ES2919572T3 - Compuestos antiproliferación y usos de los mismos - Google Patents
Compuestos antiproliferación y usos de los mismos Download PDFInfo
- Publication number
- ES2919572T3 ES2919572T3 ES19726792T ES19726792T ES2919572T3 ES 2919572 T3 ES2919572 T3 ES 2919572T3 ES 19726792 T ES19726792 T ES 19726792T ES 19726792 T ES19726792 T ES 19726792T ES 2919572 T3 ES2919572 T3 ES 2919572T3
- Authority
- ES
- Spain
- Prior art keywords
- ring
- nitrogen
- compound
- oxygen
- cancer
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 150000001875 compounds Chemical class 0.000 title claims abstract description 316
- 230000001028 anti-proliverative effect Effects 0.000 title description 8
- 206010028980 Neoplasm Diseases 0.000 claims abstract description 133
- 150000003839 salts Chemical class 0.000 claims abstract description 100
- 201000011510 cancer Diseases 0.000 claims abstract description 90
- 238000000034 method Methods 0.000 claims abstract description 55
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 40
- 230000002062 proliferating effect Effects 0.000 claims abstract description 24
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 15
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 210
- 229910052757 nitrogen Inorganic materials 0.000 claims description 166
- 125000005842 heteroatom Chemical group 0.000 claims description 140
- 229910052760 oxygen Inorganic materials 0.000 claims description 138
- 239000001301 oxygen Chemical group 0.000 claims description 137
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 136
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 claims description 127
- 229910052717 sulfur Chemical group 0.000 claims description 127
- 239000011593 sulfur Chemical group 0.000 claims description 127
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 claims description 119
- 229920006395 saturated elastomer Polymers 0.000 claims description 117
- 125000001931 aliphatic group Chemical group 0.000 claims description 92
- 239000000203 mixture Substances 0.000 claims description 81
- 125000004429 atom Chemical group 0.000 claims description 64
- 125000000623 heterocyclic group Chemical group 0.000 claims description 50
- 210000004027 cell Anatomy 0.000 claims description 48
- 229910052736 halogen Inorganic materials 0.000 claims description 47
- 150000002367 halogens Chemical class 0.000 claims description 41
- 229910052739 hydrogen Inorganic materials 0.000 claims description 40
- 239000001257 hydrogen Substances 0.000 claims description 40
- 125000001072 heteroaryl group Chemical group 0.000 claims description 33
- 125000002837 carbocyclic group Chemical group 0.000 claims description 29
- 208000035475 disorder Diseases 0.000 claims description 26
- 125000002619 bicyclic group Chemical group 0.000 claims description 25
- 125000002950 monocyclic group Chemical group 0.000 claims description 25
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 24
- 206010073071 hepatocellular carcinoma Diseases 0.000 claims description 23
- 125000002618 bicyclic heterocycle group Chemical group 0.000 claims description 22
- 206010009944 Colon cancer Diseases 0.000 claims description 21
- 210000002472 endoplasmic reticulum Anatomy 0.000 claims description 21
- 231100000844 hepatocellular carcinoma Toxicity 0.000 claims description 21
- 201000001441 melanoma Diseases 0.000 claims description 21
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 20
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 19
- 208000002154 non-small cell lung carcinoma Diseases 0.000 claims description 18
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 claims description 18
- 208000001333 Colorectal Neoplasms Diseases 0.000 claims description 15
- 230000004906 unfolded protein response Effects 0.000 claims description 15
- 206010061902 Pancreatic neoplasm Diseases 0.000 claims description 14
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 claims description 14
- 201000002528 pancreatic cancer Diseases 0.000 claims description 14
- 208000008443 pancreatic carcinoma Diseases 0.000 claims description 14
- 125000000475 sulfinyl group Chemical group [*:2]S([*:1])=O 0.000 claims description 14
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 13
- 206010035226 Plasma cell myeloma Diseases 0.000 claims description 12
- 150000002431 hydrogen Chemical group 0.000 claims description 12
- 208000026310 Breast neoplasm Diseases 0.000 claims description 11
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 10
- 208000006265 Renal cell carcinoma Diseases 0.000 claims description 10
- 230000001939 inductive effect Effects 0.000 claims description 10
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 10
- 206010006187 Breast cancer Diseases 0.000 claims description 9
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 claims description 9
- 239000011575 calcium Substances 0.000 claims description 9
- 229910052791 calcium Inorganic materials 0.000 claims description 9
- 125000004043 oxo group Chemical group O=* 0.000 claims description 9
- WFKWXMTUELFFGS-UHFFFAOYSA-N tungsten Chemical compound [W] WFKWXMTUELFFGS-UHFFFAOYSA-N 0.000 claims description 9
- 229910052721 tungsten Inorganic materials 0.000 claims description 9
- 239000010937 tungsten Substances 0.000 claims description 9
- 239000003981 vehicle Substances 0.000 claims description 8
- 239000003937 drug carrier Substances 0.000 claims description 7
- 239000002671 adjuvant Substances 0.000 claims description 6
- 150000001338 aliphatic hydrocarbons Chemical class 0.000 claims description 6
- 201000000050 myeloid neoplasm Diseases 0.000 claims description 5
- 125000000714 pyrimidinyl group Chemical group 0.000 claims description 4
- 206010014733 Endometrial cancer Diseases 0.000 claims description 3
- 206010014759 Endometrial neoplasm Diseases 0.000 claims description 3
- 208000000461 Esophageal Neoplasms Diseases 0.000 claims description 3
- 206010030155 Oesophageal carcinoma Diseases 0.000 claims description 3
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 3
- 201000004101 esophageal cancer Diseases 0.000 claims description 3
- 230000014509 gene expression Effects 0.000 claims description 3
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 claims description 2
- 239000003814 drug Substances 0.000 description 125
- 235000002639 sodium chloride Nutrition 0.000 description 84
- 229940124597 therapeutic agent Drugs 0.000 description 83
- -1 monocyclic hydrocarbon Chemical class 0.000 description 80
- 239000003112 inhibitor Substances 0.000 description 73
- 230000000694 effects Effects 0.000 description 53
- 229940076838 Immune checkpoint inhibitor Drugs 0.000 description 44
- 239000012274 immune-checkpoint protein inhibitor Substances 0.000 description 44
- 239000000543 intermediate Substances 0.000 description 40
- 239000000556 agonist Substances 0.000 description 32
- 238000011282 treatment Methods 0.000 description 27
- 206010033128 Ovarian cancer Diseases 0.000 description 24
- 108090000623 proteins and genes Proteins 0.000 description 24
- 235000018102 proteins Nutrition 0.000 description 21
- 102000004169 proteins and genes Human genes 0.000 description 21
- 125000001424 substituent group Chemical group 0.000 description 19
- 102000037984 Inhibitory immune checkpoint proteins Human genes 0.000 description 17
- 108091008026 Inhibitory immune checkpoint proteins Proteins 0.000 description 17
- 125000003118 aryl group Chemical group 0.000 description 17
- 206010025323 Lymphomas Diseases 0.000 description 15
- 206010061535 Ovarian neoplasm Diseases 0.000 description 15
- 239000003795 chemical substances by application Substances 0.000 description 15
- 230000002401 inhibitory effect Effects 0.000 description 15
- 229960003301 nivolumab Drugs 0.000 description 15
- 239000000427 antigen Substances 0.000 description 14
- 108091007433 antigens Proteins 0.000 description 14
- 102000036639 antigens Human genes 0.000 description 14
- 102100040678 Programmed cell death protein 1 Human genes 0.000 description 13
- 239000012472 biological sample Substances 0.000 description 13
- 201000010099 disease Diseases 0.000 description 13
- 230000003308 immunostimulating effect Effects 0.000 description 13
- 208000029974 neurofibrosarcoma Diseases 0.000 description 13
- 210000001744 T-lymphocyte Anatomy 0.000 description 12
- 102100036022 Wolframin Human genes 0.000 description 12
- 229910052799 carbon Inorganic materials 0.000 description 12
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 12
- 229960002621 pembrolizumab Drugs 0.000 description 12
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 12
- 229940045513 CTLA4 antagonist Drugs 0.000 description 11
- 230000027455 binding Effects 0.000 description 11
- 208000018463 endometrial serous adenocarcinoma Diseases 0.000 description 11
- 208000005017 glioblastoma Diseases 0.000 description 11
- 102000005962 receptors Human genes 0.000 description 11
- 108020003175 receptors Proteins 0.000 description 11
- 102000003964 Histone deacetylase Human genes 0.000 description 10
- 108090000353 Histone deacetylase Proteins 0.000 description 10
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 10
- 208000000172 Medulloblastoma Diseases 0.000 description 10
- 239000002552 dosage form Substances 0.000 description 10
- 125000005843 halogen group Chemical group 0.000 description 10
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 10
- 239000000243 solution Substances 0.000 description 10
- 101710089372 Programmed cell death protein 1 Proteins 0.000 description 9
- 206010060862 Prostate cancer Diseases 0.000 description 9
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 9
- 125000000217 alkyl group Chemical group 0.000 description 9
- 150000001721 carbon Chemical group 0.000 description 9
- 238000009472 formulation Methods 0.000 description 9
- 208000014829 head and neck neoplasm Diseases 0.000 description 9
- 239000003446 ligand Substances 0.000 description 9
- 239000012071 phase Substances 0.000 description 9
- 125000006239 protecting group Chemical group 0.000 description 9
- 239000002904 solvent Substances 0.000 description 9
- 239000000725 suspension Substances 0.000 description 9
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 8
- 206010003571 Astrocytoma Diseases 0.000 description 8
- 108010021064 CTLA-4 Antigen Proteins 0.000 description 8
- 102000008203 CTLA-4 Antigen Human genes 0.000 description 8
- 101150015280 Cel gene Proteins 0.000 description 8
- 208000013452 Fallopian tube neoplasm Diseases 0.000 description 8
- 206010018338 Glioma Diseases 0.000 description 8
- 208000008839 Kidney Neoplasms Diseases 0.000 description 8
- 102100024216 Programmed cell death 1 ligand 1 Human genes 0.000 description 8
- 239000012190 activator Substances 0.000 description 8
- 239000003276 histone deacetylase inhibitor Substances 0.000 description 8
- 229940043441 phosphoinositide 3-kinase inhibitor Drugs 0.000 description 8
- DXDLVWBYAYXINP-FWJOYPJLSA-N tert-butyl 3-[2-chloro-4-[(2R)-1-hydroxypropan-2-yl]oxyphenyl]-1,4-oxazepane-4-carboxylate Chemical compound C(C)(C)(C)OC(=O)N1C(COCCC1)C1=C(C=C(C=C1)O[C@@H](CO)C)Cl DXDLVWBYAYXINP-FWJOYPJLSA-N 0.000 description 8
- 238000002560 therapeutic procedure Methods 0.000 description 8
- WAEXFXRVDQXREF-UHFFFAOYSA-N vorinostat Chemical group ONC(=O)CCCCCCC(=O)NC1=CC=CC=C1 WAEXFXRVDQXREF-UHFFFAOYSA-N 0.000 description 8
- 206010052747 Adenocarcinoma pancreas Diseases 0.000 description 7
- 208000001446 Anaplastic Thyroid Carcinoma Diseases 0.000 description 7
- 206010002240 Anaplastic thyroid cancer Diseases 0.000 description 7
- 102100029822 B- and T-lymphocyte attenuator Human genes 0.000 description 7
- 206010005003 Bladder cancer Diseases 0.000 description 7
- 201000001342 Fallopian tube cancer Diseases 0.000 description 7
- 208000032612 Glial tumor Diseases 0.000 description 7
- 101000868279 Homo sapiens Leukocyte surface antigen CD47 Proteins 0.000 description 7
- 108090000172 Interleukin-15 Proteins 0.000 description 7
- 102000002698 KIR Receptors Human genes 0.000 description 7
- 108010043610 KIR Receptors Proteins 0.000 description 7
- 208000034578 Multiple myelomas Diseases 0.000 description 7
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 7
- 206010038389 Renal cancer Diseases 0.000 description 7
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 7
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 description 7
- 208000033559 Waldenström macroglobulinemia Diseases 0.000 description 7
- 125000002947 alkylene group Chemical group 0.000 description 7
- 239000005557 antagonist Substances 0.000 description 7
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 7
- 210000000988 bone and bone Anatomy 0.000 description 7
- 239000002775 capsule Substances 0.000 description 7
- 125000004122 cyclic group Chemical group 0.000 description 7
- 229940079593 drug Drugs 0.000 description 7
- 229950009791 durvalumab Drugs 0.000 description 7
- 229960005386 ipilimumab Drugs 0.000 description 7
- 201000010982 kidney cancer Diseases 0.000 description 7
- 150000002632 lipids Chemical class 0.000 description 7
- 230000001394 metastastic effect Effects 0.000 description 7
- 206010061289 metastatic neoplasm Diseases 0.000 description 7
- 201000008968 osteosarcoma Diseases 0.000 description 7
- 201000002094 pancreatic adenocarcinoma Diseases 0.000 description 7
- 201000008129 pancreatic ductal adenocarcinoma Diseases 0.000 description 7
- 229920001223 polyethylene glycol Polymers 0.000 description 7
- 201000009410 rhabdomyosarcoma Diseases 0.000 description 7
- 239000007787 solid Substances 0.000 description 7
- 206010042863 synovial sarcoma Diseases 0.000 description 7
- 239000003826 tablet Substances 0.000 description 7
- 208000019179 thyroid gland undifferentiated (anaplastic) carcinoma Diseases 0.000 description 7
- 229960000237 vorinostat Drugs 0.000 description 7
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 7
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 6
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 6
- 101710144268 B- and T-lymphocyte attenuator Proteins 0.000 description 6
- 229940123711 Bcl2 inhibitor Drugs 0.000 description 6
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 6
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 6
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 6
- 101001137987 Homo sapiens Lymphocyte activation gene 3 protein Proteins 0.000 description 6
- 101000863873 Homo sapiens Tyrosine-protein phosphatase non-receptor type substrate 1 Proteins 0.000 description 6
- 102000003812 Interleukin-15 Human genes 0.000 description 6
- 239000005517 L01XE01 - Imatinib Substances 0.000 description 6
- 102100032913 Leukocyte surface antigen CD47 Human genes 0.000 description 6
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 6
- 208000003019 Neurofibromatosis 1 Diseases 0.000 description 6
- 208000024834 Neurofibromatosis type 1 Diseases 0.000 description 6
- 208000007571 Ovarian Epithelial Carcinoma Diseases 0.000 description 6
- 208000008900 Pancreatic Ductal Carcinoma Diseases 0.000 description 6
- 102000012338 Poly(ADP-ribose) Polymerases Human genes 0.000 description 6
- 108010061844 Poly(ADP-ribose) Polymerases Proteins 0.000 description 6
- 229920000776 Poly(Adenosine diphosphate-ribose) polymerase Polymers 0.000 description 6
- 102100024213 Programmed cell death 1 ligand 2 Human genes 0.000 description 6
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 6
- 208000015634 Rectal Neoplasms Diseases 0.000 description 6
- 208000000102 Squamous Cell Carcinoma of Head and Neck Diseases 0.000 description 6
- 102100029948 Tyrosine-protein phosphatase non-receptor type substrate 1 Human genes 0.000 description 6
- 239000004480 active ingredient Substances 0.000 description 6
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 6
- 206010006007 bone sarcoma Diseases 0.000 description 6
- 208000029742 colonic neoplasm Diseases 0.000 description 6
- 208000011588 combined hepatocellular carcinoma and cholangiocarcinoma Diseases 0.000 description 6
- 150000002148 esters Chemical group 0.000 description 6
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 6
- 210000003128 head Anatomy 0.000 description 6
- 201000000459 head and neck squamous cell carcinoma Diseases 0.000 description 6
- 239000008101 lactose Substances 0.000 description 6
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 6
- BMGQWWVMWDBQGC-IIFHNQTCSA-N midostaurin Chemical compound CN([C@H]1[C@H]([C@]2(C)O[C@@H](N3C4=CC=CC=C4C4=C5C(=O)NCC5=C5C6=CC=CC=C6N2C5=C43)C1)OC)C(=O)C1=CC=CC=C1 BMGQWWVMWDBQGC-IIFHNQTCSA-N 0.000 description 6
- 229950010895 midostaurin Drugs 0.000 description 6
- 231100000252 nontoxic Toxicity 0.000 description 6
- 230000003000 nontoxic effect Effects 0.000 description 6
- 239000002777 nucleoside Substances 0.000 description 6
- 150000003833 nucleoside derivatives Chemical class 0.000 description 6
- 208000024641 papillary serous cystadenocarcinoma Diseases 0.000 description 6
- 239000000546 pharmaceutical excipient Substances 0.000 description 6
- 229910052697 platinum Inorganic materials 0.000 description 6
- 102000016914 ras Proteins Human genes 0.000 description 6
- 206010038038 rectal cancer Diseases 0.000 description 6
- 201000001275 rectum cancer Diseases 0.000 description 6
- 210000004872 soft tissue Anatomy 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- 208000024891 symptom Diseases 0.000 description 6
- 238000003786 synthesis reaction Methods 0.000 description 6
- 230000001225 therapeutic effect Effects 0.000 description 6
- 201000005112 urinary bladder cancer Diseases 0.000 description 6
- 229910001868 water Inorganic materials 0.000 description 6
- STUWGJZDJHPWGZ-LBPRGKRZSA-N (2S)-N1-[4-methyl-5-[2-(1,1,1-trifluoro-2-methylpropan-2-yl)-4-pyridinyl]-2-thiazolyl]pyrrolidine-1,2-dicarboxamide Chemical compound S1C(C=2C=C(N=CC=2)C(C)(C)C(F)(F)F)=C(C)N=C1NC(=O)N1CCC[C@H]1C(N)=O STUWGJZDJHPWGZ-LBPRGKRZSA-N 0.000 description 5
- 108010029445 Agammaglobulinaemia Tyrosine Kinase Proteins 0.000 description 5
- 239000012664 BCL-2-inhibitor Substances 0.000 description 5
- 102100024263 CD160 antigen Human genes 0.000 description 5
- 102100038078 CD276 antigen Human genes 0.000 description 5
- 101710185679 CD276 antigen Proteins 0.000 description 5
- 239000004215 Carbon black (E152) Substances 0.000 description 5
- 201000009030 Carcinoma Diseases 0.000 description 5
- 102000004190 Enzymes Human genes 0.000 description 5
- 108090000790 Enzymes Proteins 0.000 description 5
- 102100034458 Hepatitis A virus cellular receptor 2 Human genes 0.000 description 5
- 101000761938 Homo sapiens CD160 antigen Proteins 0.000 description 5
- 101001068133 Homo sapiens Hepatitis A virus cellular receptor 2 Proteins 0.000 description 5
- 101000666896 Homo sapiens V-type immunoglobulin domain-containing suppressor of T-cell activation Proteins 0.000 description 5
- 102000017578 LAG3 Human genes 0.000 description 5
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 5
- 239000012828 PI3K inhibitor Substances 0.000 description 5
- 108090000608 Phosphoric Monoester Hydrolases Proteins 0.000 description 5
- 102000004160 Phosphoric Monoester Hydrolases Human genes 0.000 description 5
- 108091000080 Phosphotransferase Proteins 0.000 description 5
- 229940079156 Proteasome inhibitor Drugs 0.000 description 5
- 208000005718 Stomach Neoplasms Diseases 0.000 description 5
- 108010016672 Syk Kinase Proteins 0.000 description 5
- 229940123237 Taxane Drugs 0.000 description 5
- BPEGJWRSRHCHSN-UHFFFAOYSA-N Temozolomide Chemical compound O=C1N(C)N=NC2=C(C(N)=O)N=CN21 BPEGJWRSRHCHSN-UHFFFAOYSA-N 0.000 description 5
- 102000004887 Transforming Growth Factor beta Human genes 0.000 description 5
- 108090001012 Transforming Growth Factor beta Proteins 0.000 description 5
- 102100029823 Tyrosine-protein kinase BTK Human genes 0.000 description 5
- 102100038183 Tyrosine-protein kinase SYK Human genes 0.000 description 5
- 108010079206 V-Set Domain-Containing T-Cell Activation Inhibitor 1 Proteins 0.000 description 5
- 102100038929 V-set domain-containing T-cell activation inhibitor 1 Human genes 0.000 description 5
- 102100038282 V-type immunoglobulin domain-containing suppressor of T-cell activation Human genes 0.000 description 5
- 239000002253 acid Substances 0.000 description 5
- 229940100198 alkylating agent Drugs 0.000 description 5
- 239000002168 alkylating agent Substances 0.000 description 5
- 239000003886 aromatase inhibitor Substances 0.000 description 5
- 229950002916 avelumab Drugs 0.000 description 5
- BMQGVNUXMIRLCK-OAGWZNDDSA-N cabazitaxel Chemical compound O([C@H]1[C@@H]2[C@]3(OC(C)=O)CO[C@@H]3C[C@@H]([C@]2(C(=O)[C@H](OC)C2=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=3C=CC=CC=3)C[C@]1(O)C2(C)C)C)OC)C(=O)C1=CC=CC=C1 BMQGVNUXMIRLCK-OAGWZNDDSA-N 0.000 description 5
- 229960004562 carboplatin Drugs 0.000 description 5
- 239000000969 carrier Substances 0.000 description 5
- 238000000576 coating method Methods 0.000 description 5
- 239000003085 diluting agent Substances 0.000 description 5
- 239000002270 dispersing agent Substances 0.000 description 5
- 229940088598 enzyme Drugs 0.000 description 5
- 206010017758 gastric cancer Diseases 0.000 description 5
- 208000006359 hepatoblastoma Diseases 0.000 description 5
- 229930195733 hydrocarbon Natural products 0.000 description 5
- IFSDAJWBUCMOAH-HNNXBMFYSA-N idelalisib Chemical group C1([C@@H](NC=2C=3N=CNC=3N=CN=2)CC)=NC2=CC=CC(F)=C2C(=O)N1C1=CC=CC=C1 IFSDAJWBUCMOAH-HNNXBMFYSA-N 0.000 description 5
- 229960002411 imatinib Drugs 0.000 description 5
- KTUFNOKKBVMGRW-UHFFFAOYSA-N imatinib Chemical compound C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 KTUFNOKKBVMGRW-UHFFFAOYSA-N 0.000 description 5
- 229940043355 kinase inhibitor Drugs 0.000 description 5
- YGPSJZOEDVAXAB-UHFFFAOYSA-N kynurenine Chemical compound OC(=O)C(N)CC(=O)C1=CC=CC=C1N YGPSJZOEDVAXAB-UHFFFAOYSA-N 0.000 description 5
- 208000032839 leukemia Diseases 0.000 description 5
- 208000020816 lung neoplasm Diseases 0.000 description 5
- 102000020233 phosphotransferase Human genes 0.000 description 5
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 5
- LHNIIDJUOCFXAP-UHFFFAOYSA-N pictrelisib Chemical compound C1CN(S(=O)(=O)C)CCN1CC1=CC2=NC(C=3C=4C=NNC=4C=CC=3)=NC(N3CCOCC3)=C2S1 LHNIIDJUOCFXAP-UHFFFAOYSA-N 0.000 description 5
- 229950010773 pidilizumab Drugs 0.000 description 5
- 239000006187 pill Substances 0.000 description 5
- 238000002360 preparation method Methods 0.000 description 5
- 239000003207 proteasome inhibitor Substances 0.000 description 5
- 238000000746 purification Methods 0.000 description 5
- 230000010076 replication Effects 0.000 description 5
- OHRURASPPZQGQM-GCCNXGTGSA-N romidepsin Chemical compound O1C(=O)[C@H](C(C)C)NC(=O)C(=C/C)/NC(=O)[C@H]2CSSCC\C=C\[C@@H]1CC(=O)N[C@H](C(C)C)C(=O)N2 OHRURASPPZQGQM-GCCNXGTGSA-N 0.000 description 5
- OHRURASPPZQGQM-UHFFFAOYSA-N romidepsin Natural products O1C(=O)C(C(C)C)NC(=O)C(=CC)NC(=O)C2CSSCCC=CC1CC(=O)NC(C(C)C)C(=O)N2 OHRURASPPZQGQM-UHFFFAOYSA-N 0.000 description 5
- 108010091666 romidepsin Proteins 0.000 description 5
- 208000000587 small cell lung carcinoma Diseases 0.000 description 5
- 150000003384 small molecules Chemical class 0.000 description 5
- 201000011549 stomach cancer Diseases 0.000 description 5
- 230000002194 synthesizing effect Effects 0.000 description 5
- AYUNIORJHRXIBJ-TXHRRWQRSA-N tanespimycin Chemical compound N1C(=O)\C(C)=C\C=C/[C@H](OC)[C@@H](OC(N)=O)\C(C)=C\[C@H](C)[C@@H](O)[C@@H](OC)C[C@H](C)CC2=C(NCC=C)C(=O)C=C1C2=O AYUNIORJHRXIBJ-TXHRRWQRSA-N 0.000 description 5
- DKPFODGZWDEEBT-QFIAKTPHSA-N taxane Chemical class C([C@]1(C)CCC[C@@H](C)[C@H]1C1)C[C@H]2[C@H](C)CC[C@@H]1C2(C)C DKPFODGZWDEEBT-QFIAKTPHSA-N 0.000 description 5
- 239000005483 tyrosine kinase inhibitor Substances 0.000 description 5
- AQTQHPDCURKLKT-JKDPCDLQSA-N vincristine sulfate Chemical compound OS(O)(=O)=O.C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C=O)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 AQTQHPDCURKLKT-JKDPCDLQSA-N 0.000 description 5
- NMUSYJAQQFHJEW-KVTDHHQDSA-N 5-azacytidine Chemical compound O=C1N=C(N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 NMUSYJAQQFHJEW-KVTDHHQDSA-N 0.000 description 4
- XKJMBINCVNINCA-UHFFFAOYSA-N Alfalone Chemical compound CON(C)C(=O)NC1=CC=C(Cl)C(Cl)=C1 XKJMBINCVNINCA-UHFFFAOYSA-N 0.000 description 4
- BFYIZQONLCFLEV-DAELLWKTSA-N Aromasine Chemical compound O=C1C=C[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CC(=C)C2=C1 BFYIZQONLCFLEV-DAELLWKTSA-N 0.000 description 4
- 208000003174 Brain Neoplasms Diseases 0.000 description 4
- 229940124297 CDK 4/6 inhibitor Drugs 0.000 description 4
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 4
- 206010008342 Cervix carcinoma Diseases 0.000 description 4
- 108091006146 Channels Proteins 0.000 description 4
- 108010019670 Chimeric Antigen Receptors Proteins 0.000 description 4
- 208000005243 Chondrosarcoma Diseases 0.000 description 4
- 208000009798 Craniopharyngioma Diseases 0.000 description 4
- 230000006820 DNA synthesis Effects 0.000 description 4
- 108050002772 E3 ubiquitin-protein ligase Mdm2 Proteins 0.000 description 4
- 102000012199 E3 ubiquitin-protein ligase Mdm2 Human genes 0.000 description 4
- 102000001301 EGF receptor Human genes 0.000 description 4
- 108060006698 EGF receptor Proteins 0.000 description 4
- 206010014967 Ependymoma Diseases 0.000 description 4
- 102100031351 Galectin-9 Human genes 0.000 description 4
- 101100229077 Gallus gallus GAL9 gene Proteins 0.000 description 4
- 101710113864 Heat shock protein 90 Proteins 0.000 description 4
- 102100034051 Heat shock protein HSP 90-alpha Human genes 0.000 description 4
- 101000834898 Homo sapiens Alpha-synuclein Proteins 0.000 description 4
- 101000916644 Homo sapiens Macrophage colony-stimulating factor 1 receptor Proteins 0.000 description 4
- 101001117317 Homo sapiens Programmed cell death 1 ligand 1 Proteins 0.000 description 4
- 101001117312 Homo sapiens Programmed cell death 1 ligand 2 Proteins 0.000 description 4
- 101000611936 Homo sapiens Programmed cell death protein 1 Proteins 0.000 description 4
- 101000652359 Homo sapiens Spermatogenesis-associated protein 2 Proteins 0.000 description 4
- GQYIWUVLTXOXAJ-UHFFFAOYSA-N Lomustine Chemical compound ClCCN(N=O)C(=O)NC1CCCCC1 GQYIWUVLTXOXAJ-UHFFFAOYSA-N 0.000 description 4
- 102100028198 Macrophage colony-stimulating factor 1 receptor Human genes 0.000 description 4
- 108010061593 Member 14 Tumor Necrosis Factor Receptors Proteins 0.000 description 4
- 206010027406 Mesothelioma Diseases 0.000 description 4
- 241001465754 Metazoa Species 0.000 description 4
- 101710181812 Methionine aminopeptidase Proteins 0.000 description 4
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 4
- 108091007960 PI3Ks Proteins 0.000 description 4
- 229910019142 PO4 Inorganic materials 0.000 description 4
- 229930012538 Paclitaxel Natural products 0.000 description 4
- 108090000430 Phosphatidylinositol 3-kinases Proteins 0.000 description 4
- 102000003993 Phosphatidylinositol 3-kinases Human genes 0.000 description 4
- 206010039491 Sarcoma Diseases 0.000 description 4
- 206010041067 Small cell lung cancer Diseases 0.000 description 4
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 description 4
- 208000024313 Testicular Neoplasms Diseases 0.000 description 4
- 206010057644 Testis cancer Diseases 0.000 description 4
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 4
- 102100028785 Tumor necrosis factor receptor superfamily member 14 Human genes 0.000 description 4
- 108010053096 Vascular Endothelial Growth Factor Receptor-1 Proteins 0.000 description 4
- 102000016548 Vascular Endothelial Growth Factor Receptor-1 Human genes 0.000 description 4
- 238000010521 absorption reaction Methods 0.000 description 4
- 208000020990 adrenal cortex carcinoma Diseases 0.000 description 4
- 208000007128 adrenocortical carcinoma Diseases 0.000 description 4
- 208000037844 advanced solid tumor Diseases 0.000 description 4
- 229950010482 alpelisib Drugs 0.000 description 4
- 229960003852 atezolizumab Drugs 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- 229940126587 biotherapeutics Drugs 0.000 description 4
- 230000037396 body weight Effects 0.000 description 4
- GXJABQQUPOEUTA-RDJZCZTQSA-N bortezomib Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)B(O)O)NC(=O)C=1N=CC=NC=1)C1=CC=CC=C1 GXJABQQUPOEUTA-RDJZCZTQSA-N 0.000 description 4
- 229950009221 chidamide Drugs 0.000 description 4
- SZMJVTADHFNAIS-BJMVGYQFSA-N chidamide Chemical compound NC1=CC(F)=CC=C1NC(=O)C(C=C1)=CC=C1CNC(=O)\C=C\C1=CC=CN=C1 SZMJVTADHFNAIS-BJMVGYQFSA-N 0.000 description 4
- 229960004316 cisplatin Drugs 0.000 description 4
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 4
- 125000000753 cycloalkyl group Chemical group 0.000 description 4
- 229940127089 cytotoxic agent Drugs 0.000 description 4
- 235000014113 dietary fatty acids Nutrition 0.000 description 4
- 229960003668 docetaxel Drugs 0.000 description 4
- INVTYAOGFAGBOE-UHFFFAOYSA-N entinostat Chemical compound NC1=CC=CC=C1NC(=O)C(C=C1)=CC=C1CNC(=O)OCC1=CC=CN=C1 INVTYAOGFAGBOE-UHFFFAOYSA-N 0.000 description 4
- 229930195729 fatty acid Natural products 0.000 description 4
- 239000000194 fatty acid Substances 0.000 description 4
- 229910052731 fluorine Inorganic materials 0.000 description 4
- IJJVMEJXYNJXOJ-UHFFFAOYSA-N fluquinconazole Chemical compound C=1C=C(Cl)C=C(Cl)C=1N1C(=O)C2=CC(F)=CC=C2N=C1N1C=NC=N1 IJJVMEJXYNJXOJ-UHFFFAOYSA-N 0.000 description 4
- 230000004927 fusion Effects 0.000 description 4
- 210000001035 gastrointestinal tract Anatomy 0.000 description 4
- XGALLCVXEZPNRQ-UHFFFAOYSA-N gefitinib Chemical compound C=12C=C(OCCCN3CCOCC3)C(OC)=CC2=NC=NC=1NC1=CC=C(F)C(Cl)=C1 XGALLCVXEZPNRQ-UHFFFAOYSA-N 0.000 description 4
- XLXSAKCOAKORKW-AQJXLSMYSA-N gonadorelin Chemical compound C([C@@H](C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)NCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)C1=CC=C(O)C=C1 XLXSAKCOAKORKW-AQJXLSMYSA-N 0.000 description 4
- 150000002430 hydrocarbons Chemical group 0.000 description 4
- 229960003445 idelalisib Drugs 0.000 description 4
- 230000002519 immonomodulatory effect Effects 0.000 description 4
- 229940124622 immune-modulator drug Drugs 0.000 description 4
- 239000003701 inert diluent Substances 0.000 description 4
- 230000005865 ionizing radiation Effects 0.000 description 4
- HPJKCIUCZWXJDR-UHFFFAOYSA-N letrozole Chemical compound C1=CC(C#N)=CC=C1C(N1N=CN=C1)C1=CC=C(C#N)C=C1 HPJKCIUCZWXJDR-UHFFFAOYSA-N 0.000 description 4
- 239000007788 liquid Substances 0.000 description 4
- 208000014018 liver neoplasm Diseases 0.000 description 4
- 201000005202 lung cancer Diseases 0.000 description 4
- 229940124302 mTOR inhibitor Drugs 0.000 description 4
- 208000026037 malignant tumor of neck Diseases 0.000 description 4
- 239000003628 mammalian target of rapamycin inhibitor Substances 0.000 description 4
- HAWPXGHAZFHHAD-UHFFFAOYSA-N mechlorethamine Chemical compound ClCCN(C)CCCl HAWPXGHAZFHHAD-UHFFFAOYSA-N 0.000 description 4
- 239000002609 medium Substances 0.000 description 4
- 239000002674 ointment Substances 0.000 description 4
- 229960003278 osimertinib Drugs 0.000 description 4
- DUYJMQONPNNFPI-UHFFFAOYSA-N osimertinib Chemical compound COC1=CC(N(C)CCN(C)C)=C(NC(=O)C=C)C=C1NC1=NC=CC(C=2C3=CC=CC=C3N(C)C=2)=N1 DUYJMQONPNNFPI-UHFFFAOYSA-N 0.000 description 4
- 229960001756 oxaliplatin Drugs 0.000 description 4
- DWAFYCQODLXJNR-BNTLRKBRSA-L oxaliplatin Chemical compound O1C(=O)C(=O)O[Pt]11N[C@@H]2CCCC[C@H]2N1 DWAFYCQODLXJNR-BNTLRKBRSA-L 0.000 description 4
- 229960001592 paclitaxel Drugs 0.000 description 4
- FPOHNWQLNRZRFC-ZHACJKMWSA-N panobinostat Chemical compound CC=1NC2=CC=CC=C2C=1CCNCC1=CC=C(\C=C\C(=O)NO)C=C1 FPOHNWQLNRZRFC-ZHACJKMWSA-N 0.000 description 4
- 230000036961 partial effect Effects 0.000 description 4
- 230000037361 pathway Effects 0.000 description 4
- 239000000816 peptidomimetic Substances 0.000 description 4
- 235000021317 phosphate Nutrition 0.000 description 4
- 229940002612 prodrug Drugs 0.000 description 4
- 239000000651 prodrug Substances 0.000 description 4
- 238000001243 protein synthesis Methods 0.000 description 4
- 108010014186 ras Proteins Proteins 0.000 description 4
- 125000006413 ring segment Chemical group 0.000 description 4
- 229960004641 rituximab Drugs 0.000 description 4
- 229960003452 romidepsin Drugs 0.000 description 4
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 4
- 239000011780 sodium chloride Substances 0.000 description 4
- 206010041823 squamous cell carcinoma Diseases 0.000 description 4
- 239000000375 suspending agent Substances 0.000 description 4
- 230000008685 targeting Effects 0.000 description 4
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 4
- 229960004964 temozolomide Drugs 0.000 description 4
- 201000003120 testicular cancer Diseases 0.000 description 4
- 238000012360 testing method Methods 0.000 description 4
- 210000001519 tissue Anatomy 0.000 description 4
- 230000000699 topical effect Effects 0.000 description 4
- UCFGDBYHRUNTLO-QHCPKHFHSA-N topotecan Chemical compound C1=C(O)C(CN(C)C)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UCFGDBYHRUNTLO-QHCPKHFHSA-N 0.000 description 4
- 230000009466 transformation Effects 0.000 description 4
- 230000014616 translation Effects 0.000 description 4
- 229940121358 tyrosine kinase inhibitor Drugs 0.000 description 4
- 241000701161 unidentified adenovirus Species 0.000 description 4
- 229950005972 urelumab Drugs 0.000 description 4
- 229960004528 vincristine Drugs 0.000 description 4
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 4
- 239000001993 wax Substances 0.000 description 4
- 239000000080 wetting agent Substances 0.000 description 4
- XRASPMIURGNCCH-UHFFFAOYSA-N zoledronic acid Chemical compound OP(=O)(O)C(P(O)(O)=O)(O)CN1C=CN=C1 XRASPMIURGNCCH-UHFFFAOYSA-N 0.000 description 4
- ZADWXFSZEAPBJS-SNVBAGLBSA-N (2r)-2-amino-3-(1-methylindol-3-yl)propanoic acid Chemical compound C1=CC=C2N(C)C=C(C[C@@H](N)C(O)=O)C2=C1 ZADWXFSZEAPBJS-SNVBAGLBSA-N 0.000 description 3
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 description 3
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 3
- IHWDSEPNZDYMNF-UHFFFAOYSA-N 1H-indol-2-amine Chemical compound C1=CC=C2NC(N)=CC2=C1 IHWDSEPNZDYMNF-UHFFFAOYSA-N 0.000 description 3
- PDGKHKMBHVFCMG-UHFFFAOYSA-N 2-[[5-(4-methylpiperazin-1-yl)pyridin-2-yl]amino]spiro[7,8-dihydropyrazino[5,6]pyrrolo[1,2-d]pyrimidine-9,1'-cyclohexane]-6-one Chemical compound C1CN(C)CCN1C(C=N1)=CC=C1NC1=NC=C(C=C2N3C4(CCCCC4)CNC2=O)C3=N1 PDGKHKMBHVFCMG-UHFFFAOYSA-N 0.000 description 3
- BEUQXVWXFDOSAQ-UHFFFAOYSA-N 2-methyl-2-[4-[2-(5-methyl-2-propan-2-yl-1,2,4-triazol-3-yl)-5,6-dihydroimidazo[1,2-d][1,4]benzoxazepin-9-yl]pyrazol-1-yl]propanamide Chemical compound CC(C)N1N=C(C)N=C1C1=CN(CCOC=2C3=CC=C(C=2)C2=CN(N=C2)C(C)(C)C(N)=O)C3=N1 BEUQXVWXFDOSAQ-UHFFFAOYSA-N 0.000 description 3
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 3
- RHXHGRAEPCAFML-UHFFFAOYSA-N 7-cyclopentyl-n,n-dimethyl-2-[(5-piperazin-1-ylpyridin-2-yl)amino]pyrrolo[2,3-d]pyrimidine-6-carboxamide Chemical compound N1=C2N(C3CCCC3)C(C(=O)N(C)C)=CC2=CN=C1NC(N=C1)=CC=C1N1CCNCC1 RHXHGRAEPCAFML-UHFFFAOYSA-N 0.000 description 3
- SJVQHLPISAIATJ-ZDUSSCGKSA-N 8-chloro-2-phenyl-3-[(1S)-1-(7H-purin-6-ylamino)ethyl]-1-isoquinolinone Chemical compound C1([C@@H](NC=2C=3N=CNC=3N=CN=2)C)=CC2=CC=CC(Cl)=C2C(=O)N1C1=CC=CC=C1 SJVQHLPISAIATJ-ZDUSSCGKSA-N 0.000 description 3
- 102100033793 ALK tyrosine kinase receptor Human genes 0.000 description 3
- 108091006112 ATPases Proteins 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- 102000057290 Adenosine Triphosphatases Human genes 0.000 description 3
- 101150051188 Adora2a gene Proteins 0.000 description 3
- 102100021569 Apoptosis regulator Bcl-2 Human genes 0.000 description 3
- 229940122815 Aromatase inhibitor Drugs 0.000 description 3
- 102000015790 Asparaginase Human genes 0.000 description 3
- 108010024976 Asparaginase Proteins 0.000 description 3
- 108010074708 B7-H1 Antigen Proteins 0.000 description 3
- MLDQJTXFUGDVEO-UHFFFAOYSA-N BAY-43-9006 Chemical compound C1=NC(C(=O)NC)=CC(OC=2C=CC(NC(=O)NC=3C=C(C(Cl)=CC=3)C(F)(F)F)=CC=2)=C1 MLDQJTXFUGDVEO-UHFFFAOYSA-N 0.000 description 3
- 206010004146 Basal cell carcinoma Diseases 0.000 description 3
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 3
- 229940122361 Bisphosphonate Drugs 0.000 description 3
- 206010006143 Brain stem glioma Diseases 0.000 description 3
- GAGWJHPBXLXJQN-UORFTKCHSA-N Capecitabine Chemical compound C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1[C@H]1[C@H](O)[C@H](O)[C@@H](C)O1 GAGWJHPBXLXJQN-UORFTKCHSA-N 0.000 description 3
- ZBNZXTGUTAYRHI-UHFFFAOYSA-N Dasatinib Chemical compound C=1C(N2CCN(CCO)CC2)=NC(C)=NC=1NC(S1)=NC=C1C(=O)NC1=C(C)C=CC=C1Cl ZBNZXTGUTAYRHI-UHFFFAOYSA-N 0.000 description 3
- XXPXYPLPSDPERN-UHFFFAOYSA-N Ecteinascidin 743 Natural products COc1cc2C(NCCc2cc1O)C(=O)OCC3N4C(O)C5Cc6cc(C)c(OC)c(O)c6C(C4C(S)c7c(OC(=O)C)c(C)c8OCOc8c37)N5C XXPXYPLPSDPERN-UHFFFAOYSA-N 0.000 description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 3
- HKVAMNSJSFKALM-GKUWKFKPSA-N Everolimus Chemical compound C1C[C@@H](OCCO)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 HKVAMNSJSFKALM-GKUWKFKPSA-N 0.000 description 3
- 208000006168 Ewing Sarcoma Diseases 0.000 description 3
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 3
- OVBPIULPVIDEAO-LBPRGKRZSA-N Folic acid Natural products C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-LBPRGKRZSA-N 0.000 description 3
- 208000022072 Gallbladder Neoplasms Diseases 0.000 description 3
- 108090000288 Glycoproteins Proteins 0.000 description 3
- 102000003886 Glycoproteins Human genes 0.000 description 3
- 108010069236 Goserelin Proteins 0.000 description 3
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 3
- 102100039620 Granulocyte-macrophage colony-stimulating factor Human genes 0.000 description 3
- 208000017604 Hodgkin disease Diseases 0.000 description 3
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 3
- 101000971171 Homo sapiens Apoptosis regulator Bcl-2 Proteins 0.000 description 3
- 101000777277 Homo sapiens Serine/threonine-protein kinase Chk2 Proteins 0.000 description 3
- 241000714260 Human T-lymphotropic virus 1 Species 0.000 description 3
- XDXDZDZNSLXDNA-TZNDIEGXSA-N Idarubicin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XDXDZDZNSLXDNA-TZNDIEGXSA-N 0.000 description 3
- XDXDZDZNSLXDNA-UHFFFAOYSA-N Idarubicin Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XDXDZDZNSLXDNA-UHFFFAOYSA-N 0.000 description 3
- 102000014150 Interferons Human genes 0.000 description 3
- 108010050904 Interferons Proteins 0.000 description 3
- 102000015696 Interleukins Human genes 0.000 description 3
- 108010063738 Interleukins Proteins 0.000 description 3
- 102000012011 Isocitrate Dehydrogenase Human genes 0.000 description 3
- 108010075869 Isocitrate Dehydrogenase Proteins 0.000 description 3
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- 102000042838 JAK family Human genes 0.000 description 3
- 108091082332 JAK family Proteins 0.000 description 3
- 239000002144 L01XE18 - Ruxolitinib Substances 0.000 description 3
- 239000002177 L01XE27 - Ibrutinib Substances 0.000 description 3
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 description 3
- 241000124008 Mammalia Species 0.000 description 3
- 102000029749 Microtubule Human genes 0.000 description 3
- 108091022875 Microtubule Proteins 0.000 description 3
- 229940122255 Microtubule inhibitor Drugs 0.000 description 3
- 201000003793 Myelodysplastic syndrome Diseases 0.000 description 3
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 3
- 206010029260 Neuroblastoma Diseases 0.000 description 3
- 201000010133 Oligodendroglioma Diseases 0.000 description 3
- 108091008606 PDGF receptors Proteins 0.000 description 3
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 3
- 208000007641 Pinealoma Diseases 0.000 description 3
- 108010038512 Platelet-Derived Growth Factor Proteins 0.000 description 3
- 102000010780 Platelet-Derived Growth Factor Human genes 0.000 description 3
- 102000011653 Platelet-Derived Growth Factor Receptors Human genes 0.000 description 3
- 239000002202 Polyethylene glycol Substances 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- 108010014608 Proto-Oncogene Proteins c-kit Proteins 0.000 description 3
- 102000016971 Proto-Oncogene Proteins c-kit Human genes 0.000 description 3
- 201000000582 Retinoblastoma Diseases 0.000 description 3
- 208000004337 Salivary Gland Neoplasms Diseases 0.000 description 3
- 206010061934 Salivary gland cancer Diseases 0.000 description 3
- 102100031075 Serine/threonine-protein kinase Chk2 Human genes 0.000 description 3
- 229920002472 Starch Polymers 0.000 description 3
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 3
- 229930006000 Sucrose Natural products 0.000 description 3
- 229940126547 T-cell immunoglobulin mucin-3 Drugs 0.000 description 3
- 108010017842 Telomerase Proteins 0.000 description 3
- CBPNZQVSJQDFBE-FUXHJELOSA-N Temsirolimus Chemical compound C1C[C@@H](OC(=O)C(C)(CO)CO)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 CBPNZQVSJQDFBE-FUXHJELOSA-N 0.000 description 3
- 102000002689 Toll-like receptor Human genes 0.000 description 3
- 108020000411 Toll-like receptor Proteins 0.000 description 3
- 102000004243 Tubulin Human genes 0.000 description 3
- 108090000704 Tubulin Proteins 0.000 description 3
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 description 3
- 241000700618 Vaccinia virus Species 0.000 description 3
- 208000014070 Vestibular schwannoma Diseases 0.000 description 3
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 3
- 208000004064 acoustic neuroma Diseases 0.000 description 3
- 208000009956 adenocarcinoma Diseases 0.000 description 3
- 235000010443 alginic acid Nutrition 0.000 description 3
- 229920000615 alginic acid Polymers 0.000 description 3
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 3
- 125000003342 alkenyl group Chemical group 0.000 description 3
- 125000004450 alkenylene group Chemical group 0.000 description 3
- YBBLVLTVTVSKRW-UHFFFAOYSA-N anastrozole Chemical compound N#CC(C)(C)C1=CC(C(C)(C#N)C)=CC(CN2N=CN=C2)=C1 YBBLVLTVTVSKRW-UHFFFAOYSA-N 0.000 description 3
- 230000001772 anti-angiogenic effect Effects 0.000 description 3
- 229940046836 anti-estrogen Drugs 0.000 description 3
- 230000001833 anti-estrogenic effect Effects 0.000 description 3
- 230000002927 anti-mitotic effect Effects 0.000 description 3
- 239000002246 antineoplastic agent Substances 0.000 description 3
- 229960002756 azacitidine Drugs 0.000 description 3
- ZAXCMPAWRCMABN-UHFFFAOYSA-N azane;2-hydroxyacetic acid;platinum Chemical compound N.N.[Pt].OCC(O)=O ZAXCMPAWRCMABN-UHFFFAOYSA-N 0.000 description 3
- NCNRHFGMJRPRSK-MDZDMXLPSA-N belinostat Chemical compound ONC(=O)\C=C\C1=CC=CC(S(=O)(=O)NC=2C=CC=CC=2)=C1 NCNRHFGMJRPRSK-MDZDMXLPSA-N 0.000 description 3
- 108010005774 beta-Galactosidase Proteins 0.000 description 3
- 239000011230 binding agent Substances 0.000 description 3
- 150000004663 bisphosphonates Chemical class 0.000 description 3
- 229960001467 bortezomib Drugs 0.000 description 3
- 239000006172 buffering agent Substances 0.000 description 3
- 235000019437 butane-1,3-diol Nutrition 0.000 description 3
- 229960001573 cabazitaxel Drugs 0.000 description 3
- 239000001768 carboxy methyl cellulose Substances 0.000 description 3
- 230000004663 cell proliferation Effects 0.000 description 3
- 201000010881 cervical cancer Diseases 0.000 description 3
- HWGQMRYQVZSGDQ-HZPDHXFCSA-N chembl3137320 Chemical compound CN1N=CN=C1[C@H]([C@H](N1)C=2C=CC(F)=CC=2)C2=NNC(=O)C3=C2C1=CC(F)=C3 HWGQMRYQVZSGDQ-HZPDHXFCSA-N 0.000 description 3
- 208000024207 chronic leukemia Diseases 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 238000011260 co-administration Methods 0.000 description 3
- 230000003111 delayed effect Effects 0.000 description 3
- 239000003534 dna topoisomerase inhibitor Substances 0.000 description 3
- 239000003995 emulsifying agent Substances 0.000 description 3
- 230000001804 emulsifying effect Effects 0.000 description 3
- 239000000839 emulsion Substances 0.000 description 3
- QAMYWGZHLCQOOJ-WRNBYXCMSA-N eribulin mesylate Chemical compound CS(O)(=O)=O.C([C@H]1CC[C@@H]2O[C@@H]3[C@H]4O[C@@H]5C[C@](O[C@H]4[C@H]2O1)(O[C@@H]53)CC[C@@H]1O[C@H](C(C1)=C)CC1)C(=O)C[C@@H]2[C@@H](OC)[C@@H](C[C@H](O)CN)O[C@H]2C[C@@H]2C(=C)[C@H](C)C[C@H]1O2 QAMYWGZHLCQOOJ-WRNBYXCMSA-N 0.000 description 3
- AAKJLRGGTJKAMG-UHFFFAOYSA-N erlotinib Chemical compound C=12C=C(OCCOC)C(OCCOC)=CC2=NC=NC=1NC1=CC=CC(C#C)=C1 AAKJLRGGTJKAMG-UHFFFAOYSA-N 0.000 description 3
- 229940011871 estrogen Drugs 0.000 description 3
- 239000000262 estrogen Substances 0.000 description 3
- 239000000328 estrogen antagonist Substances 0.000 description 3
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 3
- 229960005420 etoposide Drugs 0.000 description 3
- 229960000255 exemestane Drugs 0.000 description 3
- 239000000284 extract Substances 0.000 description 3
- 150000004665 fatty acids Chemical class 0.000 description 3
- 239000000945 filler Substances 0.000 description 3
- 229960002949 fluorouracil Drugs 0.000 description 3
- 235000013305 food Nutrition 0.000 description 3
- 239000012634 fragment Substances 0.000 description 3
- 201000010175 gallbladder cancer Diseases 0.000 description 3
- 201000011243 gastrointestinal stromal tumor Diseases 0.000 description 3
- 239000008187 granular material Substances 0.000 description 3
- 201000010536 head and neck cancer Diseases 0.000 description 3
- 229940121372 histone deacetylase inhibitor Drugs 0.000 description 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 3
- 229960001507 ibrutinib Drugs 0.000 description 3
- XYFPWWZEPKGCCK-GOSISDBHSA-N ibrutinib Chemical compound C1=2C(N)=NC=NC=2N([C@H]2CN(CCC2)C(=O)C=C)N=C1C(C=C1)=CC=C1OC1=CC=CC=C1 XYFPWWZEPKGCCK-GOSISDBHSA-N 0.000 description 3
- 229960000908 idarubicin Drugs 0.000 description 3
- 230000005764 inhibitory process Effects 0.000 description 3
- 230000003993 interaction Effects 0.000 description 3
- 229960004768 irinotecan Drugs 0.000 description 3
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 description 3
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 3
- FABUFPQFXZVHFB-PVYNADRNSA-N ixabepilone Chemical compound C/C([C@@H]1C[C@@H]2O[C@]2(C)CCC[C@@H]([C@@H]([C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)N1)O)C)=C\C1=CSC(C)=N1 FABUFPQFXZVHFB-PVYNADRNSA-N 0.000 description 3
- MXAYKZJJDUDWDS-LBPRGKRZSA-N ixazomib Chemical compound CC(C)C[C@@H](B(O)O)NC(=O)CNC(=O)C1=CC(Cl)=CC=C1Cl MXAYKZJJDUDWDS-LBPRGKRZSA-N 0.000 description 3
- 229960003881 letrozole Drugs 0.000 description 3
- 201000007270 liver cancer Diseases 0.000 description 3
- 210000004698 lymphocyte Anatomy 0.000 description 3
- 235000019359 magnesium stearate Nutrition 0.000 description 3
- 230000036210 malignancy Effects 0.000 description 3
- 230000003211 malignant effect Effects 0.000 description 3
- 238000004519 manufacturing process Methods 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 230000007246 mechanism Effects 0.000 description 3
- 206010027191 meningioma Diseases 0.000 description 3
- GLVAUDGFNGKCSF-UHFFFAOYSA-N mercaptopurine Chemical compound S=C1NC=NC2=C1NC=N2 GLVAUDGFNGKCSF-UHFFFAOYSA-N 0.000 description 3
- 239000002207 metabolite Substances 0.000 description 3
- 210000004688 microtubule Anatomy 0.000 description 3
- 231100000782 microtubule inhibitor Toxicity 0.000 description 3
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 3
- 229960001156 mitoxantrone Drugs 0.000 description 3
- 210000000822 natural killer cell Anatomy 0.000 description 3
- 229950004847 navitoclax Drugs 0.000 description 3
- JLYAXFNOILIKPP-KXQOOQHDSA-N navitoclax Chemical compound C([C@@H](NC1=CC=C(C=C1S(=O)(=O)C(F)(F)F)S(=O)(=O)NC(=O)C1=CC=C(C=C1)N1CCN(CC1)CC1=C(CCC(C1)(C)C)C=1C=CC(Cl)=CC=1)CSC=1C=CC=CC=1)CN1CCOCC1 JLYAXFNOILIKPP-KXQOOQHDSA-N 0.000 description 3
- IXOXBSCIXZEQEQ-UHTZMRCNSA-N nelarabine Chemical compound C1=NC=2C(OC)=NC(N)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@@H]1O IXOXBSCIXZEQEQ-UHTZMRCNSA-N 0.000 description 3
- 208000007538 neurilemmoma Diseases 0.000 description 3
- HHZIURLSWUIHRB-UHFFFAOYSA-N nilotinib Chemical compound C1=NC(C)=CN1C1=CC(NC(=O)C=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)=CC(C(F)(F)F)=C1 HHZIURLSWUIHRB-UHFFFAOYSA-N 0.000 description 3
- 229950011068 niraparib Drugs 0.000 description 3
- PCHKPVIQAHNQLW-CQSZACIVSA-N niraparib Chemical compound N1=C2C(C(=O)N)=CC=CC2=CN1C(C=C1)=CC=C1[C@@H]1CCCNC1 PCHKPVIQAHNQLW-CQSZACIVSA-N 0.000 description 3
- 239000000346 nonvolatile oil Substances 0.000 description 3
- 239000003921 oil Substances 0.000 description 3
- 235000019198 oils Nutrition 0.000 description 3
- FDLYAMZZIXQODN-UHFFFAOYSA-N olaparib Chemical group FC1=CC=C(CC=2C3=CC=CC=C3C(=O)NN=2)C=C1C(=O)N(CC1)CCN1C(=O)C1CC1 FDLYAMZZIXQODN-UHFFFAOYSA-N 0.000 description 3
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 3
- HYFHYPWGAURHIV-JFIAXGOJSA-N omacetaxine mepesuccinate Chemical compound C1=C2CCN3CCC[C@]43C=C(OC)[C@@H](OC(=O)[C@@](O)(CCCC(C)(C)O)CC(=O)OC)[C@H]4C2=CC2=C1OCO2 HYFHYPWGAURHIV-JFIAXGOJSA-N 0.000 description 3
- 230000002246 oncogenic effect Effects 0.000 description 3
- 230000000174 oncolytic effect Effects 0.000 description 3
- 244000309459 oncolytic virus Species 0.000 description 3
- AHJRHEGDXFFMBM-UHFFFAOYSA-N palbociclib Chemical group N1=C2N(C3CCCC3)C(=O)C(C(=O)C)=C(C)C2=CN=C1NC(N=C1)=CC=C1N1CCNCC1 AHJRHEGDXFFMBM-UHFFFAOYSA-N 0.000 description 3
- 229960005184 panobinostat Drugs 0.000 description 3
- WBXPDJSOTKVWSJ-ZDUSSCGKSA-N pemetrexed Chemical compound C=1NC=2NC(N)=NC(=O)C=2C=1CCC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 WBXPDJSOTKVWSJ-ZDUSSCGKSA-N 0.000 description 3
- 229960002087 pertuzumab Drugs 0.000 description 3
- 235000019271 petrolatum Nutrition 0.000 description 3
- 229910052698 phosphorus Inorganic materials 0.000 description 3
- 239000011574 phosphorus Substances 0.000 description 3
- 208000024724 pineal body neoplasm Diseases 0.000 description 3
- 229920000642 polymer Polymers 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 239000003755 preservative agent Substances 0.000 description 3
- 208000029340 primitive neuroectodermal tumor Diseases 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 3
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 3
- 229950004043 radotinib Drugs 0.000 description 3
- DUPWHXBITIZIKZ-UHFFFAOYSA-N radotinib Chemical compound C1=NC(C)=CN1C1=CC(NC(=O)C=2C=C(NC=3N=C(C=CN=3)C=3N=CC=NC=3)C(C)=CC=2)=CC(C(F)(F)F)=C1 DUPWHXBITIZIKZ-UHFFFAOYSA-N 0.000 description 3
- GZUITABIAKMVPG-UHFFFAOYSA-N raloxifene Chemical compound C1=CC(O)=CC=C1C1=C(C(=O)C=2C=CC(OCCN3CCCCC3)=CC=2)C2=CC=C(O)C=C2S1 GZUITABIAKMVPG-UHFFFAOYSA-N 0.000 description 3
- 229950003687 ribociclib Drugs 0.000 description 3
- 229950004707 rucaparib Drugs 0.000 description 3
- 239000000523 sample Substances 0.000 description 3
- 206010039667 schwannoma Diseases 0.000 description 3
- 239000000333 selective estrogen receptor modulator Substances 0.000 description 3
- 230000011664 signaling Effects 0.000 description 3
- 239000007909 solid dosage form Substances 0.000 description 3
- 239000008247 solid mixture Substances 0.000 description 3
- VZZJRYRQSPEMTK-CALCHBBNSA-N sonidegib Chemical compound C1[C@@H](C)O[C@@H](C)CN1C(N=C1)=CC=C1NC(=O)C1=CC=CC(C=2C=CC(OC(F)(F)F)=CC=2)=C1C VZZJRYRQSPEMTK-CALCHBBNSA-N 0.000 description 3
- 208000017572 squamous cell neoplasm Diseases 0.000 description 3
- 235000019698 starch Nutrition 0.000 description 3
- 239000005720 sucrose Substances 0.000 description 3
- 150000003871 sulfonates Chemical class 0.000 description 3
- 239000000829 suppository Substances 0.000 description 3
- 229950001269 taselisib Drugs 0.000 description 3
- NRUKOCRGYNPUPR-QBPJDGROSA-N teniposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@@H](OC[C@H]4O3)C=3SC=CC=3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 NRUKOCRGYNPUPR-QBPJDGROSA-N 0.000 description 3
- 229960001278 teniposide Drugs 0.000 description 3
- ZRKFYGHZFMAOKI-QMGMOQQFSA-N tgfbeta Chemical compound C([C@H](NC(=O)[C@H](C(C)C)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CC(C)C)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CCSC)C(C)C)[C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O)C1=CC=C(O)C=C1 ZRKFYGHZFMAOKI-QMGMOQQFSA-N 0.000 description 3
- 230000009258 tissue cross reactivity Effects 0.000 description 3
- 229940044693 topoisomerase inhibitor Drugs 0.000 description 3
- 229960000303 topotecan Drugs 0.000 description 3
- PKVRCIRHQMSYJX-AIFWHQITSA-N trabectedin Chemical compound C([C@@]1(C(OC2)=O)NCCC3=C1C=C(C(=C3)O)OC)S[C@@H]1C3=C(OC(C)=O)C(C)=C4OCOC4=C3[C@H]2N2[C@@H](O)[C@H](CC=3C4=C(O)C(OC)=C(C)C=3)N(C)[C@H]4[C@@H]21 PKVRCIRHQMSYJX-AIFWHQITSA-N 0.000 description 3
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 3
- 229960000575 trastuzumab Drugs 0.000 description 3
- 229950007217 tremelimumab Drugs 0.000 description 3
- 210000004881 tumor cell Anatomy 0.000 description 3
- VBEQCZHXXJYVRD-GACYYNSASA-N uroanthelone Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CS)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C(C)C)[C@@H](C)O)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCSC)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)CNC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CS)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CS)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC(N)=O)C(C)C)[C@@H](C)CC)C1=CC=C(O)C=C1 VBEQCZHXXJYVRD-GACYYNSASA-N 0.000 description 3
- 229950011257 veliparib Drugs 0.000 description 3
- JNAHVYVRKWKWKQ-CYBMUJFWSA-N veliparib Chemical compound N=1C2=CC=CC(C(N)=O)=C2NC=1[C@@]1(C)CCCN1 JNAHVYVRKWKWKQ-CYBMUJFWSA-N 0.000 description 3
- LQBVNQSMGBZMKD-UHFFFAOYSA-N venetoclax Chemical compound C=1C=C(Cl)C=CC=1C=1CC(C)(C)CCC=1CN(CC1)CCN1C(C=C1OC=2C=C3C=CNC3=NC=2)=CC=C1C(=O)NS(=O)(=O)C(C=C1[N+]([O-])=O)=CC=C1NCC1CCOCC1 LQBVNQSMGBZMKD-UHFFFAOYSA-N 0.000 description 3
- 229960001183 venetoclax Drugs 0.000 description 3
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 3
- BPQMGSKTAYIVFO-UHFFFAOYSA-N vismodegib Chemical compound ClC1=CC(S(=O)(=O)C)=CC=C1C(=O)NC1=CC=C(Cl)C(C=2N=CC=CC=2)=C1 BPQMGSKTAYIVFO-UHFFFAOYSA-N 0.000 description 3
- 229960004276 zoledronic acid Drugs 0.000 description 3
- SVNJBEMPMKWDCO-KCHLEUMXSA-N (2s)-2-[[(2s)-3-carboxy-2-[[2-[[(2s)-5-(diaminomethylideneamino)-2-[[4-oxo-4-[[4-(4-oxo-8-phenylchromen-2-yl)morpholin-4-ium-4-yl]methoxy]butanoyl]amino]pentanoyl]amino]acetyl]amino]propanoyl]amino]-3-hydroxypropanoate Chemical compound C=1C(=O)C2=CC=CC(C=3C=CC=CC=3)=C2OC=1[N+]1(COC(=O)CCC(=O)N[C@@H](CCCNC(=N)N)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CO)C([O-])=O)CCOCC1 SVNJBEMPMKWDCO-KCHLEUMXSA-N 0.000 description 2
- YPBKTZBXSBLTDK-PKNBQFBNSA-N (3e)-3-[(3-bromo-4-fluoroanilino)-nitrosomethylidene]-4-[2-(sulfamoylamino)ethylamino]-1,2,5-oxadiazole Chemical group NS(=O)(=O)NCCNC1=NON\C1=C(N=O)/NC1=CC=C(F)C(Br)=C1 YPBKTZBXSBLTDK-PKNBQFBNSA-N 0.000 description 2
- NBRQRXRBIHVLGI-OWXODZSWSA-N (4as,5ar,12ar)-1,10,11,12a-tetrahydroxy-3,12-dioxo-4a,5,5a,6-tetrahydro-4h-tetracene-2-carboxamide Chemical compound C1C2=CC=CC(O)=C2C(O)=C(C2=O)[C@@H]1C[C@@H]1[C@@]2(O)C(O)=C(C(=O)N)C(=O)C1 NBRQRXRBIHVLGI-OWXODZSWSA-N 0.000 description 2
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 2
- IAKHMKGGTNLKSZ-INIZCTEOSA-N (S)-colchicine Chemical compound C1([C@@H](NC(C)=O)CC2)=CC(=O)C(OC)=CC=C1C1=C2C=C(OC)C(OC)=C1OC IAKHMKGGTNLKSZ-INIZCTEOSA-N 0.000 description 2
- UMGQVUWXNOJOSJ-KMHUVPDISA-N (e)-2-cyano-3-(3,4-dihydroxyphenyl)-n-[(1r)-1-phenylethyl]prop-2-enamide Chemical compound N([C@H](C)C=1C=CC=CC=1)C(=O)C(\C#N)=C\C1=CC=C(O)C(O)=C1 UMGQVUWXNOJOSJ-KMHUVPDISA-N 0.000 description 2
- SOJJMSYMCLIQCZ-CYBMUJFWSA-N 1-[(2r)-4-[2-(2-aminopyrimidin-5-yl)-6-morpholin-4-yl-9-(2,2,2-trifluoroethyl)purin-8-yl]-2-methylpiperazin-1-yl]ethanone Chemical compound C1CN(C(C)=O)[C@H](C)CN1C1=NC2=C(N3CCOCC3)N=C(C=3C=NC(N)=NC=3)N=C2N1CC(F)(F)F SOJJMSYMCLIQCZ-CYBMUJFWSA-N 0.000 description 2
- YJZSUCFGHXQWDM-UHFFFAOYSA-N 1-adamantyl 4-[(2,5-dihydroxyphenyl)methylamino]benzoate Chemical compound OC1=CC=C(O)C(CNC=2C=CC(=CC=2)C(=O)OC23CC4CC(CC(C4)C2)C3)=C1 YJZSUCFGHXQWDM-UHFFFAOYSA-N 0.000 description 2
- UHDGCWIWMRVCDJ-UHFFFAOYSA-N 1-beta-D-Xylofuranosyl-NH-Cytosine Natural products O=C1N=C(N)C=CN1C1C(O)C(O)C(CO)O1 UHDGCWIWMRVCDJ-UHFFFAOYSA-N 0.000 description 2
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- CKTSBUTUHBMZGZ-SHYZEUOFSA-N 2'‐deoxycytidine Chemical compound O=C1N=C(N)C=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 CKTSBUTUHBMZGZ-SHYZEUOFSA-N 0.000 description 2
- UEJJHQNACJXSKW-UHFFFAOYSA-N 2-(2,6-dioxopiperidin-3-yl)-1H-isoindole-1,3(2H)-dione Chemical compound O=C1C2=CC=CC=C2C(=O)N1C1CCC(=O)NC1=O UEJJHQNACJXSKW-UHFFFAOYSA-N 0.000 description 2
- IUVCFHHAEHNCFT-INIZCTEOSA-N 2-[(1s)-1-[4-amino-3-(3-fluoro-4-propan-2-yloxyphenyl)pyrazolo[3,4-d]pyrimidin-1-yl]ethyl]-6-fluoro-3-(3-fluorophenyl)chromen-4-one Chemical compound C1=C(F)C(OC(C)C)=CC=C1C(C1=C(N)N=CN=C11)=NN1[C@@H](C)C1=C(C=2C=C(F)C=CC=2)C(=O)C2=CC(F)=CC=C2O1 IUVCFHHAEHNCFT-INIZCTEOSA-N 0.000 description 2
- RGHYDLZMTYDBDT-UHFFFAOYSA-N 2-amino-8-ethyl-4-methyl-6-(1H-pyrazol-5-yl)-7-pyrido[2,3-d]pyrimidinone Chemical compound O=C1N(CC)C2=NC(N)=NC(C)=C2C=C1C=1C=CNN=1 RGHYDLZMTYDBDT-UHFFFAOYSA-N 0.000 description 2
- QSPOQCXMGPDIHI-UHFFFAOYSA-N 2-amino-n,n-dipropyl-8-[4-(pyrrolidine-1-carbonyl)phenyl]-3h-1-benzazepine-4-carboxamide Chemical compound C1=C2N=C(N)CC(C(=O)N(CCC)CCC)=CC2=CC=C1C(C=C1)=CC=C1C(=O)N1CCCC1 QSPOQCXMGPDIHI-UHFFFAOYSA-N 0.000 description 2
- QINPEPAQOBZPOF-UHFFFAOYSA-N 2-amino-n-[3-[[3-(2-chloro-5-methoxyanilino)quinoxalin-2-yl]sulfamoyl]phenyl]-2-methylpropanamide Chemical compound COC1=CC=C(Cl)C(NC=2C(=NC3=CC=CC=C3N=2)NS(=O)(=O)C=2C=C(NC(=O)C(C)(C)N)C=CC=2)=C1 QINPEPAQOBZPOF-UHFFFAOYSA-N 0.000 description 2
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 2
- 125000002774 3,4-dimethoxybenzyl group Chemical group [H]C1=C([H])C(=C([H])C(OC([H])([H])[H])=C1OC([H])([H])[H])C([H])([H])* 0.000 description 2
- XMIIGOLPHOKFCH-UHFFFAOYSA-M 3-phenylpropionate Chemical compound [O-]C(=O)CCC1=CC=CC=C1 XMIIGOLPHOKFCH-UHFFFAOYSA-M 0.000 description 2
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 description 2
- CLPFFLWZZBQMAO-UHFFFAOYSA-N 4-(5,6,7,8-tetrahydroimidazo[1,5-a]pyridin-5-yl)benzonitrile Chemical compound C1=CC(C#N)=CC=C1C1N2C=NC=C2CCC1 CLPFFLWZZBQMAO-UHFFFAOYSA-N 0.000 description 2
- BGLPECHZZQDNCD-UHFFFAOYSA-N 4-(cyclopropylamino)-2-[4-(4-ethylsulfonylpiperazin-1-yl)anilino]pyrimidine-5-carboxamide Chemical compound C1CN(S(=O)(=O)CC)CCN1C(C=C1)=CC=C1NC1=NC=C(C(N)=O)C(NC2CC2)=N1 BGLPECHZZQDNCD-UHFFFAOYSA-N 0.000 description 2
- ADZBMFGQQWPHMJ-RHSMWYFYSA-N 4-[[2-[[(1r,2r)-2-hydroxycyclohexyl]amino]-1,3-benzothiazol-6-yl]oxy]-n-methylpyridine-2-carboxamide Chemical compound C1=NC(C(=O)NC)=CC(OC=2C=C3SC(N[C@H]4[C@@H](CCCC4)O)=NC3=CC=2)=C1 ADZBMFGQQWPHMJ-RHSMWYFYSA-N 0.000 description 2
- ADGGYDAFIHSYFI-UHFFFAOYSA-N 5-(4,6-dimorpholin-4-yl-1,3,5-triazin-2-yl)-4-(trifluoromethyl)pyridin-2-amine Chemical compound C1=NC(N)=CC(C(F)(F)F)=C1C1=NC(N2CCOCC2)=NC(N2CCOCC2)=N1 ADGGYDAFIHSYFI-UHFFFAOYSA-N 0.000 description 2
- AILRADAXUVEEIR-UHFFFAOYSA-N 5-chloro-4-n-(2-dimethylphosphorylphenyl)-2-n-[2-methoxy-4-[4-(4-methylpiperazin-1-yl)piperidin-1-yl]phenyl]pyrimidine-2,4-diamine Chemical compound COC1=CC(N2CCC(CC2)N2CCN(C)CC2)=CC=C1NC(N=1)=NC=C(Cl)C=1NC1=CC=CC=C1P(C)(C)=O AILRADAXUVEEIR-UHFFFAOYSA-N 0.000 description 2
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 2
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 2
- 229940126253 ADU-S100 Drugs 0.000 description 2
- 206010000830 Acute leukaemia Diseases 0.000 description 2
- 208000009888 Adrenocortical Adenoma Diseases 0.000 description 2
- 108010012934 Albumin-Bound Paclitaxel Proteins 0.000 description 2
- OGSPWJRAVKPPFI-UHFFFAOYSA-N Alendronic Acid Chemical compound NCCCC(O)(P(O)(O)=O)P(O)(O)=O OGSPWJRAVKPPFI-UHFFFAOYSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 2
- 206010061424 Anal cancer Diseases 0.000 description 2
- 208000007860 Anus Neoplasms Diseases 0.000 description 2
- 102000004452 Arginase Human genes 0.000 description 2
- 108700024123 Arginases Proteins 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 206010065869 Astrocytoma, low grade Diseases 0.000 description 2
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 description 2
- 102100024222 B-lymphocyte antigen CD19 Human genes 0.000 description 2
- CWHUFRVAEUJCEF-UHFFFAOYSA-N BKM120 Chemical compound C1=NC(N)=CC(C(F)(F)F)=C1C1=CC(N2CCOCC2)=NC(N2CCOCC2)=N1 CWHUFRVAEUJCEF-UHFFFAOYSA-N 0.000 description 2
- 229940125565 BMS-986016 Drugs 0.000 description 2
- DWRXFEITVBNRMK-UHFFFAOYSA-N Beta-D-1-Arabinofuranosylthymine Natural products O=C1NC(=O)C(C)=CN1C1C(O)C(O)C(CO)O1 DWRXFEITVBNRMK-UHFFFAOYSA-N 0.000 description 2
- 208000006386 Bone Resorption Diseases 0.000 description 2
- 101710149863 C-C chemokine receptor type 4 Proteins 0.000 description 2
- 102100032976 CCR4-NOT transcription complex subunit 6 Human genes 0.000 description 2
- 102100027207 CD27 antigen Human genes 0.000 description 2
- 210000001266 CD8-positive T-lymphocyte Anatomy 0.000 description 2
- 101150050673 CHK1 gene Proteins 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- KLWPJMFMVPTNCC-UHFFFAOYSA-N Camptothecin Natural products CCC1(O)C(=O)OCC2=C1C=C3C4Nc5ccccc5C=C4CN3C2=O KLWPJMFMVPTNCC-UHFFFAOYSA-N 0.000 description 2
- GAGWJHPBXLXJQN-UHFFFAOYSA-N Capecitabine Natural products C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1C1C(O)C(O)C(C)O1 GAGWJHPBXLXJQN-UHFFFAOYSA-N 0.000 description 2
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 2
- DLGOEMSEDOSKAD-UHFFFAOYSA-N Carmustine Chemical compound ClCCNC(=O)N(N=O)CCCl DLGOEMSEDOSKAD-UHFFFAOYSA-N 0.000 description 2
- 201000009047 Chordoma Diseases 0.000 description 2
- 229920002261 Corn starch Polymers 0.000 description 2
- 108010025464 Cyclin-Dependent Kinase 4 Proteins 0.000 description 2
- 108010025468 Cyclin-Dependent Kinase 6 Proteins 0.000 description 2
- 102100036252 Cyclin-dependent kinase 4 Human genes 0.000 description 2
- 102100026804 Cyclin-dependent kinase 6 Human genes 0.000 description 2
- 102000003903 Cyclin-dependent kinases Human genes 0.000 description 2
- 108090000266 Cyclin-dependent kinases Proteins 0.000 description 2
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 2
- UHDGCWIWMRVCDJ-PSQAKQOGSA-N Cytidine Natural products O=C1N=C(N)C=CN1[C@@H]1[C@@H](O)[C@@H](O)[C@H](CO)O1 UHDGCWIWMRVCDJ-PSQAKQOGSA-N 0.000 description 2
- 108020004414 DNA Proteins 0.000 description 2
- 229940123780 DNA topoisomerase I inhibitor Drugs 0.000 description 2
- 229940124087 DNA topoisomerase II inhibitor Drugs 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- 241000588700 Dickeya chrysanthemi Species 0.000 description 2
- 102400001368 Epidermal growth factor Human genes 0.000 description 2
- 101800003838 Epidermal growth factor Proteins 0.000 description 2
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 description 2
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 2
- 108091008794 FGF receptors Proteins 0.000 description 2
- 102000044168 Fibroblast Growth Factor Receptor Human genes 0.000 description 2
- 201000008808 Fibrosarcoma Diseases 0.000 description 2
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 2
- VWUXBMIQPBEWFH-WCCTWKNTSA-N Fulvestrant Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3[C@H](CCCCCCCCCS(=O)CCCC(F)(F)C(F)(F)F)CC2=C1 VWUXBMIQPBEWFH-WCCTWKNTSA-N 0.000 description 2
- 229940126656 GS-4224 Drugs 0.000 description 2
- 206010017993 Gastrointestinal neoplasms Diseases 0.000 description 2
- 206010051066 Gastrointestinal stromal tumour Diseases 0.000 description 2
- 201000010915 Glioblastoma multiforme Diseases 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- 244000060234 Gmelina philippensis Species 0.000 description 2
- 102400000932 Gonadoliberin-1 Human genes 0.000 description 2
- 239000000579 Gonadotropin-Releasing Hormone Substances 0.000 description 2
- BLCLNMBMMGCOAS-URPVMXJPSA-N Goserelin Chemical compound C([C@@H](C(=O)N[C@H](COC(C)(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N1[C@@H](CCC1)C(=O)NNC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H]1NC(=O)CC1)C1=CC=C(O)C=C1 BLCLNMBMMGCOAS-URPVMXJPSA-N 0.000 description 2
- 101150112082 Gpnmb gene Proteins 0.000 description 2
- ZRALSGWEFCBTJO-UHFFFAOYSA-N Guanidine Chemical compound NC(N)=N ZRALSGWEFCBTJO-UHFFFAOYSA-N 0.000 description 2
- 208000002250 Hematologic Neoplasms Diseases 0.000 description 2
- 239000004705 High-molecular-weight polyethylene Substances 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- 101000980825 Homo sapiens B-lymphocyte antigen CD19 Proteins 0.000 description 2
- 101000914511 Homo sapiens CD27 antigen Proteins 0.000 description 2
- 101500026183 Homo sapiens Gonadoliberin-1 Proteins 0.000 description 2
- 101000844245 Homo sapiens Non-receptor tyrosine-protein kinase TYK2 Proteins 0.000 description 2
- 101000692455 Homo sapiens Platelet-derived growth factor receptor beta Proteins 0.000 description 2
- 101000716693 Homo sapiens Sodium/iodide cotransporter Proteins 0.000 description 2
- 101000831007 Homo sapiens T-cell immunoreceptor with Ig and ITIM domains Proteins 0.000 description 2
- 241000725303 Human immunodeficiency virus Species 0.000 description 2
- 241000701806 Human papillomavirus Species 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- 108010031794 IGF Type 1 Receptor Proteins 0.000 description 2
- MPBVHIBUJCELCL-UHFFFAOYSA-N Ibandronate Chemical compound CCCCCN(C)CCC(O)(P(O)(O)=O)P(O)(O)=O MPBVHIBUJCELCL-UHFFFAOYSA-N 0.000 description 2
- 102000037982 Immune checkpoint proteins Human genes 0.000 description 2
- 108091008036 Immune checkpoint proteins Proteins 0.000 description 2
- 102000037978 Immune checkpoint receptors Human genes 0.000 description 2
- 108091008028 Immune checkpoint receptors Proteins 0.000 description 2
- 108060003951 Immunoglobulin Proteins 0.000 description 2
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 2
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 2
- 102100021317 Inducible T-cell costimulator Human genes 0.000 description 2
- VDJHFHXMUKFKET-UHFFFAOYSA-N Ingenol mebutate Natural products CC1CC2C(C)(C)C2C2C=C(CO)C(O)C3(O)C(OC(=O)C(C)=CC)C(C)=CC31C2=O VDJHFHXMUKFKET-UHFFFAOYSA-N 0.000 description 2
- 102100039688 Insulin-like growth factor 1 receptor Human genes 0.000 description 2
- 108010002586 Interleukin-7 Proteins 0.000 description 2
- 239000005411 L01XE02 - Gefitinib Substances 0.000 description 2
- 239000005551 L01XE03 - Erlotinib Substances 0.000 description 2
- 239000002147 L01XE04 - Sunitinib Substances 0.000 description 2
- 239000005511 L01XE05 - Sorafenib Substances 0.000 description 2
- 239000002136 L01XE07 - Lapatinib Substances 0.000 description 2
- 239000005536 L01XE08 - Nilotinib Substances 0.000 description 2
- 239000003798 L01XE11 - Pazopanib Substances 0.000 description 2
- 239000002118 L01XE12 - Vandetanib Substances 0.000 description 2
- 239000002145 L01XE14 - Bosutinib Substances 0.000 description 2
- 239000002146 L01XE16 - Crizotinib Substances 0.000 description 2
- 208000031671 Large B-Cell Diffuse Lymphoma Diseases 0.000 description 2
- 241000712899 Lymphocytic choriomeningitis mammarenavirus Species 0.000 description 2
- 229940124761 MMP inhibitor Drugs 0.000 description 2
- MVBPAIHFZZKRGD-UHFFFAOYSA-N MTIC Chemical compound CNN=NC=1NC=NC=1C(N)=O MVBPAIHFZZKRGD-UHFFFAOYSA-N 0.000 description 2
- 208000002030 Merkel cell carcinoma Diseases 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- 101100335081 Mus musculus Flt3 gene Proteins 0.000 description 2
- 101100407308 Mus musculus Pdcd1lg2 gene Proteins 0.000 description 2
- OVBPIULPVIDEAO-UHFFFAOYSA-N N-Pteroyl-L-glutaminsaeure Natural products C=1N=C2NC(N)=NC(=O)C2=NC=1CNC1=CC=C(C(=O)NC(CCC(O)=O)C(O)=O)C=C1 OVBPIULPVIDEAO-UHFFFAOYSA-N 0.000 description 2
- 102000000834 NK Cell Lectin-Like Receptor Subfamily C Human genes 0.000 description 2
- 108010001880 NK Cell Lectin-Like Receptor Subfamily C Proteins 0.000 description 2
- 229910002651 NO3 Inorganic materials 0.000 description 2
- 206010029266 Neuroendocrine carcinoma of the skin Diseases 0.000 description 2
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 2
- 102100032028 Non-receptor tyrosine-protein kinase TYK2 Human genes 0.000 description 2
- YGACXVRLDHEXKY-WXRXAMBDSA-N O[C@H](C[C@H]1c2c(cccc2F)-c2cncn12)[C@H]1CC[C@H](O)CC1 Chemical compound O[C@H](C[C@H]1c2c(cccc2F)-c2cncn12)[C@H]1CC[C@H](O)CC1 YGACXVRLDHEXKY-WXRXAMBDSA-N 0.000 description 2
- 239000005642 Oleic acid Substances 0.000 description 2
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 2
- 206010073338 Optic glioma Diseases 0.000 description 2
- 239000012661 PARP inhibitor Substances 0.000 description 2
- 229940124060 PD-1 antagonist Drugs 0.000 description 2
- 229940123751 PD-L1 antagonist Drugs 0.000 description 2
- HZLFFNCLTRVYJG-WWGOJCOQSA-N Patidegib Chemical compound C([C@@]1(CC(C)=C2C3)O[C@@H]4C[C@H](C)CN[C@H]4[C@H]1C)C[C@H]2[C@H]1[C@H]3[C@@]2(C)CC[C@@H](NS(C)(=O)=O)C[C@H]2CC1 HZLFFNCLTRVYJG-WWGOJCOQSA-N 0.000 description 2
- 208000027190 Peripheral T-cell lymphomas Diseases 0.000 description 2
- 239000004264 Petrolatum Substances 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 208000007913 Pituitary Neoplasms Diseases 0.000 description 2
- 102100026547 Platelet-derived growth factor receptor beta Human genes 0.000 description 2
- 229940121906 Poly ADP ribose polymerase inhibitor Drugs 0.000 description 2
- 108700030875 Programmed Cell Death 1 Ligand 2 Proteins 0.000 description 2
- 102000004245 Proteasome Endopeptidase Complex Human genes 0.000 description 2
- 108090000708 Proteasome Endopeptidase Complex Proteins 0.000 description 2
- 108010029485 Protein Isoforms Proteins 0.000 description 2
- 102000001708 Protein Isoforms Human genes 0.000 description 2
- 108090000315 Protein Kinase C Proteins 0.000 description 2
- 102000003923 Protein Kinase C Human genes 0.000 description 2
- 102000009516 Protein Serine-Threonine Kinases Human genes 0.000 description 2
- 108010009341 Protein Serine-Threonine Kinases Proteins 0.000 description 2
- 229940125566 REGN3767 Drugs 0.000 description 2
- 239000005464 Radotinib Substances 0.000 description 2
- 108091005682 Receptor kinases Proteins 0.000 description 2
- 229940127395 Ribonucleotide Reductase Inhibitors Drugs 0.000 description 2
- IIDJRNMFWXDHID-UHFFFAOYSA-N Risedronic acid Chemical compound OP(=O)(O)C(P(O)(O)=O)(O)CC1=CC=CN=C1 IIDJRNMFWXDHID-UHFFFAOYSA-N 0.000 description 2
- 108010040181 SF 1126 Proteins 0.000 description 2
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 2
- 208000000453 Skin Neoplasms Diseases 0.000 description 2
- 102000013380 Smoothened Receptor Human genes 0.000 description 2
- 108010090739 Smoothened Receptor Proteins 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- 208000021712 Soft tissue sarcoma Diseases 0.000 description 2
- 101000857870 Squalus acanthias Gonadoliberin Proteins 0.000 description 2
- 102100035533 Stimulator of interferon genes protein Human genes 0.000 description 2
- 101710196623 Stimulator of interferon genes protein Proteins 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 108091008874 T cell receptors Proteins 0.000 description 2
- 102000016266 T-Cell Antigen Receptors Human genes 0.000 description 2
- 208000031672 T-Cell Peripheral Lymphoma Diseases 0.000 description 2
- 102100024834 T-cell immunoreceptor with Ig and ITIM domains Human genes 0.000 description 2
- 108010065917 TOR Serine-Threonine Kinases Proteins 0.000 description 2
- 102000013530 TOR Serine-Threonine Kinases Human genes 0.000 description 2
- MUMGGOZAMZWBJJ-DYKIIFRCSA-N Testostosterone Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 MUMGGOZAMZWBJJ-DYKIIFRCSA-N 0.000 description 2
- DKJJVAGXPKPDRL-UHFFFAOYSA-N Tiludronic acid Chemical compound OP(O)(=O)C(P(O)(O)=O)SC1=CC=C(Cl)C=C1 DKJJVAGXPKPDRL-UHFFFAOYSA-N 0.000 description 2
- 239000000365 Topoisomerase I Inhibitor Substances 0.000 description 2
- 239000000317 Topoisomerase II Inhibitor Substances 0.000 description 2
- 102100022153 Tumor necrosis factor receptor superfamily member 4 Human genes 0.000 description 2
- 101710165473 Tumor necrosis factor receptor superfamily member 4 Proteins 0.000 description 2
- 208000002495 Uterine Neoplasms Diseases 0.000 description 2
- 241000711975 Vesicular stomatitis virus Species 0.000 description 2
- 241000700605 Viruses Species 0.000 description 2
- IEDXPSOJFSVCKU-HOKPPMCLSA-N [4-[[(2S)-5-(carbamoylamino)-2-[[(2S)-2-[6-(2,5-dioxopyrrolidin-1-yl)hexanoylamino]-3-methylbutanoyl]amino]pentanoyl]amino]phenyl]methyl N-[(2S)-1-[[(2S)-1-[[(3R,4S,5S)-1-[(2S)-2-[(1R,2R)-3-[[(1S,2R)-1-hydroxy-1-phenylpropan-2-yl]amino]-1-methoxy-2-methyl-3-oxopropyl]pyrrolidin-1-yl]-3-methoxy-5-methyl-1-oxoheptan-4-yl]-methylamino]-3-methyl-1-oxobutan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]-N-methylcarbamate Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C)[C@@H](O)c1ccccc1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)OCc1ccc(NC(=O)[C@H](CCCNC(N)=O)NC(=O)[C@@H](NC(=O)CCCCCN2C(=O)CCC2=O)C(C)C)cc1)C(C)C IEDXPSOJFSVCKU-HOKPPMCLSA-N 0.000 description 2
- IBXPAFBDJCXCDW-MHFPCNPESA-A [Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].Cc1cn([C@H]2C[C@H](O)[C@@H](COP([S-])(=O)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3CO)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c3nc(N)[nH]c4=O)n3ccc(N)nc3=O)n3cnc4c3nc(N)[nH]c4=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3ccc(N)nc3=O)n3cnc4c3nc(N)[nH]c4=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O Chemical compound [Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].Cc1cn([C@H]2C[C@H](O)[C@@H](COP([S-])(=O)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3COP([O-])(=S)O[C@H]3C[C@@H](O[C@@H]3CO)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c3nc(N)[nH]c4=O)n3ccc(N)nc3=O)n3cnc4c3nc(N)[nH]c4=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3ccc(N)nc3=O)n3cnc4c3nc(N)[nH]c4=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O IBXPAFBDJCXCDW-MHFPCNPESA-A 0.000 description 2
- 229950001573 abemaciclib Drugs 0.000 description 2
- GZOSMCIZMLWJML-VJLLXTKPSA-N abiraterone Chemical compound C([C@H]1[C@H]2[C@@H]([C@]3(CC[C@H](O)CC3=CC2)C)CC[C@@]11C)C=C1C1=CC=CN=C1 GZOSMCIZMLWJML-VJLLXTKPSA-N 0.000 description 2
- 230000002159 abnormal effect Effects 0.000 description 2
- 230000004913 activation Effects 0.000 description 2
- 208000015234 adrenal cortex adenoma Diseases 0.000 description 2
- 201000003354 adrenal cortical adenoma Diseases 0.000 description 2
- 108010081667 aflibercept Proteins 0.000 description 2
- 229960004343 alendronic acid Drugs 0.000 description 2
- 150000005215 alkyl ethers Chemical group 0.000 description 2
- KUFRQPKVAWMTJO-LMZWQJSESA-N alvespimycin Chemical compound N1C(=O)\C(C)=C\C=C/[C@H](OC)[C@@H](OC(N)=O)\C(C)=C\[C@H](C)[C@@H](O)[C@@H](OC)C[C@H](C)CC2=C(NCCN(C)C)C(=O)C=C1C2=O KUFRQPKVAWMTJO-LMZWQJSESA-N 0.000 description 2
- 150000001408 amides Chemical group 0.000 description 2
- ROBVIMPUHSLWNV-UHFFFAOYSA-N aminoglutethimide Chemical compound C=1C=C(N)C=CC=1C1(CC)CCC(=O)NC1=O ROBVIMPUHSLWNV-UHFFFAOYSA-N 0.000 description 2
- 229960003437 aminoglutethimide Drugs 0.000 description 2
- 229960002932 anastrozole Drugs 0.000 description 2
- 230000002280 anti-androgenic effect Effects 0.000 description 2
- 239000000051 antiandrogen Substances 0.000 description 2
- 239000002814 antineoplastic antimetabolite Substances 0.000 description 2
- 201000011165 anus cancer Diseases 0.000 description 2
- 238000013459 approach Methods 0.000 description 2
- 239000007900 aqueous suspension Substances 0.000 description 2
- 150000008209 arabinosides Chemical class 0.000 description 2
- 229940046844 aromatase inhibitors Drugs 0.000 description 2
- 229960003272 asparaginase Drugs 0.000 description 2
- DCXYFEDJOCDNAF-UHFFFAOYSA-M asparaginate Chemical compound [O-]C(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-M 0.000 description 2
- RITAVMQDGBJQJZ-FMIVXFBMSA-N axitinib Chemical compound CNC(=O)C1=CC=CC=C1SC1=CC=C(C(\C=C\C=2N=CC=CC=2)=NN2)C2=C1 RITAVMQDGBJQJZ-FMIVXFBMSA-N 0.000 description 2
- 210000003719 b-lymphocyte Anatomy 0.000 description 2
- 239000002585 base Substances 0.000 description 2
- 229960003094 belinostat Drugs 0.000 description 2
- YTKUWDBFDASYHO-UHFFFAOYSA-N bendamustine Chemical compound ClCCN(CCCl)C1=CC=C2N(C)C(CCCC(O)=O)=NC2=C1 YTKUWDBFDASYHO-UHFFFAOYSA-N 0.000 description 2
- 229960002707 bendamustine Drugs 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 229940077388 benzenesulfonate Drugs 0.000 description 2
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- SESFRYSPDFLNCH-UHFFFAOYSA-N benzyl benzoate Chemical compound C=1C=CC=CC=1C(=O)OCC1=CC=CC=C1 SESFRYSPDFLNCH-UHFFFAOYSA-N 0.000 description 2
- WQZGKKKJIJFFOK-FPRJBGLDSA-N beta-D-galactose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@H]1O WQZGKKKJIJFFOK-FPRJBGLDSA-N 0.000 description 2
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 description 2
- XMIIGOLPHOKFCH-UHFFFAOYSA-N beta-phenylpropanoic acid Natural products OC(=O)CCC1=CC=CC=C1 XMIIGOLPHOKFCH-UHFFFAOYSA-N 0.000 description 2
- 229960000397 bevacizumab Drugs 0.000 description 2
- 229920002988 biodegradable polymer Polymers 0.000 description 2
- 239000004621 biodegradable polymer Substances 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- 239000000090 biomarker Substances 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 230000024279 bone resorption Effects 0.000 description 2
- UBPYILGKFZZVDX-UHFFFAOYSA-N bosutinib Chemical compound C1=C(Cl)C(OC)=CC(NC=2C3=CC(OC)=C(OCCCN4CCN(C)CC4)C=C3N=CC=2C#N)=C1Cl UBPYILGKFZZVDX-UHFFFAOYSA-N 0.000 description 2
- 229960000455 brentuximab vedotin Drugs 0.000 description 2
- 229950004272 brigatinib Drugs 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- 229950003628 buparlisib Drugs 0.000 description 2
- 229940127093 camptothecin Drugs 0.000 description 2
- VSJKWCGYPAHWDS-FQEVSTJZSA-N camptothecin Chemical compound C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-FQEVSTJZSA-N 0.000 description 2
- 229940022399 cancer vaccine Drugs 0.000 description 2
- 238000009566 cancer vaccine Methods 0.000 description 2
- 229960004117 capecitabine Drugs 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 2
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 2
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 2
- BLMPQMFVWMYDKT-NZTKNTHTSA-N carfilzomib Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC(C)C)C(=O)[C@]1(C)OC1)NC(=O)CN1CCOCC1)CC1=CC=CC=C1 BLMPQMFVWMYDKT-NZTKNTHTSA-N 0.000 description 2
- 108010021331 carfilzomib Proteins 0.000 description 2
- 229960002438 carfilzomib Drugs 0.000 description 2
- 239000004359 castor oil Substances 0.000 description 2
- 235000019438 castor oil Nutrition 0.000 description 2
- 230000030833 cell death Effects 0.000 description 2
- 230000024245 cell differentiation Effects 0.000 description 2
- 229950006295 cerdulatinib Drugs 0.000 description 2
- VERWOWGGCGHDQE-UHFFFAOYSA-N ceritinib Chemical compound CC=1C=C(NC=2N=C(NC=3C(=CC=CC=3)S(=O)(=O)C(C)C)C(Cl)=CN=2)C(OC(C)C)=CC=1C1CCNCC1 VERWOWGGCGHDQE-UHFFFAOYSA-N 0.000 description 2
- 229960005395 cetuximab Drugs 0.000 description 2
- XDLYKKIQACFMJG-WKILWMFISA-N chembl1234354 Chemical compound C1=NC(OC)=CC=C1C(C1=O)=CC2=C(C)N=C(N)N=C2N1[C@@H]1CC[C@@H](OCCO)CC1 XDLYKKIQACFMJG-WKILWMFISA-N 0.000 description 2
- 238000006243 chemical reaction Methods 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- 230000001684 chronic effect Effects 0.000 description 2
- 208000032852 chronic lymphocytic leukemia Diseases 0.000 description 2
- 229960002286 clodronic acid Drugs 0.000 description 2
- 229940110456 cocoa butter Drugs 0.000 description 2
- 235000019868 cocoa butter Nutrition 0.000 description 2
- 201000010989 colorectal carcinoma Diseases 0.000 description 2
- 229950002550 copanlisib Drugs 0.000 description 2
- PZBCKZWLPGJMAO-UHFFFAOYSA-N copanlisib Chemical compound C1=CC=2C3=NCCN3C(NC(=O)C=3C=NC(N)=NC=3)=NC=2C(OC)=C1OCCCN1CCOCC1 PZBCKZWLPGJMAO-UHFFFAOYSA-N 0.000 description 2
- 239000008120 corn starch Substances 0.000 description 2
- 229940099112 cornstarch Drugs 0.000 description 2
- 239000006071 cream Substances 0.000 description 2
- KTEIFNKAUNYNJU-GFCCVEGCSA-N crizotinib Chemical compound O([C@H](C)C=1C(=C(F)C=CC=1Cl)Cl)C(C(=NC=1)N)=CC=1C(=C1)C=NN1C1CCNCC1 KTEIFNKAUNYNJU-GFCCVEGCSA-N 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- VFLDPWHFBUODDF-FCXRPNKRSA-N curcumin Chemical compound C1=C(O)C(OC)=CC(\C=C\C(=O)CC(=O)\C=C\C=2C=C(OC)C(O)=CC=2)=C1 VFLDPWHFBUODDF-FCXRPNKRSA-N 0.000 description 2
- 208000017763 cutaneous neuroendocrine carcinoma Diseases 0.000 description 2
- 239000002875 cyclin dependent kinase inhibitor Substances 0.000 description 2
- 229940043378 cyclin-dependent kinase inhibitor Drugs 0.000 description 2
- 229960004397 cyclophosphamide Drugs 0.000 description 2
- UHDGCWIWMRVCDJ-ZAKLUEHWSA-N cytidine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O1 UHDGCWIWMRVCDJ-ZAKLUEHWSA-N 0.000 description 2
- OPTASPLRGRRNAP-UHFFFAOYSA-N cytosine Chemical compound NC=1C=CNC(=O)N=1 OPTASPLRGRRNAP-UHFFFAOYSA-N 0.000 description 2
- BFSMGDJOXZAERB-UHFFFAOYSA-N dabrafenib Chemical compound S1C(C(C)(C)C)=NC(C=2C(=C(NS(=O)(=O)C=3C(=CC=CC=3F)F)C=CC=2)F)=C1C1=CC=NC(N)=N1 BFSMGDJOXZAERB-UHFFFAOYSA-N 0.000 description 2
- 229950006418 dactolisib Drugs 0.000 description 2
- JOGKUKXHTYWRGZ-UHFFFAOYSA-N dactolisib Chemical compound O=C1N(C)C2=CN=C3C=CC(C=4C=C5C=CC=CC5=NC=4)=CC3=C2N1C1=CC=C(C(C)(C)C#N)C=C1 JOGKUKXHTYWRGZ-UHFFFAOYSA-N 0.000 description 2
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 2
- 229960000975 daunorubicin Drugs 0.000 description 2
- 230000034994 death Effects 0.000 description 2
- 230000002950 deficient Effects 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 230000018109 developmental process Effects 0.000 description 2
- 238000003745 diagnosis Methods 0.000 description 2
- 235000005911 diet Nutrition 0.000 description 2
- 230000037213 diet Effects 0.000 description 2
- 230000004069 differentiation Effects 0.000 description 2
- 206010012818 diffuse large B-cell lymphoma Diseases 0.000 description 2
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 2
- KPUWHANPEXNPJT-UHFFFAOYSA-N disiloxane Chemical group [SiH3]O[SiH3] KPUWHANPEXNPJT-UHFFFAOYSA-N 0.000 description 2
- AUZONCFQVSMFAP-UHFFFAOYSA-N disulfiram Chemical compound CCN(CC)C(=S)SSC(=S)N(CC)CC AUZONCFQVSMFAP-UHFFFAOYSA-N 0.000 description 2
- VSJKWCGYPAHWDS-UHFFFAOYSA-N dl-camptothecin Natural products C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)C5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-UHFFFAOYSA-N 0.000 description 2
- MOTZDAYCYVMXPC-UHFFFAOYSA-N dodecyl hydrogen sulfate Chemical compound CCCCCCCCCCCCOS(O)(=O)=O MOTZDAYCYVMXPC-UHFFFAOYSA-N 0.000 description 2
- 229940043264 dodecyl sulfate Drugs 0.000 description 2
- 229960004679 doxorubicin Drugs 0.000 description 2
- 239000008298 dragée Substances 0.000 description 2
- 229950004949 duvelisib Drugs 0.000 description 2
- 239000012636 effector Substances 0.000 description 2
- 229940056913 eftilagimod alfa Drugs 0.000 description 2
- 239000002702 enteric coating Substances 0.000 description 2
- 238000009505 enteric coating Methods 0.000 description 2
- 229950005837 entinostat Drugs 0.000 description 2
- WXCXUHSOUPDCQV-UHFFFAOYSA-N enzalutamide Chemical compound C1=C(F)C(C(=O)NC)=CC=C1N1C(C)(C)C(=O)N(C=2C=C(C(C#N)=CC=2)C(F)(F)F)C1=S WXCXUHSOUPDCQV-UHFFFAOYSA-N 0.000 description 2
- 229950006370 epacadostat Drugs 0.000 description 2
- 229940116977 epidermal growth factor Drugs 0.000 description 2
- 229960001904 epirubicin Drugs 0.000 description 2
- 229940082789 erbitux Drugs 0.000 description 2
- 229960000439 eribulin mesylate Drugs 0.000 description 2
- 229960001433 erlotinib Drugs 0.000 description 2
- 108010038795 estrogen receptors Proteins 0.000 description 2
- 235000019441 ethanol Nutrition 0.000 description 2
- 150000002170 ethers Chemical group 0.000 description 2
- 229960005167 everolimus Drugs 0.000 description 2
- 229950011548 fadrozole Drugs 0.000 description 2
- 201000001343 fallopian tube carcinoma Diseases 0.000 description 2
- 239000000796 flavoring agent Substances 0.000 description 2
- GIUYCYHIANZCFB-FJFJXFQQSA-N fludarabine phosphate Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O GIUYCYHIANZCFB-FJFJXFQQSA-N 0.000 description 2
- 239000011737 fluorine Substances 0.000 description 2
- 229960000304 folic acid Drugs 0.000 description 2
- 235000019152 folic acid Nutrition 0.000 description 2
- 239000011724 folic acid Substances 0.000 description 2
- 229960004421 formestane Drugs 0.000 description 2
- OSVMTWJCGUFAOD-KZQROQTASA-N formestane Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CCC2=C1O OSVMTWJCGUFAOD-KZQROQTASA-N 0.000 description 2
- 229960002258 fulvestrant Drugs 0.000 description 2
- 108020001507 fusion proteins Proteins 0.000 description 2
- 102000037865 fusion proteins Human genes 0.000 description 2
- 230000002496 gastric effect Effects 0.000 description 2
- 229960002584 gefitinib Drugs 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 239000007903 gelatin capsule Substances 0.000 description 2
- QTQAWLPCGQOSGP-GBTDJJJQSA-N geldanamycin Chemical class N1C(=O)\C(C)=C/C=C\[C@@H](OC)[C@H](OC(N)=O)\C(C)=C/[C@@H](C)[C@@H](O)[C@H](OC)C[C@@H](C)CC2=C(OC)C(=O)C=C1C2=O QTQAWLPCGQOSGP-GBTDJJJQSA-N 0.000 description 2
- 229960005277 gemcitabine Drugs 0.000 description 2
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 2
- 208000002409 gliosarcoma Diseases 0.000 description 2
- 125000005456 glyceride group Chemical group 0.000 description 2
- 229960001442 gonadorelin Drugs 0.000 description 2
- 229940035638 gonadotropin-releasing hormone Drugs 0.000 description 2
- 229960002913 goserelin Drugs 0.000 description 2
- 239000003102 growth factor Substances 0.000 description 2
- 230000036541 health Effects 0.000 description 2
- 239000003481 heat shock protein 90 inhibitor Substances 0.000 description 2
- 201000002222 hemangioblastoma Diseases 0.000 description 2
- 125000004475 heteroaralkyl group Chemical group 0.000 description 2
- 208000029824 high grade glioma Diseases 0.000 description 2
- HYFHYPWGAURHIV-UHFFFAOYSA-N homoharringtonine Natural products C1=C2CCN3CCCC43C=C(OC)C(OC(=O)C(O)(CCCC(C)(C)O)CC(=O)OC)C4C2=CC2=C1OCO2 HYFHYPWGAURHIV-UHFFFAOYSA-N 0.000 description 2
- 229940040731 human interleukin-12 Drugs 0.000 description 2
- 230000036571 hydration Effects 0.000 description 2
- 238000006703 hydration reaction Methods 0.000 description 2
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 2
- FDGQSTZJBFJUBT-UHFFFAOYSA-N hypoxanthine Chemical compound O=C1NC=NC2=C1NC=N2 FDGQSTZJBFJUBT-UHFFFAOYSA-N 0.000 description 2
- 229960005236 ibandronic acid Drugs 0.000 description 2
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 2
- 229960001101 ifosfamide Drugs 0.000 description 2
- YLMAHDNUQAMNNX-UHFFFAOYSA-N imatinib methanesulfonate Chemical compound CS(O)(=O)=O.C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 YLMAHDNUQAMNNX-UHFFFAOYSA-N 0.000 description 2
- 229940091204 imlygic Drugs 0.000 description 2
- 210000000987 immune system Anatomy 0.000 description 2
- 102000018358 immunoglobulin Human genes 0.000 description 2
- 238000009169 immunotherapy Methods 0.000 description 2
- 229950009034 indoximod Drugs 0.000 description 2
- VDJHFHXMUKFKET-WDUFCVPESA-N ingenol mebutate Chemical compound C[C@@H]1C[C@H]2C(C)(C)[C@H]2[C@@H]2C=C(CO)[C@@H](O)[C@]3(O)[C@@H](OC(=O)C(\C)=C/C)C(C)=C[C@]31C2=O VDJHFHXMUKFKET-WDUFCVPESA-N 0.000 description 2
- 108091008042 inhibitory receptors Proteins 0.000 description 2
- 229940102223 injectable solution Drugs 0.000 description 2
- 229940102213 injectable suspension Drugs 0.000 description 2
- 229940079322 interferon Drugs 0.000 description 2
- 230000004068 intracellular signaling Effects 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- 229940084651 iressa Drugs 0.000 description 2
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 2
- 229960002014 ixabepilone Drugs 0.000 description 2
- 229960003648 ixazomib Drugs 0.000 description 2
- 229940025735 jevtana Drugs 0.000 description 2
- 201000000573 juvenile pilocytic astrocytoma Diseases 0.000 description 2
- BCFGMOOMADDAQU-UHFFFAOYSA-N lapatinib Chemical compound O1C(CNCCS(=O)(=O)C)=CC=C1C1=CC=C(N=CN=C2NC=3C=C(Cl)C(OCC=4C=C(F)C=CC=4)=CC=3)C2=C1 BCFGMOOMADDAQU-UHFFFAOYSA-N 0.000 description 2
- GOTYRUGSSMKFNF-UHFFFAOYSA-N lenalidomide Chemical compound C1C=2C(N)=CC=CC=2C(=O)N1C1CCC(=O)NC1=O GOTYRUGSSMKFNF-UHFFFAOYSA-N 0.000 description 2
- WOSKHXYHFSIKNG-UHFFFAOYSA-N lenvatinib Chemical compound C=12C=C(C(N)=O)C(OC)=CC2=NC=CC=1OC(C=C1Cl)=CC=C1NC(=O)NC1CC1 WOSKHXYHFSIKNG-UHFFFAOYSA-N 0.000 description 2
- 239000008297 liquid dosage form Substances 0.000 description 2
- 229950011263 lirilumab Drugs 0.000 description 2
- 229960002247 lomustine Drugs 0.000 description 2
- 239000006210 lotion Substances 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- KHPKQFYUPIUARC-UHFFFAOYSA-N lumiracoxib Chemical compound OC(=O)CC1=CC(C)=CC=C1NC1=C(F)C=CC=C1Cl KHPKQFYUPIUARC-UHFFFAOYSA-N 0.000 description 2
- 210000004072 lung Anatomy 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- 201000009020 malignant peripheral nerve sheath tumor Diseases 0.000 description 2
- 239000011159 matrix material Substances 0.000 description 2
- 229940121386 matrix metalloproteinase inhibitor Drugs 0.000 description 2
- 239000003771 matrix metalloproteinase inhibitor Substances 0.000 description 2
- 208000020968 mature T-cell and NK-cell non-Hodgkin lymphoma Diseases 0.000 description 2
- 229960004961 mechlorethamine Drugs 0.000 description 2
- 229960001428 mercaptopurine Drugs 0.000 description 2
- 239000004530 micro-emulsion Substances 0.000 description 2
- 239000002480 mineral oil Substances 0.000 description 2
- 235000010446 mineral oil Nutrition 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- 239000002829 mitogen activated protein kinase inhibitor Substances 0.000 description 2
- 229950001907 monalizumab Drugs 0.000 description 2
- UZWDCWONPYILKI-UHFFFAOYSA-N n-[5-[(4-ethylpiperazin-1-yl)methyl]pyridin-2-yl]-5-fluoro-4-(7-fluoro-2-methyl-3-propan-2-ylbenzimidazol-5-yl)pyrimidin-2-amine Chemical compound C1CN(CC)CCN1CC(C=N1)=CC=C1NC1=NC=C(F)C(C=2C=C3N(C(C)C)C(C)=NC3=C(F)C=2)=N1 UZWDCWONPYILKI-UHFFFAOYSA-N 0.000 description 2
- XGXNTJHZPBRBHJ-UHFFFAOYSA-N n-phenylpyrimidin-2-amine Chemical class N=1C=CC=NC=1NC1=CC=CC=C1 XGXNTJHZPBRBHJ-UHFFFAOYSA-N 0.000 description 2
- 229940097496 nasal spray Drugs 0.000 description 2
- 239000007922 nasal spray Substances 0.000 description 2
- 229960000513 necitumumab Drugs 0.000 description 2
- 229950007221 nedaplatin Drugs 0.000 description 2
- 229960000801 nelarabine Drugs 0.000 description 2
- 229950008835 neratinib Drugs 0.000 description 2
- JWNPDZNEKVCWMY-VQHVLOKHSA-N neratinib Chemical compound C=12C=C(NC(=O)\C=C\CN(C)C)C(OCC)=CC2=NC=C(C#N)C=1NC(C=C1Cl)=CC=C1OCC1=CC=CC=N1 JWNPDZNEKVCWMY-VQHVLOKHSA-N 0.000 description 2
- 208000023833 nerve sheath neoplasm Diseases 0.000 description 2
- 125000004433 nitrogen atom Chemical group N* 0.000 description 2
- 231100000344 non-irritating Toxicity 0.000 description 2
- 229960003347 obinutuzumab Drugs 0.000 description 2
- 229960000435 oblimersen Drugs 0.000 description 2
- 229960002450 ofatumumab Drugs 0.000 description 2
- 229960000572 olaparib Drugs 0.000 description 2
- 229950008516 olaratumab Drugs 0.000 description 2
- JLPDBLFIVFSOCC-XYXFTTADSA-N oleandrin Chemical compound O1[C@@H](C)[C@H](O)[C@@H](OC)C[C@@H]1O[C@@H]1C[C@@H](CC[C@H]2[C@]3(C[C@@H]([C@@H]([C@@]3(C)CC[C@H]32)C=2COC(=O)C=2)OC(C)=O)O)[C@]3(C)CC1 JLPDBLFIVFSOCC-XYXFTTADSA-N 0.000 description 2
- 239000004006 olive oil Substances 0.000 description 2
- 235000008390 olive oil Nutrition 0.000 description 2
- 229960002230 omacetaxine mepesuccinate Drugs 0.000 description 2
- CGBJSGAELGCMKE-UHFFFAOYSA-N omipalisib Chemical compound COC1=NC=C(C=2C=C3C(C=4C=NN=CC=4)=CC=NC3=CC=2)C=C1NS(=O)(=O)C1=CC=C(F)C=C1F CGBJSGAELGCMKE-UHFFFAOYSA-N 0.000 description 2
- 231100000590 oncogenic Toxicity 0.000 description 2
- 208000008511 optic nerve glioma Diseases 0.000 description 2
- 210000000056 organ Anatomy 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 210000002997 osteoclast Anatomy 0.000 description 2
- 230000002018 overexpression Effects 0.000 description 2
- 230000003647 oxidation Effects 0.000 description 2
- 238000007254 oxidation reaction Methods 0.000 description 2
- 125000006503 p-nitrobenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1[N+]([O-])=O)C([H])([H])* 0.000 description 2
- 229960004390 palbociclib Drugs 0.000 description 2
- WRUUGTRCQOWXEG-UHFFFAOYSA-N pamidronate Chemical compound NCCC(O)(P(O)(O)=O)P(O)(O)=O WRUUGTRCQOWXEG-UHFFFAOYSA-N 0.000 description 2
- 229960003978 pamidronic acid Drugs 0.000 description 2
- 229960001972 panitumumab Drugs 0.000 description 2
- 201000010198 papillary carcinoma Diseases 0.000 description 2
- 239000002245 particle Substances 0.000 description 2
- CUIHSIWYWATEQL-UHFFFAOYSA-N pazopanib Chemical compound C1=CC2=C(C)N(C)N=C2C=C1N(C)C(N=1)=CC=NC=1NC1=CC=C(C)C(S(N)(=O)=O)=C1 CUIHSIWYWATEQL-UHFFFAOYSA-N 0.000 description 2
- 229960005547 pelareorep Drugs 0.000 description 2
- 229960005079 pemetrexed Drugs 0.000 description 2
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 2
- 229940066842 petrolatum Drugs 0.000 description 2
- JGWRKYUXBBNENE-UHFFFAOYSA-N pexidartinib Chemical compound C1=NC(C(F)(F)F)=CC=C1CNC(N=C1)=CC=C1CC1=CNC2=NC=C(Cl)C=C12 JGWRKYUXBBNENE-UHFFFAOYSA-N 0.000 description 2
- 125000004934 phenanthridinyl group Chemical group C1(=CC=CC2=NC=C3C=CC=CC3=C12)* 0.000 description 2
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 2
- 229950005566 picoplatin Drugs 0.000 description 2
- IIMIOEBMYPRQGU-UHFFFAOYSA-L picoplatin Chemical compound N.[Cl-].[Cl-].[Pt+2].CC1=CC=CC=N1 IIMIOEBMYPRQGU-UHFFFAOYSA-L 0.000 description 2
- 229950004941 pictilisib Drugs 0.000 description 2
- 201000004123 pineal gland cancer Diseases 0.000 description 2
- 229960004403 pixantrone Drugs 0.000 description 2
- 150000003058 platinum compounds Chemical class 0.000 description 2
- UVSMNLNDYGZFPF-UHFFFAOYSA-N pomalidomide Chemical compound O=C1C=2C(N)=CC=CC=2C(=O)N1C1CCC(=O)NC1=O UVSMNLNDYGZFPF-UHFFFAOYSA-N 0.000 description 2
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 2
- 108090000765 processed proteins & peptides Proteins 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- 238000004393 prognosis Methods 0.000 description 2
- 229960004063 propylene glycol Drugs 0.000 description 2
- 239000002599 prostaglandin synthase inhibitor Substances 0.000 description 2
- 239000003528 protein farnesyltransferase inhibitor Substances 0.000 description 2
- 239000002718 pyrimidine nucleoside Substances 0.000 description 2
- 238000001959 radiotherapy Methods 0.000 description 2
- 229960004622 raloxifene Drugs 0.000 description 2
- BKXVVCILCIUCLG-UHFFFAOYSA-N raloxifene hydrochloride Chemical compound [H+].[Cl-].C1=CC(O)=CC=C1C1=C(C(=O)C=2C=CC(OCCN3CCCCC3)=CC=2)C2=CC=C(O)C=C2S1 BKXVVCILCIUCLG-UHFFFAOYSA-N 0.000 description 2
- 229960002119 raloxifene hydrochloride Drugs 0.000 description 2
- 229960002633 ramucirumab Drugs 0.000 description 2
- 229940099538 rapamune Drugs 0.000 description 2
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 2
- 102000027426 receptor tyrosine kinases Human genes 0.000 description 2
- 108091008598 receptor tyrosine kinases Proteins 0.000 description 2
- 210000000664 rectum Anatomy 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 238000006722 reduction reaction Methods 0.000 description 2
- FNHKPVJBJVTLMP-UHFFFAOYSA-N regorafenib Chemical compound C1=NC(C(=O)NC)=CC(OC=2C=C(F)C(NC(=O)NC=3C=C(C(Cl)=CC=3)C(F)(F)F)=CC=2)=C1 FNHKPVJBJVTLMP-UHFFFAOYSA-N 0.000 description 2
- 229960000759 risedronic acid Drugs 0.000 description 2
- QXKJWHWUDVQATH-UHFFFAOYSA-N rogletimide Chemical compound C=1C=NC=CC=1C1(CC)CCC(=O)NC1=O QXKJWHWUDVQATH-UHFFFAOYSA-N 0.000 description 2
- HMABYWSNWIZPAG-UHFFFAOYSA-N rucaparib Chemical compound C1=CC(CNC)=CC=C1C(N1)=C2CCNC(=O)C3=C2C1=CC(F)=C3 HMABYWSNWIZPAG-UHFFFAOYSA-N 0.000 description 2
- 229960000215 ruxolitinib Drugs 0.000 description 2
- HFNKQEVNSGCOJV-OAHLLOKOSA-N ruxolitinib Chemical compound C1([C@@H](CC#N)N2N=CC(=C2)C=2C=3C=CNC=3N=CN=2)CCCC1 HFNKQEVNSGCOJV-OAHLLOKOSA-N 0.000 description 2
- 229960005569 saridegib Drugs 0.000 description 2
- 229960005399 satraplatin Drugs 0.000 description 2
- 190014017285 satraplatin Chemical compound 0.000 description 2
- 229940095743 selective estrogen receptor modulator Drugs 0.000 description 2
- CYOHGALHFOKKQC-UHFFFAOYSA-N selumetinib Chemical compound OCCONC(=O)C=1C=C2N(C)C=NC2=C(F)C=1NC1=CC=C(Br)C=C1Cl CYOHGALHFOKKQC-UHFFFAOYSA-N 0.000 description 2
- 229910052710 silicon Inorganic materials 0.000 description 2
- 239000010703 silicon Substances 0.000 description 2
- 229960002930 sirolimus Drugs 0.000 description 2
- 210000003491 skin Anatomy 0.000 description 2
- 201000000849 skin cancer Diseases 0.000 description 2
- 230000003381 solubilizing effect Effects 0.000 description 2
- 229960005325 sonidegib Drugs 0.000 description 2
- 229960003787 sorafenib Drugs 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 238000001228 spectrum Methods 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- HKSZLNNOFSGOKW-FYTWVXJKSA-N staurosporine Chemical class C12=C3N4C5=CC=CC=C5C3=C3CNC(=O)C3=C2C2=CC=CC=C2N1[C@H]1C[C@@H](NC)[C@@H](OC)[C@]4(C)O1 HKSZLNNOFSGOKW-FYTWVXJKSA-N 0.000 description 2
- 150000003431 steroids Chemical class 0.000 description 2
- 210000002784 stomach Anatomy 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-N succinic acid Chemical compound OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 2
- VDLGAZDAHPLOIR-VAZUXJHFSA-N sulanemadlin Chemical compound CC(C)C[C@H](NC(C)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC1=CC=CC=C1)C(=O)N[C@]2(C)CCCCCC\C=C\CCC[C@](C)(NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](CCC(N)=O)NC(=O)[C@H](C)NC(=O)[C@H](CC3=CNC4=CC=CC=C34)NC(=O)[C@H](CC5=CC=C(O)C=C5)NC(=O)[C@H](CCC(O)=O)NC2=O)C(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@H](C)C(N)=O VDLGAZDAHPLOIR-VAZUXJHFSA-N 0.000 description 2
- WINHZLLDWRZWRT-ATVHPVEESA-N sunitinib Chemical compound CCN(CC)CCNC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C WINHZLLDWRZWRT-ATVHPVEESA-N 0.000 description 2
- 239000003765 sweetening agent Substances 0.000 description 2
- 229950004550 talazoparib Drugs 0.000 description 2
- 229950008461 talimogene laherparepvec Drugs 0.000 description 2
- 229960001603 tamoxifen Drugs 0.000 description 2
- 239000003277 telomerase inhibitor Substances 0.000 description 2
- 229960000235 temsirolimus Drugs 0.000 description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 125000003039 tetrahydroisoquinolinyl group Chemical group C1(NCCC2=CC=CC=C12)* 0.000 description 2
- 125000000147 tetrahydroquinolinyl group Chemical group N1(CCCC2=CC=CC=C12)* 0.000 description 2
- 229940104230 thymidine Drugs 0.000 description 2
- 229960005324 tiludronic acid Drugs 0.000 description 2
- QQHMKNYGKVVGCZ-UHFFFAOYSA-N tipiracil Chemical compound N1C(=O)NC(=O)C(Cl)=C1CN1C(=N)CCC1 QQHMKNYGKVVGCZ-UHFFFAOYSA-N 0.000 description 2
- 229960002952 tipiracil Drugs 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 229960000977 trabectedin Drugs 0.000 description 2
- LIRYPHYGHXZJBZ-UHFFFAOYSA-N trametinib Chemical compound CC(=O)NC1=CC=CC(N2C(N(C3CC3)C(=O)C3=C(NC=4C(=CC(I)=CC=4)F)N(C)C(=O)C(C)=C32)=O)=C1 LIRYPHYGHXZJBZ-UHFFFAOYSA-N 0.000 description 2
- 238000000844 transformation Methods 0.000 description 2
- 229960001612 trastuzumab emtansine Drugs 0.000 description 2
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 description 2
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 2
- VSQQQLOSPVPRAZ-RRKCRQDMSA-N trifluridine Chemical compound C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(C(F)(F)F)=C1 VSQQQLOSPVPRAZ-RRKCRQDMSA-N 0.000 description 2
- 229960003962 trifluridine Drugs 0.000 description 2
- 229950007127 trilaciclib Drugs 0.000 description 2
- 206010046766 uterine cancer Diseases 0.000 description 2
- 229960005486 vaccine Drugs 0.000 description 2
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical compound CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 2
- UHTHHESEBZOYNR-UHFFFAOYSA-N vandetanib Chemical compound COC1=CC(C(/N=CN2)=N/C=3C(=CC(Br)=CC=3)F)=C2C=C1OCC1CCN(C)CC1 UHTHHESEBZOYNR-UHFFFAOYSA-N 0.000 description 2
- 229950001067 varlilumab Drugs 0.000 description 2
- GPXBXXGIAQBQNI-UHFFFAOYSA-N vemurafenib Chemical compound CCCS(=O)(=O)NC1=CC=C(F)C(C(=O)C=2C3=CC(=CN=C3NC=2)C=2C=CC(Cl)=CC=2)=C1F GPXBXXGIAQBQNI-UHFFFAOYSA-N 0.000 description 2
- 229960003048 vinblastine Drugs 0.000 description 2
- KDQAABAKXDWYSZ-PNYVAJAMSA-N vinblastine sulfate Chemical compound OS(O)(=O)=O.C([C@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 KDQAABAKXDWYSZ-PNYVAJAMSA-N 0.000 description 2
- 229960004982 vinblastine sulfate Drugs 0.000 description 2
- 229960002110 vincristine sulfate Drugs 0.000 description 2
- 230000003612 virological effect Effects 0.000 description 2
- 229960004449 vismodegib Drugs 0.000 description 2
- 229940055760 yervoy Drugs 0.000 description 2
- DNXHEGUUPJUMQT-UHFFFAOYSA-N (+)-estrone Natural products OC1=CC=C2C3CCC(C)(C(CC4)=O)C4C3CCC2=C1 DNXHEGUUPJUMQT-UHFFFAOYSA-N 0.000 description 1
- LSPHULWDVZXLIL-UHFFFAOYSA-N (+/-)-Camphoric acid Chemical compound CC1(C)C(C(O)=O)CCC1(C)C(O)=O LSPHULWDVZXLIL-UHFFFAOYSA-N 0.000 description 1
- XMAYWYJOQHXEEK-OZXSUGGESA-N (2R,4S)-ketoconazole Chemical compound C1CN(C(=O)C)CCN1C(C=C1)=CC=C1OC[C@@H]1O[C@@](CN2C=NC=C2)(C=2C(=CC(Cl)=CC=2)Cl)OC1 XMAYWYJOQHXEEK-OZXSUGGESA-N 0.000 description 1
- RNEACARJKXYVND-KQGZCTBQSA-N (2r)-2-[[(5z)-5-[(5-ethylfuran-2-yl)methylidene]-4-oxo-1,3-thiazol-2-yl]amino]-2-(4-fluorophenyl)acetic acid Chemical compound O1C(CC)=CC=C1\C=C/1C(=O)N=C(N[C@@H](C(O)=O)C=2C=CC(F)=CC=2)S\1 RNEACARJKXYVND-KQGZCTBQSA-N 0.000 description 1
- ASUGUQWIHMTFJL-QGZVFWFLSA-N (2r)-2-methyl-2-[[2-(1h-pyrrolo[2,3-b]pyridin-3-yl)pyrimidin-4-yl]amino]-n-(2,2,2-trifluoroethyl)butanamide Chemical compound FC(F)(F)CNC(=O)[C@@](C)(CC)NC1=CC=NC(C=2C3=CC=CN=C3NC=2)=N1 ASUGUQWIHMTFJL-QGZVFWFLSA-N 0.000 description 1
- CVCLJVVBHYOXDC-IAZSKANUSA-N (2z)-2-[(5z)-5-[(3,5-dimethyl-1h-pyrrol-2-yl)methylidene]-4-methoxypyrrol-2-ylidene]indole Chemical compound COC1=C\C(=C/2N=C3C=CC=CC3=C\2)N\C1=C/C=1NC(C)=CC=1C CVCLJVVBHYOXDC-IAZSKANUSA-N 0.000 description 1
- ZZJLMZYUGLJBSO-LAEOZQHASA-N (3R,4S)-3-amino-1-[(2S)-2-aminopropanoyl]-4-(3-boronopropyl)pyrrolidine-3-carboxylic acid Chemical compound N[C@]1(CN(C[C@@H]1CCCB(O)O)C([C@H](C)N)=O)C(=O)O ZZJLMZYUGLJBSO-LAEOZQHASA-N 0.000 description 1
- KCOYQXZDFIIGCY-CZIZESTLSA-N (3e)-4-amino-5-fluoro-3-[5-(4-methylpiperazin-1-yl)-1,3-dihydrobenzimidazol-2-ylidene]quinolin-2-one Chemical compound C1CN(C)CCN1C1=CC=C(N\C(N2)=C/3C(=C4C(F)=CC=CC4=NC\3=O)N)C2=C1 KCOYQXZDFIIGCY-CZIZESTLSA-N 0.000 description 1
- SODPIMGUZLOIPE-UHFFFAOYSA-N (4-chlorophenoxy)acetic acid Chemical compound OC(=O)COC1=CC=C(Cl)C=C1 SODPIMGUZLOIPE-UHFFFAOYSA-N 0.000 description 1
- SRSHBZRURUNOSM-DEOSSOPVSA-N (4-chlorophenyl) (1s)-6-chloro-1-(4-methoxyphenyl)-1,3,4,9-tetrahydropyrido[3,4-b]indole-2-carboxylate Chemical compound C1=CC(OC)=CC=C1[C@H]1C(NC=2C3=CC(Cl)=CC=2)=C3CCN1C(=O)OC1=CC=C(Cl)C=C1 SRSHBZRURUNOSM-DEOSSOPVSA-N 0.000 description 1
- DCGDPJCUIKLTDU-SUNYJGFJSA-N (4r)-4-[(1s)-1-fluoroethyl]-3-[2-[[(1s)-1-[4-methyl-5-[2-(trifluoromethyl)pyridin-4-yl]pyridin-2-yl]ethyl]amino]pyrimidin-4-yl]-1,3-oxazolidin-2-one Chemical compound C[C@H](F)[C@H]1COC(=O)N1C1=CC=NC(N[C@@H](C)C=2N=CC(=C(C)C=2)C=2C=C(N=CC=2)C(F)(F)F)=N1 DCGDPJCUIKLTDU-SUNYJGFJSA-N 0.000 description 1
- DEQANNDTNATYII-OULOTJBUSA-N (4r,7s,10s,13r,16s,19r)-10-(4-aminobutyl)-19-[[(2r)-2-amino-3-phenylpropanoyl]amino]-16-benzyl-n-[(2r,3r)-1,3-dihydroxybutan-2-yl]-7-[(1r)-1-hydroxyethyl]-13-(1h-indol-3-ylmethyl)-6,9,12,15,18-pentaoxo-1,2-dithia-5,8,11,14,17-pentazacycloicosane-4-carboxa Chemical compound C([C@@H](N)C(=O)N[C@H]1CSSC[C@H](NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCCCN)NC(=O)[C@@H](CC=2C3=CC=CC=C3NC=2)NC(=O)[C@H](CC=2C=CC=CC=2)NC1=O)C(=O)N[C@H](CO)[C@H](O)C)C1=CC=CC=C1 DEQANNDTNATYII-OULOTJBUSA-N 0.000 description 1
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 1
- LKJPYSCBVHEWIU-KRWDZBQOSA-N (R)-bicalutamide Chemical compound C([C@@](O)(C)C(=O)NC=1C=C(C(C#N)=CC=1)C(F)(F)F)S(=O)(=O)C1=CC=C(F)C=C1 LKJPYSCBVHEWIU-KRWDZBQOSA-N 0.000 description 1
- GSQOBTOAOGXIFL-LFIBNONCSA-N (e)-2-cyano-3-(3,4-dihydroxyphenyl)-n-(3-phenylpropyl)prop-2-enamide Chemical compound C1=C(O)C(O)=CC=C1\C=C(/C#N)C(=O)NCCCC1=CC=CC=C1 GSQOBTOAOGXIFL-LFIBNONCSA-N 0.000 description 1
- GWCNJMUSWLTSCW-SFQUDFHCSA-N (e)-2-cyano-3-(3,4-dihydroxyphenyl)-n-(4-phenylbutyl)prop-2-enamide Chemical compound C1=C(O)C(O)=CC=C1\C=C(/C#N)C(=O)NCCCCC1=CC=CC=C1 GWCNJMUSWLTSCW-SFQUDFHCSA-N 0.000 description 1
- HKHOVJYOELRGMV-XYOKQWHBSA-N (e)-2-cyano-3-(3,4-dihydroxyphenyl)-n-phenylprop-2-enamide Chemical compound C1=C(O)C(O)=CC=C1\C=C(/C#N)C(=O)NC1=CC=CC=C1 HKHOVJYOELRGMV-XYOKQWHBSA-N 0.000 description 1
- ZOJKRWXDNYZASL-NSCUHMNNSA-N (e)-4-methoxybut-2-enoic acid Chemical compound COC\C=C\C(O)=O ZOJKRWXDNYZASL-NSCUHMNNSA-N 0.000 description 1
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 1
- 229940058015 1,3-butylene glycol Drugs 0.000 description 1
- SPMVMDHWKHCIDT-UHFFFAOYSA-N 1-[2-chloro-4-[(6,7-dimethoxy-4-quinolinyl)oxy]phenyl]-3-(5-methyl-3-isoxazolyl)urea Chemical compound C=12C=C(OC)C(OC)=CC2=NC=CC=1OC(C=C1Cl)=CC=C1NC(=O)NC=1C=C(C)ON=1 SPMVMDHWKHCIDT-UHFFFAOYSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- VOXZDWNPVJITMN-ZBRFXRBCSA-N 17β-estradiol Chemical compound OC1=CC=C2[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 VOXZDWNPVJITMN-ZBRFXRBCSA-N 0.000 description 1
- YKBGVTZYEHREMT-KVQBGUIXSA-N 2'-deoxyguanosine Chemical class C1=NC=2C(=O)NC(N)=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)O1 YKBGVTZYEHREMT-KVQBGUIXSA-N 0.000 description 1
- UTQNKKSJPHTPBS-UHFFFAOYSA-N 2,2,2-trichloroethanone Chemical group ClC(Cl)(Cl)[C]=O UTQNKKSJPHTPBS-UHFFFAOYSA-N 0.000 description 1
- 125000000453 2,2,2-trichloroethyl group Chemical group [H]C([H])(*)C(Cl)(Cl)Cl 0.000 description 1
- FFFIRKXTFQCCKJ-UHFFFAOYSA-M 2,4,6-trimethylbenzoate Chemical compound CC1=CC(C)=C(C([O-])=O)C(C)=C1 FFFIRKXTFQCCKJ-UHFFFAOYSA-M 0.000 description 1
- CHHHXKFHOYLYRE-UHFFFAOYSA-M 2,4-Hexadienoic acid, potassium salt (1:1), (2E,4E)- Chemical compound [K+].CC=CC=CC([O-])=O CHHHXKFHOYLYRE-UHFFFAOYSA-M 0.000 description 1
- GFMMXOIFOQCCGU-UHFFFAOYSA-N 2-(2-chloro-4-iodoanilino)-N-(cyclopropylmethoxy)-3,4-difluorobenzamide Chemical compound C=1C=C(I)C=C(Cl)C=1NC1=C(F)C(F)=CC=C1C(=O)NOCC1CC1 GFMMXOIFOQCCGU-UHFFFAOYSA-N 0.000 description 1
- HUHXLHLWASNVDB-UHFFFAOYSA-N 2-(oxan-2-yloxy)oxane Chemical class O1CCCCC1OC1OCCCC1 HUHXLHLWASNVDB-UHFFFAOYSA-N 0.000 description 1
- VHVPQPYKVGDNFY-DFMJLFEVSA-N 2-[(2r)-butan-2-yl]-4-[4-[4-[4-[[(2r,4s)-2-(2,4-dichlorophenyl)-2-(1,2,4-triazol-1-ylmethyl)-1,3-dioxolan-4-yl]methoxy]phenyl]piperazin-1-yl]phenyl]-1,2,4-triazol-3-one Chemical compound O=C1N([C@H](C)CC)N=CN1C1=CC=C(N2CCN(CC2)C=2C=CC(OC[C@@H]3O[C@](CN4N=CN=C4)(OC3)C=3C(=CC(Cl)=CC=3)Cl)=CC=2)C=C1 VHVPQPYKVGDNFY-DFMJLFEVSA-N 0.000 description 1
- VTJXFTPMFYAJJU-UHFFFAOYSA-N 2-[(3,4-dihydroxyphenyl)methylidene]propanedinitrile Chemical compound OC1=CC=C(C=C(C#N)C#N)C=C1O VTJXFTPMFYAJJU-UHFFFAOYSA-N 0.000 description 1
- TXGKRVFSSHPBAJ-JKSUJKDBSA-N 2-[[(1r,2s)-2-aminocyclohexyl]amino]-4-[3-(triazol-2-yl)anilino]pyrimidine-5-carboxamide Chemical compound N[C@H]1CCCC[C@H]1NC1=NC=C(C(N)=O)C(NC=2C=C(C=CC=2)N2N=CC=N2)=N1 TXGKRVFSSHPBAJ-JKSUJKDBSA-N 0.000 description 1
- RTQWWZBSTRGEAV-PKHIMPSTSA-N 2-[[(2s)-2-[bis(carboxymethyl)amino]-3-[4-(methylcarbamoylamino)phenyl]propyl]-[2-[bis(carboxymethyl)amino]propyl]amino]acetic acid Chemical compound CNC(=O)NC1=CC=C(C[C@@H](CN(CC(C)N(CC(O)=O)CC(O)=O)CC(O)=O)N(CC(O)=O)CC(O)=O)C=C1 RTQWWZBSTRGEAV-PKHIMPSTSA-N 0.000 description 1
- LIOLIMKSCNQPLV-UHFFFAOYSA-N 2-fluoro-n-methyl-4-[7-(quinolin-6-ylmethyl)imidazo[1,2-b][1,2,4]triazin-2-yl]benzamide Chemical compound C1=C(F)C(C(=O)NC)=CC=C1C1=NN2C(CC=3C=C4C=CC=NC4=CC=3)=CN=C2N=C1 LIOLIMKSCNQPLV-UHFFFAOYSA-N 0.000 description 1
- 125000000954 2-hydroxyethyl group Chemical group [H]C([*])([H])C([H])([H])O[H] 0.000 description 1
- 229940080296 2-naphthalenesulfonate Drugs 0.000 description 1
- LEACJMVNYZDSKR-UHFFFAOYSA-N 2-octyldodecan-1-ol Chemical compound CCCCCCCCCCC(CO)CCCCCCCC LEACJMVNYZDSKR-UHFFFAOYSA-N 0.000 description 1
- GPVOTFQILZVCFP-UHFFFAOYSA-N 2-trityloxyacetic acid Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(OCC(=O)O)C1=CC=CC=C1 GPVOTFQILZVCFP-UHFFFAOYSA-N 0.000 description 1
- HXHAJRMTJXHJJZ-UHFFFAOYSA-N 3-[(4-bromo-2,6-difluorophenyl)methoxy]-5-(4-pyrrolidin-1-ylbutylcarbamoylamino)-1,2-thiazole-4-carboxamide Chemical compound S1N=C(OCC=2C(=CC(Br)=CC=2F)F)C(C(=O)N)=C1NC(=O)NCCCCN1CCCC1 HXHAJRMTJXHJJZ-UHFFFAOYSA-N 0.000 description 1
- NHFDRBXTEDBWCZ-ZROIWOOFSA-N 3-[2,4-dimethyl-5-[(z)-(2-oxo-1h-indol-3-ylidene)methyl]-1h-pyrrol-3-yl]propanoic acid Chemical compound OC(=O)CCC1=C(C)NC(\C=C/2C3=CC=CC=C3NC\2=O)=C1C NHFDRBXTEDBWCZ-ZROIWOOFSA-N 0.000 description 1
- RNMAUIMMNAHKQR-QFBILLFUSA-N 3-[2-[4-(trifluoromethoxy)anilino]-1-[(1R,5R)-3,3,5-trimethylcyclohexyl]benzimidazol-5-yl]propanoic acid Chemical compound FC(OC1=CC=C(C=C1)NC1=NC2=C(N1[C@H]1CC(C[C@H](C1)C)(C)C)C=CC(=C2)CCC(=O)O)(F)F RNMAUIMMNAHKQR-QFBILLFUSA-N 0.000 description 1
- WEVYNIUIFUYDGI-UHFFFAOYSA-N 3-[6-[4-(trifluoromethoxy)anilino]-4-pyrimidinyl]benzamide Chemical compound NC(=O)C1=CC=CC(C=2N=CN=C(NC=3C=CC(OC(F)(F)F)=CC=3)C=2)=C1 WEVYNIUIFUYDGI-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- ZRPLANDPDWYOMZ-UHFFFAOYSA-N 3-cyclopentylpropionic acid Chemical compound OC(=O)CCC1CCCC1 ZRPLANDPDWYOMZ-UHFFFAOYSA-N 0.000 description 1
- PBJKWGWHZVXBGU-UHFFFAOYSA-N 3-methyl-5-propan-2-yl-2-(1,6,7-trihydroxy-3-methyl-5-propan-2-ylnaphthalen-2-yl)naphthalene-1,6,7-triol Chemical compound CC(C)C1=C(O)C(O)=CC2=C(O)C(C=3C(O)=C4C=C(O)C(O)=C(C4=CC=3C)C(C)C)=C(C)C=C21 PBJKWGWHZVXBGU-UHFFFAOYSA-N 0.000 description 1
- QSFREBZMBNRGOK-UHFFFAOYSA-N 4-[(2,5-dihydroxyphenyl)methylamino]benzoic acid methyl ester Chemical compound C1=CC(C(=O)OC)=CC=C1NCC1=CC(O)=CC=C1O QSFREBZMBNRGOK-UHFFFAOYSA-N 0.000 description 1
- ZHSKUOZOLHMKEA-UHFFFAOYSA-N 4-[5-[bis(2-chloroethyl)amino]-1-methylbenzimidazol-2-yl]butanoic acid;hydron;chloride Chemical compound Cl.ClCCN(CCCl)C1=CC=C2N(C)C(CCCC(O)=O)=NC2=C1 ZHSKUOZOLHMKEA-UHFFFAOYSA-N 0.000 description 1
- FPHVRPCVNPHPBH-UHFFFAOYSA-N 4-benzylbenzoic acid Chemical compound C1=CC(C(=O)O)=CC=C1CC1=CC=CC=C1 FPHVRPCVNPHPBH-UHFFFAOYSA-N 0.000 description 1
- PXACTUVBBMDKRW-UHFFFAOYSA-M 4-bromobenzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=C(Br)C=C1 PXACTUVBBMDKRW-UHFFFAOYSA-M 0.000 description 1
- MDOJTZQKHMAPBK-UHFFFAOYSA-N 4-iodo-3-nitrobenzamide Chemical compound NC(=O)C1=CC=C(I)C([N+]([O-])=O)=C1 MDOJTZQKHMAPBK-UHFFFAOYSA-N 0.000 description 1
- SPXOTSHWBDUUMT-UHFFFAOYSA-M 4-nitrobenzenesulfonate Chemical compound [O-][N+](=O)C1=CC=C(S([O-])(=O)=O)C=C1 SPXOTSHWBDUUMT-UHFFFAOYSA-M 0.000 description 1
- JOOXCMJARBKPKM-UHFFFAOYSA-M 4-oxopentanoate Chemical compound CC(=O)CCC([O-])=O JOOXCMJARBKPKM-UHFFFAOYSA-M 0.000 description 1
- 125000002471 4H-quinolizinyl group Chemical group C=1(C=CCN2C=CC=CC12)* 0.000 description 1
- 102100022464 5'-nucleotidase Human genes 0.000 description 1
- NMUSYJAQQFHJEW-UHFFFAOYSA-N 5-Azacytidine Natural products O=C1N=C(N)N=CN1C1C(O)C(O)C(CO)O1 NMUSYJAQQFHJEW-UHFFFAOYSA-N 0.000 description 1
- XAUDJQYHKZQPEU-KVQBGUIXSA-N 5-aza-2'-deoxycytidine Chemical compound O=C1N=C(N)N=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 XAUDJQYHKZQPEU-KVQBGUIXSA-N 0.000 description 1
- PDOQBOJDRPLBQU-QMMMGPOBSA-N 5-chloro-2-n-[(1s)-1-(5-fluoropyrimidin-2-yl)ethyl]-4-n-(5-methyl-1h-pyrazol-3-yl)pyrimidine-2,4-diamine Chemical compound N([C@@H](C)C=1N=CC(F)=CN=1)C(N=1)=NC=C(Cl)C=1NC=1C=C(C)NN=1 PDOQBOJDRPLBQU-QMMMGPOBSA-N 0.000 description 1
- PLIXOHWIPDGJEI-OJSHLMAWSA-N 5-chloro-6-[(2-iminopyrrolidin-1-yl)methyl]-1h-pyrimidine-2,4-dione;1-[(2r,4s,5r)-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-5-(trifluoromethyl)pyrimidine-2,4-dione;hydrochloride Chemical compound Cl.N1C(=O)NC(=O)C(Cl)=C1CN1C(=N)CCC1.C1[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C(C(F)(F)F)=C1 PLIXOHWIPDGJEI-OJSHLMAWSA-N 0.000 description 1
- SVAGFBGXEWPNJC-SPIKMXEPSA-N 6,9-bis(2-aminoethylamino)benzo[g]isoquinoline-5,10-dione;(z)-but-2-enedioic acid Chemical compound OC(=O)\C=C/C(O)=O.OC(=O)\C=C/C(O)=O.O=C1C2=CN=CC=C2C(=O)C2=C1C(NCCN)=CC=C2NCCN SVAGFBGXEWPNJC-SPIKMXEPSA-N 0.000 description 1
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 1
- VVIAGPKUTFNRDU-UHFFFAOYSA-N 6S-folinic acid Natural products C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)NC(CCC(O)=O)C(O)=O)C=C1 VVIAGPKUTFNRDU-UHFFFAOYSA-N 0.000 description 1
- PBCZSGKMGDDXIJ-HQCWYSJUSA-N 7-hydroxystaurosporine Chemical compound N([C@H](O)C1=C2C3=CC=CC=C3N3C2=C24)C(=O)C1=C2C1=CC=CC=C1N4[C@H]1C[C@@H](NC)[C@@H](OC)[C@]3(C)O1 PBCZSGKMGDDXIJ-HQCWYSJUSA-N 0.000 description 1
- PBCZSGKMGDDXIJ-UHFFFAOYSA-N 7beta-hydroxystaurosporine Natural products C12=C3N4C5=CC=CC=C5C3=C3C(O)NC(=O)C3=C2C2=CC=CC=C2N1C1CC(NC)C(OC)C4(C)O1 PBCZSGKMGDDXIJ-UHFFFAOYSA-N 0.000 description 1
- JJTNLWSCFYERCK-UHFFFAOYSA-N 7h-pyrrolo[2,3-d]pyrimidine Chemical class N1=CN=C2NC=CC2=C1 JJTNLWSCFYERCK-UHFFFAOYSA-N 0.000 description 1
- HPLNQCPCUACXLM-PGUFJCEWSA-N ABT-737 Chemical compound C([C@@H](CCN(C)C)NC=1C(=CC(=CC=1)S(=O)(=O)NC(=O)C=1C=CC(=CC=1)N1CCN(CC=2C(=CC=CC=2)C=2C=CC(Cl)=CC=2)CC1)[N+]([O-])=O)SC1=CC=CC=C1 HPLNQCPCUACXLM-PGUFJCEWSA-N 0.000 description 1
- 101710168331 ALK tyrosine kinase receptor Proteins 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 description 1
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 description 1
- 206010000871 Acute monocytic leukaemia Diseases 0.000 description 1
- 206010000890 Acute myelomonocytic leukaemia Diseases 0.000 description 1
- 208000036762 Acute promyelocytic leukaemia Diseases 0.000 description 1
- 208000006468 Adrenal Cortex Neoplasms Diseases 0.000 description 1
- 208000009746 Adult T-Cell Leukemia-Lymphoma Diseases 0.000 description 1
- 208000016683 Adult T-cell leukemia/lymphoma Diseases 0.000 description 1
- ULXXDDBFHOBEHA-ONEGZZNKSA-N Afatinib Chemical compound N1=CN=C2C=C(OC3COCC3)C(NC(=O)/C=C/CN(C)C)=CC2=C1NC1=CC=C(F)C(Cl)=C1 ULXXDDBFHOBEHA-ONEGZZNKSA-N 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- 229940097396 Aminopeptidase inhibitor Drugs 0.000 description 1
- 229940127512 Androgen Synthesis Inhibitors Drugs 0.000 description 1
- 102100032187 Androgen receptor Human genes 0.000 description 1
- 201000003076 Angiosarcoma Diseases 0.000 description 1
- 108010063104 Apoptosis Regulatory Proteins Proteins 0.000 description 1
- 102000010565 Apoptosis Regulatory Proteins Human genes 0.000 description 1
- 235000017060 Arachis glabrata Nutrition 0.000 description 1
- 244000105624 Arachis hypogaea Species 0.000 description 1
- 235000010777 Arachis hypogaea Nutrition 0.000 description 1
- 235000018262 Arachis monticola Nutrition 0.000 description 1
- 229940080328 Arginase inhibitor Drugs 0.000 description 1
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 description 1
- 208000003950 B-cell lymphoma Diseases 0.000 description 1
- 229940124290 BCR-ABL tyrosine kinase inhibitor Drugs 0.000 description 1
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 1
- QULDDKSCVCJTPV-UHFFFAOYSA-N BIIB021 Chemical compound COC1=C(C)C=NC(CN2C3=NC(N)=NC(Cl)=C3N=C2)=C1C QULDDKSCVCJTPV-UHFFFAOYSA-N 0.000 description 1
- 229940125557 BMS-986207 Drugs 0.000 description 1
- 229940124291 BTK inhibitor Drugs 0.000 description 1
- 229940122035 Bcl-XL inhibitor Drugs 0.000 description 1
- 102100026189 Beta-galactosidase Human genes 0.000 description 1
- 206010004593 Bile duct cancer Diseases 0.000 description 1
- 102000004506 Blood Proteins Human genes 0.000 description 1
- 108010017384 Blood Proteins Proteins 0.000 description 1
- 206010005949 Bone cancer Diseases 0.000 description 1
- 208000018084 Bone neoplasm Diseases 0.000 description 1
- 229940078581 Bone resorption inhibitor Drugs 0.000 description 1
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 1
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 1
- 206010055113 Breast cancer metastatic Diseases 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-M Butyrate Chemical compound CCCC([O-])=O FERIUCNNQQJTOY-UHFFFAOYSA-M 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 1
- 102000018202 CC chemokine receptor 4 Human genes 0.000 description 1
- 108010017317 CCR4 Receptors Proteins 0.000 description 1
- 229940127272 CD73 inhibitor Drugs 0.000 description 1
- 101150071146 COX2 gene Proteins 0.000 description 1
- 101100114534 Caenorhabditis elegans ctc-2 gene Proteins 0.000 description 1
- 101100463133 Caenorhabditis elegans pdl-1 gene Proteins 0.000 description 1
- 101100510617 Caenorhabditis elegans sel-8 gene Proteins 0.000 description 1
- 108090000312 Calcium Channels Proteins 0.000 description 1
- 102000003922 Calcium Channels Human genes 0.000 description 1
- 102000014914 Carrier Proteins Human genes 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- 208000006332 Choriocarcinoma Diseases 0.000 description 1
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 102000008186 Collagen Human genes 0.000 description 1
- 108010035532 Collagen Proteins 0.000 description 1
- 208000037845 Cutaneous squamous cell carcinoma Diseases 0.000 description 1
- 229940122204 Cyclooxygenase inhibitor Drugs 0.000 description 1
- QASFUMOKHFSJGL-LAFRSMQTSA-N Cyclopamine Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H](CC2=C3C)[C@@H]1[C@@H]2CC[C@@]13O[C@@H]2C[C@H](C)CN[C@H]2[C@H]1C QASFUMOKHFSJGL-LAFRSMQTSA-N 0.000 description 1
- 102000000311 Cytosine Deaminase Human genes 0.000 description 1
- 108010080611 Cytosine Deaminase Proteins 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- HMFHBZSHGGEWLO-SOOFDHNKSA-N D-ribofuranose Chemical compound OC[C@H]1OC(O)[C@H](O)[C@@H]1O HMFHBZSHGGEWLO-SOOFDHNKSA-N 0.000 description 1
- 230000000970 DNA cross-linking effect Effects 0.000 description 1
- 230000033616 DNA repair Effects 0.000 description 1
- CKTSBUTUHBMZGZ-UHFFFAOYSA-N Deoxycytidine Natural products O=C1N=C(N)C=CN1C1OC(CO)C(O)C1 CKTSBUTUHBMZGZ-UHFFFAOYSA-N 0.000 description 1
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 1
- 206010012689 Diabetic retinopathy Diseases 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- 206010061818 Disease progression Diseases 0.000 description 1
- CUDVHEFYRIWYQD-UHFFFAOYSA-N E-3810 free base Chemical compound C=1C=C2C(C(=O)NC)=CC=CC2=CC=1OC(C1=CC=2OC)=CC=NC1=CC=2OCC1(N)CC1 CUDVHEFYRIWYQD-UHFFFAOYSA-N 0.000 description 1
- 229940122558 EGFR antagonist Drugs 0.000 description 1
- 101150029707 ERBB2 gene Proteins 0.000 description 1
- 201000009051 Embryonal Carcinoma Diseases 0.000 description 1
- 201000009273 Endometriosis Diseases 0.000 description 1
- 241000792859 Enema Species 0.000 description 1
- QXRSDHAAWVKZLJ-OXZHEXMSSA-N Epothilone B Natural products O=C1[C@H](C)[C@H](O)[C@@H](C)CCC[C@@]2(C)O[C@H]2C[C@@H](/C(=C\c2nc(C)sc2)/C)OC(=O)C[C@H](O)C1(C)C QXRSDHAAWVKZLJ-OXZHEXMSSA-N 0.000 description 1
- 102000056372 ErbB-3 Receptor Human genes 0.000 description 1
- 102000044591 ErbB-4 Receptor Human genes 0.000 description 1
- 208000031637 Erythroblastic Acute Leukemia Diseases 0.000 description 1
- 208000036566 Erythroleukaemia Diseases 0.000 description 1
- 102100038595 Estrogen receptor Human genes 0.000 description 1
- DNXHEGUUPJUMQT-CBZIJGRNSA-N Estrone Chemical compound OC1=CC=C2[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CCC2=C1 DNXHEGUUPJUMQT-CBZIJGRNSA-N 0.000 description 1
- LYCAIKOWRPUZTN-UHFFFAOYSA-N Ethylene glycol Chemical class OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 1
- 229940124226 Farnesyltransferase inhibitor Drugs 0.000 description 1
- KRHYYFGTRYWZRS-UHFFFAOYSA-M Fluoride anion Chemical compound [F-] KRHYYFGTRYWZRS-UHFFFAOYSA-M 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- WMBWREPUVVBILR-UHFFFAOYSA-N GCG Natural products C=1C(O)=C(O)C(O)=CC=1C1OC2=CC(O)=CC(O)=C2CC1OC(=O)C1=CC(O)=C(O)C(O)=C1 WMBWREPUVVBILR-UHFFFAOYSA-N 0.000 description 1
- 108010082772 GFB 111 Proteins 0.000 description 1
- 101710113436 GTPase KRas Proteins 0.000 description 1
- 102100039788 GTPase NRas Human genes 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- JRZJKWGQFNTSRN-UHFFFAOYSA-N Geldanamycin Natural products C1C(C)CC(OC)C(O)C(C)C=C(C)C(OC(N)=O)C(OC)CCC=C(C)C(=O)NC2=CC(=O)C(OC)=C1C2=O JRZJKWGQFNTSRN-UHFFFAOYSA-N 0.000 description 1
- 241000206672 Gelidium Species 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 108010060309 Glucuronidase Proteins 0.000 description 1
- 102000053187 Glucuronidase Human genes 0.000 description 1
- 102000009127 Glutaminase Human genes 0.000 description 1
- 108010073324 Glutaminase Proteins 0.000 description 1
- 229940121727 Glutaminase inhibitor Drugs 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Polymers OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 229940125497 HER2 kinase inhibitor Drugs 0.000 description 1
- 108090000031 Hedgehog Proteins Proteins 0.000 description 1
- 102000003693 Hedgehog Proteins Human genes 0.000 description 1
- 208000001258 Hemangiosarcoma Diseases 0.000 description 1
- 229920002971 Heparan sulfate Polymers 0.000 description 1
- 102100024025 Heparanase Human genes 0.000 description 1
- 229940122588 Heparanase inhibitor Drugs 0.000 description 1
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 1
- 101000600756 Homo sapiens 3-phosphoinositide-dependent protein kinase 1 Proteins 0.000 description 1
- 101000678236 Homo sapiens 5'-nucleotidase Proteins 0.000 description 1
- 101000744505 Homo sapiens GTPase NRas Proteins 0.000 description 1
- 101001021491 Homo sapiens HERV-H LTR-associating protein 2 Proteins 0.000 description 1
- 101001055157 Homo sapiens Interleukin-15 Proteins 0.000 description 1
- 101000945490 Homo sapiens Killer cell immunoglobulin-like receptor 3DL2 Proteins 0.000 description 1
- 101000624643 Homo sapiens M-phase inducer phosphatase 3 Proteins 0.000 description 1
- 101000605630 Homo sapiens Phosphatidylinositol 3-kinase catalytic subunit type 3 Proteins 0.000 description 1
- 101000579425 Homo sapiens Proto-oncogene tyrosine-protein kinase receptor Ret Proteins 0.000 description 1
- 101000580039 Homo sapiens Ras-specific guanine nucleotide-releasing factor 1 Proteins 0.000 description 1
- 101001059454 Homo sapiens Serine/threonine-protein kinase MARK2 Proteins 0.000 description 1
- 101000914514 Homo sapiens T-cell-specific surface glycoprotein CD28 Proteins 0.000 description 1
- 101000914484 Homo sapiens T-lymphocyte activation antigen CD80 Proteins 0.000 description 1
- 101000997835 Homo sapiens Tyrosine-protein kinase JAK1 Proteins 0.000 description 1
- 101000997832 Homo sapiens Tyrosine-protein kinase JAK2 Proteins 0.000 description 1
- 101000934996 Homo sapiens Tyrosine-protein kinase JAK3 Proteins 0.000 description 1
- 101000889732 Homo sapiens Tyrosine-protein kinase Tec Proteins 0.000 description 1
- 101001117146 Homo sapiens [Pyruvate dehydrogenase (acetyl-transferring)] kinase isozyme 1, mitochondrial Proteins 0.000 description 1
- 102000008100 Human Serum Albumin Human genes 0.000 description 1
- 108091006905 Human Serum Albumin Proteins 0.000 description 1
- UGQMRVRMYYASKQ-UHFFFAOYSA-N Hypoxanthine nucleoside Natural products OC1C(O)C(CO)OC1N1C(NC=NC2=O)=C2N=C1 UGQMRVRMYYASKQ-UHFFFAOYSA-N 0.000 description 1
- 102100034980 ICOS ligand Human genes 0.000 description 1
- 101710093458 ICOS ligand Proteins 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 108091008029 Immune checkpoint ligands Proteins 0.000 description 1
- 102000037977 Immune checkpoint ligands Human genes 0.000 description 1
- 101710205775 Inducible T-cell costimulator Proteins 0.000 description 1
- 102000004556 Interleukin-15 Receptors Human genes 0.000 description 1
- 108010017535 Interleukin-15 Receptors Proteins 0.000 description 1
- 108010002350 Interleukin-2 Proteins 0.000 description 1
- 102000000588 Interleukin-2 Human genes 0.000 description 1
- 206010061252 Intraocular melanoma Diseases 0.000 description 1
- 108090000862 Ion Channels Proteins 0.000 description 1
- 102000004310 Ion Channels Human genes 0.000 description 1
- 241000764238 Isis Species 0.000 description 1
- 102100034840 Killer cell immunoglobulin-like receptor 3DL2 Human genes 0.000 description 1
- 102000010638 Kinesin Human genes 0.000 description 1
- 108010063296 Kinesin Proteins 0.000 description 1
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 1
- 239000002067 L01XE06 - Dasatinib Substances 0.000 description 1
- 239000002138 L01XE21 - Regorafenib Substances 0.000 description 1
- 239000002137 L01XE24 - Ponatinib Substances 0.000 description 1
- 239000002176 L01XE26 - Cabozantinib Substances 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- 239000004166 Lanolin Substances 0.000 description 1
- 208000018142 Leiomyosarcoma Diseases 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 102100020862 Lymphocyte activation gene 3 protein Human genes 0.000 description 1
- 102100023330 M-phase inducer phosphatase 3 Human genes 0.000 description 1
- 108091054455 MAP kinase family Proteins 0.000 description 1
- 102000043136 MAP kinase family Human genes 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- 240000003183 Manihot esculenta Species 0.000 description 1
- 235000016735 Manihot esculenta subsp esculenta Nutrition 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 102100027754 Mast/stem cell growth factor receptor Kit Human genes 0.000 description 1
- 101710087603 Mast/stem cell growth factor receptor Kit Proteins 0.000 description 1
- 208000007054 Medullary Carcinoma Diseases 0.000 description 1
- 102100028905 Megakaryocyte-associated tyrosine-protein kinase Human genes 0.000 description 1
- 206010027452 Metastases to bone Diseases 0.000 description 1
- 206010051676 Metastases to peritoneum Diseases 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 208000035489 Monocytic Acute Leukemia Diseases 0.000 description 1
- 101000597780 Mus musculus Tumor necrosis factor ligand superfamily member 18 Proteins 0.000 description 1
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 description 1
- 208000033835 Myelomonocytic Acute Leukemia Diseases 0.000 description 1
- FBKMWOJEPMPVTQ-UHFFFAOYSA-N N'-(3-bromo-4-fluorophenyl)-N-hydroxy-4-[2-(sulfamoylamino)ethylamino]-1,2,5-oxadiazole-3-carboximidamide Chemical compound NS(=O)(=O)NCCNC1=NON=C1C(=NO)NC1=CC=C(F)C(Br)=C1 FBKMWOJEPMPVTQ-UHFFFAOYSA-N 0.000 description 1
- CHJJGSNFBQVOTG-UHFFFAOYSA-N N-methyl-guanidine Natural products CNC(N)=N CHJJGSNFBQVOTG-UHFFFAOYSA-N 0.000 description 1
- 150000001204 N-oxides Chemical class 0.000 description 1
- JOOXLOJCABQBSG-UHFFFAOYSA-N N-tert-butyl-3-[[5-methyl-2-[4-[2-(1-pyrrolidinyl)ethoxy]anilino]-4-pyrimidinyl]amino]benzenesulfonamide Chemical compound N1=C(NC=2C=C(C=CC=2)S(=O)(=O)NC(C)(C)C)C(C)=CN=C1NC(C=C1)=CC=C1OCCN1CCCC1 JOOXLOJCABQBSG-UHFFFAOYSA-N 0.000 description 1
- CXQHYVUVSFXTMY-UHFFFAOYSA-N N1'-[3-fluoro-4-[[6-methoxy-7-[3-(4-morpholinyl)propoxy]-4-quinolinyl]oxy]phenyl]-N1-(4-fluorophenyl)cyclopropane-1,1-dicarboxamide Chemical compound C1=CN=C2C=C(OCCCN3CCOCC3)C(OC)=CC2=C1OC(C(=C1)F)=CC=C1NC(=O)C1(C(=O)NC=2C=CC(F)=CC=2)CC1 CXQHYVUVSFXTMY-UHFFFAOYSA-N 0.000 description 1
- GPVKLYONJSSZFL-UHFFFAOYSA-N NSC 750259 Natural products CCC(C)C=CC(O)C(O)C(O)C(OC)C(=O)NC1CCCCNC1=O GPVKLYONJSSZFL-UHFFFAOYSA-N 0.000 description 1
- 208000002454 Nasopharyngeal Carcinoma Diseases 0.000 description 1
- 206010061306 Nasopharyngeal cancer Diseases 0.000 description 1
- 102100029527 Natural cytotoxicity triggering receptor 3 ligand 1 Human genes 0.000 description 1
- 101710201161 Natural cytotoxicity triggering receptor 3 ligand 1 Proteins 0.000 description 1
- 208000034176 Neoplasms, Germ Cell and Embryonal Diseases 0.000 description 1
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 1
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 description 1
- 102100022673 Nuclear receptor subfamily 4 group A member 3 Human genes 0.000 description 1
- MSHZHSPISPJWHW-UHFFFAOYSA-N O-(chloroacetylcarbamoyl)fumagillol Chemical compound O1C(CC=C(C)C)C1(C)C1C(OC)C(OC(=O)NC(=O)CCl)CCC21CO2 MSHZHSPISPJWHW-UHFFFAOYSA-N 0.000 description 1
- 108010064641 ONX 0912 Proteins 0.000 description 1
- 208000008589 Obesity Diseases 0.000 description 1
- 108010016076 Octreotide Proteins 0.000 description 1
- 240000007817 Olea europaea Species 0.000 description 1
- 102000038030 PI3Ks Human genes 0.000 description 1
- 101150000187 PTGS2 gene Proteins 0.000 description 1
- 208000000821 Parathyroid Neoplasms Diseases 0.000 description 1
- 208000002471 Penile Neoplasms Diseases 0.000 description 1
- 208000012896 Peritoneal disease Diseases 0.000 description 1
- 102100038329 Phosphatidylinositol 3-kinase catalytic subunit type 3 Human genes 0.000 description 1
- 201000007286 Pilocytic astrocytoma Diseases 0.000 description 1
- 201000005746 Pituitary adenoma Diseases 0.000 description 1
- 206010061538 Pituitary tumour benign Diseases 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 229920002732 Polyanhydride Polymers 0.000 description 1
- 229920000954 Polyglycolide Polymers 0.000 description 1
- 229920001710 Polyorthoester Polymers 0.000 description 1
- 229920001214 Polysorbate 60 Polymers 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 description 1
- 206010057846 Primitive neuroectodermal tumour Diseases 0.000 description 1
- 101710094000 Programmed cell death 1 ligand 1 Proteins 0.000 description 1
- 208000033826 Promyelocytic Acute Leukemia Diseases 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 108010007568 Protamines Proteins 0.000 description 1
- 102000007327 Protamines Human genes 0.000 description 1
- 102000001253 Protein Kinase Human genes 0.000 description 1
- 229940123573 Protein synthesis inhibitor Drugs 0.000 description 1
- 102100028286 Proto-oncogene tyrosine-protein kinase receptor Ret Human genes 0.000 description 1
- 201000004681 Psoriasis Diseases 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- 102000014128 RANK Ligand Human genes 0.000 description 1
- 108010025832 RANK Ligand Proteins 0.000 description 1
- 101710100969 Receptor tyrosine-protein kinase erbB-3 Proteins 0.000 description 1
- 101710100963 Receptor tyrosine-protein kinase erbB-4 Proteins 0.000 description 1
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 description 1
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 description 1
- 229940123934 Reductase inhibitor Drugs 0.000 description 1
- 241000702263 Reovirus sp. Species 0.000 description 1
- 208000008938 Rhabdoid tumor Diseases 0.000 description 1
- 206010073334 Rhabdoid tumour Diseases 0.000 description 1
- 108010041388 Ribonucleotide Reductases Proteins 0.000 description 1
- 102000000505 Ribonucleotide Reductases Human genes 0.000 description 1
- PYMYPHUHKUWMLA-LMVFSUKVSA-N Ribose Natural products OC[C@@H](O)[C@@H](O)[C@@H](O)C=O PYMYPHUHKUWMLA-LMVFSUKVSA-N 0.000 description 1
- 208000035389 Ring chromosome 6 syndrome Diseases 0.000 description 1
- 229940044665 STING agonist Drugs 0.000 description 1
- 201000010208 Seminoma Diseases 0.000 description 1
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 1
- 102100028904 Serine/threonine-protein kinase MARK2 Human genes 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- 241000700584 Simplexvirus Species 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 235000002595 Solanum tuberosum Nutrition 0.000 description 1
- 244000061456 Solanum tuberosum Species 0.000 description 1
- 102400000046 Soluble interleukin-15 receptor subunit alpha Human genes 0.000 description 1
- 101800000582 Soluble interleukin-15 receptor subunit alpha Proteins 0.000 description 1
- 102000004584 Somatomedin Receptors Human genes 0.000 description 1
- 108010017622 Somatomedin Receptors Proteins 0.000 description 1
- 108050001286 Somatostatin Receptor Proteins 0.000 description 1
- 102000011096 Somatostatin receptor Human genes 0.000 description 1
- 229940121856 Somatostatin receptor antagonist Drugs 0.000 description 1
- HVUMOYIDDBPOLL-XWVZOOPGSA-N Sorbitan monostearate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O HVUMOYIDDBPOLL-XWVZOOPGSA-N 0.000 description 1
- SSZBUIDZHHWXNJ-UHFFFAOYSA-N Stearinsaeure-hexadecylester Natural products CCCCCCCCCCCCCCCCCC(=O)OCCCCCCCCCCCCCCCC SSZBUIDZHHWXNJ-UHFFFAOYSA-N 0.000 description 1
- 208000001662 Subependymal Glioma Diseases 0.000 description 1
- 230000005867 T cell response Effects 0.000 description 1
- 108010092262 T-Cell Antigen Receptors Proteins 0.000 description 1
- 208000000389 T-cell leukemia Diseases 0.000 description 1
- 208000028530 T-cell lymphoblastic leukemia/lymphoma Diseases 0.000 description 1
- 102100027213 T-cell-specific surface glycoprotein CD28 Human genes 0.000 description 1
- 102100027222 T-lymphocyte activation antigen CD80 Human genes 0.000 description 1
- 229940124614 TLR 8 agonist Drugs 0.000 description 1
- 108091021474 TMEM173 Proteins 0.000 description 1
- 229940126302 TTI-621 Drugs 0.000 description 1
- JXAGDPXECXQWBC-LJQANCHMSA-N Tanomastat Chemical compound C([C@H](C(=O)O)CC(=O)C=1C=CC(=CC=1)C=1C=CC(Cl)=CC=1)SC1=CC=CC=C1 JXAGDPXECXQWBC-LJQANCHMSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 229940123582 Telomerase inhibitor Drugs 0.000 description 1
- 108091033399 Telomestatin Proteins 0.000 description 1
- 239000004098 Tetracycline Substances 0.000 description 1
- ZMZDMBWJUHKJPS-UHFFFAOYSA-M Thiocyanate anion Chemical compound [S-]C#N ZMZDMBWJUHKJPS-UHFFFAOYSA-M 0.000 description 1
- 229940122020 Thymidine phosphorylase inhibitor Drugs 0.000 description 1
- 229940122149 Thymidylate synthase inhibitor Drugs 0.000 description 1
- 208000024770 Thyroid neoplasm Diseases 0.000 description 1
- 239000004012 Tofacitinib Substances 0.000 description 1
- 229940123384 Toll-like receptor (TLR) agonist Drugs 0.000 description 1
- 108091023040 Transcription factor Proteins 0.000 description 1
- 102000040945 Transcription factor Human genes 0.000 description 1
- RTKIYFITIVXBLE-UHFFFAOYSA-N Trichostatin A Natural products ONC(=O)C=CC(C)=CC(C)C(=O)C1=CC=C(N(C)C)C=C1 RTKIYFITIVXBLE-UHFFFAOYSA-N 0.000 description 1
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 1
- 241000245032 Trillium Species 0.000 description 1
- 208000003721 Triple Negative Breast Neoplasms Diseases 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- 108060008683 Tumor Necrosis Factor Receptor Proteins 0.000 description 1
- 102100033438 Tyrosine-protein kinase JAK1 Human genes 0.000 description 1
- 102100033444 Tyrosine-protein kinase JAK2 Human genes 0.000 description 1
- 102100025387 Tyrosine-protein kinase JAK3 Human genes 0.000 description 1
- 102100037236 Tyrosine-protein kinase receptor UFO Human genes 0.000 description 1
- 208000023915 Ureteral Neoplasms Diseases 0.000 description 1
- 206010046458 Urethral neoplasms Diseases 0.000 description 1
- 201000005969 Uveal melanoma Diseases 0.000 description 1
- 108091008605 VEGF receptors Proteins 0.000 description 1
- 229940091171 VEGFR-2 tyrosine kinase inhibitor Drugs 0.000 description 1
- 101800003344 Vaccinia growth factor Proteins 0.000 description 1
- 201000003761 Vaginal carcinoma Diseases 0.000 description 1
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 description 1
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 1
- 102100033177 Vascular endothelial growth factor receptor 2 Human genes 0.000 description 1
- 229940122803 Vinca alkaloid Drugs 0.000 description 1
- 206010047741 Vulval cancer Diseases 0.000 description 1
- 208000004354 Vulvar Neoplasms Diseases 0.000 description 1
- 208000008383 Wilms tumor Diseases 0.000 description 1
- 108050007567 Wolframin Proteins 0.000 description 1
- VWQVUPCCIRVNHF-OUBTZVSYSA-N Yttrium-90 Chemical compound [90Y] VWQVUPCCIRVNHF-OUBTZVSYSA-N 0.000 description 1
- HGVNLRPZOWWDKD-UHFFFAOYSA-N ZSTK-474 Chemical compound FC(F)C1=NC2=CC=CC=C2N1C(N=1)=NC(N2CCOCC2)=NC=1N1CCOCC1 HGVNLRPZOWWDKD-UHFFFAOYSA-N 0.000 description 1
- 240000008042 Zea mays Species 0.000 description 1
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 description 1
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 1
- 239000001089 [(2R)-oxolan-2-yl]methanol Substances 0.000 description 1
- ODEDPKNSRBCSDO-UHFFFAOYSA-N [2-(hexadecylsulfanylmethyl)-3-methoxypropyl] 2-(trimethylazaniumyl)ethyl phosphate Chemical compound CCCCCCCCCCCCCCCCSCC(COC)COP([O-])(=O)OCC[N+](C)(C)C ODEDPKNSRBCSDO-UHFFFAOYSA-N 0.000 description 1
- MQTBAGAVFDZXKF-UHFFFAOYSA-N [2-fluoro-4-(trifluoromethyl)phenyl]methanamine Chemical compound NCC1=CC=C(C(F)(F)F)C=C1F MQTBAGAVFDZXKF-UHFFFAOYSA-N 0.000 description 1
- 102100024148 [Pyruvate dehydrogenase (acetyl-transferring)] kinase isozyme 1, mitochondrial Human genes 0.000 description 1
- AIWRTTMUVOZGPW-HSPKUQOVSA-N abarelix Chemical compound C([C@@H](C(=O)N[C@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCNC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N[C@H](C)C(N)=O)N(C)C(=O)[C@H](CO)NC(=O)[C@@H](CC=1C=NC=CC=1)NC(=O)[C@@H](CC=1C=CC(Cl)=CC=1)NC(=O)[C@@H](CC=1C=C2C=CC=CC2=CC=1)NC(C)=O)C1=CC=C(O)C=C1 AIWRTTMUVOZGPW-HSPKUQOVSA-N 0.000 description 1
- 108010023617 abarelix Proteins 0.000 description 1
- 229960002184 abarelix Drugs 0.000 description 1
- 229960000853 abiraterone Drugs 0.000 description 1
- 229940028652 abraxane Drugs 0.000 description 1
- 239000002250 absorbent Substances 0.000 description 1
- 230000002745 absorbent Effects 0.000 description 1
- 239000003655 absorption accelerator Substances 0.000 description 1
- 229940124532 absorption promoter Drugs 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- 150000001241 acetals Chemical class 0.000 description 1
- 150000001242 acetic acid derivatives Chemical class 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- 108010052004 acetyl-2-naphthylalanyl-3-chlorophenylalanyl-1-oxohexadecyl-seryl-4-aminophenylalanyl(hydroorotyl)-4-aminophenylalanyl(carbamoyl)-leucyl-ILys-prolyl-alaninamide Proteins 0.000 description 1
- 208000017733 acquired polycythemia vera Diseases 0.000 description 1
- 125000000641 acridinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3C=C12)* 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 208000021841 acute erythroid leukemia Diseases 0.000 description 1
- 208000011912 acute myelomonocytic leukemia M4 Diseases 0.000 description 1
- WNLRTRBMVRJNCN-UHFFFAOYSA-L adipate(2-) Chemical compound [O-]C(=O)CCCCC([O-])=O WNLRTRBMVRJNCN-UHFFFAOYSA-L 0.000 description 1
- 238000009098 adjuvant therapy Methods 0.000 description 1
- 208000024447 adrenal gland neoplasm Diseases 0.000 description 1
- 229940009456 adriamycin Drugs 0.000 description 1
- 201000006966 adult T-cell leukemia Diseases 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 229960001686 afatinib Drugs 0.000 description 1
- ULXXDDBFHOBEHA-CWDCEQMOSA-N afatinib Chemical compound N1=CN=C2C=C(O[C@@H]3COCC3)C(NC(=O)/C=C/CN(C)C)=CC2=C1NC1=CC=C(F)C(Cl)=C1 ULXXDDBFHOBEHA-CWDCEQMOSA-N 0.000 description 1
- 229940042992 afinitor Drugs 0.000 description 1
- 235000010419 agar Nutrition 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 229960001611 alectinib Drugs 0.000 description 1
- KDGFLJKFZUIJMX-UHFFFAOYSA-N alectinib Chemical compound CCC1=CC=2C(=O)C(C3=CC=C(C=C3N3)C#N)=C3C(C)(C)C=2C=C1N(CC1)CCC1N1CCOCC1 KDGFLJKFZUIJMX-UHFFFAOYSA-N 0.000 description 1
- 229940072056 alginate Drugs 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 229940110282 alimta Drugs 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 150000008055 alkyl aryl sulfonates Chemical class 0.000 description 1
- 150000008052 alkyl sulfonates Chemical class 0.000 description 1
- 230000002152 alkylating effect Effects 0.000 description 1
- 125000000304 alkynyl group Chemical group 0.000 description 1
- 125000004419 alkynylene group Chemical group 0.000 description 1
- SHGAZHPCJJPHSC-YCNIQYBTSA-N all-trans-retinoic acid Chemical compound OC(=O)\C=C(/C)\C=C\C=C(/C)\C=C\C1=C(C)CCCC1(C)C SHGAZHPCJJPHSC-YCNIQYBTSA-N 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- HMFHBZSHGGEWLO-UHFFFAOYSA-N alpha-D-Furanose-Ribose Natural products OCC1OC(O)C(O)C1O HMFHBZSHGGEWLO-UHFFFAOYSA-N 0.000 description 1
- AWUCVROLDVIAJX-UHFFFAOYSA-N alpha-glycerophosphate Natural products OCC(O)COP(O)(O)=O AWUCVROLDVIAJX-UHFFFAOYSA-N 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- CEGOLXSVJUTHNZ-UHFFFAOYSA-K aluminium tristearate Chemical compound [Al+3].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CEGOLXSVJUTHNZ-UHFFFAOYSA-K 0.000 description 1
- 229940063655 aluminum stearate Drugs 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 206010002224 anaplastic astrocytoma Diseases 0.000 description 1
- 229950004189 andecaliximab Drugs 0.000 description 1
- 239000003098 androgen Substances 0.000 description 1
- 108010080146 androgen receptors Proteins 0.000 description 1
- 230000001548 androgenic effect Effects 0.000 description 1
- AEMFNILZOJDQLW-QAGGRKNESA-N androst-4-ene-3,17-dione Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CCC2=C1 AEMFNILZOJDQLW-QAGGRKNESA-N 0.000 description 1
- 229960005471 androstenedione Drugs 0.000 description 1
- AEMFNILZOJDQLW-UHFFFAOYSA-N androstenedione Natural products O=C1CCC2(C)C3CCC(C)(C(CC4)=O)C4C3CCC2=C1 AEMFNILZOJDQLW-UHFFFAOYSA-N 0.000 description 1
- 230000033115 angiogenesis Effects 0.000 description 1
- 229940045799 anthracyclines and related substance Drugs 0.000 description 1
- 150000004056 anthraquinones Chemical class 0.000 description 1
- 238000011122 anti-angiogenic therapy Methods 0.000 description 1
- 230000002424 anti-apoptotic effect Effects 0.000 description 1
- 230000000719 anti-leukaemic effect Effects 0.000 description 1
- 230000001399 anti-metabolic effect Effects 0.000 description 1
- 230000005809 anti-tumor immunity Effects 0.000 description 1
- 230000006023 anti-tumor response Effects 0.000 description 1
- 229940030495 antiandrogen sex hormone and modulator of the genital system Drugs 0.000 description 1
- 229940045985 antineoplastic platinum compound Drugs 0.000 description 1
- 229940027983 antiseptic and disinfectant quaternary ammonium compound Drugs 0.000 description 1
- 230000005975 antitumor immune response Effects 0.000 description 1
- 230000001640 apoptogenic effect Effects 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 150000003974 aralkylamines Chemical group 0.000 description 1
- 229940078010 arimidex Drugs 0.000 description 1
- 229940087620 aromasin Drugs 0.000 description 1
- 229940014583 arranon Drugs 0.000 description 1
- 125000003710 aryl alkyl group Chemical group 0.000 description 1
- 125000005160 aryl oxy alkyl group Chemical group 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229960001230 asparagine Drugs 0.000 description 1
- 235000009582 asparagine Nutrition 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- 229950004810 atamestane Drugs 0.000 description 1
- PEPMWUSGRKINHX-TXTPUJOMSA-N atamestane Chemical compound C1C[C@@H]2[C@@]3(C)C(C)=CC(=O)C=C3CC[C@H]2[C@@H]2CCC(=O)[C@]21C PEPMWUSGRKINHX-TXTPUJOMSA-N 0.000 description 1
- 230000002238 attenuated effect Effects 0.000 description 1
- 229940120638 avastin Drugs 0.000 description 1
- 229960003005 axitinib Drugs 0.000 description 1
- 108010023337 axl receptor tyrosine kinase Proteins 0.000 description 1
- 229950000971 baricitinib Drugs 0.000 description 1
- XUZMWHLSFXCVMG-UHFFFAOYSA-N baricitinib Chemical compound C1N(S(=O)(=O)CC)CC1(CC#N)N1N=CC(C=2C=3C=CNC=3N=CN=2)=C1 XUZMWHLSFXCVMG-UHFFFAOYSA-N 0.000 description 1
- XFILPEOLDIKJHX-QYZOEREBSA-N batimastat Chemical compound C([C@@H](C(=O)NC)NC(=O)[C@H](CC(C)C)[C@H](CSC=1SC=CC=1)C(=O)NO)C1=CC=CC=C1 XFILPEOLDIKJHX-QYZOEREBSA-N 0.000 description 1
- 229950001858 batimastat Drugs 0.000 description 1
- 235000013871 bee wax Nutrition 0.000 description 1
- 239000012166 beeswax Substances 0.000 description 1
- 229940077840 beleodaq Drugs 0.000 description 1
- 229930195545 bengamide Natural products 0.000 description 1
- 239000000440 bentonite Substances 0.000 description 1
- 229910000278 bentonite Inorganic materials 0.000 description 1
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 1
- 229960000686 benzalkonium chloride Drugs 0.000 description 1
- ABSXPNGWJFAPRT-UHFFFAOYSA-N benzenesulfonic acid;n-[3-[[5-fluoro-2-[4-(2-methoxyethoxy)anilino]pyrimidin-4-yl]amino]phenyl]prop-2-enamide Chemical compound OS(=O)(=O)C1=CC=CC=C1.C1=CC(OCCOC)=CC=C1NC1=NC=C(F)C(NC=2C=C(NC(=O)C=C)C=CC=2)=N1 ABSXPNGWJFAPRT-UHFFFAOYSA-N 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 229940050390 benzoate Drugs 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 1
- 235000019445 benzyl alcohol Nutrition 0.000 description 1
- 229960002903 benzyl benzoate Drugs 0.000 description 1
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 1
- 125000000649 benzylidene group Chemical group [H]C(=[*])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 229960000997 bicalutamide Drugs 0.000 description 1
- 230000001588 bifunctional effect Effects 0.000 description 1
- 210000000013 bile duct Anatomy 0.000 description 1
- 201000007180 bile duct carcinoma Diseases 0.000 description 1
- 108091008324 binding proteins Proteins 0.000 description 1
- 238000004166 bioassay Methods 0.000 description 1
- 230000008512 biological response Effects 0.000 description 1
- 238000001574 biopsy Methods 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 201000001531 bladder carcinoma Diseases 0.000 description 1
- 229960003008 blinatumomab Drugs 0.000 description 1
- 229940101815 blincyto Drugs 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 210000001124 body fluid Anatomy 0.000 description 1
- 230000036760 body temperature Effects 0.000 description 1
- 229910052796 boron Inorganic materials 0.000 description 1
- 229940083476 bosulif Drugs 0.000 description 1
- 229960003736 bosutinib Drugs 0.000 description 1
- 210000000481 breast Anatomy 0.000 description 1
- 208000003362 bronchogenic carcinoma Diseases 0.000 description 1
- 229960005539 bryostatin 1 Drugs 0.000 description 1
- MJQUEDHRCUIRLF-TVIXENOKSA-N bryostatin 1 Chemical compound C([C@@H]1CC(/[C@@H]([C@@](C(C)(C)/C=C/2)(O)O1)OC(=O)/C=C/C=C/CCC)=C\C(=O)OC)[C@H]([C@@H](C)O)OC(=O)C[C@H](O)C[C@@H](O1)C[C@H](OC(C)=O)C(C)(C)[C@]1(O)C[C@@H]1C\C(=C\C(=O)OC)C[C@H]\2O1 MJQUEDHRCUIRLF-TVIXENOKSA-N 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- HFCFMRYTXDINDK-WNQIDUERSA-N cabozantinib malate Chemical compound OC(=O)[C@@H](O)CC(O)=O.C=12C=C(OC)C(OC)=CC2=NC=CC=1OC(C=C1)=CC=C1NC(=O)C1(C(=O)NC=2C=CC(F)=CC=2)CC1 HFCFMRYTXDINDK-WNQIDUERSA-N 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 235000010216 calcium carbonate Nutrition 0.000 description 1
- 229940043430 calcium compound Drugs 0.000 description 1
- 150000001674 calcium compounds Chemical class 0.000 description 1
- FATUQANACHZLRT-KMRXSBRUSA-L calcium glucoheptonate Chemical compound [Ca+2].OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)C([O-])=O.OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)C([O-])=O FATUQANACHZLRT-KMRXSBRUSA-L 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- 230000004611 cancer cell death Effects 0.000 description 1
- 229940056434 caprelsa Drugs 0.000 description 1
- 150000004657 carbamic acid derivatives Chemical group 0.000 description 1
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 1
- 125000006243 carbonyl protecting group Chemical group 0.000 description 1
- 150000007942 carboxylates Chemical class 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 239000012876 carrier material Substances 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 229960000590 celecoxib Drugs 0.000 description 1
- RZEKVGVHFLEQIL-UHFFFAOYSA-N celecoxib Chemical compound C1=CC(C)=CC=C1C1=CC(C(F)(F)F)=NN1C1=CC=C(S(N)(=O)=O)C=C1 RZEKVGVHFLEQIL-UHFFFAOYSA-N 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 230000006369 cell cycle progression Effects 0.000 description 1
- 230000032823 cell division Effects 0.000 description 1
- 230000003833 cell viability Effects 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 235000010980 cellulose Nutrition 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- AEULIVPVIDOLIN-UHFFFAOYSA-N cep-11981 Chemical compound C1=C2C3=C4CNC(=O)C4=C4C5=CN(C)N=C5CCC4=C3N(CC(C)C)C2=CC=C1NC1=NC=CC=N1 AEULIVPVIDOLIN-UHFFFAOYSA-N 0.000 description 1
- DSRNKUZOWRFQFO-UHFFFAOYSA-N cephalotaxine Natural products COC1=CC23CCCN2CCc4cc5OCOc5cc4C3=C1O DSRNKUZOWRFQFO-UHFFFAOYSA-N 0.000 description 1
- 229960001602 ceritinib Drugs 0.000 description 1
- 208000019065 cervical carcinoma Diseases 0.000 description 1
- 229940081733 cetearyl alcohol Drugs 0.000 description 1
- 229960000541 cetyl alcohol Drugs 0.000 description 1
- NNXDIGHYPZHXTR-ONEGZZNKSA-N chembl2035185 Chemical compound C=1C=C(C=2)NC(N=3)=NC=CC=3C(O3)=CC=C3COC\C=C\COCC=2C=1OCCN1CCCC1 NNXDIGHYPZHXTR-ONEGZZNKSA-N 0.000 description 1
- PIQCTGMSNWUMAF-UHFFFAOYSA-N chembl522892 Chemical compound C1CN(C)CCN1C1=CC=C(NC(=N2)C=3C(NC4=CC=CC(F)=C4C=3N)=O)C2=C1 PIQCTGMSNWUMAF-UHFFFAOYSA-N 0.000 description 1
- SFUWDBKYNKBXQU-UHFFFAOYSA-N chembl586701 Chemical compound C1=CC(C)=CC=C1C1C2=C(O)C3=CC=CC=C3C2=NC2=CC=CC=C2S1 SFUWDBKYNKBXQU-UHFFFAOYSA-N 0.000 description 1
- ZGHQGWOETPXKLY-XVNBXDOJSA-N chembl77030 Chemical compound NC(=S)C(\C#N)=C\C1=CC=C(O)C(O)=C1 ZGHQGWOETPXKLY-XVNBXDOJSA-N 0.000 description 1
- 229910052729 chemical element Inorganic materials 0.000 description 1
- 229940089960 chloroacetate Drugs 0.000 description 1
- FOCAUTSVDIKZOP-UHFFFAOYSA-M chloroacetate Chemical compound [O-]C(=O)CCl FOCAUTSVDIKZOP-UHFFFAOYSA-M 0.000 description 1
- 125000002668 chloroacetyl group Chemical group ClCC(=O)* 0.000 description 1
- ZPEIMTDSQAKGNT-UHFFFAOYSA-N chlorpromazine Chemical compound C1=C(Cl)C=C2N(CCCN(C)C)C3=CC=CC=C3SC2=C1 ZPEIMTDSQAKGNT-UHFFFAOYSA-N 0.000 description 1
- 229960001076 chlorpromazine Drugs 0.000 description 1
- 208000006990 cholangiocarcinoma Diseases 0.000 description 1
- 125000003016 chromanyl group Chemical group O1C(CCC2=CC=CC=C12)* 0.000 description 1
- GULSIMHVQYBADX-FQEVSTJZSA-N cintirorgon Chemical compound CC(C)(C[C@H]1CN(c2cc(ccc2O1)-c1cc(F)cc(OC(F)F)c1)S(=O)(=O)c1cccc(c1)C(F)(F)F)C(O)=O GULSIMHVQYBADX-FQEVSTJZSA-N 0.000 description 1
- 239000004927 clay Substances 0.000 description 1
- ACSIXWWBWUQEHA-UHFFFAOYSA-N clodronic acid Chemical compound OP(O)(=O)C(Cl)(Cl)P(O)(O)=O ACSIXWWBWUQEHA-UHFFFAOYSA-N 0.000 description 1
- 229960002271 cobimetinib Drugs 0.000 description 1
- RESIMIUSNACMNW-BXRWSSRYSA-N cobimetinib fumarate Chemical compound OC(=O)\C=C\C(O)=O.C1C(O)([C@H]2NCCCC2)CN1C(=O)C1=CC=C(F)C(F)=C1NC1=CC=C(I)C=C1F.C1C(O)([C@H]2NCCCC2)CN1C(=O)C1=CC=C(F)C(F)=C1NC1=CC=C(I)C=C1F RESIMIUSNACMNW-BXRWSSRYSA-N 0.000 description 1
- 229960001338 colchicine Drugs 0.000 description 1
- 229920001436 collagen Polymers 0.000 description 1
- 239000008119 colloidal silica Substances 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 229940034568 cometriq Drugs 0.000 description 1
- 230000002860 competitive effect Effects 0.000 description 1
- 230000001276 controlling effect Effects 0.000 description 1
- 235000005822 corn Nutrition 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 229960005061 crizotinib Drugs 0.000 description 1
- LDHQCZJRKDOVOX-NSCUHMNNSA-N crotonic acid Chemical compound C\C=C\C(O)=O LDHQCZJRKDOVOX-NSCUHMNNSA-N 0.000 description 1
- 229940109262 curcumin Drugs 0.000 description 1
- 239000004148 curcumin Substances 0.000 description 1
- 235000012754 curcumin Nutrition 0.000 description 1
- 208000035250 cutaneous malignant susceptibility to 1 melanoma Diseases 0.000 description 1
- 208000030381 cutaneous melanoma Diseases 0.000 description 1
- 125000000392 cycloalkenyl group Chemical group 0.000 description 1
- QASFUMOKHFSJGL-UHFFFAOYSA-N cyclopamine Natural products C1C=C2CC(O)CCC2(C)C(CC2=C3C)C1C2CCC13OC2CC(C)CNC2C1C QASFUMOKHFSJGL-UHFFFAOYSA-N 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 208000031513 cyst Diseases 0.000 description 1
- 208000002445 cystadenocarcinoma Diseases 0.000 description 1
- 229960000684 cytarabine Drugs 0.000 description 1
- 229940034075 cytarabine injection Drugs 0.000 description 1
- 210000005220 cytoplasmic tail Anatomy 0.000 description 1
- 229940104302 cytosine Drugs 0.000 description 1
- 231100000433 cytotoxic Toxicity 0.000 description 1
- 239000002254 cytotoxic agent Substances 0.000 description 1
- 230000001472 cytotoxic effect Effects 0.000 description 1
- 229960002465 dabrafenib Drugs 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 229960002204 daratumumab Drugs 0.000 description 1
- 229940094732 darzalex Drugs 0.000 description 1
- 229960002448 dasatinib Drugs 0.000 description 1
- 125000004856 decahydroquinolinyl group Chemical group N1(CCCC2CCCCC12)* 0.000 description 1
- 229960003603 decitabine Drugs 0.000 description 1
- MEUCPCLKGZSHTA-XYAYPHGZSA-N degarelix Chemical compound C([C@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCNC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N[C@H](C)C(N)=O)NC(=O)[C@H](CC=1C=CC(NC(=O)[C@H]2NC(=O)NC(=O)C2)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@@H](CC=1C=NC=CC=1)NC(=O)[C@@H](CC=1C=CC(Cl)=CC=1)NC(=O)[C@@H](CC=1C=C2C=CC=CC2=CC=1)NC(C)=O)C1=CC=C(NC(N)=O)C=C1 MEUCPCLKGZSHTA-XYAYPHGZSA-N 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- 230000018044 dehydration Effects 0.000 description 1
- 238000006297 dehydration reaction Methods 0.000 description 1
- SJFBTAPEPRWNKH-CCKFTAQKSA-N delanzomib Chemical compound CC(C)C[C@@H](B(O)O)NC(=O)[C@H]([C@@H](C)O)NC(=O)C1=CC=CC(C=2C=CC=CC=2)=N1 SJFBTAPEPRWNKH-CCKFTAQKSA-N 0.000 description 1
- 230000001335 demethylating effect Effects 0.000 description 1
- 229940029030 dendritic cell vaccine Drugs 0.000 description 1
- 229960001251 denosumab Drugs 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 229910052805 deuterium Inorganic materials 0.000 description 1
- 125000002576 diazepinyl group Chemical group N1N=C(C=CC=C1)* 0.000 description 1
- 239000012954 diazonium Substances 0.000 description 1
- IJGRMHOSHXDMSA-UHFFFAOYSA-O diazynium Chemical compound [NH+]#N IJGRMHOSHXDMSA-UHFFFAOYSA-O 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- VFLDPWHFBUODDF-UHFFFAOYSA-N diferuloylmethane Natural products C1=C(O)C(OC)=CC(C=CC(=O)CC(=O)C=CC=2C=C(OC)C(O)=CC=2)=C1 VFLDPWHFBUODDF-UHFFFAOYSA-N 0.000 description 1
- OTKJDMGTUTTYMP-UHFFFAOYSA-N dihydrosphingosine Natural products CCCCCCCCCCCCCCCC(O)C(N)CO OTKJDMGTUTTYMP-UHFFFAOYSA-N 0.000 description 1
- GXGAKHNRMVGRPK-UHFFFAOYSA-N dimagnesium;dioxido-bis[[oxido(oxo)silyl]oxy]silane Chemical compound [Mg+2].[Mg+2].[O-][Si](=O)O[Si]([O-])([O-])O[Si]([O-])=O GXGAKHNRMVGRPK-UHFFFAOYSA-N 0.000 description 1
- SWSQBOPZIKWTGO-UHFFFAOYSA-N dimethylaminoamidine Natural products CN(C)C(N)=N SWSQBOPZIKWTGO-UHFFFAOYSA-N 0.000 description 1
- 229960004497 dinutuximab Drugs 0.000 description 1
- 125000000532 dioxanyl group Chemical group 0.000 description 1
- 125000005879 dioxolanyl group Chemical group 0.000 description 1
- ZPWVASYFFYYZEW-UHFFFAOYSA-L dipotassium hydrogen phosphate Chemical compound [K+].[K+].OP([O-])([O-])=O ZPWVASYFFYYZEW-UHFFFAOYSA-L 0.000 description 1
- 229910000396 dipotassium phosphate Inorganic materials 0.000 description 1
- 235000019797 dipotassium phosphate Nutrition 0.000 description 1
- 230000005750 disease progression Effects 0.000 description 1
- BNIILDVGGAEEIG-UHFFFAOYSA-L disodium hydrogen phosphate Chemical compound [Na+].[Na+].OP([O-])([O-])=O BNIILDVGGAEEIG-UHFFFAOYSA-L 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 229960002563 disulfiram Drugs 0.000 description 1
- 229940065910 docefrez Drugs 0.000 description 1
- POULHZVOKOAJMA-UHFFFAOYSA-M dodecanoate Chemical compound CCCCCCCCCCCC([O-])=O POULHZVOKOAJMA-UHFFFAOYSA-M 0.000 description 1
- 229950005778 dovitinib Drugs 0.000 description 1
- 239000006196 drop Substances 0.000 description 1
- 239000000890 drug combination Substances 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 230000002183 duodenal effect Effects 0.000 description 1
- 239000003221 ear drop Substances 0.000 description 1
- 229940047652 ear drops Drugs 0.000 description 1
- FSIRXIHZBIXHKT-MHTVFEQDSA-N edatrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CC(CC)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FSIRXIHZBIXHKT-MHTVFEQDSA-N 0.000 description 1
- 229950006700 edatrexate Drugs 0.000 description 1
- 229940121647 egfr inhibitor Drugs 0.000 description 1
- 239000003792 electrolyte Substances 0.000 description 1
- 229960004137 elotuzumab Drugs 0.000 description 1
- 229940073038 elspar Drugs 0.000 description 1
- 239000008387 emulsifying waxe Substances 0.000 description 1
- 229950002830 enadenotucirev Drugs 0.000 description 1
- 210000000750 endocrine system Anatomy 0.000 description 1
- 201000003914 endometrial carcinoma Diseases 0.000 description 1
- 239000007920 enema Substances 0.000 description 1
- 229940095399 enema Drugs 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 229960004671 enzalutamide Drugs 0.000 description 1
- 108060002566 ephrin Proteins 0.000 description 1
- 102000012803 ephrin Human genes 0.000 description 1
- 208000037828 epithelial carcinoma Diseases 0.000 description 1
- 229930013356 epothilone Natural products 0.000 description 1
- HESCAJZNRMSMJG-HGYUPSKWSA-N epothilone A Natural products O=C1[C@H](C)[C@H](O)[C@H](C)CCC[C@H]2O[C@H]2C[C@@H](/C(=C\c2nc(C)sc2)/C)OC(=O)C[C@H](O)C1(C)C HESCAJZNRMSMJG-HGYUPSKWSA-N 0.000 description 1
- HESCAJZNRMSMJG-KKQRBIROSA-N epothilone A Chemical class C/C([C@@H]1C[C@@H]2O[C@@H]2CCC[C@@H]([C@@H]([C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)O1)O)C)=C\C1=CSC(C)=N1 HESCAJZNRMSMJG-KKQRBIROSA-N 0.000 description 1
- QXRSDHAAWVKZLJ-PVYNADRNSA-N epothilone B Chemical compound C/C([C@@H]1C[C@@H]2O[C@]2(C)CCC[C@@H]([C@@H]([C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)O1)O)C)=C\C1=CSC(C)=N1 QXRSDHAAWVKZLJ-PVYNADRNSA-N 0.000 description 1
- 229940014684 erivedge Drugs 0.000 description 1
- GTTBEUCJPZQMDZ-UHFFFAOYSA-N erlotinib hydrochloride Chemical compound [H+].[Cl-].C=12C=C(OCCOC)C(OCCOC)=CC2=NC=NC=1NC1=CC=CC(C#C)=C1 GTTBEUCJPZQMDZ-UHFFFAOYSA-N 0.000 description 1
- 229940051398 erwinaze Drugs 0.000 description 1
- 230000032050 esterification Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- 229960005309 estradiol Drugs 0.000 description 1
- 229930182833 estradiol Natural products 0.000 description 1
- 102000015694 estrogen receptors Human genes 0.000 description 1
- 229960003399 estrone Drugs 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- VFRSADQPWYCXDG-LEUCUCNGSA-N ethyl (2s,5s)-5-methylpyrrolidine-2-carboxylate;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.CCOC(=O)[C@@H]1CC[C@H](C)N1 VFRSADQPWYCXDG-LEUCUCNGSA-N 0.000 description 1
- 235000019439 ethyl acetate Nutrition 0.000 description 1
- 229960004945 etoricoxib Drugs 0.000 description 1
- MNJVRJDLRVPLFE-UHFFFAOYSA-N etoricoxib Chemical compound C1=NC(C)=CC=C1C1=NC=C(Cl)C=C1C1=CC=C(S(C)(=O)=O)C=C1 MNJVRJDLRVPLFE-UHFFFAOYSA-N 0.000 description 1
- 229940085363 evista Drugs 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 239000003889 eye drop Substances 0.000 description 1
- 229940012356 eye drops Drugs 0.000 description 1
- 210000003608 fece Anatomy 0.000 description 1
- 229940087476 femara Drugs 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 229960000556 fingolimod Drugs 0.000 description 1
- KKGQTZUTZRNORY-UHFFFAOYSA-N fingolimod Chemical compound CCCCCCCCC1=CC=C(CCC(N)(CO)CO)C=C1 KKGQTZUTZRNORY-UHFFFAOYSA-N 0.000 description 1
- 229940002006 firmagon Drugs 0.000 description 1
- XRECTZIEBJDKEO-UHFFFAOYSA-N flucytosine Chemical compound NC1=NC(=O)NC=C1F XRECTZIEBJDKEO-UHFFFAOYSA-N 0.000 description 1
- 229960004413 flucytosine Drugs 0.000 description 1
- 229960000390 fludarabine Drugs 0.000 description 1
- 229960005304 fludarabine phosphate Drugs 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 230000004907 flux Effects 0.000 description 1
- 239000004052 folic acid antagonist Substances 0.000 description 1
- 125000003929 folic acid group Chemical group 0.000 description 1
- VVIAGPKUTFNRDU-ABLWVSNPSA-N folinic acid Chemical compound C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 VVIAGPKUTFNRDU-ABLWVSNPSA-N 0.000 description 1
- 235000008191 folinic acid Nutrition 0.000 description 1
- 239000011672 folinic acid Substances 0.000 description 1
- 230000003325 follicular Effects 0.000 description 1
- 229950008692 foretinib Drugs 0.000 description 1
- 150000004675 formic acid derivatives Chemical class 0.000 description 1
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 1
- 229950005309 fostamatinib Drugs 0.000 description 1
- GKDRMWXFWHEQQT-UHFFFAOYSA-N fostamatinib Chemical compound COC1=C(OC)C(OC)=CC(NC=2N=C(NC=3N=C4N(COP(O)(O)=O)C(=O)C(C)(C)OC4=CC=3)C(F)=CN=2)=C1 GKDRMWXFWHEQQT-UHFFFAOYSA-N 0.000 description 1
- 229950004003 fresolimumab Drugs 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 238000007429 general method Methods 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 201000003115 germ cell cancer Diseases 0.000 description 1
- 229940087158 gilotrif Drugs 0.000 description 1
- 229950009073 gimatecan Drugs 0.000 description 1
- UIVFUQKYVFCEKJ-OPTOVBNMSA-N gimatecan Chemical compound C1=CC=C2C(\C=N\OC(C)(C)C)=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 UIVFUQKYVFCEKJ-OPTOVBNMSA-N 0.000 description 1
- 229940080856 gleevec Drugs 0.000 description 1
- 229950000918 glembatumumab Drugs 0.000 description 1
- 229940110231 gleostine Drugs 0.000 description 1
- 229940084910 gliadel Drugs 0.000 description 1
- 239000003862 glucocorticoid Substances 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 235000001727 glucose Nutrition 0.000 description 1
- 230000004190 glucose uptake Effects 0.000 description 1
- YQEMORVAKMFKLG-UHFFFAOYSA-N glycerine monostearate Natural products CCCCCCCCCCCCCCCCCC(=O)OC(CO)CO YQEMORVAKMFKLG-UHFFFAOYSA-N 0.000 description 1
- SVUQHVRAGMNPLW-UHFFFAOYSA-N glycerol monostearate Natural products CCCCCCCCCCCCCCCCC(=O)OCC(O)CO SVUQHVRAGMNPLW-UHFFFAOYSA-N 0.000 description 1
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 1
- 229960002449 glycine Drugs 0.000 description 1
- 229960003690 goserelin acetate Drugs 0.000 description 1
- 208000035474 group of disease Diseases 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 229940118951 halaven Drugs 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 125000001188 haloalkyl group Chemical group 0.000 description 1
- 230000026030 halogenation Effects 0.000 description 1
- 238000005658 halogenation reaction Methods 0.000 description 1
- 208000025750 heavy chain disease Diseases 0.000 description 1
- 108010037536 heparanase Proteins 0.000 description 1
- MNWFXJYAOYHMED-UHFFFAOYSA-N heptanoic acid Chemical compound CCCCCCC(O)=O MNWFXJYAOYHMED-UHFFFAOYSA-N 0.000 description 1
- 229940022353 herceptin Drugs 0.000 description 1
- 125000004415 heterocyclylalkyl group Chemical group 0.000 description 1
- BXWNKGSJHAJOGX-UHFFFAOYSA-N hexadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO BXWNKGSJHAJOGX-UHFFFAOYSA-N 0.000 description 1
- UBHWBODXJBSFLH-UHFFFAOYSA-N hexadecan-1-ol;octadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO.CCCCCCCCCCCCCCCCCCO UBHWBODXJBSFLH-UHFFFAOYSA-N 0.000 description 1
- IPCSVZSSVZVIGE-UHFFFAOYSA-M hexadecanoate Chemical compound CCCCCCCCCCCCCCCC([O-])=O IPCSVZSSVZVIGE-UHFFFAOYSA-M 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 108091008039 hormone receptors Proteins 0.000 description 1
- 102000044459 human CD47 Human genes 0.000 description 1
- 102000056003 human IL15 Human genes 0.000 description 1
- 239000003906 humectant Substances 0.000 description 1
- 229940088013 hycamtin Drugs 0.000 description 1
- ZMZDMBWJUHKJPS-UHFFFAOYSA-N hydrogen thiocyanate Natural products SC#N ZMZDMBWJUHKJPS-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 1
- 229940061301 ibrance Drugs 0.000 description 1
- 229960001001 ibritumomab tiuxetan Drugs 0.000 description 1
- 229950006905 ilmofosine Drugs 0.000 description 1
- 229960003685 imatinib mesylate Drugs 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 230000003100 immobilizing effect Effects 0.000 description 1
- 210000002865 immune cell Anatomy 0.000 description 1
- 230000008105 immune reaction Effects 0.000 description 1
- 102000027596 immune receptors Human genes 0.000 description 1
- 108091008915 immune receptors Proteins 0.000 description 1
- 230000028993 immune response Effects 0.000 description 1
- 230000036039 immunity Effects 0.000 description 1
- 229940045207 immuno-oncology agent Drugs 0.000 description 1
- 238000003364 immunohistochemistry Methods 0.000 description 1
- 239000000367 immunologic factor Substances 0.000 description 1
- 239000002584 immunological anticancer agent Substances 0.000 description 1
- 239000002955 immunomodulating agent Substances 0.000 description 1
- 229960003444 immunosuppressant agent Drugs 0.000 description 1
- 239000003018 immunosuppressive agent Substances 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 230000001965 increasing effect Effects 0.000 description 1
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 229910052738 indium Inorganic materials 0.000 description 1
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 229960002993 ingenol mebutate Drugs 0.000 description 1
- 229950002133 iniparib Drugs 0.000 description 1
- 239000007972 injectable composition Substances 0.000 description 1
- 229940090044 injection Drugs 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 229940005319 inlyta Drugs 0.000 description 1
- 230000010354 integration Effects 0.000 description 1
- 229940047124 interferons Drugs 0.000 description 1
- 238000007917 intracranial administration Methods 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000007919 intrasynovial administration Methods 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 238000005342 ion exchange Methods 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- KLEAIHJJLUAXIQ-JDRGBKBRSA-N irinotecan hydrochloride hydrate Chemical compound O.O.O.Cl.C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 KLEAIHJJLUAXIQ-JDRGBKBRSA-N 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 125000002183 isoquinolinyl group Chemical class C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 125000005956 isoquinolyl group Chemical group 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 229940011083 istodax Drugs 0.000 description 1
- 229960004130 itraconazole Drugs 0.000 description 1
- 229940111707 ixempra Drugs 0.000 description 1
- 229940045773 jakafi Drugs 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 229960004125 ketoconazole Drugs 0.000 description 1
- 210000000244 kidney pelvis Anatomy 0.000 description 1
- 229940000764 kyprolis Drugs 0.000 description 1
- 229940001447 lactate Drugs 0.000 description 1
- 150000003893 lactate salts Chemical class 0.000 description 1
- 229940099584 lactobionate Drugs 0.000 description 1
- JYTUSYBCFIZPBE-AMTLMPIISA-N lactobionic acid Chemical compound OC(=O)[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O JYTUSYBCFIZPBE-AMTLMPIISA-N 0.000 description 1
- 235000019388 lanolin Nutrition 0.000 description 1
- 229940039717 lanolin Drugs 0.000 description 1
- 229960004891 lapatinib Drugs 0.000 description 1
- 229940070765 laurate Drugs 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- VHOGYURTWQBHIL-UHFFFAOYSA-N leflunomide Chemical compound O1N=CC(C(=O)NC=2C=CC(=CC=2)C(F)(F)F)=C1C VHOGYURTWQBHIL-UHFFFAOYSA-N 0.000 description 1
- 229960004942 lenalidomide Drugs 0.000 description 1
- 229960003784 lenvatinib Drugs 0.000 description 1
- 229940064847 lenvima Drugs 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- 229960001691 leucovorin Drugs 0.000 description 1
- 229950002216 linifanib Drugs 0.000 description 1
- MPVGZUGXCQEXTM-UHFFFAOYSA-N linifanib Chemical compound CC1=CC=C(F)C(NC(=O)NC=2C=CC(=CC=2)C=2C=3C(N)=NNC=3C=CC=2)=C1 MPVGZUGXCQEXTM-UHFFFAOYSA-N 0.000 description 1
- 206010024627 liposarcoma Diseases 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 239000006194 liquid suspension Substances 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 230000005923 long-lasting effect Effects 0.000 description 1
- 229940024740 lonsurf Drugs 0.000 description 1
- 229950008745 losoxantrone Drugs 0.000 description 1
- YROQEQPFUCPDCP-UHFFFAOYSA-N losoxantrone Chemical compound OCCNCCN1N=C2C3=CC=CC(O)=C3C(=O)C3=C2C1=CC=C3NCCNCCO YROQEQPFUCPDCP-UHFFFAOYSA-N 0.000 description 1
- 208000022080 low-grade astrocytoma Diseases 0.000 description 1
- 229950004231 lucitanib Drugs 0.000 description 1
- 229960000994 lumiracoxib Drugs 0.000 description 1
- 201000005296 lung carcinoma Diseases 0.000 description 1
- 208000037829 lymphangioendotheliosarcoma Diseases 0.000 description 1
- 208000012804 lymphangiosarcoma Diseases 0.000 description 1
- 229940100352 lynparza Drugs 0.000 description 1
- 210000002540 macrophage Anatomy 0.000 description 1
- 208000002780 macular degeneration Diseases 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 239000000391 magnesium silicate Substances 0.000 description 1
- 229910000386 magnesium trisilicate Inorganic materials 0.000 description 1
- 235000019793 magnesium trisilicate Nutrition 0.000 description 1
- 229940099273 magnesium trisilicate Drugs 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 201000010893 malignant breast melanoma Diseases 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- OCSMOTCMPXTDND-OUAUKWLOSA-N marimastat Chemical compound CNC(=O)[C@H](C(C)(C)C)NC(=O)[C@H](CC(C)C)[C@H](O)C(=O)NO OCSMOTCMPXTDND-OUAUKWLOSA-N 0.000 description 1
- 229950002736 marizomib Drugs 0.000 description 1
- 229940034322 marqibo Drugs 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 208000023356 medullary thyroid gland carcinoma Diseases 0.000 description 1
- 229940083118 mekinist Drugs 0.000 description 1
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 1
- 229960001924 melphalan Drugs 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 229960005558 mertansine Drugs 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 208000037819 metastatic cancer Diseases 0.000 description 1
- 208000011575 metastatic malignant neoplasm Diseases 0.000 description 1
- 208000010658 metastatic prostate carcinoma Diseases 0.000 description 1
- 208000037843 metastatic solid tumor Diseases 0.000 description 1
- 229960000485 methotrexate Drugs 0.000 description 1
- RMIODHQZRUFFFF-UHFFFAOYSA-M methoxyacetate Chemical compound COCC([O-])=O RMIODHQZRUFFFF-UHFFFAOYSA-M 0.000 description 1
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 1
- 125000004092 methylthiomethyl group Chemical group [H]C([H])([H])SC([H])([H])* 0.000 description 1
- 238000002493 microarray Methods 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 229960005225 mifamurtide Drugs 0.000 description 1
- 108700007621 mifamurtide Proteins 0.000 description 1
- 229940042472 mineral oil Drugs 0.000 description 1
- ZAHQPTJLOCWVPG-UHFFFAOYSA-N mitoxantrone dihydrochloride Chemical compound Cl.Cl.O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO ZAHQPTJLOCWVPG-UHFFFAOYSA-N 0.000 description 1
- 201000004058 mixed glioma Diseases 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 239000003607 modifier Substances 0.000 description 1
- 229950007699 mogamulizumab Drugs 0.000 description 1
- 229950008814 momelotinib Drugs 0.000 description 1
- ZVHNDZWQTBEVRY-UHFFFAOYSA-N momelotinib Chemical compound C1=CC(C(NCC#N)=O)=CC=C1C1=CC=NC(NC=2C=CC(=CC=2)N2CCOCC2)=N1 ZVHNDZWQTBEVRY-UHFFFAOYSA-N 0.000 description 1
- CQDGTJPVBWZJAZ-UHFFFAOYSA-N monoethyl carbonate Chemical compound CCOC(O)=O CQDGTJPVBWZJAZ-UHFFFAOYSA-N 0.000 description 1
- VYGYNVZNSSTDLJ-HKCOAVLJSA-N monorden Natural products CC1CC2OC2C=C/C=C/C(=O)CC3C(C(=CC(=C3Cl)O)O)C(=O)O1 VYGYNVZNSSTDLJ-HKCOAVLJSA-N 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- 229950003968 motesanib Drugs 0.000 description 1
- RAHBGWKEPAQNFF-UHFFFAOYSA-N motesanib Chemical compound C=1C=C2C(C)(C)CNC2=CC=1NC(=O)C1=CC=CN=C1NCC1=CC=NC=C1 RAHBGWKEPAQNFF-UHFFFAOYSA-N 0.000 description 1
- 229950007627 motolimod Drugs 0.000 description 1
- 229940124303 multikinase inhibitor Drugs 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- 208000025113 myeloid leukemia Diseases 0.000 description 1
- 208000001611 myxosarcoma Diseases 0.000 description 1
- SWZXEVABPLUDIO-WSZYKNRRSA-N n-[(2s)-3-methoxy-1-[[(2s)-3-methoxy-1-[[(2s)-1-[(2r)-2-methyloxiran-2-yl]-1-oxo-3-phenylpropan-2-yl]amino]-1-oxopropan-2-yl]amino]-1-oxopropan-2-yl]-2-methyl-1,3-thiazole-5-carboxamide Chemical compound N([C@@H](COC)C(=O)N[C@@H](COC)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)[C@]1(C)OC1)C(=O)C1=CN=C(C)S1 SWZXEVABPLUDIO-WSZYKNRRSA-N 0.000 description 1
- BLCLNMBMMGCOAS-UHFFFAOYSA-N n-[1-[[1-[[1-[[1-[[1-[[1-[[1-[2-[(carbamoylamino)carbamoyl]pyrrolidin-1-yl]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-4-methyl-1-oxopentan-2-yl]amino]-3-[(2-methylpropan-2-yl)oxy]-1-oxopropan-2-yl]amino]-3-(4-hydroxyphenyl)-1-oxopropan-2-yl]amin Chemical compound C1CCC(C(=O)NNC(N)=O)N1C(=O)C(CCCN=C(N)N)NC(=O)C(CC(C)C)NC(=O)C(COC(C)(C)C)NC(=O)C(NC(=O)C(CO)NC(=O)C(CC=1C2=CC=CC=C2NC=1)NC(=O)C(CC=1NC=NC=1)NC(=O)C1NC(=O)CC1)CC1=CC=C(O)C=C1 BLCLNMBMMGCOAS-UHFFFAOYSA-N 0.000 description 1
- LBWFXVZLPYTWQI-IPOVEDGCSA-N n-[2-(diethylamino)ethyl]-5-[(z)-(5-fluoro-2-oxo-1h-indol-3-ylidene)methyl]-2,4-dimethyl-1h-pyrrole-3-carboxamide;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.CCN(CC)CCNC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C LBWFXVZLPYTWQI-IPOVEDGCSA-N 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-M naphthalene-2-sulfonate Chemical compound C1=CC=CC2=CC(S(=O)(=O)[O-])=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-M 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- 201000011216 nasopharynx carcinoma Diseases 0.000 description 1
- CTMCWCONSULRHO-UHQPFXKFSA-N nemorubicin Chemical compound C1CO[C@H](OC)CN1[C@@H]1[C@H](O)[C@H](C)O[C@@H](O[C@@H]2C3=C(O)C=4C(=O)C5=C(OC)C=CC=C5C(=O)C=4C(O)=C3C[C@](O)(C2)C(=O)CO)C1 CTMCWCONSULRHO-UHQPFXKFSA-N 0.000 description 1
- 229950010159 nemorubicin Drugs 0.000 description 1
- 229940080607 nexavar Drugs 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- 229960001346 nilotinib Drugs 0.000 description 1
- 229940030115 ninlaro Drugs 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- 150000002829 nitrogen Chemical class 0.000 description 1
- OSTGTTZJOCZWJG-UHFFFAOYSA-N nitrosourea Chemical compound NC(=O)N=NO OSTGTTZJOCZWJG-UHFFFAOYSA-N 0.000 description 1
- 125000006574 non-aromatic ring group Chemical group 0.000 description 1
- 229950006584 obatoclax Drugs 0.000 description 1
- 235000020824 obesity Nutrition 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- 229960002700 octreotide Drugs 0.000 description 1
- 201000002575 ocular melanoma Diseases 0.000 description 1
- 229940024847 odomzo Drugs 0.000 description 1
- QNDVLZJODHBUFM-WFXQOWMNSA-N okadaic acid Chemical compound C([C@H](O1)[C@H](C)/C=C/[C@H]2CC[C@@]3(CC[C@H]4O[C@@H](C([C@@H](O)[C@@H]4O3)=C)[C@@H](O)C[C@H](C)[C@@H]3[C@@H](CC[C@@]4(OCCCC4)O3)C)O2)C(C)=C[C@]21O[C@H](C[C@@](C)(O)C(O)=O)CC[C@H]2O QNDVLZJODHBUFM-WFXQOWMNSA-N 0.000 description 1
- VEFJHAYOIAAXEU-UHFFFAOYSA-N okadaic acid Natural products CC(CC(O)C1OC2CCC3(CCC(O3)C=CC(C)C4CC(=CC5(OC(CC(C)(O)C(=O)O)CCC5O)O4)C)OC2C(O)C1C)C6OC7(CCCCO7)CCC6C VEFJHAYOIAAXEU-UHFFFAOYSA-N 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- 230000000771 oncological effect Effects 0.000 description 1
- 229940048191 onivyde Drugs 0.000 description 1
- 229950005750 oprozomib Drugs 0.000 description 1
- 229940041678 oral spray Drugs 0.000 description 1
- 239000000668 oral spray Substances 0.000 description 1
- 230000002611 ovarian Effects 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 125000000160 oxazolidinyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 125000006505 p-cyanobenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1C#N)C([H])([H])* 0.000 description 1
- 229950011410 pacritinib Drugs 0.000 description 1
- HWXVIOGONBBTBY-ONEGZZNKSA-N pacritinib Chemical compound C=1C=C(C=2)NC(N=3)=NC=CC=3C(C=3)=CC=CC=3COC\C=C\COCC=2C=1OCCN1CCCC1 HWXVIOGONBBTBY-ONEGZZNKSA-N 0.000 description 1
- 208000004019 papillary adenocarcinoma Diseases 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 210000002990 parathyroid gland Anatomy 0.000 description 1
- 108700017947 pasireotide Proteins 0.000 description 1
- 229960005415 pasireotide Drugs 0.000 description 1
- NEEFMPSSNFRRNC-HQUONIRXSA-N pasireotide aspartate Chemical compound OC(=O)[C@@H](N)CC(O)=O.OC(=O)[C@@H](N)CC(O)=O.C([C@H]1C(=O)N2C[C@@H](C[C@H]2C(=O)N[C@H](C(=O)N[C@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@H](C(N[C@@H](CC=2C=CC(OCC=3C=CC=CC=3)=CC=2)C(=O)N1)=O)CCCCN)C=1C=CC=CC=1)OC(=O)NCCN)C1=CC=CC=C1 NEEFMPSSNFRRNC-HQUONIRXSA-N 0.000 description 1
- 239000006072 paste Substances 0.000 description 1
- 230000007170 pathology Effects 0.000 description 1
- 229960000639 pazopanib Drugs 0.000 description 1
- 235000020232 peanut Nutrition 0.000 description 1
- WVUNYSQLFKLYNI-AATRIKPKSA-N pelitinib Chemical compound C=12C=C(NC(=O)\C=C\CN(C)C)C(OCC)=CC2=NC=C(C#N)C=1NC1=CC=C(F)C(Cl)=C1 WVUNYSQLFKLYNI-AATRIKPKSA-N 0.000 description 1
- XDRYMKDFEDOLFX-UHFFFAOYSA-N pentamidine Chemical compound C1=CC(C(=N)N)=CC=C1OCCCCCOC1=CC=C(C(N)=N)C=C1 XDRYMKDFEDOLFX-UHFFFAOYSA-N 0.000 description 1
- 229960004448 pentamidine Drugs 0.000 description 1
- 239000002304 perfume Substances 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 208000010918 peritoneal neoplasm Diseases 0.000 description 1
- JRKICGRDRMAZLK-UHFFFAOYSA-L peroxydisulfate Chemical compound [O-]S(=O)(=O)OOS([O-])(=O)=O JRKICGRDRMAZLK-UHFFFAOYSA-L 0.000 description 1
- 229950001457 pexidartinib Drugs 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 125000001791 phenazinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3N=C12)* 0.000 description 1
- 125000001484 phenothiazinyl group Chemical group C1(=CC=CC=2SC3=CC=CC=C3NC12)* 0.000 description 1
- 125000001644 phenoxazinyl group Chemical group C1(=CC=CC=2OC3=CC=CC=C3NC12)* 0.000 description 1
- FAQJJMHZNSSFSM-UHFFFAOYSA-N phenylglyoxylic acid Chemical compound OC(=O)C(=O)C1=CC=CC=C1 FAQJJMHZNSSFSM-UHFFFAOYSA-N 0.000 description 1
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 1
- XKJCHHZQLQNZHY-UHFFFAOYSA-N phthalimide Chemical compound C1=CC=C2C(=O)NC(=O)C2=C1 XKJCHHZQLQNZHY-UHFFFAOYSA-N 0.000 description 1
- 125000005545 phthalimidyl group Chemical group 0.000 description 1
- 229940107670 picato Drugs 0.000 description 1
- 229940075930 picrate Drugs 0.000 description 1
- OXNIZHLAWKMVMX-UHFFFAOYSA-M picrate anion Chemical compound [O-]C1=C([N+]([O-])=O)C=C([N+]([O-])=O)C=C1[N+]([O-])=O OXNIZHLAWKMVMX-UHFFFAOYSA-M 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 208000021310 pituitary gland adenoma Diseases 0.000 description 1
- IUGYQRQAERSCNH-UHFFFAOYSA-M pivalate Chemical compound CC(C)(C)C([O-])=O IUGYQRQAERSCNH-UHFFFAOYSA-M 0.000 description 1
- 229950010765 pivalate Drugs 0.000 description 1
- 125000005547 pivalate group Chemical group 0.000 description 1
- PEZPMAYDXJQYRV-UHFFFAOYSA-N pixantrone Chemical compound O=C1C2=CN=CC=C2C(=O)C2=C1C(NCCN)=CC=C2NCCN PEZPMAYDXJQYRV-UHFFFAOYSA-N 0.000 description 1
- 229940063179 platinol Drugs 0.000 description 1
- 229920000747 poly(lactic acid) Polymers 0.000 description 1
- 229920000058 polyacrylate Polymers 0.000 description 1
- 208000037244 polycythemia vera Diseases 0.000 description 1
- 238000006116 polymerization reaction Methods 0.000 description 1
- 239000001818 polyoxyethylene sorbitan monostearate Substances 0.000 description 1
- 235000010989 polyoxyethylene sorbitan monostearate Nutrition 0.000 description 1
- 229920001451 polypropylene glycol Polymers 0.000 description 1
- 229920000136 polysorbate Polymers 0.000 description 1
- 229940113124 polysorbate 60 Drugs 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 229960000688 pomalidomide Drugs 0.000 description 1
- 229940008606 pomalyst Drugs 0.000 description 1
- 229960001131 ponatinib Drugs 0.000 description 1
- PHXJVRSECIGDHY-UHFFFAOYSA-N ponatinib Chemical compound C1CN(C)CCN1CC(C(=C1)C(F)(F)F)=CC=C1NC(=O)C1=CC=C(C)C(C#CC=2N3N=CC=CC3=NC=2)=C1 PHXJVRSECIGDHY-UHFFFAOYSA-N 0.000 description 1
- 230000004481 post-translational protein modification Effects 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000004302 potassium sorbate Substances 0.000 description 1
- 235000010241 potassium sorbate Nutrition 0.000 description 1
- 229940069338 potassium sorbate Drugs 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 208000016800 primary central nervous system lymphoma Diseases 0.000 description 1
- 229950003608 prinomastat Drugs 0.000 description 1
- YKPYIPVDTNNYCN-INIZCTEOSA-N prinomastat Chemical compound ONC(=O)[C@H]1C(C)(C)SCCN1S(=O)(=O)C(C=C1)=CC=C1OC1=CC=NC=C1 YKPYIPVDTNNYCN-INIZCTEOSA-N 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- 235000013772 propylene glycol Nutrition 0.000 description 1
- 210000002307 prostate Anatomy 0.000 description 1
- 229950008679 protamine sulfate Drugs 0.000 description 1
- 229940121649 protein inhibitor Drugs 0.000 description 1
- 239000012268 protein inhibitor Substances 0.000 description 1
- 108060006633 protein kinase Proteins 0.000 description 1
- 239000000007 protein synthesis inhibitor Substances 0.000 description 1
- 229940034080 provenge Drugs 0.000 description 1
- 125000001042 pteridinyl group Chemical group N1=C(N=CC2=NC=CN=C12)* 0.000 description 1
- 150000003834 purine nucleoside derivatives Chemical class 0.000 description 1
- 150000003212 purines Chemical class 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- HAMAGKWXRRTWCJ-UHFFFAOYSA-N pyrido[2,3-b][1,4]oxazin-3-one Chemical compound C1=CN=C2OC(=O)C=NC2=C1 HAMAGKWXRRTWCJ-UHFFFAOYSA-N 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 150000003230 pyrimidines Chemical class 0.000 description 1
- 125000001422 pyrrolinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 150000003856 quaternary ammonium compounds Chemical class 0.000 description 1
- 125000001453 quaternary ammonium group Chemical group 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 125000004621 quinuclidinyl group Chemical group N12C(CC(CC1)CC2)* 0.000 description 1
- 229950001626 quizartinib Drugs 0.000 description 1
- CVWXJKQAOSCOAB-UHFFFAOYSA-N quizartinib Chemical compound O1C(C(C)(C)C)=CC(NC(=O)NC=2C=CC(=CC=2)C=2N=C3N(C4=CC=C(OCCN5CCOCC5)C=C4S3)C=2)=N1 CVWXJKQAOSCOAB-UHFFFAOYSA-N 0.000 description 1
- 150000003254 radicals Chemical class 0.000 description 1
- AECPBJMOGBFQDN-YMYQVXQQSA-N radicicol Chemical compound C1CCCC(=O)C[C@H]2[C@H](Cl)C(=O)CC(=O)[C@H]2C(=O)O[C@H](C)C[C@H]2O[C@@H]21 AECPBJMOGBFQDN-YMYQVXQQSA-N 0.000 description 1
- 229930192524 radicicol Natural products 0.000 description 1
- 229940044551 receptor antagonist Drugs 0.000 description 1
- 239000002464 receptor antagonist Substances 0.000 description 1
- 108091006082 receptor inhibitors Proteins 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 229940100618 rectal suppository Drugs 0.000 description 1
- 239000006215 rectal suppository Substances 0.000 description 1
- 208000019465 refractory cytopenia of childhood Diseases 0.000 description 1
- 229960004836 regorafenib Drugs 0.000 description 1
- 210000003289 regulatory T cell Anatomy 0.000 description 1
- 208000015347 renal cell adenocarcinoma Diseases 0.000 description 1
- 230000000241 respiratory effect Effects 0.000 description 1
- 239000003340 retarding agent Substances 0.000 description 1
- OAKGNIRUXAZDQF-TXHRRWQRSA-N retaspimycin Chemical compound N1C(=O)\C(C)=C\C=C/[C@H](OC)[C@@H](OC(N)=O)\C(C)=C\[C@H](C)[C@@H](O)[C@@H](OC)C[C@H](C)CC2=C(O)C1=CC(O)=C2NCC=C OAKGNIRUXAZDQF-TXHRRWQRSA-N 0.000 description 1
- 229930002330 retinoic acid Natural products 0.000 description 1
- 102000003702 retinoic acid receptors Human genes 0.000 description 1
- 108090000064 retinoic acid receptors Proteins 0.000 description 1
- 229940120975 revlimid Drugs 0.000 description 1
- 206010039073 rheumatoid arthritis Diseases 0.000 description 1
- 229960000371 rofecoxib Drugs 0.000 description 1
- RZJQGNCSTQAWON-UHFFFAOYSA-N rofecoxib Chemical compound C1=CC(S(=O)(=O)C)=CC=C1C1=C(C=2C=CC=CC=2)C(=O)OC1 RZJQGNCSTQAWON-UHFFFAOYSA-N 0.000 description 1
- 229950005230 rogletimide Drugs 0.000 description 1
- VHXNKPBCCMUMSW-FQEVSTJZSA-N rubitecan Chemical compound C1=CC([N+]([O-])=O)=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VHXNKPBCCMUMSW-FQEVSTJZSA-N 0.000 description 1
- 229950009213 rubitecan Drugs 0.000 description 1
- INBJJAFXHQQSRW-STOWLHSFSA-N rucaparib camsylate Chemical compound CC1(C)[C@@H]2CC[C@@]1(CS(O)(=O)=O)C(=O)C2.CNCc1ccc(cc1)-c1[nH]c2cc(F)cc3C(=O)NCCc1c23 INBJJAFXHQQSRW-STOWLHSFSA-N 0.000 description 1
- JFMWPOCYMYGEDM-XFULWGLBSA-N ruxolitinib phosphate Chemical compound OP(O)(O)=O.C1([C@@H](CC#N)N2N=CC(=C2)C=2C=3C=CNC=3N=CN=2)CCCC1 JFMWPOCYMYGEDM-XFULWGLBSA-N 0.000 description 1
- 229950008902 safingol Drugs 0.000 description 1
- NGWSFRIPKNWYAO-SHTIJGAHSA-N salinosporamide A Chemical compound C([C@@H]1[C@H](O)[C@]23C(=O)O[C@]2([C@H](C(=O)N3)CCCl)C)CCC=C1 NGWSFRIPKNWYAO-SHTIJGAHSA-N 0.000 description 1
- NGWSFRIPKNWYAO-UHFFFAOYSA-N salinosporamide A Natural products N1C(=O)C(CCCl)C2(C)OC(=O)C21C(O)C1CCCC=C1 NGWSFRIPKNWYAO-UHFFFAOYSA-N 0.000 description 1
- 210000003296 saliva Anatomy 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 201000008407 sebaceous adenocarcinoma Diseases 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 210000000582 semen Anatomy 0.000 description 1
- 208000004548 serous cystadenocarcinoma Diseases 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 150000004760 silicates Chemical class 0.000 description 1
- RMAQACBXLXPBSY-UHFFFAOYSA-N silicic acid Chemical compound O[Si](O)(O)O RMAQACBXLXPBSY-UHFFFAOYSA-N 0.000 description 1
- 235000012239 silicon dioxide Nutrition 0.000 description 1
- 238000009097 single-agent therapy Methods 0.000 description 1
- 229960000714 sipuleucel-t Drugs 0.000 description 1
- 201000003708 skin melanoma Diseases 0.000 description 1
- 201000010106 skin squamous cell carcinoma Diseases 0.000 description 1
- 208000000649 small cell carcinoma Diseases 0.000 description 1
- 210000000813 small intestine Anatomy 0.000 description 1
- AWUCVROLDVIAJX-GSVOUGTGSA-N sn-glycerol 3-phosphate Chemical compound OC[C@@H](O)COP(O)(O)=O AWUCVROLDVIAJX-GSVOUGTGSA-N 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- MFBOGIVSZKQAPD-UHFFFAOYSA-M sodium butyrate Chemical compound [Na+].CCCC([O-])=O MFBOGIVSZKQAPD-UHFFFAOYSA-M 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 235000017550 sodium carbonate Nutrition 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 1
- SARBMGXGWXCXFW-GJHVZSAVSA-M sodium;2-[[(2s)-2-[[(4r)-4-[[(2s)-2-[[(2r)-2-[(3r,4r,5s,6r)-3-acetamido-2,5-dihydroxy-6-(hydroxymethyl)oxan-4-yl]oxypropanoyl]amino]propanoyl]amino]-5-amino-5-oxopentanoyl]amino]propanoyl]amino]ethyl [(2r)-2,3-di(hexadecanoyloxy)propyl] phosphate;hydrate Chemical compound O.[Na+].CCCCCCCCCCCCCCCC(=O)OC[C@@H](OC(=O)CCCCCCCCCCCCCCC)COP([O-])(=O)OCCNC(=O)[C@H](C)NC(=O)CC[C@H](C(N)=O)NC(=O)[C@H](C)NC(=O)[C@@H](C)O[C@H]1[C@H](O)[C@@H](CO)OC(O)[C@@H]1NC(C)=O SARBMGXGWXCXFW-GJHVZSAVSA-M 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 239000001587 sorbitan monostearate Substances 0.000 description 1
- 235000011076 sorbitan monostearate Nutrition 0.000 description 1
- 229940035048 sorbitan monostearate Drugs 0.000 description 1
- 229950007213 spartalizumab Drugs 0.000 description 1
- OTKJDMGTUTTYMP-ZWKOTPCHSA-N sphinganine Chemical compound CCCCCCCCCCCCCCC[C@@H](O)[C@@H](N)CO OTKJDMGTUTTYMP-ZWKOTPCHSA-N 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 239000003206 sterilizing agent Substances 0.000 description 1
- 229940090374 stivarga Drugs 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 208000030819 subependymoma Diseases 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 239000001384 succinic acid Substances 0.000 description 1
- 150000003457 sulfones Chemical class 0.000 description 1
- 229960001796 sunitinib Drugs 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 229940034785 sutent Drugs 0.000 description 1
- 201000010965 sweat gland carcinoma Diseases 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 229940022873 synribo Drugs 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000002278 tabletting lubricant Substances 0.000 description 1
- 229940081616 tafinlar Drugs 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- UXXQOJXBIDBUAC-UHFFFAOYSA-N tandutinib Chemical compound COC1=CC2=C(N3CCN(CC3)C(=O)NC=3C=CC(OC(C)C)=CC=3)N=CN=C2C=C1OCCCN1CCCCC1 UXXQOJXBIDBUAC-UHFFFAOYSA-N 0.000 description 1
- 229950007866 tanespimycin Drugs 0.000 description 1
- 229940120982 tarceva Drugs 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 229940069905 tasigna Drugs 0.000 description 1
- 229940126625 tavolimab Drugs 0.000 description 1
- RCINICONZNJXQF-XAZOAEDWSA-N taxol® Chemical compound O([C@@H]1[C@@]2(CC(C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3(C21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-XAZOAEDWSA-N 0.000 description 1
- 229940063683 taxotere Drugs 0.000 description 1
- 210000001138 tear Anatomy 0.000 description 1
- 229940066453 tecentriq Drugs 0.000 description 1
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical compound C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 description 1
- PRAAPINBUWJLGA-UHFFFAOYSA-N telaglenastat Chemical compound FC(F)(F)OC1=CC=CC(CC(=O)NC=2N=NC(CCCCC=3SC(NC(=O)CC=4N=CC=CC=4)=NN=3)=CC=2)=C1 PRAAPINBUWJLGA-UHFFFAOYSA-N 0.000 description 1
- YVSQVYZBDXIXCC-INIZCTEOSA-N telomestatin Chemical compound N=1C2=COC=1C(N=1)=COC=1C(N=1)=COC=1C(N=1)=COC=1C(N=1)=COC=1C(=C(O1)C)N=C1C(=C(O1)C)N=C1[C@@]1([H])N=C2SC1 YVSQVYZBDXIXCC-INIZCTEOSA-N 0.000 description 1
- 229940061353 temodar Drugs 0.000 description 1
- QFJCIRLUMZQUOT-UHFFFAOYSA-N temsirolimus Natural products C1CC(O)C(OC)CC1CC(C)C1OC(=O)C2CCCCN2C(=O)C(=O)C(O)(O2)C(C)CCC2CC(OC)C(C)=CC=CC=CC(C)CC(C)C(=O)C(OC)C(O)C(C)=CC(C)C(=O)C1 QFJCIRLUMZQUOT-UHFFFAOYSA-N 0.000 description 1
- ILMRJRBKQSSXGY-UHFFFAOYSA-N tert-butyl(dimethyl)silicon Chemical group C[Si](C)C(C)(C)C ILMRJRBKQSSXGY-UHFFFAOYSA-N 0.000 description 1
- 229960005353 testolactone Drugs 0.000 description 1
- BPEWUONYVDABNZ-DZBHQSCQSA-N testolactone Chemical compound O=C1C=C[C@]2(C)[C@H]3CC[C@](C)(OC(=O)CC4)[C@@H]4[C@@H]3CCC2=C1 BPEWUONYVDABNZ-DZBHQSCQSA-N 0.000 description 1
- 229960003604 testosterone Drugs 0.000 description 1
- 229930101283 tetracycline Natural products 0.000 description 1
- 229960002180 tetracycline Drugs 0.000 description 1
- 235000019364 tetracycline Nutrition 0.000 description 1
- 150000003522 tetracyclines Chemical class 0.000 description 1
- MHXBHWLGRWOABW-UHFFFAOYSA-N tetradecyl octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCCCCCCCCCCCCCC MHXBHWLGRWOABW-UHFFFAOYSA-N 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- BSYVTEYKTMYBMK-UHFFFAOYSA-N tetrahydrofurfuryl alcohol Chemical compound OCC1CCCO1 BSYVTEYKTMYBMK-UHFFFAOYSA-N 0.000 description 1
- 125000001712 tetrahydronaphthyl group Chemical group C1(CCCC2=CC=CC=C12)* 0.000 description 1
- 125000003507 tetrahydrothiofenyl group Chemical group 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 229960003433 thalidomide Drugs 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000005308 thiazepinyl group Chemical group S1N=C(C=CC=C1)* 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 239000003734 thymidylate synthase inhibitor Substances 0.000 description 1
- 210000001685 thyroid gland Anatomy 0.000 description 1
- WYWHKKSPHMUBEB-UHFFFAOYSA-N tioguanine Chemical compound N1C(N)=NC(=S)C2=C1N=CN2 WYWHKKSPHMUBEB-UHFFFAOYSA-N 0.000 description 1
- 229960000940 tivozanib Drugs 0.000 description 1
- 239000011732 tocopherol Substances 0.000 description 1
- 229960001295 tocopherol Drugs 0.000 description 1
- 239000011731 tocotrienol Substances 0.000 description 1
- 229960001350 tofacitinib Drugs 0.000 description 1
- UJLAWZDWDVHWOW-YPMHNXCESA-N tofacitinib Chemical compound C[C@@H]1CCN(C(=O)CC#N)C[C@@H]1N(C)C1=NC=NC2=C1C=CN2 UJLAWZDWDVHWOW-YPMHNXCESA-N 0.000 description 1
- 239000003970 toll like receptor agonist Substances 0.000 description 1
- 229940044655 toll-like receptor 9 agonist Drugs 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 239000012049 topical pharmaceutical composition Substances 0.000 description 1
- 229940100411 torisel Drugs 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 229960004066 trametinib Drugs 0.000 description 1
- 206010044412 transitional cell carcinoma Diseases 0.000 description 1
- 229940066958 treanda Drugs 0.000 description 1
- 238000011269 treatment regimen Methods 0.000 description 1
- 229960001727 tretinoin Drugs 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- RTKIYFITIVXBLE-QEQCGCAPSA-N trichostatin A Chemical compound ONC(=O)/C=C/C(/C)=C/[C@@H](C)C(=O)C1=CC=C(N(C)C)C=C1 RTKIYFITIVXBLE-QEQCGCAPSA-N 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 1
- 125000004044 trifluoroacetyl group Chemical group FC(C(=O)*)(F)F 0.000 description 1
- 125000000025 triisopropylsilyl group Chemical group C(C)(C)[Si](C(C)C)(C(C)C)* 0.000 description 1
- KVJXBPDAXMEYOA-CXANFOAXSA-N trilostane Chemical compound OC1=C(C#N)C[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CC[C@@]32O[C@@H]31 KVJXBPDAXMEYOA-CXANFOAXSA-N 0.000 description 1
- 229960001670 trilostane Drugs 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 208000022679 triple-negative breast carcinoma Diseases 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 102000003298 tumor necrosis factor receptor Human genes 0.000 description 1
- 229940094060 tykerb Drugs 0.000 description 1
- TUCIOBMMDDOEMM-RIYZIHGNSA-N tyrphostin B42 Chemical compound C1=C(O)C(O)=CC=C1\C=C(/C#N)C(=O)NCC1=CC=CC=C1 TUCIOBMMDDOEMM-RIYZIHGNSA-N 0.000 description 1
- 230000006663 ubiquitin-proteasome pathway Effects 0.000 description 1
- 229940125117 ulevostinag Drugs 0.000 description 1
- ZDPHROOEEOARMN-UHFFFAOYSA-N undecanoic acid Chemical compound CCCCCCCCCCC(O)=O ZDPHROOEEOARMN-UHFFFAOYSA-N 0.000 description 1
- 229940022919 unituxin Drugs 0.000 description 1
- 210000000626 ureter Anatomy 0.000 description 1
- 208000010570 urinary bladder carcinoma Diseases 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- 229950003520 utomilumab Drugs 0.000 description 1
- 206010046885 vaginal cancer Diseases 0.000 description 1
- 208000013139 vaginal neoplasm Diseases 0.000 description 1
- 229940050482 valchlor Drugs 0.000 description 1
- 229960002004 valdecoxib Drugs 0.000 description 1
- LNPDTQAFDNKSHK-UHFFFAOYSA-N valdecoxib Chemical compound CC=1ON=C(C=2C=CC=CC=2)C=1C1=CC=C(S(N)(=O)=O)C=C1 LNPDTQAFDNKSHK-UHFFFAOYSA-N 0.000 description 1
- 229940070710 valerate Drugs 0.000 description 1
- 229960000241 vandetanib Drugs 0.000 description 1
- 239000002525 vasculotropin inhibitor Substances 0.000 description 1
- YCOYDOIWSSHVCK-UHFFFAOYSA-N vatalanib Chemical compound C1=CC(Cl)=CC=C1NC(C1=CC=CC=C11)=NN=C1CC1=CC=NC=C1 YCOYDOIWSSHVCK-UHFFFAOYSA-N 0.000 description 1
- 229950000578 vatalanib Drugs 0.000 description 1
- 235000013311 vegetables Nutrition 0.000 description 1
- 229940099039 velcade Drugs 0.000 description 1
- 229960003862 vemurafenib Drugs 0.000 description 1
- 229940065658 vidaza Drugs 0.000 description 1
- JXLYSJRDGCGARV-CFWMRBGOSA-N vinblastine Chemical compound C([C@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-CFWMRBGOSA-N 0.000 description 1
- JXLYSJRDGCGARV-XQKSVPLYSA-N vincaleukoblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1NC1=CC=CC=C21 JXLYSJRDGCGARV-XQKSVPLYSA-N 0.000 description 1
- 229940028393 vincasar Drugs 0.000 description 1
- 229960002066 vinorelbine Drugs 0.000 description 1
- GBABOYUKABKIAF-GHYRFKGUSA-N vinorelbine Chemical compound C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC GBABOYUKABKIAF-GHYRFKGUSA-N 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 208000012498 virus associated tumor Diseases 0.000 description 1
- 229940121351 vopratelimab Drugs 0.000 description 1
- 229960001771 vorozole Drugs 0.000 description 1
- XLMPPFTZALNBFS-INIZCTEOSA-N vorozole Chemical compound C1([C@@H](C2=CC=C3N=NN(C3=C2)C)N2N=CN=C2)=CC=C(Cl)C=C1 XLMPPFTZALNBFS-INIZCTEOSA-N 0.000 description 1
- 229940069559 votrient Drugs 0.000 description 1
- 201000005102 vulva cancer Diseases 0.000 description 1
- 208000013013 vulvar carcinoma Diseases 0.000 description 1
- 239000003871 white petrolatum Substances 0.000 description 1
- 102000018441 wolframin Human genes 0.000 description 1
- 229940049068 xalkori Drugs 0.000 description 1
- 229940053867 xeloda Drugs 0.000 description 1
- 229940014556 xgeva Drugs 0.000 description 1
- 229940085728 xtandi Drugs 0.000 description 1
- 229940004212 yondelis Drugs 0.000 description 1
- 229940036061 zaltrap Drugs 0.000 description 1
- 229940034727 zelboraf Drugs 0.000 description 1
- 150000003751 zinc Chemical class 0.000 description 1
- 229960002760 ziv-aflibercept Drugs 0.000 description 1
- 229940061261 zolinza Drugs 0.000 description 1
- 229940002005 zometa Drugs 0.000 description 1
- CGTADGCBEXYWNE-JUKNQOCSSA-N zotarolimus Chemical compound N1([C@H]2CC[C@@H](C[C@@H](C)[C@H]3OC(=O)[C@@H]4CCCCN4C(=O)C(=O)[C@@]4(O)[C@H](C)CC[C@H](O4)C[C@@H](/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C3)OC)C[C@H]2OC)C=NN=N1 CGTADGCBEXYWNE-JUKNQOCSSA-N 0.000 description 1
- 229950009819 zotarolimus Drugs 0.000 description 1
- 229940095188 zydelig Drugs 0.000 description 1
- 229940052129 zykadia Drugs 0.000 description 1
- 229940051084 zytiga Drugs 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/553—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having at least one nitrogen and one oxygen as ring hetero atoms, e.g. loxapine, staurosporine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
- C07D491/044—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
- C07D491/048—Ortho-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring the oxygen-containing ring being five-membered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/10—Spiro-condensed systems
- C07D491/107—Spiro-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/10—Spiro-condensed systems
- C07D491/113—Spiro-condensed systems with two or more oxygen atoms as ring hetero atoms in the oxygen-containing ring
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
La presente invención proporciona compuestos de fórmula (i'), o sales farmacéuticamente aceptables de las mismas, composiciones farmacéuticas de las mismas y los métodos de uso de los mismos para tratar los trastornos proliferativos celulares (por ejemplo, cáncer). (Traducción automática con Google Translate, sin valor legal)
Description
DESCRIPCIÓN
Compuestos antiproliferación y usos de los mismos
Campo técnico de la invención
La presente invención se refiere a compuestos y compuestos para su uso en métodos útiles para tratar trastornos proliferativos celulares (por ejemplo, cáncer). La invención también proporciona composiciones farmacéuticamente aceptables que comprenden compuestos de la presente invención y métodos para usar dichas composiciones en el tratamiento de diversos trastornos proliferativos.
Antecedentes de la invención
Los trastornos proliferativos celulares comprenden poblaciones de células malignas y no malignas que difieren del tejido circundante morfológica y/o genotípicamente. Los ejemplos de trastornos proliferativos celulares incluyen, por ejemplo, tumores sólidos, cáncer, retinopatía diabética, síndromes neovasculares intraoculares, degeneración macular, artritis reumatoide, psoriasis y endometriosis. El cáncer es un grupo de enfermedades que implican una proliferación celular anormal con el potencial de invadir o propagarse a otras partes del organismo. Según los centros de control y prevención de enfermedades (CDC, por sus siglas en inglés), el cáncer es la segunda causa principal de muerte en los Estados Unidos. Por lo tanto, se desean tratamientos adicionales para los trastornos proliferativos celulares para proporcionar a los pacientes más opciones.
Sumario de la invención
Se ha descubierto que los compuestos de la presente invención, y composiciones farmacéuticamente aceptables de los mismos, son útiles para tratar trastornos proliferativos (por ejemplo, cáncer). En un aspecto, la presente invención proporciona un compuesto de fórmula I:

