WO2020131282A1 - Mitochondria-targeting peptides - Google Patents
Mitochondria-targeting peptides Download PDFInfo
- Publication number
- WO2020131282A1 WO2020131282A1 PCT/US2019/062283 US2019062283W WO2020131282A1 WO 2020131282 A1 WO2020131282 A1 WO 2020131282A1 US 2019062283 W US2019062283 W US 2019062283W WO 2020131282 A1 WO2020131282 A1 WO 2020131282A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- group
- amino
- mmol
- aaa
- Prior art date
Links
- 0 C*N[C@](CCCCN)C(C)=O Chemical compound C*N[C@](CCCCN)C(C)=O 0.000 description 49
- ZSYQVVKVKBVHIL-UHFFFAOYSA-N CC(C)(C)c(cc1)ccc1C#C Chemical compound CC(C)(C)c(cc1)ccc1C#C ZSYQVVKVKBVHIL-UHFFFAOYSA-N 0.000 description 1
- QKLZSVLQZHSHTD-XQUALCHDSA-N CC(C)CCC[C@@H](C(N[C@@H](Cc1c[nH]cn1)C(N)=O)=O)NC([C@@H](CCCNC(N)=N)NC([C@H](Cc1ccccc1)N)=O)=O Chemical compound CC(C)CCC[C@@H](C(N[C@@H](Cc1c[nH]cn1)C(N)=O)=O)NC([C@@H](CCCNC(N)=N)NC([C@H](Cc1ccccc1)N)=O)=O QKLZSVLQZHSHTD-XQUALCHDSA-N 0.000 description 1
- FAOBVCBRIJALME-JTQLQIEISA-N CC(C)N[C@@H](Cc1c[s]cn1)C(C)=O Chemical compound CC(C)N[C@@H](Cc1c[s]cn1)C(C)=O FAOBVCBRIJALME-JTQLQIEISA-N 0.000 description 1
- LEJWRHOBZIKTMP-ASDGIDEWSA-N CCCCCCCC[C@@H](C(N[C@@H](CCCCN)C(N)=O)=O)NC([C@@H](CCCNC(N)=N)NC([C@H](Cc1ccccc1)N)=O)=O Chemical compound CCCCCCCC[C@@H](C(N[C@@H](CCCCN)C(N)=O)=O)NC([C@@H](CCCNC(N)=N)NC([C@H](Cc1ccccc1)N)=O)=O LEJWRHOBZIKTMP-ASDGIDEWSA-N 0.000 description 1
- KSZVOXHGCKKOLL-UHFFFAOYSA-N Cc(cc1)ccc1C#C Chemical compound Cc(cc1)ccc1C#C KSZVOXHGCKKOLL-UHFFFAOYSA-N 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N Cc1c(C)cccc1 Chemical compound Cc1c(C)cccc1 CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- RNZKDWDZYMEWMS-BPXGVECKSA-N Cc1cc(C)c(C[C@@H](C(N[C@@H](CCCCN)C(N)=O)=O)NC([C@@H](CCCNC(N)=N)NC([C@H](Cc2ccccc2)N)=O)=O)c(C)c1 Chemical compound Cc1cc(C)c(C[C@@H](C(N[C@@H](CCCCN)C(N)=O)=O)NC([C@@H](CCCNC(N)=N)NC([C@H](Cc2ccccc2)N)=O)=O)c(C)c1 RNZKDWDZYMEWMS-BPXGVECKSA-N 0.000 description 1
- RVHRALONIUGARZ-KIHHCIJBSA-N NCCCC[C@@H](C(N)=O)NC([C@H](Cc1ccccc1)NC([C@@H](CCCNC(N)=N)NC([C@H](c1ccccc1)N)=O)=O)=O Chemical compound NCCCC[C@@H](C(N)=O)NC([C@H](Cc1ccccc1)NC([C@@H](CCCNC(N)=N)NC([C@H](c1ccccc1)N)=O)=O)=O RVHRALONIUGARZ-KIHHCIJBSA-N 0.000 description 1
- OGGJFDQRPVEQHK-BJESRGMDSA-N NCCCC[C@@H](C(N)=O)NC([C@H](c1ccccc1)NC([C@@H](CCCNC(N)=N)NC([C@H](c1ccccc1)N)=O)=O)=O Chemical compound NCCCC[C@@H](C(N)=O)NC([C@H](c1ccccc1)NC([C@@H](CCCNC(N)=N)NC([C@H](c1ccccc1)N)=O)=O)=O OGGJFDQRPVEQHK-BJESRGMDSA-N 0.000 description 1
- HHGOEJKHCFLLJW-NDBXHCKUSA-N N[C@@H](Cc1ccccc1)C(N[C@H](CCCNC(N)=N)C(N[C@@H](Cc1ccccc1)C(N[C@@H](Cc1c[nH]cn1)C(N)=O)=O)=O)=O Chemical compound N[C@@H](Cc1ccccc1)C(N[C@H](CCCNC(N)=N)C(N[C@@H](Cc1ccccc1)C(N[C@@H](Cc1c[nH]cn1)C(N)=O)=O)=O)=O HHGOEJKHCFLLJW-NDBXHCKUSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/10—Tetrapeptides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/10—Tetrapeptides
- C07K5/1002—Tetrapeptides with the first amino acid being neutral
- C07K5/1016—Tetrapeptides with the first amino acid being neutral and aromatic or cycloaliphatic
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/10—Tetrapeptides
- C07K5/1019—Tetrapeptides with the first amino acid being basic
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/10—Tetrapeptides
- C07K5/1024—Tetrapeptides with the first amino acid being heterocyclic
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/04—Linear peptides containing only normal peptide links
- C07K7/06—Linear peptides containing only normal peptide links having 5 to 11 amino acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
Definitions
- SBT-20 is a mitochondria-targeting peptide compound with therapeutic potential for treating diseases associated with mitochondrial dysfunction. Because of the potential therapeutic applications of SBT-20, there exists a need to develop analogs of the compound with an improved therapeutic profile.
- An aspect of the invention is an analog of SBT-20.
- the invention provides a compound of formula (I), or a pharmaceutically acceptable salt thereof:
- Aaa 1 is an amino acid residue selected from the group consisting of:
- R is an optionally substituted alkyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, aralkyl, or heteroaralkyl; and Ra and Rb are each independently selected from H, methyl, ethyl, propyl, cyclopropyl, cyclobutyl; or Ra and Rb taken together with the nitrogen atom to which they are attached form a four-, five- or six-membered heterocyclic ring;
- Aaa 2 is an amino acid residue selected from the group consisting of:
- Aaa 3 is an amino acid residue selected from the group consisting of:
- Aaa 4 is an amino acid residue selected from the group consisting of:
- R 1a and R 4d are each independently (C 1 -C 6 )alkyl
- R 2a , R 2b , R 2e , R 3a , R 3b , R 4a , R 4b , R 4c are each independently selected from the group consisting of H and (C1-C6)alkyl; and R a , R b , R 2c , and R 2d are each independently selected from the group consisting of H, (C 1 -C 6 )alkyl, C(O)((C 1 -C 6 )alkyl), C(O)((C 1 -C 6 )haloalkyl), C(O)O((C 1 -C 6 )alkyl), and
- Another aspect of the invention is a pharmaceutical composition, comprising a compound of the invention; and a pharmaceutically acceptable carrier.
- the invention also provides methods of treating or preventing ischemia-reperfusion injury, comprising administering to a subject in need thereof a therapeutically effective amount of a compound of the invention.
- the invention also provides methods of treating or preventing myocardial infarction, comprising administering to a subject in need thereof a therapeutically effective amount of a compound of the invention.
- Figure 1 depicts various amino acid residues useful in the present invention.
- Figure 2 depicts rat permeabilized cardiac fiber A/R. See Example 30.
- Figure 3 depticts white necrotic tissue area as % of Area at Risk (only area of the white necrotic tissue is used in the analysis) and Infarct Size (%), mean ⁇ SD. See Example 31.
- Figure 4 depicts dose response in rat Myocardial Infarction (MI) model, mean ⁇ SD. See Example 32.
- Figure 5 depicts Plasma Creatinine, % Protection, mean ⁇ SEM. See Example 33.
- Figure 6 depicts BUN, % Protection, mean ⁇ SEM. See Example 33. DETAILED DESCRIPTION OF THE INVENTION
- SBT-20 (Phe-D-Arg-Phe-Lys-NH 2 ) is a mitochondria-targeting compound with therapeutic potential for treating ischemia-reperfusion injury (e.g., cardiac ischemia- reperfusion injury), and myocardial infarction. Analogs of this compound may have improved therapeutic profiles, including improved metabolic properties, selectivity, or potency.
- the invention provides a compound of formula (I), or a pharmaceutically acceptable salt thereof:
- Aaa 1 is an amino acid residue selected from the group consisting of:
- Aaa 1 is wherein R is an optionally substituted alkyl, cycloalkyl, heterocyclyl, aryl, heteroaryl, aralkyl, or heteroaralkyl; and Ra and Rb are each independently selected from H, methyl, ethyl, propyl, cyclopropyl, cyclobutyl; or Ra and Rb taken together with the nitrogen atom to which they are attached form a four-, five- or six-membered heterocyclic ring;
- Aaa 2 is an amino acid residue selected from the group consisting of:
- Aaa is an amino acid residue selected from the group consisting of:
- Aaa 4 is an amino acid residue selected from the group consisting of:
- R 1a and R 4d are each independently (C 1 -C 6 )alkyl
- R 2a , R 2b , R 2e , R 3a , R 3b , R 4a , R 4b , R 4c are each independently selected from the group consisting of H and (C1-C6)alkyl;
- R a , R b , R 2c , and R 2d are each independently selected from the group consisting of H, (C1-C6)alkyl, C(O)((C1-C6)alkyl), C(O)((C1-C6)haloalkyl), C(O)O((C1-C6)alkyl), and C(O)O(aryl(C1-C6)alkyl); and provided that the compound of formula (I) is not
- Aaa 1 is selected from the group consisting of:
- Aaa 1 is an amino acid residue selected from the group consisting of:
- Aaa 2 is an amino acid residue selected from the group consisting of:
- Aaa 2 is
- Aaa 3 is selected from the group consisting of:
- Aaa is selected from the group consisting of:
- Aaa 3 is selected from the group consisting of:
- Aaa 4 is selected from the group consisting of:
- Aaa 4 is selected from the group consisting of:
- Aaa 4 is selected from the group consisting of:
- R a and R b are each independently H or methyl, preferably H.
- the compound of formula (I) is selected from the following table:
- the peptidic compounds of the invention may be prepared using a peptide synthesis method, such as conventional liquid-phase peptide synthesis or solid-phase peptide synthesis, or by peptide synthesis by means of an automated peptide synthesizer (Kelley et al., Genetics Engineering Principles and Methods, Setlow, J. K. eds., Plenum Press NY. (1990) Vol. 12, pp.l to 19; Stewart et al., Solid-Phase Peptide Synthesis (1989) W. H.; Houghten, Proc. Natl. Acad. Sci. USA (1985) 82: p.5132).
- a peptide synthesis method such as conventional liquid-phase peptide synthesis or solid-phase peptide synthesis, or by peptide synthesis by means of an automated peptide synthesizer (Kelley et al., Genetics Engineering Principles and Methods, Setlow, J. K. eds., Plenum Press NY. (1990
- the peptide thus produced can be collected or purified by a routine method, for example, chromatography, such as gel filtration chromatography, ion exchange column chromatography, affinity chromatography, reverse phase column chromatography, and HPLC, ammonium sulfate fractionation, ultrafiltration, and
- peptides are typically synthesized from the carbonyl group side (C -terminus) to amino group side (N-terminus) of the amino acid chain.
- an amino-protected amino acid is covalently bound to a solid support material through the carboxyl group of the amino acid, typically via an ester or amido bond and optionally via a linking group.
- the amino group may be deprotected and reacted with (i.e.,“coupled” with) the carbonyl group of a second amino-protected amino acid using a coupling reagent, yielding a dipeptide bound to a solid support.
- the protecting groups used on the amino groups of the amino acid residues include 9-fluorenylmethyloxycarbonyl group (Fmoc) and t-butyloxycarbonyl (Boc).
- Fmoc 9-fluorenylmethyloxycarbonyl group
- Boc t-butyloxycarbonyl
- the amino protecting group may be formyl, acrylyl (Acr), benzoyl (Bz), acetyl (Ac), trifluoroacetyl, substituted or unsubstituted groups of aralkyloxycarbonyl type, such as the benzyloxycarbonyl (Z), p- chlorobenzyloxycarbonyl, p-bromobenzyloxycarbonyl, p-nitrobenzyloxycarbonyl, p- methoxybenzyloxycarbonyl, benzhydryloxycarbonyl, 2(p- biphenylyl)isopropyloxycarbonyl, 2-(3,5-dimethoxyphenyl)isopropyloxycarbonyl, p-phenylazobenzyloxycarbonyl,
- aralkyloxycarbonyl type such as the benzyloxycarbonyl (Z), p- chlorobenzyloxycarbonyl, p-bromobenzyloxycarbonyl
- triphenylphosphonoethyloxycarbonyl or 9-fluorenylmethyloxycarbonyl group Fmoc
- substituted or unsubstituted groups of alkyloxycarbonyl type such as the tert- butyloxycarbonyl (BOC), tert-amyloxycarbonyl, diisopropylmethyloxycarbonyl,
- methylsulphonylethyloxycarbonyl or 2,2,2-trichloroethyloxycarbonyl group groups of cycloalkyloxycarbonyl type, such as the cyclopentyloxycarbonyl, cyclohexyloxycarbonyl, adamantyloxycarbonyl or isobornyloxycarbonyl group, and groups containing a hetero atom, such as the benzenesulphonyl, p-toluenesulphonyl, mesitylenesulphonyl,
- amino acids bear reactive functional groups in the side chain.
- such functional groups are protected in order to prevent the functional groups from reacting with the incoming amino acid.
- the protecting groups used with these functional groups must be stable to the conditions of peptide synthesis, but may be removed before, after, or concomitantly with cleavage of the peptide from the solid support.
- the solid support material used in the solid-phase peptide synthesis method is a gel-type support such as polystyrene, polyacrylamide, or polyethylene glycol.
- materials such as pore glass, cellulose fibers, or polystyrene may be functionalized at their surface to provide a solid support for peptide synthesis.
- Coupling reagents that may be used in the solid-phase peptide synthesis described herein are typically carbodiimide reagents.
- carbodiimide reagents include, but are not limited to, N,N’-dicyclohexylcarbodiimide (DCC), 1-(3-dimethylaminopropyl)-3- ethylcarbodiimide (EDC), N-cyclohexyl-N’-isopropylcarbodiimide (CIC), N,N’- diisopropylcarbodiimide (DIC), N-tert-butyl-N’-methylcarbodiimide (BMC), N-tert-butyl- N’-ethylcarbodiimide (BEC), bis[[4-(2,2-dimethyl-1,3-dioxolyl)]-methyl]carbodiimide (BDDC), and N,N-dicyclopentylcarbodiimide.
