods 0.000 description 2
- 230000007813 immunodeficiency Effects 0.000 description 2
- 102000018358 immunoglobulin Human genes 0.000 description 2
- 239000003018 immunosuppressive agent Substances 0.000 description 2
- 229940125721 immunosuppressive agent Drugs 0.000 description 2
- 230000001939 inductive effect Effects 0.000 description 2
- 230000004054 inflammatory process Effects 0.000 description 2
- 108010061181 influenza matrix peptide (58-66) Proteins 0.000 description 2
- 238000003780 insertion Methods 0.000 description 2
- 230000037431 insertion Effects 0.000 description 2
- 230000008611 intercellular interaction Effects 0.000 description 2
- 229960001388 interferon-beta Drugs 0.000 description 2
- 230000002147 killing effect Effects 0.000 description 2
- 229920006008 lipopolysaccharide Polymers 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 210000004877 mucosa Anatomy 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 230000001254 nonsecretory effect Effects 0.000 description 2
- 238000004091 panning Methods 0.000 description 2
- 244000045947 parasite Species 0.000 description 2
- 210000005105 peripheral blood lymphocyte Anatomy 0.000 description 2
- 230000035699 permeability Effects 0.000 description 2
- 229910052698 phosphorus Inorganic materials 0.000 description 2
- 230000001885 phytohemagglutinin Effects 0.000 description 2
- 229920001223 polyethylene glycol Polymers 0.000 description 2
- 230000035755 proliferation Effects 0.000 description 2
- 238000003133 propidium iodide exclusion Methods 0.000 description 2
- 235000004252 protein component Nutrition 0.000 description 2
- 238000001959 radiotherapy Methods 0.000 description 2
- 230000022532 regulation of transcription, DNA-dependent Effects 0.000 description 2
- 230000000284 resting effect Effects 0.000 description 2
- 239000000523 sample Substances 0.000 description 2
- DAEPDZWVDSPTHF-UHFFFAOYSA-M sodium pyruvate Chemical compound [Na+].CC(=O)C([O-])=O DAEPDZWVDSPTHF-UHFFFAOYSA-M 0.000 description 2
- 239000000243 solution Substances 0.000 description 2
- 210000004989 spleen cell Anatomy 0.000 description 2
- KZNICNPSHKQLFF-UHFFFAOYSA-N succinimide Chemical class O=C1CCC(=O)N1 KZNICNPSHKQLFF-UHFFFAOYSA-N 0.000 description 2
- 231100000617 superantigen Toxicity 0.000 description 2
- 230000004083 survival effect Effects 0.000 description 2
- ZRKFYGHZFMAOKI-QMGMOQQFSA-N tgfbeta Chemical compound C([C@H](NC(=O)[C@H](C(C)C)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CC(C)C)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CCSC)C(C)C)[C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O)C1=CC=C(O)C=C1 ZRKFYGHZFMAOKI-QMGMOQQFSA-N 0.000 description 2
- 229940104230 thymidine Drugs 0.000 description 2
- 230000007704 transition Effects 0.000 description 2
- 238000013519 translation Methods 0.000 description 2
- 241000712461 unidentified influenza virus Species 0.000 description 2
- 229960005486 vaccine Drugs 0.000 description 2
- 239000013598 vector Substances 0.000 description 2
- 239000013603 viral vector Substances 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- 238000001262 western blot Methods 0.000 description 2
- DFUSDJMZWQVQSF-XLGIIRLISA-N (2r)-2-methyl-2-[(4r,8r)-4,8,12-trimethyltridecyl]-3,4-dihydrochromen-6-ol Chemical class OC1=CC=C2O[C@@](CCC[C@H](C)CCC[C@H](C)CCCC(C)C)(C)CCC2=C1 DFUSDJMZWQVQSF-XLGIIRLISA-N 0.000 description 1
- YQNRVGJCPCNMKT-LFVJCYFKSA-N 2-[(e)-[[2-(4-benzylpiperazin-1-ium-1-yl)acetyl]hydrazinylidene]methyl]-6-prop-2-enylphenolate Chemical compound [O-]C1=C(CC=C)C=CC=C1\C=N\NC(=O)C[NH+]1CCN(CC=2C=CC=CC=2)CC1 YQNRVGJCPCNMKT-LFVJCYFKSA-N 0.000 description 1
- 208000010543 22q11.2 deletion syndrome Diseases 0.000 description 1
- GPUBPSKTFNYPHI-AYGRAXKESA-N 3-[5-[(3as,4s,6ar)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoyl]-1-hydroxypyrrolidine-2,5-dione Chemical compound O=C1N(O)C(=O)CC1C(=O)CCCC[C@H]1[C@H]2NC(=O)N[C@H]2CS1 GPUBPSKTFNYPHI-AYGRAXKESA-N 0.000 description 1
- CIVGYTYIDWRBQU-UFLZEWODSA-N 5-[(3as,4s,6ar)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoic acid;pyrrole-2,5-dione Chemical compound O=C1NC(=O)C=C1.N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 CIVGYTYIDWRBQU-UFLZEWODSA-N 0.000 description 1
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 1
- HJCMDXDYPOUFDY-WHFBIAKZSA-N Ala-Gln Chemical compound C[C@H](N)C(=O)N[C@H](C(O)=O)CCC(N)=O HJCMDXDYPOUFDY-WHFBIAKZSA-N 0.000 description 1
- 241000221688 Aleuria aurantia Species 0.000 description 1
- 108700028369 Alleles Proteins 0.000 description 1
- 102000006306 Antigen Receptors Human genes 0.000 description 1
- 108010083359 Antigen Receptors Proteins 0.000 description 1
- XFTWUNOVBCHBJR-UHFFFAOYSA-N Aspergillomarasmine A Chemical compound OC(=O)C(N)CNC(C(O)=O)CNC(C(O)=O)CC(O)=O XFTWUNOVBCHBJR-UHFFFAOYSA-N 0.000 description 1
- 206010003645 Atopy Diseases 0.000 description 1
- 230000003844 B-cell-activation Effects 0.000 description 1
- 102100022005 B-lymphocyte antigen CD20 Human genes 0.000 description 1
- 208000035143 Bacterial infection Diseases 0.000 description 1
- 102100024167 C-C chemokine receptor type 3 Human genes 0.000 description 1
- 101710149862 C-C chemokine receptor type 3 Proteins 0.000 description 1
- 101710149863 C-C chemokine receptor type 4 Proteins 0.000 description 1
- 102100035875 C-C chemokine receptor type 5 Human genes 0.000 description 1
- 101710149870 C-C chemokine receptor type 5 Proteins 0.000 description 1
- 102100028990 C-X-C chemokine receptor type 3 Human genes 0.000 description 1
- 102100032976 CCR4-NOT transcription complex subunit 6 Human genes 0.000 description 1
- 101150013553 CD40 gene Proteins 0.000 description 1
- 102100032912 CD44 antigen Human genes 0.000 description 1
- 101100462537 Caenorhabditis elegans pac-1 gene Proteins 0.000 description 1
- BHPQYMZQTOCNFJ-UHFFFAOYSA-N Calcium cation Chemical compound [Ca+2] BHPQYMZQTOCNFJ-UHFFFAOYSA-N 0.000 description 1
- 102000009410 Chemokine receptor Human genes 0.000 description 1
- 108050000299 Chemokine receptor Proteins 0.000 description 1
- 102000019034 Chemokines Human genes 0.000 description 1
- 108010012236 Chemokines Proteins 0.000 description 1
- 108091026890 Coding region Proteins 0.000 description 1
- 102000008186 Collagen Human genes 0.000 description 1
- 108010035532 Collagen Proteins 0.000 description 1
- 206010010356 Congenital anomaly Diseases 0.000 description 1
- 239000004971 Cross linker Substances 0.000 description 1
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 1
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 description 1
- 229930105110 Cyclosporin A Natural products 0.000 description 1
- 108010036949 Cyclosporine Proteins 0.000 description 1
- ZMAODHOXRBLOQO-TZVKRXPSSA-N Cytochalasin A Chemical compound C([C@H]1[C@@H]2[C@@H](C([C@@H](O)[C@@H]3/C=C/C[C@H](C)CCCC(=O)/C=C/C(=O)O[C@@]23C(=O)N1)=C)C)C1=CC=CC=C1 ZMAODHOXRBLOQO-TZVKRXPSSA-N 0.000 description 1
- 241000450599 DNA viruses Species 0.000 description 1
- 206010011968 Decreased immune responsiveness Diseases 0.000 description 1
- 208000000398 DiGeorge Syndrome Diseases 0.000 description 1
- 206010061818 Disease progression Diseases 0.000 description 1
- 241000255581 Drosophila <fruit fly, genus> Species 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 108010087819 Fc receptors Proteins 0.000 description 1
- 102000009109 Fc receptors Human genes 0.000 description 1
- 229920001917 Ficoll Polymers 0.000 description 1
- 238000012413 Fluorescence activated cell sorting analysis Methods 0.000 description 1
- 230000035519 G0 Phase Effects 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- SXRSQZLOMIGNAQ-UHFFFAOYSA-N Glutaraldehyde Chemical compound O=CCCCC=O SXRSQZLOMIGNAQ-UHFFFAOYSA-N 0.000 description 1
- 229930186217 Glycolipid Natural products 0.000 description 1
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 1
- 102100039620 Granulocyte-macrophage colony-stimulating factor Human genes 0.000 description 1
- 108010043121 Green Fluorescent Proteins Proteins 0.000 description 1
- 108010075704 HLA-A Antigens Proteins 0.000 description 1
- 102000011786 HLA-A Antigens Human genes 0.000 description 1
- 108010074032 HLA-A2 Antigen Proteins 0.000 description 1
- 102000025850 HLA-A2 Antigen Human genes 0.000 description 1
- 241000237369 Helix pomatia Species 0.000 description 1
- 208000006968 Helminthiasis Diseases 0.000 description 1
- 241000238631 Hexapoda Species 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000897405 Homo sapiens B-lymphocyte antigen CD20 Proteins 0.000 description 1
- 101000916050 Homo sapiens C-X-C chemokine receptor type 3 Proteins 0.000 description 1
- 101000868273 Homo sapiens CD44 antigen Proteins 0.000 description 1
- 101000599852 Homo sapiens Intercellular adhesion molecule 1 Proteins 0.000 description 1
- 101001002657 Homo sapiens Interleukin-2 Proteins 0.000 description 1
- 101001018097 Homo sapiens L-selectin Proteins 0.000 description 1
- 101000578784 Homo sapiens Melanoma antigen recognized by T-cells 1 Proteins 0.000 description 1
- 101000946889 Homo sapiens Monocyte differentiation antigen CD14 Proteins 0.000 description 1
- 101001094545 Homo sapiens Retrotransposon-like protein 1 Proteins 0.000 description 1
- 108090000144 Human Proteins Proteins 0.000 description 1
- 102000003839 Human Proteins Human genes 0.000 description 1
- 101150083678 IL2 gene Proteins 0.000 description 1
- 208000001718 Immediate Hypersensitivity Diseases 0.000 description 1
- 102000017727 Immunoglobulin Variable Region Human genes 0.000 description 1
- 108010067060 Immunoglobulin Variable Region Proteins 0.000 description 1
- 102000003746 Insulin Receptor Human genes 0.000 description 1
- 108010001127 Insulin Receptor Proteins 0.000 description 1
- 108010022222 Integrin beta1 Proteins 0.000 description 1
- 102000012355 Integrin beta1 Human genes 0.000 description 1
- 108010050904 Interferons Proteins 0.000 description 1
- 102000014150 Interferons Human genes 0.000 description 1
- 102000013462 Interleukin-12 Human genes 0.000 description 1
- 108010065805 Interleukin-12 Proteins 0.000 description 1
- 101710190483 Interleukin-2 receptor subunit alpha Proteins 0.000 description 1
- 108010002386 Interleukin-3 Proteins 0.000 description 1
- 102000015696 Interleukins Human genes 0.000 description 1
- 108010063738 Interleukins Proteins 0.000 description 1
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 1
- 102100033467 L-selectin Human genes 0.000 description 1
- 240000004322 Lens culinaris Species 0.000 description 1
- 235000010666 Lens esculenta Nutrition 0.000 description 1
- 241000186779 Listeria monocytogenes Species 0.000 description 1
- 102100035304 Lymphotactin Human genes 0.000 description 1
- PEEHTFAAVSWFBL-UHFFFAOYSA-N Maleimide Chemical compound O=C1NC(=O)C=C1 PEEHTFAAVSWFBL-UHFFFAOYSA-N 0.000 description 1
- 102100028389 Melanoma antigen recognized by T-cells 1 Human genes 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- 102100035877 Monocyte differentiation antigen CD14 Human genes 0.000 description 1
- 101100117764 Mus musculus Dusp2 gene Proteins 0.000 description 1
- 101710135898 Myc proto-oncogene protein Proteins 0.000 description 1
- 102100038895 Myc proto-oncogene protein Human genes 0.000 description 1
- SQVRNKJHWKZAKO-PFQGKNLYSA-N N-acetyl-beta-neuraminic acid Chemical compound CC(=O)N[C@@H]1[C@@H](O)C[C@@](O)(C(O)=O)O[C@H]1[C@H](O)[C@H](O)CO SQVRNKJHWKZAKO-PFQGKNLYSA-N 0.000 description 1
- 108010057466 NF-kappa B Proteins 0.000 description 1
- 102000003945 NF-kappa B Human genes 0.000 description 1
- 235000021314 Palmitic acid Nutrition 0.000 description 1
- 208000030852 Parasitic disease Diseases 0.000 description 1
- 102000045595 Phosphoprotein Phosphatases Human genes 0.000 description 1
- 108700019535 Phosphoprotein Phosphatases Proteins 0.000 description 1
- 241000276498 Pollachius virens Species 0.000 description 1
- 108010039918 Polylysine Proteins 0.000 description 1
- 239000004372 Polyvinyl alcohol Substances 0.000 description 1
- 102000001253 Protein Kinase Human genes 0.000 description 1
- 102000007568 Proto-Oncogene Proteins c-fos Human genes 0.000 description 1
- 108010071563 Proto-Oncogene Proteins c-fos Proteins 0.000 description 1
- 201000004681 Psoriasis Diseases 0.000 description 1
- 230000018199 S phase Effects 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- 235000002634 Solanum Nutrition 0.000 description 1
- 241000207763 Solanum Species 0.000 description 1
- 241000256248 Spodoptera Species 0.000 description 1
- 230000006044 T cell activation Effects 0.000 description 1
- 210000000662 T-lymphocyte subset Anatomy 0.000 description 1
- QJJXYPPXXYFBGM-LFZNUXCKSA-N Tacrolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1\C=C(/C)[C@@H]1[C@H](C)[C@@H](O)CC(=O)[C@H](CC=C)/C=C(C)/C[C@H](C)C[C@H](OC)[C@H]([C@H](C[C@H]2C)OC)O[C@@]2(O)C(=O)C(=O)N2CCCC[C@H]2C(=O)O1 QJJXYPPXXYFBGM-LFZNUXCKSA-N 0.000 description 1
- 108010055044 Tetanus Toxin Proteins 0.000 description 1
- 210000004241 Th2 cell Anatomy 0.000 description 1
- GYDJEQRTZSCIOI-UHFFFAOYSA-N Tranexamic acid Chemical compound NCC1CCC(C(O)=O)CC1 GYDJEQRTZSCIOI-UHFFFAOYSA-N 0.000 description 1
- 101710150448 Transcriptional regulator Myc Proteins 0.000 description 1
- 102000007238 Transferrin Receptors Human genes 0.000 description 1
- 108010033576 Transferrin Receptors Proteins 0.000 description 1
- 102100022153 Tumor necrosis factor receptor superfamily member 4 Human genes 0.000 description 1
- 101710165473 Tumor necrosis factor receptor superfamily member 4 Proteins 0.000 description 1
- 102100040245 Tumor necrosis factor receptor superfamily member 5 Human genes 0.000 description 1
- 206010047115 Vasculitis Diseases 0.000 description 1
- 241000251539 Vertebrata <Metazoa> Species 0.000 description 1
- 206010052428 Wound Diseases 0.000 description 1
- HMNZFMSWFCAGGW-XPWSMXQVSA-N [3-[hydroxy(2-hydroxyethoxy)phosphoryl]oxy-2-[(e)-octadec-9-enoyl]oxypropyl] (e)-octadec-9-enoate Chemical compound CCCCCCCC\C=C\CCCCCCCC(=O)OCC(COP(O)(=O)OCCO)OC(=O)CCCCCCC\C=C\CCCCCCCC HMNZFMSWFCAGGW-XPWSMXQVSA-N 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- 239000011543 agarose gel Substances 0.000 description 1
- 229940072056 alginate Drugs 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 230000037446 allergic sensitization Effects 0.000 description 1
- 230000000961 alloantigen Effects 0.000 description 1
- 230000003321 amplification Effects 0.000 description 1
- 238000004873 anchoring Methods 0.000 description 1
- 210000004102 animal cell Anatomy 0.000 description 1
- 230000000259 anti-tumor effect Effects 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 238000002617 apheresis Methods 0.000 description 1
- 230000002238 attenuated effect Effects 0.000 description 1
- 230000006472 autoimmune response Effects 0.000 description 1
- 230000005784 autoimmunity Effects 0.000 description 1
- 229960002170 azathioprine Drugs 0.000 description 1
- LMEKQMALGUDUQG-UHFFFAOYSA-N azathioprine Chemical compound CN1C=NC([N+]([O-])=O)=C1SC1=NC=NC2=C1NC=N2 LMEKQMALGUDUQG-UHFFFAOYSA-N 0.000 description 1
- 208000022362 bacterial infectious disease Diseases 0.000 description 1
- 210000003651 basophil Anatomy 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- MSWZFWKMSRAUBD-QZABAPFNSA-N beta-D-glucosamine Chemical group N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O MSWZFWKMSRAUBD-QZABAPFNSA-N 0.000 description 1
- SQVRNKJHWKZAKO-UHFFFAOYSA-N beta-N-Acetyl-D-neuraminic acid Natural products CC(=O)NC1C(O)CC(O)(C(O)=O)OC1C(O)C(O)CO SQVRNKJHWKZAKO-UHFFFAOYSA-N 0.000 description 1
- 230000001588 bifunctional effect Effects 0.000 description 1
- 230000008827 biological function Effects 0.000 description 1
- 238000001574 biopsy Methods 0.000 description 1
- 230000015572 biosynthetic process Effects 0.000 description 1
- 210000000601 blood cell Anatomy 0.000 description 1
- 238000010241 blood sampling Methods 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- 229910001424 calcium ion Inorganic materials 0.000 description 1
- 230000009702 cancer cell proliferation Effects 0.000 description 1
- 230000021164 cell adhesion Effects 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 230000003915 cell function Effects 0.000 description 1
- 239000002771 cell marker Substances 0.000 description 1
- 238000003320 cell separation method Methods 0.000 description 1
- 210000002421 cell wall Anatomy 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 229960001265 ciclosporin Drugs 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 229920001436 collagen Polymers 0.000 description 1
- 150000001875 compounds Chemical class 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 210000002808 connective tissue Anatomy 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 239000013068 control sample Substances 0.000 description 1
- 238000011443 conventional therapy Methods 0.000 description 1
- 239000003246 corticosteroid Substances 0.000 description 1
- 229960001334 corticosteroids Drugs 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 238000012864 cross contamination Methods 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 239000012228 culture supernatant Substances 0.000 description 1
- 229960004397 cyclophosphamide Drugs 0.000 description 1
- 229930182912 cyclosporin Natural products 0.000 description 1
- GBOGMAARMMDZGR-JREHFAHYSA-N cytochalasin B Natural products C[C@H]1CCC[C@@H](O)C=CC(=O)O[C@@]23[C@H](C=CC1)[C@H](O)C(=C)[C@@H](C)[C@@H]2[C@H](Cc4ccccc4)NC3=O GBOGMAARMMDZGR-JREHFAHYSA-N 0.000 description 1
- GBOGMAARMMDZGR-TYHYBEHESA-N cytochalasin B Chemical compound C([C@H]1[C@@H]2[C@@H](C([C@@H](O)[C@@H]3/C=C/C[C@H](C)CCC[C@@H](O)/C=C/C(=O)O[C@@]23C(=O)N1)=C)C)C1=CC=CC=C1 GBOGMAARMMDZGR-TYHYBEHESA-N 0.000 description 1
- ZMAODHOXRBLOQO-UHFFFAOYSA-N cytochalasin-A Natural products N1C(=O)C23OC(=O)C=CC(=O)CCCC(C)CC=CC3C(O)C(=C)C(C)C2C1CC1=CC=CC=C1 ZMAODHOXRBLOQO-UHFFFAOYSA-N 0.000 description 1
- 230000002380 cytological effect Effects 0.000 description 1
- 229940127089 cytotoxic agent Drugs 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 238000000432 density-gradient centrifugation Methods 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 230000005750 disease progression Effects 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 241001493065 dsRNA viruses Species 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 238000004520 electroporation Methods 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- 238000005538 encapsulation Methods 0.000 description 1