o una sal farmacéuticamente aceptable del mismo, en donde cada variable es como se define y se describe en el presente documento.
Los compuestos de la presente invención, y las composiciones farmacéuticamente aceptables de los mismos, son útiles para tratar una variedad de trastornos proliferativos (por ejemplo, cáncer) como se describe en el presente documento.
Descripción detallada de determinadas realizaciones
1. Descripción general de determinadas realizaciones de la invención:
Se ha descubierto que los compuestos de la presente invención, o las sales de los mismos, presentan una eficacia pronunciada en múltiples modelos de xenoinjertos derivados de líneas celulares y derivados de pacientes. Por ejemplo, los compuestos de la invención, o sales de los mismos, se observa que dan lugar a una regresión completa y duradera en modelos de cáncer de pulmón no microcítico (NSCLC, por sus siglas en inglés), mieloma, carcinoma hepatocelular (HCC, por sus siglas en inglés), cáncer de mama y melanoma. También se ha descubierto que los compuestos de la invención dan como resultado una inhibición potenciada de la viabilidad celular, particularmente las células donde se sobreexpresa wolframina (WFS1). Sin desear quedar ligados a ninguna teoría específica, se cree que los compuestos de la invención provocan la liberación de calcio del retículo endoplásmico (RE) a través de un supuesto canal iónico de Ca2+ conocido como wolframina (WFS1), que induce estrés al Re y "respuesta a proteínas desplegadas" (UPR, por sus siglas en inglés) y da lugar a muerte celular.
En un aspecto, la presente invención proporciona un compuesto de fórmula I:

o una sal farmacéuticamente aceptable del mismo, en donde:
el anillo A es un anillo seleccionado entre fenilo, un anillo carbocíclico saturado o parcialmente insaturado de 5-7 miembros, un anillo heterocíclico bicíclico saturado o parcialmente insaturado de 8 -12 miembros que tiene 1 -2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, un anillo heteroaromático de 5-6 miembros que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno, o azufre, o un anillo heteroaromático bicíclico de 8-10 miembros que tiene de 1-5 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre;
cada R1 es independientemente hidrógeno o alifático C1-3; o
dos grupos R1 se toman opcionalmente junto con sus átomos intermedios para formar un anillo carbocíclico condensado parcialmente insaturado de 5-8 miembros;
cada uno de R2 es independientemente hidrógeno, halógeno, -CN, -NO2, -C(O)OR, -C(O)NR2, -NR2, -NRC(O)R, -NRC(O)OR, -NRS(O)2R, -OR, -P(O)R2, -SR, -S(O)R, -S(O)2R, -S(O)(NH)R o R; o
dos grupos R2 se toman opcionalmente juntos para formar =O;
cada R3 es independientemente hidrógeno o alifático C1-3; o:
dos grupos R3 se toman opcionalmente juntos para formar =O;
dos grupos R3 se toman opcionalmente juntos para formar =CH2;
dos grupos R3 se toman opcionalmente junto con sus átomos intermedios para formar un anillo espirocíclico saturado de 5-8 miembros que tiene 0-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre; o
dos grupos R3 se toman opcionalmente junto con sus átomos intermedios para formar un anillo bicíclico con puente saturado de 5-8 miembros que tiene 0-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre;
cada R es independientemente hidrógeno o un grupo opcionalmente sustituido seleccionado entre hidrocarburo alifático C1-6, un anillo carbocíclico monocíclico saturado o parcialmente insaturado de 3-8 miembros, fenilo, un anillo heterocíclico espirobicíclico saturado de 7-10 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, un anillo heterocíclico bicíclico condensado saturado o parcialmente insaturado de 7-10 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, un anillo heterocíclico monocíclico saturado o parcialmente insaturado de 4-8 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, o un anillo heteroaromático monocíclico de 5-6 miembros que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre; o:
dos grupos R en el mismo nitrógeno se toman opcionalmente junto con sus átomos intermedios para formar un anillo de 4-7 miembros saturado, parcialmente insaturado o heteroarilo que tiene 0-3 heteroátomos, además del nitrógeno, seleccionado independientemente entre nitrógeno, oxígeno y azufre, opcionalmente sustituido con 1-2 grupos oxo;
X es -O-, -N(R)-, -N(S(O)2(R))-, -S-, -S(O)-, -S(O)2-, -CH2-, -CH(R3)- o -C(R3)2-;
m es 0, 1 o 2;
n es 0, 1,2, 3, 4 o 5; y
p es 0, 1 o 2.
En un aspecto, la presente invención proporciona un compuesto de fórmula I':

o una sal farmacéuticamente aceptable del mismo, en donde:
el anillo A es un anillo seleccionado entre fenilo, un anillo carbocíclico saturado o parcialmente insaturado de 5-7 miembros, un anillo heterocíclico bicíclico saturado o parcialmente insaturado de 8 -12 miembros que tiene 1 -2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, un anillo heteroaromático de 5-6 miembros que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre; o un anillo heteroaromático bicíclico de 8-10 miembros que tiene 1-5 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre;
cada R1 es independientemente hidrógeno o alifático C1-3 opcionalmente sustituido con 1-6 halógeno; o
dos grupos R1 se toman opcionalmente junto con sus átomos intermedios para formar un anillo carbocíclico condensado parcialmente insaturado de 5-8 miembros;
cada uno de R2 es independientemente hidrógeno, halógeno, -CN, -NO2, -C(O)OR, -C(O)NR2, -NR2, -NRC(O)R, -NRC(O)OR, -NRS(O)2R, -OR, -P(O)R2, -SR, -S(O)R, -S(O)2R, -S(O)(NH)R, -S(O)2NR2 o R; o
dos grupos R2 se toman opcionalmente juntos para formar =O; o
dos grupos R2 se toman opcionalmente junto con sus átomos intermedios para formar un anillo espirocíclico saturado de 3-8 miembros que tiene 0-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre;
cada R3 es independientemente hidrógeno, -OH o alifático C1-3; o
dos grupos R3 se toman opcionalmente juntos para formar =O; o
dos grupos R3 se toman opcionalmente juntos para formar =CH2; o
dos grupos R3 se toman opcionalmente junto con sus átomos intermedios para formar un anillo espirocíclico saturado de 3-8 miembros que tiene 0-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre; o
dos grupos R3 se toman opcionalmente junto con sus átomos intermedios para formar un anillo bicíclico con puente saturado de 5-8 miembros que tiene 0-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre;
cada R es independientemente hidrógeno o un grupo opcionalmente sustituido seleccionado entre hidrocarburo alifático C1-6, un anillo carbocíclico monocíclico saturado o parcialmente insaturado de 3-8 miembros, fenilo, un anillo heterocíclico espirobicíclico saturado de 7-10 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, un anillo heterocíclico bicíclico condensado saturado o parcialmente insaturado de 7-10 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, un anillo heterocíclico monocíclico saturado o parcialmente insaturado de 4-8 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, o un anillo heteroaromático monocíclico de 5-6 miembros que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre; o:
dos grupos R en el mismo nitrógeno se toman opcionalmente junto con sus átomos intermedios para formar un anillo de 4-7 miembros saturado, parcialmente insaturado o heteroarilo que tiene 0-3 heteroátomos, además del nitrógeno, seleccionado independientemente entre nitrógeno, oxígeno y azufre, opcionalmente sustituido con 1-2 grupos oxo;
------es un enlace sencillo o un doble enlace;
X es -O-, -N(R)-, -N(S(O)2(R))-, -S-, -S(O)-, -S(O)2-, -CH2-, -CH(R3)- o -C(R3)2-;
m es 0, 1 o 2;
n es 0, 1,2, 3, 4 o 5; y
p es 0, 1 o 2.
2. Compuestos y definiciones:
Los compuestos de la presente invención incluyen los descritos generalmente en el presente documento, y se ilustran adicionalmente mediante las clases, subclases y especies divulgadas en el presente documento. Como se usa en el presente documento, se aplicarán las siguientes definiciones a menos que se indique lo contrario. Para los fines de esta invención, los elementos químicos se identifican de acuerdo con la Tabla Periódica de los Elementos, versión CAS, Handbook of Chemistry and Physics, 75a ed. Adicionalmente, se describen principios generales de química
orgánica en "Organic Chemistry", Thomas Sorrell, University Science Books, Sausalito: 1999 y "March's Advanced Organic Chemistry", 5a ed., Ed.: Smith, M. B. y March, J., John Wiley & Sons, Nueva York: 2001, cuyos contenidos al completo se incorporan en el presente documento por referencia.
El término "alifático" o la expresión "grupo alifático", como se usa en el presente documento, significa una cadena de hidrocarburos sustituida o sin sustituir, de cadena lineal (es decir, no ramificada) o ramificada, que está completamente saturada o que contiene una o más unidades de insaturación, o un hidrocarburo monocíclico o hidrocarburo bicíclico que está completamente saturado o que contiene una o más unidades de insaturación, pero que no es aromático (también denominado en el presente documento "carbociclo", "cicloalifático" o "cicloalquilo"), que tiene un único punto de unión al resto de la molécula. A menos que se especifique de otra manera, los grupos alifáticos contienen 1-6 átomos de carbono alifáticos. En algunas realizaciones, los grupos alifáticos contienen 1-5 átomos de carbono alifáticos. En otras realizaciones, los grupos alifáticos contienen 1-4 átomos de carbono alifáticos. Aún en otras realizaciones, los grupos alifáticos contienen 1-3 átomos de carbono alifáticos y, en otras realizaciones más, los grupos alifáticos contienen 1-2 átomos de carbono alifáticos. En algunas realizaciones, "cicloalifático" (o "carbociclo" o "cicloalquilo") se refiere a un hidrocarburo monocíclico C3-C6 que está completamente saturado o que contiene una o más unidades de insaturación, pero que no es aromático, que tiene un único punto de unión al resto de la molécula. Los grupos alifáticos adecuados incluyen, pero no se limitan a, grupos alquilo, alquenilo, alquinilo lineales o ramificados, sustituidos o sin sustituir e híbridos de los mismos, tales como (cicloalquil)alquilo, (cicloalquenil)alquilo o (cicloalquil)alquenilo.
Como se usa en el presente documento, la expresión "anillo bicíclico" o "sistema de anillo bicíclico" se refiere a cualquier sistema de anillo bicíclico, es decir, carbocíclico o heterocíclico, saturado o que tiene una o más unidades de insaturación, que tiene uno o más átomos en común entre los dos anillos del sistema de anillo. Por lo tanto, el término incluye cualquier fusión de anillo permisible, tal como orfo-condensada o espirocíclica. Como se usa en el presente documento, el término "heterobicíclico" es un subconjunto de "bicíclico" que requiere que uno o más heteroátomos estén presentes en uno o ambos anillos de la bicicleta. Tales heteroátomos pueden estar presentes en las uniones del anillo y están opcionalmente sustituidos, y pueden seleccionarse de nitrógeno (incluidos los N-óxidos), oxígeno, azufre (incluidas las formas oxidadas como sulfonas y sulfonatos), fósforo (incluidas las formas oxidadas como fosfatos), boro, etc. En algunas realizaciones, un grupo bicíclico tiene 7-12 miembros en el anillo y 0-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre. Como se usa en el presente documento, la expresión "bicíclico con puente" se refiere a cualquier sistema de anillo bicíclico, es decir, carbocíclico o heterocíclico, saturado o parcialmente insaturado, que tiene al menos un puente. Como lo define la IUPAC, un "puente" es una cadena no ramificada de átomos o un átomo o un enlace de valencia que conecta dos cabezas de puente, donde una "cabeza de puente" es cualquier átomo esquelético del sistema de anillos que está unido a tres o más átomos esqueléticos (excluyendo hidrógeno). En algunas realizaciones, un grupo bicíclico con puente tiene 7-12 miembros en el anillo y 0-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre. Tales grupos bicíclicos con puente son bien conocidos en la técnica e incluyen los grupos que se exponen a continuación, en los que cada grupo está unido al resto de la molécula en cualquier átomo de carbono o nitrógeno sustituible. A menos que se especifique de otra manera, un grupo bicíclico con puente está opcionalmente sustituido con uno o más sustituyentes como se establece para los grupos alifáticos. Adicionalmente, o como alternativa, cualquier nitrógeno sustituible de un grupo bicíclico con puente está opcionalmente sustituido. Los ejemplos de anillos bicíclicos incluyen:

Los ejemplos de bicíclicos con puente incluyen:


La expresión "alquilo inferior" se refiere a un grupo alquilo C1-4 lineal o ramificado. Los ejemplos de grupos alquilo inferior son metilo, etilo, propilo, isopropilo, butilo, isobutilo y ferc-butilo.
El término "haloalquilo inferior" se refiere a un grupo alquilo C1-4 lineal o ramificado que está sustituido con uno o más átomos de halógeno.
El término "heteroátomo" significa uno o más de oxígeno, azufre, nitrógeno, fósforo o silicio (incluyendo, cualquier forma oxidada de nitrógeno, azufre, fósforo o silicio; la forma cuaternizada de cualquier nitrógeno básico o; un nitrógeno sustituible de un anillo heterocíclico, por ejemplo N (como en 3,4-dihidro-2H-pirrolilo), NH (como en pirrolidinilo) o NR+ (como en pirrolidinilo N-sustituido)).
El término "insaturado", como se usa en el presente documento, significa que un resto tiene una o más unidades de insaturación.
Como se usa en el presente documento, la expresión "cadena de hidrocarburo C1-8 (o C1-6) bivalente saturada o insaturada, lineal o ramificada", se refiere cadenas bivalentes de alquileno, alquenileno, y alquinileno que son lineales o ramificadas como se define en el presente documento.
El término "alquileno" se refiere a un grupo alquilo bivalente. Una "cadena de alquileno" es un grupo polimetileno, es decir, -(CH2)n-, en la que n es un número entero positivo, preferentemente de 1 a 6, de 1 a 4, de 1 a 3, de 1 a 2, o de 2 a 3. Una cadena de alquileno sustituido es un grupo polimetileno en el que uno o más átomos de hidrógeno de metileno están sustituidos por un sustituyente. Los sustituyentes adecuados incluyen los descritos a continuación para un grupo alifático sustituido.
El término "alquenileno" se refiere a un grupo alquenilo bivalente. Una cadena de alquenileno sustituido es un grupo polimetileno que contiene al menos un doble enlace en que uno o más átomos de hidrógeno están sustituidos con un sustituyente. Los sustituyentes adecuados incluyen los descritos a continuación para un grupo alifático sustituido.
Como se usa en el presente documento, el término "ciclopropilenilo" se refiere a un grupo ciclopropilo bivalente de la
siguiente estructura:
El término "halógeno" significa F, Cl, Br o I.
El término "arilo" usado solo o como parte de un resto mayor como en "aralquilo", "aralcoxi", o "ariloxialquilo", se refiere a sistemas de anillos monocíclicos o bicíclicos que tienen un total de cinco a catorce miembros del anillo, en los que al menos un anillo del sistema es aromático y en los que cada anillo del sistema contiene de 3 a 7 miembros de anillo. El término "arilo" puede usarse indistintamente con la expresión "anillo de arilo". En determinadas realizaciones de la presente invención, "arilo" se refiere a un sistema de anillo aromático que incluye, pero no se limita a, fenilo, bifenilo, naftilo, antracilo y similares, que pueden llevar uno o más sustituyentes. También incluido dentro del alcance del término "arilo", como se usa en el presente documento, se encuentra un grupo en que un anillo aromático está
condensado con uno o más anillos no aromáticos, tales como indanilo, ftalimidilo, naftimidilo, fenantridinilo, o tetrahidronaftilo, y similares.
Los términos "heteroarilo" y "heteroar-", usados solos o como parte de un resto mayor, por ejemplo, "heteroaralquilo" o "heteroaralcoxi", se refieren a grupos que tienen de 5 a 10 átomos en el anillo, preferentemente 5, 6 o 9 átomos en el anillo; que tiene 6, 10, o 14 electrones n compartidos en una matriz cíclica; y que tienen, además de átomos de carbono, de uno a cinco heteroátomos. El término "heteroátomo" se refiere a nitrógeno, oxígeno o azufre, e incluye cualquier forma oxidada de nitrógeno o azufre, y cualquier forma cuaternizada de un nitrógeno básico. Los grupos heteroarilo incluyen, sin limitación, tienilo, furanilo, pirrolilo, imidazolilo, pirazolilo, triazolilo, tetrazolilo, oxazolilo, isoxazolilo, oxadiazolilo, tiazolilo, isotiazolilo, tiadiazolilo, piridilo, piridazinilo, pirimidinilo, pirazinilo, indolizinilo, purinilo, naftiridinilo, y pteridinilo. Los términos "heteroarilo" y "heteroar-", como se usa en el presente documento, también incluyen grupos en los que un anillo heteroaromático se fusiona con uno o más anillos de arilo, cicloalifático o heterociclilo, en donde el radical o punto de unión está en el anillo heteroaromático. Los ejemplos no limitantes ejemplos incluyen indolilo, isoindolilo, benzotienilo, benzofuranilo, dibenzofuranilo, indazolilo, bencimidazolilo, benzotiazolilo, quinolilo, isoquinolilo, cinolinilo, ftalazinilo, quinazolinilo, quinoxalinilo, 4H-quinolizinilo, carbazolilo, acridinilo, fenazinilo, fenotiazinilo, fenoxazinilo, tetrahidroquinolinilo, tetrahidroisoquinolinilo y pirido[2,3-b]-1,4-oxazin-3(4H)-ona. Un grupo heteroarilo puede ser mono o bicíclico. El término "heteroarilo" se puede usar de forma indistinta con los términos "anillo de heteroarilo", "grupo heteroarilo", o "heteroaromático", cualquiera de cuyas expresiones incluye anillos que están opcionalmente sustituidos. El término "heteroaralquilo" se refiere a un grupo alquilo sustituido con heteroarilo, en donde las porciones de alquilo y heteroarilo están opcionalmente sustituidas independientemente.
Como se usa en el presente documento, los términos "heterociclo", "heterociclilo", "radical heterocíclico", y "anillo heterocíclico" se usan indistintamente y se refieren a un resto heterocíclico monocíclico de 5 a 7 miembros o bicíclico de 7-10 miembros que está saturado o parcialmente insaturado y que tiene, además de átomos de carbono, uno o más, preferentemente de uno a cuatro, heteroátomos, como se ha definido anteriormente. Cuando se usa en referencia a un átomo del anillo de un heterociclo, el término "nitrógeno" incluye un nitrógeno sustituido. A modo de ejemplo, en un anillo saturado o parcialmente insaturado que tiene 0-3 heteroátomos seleccionados entre oxígeno, azufre o nitrógeno, el nitrógeno puede ser N (como en 3,4-dihidro-2H-pirrolilo), NH (como en pirrolidinilo) o NR (como en pirrolidinilo N sustituido).
Un anillo heterocíclico se puede unir a su grupo colgante en cualquier heteroátomo o átomo de carbono que dé como resultado una estructura estable y cualquiera de los átomos del anillo puede estar opcionalmente sustituido. Ejemplos de dichos radicales heterocíclicos saturados o parcialmente insaturados incluyen, sin limitación, tetrahidrofuranilo, tetrahidrotiofenilo, pirrolidinilo, piperidinilo, pirrolinilo, tetrahidroquinolinilo, tetrahidroisoquinolinilo, decahidroquinolinilo, oxazolidinilo, piperazinilo, dioxanilo, dioxolanilo, diazepinilo, oxazepinilo, tiazepinilo, morfolinilo, y quinuclidinilo. Los términos "heterociclo", "heterociclilo", "anillo de heterociclilo", "grupo heterocíclico", "resto heterocíclico", y "radical heterocíclico", se usan indistintamente en el presente documento, y también incluyen grupos en los que un anillo heterocíclilo se condensa con uno o más anillos de arilo, heteroarilo o cicloalifático, tales como indolinilo, 3H-indolilo, cromanilo, fenantridinilo o tetrahidroquinolinilo. Un grupo heterociclilo puede ser mono o bicíclico. El término "heterociclilalquilo" se refiere a un grupo alquilo sustituido por un heterociclilo, en donde las porciones de alquilo y heterociclilo están opcionalmente sustituidas independientemente.
Como se usa en el presente documento, el término "parcialmente insaturado" se refiere a un resto de anillo que incluye al menos un doble o triple enlace. La expresión "parcialmente insaturado" pretende abarcar anillos que tienen sitios múltiples de insaturación, pero no pretende incluir restos arilo o heteroarilo, como se define en el presente documento.
Como se describe en el presente documento, los compuestos de la invención pueden contener restos "opcionalmente sustituidos". En general, el término "sustituido", esté precedido o no por el término "opcionalmente", significa que se sustituyen uno o más hidrógenos del resto designado por un sustituyente adecuado. A menos que se indique otra cosa, un grupo "opcionalmente sustituido" puede tener un sustituyente adecuado en cada posición sustituible del grupo, y cuando más de una posición en cualquier estructura dada puede estar sustituida por más de un sustituyente seleccionado de un grupo específico, el sustituyente puede ser igual o diferente en cada posición. Las combinaciones de sustituyentes previstas por la presente invención son preferentemente aquellas que dan como resultado la formación de compuestos estables o químicamente factibles. El término "estable", como se usa en el presente documento, se refiere a compuestos que no se alteran de forma sustancial cuando se someten a condiciones para permitir su producción, detección y, en determinadas realizaciones, su recuperación, purificación y uso para uno o más de los propósitos desvelados en el presente documento.
Cada sustituyente opcional en un carbono sustituible es un sustituyente monovalente seleccionado independientemente entre halógeno; -(CH2)0-4R°; -(CH2)cmOR°; -O(CH2)0-4R°, -O-(CH2)0-4C(O)OR°; -(CH2)0-4CH(OR°)2; -(CH2)0-4SR°; -(CH2)0-4Ph, que puede sustituirse por R°; -(CH2)0-4O(CH2)0--iPh que puede sustituirse por R°; -CH=CHPh, que puede sustituirse por R°; -(CH2)0-4O(CH2)0-1-piridilo que puede sustituirse por R°; -NO2; -CN; -N3; -(CH2)0-4N(R°)2; -(CH2)0-4N(R°)C(O)R°; -N(R°)C(S)R°; -(CH2)0-4N(R°)C(O)NR°2; -N(R°)C(S)NR°2; -(CH2)0-4N(R°)C(O)OR°; -N(R°)N(R°)C(O)R°; -N(R°)N(R°)C(O)NR°2; -N(R°)N(R°)C(O)OR°; -(CH2)0-4C(O)R°; -C(S)R°; -(CH2)0-4C(O)OR°; -(CH2)0-4C(O)SR°; -(CH2)0-4C(O)OSiR°3; -(CH2)0-4OC(O)R°; -OC(O)(CH2)0-4SR-, SC(S)SR°; -(CH2)0-4SC(O)R°; -(CH2)0-4C(O)NR°2; -C(S)NR°2; -C(S)SR°; -SC(S)SR°, -(CH2)0-4OC(O)NR°2; -C(O)N(OR°)R°; -C(O)C(O)R°; -C(O)CH2C(O)R°;
-C(NOR°)R°; -(CH2)o-4SSR°; -(CH2)o-4S(O)2R°; -(CH2)o-4S(O)2OR°; -(CH2)o-4OS(O)2R°; -S(O)2NR°2; -S(O)(NR°)R°; -S(O)2N=C(NR°2)2í -(CH2)o-4S(0)R°; -N(R0)S(O)2NR02; -N(R0)S(O)2R0; -N(OR°)R°; -C(NH)NR02; -P(O)2R0; -P(O)R02; -OP(O)R02; -OP(0 )(o R0)2; SÍR03; -(alquileno lineal o ramificado Ci -4)0 -N(R0)2; o -(alquilen C1-4 lineal o ramificado)C(O)O-N(R0)2.
Cada R0 es independientemente hidrógeno, alifático C1-6, -CH2Ph, -O(CH2)o-iPh, -CH2-(anillo heteroarilo de 5-6 miembros) o un saturado de 5-6 miembros, que tiene de 0-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno, o azufre, o, a pesar de la definición anterior, dos apariciones independientes de R0, tomadas junto con su átomo o átomos intermedios, forman un saturado de 3-12 miembros, que tiene 0-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, que puede estar sustituido por un sustituyente divalente en un átomo de carbono saturado de R0 seleccionado entre =O y =S; o cada R0 está opcionalmente sustituido con un sustituyente monovalente seleccionado independientemente entre halógeno, -(CH2)o-2R^, -(haloR )^, -(CH2)o-20H, -(CH2)o-20 R ^, -(CH2)o-2CH(OR^ ; -O(haloR )^, -CN, -N3, -(CH2)o-2C(O)R^, -(CH2)o-2C(O)OH, -(CH2)o-2C(O)OR ,^ -(CH2)o-2SR^, -(CH2)o-2SH, -(CH2)o-2NH2, -(CH2)o-2NHR^, -(CH2)o-2NR^2, -NO2, -SiRv¡, -OSiRv¡, -C(O)SR ^ -(alquilen C1.4 lineal o ramificado)C(O)OR ,^ o -Ss RV
Cada R ^ se selecciona independientemente entre alifático C1-4, -CH2Ph, -O(CH2)o-1Ph o un anillo arilo saturado o parcialmente insaturado de 5-6 miembros, que tiene de 0-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, y donde cada R ^ no está sustituido o cuando está precedido por halo está sustituido solo con uno o más halógenos; o en donde un sustituyente opcional en un carbono saturado es un sustituyente divalente seleccionado independientemente entre =O, =S, =NNR% =NNHC(O)R^, =NNHC(O)OR ,^ =NNHS(O)2R ,^ =NR^, =NOR^, -O(C(R^ ))2-3O- o -S(C(R^))2-3S- o un sustituyente divalente unido a carbonos sustituibles vecinales de un grupo "opcionalmente sustituido" es -O(CR^ )2-3O-, en donde cada aparición independiente de R ^ se selecciona entre hidrógeno, alifático C1-6 o un saturado de 5-6 sin sustituir, parcialmente insaturado o anillo arilo que tiene de 0-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre.
Cuando R ^ es alifático C1-6, R ^ está opcionalmente sustituido con halógeno, -R ,^ -(haloR^), -OH, -OR^, -O(haloR )^, -CN, -C(O)OH, -C(O)OR ,^ -NH2, -NHR^, -NR 2^ o -NO2, en donde cada R ^ se selecciona independientemente entre alifático C1-4, -CH2Ph, -O(CH2)o-1Ph o un anillo arilo saturado o parcialmente insaturado de 5-6 miembros, que tiene de 0-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, y donde cada R ^ no está sustituido o cuando está precedido por halo está sustituido solo con uno o más halógenos.
Un sustituyente opcional en un nitrógeno sustituible es independientemente -Rf -NRf2, -C(O)Rf , -C(O)ORf , -C(O)C(O)Rt -C(O)CH2C(O)R í , -S(O)2Rf -SfO^ NR^, -C(S)NR^, -C(NH)NRt2 o -N(Rt)S(O)2Rf ; en donde cada Rt es independientemente hidrógeno, alifático C1-6, -OPh sin sustituir, o un anillo saturado, parcialmente insaturado de 5-6 miembros sin sustituir que tiene de 0-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno, o azufre, o, dos apariciones independientes de Rf , tomadas junto con su átomo o átomos intermedio (s) forman un anillo saturado o parcialmente insaturado o aril mono o bicíclico de 3-12 miembros, que tiene 0-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre; en donde cuando Rf es alifático C1-6, Rf está opcionalmente sustituido con halógeno, -R^, -(haloR^), -OH, -OR^, -O(haloR^), -CN, -C(O)OH, -C(O)OR ,^ -NH2, -NHR ,^ -Nr 2^ o -NO2, en donde cada R ^ se selecciona independientemente entre alifático C1-4, -CH2Ph, -O(CH2)o-1Ph o un anillo arilo saturado o parcialmente insaturado de 5-6 miembros, que tiene de 0-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, y donde cada R ^ no está sustituido o cuando está precedido por halo está sustituido solo con uno o más halógenos.
Como se usa en el presente documento, la expresión "sal farmacéuticamente aceptable" se refiere a las sales que son, dentro del alcance del buen criterio médico, adecuadas para su uso en contacto con los tejidos de seres humanos o animales inferiores sin excesiva toxicidad, irritación, respuesta alérgica y similares, y son proporcionadas con una relación beneficio/riesgo razonable. Las sales farmacéuticamente aceptables son bien conocidas en la técnica. Por ejemplo, S. M. Berge et al., describen detalladamente sales farmacéuticamente aceptables en J. Pharmaceutical Sciences, 1977, 66, 1-19, incorporados como referencia en el presente documento. Las sales farmacéuticamente aceptables de los compuestos de la presente invención incluyen las obtenidas a partir de ácidos y bases inorgánicas y orgánicas adecuadas. Ejemplos de sales de adición de ácido no tóxicas y farmacéuticamente aceptables son las sales de un grupo amino formado con ácidos inorgánicos como el ácido clorhídrico, ácido bromhídrico, ácido fosfórico, ácido sulfúrico y ácido perclórico o con ácidos orgánicos, tales como ácido acético, ácido oxálico, ácido maleico, ácido tartárico, ácido cítrico, ácido succínico o ácido malónico o usando otros métodos usados en la técnica, tales como intercambio iónico. Otras sales farmacéuticamente aceptables incluyen sales adipato, alginato, ascorbato, aspartato, bencenosulfonato, benzoato, bisulfato, borato, butirato, alcanforato, alcanforsulfonato, citrato, ciclopentanopropionato, digluconato, dodecilsulfato, etanosulfonato, formiato, fumarato, glucoheptonato, glicerofosfato, gluconato, hemisulfato, heptanoato, hexanoato, yodhidrato, 2-hidroxi-etanosulfonato, lactobionato, lactato, laurato, lauril sulfato, malato, maleato, malonato, metanosulfonato, 2-naftalenosulfonato, nicotinato, nitrato, oleato, oxalato, palmitato, pamoato, pectinato, persulfato, 3-fenilpropionato, fosfato, picrato, pivalato, propionato, estearato, succinato, sulfato, tartrato, tiocianato, p-toluenosulfonato, undecanoato, valerato y similares.
Las sales derivadas de bases adecuadas incluyen sales de metales alcalinos, metales alcalinotérreos, de amonio y N+(alquilo C1-4)4. Las sales de metales alcalinos o alcalinotérreos representativas incluyen sodio, litio, potasio, calcio,
magnesio y similares. Las sales farmacéuticamente aceptables adicionales incluyen, cuando sea adecuado, amonio no tóxico, amonio cuaternario y cationes de amina formados usando contraiones, tales como haluro, hidróxido, carboxilato, sulfato, fosfato, nitrato, sulfonato de alquilo inferior y sulfonato de arilo.
A menos que se indique otra cosa, se entiende que las estructuras representadas en el presente documento también pretenden incluir todas las formas isoméricas (por ejemplo, enantioméricas, diastereoméricas y geométricas (o conformacionales)) de la estructura; por ejemplo, las configuraciones R y S para cada centro asimétrico, isómeros de doble enlace Z y E e isómeros conformacionales Z y E. Por tanto, los isómeros estereoquímicos individuales, así como las mezclas enantioméricas, diastereoméricas y geométricas (o conformacionales) de los presentes compuestos están dentro del alcance de la invención. A menos que se indique otra cosa, todas las formas tautoméricas de los compuestos de la invención están dentro del alcance de la invención. Adicionalmente, se desvelan compuestos que difieren únicamente en la presencia de uno o más átomos enriquecidos isotópicamente; por ejemplo, los compuestos que tienen las presentes estructuras incluyendo la sustitución de hidrógeno por deuterio o tritio, o la sustitución de un carbono por un carbono enriquecido en 13C o 14C. Tales compuestos son útiles, por ejemplo, como herramientas de análisis, como sondas en ensayos biológicos o como agentes terapéuticos de acuerdo con la presente invención.
3. Descripción de las realizaciones ilustrativas:
En un aspecto, la presente invención proporciona un compuesto de fórmula I:

o una sal farmacéuticamente aceptable del mismo, en donde:
el anillo A es un anillo seleccionado entre fenilo, un anillo carbocíclico saturado o parcialmente insaturado de 5-7 miembros, un anillo heterocíclico bicíclico saturado o parcialmente insaturado de 8 -12 miembros que tiene 1 -2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, un anillo heteroaromático de 5-6 miembros que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno, o azufre, o un anillo heteroaromático bicíclico de 8-10 miembros que tiene de 1-5 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre;
cada R1 es independientemente hidrógeno o alifático C1-3; o
dos grupos R1 se toman opcionalmente junto con sus átomos intermedios para formar un anillo carbocíclico condensado parcialmente insaturado de 5-8 miembros;
cada uno de R2 es independientemente hidrógeno, halógeno, -CN, -NO2, -C(O)OR, -C(O)NR2, -NR2, -NRC(O)R, -NRC(O)OR, -NRS(O)2R, -OR, -P(O)R2, -SR, -S(O)R, -S(O)2R, -S(O)(NH)R o R; o
dos grupos R2 se toman opcionalmente juntos para formar =O;
cada R3 es independientemente hidrógeno o alifático C1-3; o:
dos grupos R3 se toman opcionalmente juntos para formar =O;
dos grupos R3 se toman opcionalmente juntos para formar =CH2;
dos grupos R3 se toman opcionalmente junto con sus átomos intermedios para formar un anillo espirocíclico saturado de 5-8 miembros que tiene 0-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre; o
dos grupos R3 se toman opcionalmente junto con sus átomos intermedios para formar un anillo bicíclico con puente saturado de 5-8 miembros que tiene 0-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre;
cada R es independientemente hidrógeno o un grupo opcionalmente sustituido seleccionado entre hidrocarburo alifático C1-6, un anillo carbocíclico monocíclico saturado o parcialmente insaturado de 3-8 miembros, fenilo, un anillo heterocíclico espirobicíclico saturado de 7-10 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, un anillo heterocíclico bicíclico condensado saturado o parcialmente insaturado de 7-10 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, un anillo heterocíclico monocíclico saturado o parcialmente insaturado de 4-8 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, o un anillo heteroaromático monocíclico de 5-6 miembros que tiene 1-4 heteroátomos seleccionados
independientemente entre nitrógeno, oxígeno o azufre; o:
dos grupos R en el mismo nitrógeno se toman opcionalmente junto con sus átomos intermedios para formar un anillo de 4-7 miembros saturado, parcialmente insaturado o heteroarilo que tiene 0-3 heteroátomos, además del nitrógeno, seleccionado independientemente entre nitrógeno, oxígeno y azufre, opcionalmente sustituido con 1-2 grupos oxo;
X es -O-, -N(R)-, -N(S(O)2(R))-, -S-, -S(O)-, -S(O)2-, -CH2-, -CH(R3)- o -C(R3)2-;
m es 0, 1 o 2;
n es 0, 1,2, 3, 4 o 5; y
p es 0, 1 o 2.
En un aspecto, la presente invención proporciona un compuesto de fórmula I':

o una sal farmacéuticamente aceptable del mismo, en donde:
el anillo A es un anillo seleccionado entre fenilo, un anillo carbocíclico saturado o parcialmente insaturado de 5-7 miembros, un anillo heterocíclico bicíclico saturado o parcialmente insaturado de 8 -12 miembros que tiene 1 -2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, un anillo heteroaromático de 5-6 miembros que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre; o un anillo heteroaromático bicíclico de 8-10 miembros que tiene 1-5 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre;
cada R1 es independientemente hidrógeno o alifático C1-3 opcionalmente sustituido con 1-6 halógeno; o dos grupos R1 se toman opcionalmente junto con sus átomos intermedios para formar un anillo carbocíclico condensado parcialmente insaturado de 5-8 miembros;
cada uno de R2 es independientemente hidrógeno, halógeno, -CN, -NO2, -C(O)OR, -C(O)NR2, -NR2, -NRC(O)R, -NRC(O)OR, -NRS(O)2R, -OR, -P(O)R2, -SR, -S(O)R, -S(O)2R, -S(O)(NH)R, -S(O)2NR2 o R; o
dos grupos R2 se toman opcionalmente juntos para formar =O; o
dos grupos R2 se toman opcionalmente junto con sus átomos intermedios para formar un anillo espirocíclico saturado de 3-8 miembros que tiene 0-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre;
cada R3 es independientemente hidrógeno, -OH o alifático C1-3; o
dos grupos R3 se toman opcionalmente juntos para formar =O; o
dos grupos R3 se toman opcionalmente juntos para formar =CH2; o
dos grupos R3 se toman opcionalmente junto con sus átomos intermedios para formar un anillo espirocíclico saturado de 3-8 miembros que tiene 0-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre; o
dos grupos R3 se toman opcionalmente junto con sus átomos intermedios para formar un anillo bicíclico con puente saturado de 5-8 miembros que tiene 0-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre;
cada R es independientemente hidrógeno o un grupo opcionalmente sustituido seleccionado entre hidrocarburo alifático C1-6, un anillo carbocíclico monocíclico saturado o parcialmente insaturado de 3-8 miembros, fenilo, un anillo heterocíclico espirobicíclico saturado de 7-10 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, un anillo heterocíclico bicíclico condensado saturado o parcialmente insaturado de 7-10 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, un anillo heterocíclico monocíclico saturado o parcialmente insaturado de 4-8 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, o un anillo heteroaromático monocíclico de 5-6 miembros que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre; o:
dos grupos R en el mismo nitrógeno se toman opcionalmente junto con sus átomos intermedios para formar un anillo de 4-7 miembros saturado, parcialmente insaturado o heteroarilo que tiene 0-3 heteroátomos, además del nitrógeno, seleccionado independientemente entre nitrógeno, oxígeno y azufre, opcionalmente sustituido con 1-2 grupos oxo;
------es un enlace sencillo o un doble enlace;
X es -O-, -N(R)-, -N(S(O)2(R))-, -S-, -S(O)-, -S(O)2-, -CH2-, -CH(R3)- o -C(R3)2-;
m es 0, 1 o 2;
n es 0, 1,2, 3, 4 o 5; y
p es 0, 1 o 2.
Como se ha definido anteriormente de una manera general,------es un enlace sencillo o un doble enlace.
En algunas realizaciones,------es un enlace sencillo. En algunas realizaciones,-------es un doble enlace.
En algunas realizaciones,------se selecciona entre los presentados en las Tablas 1,2 y 2A, a continuación.
Como se ha definido anteriormente de una manera general, el anillo A es un anillo seleccionado entre fenilo, un anillo carbocíclico saturado o parcialmente insaturado de 5-7 miembros, un anillo heterocíclico bicíclico saturado o parcialmente insaturado de 8-12 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, un anillo heteroaromático de 5-6 miembros que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno, o azufre, o un anillo heteroaromático bicíclico de 8-10 miembros que tiene de 1-5 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre.
En algunas realizaciones, el anillo A es fenilo. En algunas realizaciones, el anillo A es un anillo carbocíclico saturado o parcialmente insaturado de 5-7 miembros, un anillo heterocíclico bicíclico saturado o parcialmente insaturado de 8 12 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, un anillo heteroaromático de 5-6 miembros que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno, o azufre, o un anillo heteroaromático bicíclico de 8-10 miembros que tiene de 1-5 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre.
En algunas realizaciones, el anillo A es un anillo carbocíclico saturado o parcialmente insaturado de 5-7 miembros. En algunas realizaciones, el anillo A es un anillo heterocíclico bicíclico saturado o parcialmente insaturado de 8-12 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre. En algunas realizaciones, el anillo A es un anillo heteroaromático de 5-6 miembros que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre. En algunas realizaciones, el anillo A es un anillo heteroaromático bicíclico de 8-10 miembros que tiene 1-5 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre.
En algunas realizaciones, el anillo A es fenilo o  , en donde el Anillo B es un anillo heterocíclico parcialmente insaturado de 5-7 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, o el anillo B es un anillo heteroaromático de 5-6 miembros que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre. En algunas realizaciones, el anillo B se
, en donde el Anillo B es un anillo heterocíclico parcialmente insaturado de 5-7 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, o el anillo B es un anillo heteroaromático de 5-6 miembros que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre. En algunas realizaciones, el anillo B se

selecciona entre: y


En algunas realizaciones, el anillo A se selecciona entre


En algunas realizaciones, el anillo

En algunas realizaciones, el anillo A es
En algunas realizaciones, el anillo A se selecciona entre:


En algunas realizaciones, el anillo A es
En algunas realizaciones, el anillo A se selecciona entre:

En algunas realizaciones, el anillo A es 
En algunas realizaciones, el anillo A se selecciona entre:

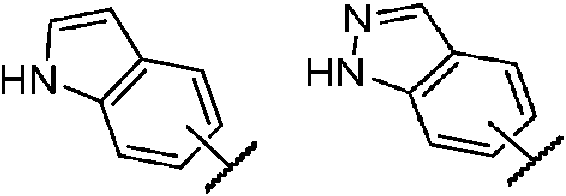
En algunas realizaciones, el anillo A es o
En algunas realizaciones, el anillo A se selecciona entre:

y
En algunas realizaciones, el anillo A se selecciona entre:

En algunas realizaciones, el anillo A se selecciona entre:

En algunas realizaciones, el anillo A se selecciona entre:

En algunas realizaciones, el anillo A se selecciona entre:


