EP0074792B1 - Isomer enrichment process for cyclopropane carboxylates - Google Patents
Isomer enrichment process for cyclopropane carboxylates Download PDFInfo
- Publication number
- EP0074792B1 EP0074792B1 EP19820304711 EP82304711A EP0074792B1 EP 0074792 B1 EP0074792 B1 EP 0074792B1 EP 19820304711 EP19820304711 EP 19820304711 EP 82304711 A EP82304711 A EP 82304711A EP 0074792 B1 EP0074792 B1 EP 0074792B1
- Authority
- EP
- European Patent Office
- Prior art keywords
- vicinal
- substituted cyclopropane
- cis
- substituted
- trans
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C51/00—Preparation of carboxylic acids or their salts, halides or anhydrides
- C07C51/42—Separation; Purification; Stabilisation; Use of additives
- C07C51/487—Separation; Purification; Stabilisation; Use of additives by treatment giving rise to chemical modification
Definitions
- Natural and synthetic cyclopropane derivatives have been of considerable interest in the pesticide field.
- various esters or analogues of chrysanthemic or pyrethric acids have achieved commercial acceptance as insecticides on a wide scale.
- Specific examples of such compounds are those of the following Formula (I): wherein X may be hydrogen to define the structure of permethrin and cyano to define cypermethrin as described in U.S. Patent 4,024,163.
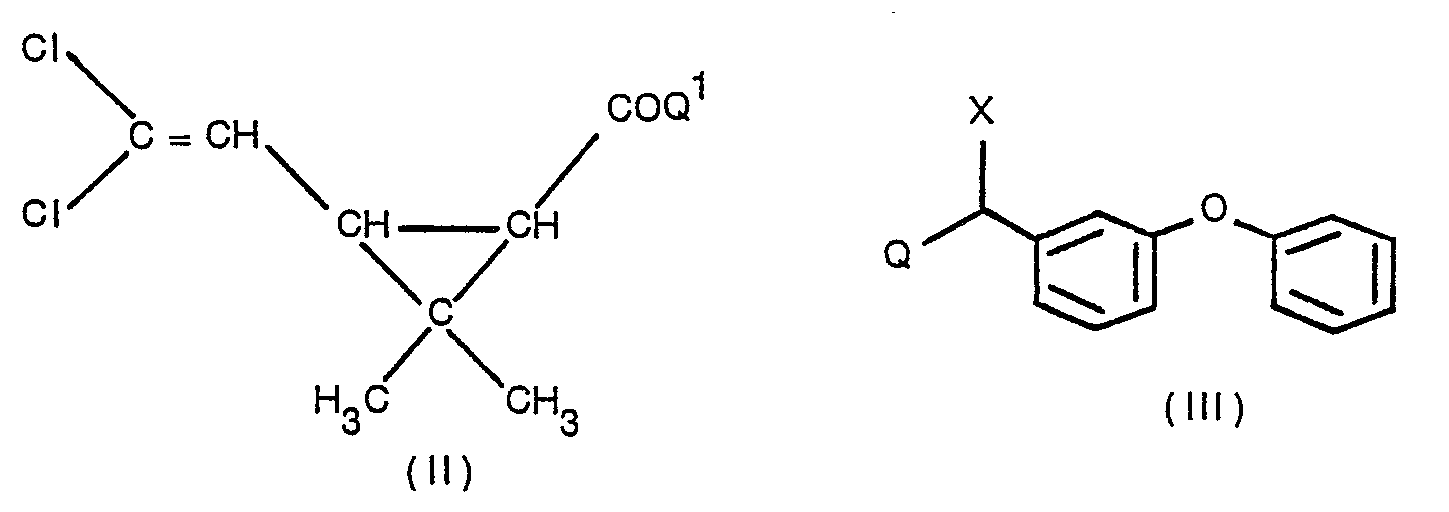
- Insecticidal compounds of the Formula (I) may be prepared by esterification of a compound of the following Formula (II) with a compound of the following Formula (III): wherein Q and COQ' are functional groups or atoms which will react together to form an ester linkage and X is as described for Formula (1). It is usually convenient to react the acid or acid halide with the alcohol, e.g.