- 239000012645 endogenous antigen Substances 0.000 description 1
- 230000006862 enzymatic digestion Effects 0.000 description 1
- 210000003979 eosinophil Anatomy 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 239000013604 expression vector Substances 0.000 description 1
- 239000011554 ferrofluid Substances 0.000 description 1
- 239000012894 fetal calf serum Substances 0.000 description 1
- 238000001943 fluorescence-activated cell sorting Methods 0.000 description 1
- 230000005714 functional activity Effects 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 108020001507 fusion proteins Proteins 0.000 description 1
- 102000037865 fusion proteins Human genes 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 238000012239 gene modification Methods 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 229930182478 glucoside Natural products 0.000 description 1
- 230000013595 glycosylation Effects 0.000 description 1
- 238000006206 glycosylation reaction Methods 0.000 description 1
- 230000005484 gravity Effects 0.000 description 1
- 239000003102 growth factor Substances 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- 230000003394 haemopoietic effect Effects 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 210000003958 hematopoietic stem cell Anatomy 0.000 description 1
- 210000003630 histaminocyte Anatomy 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 230000008348 humoral response Effects 0.000 description 1
- 210000004408 hybridoma Anatomy 0.000 description 1
- 125000001165 hydrophobic group Chemical group 0.000 description 1
- 230000009610 hypersensitivity Effects 0.000 description 1
- 230000002519 immonomodulatory effect Effects 0.000 description 1
- 230000003832 immune regulation Effects 0.000 description 1
- 238000010166 immunofluorescence Methods 0.000 description 1
- 230000005847 immunogenicity Effects 0.000 description 1
- 230000000415 inactivating effect Effects 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 208000000509 infertility Diseases 0.000 description 1
- 230000036512 infertility Effects 0.000 description 1
- 231100000535 infertility Toxicity 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 230000028709 inflammatory response Effects 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 208000014674 injury Diseases 0.000 description 1
- 229940079322 interferon Drugs 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- PGHMRUGBZOYCAA-UHFFFAOYSA-N ionomycin Natural products O1C(CC(O)C(C)C(O)C(C)C=CCC(C)CC(C)C(O)=CC(=O)C(C)CC(C)CC(CCC(O)=O)C)CCC1(C)C1OC(C)(C(C)O)CC1 PGHMRUGBZOYCAA-UHFFFAOYSA-N 0.000 description 1
- PGHMRUGBZOYCAA-ADZNBVRBSA-N ionomycin Chemical compound O1[C@H](C[C@H](O)[C@H](C)[C@H](O)[C@H](C)/C=C/C[C@@H](C)C[C@@H](C)C(/O)=C/C(=O)[C@@H](C)C[C@@H](C)C[C@@H](CCC(O)=O)C)CC[C@@]1(C)[C@@H]1O[C@](C)([C@@H](C)O)CC1 PGHMRUGBZOYCAA-ADZNBVRBSA-N 0.000 description 1
- 210000000265 leukocyte Anatomy 0.000 description 1
- 230000021633 leukocyte mediated immunity Effects 0.000 description 1
- 150000002634 lipophilic molecules Chemical class 0.000 description 1
- 108010019677 lymphotactin Proteins 0.000 description 1
- 230000002101 lytic effect Effects 0.000 description 1
- 238000007898 magnetic cell sorting Methods 0.000 description 1
- 239000000696 magnetic material Substances 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 210000004962 mammalian cell Anatomy 0.000 description 1
- 238000013507 mapping Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 210000003071 memory t lymphocyte Anatomy 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- 230000009401 metastasis Effects 0.000 description 1
- 229960000485 methotrexate Drugs 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 239000003094 microcapsule Substances 0.000 description 1
- 230000003278 mimic effect Effects 0.000 description 1
- 229960004857 mitomycin Drugs 0.000 description 1
- 230000011278 mitosis Effects 0.000 description 1
- 230000000394 mitotic effect Effects 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 108091005601 modified peptides Proteins 0.000 description 1
- 238000010369 molecular cloning Methods 0.000 description 1
- 239000003068 molecular probe Substances 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 201000006417 multiple sclerosis Diseases 0.000 description 1
- 231100000219 mutagenic Toxicity 0.000 description 1
- 230000003505 mutagenic effect Effects 0.000 description 1
- WQEPLUUGTLDZJY-UHFFFAOYSA-N n-Pentadecanoic acid Natural products CCCCCCCCCCCCCCC(O)=O WQEPLUUGTLDZJY-UHFFFAOYSA-N 0.000 description 1
- 239000013642 negative control Substances 0.000 description 1
- 210000000440 neutrophil Anatomy 0.000 description 1
- 239000002736 nonionic surfactant Substances 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 238000003199 nucleic acid amplification method Methods 0.000 description 1
- 239000002773 nucleotide Substances 0.000 description 1
- 125000003729 nucleotide group Chemical group 0.000 description 1
- 238000002515 oligonucleotide synthesis Methods 0.000 description 1
- 238000005580 one pot reaction Methods 0.000 description 1
- 230000036281 parasite infection Effects 0.000 description 1
- 208000014837 parasitic helminthiasis infectious disease Diseases 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 239000004031 partial agonist Substances 0.000 description 1
- 230000001717 pathogenic effect Effects 0.000 description 1
- 230000007310 pathophysiology Effects 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 210000005259 peripheral blood Anatomy 0.000 description 1
- 239000011886 peripheral blood Substances 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 150000004633 phorbol derivatives Chemical class 0.000 description 1
- 239000002644 phorbol ester Substances 0.000 description 1
- 210000002381 plasma Anatomy 0.000 description 1
- 239000013612 plasmid Substances 0.000 description 1
- 210000004180 plasmocyte Anatomy 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 229920002704 polyhistidine Polymers 0.000 description 1
- 229920000656 polylysine Polymers 0.000 description 1
- 238000003752 polymerase chain reaction Methods 0.000 description 1
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 1
- 229920001451 polypropylene glycol Polymers 0.000 description 1
- 229920000053 polysorbate 80 Polymers 0.000 description 1
- 229920002451 polyvinyl alcohol Polymers 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 239000011148 porous material Substances 0.000 description 1
- 230000008092 positive effect Effects 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 230000002028 premature Effects 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 108060006633 protein kinase Proteins 0.000 description 1
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 230000007115 recruitment Effects 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000001177 retroviral effect Effects 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 206010039073 rheumatoid arthritis Diseases 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 229960002930 sirolimus Drugs 0.000 description 1
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 1
- 208000017520 skin disease Diseases 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 229940054269 sodium pyruvate Drugs 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 238000007447 staining method Methods 0.000 description 1
- 210000000130 stem cell Anatomy 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 239000000021 stimulant Substances 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 238000012385 systemic delivery Methods 0.000 description 1
- QJJXYPPXXYFBGM-SHYZHZOCSA-N tacrolimus Natural products CO[C@H]1C[C@H](CC[C@@H]1O)C=C(C)[C@H]2OC(=O)[C@H]3CCCCN3C(=O)C(=O)[C@@]4(O)O[C@@H]([C@H](C[C@H]4C)OC)[C@@H](C[C@H](C)CC(=C[C@@H](CC=C)C(=O)C[C@H](O)[C@H]2C)C)OC QJJXYPPXXYFBGM-SHYZHZOCSA-N 0.000 description 1
- 229940118376 tetanus toxin Drugs 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 150000003573 thiols Chemical class 0.000 description 1
- 210000001685 thyroid gland Anatomy 0.000 description 1
- 230000000451 tissue damage Effects 0.000 description 1
- 231100000827 tissue damage Toxicity 0.000 description 1
- 230000024664 tolerance induction Effects 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 238000010361 transduction Methods 0.000 description 1
- 230000026683 transduction Effects 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 238000002054 transplantation Methods 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- 244000052613 viral pathogen Species 0.000 description 1
Images

















































Classifications
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/569—Immunoassay; Biospecific binding assay; Materials therefor for microorganisms, e.g. protozoa, bacteria, viruses
- G01N33/56966—Animal cells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/461—Cellular immunotherapy characterised by the cell type used
- A61K39/4611—T-cells, e.g. tumor infiltrating lymphocytes [TIL], lymphokine-activated killer cells [LAK] or regulatory T cells [Treg]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/46—Cellular immunotherapy
- A61K39/464—Cellular immunotherapy characterised by the antigen targeted or presented
- A61K39/464838—Viral antigens
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/06—Animal cells or tissues; Human cells or tissues
- C12N5/0602—Vertebrate cells
- C12N5/0634—Cells from the blood or the immune system
- C12N5/0636—T lymphocytes
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/5005—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/5005—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells
- G01N33/5008—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics
- G01N33/5044—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving human or animal cells for testing or evaluating the effect of chemical or biological compounds, e.g. drugs, cosmetics involving specific cell types
- G01N33/5047—Cells of the immune system
- G01N33/505—Cells of the immune system involving T-cells
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Immunology (AREA)
- Engineering & Computer Science (AREA)
- Biomedical Technology (AREA)
- Chemical & Material Sciences (AREA)
- Cell Biology (AREA)
- Hematology (AREA)
- Urology & Nephrology (AREA)
- Molecular Biology (AREA)
- Microbiology (AREA)
- General Health & Medical Sciences (AREA)
- Biotechnology (AREA)
- Medicinal Chemistry (AREA)
- Biochemistry (AREA)
- Pathology (AREA)
- Zoology (AREA)
- General Physics & Mathematics (AREA)
- Analytical Chemistry (AREA)
- Physics & Mathematics (AREA)
- Tropical Medicine & Parasitology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Food Science & Technology (AREA)
- Genetics & Genomics (AREA)
- Wood Science & Technology (AREA)
- Organic Chemistry (AREA)
- Virology (AREA)
- Pharmacology & Pharmacy (AREA)
- General Engineering & Computer Science (AREA)
- Mycology (AREA)
- Toxicology (AREA)
- Epidemiology (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
Description
(技術的分野)
本発明は、細胞集団の分析および細胞分離ならびにそれよって得られた組成物の分野におけるものである。より具体的には、本発明は、細胞表面に固定または結合した、産物に特異的な結合パートナーによってこれらの産物を捕獲することによって、細胞をその細胞が分泌した産物で一次標識することに基づく、抗原特異的T細胞の分析および分離に関する。
【0002】
(背景技術)
細胞が産生する産物に基づいて細胞の集団を分析し、そして細胞を分離する多くの試みがなされてきた。細胞分析および分離に対するそのようなアプローチは、所望の産物(「産物」)を分泌し得る細胞、または産物を比較的多く分泌する細胞を評価するのに特に有用である。これらの方法は、マイクロタイタープレートにおけるクローニングそして培養上清の産物についての分析、寒天におけるクローニングそして局在性の細胞の産物の同定方法による分析を含む。同定方法は、例えば、プラークアッセイおよびウェスタンブロッティングを含む。産物の分泌に基づく細胞の分析方法および分離方法のほとんどは、細胞の物理的単離、続いて産物の分泌が可能な条件下でのインキュベーション、および細胞の位置をスクリーニングして産物を産生する細胞または細胞クローンを検出することを含む。細胞が懸濁液中にある場合、細胞が産物を分泌した後、その産物は、その産物が分泌された細胞を同定し得るマーカーを残すことなく細胞から拡散する。従って、この型の系では、分泌細胞は非分泌細胞から分離され得ない。
【0003】
別の場合には、分泌細胞および非分泌細胞の両方が産物を細胞膜に結合し得る。この型の系の例は、モノクローナル抗体を産生するB細胞由来の細胞株である。これらの型の細胞株は、蛍光活性化セルソーター(FACS)および抗体細胞表面マーカーの存在に依存する他の方法によって分離されたことが報告された。しかし、細胞表面に自然に結合するマーカーによって細胞を分析および分離する手順は、正確に同定し得ず、および/または非分泌細胞から分泌細胞を分離するのに使用し得ない。加えて、これらのような系は分泌細胞の量的な相違(すなわち、低レベルの分泌細胞と高レベルの分泌細胞)を同定するのに有用でない。
【0004】
産物の細胞からの拡散に伴う問題を克服するために使用された方法は、細胞からの拡散速度を阻害する培地に細胞を配置することであった。代表的な方法は、細胞をゲル様培地(寒天)に固定化し、そして次いでブロッティング(例えば、ウェスタンブロット)による系を用いて、寒天プレートを産物の産生についてスクリーニングすることであった。これらの系は、多くの細胞を分泌、非分泌の性質、または分泌量について分析する場合には、煩雑かつ高価である。
【0005】
Kohlerらは、ネガティブ選択系を記載した。ここでは、抗トリニトロフェニル(抗TNP)特異性を有するIgMを分泌するハイブリドーマ株の変異体が、ハプテン(すなわちTNP)を細胞表面にカップリングさせ、そしてその細胞を補体の存在下でインキュベートすることによって濃縮された。この方法では、野生型Igを分泌する細胞が溶解されたのに対し、減少された溶解活性を有するIgMを分泌する細胞またはTNPに結合しないIgMを分泌する細胞は、優先的に生存した。KohlerおよびSchulman(1980)Eur.J.Immunol.10:467−476.
より最近には、分泌産物に基づく細胞の標識および分離のための系が記載された。PCT/US93/10126。この系では、分泌産物に対する特異的結合パートナーを細胞の表面に結合させる。産物は分泌され、放出され、そして特異的結合パートナーによって細胞に結合する。次いで、細胞を結合産物で標識された細胞の程度に基づいて分離する。
【0006】
他の系は、細胞がその産物をアガロースゲルの微小滴のコンテクスト(context)に分泌することを可能にする。これは分泌産物に結合する試薬を含み、そしてその細胞をカプセル化する。そのような方法は、Nirら(1990)Applied and Environ.Microbiol.56:2870−2875;およびNirら(1991)Applied and Environ.Microbiol.56:3861−3866による出版物で開示された。これらの方法は、種々の理由で不満足なものである。マイクロカプセル化(microcapsulation)の過程で、カプセル中の細胞数の統計的な封入(trapping)が起こり、低細胞濃度でカプセル化が起こった場合多くの空のカプセルが生じ、または高細胞濃度でカプセル化が起こった場合1カプセルあたり複数の細胞を含むカプセルが生じる。分泌産物は捕獲抗体によってアガロース滴に捕らえられ、そして第二の蛍光色素化抗体によって検出される。このプロセスは、分泌産物に基づく細胞の検出および単離を可能にするが、複雑であり、特別な装置を必要とし、そして全ての型のソーティング方法に適していない。
【0007】
この技術によって単一の細胞または単一の細胞塊を分析および分離するためには、空のカプセルの数およびマイクロカプセルの大きさ(50〜100μm)のために、比較的少数の細胞で作業するのに大量の容積を扱わなければならない。大量の容積の小滴は、フローサイトメトリー分析および分離を用いた場合バックグラウンドの問題を引き起こす。加えて、カプセルは磁気ビーズを用いた分離または細胞分離のためのパニングを許容しない。
【0008】
標識を細胞表面に結合させるのに種々の方法が使用されてきた。ここでは、蛍光色素のような標識は直接的な検出を意図する。例えば、蛍光標識を細胞に結合させるために細胞膜に挿入された疎水性リンカーが、1990年3月8日に発行されたPCT WO90/02334に記載された。HLAに対する抗体もまた、標識を細胞表面に結合させるために使用された。そのような結合は、上記で記載したカプセル化小滴より少量の容積になり、そしてそのような細胞は、フローサイトメトリーおよび磁気分離を含む標準的な分離手順で簡便に使用され得る。
【0009】
ELISpotアッセイおよび細胞内サイトカイン染色方法が、抗原特異的CD4+およびCD8+T細胞の計数および特徴づけに使用された。Lalvaniら(1997)J.Exp.Med.186:859−865;およびWaldropら(1997)J.Clin.Invest.99:1739−1750。これらの方法はT細胞エピトープマッピングまたはワクチン試行で免疫原性をモニターするのに非常に有用であり得るが、例えば、ガンまたは感染に対する特異的養子免疫療法の臨床試行のために、生の抗原特異的T細胞を単離することができない。Kernら(1998)Nat.Med.4:975−978;El Ghazaliら(1993)Curr.Opin.Immunol.23:2740−2745;およびYeeら(1997)Curr.Opin.Immunol.9:702−708。
【0010】
ペプチドを含んだ主要組織適合遺伝子複合体(MHC)分子の可溶性多価複合体が、抗原特異的T細胞を検出し、そして単離もするために最近開発された。Altmanら(1996)Science 274:94−96;Dunbarら(1998)Curr.Biol.8:413−416;Oggら(1998)279:2103−2106;Luxembourgら(1998)Nat.Biotechnol.16:281−285;Murali−Krishnaら(1998)Immunity 8:177−187;Gallimoreら(1998)J.Exp.Med.187:1383−1393;およびFlynnら(1998)Immunity 8:683−691。これらの試薬は高度に特異的であるが、そのアプローチは良く定義された抗原性ペプチドおよび制限HLA対立遺伝子の組み合せに限られる。
【0011】
免疫系は2つの型の抗原特異的細胞を含む(B細胞およびT細胞)。T細胞は、それらが抗原を認識する方法によって、それらの細胞表面マーカーによって、およびそれらの分泌産物によって、表現型的に特徴付けられ得る。可溶性抗原を認識するB細胞とは異なり、T細胞は、抗原が他の細胞の表面に主要組織適合遺伝子複合体(MHC)分子と結合した小フラグメントの形態でT細胞に提示された場合のみ抗原を認識する。表面に抗原フラグメントと結合したMHC分子を発現するあらゆる細胞は、抗原提示細胞(APC)と考えられ得る。しかし、ほとんどの場合、細胞表面のMHC結合抗原フラグメントの単なる提示以上のものが、Tリンパ球を活性化するのに必要である。抗原を含むMHC分子によって作動するT細胞レセプター(TCR)によって伝達されるシグナルに加えて、T細胞はまたAPCからの補助刺激シグナルも受け取らなくてはならない。代表的にはAPCは樹状細胞、マクロファージまたは活性化Bリンパ球である。
【0012】
T細胞は特有の膜分子を発現する。これらには、CD3と結合して細胞表面に現れるT細胞抗原レセプター(TCR);ならびにCD5、CD28、およびCD45Rのようなアクセサリー分子が含まれる。T細胞の部分集団はさらなる膜分子の存在によって区別され得る。従って、例えば、CD4を発現するT細胞はクラスII MHC分子と結合した抗原を認識し、そして一般的にはヘルパー細胞として機能する。一方、CD8を発現するT細胞は、クラスI MHC分子と結合した抗原を認識し、そして一般的には細胞傷害性細胞として機能する。T細胞のCD4+部分集団は、細胞によって分泌されるサイトカインの型に基づいて、さらに少なくとも2つのサブセットに分類され得る。従って、両方のサブセットはIL−3およびGM−CSFを分泌するが、TH1細胞は一般的にIL−2、IFN−γおよびTNF−αを分泌し、一方TH2細胞は一般的にIL−4、IL−5、IL−10およびIL−13を分泌する。
【0013】
MHC分子に結合するペプチドの小さな変更は、ペプチド−MHC分子相互作用の親和性に影響を与え得ない。しかし、それらは増殖および完全なT細胞応答の間に産生されるサイトカインの一部分のみの分泌によって特徴付けられる、中間の応答を起こす部分的なシグナルを生成し得る。いくつかの修飾ペプチドは増殖およびサイトカインの分泌を全く阻害し、そしてT細胞アネルギーまたは無応答性の状態を誘導さえし得る。従って、3つの異なる型のペプチドが存在する:アゴニスト(完全な応答を刺激する)、部分的アゴニスト(部分的な応答を刺激する)、およびアンタゴニスト(無応答性を誘導する)である。1つのAPCがその表面にアゴニストおよびアンタゴニストの混合物を提示する場合、アンタゴニストがアゴニストより非常に少量しか存在しなくても、後者の負の効果は前者の正の効果を圧倒し得る。いくつかのウイルスは、そのタンパク質の変異を使用して、本来の野生型ウイルス由来のアゴニストペプチドを認識するT細胞クローンの活性を抑制し得るアンタゴニストペプチドを産生するようである。
【0014】
T細胞による特定のサイトカインの分泌は、一般的に特定の機能と関連する。例えば、CD4+T細胞のTH1およびTH2サブセットによって分泌されるサイトカインの相違は、これら2つのサブセットの異なる生物学的機能を反映すると考えられる。TH1サブセットは遅延型過敏症および細胞傷害性T細胞の活性化のような、古典的な細胞媒介機能を担い、一方TH2サブセットはB細胞活性化のヘルパーとしてより有効に機能する。TH1サブセットは、細胞傷害性T細胞を活性化するIL−2およびIFN−γを分泌するので、特にウイルス感染および細胞内病原体に反応するのに適当であり得る。IL−4およびIL−5はそれぞれIgEの産生および好酸球の活性化を誘導することが公知であるので、TH2サブセットは細胞外の細菌および寄生虫性寄生生物(helminthic parasite)に反応するのにより適当であり得、およびアレルギー反応を媒介し得る。TH1細胞の選択的な活性化が多くの自己免疫疾患の病因において中心的な役割を果たしていることを示唆する多くの証拠も存在する。TH2細胞によるIL−10の分泌は、間接的な様式でTH1細胞によるサイトカインの産生を抑制すると考えられ、そして従って、一般的な免疫抑制効果を有する。TH1/TH2バランスのシフトが、例えば、アレルギー反応、または細胞傷害性T細胞応答の増加を生じ得る。
【0015】
TCRによって始まる、活性化の最初の数分から数時間での変化は、細胞を細胞周期のG0期からG1期へと移行させる。刺激の数時間後、T細胞はIL−2および高親和性IL−2レセプターを発現し始める。IL2遺伝子の発現は、TCR、CD28、およびおそらく他のT細胞表面分子の連結によって引き起こされる、収束するシグナル伝達経路により活性化される1組の転写因子により影響される。
【0016】
転写因子はまた、高親和性IL−2レセプターのαサブユニットをコードするCD25遺伝子の発現を誘導する。IL−2と高親和性レセプターとの相互作用は、T細胞を細胞周期のG1期からS期へ移行させ、そして細胞分裂へと進行させるシグナル伝達経路を開始する。シグナル伝達経路は、細胞分裂に必要ないくつかの重要なタンパク質の発現および活性を調節する。これらのいくつかはまた、TCR−およびCD28−依存性シグナルによって直接的に活性化され、一方、他はIL−2レセプターを介して提供されるシグナルによってのみ活性化される。
【0017】
刺激されたT細胞は、休止期から有糸分裂、そして後にエフェクター細胞および記憶細胞への分化への進行に始まる、一連の表現型の変化を受ける。最も早い(即時)の変化の中で、刺激から15〜30分以内に観察可能であるのは、c−Fos、NF−AT、c−Myc、およびNF−κBのような転写因子、Jak−3のようなタンパク質キナーゼ、およびPac−1のようなタンパク質ホスファターゼをコードする遺伝子の発現である。それに続く初期の変化は、刺激から数時間以内に起こり、活性化抗原をコードする遺伝子の発現の開始を特徴付ける。これらとしては、いくつかのサイトカイン(IL−2および他)、IL−2レセプターサブユニットα(CD25)、インスリンレセプター、トランスフェリンレセプター、およびCD26、CD30、CD54、CD69およびCD70のような他のいくつかの表面分子が挙げられる。
【0018】
活性化抗原は最初の分裂の直前、刺激から24時間後に最大の発現レベルに達する。この期間、休止期のT細胞にすでに発現しているいくつかの他の分子の発現レベルが上昇する。後に、活性化開始の数日後、後期活性化抗原がT細胞上に発現されるようになる。これらはMHCクラスII分子およびβ1インテグリンファミリーのいくつかのメンバーを含む。後期活性化抗原の発現は、活性化細胞のエフェクター細胞または記憶T細胞への分化を特徴付ける。
【0019】
T細胞は、自己免疫、炎症、細胞傷害性、移植片拒絶、アレルギー、遅延型過敏症、IgE媒介性過敏症、および体液性応答の調節に重要な役割を果たす。自己反応性T細胞の活性化、アレルギー反応を起こすT細胞の活性化、またはヒトタンパク質を模倣する抗原(これらのタンパク質は「自己抗原」になる)を産生し得る特定の細菌および寄生生物の感染に続く、自己反応性T細胞の活性化によって病的状態が起こり得る。これらの疾患は例えば、自己免疫疾患、特定の細菌または寄生生物の感染の2次的な結果として起こる自己免疫障害、T細胞媒介性アレルギー、および乾癬および脈管炎のような特定の皮膚疾患を含む。さらに、外来性抗原の望ましくない拒絶が移植片拒絶または不妊さえも引き起こし得、そしてそのような拒絶は特定のTリンパ球集団の活性化によるものであり得る。病的状態はまた、腫瘍またはウイルス感染に対する不適当なT細胞の反応によって起こり得る。これらの場合、腫瘍を抑制または排除するために、または感染を根絶するために、抗原特異的T細胞応答を増加させることが望ましい。
【0020】
自己免疫疾患は様々な原因を有する。例えば、自己免疫反応は損傷またはコラーゲンによる免疫化によって、スーパー抗原によって、遺伝的要因によって、または免疫調節の異常(error)によって起こり得る。スーパー抗原は、他のものの中でも、自己抗原と出会うことによって前にアネルギー化されたクローン、またはそれらの低い発現または利用可能性のために潜在的な自己抗原を無視したクローンを刺激し得る、ポリクローナルアクチベーターである。特定の自己免疫疾患は主に自己抗体によって起こり、他はT細胞によって媒介される。自己反応性T細胞は、慢性関節リウマチおよび多発性硬化症を含む多くの自己免疫疾患において、組織の損傷を引き起こす。
【0021】
自己免疫障害の処置において、非特異的な免疫抑制剤が、疾患を遅らせるために使用されてきた;これらの治療はしばしば、免疫適格細胞を無作為に殺傷または阻害することによって一般的な免疫抑制を引き起こす。自己反応性T細胞の活性を調節することによって自己免疫障害を処置する試みは、TCRペプチドによる免疫、インターフェロン−β(IFN−β)による処置およびTリンパ球ワクチン接種を含んでいた。Ebers(1994)Lancet 343:275−278;Hohlfeld(1997)Brain 120:865−916;およびHaflerら(1992)Clin.Immunol.Immunopathol.62:307−313.