En algunas realizaciones, el anillo A es
En algunas realizaciones, el anillo A es  es
es

que también puede estar en forma tautomérica:
En algunas realizaciones, el anillo A se selecciona entre los presentados en las Tablas 1 y 2, a continuación.
En algunas realizaciones, el anillo A se selecciona entre los presentados en la Tabla 2A, a continuación.
Como se ha definido anteriormente de una manera general, cada R1 es independientemente hidrógeno o alifático C i-3; o dos grupos R1 se toman opcionalmente junto con sus átomos intermedios para formar un anillo carbocíclico condensado parcialmente insaturado de 5-8 miembros.
En algunas realizaciones, R1 es hidrógeno. En algunas realizaciones, R1 es alifático C1-3. En algunas realizaciones, dos grupos R1 se toman opcionalmente junto con sus átomos intermedios para formar un anillo carbocíclico condensado parcialmente insaturado de 5-8 miembros.
En algunas realizaciones, R1 es metilo. En algunas realizaciones, R1 es etilo. En algunas realizaciones, R1 es propilo. En algunas realizaciones, R1 es isopropilo.
En algunas realizaciones, R1 se une a la posición 5 de la pirimidina. En algunas realizaciones, R1 se une a la posición 6 de la pirimidina.
En algunas realizaciones, dos grupos R1 se toman opcionalmente junto con sus átomos intermedios para formar un anillo carbocíclico condensado parcialmente insaturado de 5-8 miembros. En algunas realizaciones, dos grupos R1 se toman opcionalmente junto con sus átomos intermedios para formar un anillo carbocíclico condensado parcialmente insaturado de 5 miembros. En algunas realizaciones, dos grupos R1 se toman opcionalmente junto con sus átomos intermedios para formar un anillo carbocíclico condensado parcialmente insaturado de 6 miembros. En algunas realizaciones, dos grupos R1 se toman opcionalmente junto con sus átomos intermedios para formar un anillo carbocíclico condensado parcialmente insaturado de 7 miembros. En algunas realizaciones, dos grupos R1 se toman opcionalmente junto con sus átomos intermedios para formar un anillo carbocíclico condensado parcialmente insaturado de 8 miembros.
En algunas realizaciones, R1 es alifático C1-3 opcionalmente sustituido 1-6 veces por halógeno. En algunas realizaciones, R1 es alquilo C1-3 opcionalmente sustituido 1-6 veces por halógeno. En algunas realizaciones, R1 es alquilo C1-3 opcionalmente sustituido 1-6 veces por flúor. En algunas realizaciones, R1 es alquilo C1-3 opcionalmente sustituido 1-3 veces por flúor. En algunas realizaciones, R1 es -CF3.
En algunas realizaciones, R1 se selecciona entre los presentados en las Tablas 1 y 2, a continuación.
En algunas realizaciones, R1 se selecciona entre los presentados en la Tabla 2A, a continuación.
Como se ha definido anteriormente de una manera general, cada uno de R2 es independientemente hidrógeno, halógeno (F, Cl, Br o I), -CN, -NO2, -C(O)OR, -C(O)NR2, -NR2, -NRC(O)R, -NRC(O)OR, -NRS(O)2R, -OR, -P(O)R2, -SR, -S(O)R, -S(O)2R, -S(O)(NH)R o R; o dos grupos R2 se toman opcionalmente juntos para formar =O.
En algunas realizaciones, R2 es hidrógeno. En algunas realizaciones, cada uno de R2 es independientemente halógeno, -CN, -NO2, -C(O)OR, -C(O)NR2, -NR2, -NRC(O)R, -NRC(O)OR, -NRS(O)2R, -OR, -P(O)R2, -SR, -S(O)R, -S(O)2R, -S(O)(NH)R o R; o dos grupos R2 se toman opcionalmente juntos para formar =O.
En algunas realizaciones, R2 es halógeno. En algunas realizaciones, R2 es Cl. En algunas realizaciones, R2 es -CN. En algunas realizaciones, R2 es -NO2. En algunas realizaciones, R2 es -C(O)OR. En algunas realizaciones, R2 es -C(O)NR2. En algunas realizaciones, R2 es -NR2. En algunas realizaciones, R2 es -NRC(O)R. En algunas realizaciones, R2 es -NRC(O)OR. En algunas realizaciones, R2 es -NRS(O)2R. En algunas realizaciones, R2 es independientemente -OR. En algunas realizaciones, R2 es -P(O)R2. En algunas realizaciones, R2 es -SR. En algunas realizaciones, R2 es -S(O)R. En algunas realizaciones, R2 es -S(O)2R. En algunas realizaciones, R2 es -S(O)(NH)R. En algunas realizaciones, R2 es R. En algunas realizaciones, dos grupos R2 se toman opcionalmente juntos para formar =O.
En algunas realizaciones, R2 es -S(O)2NR2. En algunas realizaciones, R2 es -S(O)2NH2.
En algunas realizaciones, R2 es alifático C1-6. En algunas realizaciones, R2 es un anillo carbocíclico monocíclico saturado de 3-8 miembros. En algunas realizaciones, R2 es un anillo heterocíclico espirobicíclico saturado de 7-10 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre. En algunas realizaciones, R2 es un anillo heterocíclico bicíclico condensado saturado de 7-10 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre. En algunas realizaciones, R2 es un anillo heterocíclico monocíclico saturado de 4-8 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre. En algunas realizaciones, R2 es un anillo heteroaromático monocíclico de 5-6 miembros que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre.
En algunas realizaciones cada uno de R2 se selecciona independientemente entre: halógeno (por ejemplo, Cl), -NH2,


=O, -OH, -COOH,


En algunas realizaciones, R2 se selecciona entre -S(O)2NH2, -OCHF2, -OCF3, -C=CH, -0-CH2-CeCH,

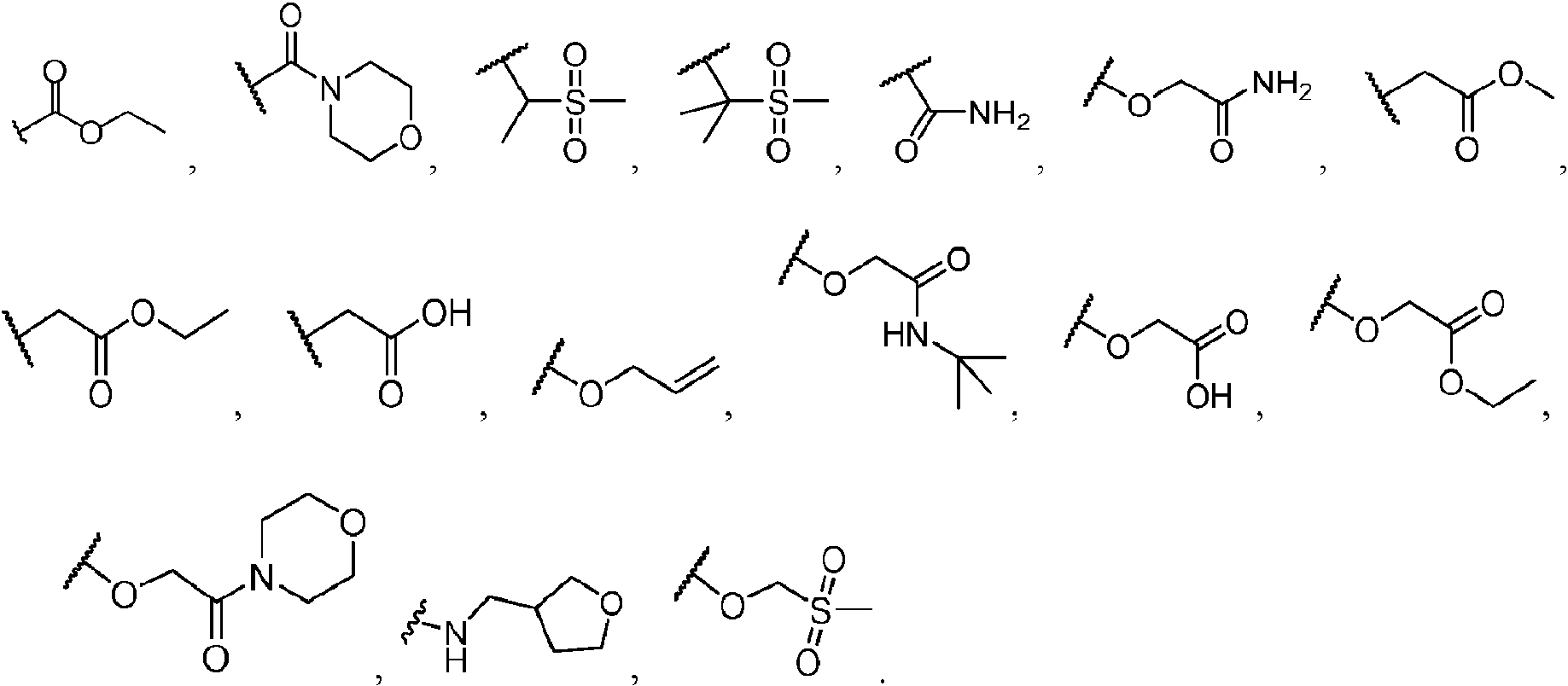
y
En algunas realizaciones, R2 es alifático C1-6, opcionalmente sustituido 1-4 veces por halógeno, -OH, NH2, -OCH3, -NHC(O)CH3, -S(O)2CH3 o -N(CH3)C(O)CH3. En algunas realizaciones, R2 se selecciona entre CH3, -CF3, -CH2CH3,

En algunas realizaciones, R2 es alifático C1-6, opcionalmente sustituido 1-4 veces por halógeno, -OH, NH2, -OCH3, -NHC(O)CH3, -S(O)2CH algunas realizaciones, R2 es
algunas realizaciones, R2 es

En algunas realizaciones, R2 es alifático C1-6, opcionalmente sustituido con un grupo -S(O)2-(CH2)ü-6, en donde (CH2)o-6 está opcionalmente sustituido 1-4 veces por halógeno, -OH, NH2 u -OCH3. En algunas realizaciones, R2 es alifático C1-6, opcionalmente sustituido con un grupo -S(O)2-(CH2)o_6 , en donde (CH2)o_6 está sin sustituir. En algunas realizaciones, R2 es alifático C1-6, opcionalmente sustituido con -S(O)2-CH3 o -S(O)2-CH2-CH3. En algunas realizaciones, R2 es alifático C1-6, opcionalmente sustituido con -S(O)2-CH3. En algunas realizaciones, R2 es -CH2-S(O)2-CH3. En algunas realizaciones, R2 es -CH2-S(O)2-CH2-CH3. En algunas realizaciones, R2 es -CH2-CH2-S(O)2-CH3. En algunas realizaciones, R2 es -CH2-CH2-S(O)2-CH2-CH3. En algunas realizaciones, R2 se selecciona entre

En algunas realizaciones, R2 es alifático C1-6 sin sustituir. En algunas realizaciones, R2 es -C=CH.
En algunas realizaciones, R2 es un anillo carbocíclico monocíclico saturado de 3-6 miembros. En algunas
realizaciones,
En algunas realizaciones, R2es un anillo heterocíclico espirobicíclico saturado de 7-10 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno u oxígeno. En algunas realizaciones, R2 se
selecciona entre
En algunas realizaciones, R2 es un anillo heterocíclico bicíclico condensado saturado de 7-10 miembros que tiene 1 2 heteroátomos seleccionados independientemente entre nitrógeno u oxígeno. En algunas realizaciones, R2 es

En algunas realizaciones, R2 es un anillo heterocíclico monocíclico saturado de 4-6 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, opcionalmente sustituido 1-4


En algunas realizaciones, R2 es un anillo heterocíclico monocíclico saturado de 4-6 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, opcionalmente sustituido 1-4

veces por halógeno, -OH, -CH3, -OCH3, =O, En algunas realizaciones,
R2 se selecciona entre 
En algunas realizaciones, R2 es un anillo heteroaromático monocíclico de 5-6 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre. En algunas realizaciones, R2 se selecciona
entre 
En algunas realizaciones, R2 es -C(O)OR, en donde R es hidrógeno o alifático C1-6. En algunas realizaciones, R2 es -C(O)OH. En algunas realizaciones, R2 es -C(O)Oalifático C1-6, en donde el alifático C1-6 está sin sustituir. En algunas


realizaciones, R2 es En algunas realizaciones, R2 es
En algunas realizaciones, R2 es -C(O)NR2, en donde cada uno de R es independientemente hidrógeno, alifático Ci_6 que está opcionalmente sustituido con un -N(CH)2, anillo carbocíclico monocíclico saturado de 3-6 miembros sin sustituir, o anillo heterocíclico monocíclico saturado de 4-6 miembros sin sustituir que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno u oxígeno, o dos R tomados junto con sus átomos intermedios para formar un anillo saturado y heterocíclico de 4-7 miembros anillo de heteroarilo sin sustituir. En algunas

realizaciones, R 2 se selecciona entre
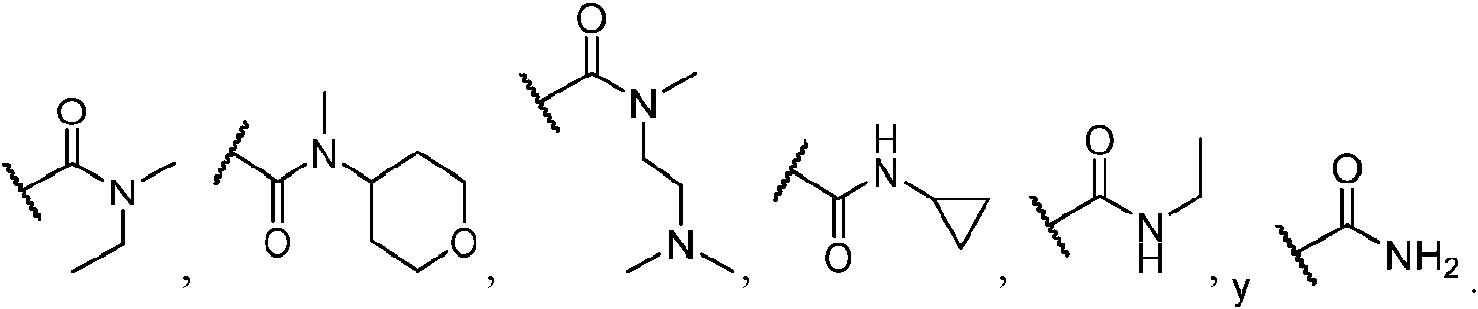
En algunas realizaciones, R2 es -C(O)NR2, en donde dos R tomados junto con sus átomos intermedios para formar un anillo saturado de 4-7 miembros que tiene 0-3 heteroátomos, además del nitrógeno, seleccionado independientemente entre nitrógeno, oxígeno y azufre, opcionalmente sustituido con 1-2 grupos oxo. En algunas realizaciones, R2 es -C(O)NR2, en donde dos R tomados junto con sus átomos intermedios para formar un anillo saturado y no sustituido de 4-7 miembros que tiene 0-3 heteroátomos, además del nitrógeno, seleccionado independientemente entre
nitrógeno, oxígeno y azufre. En algunas realizaciones, R2 es 
En algunas realizaciones, R2 es -NR2, en donde cada uno de R es independientemente:
hidrógeno;

alifático C1-6 que está opcionalmente sustituido 1-2 veces por -OH, o

anillo carbocíclico monocíclico saturado de 3-6 miembros sin sustituir;
anillo heterocíclico monocíclico saturado de 4-6 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno u oxígeno, que está opcionalmente sustituido 1-2 veces por CH3, -OH, -C(O)OC(CH)3 o -C(O)CH3; o
anillo heteroaromático monocíclico de 6 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre que está opcionalmente sustituido 1-2 veces por -CH3 o -NH2.
En algunas realizaciones, se selecciona entre


En algunas realizaciones, R2 es -NHC(O)R, en donde R es alifático C i-6 opcionalmente sustituido 1-3 veces por halógeno, -OCH3, -N(CH3)2 u -OH, anillo carbocíclico monocíclico saturado de 3-6 miembros opcionalmente sustituido 1-2 veces con halógeno u -OH, o anillo heterocíclico monocíclico saturado de 4-6 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre opcionalmente sustituido 1-2 veces por halógeno, -OH o -C H 3. En algunas realizaciones, R2 se selecciona entre

En algunas realizaciones, R2 es -NHC(O)R, en donde R es alifático C1-6 opcionalmente sustituido 1-3 veces por halógeno, -OCH3, -N(CH3)2 u -OH. En algunas realizaciones, alifático C1-6 es una cadena de hidrocarburo saturada de cadena lineal (es decir, no ramificada) o ramificada, sustituida o sin sustituir. En algunas realizaciones, alifático C1-6 es una cadena de hidrocarburo de cadena lineal (es decir, no ramificada) o ramificada, sustituida o sin sustituir que comprende un hidrocarburo monocíclico. En algunas realizaciones, alifático C1-6 se selecciona entre


En algunas realizaciones, R2 es -NHC(O)0R, en donde R es alifático C-ue sin sustituir. En algunas realizaciones, R2

es
En algunas realizaciones, R2 es -NHS(0 )2R, en donde R es alifático C-ue sin sustituir. En algunas realizaciones, R2 es

En algunas realizaciones, R2 es -OR, en donde R es H; alifático C-ue opcionalmente sustituido con un halógeno, -OH,
o o anillo heterocíclico monocíclico saturado de 4-6 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno u oxígeno. En algunas realizaciones, R2 se

selecciona 

En algunas realizaciones, R2 es -OR, en donde R es alifático C-ue opcionalmente sustituido con un halógeno, -OH,

-C(O)NHalifático C-m , -COOH, -C(O)Oalifático C-m , -CN, -
SO2alifático C1.4, o 

En algunas realizaciones, R2 se selecciona entre

En algunas realizaciones, R2 es -OR, en donde R es alifático C1-6 sin sustituir. En algunas realizaciones, R2 es -O-O-h-CECH.
En algunas realizaciones, R2 es -P(0)R2, en donde cada uno de R es independientemente alifático C1-6 sin sustituir.
O
i 11
n
En algunas realizaciones, R2 es '
En algunas realizaciones, R2 es -SR, en donde R es alifático C1-6 sin sustituir. En algunas realizaciones, R2 es

En algunas realizaciones, R2 es -S(O)R, en donde R es alifático Ci-6 sin sustituir. En algunas realizaciones, R2 es

En algunas realizaciones, R2 es -S(O)2R, en donde R es alifático C1-6 sin sustituir o anillo carbocíclico monocíclico

saturado de 3-6 miembros. En algunas realizaciones, R2 se selecciona entre y

En algunas realizaciones, R2 es-S(O)(NH)R, en donde R es alifático C-i-6 sin sustituir. En algunas realizaciones, R2 es

En algunas realizaciones, R2 es un grupo que aumenta la hidrofilia. En algunas realizaciones, R2 se selecciona entre el grupo que consiste en -NO2, -C(O)OR, -C(O)NR2, -NR2, -NRC(O)R, -NRC(O)OR, -NRS(O)2R, -OR, -P(O)R2, -SR, -S(O)R, -S(O)2R, -S(O)(NH)R o un grupo alifático C1-6 en donde una o más unidades de metileno están reemplazadas por -C(O)-, -S(O)-, -S(O)2-, -P(O)- o -P(O)2-. En algunas realizaciones, R2 es un grupo alifático C1-6 en donde una o más unidades de metileno están reemplazadas por -C(O)-, -S(O)-, -S(O)2-, -P(O)- o -P(O)2-. En algunas realizaciones, R2 es un grupo alifático C1-6 en donde una o más unidades de metileno están reemplazadas por -S(O)2-. En algunas


’ y En algunas realizaciones, R2 se

selecciona entre el grupo que consiste en y 
En algunas realizaciones, R2 se selecciona entre el grupo que consiste en

En algunas realizaciones, R2 se selecciona entre el grupo que consiste en y

En algunas realizaciones, R2 es En algunas realizaciones, R2 es



En algunas realizaciones, R2 es En algunas realizaciones, R2 es
En algunas realizaciones, dos grupos R2 se toman opcionalmente junto con sus átomos intermedios para formar un anillo espirocíclico saturado de 3-8 miembros que tiene 0-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre. En algunas realizaciones, dos grupos R2 se toman opcionalmente junto con sus átomos intermedios para formar un anillo carbocíclico espirocíclico saturado de 3-8 miembros. En algunas realizaciones, dos grupos R2 se toman opcionalmente junto con sus átomos intermedios para formar un anillo carbocíclico espirocíclico saturado de 3 miembros. En algunas realizaciones, dos grupos R2 se toman opcionalmente junto con sus átomos intermedios para formar un anillo carbocíclico espirocíclico saturado de 4, 5 o 6 miembros.
En algunas realizaciones, dos grupos R2 se unen en la misma posición. En algunas realizaciones, dos grupos R2 se unen a un átomo de carbono. En algunas realizaciones, cada uno de los dos grupos R2 unidos a un átomo de carbono es independientemente un grupo alifático C1-6 opcionalmente sustituido, como se describe en el presente documento. En algunas realizaciones, cada uno de los dos grupos R2 unidos a un átomo de carbono es alifático C1-6 independientemente no sustituido. En algunas realizaciones, cada uno de los dos grupos R2 unidos a un átomo de carbono es alquilo C1-6 independientemente no sustituido. En algunas realizaciones, cada uno de los dos grupos R2 unidos a un átomo de carbono es metilo.
En algunas realizaciones, R2 se selecciona entre los presentados en Tablas 1 y 2, a continuación.
En algunas realizaciones, R2 se selecciona entre los presentados en la Tabla 2A, a continuación.
Como se ha definido anteriormente de una manera general, cada R3 es independientemente hidrógeno o alifático C1-3; o:
dos grupos R3 se toman opcionalmente juntos para formar =O;
dos grupos R3 se toman opcionalmente juntos para formar =CH2;
dos grupos R3 se toman opcionalmente junto con sus átomos intermedios para formar un anillo espirocíclico saturado de 5-8 miembros que tiene 0-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre; o
dos grupos R3 se toman opcionalmente junto con sus átomos intermedios para formar un anillo bicíclico con puente saturado de 5-8 miembros que tiene 0-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre.
En algunas realizaciones, R3 es hidrógeno. En algunas realizaciones, R3 es alifático C1-3; o:
dos grupos R3 se toman opcionalmente juntos para formar =O;
dos grupos R3 se toman opcionalmente juntos para formar =CH2;
dos grupos R3 se toman opcionalmente junto con sus átomos intermedios para formar un anillo espirocíclico saturado de 5-8 miembros que tiene 0-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre; o
dos grupos R3 se toman opcionalmente junto con sus átomos intermedios para formar un anillo bicíclico con puente saturado de 5-8 miembros que tiene 0-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre.
En algunas realizaciones, R3 es alifático C1-3. En algunas realizaciones, dos grupos R3 se toman opcionalmente juntos para formar =O. En algunas realizaciones, dos grupos R3 se toman opcionalmente juntos para formar =CH2. En algunas realizaciones, dos grupos R3 se toman opcionalmente junto con sus átomos intermedios para formar un anillo espirocíclico saturado de 5-8 miembros que tiene 0-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre. En algunas realizaciones, dos grupos R3 se toman opcionalmente junto con sus átomos intermedios para formar un anillo bicíclico con puente saturado de 5-8 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre.
En algunas realizaciones, R3 es metilo. En algunas realizaciones, R3 es etilo. En algunas realizaciones, R3 es propilo. En algunas realizaciones, R3 es isopropilo.
En algunas realizaciones, dos grupos R3 se toman opcionalmente junto con sus átomos intermedios para formar un anillo espirocíclico saturado de 5 miembros que tiene 0-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre. En algunas realizaciones, dos grupos R3 se toman opcionalmente junto con sus átomos intermedios para formar un anillo espirocíclico saturado de 5 miembros que tiene 0-2 átomos de oxígeno. En algunas
realizaciones, dos grupos R3 se toman opcionalmente junto con sus átomos intermedios para formar  j un anillo espirocíclico en el átomo de carbono en la posición 2.
j un anillo espirocíclico en el átomo de carbono en la posición 2.
En algunas realizaciones, dos grupos R3 se toman opcionalmente junto con sus átomos intermedios para formar un anillo bicíclico con puente saturado de 5-8 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno y oxígeno. En algunas realizaciones, dos grupos R3 se toman opcionalmente junto con sus átomos intermedios para formar un anillo bicíclico con puente saturado que tiene 1-2 átomos de nitrógeno, en donde el anillo bicíclico con puente saturado comprende un anillo de 6 miembros y un anillo de 7 miembros. En algunas realizaciones,
dos grupos R3 junto con opcionalmente forman
opcionalmente forman
En algunas realizaciones, R3 es -OH.
En algunas realizaciones, dos grupos R3 se toman opcionalmente junto con sus átomos intermedios para formar un anillo espirocíclico saturado de 3-8 miembros que tiene 0-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre. En algunas realizaciones, dos grupos R3 se toman opcionalmente junto con sus átomos intermedios para formar un anillo espirocíclico saturado de 3 o 4 miembros que tiene 0-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre. En algunas realizaciones, dos grupos R3 se toman opcionalmente junto con sus átomos intermedios para formar un anillo carbocíclico espirocíclico saturado de 3 o 4 miembros. En algunas realizaciones, dos grupos R3 se toman opcionalmente junto con sus átomos intermedios para formar un anillo carbocíclico espirocíclico saturado de 3 miembros.
En algunas realizaciones, R3 se selecciona entre los presentados en Tablas 1 y 2, a continuación.
En algunas realizaciones, R3 se selecciona entre los presentados en la Tabla 2A, a continuación.
Como se ha definido anteriormente de una manera general, cada R es independientemente hidrógeno o un grupo opcionalmente sustituido seleccionado entre hidrocarburo alifático C1-6, un anillo carbocíclico monocíclico saturado o parcialmente insaturado de 3-8 miembros, fenilo, un anillo heterocíclico espirobicíclico saturado de 7-10 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, un anillo heterocíclico bicíclico condensado saturado o parcialmente insaturado de 7-10 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, un anillo heterocíclico monocíclico saturado o parcialmente insaturado de 4-8 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, o un anillo heteroaromático monocíclico de 5-6 miembros que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre; o: dos grupos R en el mismo nitrógeno se toman opcionalmente junto con sus átomos intermedios para formar un anillo de 4-7 miembros saturado, parcialmente insaturado o heteroarilo que tiene 0-3 heteroátomos, además del nitrógeno, seleccionado independientemente entre nitrógeno, oxígeno y azufre, opcionalmente sustituido con 1-2 grupos oxo.
En algunas realizaciones, R es hidrógeno. En algunas realizaciones, cada R es independientemente un grupo opcionalmente sustituido seleccionado entre alifático C1-6, un anillo carbocíclico monocíclico saturado o parcialmente insaturado de 3-8 miembros, fenilo, un anillo heterocíclico espirobicíclico saturado de 7-10 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, un anillo heterocíclico bicíclico condensado saturado o parcialmente insaturado de 7-10 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, un anillo heterocíclico monocíclico saturado o parcialmente insaturado de 4-8 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, o un anillo heteroaromático monocíclico de 5-6 miembros que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre; o: dos grupos R en el mismo nitrógeno se toman opcionalmente junto con sus átomos intermedios para formar un anillo de 4-7 miembros saturado, parcialmente insaturado o heteroarilo que tiene 0-3 heteroátomos, además del nitrógeno, seleccionado independientemente entre nitrógeno, oxígeno y azufre, opcionalmente sustituido con 1-2 grupos oxo.
En algunas realizaciones, R es un alifático C1-6 opcionalmente sustituido. En algunas realizaciones, R es un anillo carbocíclico monocíclico saturado o parcialmente insaturado de 3-8 miembros opcionalmente sustituido. En algunas realizaciones, R es un fenilo opcionalmente sustituido. En algunas realizaciones, R es un anillo heterocíclico espirobicíclico saturado de 7-10 miembros opcionalmente sustituido que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre. En algunas realizaciones, R es un anillo heterocíclico bicíclico condensado saturado o parcialmente insaturado de 7-10 miembros opcionalmente sustituido que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre. En algunas realizaciones, R es un anillo heterocíclico monocíclico saturado o parcialmente insaturado de 4-8 miembros opcionalmente sustituido que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre. En algunas realizaciones, R es un anillo heteroaromático monocíclico de 5-6 miembros opcionalmente sustituido que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre. En algunas realizaciones, dos grupos R en el mismo nitrógeno se toman opcionalmente junto con sus átomos intermedios para formar un anillo de 4-7 miembros saturado, parcialmente insaturado o heteroarilo que tiene 0-3 heteroátomos, además del nitrógeno, seleccionado independientemente entre nitrógeno, oxígeno y azufre, opcionalmente sustituido con 1-2 grupos oxo. En algunas realizaciones, R se selecciona entre los presentados en Tablas 1 y 2, a continuación.
En algunas realizaciones, R se selecciona entre los presentados en la Tabla 2A, a continuación.
Como se ha definido anteriormente de una manera general, X es -O-, -N(R)-, -N(S(O)2(R))-, -S-, -S(O)-, -S(O)2-, -CH2-, -CH(R3)- o -C(R3)2-.
En algunas realizaciones, X es -O-. En algunas realizaciones, X es -N(R)-. En algunas realizaciones, X es -N(S(O)2(R))-. En algunas realizaciones, X es S. En algunas realizaciones, X es -S(O)-. En algunas realizaciones, X es -S(O)2-. En algunas realizaciones, X es -CH2-. En algunas realizaciones, X es -CH(R3)-. En algunas realizaciones, X es -C(R3)2-. En algunas realizaciones, X es -N(S(O)2(R))-, en donde R es alifático C1-6. En algunas realizaciones, X es -N(S(O)2CH3)-. En algunas realizaciones, X es -CH(R3)-, en donde R3 es alifático C1-6. En algunas realizaciones, X es -CH(CH3)-. En algunas realizaciones, X es -C(R3)2-, en donde R3 es alifático C1-6. En algunas realizaciones, X es -C(CH3)2-.
En algunas realizaciones, X se selecciona entre los presentados en Tablas 1 y 2, a continuación.
En algunas realizaciones, X se selecciona entre los presentados en la Tabla 2A, a continuación.
Como se ha definido anteriormente de una manera general, m es 0, 1 o 2.
En algunas realizaciones, m es 0. En algunas realizaciones, m es 1. En algunas realizaciones, m es 2.
En algunas realizaciones, m se selecciona entre los presentados en Tablas 1 y 2, a continuación.
En algunas realizaciones, m se selecciona entre los presentados en la Tabla 2A, a continuación.
Como se ha definido anteriormente de una manera general, n es 0, 1,2, 3, 4 o 5.
En algunas realizaciones, n es 0. En algunas realizaciones, n es 1. En algunas realizaciones, n es 2. En algunas realizaciones, n es 3. En algunas realizaciones, n es 4. En algunas realizaciones, n es 5.
En algunas realizaciones, n se selecciona entre los presentados en Tablas 1 y 2, a continuación.
En algunas realizaciones, n se selecciona entre los presentados en la Tabla 2A, a continuación.
Como se ha definido anteriormente de una manera general, p es 0, 1 o 2.
En algunas realizaciones, p es 0. En algunas realizaciones, p es 1. En algunas realizaciones, p es 2.
En algunas realizaciones, p se selecciona entre los presentados en Tablas 1 y 2, a continuación.
En algunas realizaciones, p se selecciona entre los presentados en la Tabla 2A, a continuación.
En algunas realizaciones, la presente invención proporciona un compuesto de Fórmulas I-a o I-b:

o una sal farmacéuticamente aceptable del mismo, en donde cada uno del Anillo A, R1, R2, R3, R, X, m, n y p es como se ha definido anteriormente y se describe en realizaciones en el presente documento, tanto de forma individual como combinada.
En algunas realizaciones, la presente invención proporciona un compuesto de Fórmula II:

o una sal farmacéuticamente aceptable del mismo, en donde cada uno del Anillo A, R1, R2, R3, R, n y p es como se ha definido anteriormente y se describe en realizaciones en el presente documento, tanto de forma individual como combinada.
En algunas realizaciones, la presente invención proporciona un compuesto de Fórmulas Il-a o Il-b:

o una sal farmacéuticamente aceptable del mismo, en donde cada uno del Anillo A, R1, R2, R3, R, n y p es como se ha definido anteriormente y se describe en realizaciones en el presente documento, tanto de forma individual como combinada.
En algunas realizaciones, la presente invención proporciona un compuesto de Fórmula III

o una sal farmacéuticamente aceptable del mismo, en donde cada uno de R1, R2, R3, R, m, n y p es como se ha definido anteriormente y se describe en realizaciones en el presente documento, tanto de forma individual como combinada.
En algunas realizaciones, la presente invención proporciona un compuesto de Fórmulas lll-a o lll-b:

o una sal farmacéuticamente aceptable del mismo, en donde cada uno de R1, R2, R3, R, m, n y p es como se ha definido anteriormente y se describe en realizaciones en el presente documento, tanto de forma individual como combinada.
En algunas realizaciones, la presente invención proporciona un compuesto de Fórmula IV

o una sal farmacéuticamente aceptable del mismo, en donde cada uno del anillo B, R1, R2, R3, R, m, n y p es como se ha definido anteriormente y se describe en realizaciones en el presente documento, tanto de forma individual como combinada.
En algunas realizaciones, la presente invención proporciona un compuesto de Fórmulas IV-a o IV-b:

o una sal farmacéuticamente aceptable del mismo, en donde cada uno del anillo B, R1, R2, R3, R, m, n y p es como se ha definido anteriormente y se describe en realizaciones en el presente documento, tanto de forma individual como combinada.
En algunas realizaciones, la presente invención proporciona un compuesto de Fórmula V
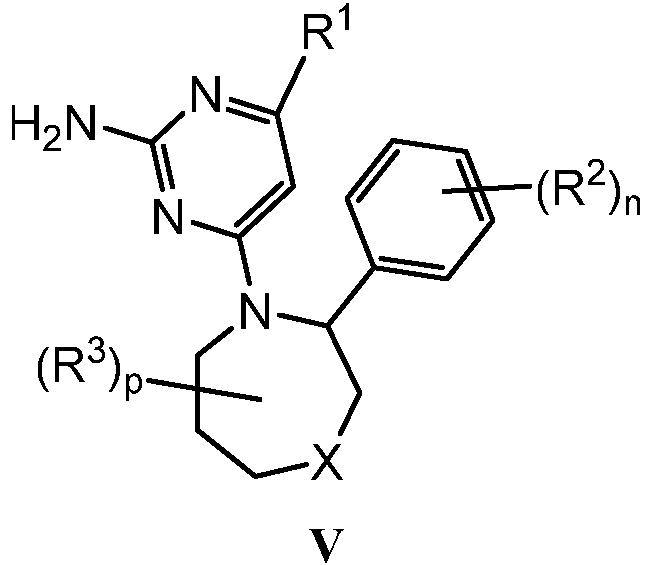
o una sal farmacéuticamente aceptable del mismo, en donde cada uno de R1, R2, R3, R, X, n y p es como se ha definido anteriormente y se describe en realizaciones en el presente documento, tanto de forma individual como combinada. En algunas realizaciones, la presente invención proporciona un compuesto de Fórmulas V-a o V-b:

o una sal farmacéuticamente aceptable del mismo, en donde cada uno de R1, R2, R3, R, X, n y p es como se ha definido anteriormente y se describe en realizaciones en el presente documento, tanto de forma individual como combinada. En algunas realizaciones, la presente invención proporciona un compuesto de Fórmula VI

o una sal farmacéuticamente aceptable del mismo, en donde cada uno del anillo B, R1, R2, R3, R, X, n y p es como se ha definido anteriormente y se describe en realizaciones en el presente documento, tanto de forma individual como combinada.
En algunas realizaciones, la presente invención proporciona un compuesto de Fórmulas Vl-a o Vl-b:

o una sal farmacéuticamente aceptable del mismo, en donde cada uno del anillo B, R1, R2, R3, R, X, n y p es como se ha definido anteriormente y se describe en realizaciones en el presente documento, tanto de forma individual como combinada.
En algunas realizaciones, la presente invención proporciona un compuesto de Fórmula Vll
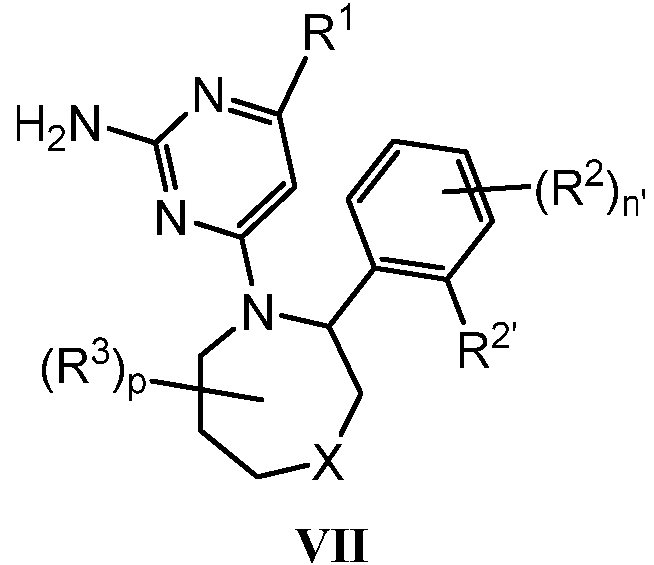
o una sal farmacéuticamente aceptable del mismo, en donde n' es 1 o 2, y R2' es halógeno u -Oalifático C1-3 y en donde cada uno de R1, R2, R3, R, X y p es como se ha definido anteriormente y se describe en realizaciones en el presente documento, tanto de forma individual como combinada. En algunas realizaciones, n' es 1. En algunas realizaciones, n' es 2. En algunas realizaciones, R2' es F. En algunas realizaciones, R2' es Cl. En algunas realizaciones, R2' es Br. En algunas realizaciones, R2' es I. En algunas realizaciones, R2' es -OCH3. En algunas realizaciones, R2' es -OC2H5. En algunas realizaciones, R2' es -OCH2CH2CH3. En algunas realizaciones, R2' es -OCH(CH3)2.
En algunas realizaciones, n' se selecciona entre los presentados en las tablas 1, 2 y 2A, a continuación.
En algunas realizaciones, R2' se selecciona entre los presentados en las tablas 1, 2 y 2A, a continuación.
En algunas realizaciones, la presente invención proporciona un compuesto de Fórmulas Vll-a o Vll-b:

o una sal farmacéuticamente aceptable del mismo, en donde cada uno de R1, R2, R2', R3, R, X, p y n' es como se ha definido anteriormente y se describe en realizaciones en el presente documento, tanto de forma individual como combinada.
En algunas realizaciones, la presente invención proporciona un compuesto de Fórmula VIII:

o una sal farmacéuticamente aceptable del mismo, en donde n" es 0, 1, 2, 3 o 4, y en donde cada uno del anillo B, R1, R2, R2', R3, R, X y p es como se ha definido anteriormente y se describe en realizaciones en el presente documento, tanto de forma individual como combinada.
En algunas realizaciones, n" es 0. En algunas realizaciones, n" es 1. En algunas realizaciones, n" es 2. En algunas realizaciones, n" es 3. En algunas realizaciones, n" es 4.
En algunas realizaciones, n" se selecciona entre los presentados en las tablas 1, 2 y 2A, a continuación.
En algunas realizaciones, la presente invención proporciona un compuesto de Fórmulas VlII-a o VlII-b:
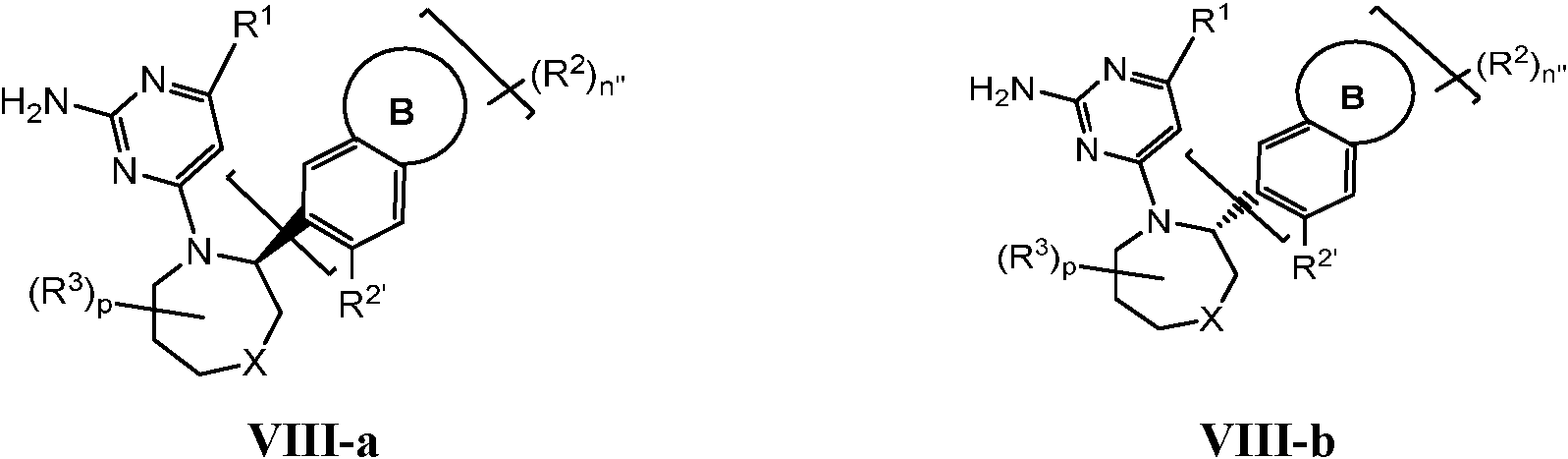
o una sal farmacéuticamente aceptable del mismo, en donde cada uno del anillo B, R1, R2, R2', R3, R, X, p y n" es como se ha definido anteriormente y se describe en realizaciones en el presente documento, tanto de forma individual
como combinada.
En algunas realizaciones, la presente invención proporciona un compuesto de Fórmula IX:

o una sal farmacéuticamente aceptable del mismo, en donde R2' es halógeno, y cada uno de R2 y n' es como se ha definido anteriormente y se describe en realizaciones en el presente documento, tanto de forma individual como combinada.
En algunas realizaciones, la presente invención proporciona un compuesto de Fórmulas IX-a o IX-b:

o una sal farmacéuticamente aceptable del mismo, en donde R2' es halógeno, y cada uno de R2 y n' es como se ha definido anteriormente y se describe en realizaciones en el presente documento, tanto de forma individual como combinada.
En algunas realizaciones, la presente invención proporciona un compuesto de Fórmulas X, XI o XII:
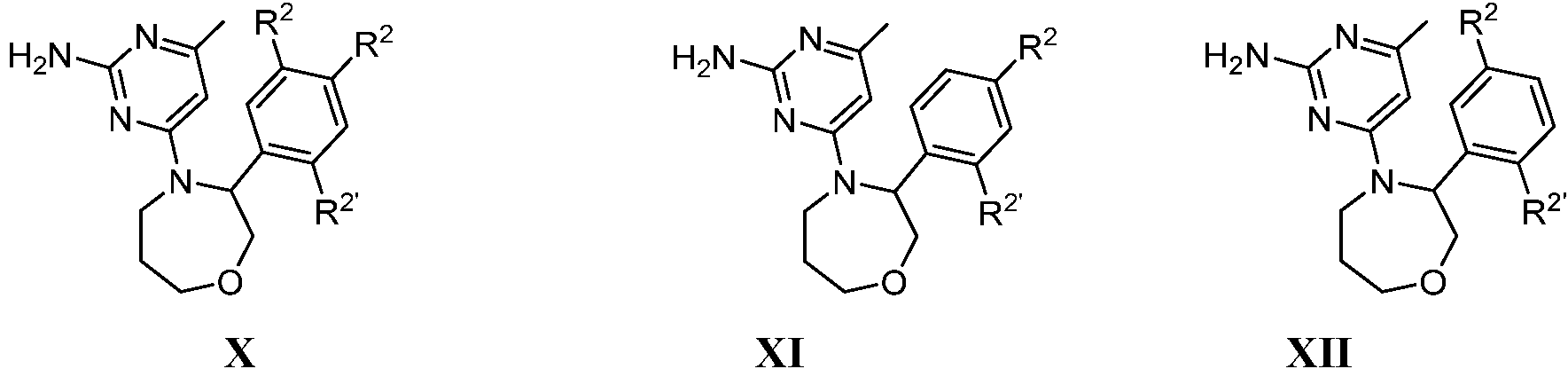
o una sal farmacéuticamente aceptable del mismo, en donde R2' es halógeno, y cada uno de R2 es como se ha definido anteriormente y se describe en realizaciones en el presente documento, tanto de forma individual como combinada. En algunas realizaciones, la presente invención proporciona un compuesto de Fórmulas X-a, X-b, XI-a, XI-b, XII-a o XII-b:


o una sal farmacéuticamente aceptable del mismo, en donde R2' es halógeno, y cada uno de R2 es como se ha definido anteriormente y se describe en realizaciones en el presente documento, tanto de forma individual como combinada. En algunas realizaciones, la presente invención proporciona un compuesto de Fórmulas X, Xl, Xll, X-a, X-b, Xl-a, Xlb, Xll-a o Xll-b, como se muestra anteriormente, en donde R2' es Cl y cada uno de R2 se selecciona entre el grupo
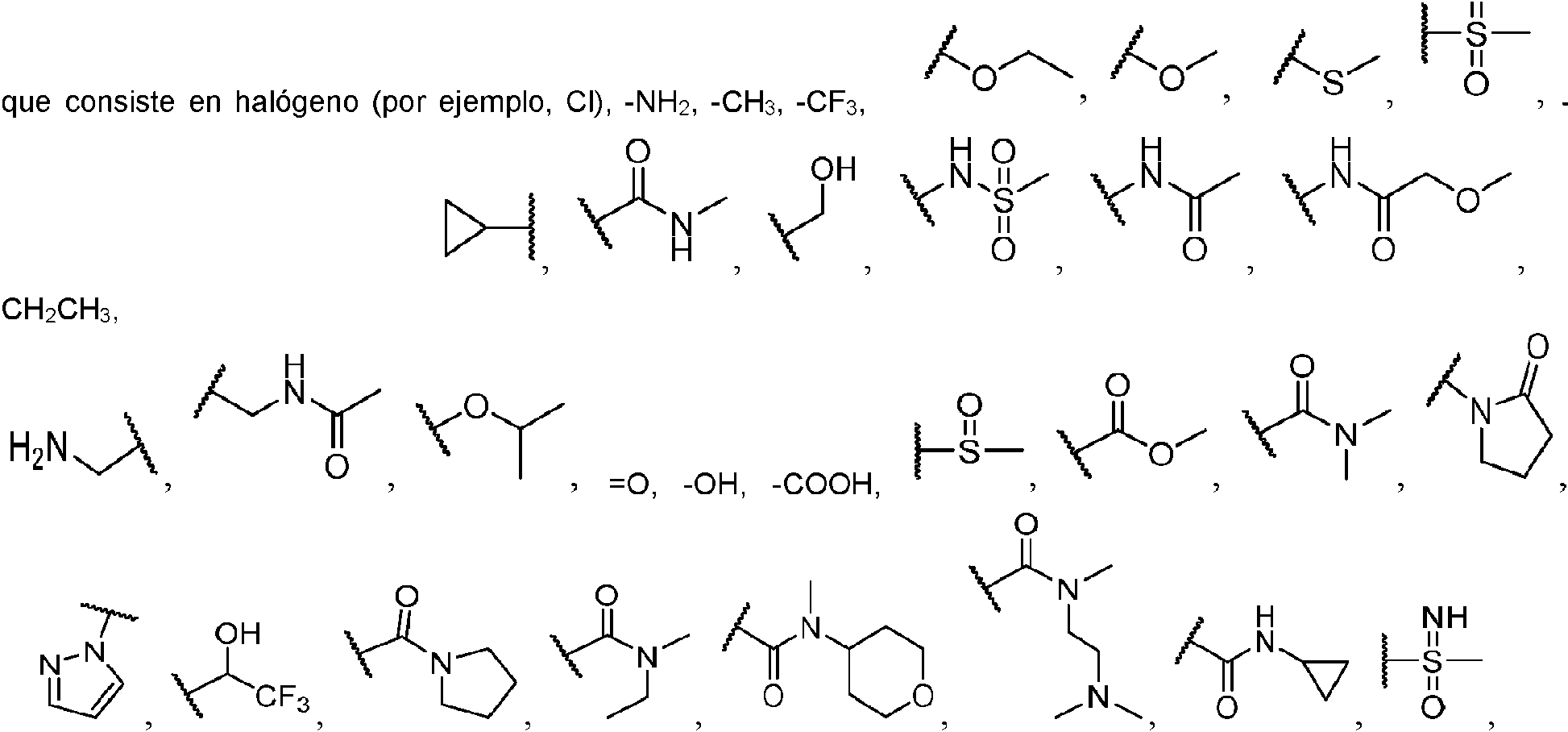


-S(O)2NH2, -OCHF2, -OCF3, -C=CH, -O-CH2-CECH,

En algunas realizaciones, los compuestos ejemplares de la invención se exponen en las Tablas 1 y 2, a continuación. En algunas realizaciones, los compuestos ejemplares de la invención se exponen en la Tabla 2A, a continuación. En algunas realizaciones, la presente invención proporciona un compuesto seleccionado entre los enumerados en la Tabla 1, o una sal farmacéuticamente aceptable del mismo.
En algunas realizaciones, la presente invención proporciona un compuesto seleccionado entre los enumerados en la Tabla 2, o una sal farmacéuticamente aceptable del mismo.
En algunas realizaciones, la presente invención proporciona un compuesto seleccionado entre los enumerados en la Tabla 2A, o una sal farmacéuticamente aceptable del mismo.
En algunas realizaciones, la presente invención proporciona una sal trifluoroacetato del compuesto D-150.
Tabla 1: Compuestos ejemplares


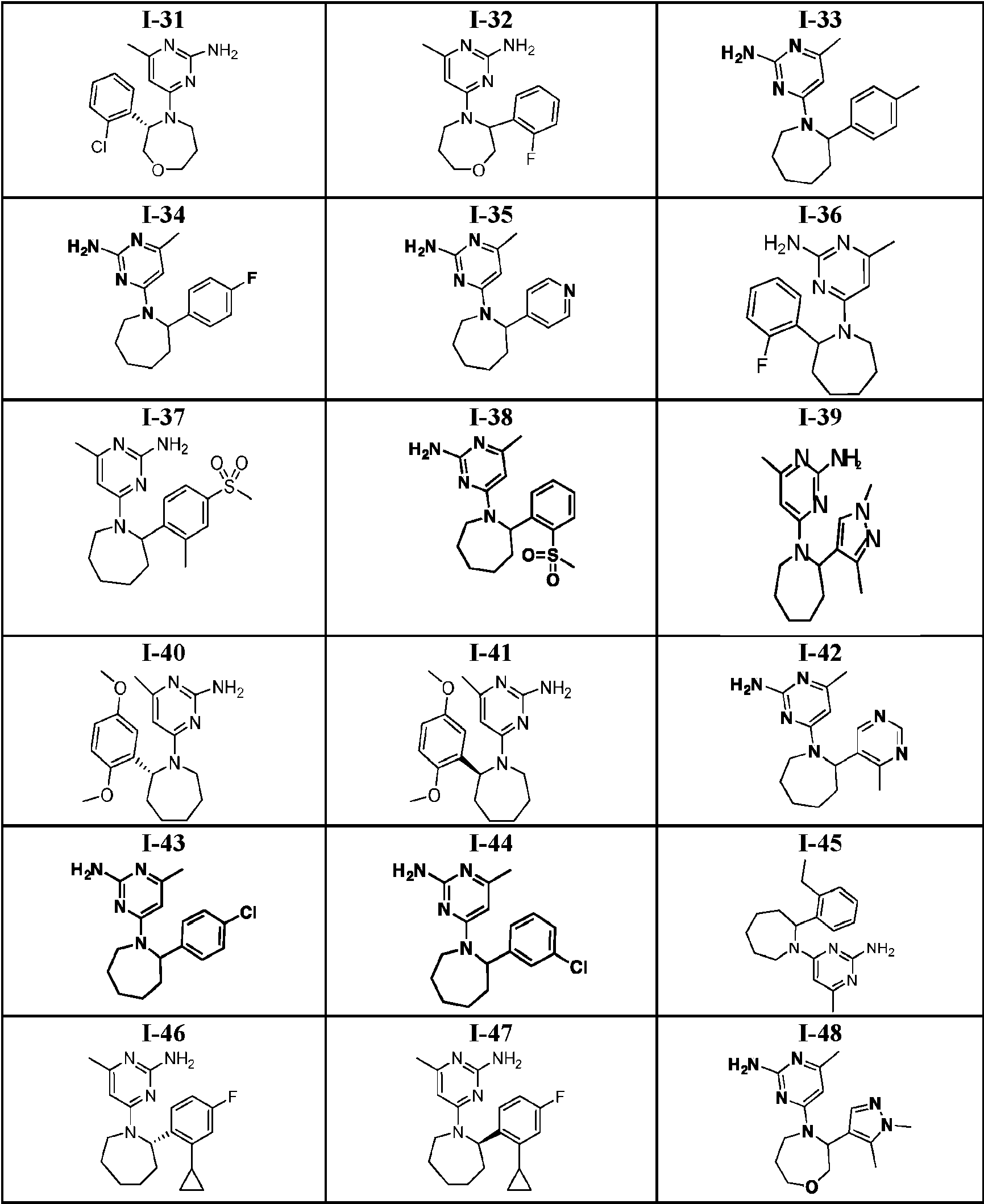
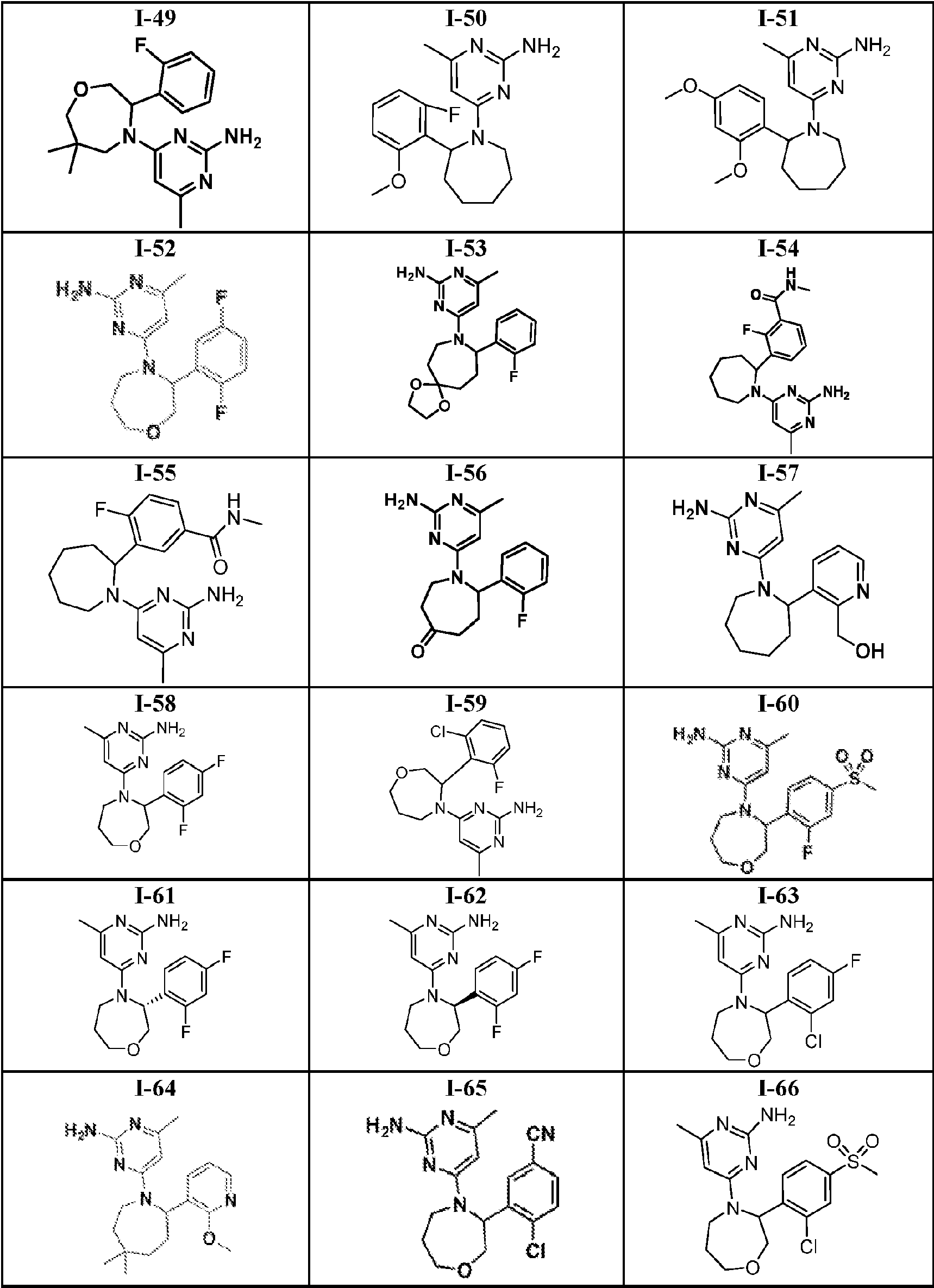




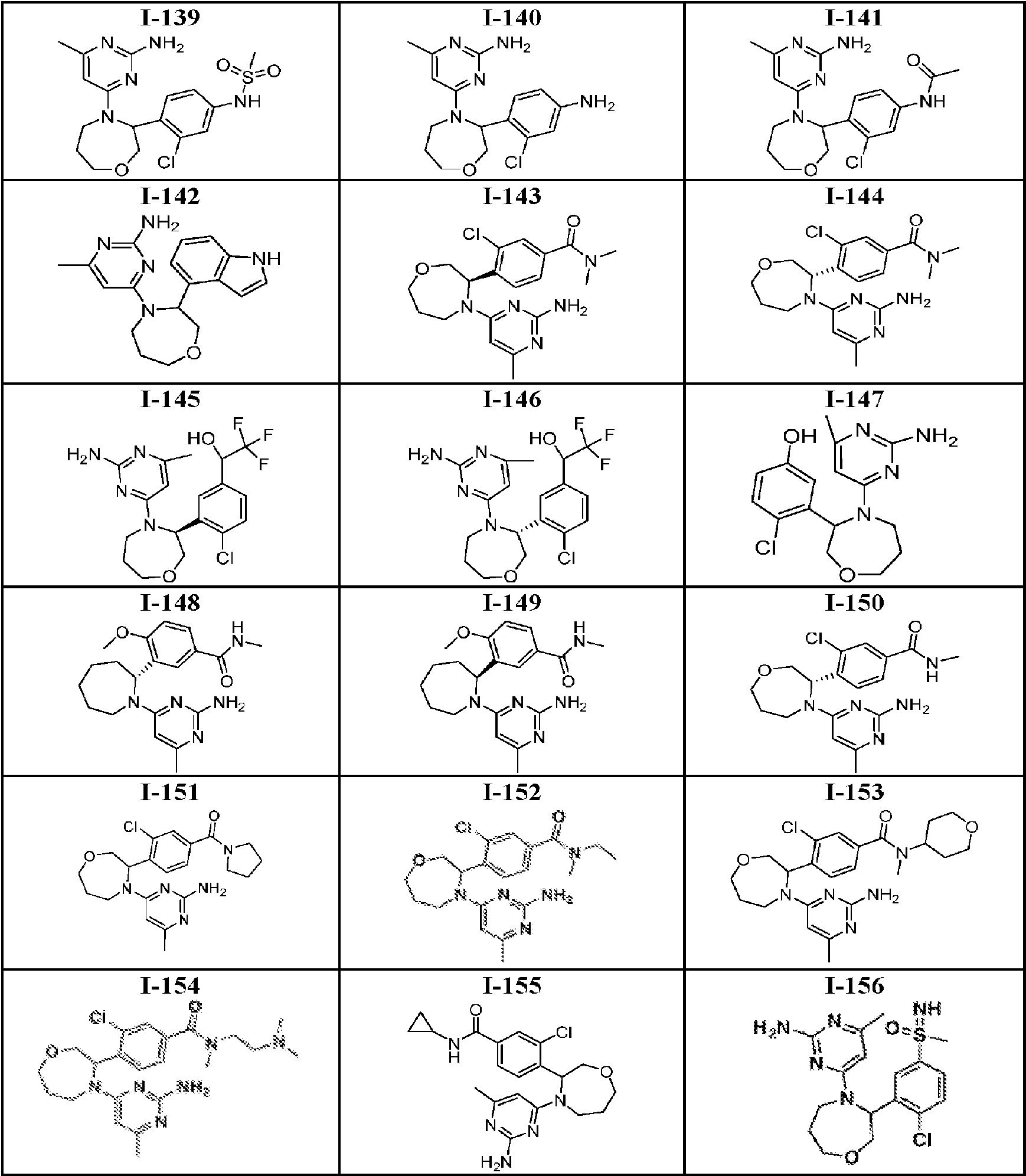




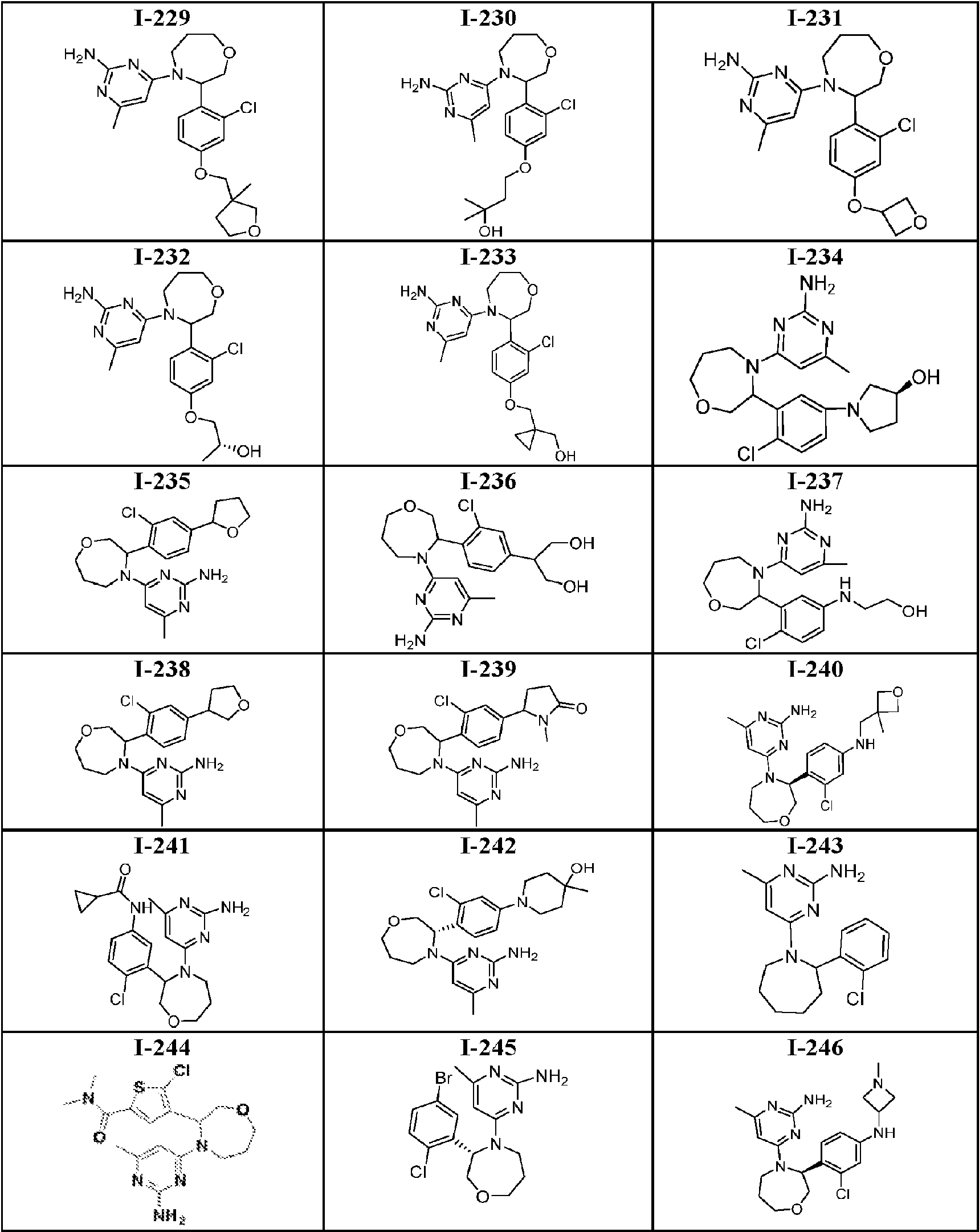


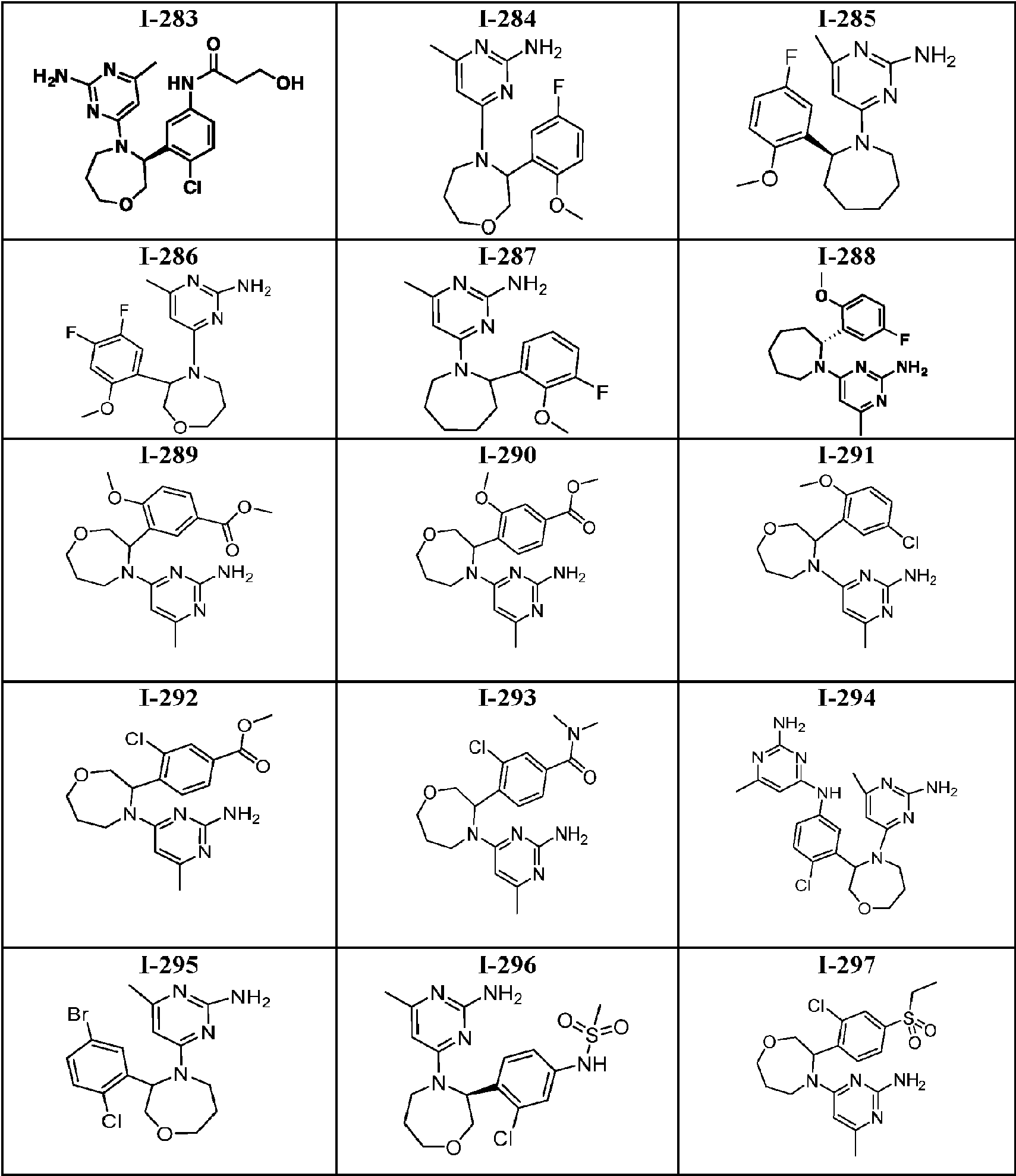



los compuestos I-120 a I-123 se pueden separar mediante purificación quiral (véase, por ejemplo, Ejemplo 18). Por consiguiente, en algunas realizaciones, la presente invención proporciona un estereoisómero seleccionado entre los compuestos I-120 a I-123, o una de sus sales farmacéuticamente aceptable.
Los compuestos I-164 a I-167 son estereoisómeros de las siguientes fórmulas:

los compuestos I-164 a I-167 se pueden separar mediante purificación quiral (véase, por ejemplo, Ejemplo 41). Por consiguiente, en algunas realizaciones, la presente invención proporciona un estereoisómero seleccionado entre los compuestos I-164 a I-167, o una de sus sales farmacéuticamente aceptable.
Los compuestos I-209 y I-210 son estereoisómeros de las siguientes fórmulas:

• compuestos 1-209 y 1-210 se pueden separar mediante purificación quiral (véase, por ejemplo, Ejemplo 52). Por consiguiente, en algunas realizaciones, la presente invención proporciona un estereoisómero seleccionado entre los compuestos I-209 a I-210, o una de sus sales farmacéuticamente aceptable.
Los compuestos I-211 y I-212 son estereoisómeros de las siguientes fórmulas:

purificación quiral (véase, por ejemplo, Ejemplo 52). Por consiguiente, en algunas realizaciones, la presente invención proporciona un estereoisómero seleccionado entre los compuestos I-211 a I-212, o una de sus sales farmacéuticamente aceptable.
Tabla 2. Compuestos ejemplares













Tabla 2A: Compuestos ejemplares


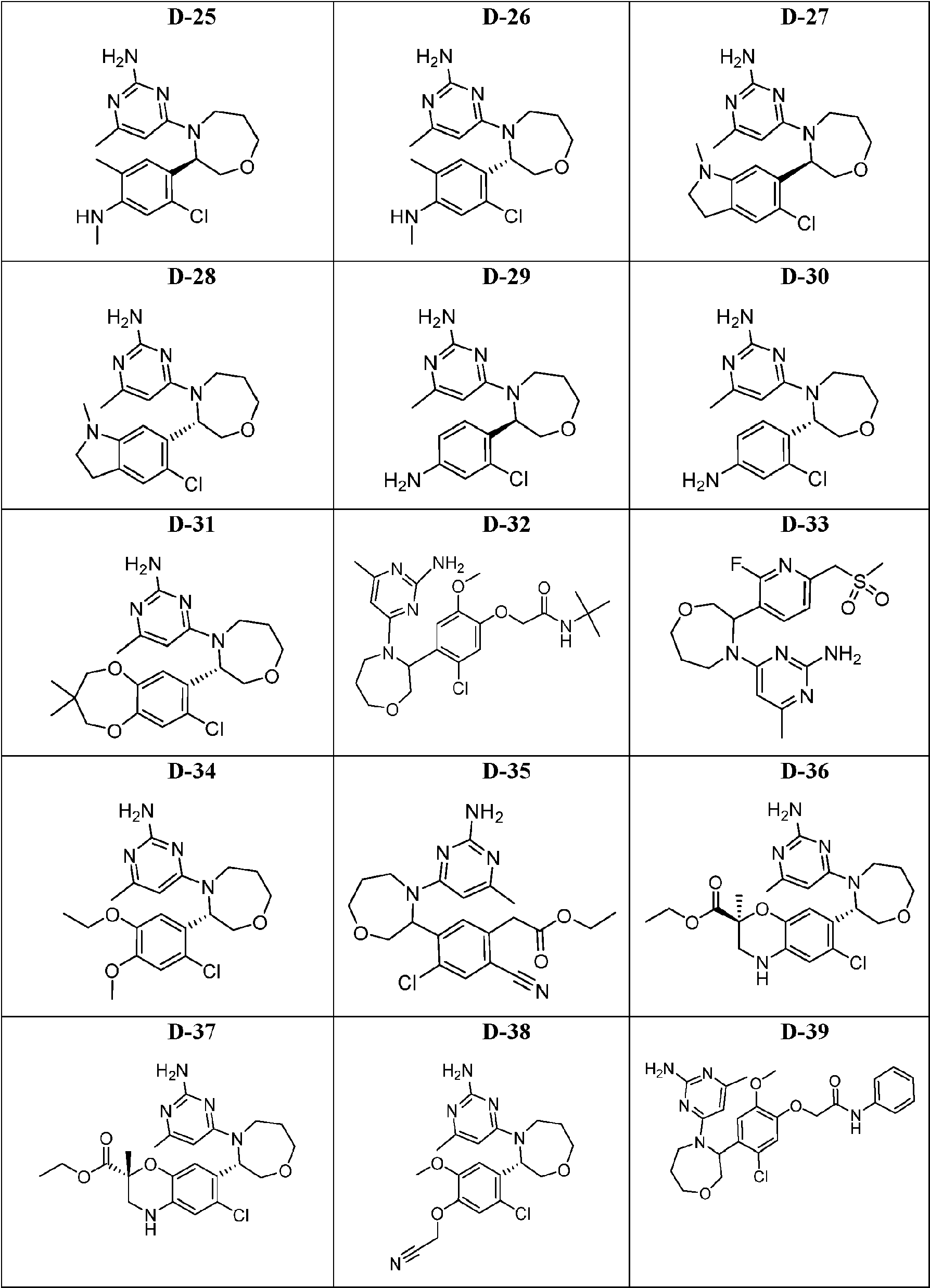





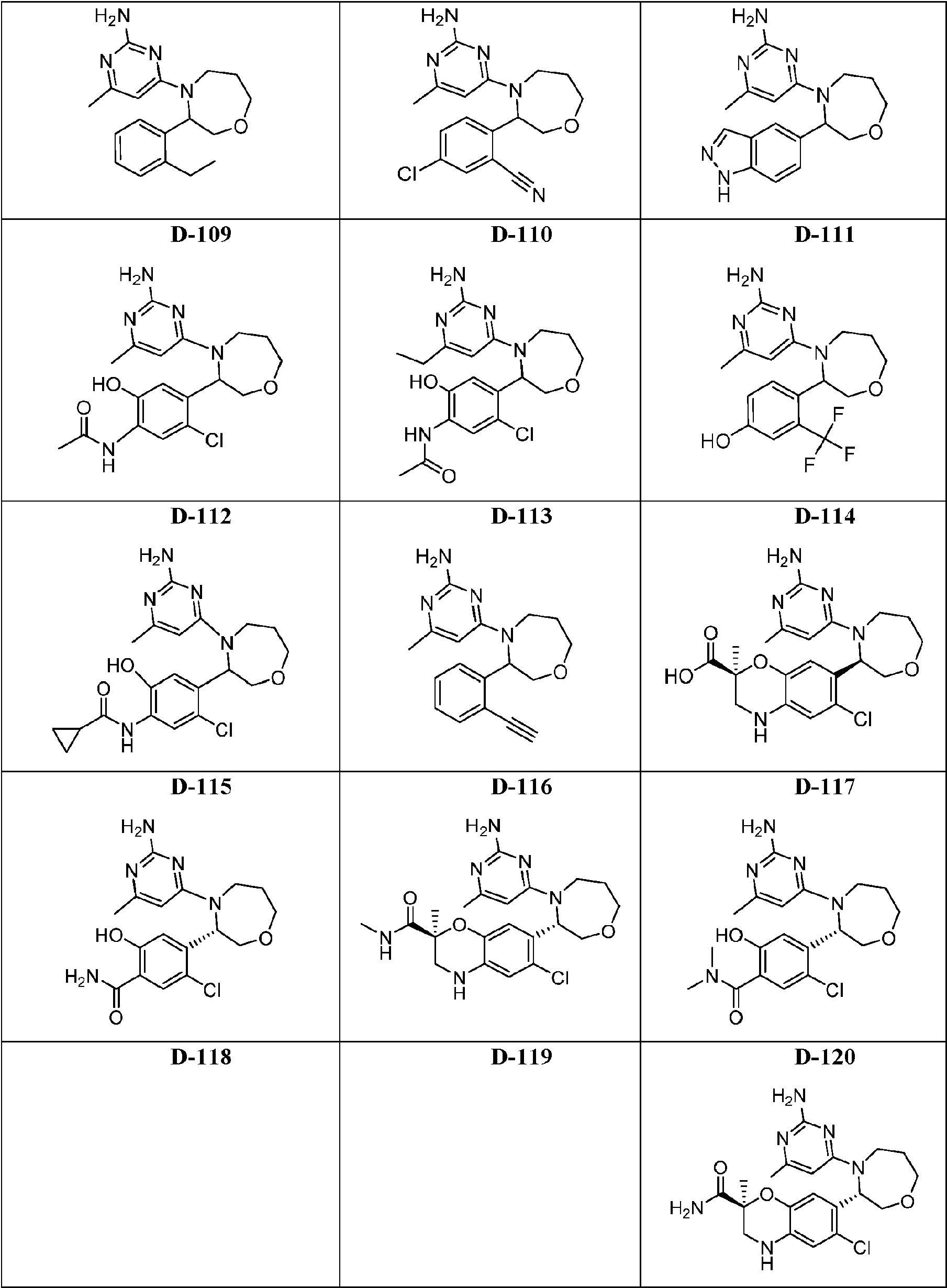


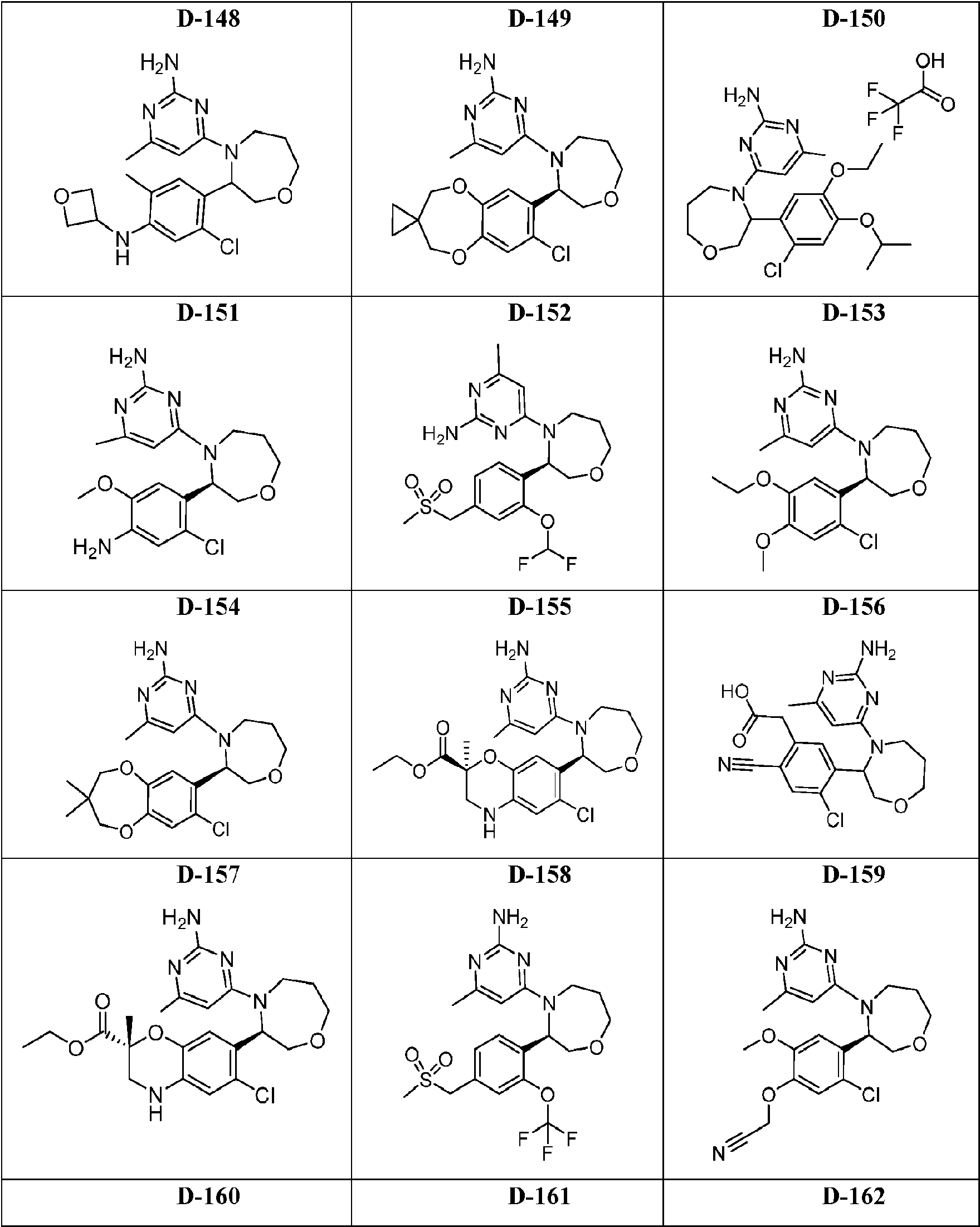



4. Métodos generales para proporcionar los presentes compuestos:
Los compuestos de esta invención pueden prepararse o aislarse en general por métodos sintéticos y/o semisintéticos conocidos por los expertos en la técnica para compuestos análogos y por métodos descritos en detalle en los Ejemplos, en el presente documento.
En los Esquemas a continuación, donde se representa un grupo protector ("PG"), un grupo saliente ("LG") o una condición de transformación particular, un experto en la materia apreciará que otros grupos protectores, grupos salientes y condiciones de transformación también son adecuados y están contemplados. Tales grupos y transformaciones se describen en detalle en March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, M. B. Smith y J. March, 5a edición, John Wiley & Sons, 2001, Comprehensive Organic Transformations, R. C. Larock, 2a edición, John Wiley & Sons, 1999, y Protecting Groups in Organic Synthesis, T. W. Greene y P. G. M. Wuts, 3a edición, John Wiley & Sons, 1999, la totalidad de cada uno de los cuales se incorpora en el presente documento como referencia.
Como se usa en el presente documento, la frase "grupo saliente" (LG) incluye, pero no se limita a, halógenos (por ejemplo, fluoruro, cloruro, bromuro, yoduro), sulfonatos (por ejemplo, mesilato, tosilato, bencenosulfonato, brosilato, nosilato, triflato), diazonio y similares.
Como se usa en el presente documento, la frase "grupo protector de oxígeno" incluye, por ejemplo, grupos protectores de carbonilo, grupos protectores de hidroxilo, etc. Los grupos protectores de hidroxilo son bien conocidos en la técnica e incluyen los descritos en detalle en Protecting Groups in Organic Synthesis, T. W. Greene y P. G. M. Wuts, 3a edición, John Wiley & Sons, 1999, y Philip Kocienski, en "Protecting Groups", Georg Thieme Verlag Stuttgart, Nueva York, 1994, cuya totalidad se incorpora en el presente documento por referencia. Los ejemplos de grupos protectores de hidroxilo adecuados incluyen, pero no se limitan a, ésteres, alil éteres, éteres, éteres de sililo, éteres de alquilo, éteres de arilalquilo y éteres de alcoxialquilo. Ejemplos de tales ésters incluyen formiatos, acetatos, carbonatos, y sulfonatos. Ejemplos específicos incluyen formiato, formiato de benzoílo, cloroacetato, trifluoroacetato, metoxiacetato, trifenilmetoxiacetato, p-clorofenoxiacetato, 3-fenilpropionato, 4-oxopentanoato, 4,4-(etilenoditio)pentanoato, pivaloato (trimetilacetilo), crotonato, 4-metoxi-crotonato, benzoato, p-bencilbenzoato, 2,4,6-trimetilbenzoato, carbonatos, tales como metilo, 9-fluorenilmetilo, etilo, 2,2,2-tricloroetilo, 2-(trimetilsilil)etilo, 2-(fenilsulfonil)etilo, vinilo, alilo y pnitrobencilo. Los ejemplos de tales éteres de sililo incluyen trimetilsililo, trietilsililo, t-butildimetilsililo, t-butildifenilsililo, triisopropilsililo y otros éteres de trialquilsililo. Los éteres de alquilo incluyen éteres de metilo, bencilo, p-metoxibencilo, 3,4-dimetoxibencilo, tritilo, t-butilo, alilo y aliloxicarbonilo o derivados. Los éteres de alcoxialquilo incluyen acetales tales como éteres de metoximetilo, metiltiometilo, (2-metoxietoxi)metilo, benciloximetilo, beta-(trimetilsilil)etoximetilo y tetrahidropiranilo. Ejemplos de éteres de arilalquilo incluyen bencilo, p-metoxibencilo (MPM), 3,4-dimetoxibencilo, O-nitrobencilo, p-nitrobencilo, p-halobencilo, 2,6-diclorobencilo, p-cianobencilo y 2 -y 4-picolilo.
Los grupos protectores de amino son bien conocidos en la técnica e incluyen los descritos en detalle en Protecting Groups in Organic Synthesis, T. W. Greene y P. G. M. Wuts, 3a edición, John Wiley & Sons, 1999, y Philip Kocienski, en "Protecting Groups", Georg Thieme Verlag Stuttgart, Nueva York, 1994, cuya totalidad se incorpora en el presente documento por referencia. Los grupos protectores de amino adecuados incluyen, pero no se limitan a, aralquilaminas, carbamatos, imidas cíclicas, alil aminas, amidas y similares. Los ejemplos de tales grupos incluyen t-butiloxicarbonilo (BOC), etiloxicarbonilo, metiloxicarbonilo, tricloroetiloxicarbonilo, aliloxicarbonilo (Alloc), benciloxocarbonilo (CBZ), alilo, ftalimida, bencilo (Bn), fluorenilmetilcarbonilo (Fmoc), formilo, acetilo, cloroacetilo, dicloroacetilo, tricloroacetilo, fenilacetilo, trifluoroacetilo, benzoílo y similares.
Un experto en la materia apreciará que varios grupos funcionales presentes en los compuestos de la invención tales como grupos alifáticos, alcoholes, ácidos carboxílicos, ésteres, amidas, aldehídos, los halógenos y los nitrilos se pueden interconvertir mediante técnicas bien conocidas en la técnica que incluyen, pero no limitado a la reducción, oxidación, esterificación, hidrólisis, oxidación parcial, reducción parcial, halogenación, deshidratación, hidratación parcial e hidratación. Véase, por ejemplo, "March's Advanced Organic Chemistry", 5a ed., Ed.: Smith, M. B. y March, J., John Wiley & Sons, Nueva York: 2001, la totalidad de la cual se incorpora en el presente documento por referencia. Tales interconversiones pueden requerir una o más de las técnicas antes mencionadas, ya continuación se describen ciertos métodos para sintetizar compuestos de la invención.
En un aspecto, la presente invención proporciona un método para sintetizar un compuesto de Fórmula I, o subfórmulas

del mismo, o una sal del mismo, que comprende hacer reaccionar un compuesto de fórmula: o

una sal del mismo y un compuesto de fórmula: > o una sal del mismo, en donde cada uno del Anillo A, R1, R2, R3, R, X, m, n y p es como se ha definido anteriormente y se describe en realizaciones en el presente documento, tanto de forma individual como combinada.
En algunas realizaciones, la presente invención proporciona un compuesto de fórmula:

o una sal del mismo, en donde cada uno de R1, R y m es como se define anteriormente y se describe en las realizaciones en el presente documento, tanto de forma individual como combinada.
En algunas realizaciones, la presente invención proporciona un compuesto de fórmula:

en donde cada uno del Anillo A, R2, R3, R, X, n y p es como se ha definido anteriormente y se describe en realizaciones en el presente documento, tanto de forma individual como combinada.
En un aspecto, la presente invención proporciona un método para sintetizar un compuesto de Fórmula I', o subfórmulas del mismo, o una sal del mismo, que comprende hacer reaccionar un compuesto de fórmula:
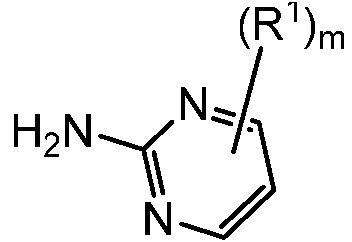
■ o una sal del mismo y un compuesto de fórmula: una sal del mismo, en donde cada uno del Anillo A, R1, R2, R3, ------, R, X, m, n y p es como se ha definido anteriormente y se describe en realizaciones en el presente documento, tanto de forma individual como combinada.
una sal del mismo, en donde cada uno del Anillo A, R1, R2, R3, ------, R, X, m, n y p es como se ha definido anteriormente y se describe en realizaciones en el presente documento, tanto de forma individual como combinada.
En algunas realizaciones, la presente invención proporciona un compuesto de fórmula:

es como se ha definido anteriormente y se describe en realizaciones en el presente documento, tanto de forma individual como combinada.
En algunas realizaciones, la presente invención proporciona un método para sintetizar un compuesto de Fórmula VII, o una de sus sales, que comprende hacer reaccionar un compuesto de fórmula:

sal del mismo, en donde cada uno de R1, R2, R2', R3, R, X, p y n' es como se ha definido anteriormente y se describe en realizaciones en el presente documento, tanto de forma individual como combinada.
En algunas realizaciones, la presente invención proporciona un método para sintetizar un compuesto de Fórmula VIII, o una de sus sales, que comprende hacer reaccionar un compuesto de fórmula:


o una sal del mismo y un compuesto de fórmula:
sal del mismo, en donde cada uno del anillo B, R1, R2, R2', R3, R, X, p y n" es como se ha definido anteriormente y se describe en realizaciones en el presente documento, tanto de forma individual como combinada.
En algunas realizaciones, la presente invención proporciona un compuesto de fórmula:

o una sal del mismo, en donde cada uno de R1 y R es como se ha definido anteriormente y se describe en realizaciones en el presente documento, tanto de forma individual como combinada.
En algunas realizaciones, la presente invención proporciona un compuesto de fórmula:

, o una sal del mismo, en donde cada uno de R2, R2', R3, R, X, p y n' es como se ha definido anteriormente y se describe en realizaciones en el presente documento, tanto de forma individual como combinada.
En algunas realizaciones, la presente invención proporciona un compuesto de fórmula:

