WO2021163546A1 - Methods and materials for assessing nucleic acids - Google Patents
Methods and materials for assessing nucleic acids Download PDFInfo
- Publication number
- WO2021163546A1 WO2021163546A1 PCT/US2021/017937 US2021017937W WO2021163546A1 WO 2021163546 A1 WO2021163546 A1 WO 2021163546A1 US 2021017937 W US2021017937 W US 2021017937W WO 2021163546 A1 WO2021163546 A1 WO 2021163546A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- sequence
- target
- watson
- stranded dna
- crick
- Prior art date
Links
- 238000000034 method Methods 0.000 title claims abstract description 355
- 150000007523 nucleic acids Chemical class 0.000 title claims description 201
- 102000039446 nucleic acids Human genes 0.000 title claims description 168
- 108020004707 nucleic acids Proteins 0.000 title claims description 168
- 239000000463 material Substances 0.000 title description 21
- 238000012163 sequencing technique Methods 0.000 claims abstract description 307
- 230000035772 mutation Effects 0.000 claims abstract description 254
- 238000009396 hybridization Methods 0.000 claims abstract description 15
- 238000002360 preparation method Methods 0.000 claims abstract description 15
- 108020004414 DNA Proteins 0.000 claims description 731
- 239000012634 fragment Substances 0.000 claims description 485
- 102000053602 DNA Human genes 0.000 claims description 465
- 230000003321 amplification Effects 0.000 claims description 241
- 238000003199 nucleic acid amplification method Methods 0.000 claims description 241
- 239000000523 sample Substances 0.000 claims description 150
- 230000000295 complement effect Effects 0.000 claims description 128
- 125000003729 nucleotide group Chemical group 0.000 claims description 122
- 239000002773 nucleotide Substances 0.000 claims description 119
- 239000012491 analyte Substances 0.000 claims description 94
- 102000040430 polynucleotide Human genes 0.000 claims description 91
- 108091033319 polynucleotide Proteins 0.000 claims description 91
- 239000002157 polynucleotide Substances 0.000 claims description 91
- 238000012408 PCR amplification Methods 0.000 claims description 87
- 108091034117 Oligonucleotide Proteins 0.000 claims description 85
- 108091093088 Amplicon Proteins 0.000 claims description 82
- 239000011324 bead Substances 0.000 claims description 73
- 238000006243 chemical reaction Methods 0.000 claims description 61
- 238000000137 annealing Methods 0.000 claims description 59
- 102000003960 Ligases Human genes 0.000 claims description 46
- 108090000364 Ligases Proteins 0.000 claims description 46
- 239000012472 biological sample Substances 0.000 claims description 41
- 230000000903 blocking effect Effects 0.000 claims description 36
- 239000007787 solid Substances 0.000 claims description 35
- 108020004682 Single-Stranded DNA Proteins 0.000 claims description 34
- 206010028980 Neoplasm Diseases 0.000 claims description 27
- 210000002381 plasma Anatomy 0.000 claims description 26
- 108010072685 Uracil-DNA Glycosidase Proteins 0.000 claims description 23
- 102000006943 Uracil-DNA Glycosidase Human genes 0.000 claims description 23
- 238000011049 filling Methods 0.000 claims description 22
- 230000015654 memory Effects 0.000 claims description 21
- 241000124008 Mammalia Species 0.000 claims description 19
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 claims description 18
- 229910019142 PO4 Inorganic materials 0.000 claims description 16
- 239000003153 chemical reaction reagent Substances 0.000 claims description 16
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 claims description 16
- 239000010452 phosphate Substances 0.000 claims description 16
- 238000011144 upstream manufacturing Methods 0.000 claims description 16
- 230000000593 degrading effect Effects 0.000 claims description 14
- 108010014303 DNA-directed DNA polymerase Proteins 0.000 claims description 13
- 102000016928 DNA-directed DNA polymerase Human genes 0.000 claims description 13
- 108060002716 Exonuclease Proteins 0.000 claims description 13
- 102000013165 exonuclease Human genes 0.000 claims description 13
- 230000002441 reversible effect Effects 0.000 claims description 13
- 108010090804 Streptavidin Proteins 0.000 claims description 12
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 claims description 9
- 229960002685 biotin Drugs 0.000 claims description 9
- 235000020958 biotin Nutrition 0.000 claims description 9
- 239000011616 biotin Substances 0.000 claims description 9
- 241000588724 Escherichia coli Species 0.000 claims description 8
- 102000012410 DNA Ligases Human genes 0.000 claims description 7
- 108010061982 DNA Ligases Proteins 0.000 claims description 7
- 239000012530 fluid Substances 0.000 claims description 7
- 238000013519 translation Methods 0.000 claims description 7
- 230000000692 anti-sense effect Effects 0.000 claims description 6
- 230000005540 biological transmission Effects 0.000 claims description 6
- 230000000694 effects Effects 0.000 claims description 6
- 239000012528 membrane Substances 0.000 claims description 6
- 238000000638 solvent extraction Methods 0.000 claims description 6
- -1 well Substances 0.000 claims description 6
- 229920002684 Sepharose Polymers 0.000 claims description 5
- 238000013507 mapping Methods 0.000 claims description 5
- 210000002966 serum Anatomy 0.000 claims description 5
- 206010003445 Ascites Diseases 0.000 claims description 4
- 206010036790 Productive cough Diseases 0.000 claims description 4
- 238000004925 denaturation Methods 0.000 claims description 4
- 230000036425 denaturation Effects 0.000 claims description 4
- 239000011541 reaction mixture Substances 0.000 claims description 4
- 210000000582 semen Anatomy 0.000 claims description 4
- 229940075620 somatostatin analogue Drugs 0.000 claims description 4
- 210000003802 sputum Anatomy 0.000 claims description 4
- 208000024794 sputum Diseases 0.000 claims description 4
- 210000004243 sweat Anatomy 0.000 claims description 4
- 210000001138 tear Anatomy 0.000 claims description 4
- 210000002700 urine Anatomy 0.000 claims description 4
- 108010006785 Taq Polymerase Proteins 0.000 claims description 3
- 210000004369 blood Anatomy 0.000 claims description 3
- 239000008280 blood Substances 0.000 claims description 3
- 238000007405 data analysis Methods 0.000 claims description 3
- 238000003505 heat denaturation Methods 0.000 claims description 3
- 230000002829 reductive effect Effects 0.000 claims description 3
- 230000001225 therapeutic effect Effects 0.000 claims description 3
- 230000029087 digestion Effects 0.000 claims description 2
- 230000026731 phosphorylation Effects 0.000 claims description 2
- 238000006366 phosphorylation reaction Methods 0.000 claims description 2
- 239000000203 mixture Substances 0.000 abstract description 37
- 239000000047 product Substances 0.000 description 126
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 42
- 239000012071 phase Substances 0.000 description 23
- 238000011084 recovery Methods 0.000 description 21
- 238000003860 storage Methods 0.000 description 20
- 102100025064 Cellular tumor antigen p53 Human genes 0.000 description 19
- 101000721661 Homo sapiens Cellular tumor antigen p53 Proteins 0.000 description 19
- 238000007857 nested PCR Methods 0.000 description 19
- 108090000623 proteins and genes Proteins 0.000 description 18
- 201000011510 cancer Diseases 0.000 description 16
- 230000008439 repair process Effects 0.000 description 15
- 238000004458 analytical method Methods 0.000 description 13
- 238000003260 vortexing Methods 0.000 description 13
- 102000004190 Enzymes Human genes 0.000 description 11
- 108090000790 Enzymes Proteins 0.000 description 11
- 238000013459 approach Methods 0.000 description 11
- 230000004048 modification Effects 0.000 description 11
- 238000012986 modification Methods 0.000 description 11
- 238000007481 next generation sequencing Methods 0.000 description 11
- 239000008188 pellet Substances 0.000 description 10
- 230000008685 targeting Effects 0.000 description 9
- 238000004891 communication Methods 0.000 description 8
- 238000010276 construction Methods 0.000 description 8
- 238000011109 contamination Methods 0.000 description 8
- 238000001514 detection method Methods 0.000 description 8
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 8
- 238000011065 in-situ storage Methods 0.000 description 8
- 238000003752 polymerase chain reaction Methods 0.000 description 8
- 238000010008 shearing Methods 0.000 description 8
- 239000006228 supernatant Substances 0.000 description 8
- 201000010099 disease Diseases 0.000 description 7
- 238000002474 experimental method Methods 0.000 description 7
- 238000010438 heat treatment Methods 0.000 description 7
- 239000007788 liquid Substances 0.000 description 7
- 108700028369 Alleles Proteins 0.000 description 6
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 6
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 6
- 238000003556 assay Methods 0.000 description 6
- 230000006872 improvement Effects 0.000 description 6
- 238000011528 liquid biopsy Methods 0.000 description 6
- 229920001184 polypeptide Polymers 0.000 description 6
- 102000004196 processed proteins & peptides Human genes 0.000 description 6
- 108090000765 processed proteins & peptides Proteins 0.000 description 6
- 102100022887 GTP-binding nuclear protein Ran Human genes 0.000 description 5
- 101000774835 Heteractis crispa PI-stichotoxin-Hcr2o Proteins 0.000 description 5
- 101000620756 Homo sapiens GTP-binding nuclear protein Ran Proteins 0.000 description 5
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 5
- 230000015572 biosynthetic process Effects 0.000 description 5
- 210000004027 cell Anatomy 0.000 description 5
- 238000009833 condensation Methods 0.000 description 5
- 230000005494 condensation Effects 0.000 description 5
- 230000003287 optical effect Effects 0.000 description 5
- 230000002093 peripheral effect Effects 0.000 description 5
- 238000012546 transfer Methods 0.000 description 5
- 206010069754 Acquired gene mutation Diseases 0.000 description 4
- 241000282412 Homo Species 0.000 description 4
- 239000005547 deoxyribonucleotide Substances 0.000 description 4
- 210000000265 leukocyte Anatomy 0.000 description 4
- 238000012545 processing Methods 0.000 description 4
- 230000037439 somatic mutation Effects 0.000 description 4
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 3
- 230000005778 DNA damage Effects 0.000 description 3
- 231100000277 DNA damage Toxicity 0.000 description 3
- 101710163270 Nuclease Proteins 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- 238000013500 data storage Methods 0.000 description 3
- 125000002637 deoxyribonucleotide group Chemical group 0.000 description 3
- 238000006209 dephosphorylation reaction Methods 0.000 description 3
- 238000005516 engineering process Methods 0.000 description 3
- 238000011156 evaluation Methods 0.000 description 3
- 230000000670 limiting effect Effects 0.000 description 3
- 230000036438 mutation frequency Effects 0.000 description 3
- 238000005192 partition Methods 0.000 description 3
- 238000011002 quantification Methods 0.000 description 3
- 238000013515 script Methods 0.000 description 3
- 239000011780 sodium chloride Substances 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- GUAHPAJOXVYFON-ZETCQYMHSA-N (8S)-8-amino-7-oxononanoic acid zwitterion Chemical compound C[C@H](N)C(=O)CCCCCC(O)=O GUAHPAJOXVYFON-ZETCQYMHSA-N 0.000 description 2
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 2
- 102000036365 BRCA1 Human genes 0.000 description 2
- 108700020463 BRCA1 Proteins 0.000 description 2
- 108091061744 Cell-free fetal DNA Proteins 0.000 description 2
- 102000004099 Deoxyribonuclease (Pyrimidine Dimer) Human genes 0.000 description 2
- 108010082610 Deoxyribonuclease (Pyrimidine Dimer) Proteins 0.000 description 2
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 2
- 206010064571 Gene mutation Diseases 0.000 description 2
- 102100032611 Guanine nucleotide-binding protein G(s) subunit alpha isoforms short Human genes 0.000 description 2
- 108091092195 Intron Proteins 0.000 description 2
- 108091028043 Nucleic acid sequence Proteins 0.000 description 2
- 238000002944 PCR assay Methods 0.000 description 2
- 102100032543 Phosphatidylinositol 3,4,5-trisphosphate 3-phosphatase and dual-specificity protein phosphatase PTEN Human genes 0.000 description 2
- 101710132081 Phosphatidylinositol 3,4,5-trisphosphate 3-phosphatase and dual-specificity protein phosphatase PTEN Proteins 0.000 description 2
- 108091081021 Sense strand Proteins 0.000 description 2
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 2
- ISAKRJDGNUQOIC-UHFFFAOYSA-N Uracil Chemical compound O=C1C=CNC(=O)N1 ISAKRJDGNUQOIC-UHFFFAOYSA-N 0.000 description 2
- 230000002411 adverse Effects 0.000 description 2
- 230000037429 base substitution Effects 0.000 description 2
- 210000000601 blood cell Anatomy 0.000 description 2
- 238000012937 correction Methods 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 230000018109 developmental process Effects 0.000 description 2
- 239000000539 dimer Substances 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- 125000000623 heterocyclic group Chemical group 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 238000010348 incorporation Methods 0.000 description 2
- 238000007726 management method Methods 0.000 description 2
- 108020004999 messenger RNA Proteins 0.000 description 2
- 230000003278 mimic effect Effects 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 238000006116 polymerization reaction Methods 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 150000003212 purines Chemical class 0.000 description 2
- 150000003230 pyrimidines Chemical class 0.000 description 2
- 210000003296 saliva Anatomy 0.000 description 2
- 238000012216 screening Methods 0.000 description 2
- 108010068698 spleen exonuclease Proteins 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- RYYWUUFWQRZTIU-UHFFFAOYSA-K thiophosphate Chemical compound [O-]P([O-])([O-])=S RYYWUUFWQRZTIU-UHFFFAOYSA-K 0.000 description 2
- 102000052609 BRCA2 Human genes 0.000 description 1
- 108700020462 BRCA2 Proteins 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 101150008921 Brca2 gene Proteins 0.000 description 1
- 241000282465 Canis Species 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- 108090000994 Catalytic RNA Proteins 0.000 description 1
- 102000053642 Catalytic RNA Human genes 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- 108091026890 Coding region Proteins 0.000 description 1
- 208000035473 Communicable disease Diseases 0.000 description 1
- 108091028732 Concatemer Proteins 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 241000699800 Cricetinae Species 0.000 description 1
- 108010009392 Cyclin-Dependent Kinase Inhibitor p16 Proteins 0.000 description 1
- 238000007400 DNA extraction Methods 0.000 description 1
- 238000001712 DNA sequencing Methods 0.000 description 1
- 102000007260 Deoxyribonuclease I Human genes 0.000 description 1
- 108010008532 Deoxyribonuclease I Proteins 0.000 description 1
- 206010061819 Disease recurrence Diseases 0.000 description 1
- 241000283073 Equus caballus Species 0.000 description 1
- 108700024394 Exon Proteins 0.000 description 1
- 101710105178 F-box/WD repeat-containing protein 7 Proteins 0.000 description 1
- 102100028138 F-box/WD repeat-containing protein 7 Human genes 0.000 description 1
- 241000282324 Felis Species 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- 102100023600 Fibroblast growth factor receptor 2 Human genes 0.000 description 1
- 101710182389 Fibroblast growth factor receptor 2 Proteins 0.000 description 1
- 101150092780 GSP1 gene Proteins 0.000 description 1
- 102100029974 GTPase HRas Human genes 0.000 description 1
- 102100039788 GTPase NRas Human genes 0.000 description 1
- 101000584633 Homo sapiens GTPase HRas Proteins 0.000 description 1
- 101000744505 Homo sapiens GTPase NRas Proteins 0.000 description 1
- 101001014590 Homo sapiens Guanine nucleotide-binding protein G(s) subunit alpha isoforms XLas Proteins 0.000 description 1
- 101001014594 Homo sapiens Guanine nucleotide-binding protein G(s) subunit alpha isoforms short Proteins 0.000 description 1
- 101001014610 Homo sapiens Neuroendocrine secretory protein 55 Proteins 0.000 description 1
- 101000605639 Homo sapiens Phosphatidylinositol 4,5-bisphosphate 3-kinase catalytic subunit alpha isoform Proteins 0.000 description 1
- 101000984753 Homo sapiens Serine/threonine-protein kinase B-raf Proteins 0.000 description 1
- 241001529936 Murinae Species 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- 108700015679 Nested Genes Proteins 0.000 description 1
- 108020004711 Nucleic Acid Probes Proteins 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 208000007660 Residual Neoplasm Diseases 0.000 description 1
- 108091028664 Ribonucleotide Proteins 0.000 description 1
- 108020004566 Transfer RNA Proteins 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 230000032683 aging Effects 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 238000004873 anchoring Methods 0.000 description 1
- 238000005422 blasting Methods 0.000 description 1
- 210000002421 cell wall Anatomy 0.000 description 1
- 230000004640 cellular pathway Effects 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 238000007385 chemical modification Methods 0.000 description 1
- 239000003795 chemical substances by application Substances 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 239000002299 complementary DNA Substances 0.000 description 1
- 230000001010 compromised effect Effects 0.000 description 1
- 230000002153 concerted effect Effects 0.000 description 1
- 230000021615 conjugation Effects 0.000 description 1
- 239000000356 contaminant Substances 0.000 description 1
- 239000013068 control sample Substances 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- SUYVUBYJARFZHO-RRKCRQDMSA-N dATP Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@H]1C[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O1 SUYVUBYJARFZHO-RRKCRQDMSA-N 0.000 description 1
- SUYVUBYJARFZHO-UHFFFAOYSA-N dATP Natural products C1=NC=2C(N)=NC=NC=2N1C1CC(O)C(COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O1 SUYVUBYJARFZHO-UHFFFAOYSA-N 0.000 description 1
- RGWHQCVHVJXOKC-SHYZEUOFSA-J dCTP(4-) Chemical compound O=C1N=C(N)C=CN1[C@@H]1O[C@H](COP([O-])(=O)OP([O-])(=O)OP([O-])([O-])=O)[C@@H](O)C1 RGWHQCVHVJXOKC-SHYZEUOFSA-J 0.000 description 1
- HAAZLUGHYHWQIW-KVQBGUIXSA-N dGTP Chemical compound C1=NC=2C(=O)NC(N)=NC=2N1[C@H]1C[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)O1 HAAZLUGHYHWQIW-KVQBGUIXSA-N 0.000 description 1
- NHVNXKFIZYSCEB-XLPZGREQSA-N dTTP Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)[C@@H](O)C1 NHVNXKFIZYSCEB-XLPZGREQSA-N 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 238000012350 deep sequencing Methods 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000006471 dimerization reaction Methods 0.000 description 1
- 208000035475 disorder Diseases 0.000 description 1
- 239000003814 drug Substances 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 description 1
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 description 1
- 238000013401 experimental design Methods 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 239000000835 fiber Substances 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 238000013467 fragmentation Methods 0.000 description 1
- 238000006062 fragmentation reaction Methods 0.000 description 1
- 238000007710 freezing Methods 0.000 description 1
- 230000008014 freezing Effects 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- 238000001641 gel filtration chromatography Methods 0.000 description 1
- 231100000118 genetic alteration Toxicity 0.000 description 1
- 230000004077 genetic alteration Effects 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 210000003958 hematopoietic stem cell Anatomy 0.000 description 1
- 230000003116 impacting effect Effects 0.000 description 1
- 238000000126 in silico method Methods 0.000 description 1
- 238000007689 inspection Methods 0.000 description 1
- 238000002372 labelling Methods 0.000 description 1
- 238000012417 linear regression Methods 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 238000007403 mPCR Methods 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 230000000873 masking effect Effects 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 description 1
- 238000002663 nebulization Methods 0.000 description 1
- 239000002853 nucleic acid probe Substances 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 238000003359 percent control normalization Methods 0.000 description 1
- 210000005259 peripheral blood Anatomy 0.000 description 1
- 239000011886 peripheral blood Substances 0.000 description 1
- XEBWQGVWTUSTLN-UHFFFAOYSA-M phenylmercury acetate Chemical compound CC(=O)O[Hg]C1=CC=CC=C1 XEBWQGVWTUSTLN-UHFFFAOYSA-M 0.000 description 1
- 239000013612 plasmid Substances 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 102000054765 polymorphisms of proteins Human genes 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 238000009598 prenatal testing Methods 0.000 description 1
- 238000004321 preservation Methods 0.000 description 1
- 238000004393 prognosis Methods 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 238000003908 quality control method Methods 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 108091008146 restriction endonucleases Proteins 0.000 description 1
- 239000002336 ribonucleotide Substances 0.000 description 1
- 125000002652 ribonucleotide group Chemical group 0.000 description 1
- 150000003291 riboses Chemical class 0.000 description 1
- 108020004418 ribosomal RNA Proteins 0.000 description 1
- 108091092562 ribozyme Proteins 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 239000004065 semiconductor Substances 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 238000004088 simulation Methods 0.000 description 1
- 239000007790 solid phase Substances 0.000 description 1
- 230000000392 somatic effect Effects 0.000 description 1
- 238000000527 sonication Methods 0.000 description 1
- 125000006850 spacer group Chemical group 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 238000007619 statistical method Methods 0.000 description 1
- 238000000528 statistical test Methods 0.000 description 1
- 238000010257 thawing Methods 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 239000001226 triphosphate Substances 0.000 description 1
- 235000011178 triphosphate Nutrition 0.000 description 1
- 229940035893 uracil Drugs 0.000 description 1
- 238000012418 validation experiment Methods 0.000 description 1
- 239000013598 vector Substances 0.000 description 1
- 238000012800 visualization Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6806—Preparing nucleic acids for analysis, e.g. for polymerase chain reaction [PCR] assay
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6813—Hybridisation assays
- C12Q1/6827—Hybridisation assays for detection of mutation or polymorphism
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6844—Nucleic acid amplification reactions
- C12Q1/6853—Nucleic acid amplification reactions using modified primers or templates
- C12Q1/6855—Ligating adaptors
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6844—Nucleic acid amplification reactions
- C12Q1/686—Polymerase chain reaction [PCR]
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6869—Methods for sequencing
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q1/00—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions
- C12Q1/68—Measuring or testing processes involving enzymes, nucleic acids or microorganisms; Compositions therefor; Processes of preparing such compositions involving nucleic acids
- C12Q1/6876—Nucleic acid products used in the analysis of nucleic acids, e.g. primers or probes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2521/00—Reaction characterised by the enzymatic activity
- C12Q2521/50—Other enzymatic activities
- C12Q2521/501—Ligase
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2521/00—Reaction characterised by the enzymatic activity
- C12Q2521/50—Other enzymatic activities
- C12Q2521/531—Glycosylase
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2525/00—Reactions involving modified oligonucleotides, nucleic acids, or nucleotides
- C12Q2525/10—Modifications characterised by
- C12Q2525/155—Modifications characterised by incorporating/generating a new priming site
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2525/00—Reactions involving modified oligonucleotides, nucleic acids, or nucleotides
- C12Q2525/10—Modifications characterised by
- C12Q2525/186—Modifications characterised by incorporating a non-extendable or blocking moiety
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2525/00—Reactions involving modified oligonucleotides, nucleic acids, or nucleotides
- C12Q2525/10—Modifications characterised by
- C12Q2525/191—Modifications characterised by incorporating an adaptor
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2531/00—Reactions of nucleic acids characterised by
- C12Q2531/10—Reactions of nucleic acids characterised by the purpose being amplify/increase the copy number of target nucleic acid
- C12Q2531/113—PCR
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2535/00—Reactions characterised by the assay type for determining the identity of a nucleotide base or a sequence of oligonucleotides
- C12Q2535/122—Massive parallel sequencing
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2563/00—Nucleic acid detection characterized by the use of physical, structural and functional properties
- C12Q2563/179—Nucleic acid detection characterized by the use of physical, structural and functional properties the label being a nucleic acid
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Q—MEASURING OR TESTING PROCESSES INVOLVING ENZYMES, NUCLEIC ACIDS OR MICROORGANISMS; COMPOSITIONS OR TEST PAPERS THEREFOR; PROCESSES OF PREPARING SUCH COMPOSITIONS; CONDITION-RESPONSIVE CONTROL IN MICROBIOLOGICAL OR ENZYMOLOGICAL PROCESSES
- C12Q2563/00—Nucleic acid detection characterized by the use of physical, structural and functional properties
- C12Q2563/185—Nucleic acid dedicated to use as a hidden marker/bar code, e.g. inclusion of nucleic acids to mark art objects or animals
Definitions
- This invention is related to the area of nucleotide sequencing. In particular, it relates to sequencing library preparation and sequencing workflow for the identification of mutations.
