WO2022055351A1 - Conjugate of saponin, oligonucleotide and galnac - Google Patents
Conjugate of saponin, oligonucleotide and galnac Download PDFInfo
- Publication number
- WO2022055351A1 WO2022055351A1 PCT/NL2021/050549 NL2021050549W WO2022055351A1 WO 2022055351 A1 WO2022055351 A1 WO 2022055351A1 NL 2021050549 W NL2021050549 W NL 2021050549W WO 2022055351 A1 WO2022055351 A1 WO 2022055351A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- saponin
- oligonucleotide
- xyl
- rha
- conjugate
- Prior art date
Links
- 150000007949 saponins Chemical class 0.000 title claims abstract description 498
- 108091034117 Oligonucleotide Proteins 0.000 title claims abstract description 419
- 229930182490 saponin Natural products 0.000 title claims abstract description 408
- 239000001397 quillaja saponaria molina bark Substances 0.000 title claims abstract description 393
- OVRNDRQMDRJTHS-KEWYIRBNSA-N N-acetyl-D-galactosamine Chemical group CC(=O)N[C@H]1C(O)O[C@H](CO)[C@H](O)[C@@H]1O OVRNDRQMDRJTHS-KEWYIRBNSA-N 0.000 claims abstract description 208
- 239000003446 ligand Substances 0.000 claims abstract description 118
- 108090000623 proteins and genes Proteins 0.000 claims abstract description 97
- 101150102415 Apob gene Proteins 0.000 claims abstract description 61
- 230000014509 gene expression Effects 0.000 claims abstract description 60
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 49
- 239000003814 drug Substances 0.000 claims abstract description 42
- 238000000034 method Methods 0.000 claims abstract description 36
- 101150075175 Asgr1 gene Proteins 0.000 claims abstract description 34
- 102100030755 5-aminolevulinate synthase, nonspecific, mitochondrial Human genes 0.000 claims abstract description 33
- 201000010099 disease Diseases 0.000 claims abstract description 32
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 32
- 101000843649 Homo sapiens 5-aminolevulinate synthase, nonspecific, mitochondrial Proteins 0.000 claims abstract description 31
- 238000011282 treatment Methods 0.000 claims abstract description 29
- 230000001404 mediated effect Effects 0.000 claims abstract description 25
- 238000011321 prophylaxis Methods 0.000 claims abstract description 25
- 230000000295 complement effect Effects 0.000 claims abstract description 19
- 230000005802 health problem Effects 0.000 claims abstract description 19
- 101150010882 S gene Proteins 0.000 claims abstract description 18
- 101150003160 X gene Proteins 0.000 claims abstract description 18
- 206010028980 Neoplasm Diseases 0.000 claims abstract description 12
- 238000000338 in vitro Methods 0.000 claims abstract description 12
- 102100038955 Proprotein convertase subtilisin/kexin type 9 Human genes 0.000 claims abstract description 11
- 208000035150 Hypercholesterolemia Diseases 0.000 claims abstract description 10
- 201000011510 cancer Diseases 0.000 claims abstract description 10
- 208000015181 infectious disease Diseases 0.000 claims abstract description 7
- 201000007905 transthyretin amyloidosis Diseases 0.000 claims abstract description 7
- 108700016481 Acute Hepatic Porphyria Proteins 0.000 claims abstract description 6
- 208000003914 Acute hepatic porphyria Diseases 0.000 claims abstract description 6
- 208000023275 Autoimmune disease Diseases 0.000 claims abstract description 6
- 208000035473 Communicable disease Diseases 0.000 claims abstract description 6
- 201000003542 Factor VIII deficiency Diseases 0.000 claims abstract description 6
- 208000004777 Primary Hyperoxaluria Diseases 0.000 claims abstract description 6
- 208000009429 hemophilia B Diseases 0.000 claims abstract description 6
- 208000033552 hepatic porphyria Diseases 0.000 claims abstract description 6
- 208000002672 hepatitis B Diseases 0.000 claims abstract description 6
- 208000034846 Familial Amyloid Neuropathies Diseases 0.000 claims abstract description 5
- 208000036142 Viral infection Diseases 0.000 claims abstract description 5
- 208000019423 liver disease Diseases 0.000 claims abstract description 5
- 230000009385 viral infection Effects 0.000 claims abstract description 5
- 235000017709 saponins Nutrition 0.000 claims description 405
- 210000004027 cell Anatomy 0.000 claims description 191
- MBLBDJOUHNCFQT-UHFFFAOYSA-N N-acetyl-D-galactosamine Natural products CC(=O)NC(C=O)C(O)C(O)C(O)CO MBLBDJOUHNCFQT-UHFFFAOYSA-N 0.000 claims description 143
- 102000005427 Asialoglycoprotein Receptor Human genes 0.000 claims description 129
- 108010006523 asialoglycoprotein receptor Proteins 0.000 claims description 129
- 150000001720 carbohydrates Chemical group 0.000 claims description 115
- 108020004459 Small interfering RNA Proteins 0.000 claims description 93
- 239000004055 small Interfering RNA Substances 0.000 claims description 85
- -1 QS-21A Chemical compound 0.000 claims description 79
- LHFTZXBOKDXUHD-TUZSFQOTSA-N (2s,3s,4s,5r,6s)-6-[[(3s,4s,4ar,6ar,6bs,8r,8ar,12as,14ar,14br)-8a-[(2s,3r,4s,5r,6r)-5-[(2s,3r,4r,5r,6r)-5-acetyloxy-3-hydroxy-6-methyl-4-[(2s,3r,4s,5r)-3,4,5-trihydroxyoxan-2-yl]oxyoxan-2-yl]oxy-3-[(2s,3r,4s,5r,6s)-5-[(2s,3r,4s,5r)-3,5-dihydroxy-4-[(2s,3r Chemical compound O([C@H]1[C@H](OC(C)=O)[C@@H](C)O[C@H]([C@@H]1O)O[C@@H]1[C@H](O)[C@H]([C@@H](O[C@@H]1C)OC(=O)[C@@]12[C@@H](CC(C)(C)CC1)C=1[C@@]([C@]3(C)CC[C@H]4[C@](C)(C=O)[C@@H](O[C@@H]5[C@@H]([C@@H](O[C@H]6[C@@H]([C@@H](O)[C@H](O)CO6)O)[C@H](O)[C@H](O5)C(O)=O)O[C@H]5[C@@H]([C@@H](O)[C@@H](O)[C@@H](CO)O5)O)CC[C@]4(C)[C@H]3CC=1)(C)C[C@H]2O)O[C@@H]1O[C@H]([C@@H]([C@@H](O)[C@H]1O)O[C@H]1[C@@H]([C@@H](O[C@H]2[C@@H]([C@@H](O)[C@H](O)CO2)O)[C@H](O)CO1)O)C)[C@@H]1OC[C@@H](O)[C@H](O)[C@H]1O LHFTZXBOKDXUHD-TUZSFQOTSA-N 0.000 claims description 77
- 230000015572 biosynthetic process Effects 0.000 claims description 61
- 241000700721 Hepatitis B virus Species 0.000 claims description 60
- 230000009466 transformation Effects 0.000 claims description 58
- TWCMVXMQHSVIOJ-UHFFFAOYSA-N Aglycone of yadanzioside D Natural products COC(=O)C12OCC34C(CC5C(=CC(O)C(O)C5(C)C3C(O)C1O)C)OC(=O)C(OC(=O)C)C24 TWCMVXMQHSVIOJ-UHFFFAOYSA-N 0.000 claims description 57
- PLMKQQMDOMTZGG-UHFFFAOYSA-N Astrantiagenin E-methylester Natural products CC12CCC(O)C(C)(CO)C1CCC1(C)C2CC=C2C3CC(C)(C)CCC3(C(=O)OC)CCC21C PLMKQQMDOMTZGG-UHFFFAOYSA-N 0.000 claims description 57
- PFOARMALXZGCHY-UHFFFAOYSA-N homoegonol Natural products C1=C(OC)C(OC)=CC=C1C1=CC2=CC(CCCO)=CC(OC)=C2O1 PFOARMALXZGCHY-UHFFFAOYSA-N 0.000 claims description 57
- 230000027455 binding Effects 0.000 claims description 55
- FHICGHSMIPIAPL-HDYAAECPSA-N [2-[3-[6-[3-[(5R,6aS,6bR,12aR)-10-[6-[2-[2-[4,5-dihydroxy-3-(3,4,5-trihydroxyoxan-2-yl)oxyoxan-2-yl]ethoxy]ethyl]-3,4,5-trihydroxyoxan-2-yl]oxy-5-hydroxy-2,2,6a,6b,9,9,12a-heptamethyl-1,3,4,5,6,6a,7,8,8a,10,11,12,13,14b-tetradecahydropicene-4a-carbonyl]peroxypropyl]-5-[[5-[8-[3,5-dihydroxy-4-(3,4,5-trihydroxyoxan-2-yl)oxyoxan-2-yl]octoxy]-3,4-dihydroxy-6-methyloxan-2-yl]methoxy]-3,4-dihydroxyoxan-2-yl]propoxymethyl]-5-hydroxy-3-[(6S)-6-hydroxy-2,6-dimethylocta-2,7-dienoyl]oxy-6-methyloxan-4-yl] (2E,6S)-6-hydroxy-2-(hydroxymethyl)-6-methylocta-2,7-dienoate Chemical compound C=C[C@@](C)(O)CCC=C(C)C(=O)OC1C(OC(=O)C(\CO)=C\CC[C@](C)(O)C=C)C(O)C(C)OC1COCCCC1C(O)C(O)C(OCC2C(C(O)C(OCCCCCCCCC3C(C(OC4C(C(O)C(O)CO4)O)C(O)CO3)O)C(C)O2)O)C(CCCOOC(=O)C23C(CC(C)(C)CC2)C=2[C@@]([C@]4(C)CCC5C(C)(C)C(OC6C(C(O)C(O)C(CCOCCC7C(C(O)C(O)CO7)OC7C(C(O)C(O)CO7)O)O6)O)CC[C@]5(C)C4CC=2)(C)C[C@H]3O)O1 FHICGHSMIPIAPL-HDYAAECPSA-N 0.000 claims description 49
- 102000004169 proteins and genes Human genes 0.000 claims description 49
- 230000008685 targeting Effects 0.000 claims description 49
- 230000030279 gene silencing Effects 0.000 claims description 48
- 238000006243 chemical reaction Methods 0.000 claims description 42
- 108091032973 (ribonucleotides)n+m Proteins 0.000 claims description 41
- 108010045100 HSP27 Heat-Shock Proteins Proteins 0.000 claims description 40
- 108020004999 messenger RNA Proteins 0.000 claims description 40
- 108010071690 Prealbumin Proteins 0.000 claims description 39
- 102000009190 Transthyretin Human genes 0.000 claims description 39
- 102100039165 Heat shock protein beta-1 Human genes 0.000 claims description 37
- 108010050122 alpha 1-Antitrypsin Proteins 0.000 claims description 36
- 102000015395 alpha 1-Antitrypsin Human genes 0.000 claims description 36
- 229940024142 alpha 1-antitrypsin Drugs 0.000 claims description 36
- 102000039446 nucleic acids Human genes 0.000 claims description 36
- 108020004707 nucleic acids Proteins 0.000 claims description 36
- 210000001163 endosome Anatomy 0.000 claims description 35
- 150000007523 nucleic acids Chemical class 0.000 claims description 35
- 239000000074 antisense oligonucleotide Substances 0.000 claims description 33
- 238000012230 antisense oligonucleotides Methods 0.000 claims description 33
- 125000003172 aldehyde group Chemical group 0.000 claims description 32
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 31
- 210000000172 cytosol Anatomy 0.000 claims description 30
- 239000002679 microRNA Substances 0.000 claims description 30
- 210000004962 mammalian cell Anatomy 0.000 claims description 29
- 108091007780 MiR-122 Proteins 0.000 claims description 28
- 125000003668 acetyloxy group Chemical group [H]C([H])([H])C(=O)O[*] 0.000 claims description 27
- 108091051828 miR-122 stem-loop Proteins 0.000 claims description 27
- 102100034808 CCAAT/enhancer-binding protein alpha Human genes 0.000 claims description 26
- 101000945515 Homo sapiens CCAAT/enhancer-binding protein alpha Proteins 0.000 claims description 26
- 101001090713 Homo sapiens L-lactate dehydrogenase A chain Proteins 0.000 claims description 26
- 101000798696 Homo sapiens Transmembrane protease serine 6 Proteins 0.000 claims description 26
- 102100034671 L-lactate dehydrogenase A chain Human genes 0.000 claims description 26
- 108700011259 MicroRNAs Proteins 0.000 claims description 26
- 150000001299 aldehydes Chemical group 0.000 claims description 26
- AEMOLEFTQBMNLQ-AQKNRBDQSA-N D-glucopyranuronic acid Chemical group OC1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-AQKNRBDQSA-N 0.000 claims description 25
- 102100032452 Transmembrane protease serine 6 Human genes 0.000 claims description 25
- 238000001212 derivatisation Methods 0.000 claims description 25
- 229940126836 transmembrane protease serine 6 synthesis reducer Drugs 0.000 claims description 25
- 210000005229 liver cell Anatomy 0.000 claims description 24
- 238000003776 cleavage reaction Methods 0.000 claims description 21
- 230000007017 scission Effects 0.000 claims description 21
- 102100038837 2-Hydroxyacid oxidase 1 Human genes 0.000 claims description 20
- 102000003855 L-lactate dehydrogenase Human genes 0.000 claims description 20
- 108700023483 L-lactate dehydrogenases Proteins 0.000 claims description 20
- MQUFAARYGOUYEV-UWEXFCAOSA-N Quillaic acid Natural products CC1(C)CC[C@@]2([C@H](O)C[C@]3(C)C(=CC[C@H]4[C@@]5(C)CC[C@H](O)[C@](C)(C=O)[C@H]5CC[C@@]34C)[C@H]2C1)C(=O)O MQUFAARYGOUYEV-UWEXFCAOSA-N 0.000 claims description 20
- 108010062584 glycollate oxidase Proteins 0.000 claims description 20
- 239000007983 Tris buffer Substances 0.000 claims description 19
- 210000005260 human cell Anatomy 0.000 claims description 18
- MQUFAARYGOUYEV-UAWZMHPWSA-N quillaic acid Chemical compound C1C[C@H](O)[C@@](C)(C=O)[C@@H]2CC[C@@]3(C)[C@]4(C)C[C@@H](O)[C@@]5(C(O)=O)CCC(C)(C)C[C@H]5C4=CC[C@@H]3[C@]21C MQUFAARYGOUYEV-UAWZMHPWSA-N 0.000 claims description 18
- 150000008130 triterpenoid saponins Chemical class 0.000 claims description 18
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 17
- UAEZPQXQAIYKCX-VDDIDERSSA-N (2s,3s,4s,5r,6s)-6-[[(3s,4s,4ar,6ar,6bs,8as,12as,14ar,14br)-8a-[(2s,3r,4s,5r,6r)-3-[(2s,3r,4s,5r,6s)-5-[(2s,3r,4s,5r)-3,5-dihydroxy-4-[(2s,3r,4s,5r)-3,4,5-trihydroxyoxan-2-yl]oxyoxan-2-yl]oxy-3,4-dihydroxy-6-methyloxan-2-yl]oxy-4-hydroxy-6-methyl-5-[(2s,3 Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](C)O[C@H]1O[C@@H]1[C@H](O)[C@@H](O[C@H]2[C@@H]([C@H](O)[C@@H](O[C@H]3[C@@H]([C@@H](O[C@H]4[C@@H]([C@@H](O)[C@H](O)CO4)O)[C@H](O)CO3)O)[C@H](C)O2)O)[C@H](OC(=O)[C@@]23[C@@H](CC(C)(C)CC2)C=2[C@@]([C@]4(C)CC[C@H]5[C@](C)(C=O)[C@@H](O[C@@H]6[C@@H]([C@@H](O[C@H]7[C@@H]([C@@H](O)[C@H](O)CO7)O)[C@H](O)[C@H](O6)C(O)=O)O[C@H]6[C@@H]([C@@H](O)[C@@H](O)[C@@H](CO)O6)O)CC[C@]5(C)[C@H]4CC=2)(C)CC3)O[C@@H]1C UAEZPQXQAIYKCX-VDDIDERSSA-N 0.000 claims description 16
- PAIBKVQNJKUVCE-UHFFFAOYSA-N Gypsogeninsaeure Natural products C1CC(O)C(C)(C(O)=O)C2CCC3(C)C4(C)CCC5(C(O)=O)CCC(C)(C)CC5C4=CCC3C21C PAIBKVQNJKUVCE-UHFFFAOYSA-N 0.000 claims description 16
- GCGBHJLBFAPRDB-KCVAUKQGSA-N Scutellaric acid Natural products CC1(C)CC[C@@]2(CC[C@@]3(C)[C@@H]4CC[C@H]5[C@@](C)(CO)[C@H](O)CC[C@]5(C)[C@H]4CC=C3[C@@H]2C1)C(=O)O GCGBHJLBFAPRDB-KCVAUKQGSA-N 0.000 claims description 16
- 102000040650 (ribonucleotides)n+m Human genes 0.000 claims description 14
- QMHCWDVPABYZMC-UHFFFAOYSA-N 3-Hydroxy-23-oxo-olean-12-en-28-saeure Natural products C1CC(O)C(C)(C=O)C2CCC3(C)C4(C)CCC5(C(O)=O)CCC(C)(C)CC5C4=CCC3C21C QMHCWDVPABYZMC-UHFFFAOYSA-N 0.000 claims description 14
- DNJIEGIFACGWOD-UHFFFAOYSA-N ethyl mercaptane Natural products CCS DNJIEGIFACGWOD-UHFFFAOYSA-N 0.000 claims description 14
- QMHCWDVPABYZMC-RFVOPWELSA-N gypsogenin Natural products CC1(C)CC[C@@]2(CC[C@]3(C)C(=CC[C@@H]4[C@@]5(C)CC[C@H](O)[C@](C)(C=O)[C@H]5CC[C@@]34C)[C@H]2C1)C(=O)O QMHCWDVPABYZMC-RFVOPWELSA-N 0.000 claims description 14
- 125000005439 maleimidyl group Chemical group C1(C=CC(N1*)=O)=O 0.000 claims description 14
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 claims description 14
- 230000009467 reduction Effects 0.000 claims description 13
- UXFQFBNBSPQBJW-UHFFFAOYSA-N 2-amino-2-methylpropane-1,3-diol Chemical compound OCC(N)(C)CO UXFQFBNBSPQBJW-UHFFFAOYSA-N 0.000 claims description 12
- QMHCWDVPABYZMC-MYPRUECHSA-N 3beta-hydroxy-23-oxoolean-12-en-28-oic acid Chemical compound C1C[C@H](O)[C@@](C)(C=O)[C@@H]2CC[C@@]3(C)[C@]4(C)CC[C@@]5(C(O)=O)CCC(C)(C)C[C@H]5C4=CC[C@@H]3[C@]21C QMHCWDVPABYZMC-MYPRUECHSA-N 0.000 claims description 12
- OVRNDRQMDRJTHS-CBQIKETKSA-N N-Acetyl-D-Galactosamine Chemical compound CC(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@H](O)[C@@H]1O OVRNDRQMDRJTHS-CBQIKETKSA-N 0.000 claims description 12
- 108020004414 DNA Proteins 0.000 claims description 11
- 238000008214 LDL Cholesterol Methods 0.000 claims description 11
- 238000012546 transfer Methods 0.000 claims description 11
- 210000004881 tumor cell Anatomy 0.000 claims description 11
- KZMAWJRXKGLWGS-UHFFFAOYSA-N 2-chloro-n-[4-(4-methoxyphenyl)-1,3-thiazol-2-yl]-n-(3-methoxypropyl)acetamide Chemical compound S1C(N(C(=O)CCl)CCCOC)=NC(C=2C=CC(OC)=CC=2)=C1 KZMAWJRXKGLWGS-UHFFFAOYSA-N 0.000 claims description 10
- 101710180553 Proprotein convertase subtilisin/kexin type 9 Proteins 0.000 claims description 10
- 238000001727 in vivo Methods 0.000 claims description 10
- 229930182493 triterpene saponin Natural products 0.000 claims description 10
- 101710095342 Apolipoprotein B Proteins 0.000 claims description 9
- 102100040202 Apolipoprotein B-100 Human genes 0.000 claims description 9
- 230000002378 acidificating effect Effects 0.000 claims description 9
- 210000003712 lysosome Anatomy 0.000 claims description 9
- 230000001868 lysosomic effect Effects 0.000 claims description 9
- 230000002829 reductive effect Effects 0.000 claims description 9
- ODVRLSOMTXGTMX-UHFFFAOYSA-N 1-(2-aminoethyl)pyrrole-2,5-dione Chemical compound NCCN1C(=O)C=CC1=O ODVRLSOMTXGTMX-UHFFFAOYSA-N 0.000 claims description 8
- 108091033760 Oncomir Proteins 0.000 claims description 8
- 108091027967 Small hairpin RNA Proteins 0.000 claims description 8
- 230000006196 deacetylation Effects 0.000 claims description 8
- 238000003381 deacetylation reaction Methods 0.000 claims description 8
- 239000003085 diluting agent Substances 0.000 claims description 8
- 239000001257 hydrogen Substances 0.000 claims description 8
- 229910052739 hydrogen Inorganic materials 0.000 claims description 8
- 125000004435 hydrogen atom Chemical class [H]* 0.000 claims description 8
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 8
- 150000002482 oligosaccharides Chemical class 0.000 claims description 8
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 8
- 208000024172 Cardiovascular disease Diseases 0.000 claims description 7
- 208000005176 Hepatitis C Diseases 0.000 claims description 7
- 108010028554 LDL Cholesterol Proteins 0.000 claims description 7
- 230000001594 aberrant effect Effects 0.000 claims description 7
- 230000006870 function Effects 0.000 claims description 7
- 208000010710 hepatitis C virus infection Diseases 0.000 claims description 7
- JKLISIRFYWXLQG-UHFFFAOYSA-N Epioleonolsaeure Natural products C1CC(O)C(C)(C)C2CCC3(C)C4(C)CCC5(C(O)=O)CCC(C)(C)CC5C4CCC3C21C JKLISIRFYWXLQG-UHFFFAOYSA-N 0.000 claims description 6
- YBRJHZPWOMJYKQ-UHFFFAOYSA-N Oleanolic acid Natural products CC1(C)CC2C3=CCC4C5(C)CCC(O)C(C)(C)C5CCC4(C)C3(C)CCC2(C1)C(=O)O YBRJHZPWOMJYKQ-UHFFFAOYSA-N 0.000 claims description 6
- MIJYXULNPSFWEK-UHFFFAOYSA-N Oleanolinsaeure Natural products C1CC(O)C(C)(C)C2CCC3(C)C4(C)CCC5(C(O)=O)CCC(C)(C)CC5C4=CCC3C21C MIJYXULNPSFWEK-UHFFFAOYSA-N 0.000 claims description 6
- 206010002022 amyloidosis Diseases 0.000 claims description 6
- 229940100243 oleanolic acid Drugs 0.000 claims description 6
- 229920001542 oligosaccharide Polymers 0.000 claims description 6
- HZLWUYJLOIAQFC-UHFFFAOYSA-N prosapogenin PS-A Natural products C12CC(C)(C)CCC2(C(O)=O)CCC(C2(CCC3C4(C)C)C)(C)C1=CCC2C3(C)CCC4OC1OCC(O)C(O)C1O HZLWUYJLOIAQFC-UHFFFAOYSA-N 0.000 claims description 6
- 206010043391 Thalassaemia beta Diseases 0.000 claims description 5
- 208000006682 alpha 1-Antitrypsin Deficiency Diseases 0.000 claims description 5
- 150000002772 monosaccharides Chemical class 0.000 claims description 5
- AXNVHPCVMSNXNP-GKTCLTPXSA-N Aescin Natural products O=C(O[C@H]1[C@@H](OC(=O)C)[C@]2(CO)[C@@H](O)C[C@@]3(C)[C@@]4(C)[C@@H]([C@]5(C)[C@H]([C@](CO)(C)[C@@H](O[C@@H]6[C@@H](O[C@H]7[C@@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O7)[C@@H](O)[C@H](O[C@H]7[C@H](O)[C@@H](O)[C@H](O)[C@H](CO)O7)[C@@H](C(=O)O)O6)CC5)CC4)CC=C3[C@@H]2CC1(C)C)/C(=C/C)/C AXNVHPCVMSNXNP-GKTCLTPXSA-N 0.000 claims description 4
- 108020004491 Antisense DNA Proteins 0.000 claims description 4
- 230000003042 antagnostic effect Effects 0.000 claims description 4
- 239000003816 antisense DNA Substances 0.000 claims description 4
- 230000002255 enzymatic effect Effects 0.000 claims description 4
- 230000002401 inhibitory effect Effects 0.000 claims description 4
- 231100000590 oncogenic Toxicity 0.000 claims description 4
- 230000002246 oncogenic effect Effects 0.000 claims description 4
- 230000017854 proteolysis Effects 0.000 claims description 4
- QLPRYZXNWYTFCI-UHFFFAOYSA-N saikosaponin D Natural products CC1OC(OC2CCC3(C)C(CCC4(C)C3C=CC56OCC7(CCC(C)(C)CC57)C(O)CC46C)C2(C)CO)C(O)C(O)C1OC8OC(CO)C(O)C(O)C8O QLPRYZXNWYTFCI-UHFFFAOYSA-N 0.000 claims description 4
- PQPVAGWUNWFCJE-UHFFFAOYSA-N saikosaponin a Natural products CC1OC(OC2CCC3(C)C(C2)C(C)(CO)CC4(C)C3C=CC56OCC7(CCC(C)(C)CC57)C(O)CC46C)C(O)C(OC8OC(CO)C(O)C(O)C8O)C1O PQPVAGWUNWFCJE-UHFFFAOYSA-N 0.000 claims description 4
- 230000001629 suppression Effects 0.000 claims description 4
- 101710184696 5-aminolevulinate synthase, nonspecific, mitochondrial Proteins 0.000 claims description 3
- 108091023037 Aptamer Proteins 0.000 claims description 3
- 239000002253 acid Substances 0.000 claims description 3
- 239000004019 antithrombin Substances 0.000 claims description 3
- 206010073071 hepatocellular carcinoma Diseases 0.000 claims description 3
- 231100000844 hepatocellular carcinoma Toxicity 0.000 claims description 3
- PIGTXFOGKFOFTO-FVFWYJKVSA-N (2S,3S,4S,5R,6R)-6-[[(3S,4S,4aR,6aR,6bS,8R,8aR,12aS,14aR,14bR)-8a-carboxy-4-formyl-8-hydroxy-4,6a,6b,11,11,14b-hexamethyl-1,2,3,4a,5,6,7,8,9,10,12,12a,14,14a-tetradecahydropicen-3-yl]oxy]-3,4,5-trihydroxyoxane-2-carboxylic acid Chemical compound O([C@H]1CC[C@]2(C)[C@H]3CC=C4[C@@]([C@@]3(CC[C@H]2[C@@]1(C=O)C)C)(C)C[C@@H](O)[C@]1(CCC(C[C@H]14)(C)C)C(O)=O)[C@@H]1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O PIGTXFOGKFOFTO-FVFWYJKVSA-N 0.000 claims description 2
- CYJWWQALTIKOAG-FLORRLIPSA-N (2S,4aR,6aR,6aS,6bR,8aR,9R,10R,11S,12aR,14bS)-10,11-dihydroxy-9-(hydroxymethyl)-2-methoxycarbonyl-2,6a,6b,9,12a-pentamethyl-1,3,4,5,6,6a,7,8,8a,10,11,12,13,14b-tetradecahydropicene-4a-carboxylic acid Chemical compound C1[C@H](O)[C@H](O)[C@@](C)(CO)[C@@H]2CC[C@@]3(C)[C@]4(C)CC[C@@]5(C(O)=O)CC[C@@](C(=O)OC)(C)C[C@H]5C4=CC[C@@H]3[C@]21C CYJWWQALTIKOAG-FLORRLIPSA-N 0.000 claims description 2
- YFESOSRPNPYODN-RSMWSHJLSA-N (2s,3s,4s,5r,6r)-6-[[(4s,6ar,6bs,8r,8ar,9r,10r,14br)-9-acetyloxy-8-hydroxy-4,8a-bis(hydroxymethyl)-4,6a,6b,11,11,14b-hexamethyl-10-[(z)-2-methylbut-2-enoyl]oxy-1,2,3,4a,5,6,7,8,9,10,12,12a,14,14a-tetradecahydropicen-3-yl]oxy]-4-hydroxy-3,5-bis[[(2s,3r,4s, Chemical compound O([C@@H]1[C@H](O[C@H]([C@@H]([C@H]1O)O[C@H]1[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O1)O)OC1CC[C@]2(C)C3CC=C4[C@@]([C@@]3(CCC2[C@]1(CO)C)C)(C)C[C@@H](O)[C@@]1(CO)[C@@H](OC(C)=O)[C@@H](C(CC14)(C)C)OC(=O)C(\C)=C/C)C(O)=O)[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O.O([C@@H]1[C@H](O[C@H]([C@@H]([C@H]1O)O[C@H]1[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O1)O)OC1CC[C@]2(C)C3CC=C4[C@@]([C@@]3(CCC2[C@]1(CO)C)C)(C)C[C@@H](O)[C@@]1(CO)[C@@H](OC(C)=O)[C@@H](C(CC14)(C)C)OC(=O)C(/C)=C/C)C(O)=O)[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O YFESOSRPNPYODN-RSMWSHJLSA-N 0.000 claims description 2
- NTWLPZMPTFQYQI-UHFFFAOYSA-N (3alpha)-olean-12-ene-3,23-diol Natural products C1CC(O)C(C)(CO)C2CCC3(C)C4(C)CCC5(C)CCC(C)(C)CC5C4=CCC3C21C NTWLPZMPTFQYQI-UHFFFAOYSA-N 0.000 claims description 2
- MIJYXULNPSFWEK-GTOFXWBISA-N 3beta-hydroxyolean-12-en-28-oic acid Chemical compound C1C[C@H](O)C(C)(C)[C@@H]2CC[C@@]3(C)[C@]4(C)CC[C@@]5(C(O)=O)CCC(C)(C)C[C@H]5C4=CC[C@@H]3[C@]21C MIJYXULNPSFWEK-GTOFXWBISA-N 0.000 claims description 2
- AXNVHPCVMSNXNP-OXPBSUTMSA-N Aescin Chemical compound O([C@@H]1[C@H](O[C@H]([C@@H]([C@H]1O)O[C@H]1[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O1)O)O[C@H]1CC[C@]2(C)[C@H]3CC=C4[C@@]([C@@]3(CC[C@H]2[C@]1(CO)C)C)(C)C[C@@H](O)[C@@]1(CO)[C@@H](OC(C)=O)[C@@H](C(C[C@H]14)(C)C)OC(=O)C(\C)=C/C)C(O)=O)[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O AXNVHPCVMSNXNP-OXPBSUTMSA-N 0.000 claims description 2
- BJFNIGZQPQQAFL-UTPDYZQNSA-N Assamsaponin F Chemical compound O([C@@H]1[C@@H](O)[C@@H](O)CO[C@H]1O[C@H]1[C@H](O)[C@H](O[C@H]([C@@H]1O[C@H]1[C@@H]([C@@H](O)[C@@H](O)[C@@H](CO)O1)O)O[C@H]1CC[C@]2(C)[C@H]3CC=C4[C@@]([C@@]3(CC[C@H]2[C@@]1(C=O)C)C)(C)C[C@H]([C@@]1(CO)[C@@H](OC(C)=O)[C@@H](C(C[C@H]14)(C)C)OC(=O)C(\C)=C/C)OC(C)=O)C(O)=O)[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O BJFNIGZQPQQAFL-UTPDYZQNSA-N 0.000 claims description 2
- 206010006187 Breast cancer Diseases 0.000 claims description 2
- 208000026310 Breast neoplasm Diseases 0.000 claims description 2
- 102000004225 Cathepsin B Human genes 0.000 claims description 2
- 108090000712 Cathepsin B Proteins 0.000 claims description 2
- COVOPPXLDJVUSC-UHFFFAOYSA-N Digitogenin Natural products CC1C(C2(CCC3C4(C)CC(O)C(O)CC4CCC3C2C2O)C)C2OC11CCC(C)CO1 COVOPPXLDJVUSC-UHFFFAOYSA-N 0.000 claims description 2
- QRLVDLBMBULFAL-UHFFFAOYSA-N Digitonin Natural products CC1CCC2(OC1)OC3C(O)C4C5CCC6CC(OC7OC(CO)C(OC8OC(CO)C(O)C(OC9OCC(O)C(O)C9OC%10OC(CO)C(O)C(OC%11OC(CO)C(O)C(O)C%11O)C%10O)C8O)C(O)C7O)C(O)CC6(C)C5CCC4(C)C3C2C QRLVDLBMBULFAL-UHFFFAOYSA-N 0.000 claims description 2
- 206010014733 Endometrial cancer Diseases 0.000 claims description 2
- 206010014759 Endometrial neoplasm Diseases 0.000 claims description 2
- LAHSXXNOJMWHBH-WVPBMNGESA-N Gypsoside Natural products O=C(O)[C@H]1[C@H](O[C@H]2[C@@H](O)[C@H](O)[C@H](O[C@@H]3[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O3)[C@H](CO)O2)[C@H](O[C@H]2[C@@H](O)[C@@H](O)[C@@H](O)CO2)[C@@H](O)[C@@H](O[C@@H]2[C@](C=O)(C)[C@@H]3[C@](C)([C@H]4[C@](C)([C@]5(C)C([C@H]6[C@](C(=O)O[C@H]7[C@H](O[C@@H]8[C@H](O[C@@H]9[C@H](O)[C@H](O)[C@H](O)CO9)[C@H](O)[C@H](O)CO8)[C@@H](O)[C@@H](O[C@@H]8[C@@H](O)[C@@H](O[C@H]9[C@@H](O)[C@@H](O)[C@@H](O)CO9)[C@H](O)[C@@H](C)O8)[C@@H](C)O7)(CC5)CCC(C)(C)C6)=CC4)CC3)CC2)O1 LAHSXXNOJMWHBH-WVPBMNGESA-N 0.000 claims description 2
- GCGBHJLBFAPRDB-UHFFFAOYSA-N Hederagenin Natural products CC1(C)CCC2(CCC3(C)C4CCC5C(C)(CO)C(O)CCC5(C)C4CC=C3C2C1)C(=O)O GCGBHJLBFAPRDB-UHFFFAOYSA-N 0.000 claims description 2
- 206010058467 Lung neoplasm malignant Diseases 0.000 claims description 2
- ZMXKPCHQLHYTHY-UHFFFAOYSA-N Phytolaccasaponin E Natural products C12CC(C(=O)OC)(C)CCC2(C(O)=O)CCC(C2(CCC3C4(CO)C)C)(C)C1=CCC2C3(C)CC(O)C4OC(C(C1O)O)OCC1OC1OC(CO)C(O)C(O)C1O ZMXKPCHQLHYTHY-UHFFFAOYSA-N 0.000 claims description 2
- YRHWKFMGEDDGIJ-UHFFFAOYSA-N Phytolaccoside E Chemical compound C12CC(C(=O)OC)(C)CCC2(C(O)=O)CCC(C2(CCC3C4(CO)C)C)(C)C1=CCC2C3(C)CC(O)C4OC(C(C1O)O)COC1OC1OC(CO)C(O)C(O)C1O YRHWKFMGEDDGIJ-UHFFFAOYSA-N 0.000 claims description 2
- 241000245063 Primula Species 0.000 claims description 2
- 235000000497 Primula Nutrition 0.000 claims description 2
- 241001092473 Quillaja Species 0.000 claims description 2
- 235000009001 Quillaja saponaria Nutrition 0.000 claims description 2
- KYWSCMDFVARMPN-MSSMMRRTSA-N Saikosaponin A Chemical compound O([C@@H]1[C@@H](O)[C@H](O[C@@H]2[C@@]([C@H]3[C@]([C@@H]4[C@@]([C@@]5(C[C@H](O)[C@]67CO[C@]5([C@@H]6CC(C)(C)CC7)C=C4)C)(C)CC3)(C)CC2)(C)CO)O[C@@H]([C@@H]1O)C)[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O KYWSCMDFVARMPN-MSSMMRRTSA-N 0.000 claims description 2
- KYWSCMDFVARMPN-LCSVLAELSA-N Saikosaponin D Chemical compound O([C@@H]1[C@@H](O)[C@H](O[C@@H]2[C@@]([C@H]3[C@]([C@@H]4[C@@]([C@@]5(C[C@@H](O)[C@]67CO[C@]5([C@@H]6CC(C)(C)CC7)C=C4)C)(C)CC3)(C)CC2)(C)CO)O[C@@H]([C@@H]1O)C)[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O KYWSCMDFVARMPN-LCSVLAELSA-N 0.000 claims description 2
- KEOITPILCOILGM-FCWYPSSHSA-N Sapindoside A Natural products O=C(O)[C@]12[C@H](C=3[C@](C)([C@@]4(C)[C@@H]([C@]5(C)[C@H]([C@@](CO)(C)[C@@H](O[C@@H]6[C@@H](O[C@H]7[C@H](O)[C@@H](O)[C@@H](O)[C@H](C)O7)[C@H](O)[C@H](O)CO6)CC5)CC4)CC=3)CC1)CC(C)(C)CC2 KEOITPILCOILGM-FCWYPSSHSA-N 0.000 claims description 2
- VUEGHZSQVJADCO-UGZFTLGKSA-N [(2S,3R,4S,5S,6R)-3,4,5-trihydroxy-6-[[(2S,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxymethyl]oxan-2-yl] (4aS,6aR,6aS,6bR,8aR,9R,10S,12aR,14bS)-10-[(2S,3R,4S,5S)-3-[(2S,3R,4R,5S,6S)-3,5-dihydroxy-6-methyl-4-[(2S,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyoxan-2-yl]oxy-4,5-dihydroxyoxan-2-yl]oxy-9-(hydroxymethyl)-2,2,6a,6b,9,12a-hexamethyl-1,3,4,5,6,6a,7,8,8a,10,11,12,13,14b-tetradecahydropicene-4a-carboxylate Chemical compound C([C@H]1O[C@H]([C@@H]([C@@H](O)[C@@H]1O)O)OC(=O)[C@]12CCC(C)(C)C[C@H]1C1=CC[C@H]3[C@@]([C@@]1(CC2)C)(C)CC[C@@H]1[C@]3(C)CC[C@@H]([C@@]1(C)CO)O[C@@H]1OC[C@H](O)[C@H](O)[C@H]1O[C@@H]1O[C@H]([C@@H]([C@@H](O[C@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O2)O)[C@H]1O)O)C)O[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O VUEGHZSQVJADCO-UGZFTLGKSA-N 0.000 claims description 2
- UZQJVUCHXGYFLQ-AYDHOLPZSA-N [(2s,3r,4s,5r,6r)-4-[(2s,3r,4s,5r,6r)-4-[(2r,3r,4s,5r,6r)-4-[(2s,3r,4s,5r,6r)-3,5-dihydroxy-6-(hydroxymethyl)-4-[(2s,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyoxan-2-yl]oxy-3,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-3,5-dihydroxy-6-(hy Chemical compound O([C@H]1[C@H](O)[C@@H](CO)O[C@H]([C@@H]1O)O[C@H]1[C@H](O)[C@@H](CO)O[C@H]([C@@H]1O)O[C@H]1CC[C@]2(C)[C@H]3CC=C4[C@@]([C@@]3(CC[C@H]2[C@@]1(C=O)C)C)(C)CC(O)[C@]1(CCC(CC14)(C)C)C(=O)O[C@H]1[C@@H]([C@@H](O[C@H]2[C@@H]([C@@H](O[C@H]3[C@@H]([C@@H](O[C@H]4[C@@H]([C@@H](O[C@H]5[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O5)O)[C@H](O)[C@@H](CO)O4)O)[C@H](O)[C@@H](CO)O3)O)[C@H](O)[C@@H](CO)O2)O)[C@H](O)[C@@H](CO)O1)O)[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O UZQJVUCHXGYFLQ-AYDHOLPZSA-N 0.000 claims description 2
- GFPLPBCJRRNZHM-ICUGHKHQSA-N [(2s,3r,4s,5s,6r)-3,4,5-trihydroxy-6-[[(2r,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxymethyl]oxan-2-yl] (4as,6ar,6as,6br,8ar,9r,10s,12ar,14bs)-10-[(2s,3r,4s,5s)-4,5-dihydroxy-3-[(2s,3r,4r,5r,6s)-3,4,5-trihydroxy-6-methyloxan-2-yl]oxyoxan- Chemical compound O[C@@H]1[C@H](O)[C@@H](O)[C@H](C)O[C@H]1O[C@H]1[C@H](O[C@@H]2[C@@]([C@H]3[C@]([C@@H]4[C@@]([C@@]5(CC[C@]6(CCC(C)(C)C[C@H]6C5=CC4)C(=O)O[C@H]4[C@@H]([C@@H](O)[C@H](O)[C@@H](CO[C@H]5[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O5)O)O4)O)C)(C)CC3)(C)CC2)(C)CO)OC[C@H](O)[C@@H]1O GFPLPBCJRRNZHM-ICUGHKHQSA-N 0.000 claims description 2
- KBYYTUYPCGPQNK-UHFFFAOYSA-N alpha-hederin Natural products CC1OC(OC2C(O)C(CO)OC2OC3CCC4(C)C(CCC5(C)C4CC=C6C7CC(C)(C)CCC7(CCC56C)C(=O)O)C3(C)CO)C(O)C(O)C1O KBYYTUYPCGPQNK-UHFFFAOYSA-N 0.000 claims description 2
- 101150078331 ama-1 gene Proteins 0.000 claims description 2
- VUEGHZSQVJADCO-UHFFFAOYSA-N aralia saponin 3 Natural products OC1C(OC2C(C(O)C(O)C(CO)O2)O)C(O)C(C)OC1OC1C(O)C(O)COC1OC(C1(C)CO)CCC2(C)C1CCC(C1(CC3)C)(C)C2CC=C1C1CC(C)(C)CCC13C(=O)OC(C(C(O)C1O)O)OC1COC1OC(CO)C(O)C(O)C1O VUEGHZSQVJADCO-UHFFFAOYSA-N 0.000 claims description 2
- LCGQSISMCSVPJV-UHFFFAOYSA-N assamsaponin F Natural products CC=C(C)/C(=O)OC1C(OC(=O)C)C2(CO)C(CC3(C)C(=CCC4C5(C)CCC(OC6OC(C(O)C(OC7OC(O)C(O)CC7OC8OC(CO)C(O)C(O)C8O)C6OC9OC(CO)C(O)C(O)C9O)C(=O)O)C(C)(C=O)C5CCC34C)C2CC1(C)C)OC(=O)C LCGQSISMCSVPJV-UHFFFAOYSA-N 0.000 claims description 2
- MDZKJHQSJHYOHJ-UHFFFAOYSA-N crataegolic acid Natural products C1C(O)C(O)C(C)(C)C2CCC3(C)C4(C)CCC5(C(O)=O)CCC(C)(C)CC5C4=CCC3C21C MDZKJHQSJHYOHJ-UHFFFAOYSA-N 0.000 claims description 2
- COVOPPXLDJVUSC-JPYPKGSXSA-N digitogenin Chemical compound O([C@@H]1[C@@H]([C@]2(CC[C@@H]3[C@@]4(C)C[C@@H](O)[C@H](O)C[C@@H]4CC[C@H]3[C@@H]2[C@@H]1O)C)[C@@H]1C)[C@]11CC[C@@H](C)CO1 COVOPPXLDJVUSC-JPYPKGSXSA-N 0.000 claims description 2
- UVYVLBIGDKGWPX-KUAJCENISA-N digitonin Chemical compound O([C@@H]1[C@@H]([C@]2(CC[C@@H]3[C@@]4(C)C[C@@H](O)[C@H](O[C@H]5[C@@H]([C@@H](O)[C@@H](O[C@H]6[C@@H]([C@@H](O[C@H]7[C@@H]([C@@H](O)[C@H](O)CO7)O)[C@H](O)[C@@H](CO)O6)O[C@H]6[C@@H]([C@@H](O[C@H]7[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O7)O)[C@@H](O)[C@@H](CO)O6)O)[C@@H](CO)O5)O)C[C@@H]4CC[C@H]3[C@@H]2[C@@H]1O)C)[C@@H]1C)[C@]11CC[C@@H](C)CO1 UVYVLBIGDKGWPX-KUAJCENISA-N 0.000 claims description 2
- UVYVLBIGDKGWPX-UHFFFAOYSA-N digitonine Natural products CC1C(C2(CCC3C4(C)CC(O)C(OC5C(C(O)C(OC6C(C(OC7C(C(O)C(O)CO7)O)C(O)C(CO)O6)OC6C(C(OC7C(C(O)C(O)C(CO)O7)O)C(O)C(CO)O6)O)C(CO)O5)O)CC4CCC3C2C2O)C)C2OC11CCC(C)CO1 UVYVLBIGDKGWPX-UHFFFAOYSA-N 0.000 claims description 2
- GFPLPBCJRRNZHM-UHFFFAOYSA-N dipsacoside B Natural products CC1OC(OC2C(O)C(O)COC2OC2CCC3(C)C(CCC4(C)C3CC=C3C5CC(C)(C)CCC5(CCC43C)C(=O)OC3OC(COC4OC(CO)C(O)C(O)C4O)C(O)C(O)C3O)C2(C)CO)C(O)C(O)C1O GFPLPBCJRRNZHM-UHFFFAOYSA-N 0.000 claims description 2
- 201000003914 endometrial carcinoma Diseases 0.000 claims description 2
- PAIBKVQNJKUVCE-JUENUIDLSA-N gypsogenic acid Chemical compound C1C[C@H](O)[C@@](C)(C(O)=O)[C@@H]2CC[C@@]3(C)[C@]4(C)CC[C@@]5(C(O)=O)CCC(C)(C)C[C@H]5C4=CC[C@@H]3[C@]21C PAIBKVQNJKUVCE-JUENUIDLSA-N 0.000 claims description 2
- PAIBKVQNJKUVCE-HDHBZPKHSA-N gypsogenic acid Natural products CC1(C)CC[C@@]2(CC[C@]3(C)C(=CC[C@@H]4[C@@]5(C)CC[C@H](O)[C@@](C)([C@H]5CC[C@@]34C)C(O)=O)[C@H]2C1)C(O)=O PAIBKVQNJKUVCE-HDHBZPKHSA-N 0.000 claims description 2
- PGOYMURMZNDHNS-MYPRUECHSA-N hederagenin Chemical compound C1C[C@H](O)[C@@](C)(CO)[C@@H]2CC[C@@]3(C)[C@]4(C)CC[C@@]5(C(O)=O)CCC(C)(C)C[C@H]5C4=CC[C@@H]3[C@]21C PGOYMURMZNDHNS-MYPRUECHSA-N 0.000 claims description 2
- KEOITPILCOILGM-LLJOFIFVSA-N kalopanaxsaponin A Chemical compound O[C@@H]1[C@H](O)[C@@H](O)[C@H](C)O[C@H]1O[C@H]1[C@H](O[C@@H]2[C@@]([C@H]3[C@]([C@@H]4[C@@]([C@@]5(CC[C@]6(CCC(C)(C)C[C@H]6C5=CC4)C(O)=O)C)(C)CC3)(C)CC2)(C)CO)OC[C@H](O)[C@@H]1O KEOITPILCOILGM-LLJOFIFVSA-N 0.000 claims description 2
- 239000007788 liquid Substances 0.000 claims description 2
- 201000005202 lung cancer Diseases 0.000 claims description 2
- 208000020816 lung neoplasm Diseases 0.000 claims description 2
- CYJWWQALTIKOAG-UHFFFAOYSA-N phytolaccagenin Natural products C1C(O)C(O)C(C)(CO)C2CCC3(C)C4(C)CCC5(C(O)=O)CCC(C(=O)OC)(C)CC5C4=CCC3C21C CYJWWQALTIKOAG-UHFFFAOYSA-N 0.000 claims description 2
- NEEQJTVMZAIFID-OBTMRSOISA-N teaseedsaponin I Natural products CC=C(C)C(=O)O[C@H]1[C@H](OC(=O)C(=CC)C)[C@]2(CO)[C@H](O)C[C@]3(C)C(=CC[C@@H]4[C@@]5(C)CC[C@H](O[C@@H]6O[C@@H]([C@@H](O)[C@H](O[C@@H]7OC[C@H](O)[C@H](O)[C@H]7O[C@@H]8O[C@H](CO)[C@@H](O)[C@H](O)[C@H]8O)[C@H]6O[C@@H]9O[C@H](CO)[C@H](O)[C@H](O)[C@H]9O)C(=O)O)[C@@](C)(C=O)[C@@H]5CC[C@@]34C)[C@@H]2CC1(C)C NEEQJTVMZAIFID-OBTMRSOISA-N 0.000 claims description 2
- QBNQXNXXIWAIMM-VMHSQIKKSA-N teaseedsaponin J Natural products CC=C(C)/C(=O)O[C@H]1[C@H](OC(=O)C(=C/C)C)[C@]2(CO)[C@H](O)C[C@]3(C)C(=CC[C@@H]4[C@@]5(C)CC[C@H](O[C@@H]6O[C@@H]([C@@H](O)[C@H](O[C@@H]7OC[C@H](O)[C@H](O)[C@H]7O[C@@H]8OC[C@@H](O)[C@H](O)[C@H]8O)[C@H]6O[C@@H]9O[C@H](CO)[C@H](O)[C@H](O)[C@H]9O)C(=O)O)[C@@](C)(C=O)[C@@H]5CC[C@@]34C)[C@@H]2CC1(C)C QBNQXNXXIWAIMM-VMHSQIKKSA-N 0.000 claims description 2
- UIERETOOQGIECD-ONEGZZNKSA-N tiglic acid Chemical compound C\C=C(/C)C(O)=O UIERETOOQGIECD-ONEGZZNKSA-N 0.000 claims description 2
- 238000004519 manufacturing process Methods 0.000 abstract description 2
- 102100022977 Antithrombin-III Human genes 0.000 abstract 1
- 101001098868 Homo sapiens Proprotein convertase subtilisin/kexin type 9 Proteins 0.000 abstract 1
- 101710146810 Type-1B angiotensin II receptor Proteins 0.000 abstract 1
- 239000000562 conjugate Substances 0.000 description 422
- 239000012636 effector Substances 0.000 description 124
- 125000005647 linker group Chemical group 0.000 description 115
- 239000002243 precursor Substances 0.000 description 54
- 238000005859 coupling reaction Methods 0.000 description 48
- 239000003053 toxin Substances 0.000 description 41
- 231100000765 toxin Toxicity 0.000 description 41
- 108700012359 toxins Proteins 0.000 description 41
- 235000018102 proteins Nutrition 0.000 description 39
- 150000001875 compounds Chemical class 0.000 description 38
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 36
- 238000003786 synthesis reaction Methods 0.000 description 30
- 230000001225 therapeutic effect Effects 0.000 description 27
- 229940079593 drug Drugs 0.000 description 26
- 238000012226 gene silencing method Methods 0.000 description 26
- 150000007857 hydrazones Chemical class 0.000 description 26
- 238000010168 coupling process Methods 0.000 description 24
- 230000008878 coupling Effects 0.000 description 23
- QWENRTYMTSOGBR-UHFFFAOYSA-N 1H-1,2,3-Triazole Chemical compound C=1C=NNN=1 QWENRTYMTSOGBR-UHFFFAOYSA-N 0.000 description 20
- 239000000611 antibody drug conjugate Substances 0.000 description 20
- 229940049595 antibody-drug conjugate Drugs 0.000 description 20
- 230000001976 improved effect Effects 0.000 description 20
- 230000000694 effects Effects 0.000 description 19
- 125000004432 carbon atom Chemical group C* 0.000 description 17
- 102000004190 Enzymes Human genes 0.000 description 16
- 108090000790 Enzymes Proteins 0.000 description 16
- 238000012228 RNA interference-mediated gene silencing Methods 0.000 description 16
- 229940088598 enzyme Drugs 0.000 description 16
- 230000009368 gene silencing by RNA Effects 0.000 description 16
- 239000000047 product Substances 0.000 description 16
- 210000003494 hepatocyte Anatomy 0.000 description 15
- 102000005962 receptors Human genes 0.000 description 15
- 108020003175 receptors Proteins 0.000 description 15
- 101000600434 Homo sapiens Putative uncharacterized protein encoded by MIR7-3HG Proteins 0.000 description 14
- 102100037401 Putative uncharacterized protein encoded by MIR7-3HG Human genes 0.000 description 14
- 108090000765 processed proteins & peptides Proteins 0.000 description 14
- 108010016790 RNA-Induced Silencing Complex Proteins 0.000 description 13
- 102000000574 RNA-Induced Silencing Complex Human genes 0.000 description 13
- 230000002708 enhancing effect Effects 0.000 description 13
- 239000000126 substance Substances 0.000 description 13