o una sal del mismo, en donde cada uno del anillo B, R2, R2', R3, R, X, p y n" es como se ha definido anteriormente y se describe en realizaciones en el presente documento, tanto de forma individual como combinada.
5. Usos, formulación y administración:
composiciones farmacéuticamente aceptables
Según otra realización, la invención proporciona una composición que comprende un compuesto de la presente invención o un derivado farmacéuticamente aceptable del mismo, y un transportador, adyuvante o vehículo farmacéuticamente aceptable. En determinadas realizaciones, la cantidad de compuesto en una composición de la presente invención es tal que es eficaz para provocar la muerte de las células cancerosas en una muestra biológica o en un paciente. En determinadas realizaciones, la cantidad de compuesto en las composiciones de la presente
invención es tal que es eficaz para inducir UPR en células cancerosas en una muestra biológica o en un paciente. En determinadas realizaciones, la cantidad de compuesto en las composiciones de la presente invención es tal que es eficaz para inducir estrés al RE en células cancerosas en una muestra biológica o en un paciente. En determinadas realizaciones, la cantidad de compuesto en las composiciones de la presente invención es tal que es eficaz para inducir la liberación de calcio desde el r E a través de WFS1 en células cancerosas en una muestra biológica o en un paciente. En determinadas realizaciones, una composición de la presente invención se formula para su administración a un paciente que necesita dicha composición. En algunas realizaciones, una composición de la presente invención se formula para su administración oral a un paciente.
El término "paciente", como se usa en el presente documento, significa un animal, preferentemente un mamífero y lo más preferentemente, un ser humano.
La expresión "transportador, adyuvante o vehículo farmacéuticamente aceptable" se refiere a un transportador, adyuvante, o vehículo no tóxico que no destruya la actividad farmacológica del compuesto con el que se formula. Los transportadores, adyuvantes o vehículos farmacéuticamente aceptables que pueden usarse en las composiciones de la presente invención incluyen, pero sin limitación, intercambiadores iónicos, alúmina, estearato de aluminio, lecitina, proteínas séricas, tales como albúmina sérica humana, sustancias tamponantes tales como fosfatos, glicina, ácido sórbico, sorbato de potasio, mezclas de glicéridos parciales de ácidos grasos vegetales saturados, agua, sales o electrolitos, tales como sulfato de protamina, hidrogenofosfato de disodio, hidrogenofosfato de potasio, cloruro de sodio, sales de cinc, sílice coloidal, trisilicato de magnesio, polivinilpirrolidona, sustancias basadas en celulosa, polietilenglicol, carboximetilcelulosa de sodio, poliacrilatos, ceras, polímeros en bloque de polietilenpolioxipropileno, polietilenglicol y lanolina.
Un "derivado farmacéuticamente aceptable" significa cualquier sal, éster, sal de un éster u otro derivado no tóxico de un compuesto de la presente invención que, tras su administración a un receptor, puede proporcionar, directa o indirectamente, un compuesto de la presente invención o un resto o metabolito activo del mismo.
Como se usa en el presente documento, la expresión "resto o metabolito activo del mismo" significa que un metabolito o resto del mismo también da como resultado la muerte celular.
Las composiciones de la presente invención pueden administrarse por vía oral, por vía parenteral, mediante pulverización para inhalación, por vía tópica, por vía rectal, por vía nasal, por vía bucal, por vía vaginal o a través de un depósito implantado. El término "parenteral", como se usa en el presente documento, incluye inyección subcutánea, intravenosa, intramuscular, intraarticular, intrasinovial, intraesternal, intratecal, intrahepática, intralesional e intracraneal o técnicas de infusión. Preferentemente, las composiciones se administran por vía oral, intraperitoneal o intravenosa. Las formas inyectables estériles de las composiciones de la presente invención pueden ser una suspensión acuosa u oleosa. Estas suspensiones pueden formularse según técnicas conocidas en la técnica usando agentes de dispersión o humectación y agentes de suspensión adecuados. La preparación inyectable estéril también puede ser una solución o suspensión inyectable estéril en un diluyente o disolvente no tóxico y parenteralmente aceptable, por ejemplo, en forma de una solución en 1,3-butanodiol. Entre los vehículos y disolventes aceptables que pueden emplearse están el agua, solución de Ringer y solución isotónica de cloruro sódico. Además, normalmente se emplean aceites no volátiles estériles como disolvente o medio de suspensión.
Para este fin, puede emplearse cualquier aceite suave no volátil, incluyendo monoglicéridos o diglicéridos sintéticos. Los ácidos grasos, tales como ácido oleico y sus derivados de glicéridos, son útiles en la preparación de inyectables, al igual que los aceites naturales farmacéuticamente aceptables, tales como aceite de oliva o aceite de ricino, especialmente en sus versiones polioxietiladas. Estas soluciones o suspensiones oleaginosas también pueden contener un diluyente o dispersante alcohólico de cadena larga, tal como carboximetilcelulosa o agentes dispersantes similares que se utilizan comúnmente en la formulación de formas farmacéuticas farmacéuticamente aceptables, incluyendo emulsiones y suspensiones. También pueden usarse otros tensioactivos comúnmente usados, tales como Tween, Span y otros agentes emulsionantes o potenciadores de la biodisponibilidad que se usan comúnmente en la fabricación de formas farmacéuticas sólidas, líquidas u otras farmacéuticamente aceptables con fines de formulación.
Las composiciones farmacéuticamente aceptables de la presente invención pueden administrarse por vía oral en cualquier forma farmacéutica oralmente aceptable, incluyendo, pero sin limitación, cápsulas, comprimidos, suspensiones o soluciones acuosas. En el caso de los comprimidos para uso oral, los transportadores comúnmente usados incluyen lactosa y almidón de maíz. Los agentes lubricantes, tales como estearato de magnesio, también se añaden normalmente. Para la administración oral en forma de cápsula, los diluyentes útiles incluyen lactosa y almidón de maíz deshidratado. Cuando se necesitan suspensiones acuosas para uso oral, el principio activo se combina con agentes emulsionantes y de suspensión. Si se desea, también pueden añadirse determinados agentes edulcorantes, aromatizantes o colorantes.
Como alternativa, las composiciones farmacéuticamente aceptables de la presente invención pueden administrarse en forma de supositorios para administración rectal. Estos pueden prepararse mezclando el agente con un excipiente no irritante adecuado que es sólido a temperatura ambiente, pero líquido a temperatura rectal y, por lo tanto, se derretirá en el recto para liberar el fármaco. Dichos materiales incluyen manteca de cacao, cera de abeja y
polietilenglicoles.
Las composiciones farmacéuticamente aceptables de la presente invención también pueden administrarse por vía tópica, especialmente cuando la diana de tratamiento incluye áreas u órganos fácilmente accesibles mediante aplicación tópica, incluyendo enfermedades de los ojos, la piel o el tracto intestinal inferior. Las formulaciones tópicas adecuadas se preparan fácilmente para cada una de estas áreas u órganos.
La aplicación tópica para el tracto intestinal inferior puede lograrse en una formulación de supositorio rectal (véase anteriormente) o en una formulación de enema adecuada. También pueden usarse parches transdérmicos por vía tópica.
Para aplicaciones tópicas, las composiciones farmacéuticamente aceptables proporcionadas pueden formularse en un ungüento adecuado que contiene el componente activo suspendido o disuelto en uno o más transportadores. Los transportadores para administración tópica de los compuestos de la presente invención incluyen, pero sin limitación, aceite mineral, vaselina líquida, vaselina blanca, propilenglicol, polioxietileno, compuesto de polioxipropileno, cera emulsionante y agua. Como alternativa, las composiciones farmacéuticamente aceptables proporcionadas se pueden formular en una loción o crema adecuados que contiene los principios activos suspendidos o disueltos en uno o más transportadores farmacéuticamente aceptables. Los transportadores adecuados incluyen, pero sin limitación, aceite mineral, monoestearato de sorbitán, polisorbato 60, cera de ésteres cetílicos, alcohol cetearílico, 2-octildodecanol, alcohol bencílico y agua.
Para uso oftálmico, las composiciones farmacéuticamente aceptables proporcionadas pueden formularse como suspensiones micronizadas en suero salino isotónico estéril con pH ajustado o, preferentemente, como soluciones en suero salino isotónico estéril con pH ajustado, tanto con un conservante como sin él, tal como cloruro de benzalconio. Como alternativa, para usos oftálmicos, las composiciones farmacéuticamente aceptables pueden formularse en un ungüento, tal como vaselina.
Las composiciones farmacéuticamente aceptables de la presente invención también pueden administrarse por aerosol nasal o inhalación. Dichas composiciones se preparan según técnicas bien conocidas en la técnica de la formulación farmacéutica y pueden prepararse como soluciones en solución salina, empleando alcohol bencílico u otros conservantes adecuados, promotores de la absorción para potenciar la biodisponibilidad, fluorocarbonos y/u otros agentes solubilizantes o dispersantes convencionales.
Lo más preferentemente, las composiciones farmacéuticamente aceptables de la presente invención se formulan para administración oral. Dichas formulaciones pueden administrarse con o sin alimento. En algunas realizaciones, las composiciones farmacéuticamente aceptables de la presente invención se administran sin alimento. En otras realizaciones, las composiciones farmacéuticamente aceptables se administran con alimento.
La cantidad de compuestos de la presente invención que puede combinarse con los materiales transportadores para producir una composición en una forma monodosis variará dependiendo del hospedador tratado, del modo particular de administración. Preferentemente, las composiciones proporcionadas deben formularse de tal manera que se pueda administrar una dosificación de entre 0,01 - 100 mg/kg de peso corporal/día del inhibidor a un paciente que recibe estas composiciones.
Debe entenderse también que un régimen de dosificación y tratamiento específico para cualquier paciente particular dependerán de una diversidad de factores, incluyendo la actividad del compuesto específico empleado, la edad, el peso corporal, el estado de salud general, el sexo, la dieta, el tiempo de administración, la tasa de excreción, la combinación farmacológica, y el criterio del médico a cargo del tratamiento, y la gravedad de la enfermedad particular que se esté tratando. La cantidad de un compuesto de la presente invención en la composición también variará dependiendo del compuesto concreto en composición.
Usos de compuestos y composiciones farmacéuticamente aceptables
Los compuestos y composiciones descritos en el presente documento son generalmente útiles para el tratamiento de trastornos proliferativos celulares. Según lo dispuesto anteriormente, se ha descubierto que los compuestos descritos en el presente documento son capaces de provocar la liberación de calcio del retículo endoplásmico (RE) a través de un supuesto canal de Ca2+ conocido como wolframina (WFS1), que induce estrés al RE y "respuesta a proteínas desplegadas" (UPR), y da lugar a la muerte.
En algunas realizaciones, la presente invención proporciona compuestos para su uso en un método para tratar un trastorno proliferativo celular en un paciente que comprende administrar a dicho paciente un compuesto de la presente invención, o una composición que comprende dicho compuesto. En algunas realizaciones, la presente invención proporciona un compuesto de la presente invención, o una composición que comprende dicho compuesto, para su uso en el tratamiento de un trastorno proliferativo celular. Dichos trastornos se describen detalladamente en el presente documento. En algunas realizaciones, un trastorno proliferativo celular es un cáncer caracterizado por la sobreexpresión de wolframina (WFS1) en las células cancerosas. En algunas realizaciones, un cáncer caracterizado
por la sobreexpresión de wolframina (WFS1) se selecciona de cáncer de pulmón no microcítico (NSCLC), mieloma, mieloma múltiple, carcinoma hepatocelular (HCC), cáncer de mama, cáncer de vejiga, cáncer de riñón y melanoma. En algunas realizaciones, un método para tratar un trastorno proliferativo celular como se describe en el presente documento comprende además determinar el nivel de expresión de wolframina (WFS1). En algunas realizaciones, el nivel de expresión de wolframina (WFS1) se determina mediante inmunohistoquímica y/o intensidad de sonda de micromatriz.
Como se usa en el presente documento, los términos "tratamiento", "tratar", y la expresión "que trata" se refieren a revertir, aliviar, retrasar el inicio o inhibir la evolución de una enfermedad o trastorno, o de uno o más síntomas de los mismos, como se describe en el presente documento. En algunas realizaciones, el tratamiento se puede administrar después de que hayan aparecido uno o más síntomas. En otras realizaciones, el tratamiento se puede administrar en ausencia de síntomas. Por ejemplo, el tratamiento se puede administrar a un individuo susceptible antes de la aparición de los síntomas (por ejemplo, a la vista de antecedentes de síntomas y/o a la vista de factores genéticos u otros factores de susceptibilidad). El tratamiento también puede continuar después de que los síntomas se hayan resuelto, por ejemplo, para prevenir o retrasar la recaída.
En algunas realizaciones, la presente invención proporciona un método para inducir estrés al RE en un paciente que lo necesita, que comprende administrar un compuesto de la presente invención, o una composición que comprende dicho compuesto. En algunas realizaciones, la presente invención proporciona un método para inducir la "respuesta a proteínas desplegadas" (UPR) en un paciente que lo necesita, que comprende administrar un compuesto de la presente invención, o una composición que comprende dicho compuesto. En algunas realizaciones, la presente invención proporciona un método para provocar la liberación de calcio desde el retículo endoplásmico (RE) a través de un supuesto canal de Ca2+ conocido como wolframina (WFS1) en un paciente que lo necesite, que comprende administrar un compuesto de la presente invención, o una composición que comprende dicho compuesto.
En algunas realizaciones, la presente invención proporciona un compuesto de una cualquiera de las Fórmulas I-VIII, o una composición que comprende dicho compuesto, para su uso para provocar la liberación de calcio del retículo endoplásmico (RE) a través de un supuesto canal de Ca2+ conocido como wolframina (WFS1) en un sujeto que lo necesite. En algunas realizaciones, la presente invención proporciona un compuesto de una cualquiera de las Fórmulas I-VIII, o una composición que comprende dicho compuesto, para su uso en la inducción de estrés al RE en un sujeto que lo necesite. En algunas realizaciones, la presente invención proporciona un compuesto de una cualquiera de las Fórmulas I-VIII, o una composición que comprende dicho compuesto, para su uso en la inducción de la "respuesta a proteínas desplegadas" (UPR) en un sujeto que lo necesite.
En algunas realizaciones, la presente invención proporciona un compuesto de Fórmula I', o una composición que comprende dicho compuesto, para su uso para provocar la liberación de calcio del retículo endoplásmico (RE) a través de un supuesto canal de Ca2+ conocido como wolframina (WFS1) en un sujeto que lo necesite. En algunas realizaciones, la presente invención proporciona un compuesto de Fórmula I', o una composición que comprende dicho compuesto, para su uso en la inducción de estrés al RE en un sujeto que lo necesite. En algunas realizaciones, la presente invención proporciona un compuesto de Fórmula I, o una composición que comprende dicho compuesto, para su uso en la inducción de la "respuesta a proteínas desplegadas" (UPR) en un sujeto que lo necesita
La actividad de un compuesto utilizado en la presente invención como inhibidor de la proliferación celular puede someterse a ensayo in vitro o in vivo. Las condiciones detalladas para someter a ensayo un compuesto en la presente invención se exponen en los ejemplos a continuación.
Trastornos proliferativos celulares
La presente invención presenta métodos y composiciones para el diagnóstico y pronóstico de trastornos proliferativos celulares (por ejemplo, cáncer) y el tratamiento de estos trastornos. Los trastornos proliferativos celulares descritos en el presente documento incluyen, por ejemplo, cáncer, obesidad y enfermedades dependientes de la proliferación. Dichos trastornos pueden diagnosticarse usando métodos conocidos en la técnica.
Cáncer
El cáncer incluye, en una realización, sin limitación, leucemias (por ejemplo, leucemia aguda, leucemia linfocítica aguda, leucemia mielocítica aguda, leucemia mieloblástica aguda, leucemia promielocítica aguda, leucemia mielomonocítica aguda, leucemia monocítica aguda, eritroleucemia aguda, leucemia crónica, leucemia mielocítica crónica, leucemia linfocítica crónica), policitemia vera, linfoma (por ejemplo, enfermedad de Hodgkin o enfermedad no Hodgkiniana), macroglobulinemia de Waldenstrom, mieloma múltiple, enfermedad de la cadena pesada y tumores sólidos tales como sarcomas y carcinomas (por ejemplo, fibrosarcoma, mixosarcoma, liposarcoma, condrosarcoma, sarcoma osteogénico, cordoma, angiosarcoma, endoteliosarcoma, linfangiosarcoma, linfangioendoteliosarcoma, sinovioma, mesotelioma, tumor de Ewing, leiomiosarcoma, rabdomiosarcoma, carcinoma de colon, cáncer pancreático, cáncer de mama, cáncer de ovario, cáncer de próstata, carcinoma de células escamosas, carcinoma basocelular, adenocarcinoma, carcinoma de glándulas sudoríparas, carcinoma de glándulas sebáceas, carcinoma papilar, adenocarcinomas papilares, cistadenocarcinoma, carcinoma medular, carcinoma broncógeno, carcinoma de
células renales, hepatoma, carcinoma de las vías biliares, coriocarcinoma, seminoma, carcinoma embrionario, tumor de Wilm, cáncer de cuello uterino, cáncer uterino, cáncer testicular, carcinoma de pulmón, carcinoma de pulmón microcítico, carcinoma de vejiga, carcinoma epitelial, glioma, astrocitoma, glioblastoma multiforme (GBM, también conocido como glioblastoma), meduloblastoma, craneofaringioma, ependimoma, pinealoma, hemangioblastoma, neuroma acústico, oligodendroglioma, schwannoma, neurofibrosarcoma, meningioma, melanoma, neuroblastoma y retinoblastoma).
En algunas realizaciones, el cáncer es glioma, astrocitoma, glioblastoma multiforme (GBM, también conocido como glioblastoma), meduloblastoma, craneofaringioma, ependimoma, pinealoma, hemangioblastoma, neuroma acústico, oligodendroglioma, schwannoma, neurofibrosarcoma, meningioma, melanoma, neuroblastoma o retinoblastoma.
En algunas realizaciones, el cáncer es neuroma acústico, astrocitoma (por ejemplo, grado I - astrocitoma pilocítico, grado II - astrocitoma de grado bajo, grado III - astrocitoma anaplásico, o grado IV - glioblastoma (GBM)), cordoma, linfoma del SNC, craneofaringioma, glioma del tronco encefálico, ependimoma, glioma mixto, glioma del nervio óptico, subependimoma, meduloblastoma, meningioma, tumor cerebral metastásico, oligodendroglioma, tumores de hipófisis, tumor neuroectodérmico primitivo (PNET, por sus siglas en inglés) o schwannoma. En algunas realizaciones, el cáncer es un tipo que se encuentra más comúnmente en niños que en adultos, tal como glioma de tronco encefálico, craneofaringioma, ependimoma, astrocitoma pilocítico juvenil (JPA, por sus siglas en inglés), meduloblastoma, glioma del nervio óptico, tumor pineal, tumores neuroectodérmicos primitivos (PNET), o tumor rabdoide. En algunas realizaciones, el paciente es un ser humano adulto. En algunas realizaciones, el paciente es un niño o paciente pediátrico.
El cáncer incluye, en otra realización, sin limitación, mesotelioma, hepatobiliar (conducto hepático y biliar), cáncer de huesos, cáncer pancreático, cáncer de piel, cáncer de cabeza o cuello, melanoma cutáneo o intraocular, cáncer de ovario, cáncer de colon, cáncer rectal, cáncer de la región anal, cáncer de estómago, gastrointestinal (gástrico, colorrectal y duodenal), cáncer uterino, carcinoma de las trompas de Falopio, carcinoma del endometrio, carcinoma del cuello uterino, carcinoma de la vagina, carcinoma de la vulva, enfermedad de Hodgkin, cáncer de esófago, cáncer del intestino delgado, cáncer del sistema endocrino, cáncer de la glándula tiroides, cáncer de la glándula paratiroidea, cáncer de la glándula suprarrenal, sarcoma de tejido blando, cáncer de la uretra, cáncer del pene, cáncer de próstata, cáncer testicular, leucemia crónica o aguda, leucemia mieloide crónica, linfomas linfocíticos, cáncer de la vejiga, cáncer del riñón o del uréter, carcinoma de células renales, carcinoma de la pelvis renal, linfoma no Hodgkiniano, tumores del eje espinal, glioma del tronco encefálico, adenoma hipofisario, cáncer adrenocortical, cáncer de vesícula biliar, mieloma múltiple, colangiocarcinoma, fibrosarcoma, neuroblastoma, retinoblastoma, o una combinación de uno o más de los cánceres anteriores.
En algunas realizaciones, el cáncer se selecciona de carcinoma hepatocelular, cáncer de ovario, cáncer epitelial de ovario o cáncer de trompa de Falopio; cistoadenocarcinoma seroso papilar o carcinoma seroso papilar uterino (UPSC, por sus siglas en inglés); cáncer de próstata; cáncer testicular; cáncer de vesícula biliar; hepatocolangiocarcinoma; sarcoma sinovial de tejido blando y hueso; rabdomiosarcoma; osteosarcoma; condrosarcoma; sarcoma de Ewing; cáncer de tiroides anaplásico; adenoma adrenocortical; cáncer pancreático; carcinoma ductal pancreático o adenocarcinoma pancreático; cáncer gastrointestinal/de estómago (GIST, por sus siglas en inglés); linfoma; carcinoma de células escamosas de cabeza y cuello (SCCHN, por sus siglas en inglés); cáncer de glándula salival; glioma o cáncer cerebral; tumores malignos de la vaina nerviosa periférica (MPNST, por sus siglas en inglés) asociados a neurofibromatosis-1; macroglobulinemia de Waldenstrom; o meduloblastoma.
En algunas realizaciones, el cáncer se selecciona de carcinoma hepatocelular (HCC), hepatoblastoma, cáncer de colon, cáncer rectal, cáncer de ovario, cáncer epitelial de ovario, cáncer de las trompas de Falopio, cistadenocarcinoma seroso papilar, carcinoma seroso papilar uterino (UPSC), hepatocolangiocarcinoma, sarcoma sinovial de tejido blando y hueso, rabdomiosarcoma, osteosarcoma, cáncer de tiroides anaplásico, adenoma adrenocortical, cáncer pancreático, carcinoma ductal pancreático, adenocarcinoma pancreático, glioma, tumores malignos de la vaina nerviosa periférica (MPNST) asociados a neurofibromatosis-1, macroglobulinemia de Waldenstrom o meduloblastoma.
En algunas realizaciones, la presente invención proporciona compuestos para su uso en un método para tratar un cáncer que se presenta como un tumor sólido, como un sarcoma, carcinoma o linfoma, que comprende la etapa de administrar un compuesto divulgado, o una sal farmacéuticamente aceptable del mismo, a un paciente que lo necesita. Los tumores sólidos generalmente comprenden una masa anormal de tejido que normalmente no incluye quistes o áreas líquidas. En algunas realizaciones, el cáncer se selecciona de carcinoma de células renales o cáncer de riñón; carcinoma hepatocelular (HCC) o hepatoblastoma, o cáncer de hígado; melanoma; cáncer de mama; carcinoma colorrectal o cáncer colorrectal; cáncer de colon; cáncer de recto; cáncer anal; cáncer de pulmón, tales como cáncer de pulmón no microcítico (NSCLC) o cáncer de pulmón microcítico (SCLC); cáncer de ovario, cáncer epitelial de ovario, carcinoma de ovario o cáncer de trompa de Falopio; cistoadenocarcinoma seroso papilar o carcinoma seroso papilar uterino (UPSC, por sus siglas en inglés); cáncer de próstata; cáncer testicular; cáncer de vesícula biliar; hepatocolangiocarcinoma; sarcoma sinovial de tejido blando y hueso; rabdomiosarcoma; osteosarcoma; condrosarcoma; sarcoma de Ewing; cáncer de tiroides anaplásico; carcinoma adrenocortical; cáncer pancreático; carcinoma ductal pancreático o adenocarcinoma pancreático; cáncer gastrointestinal/de estómago (GIST, por sus siglas en inglés); linfoma; carcinoma de células escamosas de cabeza y cuello (SCCHN, por sus siglas en inglés);
cáncer de glándula salival; glioma o cáncer cerebral; tumores malignos de la vaina nerviosa periférica (MPNST) asociados a neurofibromatosis-1; macroglobulinemia de Waldenstrom; o meduloblastoma.
En algunas realizaciones, el cáncer se selecciona de carcinoma de células renales, carcinoma hepatocelular (HCC, por sus siglas en inglés), hepatoblastoma, carcinoma colorrectal, cáncer colorrectal, cáncer de colon, cáncer rectal, cáncer anal, cáncer de ovario, cáncer epitelial de ovario, carcinoma de ovario, cáncer de las trompas de Falopio, cistadenocarcinoma seroso papilar, carcinoma seroso papilar uterino (UPSC), hepatocolangiocarcinoma, sarcoma sinovial de tejido blando y hueso, rabdomiosarcoma, osteosarcoma, condrosarcoma, cáncer de tiroides anaplásico, carcinoma adrenocortical, cáncer pancreático, carcinoma ductal pancreático, adenocarcinoma pancreático, glioma, cáncer de cerebro, tumores malignos de la vaina nerviosa periférica (MPNST) asociados a neurofibromatosis-1, macroglobulinemia de Waldenstrom o meduloblastoma.
En algunas realizaciones, el cáncer se selecciona de carcinoma hepatocelular (HCC), hepatoblastoma, cáncer de colon, cáncer rectal, cáncer de ovario, cáncer epitelial de ovario, carcinoma de ovario, cáncer de las trompas de Falopio, cistadenocarcinoma seroso papilar, carcinoma seroso papilar uterino (UPSC), hepatocolangiocarcinoma, sarcoma sinovial de tejido blando y hueso, rabdomiosarcoma, osteosarcoma, cáncer de tiroides anaplásico, carcinoma adrenocortical, cáncer pancreático, carcinoma ductal pancreático, adenocarcinoma pancreático, glioma, tumores malignos de la vaina nerviosa periférica (MPNST) asociados a neurofibromatosis-1, macroglobulinemia de Waldenstrom o meduloblastoma.
En algunas realizaciones, el cáncer es carcinoma hepatocelular (HCC). En algunas realizaciones, el cáncer es hepatoblastoma. En algunas realizaciones, el cáncer es cáncer de colon. En algunas realizaciones, el cáncer es cáncer de recto. En algunas realizaciones, el cáncer es cáncer de ovario o carcinoma de ovario. En algunas realizaciones, el cáncer es cáncer epitelial de ovario. En algunas realizaciones, el cáncer es cáncer de las trompas de Falopio. En algunas realizaciones, el cáncer es cistoadenocarcinoma seroso papilar. En algunas realizaciones, el cáncer es carcinoma seroso papilar uterino (UPSC). En algunas realizaciones, el cáncer es hepatocolangiocarcinoma. En algunas realizaciones, el cáncer es sarcoma sinovial de tejido blando y hueso. En algunas realizaciones, el cáncer es rabdomiosarcoma. En algunas realizaciones, el cáncer es osteosarcoma. En algunas realizaciones, el cáncer es cáncer de tiroides anaplásico. En algunas realizaciones, el cáncer es carcinoma adrenocortical. En algunas realizaciones, el cáncer es cáncer pancreático o carcinoma ductal pancreático. En algunas realizaciones, el cáncer es adenocarcinoma pancreático. En algunas realizaciones, el cáncer es glioma. En algunas realizaciones, el cáncer son tumores malignos de la vaina nerviosa periférica (MPNST). En algunas realizaciones, el cáncer es MPNST asociado a neurofibromatosis-1. En algunas realizaciones, el cáncer es macroglobulinemia de Waldenstrom. En algunas realizaciones, el cáncer es meduloblastoma.
La presente invención presenta además métodos y composiciones para el diagnóstico, pronóstico y tratamiento de cánceres asociados a virus, incluidos los tumores sólidos asociados al virus de la inmunodeficiencia humana (HIV), tumores sólidos incurables virus del papiloma humano (HPV)-16 positivos y leucemia de linfocitos T del adulto, que está causada por el virus de la leucemia de linfocitos T humanos de tipo I (HTLV-I) y es una forma altamente agresiva de leucemia de linfocitos T CD4+ caracterizada por la integración clonal de HTLV-I en células leucémicas (véase https://clinicaltrials.gov/ct2/show/study/NCT02631746); así como tumores asociados a virus en cáncer gástrico, carcinoma nasofaríngeo, cáncer de cuello uterino, cáncer vaginal, cáncer de vulva, carcinoma de células escamosas de cabeza y cuello y carcinoma de células de Merkel. (Véase https://clinicaltrials.gov/ct2/show/study/NCT02488759; véanse también https://clinicaltrials.gov/ct2/show/study/NCT0240886; https://clinicaltrials.gov/ct2/show/ NCT02426892)
En algunas realizaciones, la presente invención proporciona compuestos para su uso en un método para tratar un cáncer en un paciente que lo necesita, que comprende administrar al paciente cualquiera de los compuestos, sales o composiciones farmacéuticas descritos en el presente documento. En algunas realizaciones, el cáncer se selecciona entre los cánceres descritos en el presente documento. En algunas realizaciones, el cáncer es cáncer de tipo melanoma. En algunas realizaciones, el cáncer es cáncer de mama. En algunas realizaciones, el cáncer es cáncer de pulmón. En algunas realizaciones, el cáncer es cáncer de pulmón microcítico (SCLC). En algunas realizaciones, el cáncer es cáncer de pulmón no microcítico (NSCLC). En algunas realizaciones, el cáncer es mieloma. En algunas realizaciones, el cáncer es mieloma múltiple. En algunas realizaciones, el cáncer es cáncer de vejiga. En algunas realizaciones, el cáncer es cáncer de riñón. En algunas realizaciones, el cáncer es carcinoma hepatocelular (HCC). En algunas realizaciones, el cáncer es melanoma. En algunas realizaciones, el cáncer es cáncer colorrectal. En algunas realizaciones, el cáncer es cáncer de endometrio. En algunas realizaciones, el cáncer es cáncer de esófago. En algunas realizaciones, el cáncer es cáncer de páncreas. En algunas realizaciones, el cáncer es carcinoma de células renales.
En algunas realizaciones, el tumor se trata deteniendo el crecimiento adicional del tumor. En algunas realizaciones, el tumor se trata reduciendo el tamaño (por ejemplo, volumen o masa) del tumor en al menos un 5 %, 10 %, 25 %, 50 %, 75 %, 90 % o 99 % con respecto al tamaño del tumor antes del tratamiento. En algunas realizaciones, los tumores se tratan reduciendo la cantidad de tumores en el paciente en al menos un 5 %, 10 %, 25 %, 50 %, 75 %, 90 % o 99 % con respecto a la cantidad de tumores antes del tratamiento.
Los compuestos y composiciones, según el método de la presente invención, pueden administrarse usando cualquier cantidad y cualquier vía de administración eficaz para tratar o atenuar la gravedad de un trastorno proliferativo celular. La cantidad exacta necesaria variará entre sujetos, dependiendo de la especie, la edad y el estado general del sujeto, la gravedad de la enfermedad o afección, el agente particular, su modo de administración y similares. Los compuestos de la invención se formulan preferentemente en forma farmacéutica unitaria para facilitar la administración y la uniformidad de la dosificación. La expresión "forma farmacéutica unitaria", como se usa en el presente documento, se refiere a una unidad físicamente concreta de agente adecuada para el paciente que se va a tratar. Se entenderá, sin embargo, que el uso diario total de los compuestos y composiciones de la presente invención se decidirá por el médico a cargo del tratamiento dentro del alcance del criterio médico razonable. El nivel específico de dosis eficaz para cualquier paciente u organismo en particular dependerá de diversos factores, incluyendo el trastorno que se trate y la gravedad del trastorno; la actividad del compuesto específico empleado; la composición específica empleada; la edad, el peso corporal, el estado de salud general, el género y la dieta del paciente; el momento de la administración, la vía de administración y la tasa de secreción del compuesto específico empleado; la duración del tratamiento; los fármacos usados en combinación o casuales con el compuesto específico empleado y factores similares bien conocidos en las técnicas médicas. El término "paciente", como se usa en el presente documento, significa un animal, preferentemente un mamífero y lo más preferentemente, un ser humano.
Las composiciones farmacéuticamente aceptables de la presente invención pueden administrarse a seres humanos y otros animales por vía oral, por vía rectal, por vía parenteral, por vía intracisternal, por vía intravaginal, por vía intraperitoneal, por vía tópica (en forma de polvos, ungüentos o gotas), por vía bucal, como un pulverizador oral o nasal, o similares, dependiendo de la gravedad de la enfermedad o trastorno que se esté tratando. En determinadas realizaciones, los compuestos de la invención pueden administrarse por vía oral o parenteral a niveles de dosificación de aproximadamente 0,01 mg/kg a aproximadamente 50 mg/kg y, preferentemente, de aproximadamente 1 mg/kg a aproximadamente 25 mg/kg, de peso corporal del sujeto al día, una o más veces al día, para obtener el efecto terapéutico deseado.
Las formas farmacéuticas líquidas para administración oral incluyen, pero sin limitación, emulsiones, microemulsiones, soluciones, suspensiones, jarabes y elixires farmacéuticamente aceptables. Además de los compuestos activos, las formas farmacéuticas líquidas pueden contener diluyentes inertes usados habitualmente en la técnica, tales como, por ejemplo, agua u otros disolventes, agentes solubilizantes y emulsionantes tales como alcohol etílico, alcohol isopropílico, carbonato de etilo, acetato de etilo, alcohol bencílico, benzoato de bencilo, propilenglicol, 1,3-butilenglicol, dimetilformamida, aceites (en particular, aceite de semilla de algodón, cacahuete, maíz, germen, oliva, ricino y sésamo), glicerol, alcohol tetrahidrofurfurílico, polietilenglicoles y ésteres de ácidos grasos de sorbitán, y mezclas de los mismos. Además de diluyentes inertes, las composiciones orales también pueden incluir adyuvantes, tales como agentes humectantes, agentes emulsionantes y de suspensión, agentes edulcorantes, saborizantes y perfumantes.
Las preparaciones inyectables, por ejemplo, suspensiones acuosas u oleosas inyectables estériles pueden formularse según la técnica conocida utilizando agentes dispersantes o humectantes y agentes de suspensión adecuados. La preparación inyectable estéril también puede ser una solución, suspensión o emulsión inyectable estéril en un diluyente o disolvente atóxico por vía parenteral aceptable, por ejemplo, como una solución en 1,3-butanodiol. Entre los vehículos y disolventes aceptables que pueden emplearse están el agua, solución de Ringer, U.S.P. y solución de cloruro de sodio isotónica. Además, normalmente se emplean aceites no volátiles estériles como disolvente o medio de suspensión. Para este fin, puede emplearse cualquier aceite no volátil suave, incluyendo mono o diglicéridos sintéticos. Además, se usan ácidos grasos tales como ácido oleico en la preparación de inyectables.
Las formulaciones inyectables pueden esterilizarse, por ejemplo, mediante filtración a través de un filtro de retención de bacterias o mediante la incorporación de agentes esterilizantes en forma de composiciones sólidas estériles que pueden disolverse o dispersarse en agua estéril u otro medio inyectable estéril antes de su uso.
Para prolongar el efecto de un compuesto de la presente invención, es deseable con frecuencia ralentizar la absorción del compuesto de inyección subcutánea o intramuscular. Esto puede lograrse mediante el uso de una suspensión líquida de material cristalino o amorfo poco hidrosoluble. La velocidad de absorción del compuesto depende entonces de su velocidad de disolución que, a su vez, puede depender del tamaño del cristal y de la forma cristalina. Como alternativa, se realiza absorción retardada de una forma de compuesto administrado por vía parenteral disolviendo o suspendiendo el compuesto en un vehículo oleoso. Las formas de depósito inyectables se preparan mediante la formación de matrices microencapsuladas de los polímeros biodegradables tales como polilactidapoliglicólido. Dependiendo de la proporción de compuesto con respecto a polímero y de la naturaleza del polímero concreto empleado, se puede controlar la velocidad de liberación del compuesto. Los ejemplos de otros polímeros biodegradables incluyen poli(ortoésteres) y poli(anhídridos). También se preparan formulaciones de depósito inyectables inmovilizando el compuesto en liposomas o microemulsiones que son compatibles con los tejidos corporales.
Las composiciones para administración rectal o vaginal son, preferentemente, supositorios que pueden prepararse mezclando los compuestos de la presente invención con excipientes o transportadores adecuados no irritantes, tales como manteca de cacao, polietilenglicol o una cera para supositorios que son sólidos a temperatura ambiente pero líquidos a temperatura corporal y, por lo tanto, se derriten en el recto o en la cavidad vaginal y liberan el compuesto
activo.
Las formas farmacéuticas sólidas para administración oral incluyen cápsulas, comprimidos, pastillas, polvos y gránulos. En dichas formas farmacéuticas sólidas, el compuesto activo se mezcla con al menos un excipiente o transportador inerte, farmacéuticamente aceptable tal como citrato de sodio o fosfato de dicalcio y/o a) cargas o diluyentes tales como almidones, lactosa, sacarosa, glucosa, manitol y ácido silícico, b) aglutinantes, tales como, por ejemplo, carboximetilcelulosa, alginatos, gelatina, polvinilpirrolidona, sacarosa y goma arábiga, c) humectantes, tales como glicerol, d) agentes disgregantes, tales como agar-agar, carbonato de calcio, almidón de patata o tapioca, ácido algínico, determinados silicatos y carbonato de sodio, e) agentes retardantes de la solución, tales como parafina, f) aceleradores de la absorción, tales como compuestos de amonio cuaternario, g) agentes humectantes, tales como, por ejemplo, alcohol cetílico y monoestearato de glicerol, h) absorbentes, tales como caolín y arcilla bentonítica e i) lubricantes, tales como talco, estearato de calcio, estearato de magnesio, polietilenglicoles sólidos, lauril sulfato de sodio y mezclas de los mismos. En el caso de cápsulas, comprimidos y píldoras, la forma farmacéutica también puede comprender agentes tamponantes.
También pueden emplearse composiciones sólidas de tipo similar como cargas en cápsulas de gelatina de relleno blando y duro usando excipientes tales como lactosa o azúcar de la leche, así como polietilenglicoles de alto peso molecular y similares. Las formas farmacéuticas sólidas de comprimidos, grageas, cápsulas, píldoras y gránulos pueden prepararse con recubrimientos y cubiertas, tales como recubrimientos entéricos y otros recubrimientos bien conocidos en la técnica de la formulación farmacéutica. Opcionalmente, pueden contener agentes opacificantes y también pueden tener una composición que libere el principio o los principios activos solo, o preferentemente, en una parte determinada del tracto intestinal, opcionalmente, de manera retardada. Los ejemplos de composiciones de inclusión que pueden usarse incluyen sustancias poliméricas y ceras. También pueden emplearse composiciones sólidas de tipo similar como cargas en cápsulas rellenas de gelatina blanda y dura usando excipientes tales como lactosa o azúcar de la leche, así como polietilenglicoles de alto peso molecular y similares.
Los compuestos activos también pueden estar en forma microencapsulada con uno o más excipientes, como se ha indicado anteriormente. Las formas farmacéuticas sólidas de comprimidos, grageas, cápsulas, píldoras y gránulos pueden prepararse con recubrimientos y cubiertas, tales como recubrimientos entéricos, recubrimientos de control de la liberación y otros recubrimientos bien conocidos en la técnica de la formulación farmacéutica. En dichas formas farmacéuticas sólidas, el compuesto activo puede mezclarse con al menos un diluyente inerte tal como sacarosa, lactosa o almidón. Dichas formas farmacéuticas también pueden comprender, como es práctica normal, sustancias adicionales distintas de los diluyentes inertes, por ejemplo, lubricantes para la formación de comprimidos y otros adyuvantes para la formación de comprimidos, tales como estearato de magnesio y celulosa microcristalina. En el caso de cápsulas, comprimidos y píldoras, las formas farmacéuticas también pueden comprender agentes tamponantes. Opcionalmente, pueden contener agentes opacificantes y también pueden tener una composición que libere el principio o los principios activos solo, o preferentemente, en una parte determinada del tracto intestinal, opcionalmente, de manera retardada. Los ejemplos de composiciones de inclusión que pueden usarse incluyen sustancias poliméricas y ceras.
Las formas farmacéuticas para administración tópica y/o transdérmica de un compuesto de la presente invención incluyen ungüentos, pastas, cremas, lociones, geles, polvos, soluciones, aerosoles, inhalantes o parches. El componente activo se mezcla en condiciones estériles con un transportador farmacéuticamente aceptable y cualquier conservante o tampón necesario, según sea necesario. Las formulaciones oftálmicas, gotas óticas y/o colirios también están incluidos en el alcance de la presente invención. Adicionalmente, la presente invención contempla el uso de parches transdérmicos, que tienen la ventaja añadida de proporcionar un suministro controlado de un compuesto al organismo. Dichas formas farmacéuticas pueden prepararse disolviendo o dispensando el compuesto en el medio adecuado. También pueden usarse potenciadores de la absorción para aumentar el flujo del compuesto a través de la piel. La velocidad puede controlarse proporcionando una membrana de control de la velocidad o dispersando el compuesto en un matriz polimérica o gel.
En alguna realización, la invención se refiere a un método para inducir estrés al RE en una muestra biológica que comprende la etapa de poner en contacto dicha muestra biológica con un compuesto de la presente invención, o una composición que comprende dicho compuesto.
En alguna realización, la invención se refiere a un método para inducir "respuesta a proteínas desplegadas" (UPR) en una muestra biológica que comprende la etapa de poner en contacto dicha muestra biológica con un compuesto de la presente invención, o una composición que comprende dicho compuesto.
En determinadas realizaciones, la invención se refiere a un método para provocar la liberación de calcio del retículo endoplásmico (RE) a través de un supuesto canal de Ca2+ conocido como wolframina (WFS1) en una muestra biológica que comprende la etapa de poner en contacto dicha muestra biológica con un compuesto de la presente invención, o una composición que comprende dicho compuesto.
La expresión "muestra biológica", como se usa en el presente documento, incluye, sin limitación, cultivos celulares o extractos de los mismos; material de biopsia obtenido de un mamífero o extractos del mismo; y sangre, saliva, orina,
heces, semen, lágrimas u otros fluidos corporales o extractos de los mismos.
Administración conjunta de agentes terapéuticos adicionales
Dependiendo de la afección o enfermedad particular, que se va a tratar, también pueden estar presentes en las composiciones de la invención agentes terapéuticos adicionales que normalmente se administran para tratar esa afección. Como se usa en el presente documento, los agentes terapéuticos adicionales que se administran normalmente para tratar una afección o enfermedad particular, se conocen como "adecuados para la enfermedad, o afección, que se va a tratar".
En algunas realizaciones, la presente invención proporciona un método para tratar una enfermedad o afección divulgada que comprende administrar a un paciente que lo necesita una cantidad eficaz de un compuesto divulgado en el presente documento o una sal farmacéuticamente aceptable del mismo y administrar conjuntamente de manera simultánea o secuencial una cantidad eficaz de uno o más compuestos terapéuticos adicionales, tales como los descritos en el presente documento. En algunas realizaciones, el método incluye la administración conjunta de un agente terapéutico adicional. En algunas realizaciones, el método incluye la administración conjunta de dos agentes terapéuticos adicionales. En algunas realizaciones, la combinación del compuesto divulgado y el agente o agentes terapéuticos adicionales actúa sinérgicamente.
En algunas realizaciones, el agente terapéutico adicional se selecciona de un compuesto terapéutico inmunoestimulador. En algunas realizaciones, el compuesto terapéutico inmunoestimulador se selecciona de elotuzumab, mifamurtida, un agonista o activador de un receptor de tipo toll, o un activador de RORyt.
En algunas realizaciones, el método comprende además administrar a dicho paciente un tercer agente terapéutico, tal como un inhibidor de puntos de control inmunitario. En algunas realizaciones, el método comprende administrar al paciente que lo necesita tres agentes terapéuticos seleccionados de un compuesto divulgado en el presente documento o una sal farmacéuticamente aceptable del mismo, un compuesto terapéutico inmunoestimulador y un inhibidor de puntos de control inmunitario.
Otros inhibidores de puntos de control que pueden usarse en la presente invención incluyen agonistas de OX40. Los agonistas de OX40 que se están estudiando en ensayos clínicos incluyen PF-04518600/PF-8600 (Pfizer), un anticuerpo anti-OX40 agonista, en cáncer de riñón metastásico (NCT03092856) y cánceres y neoplasias avanzados (NCT02554812; NCT05082566); GSK3174998 (Merck & Co.), un anticuerpo anti-OX40 agonista, en ensayos de cáncer de fase 1 (NCT02528357); MEDI0562 (Medimmune/AstraZeneca), un anticuerpo anti-OX40 agonista, en tumores sólidos avanzados (NCT02318394 y NCT02705482); MEDI6469, un anticuerpo anti-OX40 agonista (Medimmune/AstraZeneca), en pacientes con cáncer colorrectal (NCT02559024), cáncer de mama (NCT01862900), cáncer de cabeza y cuello (NCT02274155) y cáncer de próstata metastásico (NCT01303705); y BMS-986178 (Bristol-Myers Squibb) un anticuerpo anti-OX40 agonista, en cánceres avanzados (NCT02737475).
Otros inhibidores de puntos de control que pueden usarse en la presente invención incluyen agonistas de CD137 (también denominado 4-1BB). Los agonistas de CD137 que se están estudiando en ensayos clínicos incluyen utomilumab (PF-05082566, Pfizer), un anticuerpo anti-CD137 agonista, en linfoma difuso de linfocitos B grandes (NCT02951156) y en cánceres y neoplasias avanzados (NCT02554812 y NCT05082566); urelumab (BMS-663513, Bristol-Myers Squibb), un anticuerpo anti-CD137 agonista, en melanoma y cáncer de piel (NCT02652455) y glioblastoma y gliosarcoma (NCT02658981).
Otros inhibidores de puntos de control que pueden usarse en la presente invención incluyen agonistas de CD27. Los agonistas de CD27 que se están estudiando en ensayos clínicos incluyen varlilumab (CDX-1127, Celldex Therapeutics) un anticuerpo anti-CD27 agonista, en cáncer de cabeza y cuello de células escamosas, carcinoma de ovario, cáncer colorrectal, cáncer de células renales y glioblastoma (NCT02335918); linfomas (NCT01460134); y glioma y astrocitoma (NCT02924038).
Otros inhibidores de puntos de control que pueden usarse en la presente invención incluyen agonistas del receptor del factor de necrosis tumoral (GITR, por sus siglas en inglés) inducido por glucocorticoides. Los agonistas de GITR que se están estudiando en ensayos clínicos incluyen TRX518 (Leap Therapeutics), un anticuerpo anti-GITR agonista, en melanoma maligno y otros tumores sólidos malignos (NCT01239134 y NCT02628574); GWN323 (Novartis), un anticuerpo anti-GITR agonista, en tumores sólidos y linfoma (NCT 02740270); INCAGN01876 (Incyte/Agenus), un anticuerpo anti-GITR agonista, en cánceres avanzados (NCT02697591 y NCT03126110); MK-4166 (Merck & Co.), un anticuerpo anti-GITR agonista, en tumores sólidos (NCT02132754) y MED11873 (Medimmune/AstraZeneca), una molécula ligando de GITR hexamérica agonista con un dominio Fc de IgG1 humana, en tumores sólidos avanzados (NCT02583165).
Otros inhibidores de puntos de control que pueden usarse en la presente invención incluyen agonistas del coestimulador de linfocitos T inducible (ICOS, también conocido como CD278). Los agonistas de ICOS que se están estudiando en ensayos clínicos incluyen MEDI-570 (Medimmune), un anticuerpo anti-ICOS agonista, en linfomas (NCT02520791); GSK3359609 (Merck & Co.), un anticuerpo anti-ICOS agonista, en fase 1 (NCT02723955); JTX-2011
(Jounce Therapeutics), un anticuerpo anti-ICOS agonista, en fase 1 (NCT02904226).
Otros inhibidores de puntos de control que pueden usarse en la presente invención incluyen inhibidores del receptor de tipo inmunoglobulina de células citolíticas naturales (KIR, por sus siglas en inglés). Los inhibidores de KIR que se están estudiando en ensayos clínicos incluyen lirilumab (IPH2102/BMS-986015, Innate Pharma/Bristol-Myers Squibb), un anticuerpo anti-KIR, en leucemias (NCT01687387, NCT02399917, NCT02481297, NCT02599649), mieloma múltiple (NCT02252263) y linfoma (NCT01592370); IPH2101 (1-7F9, Innate Pharma) en mieloma (NCT01222286 y NCT01217203); e IPH4102 (Innato Pharma), un anticuerpo anti-KIR que se une a tres dominios de la cola citoplasmática larga (KIR3DL2), en linfoma (n Ct 02593045).
Otros inhibidores de puntos de control que pueden usarse en la presente invención incluyen inhibidores de CD47 de la interacción entre CD47 y la proteína reguladora de señales alfa (SIRPa, por sus siglas en inglés). Los inhibidores de CD47/SIRPa que se están estudiando en ensayos clínicos incluyen ALX-148 (Alexo Therapeutics), una variante antagonista de (SIRPa) que se une a CD47 y evita la señalización mediada por CD47/SIRPa, en fase 1 (NCT03013218); TTI-621 (SIRPa-Fc, Terapéutica Trillium), una proteína de fusión recombinante soluble creada al unir el dominio de unión a CD47 aminoterminal de SIRPa con el dominio Fc de IgG1 humana, actúa al unirse al CD47 humano y evitar que suministre su señal de "no fagocitar" a los macrófagos, se encuentra en ensayos clínicos en fase 1 (NCT02890368 y NCT02663518); CC-90002 (Cel gene), un anticuerpo anti-CD47, en leucemias (NCT02641002); y Hu5F9-G4 (Forty Seven, Inc.), en neoplasias colorrectales y tumores sólidos (NCT02953782), leucemia mieloide aguda (NCT02678338) y linfoma (NCT02953509).
Otros inhibidores de puntos de control que pueden usarse en la presente invención incluyen inhibidores de CD73. Los inhibidores de CD73 que se están estudiando en ensayos clínicos incluyen MEDI9447 (Medimmune), un anticuerpo anti-CD73, en tumores sólidos (NCT02503774); y BMS-986179 (Bristol-Myers Squibb), un anticuerpo anti-CD73, en tumores sólidos (NCT02754141).
Otros inhibidores de puntos de control que pueden usarse en la presente invención incluyen agonistas de la proteína estimuladora de genes de interferón (STING, también conocida como proteína transmembrana 173 o TMEM173). Los agonistas de STING que se están estudiando en ensayos clínicos incluyen MK-1454 (Merck & Co.), un dinucleótido cíclico sintético agonista, en linfoma (NCT03010176); y ADU-S100 (MIW815, Aduro Biotech/Novartis), un dinucleótido cíclico sintético agonista, en fase 1 (NCT02675439 y NCT03172936).
Otros inhibidores de puntos de control que pueden usarse en la presente invención incluyen inhibidores de CSF1R. Los inhibidores de CSF1R que se están estudiando en ensayos clínicos incluyen pexidartinib (PLX3397, Plexxikon), un inhibidor de molécula pequeña CSF1R, en cáncer colorrectal, cáncer pancreático, cánceres metastásicos y avanzados (NCT02777710) y melanoma, cáncer de pulmón no microcítico, cáncer de células escamosas de cabeza y cuello, tumor del estroma gastrointestinal (GIST) y cáncer de ovario (NCT02452424); y IMC-CS4 (LY3022855, Lilly), un anticuerpo anti-CSF-1R, en cáncer de páncreas (NCT03153410), melanoma (NCT03101254) y tumores sólidos (NCT02718911); y BLZ945 (metilamida del ácido 4-[2((1R,2R)-2-hidroxiciclohexilamino)-benzotiazol-6-iloxilo]-piridin-2-carboxílico, Novartis), un inhibidor disponible por vía oral de CSF1R, en tumores sólidos avanzados (NCT02829723).
Otros inhibidores de puntos de control que pueden usarse en la presente invención incluyen inhibidores del receptor NKG2A. Los inhibidores del receptor NKG2A que se están estudiando en ensayos clínicos incluyen monalizumab (IPH2201, Innate Pharma), un anticuerpo anti-NKG2A, en neoplasias de cabeza y cuello (NCT02643550) y leucemia linfocítica crónica (NCT02557516).
En algunas realizaciones, el inhibidor de puntos de control inmunitario se selecciona de nivolumab, pembrolizumab, ipilimumab, avelumab, durvalumab, atezolizumab o pidilizumab.
En otro aspecto, la presente invención proporciona un método de tratamiento del cáncer en un paciente que lo necesita, en donde dicho método comprende administrar a dicho paciente un compuesto divulgado en el presente documento o una sal farmacéuticamente aceptable del mismo en combinación con uno o más agentes terapéuticos adicionales seleccionados de un inhibidor de indolamina (2,3)-dioxigenasa (IDO), un inhibidor de poli-ADP ribosa polimerasa (PARP), un inhibidor de histona desacetilasa (HDAC, por sus siglas en inglés), un inhibidor de CDK4/CDK6, o un inhibidor de fosfatidilinositol 3 cinasa (PI3K).
En algunas realizaciones, el inhibidor de IDO se selecciona de epacadostat, indoximod, capmanitib, GDC-0919, PF-06840003, BMS:F001287, Phy906/KD108, o una enzima que descompone la quinurenina.
En algunas realizaciones, el inhibidor de PARP se selecciona de olaparib, rucaparib, niraparib, iniparib, talazoparib o veliparib.
En algunas realizaciones, el inhibidor de HDAC se selecciona de vorinostat, romidepsina, panobinostat, belinostat, entinostat o chidamida.
En algunas realizaciones, el inhibidor de CDK 4/6 se selecciona de palbociclib, ribociclib, abemaciclib o trilaciclib.
En algunas realizaciones, el método comprende además administrar a dicho paciente un tercer agente terapéutico, tal como un inhibidor de puntos de control inmunitario. En algunas realizaciones, el método comprende administrar al paciente que lo necesita tres agentes terapéuticos seleccionados de un compuesto divulgado en el presente documento o una sal farmacéuticamente aceptable del mismo, un segundo agente terapéutico seleccionado de un inhibidor de indolamina (2,3)-dioxigenasa (IDO), un inhibidor de poli-ADP ribosa polimerasa (PARP), un inhibidor de histona desacetilasa (HDAC, por sus siglas en inglés), un inhibidor de CDK4/CDK6, o un inhibidor de fosfatidilinositol 3 cinasa (PI3K), y un tercer agente terapéutico seleccionado de un inhibidor de puntos de control inmunitario. En algunas realizaciones, el inhibidor de puntos de control inmunitario se selecciona de nivolumab, pembrolizumab, ipilimumab, avelumab, durvalumab, atezolizumab o pidilizumab.
Otro agente terapéutico inmunoestimulador que puede usarse en la presente invención es la interleucina 15 humana recombinante (rhIL-15). La rhIL-15 se ha probado en la práctica clínica como terapia para el melanoma y el carcinoma de células renales (NCT01021059 y NCT01369888) y las leucemias (NCT02689453). Otro agente terapéutico inmunoestimulador que puede usarse en la presente invención es la interleucina 12 humana recombinante (rhIL-12). Otro agente inmunoterapéutico basado en IL-15 adecuado es la IL-15 heterodimérica (hetIL-15, Novartis/Admune), un complejo de fusión compuesto por una forma sintética de IL-15 endógena en forma de complejo con la cadena alfa del receptor de IL-15 de la proteína de unión a IL-15 soluble (IL15:sIL-15RA), que se ha probado en ensayos clínicos de fase 1 para melanoma, carcinoma de células renales, cáncer de pulmón de células no microcítico y carcinoma de células escamosas de cabeza y cuello (NCT02452268). La interleucina 12 humana recombinante (rhIL-12) se ha probado en la práctica clínica para muchas indicaciones oncológicas, por ejemplo, como terapia para el linfoma (NM-IL-12, Neumedicines, Inc.), (NCT02544724 y NCT02542124).
En algunas realizaciones, el inhibidor de PI3K se selecciona de idelalisib, alpelisib, taselisib, pictilisib, copanlisib, duvelisib, PQR309 o TGR1202.
En otro aspecto, la presente invención proporciona compuestos para su uso en un método para el tratamiento de un cáncer en un paciente que lo necesita, en donde dicho método comprende administrar a dicho paciente un compuesto divulgado en el presente documento o una sal farmacéuticamente aceptable del mismo en combinación con uno o más agentes terapéuticos adicionales seleccionados de un agente terapéutico basado en platino, un taxano, un inhibidor de nucleósidos, o un agente terapéutico que interfiere con la síntesis normal de ADN, síntesis de proteínas, replicación celular, o inhibirá de otro modo las células que proliferan rápidamente.
En algunas realizaciones, el agente terapéutico basado en platino se selecciona de cisplatino, carboplatino, oxaliplatino, nedaplatino, picoplatino o satraplatino.
En algunas realizaciones, el taxano se selecciona de paclitaxel, docetaxel, paclitaxel unido a albúmina, cabazitaxel, o SID530.
En algunas realizaciones, el agente terapéutico que interfiere con la síntesis normal de ADN, síntesis de proteínas, replicación celular, o de otro modo interferirá con la replicación de células que proliferan rápidamente se selecciona de trabectedina, mecloretamina, vincristina, temozolomida, citarabina, lomustina, azacitidina, mepesuccinato de omacetaxina, asparaginasa de Erwinia chrysanthemi, mesilato de eribulina, capacetrina, bendamustina, ixabepilona, nelarabina, clorafabina, trifluridina o tipiracilo.
En algunas realizaciones, el método comprende además administrar a dicho paciente un tercer agente terapéutico, tal como un inhibidor de puntos de control inmunitario. En algunas realizaciones, el método comprende administrar al paciente que lo necesita tres agentes terapéuticos seleccionados de un compuesto divulgado en el presente documento o una sal farmacéuticamente aceptable del mismo, un segundo agente terapéutico seleccionado de un agente terapéutico basado en platino, un taxano, un inhibidor de nucleósidos, o un agente terapéutico que interfiere con la síntesis normal de ADN, síntesis de proteínas, replicación celular, o inhibirá de otro modo las células que proliferan rápidamente, y un tercer agente terapéutico seleccionado de un inhibidor de puntos de control inmunitario.
En algunas realizaciones, el inhibidor de puntos de control inmunitario se selecciona de nivolumab, pembrolizumab, ipilimumab, avelumab, durvalumab, atezolizumab o pidilizumab.
En algunas realizaciones, cualquiera de los métodos anteriores comprende además la etapa de obtener una muestra biológica del paciente y medir la cantidad de un biomarcador relacionado con la enfermedad.
En algunas realizaciones, la muestra biológica es una muestra de sangre.
En algunas realizaciones, el biomarcador relacionado con la enfermedad se selecciona de linfocitos T CD8+ circulantes o la proporción de linfocitos T Cd8+:linfocitos Treg.
En un aspecto, la presente invención proporciona compuestos para su uso en un método para tratar un cáncer avanzado, que comprende administrar un compuesto divulgado en el presente documento o una sal
farmacéuticamente aceptable del mismo o una composición farmacéutica del mismo, ya sea como agente único (monoterapia), o en combinación con un agente quimioterapéutico, un agente terapéutico dirigido, tal como un inhibidor de cinasa y/o una terapia inmunomoduladora, tal como un inhibidor de puntos de control inmunitario. En algunas realizaciones, el inhibidor de puntos de control inmunitario es un anticuerpo contra PD-1. PD-1 se une al receptor de muerte celular programada 1 (PD-1) para evitar que el receptor se una al ligando inhibidor PDL-1, anulando así la capacidad de los tumores para suprimir la respuesta inmunitaria antitumoral del hospedador.
En algunas realizaciones, el agente terapéutico adicional es un inhibidor de cinasa o un antagonista de VEGF-R. Los inhibidores de VEGF y los inhibidores de cinasa aprobados útiles en la presente invención incluyen: bevacizumab (Avastin®, Genentech/Roche) un anticuerpo monoclonal anti-VEGF; ramucirumab (Cyramza®, Eli Lilly), un anticuerpo anti-VEGFR-2 y ziv-aflibercept, también conocido como VEGF Trap (Zaltrap®; Regeneron/Sanofi). inhibidores de VEGFR, tal como regorafenib (Stivarga®, Bayer); vandetanib (Caprelsa®, AstraZeneca); axitinib (Inlyta®, Pfizer); y lenvatinib (Lenvima®, Eisai); inhibidores de Raf, tal como sorafenib (Nexavar®, Bayer AG y Onyx); dabrafenib (Tafinlar®, Novartis); y vemurafenib (Zelboraf®, Genentech/Roche); inhibidores de MEK, tal como cobimetanib (Cotellic®, Exelexis/Genentech/Roche); trametinib (Mekinist®, Novartis); inhibidores de la tirosina cinasa Bcr-Abl, tal como imatinib (Gleevec®, Novartis); nilotinib (Tasigna®, Novartis); dasatinib (Sprycel®, BristolMyersSquibb); bosutinib (Bosulif®, Pfizer); y ponatinib (Inclusig®, Ariad Pharmaceuticals); inhibidores de Her2 y EGFR, tal como gefitinib (Iressa®, AstraZeneca); erlotinib (Tarceeva®, Genentech/Roche/Astellas); lapatinib (Tykerb®, Novartis); afatinib (Gilotrif®, Boehringer Ingelheim); osimertinib (dirigido a EGFR activado, Tagrisso®, AstraZeneca); y brigatinib (Alunbrig®, Ariad Pharmaceuticals); inhibidores de c-Met y VEGFR2, tal como cabozanitib (Cometriq®, Exelexis); e inhibidores multicinasa, tal como sunitinib (Sutent®, Pfizer); pazopanib (Votrient®, Novartis); inhibidores de ALK, tal como crizotinib (Xalkori®, Pfizer); ceritinib (Zykadia®, Novartis); y alectinib (Alecenza®, Genentech/Roche); inhibidores de la tirosina cinasa de Bruton, tal como ibrutinib (Imbruvica®, Pharmacyclics/Janssen); e inhibidores del receptor Flt3, tal como midostaurina (Rydapt®, Novartis).
Otros inhibidores de cinasas y antagonistas de VEGF-R que están en desarrollo y pueden usarse en la presente invención incluyen tivozanib (Aveo Pharmaecuticals); vatalanib (Bayer/Novartis); lucitanib (Clovis Oncology); dovitinib (TKI258, Novartis); Chiauanib (Chipscreen Biosciences); CEP-11981 (Cephalon); linifanib (Abbott Laboratories); neratinib (HKI-272, Puma Biotechnology); radotinib (Supect®, IY5511, Il-Yang Pharmaceuticals, S. Korea); ruxolitinib (Jakafi®, Incyte Corporation); PTC299 (PTC Therapeutics); CP-547.632 (Pfizer); foretinib (Exelexis, GlaxoSmithKline); quizartinib (Daiichi Sankyo) y motesanib (Amgen/Takeda).
En algunas realizaciones, el agente terapéutico adicional es un inhibidor de mTOR, que inhibe la proliferación celular, angiogénesis y captación de glucosa. Los inhibidores de mTOR aprobados útiles en la presente invención incluyen everolimus (Afinitor®, Novartis); temsirolimus (Torisel®, Pfizer); y sirolimus (Rapamune®, Pfizer).
En algunas realizaciones, el agente terapéutico adicional es un inhibidor de poli ADP ribosa polimerasa (PARP). Los inhibidores de PARP aprobados útiles en la presente invención incluyen olaparib (Lynparza®, AstraZeneca); rucaparib (Rubraca®, Clovis Oncology); y niraparib (Zejula®, Tesaro). Otros inhibidores de PARp que se están estudiando que pueden usarse en la presente invención incluyen talazoparib (MDV3800/BMN 673/LT00673, Medivation/Pfizer/Biomarin); veliparib (ABT-888, AbbVie); y b Gb -290 (BeiGene, Inc.).
En algunas realizaciones, el agente terapéutico adicional es un inhibidor de la fosfatidilinositol 3 cinasa (PI3K). Los inhibidores de PI3K aprobados útiles en la presente invención incluyen idelalisib (Zydelig®, Gilead). Otros inhibidores de PI3K que se están estudiando que pueden usarse en la presente invención incluyen alpelisib (BYL719, Novartis); taselisib (GDC-0032, Genentech/Roche); pictilisib (GDC-0941, Genentech/Roche); copanlisib (BAY806946, Bayer); duvelisib (anteriormente IPI-145, Infinity Pharmaceuticals); PQR309 (Piqur Therapeutics, Suiza); y TGR1202 (anteriormente RP5230, TG Therapeutics).
En algunas realizaciones, el agente terapéutico adicional es un inhibidor del proteasoma. Los inhibidores del proteasoma aprobados útiles en la presente invención incluyen bortezomib (Velcade®, Takeda); carfilzomib (Kyprolis®, Amgen); e ixazomib (Ninlaro®, Takeda).
En algunas realizaciones, el agente terapéutico adicional es un inhibidor de histona desacetilasa (HDAC). Los inhibidores de HDAC aprobados útiles en la presente invención incluyen vorinostat (Zolinza®, Merck & Co.); romidepsina (Istodax®, Celgene); panobinostat (Farydak®, Novartis); y belinostat (Beleodaq®, Spectrum Pharmaceuticals). Otros inhibidores de HDAC que se están estudiando que pueden usarse en la presente invención incluyen entinostat (SNDX-275, Syndax Pharmaceuticals) (NCT00866333); y chidamida (Epidaza®, HBI-8000, Chipscreen Biosciences, China).
En algunas realizaciones, el agente terapéutico adicional es un inhibidor de CDK, tal como un inhibidor de CDK 4/6. Los inhibidores de CDK 4/6 aprobados útiles en la presente invención incluyen palbociclib (Ibrance®, Pfizer); y ribociclib (Kisqali®, Novartis). Otros inhibidores de CDK 4/6 que se están estudiando que pueden usarse en la presente invención incluyen abemaciclib (Ly2835219, Eli Lilly); y trilaciclib (G1T28, G1 Therapeutics).
En algunas realizaciones, el agente terapéutico adicional es un inhibidor de la indolamina (2,3)-dioxigenasa (IDO). Los
inhibidores de IDO que se están estudiando que pueden usarse en la presente invención incluyen epacadostat (INCB024360, Incyte); indoximod (NLG-8189, NewLink Genetics Corporation); capmanitib (INC280, Novartis); GDC-0919 (Genentech/Roche); PF-06840003 (Pfizer); BMS:F001287 (Bristol-Myers Squibb); Phy906/KD108 (Phytoceutica); y una enzima que descompone la quinurenina (Kynase, Kyn Therapeutics).
En algunas realizaciones, el agente terapéutico adicional es un antagonista del factor de crecimiento, tal como un antagonista del factor de crecimiento derivado de plaquetas (PDGF) o del factor de crecimiento epidérmico (EGF) o su receptor (EGFR). Los antagonistas de PDGF aprobados que pueden usarse en la presente invención incluyen olaratumab (Lartruvo®; Eli Lilly). Los antagonistas de EGFR aprobados que pueden usarse en la presente invención incluyen cetuximab (Erbitux®, Eli Lilly); necitumumab (Portrazza®, Eli Lilly), panitumumab (Vectibix®, Amgen); y osimertinib (dirigido a EGFR activado, Tagrisso®, AstraZeneca).
En algunas realizaciones, el agente terapéutico adicional es un inhibidor de aromatasa. Los inhibidores de aromatasa aprobados que pueden usarse en la presente invención incluyen exemestano (Aromasin®, Pfizer); anastazole (Arimidex®, AstraZeneca) y letrozol (Femara®, Novartis).
En algunas realizaciones, el agente terapéutico adicional es un antagonista de la vía de hedgehog. Los inhibidores de la vía de hedgehog aprobados que se pueden usar en la presente invención incluyen sonidegib (Odomzo®, Sun Pharmaceuticals); y vismodegib (Erivedge®, Genentech), ambos para el tratamiento del carcinoma de células basales.
En algunas realizaciones, el agente terapéutico adicional es un inhibidor del ácido fólico. Los inhibidores del ácido fólico aprobados útiles en la presente invención incluyen pemetrexed (Alimta®, Eli Lilly).
En algunas realizaciones, el agente terapéutico adicional es un inhibidor del receptor 4 de quimiocinas CC (CCR4). Los inhibidores de CCR4 que se están estudiando que pueden ser útiles en la presente invención incluyen mogamulizumab (Poteligeo®, Kyowa Hakko Kirin, Japón).
En algunas realizaciones, el agente terapéutico adicional es un inhibidor de isocitrato deshidrogenasa (IDH). Los inhibidores de IDH que se están estudiando que pueden usarse en la presente invención incluyen AG120 (Celgene; NCT02677922); AG221 (Celgene, NCT02677922; NCT02577406); BAY1436032 (Bayer, NCT02746081); IDH305 (Novartis, NCT02987010).
En algunas realizaciones, el agente terapéutico adicional es un inhibidor de arginasa. Los inhibidores de arginasa que se están estudiando que pueden usarse en la presente invención incluyen AEB1102 (arginasa recombinante pegilada, Aeglea Biotherapeutics), que se está estudiando en ensayos clínicos de fase 1 para leucemia mieloide aguda y síndrome mielodisplásico (NCT02732184) y tumores sólidos (NCT02561234); y CB-1158 (Calithera Biosciences).
En algunas realizaciones, el agente terapéutico adicional es un inhibidor de glutaminasa. Los inhibidores de glutaminasa que se están estudiando que pueden usarse en la presente invención incluyen CB-839 (Calithera Biosciences).
En algunas realizaciones, el agente terapéutico adicional es un anticuerpo que se une a antígenos tumorales, es decir, proteínas expresadas en la superficie celular de las células tumorales. Los anticuerpos aprobados que se unen a antígenos tumorales que pueden usarse en la presente invención incluyen rituximab (Rituxan®, Genentech/BiogenIdec); ofatumumab (anti-CD20, Arzerra®, GlaxoSmithKline); obinutuzumab (anti-CD20, Gazyva®, Genentech), ibritumomab (anti-CD20 e Yttrium-90, Zevalin®, Spectrum Pharmaceuticals); daratumumab (anti-CD38, Darzalex®, Janssen Biotech), dinutuximab (antiglicolípido GD2, Unituxin®, United Therapeutics); trastuzumab (anti-HER2, Herceptin®, Genentech); ado-trastuzumab emtansina (anti-HER2, fusionada a emtansina, Kadcyla®, Genentech); y pertuzumab (anti-HER2, Perjeta®, Genentech); y brentuximab vedotina (conjugado anti-CD30-fármaco, Adcetris®, Seattle Genetics).
En algunas realizaciones, el agente terapéutico adicional es un inhibidor de topoisomerasa. Los inhibidores de topoisomerasa aprobados útiles en la presente invención incluyen irinotecán (Onivyde®, Merrimack Pharmaceuticals); topotecán (Hycamtin®, GlaxoSmithKline). Los inhibidores de topoisomerasa que se están estudiando que pueden usarse en la presente invención incluyen pixantrona (Pixuvri®, c T i Biopharma).
En algunas realizaciones, el agente terapéutico adicional es un inhibidor de nucleósidos u otro agente terapéutico que interfieren con la síntesis normal de ADN, síntesis de proteínas, replicación celular o inhibirá de otro modo las células que proliferan rápidamente. Dichos inhibidores de nucleósidos u otros agentes terapéuticos incluyen trabectedina (agente alquilante de guanidina, Yondelis®, Janssen Oncology), mecloretamina (agente alquilante, Valchlor®, Aktelion Pharmaceuticals); vincristina (Oncovin®, Eli Lilly; Vincasar®, Teva Pharmaceuticals; Marqibo®, Talon Therapeutics); temozolomida (profármaco del agente alquilante 5-(3-metiltriazen-1-il)-imidazol-4-carboxamida (MTIC) Temodar®, Merck & Co.); inyección de citarabina (ara-C, análogo anti metabólico de citidina, Pfizer); lomustina (agente alquilante, CeeNU®, Bristol-Myers Squibb; Gleostine®, NextSource Biotechnology); azacitidina (análogo de nucleósido de pirimidina de citidina, Vidaza®, Celgene); mepesuccinato de omacetaxina (éster de cefalotaxina) (inhibidor de la síntesis de proteínas, Synribo®); Teva Pharmaceuticals); asparaginasa de Erwinia chrysanthemi (enzima para el
agotamiento de asparagina, Elspar®, Lundbeck; Erwinaze®, EUSA Pharma); mesilato de eribulina (inhibidor de microtúbulos, antimitótico basado en tubulina, Halaven®, Eisai); cabazitaxel (inhibidor de microtúbulos, antimitótico basado en tubulina, Jevtana®, Sanofi-Aventis); capacetrina (inhibidor de la timidilato sintasa, Xeloda®, Genentech); bendamustina (derivado bifuncional de mecloretamina, que se cree que forma entrecruzamientos de ADN entre cadenas, Treanda®, Cephalon/Teva); ixabepilona (análogo semisintético de la epotilona B, inhibidor de microtúbulos, antimitótico basado en tubulina, Ixempra®, Bristol-Myers Squibb); nelarabina (profármaco del análogo de desoxiguanosina, inhibidor metabólico de nucleósidos, Arranon®, Novartis); clorafabina (profármaco del inhibidor de la ribonucleótido reductasa, inhibidor competitivo de la desoxicitidina, Cloar®, Sanofi-Aventis); y trifluridina y tipiracilo (análogo de nucleósido basado en timidina e inhibidor de timidina fosforilasa, Lonsurf®, Taiho Oncology).
En algunas realizaciones, el agente terapéutico adicional es un agente terapéutico basado en platino, también conocido como platinos. Los platinos provocan el entrecruzamiento del ADN, de manera que inhiben la reparación del ADN y/o la síntesis del a Dn , principalmente en células que se reproducen rápidamente, tales como células cancerosas. Los agentes terapéuticos basados en platino aprobados que se pueden usar en la presente invención incluyen cisplatino (Platinol®, Bristol-Myers Squibb); carboplatino (Paraplatin®, Bristol-Myers Squibb; además, Teva; Pfizer); oxaliplatino (Eloxitin®Sanofi Aventis); y nedaplatino (Aqupla®, Shionogi). Otros productos terapéuticos basados en platino que se han sometido a pruebas clínicas y que pueden usarse en la presente invención incluyen picoplatino (Poniard Pharmaceuticals); y satraplatino (JM-216, Agennix).
En algunas realizaciones, el agente terapéutico adicional es un compuesto de taxano, que causa la ruptura de los microtúbulos, que son esenciales para la división celular. Los compuestos de taxano aprobados que se pueden usar en la presente invención incluyen paclitaxel (Taxol®, Bristol-Myers Squibb), docetaxel (Taxotere®, Sanofi-Aventis; Docefrez®, Sun Pharmaceutical), paclitaxel unido a albúmina (Abraxane®; Abraxis/Celgene) y cabazitaxel (Jevtana®, Sanofi-Aventis). Otros compuestos de taxano que se han sometido a pruebas clínicas y que pueden usarse en la presente invención incluyen SID530 (SK Chemicals, Co.) (NCT00931008).
En algunas realizaciones, el agente terapéutico adicional es un inhibidor de proteínas antiapoptóticas, tal como BCL-2. Los antiapoptóticos aprobados que pueden usarse en la presente invención incluyen venetoclax (Venclexta®, AbbVie/Genentech); y blinatumomab (Blincyto®, Amgen). Otros agentes terapéuticos dirigidos a proteínas apoptóticas que se han sometido a pruebas clínicas y que pueden usarse en la presente invención incluyen navitoclax (ABT-263, Abbott), un inhibidor de BCL-2 (NCT02079740).
En algunas realizaciones, la presente invención proporciona compuestos para su uso en un método para tratar el cáncer de próstata que comprende administrar a un paciente que lo necesita una cantidad eficaz de un compuesto divulgado en el presente documento o una sal farmacéuticamente aceptable del mismo o una composición farmacéutica del mismo en combinación con un agente terapéutico adicional que interfiere con la síntesis o actividad de los andrógenos. Los inhibidores de los receptores de andrógenos aprobados útiles en la presente invención incluyen enzalutamida (Xtandi®, Astellas/Medivation); inhibidores aprobados de la síntesis de andrógenos incluyen abiraterona (Zytiga®, Centocor/Ortho); antagonista del receptor de la hormona liberadora de gonadotropina aprobado (GnRH) (degaralix, Firmagon®, Ferring Pharmaceuticals).
En algunas realizaciones, el agente terapéutico adicional es un modulador selectivo de receptor de estrógenos (SERM, por sus siglas en inglés), que interfiere con la síntesis o actividad de los estrógenos. Los SERM aprobados útiles en la presente invención incluyen raloxifeno (Evista®, Eli Lilly).
En algunas realizaciones, el agente terapéutico adicional es un inhibidor de la resorción ósea. Un agente terapéutico aprobado que inhibe la resorción ósea es denosumab (Xgeva®, Amgen), un anticuerpo que se une a RANKL, evita la unión a su receptor RANK, se encuentra en la superficie de los osteoclastos, sus precursores, y células gigantes similares a osteoclastos, que media la patología ósea en tumores sólidos con metástasis óseas. Otros agentes terapéuticos aprobados que inhiben la resorción ósea incluyen bisfosfonatos, tal como el ácido zoledrónico (Zometa®, Novartis).
En algunas realizaciones, el agente terapéutico adicional es un inhibidor de la interacción entre las dos proteínas supresoras p53 primarias, MDMX y MDM2. Los inhibidores de las proteínas supresoras p53 que se están estudiando y que se pueden usar en la presente invención incluyen ALRN-6924 (Aileron), un péptido básico que se une de manera equipotente y altera la interacción de MDMX y MDM2 con p53. ALRN-6924 se está evaluando actualmente en ensayos clínicos para el tratamiento de AML, síndrome mielodisplásico avanzado (MDS) y linfoma periférico de linfocitos T (PTCL) (NCT02909972; NCT02264613).
En algunas realizaciones, el agente terapéutico adicional es un inhibidor del factor de crecimiento transformante beta (TGF-beta o TGFp). Los inhibidores de las proteínas TGF-beta que se están estudiando que pueden usarse en la presente invención incluyen NIS793 (Novartis), un anticuerpo anti-TGF-beta que se está probando en la práctica clínica para el tratamiento de varios tipos de cáncer, incluyendo cáncer de mama, pulmón, hepatocelular, colorrectal, pancreático, de próstata y renal (NCT 02947165). En algunas realizaciones, el inhibidor de las proteínas TGF-beta es fresolimumab (GC1008; Sanofi-Genzyme), que se están estudiando para melanoma (NCT00923169); carcinoma de células renales (NCT00356460); y cáncer de pulmón no microcítico (NCT02581787). Adicionalmente, en algunas
realizaciones, el agente terapéutico adicional es una trampa TGF-beta, tal como se describe en Connolly et al. (2012) Int'l J. Biological Sciences 8:964-978.
A gentes terapéuticos adm inistrados conjuntam ente adicionales - Agentes terapéuticos dirig idos y fárm acos inm unom oduladores
En algunas realizaciones, el agente terapéutico adicional se selecciona de un agente terapéutico dirigido o fármaco inmunomodulador. Las terapias adyuvantes con agentes terapéuticos dirigidos o fármacos inmunomoduladores han mostrado una eficacia prometedora cuando se administran solas, pero están limitadas por la aparición de inmunidad tumoral a lo largo del tiempo o la evasión de la respuesta inmunitaria.
En algunas realizaciones, la presente invención también proporciona un método para tratar cáncer, tal como un cáncer descrito en el presente documento, que comprende administrar a un paciente que lo necesita una cantidad eficaz de un compuesto divulgado en el presente documento o una sal farmacéuticamente aceptable del mismo o una composición farmacéutica del mismo en combinación con un agente terapéutico adicional tal como un agente terapéutico dirigido o fármaco inmunomodulador. En algunas realizaciones, el agente terapéutico inmunomodulador induce específicamente la apoptosis de las células tumorales. Los agentes terapéuticos inmunomoduladores aprobades que pueden usarse en la presente invención incluyen pomalidomida (Pomalyst®, Celgene); lenalidomida (Revlimid®, Celgene); ingenol mebutato (Picato®, LEO Pharma).
En otras realizaciones, el agente terapéutico inmunomodulador es una vacuna contra el cáncer. En algunas realizaciones, la vacuna contra el cáncer se selecciona de sipuleucel-T (Provenge®, Dendreon/Valeant Pharmaceuticals), que se ha aprobado para el tratamiento del cáncer de próstata metastásico resistente a la castración (refractario a las hormonas) asintomático o mínimamente sintomático; y talimogén laherparepvec (Imlygic®, BioVex/Amgen, anteriormente conocido como T-VEC), una terapia vírica oncolítica genéticamente modificada aprobada para el tratamiento de lesiones cutáneas, subcutáneas y ganglionares no resecables en melanoma. En algunas realizaciones, el agente terapéutico adicional se selecciona de una terapia vírica oncolítica tal como pexastimogene devacirpvec (PexaVec/JX-594, SillaJen/anteriormente Jennerex Biotherapeutics), un virus vaccinia deficiente en timidina cinasa-(TK-) modificado por ingeniería para expresar GM-CSF, para carcinoma hepatocelular (NCT02562755) y melanoma (NCT00429312); pelareorep (Reolysin®, Oncolytics Biotech), una variante del virus respiratorio entérico huérfano (reovirus) que no se replica en células que no están activadas por RAS, en numerosos cánceres, incluido el cáncer colorrectal (NCT01622543); cáncer de próstata (NCT01619813); cáncer de células escamosas de cabeza y cuello (NCT01166542); adenocarcinoma pancreático (NCT00998322); y cáncer de pulmón no microcítico (NSCLC) (NCT 00861627); enadenotucirev (NG-348, PsiOxus, anteriormente conocido como ColoAdl), un adenovirus modificadas por ingeniería para expresar un CD80 de longitud completa y un fragmento de anticuerpo específico para la proteína CD3 del receptor de linfocitos T, en cáncer de ovario (NCT02028117); tumores epiteliales metastásicos o avanzados, tal como en cáncer colorrectal, cáncer de vejiga, carcinoma de células escamosas de cabeza y cuello y cáncer de glándulas salivales (NCT02636036); ONCOS-102 (Targovax/anteriormente Oncos), un adenovirus modificado por ingeniería para expresar GM-CSF, en melanoma (NCT03003676); y enfermedad peritoneal, cáncer colorrectal o cáncer de ovario (NCT02963831); GL-ONC1 (GLV-1h68/GLV-1h153, Genelux GmbH), virus vaccinia modificados por ingeniería para expresar betagalactosidasa (beta-gal)/beta-glucoronidasa o betagal/simportador de yoduro de sodio humano (hNIS), respectivamente, se estudiaron en carcinomatosis peritoneal (NCT01443260); cáncer de las trompas de Falopio, cáncer de ovario (NCT 02759588); o CG0070 (Cold Genesys), un adenovirus modificado por ingeniería para expresar GM-CSF, en cáncer de vejiga (NCT02365818).
En algunas realizaciones, el agente terapéutico adicional se selecciona de JX-929 (SillaJen/anteriormente Jennerex Biotherapeutics), un virus vaccinia deficiente en factor de crecimiento de vaccinia y TK modificado por ingeniería para expresar citosina desaminasa, que es capaz de convertir el profármaco 5-fluorocitosina en el fármaco citotóxico 5-fluorouracilo; TG01 y TG02 (Targovax/anteriormente Oncos), agentes de inmunoterapia basados en péptidos dirigidos a mutaciones RAS difíciles de tratar; y TILT-123 (TILT Biotherapeutics), un adenovirus modificado por ingeniería denominado: Ad5/3-E2F-delta24-hTNFa-IRES-hIL20; y VSV-GP (ViraTherapeutics), un virus de la estomatitis vesicular (VSV) modificado por ingeniería para expresar la glicoproteína (GP) del virus de la coriomeningitis linfocítica (LCMV, por sus siglas en inglés), que puede modificarse por ingeniería adicionalmente para expresar antígenos diseñados para generar una respuesta de linfocitos T CD8+ específica de antígeno.
En algunas realizaciones, la presente invención comprende administrar a dicho paciente un compuesto divulgado en el presente documento o una sal farmacéuticamente aceptable del mismo en combinación con un linfocito T modificado por ingeniería para expresar un receptor de antígenos quimérico, o CAR. Los linfocitos T modificados por ingeniería para expresar dicho receptor de antígenos quimérico se denominan linfocitos CAR-T.
Se han construido CAR que consisten en dominios de unión, que pueden proceder de ligandos naturales, fragmentos variables monocatenarios (scFv) derivados de anticuerpos monoclonales específicos para antígenos de la superficie celular, fusionados con endodominios que son el extremo funcional del receptor de linfocitos T (TCR), como el dominio de señalización CD3-zeta de los TCR, que es capaz de generar una señal de activación en los linfocitos T. Al unirse al antígeno, dichos CAR se unen a vías de señalización endógenas en la célula efectora y generan señales de activación similares a las iniciadas por el complejo TCR.
Por ejemplo, en algunas realizaciones, el linfocito CAR-T es uno de las descritos en la patente de Estados Unidos 8.906.682 (junio; incorporada en el presente documento por referencia en su totalidad), que divulga linfocitos CAR-T diseñados para comprender un dominio extracelular que tiene un dominio de unión a antígeno (tal como un dominio que se une a CD19), fusionado a un dominio de señalización intracelular de la cadena zeta del complejo receptor de antígenos de linfocitos T (tal como CD3 zeta). Cuando se expresa en el linfocito T, el CAR es capaz de redirigir el reconocimiento del antígeno en función de la especificidad de unión al antígeno. En el caso de CD19, el antígeno se expresa en linfocitos B malignos. Actualmente se están realizando más de 200 ensayos clínicos que emplean CAR-T en una amplia gama de indicaciones. [https://clinicaltrials.gov/ct2/results7teriT F chimeri c+antigen+receptors&pg=1].
A gentes terapéuticos adicionales adm inistrados conjuntam ente - Fárm acos inm unoestim uladores
En algunas realizaciones, el agente terapéutico adicional es un fármaco inmunoestimulador. Por ejemplo, los anticuerpos que bloquean el eje inhibidor de PD-1 y PD-L1 pueden desencadenar linfocitos T reactivos a tumores activados y se ha demostrado en ensayos clínicos que inducen respuestas antitumorales duraderas en un número cada vez mayor de histologías tumorales, incluyendo algunos tipos de tumores que no se han considerado convencionalmente sensibles a inmunoterapia. Véanse, por ejemplo, Okazaki, T. et al. (2013) Nat. Immunol. 14, 1212 1218; Zou et al. (2016) Sci. Transl. Med. 8. El anticuerpo anti-pD-1 nivolumab (Opdivo®, Bristol-Myers Squibb, también conocido como ONO-4538, MDX1106 y BMS-936558), ha mostrado potencial para mejorar la supervivencia general en pacientes con RCC que habían experimentado progresión de la enfermedad durante o después de una terapia antiangiogénica previa.
En algunas realizaciones, la presente invención proporciona compuestos para su uso en un método para tratar cáncer, tal como un cáncer descrito en el presente documento, que comprende administrar a un paciente que lo necesita una cantidad eficaz de un compuesto divulgado en el presente documento o una sal farmacéuticamente aceptable del mismo o una composición farmacéutica del mismo en combinación con un agente terapéutico adicional tal como un fármaco inmunoestimulador, tal como un inhibidor de puntos de control inmunitario. En algunas realizaciones, el compuesto y el inhibidor de puntos de control se administran de manera simultánea o secuencial. En algunas realizaciones, un compuesto divulgado en el presente documento se administra antes de la dosificación inicial con el inhibidor de puntos de control inmunitario. En determinadas realizaciones, el inhibidor de puntos de control inmunitario se administra antes de la dosificación inicial con el compuesto divulgado en el presente documento.
En determinadas realizaciones, el inhibidor de puntos de control inmunitario se selecciona de un antagonista de PD-1, un antagonista de PD-L1 o un antagonista de CTLA-4. En algunas realizaciones, un compuesto divulgado en el presente documento o una sal farmacéuticamente aceptable del mismo se administra en combinación con nivolumab (anticuerpo anti-PD-1, Opdivo®, Bristol-Myers Squibb); pembrolizumab (anticuerpo anti-PD-1, Keytruda®, Merck & Co.); ipilimumab (anticuerpo anti-CTLA-4, Yervoy®, Bristol-Myers Squibb); durvalumab (anticuerpo anti-PD-L1, Imfinzi®, AstraZeneca); o atezolizumab (anticuerpo anti-PD-L1, Tecentriq®, Genentech).
Otros inhibidores de puntos de control inmunitarios adecuados para su uso en la presente invención incluyen REGN2810 (Regeneron), un anticuerpo anti-PD-1 probado en pacientes con carcinoma basocelular (NCT03132636); NSCLC (NCT03088540); carcinoma cutáneo de células escamosas (NCT02760498); linfoma (NCT02651662); y melanoma (NCT03002376); pidilizumab (CureTech), también conocido como CT-011, un anticuerpo que se une a PD-1, en ensayos clínicos para el linfoma difuso de linfocitos B grandes y el mieloma múltiple; avelumab (Bavencio®, Pfizer/Merck KGaA), también conocido como MSB0010718C), un anticuerpo IgG1 anti-PD-L1 totalmente humano, en ensayos clínicos para el cáncer de pulmón no microcítico, carcinoma de células de Merkel, mesotelioma, tumores sólidos, cáncer renal, cáncer de ovario, cáncer de vejiga, cáncer de cabeza y cuello y cáncer gástrico; y PDR001 (Novartis), un anticuerpo inhibidor que se une a PD-1, en ensayos clínicos para el cáncer de pulmón no microcítico, melanoma, cáncer de mama triple negativo y tumores sólidos avanzados o metastásicos. Tremelimumab (CP-675.206; Astrazeneca) es un anticuerpo monoclonal totalmente humano contra CTLA-4 que se ha estudiado en ensayos clínicos para varias indicaciones, incluyendo: mesotelioma, cáncer colorrectal, cáncer de riñón, cáncer de mama, cáncer de pulmón y cáncer de pulmón no microcítico, adenocarcinoma ductal pancreático, cáncer pancreático, cáncer de células germinales, cáncer de células escamosas de la cabeza y cuello, carcinoma hepatocelular, cáncer de próstata, cáncer de endometrio, cáncer metastásico en el hígado, cáncer de hígado, linfoma de linfocitos B grandes, cáncer de ovario, cáncer de cuello uterino, cáncer de tiroides anaplásico metastásico, cáncer urotelial, cáncer de las trompas de Falopio, mieloma múltiple, cáncer de vejiga, sarcoma de tejidos blandos y melanoma. AGEN-1884 (Agenus) es un anticuerpo anti-CTLA4 que se está estudiando en ensayos clínicos de fase 1 para tumores sólidos avanzados (NCT02694822).
Otro paradigma para la inmunoestimulación es el uso de virus oncolíticos. En algunas realizaciones, la presente invención proporciona un método para tratar a un paciente mediante la administración de un compuesto divulgado en el presente documento o una sal farmacéuticamente aceptable del mismo o una composición farmacéutica del mismo en combinación con una terapia inmunoestimuladora tal como virus oncolíticos. Los virus oncolíticos inmunoestimuladores aprobados que se pueden usar en la presente invención incluyen talimogén laherparepvec (virus del herpes simple vivo, atenuado, Imlygic®, Amgen).
En algunas realizaciones, el agente terapéutico adicional es un activador del receptor y huérfano relacionado con el
receptor de ácido retinoico (RORyt). RORyt es un factor de transcripción con funciones clave en la diferenciación y el mantenimiento de los subconjuntos efectores de tipo 17 de linfocitos T CD4+ (Th17) y CD8+ (Tc17), así como la diferenciación de subpoblaciones de células inmunitarias innatas que expresan IL-17, tal como las células NK. Un activador de RORyt, que se está estudiando y que se puede utilizar en la presente invención es LYC-55716 (Lycera), que actualmente se está evaluando en ensayos clínicos para el tratamiento de tumores sólidos (NCT02929862).
En algunas realizaciones, el agente terapéutico adicional es un agonista o activador de un receptor de tipo toll (TLR). Los activadores adecuados de TLR incluyen un agonista o activador de TLR9 tal como SD-101 (Dynavax). SD-101 es un CpG inmunoestimulador que se está estudiando para linfomas de linfocitos B, foliculares y otros linfomas (NCT02254772). Los agonistas o activadores de TLR8 que pueden usarse en la presente invención incluyen motolimod (VTX-2337, VentiRx Pharmaceuticals), que se está estudiando para el cáncer de células escamosas de cabeza y cuello (NCT02124850) y el cáncer de ovario (NCT02431559).
Otros inhibidores de puntos de control que pueden usarse en la presente invención incluyen inhibidores de la proteína 3 que contiene mucina e inmunoglobulina de linfocitos T (TIM-3). Los inhibidores de TIM-3 que pueden usarse en la presente invención incluyen TSR-022, LY3321367 y MBG453. TSR-022 (Tesaro) es un anticuerpo anti-TIM-3 que se está estudiando en tumores sólidos (NCT02817633). LY3321367 (Eli Lilly) es un anticuerpo anti-TIM-3 que se está estudiando en tumores sólidos (NCT03099109). MBG453 (Novartis) es un anticuerpo anti-TIM-3 que se está estudiando en neoplasias malignas avanzadas (NCT02608268).
Otros inhibidores de puntos de control que pueden usarse en la presente invención incluyen inhibidores de inmunorreceptores de linfocitos T con dominios Ig e ITIM, o TIGIT, un receptor inmunitario en determinados linfocitos T y células NK. Los inhibidores de TIGIT que pueden usarse en la presente invención incluyen BMS-986207 (Bristol-Myers Squibb), un anticuerpo monoclonal anti-TIGIT (NCT02913313); OMP-313M32 (Oncomed); y anticuerpo monoclonal anti-TIGIT (NCT03119428).
Los inhibidores de puntos de control que pueden usarse en la presente invención también incluyen inhibidores del gen 3 de activación de linfocitos (LAG-3). Los inhibidores de LAG-3 que pueden usarse en la presente invención incluyen BMS-986016 y REGN3767 e IMP321. BMS-986016 (Bristol-Myers Squibb), un anticuerpo anti-LAG-3, se está estudiando en glioblastoma y gliosarcoma (NCT02658981). REGN3767 (Regeneron), también es un anticuerpo anti-LAG-3 y se está estudiando en neoplasias malignas (NCT03005782). IMP321 (Immutep S.A.) es una proteína de fusión LAG-3-Ig, que se están estudiando en melanoma (NCT02676869); adenocarcinoma (NCT02614833); y cáncer de mama metastásico (NCT00349934).
Otros agentes inmunooncológicos que se pueden usar en la presente invención en combinación con un compuesto divulgado en el presente incluyen urelumab (BMS-663513, Bristol-Myers Squibb), un anticuerpo monoclonal anti-CD137; varlilumab (CDX-1127, Celldex Therapeutics), un anticuerpo monoclonal anti-CD27; BMS-986178 (Bristol-Myers Squibb), un anticuerpo monoclonal anti-OX40; lirilumab (IPH2102/BMS-986015, Innate Pharma, Bristol-Myers Squibb), un anticuerpo monoclonal anti-KIR; monalizumab (IPH2201, Innate Pharma, AstraZeneca) un anticuerpo monoclonal anti-NKG2A; andecaliximab (GS-5745, Gilead Sciences), un anticuerpo anti-MMP9; MK-4166 (Merck & Co.), un anticuerpo monoclonal anti-GITR.
Otros agentes terapéuticos adicionales que pueden usarse en la presente invención incluyen glembatumumab vedotina-monometil auristatina E (MMAE) (Celldex), un anticuerpo antiglucoproteína NMB (gpNMB) (CR011) unido a MMAE citotóxico. gpNMB es una proteína sobreexpresada por múltiples tipos de tumores asociados con la capacidad de metástasis de las células cancerosas.
Un compuesto de la presente invención también se puede usar ventajosamente en combinación con otros compuestos antiproliferativos. Dichos compuestos antiproliferativos incluyen, pero sin limitación, inhibidores de puntos de control; inhibidores de aromatasa; antiestrógenos; inhibidores de topoisomerasa I; inhibidores de topoisomerasa II; compuestos activos de microtúbulos; compuestos alquilantes; inhibidores de histona desacetilasa; compuestos que inducen procesos de diferenciación celular; inhibidores de ciclooxigenasa; inhibidores de MMP; inhibidores de mTOR; antimetabolitos antineoplásicos; compuestos de platino; compuestos que se dirigen a/disminuyen la actividad proteína o lípido cinasa y otros compuestos antiangiogénicos; compuestos que se dirigen a, disminuyen o inhiben la actividad de una fosfatasa de proteínas o lípidos; agonistas de gonadorelina; antiandrógenos; inhibidores de metionina aminopeptidasa; inhibidores de metaloproteinasa de matriz; bisfosfonatos; modificadores de la respuesta biológica; anticuerpos antiproliferativos; inhibidores de heparanasa; inhibidores de las isoformas oncogénicas de Ras; inhibidores de telomerasa; inhibidores del proteasoma; compuestos utilizados en el tratamiento de neoplasias malignas hematológicas; compuestos que se dirigen a, disminuyen o inhiben la actividad de Flt-3; inhibidores de Hsp90 tal como 17-AAG (17-alilaminogeldanamicina, NSC330507), 17-DMAG (17-dimetilaminoetilamino-17-desmetoxigeldanamicina, NSC707545), IPI-504, CNF1010, CNF2024, CNF1010 de Conforma Therapeutics; temozolomida (Temodal®); inhibidores de la proteína del huso de quinesina, tal como SB715992 o SB743921 de GlaxoSmithKline, o pentamidina/clorpromazina de CombinatoRx; inhibidores de MEK tal como ARRY142886 de Array BioPharma, AZd6244 de AstraZeneca, PD181461 de Pfizer y leucovorina.
La expresión "inhibidor de puntos de control", como se usa en el presente documento, se refiere a agentes útiles para
prevenir que las células cancerosas eviten el sistema inmunitario del paciente. Uno de los principales mecanismos de subversión de la inmunidad antitumoral se conoce como "agotamiento de linfocitos T", que es el resultado de la exposición crónica a antígenos que ha dado lugar al aumento de los receptores inhibidores. Estos receptores inhibidores sirven como puntos de control inmunitarios para prevenir reacciones inmunitarias descontroladas.
PD-1 y receptores coinhibidores como el antígeno 4 de linfocitos T citotóxicos (CTLA-4, el atenuador de linfocitos B y T (BTLA), CD272), el dominio 3 de mucina e inmunoglobulina de linfocitos T (Tim-3), el gen 3 de activación de linfocitos (Lag-3; CD223), y otros con frecuencia se denominan reguladores de puntos de control. Actúan como "guardianes" moleculares que permiten que la información extracelular dicte si la progresión del ciclo celular y otros procesos de señalización intracelular deben continuar.
En un aspecto, el inhibidor de puntos de control es un agente terapéutico biológico o una molécula pequeña. En otro aspecto, el inhibidor de puntos de control es un anticuerpo monoclonal, un anticuerpo humanizado, un anticuerpo completamente humano, una proteína de fusión o una combinación de los mismos. En un aspecto más, el inhibidor de puntos de control inhibe una proteína de puntos de control seleccionada de CTLA-4, PDL1, PDL2, PD1, B7-H3, B7-H4, BTLA, HVEM, TIM3, GAL9, LAG3, VISTA, KIR, 2B4, CD160, CGEN-15049, CHK 1, CHK2, A2aR, ligandos de la familia B-7 o una combinación de los mismos. En un aspecto adicional, el inhibidor de puntos de control interactúa con un ligando de una proteína del punto de control seleccionada de CTLA-4, PDL1, PDL2, PD1, B7-H3, B7-H4, BTLA, HVEM, TIM3, GAL9, LAG3, VISTA, KIR, 2B4, CD160, CGEN-15049, CHK 1, CHK2, A2aR, ligandos de la familia B-7 o una combinación de los mismos. En un aspecto, el inhibidor de puntos de control es un agente inmunoestimulador, un factor de crecimiento de linfocitos T, una interleucina, un anticuerpo, una vacuna o una combinación de los mismos. En un aspecto más, la interleucina es IL-7 o IL-15. En un aspecto específico, la interleucina es IL-7 glicosilada. En un aspecto adicional, la vacuna es una vacuna de células dendríticas (DC, por sus siglas en inglés).
Los inhibidores de puntos de control incluyen cualquier agente que bloquee o inhiba de manera estadísticamente significativa, las vías inhibidoras del sistema inmunitario. Dichos inhibidores pueden incluir inhibidores de molécula pequeña o pueden incluir anticuerpos, o fragmentos de unión a antígeno de los mismos, que se unen a y bloquean o inhiben los receptores de puntos de control inmunitario o los anticuerpos que se unen a y bloquean o inhiben los ligandos de receptores de puntos de control inmunitario. Las moléculas de puntos de control ilustrativas que se pueden dirigir para el bloqueo o la inhibición incluyen, pero sin limitación, CTLA-4, PDL1, PDL2, PD1, B7-H3, B7-H4, BTLA, HVEM, GAL9, LAG3, TIM3, VISTA, KIR, 2B4 (pertenece a la familia de moléculas de CD2 y se expresa en todas las NK, y6 , y linfocitos T CD8+ (ap) de memoria), CD160 (también denominado BY55), CGEN-15049, cinasas CHK 1 y CHK2, A2aR y varios ligandos de la familia B-7. Los ligandos de la familia B7 incluyen, pero sin limitación, B7-1, B7-2, B7-DC, B7-H1, B7-H2, B7-H3, B7-H4, B7-H5, B7-H6 y B7-H7. Dichos inhibidores de puntos de control pueden incluir anticuerpos, o fragmentos de unión a antígeno de los mismos, otras proteínas de unión, agentes terapéuticos biológicos o moléculas pequeñas, que se unen y bloquean o inhiben la actividad de uno o más de CTLA-4, PDL1, PDL2, PD1, BTLA, HVEM, TIM3, GAL9, LAG3, VISTA, KIR, 2B4, CD 160 y CGEN-15049. Los inhibidores de puntos de control inmunitario ilustrativos incluyen tremelimumab (anticuerpo que bloquea CTLA-4), anti-OX40, anticuerpo monoclonal PD-L1 (anti-B7-H1; MEDI4736), MK-3475 (bloqueante de PD-1), nivolumab (anticuerpo anti-PD1), CT-011 (anticuerpo anti-PD1), anticuerpo monoclonal BY55, A m p 224 (anticuerpo anti-PDL1), Bm S-936559 (anticuerpo anti-PDL1), MPLDL3280A (anticuerpo anti-PDL1), MSB0010718C (anticuerpo anti-PDL1) e ipilimumab (inhibidor de puntos de control anti-CTLA-4). Los ligandos de proteínas de puntos de control incluyen, pero sin limitación, PD-L1, PD-L2, B7-H3, B7-H4, CD28, CD86 y TIM-3.
En determinadas realizaciones, el inhibidor de puntos de control inmunitario se selecciona de un antagonista de PD-1, un antagonista de PD-L1 y un antagonista de CTLA-4. En algunas realizaciones, el inhibidor de puntos de control se selecciona del grupo que consiste en nivolumab (Opdivo®), ipilimumab (Yervoy®), y pembrolizumab (Keytruda®).
En algunas realizaciones, el inhibidor de puntos de control se selecciona del grupo que consiste en lambrolizumab (MK-3475), nivolumab (BMS-936558), pidilizumab (CT-011), AMP-224, MDX-1105, MEDI4736, MPDL3280A, BMS-936559, ipilimumab, lirlumab, IPH2101, pembrolizumab (Keytruda®) y tremelimumab.
La expresión "inhibidor de aromatasa", como se usa en el presente documento, se refiere a un compuesto que inhibe la producción de estrógenos, por ejemplo, la conversión de los sustratos androstenodiona y testosterona en estrona y estradiol, respectivamente. La expresión incluye, pero sin limitación, esteroides, especialmente atamestano, exemestano y formestano y, en particular, no esteroides, especialmente aminoglutetimida, rogletimida, piridoglutetimida, trilostano, testolactona, ketoconazol, vorozol, fadrozol, anastrozol y letrozol. El exemestano se comercializa con el nombre comercial Aromasin™. El formestano se comercializa con el nombre comercial Lentaron™. El fadrozol se comercializa con el nombre comercial Afema™. El anastrozol se comercializa con el nombre comercial Arimidex™. El letrozol se comercializa con los nombres comerciales Femara™ o Femar™. La aminoglutetimida se comercializa con el nombre comercial Orimeten™. Una combinación de la invención que comprende un agente quimioterapéutico que es un inhibidor de aromatasa es particularmente útil para el tratamiento de tumores con receptores hormonales positivos, como tumores de mama.
El término "antiestrógeno", como se usa en el presente documento, se refiere a un compuesto que antagoniza el efecto de los estrógenos a nivel de receptores de estrógenos. La expresión incluye, pero sin limitación, tamoxifeno,
fulvestrant, raloxifeno y clorhidrato de raloxifeno. El tamoxifeno se comercializa con el nombre comercial Nolvadex™. El clorhidrato de raloxifeno se comercializa con el nombre comercial Evista™. El fulvestrant se puede administrar con el nombre comercial Faslodex™. Una combinación de la invención que comprende un agente quimioterapéutico que es un antiestrógeno es particularmente útil para el tratamiento de tumores con receptores de estrógenos positivos, como tumores de mama.
El término "antiandrógeno", como se usa en el presente documento, se refiere a cualquier sustancia que sea capaz de inhibir los efectos biológicos de las hormonas androgénicas e incluye, pero sin limitación, bicalutamida (Casodex™). La expresión "agonista de la gonadorelina", como se usa en el presente documento, incluye, pero sin limitación, abarelix, goserelina y acetato de goserelina. La goserelina se puede administrar con el nombre comercial Zoladex™.
La expresión "inhibidor de topoisomerasa I", como se usa en el presente documento, incluye, pero sin limitación, topotecán, gimatecán, irinotecán, camptotecina y sus análogos, 9-nitrocamptotecina y el conjugado macromolecular de camptotecina PNU-166148. Se puede administrar irinotecán, por ejemplo, en la forma en que se comercializa, por ejemplo, con la marca comercial Camptosar™. El topotecán se comercializa con el nombre comercial Hycamptin™.
La expresión "inhibidor de topoisomerasa II", como se usa en el presente documento, incluye, pero sin limitación, las antraciclinas tales como la doxorrubicina (incluida la formulación liposómica, tal como Caelyx™), daunorrubicina, epirrubicina, idarrubicina y nemorrubicina, las antraquinonas mitoxantrona y losoxantrona, y las podofilotoxinas etopósido y tenipósido. El etopósido se comercializa con el nombre comercial Etopophos™. El tenipósido se comercializa con el nombre comercial VM 26-Bristol La doxorrubicina se comercializa con el nombre comercial Acriblastin™ o Adriamycin™. La epirrubicina se comercializa con el nombre comercial Farmorubicin™. La idarrubicina se comercializa con el nombre comercial Zavedos™. La mitoxantrona se comercializa con el nombre comercial Novantron.
La expresión "agente activo de microtúbulos" se refiere a compuestos estabilizadores de microtúbulos, desestabilizadores de microtúbulos e inhibidores de polimerización de microtubulinas que incluyen, pero sin limitación, taxanos, tales como paclitaxel y docetaxel; alcaloides de la vinca, tales como vinblastina o sulfato de vinblastina, vincristina o sulfato de vincristina y vinorelbina; discodermolidas; colchicina y epotilonas y derivados de los mismos. El paclitaxel se comercializa con el nombre comercial Taxol™. El docetaxel se comercializa con el nombre comercial Taxotere™. El sulfato de vinblastina se comercializa con el nombre comercial Vinblastin R.P™. El sulfato de vincristina se comercializa con el nombre comercial Farmistin™.
La expresión "agente alquilante" como se usa en el presente documento incluye, pero sin limitación, ciclofosfamida, ifosfamida, melfalán o nitrosourea (BCNU o Gliadel). La ciclofosfamida se comercializa con el nombre comercial Cyclostin™. La ifosfamida se comercializa con el nombre comercial Holoxan™.
La expresión "inhibidores de histona desacetilasa" o "inhibidores de HDAC" se refiere a compuestos que inhiben la histona desacetilasa y que poseen actividad antiproliferativa. Esto incluye, pero sin limitación, ácido hidroxámico de suberoilanilida (SAHA).
La expresión "antimetabolito antineoplásico" incluye, pero sin limitación, 5-fluorouracilo o 5-FU, capecitabina, gemcitabina, compuestos desmetilantes del ADN, tales como 5-azacitidina y decitabina, metotrexato y edatrexato, y antagonistas del ácido fólico tal como pemetrexed. La capecitabina se comercializa con el nombre comercial Xeloda™. La gemcitabina se comercializa con el nombre comercial Gemzar™.
La expresión "compuesto de platino" como se usa en el presente documento incluye, pero sin limitación, carboplatino, cisplatino, cisplatino y oxaliplatino. Se puede administrar carboplatino, por ejemplo, en la forma en que se comercializa, por ejemplo, con la marca comercial Carboplat™. Se puede administrar oxaliplatino, por ejemplo, en la forma en que se comercializa, por ejemplo, con marca comercial Eloxatin™.
La expresión "compuestos que se dirigen a/disminuyen una actividad proteína o lípido cinasa; o una actividad fosfatasa de proteínas o lípidos; o compuestos antiangiogénicos adicionales" como se usa en el presente documento incluye, pero sin limitación, inhibidores de la proteína tirosina cinasa y/o serina y/o treonina cinasa o inhibidores de la lípido cinasa, tales como a) compuestos que se dirigen a, disminuyen o inhiben la actividad de los receptores del factor de crecimiento derivado de plaquetas (PDGFR, por sus siglas en inglés), tales como compuestos que se dirigen a, disminuyen o inhiben la actividad de PDGFR, especialmente compuestos que inhiben el receptor de PDGF, tal como un derivado de N-fenil-2-pirimidina-amina, tales como imatinib, SU101, SU6668 y GFB-111; b) compuestos que se dirigen a, disminuyen o inhiben la actividad de los receptores del factor de crecimiento de fibroblastos (FGFR, por sus siglas en inglés); c) compuestos que se dirigen a, disminuyen o inhiben la actividad del receptor I del factor de crecimiento similar a insulina (IGF-IR), tales como compuestos que se dirigen a, disminuyen o inhiben la actividad de IGF-IR, especialmente compuestos que inhiben la actividad cinasa del receptor de IGF-I, o anticuerpos que se dirigen al dominio extracelular del receptor de IGF-I o sus factores de crecimiento; d) compuestos que se dirigen a, disminuyen o inhiben la actividad de la familia de receptores tirosina cinasa Trk o inhibidores de efrina B4; e) compuestos que se dirigen a, disminuyen o inhiben la actividad de la familia de receptores tirosina cinasa Axl; f) compuestos que se dirigen a, disminuyen o inhiben la actividad del receptor tirosina cinasa Ret; g) compuestos que se dirigen a, disminuyen o
inhiben la actividad de la familia de receptores tirosina cinasa Kit/SCFR, tales como imatinib; h) compuestos que se dirigen a, disminuyen o inhiben la actividad de la familia de receptores tirosina cinasa c-Kit, que forman parte de la familia PDGFR, tales como compuestos que se dirigen a, disminuyen o inhiben la actividad de la familia de receptores tirosina quinasa c-Kit, especialmente compuestos que inhiben el receptor c-Kit, tales como imatinib; i) compuestos que se dirigen a, disminuyen o inhiben la actividad de los miembros de la familia c-Abl, sus productos de fusión génica (por ejemplo, BCR-Abl cinasa) y mutantes, tales como compuestos que se dirigen a, disminuyen o inhiben la actividad de los miembros de la familia c-Abl y sus productos de fusión génica, tal como un derivado de N-fenil-2-pirimidina-amina, tal como imatinib o nilotinib (AMN107); PD180970; AG957; NSC 680410; PD173955 de Parke Davis; o dasatinib (BMS-354825); j) compuestos que se dirigen a, disminuyen o inhiben la actividad de los miembros de la familia de serina/treonina cinasas de proteína cinasa C (PKC) y Raf, miembros de las familias MEK, SRC, JAK/pan-JAK, FAK, PDK1, PKB/Akt, Ras/MAPK, PI3K, SYK, TYK2, BTK y TEC, y/o miembros de la familia de cinasas dependientes de ciclinas (CDK), incluidos los derivados de estaurosporina, tal como midostaurina; ejemplos de otros compuestos incluyen UCN-01, safingol, BAY 43-9006, briostatina 1, perifosina; llmofosina; RO 318220 y RO 320432; GO 6976; lsis 3521; LY333531/LY379196; compuestos de isoquinolina; FTI; PD184352 o QAN697 (un inhibidor de P13K) o AT7519 (inhibidor de CDK); k) compuestos que se dirigen a, disminuyen o inhiben la actividad de los inhibidores de proteínas tirosina cinasa, tales como compuestos que se dirigen a, disminuyen o inhiben la actividad de inhibidores de proteínas tirosina cinasa incluyen mesilato de imatinib (Gleevec™) o tirfostina tal como tirfostina A23/RG-50810; AG 99; tirfostina AG 213; tirfostina AG 1748; tirfostina AG 490; tirfostina B44; enantiómero (+) de tirfostina B44; tirfostina AG 555; AG 494; tirfostina AG 556, AG957 y adafostina (éster de adamantilo de ácido 4-{[(2,5-dihidroxifenil)metil]amino}-benzoico; NSC 680410, adafostina); 1) compuestos que se dirigen a, disminuyen o inhiben la actividad de la familia de receptores tirosina cinasa del factor de crecimiento epidérmico (EGFR1 ErbB2, ErbB3, ErbB4 como homo o heterodímeros) y sus mutantes, tales como compuestos que se dirigen a, disminuyen o inhiben la actividad de la familia del receptor del factor de crecimiento epidérmico son, especialmente, compuestos, proteínas o anticuerpos que inhiben los miembros de la familia del receptor tirosina cinasa de EGF, tales como el receptor de EGF, ErbB2, ErbB3 y ErbB4 o se unen a EGF o ligandos relacionados con EGF, CP 358774, ZD 1839, ZM 105180; trastuzumab (Herceptin™), cetuximab (Erbitux™), Iressa, Tarceva, OSI-774, C1-1033, EKB-569, GA/-2016, E1.1, E2.4, E2.5, E6.2, E6.4, E2.11, E6.3 o E7.6.3 y derivados de 7H-pirrolo-[2,3-d]pirimidina; m) compuestos que se dirigen a, disminuyen o inhiben la actividad del receptor c-Met, tales como compuestos que se dirigen a, disminuyen o inhiben la actividad de c-Met, especialmente compuestos que inhiben la actividad cinasa del receptor c-Met, o anticuerpos que se dirigen al dominio extracelular de c-Met o se unen a HGF, n) compuestos que se dirigen a, disminuyen o inhiben la actividad cinasa de uno o más miembros de la familia JAK (JAK1/JAK2/JAK3/TYK2 y/o pan-JAK), que incluyen, pero sin limitación, PRT-062070, SB-1578, baricitinib, pacritinib, momelotinib, VX-509, AZD-1480, TG-101348, tofacitinib y ruxolitinib; o) compuestos que se dirigen a, disminuyen o inhiben la actividad cinasa de la cinasa PI3 (PI3K) que incluyen, pero sin limitación, a TU-027, SF-1126, DS-7423, PBI-05204, GSK-2126458, ZSTK-474, buparlisib, pictrelisib, PF-4691502, BYL-719, dactolisib, XL-147, XL-765 e idelalisib; y; y q) compuestos que se dirigen a, disminuyen o inhiben los efectos de señalización de la proteína hedgehog (Hh) o las vías del receptor smoothened (SMO), incluyendo, pero sin limitación, ciclopamina, vismodegib, itraconazol, erismodegib e IPI-926 (saridegib).
La expresión "inhibidor de PI3K", como se usa en el presente documento, incluye, pero sin limitación compuestos que tienen actividad inhibidora contra una o más enzimas de la familia de las fosfatidilinositol-3-cinasas, incluyendo, pero sin limitación, PI3Ka, PI3Ky, PI3K6, PI3Kp, PI3K-C2a, PI3K-C2P, PI3K-C2y, Vps34, p110-a, p110-p, p110-Y, p110-5, p85-a, p85-p, p55-Y, p150, p101 y p87. Los ejemplos de inhibidores de PI3K útiles en la presente invención incluyen, pero sin limitación, ATU-027, SF-1126, DS-7423, PBI-05204, GSK-2126458, ZSTK-474, buparlisib, pictrelisib, PF-4691502, BYL-719, dactolisib, XL-147, XL-765 e idelalisib.
La expresión "inhibidor de Bcl-2", como se usa en el presente documento, incluye, pero sin limitación, compuestos que tienen actividad inhibidora contra la proteína 2 del linfoma de linfocitos B (Bcl-2), que incluyen, pero sin limitación, a BT-199, ABT-731, ABT-737, apogosipol, inhibidores de pan-Bcl-2 de Ascenta, curcumina (y análogos de la misma), inhibidores duales de Bcl-2/Bcl-xL (Infinity Pharmaceuticals/Novartis Pharmaceuticals), Genasense (G3139), HA14-1 (y análogos del mismo; véase el documento WO2008118802), navitoclax (y análogos del mismo, véase el documento US7390799), NH-1 (Universidad Farmacéutica de Shenayng), obatoclax (y análogos del mismo, véase el documento WO2004106328), S-001 (Gloria Pharmaceuticals), compuestos de la serie TW (Univ. de Michigan), y venetoclax. En algunas realizaciones, el inhibidor de Bcl-2 es un agente terapéutico de molécula pequeña. En algunas realizaciones, el inhibidor de Bcl-2 es un peptidomimético.
La expresión "inhibidor de BTK", como se usa en el presente documento, incluye, pero sin limitación, compuestos que tienen actividad inhibidora contra la tirosina cinasa de Bruton (BTK), incluyendo, pero sin limitación, AVL-292 e ibrutinib.
La expresión "inhibidor de SYK", como se usa en el presente documento, incluye, pero sin limitación, compuestos que tienen actividad inhibidora contra la tirosina cinasa del bazo (SYK), que incluyen, pero sin limitación, PRT-062070, R-343, R-333, Excellair, PRT-062607 y fostamatinib.
En los documentos WO2008039218 y WO2011090760, se pueden encontrar más ejemplos de compuestos inhibidores de BTK y afecciones que se pueden tratar con dichos compuestos en combinación con compuestos de la presente invención, cuya totalidad se incorpora en el presente documento por referencia.
En los documentos WO2003063794, WO2005007623 y WO2006078846, se pueden encontrar más ejemplos de compuestos inhibidores de SYK y afecciones que se pueden tratar con dichos compuestos en combinación con compuestos de la presente invención, cuya totalidad se incorpora en el presente documento por referencia.
En los documentos WO2004019973, WO2004089925, WO2007016176, US8138347, WO2002088112, WO2007084786, WO2007129161, WO2006122806, WO2005113554 y WO2007044729 se pueden encontrar más ejemplos de compuestos inhibidores de PI3K y afecciones que se pueden tratar con dichos compuestos en combinación con compuestos de la presente invención, cuya totalidad se incorpora en el presente documento por referencia.
En los documentos WO2009114512, WO2008109943, WO2007053452, WO2000142246 y WO2007070514, se pueden encontrar más ejemplos de compuestos inhibidores de JAK y afecciones que se pueden tratar con dichos compuestos en combinación con compuestos de la presente invención, cuya totalidad se incorpora en el presente documento por referencia.
Otros compuestos antiangiogénicos incluyen compuestos que tienen otro mecanismo para su actividad, por ejemplo, no relacionado con la inhibición de proteína o lípido cinasas, p. ej. talidomida (Thalomid™) y TNP-470.
Los ejemplos de inhibidores del proteasoma útiles para su uso en combinación con compuestos de la invención incluyen, pero sin limitación, bortezomib, disulfiram, epigalocatequin-3-galato (EGCG), salinosporamida A, carfilzomib, ONX-0912, CEP-18770 y MLN9708.
Los compuestos que se dirigen a, disminuyen o inhiben la actividad de una fosfatasa de proteínas o lípidos p. ej. inhibidores de la fosfatasa 1, fosfatasa 2A, o CDC25, tal como ácido okadaico o un derivado del mismo.
Los compuestos que inducen procesos de diferenciación celular incluyen, pero sin limitación, ácido retinoico, a-y- o 8-tocoferol o a-y- o 8-tocotrienol.
La expresión inhibidor de la ciclooxigenasa como se usa en el presente documento incluye, pero sin limitación, inhibidores de Cox-2, ácido 2-arilaminofenilacético sustituido con 5-alquilo y derivados, tal como celecoxib (Celebrex™), rofecoxib (Vioxx™), etoricoxib, valdecoxib o un ácido 5-alquil-2-arilaminofenilacético, tal como el ácido 5-metil-2-(2'-cloro-6'-fluoroanilino)fenilacético, lumiracoxib.
El término "bifosfonatos" como se usa en el presente documento incluye, pero sin limitación, ácido etridónico, clodrónico, tiludrónico, pamidrónico, alendrónico, ibandrónico, risedrónico y zoledrónico. El ácido etridónico se comercializa con el nombre comercial Didronel™. El ácido clodrónico se comercializa con el nombre comercial Bonefos™. El ácido tiludrónico se comercializa con el nombre comercial Skelid™. El ácido pamidrónico se comercializa con el nombre comercial Aredia™. El ácido alendrónico se comercializa con el nombre comercial Fosamax™. El ácido ibandrónico se comercializa con el nombre comercial Bondranat™. El ácido risedrónico se comercializa con el nombre comercial Actonel™. El ácido zoledrónico se comercializa con el nombre comercial Zometa™. La expresión "inhibidores de mTOR" se refiere a compuestos que inhiben la diana de rapamicina (mTOR) de mamíferos y que poseen actividad antiproliferativa tal como sirolimus (Rapamune®), everolimus (Certican™), CCI-779 y ABT578.
La expresión "inhibidor de heparanasa" como se usa en el presente documento se refiere a compuestos que se dirigen a, disminuyen o inhiben la degradación de heparín sulfato. La expresión incluye, pero sin limitación, PI-88. La expresión "modificador de la respuesta biológica", como se usa en el presente documento, se refiere a una linfocina o interferones.
La expresión "inhibidor de las isoformas oncogénicas de Ras", tal como H-Ras, K-Ras o N-Ras, como se usa en el presente documento se refiere a compuestos que se dirigen a, disminuyen o inhiben la actividad oncogénica de Ras; por ejemplo, un "inhibidor de farnesil transferasa" tal como L-744832, DK8G557 o R115777 (Zarnestra™). La expresión "inhibidor de telomerasa" como se usa en el presente documento se refiere a compuestos que se dirigen a, disminuyen o inhiben la actividad de telomerasa. Los compuestos que se dirigen a, disminuyen o inhiben la actividad de la telomerasa son especialmente compuestos que inhiben el receptor de la telomerasa, tal como la telomestatina.
La expresión "inhibidor de la metionina aminopeptidasa", como se usa en el presente documento, se refiere a compuestos que se dirigen a, disminuyen o inhiben la actividad de la metionina aminopeptidasa. Los compuestos que se dirigen a, disminuyen o inhiben la actividad de la metionina aminopeptidasa incluyen, pero sin limitación, bengamida o un derivado de la misma.
La expresión "inhibidor del proteasoma" como se usa en el presente documento se refiere a compuestos que se dirigen a, disminuyen o inhiben la actividad del proteasoma. Los compuestos que se dirigen a, disminuyen o inhiben la actividad del proteasoma incluyen, pero sin limitación, bortezomib (Velcade™) y MLN 341.
La expresión "inhibidor de metaloproteinasas de matriz" o (inhibidor de "MMP") como se usa en el presente documento incluye, pero sin limitación, inhibidores peptidomiméticos y no peptidomiméticos de colágeno, derivados de tetraciclina,
por ejemplo, el inhibidor peptidomimético de hidroxamato batimastat y su análogo marimastat biodisponible por vía oral (BB-2516), prinomastat (AG3340), metastat (NSC 683551) BMS-279251, BAY 12-9566, TAA211, MMI270B o AAJ996.
La expresión "compuestos usados en el tratamiento de neoplasias malignas hematológicas" como se usa en el presente documento incluye, pero sin limitación, inhibidores de tirosina cinasas de tipo FMS, que son compuestos que se dirigen a, disminuyen o inhiben la actividad de los receptores tirosina cinasa de tipo FMS (Flt-3R); interferón, 1-p-D-arabinofuransilcitosina (ara-c) y bisulfán; e inhibidores de ALK, que son compuestos que se dirigen a, disminuyen o inhiben la linfoma cinasa anaplásica.
Los compuestos que se dirigen a, disminuyen o inhiben la actividad de los receptores tirosina cinasa de tipo FMS (Flt-3R) son, especialmente compuestos, proteínas o anticuerpos que inhiben los miembros de la familia del receptor cinasa Flt-3R, tales como PKC412, midostaurina, un derivado de estaurosporina, SU11248 y MLN518.
La expresión "inhibidores de HSP90", como se usa en el presente documento, incluye, pero sin limitación, compuestos que se dirigen a, disminuyen o inhiben la actividad ATPasa intrínseca de HSP90; degradan, se dirigen a, disminuyen o inhiben las proteínas cliente de HSP90 a través de la vía del proteasoma de ubiquitina. Los compuestos que se dirigen a, disminuyen o inhiben la actividad ATPasa intrínseca de HSP90 son, especialmente, compuestos, proteínas o anticuerpos que inhiben la actividad ATPasa de HSP90, tales como 17-alilamino,17-desmetoxigeldanamicina (17AAG), un derivado de geldanamicina; otros compuestos relacionados con geldanamicina; inhibidores de radicicol y HDAC.
La expresión "anticuerpos antiproliferativos", como se usa en el presente documento, incluye, pero sin limitación, trastuzumab (Herceptin™), Trastuzumab-DMI erbitux, bevacizumab (Avastin™), rituximab (Rituxan®), PRO64553 (anti-CD40) y anticuerpo 2C4. Por anticuerpos se entiende anticuerpos monoclonales, anticuerpos policlonales, anticuerpos multiespecíficos inalterados formados a partir de al menos dos anticuerpos inalterados, y fragmentos de anticuerpos siempre que presenten la actividad biológica deseada.
Para el tratamiento de la leucemia mieloide aguda (LMA), los compuestos de la presente invención se pueden usar en combinación con terapias convencionales contra la leucemia, especialmente en combinación con terapias utilizadas para el tratamiento de la LMA. En particular, los compuestos de la presente invención se pueden administrar en combinación con, por ejemplo, inhibidores de la farnesil transferasa y/u otros fármacos útiles para el tratamiento de la LMA, tal como daunorrubicina, adriamicina, ara-C, VP-16, tenipósido, mitoxantrona, idarrubicina, carboplatino y PKC412.
Otros compuestos antileucémicos incluyen, por ejemplo, ara-C, un análogo de pirimidina, que es el derivado 2'-alfahidroxi ribosa (arabinósido) de la desoxicitidina. También se incluye el análogo purínico de hipoxantina, 6-mercaptopurina (6-MP) y fosfato de fludarabina. Los compuestos que se dirigen a, disminuyen o inhiben la actividad de inhibidores de histona desacetilasas (HDAC, por sus siglas en inglés), tales como el butirato de sodio y el ácido suberoilanilida hidroxámico (SAHA, por sus siglas en inglés), inhiben la actividad de las enzimas conocidas como histona desacetilasas. Los inhibidores específicos de HDAC incluyen MS275, SAHA, FK228 (anteriormente FR901228), tricostatina A y compuestos divulgados en el documento US 6.552.065 que incluyen, pero sin limitación, N-hidroxi-3-[4-[[[2-(2-metil-1H-indol-3-il)-etil]-amino]metil]fenil]-2E-2-propenamida, o una sal farmacéuticamente aceptable de la misma y N-hidroxi-3-[4-[(2-hidroxietil){2-(lH-indol-3-il)etil]-amino]metil]fenil]-2E-2-propenamida, o una sal farmacéuticamente aceptable de la misma, especialmente la sal de lactato. Los antagonistas de los receptores de somatostatina como se usan en el presente documento se refieren a compuestos que se dirigen a, tratan o inhiben el receptor de somatostatina tal como octreótido y SOM230. Los enfoques que dañan las células tumorales se refieren a enfoques tales como la radiación ionizante. La expresión "radiación ionizante" mencionada anteriormente y en lo sucesivo en el presente documento, significa radiación ionizante que se produce como rayos (tales como rayos X y rayos gamma) o partículas (tales como partículas alfa y beta) electromagnéticos. La radiación ionizante se suministra en, pero sin limitación, radioterapia y se conoce en la técnica. Véanse Hellman, Principles of Radiation Therapy, Cancer, in Principles and Practice of Oncology, Devita et al., Eds., 4.a edición, Vol. 1, págs. 248-275 (1993).
También se incluyen aglutinantes de EDG e inhibidores de ribonucleótido reductasa. La expresión "aglutinantes de EDG", como se usa en el presente documento, se refiere a una clase de inmunosupresores que modulan la recirculación de linfocitos, tal como FTY720. La expresión "inhibidores de ribonucleótido reductasa" se refiere a análogos de nucleósidos de pirimidina o purina que incluyen, pero sin limitación, fludarabina y/o arabinósido de citosina (ara-C), 6-tioguanina, 5-fluorouracilo, cladribina, 6-mercaptopurina (especialmente en combinación con ara-C contra la LLA) y/o pentostatina. Los inhibidores de ribonucleótido reductasa son especialmente derivados de hidroxiurea o 2-hidroxi-1H-isoindol-1,3-diona.
También se incluyen en particular aquellos compuestos, proteínas o anticuerpos monoclonales de VEGF tales como 1-(4-cloroanilino)-4-(4-piridilmetil)ftalazina o una sal farmacéuticamente aceptable de la misma, succinato de 1-(4-cloroanilino)-4-(4-piridilmetil)ftalazina; Angiostatin™; Endostatin™; amidas de ácido antranílico; ZD4190; Zd6474; SU5416; SU6668 ; bevacizumab; o anticuerpos anti-VEGF o anticuerpos antirreceptor de VEGF, tal como rhuMAb y RHUFab, aptámero de VEGF tal como Macugen; inhibidores de FLT-4, inhibidores de FLT-3, anticuerpo IgGI contra
VEGFR-2, angiozima (RPI 4610) y bevacizumab (Avastin™).
La terapia fotodinámica como se usa en el presente documento se refiere a la terapia que usa determinados productos químicos conocidas como compuestos fotosensibilizadores para tratar o prevenir cánceres. Los ejemplos de terapia fotodinámica incluyen el tratamiento con compuestos, tales como Visudyne™ y porfímero sódico.
Los esteroides angiostáticos como se usan en el presente documento se refieren a compuestos que bloquean o inhiben la angiogénesis, tales como, por ejemplo, anecortave, triamcinolona, hidrocortisona, 11 -a-epihidrocotisol, cortexolona, 17a-hidroxiprogesterona, corticosterona, desoxicorticosterona, testosterona, estrona y dexametasona.
Los implantes que contienen corticosteroides se refieren a compuestos, tales como fluocinolona y dexametasona.
Otros compuestos quimioterapéuticos incluyen, pero sin limitación, alcaloides vegetales, compuestos hormonales y antagonistas; modificadores de la respuesta biológica, preferentemente linfocinas o interferones; oligonucleótidos o derivados de oligonucleótidos antisentido; ARNhc o ARNip; o diversos compuestos o compuestos con otro mecanismo de acción o desconocido.
La estructura de los compuestos activos identificaos por los números de código, genéricos o nombres comerciales se puede adoptar a partir de la edición actual del compendio convencional "El índice Merck" o a partir de bases de datos, por ejemplo, internacional de patente (por ejemplo IMS World Publications).
Un compuesto de la presente invención también se puede usar en combinación con procesos terapéuticos conocidos, por ejemplo, la administración de hormonas o radiación. En determinadas realizaciones, un compuesto proporcionado se utiliza como radiosensibilizador, especialmente para el tratamiento de tumores que presentan poca sensibilidad a radioterapia.
Un compuesto de la presente invención se puede administrar solo o en combinación con uno o más compuestos terapéuticos diferentes, adoptando la posible terapia de combinación la forma de combinaciones fijas o la administración de un compuesto de la invención y uno o más compuestos terapéuticos diferentes que se escalonan o se administran independientemente entre sí, o la administración combinada de combinaciones fijas y uno o más compuestos terapéuticos diferentes. Un compuesto de la presente invención puede administrarse además especialmente para terapia tumoral en combinación con quimioterapia, radioterapia, inmunoterapia, fototerapia, intervención quirúrgica, o una combinación de estas. La terapia a largo plazo es igualmente posible como lo es la terapia adyuvante en el contexto de otras estrategias de tratamiento, como se describe anteriormente. Otros tratamientos posibles son la terapia para mantener el estado del paciente después de la regresión tumoral, o incluso quimioterapia preventiva, por ejemplo, en pacientes en riesgo.
Esos agentes adicionales pueden administrarse por separado a partir de una composición que contiene el compuesto de la invención, como parte de un régimen de dosificación múltiple. Como alternativa, estos agentes pueden ser parte de una forma farmacéutica unitaria, mezclados junto con un compuesto de la presente invención en una única composición. Si se administran como parte de un régimen de dosificación múltiple, los dos principios activos pueden suministrarse de forma simultánea, secuencialmente, o dentro de un periodo de tiempo entre sí, normalmente dentro de cinco horas entre sí.
Como se usa en el presente documento, el término "combinación", "combinado", y términos relacionados se refieren a la administración simultánea o secuencial de agentes terapéuticos según la presente invención. Por ejemplo, un compuesto de la presente invención se puede administrar con otro agente terapéutico de forma simultáneamente o secuencial en formas farmacéuticas unitarias individuales o conjuntamente en una única forma farmacéutica unitaria. En consecuencia, la presente invención proporciona una forma farmacéutica unitaria única que comprende un compuesto de la presente invención, un agente terapéutico adicional y un transportador, adyuvante o vehículo farmacéuticamente aceptable.
La cantidad tanto de un compuesto de la invención como de agente terapéutico adicional (en las composiciones que comprenden un agente terapéutico adicional como se describe anteriormente) que puede combinarse con los materiales transportadores para producir una forma farmacéutica única variará dependiendo del huésped tratado y del modo particular de administración. Preferentemente, las composiciones de la presente invención deben formularse de tal manera que se pueda administrar una dosis de entre 0,01 - 100 mg/kg de peso corporal/día de un compuesto de la invención.
En las composiciones que comprenden un agente terapéutico adicional, ese agente terapéutico adicional y el compuesto de la presente invención pueden actuar de manera sinérgica. Por lo tanto, la cantidad de agente terapéutico adicional en dichas composiciones será inferior al necesario en una monoterapia que utilice solamente dicho agente terapéutico. En dichas composiciones, se puede administrar una dosis de entre 0,01 - 1.000 pg/kg de peso corporal/día del compuesto terapéutico adicional.
La cantidad de agente terapéutico adicional presente en las composiciones de la presente invención no será mayor
que la cantidad que normalmente se administraría en una composición que comprende ese agente terapéutico como el único agente activo. Preferentemente, la cantidad de agente terapéutico adicional en las composiciones divulgadas en el presente documento variará de aproximadamente el 50 % al 100 % de la cantidad normalmente presente en una composición que comprende ese agente como el único agente terapéuticamente activo.
Los compuestos de la presente invención, o composiciones farmacéuticas de los mismos, también se pueden incorporar en composiciones para recubrir un dispositivo médico implantable, tal como prótesis, válvulas artificiales, injertos vasculares, endoprótesis y catéteres. Las endoprótesis vasculares, por ejemplo, se han usado para superar la reestenosis (nuevo estrechamiento de la pared del vaso tras una lesión). Sin embargo, los pacientes que usan endoprótesis u otros dispositivos implantables están en riesgo de formación de coágulos o activación de plaquetas. Estos efectos indeseados pueden prevenirse o mitigarse recubriendo previamente el dispositivo con una composición farmacéuticamente aceptable que comprende un inhibidor de cinasas. Los dispositivos implantables recubiertos con un compuesto de la presente invención son otra realización de la presente invención.
Ejemplos
Métodos de síntesis generales
Los siguientes ejemplos están destinados a ilustrar la invención y no deben interpretarse como que son limitaciones a la misma. A menos que se indique otra cosa, una o más formas tautoméricas de compuestos de los ejemplos descritos más adelante pueden prepararse in situ y/o aislarse. Todas las formas tautómeras de los compuestos de los ejemplos que se describen a continuación deben considerarse divulgadas. Las temperaturas se dan en grados centígrados. Si no se menciona lo contrario, todas las evaporaciones se realizan a presión reducida, preferentemente entre aproximadamente 15 mm de Hg y 100 mm de Hg (= 2 kPa-13,3 kPa (20-133 mbar)). La estructura de los productos finales, intermedios y materiales de partida se confirma por métodos analíticos convencionales, por ejemplo, microanálisis y características espectroscópicas, por ejemplo, EM, IR, RMN. Las abreviaturas usadas son las convencionales en la técnica.
Todos los materiales de partida, componentes básicos, reactivos, ácidos, bases, agentes deshidratantes, disolventes y catalizadores utilizados para sintetizar los compuestos de la presente invención están comercialmente disponibles o se pueden producir mediante métodos de síntesis orgánica conocidos por un experto en la materia (Houben-Weyl 4a Ed. 1952, Methods of Organic Synthesis, Thieme, Volumen 21). Además, los compuestos de la presente invención pueden producirse por métodos de síntesis orgánica conocidos para el experto en la materia que se muestran en los siguientes ejemplos.
Abreviaturas
equiv. o eq: equivalentes molares
o/n: durante la noche
ta: temperatura ambiente
UV: ultravioleta
HPLC: cromatografía líquida de alto rendimiento
Tr: tiempo de retención
CLEM o CL-EM: cromatografía líquida-espectrometría de masas
RMN: resonancia magnética nuclear
CC: cromatografía en columna
TLC: cromatografía de capa fina
sat: saturado
ac: acuoso Ac: acetilo
DCM: diclorometano
DCE: dicloroetano
DEA: dietilamina
DMF: dimetilformamida
DMSO: dimetilsulfóxido
ACN o MeCN: acetonitrilo
DIPEA: diisopropiletilamina
EA o EtOAc: acetato de etilo
BINAP: (±)-2,2'-Bis(difenilfosfino)-1,1'-binaftaleno
TEA: trietilamina
THF: tetrahidrofurano
TBS: terc-butildimetilsililo
KHMDS: hexametil disililazida potásica
Tf: trifluorometanosulfonato
Ms: metanosulfonilo
NBS: N-bromosuccinimida
EP: éter de petróleo
TFA: ácido trifluoroacético
FA: ácido fórmico
MMPP: monoperoxiftalato de magnesio
HATU: Hexafluorofosfato de 3-óxido de 1-[bis(dimetilamino)metilen]-1H-1,2,3-triazolo[4,5-b]piridinio
Cy: ciclohexilo
Tol: tolueno
DMP: Peryodinano de Dess-Martin
IBX: ácido 2-yodoxibenzoico
PMB: p-metoxibencilo
SEM: [2-(Trimetilsilil)etoxi]metilo
XPhos o X-Phos: 2-Diciclohexilfosfino-2',4',6'-triisopropilbifenilo
Información general: Todas las evaporaciones se llevaron a cabo al vacío con un evaporador rotatorio. Las muestras analíticas se secaron al vacío (1-5 mm de Hg) a ta. La cromatografía en capa fina (TLC) se realizó en placas de gel de sílice, las manchas se visualizaron mediante luz ultravioleta (214 y 254 nm). La purificación por columna y cromatografía ultrarrápida se llevó a cabo usando gel de sílice (malla 200-300). Los sistemas de solventes se reportan como mezclas por volumen. Todos los espectros de RMN 1H se registraron en un espectrómetro Bruker 400 (400 MHz). Los desplazamientos químicos 1H se informan en valores de 8 en partes por millón (ppm) con el disolvente deuterado como patrón interno. Los datos se informan de la siguiente manera: desplazamiento químico, multiplicidad (s = singlete, d = doblete, t = triplete, c = cuadruplete, a = ancho, m = multiplete), constante de acoplamiento (Hz), integración (es decir, número de protones). Los espectros CLEM se obtuvieron en un espectrómetro de masas Agilent 1200 series 6110 o 6120 con ionización por electropulverización y, excepto que se indique lo contrario, la condición general de CLEM fue la siguiente: columna Waters X Bridge C18 (50 mm*4,6 mm*3,5 pm), Caudal: 2,0 ml/min, la temperatura de la columna: 40 °C.
Ejemplo 1
Esquema sintético 1: (R)-W-(3-(4-(2-ammo-6-met¡lp¡r¡m¡dm-4-¡l)-1,4-oxazepan-3-M)-4-clorofeml)acetam¡da (6) I-10 y (S)-W-(3-(4-(2-amino-6-metilpirimidin-4-il)-1,4-oxazepan-3-il)-4-clorofenil)acetamida (7) I-15

(a) tamices mol. de 4 A, 2-cloro-5-nitrobenzaldehído, C ^C h ; (b) 2,6-lutidina, Cu(OTf)2, hexafluoroisopropanol, CH2O 2; c) Boc2O, EtaN, THF; (d) cinc, NH4C TPGS-750-M al 2 % en agua, 75 °C; (e) AcCl, EtaN, CH2Ch; (f) TFA, CH2CU (g) 2-amino-4-cloro-6-metilpirimidina, NMP, 150 °C; (h) separación quiral SFC
Formac¡ón de (+/-)-3-(2-cloro-5-n¡trofen¡l)-1,4-oxazepano (1)
A una solución de 3-(tributilestanilmetoxi)propan-1-amina (3,06 g, 8,09 mmol) en diclorometano anhidro (15 ml), se le añadió 2-cloro-5-nitrobenzaldehído (1,50 g, 8,08 mmol) seguido de tamices moleculares de 4 A. La mezcla se agitó durante una noche a temperatura ambiente, se retiró para retirar los tamices y se diluyó con diclorometano (75 ml). En un matraz separado que contenía hexafluoroisopropanol (22 ml) se añadió 2,6-lutidina (0,94 ml, 8,10 mmol) seguido de Cu(OTf)2 (2,93 g, 8,10 mmol). La mezcla se agitó durante 1 hora, después se añadió la solución de imina preparada anteriormente en una porción. La reacción se agitó durante una noche a temperatura ambiente. La mezcla se diluyó con 150 ml de 2:1 de solución acuosa saturada de NaHCO3 e hidróxido de amonio al 10 %. Después de agitar durante 20 minutos, la capa orgánica se retiró y se lavó con una solución acuosa saturada de NaHCO3, después salmuera. La capa orgánica se pasó a través de un embudo separador de fase y se concentró al vacío. La purificación por cromatografía en gel de sílice de fase inversa usando una columna ISCO - 100 gramos c18-aq - funcionando con ácido fórmico al 0,2 %/H2O y ácido fórmico al 0,2 %/CH3CN para proporcionar 700 mg del producto deseado en forma de un residuo de color naranja-rojo que se usó sin purificación adicional: RMN 1H (d6 - DMSO) 8 8,48 (d, J = 2,9 Hz, 1H), 8,11 (dd, J = 8 ,8 , 2,9 Hz, 1H), 7,73 (d, 1H), 4,38 - 4,21 (m, 1H), 3,90 - 3,66 (m, 3H), 3,34 (dd, J = 12,4, 8,5 Hz, 1H), 3,18 -2,86 (m, 2H), 1,95-1,84 (m, 2H); IEN-EM m/z calc. 256,06146, encontrado 257,13 (M+1)+; Tiempo de retención: 0,52 minutos.
Formación de 3-(2-cloro-5-nitrofenil)-1,4-oxazepano-4-carboxilato de (+ /-)-terc-butilo (2)
Una mezcla de 3-(2-cloro-5-nitro-fenil)-1,4-oxazepano (0,50 g, 1,93 mmol) y trietilamina (0,27 ml, 1,94 mmol) en THF (7 ml) se añadió di-ferc-butildicarbonato (0,42 g, 1,93 mmol). La mezcla de reacción se agitó a temperatura ambiente durante la noche. La mezcla de reacción se diluyó en una solución acuosa saturada de NH4Cl y se extrajo con diclorometano. La fase orgánica se pasó a través de un embudo separador de fase y se concentró al vacío. El residuo en bruto se purificó por cromatografía sobre gel de sílice usando una columna ISCO Gold de 40 g (gradiente de EtOAc al 0-20 %/CH2Cl2) para dar 376 mg del producto deseado en forma de un sólido de color blanquecino: IEN-EM m/z calc. 356,1139, encontrado 356,82 (M+1) ; Tiempo de retención: 0,92 minutos.
Formación de 3-(5-amino-2-clorofenil)-1,4-oxazepano-4-carboxilato de terc-butilo (3)
Una suspensión de 3-(2-cloro-5-nitro-fenil)-1,4-oxazepano-4-carboxilato de ferc-butilo (1,98 g, 5,55 mmol), NH4Cl (1,20 g, 22,43 mmol) y cinc (2,00 g, 30,58 mmol) se agitó TPGS-750-M al en 2 % en agua (50 ml). La mezcla de reacción se agitó vigorosamente y se calentó a 75 °C durante 23 horas. La mezcla se diluyó en una solución acuosa saturada de NaHCO3 y se extrajo con diclorometano. La fase orgánica se secó (MgSO4), se filtró y se concentró al vacío. El residuo en bruto se purificó por cromatografía sobre gel de sílice con una columna isco de 80 usando 10-50% (gradiente de MeOH-CH2Cl2 al 20 %/CH2Cl2) para proporcionar 2 gramos del producto deseado en forma de un sólido de color amarillo claro que se usó sin purificación adicional; IEN-EM m/z calc. 356,1139, encontrado 227,14 (M-Boc)+; Tiempo de retención: 0,64 minutos.
Formación de 3-(5-acetamido-2-clorofenil)-1,4-oxazepano-4-carboxilato de (+/-)-terc-butilo (4)
A una solución de 3-(5-amino-2-cloro-fenil)-1,4-oxazepano-4-carboxilato de ferc-butilo, 3, (0,25 g, 0,69 mmol) y trietilamina (0,15 ml, 1,04 mmol) en diclorometano (3 ml) se le añadió gota a gota una solución de cloruro de acetilo (0,05 ml, 0,75 mmol) en diclorometano (1 ml). La mezcla de reacción se agitó a temperatura ambiente durante 1 hora. La mezcla se inactivó mediante la adición de solución acuosa saturada de NaHCO3 y se extrajo dos veces con diclorometano. Las fases orgánicas combinadas se filtraron a través de un separador de fase y se concentró al vacío. El residuo en bruto se purificó por cromatografía sobre gel de sílice con una columna isco GOLD de 40 g usando un gradiente al 0-30% (MeOH-CH2Cl2 al 20 %/CH2Cl2) para proporcionar 185 mg de un sólido de color blanco, transparente mediante CLEM IEN-EM m/z calc. 368,15, encontrado 369,42 (M+1)+; Tiempo de retención: 0,8 minutos.
Formación de (+/-)-W-(4-cloro-3-(1,4-oxazepan-3-N)fenM)acetamida (5)
A una solución de 3-(5-acetamido-2-cloro-fenil)-1,4-oxazepano-4-carboxilato de ferc-butilo, 4, (0,18 g, 0,47 mmol) en diclorometano (2 ml), se le añadió ácido trifluoroacético (1,5 ml). Se agitó la mezcla de reacción a temperatura ambiente durante 30 minutos y después se concentró al vacío. El residuo se diluyó con diclorometano y se neutralizó con una solución acuosa saturada de NaHCO3. La fase orgánica se pasó a través de un separador de fase y el filtrado resultante se concentró al vacío para proporcionar 70 mg del producto en forma de un sólido de color blanco que se usó sin purificación adicional: IEN-EM m/z calc. 268,09, encontrado 269,20 (M+1)+; Tiempo de retención: 0,5 minutos; RMN 1H (400 MHz, DMSO-d6 ) 6 10,04 (s, 1H), 7,72 (d, J = 2,6 Hz, 1H), 7,64 (dd, J = 8,7, 2,7 Hz, 1H), 7,29 (d, J = 8,7 Hz, 1H), 4,18 (dd, J = 9,2, 3,1 Hz, 1H), 3,90 - 3,73 (m, 2H), 3,67 (dt, J = 11,9, 6,5 Hz, 1H), 3,20 (dd, J = 12,2, 9.1 Hz, 1H), 3,07 (s, 1H), 2,94 -2,80 (m, 1H), 2,70 -2,57 (m, 1H), 2,02 (s, 3H), 1,91 -1,80 (m, 2H).
Formación de W-(3-(4-(2-ammo-6-metMpinmidm-4-M)-1,4-oxazepan-3-M)-4-clorofenN)acetamida(R)-isómero (6) y (S)-isómero (7)
A una solución de W-[4-cloro-3-(1,4-oxazepan-3-il)fenil]acetamida, 5, (0,067 g, 0,224 mmol) en NMP (3 ml), se le añadió 4-cloro-6-metil-pirimidin-2-amina (0,040 g, 0,279 mmol). La mezcla de reacción se calentó a 150 °C durante 18 horas. La mezcla se enfrió a temperatura ambiente y se cargó directamente en una columna ISCO c18-aq de 50 g y se purificó por fase inversa ejecutándose con TFA al 0,1 %/H2O y TFA al 0,1 %/CH3CN. Las fracciones puras se concentraron al vacío. El residuo se diluyó con diclorometano, se neutralizó con una solución acuosa saturada de NaHCO3. La fase orgánica se pasó a través de un separador de fase y se concentró al vacío para proporcionar 69 mg de sólido de color naranja claro: alta temperatura (360 K) RMN 1H (d6-DMSO) 69,75 (s, 1H), 7,72 -7,39 (m, 3H), 7,43 -7,18 (m, 1H), 5,64 - 5,19 (m, 3H), 4,85 - 4,45 (m, 1H), 4,31 - 4,07 (m, 1H), 4,04 - 3,84 (m, 1H), 3,80 - 3,28 (m, 3H), 3.02 (s, 3H), 2,00 (s, 3H), 1,91-1,72 (m, 2H); IEN-EM m/z calc. 375,15, encontrado 376,31 (M+1)+; Tiempo de retención: 0,56 minutos. La mezcla racémica se sometió a purificación SFC quiral para obtener los enantiómeros individuales.
Pico A (jR)-W-[3-[4-(2-ammo-6-metM-pinmidm-4-M)-1,4-oxazepan-3-M]-4-cloro-feml]acetamida: alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6 ) 69,70 (s, 1H), 7,56 (dd, J = 8 ,6 , 2,6 Hz, 1H), 7,50 (d, J = 2,6 Hz, 1H), 7,31 (d, J = 8,7 Hz, 1H, 5,55 (s, 1H), 5,44 (s, 2H), 5,34 (s, 1H), 4,67 (d, J = 15,1 Hz, 1H), 4,13 (dd, J = 13,5, 5,0 Hz, 1H), 3,92 - 3,85 (m, 1H), 3,67 (dd, J = 13,5, 10,2 Hz, 1H), 3,63 - 3,49 (m, 1H), 3,40 (c, J = 7,0 Hz, 1H), 1,99 (d, J = 4,4 Hz, 6 H), 1,84-1,73 (m, 2H); IEN-EM m/z calc. 375,14, encontrado 376,27 (M+1)+; Tiempo de retención: 0,56 minutos.
Pico B (S)-W-[3-[4-(2-ammo-6-metM-pinmidm-4-M)-1,4-oxazepan-3-M]-4-cloro-fenil]acetamida: alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6 ) 69,71 (s, 1H), 7,56 (dd, J = 8 ,6 , 2,6 Hz, 1H), 7,50 (d, J = 2,6 Hz, 1H), 7,31 (d, J =
8,7 Hz, 1H, 5,55 (s, 1H), 5,44 (s, 2H), 5,34 (s, 1H), 4,67 (d, J = 15,1 Hz, 1H), 4,13 (dd, J = 13,5, 5,0 Hz, 1H), 3,95 -3,86 (m, 1H), 3,67 (dd, J = 13,5, 10,2 Hz, 1H), 3,63 - 3,49 (m, 1H), 3,40 (c, J = 7,0 Hz, 1H), 1,99 (d, J = 4,4 Hz, 6 H), 1,84-1,73 (m, 2H); IEN-EM m/z calc. 375,14, encontrado 376,27 (M+1)+; Tiempo de retención: 0,56 minutos. [a]D = 41,2°.
Los siguientes análogos se prepararon de acuerdo con el Esquema sintético 1:

(+/-)-W-(4-(4-(2-ammo-6-metNpirimidm-4-N)-1,4-oxazepan-3-M)-3-clorofeml)acetamida I-141
alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6 ) 69,77 (s, 1H), 7,77 (s, 1H), 7,35 (m, 1H), 7,21 (d, J = 8,0 Hz, 1H), 5,56 (s, 1H), 5,43 (s, 2H), 5,36-5,30 (m, 1H), 4,63-4,59 (m, 1H), 4,05 (m, 1H), 3,87 (m, 1H), 3,75-3,48 (m, 3H), 2,02 (s, 3H), 2,00 (s, 3H), 1,78-1,74 (m, 2H). IEN-EM m/z calc. 375,15, encontrado 376,27 (M+1)+; Tiempo de retención: 0,55 minutos.
El material racémico se sometió a separación quiral SFC.
Pico A: (R)-A/-(4-(4-(2-amino-6-metilpirimidin-4-il)-1,4-oxazepan-3-il)-3-clorofenil)acetamida (8) [a]D = -19,49 (c = 4,1 mg/0,8 ml de MeOH); alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6 ) 69,83 (s, 1H), 7,99 -7,73 (m, 1H), 7,54 - 7,33 (m, 1H), 7,34 - 7,12 (m, 1H), 5,83 - 5,23 (m, 4H), 4,88 - 4,54 (m, 1H), 4,28 - 4,02 (m, 1H), 4,07 - 3,85 (m, 1H), 3,85 - 3,45 (m, 3H), 2,20 - 1,93 (m, 6 H), 1,95-1,65 (m, 2H); IEN-EM m/z calc. 375,15, encontrado 376,31 (M+1)+; Tiempo de retención: 0,55 minutos. I-310
Pico B: (S)-A/-(4-(4-(2-amino-6-metilpirimidin-4-il)-1,4-oxazepan-3-il)-3 clorofenil)acetamida (9) [a]D = 13,75 (c = 4,3 mg/0,8 ml de MeOH); alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6 ) 69,79 (s, 1H), 7,77 (d, J = 2,2 Hz, 1H), 7,35 (dd, J = 8,7, 2,1 Hz, 1H), 7,21 (d, J = 8,5 Hz, 1H), 5,61 (d, J = 39,5 Hz, 1H), 5,46 (s, 2H), 5,34 (s, 1H), 4,62 (s, 1H), 4,08 (dd, J = 13,4, 5,2 Hz, 1H), 3,89 (d, J = 12,3 Hz, 1H), 3,78 - 3,47 (m, 3H), 2,01 (d, J = 8,1 Hz, 6 H), 1,83-1,46 (m, 2H); iEn -EM m/z calc. 375,15, encontrado 376,31 (M+1)+; Tiempo de retención: 0,55 minutos. I-162

(S)-W-(4-(4-(2-ammo-6-metMp¡r¡m¡dm-4-M)-1,4-oxazepan-3-M)-3-clorofeml)propionam¡da (10) I-204
Pico B a partir de la separación quiral SFC: 99,8 % ee; RMN 1H (400 MHz, MeOD) 6 7,83 (s, 1H), 7,40 (dd, J = 8,5, 2,1 Hz, 1H), 7,23 (d, J = 8,5 Hz, 1H), 6,01-4,90 (s a, 3H), 4,31 -4,23 (m, 1H), 4,04 (d, J = 8,4 Hz, 1H), 3,79 -3,58 (m, 3H), 2,39 (c, J = 7,6 Hz, 2H), 2,10 (s, 3H), 2,00 - 1,78 (m, 2H), 1,24 -1,16 (m, 3H). IEN-EM m/z calc. 389,2, encontrado 390,4 (M+1)+; Tiempo de retención: 0,58 minutos.

(S)-N-(4-(4-(2-ammo-6-metilpirímidm-4-M)-1,4-oxazepan-3-M)-3-clorofeml)propionamida (11) I-207
Pico B a partir de la separación quiral SFC: 99,8 % ee; se calentó (360K) RMN 1H (400 MHz, d6-DMSO) 6 10,03 (s, 1H), 7,80 (d, J = 2,0 Hz, 1H), 7,37 (dd, J = 8,5, 2,1 Hz, 1H), 7,21 (d, J = 8,5 Hz, 1H), 5,56 (s, 1H), 5,46 (s, 2H), 5,33 (s, 1H), 4,63 (d, J = 14,1 Hz, 1H), 4,08 (dd, J = 13,5, 5,0 Hz, 1H), 3,89 (d, J = 12,0 Hz, 1H), 3,70 (dd, J = 13,4, 10,2 Hz, 1H), 3,66 - 3,50 (m, 2H), 2,00 (s, 3H), 1,76-1,72 (m, 3H), 0,83-0,75 (m, 4H); IEN-EM m/z calc. 401,2, encontrado 402,3 (M+1)+; Tiempo de retención: 0,59 minutos.

(+/-)-W-[4-[4-(2-ammo-6-metN-pinmidm-4-M)-1,4-oxazepan-3-M]-3-cloro-feml]-2-metoxi-acetamida (12) I-192
Se calentó (360K) RMN 1H (400 MHz, MeOD) 87,74 (s, 1H), 7,35 (d, J = 8,5 Hz, 1H), 7,12 (d, J = 8,5 Hz, 1H), 5,50 (s a, 3H), 4,14 (dd, J = 13,6, 5,1 Hz, 1H), 3,91 (s, 3H), 3,68 - 3,43 (m, 3H), 3,35 (s, 3H), 1,97 (s, 3H), 1,75 (m, 2H); IEN-EM m/z calc. 405,2, encontrado 406,3 (M+1)+; Tiempo de retención: 0,56 minutos.

(S)-W-(4-(4-(2-ammo-6-metMpinmidm-4-M)-1,4-oxazepan-3-M)-3-clorofeml)propionamida (13) I-197
Pico B a partir de la separación quiral SFC: 99,4 % ee; RMN 1H (400 MHz, MeOD) 87,75 (s, 1H), 7,38 -7,31 (m, 1H), 7,12 (d, J = 8,5 Hz, 1H), 5,45 (s, 3H), 4,15 (dd, J = 13,8, 4,9 Hz, 1H), 3,91 (d, J = 9,0 Hz, 1H), 3,68 -3,45 (m, 3H), 1,97 (s, 3H), 1,75 (dd, J = 41,1, 11,5 Hz, 2H), 1,17 (dd, J = 7,7, 4,5 Hz, 2H), 0,94 (dd, J = 7,7, 4,5 Hz, 2H); IEN-EM m/z calc.
417,2, encontrado 418,3 (M+1)+; Tiempo de retención: 0,56 minutos.

(+/-)-W-[5-[4-(2-ammo-6-metN-pinmidm-4-M)-1,4-oxazepan-3-M]-4-cloro-2-fluorofeml]acetamida (14) I-85
RMN 1H (300 MHz, CDCla) 8 8,36 (d, J = 8,2 Hz, 1H), 7,51 (s, 1H), 7,18 (dd, J = 19,2, 10,3 Hz, 1H), 6,02 - 5,77 (m, 1H), 5,28 - 5,09 (m, 1H), 4,28 (dt, J = 13,7, 5,1 Hz, 1H), 4,20 - 3,95 (m, 2H), 3,84 - 3,49 (m, 4H), 2,42 - 2,27 (m, 3H), 2,22 (d, J = 0,9 Hz, 3H), 2,03-1,82 (m, 2H); IEN-EM m/z calc. 393,1, encontrado 394,1 (M+1)+; Tiempo de retención: 0,59 minutos. El material racémico se sometió a separación quiral SFC. Condiciones: Columna 20 x250 mm IC, fase móvil: MeOH al 40 % (amoniaco 5 mM), CO2 al 60 %
Pico A: A/-[5-[(3R)-4-(2-amino-6-metil-pinmidin-4-il)-1,4-oxazepan-3-il]-4-cloro-2-fluoro-fenil]acetamida (15): RMN 1H (300 MHz, Metanol-d4) 8 8,00 (d, J = 8,2 Hz, 1H), 7,33 (d, J = 10,4 Hz, 1H), 5,65 (s, 2H), 4,28 (dd, J = 13,6, 5,1 Hz, 1H), 4,05 (dd, J = 12,0, 4,5 Hz, 1H), 3,88 - 3,54 (m, 3H), 2,15 (s, 3H), 2,12 (s, 3H), 1,90 (d, J = 18,8 Hz, 2H); IEN-EM m/z calc. 393,1, encontrado 394,1 (M+1)+; Tiempo de retención: 0,59 minutos; Rotación óptica: 5 mg/1 ml de MeOH, C=1, [a] = -62,24°. I-270
Pico B: A/-[5-[(3S)-4-(2-amino-6-metil-pinmidin-4-il)-1,4-oxazepan-3-il]-4-cloro-2-fluoro-fenil]acetamida (16): RMN 1H (300 MHz, Metanol-d4) 8 8,00 (d, J = 8,2 Hz, 1H), 7,33 (d, J = 10,4 Hz, 1H), 5,65 (s, 2H), 4,28 (dd, J = 13,6, 5,1 Hz, 1H), 4,05 (dd, J = 12,0, 4,5 Hz, 1H), 3,88 - 3,54 (m, 3H), 2,15 (s, 3H), 2,12 (s, 3H), 1,90 (d, J = 18,8 Hz, 2H); IEN-EM m/z calc. 393,1, encontrado 394,2 (M+1)+; Rotación óptica: 5 mg/1 ml de MeOH, C=1, [a] = 59,6°. I-271
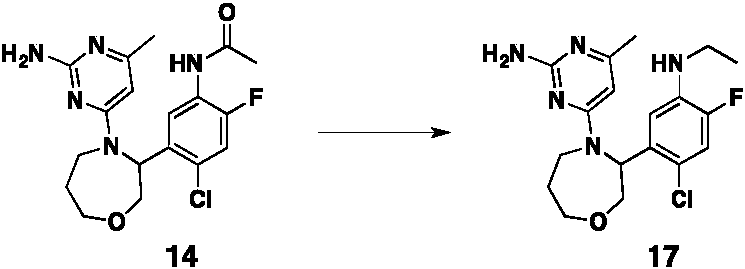
(+/-)-4-[3-[2-cloro-5-(etilamino)-4-fluoro-fenil]-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (17) I-272
A una solución de W-[5-[4-(2-amino-6-metil-pirimidin-4-il)-1,4-oxazepan-3-il]-4-cloro-2-fluoro-fenil]acetamida, 14, (0,05 g, 0,12 mmol) en tetrahidrofurano (5 ml), se le añadió hidruro de litio y aluminio (0,08 ml de 2 M, 0,16 mmol) en THF. La solución turbia se agitó a temperatura ambiente durante una noche. Se añadió más hidruro de litio y aluminio (0,10 ml) y la mezcla de reacción se calentó a 60 °C durante una noche. La mezcla se diluyó con agua (0,25 ml) y se agitó durante 10 minutos. Se añadió diclorometano (10 ml) y el sólido resultante de color blanco se filtró y se lavó con
diclorometano. Las fases orgánicas combinadas se concentraron al vacío. El residuo resultante se purificó por cromatografía sobre gel de sílice usando una columna ISCO de 4 g eluyendo un gradiente de MeOH al 0 10 %/diclorometano para proporcionar el producto deseado en forma de un sólido de color blanco: RMN 1H (300 MHz, cloroformo-d) 57,02 (d, J = 10,9 Hz, 1H), 6,45 (d, J = 9,0 Hz, 1H), 5,56 (s, 1H), 4,95 (s, 3H), 4,30 (dd, J = 13,6, 5,0 Hz, 1H), 4,10 (dd, J = 10,8, 6,6 Hz, 1H), 3,83 - 3,39 (m, 5H), 3,10 (cd, J = 7,1, 5,2 Hz, 2H), 2,17 (s, 3H), 2,09 - 1,91 (m, 1H), 1,89 - 1,76 (m, 1H), 1,24 (d, J = 7,2 Hz, 3H); IEN-EM m/z calc. 379,2, encontrado 379,8 (M+1)+; Tiempo de retención: 0,66 minutos.
Ejemplo 2
Esquema sintético 2: (+/-)-4-(3-(2-cloro-4-(metilsulfonil)fenil)-1,4-oxazepan-4-il)-6-metilpirimidin-2-amina (19) I-66

(a) tamices mol. de 4 A, 2-cloro-4-(metilsulfonil)benzaldehído, CH2Ch; (b) 2,6-lutidina, Cu(OTf)2, hexafluoroisopropanol, CH2G 2; c) 2-amino-4-cloro-6-metilpirimidina, NMP, 150 °C; (d) separación quiral SFC
Formación de (+/-)-3-(2-cloro-4-(metilsulfonil)fenil)-1,4-oxazepano (18)
A una solución de 3-(tributilestanilmetoxi)propan-1-amina (2,69 g, 7,11 mmol) en diclorometano (11 ml), se le añadió 2-cloro-5-metilsulfonil-benzaldehído (1,00 g, 4,57 mmol) seguido de tamices moleculares de 4 angstrom. La mezcla se agitó durante 14 h, se filtró para retirar los tamices y se filtró y se diluyó con diclorometano (50 ml).
En un matraz separado que contenía hexafluoroisopropanol (15 ml), se añadió 2,6-lutidina (0,53 ml, 4,58 mmol) seguido de Cu(OTf)2 (1,65 g, 4,56 mmol). La mezcla se agitó durante 1 h, después se añadió la solución de imina preparada anteriormente en una porción. La reacción se agitó durante una noche a temperatura ambiente. La mezcla se diluyó con una mezcla 2:1 de una solución acuosa saturada de NaHCO3 e hidróxido de amonio al 10 %. Después de agitar durante 10 minutos, la capa orgánica se retiró y se lavó con una solución acuosa saturada de NaHCO3, después salmuera. La capa orgánica se pasó a través de un embudo separador de fase y el filtrado se concentró al vacío. El residuo se purificó por cromatografía de fase inversa usando una columna ISCO -100 gramos c18-aq -ejecutándose con un gradiente de ácido fórmico/H2O y ácido fórmico/CH3CN. El residuo se diluyó con diclorometano, se neutralizó con una solución acuosa saturada de NaHCO3. La fase orgánica se pasó a través de un separador de fase y se concentró al vacío para proporcionar 688 mg del producto deseado: RMN 1H (400 MHz, DMSO-d6 ) 57,93 (dd, J = 1,6, 0,7 Hz, 1H), 7,90 - 7,87 (m, 2H), 4,31 (dd, J = 8,7, 3,3 Hz, 1H), 3,92 - 3,77 (m, 2H), 3,71 (dt, J = 12,2, 6,2 Hz, 1H), 3,35 -3,27 (m, 1H), 3,26 (s, 3H), 3,10 (dt, J = 13,7, 5,1 Hz, 1H), 2,89 (dt, J = 13,3, 6,4 Hz, 2H), 1,93 -1,81 (m, 2 H). Ie N-Em m/z calc. 289,05396, encontrado 290,05 (M+1 )+; Tiempo de retención: 0,5 minutos
Formación de (R)-4-(3-(2-cloro-4-(metilsulfonil)fenil)-1,4-oxazepan-4-il)-6-metilpirimidin-2-amina (20) I-67 y (S)-4-(3-(2-cloro-4-(metilsulfonil)fenil)-1,4-oxazepan-4-il)-6-metilpirimidin-2-amina (21) I-68
A una solución de 3-(2-cloro-4-(metilsulfonil)fenil)-1,4-oxazepano, 18, (0,67 g, 2,31 mmol) en NMP (7,5 ml), se le añadió 4-cloro-6-metil-pirimidin-2-amina (0,40 g, 2,79 mmol). La mezcla de reacción se calentó a 150 °C durante una noche. La mezcla se enfrió a temperatura ambiente y se cargó directamente en una columna ISCO c18-aq de 100 g y se purificó por fase inversa ejecutándose con TFA al 0,1 %/H2O y TFA al 0,1 %/CH3CN. Las fracciones puras se concentraron al vacío. El residuo resultante se diluyó con diclorometano, se neutralizó con una solución acuosa saturada de NaHCO3. La mezcla se pasó a través de un separador de fase y la fase orgánica se concentró al vacío para proporcionar 550 mg del producto deseado. La mezcla racémica se sometió a purificación quiral SFC para proporcionar 155 mg del estereoisómero A y 153 mg del estereoisómero B:
Pico A: (R)-4-(3-(2-cloro-4-(metilsulfonil)fenil)-1,4-oxazepan-4-il)-6-metilpirimidin-2-amina (20), se calentó RMN 1H (360K) (400 MHz, DMSO-d6 ) 57,97 (t, J = 1,9 Hz, 1H), 7,84 (dt, J = 8,2, 1,9 Hz, 1H), 7,60 (dd, J = 8,1, 2,0 Hz, 1H), 5,78 -5,65 (m, 1H), 5,65 -5,51 (m, 1H), 5,46 (s, 2H), 4,60 -4,41 (m, 1H), 4,25 -4,07 (m, 1H), 4,02 -3,87 (m, 1H), 3,85 - 3,68 (m, 2H), 3,68 - 3,44 (m, 1H), 3,24 (s, 3H), 2,10 - 1,99 (m, 3H), 1,89-1,70 (m, 2H); IEN-EM m/z calc. 396,10, encontrado 397,16 (M+1 )+; Tiempo de retención: 0,57 minutos; [□ p = -71,67 (c = 5,4 mg/1,5 ml de MeOH). I-67
Pico B: (S)-4-(3-(2-cloro-4-(metilsulfonil)fenil)-1,4-oxazepan-4-il)-6-metilpirimidin-2-amina (21), se calentó RMN 1H (360K) (400 MHz, DMSO-d6 ) 57,94 (d, J = 2,0 Hz, 1H), 7,80 (dd, J = 8,1, 2,0 Hz, 1H), 7,56 (d, J = 8,2 Hz, 1H), 5,72 -5,63 (m, 1H), 5,63 -5,52 (m, 1H), 5,43 (s, 2H), 4,58 -4,37 (m, 1H), 4,14 (dd, J = 13,5, 4,8 Hz, 1H), 3,97 -3,85 (m, 1H), 3,85 - 3,64 (m, 2H), 3,57 (dt, J = 12,4, 7,4 Hz, 1H), 3,21 (s, 3H), 2,03 (s, 3H), 1,90-1,70 (m, 2H); IEN-EM m/z calc.
396,10, encontrado 397,16 (M+1 )+; Tiempo de retención: 0,56 minutos; [□ p = 58,36 (c = 5,3 mg/1,5 ml de MeOH). I-68
Los siguientes análogos estaban de acuerdo con el Esquema sintético 2:

(R)-4-(2-(2-fluoro-5-metoxifenM)azepan-1-M)-6-metMpirimidm-2-amma (22) I-26 y (S)-4-(2-(2-fluoro-5-metoxifenil)azepan-1-il)-6-metilpirimidin-2-amina (23) I-27
La mezcla racémica se sintetizó de la misma manera y después se sometió a purificación SFC quiral para obtener los enantiómeros individuales:
Pico A; 98,6 % ee; alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6 ) 67,10 - 6,94 (m, 2H), 6,84 (dd, J = 9,5, 2.8 Hz, 1H), 5,59 (s, 1H), 5,54 -5,29 (m, 3H), 4,70 -4,47 (m, 1H), 4,16 (dd, J = 13,2, 5,1 Hz, 1H), 3,99 -3,77 (m, 4H ), 3,72 -3,40 (m, 3H ), 2,00 (s, 3H), 1,86-1,61 (m, 2H); [□ f = -34,12 (c = 19 mg/ 3 ml de MeOH). I-26
Pico B; 97,4 % ee; alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6 ) 67,08 - 6,93 (m, 2H), 6,84 (dd, J = 9,4, 2.8 Hz, 1H), 5,59 (s, 1H), 5,48 (s, 2H), 5,44 - 5,31 (m, 1H), 4,68 - 4,50 (m, 1H), 4,16 (dd, J = 13,3, 5,1 Hz, 1H), 3,96 -3,77 (m, 4H), 3,72 - 3,38 (m, 3H), 2,00 (s, 3H), 1,89-1,60 (m, 2H); [□ f = 40,44 (c = 19 mg/ 3 ml de MeOH); IEN-EM m/z calc. 333,21, encontrado 333,18 (M+1)+; Tiempo de retención: 0,61 minutos. I-27

4-(3-(2-cloro-5-nitrofenil)-1,4-oxazepan-4-il)-6-metilpirimidin-2-amina (24) I-72
alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6 ) 68,14 - 8,01 (m, 2H), 7,73 (d, J = 8,7 Hz, 1H), 5,73 (s, 1H), 5,68 - 5,53 (m, 1H), 5,44 (s, 2H), 4,56 -4,39 (m, 1H), 4,12 (dd, J = 13,6, 4,8 Hz, 1H), 3,95 - 3,69 (m, 3H), 3,68 - 3,53 (m, 1H), 2,04 (s, 3H), 1,85-1,73 (m, 2H); IEN-EM m/z calc. 363,11, encontrado 364,16 (M+1)+; Tiempo de retención: 0,61 minutos.

(+/-)-4-[3-(2-cloro-4-nitro-fenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (25) I-125
alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6 ) 68,22 (d, J = 2,3 Hz, 1H), 8,08 (dd, J = 8 ,6 , 2,4 Hz, 1H), 7,58 (d, J = 8,6 Hz, 1H), 5,69 (s, 1H), 5,59 (dd, J = 9,9, 4,7 Hz, 1H), 5,41 (s, 2H), 4,46 (d, J = 15,7 Hz, 1H), 4,14 (dd, J = 13,5, 4,8 Hz, 1H), 3,90 (dt, J = 11,5, 3,6 Hz, 1H), 3,77 (ddd, J = 16,3, 13,0, 8,2 Hz, 3H), 3,63 - 3,53 (m, 1H), 2,03 (s, 3H), 1,85-1,77 (m, 2H); IEN-EM m/z calc. 363,11, encontrado 364,25 (M+1)+; Tiempo de retención: 0,6 minutos.

(+/-)-4-[3-(6-cloro-1,3-benzodioxol-5-il)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (26) I-113
Se calentó (360K) RMN 1H (400 MHz, DMSO-d6 ) 67,00 (s, 1H), 6,79 (s, 1H), 6,09 (s, 2H), 6,01 (s, 2H), 5,73 (s, 1H), 5,37 (s, 1H), 4,68 - 4,50 (m, 1H), 4,05 (dd, J = 13,6, 4,9 Hz, 1H), 3,93 - 3,51 (m, 4H), 2,09 (s, 3H), 1,78 (p, J = 4,5,
3,9 Hz, 2H); IEN-EM m/z calc. 362,1, encontrado 363,0 (M+1)+; Tiempo de retención: 0,71 minutos.

(+/-)-4-(3-(2-cloro-6-fluorofenil)-1,4-oxazepan-4-il)-6-metilpirimidin-2-amina (27) I-59
RMN 1H (400 MHz, DMSO-d6 ) (se calentó 360K) 67,30 (td, J = 3,8, 2,7 Hz, 2H), 7,10 (ddd, J = 11,3, 6,1, 3,4 Hz, 1H), 5,71 (s, 1H), 5,54 (dd, J = 10,6, 5,6 Hz, 1H), 5,41 (s, 2H), 4,48 (d, J = 15,6 Hz, 1H), 4,00 - 3,88 (m, 3H), 3,71 (dd, J = 15,6, 11,2 Hz, 1H), 3,53 (td, J = 12,1, 3,1 Hz, 1H), 2,02 (s, 3H), 1,82-1,57 (m, 2H); IEN-EM m/z calc. 336,12, encontrado 337,0 (M+1)+; Tiempo de retención: 0,7 minutos.

(+/-)-4-(3-(2-cloro-4-(1H-pirazoM-N)fenM)-1,4-oxazepan-4-M)-6-metMpmmidm-2-amma (28) I-133
RMN 1H (400 MHz, DMSO-d6 ) (se calentó 360K) 68,42 (d, J = 2,5 Hz, 1H), 7,91 (d, J = 2,3 Hz, 1H), 7,79 - 7,67 (m, 2H), 7,43 (d, J = 8,5 Hz, 1H), 6,51 (dd, J = 2,6, 1,8 Hz, 1H), 6,06 (s, 2H), 5,80 (s, 1H), 5,54 (d, J = 7,6 Hz, 1H), 4,58 (d, J = 15,2 Hz, 1H), 4,15 (dd, J = 13,5, 4,9 Hz, 1H), 3,96 -3,80 (m, 2H), 3,80 - 3,69 (m, 1H), 3,68 -3,55 (m, 1H), 2,09 (s, 3H), 1,88-1,75 (m, 2H); IEN-EM m/z calc. 384,15, encontrado 385,0 (M+1)+; Tiempo de retención: 0,72 minutos.
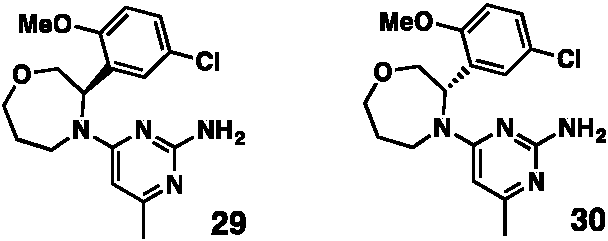
(R)-4-(3-(5-cloro-2-metoxifenM)-1,4-oxazepan-4-M)-6-metMpmmidm-2-amma (29) I-131
RMN 1H (400 MHz, DMSO-d6 ) (se calentó 360K) 67,29 (dd, J = 8 ,8 , 2,7 Hz, 1H), 7,11 (d, J = 2,7 Hz, 1H), 7,07 (d, J = 8,8 Hz, 1H), 6,97 (s, 2H), 5,95 (d, J = 35,3 Hz, 1H), 5,55 (s, 1H), 4,55 (s, 1H), 4,17 (dd, J = 13,4, 5,2 Hz, 1H), 3,87 (s, 4H), 3,82 -3,68 (m, 2H), 3,57 (ddd, J = 12,2, 8,3, 5,7 Hz, 1H), 2,19 (s, 3H), 1,79 (h, J = 3,9 Hz, 2H). IEN-EM m/z calc.
348,14, encontrado 349,0 (M+1)+; Tiempo de retención: 0,72 minutos.
(S)-4-(3-(5-cloro-2-metoxifenil)-1,4-oxazepan-4-il)-6-metilpirimidin-2-amina (30) I-132
RMN 1H (400 MHz, DMSO-d6 ) (se calentó 360K) 67,29 (dd, J = 8 ,8 , 2,7 Hz, 1H), 7,11 (d, J = 2,7 Hz, 1H), 7,07 (d, J = 8,8 Hz, 1H), 6,97 (s, 2H), 5,95 (d, J = 35,3 Hz, 1H), 5,55 (s, 1H), 4,55 (s, 1H), 4,17 (dd, J = 13,4, 5,2 Hz, 1H), 3,87 (s, 4H), 3,82 -3,68 (m, 2H), 3,57 (ddd, J = 12,2, 8,3, 5,7 Hz, 1H), 2,19 (s, 3H), 1,79 (h, J = 3,9 Hz, 2H); IEN-EM m/z calc.
348,14, encontrado 349,0 (M+1)+; Tiempo de retención: 0,72 minutos.

(+/-)-4-(3-(4-bromo-2-clorofenil)-1,4-oxazepan-4-il)-6-metilpirimidin-2-amina (31) I-194
alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6 ) 67,70 (d, J = 2,1 Hz, 1H), 7,52 (dd, J = 8,4, 2,1 Hz, 1H), 7,31 (t, J = 6,8 Hz, 3H), 4,15 (dd, J = 13,6, 5,0 Hz, 1H), 3,96 -3,72 (m, 3H), 3,64 (s, 1H), 2,25 (s, 3H), 1,85 (s, 2H); IEN-EM m/z calc. 396,0, encontrado 397,0 (M+1)+; Tiempo de retención: 0,64 minutos.
El material racémico se sometió a separación quiral SFC.
Pico A: IEN-EM m/z calc. 396,0, encontrado 399,0 (M+1)+; Tiempo de retención: 0,8 minutos; (R)-4-(3-(4-bromo-2dorofenil)-1,4-oxazepan-4-il)-6-metilpirimidin-2-amina (32). I-200
Pico B: RMN 1H (400 MHz, DMSO-d6 ) 6 7,69 (d, J = 2,0 Hz, 1H), 7,50 (dd, J = 8,4, 2,1 Hz, 1H), 7,29 (d, J = 8,4 Hz, 1H), 6,87 (s, 3H), 6,01 (s, 1H), 5,59 (s, 1H), 4,49 (d, J = 14,9 Hz, 1H), 4,13 (dd, J = 13,6, 4,9 Hz, 1H), 3,91 - 3,78 (m, 3H), 3,62 (ddd, J = 12,2, 9,4, 4,9 Hz, 1H), 2,19 (s, 4H), 1,82 (dp, J = 10,1, 3,6, 3,1 Hz, 2H); IEN-EM m/z calc. 396,0, encontrado 397,0 (M+1)+; Tiempo de retención: 0,8 minutos; [a] = 79,9 (c = 1, MeOH) 7,1 mg/ ml; (S)-4-(3-(4-bromo-2-dorofenil)-1,4-oxazepan-4-il)-6-metilpirimidin-2-amina (33). I-201

4-[(3S)-3-(5-bromo-2-doro-fenM)-1,4-oxazepan-4-M]-6-metN-pinmidm-2-amma I-245
RMN 1H (300 MHz, DMSO-c/6 ) 67,47 (m, 3H), 6,52 (s a, 1H), 5,99 (s a, 1H), 3,90 (m, 4H), 3,66 (a, 2H), 2,30 (s, 3H), 1,89 (m, 2h ); IEN-EM m/z calc. 396,03, encontrado 397,01 (M+1 )+; Tiempo de retención: 0,65 minutos; [a]D = 66 ,68° (c = 0,5, MeOH).
Ejemplo 3
Esquema sintético 3: (R)-4-(2-(2-clorofeml)azepan-1-M)-6-metMpir¡m¡dm-2-amma and (S)-4-(2-(2-clorofenil)azepan-1-il)-6-metilpirimidin-2-amina)
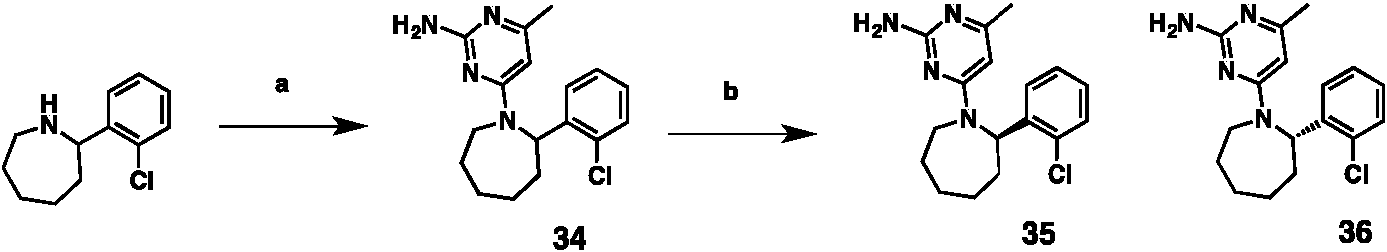
(a) 2-amino-4-cloro-6-metilpirimidina, nBuOH, 200 °C, microondas; (b) Separación HPLC quiral
Formación de (+/-)-(2-(2-clorofeml)azepan-1-M)-6-metilpmmidm-2-amma (34)
Una suspensión de 4-cloro-6-metil-pirimidin-2-amina (3,02 g, 21,03 mmol), 2-(2-clorofenil)azepano (3,99 g, 19,01 mmol) en n-butanol (15 ml) se cerró herméticamente en un tubo de microondas y se irradió a 200 °C durante 2 horas. La mezcla en bruto se concentró al vacío y se diluyó con una solución acuosa saturada de KHCO3 y se extrajo dos veces con diclorometano. La fase orgánica se concentró al vacío. Después, el residuo se volvió a cristalizar a partir de isopropanol y éter para proporcionar 3,61 g del producto racémico: RMN 1H (400 MHz, CDCh) 67,37 (d, J = 8,5 Hz, 1H), 7,16 (d, J = 13,9 Hz, 3H), 5,47 (s, 1H), 4,91 (s, 1H), 4,57 (s, 2H), 3,32 (s, 1H), 2,58 - 2,45 (m, 1H), 2,17 (d, J = 22,1 Hz, 2H), 2,03 (s, 1H), 1,91 (d, J = 10,7 Hz, 2H), 1,66 (s, 1H), 1,63 (s, 3H), 1,50-1,33 (m, 2H); IEN-EM m/z calc.
316,15, encontrado 317,2 (M+1)+; Tiempo de retención: 0,8 minutos.
La mezcla racémica se separó por HPLC quiral analítica (AD-H, 4,6* 100mm, MeOH al 40 %, amoniaco 5 mM, CO2 al 60 %,a 5 ml/min inyección isocrática 10uM en 1 mg/ ml de metanol. 12 MPa (120 bar). UV 254 nM). Tr Pico A 0,432 min, ee 97,2 %, Pico B a 0,479 min.
Pico A: 2,10 g, 97,2 % ee; rotación óptica: [□]□ = -0,096 (c= 1,04, MeOH); RMN 1H (400 MHz, CDCla) 67,36 (s, 1H), 7,17 (s, 3H), 5,81 (s, 1H), 5,44 (s, 1H), 4,90 (s, 1H), 4,68 (s, 2H), 4,03 -3,20 (m, 1H), 2,63 -2,43 (m, 1H), 2,11 (s, 3H), 2,02 (s, 1H), 1,97 - 1,82 (m, 2H), 1,76 -1,51 (m, 2H), 1,51-1,31 (m, 2H); IEN-EM m/z calc. 316,15, encontrado 317,24 (M+1)+; Tiempo de retención: 0,83 minutos. (R)-4-(2-(2-clorofenil)azepan-1-il)-6-metilpirimidin-2-amina (35) I-13
Pico B: 2,09 g, 96 % ee; rotación óptica: [□]□ = 1,373 (c= 1,02, MeOH); RMN 1H (400 MHz, CDCla) 6 1,46-1,31 (m, 2H), 1,72 - 1,53 (m, 2H), 1,96 - 1,82 (m, 2H), 2,04 (d, J = 18,8 Hz, 1H), 2,39 -2,05 (m, 3H), 2,61 -2,42 (m, 1H), 4,08 -3,22 (m, 1H), 4,72 (s, 2H), 4,89 (s, 1H), 5,43 (s, 1H), 5,82 (s, 1H), 7,16 (s, 3H), 7,35 (s, 1H); IEN-EM m/z calc. 316,15, encontrado 317,2 (M+1)+; Tiempo de retención: 0,82 minutos. (S)-4-(2-(2-clorofenil)azepan-1-il)-6-metilpirimidin-2-amina (36) I-14
Los siguientes análogos se prepararon de acuerdo con el Esquema sintético 3:

(R)-4-(2-(2-metox¡feml)azepan-1-¡l)-6-met¡lp¡r¡m¡dm-2-amma (37) I-11 y (S)-4-(2-(2-metox¡feml)azepan-1-¡l)-6-metilpirimidin-2-amina (38) I-12
Pico A a partir de la separación quiral SFC: RMN 1H (400 MHz, CDCI3) 87,01 (dd, J = 118,6, 38,8 Hz, 4H), 5,60 (2s, 1H), 5,24 - 4,44 (m, 3H), 3,82 (s, 3H), 3,32 (dd, J = 63,5, 46,9 Hz, 1H), 2,49 (2s, 1H), 2,30 - 1,91 (m, 3H), 1,94-0,56 (m, 9H); IEN-EM m/z calc. 312,20, encontrado 313,13 (M+1)+; Tiempo de retención: 0,78 minutos. (38) I-12
Pico B a partir de la separación quiral SFC: RMN 1H (400 MHz, CDCI3) 89,03-6,27 (m, 4H), 5,59 (2s, 1H), 5,29 -4,36 (m, 3H), 3,82 (s, 3H), 3,54 -2,79 (m, 1H), 2,33 (d, J = 61,8 Hz, 1H), 2,00 (s, 3H), 1,93-0,44 (m, 10H); IEN-EM m/z calc.
312,12, encontrado 313,13 (M+1)+; Tiempo de retención: 0,8 minutos. (37) I-l1
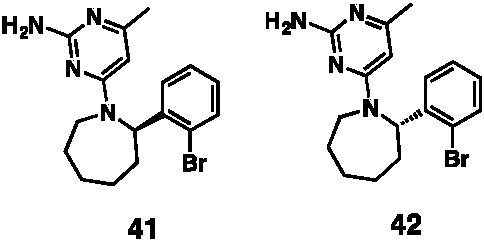
(R)-4-(2-(2-bromo)azepan-1-¡l)-6-met¡lp¡r¡m¡dm-2-amma (41) 1-2 y (S)-4-(2-(2-bromo)azepan-1-¡l)-6-met¡lp¡r¡m¡d¡n-2-am¡na (42) I-3
Pico A a partir de la separación quiral SFC: RMN 1H (400 MHz, CDCh) 87,46 (d, J = 7,4 Hz, 1H), 7,08 (d, J = 42,6 Hz, 3H), 5,95 - 5,43 (m, 1H), 5,34 (s, 1H), 4,77 (d, J = 37,2 Hz, 3H), 3,98 - 3,08 (m, 2H), 2,46 -2,32 (m, 1H), 2,08 (d, J = 40,4 Hz, 3H), 1,94 (s, 1H), 1,89 - 1,75 (m, 2H), 1,65 - 1,44 (m, 2H), 1,42-1,26 (m, 2H); IEN-EM m/z calc. 360,09, encontrado 361,12 (M+1)+; Tiempo de retención: 0,91 minutos. (41) I-2
Pico B a partir de la separación quiral SFC: RMN 1H (400 MHz, CDCh) 87,46 (s, 1H), 6,99 (t, J = 70,7 Hz, 3H), 6,02 -5,47 (m, 1H), 5,21 (t, J = 62,8 Hz, 4H), 4,78 (d, J = 17,2 Hz, 1H), 3,96 - 3,17 (m, 2H), 2,48 - 2,33 (m, 1H), 2,27 - 2,03 (m, 3H), 1,96 - 1,72 (m, 3H), 1,68 - 1,42 (m, 2H), 1,42-1,23 (m, 2H); IEN-EM m/z calc. 360,09, encontrado 361,12 (m 1)+; Tiempo de retención: 0,93 minutos. (42) I-3

(+/-)- 4-met¡l-6-(2-(2-(met¡lt¡o)fen¡l)azepan-1-¡l)p¡r¡m¡d¡n-2-am¡na (43) I-5
RMN 1H (400 MHz, MeOD) 87,33 (d, J = 7,8 Hz, 1H), 7,23 (s, 1H), 7,08 (d, J = 5,7 Hz, 2H), 5,45 (s, 1H), 3,41 (s, 1H), 2,58 (s, 3H), 2,38 (d, J = 17,3 Hz, 1H), 2,02 (d, J = 6,2 Hz, 3H), 1,96 - 1,81 (m, 2H), 1,72 (d, J = 6,7 Hz, 1H), 1,62 -1,50 (m, 1H), 1,50-1,27 (m, 2H); IEN-EM m/z calc. 328,17, encontrado 329,11 (M+1)+; Tiempo de retención: 0,75 minutos.

(+/-)- 4-met¡l-6-(2-(2-(met¡l)fen¡l)azepan-1-¡l)p¡r¡m¡d¡n-2-am¡na (44) I-6
RMN 1H (400 MHz, Metanol-d4) 87,19 - 6,99 (m, 4H), 2,46 (s, 3H), 2,27 (ddd, J = 14,3, 8,4, 5,1 Hz, 1H), 2,22 - 1,96 (m, 4H), 1,95 - 1,82 (m, 3H), 1,81 - 1,68 (m, 1H), 1,59 (d, J = 12,0 Hz, 1H), 1,42 (dtt, J = 23,7, 12,2, 6,3 Hz, 2H); IEN-EM m/z calc. 296,20, encontrado 297,14 (M+1)+; Tiempo de retención: 0,74 minutos.

(+/-)-4-(2-(2,4-diclorofenil)azepan-1-il)-6-metilpirimidin-2-amina (45) I-1
RMN 1H (400 MHz, MeOD) 87,71-7,07 (m, 3H), 6,53 -5,02 (m, 2H), 4,28 -3,43 (m, 2H), 2,66 -2,37(m , 1H), 2,25 (3H de 2s), 2,17-1,28 (m, 7H); IEN-EM m/z calc. 350,11, encontrado 351,11 (M+1)+; Tiempo de retención: 3,13 minutos.

(+/-)-4-metil-6-(2-fenilazepan-1-il)pirimidin-2-amina (45) I-4
RMN 1H (400 MHz, MeOD) 87,48-7,17 (m, 5H), 6,39-5,94 (2s, 1H), 4,04 -3,36 (m, 1H), 2,52 (td, J = 14,5, 6,2 Hz, 1H), 2,27 (d, J = 61,1 Hz, 3H), 2,04 - 1,79 (m, 4H), 1,76 - 1,05 (m, 3H).