アレルギー感作、接触過敏症および炎症の発達は、プロアレルギー性機能を示すT細胞の活性化および刺激に依存する。アレルゲン特異的T細胞は、アトピー性アレルギーの病態生理学において重要な役割を果たしていると考えられる。アレルゲン特異的T細胞の排除または抑制は、そのようなT細胞によって起こるアレルギー性疾患の改善に役立ち得る。
【0022】
アレルギー反応の初期相で、身体に入った抗原(アレルゲン)はAPCによって取り込まれ、クラスII MHC分子のからみでAPCによって提示され、そしてヘルパーT細胞前駆体によって認識される。これらは刺激されて増殖し、そして主にTH2細胞に分化する。TH2細胞はBリンパ球が抗原産生プラズマ細胞に分化するのを補助する。他のあらゆる抗体媒介性反応と同様に、TH細胞から特異的な補助を受けたB細胞は、それらの表面レセプターを介してアレルゲンを認識する細胞である。TH2細胞によって産生されるサイトカインのいくつか、特にIL−4およびIL−13は、B細胞を刺激して免疫グロブリンアイソタイプの切換えを引き起こし、そしてIgE抗体を産生させる。抗体は結合組織および粘膜にある肥満細胞表面のレセプター、ならびに循環および粘膜にある好塩基球表面の高親和性Fcレセプターに結合し、そしてアレルギー反応の発現を開始する。
【0023】
同種移植片拒絶は主に、移植片細胞上に発現する同種抗原(主にMHC分子)に応答する細胞媒介性免疫応答によって起こる。同種移植片拒絶に関わるTリンパ球部分集団の分析は、CD4+およびCD8+の両集団を含んでいた。TH1細胞は遅延型過敏症の炎症性反応を開始し、単球およびマクロファージの移植片への漸増を引き起こす。おそらく、レシピエントに存在するMHC分子が移植片に存在しないことにより警戒態勢にあるナチュラルキラー(NK)細胞がまた、応答の初期に移植片を攻撃し得る。好中球は主に、同種移植片拒絶の後期において、創傷の浄化(cleaning)、または損傷した細胞および細胞断片の除去を担う。
【0024】
開発されたほとんどの免疫抑制処置は、非特異的であるという不利な点を有する。すなわち、それらは一般化した免疫抑制を引き起こし、このことはレシピエントを増加した感染の危険にさらす。臓器拒絶を予防するために利用される免疫抑制剤は、アザチオプリン、シクロホスファミドおよびメトトレキサートのような有糸分裂インヒビター;コルチコステロイド;ならびにシクロスポリン、FK506、およびラパマイシンのような、IL−2およびIL−2に対する高親和性レセプターをコードする遺伝子の転写を阻害する薬剤を含む。
【0025】
ガンの処置において、細胞性免疫療法が、化学療法および放射線療法のような従来の治療の代わりに、またはその補助として利用されてきた。例えば、細胞傷害性Tリンパ球(CTL)応答が、腫瘍細胞によって特異的にまたは優先的に提示される抗原に対して指向され得る。適切に提示された腫瘍抗原の存在下での、T細胞サイトカインによる活性化に続いて、腫瘍浸潤リンパ球(TIL)が培養中で増殖し、そして強力な抗腫瘍細胞溶解性の特性を獲得する。Weidmannら(1994)Cancer Immunol.Immunother.39:1−14。
【0026】
インビトロで活性化したリンパ球集団をガン患者へ導入することは、いくらか成功を収めた。腫瘍を後退させるために、ガン患者に免疫学的活性な細胞を注入する養子免疫療法は、数十年間ガン治療の魅力的なアプローチであった。2つの一般的なアプローチが探求された。1つ目は、組織適合性抗原の発現の相違に基づいて宿主の腫瘍に対して自然に反応性であるドナー細胞か、または種々の「免疫」技術を用いて反応性にしたドナー細胞を採取する。次いで、これらの活性化ドナー細胞を、腫瘍保有宿主に輸注する。2つ目の一般的なアプローチでは、ガン患者からリンパ球を採取し、エキソビボで腫瘍に対して活性化し、次いで、この患者に再び注入する。Triozzi(1993)Stem Cells 11:204−211;およびSussmanら(1994)Annals Surg.Oncol.1:296.
ガン処置の現在の方法は、比較的非選択的である。手術は冒された組織を除去し、放射線療法は固形腫瘍を縮小し、そして化学療法は速く分裂する細胞を殺傷する。特に、化学療法剤の全身性送達は、多くの副作用を引き起こし、いくつかの場合においては、潜在的に有効な薬剤の使用を排除するほど重篤である。
【0027】
ウイルス性疾患もまた、免疫療法の候補である。Heslopら(1996)Nature Med.2:551−555。ウイルス病原体に応答する免疫学的応答は、時にはウイルスを根絶または十分に激減させるのに無効である。さらに、ヒト免疫不全ウイルスのような、特定のウイルスの高度に突然変異を起こしやすい性質は、それらが免疫系から逃れることを可能にする。
【0028】
明らかに、上記で述べた病因に関与するT細胞の集団を同定し、分析しそして富化(enrich)する必要性がある。現在、抗原特異的T細胞および/またはサイトカイン分泌T細胞の分析および富化のためのいくつかの方法が存在する。抗原特異的T細胞の富化は、T細胞株およびT細胞クローンを得るための選択培養技術を用いて達成され得る。これらの技術は一般的に、T細胞をインビトロで数週間以上の期間培養すること、そしてサイトカイン分泌のような望ましい表現形を示す株またはクローンを選択するためのより煩雑な方法を使用することを含む。抗原特異的T細胞を検出および富化する他の試みは、規定された多量体のMHC抗原およびMHC−ペプチド複合体を利用した。米国特許第5,635,363号。しかし、そのような技術が成功するためには、正しいMHCアロタイプのMHC−抗原複合体が必要であり、そして選択は抗原特異性に限定される(すなわち、この技術によって生じるサイトカイン分泌についての選択ではない)。
【0029】
抗原活性化後の細胞内サイトカイン染色、続くFACS分析は、抗原特異性およびサイトカイン産生の動態に関する情報を得るために使用される方法である。Waldropら(1997)J.Clin.Invest.99:1739−1750。この手順の後の細胞は生存能力がないので、この方法は分析にのみ有用である。同様に、サイトカインELISPOTアッセイは分析にのみ有用である。Miyahiraら(1995)J.Immunol.Met.181:45−54;およびLalvaniら(1997)J.Exp.Med.186:859−865。これらのアッセイでは、分泌されたサイトカインは分析のために周囲のマトリックスに捕獲されるが、サイトカインを分泌した細胞を同定および回収するための機構は存在しない。ゲル微小滴技術(gel microdrop technology)は、上記で述べた適応症の処置のために必要であるような大量の細胞の処理に適さない。
【0030】
多くの治療的目的および診断的目的のために、分泌産物に基づいてT細胞の集団を分析および分離するのに確実な技術の必要性が存在することは、前述の議論から明らかである。本発明は、抗原特異的T細胞集団の分析、分離、そして富化のための方法を提供することによってこの必要性に取り組む。
【0031】
(発明の開示)
本発明は、抗原刺激に応答してこれらの細胞によって分泌される1つ以上の産物に基づく、抗原特異的T細胞の簡便な分析および細胞分離の方法を提供する。T細胞は、産物に特異的な捕獲部分(または「特異的結合パートナー」)と共に提供される。次いでこれは、標識として直接使用され得る。産物の捕獲部分への結合は、「捕獲産物」を生じる。あるいは、この細胞は、捕獲部分を介して産物に結合し、そして産物に特異的に結合し、次に発蛍光団、放射性同位元素、発色団、または磁気粒子のような従来の標識物質で、直接的かまたは間接的のいずれかで標識される標識部分を介してさらに標識され得る。
【0032】
次いで、標識細胞をこれらの標識に基づいて、標準的なセルソーティング技術を用いて分離し得る。このような技術は、フローサイトメトリー、FACS、高勾配磁気勾配分離(high gradient magnetic gradient separation)、遠心分離を含むがこれに限定されない。
【0033】
従って、1つの局面では、本発明は、刺激に応答して抗原特異的T細胞によって分泌および放出される産物によって、抗原特異的T細胞を刺激する、および細胞の集団から分離する方法を含む。本方法は、T細胞を含む細胞の混合物を抗原で刺激すること、およびそれらが産物で標識される程度によって抗原刺激細胞の分離をもたらすことを含む。抗原刺激は、少なくとも1つのT細胞の抗原特異的刺激を誘発するのに有効な条件下で、細胞を少なくとも1つの抗原に曝露することによって達成される。この産物による標識は、細胞の表面を少なくとも1つの捕獲部分を含むように改変すること、細胞を産物が分泌、放出、そして特異的に上記捕獲部分に結合する(「捕獲される」または「包括される」)条件下で培養すること、そして捕獲産物を標識部分で標識することによって達成される。ここで標識細胞は、標識手順の一部としてか、または分離手順の一部として溶解されない。
【0034】
本発明の別の局面は、抗原刺激に応答してこれらの細胞から分泌および放出された産物を捕獲し得る抗原特異的T細胞を含む物質の組成物である。ここで、細胞の表面は産物の捕獲部分を含むように改変される。捕獲産物は、標識部分によって別個に標識され得る。
【0035】
本発明のなお別の局面は、上記の方法で分離された抗原特異的T細胞およびその子孫である。
【0036】
本発明のさらに別の局面は、これらの細胞の表面を細胞表面に結合した産物に特異的な結合パートナーを含むように改変すること、および細胞を産物が分泌および放出される条件下で培養することによる、抗原刺激に応答して細胞によって分泌および放出された産物で抗原特異的T細胞を標識する方法である。
【0037】
本発明のさらなる局面は、抗原特異的T細胞の集団を分析して、集団の他の細胞と比較してある量の産物を分泌する細胞の割合を決定する方法である。ここで産物は、抗原刺激に応答して分泌される。本方法は、細胞を上記の方法で標識すること、さらに細胞を、捕獲産物を標識しない二次標識で標識すること、そして二次細胞標識と比較した産物標識の量を検出することを含む。そのような方法は、例えば個体における免疫状態を評価するのに有用である。
【0038】
本発明のさらなる局面は、抗原特異的T細胞を富化したT細胞集団を使用する方法である。本方法は、治療の必要がある個体に、抗原特異的T細胞を富化したT細胞集団を含む組成物を投与することを含む。そのような方法は、ガン、アレルギー、免疫不全症、自己免疫疾患、およびウイルス疾患を含む種々の病的状態を治療するのに有用である。
【0039】
本発明のさらに別の局面は、抗原特異的T細胞を、エフェクター細胞を含む混合集団から分離するのに使用するためのキットである。そのキットは、全て適当な容器に包装された、ゲル様の軟度までの異なる程度の粘性であり得る生理的に許容可能な培地、アンカーおよび捕獲部分を含む産物捕獲系、捕獲産物を検出する標識システム、および試薬を使用するための説明書を含み得る。必要に応じて、このキットはさらに、磁気標識システムおよび/または1つ以上の生物学的修飾因子を含む。
【0040】
本発明のなお別の局面は、抗原刺激に応答して所望の産物を分泌する抗原特異的T細胞の検出/分離に使用するためのキットである。このキットは、全て適当な容器に包装された、アンカーおよび捕獲部分を含む産物捕獲系、捕獲産物を検出する標識システム、および試薬を使用するための説明書を含む。必要に応じて、このキットはさらに磁気標識システム、および/または抗原、および/または1つ以上の生物学的修飾因子を含む。
【0041】
(本発明を実施する方法)
本発明は、分泌産物に基づいて抗原刺激T細胞を検出、分析および分離する方法を提供する。ここで、この産物は、抗原刺激の結果として分泌される。本方法は、分泌産物の捕獲および細胞表面への移転に基づく。捕獲産物は、産物の存在、非存在、または存在する量によって細胞を検出、分析、そしてもし所望のならば分類することを可能にする。捕獲の手段は、分類される細胞に適当な手段によって細胞表面に固定した産物特異的結合パートナー(「捕獲部分」)を含む。
【0042】
ここで提示されるアプローチは、とりわけ以下の利点を兼ね備える:(a)T細胞を、抗原およびAPCで周期的に活性化する必要なく、生抗原特異的T細胞の迅速な単離、計測、表現型決定および増殖を可能にする;(b)一般的に、合成ペプチド、天然タンパク質、細胞抽出物、不活化病原体を適用した、レトロウイルスベクターで形質導入した、組換えウイルスベクターで感染させた、RNAまたはDNAでトランスフェクションした等のAPCに反応性のT細胞の単離に適用できる;(c)CD4+抗原特異的Th細胞およびCD8+抗原特異的CTLの両方の単離に使用し得る;および(d)例えば、抗原特異的Th1−、Th2−またはTh3−様リンパ球の、特定のサイトカインに媒介されたエフェクター機能を有する抗原特異的T細胞の選択的な単離を可能にする。
【0043】
本発明の実施は、他に示されなければ、当該分野の技術の範囲内である、分子生物学(組換え技術を含む)、微生物学、細胞生物学、生化学、および免疫学の従来技術を採用する。そのような技術は、「Molecular Cloning:A Laboratory Manual」第2版(Sambrookら、1989);「Oligonucleotide Synthesis」(M.J.Gait編、1984);「Animal Cell Culture」(R.I.Freshney編、1987);「Methods in Enzymology」(Academic Press,Inc.);「Handbook of Experimental Immunology」(D.M.Weir&C.C.Blackwell編);「Gene Transfer Vectors for Mammalian Cells」(J.M.Miller&M.P.Calos編、1987);「Current Protocols in Molecular Biology」(F.M.Ausubelら、編、1987、および定期的に改訂);「PCR:The Polymerase Chain Reaction」、(Mullisら、編、1994);および「Current Protocols in Immunology」(J.E.Coliganら、編、1991)のような文献において完全に説明されている。
【0044】
セルソーティングおよび細胞分析の方法は、当該分野で公知であり、そして例えば、The Handbook of Experimental Immunology、1から4巻(D.N.Weir、編者)およびFlow Cytometry and Cell Sorting(A.Radbruch、編者、Springer Verlag、1992)において記載されている。
【0045】
本明細書中で使用される場合、「特異的結合パートナー」または「捕獲部分」は、関連する分子の三次元構造に依存する特異的な非共有結合相互作用によって相互作用する1対の分子(「特異的結合対」)のメンバーを意味する。「標識部分」は、直接的かまたは間接的のいずれかで検出され得る。捕獲部分が抗体である場合、「捕獲(capture)抗体」または「捕獲(catch)抗体」と呼ばれ得る。捕獲部分は、直接的かまたは間接的のいずれかで細胞および産物の両方に結合するものである。標識部分は、産物に付着するものであり、そして直接的かまたは間接的に標識され得る。
【0046】
本明細書中で使用される時、用語「抗体」は、ポリクローナル抗体およびモノクローナル抗体、キメラ抗体、ハプテンおよび抗体フラグメント、および産物抗原上のエピトープに特異的に結合する抗体等価物である分子を含むことを意味する。用語「抗体」は、任意のアイソタイプ(IgA、IgG、IgE、IgD、IgM)のポリクローナル抗体およびモノクローナル抗体、またはその抗原結合部分を含み、F(ab)およびscFvのようなFv断片、単鎖抗体、キメラ抗体、ヒト化抗体、およびFab発現ライブラリーを含むがこれに限定されない。抗体はまた、例えばポリマーまたは粒子に固定化され得る。
【0047】
二重機能性(bifunctional)抗体としても知られる、「二重特異性(bispecific)抗体」は、第1の抗原またはハプテンに特異的な少なくとも1つの第1の抗原結合部位、および2番目の抗原またはハプテンに特異的な少なくとも1つの第2の抗原結合部位を有することによって、2つの異なる抗原を認識する抗体を意味する。そのような抗体は、組換えDNA方法によって産生され得、または当該分野で公知の方法によって化学的に産生された抗体を含むがこれに限定されない。その二価の特性を保持するように還元および再編成された二重特異性抗体、およびそれぞれの抗原に対して少なくとも2つの抗原認識部位を有するように化学的に結合させた抗体を化学的に作成した。二重特異性抗体は、2つの異なる抗原を認識し得る全ての抗体または抗体の結合体、または抗体の重合体形態を含む。標識部分は、蛍光色素化した抗産物抗体であり得る。それは磁気ビーズ結合抗体、コロイド状ビーズ結合抗体、FITC抗体、フィコエリトリン抗体、PerCP抗体、AMCA抗体、蛍光粒子抗体、またはリポソーム結合抗体を含み得るがこれらに限定されない。あるいは、標識部分は、本明細書中で記載されるものを含むがこれらに限定されない任意の適切な標識であり得る。二重特異性抗体は、その二価の特性を保持するように還元および再編成された抗体、およびそれぞれの抗原に対するいくつかの抗原認識部位を有し得るように化学的に結合させた抗体を含む。
【0048】
本明細書中で使用される場合、用語「エフェクター細胞集団」は、少なくとも1つのT細胞を含む細胞集団を意味する。エフェクター細胞集団は、抗原特異的T細胞が富化される開始細胞集団から得ることができる。
【0049】
交換可能に使用される用語「細胞」および「細胞(複数)」および「細胞集団」は、1つ以上の哺乳動物細胞を意味する。その用語は、細胞または細胞集団の子孫を含む。当業者は、「細胞」は1つの細胞の子孫を含むこと、そしてその子孫は、自然の、偶発性の、または故意の変異および/または変化によって、必ずしも本来の親細胞と完全に同一(形態学的に、または総DNA相補体)である必要は無くてもよいことを認識する。
【0050】
交換可能に使用される用語「Tリンパ球」、「T細胞」、「T細胞(複数)」、および「T細胞集団」は、その表面に、例えば、CD2およびCD3のような1つ以上のT細胞に特徴的な抗原を提示する細胞または細胞(複数)を意味する。その用語はT細胞またはT細胞集団の子孫を含む。「Tリンパ球」または「T細胞」は、その細胞表面上にCD3、および自己細胞または任意の抗原提示マトリックスの表面上に、1つ以上のMHC分子または1つ以上の非古典的MHC分子と共に提示された場合に、抗原を認識し得るT細胞抗原受容体(TCR)を発現する細胞である。本明細書中で使用される用語「T細胞」は、当該分野で公知の任意のT細胞、代表的には、抗CD3モノクローナル抗体を適切な標識技術と組み合せて用いて検出される、例えば表現型がCD3+である、すなわち細胞表面上にCD3を発現するリンパ球を示す。本発明の方法によって富化されたT細胞は、一般的にCD3+である。本発明の方法によって富化されたT細胞はまた、必ずしもそうでないが、一般的にCD4、CD8、または両方に陽性である。
【0051】
本明細書中で使用される用語「本質的に富化された」は、所望の細胞集団を含む本来の混合細胞集団から、細胞集団が少なくとも約50倍、より好ましくは少なくとも約500倍、そしてさらにより好ましくは少なくとも約5000倍以上に富化されたことを示す。
【0052】
本明細書中で使用される用語「抗原提示マトリックス」は、抗原が、T細胞の表面上にあるT細胞抗原受容体によって結合され得る方法で、抗原を提示し得る分子または複数の分子を意味する。抗原提示マトリックスは、抗原提示細胞(APC)の表面上、APCの小胞調製物上、またはビーズもしくはプレート上の合成マトリックスの形であり得る。本明細書中で使用される用語「抗原提示細胞」は、その表面上に抗原を、MHCまたはその一部、または1つ以上の非古典的MHC分子またはその一部と共に提示する任意の細胞を意味する。
【0053】
本明細書中で使用される用語「自原性」、「自己(autologous)」または「自身(self)」という用語は、細胞の起源を示す。従って、細胞が、個体(「ドナー」)または遺伝的に同一の個体に由来し、そしてその個体に再投与される場合、細胞は自原性である。自原性細胞はまた、自原性細胞の子孫であり得る。その用語はまた、異なる細胞型の細胞が同じドナー、または遺伝的に同一のドナーに由来することを示す。従って、エフェクター細胞および抗原提示細胞は、それらが、同じドナーまたはドナーと遺伝的に同一の個体由来である場合、またはそれらが、同じドナーまたはドナーと遺伝的に同一の個体由来の細胞の子孫である場合、自原性であると言われる。
【0054】
同様に、本明細書中で使用される用語「同種」または「非自己」は、細胞の起源を示す。従って、細胞が、それが投与されるレシピエントと遺伝的に同一でない個体に由来した場合、細胞またはその子孫は同種である。この用語は、発現されたMHC分子の非同一性に関連する。この用語はまた、異なる細胞型の細胞が、遺伝的に同一でないドナーに由来すること(すなわち、それらが遺伝的に同一でないドナーに由来する細胞の子孫である否か)を示す。例えば、それらが遺伝的に同一でないドナーに由来する場合、APCはエフェクター細胞に対して同種であると言われる。
【0055】
「抗原特異的T細胞の集団に関連する疾患または状態」は、抗原特異的T細胞の集団またはその適切な数の欠如に関連し得るものであり、そして例えば抗原特異的T細胞が、疾患の病因を主に担う自己免疫疾患、ガン細胞の増殖が、腫瘍特異的細胞障害性T細胞によって適切に制御されないガン、ウイルス感染細胞が、細胞障害性T細胞によって溶解されないウイルス疾患、アレルゲンに特異的なT細胞が、望ましくない効果を媒介するアレルギー、感染(例えば、HIV)または先天的(例えば、ディジョージ症候群)のいずれかのために、個体に不適切な数のT細胞が存在する免疫不全症を含む。これはまた、抗原特異的T細胞が、疾患状態を主に担う別の細胞または細胞集団の活性を調節または制御するものであり;抗原特異的T細胞の集団の存在が、疾患の主な原因ではないが、疾患の病因において重要な役割を果たすものでもあり;抗原特異的T細胞の集団が、外来性抗原の望ましくない拒絶を媒介するものでもある。
【0056】
「個体」は脊椎動物、好ましくは哺乳類、より好ましくはヒトである。哺乳類は、ヒト、家畜、スポーツ動物(sport animal)、およびペットを含むがこれらに限定されない。
【0057】
「有効量」は、有用なまたは所望の臨床結果をもたらすのに十分な量である。有効量は、1回以上の投与で投与され得る。本発明の目的のために、抗原特異的T細胞の有効量は、疾患状態を診断、寛解、回復、安定化、後退、進行の緩徐(slow)、または遅延(delay)に十分な量である。
【0058】