- 108020000948 Antisense Oligonucleotides Proteins 0.000 description 12
- 125000000539 amino acid group Chemical group 0.000 description 12
- 210000004185 liver Anatomy 0.000 description 12
- 239000002773 nucleotide Substances 0.000 description 12
- AUDYZXNUHIIGRB-UHFFFAOYSA-N 3-thiophen-2-ylpyrrole-2,5-dione Chemical compound O=C1NC(=O)C(C=2SC=CC=2)=C1 AUDYZXNUHIIGRB-UHFFFAOYSA-N 0.000 description 11
- 108010084592 Saporins Proteins 0.000 description 11
- 230000004071 biological effect Effects 0.000 description 11
- 230000021615 conjugation Effects 0.000 description 11
- 125000003729 nucleotide group Chemical group 0.000 description 11
- 238000006722 reduction reaction Methods 0.000 description 11
- 150000003384 small molecules Chemical class 0.000 description 11
- 150000001540 azides Chemical class 0.000 description 10
- 230000001965 increasing effect Effects 0.000 description 10
- 230000003834 intracellular effect Effects 0.000 description 10
- 238000010461 azide-alkyne cycloaddition reaction Methods 0.000 description 9
- 238000010195 expression analysis Methods 0.000 description 9
- 230000003389 potentiating effect Effects 0.000 description 9
- 238000002560 therapeutic procedure Methods 0.000 description 9
- 238000011746 C57BL/6J (JAX™ mouse strain) Methods 0.000 description 8
- 150000001345 alkine derivatives Chemical class 0.000 description 8
- 150000001408 amides Chemical class 0.000 description 8
- 125000000524 functional group Chemical group 0.000 description 8
- 230000001988 toxicity Effects 0.000 description 8
- 231100000419 toxicity Toxicity 0.000 description 8
- LQRNAUZEMLGYOX-LZVIIAQDSA-N CC(=O)N[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1OCCCCC(=O)NCCCNC(=O)CCOCC(COCCC(=O)NCCCNC(=O)CCCCO[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1NC(C)=O)(COCCC(=O)NCCCNC(=O)CCCCO[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1NC(C)=O)NC(=O)CCCCCCCCCCC(=O)N1C[C@H](O)C[C@H]1COP(O)(O)=O Chemical compound CC(=O)N[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1OCCCCC(=O)NCCCNC(=O)CCOCC(COCCC(=O)NCCCNC(=O)CCCCO[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1NC(C)=O)(COCCC(=O)NCCCNC(=O)CCCCO[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1NC(C)=O)NC(=O)CCCCCCCCCCC(=O)N1C[C@H](O)C[C@H]1COP(O)(O)=O LQRNAUZEMLGYOX-LZVIIAQDSA-N 0.000 description 7
- 230000015556 catabolic process Effects 0.000 description 7
- 238000006731 degradation reaction Methods 0.000 description 7
- 229950005863 inclisiran Drugs 0.000 description 7
- 230000007246 mechanism Effects 0.000 description 7
- 239000000203 mixture Substances 0.000 description 7
- 229920000642 polymer Polymers 0.000 description 7
- 231100000654 protein toxin Toxicity 0.000 description 7
- 150000007659 semicarbazones Chemical class 0.000 description 7
- 210000002966 serum Anatomy 0.000 description 7
- HSJKGGMUJITCBW-UHFFFAOYSA-N 3-hydroxybutanal Chemical compound CC(O)CC=O HSJKGGMUJITCBW-UHFFFAOYSA-N 0.000 description 6
- 102100034343 Integrase Human genes 0.000 description 6
- 101710203526 Integrase Proteins 0.000 description 6
- 238000003800 Staudinger reaction Methods 0.000 description 6
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 6
- 238000004458 analytical method Methods 0.000 description 6
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 6
- 239000012039 electrophile Substances 0.000 description 6
- 230000012202 endocytosis Effects 0.000 description 6
- 125000000623 heterocyclic group Chemical group 0.000 description 6
- 230000000269 nucleophilic effect Effects 0.000 description 6
- 229910052760 oxygen Inorganic materials 0.000 description 6
- 239000001301 oxygen Substances 0.000 description 6
- 230000037361 pathway Effects 0.000 description 6
- 150000003138 primary alcohols Chemical class 0.000 description 6
- 238000007142 ring opening reaction Methods 0.000 description 6
- 108010051109 Cell-Penetrating Peptides Proteins 0.000 description 5
- 102000020313 Cell-Penetrating Peptides Human genes 0.000 description 5
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 5
- 108010001831 LDL receptors Proteins 0.000 description 5
- 102000000853 LDL receptors Human genes 0.000 description 5
- 238000013459 approach Methods 0.000 description 5
- 125000004429 atom Chemical group 0.000 description 5
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 5
- 238000003570 cell viability assay Methods 0.000 description 5
- 150000002148 esters Chemical class 0.000 description 5
- 239000002539 nanocarrier Substances 0.000 description 5
- 230000009437 off-target effect Effects 0.000 description 5
- 229940126586 small molecule drug Drugs 0.000 description 5
- 210000001519 tissue Anatomy 0.000 description 5
- 101000780643 Homo sapiens Protein argonaute-2 Proteins 0.000 description 4
- 101000835093 Homo sapiens Transferrin receptor protein 1 Proteins 0.000 description 4
- 108010007622 LDL Lipoproteins Proteins 0.000 description 4
- 102000007330 LDL Lipoproteins Human genes 0.000 description 4
- 102100034207 Protein argonaute-2 Human genes 0.000 description 4
- 102100026144 Transferrin receptor protein 1 Human genes 0.000 description 4
- 230000004913 activation Effects 0.000 description 4
- 230000000692 anti-sense effect Effects 0.000 description 4
- 230000008901 benefit Effects 0.000 description 4
- 235000014633 carbohydrates Nutrition 0.000 description 4
- 210000000170 cell membrane Anatomy 0.000 description 4
- TZRFSLHOCZEXCC-HIVFKXHNSA-N chembl2219536 Chemical compound N1([C@H]2C[C@@H]([C@H](O2)COP(O)(=O)S[C@@H]2[C@H](O[C@H](C2)N2C3=C(C(NC(N)=N3)=O)N=C2)COP(O)(=O)S[C@@H]2[C@H](O[C@H](C2)N2C(NC(=O)C(C)=C2)=O)COP(O)(=O)S[C@@H]2[C@H](O[C@H](C2)N2C(N=C(N)C(C)=C2)=O)COP(O)(=O)S[C@@H]2[C@H](O[C@H](C2)N2C(NC(=O)C(C)=C2)=O)COP(O)(=O)S[C@@H]2[C@H](O[C@H](C2)N2C3=C(C(NC(N)=N3)=O)N=C2)COP(O)(=O)S[C@@H]2[C@H](O[C@H](C2)N2C3=NC=NC(N)=C3N=C2)COP(O)(=O)S[C@H]2[C@H]([C@@H](O[C@@H]2COP(O)(=O)S[C@H]2[C@H]([C@@H](O[C@@H]2COP(O)(=O)S[C@H]2[C@H]([C@@H](O[C@@H]2COP(O)(=O)S[C@H]2[C@H]([C@@H](O[C@@H]2COP(O)(=O)S[C@H]2[C@H]([C@@H](O[C@@H]2CO)N2C3=C(C(NC(N)=N3)=O)N=C2)OCCOC)N2C(N=C(N)C(C)=C2)=O)OCCOC)N2C(N=C(N)C(C)=C2)=O)OCCOC)N2C(NC(=O)C(C)=C2)=O)OCCOC)N2C(N=C(N)C(C)=C2)=O)OCCOC)SP(O)(=O)OC[C@H]2O[C@H](C[C@@H]2SP(O)(=O)OC[C@H]2O[C@H](C[C@@H]2SP(O)(=O)OC[C@H]2O[C@H](C[C@@H]2SP(O)(=O)OC[C@@H]2[C@H]([C@H]([C@@H](O2)N2C3=C(C(NC(N)=N3)=O)N=C2)OCCOC)SP(O)(=O)OC[C@H]2[C@@H]([C@@H]([C@H](O2)N2C(N=C(N)C(C)=C2)=O)OCCOC)SP(O)(=O)OC[C@H]2[C@@H]([C@@H]([C@H](O2)N2C3=NC=NC(N)=C3N=C2)OCCOC)SP(O)(=O)OC[C@H]2[C@@H]([C@@H]([C@H](O2)N2C(N=C(N)C(C)=C2)=O)OCCOC)SP(O)(=O)OC[C@H]2[C@H](O)[C@@H]([C@H](O2)N2C(N=C(N)C(C)=C2)=O)OCCOC)N2C(N=C(N)C(C)=C2)=O)N2C(NC(=O)C(C)=C2)=O)N2C(NC(=O)C(C)=C2)=O)C=C(C)C(N)=NC1=O TZRFSLHOCZEXCC-HIVFKXHNSA-N 0.000 description 4
- 238000007385 chemical modification Methods 0.000 description 4
- 238000011260 co-administration Methods 0.000 description 4
- 230000001268 conjugating effect Effects 0.000 description 4
- 230000001086 cytosolic effect Effects 0.000 description 4
- 239000012634 fragment Substances 0.000 description 4
- 150000004676 glycans Chemical class 0.000 description 4
- 230000005764 inhibitory process Effects 0.000 description 4
- 108091060283 mipomersen Proteins 0.000 description 4
- 229960004778 mipomersen Drugs 0.000 description 4
- 230000035772 mutation Effects 0.000 description 4
- 239000002105 nanoparticle Substances 0.000 description 4
- 150000003333 secondary alcohols Chemical class 0.000 description 4
- 239000000243 solution Substances 0.000 description 4
- 230000004936 stimulating effect Effects 0.000 description 4
- 150000003573 thiols Chemical class 0.000 description 4
- UMGDCJDMYOKAJW-UHFFFAOYSA-N thiourea Chemical compound NC(N)=S UMGDCJDMYOKAJW-UHFFFAOYSA-N 0.000 description 4
- 150000003923 2,5-pyrrolediones Chemical class 0.000 description 3
- SRBFZHDQGSBBOR-IOVATXLUSA-N D-xylopyranose Chemical compound O[C@@H]1COC(O)[C@H](O)[C@H]1O SRBFZHDQGSBBOR-IOVATXLUSA-N 0.000 description 3
- 102100023387 Endoribonuclease Dicer Human genes 0.000 description 3
- 108091027305 Heteroduplex Proteins 0.000 description 3
- 101000907904 Homo sapiens Endoribonuclease Dicer Proteins 0.000 description 3
- 101000617738 Homo sapiens Survival motor neuron protein Proteins 0.000 description 3
- PEEHTFAAVSWFBL-UHFFFAOYSA-N Maleimide Chemical compound O=C1NC(=O)C=C1 PEEHTFAAVSWFBL-UHFFFAOYSA-N 0.000 description 3
- 239000002202 Polyethylene glycol Substances 0.000 description 3
- IEDXPSOJFSVCKU-HOKPPMCLSA-N [4-[[(2S)-5-(carbamoylamino)-2-[[(2S)-2-[6-(2,5-dioxopyrrolidin-1-yl)hexanoylamino]-3-methylbutanoyl]amino]pentanoyl]amino]phenyl]methyl N-[(2S)-1-[[(2S)-1-[[(3R,4S,5S)-1-[(2S)-2-[(1R,2R)-3-[[(1S,2R)-1-hydroxy-1-phenylpropan-2-yl]amino]-1-methoxy-2-methyl-3-oxopropyl]pyrrolidin-1-yl]-3-methoxy-5-methyl-1-oxoheptan-4-yl]-methylamino]-3-methyl-1-oxobutan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]-N-methylcarbamate Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C)[C@@H](O)c1ccccc1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)OCc1ccc(NC(=O)[C@H](CCCNC(N)=O)NC(=O)[C@@H](NC(=O)CCCCCN2C(=O)CCC2=O)C(C)C)cc1)C(C)C IEDXPSOJFSVCKU-HOKPPMCLSA-N 0.000 description 3
- 150000001412 amines Chemical class 0.000 description 3
- PYMYPHUHKUWMLA-UHFFFAOYSA-N arabinose Natural products OCC(O)C(O)C(O)C=O PYMYPHUHKUWMLA-UHFFFAOYSA-N 0.000 description 3
- 230000004888 barrier function Effects 0.000 description 3
- SRBFZHDQGSBBOR-UHFFFAOYSA-N beta-D-Pyranose-Lyxose Natural products OC1COC(O)C(O)C1O SRBFZHDQGSBBOR-UHFFFAOYSA-N 0.000 description 3
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 3
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 3
- 235000012000 cholesterol Nutrition 0.000 description 3
- 239000000470 constituent Substances 0.000 description 3
- 229930191339 dianthin Natural products 0.000 description 3
- 229940000406 drug candidate Drugs 0.000 description 3
- 238000005516 engineering process Methods 0.000 description 3
- 230000037440 gene silencing effect Effects 0.000 description 3
- 229950010941 givosiran Drugs 0.000 description 3
- 150000002338 glycosides Chemical class 0.000 description 3
- 238000009396 hybridization Methods 0.000 description 3
- 230000006872 improvement Effects 0.000 description 3
- 238000010348 incorporation Methods 0.000 description 3
- 230000003993 interaction Effects 0.000 description 3
- 150000002632 lipids Chemical class 0.000 description 3
- 239000002502 liposome Substances 0.000 description 3
- 239000000178 monomer Substances 0.000 description 3
- 210000000056 organ Anatomy 0.000 description 3
- 229920001223 polyethylene glycol Polymers 0.000 description 3
- 102000004196 processed proteins & peptides Human genes 0.000 description 3
- DUIOPKIIICUYRZ-UHFFFAOYSA-N semicarbazide group Chemical group NNC(=O)N DUIOPKIIICUYRZ-UHFFFAOYSA-N 0.000 description 3
- 238000013518 transcription Methods 0.000 description 3
- 230000035897 transcription Effects 0.000 description 3
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 3
- 230000003612 virological effect Effects 0.000 description 3
- MFRNYXJJRJQHNW-DEMKXPNLSA-N (2s)-2-[[(2r,3r)-3-methoxy-3-[(2s)-1-[(3r,4s,5s)-3-methoxy-5-methyl-4-[methyl-[(2s)-3-methyl-2-[[(2s)-3-methyl-2-(methylamino)butanoyl]amino]butanoyl]amino]heptanoyl]pyrrolidin-2-yl]-2-methylpropanoyl]amino]-3-phenylpropanoic acid Chemical compound CN[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N(C)[C@@H]([C@@H](C)CC)[C@H](OC)CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 MFRNYXJJRJQHNW-DEMKXPNLSA-N 0.000 description 2
- KLEGMTRDCCDFJK-XDQSQZFTSA-N 1-[(2R,4S,5R)-4-[[(2R,3S,5R)-3-[[(2R,3S,5R)-5-(4-amino-5-methyl-2-oxopyrimidin-1-yl)-3-[[(2R,3S,5R)-3-[[(2R,3S,5R)-3-[[(2R,3S,5R)-3-[[(2R,3S,5R)-3-[[(2R,3S,5R)-3-[[(2R,3R,4R,5R)-3-[[(2R,3R,4R,5R)-3-[[(2R,3R,4R,5R)-5-(4-amino-5-methyl-2-oxopyrimidin-1-yl)-3-[[(2R,3R,4R,5R)-5-(4-amino-5-methyl-2-oxopyrimidin-1-yl)-3-[[(2R,3R,4R,5R)-5-(4-amino-5-methyl-2-oxopyrimidin-1-yl)-3-hydroxy-4-(2-methoxyethoxy)oxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxy-4-(2-methoxyethoxy)oxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxy-4-(2-methoxyethoxy)oxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxy-4-(2-methoxyethoxy)-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxy-5-(6-aminopurin-9-yl)-4-(2-methoxyethoxy)oxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxy-5-(2-amino-6-oxo-1H-purin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxyoxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphinothioyl]oxy-5-[[[(2R,3S,5R)-2-[[[(2R,3S,5R)-2-[[[(2R,3R,4R,5R)-2-[[[(2R,3R,4R,5R)-2-[[[(2R,3R,4R,5R)-2-[[[(2R,3R,4R,5R)-5-(4-amino-5-methyl-2-oxopyrimidin-1-yl)-2-[[hydroxy-[(2R,3R,4R,5R)-2-(hydroxymethyl)-4-(2-methoxyethoxy)-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-3-yl]oxyphosphinothioyl]oxymethyl]-4-(2-methoxyethoxy)oxolan-3-yl]oxy-sulfanylphosphoryl]oxymethyl]-4-(2-methoxyethoxy)-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-3-yl]oxy-hydroxyphosphinothioyl]oxymethyl]-4-(2-methoxyethoxy)-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-3-yl]oxy-hydroxyphosphinothioyl]oxymethyl]-5-(2-amino-6-oxo-1H-purin-9-yl)-4-(2-methoxyethoxy)oxolan-3-yl]oxy-hydroxyphosphinothioyl]oxymethyl]-5-(2-amino-6-oxo-1H-purin-9-yl)oxolan-3-yl]oxy-hydroxyphosphinothioyl]oxymethyl]-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-3-yl]oxy-hydroxyphosphinothioyl]oxymethyl]oxolan-2-yl]-5-methylpyrimidine-2,4-dione Chemical compound COCCO[C@@H]1[C@H](O)[C@@H](COP(O)(=S)O[C@@H]2[C@@H](COP(O)(=S)O[C@@H]3[C@@H](COP(O)(=S)O[C@@H]4[C@@H](COP(O)(=S)O[C@@H]5[C@@H](COP(O)(=S)O[C@H]6C[C@@H](O[C@@H]6COP(O)(=S)O[C@H]6C[C@@H](O[C@@H]6COP(O)(=S)O[C@H]6C[C@@H](O[C@@H]6COP(O)(=S)O[C@H]6C[C@@H](O[C@@H]6COP(O)(=S)O[C@H]6C[C@@H](O[C@@H]6COP(O)(=S)O[C@H]6C[C@@H](O[C@@H]6COP(O)(=S)O[C@H]6C[C@@H](O[C@@H]6COP(O)(=S)O[C@H]6C[C@@H](O[C@@H]6COP(O)(=S)O[C@H]6C[C@@H](O[C@@H]6COP(O)(=S)O[C@H]6C[C@@H](O[C@@H]6COP(O)(=S)O[C@@H]6[C@@H](COP(O)(=S)O[C@@H]7[C@@H](COP(O)(=S)O[C@@H]8[C@@H](COP(S)(=O)O[C@@H]9[C@@H](COP(O)(=S)O[C@@H]%10[C@@H](CO)O[C@H]([C@@H]%10OCCOC)n%10cc(C)c(=O)[nH]c%10=O)O[C@H]([C@@H]9OCCOC)n9cc(C)c(N)nc9=O)O[C@H]([C@@H]8OCCOC)n8cc(C)c(=O)[nH]c8=O)O[C@H]([C@@H]7OCCOC)n7cc(C)c(=O)[nH]c7=O)O[C@H]([C@@H]6OCCOC)n6cnc7c6nc(N)[nH]c7=O)n6cnc7c6nc(N)[nH]c7=O)n6cc(C)c(=O)[nH]c6=O)n6cc(C)c(=O)[nH]c6=O)n6cnc7c(N)ncnc67)n6cc(C)c(N)nc6=O)n6cnc7c(N)ncnc67)n6cc(C)c(=O)[nH]c6=O)n6cnc7c6nc(N)[nH]c7=O)n6cnc7c(N)ncnc67)n6cnc7c(N)ncnc67)O[C@H]([C@@H]5OCCOC)n5cnc6c(N)ncnc56)O[C@H]([C@@H]4OCCOC)n4cc(C)c(=O)[nH]c4=O)O[C@H]([C@@H]3OCCOC)n3cc(C)c(N)nc3=O)O[C@H]([C@@H]2OCCOC)n2cc(C)c(N)nc2=O)O[C@H]1n1cc(C)c(N)nc1=O KLEGMTRDCCDFJK-XDQSQZFTSA-N 0.000 description 2
- OTLLEIBWKHEHGU-UHFFFAOYSA-N 2-[5-[[5-(6-aminopurin-9-yl)-3,4-dihydroxyoxolan-2-yl]methoxy]-3,4-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-3,5-dihydroxy-4-phosphonooxyhexanedioic acid Chemical compound C1=NC=2C(N)=NC=NC=2N1C(C(C1O)O)OC1COC1C(CO)OC(OC(C(O)C(OP(O)(O)=O)C(O)C(O)=O)C(O)=O)C(O)C1O OTLLEIBWKHEHGU-UHFFFAOYSA-N 0.000 description 2
- 108010066676 Abrin Proteins 0.000 description 2
- 102100036475 Alanine aminotransferase 1 Human genes 0.000 description 2
- 108010082126 Alanine transaminase Proteins 0.000 description 2
- 231100000699 Bacterial toxin Toxicity 0.000 description 2
- 102100024217 CAMPATH-1 antigen Human genes 0.000 description 2
- 108010065524 CD52 Antigen Proteins 0.000 description 2
- 102100025475 Carcinoembryonic antigen-related cell adhesion molecule 5 Human genes 0.000 description 2
- 108010001857 Cell Surface Receptors Proteins 0.000 description 2
- 102000016607 Diphtheria Toxin Human genes 0.000 description 2
- 108010053187 Diphtheria Toxin Proteins 0.000 description 2
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 2
- 206010013801 Duchenne Muscular Dystrophy Diseases 0.000 description 2
- 102100024108 Dystrophin Human genes 0.000 description 2
- 241000711549 Hepacivirus C Species 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- 101000914324 Homo sapiens Carcinoembryonic antigen-related cell adhesion molecule 5 Proteins 0.000 description 2
- 101001012157 Homo sapiens Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 description 2
- 108060003951 Immunoglobulin Proteins 0.000 description 2
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 2
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 2
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 2
- 231100000742 Plant toxin Toxicity 0.000 description 2
- 206010036105 Polyneuropathy Diseases 0.000 description 2
- 241000589517 Pseudomonas aeruginosa Species 0.000 description 2
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 description 2
- 108091028664 Ribonucleotide Proteins 0.000 description 2
- 108090000829 Ribosome Inactivating Proteins Proteins 0.000 description 2
- 108010039491 Ricin Proteins 0.000 description 2
- 102100021947 Survival motor neuron protein Human genes 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 235000001014 amino acid Nutrition 0.000 description 2
- 150000001413 amino acids Chemical class 0.000 description 2
- 125000000129 anionic group Chemical group 0.000 description 2
- 229940125644 antibody drug Drugs 0.000 description 2
- 150000001541 aziridines Chemical class 0.000 description 2
- 239000000688 bacterial toxin Substances 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 239000004202 carbamide Substances 0.000 description 2
- 125000002091 cationic group Chemical group 0.000 description 2
- 229920006317 cationic polymer Polymers 0.000 description 2
- 230000022534 cell killing Effects 0.000 description 2
- 230000003833 cell viability Effects 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 125000003636 chemical group Chemical group 0.000 description 2
- 239000003184 complementary RNA Substances 0.000 description 2
- 230000003247 decreasing effect Effects 0.000 description 2
- 239000005547 deoxyribonucleotide Substances 0.000 description 2
- 125000002637 deoxyribonucleotide group Chemical group 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 230000018109 developmental process Effects 0.000 description 2
- KZNICNPSHKQLFF-UHFFFAOYSA-N dihydromaleimide Natural products O=C1CCC(=O)N1 KZNICNPSHKQLFF-UHFFFAOYSA-N 0.000 description 2
- 238000005906 dihydroxylation reaction Methods 0.000 description 2
- 150000002009 diols Chemical class 0.000 description 2
- 229960003668 docetaxel Drugs 0.000 description 2
- 238000007876 drug discovery Methods 0.000 description 2
- VQNATVDKACXKTF-XELLLNAOSA-N duocarmycin Chemical compound COC1=C(OC)C(OC)=C2NC(C(=O)N3C4=CC(=O)C5=C([C@@]64C[C@@H]6C3)C=C(N5)C(=O)OC)=CC2=C1 VQNATVDKACXKTF-XELLLNAOSA-N 0.000 description 2
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 description 2
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 description 2
- 238000006735 epoxidation reaction Methods 0.000 description 2
- 150000002118 epoxides Chemical class 0.000 description 2
- ZVYVPGLRVWUPMP-FYSMJZIKSA-N exatecan Chemical compound C1C[C@H](N)C2=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC3=CC(F)=C(C)C1=C32 ZVYVPGLRVWUPMP-FYSMJZIKSA-N 0.000 description 2
- 229950009429 exatecan Drugs 0.000 description 2
- 230000029142 excretion Effects 0.000 description 2
- 239000002095 exotoxin Substances 0.000 description 2
- 231100000776 exotoxin Toxicity 0.000 description 2
- 230000002068 genetic effect Effects 0.000 description 2
- 150000004820 halides Chemical class 0.000 description 2
- 125000005597 hydrazone group Chemical group 0.000 description 2
- 102000018358 immunoglobulin Human genes 0.000 description 2
- 239000002596 immunotoxin Substances 0.000 description 2
- 230000001939 inductive effect Effects 0.000 description 2
- 229950002218 inotersen Drugs 0.000 description 2
- 150000002500 ions Chemical class 0.000 description 2
- 150000002576 ketones Chemical class 0.000 description 2
- 210000005228 liver tissue Anatomy 0.000 description 2
- 230000002132 lysosomal effect Effects 0.000 description 2
- 102000006240 membrane receptors Human genes 0.000 description 2
- 229960005558 mertansine Drugs 0.000 description 2
- OSOOBMBDIGGTCP-UHFFFAOYSA-N miravirsen Chemical compound O=C1NC(=O)C(C)=CN1C1OC(COP(O)(=S)OC2C3(COP(O)(=S)OC4C(OC(C4)N4C(N=C(N)C=C4)=O)COP(O)(=S)OC4C5(CO)COC4C(O5)N4C(N=C(N)C=C4)=O)COC2C(O3)N2C3=NC=NC(N)=C3N=C2)C(OP(O)(=S)OCC2C(CC(O2)N2C(NC(=O)C(C)=C2)=O)OP(O)(=S)OCC23OC(C(OC2)C3OP(O)(=S)OCC23OC(C(OC2)C3OP(O)(=S)OCC2C(CC(O2)N2C(N=C(N)C=C2)=O)OP(O)(=S)OCC2C(CC(O2)N2C3=NC=NC(N)=C3N=C2)OP(O)(=S)OCC23OC(C(OC2)C3OP(O)(=S)OCC2C(CC(O2)N2C3=NC=NC(N)=C3N=C2)OP(O)(=S)OCC23OC(C(OC2)C3OP(O)(=S)OCC2C(CC(O2)N2C(NC(=O)C(C)=C2)=O)OP(O)(=S)OCC23OC(C(OC2)C3OP(O)(=S)OCC23C(C(OC2)C(O3)N2C(N=C(N)C=C2)=O)O)N2C(N=C(N)C=C2)=O)N2C(N=C(N)C=C2)=O)N2C(N=C(N)C=C2)=O)N2C(NC(=O)C(C)=C2)=O)N2C3=C(C(NC(N)=N3)=O)N=C2)C1 OSOOBMBDIGGTCP-UHFFFAOYSA-N 0.000 description 2
- 229950008922 miravirsen Drugs 0.000 description 2
- 108010022050 mistletoe lectin I Proteins 0.000 description 2
- 108010010621 modeccin Proteins 0.000 description 2
- 201000006938 muscular dystrophy Diseases 0.000 description 2
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 description 2
- SQDFHQJTAWCFIB-UHFFFAOYSA-N n-methylidenehydroxylamine Chemical compound ON=C SQDFHQJTAWCFIB-UHFFFAOYSA-N 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 125000004433 nitrogen atom Chemical group N* 0.000 description 2
- 108091027963 non-coding RNA Proteins 0.000 description 2
- 102000042567 non-coding RNA Human genes 0.000 description 2
- 230000003647 oxidation Effects 0.000 description 2
- 238000007254 oxidation reaction Methods 0.000 description 2
- 125000004430 oxygen atom Chemical group O* 0.000 description 2
- 230000001717 pathogenic effect Effects 0.000 description 2
- 229950005564 patisiran Drugs 0.000 description 2
- 239000003123 plant toxin Substances 0.000 description 2
- 230000007824 polyneuropathy Effects 0.000 description 2
- 229920001184 polypeptide Polymers 0.000 description 2
- 230000032361 posttranscriptional gene silencing Effects 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- 108020001580 protein domains Proteins 0.000 description 2
- 230000004853 protein function Effects 0.000 description 2
- YUOCYTRGANSSRY-UHFFFAOYSA-N pyrrolo[2,3-i][1,2]benzodiazepine Chemical compound C1=CN=NC2=C3C=CN=C3C=CC2=C1 YUOCYTRGANSSRY-UHFFFAOYSA-N 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 238000012552 review Methods 0.000 description 2
- 239000002336 ribonucleotide Substances 0.000 description 2
- 125000002652 ribonucleotide group Chemical group 0.000 description 2
- 125000000548 ribosyl group Chemical group C1([C@H](O)[C@H](O)[C@H](O1)CO)* 0.000 description 2
- 239000002924 silencing RNA Substances 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 239000003381 stabilizer Substances 0.000 description 2
- 229960002317 succinimide Drugs 0.000 description 2
- 229910052717 sulfur Inorganic materials 0.000 description 2
- ORFNVPGICPYLJV-YTVPMEHESA-N (2s)-2-[[(2r,3r)-3-[(2s)-1-[(3r,4s,5s)-4-[[(2s)-2-[[(2s)-2-[6-(2,5-dioxopyrrol-1-yl)hexanoyl-methylamino]-3-methylbutanoyl]amino]-3-methylbutanoyl]-methylamino]-3-methoxy-5-methylheptanoyl]pyrrolidin-2-yl]-3-methoxy-2-methylpropanoyl]amino]-3-phenylpropan Chemical compound C([C@H](NC(=O)[C@H](C)[C@@H](OC)[C@@H]1CCCN1C(=O)C[C@H]([C@H]([C@@H](C)CC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)CCCCCN1C(C=CC1=O)=O)C(C)C)OC)C(O)=O)C1=CC=CC=C1 ORFNVPGICPYLJV-YTVPMEHESA-N 0.000 description 1
- DLKUYSQUHXBYPB-NSSHGSRYSA-N (2s,4r)-4-[[2-[(1r,3r)-1-acetyloxy-4-methyl-3-[3-methylbutanoyloxymethyl-[(2s,3s)-3-methyl-2-[[(2r)-1-methylpiperidine-2-carbonyl]amino]pentanoyl]amino]pentyl]-1,3-thiazole-4-carbonyl]amino]-2-methyl-5-(4-methylphenyl)pentanoic acid Chemical compound N([C@@H]([C@@H](C)CC)C(=O)N(COC(=O)CC(C)C)[C@H](C[C@@H](OC(C)=O)C=1SC=C(N=1)C(=O)N[C@H](C[C@H](C)C(O)=O)CC=1C=CC(C)=CC=1)C(C)C)C(=O)[C@H]1CCCCN1C DLKUYSQUHXBYPB-NSSHGSRYSA-N 0.000 description 1
- GPXMKARIFPDQNT-UHFFFAOYSA-N (4-nitrophenyl) 3-acetylsulfanylpropanoate Chemical compound CC(=O)SCCC(=O)OC1=CC=C([N+]([O-])=O)C=C1 GPXMKARIFPDQNT-UHFFFAOYSA-N 0.000 description 1
- SVUOLADPCWQTTE-UHFFFAOYSA-N 1h-1,2-benzodiazepine Chemical compound N1N=CC=CC2=CC=CC=C12 SVUOLADPCWQTTE-UHFFFAOYSA-N 0.000 description 1
- FBUTXZSKZCQABC-UHFFFAOYSA-N 2-amino-1-methyl-7h-purine-6-thione Chemical compound S=C1N(C)C(N)=NC2=C1NC=N2 FBUTXZSKZCQABC-UHFFFAOYSA-N 0.000 description 1
- RMZNXRYIFGTWPF-UHFFFAOYSA-N 2-nitrosoacetic acid Chemical group OC(=O)CN=O RMZNXRYIFGTWPF-UHFFFAOYSA-N 0.000 description 1
- FJHBVJOVLFPMQE-QFIPXVFZSA-N 7-Ethyl-10-Hydroxy-Camptothecin Chemical compound C1=C(O)C=C2C(CC)=C(CN3C(C4=C([C@@](C(=O)OC4)(O)CC)C=C33)=O)C3=NC2=C1 FJHBVJOVLFPMQE-QFIPXVFZSA-N 0.000 description 1
- 102100031585 ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 Human genes 0.000 description 1
- 108700028369 Alleles Proteins 0.000 description 1
- 101800002638 Alpha-amanitin Proteins 0.000 description 1
- 108010027164 Amanitins Proteins 0.000 description 1
- 208000037259 Amyloid Plaque Diseases 0.000 description 1
- 102100022987 Angiogenin Human genes 0.000 description 1
- 102000006306 Antigen Receptors Human genes 0.000 description 1
- 108010083359 Antigen Receptors Proteins 0.000 description 1
- 108020005544 Antisense RNA Proteins 0.000 description 1
- 102100040214 Apolipoprotein(a) Human genes 0.000 description 1
- 108010012927 Apoprotein(a) Proteins 0.000 description 1
- 101710094856 Apoptin Proteins 0.000 description 1
- 102000008682 Argonaute Proteins Human genes 0.000 description 1
- 108010088141 Argonaute Proteins Proteins 0.000 description 1
- 102100038080 B-cell receptor CD22 Human genes 0.000 description 1
- 102100024222 B-lymphocyte antigen CD19 Human genes 0.000 description 1
- 102100022005 B-lymphocyte antigen CD20 Human genes 0.000 description 1
- 108010074708 B7-H1 Antigen Proteins 0.000 description 1
- 108091032955 Bacterial small RNA Proteins 0.000 description 1
- 102100028239 Basal cell adhesion molecule Human genes 0.000 description 1
- 102000004506 Blood Proteins Human genes 0.000 description 1
- 108010017384 Blood Proteins Proteins 0.000 description 1
- ZUHQCDZJPTXVCU-UHFFFAOYSA-N C1#CCCC2=CC=CC=C2C2=CC=CC=C21 Chemical group C1#CCCC2=CC=CC=C2C2=CC=CC=C21 ZUHQCDZJPTXVCU-UHFFFAOYSA-N 0.000 description 1
- 102100027207 CD27 antigen Human genes 0.000 description 1
- 101710115912 CD27 antigen Proteins 0.000 description 1
- 102000017420 CD3 protein, epsilon/gamma/delta subunit Human genes 0.000 description 1
- 108050005493 CD3 protein, epsilon/gamma/delta subunit Proteins 0.000 description 1
- 108010058905 CD44v6 antigen Proteins 0.000 description 1
- 102100025222 CD63 antigen Human genes 0.000 description 1
- 102100025221 CD70 antigen Human genes 0.000 description 1
- 241000244203 Caenorhabditis elegans Species 0.000 description 1
- 102100036369 Carbonic anhydrase 6 Human genes 0.000 description 1
- 102100025473 Carcinoembryonic antigen-related cell adhesion molecule 6 Human genes 0.000 description 1
- 102100023126 Cell surface glycoprotein MUC18 Human genes 0.000 description 1
- 102000009016 Cholera Toxin Human genes 0.000 description 1
- 108010049048 Cholera Toxin Proteins 0.000 description 1
- 208000006154 Chronic hepatitis C Diseases 0.000 description 1
- 108020004394 Complementary RNA Proteins 0.000 description 1
- 108010051219 Cre recombinase Proteins 0.000 description 1
- 229930188224 Cryptophycin Natural products 0.000 description 1
- 229920000858 Cyclodextrin Polymers 0.000 description 1
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- 102100039498 Cytotoxic T-lymphocyte protein 4 Human genes 0.000 description 1
- AVGPOAXYRRIZMM-UHFFFAOYSA-N D-Apiose Natural products OCC(O)(CO)C(O)C=O AVGPOAXYRRIZMM-UHFFFAOYSA-N 0.000 description 1
- ASNHGEVAWNWCRQ-LJJLCWGRSA-N D-apiofuranose Chemical compound OC[C@@]1(O)COC(O)[C@@H]1O ASNHGEVAWNWCRQ-LJJLCWGRSA-N 0.000 description 1
- ASNHGEVAWNWCRQ-UHFFFAOYSA-N D-apiofuranose Natural products OCC1(O)COC(O)C1O ASNHGEVAWNWCRQ-UHFFFAOYSA-N 0.000 description 1
- 102100036466 Delta-like protein 3 Human genes 0.000 description 1
- 108010069091 Dystrophin Proteins 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 108010066687 Epithelial Cell Adhesion Molecule Proteins 0.000 description 1
- 102000018651 Epithelial Cell Adhesion Molecule Human genes 0.000 description 1
- 108700024394 Exon Proteins 0.000 description 1
- 101710082714 Exotoxin A Proteins 0.000 description 1
- 102000010834 Extracellular Matrix Proteins Human genes 0.000 description 1
- 108010037362 Extracellular Matrix Proteins Proteins 0.000 description 1
- 108090000379 Fibroblast growth factor 2 Proteins 0.000 description 1
- 102000003974 Fibroblast growth factor 2 Human genes 0.000 description 1
- 102100027842 Fibroblast growth factor receptor 3 Human genes 0.000 description 1
- 101710182396 Fibroblast growth factor receptor 3 Proteins 0.000 description 1
- 102000010451 Folate receptor alpha Human genes 0.000 description 1
- 108050001931 Folate receptor alpha Proteins 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- 101710088083 Glomulin Proteins 0.000 description 1
- 102100041003 Glutamate carboxypeptidase 2 Human genes 0.000 description 1
- 102000003886 Glycoproteins Human genes 0.000 description 1
- 108090000288 Glycoproteins Proteins 0.000 description 1
- 102000001398 Granzyme Human genes 0.000 description 1
- 108060005986 Granzyme Proteins 0.000 description 1
- 102100030595 HLA class II histocompatibility antigen gamma chain Human genes 0.000 description 1
- 101000777636 Homo sapiens ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 Proteins 0.000 description 1
- 101000884305 Homo sapiens B-cell receptor CD22 Proteins 0.000 description 1
- 101000980825 Homo sapiens B-lymphocyte antigen CD19 Proteins 0.000 description 1
- 101000897405 Homo sapiens B-lymphocyte antigen CD20 Proteins 0.000 description 1
- 101000935638 Homo sapiens Basal cell adhesion molecule Proteins 0.000 description 1
- 101000934368 Homo sapiens CD63 antigen Proteins 0.000 description 1
- 101000934356 Homo sapiens CD70 antigen Proteins 0.000 description 1
- 101000714525 Homo sapiens Carbonic anhydrase 6 Proteins 0.000 description 1
- 101000914326 Homo sapiens Carcinoembryonic antigen-related cell adhesion molecule 6 Proteins 0.000 description 1
- 101000914321 Homo sapiens Carcinoembryonic antigen-related cell adhesion molecule 7 Proteins 0.000 description 1
- 101000623903 Homo sapiens Cell surface glycoprotein MUC18 Proteins 0.000 description 1
- 101000889276 Homo sapiens Cytotoxic T-lymphocyte protein 4 Proteins 0.000 description 1
- 101000928513 Homo sapiens Delta-like protein 3 Proteins 0.000 description 1
- 101000892862 Homo sapiens Glutamate carboxypeptidase 2 Proteins 0.000 description 1
- 101001082627 Homo sapiens HLA class II histocompatibility antigen gamma chain Proteins 0.000 description 1
- 101000606465 Homo sapiens Inactive tyrosine-protein kinase 7 Proteins 0.000 description 1
- 101001057504 Homo sapiens Interferon-stimulated gene 20 kDa protein Proteins 0.000 description 1
- 101001055144 Homo sapiens Interleukin-2 receptor subunit alpha Proteins 0.000 description 1
- 101000998120 Homo sapiens Interleukin-3 receptor subunit alpha Proteins 0.000 description 1
- 101001038507 Homo sapiens Ly6/PLAUR domain-containing protein 3 Proteins 0.000 description 1
- 101001133056 Homo sapiens Mucin-1 Proteins 0.000 description 1
- 101000623901 Homo sapiens Mucin-16 Proteins 0.000 description 1
- 101000934338 Homo sapiens Myeloid cell surface antigen CD33 Proteins 0.000 description 1
- 101000581981 Homo sapiens Neural cell adhesion molecule 1 Proteins 0.000 description 1
- 101001024605 Homo sapiens Next to BRCA1 gene 1 protein Proteins 0.000 description 1
- 101001126417 Homo sapiens Platelet-derived growth factor receptor alpha Proteins 0.000 description 1
- 101000617725 Homo sapiens Pregnancy-specific beta-1-glycoprotein 2 Proteins 0.000 description 1
- 101000932478 Homo sapiens Receptor-type tyrosine-protein kinase FLT3 Proteins 0.000 description 1
- 101000633786 Homo sapiens SLAM family member 6 Proteins 0.000 description 1
- 101000874179 Homo sapiens Syndecan-1 Proteins 0.000 description 1
- 101000914496 Homo sapiens T-cell antigen CD7 Proteins 0.000 description 1
- 101000851376 Homo sapiens Tumor necrosis factor receptor superfamily member 8 Proteins 0.000 description 1
- 101000851018 Homo sapiens Vascular endothelial growth factor receptor 1 Proteins 0.000 description 1
- 101000851007 Homo sapiens Vascular endothelial growth factor receptor 2 Proteins 0.000 description 1
- 102100039813 Inactive tyrosine-protein kinase 7 Human genes 0.000 description 1
- 108010040765 Integrin alphaV Proteins 0.000 description 1
- 108010047852 Integrin alphaVbeta3 Proteins 0.000 description 1
- 102100026878 Interleukin-2 receptor subunit alpha Human genes 0.000 description 1
- 102100033493 Interleukin-3 receptor subunit alpha Human genes 0.000 description 1
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 1
- 239000000232 Lipid Bilayer Substances 0.000 description 1
- 102100040281 Ly6/PLAUR domain-containing protein 3 Human genes 0.000 description 1
- 108090000015 Mesothelin Proteins 0.000 description 1
- 102000003735 Mesothelin Human genes 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- 102100034256 Mucin-1 Human genes 0.000 description 1
- 102100023123 Mucin-16 Human genes 0.000 description 1
- 101100369076 Mus musculus Tdgf1 gene Proteins 0.000 description 1
- 206010028289 Muscle atrophy Diseases 0.000 description 1
- 102100025243 Myeloid cell surface antigen CD33 Human genes 0.000 description 1
- 102100027347 Neural cell adhesion molecule 1 Human genes 0.000 description 1
- 102000001760 Notch3 Receptor Human genes 0.000 description 1
- 108010029756 Notch3 Receptor Proteins 0.000 description 1
- 101710163270 Nuclease Proteins 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- 102000002508 Peptide Elongation Factors Human genes 0.000 description 1
- 108010068204 Peptide Elongation Factors Proteins 0.000 description 1
- 102100030485 Platelet-derived growth factor receptor alpha Human genes 0.000 description 1
- 241000243142 Porifera Species 0.000 description 1
- 102100024216 Programmed cell death 1 ligand 1 Human genes 0.000 description 1
- 108010044159 Proprotein Convertases Proteins 0.000 description 1
- 102000006437 Proprotein Convertases Human genes 0.000 description 1
- 102100029986 Receptor tyrosine-protein kinase erbB-3 Human genes 0.000 description 1
- 101710100969 Receptor tyrosine-protein kinase erbB-3 Proteins 0.000 description 1
- 102100020718 Receptor-type tyrosine-protein kinase FLT3 Human genes 0.000 description 1
- OWPCHSCAPHNHAV-UHFFFAOYSA-N Rhizoxin Natural products C1C(O)C2(C)OC2C=CC(C)C(OC(=O)C2)CC2CC2OC2C(=O)OC1C(C)C(OC)C(C)=CC=CC(C)=CC1=COC(C)=N1 OWPCHSCAPHNHAV-UHFFFAOYSA-N 0.000 description 1
- 108010057163 Ribonuclease III Proteins 0.000 description 1
- 102000003661 Ribonuclease III Human genes 0.000 description 1
- 102000006382 Ribonucleases Human genes 0.000 description 1
- 108010083644 Ribonucleases Proteins 0.000 description 1
- PYMYPHUHKUWMLA-LMVFSUKVSA-N Ribose Natural products OC[C@@H](O)[C@@H](O)[C@@H](O)C=O PYMYPHUHKUWMLA-LMVFSUKVSA-N 0.000 description 1
- WPDOZYZAJKUVRZ-NRYSMURASA-N S-[(2R,3S,4S,6S)-6-[[(2R,3S,4S,5R,6R)-5-[(2S,4S,5S)-5-[acetyl(ethyl)amino]-4-methoxyoxan-2-yl]oxy-4-hydroxy-6-[[(2S,5Z,9R)-9-hydroxy-12-(methoxycarbonylamino)-13-[2-(methyltrisulfanyl)ethylidene]-11-oxo-2-bicyclo[7.3.1]trideca-1(12),5-dien-3,7-diynyl]oxy]-2-methyloxan-3-yl]amino]oxy-4-hydroxy-2-methyloxan-3-yl] 4-[(2S,3R,4R,5S,6S)-3,5-dihydroxy-4-methoxy-6-methyloxan-2-yl]oxy-5-iodo-2,3-dimethoxy-6-methylbenzenecarbothioate Chemical compound CCN([C@H]1CO[C@H](C[C@@H]1OC)O[C@@H]1[C@@H](O)[C@H](NO[C@H]2C[C@H](O)[C@H](SC(=O)c3c(C)c(I)c(O[C@@H]4O[C@@H](C)[C@H](O)[C@@H](OC)[C@H]4O)c(OC)c3OC)[C@@H](C)O2)[C@@H](C)O[C@H]1O[C@H]1C#C\C=C/C#C[C@]2(O)CC(=O)C(NC(=O)OC)=C1C2=CCSSSC)C(C)=O WPDOZYZAJKUVRZ-NRYSMURASA-N 0.000 description 1
- RXGJTYFDKOHJHK-UHFFFAOYSA-N S-deoxo-amaninamide Natural products CCC(C)C1NC(=O)CNC(=O)C2Cc3c(SCC(NC(=O)CNC1=O)C(=O)NC(CC(=O)N)C(=O)N4CC(O)CC4C(=O)NC(C(C)C(O)CO)C(=O)N2)[nH]c5ccccc35 RXGJTYFDKOHJHK-UHFFFAOYSA-N 0.000 description 1