(+/-)-4-(2-(2-clorofeml)azepan-1-il)-6-isopropilpirimidm-2-amma I-16
RMN 1H (400 MHz, DMSO-d6 ) 87,47 - 7,34 (m, 1H), 7,34 - 7,17 (m, 3H), 5,57 (s, 1H), 5,46 (s, 2H), 5,21 (s, 1H), 4,55 (d, J = 14,9 Hz, 1H), 3,55 -3,40 (m, 1H), 2,57 -2,52 (m, 1H), 2,34 (ddd, J = 13,8, 8,1, 5,0 Hz, 1H), 1,98 (t, J = 11,2 Hz, 1H), 1,92 - 1,64 (m, 3H), 1,61 -1,18 (m, 3H), 1,08 (d, J = 6,9 Hz, 3H), 1,04 (d, J = 6,9 Hz, 3H); IEN-EM m/z encontrado 345.
(+/-)-4-(2-(2-clorofenil)azepan-1-il)-6-etilpirimidin-2-amina I-17
RMN 1H (400 MHz, DMSO-d6 ) 87,45 -7,32 (m, 1H), 7,28 -7,13 (m, 3H), 5,56 (s, 1H), 5,41 (s, 2H), 5,21 (d, J = 12,0 Hz, 1H), 4,49 (d, J = 14,7 Hz, 1H), 3,51 - 3,38 (m, 1H), 2,52 -2,49 (m, 1H), 2,38 -2,22 (m, 3H), 2,01 - 1,64 (m, 2H), 1,59 -1,21 (m, 4H), 1,03 (t, J = 7,5 Hz, 3H); IEN-EM m/z encontrado 331.
Ejemplo 4
Esquema sintético 4: (+/-)-4-(3-(5-amino-2-clorofenil)-1,4-oxazepan-4-il)-6-metilpirimidin-2-amina (46) I-71

(a) cinc, NH4CI, TPGS-750-M al 2 % en agua, 75 °C; (b) separación quiral SFC
Formación de (R)-4-(3-(5-ammo-2-clorofeml)-1,4-oxazepan-4-il)-6-metilpirimidm-2-amma (47) y (S)- 4-(3-(5-ammo-2-clorofeml)-1,4-oxazepan-4-il)-6-metilpirimidm-2-amma (48)
4-[3-(2-doro-5-nitro-fenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina, 24, (1,00 g, 2,75 mmol), NH4Cl (0,31 g, 5,85 mmol) y Zn (0,87 g, 13,36 mmol) se agitaron en TPGS-750-M al 2 % en agua (28 ml). La mezcla de reacción se agitó vigorosamente y se calentó a 75 °C durante 24 horas. La mezcla se enfrió a temperatura ambiente y se diluyó en una solución acuosa saturada de NaHCO3 y diclorometano. La fase orgánica se secó (MgSO4), se filtró y se concentró al vacío. El residuo en bruto se purificó por cromatografía sobre gel de sílice con una columna isco GOLD de 40 g usando 0-50 % (MeOH-CH2Cl2 al 20 %/CH2Cl2) para proporcionar 390 mg del compuesto 46 en forma de una mezcla racémica. La mezcla racémica se sometió a separación SFC quiral: preparado a IPA al 50 %, Hexanos al 50 %, dietilamina al 0,2 % en AD-H para proporcionar los estereoisómeros individuales.
Pico A - 99 % puro por HPLC quiral; (R)-4-[3-(5-amino-2-cloro-fenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (47): alta temperatura (360 K) RMN 1H (400 MHz, DMSO-c/6 ) 87,01 (d, J = 8,5 Hz, 1H), 6,52 (d, J = 2,7 Hz, 1H), 6,47 (dd, J = 8,5, 2,7 Hz, 1H), 5,51 (s, 1H), 5,43 (s, 2H), 5,26 -5,10 (m, 1H), 4,94 (s, 2H), 4,82 -4,64 (m, 1H), 4,10 (dd, J = 13,5, 5,0 Hz, 1H), 3,95 - 3,84 (m, 1H), 3,69 - 3,46 (m, 3H), 1,99 (s, 2H), 1,82 - 1,65 (m, 2H). IEN-EM m/z calc. 333,14, encontrado 334,26 (M+1)+; Tiempo de retención: 0,51 minutos; [^ ]D = -118,67 (c = 12 mg/4 ml de MeOH). I-86
Pico B - 99,9% puro por HPLC quiral; (S)-4-[3-(5-amino-2-cloro-fenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (48): alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6 ) 87,01 (dd, J = 8,5, 1,5 Hz, 1H), 6,52 (d, J = 2,5 Hz, 1H), 6,47 (dt, J = 8,5, 2,1 Hz, 1H), 5,51 (s, 1H), 5,44 (s, 2H), 5,26 - 5,07 (m, 1H), 4,94 (s, 2H), 4,84 - 4,66 (m, 1H), 4,11 (ddd, J = 13,4, 5,0, 1,5 Hz, 1H), 3,97 -3,84 (m, 1H), 3,69 -3,42 (m, 3H), 2,05 - 1,92 (m, 3H), 1,83 - 1,66 (m, 2H). IEN-EM m/z calc. 333,14, encontrado 334,26 (M+1 )+; Tiempo de retención: 0,51 minutos; [^ ]D = 175 (c = 8 mg/4 ml de MeOH). I-87
El siguiente análogo se preparó de acuerdo con el Esquema sintético 4:

(+/-)-4-[3-(4-amino-2-cloro-fenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (49) I-140
alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6 ) 8 7,01 (m, 3H), 6,65 (d, J = 2,3 Hz, 1H), 6,52 (dd, J = 8,5, 2,3 Hz, 1H), 5,95 (s a, 1H), 5,49 -5,36 (m, 2H), 4,70 -4,49 (m, 1H), 4,04 (dd, J = 13,5, 5,0 Hz, 1H), 3,83 (dd, J = 10,4, 5,8 Hz, 2H), 3,75 -3,58 (m, 3H), 2,19 (s, 3H), 1,86 - 1,75 (m, 2H). IEN-EM m/z calc. 333,14, encontrado 334,26 (M+1)+; Tiempo de retención: 0,51 minutos.
Ejemplo 5
Esquema sintético 5: (+/-)-W-[4-[4-(2-ammo-6-metil-pirimidm-4-il)-1,4-oxazepan-3-il]-3-clorofenil]metanosulfonamida I-139, I-179 y I-296

(a) Cloruro de metanosulfonilo, NEt3, THF; (b) separación quiral SFC
Formación de (+/-)-3-(2-cloro-5-nitrofenil)-1,4-oxazepano (50) I-139
A una solución de 4-[3-(4-amino-2-doro-fenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina, 49, (0,034 g, 0,103 mmol) y trietilamina (0,050 ml, 0,360 mmol) en THF (1,5 ml), se le añadió cloruro de metanosulfonilo (0,009 ml, 0,113 mmol). La mezcla de reacción se agitó a temperatura ambiente durante la noche. Se añadió un adicional de 5 ul de cloruro de metanosulfonilo. Después de 20 minutos, la mezcla de reacción se concentró al vacío. La purificación se realizó en una columna ISCO c18-aq de 50 g de fase inversa, ejecutándose con TFA al 0,1 %/H2O y t Fa al 0,1 %/CH3CN. Las fracciones puras se concentraron al vacío y después se disolvieron en MeOH y se pasaron a través de dos cartuchos bicarbonato SPE (Agilent Stratospheres 100 mg/6 ml) dispuestos en serie y se concentraron para dar 7,3 mg del producto deseado: alta temperatura (360 K) RMN 1H (400 MHz, DMSO-c/6 ) 89,97 - 9,55 (s a, 1H), 7,25-7,21 (m, 2H), 7,09 (dd, J = 8,5, 2,1 Hz, 1H), 5,59 (s, 1H), 5,42 (s, 2H), 5,36 (s, 1H), 4,60 (d, J = 13,2 Hz, 1H), 4,08 (dd, J = 13,5, 5,0 Hz, 1H), 3,88 (d, J = 12,3 Hz, 1H), 3,70 (dd, J = 13,5, 10,2 Hz, 1H), 3,66 -3,50 (m, 3H), 2,01 (s, 3H), 1,76 (s, 3H); IEN-EM m/z calc. 411,11, encontrado 412,24 (M+1)+; Tiempo de retención: 0,56 minutos.
La separación HPLC quiral proporcionó enantiómeros individuales
Pico A: RMN 1H (400 MHz, DMSO-d6) 89,66 (s, 1H), 7,28 - 7,23 (m, 2H), 7,13 (dd, J = 8,5, 2,2 Hz, 1H), 5,59 (s, 1H), 5,46 (s, 2H), 5,39 (s, 1H), 4,59 (d, J = 14,9 Hz, 1H), 4,09 (dd, J = 13,5, 5,0 Hz, 1H), 3,93 - 3,85 (m, 1H), 3,71 (dd, J = 13,5, 10,1 Hz, 1H), 3,66 - 3,49 (m, 2H), 2,99 (s, 3H), 2,01 (s, 3H), 1,81 -1,70 (m, 2H). I-179
Pico B: RMN 1H (400 MHz, DMSO-d6 ) 87,33 -7,22 (m, 2H), 7,13 (dd, J = 8,5, 2,2 Hz, 1H), 5,59 (s, 1H), 5,46 (s, 2H), 5,44 - 5,34 (m, 1H), 4,59 (d, J = 15,2 Hz, 1H), 4,09 (dd, J = 13,4, 5,0 Hz, 1H), 3,93 - 3,84 (m, 1H), 3,71 (dd, J = 13,5, 10,1 Hz, 1H), 3,67 -3,49 (m, 2H), 2,99 (s, 3H), 2,01 (s, 3H), 1,77 (ddt, J = 10,2, 8,0, 3,4 Hz, 2H). I-296
Ejemplo 6
Esquema sintético 6: (+/-)-W-[4-[4-(2-ammo-6-metil-pirimidm-4-il)-1,4-oxazepan-3-il]-3-clorofenil]metanosulfonamida (51) I-282

(a) ácido 3-metiloxetano-3-carboxílico, iPr2NEt, HATU, DMF.
Formación de (+/-)-W-(4-(4-(2-ammo-6-metilpirimidm-4-il)-1,4-oxazepan-3-il)-3-clorofeml)-3-metiloxetano-3-carboxamida (51) I-282
A una solución de 4-[3-(4-amino-2-cloro-fenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (0,05 g, 0,15 mmol), ácido 3-metiloxetano-3-carboxílico (0,02 g, 0,16 mmol) y N,N-diisopropiletilamina (0,05 ml, 0,30 mmol) en DMF (1 ml), se le añadió hexafluorofosfato de N-óxido de N-[(dimetilamino)-1H-1,2,3-triazolo-[4,5-b]piridin-1-ilmetileno]-N-metilmetanaminio (HATU) (0,08 g, 0,21 mmol). La mezcla de reacción se agitó a temperatura ambiente durante una noche. El residuo resultante se purificó por HPLC preparativa de fase inversa (CH3CN/TFA ac. al 0,1 %). Las fracciones que contenían el producto deseado se basificaron con un lavado con una solución acuosa saturada de NaHCO3 y se extrajeron con diclorometano. La fase orgánica se pasó a través de un separador de fase, se concentró al vacío para proporcionar el producto deseado.
Los siguientes análogos se prepararon de acuerdo con el Esquema sintético 6 :

(3R)-W-(4-(4-(2-ammo-6-metilpirimidm-4-il)-1,4-oxazepan-3-il)-3-clorofeml)-tetrahidrofurano-3-carboxamida (52) I-280
(mezcla racémica en la posición 3 del oxazepano) RMN 1H (400 MHz, CDCh) 87,74 (s, 1H), 7,54 (s, 1H), 7,30 - 7,21 (m, 1H), 7,15 (d, J = 8,4 Hz, 1H), 5,53 (s, 1H), 4,66 (s, 2H), 4,29 (dd, J = 13,6, 5,0 Hz, 1H), 4,10 - 3,97 (m, 3H), 3,99 -3,75 (m, 2H), 3,66 -3,43 (m, 3H), 3,11 -2,95 (m, 1H), 2,32 -2,22 (m, 2H), 2,12 (s, 3H), 2,02 - 1,89 (m, 1H), 1,80 (d, J = 14,3 Hz, 3H).- IEN-EM m/z calc. 431,17, encontrado 432,18 (M+1)+; Tiempo de retención: 0,57 minutos

(3S)-W-(4-(4-(2-ammo-6-metNpirimidm-4-M)-1,4-oxazepan-3-M)-3-clorofenM)-tetrahidrofurano-3-carboxamida (53) I-281
(mezcla racémica en posición 3 del oxazepano) 7,74 (s, 1H), 7,54 (s, 1H), 7,30 - 7,21 (m, 1H), 7,15 (d, J = 8,4 Hz, 1H), 5,53 (s, 1H), 4,64 (s, 2H), 4,30 (dd, J = 13,6, 5,0 Hz, 1H), 4,11 -3,99 (m, 3H), 3,95 -3,79 (m, 2H), 3,65 -3,43 (m, 3H), 3,11 - 2,95 (m, 1H), 2,32 - 2,19 (m, 2H), 2,12 (s, 3H), 2,04 - 1,89 (m, 1H), 1,87 - 1,72 (m, 3H).- IEN-EM m/z calc.
431,17, encontrado 432,14 (M+1)+; Tiempo de retención: 0,57 minutos

(+/-)-W-(4-(4-(2-ammo-6-metNpirimidm-4-N)-1,4-oxazepan-3-M)-3-clorofenM)-2,2-difluoropropanamida(54) I-228
RMN 1H (400 MHz, CDCI3) 87,91 (s, 1H), 7,78 (s, 1H), 7,36 - 7,31 (m, 1H), 7,22 (d, J = 8,4 Hz, 1H), 5,53 (s, 1H), 4,60 (s, 2H), 4,31 (dd, J = 13,6, 5,0 Hz, 2H), 4,06 (d, J = 12,5 Hz, 1H), 3,65 -3,47 (m, 4H), 2,13 (s, 3H), 1,89 (t, J = 19,3 Hz, 5h ); iEn-EM m/z calc. 425,1, encontrado 426,2 (M+1 )+; Tiempo de retención: 0,64 minutos.

(+/-)-W-(4-(4-(2-ammo-6-metNpirimidm-4-N)-1,4-oxazepan-3-M)-3-clorofenM)-2,2-difluoroacetamida (55) I-278
RMN 1H (400 MHz, CDCl3) 87,90 (s, 1H), 7,77 (s, 1H), 7,35 (dd, J = 8,5, 2,2 Hz, 1H), 7,23 (d, J = 8,4 Hz, 1H), 6,01 (t, J = 54,2 Hz, 1H), 5,53 (s, 1H), 4,64 (s, 2H), 4,31 (dd, J = 13,6, 4,9 Hz, 1H), 4,15 - 4,01 (m, 1H), 3,68 - 3,46 (m, 3H), 2,13 (s, 3H), 2. 06 - 1,91 (m, 2H), 1,88-1,76 (m, 2H); IEN-EM m/z calc. 411,1, encontrado 412,1 (M+1)+; Tiempo de retención: 0,6 minutos.

(+/-)-W-(4-(4-(2-ammo-6-metNpirimidm-4-N)-1,4-oxazepan-3-M)-3-clorofenM)-3-fluorotetrahidrofurano-3-carboxamida (56) I-279
RMN 1H (400 MHz, CDCl3) 88,17 (d, J = 7,9 Hz, 1H), 7,81 (d, J = 7,8 Hz, 1H), 7,32 (td, J = 8,3; 2,2 Hz, 1H), 7,20 (d, J = 8,5 Hz, 1H), 5,53 (s, 1H), 4,61 (s, 2H), 4,30 (dd, J = 13,6, 5,0 Hz, 1H), 4,19 -4,02 (m, 6 H), 3,68 - 3,49 (m, 3H), 2,77 - 2,56 (m, 1H), 2,44 - 2,27 (m, 1H), 2,12 (s, 3H), 2,03 - 1,90 (m, 1H), 1,88-1,74 (m, 2H); IEN-EM m/z calc. 449,2, encontrado 450,1 (M+1)+; Tiempo de retención: 0,6 minutos.
Los siguientes análogos se prepararon de acuerdo con el Esquema 6 usando 4-(3-(5-amino-2-clorofenil)-1,4-oxazepan-4-il)-6-metilpirimidin-2-amina 46 como material de partida:

(+/-)-W-[3-[4-(2-ammo-6-metN-pirimidm-4-M)-1,4-oxazepan-3-M]-4-cloro-feml]-2-(oxetan-3-N)acetamida (57) I-185 RMN 1H (300 MHz, Metanol-d4) 56,07 - 5,93 (m, 1H), 5,75 (a, 2H), 5,26 (s, 1H), 4,27 (s, 1H), 3,87 (a, 2H), 2,80 (m, 4H), 2,52 - 2,21 (m, 5H), 1,74 - 1,35 (m, 2H), 0,83 (s, 3H), 0,56 (m, 2H); IEN-EM m/z calc. 431,2, encontrado 432,1 (M+1)+; Tiempo de retención: 0,55 minutos.

(+/-)-W-[3-[4-(2-ammo-6-metN-pinmidm-4-M)-1,4-oxazepan-3-M]-4-clorofeml]ciclopropanocarboxamida (58) I-241
Se calentó (360K) RMN 1H (300 MHz, DMSO-d6 ) 510,38 (s, 1H), 7,96 (s, 2H), 7,74 - 7,30 (m, 8H), 6,65 (s, 1H), 5,95 (dd, J = 10,3, 5,4 Hz, 1H), 5,56 (s, 1H), 5,18 (dd, J = 10,1, 4,9 Hz, 1H), 5,10 -4,93 (m, 1H), 4,34 -4,10 (m, 3H), 4,02 -3,55 (m, 7H), 2,29 (s, 3H), 2,00 - 1,63 (m, 6 H), 0,79 (d, J = 7,3 Hz, 6 H); IEN-EM m/z calc. 401,2, encontrado 402,2 (M+1)+; Tiempo de retención: 0,62 minutos.

(S)-W-[3-[(3S)-4-(2-ammo-6-metM-pinmidm-4-M)-1,4-oxazepan-3-M]-4-cloro-feml]-3-hidroxi-propanamida (59) I-283
Material racémico obtenido utilizando un procedimiento similar y luego enviado para purificación por HPLC quiral (columna (OJ-H 20x250m), fase móvil (hexanos al 80 %/IPA al 20 %/dietilamina al 0,2 %), flujo 20 ml/min).
Pico B: ee: 91 %; [a] d (c=0,5, MeOH) 32,4; RMN 1H (300 MHz, Metanol-d4) 57,63 (d, J = 2,6 Hz, 1H), 7,49 (d, J = 8,8 Hz, 1H), 7,35 (d, J = 8,7 Hz, 1H), 5,46 (a, 3H), 4,30 (dd, J = 13,6, 5,0 Hz, 1H), 4,11 - 3,97 (m, 1H), 3,85 (t, J = 6.2 Hz, 2H), 3,79 - 3,48 (m, 3H), 2,53 (t, J = 6,1 Hz, 2H), 2,07 (s, 3H), 1,87 (m, 2H); IEN-EM m/z calc. 405,2, encontrado 406.2 (M+1)+; Tiempo de retención: 0,58 minutos.

N-(3-(4-(2-amino-6-metilpirimidin-4-il)-1,4-oxazepan-3-il)-4-clorofenil)-2,2,2-trifluoroacetamida (60) I-177
Material racémico obtenido utilizando un procedimiento similar y luego enviado para purificación por HPLC quiral (columna (OJ-H 20x250m), fase móvil (hexanos al 80 %/IPA al 20 %/dietilamina al 0,2 %), flujo 20 ml/min).
Pico B: ee: 99 %; [a] d (c=0,5, MeOH) 157,3; RMN 1H (300 MHz, Metanol-d4) 56,35 (d, J = 2,5 Hz, 2H), 6,16 (d, J = 9,2 Hz, 1H), 4,22 (a, 1H), 3,02 (dd, J = 13,6, 5,1 Hz, 1H), 2,88 - 2,67 (m, 1H), 2,56 - 2,23 (m, 3H), 2,03 (m, 2H), 0,59 (m, 2H); IEN-EM m/z calc. 429,1, encontrado 429,9 (M+1)+; Tiempo de retención: 0,65 minutos.

(+/-)-W-[3-[4-(2-ammo-6-metN-pinmidm-4-M)-1,4-oxazepan-3-M]-4-cloro-feml]-2-(dimetMammo)acetamida (61) I-168
Se calentó (360K) RMN 1H (300 MHz, DMSO-d6 ) 59,90 (s, 1H), 7,73 (d, J = 8,7 Hz, 1H), 7,63 (d, J = 2,6 Hz, 1H), 7,38 (d, J = 8,7 Hz, 1H), 5,96 (s a, 2H), 5,03 (a, 2H), 4,11 (s, 1H), 3,94 (m, 1H), 3,74 - 3,47 (m, 3H), 3,33 (s, 6 H), 3,04 (s, 2h ), 2,25 (s, 3H), 1,98 (a, 2H); IEN-EM m/z calc. 418,2, encontrado 419,1 (M+1)+; Tiempo de retención: 0,58 minutos.
(+/-)-W-[3-[4-(2-ammo-6-metil-pirimidm-4-il)-1,4-oxazepan-3-il]-4-clorofeml]oxetano-2-carboxamida (62) I-184
RMN 1H (300 MHz, Metanol-d4) 88,60 (s, 1H), 6,59 -6,42 (m, 1H), 6,37 -6,19 (m, 1H), 6,10 (dd, J = 22,6, 8,7 Hz, 1H), 5,20 (s, 0,5H), 4,76 (dd, J = 10,2, 5,2 Hz, 0.5H), 4,34 (s, 0,5H), 4,04 (dd, J = 10,2, 5,0 Hz, 0.5H), 3,78 (dd, J = 9,1, 6,7 Hz, 1H), 3,48 -3,29 (m, 2H), 3,16 -2,91 (m, 2H), 2,81 -2,26 (m, 4H), 1,85 - 1,25 (m, 2H), 1,02 (s, 1,5H), 0,89 (d, J = 0,8 Hz, 1.5H), 0,61 (m, 2H); IEN-EM m/z calc. 417,2, encontrado 418,0 (M+1)+; Tiempo de retención: 0,6 minutos.

(+/-)-W-[3-[4-(2-ammo-6-metil-pirimidm-4-il)-1,4-oxazepan-3-il]-4-cloro-feml]-3-hidroxi-3-metil-butan-amida (63) I-251
RMN 1H (300 MHz, Metanol-c/4) 8 7,75 (t, J = 10,0 Hz, 1H), 7,48 - 7,40 (m, 1H), 7,39 - 7,29 (m, 1H), 6,52 (s, 0,5H), 6,08 (dd, J = 10,3, 5,3 Hz, 0.5H), 5,65 (d, J = 3,3 Hz, 0.5H), 5,35 (dd, J = 10,5, 5,1 Hz, 0.5H), 5,20 (d, J = 14,4 Hz, 0.5H), 4,46 -4,19 (m, 1.5H), 4,14 - 3,55 (m, 4H), 2,48 (d, J = 1,4 Hz, 1.5H), 2,34 (d, J = 0,8 Hz, 1H), 2,21 (t, J = 0,9 Hz, 1.5H), 2,09 (d, J =1,1 Hz, 1H), 1,47-1,21 (m, 6 H); IEN-EM m/z calc. 433,2, encontrado 434,2 (M+1)+; Tiempo de retención: 0,59 minutos.
Ejemplo 7
Esquema sintético 7: (+/-)-W-(4-(4-(2-ammo-6-metilpirimidm-4-il)-1,4-oxazepan-3-il)-3-clorofeml)-2-hidroxi-2-metilpropanamida I-277

(a) 2-hidroxi-2-metilpropanamida, NaOtBu, tBuXPhos Pd G3, tBuOH, 60 °C; (b) separación quiral SFC
Formación de (S)-W-(4-(4-(2-ammo-6-metilpirimidm-4-il)-1,4-oxazepan-3-il)-3-clorofeml)-2-hidroxi-2-metilpropanamida (64) I-276
En un tubo de microondas, una mezcla de 4-[3-(4-bromo-2-cloro-fenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina, 31, (0,77 g, 1,86 mmol), 2-hidroxi-2-metil-propanamida (0,45 g, 4,36 mmol), ferc-butóxido sódico (0,56 g, 5,78 mmol) y [2-(2-aminofenil)fenil]-metilsulfoniloxi-paladio; diterc-butil-[2-(2,4,6-triisopropilfenil)fenil]fosfano (tBuXPhos Pd G3)(0,06 g, 0,08 mmol) en 2-metil-2-propanol (14 ml) se evacuó y se volvió a llenar con nitrógeno tres veces. Después, el tubo se calentó a 60 °C durante 3 horas. La mezcla de reacción se diluyó con diclorometano y se lavó con agua y salmuera, se secó sobre sulfato sódico, se filtró y se concentró al vacío. El residuo resultante se purificó a través de cromatografía de fase inversa sobre gel de sílice usando una columna ISCO C-18 (150 g) eluyendo con CH3CN al 0 90 %/H2O (modificador de formiato amónico). Las fracciones puras se concentraron al vacío, se diluyeron con diclorometano y se lavaron con agua. Las fases orgánicas se pasaron a través de un separador de fase y se concentraron al vacío para proporcionar 207 mg del producto deseado. La mezcla racémica se sometió a purificación SFC quiral (columna IA, 20x250mm fase móvil - MeOH al 20 % (amoniaco 5mM), flujo de CO2 al 80 % - 80 ml/min).
Pico B: RMN 1H (400 MHz, CDCla) 88,68 (s, 1H), 7,83 (s, 1H), 7,31 (dd, J = 8,4, 2,2 Hz, 1H), 7,16 (d, J = 8,5 Hz, 1H), 5,53 (s, 1H), 4,60 (s, 2H), 4,30 (dd, J = 13,6, 5,0 Hz, 1H), 4,08 (d, J = 15,8 Hz, 1H), 3,64 - 3,45 (m, 4H), 2,12 (s, 3H), 1,80 (d, J = 14,3 Hz, 4H), 1,54 (s, 6 H); IEN-EM m/z calc. 419,2, encontrado 420,2 (M+1)+; Tiempo de retención: 0,58
minutos.
Ejemplo 8
Esquema sintético 8: (+/-)-4-(3-(2-cloro-5-(metilsulfonil)fenil)-1,4-oxazepan-4-il)-6-metilpirimidin-2-amina (69) I-311

(a) TMS-diazometano, tolueno, metanol: (b) NaBH4, MeOH; (c) Peryodinano de Dess-Martin, diclorometano; (d) tamices mol. de 4 A, 3-((tributilestannil)metoxi)propan-1-amina, CH2Cl2; después 2,6-lutidina, Cu(OTf)2, hexafluoroisopropanol, CH2Cl2; (e) 2-amino-4-cloro-6-metilpirimidina, NMP, 150 °C; (f) separación quiral SFC
Formación de 2-cloro-5-(metilsulfonil)benzoato de metilo (65)
A una solución de ácido 2-cloro-5-metilsulfonil-benzoico (3,0 g, 12,8 mmol) en tolueno (45 ml) y MeOH (10 ml), se le añadió gota a gota TMS-diazometano (10,7 ml de 2 M en hexano, 21,4 mmol). La mezcla de reacción se agitó durante 3 horas y el disolvente se concentró al vacío para dar 3 gramos del producto deseado en forma de un sólido esponjoso de color castaño que se usó sin más purificación: RMN 1H (400 m Hz , DMSO-d6J 68,32 (d, J = 2,3 Hz, 1H), 8,10 (dd, J = 8,4, 2,4 Hz, 1H), 7,90 (d, J = 8,5 Hz, 1H), 3,92 (s, 3H), 3,30 (s, 6 H); IEN-EM m/z calc. 247,99, encontrado 249,12 (M+1)+; Tiempo de retención: 0,71 minutos.
Formación de (2-cloro-5-(metilsulfonil)fenil)metanol (66 )
A una suspensión de 2-cloro-5-metilsulfonil-benzoato de metilo, 65, (3,0 g, 12,1 mmol) en EtOH (45 ml), se le añadió NaBH4 (1,83 g, 48,4 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 1 hora, después se calentó a 50 °C para solubilizar la mezcla. Después de 3 horas, la mezcla se inactivó mediante la adición lenta en una adición lenta en una solución saturada de NH4CL La fase acuosa se extrajo dos veces con EtOAc. Las fases orgánicas combinadas se lavaron con una solución acuosa saturada de NaHCO3, se secaron (MgSO4), se filtraron y se concentraron al vacío para dar 2,5 gramos del producto deseado en forma de un aceite de color naranja. El residuo en bruto se purificó por cromatografía sobre gel de sílice con una columna isco de 40 g usando un gradiente de EtOAc al 0-30 %/CH2Cl2 para proporcionar 2,0 gramos de producto en forma de un sólido de color blanco: IEN-EM m/z calc.
219,99, encontrado 221,06 (M+1)+; Tiempo de retención: 0,61 minutos.
Formación de 2-cloro-5-(metilsulfonil)benzaldehído (67)
Se disolvió (2-cloro-5-metilsulfonil-fenil)metanol, 66 , (1,00 g, 4,50 mmol) en cloruro de metileno (23 ml). Se añadió peryodinano de Dess-Martin (2,49 g, 5,87 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 16 horas. La solución se diluyó en una solución acuosa saturada de NaHCO3 y se extrajo dos veces con EtOAc. Las fases orgánicas combinadas se lavaron con una solución acuosa saturada de NaHCO3, se secaron (MgSO4), se filtraron y se concentraron al vacío. El residuo en bruto se purificó por cromatografía sobre gel de sílice con una columna isco de 40 g usando un gradiente de EtOAc al 0-20 %/CH2Cl2 para proporcionar 760 mg del producto deseado: RMN 1H (400 MHz, DMSO-cfe) 6 10,36 (s, 1H), 8,31 (d, J = 2,4 Hz, 1H), 8,20 (dd, J = 8,4, 2,4 Hz, 1H), 7,94 (d, J = 8,4 Hz, 1H), 3,32 (s, 3H).
Formación de 3-(2-cloro-5-(metilsulfonil)fenil)-1,4-oxazepano (68)
A una solución de 3-(tributilestanilmetoxi)propan-1-amina (0,88 g, 2,33 mmol) en diclorometano (6 ml), se le añadió 2-cloro-5-metilsulfonil-benzaldehído, 67, (0,51 g, 2,33 mmol) seguido de tamices moleculares de 4 A. La mezcla se agitó durante una noche, se filtró para retirar los tamices y se diluyó con diclorometano (25 ml).
En un matraz separado que contenía hexafluoroisopropanol (7 ml), se añadió 2,6-lutidina (0,28 ml, 2,39 mmol) seguido de Cu(OTf)2 (0,85 g, 2,34 mmol). La mezcla se agitó durante 1 hora, después se añadió la solución de imina preparada
anteriormente en una porción. La reacción se agitó durante 3 días a temperatura ambiente. La mezcla se diluyó con 60 ml de una mezcla 2:1 de una solución acuosa saturada de NaHCO3 e hidróxido de amonio al 10 %. Después de agitar durante 30 minutos, la capa orgánica se retiró y se lavó dos veces con una solución acuosa saturada de NaHCO3, después salmuera. La capa orgánica se pasó a través de un embudo separador de fase y se concentró al vacío. El residuo resultante se purificó por cromatografía de fase inversa sobre gel de sílice usando una columna ISCO - c18-aq de 100 gramos ejecutándose con TFA al 0,1 %/H2O y TFA al 0,1 %/CH3CN. Las fracciones que contenían el producto se concentraron al vacío y el residuo se diluyó con diclorometano y se neutralizó con una solución acuosa saturada de NaHCO3. La fase orgánica se pasó a través de un separador de fase y se concentró al vacío. La RMN 1H muestra el producto deseado más una impureza adicional. Se usó producto sin purificación adicional: RMN 1H (400 MHz, DMSO-d6 ) 68,16 (d, J = 2,4 Hz, 1H), 7,81 (dd, J = 8,4, 2,4 Hz, 1H), 7,70 (d, J = 8,4 Hz, 1H), 7,54 (t, J = 7.6 Hz, 1H), 4,29 (dd, J = 9,0, 3,1 Hz, 1H), 3,91 - 3,76 (m, 2H), 3,76 - 3,63 (m, 1H), 3,22 (s, 3H), 3,12 (dd, J = 12,7, 8.7 Hz, 1H), 2,89 (dt, J = 13,6, 6,8 Hz, 2H), 1,93-1,78 (m, 2H); IEN-EM m/z calc. 390,05, encontrado 390,09 (M+1)+; Tiempo de retención: 0,50 minutos.
Formación de 4-(3-(2-cloro-5-(metilsulfoml)feml)-1,4-oxazepan-4-il)-6-metilpirimidm-2-amma (69)
A una solución de 3-(2-cloro-5-metilsulfonil-fenil)-1,4-oxazepano, 68, (0,200 g, 0,690 mmol) en NMP (6 ml), se le añadió 4-cloro-6-metil-pirimidin-2-amina (0,123 g, 0,857 mmol). La mezcla de reacción se calentó a 150 °C durante 18 horas. La mezcla de reacción se enfrió a temperatura ambiente y se cargó material directamente en una columna ISCO c18-aq de 50 g y se purificó por cromatografía de fase inversa sobre gel de sílice usando una columna ISCO de 50 gramos ejecutándose con TFA al 0,1 %/H2O y TFA al 0,1 %/CH3CN. Las fracciones puras se combinaron y se concentraron al vacío. El residuo se diluyó con diclorometano, se neutralizó con una solución acuosa saturada de NaHCO3, se pasó a través de un separador de fase y la fase orgánica resultante se concentró al vacío para proporcionar 84 mg de un sólido de color pardo: alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6 ) 67,85 -7,73 (m, 2H), 7,71 (d, J = 8,3 Hz, 1H), 5,75 -5,54 (m, 2H), 5,50 -5,34 (m, 2H), 4,48 (d, J = 14,9 Hz, 1H), 4,11 (dd, J = 13,5, 4,9 Hz, 1H), 3,92 -3,65 (m, 3H), 3,67 -3,50 (m, 1H), 3,18-3,11 (m, 3H), 2,03 (d, J = 5,4 Hz, 3H), 1,80 (s, 2H).
La mezcla racémica se sometió a separación SFC quiral: preparado a IPA al 50 %, Hexanos al 50 %, dietilamina al 0,2 % en AD-H
Pico A: (R)-4-(3-(2-cloro-5-(metilsulfonil)fenil)-1,4-oxazepan-4-il)-6-metilpirimidin-2-amina (70), 96,4 % ee mediante HPLC; se calentó (360K) RMN 1H (400 MHz, DMSO-d6j 67,81 (ddd, J = 8,3, 2,3, 1,1 Hz, 1H), 7,78 (d, J = 2,3 Hz, 1H), 7,71 (dd, J = 8,3, 1,2 Hz, 1H), 5,70 (s, 1H), 5,62 (dd, J = 9,9, 4,8 Hz, 1H), 5,43 (s, 2H), 4,48 (d, J = 15,4 Hz, 1H), 4,11 (dd, J = 13,4, 4,9 Hz, 1H), 3,97 - 3,68 (m, 3H), 3,66 - 3,52 (m, 1H), 3,16 (d, J = 1,2 Hz, 3H), 2,04 (d, J = 1,0 Hz, 3H), 1,89-1,70 (m, 2H); IEN-EM m/z calc. 396,10, encontrado 397,25 (M+1)+; Tiempo de retención: 0,56 minutos; [□]□ = -42,40 (c = 5 mg/2ml de MeOH). I-102
Pico B (S)-4-(3-(2-cloro-5-(metilsulfonil)fenil)-1,4-oxazepan-4-il)-6-metilpirimidin-2-amina (71), +98% ee mediante HPLC; se calentó (360K) RMN 1H (400 MHz, DMSO-d6j 67,81 (dd, J = 8,3, 2,3 Hz, 1H), 7,78 (d, J = 2,2 Hz, 1H), 7,71 (d, J = 8,3 Hz, 1H), 5,70 (s, 1H), 5,62 (dd, J = 9,9, 4,9 Hz, 1H), 5,42 (s, 2H), 4,48 (d, J = 15,2 Hz, 1H), 4,11 (dd, J = 13,5, 4,9 Hz, 1H), 3,94 - 3,70 (m, 3H), 3,60 (ddd, J = 12,0, 9,5, 4,7 Hz, 1H), 3,15 (s, 3H), 2,04 (s, 3H), 1,80 (dt, J = 8,3, 4,2 Hz, 2H); IEN-EM m/z calc. 396,10, encontrado 397,20 (M+1)+; Tiempo de retención: 0,55 minutos; [□]□ = 77,82 (c = 5,5 mg/2ml de MeOH). I-103
El siguiente análogo se preparó de acuerdo con el Esquema sintético 8 :

1-(3-(4-(2-amino-6-metilpirimidin-4-il)-1,4-oxazepan-3-il)-4-clorofenil)pirrolidin-2-ona (72) I-119
RMN 1H (400 MHz, DMSO-d6 ) 67,76 (d, J = 2,7 Hz, 1H), 7,45 (dd, J = 8,7, 2,7 Hz, 1H), 7,39 (d, J = 8,8 Hz, 1H), 5,59 (s, 1H), 5,42 (s, 3H), 4,63 (d, J = 15,1 Hz, 1H), 4,12 (dd, J = 13,4, 5,0 Hz, 1H), 3,91 (dt, J = 11,5, 3,8 Hz, 1H), 3,81 -3,73 (m, 2H), 3,72 -3,52 (m, 3H), 2,47 -2,40 (m, 2H), 2,08 -2,01 (m, 2H), 2,00 (s, 3H), 1,80 (ddt, J = 10,9, 7,5, 4,2 Hz, 2 H); IEN-e M m/z calc. 401,16, encontrado 402,0 (M+1 )+; Tiempo de retención: 0,65 minutos.
Ejemplo 9
Esquema sintético 9: (+/-)-4-(2-(2,5-dimetoxifenil)azepan-l-il)-6-metilpirimidin-2-amina (77) I-25

(a) DMF, POCI3, CH2CI2, 0 °C a 40 °C; (b) ácido 2,5-dimetoxifenilborónico, Pd(PhaP)2Ch, DME, 50 °C; (c) Pd/C, HOAc, EtOAc, MeOH; (d) EtMgBr, THF, 0 °C; (e) 2-amino-4-doro-6-metilpirimidina, NMP, 150 °C; (f) separación quiral SFC
Formación de 7-cloro-2,3,4,5-tetrahidro-1H-azepina-1-carbaldehído (73)
Un matraz de fondo redondo de 3 l de 3 bocas equipado con un agitador superior, sonda de temperatura, un embudo de adición, entrada de nitrógeno y condensador de reflujo se cargó con DMF (360 ml, 4,65 mol) en diclorometano (500 ml) y se agitó durante 5 minutos y luego se enfrió a 0 °C. Se añadió POCl3 (220 ml, 2,36 mol) en diclorometano (300 ml) durante 60 minutos mientras se mantenía la temperatura interna por debajo de 7 °C. La mezcla de reacción se calentó a 40 °C (la solución incolora observada se volvió de color naranja pálido) y se agitó a esta temperatura durante 45 minutos. Se añadió azepan-2-ona (85 g, 751,2 mmol) en diclorometano (450 ml) durante 45 minutos a reflujo (Tmáx observada 45 °C). La mezcla de reacción resultante se agitó a esta temperatura durante 3 h, momento en el que el análisis por HPLC reveló el consumo del material de partida. La mezcla de reacción se enfrió a temperatura ambiente, se vertió en hielo picado (3 l) y después se dejó a temperatura ambiente durante 12 h. La capa acuosa se separó, se alcalinizó con K2CO3 sólido hasta pH 9, se dejó calentar a temperatura ambiente y se agitó a esta temperatura durante 18 h. La mezcla se diluyó con diclorometano (2 l) y la capa orgánica se separó. La capa acuosa se extrajo con diclorometano (1 l) y los extractos orgánicos combinados se lavaron con agua (100 ml), salmuera (200 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El residuo se purificó mediante un tapón de gel de sílice usando acetato de etilo al 0 % ^ 30 %/heptano, que contenía Et3N al 1 %, se recogieron las fracciones que contenían el producto deseado, se concentraron a presión reducida para producir 7-cloro-2,3,4,5-tetrahidroazepina-1-carbaldehído (110 g, 92 %) como un aceite transparente e incoloro.
Formación de 7-(2,5-dimetoxifenil)-2,3,4,5-tetrahidro-1H-azepina-1-carbaldehído (74)
Se cargó un matraz rb de 2 bocas en atmósfera de nitrógeno con 7-cloro-2,3,4,5-tetrahidroazepina-1-carbaldehído, 73, (3,00 g, 18,80 mmol), ácido (2,6-dimetoxifenil)borónico (4,45 g, 24,44 mmol), DME (24,67 ml) y cloruro de bis(trifenilfosfina)paladio(II) (0,53 g, 0,75 mmol). La mezcla de reacción se agitó durante una noche a 50 °C. La mezcla se diluyó con agua y diclorometano. Las capas se separaron a través de un separador de fase y la fase orgánica se concentró al vacío. El residuo en bruto se purificó por cromatografía sobre gel de sílice usando una columna ISCO GOLD de 12 g; EtOAc al 10-100 % en heptano) en forma de un aceite de color amarillo pálido. RMN 1H (300 MHz, DMSO-cfe) 87,79 (s, 1H), 7,27 (t, J = 8,4 Hz, 1H), 6,67 (d, J = 8,4 Hz, 2H), 5,41 (t, J = 6,1 Hz, 1H), 3,74 (s, 6 H), 3,69 -3,48 (m, 2H), 2,31 - 2,12 (m, 2H), 1,83-1,48 (m, 4H); IEN-EM m/z calc. 261,14, encontrado 262,15 (M+1)+; Tiempo de retención: 0,84 minutos.
Formación de 2-(2,5-dimetoxifenil)azepano-1-carbaldehído (75)
A una solución de 7-(2,6-dimetoxifenil)-2,3,4,5-tetrahidroazepina-1-carbaldehído, 74, (3,00 g, 11,48 mmol) en MeOH (30 ml) y EtOAc (30 ml), se le añadieron HOAc (9 ml) y Pd/C (0,24 g, 2,30 mmol). El matraz se cargó con un globo de hidrógeno después de purgar tres veces al vacío. La mezcla se agitó a temperatura ambiente durante una noche. La mezcla se filtró a través de celite y se evaporó el disolvente. El aceite en bruto resultante se usó sin purificación adicional.
Formación de (+/-)-2-(2,5-dimetoxifenil)azepano (76)
A una solución de 2-(2,4-dimetoxifenil)azepano-1-carbaldehído, 75, (1,80 g, 6,51 mmol) en THF (50 ml), se le añadió bromuro de etilmagnesio (2,21 g, 2,17 ml de solución 3 M en éter, 6,51 mmol) a 0 °C. La mezcla se agitó a 0 °C durante 3 horas. La mezcla se inactivó cuidadosamente mediante la adición de una solución 2 N de NaOH y después se extrajo con EtOAc. Las capas orgánicas combinadas se lavaron con salmuera y se secaron sobre MgSO4, se filtraron y se concentraron al vacío. El residuo en bruto se purificó por ISCO eluyendo con un gradiente de metanol/diclorometano.
Formación de (R)-4-(2-(2,5-dimetoxifeml)azepan-1-M)-6-metilpirimidm-2-amma (78) y (S)-4-(2-(2,5-dimetoxifenil)azepan-1-il)-6-metilpirimidin-2-amina (79)
A una mezcla de 4-doro-6-metil-pirimidin-2-amina (0,15 g, 1,02 mmol) y 2-(2,4-dimetoxifenil)azepano, 76, (0,24 g, 1,02 mmol) sólidos, en un vial, se le añadió EtOH (2 ml). El vial se puso en la placa caliente y se calentó a 180 °C sin cubrir durante 2 h. El residuo en bruto se purificó por cromatografía sobre gel de sílice en columna ISCO de 40 g eluyendo con un gradiente de MeOHdiclorometano al 20 %/diclorometano para proporcionar 32 mg del producto deseado: RMN 1H (300 MHz, DMSO-cfe) 87,19 (s, 2H), 6,91 (dd, J = 36,7, 8,5 Hz, 1H), 6,61 (s, 1H), 6,52 -6,36 (m, 2H), 5,70 (s, 1H), 4,78 (dd, J = 67,0, 11,7 Hz, 2H), 3,88 (s, 3H), 3,73 (d, J = 2,4 Hz, 5H), 3,62 - 3,14 (m, 6 H), 2,18 (d, J = 39,5 Hz, 5H), 1,95 - 1,70 (m, 5H), 1,34 (dd, J = 49,7, 10,8 Hz, 3H); IEN-EM m/z calc. 342,21, encontrado 343,32 (M+1)+; Tiempo de retención: 0,72 minutos.
El racemato (4,0 g) se sometió a separación SFC (Columna: CI, 4,6*100 mm IC, 20x250 mm Fase móvil: EtOH al 40 % (amoniaco 5 mM), CO2 al 60 % EtOH al 40 % (amoniaco 5 mM), CO2 al 60 % para proporcionar:
Pico A: 1,61 gramos de (R)-4-(2-(2,5-dimetoxifenil)azepan-1-il)-6-metilpirimidin-2-amina (78): ee = 98 %; [a]D (c = 1,0, MeOH) 111,98; RMN 1H (300 MHz, DMSO-d6 ) 86,94 (a, 1H), 6,75 (a, 1H), 6,48 (s, 1H), 5,89 (s, 3H), 4,76 (s a, 1H), 3,83 (s, 3H), 3,64 (s, 3H), 3,32 (s a, 2H), 1,94 (a, 3H), 1,81-0,95 (m, 8H); IEN-EM m/z calc. 342,21, encontrado 343,27 (M+1)+; Tiempo de retención: 0,74 minutos. I-40
Pico B: 1,21 gramos de (S)-4-(2-(2,5-dimetoxifenil)azepan-1-il)-6-metilpirimidin-2-amina (79): ee = 96 %; [a]D (c = 1,0, MeOH) -147,32; RMN 1H (300 MHz, DMSO-c/6 ) 87,56 -7,10 (a, 2H), 6,89 (a, 2H), 6,64 -6,39 (m, 1H), 5,86 -5,65 (m, 1H), 4,83 (a, 1H), 4,05 (d, J = 15,3 Hz, 0,5H), 3,83 (s, 3H), 3,67 (s, 3H), 3,59 - 3,41 (m, 1.5H), 2,22 (d, J = 37,7 Hz, 3h ), 2,00-0,94 (m, 8H); IEN-EM m/z calc. 342,21, encontrado 343,32 (M+1)+; Tiempo de retención: 0,76 minutos. I-41
Los siguientes análogos se prepararon de acuerdo con el Esquema sintético 9:

(+/-)-4-(2-(2-cloro-5-metoxifenil)azepan-1-il)-6-metilpirimidin-2-amina (80) I-117
RMN 1H (300 MHz, DMSO-cfe) 87,77 (s, 1H), 7,52 - 7,28 (m, 2H), 6,90 (ddd, J = 20,8, 8 ,8 , 2,8 Hz, 1H), 6,58 (s, 0H), 5,82 - 5,46 (m, 1H), 5,03 - 4,69 (m, 1H), 4,13 (d, J = 15,2 Hz, 1H), 3,58 - 3,25 (m, 12H), 2,28 (s, 2H), 2,04-1,08 (m, 9H); IEN-e M m/z calc. 346,16, encontrado 347,17 (M+1 )+; Tiempo de retención: 0,72 minutos.

(R)-4-(2-(2-cloro-5-metoxifenM)azepan-1-M)-6-metNpinmidm-2-amma (81) I-135 y (S)-4-(2-(2-cloro-5-metoxifenil)azepan-1-il)-6-metilpirimidin-2-amina (81) I-136
Se sometió 4-[2-(2-cloro-5-metoxi-fenil)azepan-1-il]-6-metil-pirimidin-2-amina (450 mg, 1,289 mmol) a separación SFC. Condiciones de Sf C: Columna: CI, 20x250 mm; Fase móvil: MeOH al 30 % (amoniaco 5 mM), CO2 al 70 %; Flujo: 75 ml/min; Concentraciones: ~40 mg/ ml (MeOH); Volumen de inyección: 500 pl; Longitud de onda: 214 nm; Método tipo isocrático
Pico A: [a]D (c = 0,5, MeOH) 74,56; 99,4 % ee
(R) -4-[2-(2-cloro-5-metoxi-fenil)azepan-1-il]-6-metil-pirimidin-2-amina (81): RMN 1H (300 MHz, DM-SO-d6 ) 87,36 (d, J = 8,8 Hz, 1H), 6,85 (d, J = 8,2 Hz, 1H), 6,62 (s, 1H), 6,07 (a, 2H), 4,78 (s a, 1H), 3,70 (s, 3H), 3,29 (a, 2H), 1,99 (s, 3H), 1,88-1,09 (m, 8H); IEN-EM m/z calc. 346,16, encontrado 347,2 (M+1)+; Tiempo de retención: 0,72 minutos. I-l35
Pico B: [a]D (c = 0,5, MeOH) -76,80; 99 % ee
(S) -4-[2-(2-cloro-5-metoxi-fenil)azepan-1-il]-6-metil-pirimidin-2-amina (82) (200 mg, 89 %): RMN 1H (300 MHz, DMSO-d6) 8 7,78 (s, 1H), 7,39 (m, 2H), 7,01 - 6,77 (m, 1H), 6,62 (d, 3,0 Hz, 1H), 6,49 (s, 0,5H), 5,72 (dd, J = 12,5, 5,1 Hz, 0.5H), 5,55 (s, 0,5H), 5,07 -4,89 (m, 0.5H), 4,79 (d, J = 13,8 Hz, 0.5H), 4,13 (d, J = 15,3 Hz, 0.5H), 3,73 (d, J = 3,3 Hz, 3H), 3,57 (t, J = 11,9 Hz, 1H), 3,18 (s, 1H), 2,28 (s, 1,5H), 2,15 (s, 1,5H), 2,05-0,97 (m, 8H); IEN-EM m/z calc. 346,16, encontrado 347,15 (M+1 )+; Tiempo de retención: 0,72 minutos. I-136
(+/-)-4-[2-(2-fluoro-6-metoxi-fenil)azepan-1-il]-6-metil-pirimidin-2-amina (83) I-50
RMN 1H (300 MHz, DMSO-d6 ) 6 7,44 - 6,59 (m, 5H), 5,68 (s, 1H), 5,04 (d, J = 11,0 Hz, 0.5H), 4,67 (d, J = 14,2 Hz, 0.5H), 3,94 (s, 3H), 3,60 - 3,10 (m, 2H), 2,29 - 1,69 (m, 8H), 1,53-0,87 (m, 3H); IEN-EM m/z calc. 330,19, encontrado 331,29 (M+1)+; Tiempo de retención: 0,72 minutos.

(+/-)-4-[2-(4-clorofenil)azepan-1-il]-6-metil-pirimidin-2-amina (I-43)
RMN 1H (300 MHz, DMSO-d6 ) 67,37 (d, J = 8,2 Hz, 2H), 7,28 (d, J = 8,3 Hz, 2H), 6,84 (s, 2H), 5,81 (s, 1H), 4,76 (a, 1H), 4,00 -2,99 (m, 2H), 2,20 (s, 3H), 1,95-0,97 (m, 8H); IEN-EM m/z calc. 316,15, encontrado 317,24 (M+1)+; Tiempo de retención: 0,72 minutos.
(+/-)-4-[2-(3-clorofenil)azepan-1-il]-6-metil-pirimidin-2-amina (I-44)
RMN 1H (300 MHz, DMSO-d6 ) 67,78 -7,12 (m, 5H), 6,29 (s, 2H), 6,10 -5,47 (m, 1H), 4,43 -3,61 (m, 1H), 3,34 -2,92 (m, 2H), 2,14 (s, 3H), 1,98-0,89 (m, 8H); IEN-EM m/z calc. 316,15, encontrado 317,19 (M+1)+; Tiempo de retención: 0,72 minutos.

(+/-)-4-[2-(3-fluorofenil)azepan-1-il]-6-metil-pirimidin-2-amina I-29
alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6 ) 67,31 (dd, J = 14,4, 7,7 Hz, 1H), 7,06 (d, J = 7,8 Hz, 1H), 6,95 (dd, J = 14,0, 5,8 Hz, 2H), 5,74 (s, 1H), 5,43 (s a, 3H), 4,07 (s a, 1H), 3,16 (dd, J = 13,4, 11,5 Hz, 1H), 2,46 -2,35 (m, 1H), 2,04 (s, 3H), 1,88-1,62 (m, 4H), 1,62 - 1,50 (m, 1H), 1,41 - 1,21 (m, 2H). IEN-EM m/z calc. 300,18, encontrado 301,21 (M+1)+; Tiempo de retención: 0,64 minutos.

(+/-)-4-[2-(2,4-dimetoxifenil)azepan-1-il]-6-metil-pirimidin-2-amina (84) I-51
RMN 1H (300 MHz, DMSO-d6) 67,19 (s, 2H), 6,91 (dd, J = 36,7, 8,5 Hz, 1H), 6,61 (s, 1H), 6,52 -6,36 (m, 2H), 5,70 (s, 1H), 4,78 (dd, J = 67,0, 11,7 Hz, 2H), 3,88 (s, 3H), 3,73 (d, J = 2,4 Hz, 5H), 3,62 - 3,14 (m, 6 H), 2,18 (d, J = 39,5 Hz, 5H), 1,95 - 1,70 (m, 5H), 1,34 (dd, J = 49,7, 10,8 Hz, 3H); IEN-EM m/z calc. 342,20, encontrado 343,32 (M+1)+; Tiempo de retención: 0,72 minutos.

(+/-)-4-(2-cidopentilazepan-1-il)-6-metilpirimidin-2-amina I-9
Una suspensión de 4-doro-6-metil-pirimidin-2-amina (0,094 g, 0,657 mmol), 2-ciclopentilazepano (0,100 g, 0,598 mmol) y P r2NEt (0,230 ml, 1,320 mmol) en IPA (0,6 ml) se cerró herméticamente en un tubo de microondas y se irradió a 160 °C durante 2 horas. La mezcla se concentró al vacío y se purificó por cromatografía de fase inversa (TFA al 0,1 %/acetonitrilo). El material se convirtió en sal HCl para proporcionar 46 mg del producto deseado: RMN 1H (400 MHz, MeOD) 86,31 (2s, 1H), 5,06 - 4,91 (m, 1H), 4,48 - 3,34 (m, 2H), 2,35 - 2,27 (m, 3H), 2,27 - 2,15 (m, 1H), 2,02 (cd, J = 16,7, 8,3 Hz, 1H), 1,92 -1,15 (m, 14H), 1,14-0,96 (m, 1H); IEN-EM m/z calc. 274,22, encontrado 275,18 (M+1)+; Tiempo de retención: 2,84 minutos

(+/-)-4-metil-6-[2-(4-piridil)azepan-1-il]pirimidin-2-amina I-35
IEN-EM m/z calc. 283,18, encontrado 284,22 (M+1)+; Tiempo de retención: 2,14 minutos.

(+/-)-4-[2-(4-fluorofenil)azepan-1-il]-6-metil-pirimidin-2-amina I-34
alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6 ) 87,28 - 7,21 (m, 2H), 7,05 (m, 2H), 5,72 (s, 1H), 5,65 (s, 2H), 5,42 (m, 3H), 4,03 (s, 1H), 3,15 (dd, J = 13,3, 11,6 Hz, 1H), 2,44 -2,34 (m, 1H), 2,03 (s, 3H), 1,90 - 1,65 (m, 4H), 1,63 - 1,48 (m, 1H), 1,42 - 1,21 (m, 2H). IEN-EM m/z calc. 300,18, encontrado 301,22 (M+1)+; Tiempo de retención: 2,97 minutos.

(+/-)-4-metil-6-[2-(p-tolil)azepan-1-il]pirimidin-2-amina I-33
alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6 ) 87,12 - 7,04 (m, 4H), 5,70 (s, 1H), 5,65 (s, 2H), 5,39 (s, 1H), 5,39 - 5,20 (m, 1H), 4,08 (s, 1H), 3,18 - 3,07 (m, 1H), 2,42 - 2,32 (m, 1H), 2,24 (s, 3H), 2,02 (s, 3H), 1,88 - 1,66 (m, 4H), 1,63 - 1,50 (m, 1H), 1,31 (m, 2H). IEN-EM m/z calc. 296,20, encontrado 297,25 (M+1)+; Tiempo de retención: 3,07 minutos.

(+/-)-4-[2-(4-metoxifenil)azepan-1-il]-6-metil-pirimidin-2-amina I-23
alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6 ) 87,12 - 7,04 (m, 4H), 5,70 (s, 1H), 5,65 (s, 2H), 5,39 (s, 1H), 5,39 - 5,20 (m, 1H), 4,08 (s, 1H), 3,18 - 3,07 (m, 1H), 2,42 - 2,32 (m, 1H), 2,24 (s, 3H), 2,02 (s, 3H), 1,88 - 1,66 (m, 4H), 1,63 - 1,50 (m, 1H), 1,31 (m, 2H); IEN-EM m/z calc. 296,20, encontrado 297,25 (M+1)+; Tiempo de retención: 3,07 minutos.
Ejemplo 10
Esquema sintético 10: (+/-)-4-[2-(2,5-dimetoxi-4-piridil)azepan-1-il]-6-metil-pirimidin-2-amina (88) I-116

(a) ácido (2,5-dimetoxi-4-piridil)borónico, Pd(Ph3P)2Cl2, NaHCO3, DME, agua, 60 °C; (c) Pd/C, ácido fórmico, EtOAc, MeOH; (d) nBuLi, THF, -78 °C; (e) 2-amino-4-doro-6-metilpirimidina, NMP, 150 °C; (f) separación quiral SFC
Formación de 7-(2,5-dimetoxi-4-piridil)-2,3,4,5-tetrahidroazepina-1-carbaldehído (85)
Se cargó un matraz rb de 2 bocas en atmósfera de nitrógeno con 7-cloro-2,3,4,5-tetrahidroazepina-1-carbaldehído, 73, (0,79 g, 4,95 mmol), ácido (2,5-dimetoxi-4-piridilo)borónico (1,00 g, 5,46 mmol) en DME (10 ml), seguido de NaHCO3 (8 ml de solución 1,2 M, 9,6 mmol) y cloruro de bis(trifenilfosfina)paladio (II) (0,14 g, 0,20 mmol). Se agitó durante una noche a 60 °C. Se añadieron agua y diclorometano. Las capas se separaron a través de un separador de fase y los orgánicos se concentraron al vacío después de una segunda extracción. La purificación por cromatografía sobre gel de sílice (columna GOLD de 40g; gradiente de EtOAc al 10-100 %/heptanos) proporcionó 1 g (47 %) del producto deseado: RMN 1H (300 MHz, CDCla) 87,94 (s, 1H), 7,75 (s, 1H), 7,28 (d, J = 1,1 Hz, 1H), 6,64 (s, 1H), 3,91 (s, 3H), 3,84 (s, 3H), 3,80 - 3,69 (m, 2H), 2,43 - 2,29 (m, 2H), 1,87 (dd, J = 8,5, 3,8 Hz, 2H), 1,71-1,64 (m, 2H); IEN-EM m/z calc. 262,13, encontrado 263,07 (M+1)+; Tiempo de retención: 0,75 minutos.
Formación de 2-(2,5-dimetoxi-4-piridil)azepano-1-carbaldehído (86)
A una solución de 7-(2,5-dimetoxi-4-piridil)-2,3,4,5-tetrahidroazepina-1-carbaldehído, 85, (1,0 g, 3,8 mmol) en MeOH (20 ml) y EtOAc (20 ml), se le añadieron ácido fórmico (1,7 g, 37,0 mmol) y Pd/C (40 mg, 0,4 mmol) en atmósfera de N2. Después, se cargó la mezcla de reacción con H2 (globo) y se agitó a temperatura ambiente durante una noche. Se filtró a través de celite, el disolvente se evaporó. el residuo se purificó por gel de sílice en columna ISCO (40 g) eluyendo con un gradiente de EtOAc del 0 % al 50 %/heptanos. Las fracciones deseadas se recogieron y se evaporaron para proporcionar 1,0 gramos (51 %) del producto deseado: IEN-EM m/z calc. 264,15, encontrado 265,14 (M+1)+; Tiempo de retención: 0,72 minutos.
Formación de 2-(2,5-dimetoxi-4-piridil)azepano (87)
A una solución de 2-(2,5-dimetoxi-4-piridil)azepano-1-carbaldehído, 86, (1,00 g, 3,78 mmol) en THF (20 ml), se le añadió n-butillitio (5,0 ml de 1,6 M, 8,00 mmol) a -78 °C. La mezcla se agitó a -78 °C durante 2 horas. La mezcla de reacción se inactivó cuidadosamente mediante la adición de MeOH. A la mezcla se le añadió una solución 2 N de HCl hasta que se alcanzó un pH = 2. Después, la solución resultante se basificó mediante la adición de NaOH al 6 N hasta que se alcanzó pH = 10. La solución acuosa se extrajo con EtOAc y las capas orgánicas combinadas se lavaron con salmuera, se secaron sobre MgSO4, se filtraron y se concentraron al vacío para proporcionar 230 mg del producto deseado: IEN-EM m/z calc. 236,15, encontrado 237,15 (M+1)+; Tiempo de retención: 0,58 minutos.
Formación de 4-[2-(2,5-dimetoxi-4-piridil)azepan-1-il]-6-metil-pirimidin-2-amina (88) I-116
A una mezcla de 4-cloro-6-metil-pirimidin-2-amina (0,12 g, 0,81 mmol) y 2-(2,5-dimetoxi-4-piridil)azepano (0,23 g, 0,90 mmol) sólidos, en un vial, se le añadió EtOH (2 ml). El vial se puso en una placa caliente y se calentó a 160 °C sin cubrir durante 2 horas. El sólido en bruto se purificó por cromatografía sobre gel de sílice (40 g) en ISCO eluyendo con MeOH al 20 %/diclorometano - diclorometano para proporcionar 6,5 mg del producto deseado: RMN 1H (300 MHz, DMSO-cfe) 87,85 (s, 1H), 7,56 (a, 2H), 6,48 (s, 0,5H), 6,30 (s, 0,5H), 5,69 (s, 1H), 4,81 (m, 1H), 3,88 (s, 3H), 3,75 (s, 3H), 3,61 - 3,25 (m, 2H), 2,28 (s, 3H), 1,99-1,07 (m, 8H); IEN-EM m/z calc. 343,20, encontrado 344,16 (M+1)+; Tiempo de retención: 0,65 minutos.
Los siguientes análogos se prepararon de acuerdo con el Esquema sintético 10:

4-[2-(2-cloro-5-isopropoxi-fenil)azepan-1-il]-6-metil-pirimidin-2-amina (89) I-104
RMN 1H (300 MHz, DMSO-cfe) 87,78 (s, 1H), 7,50 -7,25 (m, 2H), 6,88 (ddd, J = 20,5, 8 ,8 , 2,8 Hz, 1H), 6,68 -6,39 (m, 1H), 5,81 - 5,46 (m, 1H), 5,09 - 4,72 (m, 1H), 4,56 (m, 1H), 4,13 (d, J = 14,6 Hz, 1H), 3,87 - 3,46 (m, 1H), 2,22 (s, 3H),
2,05 -1,34 (m, 8H), 1,23 (d, J = 5,4 Hz, 6 H); IEN-EM m/z calc. 374,19, encontrado 375,07 (M+1)+; Tiempo de retención: 0,76 minutos.

(R)-4-[2-(2-cloro-5-isopropoxi-feml)azepan-1-M]-6-metN-pinmidm-2-amma (90) I-108
Condiciones de SFC: columna: CI, 4,6*100 mm IC, 20x250 mm; Fase móvil: EtOH al 40 % (amoniaco 5 mM), CO2 al 60 %.
Pico A: [a]D (c = 0,5, MeOH) 58,56; 99 % ee; (R)-4-[2-(2-cloro-5-isopropoxifenil)azepan-1-il]-6-metil-pirimidin-2-amina (90) : RMN 1H (300 MHz, DMSO-da) 87,31 (s, 1H), 6,82 (d, J = 8,9 Hz, 1H), 6,54 (s, 1H), 5,76 (a, 3H), 5,00 - 4,37 (m, 3H), 4,01 - 3,81 (m, 1H), 1,95 (s, 3H), 1,86 - 1,29 (m, 8H), 1,21 (m, 6 H); IEN-EM m/z calc. 374,19, encontrado 375,07 (M+1)+; Tiempo de retención: 0,75 minutos. I-108
Pico B: [a]D(c = 0,5, MeOH) -70,52; 98,4 % ee; (S)-4-[2-(2-cloro-5-isopropoxifenil)azepan-1-il]-6-metilpirimidin-2-amina (91) : RMN 1H (300 MHz, DMSO-d6 ) 8 7,32 (s a, 1H), 6,83 (s a, 1H), 6,55 (s, 1H), 5,83 (a, 3H), 4,96 - 4,43 (m, 2H), 4,02 (a, 2H), 3,17 (s, 3H), 1,95 (s, 3H), 1,86 - 1,31 (m, 8H), 1,26-1,15 (m, 6 H); IEN-EM m/z calc. 374,19, encontrado 375,12 (M+1)+; Tiempo de retención: 0,77 minutos. I-109

3-[1-(2-amino-6-metil-pirimidin-4-il)azepan-2-il]-4-metoxi-fenol (92) I-107
RMN 1H (300 MHz, DMSO-d6 ) 8 8,99 (a, 1H), 7,79 (s, 1H), 7,54 (s, 1H), 6,85 (dd, J = 20,5, 8,7 Hz, 1H), 6,69 - 6,57 (m, 1H), 6,53 -6,43 (m, 1H), 6,31 (d, J = 2,8 Hz, 0.5H), 5,77 (s, 0,5H), 5,70 (s, 1H), 5,09 -4,57 (m, 1H), 4,00 (m, 1H), 3,82 (s, 3H), 3,49 (m, 1H), 2,15 (s, 3H), 1,98-1,01 (m, 8H); IEN-EM m/z calc. 328,19, encontrado 329,17 (M+1)+; Tiempo de retención: 0,67 minutos.

(R)-3-[1-(2-ammo-6-metN-pinmidm-4-N)azepan-2-N]-4-metoxi-fenol (93) I-127
Condiciones de SFC: columna: CI, 4,6*100 mm IC, 20x250 mm; Fase móvil: MeOH al 30 % (amoniaco 5 mM), CO2 al 70 %.
Pico A: [a]D (c = 0,5, MeOH) 100,16; 87,6 % ee
3-[1-(2-amino-6-metil-pirimidin-4-il)azepan-2-il]-4-metoxi-fenol (93): RMN 1H (300 MHz, DMSO-d6 ) 88,84 (a, 1H), 6,81 (s, 1H), 6,55 (s, 1H), 6,39 (s, 1H), 5,80 (s, 2H), 5,32 (s, 1H), 4,74 (s, 1H), 3,79 (m, 3H), 3,30 (m, 2H), 1,92 (s, 3H), 1,82 0,64 (m, 8H); IEN-Em m/z calc. 328,19, encontrado 329,25 (M+1)+; Tiempo de retención: 0,65 minutos. I-127 Pico B: [a]D (c=0,5, MeOH) -98,32; 94 % ee
3-[1-(2-amino-6-metil-pirimidin-4-il)azepan-2-il]-4-metoxi-fenol (94): RMN 1H (300 MHz, DMSO-d6 ) 88,85 (a, 1H), 6,82 (s, 1H), 6,56 (s, 1H), 6,40 (s, 1H), 5,80 (s, 2H), 5,32 (s, 1H), 4,74 (s, 1H), 3,79 (s, 3H), 3,30 (m, 2H), 1,92 (s, 3H), 1,83 0,84 (m, 8H); IEN-Em m/z calc. 328,19, encontrado 329,1 (M+1)+; Tiempo de retención: 0,65 minutos. I-128

(+/-)-4-metil-6-(2-(2-metilpiridin-3-il)azepan-1-il)pirimidin-2-amina (95) I-24
RMN 1H (400 MHz, DMSO-da) 88,30 (d, J = 4,6 Hz, 1H), 7,42 (d, J = 7,9 Hz, 1H), 7,12 (dd, J = 7,8, 4,7 Hz, 1H), 6,94 (s, 2H), 6,18 -6,00 (m, 1H), 5,38 (s, 1H), 4,29 (s, 2H), 3,63 (s, 1H), 2,63 (s, 3H), 2,32 -2,22 (m, 1H), 2,21 (s, 3H), 2,00 1,73 (m, 3H), 1,56-1,28 (m, 4H); iEn -e M m/z calc. 297,2, encontrado 298,2 (M+1)+; Tiempo de retención: 0,49 minutos.

4-(2-(2-fluorofenil)azepan-1-il)-6-metilpirimidin-2-amina (96) I-36
RMN 1H (300 MHz, DMSO-d6 ) 8 7,28 (m, 2H), 7,20 - 7,06 (m, 3H), 6,30 (s, 2H), 6,20 - 5,62 (m, 1H), 3,48 - 3,07 (m, 2H), 2,10 (s a, 3H), 1,98-1,18 (m, 8H); IEN-EM m/z calc. 300,2, encontrado 301,2 (M+1)+; Tiempo de retención: 0,7 minutos.

4-(2-(5-cloro-2-metoxifenil)azepan-1-il)-6-metilpirimidin-2-amina I-21
RMN 1H (300 MHz, DMSO-c/6 ) 87,46 (s a, 1H), 7,37 -7,23 (m, 2H), 7,17 -6,96 (m, 2H), 6,87 (s, 0,5H), 6,43 (s, 0,5H), 5,84 - 5,59 (m, 1H), 4,96 (d, J = 10,2 Hz, 1H), 4,06 (d, J = 15,4 Hz, 1H), 3,88 (d, J = 15,1 Hz, 3H), 3,55 (dt, J = 42,6; 12,7 Hz, 1H), 2,20 (d, J = 38,7 Hz, 3H), 2,01-1,03 (m, 8H); IEN-EM m/z calc. 346,16, encontrado 347,23 (M+1)+; Tiempo de retención: 0,73 minutos.
4-(2-(4-cloro-2-metoxifenil)azepan-1-il)-6-metilpirimidin-2-amina I-20
RMN 1H (300 MHz, DMSO-d6 ) 87,44 - 6,85 (m, 6 H), 6,39 (s, 0,5H), 5,62 (s, 0,5H), 4,81 (dd, J = 71,0, 12,5 Hz, 1H), 4,18 - 3,80 (m, 4H), 3,68 - 3,23 (m, 4H), 2,18 (d, J = 39,0 Hz, 4H), 2,01-1,07 (m, 7H); IEN-EM m/z calc. 346,16, encontrado 347,23 (M+1)+; Tiempo de retención: 0,74 minutos.

4-(2-(4-fluoro-2-metoxifenil)azepan-1-il)-6-metilpirimidin-2-amina I-22
RMN 1H (400 MHz, DMSO-d6 ) 86,98 (dd, J = 8,4, 6,9 Hz, 1H), 6,84 (dd, J = 11,2, 2,5 Hz, 1H), 6,61 (td, J = 8,5; 2,5 Hz, 1H), 5,56 (s, 1H), 5,35 (s, 2H), 5,14 (s, 1H), 4,38 (s, 1H), 3,36 -3,26 (m, 1H), 2,39 -2,29 (m, 1H), 1,98 (s, 3H), 1,81 (d, J = 47,9 Hz, 3H), 1,69 - 1,44 (m, 2H), 1,40-1,18 (m, 2H); IEN-EM m/z calc. 330,2, encontrado 331,2 (M+1)+; Tiempo de retención: 0,67 minutos.
Ejemplo 11
Esquema sintético 11: (+/)-4-(2-(2-metoxi-4-(metilsulfonil)fenil)azepan-1-il)-6-metilpirimidin-2-amina (100) I-88

(a) 1-bromo-2-metoxi-4-metilsulfonil-benceno, bis(pinocalatodiboro), PdCl2(Ph3P)2, KOAc, dioxano, agua, 85 °C; (b) (c) Pd/C, ácido fórmico, EtOAc, MeOH; (d) nBuLi, Th F, -78 °C; (e) 2-amino-4-doro-6-metilpirimidina, NMP, 150 °C; (f) separación quiral SFC
Formación de 7-(2-metoxi-4-metilsulfonil-fenil)-2,3,4,5-tetrahidroazepina-1-carbaldehído (97)
Etapa 1: A una solución de 1-bromo-2-metoxi-4-metilsulfonil-benceno (1,00 g, 3,77 mmol) en dioxano (50 ml), se le añadieron bis(pinocalatodiboro) (1,44 g, 5,66 mmol), acetato potásico (1,11 g, 11,32 mmol). La mezcla se purgó con nitrógeno durante 15 minutos y se añadió dicloro-bis(trifenilfosforanil)-paladio (0,27 g, 0,37 mmol). La reacción se calentó a 85 °C durante 18 horas. La mezcla se diluyó con EtOAc y se filtró a través de celite lavado con EtOAc (60 ml). La fase orgánica se concentró al vacío. El sólido resultante de color pardo oscuro se usó sin purificación adicional.
Etapa 2: El producto en bruto anterior se disolvió en DME (30 ml). Se añadió 7-cloro-2,3,4,5-tetrahidroazepina-1-carbaldehído, 73, (0,60 g, 3,77 mmol) seguido de NaHCO3 (6,3 ml de solución 1,2 M, 7,54 mmol). La mezcla se burbujeó con nitrógeno y se añadió un catalizador de Pd(dppf)Cl2. El matraz se cubrió y se calentó a 80 °C durante 12 horas. El residuo se purificó por cromatografía sobre gel de sílice usando (columna ISCO de 40 g) un gradiente de MeOH-diclorometano al 20 %/diclorometano. Las fracciones deseadas se recogieron y se evaporaron. Las fracciones se recogieron y se usaron durante la siguiente etapa directamente.
Formación de 2-(2-metoxi-4-metilsulfonil-fenil)azepano-1-carbaldehído (98)
A una solución de 7-(2-metoxi-4-metilsulfonil-fenil)-2,3,4,5-tetrahidroazepina-1-carbaldehído, 97, (1,00 g, 3,23 mmol) en MeOH (10 ml) y EtOAc (10 ml), se le añadió ácido acético (1 ml). En una atmósfera de nitrógeno, se añadió Pd al 10 %/C (10 % mol). La mezcla de reacción se purgó 3 veces con hidrógeno y después se agitó en una atmósfera de hidrógeno durante 14 horas. La CLEM indicó mala conversión al producto deseado. La mezcla se filtró a través de celite y el disolvente se concentró parcialmente al vacío. El procedimiento anterior se repitió, excepto que se usó ácido fórmico para reemplazar ácido acético. Después de agitación durante una noche, el material de partida se convirtió en el producto deseado. La mezcla se filtró a través de celite y el filtrado se concentró al vacío. El residuo resultante se purificó por cromatografía sobre gel de sílice (columna ISCO de 40 g) eluyendo con un gradiente de EtOAc/heptanos (0-75 %) para proporcionar 760 mg del producto deseado: RMN 1H (300 MHz, CDCh) 88,20 (s, s, 1H), 7,63 - 7,38 (m, 2H), 7,35 -7,16 (m, 1H), 5,41 (dd, J = 12,3, 4,7 Hz, 0,6H), 5,00 (dd, J = 11,7, 5,6 Hz, 0.4H), 4,35 (d, J = 13,7 Hz, 0.4H), 3,96 (s, 3H), 3,91 -3,79 (m, 0.6H), 3,56 (dd, J = 14,9, 11,3 Hz, 0.6H), 3,06 (s, 3H), 3,01 -2,85 (m, 0.4H), 2,46 (m, 1H), 2,19 -1,17 (m, 7H).
Formación de 2-(2-metoxi-4-metilsulfonil-fenil)azepano (99)
A una solución de 2-(2-metoxi-4-metilsulfonil-fenil)azepano-1-carbaldehído, 98, (0,76 g, 2,44 mmol) en MeOH (20 ml), se le añadió HCl (10 ml de 12 M, 120 mmol). La mezcla se calentó a 100 °C durante 4 horas. El disolvente se concentró al vacío para proporcionar 600 mg del producto deseado que se usó sin purificación adicional: RMN 1H (300 MHz, DMSO-d6 ) 89,49 (a, 2H), 7,82 (d, J = 8,0 Hz, 1H), 7,65 - 7,48 (m, 2H), 4,59 (t, J = 9,2 Hz, 1H), 3,97 (s, 3H), 3,27 (s, 3H), 3,19 -3,08 (m, 2H), 2,34-1,51 (m, 8H); IEN-EM m/z calc.283,12, encontrado 284,27 (M+1)+; Tiempo de retención: 0,56 minutos.
Formación de (+/-)-4-[2-(2-metoxi-4-metilsulfonil-fenil)azepan-1-il]-6-metil-pirimidin-2-amina (100) I-88
A una mezcla de 4-cloro-6-metil-pirimidin-2-amina (0,10 g, 0,68 mmol) y 2-(2-metoxi-4-metilsulfonil-fenil)azepano-HCl, 99, (0,25 g, 0,78 mmol) sólidos, en un vial, se le añadió EtOH (2 ml). El vial se puso en una placa caliente y se calentó
a 170 °C sin cubrir durante 2 horas. El sólido en bruto se purificó por cromatografía sobre gel de sílice (columna ISCO de 40 g) eluyendo con MeOH-diclorometano al 20 %/diclorometano para proporcionar 182 mg del producto deseado: RMN 1H (300 MHz, DMSO-d6 ) 67,59 -7,38 (m, 2H), 7,22 (s, 1H), 6,06 -5,23 (m, 3H), 4,82 (s a, 1H), 4,00 (s, 3H), 3,31 (s a, 2H), 3,22 (s, 3H), 1,94 (s, 3H), 1,87-1,01 (m, 8H); IEN-EM m/z calc. 390,17, encontrado 391,09 (M+1)+; Tiempo de retención: 0,66 minutos.
La mezcla racémica (182 mg) se sometió a separación quiral SFC.
Condiciones de SFC: Columna: AD-H, 4,6x100 mm AD-H, 10x250 mm; Fase móvil: EtOH al 40 % (amoniaco 5 mM), CO2 al 60 %
I-98 Pico A: [a]D (c = 0,5, MeOH) -72,39; ee = 99 %
4-[2-(2-metoxi-4-metilsulfonil-fenil)azepan-1-il]-6-metil-pirimidin-2-amina (101): RMN 1H (400 MHz, DMSO-d6 ) 67,59 -7,40 (m, 2H), 7,26 (d, J = 8,0 Hz, 1H), 5,64 (s, 1H), 5,40 (a, 2H), 4,39 (s a, 1H), 4,01 (s, 3H), 3,57 - 3,34 (m, 1H), 3,17 (s, 3H), 2,48 - 2,29 (m, 1H), 2,03 (s, 3H), 1,93 - 1,04 (m, 8H). IEN-EM m/z calc. 390,17, encontrado 391,09 (M+1)+; Tiempo de retención: 0,67 minutos.
I-99 Pico B: [a]D (c = 0,5, MeOH) 86,51; ee = 99,6 %
4-[2-(2-metoxi-4-metilsulfonil-fenil)azepan-1-il]-6-metil-pirimidin-2-amina (102): RMN 1H (400 MHz, DMSO-d6 ) 6 7,39 (d, J = 1,6 Hz, 1H), 7,36 -7,26 (m, 1H), 7,16 (d, J = 7,9 Hz, 1H), 5,54 (s, 1H), 5,30 (s, 2H), 4,28 (s a, 1H), 3,92 (s, 3H), 3,35 - 3,20 (m, 1H), 3,07 (s, 3H), 2,40 - 2,26 (m, 1H), 1,93 (s, 3H), 1,87-1,10 (m, 8H); IEN-EM m/z calc. 390,17, encontrado 391,05 (M+1)+; Tiempo de retención: 0,66 minutos.
Los siguientes análogos se prepararon de acuerdo con el Esquema sintético 11:

(+/-)-4-[2-(3-metoxi-4-piridil)azepan-1-il]-6-metil-pirimidin-2-amina (103) I-80
RMN 1H (400 MHz, DMSO-d6 ) 68,57 (d, J = 5,9 Hz, 1H), 8,34 (d, J = 5,3 Hz, 1H), 7,62 (s, 2H), 7,36 (d, J = 5,3 Hz, 1H), 6,52 (s, 1H), 5,69 (m, 1H), 4,06 (s, 3H), 3,78 - 3,37 (m, 2H), 2,28 (s, 3H), 2,03-1,14 (m, 8H); IEN-EM m/z calc.
313,19, encontrado 314,14 (M+1)+; Tiempo de retención: 0,56 minutos.
(+/-)-4-(2-(2-metoxipiridin-3-il)azepan-1-il)-6-metilpirimidin-2-amina I-18
RMN 1H (400 MHz, DMSO-d6 ) 68,00 (dd, J = 4,9, 1,8 Hz, 1H), 7,34 (dd, J = 7,2, 1,6 Hz, 1H), 6,86 (dd, J = 7,3, 4,9 Hz, 1H), 5,61 (s, 1H), 5,36 (s, 2H), 5,19 (s, 1H), 4,32 (s, 1H), 3,96 (s, 3H), 3,37 -3,28 (m, 1H), 2,96 (s, 5H), 2,45 -2,35 (m, 1H), 2,00 (s, 3H), 1,88 (s, 1H), 1,75 (s, 2H), 1,70 -1,44 (m, 3H), 1,41-1,20 (m, 2H); IEN-EM m/z calc. 313,2, encontrado 314,2 (M+1)+; Tiempo de retención: 0,61 minutos.
Ejemplo 12
Esquema sintético 12: (+/-)-4-(2-(2-etilfenil)azepan-1-il)-6-metilpirimidin-2-amina (107) I-45
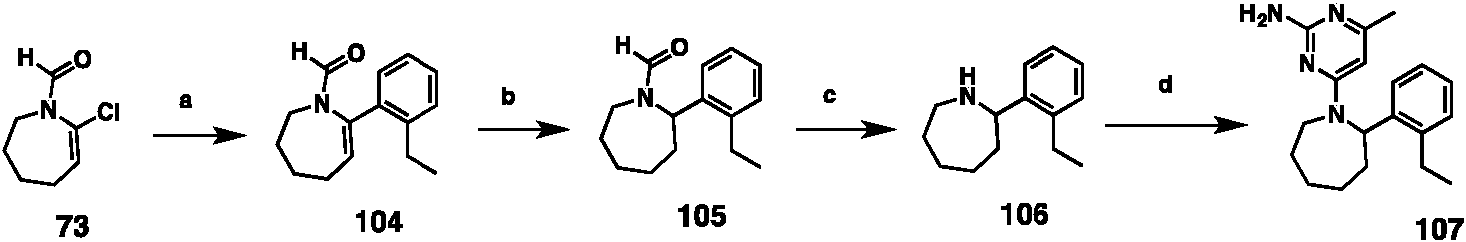
(a) ácido (2-etilfenil)borónico, PdCh(dppf), NaHCO3, DMF, agua, 85 °C; (b) Pd/C, ácido fórmico, EtOAc, MeOH; (c) nBuLi, THF, -78 °C; (d) 2-amino-4-cloro-6-metilpirimidina, NMP, 150 °C; (e) separación SFC quiral
Formación de 7-(2-etilfeml)-2,3,4,5-tetrahidro-1H-azepina-1-carbaldehído (104)
Una mezcla de 7-cloro-2,3,4,5-tetrahidroazepina-1-carbaldehído, 73, (1,5 g, 9,4 mmol), ácido (2-etilfenil)borónico (1,4 g, 9,4 mmol) y PdCh(dppf) (0,4 g, 0,5 mmol) en DMF (30 ml) y una solución acuosa saturada de NaHCO3 (10 ml) se calentó con irradiación de microondas a 80 °C durante 30 minutos. La mezcla se filtró sobre Celite, se diluyó con EtOAc y se lavó con agua. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró al vacío. El residuo resultante se purificó por cromatografía sobre gel de sílice eluyendo con EtOAc al 0-50 %/heptano. Las fracciones puras se combinaron y se concentraron para proporcionar 1,68 g del producto deseado en forma de un aceite incoloro: RMN 1H (400 MHz, CDCla) 87,94 (s, 1H), 7,29 (ddd, J = 7,7, 6,3, 2,2 Hz, 1H), 7,23 -7,14 (m, 3H), 5,39 (t, J = 5,7 Hz, 1H), 3,96 - 3,87 (m, 2H), 2,57 (c, J = 7,5 Hz, 2H), 2,39 - 2,32 (m, 2H), 1,92 (tt, J = 6,2, 5,2 Hz, 2H), 1,84 -1,74 (m, 2H), 1,20 (t, J = 7,6 Hz, 3H); IEN-EM m/z calc. 229,1, encontrado 230,0 (M+1)+; Tiempo de retención: 1,05 minutos.
Formación de (+/-)-2-(2-etilpbenil)azepano-1-carbaldehído (105)
Una mezcla de 7-(2-etilfenil)-2,3,4,5-tetrahidro-1H-azepina-1-carbaldehído, 104, (1,68 g, 7,33 mmol) y Pd húmedo/C (0,79 g, 0,37 mmol) en acetato de etilo (25 ml) y MeOH (25 ml) se agitó durante una noche a 0,38 MPa (55 psi) de hidrógeno. La mezcla de reacción se filtró a través de Celite y el lecho de filtro se aclaró con EtOAc. El filtrado se secó sobre sulfato de magnesio, se filtró y se concentró al vacío para dar 1,47 g de un aceite de color amarillo claro: RMN 1H (400 MHz, DMSO-d6) 88,06 (d, J = 55,7 Hz, 1H), 7,22 -7,05 (m, 4H), 5,04 (ddd, J = 106,0, 12,0, 4,9 Hz, 1H), 4,13 - 4,00 (m, 0.5H), 3,90 - 3,78 (m, 1H), 3,63 - 3,50 (m, 1H), 3,25 - 3,15 (m, 0.5H), 2,84 - 2,63 (m, 2H), 2,22 - 1,66 (m, 5H), 1,49-1,11 (m, 6 H); IEN-EM m/z calc. 231,2, encontrado 232,0 (M+1)+; Tiempo de retención: 1,03 minutos.
Formación de (+/-)-2-(2-etilfenil)azepano (106)
Una solución de 2-(2-etilfenil)azepano-1-carbaldehído, 105, (1,47 g, 6,35 mmol) en MeOH (5 ml) y HCl concentrado (5 ml de solución 12,1 M, 60,50 mmol) se calentó a reflujo durante una noche. La mezcla resultante se concentró a sequedad, se disolvió en una cantidad mínima de MeOH y se vertió en éter dietílico frío mientras se agitaba vigorosamente. El precipitado de color blanco resultante se filtró y se secó para dar 1,15 g del producto deseado como una sal HCl: RMN 1H (300 MHz, DMSO-d6) 89,72 (s, 1H), 9,26 (s, 1H), 7,76 - 7,61 (m, 1H), 7,41 - 7,18 (m, 3H), 4,43 (d, J = 10,6 Hz, 1H), 3,42 -3,02 (m, 2H), 2,72 (ddt, J = 19,3, 14,6, 7,3 Hz, 2H), 2,33 - 1,45 (m, 6 H), 1,16 (t, J = 7,5 Hz, 3h ); IEN-EM m/z calc. 203,2, encontrado 204,0 (M+1)+; Tiempo de retención: 0,67 minutos.
Formación de (+/-)-4-(2-(2-etilfenil)azepan-1-il)-6-metilpirimidin-2-amina (107) I-45
Una mezcla de 2-(2-etilfenil)azepano-HCl, 106, (0,15 g, 0,63 mmol), 4-cloro-6-metil-pirimidin-2-amina (0,09 g, 0,63 mmol) y trietilamina (0,17 ml, 1,25 mmol) en NMP (2 ml) se agitó durante 5 horas en un tubo cerrado herméticamente a 150 °C. La mezcla de reacción en bruto se purificó por cromatografía de fase inversa sobre gel de sílice inyectándose directamente en una columna C18 acuosa iSCO de 50 g y eluyendo con MeCN al 5-50 % en agua con TFA al 0,1 %. Las fracciones puras se combinaron, se neutralizaron con bicarbonato sódico saturado y se extrajeron con EtOAc. La capa orgánica se secó sobre sulfato de magnesio, se filtró, se concentró al vacío y se liofilizó para proporcionar 45 mg del producto deseado: RMN 1H (400 MHz, DMSO-d6) 57,18 (d, J = 7,5 Hz, 1H), 7,15 - 7,09 (m, 1H), 7,09 - 7,04 (m, 2H), 5,67 (s, 1H), 5,54 (s, 2H), 5,27 (s, 1H), 4,33 (d, J = 14,8 Hz, 1H), 3,54 - 3,40 (m, 1H), 2,88 - 2,69 (m, 2H), 2,16 (ddd, J = 14,2, 8,2, 5,1 Hz, 1H), 2,01 (s, 3H), 1,96 - 1,67 (m, 3H), 1,52 (c, J = 12,9, 12,4 Hz, 1H), 1,46 - 1,33 (m, 2H), 1,33-1,24 (m, 4H); IEN-EM m/z calc. 310,2, encontrado 311,0 (M+1)+; Tiempo de retención: 0,84 minutos.
El siguiente análogo se preparó de acuerdo con el Esquema sintético 12:

3-(1-(2-ammo-6-metilpirimidm-4-il)azepan-2-il)-4-fluoro-N-metilbenzamida (108) I-55
RMN 1H (400 MHz, DMSO-d6 ) 88,14 -8,06 (m, 1H), 7,71 (ddd, J = 8,5, 5,0, 2,3 Hz, 1H), 7,62 (dd, J = 7,4, 2,3 Hz, 1H), 7,19 -7,13 (m, 1H), 5,80 (s, 1H), 5,68 (s, 2H), 5,41 (d, J = 35,0 Hz, 1H), 4,25 (m, 1H), 3,49 -3,42 (m, 1H), 2,76 (d, J = 4,5 Hz, 3H), 2,32 (dt, J = 14,3, 7,1 Hz, 1H), 2,06 (s, 3H), 1,95 - 1,69 (m, 4H), 1,55 (p, J = 11,5 Hz, 1H), 1,45-1,22 (m, 2H); IEN-EM m/z calc. 357,2, encontrado 358,0 (M+1)+; Tiempo de retención: 0,67 minutos.
Ejemplo 13
Esquema sintético 13: (+/-)-3-[4-(2-amino-6-metil-pirimidin-4-il)-1,4-oxazepan-3-il]-4-cloro-fenol (112) I-147

(a) ácido 2,5-dimetoxifenilborónico, Pd(PhaP)2Cl2, DME, 50 °C; (b) nBuLi, THF, -78 °C; (c) NaBH4, MeOH; (d) 2-amino-4-doro-6-metilpirimidina, EtOH, 160 °C; (e) separación SFC quiral
Formación de 3-cloro-6,7-dihidro-1,4-oxazepina-4(5H)-carbaldehído (109)
Se preparó el Intermedio, 109, de acuerdo con el Esquema sintético 9 usando 1,4-oxazepan-3-ona en lugar de azepan-2 -ona.
Formación de 3-(2-cloro-5-hidroxifenN)-6,7-dihidro-1,4-oxazepma-4(5H)-carbaldehído (110)
Se cargó un matraz rb de 2 bocas en atmósfera de nitrógeno con 3-cloro-6,7-dihidro-5H-1,4-oxazepina-4-carbaldehído, 109, (0,46 g, 2,80 mmol), NaHCO3 (6,0 ml de solución 1,2 M, 7,2 mmol) y ácido (2-cloro-5-hidroxi-fenil)borónico (0,50 g, 2,90 mmol) en dimetoxietano (10 ml). Después, se añadió cloruro de bis(trifenil-fosfina)paladio (II) (0,10 g, 0,14 mmol), la mezcla de reacción se calentó durante una noche a 60 °C. La mezcla se diluyó en agua y diclorometano. Las capas se separaron a través de un separador de fase y los orgánicos se concentraron al vacío después de una segunda extracción. La purificación por cromatografía sobre gel de sílice (columna GOLD de 40g; gradiente de EtOAc al 10-100 %/heptanos) proporcionó 650 mg del producto deseado: RMN 1H (300 MHz, CDCfe) 87,93 (s, 1H), 7,28 (s, 0H), 7,20 (d, J = 8,4 Hz, 1H), 6,81 (d, J = 2,7 Hz, 1H), 6,13 (s, 1H), 4,25 (t, J = 5,8 Hz, 2H), 4,11 (t, J = 6,3 Hz, 3H), 2,15 (t, J = 6,2 Hz, 2H); IEN-EM m/z calc. 253,05, encontrado 252,29 (M+1)+; Tiempo de retención: 0,55 minutos.
Formación de (+/-)-4-cloro-3-(1,4-oxazepan-3-il)fenol (111)
A una solución fría (-78 °C) de 3-(2-cloro-5-hidroxi-fenil)-6,7-dihidro-5H-1,4-oxazepina-4-carbaldehído, 110, (0,65 g, 1,55 mmol) en THF (10 ml), se le añadió n-butillitio (3 ml de solución 1,6 M, 4,80 mmol). La mezcla se agitó a esta temp. durante 50 minutos. La mezcla de reacción se inactivó cuidadosamente mediante la adición de metanol. Se añadió más MeOH (30 ml) y la solución se calentó a temperatura ambiente. La solución resultante se usó directamente durante la siguiente etapa; IEN-EM m/z calc. 225,06, encontrado 226,08 (M+1)+; Tiempo de retención: 0,54 minutos.
A la solución anterior se le añadió NaBH4 (0,09 g, 2,38 mmol). La mezcla se agitó a temperatura ambiente durante una noche. La reacción se interrumpió mediante la adición de MeOH y después una solución HCl 2 N. Después, la solución ácida se basificó con NaOH 6 N y la fase acuosa se extrajo con EtOAc tres veces. Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre MgSO4, se filtraron y se evaporaron. El producto en bruto (300 mg, 77 %) se obtuvo y se usó directamente; IEN-EM m/z calc. 227,07, encontrado 228,09 (M+1)+; Tiempo de retención: 0,54 minutos.
Formación de (+/-)-3-(4-(2-amino-6-metilpirimidin-4-il)-1,4-oxazepan-3-il)-4-clorofenol (112) I-147
A una mezcla de 4-cloro-6-metil-pirimidin-2-amina (0,30 g, 2,04 mmol) y 4-cloro-3-(1,4-oxazepan-3-il)fenol, 111, (1,43 g, 2,22 mmol) sólidos, en un vial, se le añadió EtOH (2 ml). El vial se puso en una placa caliente y se calentó a 160 °C sin cubrir durante 2 horas. El sólido en bruto se purificó por columna ISCO sobre gel de sílice (40 g) eluyendo con un gradiente de MeOH al 20 %-DCM/DCM (B al 0 % a B al 50 %) para proporcionar 192 mg del producto deseado: RMN 1H (300 MHz, DMSO-cfe) 89,80 (s, 1H), 7,72 (s a, 1H), 7,48 -7,18 (m, 2H), 6,90 -6,54 (m, 2.5H), 5,93 (a, 0,5H), 5,59 (s, 0,5H), 5,26 - 5,07 (m, 0.5H), 4,99 (m, 0,5H), 4,43 - 4,04 (m, 1.5H), 3,86 (m, 3H), 3,62 (t, J = 10,2 Hz, 1H), 2,18 (s, 3h ), 1,84 (m, 2H); IEN-EM m/z calc. 334,12, encontrado 335,10 (M+1)+; Tiempo de retención: 0,6 minutos. La mezcla racémica (180 mg) se sometió a purificación SFC quiral para obtener los enantiómeros individuales. Condiciones de SFC: Columna: Cellulose-2, 20x250 mm; Fase móvil: EtOH al 30 % (amoniaco 5 mM), CO2 al 70 %; Flujo: 80 ml/min; Concentraciones: ~18 mg/ ml (MeOH); Volumen de inyección: 250 pl; Longitud de onda: 220 nm
(I-169) Pico A: 96,8 % ee [a]p (c = 0,5, MeOH) -85,10
(R) -3-[4-(2-amino-6-metil-pirimidin-4-il)-1,4-oxazepan-3-il]-4-cloro-fenol (113): RMN 1H (300 MHz, DMSO-cfe) 89,73 (s, 1H), 7,25 (d, J = 8,6 Hz, 1H), 6,75 - 6,60 (m, 2H), 6,40 (s, 2H), 5,01 (s, 1H), 4,68 - 4,54 (m, 1H), 4,12 (a, 1H), 3,99 -3,86 (m, 2H), 3,83 - 3,48 (m, 3H), 2,04 (s a, 3H), 1,76 (m, 2H); IEN-EM m/z calc. 334,12, encontrado 335,11 (M+1)+; Tiempo de retención: 0,61 minutos.
(I-170) Pico B: 95,4 % ee [a]p (c = 0,5, MeOH) 79,40
(S) -3-[4-(2-amino-6-metil-pirimidin-4-il)-1,4-oxazepan-3-il]-4-cloro-fenol (114): RMN 1H (300 MHz, DMSO-cfe) 89,75 (s, 1H), 7,25 (d, J = 8,5 Hz, 1H), 6,79 - 6,63 (m, 2H), 6,57 (s, 1,5H), 5,34 (s a, 0,5H), 5,01 (s a, 1H), 4,71 - 4,53 (m, 1H), 4,13 (a, 1H), 3,91 (m, 2H), 3,83 - 3,48 (m, 3H), 2,06 (a, 3H), 1,77 (m, 2H); IEN-EM m/z calc. 334,12, encontrado 335,10 (M+1)+; Tiempo de retención: 0,63 minutos.
Los siguientes análogos se prepararon de acuerdo con el Esquema sintético 13:

(+/-)-4-(3-(2-cloro-5-metoxifenil)-1,4-oxazepan-4-il)-6-metilpirimidin-2-amina (115) I-118
RMN 1H (300 MHz, DMSO-d6 ) 67,95 (s a, 1H), 7,59 (s, 1H), 7,43 (dd, J = 24,4, 8,7 Hz, 1H), 7,07 - 6,88 (m, 1H), 6,84 - 6,72 (m, 1H), 6,07 - 5,47 (m, 1H), 5,09 (dd, J = 58,8, 11,2 Hz, 1H), 4,38 -4,05 (m, 2H), 4,01 - 3,48 (m, 7H), 2,29 (s, 3H), 1,84 (a, 2h ). IEN-EM m/z calc. 348,13, encontrado 349,11 (M+1)+; Tiempo de retención: 0,67 minutos.

116 117
(+/-)-4-(3-(2-cloro-5-metoxifenil)-1,4-oxazepan-4-il)-6-metilpirimidin-2-amina (115) I-118
La mezcla racémica 115 se sometió a purificación SFC quiral para obtener los enantiómeros individuales.
Condiciones de SFC: Columna: AD-H, 20x250 mm; Fase móvil: IPA al 30 % (amoniaco 5 mM), CO2 al 70 %; Flujo: 75 ml/min; Concentraciones: ~75 mg/ ml (MeOH); Volumen de inyección: 500 pl; Longitud de onda: 214 nm.
Pico A: sólido de color blanco, ee 99,4 %; [a]o (c = 0,5, MeOH) -67,96 (R)-4-[3-(2-cloro-5-metoxi-fenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (116): RMN 1H (300 MHz, DMSO-d6j 57,94 (s, 1H), 7,59 (s, 1H), 7,52 - 7,16 (m, 1H), 7,09 - 6,88 (m, 1H), 6,86 - 6,75 (m, 1H), 6,58 (s, 0,5H), 5,95 (a, 0,5H), 5,60 (s, 0,5H), 5,17 (s, 0,5H), 4,99 (a, 0,5H), 4,22 (m, 1H), 4,17 - 4,03 (m, 0.5H), 3,88 (m, 2H), 3,75 (s, 3H), 3,61 (m, 2H), 2,23 (s, s, 3H), 1,84 (a, 2H); IEN-EM m/z calc. 348,13, encontrado 349,15 (M+1 )+; Tiempo de retención: 0,64 minutos. I-137
Pico B: sólido de color blanco, ee 99,2 %; [a]o (c = 0,5, MeOH) 60,08
(S)-4-[3-(2-cloro-5-metoxi-fenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (117) RMN 1H (300 MHz, DMSO-d6J 6 7,39 (d, J = 8,5 Hz, 1H), 6,91 (dd, J = 8,7, 3,0 Hz, 1H), 6,75 (t, J = 2,3 Hz, 1H), 6,27 (s, 2H), 4,32 (s, 1H), 4,11 (d, J = 13,3 Hz, 1H), 3,92 (d, J = 11,8 Hz, 1H), 3,85 - 3,66 (m, 5H), 3,57 (d, J = 12,3 Hz, 1H), 3,25 (d, J = 43,7 Hz, 1H), 2,05 (s, 3H), 1,77 (s, 2h ); IEN-EM m/z calc. 348,14, encontrado 349,15 (M+1)+; Tiempo de retención: 0,64 minutos. I-138

118
(+/-)-4-[3-(2,5-dimetoxifenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (118) I-93
RMN 1H (300 MHz, DMSO-d6 ) 67,61 - 7,24 (a, 2H), 7,10 - 6,77 (m, 2H), 6,77-6,42 (a, 1H), 5,84 (a, 1H), 5,04 (a, 1H), 4,22 (a, 1H), 4,00 - 3,44 (m, 12H), 2,34 - 1,62 (m, 3H), 1,75 (a, 2H); IEN-EM m/z calc. 344,18, encontrado 345,06 (M+1)+; Tiempo de retención: 0,65 minutos.

La mezcla racémica se sometió a purificación SFC quiral para obtener los enantiómeros individuales. Condiciones de SFC: Columna: AD-H, 10x250 mm; Fase móvil: EtOH al 30 % (amoniaco 5 mM), CO2 al 70 %; Flujo: 15 ml/min; Concentraciones: ~40 mg/ ml (MeOH); Volumen de inyección: 100 pl; Longitud de onda: 214 nm.
Pico A: sólido de color blanco, ee 97,6 %; [a]o (c = 0,5, MeOH) -96,56
(R) -4-[3-(2,5-dimetoxifenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (119): RMN 1H (400 MHz, DMSO-d6 ) 6 6,99
(d, J = 8,9 Hz, 1H), 6,84 (dd, J = 8,9, 3,1 Hz, 1H), 6,69 (s, 1H), 6,24 (s, 3H), 5,81 (s, 2H), 5,46 (s, 1H), 4,67 (s, 1H), 4,18 (dd, J = 13,1, 5,2 Hz, 1H), 3,94 (d, J = 12,5 Hz, 2H), 3,85 (s, 1H), 3,77 - 3,66 (m, 7H), 3,60 - 3,48 (m, 2H), 2,11 (s, 2 H), 1,89-1,70 (m, 3H); Ie N-EM m/z calc. 344,18, encontrado 345,06 (M+1 )+; Tiempo de retención: 0,65 minutos.
I-100
Pico B: sólido de color blanco, ee 99,6 %; [a]D (c = 0,5, MeOH) 98,16;
(S) -4-[3-(2,5-dimetoxifenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (120): RMN 1H (400 MHz, DMSO-d6 ) 6 6,95 (d, J = 8,9 Hz, 1H), 6,79 (dd, J = 8,9, 3,1 Hz, 1H), 6,64 (d, J = 3,1 Hz, 1H), 5,68 (s, 2H), 5,64 (s, 1H), 5,37 (s, 1H), 4,65 (d, J = 14,8 Hz, 1H), 4,15 (dd, J = 13,3, 5,2 Hz, 1H), 3,98 -3,87 (m, 1H), 3,82 (s, 3H), 3,71 -3,58 (m, 2H), 3,65 (s, 3H), 3,50 (td, J = 11,5, 4,0 Hz, 1H), 2,02 (s, 3H), 1,75 (m, 2H). IEN-EM m/z calc. 344,18, encontrado 345,1 (M+1)+; Tiempo de retención: 0,65 minutos. I-101
Ejemplo 14
Esquema sintético 14: (+/-)-4-[3-(2-clorofenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (121) I-19

(a) 4-cloro-6-metil-pirimidin-2-amina, EtOH, 160 °C; (b) purificación quiral SFC
Formación de (+/-)-4-[3-(2-clorofenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (121) I-19
A una mezcla de 4-cloro-6-metil-pirimidin-2-amina (0,45 g, 3,07 mmol) y 3-(2-clorofenil)-1,4- oxazepano (0,65 g, 3,07 mmol) sólidos en un vial, se le añadió EtOH (9 ml). El vial se puso en una placa caliente y se calentó a 160 °C sin cubrir durante 2 horas. El sólido en bruto se purificó por cromatografía sobre gel de sílice (40 g) en ISCO eluyendo con MeOH-diclorometano al 20 %/diclorometano. Las fracciones deseadas se recogieron y se concentraron al vacío. La mezcla racémica se sometió a purificación SFC quiral para obtener los enantiómeros individuales. Condiciones de SFC: Columna: Cellulose-2, 20x250 mm; Fase móvil: EtOH al 40 % (amoniaco 5 mM), CO2 al 60 %; Flujo: 80 ml/min; Concentraciones: ~30 mg/ ml (MeOH); Longitud de onda: 254 nm; Método tipo isocrático.
Pico A: 97% ee; [a]D (c = 1,0, MeOH) -16,26.
(R) -4-[3-(2-clorofenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (122): RMN 1H (300 MHz, DMSO-d6 ) 67,48 (dd, J = 6,2, 3,1 Hz, 1H), 7,32 (m, 3H), 6,58 (a, 2H), 4,61 (a, 0,5H), 4,15 (a, 1H), 3,93 (a, 1H), 3,84 -3,69 (m, 2H), 3,66 -3,53 (m, 1H), 3,44 (m, 0,5H), 2,09 (s, 3H), 1,78 (a, 2H);
IEN-e M m/z calc. 318,12, encontrado 319,13 (M+1 )+; Tiempo de retención: 0,64 minutos. I-30
Pico B: 89 % ee; [a]D (c = 1,0, MeOH) 39,92.
(S) -4-[3-(2-clorofenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (123) RMN 1H (300 MHz, DMSO-d6 ) 6 7,83 - 7,23 (m, 6 H), 6,59 (s, 0,5H), 6,00 (s a, 0,5H), 5,59 (s, 0,5H), 5,24 (s a, 0,5H), 4,98 (a, 0,5H), 4,58 (a, 0,5H), 4,04 (m, 4H), 3,61 (td, J = 11,3, 3,9 Hz, 1H), 2,22 (a, 3H), 1,84 (s, 2H).
IEN-Em m/z calc. 318,12, encontrado 319,13 (M+1 )+; Tiempo de retención: 0,64 minutos. I-31
Los siguientes análogos se prepararon de acuerdo con el Esquema sintético 14:

(+/-)-4-[3-(2-cloro-5-pirrolidin-1-il-fenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (124) I-199
RMN 1H (300 MHz, Metanol-d4) 67,16 (d, J = 8,8 Hz, 1H), 6,46 (dd, J = 8 ,8 , 2,9 Hz, 1H), 6,35 (d, J = 2,9 Hz, 1H), 5,52 (s a, 1H), 5,20 (a, 1H), 4,28 (dd, J = 13,6, 4,9 Hz, 1H), 4,03 (dd, J = 12,4, 4,8 Hz, 1H), 3,80 - 3,50 (m, 3H), 3,30 - 3,04 (m, 5H), 2,05 (s, 3H), 2,02 - 1,94 (m, 4H), 1,91-1,64 (m, 2H); IEN-EM m/z calc. 387,2, encontrado 388,33 (M+1)+; Tiempo de retención: 0,71 minutos.

(+/-)-4-(3-(2-cloro-5-morfolinofenil)-1,4-oxazepan-4-il)-6-metilpirimidin-2-amina I-226
RMN 1H (300 MHz, Metanol-c/4) 67,29 (dd, J = 18,3, 8,8 Hz, 2H), 6,91 (ddd, J = 16,6, 8,9, 2,9 Hz, 1H), 6,78 (dd, J = 26,7, 3,0 Hz, 0.5H), 6,48 (s, 1H), 6,10 (dd, J = 9,5, 5,1 Hz, 0.5H), 5,68 (s, 1H), 5,39 -5,10 (m, 2H), 4,42 -4,17 (m, 1H), 4,09 - 3,59 (m, 11H), 3,09 (t, J = 4,9 Hz, 6 H), 2,28 (dd, J = 40,8, 0,8 Hz, 5H), 2,09-1,84 (m, 4H); IEN-EM m/z calc.
403,18, encontrado 404,21 (m 1)+; Tiempo de retención: 0,62 minutos.
Ejemplo 15
Esquema sintético 15: (+/-)-4-[3-[2-cloro-5-(metilamino)fenil]-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (129) I-175

(a) NaH, DMF, 0 °C; (b) bis(pinocalatodiboro), Pd(PhaP)2Cl2, KOAc, dioxano, 85 °C; (c) nBuLi, THF, -78 °C; (d) 2-amino-4-cloro-6-metil pirimidina, MeOH, 125 °C; (e) TFA, CH2Cl2
Formación de (3-bromo-4-clorofenil)(metil)carbamato de ferc-butilo (125)
A una solución de W-(3-bromo-4-cloro-fenil)carbamato de ferc-butilo (2,00 g, 6,20 mmol) en DMF (20 ml), se le añadió NaH (0,30 g, 7,50 mmol) a 0 °C. La mezcla se agitó 0 °C durante 30 minutos. Se añadió yoduro de metilo (0,47 ml, 7,55 mmol) a la mezcla de reacción. La mezcla de reacción se diluyó en agua y se extrajo con EtOAc. La fase orgánica se secó (MgSO4), se filtró y se concentró al vacío para proporcionar 1,5 gramos del producto deseado: RMN 1H (300 MHz, CDCla) 67,55 (d, J = 2,5 Hz, 1H), 7,40 (d, J = 8,7 Hz, 1H), 7,24 - 7,12 (m, 1H), 3,25 (s, 3H), 1,48 (s, 9H); IEN-EM m/z calc. 319,0, encontrado 320,0 (M+1)+; Tiempo de retención: 1,06 minutos.
Formación de (4-cloro-3-(4-formil-4,5,6,7-tetrahidro-1,4-oxazepin-3-il)fenil)-(metil)carbamato de ferc-butilo (126)
Etapa 1: A una solución de W-(3-bromo-4-cloro-fenil)-W-metil-carbamato de ferc-butilo, 125, (1,5 g, 4,7 mmol) en dioxano (75 ml), se le añadieron bis(pinocalatodiboro) (1,5 g, 5,9 mmol), acetato potásico (1,4 g, 14,0 mmol). A esta mezcla se le purgó con atmósfera de nitrógeno durante 15 minutos y se le añadió dicloro-bis(trifenilfosforanil)paladio (0,3 g, 0,5 mmol). La reacción se calentó a 85 °C durante 18 horas. La mezcla de reacción se diluyó con EtOAc y se filtró a través de celite con EtOAc lavado (60 ml). Las fases orgánicas se concentraron al vacío. El sólido resultante de color pardo oscuro se usó sin purificación adicional: IEN-EM m/z calc. 367,2, encontrado 367,6 (M+1)+; Tiempo de retención: 1,0 minutos.
Etapa 2: Al producto en bruto anterior disuelto en DME (45 ml), se le añadió 3-cloro-6,7-dihidro-5H-1,4-oxazepina-4-carbaldehído, 109, (0,76 g, 4,70 mmol) seguido de NaHCO3 acuoso (8 ml de 1,2 M, 9,60 mmol). La mezcla se burbujeó con atmósfera de nitrógeno y se añadió PdCh(dppf) a la mezcla de reacción. El matraz se tapó y se calentó a 80 °C durante 12 horas. La mezcla se concentró al vacío y se purificó por cromatografía en gel de sílice (columna ISCO de 40 g eluyendo con MeOH al 20 %/diclorometano. Las fracciones deseadas se recogieron y se concentraron al vacío para proporcionar 500 mg del producto deseado: IEN-EM m/z calc. 366,1, encontrado 367,1 (M+1)+; Tiempo de retención: 0,92 minutos.
Formación de (4-doro-3-(1,4-oxazepan-3-il)feml)(metil)carbamato de (+/-)-terc-butilo (127)
A una solución de A/-[4-cloro-3-(4-form¡l-6,7-d¡h¡dro-5H-1,4-oxazep¡n-3-¡l)fen¡l]-A/-met¡l-carbamato de terc-but¡lo, 126, (0,40 g, 1,09 mmol) en THF (10 ml), se le nBuL¡ (2,1 ml de soluc¡ón 1,6 M, 3,3 mmol) a -78 °C. La mezcla de reacdón se ag¡tó a -78 °C durante 50 m¡nutos. A esta mezcla se le añad¡ó cu¡dadosamente MeOH para ¡nterrump¡r la reacc¡ón. Se añad¡ó MeOH ad¡c¡onal (30 ml) y la soluc¡ón se calentó a temperatura amb¡ente: IEN-EM m/z calc. 338,1, encontrado 339,1 (M+1)+; T¡empo de retenc¡ón: 0,63 m¡nutos. A esta soluc¡ón se le añad¡ó NaBH4 (0,07 g, 1,81 mmol). La mezcla de reacc¡ón se ag¡tó a temperatura amb¡ente durante una noche. La mezcla se d¡luyó en agua y se extrajo dos veces con EtOAc. Las fases orgán¡cas comb¡nadas se secaron (MgSO4), se f¡ltraron y se concentraron al vacío para proporc¡onar 200 mg del producto deseado que se usó s¡n pur¡f¡cac¡ón ad¡c¡onal: IEN-EM m/z calc. 340,1, encontrado 341,2 (M+1)+; T¡empo de retenc¡ón: 0,64 m¡nutos.
Formación de (3-(4-(2-ammo-6-metilpirimidm-4-il)-1,4-oxazepan-3-il)-4-dorofeml)(metil)carbamato de (+/-)-tercbutilo (128)
Una mezcla de 4-cloro-6-met¡l-p¡r¡m¡d¡n-2-am¡na (0,08 g, 0,54 mmol) y A/-[4-cloro-3-(1,4-oxazepan-3-¡l)fen¡l]-W-met¡lcarbamato de terc-but¡lo, 127, (0,30 g, 0,77 mmol) se mezcló en una pequeña cant¡dad de metanol. La suspens¡ón resultante se calentó a 125 °C en un matraz ab¡erto durante 16 horas para perm¡t¡r que se evaporara el d¡solvente. El sól¡do resultante se d¡solv¡ó en MeOH y se pur¡f¡có por cromatografía sobre gel de síl¡ce (columna ISCO de 40 g) eluyendo con MeOH-d¡clorometano al 20 %/d¡clorometano. Las fracc¡ones deseadas se recog¡eron y se evaporaron para proporc¡onar 220 mg del producto deseado: RMN 1H (300 MHz, DMSO-d6 ) 67,45 (d, J = 8,5 Hz, 1H), 7,25 (d, J = 8,7 Hz, 1H), 7,16 (s, 1H), 6,95 (a, 2H), 6,46 (a, 1H), 5,95 (a, 1H), 5,11 (a, 1H), 4,15 (a, 2H), 4,02 -3,70 (m, 3H), 3,57 (td, J = 11,4, 4,0 Hz, 1H), 3,15 (s, 3H), 2,15 (a, 3H), 1,80 (m, 2H), 1,28 (s, 9H); IEN-EM m/z calc. 447,2, encontrado 448.1 (M+1)+; T¡empo de retenc¡ón: 0,68 m¡nutos.
Formación de (+/-)-4-[3-[2-doro-5-(metilamino)fenil]-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (129)
A una soluc¡ón de W-[3-[4-(2-am¡no-6-met¡l-p¡r¡m¡d¡n-4-¡l)-1,4-oxazepan-3-¡l]-4-cloro-fen¡l]-W-met¡l-carbamato de terc-but¡lo, 128, (0,25 g, 0,53 mmol) en d¡clorometano (10 ml), se le añad¡ó ác¡do tr¡fluoroacét¡co (0,74 g, 6,49 mmol). La mezcla se ag¡tó a temperatura amb¡ente durante 12 horas y el d¡solvente se evaporó al vacío. El res¡duo resultante se pur¡f¡có por ISCO ¡nverso eluyendo con TFA al 0,1 %-aceton¡tr¡lo/TFA al 0,1 %-agua para proporc¡onar 183 mg del producto deseado: RMN 1H (300 MHz, DMSO-cfe) 67,82 (s, 1H), 7,49 (s, 1H), 7,17 (dd, J = 19,2, 8,6 Hz, 1H), 6,73 -6,34 (m, 2H), 5,93 (dd, J = 10,1, 5,4 Hz, 0H), 5,16 -5,07 (m, 0H), 5,06 -4,90 (m, 10H), 4,35 -4,05 (m, 1H), 4,00 -3,47 (m, 3H), 2,62 (d, J = 1,9 Hz, 3H), 2,23 (d, J = 35,1 Hz, 2H), 1,94-1,67 (m, 2H); IEN-EM m/z calc. 347,2, encontrado 348.2 (M+1)+; T¡empo de retenc¡ón: 0,57 m¡nutos. I-175
La separac¡ón qu¡ral SFC proporc¡onó enant¡ómeros ¡nd¡v¡duales. Columna: Cellulose-2, 20x250 mm Fase móv¡l: MeOH al 30 % (amon¡aco 5 mM), CO2 al 70 %; Flujo: 80 ml/m¡n. Concentrac¡ones: ~40 mg/ ml (MeOH) Volumen de ¡nyecc¡ón: 250 pl, Long¡tud de onda: 220 nm; Método t¡po ¡socrát¡co
P¡co A: ee: 97 % [a]D (c=0,5, MeOH) -296,96 I-312
4-[3-[2-cloro-5-(met¡lam¡no)fen¡l]-1,4-oxazepan-4-¡l]-6-met¡l-p¡r¡m¡d¡n-2-am¡na (38,5 mg, 47% ) RMN 1H (300 MHz, Metanol-d4) 6 5,64 (d, J = 8,7 Hz, 1H), 5,05 - 4,96 (m, 2H), 4,89 (d, J = 2,8 Hz, 1H), 4,13 (d, J = 1,0 Hz, 1H), 3,78 -3,61 (m, 2H), 2,78 (ddd, J = 27,1, 13,7, 5,1 Hz, 2H), 2,57 -2,03 (m, 4H), 1,16 (s, 3H), 0,67 (d, J = 0,8 Hz, 3H), 0,37 (td, J = 9,1, 7,4, 4,1 Hz, 3H). IEN-EM m/z calc. 347,15128, encontrado 348,15 (M+1)+; T¡empo de retenc¡ón: 0,57 m¡nutos
P¡co B: ee: 95,4 % [a]D (c=0,5, MeOH) 254,02 I-178
4-[3-[2-cloro-5-(met¡lam¡no)fen¡l]-1,4-oxazepan-4-¡l]-6-met¡l-p¡r¡m¡d¡n-2-am¡na (36,0 mg, 44% ) RMN 1H (300 MHz, Metanol-d4) 65,84 (d, J = 8,6 Hz, 1H), 5,29 - 5,07 (m, 2H), 4,21 (a, 1H), 3,78 (a, 2H), 2,99 (dd, J = 13,6, 5,0 Hz, 1H), 2,75 (dd, J = 12,2, 4,8 Hz, 1H), 2,54 - 2,20 (m, 3H), 1,39 (s, 3H), 0,78 (s, 3H), 0,68 - 0,41 (m, 2H). IEN-EM m/z calc.
347,15128, encontrado 348,15 (M+1) ; T¡empo de retenc¡ón: 0,57 m¡nutos
Ejemplo 16
Esquema sintético 16: (+/-)-4-metil-6-(2-(2-metil-4-(metilsulfonil)fenil)azepan-1-il)pirimidin-2-amina (132) I-37

(a) ác¡do (2-met¡l-4-met¡lsulfon¡l-fen¡l)borón¡co, Pd(PhaP)2Cl2, EtaN, DMF; (b) Pd/C, HOAc, EtOAc, MeOH; (c) nBuL¡, THf , -78 °C; (d) 2-am¡no-4-cloro-6-met¡lp¡r¡m¡d¡na, NMP, 150 °C
Formación de 7-(2-metil-4-(metilsulfoml)feml)-2,3,4,5-tetrahidro-1H-azepma-1-carbaldehído (129)
Se cargó un matraz r b en atmósfera de nitrógeno con 7-doro-2,3,4,5-tetrahidroazepina-1-carbaldehído, 73, (0,60 g, 3,76 mmol), ácido (2-metil-4-metilsulfonil-fenil)borónico (1,00 g, 4,67 mmol), DMF (3,5 ml), trietilamina (2,5 ml, 17,9 mmol) y después cloruro de bis(trifenilfosfina)paladio II (0,11 g, 0,15 mmol). La mezcla se lavó abundantemente con corriente de nitrógeno y se calentó a 50 °C durante 7 horas. Se añadió agua y se extrajo dos veces con EtOAc. Las fases orgánicas combinadas se lavaron dos veces con salmuera, se secaron (MgSO4), se filtraron y se concentraron al vacío. El residuo resultante se purificó por cromatografía en columna (columna ISCO gold de 40 g; gradiente de EtOAc al 0-30 %/CH2Cl2) para proporcionar 161 mg del producto deseado: RMN 1H (400 MHz, DMSO-d6 ) 6 7,83 -7,58 (m, 3H), 7,57 - 7,33 (m, 1H), 5,82 - 5,50 (m, 1H), 3,83 - 3,60 (m, 2H), 3,20 (s, 3H), 2,39 - 2,31 (m, 2H), 2,28 (s, 3H), 1,87 - 1,73 (m, 2H), 1,73-1,62 (m, 2H); IEN-EM m/z calc. 293,1, encontrado 294,1 (M+1)+; Tiempo de retención: 0,71 minutos.
Formación de (+/-)-2-(2-metil-4-(metilsulfonil)fenil)azepano-1-carbaldehído (130)
Una mezcla de 7-(2-metil-4-metilsulfonil-fenil)-2,3,4,5-tetrahidroazepina-1-carbaldehído, 129, (0,162 g, 0,552 mmol) y paladio sobre carbono (0,056 g, 0,526 mmol) en acetato de etilo (3 ml), metanol (3 ml) y ácido acético (1 ml) se agitó en una atmósfera de gas hidrógeno. Después de 21 horas, la mezcla se filtró a través de un lecho pequeño de fluorosilo y se lavó con EtOAc. El filtrado se concentró al vacío para proporcionar 190 mg del producto deseado en forma de un aceite que se usó sin purificación adicional: IEN-EM m/z calc.295,12, encontrado 296,17 (M+1)+; Tiempo de retención: 0,69 minutos pico TR = 0,69 (M+H) 296 es el producto deseado.
Formación de (+/-)-2-(2-metil-4-(metilsulfonil)fenil)azepano (131)
A una solución fría (-78 °C) de 2-(2-metil-4-metilsulfonil-fenil)azepano-1-carbaldehído, 130, (0,170 g, 0,576 mmol) en THF (4 ml), se le añadió n-butillitio (0,450 ml de solución 1,6 M en hexanos, 0,720 mmol). Después de 30 minutos, se añadió otros 0,4 ml de solución de nBuLi. Después de un adicional de 10 minutos, se paró la reacción mediante la adición lenta de una mezcla de reacción en agua. La fase acuosa se extrajo dos veces con EtOAc. Las fases orgánicas combinadas se secaron (MgSO4), se filtraron y se concentraron al vacío para proporcionar 90 mg de aceite en bruto de color naranja que se usó en la siguiente etapa sin purificación adicional: IEN-EM m/z calc. 267,13, encontrado 268,16 (M+1)+; Tiempo de retención: 0,69 minutos pico Tr = 0,69 (M+H) 296 es el producto deseado.
Formación de (+/-)-4-metil-6-(2-(2-metil-4-(metilsulfonil)fenil)azepan-1-il)pirimidin-2-amina (132) I-37
A una solución de 2-(2-metil-4-metilsulfonil-fenil)azepano, 131, (0,090 g, 0,337 mmol) en NMP (2 ml), se le añadió 4-cloro-6-metil-pirimidin-2-amina (0,048 g, 0,337 mmol). La mezcla de reacción se calentó a 150 °C durante una noche en vial cerrado herméticamente con un tabique de teflon. Después de 15 horas, la mezcla se enfrió a temperatura ambiente y se cargó directamente en una columna ISCO c18-aq de 15 g y se purificó por cromatografía de fase inversa sobre gel de sílice ejecutándose con TFA al 0,1 %/H2O y t Fa al 0,1 %/CH3CN. Las fracciones combinadas que contenían el producto deseado se concentraron al vacío y el residuo resultante se diluyó con diclorometano y se neutralizó con una solución acuosa saturada de NaHCO3. La mezcla se pasó a través de un separador de fase y la fase orgánica resultante se concentró al vacío para proporcionar 46 mg del producto deseado en forma de un sólido de color pardo claro: alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6 ) 67,71 - 7,53 (m, 3H), 7,31 (d, J = 8,2 Hz, 1H), 5,34 (s, 2H), 4,24 (s, 1H), 3,57 - 3,44 (m, 1H), 3,15-3,05 (m, 1H), 3,10 (s, 3H), 2,55 (s, 3H), 2,51-2,40 (m, 1H), 2,29 -2,08 (m, 1H), 2,01 (s, 3H), 1,98 - 1,88 (m, 1H), 1,88 - 1,59 (m, 2H), 1,59-1,13 (m, 3H); IEN-EM m/z calc. 374,18, encontrado 375,23 (M+1)+; Tiempo de retención: 0,61 minutos.
El siguiente análogo se preparó del mismo modo:

(R)-4-(2-(2-ciclopropil-4-fluorofeml)azepan-1-M)-6-metilpirimidm-2-amma I-47 and (S)-4-(2-(2-ciclopropil-4-fluorofenil)azepan-1-il)-6-metilpirimidin-2-amina I-46
Pico A: RMN 1H (400 MHz, DMSO-d6 ) 67,06 (dd, J = 8,7, 6,1 Hz, 1H), 6,83 (td, J = 8,6 ; 2,8 Hz, 1H), 6,69 (dd, J = 10,7, 2,8 Hz, 1H), 5,63 (d, J = 15,1 Hz, 1H), 5,46 - 5,19 (m, 3H), 4,39 (s, 1H), 3,42 (dd, J = 14,6, 11,1 Hz, 1H), 2,33 (dt, J = 14,1, 6,4 Hz, 1H), 2,22 (p, J = 8,3 Hz, 1H), 1,99 (s, 3H), 1,96 - 1,84 (m, 1H), 1,84 - 1,60 (m, 3H), 1,52 (d, J = 12,7 Hz, 1H), 1,44 - 1,18 (m, 2H), 1,13 - 0,91 (m, 2H), 0,78 (dtd, J = 13,8, 10,6, 10,0, 4,6 Hz, 2H); IEN-EM m/z encontrado 341,24 (M+1)+; Tiempo de retención: 0,70 minutos; [a]o = -12,57 (c = 21 mg/3 ml).
Pico B: RMN 1H (400 MHz, DMSO-d6 ) 67,13 - 6,98 (m, 1H), 6,83 (td, J = 8 ,6 ; 3,0 Hz, 1H), 6,75 - 6,64 (m, 1H), 5,62 (s, 1H), 5,49 -5,25 (m, 3H), 4,40 (s, 1H), 3,70 (s, 1H), 3,42 (dd, J = 14,7, 11,0 Hz, 1H), 2,41 -2,25 (m, 1H), 2,28 -2,11
(m, 1H), 2,04 - 1,95 (m, 3H), 1,92 (s, 1H), 1,88 - 1,62 (m, 3H), 1,61 - 1,44 (m, 1H), 1,44 -1,18 (m, 2H), 1,14 -0,94 (m, 2h ), 0,87-0,64 (m, 2H); IEN-EM m/z encontrado 341,24 (M+1)+; Tiempo de retención: 0,70 minutos; [a]D = 31,94 (c = 23 mg/3 ml).
Ejemplo 17
Esquema sintético 17: (+/-)-4-(3-(2-fluoro-4-(metilsulfonil)fenil)-1,4-oxazepan-4-il)-6-metil-pirimidin-2-amina (135) I-60

(a) Pd/C, HOAc, EtOAc, MeOH; (b) HCl, MeOH, reflujo; (c) 2-amino-4-cloro-6-metilpirimidina, NMP, 150 °C Formación de 3-(2-fluoro-4-(metilsulfoml)feml)-6,7-dihidro-1,4-oxazepma-4(5H)-carbaldehído (133)
Se preparó el Intermedio 132 para el Esquema sintético 13 usando 2-fluoro-4-(metilsulfonil)-benzaldehído en lugar de 2-cloro-5-hidroxibenzaldehído.
Formación de (+/-)-3-(2-fluoro-4-(metilsulfonil)fenil)-1,4-oxazepano-4-carbaldehído (133)
Una mezcla de 3-(2-fluoro-4-metilsulfonil-fenil)-6,7-dihidro-5H-1,4-oxazepina-4-carbaldehído, 132, (0,73 g, 2,30 mmol), paladio sobre carbono (0,23 g, 2,18 mmol) en EtOAc (6 ml) y MeOH (6 ml) y ácido acético (3 ml) se agitó en una atmósfera de gas hidrógeno. Después de 8 días, se detuvo la reacción. La mezcla se filtró a través de un lecho pequeño de florosilo y se lavó con EtOAc. La fase orgánica se neutralizó lavándose con una solución acuosa saturada de NaHCO3, se secó (MgSO4), se filtró y se concentró el filtrado al vacío para proporcionar 280 mg de aceite incoloro. El residuo resultante se usó sin purificación adicional.
Formación de (+/-)-3-(2-fluoro-4-(metilsulfonil)fenil)-1,4-oxazepano (134)
Una solución de 3-(2-fluoro-4-metilsulfonil-fenil)-1,4-oxazepano-4-carbaldehído, 133, (0,28 g, 0,93 mmol) en metanol (6 ml) y HCl concentrado (5,5 ml de 12,1 M, 66,55 mmol) se calentó a reflujo durante 2,5 horas. La mezcla se enfrió a temperatura ambiente y se diluyó en agua. La mezcla se neutralizó mediante la adición de una solución acuosa saturada de NaHCO3 y después se extrajo tres veces con EtOAc. La fase orgánica se secó (MgSO4), se filtró y se concentró al vacío para proporcionar el producto deseado. IEN-EM m/z calc. 274, encontrado 274 (M+1)+; Tiempo de retención: 0,48 minutos.
Formación de (+/-)-4-(3-(2-fluoro-4-(metilsulfonil)fenil)-1,4-oxazepan-4-il)-6-metil-pirimidin-2-amina (135) I-60 A una solución de 3-(2-fluoro-4-metilsulfonil-fenil)-1,4-oxazepano (0,25 g, 0,81 mmol) en NMP (10 ml), se le añadieron trietilamina (1,00 ml, 8,89 mmol) y 4-cloro-6-metil-pirimidin-2-amina (0,13 g, 0,87 mmol). La mezcla de reacción se calentó a 150 °C. Después de 17 horas, se enfrió la mezcla a temperatura ambiente y se cargó material directamente en una columna ISCO c18-aq de 100 g y se purificó por ejecución de fase inversa con TFA al 0,1 %/H2O y TFA al 0,1 %/CH3CN. Las fracciones que contenían el producto deseado se concentraron al vacío se diluyeron con diclorometano. La mezcla se pasó a través de un separador de fase y la fase orgánica se concentró al vacío para proporcionar 69 mg del producto deseado en forma de un sólido de color pardo claro: alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6 ) 67,71 -7,56 (m, 2H), 7,52 -7,40 (m, 1H), 5,71 (s, 1H), 5,66 -5,59 (m, 1H), 5,40 (s, 2H), 4,32 - 4,16 (m, 1H), 4,09 (dd, J = 13,4, 5,2 Hz, 1H), 3,88 - 3,78 (m, 1H), 3,74 (dd, J = 13,3, 10,0 Hz, 1H), 3,63 - 3,38 (m, 2H), 3,11 (s, 3H), 1,97 (s, 3H), 1,84 - 1,60 (m, 2H).
El siguiente análogo se preparó de acuerdo con el Esquema sintético 17:

4-[3-(2,4-difluorofenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (136) I-58
alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6) 6 7,36 - 7,27 (m, 1H), 7,13 - 7,05 (m, 1H), 6,97 (td, J = 8,2,
1,9 Hz, 1H), 5,73 (s, 1H), 5,58 - 5,50 (m, 1H), 5,45 (s, 2H), 4,37 (d, J = 15,7 Hz, 1H), 4,10 (dd, J = 12,9, 5,7 Hz, 1H), 3,92 -3,85 (m, 1H), 3,78 (dd, J = 13,3, 10,0 Hz, 1H), 3,62 -3,51 (m, 2H), 2,04 (s, 3H), 1,89-1,68 (m, 2H); IEN-EM m/z calc. 320,14, encontrado 321,11 (M+1)+; Tiempo de retención: 0,6 minutos. La mezcla racémica se sometió a separación SFC quiral.
Pico A; (R)-4-[3-(2,4-difluorofenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (137); 99,5 % ee; alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6 ) 67,36 -7,27 (m, 1H), 7,13 -7,05 (m, 1H), 6,97 (td, J = 8,2, 1,9 Hz, 1H), 5,73 (s, 1H), 5.58 - 5,50 (m, 1H), 5,45 (s, 2H), 4,37 (d, J = 15,7 Hz, 1H), 4,10 (dd, J = 12,9, 5,7 Hz, 1H), 3,92 - 3,85 (m, 1H), 3,78 (dd, J = 13,3, 10,0 Hz, 1H), 3,62 -3,51 (m, 2H), 2,04 (s, 3H), 1,89-1,68 (m, 2H); IEN-EM m/z calc. 320,14, encontrado 321,13 (M+1)+; Tiempo de retención: 0,6 minutos. I-61
Pico B; (S)-4-[3-(2,4-difluorofenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (138); 99,5 % ee; alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6 ) 67,36 -7,27 (m, 1H), 7,13 -7,05 (m, 1H), 6,97 (td, J = 8,2, 1,9 Hz, 1H), 5,73 (s, 1H), 5.58 - 5,50 (m, 1H), 5,45 (s, 2H), 4,37 (d, J = 15,7 Hz, 1H), 4,10 (dd, J = 12,9, 5,7 Hz, 1H), 3,92 - 3,85 (m, 1H), 3,78 (dd, J = 13,3, 10,0 Hz, 1H), 3,62 -3,51 (m, 2H), 2,04 (s, 3H), 1,89 - 1,68 (m, 2H). IEN-EM m/z calc. 320,14, encontrado 321,16 (M+1)+; Tiempo de retención: 0,6 minutos. I-62
Ejemplo 18
Esquema sintético 18: (+/-)-4-(3-(2-cloro-5-(metilsulfinil)fenil)-1,4-oxazepan-4-il)-6-metilpirimidin-2-amina (143) I-110

(a) mCPBA, CH2Cl2, 0 °C; (b) NaBH4, EtOH; (c) Peryodinano de Dess-Martin, CH2Ch; (d) tamices mol. de 4 A, 3-((tributilstan-nil)metoxi)propan-1-amina, CH2O 2; (e) 2,6-lutidina, Cu(OTf)2, hexafluoroisopropanol, CH2Ch; (f) 2-amino-4-cloro-6-metilpirimidina, NMP, 150 °C; (g) separación quiral SFC
Formación de 2-cloro-5-(metilsulfinil)benzoato de metilo (139)
A una solución fría (0 °C) de 2-cloro-5-metilsulfanil-benzoato de metilo (2,0 g, 9,2 mmol) en diclorometano (40 ml), se le añadió ácido 3-cloroperoxibenzoico (2,1 g de 77 % p/p, 9,4 mmol). La mezcla de reacción se calentó lentamente a temperatura ambiente durante 3 horas. Después de 3,5 horas, la mezcla de reacción se diluyó en una solución acuosa saturada de NaHCO3 y se extrajo dos veces con diclorometano. Las fases orgánicas combinadas se lavaron dos veces con una solución acuosa saturada de NaHCO3 y después se pasó a través de un separador de fase. El filtrado resultante se concentró al vacío. El residuo en bruto se purificó por cromatografía sobre gel de sílice usando una columna isco de 80 g usando un gradiente de EtOAc al 0-30 %/CH2Cl2 para proporcionar 1,8 g en forma de un sólido de color blanco: RMN 1H (400 MHz, DMSO-d6 ) 6 8,11 (dd, J = 2,2, 0,4 Hz, 1H), 7,88 (dd, J = 8,3, 2,2 Hz, 1H), 7,80 (dd, J = 8,4, 0,4 Hz, 1H), 3,90 (s, 3H), 2,80 (s, 3H); IEN-EM m/z calc. 232,00, encontrado 233,08 (M+1)+; Tiempo de retención: 0,63 minutos.
Formación de (2-cloro-5-(metilsulfinil)fenil)metanol (140)
A una solución de 2-cloro-5-metilsulfinil-benzoato de metilo, 139, (0,88 g, 3,79 mmol) en EtOH (15 ml), se le añadió NaBH4 (0,57 g, 15,09 mmol) en porciones. La reacción se agitó a temperatura ambiente durante 30 minutos y después se calentó a 50 °C. Después de 3 horas, la mezcla de reacción se inactivó por adición lenta en una solución acuosa saturada de NaHCO3y se extrajo dos veces con EtOAc. Las fases orgánicas combinadas se lavaron con una solución acuosa saturada de NH4Cl, se secaron (MgSO4), se filtraron y se concentraron al vacío para dar 486 mg del producto deseado: RMN 1H (400 MHz, DMSO-d6j 57,92 -7,81 (m, 1H), 7,69 -7,53 (m, 2H), 5,58 (t, J = 5,6 Hz, 1H), 4,62 (dt, J = 5,6, 0,8 Hz, 2H), 2,74 (s, 3H); IEN-EM m/z calc. 204,00, encontrado 205,07 (M+1)+; Tiempo de retención: 0,55 minutos.
Formación de 2-cloro-5-(metilsulfinil)benzaldehído (141)
Se disolvió (2-doro-5-metilsulfinil-fenil)metanol, 140, (0,48 g, 2,35 mmol) en cloruro de metileno (9,6 ml). Se añadió peryodinano de Dess-Martin (1,20 g, 2,83 mmol) y la solución de reacción se agitó a temperatura ambiente durante 16 horas. La solución se diluyó en una solución acuosa saturada de NaHCO3 y se extrajo dos veces con CH2O 2. Las fases orgánicas combinadas se filtraron a través de un separador de fase y se concentró al vacío. El residuo en bruto se purificó por cromatografía sobre gel de sílice con una columna isco de 40 g usando un gradiente de EtOAc al 0 20 %/CH2Cl2 para proporcionar 429 mg del producto deseado: RMN 1H (400 MHz, DMSO-cfe) 510,37 (s, 1H), 8,17 -8,15 (m, 1H), 7,99 (dd, J = 8,3, 2,3 Hz, 1H), 7,87 - 7,83 (m, 1H), 2,81 (s, 3H); IEN-EM m/z calc. 202,0, encontrado 203,0 (M+1)+, Tiempo de retención: 0,59 minutos.
Formación de 3-(2-doro-5-(metilsulfinil)fenil)-1,4-oxazepano (142)
A una solución de 3-(tributilestanilmetoxi)propan-1-amina, 141, (0,78 g, 2,05 mmol) en diclorometano (5 ml), se le añadió 2-cloro-5-metilsulfinil-benzaldehído (0,42 g, 1,99 mmol) seguido de tamices moleculares de 4 angstrom. La mezcla se agitó durante una noche, se filtró para retirar los tamices y se lavó con diclorometano (20 ml).
En un matraz separado que contenía hexafluoroisopropanol (5,5 ml), se añadió 2,6-lutidina (0,24 ml, 2,05 mmol) seguido de Cu(oTf)2 (0,72 g, 2,00 mmol). Se agitó la mezcla durante 1 horas. Después se añadió la solución de imina preparada anteriormente en una porción. La reacción se agitó a temperatura ambiente. Después de 3 días, la mezcla se diluyó con 60 ml de 2:1 de una solución acuosa saturada de NaHCO3 e hidróxido de amonio al 10 %. Después de agitar durante 30 minutos, la capa orgánica se retiró y se lavó dos veces con una solución acuosa saturada de NaHCO3, después salmuera. La capa orgánica se pasó a través de un embudo separador de fase y se concentró al vacío. El residuo en bruto, que contiene hexafluoroisopropanol, se cargó directamente sobre una columna c18-aq de 100 gramos y purificación a través de cromatografía de fase inversa eluyendo con TFA al 0,1 %/H2O y TFA al 0,1 %/CH3CN. Las fracciones que contenían el producto se concentraron al vacío, se diluyeron con diclorometano y se neutralizaron con una solución acuosa saturada de NaHCO3. La mezcla se pasó a través de un separador de fase y el filtrado orgánico se concentró al vacío para proporcionar 110 mg del producto deseado: IEN-EM m/z calc. 270,06, encontrado 271,11 (M+1)+; Tiempo de retención: 0,46 minutos.
Formación de (+/-)-3-(2-cloro-5-(metilsulfinil)fenil)-1,4-oxazepano (143)
A una solución de 3-(2-cloro-5-metilsulfinil-fenil)-1,4-oxazepano, 142, (0,11 g, 0,40 mmol) en NMP (5 ml), se le añadió 4-cloro-6-metil-pirimidin-2-amina (0,08 g, 0,52 mmol). La mezcla de reacción se calentó a 150 °C durante 17 horas. La mezcla se enfrió a temperatura ambiente y se cargó directamente en una columna ISCO c18-aq de 50 g y el material en bruto se purificó por fase inversa eluyendo con un gradiente de TFA al 0,1 %/H2O y TFA al 0,1 %/CH3CN. Las fracciones que contenían el producto deseado se concentraron al vacío se diluyeron con diclorometano, se neutralizaron con una solución acuosa saturada de NaHCO3 y la mezcla se pasó a través de un separador de fase. La fase orgánica se concentró al vacío para proporcionar 50 mg del producto deseado como una mezcla de 4 estereoisómeros: alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6 ) 57,69 - 7,48 (m, 3H), 5,66 (s, 1H), 5,58 -5.50 (m, 1H), 5,43 (s, 2H), 4,60 -4,46 (m, 1H), 4,18 -4,04 (m, 1H), 3,95 -3,85 (m, 1H), 3,85 -3,66 (m, 2H), 3,65 -3,49 (m, 1H), 2,68 (d, J = 4,9 Hz, 3H), 2,02 (d, J = 2,4 Hz, 3H), 1,86 - 1,67 (m, 2H). IEN-EM m/z calc. 380,11, encontrado 381,22 (M+1)+; Tiempo de retención: 0,53 minutos.
La mezcla (34 mg) se sometió a separación quiral SFC para proporcionar 4 estereoisómeros:
Pico A: (144) alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6 ) 57,72 - 7,50 (m, 3H), 5,66 (s, 1H), 5,62 - 5,50 (m, 1H), 5,44 (s, 2H), 4,64 -4,45 (m, 1H), 4,12 (dd, J = 13,3, 5,2 Hz, 1H), 3,98 -3,86 (m, 1H), 3,84 -3,49 (m, 3H), 2,69 (s, 3H), 2,03 (d, J = 1,6 Hz, 3H), 1,89-1,67 (m, 2H); IEN-EM m/z calc. 380,11, encontrado 381,22 (M+1)+; Tiempo de retención: 0,53 minutos. I-120
Pico B: (145) alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6 ) 57,68 - 7,47 (m, 3H), 5,65 (s, 1H), 5,62 - 5,50 (m, 1H), 5,42 (s, 2H), 4,61 -4,46 (m, 1H), 4,13 (dd, J = 13,4, 4,9 Hz, 1H), 3,99 -3,66 (m, 3H), 3,64 -3,47 (m, 1H), 2,70 - 2,67 (m, 3H), 2,02 (s, 3H), 1,85-1,69 (m, 2H); IEN-EM m/z calc. 380,11, encontrado 381,22 (M+1)+; Tiempo de retención: 0,53 minutos. I-121
Pico C: (146) alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6 ) 57,68 - 7,47 (m, 3H), 5,65 (s, 1H), 5,62 -5.50 (m, 1H), 5,42 (s, 2H), 4,61 -4,46 (m, 1H), 4,13 (dd, J = 13,4, 4,9 Hz, 1H), 3,99 -3,66 (m, 3H), 3,64 -3,47 (m, 1H), 2,70 -2,67 (m, 3H), 2,02 (s, 3H), 1,85-1,69 (m, 2H); IEN-EM m/z calc. 380,11, encontrado 381,22 (M+1)+; Tiempo de retención: 0,53 minutos. I-122
Pico D: (147) alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6 ) 57,68 - 7,48 (m, 3H), 5,65 (s, 1H), 5,61 - 5,49 (m, 1H), 5,43 (s, 2H), 4,59 -4,42 (m, 1H), 4,12 (dd, J = 13,5, 5,0 Hz, 1H), 4,00 -3,65 (m, 3H), 3,65 -3,45 (m, 1H), 2,72 - 2,66 (m, 3H), 2,02 (s, 3H), 1,86-1,68 (m, 2H); IEN-EM m/z calc. 380,11, encontrado 381,22 (M+1)+; Tiempo de retención: 0,53 minutos. I-123
Ejemplo 19
Esquema sintético 19: (+/-)-1-(3-(4-(2-ammo-6-metilpi
N
midm-4-il)-1,4-oxazepan-3-il)-4-clorofeml)-2,2,2-trifluoroetan-1-ol (154) I-134