- a compound of the invention for example, the compound pictured below, is synthesized in a linear sequential fashion, according to the solid phase synthesis depicted in Scheme 1:
- a compound of the invention may be synthesized in a convergent fashion, for example, according to Scheme 2:
- the compounds of the invention may also be synthesized according to conventional liquid-phase peptide synthetic routes.
- the compound pictured below may be synthesized in a convergent liquid-phase synthesis, as depicted in Scheme 3.
- a compound of the invention is made via the linear sequential liquid phase synthesis depicted in Scheme 4.
- amino acid includes both a naturally occurring amino acid and a non-natural amino acid.
- amino acid includes both isolated amino acid molecules (i.e., molecules that include both, an amino-attached hydrogen and a carbonyl carbon-attached hydroxyl) and residues of amino acids (i.e., molecules in which either one or both an amino-attached hydrogen or a carbonyl carbon- attached hydroxyl are removed).
- the amino group can be alpha-amino group, beta-amino group, etc.
- amino acid alanine can refer either to an isolated alanine H-Ala-OH or to any one of the alanine residues H-Ala-, -Ala-OH, or -Ala-.
- all amino acids found in the compounds described herein can be either in D or L configuration.
- An amino acid that is in D configuration may be written such that“D” precedes the amino acid abbreviation.
- “D-Arg” represents arginine in the D configuration.
- the term“amino acid” includes salts thereof, including pharmaceutically acceptable salts. Any amino acid can be protected or unprotected.
- Protecting groups can be attached to an amino group (for example alpha-amino group), the backbone carboxyl group, or any functionality of the side chain.
- an amino group for example alpha-amino group
- the backbone carboxyl group or any functionality of the side chain.
- phenylalanine protected by a benzyloxycarbonyl group (Z) on the alpha-amino group would be represented as Z-Phe-OH.
- N-terminal amino acid all abbreviations of amino acids (for example, Phe) in this disclosure stand for the structure of—NH—C(R)(R ⁇ )—CO—, wherein R and R ⁇ each is, independently, hydrogen or the side chain of an amino acid (e.g., R ⁇ benzyl and R ⁇ H for Phe). Accordingly, phenylalanine is H-Phe-OH.
- OH OH
- peptides e.g., Lys-Val-Leu-OH
- certain R and R’, separately, or in combination as a ring structure, can include functional groups that require protection during the liquid phase synthesis.
- amino acid has isomeric forms, it is the L form of the amino acid that is represented unless otherwise explicitly indicated as D form, for example, D-Arg.
- D form for example, D-Arg.
- many amino acid residues are commercially available in both D- and L-form.
- D-Arg is a commercially available D-amino acid.
- a capital letter“D” used in conjunction with an abbreviation for an amino acid residue refers to the D-form of the amino acid residue.
- the term“peptide” refers to two or more amino acids covalently linked by at least one amide bond (i.e., a bond between an amino group of one amino acid and a carboxyl group of another amino acid selected from the amino acids of the peptide fragment).
- the term“peptide” includes salts thereof, including pharmaceutically acceptable salts.
- alkyl as used herein is a term of art and refers to saturated aliphatic groups, including straight-chain alkyl groups, branched-chain alkyl groups, cycloalkyl (alicyclic) groups, alkyl substituted cycloalkyl groups, and cycloalkyl substituted alkyl groups.
- a straight-chain or branched-chain alkyl has about 30 or fewer carbon atoms in its backbone (e.g., C 1 -C 30 for straight chain, C 3 -C 30 for branched chain), and alternatively, about 20 or fewer, 10 or fewer (i.e., C1-C10), or 6 or fewer (i.e., C1- C 6 ).
- alkyl include, but are not limited to, methyl, ethyl, n-propyl, iso-propyl, n-butyl, sec-butyl, iso-butyl, tert-butyl, n-pentyl, isopentyl, neopentyl, and n- hexyl.
- aryl refers to a radical of a monocyclic or polycyclic (e.g., bicyclic or tricyclic) 4n+2 aromatic ring system (e.g., having 6, 10, or 14 ⁇ electrons shared in a cyclic array) having 6-14 ring carbon atoms and zero heteroatoms provided in the aromatic ring system ("C6-C14 aryl”).
- an aryl group has six ring carbon atoms (“C6 aryl”; e.g., phenyl).
- an aryl group has ten ring carbon atoms ("C 10 aryl”; e.g., naphthyl such as 1-naphthyl and 2- naphthyl). In some embodiments, an aryl group has fourteen ring carbon atoms ("C 14 aryl”; e.g., anthracyl).
- An aryl group may be described as, e.g., a C6-C10-membered aryl, wherein the term “membered” refers to the non-hydrogen ring atoms within the moiety.
- Aryl groups include phenyl, naphthyl, indenyl, and tetrahydronaphthyl.
- Each instance of an aryl group may be independently optionally substituted, i.e., unsubstituted (an "unsubstituted aryl") or substituted (a "substituted aryl") with one or more substituents; e.g., for instance from 1 to 5 substituents, 1 to 4 substituents, 1 to 3 substituents, 1 to 2 substituents or just 1 substituent.
- the aromatic ring may be substituted at one or more ring positions with one or more substituents, such as halogen, azide, alkyl, aralkyl, alkenyl, alkynyl, cycloalkyl, hydroxyl, alkoxyl, amino, nitro, sulfhydryl, imino, amido, phosphonate, phosphinate, carbonyl, carboxyl, silyl, ether, alkylthio, sulfonyl, sulfonamido, ketone, aldehyde, ester, heterocyclyl, aromatic or heteroaromatic moieties, fluoroalkyl (such as trifluromethyl), cyano, or the like.
- substituents such as halogen, azide, alkyl, aralkyl, alkenyl, alkynyl, cycloalkyl, hydroxyl, alkoxyl, amino, nitro, sulfhydryl, im
- the aryl group can be an unsubstituted C 5 -C 12 aryl and in certain embodiments, the aryl group can be a substituted C 5 -C 10 aryl.
- heteroaryl refers to a radical of an aromatic heterocycle that comprises 1, 2, 3 or 4 heteroatoms selected, independently of the others, from nitrogen, sulfur and oxygen.
- heteroaryl refers to a group that may be substituted or unsubstituted.
- a heteroaryl may be fused to one or two rings, such as a cycloalkyl, an aryl, or a second heteroaryl ring.
- heteroaryl The point of attachment of a heteroaryl to a molecule may be on the heteroaryl, cycloalkyl, heterocycloalkyl or aryl ring, and the heteroaryl group may be attached through carbon or a heteroatom.
- heteroaryl groups include imidazolyl, furyl, pyrrolyl, thienyl, thiazolyl, isoxazolyl, isothiazolyl, thiadiazolyl, oxadiazolyl, pyridinyl, pyrimidyl, pyrazinyl, pyridazinyl, quinolyl, isoquinolinyl, indazolyl, benzoxazolyl, benzisooxazolyl, benzofuryl, benzothiazolyl, indolizinyl, imidazopyridinyl, pyrazolyl, triazolyl, oxazolyl, tetrazolyl, benzimidazolyl
- the aromatic heterocycle may be substituted at one or more ring positions with one or more substituents, such as halogen, azide, alkyl, aralkyl, alkenyl, alkynyl, cycloalkyl, hydroxyl, alkoxyl, amino, nitro, sulfhydryl, imino, amido, phosphonate, phosphinate, carbonyl, carboxyl, silyl, ether, alkylthio, sulfonyl, sulfonamido, ketone, aldehyde, ester, heterocyclyl, aromatic or heteroaromatic moieties, fluoroalkyl (such as trifluromethyl), cyano, or the like.
- substituents such as halogen, azide, alkyl, aralkyl, alkenyl, alkynyl, cycloalkyl, hydroxyl, alkoxyl, amino, nitro, sulfhydryl, im
- heterocyclyl refers to a radical of a non-aromatic ring system, including, but not limited to, monocyclic, bicyclic, and tricyclic rings, which can be completely saturated or which can contain one or more units of unsaturation— for the avoidance of doubt, the degree of unsaturation does not result in an aromatic ring system— and having 3 to 12 atoms including at least one heteroatom, such as nitrogen, oxygen, or sulfur.
- heterocyclyl rings aziridinyl, azirinyl, oxiranyl, thiiranyl, thiirenyl, dioxiranyl, diazirinyl, azetyl, oxetanyl, oxetyl, thietanyl, thietyl, diazetidinyl, dioxetanyl, dioxetenyl, dithietanyl, dithietyl, furyl, dioxalanyl, pyrrolyl, oxazolyl, thiazolyl, imidazolyl, oxadiazolyl, thiadiazolyl, triazolyl, triazinyl, isothiazolyl, isoxazolyl, thiophenyl, pyrazolyl, tetrazolyl, pyridyl
- heterocyclic ring or“heterocycle” refers to a ring of atoms of at least two different elements, one of which is carbon. Additional reference is made to: Oxford Dictionary of Biochemistry and Molecular Biology, Oxford University Press, Oxford, 1997 as evidence that the term“heterocyclic ring” is a term well-established in field of organic chemistry.
- arylalkyl or“aralkyl” refers to a radical of an aryl or heteroaryl group (“heteroaralkyl”) that is attached to a (Ci-Ci2)alkyl group via an alkylene linker.
- heteroarylkyl refers to a radical of an aryl or heteroaryl group that is attached to a (Ci-Ci2)alkyl group via an alkylene linker.
- arylalkyl refers to a group that may be substituted or unsubstituted.
- arylalkyl is also intended to refer to those compounds wherein one or more methylene groups in the alkyl chain of the arylalkyl group can be replaced by a heteroatom such as O, N, P, Si, and S, and wherein the nitrogen, phosphorus and sulfur atoms may optionally be oxidized and the nitrogen heteroatom may optionally be quaternized with appended alkyl and/or aryl groups.
- Arylalkyl groups include for example, benzyl.
- cycloalkyl refers to a radical of a non-aromatic cyclic hydrocarbon group having from 3 to 12 ring carbon atoms (“C3-C12 cycloalkyl").
- a cycloalkyl group has 3 to 10 ring carbon atoms ("C3-C10 cycloalkyl”). In some embodiments, a cycloalkyl group has 3 to 8 ring carbon atoms ("C 3 -C 8 cycloalkyl”). In some embodiments, a cycloalkyl group has 3 to 6 ring carbon atoms ("C3-C6 cycloalkyl”). In some embodiments, a cycloalkyl group has 5 to 7 ring carbon atoms (“C5-C7 cycloalkyl").
- a cycloalkyl group maybe described as, e.g., a C 4 -C 7 -membered cycloalkyl, wherein the term “membered” refers to the non-hydrogen ring atoms within the moiety.
- Exemplary C 3 -C 6 cycloalkyl groups include, without limitation, cyclopropyl (C3), cyclopropenyl (C3), cyclobutyl (C4), cyclobutenyl (C 4 ), cyclopentyl (C 5 ), cyclopentenyl (C 5 ), cyclohexyl (C 6 ), cyclohexenyl (C 6 ), cyclohexadienyl (C 6 ), and the like.
- Exemplary C 3 -C 7 cycloalkyl groups include, without limitation, the aforementioned C3-C5 cycloalkyl groups as well as cycloheptyl (C6), cycloheptenyl (C7), cycloheptadienyl (C7), and cycloheptatrienyl (C7), bicyclo[2.1.1]hexanyl (C 6 ), bicyclo[3.1.1 ]heptanyl (C 7 ), and the like.
- Exemplary C 3 -C 10 cycloalkyl groups include, without limitation, the aforementioned C3-C7 cycloalkyl groups as well as cyclononyl (C9), cyclononenyl (C9), cyclodecyl (C10), cyclodecenyl (C10), octahydro-1 H-indenyl (C9), decahydronaphthalenyl (C 10 ), spiro[4.5]decanyl (C 10 ), and the like.
- the cycloalkyl group is either monocyclic (“monocyclic cycloalkyl”) or contain a fused, bridged or spiro ring system such as a bicyclic system (“biscyclic cycloalkyl”) and can be saturated or can be partially unsaturated.
- monocyclic cycloalkyl or contain a fused, bridged or spiro ring system such as a bicyclic system (“biscyclic cycloalkyl”) and can be saturated or can be partially unsaturated.
- biscyclic cycloalkyl groups include 1-ethylbicyclo[1.1.1]pentane, 1- ethylbicyclo[2.2.2]octane and (3r,5r,7r)-1-ethyladamantane.
- Cycloalkyl also includes ring systems wherein the cycloalkyl ring, as defined above, is fused with one or more aryl groups wherein the point of attachment is on the cycloalkyl ring, and in such instances, the number of carbons continue to designate the number of carbons in the cycloalkyl ring system.
- Each instance of a cycloalkyl group may be independently optionally substituted, i.e., unsubstituted (an "unsubstituted cycloalkyl") or substituted (a "substituted cycloalkyl") with one or more substituents.
- the invention also provides salts of the compounds of the invention.
- salts derived from inorganic or organic acids including, for example, hydrochloric, hydrobromic, sulfuric, nitric, perchloric, phosphoric, formic, acetic, lactic, maleic, fumaric, succinic, tartaric, glycolic, salicylic, citric, methanesulfonic, benzenesulfonic, benzoic, malonic, trifluoroacetic, trichloroacetic, naphthalene-2-sulfonic, and other acids.
- Pharmaceutically acceptable salt forms can include forms wherein the ratio of molecules comprising the salt is not 1:1.
- the salt may comprise more than one inorganic or organic acid molecule per molecule of base, such as two hydrochloric acid molecules per molecule of compound.
- the salt may comprise less than one inorganic or organic acid molecule per molecule of base, such as two molecules of compound per molecule of tartaric acid.
- carrier and“pharmaceutically acceptable carrier” as used herein refer to a diluent, adjuvant, excipient, or vehicle with which a compound is administered or formulated for administration.
- pharmaceutically acceptable carriers include liquids, such as water, saline, and oils; and solids, such as gum acacia, gelatin, starch paste, talc, keratin, colloidal silica, urea, and the like.
- auxiliary, stabilizing, thickening, lubricating, flavoring, and coloring agents may be used.
- suitable pharmaceutical carriers are described in Remington’s Pharmaceutical Sciences by E.W. Martin, herein incorporated by reference in its entirety.
- inhibit or inhibiting means reduce by an objectively measureable amount or degree compared to control. In one embodiment, inhibit or inhibiting means reduce by at least a statistically significant amount compared to control. In one embodiment, inhibit or inhibiting means reduce by at least 5 percent compared to control. In various individual embodiments, inhibit or inhibiting means reduce by at least 10, 15, 20, 25, 30, 33, 40, 50, 60, 67, 70, 75, 80, 90, 95, or 99 percent compared to control.
- the terms“treating” and“treat” refer to performing an intervention that results in (a) preventing a condition or disease from occurring in a subject that may be at risk of developing or predisposed to having the condition or disease but has not yet been diagnosed as having it; (b) inhibiting a condition or disease, e.g., slowing or arresting its development or progression; or (c) relieving or ameliorating a condition or disease, e.g., causing regression of the condition or disease.
- the terms“treating” and “treat” refer to performing an intervention that results in (a) inhibiting a condition or disease, e.g., slowing or arresting its development; or (b) relieving or ameliorating a condition or disease, e.g., causing regression of the condition or disease.