本明細書中で使用される場合、「処置」は、有用なまたは所望の臨床結果を得るためのアプローチである。本発明の目的のために、有用なまたは所望の臨床結果は、検出できるかまたは検出できない、症状の緩和、疾患の拡大の減少、疾患の状態の安定化(すなわち悪化しない)、疾患の伝播(すなわち転移)の防止、疾患の進行の遅延(delay)または緩徐(slowing)、疾患の状態の回復または寛解、および緩解(部分的または全体のどちらか)を含むがこれらに限定されない。「処置」はまた、処置を受けない場合に期待される生存期間と比べた生存期間の延長を意味し得る。
【0059】
疾患の「緩和」は、本発明の富化T細胞集団を投与しないのと比べて、疾患の状態の拡大および/または望ましくない臨床的症状が減少するおよび/または進行の時間経過が緩徐されるかまたは延長されることを意味する。
【0060】
本発明は、産物を分泌する抗原特異的T細胞を富化した細胞集団を得る方法を提供する。ここで、この産物は、抗原刺激の結果として分泌される。本方法は、一般的にT細胞を含む細胞の混合集団を得ること;少なくとも1つのT細胞の抗原特異的刺激を誘発するのに効果的な条件下で細胞集団を少なくとも1つの抗原に曝露すること;上記混合集団の表面を、それに付着した産物に特異的な結合パートナーを含むように改変すること;刺激されたT細胞によって少なくとも1つの産物の発現を可能にすること(ここで、産物は刺激に応答して分泌される);産物を細胞表面に結合した捕獲部分に結合させて、細胞結合捕獲部分−産物複合体を形成し、それによって細胞を標識すること;および上記産物で標識される程度によって刺激されたT細胞を分離することを含む。
【0061】
確かに、細胞表面の特異的結合パートナーによる改変は、抗原刺激の前、間、または後に行われ得る。
【0062】
(抗原提示マトリックスおよびエフェクター細胞集団)
本発明は、抗原刺激に応答して産物を分泌する抗原特異的T細胞を富化した細胞集団を得るための方法を提供する。本方法は、細胞の混合集団(すなわち、「エフェクター細胞集団」)を得ること、および細胞集団を少なくとも1つの抗原に曝露することを含む。混合細胞集団は、当該分野で公知の任意の方法によって得ることができ、そして好ましくはT細胞が富化されている。抗原への曝露は、抗原提示マトリックスを用いて達成され得る。それは抗原提示細胞(APC)の表面上であり得る。抗原提示マトリックスおよびエフェクター細胞は、種々の供給源から得ることができる。細胞の混合集団は、インビトロまたはインビボで抗原によって刺激され得るか、または種々の任意の方法で、たとえば化学的または遺伝的に改変され得る。
【0063】
(抗原提示マトリックス)
本発明の方法を適用したT細胞集団は、抗原特異的刺激を誘発するのに有効な条件下で、少なくとも1つの抗原に曝露される。少なくとも1つの抗原で刺激されたT細胞は、抗原特異的と言われる。すなわち、それはその細胞表面に、抗原提示マトリックス(例えば合成抗原提示マトリックス)、またはAPCの表面上に存在する抗原提示マトリックス上に抗原(例えば、古典的もしくは非古典的MHC分子またはその一部)を提示し得る分子と会合する抗原を特異的に認識しそして結合する抗原レセプターを提示する。
【0064】
抗原提示分子は、クラスIもしくはクラスIIであり得るか、またはCD1のような非古典的MHC分子であり得るMHC分子;MHCエピトープ;MHCエピトープを含む融合タンパク質;あるいは合成MHCエピトープであり得る。エフェクター細胞に対して抗原を提示し得る限り、抗原提示分子の性質は重要でない。MHCエピトープを調製する方法は、当該分野で公知である。
【0065】
抗原提示マトリックスは、APCの表面上の抗原提示マトリックス、および合成抗原提示マトリックスを含む。本発明における使用に適当なAPCは、MHC分子のような抗原提示分子と会合するT細胞に対して、外来性のペプチドもしくはタンパク質、または内因性の抗原を提示し得る。APCは、マクロファージ、樹状細胞、CD40活性化B細胞、抗原特異的B細胞、腫瘍細胞、ウイルス感染細胞および遺伝的に改変された細胞を含むがこれに限定されない。
【0066】
APCは、末梢血単核球(PBMC)、混合集団を含む全血またはその画分、脾臓細胞、骨髄細胞、腫瘍浸潤リンパ球、白血球搬出法によって得た細胞、リンパ節(例えば、腫瘍から流出したリンパ節)を含むが、これらに限定されない種々の供給源から得られ得る。適当なドナーは、免疫したドナー、免疫していない(ナイーブ)ドナー、処置または未処置のドナーを含む。「処置」ドナーは1つ以上の生物学的修飾因子に曝露されたものである。「未処置」ドナーは1つ以上の生物学的修飾因子に曝露されたことがない。APCはまた、インビトロで1つ以上の生物学的修飾因子で処理され得る。
【0067】
APCは一般的に生きているが、放射線照射、マイトマイシンC処理、弱毒化、または化学的に固定もされ得る。さらに、APCは全細胞である必要はない。かわりに、APCの小胞調製物を使用し得る。
【0068】
APCは遺伝的に改変され得る。すなわち、通常発現しないか、通常より低いレベルで発現するポリペプチドまたはRNA分子を発現するように、組換えポリヌクレオチド構築物でトランスフェクトされ得る。ポリヌクレオチドの例は、MHC分子、B7のような同時刺激分子、または抗原をコードするものを含むが、これに限定されない。例えば、CMVプロモーターのような強力なプロモーターの転写調節下にあるMHC分子をコードするポリヌクレオチドの発現は、高レベルのMHC分子の細胞表面上での発現を引き起こし得、従って抗原提示の密度を増加させる。あるいは、抗原がMHC分子と共に細胞表面上で発現するように、APCは、CMVプロモーターのような強力なプロモーターの転写調節下で、抗原をコードするポリヌクレオチドを含むポリヌクレオチド構築物でトランスフェクトされ得る。
【0069】
ポリペプチドをコードするヌクレオチド配列は、転写および翻訳のために、調節配列に作動可能に連結される。調節配列が転写または翻訳を制御する場合、調節配列はコードする配列に「作動可能に連結」される。当該分野における任意の方法(例えば、精製DNA、ウイルスベクター、またはDNAもしくはRNAウイルスのいずれかを用いたリポフェクチン、形質導入、感染、またはエレクトロポレーション)が、外来性のポリヌクレオチドのAPCへの形質転換、または挿入に使用され得る。外来性のポリヌクレオチドは、非組み込みベクター、例えばプラスミドとして保持され得、または宿主細胞ゲノムに組み込まれ得る。
【0070】
哺乳動物において、インビボでは通常はAPCとして機能しない細胞を、APCとして機能するように改変し得る。広範な種類の細胞が適切に改変された場合、APCとして機能し得る。そのような細胞の例は、昆虫細胞(例えば、Drosophila、またはSpodoptera)、抗原提示経路に内因性のペプチドと細胞表面MHCクラスI分子との結合を制限する変異を有するヒトT2細胞株のようなフォスター(foster)細胞である。Zweerinkら(1993)J.Immunol.150:1763−1771。例えば、MHC分子のような1つ以上の抗原提示ポリペプチド、および必要に応じてB7のようなアクセサリー分子の合成を指示する発現ベクターをこれらの細胞に導入し、これらの細胞表面に抗原提示分子および必要に応じてアクセサリー分子、またはその機能的な部分の発現をもたらし得る。あるいは、それ自身を細胞膜に挿入し得る抗原提示ポリペプチドおよびアクセサリー分子を使用し得る。例えば、グリコシル−ホスファチジルイノシトール(phosphotidylinositol)(GPI)−改変ポリペプチドはそれ自身を細胞膜に挿入し得る。Medofら、J.Exp.Med.160:1558−1578;およびHuangら、Immunity 1:607−613。アクセサリー分子は、CD28、CD80またはCD86に特異的な抗体のような同時刺激抗体;B7.1およびB7.2を含むがこれに限定されない同時刺激分子;ICAM−1およびLFA−3のような接着分子;ならびに、FasリガンドおよびCD70のような生存分子、を含むがこれらに限定されない。例えば、PCT公開番号第WO97/46256号を参照のこと。
【0071】
あるいは、合成抗原提示マトリックスを、エフェクター細胞に対する抗原を提示するために使用し得る。合成マトリックスは、固体支持体(例えばビーズまたはプレート)に固定化された抗原提示分子、好ましくはMHCクラスIまたはMHCクラスII分子を含み得る。アクセサリー分子が提示され得、それは共に固定化されるかまたは可溶性であり得る。その分子は、CD28、CD80またはCD86に特異的な抗体のような同時刺激抗体;B7.1およびB7.2を含むがこれに限定されない同時刺激分子;ICAM−1およびLFA−3のような接着分子;ならびに、FasリガンドおよびCD70のような生存分子を含むが、これらに限定されない。アクセサリー分子の機能が維持される限り、その分子の一部もまた使用され得る。固体支持体は、金属またはプラスチック、多孔性の物質、マイクロビーズ、マイクロタイタープレート、赤血球、およびリポソームを含む。例えば、PCT公開番号第WO97/46256号、および同第WO97/35035号を参照のこと。
【0072】
抗原提示マトリックスが、細胞表面または合成支持体上にあろうとなかろうと、エフェクター細胞に対する抗原を提示し得るか否かを決定する方法は、当該分野で公知であり、そして例えばエフェクター細胞による3H−チミジンの取り込み、エフェクター細胞によるサイトカインの産生、および細胞溶解性51Cr−放出アッセイを含む。
【0073】
(エフェクター細胞集団)
抗原特異的T細胞は、エフェクター細胞集団、すなわち造血細胞集団、好ましくはT細胞が富化された集団から単離され得る。エフェクター細胞集団は、抗原特異的T細胞が単離される最初の集団である。
【0074】
本発明において使用するのに適当なエフェクター細胞集団は、自原性または同種異系、好ましくは自原性であり得る。エフェクター細胞が同種異系である場合、好ましくは、使用の前に同種反応性(alloreactive)の細胞から細胞を涸渇させる。これは、例えば同種エフェクター細胞およびレシピエント細胞集団を混合すること、ならびにそれらを適当な時間インキュベートすること、次いでCD69+細胞を涸渇させること、または同種反応性細胞を不活化すること、または同種反応性細胞集団のアネルギーを誘導することを含む、任意の公知の方法によって達成され得る。
【0075】
エフェクター細胞集団は、未分離の細胞、すなわち混合集団(例えば、PBMC集団、全血など)を含み得る。エフェクター細胞集団は、細胞表面マーカーの発現に基づくポジティブセレクション、細胞表面マーカーの発現に基づくネガティブセレクション、インビトロまたはインビボでの1つ以上の抗原による刺激、インビトロまたはインビボでの1つ以上の生物学的修飾因子による処理、1つ以上の抗原または生物学的修飾因子による減法刺激、またはこれらのいずれかまたは全ての組み合せによって操作され得る。
【0076】
エフェクター細胞は、PBMC、全血、または混合集団を含むその画分、脾臓細胞、骨髄細胞、腫瘍浸潤リンパ球、白血球搬出法によって得られた細胞、生検組織、リンパ節(例えば、腫瘍から流出したリンパ節)を含むがこれらに限定されない種々の供給源から得られ得る。適切なドナーは、免疫したドナー、免疫していない(ナイーブ)ドナー、処置または未処置ドナーを含む。「処置」ドナーは1つ以上の生物学的修飾因子に曝露されたものである。「未処置」ドナーは1つ以上の生物学的修飾因子に曝露されたことがない。
【0077】
エフェクター細胞を抽出および培養する方法は、周知である。例えば、エフェクター細胞は、白血球搬出法、連続フローセル分離機(continuous flow cell separator)を用いた機械的アフェレーシスによって得られ得る。例えば、リンパ球および単球を、軟膜から、Ficcoll−HypaqueTM勾配による分離、Percoll勾配による分離、または溶出を含むがこれに限定されない任意の公知の方法によって単離し得る。Ficcoll−HypaqueTMの濃度は、所望の集団(例えば、T細胞が富化された集団)を得るために調節され得る。細胞特異的アフィニティーカラムに基づく他の方法が公知であり、そして使用され得る。これらは、例えば蛍光標示式細胞分取(FACS)、細胞接着、磁気ビーズ分離などを含む。親和性に基づく方法は、細胞表面マーカーに特異的で、かつAmerican Type Culture Collection(Rockville、MD)を含む種々の市販の供給源から入手可能な抗体またはその一部を利用し得る。あるいは、親和性に基づく方法は、細胞表面レセプターのリガンドまたはリガンドアナログを利用し得る。
【0078】
エフェクター細胞集団は、細胞表面マーカーの発現に基づく1つ以上の分離プロトコールに供され得る。例えば、細胞は、CD2、CD3、CD4、CD8、TCR、CD45、CD45RO、CD45RA、CD11b、CD26、CD27、CD28、CD29、CD30、CD31、CD40Lのような「クラスターの分化」細胞表面マーカー;リンパ球活性化遺伝子3産物(LAG3)、シグナル伝達リンパ球活性化分子(SLAM)、T1/ST2のようなリンパ球活性化に関連する他のマーカー;CCR3、CCR4、CXCR3、CCR5のようなケモカインレセプター;CD62L、CD44、CLA、CD146、α4β7、αEβ7のようなホーミングレセプター;CD25、CD69およびOX40のような活性化マーカー;ならびに、CD1によって提示されるリポグリカン、を含むがこれらに限定されない、1つ以上の細胞表面ポリペプチドの発現に基づくポジティブセレクションに供され得る。エフェクター細胞集団は、非T細胞および/または特定のT細胞サブセットの涸渇についてのネガティブセレクションに供され得る。ネガティブセレクションは、CD19、およびCD20のようなB細胞マーカー;単球マーカーCD14;NK細胞マーカーCD56を含むがこれらに限定されない、種々の分子の細胞表面発現に基づいて実施され得る。
【0079】
エフェクター細胞集団は、インビボまたはインビトロで1つ以上の抗原にさらすことによって処理され得る。抗原は、ペプチド;タンパク質;糖タンパク質;脂質;糖脂質;細胞;細胞抽出物;組織抽出物;原生動物、細菌およびウイルスのような微生物全体を含むがこれらに限定されない。抗原は未改変、すなわちその自然の状態で使用され得る。あるいは、抗原は、例えばタンパク質を変性または病原体を不活化するための加熱;タンパク質を変性するためまたは2つの抗原分子の架橋するための化学的改変;グリコシル化;ポリエチレングリコールを含むがこれに限定されない部分による化学的改変;および酵素消化、を含むがこれらに限定されない任意の公知の方法によって改変され得る。1より多い抗原が使用される場合、曝露は同時または連続的であり得る。
【0080】
エフェクター細胞を、処置される状態に関連する少なくとも1つの抗原の存在下で培養し得る。抗原は複数の抗原決定基を有する単一の抗原であり得るか、または抗原の混合物であり得る。抗原は、処置される状態に依存して、自己抗原または外来性抗原であり得る。自己抗原は、自己免疫疾患に関連する抗原、およびガン細胞に関連する抗原を含む。抗原は、タンパク質、細胞、組織または標的器官であり得る。抗原が自己抗原である場合、自己抗原は器官(例えば、脳または甲状腺)の一部であり得、そしてそれから精製される必要はない。精製自己抗原または精製自己抗原の混合物も使用され得る。
【0081】
末梢血リンパ球(PBL)または腫瘍浸潤リンパ球(TIL)と自己腫瘍細胞との同時培養は、一般的にサイトカイン刺激を伴う。Spornら(1993)Cancer Immunol.Immunother.37:175−180;およびPeyretら(1991)Chirurgie 117:700−709。
【0082】
エフェクター細胞集団は、インビボまたはインビトロで1つ以上の生物学的修飾因子に曝露することによって処理され得る。適切な生物学的修飾因子は、IL−2、IL−4、IL−10、TNF−α、IL−12、IFN−γのようなサイトカイン;フィトヘマグルチニン(PHA)、ホルボールミリステートアセテート(PMA)のようなホルボールエステル、コンカナバリン−A、およびイオノマイシンのような非特異的修飾因子;抗CD2、抗CD3、抗IL−2レセプター、抗CD28のような細胞表面マーカーに特異的な抗体;ケモカイン(例えば、リンホタクチンを含む)を含むがこれに限定されない。生物学的修飾因子は、天然の供給源から得られた天然の因子、組換えDNA技術によって産生された因子、化学的に合成されたポリペプチドまたは他の分子、または天然の因子の機能的活性を有する、それらの任意の誘導体であり得る。1つ以上の生物学的修飾因子が使用される場合、曝露は同時または連続的であり得る。
【0083】
本発明は、抗原特異的細胞が富化された、本発明の方法によって富化されたT細胞を含む組成物を提供する。「富化された」は細胞集団が少なくとも約50倍、より好ましくは少なくとも約500倍、そしてさらにより好ましくは少なくとも約5000倍以上、所望の細胞集団を含む元の混合細胞集団から富化されたことを意味する。所望の抗原特異的細胞を含む富化細胞集団の割合は、10%未満から100%の抗原特異的細胞まで、実質的に異なり得る。抗原特異的である割合は、例えばT細胞集団が所望の抗原を提示する抗原提示マトリックスによって攻撃される、3H−チミジンの取り込みアッセイによって、容易に決定され得る。
【0084】
(細胞標識)
本明細書中の方法は、細胞を、細胞によって分泌された産物で標識することに基づく。ここで、この産物は抗原刺激に反応して分泌される。標識を達成するために、細胞集団の細胞表面は、産物に特異的に結合する部分である、「特異的結合パートナー」が直接、または固定する手段(「アンカー部分」)のいずれかによって、必要に応じて捕獲部分を形成するためのリンカーによって、細胞表面に結合するように改変される。細胞集団は、多くの型の細胞を含み得、そして一般的に混合集団からなった。好ましくは、細胞集団は造血系であり、より好ましくは、細胞集団はエフェクター細胞であり、最も好ましくは、細胞集団はT細胞またはそのサブセットである。サブセットは、例えばリンパ球に関してはCD45、細胞傷害性細胞に関してはCD8などの細胞表面マーカーによって単離され得る。
【0085】
抗原刺激に反応して分泌された産物は、当該分野で公知であり、そしてIL−2、IL−4、IL−10、TNF−α、TGF−β、およびIFN−γのようなサイトカインを含むがこれらに限定されない。
【0086】
特異的結合パートナーは、比較的高い親和性および特異性が、産物および結合パートナーの間に存在する部分、および産物:パートナー複合体の分離が比較的遅く、その結果、産物:パートナー複合体が細胞分離技術の間、検出される任意の分子を含む。特異的結合パートナーは、産物が結合する基質または基質アナログ、ペプチド、ポリサッカライド、ステロイド、ビオチン、ジギトキシン、ジギトニンおよびそれらの誘導体を含むがこれに限定されない。好ましい実施態様では、特異的結合パートナーは、抗体または抗原結合フラグメントもしくはその誘導体である。用語「抗原結合フラグメント」は、産物に特異的に結合する任意のペプチドを含む。代表的には、これらのフラグメントはFab、F(ab’)2、Fab’、scFv(単量体および多量体の両方)および単離されたH鎖およびL鎖のような免疫グロブリンフラグメントを含む。抗原結合フラグメントは、アビディティおよび/または親和性は変化し得るが、インタクトな免疫グロブリンの特異性を保持する。
【0087】
本発明の実施において、捕獲部分は、細胞膜(または細胞壁)に種々の方法で結合され得る。適切な方法は、タンパク質構成要素のアミノ基への直接的な化学的結合、タンパク質構成要素のチオールへの結合(ジスルフィド結合の還元後に形成)、抗体(抗体の組を含む)またはレクチンによる間接的な結合、疎水性アンカーによる脂質二重膜への固定、およびポリカチオンによる負に荷電している細胞表面への結合を含むがこれに限定されない。
【0088】
本発明の他の実施態様では、捕獲部分は2つ以上の工程を用いて、例えば、細胞を、直接的に例えばビオチン/アビジン複合体によってか、または間接的に適当な結合部分または複数の部分によってのいずれかで、捕獲部分のアンカー部分への結合を可能にする、少なくとも1つのアンカー部分で標識することによって導入される。
【0089】
適切なアンカー分子は、脂肪酸のような脂肪親和性の分子を含む。あるいは、抗体、またはMHC抗原または糖タンパク質のような細胞表面マーカーに対する他の特異的結合物も使用され得る。
【0090】
「捕獲部分」は、結合剤によってアンカー部分に結合し得、そしてまた、例えば改変デキストラン分子、ポリエチレングリコール、ポリプロピレングリコール、ポリビニルアルコール、およびポリビニルピロリドンを含む分枝ポリマーのような、利用可能な捕獲部分の数、および従って産物の捕獲の可能性を増すリンカーを含み得る。
【0091】
抗体の細胞表面への直接的な化学結合の方法は、当該分野で公知であり、そして例えば、グルタルアルデヒドまたはマレイミド活性化抗体を用いた結合を含む。複数の工程手順を使用する化学的結合方法は、ビオチン化、例えばこれらの化合物のスクシニミドエステルを用いたトリニトロフェノール(TNP)またはジゴキシゲニンの結合を含むがこれに限定されない。ビオチン化は、例えば、D−ビオチニル−N−ヒドロキシスクシニミドの使用によって達成され得る。スクシニミド基は、pH値が7より上で、そして好ましくは約pH8.0から約pH8.5の間で、有効にアミノ基と反応する。ビオチン化は、例えば細胞をジチオトレイトールで処理し、続いてビオチンマレイミドを加えることによって達成され得る。
【0092】
細胞への結合はまた、細胞表面抗原(「マーカー」)に対する抗体を用いて達成され得る。表面抗原に対する抗体は、一般的に107細胞あたり0.1から1μgの範囲の抗体を必要とする。しかし、この必要性は、抗体の産物に対する親和性に反応して大きく変化し、そして経験的に決定する必要がある。そのような決定は、当該分野における技術の範囲内である。従って、適当な量の抗体は、経験的に決定されるべきであり、そして当該分野における技術の範囲内である。これは、細胞型に特異的なマーカー発現について特異的な細胞に結合することを可能にする。例えば、T細胞またはそのサブセットのような細胞のクラスが特異的に標識され得る。捕獲部分として、細胞またはそれに存在するアンカー部分、および産物に対する抗原認識部位を有する二重特異性抗体が使用され得る。
【0093】
捕獲部分、特に捕獲抗体は、分泌産物の量に基づいて選択されるべきである。例えば、少数の分子のみを分泌する細胞に関しては、高い親和性の抗体が、大部分の分泌分子を捕獲する。あるいは、インキュベーション時間の間に、細胞が多くの分子を分泌する場合は、捕獲マトリックスの早すぎる飽和を防止するために、より低い親和性の抗体が好ましくあり得る。分泌されたタンパク質のレベルに適当な親和性の決定は、経験的に決定され、そして当該分野の技術の範囲内である。
【0094】
大量のN−アセチルノイラミン酸を、そのリポポリサッカライドの構成物として表面に有する細胞は、生理的なpH値で負電荷を有する。捕獲部分の結合は、電荷の相互作用により得る。例えば、ポリカチオンを有する部分は負に荷電した細胞に結合する。ポリカチオンは当該分野で公知であり、そして例えばポリリシンおよびキトサンを含む。キトサンは、α−(1−4)グルコシド結合で結合したD−グルコサミン基からなるポリマーである。