(a) tamices mol. de 4 A, 3-((tributilestannil)metoxi)propan-1-amina, CH2CI2; después 2,6-lutidina, Cu(OTf)2, hexafluoroisopropanol, CH2Ch; (b) Boc2O, Et3N, CH2Cl2; THF; (c) NaBH4, EtOH, del 50 °C al 80 °C; (d) Peryodinano de Dess-Martin, CH2Cl2; (e) TBAF, (CH3)3SiCF3, THF, 0 °C a ta; (f) ácido trifluoroacético, CH2O 2; (g) 2-amino-4-cloro-6-metilpirimidina, NMP, 150 °C; (h) separación quiral SFC
Formación de 4-cloro-3-(1,4-oxazepan-3-il)benzoato de metilo (148)
A una solución de 3-(tributilestanilmetoxi)propan-1-amina (2,09 g, 5,53 mmol) en diclorometano (14 ml), se le añadió 4-cloro-3-formil-benzoato de metilo (1,12 g, 5,36 mmol) seguido de tamices moleculares de 4 angstrom. La mezcla se agitó durante una noche, se filtró para retirar los tamices, se lavó con diclorometano (50 ml).
En un matraz separado que contenía hexafluoroisopropanol (14 ml), se añadió 2,6-lutidina (0,64 ml, 5,52 mmol) seguido de Cu(OTf)2 (1,95 g, 5,40 mmol). La mezcla se agitó durante 1 hora, después se añadió la solución de imina preparada anteriormente en una porción. La mezcla de reacción cambió de color azul a verde y la mezcla se agitó durante la noche a temperatura ambiente. La mezcla se diluyó con 120 ml de 2:1 de solución acuosa saturada de NaHCO3 e hidróxido de amonio al 10 %. Después de agitar durante 40 minutos, la mezcla se diluyó con diclorometano (50 ml) y la capa orgánica se retiró y se lavó dos veces con una solución acuosa saturada de NaHCO3. La capa orgánica se pasó a través de un embudo separador de fase y se concentró al vacío. El residuo en bruto se purificó por fase inversa usando una columna ISCO c18-aq de 100 gramos eluyendo con TFA al 0,1 %/H2O y TFA al 0,1 %/CH3Cn . Las fracciones que contenían el producto deseado se concentraron al vacío se diluyeron con diclorometano, se neutralizaron con una solución acuosa saturada de NaHCO3 y la mezcla se pasó a través de un separador de fase. La fase orgánica se concentró al vacío para proporcionar 754 mg del producto deseado en forma de un aceite de color amarillo: RMN 1H (400 MHz, DMSO-d6 ) 68,23 (d, J = 2,3 Hz, 1H), 7,82 (dd, J = 8,3, 2,2 Hz, 1H), 7,56 (d, J = 8,3 Hz, 1H), 4,26 (dd, J = 8,9, 3,2 Hz, 1H), 3,87 (s, 4H), 3,84 - 3,78 (m, 2H), 3,72 (ddd, J = 12,0, 6,7, 6,0 Hz, 1H), 3,26 (dd, J = 12,3, 8,9 Hz, 1H), 3,10 (dt, J = 13,5, 5,1 Hz, 1H), 2,88 (ddd, J = 13,0, 7,2, 5,3 Hz, 2H), 1,89 (dtd, J = 7,9, 6,1, 5,0 Hz, 2H); IEN-EM m/z calc. 269,08, encontrado 270,19 (M+1)+; Tiempo de retención: 0,53 minutos.
Formación de 3-(2-cloro-5-(metoxicarbonil)fenil)-1,4-oxazepano-4-carboxilato de íerc-butilo (149)
Una mezcla de 4-cloro-3-(1,4-oxazepan-3-il)benzoato de metilo, 148, (0,51 g, 1,80 mmol) y trietilamina (0,28 ml, 2,00 mmol) en THF (8 ml), se añadió terc-butil carbonato de terc-butoxicarbonilo (0,41 g, 1,90 mmol) y la mezcla se agitó durante una noche a temperatura ambiente. La mezcla de reacción se diluyó en una solución acuosa saturada de NH4Cl y se extrajo con diclorometano. La fase orgánica se pasó a través de un embudo separador de fase y se concentró al vacío. El residuo en bruto se purificó por cromatografía sobre gel de sílice usando una columna ISCO de 40 g (gradiente de EtOAc al 0-20 %/CH2Ch) para dar 545 mg del producto deseado en forma de un aceite incoloro: RMN 1H (400 MHz, DMSO-d6 ) 67,92 -7,78 (m, 2H), 7,61 (d, J = 8,2 Hz, 1H), 5,48 -5,23 (m, 1H), 4,37 -4,24 (m, 1H), 3,90 (s, 2H), 3,86 (s, 3H), 3,76 - 3,43 (m, 3H), 1,87 - 1,56 (m, 2H), 1,42-1,04 (m, 9H); IEN-EM m/z calc. 369,13, encontrado 370,33 (M+1)+; Tiempo de retención: 0,92 minutos.
Formación de 3-(2-cloro-5-(hidroximetil)fenil)-1,4-oxazepano-4-carboxilato de ferc-butilo (150)
A una solución de 3-(2-cloro-5-metoxicarbonil-fenil)-1,4-oxazepano-4-carboxilato de terc-butilo, 149, (0,52 g, 1,47 mmol) en EtOH (9 ml), se le añadió NaBH4 (0,22 g, 5,84 mmol) en porciones. La reacción se agitó a temperatura ambiente durante 15 minutos y después se calentó a 50 °C durante 23 horas. La temperatura se incrementó a 80 °C y se agitó a esta temperatura durante 12 horas. La reacción se interrumpió por adición lenta en una solución acuosa saturada de NaHCO3. La fase acuosa se extrajo dos veces con EtOAc. Las fases orgánicas combinadas se lavaron con una solución acuosa de NH4Cl, se secaron (MgSO4), se filtraron y se concentraron al vacío. El residuo en bruto se purificó por cromatografía sobre gel de sílice con una columna isco de 40 g usando 0-60 % (EtOAc/CH2Ch) para
proporcionar 270 mg del producto deseado en forma de un sólido de color blanco: RMN 1H (400 MHz, DMSO-d6 ) 6 7,36 (d, J = 8,1 Hz, 1H), 7,27 - 7,16 (m, 2H), 5,28 (dt, J = 11,5, 5,2 Hz, 2H), 4,46 (d, J = 5,6 Hz, 2H), 4,37 - 3,97 (m, 1H), 3,92 (d, J = 10,7 Hz, 2H), 3,52 (dc, J = 23,3, 12,7, 11,9 Hz, 3H), 1,72 (d, J = 28,4 Hz, 2H), 1,52 - 1,03 (m, 9H).
Formación de 3-(2-cloro-5-formilfenil)-1,4-oxazepano-4-carboxilato de ferc-butilo (151)
Se disolvió 3-[2-doro-5-(hidroximetil)fenil]-1,4-oxazepano-4-carboxilato de ferc-butilo, 150, (0,27 g, 0,78 mmol) en cloruro de metileno (6 ml). Se añadió peryodinano de Dess-Martin (0,40 g, 0,93 mmol) y la solución de reacción se agitó a temperatura ambiente durante 18 horas. La mezcla se diluyó en una solución acuosa saturada de NaHCO3 y se extrajo dos veces con EtOAc. Las fases orgánicas combinadas se secaron (MgSO4), se filtraron y se concentraron al vacío. El residuo en bruto se purificó por cromatografía sobre gel de sílice con una columna isco de 40 g usando un gradiente de 0-30 % EtOAc/CH2Ch) para proporcionar 187 mg del producto deseado en forma de un sólido de color blanco: RMN 1H (400 MHz, DMSO-d6) 6 10,02 (s, 1H), 7,89 - 7,75 (m, 2H), 7,69 (d, J = 8,7 Hz, 1H), 5,50 - 5,26 (m, 1H), 4,43 -4,11 (m, 1H), 4,07 - 3,85 (m, 2H), 3,79 -3,40 (m, 3H), 1,89 - 1,49 (m, 2H), 1,47-1,04 (m, 9H); IEN-EM m/z calc. 339,12, encontrado 338,54 (M+1)+; Tiempo de retención: 0,87 minutos.
Formación de 3-(2-cloro-5-(2,2,2-trifluoro-1-hidroxietil)fenil)-1,4-oxazepano-4-carboxilato de ferc-butilo (152)
A un solución agitada fría (0 °C) de 3-(2-cloro-5-formil-fenil)-1,4-oxazepano-4-carboxilato de ferc-butilo, 151, (0,17 g, 0,48 mmol) y trimetil(trifluorometil)silano (0,09 ml, 0,58 mmol) en THF (2 ml), se le añadió fluoruro de tetrabutilamonio (0,05 ml de una solución 1 M en THF, 0,05 mmol). La mezcla de reacción se agitó a 0 °C durante 40 minutos y después a temperatura ambiente durante 3 horas. Después, la mezcla se diluyó con HCl ac. 1 N (2 ml) y la agitación se continuó durante un adicional de 3 días. La mezcla se diluyó con diclorometano y se lavó con NaHCO3 acuoso saturado. La fase acuosa se extrajo de nuevo con diclorometano. Las fases orgánicas combinadas se filtraron a través de un separador de fase y se concentró al vacío. El residuo resultante se purificó por cromatografía en columna sobre gel de sílice usando una columna ISCO de 12 gramos eluyendo con un gradiente de EtOAc del 0 al 20 %/CH2Cl2 para proporcionar 80 mg del producto deseado en forma de un sólido de color blanco: RMN 1H (400 MHz, DMSO-d6) 67,56 - 7,30 (m, 3H), 6,90 (s, 1H), 5,43 - 5,13 (m, 2H), 4,42 - 4,07 (m, 1H), 3,98 - 3,80 (m, 2H), 3,65 - 3,34 (m, 3H), 1,85 -1,48 (m, 2H), 1,43 - 1,03 (m, 9H).
Formación de 1-(4-cloro-3-(1,4-oxazepan-3-il)fenil)-2,2,2-trifluoroetan-1-ol (153)
A una solución de 3-[2-cloro-5-(2,2,2-trifluoro-1-hidroxi-etil)fenil]-1,4-oxazepano-4-carboxilato de ferc-butilo, 152, (0,08 g, 0,18 mmol) en diclorometano (2 ml), se le añadió ácido trifluoroacético (2 ml). La mezcla de reacción se agitó a temperatura ambiente durante una hora y se concentró al vacío. El residuo se disolvió en diclorometano y se neutralizó lavándose con una solución acuosa saturada de NaHCO3. La fase acuosa se extrajo de nuevo con diclorometano. Las fases orgánicas combinadas se pasaron a través de un separador de fase y se concentró al vacío para proporcionar aproximadamente 40 mg del producto deseado que se usó sin purificación adicional: IEN-EM m/z calc. 309,07, encontrado 310,17 (M+1)+; Tiempo de retención: 0,55 minutos.
Formación de (+/-)-1-(3-(4-(2-amino-6-metilpirimidin-4-il)-1,4-oxazepan-3-il)-4-clorofenil)-2,2,2-trifluoroetan-1-ol (154)
A una solución de 1-[4-cloro-3-(1,4-oxazepan-3-il)fenil]-2,2,2-trifluoro-etanol (0,040 g, 0,129 mmol) en NMP (1,5 ml), se le añadió 4-cloro-6-metil-pirimidin-2-amina (0,024 g, 0,168 mmol). La mezcla de reacción se calentó a 150 °C durante 17 horas. Después de enfriar la mezcla a temperatura ambiente, la mezcla se cargó directamente sobre una columna ISCO c18- aq de 15 g y se purificó por ejecución de fase inversa con TFA al 0,1 %/H2O y TFA al 0,1 %/CH3CN. Las fracciones que contenían el producto deseado se concentraron al vacío se diluyeron con diclorometano, se neutralizaron con una solución acuosa saturada de NaHCO3 y la mezcla se pasó a través de un separador de fase. La fase orgánica se concentró al vacío para proporcionar 41 mg del producto deseado: se calentó (360K) RMN 1H (400 MHz, DMSO-d6 ) 6 7,50 - 7,29 (m, 3H), 6,65 - 6,48 (m, 1H), 5,63 (d, J = 19,0 Hz, 1H), 5,43 (s, 2H), 5,09 (t, J = 6,8 Hz, 1H), 4,60 (d, J = 15,2 Hz, 1H), 4,19 -4,05 (m, 1H), 3,97 - 3,81 (m, 0H), 3,78 - 3,47 (m, 3H), 2,01 (s, 3H), 1,78 (d, J = 5,6 Hz, 2H); IEN-EM m/z calc. 416,12, encontrado 417,28 (M+1)+; Tiempo de retención: 0,61 minutos.
El producto mezcla racémico se sometió a separación SFC quiral. Condiciones de SFC: (MeOH al 30 % (amoniaco 5 mM) en Cellulose-2) para proporcionar dos productos racémicos separados:
Pico A, (S)-1-(3-(4-(2-amino-6-metilpirimidin-4-il)-1,4-oxazepan-3-il)-4-clorofenil)-2,2,2-trifluoroetan-1-ol (155); 98,5 % puro mediante HPLC: se calentó (360K) RMN 1H (400 MHz, DMSO-d6 ) 67,54 - 7,30 (m, 3H), 6,56 (s, 1H), 5,63 (d, J = 19,0 Hz, 1H), 5,55 - 5,37 (m, 3H), 5,09 (d, J = 7,8 Hz, 1H), 4,60 (d, J = 15,4 Hz, 1H), 4,12 (dd, J = 13,4, 4,9 Hz, 1H), 3,89 (d, J = 11,9 Hz, 1H), 3,76 -3,41 (m, 3H), 2,01 (s, 3H), 1,89 -1,71 (m, 2H). IEN-EM m/z calc. 416,12268, encontrado 417,33 (M+1)+; Tiempo de retención: 0,6 minutos. I-145
Pico B; (R)-1-(3-(4-(2-amino-6-metilpirimidin-4-il)-1,4-oxazepan-3-il)-4-clorofenil)-2,2,2-trifluoroetan-1-ol (156); 99,5% puro mediante HPLC: se calentó (360K) RMN 1H (400 MHz, DMSO-d6 ) 67,57 - 7,33 (m, 3H), 5,60 (s, 1H), 5,56 - 5,36 (m, 3H), 5,09 (c, J = 7,4 Hz, 1H), 4,60 (d, J = 14,9 Hz, 1H), 4,12 (dd, J = 13,4, 4,9 Hz, 1H), 3,99 - 3,79 (m, 1H), 3,79 3,48 (m, 3H), 2,01 (s, 3H), 1,81 (d, J = 19,9 Hz, 2H). I-146
El siguiente análogo se preparó de acuerdo con el Esquema sintético 19:

3-(5-bromo-2-cloro-fenil)-1,4-oxazepano-4-carboxilato de íerc-butilo (157)
RMN 1H (400 MHz, DMSO-d6 ) 67,50 (dd, J = 8,5, 2,4 Hz, 1H), 7,46 -7,33 (m, 2H), 5,38 -5,14 (m, 1H), 4,38 -4,03 (m, 1H), 3,89 (d, J = 12,4 Hz, 2H), 3,75 - 3,52 (m, 2H), 3,49 (t, J = 11,7 Hz, 1H), 1,71 (d, J = 32,1 Hz, 2H), 1,45-1,05 (m, 9h ); IEN-Em m/z calc. 389,04, encontrado 390,27 (M+1 )+; Tiempo de retención: 1,0 minutos.
Ejemplo 20
Esquema sintético 20: (+/-)-4-(3-(2-cloro-5-(1H-pirazol-5-il)feml)-1,4-oxazepan-4-il)-6-metil-pirimidm-2-amma (159) I-157

(a) ácido (2-ferc-butoxicarbonilpirazol-3-il)borónico, Pd(dppf)Cl2-CH2Cl2, 1,4-dioxano, agua, Na2CO3, 105 °C, microondas; (b) diclorometano, ácido trifluoroacético; (g) 2-amino-4-cloro-6-metilpirimidina, NMP, 150 °C
Formación de 3-(2-cloro-5-(1H-pirazol-5-il)fenil)-1,4-oxazepano (158)
A una suspensión de 3-(5-bromo-2-cloro-fenil)-1,4-oxazepano-4-carboxilato de ferc-butilo, 157, (0,48 g, 0,89 mmol) y ácido (2-ferc-butoxicarbonilpirazol-3-il)borónico (0,53 g, 2,48 mmol) en 1,4-dioxano (7,3 ml) y agua (0,73 ml), se le añadieron Pd(dppf)Cl2.DCM (0,20 g, 0,25 mmol) y Na2CO3 (0,39 g, 3,72 mmol). La mezcla se burbujeó con nitrógeno durante 10 minutos y después se calentó en el microondas a 105 °C durante 30 minutos. La mezcla se diluyó en agua y se extrajo dos veces con EtOAc. Las fases orgánicas combinadas se lavaron con salmuera, se secaron (MgSO4), se filtraron y se concentraron al vacío. El material en bruto se diluyó con diclorometano (5 ml) y se añadió ácido trifluoroacético (5 ml) a la mezcla de reacción. La mezcla se agitó a temperatura ambiente durante 30 minutos y se concentró al vacío. El residuo resultante se cargó directamente sobre una columna ISCO c18-aq de 15 g y se purificó por ejecución de fase inversa con TFA al 0,1 %/H2O y TFA al 0,1 %/CH3CN.
Formación de (+/-)-4-(3-(2-cloro-5-(1H-pirazol-5-il)feml)-1,4-oxazepan-4-il)-6-metil-pirimidm-2-amma (159) I-157
A una solución de 3-[2-cloro-5-(1H-pirazol-5-il)fenil]-1,4-oxazepano, 158, (0,22 g, 0,50 mmol) en NMP (4,5 ml), se le añadió 4-cloro-6-metil-pirimidin-2-amina (0,09 g, 0,65 mmol). La mezcla se calentó a 150 °C durante 18 horas. El material se enfrió a temperatura ambiente y se cargó directamente en una columna ISCO c18-aq de 50 g y se purificó por ejecución de fase inversa con TFA al 0,1 %/H2O y TFA al 0,1 %/CH3CN. Las fracciones que contenían el producto deseado junto con una impureza se concentraron al vacío, se diluyeron con diclorometano, se neutralizaron con una solución acuosa saturada de NaHCO3 y la mezcla se pasó a través de un separador de fase. La fase orgánica se concentró al vacío para proporcionar 420 mg del producto deseado: se calentó (360K) RMN 1H (400 MHz, DMSO-d6 ). La mezcla se purificó de nuevo por cromatografía sobre gel de sílice con una columna isco GOLD de 40 g usando un gradiente al 5-100% (MeOH-CH2Cl2 al 20 %/CH2Cl2) para proporcionar 5 mg del producto deseado: RMN 1H (se calentó 360K) (400 MHz, DMSO-d6 ) 6 12,66 (s, 1H), 7,84 - 7,53 (m, 2H), 7,53 - 7,27 (m, 1H), 6,59 (s, 1H), 5,77 - 5,55 (m, 2H), 5,43 (s, 3H), 4,79 - 4,46 (m, 1H), 4,24 - 4,03 (m, 1H), 3,98 - 3,84 (m, 1H), 3,84 - 3,65 (m, 2H), 3,65 - 3,39 (m, 1H), 2,00 (s, 3H), 1,92-1,65 (m, 2H); IEN-EM m/z calc. 384,15, encontrado 385,32 (M+1)+; Tiempo de retención: 0,57 minutos.
Ejemplo 21
Esquema sintético 21: (+/-)-W-(3-(4-(2-ammo-6-metilpirimidm-4-il)-1,4-oxazepan-3-il)-4-clorofenil)metanosulfonamida (164) I-74

(a) CH3SO2CI, piridina, CH2CI3; (b) LÍBH4, THF, 50 °C; (c) MnO2, DMF, CH2CI2; (d) tamices mol. de 4 A, 3-((tributilestannil)metoxi)propan-1-amina, CH2Cl2; después 2,6-lutidina, Cu(OTf)2, hexafluoroisopropanol, CH2Cl2; (e) 2-amino-4-doro-6-metilpirimidina, NMP, 150 °C; (f) separación quiral SFC
Formación de 2-cloro-5-(metilsulfonamido)benzoato de etilo (160)
A una solución de 5-amino-2-cloro-benzoato de etilo (2,95 g, 14,80 mmol) en diclorometano (60 ml), se le añadió piridina (1,32 ml, 16,30 mmol) seguido de la adición gota a gota de cloruro de metanosulfonilo (1,26 ml, 16,30 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 22 horas. La mezcla se diluyó en una solución acuosa saturada de NH4Cl y se extrajo con EtOAc. La fase orgánica se secó (MgSO4), se filtró y se concentró al vacío. El residuo resultante se purificó por cromatografía sobre gel de sílice con una columna isco GOLD 80 g usando 0-20 % (EtOAc/CH2Cl2) para proporcionar 2,6 gramos del producto deseado: RMN 1H (400 MHz, DMSO-cfe) 8 10,08 (s, 1H), 7,60 (d, J = 2,7 Hz, 1H), 7,54 (d, J = 8,8 Hz, 1H), 7,38 (dd, J = 8,7, 2,8 Hz, 1H), 4,33 (c, J = 7,1 Hz, 2H), 3,04 (s, 3H), 1,31 (t, J = 7,1 Hz, 3H); IEN-EM m/z calc. 277,02, encontrado 278,16 (M+1)+; Tiempo de retención: 0,75 minutos.
Formación de W-(4-cloro-3-(hidroximetil)feml)metanosulfonamida (161)
A una solución de 2-cloro-5-(metanosulfonamido)benzoato de etilo, 160, (1,20 g, 4,10 mmol) en THF (20 ml), se le añadió borohidruro de litio (0,26 g, 11,80 mmol). La reacción se agitó a temperatura ambiente durante 10 minutos y después se calentó a 50 °C durante 8 horas y después a temperatura ambiente durante 3 días. La mezcla se diluyó en una solución acuosa saturada de NH4Cl y se extrajo dos veces con EtOAc. Las fases orgánicas combinadas se secaron (MgSO4), se filtraron y se concentraron al vacío para proporcionar 900 mg del producto deseado en forma de un sólido de color blanco: RMN 1H (400 MHz, DMSO-d6) 8 9,84 (s, 1H), 7,44 (dd, J = 2,8, 0,9 Hz, 1H), 7,35 (d, J = 8,6 Hz, 1H), 7,11 (ddd, J = 8 ,6 , 2,8, 0,7 Hz, 1H), 5,43 (t, J = 5,6 Hz, 1H), 4,52 (d, J = 5,4 Hz, 2H), 2,98 (s, 3H).
Formación de N-(4-cloro-3-formilfeml)metanosulfonamida (162)
A una solución de W-[4-cloro-3-(hidroximetil)fenil]metanosulfonamida, 161, (1,8 g, 7,6 mmol) en diclorometano (60 ml) y DMF (10 ml), se le añadió dióxido de manganeso (10,9 g, 125,0 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 7 horas. La mezcla se diluyó con diclorometano y se filtró a través de un lecho de celite y se lavó con diclorometano. El filtrado se concentró al vacío. La mezcla resultante se diluyó en salmuera y se extrajo dos veces con EtOAc. Las fases orgánicas combinadas se lavaron con salmuera, se secaron (MgSO4), se filtraron y se concentraron al vacío. El sólido bruto era mayoritariamente insoluble en diclorometano y EtOAc. El sólido se diluyó con EtOAc al 50 %/diclorometano. El filtrado se purificó por cromatografía sobre gel de sílice con una columna isco de 40 g usando un gradiente de EtOAc al 0-30 %/CH2Cl2. Las fracciones que contenían el producto transparente se combinaron con el precipitado para proporcionar 1,08 gramos del sólido de color blanquecino: IEN-EM m/z calc.
232,99, encontrado 234,03 (M+1)+; Tiempo de retención: 0,69 minutos.
Formación de N-(4-cloro-3-(1,4-oxazepan-3-il)feml)metanosulfonamida (163)
A una solución de 3-(tributilestanilmetoxi)propan-1-amina (1,79 g, 4,73 mmol) en diclorometano (12 ml), se le añadió W-(4-cloro-3-formil-fenil)metanosulfonamida, 162, (1,07 g, 4,58 mmol) seguido de tamices moleculares de 4 angstrom. La mezcla se agitó durante 2 días, se filtró para retirar los tamices y se lavó con diclorometano (45 ml).
En un matraz separado que contenía hexafluoroisopropanol (12 ml), se añadió 2,6-lutidina (0,55 ml, 4,72 mmol) seguido de bis(trifluorometilsulfoniloxi)cobre (1,77 g, 4,89 mmol). La mezcla se agitó durante 1 h, después se añadió la solución de imina preparada anteriormente en una porción. La mezcla cambió de color azul a verde. La mezcla se agitó durante 2 días a temperatura ambiente. La mezcla se diluyó con 150 ml de 2:1 de solución acuosa saturada de NaHCO3 e hidróxido de amonio al 10 %. Después de agitar durante 30 minutos, la capa orgánica se retiró y se lavó dos veces con una solución acuosa saturada de NaHCO3. La capa orgánica se pasó a través de un embudo separador de fase y se concentró al vacío. El residuo en bruto se purificó por fase inversa con columna ISCO - c18-aq de 150 gramos ejecutándose con TFA al 0,1 %/H2O y TFA al 0,1 %/CH3CN. El producto resultante se purificó de nuevo por fase inversa con columna ISCO - c18-aq de 100 gramos ejecutándose con TFA al 0,1 %/H2O y TfA al 0,1 %/CH3Cn .
Todas las fracciones que contenían el producto se concentraron al vacío y el residuo se diluyó con una solución acuosa saturada de NaHCÜ3 y se extrajo tres veces con diclorometano y después tres veces con MeOH al 10 %/diclorometano. La mezcla se pasó a través de un separador de fase y la fase orgánica se concentró al vacío para proporcionar 163 mg del producto deseado: IEN-EM m/z calc. 304,06, encontrado 305,18 (M+1)+; Tiempo de retención: 0,51 minutos. La CLEM todavía mostró producto en fase acuosa. La fase acuosa se concentró al vacío. El sólido de color blanco resultante se diluyó con acetonitrilo y se agitó vigorosamente durante 30 minutos, se filtró, se lavó con acetonitrilo y el filtrado se concentró al vacío para proporcionar 500 mg de mezcla que contenía la mayor parte del producto deseado.
Formación de (R)-W-(3-(4-(2-ammo-6-metMpinmidm-4-M)-1,4-oxazepan-3-M)-4-clorofeml) metanosulfonamida (165) y (S)-W-(3-(4-(2-ammo-6-metMp¡rim¡dm-4-M)-1,4-oxazepan-3-M)-4-clorofeml)metanosulfon-am¡da (166)
A una solución de W-[4-cloro-3-(1,4-oxazepan-3-il)fenil]metanosulfonamida, 163, (0,16 g, 0,53 mmol) en NMP (4 ml), se le añadió 4-cloro-6-metil-pirimidin-2-amina (0,10 g, 0,69 mmol). La mezcla de reacción se calentó a 150 °C durante 16 horas. La mezcla de reacción se enfrió a temperatura ambiente y se cargó directamente en una columna ISCO c18-aq de 50 g y se purificó por ejecución de fase inversa con TFA al 0,1 %/H2O y TFA al 0,1 %/CH3CN. Todas las fracciones que contenían el producto se concentraron al vacío y el residuo resultante se diluyó con diclorometano, se neutralizaron con una solución acuosa saturada de NaHCO3 y se pasó a través de un separador de fase. La fase orgánica se concentró al vacío para proporcionar 233 mg del producto deseado en forma de un sólido de color pardo claro. La mezcla racémica se sometió a separación SFC quiral.
Pico A: (R)-W-(3-(4-(2-amino-6-metilpyiimidin-4-il)-1,4-oxazepan-3-il)-4-clorofenil) metanosulfonamida (165); 74 mg de sólido de color amarillo. [□fc = -128,77 (c = 3,5 mg/0,8 ml de MeOH), 99 % mediante HPLC quiral; 98 % ee. RMN 1H se calentó (360K) (400 MHz, DMSO-c/6 ) 89,79 -9,44 (m, 1H), 7,62 - 7,33 (m, 1H), 7,36 -6,98 (m, 2H), 5,85 -5,25 (m, 4H), 4,81 - 4,46 (m, 1H), 4,38 - 4,09 (m, 1H), 3,94 (s, 4H), 3,24 (s, 3H), 2,06 (s, 3H), 1,98-1,62 (m, 2H); IEN-EM m/z calc. 411,11, encontrado 412,28 (M+1)+; Tiempo de retención: 0,57 minutos. I-160
Pico B: (S)-W-(3-(4-(2-amino-6-metilpirimidin-4-il)-1,4-oxazepan-3-il)-4-clorofenil) metanosulfonamida (166); 65 mg de sólido de color amarillo. [□fc = 124,74 (c = 3,1 mg/0,8 ml de MeOH); 99 % mediante HPLC quiral; 98 % ee. RMN 1H se calentó (360K) (400 MHz, DMSO-d6 ) 89,54 (s, 1H), 7,36 (dd, J = 8 ,8 , 4,4 Hz, 1H), 7,26 - 7,03 (m, 2H), 5,47 (t, J = 40,2 Hz, 4H), 4,58 (s, 1H), 4,19 -4,02 (m, 1H), 3,88 (s, 1H), 3,80 - 3,40 (m, 3H), 2,91 (s, 3H), 2,00 (s, 3H), 1,88 1,69 (m, 2H); IEN-EM m/z calc. 411,11, encontrado 412,28 (M+1)+; Tiempo de retención: 0,57 minutos. I-161
Ejemplo 22
Esquema sintético 22: (+/-)-4-(2-(2-clorofeml)-4-(metMsulfoml)-1,4-diazepan-1-il)-6-metilpirimidm-2-amma (170) I-28

(a) 2-clorobenzaldehído, tamices moleculares de 4 A, CH2O 2 después 2,6-lutidina, Cu(OTf)2, hexafluoroisopropanol, cH 2Cl2; (b) 2-amino-4-cloro-6-metilpirimidina, Et3N, NMP, 150 °C; (c) HCl, 1,4-dioxano; (d) cloruro de metanosulfonilo, Et3N, CH2Cl2.
Formación de 3-(2-clorofenil)-1,4-diazepano-1-carboxilato de (+/-)-terc-butilo (167)
A una solución de (3-aminopropil)((tributilestannil)metil)carbamato de ferc-butilo (SnAP-DA) (3,0 g, 6,3 mmol) en diclorometano anhidro (16 ml), se le añadió 2-clorobenzaldehído (0,71 ml) seguido de 4A MS (0,64 g). La mezcla de color amarillo turbia se agitó a temperatura ambiente durante 2 horas y después se filtró a través de Celite. El lecho de filtro se aclaró con 25 ml de diclorometano y el filtrado se concentró a sequedad.
En un matraz RB de 250 ml separado que contenía hexafluoroisopropanol (25 ml), se le añadió 2,6-lutidina (0,73 ml, 6,28 mmol) seguido de Cu(OTf)2 (0,40 g, 6,295 mmol) (Cu(OTf)2 se secó a alto vacío durante 30 minutos y se calentó con una pistola de calor). La suspensión instantáneamente se volvió de color azul oscuro tras la adición Cu(OTf)2. La mezcla se agitó a temperatura ambiente durante 1 h.
La solución de imina preparada anteriormente se disolvió en diclorometano (100 ml) y se vertió directamente en la mezcla de lutidin-Cu(OTf)2-hexafluoroisopropanol de color verdoso/azul. La reacción se volvió de color verde muy oscuro inmediatamente y se agitó durante una noche a temperatura ambiente. La reacción se interrumpió con 150 ml de una mezcla 2:1 de solución acuosa saturada de bicarbonato sódico e hidróxido de amonio al 10 %. La mezcla se
agitó durante 15 minutos y después se separó. La capa acuosa se extrajo 2 x 150 ml de diclorometano. Las fases orgánicas combinadas se lavaron con salmuera, se secaron sobre sulfato de magnesio, se filtraron y se concentraron para dar 13 g de un aceite de color ámbar. El material en bruto se purificó por cromatografía sobre gel de sílice eluyendo con EtOAc al 0-100% en heptano (columna ISCO de 40 g). Se usó TLC con tinción con ninhidrina para identificar las fracciones que contenían el producto deseado. Las fracciones que contenían el producto deseado se combinaron y se concentraron para dar 1,7 g de un aceite de color naranja claro. El material se purificó una segunda vez por cromatografía sobre gel de sílice eluyendo con EtOAc al 0-75 % en heptano (columna ISCO de 40 g). Las fracciones puras se combinaron y se concentraron para dar 700 mg del producto deseado en forma de un aceite incoloro: RMN 1H (400 MHz, CDCla) 87,54 (ddd, J = 13,2, 7,7, 1,8 Hz, 1H), 7,36 (dd, J = 7,9, 1,5 Hz, 1H), 7,31 - 7,14 (m, 2H), 4,32 (dd, J = 10,2, 3,4 Hz, 1H), 4,18 - 3,88 (m, 2H), 3,41 - 3,18 (m, 2H), 3,01 - 2,72 (m, 2H), 1,99 - 1,72 (m, 2h ), 1,50 (d, J = 4,4 Hz, 9H); IEN-EM m/z calc. 310,1, encontrado 311,0 (M+1)+; Tiempo de retención: 0,71 minutos.
Formación de 4-(2-ammo-6-metil-pirimidm-4-il)-3-(2-dorofeml)-1,4-diazepano-1-carboxilato de (+/-j-íerc-butilo (168)
Una mezcla de 3-(2-clorofenil)-1,4-diazepano-1-carboxilato de ferc-butilo, 167, (0,42 g, 1,35 mmol), 4-cloro-6- metilpirimidin-2-amina (0,19 g, 1,34 mmol) y trietilamina (0,38 ml, 2,69 mmol) en NMP (6 ml) se agitó durante 1 día a 150 °C y después 3 días a temperatura ambiente. La reacción se diluyó con agua y se extrajo con EtOAc. La capa orgánica se secó sobre sulfato de magnesio, se filtró y se concentró a sequedad. El producto en bruto se purificó por cromatografía sobre gel de sílice eluyendo con MeOH al 0-15 % en diclorometano. Las fracciones puras se combinaron y se concentraron para dar 163 mg del producto deseado en forma de un aceite de color pardo: RMN 1H (400 MHz, DMSO-d6 ) 87,46 -7,39 (m, 1H), 7,31 -7,21 (m, 3H), 5,78 (s, 1H), 5,61 (s, 1H), 5,40 (s, 2H), 4,54 (d, J = 14,9 Hz, 1H), 4,33 (dd, J = 14,8, 5,7 Hz, 1H), 3,99 (d, J = 13,7 Hz, 1H), 3,67 -3,55 (m, 1H), 3,16 (dd, J = 14,9, 11,2 Hz, 1H), 2,86 (t, J = 12,9 Hz, 1H), 2,00 (s, 3H), 1,87 - 1,52 (m, 2H), 1,38 (s, 9H); IEN-EM m/z calc. 417,2, encontrado 418,0 (M+1)+; Tiempo de retención: 0,81 minutos.
Formación de (+/-)-4-(2-(2-dorofeml)-1,4-diazepan-1-il)-6-metilpirimidm-2-amma (169)
Una solución de 4-(2-amino-6-metil-pirimidin-4-il)-3-(2-clorofenil)-1,4-diazepano-1-carboxilato de ferc-butilo, 168, (0,08 g, 0,19 mmol) en HCl (3 ml de solución 4 M, 12,00 mmol) en dioxano se agitó durante una noche a temperatura ambiente y después se concentró a sequedad. El residuo en bruto se usó sin purificación adicional: IEN-EM m/z calc.
317,1, encontrado 318,0 (M+1)+; Tiempo de retención: 0,49 minutos.
Formación de (+/-)-4-(2-(2-clorofenil)-4-(metilsulfonil)-1,4-diazepan-1-il)-6-metilpirimidin-2-amina (170) I-28
A una solución de 4-[2-(2-clorofenil)-1,4-diazepan-1-il]-6-metil-pirimidin-2-amina, 169, (0,03 g, 0,09 mmol) y trietilamina (0,53 ml, 0,38 mmol) en diclorometano (1,9 ml), se le añadió gota a gota una solución de cloruro de metano sulfonilo (0,008 ml, 0,0969 mmol) en diclorometano (1,2 ml). La reacción se agitó durante 5 minutos y después se concentró a sequedad. El residuo en bruto se purificó por cromatografía sobre gel de sílice eluyendo con MeOH al 0-10% en diclorometano (columna ISCO de 4 g). Las fracciones puras se combinaron, se concentraron y se liofilizaron para proporcionar 15 mg del producto deseado: RMN 1H (400 MHz, DMSO-d6 ) 87,50 - 7,39 (m, 1H), 7,36 - 7,20 (m, 3H), 5,96 (s, 2H), 5,74 (s, 1H), 4,60 (d, J = 15,2 Hz, 1H), 4,21 (ddd, J = 15,4, 5,3, 1,2 Hz, 1H), 3,80 -3,61 (m, 2H), 3,28 (dd, J = 15,4, 11,1 Hz, 1H), 3,08 -3,01 (m, 1H), 2,86 (s, 3H), 2,08 (s, 3H), 1,92 - 1,72 (m, 2H), 1,20 (t, J = 7,3 Hz, 1H); IEN-EM m/z calc. 395,1, encontrado 396,0 (M+1)+; Tiempo de retención: 0,65 minutos.
Ejemplo 23
Esquema sintético 23: (+/-)-W-[3-[4-(2-ammo-6-metil-pirimidm-4-il)-1,4-oxazepan-3-il]-4-doro-feml]-3-hidroxipropanamida (171) I-174

(+/-)-W-[3-[4-(2-ammo-6-metil-pirimidm-4-il)-1,4-oxazepan-3-il]-4-doro-feml]-3-hidroxi-propanamida (171) I-174
A una solución de 4-[3-(5-amino-2-cloro-fenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina, 46, (0,22 g, 0,66 mmol) en DMF (18 ml), se le añadieron ácido 3-hidroxipropanoico (0,08 g, 0,89 mmol), diisopropiletil amina (0,26 g, 2,01 mmol) y HATU (0,46 g, 1,21 mmol). La mezcla se agitó a temperatura ambiente durante 10 horas. La mezcla en bruto se diluyó con EtOAc y se lavó tres veces con una solución acuosa saturada de NaHCO3. La fase orgánica se secó (MgSO4), se filtró y se concentró al vacío. El producto en bruto se purificó por cromatografía sobre gel de sílice (columna ISCO de 40 g) eluyendo con DCM, MeOH al 10 %/DCM (gradiente del 0 % al 65 %) para proporcionar 54
mg (20 %): RMN 1H (300 MHz, DMSO-d6j 5 12,67 (a, 1H), 10,08 (d, J = 14,6 Hz, 1H), 7,83 (a, 1H), 7,74 - 7,31 (m, 4H), 6,67 (s, 0,5H), 5,95 (dd, J = 10,3, 5,4 Hz, 0.5H), 5,57 (d, J = 3,0 Hz, 0.5H), 5,18 (dd, J = 9,6, 4,7 Hz, 0.5H), 5,01 (d, J = 13,9 Hz, 1H), 4,63 (t, J = 5,9 Hz, 2H), 4,23 (ddd, J = 30,9, 13,7, 5,2 Hz, 2H), 4,01 - 3,53 (m, 4H), 2,81 (t, J = 5,9 Hz, 1H), 2,42 (t, J = 6,2 Hz, 1H), 2,29 (s, 1,5H), 2,20 (s, 1,5H), 1,83 (m, 2H); IEN-EM m/z calc. 405,16, encontrado 406,2 (M+1)+; Tiempo de retención: 0,57 minutos.
Los siguientes análogos se prepararon de acuerdo con el Esquema sintético 23:

(+/-)-W-[3-[4-(2-ammo-6-metN-pinmidm-4-M)-1,4-oxazepan-3-M]-4-cloro-fenM]-2,2,2-t
N fl
uoro-acetamida (172) I-159
Se calentó (360K) RMN 1H (400 MHz, DMSO-d6 ) 6 11,12 (s, 1H), 7,71 (d, J = 7,3 Hz, 2H), 7,52 (d, J = 9,1 Hz, 1H), 7,12 (s, 2H), 6,13 (s a, 1H), 5,66 (s a, 1H), 4,56 (a, 1H), 4,23 (dd, J = 13,6, 5,1 Hz, 1H), 4,01 -3,75 (m, 3H), 3,72-3,57 (m, 1H), 2,25 (s, 3H), 1,89 (d, J = 14,5 Hz, 2H); IEN-EM m/z calc. 429,12, encontrado 430,14 (M+1)+; Tiempo de retención: 0,68 minutos.
La mezcla racémica se sometió a separación SFC quiral. Condiciones de SFC: Columna: CI, 20x250mm, fase móvil: MeOH al 30 % (amoniaco 5 mM), flujo de CO2 al 70 %; 75 ml/min; concentraciones: ~24 mg/ ml (MeOH); volumen de inyección de 250 pl; longitud de onda: 254 nM; método tipo - isocrático.
Pico A: (R)-A/-[3-[4-(2-amino-6-metil-pirimidin-4-il)-1,4-oxazepan-3-il]-4-clorofenil]-2,2,2-trifluoroacetamida (173); [a]D (c = 0,5, MeOH) -159,04 (98 % ee); IEN-EM m/z calc. 429,1, encontrado 431,8 (M+1)+; Tiempo de retención: 0,54 minutos. I-176
Pico B: (S)-W-[3-[4-(2-amino-6-metil-pirimidin-4-il)-1,4-oxazepan-3-il]-4-clorofenil]-2,2,2-trifluoroacetamida (174): [a]D (c = 0,5, MeOH) 157,28 (99 % ee); RMN 1H (300 MHz, Metanol-d4) 66,35 (d, J = 2,5 Hz, 2H), 6,16 (d, J = 9,2 Hz, 1H), 4,22 (a, 1H), 3,02 (dd, J = 13,6, 5,1 Hz, 1H), 2,88 -2,67 (m, 1H), 2,56 -2,23 (m, 3H), 2,03 (m, 2H), 0,59 (m, 2H); Ie N-EM m/z calc. 429,12, encontrado 429,99 (M+1 )+; Tiempo de retención: 0,65 minutos. I-177

(+/-)-W-[3-[4-(2-ammo-6-metN-pinmidm-4-M)-1,4-oxazepan-3-M]-4-cloro-fenM]-2-(dimetMammo)acetamida (175) I-168
RMN 1H (300 MHz, DMSO-d6) 6 9,90 (s, 1H), 7,73 (d, J = 8,7 Hz, 1H), 7,63 (d, J = 2,6 Hz, 1H), 7,38 (d, J = 8,7 Hz, 1H), 5,96 (s a, 2H), 5,03 (a, 2H), 4,11 (s, 1H), 3,94 (m, 1H), 3,74 -3,47 (m, 3H), 3,33 (s, 6 H), 3,04 (s, 2H), 2,25 (s, 3H), 1,98 (a, 2 H); IEN-EM m/z calc. 418,19, encontrado 419,09 (M+1)+; Tiempo de retención: 0,58 minutos.

(+/-)-W-(3-(4-(2-ammo-6-metNpirimidm-4-N)-1,4-oxazepan-3-M)-4-clorofenM)-2,2-difluoroacetamida I-250
RMN 1H (300 MHz, Metanol-d4) 67,78 (dd, J = 9,2, 2,5 Hz, 1H), 7,67 - 7,35 (m, 2H), 6,63 - 6,48 (m, 0H), 6,36 - 5,91 (m, 1H), 5,65 (d, J = 1,0 Hz, 1H), 5,37 (dd, J = 10,2, 5,0 Hz, 1H), 5,20 (d, J = 14,5 Hz, 1H), 4,47 -4,21 (m, 1H), 4,12 -3,53 (m, 4H), 2,35 (d, J = 0,8 Hz, 1H), 2,21 (d, J = 0,8 Hz, 2H), 1,96 (d, J = 10,7 Hz, 2H); IEN-EM m/z calc. 411,13, encontrado 412,23 (M+1)+; Tiempo de retención: 0,62 minutos.
Ejemplo 24
Esquema sintético 24: (+/-)-4-[4-(2-amino-6-metil-pirimidin-4-il)-1,4-oxazepan-3-il]-3-cloro-benzonitrilo (179) I-97

(a) ácido 2-doro-4-cianofenilborónico, Pd(Ph3P)2Cl2, DME, 50 °C; (b) MeMgBr, DME; (c) NaBH4, MeOH; (d) 4-cloro-6-metil-pirimidin-2-amina, NMP, 150 °C; (e) cromatografía SFC quiral.
Formación de (+/-)-3-cloro-4-(2,5,6,7-tetrahidro-1,4-oxazepin-3-il)benzonitrilo (176)
Se preparó el Intermedio 176 de acuerdo con Esquema sintético 13 usando ácido 2-cloro-4-cianofenilborónico en lugar de ácido 2-cloro-5-hidroxifenilo borónico.
Formación de (+/-)-3-cloro-4-(2,5,6,7-tetrahidro-1,4-oxazepin-3-il)benzonitrilo (177)
A una solución de 3-cloro-4-(4-formil-6,7-dihidro-5H-1,4-oxazepin-3-il)benzonitrilo, 176, (1,0 g, 3,8 mmol) en DME (60 ml) de -5 a 0 °C, se le añadió MeMgBr (2 ml de solución 2 M en 2-MeTHF, 6,4 mmol) en una atmósfera de nitrógeno. Después de 15 minutos, la reacción se interrumpió con tartrato de sodio y potasio 1 M y se agitó vigorosamente durante 1 hora. La mezcla de reacción se concentró parcialmente y después se extrajo dos veces con diclorometano. Las capas se separaron con la ayuda de un separador de fase y los orgánicos combinados se concentraron al vacío para dar el producto deseado en forma de un semisólido de color amarillo. El producto en bruto se usó en la siguiente etapa sin purificación adicional: IEN-EM m/z calc. 252,07 encontrado 253,12 (M+1)+; Tiempo de retención: 0,55 minutos.
Formación de (+/-)-3-cloro-4-(1,4-oxazepan-3-il)benzonitrilo (178)
A una solución de 3-cloro-4-(2,5,6,7-tetrahidro-1,4-oxazepin-3-il)benzonitrilo, 177, (0,84 g, 3,59 mmol) en MeOH (10 ml), se le añadió NaBH4 (0,68 g, 18,00 mmol) a temperatura ambiente. Después de 1,5 horas, la mezcla se calentó a 50 °C. Después de otras 3 horas, se añadió 1,0 g adicional de NaBH4 a 50 °C y después se agitó a temperatura ambiente durante una noche. La mezcla de reacción se concentró y se extrajo dos veces con diclorometano. Las capas se separaron con la ayuda de un separador de fase y los orgánicos se concentraron al vacío. La purificación se realizó en una columna Is Co c18-aq de 100 g de fase inversa, ejecutándose con TFA al 0,1 %/H2O y TFA al 0,1 %/CH3CN. Las fracciones puras se concentraron parcialmente al vacío, se añadió algo de NaOH 1 M y la mezcla se extrajo con diclorometano tres veces y se concentró al vacío para dar 294 mg del producto deseado: RMN 1H (400 MHz, DMSO-d6 ) 58,00 (m, 1H), 7,81 (m, 1H), 4,28 (dd, J = 8,7, 3,3 Hz, 1H), 3,85 - 3,78 (m, 2H), 3,75-3,60 (m, 1H), 3,30 - 3,26 (m, 1H), 3,13 - 3,03 (m, 1H), 2,92 - 2,84 (m, 1H), 1,91 - 1,81 (m, 2H). IEN-EM m/z calc. 236,07, encontrado 237,1 (M+1)+; Tiempo de retención: 0,49 minutos.
Formación de (+/-)-4-[4-(2-amino-6-metil-pirimidin-4-il)-1,4-oxazepan-3-il]-3-cloro-benzonitrilo (179) I-97 Una mezcla de 3-cloro-4-(1,4-oxazepan-3-il)benzonitrilo (0,19 g, 0,80 mmol) y 4-cloro-6-metil-pirimidin-2-amina (0,10 g, 0,70 mmol) se calentó en NMP (1,5 ml) a 160 °C durante 3,5 h. La purificación se realizó en una columna ISCO c18-aq de 50 g de fase inversa, ejecutándose con TFA al 0,1 %/H2O y TFA al 0,1 %/CH3CN. Las fracciones puras se concentraron al vacío, se añadió trietilamina (1 ml) y se concentró al vacío de nuevo. La purificación por cromatografía en columna (columna de 40 g; MeOH al 0-10 %/diclorometano) proporcionó 124 mg del producto deseado: alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6) 57,96 (s, 1H), 7,73 (dd, J = 8,1, 1,6 Hz, 1H), 7,50 (d, J = 8,1 Hz, 1H), 6,37 (s, 2H), 5,96 (s, 1H), 5,65 (s, 1H), 4,47 (d, J = 16,3 Hz, 1H), 4,16 (dd, J = 13,6, 4,9 Hz, 1H), 3,93 - 3,76 (m, 3H), 3,65 -3,57 (m, 1H), 2,15 (s, 3H), 1,87-1,79 (m, 2H); IEN-EM m/z calc. 343,12, encontrado 344,15 (M+1)+; Tiempo de retención: 0,57 minutos. La mezcla racémica se sometió a purificación SFC quiral para obtener los enantiómeros individuales:
Pico A: (R)-4-[4-(2-amino-6-metil-pirimidin-4-il)-1,4-oxazepan-3-il]-3-cloro-benzonitrilo (180); >99% ee; alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6 ) 57,93 (d, J = 1,5 Hz, 1H), 7,69 (dd, J = 8,1, 1,6 Hz, 1H), 7,48 (d, J = 8,1 Hz, 1H), 5,66 (s, 1H), 5,54 (d, J = 5,3 Hz, 1H), 5,41 (s, 2H), 4,47 (d, J = 16,0 Hz, 1H), 4,12 (dd, J = 13,5, 4,8 Hz, 1H), 3,89 (dd, J = 8,5, 3,6 Hz, 1H), 3,73 (ddd, J = 15,2, 12,9, 7,7 Hz, 2H), 3,61 - 3,52 (m, 1H), 2,02 (s, 3H), 1,83-1,74 (m, 2 H); iEn -EM m/z calc. 343,12, encontrado 344,17 (m 1 )+; Tiempo de retención: 0,58 minutos. I-96
Pico B: (S)-4-[4-(2-amino-6-metil-pirimidin-4-il)-1,4-oxazepan-3-il]-3-cloro-benzonitrilo (181); >99% ee; alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6 ) 57,93 (d, J = 1,5 Hz, 1H), 7,69 (dd, J = 8,1, 1,6 Hz, 1H), 7,48 (d, J = 8,1 Hz, 1H), 5,66 (s, 1H), 5,54 (d, J = 5,3 Hz, 1H), 5,41 (s, 2H), 4,47 (d, J = 16,0 Hz, 1H), 4,12 (dd, J = 13,5, 4,8 Hz, 1H), 3,89 (dd, J = 8,5, 3,6 Hz, 1H), 3,73 (ddd, J = 15,2, 12,9, 7,7 Hz, 2H), 3,61 - 3,52 (m, 1H), 2,02 (s, 3H), 1,83-1,74
(m, 2H); IEN-EM m/z calc. 343,12, encontrado 344,17 (M+1)+; Tiempo de retención: 0,58 minutos. I-97
Los siguientes análogos se prepararon de acuerdo con el Esquema sintético 24 partiendo de los ácidos borónicos apropiados:
(+/-)-4-[3-(2-cloro-5-fluoro-fenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (182) I-70
alta temperatura (360 K) RMN 1H (400 MHz, DMSO-c/6 ) 87,47 (dd, J = 8 ,8 , 5,2 Hz, 1H), 7,11 (td, J = 8,2; 3,0 Hz, 1H), 7,06 (dd, J = 9,6, 3,1 Hz, 1H), 5,62 (s, 1H), 5,49-5,41 (m, 3H), 4,60-4,50 (m, 1H), 4,11 (dd, J = 13,5, 4,9 Hz, 1H), 3,92 -3,85 (m, 1H), 3,76 (dd, J = 13,5, 10,0 Hz, 1H), 3,73 -3,64 (m, 1H), 3,56 (dd, J = 14,4, 12,0 Hz, 1H), 2,02 (s, 3H), 1,80 1,74 (m, 2H); IEN-EM m/z calc. 336,12, encontrado 337,14 (M+1)+; Tiempo de retención: 0,62 minutos. La mezcla racémica se sometió a purificación SFC quiral para obtener los enantiómeros individuales:
Pico A: (R)-4-[3-(2-cloro-5-fluoro-fenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (183); 99,9 % ee, alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6 ) 87,93 (d, J = 1,5 Hz, 1H), 7,69 (dd, J = 8,1, 1,6 Hz, 1H), 7,48 (d, J = 8,1 Hz, 1H), 5,66 (s, 1H), 5,54 (d, J = 5,3 Hz, 1H), 5,41 (s, 2H), 4,47 (d, J = 16,0 Hz, 1H), 4,12 (dd, J = 13,5, 4,8 Hz, 1H), 3,89 (dd, J = 8,5, 3,6 Hz, 1H), 3,73 (ddd, J = 15,2, 12,9, 7,7 Hz, 2H), 3,61 - 3,52 (m, 1H), 2,02 (s, 3H), 1,83-1,74 (m, 2H); IEN-EM m/z calc. 343,12, encontrado 344,17 (M+1)+; Tiempo de retención: 0,58 minutos. I-78
Pico B: (S)-4-[3-(2-cloro-5-fluoro-fenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (184); 98% ee; alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6 ) 87,93 (d, J = 1,5 Hz, 1H), 7,69 (dd, J = 8,1, 1,6 Hz, 1H), 7,48 (d, J = 8,1 Hz, 1H), 5,66 (s, 1H), 5,54 (d, J = 5,3 Hz, 1H), 5,41 (s, 2H), 4,47 (d, J = 16,0 Hz, 1H), 4,12 (dd, J = 13,5, 4,8 Hz, 1H), 3,89 (dd, J = 8,5, 3,6 Hz, 1H), 3,73 (ddd, J = 15,2, 12,9, 7,7 Hz, 2H), 3,61 - 3,52 (m, 1H), 2,02 (s, 3H), 1,83-1,74 (m, 2H); IEN-EM m/z calc. 343,12, encontrado 344,17 (M+1)+; Tiempo de retención: 0,58 minutos. I-79
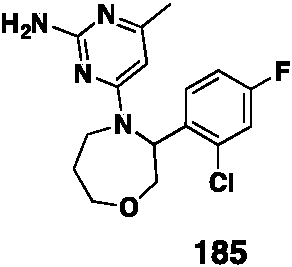
(+/-)-4-[3-(2-cloro-4-fluoro-fenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (185) I-63
alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6 ) 87,35 (dd, J = 8 ,8 , 6,4 Hz, 2H), 7,13 (d, J = 6,0 Hz, 1H), 5,59 (s, 1H), 5,47-5,41 (m, 2H), 4,58 (s, 1H), 4,08 (dd, J = 13,4, 5,1 Hz, 1H), 3,87 (s, 1H), 3,75-3,50 (m, 3H), 2,01 (s, 3H), 1,77 (s, 2H); IEN-EM m/z calc. 336,12, encontrado 337,14 (M+1)+; Tiempo de retención: 0,62 minutos.
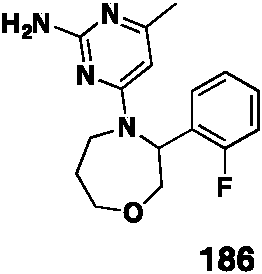
(+/-)-4-[3-(2-fluorofenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (186) I-32
alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6 ) 87,32 - 7,23 (m, 2H), 7,14 (dd, J = 13,0, 5,0 Hz, 2H), 5,71 (s, 1H), 5,55 (s, 1H), 5,45 (s, 2H), 4,43 (d, J = 15,5 Hz, 1H), 4,15 (dd, J = 13,4, 5,1 Hz, 1H), 3,90 (d, J = 12,0 Hz, 1H), 3,77 (dd, J = 13,3, 10,2 Hz, 1H), 3,63 -3,49 (m, 2H), 2,02 (s, 3H), 1,89-1,68 (m, 2H); IEN-EM m/z calc. 302,15, encontrado 303,19 (M+1)+; Tiempo de retención: 0,58 minutos.

(+/-)-1-[3-[4-(2-amino-6-metil-pirimidin-4-il)-1,4-oxazepan-3-il]-4-clorofenil]pirrolidin-2-ona (187) I-119
alta temperatura (360 K) RMN 1H (400 MHz, DMSO-c/6 ) 87,76 (d, J = 2,7 Hz, 1H), 7,45 (dd, J = 8,7, 2,7 Hz, 1H), 7,39 (d, J = 8,8 Hz, 1H), 5,59 (s, 1H), 5,42 (s, 3H), 4,63 (d, J = 15,1 Hz, 1H), 4,12 (dd, J = 13,4, 5,0 Hz, 1H), 3,91 (dt, J = 11,5, 3,8 Hz, 1H), 3,81 - 3,73 (m, 2H), 3,72 - 3,52 (m, 3H), 2,47 -2,40 (m, 2H), 2,08 -2,01 (m, 2H), 2,00 (s, 3H), 1,80 (ddt, J = 10,9, 7,5, 4,2 Hz, 2H); IEN-EM m/z calc. 401,2, encontrado 402,0 (M+1)+; Tiempo de retención: 0,65 minutos.

(+/-)-(2-(4-(2-amino-6-metilpirimidin-4-il)-1,4-oxazepan-3-il)fenil)metanoM-91
alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6) 87,77 (s, 1H), 7,51 (dd, J = 7,7, 0,7 Hz, 1H), 7,36 (td, J = 7,4, 1,8 Hz, 1H), 7,29 -7,21 (m, 2H), 6,12 (s, 1H), 5,18 (t, J = 5,5 Hz, 1H), 4,41 (d, J = 5,5 Hz, 2H), 4,18 (t, J = 5,9 Hz, 2H), 3,93 (t, J = 6,5 Hz, 2H), 2,03-1,95 (m, 2H); IEN-EM m/z calc. 233,11, encontrado 234,17 (M+1)+; Tiempo de retención: 0,62 minutos

(+/-)-4-[3-(6-metoxi-2-metil-3-piridil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina I-77
alta temperatura (360 K) RMN 1H (400 MHz, DMSO-c/6 ) 87,56 (s, 1H), 6,59 (d, J = 8,5 Hz, 1H), 6,04 - 5,62 (m, 1H), 4,08 -4,01 (m, 1H), 3,89-3,85 (m, 5H), 3,75-3,55 (m, 2H), 3,33-3,27 (m, 2H), 2,52 (s, 3H), 2,15 (s, 3H), 2,01-1,75 (m, 2 H); IEN-e M m/z calc. 329,19, encontrado 330,24 (M+1 )+; Tiempo de retención: 0,47 minutos. 888

(+/-)-4-[3-(2,5-difluorofenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina I-52
alta temperatura (360 K) RMN 1H (400 MHz, DMSO-c/6 ) 87,23 - 7,15 (m, 1H), 7,12 - 6,98 (m, 2H), 5,75 (s, 1H), 5,58 (s, 1H), 5,47 (s, 2H), 4,36 (d, J = 15,7 Hz, 1H), 4,13 (dd, J = 13,3, 5,1 Hz, 1H), 3,89 (d, J =11,4 Hz, 1H), 3,80 (dd, J = 13,3, 10,0 Hz, 1H), 3,65 - 3,50 (m, 2H), 2,04 (s, 3H), 1,87-1,68 (m, 2H); IEN-EM m/z calc. 320,14, encontrado 321,2 (M+1)+; Tiempo de retención: 0,6 minutos.
Ejemplo 25
Esquema sintético 25: (+/-)-4-[4-(2-ammo-6-metil-pirimidm-4-M)-1,4-oxazepan-3-M]-3-cloro-W-ciclopropilbenzamida (191) I-155

(a) 3-(tributilestanilmetoxi)propan-1-amina, tamices moleculares de 4 A, CH2CI2; (b) 2,6-lutidina, Cu(OTf)2, hexafluoroisopropanol, C ^C h ; c) 2-amino-4-doro-6-metilpirimidina, NMP, 150 °C; (d) LiOH, MeOH, H2O; (e) ciclopropil amina, HATU, Et3N, DMF.
Formación de 3-cloro-4-(1,4-oxazepan-3-il)benzoato de metilo (188)
A una solución de 3-(tributilestanilmetoxi)propan-1-amina (8,0 g, 21,0 mmol) en diclorometano anhidro (60 ml), se le añadió 3-cloro-4-formil-benzoato de metilo (4,2 g, 21,0 mmol) seguido de tamices moleculares de 4 A (5 g). La reacción se agitó a temperatura ambiente durante 2 horas, se filtró sobre Celite y se diluyó con diclorometano anhidro (180 ml). A un matraz separado que contenía hexafluoroisopropanol (60 ml), se le añadió 2,6-lutidina (2,5 ml, 21,7 mmol) seguido de Cu(OTf)2 (7,6 g, 21,0 mmol). La suspensión de color azul se agitó a temperatura ambiente durante 2 h, y después se añadió la solución de imina preparada anteriormente en una porción. La mezcla de reacción se agitó durante una noche y después se inactivó con 100 ml de una mezcla 2 :1 de una solución acuosa saturada de bicarbonato sódico e hidróxido de amonio al 10 %. La mezcla se agitó durante 15 minutos y después se separó. La capa orgánica se lavó con una solución acuosa saturada de bicarbonato sódico seguido de salmuera. La capa orgánica se concentró al vacío y se purificó por cromatografía sobre gel de sílice eluyendo con EtOAc al 0-70 % en heptano. Las fracciones puras se combinaron y se concentraron al vacío para dar 2,9 g (51 %) del producto deseado: RMN 1H (400 MHz, CDCh) 58,01 (d, J = 1,7 Hz, 1H), 7,91 (ddd, J = 8,1, 1,7, 0,5 Hz, 1H), 7,68 (d, J = 8,1 Hz, 1H), 4,49 (dd, J = 8,9, 3,5 Hz, 1H), 4,04 - 3,96 (m, 2H), 3,92 (s, 3H), 3,84 (dt, J = 12,3, 6,2 Hz, 1H), 3,42 (dd, J = 12,4, 9,0 Hz, 1H), 3,24 (dt, J = 13,6, 5,0 Hz, 1H), 3,07 (dt, J = 13,6, 6,8 Hz, 1H), 2,00 (cd, J = 6,4, 5,0 Hz, 2H); IEN-EM m/z calc. 269,08, encontrado 270,0 (M+1)+; Tiempo de retención: 0,58 minutos.
Formación de 4-(4-(2-amino-6-metilpirimidin-4-il)-1,4-oxazepan-3-il)-3-dorobenzoato de (+/-)-metilo 189)
Una mezcla de 3-cloro-4-(1,4-oxazepan-3-il)benzoato de metilo, 188, (0,37 g, 1,35 mmol) y 4-cloro-6-metilpirimidin-2-amina (0,19 g, 1,35 mmol) en NMP (4 ml) se agitó durante 3 horas a 150 °C. La mezcla de reacción se enfrió a temperatura ambiente, se diluyó con EtOAc y se lavó con agua. La capa orgánica se concentró a sequedad y se purificó por cromatografía sobre gel de sílice eluyendo con MeOH al 0-15 % en diclorometano para dar 252 mg (47 %) del producto deseado: RMN 1H (400 MHz, DMSO-d6 ) 57,94 (d, J = 1,7 Hz, 1H), 7,85 (dd, J = 8,1, 1,7 Hz, 1H), 7,49 (d, J = 8,1 Hz, 1H), 6,90 (s, 2H), 6,09 (d, J = 26,9 Hz, 1H), 5,69 (s, 1H), 4,51 (d, J = 15,6 Hz, 1H), 4,18 (dd, J = 13,7, 4,9 Hz, 1H), 3,97 - 3,79 (m, 6 H), 3,71 - 3,56 (m, 1H), 2,20 (s, 3H), 1,86 (d, J = 5,3 Hz, 2H); IEN-EM m/z calc. 376,13, encontrado 377,0 (M+1)+; Tiempo de retención: 0,72 minutos.
Formación de sal trifluoroacetato del ácido (+/-)-4-[4-(2-ammo-6-metM-pirimidm-4-N)-1,4-oxazepan-3-M]-3-clorobenzoico (190)
A una solución de 4-(4-(2-amino-6-metilpirimidin-4-il)-1,4-oxazepan-3-il)-3-clorobenzoato de metilo, 189, (0,23 g, 0,61 mmol) en MeOH (4 ml) y agua (4,0 ml), se le añadió LiOH (0,10 g, 4,18 mmol). La mezcla de reacción se agitó a temperatura ambiente durante una noche, se acidificó con HCl ac. 1 M y se concentró a sequedad. El residuo en bruto resultante se purificó por cromatografía de fase inversa eluyendo con MeCN al 0-60 % en agua con TFA al 0,1 %. Las fracciones que contenían el producto deseado se combinaron y se concentraron al vacío para dar 134 mg del producto deseado: RMN 1H (400 MHz, DMSO-d6 ) 57,93 (d, J = 1,7 Hz, 1H), 7,85 (dd, J = 8,1, 1,7 Hz, 1H), 7,47 (d, J = 8,1 Hz, 1H), 7,39 (d, J = 23,9 Hz, 2H), 6,46 -5,31 (m, 2H), 5,04 -4,28 (m, 1H), 4,20 (dd, J = 13,7, 5,0 Hz, 1H), 3,96 -3,80 (m, 3H), 3,65 (ddd, J = 12,2, 10,0, 4,3 Hz, 1H), 2,28 -2,19 (m, 4H), 1,91-1,85 (m, 2H); IEN-EM m/z calc. 362,11, encontrado 363,0 (M+1)+; Tiempo de retención: 0,63 minutos.
Formación de (+/-)-4-[4-(2-ammo-6-metM-pirimidm-4-N)-1,4-oxazepan-3-M]-3-doro-W-ddopropM-benzamida (191) I-155
A una solución de ácido 4-[4-(2-amino-6-metil-pirimidin-4-il)-1,4-oxazepan-3-il]-3-cloro-benzoico (sal trifluoroacetato), 190, (0,050 g, 0,100 mmol) en DMF (0,33 ml), se le añadió HATU (0,057 g, 0,150 mmol) seguido de Et3N (0,041 ml, 0,290 mmol). Después de agitar durante 15 minutos, se añadió ciclopropilamina (0,011 ml, 0,150 mmol) y la mezcla se agitó durante una noche. La mezcla de reacción se diluyó con 0,25 ml de agua y se extrajo con 1 ml de EtOAc. La capa orgánica se concentró a sequedad y el producto se purificó por cromatografía sobre gel de sílice eluyendo con MeOH al 0-12 % en diclorometano. Las fracciones puras se combinaron, se concentraron al vacío y se liofilizaron para dar 7 mg del producto deseado: RMN 1H (400 MHz, DMSO-d6 )(se calentó 360K) 58,22 (s, 1H), 7,85 (d, J = 1,7 Hz, 1H), 7,70 (dd, J = 8,1, 1,8 Hz, 1H), 7,37 (d, J = 8,2 Hz, 1H), 6,07 (s, 2H), 5,79 (s, 1H), 5,55 (s, 1H), 4,64 -4,50 (m, 1H), 4,13 (dd, J = 13,6, 4,9 Hz, 1H), 3,97 - 3,85 (m, 1H), 3,85 - 3,67 (m, 2H), 3,67 - 3,53 (m, 1H), 2,83 (tt, J = 7,7, 3,9 Hz, 1H), 2,09 (s, 3H), 1,86 - 1,74 (m, 2H), 0,68 (td, J = 7,1, 4,6 Hz, 2H), 0,60-0,50 (m, 2H); IEN-EM m/z encontrado 402,0 (M+1)+; Tiempo de retención: 0,62 minutos.
Los siguientes análogos se prepararon de acuerdo con el Esquema sintético 25:

(+/-)-4-(4-(2-ammo-6-metMpmmidm-4-N)-1,4-oxazepan-3-M)-3-doro-W-metN-W-(tetrahidro-2H-piran-4-il)benzamida (192) I-153
RMN 1H (400 MHz, DMSO-d6 ) (se calentó 360K) 6 7,48 - 7,36 (m, 2H), 7,29 (ddd, J = 8,3, 6,7, 1,7 Hz, 1H), 6,65 (s, 2H), 6,00 (s, 1H), 5,64 (s, 1H), 4,54 (d, J = 14,9 Hz, 1H), 4,19 (dd, J = 13,6, 5,0 Hz, 1H), 3,94 - 3,75 (m, 5H), 3,70 -3,58 (m, 1H), 3,43 - 3,11 (m, 2H), 2,79 (s, 2H), 2,18 (s, 3H), 1,92 - 1,72 (m, 4H), 1,58 (ddt, J = 13,5, 5,2, 2,7 Hz, 2H); IEN-Em m/z encontrado 460,0 (M+1 )+; Tiempo de retención: 0,62 minutos.

(+/-)-(4-(4-(2-amino-6-metilpirimidin-4-il)-1,4-oxazepan-3-il)-3-clorofenil)(pirrolidin-1-il)metanona (193) I-151
RMN 1H (400 MHz, DMSO-d6 ) (se calentó 360K) 6 7,54 (d, J = 1,6 Hz, 1H), 7,47-7,33 (m, 2H), 5,84 (s, 1H), 5,58 (s, 1H), 4,56 (d, J = 15,0 Hz, 1H), 4,16 (dd, J = 13,5, 5,0 Hz, 1H), 3,90 (dt, J = 12,0, 4,1 Hz, 1H), 3,86-3,71 (m, 2H), 3,60 (dt, J = 12,2, 7,4 Hz, 1H), 3,14-3,10 (m, 2H), 2,11 (s, 3H), 1,85 (dc, J = 11,9, 3,6 Hz, 8H); IEN-EM m/z encontrado 416,0 (M+1)+; Tiempo de retención: 0,75 minutos.

3-(4-(2-amino-6-metilpirimidin-4-il)-1,4-oxazepan-3-il)-4-clorobenzoato de (+/-)-metilo(194) I-126
RMN 1H (400 MHz, DMSO-d6 ) (se calentó 360K) 67,91 - 7,82 (m, 2H), 7,65 - 7,53 (m, 1H), 6,47 (s, 2H), 5,95 (s, 1H), 5,66 (s, 1H), 4,53 (d, J = 15,3 Hz, 1H), 4,14 (dd, J = 13,6, 4,9 Hz, 1H), 3,92 -3,85 (m, 2H), 3,84 (s, 3H), 3,82 -3,75 (m, 1H), 3,65 (dt, J = 12,2, 7,1 Hz, 1H), 2,15 (s, 3H), 1,85 (dc, J = 8,5, 4,2 Hz, 2H); IEN-EM m/z calc. 376,13, encontrado 377,0 (M+1)+; Tiempo de retención: 0,7 minutos.

(+/-)-3-(4-(2-ammo-6-metNpmmidm-4-N)-1,4-oxazepan-3-N)-4-cloro-W-metNbenzamida (195) I-129
RMN 1H (400 MHz, DMSO-d6 ) (se calentó 360K) 68,21 (s, 1H), 7,79 - 7,67 (m, 2H), 7,54 (d, J = 8,2 Hz, 1H), 6,89 (s, 2H), 6,07 (s, 1H), 5,67 (s, 1H), 4,57 (s, 1H), 4,16 (dd, J = 13,7, 5,0 Hz, 1H), 3,89 (cd, J = 9,5, 3,5 Hz, 3H), 3,64 (ddd, J = 12,3, 10,3, 4,2 Hz, 1H), 2,78 (d, J = 4,5 Hz, 3H), 2,21 (s, 3H), 1,85 (dt, J = 17,6, 5,7 Hz, 2H); IEN-EM m/z calc. 375,15, encontrado 376,0 (M+1)+; Tiempo de retención: 0,59 minutos.

(R) -4-[4-(2-ammo-6-metMpinmidm-4-M)-1,4-oxazepan-3-M]-3-cloro-W,W-dimetMbenzamida (196) I-143 y (S) -4-[4-
(2-ammo-6-met¡l-p¡r¡m¡dm-4-¡l)-1,4-oxazepan-3-¡l]-3-cloro-W,W-d¡met¡l-benzam¡da (197) I-144
Pico A: 4-[4-(2-am¡no-6-met¡l-p¡nm¡d¡n-4-¡l)-1,4-oxazepan-3-¡l]-3-cloro-W,W-d¡met¡lbenzam¡da (196); RMN 1H (400 MHz, DMSO-c/6 ) (se calentó 360K) 87,47 (d, J = 1,6 Hz, 1H), 7,39 (d, J = 8,0 Hz, 1H), 7,33 (dd, J = 8,0, 1,7 Hz, 1H), 6,78 (s, 2H), 6,05 (s, 1H), 5,65 (s, 1H), 4,54 (d, J = 14,9 Hz, 1H), 4,19 (dd, J = 13,7, 5,0 Hz, 1H), 3,96 - 3,75 (m, 3H), 3,64 (ddd, J = 12,2, 9,2, 4,9 Hz, 1H), 2,92 (s, 6 H), 2,20 (s, 3H), 1,84 (dp, J = 9,9, 3,7, 3,2 Hz, 2H); IEN-EM m/z calc. 389,16, encontrado 390,0 (M+1)+; T¡empo de retenc¡ón: 0,61 m¡nutos.
P¡co B: 4-[4-(2-am¡no-6-met¡l-p¡nm¡d¡n-4-¡l)-1,4-oxazepan-3-¡l]-3-cloro-W,W-d¡met¡l-benzam¡da (197) RMN 1H (400 MHz, DMSO-d6 ) 87,47 (d, J = 1,6 Hz, 1H), 7,39 (d, J = 8,0 Hz, 1H), 7,33 (dd, J = 8,0, 1,7 Hz, 1H), 6,78 (s, 2H), 6,05 (s, 1H), 5,65 (s, 1H), 4,54 (d, J = 14,9 Hz, 1H), 4,19 (dd, J = 13,7, 5,0 Hz, 1H), 3,96 - 3,75 (m, 3H), 3,64 (ddd, J = 12,2, 9,2, 4,9 Hz, 1H), 2,92 (s, 6 H), 2,20 (s, 3H), 1,84 (dp, J = 9,9, 3,7, 3,2 Hz, 2H); IEN-EM m/z calc. 389,16187, encontrado 390,0 (M+1)+; T¡empo de retenc¡ón: 0,61 m¡nutos.

(S)-4-(4-(2-am¡no-6-met¡lp¡r¡m¡d¡n-4-¡l)-1,4-oxazepan-3-¡l)-3-cloro-W-met¡lbenzam¡da (198) I-150
RMN 1H (400 MHz, DMSO-d6 ) (se calentó 360K) 88,20 (s, 1H), 7,85 (d, J = 1,8 Hz, 1H), 7,70 (dd, J = 8,1, 1,8 Hz, 1H), 7,37 (d, J = 8,1 Hz, 1H), 5,59 (s, 1H), 5,55 - 5,38 (m, 2H), 4,59 (d, J = 15,5 Hz, 1H), 4,12 (dd, J = 13,5, 4,9 Hz, 1H), 3,90 (dt, J = 11,9, 3,9 Hz, 1H), 3,81 - 3,64 (m, 2H), 3,64 - 3,51 (m, 1H), 2,77 (d, J = 4,6 Hz, 3H), 2,00 (s, 3H), 1,78 (dt, J = 7,8, 3,9 Hz, 2H); IEN-EM m/z calc. 375,15, encontrado 376,0 (M+1)+; T¡empo de retenc¡ón: 0,58 m¡nutos.

(+/-)-4-(4-(2-ammo-6-met¡lpmm¡dm-4-¡l)-1,4-oxazepan-3-¡l)-3-cloro-N-(2-(d¡met¡lammo)et¡l)-W-met¡lbenzam¡da I-154
RMN 1H (400 MHz, DMSO-d6 ) (se calentó 360K) 87,53 (d, J = 1,6 Hz, 1H), 7,42 (d, J = 8,0 Hz, 1H), 7,37 (dd, J = 8,0, 1,6 Hz, 1H), 6,72 (s, 2H), 6,03 (s, 1H), 5,67 (s, 1H), 4,53 (s, 1H), 4,18 (dd, J = 13,6, 4,9 Hz, 1H), 3,94 - 3,75 (m, 3H), 3,73 - 3,59 (m, 3H), 3,55 (td, J = 10,2, 9,3, 5,6 Hz, 1H), 3,34 - 3,25 (m, 1H), 2,93 (s, 3H), 2,73 - 2,65 (m, 6 H), 2,19 (s, 3H), 1,92-1,83 (m, 2H); IEN-EM m/z encontrado 447,0 (M+1)+; T¡empo de retenc¡ón: 0,52 m¡nutos.

(+/-)-4-(4-(2-ammo-6-metilpmmidm-4-il)-1,4-oxazepan-3-il)-3-cloro-W-etil-W-metil benzamida I-152
RMN 1H (400 MHz, DMSO-d6 ) (se calentó 360K) 87,46 - 7,37 (m, 2H), 7,34 - 7,25 (m, 1H), 6,79 (s, 2H), 6,05 (s, 1H), 5,66 (s, 1H), 4,53 (d, J = 14,7 Hz, 1H), 4,19 (dd, J = 13,6, 5,0 Hz, 1H), 3,95 ■ 3,77 (m, 3H), 3,64 (ddd, J = 12,2, 9,3, 4,9 Hz, 1H), 3,11 (c, J = 7,3 Hz, 2H), 2,92 - 2,86 (m, 3H), 2,20 (s, 3H), 1,89 - 1,79 (m, 2H), 1,21 (t, J = 7,3 Hz, 3H); IEN-EM m/z encontrado 404,0 (M+1)+; T¡empo de retenc¡ón: 0,64 m¡nutos.

(+/-)-3-(4-(2-ammo-6-metilpmmidm-4-il)-1,4-oxazepan-3-il)-4-cloro-W,W-dimetilbenzamida I-130
RMN 1H (400 MHz, DMSO-d6 ) (se calentó 360K) 87,51 (d, J = 8,1 Hz, 1H), 7,39 - 7,28 (m, 2H), 6,71 (s, 2H), 6,03 (s,
1H), 5,65 (s, 1H), 4,52 (d, J = 15,0 Hz, 1H), 4,17 (dd, J = 13,6, 4,9 Hz, 1H), 3,96 - 3,73 (m, 3H), 3,65 (ddd, J = 12,2, 9,2, 6,1 Hz, 1H), 2,89 (d, J = 3,4 Hz, 6 H), 2,19 (s, 3H), 1,85 (dt, J = 7,6, 4,3 Hz, 2H); IEN-EM m/z calc. 389,16, encontrado 390,0 (M+1)+; Tiempo de retención: 0,61 minutos.

(+/-)-4-[4-(2-ammo-6-metN-pirimidm-4-N)-1,4-oxazepan-3-il]-3-cloro-W-etil-benzamida I-171
RMN 1H (400 MHz, DMSO-d6) (se calentó 360K) 57,51 (d, J = 8,1 Hz, 1H), 7,39 - 7,28 (m, 2H), 6,71 (s, 2H), 6,03 (s, 1H), 5,65 (s, 1H), 4,52 (d, J = 15,0 Hz, 1H), 4,17 (dd, J = 13,6, 4,9 Hz, 1H), 3,96 - 3,73 (m, 3H), 3,65 (ddd, J = 12,2, 9,2, 6,1 Hz, 1H), 2,89 (d, J = 3,4 Hz, 6 H), 2,19 (s, 3H), 1,85 (dt, J = 7,6, 4,3 Hz, 2H); IEN-EM m/z calc. 389,16, encontrado 390,0 (M+1)+; Tiempo de retención: 0,61 minutos.

(+/)-3-(4-(2-ammo-6-metilpmmidm-4-M)-1,4-oxazepan-3-M)-4-cloro-N-ciclopropMbenzamida I-172
RMN 1H (400 MHz, DMSO-da) 58,25 (s, 1H), 7,76 - 7,66 (m, 2H), 7,53 (d, J = 8,2 Hz, 1H), 6,93 (s, 2H), 6,08 (s, 1H), 4,56 (s, 1H), 4,15 (dd, J = 13,7, 5,0 Hz, 1H), 3,89 (ddd, J = 16,0, 9,1, 3,7 Hz, 3H), 3,64 (ddd, J = 12,2, 10,3, 4,2 Hz, 1H), 2,89 (s, 1H), 2,80 (tq, J = 7,7, 4,0 Hz, 1H), 2,21 (s, 3H), 1,95 - 1,76 (m, 2H), 0,69 (td, J = 7,0, 4,6 Hz, 2H), 0,56 (dt, J = 6,9, 4,3 Hz, 2H); IEN-EM m/z encontrado 402 (M+1).

(+/-)-3-[4-(2-ammo-6-metN-pirimidm-4-N)-1,4-oxazepan-3-il]-4-cloro-W-etil-benzamida I-173
RMN 1H (400 MHz, DMSO-da) 58,24 (s, 1H), 7,76 - 7,69 (m, 2H), 7,52 (dd, J = 7,9, 0,7 Hz, 1H), 6,32 (s, 2H), 5,89 (s, 1H), 5,60 (s, 1H), 4,59 (d, J = 15,1 Hz, 1H), 4,13 (dd, J = 13,6, 5,0 Hz, 1H), 3,96 - 3,78 (m, 3H), 3,67 - 3,55 (m, 1H), 3,27 (dtd, J = 8,0, 7,2, 5,9 Hz, 2H), 2,13 (s, 3H), 1,89 - 1,76 (m, 2H), 1,12 (t, J = 7,2 Hz, 3H); IEN-EM m/z encontrado 390.
Ejemplo 26
Esquema sintético 26: (+/-)-3-(4-(2-ammo-6-metilpmmidm-4-M)-l,4-oxazepan-3-M)-4-metoxi-N-metMbenzamida I-313

(a) 3-formil-4-metoxibenzoato de metilo, PdCh(dppf), DMF, NaHCO3, H2O, 80 °C, irradiación por microondas; (b) H2, Pd/C, MeOH-EtOAc; (c) HCl, MeOH, 100 °C; (d) diazometil(trimetil)silano, tolueno, MeOH; (e) 2-amino-4-cloro-6-metilpirimidina, NMP, 150 °C; (f) LiOH, MeOH, H2O; (g) MeNH2, HATU, Et3N, DMF; (h) cromatografía SFC quiral.
Formación de 3-(4-formil-4,5,6,7-tetrahidro-1,4-oxazepin-3-il)-4-metoxibenzoato de metilo (199)
Una mezcla de 3-cloro-6,7-dihidro-5H-1,4-oxazepina-4-carbaldehído, 109, (2,0 g, 12,4 mmol), ácido (2-metoxi-5-metoxicarbonil-fenil)borónico (2,6 g, 12,4 mmol) y PdCh(dppf) (1,0 g, 1,3 mmol) en DMF (37 ml) y bicarbonato sódico acuoso saturado (12 ml) se calentó en un reactor de microondas a 80 °C durante 30 minutos. La mezcla de reacción se diluyó con agua, se lavó con agua, y después la fase orgánica se concentró a sequedad. El residuo resultante se purificó por cromatografía sobre gel de sílice eluyendo con EtOAc al 40-100 % en heptanos seguido de un MeOH al 10% en diclorometano enjuagado. Las fracciones que contenían el producto deseado se combinaron y se concentraron al vacío para proporcionar 2,4 g (63 %) del producto deseado en forma de un aceite incoloro: RMN 1H (400 MHz, CDCl3) 88,04 (dd, J = 8 ,6 , 2,2 Hz, 1H), 7,95 (s, 1H), 7,92 (d, J = 2,2 Hz, 1H), 6,90 (d, J = 8,6 Hz, 1H), 6,19 (s, 1H), 4,24 (dd, J = 6,3, 5,3 Hz, 2H), 4,06 (t, J = 6,6 Hz, 2H), 3,92 (s, 3H), 3,87 (s, 3H), 2,18-2,09 (m, 2H); IEN-EM m/z calc. 291,11, encontrado 290,0 (M+1 )+; Tiempo de retención: 0,9 minutos.
Formación de 3-(4-formil-1,4-oxazepan-3-il)-4-metoxibenzoato de (+/-)-metilo (200)
Una mezcla de 3-(4-formil-4,5,6,7-tetrahidro-1,4-oxazepin-3-il)-4-metoxibenzoato de metilo, 199, (2,4 g, 8,2 mmol) y Pd/C (1,5 g, 0,7 mmol) en acetato de etilo (25 ml) y MeOH (25 ml) se agitó durante una noche a 0,38 MPa (55 psi) de hidrógeno. La mezcla de reacción se filtró sobre Celite y el filtrado secundario se concentró a sequedad. El residuo resultante se purificó por cromatografía sobre gel de sílice eluyendo con EtOAc al 40-100 % en heptanos. Varias fracciones mixtas que contenían el producto deseado se llevaron a la siguiente etapa tal como está: IEN-EM m/z calc.
293,13, encontrado 294,0 (M+1 )+; Tiempo de retención: 0,77 minutos.
Formación de clorhidrato del ácido (+/-)-4-metoxi-3-(1,4-oxazepan-3-il)benzoico (201)
Una solución de 3-(4-formil-1,4-oxazepan-3-il)-4-metoxibenzoato de metilo, 200, (2,4 g, 8,2 mmol) en MeOH (40 ml) y HCl concentrado (40 ml de solución 12,1 M, 484,0 mmol) se agitó durante una noche a 100 °C. La mezcla se concentró a sequedad. El producto se recogió en MeOH y se diluyó en éter dietílico, después se filtró y se secó para dar 2,2 g (84 %) de un sólido de color blanco: RMN 1H (400 MHz, DMSO-c/6 ) 88,12 (d, J = 2,1 Hz, 1H), 7,98 (ddd, J = 8,7, 3,3, 2,1 Hz, 1H), 7,18 (d, J = 8,7 Hz, 1H), 4,77 -4,55 (m, 1H), 4,00 (dd, J = 13,5, 8,9 Hz, 1H), 3,93 (d, J = 1,9 Hz, 3H), 3,91 - 3,85 (m, 2H), 3,49 -3,41 (m, 2H), 3,26 (ddd, J = 13,4, 9,4, 3,4 Hz, 1H), 2,83 (ddt, J = 47,7, 12,7, 7,3 Hz, 0,5H), 2,31 - 2,05 (m, 1H), 2,05-1,75 (m, 0,5H); IEN-EM m/z calc. 251,12, encontrado 252,0 (M+1)+; Tiempo de retención: 0,5 minutos.
Formación de 4-metoxi-3-(1,4-oxazepan-3-il)benzoato de (+/-)-metilo (202)
A una solución de clorhidrato del ácido 4-metoxi-3-(1,4-oxazepan-3-il)benzoico, 201, (0,53 g, 1,65 mmol) en tolueno (22 ml) y MeOH (2,5 ml), se le añadió diazometil(trimetil)silano (0,84 ml de solución 2 M, 1,69 mmol) en hexanos. La mezcla se agitó durante 15 minutos, después se concentró a sequedad para proporcionar 487 mg de un aceite incoloro: IEN-EM m/z calc. 265,13, encontrado 266,0 (M+1)+; Tiempo de retención: 0,57 minutos.
Formación de 3-(4-(2-amino-6-metilpirimidin-4-il)-1,4-oxazepan-3-il)-4-metoxibenzoato de (+/-)-metilo (203)
Una mezcla de 4-metoxi-3-(1,4-oxazepan-3-il)benzoato de metilo, 202, (0,44 g, 1,65 mmol) y 4-cloro-6-metilpirimidin-2-amina (0,26 g, 1,82 mmol) en NMP (5,5 ml) se agitó durante 4 horas a 150 °C en un tubo cerrado herméticamente. La mezcla se diluyó con agua y se extrajo con EtOAc. La capa orgánica se concentró a sequedad y el residuo resultante se purificó por cromatografía sobre gel de sílice eluyendo con MeOH al 0-12 % en diclorometano. Las fracciones puras se combinaron y se concentraron al vacío para dar 89 mg (14 %) del producto deseado: RMN 1H (400 MHz, DMSO-d6 ) (se calentó 360K) 87,89 (dd, J = 8 ,6 , 2,2 Hz, 1H), 7,70 (d, J = 2,2 Hz, 1H), 7,16 (d, J = 8,7 Hz, 1H), 6,60 (s, 2H), 5,89 (s, 1H), 5,57 (s, 1H), 4,60 (d, J = 14,7 Hz, 1H), 4,19 (dd, J = 13,4, 5,2 Hz, 1H), 3,95 (s, 3H), 3,90 (dt, J = 12,0, 3,8 Hz, 1H), 3,79 (s, 3H), 3,77 - 3,68 (m, 1H), 3,63 - 3,54 (m, 1H), 2,14 (s, 3H), 1,80 (dt, J = 7,7, 4,2 Hz, 2H). IEN-EM m/z calc. 372,18, encontrado 373,0 (M+1)+; Tiempo de retención: 0,66 minutos.
Formación de sal trifluoroacetato del ácido (+/-)-3-(4-(2-amino-6-metilpirimidin-4-il)-1,4-oxazepan-3-il)-4-metoxibenzoico (204)
A una solución de 3-(4-(2-amino-6-metilpirimidin-4-il)-1,4-oxazepan-3-il)-4-metoxibenzoato de metilo, 203, (0,090 g, 0,230 mmol) en MeOH (1 ml) y agua (1 ml), se le añadió LiOH (0,025 g, 1,044 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 3 horas, se añadió HCl 1 M y la mezcla se purificó por cromatografía de fase inversa eluyendo con MeCN al 10-90 % en agua con TFA al 0,1 %. Las fracciones puras se combinaron, se concentraron y se liofilizaron para dar 50 mg (58 %) del producto deseado: RMN 1H (400 MHz, DMSO-d6 ) (se calentó 360K) 87,92 (dd, J = 8 ,6 , 2,1 Hz, 1H), 7,72 (s, 1H), 7,41 (s, 2H), 7,17 (d, J = 8,6 Hz, 1H), 4,24 (s, 1H), 3,99 - 3,90 (m, 4H), 3,80 (t, J =
7,1 Hz, 2H), 3,64 (dt, J = 12,2, 7,4 Hz, 1H), 2,33-1,65 (m, 6 H); IEN-EM m/z calc. 358,16, encontrado 359,0 (M+1)+; Tiempo de retención: 0,6 minutos.
Formación de (R)-3-(4-(2-ammo-6-metMp¡r¡m¡dm-4-M)-1,4-oxazepan-3-M)-4-metoxi-W-met¡lbenzam¡da y (S)-3-(4-(2-ammo-6-metMp¡r¡m¡dm-4-M)-1,4-oxazepan-3-M)-4-metoxi-W-met¡lbenzam¡da (205)
A una solución de ácido 3-[4-(2-amino-6-metil-pirimidin-4-il)-1,4-oxazepan-3-il]-4-doro-benzoico (sal trifluoroacetato), 204, (0,085 g, 0,180 mmol) en DMF (1 ml), se le añadió HATU (0,102 g, 0,268 mmol) seguido de EtsN (0,125 ml, 0,897 mmol). Después de agitar durante 10 minutos, se añadió metilamina (0,700 ml de 2 M, 1,40 mmol) en THF y la reacción se agitó durante una noche. La mezcla de reacción se diluyó con agua y se extrajo con EtOAc. La capa orgánica se concentró a sequedad y se purificó por cromatografía sobre gel de sílice eluyendo con MeOH al 0-10% en diclorometano. Las fracciones puras se combinaron, se concentraron al vacío y se sometieron a purificación SFC para proporcionar la mezcla racémica, 205, que después se sometió a separación quiral SFC.
Pico A: (R)-3-(4-(2-amino-6-metilpirimidin-4-il)-1,4-oxazepan-3-il)-4-metoxi-W-metilbenzamida (206); RMN 1H (400 MHz, DMSO-d6 ) (se calentó 360K) 67,90 (s, 1H), 7,68 (dd, J = 8,5, 2,3 Hz, 1H), 7,50 (d, J = 2,2 Hz, 1H), 7,04 (d, J = 8,6 Hz, 1H), 5,71 (d, J = 27,7 Hz, 3H), 5,25 (s, 1H), 4,43 (s, 1H), 3,92 (s, 3H), 3,45 (dd, J = 14,5, 11,2 Hz, 1H), 2,74 (d, J = 4,6 Hz, 3H), 2,34 (dt, J = 14,0, 6,9 Hz, 1H), 2,03 (s, 3H), 1,97 - 1,62 (m, 2H), 1,52 (c, J = 12,5 Hz, 1H), 1,43 1,21 (m, 2H); IEN-EM m/z calc. 369,22, encontrado 370,0 (M+1)+; Tiempo de retención: 0,68 minutos. I-314
Pico B: (S)-3-(4-(2-amino-6-metilpirimidin-4-il)-1,4-oxazepan-3-il)-4-metoxi-W-metilbenzamida (207); RMN 1H (400 MHz, DMSO-d6 ) (se calentó 360K) 67,90 (s, 1H), 7,68 (dd, J = 8,5, 2,3 Hz, 1H), 7,50 (d, J = 2,2 Hz, 1H), 7,04 (d, J = 8,6 Hz, 1H), 5,71 (d, J = 27,7 Hz, 3H), 5,25 (s, 1H), 4,43 (s, 1H), 3,92 (s, 3H), 3,45 (dd, J = 14,5, 11,2 Hz, 1H), 2,74 (d, J = 4,6 Hz, 3H), 2,34 (dt, J = 14,0, 6,9 Hz, 1H), 2,03 (s, 3H), 1,97 - 1,62 (m, 2H), 1,52 (c, J = 12,5 Hz, 1H), 1,43 1,21 (m, 2H); IEN-EM m/z calc. 369,22, encontrado 370,0 (M+1)+; Tiempo de retención: 0,68 minutos. I-315
Los siguientes análogos se prepararon de acuerdo con el Esquema sintético 26:

(+/-)-4-(4-(2-ammo-6-metMpmmidm-4-N)-1,4-oxazepan-3-M)-3-metoxi-W-metilbenzamida (208) I-316
RMN 1H (400 MHz, DMSO-d6 ) (se calentó 360K) 68,12 (s, 1H), 7,49 (d, J = 1,6 Hz, 1H), 7,40 (dd, J = 7,9, 1,6 Hz, 1H), 7,23 (d, J = 7,9 Hz, 1H), 7,02 (s, 2H), 6,05 (s, 1H), 5,61 (s, 1H), 4,62 (s, 1H), 4,24 (dd, J = 13,4, 5,2 Hz, 1H), 3,95 (s, 4H), 3,81 (dt, J = 13,4, 7,9 Hz, 2H), 3,60 (ddd, J = 12,1, 9,4, 5,1 Hz, 1H), 2,82 (d, J = 4,5 Hz, 3H), 2,22 (s, 3H), 1,91 1,74 (m, 2H); IEN-EM m/z calc. 371,20, encontrado 372,0 (M+1)+; Tiempo de retención: 0,55 minutos.
Se prepararon los siguientes azepanos de acuerdo con Esquema sintético 12. Preparación de amida de acuerdo con Esquema sintético 26

3-(1-(2-amino-6-met¡lp¡r¡m¡d¡n-4-¡l)azepan-2-¡l)-4-metox¡benzoato de (+/-)-metilo (209) I-111
RMN 1H (400 MHz, DMSO-d6 ) (se calentó 360K) 67,82 (dd, J = 8 ,6 , 2,2 Hz, 1H), 7,59 (d, J = 2,2 Hz, 1H), 7,11 (d, J = 8,6 Hz, 1H), 5,55 (d, J = 45,8 Hz, 3H), 5,25 (s, 1H), 4,36 (d, J = 44,7 Hz, 1H), 3,96 (s, 3H), 3,77 (s, 3H), 3,39 - 3,28 (m, 1H), 2,39 (dt, J = 14,1, 6,9 Hz, 1H), 2,00 (s, 3H), 1,95 - 1,71 (m, 3H), 1,71 - 1,44 (m, 2H), 1,45-1,16 (m, 2H); IEN-EM m/z calc. 370,20, encontrado 371,0 (M+1)+; Tiempo de retención: 0,78 minutos.

(+/-)-3-(1-(2-ammo-6-met¡lp¡r¡m¡dm-4-¡l)azepan-2-¡l)-4-metox¡-W,W-d¡met¡lbenzam¡da (210) I-115
RMN 1H (400 MHz, DMSO-d6) (se calentó 360K) 67,31 (dd, J = 8,4, 2,1 Hz, 1H), 7,15 - 6,93 (m, 4H), 6,02 (s, 1H), 3,91 (s, 3H), 3,54 (s, 1H), 2,90 (s, 6H), 2,20 (s, 3H), 2,01 -1,68 (m, 4H), 1,63-1,20 (m, 3H); IEN-EM m/z calc. 383,23, encontrado 384,0 (M+1)+; Tiempo de retención: 0,7 minutos.

(+/-)-3-(1-(2-ammo-6-met¡lp¡r¡m¡dm-4-¡l)azepan-2-¡l)-4-metox¡-W-met¡lbenzam¡da (211) I-114
RMN 1H (400 MHz, DMSO-d6) (se calentó 360K) 67,90 (s, 1H), 7,68 (dd, J = 8,5, 2,3 Hz, 1H), 7,50 (d, J = 2,2 Hz, 1H), 7,04 (d, J = 8,6 Hz, 1H), 5,71 (d, J = 27,7 Hz, 3H), 5,25 (s, 1H), 4,43 (s, 1H), 3,92 (s, 3H), 3,45 (dd, J = 14,5, 11,2 Hz, 1H), 2,74 (d, J = 4,6 Hz, 3H), 2,34 (dt, J = 14,0, 6,9 Hz, 1H), 2,03 (s, 3H), 1,97 -1,62 (m, 2H), 1,52 (c, J = 12,5 Hz, 1H), 1,43-1,21 (m, 2H); IEN-EM m/z calc. 369,22, encontrado 370,0 (M+1)+; Tiempo de retención: 0,68 minutos.

(S)-3-(1-(2-ammo-6-met¡lp¡r¡m¡dm-4-¡l)azepan-2-¡l)-4-metox¡-N-met¡lbenzamida (212) I-149; (R)-3-(1-(2- amino-6-met¡lp¡r¡m¡dm-4-¡l)azepan-2-il)-4-metox¡-W-met¡lbenzam¡da (213) I-148
(S)-3-[1-(2-amino-6-metil-pirimidin-4-il)azepan-2-il]-4-metoxi-W-metil-benzamida (212); RMN 1H (400 MHz, DMSO-d6) 6 7,90 (s, 1H), 7,68 (dd, J = 8,5, 2,3 Hz, 1H), 7,50 (d, J = 2,2 Hz, 1H), 7,04 (d, J = 8,6 Hz, 1H), 5,71 (d, J = 27,7 Hz, 3H), 5,25 (s, 1H), 4,43 (s, 1H), 3,92 (s, 3H), 3,45 (dd, J = 14,5, 11,2 Hz, 1H), 2,74 (d, J = 4,6 Hz, 3H), 2,34 (dt, J = 14,0, 6,9 Hz, 1H), 2,03 (s, 3H), 1,97 - 1,62 (m, 2H), 1,52 (c, J = 12,5 Hz, 1H), 1,43-1,21 (m, 2H); IEN-EM m/z calc.
369,21646, encontrado 370,0 (M+1)+; Tiempo de retención: 0,68 minutos. I-149
(R)-3-[1-(2-amino-6-metil-pirimidin-4-il)azepan-2-il]-4-metoxi-W-metil-benzamida (213); RMN 1H (400 MHz, DMSO-d6) (se calentó 360K) 67,90 (s, 1H), 7,68 (dd, J = 8,5, 2,3 Hz, 1H), 7,50 (d, J = 2,2 Hz, 1H), 7,04 (d, J = 8,6 Hz, 1H), 5,71 (d, J = 27,7 Hz, 3H), 5,25 (s, 1H), 4,43 (s, 1H), 3,92 (s, 3H), 3,45 (dd, J = 14,5, 11,2 Hz, 1H), 2,74 (d, J = 4,6 Hz, 3H), 2,34 (dt, J = 14,0, 6,9 Hz, 1H), 2,03 (s, 3H), 1,97 - 1,62 (m, 2H), 1,52 (c, J = 12,5 Hz, 1H), 1,43-1,21 (m, 2H); IEN-EM m/z calc. 369,22, encontrado 370,0 (M+1)+; Tiempo de retención: 0,68 minutos. I-148

(+/-)-4-[3-[2-cloro-4-(1,1-d¡oxo-1,2-t¡azol¡d¡n-2-¡l)fen¡l]-1,4-oxazepan-4-¡l]-6-met¡l-p¡r¡m¡d¡n-2-am¡na (214) I-193
Se calentó (360K) RMN 1H (400 MHz, DMSO-d6) 67,30 (d, J = 8,6 Hz, 1H), 7,26 (d, J = 2,4 Hz, 1H), 7,13 (dd, J = 8,6, 2,4 Hz, 1H), 5,59 (s, 1H), 5,47 (s, 2H), 4,60 (d, J = 15,1 Hz, 1H), 4,09 (dd, J = 13,4, 5,0 Hz, 1H), 3,93 - 3,84 (m, 1H), 3,77 - 3,71 (m, 3H), 3,70 - 3,50 (m, 2H), 3,46 (t, J = 7,4 Hz, 4H), 2,44 - 2,35 (m, 2H), 2,01 (s, 3H), 1,78 (tt, J = 8,3, 4,3 Hz, 2H); IEN-EM m/z calc. 437,1, encontrado 438,0 (M+1)+; Tiempo de retención: 0,67 minutos.
Ejemplo 27
Esquema sintético 27: (+/-)-4-(3-(2-cloro-4-((metilsulfoml)metil)feml)-1,4-oxazepan-4-il)-6-metilpirimidm-2-amina (219) I-208


(a) NaSMe, MeOH, 0 °C a TA; (b) nBuLi, THF, -78 °C, después DMF; (c) mCPBA, CH2CI2; (d) i) tamices mol de 4 A, 3-((tributilestannil)metoxi)propan-1-amina, CH2Ch; ii) 2,6-lutidina, Cu(OTf)2, hexafluoroisopropanol, CH2Cl2; (e) 2-amino-4-doro-6-metilpirimidina, nBuOH, 135 °C; (f) cromatografía SFC quiral.
Formación de (4-bromo-3-clorobencil)(metil)sulfano (215)
El 1-bromo-4-(bromometil)-2-cloro-benceno (120 g, 422 mmol) se disolvió en MeOH (1 l) en un matraz de fondo redondo de 2 l con un agitador superior, sonda de temperatura y un embudo de adición de 500 ml. La solución se enfrió a 0 °C en un baño de salmuera. La solución de NaSMe (235 g de 15 % p/p, 503 mmol) se añadió gota a gota a una velocidad para mantener la temperatura por debajo de 10 °C. Precipitó un sólido de color blanco. La solución se agitó durante el fin de semana. La reacción se vertió en NaOH 1 N y se extrajo tres veces con diclorometano. Los extractos se combinaron, se secaron (MgSO4), se filtraron y se evaporaron al vacío para proporcionar 1-bromo-2-cloro-4- (metilsulfanilmetil)benceno (105 g, 99 %) en forma de un aceite transparente ligeramente púrpura. La RMN 1H mostró que el aceite era un producto puro y consistente con lotes anteriores: RMN 1H (300 MHz, CDCh) 7,54 (d, J = 8,2 Hz, 1H), 7,40 (s, 1H), 7,07 (dd, J = 8,2, 1,6 Hz, 1H), 3,59 (s, 1H), 1,99 (s, 2H) ppm.
Formación de 2-cloro-4-((metiltio)metil)benzaldehído (216)
El 1-bromo-2-cloro-4-(metilsulfanilmetil)benceno, 215, (2,9 g, 11,4 mmol) se disolvió en THF (75 ml) en un matraz de fondo redondo de 250 ml secado a la llama equipado con una barra de agitación magnética. La solución amarilla se enfrió a -78 °C en un baño de hielo seco/acetona. El nBuLi (6,6 ml de solución 1,9 M, 12,5 mmol) se añadió gota a gota mediante una jeringa a una velocidad para mantener la temperatura por debajo de -60 °C. La reacción inicialmente se volvió de color naranja rojizo durante la adición de nBuLi pero cambió a un color pardo después de agitar durante 15 minutos. La DMF (1,2 ml, 15,5 mmol) se añadió mediante una jeringa a una velocidad para mantener la temperatura por debajo de -60 °C. La reacción se agitó durante 30 minutos a -78 °C y después se retiró del baño de enfriamiento y se dejó calentar a temperatura ambiente. La mezcla de reacción se vertió en HCl 1 N y se extrajo con MTBE. El extracto se secó (MgSO4), se filtró y se evaporó al vacío para proporcionar 2,2 g del producto en bruto en forma de un aceite de color amarillo. El producto se purificó por cromatografía sobre gel de sílice usando un cartucho de gel de sílice de ISCO de 120 g usando un gradiente isocrático de diclorometano al 50 %/heptano. Las fracciones que contenían el pico más grande se combinaron y evaporaron al vacío para producir 2-cloro-4-(metilsulfanilmetil)benzaldehído (1,95 g, 85%) en forma de un aceite de color ligeramente amarillo. RMN 1H fue consistente con el producto: RMN 1H (300 MHz, CDCla) 10,45 (s, 1H), 7,89 (d, J = 8,0 Hz, 1H), 7,43 (d, J = 1,3 Hz, 1H), 7,39 - 7,30 (m, 1H), 3,68 (s, 2H), 2,02 (s, 3H) ppm.
Formación de 2-cloro-4-((metilsulfonil)metil)benzaldehído (217)
El 2-cloro-4-(metilsulfanilmetil)benzaldehído 2-cloro-4-(metilsulfanilmetil) benzaldehído, 216, (8,75 g, 43,60 mmol) se disolvió en diclorometano (400 ml) en un matraz de fondo redondo de 1 l equipado con una barra de agitación magnética. Se añadió el mCPBA (22,2 g, 89,97 mmol) disolviéndolo completamente. Después de 15 minutos, precipitó un sólido de color blanco. Después de agitar durante 1 hora, la mezcla de reacción se vertió en una solución acuosa saturada de NaHCO3 y se extrajo con diclorometano. La fase orgánica se secó (MgSO4) y se filtró sobre un lecho de gel de sílice. El tapón se eluyó con EtOAc al 25 %/diclorometano y el filtrado se evaporó al vacío para proporcionar 2-cloro-4-(metilsulfonilmetil)benzaldehído (9,1 g, 90 %) en forma de un sólido de color blanco. RMN 1H fue consistente con el producto: RMN 1H (300 MHz, CDCla) 10,48 (s, 1H), 7,97 (d, J = 8,0 Hz, 1H), 7,56 (s, 1H), 7,50 - 7,40 (m, 2H), 4,28 (s, 2H), 2,86 (s, 4H) ppm.
Formación de 3-(2-cloro-4-((metilsulfonil)metil)fenil)-1,4-oxazepano (218)
Una mezcla de 3-(tributilestanilmetoxi)propan-1-amina (3,4 g, 8,9 mmol), 2-cloro-4-(metilsulfonilmetil)benzaldehído, 217, (2,0 g, 8,6 mmol) y tamices moleculares de 4 angstrom en diclorometano (40 ml) se agitó durante 20 horas. La mezcla se filtró. En un matraz separado que contenía hexafluoroisopropanol (10,0 ml) se añadió 2,6-lutidina (1,1 ml, 9,5 mmol) seguido de Cu(OTf)2 (3,3 g, 9,1 mmol) y diclorometano (10 ml). La mezcla se agitó durante 3 horas a temperatura ambiente. La solución de imina filtrada se añadió en una porción al segundo matraz de una sola vez. La mezcla de reacción resultante se agitó durante la noche, se filtró y luego se trató con 100 ml de una mezcla 2:1 de solución acuosa saturada de NaHCO3 e hidróxido de amonio al 10 %. La fase orgánica se separó y se lavó con una solución acuosa saturada de NaHCO3, se secó con sulfato sódico, se filtró y se concentró al vacío. El residuo resultante se purificó por cromatografía en gel de sílice usando una columna ISCO de 80 gramos eluyendo con 0-15% de
metanol/diclorometano para proporcionar 1,1 gramos del producto deseado en forma de un aceite de color amarillo. Se requirió una segunda purificación por cromatografía en gel de sílice usando una columna ISCO de 40 gramos eluyendo con metanol al 0-8 %/diclorometano para proporcionar 690 mg del producto deseado puro en forma de un aceite de color amarillo: RMN 1H (400 MHz, CDCla) 87,64 (d, J = 8,0 Hz, 1H), 7,43 (d, J = 1,8 Hz, 1H), 7,33 (dd, J = 8,0, 1,9 Hz, 1H), 4,46 (dd, J = 9,0, 3,5 Hz, 1H), 4,20 (s, 2H), 4,07 - 3,92 (m, 2H), 3,85 (dt, J = 12,3, 6,2 Hz, 1H), 3,49 (s, 2H), 3,44 (dd, J = 12,4, 9,1 Hz, 3H), 3,24 (dt, J = 13,6, 5,0 Hz, 1H), 3,06 (dt, J = 13,7, 6,8 Hz, 1H), 2,81 (d, J = 0,8 Hz, 3H), 2,00 (cd, J = 6,4, 4,9 Hz, 2H), 1,81 (s, 2H); IEN-EM m/z calc. 303,8, encontrado 304,0 (M+1)+; Tiempo de retención: 0,48 minutos.
Formación de 4-(3-(2-cloro-4-((metilsulfoml)metil)feml)-1,4-oxazepan-4-il)-6-metilpirimidm-2-amma (219)
Una solución de 3-[2-cloro-4-(metilsulfonilmetil)fenil]-1,4-oxazepano, 218, (1,02 g, 3,37 mmol) y 4-cloro-6-metilpirimidin-2-amina (0,58 g, 4,05 mmol) en n-BuOH (15 ml) se agitó durante una noche en un tubo cerrado herméticamente a 135 °C, después se concentró a sequedad. El residuo se disolvió en EtOAc y se lavó con una solución acuosa saturada de bicarbonato sódico. La capa orgánica se concentró a sequedad y se purificó por cromatografía sobre gel de sílice eluyendo con MeOH al 0-10 % en diclorometano. Las fracciones puras se combinaron y se concentraron para dar 830 mg del producto deseado: IEN-EM m/z calc. 410,1, encontrado 411,0 (M+1)+; Tiempo de retención: 0,63 minutos.
La mezcla racémica se sometió a purificación SFC: columna AD-H 20x250 mm usando MeOH al 40 % (amoniaco 5 mM) CO2 al 60 % método isocrático. Las fracciones puras se concentraron a sequedad y se pasaron a través de un lecho de sílice eluyendo con MeOH al 0-10 % en diclorometano. Las fracciones se concentraron y se liofilizaron.
Pico A: (R)-4-[3-[2-cloro-4-(metilsulfonilmetil)fenil]-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (220) RMN 1H (400 MHz, DMSO-d6 ) 87,50 (d, J = 1,2 Hz, 1H), 7,33 (d, J = 1,0 Hz, 2H), 5,59 (s, 1H), 5,46 (s, 3H), 4,59 (d, J = 14,9 Hz, 1H), 4,43 (s, 2H), 4,13 (dd, J = 13,5, 5,0 Hz, 1H), 3,94 - 3,85 (m, 1H), 3,74 (dd, J = 13,6, 10,3 Hz, 1H), 3,70 -3,62 (m, 1H), 3,55 (ddd, J = 12,0, 9,8, 4,6 Hz, 1H), 2,88 (s, 3H), 2,00 (s, 3H), 1,82-1,72 (m, 2H); IEN-EM m/z calc. 410,1, encontrado 411,0 (M+1)+; Tiempo de retención: 0,62 minutos. I-223
Pico B: (S)-4-[3-[2-cloro-4-(metilsulfonilmetil)fenil]-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (221); RMN 1H (400 MHz, DMSO-d6 ) 87,50 (d, J = 1,2 Hz, 1H), 7,33 (d, J = 1,1 Hz, 2H), 5,60 (s, 1H), 5,46 (s, 3H), 4,59 (d, J = 14,8 Hz, 1H), 4,43 (s, 2H), 4,14 (dd, J = 13,5, 5,0 Hz, 1H), 3,95 - 3,85 (m, 1H), 3,74 (dd, J = 13,5, 10,2 Hz, 1H), 3,70 -3,62 (m, 1H), 3,55 (ddd, J = 12,0, 9,9, 4,6 Hz, 1H), 2,88 (s, 3H), 2,01 (s, 3H), 1,84-1,73 (m, 2H); IEN-EM m/z calc. 410,1, encontrado 411,0 (M+1)+; Tiempo de retención: 0,64 minutos; Rotación óptica: 47,7° (3,1 mg en 1 ml de MeOH). I-224
Ejemplo 28
Esquema sintético 28: (S)-1-(4-(4-(2-amino-6-metilpirimidin-4-il)-1,4-oxazepan-3-il)-3-clorofenil)-4-metilpiperidin-4-ol (222) I-242

(a) 4-metilpiperidin-4-ol, NaOtBu, 2,6-lutidina, tBuXPhos palladacicle G3
Formación de 1-[4-[(3S)-4-(2-amino-6-metil-pirimidin-4-il)-1,4-oxazepan-3-il]-3-clorofenil]-4-metil-piperidin-4-ol (222)
Una solución de (S)-4-[(3S)-3-(4-bromo-2-cloro-fenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina, 33, (3,1 g, 7,8 mmol), 4-metilpiperidin-4-ol (3,5 g, 30,4 mmol) y NaOtBu (3,0 g, 31,2 mmol) en 2,6-lutidina (40 ml) se desgasificó con nitrógeno durante 5 minutos. Se añadió tBuXPhos palladacicle G3 (0,9 g, 1,2 mmol) y la mezcla de reacción se agitó a temperatura ambiente durante 2 horas. La mezcla se diluyó con agua y se extrajo 3 veces con EtOAc. Las fases orgánicas combinadas se secaron sobre sulfato de magnesio, se filtraron y se concentraron al vacío. El residuo resultante se purificó por cromatografía sobre gel de sílice eluyendo con MeOH al 0-15% en diclorometano. Las fracciones que contenían el producto deseado se combinaron y se concentraron para dar un aceite de color pardo claro. Una segunda purificación usando cromatografía de fase inversa sobre gel de sílice en columna C18-acuosa de 275 g eluyendo con MeCN al 5-60 % en agua con TFA al 0,1 %. Las fracciones puras se combinaron, se neutralizaron con una solución acuosa saturada de bicarbonato sódico y se extrajeron dos veces con EtOAc. Las fases orgánicas combinadas se secaron sobre sulfato de magnesio, se filtraron, se concentraron al vacío y se liofilizaron para proporcionar 850 mg del producto: se calentó (360K) RMN 1H (400 MHz, DMSO-d6 ) 87,07 (d, J = 8,6 Hz, 1H), 6,86
(d, J = 2,6 Hz, 1H), 6,79 (dd, J = 8,8, 2,6 Hz, 1H), 5,70 (s, 2H), 5,60 (s, 1H), 5,28 (s, 1H), 4,63 (d, J = 14,6 Hz, 1H), 4,11 -3,95 (m, 2H), 3,90 -3,81 (m, 1H), 3,67 (dd, J = 13,4, 10,0 Hz, 1H), 3,63 -3,47 (m, 2H), 3,31 -3,08 (m, 4H), 2,01 (s, 3H), 1,75 (dp, J = 12,3, 4,5 Hz, 2H), 1,57 - 1,45 (m, 4H), 1,13 (s, 3H); IEN-EM m/z calc. 431,2, encontrado 432,0 (M+1)+; Tiempo de retención: 0,58 minutos.
Los siguientes análogos se prepararon de acuerdo con el Esquema sintético 28:

(S)-1-(4-(4-(2-amino-6-metilpirimidin-4-il)-1,4-oxazepan-3-il)-3-clorofenil)-3-metilazetidin-3-ol (223) I-261
Se calentó (360K) RMN 1H (400 MHz, DMSO-d6) 67,07 (d, J = 8,5 Hz, 1H), 6,41 (d, J = 2,3 Hz, 1H), 6,32 (dd, J = 8,5, 2,4 Hz, 1H), 5,68 (s, 2H), 5,60 (s, 1H), 5,24 (d, J = 22,4 Hz, 2H), 4,66 (d, J = 14,8 Hz, 1H), 4,09 - 3,99 (m, 1H), 3,87 (dt, J = 11,6, 3,6 Hz, 1H), 3,72 (d, J = 7,3 Hz, 2H), 3,63 (t, J = 7,5 Hz, 2H), 3,55 (ddd, J = 12,4, 9,6, 5,0 Hz, 2H), 2,02 (s, 3H), 1,76 (tt, J = 8,3, 3,6 Hz, 2H), 1,43 (s, 3H); IEN-EM m/z calc. 403,2, encontrado 404,0 (M+1)+; Tiempo de retención: 0,7 minutos.

4-[(3S)-3-[2-cloro-4-(2-oxa-6-azaespiro[3,3]heptan-6-il)fenil]-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (224) I-259
Se calentó (360K) RMN 1H (400 MHz, DMSO-d6) 67,08 (d, J = 8,4 Hz, 1H), 6,43 (d, J = 2,4 Hz, 1H), 6,33 (dd, J = 8,5, 2,4 Hz, 1H), 5,59 (d, J = 11,6 Hz, 4H), 5,26 (s, 1H), 4,68 (s, 4H), 4,02 (dd, J = 13,5, 5,0 Hz, 1H), 3,91 - 3,83 (m, 1H), 3,67 (dd, J = 13,4, 10,1 Hz, 1H), 3,63 - 3,50 (m, 2H), 2,01 (d, J = 2,4 Hz, 3H), 1,74 (dd, J = 8,4, 4,3 Hz, 3H), 1,25 (s, 3H); IEN-EM m/z calc. 415,2, encontrado 416,0 (M+1)+; Tiempo de retención: 0,7 minutos.

4-[(3S)-3-[2-cloro-4-[(3-metiloxetan-3-il)amino[fenil|-1,4-oxazepan-4-il|-6-metil-pirimidin-2-amina (225) I-266
RMN 1H (400 MHz, MeOD) 67,00 (d, J = 8,5 Hz, 1H), 6,47 (d, J = 2,4 Hz, 1H), 6,32 (dd, J = 8,5, 2,4 Hz, 1H), 5,56 (s a, 1H), 5,27-4,97 (s a, 2H), 4,73 (dd, J = 5,9, 2,3 Hz, 2H), 4,53 (dd, J = 5,9, 1,8 Hz, 2H), 4,19 (dd, J = 13,6, 5,1 Hz, 1H), 4,00 (d, J = 8,3 Hz, 1H), 3,68 (dd, J = 13,6, 10,5 Hz, 1H), 3,60 (dd, J = 16,6, 7,8 Hz, 2H), 2,08 (s, 3H), 1,98-1,82 (m, 1H), 1,82-1,73 (m, 1H), 1,61 (s, 3H). IEN-EM m/z calc. 403,1775, encontrado 404,25 (M+1)+; Tiempo de retención: 0,6 minutos.

4-[(3S)-3-[2-cloro-4-[(3-metiloxetan-3-il)metilamino]fenil]-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (226) I-240
RMN 1H (400 MHz, MeOD) 66,99 (d, J = 8,5 Hz, 1H), 6,70 (d, J = 2,4 Hz, 1H), 6,57 (dd, J = 8,6, 2,4 Hz, 1H), 5,75 4,80 (m, 3H), 4,53 (d, J = 5,9 Hz, 2H), 4,38 (d, J = 5,9 Hz, 2H), 4,19 (dd, J = 13,6, 5,1 Hz, 1H), 4,00 (d, J = 7,2 Hz, 1H), 3,69 (dd, J = 13,6, 10,5 Hz, 1H), 3,60 (dd, J = 13,7, 10,7 Hz, 2H), 3,26 (s, 2H), 2,07 (s, 3H), 1,96 - 1,71 (m, 2H), 1,36 (s, 3H); IEN-EM m/z calc. 417,2, encontrado 418,3 (M+1)+; Tiempo de retención: 0,63 minutos.

1-[4-[(3S)-4-(2-ammo-6-metM-pirimidm-4-M)-1,4-oxazepan-3-M]-3-cloro-amlmo]-2-metN-propan-2-ol (227) I-247
RMN 1H (400 MHz, MeOD) 86,97 (d, J = 8,5 Hz, 1H), 6,70 (d, J = 2,4 Hz, 1H), 6,57 (dd, J = 8,6, 2,4 Hz, 1H), 5,57 (s a, 1H), 5,14 (s a, 2H), 4,19 (dd, J = 13,6, 5,1 Hz, 1H), 4,00 (d, J = 8,2 Hz, 1H), 3,75 - 3,52 (m, 3H), 3,03 (s, 2H), 2,07 (s, 3H), 2,00-1,70 (m, 2H), 1,23 (s, 6H); IEN-EM m/z calc. 405,2, encontrado 406,2 (M+1)+; Tiempo de retención: 0,6 minutos.

1-[[4-[(3S)-4-(2-amino-6-metil-pirimidin-4-il)-1,4-oxazepan-3-il]-3-cloro-anilino]metil]-ciclobutanol (228) I-263
RMN 1H (400 MHz, MeOD) 86,98 (d, J = 8,5 Hz, 1H), 6,72 (d, J = 2,4 Hz, 1H), 6,58 (dd, J = 8,6, 2,4 Hz, 1H), 5,56 (s a, 1H), 5,07 (s a, 2H), 4,19 (dd, J = 13,6, 5,1 Hz, 1H), 4,00 (d, J = 8,7 Hz, 1H), 3,73-3,55 (m, 1H), 3,59 (t, J = 12,3 Hz, 2H), 3,17 (s, 2H), 2,16 - 1,98 (m, 6H), 1,96 - 1,70 (m, 3H), 1,64-1,56 (m, 1H); IEN-EM m/z calc. 417,2, encontrado 418,1 (M+1)+; Tiempo de retención: 2,72 minutos.

4-[(3S)-3-[2-cloro-4-(ciclopropilamino)fenil]-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (229) I-248
RMN 1H (400 MHz, MeOD) 86,98 (d, J = 8,6 Hz, 1H), 6,80 (d, J = 2,3 Hz, 1H), 6,64 (dd, J = 8,5, 2,3 Hz, 1H), 5,57 (s a, 1H), 5,35-4,80 (s a, 2H), 4,19 (dd, J = 13,6, 5,1 Hz, 1H), 4,01 (d, J = 8,0 Hz, 1H), 3,65 (ddd, J = 24,2, 17,3, 11,3 Hz, 4H), 2,36 - 2,28 (m, 1H), 2,07 (s, 3H), 1,95 - 1,72 (m, 3H), 0,73 - 0,65 (m, 2H), 0,42 (td, J = 6,6, 4,4 Hz, 2H); IEN-EM m/z calc. 373,2, encontrado 374,2 (M+1)+; Tiempo de retención: 0,65 minutos.

4-[(3S)-3-[2-cloro-4-[(1-metilazetidin-3-il)amino]fenil]-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (230) I-246
RMN 1H (400 MHz, MeOD) 87,00 (d, J = 8,5 Hz, 1H), 6,57 (d, J = 2,4 Hz, 1H), 6,45 (dd, J = 8,5, 2,4 Hz, 1H), 5,53 (s a, 1H), 5,09 (s a, 2H), 4,18 (dd, J = 13,6, 5,1 Hz, 1H), 4,05 - 3,95 (m, 2H), 3,82 - 3,74 (m, 2H), 3,68 (dd, J = 13,6, 10,5 Hz, 1H), 3,59 (t, J = 12,1 Hz, 2H), 3,02 - 2,94 (m, 2H), 2,39 (s, 3H), 2,06 (s, 3H), 1,96-1,73 (m, 2H); IEN-EM m/z calc. 402,2, encontrado 403,3 (M+1)+; Tiempo de retención: 0,53 minutos.

(+/-)-[4-[4-[4-(2-amino-6-metil-pirimidin-4-il)-1,4-oxazepan-3-il]-3-clorofenil]piperazin-1-il]-ciclopropilmetanona (231) I-273
RMN 1H (400 MHz, CDCI3) 57,13 (d, J = 8,6 Hz, 1H), 6,92 (d, J = 2,5 Hz, 1H), 6,76 (dd, J = 8,7, 2,6 Hz, 1H), 5,58 (s, 1H), 4,60 (s, 2H), 4,31 (dd, J = 13,5, 5,0 Hz, 1H), 4,08 (d, J = 12,2 Hz, 1H), 4,01 - 3,05 (m, 11H), 2,15 (s, 3H), 1,92 -1,62 (m, 3H), 1,14 - 0,98 (m, 2H), 0,91-0,65 (m, 2H); IEN-EM m/z calc. 470,2, encontrado 471,2 (M+1)+; Tiempo de retención: 2,72 minutos.

4-[(3S)-3-(2-cloro-4-morfolin-fenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (232) I-216
Se calentó (360K) RMN 1H (400 MHz, DMSO-d6 ) 57,14 (d, J = 8,7 Hz, 1H), 6,92 (d, J = 2,6 Hz, 1H), 6,83 (dd, J = 8,7, 2,6 Hz, 1H), 5,60 (s, 3H), 5,31 (s, 1H), 4,65 (d, J = 14,9 Hz, 1H), 4,06 (dd, J = 13,4, 5,1 Hz, 1H), 3,93 - 3,83 (m, 1H), 3,74 - 3,67 (m, 5H), 3,65 - 3,50 (m, 2H), 3,14 - 3,08 (m, 4H), 2,01 (d, J = 1,8 Hz, 3H), 1,76 (dt, J = 8,3, 4,1 Hz, 2H); IEN-EM m/z calc. 403,2, encontrado 404,0 (M+1)+; Tiempo de retención: 0,7 minutos.

4-[(3S)-3-[2-doro-4-(2-oxa-7-azaespiro[4,4]nonan-7-N)feml]-1,4-oxazepan-4-M] -6-metil-pirimidin-2-amina (233) I-219
Se calentó (360K) RMN 1H (400 MHz, DMSO-d6) 57,07 (d, J = 8,6 Hz, 1H), 6,52 (d, J = 2,6 Hz, 1H), 6,45 (dd, J = 8,7, 2,5 Hz, 1H), 5,56 (s, 1H), 5,49 (s, 2H), 5,24 (s, 1H), 4,69 (d, J = 14,7 Hz, 1H), 4,03 (dd, J = 13,4, 5,0 Hz, 1H), 3,87 (dt, J = 12,0, 3,9 Hz, 1H), 3,80 (t, J = 7,0 Hz, 2H), 3,67 (dd, J = 13,4, 10,1 Hz, 1H), 3,59 -3,49 (m, 3H), 3,29 (dddd, J = 9,6, 7,3, 4,9, 2,4 Hz, 2H), 3,20 (c, J = 3,0, 2,3 Hz, 2H), 1,99 (s, 3H), 1,98 - 1,80 (m, 4H), 1,76 (tt, J = 8,3, 4,4 Hz, 2H); IEN-EM m/z calc. 443,2, encontrado 444,0 (M+1)+; Tiempo de retención: 0,74 minutos.

4-[(3S)-3-[2-cloro-4-(3-metoxiazetidin-1-il)fenil]-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (234) I-217
Se calentó (360K) RMN 1H (400 MHz, DMSO-d6 ) 57,08 (d, J = 8,5 Hz, 1H), 6,43 (d, J = 2,4 Hz, 1H), 6,33 (dd, J = 8,5, 2,4 Hz, 1H), 5,55 (d, J = 7,9 Hz, 1H), 5,47 (s, 2H), 5,29 (d, J = 33,3 Hz, 1H), 4,67 (d, J = 14,8 Hz, 1H), 4,29 (tt, J = 6,2, 4,2 Hz, 1H), 4,06 - 3,99 (m, 3H), 3,87 (dt, J = 12,0, 4,0 Hz, 1H), 3,71 - 3,48 (m, 5H), 3,24 (s, 3H), 1,99 (d, J = 2,8 Hz, 3H), 1,81-1,67 (m, 2H); IEN-EM m/z calc. 403,2, encontrado 404,0 (M+1)+; Tiempo de retención: 0,74 minutos.
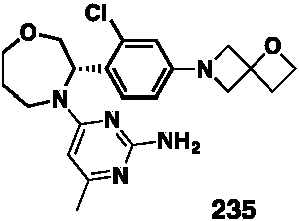
4-[(3S)-3-[2-cloro-4-(1-oxa-6-azaespiro [3,3] heptan-6-il)fenil]-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (235) I-260
Se calentó (360K) RMN 1H (400 MHz, DMSO-d6 ) 57,08 (d, J = 8,4 Hz, 1H), 6,44 (t, J = 2,1 Hz, 1H), 6,34 (dt, J = 8,5, 2,0 Hz, 1H), 5,70 (s, 2H), 5,60 (s, 1H), 5,28 (s, 1H), 4,65 (d, J = 14,6 Hz, 1H), 4,43 (td, J = 7,5, 1,5 Hz, 2H), 4,12 -4,00 (m, 3H), 3,93 - 3,83 (m, 3H), 3,74 - 3,65 (m, 1H), 3,65 - 3,50 (m, 2H), 2,83 (td, J = 7,5, 1,6 Hz, 2H), 2,02 (d, J = 1,4 Hz, 3H), 1,76 (h, J = 5,1, 4,4 Hz, 2H); IEN-EM m/z calc. 415,2, encontrado 416,0 (M+1)+; Tiempo de retención: 0,74 minutos.

4-[(3S)-3-[2-cloro-4-(3-metoxipirrolidin-1-il)fenil]-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (236) I-218 (mezcla de 2 diastereómeros)
Se calentó (360K) RMN 1H (400 MHz, DMSO-d6) 7,07 (d, J = 8,6 Hz, 1H), 6,53 (d, J = 2,5 Hz, 1H), 6,46 (dd, J = 8,6, 2,5 Hz, 1H), 5,55 (s, 1H), 5,48 (s, 2H), 5,27 (d, J = 35,5 Hz, 1H), 4,70 (d, J = 15,4 Hz, 1H), 4,10-3,99 (m, 2H), 3,87 (d, J = 12,2 Hz, 1H), 3,67 (dd, J = 13,4, 10,1 Hz, 1H), 3,55 (cd, J = 12,0, 10,9, 3,8 Hz, 2H), 3,39 (ddd, J = 10,7, 5,3, 2,5 Hz, 1H), 3,28-3,16 (m, 6H), 2,09-2,02 (m, 1H), 2,00 (d, J = 3,9 Hz, 3H), 1,81-1,69 (m, 2H); IEN-EM m/z calc. 417,2, encontrado 418,0 (M+1)+; Tiempo de retención: 0,78 minutos.

3-[4-[(3S)-4-(2-ammo-6-metil-pirimidm-4-il)-1,4-oxazepan-3-il]-3-cloro-amlmo]azetidm-l-carboxilato de terc-butilo (237) I-256
RMN 1H (400 MHz, MeOD) 67,02 (d, J = 8,5 Hz, 1H), 6,58 (d, J = 2,4 Hz, 1H), 6,46 (dd, J = 8,5, 2,4 Hz, 1H), 5,54 (s a, 1H), 5,28 -4,90 (m, 3H), 4,30 -4,14 (m, 4H), 4,02-3,96 (m, 1H), 3,72-3,58 (m, 5H), 2,07 (s, 3H), 1,94-1,73 (m, 2H), 1,43 (s, 9H); IEN-EM m/z calc. 488,2, encontrado 489,3 (M+1)+; Tiempo de retención: 0,69 minutos.
Ejemplo 29
Esquema sintético 29: (+/-)-4-[3-[2-cloro-4-[metil(oxetan-3-il)ammo]feml]-1,4-oxazepan-4-il]-6-metil-pirimidm-2-amina I-186

(a) oxetan-3-ona, Na(OAc)3BH, AcOH, CH2CI2; (b) NaH, yoduro de metilo, DMF; (c) TFA, CH2CI2; (d) 2-amino-4-cloro-6- metilpirimidina, nBuOH, 130 °C; (e) separación SFC quiral
Formación de 3-(2-cloro-4-(oxetan-3-ilamino)fenil)-1,4-oxazepano-4-carboxilato de terc-butilo (239)
A una solución de 3-(4-amino-2-cloro-fenil)-1,4-oxazepano-4-carboxilato de ferc-butilo, 238, (0,67 g, 2,04 mmol) en diclorometano (20 ml), se le añadió oxetan-3-ona (0,22 g, 3,05 mmol), después AcOH (0,15 ml, 2,65 mmol). Después de 3 minutos, se añadió triacetoxiborohidruro sódico (0,65 g, 3,05 mmol) a la mezcla de reacción. Después de 4 horas, se añadió una solución acuosa saturada de bicarbonato sódico y la fase orgánica se pasó a través de un separador de fase y el filtrado resultante se concentró al vacío. El residuo resultante se purificó por cromatografía sobre gel de sílice (columna ISCO de 12 g; EtOAc al 20-100 % en heptano) seguido de una segunda purificación por cromatografía en columna (columna C18 AQ de 40 g; TFA al 0,1 %-agua/TFA al 0,1 %-MeCN). Las fracciones que contenían el producto deseado se concentraron al vacío y después se diluyeron con MeOH y se retiraron por filtración de 300 mg (39 %) del producto deseado en forma de un sólido de color blanco: RMN 1H (400 MHz, d6-DMSO) 67,00 (s, 1H), 6,60 (s, 1H), 6,45 (d, J = 2,0 Hz, 2H), 5,18-5,14 (m, 1H), 4,82 (t, J = 6,5 Hz, 2H), 4,51 (dd, J = 12,8, 6,4 Hz, 1H), 4,36 (t, J = 6,0 Hz, 2H), 4,25 (d, J = 14,0 Hz, 1H), 3,89 (d, J = 11,7 Hz, 2H), 3,44 (m, 4H), 1,75-1,65 (m, 2H), 1,17 (s, 9H); IEN-EM m/z calc. 382,2, encontrado 383,3 (M+1)+; Tiempo de retención: 0,81 minutos.
Formación de 3-[2-cloro-4-[metil(oxetan-3-il)amino]fenil]-1,4-oxazepano-4-carboxilato de tere-butilo (240)
A una solución de 3-[2-doro-4-(oxetan-3-ilamino)fenil]-1,4-oxazepano-4-carboxilato de ferc-butilo, 239, (0,117 g, 0,306 mmol) en DMF (2 ml) en una atmósfera de nitrógeno, se le añadió NaH (0,021 g de 60 % p/p, 0,525 mmol). Después de 20 minutos, se añadió yoduro de metilo (0,070 ml, 1,124 mmol) a la mezcla de reacción, que después se agitó a temperatura ambiente. Después de 60 minutos, se añadieron NaH adicional (0,060 g) y yoduro de metilo (0,100 ml) a la mezcla. Se añadió agua y después se extrajo dos veces con diclorometano. Las fases orgánicas se pasaron a través de un separador de fase y se concentraron al vacío. La purificación por cromatografía sobre gel de sílice (columna ISCO de 40 g; gradiente de EtOAc al 20-100 %/heptanos) para proporcionar 101 mg del producto deseado en forma de un aceite incoloro: IEN-EM m/z calc. 396,2, encontrado 397,34 (M+1)+; Tiempo de retención: 0,87 minutos.
Formación de W-[3-cloro-4-(1,4-oxazepan-3-N)fenM]-N-metM-oxetan-3-amma (241)
A una solución de 3-[2-cloro-4-[metil(oxetan-3-il)amino]fenil]-1,4-oxazepano-4-carboxilato de ferc-butilo, 240, (0,10 g, 0,25 mmol) en diclorometano (2 ml), se le añadió ácido trifluoroacético (1,0 ml, 12,9 mmol). La mezcla de reacción se agitó durante 20 minutos y se concentró al vacío. El residuo resultante se diluyó con metanol y después se pasó a través de un cartucho de bicarbonato SPE (5 g/60 ml) y se concentró para dar 71 mg de un aceite incoloro que se usó sin purificación adicional: IEN-EM m/z calc. 296,1, encontrado 297,3 (M+1)+; Tiempo de retención: 0,52 minutos.
Formación de (+/-)-4-[3-[2-cloro-4-[metil(oxetan-3-il)amino|fenil]-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (242) I-186
Una mezcla de W-[3-cloro-4-(1,4-oxazepan-3-il)fenil]-W-metil-oxetan-3-amina, 241, (0,07 g, 0,24 mmol) y 4-cloro-6-metil-pirimidin-2-amina (0,03 g, 0,22 mmol) en nBuOH (2 ml) se calentó a 130 °C durante una noche. La mezcla de reacción se enfrió a temperatura ambiente seguido de purificación por cromatografía sobre gel de sílice (columna C18 AQ de 50 g; TFA al 0,1 %-agua/TFA al 0,1 %-MeCN). Las fracciones puras se concentraron al vacío y después se disolvieron en MeOH y se pasaron a través de un cartucho de bicarbonato SPE (Agilent Stratospheres 500 mg/6 ml) y se concentraron al vacío. Se añadió éter y se concentró de nuevo para dar 42 mg del producto en forma de un sólido de color blanco: RMN 1H (400 MHz, d6- DMSO) 87,06 (d, J = 8,4 Hz, 1H), 6,71 (s, 1H), 6,58 (d, J = 8,5 Hz, 1H), 5,85 (s a, 2H), 5,45-5,26 (s a, 1H), 5,05-4,88 (s a, 1H), 4,78-4,70 (m, 3H), 4,57 (d, J = 5,0 Hz, 2H), 4,10-3,95 (m, 1H), 3,90 3,82 (m, 1H), 3,70-3,46 (m, 3H), 2,86 (s, 3H), 1,97 (s, 3H), 1,72 (s, 2H); IEN-EM m/z calc. 403,2, encontrado 404,4 (M+1)+; Tiempo de retención: 0,57 minutos.
La mezcla racémica se sometió a separación SFC quiral: columna AD-H 20x250 mm usando MeOH al 40 % (amoniaco 5 mM) CO2 al 60 % método isocrático.
Pico A: (R)-4-[3-[2-cloro-4-[metil(oxetan-3-il)amino]fenil]-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (243); +99 % ee; RMN 1H (400 MHz, d6-DMSO) 87,06 (d, J = 8,4 Hz, 1H), 6,71 (s, 1H), 6,58 (d, J = 8,5 Hz, 1H), 5,85 (s a, 2H), 5,45 5,26 (s a, 1H), 5,05-4,88 (s a, 1H), 4,78-4,70 (m, 3H), 4,57 (d, J = 5,0 Hz, 2H), 4,10-3,95 (m, 1H), 3,90-3,82 (m, 1H), 3.70- 3,46 (m, 3H), 2,86 (s, 3H), 1,97 (s, 3H), 1,72 (s, 2H); IEN-EM m/z calc. 403,2, encontrado 404,2 (M+1)+; Tiempo de retención: 0,58 minutos. I-189
Pico B: (S)-4-[3-[2-cloro-4-[metil(oxetan-3-il)amino]fenil]-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (244); 99,4 % ee; RMN 1H (400 MHz, d6-DMSO) 87,06 (d, J = 8,4 Hz, 1H), 6,71 (s, 1H), 6,58 (d, J = 8,5 Hz, 1H), 5,85 (s a, 2H), 5,45 5,26 (s a, 1H), 5,05-4,88 (s a, 1H), 4,78-4,70 (m, 3H), 4,57 (d, J = 5,0 Hz, 2H), 4,10-3,95 (m, 1H), 3,90-3,82 (m, 1H), 3.70- 3,46 (m, 3H), 2,86 (s, 3H), 1,97 (s, 3H), 1,72 (s, 2H); IEN-EM m/z calc. 403,2, encontrado 404,3 (M+1)+; Tiempo de retención: 0,58 minutos. I-190
Los siguientes análogos se prepararon de acuerdo con el Esquema sintético 29:

4-[(3S)-3-[2-cloro-4-(oxetan-3-ilamino)fenil]-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (245) I-215
RMN 1H (300 MHz, CDCla) 86,91 (d, J = 8,4 Hz, 1H), 6,38 (d, J = 2,4 Hz, 1H), 6,25 (dd, J = 8,5, 2,5 Hz, 1H), 5,46 (s, 1H), 5,16 (s, 1H), 4,88 (td, J = 6,3, 2,3 Hz, 2H), 4,65 (s, 2H), 4,55 - 4,33 (m, 3H), 4,26 - 4,11 (m, 2H), 4,03 (c, J = 7,1 Hz, 2H), 3,58 - 3,26 (m, 3H), 2,04 (s, 3H), 1,83 (s, 1H), 1,73 (s, 1H); IEN-EM m/z calc. 389,2, encontrado 390,2 (M+1)+; Tiempo de retención: 0,6 minutos; Rotación óptica: 32,4 (MeOH).
Ejemplo 30
Esquema sintético 30: (S)-1-(3-((4-(4-(2-ammo-6-metilpirimidm-4-il)-1,4-oxazepan-3-il)-3-clorofenil)amino)azetidin-1-il)etan-1-ona (247) I-262

(a) TFA, CH2CI2; (b) 2-amino-4-doro-6-metilpirimidina, nBuOH, 130 °C.
Formación de 4-[(3S)-3-[4-(azetidin-3-ilamino)-2-cloro-fenil]-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (246) I-257
A una solución de 3-[4-[(3S)-4-(2-amino-6-metil-pirimidin-4-il)-1,4-oxazepan-3-il]-3-doro-anilino]azetidin-1-carboxilato de ferc-butilo, 237, (0,118 g, 0,239 mmol) en diclorometano (2 ml), se le añadió ácido trifluoroacético (1,0 ml). La mezcla de reacción se agitó a temperatura ambiente durante 30 minutos y después se concentró parcialmente al vacío. Se añadió HCl 1 M (2 ml) y la mezcla se lavó dos veces con diclorometano. La capa acuosa se basificó con NaOH 2 M (5 ml) y después se extrajo con diclorometano. Las capas se separaron con la ayuda de un separador de fase. La capa acuosa se extrajo de nuevo con diclorometano y las capas se separaron a través de un separador de fase de nuevo y las fases orgánicas combinadas se concentraron al vacío para proporcionar 51 mg del producto deseado en forma de un sólido de color blanco: RMN 1H (400 MHz, MeOD) 87,00 (d, J = 8,5 Hz, 1H), 6,56 (d, J = 2,4 Hz, 1H), 6,45 (dd, J = 8,5, 2,4 Hz, 1H), 5,53 (s a, 1H), 5,08 (s a, 2H), 4,35 -4,26 (m, 1H), 4,18 (dd, J = 13,6, 5,1 Hz, 1H), 4,00 (d, J = 6,8 Hz, 1H), 3,88 (t, J = 7,8 Hz, 2H), 3,74 - 3,45 (m, 5H), 2,06 (s, 3H), 1,94-1,68 (m, 2H); IEN-EM m/z calc.
388,2, encontrado 389,3 (M+1)+; Tiempo de retención: 0,53 minutos.
Formación de 1-[3-[4-[(3S)-4-(2-amino-6-metil-pirimidin-4-il)-1,4-oxazepan-3-il]-3-cloro-anilino]azetidin-1-il]etanona (247)
A una suspensión de 4-[(3S)-3-[4-(azetidin-3-ilamino)-2-cloro-fenil]-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina, 246, (0,040 g, 0,088 mmol) en diclorometano (4 ml) y THF (1 ml), se le añadió trietilamina (0,025 ml, 0,176 mmol) seguido de cloruro de acetilo (0,007 ml, 0,097 mmol). La mezcla de reacción se agitó a temperatura ambiente. Después de 1 hora, se añadió cloruro de acetilo adicional (0,002 ml) y la agitación se continuó a temperatura ambiente durante una noche. El precipitado resultante se filtró y se lavó con una cantidad mínima de diclorometano. El sólido de color blanco se secó para proporcionar 25 mg del producto deseado: RMN 1H (400 MHz, MeOD) 87,03 (d, J = 8,5 Hz, 1H), 6,60 (dd, J = 4,0, 2,4 Hz, 1H), 6,51 - 6,44 (m, 1H), 5,55 (s a, 1H), 4,93 (s a, 2H), 4,57 - 4,49 (m, 1H), 4,35 -4,15 (m, 3H), 4,04 - 3,90 (m, 2H), 3,79 - 3,53 (m, 4H), 2,07 (s, 3H), 1,87 (s, 3H), 1,95-1,70 (m, 2H); IEN-EM m/z calc. 430,2, encontrado 431,3 (M+1)+; Tiempo de retención: 0,57 minutos.
Ejemplo 31
Esquema sintético 31: 4-[(3S)-3-[4-(1,3,3a,4,6,6a-hexahidrofuro[3,4-c]pirrol-5-il)-2-cloro-fenil]-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (248) I-220
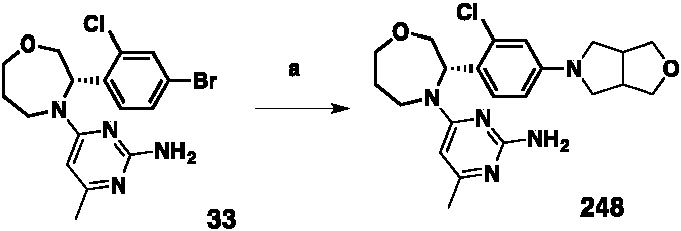
(a) Cul, (3aS,6aR)-3,3a,4,5,6,6a-hexahidro-1H-furo[3,4-c]pirrol, K2CO3, W,W-dimetilglicina
Formación de 4-[(3S)-3-[4-(1,3,3a,4,6,6a-hexahidrofuro[3,4-c]pirrol-5-il)-2-cloro-fenil]-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (248)
Un tubo de Schlenck se cargó con 4-[(3S)-3-(4-bromo-2-cloro-fenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina, 33, (0,30 g, 0,75 mmol), (3aS,6aR)-3,3a,4,5,6,6a-hexahidro-1H-furo[3,4-c]pirrol (0,36 g, 3,19 mmol), W,W-dimetilglicina (0,05 g, 0,4364 mmol), Cul (0,10 g, 0,50 mmol), K2CO3 (0,63 g, 4,53 mmol) y DMSO (4 ml) en ese orden y ciclos de vacío/nitrógeno cinco veces. El matraz se calentó a 90 °C. Después de 24 horas, se añadió MeOH al 10 % en EtOAc y la mezcla se filtró a través de Celite y se concentró (DMSO todavía presente). La purificación por cromatografía sobre gel de sílice (columna GOLD ISCO de 80 g; gradiente de MeOH al 0-10 %/diclorometano) y las fracciones que contenían el producto se concentraron al vacío. El producto se sometió a una segunda purificación por cromatografía sobre gel de sílice (columna ISCO C18 acuosa de 80 g eluyendo con TFA al 0,1 %-agua/TFA al 0,1 %-MeCN): RMN
1H (400 MHz, MeOD) 57,06 (d, J = 8,6 Hz, 1H), 6,67 (d, J = 2,5 Hz, 1H), 6,56 (dd, J = 8,6, 2,5 Hz, 1H), 5,70-4,70 (m a, 3H), 4,20 (dd, J = 13,6, 5,1 Hz, 1H), 4,06 -3,89 (m, 3H), 3,75-7,53 (m, 5H), 3,38 (dd, J = 8,8; 7,1 Hz, 2H), 3,23-3,15 (m, 2H), 3,10-3,00 (m, 2H), 2,06 (s, 3H), 1,95-1,75 (m, 2H); IEN-EM m/z calc. 429,2, encontrado 430,3 (M+1)+; Tiempo de retención: 0,63 minutos.
Los siguientes análogos se prepararon de acuerdo con el Esquema sintético 31:

(+/-)-4-[3-[2-cloro-4-(4-metilpiperazin-1-il)fenil]-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (249) I-225
Se calentó (360K) RMN 1H (400 MHz, DMSO-d6) 57,11 (d, J = 8,7 Hz, 1H), 6,90 (d, J = 2,5 Hz, 1H), 6,82 (dd, J = 8,8, 2,6 Hz, 1H), 5,56 (d, J = 12,9 Hz, 3H), 5,28 (s, 1H), 4,66 (d, J = 14,9 Hz, 1H), 4,11 -4,00 (m, 1H), 3,87 (dt, J = 12,1, 3,9 Hz, 1H), 3,68 (dd, J = 13,4, 10,1 Hz, 1H), 3,64 -3,49 (m, 2H), 3,20 -3,09 (m, 4H), 2,46 -2,41 (m, 4H), 2,22 (s, 3H), 2,00 (s, 3H), 1,76 (tt, J = 7,9, 3,9 Hz, 2H). IEN-EM m/z calc. 416,2, encontrado 417,0 (M+1)+; Tiempo de retención: 0,56 minutos.

(S)-4-[3-[2-cloro-4-(4-metilpiperazm-1-M)feml]-1,4-oxazepan-4-M]-6-metN-pmmidm-2-amma (249) I-275
RMN 1H (400 MHz, Cloroformo-d) 56,92 (d, J = 8,7 Hz, 1H), 6,74 (d, J = 2,5 Hz, 1H), 6,57 (dd, J = 8,7, 2,6 Hz, 1H), 5,40 (s, 1H), 4,46 (s, 2H), 4,13 (dd, J = 13,5, 5,0 Hz, 1H), 3,90 (d, J = 12,0 Hz, 1H), 3,51 -3,25 (m, 2H), 3,13 -2,97 (m, 4H), 2,43 - 2,33 (m, 4H), 2,18 (s, 3H), 1,91 - 1,80 (m, 1H), 1,68-1,45 (m, 1H); IEN-EM m/z calc. 416,21, encontrado 417,0 (M+1)+; Tiempo de retención: 0,51 minutos.
Los siguientes análogos se prepararon de acuerdo con el Esquema sintético 31 con la excepción que se usó N-metilglicina en lugar de N,N-dimetilglicina:

4-[(3S)-3-[2-cloro-5-(oxetan-3-ilamino)fenil]-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (250) I-258
RMN 1H (400 MHz, Metanol-d4) 57,16 (dd, J = 25,4, 8,6 Hz, 1H), 6,53 - 6,35 (m, 2H), 6,05 (dd, J = 9,5, 5,1 Hz, 0H), 5,67 (s, 1H), 5,26 (dd, J = 10,2, 4,8 Hz, 1H), 5,19 (d, J = 14,4 Hz, 1H), 4,91 (dt, J = 12,7, 6,3 Hz, 2H), 4,50 (ddt, J = 21,1, 11,9, 4,5 Hz, 2H), 4,40 - 4,13 (m, 1H), 4,11 - 3,56 (m, 4H), 2,44 - 2,15 (m, 3H), 1,92 (s, 3H); IEN-EM m/z calc.
389,16187, encontrado 390,13 (M+1)+; Tiempo de retención: 0,6 minutos; [a]D = 60,6° (c = 1, MeOH).