- a“subject” refers to a living animal.
- a subject is a mammal.
- a subject is a non-human mammal, including, without limitation, a mouse, rat, hamster, guinea pig, rabbit, sheep, goat, cat, dog, pig, horse, cow, or non-human primate.
- the subject is a human.
- administering has its usual meaning and encompasses administering by any suitable route of administration, including, without limitation, intravenous, intramuscular, intraperitoneal, subcutaneous, direct injection, mucosal, inhalation, oral, and topical.
- the phrase“effective amount” refers to any amount that is sufficient to achieve a desired biological effect.
- A“therapeutically effective amount” is an amount that is sufficient to achieve a desired therapeutic effect, e.g., to treat ischemia-reperfusion injury.
- Compounds of the invention and the salts thereof can be combined with other therapeutic agents.
- the compounds of the invention and other therapeutic agent may be administered simultaneously or sequentially.
- the other therapeutic agents When the other therapeutic agents are administered simultaneously, they can be administered in the same or separate formulations, but they are administered substantially at the same time.
- the other therapeutic agents are administered sequentially with one another and with compounds of the invention, when the administration of the other therapeutic agents and the compound of the invention is temporally separated. The separation in time between the administration of these compounds may be a matter of minutes or it may be longer.
- the invention is directed to a pharmaceutical composition, comprising a compound of the invention and a pharmaceutically acceptable carrier.
- the pharmaceutical composition comprises a plurality of compounds of the invention and a pharmaceutically acceptable carrier.
- a pharmaceutical composition of the invention further comprises at least one additional pharmaceutically active agent other than a compound of the invention.
- the at least one additional pharmaceutically active agent can be an agent useful in the treatment of ischemia-reperfusion injury.
- Pharmaceutical compositions of the invention can be prepared by combining one or more compounds of the invention with a pharmaceutically acceptable carrier and, optionally, one or more additional pharmaceutically active agents.
- an“effective amount” refers to any amount that is sufficient to achieve a desired biological effect.
- an effective prophylactic or therapeutic treatment regimen can be planned which does not cause substantial unwanted toxicity and yet is effective to treat the particular subject.
- the effective amount for any particular application can vary depending on such factors as the disease or condition being treated, the particular compound of the invention being administered, the size of the subject, or the severity of the disease or condition.
- One of ordinary skill in the art can empirically determine the effective amount of a particular compound of the invention and/or other therapeutic agent without necessitating undue experimentation.
- a maximum dose may be used, that is, the highest safe dose according to some medical judgment.
- Multiple doses per day may be contemplated to achieve appropriate systemic levels of compounds. Appropriate systemic levels can be determined by, for example, measurement of the patient’s peak or sustained plasma level of the drug.“Dose” and“dosage” are used interchangeably herein.
- intravenous administration of a compound may typically be from 0.1 mg/kg/day to 20 mg/kg/day. In one embodiment, intravenous administration of a compound may typically be from 0.1 mg/kg/day to 2 mg/kg/day. In one embodiment, intravenous administration of a compound may typically be from 0.5 mg/kg/day to 5 mg/kg/day. In one embodiment, intravenous administration of a compound may typically be from 1 mg/kg/day to 20 mg/kg/day. In one embodiment, intravenous administration of a compound may typically be from 1 mg/kg/day to 10 mg/kg/day.
- daily oral doses of a compound will be, for human subjects, from about 0.01 milligrams/kg per day to 1000 milligrams/kg per day. It is expected that oral doses in the range of 0.5 to 50 milligrams/kg, in one or more administrations per day, will yield therapeutic results. Dosage may be adjusted appropriately to achieve desired drug levels, local or systemic, depending upon the mode of administration. For example, it is expected that intravenous administration would be from one order to several orders of magnitude lower dose per day. In the event that the response in a subject is insufficient at such doses, even higher doses (or effective higher doses by a different, more localized delivery route) may be employed to the extent that patient tolerance permits. Multiple doses per day are
- the therapeutically effective amount can be initially determined from animal models.
- a therapeutically effective dose can also be determined from human data for compounds which have been tested in humans and for compounds which are known to exhibit similar pharmacological activities, such as other related active agents. Higher doses may be required for parenteral administration.
- the applied dose can be adjusted based on the relative bioavailability and potency of the administered compound. Adjusting the dose to achieve maximal efficacy based on the methods described above and other methods as are well-known in the art is well within the capabilities of the ordinarily skilled artisan.
- compositions of the invention can be administered in pharmaceutically acceptable solutions, which may routinely contain pharmaceutically acceptable concentrations of salt, buffering agents, preservatives, compatible carriers, adjuvants, and optionally other therapeutic ingredients.
- an effective amount of the compound can be administered to a subject by any mode that delivers the compound to the desired surface.
- Administering a pharmaceutical composition may be accomplished by any means known to the skilled artisan. Routes of administration include but are not limited to intravenous, intramuscular, intraperitoneal, intravesical (urinary bladder), oral, subcutaneous, direct injection (for example, into a tumor or abscess), mucosal (e.g., topical to eye), inhalation, and topical.
- a compound of the invention can be formulated as a lyophilized preparation, as a lyophilized preparation of liposome-intercalated or -encapsulated active compound, as a lipid complex in aqueous suspension, or as a salt complex.
- Lyophilized formulations are generally reconstituted in suitable aqueous solution, e.g., in sterile water or saline, shortly prior to administration.
- the compounds can be formulated readily by combining the active compound(s) with pharmaceutically acceptable carriers well known in the art.
- Such carriers enable the compounds of the invention to be formulated as tablets, pills, dragees, capsules, liquids, gels, syrups, slurries, suspensions and the like, for oral ingestion by a subject to be treated.
- Pharmaceutical preparations for oral use can be obtained as solid excipient, optionally grinding a resulting mixture, and processing the mixture of granules, after adding suitable auxiliaries, if desired, to obtain tablets or dragee cores.
- Suitable excipients are, in particular, fillers such as sugars, including lactose, sucrose, mannitol, or sorbitol; cellulose preparations such as, for example, maize starch, wheat starch, rice starch, potato starch, gelatin, gum tragacanth, methyl cellulose, hydroxypropylmethyl-cellulose, sodium carboxymethylcellulose, and/or polyvinylpyrrolidone (PVP).
- fillers such as sugars, including lactose, sucrose, mannitol, or sorbitol
- cellulose preparations such as, for example, maize starch, wheat starch, rice starch, potato starch, gelatin, gum tragacanth, methyl cellulose, hydroxypropylmethyl-cellulose, sodium carboxymethylcellulose, and/or polyvinylpyrrolidone (PVP).
- PVP polyvinylpyrrolidone
- disintegrating agents may be added, such as the cross-linked polyvinyl pyrrolidone, agar, or alginic acid or a salt thereof such as sodium alginate.
- the oral formulations may also be formulated in saline or buffers, e.g., EDTA for neutralizing internal acid conditions or may be administered without any carriers.
- oral dosage forms of the above component or components may be chemically modified so that oral delivery of the derivative is efficacious.
- the chemical modification contemplated is the attachment of at least one moiety to the component molecule itself, where said moiety permits (a) inhibition of acid hydrolysis; and (b) uptake into the blood stream from the stomach or intestine.
- the increase in overall stability of the component or components and increase in circulation time in the body examples include: polyethylene glycol, copolymers of ethylene glycol and propylene glycol, carboxymethyl cellulose, dextran, polyvinyl alcohol, polyvinyl pyrrolidone and polyproline.
- the location of release may be the stomach, the small intestine (the duodenum, the jejunum, or the ileum), or the large intestine.
- the stomach the small intestine (the duodenum, the jejunum, or the ileum), or the large intestine.
- One skilled in the art has available formulations which will not dissolve in the stomach, yet will release the material in the duodenum or elsewhere in the intestine.
- the release will avoid the deleterious effects of the stomach environment, either by protection of the compound of the invention (or derivative) or by release of the biologically active material beyond the stomach environment, such as in the intestine.
- a coating impermeable to at least pH 5.0 is essential.
- examples of the more common inert ingredients that are used as enteric coatings are cellulose acetate trimellitate (CAT), hydroxypropylmethylcellulose phthalate (HPMCP), HPMCP 50, HPMCP 55, polyvinyl acetate phthalate (PVAP), Eudragit L30D, Aquateric, cellulose acetate phthalate (CAP), Eudragit L, Eudragit S, and shellac. These coatings may be used as mixed films.
- a coating or mixture of coatings can also be used on tablets, which are not intended for protection against the stomach. This can include sugar coatings, or coatings which make the tablet easier to swallow.
- Capsules may consist of a hard shell (such as gelatin) for delivery of dry therapeutic (e.g., powder); for liquid forms, a soft gelatin shell may be used.
- the shell material of cachets could be thick starch or other edible paper. For pills, lozenges, molded tablets or tablet triturates, moist massing techniques can be used.
- the therapeutic can be included in the formulation as fine multi-particulates in the form of granules or pellets of particle size about 1 mm.
- the formulation of the material for capsule administration could also be as a powder, lightly compressed plugs or even as tablets.
- the therapeutic could be prepared by compression.
- Colorants and flavoring agents may all be included.
- the compound of the invention (or derivative) may be formulated (such as by liposome or microsphere encapsulation) and then further contained within an edible product, such as a refrigerated beverage containing colorants and flavoring agents.
- diluents could include carbohydrates, especially mannitol, a-lactose, anhydrous lactose, cellulose, sucrose, modified dextrans and starch. Certain inorganic salts may be also be used as fillers including calcium triphosphate, magnesium carbonate and sodium chloride. Some commercially available diluents are Fast-Flo, Emdex, STA-Rx 1500, Emcompress and Avicell.
- Disintegrants may be included in the formulation of the therapeutic into a solid dosage form.
- Materials used as disintegrates include but are not limited to starch, including the commercial disintegrant based on starch, Explotab. Sodium starch glycolate, Amberlite, sodium carboxymethylcellulose, ultramylopectin, sodium alginate, gelatin, orange peel, acid carboxymethyl cellulose, natural sponge and bentonite may all be used.
- Another form of the disintegrants are the insoluble cationic exchange resins.
- Powdered gums may be used as disintegrants and as binders and these can include powdered gums such as agar, Karaya or tragacanth. Alginic acid and its sodium salt are also useful as disintegrants.
- Binders may be used to hold the therapeutic agent together to form a hard tablet and include materials from natural products such as acacia, tragacanth, starch and gelatin. Others include methyl cellulose (MC), ethyl cellulose (EC) and carboxymethyl cellulose (CMC). Polyvinyl pyrrolidone (PVP) and hydroxypropylmethyl cellulose (HPMC) could both be used in alcoholic solutions to granulate the therapeutic.
- MC methyl cellulose
- EC ethyl cellulose
- CMC carboxymethyl cellulose
- PVP polyvinyl pyrrolidone
- HPMC hydroxypropylmethyl cellulose
- Lubricants may be used as a layer between the therapeutic and the die wall, and these can include but are not limited to; stearic acid including its magnesium and calcium salts, polytetrafluoroethylene (PTFE), liquid paraffin, vegetable oils and waxes. Soluble lubricants may also be used such as sodium lauryl sulfate, magnesium lauryl sulfate, polyethylene glycol of various molecular weights, Carbowax 4000 and 6000.
- the glidants may include starch, talc, pyrogenic silica and hydrated silicoaluminate.
- Surfactants may include anionic detergents such as sodium lauryl sulfate, dioctyl sodium sulfosuccinate and dioctyl sodium sulfonate.
- anionic detergents such as sodium lauryl sulfate, dioctyl sodium sulfosuccinate and dioctyl sodium sulfonate.
- Cationic detergents which can be used and can include benzalkonium chloride and benzethonium chloride.
- Non-ionic detergents that could be included in the formulation as surfactants include lauromacrogol 400, polyoxyl 40 stearate, polyoxyethylene hydrogenated castor oil 10, 50 and 60, glycerol monostearate, polysorbate 40, 60, 65 and 80, sucrose fatty acid ester, methyl cellulose and carboxymethyl cellulose. These surfactants could be present in the formulation of the compound of the invention or derivative either alone or as a mixture in different ratios.
- compositions which can be used orally include push-fit capsules made of gelatin, as well as soft, sealed capsules made of gelatin and a plasticizer, such as glycerol or sorbitol.
- the push-fit capsules can contain the active ingredients in admixture with filler such as lactose, binders such as starches, and/or lubricants such as talc or magnesium stearate and, optionally, stabilizers.
- the active compounds may be dissolved or suspended in suitable liquids, such as fatty oils, liquid paraffin, or liquid polyethylene glycols.
- stabilizers may be added.
- Microspheres formulated for oral administration may also be used. Such microspheres have been well defined in the art. All formulations for oral administration should be in dosages suitable for such administration.
- compositions may take the form of tablets or lozenges formulated in conventional manner.
- the compound may be formulated as solutions, gels, ointments, creams, suspensions, etc. as are well-known in the art.
- Systemic formulations include those designed for administration by injection, e.g., subcutaneous, intravenous, intramuscular, intrathecal or intraperitoneal injection, as well as those designed for transdermal, transmucosal oral or pulmonary administration.
- compounds for use according to the present invention may be conveniently delivered in the form of an aerosol spray presentation from pressurized packs or a nebulizer, with the use of a suitable propellant, e.g.,
- the dosage unit may be determined by providing a valve to deliver a metered amount.
- Capsules and cartridges of e.g., gelatin for use in an inhaler or insufflator may be formulated containing a powder mix of the compound and a suitable powder base such as lactose or starch.
- the compound is delivered to the lungs of a mammal while inhaling and traverses across the lung epithelial lining to the blood stream.
- Other reports of inhaled molecules include Adjei et al., Pharm Res 7:565-569 (1990); Adjei et al., Int J Pharmaceutics 63:135-144 (1990) (leuprolide acetate); Braquet et al., J Cardiovasc Pharmacol 13(suppl.
- Contemplated for use in the practice of this invention are a wide range of mechanical devices designed for pulmonary delivery of therapeutic products, including but not limited to nebulizers, metered dose inhalers, and powder inhalers, all of which are familiar to those skilled in the art.
- Ultravent nebulizer manufactured by Mallinckrodt, Inc., St. Louis,
- each formulation is specific to the type of device employed and may involve the use of an appropriate propellant material, in addition to the usual diluents, adjuvants and/or carriers useful in therapy. Also, the use of liposomes, microcapsules or microspheres, inclusion complexes, or other types of carriers is
- Chemically modified compound of the invention may also be prepared in different formulations depending on the type of chemical modification or the type of device employed.
- Formulations suitable for use with a nebulizer will typically comprise a compound of the invention (or derivative) dissolved in water at a concentration of about 0.1 to 25 mg of biologically active compound of the invention per mL of solution.
- the formulation may also include a buffer and a simple sugar (e.g., for inhibitor stabilization and regulation of osmotic pressure).
- the nebulizer formulation may also contain a surfactant, to reduce or prevent surface induced aggregation of the compound of the invention caused by atomization of the solution in forming the aerosol.
- Formulations for use with a metered-dose inhaler device will generally comprise a finely divided powder containing the compound of the invention (or derivative) suspended in a propellant with the aid of a surfactant.