- Such reactions are described in U.S. Patents 4,024,163, 4,254,050, 4,254,051, 4,254,052 and 4,280,965.
- Carboxylic acid or acid chloride compounds of Formula (II) may be obtained from the corresponding intermediate ester, e.g. COQ' is COOH 3 , by hydrolysis with strong base or hydrolysis followed by halogenation, respectively.
- the intermediate esters may be prepared as described in "Recent Advances in Synthetic Pyrethroids" by Alfred Bader in Aldrichimica Acta 9, No. 3, pages 49-51 (1976).
- An object of the present invention is a process which adjusts the cis/trans ratio of a vicinal-substituted cyclopropane carboxylate compound or changes the ratio of a product of a reaction of a vicinal-substituted cyclopropane carboxylate compared to the starting material.
- the vicinal-substituted cyclopropane carboxylates or said reaction products are known intermediates in the synthesis of pyrethroid pesticides.
- the present invention provides a method of adjusting the cis/trans ratio of a vicinally-disubstituted cyclopropane, a carboxylate being one of the substituents, by the use of a controlled hydrolysis reaction of the carboxylate functionality whereby less than 100% of the starting material is converted to the corresponding acid or salt. It has been found that the cis and trans isomers do not hydrolyse at the same rate and this factor may be used to recover the carboxylate and/or the product acid in a different cis/trans ratio than was present in the starting material.
- the starting material for the process of the invention is a cis/trans isomeric mixture of a vicinal-substituted cyclopropane carboxylic acid ester or a mixture of different vicinal-substituted cyclopropane carboxylates.
- R I represents hydrogen
- R 2 and R 3 which may be the same or different, represent halogen, e.g., fluorine, chlorine, bromine or iodine, or a halogenated alkyl group, e.g., a lower alkyl group of about 1 to 6 carbons having at least one halogen atom such as fluorine, chlorine, bromine or iodine, an example being trifluoromethyl
- R may be an alkyl group such as a lower alkyl group of about 1 to 6 carbons, e.g., a methyl or ethyl group, or an aryl group such as a phenyl group or a benzyl or substituted benzyl group.
- compounds of Formula (IV) are the 2,2-dimethyl-3-(2,2-dichlorovinyl)cyclopropane carboxylates and 2,2-dimethyl-3-(2-chloro-3,3,3-trifluoroprop-1-en-1-yl) cyclopropane carboxylates and particular carboxylates are the methyl and ethyl esters.
- cis and trans refer to the relationship between two substituents on vicinal, i.e., adjacent, carbons of a cyclopropane ring.
- An example of this usage is the vinyl and ester moieties directly attached to the cyclopropane ring in Formula (IV), see Chapter 7 of "Stereochemistry of Carbon Compounds" by E. L. Eliel, McGraw-Hill, New York (1962).
- a first embodiment of the present invention involves the steps of i) partially hydrolysing the vicinal-substituted cyclopropane carboxylate to form less than 100 mole % of the corresponding acid or salt thereof: ii) separating the vicinal-substituted cyclopropane carboxylic acid formed in step i) from the unreacted vicinal-substituted cyclopropane carboxylate; and iii) recovering the cis-enriched vicinal-substituted cyclopropane carboxylate.
- steps i) and ii) of the first embodiment are carried out but the third step is the recovery of the trans-enriched vicinal-substituted carboxylic acid reaction product.
- the trans-enriched carboxylic acid may simply be esterified with an alkanol by conventional means.
- the trans-enriched carboxylate may be used as an intermediate for a pyrethroid final product having a high trans content.
- a high trans material may be less active as a pesticide than a cis-enriched pyrethroid and lower activity may be desirable in view of a lessened effect against non-target species.