【0095】
結合パートナー(1つ以上の捕獲部分を含み得る)を細胞へ結合させる他の方法は、細胞表面のポリサッカライドに結合させることによる。ポリサッカライドに結合する基質は当該分野で公知であり、そして、例えばSigma Chemical CompanyおよびAldrich Chemical Companyを含む多くの会社によって供給されるレクチン(例えば、コンカナバリンA、solanum tuberosum、aleuria aurantia、datura stramonium、galanthus nivalis、helix pomatia、lens culinarisおよび他の既知のレクチンを含む)を含む。
【0096】
本発明のいくつかの実施態様において、産物結合パートナーを、細胞膜に疎水性に固定することにより、細胞に結合する。膜の脂質二重層と相互作用する適切な疎水基は、当該分野において公知であり、そして脂肪酸および非イオン性界面活性剤(例えば、Tween−80を含む)が挙げられるがこれらに限定されない。疎水性アンカーの挿入を介する、細胞への捕獲部分の付着の欠点は、細胞中への疎水性部分の組み込みの速度が遅いことである。従って、高濃度の疎水性アンカーを有する部分がしばしば必要とされる。この後者状況は、捕獲部分が比較的制限されている場合、または高価な物質(例えば、抗体)である場合は、しばしば非経済的である。
【0097】
膜中にそれ自体を包埋する疎水性分子の低収率は、これらの分子が比較的限定された量において利用可能である場合にのみ、問題となる。この問題は、固定化パートナーおよび捕獲部分を含むパートナーを含有する架橋系を使用することによって克服され得る。この架橋系で、このパートナーの1つは、より高度に利用可能であり、そして架橋系の2つのパートナーがお互いに高度な特異性および親和性を有する。例えば、1つの実施態様において、アビジンまたはストレプトアビジンを、疎水性アンカーを介して細胞表面に付着させ、一方、産物捕獲部分を有するパートナーは、ビオチン化された抗−産物抗体である。別の実施態様において、細胞表面をジゴキシゲニンで標識し、次に、抗ジゴキシゲニン抗体フラグメントと抗−産物抗体との結合体を用いる。このアプローチを、連結を形成し得る他の分子対(例えば、ハプテンと抗ハプテン抗体、NTAとポリヒスチジン残基、またはレクチンと多糖類が挙げられる)を用いて使用し得る。好ましい実施態様は、アンカー部分あたりの捕獲部分の数を増加することによる、この系の「増幅」を可能とする実施態様である。
【0098】
1つの例示的な実施態様において、分岐したデキストランを、パルミチン酸と結合し、そのようにして、複数の利用可能な結合部位を提供する。その結果、このデキストランをビオチンと結合し、そして産物に特異的なアビジン−結合体化抗体で処理する。
【0099】
当然のことながら、本発明の実施態様において、アンカー部分が任意の様式において細胞表面と結合する場合、架橋系を、アンカー部分と捕獲部分との間に使用し得ることが意図される。従って、例えば、アビジン(またはストレプトアビジン)ビオチンリンカー部分は、抗体アンカー部分と捕獲部分を連結し得る。二重特異性抗体系もまた、リンカー部分として作用し得る。
【0100】
産物を分泌する能力を有する細胞を分析し、そして所望される場合に選択するために、捕獲部分を含有するように上記のように改変された細胞が、捕獲された産物を含有する細胞への結合および検出を可能とするのに十分な量で、産物を産生しそして分泌することを可能にする条件下で、インキュベートされる。これらの条件は、当業者に公知であり、そして、とりわけ適切な温度、pH、ならびにインキュベーション培地中の塩濃度、増殖因子濃度、および基質濃度、ならびに気相中の適切な気体濃度が挙げられる。高産生細胞と低産生細胞とを区別することが所望される場合、インキュベーション時間は、細胞により分泌される産物がなお線形の状態である程度である。適切な条件は、経験に決定され得、そしてそのような決定は、当業者の範囲内である。
【0101】
さらに、細胞の分泌を、生物学的修飾因子を使用して、改変(すなわち、アップレギュレート、誘導、または減少)し得る。生物学的修飾因子は、任意の時点で添加され得るが、好ましくは、インキュベーション培地に添加される。あるいは、インキュベーション工程の前に、細胞をこれらの薬剤かまたは細胞で前処理し得る。適切な生物学的修飾因子としては、分子または他の細胞が挙げられるがこれらに限定されない。適切な分子としては、薬物、サイトカイン、低分子、ホルモン、インターロイキンの組み合わせ、レクチン、および他の刺激剤(例えば、PMA、LPS、二重特異性抗体および細胞機能またはタンパク質発現を改変する他の薬剤)が挙げられるが、これらに限定されない。
【0102】
適切な細胞としては、例えば、腫瘍細胞とT細胞との間のような直接的な細胞間相互作用、および例えば、生物学的修飾因子を分泌する他の細胞の近接によって誘導されるような間接的細胞間相互作用が挙げられるが、これらに限定されない。適切な細胞としては、血球細胞、末梢骨髄細胞および種々の細胞株が挙げられるがこれらに限定されない。
【0103】
インキュベーション条件はまた、産物産生細胞から非産生細胞を区別するか、または低産生細胞から高産生細胞を区別するように、産物が他の細胞によって実質的に捕獲されないか、または他の細胞よりかなり少なく捕獲されるものである。一般に、インキュベーション時間は、5分〜10時間の間であり、そしてより通常は、1時間〜5時間の間である。インキュベーション培地は、必要に応じて、産生細胞からの産物の拡散を遅くする物質を含み得る。液体培地中の産物の拡散を阻害し、そして細胞に対して非毒性である物質は、当該分野において公知であり、そして部分的または完全なゲル(例えば、アルギン酸塩、低融点アガロースおよびゼラチンを含む)である種々の物質が挙げられる。培地の粘性または透過性を変化させることにより、産生細胞による異なるサイズの産物の局所的な捕獲を、調節し得る。培地の分子サイズ排除を調節して、反応を最適化し得る。培地の最適な組成は、経験的に決定され得、そして細胞濃度、分泌のレベルおよび産物の分子量、ならびに産物についての捕獲部分の親和性による影響を受ける。そのような決定は、当業者の範囲内である。
【0104】
好ましくは、ゲルを、インキュベーション後に可溶化し、細胞分離技術によって、培地からの細胞または細胞群の単離を可能にする。従って、例えば、ゲルを、βメルカプトエタノールまたはジチオスレイトールのようなスルフヒドリル還元剤によって解離し得るジスルフィド結合によって連結し得るか、またはゲルは、イオン架橋剤(例えば、EDTAのようなキレート剤の添加によって可溶化されるカルシウムイオンを含む)を含有し得る。
【0105】
分泌期の終期において、細胞は、通常冷却され、さらなる分泌を妨げられ、そしてゲルマトリックスがある場合、可溶化される。この順番は、当然のことながら、逆にし得る。架橋に起因して、捕獲部分の添加後に、キャッピングが生じ得るので、キャッピングを減少するインキュベーション工程が、この時点で追加され得る。細胞を例えば、サイトカラシンAまたはBあるいはキャッピングを防ぐ他の任意の適切な物質中で、インキュベートし得る。次に、捕獲された産物を含有する細胞は、標識化部分を用いて標識される。標識化は当業者に公知の任意の方法によって達成され得る。例えば、抗−産物抗体を使用して、直接的にか、または間接的に、産物を含有する細胞を標識し得る。使用する標識は、産物に対する標識部分の付着に基づいて、細胞を分析するか、または分類する系における使用に適切な標識である。
【0106】
他の実施態様において、捕獲産物を含有しない捕獲部分が、検出され得る。このことは、例えば、ネガティブ分離方法(すなわち、産物で高度には飽和していない細胞の検出)を使用することによって、多量に分泌する細胞の単離を可能とする。細胞は、例えば、細胞表面マーカー、細胞型、DNA含量のような細胞のパラメーター、細胞の状態、または捕獲部分の数を認識する他の標識化物質を用いて標識され得る。
【0107】
実際の捕獲部分を数えることは、例えば、細胞の異なる結合体化の可能性に起因してこれら分子の量が変化することを補うために重要であり得る。まれな細胞の単離のためには、産物捕獲系(アンカー部分および捕獲部分を含む)の結合能が減少または増加した細胞を除くことが特に重要であり得る。あるいは、反応は、「一工程反応」において同時に進行し得る。
【0108】
(細胞分析およびセルソーティング)
標識の存在に基づく細胞集団の分析およびセルソーティングは、当該分野において公知の多数の技術によって達成され得る。細胞は、例えば、フローサイトメトリーまたはFACSによって分析または分類され得る。これらの技術は、細胞の1つ以上のパラメーターに従って、分析および分類することを可能にする。通常、1つまたは複数の分泌パラメーターを、細胞の他の測定可能なパラメーター(細胞型、細胞表面マーカー、DNA含量などを含むがこれらに限定されない)と組み合わせて、同時に分析し得る。データを分析し得、そして測定したパラメーターの任意の手法または組み合わせを使用して、細胞を分類し得る。セルソーティングおよび細胞分析方法は、当該分野において公知であり、そして例えば、The Handbook of Experimental Immunology、第1〜4巻、(D.N.Weir、編);Flow Cytometry Cell Sorting(A.Radbruch、編、Springer Verlag、1992);およびCell Separation Methods and Applications(D.RecktenwaldおよびA.Radbruch、編、1997)Marcel Dekker、Inc.N.Y.)に記載される。細胞をまた、顕微鏡技術(例えば、レーザー走査型顕微鏡、蛍光顕微鏡を含む;画像化分析系と組み合わせて使用され得るような技術)を使用して分析し得る。セルソーティングの他の方法としては、例えば、親和性技術を使用するパニングおよび分離(プレート、ビーズおよびカラムのような固体支持体を使用する分類技術を含む)が挙げられる。
【0109】
セルソーティングのいくつかの方法は、磁気分離を利用し、これらの方法のいくつかは、磁気ビーズを利用する。種々の磁気ビーズが多数の供給源(例えば、Dynal(Norway)、Advanced Magnetics(Cambrigde、MA、U.S.A.)、Immuncon(Philadelphia、U.S.A.)、Immunotec(Marseilles、France)、およびMiltenyi Biotec GmbH(Germany)が挙げられる)から利用可能である。
【0110】
好ましい磁気標識方法としては、5〜200nmの範囲のサイズ、好ましくは10〜100nmのサイズのコロイド超常磁性(superparamagnetic)粒子が挙げられる。これらの磁性粒子は、細胞の定量的磁気標識を可能にし、従って、結合した磁気標識の量は、結合した産物の量と比例し、そして磁気分離方法は、産物分泌の異なる量に対して感度がよい。種々の特異性を有するコロイド粒子が、当該分野において公知であり、そして例えば、Miltenyi Biotec GmbHより入手可能である。免疫特異的蛍光または磁気リポソームの使用もまた、捕獲産物の定量的標識のために使用され得る。これらの場合、磁気物質および/または蛍光色素を含有するリポソームは、その表面上で抗体と結合体化し、そして磁気分離を使用して、非産生細胞、低産生細胞と高産生細胞との間の最適な分離を可能にする。
【0111】
磁気分離は、磁力を引き付ける第2の力を組み合わせ、2つの対抗力の異なる強度に基づく分離をすることにより、高効率で、達成され得る。代表的な対抗力は、例えば、磁気分離チャンバー中の分離培地中に混合された磁性液体によって誘導される力、重力、および細胞に対する培地の流速によって誘導される粘性力である。任意の磁気分離方法(好ましくは定量的分離を可能とする磁気分離方法)が使用される。異なる分離方法を組み合わせ得ることもまた、意図される。例えば、磁気セルソーティングは、FACSと組み合わせられ、分離の質を増加し得るか、または複数のパラメーターによる分類を可能にし得る。
【0112】
好ましい技術としては、高勾配磁気選別法(HGMS)(磁場に配置されたチャンバーまたはカラム中に磁性物質を選択的に保持するための手順)が挙げられる。本技術の1つの適用において、産物を磁気粒子に付着させることによって標識する。この付着は、一般に、結合体化のための官能基を提供する、磁気粒子上のコーティングと結合体化される標識部分と、産物との会合を介する。従って、捕獲産物は、磁気「標識」と結合され、次にチャンバーに適用される液体中に懸濁される。チャンバーを横切って供給される磁気勾配の存在下で、磁気標識化標的細胞は、チャンバー中に保持される;チャンバーがマトリックスを含有する場合、その細胞はマトリックスと会合する。磁気標識を有さない細胞、または磁気標識の少量のみを有する細胞は、チャンバーを通過する。
【0113】
次に保持された細胞は、磁場の強度を変えるか、または磁場を取り除くことによってか、あるいは、磁性流体を導入することによって、溶出され得る。捕獲産物についての選択性は、磁気粒子に直接的にかまたは間接的にかのいずれかで、結合体化された標識部分によってか、あるいは一次抗体および一次抗体を認識する磁気粒子の使用によって、供給される。横切って磁場が適用されるチャンバーは、適切な磁場の磁化率の物質のマトリックスがしばしば提供され、マトリックスの表面に近接する容量のチャンバー中において、局所的な高度の磁場勾配を誘導する。このことは、かなり弱く磁化された粒子の保持を可能にする。種々のHGMS系を記載する刊行物が、当該分野において公知であり、そして例えば、米国特許第4,452,773号、同第4,230,685号、PCT公開第WO85/04330号、米国特許第4,770,183号、およびPCT/EP89/01602号が挙げられる;系は、米国特許第5,411,863号;同第5,543,289号;同第5,385,707号;および同第5,693,539号にもまた記載される。これらは、共有(commonly owned)であり、そして本明細書において参考として援用される。
【0114】
さらに、他の実施態様において、プロセスは、捕獲部分(存在する場合)によって捕獲された産物を含有する細胞の標識工程を含む。他の実施態様はまた、細胞集団を分析する工程を包含し、標識された細胞(存在する場合)を検出し、そして所望される場合、標識された細胞(存在する場合)を分類する。
【0115】
(抗原特異的T細胞を検出する診断方法)
本発明は、抗原特異的T細胞を検出するための診断方法を、さらに提供する。これらの方法としては、T細胞を富化した細胞の集団を分析して、抗原特異的T細胞を同定する方法または数える方法、ならびに抗原刺激に応答して、産物を分泌する抗原特異的T細胞の分布を決定する方法が挙げられる。
【0116】
T細胞を富化した細胞の集団を分析して、集団の他の細胞と比較して、ある量の産物を分泌しそして放出する抗原特異的T細胞(ここで産物は、抗原刺激に応答して分泌されそして放出される)を、同定するか、または数える方法は、本発明の方法によって細胞を標識する工程;捕獲産物を標識しない、少なくとも1つのさらなる標識で、細胞を標識する工程;および、さらなる標識と比較した、産物標識の量を検出する工程、を包含する。そのような方法は、例えば、所与の抗原について特異的な細胞の集団の割合を決定する際に、有用である。この方法を使用して、個体の免疫状態に関する情報(アレルゲン、腫瘍またはウイルスに対する免疫応答を評価すること、あるいは自己免疫疾患を検出またはモニターするために自己反応性である、個体中の細胞の集団を評価することを包含する)を提供し得る。
【0117】
(富化された抗原特異的T細胞を使用する処置方法)
本発明は、本発明の富化されたT細胞を使用して、抗原特異的T細胞の集団に関する疾患または状態を処置する方法を提供する。
【0118】
処置方法としては、抗原特異的T細胞集団を、同定し、富化し、そして個体に導入する方法;抗原特異的T細胞の集団を、同定し、富化し、そして個体への導入前にインビトロで拡大する方法;抗原特異的T細胞の集団を同定し、そして個体へ導入される細胞の集団から除去する方法;投与前のエキソビボ遺伝子改変;ならびにサイトカイン発現に従って選択される抗原特異的T細胞の選択が、挙げられる。サイトカイン発現に従って選択される抗原特異的T細胞の例としては、癌、ウイルス(例えば、CMV、EBV)および細菌(例えば、リステリア菌、マイコバクテリア)感染の処置のためのIFN−γまたはTNF−α分泌CD8+ T細胞(細胞傷害性);同じ徴候のため、ならびにまた、アレルギーの抑制および/または逆の調節もしくはアレルギーに対するワクチン接種、TH2関連自己免疫疾患の抑制もしくはこれら自己免疫疾患に対するワクチン接種のためのIFN−γ分泌CD4+ T細胞;TH1抑制のためだけでなく、またTH2関連自己免疫疾患のための、またはこれらの自己免疫疾患に対するワクチン接種(寛容誘導)のための、IL−10またはTGF−β分泌CD4+ T細胞;TH1関連自己免疫疾患の抑制、またはこれらの自己免疫疾患に対するワクチン接種のためのIL−4分泌CD4+ T細胞;ならびに蠕虫感染の処置のためのIL−4またはIL−5分泌CD4+ T細胞、が挙げられるが、これらに限定されない。
【0119】
本発明の方法に従って富化されたT細胞の集団を使用して、種々の障害を処置し得る。これらの障害に含まれるものは、癌である。腫瘍抗原に特異的なT細胞は、本発明の方法を使用して、得られ得る。腫瘍細胞は、個体から得られ得、そしてこれらの細胞は、同一の個体から得られたT細胞とインビトロで共培養され得る。適切な時間の細胞の共培養の後に、腫瘍特異的T細胞を、本発明の方法に従って富化し得る。次にこの富化された集団は、患者に再導入され得る。自系T細胞を使用する抗腫瘍免疫療法のための方法は、当該分野において公知である。例えば、WO97/05239号を参照のこと。
【0120】
あるいは、抗腫瘍免疫療法の処置において使用される細胞は、異質遺伝子型であり得る。異質遺伝子型T細胞を用いる癌の処置の種々の方法は、当該分野において記載され、そして本発明の方法において使用され得る。例えば、PCT公開第WO96/37208号を参照のこと。必要に応じて、異質遺伝子型T細胞は、個体への導入前に活性化され得る。活性化は、生物学的修飾因子、細胞表面マーカーを指向する抗体、または細胞表面レセプターについての、そのリガンドもしくはそのアナログとの接触を介して、生じ得る。
【0121】
本発明の富化されたT細胞集団の別の使用は、例えば、自己免疫疾患、炎症性障害、アレルギーおよび過敏症(例えば、遅延型過敏症および接触過敏症)の処置における免疫調節における使用である。自己反応性細胞の活性を破壊または抑制し得るT細胞は、インビトロで富化され得、必要に応じてインビトロで拡大され得、次に患者に再導入され得る。アレルギー応答の処置において、TH1細胞とTH2細胞との比を変化させ得るか、またはアレルゲン特異的細胞に対して反応性の細胞を、富化し、そして個体に導入し得る。
【0122】
T細胞アネルギーの誘導もまた使用して、同種移植拒絶を、処置、寛解または予防し得、従って、器官移植の結果を改善し、そして患者を組織適合性とし得る組織型(histotype)の範囲を増加し得る。
【0123】
富化されたT細胞集団を含有する組成物をさらに、ワクチンとして使用して、ウイルス感染、自己免疫障害、アレルギー応答、癌、または他の障害のような疾患状態の発生の可能性を防ぐか、または実質的に減少し得るか、あるいは、その後に疾患に感染するか、または疾患に罹患する場合、その疾患の重篤度または持続時間を減少する。
【0124】
細胞組成物は、任意の公知の経路(静脈内投与、非経口的投与、または局所的投与が挙げられるがこれらに限定されない)によって投与され得る。本発明の処置方法において、富化されたT細胞は、個体に投与される。全細胞数、用量数、および1用量あたりの細胞数は、処置される状態に依存する。一般に、約106〜1011の細胞が、約5ml〜1リットルの範囲の容量において投与される。細胞は、単回用量において、または選択された時間間隔にわたり数回の用量において、投与され得る。投与される細胞のうち、好ましくは少なくとも約10%、より好ましくは少なくとも約20%、より好ましくは少なくとも約50%が、産物を分泌する抗原特異的T細胞である。
【0125】
(キット)
所望される産物の分泌細胞の検出において使用される試薬が、都合のよいように、キットの形態でパッケージングされ得ることが意図される。このキットは、例えば、必要に応じて、調製されたゼラチン状の細胞培養培地において使用するための1つ以上の物質、所望の分泌産物の産生のための細胞インキュベーションのために使用される培地;アンカー部分および捕獲部分から構成される産物捕獲系;標識部分;ならびに試薬の使用のための指示書を含む。全ての試薬は、適切な容器中にパッケージングされる。
【0126】
キットはまた、以下を含むように処方され得る。この場合、全ての試薬は、好ましくは、細胞が添加される1つのバイアル中に配置される。特定の細胞表面構造またはアンカー部分、および産物について二重特異性である、少なくとも1つの抗体。少なくとも1つの標識部分、および必要に応じて、生物学的修飾因子。
【0127】
必要に応じて、キットは、生理学的に受容可能なキャリアを含有し得る。そのような緩衝液は、当該分野において公知であり、そしてBSA含有PBSまたはBSA非含有PBS、等張生理食塩水、細胞培養培地、および特定の細胞型に必要とされる任意の特定の培地が挙げられるが、これらに限定されない。交差標識を減少し、そして細胞の周囲の局所的な産物濃度を増加する緩衝液が使用され得る。緩衝液は、粘性を増加するか、または透過性を減少する薬剤を含み得る。適切な薬剤は、本明細書において記載される。培地の粘性は、当該分野において公知の任意の方法(生理学的に受容可能な緩衝液中への溶解、熱溶解、EDTA溶解、および酵素溶解が挙げられるが、これらに限定されない)によって分析前に減少され得る。添加培地の非存在下で、培地中に既に懸濁されている細胞は、直接バイアルに添加され得る。適切な細胞懸濁液としては、細胞株および生物学的サンプルが挙げられるがこれらに限定されない。生物学的サンプルとしては、血液、尿および血漿が挙げられるが、これらに限定されない。
【0128】
さらなる構造物が、非結合産物を捕獲して、細胞の交差夾雑を減少し、それにより産生細胞から産物が拡散していくことを減少するために、添加され得る。これらには、ゲルエレメント、ビーズ、磁気ビーズ、およびポリマーに固定化された抗−産物抗体が挙げられるが、これらに限定されない。
【0129】
生物学的修飾因子もまた、緩衝液または培地に添加され、特異的分泌を誘導し得る。
【0130】
抗体(磁気的に、または蛍光的に標識された)のようなさらなる標識部分もまた、存在し得る。これらには、細胞型を同定する抗細胞表面マーカー抗体、死細胞を標識するヨウ化プロピジウム、および特定の細胞型を標識する磁気ビーズが挙げられるが、これらに限定されない。