- the propellant may be any conventional material employed for this purpose, such as a chlorofluorocarbon, a hydrochlorofluorocarbon, a hydrofluorocarbon, or a hydrocarbon, including trichlorofluoromethane,
- Formulations for dispensing from a powder inhaler device will comprise a finely divided dry powder containing compound of the invention (or derivative) and may also include a bulking agent, such as lactose, sorbitol, sucrose, or mannitol in amounts which facilitate dispersal of the powder from the device, e.g., 50 to 90% by weight of the formulation.
- the compound of the invention (or derivative) should advantageously be prepared in particulate form with an average particle size of less than 10 micrometers (pm), most preferably 0.5 to 5 pm, for most effective delivery to the deep lung.
- Nasal delivery of a pharmaceutical composition of the present invention is also contemplated.
- Nasal delivery allows the passage of a pharmaceutical composition of the present invention to the blood stream directly after administering the therapeutic product to the nose, without the necessity for deposition of the product in the lung.
- Formulations for nasal delivery include those with dextran or cyclodextran.
- a useful device is a small, hard bottle to which a metered dose sprayer is attached.
- the metered dose is delivered by drawing the pharmaceutical composition of the present invention solution into a chamber of defined volume, which chamber has an aperture dimensioned to aerosolize and aerosol formulation by forming a spray when a liquid in the chamber is compressed.
- the chamber is compressed to administer the pharmaceutical composition of the present invention.
- the chamber is a piston arrangement.
- Such devices are commercially available.
- a plastic squeeze bottle with an aperture or opening dimensioned to aerosolize an aerosol formulation by forming a spray when squeezed is used.
- the opening is usually found in the top of the bottle, and the top is generally tapered to partially fit in the nasal passages for efficient administration of the aerosol formulation.
- the nasal inhaler will provide a metered amount of the aerosol formulation, for administration of a measured dose of the drug.
- the compounds when it is desirable to deliver them systemically, may be formulated for parenteral administration by injection, e.g., by bolus injection or continuous infusion.
- Formulations for injection may be presented in unit dosage form, e.g., in ampoules or in multi-dose containers, with an added preservative.
- the compositions may take such forms as suspensions, solutions or emulsions in oily or aqueous vehicles, and may contain formulatory agents such as suspending, stabilizing and/or dispersing agents.
- Pharmaceutical formulations for parenteral administration include aqueous solutions of the active compounds in water-soluble form. Additionally, suspensions of the active compounds may be prepared as appropriate oily injection suspensions.
- Suitable lipophilic solvents or vehicles include fatty oils such as sesame oil, or synthetic fatty acid esters, such as ethyl oleate or triglycerides, or liposomes.
- Aqueous injection suspensions may contain substances which increase the viscosity of the suspension, such as sodium
- the suspension may also contain suitable stabilizers or agents which increase the solubility of the compounds to allow for the preparation of highly concentrated solutions.
- the active compounds may be in powder form for constitution with a suitable vehicle, e.g., sterile pyrogen-free water, before use.
- a suitable vehicle e.g., sterile pyrogen-free water
- the compounds may also be formulated in rectal or vaginal compositions such as suppositories or retention enemas, e.g., containing conventional suppository bases such as cocoa butter or other glycerides.
- a compound may also be formulated as a depot preparation.
- Such long acting formulations may be formulated with suitable polymeric or hydrophobic materials (for example as an emulsion in an acceptable oil) or ion exchange resins, or as sparingly soluble derivatives, for example, as a sparingly soluble salt.
- compositions also may comprise suitable solid or gel phase carriers or excipients.
- suitable solid or gel phase carriers or excipients include but are not limited to calcium carbonate, calcium phosphate, various sugars, starches, cellulose derivatives, gelatin, and polymers such as polyethylene glycols.
- Suitable liquid or solid pharmaceutical preparation forms are, for example, aqueous or saline solutions for inhalation, microencapsulated, encochleated, coated onto microscopic gold particles, contained in liposomes, nebulized, aerosols, pellets for implantation into the skin, or dried onto a sharp object to be scratched into the skin.
- the pharmaceutical compositions also include granules, powders, tablets, coated tablets, (micro)capsules, suppositories, syrups, emulsions, suspensions, creams, drops or preparations with protracted release of active compounds, in whose preparation excipients and additives and/or auxiliaries such as disintegrants, binders, coating agents, swelling agents, lubricants, flavorings, sweeteners or solubilizers are customarily used as described above.
- the pharmaceutical compositions are suitable for use in a variety of drug delivery systems. For a brief review of methods for drug delivery, see Langer R, Science 249:1527-33 (1990).
- the compound of the invention and optionally other therapeutics may be administered per se (neat) or in the form of a pharmaceutically acceptable salt.
- the salts should be pharmaceutically acceptable, but non-pharmaceutically acceptable salts may conveniently be used to prepare pharmaceutically acceptable salts thereof.
- Such salts include, but are not limited to, those prepared from the following acids: hydrochloric, hydrobromic, sulphuric, nitric, phosphoric, maleic, acetic, salicylic, p-toluene sulphonic, tartaric, citric, methane sulphonic, formic, malonic, succinic, naphthalene-2-sulphonic, and benzene sulphonic.
- such salts can be prepared as alkaline metal or alkaline earth salts, such as sodium, potassium or calcium salts of the carboxylic acid group.
- Suitable buffering agents include: acetic acid and a salt (1-2% w/v); citric acid and a salt (1-3% w/v); boric acid and a salt (0.5-2.5% w/v); and phosphoric acid and a salt (0.8-2% w/v).
- Suitable preservatives include benzalkonium chloride (0.003-0.03% w/v);
- chlorobutanol (0.3-0.9% w/v); parabens (0.01-0.25% w/v) and thimerosal (0.004-0.02% w/v).
- pharmaceutically acceptable carrier means one or more compatible solid or liquid filler, diluents or encapsulating substances which are suitable for administration to a human or other vertebrate animal.
- carrier denotes an organic or inorganic ingredient, natural or synthetic, with which the active ingredient is combined to facilitate the application.
- the components of the pharmaceutical compositions also are capable of being commingled with the compounds of the present invention, and with each other, in a manner such that there is no interaction which would substantially impair the desired pharmaceutical efficiency.
- the therapeutic agent(s), including specifically but not limited to a compound of the invention, may be provided in particles.
- Particles as used herein means nanoparticles or microparticles (or in some instances larger particles) which can consist in whole or in part of the compound of the invention or the other therapeutic agent(s) as described herein.
- the particles may contain the therapeutic agent(s) in a core surrounded by a coating, including, but not limited to, an enteric coating.
- the therapeutic agent(s) also may be dispersed throughout the particles.
- the therapeutic agent(s) also may be adsorbed into the particles.
- the particles may be of any order release kinetics, including zero-order release, first-order release, second-order release, delayed release, sustained release, immediate release, and any combination thereof, etc.
- the particle may include, in addition to the therapeutic agent(s), any of those materials routinely used in the art of pharmacy and medicine, including, but not limited to, erodible, nonerodible, biodegradable, or nonbiodegradable
- the particles may be microcapsules which contain the compound of the invention in a solution or in a semi-solid state.
- the particles may be of virtually any shape.
- Both non-biodegradable and biodegradable polymeric materials can be used in the manufacture of particles for delivering the therapeutic agent(s).
- Such polymers may be natural or synthetic polymers.
- the polymer is selected based on the period of time over which release is desired.
- Bioadhesive polymers of particular interest include bioerodible hydrogels described in Sawhney H S et al. (1993 ) Macromolecules 26:581-7, the teachings of which are incorporated herein.
- polyhyaluronic acids include polyhyaluronic acids, casein, gelatin, glutin, polyanhydrides, polyacrylic acid, alginate, chitosan, poly(methyl methacrylates), poly(ethyl methacrylates), poly(butylmethacrylate), poly(isobutyl methacrylate),
- controlled release is intended to refer to any drug-containing formulation in which the manner and profile of drug release from the formulation are controlled. This refers to immediate as well as non-immediate release formulations, with non-immediate release formulations including but not limited to sustained release and delayed release formulations.
- sustained release also referred to as“extended release” is used in its
- delayed release is used in its conventional sense to refer to a drug formulation in which there is a time delay between administration of the formulation and the release of the drug there from.“Delayed release” may or may not involve gradual release of drug over an extended period of time, and thus may or may not be“sustained release.”
- Use of a long-term sustained release implant may be particularly suitable for treatment of chronic conditions.“Long-term” release, as used herein, means that the implant is constructed and arranged to deliver therapeutic levels of the active ingredient for at least 7 days, and preferably 30-60 days. Long-term sustained release implants are well-known to those of ordinary skill in the art and include some of the release systems described above.
- the present invention provides peptidic compounds that are useful for treating or preventing ischemia-reperfusion injury or myocardial infarction, or injury associated with myocardial infarction.
- the invention is directed to a method of treating or preventing ischemia-reperfusion injury, comprising administering to a subject in need thereof a therapeutically effective amount of a compound of formula (I), described herein, or a pharmaceutically acceptable salt thereof.
- the ischemia- reperfusion injury is cardiac ischemia-reperfusion injury.
- the compound is administered orally, topically, systemically, intravenously, subcutaneously, intraperitoneally, or intramuscularly.
- the present invention provides a method for treating or preventing a myocardial infarction, comprising administering to a subject in need thereof a therapeutically effective amount of compound of formula (I), or a pharmaceutically acceptable salt thereof.
- Such methods may prevent injury to the heart upon reperfusion by preventing the initiation or progression of the infarction.
- the compound is administered orally, topically, systemically, intravenously, subcutaneously, intraperitoneally, or intramuscularly
- Ischemia is reduction or decrease in blood supply to a tissue or an organ and has many different causes. Ischemia may be local, e.g., caused by thrombus or embolus, or more global, e.g., due to low perfusion pressure.
- An ischemic event can lead to hypoxia (reduced oxygen) and/or anoxia (absence of oxygen).
- Ischemia in a tissue or organ of a mammal is a multifaceted pathological condition that is caused by oxygen deprivation (hypoxia) and/or glucose (e.g., substrate) deprivation.
- Oxygen and/or glucose deprivation in cells of a tissue or organ leads to a reduction or total loss of energy generating capacity and consequent loss of function of active ion transport across the cell membranes.
- Oxygen and/or glucose deprivation also leads to pathological changes in other cell membranes, including permeability transition in the mitochondrial membranes.
- other molecules, such as apoptotic proteins normally
- Ischemia or hypoxia in a particular tissue or organ may be caused by a loss or severe reduction in blood supply to the tissue or organ.
- the loss or severe reduction in blood supply may, for example, be due to thromboembolic stroke, coronary atherosclerosis, or peripheral vascular disease.
- the tissue affected by ischemia or hypoxia is typically muscle, such as cardiac, skeletal, or smooth muscle.
- the organ affected by ischemia or hypoxia may be any organ that is subject to ischemia or hypoxia.
- cardiac muscle ischemia or hypoxia is commonly caused by atherosclerotic or thrombotic blockages, which lead to the reduction or loss of oxygen delivery to the cardiac tissues by the cardiac arterial and capillary blood supply.
- Such cardiac ischemia or hypoxia may cause pain and necrosis of the affected cardiac muscle, and ultimately may lead to cardiac failure.
- Reperfusion is the restoration of blood flow to any organ or tissue in which the flow of blood is decreased or blocked.
- blood flow can be restored to any organ or tissue affected by ischemia.
- the restoration of blood flow can occur by any method known to those in the art. For instance, reperfusion of ischemic cardiac tissues may arise from angioplasty, coronary artery bypass graft, or the use of thrombolytic drugs.
- Ischemia-reperfusion injury is the cellular or tissue damage caused when blood supply returns to the affected area after a period of ischemia.
- the lack of oxygen and nutrients during ischemia creates a condition in which the restoration of circulation results damage to the tissues.
- forms of myocardial reperfusion injury including reperfusion-induced arrhythmias, myocardial stunning, microvascular obstruction manifesting in sluggish coronary blood flow, and lethal myocardial reperfusion injury (i.e., reperfusion-induced death of cardiomyocytes that were viable at the end of the index ischemic event).
- lethal myocardial reperfusion injury accounts for about 50% of the final myocardial infarct size.
- the peptide is administered orally, intravenously, or parenterally.
- the subject is a human.
- a peptidic compound of the invention, or a pharmaceutically acceptable salt thereof, such as acetate, tartrate, or trifluoroacetate salt may be administered to a subject suspected of, or already suffering from ischemic injury in an amount sufficient to cure, or at least partially arrest, the symptoms of the disease, including its complications and intermediate pathological phenotypes in development of the disease.
- Subjects suffering from ischemic injury can be identified by any or a combination of diagnostic or prognostic assays known in the art.
- the ischemic injury is related to cardiac ischemia, brain ischemia, renal ischemia, cerebral ischemia, intestinal ischemia, hepatic ischemia, or myocardial infarction.
- typical symptoms of cardiac ischemia include, but are not limited to, angina (e.g., chest pain and pressure), shortness of breath, palpitations, weakness, dizziness, nausea, sweating, rapid heartbeat, and fatigue.
- treatment of subjects diagnosed with cardiac ischemia with at least one peptide disclosed herein ameliorates or eliminates of one or more of the following symptoms of cardiac ischemia: angina (e.g., chest pain and pressure), shortness of breath, palpitations, weakness, dizziness, nausea, sweating, rapid heartbeat, and fatigue.
- typical symptoms of renal ischemia include, but are not limited to, uremia (i.e., high blood levels of protein by-products, such as, e.g., urea), acute episodes of dyspnea (labored or difficult breathing) caused by sudden accumulation of fluid in the lungs, hypertension, pain felt near the kidneys, weakness, hypertension, nausea, a history of leg pain, a stride that reflects compromised circulation to the legs, and Sons (sound or murmurs heard with a stethoscope) caused by turbulent blood flow within the arteries may be detected in the neck (e.g., carotid artery bruit), abdomen (which may reflect narrowing of the renal artery), and groin (femoral artery bruit).
- uremia i.e., high blood levels of protein by-products, such as, e.g., urea
- dyspnea labored or difficult breathing
- nausea nausea
- a history of leg pain a stride that reflects compromised circulation to the legs
- treatment of subjects diagnosed with renal ischemia with at least one peptide disclosed herein ameliorates or eliminates of one or more of the following symptoms of renal ischemia: uremia (i.e., high blood levels of protein by-products, such as, e.g., urea), acute episodes of dyspnea (labored or difficult breathing) caused by sudden accumulation of fluid in the lungs, hypertension, pain felt near the kidneys, weakness, hypertension, nausea, a history of leg pain, a stride that reflects compromised circulation to the legs, and Sonides (sound or murmurs heard with a stethoscope) caused by turbulent blood flow within the arteries may be detected in the neck (e.g., carotid artery bruit), abdomen (which may reflect narrowing of the renal artery), and groin (femoral artery bruit).
- uremia i.e., high blood levels of protein by-products, such as, e.g., urea
- dyspnea labored or difficult breathing
- typical symptoms of cerebral (or brain) ischemia include, but are not limited to, blindness in one eye, weakness in one arm or leg, weakness in one entire side of the body, dizziness, vertigo, double vision, weakness on both sides of the body, difficulty speaking, slurred speech, and the loss of coordination.