- NGS Next generation sequencing
- molecular barcodes to tag original template molecules was designed to overcome various obstacles in the detection of rare mutations. With molecular barcoding, redundant sequencing of the PCR-generated progeny of each tagged molecule is performed and sequencing errors are easily recognized (Kinde et al., Proc Natl Acad Sci USA 108:9530-9535 (2011)). For example, if a given threshold of the progeny of the barcoded template molecule contain the same mutation, then the mutation is considered genuine (a “supermutant”). If less than a given threshold of the progeny contain the mutation of interest, then the mutation is considered an artifact. Two types of molecular barcodes have been described: exogenous and endogenous.
- Exogenous barcodes (also referred to herein as exogenous UIDs) comprise pre-specified or random nucleotides, and are appended during library preparation or during PCR.
- Endogenous barcodes (also referred to herein as endogenous UIDS) are formed by the sequences at the 5’ and 3’ ends of fragments (e.g., fragments generated by random shearing of DNA or fragments present in a cell -free fluid biological sample) of the template DNA to be assayed.
- fragments e.g., fragments generated by random shearing of DNA or fragments present in a cell -free fluid biological sample
- Forked adapters for paired end sequencing have been developed to enable “duplex sequencing”, wherein each of the two strands (Watson and Crick) of the original DNA duplex can be discerned by the 5’ to 3’ directionality revealed upon sequencing.
- Duplex sequencing reduces sequencing errors because it is extremely unlikely that both strands of DNA would contain the identical mutation if that mutation was erroneously generated during library preparation or sequencing.
- Preparation of targeted sequencing libraries generally involve adapter attachment to sequencing templates, amplification of the library, and hybridization capture to enrich the library for targets of interest. While effective for enriching large regions of interest, hybridization capture does not scale well for small target regions (Springer et ak, Elife 7:doi: 10.7554/eLife.32143 (2016)) and exhibits poor duplex recovery (Wang et ak, Proc Natl Acad Sci USA 112:9704-9709 (2015); and Wang et ak, Elife 5:doi:10.7554/eLife.15175 (2016)). Sequential rounds of capture can partially overcome these limitations, but even with such improvements, duplex recovery rates are typically ⁇ 1%. CRISPR-DS can achieve up to 15% recovery, but is not applicable to cell-free DNA.
- the present document provides methods and materials that addresses these issues by providing an approach to identically barcode both strands of templates, and by providing a method for PCR-based enrichment of each strand that does not require hybridization capture.
- methods for detecting one or more mutations present on both strands of a double stranded nucleic acid can include generating a duplex sequencing library having a duplex molecular barcode on each end (e.g., the 5’ end and the 3’ end) of each nucleic acid in the library, generating a library of single stranded Watson strand-derived sequences and a library of single stranded Crick-strand derived sequences from the duplex sequencing library, and detecting the presence of one or more mutations present on both strands of the double stranded nucleic acid in each single stranded library.
- a single stranded DNA library corresponding to the Watson strands of a double stranded nucleic acid template and a single stranded DNA library corresponding to the Crick strands of a double stranded nucleic acid template can be generated from sequencing libraries incorporating duplex molecular barcodes, each single stranded DNA library can be enriched for a target region using a strand-specific anchored PCR approach, and the target region can be sequenced to detect the presence of absence of one or more mutations within the target region of a nucleic acid.
- a double stranded nucleic acid can be termed the Sequence Ascertainment Free of Errors Sequencing System (SaferSeqS), and can include for example, library construction with in situ generation of double stranded molecular barcodes (see, e.g., Fig. 22a), target enrichment via anchored PCR (see, e.g., Fig. 22b), and in silico reconstruction of template molecules (see, e.g., Fig. 22c).
- SaferSeqS Sequence Ascertainment Free of Errors Sequencing System
- Bona fide mutations present in the original starting templates can be identified by requiring alterations to be found on both strands of the same initial nucleic acid molecule. Having the ability to detect one or more mutations present on both strands of a double stranded nucleic acid (e.g ., true somatic mutations) provides a unique and unrealized opportunity to accurately and efficiently assess multiple mutations simultaneously and at an affordable cost.
- Using methods and materials described herein to detect the presence of one or more mutations present on both strands of a double stranded nucleic acid can permit the identification of rare mutations with high confidence, while minimizing the amount of DNA damage, the amount of PCR to be performed, and/or the number of DNA damage artifacts.
- the terms "Watson strand” and "Crick strand” are used simply to distinguish between the two strands of a double-stranded starting nucleic acid sequence. Either of the strands can be denoted “Watson” or "Crick,” and then the other strand is denoted with the other name.
- methods of comprising: a) forming a reaction mixture comprising: i) a plurality of double-stranded DNA fragments that are dephosphorylated and blunt-ended, wherein each of the double-stranded DNA fragments comprises Watson and Crick strands; ii) a plurality of adaptors, wherein each of the adaptors comprises, in the 5’ to 3’ direction: A) a barcode, and B) a universal 3’ adaptor sequence; and iii) a ligase; and b) incubating the reaction mixture such that: i) adaptors are ligated to the 3' ends of the Watson and Crick strands, and ii) adaptors are not ligated to the 5' ends of either the Watson or Crick strands, thereby generating double-stranded ligation products.
- each of the plurality of adaptors comprises a unique barcode.
- each of the double-stranded ligation products comprises a Watson strand with only one barcode and a Crick strand with only one barcode that is different from the barcode on the Watson strand.
- the methods further comprise the step of: c) sequencing at least a portion of the double-stranded ligation products.
- kits and compositions comprising: a) a population of partially double-stranded 3’ adaptors (3'PDSAs) configured to be ligated to the 3’ ends of both Watson and Crick strands of a population of double-stranded DNA fragments, wherein a first strand of the 3'PDSAs comprises, in the 5’-3’ direction, (i) a first segment, (ii) an exogenous UID sequence, (iii) an annealing site for a 5’ adaptor, and (iv) a universal 3’ adaptor sequence comprising an R2 sequencing primer site, and wherein the second strand of the 3'PDSAs comprises, in the 5’ to 3’ direction, (i) a segment complementary to the first segment, and (ii) a 3’ blocking group; and b) a population of 5’ adaptors configured to anneal to the annealing site, wherein the 5’ adaptors comprise, in the 5’ to 3’ direction, (
- the systems, kits, and compositions further comprise: c) the population of double-stranded DNA fragments from a biological sample, and/or c) reagents for degrading the second strand of the 3'PDSAs to generate single-stranded 3' adaptors
- Watson target-selective primers comprising a target-selective sequence; and/or c) a first set of Crick target-selective primer pairs, comprising (i) one or more Crick target- selective primers comprising a sequence complementary to a portion of the universal 5’ adapter sequence, and (ii) one or more second Crick target-selective primers, each of the one or more second Crick target selective primers comprising the same target-selective sequence as the second Watson target-selective primer sequence.
- the methods further comprise: amplifying the plurality of adaptor-ligated, double-stranded DNA fragments with a first primer complementary to the universal 3’ adaptor sequence, and second primer complementary to the complement of the universal 5' adaptor sequence, thereby generating amplicons, wherein the amplicons comprise a plurality of double-stranded Watson templates and a plurality of double-stranded Crick templates.
- the methods further comprise: selectively amplifying said double-stranded Watson templates with a first set of Watson target-selective primer pairs, the first set of Watson target-selective primer pairs comprising: (i) a first Watson target-selective primer comprising a sequence complementary to a portion of the universal 3’ adapter sequence, and (ii) a second Watson target-selective primer comprising a target- selective sequence, thereby creating target Watson amplification products.
- the methods further comprise: selectively amplifying said double-stranded Crick templates with a first set of Crick target-selective primer pairs, the first set of Crick target- selective primer pairs comprising: (i) a first Crick target- selective primer comprising a sequence complementary to the complement of a portion of the universal 5’ adapter sequence, and (ii) a second Crick target-selective primer comprising the same target-selective sequence as the second Watson target- selective primer sequence, thereby creating target Crick amplification products.
- the population of double-stranded DNA fragments are incubated with a mixture of uracil-DNA glycosylase and DNA glycosylase-lyase Endonuclease VIII, prior to ligating any adapters.
- the polymerase employed e.g., for extending the 5' adapter sequences
- has 5' to 3' exonuclease activity e.g., which can digest the second strand of the 3'PDSAs.
- the polymerase employed e.g., for extending the 5' adapter sequences
- the methods further comprise: removing the second strand of the 3'PDSAs to generate single-stranded 3' adaptors (3'SSAs).
- the removing the second strand occurs after step b), or before step b), or during step b).
- the removing the second strand of the 3'PDSAs comprises contacting the 3’ duplex adapter with uracil-DNA glycosylase (UDG) to degrade the second strand.
- the removing the second strand is accomplished by a polymerase with exonuclease activity, wherein the polymerase extends the 5’ adaptors across the exogenous UID sequence and the first segment.
- the methods further comprise: determining sequence reads of one or more of the amplicons. In other embodiments, the methods further comprise: assigning the sequence reads into UID families, wherein each member of a UID family comprises the same exogenous UID sequence. In particular embodiments, the methods further comprise: assigning sequence reads of each UID family into a Watson subfamily and Crick subfamily based on spatial relationship of the exogenous UID sequence to the R1 and R2 read sequence. In other embodiments, the methods further comprise: identifying a nucleotide sequence as accurately representing a Watson strand of an analyte DNA fragment when at least 50% (e.g., 50 ... 75 ... 95%) of the Watson subfamily contain the sequence.
- the methods further comprise: identifying a nucleotide sequence as accurately representing a Crick strand of an analyte DNA fragment when at least 50% (e.g., 50 ... 75 ... 90%) of the Crick subfamily contain the sequence.
- the methods further comprise: identifying a mutation in the nucleotide sequence as accurately representing a the Watson strand when the sequence accurately representing the Watson strand differs from a reference sequence that lacks the mutation. In additional embodiments, the methods further comprise: identifying a mutation in the nucleotide sequence as accurately representing the Crick Strand when the sequence accurately representing the Crick Strand differs from a reference sequence that lacks the mutation. In other embodiments, the methods further comprise: identifying a mutation in the analyte DNA fragment when the mutation in the nucleotide sequence accurately representing the Watson strand and the mutation in the nucleotide sequence accurately representing the Crick strand are the same mutation.
- each member of a UID family further comprises the same endogenous UID sequence, wherein the endogenous UID sequence comprises an end of a double-stranded DNA fragment from the population.
- the population of double-stranded DNA fragments has blunt ends.
- the method comprises: A) generating double stranded DNA fragments each having a duplex molecular barcode on each end of the double stranded DNA fragment; B) amplifying the double stranded DNA fragment comprising the duplex molecular barcode on each end of the double stranded DNA fragment to generate an amplified duplex sequencing library, wherein the amplifying comprises contacting the double stranded DNA fragment comprising the duplex molecular barcode on each end of the double stranded DNA fragment with a universal primer pair under whole-genome PCR conditions; C) optionally, generating a single stranded DNA library of Watson strands from the amplified duplex sequencing library; D) optionally, generating a single stranded DNA library
- generating the double stranded DNA fragments each having the duplex molecular barcode on each end of the double stranded DNA fragment comprises: i) ligating a 3’ duplex adapter to each 3’ end of a double stranded DNA fragment obtained from the double stranded DNA template, wherein the 3’ duplex adapter comprises a) a first oligonucleotide comprising a 5’ phosphate, a first molecular barcode, and a 3’ oligonucleotide annealed to b) a second oligonucleotide comprising a degradable 3’ blocking group, wherein the 3’ oligonucleotide and the second oligonucleotide sequence are complementary; ii) degrading the degradable 3’ blocking group; iii) ligating a 5’ adapter to each dephosphorylated 5’ end of the double stranded DNA fragment obtained from the double stranded DNA template, where
- generating the DNA library (e.g., the single stranded DNA library) (e.g., the DNA library (e.g., the single stranded DNA library)) of Watson strands from the amplified duplex sequencing library comprises: i) amplifying a first aliquot of the amplified duplex sequencing library using a primer pair consisting of a first primer and a second primer, wherein the first primer is capable of hybridizing to the Watson strand, and wherein the first primer comprises a tag, to generate double stranded amplification products having tagged Watson strands; ii) denaturing the double stranded amplification products having tagged Watson strands to generate single stranded tagged Watson strands and single stranded Crick strands; and iii) recovering the single stranded tagged Watson strands to generate the DNA library (e.g., the single stranded DNA library) of Watson strands from the amplified duplex sequencing library.
- the DNA library
- the double stranded DNA template is obtained from a sample from a mammal the generating the DNA library (e.g., the single stranded DNA library) of Crick strands from the amplified duplex sequencing library comprises: i) amplifying a second aliquot of the amplified duplex sequencing library using a primer pair consisting of a first primer and a second primer, wherein the first primer is capable of hybridizing to the Crick strand, and wherein the first primer comprises a tag, to generate double stranded amplification products having tagged Crick strands; ii) denaturing the double stranded amplification products having tagged Crick strands to generate single stranded tagged Crick strands and single stranded Watson strands; and iii) recovering the single stranded tagged Crick strands to generate the DNA library (e.g., the single stranded DNA library) of Crick strands from the amplified duplex sequencing library
- the method further comprises, prior to the generating a double stranded DNA fragment having the duplex molecular barcode on each end of the double stranded DNA fragment: fragmenting the double stranded DNA to generate double stranded
- ligating a 3’ duplex adapter to each 3’ end of a double stranded DNA fragment obtained from the double stranded DNA template comprises contacting the 3’ duplex adapter and the double stranded DNA fragment obtained from the double stranded DNA template in the presence of a ligase.
- the ligase is a T4 DNA ligase.
- degrading the degradable 3’ blocking group comprises contacting the 3’ duplex adapter with uracil-DNA glycosylase (UDG).
- ligating the 5’ adapter to each dephosphorylated 5’ end of the double stranded DNA fragment obtained from the double stranded DNA template comprises contacting the 5’ adapter and the double stranded DNA fragment obtained from the double stranded DNA template in the presence of a ligase.
- the ligase is an Escherichia coli ligase.
- filling in the gap of single stranded nucleic acid between the 5’ end of the double stranded DNA fragment and the 5’ adapter comprises contacting the 5’ end of the double stranded DNA fragment and the 5’ adapter in the presence of a polymerase and dNTPs.
- the polymerase is a Taq polymerase.
- amplifying the double stranded DNA fragment comprising the duplex molecular barcode on each end of the double stranded DNA fragment to generate an amplified duplex sequencing library comprises contacting the double stranded DNA fragment comprising the duplex molecular barcode on each end of the double stranded DNA fragment with a universal primer pair under PCR conditions.
- the amplifying comprises whole-genome PCR.
- the tagged primers are biotinylated primers, and wherein the biotinylated primers can generate biotinylated single stranded Watson strands and biotinylated single stranded Crick strands.
- the denaturing steps comprise NaOH denaturation, heat denaturation, or a combination of both.
- the recovering steps comprise contacting the tagged Watson strands with streptavidin-functionalized beads and contacting the tagged Crick strands with streptavidin-functionalized beads. In some embodiments, the recovering steps further comprise denaturing non-tagged Watson strands and denaturing non-tagged Watson strands.
- the recovering steps further comprise releasing the biotinylated single stranded Watson strands from the streptavidin-functionalized beads and releasing the biotinylated single stranded Crick strands from the streptavidin-functionalized beads.
- the tagged primers are phosphorylated primers, and the phosphorylated primers can generate phosphorylated single stranded Watson strands and phosphorylated single stranded Crick strands.
- the denaturing steps comprises lambda exonuclease digestion.
- amplifying the target region from the DNA library (e.g., the single stranded DNA library) of Watson strands further comprises second amplification using a second primer pair consisting of a first primer capable of hybridizing to the target region and a second primer capable of hybridizing to the 3’ duplex adapter; and wherein the amplifying the target region from the DNA library (e.g., the single stranded DNA library) of Crick strands further comprises a second amplification using a second primer pair consisting of a first primer capable of hybridizing to the target region and a second primer capable of hybridizing to the 5’ adapter.
- the sequencing steps comprise paired- end sequencing.
- the double stranded DNA template is a genomic DNA sample and generating the double stranded DNA fragments each having the duplex molecular barcode on each end of the double stranded DNA fragment comprises: i) ligating a 3’ duplex adapter to each 3’ end of a double stranded DNA fragment obtained from the double stranded DNA template, wherein the 3’ duplex adapter comprises a) a first oligonucleotide comprising a 5’ phosphate, a first molecular barcode, and a 3’ oligonucleotide annealed to b) a second oligonucleotide comprising a degradable 3’ blocking group, wherein the 3’ oligonucleotide and the second oligonucleotide sequence are complementary; ii) degrading the degradable 3’ blocking group; iii) ligating a 5’ adapter to each dephosphorylated 5’ end of the double strande
- the double stranded DNA template is obtained from a sample from a mammal.
- the mammal is a human.
- the method further comprises, prior to the generating a double stranded DNA fragment having the duplex molecular barcode on each end of the double stranded DNA fragment: fragmenting the double stranded DNA to generate double stranded DNA fragments; dephosphorylating 5’ ends of the double stranded DNA fragments; and blunting the ends of double stranded DNA fragments.
- ligating a 3’ duplex adapter to each 3’ end of a double stranded DNA fragment obtained from the double stranded DNA template comprises contacting the 3’ duplex adapter and the double stranded DNA fragment obtained from the double stranded DNA template in the presence of a ligase.
- the ligase is a T4 DNA ligase.
- the degradable 3’ blocking group comprises contacting the 3’ duplex adapter with uracil-DNA glycosylase (UDG).
- ligating the 5’ adapter to each dephosphorylated 5’ end of the double stranded DNA fragment obtained from the double stranded DNA template comprises contacting the 5’ adapter and the double stranded DNA fragment obtained from the double stranded DNA template in the presence of a ligase.
- the ligase is an Escherichia coli ligase.
- filling in the gap of single stranded nucleic acid between the 5’ end of the double stranded DNA fragment and the 5’ adapter comprises contacting the 5’ end of the double stranded DNA fragment and the 5’ adapter in the presence of a polymerase and dNTPs.
- the polymerase is a Taq-B polymerase.
- ligating the 5’ adapter to each 5’ end of the double stranded DNA fragment, and the filling in the gap between the 5’ end of the double stranded DNA fragment and the 5’ adapter are performed concurrently.
- amplifying the double stranded DNA fragment having the duplex molecular barcode on each end of the double stranded DNA fragment comprises contacting the double stranded DNA fragment comprising the duplex molecular barcode on each end of the double stranded DNA fragment with the primer pair under PCR conditions.
- amplifying comprises whole-genome PCR.
- amplifying the target region from the DNA library (e.g., the single stranded DNA library) of Watson strands further comprises second amplification using a second primer pair consisting of a first primer capable of hybridizing to the target region and a second primer capable of hybridizing to the 3’ duplex adapter; and wherein the amplifying the target region from the DNA library (e.g., the single stranded DNA library) of Crick strands further comprises a second amplification using a second primer pair consisting of a first primer capable of hybridizing to the target region and a second primer capable of hybridizing to the 5’ adapter.
- the sequencing steps comprise paired-end sequencing or single-end sequencing.
- the double stranded DNA template is a genomic DNA sample and generating the double stranded DNA fragments each having the duplex molecular barcode on each end of the double stranded DNA fragment comprises: i) ligating a 3’ duplex adapter to each 3’ end of a double stranded DNA fragment obtained from the double stranded DNA template, wherein the 3’ duplex adapter comprises a) a first oligonucleotide comprising a 5’ phosphate, a first molecular barcode, and a 3’ oligonucleotide annealed to b) a second oligonucleotide comprising a degradable 3’ blocking group, wherein the 3’ oligonucleotide and the second oligonucleotide sequence are complementary; ii) degrading the degradable 3’ blocking group; iii) ligating a 5’ adapter to each dephosphorylated 5’ end of the double strande
- the method further comprises, prior to the generating a double stranded DNA fragment having the duplex molecular barcode on each end of the double stranded DNA fragment: fragmenting the double stranded DNA to generate double stranded DNA fragments; dephosphorylating 5’ ends of the double stranded DNA fragments; and blunting the ends of double stranded DNA fragments.
- ligating a 3’ duplex adapter to each 3’ end of a double stranded DNA fragment obtained from the double stranded DNA template comprises contacting the 3’ duplex adapter and the double stranded DNA fragment obtained from the double stranded DNA template in the presence of a ligase.