- the trans-enriched material may be accumulated and reprocessed to raise the cis content, e.g., by equilibration.
- the first step of either embodiment of the invention involves hydrolysis of less than 100 mole % of the vicinal-substituted cyclopropane carboxylate, more particularly about 30 to 70 mole %.
- the partial hydrolysis may be accomplished by acidic or basic hydrolysis.
- basic hydrolysis may be carried out utilizing a base such as an alkali metal hydroxide, e.g., NaOH or KOH, or an alkaline earth metal hydroxide or carbonate in less than 100 mole % or by using a base at a lower hydrolysis temperature or for a shorter reaction time. Variation in these parameters, i.e., the particular base, the molar amount of the base, reaction temperature and reaction time, may be used to control the rate and extent of hydrolysis.
- the hydrolysis may be carried out at a temperature less than about 100°C, e.g., from about 20°C to 100°C. High temperature may result in degradation of the starting material.
- One or more solvents may be present during the hydrolysis reaction. Particular solvents for the hydrolysis are water-miscible solvents such as lower alcohols, e.g., methanol or ethanol, glycols, e.g. propylene glycol, and lower ketones, e.g., acetone, as well as water itself.
- Basic hydrolysis has the advantage in the process of the invention of yielding the product of the hydrolysis in the form of a salt which is readily separated from an organic phase composed of the starting material ester.
- Acidic hydrolysis i.e., acid-catalyzed hydrolysis, may be advantageous if any of the substituents on the cyclopropane ring are base-sensitive.
- Acid-catalysed hydrolysis may be controlled, as the basic hydrolysis, to result in less than 100% conversion to the carboxylic acid with a difference in the cis/trans distribution between the starting ester and the carboxylic acid product.
- the second step of either embodiment of the invention is the separation of the vicinal-substituted cyclopropane carboxylic acid product from the starting material.
- Conventional techniques such as crystallization, distillation, chromatography or separation by differences in solvent solubility may be used although the prefered method in view of simplicity is separation by solvent solubility.
- a water-immiscible solvent or solvents can be added to the reaction mixture to produce a two-phase system with the acid or salt product being more soluble in the water-miscible phase and the carboxylate starting material being more soluble in the water-miscible phase.
- Particular water-immiscible solvents include aromatic solvents such as benzene, toluene or xylene, higher ketones such as methyl ethyl ketone and methyl iso-butyl ketone, chlorinated solvents such as methylene chloride and chlorobenzene and esters such as ethyl acetate and butyl acetate.
- aromatic solvents such as benzene, toluene or xylene
- higher ketones such as methyl ethyl ketone and methyl iso-butyl ketone
- chlorinated solvents such as methylene chloride and chlorobenzene and esters
- ethyl acetate and butyl acetate ethyl acetate and butyl acetate.
- the cis-enriched carboxylate or trans-enriched acid or salt product is recovered, the enrichment being relative to the cis/trans distribution of the carboxylate before the hydrolysis reaction. If a two-phase solvent system is used for the second step separation, the recovery can be accomplished by drawing off or decanting one of the two phases.
- All cis or trans contents are reported on a normalized basis, i.e., the amount of cis and trans isomer of the chemical entity being considered is considered to be 100%.
- the actual analysis may be different on a weight or molar basis since some inert or side reaction product may be present.
- all cis/trans percentage distributions are on a weight basis and determined percentage distributions are on a weight basis and determined by gas-liquid chromatography.
- the brine solution used in the Examples is an about 10% by weight sodium chloride in water solution obtained by mixing 100 g of NaCl in 1 liter of water.
- a solution of 21 g PAE having a 42.9/57.1 cis/trans distribution in 30 ml of methanol was mixed at 67°C for 2.5 hours with a solution of 1.8 g NaOH in 20 ml of water.
- the organic layer was isolated as in Examples 1 and 2 using 40 ml of toluene and 80 ml of a brine solution.
- the 11.9 g of cis-enriched ester was found to be a mixture of PAM and PAE having a 53.4% cis content by weight.