- 102100029197 SLAM family member 6 Human genes 0.000 description 1
- 108091081021 Sense strand Proteins 0.000 description 1
- 108010079723 Shiga Toxin Proteins 0.000 description 1
- 101710084578 Short neurotoxin 1 Proteins 0.000 description 1
- 108010003723 Single-Domain Antibodies Proteins 0.000 description 1
- 102000003705 Syndecan-1 Human genes 0.000 description 1
- 102100035721 Syndecan-1 Human genes 0.000 description 1
- 108090000058 Syndecan-1 Proteins 0.000 description 1
- 102100027208 T-cell antigen CD7 Human genes 0.000 description 1
- 101710182532 Toxin a Proteins 0.000 description 1
- 108090000704 Tubulin Proteins 0.000 description 1
- 102000004243 Tubulin Human genes 0.000 description 1
- 102100036857 Tumor necrosis factor receptor superfamily member 8 Human genes 0.000 description 1
- 108010061323 Type 2 Ribosome Inactivating Proteins Proteins 0.000 description 1
- 206010045261 Type IIa hyperlipidaemia Diseases 0.000 description 1
- 108010046334 Urease Proteins 0.000 description 1
- 108010069201 VLDL Cholesterol Proteins 0.000 description 1
- 102100033178 Vascular endothelial growth factor receptor 1 Human genes 0.000 description 1
- 102100033177 Vascular endothelial growth factor receptor 2 Human genes 0.000 description 1
- 101800004982 Volkensin A chain Proteins 0.000 description 1
- HKGATZAPXCCEJR-OWRSNIELSA-N [4-[[(2s)-2-[[(2s)-2-[3-[2-[2-[2-[2-[2-[2-[2-[2-[3-(2,5-dioxopyrrol-1-yl)propanoylamino]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]ethoxy]propanoylamino]-3-methylbutanoyl]amino]propanoyl]amino]phenyl]methyl (6s,6as)-3-[5-[[(6as)-2-methoxy-8-methyl-1 Chemical compound N([C@H](C(=O)N[C@@H](C)C(=O)NC1=CC=C(C=C1)COC(=O)N1C=2C=C(C(=CC=2C(=O)N2C=C(C)C[C@H]2[C@@H]1O)OC)OCCCCCOC1=CC2=C(C(N3C=C(C)C[C@H]3C=N2)=O)C=C1OC)C(C)C)C(=O)CCOCCOCCOCCOCCOCCOCCOCCOCCNC(=O)CCN1C(=O)C=CC1=O HKGATZAPXCCEJR-OWRSNIELSA-N 0.000 description 1
- JNWFIPVDEINBAI-UHFFFAOYSA-N [5-hydroxy-4-[4-(1-methylindol-5-yl)-5-oxo-1H-1,2,4-triazol-3-yl]-2-propan-2-ylphenyl] dihydrogen phosphate Chemical compound C1=C(OP(O)(O)=O)C(C(C)C)=CC(C=2N(C(=O)NN=2)C=2C=C3C=CN(C)C3=CC=2)=C1O JNWFIPVDEINBAI-UHFFFAOYSA-N 0.000 description 1
- DHKHKXVYLBGOIT-UHFFFAOYSA-N acetaldehyde Diethyl Acetal Natural products CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 1
- 150000001241 acetals Chemical class 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- 239000004007 alpha amanitin Substances 0.000 description 1
- HMFHBZSHGGEWLO-UHFFFAOYSA-N alpha-D-Furanose-Ribose Natural products OCC1OC(O)C(O)C1O HMFHBZSHGGEWLO-UHFFFAOYSA-N 0.000 description 1
- CIORWBWIBBPXCG-SXZCQOKQSA-N alpha-amanitin Chemical compound O=C1N[C@@H](CC(N)=O)C(=O)N2C[C@H](O)C[C@H]2C(=O)N[C@@H]([C@@H](C)[C@@H](O)CO)C(=O)N[C@@H](C2)C(=O)NCC(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@H]1C[S@@](=O)C1=C2C2=CC=C(O)C=C2N1 CIORWBWIBBPXCG-SXZCQOKQSA-N 0.000 description 1
- CIORWBWIBBPXCG-UHFFFAOYSA-N alpha-amanitin Natural products O=C1NC(CC(N)=O)C(=O)N2CC(O)CC2C(=O)NC(C(C)C(O)CO)C(=O)NC(C2)C(=O)NCC(=O)NC(C(C)CC)C(=O)NCC(=O)NC1CS(=O)C1=C2C2=CC=C(O)C=C2N1 CIORWBWIBBPXCG-UHFFFAOYSA-N 0.000 description 1
- 108010001818 alpha-sarcin Proteins 0.000 description 1
- CIORWBWIBBPXCG-JZTFPUPKSA-N amanitin Chemical compound O=C1N[C@@H](CC(N)=O)C(=O)N2CC(O)C[C@H]2C(=O)N[C@@H](C(C)[C@@H](O)CO)C(=O)N[C@@H](C2)C(=O)NCC(=O)N[C@@H](C(C)CC)C(=O)NCC(=O)N[C@H]1CS(=O)C1=C2C2=CC=C(O)C=C2N1 CIORWBWIBBPXCG-JZTFPUPKSA-N 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 108010072788 angiogenin Proteins 0.000 description 1
- 150000001450 anions Chemical class 0.000 description 1
- 229940045799 anthracyclines and related substance Drugs 0.000 description 1
- 108091034201 anti-miRNA oligonucleotide Proteins 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 210000001367 artery Anatomy 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 208000025341 autosomal recessive disease Diseases 0.000 description 1
- IVRMZWNICZWHMI-UHFFFAOYSA-N azide group Chemical group [N-]=[N+]=[N-] IVRMZWNICZWHMI-UHFFFAOYSA-N 0.000 description 1
- 229940049706 benzodiazepine Drugs 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 239000002981 blocking agent Substances 0.000 description 1
- 230000037058 blood plasma level Effects 0.000 description 1
- 210000004204 blood vessel Anatomy 0.000 description 1
- HXCHCVDVKSCDHU-LULTVBGHSA-N calicheamicin Chemical compound C1[C@H](OC)[C@@H](NCC)CO[C@H]1O[C@H]1[C@H](O[C@@H]2C\3=C(NC(=O)OC)C(=O)C[C@](C/3=C/CSSSC)(O)C#C\C=C/C#C2)O[C@H](C)[C@@H](NO[C@@H]2O[C@H](C)[C@@H](SC(=O)C=3C(=C(OC)C(O[C@H]4[C@@H]([C@H](OC)[C@@H](O)[C@H](C)O4)O)=C(I)C=3C)OC)[C@@H](O)C2)[C@@H]1O HXCHCVDVKSCDHU-LULTVBGHSA-N 0.000 description 1
- 229930195731 calicheamicin Natural products 0.000 description 1
- VSJKWCGYPAHWDS-FQEVSTJZSA-N camptothecin Chemical class C1=CC=C2C=C(CN3C4=CC5=C(C3=O)COC(=O)[C@]5(O)CC)C4=NC2=C1 VSJKWCGYPAHWDS-FQEVSTJZSA-N 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 230000006369 cell cycle progression Effects 0.000 description 1
- 230000015861 cell surface binding Effects 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 230000005754 cellular signaling Effects 0.000 description 1
- 230000007541 cellular toxicity Effects 0.000 description 1
- 230000004700 cellular uptake Effects 0.000 description 1
- 229960005395 cetuximab Drugs 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 1
- 108010006226 cryptophycin Proteins 0.000 description 1
- PSNOPSMXOBPNNV-VVCTWANISA-N cryptophycin 1 Chemical compound C1=C(Cl)C(OC)=CC=C1C[C@@H]1C(=O)NC[C@@H](C)C(=O)O[C@@H](CC(C)C)C(=O)O[C@H]([C@H](C)[C@@H]2[C@H](O2)C=2C=CC=CC=2)C/C=C/C(=O)N1 PSNOPSMXOBPNNV-VVCTWANISA-N 0.000 description 1
- PSNOPSMXOBPNNV-UHFFFAOYSA-N cryptophycin-327 Natural products C1=C(Cl)C(OC)=CC=C1CC1C(=O)NCC(C)C(=O)OC(CC(C)C)C(=O)OC(C(C)C2C(O2)C=2C=CC=CC=2)CC=CC(=O)N1 PSNOPSMXOBPNNV-UHFFFAOYSA-N 0.000 description 1
- 229940097362 cyclodextrins Drugs 0.000 description 1
- 229960004397 cyclophosphamide Drugs 0.000 description 1
- 230000009089 cytolysis Effects 0.000 description 1
- 210000000805 cytoplasm Anatomy 0.000 description 1
- 231100000433 cytotoxic Toxicity 0.000 description 1
- 230000001472 cytotoxic effect Effects 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 238000006114 decarboxylation reaction Methods 0.000 description 1
- 229960004120 defibrotide Drugs 0.000 description 1
- 229940124447 delivery agent Drugs 0.000 description 1
- 229940127276 delta-like ligand 3 Drugs 0.000 description 1
- 206010012601 diabetes mellitus Diseases 0.000 description 1
- 230000029087 digestion Effects 0.000 description 1
- 230000003292 diminished effect Effects 0.000 description 1
- WQLVFSAGQJTQCK-UHFFFAOYSA-N diosgenin Natural products CC1C(C2(CCC3C4(C)CCC(O)CC4=CCC3C2C2)C)C2OC11CCC(C)CO1 WQLVFSAGQJTQCK-UHFFFAOYSA-N 0.000 description 1
- 231100000676 disease causative agent Toxicity 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 125000002228 disulfide group Chemical group 0.000 description 1
- 229960004679 doxorubicin Drugs 0.000 description 1
- 238000009510 drug design Methods 0.000 description 1
- 229960005501 duocarmycin Drugs 0.000 description 1
- 229930184221 duocarmycin Natural products 0.000 description 1
- 230000002526 effect on cardiovascular system Effects 0.000 description 1
- 230000002121 endocytic effect Effects 0.000 description 1
- 230000003511 endothelial effect Effects 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 1
- 229960005420 etoposide Drugs 0.000 description 1
- 230000028023 exocytosis Effects 0.000 description 1
- 210000002744 extracellular matrix Anatomy 0.000 description 1
- 210000003414 extremity Anatomy 0.000 description 1
- 238000010363 gene targeting Methods 0.000 description 1
- 238000010362 genome editing Methods 0.000 description 1
- 229930182470 glycoside Natural products 0.000 description 1
- 150000002373 hemiacetals Chemical class 0.000 description 1
- 208000018645 hepatic veno-occlusive disease Diseases 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 150000002466 imines Chemical class 0.000 description 1
- 230000036039 immunity Effects 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 230000028709 inflammatory response Effects 0.000 description 1
- 238000003780 insertion Methods 0.000 description 1
- 230000037431 insertion Effects 0.000 description 1
- 108010044426 integrins Proteins 0.000 description 1
- 102000006495 integrins Human genes 0.000 description 1
- 230000002452 interceptive effect Effects 0.000 description 1
- 230000010468 interferon response Effects 0.000 description 1
- 230000002147 killing effect Effects 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 230000003908 liver function Effects 0.000 description 1
- 230000007774 longterm Effects 0.000 description 1
- 230000006674 lysosomal degradation Effects 0.000 description 1
- 229920002521 macromolecule Polymers 0.000 description 1
- FSQQTNAZHBEJLS-UPHRSURJSA-N maleamic acid Chemical compound NC(=O)\C=C/C(O)=O FSQQTNAZHBEJLS-UPHRSURJSA-N 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 229960000485 methotrexate Drugs 0.000 description 1
- 125000000250 methylamino group Chemical group [H]N(*)C([H])([H])[H] 0.000 description 1
- 108091070501 miRNA Proteins 0.000 description 1
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 1
- 229960001156 mitoxantrone Drugs 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000009456 molecular mechanism Effects 0.000 description 1
- 108010093470 monomethyl auristatin E Proteins 0.000 description 1
- 108010059074 monomethylauristatin F Proteins 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 230000020763 muscle atrophy Effects 0.000 description 1
- 201000000585 muscular atrophy Diseases 0.000 description 1
- 238000002703 mutagenesis Methods 0.000 description 1
- 231100000350 mutagenesis Toxicity 0.000 description 1
- 239000002636 mycotoxin Substances 0.000 description 1
- MBLBDJOUHNCFQT-OSMVPFSASA-N n-[(2r,3r,4r,5r)-3,4,5,6-tetrahydroxy-1-oxohexan-2-yl]acetamide Chemical compound CC(=O)N[C@@H](C=O)[C@@H](O)[C@@H](O)[C@H](O)CO MBLBDJOUHNCFQT-OSMVPFSASA-N 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 239000002547 new drug Substances 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 150000002923 oximes Chemical class 0.000 description 1
- 229950007318 ozogamicin Drugs 0.000 description 1
- 230000020477 pH reduction Effects 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- 239000000123 paper Substances 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 244000052769 pathogen Species 0.000 description 1
- 210000002856 peripheral neuron Anatomy 0.000 description 1
- 150000008300 phosphoramidites Chemical class 0.000 description 1
- 230000035479 physiological effects, processes and functions Effects 0.000 description 1
- 239000013612 plasmid Substances 0.000 description 1
- 108700028325 pokeweed antiviral Proteins 0.000 description 1
- 229920001481 poly(stearyl methacrylate) Polymers 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 108091007428 primary miRNA Proteins 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- UOWVMDUEMSNCAV-WYENRQIDSA-N rachelmycin Chemical class C1([C@]23C[C@@H]2CN1C(=O)C=1NC=2C(OC)=C(O)C4=C(C=2C=1)CCN4C(=O)C1=CC=2C=4CCN(C=4C(O)=C(C=2N1)OC)C(N)=O)=CC(=O)C1=C3C(C)=CN1 UOWVMDUEMSNCAV-WYENRQIDSA-N 0.000 description 1
- 230000010837 receptor-mediated endocytosis Effects 0.000 description 1
- OWPCHSCAPHNHAV-LMONGJCWSA-N rhizoxin Chemical compound C/C([C@H](OC)[C@@H](C)[C@@H]1C[C@H](O)[C@]2(C)O[C@@H]2/C=C/[C@@H](C)[C@]2([H])OC(=O)C[C@@](C2)(C[C@@H]2O[C@H]2C(=O)O1)[H])=C\C=C\C(\C)=C\C1=COC(C)=N1 OWPCHSCAPHNHAV-LMONGJCWSA-N 0.000 description 1
- HNMATTJJEPZZMM-BPKVFSPJSA-N s-[(2r,3s,4s,6s)-6-[[(2r,3s,4s,5r,6r)-5-[(2s,4s,5s)-5-[acetyl(ethyl)amino]-4-methoxyoxan-2-yl]oxy-6-[[(2s,5z,9r,13e)-13-[2-[[4-[(2e)-2-[1-[4-(4-amino-4-oxobutoxy)phenyl]ethylidene]hydrazinyl]-2-methyl-4-oxobutan-2-yl]disulfanyl]ethylidene]-9-hydroxy-12-(m Chemical compound C1[C@H](OC)[C@@H](N(CC)C(C)=O)CO[C@H]1O[C@H]1[C@H](O[C@@H]2C\3=C(NC(=O)OC)C(=O)C[C@@](C/3=C/CSSC(C)(C)CC(=O)N\N=C(/C)C=3C=CC(OCCCC(N)=O)=CC=3)(O)C#C\C=C/C#C2)O[C@H](C)[C@@H](NO[C@@H]2O[C@H](C)[C@@H](SC(=O)C=3C(=C(OC)C(O[C@H]4[C@@H]([C@H](OC)[C@@H](O)[C@H](C)O4)O)=C(I)C=3C)OC)[C@@H](O)C2)[C@@H]1O HNMATTJJEPZZMM-BPKVFSPJSA-N 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- NWMIYTWHUDFRPL-UHFFFAOYSA-N sapogenin Natural products COC(=O)C1(CO)C(O)CCC2(C)C1CCC3(C)C2CC=C4C5C(C)(O)C(C)CCC5(CCC34C)C(=O)O NWMIYTWHUDFRPL-UHFFFAOYSA-N 0.000 description 1
- 150000003335 secondary amines Chemical class 0.000 description 1
- 125000005629 sialic acid group Chemical group 0.000 description 1
- 230000022379 skeletal muscle tissue development Effects 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- XNZLMZRIGGHITK-VKXBZTRUSA-N soravtansine Chemical compound CO[C@@H]([C@@]1(O)C[C@H](OC(=O)N1)[C@@H](C)[C@@H]1O[C@@]1(C)[C@@H](OC(=O)[C@H](C)N(C)C(=O)CCC(C)(C)SSCC[C@@H](C(O)=O)S(O)(=O)=O)CC(=O)N1C)\C=C\C=C(C)\CC2=CC(OC)=C(Cl)C1=C2.CO[C@@H]([C@@]1(O)C[C@H](OC(=O)N1)[C@@H](C)[C@@H]1O[C@@]1(C)[C@@H](OC(=O)[C@H](C)N(C)C(=O)CCC(C)(C)SSCC[C@H](C(O)=O)S(O)(=O)=O)CC(=O)N1C)\C=C\C=C(C)\CC2=CC(OC)=C(Cl)C1=C2 XNZLMZRIGGHITK-VKXBZTRUSA-N 0.000 description 1
- 210000000952 spleen Anatomy 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 230000001839 systemic circulation Effects 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- CXWPTNOTDDVZHE-RNFDLKLBSA-A tetradecasodium 1-[(2R,4S,5R)-4-[[(2R,3S,5R)-3-[[(1S,3R,4R,7S)-7-[[(1S,3R,4R,7S)-7-[[(2R,3S,5R)-3-[[(2R,3S,5R)-3-[[(1S,3R,4R,7S)-3-(4-amino-5-methyl-2-oxopyrimidin-1-yl)-7-[[(2R,3S,5R)-3-[[(1S,3R,4R,7S)-3-(4-amino-5-methyl-2-oxopyrimidin-1-yl)-7-[[(2R,3S,5R)-3-[[(1S,3R,4R,7S)-3-(4-amino-5-methyl-2-oxopyrimidin-1-yl)-7-[[(1S,3R,4R,7S)-3-(4-amino-5-methyl-2-oxopyrimidin-1-yl)-7-hydroxy-2,5-dioxabicyclo[2.2.1]heptan-1-yl]methoxy-oxidophosphinothioyl]oxy-2,5-dioxabicyclo[2.2.1]heptan-1-yl]methoxy-oxidophosphinothioyl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-oxidophosphinothioyl]oxy-2,5-dioxabicyclo[2.2.1]heptan-1-yl]methoxy-oxidophosphinothioyl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-oxidophosphinothioyl]oxy-2,5-dioxabicyclo[2.2.1]heptan-1-yl]methoxy-oxidophosphinothioyl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-oxidophosphinothioyl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-oxidophosphinothioyl]oxy-3-(5-methyl-2,4-dioxopyrimidin-1-yl)-2,5-dioxabicyclo[2.2.1]heptan-1-yl]methoxy-oxidophosphinothioyl]oxy-3-(2-amino-6-oxo-1H-purin-9-yl)-2,5-dioxabicyclo[2.2.1]heptan-1-yl]methoxy-oxidophosphinothioyl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-oxidophosphinothioyl]oxy-5-[[[(1S,3R,4R,7S)-1-[[[(2R,3S,5R)-2-[[[(1R,3R,4R,7S)-3-(4-amino-5-methyl-2-oxopyrimidin-1-yl)-1-(hydroxymethyl)-2,5-dioxabicyclo[2.2.1]heptan-7-yl]oxy-oxidophosphinothioyl]oxymethyl]-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-3-yl]oxy-sulfidophosphoryl]oxymethyl]-3-(6-aminopurin-9-yl)-2,5-dioxabicyclo[2.2.1]heptan-7-yl]oxy-oxidophosphinothioyl]oxymethyl]oxolan-2-yl]-5-methylpyrimidine-2,4-dione Chemical compound [Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].[Na+].Cc1cn([C@@H]2O[C@]3(COP([O-])(=S)O[C@H]4[C@H]5OC[C@@]4(COP([O-])(=S)O[C@H]4C[C@@H](O[C@@H]4COP([O-])(=S)O[C@H]4[C@H]6OC[C@@]4(COP([O-])(=S)O[C@H]4C[C@@H](O[C@@H]4COP([O-])(=S)O[C@H]4[C@H]7OC[C@@]4(COP([O-])(=S)O[C@H]4C[C@@H](O[C@@H]4COP([O-])(=S)O[C@H]4C[C@@H](O[C@@H]4COP([O-])(=S)O[C@H]4[C@H]8OC[C@@]4(COP([O-])(=S)O[C@H]4[C@H]9OC[C@@]4(COP([O-])(=S)O[C@H]4C[C@@H](O[C@@H]4COP([O-])(=S)O[C@H]4C[C@@H](O[C@@H]4COP([O-])(=S)O[C@H]4[C@H]%10OC[C@@]4(COP([S-])(=O)O[C@H]4C[C@@H](O[C@@H]4COP([O-])(=S)O[C@H]4[C@H]%11OC[C@@]4(CO)O[C@H]%11n4cc(C)c(N)nc4=O)n4ccc(N)nc4=O)O[C@H]%10n4cnc%10c(N)ncnc4%10)n4cc(C)c(=O)[nH]c4=O)n4cc(C)c(=O)[nH]c4=O)O[C@H]9n4cnc9c4nc(N)[nH]c9=O)O[C@H]8n4cc(C)c(=O)[nH]c4=O)n4ccc(N)nc4=O)n4cnc8c(N)ncnc48)O[C@H]7n4cc(C)c(N)nc4=O)n4cnc7c(N)ncnc47)O[C@H]6n4cc(C)c(N)nc4=O)n4cc(C)c(=O)[nH]c4=O)O[C@H]5n4cc(C)c(N)nc4=O)CO[C@@H]2[C@@H]3O)c(=O)nc1N CXWPTNOTDDVZHE-RNFDLKLBSA-A 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 238000010361 transduction Methods 0.000 description 1
- 230000026683 transduction Effects 0.000 description 1
- 238000013519 translation Methods 0.000 description 1
- 230000032258 transport Effects 0.000 description 1
- 229960000575 trastuzumab Drugs 0.000 description 1
- 229930184737 tubulysin Natural products 0.000 description 1
- 230000002792 vascular Effects 0.000 description 1
- 210000003462 vein Anatomy 0.000 description 1
- 108010043904 volkensin Proteins 0.000 description 1
- KQNNSYZQMSOOQH-GLDAUDTLSA-N volkensin Chemical compound C=1([C@@H]2C[C@@H]3O[C@@H](O)C[C@@H]4[C@]5(C)[C@H]6[C@H]([C@H]([C@@]4(C)C3=C2C)O)OC[C@]6(C)[C@H](OC(C)=O)C[C@@H]5OC(=O)C(/C)=C/C)C=COC=1 KQNNSYZQMSOOQH-GLDAUDTLSA-N 0.000 description 1
- 230000036642 wellbeing Effects 0.000 description 1
- 229960005502 α-amanitin Drugs 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
- A61K47/55—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound the modifying agent being also a pharmacologically or therapeutically active agent, i.e. the entire conjugate being a codrug, i.e. a dimer, oligomer or polymer of pharmacologically or therapeutically active compounds
Definitions
- the invention relates to an oligonucleotide conjugate comprising a saponin covalently linked to a ligand for ASGPR (ASGPR ligand) and further comprising a covalently linked oligonucleotide, i.e.
- a (saponin, oligonucleotide, ASGPR ligand) conjugate comprising a saponin covalently linked to a ligand for ASGPR, the ligand comprising at least one GalNAc, wherein the saponin-oligonucleotide-ASGPR ligand conjugate further comprises a covalently bound oligonucleotide such as an siRNA or an antisense oligonucleotide (referred to as oligonucleotide conjugate of the invention).
- the invention relates to a pharmaceutical composition of the invention comprising the saponin-oligonucleotide- ASGPR ligand conjugate, for use as a medicament.
- the invention also relates to a pharmaceutical composition of the invention, for use in the treatment or prophylaxis of a disease or health problem in which an expression product is involved of for example genes: HSP27, apoB, TTR, PCSK9, TMPRSS6, ALAS1 , AT3, GO, CC5, X gene of HBV, S gene of HBV, AAT, miR-122, hepatitis B virus HbsAg, LDHA, CEBPA and LDH, and for use in the treatment or prophylaxis of for example a cancer, an infectious disease, a viral infection, hypercholesterolemia, cardiovascular disease, primary hyperoxaluria, haemophilia A, haemophilia B, AAT related liver disease, acute hepatic porphyria, TTR-mediated amyloidosis, hereditary TTR amyloidosis (hATTR), complement-mediated disease, hepatitis B infection, hepatitis C infection, ⁇ 1-antitryp
- the invention also relates to a method for producing an oligonucleotide conjugate of the invention.
- the invention relates to an in vitro or ex vivo method for transferring the the saponin-oligonucleotide- ASGPR ligand conjugate of the invention (also referred to as oligonucleotide conjugate of the invention) from outside a cell to inside said cell.
- Oligonucleotide therapy is a relatively young and fast-developing field which aims to overcome many of the issues encountered with small-molecule drugs or antibody drugs by directly manipulating the genetic transcription and translation pathways.
- the potency and versatility of oligonucleotides in particular the prospect of suppressing genes encoding proteins that are ‘undruggable’ by classical small molecule drugs, makes them attractive drug candidates.
- the first oligonucleotide drugs were based on antisense technology, whereby single-stranded nucleic acid molecules would bind with sequence specificity to their complementary mRNA target, thus triggering degradation of the duplex by the RNase H system.
- RNAi double-stranded RNAs
- dsRNAs double-stranded RNAs
- PTGS post-transcriptional gene silencing
- RNAi RNA interference
- RNAi pathway through small dsRNAs such as small interfering RNAs (siRNAs) and short hairpin RNA (shRNA) has several theoretical advantages over antisense being notably more efficient (catalytic) and giving longer inhibition of gene expression. This could translate into lower doses and lower cost together with less frequent dosing. Lower exposures could also mean fewer toxicity problems for siRNA.
- siRNAs small interfering RNAs
- shRNA short hairpin RNA
- siRNA RNA-lnduced Silencing Complex
- RISC RNA-lnduced Silencing Complex
- the siRNA Upon entering the bloodstream, siRNA is vulnerable to degradation by endogenous nucleases, and renal excretion due to its small size and highly anionic character.
- the siRNA before reaching its target cell, the siRNA must navigate the tight endothelial junctions of the blood vessels and diffuse through the extracellular matrix. Due to its numerous negative charges, siRNA does not readily bind to or cross the cell membrane, and once inside cells, it must escape from endosomes to interact with its intracellular protein targets.
- oligonucleotide such as siRNA
- delivery systems which improve oligonucleotide (such as siRNA and antisense oligonucleotides) delivery.
- siRNA or other oligonucleotides
- liposomes or cell-penetrating peptides (CPPs) and their mimics, or nanoparticles
- CPPs cell-penetrating peptides
- nanoparticles Gooding, Matt, et al. Chemical biology & drug design 80.6 (2012): 787-809.
- Safety concerns such as the possibility of insertion mutagenesis and immunogenesis, are considered to limit the future of viral approaches.
- Liposomes are the most commonly used delivery vector for oligonucleotides such as siRNA, where the oligonucleotide is encapsulated within a lipid bilayer.
- oligonucleotides such as siRNA
- Several problems are encountered with liposomal oligonucleotide delivery, such as oxygen radical-mediated toxicity (typical for of cationic liposomes), cell toxicity, effects on gene regulation and inflammatory responses.
- oxygen radical-mediated toxicity typically of cationic liposomes
- cell toxicity effects on gene regulation and inflammatory responses.
- in vivo delivery using lipids seems to predominantly target the liver and spleen.
- CPP Cell-penetrating peptides
- protein transduction domains are short peptides (usually ⁇ 30 amino-acid residues long) which have the unusual property of being able to cross the cell membrane.
- a CPP may enhance cellular uptake of the oligonucleotide.
- the field of CPP mediated oligonucleotide delivery is extremely complex since it combines the challenges posed by both oligonucleotide technology and peptide technology, two fields which are far from mature, in a single drug.
- Nanoparticles and nano-carriers are nanoscale oligonucleotide delivery systems typically comprised of a polymer, biological stabilizers and cell-targeting ligands complexed with the oligonucleotide.
- An exemplary siRNA nano-carrier system is known under the name siRNA Dynamic Poly-Conjugates. This system is based around an amphipathic polymer linked to polyethylene glycol (PEG) as a biological stabilizer and a hepatocyte-targeting ligand wherein the siRNA is covalently bound to the polymer by a disulfide bond.
- PEG polyethylene glycol
- the targeting moieties and PEG moieties are attached to the polymer via maleamate linkages, forming negatively charged nanoparticles, which do not bind to serum proteins.
- Nano-carriers are also known in the form of cationic polymers such as polyethylene-imines (PEIs) (sometimes combined with cyclodextrins), which can form electrostatic complexes with oligonucleotides such as siRNA, affording protection from degradation and aiding internalisation.
- PEIs polyethylene-imines
- oligonucleotides such as siRNA
- Oligonucleotides can be used to modulate gene expression via a range of processes including RNAi, target degradation by RNase H-mediated cleavage, splicing modulation, non-coding RNA inhibition, gene activation and programmed gene editing. Oligonucleotides are nucleic acid polymers with the potential to treat or manage a wide range of diseases. Although the majority of oligonucleotide therapeutics have focused on gene silencing, other strategies are being pursued, including splice modulation and gene activation, expanding the range of possible targets beyond what is generally accessible to conventional pharmaceutical modalities.
- oligonucleotides are single-stranded antisense oligonucleotides and double-stranded short interfering RNAs (siRNAs) which share a fundamental principle: an oligonucleotide binds a target RNA through Watson-Crick base pairing, and the resulting duplex directs degradation of the target messenger RNA (mRNA).
- mRNA target messenger RNA
- the oligonucleotides can modulate the expression of the cognate RNA.
- Antisense oligonucleotides are small ( ⁇ 18-30 nucleotides), synthetic, single-stranded nucleic acid polymers of diverse chemistries, which can be employed to modulate gene expression via various mechanisms.
- ASOs can be subdivided into two major categories: RNase H competent and steric block.
- Steric block oligonucleotides are ASOs that are designed to bind to target transcripts with high affinity but do not induce target transcript degradation as they lack RNase H competence. Steric block oligonucleotides can mask specific sequences within a target transcript and thereby interfere with transcript RNA-RNA and/or RNA-protein interactions.
- SiRNA molecules are the effector molecules of RNAi and classically consist of a characteristic 19 + 2mer structure (that is, a duplex of two 21-nucleotide RNA molecules with 19 complementary bases and terminal 2-nucleotide 3 ⁇ overhangs).
- One of the strands of the siRNA (the guide or antisense strand) is complementary to a target transcript, whereas the other strand is designated the passenger or sense strand.
- SiRNAs act to guide the Argonaute 2 protein (AGO2), as part of the RNA-induced silencing complex (RISC), to complementary target transcripts.
- AGO2 Argonaute 2 protein
- RISC RNA-induced silencing complex
- siRNA approaches include Dicer substrate siRNAs, small internally segmented siRNAs, self-delivering siRNAs (asymmetric and hydrophobic), single-stranded siRNAs and divalent siRNAs.
- Steric block ASOs competitively inhibit miRNAs via direct binding to the small RNA species within the RISC complex. Such ASOs are known as anti-miRNA oligonucleotides, anti-miRs or antagomirs.
- the first anti-miRNA drug to enter clinical trials was miravirsen (SPC3649), which is an ASO designed to treat chronic hepatitis C virus (HCV) infection via targeting of the liver-specific miRNA miR-122.
- miravirsen SPC3649
- Other classes of oligonucleotide therapeutics modulate RNA function by binding to splice sites on pre-mRNAs, which results — for example — in the skipping of mutation-containing exons in diseases such as muscular dystrophy.
- Further examples of oligonucleotides for clinical applications are microRNAs (miRNAs) which are endogenous RNAi triggers that have been implicated in e.g. cancer, cell cycle progression, infectious disease, immunity, diabetes, metabolism, myogenesis and muscular dystrophy.
- MiRNA hairpins embedded within long primary miRNA transcripts are sequentially processed by two RNase III family enzymes, DICER1 (Dicer) and DROSHA, which liberate the hairpin and then cleave the loop sequence, respectively.
- the resulting duplex RNA (analogous to an siRNA) is loaded into an Argonaute protein (for example, AGO2) and one strand discarded to generate the mature, single-stranded miRNA species.
- siRNAs guide RISC to target sequences where they initiate gene silencing.
- miRNAs typically bind with partial complementarity and induce silencing via slicer- independent mechanisms.
- Therapeutic oligonucleotides are generally 15 to 30 nucleotides in length and are designed to be complementary to a specific region of a messenger RNA (mRNA) encoding a disease-related protein or a regulatory RNA. After parenteral administration, the oligonucleotide enters a cell and binds to any complementary RNA. Once the oligonucleotide drug has bound to its complementary mRNA or pre- mRNA, a series of events ensues.
- mRNA messenger RNA
- oligonucleotide-comprising drug molecule is an siRNA–GalNAc conjugate, in which the siRNA portion targets the PCSK9 enzyme. This enzyme binds and degrades the low-density lipoprotein receptor (when bound to low-density lipoprotein) and is a target in the treatment of cardiovascular disease.
- a further example is a GalNAc conjugate comprising antisense oligonucleotide against apolipoprotein(a), which is expressed in the liver.
- gene DMD has been the target of oligonucleotide based drug molecules.
- Duchenne’s muscular dystrophy is a uniformly fatal disease caused by mutations in DMD, the gene encoding dystrophin.
- Spinal muscle atrophy is an autosomal recessive disease caused by mutations in SMN1 that result in loss of SMN1 protein function.
- a target for oligonucleotide-based therapy is gene SMN2.
- Oligonucleotide drugs that use sequence-driven cleavage mechanisms to reduce levels of a disease-related mRNA and its protein product are for example mipomersen and inclisiran, which use cleavage mechanisms to modify cholesterol disposition.
- the two drugs each target a different gene product, each of which is important in hypercholesterolemia.
- the common factor in each mechanism as a result of the hybridization of each oligonucleotide drug to the target — is the activation of endogenous enzymes, which results in cleavage of the targeted mRNA at the site of hybridization.
- Mipomersen is a single-stranded oligonucleotide with a sequence that is complementary to a portion of the RNA encoding apolipoprotein B (apoB), a component of low-density lipoprotein (LDL) cholesterol that is produced in the liver.
- apoB apolipoprotein B
- LDL low-density lipoprotein
- Inclisiran cleaves and inactivates PCSK9 mRNA, which has the effect of decreasing levels of PCSK9 and therefore increasing both, LDLR levels and the clearance of LDL cholesterol, and reducing circulating levels of LDL cholesterol.
- Inclisiran is a double-stranded, small interfering RNA (siRNA).
- siRNA small interfering RNA
- One of the RNA strands of inclisiran is complementary to a portion of PCSK9 mRNA. Once inclisiran enters the cell, the complementary strand (or guide strand) is loaded into the RNA-induced silencing complex (RISC), a protein complex that displays the strand to the intracellular milieu.
- RISC RNA-induced silencing complex
- Oligonucleotide-based drugs that trigger RNase H-mediated and RISC-mediated cleavage are being used in the treatment of transthyretin (TTR) amyloidosis.
- TTR transthyretin
- mutations in TTR induce misfolding of the protein product, which results in the formation of amyloid deposits in multiple tissues, including peripheral neurons and the heart.
- oligonucleotide therapeutics targeting the liver are mipomersen (targeting gene apoB for treating homozygous familial hypercholesterolaemia), Defibrotide (hepatic veno-occlusive disease), patisiran TTR (hereditary transthyretin amyloidosis, polyneuropathy)), inotersen TTR (hereditary transthyretin amyloidosis, polyneuropathy)), and givosiran.
- Givosiran is a GalNAc conjugate for treating acute hepatic porphyria; the target gene is ALAS1.
- Oligonucleotide drug candidates under clinical development, targeting genes in the liver are for example miravirsen targeting miR-122 (hepatitis C infection), RG-101 targeting miR-122 (hepatitis C infection), AB-729 targeting hepatitis B virus HBsAg (hepatitis B infection), ARO-AAT targeting AAT ( ⁇ 1-antitrypsin deficiency), SLN124 targeting gene TMPRSS6 ( ⁇ -thalassaemia), DCR-PHXC targeting LDHA (primary hyperoxaluria) and MTL-CEPBA targeting CEBPA (hepatocellular carcinoma).
- oligonucleotide therapeutics drug safety and delivery.
- drug safety and delivery The inability to deliver the oligonucleotide drug candidate to the organs and cells that are expressing the disease-related RNAs has proved to be the greatest obstacle to the successful development of oligonucleotide therapeutics.
- Oligonucleotides are considerably larger than traditional small-molecule drug candidates. Their size, coupled with their highly anionic nature, makes it difficult for them to diffuse across cell membranes to reach the cytoplasmic and nuclear compartments.
- Oligonucleotides are typically large, hydrophilic poly-anions (single-stranded ASOs are ⁇ 4–10 kDa, double-stranded siRNAs are ⁇ 14 kDa), properties that mean they do not readily pass through the plasma membrane.
- difficulties arise when considering sufficient release and delivery of the oligonucleotides in the cytosol and/or nucleus, when escape of the oligonucleotide from the endolysosomal system before lysosomal degradation or re-export via exocytosis is considered in order to be able to arrive at the correct intracellular site of action.
- oligonucleotide cell delivery strategies are inefficient, leading to renal excretion of the drug or delivery of the majority of the drug to cells and tissues that are not of therapeutic interest.
- improved oligonucleotide such as siRNA
- endosomal escape remains a major barrier to the application of oligonucleotide-based therapeutics such as RNAi-based therapeutics. Only a minuscule portion of endocytosed oligonucleotide escapes into the cytoplasmic space where it can exert its intended function, while the vast majority remains trapped in endocytic compartments and is inactive.
- ASOs antisense oligonucleotides
- GalNAc tri-antennary N-acetylgalactosamine
- oligonucleotide therapeutics yield 10– 30-fold increased potency in isolated hepatocytes, as well as in liver in vivo.
- GalNAc is found on damaged glycoproteins that have lost terminal sialic acid residues from their pendant oligosaccharides.
- the liver functions to clear these proteins from the systemic circulation by expressing trimeric asialoglycoprotein receptors (ASGPRs) at very high levels (order of 10 5 –10 6 per cell) on the surface of hepatocytes.
- ASGPRs trimeric asialoglycoprotein receptors
- ASGPRs bind specifically to GalNAc at neutral pH for endocytosis of circulating macromolecules from the blood and release GalNAc at acidic pH ( ⁇ 5–6) for cargo drop-off in the early endosome. Freed ASGPRs are then recycled back to the cell surface for reuse.
- RNAi drugs currently in clinical trials are single- molecule, chemically modified RNAi triggers conjugated to multivalent GalNAc ligands targeting the ASGPRs.
- the suitable physiology of the liver, the unique properties of ASGPRs, the non-toxic nature of the GalNAc ligand and the simplicity of GalNAc–siRNA conjugates make this an attractive approach for systemic RNAi delivery to hepatocytes.
- oligonucleotide therapeutics oligonucleotide bioconjugates
- ASGPR asialoglycoprotein receptor
- the ASGPR binds and internalizes tri-antennary N-acetylgalactosamine (GalNAc) conjugated with for example a therapeutic antisense oligonucleotide, which are internalized and then released inside the cell, where they can hybridize to their cognate pre–messenger RNA (mRNA) and induce cleavage of the RNA–DNA heteroduplex, or for example conjugated with a small interfering RNA (siRNA) directed against a disease-related gene.
- mRNA pre–messenger RNA
- siRNA small interfering RNA
- the now-cytoplasmic siRNA can load into the RNA-induced silencing complex (RISC).
- RISC RNA-induced silencing complex
- RNAi denotes RNA interference.
- This receptor-mediated uptake allows for dosing that is lower than that required for the therapeutic delivery of unconjugated oligonucleotides.
- Both single-stranded and double-stranded oligo- nucleotides can be delivered to hepatocytes with the use of GalNAc conjugates.
- drugs for the treatment of diseases such as haemophilia A and haemophilia B are developed based on oligonucleotide-GalNAc conjugates, as well as siRNA-GalNAc conjugate inclisiran targeting gene PCSK9 and siRNA-GalNAc conjugate givosiran targeting gene ALAS1.