【0131】
この実施態様において、すべての物質が、バイアルのような単一の容器中に配置され、そして細胞サンプルを添加し得る。内容物をインキュベートし、産物の分泌、ならびにその後の産物の捕獲および標識化部分の産物への結合を可能にする。次に、産物を分泌する細胞、および産物を結合する細胞が、捕獲産物の存在、非存在または量に基づいて、分類および/または分析され得る。分類は、当該分野において公知の任意の方法によってなされ得る。この方法には、単純な希釈、赤血球溶解、遠心分離−洗浄工程、磁気分離、FACSおよびフィコール分離が挙げられるが、これらに限定されない。細胞の分析は、種々の方法によって実行され得る。これらの方法には、FACS、画像化分析、細胞学的標識、および免疫アッセイが挙げられるが、これらに限定されない。
【0132】
以下の実施例は、単に本発明の説明の目的ために提供され、そして本発明の範囲を限定するために提供されない。本発明の開示を考慮すると、特許請求の範囲内の多数の実施態様が、当業者にとって明らかである。
【0133】
(実施例1)
末梢血単核細胞(PBMC)を、100U/mlのペニシリン、0.1mg/mlのストレプトマイシン、0.3mg/mlのグルタミン、10mMの2−メルカプトエタノール、および10%ヒトAB型血清(Sigma、St.Louis、MO)を含む完全RPMI 1640(Gibco BRL、Grand Island、NY)中で、細胞濃度2×106細胞/mlで培養した。インフルエンザウイルスマトリックスタンパク質由来のペプチドM1 58−66(GILGFVFTL;Neosystem、Strasbourg、France)を、1μMの最終濃度まで添加した。コントロール細胞をペプチドなしで培養した。
【0134】
細胞を、7.5%CO2含有雰囲気下で、37℃でインキュベートした。5時間および30分後、細胞を遠心分離により採集した。細胞を、細胞濃度5×107細胞/mlで、完全RPMI 1640中で、抗ヒトCD45mAb 5B1に結合した抗ヒトインターフェロンγ(IFN−γ)モノクローナル抗体(mAb)4SB3(30μg/ml)と共に、8℃で7分間インキュベートした。次いで、細胞を、10%FCSを含む完全RPMI 1640を用いて、2×106細胞/mlに希釈し、そして37℃で45分間インキュベートした。次いで、細胞をペレットにし、そしてフィコエリトリン(PE)結合体化抗ヒトインターフェロンγ(IFN−γ)mAb NIB42(4μg/ml)およびFITC標識化抗CD8mAbと共に、PBS/BSA/EDTA溶液(0.05%BSAおよび2mM EDTA)中で、4℃で10分間インキュベートした。次いで、細胞をPBS/BSA/EDTA中で洗浄し、そしてMicroBeads(Miltenyi Biotec)に結合体化したマウス抗PE mAb80−5で、PBS/BSA/EDTA中、8℃で15分間標識した。細胞を洗浄し、そして500μlのPBS/BSA/EDTA中に再懸濁した。
【0135】
IFN−γ分泌細胞を、磁気細胞分離システムMACSで富化させた。磁気で標識された細胞懸濁液を、MiniMACS分離ユニット中のMiniMACS分離カラム上にピペットし、その細胞懸濁液を通過させ、そしてそのカラムを500μlの緩衝液で3回洗浄した。この溶出液を負の画分(N1)として採集した。このカラムを分離機から取り出し、そして適切な管上に配置した。1mlの緩衝液をカラムの一番上にピペットし、そして磁気で標識された細胞を、プランジャーを用いて流し、そして二回目のMiniMACS分離に供した。
【0136】
もとの細胞(すなわち、MACS分離の前)、負の細胞画分(一回目および二回目のMACS分離のもの、N1およびN2とそれぞれ称する)および二回目のMACS分離の正の細胞画分(P2)を、フローサイトメトリーで分析した。FACScanおよびCELLQuest 研究用ソフトウェア(Becton Dickinson、Mountain View、CA)をフローサイトメトリー分析に使用した。死んだ細胞および細胞細片を散乱特性に従って除去し、そしてヨウ化プロピジウム(PI;0.3μg/ml)で染色した。
【0137】
結果を図1A〜Pに示す。ドットプロットA〜Hは、ペプチドなしで培養したコントロール細胞の分析を示し、一方、プロットI〜Pは、ペプチド刺激細胞の分析を示す。ドットプロットは、出発細胞集団(AおよびI)および富化細胞集団(CおよびK)の散乱特性を示し;そして出発細胞集団(BおよびJ)ならびに富化細胞集団(DおよびL)のPI蛍光対PE蛍光を示す。
【0138】
ドットプロットE〜HおよびM〜Pは、もとの細胞画分(EおよびM)、一回目の負の細胞画分(FおよびN)、二回目の負の細胞画分(GおよびO)、および最終の正の細胞画分(HおよびP)におけるゲート細胞の抗CD8−FITC染色対抗IFN−γ−PE染色を示す。
【0139】
コントロール細胞集団中で、CD8+IFN−γ+細胞は生存細胞(図1H)中11%まで富化したが、一方ペプチド刺激細胞集団中では、CD8+IFN−γ+細胞は40%まで富化した(図1P)。3.5×107コントロール細胞の出発集団から、約600のCD8+IFN−γ+細胞を単離し、3.5×107ペプチド刺激細胞の出発集団から単離した4100のCD8+IFN−γ+細胞と比較した。
【0140】
PE標識された抗IFN−γで明るく染色されたCD8細胞は、CD19+B細胞(識別試薬(おそらくPE)に特異的な最も可能生のあるB細胞)であった。これらの細胞は、ペプチドで刺激した細胞と比較して、コントロール細胞から同じ程度まで富化された。
【0141】
PE標識された抗IFN−γでうす暗く染色されたCD8-細胞(CD8+IFN−γ+細胞のような)もまた、ペプチドで刺激した細胞と比較して、コントロール細胞から同じ程度まで富化された。このような細胞は、部分的にCD4およびCD56について染色し、従って、IFN−γを分泌する最も可能性のあるTヘルパー細胞またはNK細胞である。
【0142】
従って、インビトロの内部抗原特異的刺激によらない(CD4+)Tヘルパー細胞、(CD8+)細胞傷害性T細胞および(CD56+)NK細胞による基底レベルのIFN−γの分泌が存在し、これは、血液サンプリング時に進行中の免疫応答において、インビボですでに誘導されたIFN−γ分泌を最も可能に反映する。
【0143】
しかし、HLAクラスI制限インフルエンザペプチドM1 58−66で刺激されることによって誘導されるIFN−γ+分泌CD8+細胞は、このバックグラウンドレベルを超えて有意に富化された;従って、ペプチド刺激された細胞から富化された、大部分のCD8+IFN−γ細胞は、ペプチド特異的T細胞である。富化された細胞の特異性は、さらにVβ17 TCR(これは、M1 58−66特異的細胞傷害性T細胞中の保存されたT細胞レセプター(TCR)セグメントである)の存在についての染色について確認された。Lehnerら(1995)J.Exp.Med.181:79−91;およびLalvaniら(1997)J.Exp.Med.186:859−865。コントロール細胞から単離されたIFN−γ+細胞ではほとんど発現しないが、ペプチド刺激された細胞から単離されたIFN−γ+細胞のほとんどがVβ17+TCRを発現する。
【0144】
(実施例2)
末梢血単核細胞(PBMC)を、100U/mlのペニシリン、0.1mg/mlのストレプトマイシン、0.3mg/mlのグルタミン、10mMの2−ME、および10%ヒトAB型血清(Sigma、St.Louis、MO)を含む完全RPMI 1640(Gibco BRL、Grand Island、NY)中で、2×106細胞/mlで培養した。インフルエンザウイルスマトリックスタンパク質由来のペプチドM1 58−66(GILGFVFTL;Neosystem、Strasbourg、France)を、1μMの最終濃度に添加した。コントロール細胞をペプチドなしで培養した。
【0145】
5時間30分後、細胞を遠心分離により採集した。細胞を、5×107細胞/mlで、完全RPIM 1640中で、抗ヒトCD45mAB 5B1(30μg/ml)に結合した抗ヒトインターフェロンγ(IFN−γ)mAb 4SB3と共に、8℃で7分間インキュベートした。次いで、細胞を、10%FCSを含む完全RPMI 1640を用いて、2×106細胞/mlに希釈し、そして37℃で45分間インキュベートした。次いで、細胞をスピンダウンし、そしてフィコエリトリン(PE)結合体化抗ヒトIFN−γ mAb NIB42(4μg/ml)およびFITC標識化抗CD8と共に、PBS/BSA/EDTA溶液中で、4℃で10分間インキュベートした。次いで、細胞をPBS/BSA/EDTA中で洗浄し、そしてマウス抗PE mAb80−5結合体化MicroBeads(Miltenyi Biotec)を用いて、PBS/BSA/EDTA中、8℃で15分間標識した。細胞を洗浄し、そして500μlのPBS/BSA/EDTA中に再懸濁した。
【0146】
IFN−γ分泌細胞を、磁気細胞分離システムMACSで富化させた。磁気で標識された細胞懸濁液を、MiniMACS分離ユニット中のMiniMACS分離カラムの一番上にピペットし、細胞懸濁液を通過させ、そしてカラムを500μlの緩衝液で3回洗浄した。この溶出液を負の画分として採集した。このカラムを分離器から取り出し、そして適切な管上に配置した。1mlの緩衝液をカラムの一番上にピペットし、そして磁気で標識された細胞を、プランジャーを用いて還流し、そして二回目のMiniMACS分離に供した。
【0147】
もとの細胞(すなわち、MACS分離の前)、負の細胞画分(一回目および二回目のMACS分離のもの)および二回目のMACS分離の正の細胞画分を、フローサイトメトリーで分析した。FACScanおよびCELLQuest研究用ソフトウェア(Becton Dickinson、Mountain View、CA)をフローサイトメトリーに使用した。死んだ細胞および細胞細片を、実施例1に示すように、散乱特性に従って除去し、そしてヨウ化プロピジウム(PI;0.3μg/ml)で染色した。この結果を図2に示す。
【0148】
ドットプロット2A〜Gはペプチドなしで培養したコントロール細胞の分析を示し、プロット2J〜Rはペプチド刺激細胞の分析を示す。
【0149】
ドットプロット2A〜Dおよび2J〜Mは、もとの細胞画分(A、J)、一回目の負の細胞画分(B,K)、二回目の負の細胞画分(C、L)および最終の正の細胞画分(D、M)における、ゲート細胞のFITC標識された抗CD8対PE標識された抗IFN−γ染色を示す。
【0150】
コントロール細胞において、CD8+IFN−γ細胞を生存細胞の8.2%まで富化した(2D)。ペプチド刺激細胞から、CD8+IFN−γ細胞を41.6%(2M)まで富化した。6.1×107のコントロール細胞から、約1360のCD8+IFN−γ細胞を単離して6.9×107のペプチド刺激細胞をからの11700のCD8+IFN−γ細胞と比較した。
【0151】
HLAクラスI制限インフルエンザペプチドM1 58−66を用いた刺激により誘導されるIFN−γ+分泌CD8+細胞は、有意にバックグラウンドレベルより上に、有意に富化された(すなわち、ペプチド刺激細胞から富化された大部分のCD8+IFN−γ+細胞は、ペプチド特異的T細胞であるはずである)。富化された細胞の特異性は、Vβ17 TCR(これは、M1 58−66特異的細胞傷害性T細胞中の保存されたT細胞レセプターセグメントである)に対する染色によりさらに確認された(Lehner 1995;Lalvani 1997)。ペプチド刺激細胞から単離されたIFN−γ+細胞の中でのみ、大部分がVβ17+TCRを発現し、コントロール細胞から単離されたIFN−γ+細胞の大部分では、発現しない(2F対2O)
以下の実施例は、適切な抗原特異的刺激であるCD4+およびCD8+リンパ球は、サイトカインを迅速に発現することを示す。この技術は、ここで、HLA−A0201制限インフルエンアマトリックスタンパク質(FLU)ペプチド58−66特異的CD8+細胞傷害性Tリンパ球(CTL)、インフルエンザAウイルスおよび組換え破傷風毒素C(rTT.C)フラグメント特異的Tヘルパー1型(Th1)細胞、および破傷風トキソイド(TT)特異的Tヘルパー2型(Th2)細胞について実証される。
【0152】
(実施例3)
(実施例4〜8の材料および方法)
(細胞およびエキソビボ刺激)
バフィーコートをthe Institute for Trasnfusions medicine,Hospital Merheim,Cologne,Germanyより入手した。そして必要である場合、HLA型に基づいて選択した。PBMCを、標準的なFicoll−Pacque(Pharmacia,Uppsala,Sweden)密度勾配遠心分離法により調製し、そしてPBS中で2回洗浄し、そして10%(wt/vol)ヒトAB血清(Boehringer Ingelheim,Ingelheim,Germany)、1mMのL−アラニル−グルタミン(Life Technologies)、100U/mlのペニシリン/ストレプトマイシン(Life Technologies)、0.05mMの2−メルカプトエタノール(Life Technologies)および1mMのピルビン酸ナトリウム(Life Technologies)を補充したRPMI 1640(Life Technologies、Paisley,UK)からなる細胞培養培地中に、1mlあたり2×106細胞の細胞濃度で再懸濁した。12.5mlの細胞懸濁液を、100×20mmの組織培養ディッシュ(Sarstedt,Newton,MA)中に配置し、そしてFLU 58−66ペプチド(Neosystems,Strasbourg,France)を最終濃度1μMまで添加し、そして精製インフルエンザAウイルス調製物(Biodesign,Kennebunk,ME)を最終濃度μg/mlまで添加し、rTT.C(Boehringer Mannheim,Manneheim,Germany)を最終濃度7μg/mlまで添加し、そして精製TT(Statens Serum Institut,Copenhagen,Denmark)を最終濃度1μg/mlまで添加した。細胞を加湿7.5%CO2雰囲気下、37℃で5〜10時間インキュベートした。
【0153】
(細胞親和性マトリックスによる分泌サイトカインの捕獲)
CD45に対するAb−Ab結合体およびIL−4もしくはIFN−γのいずれかを、標準的なタンパク質カップリング技術により生成した。Aslamら(1998)Bioconjugation,Macmillan Reference Ltd.,London。エキソビボ刺激の後、ディスポーサブル細胞スクレーパ(Costar,Cambridge,MA)を用いて細胞を採集し、そして1mlあたり50μgのAb−Ab結合体で、氷冷培地中で1mlあたり108細胞の細胞濃度で、7分間標識した。次いで、細胞を、最終細胞濃度が1mlあたり2×106細胞まで培地で希釈し、そして加湿7.5%CO2雰囲気下で37℃で45分間分泌させた。
【0154】
(サイトカイン分泌細胞の磁気的富化および検出)
サイトカイン捕獲期間の後、細胞を再び採集し、リン酸緩衝化生理食塩水(0.5%(w/v)ウシ血清アルブミンおよび5mM EDTA(緩衝液)を含む)中に1mlあたり108細胞の細胞濃度で再懸濁し、そして5μg/mlの抗IFN−γ−PEまたは抗IL−4−PEをそれぞれ用いて+4℃で10分間染色した。細胞を緩衝液(300×g、10分)で洗浄し、400μlの緩衝液中に再懸濁し、そして100μlの抗PE Ab−マイクロビース(Miltenyi Biotec,Bergisch,Gladbach,Germany)を用いて、+4℃で15分間、磁気的に標識した。洗浄後、この細胞をMS+カラム上に適用し、そしてMiniMACS磁石(Miltenyi Biotech)中に配置した。このカラムを緩衝液でリンスし、そしてより高い富化速度を達成するために磁界からカラムを除去した後、保持された細胞をカラムから溶出し、最初のカラムから溶出された細胞を別のMS+カラムに適用し、そして磁気分離を繰り返した。細胞サンプルを、FACScaliburフローサイトメーター(Becton Dickinson,San Jose、CA)で、CellQuestソフトウェアパッケージを用いて分析した。
【0155】
(サイトカイン分泌細胞の磁気富化および検出)
サイトカイン分泌細胞の検出、計数および表現型化について、以下の試薬を使用した:抗IFN−γ−CD45(抗IFN−γ、クローン4SB3;CD45、クローン5B1、W.Knapp,Vienna,Austria)、抗IFN−γ−PE(クローン45−15)、抗IL−4−CD45(抗IL−4、クローン1 A6−10;CD45、クローン5B1、W.Knapp,Vienna,Austria)、抗IL−4−PE(クローン7A3−3)CD8−Cy5(クローンBM135/80、Behring Diagnostics,Marburg,Germany)、CD4−Cy5(クローンM−T321、Behring)、CD4−FITC(クローンSK3、Becton Dickinson)、CD27−FITC(クローンM−T271、Pharmingen,San Diego,CA)、CD28−FITC(クローンCD28.2,Pharmingen)CD57−FITC(クローンHNK−1、Becton Dickinson)、抗Vβ17.FITC(クローンE17.5F3.15.13、Coulter−Imunotech,Marseille,France)。Meagerら(1984)Interferon Res.4:619〜625;Alkanら(1994)J.Immunoassay 15:217〜225;およびBirdら(1991)Cytokine 3:562〜567。
【0156】
(細胞傷害性活性アッセイ)
富化されたサイトカイン分泌細胞の細胞傷害性活性を、以前に記載されたフローサイトメトリーに基づくアッセイを用いて分析した。Mattisら(1997)J.Immunol.Met.204:135〜142。簡単には、1×106HLA−A.1+T2細胞を、1mlあたり4μgのグリーン蛍光色素DiO(Molecular Probes,Eugene,OR)を用いて、5mM EDTAおよび3%ウシ胎仔血清を含むリン酸緩衝化生理食塩水中で、37℃で、45分間標識した。細胞を緩衝液で3回洗浄し、細胞培養培地中に再懸濁し、そして1μM Flu58−66ペプチドまたはMelan A/MART1 27−35ペプチド(Bachem,Heidelberg,Germany)を、加湿7.5%CO2雰囲気下で37℃で一晩、ロードした。富化したサイトカイン分泌細胞を、組換えヒトIL−2(Peprotech,London,U.K.)存在下の組織培養中で18日間増殖させた。増殖させたサイトカイン分泌細胞およびペプチドロードしたDiO標識HLA−A2.1+T2細胞を、1:1の比率で、加湿7.5%CO2雰囲気下で37℃で、16時間同時培養した。培養期間後、細胞を採集し、そしてフローサイトメトリーによって分析した。生存しているDio標識化T2細胞と死んだDio標識化T2細胞との間の区別を可能にするために、サンプルを赤色蛍光排除色素ヨウ化プロピジウムで対比染色した。
【0157】
(実施例4)
合成ペプチドパルスAPCまたは天然抗原パルスAPCでの短期間の抗原性再刺激の後のIFN−γのようなエフェクターサイトカインを分泌する能力は、記憶/エフェクターCD4+(Th1型)およびCD8+T細胞の代表的な特徴である。Salmonら(1989)J.Immunol.143:907〜912;およびHamaanら(1997)186:1407〜1418。IFN−γの抗原誘導化分泌および細胞親和性マトリックス技術に基づく末梢血から直接的に、低頻度の記憶/エフェクター抗原特異的CD4+およびCD8+T細胞を単離するために、HLAが一致した成人の健康な血液ドナー由来の末梢血単核細胞(PBMC)を、5〜6時間、以下を用いて刺激した:(a)HLA−A0201制限FLUペプチド58−66、(b)精製インフルエンザAウイルス調製物、および(c)rTT.C。刺激期間後、IFN−γに対する親和性マトリックスを、CD45およびIFN−γに対する抗体(Ab)−Ab結合体を用いて細胞表面上で作製し、そして細胞を45分間、培養物中でIFN−γを分泌させた。次いで、分泌細胞の親和性マトリックスに再配置させたIFN−γを、フィコエリトリン(PE)結合体化IFN−γ特異的Abで染色し、そしてPE標識化細胞を抗PE Abマイクロビーズを用いてMACSにより富化した。Brosterhusら、10th International Congress in Immunology,New Delhi,India,1〜6 1998年11月、1469〜1473頁もまた参照のこと。
【0158】
非刺激コントロールサンプルと比較して、有意により高い割合のIFN−γ分泌CD8+細胞は、FLU58−66ペプチド刺激サンプル(図3A:38.3%対13.7%)中での富化後、検出可能であり、そして有意により高い割合のIFN−γ分泌CD4+細胞は、それぞれ、インフルエンザAウイルス調製物で刺激したサンプル(図3B:35.5%対1.1%)およびrTT.Cで刺激したサンプル(図3C:6.1%対0.3%)中での富化後、検出可能であった。富化IFN−γ分泌T細胞の絶対数および全PBMC中のそれらの頻度で見る場合、刺激サンプルと非刺激サンプルとの間の差異は、さらにより顕著である:(a)12,500のIFN−γ分泌CD8+T細胞を5.3×107FLU58−66ペプチド刺激PBMCから単離し(頻度4,200分の1)、そして1370のIFN−γ分泌細胞CD8+T細胞を、5.1×107非刺激PBMCから単離した(頻度:37,000分の1);(b)351のIFN−γ分泌CD4+T細胞を5×106のインフルエンザAウイルス刺激PBMCから単離し(頻度14,000分の1)、そして4個のIFN−γ分泌CD4+T細胞を5.0×106の非刺激PBMCから単離した(頻度1,250,000分の1);ならびに(c)132のIFN−γ分泌CD4+T細胞を1.8×107のrTT.C刺激PBMCから単離し(頻度:136,000分の1)、そして7個のIFN−γ分泌CD4+T細胞を1.9×107の非刺激PBMCから単離した(頻度:約2,710,000分の1)。これらの実験結果を考慮すると、10-6以下の頻度で存在するIFN−γ分泌T細胞が、本発明者らの技術で検出され得る。
【0159】
(実施例5)
記憶型CD8+T細胞およびエフェクター型CD8+T細胞の両方は、IFN−γを分泌し得る。Hamannら(1997)。FLU58−66ペプチド特異的CD8+T細胞の表現型を決定するために、FLU58−66ペプチド刺激サンプルおよびコントロールサンプル由来の富化IFN−γ分泌CD8+T細胞を、記憶型CD8+T細胞とエフェクター型CD8+T細胞との間を区別し得る、白血球表面マーカーのパネルの発現について三色免疫蛍光法によって分析した。Hamannら。図2に示すように、大部分のFLU58−66ペプチド特異的CD8+T細胞は、(1997)記憶表現型と一致するCD27+、CD28+およびCD57-であった。一方、FLU58−66ペプチドと独立して単離された大部分のIFN−γ分泌CD8+Tは、エフェクター表現型と一致するCD27-、CD28-、CD57+であった。後者は、インビボで誘導されてIFN−γを分泌し得、従って、進行中の免疫応答を反映したかもしれない。