- the molar ratio in the product of PAM:PAE was 0.7:1.
- Example 3 The procedure and material amounts of Example 3 were repeated using 30 ml of ethanol in the place of methanol.
- the yield of cis-enriched PAE was 12.4 g which showed a 51.2% cis content.
- a solution of a 10 g of PAM having a 41.5/58.5 cis/trans distribution in 15 ml of methanol was mixed with a solution of 0.75 g NaOH in 10 ml of water and heated at 67°C for 1.5 hours.
- the reaction product was cooled to room temperature and 20 ml of toluene and 40 ml of brine were added.
- the mixture was shaken and the toluene layer was removed after phase separation. Evaporation of the toluene layer at 65°C and 10-20 mm Hg gave a product residue of 6.8 g. Analysis showed a 49.8/50.2 cis/trans distribution.
- Example 5 The procedures of Example 5 were repeated using twice the amounts of each material with the exception of using 1.1 g of NaOH. After work-up, analysis of the 14.2 g residue showed a 47.5/52.5 cis/trans distribution.
- a solution of 20 g PAM having a 41.5/58.5 cis/trans distribution in 30 ml of methanol was mixed with a solution of 5.93 g potassium carbonate in 20 ml water and heated at 65--75°C for 2.5 hours.
- the reaction mixture was cooled and phase separation occurred after adding 40 g of toluene and 80 ml of brine and shaking.
- the toluene phase was drawn off and evaporated at 60°C and 40 mm Hg, followed by 5-7 mm Hg. Analysis of the 15.6 g of residue showed a 45/55 cis/trans distribution.
- Example 2 This example was carried out following the procedures and material amounts described in Example 2 except that the PAM had a 55.3/44.7 cis/trans distribution, the reaction time was 1.5 hours and 2.35 g of NaOH were used as opposed to 1.8 g. After the work-up described in Example 2, analysis of the 8.3 g of residue showed a 74.6/25.4 cis/trans distribution.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Description
- Natural and synthetic cyclopropane derivatives have been of considerable interest in the pesticide field. In particular, various esters or analogues of chrysanthemic or pyrethric acids have achieved commercial acceptance as insecticides on a wide scale. Specific examples of such compounds are those of the following Formula (I):

- It is known that various pyrethroid type cyclopropane derivatives exhibit a difference in biological activity depending on whether the cis or trans isomer is used as set forth in U.S. Patent 4,024,163. Interconversions between cis and trans in this series are known but involve considerable chemical effort and expense, see Elliot and James in "Pyrethrum, the Natural Insecticide" by John E. Casida, Academic Press at Page 76 (1973).
- An object of the present invention is a process which adjusts the cis/trans ratio of a vicinal-substituted cyclopropane carboxylate compound or changes the ratio of a product of a reaction of a vicinal-substituted cyclopropane carboxylate compared to the starting material. The vicinal-substituted cyclopropane carboxylates or said reaction products are known intermediates in the synthesis of pyrethroid pesticides.
- The present invention provides a method of adjusting the cis/trans ratio of a vicinally-disubstituted cyclopropane, a carboxylate being one of the substituents, by the use of a controlled hydrolysis reaction of the carboxylate functionality whereby less than 100% of the starting material is converted to the corresponding acid or salt. It has been found that the cis and trans isomers do not hydrolyse at the same rate and this factor may be used to recover the carboxylate and/or the product acid in a different cis/trans ratio than was present in the starting material.
- The starting material for the process of the invention is a cis/trans isomeric mixture of a vicinal-substituted cyclopropane carboxylic acid ester or a mixture of different vicinal-substituted cyclopropane carboxylates. Of particular interest are compounds of the following Formula (IV): -

- As used in the present invention, cis and trans refer to the relationship between two substituents on vicinal, i.e., adjacent, carbons of a cyclopropane ring. An example of this usage is the vinyl and ester moieties directly attached to the cyclopropane ring in Formula (IV), see Chapter 7 of "Stereochemistry of Carbon Compounds" by E. L. Eliel, McGraw-Hill, New York (1962).