- oligonucleotide-GalNAc conjugates as well as siRNA-GalNAc conjugate inclisiran targeting gene PCSK9 and siRNA-GalNAc conjugate givosiran targeting gene ALAS1.
- oligonucleotide-based drug platforms approaches aiming at improving oligonucleotide delivery, such as chemical modification of oligonucleotides, bio-conjugation and the use of nano-carriers for delivery of oligonucleotides, are used, but the delivery challenge still needs improvement. There still exists a need for improved delivery systems which further increase the potency, decrease the toxicity and/or reduce off-target effects of ASGPR targeted oligonucleotide systems (e.g. GalNAc-oligonucleotide conjugates).
- ASGPR targeted oligonucleotide systems e.g. GalNAc-oligonucleotide conjugates.
- An aspect of the invention relates to an oligonucleotide conjugate comprising at least one saponin covalently linked to a ligand for asialoglycoprotein receptor (ASGPR), wherein the ligand for ASGPR comprises at least one N-acetylgalactosamine (GalNAc) moiety, preferably three or four GalNAc moieties, more preferably the ligand for ASGPR comprises or consists of (GalNAc) 3 Tris, and further covalently linked to an oligonucleotide, wherein the at least one saponin is selected from monodesmosidic triterpenoid saponins and bidesmosidic triterpenoid saponins.
- ASGPR asialoglycoprotein receptor
- An aspect of the invention relates to a pharmaceutical composition comprising the oligonucleotide conjugate of the invention, and optionally a pharmaceutically acceptable excipient and/or optionally a pharmaceutically acceptable diluent.
- An aspect of the invention relates to a pharmaceutical composition comprising the oligonucleotide conjugate of the invention, for use as a medicament.
- An aspect of the invention relates to a pharmaceutical composition
- a pharmaceutical composition comprising the oligonucleotide conjugate of the invention, for use in the treatment or prophylaxis of a disease or health problem in which an expression product is involved of any one or more of genes: HSP27, apoB, TTR, PCSK9, TMPRSS6, ALAS1, AT3, GO, CC5, X gene of HBV, S gene of HBV, AAT, miR-122, hepatitis B virus HbsAg, LDHA, CEBPA and LDH, and/or for use in the treatment or prophylaxis of a disease or health problem which involves any one or more of genes: HSP27, apoB, TTR, PCSK9, TMPRSS6, ALAS1, AT3, GO, CC5, X gene of HBV, S gene of HBV, AAT, miR-122, hepatitis B virus HbsAg, LDHA, CEBPA and LDH.
- An aspect of the invention relates to a pharmaceutical composition
- a pharmaceutical composition comprising the oligonucleotide conjugate of the invention, for use in the treatment or prophylaxis of a disease or health problem in which an expression product is involved of any one or more of genes: HSP27, apoB, TTR, PCSK9, TMPRSS6, ALAS1, AAT, miR-122, hepatitis B virus HbsAg, LDHA and CEBPA, and/or for use in the treatment or prophylaxis of a disease or health problem which involves any one or more of genes: HSP27, apoB, TTR, PCSK9, TMPRSS6, ALAS1, AAT, miR-122, hepatitis B virus HbsAg, LDHA and CEBPA.
- An aspect of the invention relates to a pharmaceutical composition
- a pharmaceutical composition comprising the oligonucleotide conjugate of the invention, for use in the treatment or prophylaxis of a disease or health problem in which an expression product is involved of any one or more of genes: HSP27 and apoB, preferably apoB, and/or for use in the treatment or prophylaxis of a disease or health problem which involves any one or more of genes: HSP27 and apoB, preferably apoB.
- An aspect of the invention relates to a pharmaceutical composition
- a pharmaceutical composition comprising the oligonucleotide conjugate of the invention, for use in the treatment or prophylaxis of a cancer, an infectious disease, a viral infection, hypercholesterolemia, cardiovascular disease, primary hyperoxaluria, haemophilia A, haemophilia B, AAT related liver disease, acute hepatic porphyria, TTR- mediated amyloidosis, hereditary TTR amyloidosis (hATTR), complement-mediated disease, hepatitis B infection, hepatitis C infection, ⁇ 1-antitrypsin deficiency, ⁇ -thalassaemia, or an auto-immune disease.
- An aspect of the invention relates to a pharmaceutical composition
- a pharmaceutical composition comprising the oligonucleotide conjugate of the invention, for use in the treatment or prophylaxis of a cancer such as endometrial carcinoma, breast cancer, lung cancer or hepatocellular carcinoma, and a cardiovascular disease such as hypercholesterolemia, preferably hypercholesterolemia.
- An aspect of the invention relates to an in vitro or ex vivo method for transferring the oligonucleotide conjugate of the invention from outside a cell to inside said cell, preferably for subsequently transferring the oligonucleotide comprised by the oligonucleotide conjugate of the invention into the cytosol and/or into the nucleus of said cell, comprising the steps of: a) providing a cell which expresses ASGPR, preferably ASGPR1, on its surface, the cell preferably selected from a liver cell, a virally infected mammalian cell and a mammalian tumor cell, wherein preferably said cell is a human cell; b) providing the oligonucleotide conjugate of the invention for transferring into the cell provided in step a); c) contacting the cell of step a) in vitro or ex vivo with the oligonucleotide conjugate of step b), preferably in a liquid medium, therewith effecting the transfer of the
- An aspect of the invention relates to a saponin conjugate comprising at least one saponin covalently linked to a ligand for asialoglycoprotein receptor (ASGPR), wherein the ligand for ASGPR comprises at least one N-acetylgalactosamine (GalNAc) moiety, preferably three or four GalNAc moieties, more preferably the ligand for ASGPR comprises or consists of (GalNAc) 3 Tris, wherein the at least one saponin is selected from monodesmosidic triterpenoid saponins and bidesmosidic triterpenoid saponins.
- ASGPR asialoglycoprotein receptor
- An aspect of the invention relates to a pharmaceutical combination comprising: - a first pharmaceutical composition comprising the saponin conjugate of the invention and optionally comprising a pharmaceutically acceptable excipient and/or a pharmaceutically acceptable diluent; and - a second pharmaceutical composition comprising a second conjugate of an effector molecule and a ligand for ASGPR, wherein the ligand for ASGPR preferably comprises at least one GalNAc moiety, preferably three or four GalNAc moieties, more preferably the ligand for ASGPR comprises or consists of (GalNAc) 3 Tris, or a third conjugate of an effector molecule and a binding molecule comprising a binding site for a cell-surface molecule, and optionally comprising a pharmaceutically acceptable excipient and/or pharmaceutically acceptable diluent.
- An aspect of the invention relates to a pharmaceutical composition
- a pharmaceutical composition comprising: - the saponin conjugate of the invention; - a second conjugate of an effector molecule and a ligand for ASGPR wherein the ligand for ASGPR preferably comprises at least one GalNAc moiety, preferably three or four GalNAc moieties, more preferably the ligand for ASGPR comprises or consists of (GalNAc) 3 Tris, or a third conjugate of an effector molecule and a binding molecule comprising a binding site for a cell-surface molecule, and optionally comprising a pharmaceutically acceptable excipient and/or pharmaceutically acceptable diluent.
- An aspect of the invention relates to a pharmaceutical combination of the invention comprising the saponin conjugate of the invention or to a pharmaceutical composition of the invention comprising the saponin conjugate of the invention, for use as a medicament.
- An aspect of the invention relates to a pharmaceutical combination of the invention comprising the saponin conjugate of the invention or a pharmaceutical composition of the invention comprising the saponin conjugate of the invention, for use in the treatment or prophylaxis of a disease or health problem in which an expression product is involved of any one or more of genes: HSP27, apoB, TTR, PCSK9, TMPRSS6, ALAS1 , AT3, GO, CC5, X gene of HBV, S gene of HBV, AAT, miR-122, hepatitis B virus HbsAg, LDHA, CEBPA and LDH.
- An aspect of the invention relates to an in vitro or ex vivo method for transferring the second conjugate or the third conjugate of the invention from outside a cell to inside said cell, preferably subsequently transferring the effector molecule comprised by the second conjugate or the third conjugate of the invention into the cytosol of said cell, comprising the steps of: a) providing a cell which expresses ASGPR on its surface, and, when the third conjugate is to be transferred into the cell, which expresses the cell-surface molecule for which the third conjugate comprises a binding molecule for binding to said cell-surface molecule, the cell preferably selected from a liver cell, a virally infected cell and a tumor cell; b) providing the second conjugate or the third conjugate of any one of the invention for transferring into the cell provided in step a); c) providing the saponin conjugate of the invention; d) contacting the cell of step a) in vitro or ex vivo with the second conjugate or the third conjugate of step b)
- An aspect of the invention relates to a method for providing the oligonucleotide conjugate of the invention, comprising the steps of:
- An embodiment is the method for providing the oligonucleotide conjugate of the invention, wherein the seventh linker is a tri-functional linker, such as the tri-functional linker represented by formula (XXI):
- GalNAc has its regular scientific meaning and here refers to N-acetylgalactosamine and to the IUPAC name thereof: 2-(acetylamino)-2-deoxy-D-galactose.
- (GalNAc) 3 Tris has its regular scientific meaning in for example the field of siRNA- based therapy, and here refers to a moiety comprising three GalNAc units each separately covalently bound to the hydroxyl groups of tris(hydroxymethyl)aminomethane (Tris) (IUPAC name: 2-amino-2- (hydroxymethyl) propane-1,3-diol), preferably via at least one linker.
- Tris tris(hydroxymethyl)aminomethane
- (GalNAc) 3 Tris can exist as a free amine comprising molecule or may be further functionalized via the remaining amine binding site, for example to form the (GalNAc) 3 Tris-moiety comprising conjugates described herein.
- oligonucleotide has its regular scientific meaning and here refers to a string of two or more nucleotides, i.e. an oligonucleotides is a short oligomer composed of ribonucleotides or deoxyribonucleotides.
- RNA and DNA examples are RNA and DNA, and any modified RNA or DNA, such as a string of nucleic acids comprising a nucleotide analogue such as a bridged nucleic acid (BNA), also known as locked nucleic acid (LNA) or a 2'-O,4'-C-aminoethylene or a 2'-O,4'-C-aminomethylene bridged nucleic acid (BNA NC ), wherein the nucleotide is a ribonucleotide or a deoxyribonucleotide.
- BNA bridged nucleic acid
- LNA locked nucleic acid
- BNA NC 2'-O,4'-C-aminoethylene
- BNA NC 2'-O,4'-C-aminomethylene bridged nucleic acid
- RNAi-mediated gene-targeting has its regular scientific meaning and here refers to the in vivo, ex vivo or in vitro approach of influencing functioning of the gene in a cell by transferring into said cell an oligonucleotide, such as a small double-stranded RNA molecule, that targets mRNA involved in transcription of the gene: for example, small double-stranded RNA molecules are capable of efficiently triggering RNAi silencing of specific genes.
- bridged nucleic acid in short, or “locked nucleic acid” or “LNA” in short or 2'-O,4'-C-aminoethylene or 2'-O,4'-C-aminomethylene bridged nucleic acid (BNA NC ), has its regular scientific meaning and here refers to a modified RNA nucleotide.
- a BNA is also referred to as ‘constrained RNA molecule’ or ‘inaccessible RNA molecule’.
- a BNA monomer can contain a five- membered, six-membered or even a seven-membered bridged structure with a “fixed” C 3 ’-endo sugar puckering.
- the bridge is synthetically incorporated at the 2’, 4’-position of the ribose to afford a 2’, 4’- BNA monomer.
- a BNA monomer can be incorporated into an oligonucleotide polymeric structure using standard phosphoramidite chemistry known in the art.
- a BNA is a structurally rigid oligonucleotide with increased binding affinity and stability.
- the term antisense oligonucleotide has its regular scientific meaning and may be indicated in short in the description as “AON” or “ASO”.
- BNA-based antisense oligonucleotide or in short “BNA AON”, has its regular scientific meaning and here refers to a string of antisense nucleotides wherein at least one of said nucleotides is a BNA.
- proteinaceous has its regular scientific meaning and here refers to a molecule that is protein-like, meaning that the molecule possesses, to some degree, the physicochemical properties characteristic of a protein, is of protein, relating to protein, containing protein, pertaining to protein, consisting of protein, resembling protein, or being a protein.
- proteinaceous refers to the presence of at least a part of the molecule that resembles or is a protein, wherein ‘protein’ is to be understood to include a chain of amino-acid residues at least two residues long, thus including a peptide, a polypeptide and a protein and an assembly of proteins or protein domains.
- the at least two amino-acid residues are for example linked via (an) amide bond(s), such as (a) peptide bond(s).
- the aminoacid residues are natural amino-acid residues and/or artificial amino-acid residues such as modified natural amino-acid residues.
- a proteinaceous molecule is a molecule comprising at least two amino-acid residues, preferably between two and about 2.000 amino-acid residues. In one embodiment, a proteinaceous molecule is a molecule comprising from 2 to 20 (typical for a peptide) amino acids. In one embodiment, a proteinaceous molecule is a molecule comprising from 21 to 1 .000 (typical for a polypeptide, a protein, a protein domain, such as an antibody, a Fab, an scFv, a ligand for a receptor such as EGF) amino acids.
- the amino-acid residues are (typically) linked via (a) peptide bond(s). According to the invention, said amino-acid residues are or comprise (modified) (non-)natural amino acid residues.
- effector molecule when referring to the effector molecule as part of e.g. a covalent conjugate, has its regular scientific meaning and here refers to a molecule that can selectively bind to for example any one or more of the target molecules: a protein, a peptide, a carbohydrate, a saccharide such as a glycan, a (phospho)lipid, a nucleic acid such as DNA, RNA, an enzyme, and regulates the biological activity of such one or more target molecule(s).
- the effector molecule is for example a molecule selected from any one or more of a small molecule such as a drug molecule, a toxin such as a protein toxin, an oligonucleotide such as a BNA, a xeno nucleic acid or an siRNA, an enzyme, a peptide, a protein, or any combination thereof.
- a small molecule such as a drug molecule
- a toxin such as a protein toxin
- an oligonucleotide such as a BNA, a xeno nucleic acid or an siRNA
- an enzyme a peptide, a protein, or any combination thereof.
- an effector molecule or an effector moiety is a molecule or moiety selected from any one or more of a small molecule such as a drug molecule, a toxin such as a protein toxin, an oligonucleotide such as a BNA, a xeno nucleic acid or an siRNA, an enzyme, a peptide, a protein, or any combination thereof, that can selectively bind to any one or more of the target molecules: a protein, a peptide, a carbohydrate, a saccharide such as a glycan, a (phospho)lipid, a nucleic acid such as DNA, RNA, an enzyme, and that upon binding to the target molecule regulates the biological activity of such one or more target molecule(s).
- a small molecule such as a drug molecule
- a toxin such as a protein toxin
- an oligonucleotide such as a BNA
- an effector molecule can exert a biological effect inside a cell such as a mammalian cell such as a human cell, such as in the cytosol of said cell.
- Typical effector molecules are thus drug molecules, plasmid DNA, toxins such as toxins comprised by antibody-drug conjugates (ADCs), oligonucleotides such as siRNA, BNA, nucleic acids comprised by an antibody-oligonucleotide conjugate (AOC).
- ADCs antibody-drug conjugates
- oligonucleotides such as siRNA, BNA
- AOC antibody-oligonucleotide conjugate
- an effector molecule is a molecule which can act as a ligand that can increase or decrease (intracellular) enzyme activity, gene expression, or cell signalling.
- health problem has its regular scientific meaning and here refers to any condition of the body of a subject such as a human patient, or of a part, organ, muscle, vein, artery, skin, limb, blood, cell, etc. thereof, that is suboptimal when compared to the condition of that body or part thereof in a healthy subject, therewith hampering the proper functioning and/or well-being of the subject, e.g. impairs normal functioning of a human body.
- GalNAc-decorated oligonucleotide drug has its regular scientific meaning and here refers to an oligonucleotide for interfering in transcription of a gene, wherein one or more GalNAc units are coupled to the oligonucleotide, e.g. via at least one linker.
- locked nucleic acid in short “LNA”, which is a bridged nucleic acid, has its regular scientific meaning and here refers to an oligonucleotide that contains one or more nucleotide building blocks in which an additional methylene bridge fixes the ribose moiety either in the C3'-endo conformation (beta-D-LNA) or C2'-endo (alpha-L-LNA) conformation, as is known in the art.
- BNA refers to BNA NC or 2',4'-BNA NC (2'-O,4'-aminoethylene bridged nucleic acid) and has its regular scientific meaning and here refers to an oligonulceotide that contains one or more nucleotide building blocks with a six-member bridged structure with an N-O linkage, and with an (N-H) or (N-Me) residue.
- siRNA has its regular scientific meaning and here refers to an siRNA molecule comprising chemical modifications compared to the RNA oligonucleotide consisting of naturally occurring nucleotides, such that the modified siRNA molecule is more resistant towards metabolic degradation, digestion, enzymatic lysis, etc., i.e. more stable.
- siponin“ has its regular scientific meaning and here refers to a group of amphipatic glycosides which comprise one or more hydrophilic glycone moieties combined with a lipophilic aglycone core which is a sapogenin.
- the saponin may be naturally occurring or synthetic (i.e. non-naturally occurring).
- the term “saponin” includes naturally-occurring saponins, derivatives of naturally-occurring saponins as well as saponins synthesized de novo through chemical and/or biotechnological synthesis routes.
- the term “saponin derivative“ (also known as “modified saponin”) has its regular scientific meaning and here refers to a compound corresponding to a naturally-occurring saponin which has been derivatised by one or more chemical modifications, such as the oxidation of a functional group, the reduction of a functional group and/or the formation of a covalent bond with another molecule.
- Preferred modifications include derivatisation of an aldehyde group of the aglycone core; of a carboxyl group of a saccharide chain or of an acetoxy group of a saccharide chain.
- the saponin derivative does not have a natural counterpart, i.e. the saponin derivative is not produced naturally by e.g. plants or trees.
- the term “saponin derivative” includes derivatives obtained by derivatisation of naturally-occurring saponins as well as derivatives synthesized de novo through chemical and/or biotechnological synthesis routes resulting in a compound corresponding to a naturally-occurring saponin which has been derivatised by one or more chemical modifications.
- aglycone core structure has its regular scientific meaning and here refers to the aglycone core of a saponin without the one or two carbohydrate antenna or saccharide chains (glycans) bound thereto.
- quillaic acid is the aglycone glycoside core structure for SO1861, QS-7, and QS-21.
- saccharide chain has its regular scientific meaning and here refers to any of a glycan, a carbohydrate antenna, a single saccharide moiety (monosaccharide) or a chain comprising multiple saccharide moieties (oligosaccharide, polysaccharide).
- the saccharide chain can consist of only saccharide moieties or may also comprise further moieties such as any one of 4E-Methoxycinnamic acid, 4Z-Methoxycinnamic acid, and 5-O-[5-O-Ara/Api-3,5-dihydroxy-6-methyl-octanoyl]-3,5-dihydroxy- 6-methyl-octanoic acid), such as for example present in QS-21.
- Api/Xyl-“ or “Api- or Xyl-“ in the context of the name of a saccharide chain has its regular scientific meaning and here refers to the saccharide chain either comprising an apiose (Api) moiety, or comprising a xylose (Xyl) moiety.
- antibody-drug conjugate has its regular scientific meaning and here refers to any conjugate of an antibody such as an IgG, a Fab, an scFv, an immunoglobulin, an immunoglobulin fragment, one or multiple Vh domains, etc., and any molecule that can exert a therapeutic effect when contacted with cells of a subject such as a human patient, such as an active pharmaceutical ingredient, a toxin, an oligonucleotide, an enzyme, a small molecule drug compound, etc.
- ADC antibody-drug conjugate
- antibody-oligonucleotide conjugate has its regular scientific meaning and here refers to any conjugate of an antibody such as an IgG, a Fab, an scFv, an immunoglobulin, an immunoglobulin fragment, one or multiple Vh domains, etc., with any oligonucleotide molecule that can exert a therapeutic effect when contacted with cells of a subject such as a human patient, such as an oligonucleotide selected from a natural or synthetic string of nucleic acids encompassing DNA, modified DNA, RNA, modified RNA, synthetic nucleic acids, presented as a single-stranded molecule or a double- stranded molecule, such as a BNA, an allele-specific oligonucleotide (ASO), a short or small interfering RNA (siRNA; silencing RNA), an anti-sense DNA, anti-sense RNA, etc.
- ASO allele-specific oligonucleotide
- siRNA si
- conjugate has its regular scientific meaning and here refers to at least a first molecule that is covalently bound to at least a second molecule, therewith forming a covalently coupled assembly comprising or consisting of the first molecule and the second molecule.
- Typical conjugates are (GalNAc) 3 Tris – siRNA, an ADC, an AOC, and SO1861-EMCH (EMCH linked to the aldehyde group of the aglycone glycoside core structure of the saponin).
- moiety has its regular scientific meaning and here refers to a molecule that is bound, linked, conjugated to a further molecule, linker, assembly of molecules, etc., and therewith forming part of a larger molecule, conjugate, assembly of molecules.
- a moiety is a first molecule that is covalently bound to a second molecule, involving one or more chemical groups initially present on the first and second molecules.
- saporin is a typical effector molecule.
- the saporin is a typical effector moiety in the ADC.
- a BNA or an siRNA is a typical effector moiety in the AOC.
- S as used such as in an antibody-saponin conjugate or construct comprising a linker, represents ‘stable linker’ which remains intact in the endosome and in the cytosol.
- L as used such as in an antibody-saponin conjugate or construct comprising a linker, represents ‘labile linker’ which is cleaved under slightly acid conditions in the endosome.
- moiety has its regular scientific meaning and here refers to a molecule that is bound, linked, conjugated to a further molecule, linker, assembly of molecules, etc., and therewith forming part of a larger molecule, conjugate, assembly of molecules.
- a moiety is a first molecule that is covalently bound to a second molecule, involving one or more chemical groups initially present on the first molecule and present on the second molecule.
- saporin is a typical molecule, and is an example of an effector molecule.
- the saporin is a typical effector moiety in the ADC.
- a BNA or an siRNA is a typical effector moiety in the AOC.
- a composition comprising components A and B should not be limited to a composition consisting only of components A and B, rather with respect to the present invention, the only enumerated components of the composition are A and B, and further the claim should be interpreted as including equivalents of those components.
- reference to an element or a component by the indefinite article “a” or “an” does not exclude the possibility that more than one of the elements or components are present, unless the context clearly requires that there is one and only one of the elements or components.
- the indefinite article “a” or “an” thus usually means “at least one".
- DAR normally stands for Drug Antibody Ratio and refers to the average drug to antibody ratio for a given preparation of antibody drug conjugate (ADC) and here refers to a ratio of the number of bound SO1861 moieties, or SPT001, or bound payload, e.g. an AON such as an ApoB BNA, with respect to the conjugate molecule.
- ADC antibody drug conjugate
- Figure 1A-C synthesis of trivalent-GalNAc.
- Figure 2A-B synthesis of SO1861-DBCO.
- Figure 3 synthesis of monovalent-GalNAc-SO1861.
- Figure 4 synthesis of trivalent-GalNAc-SO1861.
- Figure 5A-C synthesis of monovalent-GalNAc-BNA.
- Figure 6A-B synthesis of trivalent-GalNAc-BNA.
- Figure 7 synthesis of trivalent-GalNAc-BNA.
- Figure 8A-G synthesis of trivalent GalNAc.
- Figure 9A and 9B General toxicity (MTS) of GalNAc-SO1861, SO1861-EMCH, and trivalent-GalNAc- SO1861 on HepG2 (A) and Huh7 (B) cell lines. The legend displayed next to Figure 9B also applies for Figure 9A.
- Figure 10A and 10 B Cell killing assay (MTS) HepG2 (A) and Huh7 (B) cell lines.
- Figure 11A and 11B Gene expression analysis on HepG2 (A) and Huh7 (B) cell lines.
- Figure 12A and 12B Gene expression analysis on HepG2 (A) and Huh7 (B) cell lines.
- Figure 13A and 13B Cell viability assay on HepG2 (A) and Huh7 (B) cell lines.
- Figure 14A and 14B Gene expression analysis on HepG2 (A) and Huh7 (B) cell lines.
- Figure 14A also applies for Figure 14A.
- Figure 15A and 15B Cell viability assay on HepG2 (A) and Huh7 (B) cell lines. The legend displayed next to Figure 15B also applies for Figure 15A.
- Figure 16A and 16B Gene expression analysis on HepG2 (A) and Huh7 (B) cell lines. The legend displayed next to Figure 16B also applies for Figure 16A.
- Figure 16C and 16D Cell viability assay on HepG2 (C) and Huh7 (D) cell lines. The legend displayed next to Figure 16D also applies for Figure 16C.
- Figure 17A and 17B Gene expression analysis on HepG2 (A) and Huh7 (B) cell lines. The legend displayed next to Figure 17B also applies for Figure 17A.
- SO1861 is also referred to as ‘SPT001’ and as ‘dSPT’, throughout the description and claims.
- Figure 19A and 19B Cell viability of primary human hepatocytes (A) and Huh7 hepatocyte cell line (B) (both compared with untreated cells (‘Untr’), which are set to 100%), upon contacting the cells with a concentration series of trivalent GalNAc conjugated with SO1861 (Trivalent GalNAc-SPT001), the combination of 10 pM monoclonal antibody anti-CD71 conjugated with saporin (CD71-saporin) and a concentration series of covalent conjugate Trivalent GalNAc-SPT001.
- ‘S’ is SPT001 SO1861 in the cartoon.
- Figure 19D Cartoon of anti-CD71 antibody – saporin conjugate (OKT-9), wherein ‘T’ is the toxin saporin covalently linked to the antibody heavy chain.
- S’ is SPT001 SO1861 in the cartoon; dSPT4 describes a dendron with four SO1861 molecules, conjugated as described elsewhere.
- Figure 20A and B Relative apoB expression in primary human hepatocytes (A) and Huh7 hepatocyte cell line (B), upon contacting the cells with a concentration series of trivalent GalNAc conjugated with antisense oligonucleotide for silencing ApoB mRNA and ApoB protein expression, i.e.
- FIG. 20C Cartoon of covalent TrivalentGalNAc-ApoBBNA conjugate. ‘A’ is antisense oligonucleotide ApoB (ApoBBNA) in the cartoon.
- Figure 20D Cartoon of TrivalentGalNAc-ApoBBNA-SPT001. ‘S’ is SPT001, also referred to as SO1861, in the cartoon; ‘E’ is the effector moiety antisense oligonucleotide targeting apoB (ApoB BNA) in the cartoon.
- S is SPT001, also referred to as SO1861, in the cartoon;
- E is the effector moiety antisense oligonucleotide targeting apoB (ApoB BNA) in the cartoon.
- Figure 21A and B Absolute apoB protein expression in primary human hepatocytes (A) and in the Huh7 hepatocyte cell line (B), under influence of contacting the cells with a concentration series of TrivalentGalNAc-ApoBBNA, the combination of 300 nM TrivalentGalNAc-SPT001 and a concentration series of TrivalentGalNAc-ApoBBNA, or with a concentration series of TrivalentGalNAc-ApoBBNA- SPT001; here four SO1861 molecules were conjugated with a single dendron moiety to the GalNAc / oligonucleotide conjugate (therefore also named (GalNAc)3-dSPT4-ApoB).
- Figure 22A-C Synthesis of a saponin-oligonucleotide-ASGPR ligand conjugate, i.e. TrivalentGalNAc- ApoB BNA-SPT001, also referred to as (GalNAc) 3 -SO1861-ApoB.
- Figure 23A Synthesis of SO1861-OEG-NHS (SPT001-L-NHS, molecule 12, wherein ‘L’ refers to a labile, cleavable linker).
- Figure 23B Synthesis of dendron(SPT001) 4 -NH 2 (molecule 15 from molecules 13 and 14).
- Figure 23C Synthesis dendron(SPT001) 4 -azide (molecule 19 from molecules 15 and 18).
- Figure 23D-E Synthesis dendron(SPT001) 4 -trivalent GalNAc (molecule 24 from molecules 19 and 23).
- Figure 24A-B Synthesis of DBCO-TCO-trivalent GalNAc (molecule 29) from molecule 27 and molecule 28 (trifunctional linker), wherein molecule 27 is GalNAc-thioacetate obtained from the reaction between molecule 22, the formic salt of trivalent GalNAc-amine, and molecule 26 (4-nitrophenyl 3- (acetylthio)propanate.
- Figure 24C Synthesis of DBCO-L-ApoB BNA oligo-trivalent GalNAc (molecule 31) by conjugating methyltetrazine-L-ApoB BNA oligo (molecule 30) to DBCO-TCO-trivalent GalNAc (molecule 29).
- Figure 24D Synthesis of dendron(-L-SO1861) 4 -L-ApoB BNA oligo-trivalent GalNAc (molecule 34; ‘Trivalent GalNAc-ApoB BNA-SPT001 conjugate’) by conjugating molecule 31 (DBCO-L-ApoB BNA oligo-trivalent GalNAc) and molecule 19 (dendron(SPT001) 4 -azide).
- Figure 25 ApoB expression analysis in liver tissue of C57BL/6J mice.
- Figure 26 Serum apoB protein analysis in C57BL/6J mice. Levels for 0.1 mg/kg ApoB#02 (196 hrs), 0.01 mg/kg (GalNAc) 3 -ApoB + 5 mg/kg (GalNAc) 3 -SO1861 (196 hrs and 336 hrs) and 0.1 mg/kg (GalNAc) 3 -ApoB + 5 mg/kg (GalNAc) 3 -SO1861 (196 hrs) are not reported.
- Figure 27 Serum LDL-cholesterol (LDL-C) analysis in C57BL/6J mice.
- Figure 28 Serum ALT analysis in C57BL/6J mice.
- Figure 29A-C ApoB expression analysis and cell viability assay on primary human hepatocytes, HepG2 and Huh7 cells for ApoB#02 conjugates.
- Figure 30 ApoB expression analysis and cell viability assay on primary human hepatocytes for ApoB#02 conjugates
- Figure 31 ApoB expression analysis in liver tissue of C57BL/6J mice.
- Figure 32 Serum ApoB protein analysis in C57BL/6J mice.
- Figure 33 Serum LDL-cholesterol (LDL-C) analysis in C57BL/6J mice.
- Figure 34 Serum ALT analysis in C57BL/6J mice.
- Figure 35A-D Trifunctional linker-(L-hydrazone-SO1861)-(L-BNA oligo)-(Trivalent-GalNAc) synthesis.
- Figure 36A-E Trifunctional linker-(dendron(-L-hydrazone-SO1861)4)-(L-BNA oligo)-(Trivalent-GalNAc) synthesis. Note that the drawing of Figure 36 E encompasses two sheets, labelled Fig.36 E and Fig. 36 E (continued), which two sheets together display the conjugation of molecule 21 with molecule 19, therewith forming conjugate molecule 22.
- Figure 37A-C Trifunctional linker-(dendron(-L-hydrazone-SO1861)8)-(BNA oligo)-(Trivalent-GalNAc) synthesis.
- Figure 37 B encompasses two sheets, labelled Fig.37 B and Fig. 37 B (continued), which two sheets together display the conjugation of molecule 24 with molecule 18, therewith forming conjugate molecule 25 (intermediate 12).
- Figure 37 C encompasses two sheets, labelled Fig. 37 C and Fig. 37 C (continued), which two sheets together display the conjugation of molecule 25 with molecule 21, therewith forming conjugate molecule 26.
- Figure 38A-C Trifunctional linker-(L-semicarbazone-SO1861)- (BNA oligo) – (Trivalent-GalNAc) synthesis.
- Figure 39A-F Trifunctional linker – (dendron(L-semicarbazone-SO1861)4) - (L-BNA oligo) - (Trivalent- GalNAc) synthesis. Note that the drawing of Figure 39 E encompasses two sheets, labelled Fig.39 E and Fig. 39 E (continued), which two sheets together display the conjugation of molecule 40 with molecule 20, therewith forming conjugate intermediate 24.
- Figure 40A-D Trifunctional linker – (dendron(L-semicarbazone-SO1861)8) - (L-BNA oligo) - (Trivalent- GalNAc) synthesis.
- Figure 40 A encompasses two sheets, labelled Fig.40 A and Fig. 40 A (continued), which two sheets together display the conjugation of molecule 43 with molecule 37, therewith forming conjugate (molecule 44) intermediate 25.
- Figure 40 B encompasses two sheets, labelled Fig.40 B and Fig.40 B (continued), which two sheets together display the conjugation of molecule 44 with molecule 18, therewith forming conjugate molecule 45 (intermediate 26).
- Figure 40 C encompasses two sheets, labelled Fig.40 C and Fig.40 C (continued), which two sheets together display the conjugation of molecule 45 with molecule 20, therewith forming conjugate molecule 46 (intermediate 27).
- Figure 41 Trivalent linker-(L-SPT001)-(blocked TCO)-(trivalent GalNAc) synthesis synthesis.
- Figure 42A-B Trivalent linker-(blocked DBCO)-(L-BNA oligo)-(trivalent GalNAc) synthesis.
- Figure 43A-B Overview of conjugates obtainable with applying TFL (trifunctional linker) methodology.
- FIG. 43A and B together displays the conjugation of Trifunctional linker labelled molecule 4 with Targeting ligand labelled molecule 41, Effector labelled molecule 12, and one of the Enhancers labelled molecule 3, 19, 25, 28, 40 and 45 or one of the Blocking agents for controls labelled molecule 5, 12a and 50, therewith forming a conjugate with the general conjugate structure ‘CONJUGATE C1’ as depicted in Figure 43 B, comprising group R1 and group R2, wherein R1 is any one of the groups R1 depicted below the conjugate CONJUGATE C1 and R2 in CONJUGATE C1 is any one of the groups R2 depicted below the conjugate CONJUGATE C1 in Figure 43 B.
- a first aspect of the invention relates to a saponin conjugate comprising at least one saponin covalently linked to a ligand for asialoglycoprotein receptor (ASGPR), wherein the ligand for ASGPR comprises at least one N-acetylgalactosamine (GalNAc) moiety, preferably three or four GalNAc moieties, more preferably the ligand for ASGPR comprises or consists of (GalNAc) 3 Tris, wherein the at least one saponin is selected from monodesmosidic triterpenoid saponins and bidesmosidic triterpenoid saponins.
- ASGPR asialoglycoprotein receptor
- An aspect of the invention relates to an oligonucleotide conjugate comprising at least one saponin covalently linked to a ligand for asialoglycoprotein receptor (ASGPR), wherein the ligand for ASGPR comprises at least one N-acetylgalactosamine (GalNAc) moiety, preferably three or four GalNAc moieties, more preferably the ligand for ASGPR comprises or consists of (GalNAc) 3 Tris, and further covalently linked to an oligonucleotide, wherein the at least one saponin is selected from monodesmosidic triterpenoid saponins and bidesmosidic triterpenoid saponins.
- ASGPR asialoglycoprotein receptor
- the ASGPR is ASGPR1.
- the triterpene saponin is typically a triterpenoid saponin of the 12,13-dehydrooleanane type such as a triterpenoid saponin or a bidesmosidic triterpene saponin belonging to the type of a 12,13-dehydrooleanane with an aldehyde function in position C-23.
- Preferred are the mono-desmosidic or bi-desmosidic penta-cyclic triterpene saponins of the 12,13-dehydrooleanane type, preferably with an aldehyde function in position C-23.
- the inventors established that the potency of a gene silencing oligonucleotide (ASO, AON, siRNA, BNA) is enhanced with at least a factor 10, that is to say at least a factor 100 or even a factor over 1000, when the mRNA expression and/or the protein expression relating to the target gene targeted by the therapeutic oligonucleotide, is considered, when the therapeutic oligonucleotide is contacted with a mammalian cell bearing ASGPR such as a human cell, such as a human liver cell, in the presence of a saponin such as SO1861 which is covalently conjugated with a ligand for the liver-cell specific receptor ASGPR1, such as trivalent-GalNAc (GN3) (i.e.
- ASO gene silencing oligonucleotide
- a conjugate comprising three GalNAc moieties comprising three GalNAc moieties.
- the therapeutic oligonucleotide such as an siRNA or an ASO relating to treating a cancer (for example by silencing HSP27 expression) or relating to treating a too high blood plasma level of (LDL-)cholesterol, by targeting the apoB gene, with a saponin, e.g. provided as a GalNAc-comprising conjugate separate from the oligonucleotide, or e.g. provided as a saponin moiety covalently linked to a conjugate of an ASGPR ligand and the oligonucleotide, widens the therapeutic window for the therapeutic oligonucleotides.
- a saponin e.g. provided as a GalNAc-comprising conjugate separate from the oligonucleotide, or e.g. provided as a saponin moiety covalently linked to a conjugate
- An effective gene silencing (any relevant extent of silencing) is achieved at lower oligonucleotide dose in the presence of an ASGPR1 targeting ligand comprising covalently linked saponin, when compared to the gene silencing obtained in the absence of (ASGPR1 targeting) saponin when contacting a target cell with the oligonucleotide coupled to a ligand for ASGPR1. That is to say, the inventors established near-complete (defined as at least 95% or higher) RNA expression inhibition, when receptor bearing cells, in vitro or in vivo, are contacted with the oligonucleotide conjugate of the invention.
- the inventors also achieved (near-)complete inhibition of protein expression, which, for example, for apoB protein subsequently results in (near-)complete clearance of LDL-cholesterol in serum.
- This surprisingly strong extent and efficacy of the gene silencing under influence of the oligonucleotide conjugate of the invention cannot be achieved when the saponin is not part of a conjugate comprising the payload and a ligand for the ASGPR1, such as GN3.
- the improved potency of the gene silencing after contacting target (liver) cells that express the ASGPR1 with saponin- comprising conjugate of the invention is apparent when mRNA expression is assessed and/or when protein expression relating to the target gene is assessed, and indirectly when LDL-cholesterol level is assessed, since an LDL particle comprises one copy of the apoB protein (more specifically, the apoB100 protein), which apoB expression is silenced upon contacting cells with the oligonucleotide conjugate of the invention.
- An improved gene silencing is achieved at a fixed oligonucleotide dose in the presence of an ASGPR1 targeting ligand comprising covalently linked saponin, when compared to the gene silencing efficacy obtained in the absence of saponin when contacting a target cell with the same dose of oligonucleotide coupled to a ligand for ASGPR1.
- the inventors also found that covalently coupling a saponin to a therapeutic oligonucleotide and a ligand for ASGPR1 such as (GalNAc) 3 resulted in the surprisingly high improvement of the gene silencing efficacy, when the combination is contacted with ASGPR1 bearing cells such as human liver cells, compared with the efficacy obtained in the absence of the saponin in the GalNAc-oligonucleotide conjugate.
- Saponins of the triterpene glycoside type have the capacity to stimulate the delivery of effector molecules from outside a target cell to inside said cell, and subsequently into the cytosol of said cell.
- Saponins facilitate for example receptor-mediated uptake of certain effector molecules, such as a protein toxin comprised in an ADC. Endocytosis of the toxin, as part of the ADC, delivers the toxin to the endosome and/or the lysosome of the target cell. Without wishing to be bound by any theory, subsequent release of the ADC, or at least of the toxin, out of the endosome or lysosome and into the cell cytosol, is stimulated or mediated by the triterpenoid saponin applied in the current invention.
- the toxin When a target cell is contacted with the toxin at a toxin dose that is too low for exerting an intracellular effect, co-administration of the toxin, such as part of an ADC, and a free saponin, such that the target cell is contacted with both the ADC and the saponin, can result in efficient toxin-mediated killing of the target cell, at the same toxin dose that is ineffective when contacted with the cell in the absence of the saponin.
- the potency of the toxin is improved.
- the toxin dose required for efficient and sufficient target cell killing can be accompanied by undesired off-target effects, for example when the tumor cell receptor targeted by the ADC is not a truly (exclusively) tumor-cell specific receptor, but also expressed in e.g. healthy cells, elsewhere in a patient’s body, or on cells that are present in the same organ where the target cells reside.
- the required dose of the free saponin needed to reach a sufficient extent of toxin activation (delivery of the toxin into the target-cell cytosol; the saponin threshold to mediate on-target delivery), may be at a level inducing undesired off- target effects.
- the free saponin is administered systemically, and non-targeted when the target (tumor) cell to be treated with the toxin is considered, a relatively high saponin dose is to be administered to the patient in need of the toxin treatment, in order to reach the local saponin dose at the level of the target cells.
- one of the further problems and shortcomings encountered when the free saponin is co-administered with e.g. an ADC is the difficulty to optimize the synchronization of the activity of the ADC + SPT001 (free saponin) at the right time and place in the target cell in the patient.
- the inventors now provide a 1-component conjugate solution which provides a solution for this problem.
- the inventors now surprisingly found that the saponin dose required for potentiating the effect of a toxin on tumor cells can be lowered by a factor of 10, or even more by about a factor of 100 to 1000, when the saponin is conjugated with a cell-targeting moiety such as a receptor ligand (e.g. EGF or a cytokine), an antibody, or a binding fragment or domain thereof.
- a cell-targeting moiety such as a receptor ligand (e.g. EGF or a cytokine), an antibody, or a binding fragment or domain thereof.
- a receptor ligand e.g. EGF or a cytokine
- Such targeted saponin conjugates indeed are able to enhance e.g. the cytotoxic effect of a toxin inside a target cell, requiring an at least 10-fold, such as a 100-fold to 1000-fold lower effective dose of the conjugate compared to the dose required for the same saponin in free form, i.e., not provided with a tumor cell-targeting moiety.
- target-cell specific delivery and subsequent endocytosis of the (targeted) saponin is also possible for non-aberrant cells or aberrant cells not related to e.g. cancer (tumor cells), auto-immune disease, virally infected cells, etc.
- the inventors found that the ASGPR provides a suitable target receptor on cells bearing this receptor, such as liver cells, for entry of saponins provided with a ligand for ASGPR, in particular ASGPR1 (see Figure 3 for an example: saponin SO1861 (also referred to as ‘SPT001’) conjugated with a single GalNAc moiety).
- ASGPR1 saponin SO1861 (also referred to as ‘SPT001’) conjugated with a single GalNAc moiety).
- ligands such as monovalent GalNAc and trivalent GalNAc (GN) 3 are successfully coupled to one or a multitude (2, 4, 8, etc.) of saponin molecules, providing saponin conjugates comprising the ASGPR ligand and comprising at least one saponin moiety, preferably a bidesmosidic pentacyclic triterpene saponin of the 12,13-dehydrooleanane type with an aldehyde function in position C-23.
- saponin conjugates comprising the ASGPR ligand and comprising at least one saponin moiety, preferably a bidesmosidic pentacyclic triterpene saponin of the 12,13-dehydrooleanane type with an aldehyde function in position C-23.
- oligonucleotide conjugates of the invention comprising at least one saponin, a ligand for ASGPR1 and an oligonucleotide, covalently linked together, such as via a trifunctional linker.
- Providing the saponin with a ligand for ASGPR results in effects in the target cell, exerted by a co-administered effector molecule such as a targeted oligonucleotide or toxin (e.g.
- the saponin conjugates of the invention and the oligonucleotide conjugates of the invention improve the potency of effector molecules such as oligonucleotides when a target cell comprising ASGPR at its surface is considered, and such target cell is contacted with both the saponin-ASGPR ligand of the invention and the (targeted) effector molecule, or with the oligonucleotide conjugate comprising saponin for enhancing endosomal escape of the effector moiety from the endosome and/or lysosome into the cytosol and/or ultimately into the nucleus of the target cell, GalNAc as the ligand for ASGPR1 and an oligonucleotide as the effector moiety.
- the therapeutic window of the effector molecule is improved: higher therapeutic effect at the same dose, under influence of co-administration of the saponin conjugate or under influence of the oligonucleotide conjugate; or similar therapeutic effect at lower dose, under influence of co- administration of the saponin conjugate or under influence of the oligonucleotide conjugate wherein the oligonucleotide is the effector moiety.
- the inventors also found that at least a 10-fold, for example about a 100-fold to 1000-fold lower dose of the saponin conjugate or of the oligonucleotide conjugate is required to reach a certain level of therapeutic effect in the target ASGPR expressing cell, such as a liver cell, induced by the co-administered ((ASGPR1) targeted) effector molecule such as an ASO, an siRNA, a BNA, a toxin such as a protein toxin, or induced by the oligonucleotide comprised as the effector moiety in the oligonucleotide conjugate, when compared to the saponin dose required for reaching the similar effect, when the saponin is not provided with a ligand for ASGPR.
- the target ASGPR expressing cell such as a liver cell
- the co-administered ((ASGPR1) targeted) effector molecule such as an ASO, an siRNA, a BNA, a toxin
- an oligonucleotide is required to reach a certain level of therapeutic effect in the target ASGPR expressing cell, such as a liver cell, induced by an ((ASGPR1) targeted) oligonucleotide as the effector molecule such as an ASO, an siRNA, a BNA, that is administered to cells, when at least one saponin moiety such as an SO1861 moiety or SO1832 moiety is covalently linked to the oligonucleotide conjugate comprising at least one GalNAc, such as trivalent GalNAc.
- the inventors now provide for improved therapeutic methods using the ASGPR as a target receptor for saponin-mediated delivery of an effector molecule such as a therapeutic oligonucleotide in the cytosol of ASGPR expressing cells, resulting in similar therapeutic effects induced by the effector molecule at lower doses than possible in the absence of the saponin conjugate of the invention or in the absence of covalently linked saponin in the oligonucleotide conjugate.
- an effector molecule such as a therapeutic oligonucleotide in the cytosol of ASGPR expressing cells
- the present invention provides for improved therapeutic use of the stimulatory effect of saponins, when the delivery of an effector molecule into the cytosol or nucleus of, e.g., liver cells is considered, by providing the saponin – ASGPR ligand conjugate of the invention and by providing the oligonucleotide conjugate of the invention (e.g. saponin, oligonucleotide, (GalNAc) 3 covalently linked together in a single conjugate).
- the oligonucleotide conjugate of the invention e.g. saponin, oligonucleotide, (GalNAc) 3 covalently linked together in a single conjugate.
- oligonucleotide such as an oligonucleotide (siRNA or ASO) by co-administration of the ((ASGPR1) targeted) effector molecule with saponin or by administration of the oligonucleotide conjugate comprising the GalNAc moiety/moieties for targeting ASGPR1, comprising the oligonucleotide (e.g.
