WO2019173804A1 - Inhibitors of protein arginine methyltransferase 5 (prmt5), pharmaceutical products thereof, and methods thereof - Google Patents
Inhibitors of protein arginine methyltransferase 5 (prmt5), pharmaceutical products thereof, and methods thereof Download PDFInfo
- Publication number
- WO2019173804A1 WO2019173804A1 PCT/US2019/021497 US2019021497W WO2019173804A1 WO 2019173804 A1 WO2019173804 A1 WO 2019173804A1 US 2019021497 W US2019021497 W US 2019021497W WO 2019173804 A1 WO2019173804 A1 WO 2019173804A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- dihydroisoquinolin
- hydroxypropyl
- amino
- oxazepin
- methyl
- Prior art date
Links
- 102100034607 Protein arginine N-methyltransferase 5 Human genes 0.000 title claims abstract description 65
- 238000000034 method Methods 0.000 title claims description 93
- 101000924530 Homo sapiens Protein arginine N-methyltransferase 5 Proteins 0.000 title claims description 63
- 239000003112 inhibitor Substances 0.000 title abstract description 11
- 239000000825 pharmaceutical preparation Substances 0.000 title abstract description 4
- 229940127557 pharmaceutical product Drugs 0.000 title abstract description 4
- 101150097768 prmt5 gene Proteins 0.000 title 1
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 61
- 208000035475 disorder Diseases 0.000 claims abstract description 31
- 206010028980 Neoplasm Diseases 0.000 claims abstract description 25
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 23
- 229910052799 carbon Inorganic materials 0.000 claims abstract description 21
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims abstract description 20
- 239000001257 hydrogen Substances 0.000 claims abstract description 18
- 229910052757 nitrogen Inorganic materials 0.000 claims abstract description 16
- 201000011510 cancer Diseases 0.000 claims abstract description 14
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims abstract description 11
- 125000004043 oxo group Chemical group O=* 0.000 claims abstract description 10
- 125000003003 spiro group Chemical group 0.000 claims abstract description 10
- 229910052760 oxygen Inorganic materials 0.000 claims abstract description 8
- 208000019838 Blood disease Diseases 0.000 claims abstract description 7
- 208000014951 hematologic disease Diseases 0.000 claims abstract description 7
- 208000018706 hematopoietic system disease Diseases 0.000 claims abstract description 7
- 230000002062 proliferating effect Effects 0.000 claims abstract description 7
- 208000030159 metabolic disease Diseases 0.000 claims abstract description 5
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims abstract description 4
- 239000001301 oxygen Substances 0.000 claims abstract description 4
- 150000001875 compounds Chemical class 0.000 claims description 199
- NBJDSMIOWPVWRW-UHFFFAOYSA-N 2h-oxazepin-5-one Chemical compound O=C1C=CNOC=C1 NBJDSMIOWPVWRW-UHFFFAOYSA-N 0.000 claims description 153
- -1 acetylpiperidinyl Chemical group 0.000 claims description 104
- 150000003839 salts Chemical class 0.000 claims description 31
- 125000004432 carbon atom Chemical group C* 0.000 claims description 30
- 201000010099 disease Diseases 0.000 claims description 30
- 239000000651 prodrug Substances 0.000 claims description 23
- 229940002612 prodrug Drugs 0.000 claims description 23
- 125000000217 alkyl group Chemical group 0.000 claims description 21
- 239000012453 solvate Substances 0.000 claims description 21
- 125000004435 hydrogen atom Chemical class [H]* 0.000 claims description 20
- 230000002401 inhibitory effect Effects 0.000 claims description 20
- 150000002148 esters Chemical class 0.000 claims description 19
- 208000007056 sickle cell anemia Diseases 0.000 claims description 18
- 230000014509 gene expression Effects 0.000 claims description 13
- 208000034737 hemoglobinopathy Diseases 0.000 claims description 12
- 239000008194 pharmaceutical composition Substances 0.000 claims description 12
- 102000004190 Enzymes Human genes 0.000 claims description 10
- 108090000790 Enzymes Proteins 0.000 claims description 10
- 125000005842 heteroatom Chemical group 0.000 claims description 10
- 208000031261 Acute myeloid leukaemia Diseases 0.000 claims description 9
- 201000003793 Myelodysplastic syndrome Diseases 0.000 claims description 9
- 206010012601 diabetes mellitus Diseases 0.000 claims description 9
- 230000000694 effects Effects 0.000 claims description 9
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 9