- A first embodiment of the present invention involves the steps of i) partially hydrolysing the vicinal-substituted cyclopropane carboxylate to form less than 100 mole % of the corresponding acid or salt thereof: ii) separating the vicinal-substituted cyclopropane carboxylic acid formed in step i) from the unreacted vicinal-substituted cyclopropane carboxylate; and iii) recovering the cis-enriched vicinal-substituted cyclopropane carboxylate. In a second embodiment of the invention, steps i) and ii) of the first embodiment are carried out but the third step is the recovery of the trans-enriched vicinal-substituted carboxylic acid reaction product. If a trans-enriched vicinal-substituted carboxylic acid ester is desired, the trans-enriched carboxylic acid may simply be esterified with an alkanol by conventional means. The trans-enriched carboxylate may be used as an intermediate for a pyrethroid final product having a high trans content. A high trans material may be less active as a pesticide than a cis-enriched pyrethroid and lower activity may be desirable in view of a lessened effect against non-target species. Alternatively, the trans-enriched material may be accumulated and reprocessed to raise the cis content, e.g., by equilibration.
- The first step of either embodiment of the invention involves hydrolysis of less than 100 mole % of the vicinal-substituted cyclopropane carboxylate, more particularly about 30 to 70 mole %. The partial hydrolysis may be accomplished by acidic or basic hydrolysis. In particular, basic hydrolysis may be carried out utilizing a base such as an alkali metal hydroxide, e.g., NaOH or KOH, or an alkaline earth metal hydroxide or carbonate in less than 100 mole % or by using a base at a lower hydrolysis temperature or for a shorter reaction time. Variation in these parameters, i.e., the particular base, the molar amount of the base, reaction temperature and reaction time, may be used to control the rate and extent of hydrolysis. The hydrolysis may be carried out at a temperature less than about 100°C, e.g., from about 20°C to 100°C. High temperature may result in degradation of the starting material. One or more solvents may be present during the hydrolysis reaction. Particular solvents for the hydrolysis are water-miscible solvents such as lower alcohols, e.g., methanol or ethanol, glycols, e.g. propylene glycol, and lower ketones, e.g., acetone, as well as water itself.
- Basic hydrolysis has the advantage in the process of the invention of yielding the product of the hydrolysis in the form of a salt which is readily separated from an organic phase composed of the starting material ester. Acidic hydrolysis, i.e., acid-catalyzed hydrolysis, may be advantageous if any of the substituents on the cyclopropane ring are base-sensitive. Acid-catalysed hydrolysis may be controlled, as the basic hydrolysis, to result in less than 100% conversion to the carboxylic acid with a difference in the cis/trans distribution between the starting ester and the carboxylic acid product.
- The second step of either embodiment of the invention is the separation of the vicinal-substituted cyclopropane carboxylic acid product from the starting material. Conventional techniques such as crystallization, distillation, chromatography or separation by differences in solvent solubility may be used although the prefered method in view of simplicity is separation by solvent solubility. Thus, a water-immiscible solvent or solvents can be added to the reaction mixture to produce a two-phase system with the acid or salt product being more soluble in the water-miscible phase and the carboxylate starting material being more soluble in the water-miscible phase. Particular water-immiscible solvents include aromatic solvents such as benzene, toluene or xylene, higher ketones such as methyl ethyl ketone and methyl iso-butyl ketone, chlorinated solvents such as methylene chloride and chlorobenzene and esters such as ethyl acetate and butyl acetate. The two-phase system is shaken to achieve proper separation of the components between the two solvent system. Further, a phase separation aid such as an aqueous solution of sodium chloride or brine, may be added.