- the ligase is a T4 DNA ligase.
- degrading the degradable 3’ blocking group comprises contacting the 3’ duplex adapter with uracil-DNA glycosylase (UDG).
- ligating the 5’ adapter to each dephosphorylated 5’ end of the double stranded DNA fragment obtained from the double stranded DNA template comprises contacting the 5’ adapter and the double stranded DNA fragment obtained from the double stranded DNA template in the presence of a ligase.
- the ligase is an Escherichia coli ligase.
- filling in the gap of single stranded nucleic acid between the 5’ end of the double stranded DNA fragment and the 5’ adapter comprises contacting the 5’ end of the double stranded DNA fragment and the 5’ adapter in the presence of a DNA polymerase and dNTPs.
- the DNA polymerase is a Taq-B polymerase.
- amplifying the double stranded DNA fragment having the duplex molecular barcode on each end of the double stranded DNA fragment comprises contacting the double stranded DNA fragment comprising the duplex molecular barcode on each end of the double stranded DNA fragment with the primer pair under PCR conditions. In some embodiments, amplifying comprises whole-genome PCR.
- amplifying the target region from the DNA library (e.g., the single stranded DNA library) of Watson strands further comprises second amplification using a second primer pair consisting of a first primer capable of hybridizing to the target region and a second primer capable of hybridizing to the 3’ duplex adapter; and wherein the amplifying the target region from the DNA library (e.g., the single stranded DNA library) of Crick strands further comprises a second amplification using a second primer pair consisting of a first primer capable of hybridizing to the target region and a second primer capable of hybridizing to the 5’ adapter.
- the sequencing steps comprise paired-end sequencing.
- the 5’ adaptors comprise, in the 5’ to 3’ direction, (i) a universal 5’ adaptor sequence that is not complementary to the universal 3’ adaptor sequence and that comprises an R1 sequencing primer site, and (ii) a sequence complementary to the annealing site for the 5’ adaptor; c.
- a nick translation-like reaction to extend the 5’ adaptors across the exogenous UID sequence of the 3’ adaptors (e.g., using a DNA polymerase) and covalently link the extended 5’ adaptor to the 5’ ends of the Watson and Crick strands of the double-stranded DNA fragments (e.g., using a ligase); d. performing an initial amplification to amplify the adaptor-ligated, double- stranded DNA fragments to produce amplicons; e. determining sequence reads of one or more amplicons of the one or more of the adaptor-ligated, double-stranded DNA fragments; f.
- each member of a UID family comprises the same exogenous UID sequence
- g assigning sequence reads of each UID family into a Watson subfamily and Crick subfamily based on spatial relationship of the exogenous UID sequence to the R1 and R2 read sequence
- h identifying a nucleotide sequence as accurately representing a Watson strand of an analyte DNA fragment when a threshold percentage of members of the Watson subfamily contain the sequence
- i identifying a nucleotide sequence as accurately representing a Crick strand of an analyte DNA fragment when a threshold percentage of members of the Crick subfamily contain the sequence
- j assigning the sequence reads into UID families, wherein each member of a UID family comprises the same exogenous UID sequence
- g assigning sequence reads of each UID family into a Watson subfamily and Crick subfamily based on spatial relationship of the exogenous UID sequence to the R1 and R2 read sequence
- h identifying a nucleotide sequence as
- identifying a mutation in the nucleotide sequence accurately representing the Watson Strand when the sequence accurately representing the Watson Strand differs from a reference sequence that lacks the mutation k. identifying a mutation in the nucleotide sequence accurately representing the Crick Strand when the sequence accurately representing the Crick Strand differs from a reference sequence that lacks the mutation; and l. identifying a mutation in the analyte DNA fragment when the mutation in the nucleotide sequence accurately representing the Watson strand and the mutation in the nucleotide sequence accurately representing the Crick strand are the same mutation.
- each member of a UID family further comprises the same endogenous UID sequence, wherein the endogenous UID sequence comprises an end of a double-stranded DNA fragment from the population.
- the endogenous UID sequence comprising the end of the double-stranded DNA fragment comprises at least 8, 10, or 15 bases.
- exogenous UID sequence is unique to each double- stranded DNA fragment.
- the exogenous UID sequence is not unique to each double-stranded DNA fragment.
- each member of a UID family comprises the same endogenous UID sequence and the same exogenous UID sequence.
- step (d) comprises no more than 11 cycles of PCR amplification.
- step (d) comprises no more than 7 cycles of PCR amplification. In some embodiments, step (d) comprises no more than 5 cycles of PCR amplification. In some embodiments, step (d) comprises at least 1 cycles of PCR amplification.
- the amplicons are enriched for one or more target polynucleotides.
- the enriching comprises: a. selectively amplifying amplicons of Watson strands comprising the target polynucleotide sequence with a first set of Watson target-selective primer pairs, the first set of Watson target- selective primer pairs comprising: (i) a first Watson target- selective primer comprising a sequence complementary to a portion of the universal 3’ adapter sequence, optionally wherein the portion of the universal 3’ adapter sequence is the R2 sequencing primer site of the universal 3’ adaptor sequence, and (ii) a second Watson target-selective iy primer comprising a target-selective sequence, thereby creating target Watson amplification products; and b.
- the first set of Crick target-selective primer pairs comprising: (i) a first Crick target-selective primer comprising a sequence complementary to a portion of the universal 5’ adapter sequence, optionally wherein the portion of the universal 5’ adapter sequence is the R1 sequencing primer site of the universal 5’ adaptor sequence, and (ii) a second Crick target-selective primer comprising the same target- selective sequence as the second Watson target- selective primer sequence, thereby creating target Crick amplification products.
- the method further comprises purifying the target Watson amplification products and the target Crick amplification products from non-target polynucleotides. In some embodiments, the method further comprises purifying comprises attaching the target Watson amplification products and the target Crick amplification products to a solid support.
- the first Watson target-selective primer and first Crick target-selective primer comprises a first member of an affinity binding pair, and wherein the solid support comprises a second member of the affinity binding pair.
- the first member is biotin and the second member is streptavidin.
- the solid support comprises a bead, well, membrane, tube, column, plate, sepharose, magnetic bead, or chip. In some embodiments, the method further comprises removing polynucleotides that are not attached to the solid support.
- the method further comprises: a. further amplifying the target Watson amplification products with a second set of Watson target-selective primers, the second set of Watson target-selective primers comprising (i) a third Watson target-selective primer comprising a sequence complementary to a portion of the universal 3’ adapter sequence, optionally wherein the portion of the universal 3’ adapter sequence is the R2 sequencing primer site of the universal 3 ’ adaptor sequence , and (ii) a fourth
- Watson target-selective primer comprising, in the 5’ to 3’ direction, an R1 sequencing primer site and a target-selective sequence selective for the same target polynucleotide, thereby creating target Watson library members; zu b. further amplifying the target Crick amplification products with a second set of Crick target-selective primers, the second set of Crick target- selective primers comprising (i) a third Crick target-selective primer comprising a sequence complementary to a portion of the universal 5’ adapter sequence, optionally wherein the portion of the universal 5’ adapter sequence is the R1 sequencing primer site of the universal 5’ adaptor sequence, and (ii) a fourth Crick target- selective primer comprising, in the 5’ to 3’ direction, an R2 sequencing primer site and the target-selective sequence selective for the same target polynucleotide of the fourth Watson target- selective primer, thereby creating target Crick library members.
- the third Watson and Crick target-selective primers further comprise a sample barcode sequence.
- the third Watson target- selective primer further comprises a first grafting sequence that enables hybridization to a first grafting primer on a sequencer and wherein the third Crick target-selective primer further comprises a second grafting sequence that enables hybridization to a second grafting primer on the sequencer.
- the fourth Watson target- selective primer further comprises the second grafting sequence and wherein the fourth Crick target-selective primer further comprises the first grafting sequence.
- the first grafting sequence is a P7 sequence and wherein the second grafting sequence is a P5 sequence.
- the target Watson library members and the target Crick library members represent at least 50% of the target polynucleotides in the population of double-stranded DNA fragments. In some embodiments, the target Watson library members and the target Crick library members represent at least 70% of the target polynucleotides in the population of double-stranded DNA fragments. In some embodiments, the target Watson library members and the target Crick library members represent at least 80% of the target polynucleotides in the population of double-stranded DNA fragments. In some embodiments, the target Watson library members and the target Crick library members represent at least 90% of the target polynucleotides in the population of double-stranded DNA fragments.
- the target Watson library members and the target Crick library members represent at least 50% of the population of total DNA fragments.
- the target Watson library members and the target Crick library members represent at least 70% of the population of total DNA fragments.
- the target Watson library members and the target Crick library members represent at least 80% of the population of total DNA fragments.
- the target Watson library members and the target Crick library members represent at least 90% of the population of total DNA fragments.
- Also provided herein are methods comprising: a. attaching adaptors to a population of double-stranded DNA fragments in an analyte DNA sample, wherein the adaptors comprise a double-stranded portion comprising an exogenous UID and a forked portion comprising (i) a single- stranded 3’ adaptor sequence comprising an R2 sequencing primer site and (ii) a single-stranded 5’ adaptor sequence comprising an R1 sequencing primer site; b. performing an initial amplification to amplify the adaptor-ligated, double- stranded DNA fragments to produce amplicons; c.
- the first set of Watson target- selective primer pairs comprising: (i) a first Watson target- selective primer comprising a sequence complementary to a portion of the universal 3’ adapter sequence, optionally wherein the portion of the universal 3’ adapter sequence is the R2 sequencing primer site of the universal 3’ adaptor sequence, and (ii) a second Watson target-selective primer comprising a target-selective sequence, thereby creating target Watson amplification products; d.
- the first set of Crick target-selective primer pairs comprising: a first Crick target-selective primer comprising a sequence a sequence complementary to a portion of the universal 5’ adapter sequence, optionally wherein the portion of the universal 5’ adapter sequence is the R1 sequencing primer site of the universal 5’ adaptor sequence, and (ii) a second Crick target-selective primer comprising the same target-selective sequence as the second Crick target- selective primer sequence, thereby creating target Crick amplification products.
- sequence reads of the target Watson amplification products and the target Crick amplification products S ' I f. assigning the sequence reads into UID families, wherein each member of a UID family comprises the same exogenous UID sequence; g. assigning sequence reads of each UID family into a Watson subfamily and Crick subfamily based on spatial relationship of the exogenous UID sequence to the R1 and R2 read sequence; h. identifying a nucleotide sequence as accurately representing a Watson strand of an analyte DNA fragment when a threshold percentage of members of the Watson family contain the sequence; i.
- the method further comprises purifying the target Watson amplification products and the target Crick amplification products from non-target polynucleotides. In some embodiments, the method further comprises attaching the target Watson amplification products and the target Crick amplification products to a solid support.
- the first Watson target-selective primer and first Crick target- selective primer comprises a first member of an affinity binding pair, and wherein the solid support comprises a second member of the affinity binding pair.
- the first member is biotin and the second member is streptavidin.
- the solid support comprises a bead, well, membrane, tube, column, plate, sepharose, magnetic bead, or chip. In some embodiments, the method further comprises removing polynucleotides that are not attached to the solid support.
- the method further comprises: a. further amplifying the target Watson amplification products with a second set of Watson target-selective primers, the second set of Watson target-selective primers comprising (i) a third Watson target-selective primer comprising a sequence complementary to the R2 sequencing primer site of the universal 3’ adaptor sequence, and (ii) a fourth Watson target-selective primer comprising, in the 5’ to 3’ direction, an R1 sequencing primer site and a target-selective
- the third Watson and Crick target-selective primers further comprise a sample barcode sequence.
- the third Watson target- selective primer further comprises a first grafting sequence that enables hybridization to a first grafting primer on a sequencer and wherein the third Crick target-selective primer further comprises a second grafting sequence that enables hybridization to a second grafting primer on the sequencer.
- the fourth Watson target- selective primer further comprises the second grafting sequence and wherein the fourth Crick target-selective primer further comprises the first grafting sequence.
- the first grafting sequence is a P7 sequence and wherein the second grafting sequence is a P5 sequence.
- the attaching comprises attaching A-tailed adaptors to the population of double-stranded DNA fragments. In some embodiments, the attaching comprises attaching A-tailed adaptors to both ends of DNA fragments in the population.
- the attaching comprises: a. attaching partially double-stranded 3’ adaptors to 3’ ends of both Watson and Crick strands of a population of double-stranded DNA fragments, wherein a first strand of the partially double-stranded 3’ adaptor comprises, in the 5’ -3’ direction, (i) a first segment, (ii) optionally, an exogenous UID sequence, (iii) an annealing site for a 5’ adaptor, and (iv) a universal 3’ adaptor sequence comprising an R2 sequencing primer site, and wherein the second strand of the partially double-stranded 3’ adaptor comprises, in the 5’ to 3’ direction, (i) a segment complementary to the first segment, and (ii) a 3’ blocking group, optionally wherein the second strand is degradable; and b.
- annealing 5’ adaptors to the 3’ adaptors via the annealing site, wherein the 5’ adaptors comprise, in the 5’ to 3’ direction, (i) a universal 5’ adaptor sequence that is not complementary to the universal 3’ adaptor sequence and that comprises an R1 sequencing primer site, and (ii) a sequence complementary to the annealing site for the 5’ adaptor; and c. performing a nick translation-like reaction to extend the 5’ adaptors across the 3’ adaptors (e.g., using a DNA polymerase) and covalently link the extended 5’ adaptor to the 5’ ends of the Watson and Crick strands of the double- stranded DNA fragments (e.g. using a ligase).
- a nick translation-like reaction to extend the 5’ adaptors across the 3’ adaptors (e.g., using a DNA polymerase) and covalently link the extended 5’ adaptor to the 5’ ends of the Watson and Crick strands
- the UID sequence comprises an endogenous UID sequence comprising an end of a double-stranded DNA fragment from the population.
- the endogenous UID sequence comprising the end of the double-stranded DNA fragment comprises at least 8, 10, or 15 bases.
- the exogenous UID sequence is unique to each double-stranded DNA fragment.
- the exogenous UID sequence is not unique to each double-stranded DNA fragment.
- each member of a UID family comprises the same endogenous UID sequence and the same exogenous UID sequence.
- amplifying the adaptor-ligated, double-stranded DNA fragments to produce amplicons comprises no more than 11 cycles of PCR amplification. In some embodiments, amplifying the adaptor-ligated, double-stranded DNA fragments to produce amplicons comprises no more than 7 cycles of PCR amplification. In some embodiments, amplifying the adaptor-ligated, double-stranded DNA fragments to produce amplicons comprises no more than 5 cycles of PCR amplification. In some embodiments, amplifying the adaptor-ligated, double-stranded DNA fragments to produce amplicons comprises at least 1 cycles of PCR amplification.
- the target Watson library members and the target Crick library members represent at least 50% of the target polynucleotides in the population of double-stranded DNA fragments. In some embodiments, the target Watson library members and the target Crick library members represent at least 70% of the target polynucleotides in the population of double-stranded DNA fragments. In some embodiments, the target Watson library members and the target Crick library members represent at least 80% of the target polynucleotides in the population of double-stranded DNA fragments. In some embodiments, the target Watson library members and the target Crick library members represent at least 90% of the target polynucleotides in the population of double-stranded DNA fragments.
- the target Watson library members and the target Crick library members represent at least 50% of the population of total DNA fragments. In some embodiments, the target Watson library members and the target Crick library members represent at least 70% of the population of total DNA fragments. In some embodiments, the target Watson library members and the target Crick library members represent at least 80% of the population of total DNA fragments. In some embodiments, the target Watson library members and the target Crick library members represent at least 90% of the population of total DNA fragments.
- the determination of the sequence reads enables sequence determination of both ends of template molecules. In some embodiments, the determination of both ends of template molecules comprises paired end sequencing. In some embodiments, the determination of the sequence reads comprises single read sequencing across the length of the templates for generating the sequence reads. In some embodiments, the determination of the sequence reads comprises sequencing with a massively parallel sequencer. In some embodiments, the massively parallel sequencer is configured to determine sequence reads from both ends of template polynucleotides. In some embodiments, the population of double- stranded DNA fragments comprises one or more fragments that are about 50-600 nt in length. In some embodiments, the population of double-stranded DNA fragments comprises one or more fragments that are less than 2000, less than 1000, less than 500, less than 400, less than 300, or less than 250 nt in length.
- methods provided herein further comprise, after the initial amplification and before the selective amplification, preparing single stranded (ss) DNA libraries corresponding to sense and antisense strands of the amplicons.
- the ss DNA library preparation comprises: a. performing an amplification reaction utilizing two primers, wherein only one of the two primers comprises a first member of an affinity binding pair, thereby creating amplification products comprising a strand comprising the first member of the affinity binding pair and a strand not comprising the first member of the affinity binding pair; b. contacting the amplification products with a solid support, wherein the solid support comprises the second member of the affinity binding pair; c.
- the first member of the affinity binding pair is biotin and the second member of the affinity binding pair is streptavidin.
- the ss DNA library preparation comprises: a. partitioning the amplicons into two amplification reactions, each amplification reaction utilizing a forward primer and a reverse primer, wherein only one of the two primers is phosphorylated, thereby creating amplification products comprising a phosphorylated strand and a non-phosphorylated strand; b. contacting the amplification products with an exonuclease that selectively digests the strands with the 5’ phosphate.
- the forward primer in the first amplification reaction, the forward primer is phosphorylated and the reverse primer is non-phosphorylated; b. in the second amplification reaction, the reverse primer is phosphorylated and the forward primer is non-phosphorylated.
- the exonuclease is a lambda exonuclease. In some embodiments, the phosphorylation is at the 5’ site.
- the initial amplification comprises: a. amplifying with a primer pair, in which only one of the two primers in the primer pair comprises a first member of an affinity binding pair, thereby creating amplification products comprising a strand comprising the first member of the affinity binding pair and a strand not comprising the first member of the affinity binding pair; b. contacting the amplification products with a solid support, wherein the solid support comprises the second member of the affinity binding pair; c. denaturing the amplification products to separate the strand comprising the first member of the affinity binding pair from the strand not comprising the first member of the affinity binding pair; and d. purifying the separated strands comprising the first member of the affinity binding pair and the separated strands not comprising the first member of the affinity binding pair.
- the first member of the affinity binding pair is biotin and the second member of the affinity binding pair is streptavidin.
- sequence reads of a UID family are assigned to a Watson subfamily when the exogenous UID sequence is downstream of the R2 sequence and upstream of the R1 sequence.
- sequence reads of a UID family are assigned to a Crick subfamily when the exogenous UID sequence is downstream of the R1 sequence and upstream of the R2 sequence.
- sequence reads of a UID family are assigned to a Watson subfamily when the exogenous UID sequence is in greater proximity to the R2 sequence and lesser proximity to the R1 sequence.
- sequence reads of a UID family are assigned to a Crick subfamily when the exogenous UID sequence is in greater proximity to the R1 sequence and in lesser proximity to the R2 sequence.
- sequence reads of a UID family are assigned to a Watson subfamily when the exogenous UID sequence is immediately downstream or within 1-300, 1-70, 1-60, 1-50, 1-40, 1-30, 1- 20, 1-10, or 1-5 nucleotides of the R2 sequence.
- sequence reads of a UID family are assigned to a Crick subfamily when the exogenous UID sequence is immediately downstream or within 1-300, 1-70, 1-60, 1-50, 1-40, 1-30, 1-20, 1-10, or 1-5 nucleotides of the R1 sequence.
- the population of double-stranded DNA fragments are from a biological sample.
- the biological sample is obtained from a subject.
- the subject is a human subject.
- the biological sample is a fluid sample.
- the fluid sample is selected from whole blood, plasma, serum sputum, urine, sweat, tears, ascites, semen, and bronchoaveolar lavage.
- the fluid sample is a cell-free or essentially cell-free sample.
- the biological sample is a solid biological sample.
- the solid biological sample is a tumor sample.
- the identified mutation is present in the population of double- stranded DNA fragments at a frequency of 0.1% or less. In some embodiments, the identified mutation is present in the population of double-stranded DNA fragments at a frequency of 0.1% to 0.00001%. In some embodiments, the identified mutation is present in the population of double-stranded DNA fragments at a frequency of 0.1% to 0.01%. In some embodiments, the determining of sequence reads comprises determining sequence reads from both Watson and Crick strands of at least 50% of the double-stranded DNA fragments comprising a target polynucleotide in the analyte DNA sample.
- the determining of sequence reads comprises determining sequence reads from both Watson and Crick strands of at least 70% of the double-stranded DNA fragments comprising a target polynucleotide in the analyte DNA sample. In some embodiments, the determining of sequence reads comprises determining sequence reads from both Watson and Crick strands of at least 80% of the double-stranded DNA fragments comprising a target polynucleotide in the analyte DNA sample. In some embodiments, the determining of sequence reads comprises determining sequence reads from both Watson and Crick strands of at least 90% of the double-stranded DNA fragments comprising a target polynucleotide in the analyte DNA sample.
- the determining of sequence reads comprises determining sequence reads from both Watson and Crick strands of at least 50% of the double-stranded DNA fragments in the analyte DNA sample. In some embodiments, the determining of sequence reads comprises determining sequence reads from both Watson and Crick strands of at least 70% of the double-stranded DNA fragments in the analyte DNA sample. In some embodiments, the determining of sequence reads comprises determining sequence reads from both Watson and Crick strands of at least 80% of the double-stranded DNA fragments in the analyte DNA sample. In some embodiments, the determining of sequence reads comprises determining sequence reads from both Watson and Crick strands of at least 90% of the double-stranded DNA fragments in the analyte DNA sample.
- the error rate associated with the identification of one or more mutations in analyte DNA fragments according to a method of any one of the preceding claims is reduced by at least 2-fold, 4-fold, 5-fold, 10-fold, 20-fold, 30-fold, 40-fold, 50-fold, 60-fold, 70-fold, 80-fold, 90-fold, or 100-fold, as compared to an alternative method of identifying mutations that does not require the mutation to be detected in both Watson and Crick strands of an analyte DNA fragment.
- the alternative method comprises standard molecular barcoding or standard PCR-based molecular barcoding.
- the alternative method comprises: a.