- 125000005843 halogen group Chemical group 0.000 claims description 8
- 230000001404 mediated effect Effects 0.000 claims description 8
- 125000003386 piperidinyl group Chemical group 0.000 claims description 8
- 206010006187 Breast cancer Diseases 0.000 claims description 7
- 208000026310 Breast neoplasm Diseases 0.000 claims description 7
- 206010058467 Lung neoplasm malignant Diseases 0.000 claims description 7
- 206010025323 Lymphomas Diseases 0.000 claims description 7
- 208000008589 Obesity Diseases 0.000 claims description 7
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 7
- 201000007270 liver cancer Diseases 0.000 claims description 7
- 208000014018 liver neoplasm Diseases 0.000 claims description 7
- 201000005202 lung cancer Diseases 0.000 claims description 7
- 208000020816 lung neoplasm Diseases 0.000 claims description 7
- 235000020824 obesity Nutrition 0.000 claims description 7
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 7
- AINNMAPIUSOXEL-UHFFFAOYSA-N 3-[3-(3,4-dihydro-1H-isoquinolin-2-yl)-2-hydroxypropyl]-6-(oxetan-3-ylamino)quinazolin-4-one Chemical compound C1N(CCC2=CC=CC=C12)CC(CN1C=NC2=CC=C(C=C2C1=O)NC1COC1)O AINNMAPIUSOXEL-UHFFFAOYSA-N 0.000 claims description 6
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 claims description 6
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 claims description 6
- 206010035226 Plasma cell myeloma Diseases 0.000 claims description 6
- 150000001721 carbon Chemical group 0.000 claims description 6
- 208000029742 colonic neoplasm Diseases 0.000 claims description 6
- 229910052731 fluorine Inorganic materials 0.000 claims description 6
- 238000000338 in vitro Methods 0.000 claims description 6
- 208000018337 inherited hemoglobinopathy Diseases 0.000 claims description 6
- 208000008443 pancreatic carcinoma Diseases 0.000 claims description 6
- USALWHPSERFAIH-UHFFFAOYSA-N 2-[3-(3,4-dihydro-1H-isoquinolin-2-yl)-2-hydroxypropyl]-7-(pyridazin-4-ylamino)phthalazin-1-one Chemical compound C1N(CCC2=CC=CC=C12)CC(CN1C(C2=CC(=CC=C2C=N1)NC1=CN=NC=C1)=O)O USALWHPSERFAIH-UHFFFAOYSA-N 0.000 claims description 5
- 206010061902 Pancreatic neoplasm Diseases 0.000 claims description 5
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 claims description 5
- ZGLDQGIONSGJCN-UHFFFAOYSA-N diazepin-5-one Chemical compound O=C1C=CN=NC=C1 ZGLDQGIONSGJCN-UHFFFAOYSA-N 0.000 claims description 5
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 claims description 5
- 125000002098 pyridazinyl group Chemical group 0.000 claims description 5
- 238000013518 transcription Methods 0.000 claims description 5
- 230000035897 transcription Effects 0.000 claims description 5
- JXLRQFLREJBDHU-YTZIBCIGSA-N 5-[(1-acetylpiperidin-4-yl)amino]-9-[(2S)-3-(3,4-dihydro-1H-isoquinolin-2-yl)-2-hydroxypropyl]-9-azatricyclo[8.1.1.02,7]dodeca-2(7),3,5-trien-8-one Chemical compound C(C)(=O)N1CCC(CC1)NC=1C=CC2=C(C(N(C3CC2C3)C[C@H](CN2CC3=CC=CC=C3CC2)O)=O)C=1 JXLRQFLREJBDHU-YTZIBCIGSA-N 0.000 claims description 4
- 239000005977 Ethylene Substances 0.000 claims description 4
- 206010060862 Prostate cancer Diseases 0.000 claims description 4
- 208000000236 Prostatic Neoplasms Diseases 0.000 claims description 4
- 230000001594 aberrant effect Effects 0.000 claims description 4
- 208000005980 beta thalassemia Diseases 0.000 claims description 4
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 4
- 239000003937 drug carrier Substances 0.000 claims description 4
- 208000027866 inflammatory disease Diseases 0.000 claims description 4
- 125000004940 pyridazin-4-yl group Chemical group N1=NC=C(C=C1)* 0.000 claims description 4
- 125000000714 pyrimidinyl group Chemical group 0.000 claims description 4
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 claims description 3
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 claims description 3
- 208000010507 Adenocarcinoma of Lung Diseases 0.000 claims description 3
- 208000023275 Autoimmune disease Diseases 0.000 claims description 3
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 claims description 3
- 206010005003 Bladder cancer Diseases 0.000 claims description 3
- 208000003174 Brain Neoplasms Diseases 0.000 claims description 3
- 206010008342 Cervix carcinoma Diseases 0.000 claims description 3
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 claims description 3
- 206010009944 Colon cancer Diseases 0.000 claims description 3
- 208000000461 Esophageal Neoplasms Diseases 0.000 claims description 3