- In the third step of the two embodiments of the invention, the cis-enriched carboxylate or trans-enriched acid or salt product is recovered, the enrichment being relative to the cis/trans distribution of the carboxylate before the hydrolysis reaction. If a two-phase solvent system is used for the second step separation, the recovery can be accomplished by drawing off or decanting one of the two phases.
- In order to describe the invention so that it may be more clearly understood, the following examples are set forth. Abbreviations used include: °C (degrees centigrade); g (grams); ml (milliliters); mm Hg (milliliters of mercury pressure); C, H, N, 0, Na, etc. (the universally-accepted symbols for the chemical elements); PAM (methyl-3-(2,2-dichlorovinyl)-2,2-dimethylcyclopropane carboxylate also known as permethrin acid methyl ester); and PAE (ethyl-3-(2,2-dichlorovinyl)-2,2-dimethylcyclopropane carboxylate also known as permethrin acid ethyl ester). All cis or trans contents are reported on a normalized basis, i.e., the amount of cis and trans isomer of the chemical entity being considered is considered to be 100%. The actual analysis may be different on a weight or molar basis since some inert or side reaction product may be present. Unless otherwise noted, all cis/trans percentage distributions are on a weight basis and determined percentage distributions are on a weight basis and determined by gas-liquid chromatography. The brine solution used in the Examples, unless otherwise noted, is an about 10% by weight sodium chloride in water solution obtained by mixing 100 g of NaCl in 1 liter of water.
- A mixture of 10 g of PAM having a 43/57 cis/trans distribution, 15 ml of methanol and a solution of 0.9 g of NaOH in 10 ml of water was heated with stirring at 64-67°C for 2 hours. In preparing this and other mixtures in the Examples, care should be taken to avoid contacting the cyclopropane carboxylate ester, such as PAM, directly with concentrated base. Thus, in the Example, the aqueous caustic was first diluted with the methanol after which the PAM was added. The NaOH represented about one half of the calculated amount necessary to fully hydrolyze the PAM. After cooling to room temperature, 20 ml of toluene and 40 ml of brine, were added and the mixture was shaken. The toluene layer was withdrawn and the solvents evaporated on a rotary vacuum evaporator to yield 6.0 g of unhydrolyzed PAM having a cis/trans distribution of about 54/46.
- To a solution of 20 g PAM having a 47.3/52.7 cis/trans distribution in 30 ml of methanol was added a solution of 1.8 g of NaOH in 20 ml water. The mixture was stirred and heated to 67°C for 2.5 hours. After cooling to room temperature, 40 ml of toluene and 80 ml of a sodium chloride brine solution were added and the mixture was shaken. After being allowed to settle, separation of the phases occurred and the toluene layer was withdrawn and vacuum evaporated at 60°C and 45 mm Hg followed by less than 1 mm Hg. Analysis of the 10.3 g of residue showed a 60.2/39.8 cis/trans distribution.
- A solution of 21 g PAE having a 42.9/57.1 cis/trans distribution in 30 ml of methanol was mixed at 67°C for 2.5 hours with a solution of 1.8 g NaOH in 20 ml of water. The organic layer was isolated as in Examples 1 and 2 using 40 ml of toluene and 80 ml of a brine solution. The 11.9 g of cis-enriched ester was found to be a mixture of PAM and PAE having a 53.4% cis content by weight. The molar ratio in the product of PAM:PAE was 0.7:1.
- The procedure and material amounts of Example 3 were repeated using 30 ml of ethanol in the place of methanol. The yield of cis-enriched PAE was 12.4 g which showed a 51.2% cis content.
- A solution of a 10 g of PAM having a 41.5/58.5 cis/trans distribution in 15 ml of methanol was mixed with a solution of 0.75 g NaOH in 10 ml of water and heated at 67°C for 1.5 hours. The reaction product was cooled to room temperature and 20 ml of toluene and 40 ml of brine were added. The mixture was shaken and the toluene layer was removed after phase separation. Evaporation of the toluene layer at 65°C and 10-20 mm Hg gave a product residue of 6.8 g. Analysis showed a 49.8/50.2 cis/trans distribution.