【0160】
コントロールサンプル由来の2.2%未満のIFN−γ分泌CD8+T細胞と比較して、FLU58−66ペプチド刺激サンプル由来の、54.8%より多いIFN−γ分泌CD8+T細胞が、Vβ17TCR鎖を発現した(図4)。このことは、HLA−A0201制限FLUペプチド58−66特異的CD8+T細胞の最初はクローン化CTLにおいて、Vβ17TCR鎖の使用への偏向、後にPBMCにおいても、FLU58−66ペプチドをロードしたHLA−A2.1分子の蛍光テトラマーの使用への偏向を示す以前の報告を確認する。Lehnerら(1995)J.Exp.Med.181:79〜91;およびDunbarら(1998)。
【0161】
(実施例6)
FLU58−66ペプチド刺激PBMC由来の富化IFN−γ分泌CD8+T細胞の特異性をさらに確認するために、そしてそれらの細胞傷害性活性を研究するために、これらの細胞をIL−2の存在下の組織培養中で18日間増殖させ、次いで1:1のエフェクター:標的の割合でのCTL活性についてアッセイした。図5に示すように、標的細胞をFLU58−66ペプチドでロードした場合、有意な殺傷が観察されたが、標的細胞をコントロールペプチド(Melan A/MART 1 27−35)でロードした場合、観察されなかった。
【0162】
(実施例7)
49のHLA−A2+個体由来のPBMCを、FLU58−66ペプチドを伴ってか、または伴わずに培養し、そして実施例3に記載されるようにIFN−γ分泌細胞についての富化手順に供した。45の場合において、コントロールサンプルと比較して、平均約80倍より多いIFN−γ分泌CD8+T細胞を、FLU58−66ペプチド刺激サンプルから単離した。3つの場合においてのみ、有意な差異は、両サンプル間で検出されなかった。PBMC間のFLU58−66ペプチド特異的CD8+T細胞の平均頻度は、FLU58−66ペプチド刺激化サンプルの頻度からコントロールサンプルの頻度を減算することによって決定されるように、30,000分の1(600,000分の1〜1000分の1との間の範囲)であった。これらの結果は、以前の報告と完全に一致する。FLU58−66ペプチド特異的CD8+T細胞の頻度を、単一細胞のIFN−γの放出またはFLU58−66ペプチドロードしたHLA−A2.1分子の四量体についての酵素結合免疫スポット(ELISPOT)アッセイを用いて決定した。Lalvaniら(1997;およびDunbarら(1998))。
【0163】
(実施例8)
本発明者らのアプローチが生存している抗原特異的Th2型CD4+T細胞を単離することを実証するために、PBMCを精製したTTで刺激し、そしてIL−4分泌CD4+T細胞を、CD45およびIL−4に対するAb−Ab結合体を用いて単離した。10時間のTT刺激の後、150のIL−4分泌CD4+T細胞を、2.2×107のPBMCから単離し得、純度は6,89%であった(図6)。これは、全CD4+T細胞におけるTT特異的Th2細胞の頻度である、94,000分の1に対応する。TTなしのコントロール培養物におけるIL−4分泌CD4+T細胞の頻度は、約10倍低かった。
【0164】
本明細書中で引用される全ての参考文献、前出および後出の両方は、本明細書により本明細書中に援用される。前述の発明が、明確さおよび理解する目的のための例示および実施例によりいくらか詳細に記載されているが、特定の変更および改変が実施され得ることは当業者に明らかである。従って、本説明および実施例は、添付の特許請求の範囲により表される本発明の範囲を制限するよう解釈されるべきでない。
【図面の簡単な説明】
【図1】図1A−Pは、実施例1で記載した分離プロトコールに供した細胞の分析を示すFACSプロットである。A−Hはペプチドなしで培養したコントロール細胞の分析を示す。I−Pはペプチド刺激細胞の分析を示す。A、C、IおよびKは、開始時の細胞集団(AおよびI)ならびに富化細胞集団(CおよびK)の分散の特性を示す。B、D、JおよびLは、開始時の細胞集団(BおよびJ)ならびに富化細胞集団(DおよびL)のPI対PE染色のプロフィールを示す。プロットE−HおよびM−Pは、開始時の細胞集団(EおよびM)、最初の陰性集団(FおよびN)、2番目の陰性集団(GおよびO)および富化細胞集団(HおよびP)の、FITC標識抗CD8対PE標識抗IFN−γ染色を示す。
【図2】図2A−Nは、実施例2で記載した分離プロトコールに供した細胞の分析を示すFACSプロットである。A−Gは、ペプチドなしで培養したコントロール細胞の分析を示す。N−Rは、ペプチド刺激細胞の分析を示す。A−DおよびH−Kは、開始時の細胞集団(AおよびJ)、最初の陰性集団(BおよびI)、2番目の陰性集団(CおよびJ)および富化細胞集団(DおよびK)の、FITC標識抗CD8対PE標識抗IFN−γ染色を示す。FおよびMは、富化細胞集団のVβ17TCRに対する染色を示す。
【図3】図3は、生抗原特異的CD4+およびCD8+T細胞の、IFN−γの分泌に基づいた富化および検出を示す一連のドットプロットである。ドットプロットは、HLA−A0201−制限FLU58−66ペプチドと共に(A、B)または無しで(C、D)、精製インフルエンザAウイルス調製物(と共に(E、F)無しで(G、H))、およびrTT.C(と共に(I、J)無しで(K、L))で刺激した健常な成人ドナー由来のPBMCの、IFN−γ分泌細胞の磁気富化前(A、C、E、G、I、K)および後(B、D、F、H、J、L)の、CD8−Cy5対抗IFN−γ−PE(A−D)またはCD4−Cy5対抗IFN−γ−PE(E−L)染色を示す。生リンパ球は、光散乱特性およびヨウ化プロピジウム排除によって制御された(gated)。
【図4】図4は、富化Flu58−66ペプチド特異的CD8+T細胞の表現型分析を示す一連のドットプロットである。FLU58−66ペプチド刺激PBMC(A、B、E、F)由来、およびコントロールとして非刺激PBMC(C、D、G、H)由来の富化IFN−γ分泌CD8+T細胞を、抗IFN−γ−PEで染色、およびCD27、CD28、CD57およびTCR Vβ17鎖に対するFITC結合抗体で対比染色した。生CD8+T細胞を制御(gating)するために、光散乱特性、ヨウ化プロピジウムおよびCD8−Cy5染色を使用した。
【図5】図5は、富化および増殖させたFlu58−66ペプチド特異的T細胞の細胞溶解活性を示すグラフである。FLU58−66ペプチド刺激PBMC由来のIFN−γ分泌CD8+T細胞を、IL−2存在下の組織培養で18日間増殖させ、次いでCTL活性アッセイに関してアッセイした。図は、Flu58−66ペプチドか、または陰性コントロールペプチドMelan A/MART 1 27−35のいずれかを用いて適用された溶解したHLA−A2.1+T2細胞の割合を示す。
【図6】図6は、TT特異的IL−4分泌CD4+T細胞の単離および検出を示す一連のドットプロットである。ドットプロットは、IL−4分泌細胞の磁気富化有り(A、C)または無し(B、D)で刺激した健常な成人ドナー由来のPBMCの、CD4−Cy5対抗IL−4−PE染色を示す。生リンパ球は、光散乱特性およびヨウ化プロピジウム排除によって制御された(gated)。
Claims (14)
- 抗原特異的T細胞を、該細胞によって分泌されそして放出された産物で標識する方法であって、ここで、該産物は抗原刺激に応答して分泌され、該方法は、以下の工程:
a)該抗原特異的T細胞を、少なくとも1つのT細胞の抗原特異的刺激を誘発するに有効な条件下にて、少なくとも1つの抗原に暴露し、該刺激されたT細胞による少なくとも1つの産物の発現を可能にする工程であって、ここで、該産物は、抗原刺激に応答して分泌される、工程;
b)該細胞の表面を、該産物に特異的な捕獲部分を含むように改変する工程であって、その結果、該捕獲部分が該細胞表面に結合される、工程;および
c)該細胞を、該産物が分泌され、放出され、そして該捕獲部分に特異的に結合される条件下で培養して、それによって該産物分泌細胞を標識する工程;
を包含し、ここで、工程(a)および(b)は、任意の順序で行われ得、そして該抗原特異的T細胞は、細胞の混合集団中に存在する、方法。 - 前記捕獲部分が、抗体または抗体の抗原結合フラグメントである、請求項1に記載の方法。
- 前記抗体が二重特異性抗体である、請求項1に記載の方法。
- 前記捕獲部分が、以下の(a)〜(d)を通じて前記細胞の表面に結合される、請求項1〜3のいずれか1項に記載の方法:
(a)該捕獲部分に結合される脂質アンカー;
(b)該捕獲部分に結合される抗体またはその抗原結合フラグメント;
(c)該細胞表面上の成分への該捕獲部分の直接的な化学結合;または
(d)該細胞への該抗体の特異的結合。 - 前記脂質アンカーが、連結部分を介して前記捕獲部分に結合される、請求項4に記載の方法。
- 前記抗体または抗体の抗原結合フラグメントが、結合剤を介して前記捕獲部分に結合される、請求項4に記載の方法。
- 前記細胞の表面が、工程(b)において、該細胞を、細胞表面分子および前記産物に特異的な結合部位を有する少なくとも1つの二重特異性抗体と結合させることによって改変される、請求項1〜3のいずれか1項に記載の方法。
- 前記細胞表面分子が、CD2、CD3、CD4、CD5、CD8、CD11b、CD26、CD27、CD28、CD29、CD30、CD31、CD38、CD40L、CD45RO、CD45RA、LAG3、T1/ST2、SLAM、クラスI MHC分子、クラスII MHC分子、T細胞抗原レセプター、またはβ2−ミクログロブリンである、請求項7に記載の方法。
- 前記産物を標識部分で標識する工程をさらに包含する、請求項1〜8のいずれか1項に記載の方法。
- 前記標識部分が、前記産物に特異的な抗体である、請求項9に記載の方法。
- 前記標識部分が、発蛍光団、放射性同位元素、発色団、磁化可能部分で直接的もしくは間接的に標識されるか、または磁気粒子を含むかのいずれかである、請求項9または10に記載の方法。
- 前記標識部分が、直径が5〜200nmのコロイド状磁気粒子を含む、請求項10に記載の方法。
- 前記細胞が、高粘性培地またはゲル形成培地で培養される、請求項1〜12のいずれか1項に記載の方法。
- 前記細胞を、該細胞が標識される程度に従って分離する工程をさらに包含する、請求項1〜13のいずれか1項に記載の方法。
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US8513698P | 1998-05-11 | 1998-05-11 | |
| US60/085,136 | 1998-05-11 | ||
| PCT/US1999/010200 WO1999058977A1 (en) | 1998-05-11 | 1999-05-10 | Method of direct selection of antigen-specific t cells |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| JP2002514765A JP2002514765A (ja) | 2002-05-21 |
| JP2002514765A5 JP2002514765A5 (ja) | 2006-06-22 |
| JP4601166B2 true JP4601166B2 (ja) | 2010-12-22 |
Family
ID=22189678
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| JP2000548729A Expired - Lifetime JP4601166B2 (ja) | 1998-05-11 | 1999-05-10 | 抗原特異的t細胞の直接的選択方法 |
Country Status (10)
| Country | Link |
|---|---|
| US (2) | US6576428B1 (ja) |
| EP (1) | EP1078263B1 (ja) |
| JP (1) | JP4601166B2 (ja) |
| AT (1) | ATE437366T1 (ja) |
| AU (1) | AU765085B2 (ja) |
| CA (1) | CA2330678C (ja) |
| DE (1) | DE69941150D1 (ja) |
| DK (1) | DK1078263T3 (ja) |
| ES (1) | ES2328650T3 (ja) |
| WO (1) | WO1999058977A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN117433867A (zh) * | 2023-10-24 | 2024-01-23 | 深圳联合医学科技有限公司 | 阴道微生物的荧光染色液及检测阴道微生物的方法 |
Families Citing this family (60)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7166423B1 (en) | 1992-10-21 | 2007-01-23 | Miltenyi Biotec Gmbh | Direct selection of cells by secretion product |
| DE69332884T2 (de) * | 1992-10-21 | 2003-11-20 | Stefan Miltenyi | Direkte auswahl von zellen durch ein aussheidungsprodukt |
| KR20010072957A (ko) * | 1998-08-24 | 2001-07-31 | 추후보정 | H2 수용체 작용제 및 기타 t-세포 활성화제를 사용한t-세포(cd4+ 및 cd8+) 활성화 및 보호 방법 |
| US7807377B2 (en) * | 1998-10-20 | 2010-10-05 | Salvatore Albani | Method of isolating antigen-specific T cells employing artificial antigen presenting cells |
| US20020044656A1 (en) * | 1999-10-13 | 2002-04-18 | Brant L. Candelore | Interfacing a conditional access circuit to a digital device using input and output stream switching |
| US7541184B2 (en) * | 2000-02-24 | 2009-06-02 | Invitrogen Corporation | Activation and expansion of cells |
| US7462486B2 (en) | 2000-05-12 | 2008-12-09 | Oregon Health & Science University | Methods of selecting T cell receptor V peptides for therapeutic use |
| AU5984701A (en) | 2000-05-12 | 2001-11-20 | Univ Oregon Health & Science | Method of treating immune pathologies with low dose estrogen |
| JP2002365286A (ja) * | 2000-11-13 | 2002-12-18 | Kyogo Ito | 細胞性免疫検出法およびその医薬への応用 |
| US6919183B2 (en) | 2001-01-16 | 2005-07-19 | Regeneron Pharmaceuticals, Inc. | Isolating cells expressing secreted proteins |
| US20090137416A1 (en) | 2001-01-16 | 2009-05-28 | Regeneron Pharmaceuticals, Inc. | Isolating Cells Expressing Secreted Proteins |
| GB0118337D0 (en) * | 2001-07-27 | 2001-09-19 | Lonza Biologics Plc | Method for selecting antibody expressing cells |
| US20060035291A1 (en) * | 2001-09-18 | 2006-02-16 | Kyogo Itoh | Method of detecting cellular immunity and application thereof to drugs |
| US20040175373A1 (en) * | 2002-06-28 | 2004-09-09 | Xcyte Therapies, Inc. | Compositions and methods for eliminating undesired subpopulations of T cells in patients with immunological defects related to autoimmunity and organ or hematopoietic stem cell transplantation |
| US20050084967A1 (en) * | 2002-06-28 | 2005-04-21 | Xcyte Therapies, Inc. | Compositions and methods for eliminating undesired subpopulations of T cells in patients with immunological defects related to autoimmunity and organ or hematopoietic stem cell transplantation |
| EP1539929B1 (en) * | 2002-06-28 | 2013-04-10 | Life Technologies Corporation | Methods for restoring immune repertoire in patients with immunological defects related to autoimmunity and organ or hematopoietic stem cell transplantation |
| ATE328279T1 (de) * | 2002-08-23 | 2006-06-15 | Deutsches Rheuma Forschungszen | Verfahren zum nachweis und zur isolierung von t- lymphozyten, die ein definiertes antigen erkennen |
| PT1570267E (pt) | 2002-12-03 | 2012-01-03 | Ucb Pharma Sa | Ensaio para a identificação de células produtoras de anticorpos |
| US20050164168A1 (en) * | 2003-03-28 | 2005-07-28 | Cullum Malford E. | Method for the rapid diagnosis of infectious disease by detection and quantitation of microorganism induced cytokines |
| US20040248205A1 (en) * | 2003-04-16 | 2004-12-09 | Stern Lawrence J. | Major histocompatibility complex (MHC)-peptide arrays |
| MXPA05012080A (es) * | 2003-05-08 | 2006-02-22 | Xcyte Therapies Inc | Generacion y aislamiento de celulas t especificas al antigeno. |
| WO2005019823A1 (en) * | 2003-08-20 | 2005-03-03 | Celltech R & D Limited | Methods for obtaining antibodies |
| WO2005031357A2 (en) * | 2003-09-24 | 2005-04-07 | Ucl Biomedica Plc. | Cell sample analysis |
| JP2008507259A (ja) * | 2004-05-24 | 2008-03-13 | ベイラー・リサーチ・インスチチユート | 免疫応答の評価方法 |
| GB0412973D0 (en) * | 2004-06-10 | 2004-07-14 | Celltech R&D Ltd | Identification of antibody producing cells |
| CA2575604A1 (en) | 2004-07-30 | 2006-02-02 | Oregon Health And Science University | Methods for detecting and treating autoimmune disorders |
| WO2006050138A2 (en) * | 2004-10-29 | 2006-05-11 | Benaroya Research Institute At Virginia Mason | Methods of generating antigen-specific cd4+cd25+ regulatory t cells, compositions and methods of use |
| WO2006124356A2 (en) * | 2005-05-16 | 2006-11-23 | The Rockefeller University | Method for isolating dermal papilla cells and uses thereof |
| DK1996931T3 (en) * | 2005-12-28 | 2014-02-17 | Gen Hospital Corp | Methods and systems for sorting of blood cells |