- treatment of subjects diagnosed with cerebral (or brain) ischemia with at least one peptide disclosed herein ameliorates or eliminates of one or more of the following symptoms of cerebral (or brain) ischemia: blindness in one eye, weakness in one arm or leg, weakness in one entire side of the body, dizziness, vertigo, double vision, weakness on both sides of the body, difficulty speaking, slurred speech, and the loss of coordination.
- the present invention relates to methods of treating ischemia reperfusion injury and/or side effects associated with existing therapeutics against ischemia reperfusion injury.
- a composition or medicament comprising at least one compound of the invention, or a pharmaceutically acceptable salt thereof, such as acetate, tartrate or trifluoroacetate, is administered to a subject suspected of, or already suffering from ischemic reperfusion injury in an amount sufficient to cure, or at least partially arrest, the symptoms of the disease, including its complications and intermediate pathological phenotypes in development of the disease.
- Subjects suffering from ischemic-reperfusion injury can be identified by any or a combination of diagnostic or prognostic assays known in the art.
- the ischemia-reperfusion injury is related to cardiac ischemia, brain ischemia, renal ischemia, cerebral ischemia, intestinal ischemia, and hepatic ischemia.
- the peptidic compounds disclosed herein are useful in the treatment of cardiac ischemia-reperfusion injury.
- the peptidic compounds disclosed herein are useful in treating myocardial infarction in a subject to prevent injury to the heart upon reperfusion.
- the invention relates to methods of coronary revascularization, comprising administering to a mammalian subject a therapeutically effective amount of a peptidic compound of the invention, or a pharmaceutically acceptable salt thereof, and performing a coronary artery bypass graft (CABG) procedure on the subject.
- CABG coronary artery bypass graft
- treatment of myocardial infarction with the peptidic compounds disclosed herein reduces infarct size, increases LVDP, and increases maximal rates of contraction and relaxation ( ⁇ dP/dt).
- the present invention provides methods for preventing or delaying the onset of ischemic injury or symptoms of ischemic injury in a subject at risk of having ischemia injury. In some embodiments, the present technology provides methods for preventing or reducing the symptoms of ischemic injury in a subject at risk of having ischemia injury.
- the present invention provides methods for preventing or delaying the onset of ischemia-reperfusion injury or symptoms of ischemia-reperfusion injury in a subject at risk of having ischemia-reperfusion injury. In some embodiments, the present invention provides methods for preventing or reducing the symptoms of ischemia reperfusion injury in a subject at risk of having ischemia-reperfusion injury.
- the ischemic injury, the ischemia-reperfusion injury, or symptoms of ischemic or ischemia-reperfusion injury is related to cardiac ischemia, brain ischemia, renal ischemia, cerebral ischemia, intestinal ischemia, and hepatic ischemia.
- the ischemic injury is myocardial infarction.
- the peptidic compounds disclosed herein are useful in the treatment or prevention of cardiac ischemia-reperfusion injury. In some embodiments, the peptidic compounds disclosed herein are useful in the prevention of cardiac ischemia- reperfusion injury.
- Subjects at risk for ischemic injury or ischemia-reperfusion injury can be identified by, e.g, any or a combination of diagnostic or prognostic assays known in the art.
- a pharmaceutical composition or medicament of a compound of the invention, or a pharmaceutically acceptable salt thereof, such as acetate, tartrate, or trifluoroacetate salt is administered to a subject susceptible to, or otherwise at risk of for ischemic injury or ischemia reperfusion injury in an amount sufficient to eliminate, reduce the risk, or delay the onset of the disease, including biochemical, histologic and/or behavioral symptoms of the disease, its complications and intermediate pathological phenotypes presenting during development of the disease or reduce the symptoms and/or complications and intermediate pathological phenotypes presenting during development of the disease.
- Administration of a prophylactic peptide can occur prior to the manifestation of symptoms characteristic of the disease or disorder, such that the disease or disorder is prevented, delayed in its progression, or the severity of the symptoms or side effects of the disease or disorder are reduced.
- subjects may be at risk for cardiac ischemia if they have coronary artery disease (atherosclerosis), blood clots, or coronary artery spasm.
- subjects may be at risk for renal ischemia if they have kidney injury (e.g ., acute kidney injury) and/or injuries or complications from surgeries in which the kidneys are deprived of normal blood flow for extended periods of time (e.g., heart-bypass surgery).
- kidney injury e.g ., acute kidney injury
- injuries or complications from surgeries in which the kidneys are deprived of normal blood flow for extended periods of time e.g., heart-bypass surgery.
- subjects may be at risk for cerebral ischemia if they have sickle cell anemia, compressed blood vessels, ventricular tachycardia, plaque buildup in the arteries, blood clots, extremely low blood pressure as a result of heart attack, had a stroke, or congenital heart defects.
- a composition comprising at least one peptidic compound described herein, or a pharmaceutically acceptable salt thereof, such as acetate, tartrate, or trifluoroacetate salt, is administered to a subject in need thereof.
- the peptide composition is administered one, two, three, four, or five times per day. In some embodiments, the peptide composition is administered more than five times per day. Additionally or alternatively, in some embodiments, the peptide composition is administered every day, every other day, every third day, every fourth day, every fifth day, or every sixth day. In some embodiments, the peptide composition is administered weekly, bi-weekly, tri-weekly, or monthly.
- the peptide composition is administered for a period of one, two, three, four, or five weeks. In some embodiments, the peptide is administered for six weeks or more. In some embodiments, the peptide is administered for twelve weeks or more. In some embodiments, the peptide is administered for a period of less than one year. In some embodiments, the peptide is administered for a period of more than one year. In some embodiments, treatment with at least one peptide disclosed herein will prevent or delay the onset of one or more of the following symptoms of cardiac ischemia: angina (e.g ., chest pain and pressure), shortness of breath, palpitations, weakness, dizziness, nausea, sweating, rapid heartbeat, and fatigue.
- angina e.g ., chest pain and pressure
- treatment with at least one peptide disclosed herein will prevent or delay the onset of one or more of the following symptoms of renal ischemia:
- uremia i.e., high blood levels of protein by-products, such as, e.g., urea
- dyspnea labored or difficult breathing
- hypertension i.e., high blood levels of protein by-products, such as, e.g., urea
- weakness hypertension
- nausea a history of leg pain
- stride that reflects compromised circulation to the legs
- bruits sound or murmurs heard with a stethoscope
- turbulent blood flow within the arteries may be detected in the neck (e.g, carotid artery bruit), abdomen (which may reflect narrowing of the renal artery), and groin (femoral artery bruit).
- treatment with at least one peptide disclosed herein will prevent or delay the onset of one or more of the following symptoms of cerebral (or brain) ischemia: blindness in one eye, weakness in one arm or leg, weakness in one entire side of the body, dizziness, vertigo, double vision, weakness on both sides of the body, difficulty speaking, slurred speech, and the loss of coordination.
- the following methods can be used to evaluate the metabolic stability of the compounds of the invention.
- 7.5 mM stock solutions of test compounds are prepared in DMSO.
- the 7.5 mM stock solutions are diluted to 12.5-50 mM in acetonitrile (ACN).
- ACN acetonitrile
- the 20 mg/mL human liver microsomes are diluted to 0.625 mg/mL in 0.1 M potassium phosphate buffer, pH 7.4, containing 3 mM MgCl2.
- the diluted microsomes are added to wells of a 96-well deep-well polypropylene plate in triplicate.
- a 10 mL aliquot of the 12.5-50 mM test compound is added to the microsomes and the mixture is pre-warmed for 10 minutes. Reactions are initiated by addition of pre-warmed NADPH solution.
- the final reaction volume is 0.5 mL and contains 0.5 mg/mL human liver microsomes, 0.25-1.0 mM test compound, and 2 mM NADPH in 0.1 M potassium phosphate buffer, pH 7.4, and 3 mM MgCl2.
- the reaction mixtures are incubated at 37° C., and 50 mL aliquots are removed at 0, 5, 10, 20, and 30 minutes and added to shallow-well 96-well plates which contain 50 mL of ice- cold ACN with internal standard to stop the reactions.
- the plates are stored at 4° C for 20 minutes after which 100 mL of water is added to the wells of the plate before centrifugation to pellet precipitated proteins.
- Supernatants are transferred to another 96-well plate and analyzed for amounts of parent remaining by LC-MS/MS using an Applied Bio-systems API 4000 mass spectrometer. Testing is done in triplicate.
- Step c Coupling 1) Add 12mmol Fmoc-Lys(Boc)-OH, 12mmol HOBT, 12mmol HBTU and 12mmol DIEA into the resin for coupling for 40min at RT.
- Step i Deprotect the peptide
- Example 12 Synthesis of (S)-6-amino-2-((S)-2-((R)-2-((S)-2-amino-3- phenylpropanamido)-5-guanidinopentanamido)-3-(4-(tert- butyl)phenyl)propanamido)hexanamide (Phe-D-Arg-(4-tert-Butyl)-Phe-Lys-NH 2 , 15l)
- Example 13 Synthesis of (S)-2-((R)-2-((S)-2-amino-3-phenylpropanamido)-5- guanidinopentanamido)-N-((S)-1,6-diamino-1-oxohexan-2-yl)decanamide (Phe-D-Arg-( ⁇ - n-heptyl)-Ala-Lys-NH 2 , 15m)
- Example 14 Synthesis of (R)-N-((S)-1-(((S)-1-amino-3-(1H-indol-3-yl)-1-oxopropan-2- yl)amino)-1-oxo-3-phenylpropan-2-yl)-2-((S)-2-amino-3-phenylpropanamido)-5- guanidinopentanamide (Phe-D-Arg-Phe-Trp-NH 2 , 15n)
- Example 15 Synthesis of (R)-N-((S)-1-(((S)-1-amino-3-(1H-imidazol-4-yl)-1-oxopropan-2- yl)amino)-1-oxo-3-phenylpropan-2-yl)-2-((S)-2-amino-3-phenylpropanamido)-5- guanidinopentanamide (Phe-D-Arg-Phe-His-NH 2 , 15o)
- Example 17 Synthesis of (S)-6-amino-2-((S)-2-((R)-2-((S)-2-amino-3- phenylpropanamido)-5-guanidinopentanamido)-3-(2,4- dimethylphenyl)propanamido)hexanamide (Phe-D-Arg-(2,4-dimethyl)-Phe-Lys-NH 2 , 15q)
- Example 18 Synthesis of (S)-N-((S)-1-amino-3-(1H-imidazol-4-yl)-1-oxopropan-2-yl)-2- ((R)-2-((S)-2-amino-3-phenylpropanamido)-5-guanidinopentanamido)-5- methylhexanamide (Phe-D-Arg-homoLeu -His-NH 2 , 15r)
- Titanium tetraethoxide 700 g, 3.06 mol was added to a stirred solution of 1-adamantyl acetaldehyde (18, 265 g, 1.49 mol) and (S)-tert-butanesulfinamide (19, 223 g, 1.84 mol) in THF (4 L) at room temperature under nitrogen atmosphere. The mixture was stirred at 15 oC for 12 h. TLC and HPLC indicated the reaction was completed. Then ethyl acetate (4 L) and water (4 L) was added. The reaction mixture was filtered through celite and the aqueous layer was extracted with ethyl acetate (2 L).
- Step h Synthesis of benzyl ((5S)-5-((2S)-3-(adamantan-1-yl)-2-((tert- butoxycarbonyl)amino)propanamido)-6-amino-6-oxohexyl)carbamate (26)
- Step j Synthesis of tert-butyl ((9S,12S,15R,18S)-12-(adamantan-1-ylmethyl)-9- carbamoyl-15-(3-guanidinopropyl)-3,11,14,17-tetraoxo-1,19-diphenyl-2-oxa- 4,10,13,16-tetraazanonadecan-18-yl)carbamate (30)
- Step k Synthesis of tert-butyl ((2S)-1-(((2R)-1-(((2S)-3-(adamantan-1-yl)-1-(((S)-1,6- diamino-1-oxohexan-2-yl)amino)-1-oxopropan-2-yl)amino)-5-guanidino-1- oxopentan-2-yl)amino)-1-oxo-3-phenylpropan-2-yl)carbamate (31)
- Step l Synthesis of (2S)-2-((2S)-3-(adamantan-1-yl)-2-((R)-2-((S)-2-amino-3- phenylpropanamido)-5-guanidinopentanamido)propanamido)-6-aminohexanamide (15v)
- Step a Synthesis of benzyl ((S)-6-amino-5-((S)-2-((tert-butoxycarbonyl)amino)-3- (perfluorophenyl)propanamido)-6-oxohexyl)carbamate (33)
- Step b Synthesis of benzyl ((S)-6-amino-5-((S)-2-amino-3- (perfluorophenyl)propanamido)-6-oxohexyl)carbamate (34)
- Step c Synthesis of benzyl ((6R,9S,12S)-12-carbamoyl-6-(3-guanidinopropyl)-2,2- dimethyl-4,7,10-trioxo-9-((perfluorophenyl)methyl)-3-oxa-5,8,11-triazahexadecan- 16-yl)carbamate (36)
- Step d Synthesis of benzyl ((S)-6-amino-5-((S)-2-((R)-2-amino-5- guanidinopentanamido)-3-(perfluorophenyl)propanamido)-6-oxohexyl)carbamate (37)
- Step e Synthesis of benzyl ((6S,9R,12S,15S)-6-benzyl-15-carbamoyl-9-(3- guanidinopropyl)-2,2-dimethyl-4,7,10,13-tetraoxo-12-((perfluorophenyl)methyl)-3- oxa-5,8,11,14-tetraazanonadecan-19-yl)carbamate (39)
- Step f Synthesis of benzyl ((S)-6-amino-5-((S)-2-((R)-2-((S)-2-amino-3- phenylpropanamido)-5-guanidinopentanamido)-3-(perfluorophenyl)propanamido)- 6-oxohexyl)carbamate (40)
- Step g Synthesis of (S)-6-amino-2-((S)-2-((R)-2-((S)-2-amino-3- phenylpropanamido)-5-guanidinopentanamido)-3- (perfluorophenyl)propanamido)hexanamide (15w)
- Step a Synthesis of benzyl ((S)-6-amino-5-((S)-2-((tert-butoxycarbonyl)amino)-3- phenylpropanamido)-6-oxohexyl)carbamate (41)
- Step c Synthesis of benzyl ((6R,9S,12S)-9-benzyl-12-carbamoyl-6-(3- guanidinopropyl)-2,2-dimethyl-4,7,10-trioxo-3-oxa-5,8,11-triazahexadecan-16- yl)carbamate (43)
- Step d Synthesis of benzyl ((S)-6-amino-5-((S)-2-((R)-2-amino-5- guanidinopentanamido)-3-phenylpropanamido)-6-oxohexyl)carbamate (44)
- Step e Synthesis of benzyl ((6S,9R,12S,15S)-6-(adamantan-1-ylmethyl)-12-benzyl- 15-carbamoyl-9-(3-guanidinopropyl)-2,2-dimethyl-4,7,10,13-tetraoxo-3-oxa- 5,8,11,14-tetraazanonadecan-19-yl)carbamate (45)
- Step f Synthesis of tert-butyl ((2S)-3-(adamantan-1-yl)-1-(((R)-1-(((S)-1-(((S)-1,6- diamino-1-oxohexan-2-yl)amino)-1-oxo-3-phenylpropan-2-yl)amino)-5-guanidino-1- oxopentan-2-yl)amino)-1-oxopropan-2-yl)carbamate (46)
- Step f Synthesis of methyl 2-amino-3-(bicyclo[2.2.2]octan-1-yl)propanoate (55)
- a solution of 54 (9.7 g, 31.2 mmol) in 4 N HCl/Dioxane (100 mL) was stirred at room temperature for 2 h, the reaction mixture was concentrated in vacuo to give the desired product as white solid (55, 7.7 g, crude).