- The procedures of Example 5 were repeated using twice the amounts of each material with the exception of using 1.1 g of NaOH. After work-up, analysis of the 14.2 g residue showed a 47.5/52.5 cis/trans distribution.
- A solution of 20 g PAM having a 41.5/58.5 cis/trans distribution in 30 ml of methanol was mixed with a solution of 5.93 g potassium carbonate in 20 ml water and heated at 65--75°C for 2.5 hours. The reaction mixture was cooled and phase separation occurred after adding 40 g of toluene and 80 ml of brine and shaking. The toluene phase was drawn off and evaporated at 60°C and 40 mm Hg, followed by 5-7 mm Hg. Analysis of the 15.6 g of residue showed a 45/55 cis/trans distribution.
- A sample of 20 g of PAM having a 55.3/44.7 cis/trans distribution was treated in the manner described in Example 2 for 1.2 hours and worked up similarly. Analysis of the 10.7 g of residue showed a 70/30 cis/ trans distribution.
- This example was carried out following the procedures and material amounts described in Example 2 except that the PAM had a 55.3/44.7 cis/trans distribution, the reaction time was 1.5 hours and 2.35 g of NaOH were used as opposed to 1.8 g. After the work-up described in Example 2, analysis of the 8.3 g of residue showed a 74.6/25.4 cis/trans distribution.
Claims (11)

Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US30279681A | 1981-09-16 | 1981-09-16 | |
| US302796 | 1981-09-16 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| EP0074792A1 EP0074792A1 (en) | 1983-03-23 |
| EP0074792B1 true EP0074792B1 (en) | 1985-11-27 |
Family
ID=23169248
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP19820304711 Expired EP0074792B1 (en) | 1981-09-16 | 1982-09-08 | Isomer enrichment process for cyclopropane carboxylates |
Country Status (6)
| Country | Link |
|---|---|
| EP (1) | EP0074792B1 (en) |
| JP (1) | JPS5862137A (en) |
| BR (1) | BR8205399A (en) |
| CA (1) | CA1192218A (en) |
| DE (1) | DE3267706D1 (en) |
| GB (1) | GB2108494A (en) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6218542B1 (en) * | 1996-10-15 | 2001-04-17 | Jenssen Pharmaceutica N.V. | Synthesis of cisapride |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS533386B2 (en) * | 1972-11-13 | 1978-02-06 | ||
| US4288610A (en) * | 1980-02-25 | 1981-09-08 | Stauffer Chemical Company | Purification of pyrethroid intermediate compounds by selective partial saponification |
-
1982
- 1982-08-30 CA CA000410426A patent/CA1192218A/en not_active Expired
- 1982-09-08 GB GB08225650A patent/GB2108494A/en not_active Withdrawn
- 1982-09-08 DE DE8282304711T patent/DE3267706D1/en not_active Expired
- 1982-09-08 EP EP19820304711 patent/EP0074792B1/en not_active Expired
- 1982-09-15 BR BR8205399A patent/BR8205399A/en unknown
- 1982-09-16 JP JP15973682A patent/JPS5862137A/en active Granted
Also Published As
| Publication number | Publication date |
|---|---|
| CA1192218A (en) | 1985-08-20 |
| BR8205399A (en) | 1983-08-23 |
| DE3267706D1 (en) | 1986-01-09 |
| GB2108494A (en) | 1983-05-18 |
| JPH0251419B2 (en) | 1990-11-07 |
| EP0074792A1 (en) | 1983-03-23 |
| JPS5862137A (en) | 1983-04-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US4061664A (en) | Preparation of phenoxybenzyl esters | |
| EP0794180A1 (en) | Process for the preparation of 2-chloro-5-chloromethyl-1,3-thiazole | |
| EP0074792B1 (en) | Isomer enrichment process for cyclopropane carboxylates | |
| US10662166B2 (en) | Process for preparing (2E,6Z)-2,6-nonadienal and a process for preparing (2E)-cis 6,7-epoxy-2-nonenal | |
| EP0221635B1 (en) | Fluoro alcohols and insecticidal esters thereof | |
| US4874887A (en) | Process for the preparation of pyrethroid type ester compounds | |
| US4551281A (en) | Process for the preparation of cyclopropane carboxylic acid esters | |