- adaptors attaching adaptors to a population of double-stranded DNA fragments in an analyte DNA sample, wherein the adaptors comprise a unique exogenous UID; b. performing an initial amplification to amplify the adaptor-ligated, double- stranded DNA fragments to produce amplicons; c. determining sequence reads of one or more amplicons of the one or more of the adaptor-ligated, double-stranded DNA fragments; d. assigning the sequence reads into UID families, wherein each member of a UID family comprises the same exogenous UID sequence; e.
- the error rate associated with the identification of one or more mutations in analyte DNA fragments according to a method of any one of the preceding claims is no more than lxlO 2 , no more than lxlO 3 , no more than lxlO 4 , no more than 1x10 5 , no more than lxlO 6 , no more than 5xl0 6 , or no more than lxlO 7 .
- the computer readable medium comprises executable instructions for a. assigning sequence reads into UID families, wherein each member of a UID family comprises the same exogenous UID sequence; b. assigning sequence reads of each UID family into Watson and Crick subfamilies based on spatial relationship of the exogenous UID sequence to the R1 and R2 read sequence; c. identifying a nucleotide sequence as accurately representing a Watson strand of an analyte DNA fragment when a threshold percentage of members of the Watson subfamily contain the sequence; d.
- the computer readable medium comprises executable instructions for assigning UID family members to the Watson subfamily when the exogenous UID sequence is immediately downstream or within 1-300 nucleotides of the R2 sequencing primer binding site. In some embodiments, the computer readable medium comprises executable instructions for assigning UID family members to the Crick subfamily when the exogenous UID sequence is immediately downstream or within 1-300 nucleotides of the R1 sequencing primer binding site. In some embodiments, the computer readable medium comprises executable instructions for mapping the sequence reads to a reference genome. In some embodiments, the reference genome is a human reference genome.
- the computer readable medium further comprises computer executable instructions to generate a report of therapeutic options based on the presence, absence, or amount of a mutation in the sample. In some embodiments, the computer readable medium further comprises computer executable code that enables transmission of the data over a network.
- Also provided herein are computer systems comprising: a. a memory unit configured to receive sequence data from a nucleic acid sample, wherein the data is generated by a method of any one of the preceding claims; b. a processor communicatively coupled to the memory unit, wherein the processor comprises a computer readable medium of any of the preceding claims.
- the computer system further comprises a sequencing system configured to communicate the data to the memory unit.
- the computer system further comprises a user interface configured to communicate or display the report to a user.
- the computer system further comprises a digital processor configured to transmit results of the data analysis over a network.
- nick translation-like reaction e.g., with a DNA polymerase, cohesive end-specific ligase, and uracil-DNA glycosylase
- reagents for enriching amplicons for one or more target polynucleotides e.g., a sequencing system.
- system further comprises a computer system of any one of the preceding claims.
- kits comprising: a. a first set of Watson target- selective primer pairs, comprising (i) one or more first Watson target- selective primers comprising a sequence complementary to a portion of the universal 3’ adapter sequence, optionally wherein the portion of the universal 3’ adapter sequence is the R2 sequencing primer site of the universal 3’ adaptor sequence, and (ii) one or more second Watson target- selective primers, each of the one or more second Watson target-selective primers comprising a target-selective sequence; b.
- a first set of Watson target- selective primer pairs comprising (i) one or more first Watson target- selective primers comprising a sequence complementary to a portion of the universal 3’ adapter sequence, optionally wherein the portion of the universal 3’ adapter sequence is the R2 sequencing primer site of the universal 3’ adaptor sequence, and (ii) one or more second Watson target- selective primers, each of the one or more second Watson target-selective primers comprising a target-selective sequence; b.
- a first set of Crick target-selective primer pairs comprising (i) one or more Crick target-selective primers comprising a sequence a sequence complementary to a portion of the universal 5’ adapter sequence, optionally wherein the portion of the universal 5’ adapter sequence is the R1 sequencing primer site of the universal 5’ adaptor sequence, and (ii) one or more second Crick target-selective primers, each of the one or more second Crick target- selective primers comprising the same target-selective sequence as the second Watson target-selective primer sequence; c.
- a second set of Watson target-selective primer pairs comprising (i) one or more third Watson target-selective primers comprising a sequence complementary to the R2 sequencing primer site of the universal 3’ adaptor sequence, and (ii) one or more fourth Watson target-selective primers, each of ii the one or more fourth Watson target-selective primers comprising, in the 5’ to 3’ direction, an R1 sequencing primer site and a target-selective sequence selective for the same target polynucleotide; and d.
- a second set of Crick target-selective primers comprising (i) one or more third Crick target-selective primers comprising a sequence complementary to the R1 sequencing primer site of the universal 3’ adaptor sequence, and (ii) one or more fourth Crick target-selective primers, each of the one or more fourth Crick target-selective primers comprising, in the 5’ to 3’ direction, an R2 sequencing primer site and a target-selective sequence selective for the same target polynucleotide.
- Figure 1 contains a schematic of an exemplary duplex anchored PCR method.
- Duplex adapters with molecular barcodes are ligated onto the ends of nucleic acid fragments having blunted ends, to generate a duplex sequencing library, and the duplex sequencing library is subjected to PCR to generate an amplified duplex sequencing library.
- the amplification products in the amplified duplex sequencing library are divided into two aliquots and each aliquot is subjected to PCR where the Watson strand is amplified from the first aliquot, and the Crick strand is amplified from the second aliquot.
- Figure 2 contains a schematic of an exemplary second round of library amplification where the Watson strands amplified from the first aliquot in Figure 1 are subjected to PCR using a primer pair where a first primer is biotinylated and a second primer is non- biotinylated to generate a single stranded DNA library that can be used to amplify and assess Watson strands.
- Figure 3 contains a schematic of an exemplary second round of library amplification where the Crick strands amplified from the first aliquot in Figure 1 are subjected to PCR using a primer pair where a first primer is non-biotinylated and a second primer is biotinylated to generate a single stranded DNA library that can be used to amplify and assess Crick strands.
- Figure 4 contains a schematic of an exemplary Watson amplification.
- Figure 5 contains a schematic of an exemplary Crick amplification.
- Figure 6 contains a schematic of an exemplary amplified Watson strand and an exemplary amplified Crick strand.
- Figure 7 contains a schematic of an exemplary nested Watson amplification.
- Figure 8 contains a schematic of an exemplary nested Crick amplification.
- Figure 9 contains a schematic of an exemplary removal of a 5’ phosphate.
- Figure 10 contains a schematic of an exemplary filling in of a 3’ end of an amplification fragment having a 5’ overhang to generate a blunt end amplification product.
- Figure 11 contains a schematic of an exemplary 3’ duplex adapter including a 3SpC3 spacer, exogenous UID sequence containing a molecular barcode, and a 3’ oligonucleotide (dT) hybridized to a 3’ blocking group which can be degraded by uracil-DNA glycosylase (UDG).
- a 3’ duplex adapter including a 3SpC3 spacer, exogenous UID sequence containing a molecular barcode, and a 3’ oligonucleotide (dT) hybridized to a 3’ blocking group which can be degraded by uracil-DNA glycosylase (UDG).
- dT oligonucleotide
- Figure 12 contains a schematic of an exemplary 3’ adapter ligation using a 3’ duplex adapter.
- the 5’ phosphate of the 3’ duplex adapter is ligated to the 3’ end of the nucleic acid template.
- Figure 13 contains a schematic of an exemplary 5’ adapter ligation.
- the blocking group of the 3’ duplex adapter is degraded and a 5’ adapter containing is ligated to the 5’ end of the nucleic acid template via a nick-translation reaction.
- Figure 14 contains a schematic of an exemplary library PCR amplification.
- Figure 15 contains a schematic of an exemplary Watson amplification.
- Figure 16 contains a schematic of an exemplary nested Watson amplification.
- Figure 17 contains a schematic of an exemplary Crick amplification.
- Figure 18 contains a schematic of an exemplary nested Crick amplification.
- Figure 19 contains a schematic of final amplification products generated by an exemplary duplex anchored PCR.
- Figure 20 contains a schematic showing how paired-end sequencing can be used to distinguish the Watson strand from the Crick strand of the input nucleic acid using the final amplification products generated by an exemplary duplex anchored PCR.
- Figure 21 contains a schematic showing how paired-end sequencing can be used to distinguish the Watson strand from the Crick strand of the input nucleic acid using the final amplification products generated by an exemplary duplex anchored PCR.
- Figure 22 contains a schematic of an overview of an exemplary SaferSeqS method.
- step 1 Library preparation begins with end repair (step 1) in which DNA template molecules are dephosphorylated and blunted.
- step 2 a 3’ adapter containing a unique identifier (UID) sequence (narrow or wide diagonal cross-hatching) is ligated to the 3’ fragment ends (step 2).
- UID unique identifier
- the UID sequences are converted into double stranded barcodes upon extension and ligation of the 5’ adapter (step 3). Finally, redundant PCR copies of each original template molecule are generated during library amplification (step 4). (b) Target enrichment is achieved with strand-specific hemi-nested PCRs.
- the amplified library is partitioned into Watson and
- step 5 which selectively amplify products derived from one of the
- DNA strands (step 6). Additional on-target specificity and incorporation of sample barcodes are achieved with a second nested PCR (step 7).
- the final PCR products (step 8) are subjected to paired-end sequencing (step 9).
- the endogenous barcode represents the end of the template fragment prior to library construction (c) Following sequencing, reads are determined to be derived from the Watson or Crick strand. Because each strand of the original template molecules is tagged with the same exogenous barcode, and has the same endogenous barcode, reads derived from each of the two strands of the same parental DNA duplex can be grouped together into a duplex family. The different cross-hatch and stipple patterns at the right ends of the strands represent different barcodes.
- each duplex family has eight members, four representing the Watson strand and four representing the Crick strand.
- each family contains at least two members from the Watson strand and two from the Crick strand, with the actual number dependent on the depth of sequencing.
- Bona fide mutations represented by the asterisks within the Bona Fide Mutation family, are present in both parental strands of a DNA duplex and are therefore found in both Watson and Crick families.
- PCR or sequencing errors represented by asterisks within the PCR of Sequencing Error family, are limited to a subset of reads from one of the two strands.
- Watson strand-specific (asterisks within Damaged Watson Strand family) and Crick strand-specific (asterisks within the Damaged Crick Strand family) artifacts are found in all copies of the Watson or Crick family, but not in both.
- FIG. 24 High duplex recovery and efficient target enrichment with SaferSeqS. Thirty-three ng of admixed cfDNA samples were assayed for one of three different mutations in TP53 (p.L264fs, p.P190L, or p.R342X). Three libraries were prepared per cfDNA sample, each containing ⁇ 11 ng of cfDNA. (a) The median number of duplex families (i.e., both Watson and Crick strands containing the same endogenous and exogenous barcodes) was 89% (range: 65% to 102%) of the number of original template molecules (b) The median fraction of on-target reads was 80% (range: 72% to 91%). Lower and upper hinges correspond to the 25 th and 75 th percentile, whiskers extend to 1.5 times the interquartile range. Individual points are overlaid with random scatter for ease of visualization.
- Figure 25 contains graphs showing the detection of exemplary mutations in liquid biopsy samples. Analysis of 33 ng of plasma cell-free DNA from healthy individuals admixed with cell-free plasma DNA from a cancer patient. The mixtures were created to generate a high frequency (-0.5-1%) of mutation, low frequency (-0.01-0.1%) of mutation, or no mutation. The admixed TP53 p.R342X sample was assayed with (a) SafeSeqs or (b) SaferSeqS.
- TP53 p.L264fs sample was assayed with (c) SafeSeqs and (d) SaferSeqS
- the admixed TP53 p.P190L sample was assayed with (e) SafeSeqs and (f) SaferSeqS.
- Mutation numbers represent each of the 153 distinct mutations observed with SafeSeqS (defined in Table 8).
- the single supercalimutant detected by SaferSeqS (Table 9) was outside the genomic region assayed by SafeSeqS and is therefore not shown.
- Figure 26 Errors in SaferSeqS as compared to those of strand-agnostic, ligation- based molecular barcoding methods. Analysis of 33 ng of plasma cell-free DNA from healthy individuals admixed with cell-free plasma DNA from a cancer patient. The mixtures were created to generate a high frequency (-0.5-1%) of mutation, low frequency (-0.01-
- the admixed TP53 p.R342X sample was assayed with SaferSeqS but (a) strand information was ignored in the analysis to mimic strand-agnostic, ligation-based molecular barcoding methods or (b) strand information was considered during mutation calling.
- the admixed TP53 p.L264fs sample was assayed (c) without consideration of strand information and (d) with SaferSeqS.
- the admixed TP53 p.P190L sample was similarly assayed (e) without consideration of strand information and (f) with SaferSeqS. Mutation numbers are defined in Supplementary Table 3. The asterisks denote the admixed mutations. The single unexpected supercalimutant detected by SaferSeqS is shown in (e).
- FIG. 27 Evaluation of plasma samples from cancer patients.
- Plasma cell-free DNA samples from five cancer patients harboring eight known mutations at frequencies between 0.01% and 0.1% were assayed with a previously described, PCR-based molecular barcoding method (“SafeSeqS” rather than “SaferSeqS”) and with SaferSeqS. Mutation numbers are defined in Table 11. Asterisks denote the expected mutations. The single unexpected supercalimutant detected by SaferSeqS (Table 11) was outside the genomic region assayed by SafeSeqS and is therefore not shown.
- Figure 28 Effects of PCR efficiency and cycle number on duplex recovery.
- the probability of recovering both strands of the original DNA duplexes (y-axis) is plotted against library amplification cycle number (x-axis).
- Each pane in the figure represents the assumed PCR efficiency denoted at the top of the pane.
- the proportion of the library amplification product used in the strand-specific PCRs are shown.
- Library amplification cycle number was varied from one to 11.
- PCR efficiency was varied from 100% to 50% in 10% increments.
- the proportion of library amplification product using in each strand- specific PCRs was varied from 50% to 1.4%. Probabilistic modeling was performed as described in Example 2.
- Figure 29 contains graphs showing a multiplex panel for detection of exemplary cancer driver gene mutations. Recovery and coverage of the 36 amplicons that successfully amplified within the multiplex panel.
- the horizontal axis displays the position downstream of the 3’ end of the second gene-specific primer (GSP2). The gradual decline in coverage with increasing distance from the 3’ primer end is a consequence of the input DNA fragmentation pattern. Details regarding the theoretical recovery of reads with specific amplicon lengths are discussed in Example 2.
- Figure 30 Performance of the 48 primer pairs used in a multiplex panel to assay regions of driver genes commonly mutated in cancer.
- the proportion of on -target reads i.e. the fraction of total reads that map to the intended target
- Primers were used at equimolar concentrations in each gene-specific PCR.
- Figure 31 Performance of 62 primer pairs.
- the proportion of on-target reads i.e. the fraction of total reads that map to the intended target
- Fifty of the 62 pairs (81%) exhibit an on-target rate of greater than 50%.
- the results presented reflect a single attempt at primer design.
- Figure 32 depicts an exemplary computer system adapted to enable a user to analyze a nucleic acid sample according to a method described herein.
- Nucleotides and “nt” are used interchangeably herein to generally refer to biological molecules that comprise nucleic acids. Nucleotides can have moieties that contain the known purine and pyrimidine bases. Nucleotides may have other heterocyclic bases that have been modified. Such modifications include, e.g., methylated purines or pyrimidines, acylated purines or pyrimidines, alkylated riboses, or other heterocycles. The terms “polynucleotides,” “nucleic acid,” and “oligonucleotides” can be used interchangeably.
- Polynucleotides can refer to a polymeric form of nucleotides of any length, either deoxyribonucleotides or ribonucleotides, or analogs thereof.
- Polynucleotides may have any three-dimensional structure, and may perform any function, known or unknown.
- polynucleotides coding or non-coding regions of a gene or gene fragment, loci (locus) defined from linkage analysis, exons, introns, messenger RNA (mRNA), transfer RNA, ribosomal RNA, ribozymes, cDNA, recombinant polynucleotides, branched polynucleotides, plasmids, vectors, isolated DNA of any sequence, isolated RNA of any sequence, nucleic acid probes, and primers.
- a polynucleotide may comprise non-naturally occurring sequences.
- a polynucleotide may comprise modified nucleotides, such as methylated nucleotides and nucleotide analogs.
- modifications to the nucleotide structure may be imparted before or after assembly of the polymer.
- the sequence of nucleotides may be interrupted by non-nucleotide components.
- a polynucleotide may be further modified after polymerization, such as by conjugation with a labeling component.
- a “primer” is generally a polynucleotide molecule comprising a nucleotide sequence (e.g., an oligonucleotide), generally with a free 3'-OH group, that hybridizes with a template sequence (such as a target polynucleotide, or a primer extension product) and is capable of promoting polymerization of a polynucleotide complementary to the template.
- mammal as used herein includes both humans and non-humans and include but is not limited to humans, non-human primates, canines, felines, murines, bovines, equines, and porcines.
- the method comprises identifying the mutation when it is present on both Watson and Crick strands of a double stranded nucleic acid template. Such methods are particularly useful for distinguishing true mutations from artifacts stemming from, e.g., DNA damage, PCR, and other sequencing artifacts, allowing for the identification of mutations with high confidence.
- the methods and materials described herein can detect one or more mutations with a low error rate.
- the methods and materials described herein can be used to detect the presence or absence of a nucleic acid mutation in a nucleic acid template with an error rate of less than about 1% (e.g., less than about 0.1%, less than about 0.05%, or less than about 0.01%).
- the methods and materials described herein can be used to detect the presence or absence of a nucleic acid mutation in a nucleic acid template with an error rate of from about 0.001% to about 0.01%.
- the error rate associated with the identification of one or more mutations in analyte DNA fragments according to a method described herein is no more than lxlO 2 , no more than lxlO 3 , no more than lxlO 4 , no more than lxlO 5 , no more than lxlO 6 , no more than 5x1 O 6 , or no more than lxlO 7 .
- the error rate associated with the identification of one or more mutations in analyte DNA fragments according to a method described herein is reduced by at least 2-fold, 4-fold, 5-fold, 10-fold, 20-fold, 30-fold, 40-fold, 50-fold, 60-fold, 70-fold, 80- fold, 90-fold, or 100-fold, as compared to an alternative method of identifying mutations that does not require the mutation to be detected in both Watson and Crick strands of an analyte DNA fragment.
- the alternative method comprises standard molecular barcoding or standard PCR-based molecular barcoding followed by sequencing.
- the alternative method comprises: (a) attaching adapters to a population of double-stranded DNA fragments in an analyte DNA sample, wherein the adapters comprise a unique exogenous UID; (b) performing an initial amplification to amplify the adapter-ligated, double-stranded DNA fragments to produce amplicons; (c) determining sequence reads of one or more amplicons of the one or more of the adapter-ligated, double-stranded DNA fragments; (d) assigning the sequence reads into UID families, wherein each member of a UID family comprises the same exogenous UID sequence; (e) identifying a nucleotide sequence as accurately representing an analyte DNA fragment when a threshold percentage of members of a UID family contain the sequence; and (f) identifying a mutation in the analyte DNA fragment when the sequence identified as
- the methods and materials described herein can be used to achieve efficient duplex recovery.
- methods described herein can be used to recover PCR amplification products derived from both the Watson strand and the Crick strand of a double stranded nucleic acid template.
- the methods described herein can be used to achieve at least 50% (e.g. , about 50%, about 60%, about 70%, about 75%, about 80%, about 82%, about 85%, about 88%, about 90%, about 93%, about 95%, about 97%, about 99%, or 100%) duplex recovery.
- the methods and materials described herein can be used to detect a mutation having low allele frequency.
- methods described herein can be used to detect a mutation having low allele frequency of less than about 1% (e.g., less than about 0.1%, less than about 0.05%, or less than about 0.01%).
- the methods described herein can be used to detect a mutation having low allele frequency of about 0.001%.
- the methods described herein can be used to detect a mutation that is present in an analyte nucleic acid sample at a frequency of 0.1% or less. In some embodiments, the methods described herein can be used to detect a mutation that is present in an analyte nucleic acid sample at a frequency of 0.1% to 0.00001%. In some embodiments, the methods described herein can be used to detect a mutation that is present in an analyte nucleic acid sample at a frequency of 0.1% to 0.01%.
- the methods and materials described herein can be used to detect a mutation with minimal (or no) background artifact mutations. In some cases, the methods described herein can be used to detect a mutation with less than 0.01% background artifact mutations. In some cases, the methods described herein can be used to detect a mutation with no background artifact mutations.
- methods for detecting one or more mutations present on both strands of a double stranded nucleic acid can include generating a duplex sequencing library having a duplex molecular barcode on each end ( e.g ., the 5’ end and the 3’ end) of each nucleic acid in the library, generating a library of single stranded Watson strand-derived sequences and a library of single stranded Crick-strand derived sequences from the duplex sequencing library, and detecting the presence of one or more mutations present on both strands of the double stranded nucleic acid in each single stranded library.
- first molecular barcode in a 3’ duplex adapter and a second molecular barcode present in a 5’ adapter can be used to distinguish amplification products derived from the Watson strand from amplification products derived from the Crick strand.
- methods for identifying a mutation comprises: (a) attaching partially double-stranded 3’ adapters to 3’ ends of both Watson and Crick strands of a population of double-stranded DNA fragments in an analyte DNA sample, wherein a first strand of the partially double-stranded 3’ adapter comprises, in the 5’ -3’ direction, (i) a first segment, (ii) an exogenous UID sequence, (iii) an annealing site for a 5’ adapter, and (iv) a universal 3’ adapter sequence comprising an R2 sequencing primer site, and wherein the second strand of the partially double-stranded 3’ adapter comprises, in the 5’ to 3’ direction, (i) a segment complementary to the first segment, and (ii) a 3’ blocking group, optionally wherein the second strand is degradable; (b) annealing 5’ adapters to the 3’ adapters via the annealing site, wherein the 5’ adapters comprise
- methods for identifying a mutation comprises: (a) attaching adapters to a population of double-stranded DNA fragments, wherein the adapters comprise a double- stranded portion comprising an exogenous UID and a forked portion comprising (i) a single- stranded 3’ adapter sequence comprising an R2 sequencing primer site and (ii) a single- stranded 5’ adapter sequence comprising an R1 sequencing primer site; (b) performing an initial amplification to amplify the adapter-ligated, double-stranded DNA fragments to produce amplicons; (c) selectively amplifying amplicons of Watson strands comprising the target polynucleotide sequence with a first set of Watson target-selective primer pairs, the first set of Watson target-selective primer pairs comprising: (i) a first Watson target-selective primer comprising a sequence complementary to the R2 sequencing primer site of the universal 3’ adapter sequence, and (ii) a second
- the methods and materials described herein can be used to independently assess each strand of a double stranded nucleic acid. For example, when a nucleic acid mutation is identified in independently assessed strands of a double stranded nucleic acid as described herein, the materials and methods described herein can used to determine from which strand of the double stranded nucleic acid the nucleic acid mutation originated.