- the ligand for ASGPR can be the same in the saponin conjugate and in the targeted effector molecule conjugate, resulting in efficient effector molecule-mediated effects in the target cell, under stimulatory influence of the saponin. That is to say, the saponin conjugate and the effector molecule conjugate, both comprising covalently linked ligand for ASGPR such as trivalent GalNAc, can be co-administered to target cells, such as liver cells exposing the ASGPR at their surface, such that the desired therapeutic effect of the effector molecule is efficiently obtained, while both conjugates target the same receptor and use the same route for endocytosis into the cell.
- target cells such as liver cells exposing the ASGPR at their surface
- liver-cell targeting therapies such as therapies involving cytosolic delivery of a nucleic acid (ASO, BNA, siRNA, to list a few), since liver cells do express the ASGPR, which ASGPR is specific for liver cells, e.g. ASGPR1 , whereas further liver cell receptors specific enough for use in liver cell targeting therapy are virtually absent.
- ASO nucleic acid
- BNA nucleic acid
- siRNA siRNA
- the current invention thus opens new ways to improve liver-cell directed therapies wherein effector molecules such as ASO and siRNA have to be intracellularly delivered and located for their mode of action.
- Targeted saponin provided as the saponin conjugates of the invention and the oligonucleotide conjugates of the invention widen the therapeutic window of such effector molecules (oligonucleotides such as siRNA, ASO), and at the same time, by providing the saponin with a liver celltargeting binding molecule, also the therapeutic window of the saponin is improved, according to the invention.
- the inventors achieved efficient oligonucleotide delivery, therewith providing a solution to the major translational limitation that is apparent in the field of therapeutic oligonucleotides.
- the inventors provide a solution resulting in improving oligonucleotide delivery, based on the bio-conjugation of the invention for improved delivery of oligonucleotides.
- the improved delivery system of the invention results in increased potency of the oligonucleotide, that may aid in decreasing the toxicity and/or may aid in reducing off-target effects of ASGPR targeted oligonucleotide systems (e.g. GalNAc-oligonucleotide conjugates).
- the current inventors managed to synthesize conjugates of non-proteinaceous nature which are composed of two or three biologically active small molecules, for example when compared to the molecular size (molecular weight) of antibodies applied in (antibody-drug) conjugates based on antibodies such as IgG, which conjugates of the invention are still biologically active when the biological activity of each of the two or three small molecules comprised by the conjugates is considered: saponin conjugate of the invention and oligonucleotide conjugate of the invention.
- the conjugates of the invention are relatively small, though the two or three small molecules that are covalently conjugated together in a single molecule are still able to exert the biological activity related to the nature of each of the small molecules.
- the saponin enhances endosomal and lysosomal escape of oligonucleotides that are taken up by a cell. That is to say, ASGPR bearing cells take up the GalNAc comprising saponin conjugate and oligonucleotide conjugate of the invention. That is to say, gene silencing effects are apparent when ASGPR bearing cells are contacted with the oligonucleotide conjugate of the invention.
- the saponin- and GalNAc-comprising conjugate of the invention provides endosomal escape enhancing activity towards oligonucleotides when co-administered to the same cells with an oligonucleotide- and GalNAc-comprising conjugate.
- Both, the saponin and the oligonucleotide are taken up by those cells, mediated by binding of GalNAc to the ASGPR on the target cells.
- the oligonucleotide that is delivered into the cytosol to a higher extent under influence of the saponin in the endosome/lysosome exerts its intracellular biological activity expressed as silencing of the gene targeted by the oligonucleotide (for example targeting the genes HSP27 and apoB).
- the oligonucleotide conjugate of the invention provides endosomal escape enhancing activity towards oligonucleotides comprised by the oligonucleotide conjugate.
- Both, the saponin and the oligonucleotide are taken up by those cells as part of the oligonucleotide conjugate of the invention, mediated by binding of GalNAc to the ASGPR on the target cells.
- each of at least one saponin such as one, four, eight saponin moieties
- a GalNAc moiety or cluster of GalNAc moieties e.g. (GN) 3
- an oligonucleotide is still biologically active compared to the biological activity measured when cells are contacted with e.g. free saponin (e.g. in combination with a molecule that exerts its activity in the cytosol), GalNAc-oligonucleotide, free oligonucleotide.
- free saponin e.g. in combination with a molecule that exerts its activity in the cytosol
- GalNAc-oligonucleotide free oligonucleotide.
- the saponin, the GalNAc and the oligonucleotide are relatively small molecules, conjugating these molecules together does not hamper their biological activity.
- the present invention provides a conjugate of a saponin and a ligand for asialoglycoprotein receptor (ASGPR), referred to herein as “saponin conjugate”.
- ASGPR ligand comprises at least one N- acetylgalactosamine (GalNAc) moiety.
- each GalNAc moiety is bound to the remainder of the ASGPR ligand or - in case the ASGPR ligand consists of a single GalNAc moiety - to the saponin moiety, via a covalent bond to the oxygen on position “1” as indicated in formula (I):
- the ASGPR ligand consists of a single GalNAc moiety bound to the saponin moiety S, preferably via a saponin moiety linker L S .
- the ASGPR ligand may comprise more than one GalNAc moiety, such as 2, 3, 4, 5 or 6 GalNAc moieties, preferably 3 or 4 GalNAc moieties, more preferably 3 GalNAc moities.
- the GalNAc moieties are each separately covalently bound via the oxygen on position “1” to a central bridging moiety B, which effectively forms a bridge between the GalNAc moieties and the saponin moiety, preferably via a saponin moiety linker L S .
- the GalNAc moieties may be directly bound to the bridging moiety B as shown in formula (III) S : wherein n is an integer larger than or equal to 2, L S is a saponin moiety linker and S is the saponin moiety. More preferably, the GalNAc moieties are bound to the bridging moiety B via GalNAc linkers L GAL as shown in formula (IV) S : wherein n is an integer larger than or equal to 2, L S is a saponin moiety linker and S is the saponin moiety. While each occurrence of L GAL may be independently chosen, the skilled person will appreciate that the synthesis of the saponin conjugate is simplified in case each occurrence of L GAL represents the same moiety.
- the saponin moiety linker L S represents any chemical moiety suitable for covalently binding a saponin to GalNAc as in formula (II) S or to the bridging moiety B as in formula (III) S and (IV) S .
- the identity and size of the linker is not particularly limited and covalently binding a saponin to GalNAc as in formula (II) S or to the bridging moiety B as in formula (III) S and (IV) S may be effected by means of a ‘regular’ chemical functional group (e.g. an ester bond) as well as through “click-chemistry” type linkers which typically have a long chain length, resulting in a linker ES comprising e.g.
- linkers L S and associated coupling reactions for binding molecules to each other are described in the handbook Hermanson, Greg T. Bioconjugate techniques. Academic press, 2013.
- the saponin moiety linker L S will typically be the result of a coupling reaction (e.g. “click-chemistry” type) between at least a first precursor L S1 covalently bound to GalNAc or the bridging moiety B and a second precursor L S2 covalently bound to the saponin moiety.
- L S is a saponin moiety linker
- L S1 is a precursor of the saponin moiety linker L S which is covalently bound to GalNAc or to the bridging moiety B
- L S2 is a precursor of the saponin moiety linker L S which is covalently bound to the saponin moiety and S is the saponin moiety.
- L S is a saponin moiety linker
- L S1 is a precursor of the saponin moiety linker L S which is covalently bound to GalNAc
- L S2 is a precursor of the saponin moiety linker L S which is covalently bound to the saponin moiety
- n is an integer larger than or equal to 2
- B is a bridging moiety
- S is the saponin moiety.
- L S is a saponin moiety linker
- L S1 is a precursor of the saponin moiety linker L S which is covalently bound to GalNAc
- L S2 is a precursor of the saponin moiety linker L S which is covalently bound to the saponin moiety
- n is an integer larger than or equal to 2
- B is a bridging moiety
- S is the saponin moiety
- L GAL is a GalNAc linker.
- the saponin moiety linker L S can be the result of a coupling reaction between at least a first precursor L S1 covalently bound to GalNAc or the bridging moiety B and a second precursor L S2 covalently bound to the saponin moiety, wherein the coupling reaction is for example an azide-alkyne cycloaddition, a thiol maleimide coupling, a Staudinger reaction, a nucleophilic ring-opening of strained heterocyclic electrophiles (such as aziridines, epoxides, cyclic sulphates, aziridinium ions, episulfonium ions), a carbonyl reaction of the non-aldol type (such as urea, thiourea, hydrazone, oxime ether, amide or aromatic heterocycle formation) or an addition to a carbon-carbon double bond (such as epoxidation, aziridination, dihydroxylation, sulfentyl halide
- the saponin moiety linker L S comprises a semicarbazone and/or a hydrazone and/or a 1, 2, 3-triazole, preferably a hydrazone and a 1, 2, 3- triazole or at least a semicarbazone.
- a 1, 2, 3-triazole in the context of the linkers of the present application, this preferably means a 1H-1, 2, 3-triazole.
- Such a hydrazone is e.g. the result of a coupling between a terminal hydrazide and an aldehyde containing compound (such as an aldehyde containing saponin).
- This hydrazide/aldehyde coupling is a known ‘click’-chemistry tool in the field of bio-conjugation and is for example described in Hermanson, Greg T. Bioconjugate techniques. Academic press, 2013.
- a semicarbazone is e.g. the result of a coupling between a terminal semicarbazide and an aldehyde containing compound (such as an aldehyde containing saponin).
- an aldehyde containing compound such as an aldehyde containing saponin.
- Such a 1, 2, 3-triazole is e.g. the result of a coupling between an azide and an alkyne containing compound.
- This azide/alkyne coupling is a commonly known ‘click’-chemistry tool in the field of bio- conjugation and is for example described in Hermanson, Greg T.
- the saponin moiety linker L S is the result of a coupling reaction between a first precursor L S1 covalently bound to GalNAc or the bridging moiety B, the first precursor L S1 comprising an azide; and a second precursor L S2 covalently bound to the saponin moiety, the second precursor L S2 comprising an alkyne and preferably comprising a hydrazone resulting from a hydrazide/aldehyde coupling to an aldehyde of the saponin moiety or preferably comprising a semicarbazone resulting from a semicarbazide/aldehyde coupling to an aldehyde of the saponin moiety.
- Embodiments of the structure for the precursor L S1 present in the compounds of formula (V) S are the following azides: wherein a represents an integer larger than or equal to 0, preferably a represents an integer selected from 1, 2, and 3, more preferably a represents 2.
- Embodiments of the structure for the precursor L S1 present in the compounds of formula (VII) S or (VIII) S are the following azides: wherein c represents an integer larger than or equal to 0, preferably c represents an integer in the range of 5-15, more preferably c represents 9.
- Embodiments of the structure for the precursor L S2 present in the compounds of formula (VI) S are the following hydrazones: wherein a represents an integer larger than or equal to 0, preferably a represents an integer in the range of 2-6, more preferably a represents 4, and wherein L S2a represents an alkyne-containing moiety.
- a represents an integer larger than or equal to 0, preferably a represents an integer in the range of 2-6, more preferably a represents 4, and wherein L S2a represents an alkyne-containing moiety.
- the hydrazone depicted in formula (XIX) results from a reaction of a hydrazide with an aldehyde of the saponin moiety.
- L S2a preferably comprises less than 20 carbon atoms, more preferably L S2a represents a moiety according to formula (XX):
- the saponin conjugate is a compound of formula (II) S , (III) S or (IV) S as described herein, wherein the saponin moiety linker L S is the result of a coupling reaction between a compound of formula (V) S , (VII) S or (VIII) S with a compound of formula (VI) S , wherein the first precursor L S1 is an azide, for example an azide of formula (XVII) or formula (XVIII), and wherein the second precursor L S2 is a compound of formula (XIX) comprising an alkyne-containing moiety L S2a , for example the alkyne of formula (XX) and a hydrazone resulting from a reaction of a hydrazide with an aldehyde of the saponin moiety.
- Saponin conjugate – saponin An aspect of the invention relates to a saponin conjugate comprising at least one saponin covalently linked to a ligand for asialoglycoprotein receptor (ASGPR), wherein the ligand for ASGPR comprises at least one N-acetylgalactosamine (GalNAc) moiety, preferably three or four GalNAc moieties, more preferably three GalNAc moieties, more preferably the ligand for ASGPR comprises or consists of (GalNAc) 3 Tris, wherein the at least one saponin is selected from monodesmosidic triterpenoid saponins and bidesmosidic triterpenoid saponins.
- ASGPR asialoglycoprotein receptor
- An aspect of the invention relates to an oligonucleotide conjugate comprising at least one saponin, preferably 1-32, more preferably 1-16, most preferably 1-8 saponin moieties, such as 1, 4 or 8 saponin moieties, covalently linked to a ligand for asialoglycoprotein receptor (ASGPR), wherein the ligand for ASGPR comprises at least one N-acetylgalactosamine (GalNAc) moiety, preferably three or four GalNAc moieties, more preferably three GalNAc moieties, more preferably the ligand for ASGPR comprises or consists of (GalNAc) 3 Tris, and further covalently linked to an oligonucleotide, wherein the at least one saponin is selected from monodesmosidic triterpenoid saponins and bidesmosidic triterpenoid saponins.
- GalNAc N-acetylgalactosamine
- the saponin is selected for its endosomal escape enhancing activity when the intracellular effect of a molecule such as a proteinaceous toxin (e.g. saporin, dianthin) or a gene- silencing oligonucleotide such as HSP27 BNA is assessed when contacted with a target cell in the presence or absence of the saponin.
- a proteinaceous toxin e.g. saporin, dianthin
- a gene- silencing oligonucleotide such as HSP27 BNA
- An embodiment is the saponin conjugate of the invention or the oligonucleotide conjugate of the invention, wherein the saponin is a pentacyclic triterpene saponin of the 12,13-dehydrooleanane type, preferably with an aldehyde function in position C-23 of the aglycone core structure of the saponin.
- An embodiment is the saponin conjugate of the invention or the oligonucleotide conjugate of the invention, wherein the saponin is a monodesmosidic or bidesmosidic pentacyclic triterpene saponin of the 12,13-dehydrooleanane type, preferably with an aldehyde function in position C-23 of the aglycone core structure of the saponin.
- An embodiment is the saponin conjugate of the invention or the oligonucleotide conjugate of the invention, wherein the saponin comprises an aglycone core structure selected from the group consisting of: 2alpha-hydroxy oleanolic acid; 16alpha-hydroxy oleanolic acid; hederagenin (23-hydroxy oleanolic acid); 16alpha,23-dihydroxy oleanolic acid; gypsogenin; quillaic acid; protoaescigenin-21(2-methylbut-2-enoate)-22-acetate; 23-oxo-barringtogenol C-21,22-bis(2-methylbut-2-enoate); 23-oxo-barringtogenol C-21(2-methylbut-2-enoate)-16,22-diacetate; digitogenin; 3,16,28-trihydroxy oleanan-12-en; gypsogenic acid, and derivatives thereof, preferably the saponin comprises an ag
- Such preferred aglycones comprise an aldehyde group at the C-23 atom or a derivative thereof as described herein elsewhere.
- an aldehyde group contributes to the endosomal escape enhancing activity of saponins of the triterpene glycoside type, comprising an aglycone selected from Group C, especially quillaic acid and gypsogenin.
- An embodiment is the saponin conjugate of the invention or the oligonucleotide conjugate of the invention, wherein the at least one saponin comprises a first saccharide chain, which is bound to the C 3 atom or the C28 atom of the aglycone core structure of the at least one saponin, preferably to the C 3 atom, and/or wherein the at least one saponin comprises a second saccharide chain, which is bound to the C28 atom of the aglycone core structure of the at least one saponin, preferably, the saponin comprises the first and second saccharide chain.
- the saponin comprised by the saponin conjugate of the invention or the oligonucleotide conjugate of the invention bears two glycans (saccharide chains), the first saccharide chain is bound at position C 3 of the aglycone core structure and the second saccharide chain is bound at position C28 of the aglycone core structure.
- An embodiment is the saponin conjugate of the invention or the oligonucleotide conjugate of the invention, wherein ⁇ the saponin comprises a saccharide chain bound to the aglycone core structure, which is selected from group A: GlcA-, Glc-, Gal-, Rha-(1 ⁇ 2)-Ara-, Gal-(1 ⁇ 2)-[Xyl-(1 ⁇ 3)]-GlcA-, Glc-(1 ⁇ 2)-[Glc-(1 ⁇ 4)]-GlcA-, Glc-(1 ⁇ 2)-Ara-(1 ⁇ 3)-[Gal-(1 ⁇ 2)]-GlcA-, Xyl-(1 ⁇ 2)-Ara-(1 ⁇ 3)-[Gal-(1 ⁇ 2)]-GlcA-, Glc-(1 ⁇ 3)-Gal-(1 ⁇ 2)-[Xyl-(1 ⁇ 3)]-Glc-(1 ⁇ 4)-Gal-, Rha-(1
- the saponin comprised by the saponin conjugate of the invention or the oligonucleotide conjugate of the invention bears two glycans (saccharide chains), the first saccharide chain is bound at position C 3 of the aglycone core structure and the second saccharide chain is bound at position C 28 of the aglycone core structure.
- aglycone an aldehyde group or a derivative thereof in the aglycone core structure
- saponin conjugates of the invention or the oligonucleotide conjugates of the invention comprising saponin which has an aglycone with an aldehyde group is preferred.
- An embodiment is the saponin conjugate of the invention or the oligonucleotide conjugate of the invention, wherein the saponin is selected from the group consisting of: Quillaja bark saponin, dipsacoside B, saikosaponin A, saikosaponin D, macranthoidin A, esculentoside A, phytolaccagenin, aescinate, AS6.2, NP-005236, AMA-1, AMR, alpha-Hederin, NP-012672, NP-017777, NP-017778, NP- 017774, NP-018110, NP-017772, NP-018109, NP-017888, NP-017889, NP-018108, SA1641, AE X55, NP-017674, NP-017810, AG1, NP-003881,
- the saponin is selected from the group consisting of QS-21, a QS-21 derivative, SO1861, a SO1861 derivative, SA1641, a SA1641 derivative, GE1741, a GE1741 derivative and combinations thereof, more preferably the saponin is selected from the group consisting of a QS-21 derivative, a SO1861 derivative, SO1861 and combinations thereof, most preferably the saponin is a SO1861 derivative or SO1861, or a SO1832 derivative or SO1832.
- Such derivatives of the saponins are endosomal escape enhancing derivatives, to the same or similar extent as their non-derivatized natural counterparts.
- Such saponins of the triterpene glycoside type are capable of enhancing the endosomal escape of (effector) molecules such as oligonucleotides comprised by the oligonucleotide conjugate of the invention, that are present in the endosome (or lysosome) of a cell, when the saponin co-localizes with such (effector) molecule inside the cell.
- the inventors established that the endosomal escape enhancing activity of these saponins is at least about 5 times more potent, that is to say at least about 10 times more potent, such as 10 – 1000 times more potent or about 100 to 1000 times more potent when the saponin is contacted with a cell when the saponin is comprised by the saponin conjugate of the invention or when the saponin is comprised by the oligonucleotide conjugate of the invention.
- the free saponin is capable of stimulating the delivery of (effector) molecules in the cytosol of cells, when such cells are contacted with the (effector) molecules and the saponin, at 100-1000 times higher saponin concentration, compared to the concentration of the same saponin which is comprised by the saponin conjugate of the invention or which is comprised by the oligonucleotide conjugate of the invention, required to achieve the same extent of delivery of the (effector) molecule, such as an oligonucleotide selected from an ASO or an siRNA, from outside the cell to inside the endosome and finally in the cytosol and/or in the nucleus of said cell.
- an oligonucleotide selected from an ASO or an siRNA from outside the cell to inside the endosome and finally in the cytosol and/or in the nucleus of said cell.
- Saponins which display such endosomal escape enhancing activity are listed in Table A1, as well as saponins with high structural similarity with saponins for which the ability to potentiate the cytosolic delivery of (effector) molecules has been established.
- the targeted delivery of the saponin upon binding of the ligand such as a single GalNAc moiety or a cluster of three covalently coupled GalNAc moieties, for ASGPR, such as ASGPR1
- ASGPR such as ASGPR1
- the covalently bound saponin in the conjugate is cleaved-off (released) from the conjugate once the conjugate is taken up by the ASGPR bearing cell and transported into the endosome.
- the chemical conditions and/or pH are such that the covalent bond between the saponin(s) and the remainder of the conjugate is broken and the saponin is released in free form in the endosome. It is preferred that the cleaved-off saponin has its natural chemical structure.
- the aldehyde group of a saponin is implied in covalent coupling of the saponin to the GalNAc moiety/moieties (typically via a linker) and/or to the oligonucleotide (typically via a linker), it is preferred that the saponin that is released from the conjugate in the endosome again comprises this aldehyde group, formed under influence of the de-coupling chemistry.
- the saponin in its free form preferably with a freely accessible aldehyde group, preferably with an aldehyde function in position C-23 of the aglycone core structure of the saponin
- the saponin exerts optimal endosomal escape enhancing activity in the endosome.
- Saponin is for example cleaved off from conjugates in which the saponin is bound via a hydrazone bond or a semicarbazone bond, under influence of the acidic pH in the endosome of cells such as mammalian cells such as human cells, e.g. tumor cells, liver cells.
- the saponin is covalently bound in the conjugates of the invention via a cleavable bond, preferably a cleavable covalent bond that is cleaved inside the endosome of cell, such as a bond that is cleaved due to the acidic conditions in the endosome.
- the saponin conjugate or the oligonucleotide conjugate comprises at least one saponin, wherein the saponin is a derivative wherein i. the aglycone core structure of the saponin comprises an aldehyde group which has been derivatised; ii. a saccharide chain of the saponin, preferably the saccharide chain selected from group A comprises a carboxyl group which has been derivatised; iii. a saccharide chain of the saponin, preferably the saccharide chain selected from group B comprises an acetoxy (Me(CO)O-) group which has been derivatised; or iv. any combination of derivatisations (i), (ii) and/or (iii) is present.
- the saponin is a derivative wherein i. the aglycone core structure of the saponin comprises an aldehyde group which has been derivatised; ii. a saccharide chain of the sap
- Exemplary derivatisations (i) for an aldehyde group of the aglycone core structure include reduction to an alcohol, such as a primary alcohol; transformation into a hydrazone (functional group) or semicarbazone (functional group); transformation into a hemiacetal; transformation into an acetal; oxidation to a carboxyl; transformation into an imine; transformation into an oxime; transformation into a ⁇ -hydroxy ketone; or transformation into an enone, preferably reduction to an alcohol, such as a primary alcohol; or transformation into a hydrazone (functional group) or into a semicarbazone (functional group).
- an alcohol such as a primary alcohol
- transformation into a hemiacetal transformation into an acetal
- oxidation to a carboxyl transformation into an imine
- transformation into an oxime transformation into a ⁇ -hydroxy ketone
- transformation into an enone
- the derivatised saponin of the conjugate of the invention still exerts its endosomal escape enhancing activity, once present in the endosome of cells that take up the saponin conjugate or the oligonucleotide conjugate of the invention.
- R a is an N-alkylated maleimide.
- the aldehyde group has been transformation into a hydrazone bond through reaction with N- ⁇ -maleimidocaproic acid hydrazide (EMCH); N-[ß-maleimidopropionic acid] hydrazide (BMPH); or N- [ ⁇ -maleimidoundecanoic acid] hydrazide (KMUH).
- EMCH N- ⁇ -maleimidocaproic acid hydrazide
- BMPH N-[ß-maleimidopropionic acid] hydrazide
- KMUH N- [ ⁇ -maleimidoundecanoic acid] hydrazide
- Exemplary derivatisations (ii) for a carboxyl group of a saccharide chain include: reduction to an alcohol, such as a primary alcohol; transformation into an amide; transformation into an ester; transformation into an amine, such as a primary or secondary amine; or decarboxylation, preferably reduction to an alcohol, such as a primary alcohol; transformation into an amide; or transformation into an ester; more preferably transformation into an amide.
- the carboxyl group which is derivatised is part of a glucuronic acid moiety and, preferably, the saccharide chain is selected from group A.
- the saponin is a derivative wherein a saccharide chain, preferably a saccharide chain selected from group A, comprises a carboxyl group, preferably a carboxyl group of a glucuronic acid moiety, which has been derivatised by: - reduction to an alcohol, such as a primary alcohol; - transformation into an amide of formula R a NH(CO)R b ; or - transformation into an ester of formula R a O(CO)R b wherein R a is group comprising less than 20 carbon atoms, preferably less than 10 carbon atoms and R b is the remainder of the saponin.
- R a is an N-alkylated maleimide or a diol.
- the carboxyl group has been derivatised by transformation into an amide bond through reaction with 2-amino-2-methyl-1,3-propanediol (AMPD) or N-(2-aminoethyl)maleimide (AEM).
- exemplary derivatisations (iii) for an acetoxy group of a saccharide chain include: transformation into an alcohol, such as a secondary alcohol, by deacetylation, optionally followed by transformation of the resulting alcohol into an ether; ester or ketone; preferably transformation into an alcohol, such as a secondary alcohol, by deacetylation.
- the acetoxy group which is derivatised is part of a saccharide chain selected from group B.
- the saponin is a derivative wherein a saccharide chain, preferably a saccharide chain selected from group B, comprises an acetoxy group which has been derivatised by: - transformation into an alcohol, such as a secondary alcohol, by deacetylation; or - transformation into an alcohol, such as a secondary alcohol, by deacetylation followed by transformation of the resulting alcohol into an ether of formula R a OR b , an ester of formula R a (CO)OR b , or a ketone of formula R a (CO)R b wherein R a is group comprising less than 20 carbon atoms, preferably less than 10 carbon atoms and R b is the remainder of the saponin.
- a saccharide chain preferably a saccharide chain selected from group B, comprises an acetoxy group which has been derivatised by: - transformation into an alcohol, such as a secondary alcohol, by deacetylation; or - transformation into an alcohol, such as a secondary alcohol
- R a is an N-alkylated maleimide or a diol.
- the saponin is a derivative wherein: i. the aglycone core structure comprises an aldehyde group which has been derivatised by: - reduction to an alcohol; - transformation into a hydrazone bond through reaction with N- ⁇ -maleimidocaproic acid hydrazide (EMCH); - transformation into a hydrazone bond through reaction with N-[ß-maleimidopropionic acid] hydrazide (BMPH); or - transformation into a hydrazone bond through reaction with N-[ ⁇ -maleimidoundecanoic acid] hydrazide (KMUH); ii.
- the saccharide chain selected from group A comprises a carboxyl group, preferably a carboxyl group of a glucuronic acid moiety, which has been derivatised by transformation into an amide bond through reaction with 2-amino-2-methyl-1,3-propanediol (AMPD) or N-(2- aminoethyl)maleimide (AEM); iii. the saccharide chain selected from group B comprises an acetoxy group (Me(CO)O-) which has been derivatised by transformation into a hydroxyl group (HO-); or iv. any combination of derivatisations (i), (ii) and/or (iii) is present.
- AMPD 2-amino-2-methyl-1,3-propanediol
- AEM N-(2- aminoethyl)maleimide
- An embodiment is the saponin conjugate of the invention or the oligonucleotide conjugate of the invention, wherein the saponin is a saponin derivative wherein i. the saponin derivative comprises an aglycone core structure comprising an aldehyde group which has been derivatised; ii. the saponin derivative comprises a saccharide chain, preferably a saccharide chain selected from group A, the saccharide chain comprising a carboxyl group which has been derivatised; iii.
- the saponin derivative comprises a saccharide chain, preferably a saccharide chain selected from group B, the saccharide chain comprising an acetoxy (Me(CO)O-) group which has been derivatised; or iv.
- the saponin derivative comprises any combination of derivatisations i., ii. and iii., preferably any combination of two derivatisations of derivatisations i., ii. and iii.
- An embodiment is the saponin conjugate of the invention or the oligonucleotide conjugate of the invention, wherein the saponin is any one or more of: SO1861, SA1657, GE1741, SA1641, QS-21, QS- 21A, QS-21 A-api, QS-21 A-xyl, QS-21B, QS-21 B-api, QS-21 B-xyl, QS-7-xyl, QS-7-api, QS-17-api, QS-17-xyl, QS1861, QS1862, Quillajasaponin, Saponinum album, QS-18, Quil-A, Gyp1, gypsoside A, AG1, AG2, SO1542, SO1584, SO1658, SO1674, SO1832, SO1904, stereoisomers thereof, derivatives thereof and combinations thereof, preferably the saponin is selected from the group consisting of QS- 21, a QS-21 derivative, SO1861, a SO1861 derivative, SO1832, SA1641, a SA16
- the saponin is selected from the group consisting of a QS-21 derivative, a SO1861 derivative, SO1861 and combinations thereof, even more preferably the saponin is a SO1861 derivative or SO1861, or a SO1832 derivative or SO1832.
- An embodiment is the saponin conjugate of the invention or the oligonucleotide conjugate of the invention, wherein the saponin is a saponin derivative of the quillaic acid saponin or the gypsogenin saponin according to the invention and is represented by Molecule 1: wherein A 1 represents hydrogen, a monosaccharide or a linear or branched oligosaccharide, preferably A 1 represents a saccharide chain selected from group A, more preferably A 1 represents a saccharide chain selected from group A and A 1 comprises or consists of a glucuronic acid moiety; A 2 represents hydrogen, a monosaccharide or a linear or branched oligosaccharide, preferably A 2 represents a saccharide chain selected from group B, more preferably A 2 represents a saccharide chain selected from group B and A 2 comprises at least one acetoxy (Me(CO)O-) group, such as one, two, three or four acetoxy groups,
- the aldehyde group at position C 23 of the quillaic acid or gypsogenin has been derivatised; ii. the carboxyl group of a glucuronic acid moiety of A 1 , when A 1 represents a saccharide chain selected from group A and A 1 comprises or consists of a glucuronic acid moiety, has been derivatised; and iii. one or more, preferably all, of acetoxy group(s) of one saccharide moiety or of two or more saccharide moieties of A 2 , when A 2 represents a saccharide chain selected from group B and A 2 comprises at least one acetoxy group, has/have been derivatised.
- An embodiment is the saponin conjugate of the invention or the oligonucleotide conjugate of the invention, wherein A 1 represents a saccharide chain selected from group A and comprises or consists of a glucuronic acid moiety and wherein the carboxyl group of a glucuronic acid moiety of A 1 has been derivatised and/or wherein A 2 represents a saccharide chain selected from group B and A 2 comprises at least one acetoxy group and wherein at least one acetoxy group of A 2 has been derivatised.
- An embodiment is the saponin conjugate of the invention or the oligonucleotide conjugate of the invention, wherein the saponin represented by Molecule 1 is a bidesmosidic triterpene saponin.
- An embodiment is the saponin conjugate of the invention or the oligonucleotide conjugate of the invention, wherein the saponin derivative corresponds to the saponin represented by Molecule 1 wherein at least one, preferably one or two, more preferably one, of the following derivatisations is present: i.
- the aldehyde group at position C 23 of the quillaic acid or gypsogenin has been derivatised by; - reduction to an alcohol; - transformation into a hydrazone bond through reaction with N- ⁇ -maleimidocaproic acid hydrazide (EMCH), therewith providing a saponin-Ald-EMCH such as a SO1861-Ald-EMCH or a QS-21-Ald- EMCH, wherein the maleimide group of the EMCH is optionally derivatised by formation of a thio-ether bond with mercaptoethanol; - transformation into a hydrazone bond through reaction with N-[ß-maleimidopropionic acid] hydrazide (BMPH) wherein the maleimide group of the BMPH is optionally derivatised by formation of a thio-ether bond with mercaptoethanol; or - transformation into a hydrazone bond through reaction with N-[ ⁇ -maleimido
- the carboxyl group of a glucuronic acid moiety of A 1 when A 1 represents a saccharide chain selected from group A and A 1 comprises or consists of a glucuronic acid moiety, has been derivatised by transformation into an amide bond through reaction with 2-amino-2-methyl- 1,3-propanediol (AMPD) or N-(2-aminoethyl)maleimide (AEM), therewith providing a saponin-Glu-AMPD such as a QS-21-Glu-AMPD or a SO1861-Glu-AMPD or a saponin-Glu- AEM such as a QS-21-Glu-AEM or a SO1861-Glu-AEM; and iii.
- AMPD 2-amino-2-methyl- 1,3-propanediol
- AEM N-(2-aminoethyl)maleimide
- a 2 represents a saccharide chain selected from group B and A 2 comprises at least one acetoxy group, has/have been derivatised by transformation into a hydroxyl group (HO-) by deacetylation.
- An embodiment is the saponin conjugate of the invention or the oligonucleotide conjugate of the invention, wherein A 1 is Gal-(1 ⁇ 2)-[Xyl-(1 ⁇ 3)]-GlcA and/or A 2 is Glc-(1 ⁇ 3)-Xyl-(1 ⁇ 4)-Rha-(1 ⁇ 2)-[Xyl- (1 ⁇ 3)-4-OAc-Qui-(1 ⁇ 4)]-Fuc, preferably the saponin represented by Molecule 1 is 3-O-beta-D- galactopyranosyl-(1 ⁇ 2)-[beta-D-xylopyranosyl-(1 ⁇ 3)]-beta-D-glucuronopyranosyl quillaic acid 28-O- beta-D-glucopyranosyl-(1 ⁇ 3)-beta-D-xylopyranosyl-(1 ⁇ 4)- alpha-L-rhamn
- An embodiment is the saponin conjugate of the invention or the oligonucleotide conjugate of the invention, wherein the saponin is a saponin derivative wherein i. the saponin derivative comprises an aglycone core structure comprising an aldehyde group which has been derivatised by: - reduction to an alcohol; - transformation into a hydrazone bond through reaction with N- ⁇ -maleimidocaproic acid hydrazide (EMCH), therewith providing a saponin-Ald-EMCH such as a SO1861-Ald-EMCH or a QS-21-Ald- EMCH, wherein the maleimide group of the EMCH is optionally derivatised by formation of a thio-ether bond with mercaptoethanol; - transformation into a hydrazone bond through reaction with N-[ß-maleimidopropionic acid] hydrazide (BMPH) wherein the maleimide group of the BMPH is optionally der
- the saponin derivative comprises a saccharide chain, preferably a saccharide chain selected from group A, the saccharide chain comprising a carboxyl group, preferably a carboxyl group of a glucuronic acid moiety which has been derivatised by transformation into an amide bond through reaction with 2-amino-2-methyl-1,3-propanediol (AMPD) or N-(2-aminoethyl)maleimide (AEM), therewith providing a saponin-Glu-AMPD such as a QS-21-Glu- AMPD or a SO1861- Glu-AMPD or a saponin-Glu-AEM such as a QS-21-Glu-AEM or a SO1861-Glu-AEM; iii.
- AMPD 2-amino-2-methyl-1,3-propanediol
- AEM N-(2-aminoethyl)maleimide
- the saponin derivative comprises a saccharide chain, preferably a saccharide chain selected from group B, the saccharide chain comprising an acetoxy (Me(CO)O-) group which has been derivatised by transformation into a hydroxyl group (HO-) by deacetylation; or iv.
- the saponin derivative comprises any combination of derivatisations i., ii. and iii., preferably any combination of two derivatisations of derivatisations i., ii.
- the saponin derivative comprises an aglycone core structure wherein the aglycone core structure comprises an aldehyde group which has been derivatised by transformation into a hydrazone bond through reaction with EMCH wherein the maleimide group of the EMCH is optionally derivatised by formation of a thio-ether bond with mercaptoethanol.
- An embodiment is the saponin conjugate of the invention or the oligonucleotide conjugate of the invention, wherein the saponin is a saponin derivative wherein i.
- the saponin derivative comprises an aglycone core structure comprising an aldehyde group which has been derivatised by transformation into a hydrazone bond through reaction with N- ⁇ -maleimidocaproic acid hydrazide (EMCH), therewith providing a saponin-Ald-EMCH such as a SO1861-Ald-EMCH or a QS-21-Ald-EMCH; ii.
- EMCH N- ⁇ -maleimidocaproic acid hydrazide
- the saponin derivative comprises a saccharide chain, preferably a saccharide chain selected from group A, the saccharide chain comprising a carboxyl group, preferably a carboxyl group of a glucuronic acid moiety which has been derivatised by transformation into an amide bond through reaction with /V-(2-aminoethyl)maleimide (AEM), therewith providing a saponin-Glu- AEM such as a QS-21-Glu-AEM or a SO1861-Glu-AEM; or iii. the saponin derivative comprises a combination of derivatisations i. and ii.
- An embodiment is the saponin conjugate of the invention or the oligonucleotide conjugate of the invention, wherein the saponin derivative comprises an aglycone core structure wherein the aglycone core structure comprises an aldehyde group and wherein the saponin derivative comprises a saccharide chain, preferably a saccharide chain selected from group A, the saccharide chain comprising a carboxyl group, preferably a carboxyl group of a glucuronic acid moiety, which glucuronic acid moiety has been derivatised by transformation into an amide bond through reaction with /V-(2-aminoethyl)maleimide (AEM).
- AEM /V-(2-aminoethyl)maleimide
- An embodiment is the saponin conjugate of the invention or the oligonucleotide conjugate of the invention, wherein the saponin derivative is represented by Molecule 2: or wherein the saponin derivative is represented by Molecule 3:
- An embodiment is the saponin conjugate of the invention or the oligonucleotide conjugate of the invention, wherein the at least one saponin and the ligand for ASGPR are covalently linked directly or via at least one linker.
- An embodiment is the oligonucleotide conjugate of the invention, wherein the at least one saponin and the ligand for ASGPR are covalently linked directly or via at least one linker, and/or wherein the at least one saponin and the oligonucleotide are covalently linked directly or via at least one linker, and/or wherein the ligand for ASGPR and the oligonucleotide are covalently linked directly or via at least one linker, preferably, the at least one saponin, the ligand for ASGPR and the oligonucleotide are linked via at least one linker.
- An embodiment is the saponin conjugate of the invention, wherein the GalNAc moiety is bound to the saponin S, preferably via a saponin linker Ls, as represented in formula (II) S :
- An embodiment is the saponin conjugate of the invention, wherein the GalNAc moieties are each separately covalently bound via the oxygen on position “1” of the GalNAc moiety to a central bridging moiety B, which effectively forms a bridge between the GalNAc moieties and the saponin moiety, preferably via a saponin moiety linker L S , as shown in formula (III) S : wherein n is an integer larger than or equal to 2, preferably n is 3, L S is a saponin moiety linker and S is the saponin moiety.
- An embodiment is the saponin conjugate of the invention, wherein the GalNAc moieties are bound to the bridging moiety B via GalNAc linkers L GAL as shown in formula (IV) S : wherein n is an integer larger than or equal to 2, preferably n is 3, L S is a saponin moiety linker and S is the saponin moiety.
- An embodiment is the saponin conjugate of the invention, wherein the saponin moiety linker Ls represents any chemical moiety suitable for covalently binding a saponin to GalNAc as in formula (II) S or to the bridging moiety B as in formula (III) S and (IV) S .
- An embodiment is the saponin conjugate of the invention, wherein the saponin moiety linker L S is the result of a coupling reaction between at least a first precursor L S1 covalently bound to GalNAc or the bridging moiety B and a second precursor L S2 covalently bound to the saponin moiety, wherein L S1 is a precursor of the saponin moiety linker L S which is covalently bound to GalNAc or to the bridging moiety B and L S2 is a precursor of the saponin moiety linker L S which is covalently bound to the saponin moiety.
- An embodiment is the saponin conjugate of the invention, wherein the saponin moiety linker L S is the result of a coupling reaction between at least a first precursor L S1 covalently bound to GalNAc or the bridging moiety B and a second precursor L S2 covalently bound to the saponin moiety, wherein the coupling reaction is selected from the group consisting of an azide-alkyne cycloaddition, a thiol maleimide coupling, a Staudinger reaction, a nucleophilic ring-opening of strained heterocyclic electrophiles, a carbonyl reaction of the non-aldol type and an addition to a carbon-carbon double bond, preferably wherein the coupling reaction is an azide-alkyne cycloaddition, a thiol maleimide coupling, a Staudinger reaction, a nucleophilic ring-opening of strained heterocyclic electrophiles, more preferably wherein the coupling reaction is an azi
- An embodiment is the saponin conjugate of the invention, wherein the saponin moiety linker L S is the result of a coupling reaction between a first precursor L S1 covalently bound to GalNAc or the bridging moiety B, the first precursor L S1 comprising an azide; and a second precursor L S2 covalently bound to the saponin moiety, the second precursor L S2 comprising an alkyne and preferably comprising a hydrazone resulting from a hydrazide/aldehyde coupling to an aldehyde of the saponin moiety.
- An embodiment is the saponin conjugate of the invention, wherein the structure for the precursor L S1 is the following azide of formula (XVII): wherein a represents an integer larger than or equal to 0, preferably a represents an integer selected from 1, 2, and 3, more preferably a represents 2, or wherein the structure for the precursor L S1 comprises the following azide of formula (XVIII): wherein c represents an integer larger than or equal to 0, preferably c represents an integer in the range of 5-15, more preferably c represents 9.
- An embodiment is the saponin conjugate of the invention, wherein the structure for the precursor L S2 comprises the following hydrazone with formula (XIX): wherein a represents an integer larger than or equal to 0, preferably a represents an integer in the range of 2-6, more preferably a represents 4, and wherein L S2a represents an alkyne-containing moiety.
- An embodiment is the saponin conjugate of the invention, wherein L S2a comprises less than 20 carbon atoms, preferably L S2a represents a moiety according to formula (XX):
- An embodiment is the saponin conjugate of the invention, wherein the bridging moiety is a compound of formula (XV): wherein the oxygen atoms of the compound of formula (XV) are bound to GalNAc or to the GalNAc linkers L GAL , and the nitrogen atom of the compound of formula (XV) is bound to the saponin moiety linker L S .
- L GAL represents any chemical moiety suitable for covalently binding GalNAc to the bridging moiety B.
- An embodiment is the saponin conjugate of the invention, wherein L GAL comprises 2-25 carbon atoms, preferably 7-15 carbon atoms, more preferably 11 carbon atoms and wherein L GAL comprises at least one, preferably two amide moieties.
- An embodiment is the saponin conjugate of the invention, wherein L GAL is a compound according to formula (XVI):
- An embodiment is the oligonucleotide conjugate of the invention, wherein the at least one GalNAc moiety, the at least one saponin and the oligonucleotide are covalently bound via a tri-functional linker, preferably with each of the GalNAc moiety, the saponin and the oligonucleotide covalently bound to a separate arm of the tri-functional linker.
- a preferred oligonucleotide conjugate of the invention is an oligonucleotide conjugate, wherein the at least one GalNAc moiety, preferably three GalNAc moieties, at least one saponin, preferably 1-16 saponin moieties, more preferably 1-8 saponin moieties such as 1 , 4 or 8 saponin moieties, and the oligonucleotide are covalently bound via a trifunctional linker, preferably with each of the GalNAc moiety/moieties, the saponin/saponin moieties and the oligonucleotide covalently bound to a separate arm of the trifunctional linker.
- a trifunctional linker that is suitable for incorporation into an oligonucleotide conjugate of the invention is the trifunctional linker represented by formula (XXI):
- An embodiment is the oligonucleotide conjugate of the invention wherein a trifunctional linker, such as the trifunctional linker represented by formula (XXI), is covalently bound to one or more saponin moieties via a first arm of the linker, preferably 1-16 saponin moieties, more preferably 1-8 saponin moieties such as 1 , 4 or 8 saponin moieties, covalently bound to at least one GalNAc moiety, preferably 1-4 GalNAc moieties, more preferably 3 GalNAc moieties, via a second arm of the linker, and covalently bound to an oligonucleotide, preferably an AON such as a BNA or an siRNA, via a third arm of the trifunctional linker.
- a trifunctional linker such as the trifunctional linker represented by formula (XXI)
- An embodiment is the saponin conjugate of the invention or the oligonucleotide conjugate of the invention, wherein the ligand for ASGPR is (GalNAc) 3 Tris represented by Molecule I’: or wherein the ligand for ASGPR is mono-GalNAc represented by Molecule II’:
- An embodiment is the saponin conjugate of the invention, wherein the at least one saponin is covalently bound to Molecule I’ or Molecule II’ of the invention via a linker represented by Molecule III’:
- An embodiment is the oligonucleotide conjugate of the invention, wherein the at least one saponin is covalently bound to the ligand for ASGPR via at least one cleavable linker, and/or wherein the at least one saponin is covalently bound to the oligonucleotide via at least one cleavable linker.
- An embodiment is the saponin conjugate of the invention or the oligonucleotide conjugate of the invention, wherein the at least one saponin is covalently bound to the ligand for ASGPR via at least one cleavable linker.
- An embodiment is the saponin conjugate of the invention or the oligonucleotide conjugate of the invention, wherein the cleavable linker is subject to cleavage under acidic conditions, reductive conditions, enzymatic conditions and/or light-induced conditions, and preferably the cleavable linker comprises a cleavable bond selected from a hydrazone bond and a hydrazide bond subject to cleavage under acidic conditions, and/or a bond susceptible to proteolysis, for example proteolysis by Cathepsin B, and/or a bond susceptible for cleavage under reductive conditions such as a disulfide bond.
- the cleavable linker comprises a cleavable bond selected from a hydrazone bond and a hydrazide bond subject to cleavage under acidic conditions, and/or a bond susceptible to proteolysis, for example proteolysis by Cathepsin B, and/or a bond susceptible for cleavage
- An embodiment is the saponin conjugate of the invention or the oligonucleotide conjugate of the invention, wherein the cleavable linker is subject to cleavage in vivo under acidic conditions such as for example present in endosomes and/or lysosomes of mammalian cells, preferably human cells, preferably the cleavable linker is subject to cleavage in vivo at pH 4.0 - 6.5, and more preferably at pH ⁇ 5.5.
- An embodiment is the saponin conjugate of the invention or the oligonucleotide conjugate of the invention, wherein the conjugate comprises 1 , 2, 3, 4, 5, 6, 8, 10, 16, 32, 64, 128 or 1-100 saponin moieties, or any number of saponin moieties therein between, such as 7, 9, 12 saponin moieties, and preferably 1 , 4 or 8 saponin moieties.
- Preferred is the saponin SO1861 or SO1832, or a functional derivative thereof, and for example also QS-21 is a suitable saponin, or a functional derivative thereof.
- An embodiment is the saponin conjugate of the invention or the oligonucleotide conjugate of the invention, wherein the conjugate comprises 1 saponin moiety.
- An embodiment is the saponin conjugate of the invention or the oligonucleotide conjugate of the invention, wherein the conjugate comprises 4 or 8 saponin moieties.