| US5047581A (en) | Isolation of cis-isomers from isomeric mixtures of cis/transcyclopropanecarboxylates | |
| US4419524A (en) | Process for the preparation of dihalovinylcyclopropanecarboxylic acids | |
| EP1056709B1 (en) | A process for the preparation of cyclopropane carboxylic acids | |
| US5770767A (en) | Process for producing 2-fluorocyclopropancecarboxlic acid | |
| EP0064781B1 (en) | Process for the preparation of cyclopropane compounds | |
| US11591282B2 (en) | Process for preparing 6-isopropenyl-3-methyl-9-decenyl acetate and intermediates thereof | |
| US4118413A (en) | Preparation of phenoxybenzyl esters | |
| SK7198A3 (en) | A process for the preparation of cyclopropane carboxylic acids and intermediates therefor | |
| EP0029621A1 (en) | Iodo-lactones, a process for their preparation and their use in separating cis acids from cis/trans acids | |
| EP0038053B1 (en) | Method for the preparation of cis-nonen-6-yl chloride | |
| JPS6252736B2 (en) | ||
| EP0007142B1 (en) | Novel intermediates in the preparation of cyclopropanecarboxylate esters and process for their manufacture | |
| HU203520B (en) | Process for producing disubstituted cyclopropane derivatives | |
| JP2720934B2 (en) | Process for producing 3- (2-chloro-2- (4-chlorophenyl) -vinyl) -2,2-dimethylcyclopropanecarboxylic acid | |
| US4360478A (en) | Preparation of α-cyanobenzyl esters | |
| KR890001917B1 (en) | Process for the preparation of pyrecthroids | |
| KR960010531B1 (en) | Process for preparation of permethrin | |
| GB2065118A (en) | 2-(2',2'-dichloro-3',3',3'-trifluoropropyl)- and 2-(2',2',3'-trichloro-3',3'-difluoropropyl)-4-chlorocyclobutan-1-ones |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| AK | Designated contracting states |
Designated state(s): BE CH DE FR GB IT LI NL |
|
| 17P | Request for examination filed |
Effective date: 19830823 |
|
| ITF | It: translation for a ep patent filed |
Owner name: ING. C. GREGORJ S.P.A. |
|
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| AK | Designated contracting states |
Designated state(s): BE CH DE FR GB IT LI NL |
|
| ET | Fr: translation filed | ||
| REF | Corresponds to: |
Ref document number: 3267706 Country of ref document: DE Date of ref document: 19860109 |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |
|
| 26N | No opposition filed | ||
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: FR Payment date: 19890807 Year of fee payment: 8 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: CH Payment date: 19890811 Year of fee payment: 8 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: BE Payment date: 19890818 Year of fee payment: 8 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: DE Payment date: 19890831 Year of fee payment: 8 |
|
| ITTA | It: last paid annual fee | ||
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: NL Payment date: 19890930 Year of fee payment: 8 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: LI Effective date: 19900930 Ref country code: CH Effective date: 19900930 Ref country code: BE Effective date: 19900930 |
|
| BERE | Be: lapsed |
Owner name: ICI AMERICAS INC Effective date: 19900930 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: NL Effective date: 19910401 |
|
| NLV4 | Nl: lapsed or anulled due to non-payment of the annual fee | ||
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: FR Effective date: 19910530 |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PL |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: DE Effective date: 19910601 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: ST |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: GB Payment date: 19930824 Year of fee payment: 12 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GB Effective date: 19940908 |
|
| GBPC | Gb: european patent ceased through non-payment of renewal fee |
Effective date: 19940908 |