- a duplex sequencing library is a plurality of nucleic acid fragments including a duplex molecular barcode on at one end ( e.g ., the 5’ end and/or the 3’ end) of each nucleic acid fragment in the library and can allow both strands of a double stranded nucleic acid to be sequenced.
- a nucleic acid sample can be fragmented to generate nucleic acid fragments, and the generated nucleic acid fragments can be used to generate a duplex sequencing library.
- Nucleic acid fragments used to generate a duplex sequencing library can also be referred to herein as input nucleic acid.
- a duplex sequencing library can include any appropriate number of nucleic acid fragments.
- generating a duplex sequencing library can include fragmenting a nucleic acid template and ligating adapters to each end of each nucleic acid fragment in the library.
- Nucleic acid templates in an analyte nucleic acid sample can comprise any type of nucleic acid (e.g ., DNA, RNA, and DNA/RNA hybrids).
- a nucleic acid template can be a double-stranded DNA template.
- Examples of nucleic acid can be used as a template for the methods described herein include, without limitation, genomic DNA, circulating free DNA (cfDNA; e.g. , circulating tumor DNA (ctDNA), and cell-free fetal DNA (cffDNA)).
- the nucleic acid templates in the nucleic acid sample are nucleic acid fragments, e.g., DNA fragments.
- the ends of a DNA fragment represent unique sequences which can be used as an endogenous unique identifier of the fragment.
- the fragments are manually produced.
- the fragments are produced by shearing, e.g., enzymatic shearing, shearing by chemical means, acoustic shearing, nebulization, centrifugal shearing, point-sink shearing, needle shearing, sonication, restriction endonucleases, non-specific nucleases (e.g., DNase I), and the like.
- the fragments are not manually produced.
- the fragments are from a cfDNA sample.
- a nucleic acid fragment in the nucleic acid sample has a length.
- the length may be about 4-1000 nucleotides.
- the length may be about 60-300 nucleotides.
- the length may be about 60-200 nucleotides.
- length can be about 140- 170 nucleotides.
- the length may be less than 500, less than 400, less than 300, less than 250 nt, or less than 200 nt.
- ends of nucleic acid templates are used as endogenous UIDs.
- a skilled artisan may determine the length of the endogenous UID needed to uniquely identify a nucleic acid template, using factors such as, e.g., overall template length, complexity of nucleic acid templates in a partition or starting nucleic acid sample, and the like.
- 10-500 nucleotides of the ends of nucleic acid templates are used as endogenous UIDs.
- 15-100 nucleotides of the ends of nucleic acid templates are used as endogenous UIDs.
- 15-40 nucleotides of the ends of nucleic acid templates are used as endogenous UIDs.
- at least 10 nucleotides of the ends of nucleic acid templates are used as endogenous UIDs.
- at least 15 nucleotides of the ends of nucleic acid templates are used as endogenous UIDs.
- only one end of a nucleic acid template is used as an endogenous UID.
- nucleic acid templates comprise one or more target polynucleotides.
- target polynucleotide refers to a polynucleotide of interest under study.
- a target polynucleotide contains one or more sequences that are of interest and under study.
- a target polynucleotide can comprise, for example, a genomic sequence.
- the target polynucleotide can comprise a target sequence whose presence, amount, and/or nucleotide sequence, or changes in these, are desired to be determined.
- the target polynucleotide can be a region of gene associated with a disease.
- the gene is a druggable target.
- the term “druggable target”, as used herein, generally refers to a gene or cellular pathway that is modulated by a disease therapy.
- the disease can be cancer. Accordingly, the gene can be a known cancer-related gene.
- the input nucleic acid also referred to herein as the nucleic acid sample
- the biological sample may be obtained from a subject.
- the subject is a mammal.
- mammals from which nucleic acid can be obtained and used as a nucleic acid template in the methods described herein include, without limitation, humans, non-human primates (e.g ., monkeys), dogs, cats, sheep, rabbits, mice, hamsters, and rats.
- the subject is a human subject.
- the subject is a plant.
- Biological samples include but are not limited to plasma, serum, blood, tissue, tumor sample, stool, sputum, saliva, urine, sweat, tears, ascites, bronchoaveolar lavage, semen, archeologic specimens and forensic samples.
- the biological sample is a solid biological sample, e.g., a tumor sample.
- the solid biological sample is processed.
- the solid biological sample may be processed by fixation in a formalin solution, followed by embedding in paraffin (e.g., is a FFPE sample). Processing can alternatively comprise freezing of the sample prior to conducting the probe-based assay.
- the sample is neither fixed nor frozen.
- the unfixed, unfrozen sample can be, by way of example only, stored in a storage solution configured for the preservation of nucleic acid.
- the biological sample is a liquid biological sample.
- Liquid biological samples include, but are not limited to plasma, serum, blood, sputum, saliva, urine, sweat, tears, ascites, bronchoaveolar lavage, and semen.
- the liquid biological sample is cell free or substantially cell free.
- the biological sample is a plasma or serum sample.
- the liquid biological sample is a whole blood sample.
- the liquid biological sample comprises peripheral mononuclear blood cells.
- a nucleic acid sample has been isolated and purified from the biological sample.
- Nucleic acid can be isolated and purified from the biological sample using any means known in the art.
- a biological sample may be processed to release nucleic acid from cells, or to separate nucleic acids from unwanted components of the biological sample (e.g., proteins, cell walls, other contaminants).
- nucleic acid can be extracted from the biological sample using liquid extraction (e.g., Trizol, DNAzol) techniques.
- Nucleic acid can also be extracted using commercially available kits (e.g.,
- the biological sample comprises low amounts of nucleic acid. In some embodiments, the biological sample comprises less than about 500 nanograms (ng) of nucleic acid. For example, the biological sample comprises from about 30 ng to about 40 ng of nucleic acid.
- Nucleic acid can be concentrated by known methods, including, by way of example only, centrifugation. Nucleic acid can be bound to a selective membrane (e.g., silica) for the purposes of purification. Nucleic acid can also be enriched for fragments of a desired length, e.g., fragments which are less than 1000, 500, 400, 300, 200 or 100 base pairs in length. Such an enrichment based on size can be performed using, e.g., PEG-induced precipitation, an electrophoretic gel or chromatography material (Huber et al. (1993) Nucleic Acids Res. 21:1061-6), gel filtration chromatography, TSK gel (Kato et al. (1984) J. Biochem, 95:83- 86), which publications are hereby incorporated by reference.
- PEG-induced precipitation an electrophoretic gel or chromatography material
- TSK gel Keratibility et al. (1984) J. Biochem, 95:83- 86
- Polynucleotides extracted from a biological sample can be selectively precipitated or concentrated using any methods known in the art.
- the nucleic acid sample comprises less than about 35 ng of nucleic acid.
- the nucleic acid sample comprises can include from about 1 ng to about 35 ng of nucleic acid (e.g., from about 1 ng to about 30 ng, from about 1 ng to about 25 ng, from about 1 ng to about 20 ng, from about 1 ng to about 15 ng, from about 1 ng to about
- a nucleic acid sample can include nucleic acid from a genome that includes more than about several hundred nucleotides of nucleic acid.
- a nucleic acid sample can be essentially free of contamination.
- the cfDNAcan be essentially free of genomic DNA contamination.
- a cfDNA sample that is essentially free of genomic DNA contamination can include minimal (or no) high molecular weight (e.g ., > 1000 bp) DNA.
- methods described herein can include determining whether a nucleic acid sample is essentially free of contamination. Any appropriate method can be used to determine whether a nucleic acid sample is essentially free of contamination.
- Examples of methods that can be used to determine whether a nucleic acid sample is essentially free of contamination include, for example, a TapeStation system, and a Bioanalyzer.
- a TapeStation system and/or a Bioanalyzer to determine whether a cfDNA sample is essentially free of genomic DNA contamination
- a prominent peak at -180 bp can be used to indicate that the nucleic acid sample is essentially free of genomic DNA contamination.
- nucleic acid fragments that can be used to generate a duplex sequencing library can be end-repaired.
- Any appropriate method can be used to end-repair a nucleic acid template.
- blunting reactions e.g, blunt end ligations
- dephosphorylation reactions can be used to end-repair a nucleic acid template.
- blunting can include filling in a single stranded region.
- blunting can include degrading a single stranded region.
- blunting and dephosphorylation reactions can be used to end-repair a nucleic acid template as shown in Figure 9 and/or Figure 10.
- the method comprises attaching adapters to a population of double-stranded DNA fragments to produce a population of adapter-attached, double- stranded DNA fragments.
- the adapters comprise a double-stranded portion comprising an exogenous UID and a forked portion comprising (i) a single-stranded 3’ adapter sequence and (ii) a single-stranded 5’ adapter sequence.
- the single-stranded 3’ adapter sequence is not complementary to the single-stranded 5’ adapter sequence.
- the 3’ adapter sequence comprises a second (e.g., R2) sequencing primer site and the 5’ adapter sequence comprises a first (e.g., Rl) sequencing primer site.
- R2 and R2 sequencing primer sites are used by sequencing systems that produce paired end reads, e.g., reads from opposite ends of a DNA fragment to be sequenced.
- the Rl sequencing primer is used to produce a first population of reads from first ends of DNA fragments
- the R2 sequencing primer is used to produce a second population of reads from the opposite ends of the DNA fragments.
- the first population is referred to herein as “Rl” or “Read 1” reads.
- the second population is referred to herein as “R2” or “Read 2” reads.
- the Rl and R2 reads can be aligned as “read pairs” or “mate pairs” corresponding to each strand of a double-stranded analyte DNA fragment.
- Certain sequencing systems utilizes what they refer to as “Rl” and “R2” primers, and “Rl” and “R2” reads.
- Rl and R2 and “Read 1” and “Read 2”, for the purposes of this application, are not limited to how they are referenced in relation to a particular sequencing platform.
- the “R2” primer and corresponding R2 read disclosed herein may refer to the Illumina “R2” primer and read, or may refer to the Illumina “Rl” primer and read, so long as the “Rl” primer and corresponding Rl read disclosed herein refers to the other Illumina primer and read.
- an “R2” primer provided herein is the Illumina “Rl” primer producing “Rl” reads
- the corresponding “Rl” primer provided herein is the Illumina “R2” primer producing “R2” reads.
- an “R2” primer provided herein is the Illumina “R2” primer providing “R2” reads
- the “Rl” primer provided herein is the Illumina “Rl” primer providing Rl reads.
- the exogenous UID is unique to each double-stranded DNA fragment in the nucleic acid sample. In some embodiments, the exogenous UID is not unique to each double-stranded DNA fragment.
- the exogenous UID has a length.
- the length can be about 2-
- the length can be about 6-100 nt.
- the length can be about 8-50 nt.
- the length can be about 10-20 nt.
- the length can be about 12-14 nt.
- the length of the exogenous UID is sufficient to uniquely barcode the molecules and the length/sequence of the exogenous UID does not interfere with the downstream amplification steps.
- the exogenous UID sequence does not exist in the nucleic acid template. In some embodiments, the exogenous UID sequence does not exist in a desired template harboring a desired locus. Such unique sequences can be randomly generated, e.g., by a computer readable medium, and selected by BLASTing against known nucleotide databases such as, e.g., EMBL, GenBank, or DDBJ. In some embodiments, an exogenous UID sequence exists in a nucleic acid template. In such cases, the position of the exogenous UID sequence in the sequence read is used to distinguish the exogenous UID sequence from a sequence within the nucleic acid template.
- the exogenous UID sequence is random. In some embodiments, the exogenous UID sequence is a random N-mer. For example, if the exogenous UID sequence has a length of six nt, then it may be a random hexamer. If the exogenous UID sequence has a length of 12 nt, then it may be a random 12-mer.
- Exogenous UIDs may be made using random addition of nucleotides to form a sequence having a length to be used as an identifier. At each position of addition, a selection from one of four deoxyribonucleotides may be used. Alternatively a selection from one of three, two, or one deoxyribonucleotides may be used. Thus the UID may be fully random, somewhat random, or non-random in certain positions.
- the exogenous UIDs are not random N-mers, but are selected from a predetermined set of exogenous UID sequences.
- Forked adapters described herein may be attached to double-stranded DNA fragments by any means known in the art.
- the forked adapters are attached to double-stranded DNA fragments by: (a) attaching partially double-stranded 3’ adapters to 3’ ends of both Watson and Crick strands of a population of double-stranded DNA fragments, wherein a first strand of the partially double-stranded 3’ adapter comprises, in the 5’ -3’ direction, (i) a first segment, (ii) an exogenous UID sequence, (iii) an annealing site for a 5’ adapter, and (iv) a universal 3’ adapter sequence comprising an R2 sequencing primer site, and wherein the second strand of the partially double-stranded 3’ adapter comprises, in the 5’ to 3’ direction,
- the forked adapters are attached to double-stranded DNA fragments by: (a) attaching a 3’ duplex adapter to 3’ ends of both Watson and Crick strands of a population of double-stranded DNA fragments.
- A3’ duplex adapter also referred to herein as a partially double stranded 3’ adapter, as described herein is an oligonucleotide complex including a molecular barcode that can have a first oligonucleotide (also referred to herein as “first strand”) annealed (hybridized) to a second oligonucleotide (also referred to herein as “second strand”) such that a portion (e.g., first portion) of the 3’ duplex adapter is double stranded and a portion (e.g, a second portion) of the 3’ duplex adapter is single stranded.
- first oligonucleotide also referred to herein as “first strand”
- a first oligonucleotide of a 3’ duplex adapter described herein comprises a first segment comprising nucleotides that are complementary to nucleotides present in a second oligonucleotide of the 3’ duplex adapter (e.g, such that the first oligonucleotide of the 3’ duplex adapter and the second oligonucleotide of the 3’ duplex adapter can anneal at the complementary region).
- An exemplary structure of a 3’ duplex adapter can be as shown in Figure 11.
- the first oligonucleotide of a 3’ duplex adapter described herein can be an oligonucleotide that includes a 5’ phosphate and a molecular barcode.
- the first oligonucleotide of a 3’ duplex adapter described herein can include any appropriate number of nucleotides.
- Any appropriate molecular barcode can be included in a first oligonucleotide of a 3’ duplex adapter described herein.
- a molecular barcode can include a random sequence.
- a molecular barcode can include a fixed sequence.
- oligonucleotides that include a 5’ phosphate and a molecular barcode and can be included in a first oligonucleotide of a 3’ duplex adapter described herein include, without limitation,
- the first oligonucleotide of the 3’ duplex adapter comprises an annealing site for a 5’ adapter.
- the first oligonucleotide of the 3’ duplex adapter comprises a universal 3’ adapter sequence.
- the universal 3’ adapter sequence comprises an R2 sequencing primer site.
- a first oligonucleotide of a 3’ duplex adapter described herein also can include one or more features to prevent or reduce extension during a PCR.
- a feature that can prevent or reduce extension during a PCR can be any type of feature ( e.g ., a chemical modification). Examples of feature that can prevent or reduce extension during a PCR and can be included in a first oligonucleotide of a 3’ duplex adapter described herein include, without limitation, 3SpC3 and 3Phos.
- a feature that can prevent or reduce extension during a PCR can be incorporated into a first oligonucleotide of a 3’ duplex adapter described herein in any appropriate position within the oligonucleotide.
- a molecule that can prevent or reduce extension during a PCR can be incorporated internally within the oligonucleotide. In some case, a molecule to prevent or reduce extension during a PCR can be incorporated at and end (e.g., the 5’ end) of the oligonucleotide.
- the first oligonucleotide of the 3’ duplex adapter comprises a 5’ phosphate, a first segment comprising nucleotides that are complementary to nucleotides present in a second oligonucleotide of the 3’ duplex adapter, an exogenous UID sequence, an annealing site for a 5’ adapter, and a universal 3’ adapter sequence.
- the second oligonucleotide of a 3’ duplex adapter described herein can be an oligonucleotide that includes a blocked 3’ group (e.g., to reduce or eliminate dimerization of two adapters).
- the second oligonucleotide of a 3’ duplex adapter described herein can include any appropriate number of nucleotides.
- the second oligonucleotide of the 3’ duplex adapter is complementary to the first segment of the first oligonucleotide of the 3’ duplex adapter.
- An exemplary oligonucleotide that includes a blocked 3’ group and can be included in a second oligonucleotide of a 3’ duplex adapter described herein includes, without limitation, GCCGUCGUUUUAdT (SEQ ID NO:3).
- the second oligonucleotide of a 3’ duplex adapter described herein can be degradable. Any appropriate method can be used to degrade a second oligonucleotide of a 3’ duplex adapter described herein.
- UDG can be used to degrade a second oligonucleotide of a 3’ duplex adapter described herein.
- a 3’ duplex adapter described herein can include a first oligonucleotide including the sequence
- ATAAAACGACGGCNNNNNNNNNNNNNNNNAGATCGGA AGAGC AC ACGTCTGAACT CCAG*T*C/3SpC3 (SEQ ID NO:l) annealed to a second oligonucleotide including the sequence GCCGUCGUUUUAdT (SEQ ID NO:3).
- a 3’ duplex adapter described herein can include a commercially available adapter.
- An exemplary commercially available adapters that can be used as (or can be used to generate) a 3’ duplex adapter described herein includes, without limitation, adapters in an Accel-NGS 2S DNA Library Kit (Swift Biosciences, cat. # 21024).
- a 3’ duplex adapter described herein can be as described in Example 1.
- the 3’ adapters can be attached ( e.g ., covalently attached) to 3’ ends of the double- stranded DNA fragments using any appropriate method.
- the 3’ adapters are attached by ligation.
- the ligation comprises use of a ligase.
- ligases that can be used to attach a 3’ adapter to the 3’ ends of each nucleic acid fragment include, without limitation, T4 DNA ligases, E. coli ligases (e.g., Enzyme Y3), CircLigase I, CircLigase II, Taq-Ligase, T3 Ligase, T7 Ligase, and 9N Ligase.
- the second oligonucleotide of a 3’ duplex adapter described herein can be degraded, and a 5’ adapter can be attached (e.g, covalently attached) to the 5’ ends of each nucleic acid fragment.
- the 5’ adapter sequence is not complementary to the first oligonucleotide of the 3’ adapter.
- the 5’ adapter sequence comprises, in the 5’ to 3’ direction, an R1 sequencing primer site and a sequence complementary to the annealing site of the 3’ adapter.
- the attaching of the 5’ adapter comprises annealing the 5’ adapter to the 3’ adapter via the annealing site.
- a 5’ adapter can anneal to a nucleic acid fragment upstream of a molecular barcode on a 3’ duplex adapter such that a gap (e.g, single stranded nucleic acid fragment) containing a portion ( e.g ., a molecular barcode) of the 3’ duplex adapter is present on the nucleic acid fragment.
- the gap containing a portion of the 3’ duplex adapter can be filled in (e.g., to generate a double stranded nucleic acid fragment). Any appropriate method can be used to fill in the single stranded gap.
- Examples of methods that can be used to fill in a single stranded gap on a nucleic acid fragment include, without limitation, polymerases such as DNA polymerases (e.g, Taq polymerases such as a Taq-B polymerase) and nick-translation reactions (e.g, including both a ligase such as an E. coli ligase and a polymerase such as a DNA polymerase).
- the method also can include providing deoxyribonucleotide triphosphates (dNTPs; e.g, dATP, dGTP, dCTP, and dTTP).
- dNTPs deoxyribonucleotide triphosphates
- attaching a 5’ adapter to the 5’ ends of each nucleic acid fragment and filling in the single stranded gap can be done concurrently (e.g, in a single reaction tube).
- nucleic acid fragments can be treated with single strand nucleases (e.g, to digest overhangs) followed by ligation can be used to prepare a duplex sequencing library.
- single nucleotide can be added to the 3’ ends of each nucleic acid fragment and adapters (e.g, containing a molecular barcode) containing a complementary base at the 5’ end can be ligated to each nucleic acid fragment to prepare a duplex sequencing library of adapter-attached templates.
- the adapter-attached templates can be amplified (e.g, PCR amplified) in an initial amplification reaction. Any appropriate method can be used to amplify the adapter-attached templates.
- An exemplary method that can be used to amplify the adapter-attached templates includes, without limitation, whole-genome PCR.
- Any appropriate primer pair can be used to amplify the adapter-attached templates.
- a universal primer pair can be used.
- a primer can include, without limitation from about 12 nucleotides to about 30 nucleotides.
- primer pairs that can be used to amplify the adapter-attached templates as described herein include, without limitation, those described in Example 1 and/or Example 2.
- PCR amplification can include a denaturing phase, an annealing phase, and an extension phase.
- Each phase of an amplification cycle can include any appropriate conditions.
- a denaturing phase can include a temperature of about 90°C to about 105°C (e.g., about 94°C to about 98°C), and a time of about 1 second to about 5 minutes (e.g., about 10 seconds to about 1 minute).
- a denaturing phase can include a temperature of about 98°C for about 10 seconds.
- an annealing phase can include a temperature of about 50°C to about 72°C, and a time of about 30 seconds to about 90 seconds.
- an extension phase can include a temperature of about 55°C to about 80°C, and a time of about 15 seconds per kb of the amplicon to be generated to about 30 seconds per kb of the amplicon to be generated.
- annealing and extension phases can be performed in a single cycle.
- an annealing and phase extension phase can include a temperature of about 65°C for about 75 seconds.
- PCR conditions used in the initial amplification can include any appropriate number of PCR amplification cycles. In some cases, PCR amplification can include from about 1 to about 50 cycles. In some embodiments, the PCR amplification comprises no more than 11 cycles. In some embodiments, the PCR amplification comprises no more than 7 cycles. In some embodiments, the PCR amplification comprises no more than 5 cycles.
- PCR amplification when PCR conditions include a heat-activated polymerase, PCR amplification also can include an initialization step.
- PCR amplification can include an initialization step prior to performing the PCR amplification cycles.
- an initialization step can include a temperature of about 94°C to about 98°C, and a time of about 15 seconds to about 1 minute.