- One of the many benefits of the conjugates of the invention and the application of the saponin conjugate and the oligonucleotide conjugate of the invention is the possibility to choose and select the number of saponin moieties in the conjugate that is suitable for achieving improved intracellular oligonucleotide activity (such as gene silencing activity), when compared to the activity of the oligonucleotide when taken up by a cell that is not co-contacted with saponin(s) together with the oligonucleotide.
- the inventors demonstrate (see the Examples section) that incorporation of a single saponin moiety in the conjugates of the invention already suffices for achieving improved intracellular oligonucleotide activity as measured as gene silencing.
- gene silencing is further improved by for example including 4 or 8 saponin moieties in the conjugate of the invention.
- the saponin conjugate and the oligonucleotide conjugate provide the freedom to couple any number between 1-64 or even more, such as 128 saponin moieties to the GalNAc moiety or moieties, preferably 3 GalNAc moieties, for the saponin conjugate, or to the GalNAc moiety/moieties and the oligonucleotide, for the oligonucleotide conjugate.
- the administered oligonucleotide as part of the oligonucleotide conjugate or separate as a GalNAc- oligonucleotide conjugate, that is present in the endosome together with the saponin, is, as a consequence of improved saponin-mediated endosomal escape activity, more efficiently transferred from the endosome into the cytosol, resulting in increased gene silencing activity.
- the possibility to incorporate one or more saponin moieties in the conjugate allows for tailormade design of the conjugate when optimal gene silencing with a selected oligonucleotide is considered.
- Oligonucleotides which transfer relatively inefficiently from the endosome to the cytosol, for example with an efficiency of 1-2%, benefit from the presence of a single saponin moiety in the conjugate (1:1 ratio in the oligonucleotide conjugate), whereas oligonucleotides which are released into the cytosol from the endosome to a lower extent, may increasingly be transferred into the cytosol by applying more than a single saponin moiety in the conjugate, such as 2-100 moieties, 2-64, 2-34, 2-16, 4-12, 4-8 moieties.
- a suitable method for incorporating more than one saponin moiety is for example the incorporation of a dendron in the conjugate of the invention, such as a G2, G3 or G4 dendron, for example for binding 4 or 8 or 16 saponin moieties together in a conjugate.
- a dendron in the conjugate of the invention such as a G2, G3 or G4 dendron
- examples of improved gene silencing are provided for, for example, oligonucleotide conjugates comprising a single saponin moiety, 4 saponin moieties and 8 saponin moieties.
- the inventors established that by contacting a cell with the oligonucleotide conjugate of the invention, the extent of gene silencing at a certain time point is improved and that also the duration in time of the gene silencing is extended, when compared to gene silencing in cells which are contacted with the oligonucleotide in the absence of saponin.
- the conjugate comprises 1 or 3 GalNAc moieties, preferably 3 GalNAc moieties.
- the saponin is typically one or more moieties of SO1861 or SO1832 (which is also referred to as SO1831), such as 1-32 moeities of SO1861 or SO1832, preferably 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15 or 16 moeities, more preferably 1, 2, 4, 8, 12 or 16 moieties, most preferably 1, 4 or 8 moeieties, such as 1, 4 or 8 SO1861 or 1, 4 or 8 SO1832.
- SO1832 is about equally active as SO1861 when endosomal escape enhancing activity towards an oligonucleotide (or protein toxin) in the endosome is considered.
- the number of saponin moieties can be optimized for the conjugates of the invention, when the extent and duration of gene silencing upon contacting a cell with an oligonucleotide is considered.
- this also applies to the number of GalNAc moieties in the conjugates of the invention.
- optimal gene silencing is established when 1 or 3 GalNAc moieties are comprised by the conjugates of the invention.
- the saponin SO1832 consists of the quillaic acid aglycone core with the carbohydrate substituent Gal-(1 ⁇ 2)-[Xyl-(1 ⁇ 3)]-GlcA- at the C-3beta-OH group and with the carbohydrate substituent Xyl-(1 ⁇ 3)-Xyl-(1 ⁇ 4)-Rha-(1 ⁇ 2)-[Xyl-(1 ⁇ 3)-4-OAc-Qui-(1 ⁇ 4)]-Fuc- at the C-28-OH group (See also Table A1).
- the chemical formula is C 82 H 128 O 45 and the exact mass is 1832,77 Dalton.
- the saponin structure of SO1832 is according to molecule (SO1832):
- the saponin comprised in the saponin conjugate of the present invention is a compound of formula (II) S , (III) S or (IV) S as described herein wherein the saponin moiety linker L S is the result of a coupling reaction between a first precursor L S1 covalently bound to GalNAc or the bridging moiety and a second precursor L S2 covalently bound to the saponin moiety, the second precursor L S2 comprising a hydrazone resulting from a hydrazide/aldehyde coupling to an aldehyde of the saponin moiety or comprising a semicarbazone functional group resulting from a semicarbazide/aldehyde coupling to an aldehyde of the saponin moiety, as described herein, this means that the aldehyde group is already occupied by the linker, such that suitable saponin derivative
- Effector molecule conjugate introduction
- Certain pharmaceutical combinations and certain pharmaceutical compositions of the present invention comprise a second conjugate of an effector molecule and a ligand for asialoglycoprotein receptor (ASGPR), referred to herein as “effector molecule conjugate”.
- ASGPR asialoglycoprotein receptor
- effector moiety When the effector molecule is bound to the remainder of the effector molecule conjugate it is referred to herein as an “effector moiety”.
- the ASGPR ligand comprises at least one N-acetylgalactosamine (GalNAc) moiety and preferably three GalNAc moieties (GN) 3 .
- each GalNAc moiety is bound to the remainder of the ASGPR ligand or - in case ASGPR ligand consists of a single GalNAc moiety - to the effector moiety, via a covalent bond to the oxygen on position “1” as indicated in formula (I): (I) As shown in formula (II) E , the ASGPR ligand consists of a single GalNAc moiety bound to the effector moiety E, preferably via an effector moiety linker L E .
- the ASGPR ligand may comprise more than one GalNAc moiety, such as 2, 3, 4, 5 or 6 GalNAc moieties, preferably 3 or 4 GalNAc moieties, most preferably three moieties.
- the GalNAc moieties are each separately covalently bound via the oxygen on position “1” to a central bridging moiety B, which effectively forms a bridge between the GalNAc moieties and the effector moiety, preferably via an effector moiety linker L E .
- the GalNAc moieties may be directly bound to the bridging moiety B as shown in formula (III) E : wherein n is an integer larger than or equal to 2, L E is an effector moiety linker and E is the effector moiety.
- the GalNAc moieties are bound to the bridging moiety B via GalNAc linkers L GAL as shown in formula (IV) E : (IV) E wherein n is an integer larger than or equal to 2, L E is an effector moiety linker and E is the effector moiety. While each occurrence of L GAL may be independently chosen, the skilled person will appreciate that the synthesis of the conjugate is simplified in case each occurrence of L GAL represents the same moiety. Effector molecule conjugate - Linker L E The effector moiety linker L E represents any chemical moiety suitable for covalently binding an effector moiety to GalNAc as in formula (II) E or to the bridging moiety B as in formula (III) E and (IV) E .
- linker is not particularly limited and covalently binding an effector molecule to GalNAc as in formula (II) E or to the bridging moiety B as in formula (III) E and (IV) E may be effected by means of a ‘regular’ chemical functional group (e.g. an ether bond) as well as through “click-chemistry” type linkers which typically have a long chain length, resulting in a linker EL comprising e.g. more than 10 or more than 20 carbon atoms.
- Suitable effector moiety linkers L E and associated coupling reactions are described in the handbook Hermanson, Greg T. Bioconjugate techniques. Academic press, 2013.
- the effector moiety linker L E will typically be the result of a coupling reaction (e.g. “click-chemistry” type) between at least a first precursor L E1 covalently bound to GalNAc or the bridging moiety B and a second precursor L E2 covalently bound to the effector moiety.
- This principle is illustrated in the following reaction scheme for the compound of formula (II): wherein L E is an effector moiety linker, L E1 is a precursor of the effector moiety linker L E which is covalently bound to GalNAc and L E2 is a precursor of the effector moiety linker L E which is covalently bound to the effector moiety and E is the effector moiety.
- the corresponding reaction scheme for the compound of formula (III) is as follows: wherein L E is an effector moiety linker, L E1 is a precursor of the effector moiety linker L E which is covalently bound to GalNAc and L E2 is a precursor of the effector moiety linker L E which is covalently bound to the effector moiety, n is an integer larger than or equal to 2 and E is the effector moiety.
- the corresponding reaction scheme for the compound of formula (IV) E is as follows:
- L E is an effector moiety linker
- L E1 is a precursor of the effector moiety linker L E which is covalently bound to GalNAc
- L E2 is a precursor of the effector moiety linker L E which is covalently bound to the effector moiety
- n is an integer larger than or equal to 2
- E is the effector moiety
- L GAL is a GalNAc linker and B is a bridging moiety.
- the effector moiety linker L E can be the result of a coupling reaction between at least a first precursor L E1 covalently bound to GalNAc or the bridging moiety and a second precursor L E2 covalently bound to the effector moiety, wherein the coupling reaction is for example an azide-alkyne cycloaddition, a thiol maleimide coupling, a Staudinger reaction, a nucleophilic ring-opening of strained heterocyclic electrophiles (such as aziridines, epoxides, cyclic sulphates, aziridinium ions, episulfonium ions), a carbonyl reaction of the non-aldol type (such as urea, thiourea, hydrazone, oxime ether, amide or aromatic heterocycle formation) or an addition to a carbon-carbon double bond (such as epoxidation, aziridination, dihydroxylation, sulfentyl halide addition,
- the effector moiety linker L E comprises a succinimide thio-ether moiety.
- a succinimide thio-ether is e.g. the result of a thiol maleimide coupling between an N-substituted maleimide and a thiol or sulfhydryl containing compound.
- This thiol maleimide coupling is a commonly known ‘click’-chemistry tool in the field of bio-conjugation and is for example described in Hermanson, Greg T. Bioconjugate techniques. Academic press, 2013 page 289.
- the effector moiety linker L E is the result of a coupling reaction between a first precursor L E1 covalently bound to GalNAc or the bridging moiety, the first precursor L E1 comprising an N-substituted maleimide; and a second precursor L E2 covalently bound to the effector moiety, the second precursor L E2 comprising a thiol or a precursor thereof.
- a suitable thiol precursor is a disulfide, which may be cleaved (e.g. in-situ) via reduction to the corresponding thiol.
- a preferred structure for the precursor L E1 present in the compounds of formula (V) E , (VII) E or (VIII) E is the following terminal N-substituted maleimide: wherein X represents any linker suitable for covalently bonding the terminal N-substituted maleimide to GalNAc or the bridging moiety B.
- X may be the result of a coupling reaction between a first moiety covalently bound to GalNAc or the bridging moiety and a second moiety covalently bound to the maleimide.
- X can comprise a hydrazone and/or a 1, 2, 3-triazole.
- Embodiments of the structure for the precursor L E1 present in the compounds of formula (V) E , (VII) E or (VIII) E are the following terminal N-substituted maleimides: (X) wherein a and b each independently represent an integer larger than or equal to 0, preferably a and b each independently represent an integer selected from 0, 1, 2, and 3, more preferably a and b represent 2, and wherein L E1a represents a hydrazone and/or a 1, 2, 3-triazole, preferably a 1, 2, 3-triazole and wherein L E1b represents a hydrazone and/or a 1, 2, 3-triazole, preferably a hydrazone; (XI) wherein b and c each independently represent an integer larger than or equal to 0, preferably b represents an integer selected from 0, 1, 2, and 3 and c represents an
- L E1a , L E1b and L E1c each preferably comprise less than 20 carbon atoms, more preferably L E1a , L E1b and L E1c each independently represent a moiety according to formula (XIII), (XIV) or (XV), preferably L E1a is the 1,2,3-triazole of formula (XIII), L E1b is the hydrazone of formula (XIV) and L E1c is the 1,2,3- triazole of formula (XV) :
- the effector molecule conjugate is a compound of formula (II) E , (III) E or (IV) E as described herein, wherein the effector moiety linker L E is the result of a coupling reaction between a compound of formula (V) E , (VII) E or (VIII) E with a compound of formula (VI) E , wherein the first precursor L E1 is a terminal N-substituted maleimide of formula (X), (XI) or
- Effector molecule conjugate - Effector moiety An aspect of the invention relates to a pharmaceutical combination comprising: - a first pharmaceutical composition comprising the saponin conjugate of the invention and optionally comprising a pharmaceutically acceptable excipient and/or a pharmaceutically acceptable diluent; and - a second pharmaceutical composition comprising a second conjugate of an effector molecule and a ligand for ASGPR, wherein the ligand for ASGPR preferably comprises at least one GalNAc moiety, preferably three or four GalNAc moieties, more preferably the ligand for ASGPR comprises or consists of (GalNAc) 3 Tris, or a third conjugate of an effector molecule and a binding molecule comprising a binding site for a cell-surface molecule, and optionally comprising a pharmaceutically acceptable excipient and/or pharmaceutically acceptable diluent.
- a pharmaceutical combination comprising: - a first pharmaceutical composition comprising the saponin conjugate of the invention and optionally comprising
- An aspect of the invention relates to a pharmaceutical composition
- a pharmaceutical composition comprising: - the saponin conjugate of the invention; - a second conjugate of an effector molecule and a ligand for ASGPR wherein the ligand for ASGPR preferably comprises at least one GalNAc moiety, preferably three or four GalNAc moieties, more preferably the ligand for ASGPR comprises or consists of (GalNAc) 3 Tris, or a third conjugate of an effector molecule and a binding molecule comprising a binding site for a cell-surface molecule, and optionally comprising a pharmaceutically acceptable excipient and/or pharmaceutically acceptable diluent.
- An embodiment is the pharmaceutical combination of the invention comprising the saponin conjugate of the invention or the pharmaceutical composition of the invention comprising the saponin conjugate of the invention, wherein the effector molecule comprises or consists of at least one of a small molecule such as a drug molecule, a toxin such as a protein toxin, an oligonucleotide such as an AON such as a BNA, a xeno nucleic acid or an siRNA, an enzyme, a peptide, a protein, or any combination thereof, preferably, the effector molecule is a toxin, an enzyme or an oligonucleotide.
- An embodiment is the pharmaceutical combination of the invention comprising the saponin conjugate of the invention or the pharmaceutical composition of the invention comprising the saponin conjugate of the invention, wherein the ligand for ASGPR comprises or is (GalNAc) 3 Tris, and/or wherein the ligand for ASGPR and the effector molecule are conjugated via a covalent bond, preferably via at least one linker.
- An embodiment is the pharmaceutical combination of the invention comprising the saponin conjugate of the invention or the pharmaceutical composition of the invention comprising the saponin conjugate of the invention, wherein the effector molecule is an oligonucleotide selected from any one or more of a(n): short interfering RNA (siRNA), short hairpin RNA (shRNA), anti-hairpin-shaped microRNA (miRNA), single-stranded RNA, aptamer RNA, double-stranded RNA (dsRNA), anti-microRNA (anti- miRNA, anti-miR), antisense oligonucleotide (ASO), DNA, antisense DNA, locked nucleic acid (LNA), bridged nucleic acid (BNA), 2’-O,4’-aminoethylene bridged nucleic Acid (BNA NC ), BNA-based siRNA, and BNA-based antisense oligonucleotide (BNA-AON).
- siRNA short interfering RNA
- An embodiment is the pharmaceutical combination of the invention comprising the saponin conjugate of the invention or the pharmaceutical composition of the invention comprising the saponin conjugate of the invention, wherein the effector molecule is an oligonucleotide selected from any one or more of a(n): anti-miRNA, a BNA-AON or an siRNA, such as BNA-based siRNA, selected from chemically modified siRNA, metabolically stable siRNA and chemically modified, metabolically stable siRNA.
- An embodiment is the pharmaceutical combination of the invention comprising the saponin conjugate of the invention or the pharmaceutical composition of the invention comprising the saponin conjugate of the invention, wherein the effector molecule is an oligonucleotide capable of, for example when present inside a mammalian cell, silencing any one of genes: apolipoprotein B (apoB), HSP27, transthyretin (TTR), proprotein convertase subtilisin/kexin type 9 (PCSK9), delta-aminolevulinate synthase 1 (ALAS1), anti-thrombin 3 (AT3), glycolate oxidase (GO), complement component C5 (CC5), X gene of hepatitis B virus (HBV), S gene of HBV, alpha-1 antitrypsin (AAT) and lactate dehydrogenase (LDH), and/or is an oligonucleotide capable of, for example when present inside a mammalian cell, targeting an aberrant miRNA.
- An embodiment is the pharmaceutical combination of the invention comprising the saponin conjugate of the invention or the pharmaceutical composition of the invention comprising the saponin conjugate of the invention, wherein the effector molecule is an oligonucleotide capable of, for example when present inside a mammalian cell, silencing any one of genes: HSP27, apoB, TTR, PCSK9, TMPRSS6, ALAS1, AT3, GO, CC5, X gene of HBV, S gene of HBV, AAT, miR- 122, hepatitis B virus HbsAg, LDHA, CEBPA and LDH, and/or for use in the treatment or prophylaxis of a disease or health problem which involves any one or more of genes: HSP27, apoB, TTR, PCSK9, TMPRSS6, ALAS1, AT3, GO, CC5, X gene of HBV, S gene of HBV, AAT, miR-122, hepatitis B virus HbsAg, LD
- Preferred target genes are HSP27, apoB, TTR, PCSK9, TMPRSS6, ALAS1, AAT, miR-122, hepatitis B virus HbsAg, LDHA and CEBPA, more preferred are genes HPS27 and apoB.
- An embodiment is the pharmaceutical combination of the invention comprising the saponin conjugate of the invention or the pharmaceutical composition of the invention comprising the saponin conjugate of the invention, wherein the effector molecule is an oligonucleotide capable of, for example when present inside a mammalian cell, targeting an mRNA involved in expression of any one of proteins: apoB, HSP27, TTR, PCSK9, ALAS1, AT3, GO, CC5, expression product of X gene of HBV, expression product of S gene of HBV, AAT and LDH, or is capable of, for example when present inside a mammalian cell, antagonizing or restoring an miRNA function such as inhibiting an oncogenic miRNA (onco-miR) or suppression of expression of an onco-miR.
- the effector molecule is an oligonucleotide capable of, for example when present inside a mammalian cell, targeting an mRNA involved in expression of any one of proteins: apoB, HSP27, TTR
- An embodiment is the pharmaceutical combination of the invention comprising the saponin conjugate of the invention or the pharmaceutical composition of the invention comprising the saponin conjugate of the invention, wherein the effector molecule is an oligonucleotide capable of, for example when present inside a mammalian cell, targeting an mRNA involved in expression of any one of proteins: HSP27, apoB, TTR, PCSK9, TMPRSS6, ALAS1, AT3, GO, CC5, X gene of HBV, S gene of HBV, AAT, miR-122, hepatitis B virus HbsAg, LDHA, CEBPA and LDH, preferably HSP27, apoB, TTR, PCSK9, TMPRSS6, ALAS1, AAT, miR-122, hepatitis B virus HbsAg, LDHA and CEBPA, or is capable of, for example when present inside a mammalian cell, antagonizing or restoring an miRNA function such as inhibiting an oncogenic mi
- An embodiment is the pharmaceutical combination of the invention comprising the saponin conjugate of the invention or the pharmaceutical composition of the invention comprising the saponin conjugate of the invention, wherein the effector molecule is or comprises a toxin which toxin comprises or consists of at least one molecule selected from any one or more of a peptide, a protein, an enzyme such as urease and Cre-recombinase, a proteinaceous toxin, a ribosome-inactivating protein, and/or a bacterial toxin, a plant toxin, more preferably selected from any one or more of a viral toxin such as apoptin; a bacterial toxin such as Shiga toxin, Shiga-like toxin, Pseudomonas aeruginosa exotoxin (PE) or exotoxin A of PE, full-length or truncated diphtheria toxin (DT), cholera toxin; a fungal tox
- the protein toxin is dianthin and/or saporin, and/or comprises or consists of at least one of a toxin targeting ribosome, a toxin targeting elongation factor, a toxin targeting tubulin, a toxin targeting DNA and a toxin targeting RNA, more preferably any one or more of emtansine,
- the bridging moiety B present in the saponin conjugates of formula (III) S or (IV) S and in the effector molecule conjugates of formula as (III) E or (IV) E described herein represents any moiety suitable for covalently binding 2 or more, preferably 3 or 4, more preferably 3 GalNAc moieties and the saponin moiety linker L S or the effector moiety linker L E .
- the bridging moiety B is a compound of formula (XV):
- the GalNAc linkers L GAL represent any chemical moiety suitable for covalently binding GalNAc to the bridging moiety as in formula (IV) S or (IV) E .
- the identity and size of the linker is not particularly limited.
- the GalNAc linkers L GAL each comprise 2-25 carbon atoms, preferably 7-15 carbon atoms, more preferably 11 carbon atoms.
- the GalNAc linkers L GAL comprise at least one, preferably two amide moieties.
- a particularly preferred GalNAc linker L GAL is a compound according to formula (XVI):
- An embodiment is the pharmaceutical combination of the invention comprising the saponin conjugate of the invention or the pharmaceutical composition of the invention comprising the saponin conjugate of the invention, wherein the binding molecule comprising a binding site for a cell-surface molecule, comprised by the third conjugate, is a ligand for a cell-surface molecule or an antibody comprising a binding site for a cell-surface molecule or at least one domain or fragment thereof comprising the binding site.
- An embodiment is the pharmaceutical combination of the invention comprising the saponin conjugate of the invention or the pharmaceutical composition of the invention comprising the saponin conjugate of the invention, wherein the third conjugate is any one or more of: an antibody-toxin conjugate, a receptor-ligand – toxin conjugate, an antibody-drug conjugate, a receptor-ligand – drug conjugate, an antibody-nucleic acid conjugate or a receptor-ligand – nucleic acid conjugate.
- An embodiment is the pharmaceutical combination of the invention comprising the saponin conjugate of the invention or the pharmaceutical composition of the invention comprising the saponin conjugate of the invention, wherein the binding molecule comprising a binding site for a cell-surface molecule, comprised by the third conjugate, is capable of binding to any one of cell-surface molecules: CD71, CD63, CA125, EpCAM(17-1A), CD52, CEA, CD44v6, FAP, EGF-IR, integrin, syndecan-1, vascular integrin alpha-V beta-3, HER2, EGFR, CD20, CD22, Folate receptor 1, CD146, CD56, CD19, CD138, CD27L receptor, PSMA, CanAg, integrin-alphaV, CA6, CD33, mesothelin, Cripto, CD3, CD30, CD239, CD70, CD123, CD352, DLL3, CD25, ephrinA4, MUC1, Trop2, CEACAM5, CEACAM6, HER3, CD74, PTK
- An embodiment is the pharmaceutical combination of the invention comprising the saponin conjugate of the invention or the pharmaceutical composition of the invention comprising the saponin conjugate of the invention, wherein the binding molecule comprising a binding site for a cell-surface molecule, comprised by the third conjugate, is or comprises any one of: an antibody, preferably a monoclonal antibody such as a human monoclonal antibody, an IgG, a molecule comprising or consisting of a single-domain antibody, at least one V HH domain, preferable a camelid V H , a variable heavy chain new antigen receptor (V NAR ) domain, a Fab, an scFv, an Fv, a dAb, an F(ab)2 and a Fcab fragment.
- an antibody preferably a monoclonal antibody such as a human monoclonal antibody, an IgG, a molecule comprising or consisting of a single-domain antibody, at least one V HH domain, preferable a camelid V H
- An aspect of the invention relates to the pharmaceutical combination of the invention comprising the saponin conjugate of the invention or to the pharmaceutical composition of the invention comprising the saponin conjugate of the invention, for use as a medicament.
- An aspect of the invention relates to the oligonucleotide conjugate of the invention, for use as a medicament.
- An aspect of the invention relates to the pharmaceutical combination of the invention comprising the saponin conjugate of the invention or to the pharmaceutical composition of the invention comprising the saponin conjugate of the invention, for use in the treatment or prophylaxis of a disease or health problem in which an expression product is involved of any one or more of genes: apoB, TTR, PCSK9, ALAS1, AT3, GO, CC5, X gene of HBV, S gene of HBV, AAT and LDH.
- An aspect of the invention relates to the pharmaceutical combination of the invention comprising the saponin conjugate of the invention or to the pharmaceutical composition of the invention comprising the saponin conjugate of the invention, for use in the treatment or prophylaxis of a disease or health problem in which an expression product is involved of any one or more of genes: HSP27, apoB, TTR, PCSK9, TMPRSS6, ALAS1, AT3, GO, CC5, X gene of HBV, S gene of HBV, AAT, miR-122, hepatitis B virus HbsAg, LDHA, CEBPA and LDH, preferably HSP27, apoB, TTR, PCSK9, TMPRSS6, ALAS1, AAT, miR-122, hepatitis B virus HbsAg, LDHA and CEBPA, more preferably HSP27, apoB.
- An embodiment is the pharmaceutical combination of the invention comprising the saponin conjugate of the invention or the pharmaceutical composition of the invention comprising the saponin conjugate of the invention, or the pharmaceutical combination comprising the saponin conjugate of the invention or pharmaceutical composition comprising the saponin conjugate of the invention for use of the invention, for use in the treatment or prophylaxis of a cancer, an infectious disease, a viral infection, hypercholesterolemia, cardiovascular disease, primary hyperoxaluria, haemophilia A, haemophilia B, AAT related liver disease, acute hepatic porphyria, TTR-mediated amyloidosis, hereditary TTR amyloidosis (hATTR), complement-mediated disease, hepatitis B infection, hepatitis C infection, ⁇ 1- antitrypsin deficiency, ⁇ -thalassaemia, or an auto-immune disease.
- a cancer an infectious disease, a viral infection, hypercholesterolemia, cardiovascular disease, primary hyperox
- An embodiment is the pharmaceutical combination comprising the saponin conjugate of the invention for use of the invention or the pharmaceutical composition comprising the saponin conjugate of the invention for use of the invention or the pharmaceutical composition comprising the oligonucleotide conjugate of the invention, wherein the saponin is a saponin derivative, preferably a QS- 21 derivative or an SO1861 derivative or an SO1832 derivative according to the invention.
- An aspect of the invention relates to an in vitro or ex vivo method for transferring the second conjugate or the third conjugate of the invention from outside a cell to inside said cell, preferably subsequently transferring the effector molecule comprised by the second conjugate or the third conjugate according to the invention into the cytosol of said cell, comprising the steps of: a) providing a cell which expresses ASGPR on its surface, and, when the third conjugate is to be transferred into the cell, which expresses the cell-surface molecule for which the third conjugate comprises a binding molecule for binding to said cell-surface molecule, the cell preferably selected from a liver cell, a virally infected cell and a tumor cell; b) providing the second conjugate or the third conjugate of the invention, for transferring into the cell provided in step a); c) providing the saponin conjugate of the invention; d) contacting the cell of step a) in vitro or ex vivo with the second conjugate or the third conjugate of step b) and
- An embodiment is the oligonucleotide conjugate of the invention, wherein the oligonucleotide is any one of a BNA, a xeno nucleic acid, an siRNA, an antisense oligonucleotide.
- An embodiment is the oligonucleotide conjugate of the invention, wherein the oligonucleotide is selected from any one or more of a(n): short interfering RNA (siRNA), short hairpin RNA (shRNA), anti- hairpin-shaped microRNA (miRNA), single-stranded RNA, aptamer RNA, double-stranded RNA (dsRNA), anti-microRNA (anti-miRNA, anti-miR), antisense oligonucleotide (ASO), DNA, antisense DNA, locked nucleic acid (LNA), bridged nucleic acid (BNA), 2’-O,4’-aminoethylene bridged nucleic acid (BNA NC ), BNA-based siRNA, and BNA-based antisense oligonucleotide (BNA-AON).
- siRNA short interfering RNA
- shRNA short hairpin RNA
- miRNA anti- hairpin-shaped microRNA
- dsRNA double
- An embodiment is the oligonucleotide conjugate of the invention, wherein the oligonucleotide is selected from any one or more of a(n): anti-miRNA, a BNA-AON or an siRNA, such as BNA-based siRNA, selected from chemically modified siRNA, metabolically stable siRNA and chemically modified, metabolically stable siRNA.
- a(n): anti-miRNA, a BNA-AON or an siRNA such as BNA-based siRNA, selected from chemically modified siRNA, metabolically stable siRNA and chemically modified, metabolically stable siRNA.
- an embodiment is the oligonucleotide conjugate of the invention, wherein the oligonucleotide is an oligonucleotide capable of, for example when present inside a mammalian cell and preferably when present inside a human cell, silencing any one of genes: apolipoprotein B (apoB), HSP27, transthyretin (TTR), proprotein convertase subtilisin/kexin type 9 (PCSK9), TMPRSS6, delta-aminolevulinate synthase 1 (ALAS1), antithrombin 3 (AT3), glycolate oxidase (GO), complement component C5 (CC5), X gene of hepatitis B virus (HBV), S gene of HBV, alpha-1 antitrypsin (AAT), miR-122, hepatitis B virus HbsAg, LDHA, CEBPA and lactate dehydrogenase (LDH), and/or is an oligonucleotide capable of,
- the gene is any one of the genes HSP27, apoB, TTR, PCSK9, TMPRSS6, ALAS1, AAT, miR-122, hepatitis B virus HbsAg, LDHA and CEBPA for expressing proteins HSP27, apoB, TTR, PCSK9, TMPRSS6, ALAS1, AAT, miR-122, hepatitis B virus HbsAg, LDHA and CEBPA.
- An embodiment is the oligonucleotide conjugate of the invention, wherein the oligonucleotide is an oligonucleotide capable of, for example when present inside a mammalian cell and preferably when present inside a human cell, silencing any one of genes: apolipoprotein B (apoB) and HSP27.
- oligonucleotide is an oligonucleotide capable of, for example when present inside a mammalian cell and preferably when present inside a human cell, silencing any one of genes: apolipoprotein B (apoB) and HSP27.
- apoB apolipoprotein B
- an embodiment is the oligonucleotide conjugate of the invention, wherein the oligonucleotide is an oligonucleotide capable of, for example when present inside a mammalian cell and preferably when present inside a human cell, targeting an mRNA involved in expression of any one of proteins: apoB, HSP27, TTR, PCSK9, TMPRSS6, ALAS1, AT3, GO, CC5, expression product of X gene of HBV, expression product of S gene of HBV, AAT, miR-122, hepatitis B virus HbsAg, LDHA, CEBPA and LDH, or is capable of, for example when present inside a mammalian cell and preferably when present inside a human cell, antagonizing or restoring an miRNA function such as inhibiting an oncogenic miRNA (onco-miR) or suppression of expression of an onco-miR.
- the oligonucleotide is an oligonucleotide capable of
- the protein is a liver protein selected from: HSP27, apoB, TTR, PCSK9, TMPRSS6, ALAS1, AAT, miR-122, hepatitis B virus HbsAg, LDHA and CEBPA.
- An embodiment is the oligonucleotide conjugate of the invention, wherein the oligonucleotide is an oligonucleotide capable of, for example when present inside a mammalian cell and preferably when present inside a human cell, targeting an mRNA involved in expression of any one of proteins: apoB and HSP27.
- An aspect of the invention relates to a method for providing the oligonucleotide conjugate of the invention, comprising the steps of: (a) providing at least one saponin moiety comprising a covalently bound first linker, wherein the first linker comprises at least one first reactive group for covalent binding to a second reactive group on a second linker or to a seventh reactive group on a seventh linker; (b) providing an oligonucleotide comprising a covalently bound third linker, wherein the third linker comprises a third reactive group for covalent binding to a fourth reactive group on a fourth linker or to an eighth reactive group on the seventh linker; (c) providing at least one GalNAc moiety comprising a covalently bound fifth linker, wherein the fifth linker comprises a fifth reactive group for covalent binding to a sixth reactive group on a sixth linker or to a ninth reactive group on the seventh linker; and either
- An embodiment is the method for providing the oligonucleotide conjugate of the invention, wherein the seventh linker is a tri-functional linker, such as the tri-functional linker represented by formula (XXI):
- Preferred is the method for providing the oligonucleotide conjugate of the invention, wherein the at least one saponin moiety are 1-16 saponin moieties, preferably 1-8 saponin moieties, such as 1 , 4 or 8 saponin moieties.
- An aspect of the invention relates to a pharmaceutical composition
- a pharmaceutical composition comprising the oligonucleotide conjugate of the invention, and optionally a pharmaceutically acceptable excipient and/or optionally a pharmaceutically acceptable diluent.
- An aspect of the invention relates to a pharmaceutical composition
- a pharmaceutical composition comprising the oligonucleotide conjugate of the invention, and optionally a pharmaceutically acceptable excipient and/or optionally a pharmaceutically acceptable diluent, for use as a medicament.
- An aspect of the invention relates to the oligonucleotide conjugate of the invention, for use as a medicament.
- An aspect of the invention relates to a pharmaceutical composition
- a pharmaceutical composition comprising the oligonucleotide conjugate of the invention, and optionally a pharmaceutically acceptable excipient and/or optionally a pharmaceutically acceptable diluent or to the oligonucleotide conjugate of the invention, for use in the treatment or prophylaxis of a disease or health problem in which an expression product is involved of any one or more of genes: HSP27, apoB, TTR, PCSK9, TMPRSS6, ALAS1 , AT3, GO, CC5, X gene of HBV, S gene of HBV, AAT, miR-122, hepatitis B virus HbsAg, LDHA, CEBPA and LDH, and/or for use in the treatment or prophylaxis of a disease or health problem which involves any one or more of genes: HSP27, apoB, TTR, PCSK9, TMPRSS6, ALAS1 , AT3, GO, CC5, X
- the disease is a cancer associated with (over-)expression of HSP27 in the tumor cell.
- Administering the oligonucleotide conjugate of the invention comprising an oligonucleotide for silencing HSP27 gene to a patient suffering from a tumor for which tumor growth is associated with (over-)expression of HSP27 results in silencing of the HSP27 gene and therewith with halting tumor growth.
- a health problem associated with apoB expression is a health problem related to elavated levels of LDL-cholesterol in the blood of a human subject, e.g.
- a patient at risk for atherosclerosis and/or hypercholesterolemia and/or a patient suffering from said atherosclerosis and/or hypercholesterolemia.
- Administering to said human subject or patient an oligonucleotide of the invention comprising an oligonucleotide for silencing the apoB gene results in inhibiting or lowering apoB expression and therewith as a consequence, lowering of LDL (- cholesterol), since apoB is the protein component of the LDL particle.
- a lowered level of circulating LDL is associated with less circulating LDL-cholesterol and therewith a diminished risk for LDL-cholesterol associated risk for health and disease symptoms as apparent in a patient suffering from atherosclerosis and/or hypercholesterolemia or in a subject at riks for developing atherosclerosis and/or hypercholesterolemia under influence of above normal blood level of LDL-cholesterol.
- An aspect of the invention relates to a pharmaceutical composition
- a pharmaceutical composition comprising the oligonucleotide conjugate of the invention or to the oligonucleotide conjugate of the invention, for use in the treatment or prophylaxis of a disease or health problem in which an expression product is involved of any one or more of genes: HSP27, apoB, TTR, PCSK9, TMPRSS6, ALAS1, AAT, miR-122, hepatitis B virus HbsAg, LDHA and CEBPA, and/or for use in the treatment or prophylaxis of a disease or health problem which involves any one or more of genes: HSP27, apoB, TTR, PCSK9, TMPRSS6, ALAS1, AAT, miR-122, hepatitis B virus HbsAg, LDHA and CEBPA.
- An embodiment is the pharmaceutical composition, for use according to the invention, comprising the oligonucleotide conjugate of the invention, or is the oligonucleotide conjugate of the invention, for use according to the invention, wherein said use is in the treatment or in the prophylaxis of a disease or health problem in which an expression product is involved of any one or more of genes: HSP27 and apoB, preferably apoB, and/or for use in the treatment or prophylaxis of a disease or health problem which involves any one or more of genes: HSP27 and apoB, preferably apoB.
- An embodiment is the pharmaceutical composition for use according to the invention, comprising the oligonucleotide conjugate of the invention, or is the oligonucleotide conjugate of the invention, for use according to the invention, wherein said use is in the treatment or in the prophylaxis of a cancer, an infectious disease, a viral infection, hypercholesterolemia, primary hyperoxaluria, haemophilia A, haemophilia B, AAT related liver disease, acute hepatic porphyria, TTR-mediated amyloidosis, hereditary TTR amyloidosis (hATTR), complement-mediated disease, hepatitis B infection, or an auto- immune disease.
- An embodiment is the pharmaceutical composition for use according to the invention, comprising the oligonucleotide conjugate of the invention, or is the oligonucleotide conjugate of the invention, for use according to the invention, wherein said use is in the treatment or in the prophylaxis of a cancer such as endometrial carcinoma, breast cancer, lung cancer and hypercholesterolemia, preferably hypercholesterolemia.
- a cancer such as endometrial carcinoma, breast cancer, lung cancer and hypercholesterolemia, preferably hypercholesterolemia.
- An aspect of the invention relates to a pharmaceutical composition
- a pharmaceutical composition comprising the oligonucleotide conjugate of the invention, or to the oligonucleotide conjugate of the invention, for use in the treatment or prophylaxis of a cancer, an infectious disease, a viral infection, hypercholesterolemia, cardiovascular disease, primary hyperoxaluria, haemophilia A, haemophilia B, AAT related liver disease, acute hepatic porphyria, TTR-mediated amyloidosis, hereditary TTR amyloidosis (hATTR), complement-mediated disease, hepatitis B infection, hepatitis C infection, ⁇ 1-antitrypsin deficiency, ⁇ -thalassaemia, or an auto- immune disease.
- An aspect of the invention relates to a pharmaceutical composition
- a pharmaceutical composition comprising the oligonucleotide conjugate of the invention, or to the oligonucleotide conjugate of the invention, for use in the treatment or prophylaxis of a cancer such as endometrial carcinoma, breast cancer, lung cancer or hepatocellular carcinoma, and/or a cardiovascular disease such as atherosclerosis and/or hypercholesterolemia, preferably atherosclerosis and/or hypercholesterolemia.
- the disease is a cancer associated with (over-)expression of HSP27 in the tumor cell.
- oligonucleotide conjugate of the invention comprising an oligonucleotide for silencing HSP27 gene to a patient suffering from a tumor for which tumor growth is associated with (over- )expression of HSP27, results in silencing of the HSP27 gene and therewith with halting tumor growth.
- a health problem associated with apoB expression is a health problem related to elavated levels of LDL-cholesterol in the blood of a human subject, e.g. a patient at risk for atherosclerosis and/or hypercholesterolemia, and/or a patient suffering from said atherosclerosis and/or hypercholesterolemia.
- Administering to said human subject or patient an oligonucleotide of the invention comprising an oligonucleotide for silencing the apoB gene results in inhibiting or lowering apoB expression and therewith as a consequence, lowering of LDL(-cholesterol), since apoB is the protein component of the LDL particle.
- a lowered level of circulating LDL is associated with less circulating LDL-cholesterol and therewith a diminished risk for LDL-cholesterol associated risk for health and disease symptoms as apparent in a patient suffering from atherosclerosis and/or hypercholesterolemia or in a subject at riks for developing atherosclerosis and/or hypercholesterolemia under influence of above normal blood level of LDL-cholesterol.
- An aspect of the invention relates to a pharmaceutical composition comprising the oligonucleotide conjugate of the invention, or to the oligonucleotide conjugate of the invention, for use in the lowering of LDL-cholesterol in a subject.
- An aspect of the invention relates to a pharmaceutical composition comprising the oligonucleotide conjugate of the invention, or to the oligonucleotide conjugate of the invention, for use in a method for lowering of LDL-cholesterol concentration in blood of a human subject.
- An aspect of the invention relates to a pharmaceutical composition comprising the oligonucleotide conjugate of the invention, or to the oligonucleotide conjugate of the invention, for use in a method for the treatment or prophylaxis of a cardiovascular disease.
- the treatment or prophylaxis is in a human patient suffering from a cardiovascular disease or in a human subject at risk for developing a cardiovascular disease.
- the cardiovascular disease is associated with an elevated blood level of LDL-cholesterol compared to the upper limit of the range of normal LDL-cholesterol levels.
- An aspect of the invention relates to an in vitro or ex vivo method for transferring the oligonucleotide conjugate of the invention from outside a cell to inside said cell, preferably for subsequently transferring the oligonucleotide comprised by the oligonucleotide conjugate of the invention into the cytosol and/or into the nucleus of said cell, comprising the steps of: a) providing a cell which expresses ASGPR, preferably ASGPR1 , on its surface, the cell preferably selected from a liver cell, a virally infected mammalian cell and a mammalian tumor cell, wherein preferably said cell is a human cell; b) providing the oligonucleotide conjugate of the invention for transferring into the cell provided in step a); c) contacting the cell of step a) in vitro or ex vivo with the oligonucleotide conjugate of step b), preferably in a liquid medium, therewith effecting the transfer
- toxin cytotoxicity or BNA-mediated gene silencing is considered (without wishing to be bound by any theory: relating to similar or improved endosomal escape enhancing activity of the modified saponin), if the aldehyde functional group is transformed to a semicarbazone functional group according to formula (11); and/or has reduced hemolytic activity, when compared with the toxicity, activity and haemolytic activity of unmodified saponin. That is to say, the saponin derivative has at least one, preferably to, more preferably all three of:
- the inventors provide saponin derivatives with an improved therapeutic window, since for the saponin derivatives, the cytotoxicity is lower than cytotoxicity determined for their naturally occurring counterparts, the haemolytic activity is lower than haemolytic activity determined for the naturally occurring counterparts of the saponin derivatives, the ratio between IC50 values for cell toxicity and e.g. IC50 values for toxin potentiation or IC50 values for gene silencing is similar or increased, and/or since the ratio between IC50 values for saponin haemolytic activity and e.g. IC50 values fortoxin potentiation or IC50 values for gene silencing is similar or increased.
- the term “endosomal escape enhancing activity” can be abbreviated as ‘activity’.
- the inventors surprisingly established (tumor) cell killing by contacting such cells with a saponin conjugate based on a saponin derivative comprising the semicarbazone functional group, together with an ADC such as an antibody - protein toxin conjugate, despite the medium to low expression of the cell-surface receptor targeted by the cell-surface molecule binding-molecule (e.g. antibody) comprised by the saponin conjugate and/or despite the medium to low expression of the cellsurface receptor targeted by the ADC.
- the saponin derivative and the saponin conjugate comprise the semicarbazone functional group.
- a more potent saponin derivative and saponin conjugate when the activation or potentiating of an effector moiety such as an effector moiety comprised by an ADC or AOC, is considered.
- This has the benefit that a lower amount of the saponin derivatives according to the invention should be administered to a patient in need of potentiation of e.g.
- an ADC or an AOC or a gene-silencing oligonucleotide such as a BNA, e.g. coupled to one or more GalNAc moieties, to obtain the same amount of native saponin comprising the “free” aldehyde to act as endosomal escape enhancers for targeted toxins or targeted oligonucleotides, compared to the required amount of saponin derivative comprising e.g. the hydrazone functional group ( N-N(H)-C(O)-).
- the hydrazone functional group is at the basis for the improved activity of the saponin conjugate comprising a saponin derivative comprising a semicarbazone functional group as established by the inventors, such as a conjugate comprising the saponin, an oligonucleotide and a cell-surface receptor ligand such as one to three GalNAc moieties (for targeting (liver) cells expressing ASGPR), wherein the activity is the potentiation of the effector moiety activity such as a gene-silencing oligonucleotide, inside the cytosol or nucleus of the targeted cell by endosomal escape enhancement.
- the effector moiety activity such as a gene-silencing oligonucleotide
- transformation (derivatisation) of the aldehyde group at C-23 of the aglycone of the saponin into a semicarbazone functional group according to formula (I1) results in a decrease in cytotoxicity when such saponin derivatives are contacted with cells, i.e. various types of cells. It is thus beneficial that these series of saponin derivatives with decreased cytotoxicity are provided, wherein the decrease in cytotoxicity is relative to the cytotoxicity as determined for the unmodified naturally occurring saponin counterparts.
- the saponin derivatives can be formed from such naturally occurring saponins, such as SO1861, equally active SO1832 and QS-21 (isoforms), preferably from SO1861 or SO1832.
- saponin derivatives comprising the semicarbazone functional group
- saponins with decreased cytotoxicity are equally suitable, when saponins with decreased cytotoxicity are to be provided.
- the inventors thus have provided saponin derivatives with an improved therapeutic window when cytotoxicity is considered and/or when haemolytic activity is considered, and when the potentiation of e.g. (gene-silencing) oligonucleotides is considered compared to the corresponding underivatised saponin.
- Such saponin derivatives comprising the semicarbazone functional group are for example in particular suitable for application in a therapeutic regimen involving a GalNAc-oligonucleotide conjugate which is combined with the saponin derivative or which conjugate is the oligonucleotide conjugate of the invention comprising the covalently linked saponin together with the GalNAc moiety/moieties and the oligonucleotide, for the prophylaxis or treatment of e.g. a cancer, hypercholesterolemia, cardiovascular disease or enhanced level of LDL-cholesterol above normal levels for healthy subjects in a (human) subject in need thereof.
- a cancer hypercholesterolemia
- cardiovascular disease or enhanced level of LDL-cholesterol above normal levels for healthy subjects in a (human) subject in need thereof.
- the safety of such saponin derivatives is improved when cytotoxicity and/or haemolytic activity is considered, especially when such saponin derivatives are administered to a patient in need of e.g. treatment with an ADC or with and AOC or with a conjugate comprising at least one GalNAc moiety and comprising an oligonucleotide, e.g. a BNA for silencing a gene such as HSP27 and apoB, or when the saponin derivative comprising the semicarbazone functional group is part of the oligonucleotide conjugate of the invention.
- a conjugate comprising at least one GalNAc moiety and comprising an oligonucleotide, e.g. a BNA for silencing a gene such as HSP27 and apoB, or when the saponin derivative comprising the semicarbazone functional group is part of the oligonucleotide conjugate of the invention.
- An embodiment is the oligonucleotide conjugate of the invention comprising one saponin moiety.
- An aspect of the invention relates to oligonucleotide conjugate according to molecule (EE):
- R 1 and R 2 are independently selected from hydrogen, a monosaccharide, a linear oligosaccharide and a branched oligosaccharide, and wherein the saponin moiety according to formula (SM) is based on a saponin comprising an aldehyde group in position C-23, and with (2) GalNAc conjugate according to molecule (FF): , wherein represents the tri-GalNAc conjugate according to molecule (DD):
- oligonucleotide provided with a linker according to molecule (GG): , wherein the molecule (GG) represents the conjugation product of the conjugation reaction between the linker (E)-1-(4-((2-(6-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1- yl)hexanoyl)hydrazineylidene)methyl)benzamido)-N-(4-(6-methyl-1,2,4,5-tetrazin-3-yl)benzyl)-3,6,9,12- tetraoxapentadecan-15-amide and the oligonucleotide-linker molecule according to molecule (HH): (HH).