- Step g Synthesis of methyl 2-(((benzyloxy)carbonyl)amino)-3-(bicyclo[2.2.2]octan-1- yl)propanoate (56)
- Step k Synthesis of tert-butyl ((S)-1-(((S)-1-amino-6-(((benzyloxy)carbonyl)amino)- 1-oxohexan-2-yl)amino)-3-(bicyclo[2.2.2]octan-1-yl)-1-oxopropan-2-yl)carbamate (60)
- Step l Synthesis of benzyl ((S)-6-amino-5-((S)-2-amino-3-(bicyclo[2.2.2]octan-1- yl)propanamido)-6-oxohexyl)carbamate (61)
- Step m Synthesis of benzyl ((6S,9S,12S,15S)-6-benzyl-12-(bicyclo[2.2.2]octan-1- ylmethyl)-15-carbamoyl-9-(3-guanidinopropyl)-2,2-dimethyl-4,7,10,13-tetraoxo-3- oxa-5,8,11,14-tetraazanonadecan-19-yl)carbamate (62)
- Step n Synthesis of benzyl ((S)-6-amino-5-((S)-2-((S)-2-((S)-2-amino-3- phenylpropanamido)-5-guanidinopentanamido)-3-(bicyclo[2.2.2]octan-1- yl)propanamido)-6-oxohexyl)carbamate (63)
- Step b Synthesis of methyl (S)-2-((tert-butoxycarbonyl)amino)-3-(3- iodobicyclo[1.1.1]pentan-1-yl)propanoate (67)
- Step f Synthesis of (9H-fluoren-9-yl)methyl ((9S,12S,15S,18S)-12-benzyl-19- (bicyclo[1.1.1]pentan-1-yl)-9-carbamoyl-15-(3-guanidinopropyl)-3,11,14,17-tetraoxo- 1-phenyl-2-oxa-4,10,13,16-tetraazanonadecan-18-yl)carbamate (71)
- Step g Synthesis of benzyl ((S)-6-amino-5-((S)-2-((S)-2-((S)-2-amino-3- (bicyclo[1.1.1]pentan-1-yl)propanamido)-5-guanidinopentanamido)-3- phenylpropanamido)-6-oxohexyl)carbamate (72) 71 (190 mg) was treated with a mixture of 20% Piperidine/DMF (3 mL).
- reaction mixture was stirred 1h at rt, then organic solvent was evaporated and crude product was purified by flash reverse phase chromatography (eluent H 2 O (0.2% AcOH)/MeOH from 5% to 70% of methanol). As a result, 110 mg of 72 was isolated as diacetate salt.
- Step a Synthesis of (9H-fluoren-9-yl)methyl ((S)-1-(((S)-1-amino-6- (((benzyloxy)carbonyl)amino)-1-oxohexan-2-yl)amino)-3-(bicyclo[1.1.1]pentan-1-yl)- 1-oxopropan-2-yl)carbamate (73)
- Step b Synthesis of benzyl ((S)-6-amino-5-((S)-2-amino-3-(bicyclo[1.1.1]pentan-1- yl)propanamido)-6-oxohexyl)carbamate (74)
- Step c Synthesis of tert-butyl ((9S,12S,15R,18S)-12-(bicyclo[1.1.1]pentan-1- ylmethyl)-9-carbamoyl-15-(3-guanidinopropyl)-3,11,14,17-tetraoxo-1,19-diphenyl-2- oxa-4,10,13,16-tetraazanonadecan-18-yl)carbamate (75)
- Step d Synthesis of benzyl ((S)-6-amino-5-((S)-2-((R)-2-((S)-2-amino-3- phenylpropanamido)-5-guanidinopentanamido)-3-(bicyclo[1.1.1]pentan-1- yl)propanamido)-6-oxohexyl)carbamate (76)
- Example 28 Synthesis of (S)-6-amino-2-((S)-2-((R)-2-((S)-2-amino-3- phenylpropanamido)-5-guanidinopentanamido)-3-(tetrahydro-2H-pyran-4- yl)propanamido)hexanamide (Phe-D-Arg-( ⁇ -tetrahydro-2H-pyran-4-yl)-Ala-Lys-NH 2 , 15ab)
- Step a Synthesis of (9H-fluoren-9-yl)methyl ((S)-1-(((S)-1-amino-6- (((benzyloxy)carbonyl)amino)-1-oxohexan-2-yl)amino)-1-oxo-3-(tetrahydro-2H- pyran-4-yl)propan-2-yl)carbamate (79)
- Step b Synthesis of benzyl ((S)-6-amino-5-((S)-2-amino-3-(tetrahydro-2H-pyran-4- yl)propanamido)-6-oxohexyl)carbamate (80)
- Step c Synthesis of tert-butyl ((9S,12S,15R,18S)-9-carbamoyl-15-(3- guanidinopropyl)-3,11,14,17-tetraoxo-1,19-diphenyl-12-((tetrahydro-2H-pyran-4- yl)methyl)-2-oxa-4,10,13,16-tetraazanonadecan-18-yl)carbamate (81)
- Step d Synthesis of benzyl ((S)-6-amino-5-((S)-2-((R)-2-((S)-2-amino-3- phenylpropanamido)-5-guanidinopentanamido)-3-(tetrahydro-2H-pyran-4- yl)propanamido)-6-oxohexyl)carbamate (82)
- the permeabilized cardiac fibers are prepared from normoxic heart as described previously (Kuka J, Vilskersts R, Cirule H, Makrecka M, Pugovics O, Kalvinsh I, Dambrova M, Liepinsh E.
- the cardioprotective effect of mildronate is diminished after co-treatment with L-carnitine. J Cardiovasc Pharmacol Ther.2012 Jun;17(2):215-22. doi: 10.1177/1074248411419502) with some modifications.
- the bundles of fibers are permeabilized using 50 ⁇ g/mL saponin and 0.5 mg/mL collagenase at 4oC in 1 mL of buffer A (20 mM imidazole, 0.5 mM dithiothreitol, 20 mM taurine, 7.1 mM MgCl 2 , 50 mM MES, 5 mM ATP, 15 mM phosphocreatine, 2.6 mM CaK2EGTA, 7.4 mM K2EGTA, pH 7.0 at 0oC).
- buffer A (20 mM imidazole, 0.5 mM dithiothreitol, 20 mM taurine, 7.1 mM MgCl 2 , 50 mM MES, 5 mM ATP, 15 mM phosphocreatine, 2.6 mM CaK2EGTA, 7.4 mM K2EGTA, pH 7.0 at 0oC).
- the fibers are washed for 15 min in 2 mL of buffer B (20 mM imidazole, 0.5 mM dithiothreitol, 20 mM taurine, 1.6 mM MgCl 2 , 100 mM MES, 3 mM KH 2 PO 4 , 2.9 mM CaK 2 EGTA, 7 mM K 2 EGTA, pH 7.1 at 37oC) supplemented with compound (e.g.100 nM) or vehicle.
- buffer B (20 mM imidazole, 0.5 mM dithiothreitol, 20 mM taurine, 1.6 mM MgCl 2 , 100 mM MES, 3 mM KH 2 PO 4 , 2.9 mM CaK 2 EGTA, 7 mM K 2 EGTA, pH 7.1 at 37oC
- compound e.g.100 nM
- H 2 O 2 flux is measured simultaneously with respirometry in the O2k-Fluorometer using the H2O2-sensitive probe AmplifluTM Red (AmR) (Makrecka-Kuka M, Krumschnabel G, Gnaiger E. High-Resolution Respirometry for Simultaneous Measurement of Oxygen and Hydrogen Peroxide Fluxes in Permeabilized Cells, Tissue Homogenate and Isolated Mitochondria. Biomolecules.2015 Jun 29;5(3):1319-38.
- AmplifluTM Red AmplifluTM Red
- the hearts are perfused with oxygenated (95% O2 - 5% CO2) Krebs-Henseleit (KH) buffer solution (118 mmol/L NaCl, 4.7 mmol/L KCl, 1.24 mmol/L CaCl2, 1.64 mmol/L MgCl2, 24.88 mmol/L NaHCO3, 1.18 mmol/L KH2PO4, and 0.05 mmol/L EDTA; pH 7.3–7.5; 36.8-37.0°C) supplemented with 10 mM glucose at a constant perfusion pressure of 60 mmHg.
- KH Krebs-Henseleit
- a water-ethanol mixture (1:1)-filled balloon connected to a physiological pressure transducer (ADInstruments) is inserted into the left ventricle, and the baseline end-diastolic pressure set at 5-10 mmHg.
- the heart rate (HR), flow, left-ventricle developed pressure (LVDP), contractility (+dp/dt) are continuously recorded using a PowerLab 8/35 system from ADInstruments.
- the isolated rat hearts are adapted for 20 min and the left anterior descending coronary artery (LAD) is subsequently occluded for 30 min followed by 120 min of reperfusion.
- KH perfusion solution with or without added compound of interest (vehicle or 1 mM concentration) will be used for the whole time of isolated heart perfusion.
- the planimetric analysis of cross-sectional images is performed using Image-Pro Plus v6.3 software to determine the area at risk (AR) and area of necrosis (AN), each expressed as a percentage of cross-sectional slice area.
- AR area at risk
- AN area of necrosis
- the obtained values are then used to calculate the infarct size (IS) as a percentage of the risk area according to the formula:
- Area of necrosis is determined by combining areas of the white necrotic and pink tissue.
- test article concentration s may be adjusted. Any changes will be recorded in the study file and the final report.
- the protocol and the number of compounds to be tested may be modified based on the experimental results and discussions with the Sponsor. Any changes to the protocol will be documented in the study file and in the protocol amendment.
- the Rat Myocardial Infarction Model was performed by IPST Therapeutique Inc, Sherbrooke, Quebec, Canada. The animals were randomized in terms of even distribution between treatment groups based on their body weight by the Study Director with the aim of scheduling animal from each treatment group for each day of surgery (when possible).
- Route of administration s.c.;
- Treatment 0.01, 0.1, 0.5 and 2 mg/kg doses, 30 min before ischemia.
- a thoracotomy will be performed through the left forth intercostal space to exposed the heart.
- a 5-0 sofsilk suture will be placed around the left anterior descending (LAD) artery, 2-3 mm below the left atrium.
- subcutaneous injection will be done for postoperative pain management.
- the heart will be excised and mounted into a Langendorff apparatus. Oxygenated Tyrode’s solution heated at 35 ⁇ 2°C will perfuse the heart in a retrograde manner at a pressure of approximately 70 mmHg and a flow rate on the order of 10 mL/min. 10) The heart will then be perfused with Evans blue dye to evaluate the size of the myocardial infarction. Following Evans blue staining, the heart will be removed from the Langendorff apparatus and immersed in cold ethanol (-50 °C). The heart will be cut in transversal slices sections of approximately 2 mm.
- the slices will be scanned to evaluate the area at risk (AAR) before to be incubated in phosphate buffer containing 1% TTC for 30 minutes at 35 ⁇ 2°C and then transfer in formalin 4% for 24 hours at 4 ⁇ 2°C. The slices will be re-scanned to measure the infracted area. Animals with an area at risk >60% will be excluded from the study.
- AAR area at risk
- a networked personal computer running either Microsoft Windows8, XP Professional or Microsoft Windows Vista Business will be used for data acquisition.
- the analysis software will be Microsoft Office Excel 2007 installed on networked personal computers running Microsoft Windows8, XP Professional or vista.
- non-audited raw data in Excel spreadsheets and a draft non-audited report containing the study design, the study’s quantitative/qualitative results, and individual data graphs will be submitted to the Sponsor.
- the final report will be provided within 1 week of receiving Sponsors comments.
- the Rat Acute Kidney Injury (AKI) Model was performed by IPST Therapeutique Inc, Sherbrooke, Quebec, Canada. The animals were randomized in terms of even distribution between treatment groups based on their body weight by the Study Director with the aim of scheduling animal from each group at each day of surgery. The rats will be given free access to food and water.
- Route of administration s.c.;
- Route of administration s.c.; Treatment dose: 2 X 2 mg/kg, 30 min before ischemia and 5 min before reperfusion.
- Rats will be anaesthetized with isoflurane USP (Abbot Laboratories, Montreal Canada) 2% in oxygen and placed on a heated pad to maintain body temperature. The ECG and oxygen saturation will be monitored for the entire surgical process. The body temperature will be monitored with a probe thermometer introduced into the abdomen, very close to the kidneys.
- USP Abbot Laboratories, Montreal Canada
- a 1 mL blood draw will be taken from the jugular vein.
- the blood will be collected into lithium heparin tubes and centrifuged at 3000 rpm for 10 min. to obtain the plasma.
- the plasma will be separated into 200 mL aliquot and stored at -20°C until dosage of biomarkers.
- the abdomen will be disinfected with providone iodine and alaparotomy will be performed.
- kidneys will be exposed and a temporary suture will be placed around renal artery of the two kidneys. Renal ischemia will be visually confirmed by a gradual changed of the kidneys colour going from red to dark purple within a couple of minutes following the start of the ischemia. During the ischemia, the kidneys will be kept moist and warm using a heat lamp and sterile gauze soaked in warm (37°C) saline. The temperature will be monitored with a probe thermometer introduced into the abdomen, very close to the kidneys.
- the analysis software will be Microsoft Office Excel 2007 installed on networked personal computers running Microsoft Windows 8, XP Professional or Vista. DATA ANALYSIS
- the vehicle group was compared to the sham group while the test article was compared to the vehicle group.
- the plasma creatinine post-I/R (% mean of vehicle) was calculated using the following formula:
- Mean D plasma creatinine in sham group Mean (plasma creatinine 24h post-isch. - plasma creatinine pre-isch.) in sham group
- Mean D plasma creatinine in vehicle group Mean ((plasma creatinine 24h post-isch. - plasma creatinine pre-isch.) - Mean D plasma creatinine in sham group) in vehicle group
- the BUN post-I/R (% mean of vehicle) was calculated using the following formula: ((BUN 24 h post-isch.) - (BUN pre-isch.)) - Mean D BUN in sham group ⁇ 100
- Mean D BUN in sham group Mean (BUN 24h post-isch. - BUN pre-isch.) in sham group
- Mean D BUN in vehicle group Mean ((BUN 24h post-isch. - BUN pre-isch.) - Mean D BUN in sham group) in vehicle group
- BUN 100% - D BUN post-I/R (% mean of veh.)