2-[3-[(3R)-4-(2-ammo-6-metM-pirimidm-4-M)-1,4-oxazepan-3-N]-4-cloro-amlmo]etanol 251) I-252 y 2-[3-[(3S)-4-(2-ammo-6-metM-pinmidm-4-M)-1,4-oxazepan-3-M]-4-cloro-amlmo]etanol (252) I-253
(mezcla racémica): RMN 1H (300 MHz, Metanol-d4) 57,21 (dd, J = 8,5, 4,9 Hz, 1H), 6,79 -6,51 (m, 2H), 6,06 (dd, J = 9,8, 5,1 Hz, 0.5H), 5,76 - 5,58 (m, 1H), 5,32 - 5,09 (m, 1.5H), 4,46 - 4,15 (m, 2H), 4,12 - 3,87 (m, 2H), 3,86 - 3,54 (m, 5H), 3,20 (dt, J = 17,0, 5,7 Hz, 2H), 2,21 (d, J = 0,8 Hz, 3H), 2,05-1,81 (m, 2H); IEN-EM m/z calc. 377,2, encontrado 378,2 (M+1)+; Tiempo de retención: 0,57 minutos. I-237
Condiciones de SFC: Columna: AD-H, 20x250 mm; Fase móvil: MeOH al 40 % (amoniaco 5 mM), CO2 al 60 %; Flujo: 80 ml/min; Concentraciones: ~50 mg/ ml (MeOH)
Pico A: ee: 99,2%; [a]o = -190,45° (c=0,5, MeOH); 2-[3-[(3R)-4-(2-amino-6-metil-pirimidin-4-il)-1,4-oxazepan-3-il]-4-cloro-anilino]etanol (251): RMN 1H (300 MHz, Metanol-d4) 67,14 (d, J = 8,6 Hz, 1H), 6,63 - 6,46 (m, 2H), 5,54 (s a, 1H), 5,17 (s a, 1H), 4,28 (dd, J = 13,7, 5,0 Hz, 1H), 4,03 (d, J = 12,1 Hz, 1H), 3,82 - 3,53 (m, 4H), 3,14 (t, J = 5,7 Hz, 2h ), 2,13 (s, 3H), 1,90 (a, 2H); IEN-EM m/z calc. 377,2, encontrado 378,2 (M+1)+; Tiempo de retención: 0,57 minutos.
I-252
Pico B: ee 99%; [a ]d = 172,58° (c=0,5, MeOH); 2-[3-[(3S)-4-(2-amino-6-metil-pirimidin-4-il)-1,4-oxazepan-3-il]-4-cloro-anilino]etanol (252): RMN 1H (300 MHz, Metanol-d4) 67,12 (d, J = 8,4 Hz, 1H), 6,68 - 6,46 (m, 2H), 5,48 (s a, 1H), 5,13 (s a, 1H), 4,27 (dd, J = 13,6, 5,0 Hz, 1H), 4,03 (dd, J = 12,1, 4,7 Hz, 1H), 3,77 - 3,52 (m, 4H), 3,13 (t, J = 5,7 Hz, 2H), 2,06 (s, 3H), 1,97-1,71 (m, 2H); IEN-EM m/z calc. 377,2, encontrado 378,2 (M+1)+; Tiempo de retención: 0,57 minutos. I-253

(+/-)-3-[3-[4-(2-amino-6-metil-pirimidin-4-il)-1,4-oxazepan-3-il]-4-cloro-anilino]propan-1-ol (253) I-249
RMN 1H (300 MHz, Metanol-d4) 6 7,18 (d, J = 8,7 Hz, 1H), 6,59 (t, J = 7,8 Hz, 1H), 6,48 (s, 1H), 6,05 (dd, J = 9,8, 5,2 Hz, 0,5H), 5,68 (s, 1H), 5,31 - 5,12 (m, 1.5H), 4,32 (ddd, J = 27,7, 13,8, 5,1 Hz, 2H), 4,09 - 3,52 (m, 7H), 2,35 (s, 1,5H), 2,21 (s, 1,5H), 2,04-1,65 (m, 4H); IEN-EM m/z calc. 391,2, encontrado 392,2 (M+1)+; Tiempo de retención: 0,56 minutos.

(3S)-1-[3-[4-(2-amino-6-metil-pirimidin-4-il)-1,4-oxazepan-3-il]-4-cloro-fenil]pirrolidin-3-ol (254) I-234
RMN 1H (300 MHz, Metanol-d4) 67,33 -7,11 (m, 2H), 6,59 -6,25 (m, 3H), 5,68 (s, 1H), 5,36 -5,11 (m, 2H), 4,57 -4,17 (m, 2H), 4,13 -3,58 (m, 5H), 3,51 -2,97 (m, 3H), 2,20 (s, 3H), 2,13-1,79 (m, 4H); IEN-EM m/z calc. 403,2, encontrado 404,21 (M+1)+; Tiempo de retención: 0,62 minutos.

(a) tBuXPhos Pd G3, morfolina, dioxano, ferc-BuOH, Cs2CO3, 75 °C
Formación de 4-(3-(5-bromo-2-clorofenil)-1,4-oxazepan-4-il)-6-metilpirimidin-2-amina (255)
El Intermedio, 255, se preparó de acuerdo con el Esquema sintético 2, usando intermedio 3-(5-bromo-2-clorofenil)-1,4-oxazepano-4-carboxilato de tere-butilo, 157.
Formación de (+/-)-4-[3-(2-doro-5-morfoMn-feml)-1,4-oxazepan-4-N]-6-metN-pinmidm-2-amma (256) I-195
Un matraz de 250 ml se cargó con 4-[3-(5-bromo-2-cloro-fenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina, 255, (0,30 g, 0,75 mmol), morfolina (0,20 ml, 2,29 mmol), dioxano (7 ml) y íere-BuOH (8 ml). La mezcla de reacción se agitó para dar una solución transparente. Después, la solución se desgasificó con una corriente de nitrógeno durante 10 minutos. Se añadió [2-(2-aminofenil)fenil]-metilsulfoniloxi-paladio;diterc-butil-[2-(2,4,6-triisopropilfenil)fenil]fosfano (0,03 g, 0,04 mmol) (tBuXPhos Pd G3), después se continuó burbujeando nitrógeno durante 5 minutos. Se puso Cs2CO3 (0,59 g, 1,81 mmol) en una atmósfera de nitrógeno y después se calentó a 75 °C. Después de 90 minutos, la mezcla de reacción se repartió entre EtOAc y agua. La fase orgánica se lavó con agua y salmuera, se secó (MgSO4), se filtró y
se concentró al vacío. El residuo resultante se purificó por cromatografía sobre gel de sílice usando una columna ISCO de 40 gramos eluyendo con un gradiente lineal de EtOAc al 0-100 %/heptanos para proporcionar 17 mg del producto deseado: RMN 1H (300 MHz, Metanold4) 67,29 (dd, J = 18,3, 8,8 Hz, 2H), 6,91 (ddd, J = 16,6, 8,9, 2,9 Hz, 1H), 6,78 (dd, J = 26,7, 3,0 Hz, 0.5H), 6,48 (s, 1H), 6,10 (dd, J = 9,5, 5,1 Hz, 0.5H), 5,68 (s, 1H), 5,39 -5,10 (m, 2H), 4,42 -4,17 (m, 1H), 4,09 - 3,59 (m, 11H), 3,09 (t, J = 4,9 Hz, 6H), 2,28 (dd, J = 40,8, 0,8 Hz, 5H), 2,09-1,84 (m, 4H); IEN-EM m/z calc. 403,2, encontrado 404,2 (M+1)+; Tiempo de retención: 0,62 minutos.
Ejemplo 32
Esquema sintético 32: (+/-)-4-[3-(2-cloro-4-pirrolidin-1-il-fenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (257) I-227

(a) Ir(dF(CF3)ppy)2(dtbpy)PF6, dibromoníquel; 1-metoxi-2-(2-metoxietoxi)etano, pirrolidina, DABCO, dimetilacetamida, reactor de flujo, 30 °C
Formación de (+/-)-4-[3-(2-cloro-4-pirrolidin-1-il-fenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (257) I-227
Una solución de 4-[3-(4-bromo-2-cloro-fenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina, 31, (0,050 g, 0,120 mmol), pirrolidina (0,015 ml, 0,180 mmol) y 1,4-diazabiciclo[2,2,2]octano (DABCO) (0,025 g, 0,223 mmol) en dimetilacetamida (2 ml) se burbujeó con una corriente de nitrógeno. Al el vial de reacción, se le añadió Ir(dF(CF3)ppy)2(dtbpy)PF6 (0,3 mg) en dimetilacetamida (0,1 ml) y dibromoníquel; 1-metoxi-2-(2-metoxietoxi)etano (2,2 mg, 0,006 mmol) en dimetilacetamida (0,1 ml). La reacción se realizó en un reactor de flujo Vapourtec, con 0,2 ml/min a 30 °C. La mezcla resultante se diluyó con EtOAc, se lavó con H2O, se secó sobre Na2SO4, se filtró y se concentró al vacío. Se purificó mediante 4 g de cartucho de gel de sílice eluyendo con MeOH al 0-10 %/diclorometano para proporcionar 2 mg del producto deseado: RMN 1H (400 MHz, CDCh) 67,04 (t, J = 7,5 Hz, 1H), 6,56 (d, J = 2,5 Hz, 1H), 6,40 (dd, J = 8,6, 2,5 Hz, 1H), 5,60 (s, 1H), 4,60 (s, 2H), 4,30 (dd, J = 13,5, 5,1 Hz, 1H), 3,34 -3,16 (m, 4H), 2,14 (s, 3H), 2,08 - 1,92 (m, 4H), 1,80 (d, J = 14,5 Hz, 1H); IEN-EM m/z calc. 387,2, encontrado 388,1 (M+1)+; Tiempo de retención: 3,13 minutos.
Ejemplo 33
Esquema sintético 33: (+/-)-1-(4-(4-(2-amino-6-metilpirimidin-4-il)-1,4-oxazepan-3-il)-3-clorofenoxi)propan-2-ol (261) I-232

(a) 3-(tributilestannil)metoxi)propan-1-amina tamices mol. de 4 A, CH2Ch; después 2,6-lutidina, Cu(OTf)2, hexafluoroisopropanol, CH2O 2; (b) BoC2O, Et3N, THF; (c) di-terc-butilXPhos, Cs2CO3, (2R)-propano-1,2-diol, 100 °C; (d) TFA, CH2Ó 2; (e) 2-amino-4-cloro-6-metilpirimidina, nBuOH, 120 °C; (f) separación quiral SFC
Formación de 3-(4-bromo-2-cloro-fenil)-1,4-oxazepano-4-carboxilato de (+/-)-terc-butilo (258)
Una mezcla de 4-bromo-2-cloro-benzaldehído (10,0 g, 45,6 mmol), 3-(tributilestanilmetoxi)-propan-1-amina (17,2 g, 45,6 mmol) y tamices moleculares de 4 angstrom (5,2 g) en diclorometano (180 ml) se agitó a temperatura ambiente
durante 2 horas, se filtró y se diluyó con más cantidad de diclorometano (540 ml). A un matraz separado que contenía hexafluoroisopropanol (180 ml), se le añadió 2,6-lutidina (5,3 ml, 45,6 mmol) seguido de Cu(OTf)2 (16,5 g, 45,7 mmol). La mezcla de reacción se agitó durante 2 horas y después se añadió la solución de imina preparada anteriormente en una porción. La mezcla de reacción se agitó durante una noche y después se trató con una mezcla 2:1 de una solución acuosa saturada de bicarbonato sódico e hidróxido de amonio al 10 %. La capa orgánica se separó y se lavó con una solución acuosa saturada de bicarbonato sódico, se filtró a través de un separador de fase y se concentró a sequedad. El residuo resultante se purificó por cromatografía sobre gel de sílice eluyendo con EtOAc al 0-75 %/heptano. Las fracciones puras se combinaron y se concentraron al vacío para proporcionar 6,2 g de 3-(4-bromo-2-clorofenil)-1,4-oxazepano en forma de un aceite de color amarillo: RMN 1H (400 MHz, CDCh) 87,50 (d, J = 2,0 Hz, 1H), 7,47 (d, J = 8,4 Hz, 1H), 7,38 (dd, J = 8,5, 2,0 Hz, 1H), 4,39 (dd, J = 9,0, 3,4 Hz, 1H), 4,01 -3,92 (m, 2H), 3,82 (dt, J = 12,3, 6,2 Hz, 1H), 3,38 (dd, J = 12,4, 9,0 Hz, 1H), 3,20 (dt, J = 13,5, 5,0 Hz, 1H), 3,04 (dt, J = 13,6, 6,8 Hz, 1H), 1,98 (cd, J = 6,4, 5,0 Hz, 2H); IEN-EM m/z calc. 289,0, encontrado 290,0 (M+1)+; Tiempo de retención 0,6 minutos.
A una solución de 3-(4-bromo-2-cloro-fenil)-1,4-oxazepano (1,97 g, 6,78 mmol) y trietilamina (1,04 ml, 7,46 mmol) en THF (40 ml), se le añadió Boc anhídrido (1,67 g, 7,65 mmol). La mezcla de reacción se agitó a temperatura ambiente durante una noche, después se diluyó con EtOAc y se lavó con HCl 1 M. La capa orgánica se concentró a sequedad y se purificó por cromatografía sobre gel de sílice eluyendo con EtOAc al 0-40 % en heptano. Las fracciones que contenían el producto deseado se combinaron y se concentraron para dar 2,28 g del producto deseado, 258, en forma de un aceite incoloro que solidificó después de un periodo de reposo: RMN 1H (400 MHz, DMSO-d6) 87,63 (dt, J = 6,8, 2,0 Hz, 1H), 7,56 -7,48 (m, 1H), 7,26 (d, J = 8,3 Hz, 1H), 5,29 (dd, J = 10,4, 4,9 Hz, 1H), 4,26 -4,12 (m, 1H), 4,00 - 3,84 (m, 2H), 3,63 - 3,44 (m, 3H), 1,83 -1,64 (m, 2H), 1,25 (s, 7H); IEN-EM m/z calc. 389,0, encontrado 390,0 (M+1)+; Tiempo de retención: 0,69 minutos.
Formación de 3-(2-doro-4-((R)-2-hidroxipropoxi)feml)-1,4-oxazepano-4-carboxNato de tere-butilo (259)
Se pusieron 3-(4-bromo-2-cloro-fenil)-1,4-oxazepano-4-carboxilato de tere-butilo, 258, (1,13 g, 2,89 mmol) y Cs2CO3 (1,40 g, 4,30 mmol) en un vial de microondas en una atmósfera de nitrógeno. Se añadieron tolueno (3,0 ml) y (2R)-propano-1,2-diol (1,10 ml, 14,98 mmol). La mezcla se agitó durante 5 minutos. Se añadió di-tere-but/l-[6-metoxi-3-metil-2-(2,4,6-triisopropilfenil)-fenil]fosfano:metanosulfonato paladio (2+);2-fenilanilina (di-tereBu-XPhos) (0,12 g, 0,14 mmol). La mezcla de reacción se calentó durante 1 hora a 100 °C. La mezcla se diluyó con agua y diclorometano. Las fases se separaron en un separador de fase y la fase orgánica se concentró al vacío. El residuo resultante se purificó por cromatografía sobre gel de sílice (Acetato de etilo/Heptanos) para proporcionar 820 mg del producto deseado: IEN-EM m/z calc. 385,2, encontrado 386,2 (M+1)+; Tiempo de retención: 0,85 minutos.
Formación de (2R)-1-(3-cloro-4-(1,4-oxazepan-3-N)fenoxi)propan-2-ol (260)
Se disolvió 3-[2-cloro-4-[(2R)-2-hidroxipropoxi]fenil]-1,4-oxazepano-4-carboxilato de tere-butilo, 259, (0,82 g, 2,13 mmol) en diclorometano (20 ml) y se añadió ácido trifluoroacético (10 ml). Los volátiles se retiraron en un evaporador rotatorio. Se añadieron bicarbonato sódico acuoso saturado y diclorometano. Las fases se separaron en un separador de fase. La fase orgánica se concentró al vacío. El residuo resultante se purificó por cromatografía sobre gel de sílice (Eluyente: Acetato de etilo/Heptanos) para proporcionar 330 mg del producto deseado: IEN-EM m/z calc. 285,1, encontrado 286,2 (M+1)+; Tiempo de retención: 0,5 minutos.
Formación de (2R)-1-(4-(4-(2-ammo-6-metMpinmidm-4-M)-1,4-oxazepan-3-M)-3-dorofenoxi)propan-2-ol (261)
Se disolvieron 4-cloro-6-metil-pirimidin-2-amina (0,12 g, 0,83 mmol) y (2R)-1-[3-cloro-4-(1,4-oxazepan-3-il)fenoxi]propan-2-ol, 260, (0,33 g, 0,83 mmol) en 1-butanol (2,4 ml). La mezcla de reacción se calentó durante 16 horas a 130 °C. Los volátiles se retiraron en un evaporador rotatorio. El residuo en bruto se purificó por cromatografía sobre gel de sílice (Eluyente: metanol/diclorometano). Se realizó una segunda purificación usando una columna ISCO amino-gel de sílice (Eluyente: Acetato de etilo/Heptanos) para proporcionar 270 mg del producto deseado como mezcla de 2 diastereómeros: RMN 1H (400 MHz, MeOD) 87,24-7,10 (m, 1H), 7,01 (t, J = 6,4 Hz, 1H), 6,91 -6,78 (m, 1H), 4,22 (dd, J = 13,6, 5,1 Hz, 1H), 4,12 - 3,96 (m, 2H), 3,90 - 3,77 (m, 2H), 3,75 - 3,54 (m, 3H), 2,06 (s, 3H), 1,89 (dddd, J = 35,0, 32,2, 11,8, 9,7 Hz, 2H), 1,24 (dd, J = 6,4, 2,5 Hz, 3H); IEN-EM m/z calc. 392,2, encontrado 393,2 (M+1)+; Tiempo de retención: 0,59 minutos.
Separación quiral SFC: Columna: AD-H, 20x250 mm; Fase móvil: MeOH al 40 % (amoniaco 5 mM), CO2 al 60 %; Flujo: 80 ml/min; Concentraciones: ~50 mg/ ml (MeOH)
Pico A: (R)-1-(4-((R)-4-(2-amino-6-metilpirimidin-4-il)-1,4-oxazepan-3-il)-3-clorofenoxi)-propan-2-ol (262) rotación óptica 5,0 mg en 0,5 ml de MeOH = -17,20°; RMN 1H (400 MHz, MeOD) 87,19 (d, J = 8,7 Hz, 1H), 7,03 (d, J = 2,5 Hz, 1H), 6,88 (dd, J = 8,6, 2,4 Hz, 1H), 4,22 (dd, J = 13,7, 5,0 Hz, 1H), 4,15 -3,95 (m, 2H), 3,85 (ddd, J = 16,2, 9,7, 5,4 Hz, 2H), 3,77 - 3,53 (m, 3H), 2,07 (s, 3H), 1,86 (d, J = 29,4 Hz, 2H), 1,24 (d, J = 6,4 Hz, 3H); IEN-EM m/z calc. 392,2, encontrado 393,2 (M+1)+; Tiempo de retención: 0,58 minutos. I-264
Pico B: (R)-1-(4-((S)-4-(2-amino-6-metilpirimidin-4-il)-1,4-oxazepan-3-il)-3-clorofenoxi)-propan-2-ol (263): rotación óptica 4,8 mg en 0,5 ml de MeOH = -22,76°; RMN 1H (400 MHz, MeOD) 87,19 (d, J = 8,7 Hz, 1H), 7,03 (d, J = 2,4 Hz, 1H), 6,87 (d, J = 8,7 Hz, 1H), 4,22 (dd, J = 13,6, 5,1 Hz, 1H), 4,13 -3,94 (m, 2H), 3,85 (ddd, J = 16,2, 9,6, 5,4 Hz, 2H),
3,76 -3,52 (m, 2H), 2,07 (s, 2H), 1,86 (d, J = 33,1 Hz, 2H); IEN-EM m/z calc. 392,2, encontrado 393,2 (M+1)+; Tiempo de retención: 0,57 minutos. I-265
Los siguientes análogos se prepararon de acuerdo con el Esquema sintético 33:

(+/-H1-[[4-[4-(2-ammo-6-metN-pirimidm-4-N)-1,4-oxazepan-3-M]-3-cloro-fenoxi]metil]-ciclopropN]metanol (264) I-233
RMN 1H (400 MHz, MeOD) 87,20 (d, J = 8,6 Hz, 1H), 7,03 (d, J = 2,6 Hz, 1H), 6,88 (dd, J = 8,6, 2,5 Hz, 1H), 4,23 (dd, J = 13,6, 5,0 Hz, 1H), 4,05 - 3,45 (m, 8H), 2,17 (s, 3H), 1,89 (d, J = 13,2 Hz, 2H), 0,58 (dt, J = 5,2, 1,9 Hz, 4H); IEN-EM m/z calc. 418,2, encontrado 419,3 (M+1)+; Tiempo de retención: 0,62 minutos.

(+/-)-4-[3-[2-cloro-4-(oxetan-3-iloxi)fenil]-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (265) I-231
RMN 1H (400 MHz, MeOD) 87,20 (d, J = 8,6 Hz, 1H), 6,86 (d, J = 2,6 Hz, 1H), 6,70 (dd, J = 8,8, 2,6 Hz, 1H), 5,32 -5,16 (m, 1H), 5,04 -4,90 (m, 2H), 4,71 -4,50 (m, 2H), 4,22 (dd, J = 13,6, 5,1 Hz, 1H), 4,01 (d, J = 11,9 Hz, 1H), 3,73 -3,53 (m, 3H), 2,07 (s, 3H), 1,86 (d, J = 34,4 Hz, 2H); IEN-EM m/z calc. 390,1, encontrado 391,2 (M+1)+; Tiempo de retención: 0,61 minutos.

(+/-)-4-[4-[4-(2-amino-6-metil-pirimidin-4-il)-1,4-oxazepan-3-il]-3-cloro-fenoxi]-2-metil-butan-2-ol (266) I-230 RMN 1H (400 MHz, MeOD) 87,19 (d, J = 8,7 Hz, 1H), 6,99 (d, J = 2,7 Hz, 1H), 6,85 (dd, J = 8,9, 2,6 Hz, 1H), 4,22 (dd, J = 13,6, 5,0 Hz, 1H), 4,10 (t, J = 6,9 Hz, 2H), 4,06 -3,90 (m, 1H), 3,67 (ddd, J = 45,9, 13,8, 10,6 Hz, 3H), 2,09 (s, 3H), 2,00 -1,71 (m, 3H), 1,26 (s, 6H); IEN-EM m/z calc. 420,2, encontrado 421,3 (M+1)+; Tiempo de retención: 0,63 minutos.

(+/-)-4-[3-[2-cloro-4-[(3-metiltetrahidrofurano-3-il)metoxi]fenil]-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (267) I-229
RMN 1H (400 MHz, MeOD) 87,19 (d, J = 8,7 Hz, 1H), 7,02 (d, J = 2,6 Hz, 1H), 6,87 (dd, J = 8,7, 2,6 Hz, 1H), 4,22 (dd, J = 13,6, 5,0 Hz, 1H), 4,01 (d, J = 11,3 Hz, 1H), 3,93 - 3,54 (m, 8H), 3,53 - 3,38 (m, 2H), 2,08 (s, 3H), 2,03 - 1,66 (m, 4H), 1,23 (s, 3H); Ie N-EM m/z calc. 432,2, encontrado 433,3 (M+1)+; Tiempo de retención: 0,67 minutos.
Ejemplo 34
Esquema sintético 34: (+/-)-6-(4-(2-ammo-6-metilpinmidm-4-M)-1,4-oxazepan-3-M)-7-cloro-2H-benzo[¿][1,4]tiazin-3(4H)-ona (273) I-268 y (+/-)-4-(3-(7-cloro-3,4-dihidro-2H-benzo[6][1,4]tiazm-6-il)-1,4-oxazepan-4-il)-6-metilpirimidin-2-amina (274) I-274

(a) 3-(tributilestannil)metoxi)propan-1-amina, tamices moleculares de 4 A, CH2CI2; después 2,6-lutidina, Cu(OTf)2, hexafluoroisopropanol, C ^C h ; b) BoC2O, CH2Cl2; c) 2-sulfanilacetato de metilo, K2CO3, DMF, 50 °C; d) TFA, CH2Cl2; e) 2-amino-4-cloro-6-metilpirimidina, nBuOH, 120 °C; f) hierro, ácido acético, 60 °C; g) LiAlH4, THF, 60 °C.
Formación de 3-(2-cloro-4-fluoro-5-nitro-fenil)-1,4-oxazepano (268)
A una solución de 2-cloro-4-fluoro-5-nitro-benzaldehído (2,0 g, 9,8 mmol) en diclorometano (50 ml), se le añadieron 3-(tributilestanilmetoxi)propan-1-amina (3,8 g, 9,9 mmol) y tamices moleculares de 4 angstrom (1,5 g). La mezcla de reacción se agitó a temperatura ambiente durante dos horas y se filtró a través de una capa corta de Celite y se enjuagó con diclorometano. El filtrado se concentró al vacío para proporcionar la imina en bruto.
A una solución separada de 2,6-lutidina (1,4 ml, 12,1 mmol) en hexafluoroisopropanol (50 ml), se le añadió Cu(OTf)2 (4,3 g, 11,9 mmol) (1,20 equiv., se precalentó a 110 °C durante 1 h a alto vacío) y se agitó a temperatura ambiente durante 1 hora. Una solución de la imina en diclorometano (160 ml) se añadió en una porción y la mezcla resultante se agitó a temperatura ambiente durante 12 horas. La reacción se interrumpió a temperatura ambiente con una mezcla de solución acuosa saturada de NaHCO3 (40 ml) e hidróxido de amonio acuoso al 10% (20 ml) y se agitó vigorosamente durante 15 minutos. Las capas se separaron y la capa acuosa se extrajo con diclorometano (3 x 50 ml). Las capas orgánicas combinadas se lavaron con agua (3 x 5 ml) y salmuera (10 ml), se secaron sobre Na2SO4, se filtraron y se concentraron al vacío. La purificación por cromatografía sobre gel de sílice (gradiente de MeOH al 0 6 %/diclorometano) proporcionó 420 mg del producto deseado: RMN 1H (300 MHz, CDCh) 88,21-8,04 (m, 1H), 7,36 -7,05 (m, 1H), 4,03 (ddd, J = 15,7, 7,3, 4,1 Hz, 1H), 3,95 -3,72 (m, 2H), 3,72 -3,47 (m, 2H), 3,50 -3,30 (m, 2H), 2,13 1,51 (m, 2H); IEN-e M m/z calc. 274,1, encontrado 275,2 (M+1); Tiempo de retención: 0,84 minutos.
Formación de 3-(2-cloro-4-fluoro-5-nitro-fenil)-1,4-oxazepano-4-carboxilato de tere-butilo (269)
Se añadió 3-(2-cloro-4-fluoro-5-nitro-fenil)-1,4-oxazepano, 268, (0,42 g, 1,53 mmol) en diclorometano (5 ml) Boc2O (0,50 g, 2,29 mmol). La mezcla de reacción se agitó a temperatura ambiente durante la noche. La mezcla se concentró al vacío y se purificó por cromatografía sobre gel de sílice (columna ISCO de 12 g, eluyendo con EtOAc al 10 100 %/Heptanos) para proporcionar 350 mg del producto deseado en forma de un sólido cristalino de color amarillo: RMN 1H (300 MHz, CDCh) 87,97 (t, J = 6,1 Hz, 1H), 7,34 (d, J = 10,1 Hz, 1H), 5,50 (ddd, J = 33,9, 10,1, 4,4 Hz, 1H), 4,55 - 3,94 (m, 3H), 3,68 - 3,23 (m, 3H), 2,17 - 1,72 (m, 2H), 1,34 (d, J = 59,9 Hz, 9H); IEN-EM m/z calc. 374,1, encontrada 375,2; Tiempo de retención: 0,93 minutos.
Formación de 3-[2-cloro-4-(2-metoxi-2-oxo-etil)sulfanil-5-nitro-fenil]-1,4-oxazepano-4-carboxilato de tere-butilo (270)
Se disolvió 3-(2-cloro-4-fluoro-5-nitro-fenil)-1,4-oxazepano-4-carboxilato de tere-butilo, 269, (0,31 g, 0,83 mmol) y K2CO3 (0,24 g, 1,77 mmol) en DMF (2 ml) en un vial pequeño. Se añadió 2-sulfanilacetato de metilo (0,09 ml, 0,95 mmol) a la mezcla. La mezcla se calentó a 50 °C durante 6 horas. Después de la retirada de DMF al vacío, el producto en bruto se diluyó con EtOAc (5 ml). La capa orgánica se filtró y el disolvente se retiró al vacío para proporcionar 380 mg del producto deseado en forma de un sólido de color amarillo: IEN-EM m/z calc. 460,1, encontrada 461,10; Tiempo de retención: 0,92 minutos.
Formación de 2-((5-cloro-2-nitro-4-(1,4-oxazepan-3-il)fenil)tio)acetato de metilo (271)
Se añadió 3-[2-cloro-4-(2-metoxi-2-oxo-etil)sulfanil-5-nitro-fenil]-1,4-oxazepano-4-carboxilato de tere-butilo, 270, (50 mg, 0,1085 mmol) en diclorometano (1,5 ml) ácido trifluoroacético (0,25 ml, 3,25 mmol) y se agitó durante 1 hora a temperatura ambiente. La CLEM iindicó que no había más material de partida presente. El disolvente se retiró al vacío y el productoen bruto (sal de TFA) se usó directamente sin purificación adicional.
Formación de 2-((4-(4-(2-amino-6-metilpirimidin-4-il)-1,4-oxazepan-3-il)-5-cloro-2-nitrofenil)tio)acetato de
metilo (272)
En un vial de microondas, se calentaron 2-[5-cloro-2-nitro-4-(1,4-oxazepan-3-il)fenil]sulfanilacetato de metilo (sal trifluoroacetato), 271, (0,05 g) y 4-cloro-6- metil-pirimidin-2-amina (0,03 g, 0,17 mmol) en n-BuOH (2 ml) a 120 °C durante una noche. La CLEM indicó la desaparición del material de partida. Se obtuvieron productos tanto de éster metílico como de éster n-bu. Se retiró el n-BuOH para dar el producto en bruto en forma de un sólido de color amarillo y se usó directamente sin más purificación.
Formación de 6-[4-(2-ammo-6-metil-pinmidm-4-M)-1,4-oxazepan-3-il]-7-cloro-4H-1,4-benzotiazm-3-ona (273) I-268
3-[2-Cloro-4-(2-metoxi-2-oxo-etil)sulfanil-5-nitro-fenil]-1,4-oxazepano-4-carboxilato de tere-butilo, 272, (0,05 g, 0,11 mmol) y hierro (0,06 g, 1,07 mmol) en un vial se añadió ácido acético (2 ml). La mezcla se calentó a 60 °C y se agitó durante 1 hora. Se retiró ácido acético y la mezcla en bruto se purificó por cromatografía sobre gel de sílice (columna ISCO de 12 g, eluyendo con un gradiente de MeOH al 0-10 %/diclorometano) para proporcionar 24 mg del producto deseado en forma de un sólido de color blanco: RMN 1H (300 MHz, CDCls) 810,01 (s, 1H), 7,29 (s, 1H), 6,93 (d, J = 52,9 Hz, 2H), 5,87 (d, J = 52,3 Hz, 1H), 5,50-4,71 (m, 2H), 4,26 (dd, J = 13,7, 5,0 Hz, 1H), 4,12 - 3,92 (m, 1H), 3,72 -3,37 (m, 3H), 3,31 (d, J = 1,8 Hz, 2H), 2,01 (d, J = 17,2 Hz, 4H), 1,81 (s, 2H); IEN-EM m/z calc. 405,1, encontrado 406,1 (M+1)+; Tiempo de retención: 0,61 minutos.
Formación de 4-[3-(7-cloro-3,4-dihidro-2H-1,4-benzotiazin-6-il)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (274) I-274
A temperatura ambiente, se añadió 6-[4-(2-amino-6-metil-pirimidin-4-il)-1,4-oxazepan-3-il]-7-cloro-4H-1,4-benzotiazin-3-ona, 273, (0,02 g, 0,05 mmol) en THF (2,0 ml) a una solución de LiAlH4 (0,05 ml de solución 2 M en THF, 0,10 mmol). La solución turbia se agitó durante una noche. Se añadió una solución adicional de LiAlH4 (0,10 ml) y la mezcla se calentó a 60 °C durante una noche. Se añadió agua enfriada con hielo (0,25 ml) y la mezcla se agitó durante 10 minutos. Se añadió diclorometano (10 ml). El sólido de color blanco resultante se filtró y se lavó con diclorometano. Las fases orgánicas combinadas se concentraron al vacío. El residuo en bruto se purificó por cromatografía sobre gel de sílice (columna ISCO de 4 g, eluyendo con un gradiente de MeOH al 0-10 %/diclorometano) para proporcionar 7,5 mg del producto deseado en forma de un sólido de color blanco: RMN 1H (300 MHz, CDCla) 86,92 (s, 1H), 6,27 (s, 1H), 5,49 (s, 3H), 5,06 (s, 2H), 4,23 (dd, J = 13,6, 5,0 Hz, 2H), 3,98 (s, 2H), 3,61 - 3,35 (m, 6H), 3,00 - 2,85 (m, 2H), 2,11 (s, 4H), 1,74 (d, J = 13,7 Hz, 2H); IEN-EM m/z calc. 391,1, encontrado 392,1 (M+1)+; Tiempo de retención: 0,67 minutos.
Ejemplo 35
Esquema sintético 35: (+/-)-4-[3-[2-cloro-4-(oxetan-3-il)fenil]-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (275) I-255

Un tubo Pirex se cargó con 3-bromooxetano (0,025 g, 0,180 mmol), NiCl2glyma(0,003 g, 0,014 mmol), 1,10-fenantrolina (0,005 g, 0,028 mmol), NaBF4 (0,007 g, 0,065 mmol), manganeso (0,013 g, 0,240 mmol). El tubo se burbujeó con nitrógeno durante 5 minutos. A la mezcla se le añadieron MeOH (0,5 ml), 4-etilpiridina (0,007 g, 0,060 mmol) y 4-[3-(4-bromo-2-cloro-fenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina, 31, (0,050 g, 0,120 mmol). La mezcla de reacción se agitó durante una noche a 60 °C. La mezcla de reacción se diluyó con EtOAc y se filtró a través de una capa de Celite. El filtrado se concentró al vacío y el residuo se purificó por cromatografía sobre gel de sílice usando una columna ISCO de 4 g eluyendo con MeOH al 0-10 %. El producto recuperado tiene impurezas menores de desbromación. El producto se purificó de nuevo mediante cromatografía HPLC de fase inversa para proporcionar 9,0 mg del producto deseado: RMN 1H (400 MHz, CDCh) 87,44 (d, J = 1,5 Hz, 1H), 7,28 - 7,22 (m, 2H), 5,71 (s, 1H), 5,58 (s, 1H), 5,07 (ddd, J = 8,3, 6,1, 0,9 Hz, 2H), 4,74 (ddd, J = 6,8, 6,1, 0,9 Hz, 2H), 4,63 (s, 2H), 4,35 (dd, J = 13,6, 5,0 Hz, 1H), 4,26 -4,02 (m, 2H), 2,15 (s, 3H), 2,10 -1,95 (m, 1H), 1,90-1,79 (m, 1H); IEN-EM m/z calc. 374,2, encontrado 375,0 (M+1)+; Tiempo de retención: 2,63 minutos.
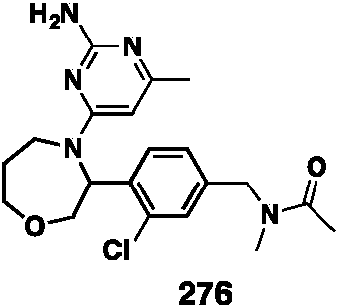
(+/-)-W-(4-(4-(2-am m o-6-metilpi N mid m -4-il)-1,4-oxazepan-3-il)-3-clorobencil)-W-metilacetamida (276) I-254 RMN 1H (400 MHz, CDCI3) 87,28-7,17 (m, 2H), 7,13 -6,99 (m, 1H), 5,57 (s, 2H), 4,79 -4,56 (m, 3H), 4,56 -4,43 (m, 2H), 4,34 (ddd, J = 13,6, 5,0, 3,6 Hz, 1H), 4,10 (d, J = 11,6 Hz, 1H), 3,70 -3,47 (m, 1H), 2,97 (d, J = 7,5 Hz, 3H), 2,17 (d, J = 13,1 Hz, 7H); IEN-EM m/z calc. 403,2, encontrado 404,0 (M+1)+; Tiempo de retención: 2,55 minutos.

(+/-)-4-(3-(2-cloro-4-(tetrahidrofurano-2-il)fenil)-1,4-oxazepan-4-il)-6-metilpirimidin-2-amina (277) I-235 RMN 1H (400 MHz, CDCI3) 87,40 (t, J = 1,8 Hz, 1H), 7,24 -7,10 (m, 2H), 5,56 (s, 1H), 4,85 (t, J = 7,2 Hz, 1H), 4,60 (s, 2H), 4,35 (ddd, J = 13,6, 5,1, 2,1 Hz, 1H), 4,10 (dtd, J = 8,7, 6,8, 2,2 Hz, 2H), 4,01 -3,89 (m, 1H), 3,70 - 3,46 (m, 3H), 2,39 - 2,28 (m, 1H), 2,14 (s, 3H), 2,02 (ttd, J = 8,1, 6,7, 6,2, 5,3 Hz, 2H), 1,90 - 1,74 (m, 2H), 1,29 (d, J = 6,0 Hz, 1H); IEN-EM m/z calc. 388,2, encontrado 389,0 (M+1)+; Tiempo de retención: 3,09 minutos.

(+/-)-5-(4-(4-(2-amino-6-metilpirimidin-4-il)-1,4-oxazepan-3-il)-3-clorofenil)-1-metilpirrolidin-2-ona (278) I-239 RMN 1H (400 MHz, CDCI3) 87,30 (s, 1H), 7,28 - 7,22 (m, 1H), 7,18 - 7,03 (m, 1H), 5,68 (s, 5H), 4,60 - 4,26 (m, 2H), 4,21 - 4,02 (m, 1H), 3,65 (dd, J = 26,2, 13,5 Hz, 4H), 3,32 (t, J = 7,1 Hz, 1H), 2,71 (d, J = 2,2 Hz, 2H), 2,64 - 2,34 (m, 2H), 2,35-1,76 (m, 6H); IEN-EM m/z calc. 415,2, encontrado 416,27 (M+1)+; Tiempo de retención: 0,66 minutos. Ejemplo 36
Esquema sintético 36: (+/-)-4-(3-(2-cloro-4-(tetrahidrofurano-3-il)fenil)-1,4-oxazepan-4-il)-6-metilpirimidin-2-amina (279) I-238

Un tubo pyrex se cargó con 3-bromotetrahidrofurano (0,027 g, 0,179 mmol) dicloroníquel;1,2-dimetoxietano (0,003 g, 0,014 mmol), 1,10-fenantrolina (0,005 g, 0,028 mmol), BF4 (sal de sodio) (0,007 g, 0,064 mmol), manganeso (0,013 g, 0,237 mmol). La mezcla de reacción se burbujeó con nitrógeno durante 5 minutos. A la mezcla se le añadieron MeOH (4 ml), 4-etilpiridina (0,007 g, 0,061 mmol) y seguido de 4-[3-(4-bromo-2-cloro-fenil)-1,4-oxazepan-4-il]-6-metilpirimidin-2-amina, 31, (0,050 g, 0,121 mmol). La mezcla se agitó a 55 °C durante una noche. La mezcla de reacción se diluyó con EtOAc, se filtró a través de una capa de celite y se concentró al vacío. El residuo resultante se purificó por cromatografía sobre gel de sílice usando un cartucho de gel de sílice ISCO de 4 g eluyendo con MeOH al 0-10 %. El producto recuperado se volvió a purificar por cromatografía de fase inversa. Para proporcionar 6,7 mg del producto deseado: RMN 1H (400 MHz, CDCh) 87,30 (d, J = 1,7 Hz, 1H), 7,24 (d, J = 3,2 Hz, 1H), 7,18 (d, J = 8,0 Hz, 1H), 7,11
(d, J = 8,1 Hz, 1H), 5,58 (s, 1H), 4,90 (s, 2H), 4,51 - 4,26 (m, 1H), 4,18 -4,05 (m, 2H), 3,92 (c, J = 7,8 Hz, 1H), 3,73 (ddd, J = 8,7, 7,0, 1,8 Hz, 1H), 3,69 -3,49 (m, 3H), 3,37 (p, J = 7,6 Hz, 1H), 2,45 -2,27 (m, 1H), 2,17 (s, 3H), 2,08-1,95 (m, 2H); IEN-EM m/z calc. 388,2, encontrado 389,0 (M+1)+; Tiempo de retención: 3,06 minutos.
Ejemplo 37
Esquema sintético 37: (+/-)-4-(3-(2-cloro-4-(pirrolidin-2-il)fenil)-1,4-oxazepan-4-il)pirimidin-2-amina (280) I-267

Un vial pyrex se cargó con N O 2-6 H2O (0,029 g, 0,121 mmol) y 4,7-dimetoxi-1,10-fenantrolina (0,125 ml de 0,1 M, 0,013 mmol) en DMSO (1 ml). El tubo se sonicó durante 5 minutos hasta que se disolvieron los materiales. En un vial de reacción cargado con 4-[3-(4-bromo-2-cloro-fenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina, 31, (0,050 g, 0,121 mmol), Ir(dF(CF3)ppy)2(dtbpy)PF6 (0,0013 g, 0,0012 mmol), pirrolidin-1-carboxilato de ferc-butilo (0,050 ml, 0,285 mmol), acetato de quinuclidin-3-ilo (0,270 ml de solución 0,5 M, 0,135 mmol) en DMSO (0,40 ml) y 4,7-dimetoxi-1,10-fenantrolina (0,125 ml de solución 0,1 M en DMSO, 0,013 mmol). El tubo se burbujeó con nitrógeno durante 5 minutos. A la mezcla se le añadió H2O (0,090 ml, 4,996 mmol). El tubo de reacción se expuso a una luz LED de color azul y se agitó durante una noche. La mezcla se diluyó con EtOAc, se lavó con H2O y salmuera. La fase orgánica se secó sobre Na2SO4, se filtró y se concentró al vacío. El residuo resultante se purificó por cromatografía sobre gel de sílice usando una columna ISCO de 4 g eluyendo con MeOH al 0-10 %/diclorometano para proporcionar la mayoría del producto deseado puro que se usó en la siguiente etapa sin purificación adicional.
A una solución del producto anterior disuelta en diclorometano (1 ml), se le añadió ácido trifluoroacético (0,500 ml, 6,490 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 20 minutos. La mezcla de reacción se sometió directamente a purificación HPLC de fase inversa para proporcionar 16 mg del producto deseado en forma de una sal TFA. El producto se convirtió en base libre pasándolo a través de un cartucho de PE PL-HCO3 MPS y el filtrado se concentró al vacío para proporcionar 5,6 mg del producto deseado: RMN 1H (400 MHz, CDCh) 87,43 (d, J = 8,5 Hz, 1H), 7,24 - 7,15 (m, 2H), 4,59 (s, 3H), 4,35 (ddd, J = 13,6, 5,0, 1,7 Hz, 1H), 4,10 (d, J = 8,0 Hz, 3H), 3,69 -3,49 (m, 4H), 3,27 - 3,14 (m, 1H), 3,04 (d, J = 8,6 Hz, 1H), 2,64 (s, 6 H), 2,30-2,09 (m, 4H); IEN-EM m/z calc. 387,2, encontrado 388,2 (M+1)+; Tiempo de retención: 0,5 minutos.
Ejemplo 38
Esquema sintético 38: (+/-)-4-(3-(2-cloro-4-(metoximetil)fenil)-1,4-oxazepan-4-il)-6-metilpirimidin-2-amina (286) I-205

(a) BoC2O, EtaN, THF; (b) NaBH4, EtOH; (c) NBS, PhaP, CH2Ch; (d) NaOMe, MeOH, 70 °C; (e) HCl, dioxano; (f) 2-amino-4-cloro-6-metilpirimidina, nBuOH, 120 °C.
Formación de 3-(2-cloro-4-(metoxicarbonil)fenil)-1,4-oxazepano-4-carboxilato de (+/-)-terc--butilo (281)
A una solución de 3-cloro-4-(1,4-oxazepan-3-il)benzoato de metilo, 188, (5,69 g, 21,10 mmol) y trietilamina (3,25 ml, 23,32 mmol) en THF (80 ml), se le añadió Boc anhídrido (5,07 g, 23,23 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 3 días, se diluyó con HCl 1 M y se extrajo dos veces con EtOAc. Los extractos orgánicos combinados se concentraron a sequedad y se purificaron por cromatografía sobre gel de sílice eluyendo con EtOAc al 0-50 % en heptano. Las fracciones puras se combinaron y se concentraron al vacío para dar 6,27 g del producto deseado en forma de un aceite de color amarillo claro: RMN 1H (400 MHz, CDCh) 88,03 (d, J = 1,7 Hz, 1H), 7,90 (dd, J = 8,1, 1,7 Hz, 1H), 7,34 (d, J = 8,2 Hz, 1H), 5,56 (dd, J = 10,6, 4,6 Hz, 1H), 4,56 (dd, J = 15,0, 5,5 Hz, 1H), 4,31 4.06 (m, 2H), 3,94 (s, 3H), 3,58 - 3,33 (m, 3H), 2,07 - 1,91 (m, 1H), 1,85 (d, J = 15,9 Hz, 1H), 1,22 (s, 10H).
Formación de 3-[2-cloro-4-(hidroximetil)fenil]-1,4-oxazepano-4-carboxilato de (+ /-)-terc-butilo (282)
A una solución de 3-(2-cloro-4-metoxicarbonil-fenil)-1,4-oxazepano-4-carboxilato de ferc-butilo, 281, (6,25 g, 16,05 mmol) en EtOH (80 ml), se le añadió NaBH4 (6,07 g, 160,4 mmol). La reacción se agitó durante una noche a temperatura ambiente y después cuidadosamente se inactivó con HCl 1 M acuoso a pH ~ 1. El producto se extrajo tres veces con EtOAc y los extractos orgánicos combinados se concentraron a sequedad y se purificaron por cromatografía sobre gel de sílice eluyendo con EtOAc al 0-65 % en heptano. Las fracciones puras se combinaron y se concentraron al vacío para dar 4,94 g del producto: RMN 1H (400 MHz, CDCla) 87,37 (s, 1H), 7,23 (s, 2H), 5,52 (dd, J = 10,8, 4,7 Hz, 1H), 4,68 (d, J = 3,3 Hz, 2H), 4,54 (dd, J = 15,1, 5,3 Hz, 1H), 4,12 (ddd, J = 28,6, 13,6, 4,6 Hz, 2H), 3,57-3,28 (m, 3H), 2,08-1,90 (m, 2H), 1,90-1,75 (m, 1H), 1,24 (s, 9H); IEN-EM m/z calc. 341,2, encontrado 342,0 (M+1)+; Tiempo de retención: 0,96 minutos.
Formación de 3-[4-(bromometil)-2-cloro-fenil]-1,4-oxazepano-4-carboxilato de (+/-)-terc-butilo (283)
A una solución de 3-[2-cloro-4-(hidroximetil)fenil]-1,4-oxazepano-4-carboxilato de ferc-butilo, 282, (4,94 g, 13,73 mmol) y trifenilfosfina (4,35 g, 16,58 mmol) en diclorometano (50 ml), se le añadió W-bromosuccinimida (3,75 g, 21,07 mmol). La mezcla de reacción se agitó durante 20 minutos a temperatura ambiente y después se diluyó con agua. La capa orgánica se concentró a sequedad y se purificó a través de cromatografía sobre gel de sílice eluyendo con EtOAc al 0-40 % en heptano. Las fracciones puras se combinaron y se concentraron para dar 5,0 g del producto deseado en forma de un aceite incoloro: RMN 1H (400 MHz, CDCla) 87,43-7,36 (m, 1H), 7,32 - 7,18 (m, 2H), 5,51 (dd, J = 10,8, 4.7 Hz, 1H), 4,60 -4,48 (m, 1H), 4,44 (s, 2H), 4,35 - 3,98 (m, 2H), 3,48 (dddd, J = 17,1, 14,3, 11,4, 3,1 Hz, 3H), 2,09 -1,90 (m, 1H), 1,90 - 1,71 (m, 1H), 1,31-1,18 (m, 9H); IEN-EM m/z calc. 403,1, encontrado 404,0 (M+1)+; Tiempo de retención: 0,63 minutos.
Formación de 3-[2-cloro-4-(metoximetil)fenil]-1,4-oxazepano-4-carboxilato de (+/-)-terc-butilo (284)
Una suspensión de 3-[4-(bromometil)-2-cloro-fenil]-1,4-oxazepano-4-carboxilato de (+/-)-ferc-butilo, 283, (0,37 g, 0,88 mmol) se agitó en metóxido sódico (3 ml de solución al 25 % p/v en MeOH, 14 mmol) a 70 °C durante una noche en un tubo cerrado herméticamente. La mezcla se diluyó con agua y se extrajo con EtOAc. La capa orgánica se concentró a sequedad y se purificó por cromatografía sobre gel de sílice eluyendo con EtOAc al 0-50 % en heptano. Las fracciones que contenían el producto deseado se combinaron y se concentraron para dar 161 mg de aceite incoloro: RMN 1H (400 MHz, CDCh) 87,39-7,30 (m, 1H), 7,30 - 7,17 (m, 2H), 5,52 (dd, J = 10,9, 4,7 Hz, 1H), 4,62 - 4,49 (m, 1H), 4,42 (s, 2H), 3,55 -3,43 (m, 2H), 3,40 (s, 3H), 1,98 (qdd, J = 11,2, 5,8, 2,2 Hz, 1H), 1,87 - 1,72 (m, 1H), 1,47 -1,24 (m, 9H); iEn -EM m/z calc. 355,2, encontrado 356,0 (M+1)+; Tiempo de retención: 0,55 minutos.
Formación de (+/-)-3-[2-cloro-4-(metoximetil)fenil]-1,4-oxazepano (285)
Una solución de 3-[2-cloro-4-(metoximetil)fenil]-1,4-oxazepano-4-carboxilato de (+/-)-ferc-butilo, 284, (0,16 g, 0,45 mmol) en HCl (3,0 ml de 4 M, 12,00 mmol) se agitó durante 1 hora a temperatura ambiente. Después, la mezcla de reacción se concentró a sequedad para proporcionar 132 mg del producto deseado como una sal HCl y se usó en la siguiente etapa sin purificación adicional: lEN-EM m/z calc. 255,1, encontrado 256,0 (M+1)+; Tiempo de retención: 0,56 minutos.
Formación de (+/-)-4-[3-[2-cloro-4-(metoximetil)fenil]-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (286) I-205
Una mezcla de (+/-)-3-[2-cloro-4-(metoximetil)fenil]-1,4-oxazepano-HCl, 285, (0,13 g, 0,45 mmol), 4-cloro-6-metilpirimidin-2-amina (0,10 g, 0,70 mmol) y trietilamina (0,19 ml, 1,36 mmol) en n-BuOH (2 ml) se calentó en un tubo cerrado herméticamente a 120 °C durante una noche. La mezcla de reacción se concentró a sequedad, después se disolvió en EtOAc y se lavó con una solución acuosa saturada de bicarbonato sódico. La fase orgánica se aisló y se concentró a sequedad. El residuo resultante se purificó por cromatografía sobre gel de sílice eluyendo con MeOH al 0-10 % en diclorometano. Las fracciones puras se combinaron, se concentraron y se liofilizaron para proporcionar 18 mg del producto deseado: RMN 1H (400 MHz, DMSO-d6) 87,36 (d, J = 1,6 Hz, 1H), 7,28 (d, J = 8,0 Hz, 1H), 7,21 (dd, J = 8,1, 1,7 Hz, 1H), 5,57 (s, 1H), 5,46 (s, 2H), 4,63 (d, J = 14,9 Hz, 1H), 4,38 (s, 2H), 4,11 (dd, J = 13,5, 5,0 Hz, 1H), 3,93 -3,86 (m, 1H), 3,76-3,60 (m, 2H), 3,60 - 3,48 (m, 1H), 3,30 (d, J = 0,7 Hz, 3H), 1,99 (s, 3H), 1,77 (p, J = 4,1 Hz, 2H); IEN-e M m/z calc. 362,2, encontrado 363,0 (M+1)+; Tiempo de retención: 0,7 minutos.
Ejemplo 39
Esquema sintético 39: (+/-)-2-(4-(4-(2-amino-6-metilpirimidin-4-il)-1,4-oxazepan-3-il)-3-clorofenil)propano-1,3-diol (288) I-236

(a) Ir(dF(CFa)ppy)2(dtbpy)PF6, NiCh glima, TTMMS, Na2CO3, DME, luz LED de color azul; (b) TFA, CH2CI2; (c) 2-amino-4-cloro-6-metilpirimidina, nBuOH, 130 °C
Formación de 3-(2-cloro-4-(oxetan-3-il)fenil)-1,4-oxazepano-4-carboxilato de (+/-)-ferc-butilo (286)
A un vial de reacción, se le añadieron NiCh glima (0,003 g, 0,014 mmol), 4-ferc-butil-2-(4-ferc-butil-2-piridil)piridina (0,004 g, 0,015 mmol) y DME (1 ml). La mezcla se burbujeó con nitrógeno durante 5 minutos hasta que el sólido se disolvió en DME (1 ml). En otro vial se añadió 3-(4-bromo-2-cloro-fenil)-1,4-oxazepano-4-carboxilato de ferc-butilo, 258, (0,200 g, 0,510 mmol), 3-bromooxetano (0,110 g, 0,800 mmol), Ir[dF(CF3)ppy]2(dtbby)PF6 (0,003 g, 0,0034 mmol), TTMSS (0,170 ml, 0,550 mmol) y Na2CO3 (0,062 g, 0,590 mmol) y d Me (2 ml). A la mezcla se le añadió una solución de NiCdhglima y se agitó durante 18 h frente a una lámpara LED de color azul. La mezcla se diluyó con H2O, se extrajo con diclorometano y la fase orgánica se concentró al vacío. El residuo se purificó por cromatografía sobre gel de sílice usando una columna ISCO de 12 g eluyendo con EtOAc al 0-50 %/heptano para proporcionar 80 mg del producto deseado: RMN 1H (400 MHz, CDCl3) 87,22 (s, 1H), 7,11 (s, 2H), 5,47 -5,27 (m, 1H), 4,92 (t, J = 7,2 Hz, 2H), 4,57 (d, J = 6,4 Hz, 1H), 4,16 -3,76 (m, 3H), 3,49 -3,12 (m, 4H), 1,74 (d, J = 46,8 Hz, 1H), 1,41 (s, 3H), 1,08 (s, 9H).
Formación de 3-(2-cloro-4-(oxetan-3-il)fenil)-1,4-oxazepano (287)
A una solución de 3-[2-cloro-4-(oxetan-3-il)fenil]-1,4-oxazepano-4-carboxilato de ferc-butilo, 286, (0,080 g, 0,220 mmol) en diclorometano (5 ml), se le añadió ácido trifluoroacético (0,20 ml, 2,60 mmol). La mezcla de reacción se agitó durante 1 hora a temperatura ambiente. La mezcla se concentró al vacío. La sal TFA en bruto se convirtió en su forma original pasándola a través de un cartucho PL-HCO3 MP SPE con MeOH como disolvente. El filtrado se concentró al vacío. El producto en bruto se llevó a la siguiente etapa sin purificación adicional: IEN-EM m/z calc. 267,1 encontrado 268,2 (M+1)+; Tiempo de retención: 0,66 minutos.
Formación de (+/-)-2-(4-(4-(2-amino-6-metilpirimidin-4-il)-1,4-oxazepan-3-il)-3-clorofenil)propano-1,3-diol (288) 1- 236
Una mezcla de 3-[2-cloro-4-(oxetan-3-il)fenil]-1,4-oxazepano, 287, (0,050 g, 0,190 mmol) y 4-cloro-6- metil-pirimidin-2- amina (0,035 g, 0,240 mmol) en n-BuOH (5 ml) se calentó durante una noche a 130 °C. La CL-EM mostró un producto de apertura del anillo de oxeteno. La mezcla se concentró al vacío. El residuo resultante se purificó por cromatografía sobre gel de sílice usando una columna ISCO de 4 g eluyendo con MeOH al 0-10 %/diclorometano para proporcionar 11,4 mg del producto deseado: RMN 1H (400 MHz, CDCl3) 87,30 (s, 1H), 7,23 - 7,10 (m, 2H), 5,58 (s, 1H), 4,89 (s, 2H), 4,35 (dd, J = 13,6, 5,1 Hz, 1H), 4,10 (dd, J = 12,3, 4,8 Hz, 1H), 4,02 - 3,84 (m, 4H), 3,67 - 3,53 (m, 2H), 3,13 -2,89 (m, 1H), 2,14 (s, 3H), 2,08 - 1,95 (m, 1H), 1,83 (d, J = 14,4 Hz, 1H), 1,38-1,14 (m, 1H); IEN-EM m/z calc. 392,2, encontrado 393,0 (M+1)+; Tiempo de retención: 2,82 minutos. Los siguientes análogos estaban de acuerdo con el Esquema sintético 3:

(+/-)-4-(2-(2-bromofeml)azepan-1-M)-6,7-dihidro-5H-ciclopenta[c/]pirimidm-2-amma I-7
RMN 1H (400 MHz, MeOD) 87,58 (s, 1H), 7,29 (d, J = 65,1 Hz, 3H), 5,61 (d, J = 166,6 Hz, 2H), 4,54 (s, 1H), 3,82 (s, 1H), 3,48 (s, 1H), 3,27 -3,21 (m, 1H), 3,20-1,31 (m, 12H); IEN-EM m/z calc. 272,16, encontrado 272,92 (M+1)+; Tiempo de retención: 0,58 minutos.

(+/-)-4-(2-(2-bromofenil)azepan-1-il)-5,6-dimetilpirimidin-2-amina I-8
RMN 1H (400 MHz, MeOD) 87,60 (d, J = 8,0 Hz, 1H), 7,33 (c, J = 7,7 Hz, 2H), 7,15 (dd, J = 11,3, 5,2 Hz, 1H), 5,51 (s, 1H), 4,49 (s, 1H), 3,90 - 3,51 (m, 1H), 2,62 (s, 1H), 2,27 (s, 3H), 2,08 (s, 3H), 1,84 (dd, J = 33,6, 27,8 Hz, 5H), 1,59 1,37 (m, 2H); Ie N-EM m/z calc. 375,0, encontrado 375,1 (M+1)+; Tiempo de retención: 2,95 minutos.

4-metil-6-(2-(2-(metilsulfonil)fenil)azepan-1-il)pirimidin-2-amina I-38
alta temperatura (360 K) RMN 1H (400 MHz, DMSO-c/6) 87,93 (dd, J = 8,0, 1,4 Hz, 1H), 7,61 (td, J = 7,6, 1,5 Hz, 1H), 7,47 (td, J = 7,6, 1,3 Hz, 1H), 7,38 (dd, J = 7,9, 1,3 Hz, 1H), 6,05 - 5,80 (m, 1H), 5,65 (d, J = 11,6 Hz, 1H), 5,56 - 5,29 (m, 2H), 4,14 - 3,80 (m, 1H), 3,60 (s, 3H), 2,66 - 2,54 (m, 1H), 2,05 (s, 3H), 1,97 - 1,76 (m, 2H), 1,81 - 1,54 (m, 2H), 1,56 -1,11 (m, 4H).

4-[2-(2,6-dimetoxi-3-piridil)azepan-1-il]-6-metil-pirimidin-2-amina, I-73
RMN 1H (300 MHz, DMSO-d6) 87,37 (a, 1H), 7,11 (s, 2H), 6,31 (s, 1H), 5,66 (a, 1H), 4,73 (a, 1H), 4,02 -3,72 (m, 6H), 3,68 - 3,01 (m, 2H), 2,17 (s a, 3H), 1,98-0,93 (m, 8H); IEN-EM m/z calc. 343,20, encontrado 344,35 (M+1)+; Tiempo de retención: 0,72 minutos.

4-(3-(5-fluoro-2-metoxifenil)-2-azabiciclo[3.2.2]nonan-2-il)-6-metilpirimidin-2-amina I-76
alta temperatura (360 K) RMN 1H (400 MHz, DMSO-c/6) 86,99 (dd, J = 9,0, 4,6 Hz, 1H), 6,95 - 6,81 (m, 2H), 5,65 (s, 1H), 5,53 -5,33 (m, 2H), 5,26 -5,11 (m, 1H), 3,88 (s, 3H), 2,45 -2,31 (m, 1H), 2,23 -2,11 (m, 1H), 2,10 -1,96 (m, 2H), 1,91 (s, 3H), 1,88 - 1,74 (m, 1H), 1,73-1,42 (m, 7H); IEN-EM m/z calc. 356,20, encontrado 357,25 (M+1)+; Tiempo de retención: 0,68 minutos.

(+/-)-4-metil-6-[2-(4-metilpirimidin-5-il)azepan-1-il]pirimidin-2-amina I-42
alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6) 88,81 (s, 1H), 8,38 (s, 1H), 5,74 (s, 1H), 5,41-5,33 (m, 3H), 4,10 (d, J = 12,0 Hz, 1H), 3,52 (dd, J = 15,0, 11,0 Hz, 2H), 2,62 (s, 3H), 2,25 -2,14 (m, 1H), 2,04 (s, 3H), 1,93 (s, 1H), 1,87 - 1,72 (m, 2H), 1,56-1,28 (m, 4H); IEN-EM m/z calc. 298,19, encontrado 299,19 (M+1)+; Tiempo de retención: 0,53 minutos.

3-(1-(2-ammo-6-met¡lp¡r¡m¡dm-4-¡l)azepan-2-¡l)-2-fluoro-W-met¡lbenzam¡da I-54
RMN 1H (400 MHz, DMSO-d6) 57,87 (s, 1H), 7,43 (td, J = 7,2, 1,9 Hz, 1H), 7,22 (td, J = 7,5, 1,9 Hz, 1H), 7,12 (t, J = 7,6 Hz, 1H), 5,72 (s, 1H), 5,40 (s, 2H), 4,20 (d, J = 14,4 Hz, 1H), 3,42 - 3,29 (m, 1H), 2,97 (s, 1H), 2,80 (d, J = 4,7 Hz, 3H), 2,43 -2,29 (m, 1H), 2,03 (s, 3H), 1,97 -1,71 (m, 4H), 1,67 - 1,23 (m, 3H). IEN-EM m/z calc. 357,1965, encontrado 358,0 (M+1)+; Tiempo de retención: 0,66 minutos.
Ejemplo 40
Esquema sintético 40: (+/-)-4-(3-(2-clorofen¡l)-6-met¡leno-1,4-oxazepan-4-¡l)-6-met¡lp¡r¡m¡d¡n-2-am¡na I-75

(a) 3-doro-2-(dorometil)prop-1-eno, LiI, NaH, DMF, ta, después 50 °C; (b) TFA, CH2CI2; (c) NH4OH, CH3CN, 70 °C Formac¡ón de 3-(2-clorofen¡l)-6-met¡leno-1,4-oxazepano-4-carbox¡lato de (+/-)-íerc-but¡lo
A una solución de W-[1-(2-clorofenil)-2-hidroxi-etil]carbamato de ferc-butilo (5,00 g, 18,40 mmol), 3-cloro-2-(clorometil)prop-1-eno (2,60 g, 20,80 mmol) y yoduro de litio (0,11 g, 0,84 mmol) en DMF (200 ml), se le añadió un equivalente de NaH en pociones (0,75 g, 18,62 mmol). La mezcla se agitó a temperatura ambiente durante 15 horas. Después se añadió el 2° equivalente de NaH (0,75 g, 18,62 mmol). Después de 24 horas, se calentó la mezcla de reacción a 50 °C durante 3 días. La mezcla de reacción se diluyó en una solución acuosa saturada de NH4Cl y se extrajo dos veces con EtOAc. Las fases orgánicas combinadas se lavaron dos veces con salmuera, se secaron (MgSO4), se filtraron y se concentró al vacío. El residuo resultante se purificó por cromatografía sobre gel de sílice con una columna ISCO de 80 g usando 0-30 % (EtOAc/heptanos): RMN 1H (400 MHz, DMSO-d6) 57,50 - 7,25 (m, 4H), 5,50 -5,19 (m, 1H), 5,00 (s, 2H), 4,72 -4,23 (m, 1H), 4,16 (s, 2H), 4,09 -3,74 (m, 3H), 1,54 - 1,02 (m, 9H).
Formac¡ón de (+/-)-3-(2-clorofen¡l)-6-met¡leno-1,4-oxazepano
A una solución de 3-(2-clorofenil)-6-metileno-1,4-oxazepano-4-carboxilato de ferc-butilo (0,76 g, 2,23 mmol) en diclorometano (8 ml), se le añadió ácido trifluoroacético (6 ml). Se agitó la mezcla de reacción a temperatura ambiente durante 1 hora y se concentró al vacío. El residuo se diluyó con diclorometano y se neutralizó con una solución acuosa saturada de NaHCO3. La fase orgánica se pasó a través de un separador de fase y el filtrado resultante se concentró al vacío para proporcionar 485 mg del producto en forma de un aceite de color amarillo.
Formac¡ón de (+/-)-4-(3-(2-clorofen¡l)-6-met¡leno-1,4-oxazepan-4-¡l)-6-met¡lp¡r¡m¡d¡n-2-am¡na I-75
A una solución de 3-(2-clorofenil)-6-metileno-1,4-oxazepano en (0,49 g, 2,17 mmol) en NMP (6 ml), se le añadió 4-cloro-6-metil-pirimidin-2-amina (0,38 g, 2,61 mmol). La mezcla de reacción se calentó a 150 °C durante 16 horas. La mezcla se enfrió a temperatura ambiente y se cargó directamente en una columna ISCO c18-aq de 100 g y el material en bruto se purificó por fase inversa eluyendo con un gradiente de TFA al 0,1 %/H2O y TFA al 0,1 %/Ch 3CN. Las fracciones que contenían el producto deseado se concentraron al vacío se diluyeron con diclorometano, se neutralizaron con una solución acuosa saturada de NaHCO3 y la mezcla se pasó a través de un separador de fase. La fase orgánica se concentró al vacío para proporcionar 575 mg de sólido de color naranja: Se calentó RMN 1H (360K) (400 MHz, DMSO-d6) 57,69 - 7,52 (m, 1H), 7,50 - 7,36 (m, 1H), 7,36 - 7,20 (m, 2H), 5,76 (s, 1H), 5,73 (d, J = 2,2 Hz, 1H), 5,52 (s, 2H), 5,05 (d, J = 51,9 Hz, 2H), 4,72 (d, J = 16,0 Hz, 1H), 4,29 -4,09 (m, 2H), 4,08 -3,97 (m, 2H), 3,92 (d, J = 16,0 Hz,
Ejemplo 41
Esquema s¡ntét¡co 41: (+/-)-(3-(4-(2-am¡no-6-met¡lp¡r¡m¡d¡n-4-¡l)-1,4-oxazepan-3-¡l)-4-clorofen¡l)(¡m¡no)(met¡l)-A6-sulfanona (I-156)
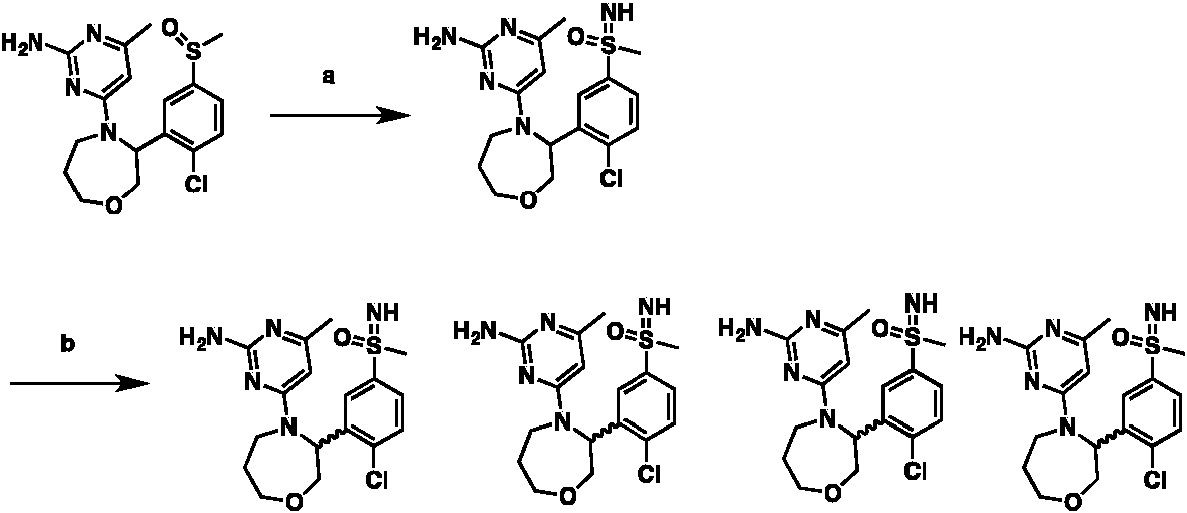
(a) PhI(OAc)2, (NH4)2CO3; MeOH; (b) separación quiral SFC
Formación de (+/-)-(3-(4-(2-ammo-6-metilpirimidm-4-il)-1,4-oxazepan-3-il)-4-clorofeml)(immo)(metil)-A6-sulfanona I-156
Se combinaron 4-[3-(2-doro-5-metilsulfinil-fenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (0,25 g, 0,59 mmol), carbamato de amonio (0,18 g, 2,34 mmol) y (diacetoxiyodo)benceno (0,57 g, 1,78 mmol) en un matraz de fondo redondo seguido de la adición de MeOH (4 ml) y se continuó agitando durante 75 minutos. La mezcla de reacción se diluyó en EtOAc y se lavó con una solución acuosa saturada de NaHCO3. La fase acuosa se extrajo con EtOAc. Las fases orgánicas combinadas se secaron (MgSO4), se filtraron y se concentraron al vacío. El residuo en bruto se purificó por cromatografía de fase inversa sobre gel de sílice con una columna ISCO c18-aq de 50 g ejecutándose con TFA al 0,1 %/H2O y TFA al 0,1 %/CH3CN. Las fracciones puras se concentraron al vacío y el residuo se diluyó con diclorometano y se neutralizó con una solución acuosa saturada de NaHCO3. La mezcla se pasó a través de un separador de fase y el filtrado se concentró al vacío para proporcionar 27 mg del producto racémico deseado en forma de un sólido de color amarillo claro: Se calentó RMN (360K) RMN 1H (400 MHz, DMSO-d6) 67,90 - 7,70 (m, 2H), 7,63 (dd, J = 14,2, 8,3 Hz, 1H), 6,35 -5,32 (m, 4H), 4,91 -4,41 (m, 1H), 4,18 -3,50 (m, 5H), 3,06 -3,00 (m, 3H), 2,12 -1,99 (m, 3H), 1,87 - 1,64 (m, 2H).
La mezcla racémica se sometió a separación SFC quiral
Pico A, 2,9 mg (89,8% ee). 4-[3-[2-cloro-5-(metilsulfonimidoil)fenil]-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (2,9 mg, 5 %): IEN-EM m/z calc. 395,12, encontrado 396,0 (M+1)+; Tiempo de retención: 0,52 minutos. I-164
Pico B, 4,3 mg (97,4% ee). 4-[3-[2-cloro-5-(metilsulfonimidoil)fenil]-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (4,3 mg, 7 %): IEN-EM m/z calc. 395,12, encontrado 396,0 (M+1)+; Tiempo de retención: 0,52 minutos. I-165
Pico C, 3,2 mg (98 % ee). 4-[3-[2-cloro-5-(metilsulfonimidoil)fenil]-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (3,2 mg, 5 %): IEN-EM m/z calc. 395,12, encontrado 396,0 (M+1)+; Tiempo de retención: 0,52 minutos. I-166
Pico D, 4,7 mg (98 % ee). 4-[3-[2-cloro-5-(metilsulfonimidoil)fenil]-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (4,7 mg, 8 %): IEN-EM m/z calc. 395,12, encontrado 396,0 (M+1)+; Tiempo de retención: 0,52 minutos. I-167
Ejemplo 42
Esquema sintético 42: (+/-)-2-[4-(2-amino-6-metil-pirimidin-4-il)-1,4-oxazepan-3-il]benzonitrilo, (+/-)-4-[3-[2-(aminometil)fenil]-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina y (+/-)-N-[[2-[4-(2-ammo-6-metil-pirimidm-4-il)-1,4-oxazepan-3-il]fenil]metil]acetamida

Formación de (+/-)-2-[4-(2-amino-6-metil-pirimidin-4-il)-1,4-oxazepan-3-il]benzonitrilo I-89
A una solución de [2-[4-(2-amino-6-metil-pirimidin-4-il)-1,4-oxazepan-3-il]fenil]metanol (0,16 g, 0,52 mmol) en THF (3 ml), se le añadió amoniaco (2 ml de solución 2 M, 4,00 mmol), después MgSO4 anhidro (1,07 g, 8,89 mmol) y finalmente
MnO2 (135 |jl, 7,78 mmol). A la mañana siguiente, se añadieron 1 g más de MgSO4, 1 g más de MnO2 y amoniaco (4 ml de 2 M, 8,000 mmol) y la mezcla se calentó a 45 °C. Después de 1 h, la mezcla de reacción se filtró a través de Celite con la ayuda de MeOH al 10 % en EtOAc y se concentró al vacío. Se añadió éter y 160 mg del producto deseado se retiraron por filtración: alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6) 67,76 (d, J = 7,7 Hz, 1H), 7,63 (t, J = 7,6 Hz, 1H), 7,49 (d, J = 7,5 Hz, 1H), 7,43 (t, J = 7,6 Hz, 1H), 5,75 (s, 1H), 5,55 (dd, J = 10,6, 5,2 Hz, 1H), 5,39 (s, 2H), 4,38 (d, J = 14,8 Hz, 1H), 4,11 (dd, J = 13,6, 5,1 Hz, 1H), 3,95 - 3,83 (m, 2H), 3,74 (dd, J = 15,3, 12,7 Hz, 1H), 3,58 (dd, J = 14,6, 12,1 Hz, 1H), 2,04 (s, 3H), 1,83-1,75 (m, 2H); IEN-EM m/z calc. 309,16, encontrado 310,22 (M+1)+; Tiempo de retención: 0,55 minutos.
Formación de (+/-)-4-[3-[2-(aminometil)fenil]-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina I-94
A una solución de 2-[4-(2-amino-6-metil-pirimidin-4-il)-1,4-oxazepan-3-il]benzonitrilo (0,055 g, 0,177 mmol) en THF (3 ml) con enfriamiento en baño de hielo, se le añadió LiAlH4 (0,034 g, 0,873 mmol) en atmósfera de nitrógeno y la mezcla se agitó durante una noche a temperatura ambiente. Adición cuidadosa gota a gota de agua y después se añadió diclorometano. La mezcla de reacción se filtró a través de Celite con la ayuda de una solución acuosa saturada de bicarbonato sódico. Las capas se separaron y la capa acuosa se extrajo de nuevo con diclorometano y se concentraron al vacío. El residuo se cargó directamente sobre una columna ISCO de ISCO c18-aq de 30 g y se purificó por fase inversa ejecutándose con TFA al 0,1 %/H2O y TFA al 0,1 %/CH3CN. Las fracciones puras se concentraron al vacío y después se disolvieron en MeOH y se pasó a través de un cartucho de bicarbonato SPE (Agilent Stratospheres 500 mg/6 ml) y se concentraron al vacío para dar un aceite de color rosa. Se añadió éter y la mezcla se sonicó y 43 mg del producto deseado se retiraron por filtración: alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6,) 57,37 (d, J = 7.1 Hz, 1H), 7,25 (d, J = 7,5 Hz, 1H), 7,17 (tt, J = 7,4, 5,9 Hz, 2H), 5,82 (s, 1H), 5,54 (d, J = 4,6 Hz, 1H), 5,37 (s, 2H), 4,47 (d, J = 14,1 Hz, 1H), 4,08 (dd, J = 13,5, 4,9 Hz, 1H), 4,00 - 3,80 (m, 3H), 3,75 (dd, J = 13,5, 9,7 Hz, 1H), 3,59 (ddd, J = 16,7, 14,1,7,3 Hz, 2H), 2,00 (s, 3H), 1,76 (s, 4H); IEN-EM m/z calc. 313,19, encontrado 314,23 (M+1)+; Tiempo de retención: 0,48 minutos.
Formación de (+/-)-N-[[2-[4-(2-amino-6-metil-pirimidin-4-il)-1,4-oxazepan-3-il]fenil]metil]acetamida I-95
A una solución de 4-[3-[2-(aminometil)fenil]-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (0,015 g, 0,049 mmol) en diclorometano (2 ml), se le añadió trietilamina (0,035 ml, 0,251 mmol), después anhídrido acético (0,006 ml, 0,064 mmol) a temperatura ambiente. Después de 20 minutos, se añadieron una solución acuosa saturada de bicarbonato sódico y diclorometano y las capas se separaron. La capa acuosa se extrajo de nuevo con diclorometano. Las capas se separaron con la ayuda de un separador de fase y los orgánicos se concentraron al vacío para dar 2,8 mg del producto deseado: alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6) 68,07 (s a, 1H), 7,28 -7,13 (m, 4H), 5,75 (s, 1H), 5,53 (s, 2H), 5,48-5,44 (m, 1H), 4,76 (dd, J = 15,2, 5,8 Hz, 1H), 4,42-4,22 (m, 2H), 3,98 (dd, J = 13,6, 4,9 Hz, 1H), 3,87 (d, J = 11,8 Hz, 1H), 3,78 - 3,64 (m, 2H), 3,61 - 3,52 (m, 1H), 2,02 (s, 3H), 1,92 (s, 3H), 1,76 (dd, J = 8,2, 4.2 Hz, 2H); IEN-EM m/z calc. 355,20, encontrado 356,28 (M+1)+; Tiempo de retención: 0,53 minutos.
Ejemplo 43
Esquema sintético 43: (+/-)-3-[4-(2-amino-6-metil-pirimidin-4-il)-1,4-oxazepan-3-il]-4-cloro-benzonitrilo. I-65

(a) DMF, POCla; (b) ácido 2-cloro-5-cianofenilo borónico, PdCh(PPh3)2, EtaN, DMF, 70 °C; (c) nBuLi, 2-MeTHF, -78 °C; (d) NaBH4, 2-MeTHF, MeOH; (e) 4-cloro-6-metil-pirimidin-2-amina, NMP, 160 °C.
Formación de 3-cloro-6,7-dihidro-1,4-oxazepina-4(5H)-carbaldehído
Un matraz de fondo redondo de 2 l y 3 bocas equipado con un agitador superior, sonda de temperatura, embudo de adición, entrada de nitrógeno y condensador de reflujo se cargó con DMF (150 ml, 1,94 mol) en diclorometano (300 ml). La mezcla se agitó durante 5 minutos y después se enfrió a 0 °C. Se añadió POCl3 (90 ml, 0,97 mol) en diclorometano (100 ml) durante 30 minutos mientras que se mantenía la temperatura interna por debajo de 6 °C. La
mezcla de reacción se calentó a 40 °C y se agitó a esta temperatura durante 45 minutos. Se añadió 1,4-oxazepan-3-ona (50 g, 0,43 mol) en diclorometano (300 ml) durante 40 minutos, se observó exotermia, se mantuvo la temperatura interna ~40 °C. La mezcla de reacción resultante se agitó a esta temperatura durante 90 minutos, momento en el cual la TLC (metanol al 10 % en diclorometano) y el análisis de CLEM revelaron el consumo del material de partida, pico principal Tr = 0,51 minutos (M+H)+189/191 que corresponde al intermedio de amidina. La mezcla de reacción se enfrió a temperatura ambiente, se vertió en hielo triturado (1,2 l) y después se dejó calentar a temperatura ambiente durante 1 hora y se agitó durante 1 hora más. Se separó la capa acuosa, se basificó con K2CO3 sólido hasta pH 9, se dejó a temperatura ambiente, se agitó a esta temperatura durante 12 horas. La mezcla de reacción se diluyó con diclorometano (300 ml) y la capa orgánica se separó. La capa acuosa se extrajo con diclorometano (2 x 100 ml). Los extractos orgánicos combinados se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida. El residuo se purificó por cromatografía sobre gel de sílice (columna isco de 330 g de gradiente lineal, 20 CV, acetato de etilo al 0% ^ 50 %/heptano-que contenía Et3N al 1%), para proporcionar 3-cloro-6,7-dihidro-5H-1,4-oxazepina-4-carbaldehído (40 g, 57 %) en forma de un aceite incoloro transparente que contenía trazas de DMF
Formación de 4-cloro-3-(4-formil-4,5,6,7-tetrahidro-1,4-oxazepin-3-il)benzonitrilo
Se cargó un vial de 20 ml con un tapón de alivio de presión en atmósfera de nitrógeno con ácido (2-cloro-5-cianofenil)borónico (2,98 g, 16,43 mmol), 3-cloro-6,7-dihidro-5H-1,4-oxazepina-4-carbaldehído (2,00 g, 11,76 mmol), PdCl2(PPh3)2 (0,40 g, 0,58 mmol), DMF (9 ml) y trietilamina (5,0 ml, 35,9 mmol). La mezcla se calentó a 70 °C durante una noche. Se añadieron agua y salmuera y EtOAc y después se separaron las capas. La capa acuosa se extrajo de nuevo con EtOAc y los extractos orgánicos combinados se lavaron con agua (x 3), se secaron (MgSO4), se filtraron y se concentraron al vacío. La purificación por cromatografía sobre gel de sílice (columna ISCO de 120 g; EtOAc al 0 100 % en heptano) para proporcionar el producto deseado en forma de un sólido de color verde pálido (1,5 g, 49 %): RMN 1H (400 MHz, ACN) 87,85 (s, 1H), 7,77 (d, J = 2,0 Hz, 1H), 7,68 (dd, J = 8,3, 2,1 Hz, 1H), 7,60 - 7,57 (m, 1H), 6,19 (s, 1H), 4,28 - 4,24 (m, 2H), 4,00 (t, J = 6,6 Hz, 2H), 2,11-2,05 (m, 2H); IEN-EM m/z calc. 262,05, encontrado 263,08 (M+1)+; Tiempo de retención: 0,74 minutos.
Formación de 4-cloro-3-(2,5,6,7-tetrahidro-1,4-oxazepin-3-il)benzonitrilo
A una solución de 4-cloro-3-(4-formil-6,7-dihidro-5H-1,4-oxazepin-3-il)benzonitrilo (1,5 g, 5,7 mmol) en 2- MeTHF (23 ml) a -78 °C, se le añadió nBuLi (4,7 ml de 1,6 M, 7,5 mmol) en una atmósfera de nitrógeno durante 2 minutos. Se añadió agua y después se retiró el baño frío. Se extrajo con diclorometano, se separaron las capas con la ayuda de un separador de fase. La capa acuosa se extrajo de nuevo con diclorometano y las capas se separaron a través de un separador de fase de nuevo y los orgánicos se concentraron para proporcionar el producto deseado (1,34 g, 100 %): IEN-EM m/z calc. 234,06, encontrado 235,11 (M+1)+; Tiempo de retención: 0,74 minutos.
Formación de (+/-)-4-cloro-3-(1,4-oxazepan-3-il)benzonitrilo y (+/-)-3-(1,4-oxazepan-3-il)benzonitrilo
A una solución de 4-cloro-3-(2,5,6,7-tetrahidro-1,4-oxazepin-3-il)benzonitrilo (1,34 g, 5,71 mmol) en 2-MeTHF (15 ml), se le añadió NaBH4 (2,16 g, 57,10 mmol) a temperatura ambiente durante una noche. La mezcla de reacción se enfrió en un baño de hielo y se añadió MeOH (5 ml). Después de 10 minutos, el baño de refrigeración se retiró y la agitación se continuó a temperatura ambiente. La reacción se calentó a 50 °C durante una noche. Se añadieron agua y K2CO3 sólido en exceso y la mezcla se concentró parcialmente. Se añadió diclorometano y la agitación se continuó durante una noche. Las capas se separaron con la ayuda de un separador de fase. La capa acuosa se extrajo de nuevo con diclorometano y las capas se separaron a través de un separador de fase de nuevo y los orgánicos combinados se concentraron al vacío. El residuo en bruto se purificó por cromatografía sobre gel de sílice con una columna isco de 40 g usando un gradiente de EtOAc al 0-100 %/heptano seguido de MeOH al 0-30 %/diclorometano para proporcionar 283 mg del producto deseado en forma de un aceite de color amarillo pálido: RMN 1H (400 MHz, d Ms O-c(6) 88,01 (d, J = 2,1 Hz, 1H), 7,75 (dt, J = 6,2,3,1 Hz, 1H), 7,64 (d, J = 8,3 Hz, 1H), 4,26 (d, J = 5,8 Hz, 1H), 3,84 - 3,78 (m, 2H), 3,71 (m, 1H), 3,33 (m, 1H), 3,08 (m, 1H), 2,92 -2,83 (m, 1H) 1,86 (m, 2H); IEN-EM m/z calc. 236,07, encontrado 237,14 (M+1)+; Tiempo de retención: 0,52 minutos.
También se aisló un producto secundario, 3-(1,4-oxazepan-3-il)benzonitrilo después de la repurificación de algunas de las fracciones: columna de fase inversa ISCO c18-aq de 100 g ejecutándose con TFA al 0,1 %/H2O y TFA al 0,1 %/CH3CN y las fracciones puras se concentraron al vacío. El residuo se disolvió en MeOH y se pasó a través de un cartucho de bicarbonato SpE (Agilent Stratospheres 500 mg/6 ml) y se concentró para dar 64 mg del producto deseado en forma de un aceite incoloro. RMN 1H (400 MHz, DMSO-d6) 87,81 (d, J = 1,7 Hz, 1H), 7,72 -7,67 (m, 2H), 7,51 (t, J = 7,7 Hz, 1H), 3,93 (dd, J = 9,0, 3,4 Hz, 1H), 3,85 -3,79 (m, 2H), 3,69 (dd, J = 12,4, 6,1 Hz, 1H), 3,35 (dd, J = 12,2, 9,1 Hz, 1H), 3,08 -3,01 (m, 1H), 2,87 -2,79 (m, 1H), 1,85 (m, 2H); IEN-EM m/z calc. 202,11, encontrado 203,14 (M+1)+; Tiempo de retención: 0,5 minutos.
Formación de (+/-)-3-[4-(2-amino-6-metil-pirimidin-4-il)-1,4-oxazepan-3-il]-4-cloro-benzonitrilo I-65
Una mezcla de 4-cloro-3-(1,4-oxazepan-3-il)benzonitrilo (0,078 g, 0,333 mmol) y 4-cloro-6-metil-pirimidin-2-amina (0,047 g, 0,330 mmol) en NMP (0,60 ml) se calentó a 160 °C en un vial de centelleo sobre un bloque calefactor. La mezcla se enfrió a temperatura ambiente y se cargó directamente en una columna ISCO c18-aq de 50 g y se purificó
por fase inversa ejecutándose con TFA al 0,1 %/H2O y TFA al 0,1 %/CH3CN. Las fracciones puras se concentraron parcialmente al vacío. Se añadió NaOH 1 M y la mezcla se extrajo con diclorometano dos veces. Las capas se separaron con la ayuda de un separador de fase y los orgánicos se concentraron al vacío. La trituración con éter dio 64 mg del producto deseado en forma de un sólido de color blanco: alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6 ) 67,72 - 7,61 (m, 3H), 5,69 (s, 1H), 5,55 (dd, J = 9,9, 4,9 Hz, 1H), 5,41 (s, 2H), 4,46 (d, J = 15,8 Hz, 1H), 4,08 (dd, J = 13,5, 4,9 Hz, 1H), 3,92 -3,85 (m, 1H), 3,84 -3,72 (m, 2H), 3,61 -3,53 (m, 1H), 2,03 (s, 3H), 1,83-1,75 (m, 2H); IEN-EM m/z calc. 343,12, encontrado 344,17 (M+1)+; Tiempo de retención: 0,59 minutos.
Ejemplo 44
Esquema sintético 44: (+/-)-4-[2-(2-metoxi-3-piridil)-5,5-dimetil-azepan-1-il]-6-metil-pirimidin-2-amina I-64

(a) POCla, CH2O 2; (b) ácido (2-metoxi-3-piridil)borónico, PdC^PPhah Et3N, DMF; (c) n-BuLi, THF, -78 °C; (d) UBH4, THf ; (e) 2-amino-4-cloro-6-metilpirimidina, NMP, 150 °C
Formación de (+/-)-7-cloro-4,4-dimetil-2,3,4,5-tetrahidro-1H-azepina-1-carbaldehído
Un matraz de fondo redondo de 250 ml con agitador magnético en una atmósfera de nitrógeno se cargó con DMF (8,2 ml, 106 mmol) en diclorometano (15 ml) y se enfrió a 0 °C en un baño de hielo. Se añadió POCh (5 ml, 53,6 mmol) en diclorometano (10 ml) durante 5 minutos. La mezcla de reacción se calentó a 40 °C y se agitó a esta temperatura durante 30 minutos. Se añadió 5,5-dimetilazepan-2-ona (2,5 g, 17,7 mmol) en diclorometano (20 ml) durante 10 minutos. La mezcla de reacción resultante se agitó a esta temperatura durante 4 horas. La mezcla de reacción se enfrió a temperatura ambiente, se vertió en hielo picado (200 ml), y después, se dejó calentar a temperatura ambiente durante 1 h y se agitó durante un adicional de 1 h. La capa acuosa se basificó con K2CO3 sólido hasta pH 9-10 y se agitó a temperatura ambiente durante el fin de semana. La mezcla de reacción se diluyó con diclorometano (200 ml) y la capa orgánica se separó. La capa acuosa se extrajo con diclorometano. Los extractos orgánicos combinados se lavaron con salmuera, se separaron a través de un separador de fase y se concentró al vacío. El residuo se purificó por cromatografía sobre gel de sílice usando una columna isco de 80 g (gradiente de EtOAc al 0-40 %/CH2Cl2) para proporcionar 1,56 gramos del producto deseado: RMN 1H (400 MHz, DMSO-d6 ) 68,53 - 8,04 (m, 1H), 5,92 - 5,77 (m, 1H), 3,56 - 3,38 (m, 2H), 2,05 - 1,82 (m, 2H), 1,63 - 1,39 (m, 2H), 0,97 - 0,83 (m, 6H).
Formación de (+/-)-7-(2-metoxi-3-piridil)-4,4-dimetil-3,5-dihidro-2H-azepina-1-carbaldehído
Una mezcla de ácido (2-metoxi-3-piridil)borónico (0,86 g, 5,61 mmol), 7-cloro-4,4-dimetil-3,5-dihidro-2H-azepina-1-carbaldehído (0,74 g, 3,74 mmol), PdCl2(PPh3)2 (128 mg, 0,182 mmol), DMF (8 ml), NEt3 (2 ml, 14,4 mmol) se calentó en un vial de 20 ml con un tapón de alivio de presión en atmósfera de nitrógeno a 70 °C durante 2 h. Se añadieron agua y EtOAc y las capas se separaron. La capa acuosa se extrajo de nuevo con EtOAc y los extractos orgánicos combinados se lavaron con agua, después salmuera, se secaron (sulfato sódico), se filtraron y se concentraron al vacío. La purificación por cromatografía sobre gel de sílice (columna GOLD de 40 g; EtOAc al 0-75 % en heptano) dio 750 mg del producto deseado en forma de un aceite de color pardo: RMN 1H (400 MHz, CD3CN) 68,14 (dd, J = 5,0, 1,9 Hz, 1H), 7,88 (s, 1H), 7,61 (dd, J = 7,4, 1,9 Hz, 1H), 6,97 (dd, J = 7,3, 5,0 Hz, 1H), 5,92 (t, J = 7,1 Hz, 1H), 3,74 -3,68 (m, 2H), 2,17 (m, 2H), 1,67 - 1,63 (m, 2H), 1,01 (s, 6 H). IEN-EM m/z calc. 260,15, encontrado 261,18 (M+1)+; Tiempo de retención: 0,85 minutos.
Formación de 7-(2-metoxi-3-piridil)-4,4-dimetil-2,3,5,6-tetrahidroazepina
A una solución de 7-(2-metoxi-3-piridil)-4,4-dimetil-3,5-dihidro-2H-azepina-1-carbaldehído (0,75 g, 2,88 mmol) en THF (5 ml) a -78 °C, se le añadió en atmósfera de nitrógeno, n-BuLi (2,3 ml de solución 1,6 M, 3,68 mmol). Después de 15 minutos, se añadió un adicional de 1 ml de n-BuLi. Después de 5 minutos, la reacción se interrumpió con agua y se lavó a temperatura ambiente. Se añadió salmuera y la mezcla se extrajo con diclorometano. La concentración al vacío proporcionó 669 mg del producto deseado en forma de un aceite de color pardo: IEN-EM m/z calc. 232,16, encontrado 233,17 (M+1)+; Tiempo de retención: 0,57 minutos.
Formación de 2-(2-metoxi-3-piridil)-5,5-dimetil-azepano
A una solución de 7-(2-metoxi-3-piridil)-4,4-dimetil-2,3,5,6-tetrahidroazepina (300 mg, 1,29 mmol) en THF (3 ml), se le añadió LiBH4 (250 mg, 11,5 mmol) a TA y la mezcla se agitó durante una noche. Se añadió HCl 1 M (1 ml) y la mezcla se extrajo con DCM dos veces. Las capas se separaron con la ayuda de un separador de fase y los orgánicos se concentraron para dar 72 mg del producto deseado en forma de un aceite de color amarillo pálido: RMN 1H (400 MHz, DMSO) 57,99 (dd, J = 4,9, 1,9 Hz, 1H), 7,82 - 7,77 (m, 1H), 6,93 (dd, J = 7,3, 4,9 Hz, 1H), 4,00 (dd, J = 8,1, 3,5 Hz, 1H), 3,86 (s, 3H), 2,85 - 2,78 (m, 1H), 2,75 - 2,65 (m, 1H), 1,74 (ddd, J = 10,4, 7,8, 3,9 Hz, 1H), 1,52 - 1,38 (m, 5H), 0,92 (s, 3H), 0,91 (s, 3H). Ie N-e M m/z calc. 234,17, encontrado 235,2 (M+1)+; Tiempo de retención: 0,57 minutos
Formación de 4-[2-(2-metoxi-3-piridil)-5,5-dimetil-azepan-1 -il]-6-metil-pirimidin-2-amina
Una mezcla de 2-(2-metoxi-3-piridil)-5,5-dimetil-azepano (0,21 g, 0,88 mmol) y 4-cloro-6-metil-pirimidin-2-amina (0,11 g, 0,77 mmol) en Nm P (1,3 ml), se calentó en un microondas a 175 °C durante 30 minutos. La purificación se realizó en una columna ISCO c18-aq de 50 g de fase inversa, ejecutándose con TFA al 0,1 %/H2O y TFA al 0,1 %/CH3CN. Las fracciones puras se concentraron parcialmente, se añadió algo de NaOH 1 M y se extrajo con diclorometano dos veces. Las capas se separaron con la ayuda de un separador de fase y los orgánicos se concentraron al vacío para proporcionar 18 mg del producto deseado en forma de un sólido de color amarillo: RMN 1H (400 MHz, DMSO) 58,00 (dd, J = 4,9, 1,8 Hz, 1H), 7,38 -7,32 (m, 1H), 6,86 (dd, J = 7,3,4,9 Hz, 1H), 5,58 (s, 1H), 5,38 (s, 2H), 5,12 (s, 1H), 4,16 (s, 1H), 3,96 (s, 3H), 3,43 - 3,32 (m, 1H), 2,22 - 2,12 (m, 1H), 1,99 (s, 3H), 1,80 (m, 1H), 1,54 - 1,23 (m, 4H), 0,96 (s, 3H), 0,90 (s, 3H); iEn -EM m/z calc. 341,22, encontrado 342,23 (M+1)+; Tiempo de retención: 0,67 minutos.
Ejemplo 45
Esquema sintético 45: (+/-)-[3-[1-(2-amino-6-metil-pirimidin-4-il)azepan-2-il]-2-piridil]metanol I-57

(a) H2, Pd al 10 %/C, MeOH, EtOAc, AcOH; (b) mCPBA, DCM; (c) TFAA, DCM después Na2CO3 ac.; (d) HCl conc., 100 °C; (e) 2-amino-4-cloro-6-metilpirimidina, NMP, 150 °C
Formación de (+/-)-2-(2-metil-3-piridil)azepano-1-carbaldehído
Una mezcla de 7-(2-metil-3-piridil)-2,3,4,5-tetrahidroazepina-1-carbaldehído (1,60 g, 7,40 mmol), Pd/C (10% tipo húmedo Degussa, 400 mg), MeOH (10 ml), EtOAc (10 ml) y AcOH (2 ml) se agitó en una atmósfera de gas hidrógeno. Después de 2 horas, la mezcla de reacción se filtró a través de Celite con la ayuda de MeOH y después se concentró al vacío para dar 1,61 g del producto deseado en forma de un aceite de color amarillo pálido: RMN 1H (400 MHz, DMSO-d6) 58,27 (dd, J = 4,8, 1,7 Hz, 1H), 8,15 (s, 1H), 7,95 (s, 1H), 7,15 (dd, J = 7,8, 4,7 Hz, 1H), 5,01 (dd, J = 12,8, 4,7 Hz, 1H), 3,87 (dd, J = 15,0, 4,9 Hz, 1H), 3,59 (dd, J = 14,8, 10,2 Hz, 1H), 2,89 (s, 3H), 2,12 - 2,02 (m, 1H), 2,00 -1,92 (m, 1H), 1,82 - 1,61 (m, 2H), 1,37 - 1,28 (m, 4H). IEN-EM m/z calc. 218,14, encontrado 219,17 (M+1)+; Tiempo de retención: 0,49 minutos.
Formación de (+/-)-2-(2-metil-3-piridil)azepano-1-carbaldehído
A una solución de 2-(2-metil-3-piridil)azepano-1-carbaldehído (1,61 g, 7,38 mmol) en diclorometano (20 ml), se le añadió mCPBA (2,54 g, 14,80 mmol) y la mezcla se agitó durante una noche a temperatura ambiente. El sólido de color blanco se retiró por filtración lavándose con algo de diclorometano. Al filtrado se le añadió cuidadosamente bicarbonato sódico ac. sat. Los extractos orgánicos se lavaron un segundo tiempo con bicarbonato sódico ac. sat. Las capas se separaron con la ayuda de un separador de fase y los orgánicos se concentraron al vacío para dar 600 mg del producto deseado en forma de un aceite de color amarillo: RMN 1H (400 MHz, DMSO-cfe) 58,18 (s, 1H), 8,15 (d, J = 6,0 Hz, 1H), 7,22-7,17 (m, 1H), 7,08 (d, J = 7,9 Hz, 1H), 5,04-4,95 (m, 1H), 3,89 (dd, J = 15,1, 5,3 Hz, 1H), 3,61
3,51 (m, 1H), 2,45 (s, 3H), 2,11 -1,83 (m, 2H), 1,82 - 1,59 (m, 2H), 1,32 (d, J = 5,8 Hz, 4H); IEN-EM m/z calc. 234,14, encontrado 235,16 (M+1)+; Tiempo de retención: 0,53 minutos.
Formación de (+/-)-2-[2-(hidroximetil)-3-piridil]azepano-1-carbaldehído
A una solución de 2-(2-metil-1-oxido-piridin-1-ium-3-il)azepano-1-carbaldehído (0,60 g, 2,56 mmol) en diclorometano (15 ml), se le añadió TFAA (0,36 ml, 2,59 mmol) a temperatura ambiente. Después de 7 horas, se añadió cuidadosamente Na2CO3 ac. sat. y la agitación se continuó durante una noche. Se añadió agua y diclorometano. Las capas se separaron con la ayuda de un separador de fase. La capa acuosa se extrajo de nuevo con diclorometano y las capas se separaron a través de un separador de fase de nuevo y los orgánicos combinados se concentraron al vacío. El residuo en bruto se purificó por cromatografía sobre gel de sílice con una columna isco GOLD de 12 g usando MeOH al 0-12,5 %/diclorometano para proporcionar 324 mg del producto deseado en forma de un sólido de color pardo: RMN 1H (400 MHz, DMSO) 58,36 (dd, J = 4,7, 1,6 Hz, 1H), 8,11 (s, 1H), 7,68 (dd, J = 7,8, 1,6 Hz, 1H), 7,27 (dd, J = 7,8, 4,7 Hz, 1H), 5,16 (dd, J = 7,0, 4,0 Hz, 1H), 4,91 (dd, J = 12,4, 7,0 Hz, 1H), 4,64 (dd, J = 12,4, 3,9 Hz, 1H), 3,84 (dd, J = 14,9, 4,5 Hz, 1H), 3,66 (dd, J = 15,0, 10,2 Hz, 1H), 2,21 -2,11 (m, 1H), 1,97 - 1,70 (m, 3H), 1,38-1,22 (m, 4H); IEN-EM m/z calc. 234,14, encontrado 235,2 (M+1)+; Tiempo de retención: 0,48 minutos.
Formación de (+/-)-[3-(azepan-2-il)-2-piridil]metanol
Una mezcla de 2-[2-(hidroximetil)-3-piridil]azepano-1-carbaldehído (0,28 g, 1,20 mmol) y HCl (1,5 ml de 38 %p/v, 15,6 mmol) se calentó a 100 °C durante una noche. La purificación se realizó en una columna ISCO c18-aq de 50 g de fase inversa, ejecutándose con TFA al 0,1 %/H2O y TFA al 0,1 %/CH3CN. Las fracciones puras se concentraron al vacío y después se disolvieron en MeOH y se pasaron a través de un cartucho de bicarbonato SPE (Agilent Stratospheres 5 g/60 ml) y se concentraron para dar 150 mg del producto deseado en forma de un aceite de color naranja: RMN 1H (400 MHz, DMSO) 58,35 (dd, J = 4,7,1,7 Hz, 1H), 7,83 (dd, J = 7,8, 1,6 Hz, 1H), 7,27 (dd, J = 7,8, 4,7 Hz, 1H), 4,62 (dd, J = 28,7, 12,7 Hz, 2H), 4,00 (dd, J = 9,4, 3,5 Hz, 1H), 2,95 (dd, J = 11,6, 6,6 Hz, 1H), 2,77-2,69 (m, 1H), 1,87 (ddd, J = 13,9, 6,6, 3,2 Hz, 1H), 1,80 - 1,72 (m, 1H), 1,72-1,45 (m, 6H); IEN-EM m/z calc. 206,14, encontrado 207,19 (M+1)+; Tiempo de retención: 0,25 minutos.
Formación de (+/-)-[3-[1-(2-amino-6-metil-pirimidin-4-il)azepan-2-il]-2-piridil]metanol
Una mezcla de [3-(azepan-2-il)-2-piridil]metanol (0,15 g, 0,72 mmol) y 4-cloro-6-metil-pirimidin-2-amina (0,09 g, 0,66 mmol) se agitó en NMP (1 ml) a 170 °C en un vial de centelleo durante 3 h. La purificación se llevó a cabo en una columna ISCO c18-aq de 50 g de fase inversa, ejecutándose con TFA al 0,1 %/H2O y TFA al 0,1 %/CH3CN. Las fracciones puras se concentraron al vacío y después se añadieron HCl (2 ml de 2 M, 4,00 mmol) y MeCN (5 ml) al vacío de nuevo para dar 119 mg del producto deseado en forma de un sólido de color blanquecino: RMN de 1H (400 MHz, D2O) 58,55 (d, J = 5,8 Hz, 1H), 8,33 (d, J = 8,1 Hz, 1H), 7,86 - 7,79 (m, 1H), 6,35 (s, 1H), 5,46 - 5,36 (m, 2H), 5,14 - 5,07 (m, 1H), 4,08 (dd, J = 15,5, 4,5 Hz, 1H), 3,83 (dd, J = 15,7, 10,8 Hz, 1H), 2,25 (s, 3H), 2,22 - 2,12 (m, 1H), 2,05 - 1,77 (m, 4H), 1,60-1,25 (m, 3H); IEN-EM m/z calc. 313,19, encontrado 314,23 (M+1)+; Tiempo de retención: 0,49 minutos.
Ejemplo 46
Esquema sintético 46: (+/-)-4-(3-(2-fluorofenil)-6,6-dimetil-1,4-oxazepan-4-il)-6-metilpirimidin-2-amina I-49

(a) tritilo cloruro, Et3N, CH2Ch; (b) NaH, DMF, tributil(yodometil)estannano; (c) 2,2,2-trifluoroetanol, AcOH, CH2Ch; (d)
2-fluorobenzaldehído; tamices moleculares de 4 A, CH2CI2; (e) 2,6-lutidina, Cu(OTf)2, hexafluoroisopropanol, CH2CI2; (f) 2-amino-4-doro-6-metilpirimidina, NMP, 150 °C.
Formación de 2,2-dimetil-3-(tritilamino)propan-1-ol
A una solución de 3-amino-2,2-dimetil-propan-1-ol (5,1 g, 49,2 mmol) y trietilamina (13,5 ml, 96,9 mmol) en CH2Ch (50 ml) a 0 °C, se le añadió gota a gota una solución de cloruro de tritilo (13,6 g, 48,8 mmol) en CH2Cl2 (50 ml). La mezcla de reacción se agitó durante 48 horas con calentamiento gradual a temperatura ambiente. La mezcla de reacción se lavó con 50 ml de agua y la capa orgánica se extrajo con 50 ml de CH2Ch. Las fases orgánicas combinadas se concentraron a sequedad y se purificaron por cromatografía sobre gel de sílice eluyendo con EtOAc al 0 25 %/heptanos. Las fracciones puras se combinaron y se concentraron al vacío para dar 14,4 g (85 %) del producto deseado en forma de un aceite incoloro: RMN 1H (400 MHz, CDCls) 87,47-7,42 (m, 6H), 7,36 - 7,29 (m, 6H), 7,27 -7,21 (m, 3H), 3,63 (s, 2H), 2,17 (s, 2H), 0,89 (s, 6H); IEN-EM m/z calc. 345,21, encontrado 346,0 (M+1)+; Tiempo de retención: 0,82 minutos.
Formación de 2,2-dimetM-3-((tributMestannM)metoxi)-W-tritMpropan-1-amma
Se lavó hidruro sódico (1,0 g, 26,0 mmol, 60 % en aceite mineral) con heptano en atmósfera de nitrógeno, después se suspendió en DMF anhidra (85 ml) y se enfrió a 0 °C. Una solución de 2,2-dimetil-3-(tritilamino)propan-1-ol (6,0 g, 17,4 mmol) en DMF (85 ml) se añadió gota a gota durante 15 minutos y después la reacción se calentó a temperatura ambiente y se agitó durante 1 hora. La mezcla se enfrió de nuevo a 0 °C, y después se añadió gota a gota tributil(yodometil)estannano (8,2 g, 19,0 mmol). La reacción se agitó a 0 °C con calentamiento gradual a temperatura ambiente durante 4 horas. La reacción se interrumpió a 0 °C mediante la adición lenta de 85 ml una solución acuosa saturada de cloruro de amonio. Las capas se separaron y la capa orgánica se lavó con agua. La capa orgánica se concentró, se disolvió en 500 ml MTBE y se lavó 3 x 150 ml de agua. La capa orgánica se concentró a sequedad, se cargó en seco sobre Celite y se purificó mediante cromatografía en gel de sílice eluyendo con 0-35 % de EtOAc en heptano. Las fracciones puras se combinaron y se concentraron para dar 4,55 g (40 %) del producto en forma de un aceite de color amarillo claro: RMN 1H (400 MHz, CDCla) 87,58-7,54 (m, 7H), 7,32 (dd, J = 8,4, 6,9 Hz, 7H), 7,25 -7,18 (m, 3H), 3,71 (s, 2H), 3,17 (s, 2H), 2,04 (d, J = 7,8 Hz, 2H), 1,85 (t, J = 8,0 Hz, 1H), 1,58 - 1,49 (m, 5H), 1,38 -1,30 (m, 10H), 0,98-0,93 (m, 18H); IEN-EM m/z calc. 649,34, encontrado 649,0 (M+1)+; Tiempo de retención: 0,95 minutos.
Formación de 2,2-dimetil-3-((tributilestannil)metoxi)propan-1-amina
Se disolvió 2,2-dimetil-3-((tributilestannil)metoxi)-W-tritilpropan-1-amina (4,55 g, 7,02 mmol) en diclorometano (150 ml), 2,2,2-trifluoroetanol (43 ml) y ácido acético (22 ml) y se agitó durante una noche a temperatura ambiente. La reacción se neutralizó mediante la adición en porciones de una solución acuosa saturada de bicarbonato sódico durante 90 minutos. La capa orgánica se retiró y la capa acuosa se extrajo tres veces con diclorometano. Las fases orgánicas combinadas se secaron sobre sulfato de magnesio, se filtraron y se concentraron al vacío para dar 4,7 g de un aceite de color amarillo claro que contenía ppt. de color blanco. El producto en bruto se purificó por cromatografía sobre gel de sílice eluyendo con MeOH al 0-5 %/diclorometano que contenía trietilamina al 0,1 %. Las fracciones puras se combinaron y se concentraron para dar 1,21 g (42 %) del producto en forma de un aceite incoloro: RMN 1H (400 MHz, CDCh) 83,68 (s, 2H), 3,08 (s, 2H), 2,52 (s, 2H), 1,60 - 1,45 (m, 6H), 1,36 - 1,28 (m, 6H), 0,95 - 0,87 (m, 15H), 0,86 (s, 6H); iEn-EM m/z calc. 407,22, encontrado 408,0 (M+1)+; Tiempo de retención: 0,76 minutos.
Formación de (+/-)-3-(2-fluorofenil)-6,6-dimetil-1,4-oxazepano
A una solución de 2,2-dimetil-3-((tributilestannil)metoxi)propan-1-amina (1,21 g, 2,979 mmol) en diclorometano anhidro (8 ml), se le añadió 2-fluorobenzaldehído (0,33 ml, 2,25 mmol) seguido de tamices moleculares de 4 A (0,32 g). La mezcla incolora turbia se agitó a temperatura ambiente durante 2 horas y después se filtró sobre Celite. El lecho del filtro se enjuagó con 55 ml de diclorometano y el filtrado se almacenó en atmósfera de nitrógeno. En un matraz de fondo redondo de 250 ml separado que contenía hexafluoroisopropanol (15 ml) en atmósfera de nitrógeno, se añadió 2,6-lutidina anhidra (0,35 ml, 3,02 mmol) seguido de Cu(OTf)2 (0,19 g, 2,99 mmol). La mezcla se agitó a temperatura ambiente durante 1 hora, después se añadió la solución de imina preparada anteriormente. La mezcla de reacción se agitó durante una noche a temperatura ambiente y se inactivó con 75 ml de una mezcla acuosa 2:1 de una solución acuosa saturada de bicarbonato sódico e hidróxido de amonio al 10%. La mezcla se agitó durante 15 minutos y después se separó. La capa acuosa se extrajo 2 x 75 ml de diclorometano. Los extractos orgánicos combinados se lavaron con salmuera, se secaron sobre sulfato de magnesio, se filtraron y se concentró al vacío. El material en bruto se purificó por cromatografía sobre gel de sílice eluyendo con MeOH al 0-8 % en diclorometano. Las fracciones puras se combinaron y se concentraron para dar 90 mg (13 %) del producto deseado: RMN 1H (400 MHz, CDCh) 87,51 (td, J = 7,5, 1,8 Hz, 1H), 7,30-7,20 (m, 1H), 7,14 (dt, J = 7,5, 1,3 Hz, 1H), 7,02 (ddd, J = 10,5, 8,1,1,3 Hz, 1H), 4,31 (dd, J = 9,9, 4,4 Hz, 1H), 4,05 (ddd, J = 12,0, 4,4, 0,7 Hz, 1H), 3,62 (d, J = 12,1 Hz, 1H), 3,50 - 3,41 (m, 2H), 2,86 (d, J = 13,7 Hz, 1H), 2,77 (d, J = 13,7 Hz, 1H), 0,98 (s, 6H). IEN-EM m/z calc. 223,14, encontrado 224,0 (M+1)+; Tiempo de retención: 0,56 minutos.
Formación de (+/-)-4-(3-(2-fluorofenil)-6,6-dimetil-1,4-oxazepan-4-il)-6-metilpirimidin-2-amina I-49
Una mezcla de 3-(2-fluorofenil)-6,6-dimetil-1,4-oxazepano (0,09 g, 0,38 mmol) y 4-doro-6-metil-pirimidin-2-amina (0,06 g, 0,38 mmol) en NMP (1,2 ml) se calentó a 150 °C durante 150 minutos en un tubo cerrado herméticamente. La mezcla de reacción se purificó a través de cromatografía de fase inversa eluyendo con MeCN al 5-50 % en agua con TFA al 0,1 %. Las fracciones puras se combinaron, se neutralizaron usando una solución de bicarbonato sódico y se extrajo con EtOAc. La capa orgánica se secó sobre sulfato de magnesio, se filtró, se concentró al vacío y se liofilizó para dar 12 mg (9 %) del producto deseado: RMN 1H (400 MHz, DMSO-d6) (se calentó 360K) 57,30 -7,18 (m, 2H), 7,18 -7,07 (m, 2H), 5,73 (s, 1H), 5,61 (s, 2H), 5,47 - 5,35 (m, 1H), 4,57 (d, J = 14,9 Hz, 1H), 4,13 -4,03 (m, 1H), 3,71 (dd, J = 13,4, 11,0 Hz, 1H), 3,53 -3,38 (m, 2H), 3,31 -3,21 (m, 1H), 2,00 (s, 3H), 0,89 (d, J = 8,1 Hz, 6H); IEN-EM m/z calc. 330,18, encontrado 331,0 (M+1)+; Tiempo de retención: 0,74 minutos.
Ejemplo 47
Esquema sintético 47: (+/-)-3-(4-(2-ammo-6-metilpirimidm-4-il)-1,4-oxazepan-3-M)-4-metoxi-W-metilbenzamida I-82

(a) 3-cloro-6,7-dihidro-5H-1,4-oxazepina-4-carbaldehído), PdCl2(dppf), DMF, NaHCO3, H2O, 80 °C, irradiación por microondas; (b) H2, Pd/C, MeOH-EtOAc; (c) HCl, MeOH, 100 °C; (d) diazometil(trimetil)silano, tolueno, MeOH; (e) 2-amino-4-cloro-6-metilpirimidina, NMP, 150 °C; (f) LiOH, MeOH, H2O; (g) MeNH2, HATU, Et3N, DMF.
Formación de 3-(4-formil-4,5,6,7-tetrahidro-1,4-oxazepin-3-il)-4-metoxibenzoato de metilo
Una mezcla de 3-cloro-6,7-dihidro-5H-1,4-oxazepina-4-carbaldehído (2,0 g, 12,4 mmol), ácido (2-metoxi-5-metoxicarbonil-fenil)borónico (2,6 g, 12,4 mmol) y PdCl2(dppf) (1,0 g, 1,3 mmol) en DMF (37 ml) y bicarbonato sódico acuoso saturado (12 ml) se calentó en un reactor de microondas a 80 °C durante 30 minutos. La mezcla de reacción se diluyó con agua, se lavó con agua, y después la fase orgánica se concentró a sequedad. El residuo resultante se purificó por cromatografía sobre gel de sílice eluyendo con EtOAc al 40-100 % en heptanos seguido de un MeOH al 10% en diclorometano enjuagado. Las fracciones que contenían el producto deseado se combinaron y se concentraron al vacío para proporcionar 2,4 g (63 %) del producto deseado en forma de un aceite incoloro: RMN 1H (400 MHz, CDCl3) 58,04 (dd, J = 8,6, 2,2 Hz, 1H), 7,95 (s, 1H), 7,92 (d, J = 2,2 Hz, 1H), 6,90 (d, J = 8,6 Hz, 1H), 6,19 (s, 1H), 4,24 (dd, J = 6,3, 5,3 Hz, 2H), 4,06 (t, J = 6,6 Hz, 2H), 3,92 (s, 3H), 3,87 (s, 3H), 2,18-2,09 (m, 2H); IEN-EM m/z calc. 291,11, encontrado 290,0 (M+1)+; Tiempo de retención: 0,9 minutos.
Formación de 3-(4-formil-1,4-oxazepan-3-il)-4-metoxibenzoato de (+/-)-metilo
Una mezcla de 3-(4-formil-4,5,6,7-tetrahidro-1,4-oxazepin-3-il)-4-metoxibenzoato de metilo (2,4 g, 8,2 mmol) y Pd/C (1,5 g, 0,7 mmol) en acetato de etilo (25 ml) y MeOH (25 ml) se agitó durante una noche 0,38 MPa (55 psi) de hidrógeno. La mezcla de reacción se filtró sobre Celite y el filtrado secundario se concentró a sequedad. El residuo resultante se purificó por cromatografía sobre gel de sílice eluyendo con EtOAc al 40-100 % en heptanos. Varias fracciones mixtas que contenían el producto deseado se llevaron a la siguiente etapa tal como está: IEN-EM m/z calc.
293,13, encontrado 294,0 (M+1)+; Tiempo de retención: 0,77 minutos.
Formación de clorhidrato de ácido (+/-)-4-metoxi-3-(1,4-oxazepan-3-il)benzoico
Una solución de 3-(4-formil-1,4-oxazepan-3-il)-4-metoxibenzoato de metilo (2,4 g, 8,2 mmol) en MeOH (40 ml) y HCl concentrado (40 ml de solución 12,1 M, 484,0 mmol) se agitó durante una noche a 100 °C. La mezcla se concentró a sequedad. El producto se recogió en MeOH y se diluyó en éter dietílico, después se filtró y se secó para dar 2,2 g (84 %) de un sólido de color blanco: RMN 1H (400 MHz, DMSO-dS) 58,12 (d, J = 2,1 Hz, 1H), 7,98 (ddd, J = 8,7, 3,3, 2,1 Hz, 1H), 7,18 (d, J = 8,7 Hz, 1H), 4,77 -4,55 (m, 1H), 4,00 (dd, J = 13,5, 8,9 Hz, 1H), 3,93 (d, J = 1,9 Hz, 3H), 3,91 - 3,85 (m, 2H), 3,49 - 3,41 (m, 2H), 3,26 (ddd, J = 13,4, 9,4, 3,4 Hz, 1H), 2,83 (ddt, J = 47,7, 12,7, 7,3 Hz, 0,5H), 2,31
- 2,05 (m, 1H), 2,05-1,75 (m, 0,5H); IEN-EM m/z calc. 251,12, encontrado 252,0 (M+1)+; Tiempo de retención: 0,5 minutos.
Formación de 4-metoxi-3-(1,4-oxazepan-3-il)benzoato de (+/-)-metilo
A una solución de clorhidrato del ácido 4-metoxi-3-(1,4-oxazepan-3-il)benzoico (0,53 g, 1,65 mmol) en tolueno (22 ml) y MeOH (2,5 ml), se le añadió diazometil(trimetil)silano (0,84 ml de solución 2 M, 1,69 mmol) en hexanos. La mezcla se agitó durante 15 minutos, después se concentró a sequedad para proporcionar 487 mg de un aceite incoloro: IEN-EM m/z calc. 265,13, encontrado 266,0 (M+1)+; Tiempo de retención: 0,57 minutos.
Formación de 3-(4-(2-amino-6-metilpirimidin-4-il)-1,4-oxazepan-3-il)-4-metoxibenzoato de (+/-)-metilo
Una mezcla de 4-metoxi-3-(1,4-oxazepan-3-il)benzoato de metilo (0,44 g, 1,65 mmol) y 4-cloro-6-metilpirimidin-2-amina (0,26 g, 1,82 mmol) en NMP (5,5 ml) se agitó durante 4 horas a 150 °C en un tubo cerrado herméticamente. La mezcla se diluyó con agua y se extrajo con EtOAc. La capa orgánica se concentró a sequedad y el residuo resultante se purificó por cromatografía sobre gel de sílice eluyendo con MeOH al 0-12 % en diclorometano. Las fracciones puras se combinaron y se concentraron al vacío para dar 89 mg (14 %) del producto deseado: RMN 1H (400 MHz, DMSO-d6) (se calentó 360K) 87,89 (dd, J = 8,6, 2,2 Hz, 1H), 7,70 (d, J = 2,2 Hz, 1H), 7,16 (d, J = 8,7 Hz, 1H), 6,60 (s, 2H), 5,89 (s, 1H), 5,57 (s, 1H), 4,60 (d, J = 14,7 Hz, 1H), 4,19 (dd, J = 13,4, 5,2 Hz, 1H), 3,95 (s, 3H), 3,90 (dt, J = 12,0, 3,8 Hz, 1H), 3,79 (s, 3H), 3,77 - 3,68 (m, 1H), 3,63 - 3,54 (m, 1H), 2,14 (s, 3H), 1,80 (dt, J = 7,7, 4,2 Hz, 2H). IEN-EM m/z calc. 372,18, encontrado 373,0 (M+1)+; Tiempo de retención: 0,66 minutos.
Formación de sal trifluoroacetato del ácido (+/-)-3-(4-(2-amino-6-metilpirimidin-4-il)-1,4-oxazepan-3-il)-4-metoxibenzoico
A una solución de 3-(4-(2-amino-6-metilpirimidin-4-il)-1,4-oxazepan-3-il)-4-metoxibenzoato de metilo (0,090 g, 0,230 mmol) en MeOH (1 ml) y agua (1 ml), se le añadió LiOH (0,025 g, 1,044 mmol). La mezcla de reacción se agitó a temperatura ambiente durante 3 horas, se añadió HCl 1 M y la mezcla se purificó por cromatografía de fase inversa eluyendo con MeCN al 10-90 % en agua con TFA al 0,1 %. Las fracciones puras se combinaron, se concentraron y se liofilizaron para dar 50 mg (58 %) del producto deseado: RMN 1H (400 MHz, DMSO-d6) (se calentó 360K) 87,92 (dd, J = 8,6, 2,1 Hz, 1H), 7,72 (s, 1H), 7,41 (s, 2H), 7,17 (d, J = 8,6 Hz, 1H), 4,24 (s, 1H), 3,99 - 3,90 (m, 4H), 3,80 (t, J = 7,1 Hz, 2H), 3,64 (dt, J = 12,2, 7,4 Hz, 1H), 2,33-1,65 (m, 6H); IEN-EM m/z calc. 358,16, encontrado 359,0 (M+1)+; Tiempo de retención: 0,6 minutos.
Formación de (+/-)-3-(4-(2-ammo-6-metilpirimidm-4-N)-1,4-oxazepan-3-N)-4-metoxi-W-metNbenzamida I-82 A una solución de ácido 3-[4-(2-amino-6-metil-pirimidin-4-il)-1,4-oxazepan-3-il]-4-cloro-benzoico (sal trifluoroacetato) (0,085 g, 0,180 mmol) en Dm F (1 ml), se le añadió HATU (0,102 g, 0,268 mmol) seguido de Et3N (0,125 ml, 0,897 mmol). Después de agitar durante 10 minutos, se añadió metilamina (0,700 ml de 2 M, 1,40 mmol) en THF y la reacción se agitó durante una noche. La mezcla de reacción se diluyó con agua y se extrajo con EtOAc. La capa orgánica se concentró a sequedad y se purificó por cromatografía sobre gel de sílice eluyendo con MeOH al 0-10% en diclorometano. Las fracciones puras se combinaron, se concentraron al vacío para proporcionar el producto deseado: RMN 1H (400 MHz, DMSO-d6) (se calentó 360K) 87,90 (s, 1H), 7,68 (dd, J = 8,5, 2,3 Hz, 1H), 7,50 (d, J = 2,2 Hz, 1H), 7,04 (d, J = 8,6 Hz, 1H), 5,71 (d, J = 27,7 Hz, 3H), 5,25 (s, 1H), 4,43 (s, 1H), 3,92 (s, 3H), 3,45 (dd, J = 14,5, 11,2 Hz, 1H), 2,74 (d, J = 4,6 Hz, 3H), 2,34 (dt, J = 14,0, 6,9 Hz, 1H), 2,03 (s, 3H), 1,97 -1,62 (m, 2H), 1,52 (c, J = 12,5 Hz, 1H), 1,43-1,21 (m, 2H); IEN-EM m/z calc. 369,22, encontrado 370,0 (M+1)+; Tiempo de retención: 0,68 minutos.
Los siguientes análogos se prepararon de acuerdo con el Esquema sintético 47:

(+/-)-4-(4-(2-ammo-6-metilpirimidm-4-N)-1,4-oxazepan-3-N)-3-metoxi-W-metNbenzamida I-83
RMN 1H (400 MHz, DMSO-d6) (se calentó 360K) 88,12 (s, 1H), 7,49 (d, J = 1,6 Hz, 1H), 7,40 (dd, J = 7,9, 1,6 Hz, 1H), 7,23 (d, J = 7,9 Hz, 1H), 7,02 (s, 2H), 6,05 (s, 1H), 5,61 (s, 1H), 4,62 (s, 1H), 4,24 (dd, J = 13,4, 5,2 Hz, 1H), 3,95 (s, 4H), 3,81 (dt, J = 13,4, 7,9 Hz, 2H), 3,60 (ddd, J = 12,1, 9,4, 5,1 Hz, 1H), 2,82 (d, J = 4,5 Hz, 3H), 2,22 (s, 3H), 1,91 1,74 (m, 2H); IEN-EM m/z calc. 371,20, encontrado 372,0 (M+1)+; Tiempo de retención: 0,55 minutos.
Ejemplo 48
Esquema sintético 48: (+/-)-4-(4-(2-amino-6-metilpirimidin-4-il)-1,4-oxazepan-3-il)indolin-2-ona I-105

(a) NBS, t-BuOH, CH2CI2; (b) HCl ac. (4 M), dioxano, 110 °C; (c) 2-amino-4-cloro-6-metilpirimidina, NMP, 150 °C.
Formación de (+/-)-4-(1,4-oxazepan-3-il)indolin-2-ona
A una suspensión de 3-(1H-indol-4-il)-1,4-oxazepano-4-carbaldehído (0,22 g, 0,91 mmol) en tBuOH (5 ml), se le añadió NBS (0,18 g, 1,00 mmol) a temperatura ambiente. Después de 2 horas, se añadió diclorometano (5 ml) para ayudar a la solubilidad. Después de 1 hora, se añadió NBS adicional (0,08 g). Después de 1 hora, se añadió una solución acuosa saturada de bicarbonato sódico y la mezcla se extrajo con dos veces con diclorometano. Los extractos orgánicos combinados se lavaron con agua y después las capas se separaron con la ayuda de un separador de fase y los orgánicos se concentraron. El residuo de color pardo se disolvió en dioxano (4 ml) y se añadió HCl ac. (4 ml, 4 M) y la mezcla se calentó en un reactor de microondas durante 10 min a 100 °C y después 50 min adicionales a 110 °C. La reacción se concentró parcialmente y después se purificó por cromatografía en columna (columna C18 AQ 50 g; TFA al 0,1 %-agua/TFA al 0,1 %-MeCN). Las fracciones puras se concentraron y después se disolvieron en MeOH y se pasaron a través de un cartucho de bicarbonato SPE (Agilent Stratospheres 5 g/60 ml) y concentraron para dar 72 mg del producto deseado. RMN 1H (400 MHz, DMSO) 810,32 (s, 1H), 7,12 (t, J = 7,7 Hz, 1H), 6,96 (d, J = 7,7 Hz, 1H), 6,69 (d, J = 7,6 Hz, 1H), 3,89 - 3,65 (m, 4H), 3,51 (s, 2H), 3,35 (m, 2H), 3,08 (dt, J = 13,3, 5,0 Hz, 1H), 2,80 (dt, J = 13,9, 7,1 Hz, 1H), 1,91-1,78 (m, 2H); IEN-EM m/z calc. 232,12, encontrado 233,26 (M+1)+; Tiempo de retención: 0,46 minutos.
Formación de (+/-)-4-[4-(2-amino-6-metil-pirimidin-4-il)-1,4-oxazepan-3-il]indolin-2-ona I-105
Una mezcla de 4-(1,4-oxazepan-3-il)indolin-2-ona (0,07 g, 0,30 mmol) y 4-cloro-6-metil-pirimidin-2-amina (0,04 g, 0,28 mmol) se calentó en NMP (1,5 ml) a 150 °C durante una noche en un vial de reacción equipado con un tapón de alivio de presión. La mezcla se enfrió a temperatura ambiente y se cargó directamente en una columna ISCO c18-aq de 50 g y se purificó por fase inversa ejecutándose con TFA al 0,1 %/H2O y TFA al 0,1 %/CH3CN. Las fracciones puras se concentraron al vacío y después se disolvieron en MeOH y se pasaron a través de un cartucho de bicarbonato SPE (Agilent Stratospheres 500 mg/6 ml) y se concentraron para dar 74,1 mg del producto deseado: alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6) 89,97 (s, 1H), 7,03 (t, J = 7,7 Hz, 1H), 6,77 (d, J = 8,0 Hz, 1H), 6,62 (d, J = 7,7 Hz, 1H), 5,62 (s, 1H), 5,57 (s, 1H), 5,39 (s, 2H), 5,30 (s, 1H), 4,28 (s, 1H), 3,98 (dd, J = 13,4, 5,1 Hz, 1H), 3,74 (dd, J = 13,5, 9,6 Hz, 2H), 3,45 (t, J = 15,5 Hz, 3H), 3,24 (d, J = 22,5 Hz, 1H), 1,95 (s, 3H), 1,66 (m, 2H); IEN-EM m/z calc.
339,17, encontrado 340,25 (M+1)+; Tiempo de retención: 0,51 minutos.
Ejemplo 49
Esquema sintético 49: (+/-)- 4-(9-(2-fluorofenil)-1,4-dioxa-8-azaespiro[4,6]undecan-8-il)-6-metilpirimidin-2-amina y (+/-)- 1-(2-amino-6-metilpirimidin-4-il)-7-(2-fluorofenil)azepan-4-ona


(a) PdCl2(dppf), NaHCOa ac., DME, 80 °C; (b) ozono, CH2CI2, -78 °C, después PhaP; (c) NH2OH-HCI, NaCNBHa, MeOH; (d) H2, Pd/C, MeOH; (e) EtOH, 180 °C; (f) HCl, acetona, 50 °C
Formación de 8-(2-fluorofenil)-1,4-dioxaespiro[4,5]dec-7-eno
A una solución de 1-bromo-2-fluoro-benceno (0,98 g, 5,64 mmol) y 2-(1,4-dioxaespiro[4,5]dec-7-en-8-il)-4,4,5,5-tetrametil-1,3,2-dioxaborolano (1,50 g, 5,64 mmol) en DME (20 ml), se le añadieron NaHCOa (9,4 ml de solución ac.
1,2 M, 11,27 mmol) y PdCh(dppf) (0,43 g, 0,52 mmol) en una atmósfera de nitrógeno. La mezcla se calentó a 80 °C durante 20 horas. La mezcla de reacción se filtró a través de una capa de celite y se concentró al vacío. El residuo resultante se purificó por cromatografía sobre gel de sílice (columna ISCO de 40 g) eluyendo con heptanos/EtOAc (gradiente del 0 % al 40 %) para proporcionar 1,0 g (76 %) del producto deseado: r Mn 1H (300 MHz, CDCla) 87,34 7,16 (m, 2H), 7,14 - 6,90 (m, 2H), 5,87 (cd, J = 2,2, 0,7 Hz, 1H), 4,05 (s, 4H), 2,66 (tdd, J = 5,5, 2,6, 1,5 Hz, 2H), 2,55 -2,43 (m, 2H), 1,99 - 1,86 (m, 2H).
Formación de 2-(2-(3-(2-fluorofenil)-3-oxopropil)-1,3-dioxolan-2-il)acetaldehído
A una solución de 8-(2-fluorofenil)-1,4-dioxaespiro[4,5]dec-7-eno (1,00 g, 4,27 mmol) en diclorometano (30 ml) se burbujeó ozono a -78 °C hasta que la solución permaneció de color azul. Se apagó el generador de ozono y se burbujeó aire a través de la mezcla durante 30 minutos. Después se añadió trifenilfosfina (1,12 g, 4,27 mmol) y la mezcla se calentó a temperatura ambiente y se agitó durante 12 horas. El producto en bruto se purificó por cromatografía sobre gel de sílice (columna ISCO de 40 g) eluyendo con EtOAc/heptanos. Las fracciones deseadas se recogieron y se evaporaron para proporcionar el producto deseado: RMN 1H (300 MHz, CDCh) 89,77 (t, J = 2,9 Hz, 1H), 7,86 (dt, J = 7,6, 1,9 Hz, 1H), 7,53 (dddd, J = 8,3, 7,1, 5,0, 1,9 Hz, 1H), 7,32 - 7,21 (m, 1H), 7,15 (ddd, J = 11,3, 8,3, 1,1 Hz, 1H), 4,03 (d, J = 0,8 Hz, 3H), 3,10 (ddd, J = 7,6, 6,8, 2,9 Hz, 2H), 2,74 (d, J = 2,9 Hz, 2H), 2,23 (ddd, J = 8,1, 6,9, 0,9 Hz, 2H).
Formación de 9-(2-fluorofenil)-1,4-dioxa-8-azaespiro[4,6]undecan-8-ol
A una solución de 2-[2-[3-(2-fluorofenil)-3-oxo-propil]-1,3-dioxolan-2-il]acetaldehído (1,00 g, 3,76 mmol) en MeOH (20 ml), se le añadieron clorhidrato de hidroxilamina (0,26 g, 3,76 mmol) y NaHCO3. La mezcla se agitó a temperatura ambiente durante 30 minutos antes de añadir cianoborohidruro sódico (1,18 g, 18,78 mmol). La reacción se agitó a temperatura ambiente durante una noche. La mezcla se inactivó mediante la adición de etilendiamina. El disolvente se evaporó y el residuo se purificó por cromatografía sobre gel de sílice (columna ISCO de 40 g) eluyendo con EtOAc/heptanos. Las fracciones deseadas se recogieron y se evaporaron. El producto deseado se usó sin purificación adicional.
Formación de (+/-)-9-(2-fluorofenil)-1,4-dioxa-8-azaespiro[4,6]undecano
A una solución de 9-(2-fluorofenil)-1,4-dioxa-8-azaespiro[4,6]undecan-8-ol (0,20 g, 0,75 mmol) en MeOH (15 ml), se le añadió Pd al 10%/C en una atmósfera de nitrógeno. El matraz se cargó con un globo de hidrógeno y se lavó abundantemente al vacío y se hidrógeno tres veces. La reacción se agitó a temperatura ambiente en una atmósfera de nitrógeno durante una noche. La mezcla se filtró a través de celite y el filtrado se concentró al vacío. El producto deseado se usó sin purificación adicional.
Formación de (+/-)-4-[8-(2-fluorofenil)-1,4-dioxa-9-azaespiro[4,6]undecan-9-il]-6-metil-pirimidin-2-amina I-53
A una mezcla de 4-cloro-6-metil-pirimidin-2-amina (0,14 g, 0,95 mmol) y 8-(2-fluorofenil)-1,4- dioxa-9-azaespiro[4,6]undecano (0,24 g, 0,95 mmol) sólidos, en un vial, se le añadió EtOH (2 ml). El vial se puso en una placa y se calentó a 180 °C sin cubrir durante 2 horas. El sólido en bruto se purificó por cromatografía sobre gel de sílice (columna ISCO de 40 g) eluyendo con DCM, MeOH al 20 %/DCM. Las fracciones deseadas se recogieron y se evaporaron para proporcionar 260,0 mg (72 %) del producto deseado: RMN 1H (300 MHz, DMSO-d6) 87,39 - 7,10 (m, 4H), 6,90 (s, 2H), 5,80 (m, 1H), 4,74 - 4,38 (m, 1H), 3,89 (m, 4H), 3,35 (m, 2H), 2,33-1,51 (m, 9H); IEN-EM m/z calc.
358,18, encontrado 359,21 (M+1)+; Tiempo de retención: 0,66 minutos.
Formación de (+/-)-1-(2-amino-6-metil-pirimidin-4-il)-7-(2-fluorofenil)azepan-4-ona I-56
A una solución de 4-[8-(2-fluorofenil)-1,4-dioxa-9-azaespiro[4,6]undecan-9-il]-6-metil-pirimidin-2-amina (0,22 g, 0,59 mmol) en acetona (10 ml), se le HCl ac. (10 ml de 6 M, 59,47 mmol) a temperatura ambiente. La mezcla se calentó a 50 °C durante 3 horas. El disolvente se evaporó y el residuo se purificó por cromatografía sobre sílice (columna ISCO de 40 g) eluyendo con DCM, MeOH al 10%/DCM. Las fracciones deseadas se recogieron y se evaporaron para proporcionar 57 mg (28%) del producto deseado: RMN 1H (300 MHz, DMSO-d6) 67,31 (m, 1H), 7,26 - 7,06 (m, 3H), 5,93 (s, 2H), 5,77 (a, 1H), 4,43 (a, 1H), 3,74 (t, J = 13,3 Hz, 1H), 3,35 (d, J = 24,5 Hz, 2H), 2,83 - 2,68 (m, 1H), 2,54 (m, 2H), 2,36 -2,21 (m, 2H), 2,04 (s, 3H); IEN-EM m/z calc. 314,15, encontrado 315,26 (M+1)+; Tiempo de retención: 0,61 minutos.
Ejemplo 50
Esquema sintético 50: (+/-)-4-(3-(1H-indol-4-il)-1,4-oxazepan-4-il)-6-metilpirimidin-2-amina

(a) ácido 1 H-indol-4-ilborónico, P d ^ P ^ C h , EtaN, DMF, 70 °C; (b) H2, Pd/C, MeOH; (c) nBuLi, THF; (d) 2-amino-4-cloro-6-metilpirimidina, NMP, 140 °C
Formación de 3-(1H-mdol-4-il)-6,7-dihidro-1,4-oxazepma-4(5H)-carbaldehído
Se cargó un vial de 40 ml con un tapón de alivio de presión en atmósfera de nitrógeno con 3-cloro-6,7-dihidro-5H-1,4-oxazepina-4-carbaldehído (3,4 g, 20,0 mmol), ácido 1H-indol-4-ilborónico (3,2 g, 20,0 mmol), PdC^PPhb^ (0,55 g, 0,78 mmol) y trietilamina (11 ml, 79 mmol) en DMF (16 ml) y se burbujeó nitrógeno a través de la mezcla durante 10 minutos. La mezcla de reacción se calentó a 70 °C durante una noche. La mezcla se diluyó en agua y EtOAc y se retiraron por filtración los sólidos de color oscuro. Se añadió salmuera al filtrado y después se separaron las capas. La capa acuosa se extrajo de nuevo con EtOAc y los extractos orgánicos combinados se concentraron al vacío. La purificación por cromatografía sobre gel de sílice (columna de 120 g; EtOAc al 20-100% en heptano) dio la mayor parte del producto deseado puro que se usó sin purificación adicional.
Formación de (+/-)-3-(1H-mdol-4-il)-1,4-oxazepano-4-carbaldehído
Una mezcla de 3-(1H-indol-4-il)-6,7-dihidro-5H-1,4-oxazepina-4-carbaldehído (4,8 g, 19,8 mmol) y Pd/C (10 % en peso Degussa, 1,6 g) en MeOH (30 ml) se agitó en una atmósfera de gas hidrógeno durante una noche en el hidrogenador Parr a 0,38 MPa (55 psi) de H2. Se filtró la mezcla de reacción a través de Florisil con la ayuda de MeOH y después por concentración, precipitó un sólido de color blanco. 1,05 g. Filtrado descartado. El sólido aislado proporcionó 1,05 g (22% - 2 etapas) del producto deseado: RMN 1H (400 MHz, DMSO-c/6) 6 (rotamérico a TA) 11,13 (s, 1H), 8,26 (s, 1H), 7,35 - 7,33 (m, 1H), 7,29 (d, J = 8,1 Hz, 1H), 7,05 - 6,99 (m, 1H), 6,85 (d, J = 7,3 Hz, 1H), 6,54 - 6,47 (m, 1H), 5,62 (dd, J = 10,5, 5,1 Hz, 1H), 4,32 (dd, J = 13,4, 5,7 Hz, 1H), 4,00 - 3,91 (m, 2H), 3,72 (m, 2H), 3,57 - 3,49 (m, 1H), 1,78 - 1,67 (m, 2H). IEN-EM m/z calc. 244,12, encontrado 245,2 (M+1)+; Tiempo de retención: 0,62 minutos.
Formación de (+/-)-3-(1H-mdol-4-il)-1,4-oxazepano
A una solución de 3-(1H-indol-4-il)-1,4-oxazepano-4-carbaldehído (0,11 g, 0,44 mmol) en THF (10 ml), se le añadió nBuLi (0,82 ml de solución 1,6 M, 1,30 mmol). La reacción se realizó a temperatura ambiente debido a problemas de insolubilidad a temperaturas más frías. Después de 30 minutos, la mezcla se diluyó en agua y se extrajo con diclorometano. La fase acuosa se extrajo de nuevo con diclorometano. Las fases orgánicas combinadas se separaron a través de un separador de fase y se concentraron al vacío para proporcionar 90 mg (95 %) del producto deseado en forma de una espuma de color amarillo pálida: RMN 1H (400 MHz, DMSO-cfe) 611,06 (s, 1H), 7,32-7,29 (m, 1H), 7,29 7,24 (m, 1H), 7,05 - 6,99 (m, 2H), 6,55 (ddd, J = 3,0, 2,0, 0,9 Hz, 1H), 4,19 (dd, J = 9,7, 3,3 Hz, 1H), 3,96 - 3,81 (m,
2H), 3,75 (dt, J = 12,0, 6,7 Hz, 1H), 3,43 (dd, J = 11,9, 9,7 Hz, 1H), 3,16 (dt, J = 13,3, 5,0 Hz, 1H), 2,93 -2,81 (m, 1H), 1.89 (td, J = 11,8, 6,4 Hz, 2H); IEN-EM m/z calc. 216,13, encontrado 217,19 (M+1)+; Tiempo de retención: 0,49 minutos.
Formación de (+/-)-4-(3-(1H-indol-4-il)-1,4-oxazepan-4-il)-6-metilpirimidin-2-aminal-142
Una mezcla de 3-(1H-indol-4-il)-1,4-oxazepano (0,09 g, 0,42 mmol) y 4-doro-6-metil-pirimidin-2-amina (0,06 g, 0,40 mmol) se calentó en NMP (1,5 ml) a 140 °C en un vial equipado con un tapón de alivio de presión durante una noche.
La reacción en bruto se cargó directamente sobre una columna C18 AQ ISCO de 50 g y se purificó por cromatografía de fase inversa eluyendo con TFA al 0,1 %/MeCN y TFA al 0,1 %/agua. Las fracciones puras se concentraron parcialmente, se añadió algo de NaOH 1 M y se extrajo con diclorometano dos veces y se concentró al vacío. Después éter y se concentró al vacío para proporcionar 79 mg (54 %) del producto deseado en forma de un sólido de color blanco: (se calentó 360K) RMN 1H (400 MHz, DMSO-d6) 610,86 (s a, 1H), 7,34 - 7,24 (m, 2H), 7,06 - 6,97 (m, 1H), 6.90 (d, J = 7,2 Hz, 1H), 6,52 (s, 1H), 5,69 (s, 1H), 5,64 (s, 1H), 5,45 (s, 2H), 4,53 (s, 1H), 4,23 (dd, J = 13,3, 5,2 Hz, 1H), 3,94 - 3,80 (m, 2H), 3,57 (ddd, J = 29,7, 18,9, 8,2 Hz, 2H), 1,94 (s, 2H), 1,87 (s, 1H), 1,73 (d, J = 15,0 Hz, 1H);
Ie N-EM m/z calc. 323,17, encontrado 324,27 (M+1)+; Tiempo de retención: 0,56 minutos.
Los siguientes análogos se prepararon:

(+/-)-4-(3-(1H-indazol-4-il)-1,4-oxazepan-4-il)-6-metilpirimidin-2-amina I-112
alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6) 612,83 (s, 1H), 8,07 (s, 1H), 7,41 (d, J = 8,4 Hz, 1H), 7,30 -7,24 (m, 1H), 7,01 (d, J = 7,1 Hz, 1H), 5,91 (s, 1H), 5,72 (s, 1H), 5,47 (s, 2H), 4,34 (s, 1H), 4,27 (dd, J = 13,2, 5,3 Hz, 1H), 3,96 (dd, J = 13,2, 8,8 Hz, 1H), 3,84 (m, 1H), 3,67 - 3,58 (m, 1H), 3,55 - 3,45 (m, 1H), 1,99 (s, 3H), 1,91 (m, 1H), 1,75 (m, 1H); IEN-EM m/z calc. 324,17, encontrado 325,26 (M+1)+; Tiempo de retención: 0,52 minutos.
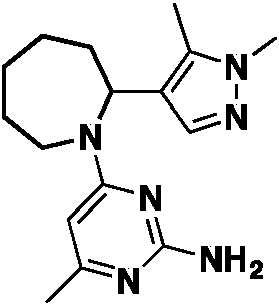
(+/-)-4-[2-(1,5-dimetilpirazol-4-il)azepan-1-il]-6-metil-pirimidin-2-amina I-48
alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6) 67,36 (s, 1H), 6,22 (s, 1H), 6,13 (d, J = 15,9 Hz, 1H), 5,81 (dt, J = 15,9, 6,8 Hz, 1H), 5,59 (s, 1H), 5,33 (s, 2H), 3,66 (s, 3H), 3,20 (dd, J = 11,5, 5,8 Hz, 2H), 2,19 (s, 3H), 2,18 -2,10 (m, 2H), 2,00 (s, 3H), 1,54 (dt, J = 13,7, 6,9 Hz, 2H), 1,49-1,40 (m, 2H); IEN-EM m/z calc. 300,21, encontrado 301,26 (M+1)+; Tiempo de retención: 0,55 minutos.