- an initialization step can include a temperature of about 98°C for about 30 seconds.
- PCR amplification also can include a hold step.
- PCR amplification can include a hold step after performing the PCR amplification cycles, an optionally after performing any final extension step.
- a hold step can include a temperature of about 4°C to about 15°C, for an indefinite amount of time.
- a duplex sequencing library generated as described herein can be purified.
- Any appropriate method can be used to purify a duplex sequencing library.
- An exemplary method that can be used to purify a duplex sequencing library includes, without limitation, magnetic beads (e.g, solid phase reversible immobilization (SPRI) magnetic beads).
- SPRI solid phase reversible immobilization
- a duplex sequencing library can be used to generate a library of single stranded Watson strand-derived sequences and a library of single stranded Crick-strand derived sequences. Generating a library of single stranded Watson strand-derived sequences and a library of single stranded Crick-strand derived sequences can minimize non-specific amplification (e.g ., from a primer complementary to a ligated sequence such as a 3’ duplex adapter or a 5’ adapter).
- a library of single stranded Watson strand-derived sequences and a library of single stranded Crick-strand derived sequences can be generated from an amplified duplex sequencing library by dividing the amplification products into at least two aliquots, and subjecting each aliquot to a PCR amplification where the Watson strand is amplified from a first aliquot, and the Crick strand is amplified from a second aliquot.
- a first aliquot of amplification products from an amplified duplex sequencing library can be subjected to a PCR amplification using a primer pair where a first primer is biotinylated and a second primer is non-biotinylated to generate a single stranded library of Watson strands
- a second aliquot of amplification products from an amplified duplex sequencing library can be subjected to a PCR amplification using a primer pair where a first primer is non- biotinylated and a second primer is biotinylated to generate a single stranded library of Crick strands.
- a library of single stranded Watson strand-derived sequences and a library of single stranded Crick-strand derived sequences can be generated as shown in Figure 2 and Figure 3.
- amplification products from an amplified duplex sequencing library can be separated into a first PCR amplification and a second PCR amplification in which only one of the two primers in the PCR primer pair is tagged.
- a first PCR amplification can use a primer pair that includes a primer (e.g, a first primer) that is tagged and a primer (e.g, a second primer) that is not tagged
- a second PCR amplification can use a primer pair that includes a primer (e.g, a first primer) that is not tagged and a primer (e.g, a second primer) that is tagged.
- a primer tag can be any tag that enables a PCR amplification product generated from the tagged primer to be recovered.
- a tagged primer can be a biotinylated primer, and a PCR amplification produce generated from the biotinylated primer can be recovered using streptavidin.
- a library of single stranded Watson strand-derived sequences and a library of single stranded Crick-strand derived sequences can be generated in a PCR amplification using a primer pair including a biotinylated primer and a non-biotinylated primer.
- a tagged primer can be a phosphorylated primer, and a PCR amplification produce generated from the phosphorylated primer can be recovered using a lambda nuclease.
- a library of single stranded Watson strand-derived sequences and a library of single stranded Crick-strand derived sequences can be generated in a PCR amplification using a primer pair including a phosphorylated primer and a non- phosphorylated primer.
- a primer can include, without limitation, from about 12 nucleotides to about 30 nucleotides.
- a primer pair can include at least one primer that can target (e.g., target and bind to) an adapter sequence (e.g, an adapter sequence containing a molecular barcode) present in an amplification product generated as described herein (e.g, by ligating a 3’ duplex adapter including a first molecular barcode and a 5’ adapter including a second molecular barcode to a nucleic acid fragment in a duplex sequencing library prior to the amplification).
- an adapter sequence e.g, an adapter sequence containing a molecular barcode
- primer pairs that can be used to generate a library of single stranded Watson strand-derived sequences and a library of single stranded Crick-strand derived sequences as described herein include, without limitation, a P5 primer and a P7 primer.
- Any appropriate PCR conditions can be used to generate a library of single stranded
- PCR amplification can include a denaturing phase, an annealing phase, and an extension phase.
- Each phase of an amplification cycle can include any appropriate conditions.
- a denaturing phase can include a temperature of about 90°C to about 105°C, and a time of about 1 second to about 5 minutes.
- a denaturing phase can include a temperature of about 98°C for about 10 seconds.
- an annealing phase can include a temperature of about 50°C to about 72°C, and a time of about 30 seconds to about 90 seconds.
- an extension phase can include a temperature of about 55°C to about 80°C, and a time of about 15 seconds per kb of the amplicon to be generated to about 30 seconds per kb of the amplicon to be generated
- an extension phase reflects the processivity of the polymerase that is used.
- annealing and extension phases can be performed in a single cycle.
- an annealing and phase extension phase can include a temperature of about 65°C for about 75 seconds.
- PCR conditions used to generate a library of single stranded Watson strand-derived sequences and a library of single stranded Crick-strand derived sequences can include any appropriate number of PCR amplification cycles.
- PCR amplification can include, without limitation, from about 1 to about 50 cycles.
- PCR amplification can include about 4 amplification cycles.
- PCR amplification when PCR conditions include a heat-activated polymerase, PCR amplification also can include an initialization step.
- PCR amplification can include an initialization step prior to performing the PCR amplification cycles.
- an initialization step can include a temperature of about 94°C to about 98°C, and a time of about 15 seconds to about 1 minute.
- an initialization step can include a temperature of about 98°C for about 30 seconds.
- PCR amplification also can include a hold step.
- PCR amplification can include a hold step after performing the PCR amplification cycles, an optionally after performing any final extension step.
- a hold step can include a temperature of about 4°C to about 15°C, for an indefinite amount of time.
- a double stranded amplification products can be denatured to separate double stranded amplification products into two single stranded amplification products.
- methods that can be used to separate a double stranded amplification product into single stranded amplification products include, without limitation, heat denaturation, chemical (e.g., NaOH) denaturation, and salt denaturation.
- the tagged PCR amplification products can be recovered. Any appropriate method can be used to recover tagged PCR amplification products generated using a tagged primer.
- a tagged primer is a biotinylated primer
- the biotinylated amplification products e.g, generated from the biotinylated primer
- streptavidin e.g ., streptavi din-functionalized beads
- an amplified duplex sequencing library is further amplified in a first PCR amplification using a primer pair that includes a first biotinylated primer and a second non-biotinylated primer, and a second PCR amplification using a primer pair that includes a first non- biotinylated primer and a second biotinylated primer
- the biotinylated amplification products generated from the first PCR amplification can be bound to streptavidin-functionalized beads (e.g., a first set of streptavidin-functionalized beads) and the biotinylated amplification products generated from the second PCR amplification can be bound to streptavidin- functionalized beads (e.g, a first second of streptavidin-functionalized beads), and the double stranded amplification products can be separated (e.g, denatured) into single strands of the amplification products.
- streptavidin-functionalized beads e.g., a first set of
- recovering biotinylated PCR amplification products also can include releasing the biotinylated PCR amplification products from the streptavidin (e.g, the streptavidin-functionalized beads).
- the streptavidin e.g, the streptavidin-functionalized beads.
- Separating the double stranded amplification products generated by a first PCR amplification using a primer pair that includes a first biotinylated primer and a second non-biotinylated primer, and a second PCR amplification using a primer pair that includes a first non-biotinylated primer and a second biotinylated primer can allow single stranded amplification products generated from the biotinylated primers to remain bound to the streptavidin-functionalized beads while single stranded amplification products generated from the non-biotinylated primers can be denatured (e.g, denatured and degraded) from the streptavidin-functionalized beads
- the phosphorylated amplification products (e.g, generated from the phosphorylated primer) can be recovered using an exonuclease (e.g, a lambda exonuclease).
- an exonuclease e.g, a lambda exonuclease
- the double stranded amplification products can be separated into single strands of the amplification products. Separating the double stranded amplification products generated by a first PCR amplification using a primer pair that includes a first phosphorylated primer and a second non-phosphorylated primer, and a second exonuclease.
- PCR amplification using a primer pair that includes a first non-phosphorylated primer and a second phosphorylated primer can allow single stranded amplification products generated from the non-phosphorylated primers to be recovered while single stranded amplification products generated from the phosphorylated primers can be degraded by a lambda exonuclease, thereby generating a library of single stranded Watson strand-derived sequences and a library of single stranded Crick-strand derived sequences of the duplex sequencing library.
- amplicons produced by the initial amplification are enriched for one or more target polynucleotides.
- single-stranded DNA libraries are prepared from amplicons produced by the initial amplification. Exemplary methods for producing the single-stranded DNA libraries are described herein.
- Any appropriate method can be used to amplify a target region from a library of amplification products (e.g. , a duplex sequencing library, a library of single stranded Watson strand-derived sequences, or a library of single stranded Crick-strand derived sequences generated as described herein).
- a library of amplification products e.g. , a duplex sequencing library, a library of single stranded Watson strand-derived sequences, or a library of single stranded Crick-strand derived sequences generated as described herein).
- a target region can be amplified from library of amplification products by subjecting the library of amplification products to a PCR amplification using a primer pair where a primer (e.g, a first primer) that can target (e.g, target and bind to) an adapter sequence (e.g, an adapter sequence containing a molecular barcode) present in an amplification product generated as described herein (e.g, by ligating a 3’ duplex adapter including a first molecular barcode and a 5’ adapter including a second molecular barcode to a nucleic acid fragment in a duplex sequencing library prior to the amplification) and a primer (e.g, a second primer) that can target (e.g, target and bind to) a target region (e.g, a region of interest).
- a primer e.g, a first primer
- an adapter sequence e.g, an adapter sequence containing a molecular barcode
- a primer e
- a library of single stranded Watson strand-derived sequences and a library of single stranded Crick-strand derived sequences can be generated as shown in Figure 4 and Figure 5.
- a library of single stranded Watson strand-derived sequences and a library of single stranded Crick-strand derived sequences can be generated as described in Example 2.
- a target region can be amplified from a library of amplification products (e.g, a duplex sequencing library, a library of single stranded Watson strand-derived sequences, or a library of single stranded Crick-strand derived sequences generated as described herein) in a single PCR amplification.
- a target region can be amplified from a library of amplification products in a single PCR amplification using a primer pair including a first primer that can target an adapter sequence (e.g .
- an adapter sequence containing a molecular barcode present in an amplification product generated as described herein (e.g., by ligating a 3’ duplex adapter including a first molecular barcode and a 5’ adapter including a second molecular barcode to a nucleic acid fragment in a duplex sequencing library prior to the amplification) and a second primer that can target a target region.
- a target region can be amplified from a library of amplification products in a single PCR amplification as shown in Figure 4, Figure 5, Figure 15, and Figure 17.
- a target region can be amplified from a library of amplification products (e.g, a duplex sequencing library, a library of single stranded Watson strand-derived sequences, or a library of single stranded Crick-strand derived sequences generated as described herein) in multiple PCR amplifications.
- Multiple PCR amplifications e.g, a first PCR amplification and a subsequent, nested PCR amplification
- multiple PCR amplifications can be used to increase the specificity of amplifying a target region.
- a target region can be amplified from a library of amplification products in a series of PCR amplifications where a first PCR amplification uses a primer pair including a first primer that can target an adapter sequence (e.g, an adapter sequence containing a molecular barcode) present in an amplification product generated as described herein (e.g, by ligating a 3’ duplex adapter including a first molecular barcode and a 5’ adapter including a second molecular barcode to a nucleic acid fragment in a duplex sequencing library prior to the amplification) and a second primer that can target a target region, and subjecting the amplification products generated in the first PCR amplification to a subsequent, nested PCR amplification that uses a primer pair including a first primer that can target an adapter sequence (e.g.
- an adapter sequence containing a molecular barcode) present in an amplification product generated as described herein e.g, by ligating a 3’ duplex adapter including a first molecular barcode and a 5’ adapter including a second molecular barcode to a nucleic acid fragment in a duplex sequencing library prior to the amplification
- a second primer that can target a nucleic acid sequence from the target region that is present in the amplification products generated in the first PCR amplification.
- a target region can be amplified from a library of amplification products in a series of PCR amplifications as shown in Figure 7, Figure 8, Figure 16, and Figure 18.
- any appropriate primer pair can be used to amplify a target region from a library of amplification products (e.g ., a duplex sequencing library, a library of single stranded Watson strand-derived sequences, or a library of single stranded Crick-strand derived sequences generated as described herein).
- a primer can include, without limitation, from about 12 nucleotides to about 30 nucleotides.
- a primer pair can include a primer (e.g., a first primer) that can target (e.g. , target and bind to) an adapter sequence (e.g.
- an adapter sequence containing a molecular barcode present in an amplification product generated as described herein (e.g, by ligating a 3’ duplex adapter including a first molecular barcode and a 5’ adapter including a second molecular barcode to a nucleic acid fragment in a duplex sequencing library prior to the amplification) and a primer (e.g, a second primer) that can target (e.g, target and bind to) a target region (e.g, a region of interest).
- a primer e.g, a second primer
- primers that can target an adapter sequence containing a molecular barcode present in an amplification product generated as described herein include, without limitation, an i5 index primer and an i7 index primer.
- Primers that can target a target region can include a sequence that is complementary to the target region.
- examples of primers that can target nucleic acid encoding TP53 include, without limitation, TP53 342 GSP1 and TP53 GSP2.
- a primer targeting the nucleic acid encoding TP53 can be as described in Example 2.
- one or both primers of a primer pair used to amplify a target region from a library of amplification products can include one or more molecular barcodes.
- one or both primers of a primer pair used to amplify a target region from a library of amplification products can include one or more graft sequences (e.g. graft sequences for next generation sequencing).
- the target enrichment comprises (a) selectively amplifying amplicons of
- Watson strands comprising the target polynucleotide sequence with a first set of Watson target- selective primer pairs
- the first set of Watson target- selective primer pairs comprising: (i) a first Watson target-selective primer comprising a sequence complementary to the R2 sequencing primer site of the universal 3’ adapter sequence, and (ii) a second Watson target- selective primer comprising a target- selective sequence, thereby creating target Watson amplification products; and (b) selectively amplifying amplicons of Crick strands comprising the same target polynucleotide sequence with a first set of Crick target-selective primer pairs, the first set of Crick target-selective primer pairs comprising: (i) a first Crick target-selective primer comprising a sequence complementary to the R1 sequencing primer site of the universal 5’ adapter sequence, and (ii) a second Crick target- selective primer comprising the same target- selective sequence as the second Watson target- selective primer sequence, thereby creating target Crick amplification
- the method further comprises purifying the target Watson amplification products and the target Crick amplification products from non-target polynucleotides.
- the purifying comprises attaching the target Watson amplification products and the target Crick amplification products to a solid support.
- the first Watson target-selective primer and first Crick target-selective primer comprises a first member of an affinity binding pair, and wherein the solid support comprises a second member of the affinity binding pair.
- the first member is biotin and the second member is streptavidin.
- the solid support comprises a bead, well, membrane, tube, column, plate, sepharose, magnetic bead, or chip.
- the method comprises removing polynucleotides that are not attached to the solid support.
- the method further comprises (a) further amplifying the target
- Watson amplification products with a second set of Watson target- selective primers comprising (i) a third Watson target-selective primer comprising a sequence complementary to the R2 sequencing primer site of the universal 3’ adapter sequence, and (ii) a fourth Watson target- selective primer comprising, in the 5’ to 3’ direction, an R1 sequencing primer site and a target-selective sequence selective for the same target polynucleotide, thereby creating target Watson library members; (b) further amplifying the target Crick amplification products with a second set of Crick target- selective primers, the second set of Crick target- selective primers comprising (i) a third Crick target- selective primer comprising a sequence complementary to the R1 sequencing primer site of the universal 3’ adapter sequence, and (ii) a fourth Crick target- selective primer comprising, in the 5’ to 3’ direction, an R2 sequencing primer site and the target-selective sequence selective for the same target polyn
- the third Watson and Crick target- selective primers further comprise a sample barcode sequence.
- the third Watson target- selective primer further comprises a first grafting sequence that enables hybridization to a first grafting primer on a sequencer and wherein the third Crick target-selective primer further comprises a second grafting sequence that enables hybridization to a second grafting primer on the sequencer.
- the fourth Watson target- selective primer further comprises the second grafting sequence and wherein the fourth Crick target-selective primer further comprises the first grafting sequence.
- the first grafting sequence is a P7 sequence and wherein the second grafting sequence is a P5 sequence.
- PCR conditions can be used to generate an amplified target region as described herein (e.g ., from a library of amplification products such as a duplex sequencing library, a library of single stranded Watson strand-derived sequences, or a library of single stranded Crick-strand derived sequences generated).
- exemplary PCR conditions are described herein.
- PCR conditions used to generate an amplified target region as described herein can include any appropriate number of PCR amplification cycles.
- PCR amplification can include, without limitation, from about 1 to about 50 cycles.
- the PCR amplification can include about 18 amplification cycles.
- the first PCR amplification can include about 18 amplification cycles
- the subsequent, nested PCR amplification can include about 10 amplification cycles.
- Any appropriate target region (e.g, a region of interest) can be amplified from a library of amplification products (e.g, a duplex sequencing library, a library of single stranded Watson strand-derived sequences, or a library of single stranded Crick-strand derived sequences generated as described herein) and assessed for the presence or absence of one or more mutations.
- a target region can be a region of nucleic acid in which one or more mutations are associated with a disease or disorder.
- target regions that can be amplified and assessed for the presence or absence of one or more mutations include, without limitation, nucleic acid encoding tumor protein p53 (TP53), nucleic acid encoding breast cancer 1 (BRCA1), nucleic acid encoding BRCA2, nucleic acid encoding a phosphatase and tensin homolog (PTEN) polypeptide, nucleic acid encoding a AKTl polypeptide, nucleic acid encoding a APC polypeptide, nucleic acid encoding a CDKN2A polypeptide, nucleic acid encoding a EGFR polypeptide, nucleic acid encoding a FBXW7 polypeptide, nucleic acid encoding a GNAS polypeptide, nucleic acid encoding a KRAS polypeptide, nucleic acid encoding a NRAS polypeptide, nucleic acid encoding a PIK3CA polypeptide, nucleic acid encoding a
- Any appropriate method can be used to assess a target region (e.g ., an amplified target region) for the presence or absence of one or more mutations.
- a target region e.g ., an amplified target region
- one or more sequencing methods can be used to assess an amplified target region for the presence or absence of one or more mutations.
- one or more sequencing methods can be used to assess an amplified target region determine whether the mutation(s) are present on both the Watson strand and the Crick strand.
- sequencing reads can be used to assess an amplified target region for the presence or absence of one or more mutations and can be used to determine whether the mutation(s) are present on both the Watson strand and the Crick strand.
- the single read sequencing comprises sequencing across the entire length of the templates to generate the sequence reads.
- the sequencing comprises paired end sequencing.
- the sequencing is performed with a massively parallel sequencer.
- the massively parallel sequencer is configured to determine sequence reads from both ends of template polynucleotides.
- the sequence reads are mapped to a reference genome.
- the sequence reads are assigned into UID families.
- a UID family can comprise sequence reads from amplicons originating from an original template, e.g., original double-stranded DNA fragment from a nucleic acid sample.
- each member of a UID family comprises the same exogenous UID sequence. In some embodiments, each member of a UID family further comprises the same endogenous UID sequence. Endogenous UIDs are described herein.
- each member of a UID family further comprises the same exogenous UID sequence and the same endogenous UID sequence.
- the combination of the exogenous UID sequence and endogenous UID sequence are unique to the UID family.
- the combination of the exogenous UID sequence and endogenous UID sequence does not exist in another UID family represented in the nucleic acid sample.
- UID family can depend on the depth of sequencing. In some embodiments, a UID family comprises at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30,
- a UID family comprises about 2-1000 members, about 2-500 members, about 2-100 members, about 2-50 members, or about 2-20 members.
- sequence reads of an individual UID family are assigned to a Watson subfamily and a Crick subfamily. In some embodiments, the sequence reads of an individual UID family are assigned to the Watson and Crick subfamilies based on the orientation of the insert relative to the adapter sequences. In some embodiments, the orientation of the insert relative to the adapter sequences is resolved by how the sequence reads were aligned as “read pairs” or “mate pairs”.
- the assignment of the sequence reads into the Watson and
- Crick subfamilies are based on spatial relationship of the exogenous UID sequence to the R1 and R2 read sequence.
- members of the Watson subfamily are characterized by the exogenous UID sequence being downstream of the R2 sequence and upstream of the R1 sequence.
- members of the Crick subfamily are characterized by the exogenous UID sequence being downstream of the R1 sequence and upstream of the R2 sequence.
- members of the Watson subfamily are characterized by the exogenous UID sequence being in greater proximity to the R2 sequence and lesser proximity to the R1 sequence.
- members of the Crick subfamily are characterized by the exogenous UID sequence being in greater proximity to the R1 sequence and in lesser proximity to the R2 sequence.
- members of the Watson subfamily are characterized by the exogenous UID sequence being immediately downstream or within 1-70, 1-60, 1-50, 1-40, 1-30, 1-20, 1-10, or 1-5 nucleotides of the R2 sequence.
- members of the Crick subfamily are characterized by the exogenous UID sequence being immediately downstream or within 1-70, 1-60, 1-50, 1-40, 1- 30, 1-20, 1-10, or 1-5 nucleotides of the R1 sequence.
- a UID subfamily (e.g., Watson subfamily and/or Crick subfamily) comprises at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200, 210, 220, 230, 240, 250, 260, 270, 280, 290, 300, 310, 320, 330, 340, 350, 360, 370, 380, 390, 400,
- Watson subfamily and/or Crick subfamily comprises at least 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 65, 70, 75, 80, 85, 90, 95, 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200, 210, 220, 230, 240, 250, 260, 270, 280, 290, 300, 310
- a UID subfamily (e.g., Watson subfamily and/or Crick subfamily) comprises about 2-500 members, about 2-100 members, about 2-50 members, about 2-20 members, or about 2-10 members.
- a nucleotide sequence is determined to accurately represent a Watson strand of an analyte DNA fragment, e.g., a double stranded DNA fragment from the nucleic acid sample, when a threshold percentage (or a percentage exceeding a threshold) of members of the Watson subfamily contain the sequence.
- a nucleotide sequence is determined to accurately represent a Crick strand of an analyte DNA fragment, e.g., a double stranded DNA fragment from the nucleic acid sample, when a threshold percentage (or a percentage exceeding a threshold) of members of the Crick subfamily contain the sequence.