Abstract
Description







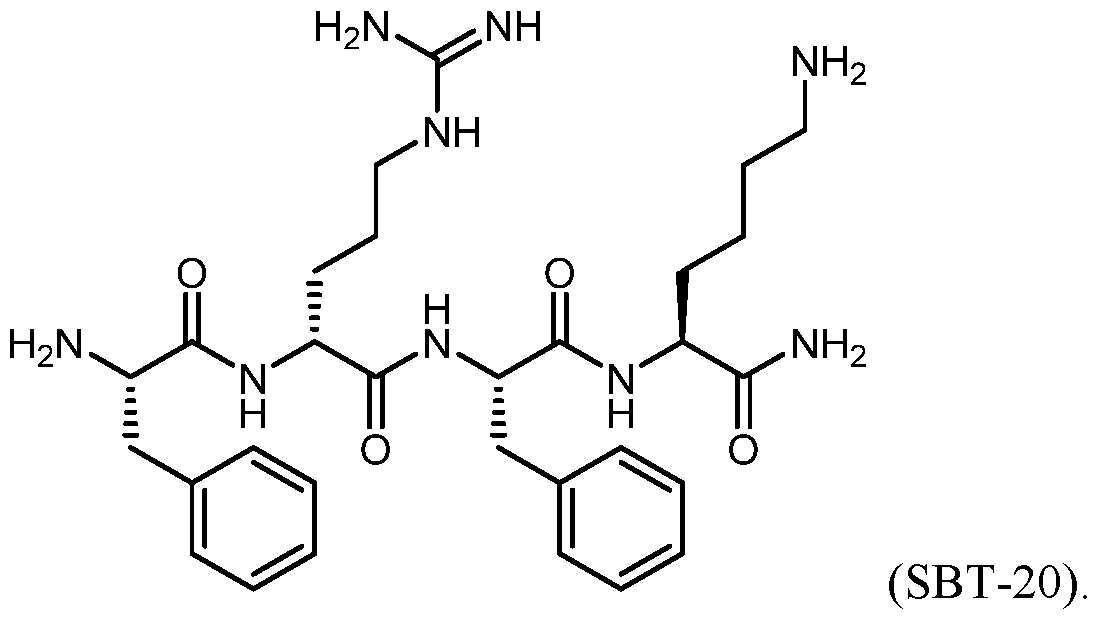













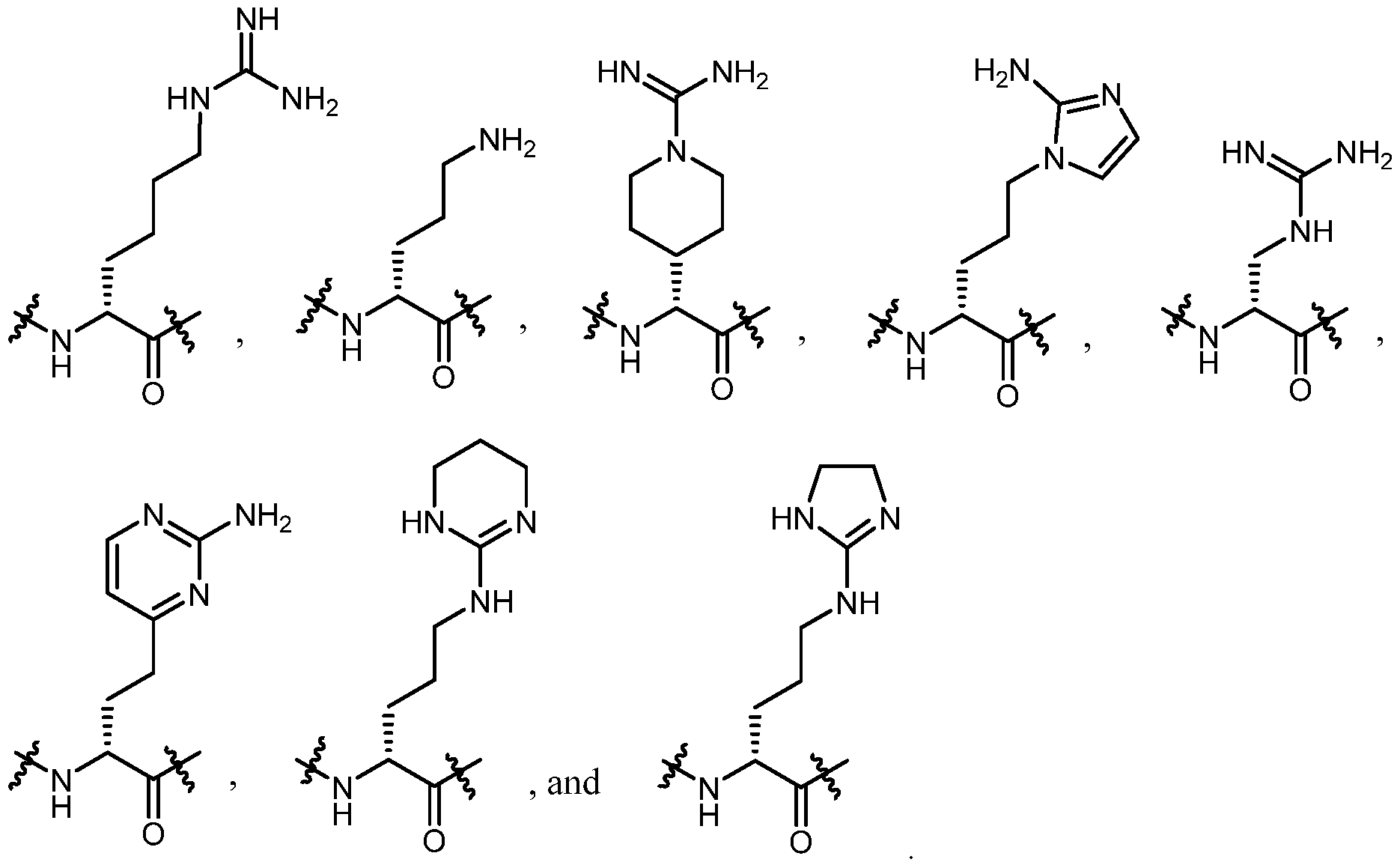


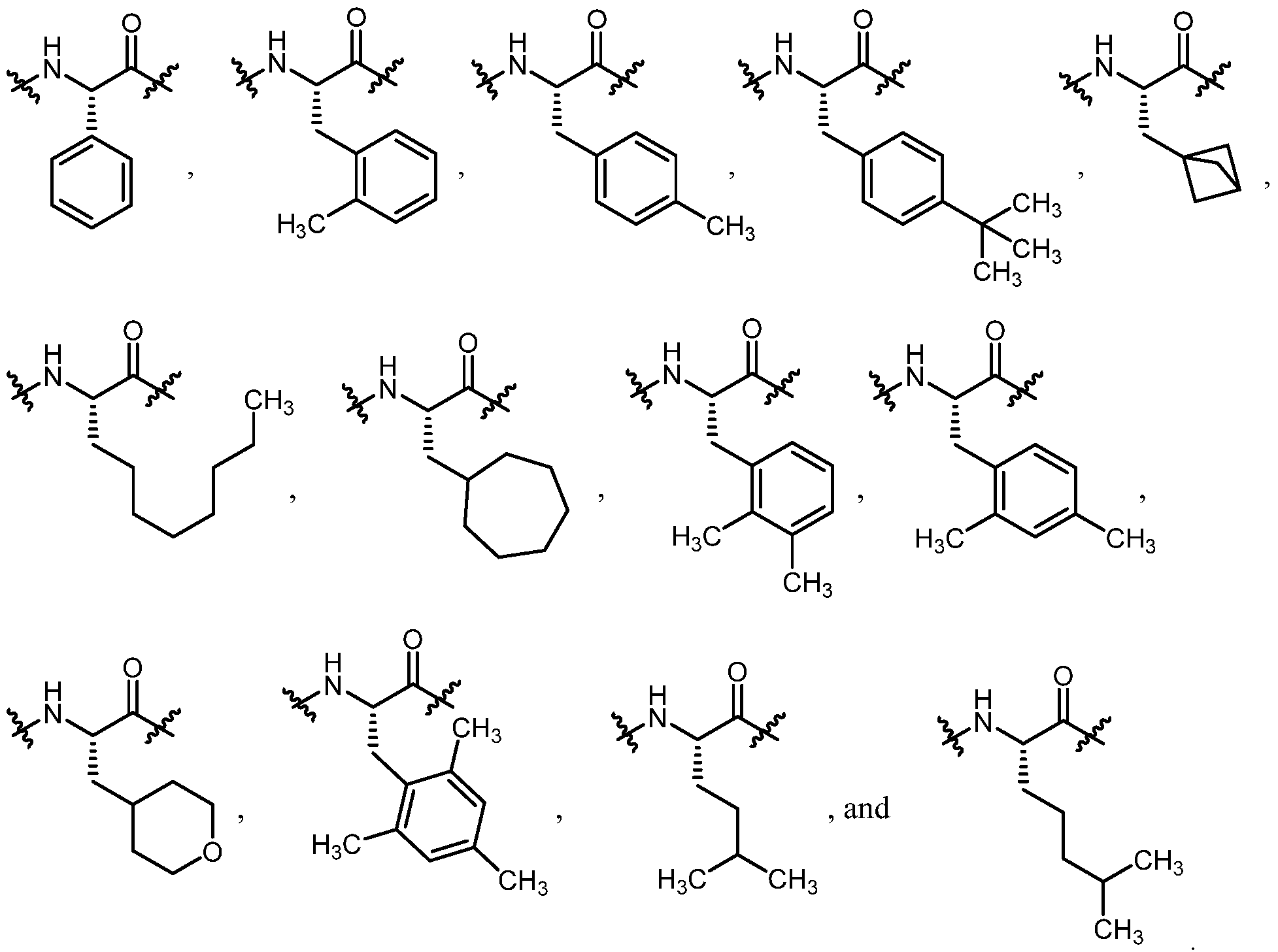






































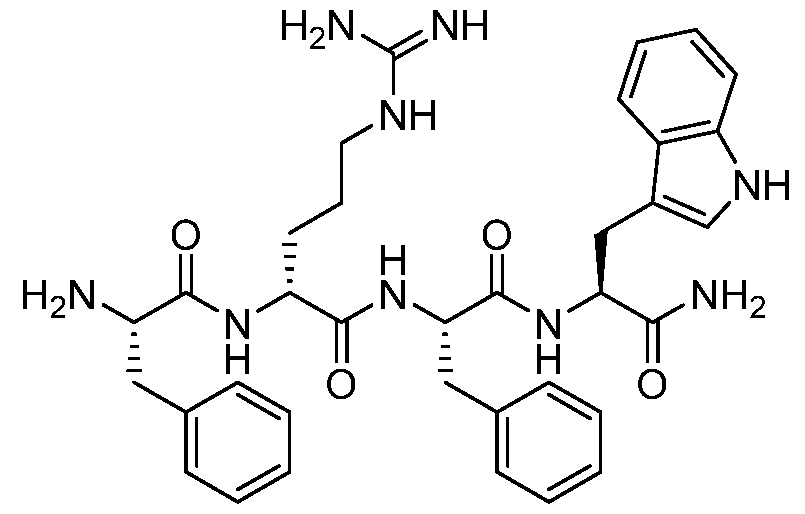






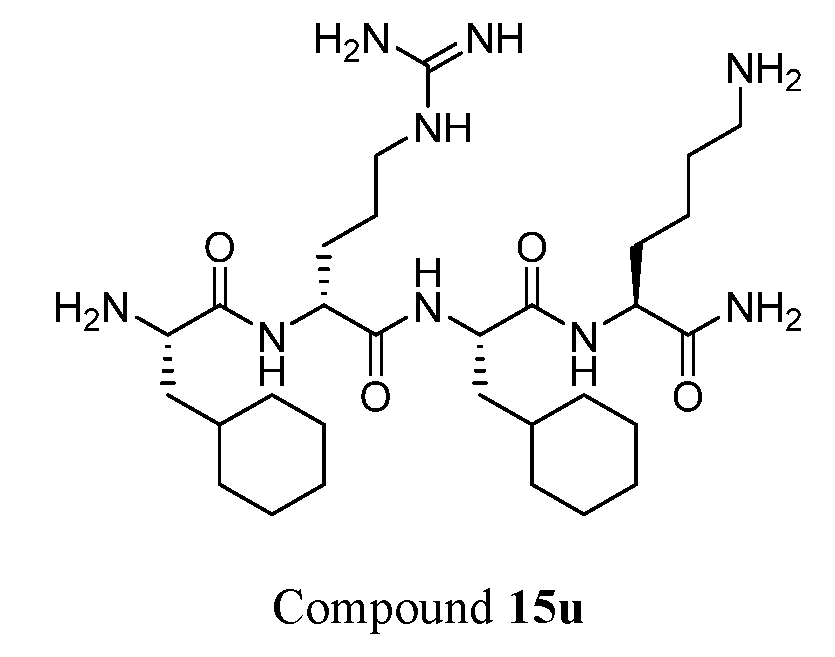







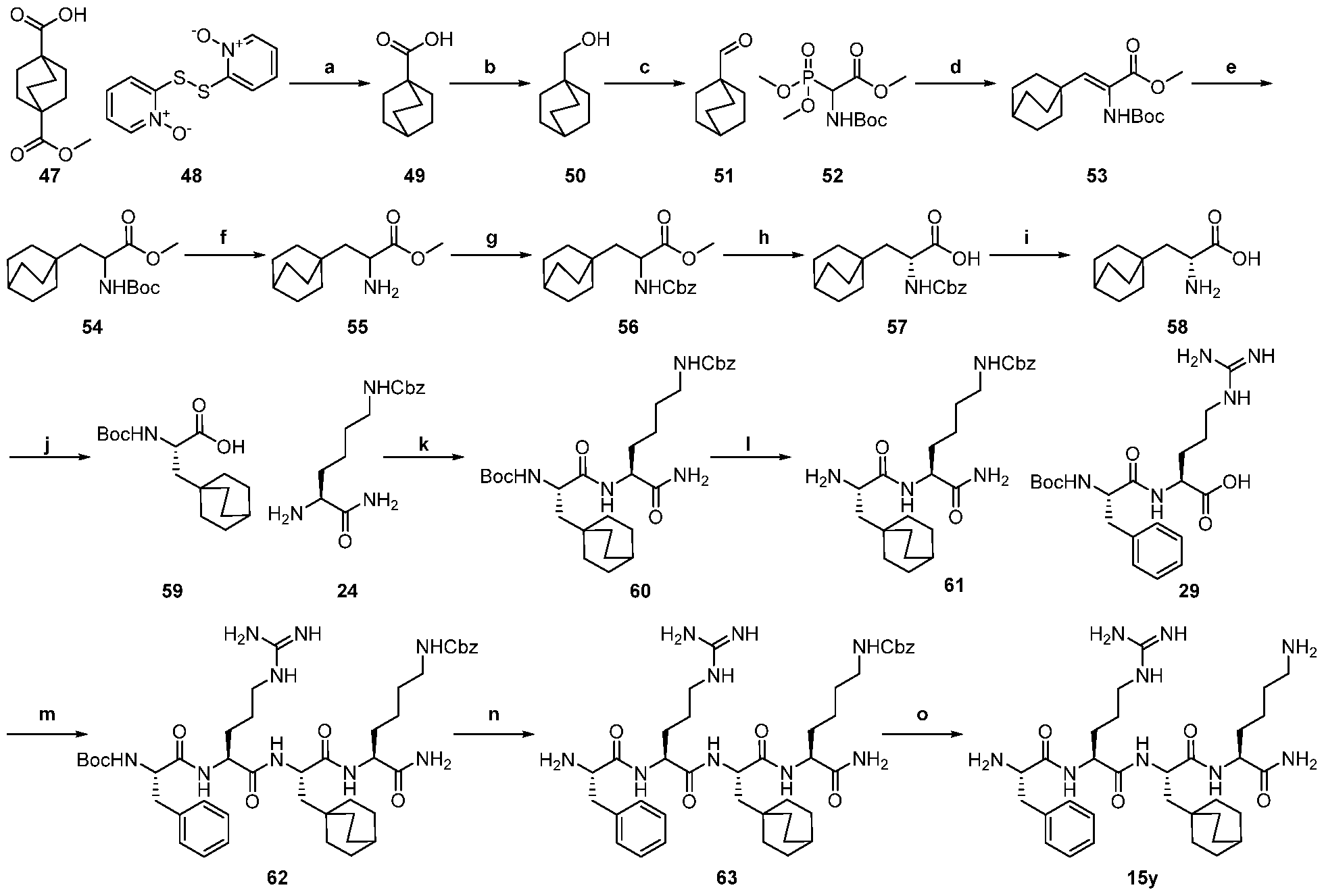





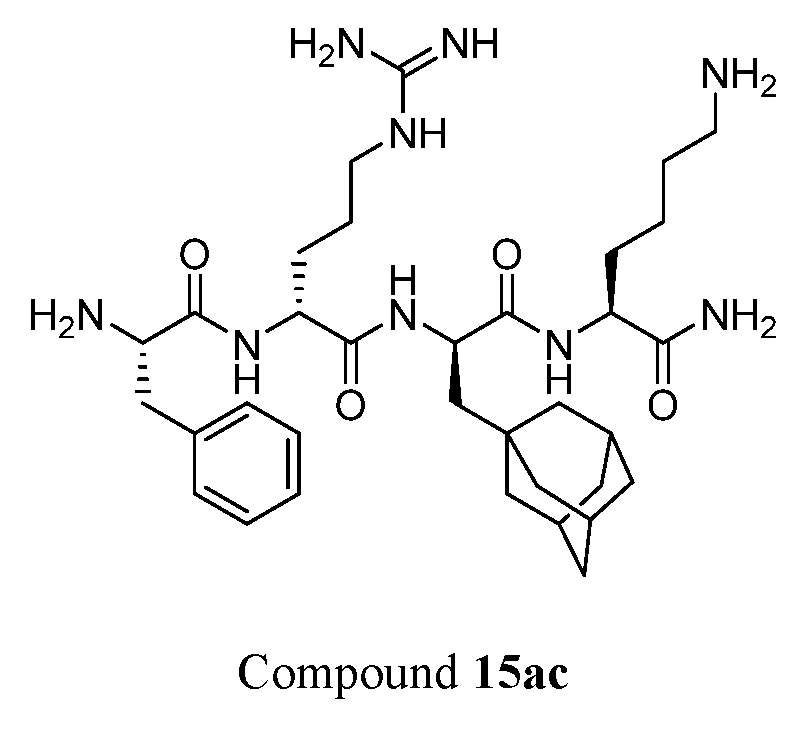
Claims



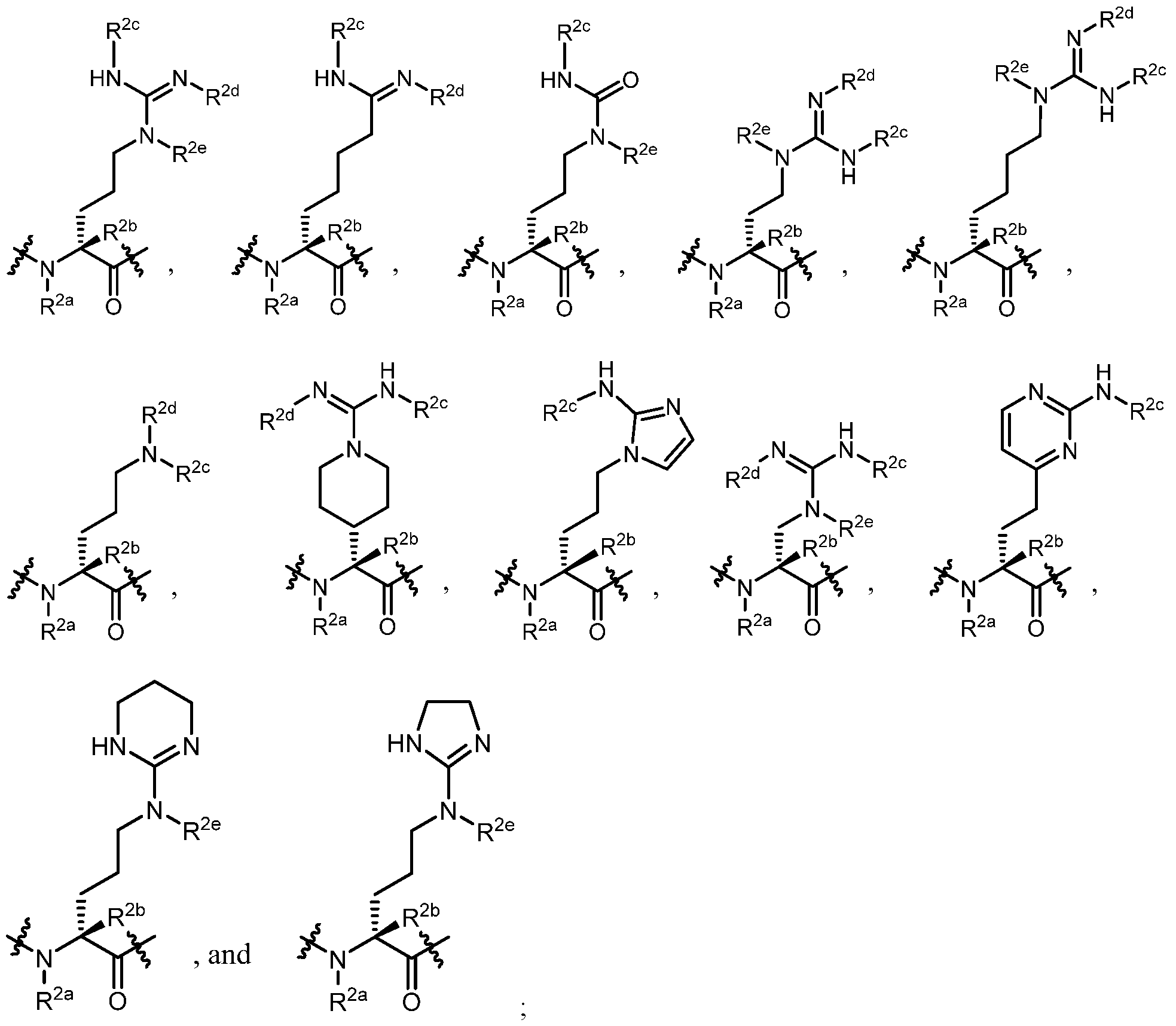










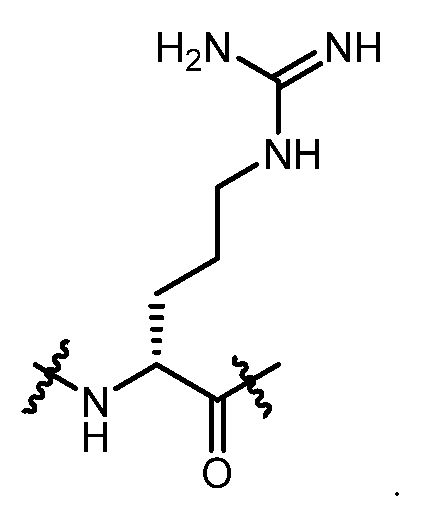















Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201980083951.0A CN113544138A (en) | 2018-12-18 | 2019-11-19 | Mitochondrial targeting peptides |
| US17/414,103 US20220041653A1 (en) | 2018-12-18 | 2019-11-19 | Mitochondria-targeting peptides |
| EP19898721.6A EP3898652A4 (en) | 2018-12-18 | 2019-11-19 | Mitochondria-targeting peptides |
| CA3123368A CA3123368A1 (en) | 2018-12-18 | 2019-11-19 | Mitochondria-targeting peptides |
| AU2019403856A AU2019403856A1 (en) | 2018-12-18 | 2019-11-19 | Mitochondria-targeting peptides |
| KR1020217018413A KR20210093957A (en) | 2018-12-18 | 2019-11-19 | Mitochondria-Targeting Peptides |
| JP2021533219A JP2022511939A (en) | 2018-12-18 | 2019-11-19 | Mitochondrial targeting peptide |
| IL284006A IL284006A (en) | 2018-12-18 | 2021-06-15 | Mitochondria-targeting peptides |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201862781153P | 2018-12-18 | 2018-12-18 | |
| US62/781,153 | 2018-12-18 | ||
| US201962892939P | 2019-08-28 | 2019-08-28 | |
| US62/892,939 | 2019-08-28 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2020131282A1 true WO2020131282A1 (en) | 2020-06-25 |
Family
ID=71101606
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2019/062283 WO2020131282A1 (en) | 2018-12-18 | 2019-11-19 | Mitochondria-targeting peptides |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US20220041653A1 (en) |
| EP (1) | EP3898652A4 (en) |
| JP (1) | JP2022511939A (en) |
| KR (1) | KR20210093957A (en) |
| CN (1) | CN113544138A (en) |
| AU (1) | AU2019403856A1 (en) |
| CA (1) | CA3123368A1 (en) |
| IL (1) | IL284006A (en) |
| WO (1) | WO2020131282A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11332499B2 (en) | 2018-08-16 | 2022-05-17 | Regents Of The University Of Minnesota | Cyclic peptides and methods of use thereof |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011116007A1 (en) * | 2010-03-15 | 2011-09-22 | Stealth Peptides International, Inc. | Combination therapies using cyclosporine and aromatic cationic peptides |
| WO2012022467A2 (en) * | 2010-08-16 | 2012-02-23 | Santhera Pharmaceuticals (Schweiz) Ag | Novel benzoquinone derivatives and use thereof as modulators of mitochondrial function |
| US20140294796A1 (en) * | 2011-12-09 | 2014-10-02 | D. Travis Wilson | Aromatic-cationic peptides and uses of same |
| US20150010588A1 (en) * | 2012-02-23 | 2015-01-08 | Cornell University | Aromatic-cationic peptides and uses of same |
| WO2017180535A1 (en) * | 2016-04-11 | 2017-10-19 | Carnot, Llc | Chiral peptides |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104225574B (en) * | 2003-02-04 | 2017-01-11 | 科内尔研究基金会 | Methods for preventing mitochondrial permeability transition |
| US8383919B2 (en) * | 2010-12-14 | 2013-02-26 | Xueyun Gao | Highly fluorescent peptide-metallic nanoclusters as bio-probes and methods of synthesis thereof |