- HH HH
- Such an oligonucleotide conjugate according to molecule (EE) of the invention displays a surprisingly improved potency when the endosomal escape enhancing activity of the saponin moiety is considered. It is thought that improved efficacy relates to improved de-coupling (release) of the saponin from the conjugate by cleavage of the semicarbazone functional group under influence of the slightly acidic pH in the endosome of cells which have taken up the conjugate mediated by binding of the conjugate to the ASGPR.
- An embodiment is the oligonucleotide conjugate of the invention comprising four saponin moieties.
- An aspect of the invention relates to oligonucleotide conjugate according to molecule (PP):
- molecule (PP) is the covalent conjugation product obtained by the covalent conjugation of the saponin-GalNAc conjugate according to molecule (LL):
- GG represents the conjugation product of the conjugation reaction between the linker (E)-1-(4-((2-(6-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1- yl)hexanoyl)hydrazineylidene)methyl)benzamido)-N-(4-(6-methyl-1,2,4,5-tetrazin-3-yl)benzyl)-3,6,9,12- tetraoxapentadecan-15-amide and the oligonucleotide-linker molecule according to molecule (HH): , which molecule (LL) is the covalent conjugation product obtained by the covalent conjugation of the saponin derivative according to molecule (JJ):
- molecule (JJ) is the conjugate product of conjugation of N,N'-((9S,19S)-14-(6- aminohexanoyl)-1-mercapto-9-(3-mercaptopropanamido)-3,10,18-trioxo-4,11,14,17-tetraazatricosane- 19,23-diyl)bis(3-mercaptopropanamide first with the saponin derivative according to molecule (KK):
- SM saponin moiety according to formula (SM): , wherein R 1 and R 2 are independently selected from hydrogen, a monosaccharide, a linear oligosaccharide and a branched oligosaccharide according to the invention, and wherein the saponin moiety according to formula (SM) is based on a saponin comprising an aldehyde group in position C-23 according to any one of the saponins of the invention, and for example listed in Table A1, such as QS- 21, SO1861, SO1832, and subsequently with 2,5-dioxopyrrolidin-1-yl 1-azido-3,6,9,12-tetraoxapentadecan-15-oate, , and wherein molecule (MM) is the conjugate of the tri-functional linker according to formula (XXI):
- An embodiment is the oligonucleotide conjugate of the invention comprising eight saponin moieties.
- An aspect of the invention relates to oligonucleotide conjugate according to molecule (SS):
- molecule (SS) is the covalent conjugation product obtained by the covalent conjugation of the saponin-GalNAc conjugate according to molecule (RR):
- GG represents the conjugation product of the conjugation reaction between the linker (E)-1-(4-((2-(6-(2,5-dioxo-2,5-dihydro-1H-pyrrol-1- yl)hexanoyl)hydrazineylidene)methyl)benzamido)-N-(4-(6-methyl-1,2,4,5-tetrazin-3-yl)benzyl)-3,6,9,12- tetraoxapentadecan-15-amide and the oligonucleotide-linker molecule according to molecule (HH): , which molecule (RR) is the covalent conjugation product obtained by the covalent conjugation of the saponin derivative according to molecule (QQ):
- molecule (QQ) is the conjugate product of conjugation of (2S)-N-[(1S)-1- ⁇ [2-(6-amino- N- ⁇ 2-[(2S)-2,6-bis[(2S)-2,6-bis(3- sulfanylpropanamido)hexanamido]hexanamido]ethyl ⁇ hexanamido)ethyl]carbamoyl ⁇ -5-[(2S)-2,6-bis(3- sulfanylpropanamido)hexanamido]pentyl]-2,6-bis(3-sulfanylpropanamido)hexanamide formate first (a) with the saponin derivative according to molecule (KK): , wherein represents a saponin moiety according to formula (SM): , wherein R 1 and R 2 are independently selected from
- An embodiment is the oligonucleotide conjugate according to the invention and according to any one of the molecules (EE), (PP) and (SS), wherein the oligonucleotide conjugate is based on a saponin according to any one of the saponins listed here above and in Table A1.
- the oligonucleotide conjugate according to the invention and according to any one of the molecules (EE), (PP) and (SS), wherein the oligonucleotide conjugate is based on a saponin selected from SO1861, SO1832 and QS-21, preferably SO1861 and SO1832, more preferably SO1861 or SO1832.
- a saponin selected from SO1861, SO1832 and QS-21, preferably SO1861 and SO1832, more preferably SO1861 or SO1832.
- oligonucleotide conjugate is conjugated with an oligonucleotide and with at least one GalNAc moiety, to provide an oligonucleotide conjugate of the invention.
- An embodiment is the oligonucleotide conjugate according to the invention and according to any one of the molecules (EE), (PP) and (SS), comprising an oligonucleotide, wherein the oligonucleotide is an AON such as any one of a BNA, a xeno nucleic acid, an siRNA.
- An embodiment is the oligonucleotide conjugate according to the invention and according to any one of the molecules (EE), (PP) and (SS), comprising an oligonucleotide, wherein the oligonucleotide is selected from any one or more of a(n): short interfering RNA (siRNA), short hairpin RNA (shRNA), anti- hairpin-shaped microRNA (miRNA), single-stranded RNA, aptamer RNA, double-stranded RNA (dsRNA), anti-microRNA (anti-miRNA, anti-miR), antisense oligonucleotide (ASO), DNA, antisense DNA, locked nucleic acid (LNA), bridged nucleic acid (BNA), 2’-O,4’-aminoethylene bridged nucleic acid (BNA NC ), BNA-based siRNA, and BNA-based antisense oligonucleotide (BNA-AON).
- siRNA short interfering
- An embodiment is the oligonucleotide conjugate according to the invention and according to any one of the molecules (EE), (PP) and (SS), comprising an oligonucleotide, wherein the oligonucleotide is selected from any one or more of a(n): anti-miRNA, a BNA-AON or an siRNA, such as BNA-based siRNA, selected from chemically modified siRNA, metabolically stable siRNA and chemically modified, metabolically stable siRNA.
- An embodiment is the oligonucleotide conjugate according to the invention and according to any one of the molecules (EE), (PP) and (SS), comprising an oligonucleotide, wherein the oligonucleotide is an oligonucleotide capable of, for example when present inside a mammalian cell and preferably when present inside a human cell, silencing any one of genes: apolipoprotein B (apoB), HSP27, transthyretin (TTR), proprotein convertase subtilisin/kexin type 9 (PCSK9), TMPRSS6, delta-aminolevulinate synthase 1 (ALAS1), anti-thrombin 3 (AT3), glycolate oxidase (GO), complement component C5 (CC5), X gene of hepatitis B virus (HBV), S gene of HBV, alpha-1 antitrypsin (AAT), miR-122, hepatitis B virus HbsAg, LDHA, CE
- An embodiment is the oligonucleotide conjugate according to the invention and according to any one of the molecules (EE), (PP) and (SS), comprising an oligonucleotide, wherein the oligonucleotide is an oligonucleotide capable of, for example when present inside a mammalian cell and preferably when present inside a human cell, silencing any one of genes: apolipoprotein B (apoB) and HSP27.
- An embodiment is the oligonucleotide conjugate according to the invention and according to any one of the molecules (EE), (PP) and (SS), comprising an oligonucleotide, wherein the oligonucleotide is an oligonucleotide capable of, for example when present inside a mammalian cell and preferably when present inside a human cell, targeting an mRNA involved in expression of any one of proteins: apoB, HSP27, TTR, PCSK9, TMPRSS6, ALAS1 , AT3, GO, CC5, expression product of X gene of HBV, expression product of S gene of HBV, AAT, miR-122, hepatitis B virus HbsAg, LDHA, CEBPA and LDH, or is capable of, for example when present inside a mammalian cell and preferably when present inside a human cell, antagonizing or restoring an miRNA function such as inhibiting an oncogenic miRNA (onco-miR) or suppression of expression
- An embodiment is the oligonucleotide conjugate according to the invention and according to any one of the molecules (EE), (PP) and (SS), comprising an oligonucleotide, wherein the oligonucleotide is an oligonucleotide capable of, for example when present inside a mammalian cell and preferably when present inside a human cell, targeting an mRNA involved in expression of any one of proteins: HSP27, apoB, TTR, PCSK9, TMPRSS6, ALAS1 , AAT, miR-122, hepatitis B virus HbsAg, LDHA and CEBPA.
- An embodiment is the oligonucleotide conjugate according to the invention and according to any one of the molecules (EE), (PP) and (SS), comprising an oligonucleotide, wherein the oligonucleotide is an oligonucleotide capable of, for example when present inside a mammalian cell and preferably when present inside a human cell, targeting an mRNA involved in expression of any one of proteins: apoB and HSP27.
- An aspect of the invention relates to a second pharmaceutical composition
- a second pharmaceutical composition comprising the oligonucleotide conjugate of the invention and according to any one of the molecules (EE), (PP) and (SS), and optionally a pharmaceutically acceptable excipient and/or optionally a pharmaceutically acceptable diluent.
- An aspect of the invention relates to the second pharmaceutical composition of the invention or to the oligonucleotide conjugate of the invention and according to any one of the molecules (EE), (PP) and (SS), for use as a medicament.
- An aspect of the invention relates to the second pharmaceutical composition of the invention or to the oligonucleotide conjugate of the invention and according to any one of the molecules (EE), (PP) and (SS), for use in the treatment or prophylaxis of a disease or health problem in which an expression product is involved of any one or more of genes: HSP27, apoB, TTR, PCSK9, TMPRSS6, ALAS1, AT3, GO, CC5, X gene of HBV, S gene of HBV, AAT, miR-122, hepatitis B virus HbsAg, LDHA, CEBPA and LDH, and/or for use in the treatment or prophylaxis of a disease or health problem which involves any one or more of genes: HSP27, apoB,
- An aspect of the invention relates to the second pharmaceutical composition of the invention or to the oligonucleotide conjugate of the invention and according to any one of the molecules (EE), (PP) and (SS), for use in the treatment or prophylaxis of a disease or health problem in which an expression product is involved of any one or more of genes: HSP27, apoB, TTR, PCSK9, TMPRSS6, ALAS1, AAT, miR-122, hepatitis B virus HbsAg, LDHA and CEBPA, and/or for use in the treatment or prophylaxis of a disease or health problem which involves any one or more of genes: HSP27, apoB, TTR, PCSK9, TMPRSS6, ALAS1, AAT, miR-122, hepatitis B virus HbsAg, LDHA and CEBPA.
- An embodiment is the second pharmaceutical composition of the invention or the oligonucleotide conjugate of the invention and according to any one of the molecules (EE), (PP) and (SS), for use as here above outlined, wherein said use is in the treatment or prophylaxis of a disease or health problem in which an expression product is involved of any one or more of genes: HSP27 and apoB, preferably apoB, and/or for use in the treatment or prophylaxis of a disease or health problem which involves any one or more of genes: HSP27 and apoB, preferably apoB.
- An embodiment is the second pharmaceutical composition of the invention or the oligonucleotide conjugate of the invention and according to any one of the molecules (EE), (PP) and (SS), for use as here above outlined, for use in the treatment or prophylaxis of a cancer, an infectious disease, a viral infection, hypercholesterolemia, cardiovascular disease, primary hyperoxaluria, haemophilia A, haemophilia B, AAT related liver disease, acute hepatic porphyria, TTR-mediated amyloidosis, hereditary TTR amyloidosis (hATTR), complement-mediated disease, hepatitis B infection, hepatitis C infection, ⁇ 1-antitrypsin deficiency, ⁇ -thalassaemia, or an auto-immune disease.
- a cancer an infectious disease, a viral infection, hypercholesterolemia, cardiovascular disease, primary hyperoxaluria, haemophilia A, haemophilia B, AAT related
- An embodiment is the second pharmaceutical composition of the invention or the oligonucleotide conjugate of the invention and according to any one of the molecules (EE), (PP) and (SS), for use as here above outlined, wherein said use is in the treatment or prophylaxis of a cancer such as endometrial carcinoma, breast cancer, lung cancer or hepatocellular carcinoma, and/or a cardiovascular disease such as hypercholesterolemia, preferably hypercholesterolemia.
- An aspect of the invention relates to the second pharmaceutical composition of the invention or to the oligonucleotide conjugate of the invention and according to any one of the molecules (EE), (PP) and (SS), for use in the lowering of LDL-cholesterol in a subject.
- Saponin conjugate comprising at least one saponin covalently linked to a ligand for asialoglycoprotein receptor (ASGPR), wherein the ligand for ASGPR comprises at least one N-acetylgalactosamine (GalNAc) moiety, preferably three or four GalNAc moieties, more preferably the ligand for ASGPR comprises or consists of (GalNAc) 3 Tris, wherein the at least one saponin is selected from monodesmosidic triterpenoid saponins and bidesmosidic triterpenoid saponins. 2.
- ASGPR asialoglycoprotein receptor
- saponin conjugate of any one of the embodiments 1-3 wherein the saponin is selected from the group consisting of: Quillaja bark saponin, dipsacoside B, saikosaponin A, saikosaponin D, macranthoidin A, esculentoside A, phytolaccagenin, aescinate, AS6.2, NP-005236, AMA-1, AMR, alpha-Hederin, NP-012672, NP-017777, NP-017778, NP-017774, NP-018110, NP-017772, NP- 018109, NP-017888, NP-017889, NP-018108, SA1641, AE X55, NP-017674, NP-017810, AG1, NP- 003881, NP-017676, NP-017677, NP-017706, NP-017705, NP-017773, NP-017775, SA1657, AG2, SO1861, GE1741, SO1542, SO
- saponin conjugate of embodiment 3 or 4 wherein the saponin is a saponin derivative wherein i. the saponin derivative comprises an aglycone core structure comprising an aldehyde group which has been derivatised; ii. the saponin derivative comprises a saccharide chain, preferably a saccharide chain selected from group A as defined in embodiment 3, the saccharide chain comprising a carboxyl group which has been derivatised; iii. the saponin derivative comprises a saccharide chain, preferably a saccharide chain selected from group B as defined in embodiment 3, the saccharide chain comprising an acetoxy (Me(CO)O-) group which has been derivatised; or iv.
- the saponin derivative comprises an aglycone core structure comprising an aldehyde group which has been derivatised; ii. the saponin derivative comprises a saccharide chain, preferably a saccharide chain selected from group A as defined in embodiment 3, the
- the saponin derivative comprises any combination of derivatisations i., ii. and iii., preferably any combination of two derivatisations of derivatisations i., ii. and iii. 6.
- Saponin conjugate of any one of the embodiments 1-5 wherein the saponin is any one or more of: SO1861, SA1657, GE1741, SA1641, QS-21, QS-21A, QS-21 A-api, QS-21 A-xyl, QS-21B, QS-21 B- api, QS-21 B-xyl, QS-7-xyl, QS-7-api, QS-17-api, QS-17-xyl, QS1861, QS1862, Quillajasaponin, Saponinum album, QS-18, Quil-A, Gyp1, gypsoside A, AG1, AG2, SO1542, SO1584, SO1658, SO1674, SO1832, SO1904, stereoisomers thereof, derivatives thereof
- the aldehyde group at position C 23 of the quillaic acid or gypsogenin has been derivatised; ii. the carboxyl group of a glucuronic acid moiety of A 1 , when A 1 represents a saccharide chain selected from group A as defined in embodiment 3 and A 1 comprises or consists of a glucuronic acid moiety, has been derivatised; and iii. one or more, preferably all, of acetoxy group(s) of one saccharide moiety or of two or more saccharide moieties of A 2 , when A 2 represents a saccharide chain selected from group B as defined in embodiment 3 and A 2 comprises at least one acetoxy group, has/have been derivatised.
- Saponin conjugate of embodiment 7 wherein A 1 represents a saccharide chain selected from group A as defined in embodiment 3 and comprises or consists of a glucuronic acid moiety and wherein the carboxyl group of a glucuronic acid moiety of A 1 has been derivatised and/or wherein A 2 represents a saccharide chain selected from group B as defined in embodiment 3 and A 2 comprises at least one acetoxy group and wherein at least one acetoxy group of A 2 has been derivatised.
- Saponin conjugate of embodiment 7 or 8 wherein the saponin represented by Molecule 1 is a bidesmosidic triterpene saponin. 10.
- the aldehyde group at position C 23 of the quillaic acid or gypsogenin has been derivatised by; - reduction to an alcohol; - transformation into a hydrazone bond through reaction with N- ⁇ -maleimidocaproic acid hydrazide (EMCH), therewith providing a saponin-Ald-EMCH such as a SO1861-Ald-EMCH or a QS-21-Ald- EMCH, wherein the maleimide group of the EMCH is optionally derivatised by formation of a thio-ether bond with mercaptoethanol; - transformation into a hydrazone bond through reaction with N-[ß-maleimidopropionic acid] hydrazide (BMPH) wherein the maleimide group of the BMPH is optionally derivatised by formation of a thio-ether bond with mercaptoethanol; or - transformation into a hydrazone bond through reaction with N-[ ⁇ -maleimido
- the carboxyl group of a glucuronic acid moiety of A 1 when A 1 represents a saccharide chain selected from group A as defined in embodiment 3 and A 1 comprises or consists of a glucuronic acid moiety, has been derivatised by transformation into an amide bond through reaction with 2-amino-2-methyl-1,3-propanediol (AMPD) or N-(2-aminoethyl)maleimide (AEM), therewith providing a saponin-Glu-AMPD such as a QS-21-Glu-AMPD or a SO1861- Glu-AMPD or a saponin-Glu-AEM such as a QS-21-Glu-AEM or a SO1861-Glu-AEM; and iii.
- AMPD 2-amino-2-methyl-1,3-propanediol
- AEM N-(2-aminoethyl)maleimide
- a 2 represents a saccharide chain selected from group B as defined in embodiment 3 and A 2 comprises at least one acetoxy group, has/have been derivatised by transformation into a hydroxyl group (HO-) by deacetylation.
- a 1 is Gal-(1 ⁇ 2)-[Xyl-(1 ⁇ 3)]-GlcA and/or A 2 is Glc-(1 ⁇ 3)-Xyl-(1 ⁇ 4)-Rha-(1 ⁇ 2)-[Xyl-(1 ⁇ 3)-4-OAc-Qui-(1 ⁇ 4)]-Fuc
- the saponin represented by Molecule 1 is 3-O-beta-D-galactopyranosyl-(1 ⁇ 2)-[beta-D-xylopyranosyl- (1 ⁇ 3)]-beta-D-glucuronopyranosyl quillaic acid 28-O-beta-D-glucopyranosyl-(1 ⁇ 3)-beta-D- xylopyranosyl-(1 ⁇ 4)- alpha-L-rhamnopyranosyl-(1 ⁇ )
- the saponin derivative comprises a saccharide chain, preferably a saccharide chain selected from group A as defined in embodiment 3, the saccharide chain comprising a carboxyl group, preferably a carboxyl group of a glucuronic acid moiety which has been derivatised by transformation into an amide bond through reaction with 2-amino-2-methyl-1,3-propanediol (AMPD) or N-(2-aminoethyl)maleimide (AEM), therewith providing a saponin-Glu-AMPD such as a QS-21-Glu- AMPD or a SO1861-Glu-AMPD or a saponin-Glu-AEM such as a QS-21-Glu- AEM or a SO1861-Glu-AEM; iii.
- AMPD 2-amino-2-methyl-1,3-propanediol
- AEM N-(2-aminoethyl)maleimide
- the saponin derivative comprises a saccharide chain, preferably a saccharide chain selected from group B as defined in embodiment 3, the saccharide chain comprising an acetoxy (Me(CO)O-) group which has been derivatised by transformation into a hydroxyl group (HO-) by deacetylation; or iv.
- the saponin derivative comprises any combination of derivatisations i., ii. and iii., preferably any combination of two derivatisations of derivatisations i., ii.
- the saponin derivative comprises an aglycone core structure wherein the aglycone core structure comprises an aldehyde group which has been derivatised by transformation into a hydrazone bond through reaction with EMCH wherein the maleimide group of the EMCH is optionally derivatised by formation of a thio-ether bond with mercaptoethanol.
- the saponin derivative comprises an aglycone core structure comprising an aldehyde group which has been derivatised by transformation into a hydrazone bond through reaction with N- ⁇ -maleimidocaproic acid hydrazide (EMCH), therewith providing a saponin-Ald-EMCH such as a SO1861-Ald-EMCH or a QS-21-Ald-EMCH; ii.
- EMCH N- ⁇ -maleimidocaproic acid hydrazide
- the saponin derivative comprises a saccharide chain, preferably a saccharide chain selected from group A as defined in embodiment 3, the saccharide chain comprising a carboxyl group, preferably a carboxyl group of a glucuronic acid moiety which has been derivatised by transformation into an amide bond through reaction with N-(2-aminoethyl)maleimide (AEM), therewith providing a saponin-Glu-AEM such as a QS-21-Glu-AEM or a SO1861-Glu-AEM; or iii. the saponin derivative comprises a combination of derivatisations i. and ii. 14.
- AEM N-(2-aminoethyl)maleimide
- AEM N-(2-aminoethyl)maleimide
- Saponin conjugate of any one of embodiments 17-20 wherein the saponin moiety linker L S is the result of a coupling reaction between at least a first precursor L S1 covalently bound to GalNAc or the bridging moiety B and a second precursor L S2 covalently bound to the saponin moiety, wherein L S1 is a precursor of the saponin moiety linker L S which is covalently bound to GalNAc or to the bridging moiety B and L S2 is a precursor of the saponin moiety linker L S which is covalently bound to the saponin moiety. 22.
- Saponin conjugate of any one of embodiments 17-21 wherein the saponin moiety linker L S is the result of a coupling reaction between at least a first precursor L S1 covalently bound to GalNAc or the bridging moiety B and a second precursor L S2 covalently bound to the saponin moiety, wherein the coupling reaction is selected from the group consisting of an azide-alkyne cycloaddition, a thiol maleimide coupling, a Staudinger reaction, a nucleophilic ring-opening of strained heterocyclic electrophiles, a carbonyl reaction of the non-aldol type and an addition to a carbon-carbon double bond, preferably wherein the coupling reaction is an azide-alkyne cycloaddition, a thiol maleimide coupling, a Staudinger reaction, a nucleophilic ring-opening of strained heterocyclic electrophiles, more preferably wherein the coupling reaction is an azi
- the saponin moiety linker L S is the result of a coupling reaction between a first precursor L S1 covalently bound to GalNAc or the bridging moiety B, the first precursor L S1 comprising an azide; and a second precursor L S2 covalently bound to the saponin moiety, the second precursor L S2 comprising an alkyne and preferably comprising a hydrazone resulting from
- Saponin conjugate of any one of embodiments 21-23 wherein the structure for the precursor L S1 is the following azide of formula (XVII): wherein a represents an integer larger than or equal to 0, preferably a represents an integer selected from 1, 2, and 3, more preferably a represents 2, or wherein the structure for the precursor L S1 comprises the following azide of formula (XVIII): wherein c represents an integer larger than or equal to 0, preferably c represents an integer in the range of 5-15, more preferably c represents 9. 25.
- XVII azide of formula
- a represents an integer larger than or equal to 0, preferably a represents an integer in the range of 2-6, more preferably a represents 4, and wherein L S2a represents an alkyne-containing moiety.
- the cleavable linker is subject to cleavage under acidic conditions, reductive conditions, enzymatic conditions and/or light-induced conditions, and preferably the cleavable linker comprises a cleavable bond selected from a hydrazone bond and a hydrazide bond subject to cleavage under acidic conditions, and/or a bond susceptible to proteolysis, for example proteolysis by Cathepsin B, and/or a bond susceptible for cleavage under reductive conditions such as a disulfide bond.
- composition comprising: - a first pharmaceutical composition comprising the saponin conjugate of any one of the embodiments 1-36 and optionally comprising a pharmaceutically acceptable excipient and/or a pharmaceutically acceptable diluent; and - a second pharmaceutical composition comprising a second conjugate of an effector molecule and a ligand for ASGPR, wherein the ligand for ASGPR preferably comprises at least one GalNAc moiety, preferably three or four GalNAc moieties, more preferably the ligand for ASGPR comprises or consists of (GalNAc) 3 Tris, or a third conjugate of an effector molecule and a binding molecule comprising a binding site for a cell-surface molecule, and optionally comprising a pharmaceutically acceptable excipient and/or pharmaceutically acceptable diluent.
- the ligand for ASGPR preferably comprises at least one GalNAc moiety, preferably three or four GalNAc moieties, more preferably the ligand for ASGPR comprises or consists
- composition comprising: - the saponin conjugate of any one of the embodiments 1-36; - a second conjugate of an effector molecule and a ligand for ASGPR wherein the ligand for ASGPR preferably comprises at least one GalNAc moiety, preferably three or four GalNAc moieties, more preferably the ligand for ASGPR comprises or consists of (GalNAc) 3 Tris, or a third conjugate of an effector molecule and a binding molecule comprising a binding site for a cell-surface molecule, and optionally comprising a pharmaceutically acceptable excipient and/or pharmaceutically acceptable diluent. 39.
- the effector molecule comprises or consists of at least one of a small molecule such as a drug molecule, a toxin such as a protein toxin, an oligonucleotide such as an AON such as a BNA, a xeno nucleic acid or an siRNA, an enzyme, a peptide, a protein, or any combination thereof, preferably, the effector molecule is a toxin, an enzyme or an oligonucleotide. 40.
- siRNA short interfering RNA
- shRNA short hairpin RNA
- miRNA anti- hairpin-shaped
- apoB apolipoprotein B
- HSP27 trans
- the effector molecule is an oligonucleotide capable of, for example when present inside a mammalian cell, targeting an mRNA involved in expression of any one of proteins: apoB, HSP27, TTR, PCSK9, ALAS1, AT3, GO,
- composition of any one of the embodiments 38-44 wherein the effector molecule is or comprises a toxin which toxin comprises or consists of at least one molecule selected from any one or more of a peptide, a protein, an enzyme such as urease and Cre-recombinase, a proteinaceous toxin, a ribosome- inactivating protein, and/or a bacterial toxin, a plant toxin, more preferably selected from any one or more of a viral toxin such as apoptin; a bacterial toxin such as Shiga toxin, Shiga-like toxin, Pseudomonas aeruginosa exotoxin (PE) or exotoxin A of PE, full-length or truncated diphtheria toxin (DT), cholera toxin; a fungal toxin such as alpha-sarcin; a
- the protein toxin is dianthin and/or saporin, and/or comprises or consists of at least one of a toxin targeting ribosome, a toxin targeting elongation factor, a toxin targeting tubulin, a toxin targeting DNA and a toxin targeting RNA, more preferably any one or more of emtansine,
- any one of the embodiments 37 or 39-47 or pharmaceutical composition of any one of the embodiments 38-47, wherein the binding molecule comprising a binding site for a cell-surface molecule, comprised by the third conjugate, is capable of binding to any one of cell-surface molecules: CD71 , CD63, CA125, EpCAM(17-1A), CD52, CEA, CD44v6, FAP, EGF-IR, integrin, syndecan-1 , vascular integrin alpha-V beta-3, HER2, EGFR, CD20, CD22, Folate receptor 1 , CD146, CD56, CD19, CD138, CD27L receptor, PSMA, CanAg, integrin-alphaV, CA6, CD33, mesothelin, Cripto, CD3, CD30, CD239, CD70, CD123, CD352, DLL3, CD25, ephrinA4, MUC1 , Trop2, CEACAM5, CEACAM6, HER3, CD74, PTK7, Notch3, F
- any one of the embodiments 37 or 39-48 or pharmaceutical composition of any one of the embodiments 38-48, wherein the binding molecule comprising a binding site for a cell-surface molecule, comprised by the third conjugate, is or comprises any one of: an antibody, preferably a monoclonal antibody such as a human monoclonal antibody, an IgG, a molecule comprising or consisting of a single-domain antibody, at least one V HH domain, preferable a camelid V H , a variable heavy chain new antigen receptor (V NAR ) domain, a Fab, an scFv, an Fv, a dAb, an F(ab)2 and a Fcab fragment.
- an antibody preferably a monoclonal antibody such as a human monoclonal antibody, an IgG, a molecule comprising or consisting of a single-domain antibody, at least one V HH domain, preferable a camelid V H , a variable heavy chain new antigen receptor (V NAR
- composition of any one of the embodiments 38-49 for use in the treatment or prophylaxis of a disease or health problem in which an expression product is involved of any one or more of genes: apoB, TTR, PCSK9, ALAS1 , AT3, GO, CC5, X gene of HBV, S gene of HBV, AAT and LDH.
- compositions 37, 39-49 or pharmaceutical composition of any one of the embodiments 38-49, or pharmaceutical combination or pharmaceutical composition for use of embodiment 51 for use in the treatment or prophylaxis of a cancer, an infectious disease, a viral infection, hypercholesterolemia, primary hyperoxaluria, haemophilia A, haemophilia B, AAT related liver disease, acute hepatic porphyria, TTR-mediated amyloidosis, hereditary TTR amyloidosis (hATTR), complement-mediated disease, hepatitis B infection, or an auto-immune disease.
- composition for use of embodiment 51 or 52 wherein the saponin is a saponin derivative, preferably a QS-21 derivative or an SO1861 derivative of any one of the embodiments 4-15.
- Oligonucleotide conjugate comprising at least one saponin covalently linked to a ligand for asialoglycoprotein receptor (ASGPR), wherein the ligand for ASGPR comprises at least one N- acetylgalactosamine (GalNAc) moiety, preferably three or four GalNAc moieties, more preferably the ligand for ASGPR comprises or consists of (GalNAc) 3 Tris, and further covalently linked to an oligonucleotide, wherein the at least one saponin is selected from monodesmosidic triterpenoid saponins and bidesmosidic triterpenoid saponins.
- ASGPR asialoglycoprotein receptor
- the ligand for ASGPR comprises at least one N- acetylgalactosamine (GalNAc) moiety, preferably three or four GalNAc moieties, more preferably the ligand for ASGPR comprises or consists of (GalNAc)
- Oligonucleotide conjugate of embodiment I wherein the saponin is a penta-cyclic triterpene saponin of the 12,13-dehydrooleanane type, preferably with an aldehyde function in position C-23 of the aglycone core structure of the saponin.
- the saponin comprises an aglycone core structure selected from the group consisting of: 2alpha-hydroxy oleanolic acid; 16alpha-hydroxy oleanolic acid; hederagenin (23-hydroxy oleanolic acid); 16alpha,23-dihydroxy oleanolic acid; gypsogenin; quillaic acid; protoaescigenin-21(2-methylbut-2-enoate)-22-acetate; 23-oxo-barringtogenol C-21,22-bis(2-methylbut-2-enoate); 23-oxo-barringtogenol C-21(2-methylbut-2-enoate)-16,22-diacetate; digitogenin; 3,16,28-trihydroxy oleanan-12-en; gypsogenic acid, and derivatives thereof, preferably the saponin comprises an aglycone core structure selected from quill
- ⁇ the saponin comprises a saccharide chain bound to the aglycone core structure, which is selected from group A: GlcA-, Glc-, Gal-, Rha-(1 ⁇ 2)-Ara-, Gal-(1 ⁇ 2)-[Xyl-(1 ⁇ 3)]-GlcA-, Glc-(1 ⁇ 2)-[Glc-(1 ⁇ 4)]-GlcA-, Glc-(1 ⁇ 2)-Ara-(1 ⁇ 3)-[Gal-(1 ⁇ 2)]-GlcA-, Xyl-(1 ⁇ 2)-Ara-(1 ⁇ 3)-[Gal-(1 ⁇ 2)]-GlcA-, Glc-(1 ⁇ 3)-Gal-(1 ⁇ 2)-[Xyl-(1 ⁇ 3)]-Glc-(1 ⁇ 4)-Gal-, Rha-(1 ⁇ 2)-Gal-(
- the saponin is a saponin derivative wherein i. the saponin derivative comprises an aglycone core structure comprising an aldehyde group which has been derivatised; ii. the saponin derivative comprises a saccharide chain, preferably a saccharide chain selected from group A as defined in embodiment V, the saccharide chain comprising a carboxyl group which has been derivatised; iii.
- the saponin derivative comprises a saccharide chain, preferably a saccharide chain selected from group B as defined in embodiment V, the saccharide chain comprising an acetoxy (Me(CO)O-) group which has been derivatised; or iv.
- the saponin derivative comprises any combination of derivatisations i., ii. and iii., preferably any combination of two derivatisations of derivatisations i., ii. and iii. VIII.
- the saponin is any one or more of: SO1861, SA1657, GE1741, SA1641, QS-21, QS-21A, QS-21 A-api, QS-21 A-xyl, QS-21B, QS-21 B-api, QS-21 B-xyl, QS-7-xyl, QS-7-api, QS-17-api, QS-17-xyl, QS1861, QS1862, Quillajasaponin, Saponinum album, QS-18, Quil-A, Gyp1, gypsoside A, AG1, AG2, SO1542, SO1584, SO1658, SO1674, SO1832, SO1904, stereoisomers thereof, derivatives thereof and combinations thereof, preferably the saponin is selected from the group consisting of QS-21, a QS-21 derivative, SO1861, a SO1861 derivative, SA1641, a SA1641 derivative, GE1741, a GE17
- the aldehyde group at position C 23 of the quillaic acid or gypsogenin has been derivatised; ii. the carboxyl group of a glucuronic acid moiety of A 1 , when A 1 represents a saccharide chain selected from group A as defined in embodiment V and A 1 comprises or consists of a glucuronic acid moiety, has been derivatised; and iii. one or more, preferably all, of acetoxy group(s) of one saccharide moiety or of two or more saccharide moieties of A 2 , when A 2 represents a saccharide chain selected from group B as defined in embodiment V and A 2 comprises at least one acetoxy group, has/have been derivatised.
- Oligonucleotide conjugate of embodiment IX wherein A 1 represents a saccharide chain selected from group A as defined in embodiment V and comprises or consists of a glucuronic acid moiety and wherein the carboxyl group of a glucuronic acid moiety of A 1 has been derivatised and/or wherein A 2 represents a saccharide chain selected from group B as defined in embodiment V and A 2 comprises at least one acetoxy group and wherein at least one acetoxy group of A 2 has been derivatised.
- XI Oligonucleotide conjugate of embodiment IX or X, wherein the saponin represented by Molecule 1 is a bidesmosidic triterpene saponin. XII.
- the aldehyde group at position C 23 of the quillaic acid or gypsogenin has been derivatised by; - reduction to an alcohol; - transformation into a hydrazone bond through reaction with N- ⁇ -maleimidocaproic acid hydrazide (EMCH), therewith providing a saponin-Ald-EMCH such as a SO1861-Ald-EMCH or a QS-21-Ald- EMCH, wherein the maleimide group of the EMCH is optionally derivatised by formation of a thio-ether bond with mercaptoethanol; - transformation into a hydrazone bond through reaction with N-[ß-maleimidopropionic acid] hydrazide (BMPH) wherein the maleimide group of the BMPH is optionally derivatised by formation of a thio-ether bond with mercaptoethanol; or - transformation into a hydrazone bond through reaction with N-[ ⁇ -maleimido
- the carboxyl group of a glucuronic acid moiety of A 1 when A 1 represents a saccharide chain selected from group A as defined in embodiment V and A 1 comprises or consists of a glucuronic acid moiety, has been derivatised by transformation into an amide bond through reaction with 2-amino-2-methyl-1,3-propanediol (AMPD) or N-(2-aminoethyl)maleimide (AEM), therewith providing a saponin-Glu-AMPD such as a QS-21-Glu-AMPD or a SO1861- Glu-AMPD or a saponin-Glu-AEM such as a QS-21-Glu-AEM or a SO1861-Glu-AEM; and iii.
- AMPD 2-amino-2-methyl-1,3-propanediol
- AEM N-(2-aminoethyl)maleimide
- a 2 represents a saccharide chain selected from group B as defined in embodiment V and A 2 comprises at least one acetoxy group, has/have been derivatised by transformation into a hydroxyl group (HO-) by deacetylation.
- the saponin is a saponin derivative wherein i. the saponin derivative comprises an aglycone core structure comprising an aldehyde group which has been derivatised by: - reduction to an alcohol; - transformation into a hydrazone bond through reaction with N- ⁇ -maleimidocaproic acid hydrazide (EMCH), therewith providing a saponin-Ald-EMCH such as a SO1861-Ald-EMCH or a QS-21-Ald- EMCH, wherein the maleimide group of the EMCH is optionally derivatised by formation of a thio-ether bond with mercaptoethanol; - transformation into a hydrazone bond through reaction with N-[ß-maleimidopropionic acid] hydrazide (BMPH) wherein the maleimide group of the BMPH is optionally derivatised by
- the saponin derivative comprises a saccharide chain, preferably a saccharide chain selected from group A as defined in embodiment V, the saccharide chain comprising a carboxyl group, preferably a carboxyl group of a glucuronic acid moiety which has been derivatised by transformation into an amide bond through reaction with 2-amino-2-methyl-1,3-propanediol (AMPD) or N-(2-aminoethyl)maleimide (AEM), therewith providing a saponin-Glu-AMPD such as a QS-21-Glu- AMPD or a SO1861-Glu-AMPD or a saponin-Glu-AEM such as a QS-21-Glu- AEM or a SO1861-Glu-AEM; iii.
- AMPD 2-amino-2-methyl-1,3-propanediol
- AEM N-(2-aminoethyl)maleimide
- the saponin derivative comprises a saccharide chain, preferably a saccharide chain selected from group B as defined in embodiment V, the saccharide chain comprising an acetoxy (Me(CO)O-) group which has been derivatised by transformation into a hydroxyl group (HO-) by deacetylation; or iv.
- the saponin derivative comprises any combination of derivatisations i., ii. and iii., preferably any combination of two derivatisations of derivatisations i., ii.
- the saponin derivative comprises an aglycone core structure wherein the aglycone core structure comprises an aldehyde group which has been derivatised by transformation into a hydrazone bond through reaction with EMCH wherein the maleimide group of the EMCH is optionally derivatised by formation of a thio-ether bond with mercaptoethanol.
- the saponin is a saponin derivative wherein a) the saponin derivative comprises an aglycone core structure comprising an aldehyde group which has been derivatised by transformation into a hydrazone bond through reaction with N- ⁇ - maleimidocaproic acid hydrazide (EMCH), therewith providing a saponin-Ald-EMCH such as a SO1861-Ald-EMCH or a QS-21-Ald-EMCH; b) the saponin derivative comprises a saccharide chain, preferably a saccharide chain selected from group A as defined in embodiment V, the saccharide chain comprising a carboxyl group, preferably a carboxyl group of a glucuronic acid moiety which has been derivatised by transformation into an amide bond through reaction with N-(2-aminoethyl)maleimide (AEM), therewith providing a saponin-Glu-AEM
- AEM N-(2-aminoethyl)
- AEM N-(2-aminoethyl)maleimide
- Oligonucleotide conjugate of any one of the embodiments l-XVIl wherein the at least one saponin and the ligand for ASGPR are covalently linked directly or via at least one linker, and/or wherein the at least one saponin and the oligonucleotide are covalently linked directly or via at least one linker, and/or wherein the ligand for ASGPR and the oligonucleotide are covalently linked directly or via at least one linker, preferably, the at least one saponin, the ligand for ASGPR and the oligonucleotide are linked via at least one linker.
- XX Oligonucleotide conjugate of any one of the embodiments l-XIX, wherein the ligand for ASGPR is the (GalNAc) 3 Tris represented by Molecule I’: or wherein the ligand for ASGPR is mono-GalNAc represented by Molecule II’: XXI. Oligonucleotide conjugate of any one of the embodiments I-XX, wherein the at least one saponin is covalently bound to the ligand for ASGPR via at least one cleavable linker, and/or wherein the at least one saponin is covalently bound to the oligonucleotide via at least one cleavable linker. XXII.
- Oligonucleotide conjugate of embodiment XIX wherein the at least one saponin is covalently bound to an arm of the tri-functional linker via at least one cleavable linker.
- XXIII Oligonucleotide conjugate of embodiment XXI or XXII, wherein the cleavable linker is subject to cleavage under acidic conditions, reductive conditions, enzymatic conditions and/or light-induced conditions, and preferably the cleavable linker comprises a cleavable bond selected from a hydrazone bond and a hydrazide bond subject to cleavage under acidic conditions, and/or a bond susceptible to proteolysis, for example proteolysis by Cathepsin B, and/or a bond susceptible for cleavage under reductive conditions such as a disulfide bond.
- XXIV Oligonucleotide conjugate of any one of the embodiments XXI-XXIII, wherein the cleavable linker is subject to cleavage in vivo under acidic conditions such as for example present in endosomes and/or lysosomes of mammalian cells, preferably human cells, preferably the cleavable linker is subject to cleavage in vivo at pH 4.0 – 6.5, and more preferably at pH ⁇ 5.5.
- XXV Oligonucleotide conjugate of any one of the embodiments XXI-XXIII, wherein the cleavable linker is subject to cleavage in vivo under acidic conditions such as for example present in endosomes and/or lysosomes of mammalian cells, preferably human cells, preferably the cleavable linker is subject to cleavage in vivo at pH 4.0 – 6.5, and more preferably at pH ⁇ 5.5.
- XXVI. Oligonucleotide conjugate of any one of the embodiments I-XXIV, wherein the oligonucleotide conjugate comprises 1 saponin moiety.
- XXVII Oligonucleotide conjugate of any one of the embodiments I-XXIV, wherein the oligonucleotide conjugate comprises 1 saponin moiety.
- oligonucleotide is any one of a BNA, a xeno nucleic acid, an siRNA, an antisense oligonucleotide.
- Oligonucleotide conjugate of any one of the embodiments I-XXVI wherein the oligonucleotide is selected from any one or more of a(n): short interfering RNA (siRNA), short hairpin RNA (shRNA), anti-hairpin-shaped microRNA (miRNA), single-stranded RNA, aptamer RNA, double-stranded RNA (dsRNA), anti-microRNA (anti-miRNA, anti-miR), antisense oligonucleotide (ASO), DNA, antisense DNA, locked nucleic acid (LNA), bridged nucleic acid (BNA), 2’-O,4’-aminoethylene bridged nucleic acid (BNA NC ), BNA-based siRNA, and BNA-based antisense oligonucleotide (BNA-AON).
- siRNA short interfering RNA
- shRNA short hairpin RNA
- miRNA anti-hairpin-shaped microRNA
- XXIX Oligonucleotide conjugate of any one of the embodiments I-XXVI, wherein the oligonucleotide is selected from any one or more of a(n): anti-miRNA, a BNA-AON or an siRNA, such as BNA-based siRNA, selected from chemically modified siRNA, metabolically stable siRNA and chemically modified, metabolically stable siRNA.
- Oligonucleotide conjugate of any one of the embodiments I-XXIX wherein the oligonucleotide is an oligonucleotide capable of, for example when present inside a mammalian cell and preferably when present inside a human cell, silencing any one of genes: apolipoprotein B (apoB), HSP27, transthyretin (TTR), proprotein convertase subtilisin/kexin type 9 (PCSK9), delta-aminolevulinate synthase 1 (ALAS1), anti-thrombin 3 (AT3), glycolate oxidase (GO), complement component C5 (CC5), X gene of hepatitis B virus (HBV), S gene of HBV, alpha-1 antitrypsin (AAT) and lactate dehydrogenase (LDH), and/or is an oligonucleotide capable of, for example when present inside a mammalian cell, targeting an aberrant miRNA.
- apoB
- XXXI Oligonucleotide conjugate of any one of the embodiments I-XXIX, wherein the oligonucleotide is an oligonucleotide capable of, for example when present inside a mammalian cell and preferably when present inside a human cell, silencing any one of genes: apolipoprotein B (apoB) and HSP27. XXXII.
- Oligonucleotide conjugate of any one of the embodiments I-XXXI wherein the oligonucleotide is an oligonucleotide capable of, for example when present inside a mammalian cell and preferably when present inside a human cell, targeting an mRNA involved in expression of any one of proteins: apoB, HSP27, TTR, PCSK9, ALAS1, AT3, GO, CC5, expression product of X gene of HBV, expression product of S gene of HBV, AAT and LDH, or is capable of, for example when present inside a mammalian cell and preferably when present inside a human cell, antagonizing or restoring an miRNA function such as inhibiting an oncogenic miRNA (onco-miR) or suppression of expression of an onco-miR.
- an oncogenic miRNA onco-miR
- XXXIV Oligonucleotide conjugate of any one of the claims I-XXXI, wherein the oligonucleotide is an oligonucleotide capable of, for example when present inside a mammalian cell and preferably when present inside a human cell, targeting an mRNA involved in expression of any one of proteins: HSP27, apoB, TTR, PCSK9, TMPRSS6, ALAS1, AAT, miR-
- Oligonucleotide conjugate of any one of the embodiments I-XXXII wherein the oligonucleotide is an oligonucleotide capable of, for example when present inside a mammalian cell and preferably when present inside a human cell, targeting an mRNA involved in expression of any one of proteins: apoB and HSP27.
- XXXV Pharmaceutical composition comprising the oligonucleotide conjugate of any one of the embodiments I-XXXIV, and optionally a pharmaceutically acceptable excipient and/or optionally a pharmaceutically acceptable diluent.
- XXXVI Pharmaceutical composition of embodiment XXV, for use as a medicament.
- XXXVII Pharmaceutical composition of embodiment XXV, for use as a medicament.
- composition of claim XXXV for use in the treatment or prophylaxis of a disease or health problem in which an expression product is involved of any one or more of genes: HSP27, apoB, TTR, PCSK9, TMPRSS6, ALAS1 , AT3, GO, CC5, X gene of HBV, S gene of HBV, AAT, miR- 122, hepatitis B virus HbsAg, LDHA, CEBPA and LDH, and/or for use in the treatment or prophylaxis of a disease or health problem which involves any one or more of genes: HSP27, apoB, TTR, PCSK9, TMPRSS6, ALAS1 , AT3, GO, CC5, X gene of HBV, S gene of HBV, AAT, miR-122, hepatitis B virus HbsAg, LDHA, CEBPA and LDH.
- composition of claim XXXV for use in the treatment or prophylaxis of a disease or health problem in which an expression product is involved of any one or more of genes: HSP27, apoB, TTR, PCSK9, TMPRSS6, ALAS1 , AAT, miR-122, hepatitis B virus HbsAg, LDHA and CEBPA, and/or for use in the treatment or prophylaxis of a disease or health problem which involves any one or more of genes: HSP27, apoB, TTR, PCSK9, TMPRSS6, ALAS1 , AAT, miR-122, hepatitis B virus HbsAg, LDHA and CEBPA.