- Thresholds can be determined by a skilled artisan based on, e.g., number of the members of the subfamily, the particular purpose of the sequencing experiment, and the particular parameters of the sequencing experiment.
- the threshold is set at 1%, 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 75%, 80%, 85%, 90%, 95%, 96%, 97%, 98%, 99%, or 100%.
- the threshold is set at 50%.
- a nucleotide sequence is determined to accurately represent a Watson or Crick strand of an analyte DNA fragment, e.g., a double stranded DNA fragment from the nucleic acid sample, when at least 50% of the subfamily members contain the sequence.
- a nucleotide sequence is determined to accurately represent a Watson or Crick strand of an analyte DNA fragment, e.g., a double stranded DNA fragment from the nucleic acid sample, when more than 50% of the subfamily members contain the sequence.
- the sequence accurately representing the Watson strand of the analyte DNA fragment is determined to have a mutation. In some embodiments, the sequence accurately representing the Watson strand of the analyte DNA fragment is determined to have a mutation when the sequence differs from a reference sequence that lacks the mutation.
- the sequence accurately representing the Crick strand of the analyte DNA fragment is determined to have a mutation. In some embodiments, the sequence accurately representing the Crick strand of the analyte DNA fragment is determined to have a mutation when the sequence differs from a reference sequence that lacks the mutation.
- the analyte DNA fragment is determined to have the mutation when sequence accurately representing the Watson strand the sequence accurately representing the Crick strand comprise the same mutation.
- the location of the molecular barcode within the paired-end sequencing reads of the amplified target region can be used to distinguish which strand of the double stranded nucleic acid template the amplified target region was derived from. For example, when a first a paired-end sequencing read of an amplified target region indicates that a molecular barcode is read last, the amplified target region can be identified as being derived from the sense strand of the nucleic acid template, and when a first a paired-end sequencing read of an amplified target region indicates that a molecular barcode is read first, the amplified target region can be identified as being derived from the anti-sense strand of the nucleic acid template.
- the amplified target region when a second a paired-end sequencing read of an amplified target region indicates that a molecular barcode is read first, the amplified target region can be identified as being derived from the anti-sense strand of the nucleic acid template, and when a second a paired-end sequencing read of an amplified target region indicates that a molecular barcode is read last, the amplified target region can be identified as being derived from the sense strand of the nucleic acid template.
- paired-end sequencing can be used to distinguish amplification products derived from the Watson strand from amplification products derived from the Crick strand as shown in Figure 20 and Figure 21
- sequencing reads can be aligned to a reference genome and grouped by the molecular barcode present in each sequencing read.
- sequencing reads that include the same molecular barcode and map to both the Watson strand and the Crick strand of the double stranded nucleic acid template e.g., both the Watson strand and the Crick strand of the target region
- the mutation(s) can be identified as having duplex support.
- kits may comprise sets of primer pairs for amplification of one or more target polynucleotides.
- the kit comprises (a) a first set of Watson target- selective primer pairs, comprising (i) one or more first Watson target-selective primers comprising a sequence complementary to the R2 sequencing primer site of the universal 3’ adapter sequence, and (ii) one or more second Watson target-selective primers, each of the one or more second Watson target-selective primers comprising a target-selective sequence; (b) a first set of Crick target- selective primer pairs, comprising (i) one or more Crick target- selective primers comprising a sequence complementary to the R1 sequencing primer site of the universal 5’ adapter sequence, and (ii) one or more second Crick target- selective primers, each of the one or more second Crick target- selective primers comprising the same target- selective sequence as the second Watson target-selective primer sequence; (c) a second set of Watson target-selective primer pairs, comprising (i) one or more third Watson target- selective primers comprising a sequence complementary to
- the kit may comprise sets of primer pairs for multiplex amplification of a plurality of target polynucleotides.
- a computer readable medium comprising computer executable instructions configured to implement any of the methods described herein.
- the computer readable medium can comprise computer executable instructions for analyzing sequence data from a nucleic acid sample, wherein the data is generated by a method of any one of the preceding claims.
- the computer readable medium can implement a method for semi-automated or automated sequence data analysis.
- the computer readable medium comprises executable instructions for (a) assigning sequence reads into UID families, wherein each member of a UID family comprises the same exogenous UID sequence; (b) assigning sequence reads of each UID family into Watson and Crick subfamilies; (c) identifying a nucleotide sequence as accurately representing a Watson strand of an analyte DNA fragment when a threshold percentage of members of the Watson subfamily contain the sequence; (d) identifying a nucleotide sequence as accurately representing a Crick strand of an analyte DNA fragment when a threshold percentage of members of the Crick subfamily contain the sequence; (e) identifying a mutation in the nucleotide sequence accurately representing the Watson Strand when the sequence accurately representing the Watson Strand differs from a reference sequence that lacks the mutation; (f) identifying a mutation in the nucleotide sequence accurately representing the Crick Strand when the sequence accurately representing the Crick Strand differs from a reference sequence that lacks the mutation; and (g)
- the computer readable medium comprises executable code for assigning UID family members to the Watson subfamily or Crick subfamily based on spatial relationship of the exogenous UID sequence to the R1 and R2 read sequence. In some embodiments, the computer executable code assigns UID family members to the Watson subfamily when the exogenous UID sequence is downstream of the R2 sequence and upstream of the R1 sequence. In some embodiments, the computer executable code assigns UID family members to the Crick subfamily when the exogenous UID sequence is downstream of the R1 sequence and upstream of the R2 sequence. In some embodiments, the computer executable code assigns UID family members to the Watson subfamily when the exogenous UID sequence is in greater proximity to the R2 sequence and lesser proximity to the R1 sequence.
- the computer executable code assigns UID family members to the Crick subfamily when the exogenous UID sequence is in greater proximity to the R1 sequence and in lesser proximity to the R2 sequence. In some embodiments, the computer executable code assigns UID family members to the Watson subfamily when the exogenous UID sequence is immediately downstream or within 1-70, 1-60, 1-50, 1-40, 1-30, 1-20, 1-10, or 1-5 nucleotides of the R2 sequence. In some embodiments, the computer executable code assigns UID family members to the Crick subfamily when the exogenous UID sequence is immediately downstream or within 1-70, 1-60, 1-50, 1-40, 1-30, 1-20, 1-10, or 1-5 nucleotides of the R1 sequence.
- the computer readable medium comprises executable code for mapping the sequence reads to a reference genome.
- the reference genome is a human reference genome.
- the computer readable medium comprises executable code for generating a report of disease status, prognosis, or theranosis based on the presence, absence, or amount of a mutation in the sample.
- the disease is cancer.
- the computer readable medium comprises executable code for generating a report of therapeutic options based on the presence, absence, or amount of a mutation in the sample.
- the computer readable medium comprises executable code for transmission of the data over a network.
- the computer system comprises a memory unit configured to receive and store sequence data from a nucleic acid sample, the data generated by a method described herein; and a processor communicatively coupled to the storage memory, wherein the processor comprises a computer readable medium disclosed herein.
- FIG 32 depicts an exemplary computer system 900 adapted to enable a user to analyze a nucleic acid sample according to any of the methods described herein.
- the system 900 includes a central computer server 901 that is programmed to implement exemplary methods described herein.
- the server 901 includes a central processing unit (CPU, also "processor") 905 which can be a single core processor, a multi core processor, or plurality of processors for parallel processing.
- the server 901 also includes memory 910 (e.g. random access memory, read-only memory, flash memory); electronic storage unit 915 (e.g. hard disk); communications interface 920 (e.g.
- the network adapter for communicating with one or more other systems, e.g., a sequencing system; and peripheral devices 925 which may include cache, other memory, data storage, and/or electronic display adapters.
- the memory 910, storage unit 915, interface 920, and peripheral devices 925 are in communication with the processor 905 through a communications bus (solid lines), such as a motherboard.
- the storage unit 915 can be a data storage unit for storing data.
- the server 901 is operatively coupled to a computer network (“network") 930 with the aid of the communications interface 920.
- the network 930 can be the Internet, an intranet and/or an extranet, an intranet and/or extranet that is in communication with the Internet, a telecommunication or data network.
- the network 930 in some cases, with the aid of the server 901, can implement a peer-to-peer network, which may enable devices coupled to the server 901 to behave as a client or a server.
- the storage unit 915 can store files, such as sequence data, barcode sequence data, or any aspect of data associated with the invention.
- the data storage unit 915 may be coupled with data relating to locations of cells in a virtual grid.
- the server can communicate with one or more remote computer systems through the network 930.
- the one or more remote computer systems may be, for example, personal computers, laptops, tablets, telephones, Smart phones, or personal digital assistants.
- the system 900 includes a single server 901. In other situations, the system includes multiple servers in communication with one another through an intranet, extranet and/or the Internet.
- the server 901 can be adapted to store sequence data, data regarding the nucleic acid sample, data regarding the biological sample, data regarding the subject, and/or other information of potential relevance. Such information can be stored on the storage unit 915 or the server 901 and such data can be transmitted through a network.
- Methods as described herein can be implemented by way of machine (e.g., computer processor) computer readable medium (or software) stored on an electronic storage location of the server 901, such as, for example, on the memory 910, or electronic storage unit 915. During use, the code can be executed by the processor 905.
- machine e.g., computer processor
- computer readable medium or software stored on an electronic storage location of the server 901, such as, for example, on the memory 910, or electronic storage unit 915.
- the code can be executed by the processor 905.
- the code can be retrieved from the storage unit 915 and stored on the memory 910 for ready access by the processor 905. In some situations, the electronic storage unit 915 can be precluded, and machine-executable instructions are stored on memory 910. Alternatively, the code can be executed on a second computer system 940.
- aspects of the systems and methods provided herein can be embodied in programming.
- Various aspects of the technology may be thought of as “products” or “articles of manufacture” typically in the form of machine (or processor) executable code and/or associated data that is carried on or embodied in a type of machine readable medium (e.g., computer readable medium).
- Machine-executable code can be stored on an electronic storage unit, such memory (e.g., read-only memory, random access memory, and flash memory) or a hard disk.
- Storage type media can include any or all of the tangible memory of the computers, processors or the like, or associated modules thereof, such as various semiconductor memories, tape drives, disk drives and the like, which may provide non-transitory storage at any time for the software programming. All or portions of the software may at times be communicated through the Internet or various other telecommunication networks. Such communications, for example, may enable loading of the software from one computer or processor into another, for example, from a management server or host computer into the computer platform of an application server.
- another type of media that may bear the software elements includes optical, electrical, and electromagnetic waves, such as used across physical interfaces between local devices, through wired and optical landline networks and over various air-links.
- a machine readable medium such as computer-executable code
- Non-volatile storage media can include, for example, optical or magnetic disks, such as any of the storage devices in any computer(s) or the like, such may be used to implement the system.
- Tangible transmission media can include: coaxial cables, copper wires, and fiber optics (including the wires that comprise a bus within a computer system).
- Carrier-wave transmission media may take the form of electric or electromagnetic signals, or acoustic or light waves such as those generated during radio frequency (RF) and infrared (IR) data communications.
- RF radio frequency
- IR infrared
- Computer- readable media therefore include, for example: a floppy disk, a flexible disk, hard disk, magnetic tape, any other magnetic medium, a CD-ROM, DVD, DVD-ROM, any other optical medium, punch cards, paper tame, any other physical storage medium with patterns of holes, a RAM, a ROM, a PROM and EPROM, a FLASH-EPROM, any other memory chip or cartridge, a carrier wave transporting data or instructions, cables, or links transporting such carrier wave, or any other medium from which a computer may read programming code and/or data. Many of these forms of computer readable media may be involved in carrying one or more sequences of one or more instructions to a processor for execution.
- results of the analysis can be presented to a user with the aid of a user interface, such as a graphical user interface.
- This protocol can prepare duplex libraries using a Swift Accel-NGS 2S PCR-Free Library Kit (Cat. # 20024 and 20096) and specific truncated adapters and primers. In some cases, full-length P5 and P7 graft sequences can be added to the library by a separate PCR for sequencing on an Illumina instrument.
- This protocol is for PCR tubes but can be scaled to PCR plates.
- Truncated P5 Primer a ACACTCTTTCCCTACACGACGCTCTTCCGATCT (SEQ ID NO:4) b. No modifications required, desalted from IDT, 100 mM in IDTE 7.
- Truncated P7 Primer a GACTGGAGTTCAGACGTGTGCTCTTCCGATCT (SEQ ID NO: 5) b. No modifications required, desalted from IDT, 100 mM in IDTE
- PCR tube strips e.g ., GeneMate VWR Cat. No. 490003-710)
- Magnetic rack e.g., Permagen MSRLV08
- USER Enzyme (NEB Cat. No. M5505L) - which is a mixture of uracil-DNA glycosylase and DNA glycosylase-lyase Endonuclease VIII.
- Enzyme tubes can be removed from -20°C storage and placed on ice, for about 10 minutes to allow enzymes to reach 4°C prior to pipetting. Pipetting enzymes at -20°C may result in a shortage of enzyme reagents.
- reagents except the enzymes
- Reagents should be added to the master mix in the specified order as stated throughout the protocol. Reagents can be prepared in advance ( e.g ., to ensure that the magnetic beads do not dry out during size selection steps).
- thermocycler Place the samples in the thermocycler, programmed at 37°C for 15 minutes with lid heating off.
- Step 2 End Repair 1 1. Gently spin down samples to collect any condensation.
- thermocycler Place the samples in the thermocycler, programmed at 25°C for 15 minutes with lid heating off.
- Ligation II Master Mix 1. Add 50 m ⁇ of pre-mixed Ligation II Master Mix (see Table 4) to the beads for each sample and mix by vortexing until homogeneous. Table 4. Ligation II Master Mix 2. Place the samples in the thermocycler, programmed at 40°C for 10 minutes with lid heating off.
- Step 6 PCR-Library Amplification
- PCR-library Amplification Master Mix 2. Carefully transfer the supernatant containing the final post-ligation library to the PCR- Library Amplification Master Mix.
- Sequencing libraries incorporating duplex molecular barcodes were generated by sequentially ligating two adapter molecules to double stranded input DNA.
- input DNA was end-repaired via blunting and dephosphorylation reactions (Figs. 9 and 10).
- a 3’ adapter containing a 5’ phosphate (3’ oligo #1) annealed to a short oligonucleotide with a blocked 3’ group (3’ oligo #2) was ligated to each 3’ end of the input DNA (Fig. 12).
- the oligonucleotides contains a 3’ blocking group
- only the oligonucleotide containing the 5’ phosphate (3’ oligo #1) was covalently attached to the input DNA at the 3’ ends.
- the attached 3’ oligonucleotide also contains a molecular barcode which uniquely labels each strand (Fig. 11).
- the 3’ oligo containing a 3’ blocking group was degraded and a 5’ adapter oligo was ligated to each 5’ end via a nick translation like reaction.
- the 5’ adapter oligo anneals immediately upstream of the molecular barcode on 3’ adapter oligo #1 leaving a gap.
- the product can optionally be purified and single stranded (ss) NA libraries corresponding to sense and anti-sense strands are generated (Figs. 2 and 3).
- ss single stranded
- the amplified DNA libraries were enriched for the desired targets using a strand- specific anchored PCR approach.
- This PCR enrichment utilized a single primer targeting the desired region of interest and a second primer targeting the ligated adapter sequence (Figs. 4, 5, 15, 17).
- a second nested PCR can be performed using a single primer targeting the desired region of interest and a second primer targeting the ligated adapter sequence (Figs. 7, 8, 16, 18).
- a second nested PCR can be used to incorporate sample barcodes as well as the requisite graft sequences necessary for next generation sequencing.
- the resulting libraries are then quantified, normalized, and sequenced.
- This example describes a method (termed SaferSeqS) that addresses this challenge by (i) efficiently introducing identical molecular barcodes in the Watson and Crick strands of template molecules and (ii) enriching for genomic regions of interest with a novel strand-specific PCR assay. It can be applied to evaluate mutations within a single amplicon or simultaneously within multiple amplicons, can assess limited quantities of DNA such as those found in plasma, and reduces the error rate of existing PCR-based molecular barcoding approaches by at least two orders of magnitude.
- a strategy was designed that involves the sequential ligation of adapter sequences to the 3’ and 5’ DNA fragment ends and the generation of double stranded molecular barcodes in situ (Fig. 22a).
- the in situ generation of molecular barcodes is a key innovation of the new library preparation method.
- the enzymes used for the in situ generation of double stranded molecular barcodes uniquely barcoded each DNA fragment and obviated the need to enzymatically prepare duplex adapters (Fig. 22a, steps 2 and 3).
- the adapter contained a stretch of 14 random nucleotides as the exogenous molecular barcode (unique identifier sequence [UID]).
- the adapter-ligated fragments were subjected to a limited of number of PCR cycles to create redundant copies of the two original DNA strands (Fig. 22a, step 4).
- a UCSC reference sequence available at genome.ucsc.edu/ was arbitrarily defined as the “Watson” strand and its reverse complement as the “Crick” strand.
- hemi-nested PCR has previously been used for target enrichment (see, e.g., Zheng et ak, 2014, Nat Med 20:1479-1484), major changes were required to apply it to duplex sequencing.
- two separate PCRs were performed - one for the Watson strand and one for the Crick strand.
- Both PCRs employed the same gene-specific primer, but each employed a different anchoring primer.
- PCR duplicates derived from each strand could be distinguished by the orientation of the insert relative to the exogenous UID (Fig. 22b).
- Fig. 22c Those present in >80% of both the Watson and Crick families with the same UID (a “duplex family”) were called “supercalifragilisticexpialidocious mutants”, referred to herein as “supercalimutants” (Fig. 22c).
- Fig. 22c Those present in >80% of both the Watson and Crick families with the same UID (a “duplex family”) were called “supercalifragilisticexpialidocious mutants”, referred to herein as “supercalimutants” (Fig. 22c).
- Fig. 22c As an initial demonstration of SaferSeqS, a mixing experiment was conducted in which DNA with a known mutation was spiked into DNA from a normal individual’s leukocytes at ratios varying from 10% to 0%. These admixtures were predicted to result in 15,400, 150, 15, 15, 8 or 0 supercalimutants per assay.
- SaferSeqS could simultaneously assay multiple targets, which can be useful for a variety of sequencing applications.
- SaferSeqS permits two types of multiplexing, one in which multiple targets are assayed in separate PCR reactions, and another in which multiple targets are assayed in the same PCR reaction. Because redundant Watson and Crick strand-derived copies are created during library amplification, the library can be partitioned into multiple PCR reactions without adversely impacting sample recovery. For example, assuming a PCR efficiency of 70%, up to 22 targets can be separately assayed with ⁇ 10% loss in recovery if a DNA library is amplified with 11 PCR cycles (Fig. 28).
- the median on-target rate for Watson-derived reads was 95% (range: 39% to 97%), and the median on-target rate for Crick-derived reads was 95% (range: 39% to 98%).
- the targets demonstrated relatively uniform recovery of the input molecules, with a coefficient of variation of only 17% (Fig. 29).
- the lengths of the amplicons sequenced were also similar in all amplicons and consistent with the initial size of cell-free plasma DNA being -167 bp ⁇ 10.4 bp (Fig. 29).
- Table 11 Composition and GSP primer sequences of multiplex panel.
- Two exemplary methods can be used to assess multiple amplicons.
- the first involves parallel amplicon-specific PCR's in different wells.
- this strategy can be readily applied without concern about cross-hybridization among primers or other issues commonly encountered in multiplex PCR reactions.
- the evaluation of many more amplicons is useful; e.g., a combination of multiple primer pairs in each PCR well.
- SaferSeqS can enable the development of highly sensitive and specific DNA-based molecular diagnostics as well as help answer a variety of important basic scientific questions.
- DNA was purified from 10 mL plasma using cfPure MAX Cell-Free DNA Extraction Kit (BioChain, cat. # K5011625MA) as specified by the manufacturers. DNA from peripheral WBCs was purified with the QIAsymphony DSP DNA Midi Kit (Qiagen, cat. # 937255) as specified by the manufacturer. Purified DNA from all samples was quantified as described elsewhere (see, e.g., Douville et al., 2019 bioRxiv , 660258).
- a custom library preparation workflow was developed that could efficiently recover input DNA fragments and simultaneously incorporate double stranded molecular barcodes.
- duplex sequencing libraries were prepared with cell-free DNA or peripheral WBC
- DNA using an Accel -NGS 2S DNA Library Kit (Swift Biosciences, cat. # 21024) with the following modifications: 1) DNA was pre-treated with three units of USER enzyme (New
- Libraries were amplified with 5, 7, or 11 cycles of PCR, depending on how many experiments were planned, according the following protocol: 98°C for 30 seconds, cycles of 98°C for 10 seconds, 65°C for 75 seconds, and 4°C hold. If 5 or 7 cycles were used, the libraries were amplified in single 50 pL reactions. If 11 cycles were used, the libraries were divided into eight aliquots and amplified in eight 50 pL reactions each supplemented with an additional 0.5 units of Q5® Hot Start High-Fidelity DNA Polymerase (New England BioLabs, cat. # M0493L), 1 pL of 10 mM dNTPs (New England BioLabs, cat.
- a strategy that involves the sequential ligation of adapter sequences to the 3’ and 5’ DNA fragment ends and the generation of double stranded molecular barcodes in situ was designed (Fig. 22a). After DNA ends were dephosphorylated and repaired (Fig. 22a, step 1), an adapter was attached to the 3’ end of DNA fragments (Fig. 22a, step 2). The adapter was a partially double-stranded DNA fragment with end modifications that selectively ligated to the 3’ DNA ends and prevented adapter-dimer formation.
- this adapter consisted of one oligonucleotide containing a 5’ phosphate end modification (Table 12, 3’ N14 Adapter Oligo #1) which was hybridized to another oligonucleotide containing a 3’ blocking group and deoxyuridines substituted for deoxythymidines (Table 12, 3’ N14 Adapter Oligo #2).
- This design permitted of the use of adapters at high concentration in the ligation reaction which promoted efficient attachment to the 3’ ends without the risk of significant dimer or concatemer formation.
- the adapter contained a stretch of 14 random nucleotides in one of the two oligonucleotides which compromised one strand of the duplex UID.
- a second adapter (Table 12, 5’ Adapter) was ligated to the 5’ DNA fragment ends via a nick translation-like reaction consisting of a DNA polymerase, cohesive end-specific ligase, and uracil-DNA glycosylase (Fig. 22a, step 3).