| EP1878744A1 (en) * | 2006-07-13 | 2008-01-16 | Max-Delbrück-Centrum für Molekulare Medizin (MDC) | Epitope-tag for surface-expressed T-cell receptor proteins, uses thereof and method of selecting host cells expressing them |
| WO2008016431A2 (en) * | 2006-07-29 | 2008-02-07 | Robert Lamar Bjork | Bi-specific monoclonal antibody (specific for both cd3 and cd11b) therapeutic drug |
| CN101221171B (zh) * | 2007-12-11 | 2011-07-20 | 广州益善生物技术有限公司 | 用于骨代谢生化标志物检测的液相芯片及其制备方法 |
| WO2009103753A1 (en) * | 2008-02-20 | 2009-08-27 | Ablynx Nv | Methods for identifying and/or sorting cells by secreted molecule and kits for performing such methods |
| US20110177592A1 (en) * | 2008-07-11 | 2011-07-21 | Faustman Denise L | Magnetic apparatus for blood separation |
| US8309306B2 (en) * | 2008-11-12 | 2012-11-13 | Nodality, Inc. | Detection composition |
| GB0823553D0 (en) | 2008-12-24 | 2009-01-28 | Immunosolv Ltd | Cell separation technique |
| US20120045827A1 (en) * | 2009-03-09 | 2012-02-23 | Biofactura, Inc. | Separation of antigen-specific memory b cells with a conjugated biopolymer surface |
| US8790916B2 (en) | 2009-05-14 | 2014-07-29 | Genestream, Inc. | Microfluidic method and system for isolating particles from biological fluid |
| CN102033129A (zh) * | 2009-09-29 | 2011-04-27 | 上海英伯肯医学生物技术有限公司 | 用抗原刺激的细胞免疫反应来检测病原微生物的方法及测试笔 |
| EP2390661B1 (en) | 2010-05-02 | 2015-06-24 | Miltenyi Biotec GmbH | An anchoring/capturing means for selecting or analyzing a CHO cell according to a product secreted by the CHO cell |
| WO2012048298A2 (en) | 2010-10-08 | 2012-04-12 | Caridianbct, Inc. | Methods and systems of growing and harvesting cells in a hollow fiber bioreactor system with control conditions |
| SG10202012534VA (en) | 2011-05-26 | 2021-01-28 | Geneius Biotechnology Inc | Modulated Immunodominance Therapy |
| AU2012355990B2 (en) | 2011-12-22 | 2017-10-12 | Yeda Research And Development Co. Ltd. | A combination therapy for a stable and long term engraftment |
| US10379026B2 (en) | 2012-08-29 | 2019-08-13 | Inguran, Llc | Cell processing using magnetic particles |
| EP2890498B1 (en) | 2012-08-29 | 2018-02-21 | Inguran, LLC | Magnetic removal or identification of damaged or compromised cells or cellular structures |
| TW202206465A (zh) | 2012-11-14 | 2022-02-16 | 美商再生元醫藥公司 | 重組細胞表面捕捉蛋白質 |
| CN105793411B (zh) | 2013-11-16 | 2018-04-17 | 泰尔茂比司特公司 | 生物反应器中的细胞扩增 |
| WO2015148704A1 (en) | 2014-03-25 | 2015-10-01 | Terumo Bct, Inc. | Passive replacement of media |
| CN103901188B (zh) * | 2014-04-11 | 2016-08-24 | 苏州浩欧博生物医药有限公司 | 一种吸入过敏原的化学发光定量检测试剂盒及其制备方法和检测方法 |
| WO2016049421A1 (en) | 2014-09-26 | 2016-03-31 | Terumo Bct, Inc. | Scheduled feed |
| WO2017004592A1 (en) | 2015-07-02 | 2017-01-05 | Terumo Bct, Inc. | Cell growth with mechanical stimuli |
| EP3464565A4 (en) | 2016-05-25 | 2020-01-01 | Terumo BCT, Inc. | CELL EXPANSION |
| US11685883B2 (en) | 2016-06-07 | 2023-06-27 | Terumo Bct, Inc. | Methods and systems for coating a cell growth surface |
| US11104874B2 (en) | 2016-06-07 | 2021-08-31 | Terumo Bct, Inc. | Coating a bioreactor |
| MX2019000180A (es) * | 2016-06-28 | 2019-11-05 | Geneius Biotechnology Inc | Composiciones de células t para la inmunoterapia. |
| EP3656841A1 (en) | 2017-03-31 | 2020-05-27 | Terumo BCT, Inc. | Cell expansion |
| US11624046B2 (en) | 2017-03-31 | 2023-04-11 | Terumo Bct, Inc. | Cell expansion |
| EP3662057A4 (en) * | 2017-08-01 | 2021-04-28 | Benaroya Research Institute at Virginia Mason | PRO-ALLERGIC SPECIFIC T-LYMPHOCYTES IDENTIFICATION AND SEPARATION PROCESSES |
| AU2019362400A1 (en) * | 2018-10-19 | 2021-04-22 | ETH Zürich | Chimeric molecules |
| WO2021030606A1 (en) * | 2019-08-14 | 2021-02-18 | Sanford Burnham Prebys Medical Discovery Institute | Methods and devices for rare cell capture |
Family Cites Families (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4230685A (en) | 1979-02-28 | 1980-10-28 | Northwestern University | Method of magnetic separation of cells and the like, and microspheres for use therein |
| US4452773A (en) | 1982-04-05 | 1984-06-05 | Canadian Patents And Development Limited | Magnetic iron-dextran microspheres |
| GB8408127D0 (en) | 1984-03-29 | 1984-05-10 | Nyegaard & Co As | Contrast agents |
| US4770183A (en) | 1986-07-03 | 1988-09-13 | Advanced Magnetics Incorporated | Biologically degradable superparamagnetic particles for use as nuclear magnetic resonance imaging agents |
| IL91414A0 (en) | 1988-08-31 | 1990-04-29 | Zynaxis Technologies Inc | Reagents and methods for the determination of analytes |
| US5385707A (en) | 1988-12-28 | 1995-01-31 | Stefan Miltenyi | Metal matrices for use in high gradient magnetic separation of biological materials and method for coating the same |
| WO1990007380A2 (en) | 1988-12-28 | 1990-07-12 | Stefan Miltenyi | Methods and materials for high gradient magnetic separation of biological materials |
| DE69332884T2 (de) * | 1992-10-21 | 2003-11-20 | Stefan Miltenyi | Direkte auswahl von zellen durch ein aussheidungsprodukt |
| US5635363A (en) | 1995-02-28 | 1997-06-03 | The Board Of Trustees Of The Leland Stanford Junior University | Compositions and methods for the detection, quantitation and purification of antigen-specific T cells |
| CA2220971A1 (en) | 1995-05-25 | 1996-11-28 | Baxter International Inc. | Allogeneic cell therapy for cancer following allogeneic stem cell transplantation |
| JP2001520509A (ja) | 1995-07-25 | 2001-10-30 | セルセラピー・インコーポレイテツド | 自己免疫細胞療法:細胞組成物、方法およびヒト疾患の治療への応用 |
| WO1997035035A1 (en) | 1996-03-20 | 1997-09-25 | Charles Nicolette | A method for identifying cytotoxic t-cell epitopes |
| ATE347588T1 (de) | 1996-05-23 | 2006-12-15 | Scripps Research Inst | Systeme zur präsentation von mhc-antigenen der klasse ii und verfahren zur aktivierung von cd4?+-t-lymphozyten |
| US5750356A (en) * | 1996-05-31 | 1998-05-12 | Anergen, Inc. | Method for monitoring T cell reactivity |
-
1999
- 1999-05-10 DE DE69941150T patent/DE69941150D1/de not_active Expired - Lifetime
- 1999-05-10 CA CA002330678A patent/CA2330678C/en not_active Expired - Lifetime
- 1999-05-10 WO PCT/US1999/010200 patent/WO1999058977A1/en active IP Right Grant
- 1999-05-10 US US09/309,199 patent/US6576428B1/en not_active Expired - Lifetime
- 1999-05-10 AT AT99924167T patent/ATE437366T1/de not_active IP Right Cessation
- 1999-05-10 ES ES99924167T patent/ES2328650T3/es not_active Expired - Lifetime
- 1999-05-10 JP JP2000548729A patent/JP4601166B2/ja not_active Expired - Lifetime
- 1999-05-10 EP EP99924167A patent/EP1078263B1/en not_active Expired - Lifetime
- 1999-05-10 DK DK99924167T patent/DK1078263T3/da active
- 1999-05-10 AU AU40736/99A patent/AU765085B2/en not_active Ceased
-
2003
- 2003-05-23 US US10/445,391 patent/US20040023377A1/en not_active Abandoned
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN117433867A (zh) * | 2023-10-24 | 2024-01-23 | 深圳联合医学科技有限公司 | 阴道微生物的荧光染色液及检测阴道微生物的方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20040023377A1 (en) | 2004-02-05 |
| AU765085B2 (en) | 2003-09-11 |
| US6576428B1 (en) | 2003-06-10 |
| CA2330678A1 (en) | 1999-11-18 |
| CA2330678C (en) | 2009-04-07 |
| EP1078263A1 (en) | 2001-02-28 |
| AU4073699A (en) | 1999-11-29 |
| ES2328650T3 (es) | 2009-11-16 |
| DE69941150D1 (de) | 2009-09-03 |
| EP1078263B1 (en) | 2009-07-22 |
| DK1078263T3 (da) | 2009-11-02 |
| JP2002514765A (ja) | 2002-05-21 |
| ATE437366T1 (de) | 2009-08-15 |
| WO1999058977A1 (en) | 1999-11-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4601166B2 (ja) | 抗原特異的t細胞の直接的選択方法 | |
| JP7084304B2 (ja) | 細胞表面シグナル及びシグナル比を変えることによる、異なるt細胞亜集団の選択的増殖の方法 | |
| US20150210982A1 (en) | Isolation and Use of Human Regulatory T Cells | |
| WO2006050138A2 (en) | Methods of generating antigen-specific cd4+cd25+ regulatory t cells, compositions and methods of use | |
| JP2004512030A (ja) | 特異的細胞溶解性t細胞応答を誘導するための組成物および方法 | |
| JP2016192970A (ja) | Tr1細胞を単離する新規な方法 | |
| JP2017148042A (ja) | Il−13産生tr1−様細胞及びその使用 | |
| US20070128670A1 (en) | Methods for the identification and preparation of regulator/suppressor t lymphocytes, compositions and use thereof | |
| JP7277255B2 (ja) | インビトロでの制御性t細胞の特異性評価方法 | |
| JP2003033175A (ja) | 選択的免疫応答抑制を誘導する末梢血樹状細胞サブセット | |
| US20070141034A1 (en) | Methods for isolating t cells and uses thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20060426 |
|
| A621 | Written request for application examination |
Free format text: JAPANESE INTERMEDIATE CODE: A621 Effective date: 20060426 |
|
| A977 | Report on retrieval |
Free format text: JAPANESE INTERMEDIATE CODE: A971007 Effective date: 20090128 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20090323 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20090622 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20090629 |
|
| A601 | Written request for extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A601 Effective date: 20090713 |
|
| A602 | Written permission of extension of time |
Free format text: JAPANESE INTERMEDIATE CODE: A602 Effective date: 20090721 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20090820 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20100323 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20100623 |
|
| A131 | Notification of reasons for refusal |
Free format text: JAPANESE INTERMEDIATE CODE: A131 Effective date: 20100715 |
|
| A521 | Request for written amendment filed |
Free format text: JAPANESE INTERMEDIATE CODE: A523 Effective date: 20100823 |
|
| TRDD | Decision of grant or rejection written | ||
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 Effective date: 20100913 |
|
| A01 | Written decision to grant a patent or to grant a registration (utility model) |
Free format text: JAPANESE INTERMEDIATE CODE: A01 |
|
| A61 | First payment of annual fees (during grant procedure) |
Free format text: JAPANESE INTERMEDIATE CODE: A61 Effective date: 20100928 |
|
| FPAY | Renewal fee payment (event date is renewal date of database) |
Free format text: PAYMENT UNTIL: 20131008 Year of fee payment: 3 |
|
| R150 | Certificate of patent or registration of utility model |
Free format text: JAPANESE INTERMEDIATE CODE: R150 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| R250 | Receipt of annual fees |
Free format text: JAPANESE INTERMEDIATE CODE: R250 |
|
| EXPY | Cancellation because of completion of term |