- infectious disease a viral infection, hypercholesterolemia, cardiovascular disease, primary hyperoxaluria, haemophilia A, haemophilia B, AAT related liver disease, acute hepatic porphyria, TTR-mediated amyloidosis, hereditary TTR amyloidosis (
- a cancer such as endometrial carcinoma, breast cancer, lung cancer or hepatocellular carcinoma
- a cardiovascular disease such as hypercholesterolemia, preferably hypercholesterolemia.
- XLI I In vitro or ex vivo method for transferring the oligonucleotide conjugate of any one of the embodiments l-XXXIV from outside a cell to inside said cell, preferably for subsequently transferring the oligonucleotide comprised by the oligonucleotide conjugate of any one of the embodiments l-XXXIV into the cytosol and/or into the nucleus of said cell, comprising the steps of: a) providing a cell which expresses ASGPR, preferably ASGPR1 , on its surface, the cell preferably selected from a liver cell, a virally infected mammalian cell and a mammalian tumor cell, wherein preferably said cell is a human cell; b) providing the oligonucleotide conjugate of any one of the embodiments l-XXXIV for transferring into the cell provided in step a); c) contacting the cell of step a) in vitro or ex vivo with the
- An aspect of the invention relates to a method for providing the oligonucleotide conjugate of the invention, comprising the steps of:
- An embodiment is the method for providing the oligonucleotide conjugate of the invention, wherein the seventh linker is a tri-functional linker, such as the tri-functional linker represented by formula (XXI):
- Trivalent-GalNAc is a targeting ligand that recognizes and binds the ASGPR1 receptor on hepatocytes. Trivalent GalNAc was produced ( Figure 1A-C and Figure 8A-G) and SO1861-EMCH was conjugated (labile, in a similar manner as described in Figure 2A-B and Figure 4 for underivatised SO1861, wherein SPT001 is synonymous to SO1861) to trivalent-GalNAc, with a DAR 1, (trivalent-GalNAc-SO1861).
- SO1861-EMCH refers to SO1861 wherein the aldehyde group is derivatised by transformation into a hydrazone bond through reaction with N- ⁇ -maleimidocaproic acid hydrazide (EMCH), therewith providing SO1861-Ald-EMCH.
- the ‘trivalent-GalNAc’ as depicted in Figures 1A-C and 8A-G is a typical (GalNAc) 3 Tris conjugate suitable for coupling to an effector molecule or to saponin.
- SO1861-EMCH was also conjugated (labile) to monovalent-GalNAc ( Figure 3), with a DAR 1, (GalNAc- SO1861).
- HepG2 (ASGPR1 + ; CD71 + , Table A3) and Huh7 (ASGPR1 +/- ; CD71 + , Table A3) cells were treated with a concentration range of trivalent-GalNAc-L-SO1861 and GalNAc-L-SO1861 in the presence and absence of 10 pM CD71-saporin (monoclonal antibody clone OKT-9 targeting CD71 conjugated to the protein toxin saporin).
- CD71-saporin monoclonal antibody clone OKT-9 targeting CD71 conjugated to the protein toxin saporin.
- Cell treatment in absence of CD71-saporin show that the trivalent-GalNAc-L-SO1861 (or trivalent-GalNAc) as single compound is not toxic up to 15000 nM ( Figure 9).
- HepG2 and Huh7 cells were also treated with a range of concentrations of CD71-saporin in combination with a fixed concentration of 1000 nM or 4000 nM trivalent-GalNAc-SO1861 (DAR1).
- DAR1 trivalent-GalNAc-SO1861
- Targeted protein toxin mediated cell killing of ASGPR1/CD71 expressing cells was determined.
- Example 1A CD71-saporin + concentration series of trivalent GalNAc-L-SO1861, trivalent GalNAc-(L- SO1861)4, SO1861-EMCH Trivalent GalNAc was produced ( Figure 1A-C and Figure 8A-G) and SO1861-EMCH was conjugated (labile, in a similar manner as described in Figure 2A-B and Figure 4 for underivatised SO1861, wherein SPT and SPT001 are synonymous to saponin SO1861) to trivalent-GalNAc, with a DAR 1, (trivalent-GalNAc-SO1861; (GN)3-SPT)) or with a DAR 4 by first coupling the SO1861-EMCH to a dendron (trivalent-GalNAc-dendron(SO1861) 4 ; (GN)3-dSPT4; Figure 23D-E).
- SO1861- EMCH refers to SO1861 wherein the aldehyde group is derivatised by transformation into a hydrazone bond through reaction with N- ⁇ -maleimidocaproic acid hydrazide (EMCH), therewith providing SO1861- Ald-EMCH.
- EMCH N- ⁇ -maleimidocaproic acid hydrazide
- the ‘trivalent-GalNAc’ as depicted in Figure 1A-C and Figure 8A-G is a typical (GalNAc) 3 Tris conjugate suitable for coupling to an effector molecule or to saponin.
- SO1861-EMCH was also conjugated (labile) to monovalent-GalNAc ( Figure 3), with a DAR 1 or with a DAR 4 (using a dendron), (GalNAc-SO1861 and GalNAc-(SO1861) 4 ).
- trivalent-GalNAc-SO1861 and trivalent-GalNAc-(SO1861)4 in combination with low concentrations of CD71-Saporin effectively induce cell killing in ASGPR1/CD71 expressing cells.
- trivalent-GalNAc-SO1861 effectively induces endosomal escape of a protein toxin in ASGPR1 expressing cells.
- SPT001 and GalNAc targeted SPT001 both increase potency of industry standard GalNAc targeted ASO; SO1861-EMCH with about a factor 10, and both (GN)3-SPT and (GN)3-dSPT4 about 100x, when cell viability of liver cells is considered.
- Example 1B Example 1B.
- Trivalent GalNAc was produced ( Figure 1A-C and Figure 8A-G) and SO1861-EMCH was conjugated (labile, in a similar manner as described in Figure 2A-B and Figure 4 for underivatised SO1861, wherein SPT and SPT001 are synonymous to saponin SO1861) to trivalent-GalNAc, with a DAR 1, (trivalent- GalNAc-SO1861; (GalNAc) 3 -SPT001); Figure 19C).
- SO1861-EMCH refers to SO1861 wherein the aldehyde group is derivatised by transformation into a hydrazone bond through reaction with N- ⁇ - maleimidocaproic acid hydrazide (EMCH), therewith providing SO1861-Ald-EMCH.
- EMCH N- ⁇ - maleimidocaproic acid hydrazide
- the covalent conjugate of trivalent GalNAc coupled to both the ApoB antisense BNA oligonucleotide ApoBBNA (SEQ ID NO: 2) and a dendron comprising four covalently coupled SO1861-EMCH was prepared (Figure 20E).
- the primary hepatocytes were contacted with a concentration series of a saponin- oligonucleotide-ASGPR ligand conjugate, i.e. trivalent GalNAc conjugated with both SO1861 (DAR is 4) and ApoB BNA (SEQ ID NO.2) ((GalNAc) 3 -dSPT 4 -ApoB; Figure 24D), and apoB gene expression was assessed (Figure 18B and Figure 20A).
- the apoB gene silencing potency of the (GalNAc) 3 -dSPT 4 -ApoB is about a factor 100 higher compared with the (GalNAc) 3 -ApoB conjugate lacking the covalently linked saponin.
- a saponin- oligonucleotide-ASGPR ligand conjugate i.e. trivalent GalNAc conjugated with both SO1861 (DAR is 4) and ApoB BNA (SEQ ID NO.2) ((GalNAc
- SO1861-EMCH refers to SO1861 wherein the aldehyde group is derivatised by transformation into a hydrazone bond through reaction with N- ⁇ - maleimidocaproic acid hydrazide (EMCH), therewith providing SO1861-Ald-EMCH.
- EMCH N- ⁇ - maleimidocaproic acid hydrazide
- Trivalent-GalNAc- SO1861 effectively induces endosomal escape of a protein toxin in ASGPR1 expressing cells.
- low nM TrivalentGalNAc-SPT001 enhances cytoplasmic delivery of low pM ADC anti-CD71 MoAb- saporin in primary hepatocytes; and hepatocyte cell lines with low ASGPR1 expression (Huh7) are not responsive at low nM concentrations of TrivalentGalNAc-SPT001.
- Example 1D Example 1D.
- TrivalentGalNAc-SPT001 is highly effective ( ⁇ factor 100 more potent) in reducing the apoB mRNA expression in the primary hepatocytes compared to apoB mRNA expression in the primary liver cells treated with TrivalentGalNAc-ApoBBNA only ( Figure 20A). Since the Huh7 cells express very low levels of ASGPR1, neither TrivalentGalNAc-ApoBBNA, nor the combination of TrivalentGalNAc- ApoBBNA and TrivalentGalNAc-SPT001 induce lowering of the apoB mRNA expression in these liver cells.
- TrivalentGalNAc-SPT001 in combination with TrivalentGalNAc-ApoBBNA effectively induces silencing of apoB mRNA expression in ASGPR1 expressing cells.
- TrivalentGalNAc-SPT001 effectively induces endosomal escape of a BNA in ASGPR1 expressing cells.
- the primary hepatocytes and the liver cells of cell line Huh7 were contacted with a concentration series of a saponin-oligonucleotide-ASGPR ligand conjugate, i.e. trivalent GalNAc conjugated with both SO1861 (DAR is 4, in a dendron with four SO1861 molecules) and ApoB BNA (SEQ ID NO.
- TrivalentGalNAc-SPT001 is highly effective (with a factor of over 1000 more potency) in reducing the apoB protein expression in the primary hepatocytes compared to apoB protein expression in the primary liver cells treated with TrivalentGalNAc-ApoBBNA only ( Figure 21A). Since the Huh7 cells express very low levels of ASGPR1, neither TrivalentGalNAc-ApoBBNA, nor the combination of TrivalentGalNAc-ApoBBNA and TrivalentGalNAc-SPT001 induced marked lowering of the apoB protein expression in these liver cells (Figure 21B).
- TrivalentGalNAc-SPT001 in combination with TrivalentGalNAc-ApoBBNA effectively induces silencing of apoB protein expression in high ASGPR1 expressing cells.
- TrivalentGalNAc-SPT001 effectively induces endosomal escape of a BNA in high ASGPR1 expressing cells.
- low nM trivalentGalNAc-ApoBBNA + trivalentGalNAc-SPT001 can enhance endosomal escape and cytoplasmic delivery of ApoBBNA resulting in apoB mRNA silencing in primary hepatocytes; low nM ((GalNAc)3-dSPT4-ApoB can enhance endosomal escape and cytoplasmic delivery of ApoBBNA resulting in apoB mRNA silencing in primary hepatocytes; hepatocytes that have very low ASGPR1 expression are not responsive for both therapies; low nM trivalentGalNAc-ApoBBNA + trivalentGalNAc-SPT001 can enhance endosomal escape and cytoplasmic delivery of ApoBBNA resulting in apoB protein decay in primary hepatocytes; low nM ((GalNAc)3-dSPT4-ApoB can enhance endosomal escape and cytoplasmic delivery of ApoBBNA resulting in apoB
- Example 2 trivalent-GalNAc-L/S-BNA + SO1861-EMCH HSP27BNA was conjugated to trivalent-GalNAc ( Figure 5A, B, Figure 6, Figure 7) and HepG2 (ASGPR1 + ) cells or Huh7 (ASGPR1 +/- ) were treated with a range of concentrations of trivalent-GalNAc- L-HSP27BNA (labile conjugation, Figure 5A-B, Figure 6A-B) or trivalent-GalNAc-S-HSP27BNA (stable conjugation, Figure 7) in combination with a fixed concentration of 4000 nM SO1861-EMCH. HSP27 gene silencing in HepG2 and Huh7 cells was determined.
- ApoBBNA was conjugated in a similar manner ( Figure 5 A-B, Figure 6A-B, Figure 7) to trivalent-GalNAc and HepG2 (ASGPR1 + ) cells or Huh7 (ASGPR1 +/- ) were treated with a range of concentrations of trivalent-GalNAc-L-ApoBBNA (labile conjugation, Figure 5A-B; Figure 6A-B, Figure 12, Figure 13) or trivalent GalNAc-S-ApoBBNA (stable conjugation, Figure 7, Figure 14, Figure 15) or a trivalent GalNAc-L-ApoB scrambled BNA or trivalent GalNAc-S-ApoB scrambled BNA (where the scrambled ApoBBNA is an antisense scrambled oligonucleotide sequence that does not bind the apoB mRNA; off- target control BNA) in combination with a fixed concentration of 4000 nM SO1861-EMCH.
- the treatments did not affect the viability of the cell lines (( Figure 13, Figure 15).
- the ‘L’ refers to a ‘labile’ linker, which indicates that the linker is cleaved in the cell, i.e. at pH as apparent in the endosome and the lysosome.
- the ‘S’ refers to a ‘stable’ linker, which indicates that the linker is not cleaved in the cell, i.e. at pH as apparent in the endosome and the lysosome.
- a conjugate comprising a labile bond between any two molecules forming the conjugate or comprised by the conjugate will be cleaved at a position in the labile linker, therewith dissociating the two linked or bound molecules.
- An example is the cleavage of a hydrazone bond under acidic conditions as apparent in the endosome and lysosome of mammalian cells such as human cells. All this shows that SO1861-EMCH can enhance endosomal escape and target gene silencing of trivalent-GalNAc-L-BNA or trivalent-GalNAc-S-BNA for two independent gene targets.
- the gene silencing is more efficient in HepG2 cells compared to Huh7 cells, suggesting that higher levels of ASGPR1 expression at the plasma membrane of the cell facilitates increased uptake of the GalNAc- BNA conjugates.
- Example 3 trivalent-GalNAc-L/S-BNA + trivalent-GalNAc-L-SO1861 SO1861-EMCH (also referred to as SPT) was conjugated (labile, in a similar manner as described in Figure 2A-B and Figure 4 for underivatised SO1861) to trivalent-GalNAc ((GalNAc)3 or (GN)3), with a DAR 1, (trivalent-GalNAc-SO1861).
- HepG2 (ASGPR1 + ) cells or Huh7 (ASGPR1 +/- ) cells were treated with a range of concentrations of trivalent-GalNAc-L-ApoBBNA (labile conjugation Figure 16, Figure 17), trivalent-GalNAc-S-ApoBBNA (stable conjugation ( Figure 16, Figure 17) or the scrambled (Scr.) off- target control conjugates (trivalent-GalNAc-L-ApoB scrambled BNA or trivalent-GalNAc-S-ApoB scrambled BNA) in combination with a fixed concentration of 4000 nM trivalent-GalNAc-L-SO1861 (DAR1).
- DAR1 trivalent-GalNAc-L-SO1861
- LC-MS method 1 Apparatus: Waters IClass; Bin. Pump: UPIBSM, SM: UPISMFTN with SO; UPCMA, PDA: UPPDATC, 210-320 nm, SQD: ACQ-SQD2 ESI, mass ranges depending on the molecular weight of the product: neg or neg/pos within in a range of 1500-2400 or 2000-3000; ELSD: gas pressure 40 psi, drift tube temp: 50°C; column: Acquity C18, 50 ⁇ 2.1 mm, 1.7 ⁇ m Temp: 60oC, Flow: 0.6 mL/min, lin.
- UPIBSM UPISMFTN with SO
- UPCMA PDA: UPPDATC, 210-320 nm
- SQD ACQ-SQD2 ESI
- ELSD gas pressure 40 psi
- drift tube temp 50 °C
- column Waters XSelect TM CSH C18, 50 ⁇ 2.1 mm, 2.5 ⁇ m
- Temp 25°C
- Flow 0.5 mL/min
- Gradient: t 0min 5% A
- t 2.0min 98% A
- t 2.7min 98% A
- Eluent A acetonitrile
- SO1861-EMCH synthesis (SO1861-EMCH is also referred to as SO1861-Ald-EMCH and SPT- EMCH)
- SO1861-EMCH is also referred to as SO1861-Ald-EMCH and SPT- EMCH
- To SO1861 121 mg, 0.065 mmol
- EMCH.TFA 110 mg, 0.325 mmol
- TFA 0.020 mL, 0.260 mmol
- the reaction mixture stirred at room temperature. After 1.5 hours the reaction mixture was subjected to preparative MP-LC. Fractions corresponding to the product were immediately pooled together, frozen and lyophilized overnight to give the title compound (120 mg, 90%) as a white fluffy solid. Purity based on LC-MS 96%.
- reaction mixture was stirred at room temperature. After 1.5 hours the reaction mixture was evaporated in vacuo, co-evaporated with toluene (3 ⁇ 10 mL) and DCM (3 ⁇ 10 mL) to give the crude title product as a colorless oil.
- reaction mixture was stirred at room temperature. After 2 hours the reaction mixture was evaporated in vacuo, co-evaporated with toluene (3 ⁇ 10 mL) and DCM (3 ⁇ 10 mL) to give the crude title product as a yellowish viscous oil.
- the reaction mixture was filtered by using a centrifugal filter with a molecular weight cut-off of 3000 Da (5000 ⁇ g for 30 min, 2 ⁇ 0.50 mL).
- the residue solution was washed twice with a solution of 20 mM ammonium bicarbonate with 2.5 mM TCEP (0.50 mL), each time filtered under the same conditions described above.
- the residue solution was diluted with 20 mM ammonium bicarbonate/acetonitrile (3:1, v/v, 1.00 mL) and the resulting solution was directly added to trivalent GalNAc-S-maleimide (3.02 mg, 1.43 ⁇ mol).
- the reaction mixture was shaken for 1 min and left standing at room temperature.
- the residue solution was washed twice with a solution of 20 mM ammonium bicarbonate with 2.5 mM TCEP (0.50 mL), each time filtered under the same conditions described above.
- the residue solution was diluted with 20 mM ammonium bicarbonate/acetonitrile (3:1, v/v, 1.00 mL) and the resulting solution was directly added to trivalent GalNAc-S-maleimide (3.09 mg, 1.46 ⁇ mol).
- the reaction mixture was shaken for 1 min and left standing at room temperature. After 1.5 hours the reaction mixture was subjected to preparative LC-MS.
- the reaction mixture was shaken for 1 min and left standing at room temperature. After 1 hour the reaction mixture was filtered by using a centrifugal filter with a molecular weight cut-off of 3000 Da (5000 ⁇ g for 30 min, 2 ⁇ 0.50 mL). Next, the residue solution was washed twice with a solution of 20 mM ammonium bicarbonate with 2.5 mM TCEP (0.50 mL), each time filtered under the same conditions described above. As next, the residue solution was diluted with 20 mM ammonium bicarbonate/acetonitrile (3:1, v/v, 1.00 mL) and the resulting solution was directly added to trivalent GalNAc-L-maleimide (3.63 mg, 1.45 ⁇ mol).
- the residue solution was washed twice with a solution of 20 mM ammonium bicarbonate with 2.5 mM TCEP (0.50 mL), each time filtered under the same conditions described above.
- the residue solution was diluted with 20 mM ammonium bicarbonate/acetonitrile (3:1, v/v, 1.00 mL) and the resulting solution was directly added to trivalent GalNAc-L-maleimide (3.68 mg, 1.47 ⁇ mol).
- the reaction mixture was shaken for 1 min and left standing at room temperature. After 1.5 hours the reaction mixture was subjected to preparative LC-MS.
- the trifunctional linker has the IUPAC name: 5,8,11,18,21,24,27-heptaoxa-2,14,30-triazatritriacontanoic acid, 14-[16-(11,12-didehydrodibenz[b,f]azocin-5(6H)-yl)-13,16-dioxo-3,6,9-trioxa-12-azahexadec-1- yl]-33-(2,5-dihydro-2,5-dioxo-1H-pyrrol-1-yl)-15,31-dioxo-, (1R,4E)-4-cycloocten-1-yl ester.
- the trifunctional linker has the following formula (XXI):
- the DBCO-L-ApoB BNA oligo-trivalent GalNAc (molecule 31) was synthesized by conjugating methyltetrazine-L-ApoB BNA oligo (molecule 30) to DBCO-TCO-trivalent GalNAc (molecule 29) ( Figure 24A-B and 24C), wherein the DBCO-TCO-trivalent GalNAc (molecule 29) was synthesized with molecule 27 and molecule 28 (trifunctional linker), wherein molecule 27 is GalNAc-thioacetate obtained from the reaction between molecule 22, the formic salt of trivalent GalNAc-amine, and molecule 26 (4- nitrophenyl 3-(acetylthio)propanate (Figure 24A-B).
- the ApoB BNA, the trivalent GalNAc and the SPT001 (which is saponin SO1861) are conjugated into a single covalent conjugate using tri-functional linker Figure 24D).
- a conjugate in which the ApoB BNA was replaced for a BNA with a scrambled ApoB nucleic acid sequence was synthesized, for control testing purposes.
- Dendron(-L-SO1861) 8 -L-ApoB BNA oligo-trivalent GalNAc synthesis To DBCO-L-ApoB BNA oligo-trivalent GalNAc (1.38 mg, 0.127 ⁇ mol) and dendron(-L-SO1861) 8 -azide (2.51 mg, 0.135 ⁇ mol) was added 20 mM ammonium bicarbonate/acetonitrile (3:1, v/v, 0.5 mL). The reaction mixture was shaken for 1 min and left standing at room temperature. After 4 hours the reaction mixture was subjected to preparative LC-MS.
- CD71 monoclonal antibody was purchased from InVivoMab, anti-human CD71 (OKT- 9), BioXCell.
- the antisense BNA (HSP27) was an BNA( NC ) with oligo nucleic acid sequence 5’-GGCacagccagtgGCG-3’ modified from Zhang et al.
- Plating medium was replaced by 315 ⁇ l or 108 ⁇ l maintenance media (Cytes Biotechnologies S.L., Spain), after which conjugates were added from a 10x concentrated stock solution in PBS. Plates were incubated for 72 hr at 37°C being harvested for gene expression and cell viability analysis. RNA isolation and gene expression analysis from human cell culture RNA from cells was isolated and analysed according to standard protocols (Biorad). qPCR primers that were used are indicated in Table A2. Table A2.
- MTS Cell viability assay
- the OD at 492 nm was measured on a Thermo Scientific Multiskan FC plate reader (Thermo Scientific).
- Thermo Scientific Multiskan FC plate reader Thermo Scientific.
- the background signal of ‘medium only‘ wells was subtracted from all other wells, before the cell viability percentage of treated/untreated cells was calculated, by dividing the background corrected signal of treated wells over the background corrected signal of the untreated wells (x 100).
- PE anti-human CD71 (#334106, Biolegend) was used to stain the transferrin receptor, PE Mouse IgG2a, ⁇ Isotype Ctrl FC (#400212, Biolegend) was used as its matched isotype control.
- PE anti-human ASGPR1 (#130-122- 963, Miltenyi) was used to stain the ASGPR1 receptor and PE Mouse IgG1, Isotype Ctrl (#130-113-762, Miltenyi) was used as its matched isotype control. Samples were incubated for 30 min. at 4°C.
- UPIBSM UPISMFTN with SO
- UPCMA PDA: UPPDATC, 210-320 nm
- SQD ACQ-SQD2 ESI
- ELSD gas pressure 40 psi
- drift tube temp 50°C
- column Acquity C18, 50 ⁇ 2.1 mm, 1.7 ⁇ m Temp: 60oC
- Flow 0.6 mL/min, lin.
- UPIBSM UPISMFTN with SO
- UPCMA PDA: UPPDATC, 210-320 nm
- SQD ACQ-SQD2 ESI
- ELSD gas pressure 40 psi
- drift tube temp 50 °C
- column Waters XSelect TM CSH C18, 50 ⁇ 2.1 mm, 2.5 ⁇ m
- Temp 25°C
- Flow 0.5 mL/min
- Gradient: t 0min 5% A
- t 2.0min 98% A
- t 2.7min 98% A
- Eluent A acetonitrile
- the resulting gel was imaged and processed using ImageJ (Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA).
- ImageJ Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA.
- TBEU-PAGE Native protein, conjugates and BNA standard were analysed under heat denaturing non-reducing and reducing conditions by TBEU-PAGE against an oligo ladder using a 15% TBE-Urea gel and TBE as running buffer (180V, ⁇ 60 minutes). Samples were prepared to 0.5 mg/ml, and BNA standard was prepared to 20 ⁇ g/ml, respectively, all comprising TBE Urea sample buffer and purified H 2 O as diluent.
- MALDI-TOF-MS MALDI-TOF spectra were recorded on a MALDI-Mass Spectrometer (Bruker Ultrafex III). Typically, the sample dissolved in MilliQ water in nanomolar to micromolar range was spotted on the target (MTP 384 target plate polished steel T F, Bruker Daltons) using either super-DHB (99%, Fluka) or sinapinic acid (SA, 99%, Sigma-Aldrich) as the matrix dissolved in acetonitrile (MADLI-TOF-MS tested, Sigma) / 0.1 % TFA (7:3 v/v) via the dried-droplet-method.
- Trifunctional linker-(L-hydrazone-SO1861)- (L-BNA oligo) – (Trivalent-GalNAc) synthesis ( Figure 35)
- the trifunctional linker (TFL) has the IUPAC name: 5,8,11,18,21,24,27-Heptaoxa-2,14,30- triazatritriacontanoic acid, 14-[16-(11,12-didehydrodibenz[b,f]azocin-5(6H)-yl)-13,16-dioxo-3,6,9-trioxa- 12-azahexadec-1-yl]-33-(2,5-dihydro-2,5-dioxo-1H-pyrrol-1-yl)-15,31-dioxo-, (1R,4E)-4-cyclo
- reaction mixture was added to a freshly prepared solution of TFL-(DBCO)-(TCO)-(Maleimide) (20.6 mg, 6.13 ⁇ mol, molecule 5) in 20 mM ammonium bicarbonate/acetonitrile (3:1, v/v, 2.00 mL). The resulting mixture was shaken for 1 min and left standing at room temperature. After 2 hours the reaction mixture was subjected to preparative MP-LC. 2A Fractions corresponding to the product were immediately pooled together, frozen and lyophilized overnight to give the title compound (3.83 mg, 21%) as a white solid. Purity based on LC-MS 89%.
- reaction mixture was shaken for 1 min and left standing at room temperature. After 30 min the reaction mixture was frozen and lyophilized overnight to yield the crude title product as a pink fluffy solid.
- To the crude product was added a solution of 20 mM ammonium bicarbonate (1.50 mL) and the resulting suspension was filtered over a 0.45 ⁇ m syringe filter. The filtrate was lyophilized overnight to yield the title product (3.4 mg, 89%) as a pink fluffy solid. Purity based on LC-MS 90%.
- reaction mixture was shaken for 1 min and left standing at room temperature. After 30 min the reaction mixture was added to a freshly prepared solution of trifunctional linker (7.39 mg, 6.13 ⁇ mol, molecule 4) in 20 mM ammonium bicarbonate/acetonitrile (3:1, v/v, 2.00 mL). The resulting mixture was shaken for 1 min and left standing at room temperature. After 2 hours the reaction mixture was subjected to preparative MP- LC. 2A Fractions corresponding to the product were immediately pooled together, frozen and lyophilized overnight to give the title compound (3.83 mg, 21%) as a white solid. Purity based on LC-MS 89%.
- reaction mixture was shaken for 1 min and left standing at room temperature. After 30 min the reaction mixture was added to a freshly prepared solution of TFL-(L-semicarbazone-SO1861)-(TCO)-(maleimide) (20.4 mg, 6.13 ⁇ mol, molecule 29) in 20 mM ammonium bicarbonate/acetonitrile (3:1, v/v, 2.00 mL). The resulting mixture was shaken for 1 min and left standing at room temperature. After 2 hours the reaction mixture was subjected to preparative MP-LC. 2A Fractions corresponding to the product were immediately pooled together, frozen and lyophilized overnight to give the title compound (16.7 mg, 54%) as a white solid. Purity based on LC-MS 94%.
- reaction mixture was stirred at room temperature. After 30 min the reaction mixture was evaporated in vacuo and co-evaporated with dichloromethane (2 ⁇ 50 mL). Next, the residue was dissolved in dichloromethane (100 mL) and resulting solution was stirred at 0 °C. A solution of Boc hydrazine (2.33 mL, 15.0 mmol) in dichloromethane (20 mL) and DiPEA (3.83 mL, 22.0 mL) was added and the reaction mixture was stirred overnight at room temperature.
- Trifunctional linker-(dendron(-L- semicarbazone -SO1861)8)- BNA oligo - Trivalent-GalNAc synthesis ( Figure 40) Intermediate 25: dendron(L-semicarbazone-SO1861) 8 -amine (molecule 44) (2S)-N-[(1S)-1- ⁇ [2-(6-amino-N- ⁇ 2-[(2S)-2,6-bis[(2S)-2,6-bis(3- sulfanylpropanamido)hexanamido]hexanamido]ethyl ⁇ hexanamido)ethyl]carbamoyl ⁇ -5-[(2S)-2,6-bis(3- sulfanylpropanamido)hexanamido]pentyl]-2,6-bis(3-sulfanylpropanamido)hexanamide formate (0.61 mg, 0.299 ⁇ mol, molecule 43) was dissolved in a
- TCO blocking on TFL has been performed on the following conjugetes: TFL-(L-hydrazone-SPT001)-(blocked TCO)-(trivalent GalNAc) (molecule 51) TFL-(dendron (L-hydrazone-SPT001)4)-(blocked TCO)-(trivalent GalNAc) (molecule 52) TFL-(dendron-(L-hydrazone-SPT001)8)-(blocked TCO)-(trivalent GalNAc) (molecule 53) TFL-(L-semicarbazone-SPT001)-(blocked TCO)-(trivalent GalNAc) (molecule 54) TFL-(dendron((L-semicarbazone-SPT001)4)-(blocked TCO)-(trivalent GalNAc) (molecule 55) TFL-(dendron(L-semicarbazone-SPT001)8)-(blocked TCO)-(trivalent GalNAc) (molecule 56) The procedure is exemplary described for TFL-
- CTG Cell viability assay
- the background signal of ‘medium only‘ wells was subtracted from all other wells before the cell viability percentage of treated/untreated cells was calculated by dividing the background corrected signal of treated wells over the background corrected signal of the untreated wells (x 100).
- Human primary hepatocyte treatment Cryopreserved primary human hepatocytes (Cytes Biotechnologies S.L., Spain) were thawed in hepatocyte thawing media (Cytes Biotechnologies S.L., Spain). Cells were re-suspended in hepatocyte plating media (Cytes Biotechnologies S.L., Spain).
- Cells were seeded onto collagen-I coated plates at a density of 215.600 cells/well or 66.600 cells/well for the 48 or 96-well plates (Greiner BioOne). Cells were pre-cultured for 4-6 hrs at 37°C allowing for cell attachment to cell culture plates before the start of treatment. Plating medium was replaced by 315 ⁇ l or 108 ⁇ l maintenance media (Cytes Biotechnologies S.L., Spain), after which conjugates were added from a 10x concentrated stock solution in PBS. Plates were incubated for 72 hr at 37°C being harvested for gene expression and cell viability analysis.
- ApoB#02 oligo sequences Murine and human sequence-compatible ApoB#02 (5′-GCattggtatTCA-3′) [SEQ ID NO: 12] was a BNA NC oligonucleotide and synthesized with 5’-Thiol C6 linker at Bio-Synthesis Inc, with BNA NC bases in capitals, and fully phosphorothioated backbones (Lewisville, Texas).
- RNA isolation and gene expression analysis from primary mouse and human hepatocytes was isolated using TRI Reagent® Solution (Thermo Scientific) according to the manufacturer’s instruction. Conversion into cDNA was performed using iScriptTM cDNA Synthesis Kit (BioRad) using standard protocols.
- ApoB expression levels and levels of specific hepatocyte housekeeping genes were determined using quantitative real-time PCR assays (qRT-PCR) using iTaqTM Universal SYBR® Green Supermix (BioRad) and the Light Cycler 480 (Roche Diagnostics, Rotnch, Switzerland) with specific DNA primers, listed in Table A4 (study A) and Table A7 (study B), respectively. Analysis was done by the ⁇ Ct method to determine apoB expression relative to 2 hepatocyte-specific housekeeping control mRNAs. Each analysis reaction was performed in triplicate. Table A4.
- Primers used in qPCR analysis of mouse primary hepatocytes/liver tissue analysis FACS analysis Cells were seeded in DMEM (PAN-Biotech GmbH) supplemented with 10% fetal bovine serum (PAN- Biotech GmbH) and 1% penicillin/streptomycin (PAN-Biotech GmbH), in T75 flasks at appropriate density for each cell-line and incubated for 72-96 hrs (5% CO2, 37°C), until a confluency of 90% was reached. Next, the cells were trypsinized (TryplE Express, Gibco Thermo Scientific) to single cells, transferred to a 15 mL falcon tube, and centrifuged (1,400 rpm, 3 min).
- PE anti-human CD71 (#334106, Biolegend) was used to stain the transferrin receptor, PE Mouse IgG2a, ⁇ Isotype Ctrl FC (#400212, Biolegend) was used as its matched isotype control.
- PE anti-human ASGPR1 (#130-122- 963, Miltenyi) was used to stain the ASGPR1 receptor and PE Mouse IgG1, Isotype Ctrl (#130-113-762, Miltenyi) was used as its matched isotype control. Samples were incubated for 30 min. at 4°C.
- 3 animals per timepoint per treatment group (6 animals in the vehicle control group in Study A) were sacrificed to collect terminal bleeds and collect relevant organs, such as liver and kidney tissues for further analyses, at end of study, the remaining 2 animals (4 in the control group in Study A) per group were sacrificed. One half of the liver and one kidney was frozen and used for tissue RNA analysis.
- ApoB expression levels and levels of specific liver housekeeping genes were determined using quantitative real-time PCR assays (qRT-PCR) using iTaqTM Universal SYBR® Green Supermix (BioRad) and the Light Cycler 480 (Roche Diagnostics, Rotnch, Switzerland) with specific DNA primers, listed in Table A4 (Study A) and Table A7 (Study B), respectively. Analysis was done by the ⁇ Ct method to determine apoB expression relative to 2 liver-specific housekeeping control mRNAs. Each analysis reaction was performed in triplicate. Table A7.
- NB levels for 0.1 mg/kg ApoB#02 (196 hrs), 0.01 mg/kg (GalNAc) 3 -ApoB + 5 mg/kg (GalNAc) 3 -SO1861 (196 hrs and 336 hrs) and 0.1 mg/kg (GalNAc) 3 -ApoB + 5 mg/kg (GalNAc) 3 -SO1861 (196 hrs) are not reported.
- Targeted (GalNAc) 3 -ApoB#02 at a dose of 1 mg/kg reduced LDL-C to an average of 2.7 mg/dl within 24 hrs already (n 3 animals), i.e., a 53% reduction compared to vehicle and ApoB#02 BNA groups.
- the same reduction of LDL-C to an average of 2.7 mg/dl within 24 hrs (n 3 animals), i.e.
- RNA levels when increasing the saponin-per-payload, by dosing 0.173 mg/kg (GalNAc) 3 -d(SO1861)4-ApoB#02 (sc) or 0.265 mg/kg (GalNAc) 3 -d(SO1861)8-ApoB#02 (sc), respectively, serum apoB protein was reduced to 89 ⁇ g/ml and 50 ⁇ g/ml, respectively, while it was 141 ⁇ g/ml for 0.1 mg/kg (GalNAc) 3 - SO1861-ApoB#02 (sc), showing that an increase of saponin markedly increases potency of the conjugate in reducing apoB protein.
- a decrease of saponin amount (0.1 mg/kg (GalNAc) 3 -SO1861 (sc) + 0.1 mg/kg (GalNAc) 3 -ApoB#02) yielded less potency (122 ⁇ g/ml apoB protein).
- RNA and apoB protein levels when increasing the saponin-per-payload, by dosing 0.173 mg/kg (GalNAc) 3 -d(SO1861)4-ApoB#02 (sc) or 0.265 mg/kg (GalNAc) 3 -d(SO1861)8-ApoB#02 (sc), respectively, LDL-C was reduced, to 2.3 mg/dl and 1.3 mg/dl, respectively, while it was 4.3 mg/dl for 0.1 mg/kg (GalNAc) 3 - SO1861-ApoB#02 (sc), showing that an increase of saponin markedly increases potency of the conjugate in reducing LDL-C.
- a decrease of saponin amount (0.1 mg/kg (GalNAc) 3 - SO1861 (sc) + 0.1 mg/kg (GalNAc) 3 -ApoB#02) yielded less potency (3.7 mg/dl LDL-C).
- mice in treatment groups gained weight comparable to the mice receiving vehicle. Serum ALT levels were determined and results are shown in Figure 34.
- this data shows that the saponin conjugates are well tolerated in the doses and dose regimen applied, and particularly, that administration of SO1861-containing conjugates (GalNAc) 3 - SO1861-ApoB#02 (sc), (GalNAc) 3 -d(SO1861)4-ApoB#02 (sc), and (GalNAc) 3 -d(SO1861)8-ApoB#02 (sc), as well as co-administration of (GalNAc) 3 -SO1861 with (GalNAc) 3 -ApoB#02 has no immediate tolerability issues, while showing high potency in the C57BL/6J mouse model.
Landscapes
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Epidemiology (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description


































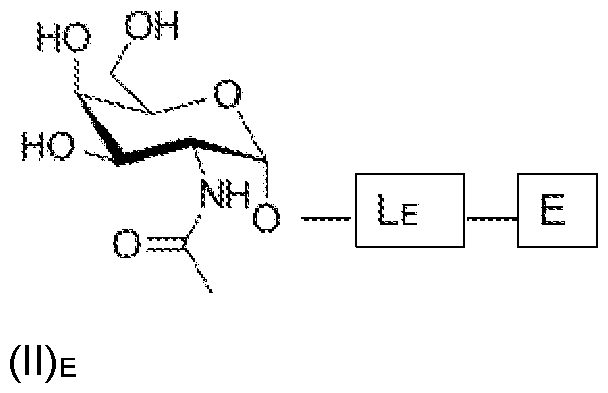
















































































Claims






Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA3192469A CA3192469A1 (en) | 2020-09-10 | 2021-09-09 | Conjugate of saponin, oligonucleotide and galnac |
| IL301214A IL301214A (en) | 2020-09-10 | 2021-09-09 | Conjugate of saponin, oligonucleotide and galnac |
| EP21794416.4A EP4210760A1 (en) | 2020-09-10 | 2021-09-09 | Conjugate of saponin, oligonucleotide and galnac |
| CN202180070494.9A CN116437965A (en) | 2020-09-10 | 2021-09-09 | Conjugates of saponins, oligonucleotides and GALNAC |
| JP2023516250A JP2023542500A (en) | 2020-09-10 | 2021-09-09 | Conjugate of saponin, oligonucleotide, and GALNAC |
| AU2021339332A AU2021339332A1 (en) | 2020-09-10 | 2021-09-09 | Conjugate of saponin, oligonucleotide and GalNAc |
| KR1020237011435A KR20230066384A (en) | 2020-09-10 | 2021-09-09 | Conjugates of saponins, oligonucleotides and GALNAC |
| EP22711105.1A EP4355369A1 (en) | 2021-06-18 | 2022-03-08 | Conjugate of saponin, oligonucleotide and galnac |
| PCT/NL2022/050127 WO2022265493A1 (en) | 2021-06-18 | 2022-03-08 | Conjugate of saponin, oligonucleotide and galnac |
| CN202280056303.8A CN117881427A (en) | 2021-06-18 | 2022-03-08 | Conjugates of saponins, oligonucleotides and GALNAC |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| NL2026442 | 2020-09-10 | ||
| NL2026442 | 2020-09-10 | ||
| NL2027405A NL2027405B1 (en) | 2021-01-26 | 2021-01-26 | Semicarbazone-based saponin conjugate |
| NL2027405 | 2021-01-26 | ||
| NLPCT/NL2021/050384 | 2021-06-18 | ||
| PCT/NL2021/050384 WO2021261992A1 (en) | 2020-06-24 | 2021-06-18 | Conjugate of galnac and saponin, therapeutic composition comprising said conjugate and a galnac-oligonucleotide conjugate |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2022055351A1 true WO2022055351A1 (en) | 2022-03-17 |
Family
ID=78269666
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/NL2021/050549 WO2022055351A1 (en) | 2020-09-10 | 2021-09-09 | Conjugate of saponin, oligonucleotide and galnac |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2022055351A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022265493A1 (en) * | 2021-06-18 | 2022-12-22 | Sapreme Technologies B.V. | Conjugate of saponin, oligonucleotide and galnac |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011054811A1 (en) * | 2009-11-03 | 2011-05-12 | Santaris Pharma A/S | Rna antagonists targeting hsp27 combination therapy |
| WO2015071388A1 (en) * | 2013-11-14 | 2015-05-21 | Roche Innovation Center Copenhagen A/S | Apob antisense conjugate compounds |
| WO2020126620A2 (en) * | 2018-12-21 | 2020-06-25 | Sapreme Technologies B.V. | Improved antibody-oligonucleotide conjugate |
-
2021
- 2021-09-09 WO PCT/NL2021/050549 patent/WO2022055351A1/en unknown
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011054811A1 (en) * | 2009-11-03 | 2011-05-12 | Santaris Pharma A/S | Rna antagonists targeting hsp27 combination therapy |
| WO2015071388A1 (en) * | 2013-11-14 | 2015-05-21 | Roche Innovation Center Copenhagen A/S | Apob antisense conjugate compounds |
| WO2020126620A2 (en) * | 2018-12-21 | 2020-06-25 | Sapreme Technologies B.V. | Improved antibody-oligonucleotide conjugate |
| WO2020126627A1 (en) * | 2018-12-21 | 2020-06-25 | Sapreme Technologies B.V. | Improved cell-targeting binding molecule |
Non-Patent Citations (9)
| Title |
|---|
| BACHRAN CHRISTOPHER ET AL: "Saponins in Tumor Therapy", MINI-REVIEWS IN MEDICINAL CHEMISTRY, vol. 8, 1 August 2008 (2008-08-01), pages 575 - 584, XP055775950 * |
| GOODING, MATT ET AL., CHEMICAL BIOLOGY & DRUG DESIGN, vol. 80, no. 6, 2012, pages 787 - 809 |
| HENDRIK FUCHS ET AL: "Glycosylated Triterpenoids as Endosomal Escape Enhancers in Targeted Tumor Therapies", BIOMEDICINES, vol. 5, no. 2, 29 March 2017 (2017-03-29), pages 14, XP055429235, DOI: 10.3390/biomedicines5020014 * |
| HERMANSON, GREG T: "Bioconjugate techniques.", 2013, ACADEMIC PRESS, pages: 289 |
| RASBAND, W.S.: "ImageJ", U. S. NATIONAL INSTITUTES OF HEALTH |
| SABINE SEWING ET AL: "GalNAc Conjugation Attenuates the Cytotoxicity of Antisense Oligonucleotide Drugs in Renal Tubular Cells", MOLECULAR THERAPY: NUCLEIC ACIDS., vol. 14, 1 March 2019 (2019-03-01), US, pages 67 - 79, XP055762603, ISSN: 2162-2531, DOI: 10.1016/j.omtn.2018.11.005 * |
| SETTEN, RYAN L.JOHN J. ROSSISI-PING HAN, NATURE REVIEWS DRUG DISCOVERY, vol. 18, no. 6, 2019, pages 421 - 446 |
| XU RAN ET AL: "On the origins of triterpenoid skeletal diversity", PHYTOCHEMISTRY, ELSEVIER, AMSTERDAM , NL, vol. 65, no. 3, 9 May 2017 (2017-05-09), pages 261 - 291, XP085002429, ISSN: 0031-9422, DOI: 10.1016/J.PHYTOCHEM.2003.11.014 * |
| YZHANGZ QUS KIMV SHIB LIAO 1P KRAFTR BANDARUY WU,LM GREENBERGERID HORAK: "Down-modulation of cancer targets using locked nucleic acid (LNA)-based antisense oligonucleotides without transfection", GENE THERAPY, vol. 18, 2011, pages 326 - 333, XP055412231, DOI: 10.1038/gt.2010.133 |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022265493A1 (en) * | 2021-06-18 | 2022-12-22 | Sapreme Technologies B.V. | Conjugate of saponin, oligonucleotide and galnac |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR20210110323A (en) | Improved Cell Targeting Binding Molecules | |
| WO2021261992A1 (en) | Conjugate of galnac and saponin, therapeutic composition comprising said conjugate and a galnac-oligonucleotide conjugate | |
| US20230263897A1 (en) | Conjugate of galnac and saponin, therapeutic composition comprising said conjugate and a galnac-oligonucleotide conjugate | |
| WO2022055352A1 (en) | Semicarbazone-based saponin conjugate | |
| WO2022055351A1 (en) | Conjugate of saponin, oligonucleotide and galnac | |
| EP4210760A1 (en) | Conjugate of saponin, oligonucleotide and galnac | |
| KR20230043111A (en) | A combination comprising an ADC or AOC comprising VHH and a saponin or ligand-saponin conjugate | |
| EP4355369A1 (en) | Conjugate of saponin, oligonucleotide and galnac | |
| US20230293562A1 (en) | Therapeutic combination of galnac-oligonucleotide conjugate and saponin, and uses thereof | |
| KR20230043117A (en) | Conjugates of single domain antibodies, saponins and effector molecules, pharmaceutical compositions comprising the same, therapeutic uses of the pharmaceutical compositions | |
| CN117881427A (en) | Conjugates of saponins, oligonucleotides and GALNAC | |
| US20240066045A1 (en) | Semicarbazone-based saponin conjugate | |
| US20240115712A1 (en) | Semicarbazone-based saponin conjugate | |
| WO2023038517A1 (en) | Semicarbazone-based saponin conjugate | |
| WO2023121447A1 (en) | Conjugate of a single domain antibody, a saponin and an effector molecule, pharmaceutical composition comprising the same, therapeutic use of said pharmaceutical composition | |
| WO2022164316A1 (en) | Semicarbazone-based saponin conjugate |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 21794416 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2023516250 Country of ref document: JP Kind code of ref document: A Ref document number: 3192469 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2021339332 Country of ref document: AU Date of ref document: 20210909 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20237011435 Country of ref document: KR Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2021794416 Country of ref document: EP Effective date: 20230411 |