- the number of PCR cycles and the efficiency of duplication during library amplification can be adjusted to optimize SaferSeqS parameters. Because SaferSeqS can involve partitioning of redundant Watson and Crick strand-derived copies into specific strand-specific PCRs for target enrichment, in preferred embodiments a requisite number of copies should be generated to ensure a high probability of duplex recovery. For example, assuming 100% efficiency, after one PCR cycle, each template DNA duplex is converted into two double stranded copies (one representing each strand), and there is only a 25% probability of properly distributing these two copies such that the one Watson strand-derived copy is partitioned into the Watson-specific PCR and the one Crick strand-derived copy is partitioned into the Crick-specific PCR. Increasing the number of PCR cycles, or increasing the amplification efficiency, generates more redundant copies which in turn increases the probability of recovering the original DNA duplex.
- a probabilistic model was developed to estimate the number of PCR cycles and amplification efficiency necessary for efficient duplex recovery. This model consisted of three steps: 1) simulate the number of PCR progeny generated during library amplification;
- duplex recovery that is, the proportion of original DNA duplexes which have at least one Watson strand-derived copy partitioned into the Watson strand-specific reaction(s) and at least one Crick strand-derived copy partitioned into the Crick strand- specific reaction(s).
- the number of PCR copies of the original template strands generated during each library amplification cycle follows a binomial distribution.
- the number of strand-specific copies were initialized to one. It should be noted that the counts were initialized to one (instead of two) because the first library amplification cycle merely serves to denature the two original template strands and convert them into physically distinct double stranded forms.
- each of the n, PCR copies can replicate with probability p (i.e. the efficiency of amplification) to generate a total of m+i PCR copies equal to m + Binom(/3 ⁇ 4, p).
- This process was iteratively repeated to simulate the number of progeny generated after i PCR cycles.
- each original DNA duplex has been amplified to generate n, w copies of the Watson strand and th e copies of the Crick strand as described above.
- Each of the n, w and n, c copies are randomly partitioned into Watson and Crick strand-specific PCR reactions with a probability q that is equal to the fraction of the library used for each reaction.
- q the probability that is equal to the fraction of the library used for each reaction.
- the number of PCR copies that are partitioned into the appropriate strand-specific PCR (Nk.w or Nk, c for the Mi Watson-specific or Crick-specific PCR, respectively) is drawn from a Binomial distribution with m, w or m, c “trials” and probability q of “success” for the Watson and Crick copies, respectively. Therefore, the probability of partitioning at least one Watson-derived PCR copy into the Mi Watson-specific PCR reaction is:
- the probability of partitioning at least one Crick-derived PCR copy into the Mi Crick-specific PCR reaction is:
- Anchored hemi-nested PCR theoretically demonstrates a higher recovery of template molecules than traditional amplicon PCR.
- a template molecule In traditional amplicon PCR, a template molecule must contain the both forward and reverse primer binding sites and the intervening sequence that defines the amplicon.
- the template molecules In contrast, in anchored hemi-nested PCR, the template molecules need only harbor the union of the two gene-specific primer binding sites in order to be recovered.
- the combined footprints of the nested gene-specific primers used in SaferSeqS are approximately 30 bp, whereas the amplicon lengths employed by SafeSeqS for profiling cfDNA are typically 70-80 bp.
- the probability of recovering a template molecule of length L is where r is the amplicon length in the case of traditional PCR or the length of the combined footprint of the gene specific primers in the case of anchored hemi-nested PCR.
- r is the amplicon length in the case of traditional PCR or the length of the combined footprint of the gene specific primers in the case of anchored hemi-nested PCR.
- anchored hemi-nested PCR can theoretically recover -25% more of the original template fragments than traditional amplicon PCR.
- anchored hemi-nested produces fragments of varying lengths with only one of the fragment ends dictated by the positions of the gene specific primers. Assuming template molecules of length L with uniformly random start/end
- the observed fragment length after anchored hemi-nested PCR will be “ where r is the length of the combined footprint of the gene specific primers.
- the Watson and Crick reads for each sample were merged into a single BAM file and sorted by read name using SAMtools so that mate pairs could be readily extracted.
- Custom Python scripts were used for subsequent reconstruction of the duplex families and identification of Watson supermutants, Crick supermutants, and supercalimutants.
- Bitwise flags are numerical values that are assigned to read pairs during mapping. Their value indicates how the read mates align to the genome relative to one another. For example, if a read maps to the reference strand and its mate maps to the reverse (complementary) strand, then this read pair was derived from the Watson strand. Similarly, if a read maps to the reverse (complementary) strand and its mate maps to the reference strand, then this read pair was derived from the Crick strand.
- the expected number of barcode collisions can be calculated from the classical “birthday problem” and is: where n is equal to the number of template molecules and N is equal to the number of possible barcodes.
- n is equal to the number of template molecules
- N is equal to the number of possible barcodes.
- non-unique exogenous UIDs may be used, and non-unique exogenous UID can be used in combination with endogenous UIDs for assignment into UID families.
- the DNA used in this study was obtained from a set of individuals of average age 30.
- the expected frequency of non-clonal somatic single base substitutions in these samples is 426 per diploid genome, or approximately 7 x 10 8 mutations per bp.
- this study we evaluated a total of 42,695,395 bases with SaferSeqS from DNA derived from healthy control subjects. Among these 42,695,395 bases, 5 single base substitution supercalimutants, representing a mutation frequency of 12 c 10 8 were detected. To determine whether the frequency of supercalimutants observed is in accordance with previous estimates of non- clonal somatic mutation rates in healthy blood cells, the following exact one-sided binomial p-value was calculate:
- Target enrichment of the regions of interest was achieved using critical modifications of anchored hemi-nested PCR necessary for duplex sequencing.
- various reaction conditions were optimized, including the number of cycles, the primer concentrations, and the polymerase formulation.
- the final optimized protocol was as follows: the first round of PCR was performed in a 50 pL reaction with following conditions: IX NEBNext Ultra II Q5 Master Mix (New England BioLabs, cat. # M0544L), 2 pM GSP1 primer, and 2 pM P7 short anchor primer for amplification of the Watson strand.
- the GSP1 primer was specific for each amplicon, and the P7 short anchor primer was used as the anchor primer for the Watson strand of all amplicons (Tables 11 and 12).
- the Crick strand was amplified the same way, in a separate well, except that the P5 short primer anchor primer was substituted for the P7 short primer.
- the GSP1 primer used for amplification of the Watson strand was identical to the GSP1 primer used for the Crick strand; the only difference between the Watson and Crick strand PCRs was the anchor primers. Both reactions (Watson and Crick strands) were amplified with 19 cycles according to the thermocycling protocol described above.
- a second round of PCR was formed in 50 pi reactions using the identical reaction conditions used for the first round of PCR. The differences were (i) Template: 1% of product from the first anchored Watson strand PCR was used as the template (instead of the library used as template for the first PCR), and (ii) Primers: the gene- specific primers GSP2 were substituted for the GSP1 gene-specific primers and anchor P5 indexing primer was substituted for the P7 short anchor primer.
- the second round of PCR for the Crick strand was performed identically except that (i) Template: the first Crick strand PCR was used as the template and (ii) Primers: anchor P7 indexing primer was substituted for anchor P5 indexing primer.
- each gene-specific primer was included at a final concentration of 0.25 mM
- the concentration of the anchor primer was included at a final concentration of 0.25 pM per target (e.g. at a final concentration of 6.25 pM if 25 targets were co-amplified).
- Sequencing reads underwent initial processing by extracting the first 14 nucleotides as the UID sequence and masking adapter sequences using Picard’s IlluminaBasecallsToSam (broadinstitute.github.io/picard). Reads were then mapped to the hgl9 reference genome using BWA-MEM (version 0.7.17) and sorted by UID sequence using SAMtools. UID families were scored if they consisted of 2 or more reads and if >90% of the reads mapped to the reference genome with the expected primer sequences. “Supermutants” were identified as mutations that were present in >95% of the mapped reads and had an average Phred score greater than 25.
- Python scripts were used for subsequent reconstruction of the duplex families and identification of Watson supermutants, Crick supermutants, and supercalimutants. After correcting for PCR and sequencing errors within the molecular barcode sequences as described elsewhere (see, e.g., Smith et ak, 2017 Genome Res 27:491-499), Watson and Crick reads belonging to the same duplex family were grouped together to reconstruct the sequence of the original template molecule. To exclude artifacts stemming from the end repair step of library construction, bases fewer than 10 bases from the 3’ adapter sequence were not considered for mutation analysis.
- Watson and Crick supermutants were defined as mutations present in > 80% of the Watson or Crick reads of a duplex family, respectively.
- Supercalimutants were defined as mutations present in >80% of both the Watson and Crick families with the same UID.
- SaferSeqS can detect rare mutations with extremely high specificity.
- the technique is highly scalable, cost effective, and amenable to high- throughput automation.
- SaferSeqS achieved up to a 5- to 75-fold improvement in input recovery over existing duplex sequencing techniques and can be applied to limited amounts of starting material, and resulted in a >50-fold improvement in error correction over standard PCR-based approaches employing molecular barcodes (Fig. 23, Table 8). It also offers a >50-fold improvement in error correction over optimal ligation-based techniques that employ only Watson or Crick supermutants rather than supercalimutants (Fig. 26, Table 9). Both reductions are useful for to the detection of mutations present at single or very low copy number, such as in cancer screening and minimal residual disease settings.
- SaferSeqS is considerably more sensitive than digital droplet PCR for the analysis of single amplicons, and, unlike digital droplet PCR, can be highly multiplexed.
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Analytical Chemistry (AREA)
- Microbiology (AREA)
- Immunology (AREA)
- Molecular Biology (AREA)
- Biotechnology (AREA)
- Biophysics (AREA)
- Physics & Mathematics (AREA)
- Biochemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
Abstract
Description











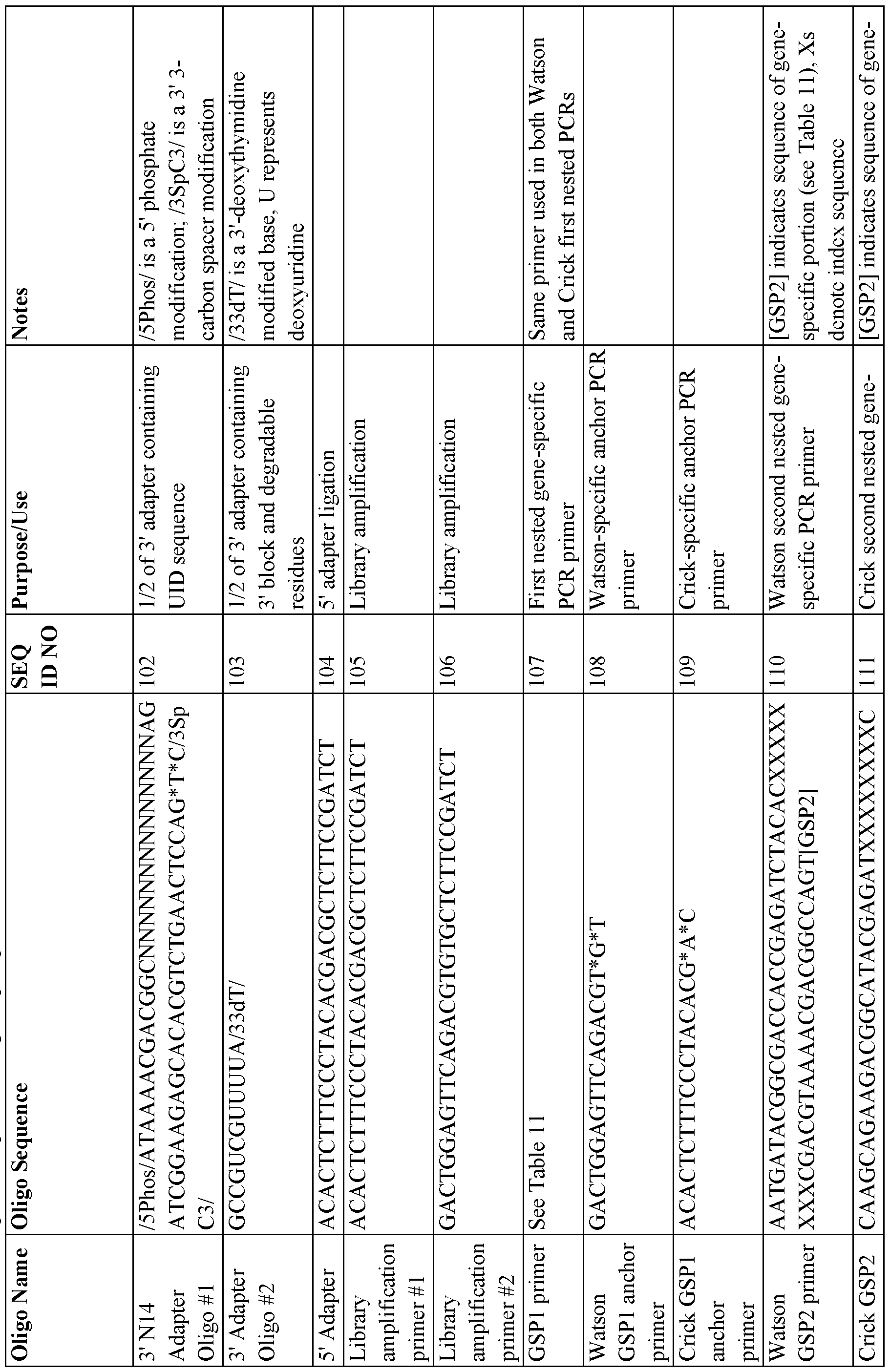

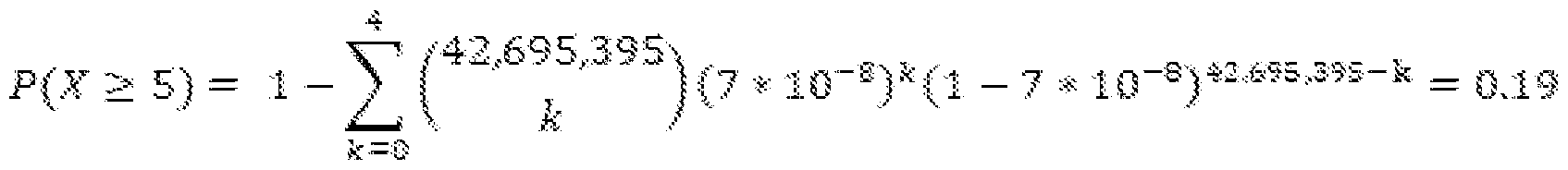
Claims
Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| MX2022010038A MX2022010038A (en) | 2020-02-14 | 2021-02-12 | Methods and materials for assessing nucleic acids. |
| KR1020227031690A KR20220154690A (en) | 2020-02-14 | 2021-02-12 | Nucleic Acid Evaluation Methods and Materials |
| CA3170345A CA3170345A1 (en) | 2020-02-14 | 2021-02-12 | Methods and materials for assessing nucleic acids |
| EP21753424.7A EP4103748A4 (en) | 2020-02-14 | 2021-02-12 | Methods and materials for assessing nucleic acids |
| IL295297A IL295297A (en) | 2020-02-14 | 2021-02-12 | Methods and materials for assessing nucleic acids |
| CN202180028441.0A CN115516108A (en) | 2020-02-14 | 2021-02-12 | Methods and materials for assessing nucleic acids |
| BR112022015909A BR112022015909A2 (en) | 2020-02-14 | 2021-02-12 | METHODS AND MATERIALS FOR EVALUATION OF NUCLEIC ACIDS |
| JP2022549104A JP2023513606A (en) | 2020-02-14 | 2021-02-12 | Methods and Materials for Assessing Nucleic Acids |
| AU2021219794A AU2021219794A1 (en) | 2020-02-14 | 2021-02-12 | Methods and materials for assessing nucleic acids |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US202062977066P | 2020-02-14 | 2020-02-14 | |
| US62/977,066 | 2020-02-14 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2021163546A1 true WO2021163546A1 (en) | 2021-08-19 |
| WO2021163546A9 WO2021163546A9 (en) | 2023-09-07 |
Family
ID=77292957
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2021/017937 WO2021163546A1 (en) | 2020-02-14 | 2021-02-12 | Methods and materials for assessing nucleic acids |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US20220073977A1 (en) |
| EP (1) | EP4103748A4 (en) |
| JP (1) | JP2023513606A (en) |
| KR (1) | KR20220154690A (en) |
| CN (1) | CN115516108A (en) |
| AU (1) | AU2021219794A1 (en) |
| BR (1) | BR112022015909A2 (en) |
| CA (1) | CA3170345A1 (en) |
| IL (1) | IL295297A (en) |
| MX (1) | MX2022010038A (en) |
| WO (1) | WO2021163546A1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2023150277A1 (en) * | 2022-02-03 | 2023-08-10 | The Johns Hopkins University | Methods for sequencing an immune cell receptor |
| WO2024073034A1 (en) * | 2022-09-29 | 2024-04-04 | The Board Of Trustees Of The Leland Stanford Junior University | Simplified sequencing library preparation for dna |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2024091545A1 (en) * | 2022-10-25 | 2024-05-02 | Cornell University | Nucleic acid error suppression |
| CN115928225A (en) * | 2023-01-03 | 2023-04-07 | 京东方科技集团股份有限公司 | Free DNA methylation library construction method, library, application and kit |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016181128A1 (en) * | 2015-05-11 | 2016-11-17 | Genefirst Ltd | Methods, compositions, and kits for preparing sequencing library |
| US20190218605A1 (en) * | 2014-03-03 | 2019-07-18 | Swift Biosciences, Inc. | Enhanced Adaptor Ligation |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| HUE059100T2 (en) * | 2017-01-27 | 2022-10-28 | Integrated Dna Tech Inc | Construction of next generation sequencing (ngs) libraries using competitive strand displacement |
| EP3601598B1 (en) * | 2017-03-23 | 2022-08-03 | University of Washington | Methods for targeted nucleic acid sequence enrichment with applications to error corrected nucleic acid sequencing |
-
2021
- 2021-02-12 AU AU2021219794A patent/AU2021219794A1/en active Pending
- 2021-02-12 KR KR1020227031690A patent/KR20220154690A/en unknown
- 2021-02-12 US US17/174,838 patent/US20220073977A1/en active Pending
- 2021-02-12 WO PCT/US2021/017937 patent/WO2021163546A1/en active Application Filing
- 2021-02-12 CA CA3170345A patent/CA3170345A1/en active Pending
- 2021-02-12 BR BR112022015909A patent/BR112022015909A2/en unknown
- 2021-02-12 IL IL295297A patent/IL295297A/en unknown
- 2021-02-12 CN CN202180028441.0A patent/CN115516108A/en active Pending
- 2021-02-12 MX MX2022010038A patent/MX2022010038A/en unknown
- 2021-02-12 JP JP2022549104A patent/JP2023513606A/en active Pending
- 2021-02-12 EP EP21753424.7A patent/EP4103748A4/en active Pending
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20190218605A1 (en) * | 2014-03-03 | 2019-07-18 | Swift Biosciences, Inc. | Enhanced Adaptor Ligation |
| WO2016181128A1 (en) * | 2015-05-11 | 2016-11-17 | Genefirst Ltd | Methods, compositions, and kits for preparing sequencing library |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP4103748A4 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2023150277A1 (en) * | 2022-02-03 | 2023-08-10 | The Johns Hopkins University | Methods for sequencing an immune cell receptor |
| WO2024073034A1 (en) * | 2022-09-29 | 2024-04-04 | The Board Of Trustees Of The Leland Stanford Junior University | Simplified sequencing library preparation for dna |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2021163546A9 (en) | 2023-09-07 |
| US20220073977A1 (en) | 2022-03-10 |
| AU2021219794A1 (en) | 2022-09-08 |
| JP2023513606A (en) | 2023-03-31 |
| CA3170345A1 (en) | 2021-08-19 |
| BR112022015909A2 (en) | 2022-10-04 |
| EP4103748A1 (en) | 2022-12-21 |
| CN115516108A (en) | 2022-12-23 |
| EP4103748A4 (en) | 2024-03-13 |
| IL295297A (en) | 2022-10-01 |
| MX2022010038A (en) | 2022-09-05 |
| KR20220154690A (en) | 2022-11-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7542672B2 (en) | Methods and compositions for analyzing nucleic acids | |
| CN110036118B (en) | Compositions and methods for identifying nucleic acid molecules | |
| CN110036117B (en) | Method for increasing throughput of single molecule sequencing by multiple short DNA fragments | |
| JP7008407B2 (en) | Methods for Identifying and Counting Methylation Changes in Nucleic Acid Sequences, Expressions, Copies, or DNA Using Combinations of nucleases, Ligses, Polymerases, and Sequencing Reactions | |
| JP6982087B2 (en) | Building a Next Generation Sequencing (NGS) Library Utilizing Competitive Chain Substitution | |
| JP7379418B2 (en) | Deep sequencing profiling of tumors | |
| CA3062174A1 (en) | Universal short adapters for indexing of polynucleotide samples | |
| US20180363039A1 (en) | Methods and compositions for forming ligation products | |
| US20220073977A1 (en) | Methods and materials for assessing nucleic acids | |
| WO2018195217A1 (en) | Compositions and methods for library construction and sequence analysis | |
| MX2013003349A (en) | Direct capture, amplification and sequencing of target dna using immobilized primers. | |
| EP4172357B1 (en) | Methods and compositions for analyzing nucleic acid | |
| CN114450420A (en) | Compositions and methods for accurate determination of oncology | |
| US20240352518A1 (en) | Methods for simultaneous mutation detection and methylation analysis | |
| US20220145368A1 (en) | Methods for noninvasive prenatal testing of fetal abnormalities | |
| WO2023150277A1 (en) | Methods for sequencing an immune cell receptor | |
| WO2024054517A1 (en) | Methods and compositions for analyzing nucleic acid | |
| AU2021281234A1 (en) | Isolation and immobilization of nucleic acids and uses thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 21753424 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 3170345 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2022549104 Country of ref document: JP Kind code of ref document: A |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112022015909 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 2021219794 Country of ref document: AU Date of ref document: 20210212 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202217052020 Country of ref document: IN |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2021753424 Country of ref document: EP Effective date: 20220914 |
|
| ENP | Entry into the national phase |
Ref document number: 112022015909 Country of ref document: BR Kind code of ref document: A2 Effective date: 20220811 |