| CA3006698A1 (en) * | 2015-11-30 | 2017-06-08 | Peter Schiller | Aromatic-cationic peptides conjugated to antioxidants and their use in treating complex regional pain syndrome |
-
2019
- 2019-11-19 AU AU2019403856A patent/AU2019403856A1/en active Pending
- 2019-11-19 US US17/414,103 patent/US20220041653A1/en active Pending
- 2019-11-19 EP EP19898721.6A patent/EP3898652A4/en active Pending
- 2019-11-19 JP JP2021533219A patent/JP2022511939A/en active Pending
- 2019-11-19 WO PCT/US2019/062283 patent/WO2020131282A1/en unknown
- 2019-11-19 KR KR1020217018413A patent/KR20210093957A/en active Search and Examination
- 2019-11-19 CA CA3123368A patent/CA3123368A1/en active Pending
- 2019-11-19 CN CN201980083951.0A patent/CN113544138A/en active Pending
-
2021
- 2021-06-15 IL IL284006A patent/IL284006A/en unknown
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011116007A1 (en) * | 2010-03-15 | 2011-09-22 | Stealth Peptides International, Inc. | Combination therapies using cyclosporine and aromatic cationic peptides |
| WO2012022467A2 (en) * | 2010-08-16 | 2012-02-23 | Santhera Pharmaceuticals (Schweiz) Ag | Novel benzoquinone derivatives and use thereof as modulators of mitochondrial function |
| US20140294796A1 (en) * | 2011-12-09 | 2014-10-02 | D. Travis Wilson | Aromatic-cationic peptides and uses of same |
| US20150010588A1 (en) * | 2012-02-23 | 2015-01-08 | Cornell University | Aromatic-cationic peptides and uses of same |
| WO2017180535A1 (en) * | 2016-04-11 | 2017-10-19 | Carnot, Llc | Chiral peptides |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3898652A4 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11332499B2 (en) | 2018-08-16 | 2022-05-17 | Regents Of The University Of Minnesota | Cyclic peptides and methods of use thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2022511939A (en) | 2022-02-01 |
| EP3898652A1 (en) | 2021-10-27 |
| US20220041653A1 (en) | 2022-02-10 |
| EP3898652A4 (en) | 2022-08-24 |
| CN113544138A (en) | 2021-10-22 |
| CA3123368A1 (en) | 2020-06-25 |
| KR20210093957A (en) | 2021-07-28 |
| IL284006A (en) | 2021-08-31 |
| AU2019403856A1 (en) | 2021-06-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP3723783B1 (en) | Mitochondria-targeting peptides | |
| JP2003524001A (en) | Novel malonic acid derivatives, their preparation, their use as inhibitors of factor Xa activity, and pharmaceutical compositions containing them | |
| US20240092832A1 (en) | Deuterated tetrapeptides that target mitochondria | |
| HRP20010912A2 (en) | FACTOR VIIa INHIBITORS | |
| JP2005526762A (en) | Inhibitors of urokinase, their production and use | |
| WO2020131282A1 (en) | Mitochondria-targeting peptides | |
| WO2021262710A1 (en) | Prodrugs of mitochodria-targeting oligopeptides | |
| WO2020131283A1 (en) | Analogs that target mitochondrial diseases | |
| US20240059734A1 (en) | Mitochondria-targeting peptides | |
| EP4168424A1 (en) | Prodrugs of mitochodria-targeting oligopeptides |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 19898721 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2021533219 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 3123368 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 20217018413 Country of ref document: KR Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2019403856 Country of ref document: AU Date of ref document: 20191119 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2019898721 Country of ref document: EP Effective date: 20210719 |