(+/-)-4-[2-(1,3-dimetilpirazol-4-il)azepan-1-il]-6-metil-pirimidin-2-amina I-39
alta temperatura (360 K) RMN 1H (400 MHz, DMSO-d6) 67,50 (s, 1H), 6,22 (s, 1H), 6,13 (d, J = 16,0 Hz, 1H), 5,81 -5,73 (m, 1H), 5,59 (s, 1H), 5,33 (s, 2H), 3,68 (s, 3H), 3,23 - 3,16 (m, 2H), 2,26 -2,12 (m, 3H), 2,00 (s, 3H), 1,79-1,14 (m, 8H); IEN-EM m/z calc. 300,21, encontrado 301,26 (M+1)+; Tiempo de retención: 0,54 minutos.
Ejemplo 51
Esquema sintético 51: 4-(3-(5-amino-2-cloro-4-fluorofenil)-1,4-oxazepan-4-il)-6-metilpirimidin-2-amina

(a) 3-(tributilestanilmetoxi)propan-1-amina, Cu(OTf)2, 2,6-lutidina, CH2CI2; (b) nBuOH, ju> 170 °C, 45 min; C. HCl, MeOH, reflujo
Formación de W-[4-cloro-2-fluoro-5-(1,4-oxazepan-3-il)feml] acetamida
A una solución de N-(4-cloro-2-fluoro-5-formil-fenil)acetamida (0,66 g, 3,06 mmol) en CH2Cl2 (20 ml), se le añadieron el amino tributilestannano - SnAP reactivo 3-(tributilestanilmetoxi) propan-1-amina (1,20 g, 3,17 mmol)(1,00 equiv.) y tamices moleculares (0,9 g). La mezcla de reacción se agitó a temperatura ambiente durante 2 h y se filtró a través de una capa corta de Celite (enjuague con CH2O 2). El filtrado se usó directamente.
Por separado, a una solución de 2,6-lutidina (440 jl, 3,798 mmol) en HFIP (15 ml) (4 ml/mmol, se secó sobre MgSO4 anhidro), se le añadió Cu(OTf)2 (1,35 g, 3,73 mmol) (1,20 equiv, precalentó a 110 °C durante 1 h a alto vacío)) y se agitó a temperatura ambiente durante 1 h, tiempo durante el cual se formó una suspensión homogénea con algo de sólido de color blanco todavía existente.
Se añadió una solución de la imina en CH2Cl2 (40 ml) (160 ml, 16 ml/mmol total) en una porción y la mezcla resultante se agitó a temperatura ambiente durante 12 h y se convirtió en una solución homogénea transparente. La reacción se interrumpió a temperatura ambiente con una mezcla de una solución acuosa saturada de NaHCO3 (40 ml) y NH4OH ac. al 10 % (20 ml) y se agitó vigorosamente durante 15 min. Las capas se separaron y la capa acuosa se extrajo con CH2Cl2 (3 x 50 ml). Las capas orgánicas combinadas se lavaron con H2O (3 x 5 ml) y salmuera (10 ml), se secaron sobre Na2SO4, se filtraron y se concentraron al vacío. La purificación por cromatografía sobre gel de sílice en Teledyne ISCO (MeOH/ CH2O 20- 8 % en 20 min) proporcionó 435 mg del producto deseado en forma de un líquido de color amarillo, después se volvió un sólido al vacío (50 %): RMN 1H (300 MHz, CDCh) 88,39 (d, J = 8,6 Hz, 1H), 7,67 (s, 1H), 7,06 (d, J = 10,4 Hz, 1H), 4,34 (dd, J = 9,3, 3,4 Hz, 1H), 4,02 -3,87 (m, 2H), 3,79 (ddd, J = 12,3, 6,7, 5,9 Hz, 1H), 3,40 (dd, J = 12,4, 9,3 Hz, 1H), 3,21 (dt, J = 13,7, 5,0 Hz, 1H), 3,01 (ddd, J = 13,6, 8,2, 5,4 Hz, 1H), 2,51 (s, 2H), 2,18 (s, 3 H), 2,07 -1,82 (m, 2H); IEN-EM m/z calc. 286,09, encontrado 287,09 (M+1)+; Tiempo de retención: 0,53 minutos.
Formación de W-[5-[4-(2-ammo-6-metil-pinmidm-4-M)-1,4-oxazepan-3-il]-4-cloro-2-fluoro-feml]acetamida I-85
Se irradiaron 4-cloro-6-metil-pirimidin-2-amina (0,11 g, 0,77 mmol) y N-[4-cloro-2-fluoro-5-(1,4-oxazepan-3-il)fenil]acetamida (0,20 g, 0,70 mmol) en nBuOH (5 ml) en microondas a 170 °C durante 45 minutos. Se retiró nBuOH al vacío y el residuo en bruto se purificó por cromatografía sobre gel de sílice: columna ISCO de 12 g, eluyendo con MeOH al 0-10 % en CH2Cl2 para proporcionar 186 mg del producto deseado en forma de un sólido de color amarillo. La RMN mostró dos rotámeros. (67 %): RMN 1H (300 MHz, CDCls) 88,36 (d, J = 8,2 Hz, 1H), 7,51 (s, 1H), 7,18 (dd, J = 19,2, 10,3 Hz, 1H), 6,02 -5,77 (m, 1H), 5,28 -5,09 (m, 1H), 4,28 (dt, J = 13,7, 5,1 Hz, 1H), 4,20 - 3,95 (m, 2H), 3,84 - 3,49 (m, 4H), 2,42 - 2,27 (m, 3H), 2,22 (d, J = 0,9 Hz, 3H), 2,03-1,82 (m, 2H); IEN-EM m/z calc. 393,14, encontrado 394,09 (M+1)+; Tiempo de retención: 0,59 minutos.
Formación de 4-[3-(5-amino-2-cloro-4-fluoro-fenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina I-269
A una solución de N-[5-[4-(2-amino-6-metil-pirimidin-4-il)-1,4-oxazepan-3-il]-4-cloro-2-fluoro-fenil]acetamida (0,065 g, 0,162 mmol) en metanol (0,25 ml), se le añadió HCl (1 ml de 2 M, 2,000 mmol). La solución se calentó a 100 °C hasta que la CLEM indicó que no había más amina de partida (2h). La mayoría del disolvente se retiró al vacío y la solución restante se neutralizó con una solución acuosa saturada de NaHCO3. Después, la fase acuosa se extrajo con CH2O 2 (3 x 5 ml). Las capas orgánicas combinadas se secaron (MgSO4), se filtraron y se concentraron al vacío para proporcionar 42 mg del producto deseado en forma de un sólido de color amarillo (73 %): RMN 1H (300 MHz, CDCla) 8 7,06 (d, J = 10,4 Hz, 1H), 6,65 (d, J = 9,0 Hz, 1H), 5,53 (s, 4H), 4,30 (dd, J = 13,7, 5,0 Hz, 1H), 4,17-4,01 (m, 1H), 3,82 (d, J = 14,4 Hz, 2H), 3,71 - 3,51 (m, 3H), 3,50 (s, 1H), 2,21 (s, 3H), 2,04-1,75 (m, 2H); IEN-EM m/z calc. 351,13, encontrado 352,15 (M+1)+; Tiempo de retención: 0,61 minutos.
Ejemplo 52
Esquema sintético 52: (+/-)-4-(3-(2-clorotiofen-3-il)-1,4-oxazepan-4-il)-6-metilpirimidin-2-amina I-163

(a) 3-(tributilestanilmetoxi)propan-1-amina, Cu(OTf)2, 2,6-lutidina, CH2CI2, HFIP; (b) nBuOH, ju> 170 °C, 45 min; (c) separación HPLC quiral
Formación de 3-(2-clorotiofen-3-il)-1,4-oxazepano
A una solución de 3-(tributilestanilmetoxi)propan-1-amina (reactivo SnAP) (13,0 g, 34,4 mmol) en CH2Cl2 (170 ml) a temperatura ambiente, se le añadieron 2-clorotiofeno-3-carbaldehído (5,0 g, 34,1 mmol) y tamices moleculares (5,0 g). La mezcla de reacción se agitó a temperatura ambiente durante 2 horas y se filtraron a través de una capa corta de Celite (enjuague con CH2O 2). El filtrado se concentró a presión reducida para proporcionar la imina (contiene 10 % de aldehido SM, 8, 9,96). RMN 1H (300 MHz, CDCls) 88,26 (s, 1H), 7,31 (t, J = 6,7 Hz, 1H), 7,08 -6,92 (m, 1H), 3,65 (s, 2H), 3,58 (t, J = 6,9 Hz, 2H), 3,31 (t, J = 6,2 Hz, 2H), 1,85 (p, J = 6,5 Hz, 2H), 1,44 (cd, J = 9,0, 8,0,6,1 Hz, 6H), 1,23 (h, J = 7,1 Hz, 8H), 0,82 (td, J = 8,0, 7,3, 3,7 Hz, 15H).
Por separado, a una solución de 2,6-lutidina (4,8 ml, 41,4 mmol) en hexafluoroisopropanol (150 ml), se le añadió Cu(OTf)2 (bis(trifluorometilsulfoniloxi)cobre) (15,0 g, 41,5 mmol) y se agitó a temperatura ambiente durante 1 hora, tiempo durante el cual se formó la mayor parte de una suspensión homogénea con algo de sólido de color blanco todavía existente. Una solución de la imina en CH2O 2 (500 ml) se añadió en una porción y la mezcla resultante se agitó a temperatura ambiente durante 12 horas. La reacción se interrumpió a temperatura ambiente con una mezcla de NaHCO3 acuoso saturado (60 ml) y NH4OH ac. al 10 % (40 ml) y se agitó vigorosamente durante 15 min. Las capas se separaron y la capa acuosa se extrajo con CH2Cl2 (3 x 50 ml). Las capas orgánicas combinadas se lavaron con H2O (3 x 5 ml) y salmuera (10 ml), se secaron sobre Na2SO4, se filtraron y se concentraron al vacío. La purificación por cromatografía en columna sobre gel de sílice (gradiente de 0-10 % de MeOH/ CH2O 2) proporcionó 2,4 gramos del producto deseado en forma de un líquido de color pardo claro (32 %); IEN-EM m/z calc. 217,03, encontrado 215,13 (M+1)+; Tiempo de retención: 0,36 minutos.
Formación de 4-[3-(2-cloro-3-tienM)-1,4-oxazepan-4-M]-6-metM-pinmidm-2-amma
Una mezcla de 4-cloro-6-metil-pirimidin-2-amina (0,21 g, 1,46 mmol) y 3-(2-cloro-3-tienil)-1,4-oxazepano (0,32 g, 1,41 mmol) en n-butanol (3 ml) se irradió en un microondas durante 1 hora en un tubo cerrado herméticamente a 170 °C. La mezcla se concentró a sequedad y se purificó por cromatografía sobre gel de sílice eluyendo con MeOH al 0 10 %/CH2Cl2. Las fracciones puras se combinaron y se concentraron para dar 455 mg del producto deseado en forma de un sólido de color amarillo claro (99 %): RMN 1H (300 MHz, cloroformo-d) 87,16 (d, J = 5,6 Hz, 1H), 6,83 (s, 1H), 6,33 (s, 2H), 5,75 (s, 1H), 4,34 - 4,17 (m, 1H), 4,06 (s, 1H), 3,77 (s, 2H), 3,74 - 3,42 (m, 2H), 2,36 (s, 3H), 2,18-1,80 (m, 2H); IEN-EM m/z calc. 324,08, encontrado 325,05 (M+1)+; Tiempo de retención: 0,67 minutos; IEN-EM m/z calc.
324,08115, encontrado 325,05 (M+1)+; Tiempo de retención: 0,67 minutos.
El compuesto del título se sometió a separación HPLC quiral de enantiómeros:
Pico A: I-211
4-[3-(2-cloro-3-tienil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (128 mg): RMN 1H (300 MHz, CDCla) 86,98 (d, J = 5,8 Hz, 1H), 6,72 (d, J = 5,8 Hz, 1H), 5,58 (s, 1H), 5,25 (s, 1H), 4,89 (s, 2H), 4,12 (dd, J = 13,5, 5,4 Hz, 1H), 4,05 -3.84 (m, 1H), 3,75-3,57 (m, 1H), 3,57-3,25 (m, 2H), 2,09 (s, 3H), 2,01-1,52 (m, 2H); IEN-EM m/z calc. 324,08, encontrado 325,15 (M+1)+; Tiempo de retención: 0,62 minutos Hp Lc quiral: >98 % ee, Ac. Método: MeOH al 20 %-EtOH al 30 %-HEX al 50 % en 20 min en columna PAK IC quiral
Rotación óptica: T = 20,6 °C, 5 mg en 1 ml de CHCh, C=1, [a] = 0,92°
Pico B: I-212
4-[3-(2-cloro-3-tienil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (142 mg): RMN 1H (300 MHz, CDCh) 86,98 (d, J = 5,8 Hz, 1H), 6,72 (d, J = 5,8 Hz, 1H), 5,58 (s, 1H), 5,25 (s, 1H), 4,89 (s, 2H), 4,12 (dd, J = 13,5, 5,4 Hz, 1H), 4,05 -3.84 (m, 1H), 3,75 - 3,57 (m, 1H), 3,57 - 3,25 (m, 2H), 2,09 (s, 3H), 2,01-1,52 (m, 2H); IEN-EM m/z calc. 324,08, encontrado 325,15 (M+1)+; Tiempo de retención: 0,62 minutos; Tiempo de retención: 0,62 minutos HPLC quiral: >98 % ee, Método Ac.: MeOH al 20 %, EtOH al 30 %, HEX al 50 %en 20 min en columna PAK IC quiral
Rotación óptica: T = 23,2 °C, 5 mg en 1 ml de CHCh, C=1, [a] = 2,0°
Los siguientes análogos se prepararon de acuerdo con el Esquema sintético 52: I-209 e I-210:

Separación SFC de E29862-1390: 4-[3-(3-doro-2-tienil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (295 mg, 0,9082 mmol) Columna: Cellulose-2, 20x250 mm Fase móvil: MeOH al 40 % (amoniaco 5 mM), CO2 al 60 % isocrático
Pico A: I-210
4-[3-(3-cloro-2-tienil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (122 mg, 82 %) RMN 1H (300 MHz, Cloroformo-d) 8 7.19 (d, J = 5,3 Hz, 1H), 6,91 (d, J = 5,3 Hz, 1H), 5,75 (s, 1H), 4,78 (s, 2H), 4,33 (dd, J = 13,5, 5,5 Hz, 1H), 4,08 (dd, J = 12,6, 5,1 Hz, 1H), 3,74 (dd, J = 13,5, 10,1 Hz, 1H), 3,68 - 3,47 (m, 2H), 2,21 (s, 3H), 2,00 (dddd, J = 14,0, 11,4, 5,4, 2.7 Hz, 1H), 1,81 (dd, J = 14,3, 2,5 Hz, 1H). IEN-EM m/z calc. 324,08115, encontrado 325,1 (M+1)+; Tiempo de retención: 0,63 minutos
HPLC quiral: >98 % ee, Ac. Método: MEOH al 20 %-ETOH al 30 %-HEX al 50 %en 20 min en columna PAK IC quiral rotación óptica: T = 24,2 °C, 5 mg en 1 ml de CHCl3, C=1, [a] = 6,8°
Pico B: I-209
4-[3-(3-cloro-2-tienil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (121 mg, 82 %) RMN 1H (300 MHz, Cloroformo-d) 8 7.19 (d, J = 5,3 Hz, 1H), 6,91 (d, J = 5,3 Hz, 1H), 5,75 (s, 1H), 4,78 (s, 2H), 4,33 (dd, J = 13,5, 5,5 Hz, 1H), 4,08 (dd, J = 12,6, 5,1 Hz, 1H), 3,74 (dd, J = 13,5, 10,1 Hz, 1H), 3,68 - 3,47 (m, 2H), 2,21 (s, 3H), 2,00 (dddd, J = 14,0, 11,4, 5,4, 2.7 Hz, 1H), 1,81 (dd, J = 14,3, 2,5 Hz, 1H). IEN-EM m/z calc. 324,08115, encontrado 325,1 (M+1)+; Tiempo de retención: 0,62 minutos HPLC quiral: >98 % ee, Ac. Método: MEOH al 20 %-ETOH al 30 %-HEX al 50 %en 20 min en columna PAK IC quiral rotación óptica: T = 24,3 °C, 5 mg en 1 ml de CHCl3, C=1, [a] = -7,4°
Ejemplo 53
Esquema sintético 53: 4-[4-(2-ammo-6-metil-pirimidm-4-il)-1,4-oxazepan-3-il]-5-cloro-W,W-dimetil-tiofeno-2-carboxamida I-244

(a) NBS, CH3CN; (b) nBuLi, CO2, THF, -78 °C; (c) Me2NH, iPr2NEt, T3P, EtOAc; (d) HC1, MeOH, Reflujo; (e) nBuOH, ju>, 170 °C, 45 min
Formación de 3-(5-bromo-2-cloro-3-tienil)-1,4-oxazepano-4-carboxilato de íerc-butilo
A una solución de 3-(2-cloro-3-tienil)-l,4-oxazepano-4-carboxilato de íerc-butilo (0,42 g, 1,30 mmol) en CH3CN (5 ml), se le añadió NBS (0,25 g, 1,40 mmol) a temperatura ambiente. La reacción se agitó a temperatura ambiente durante 1 hora. La solución se evaporó y se purificó por cromatografía sobre gel de sílice usando columna ISCO de 40 g eluyendo con EtOAc/Hexano (0-30 %) para proporcionar 495 mg del producto en forma de un aceite de color amarillo transparente (95 %): RMN 1H (300 MHz, CDCl3) 86,73 (s, 1H), 5,45 - 5,00 (m, 1H), 4,39 - 3,78 (m, 3H), 3,35 (dt, J = 60,4, 12,8 Hz, 3H), 1,86 (dtdd, J = 13,7, 10,9, 5,0, 2,7 Hz, 1H), 1,77 - 1,68 (m, 1H), 1,51 - 1,22 (m, 9H) Rotámeros, relación: 1:2,5; Ie N-EM m/z calc. 395,00, encontrado 395,75 (M+1)+; Tiempo de retención: 1,07 minutos.
Formación de ácido 4-(4-ferc-butoxicarboml-1,4-oxazepan-3-il)-5-cloro-tiofeno-2-carboxflico A una solución fría (-70 °C) de 3-(5-bromo-2-cloro-3-tienil)-1,4-oxazepano-4-carboxilato de ferc-butilo (0,186 g, 0,468 mmol) en THF (4 ml), se le añadió gota a gota n-BuLi (0,220 ml de 2,5 M, 0,550 mmol). El color de la solución cambió de color amarillo claro a pardo oscuro de inmediato. Después de 15 minutos, se añadió hielo seco CO2 (0,5 g, 10 mmol) y la reacción se agitó durante 30 minutos y después se dejó calentar a temperatura ambiente seguido de tratamiento con solución acuosa saturada de NH4Cl y EtOAc. La fase orgánica se secó (MgSO4), se filtró y se concentró al vacío para proporcionar 160 mg del producto en bruto, (93 %): RMN 1H (300 MHz, Cd CI3) 87,41 (s, 1H), 5,19 (d, J = 40,5 Hz, 1H), 4,16 (s, 1H), 3,95 (d, J = 37,3 Hz, 2H), 3,68 -2,98 (m, 3H), 1,93 - 1,51 (m, 2H), 1,20 (cd, J = 6,9, 6,2, 2,7 Hz, 9H); iEn-EM m/z calc. 361,08, encontrado 362,1 (M+1)+; Tiempo de retención: 0,87 minutos.
Formación de 3-[2-cloro-5-(dimetilcarbamol.)-3-tienil]-1,4-oxazepano-4-carboxilato de ferc-butilo
A una solución de ácido 4-(4-ferc-butoxicarbonil-1,4-oxazepan-3-il)-5-cloro-tiofeno-2-carboxílico (0,150 g, 0,415 mmol) en EtOAc (2 ml), se le añadieron secuencialmente dimetilamina (1,0 ml de 2 M, 2,0 mmol), diisopropiletil amina (0,150 ml, 0,861 mmol) y T3P (0,50 ml de 50 % p/p, 0,84 mmol) en EtOAc. La reacción se agitó durante una noche. La CLEM indicó solo producto. Lavado acuoso con solución acuosa saturada de NH4CI y salmuera. La fase orgánica se secó sobre MgSO4, se filtró y se concentró al vacío para proporcionar 157 mg del producto en bruto en forma de un sólido de color amarillo (97 %): RMN 1H (300 MHz, CDCI3) 87,09 (d, J = 37,5 Hz, 1H), 5,26 (d, J = 49,8 Hz, 1H), 4,43 -3,78 (m, 4H), 3,66 -3,38 (m, 2H), 3,09 (s, 6H), 1,94 -1,54 (m, 2H), 1,45-1,21 (m, 9H); IEN-EM m/z calc. 388,12, encontrado 389,27 (M+1)+; Tiempo de retención: 0,85 minutos.
Formación de 5-cloro-W,W-dimetil-4-(1,4-oxazepan-3-il)tiofeno-2-carboxamida
A una solución de 3-[2-cloro-5-(dimetilcarbamol.)-3-tienil]-1,4-oxazepano-4-carboxilato de ferc-butilo (0,16 g, 0,41 mmol) en 1,4-dioxano (3 ml), se le añadió HCl (1,0 ml de 4 M, 4,0 mmol). Después de agitar durante 1 hora a temperatura ambiente los volátiles se retiraron para proporcionar 120 mg del producto deseado en forma de una sal TFA, que se usó sin purificación adicional: IEN-Em m/z calc. 288,07, encontrado 289,16 (M+1)+; Tiempo de retención: 0,53 minutos.
Formación de 4-[4-(2-ammo-6-metil-pirimidm-4-il)-1,4-oxazepan-3-il]-5-cloro-W,W-dimetil-tiofeno-2-carboxamida I-244
Una solución de 5-cloro-W,W-dimetil-4-(1,4-oxazepan-3-il)tiofeno-2-carboxamida-sal TFA (0,120 g) y 4-cloro-6-metilpirimidin-2-amina (0,075 g, 0,522 mmol) en n-BuOH (3 ml) se irradió en un reactor de microondas a 170 °C durante 45 minutos. Se retiró nBuOH a presión reducida y el residuo en bruto se purificó por cromatografía sobre gel de sílice usando una columna ISCO de 12 g, eluyendo con MeOH al 0-10 % en CH2CI2 para proporcionar 40 mg del producto deseado en forma de un sólido de color blanco (25 %): RMN 1H (300 MHz, CDCI3) 87,15 (s, 1H), 5,70 (s, 1H), 5,24 (s, 2H), 4,42 (s, 2H), 4,19 (dd, J = 13,5, 5,2 Hz, 1H), 4,00 (d, J = 11,7 Hz, 1H), 3,76 (dd, J = 13,4, 9,5 Hz, 1H), 3,64 (d, J = 10,3 Hz, 1H), 3,50 (t, J = 13,1 Hz, 1H), 3,15 (s, 6H), 2,25 (s, 3H), 2,04 (s, 1H), 1,83 (d, J = 14,4 Hz, 1H); IEN-EM m/z calc. 395,11, encontrado 396,16 (M+1)+; Tiempo de retención: 0,59 minutos.
Los siguientes análogos se prepararon de acuerdo con el Esquema sintético 53:
4-(4-(2-ammo-6-metilpirimidm-4-il)-1,4-oxazepan-3-il)-5-cloro-W-metiltiofeno-2-carboxamida I-182

RMN 1H (300 MHz, Metanol-d4) 87,53 (d, J = 15,5 Hz, 1H), 6,46-5,88 (s, 1H), 6,02-5,19 (dd, J = 9,3, 5,4 Hz, 1H), 4,29 3,60 (m, 6H), 2,84 (d, J = 2,4 Hz, 3H), 2,30 (d, J = 24,3 Hz, 3H), 1,56-1,34 (m, 3H).
Ejemplo 54
Esquema sintético 54: (+/-)-4-[3-(2-cloro-4-dimetilfosforil-fenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina I-202

(a) metilfosfonilmetano, K3PO4, Pd(OAc)2, Xantphos, DMF; (b) TFA, CH2CI2; (c) 2-amino-4-cloro-6-metilpirimidina, nBuOH, 120 °C; (d) separación HPLC quiral
Formación de 3-(2-cloro-4-dimetilfosforil-fenil)-1,4-oxazepano-4-carboxilato de (+/-)-íerc-butilo
Un tubo de Schlenk se cargó con 3-(4-bromo-2-cloro-fenil)-1,4-oxazepano-4-carboxilato de ferc-butilo (223 mg, 0,542 mmol), Xantphos (37 mg, 0,065 mmol), Pd(OAc)2 (12,2 mg, 0,054 mmol), metilfosfonoilmetano (70 mg, 0,897 mmol), K3PO4 (230 mg, 1,08 mmol), DMF (3 ml) y ciclos de vacío/nitrógeno tres veces, después se sumergió en un baño caliente a 120 °C durante la noche. Se añadieron DCM y agua a la mezcla de reacción y las capas se separaron con la ayuda de un separador de fase. La capa acuosa se extrajo de nuevo con DCM, se separó con la ayuda de un separador de fase y los orgánicos combinados se concentraron. El residuo resultante se purificó por cromatografía sobre gel de sílice eluyendo con EtOAc al 30-100 %/heptano, después MeOH al 0-10 %/DCM. Las fracciones puras se combinaron y se concentraron al vacío para proporcionar 63 mg de 3-(2-cloro-4-dimetilfosforil-fenil)-1,4-oxazepano-4-carboxilato de ferc-butilo en forma de un aceite de color pajizo. RMN 1H (400 MHz, MeOH-d4) 87,84 (d, J = 11,7 Hz, 1H), 7,78 - 7,68 (m, 1H), 7,52 (dd, J = 7,9, 3,0 Hz, 1H), 5,52 (dd, J = 10,7, 4,2 Hz, 1H), 4,42 (d, J = 15,7 Hz, 1H), 4,29 (s, 1H), 4,08 - 4,00 (m, 1H), 3,71 - 3,52 (m, 3H), 1,86 (s, 2H), 1,83 (s, 3H), 1,79 (s, 3H), 1,24 (s, 6H); IEN-EM m/z calc.
387,1, encontrado 388,3 (M+1)+; Tiempo de retención 0,67 minutos.
Formación de (+/-)-3-(2-cloro-4-dimetilfosforil-fenil)-1,4-oxazepano
Se disolvió 3-(2-cloro-4-dimetilfosforil-fenil)-1,4-oxazepano-4-carboxilato de ferc-butilo (63 mg, 0,162 mmol) en diclorometano (1 ml) y se añadió ácido trifluoroacético (0,5 ml). Después de 15 min, los volátiles se retiraron en un evaporador rotatorio. La mezcla de reacción se concentró y después se disolvió en MeOH y se pasó a través de un cartucho de bicarbonato SPE (Agilent Stratospheres 500 mg/6 ml) y se concentró para proporcionar 38 mg del producto deseado en forma de un aceite de color pajizo. RMN 1H (400 MHz, MeOH-d4) 87,87 - 7,82 (m, 1H), 7,80 - 7,71 (m, 2H), 4,49 (dd, J = 9,2, 3,5 Hz, 1H), 4,02 - 3,94 (m, 2H), 3,90 - 3,82 (m, 1H), 3,49 (dd, J = 12,5, 9,2 Hz, 1H), 3,25 (dt, J = 13,9, 5,0 Hz, 1H), 3,05 -2,96 (m, 1H), 2,08 - 1,99 (m, 2H), 1,82 (d, J = 2,9 Hz, 3H), 1,79 (d, J = 2,9 Hz, 3H). IEN-EM m/z calc. 287,1, encontrado 288,3 (M+1)+; Tiempo de retención: 0,46 minutos.
Formación de (+/-)-4-[3-(2-cloro-4-dimetilfosforil-fenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina
Una mezcla de 4-cloro-6-metil-pirimidin-2-amina (18,4 mg, 0,128 mmol) y 3-(2-cloro-4-dimetilfosforilfenil)-1,4-oxazepano (38 mg, 0,132 mmol) en nBuOH (1,3 ml) se calentó a 125 °C durante una noche. La mezcla de reacción se concentró y el residuo resultante se purificó por cromatografía sobre gel de sílice eluyendo con MeOH al 0 30 %/DCM para proporcionar 14 mg de un sólido de color blanco. RMN 1H (400 MHz, MeOH-d4) 87,89 (d, J = 11,5 Hz, 1H), 7,75 - 7,67 (m, 1H), 7,51 (dd, J =8,0, 3,0 Hz, 1H), 7,00-4,90 (s a, 3H), 4,35 (dd, J = 13,7, 5,0 Hz, 1H), 4,06 (d, J = 11,8 Hz, 1H), 3,83 (dd, J = 13,7, 10,3 Hz, 2H), 3,68 (dd, J = 18,9, 8,2 Hz, 1H), 2,22 (s, 3H), 2,00-1,82 (m, 2H), 1,82 (s, 3H), 1,79 (s, 3H). IEN-EM m/z calc. 394,1, encontrado 395,4 (M+1)+; Tiempo de retención: 0,51 minutos.
Separación HPLC quiral: Columna: AD-H, 20x250 mm; Fase móvil: Hexanos al 70 %, EtOH al 30 %/MeOH (Dietilamina al 0,2 %); Flujo: 20 ml/min; Concentraciones: ~15 mg/ ml (MeOH). Estereoquímica absoluta de cada pico sin asignar.
Pico A: 4-[3-(2-cloro-4-dimetilfosforil-fenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina; 99 % ee; RMN 1H (400 MHz, DMSO-d6) 87,77 (dd, J = 11,3, 1,3 Hz, 1H), 7,67 - 7,60 (m, 1H), 7,43 (dd, J = 7,9, 2,9 Hz, 1H), 5,64 (s, 1H), 5,54 (s, 1H), 5,44 (s, 2H), 4,55 (s, 1H), 4,14 (dd, J = 13,4, 5,0 Hz, 1H), 3,90 (d, J = 11,7 Hz, 1H), 3,78 -3,66 (m, 2H), 3,56 (dd, J = 14,6, 12,2 Hz, 1H), 2,02 (s, 3H), 1,81-1,75 (m, 2H), 1,65 (s, 3H), 1,62 (s, 3H). IEN-EM m/z calc. 394,1, encontrado
395,2 (M+1)+; Tiempo de retención: 0,5 minutos. I-213
Pico B: 4-[3-(2-cloro-4-dimetilfosforil-fenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina; 99 % ee; RMN 1H (400 MHz, DMSO-da) 57,77 (dd, J = 11,3, 1,3 Hz, 1H), 7,67 - 7,60 (m, 1H), 7,43 (dd, J = 7,9, 2,9 Hz, 1H), 5,64 (s, 1H), 5,54 (s, 1H), 5,44 (s, 2H), 4,55 (s, 1H), 4,14 (dd, J = 13,4, 5,0 Hz, 1H), 3,90 (d, J = 11,7 Hz, 1H), 3,78 - 3,66 (m, 2H), 3,56 (dd, J = 14,6, 12,2 Hz, 1H), 2,02 (s, 3H), 1,81-1,75 (m, 2H), 1,65 (s, 3H), 1,62 (s, 3H). IEN-EM m/Z calc. 394,1, encontrado 395,1 (M+1)+; Tiempo de retención: 0,5 minutos. I-214
Ejemplo 55
Esquema sintético 55: 4-(3-(6-cloroimidazo[1,2-d]piridm-7-il)-1,4-oxazepan-4-il)-6-metilpirimidm-2-amma
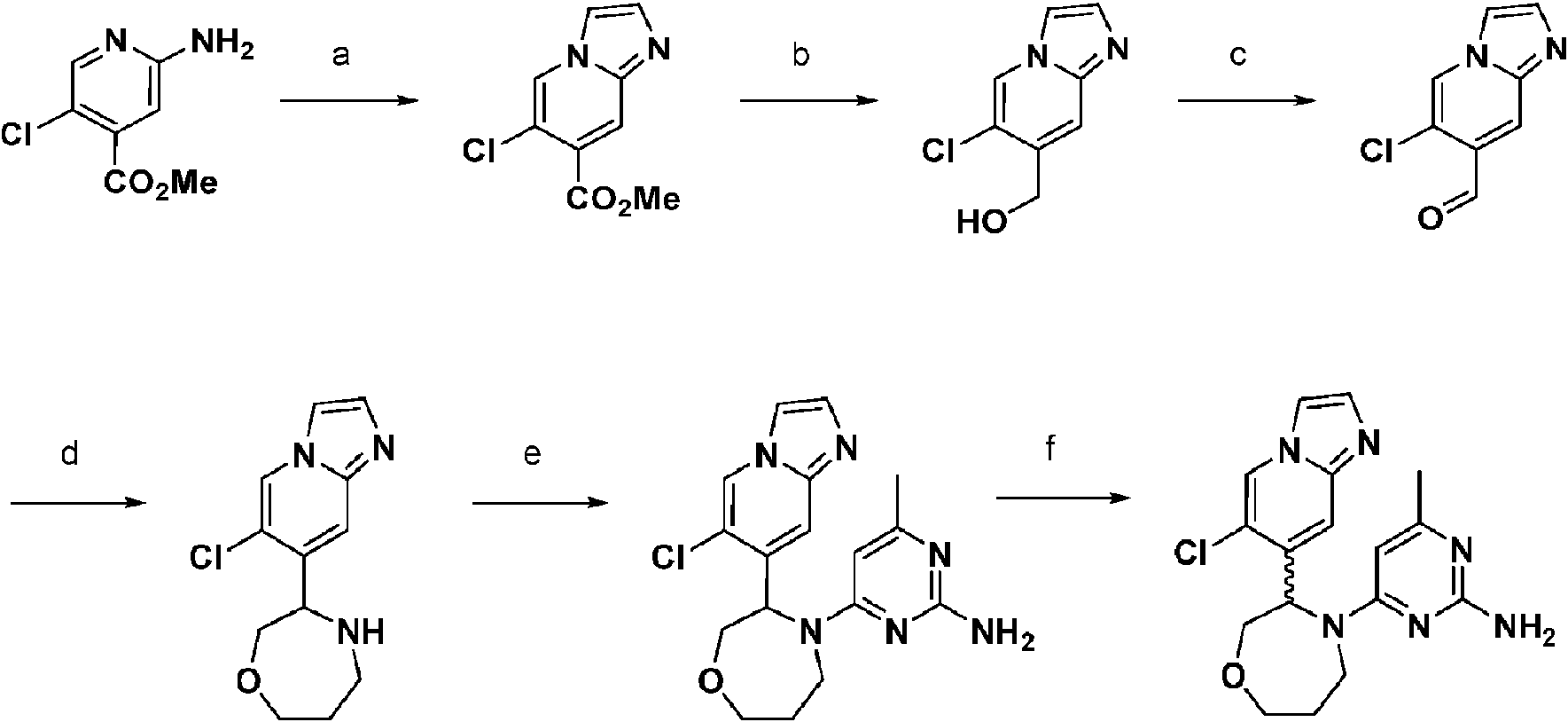
(a) 2-cloroacetaldehído, EtOH; (b) DIBAL-H, DCM, THF; (c) MnO2, DCM, 2-MeTHF, acetona; (d) 3-(tributilestannil)metoxi)propan-1-amina tamices mol. de 4 A, CH2Cl2; después 2,6-lutidina, Cu(OTf)2, hexafluoroisopropanol; (e) 2-amino-4-cloro-6-metilpirimidina, NMP, 150 °C; (f) separación HPLC quiral
Formación de 6-cloroimidazo[1,2-a]piridin-7-carboxilato de metilo
Una mezcla de 2-amino-5-cloro-piridin-4-carboxilato de metilo (7,15 g, 38,3 mmol), 2-cloroacetaldehído (7,3 ml, 115 mmol) y EtOH (60 ml) se calentó a reflujo. Después de 3 h, se añadió un adicional de 3 ml de cloroacetaldehído y la agitación se continuó durante una noche. La mezcla de reacción se concentró parcialmente. Se añadieron agua y NaOH 6 M y la mezcla se extrajo con EtOAc dos veces. Los extractos orgánicos combinados se secaron (Na2SO4), se filtraron y se concentraron dando 8 g de 6-cloroimidazo[1,2-a]piridin-7-carboxilato de metilo en forma de un sólido de color grisáceo pardo: RMN 1H (400 MHz, DMSO-da) 58,96 (d, J = 0,6 Hz, 1H), 8,13 (d, J = 4,2 Hz, 1H), 8,07 (d, J = 0,9 Hz, 1H), 7,81 (d, J = 1,1 Hz, 1H), 3,89 (s, 3H). IEN-EM m/z calc. 210,0, encontrado 211,1 (M+1)+; Tiempo de retención: 0,48 minutos.
Formación de (6-cloroimidazo[1,2-a]piridin-7-il)metanol
A una solución de 6-cloroimidazo[1,2-a]piridin-7-carboxilato de metilo (8 g, 37,98 mmol) en DCM (60 ml) y THF (100 ml, para ayudar la solubilidad), se le añadió a DIBAL-H (1 M, 45,6 mmol) a -78 °C durante 1 h. La mezcla de reacción se dejó calentar a TA durante una noche. Se añadieron otros 15 ml de DIBAL-H enfriando con baño de hielo. Después de 2 h a la misma temperatura, Se añadió sal Rochelle (1,5 M, 200 ml) y la agitación se continuó durante un adicional de 2 h. Se retiraron 3,7 g del producto deseado por filtración en forma de un sólido de color blanco después de lavarse con agua: RMN 1H (400 MHz, D M S O ^) 58,82 (s, 1H), 7,88 (s, 1H), 7,58 (d, J = 1,2 Hz, 2H), 5,55 (t, J = 5,6 Hz, 1H), 4,57 (dd, J = 5,6, 1,3 Hz, 2H). IEN-EM m/Z calc. 182,0, encontrado 183,0 (M+1)+; Tiempo de retención: 0,44 minutos.
Formación de 6-cloroimidazo[1,2-a]piridin-7-carbaldehído
A una suspensión de (6-cloroimidazo[1,2-a]piridin-7-il)metanol (3,7 g, 20,3 mmol) en DCM (60 ml), 2-MeTHF (50 ml) y acetona (50 ml), se le añadió MnO2 activado (10 g, 115 mmol). Después de tres días a 50 °C, la mezcla de reacción se filtró a través de Celite con la ayuda de EtOAc y se concentró. Se añadió EtOAc, la mezcla se sonicó y 2,93 g del producto deseado se retiraron por filtración en forma de un sólido de color amarillo: RMN 1H (400 MHz, DMSO-d6) 5 10,18 (s, 1H), 8,97 (d, J = 0,5 Hz, 1H), 8,20 (s, 1H), 8,14 (d, J = 0,8 Hz, 1H), 7,90 (d, J = 1,1 Hz, 1H). IEN-EM m/Z calc.
180,0, encontrado 181,0 (M+1)+; Tiempo de retención: 0,45 minutos.
Formación de 3-(6-doroimidazo[1,2-a]piridm-7-il)-1,4-oxazepano y 4-(3-(6-doroimidazo[1,2-a]piridm-7-il)- 1,4-oxazepan-4-il)-6-metilpirimidin-2-amina (I-191)
La formación del producto del sistema de anillo de oxazepano seguido de la adición a 2-amino-4-cloro-6-metilpirimidina se llevó a cabo de la misma manera que se muestra en el Esquema sintético 55 para producir 4-[3-(6-cloroimidazo[1,2-a]piridin-7-il)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina I-191.
Separación HPLC quiral: Columna: AD-H, 20x250 mm; Fase móvil: Hexanos al 70 %, EtOH al 30 %/MeOH (Dietilamina al 0,2 %); Flujo: 20 ml/min; Concentraciones: ~15 mg/ ml (MeOH) proporcionó el enantiómero individual (I-221): RMN 1H (400 MHz, DMSO-da) 58,78 (s, 1H), 7,86 (s, 1H), 7,56 (d, J = 1,1 Hz, 1H), 7,40 (s, 1H), 5,71 (s, 1H), 5,45 (s, 3H), 4,51 (d, J = 15,1 Hz, 1H), 4,18 (dd, J = 13,5, 4,8 Hz, 1H), 3,90 - 3,80 (m, 2H), 3,73 (ddd, J = 15,2, 9,5, 3,5 Hz, 1H), 3,60 (ddd, J = 12,1,9,7, 4,7 Hz, 1H), 2,03 (s, 3H), 1,83-1,79 (m, 2H); IEN-EM m/z calc. 358,1, encontrado 359,1 (M+1)+; Tiempo de retención: 0,45 minutos.
Ejemplo 56
Esquema sintético 56: 4-(3-(2-doro-4-(ddobutilsulfoml)feml)-1,4-oxazepan-4-il)-6-metilpirimidm-2-amma I-222

(a) ciclobutanotiol, Et3N, CH3CN, 60 °C; (b) Oxone, MeOH, H2O; (c) NaOH, H2O, después HCl; (d) TMS-diazometano, tolueno, metanol: (e) NaBH4, MeOH; (f) Peryodinano de Dess-Martin, diclorometano; (g) tamices mol. de 4 A, 3-((tributilestannil)metoxi)propan-1-amina, CH2Ch; después 2,6-lutidina, Cu(OTf)2, hexafluoroisopropanol, CH2Ch; (h) 2-amino-4-cloro-6-metilpirimidina, NMP, 150 °C;
Formación de 2-cloro-4-(ciclobutiltio)benzonitrilo
Una mezcla de 2-cloro-4-fluoro-benzonitrilo (1,00 g, 6,43 mmol), ciclobutanotiol (1,15 g, 13,04 mmol) y trietilamina (1,79 ml, 12,84 mmol) en acetonitrilo (8 ml) se agitó en un tubo cerrado herméticamente a 60 °C durante una noche. La mezcla se diluyó con agua y se extrajo con EtOAc. La capa orgánica se concentró a sequedad, se cargó en seco sobre gel de sílice suelto y se purificó mediante cromatografía en gel de sílice eluyendo con 0-25 % de EtOAc en heptano. Las fracciones que contenían el producto deseado se combinaron y se concentraron para proporcionar 1,0 g del producto deseado en forma de un aceite incoloro: RMN 1H (400 MHz, cloroformo-d) 57,49 (d, J = 8,3 Hz, 1H), 7,20 (d, J = 1,8 Hz, 1H), 7,07 (dd, J = 8,3, 1,8 Hz, 1H), 3,98 (dc, J = 9,5, 7,1, 6,3 Hz, 1H), 2,65 - 2,50 (m, 2H), 2,21 2,02 (m, 4H)
Formación de 2-cloro-4-(ciclobutilsulfonil)benzonitrilo
A una solución de 2-cloro-4-(ciclobutiltio)benzonitrilo (2,8 g, 11,9 mmol) en MeOH (150 ml), se le añadió una solución de Oxone (14,6 g, 23,8 mmol) en agua (75 ml). La mezcla de reacción se agitó durante 2 días a temperatura ambiente y después se concentró a sequedad. El precipitado de color blanco resultante se repartió entre agua y EtOAc. La capa orgánica se concentró a sequedad y se purificó por cromatografía sobre gel de sílice eluyendo con EtOAc al 0-50 % en heptano. Las fracciones puras se combinaron y se concentraron para dar 1,07 g del producto deseado en forma de un sólido de color blanco: RMN 1H (400 MHz, Cloroformo-d) 58,02 (t, J = 1,1 Hz, 1H), 7,86 (d, J = 1,1 Hz, 2H), 3,83 (pd, J = 8,2, 0,7 Hz, 1H), 2,66 - 2,50 (m, 2H), 2,28 - 2,17 (m, 2H), 2,12 - 1,96 (m, 2H).
Formación de ácido 2-doro-4-(ddobutilsulfoml)benzoico
Una solución de gránulos de 2-cloro-4-(ciclobutilsulfonil)benzonitrilo (1,14 g, 4,46 mmol) y NaOH (0,40 g, 10,00 mmol) en agua (30 ml) se calentó a reflujo durante 4 horas, se enfrió a temperatura ambiente y se acidificó a pH ~ 3 usando
HCl (10 ml de 6 M, 60 mmol). El precipitado de color blanco resultante se filtró, se lavó con agua y se secó al vacío durante una noche para proporcionar 1,21 g del producto deseado en forma de un polvo de color blanco: RMN 1H (400 MHz, Cloroformo-d) 87,93 (d, J = 8,1 Hz, 1H), 7,81 (d, J = 1,7 Hz, 1H), 7,65 (dd, J = 8,2, 1,7 Hz, 1H), 3,71 -3,59 (m, 1H), 2,47 -2,33 (m, 2H), 2,09 - 1,97 (m, 2H), 1,91 -1,77 (m, 2H).
Formación de 4-(3-(2-doro-4-(ddobutilsulfoml)feml)-1,4-oxazepan-4-il)-6-metilpirimidm-2-amma
La conversión de ácido 2-cloro-4-(ciclobutilsulfonil)benzoico al compuesto del título se preparó de acuerdo con el procedimiento enumerado en el Esquema sintético 8: RMN 1H (400 Mh z , DMSO-d6) 87,83 (d, J = 1,9 Hz, 1H), 7,76 -7,68 (m, 1H), 7,56 (d, J = 8,2 Hz, 1H), 5,69 - 5,51 (m, 2H), 5,46 (s, 2H), 4,48 (d, J = 15,3 Hz, 1H), 4,12 (dc, J = 12,3, 7,8, 6,8 Hz, 2H), 3,95 - 3,84 (m, 1H), 3,84 - 3,64 (m, 2H), 3,57 (c, J = 10,1, 8,6 Hz, 1H), 2,34 (p, J = 9,6, 9,0 Hz, 2H), 2,22 -2,07 (m, 2H), 2,02 (d, J = 3,7 Hz, 3H), 1,93 -1,87 (m, 2H), 1,84-1,74 (m, 2H); IEN-EM m/z calc. 436,1, encontrado 437,0 (M+1)+; Tiempo de retención: 0,68 minutos.
Los siguientes análogos se prepararon de acuerdo con el Esquema sintético 56:
4-(3-(2-doro-4-(etilsulfonil)fenil)-1,4-oxazepan-4-il)-6-metilpirimidin-2-amina I-183 y I-317

Pico A: 4-[3-(2-cloro-4-etilsulfonil-fenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina: RMN 1H (400 MHz, DMSO-cfe) 87,89 (d, J = 1,9 Hz, 1H), 7,77 (dd, J = 8,1, 1,9 Hz, 1H), 7,57 (d, J = 8,2 Hz, 1H), 5,66 (s, 1H), 5,58 (dd, J = 10,2, 4.8 Hz, 1H), 5,45 (s, 2H), 4,50 (d, J = 15,3 Hz, 1H), 4,15 (dd, J = 13,5, 4,9 Hz, 1H), 3,90 (dt, J = 12,3, 3,9 Hz, 1H), 3,83 3,69 (m, 2H), 3,62 - 3,52 (m, 1H), 3,30 (c, J = 7,4 Hz, 2H), 2,02 (s, 3H), 1,80 (dc, J = 11,0, 6,7, 5,4 Hz, 2H), 1,12 (d, J = 7,4 Hz, 3h ); IEN-EM m/z calc. 410,12, encontrado 411,0 (M+1)+; Tiempo de retención: 0,64 minutos.
Pico B: 4-[3-(2-cloro-4-etilsulfonil-fenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina RMN 1H (400 MHz, DMSO-d6) 87,88 (d, J = 1,8 Hz, 1H), 7,77 (dd, J = 8,2, 1,9 Hz, 1H), 7,57 (d, J = 8,2 Hz, 1H), 5,66 (s, 1H), 5,58 (dd, J = 10,0, 4.8 Hz, 1H), 5,44 (s, 2H), 4,50 (d, J = 15,3 Hz, 1H), 4,15 (dd, J = 13,5, 4,9 Hz, 1H), 3,90 (dt, J = 11,9, 3,9 Hz, 1H), 3,75 (ddd, J = 18,4, 14,4, 8,0 Hz, 2H), 3,57 (dt, J = 12,1, 7,2 Hz, 1H), 3,30 (c, J = 7,3 Hz, 2H), 2,02 (s, 3H), 1,80 (dc, J = 7,2, 4,2 Hz, 2H), 1,12 (dd, J = 7,4, 0,9 Hz, 3H); IEN-EM m/z calc. 410,12, encontrado 411,0 (M+1)+; Tiempo de retención: 0,64 minutos.
Ejemplo 57
La RMN 1H se registró en un espectrómetro Bruker de 400 MHz, usando una señal residual de disolvente deuterado como patrón interno. Los desplazamientos químicos (8) se informan en ppm en relación con la señal del disolvente residual (8 = 2,49 ppm para RMN 1H en DMSO-d6). Los datos de RMN 1H se informan de la siguiente manera: desplazamiento químico (multiplicidad, constantes de acoplamiento, y número de hidrógenos). La multiplicidad se abrevia como se indica a continuación: s (singlete), d (doblete), t (triplete), c (cuadruplete), m (multiplete), a (ancho).
El análisis CLEM se realizó en las siguientes condiciones:
Método: A: TFA al 0,1 % en H2O, B: TFA al 0,1 % en ACN:
Tiempo de ejecución: 6,5 min
Caudal: 1,0 ml/min
Gradiente: B al 5-95 % en 4,5 min, longitud de onda 254 y 215 nM.
Columna: Waters Sunfire C18, 3,0x50 mm, 3,5 um, modo positivo
Barrido de masa: 100-900 Da
Esquema 1: Síntesis del Compuesto C-60

Reactivos: a) i) tamices mol. de 4 A, 3-((tributilestannil)metoxi)propan-1-amina, CH2CI2; ii) 2,6-lutidina, Cu(OTf)2, hexafluoroisopropanol, CH2Cl2; b) decarbonato de di-ferc-butilo, TEA, DCM; c) (r)-(-)-2-cloropropan-1-ol, KOH, DMF; d) TFA, DCM; e) 2-amino-4-cloro-6-metilpirimidina, nBuOH, 135 °C.
Formación de 3-cloro-4-[1,4]oxazepan-3-il-fenol (1)
Una mezcla de 3-(tributilestanilmetoxi)propan-1-amina (2,42 g, 6,4 mmol), 2-cloro-4-hidroxibenzaldehído (1,0 g, 6,4 mmol) y tamices moleculares de 4 angstrom en diclorometano (10 ml) se agitó durante 20 horas. La mezcla se filtró. En un matraz separado que contenía hexafluoroisopropanol (20,0 ml) se añadió 2,6-lutidina (0,9 ml, 7,6 mmol) seguido de Cu(OTf)2 (2,78 g, 7,6 mmol) y diclorometano (3 ml). La mezcla se agitó durante 3 horas a temperatura ambiente. La solución de imina filtrada se añadió en una porción al segundo matraz de una sola vez. La mezcla de reacción resultante se agitó durante la noche, se filtró y luego se trató con 100 ml de una mezcla 2:1 de solución acuosa saturada de NaHCO3 e hidróxido de amonio al 10 %. La fase orgánica se separó y se lavó con una solución acuosa saturada de NaHCO3, se secó con sulfato sódico, se filtró y se concentró al vacío. La mezcla en bruto se purificó usando un sistema Biotage, columna de 100 g usando MeOH al 2-15 %:DCM para proporcionar 1000 mg (rendimiento del 55 %) del producto en forma de un aceite incoloro. CL-EM (M+H)+: 228.
Formación de éster ferc-butílico del ácido 3-(2-cloro-4-hidroxi-fenil)-[1,4]oxazepano-4-carboxílico (2)
A una solución agitada de 3-cloro-4-[1,4]oxazepan-3-il-fenol (1,6 g; 7,03 mmol; 1,00 equiv.) en DCM (15,00 ml; 234,01 mmol; 33,30 equiv.), se le añadieron dicarbonato de di-ferc-butilo (1,68 g; 7,73 mmol; 1,10 equiv.) y TEA (2,94 ml; 21,08 mmol; 3,00 equiv.). La reacción se agitó a ta durante 1 h. El disolvente se evaporó y la mezcla en bruto se secó para proporcionar el compuesto del título (2,3 g: Rendimiento: 100 %) en forma de un sólido de color blanquecino. CL-EM (M-Boc)+: 228.
Formación de éster ferc-butílico del ácido 3-[2-cloro-4-((R)-2-hidroxi-1-metil-etoxi)-fenil]-[1,4]oxazepano-4-carboxílico (3)
A una solución agitada de éster ferc-butílico del ácido 3-(2-cloro-4-hidroxi-fenil)-[1,4]oxazepano-4-carboxílico (50,00 mg; 0,15 mmol; 1,00 equiv.) en DMF (1,50 ml; 19,45 mmol; 127,54 equiv.) en un vial de microondas, se le añadió hidróxido potásico (34,23 mg; 0,61 mmol; 4,00 equiv.) y. La mezcla de reacción se agitó a 100 °C durante una noche. La reacción se interrumpió usando agua y se extrajo con DCM. La capa orgánica se concentró al vacío y la mezcla en bruto se purificó usando un sistema Biotage, columna de 10 g usando AcOEt al 5-50 %:PS para proporcionar 55 mg (rendimiento del 93 %) del compuesto del título. CL-EM (M-Boc)+: 286.
Formación de (R)-2-(3-Cloro-4-[1,4]oxazepan-3-il-fenoxi)-propan-1-ol (4)
A una solución agitada de éster ferc-butílico del ácido 3-[2-cloro-4-((R)-2-hidroxi-1-metil-etoxi)-fenil]-[1,4]oxazepano-4-carboxílico (55,00 mg; 0,14 mmol; 1,00 equiv.) en DCM (2,00 ml; 31,20 mmol; 218,91 equiv.) a ta, se le añadió TFA (0,50 ml; 6,53 mmol; 45,81 equiv.). La reacción continuó durante 30 min. El disolvente se evaporó y la mezcla en bruto se secó para proporcionar el compuesto del título (40 mg: Rendimiento: 100%) en forma de un sólido de color blanquecino. CL-EM (M-Boc)+: 286.
Formación de (R)-2-14-[4-(2-Amino-6-metil-pirimidin-4-il)-[1,4]oxazepan-3-il]-3-cloro-fenoxi}-propan-1-ol (5) Compuesto C-60
A una solución agitada de (R)-2-(3-Cloro-4-[1,4]oxazepan-3-il-fenoxi)-propan-1-ol (15,00 mg; 0,05 mmol; 1,00 equiv.) en 1-butanol (1,50 ml), se le añadieron 2-amino-4-doro-6-metilpirimidina (15,07 mg; 0,10 mmol; 2,00 equiv.) y TEA (0,02 ml; 0,16 mmol; 3,00 equiv.). La reacción se calentó a 135 °C durante una noche. Se registró la CLEM al día siguiente para confirmar la finalización de la reacción. La mezcla de reacción se cargó tal cual en el sistema de preparación Intershim usando condiciones básicas, acetonitrilo al 10-90 %:H2O para proporcionar 3,7 mg (rendimiento del 18 %) del producto del título. CL-EM (M+H)+: 393. RMN 1H (400 MHz, DMSO-d6) d 8,34 (s, 1H), 7,18 (d, J = 8,7 Hz, 1H), 7,03 (d, J = 2,5 Hz, 1H), 6,88 (dd, J = 8,7, 2,6 Hz, 1H), 5,86 (s, 3H), 4,05 (dd, J = 13,6, 5,0 Hz, 1H), 3,91 (dt, J = 11,6, 5,6 Hz, 2H), 3,80 (dd, J = 6,0, 3,0 Hz, 1H), 3,68 (dd, J = 13,4, 10,6 Hz, 2H), 3,55 - 3,49 (m, 2H), 3,37 (s, 5H, se superpone con el pico de H2O), 1,98 (s, 3H), 1,73 (s, 2h ).
Compuestos preparados de manera similar al Compuesto C-60:

(6) Compuesto C-42
Se proporcionó (S)-3-{4-[4-(2-amino-6-metil-pirimidin-4-il)-[1,4]oxazepan-3-il]-3-cloro-fenoxi}-propano-1,2-diol en forma de un sólido de color blanco (51 mg, rendimiento del 63 %). CL-EM (M+H)+: 409. RMN 1H (400 MHz, DMSO-d6) d 7,18 (d, J = 8,7 Hz, 1H), 7,04 (d, J = 2,5 Hz, 1H), 6,89 (dd, J = 8,7, 2,6 Hz, 1H), 5,91 (s, 3H), 4,92 (dd, J = 5,1, 1,6 Hz, 1H), 4,63 (dd, J = 6,4, 4,9 Hz, 1H), 4,11 -3,96 (m, 2H), 3,94 -3,82 (m, 2H), 3,79 -3,58 (m, 3H), 3,58 -3,48 (m, 1H), 3,42 (t, J = 5,7 Hz, 2H), 1,99 (s, 3H), 1,73 (s, 2H).

(7) Compuesto C-61
Se proporcionó (R)-3-{4-[4-(2-a-6-metil-pirimidin-4-il)-[1,4]oxazepan-3-il]-3-cloro-fenoxi}-propano-1,2-diol en forma de un sólido de color blanco (6 mg, rendimiento del 11 %). CL-EM (M+H)+: 409. RMN 1H (400 MHz, DMSO-d6) d 7,18 (d, J = 8,7 Hz, 1H), 7,03 (d, J = 2,5 Hz, 1H), 6,89 (dd, J = 8,7, 2,6 Hz, 1H), 5,84 (s, 3H), 4,93 (d, J = 5,1 Hz, 1H), 4,63 (d, J = 6,0 Hz, 1H), 4,11 - 3,96 (m, 2H), 3,95 - 3,82 (m, 2H), 3,79 - 3,58 (m, 3H), 3,57 - 3,48 (m, 1H), 3,41 (t, J = 5,1 Hz, 2H), 1,98 (s, 3H), 1,74 (d, J = 9,9 Hz, 2H).

Formación de 2-{4-[4-(2-Amino-6-metil-pirimidin-4-il)-[1,4]oxazepan-3-il]-3-cloro-fenoxi}-acetamida (8) Compuesto C-53
Se proporcionó 2-{4-[4-(2-amino-6-metil-pirimidin-4-il)-[1,4]oxazepan-3-il]-3-doro-fenoxi}-acetamida en forma de un sólido de color blanco (2,8 mg, rendimiento del 4,8 %). CL-EM (M+H)+: 392. RMN 1H (400 MHz, metanol-d4) 88,49 (s, 1H), 7,28 (d, J = 8,7 Hz, 1H), 7,14 (d, J = 2,6 Hz, 1H), 6,97 (dd, J = 8,7, 2,6 Hz, 1H), 6,22-5,19 (s a, 3H), 4,52 (s, 2H), 4,26 (dd, J = 13,7, 5,1 Hz, 1H), 4,02 (d, J = 12,0 Hz, 1H), 3,82 (dd, J = 13,7, 10,1 Hz, 2H), 3,67 (t, J = 11,1 Hz, 1H), 2,67 (s, 1H), 2,20 (s, 3H), 1,96-1,90 (m, 2H).

Compuesto C-105
Formación de 4-[3-(2-Cloro-4-metilsulfanilmetoxi-fenil)-[1,4]oxazepan-4-il]-6-metil-pirimidin-2-ilamina (9) Compuesto C-105
Se proporcionó 4-[3-(2-cloro-4-metilsulfanilmetoxi-fenil)-[1,4]oxazepan-4-il]-6-metil-pirimidin-2-ilamina en forma de un sólido de color blanquecino (25 mg, rendimiento del 6,1 %). CL-EM (M+H)+: 395. RMN 1H (400 MHz, M etano l^) 8 7,22 (d, J = 8,7 Hz, 1H), 7,08 (d, J = 2,5 Hz, 1H), 6,92 (dd, J = 8,7, 2,6 Hz, 1H), 6,02 - 5,32 (m, 3H), 5,22 (s, 2H), 4,26 (dd, J = 13,6, 5,1 Hz, 1H), 4,03 (dd, J = 12,3, 4,7 Hz, 1H), 3,74 (dd, J = 13,6, 10,5 Hz, 1H), 3,63 (td, J = 12,2, 3,7 Hz, 2H), 2,22 (s, 3H), 2,09 (s, 3H), 1,93 (ddd, J = 11,2, 5,2, 2,5 Hz, 1H), 1,88 - 1,75 (m, 1H).

Compuesto C-120
Formación de 4-[3-(2-Cloro-4-metanosulfonilmetoxi-fenil)-[1,4]oxazepan-4-il]-6-metil-pirimidin-2-ilamina (10) Compuesto C-120
Se proporcionó 4-[3-(2-cloro-4-metanosulfonilmetoxi-fenil)-[1,4]oxazepan-4-il]-6-metil-pirimidin-2-ilamina en forma de un sólido de color blanquecino (20 mg, rendimiento del 13 %). CL-EM (M+H)+: 427. RMN 1H (400 MHz, M etano l^) 8 7,29-7,27 (m, 2H), 7,08 (d, J = 8,9 Hz, 1H), 6,00 -5,29 (m, 3H), 5,21 (s, 2H), 4,25 (dd, J = 13,6, 5,1 Hz, 1H), 4,12-3,95
(m, 1H), 3,82-3,54 (m, 3H), 3,04 (s, 3H), 2,09 (s, 3H), 1,98- 1,87 (m, 1H), 1,82 (d, J = 14,9 Hz, 1H). Purificación SFC quiral para obtener los enantiómeros individuales: Compuestos C-64 y C-65

(S)-3-{4-[(S)-4-(2-Ammo-6-metM-pirimidm-4-MH1,4|oxazepan-3-M]-3-doro-fenoxi}-propano-1,2-diol (11) Compuesto C-64
La mezcla racémica (6) se sometió a purificación SFC: columna AD-H 10x250 mm usando método de gradiente de MeOH al 5-60 % (DMEA al 0,5 %) durante 5 min. CL-EM (M+H)+: 409. RMN 1H (400 MHz, DMSO-d6) d 7,18 (d, J = 8,7 Hz, 1H), 7,03 (d, J = 2,5 Hz, 1H), 6,89 (dd, J = 8,7, 2,6 Hz, 1H), 5,83 (s, 3H), 4,93 (d, J = 5,1 Hz, 1H), 4,64 (t, J = 5.6 Hz, 1H), 4,09 - 3,95 (m, 2H), 3,93 - 3,82 (m, 2H), 3,81 - 3,61 (m, 3H), 3,57 - 3,48 (m, 1H), 3,41 (t, J = 5,6 Hz, 2H), 1,98 (s, 3H), 1,73 (s, 2H). Tiempo de retención: 3,58 minutos.
(S)-3-{4-[(R)-4-(2-Amino-6-metil-pirimidin-4-il)-[1,4]oxazepan-3-il]-3-doro-fenoxi}-propano-1,2-diol (12) Compuesto C-65
La mezcla racémica (6) se sometió a purificación SFC: columna AD-H 10x250 mm usando método de gradiente de MeOH al 5-60 % (DMEA al 0,5 %) durante 5 min. CL-EM (M+H)+: 409. RMN 1H (400 MHz, DMSO-d6) d 7,18 (d, J = 8.7 Hz, 1H), 7,03 (d, J = 2,5 Hz, 1H), 6,89 (dd, J = 8,7, 2,6 Hz, 1H), 5,84 (s, 3H), 4,97 (s, 1H), 4,69 (s, 1H), 4,05 (dd, J = 13,5, 5,0 Hz, 1H), 3,99 (dt, J = 10,0, 4,1 Hz, 1H), 3,94 - 3,82 (m, 2H), 3,80 - 3,58 (m, 3H), 3,57 - 3,48 (m, 1H), 3,42 (d, J = 5,7 Hz, 2H), 1,98 (s, 3h ), 1,73 (s, 2H). Tiempo de retención: 4,16 minutos.

121
La mezcla racémica (9) se sometió a purificación SFC: columna IA-H 4,6x100 mm usando método de gradiente de MeOH al 5-50 % (DMEA al 0,5 %) durante 5 min seguido de método isocrático de MeOH al 50 % (DMEA al 0,5 %) durante 3 min. CL-EM (M+H)+: 395. RMN 1H (400 MHz, Metanol-d>) 57,22 (d, J = 8,6 Hz, 1H), 7,07 (d, J = 2,1 Hz, 1H), 6,92 (d, J = 8,4 Hz, 1H), 6,07 -5,43 (m, 3H), 5,21 (s, 2H), 4,25 (dd, J = 13,6, 5,1 Hz, 1H), 4,12-3,98 (m, 1H), 3,80-3,69 (m, 1H), 3,63 (dd, J = 13,9, 10,5 Hz, 2H), 2,22 (s, 3H), 2,09 (s, 3H), 2,02 - 1,87 (m, 1H), 1,82 (d, J = 13,0 Hz, 1H). Tiempo de retención: 3,20 minutos.
4-[(R)-3-(2-Cloro-4-metilsulfanilmetoxi-fenil)-[1,4]oxazepan-4-il]-6-metil-pirimidin-2-ilamina (14) Compuesto C-122
La mezcla racémica (9) se sometió a purificación SFC: columna IA-H 4,6x100 mm usando método de gradiente de MeOH al 5-50 % (DMEA al 0,5 %) durante 5 min seguido de método isocrático de MeOH al 50 % (DMEA al 0,5 %) durante 3 min. CL-EM (M+H)+: 395. RMN 1H (400 MHz, Metanol-d4) 57,21 (d, J = 8,6 Hz, 1H), 7,07 (d, J = 2,4 Hz, 1H), 7,00 - 6,84 (m, 1H), 6,09 - 5,22 (m, 3H), 5,21 (s, 2H), 4,25 (dd, J = 13,6, 5,1 Hz, 1H), 4,03 (dd, J = 12,9, 4,4 Hz, 1H), 3,78 - 3,69 (m, 1H), 3,70 - 3,57 (m, 2H), 2,22 (d, J = 1,6 Hz, 3H), 2,09 (s, 3H), 1,93 (dd, J = 12,3, 4,1 Hz, 1H), 1,87 -1,74 (m, 1H). Tiempo de retención: 3,34 minutos.

4-[(S)-3-(2-Cloro-4-metanosulfonilmetoxi-fenil)-[1,4]oxazepan-4-il]-6-metil-pirimidin-2-ilamina (15) Compuesto C-153
La mezcla racémica (10) se sometió a purificación SFC: columna IA-H 4,6x250 mm usando método de gradiente de MeOH al 5-60 % (d MeA al 0,5 %) durante 5 min seguido de método isocrático de MeOH al 50 % (DMEA al 0,5 %) durante 3 min. CL-EM (M+H)+: 427. RMN 1H (400 MHz, DMSO-da) 57,32 (s, 1H), 7,24 (d, J = 8,8 Hz, 1H), 7,09 (d, J = 8,7 Hz, 1H), 5,83 (s a, 3H), 5,34 (s, 2H), 4,07 (d, J = 12,6 Hz, 1H), 3,91 (d, J = 13,3 Hz, 1H), 3,77 - 3,60 (m, 2H), 3,52 (d, J = 14,3 Hz, 1H), 3,05 (s, 3H), 1,99 (s, 3H), 1,74 (s a, 2H). Tiempo de retención: 4,72 minutos.
4-[(R)-3-(2-Cloro-4-metanosulfonilmetoxi-fenil)-[1,4]oxazepan-4-il]-6-metil-pirimidin-2-ilamina (16) Compuesto C-154
La mezcla racémica (10) se sometió a purificación SFC: columna IA-H 4,6x250 mm usando método de gradiente de MeOH al 5-60 % (d MeA al 0,5 %) durante 5 min seguido de método isocrático de MeOH al 50 % (DMEA al 0,5 %) durante 3 min. CL-EM (M+H)+: 427. RMN 1H (400 MHz, DMSO-d6) 57,32 (s, 1H), 7,24 (d, J = 8,6 Hz, 1H), 7,17 - 7,03 (m, 1H), 6,06 -5,64 (m, 3H), 5,34 (s, 2H), 4,07 (d, J = 11,8 Hz, 1H), 3,91 (d, J = 12,7 Hz, 1H), 3,77 -3,43 (m, 3H), 3,05 (s, 3H), 1,99 (s, 3H), 1,74 (s a, 2H). Tiempo de retención: 5,38 minutos.
Esquema 2: Síntesis del Compuesto C-63

Reactivos: (a) NaSMe, MeOH, 0 °C a TA; (b) nBuLi, THF, -78 °C, después DMF; (c) mCPBA, CH2Ch; (d) i) tamices mol de 4 A, 2-Metil-3-tributilestannanilmetoxi-propilamina, CH2Ch; ii) 2,6-lutidina, Cu(OTf)2, hexafluoroisopropanol, CH2Cl2; (e) 2-amino-4-cloro-6-metilpirimidina, nBuOH, 135 °C.
Formación de (4-bromo-3-clorobencil)(metil)sulfano (17)
El 1-bromo-4-(bromometil)-2-cloro-benceno (3,5 g, 12,3 mmol) se disolvió en MeOH (40 ml) en un matraz de fondo redondo de 250 ml con un agitador superior, sonda de temperatura y un embudo de adición de 25 ml. La solución se enfrió a 0 °C en un baño de salmuera. La solución de NaSMe (2,1 g, 29,5 mmol en 5 ml de MeOH) se añadió gota a gota a una velocidad para mantener la temperatura por debajo de 10 °C. Precipitó un sólido de color blanco. La solución se ejecutó durante 24 h a ta. La reacción se vertió en NaOH 1 N y se extrajo tres veces con diclorometano. Los extractos se combinaron, se secaron (MgSO4), se filtraron y se evaporaron al vacío para proporcionar 1-bromo-2-cloro-4-(metilsulfanilmetil)benceno (2,8 g, rendimiento del 86% ) en forma de un aceite transparente. RMN 1H (300 MHz, CDCh) 7,54 (d, J = 8,2 Hz, 1H), 7,40 (s, 1H), 7,07 (dd, J = 8,2, 1,6 Hz, 1H), 3,59 (s, 1H), 1,99 (s, 2H) ppm.
Formación de 2-Cloro-4-metilsulfanilmetil-benzaldehído (18)
A una solución agitada de 1-bromo-2-cloro-4-metilsulfanilmetil-benceno (4800,00 mg; 19,08 mmol; 1,00 equiv.) en THF (130,00 ml) a -78 °C, se le añadió n-butillitio (8,40 ml; 20,99 mmol; 1,10 equiv.). La reacción se agitó durante 30 min
seguido de la adición de DMF (2,00 ml). La agitación continuó durante la adición durante 15 min a -78 °C seguido de agitación a ta durante 3 h. La mezcla de reacción se vertió en HCl 1 N y se extrajo con MTBE. El extracto se secó (MgSO4), se filtró y se evaporó al vacío para proporcionar un producto en bruto en forma de un aceite de color amarillo. El producto se purificó usando un sistema Biotage, columna de 100 g usando diclorometano al 10-50 %/hexano. Se obtuvieron 3000 mg (rendimiento del 76 %) del compuesto deseado. CL-EM (M+H)+: 201.
Formación de 2-Cloro-4-metanosulfonilmetil-benzaldehído (19)
A una solución agitada de 2-cloro-4-metilsulfanilmetil-benzaldehído (2000,00 mg; 9,97 mmol; 1,00 equiv.) en DCM (80,00 ml), se le añadió ácido 3-cloroperoxibenzoico (3783,47 mg; 21,92 mmol; 2,20 equiv.) a ta. Después de 15 min, precipitó un sólido de color blanco. Después de agitar durante 1 hora, la mezcla de reacción se vertió en una solución acuosa saturada de NaHCO3 y se extrajo con diclorometano. La mezcla en bruto se purificó usando un sistema Biotage, columna de 100 g usando AcOEt al 5-40 %:DCM. Se obtuvieron 2000 mg (rendimiento del 78 %) del producto. CL-EM (M+H)+: 233.
Formación de 3-(2-Cloro-4-metanosulfonilmetil-fenil)-6-metil-[1,4]oxazepano (20)
Esta es una reacción de 2 etapas. Etapa 1: Una mezcla de 2-cloro-4-metanosulfonilmetil-benzaldehído (75 mg; 0,32 mmol; 1,00 equiv.), 2-metil-3-tributilestannanilmetoxi-propilamina (126 mg; 0,32 mmol; 1,00 equiv.) y tamices moleculares de 4 angstrom en DCM (1 ml) se agitó durante 20 horas. La mezcla se filtró y se llevó a la siguiente reacción. Etapa 2: A una solución agitada de trifluorometanosulfonato de cobre (II) (139,50 mg; 0,39 mmol; 1,50 equiv.) en 1,1,1,3,3,3- hexafluoro-2-propanol (4 ml) a ta, se le añadió 2,6-dimetilpiridina (0,04 ml; 0,39 mmol; 1,50 equiv.). La mezcla de reacción se agitó durante 1,5 horas seguido de la adición de [1-(2-cloro-4-metanosulfonilmetil-fenil)-met-(E)-ilideno]-(3- tributilestannanilmetoxi-propil)-amina (195 mg; 0,32 mmol; 1,00 equiv.). La mezcla de reacción resultante se agitó a ta durante una noche. La mezcla de reacción se diluye con CH2Cl2, se trató con una solución de NH4OH ac. al 12 % y salmuera (1:1) y se agitó vigorosamente durante 15 min a ta. Las capas se separan y la capa acuosa se extrajo con CH2Cl2. Las capas orgánicas combinadas se lavaron con H2O y salmuera, se secaron sobre Na2SO4 anhidro, se filtraron y se concentraron. La purificación por un sistema Biotage, columna de 330 g usando AcOEt al 10-40 %:DCM seguido de MeOH al 5-20 %:DCM. Se aislaron 75 mg (rendimiento del 51 %) del compuesto del título. CL-EM (M+H)+: 318.
Formación de 4-[3-(2-Cloro-4-metanosulfonilmetil-fenil)-6-metil-[1,4]oxazepan-4-il]-6-metil-pirimidin-2-ilamina (21) Compuesto C-63
A una mezcla de 3-(2-cloro-4-metanosulfonilmetil-fenil)-6-metil-[1,4]oxazepano (75,00 mg; 0,24 mmol; 1,00 equiv.) y 2-amino-4-cloro-6-metilpirimidina (67,76 mg; 0,47 mmol; 2,00 equiv.), se le añadió 1-butanol (2,00 ml). La mezcla de reacción se calentó a 135 °C durante 16 horas.
Se registró la CLEM al día siguiente para confirmar la finalización de la reacción. La mezcla de reacción se cargó tal cual en el sistema de preparación Intershim usando condiciones básicas, Acetonitrilo al 10-90 %:H2O para proporcionar 44 mg (rendimiento del 44 %) del producto deseado. CL-EM (M+H)+: 425. RMN 1H (400 MHz, DMSO-d6) d 7,50 (d, J = 8,2 Hz, 1H), 7,33 (d, J = 2,1 Hz, 1H), 7,30 (dd, J = 8,2, 2,1 Hz, 1H), 5,79 (s, 2H), 5,43 (s, 1H), 5,22 (s, 1H), 4,51 - 4,38 (m, 2H), 4,03 (dd, J = 13,6, 4,4 Hz, 1H), 3,82 (d, J = 15,0 Hz, 1H), 3,72 - 3,51 (m, 3H), 2,76 (s, 3H), 2,07 (s, 2H), 1,93 (s, 3H), 0,87 (d, J = 7,0 Hz, 3H).
Compuestos preparados de manera similar al (21):

Formación de 4-[7-(2-Cloro-4-metanosulfonilmetil-fenil)-5-oxa-8-aza-espiro[2,6]non-8-il]-6-metil-pirimidin-2-ilamina (22) Compuesto C-62
Se proporcionó 4-[7-(2-cloro-4-metanosulfonilmetil-fenil)-5-oxa-8-aza-espiro[2,6]non-8-il]-6-metil-pirimidin-2-illamina en forma de un sólido de color blanco (30 mg, rendimiento del 30 %). CL-EM (M+H)+: 437. RMN 1H (400 MHz, DMSO-d6) d 7,52 (d, J = 8,2 Hz, 1H), 7,38 (d, J = 2,2 Hz, 1H), 7,32 (dd, J = 8,2, 2,1 Hz, 1H), 5,81 (s, 2H), 5,33 (s, 2H), 4,56 -4,38 (m, 3H), 4,10 (dd, J = 13,7, 4,6 Hz, 1H), 4,01 (d, J = 15,0 Hz, 1H), 3,85 (dd, J = 13,4, 10,3 Hz, 2H), 3,09 (d, J =
12,1 Hz, 1H), 2,78 (s, 3H), 1,94 (s, 3H), 0,89 (d, J = 22,3 Hz, 1H), 0,57 (dt, J = 9,3, 4,7 Hz, 1H), 0,39 (ddt, J = 33,0, 9,5, 4,8 Hz, 2H).

Formación de 4-[3-(2-Cloro-4-metanosulfonNmetN-fenN)-6,6-dimetN-[1,4]oxazepan-4-M]-6-metM-pirimidm-2-ilamina (23) Compuesto C-73
Se proporcionó 4-[3-(2-doro-4-metanosulfonilmetil-fenil)-6,6-dimetil-[1,4]oxazepan-4-il]-6-metil-pirimidin-2-ilamina en forma de un sólido de color blanco (50 mg, rendimiento del 50 %). Cl-Em (M+H)+: 439. RMN 1H (400 MHz, DMSO-d6) d 7,50 (d, J = 8,6 Hz, 1H), 7,30 (d, J = 2,0 Hz, 2H), 5,85 (s, 1H), 5,14 (s, 2H), 4,44 (s, 2H), 4,04 (dd, J = 13,6, 4,9 Hz, 1H), 3,60 (dd, J = 13,5, 11,1 Hz, 1H), 3,48 -3,38 (m, 1H), 3,32-3,22 (m, 4H, se superpone con el pico de H2O), 2,75 (s, 3H), 1,93 (s, 3H), 0,88 (d, J = 4,3 Hz, 6H).

Formación de 4-[3-(2-Cloro-4-metanosulfonilmetil-fenil)-6-etil-[1,4]oxazepan-4-il]-6-metil-pirimidin-2-ilamina (24) Compuesto C-74
Se proporcionó 4-[3-(2-cloro-4-metanosulfonilmetil-fenil)-6-etil-[1,4]oxazepan-4-il]-6-metil-pirimidin-2-ilamina en forma de un sólido de color blanco (40 mg, rendimiento del 40 %). CL-EM (M+H)+: 439. RMN 1H (400 MHz, DMSO-d6) d 7,50 (d, J = 8,2 Hz, 1H), 7,33 (d, J = 2,2 Hz, 1H), 7,30 (dd, J = 8,2, 2,2 Hz, 1H), 5,81 (s, 2H), 5,18 (s, 1H), 4,50 -4,38 (m, 2H), 4,00 (dd, J = 13,6, 4,4 Hz, 1H), 3,83 -3,75 (m, 2H), 3,69 (dd, J = 13,6, 10,0 Hz, 1H), 3,54 (dd, J = 12,1, 2,8 Hz, 1H), 3,33 (d, J = 2,7 Hz, 3H, se superpone con el pico de H2O), 2,75 (s, 3H), 1,92 (s, 3H), 1,40 (tq, J = 12,7, 7,4, 6,5 Hz, 1H), 1,29 -1,14 (m, 1H), 0,88 (t, J = 7,4 Hz, 3H).

Formación de 4-[3-(2-Fluoro-4-metanosulfonilmetil-fenil)-[1,4]oxazepan-4-il]-6-metil-pirimidin-2-ilamina (25) Compuesto C-11
Se proporcionó 4-[3-(2-fluoro-4-metanosulfonilmetil-fenil)-[1,4]oxazepan-4-il]-6-metil-pirimidin-2-ilamina en forma de un sólido de color blanco (15 mg, rendimiento del 44 %). CL-EM (M+H)+: 395. RMN de 1H (400 MHz, DMSO-d6) 87,317,18 (m, 3H), 5,97 (s, 3H), 4,49 (s, 2H), 4,25 -4,14 (m, 1H), 3,96 - 3,90 (m, 1H), 3,74 (t, J = 12,0 Hz, 1H), 3,63 (d, J = 11,9 Hz, 1H), 3,52 (td, J = 11,7, 3,9 Hz, 1H), 3,32 (s, 2H, se superpone con el pico de H20), 2,92 (s, 3H), 2,04 (s, 3H), 1,77 - 1,69 (m, 2H).

Formación de 4-[3-(2-Cloro-5-metanosulfonilmetil-fenil)-[1,4]oxazepan-4-il]-6-metil-pirimidin-2- ilamina (26) Compuesto C-12
Se proporcionó 4-[3-(2-doro-5-metanosulfonilmetil-fenil)-[1,4]oxazepan-4-il]-6-metil-pirimidin-2-ilamina en forma de un sólido de color blanco (55 mg, rendimiento del 54%). CL-EM (M+H)+: 411. RMN 1H (400 MHz, DMSO-d6) d 7,51 (d, J = 8,1 Hz, 1H), 7,36 (d, J = 2,1 Hz, 1H), 7,32 (dd, J = 8,2, 2,1 Hz, 1H), 5,66 (d, J = 144,9 Hz, 3H), 4,55 -4,39 (m, 2H), 4,15 (dd, J = 13,4, 5,0 Hz, 1H), 3,92 (dt, J = 12,2, 3,8 Hz, 1H), 3,72 - 3,48 (m, 3H), 3,33 (s, 2H, se superpone con el pico de H2O), 2,83 (s, 3H), 1,99 (s, 3H), 1,77 (c, J = 3,7 Hz, 2H).
Purificación SFC quiral para obtener los enantiómeros individuales: Compuestos C-49 y C-50

Formación de 4-[(R)-3-(2-Fluoro-4-metanosulfonilmetil-fenil)-[1,4]oxazepan-4-il]-6-metil-pirimidin-2-ilamina (27) Compuesto C-49
La mezcla racémica (25) se sometió a purificación SFC: columna IC 4,6x100 mm usando método de gradiente de MeOH al 5-60 % (DMEA al 0,5 %) durante 5 min seguido de método isocrático de MeOH al 50 % (DMEA al 0,5 %) durante 3 min. CL-EM (M+H)+: 395. RMN de 1H (400 MHz, DMSO-d6) 87,31-7,18 (m, 3H), 5,97 (s, 3H), 4,49 (s, 2H), 4.25 -4,14 (m, 1H), 3,96 -3,90 (m, 1H), 3,74 (t, J = 12,0 Hz, 1H), 3,63 (d, J = 11,9 Hz, 1H), 3,52 (td, J = 11,7, 3,9 Hz, 1H), 3,32 (s, 2H, se superpone con el pico de H20), 2,92 (s, 3H), 2,04 (s, 3H), 1,77 -1,69 (m, 2H). Tiempo de retención: 5,06 minutos.
Formación de 4-[(S)-3-(2-Fluoro-4-metanosulfonilmetil-fenil)-[1,4]oxazepan-4-il]-6-metil-pirimidin-2-ilamina (27) Compuesto C-50
La mezcla racémica (25) se sometió a purificación SFC: columna IC 4,6x100 mm usando método de gradiente de MeOH al 5-60 % (DMEA al 0,5 %) durante 5 min seguido de método isocrático de MeOH al 50 % (DMEA al 0,5 %) durante 3 min. CL-EM (M+H)+: 395. RMN de 1H (400 MHz, DMSO-d6) 87,31-7,18 (m, 3H), 5,97 (s, 3H), 4,49 (s, 2H), 4.25 -4,14 (m, 1H), 3,96 -3,90 (m, 1H), 3,74 (t, J = 12,0 Hz, 1H), 3,63 (d, J = 11,9 Hz, 1H), 3,52 (td, J = 11,7, 3,9 Hz, 1H), 3,32 (s, 2H, se superpone con el pico de H20), 2,92 (s, 3H), 2,04 (s, 3H), 1,77 -1,69 (m, 2H). Tiempo de retención: 5,50 minutos.

4-[(R)-3-(2-Cloro-5-metanosulfonilmetil-fenil)-[1,4]oxazepan-4-il]-6-metil-pirimidin-2-ilamina (29) Compuesto C-51
La mezcla racémica (26) se sometió a purificación SFC: columna OD-H 4,6x100 mm usando método de gradiente de MeOH al 5-60 % (d MeA al 0,5 %) durante 5 min seguido de método isocrático de MeOH al 50 % (DMEA al 0,5 %) durante 3 min. CL-EM (M+H)+: 411. RMN 1H (400 MHz, DMSO-d6) d 7,51 (d, J = 8,1 Hz, 1H), 7,36 (d, J = 2,1 Hz, 1H), 7.32 (dd, J = 8,2, 2,1 Hz, 1H), 5,66 (d, J = 144,9 Hz, 3H), 4,55 -4,39 (m, 2H), 4,15 (dd, J = 13,4, 5,0 Hz, 1H), 3,92 (dt, J = 12,2, 3,8 Hz, 1H), 3,72 - 3,48 (m, 3H), 3,33 (s, 2H, se superpone con el pico de H2O), 2,83 (s, 3H), 1,99 (s, 3H), 1.77 (c, J = 3,7 Hz, 2H). Tiempo de retención: 2,37 minutos.
4-[(R)-3-(2-Cloro-5-metanosulfonilmetil-fenil)-[1,4]oxazepan-4-il]-6-metil-pirimidin-2-ilamina (30) Compuesto C-52
La mezcla racémica (26) se sometió a purificación SFC: columna OD-H 4,6x100 mm usando método de gradiente de MeOH al 5-60 % (d MeA al 0,5 %) durante 5 min seguido de método isocrático de MeOH al 50 % (DMEA al 0,5 %) durante 3 min. CL-EM (M+H)+: 411. RMN 1H (400 MHz, DMSO-d6) d 7,51 (d, J = 8,1 Hz, 1H), 7,36 (d, J = 2,1 Hz, 1H), 7.32 (dd, J = 8,2, 2,1 Hz, 1H), 5,66 (d, J = 144,9 Hz, 3H), 4,55 -4,39 (m, 2H), 4,15 (dd, J = 13,4, 5,0 Hz, 1H), 3,92 (dt, J = 12,2, 3,8 Hz, 1H), 3,72 - 3,48 (m, 3H), 3,33 (s, 2H, se superpone con el pico de H2O), 2,83 (s, 3H), 1,99 (s, 3H), 1.77 (c, J = 3,7 Hz, 2H). Tiempo de retención: 2,77 minutos.
Ejemplo 58
Ensayo indicador en Colo 205
Los compuestos de la invención descritos en el presente documento se seleccionaron usando el procedimiento de ensayo para determinar la actividad de transcripción de indicador mediada por p-catenina-TCF que se describe a continuación.
En células con señalización WNT activada, se ha descubierto que la inducción de estrés al RE por el mecanismo de estos compuestos da como resultado una reducción rápida en la actividad de este gen indicador y que la actividad en el ensayo se correlaciona con la actividad de estos compuestos como inductores de estrés al r E y UPR, y todas las demás medidas de actividad específica de estos compuestos, incluyendo la liberación de calcio, viabilidad y desplazamiento de la versión radiomarcada de estos compuestos de su sitio de unión específico en las células.
Las líneas celulares indicadoras se generaron mediante la transfección estable de células de líneas celulares cancerosas (por ejemplo, cáncer de colon) con una construcción plasmídica indicadora (de SABiosciences, una empresa de QlAGEN) que incluye el promotor TCF/LEF que impulsa la expresión del gen de luciferasa de luciérnaga. Se realizaron construcciones indicadoras con TCF/LEF en las que el promotor TCF/LEF, un promotor con un número óptimo de sitios de unión a TCF/LEF diseñado por SABiosceinces, estaba unido cadena arriba del gen de luciferasa de luciérnaga. Esta construcción también podría incluir un gen de resistencia a puromicina como marcador seleccionable. Esta construcción también podría usarse para transfectar de manera estable células Colo 205, una línea celular de cáncer de colon que tiene un gen APC mutado que provoca una p-catenina constitutivamente activa. Se generó una línea celular de control usando otra construcción plasmídica que contenía el gen de luciferasa bajo el control de un promotor basal de CMV que no se activa con p-catenina.
Las células Colo 205 cultivadas con una construcción indicadora transfectada de forma estable se sembraron en placa a aproximadamente 10.000 células por pocillo en placas multipocillo de 384 pocillos durante veinticuatro horas. A continuación, se añadieron los compuestos de prueba a los pocillos en diluciones en serie de 2 veces utilizando una concentración máxima de veinte micromolar. Una serie de pocillos de control para cada tipo celular recibió solamente disolvente compuesto. Cinco horas después de la adición del compuesto, se sometió a ensayó la actividad indicadora de la luciferasa, mediante la adición del reactivo de luminiscencia SteadyGlo (Promega). La actividad de luminiscencia indicadora se midió utilizando un lector de placas Pherastar (BMG Labtech). Las lecturas se normalizaron a las células tratadas con DMSO solamente, y después se usaron las actividades normalizadas en los cálculos de CI50. Los datos del ensayo indicador en Colo 205 se resumen en la tabla 3: A < 0,3 pM; 0,3 pM < B < 1,0 pM; 1,0 pM < C < 5,0 pM; D > 5,0 pM.
Ensayo indicador en HepG2 XBP1
Las células de hepatoma HepG2 se transdujeron con un retrovirus que codifica el ADNc para (u) XBP1 sin corte y empalme, que contiene un intrón sin procesar, fusionado al ADNc para luciferasa de luciérnaga. Tras la inducción de estrés al RE, el intrón sin procesar de XBP1(u) se corta y empalma por la endonucleasa IRE1alfa activa. El uno o más XBP1 con cortados y empalmados resultantes ahora está en marco con luciferasa que provoca la producción de proteína luciferasa activa, dando como resultado bioluminiscencia.
Se sembraron en placas células HepG2 XBP1(u)-Luc. La células Colo 205 cultivadas con una construcción indicadora transfectada de forma estable se sembraron en placa a aproximadamente 30.000 células por pocillo en placas multipocillo de 96 pocillos durante veinticuatro horas. A continuación, se añadieron los compuestos de prueba a los pocillos en diluciones en serie de 3 veces utilizando una concentración máxima de veintisiete micromolar. Una serie de pocillos de control para cada tipo celular recibió solamente disolvente compuesto. Seis horas después de la adición del compuesto, se sometió a ensayó la actividad indicadora de la luciferasa, mediante la adición del reactivo de luminiscencia SteadyGlo (Promega). La actividad de luminiscencia indicadora se midió utilizando un lector de placas Pherastar (BMG Labtech). Las lecturas se normalizaron a las células tratadas con DMSO solamente, y después se usaron las actividades normalizadas en los cálculos de CI50. Los datos del ensayo indicador en HepG2 XBP1 se resumen en las tablas 3-5: A < 0,6 pM; 0,6 pM < B < 2,0 pM; 2,0 pM < C < 5,0 pM; D > 5,0 pM.
Ensayo de flujo de calcio
Los compuestos descritos en el presente documento indujeron estrés en el RE provocando un flujo de calcio intracelular. El flujo de calcio se midió en células Colo-205 usando el kit de ensayo FLIPR® Calcium 5 según el protocolo del fabricante (Molecular Devices, número de cat. R8186) en un sistema FLIPR3 (Molecular Devices). El flujo de calcio se mide durante 36 minutos. Los datos del ensayo de flujo de calcio en Colo-205 se resumen en la tabla 3 : A < 0,6 pM; 0,6 pM < B < 2,0 pM; 2,0 pM < C < 10,0 pM; D > 10,0 pM.
Métodos de cultivo celular
Las células se retiraron del almacenamiento en nitrógeno líquido, se descongelaron y se dejaron crecer en medios de crecimiento apropiados. Una vez que habían crecido, las células se sembraron en placas tratadas con cultivo de tejidos de 384 pocillos a razón de 500 células por pocillo. Después de 24 horas, las células se trataron durante 0 horas o se trataron durante 96 horas con un compuesto de prueba (a las concentraciones de 100 nM y 2 uM). Al final de las 0 horas o de las 96 horas, el estado de las células se analizó usando ATPLite (Perkin Elmer) para evaluar la respuesta biológica de las células al compuesto de prueba. Los datos de CI50 en NCI-H929 (células de mieloma múltiple) y DU4475 (células de cáncer de mama) se resumen en las tablas 4 y 5 : A < 1 pM; 1 pM < B < 10,0 pM; 10,0 pM < C < 25,0 pM; D > 25,0 pM.
Tabla 3

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

(continuación)

Tabla 4.

(continuación)

(continuación)

(continuación)

(continuación)
Tabla 5.

(continuación)

(continuación)

(continuación)

Ejemplo 59
Síntesis de (+/-)- 4-(3-(2-cloro-4-(metilsulfoml)feml)-1,4-oxazepan-4-M)-6-metilpmmidm-2-amma (I-66) y (R)-4-(3-(2-cloro-4-(metilsulfoml)feml)-1,4-oxazepan-4-il)-6-metilpmmidm-2-amma (I-67) y (S)- 4-(3-(2-cloro-4-(metilsulfoml)feml)-1,4-oxazepan-4-il)-6-metilpmmidm-2-amma (I-68)

Formación de 2-cloro-4-metilsulfonil-benzoato de metilo
Se cargó ácido 2-doro-4-metilsulfonilbenzoico (100 g, 426,2 mmol) en metanol (1,5 l) en un matraz RB de 3 bocas de 5 l con agitador superior, sonda de temperatura, condensador de reflujo y embudo adicional, se agitó durante 10 minutos y después se enfrió a 0°C con un baño de hielo. Se añadió cloruro de tionilo (40 ml, 548,4 mmol) durante 20 minutos, se dejó a temperatura ambiente durante 1 h y luego se calentó a 60 °C, se agitó a esta temperatura durante 12 h (durante la noche), momento en el que el análisis de CLEM y HPLC reveló el consumo del material de partida. La HPLC muestra el tiempo de retención máximo del material de partida 1 a 1,23 minutos, el tiempo de retención máximo del producto deseado 2 a 2,37 minutos.
La mezcla de reacción se enfrió a temperatura ambiente, se concentró a presión reducida, el material en bruto se
repartió entre acetato de etilo (1 l) y una solución acuosa sat. de NaHCO3 (500 ml), se agitó durante 20 minutos y después se separó la fase orgánica. La capa acuosa se extrajo con acetato de etilo (500 ml), la fase orgánica combinada se lavó con agua (100 ml), salmuera (100 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida para producir 2-cloro-4-metilsulfonilo-benzoato de metilo (106 g, 99 %) en forma de un sólido de color blanco. La RMN 1H se ajustó a la estructura.
RMN 1H (400 MHz, CDCl3) 88,04 (d, J = 1,6 Hz, 1H), 8,00 -7,93 (m, 1H), 7,88 (dd, J = 8,1, 1,8 Hz, 1H), 3,98 (s, 3H), 3,08 (s, 3H).
Formación de (2-cloro-4-metilsulfonil-fenil)metanol
Se cargó 2-cloro-4-metilsulfonil-benzoato de metilo (110 g, 437,9 mmol) en un matraz RB de 3 bocas de 5 l con agitador superior, sonda de temperatura y condensador de reflujo en una mezcla de THF (1,3 l) y metanol (450 ml). ), se agitó durante 10 minutos y después se enfrió a 0 °C con un baño de hielo. Se añadió NaBH4 (85 g, 2,247 mol) en cuatro porciones durante 30 minutos. Después de que se retiró el baño de enfriamiento adicional y se dejó a temperatura ambiente (exotermia observada, Tmáx 50 °C), se agitó a esta temperatura (40 a 50 °C) durante 4 h, momento en el cual el análisis por TLC y HPLC reveló el consumo del material de partida.
La HPLC muestra el tiempo de retención máximo del material de partida 1 a 2,47 minutos, el tiempo de retención máximo del producto deseado 3 a 1,48 minutos. La TLC (acetato de etilo al 50 % en heptano) muestra Fr del material de partida = 0,5 y Fr del producto = 0,4.
La mezcla de reacción se enfrió a temperatura ambiente, se inactivó con la adición lenta de metanol (100 ml), seguido de una solución acuosa de HCl 1 N (~1 l) hasta un pH de ~7 a 8. La mezcla de reacción se extrajo con acetato de etilo (2 x 500 ml). La fase orgánica combinada se lavó con salmuera (~300 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida para proporcionar (2-cloro-4-metilsulfonil-fenil)metanol (90 g, 92 %) en forma de un sólido de color blanco. La RMN 1H se ajustó a la estructura.
RMN 1H (400 MHz, CDCl3) 87,92 (d, J = 1,8 Hz, 1H), 7,85 (dd, J = 8,1, 1,8 Hz, 1H), 7,78 (d, J = 8,1 Hz, 1H), 4,87 (d, J = 5,9 Hz, 2H), 3,06 (s, 3H).
Este material se recogió en la siguiente etapa sin purificación adicional.
Formación de 2-cloro-4-metilsulfonil-benzaldehído
Se cargó (2-cloro-4-metilsulfonil-fenil)metanol (96 g, 435,0 mmol) en CHCh (2,5 l) un matraz RB de 3 bocas de 5 l con agitador superior, sonda de temperatura y condensador de reflujo, se agitó durante 15 minutos y después se añadió dióxido de manganeso (250 g, 2,876 mol). La mezcla de reacción resultante se calentó a 45 °C (exotermia Tmáx 50 °C), se agitó a esta temperatura durante 3 h, momento en el que el análisis por TLC reveló el consumo del material de partida. La TLC (EtOAc al 50 %/heptano) no muestra más material de partida (Fr = 0,2).
La mezcla de reacción se filtró a través de un lecho de celite, el lecho se lavó con DCM (3 x 100 ml), los filtrados combinados se concentraron a presión reducida para producir 2-cloro-4-metilsulfonil-benzaldehído (82,5 g, 86 %) en forma de un sólido blanco. La RMN 1H se ajustó a la estructura.
RMN 1H (400 MHz, CDCl3) 8 10,54 (d, J = 0,8 Hz, 1H), 8,13 - 8,09 (m, 1H), 8,07 (d, J = 1,7 Hz, 1H), 7,95 (ddd, J = 8,1, 1,7, 0,8 Hz, 1H), 3,11 (s, 3H).
Formación de 2-cloro-metilsulfonilfeniloxazepano
A una solución de 3-(tributilestanilmetoxi)propan-1-amina (70 g, 185,1 mmol) en diclorometano anhidro (1,4 l), se le añadió 2-cloro-4-metilsulfonil-benzaldehído (40 g, 182,9 mmol) seguido de tamices moleculares de 4 angstrom (130 g). La mezcla se agitó durante 12 h momento en el que la RMN 1H de la alícuota reveló el consumo de los materiales de partida.
RMN 1H (300 MHz, CDCl3) 88,72 (d, J = 0,5 Hz, 1H), 8,23 (d, J = 8,2 Hz, 1H), 7,97 (d, J = 1,6 Hz, 1H), 7,83 (ddd, J = 8,2, 1,8, 0,7 Hz, 1H), 3,89-3,63 (m, 4H), 3,40 (t, J = 6,1 Hz, 2H), 3,07 (s, 3H), 1,96 (p, J = 6,7 Hz, 2H), 1,70 - 1,39 (m, 8H), 1,40 -1,19 (m, 6H), 0,98 - 0,77 (m, 15H).
La mezcla de reacción se filtró a través de un lecho de celite, el lecho se lavó con diclorometano (1,5 l). En un matraz separado que contenía hexafluoroisopropanol (700 ml), se le añadió 2,6-lutidina (25 ml, 215,8 mmol) seguido de Cu(OTf)2 (70 g, 193,5 mmol) [Cu(OTf)2 se secó a presión reducida durante 8 h a 100 °C]. La suspensión de color azul se agitó durante 1 h, después se añadió en una porción la solución de imina (producto 3) preparada anteriormente. La mezcla de reacción de color verde se agitó durante la noche a temperatura ambiente.
La CLEM muestra un pequeño pico correspondiente al producto deseado - Tr = 0,48 minutos (M+H) 289,95. La mezcla 2:1 se diluyó con 1,7 l de solución acuosa saturada de NaHCO3 e hidróxido de amonio al 10%. Después de agitar
durante 30 minutos, la fase orgánica se separó, se lavó dos veces con solución acuosa saturada de NaHCO3 (2 x 200 ml), salmuera (~200 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida.
El residuo se diluyó con acetonitrilo (600 ml), se lavó con heptano (4 x 100 ml) para retirar las impurezas de estanano. La fase de acetonitrilo (capa inferior) se evaporó a presión reducida para producir un sólido de color amarillo que se trituró con MTBE (400 ml), se filtró a través de un embudo fritado medio, se enjuagó con MTBE (100 ml) para producir el producto deseado 4 (28 g, rendimiento del 52 %) en forma de un sólido de color amarillo. La RMN 1H y la CLEM se ajustan a la estructura.
RMN 1H (400 MHz, DMSO) 87,96 - 7,92 (m, 1H), 7,92 - 7,83 (m, 2H), 4,31 (dd, J = 8,7, 3,3 Hz, 1H), 3,94 - 3,78 (m, 2H), 3,78 -3,62 (m, 1H), 3,36 -3,28 (m, 2H), 3,26 (s, 3H), 3,18 -3,04 (m, 1H), 3,00 -2,82 (m, 2H), 1,98 -1,71 (m, 2H). Ie N-EM m/z calc. 289,05396, encontrado 290,1 (M+1)+; Tiempo de retención: 0,49 minutos.
El filtrado se concentró a presión reducida para producir un aceite de color marrón claro que se purificó por cromatografía en gel de sílice (330 g de gradiente lineal de columna isco, 20 CV, 0 % ^ 100 % de acetato de etilo que contiene Et3N al 1 %/CH2Cl2), fracciones que contenían el producto deseado se recogieron, se concentraron a presión reducida, seguido de trituración con MTBE (~100 ml) para proporcionar el producto deseado 4 (5,8 g) en forma de un sólido de color amarillo. La RMN 1H se ajustó a la estructura.
Formación de I-66
Se cargó 3-(2-cloro-4-metilsulfonil-fenil)-1,4-oxazepano (28 g, 92,76 mmol) en un matraz RB de 2 l y 3 bocas con agitador superior, sonda de temperatura, condensador de reflujo y entrada de nitrógeno. -BuOH (550 ml), se agitó durante 15 minutos y después se añadió 4-cloro-6-metil-pirimidin-2-amina (17 g, 118,4 mmol). La mezcla de reacción resultante se calentó a 118 °C, se agitó a esta temperatura durante 14 h (durante una noche), momento en el cual el análisis HPLC y CLEM reveló el consumo del material de partida. La mezcla de reacción se enfrió a temperatura ambiente (precipitación observada), se diluyó con MTBE (500 ml). se agitó durante 30 minutos, se formó un precipitado de color blanco que se filtró a través de un embudo fritado medio, se enjuagó con MTBE (2 x 100 ml).
El precipitado (sal HCl del producto 3) se repartió entre acetato de etilo (500 ml) y solución acuosa sat. de NaHCO3 (~700 ml), agitada durante 30 minutos (pH ~8), se separó la fase orgánica. La fase acuosa se extrajo con acetato de etilo (2 x 100 ml), la fase orgánica combinada se lavó con salmuera (~100 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida.
El residuo se purificó por trituración con acetato de etilo al 10 % en MTBE (500 ml) para proporcionar 4-[3-(2-cloro-4-metilsulfonil-fenil)-1,4-oxazepan4-il]-6-metil-pirimidin-2-amina (32 g, 86 %) en forma de un sólido de color blanco. La RMN 1H y la Cl Em se ajustan a la estructura.
RMN 1H (400 MHz, DMSO) 87,96 (d, J = 1,7 Hz, 1H), 7,82 (dd, J = 8,2, 1,8 Hz, 1H), 7,58 (d, J = 8,2 Hz, 1H), 5,69 (d, J = 8,6 Hz, 1H), 5,61 (dd, J = 9,7, 4,6 Hz, 1H), 5,46 (s, 2H), 4,52 (d, J = 15,6 Hz, 1H), 4,16 (dd, J = 13,5, 4,9 Hz, 1H), 3,92 (dt, J = 12,0, 3,8 Hz, 1H), 3,85 - 3,69 (m, 2H), 3,66 - 3,53 (m, 1H), 3,23 (s, 3H), 2,05 (s, 3H), 1,88 - 1,76 (m, 2H). IEN-Em m/z calc. 396,1023, encontrado 397,1 (M+1)+; Tiempo de retención: 0,55 minutos.
Formación de I-67 y I-68
Se separó I-66 (66 g, 166,3 mmol) a través de SFC usando una columna AD-H con MeOH al 30 % y amoniaco al 0,2 %.
El pico B obtenido por SFC es el enantiómero deseado (conformador S) que se disolvió en CH2Cl2 (1 l), se lavó con solución acuosa sat. de NaHCO3 (2 x 200 ml), agua (50 ml), salmuera (100 ml), se secó sobre Na2SO4, se filtró a través de un lecho de gel de sílice (~80 g), el lecho se lavó con DCM (60 ml). Los filtrados combinados se concentraron a presión reducida para proporcionar el producto deseado en forma de un material amorfo ~32 g, que se disolvió en acetato de etilo (200 ml), se concentró a presión reducida. Este material se trituró con MTBE (2 x 200 ml) para producir un sólido de color blanco, seguido de azeotropía con acetato de etilo (200 ml) y una mezcla 1:1 de acetato de etilo y heptano (2 x 200 ml), se secó en estufa de vacío a 50 °C durante 14 h para producir el producto deseado que contenía acetato de etilo solvente residual ~ 4,6 % por moles, que se secó adicionalmente en un horno de vacío a 70 °C durante 14 h para proporcionar 4-[(3S)-3-(2-cloro-4-metilsulfonil-fenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (28,7 g, 85 %) ~2,6 % por moles de acetato de etilo residual.
RMN 1H (400 MHz, DMSO) 87,96 (s, 1H), 7,83 (dd, J = 8,2, 1,4 Hz, 1H), 7,58 (d, J = 8,2 Hz, 1H), 5,70 (s, 1H), 5,66 -5,55 (m, 1H), 5,46 (s, 2H), 4,52 (d, J = 14,9 Hz, 1H), 4,16 (dd, J = 13,5, 4,9 Hz, 1H), 4,00 - 3,88 (m, 1H), 3,85 - 3,70 (m, 2H), 3,65 - 3,52 (m, 1H), 3,23 (s, 3H), 2,05 (s, 3H), 1,88 - 1,72 (m, 2H).
IEN-e M m/z calc. 396,1023, encontrado 397,15 (M+1)+; Tiempo de retención: 0,55 minutos
- XRPD confirma que el material es cristalino.
- DSC muestra un punto de fusión de 194 °C.
- La estructura cristalina de rayos X de molécula pequeña para el pico B confirma que este es un conformador S. [a]D23 = 29,47 (c = 1,1, MeOH) durante 99,4 % ee (pico B)
Pico A (R-confórmero)
4-[(3R)-3-(2-cloro-4-metilsulfonil-fenil)-1,4-oxazepan-4-il]-6-metil-pirimidin-2-amina (25 g, 74 %) en forma de un sólido
de color blanquecino.
IEN-EM m/z calc. 396,1023, encontrado 397,1 (M+1)+; Tiempo de retención: 0,55 minutos [a]D23 = -23,33 (c = 1,0, MeOH) durante 99 % ee (pico A)
Ejemplo 60
Síntesis de 3-cloro-6,7-dihidro-1,4-oxazepina-4(5H)-carbaldehído

Formación de N-(3-hidroxipropil)carbamato de tere-butilo
Un matraz RB de 3 bocas de 2 l con agitador magnético y sonda de temperatura se cargó con 3-aminopropan-1-ol (43 g, 572,5 mmol) en una mezcla de THF (450 ml) y agua (450 ml), se agitó durante 10 minutos. y después se enfrió a 0 °C con un baño de hielo. Se añadió terc-butilcarbonato de terc-butoxicarbonilo (131 g, 600,2 mmol) en porciones durante 10 minutos y después la mezcla de reacción resultante se dejó lentamente a temperatura ambiente durante 12 h (durante la noche), momento en el cual se realizó análisis TLC (acetato de etilo al 50 % en heptano) que reveló el consumo del material de partida. La mezcla de reacción se concentró a ~50 % del volumen (para retirar el THF) y después se extrajo con DCM (2 x 500 ml). Los extractos orgánicos combinados se lavaron con agua (100 ml), salmuera (100 ml), se secaron sobre Na2SO4, se filtraron y se concentraron a presión reducida para producir N-(3-hidroxipropil)carbamato de terc-butilo (98 g, 98 %) en forma de un aceite transparente e incoloro. La RMN 1H se ajustó a la estructura. RMN 1H (300 MHz, cloroformo-d) 54,73 (s, 1H), 3,66 (t, J = 5,7 Hz, 2H), 3,28 (t, J = 6,2 Hz, 2H), 2,55 (s, 1H), 1,77 - 1,58 (m, 2H), 1,44 (s, 9H).
Formación de ácido 2-[3-(tere-butoxicarbonilamino)propoxi]acético
Se cargó un matraz RB de 2 L de 3 bocas con agitador superior, sonda de temperatura y condensador de reflujo con N-(3-hidroxipropil)carbamato de terc-butilo (42 g, 239,7 mmol) y tetrabutilamonio (ion bromuro (1)) (4 g, 12,41 mmol) en tolueno (300 ml), se agitó durante 5 minutos y después se enfrió a 0 °C con un baño de hielo. Se añadió NaOH (200 ml de 6 M, 1200 mol) mientras se mantenía la temperatura interna por debajo de 10 °C y se agitó durante 20 minutos más. Se añadió 2-bromoacetato de terc-butilo (55 g, 282,0 mmol) en tolueno (50 ml) durante 5 minutos. La mezcla de reacción resultante se calentó a 60 °C (Tmáx 65 °C), se agitó a esta temperatura durante 14 h. La mezcla de reacción se enfrió a temperatura ambiente y las capas se separaron, la capa acuosa se extrajo con tolueno (60 ml). La fase acuosa se enfrió a 0 °C con un baño de hielo y se acidificó con HCl 12 N hasta que el pH fue de 3. Se añadió acetato de etilo (150 ml) y las capas se separaron, la capa acuosa se extrajo con acetato de etilo (50 ml). La capa orgánica combinada se secó sobre Na2SO4, se filtró y se concentró a presión reducida para producir ácido 2-[3-(tercbutoxicarbonilamino)propoxi]acético (36 g, 61 %) en forma de un aceite transparente e incoloro que contenía pequeñas cantidades de acetato de etilo.
RMN 1H (300 MHz, MeOD) 54,07 (s, 2H), 3,60-3,54 (m, 2H), 3,32-3,30 (m, 2H), 1,79-1,70 (m, 2H), 1,47 (s, 9H) Este material se recogió en la siguiente etapa sin purificación adicional.
Formación de 2-(3-aminopropoxi)acetato de metilo
Un matraz RB de 3 bocas de 1 l con agitador magnético, sonda de temperatura, se cargó con ácido 2-[3-(tercbutoxicarbonilamino)propoxi]acético (12 g, 51,44 mmol) en metanol (120,0 ml), se agitó durante 5 minutos y se luego se enfrió a 0 °C con un baño de hielo. Se añadió cloruro de tionilo (10 ml, 137,1 mmol) durante 10 minutos y después se dejó lentamente a temperatura ambiente durante 12 h (durante una noche). La mezcla de reacción se concentró a presión reducida. El residuo se destiló azeotrópicamente con DCM (2 x 60 ml) para proporcionar 2-(3-aminopropoxi)acetato de metilo (sal clorhidrato) (10 g, 95%), ~90 % de pureza en forma de un aceite viscoso transparente e incoloro. La RMN 1h se ajustó a la estructura. Rm N 1H (300 MHz, DMSO-d6) 88,05 (s, 3H), 4,12 (s, 2H), 3,66 (s, 3H), 3,54 (t, J = 6,0 Hz, 2H), 2,84 (t, J = 7,4 Hz, 2H), 1,95 - 1,75 (m, 2H).
Este material se recogió en la siguiente etapa sin purificación adicional.
Formación de 1,4-oxazepan-3-ona
Se cargó un matraz RB de 3 bocas de 1 l con agitador magnético, sonda de temperatura y condensador de reflujo con 2-(3-aminopropoxi)acetato de metilo (sal clorhidrato) (10 g, 49,01 mmol) en metanol (100 ml), se agitó durante 5 minutos y después se añadió K2CO3 (14 g, 101,3 mmol). La mezcla de reacción resultante se calentó a 60 °C, se agitó a esta temperatura durante 2 h, se observó la mancha del producto deseado 2 (TLC, metanol al 10 % en DCM) y la mancha de referencia podría ser el material de partida). La mezcla de reacción se enfrió a temperatura ambiente, se filtró a través de un lecho de celite y el lecho se lavó con metanol (2 x 25 ml). Los filtrados combinados se concentraron a presión reducida. El residuo se repartió entre DCM (150 ml) y agua (50 ml), la fase orgánica se separó, se lavó con salmuera (50 ml), se secó sobre Na2SO4, se filtró y se concentró a presión reducida. El residuo se purificó por cromatografía sobre gel de sílice (columna isco de 40 g de gradiente lineal, 20 CV, metanol al 0 % ^ 20 %/DC), las fracciones que contenían el producto deseado se recogieron, se concentraron a presión reducida para proporcionar 1,4-oxazepan-3-ona (1,8 g, 32 %) en forma de un sólido de color blanco. La RMN 1H se ajustó a la estructura.
RMN 1H (300 MHz, DMSO-d6) 87,65 (s, 1H), 3,99 (s, 2H), 3,83 - 3,61 (m, 2H), 3,25 - 3,06 (m, 2H), 1,87 - 1,62 (m, 2H).
Formación de 3-cloro-6,7-dihidro-5H-1,4-oxazepina-4-carbaldehído
Se cargó DMF (141,6 g, 150,0 ml, 1,937 mol) en DCM (300,0 ml) en un matraz RB de 2 l y 3 bocas con agitador superior, sonda de temperatura, embudo adicional, entrada de nitrógeno y condensador de reflujo, se agitó durante 5 minutos y después se enfrió a 0 °C con baño de hielo. Se añadió POCl3 (90 ml, 965,6 mmol) en DCM (100,0 ml) durante 30 minutos mientras se mantenía la temperatura interna por debajo de 6 °C. La mezcla de reacción se calentó a 40 °C (nota: la mezcla de reacción se convirtió en una solución transparente de color rojo y se observó exotermia, se mantuvo la temperatura interna a ~40 °C) y se agitó a esta temperatura durante 45 minutos. Se añadió 1,4-oxazepan-3-ona (50 g, 434,3 mmol) en DCM (300 ml) durante 40 minutos, se observó exotermia, se mantuvo la temperatura interna ~40 °C. La mezcla de reacción resultante se agitó a esta temperatura durante 90 minutos, momento en el cual la TLC (metanol al 10 % en DCM) y el análisis de CLEM revelaron el consumo del material de partida 1, pico principal Tr = 0,51 minutos (M+H)+ 189/191 que corresponde al intermedio de amidina. La mezcla de reacción se enfrió a temperatura ambiente, se vertió en hielo triturado (1,2 l) y después se dejó a temperatura ambiente durante 1 hora y se agitó durante 1 hora más. Se separó la capa acuosa, se basificó con K2CO3 sólido hasta pH 9, se dejó a temperatura ambiente, se agitó a esta temperatura durante 12 h (durante la noche), momento en el cual el análisis CLEM reveló un pico principal a los 0,69 minutos (M+1)+ 162,04 que corresponde a producto deseado 2. La mezcla de reacción se diluyó con d Cm (300 ml), la capa orgánica se separó. La capa acuosa se extrajo con DCM (100 ml), los extractos orgánicos combinados se secaron sobre Na2SO4, se filtraron y concentraron a presión reducida. El residuo se purificó por cromatografía sobre gel de sílice (columna isco de 330 g de gradiente lineal, 20 CV, acetato de etilo al 0 % ^ 50 %/heptano-que contenía Et3N al 1 %), las fracciones que contenían el producto deseado se recogieron, se concentraron a presión reducida para proporcionar 3-cloro-6,7-dihidro-5H-1,4-oxazepina-4-carbaldehído (41 g, 58 %) en forma de un aceite transparente e incoloro.
RMN 1H (400 MHz, DMSO-d6) 88,32 (rotámero; d, J = 157,3 Hz, 1H), 6,59 (d, J = 4,2 Hz, 1H), 4,19 - 3,87 (m, 2H), 3,85 - 3,51 (m, 2H), 2,02 - 1,71 (m, 2H).
La RMN 1H muestra una mezcla de rotámeros.
Los compuestos de la Tabla 2A se preparan mediante métodos de síntesis similares a los descritos anteriormente. Los datos analíticos de algunos de los compuestos se enumeran a continuación.

continuación

continuación

continuación

continuación

continuación

continuación

continuación

continuación

continuación

continuación

continuación

continuación

continuación

continuación

continuación

continuación

continuación
continuación

continuación

continuación

Claims (20)
1. Un compuesto de fórmula I':

I’
o una sal farmacéuticamente aceptable del mismo, en donde:
el anillo A es un anillo seleccionado entre fenilo, un anillo carbocíclico saturado o parcialmente insaturado de 5-7 miembros, un anillo heterocíclico bicíclico saturado o parcialmente insaturado de 8 -12 miembros que tiene 1 -2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, un anillo heteroaromático de 5-6 miembros que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre; o un anillo heteroaromático bicíclico de 8-10 miembros que tiene 1-5 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre;
cada R1 es independientemente hidrógeno o alifático C1-3 opcionalmente sustituido con 1-6 halógeno; o
dos grupos R1 se toman opcionalmente junto con sus átomos intermedios para formar un anillo carbocíclico condensado parcialmente insaturado de 5-8 miembros;
cada uno de R2 es independientemente hidrógeno, halógeno, -CN, -NO2, -C(O)OR, -C(O)NR2, -NR2, -NRC(O)R, -NRC(O)OR, -NRS(O)2R, -OR, -P(O)R2, -SR, -S(O)R, -S(O)2R, -S(O)(NH)R, -S(O)2NR2 o R; o
dos grupos R2 se toman opcionalmente juntos para formar =O; o
dos grupos R2 se toman opcionalmente junto con sus átomos intermedios para formar un anillo espirocíclico saturado de 3-8 miembros que tiene 0-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre; cada R3 es independientemente hidrógeno, -OH o alifático C1-3; o
dos grupos R3 se toman opcionalmente juntos para formar =O; o
dos grupos R3 se toman opcionalmente juntos para formar =CH2; o
dos grupos R3 se toman opcionalmente junto con sus átomos intermedios para formar un anillo espirocíclico saturado de 3-8 miembros que tiene 0-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre; o
dos grupos R3 se toman opcionalmente junto con sus átomos intermedios para formar un anillo bicíclico con puente saturado de 5-8 miembros que tiene 0-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre;
cada R es independientemente hidrógeno o un grupo opcionalmente sustituido seleccionado entre hidrocarburo alifático C1-6, un anillo carbocíclico monocíclico saturado o parcialmente insaturado de 3-8 miembros, fenilo, un anillo heterocíclico espirobicíclico saturado de 7-10 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, un anillo heterocíclico bicíclico condensado saturado o parcialmente insaturado de 7-10 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, un anillo heterocíclico monocíclico saturado o parcialmente insaturado de 4-8 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, o un anillo heteroaromático monocíclico de 5-6 miembros que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre; o:
dos grupos R en el mismo nitrógeno se toman opcionalmente junto con sus átomos intermedios para formar un anillo de 4-7 miembros saturado, parcialmente insaturado o heteroarilo que tiene 0-3 heteroátomos, además del nitrógeno, seleccionado independientemente entre nitrógeno, oxígeno y azufre, opcionalmente sustituido con 1-2 grupos oxo;
-----es un enlace sencillo o un enlace doble;
X es -O-, -N(R)-, -N(S(O)2(R))-, -S-, -S(O)-, -S(O)2-, -CH2-, -CH(R3)- o -C(R3)2-;
m es 0, 1 o 2;
n es 0, 1, 2, 3, 4 o 5; y
p es 0, 1 o 2.
2. El compuesto de la reivindicación 1 o una sal farmacéuticamente aceptable de este, en donde el anillo A es fenilo; o:
en donde el anillo A es un anillo heterocíclico bicíclico saturado o parcialmente insaturado de 8-12 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre; o:
en donde el anillo A es un anillo heteroaromático bicíclico de 8-10 miembros que tiene 1-5 heteroátomos seleccionados
independientemente entre nitrógeno, oxígeno o azufre; o: en donde el anillo A es > en donde el Anillo B es un anillo heterocíclico parcialmente insaturado de 5-7 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, o el anillo B es un anillo heteroaromático de 5-6 miembros que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre; o:
> en donde el Anillo B es un anillo heterocíclico parcialmente insaturado de 5-7 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, o el anillo B es un anillo heteroaromático de 5-6 miembros que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre; o:
en donde el anillo A se selecciona entre:

y
3. El compuesto de la reivindicación 1 o la reivindicación 2, o una sal farmacéuticamente aceptable del mismo, en donde al menos uno de R1 es alifático C1-3; o:
en donde al menos uno de R1 es -CH3; y/o:
en donde al menos uno de R1 se une a la posición 6 de la pirimidina.
4. El compuesto de una cualquiera de las reivindicaciones anteriores, o una sal farmacéuticamente aceptable del mismo, en donde al menos uno de R2 es alifático C1-6, opcionalmente sustituido 1-4 veces por halógeno, -OH, NH2, -OCH3, -NHC(O)CH3, -S(O)2CH3, -COOH, -CO2CH3, -CO2C2H5 o -N(CH3)C(O)CH3; opcionalmente:

y

o:
en donde al menos uno de R2 es un anillo carbocíclico monocíclico saturado de 3-6 miembros; opcionalmente:

en donde al menos uno de R2 es o:
en donde al menos uno de R2 es un anillo heterocíclico espirobicíclico saturado de 7-10 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno u oxígeno; opcionalmente:

en donde al menos uno de R2 se selecciona entre y
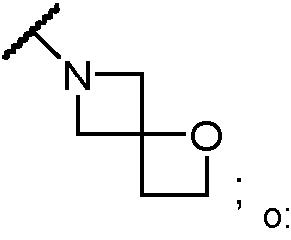
en donde al menos uno de R2 es un anillo heterocíclico bicíclico condensado saturado de 7-10 miembros que tiene 1 2 heteroátomos seleccionados independientemente entre nitrógeno u oxígeno; opcionalmente:
en donde al menos uno d :
:
en donde al menos uno de R2 es un anillo heterocíclico monocíclico saturado de 4-6 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, opcionalmente sustituido 1-4
veces por halógeno, -OH, -CH3, -OCH3, =O 

en donde al menos uno de R2 se selecciona entre


en donde al menos uno de R2 es un anillo heteroaromático monocíclico de 5-6 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre; opcionalmente:

en donde al menos uno de R2 se selecciona entre y

o:
en donde al menos uno de R2 es -C(O)OR, en donde R es hidrógeno o alifático C-i-a; opcionalmente:
en donde al menos uno de R2 es-C(O)0H,  o:
o:
en donde al menos uno de R2 es -C(O)NR2, en donde cada uno de R es independientemente hidrógeno, alifático C1-6 que está opcionalmente sustituido con un -N(CH3)2, anillo carbocíclico monocíclico saturado de 3-6 miembros sin sustituir, o anillo heterocíclico monocíclico saturado de 4-6 miembros sin sustituir que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno u oxígeno, o dos R tomados junto con sus átomos intermedios para formar un anillo saturado y sin sustituir de 4-7 miembros que tiene de 0-3 heteroátomos, además del nitrógeno, seleccionado independientemente entre nitrógeno, oxígeno y azufre; opcionalmente:
en donde al menos uno de R2 se selecciona entre 

o:
en donde al menos uno de R2 es -NR2, en donde cada uno de R es independientemente:
hidrógeno;


anillo carbocíclico monocíclico saturado de 3-6 miembros sin sustituir;
anillo heterocíclico monocíclico saturado de 4-6 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno u oxígeno, que está opcionalmente sustituido 1-2 veces por CH3, -OH, -C(O)OC(CHa)a o -C(C)CH3; o
un anillo heteroaromático monocíclico de 6 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre que está opcionalmente sustituido 1-2 veces por -CH3 o -NH2; opcionalmente:
en donde al menos uno de R2 se selecciona entre:

5. El compuesto de una cualquiera de las reivindicaciones anteriores, o una sal farmacéuticamente aceptable del mismo, en donde al menos uno de R2 es -NHC(O)R, en donde R es alifático C1-6 opcionalmente sustituido 1-3 veces por halógeno, -OCH3, -N(CH3)2 u -OH, anillo carbocíclico monocíclico saturado de 3-6 miembros opcionalmente sustituido 1-2 veces con halógeno u -OH, o anillo heterocíclico monocíclico saturado de 4-6 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre opcionalmente sustituido 1-2 veces por halógeno, -OH o -CH3; opcionalmente:

en donde al menos uno de R2 se selecciona entre


en donde al menos uno de R2 es -NHC(O)OR, en donde R es alifático Ci-6 sin sustituir; opcionalmente:
en donde al menos uno d
 :
:
en donde al menos uno de R2 es -NHS(O)2R, en donde R es alifático C1-6 sin sustituir; opcionalmente:
en donde al menos uno d
 :
:
en donde al menos uno de R2 es -OR, en donde R es:
hidrógeno;

anillo heterocíclico monocíclico saturado de 4-6 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno u oxígeno; opcionalmente:
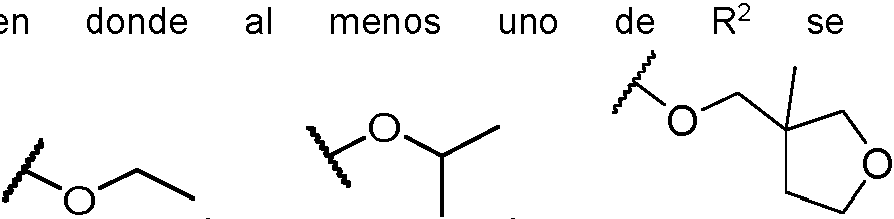



y o: en donde al menos uno de R2 es -P(O)R2, en donde cada uno de R es independientemente alifático C1-6 sin sustituir; opcionalmente:
O
i M
en donde al menos uno de R2 es
h ~
’ o:
en donde al menos uno de R2 es -SR, en donde R es alifático C1-6 sin sustituir; opcionalmente:
en donde al menos uno de R2 es
 opcionalmente al mismo:
opcionalmente al mismo:
O
I— S — .
en donde al menos uno de R2 es * > o:
en donde al menos uno de R2 es -S(O)R, en donde R es alifático C1-6 sin sustituir; o:
en donde al menos uno de R2 es -S(O)2R, en donde R es alifático C1.6 sin sustituir o anillo carbocíclico monocíclico saturado de 3-6 miembros; opcionalmente:
en donde al menos uno de R2 se selecciona entre
 o:
o:
en donde al menos uno de R2 es -S(O)(NH)R, en donde R es alifático C1-6 sin sustituir; opcionalmente:
en donde al menos uno
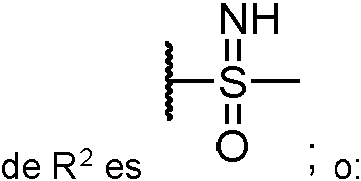
en donde al menos uno de R2 es -S(O)2NR2; opcionalmente:
en donde al menos uno de R2 es -S(O)2NH2.
6. El compuesto de una cualquiera de las reivindicaciones anteriores, o una sal farmacéuticamente aceptable del mismo, en donde al menos uno de R3 es alifático C1-3; y/o:
en donde X es -O-; y/o:
en donde m es 1; y/o:
en donde p es 0; y/o:
en donde n es 1, 2, 3, 4 o 5; y/o:
en donde------es un enlace sencillo.
7. El compuesto de una cualquiera de las reivindicaciones anteriores, o una sal farmacéuticamente aceptable del mismo, en donde el compuesto es de Fórmula VII

o una sal farmacéuticamente aceptable del mismo, en donde:
n' es 1 o 2 ; y
R2' es halógeno u -Oalifático C1-3; o:
en donde el compuesto es de Fórmula VIII

o una sal farmacéuticamente aceptable del mismo, en donde:
el anillo B es un anillo heterocíclico parcialmente insaturado de 5-7 miembros que tiene 1-2 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre, o el anillo B es un anillo heteroaromático de 5 6 miembros que tiene 1-4 heteroátomos seleccionados independientemente entre nitrógeno, oxígeno o azufre; n" es 0, 1, 2, 3 o 4; y
R2' es halógeno u -Oalifático C1-3.
8. El compuesto de la reivindicación 7 o una sal farmacéuticamente aceptable del mismo, en donde R2' es halógeno u -Oalifático C1-3; o:
en donde R2' es Cl; o:
en donde R2' es -OCH3.
9. El compuesto de una cualquiera de las reivindicaciones 7 u 8 o una sal farmacéuticamente aceptable del mismo, en donde
el compuesto es de Fórmula VII-a

; y
R2' es Cl.
10. El compuesto de cualquiera de las reivindicaciones precedentes o una sal farmacéuticamente aceptable del mismo, en donde el compuesto es de Fórmula XI o Xl-a,

11. El compuesto de la reivindicación 10 o una sal farmacéuticamente aceptable de este, en donde R2' es Cl y cada




12. El compuesto una cualquiera de las reivindicaciones anteriores, o una sal farmacéuticamente aceptable del mismo, en donde el compuesto se selecciona entre los compuestos representados en las Tablas 1, 2 y 2A.
13. El compuesto de la reivindicación 12 o una sal farmacéuticamente aceptable de este, en donde el compuesto se selecciona entre los compuestos


IJ63

14. El compuesto de una cualquiera de las reivindicaciones 12 y 13, o una sal farmacéuticamente aceptable del mismo, en donde el compuesto es
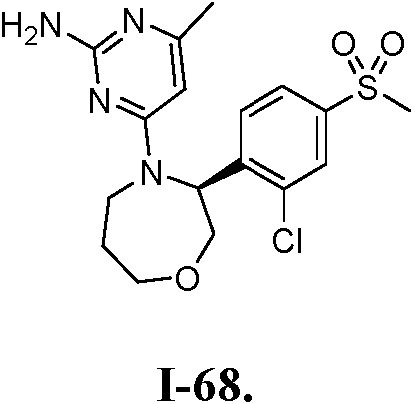
15. Una composición farmacéutica que comprende un compuesto según una cualquiera de las reivindicaciones 1 a 14, o una sal farmacéuticamente aceptable del mismo y un transportador, adyuvante o vehículo farmacéuticamente aceptable.
16. Un compuesto según una cualquiera de las reivindicaciones 1 a 14, o una sal farmacéuticamente aceptable del mismo, o una composición farmacéutica del mismo, para su uso en un método para tratar un trastorno proliferativo
celular en un paciente, que comprende administrar a dicho paciente dicho compuesto o composición farmacéutica.
17. El compuesto o la composición para el uso de la reivindicación 16, comprendiendo además dicho uso determinar el nivel de expresión de wolframina (WFS1).
18. El compuesto o composición para el uso de las reivindicaciones 16 o 17, en donde el trastorno proliferativo celular es cáncer; o:
en donde el trastorno proliferativo celular se selecciona de cáncer de pulmón no microcítico (NSCLC), mieloma, carcinoma hepatocelular (HCC), cáncer de mama, cáncer colorrectal, cáncer de endometrio, cáncer de esófago, cáncer pancreático, carcinoma de células renales y melanoma.
19. Un compuesto según una cualquiera de las reivindicaciones 1 a 14, o una sal farmacéuticamente aceptable del mismo, o una composición farmacéutica del mismo, para su uso en un método para inducir estrés al RE en un paciente que lo necesite, que comprende administrar dicho compuesto o composición a dicho paciente; o:
Un compuesto según una cualquiera de las reivindicaciones 1 a 14 para su uso en un método para inducir una respuesta a proteínas desplegadas (UPR) en un paciente que lo necesite, que comprende administrar a dicho paciente dicho compuesto o composición.
20. Un compuesto según una cualquiera de las reivindicaciones 1 a 14, o una sal farmacéuticamente aceptable del mismo, o una composición farmacéutica del mismo, para su uso en un método para provocar la liberación de calcio del retículo endoplásmico (RE) a través de un supuesto canal de Ca2+ conocido como wolframina (WFS1) en un paciente que lo necesite, que comprende administrar a dicho paciente dicho compuesto o composición.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201862661719P | 2018-04-24 | 2018-04-24 | |
| PCT/US2019/028607 WO2019209759A1 (en) | 2018-04-24 | 2019-04-23 | Antiproliferation compounds and uses thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2919572T3 true ES2919572T3 (es) | 2022-07-27 |
Family
ID=66655429
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES19726792T Active ES2919572T3 (es) | 2018-04-24 | 2019-04-23 | Compuestos antiproliferación y usos de los mismos |
Country Status (24)
| Country | Link |
|---|---|
| US (3) | US10815225B2 (es) |
| EP (2) | EP3784666B1 (es) |
| JP (2) | JP7304892B2 (es) |
| KR (1) | KR20210005151A (es) |
| CN (1) | CN112236428A (es) |
| AR (1) | AR114496A1 (es) |
| AU (1) | AU2019261308B2 (es) |
| BR (1) | BR112020020940A2 (es) |
| CA (1) | CA3098335A1 (es) |
| DK (1) | DK3784666T3 (es) |
| ES (1) | ES2919572T3 (es) |
| HR (1) | HRP20220710T1 (es) |
| HU (1) | HUE058746T2 (es) |
| IL (2) | IL301501A (es) |
| LT (1) | LT3784666T (es) |
| MX (1) | MX2020011089A (es) |
| PL (1) | PL3784666T3 (es) |
| PT (1) | PT3784666T (es) |
| RS (1) | RS63281B1 (es) |
| SG (1) | SG11202009500QA (es) |
| SI (1) | SI3784666T1 (es) |
| TW (1) | TWI813673B (es) |
| WO (1) | WO2019209759A1 (es) |
| ZA (1) | ZA202006071B (es) |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RS63281B1 (sr) | 2018-04-24 | 2022-06-30 | Merck Patent Gmbh | Antiproliferativna jedinjenja i njihova upotreba |
| WO2020010451A1 (en) * | 2018-07-10 | 2020-01-16 | Trillium Therapeutics Inc. | Heteroaromatic-fused imidazolyl amides, compositions and uses thereof as sting agonists |
| WO2020236834A1 (en) * | 2019-05-20 | 2020-11-26 | Seshadri Raju | Modified z stents for iliac vein stenting |
| CA3165267A1 (en) | 2019-12-20 | 2021-06-24 | Bayer Aktiengesellschaft | Substituted thiophene carboxamides, thiophene carboxylic acids and derivatives thereof |
| WO2024073502A1 (en) * | 2022-09-28 | 2024-04-04 | Accutar Biotechnology Inc. | Heterocyclic compounds as e3 ligase inhibitors |
Family Cites Families (32)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR901228A (fr) | 1943-01-16 | 1945-07-20 | Deutsche Edelstahlwerke Ag | Système d'aimant à entrefer annulaire |
| DE19840028A1 (de) | 1998-09-02 | 2000-03-09 | Max Planck Gesellschaft | Nucleinsäuremoleküle codierend Enzyme, die Fructosyltransferaseaktivität besitzen, und deren Verwendung |
| MXPA02005675A (es) | 1999-12-10 | 2002-09-02 | Pfizer Prod Inc | Compuestos de pirrolo(2,3-d)pirimidina. |
| PE20020354A1 (es) | 2000-09-01 | 2002-06-12 | Novartis Ag | Compuestos de hidroxamato como inhibidores de histona-desacetilasa (hda) |
| KR100833371B1 (ko) | 2001-04-27 | 2008-05-28 | 젠야쿠코교가부시키가이샤 | 헤테로시클릭 화합물 및 이를 유효 성분으로 하는 항종양제 |
| TWI329105B (en) | 2002-02-01 | 2010-08-21 | Rigel Pharmaceuticals Inc | 2,4-pyrimidinediamine compounds and their uses |
| PT1536827E (pt) | 2002-08-14 | 2009-03-20 | Silence Therapeutics Ag | Utilização de proteína cinase n beta |
| CN1826331A (zh) | 2003-04-03 | 2006-08-30 | 塞马福尔药业公司 | Pi-3激酶抑制剂前药 |
| CA2527583C (en) | 2003-05-30 | 2013-12-17 | Giorgio Attardo | Triheterocyclic compounds, compositions, and methods for treating cancer or viral diseases |
| EP1692153A4 (en) | 2003-07-03 | 2007-03-21 | Univ Pennsylvania | INHIBITION OF EXPRESSION OF SYK-KINASE |
| RS55551B1 (sr) | 2004-05-13 | 2017-05-31 | Icos Corp | Hinazolinoni kao inhibitori humane fosfatidilinozitol 3-kinaze delta |
| ATE451381T1 (de) | 2005-01-19 | 2009-12-15 | Rigel Pharmaceuticals Inc | Prodrugs aus 2,4-pyrimidindiamin-verbindungen und ihre verwendungen |
| MX2007014049A (es) | 2005-05-12 | 2008-02-11 | Abbott Lab | Activadores de apoptosis. |
| GB0510390D0 (en) | 2005-05-20 | 2005-06-29 | Novartis Ag | Organic compounds |
| US7402325B2 (en) | 2005-07-28 | 2008-07-22 | Phoenix Biotechnology, Inc. | Supercritical carbon dioxide extract of pharmacologically active components from Nerium oleander |
| WO2007044729A2 (en) | 2005-10-07 | 2007-04-19 | Exelixis, Inc. | N- (3-amino-quinoxalin-2-yl) -sulfonamide derivatives and their use as phosphatidylinositol 3-kinase inhibitors |
| HUE028987T2 (en) | 2005-11-01 | 2017-01-30 | Targegen Inc | BI-aryl-meta-pyrimidine inhibitors of kinases |
| ES2612196T3 (es) | 2005-12-13 | 2017-05-12 | Incyte Holdings Corporation | Pirrolo[2,3-b]piridinas y pirrolo[2,3-b]pirimidinas sustituidas con heteroarilo como inhibidores de quinasas Janus |
| JO2660B1 (en) | 2006-01-20 | 2012-06-17 | نوفارتيس ايه جي | Pi-3 inhibitors and methods of use |
| ATE471940T1 (de) | 2006-04-26 | 2010-07-15 | Hoffmann La Roche | Thienoä3,2-düpyrimidin-derivat geeignet als pi3k inhibitor |
| LT2530083T (lt) | 2006-09-22 | 2016-09-26 | Pharmacyclics Llc | Brutono tirozinkinazės inhibitoriai |
| CN101861313B (zh) | 2007-03-12 | 2014-06-04 | Ym生物科学澳大利亚私人有限公司 | 苯基氨基嘧啶化合物及其用途 |
| US8394794B2 (en) | 2007-03-23 | 2013-03-12 | Regents Of The University Of Minnesota | Therapeutic compounds |
| PE20090717A1 (es) | 2007-05-18 | 2009-07-18 | Smithkline Beecham Corp | Derivados de quinolina como inhibidores de la pi3 quinasa |
| KR20120108042A (ko) | 2008-03-11 | 2012-10-04 | 인사이트 코포레이션 | Jak 억제제로서의 아제티딘 및 시클로부탄 유도체 |
| US8338439B2 (en) | 2008-06-27 | 2012-12-25 | Celgene Avilomics Research, Inc. | 2,4-disubstituted pyrimidines useful as kinase inhibitors |
| DE102008031517A1 (de) * | 2008-07-03 | 2010-01-07 | Merck Patent Gmbh | Pyrrolopyridinyl-pyrimidin-2-yl-amin-derivate |
| AR076014A1 (es) * | 2009-04-02 | 2011-05-11 | Sanofi Aventis | Derivados de 3- (1,4) oxazepan -4-pirimidona |
| CN102753528A (zh) * | 2009-12-04 | 2012-10-24 | 霍夫曼-拉罗奇有限公司 | 作为单胺重摄取抑制剂的二苯基氮杂*衍生物 |
| PL3214091T3 (pl) | 2010-12-09 | 2019-03-29 | The Trustees Of The University Of Pennsylvania | Zastosowanie komórek T modyfikowanych chimerycznymi receptorami antygenowymi do leczenia nowotworów |
| AU2016218569A1 (en) * | 2015-02-13 | 2017-09-28 | Merck Patent Gmbh | Pyrimidine derivatives for use in the treatment of cancer |
| RS63281B1 (sr) | 2018-04-24 | 2022-06-30 | Merck Patent Gmbh | Antiproliferativna jedinjenja i njihova upotreba |
-
2019
- 2019-04-23 RS RS20220537A patent/RS63281B1/sr unknown
- 2019-04-23 BR BR112020020940-6A patent/BR112020020940A2/pt unknown
- 2019-04-23 HU HUE19726792A patent/HUE058746T2/hu unknown
- 2019-04-23 TW TW108114146A patent/TWI813673B/zh active
- 2019-04-23 EP EP19726792.5A patent/EP3784666B1/en active Active
- 2019-04-23 HR HRP20220710TT patent/HRP20220710T1/hr unknown
- 2019-04-23 LT LTEPPCT/US2019/028607T patent/LT3784666T/lt unknown
- 2019-04-23 SG SG11202009500QA patent/SG11202009500QA/en unknown
- 2019-04-23 IL IL301501A patent/IL301501A/en unknown
- 2019-04-23 DK DK19726792.5T patent/DK3784666T3/da active
- 2019-04-23 JP JP2020559394A patent/JP7304892B2/ja active Active
- 2019-04-23 AU AU2019261308A patent/AU2019261308B2/en active Active
- 2019-04-23 WO PCT/US2019/028607 patent/WO2019209759A1/en unknown
- 2019-04-23 EP EP22161962.0A patent/EP4043460A1/en active Pending
- 2019-04-23 IL IL278142A patent/IL278142B2/en unknown
- 2019-04-23 ES ES19726792T patent/ES2919572T3/es active Active
- 2019-04-23 SI SI201930251T patent/SI3784666T1/sl unknown
- 2019-04-23 MX MX2020011089A patent/MX2020011089A/es unknown
- 2019-04-23 CA CA3098335A patent/CA3098335A1/en active Pending
- 2019-04-23 PL PL19726792.5T patent/PL3784666T3/pl unknown
- 2019-04-23 KR KR1020207033873A patent/KR20210005151A/ko unknown
- 2019-04-23 AR ARP190101062A patent/AR114496A1/es unknown
- 2019-04-23 US US16/391,419 patent/US10815225B2/en active Active
- 2019-04-23 PT PT197267925T patent/PT3784666T/pt unknown
- 2019-04-23 CN CN201980036896.XA patent/CN112236428A/zh active Pending
-
2020
- 2020-08-27 US US17/004,109 patent/US11440907B1/en active Active
- 2020-09-30 ZA ZA2020/06071A patent/ZA202006071B/en unknown
-
2022
- 2022-09-07 US US17/930,352 patent/US20230150993A1/en active Pending
-
2023
- 2023-06-05 JP JP2023092450A patent/JP2023101824A/ja active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11780837B2 (en) | CXCR4 inhibitors and uses thereof | |
| US20210363139A1 (en) | Cxcr4 inhibitors and uses thereof | |
| ES2919572T3 (es) | Compuestos antiproliferación y usos de los mismos | |
| TW202108559A (zh) | Tead抑制劑及其用途 | |
| EP3846793B1 (en) | Eif4e inhibitors and uses thereof | |
| US20230110180A1 (en) | Cdk2 degraders and uses thereof | |
| RU2811969C2 (ru) | Антипролиферативные соединения и их применения | |
| US20240174665A1 (en) | Cdk2 inhibitors and uses thereof | |
| WO2024054603A1 (en) | Heterobifunctional compounds and methods of treating disease | |
| CN114206869A (zh) | Cxcr4抑制剂和其用途 |