- 208000000172 Medulloblastoma Diseases 0.000 claims description 3
- 208000034578 Multiple myelomas Diseases 0.000 claims description 3
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 claims description 3
- 208000014767 Myeloproliferative disease Diseases 0.000 claims description 3
- 206010030155 Oesophageal carcinoma Diseases 0.000 claims description 3
- 201000010133 Oligodendroglioma Diseases 0.000 claims description 3
- 206010033128 Ovarian cancer Diseases 0.000 claims description 3
- 206010061535 Ovarian neoplasm Diseases 0.000 claims description 3
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 claims description 3
- 208000006265 Renal cell carcinoma Diseases 0.000 claims description 3
- 208000000453 Skin Neoplasms Diseases 0.000 claims description 3
- 208000000102 Squamous Cell Carcinoma of Head and Neck Diseases 0.000 claims description 3
- 208000005718 Stomach Neoplasms Diseases 0.000 claims description 3
- 208000024313 Testicular Neoplasms Diseases 0.000 claims description 3
- 206010057644 Testis cancer Diseases 0.000 claims description 3
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 claims description 3
- 208000006105 Uterine Cervical Neoplasms Diseases 0.000 claims description 3
- 208000002495 Uterine Neoplasms Diseases 0.000 claims description 3
- 208000002517 adenoid cystic carcinoma Diseases 0.000 claims description 3
- 125000002393 azetidinyl group Chemical group 0.000 claims description 3
- 201000010881 cervical cancer Diseases 0.000 claims description 3
- 201000010897 colon adenocarcinoma Diseases 0.000 claims description 3
- 201000004101 esophageal cancer Diseases 0.000 claims description 3
- 206010017758 gastric cancer Diseases 0.000 claims description 3
- 201000009277 hairy cell leukemia Diseases 0.000 claims description 3
- 201000000459 head and neck squamous cell carcinoma Diseases 0.000 claims description 3
- 201000005787 hematologic cancer Diseases 0.000 claims description 3
- 208000024200 hematopoietic and lymphoid system neoplasm Diseases 0.000 claims description 3
- 206010073071 hepatocellular carcinoma Diseases 0.000 claims description 3
- 231100000844 hepatocellular carcinoma Toxicity 0.000 claims description 3
- 208000032839 leukemia Diseases 0.000 claims description 3
- 201000005249 lung adenocarcinoma Diseases 0.000 claims description 3
- 208000008585 mastocytosis Diseases 0.000 claims description 3
- 201000000050 myeloid neoplasm Diseases 0.000 claims description 3
- 201000003707 ovarian clear cell carcinoma Diseases 0.000 claims description 3
- 201000003733 ovarian melanoma Diseases 0.000 claims description 3
- 201000010302 ovarian serous cystadenocarcinoma Diseases 0.000 claims description 3
- 125000003566 oxetanyl group Chemical group 0.000 claims description 3
- 125000003226 pyrazolyl group Chemical group 0.000 claims description 3
- 125000000719 pyrrolidinyl group Chemical group 0.000 claims description 3
- 201000001281 rectum adenocarcinoma Diseases 0.000 claims description 3
- 201000007416 salivary gland adenoid cystic carcinoma Diseases 0.000 claims description 3
- 201000000849 skin cancer Diseases 0.000 claims description 3
- 201000011549 stomach cancer Diseases 0.000 claims description 3
- 201000003120 testicular cancer Diseases 0.000 claims description 3
- 125000001412 tetrahydropyranyl group Chemical group 0.000 claims description 3
- 201000005112 urinary bladder cancer Diseases 0.000 claims description 3
- 206010046766 uterine cancer Diseases 0.000 claims description 3
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 2
- 208000032612 Glial tumor Diseases 0.000 claims description 2
- 206010018338 Glioma Diseases 0.000 claims description 2
- 101000600434 Homo sapiens Putative uncharacterized protein encoded by MIR7-3HG Proteins 0.000 claims description 2
- 102100037401 Putative uncharacterized protein encoded by MIR7-3HG Human genes 0.000 claims description 2
- 125000004567 azetidin-3-yl group Chemical group N1CC(C1)* 0.000 claims description 2
- 125000006299 oxetan-3-yl group Chemical group [H]C1([H])OC([H])([H])C1([H])* 0.000 claims description 2
- 125000005476 oxopyrrolidinyl group Chemical group 0.000 claims description 2
- 125000004430 oxygen atom Chemical group O* 0.000 claims description 2
- 201000002528 pancreatic cancer Diseases 0.000 claims description 2
- 125000004307 pyrazin-2-yl group Chemical group [H]C1=C([H])N=C(*)C([H])=N1 0.000 claims description 2
- 125000002206 pyridazin-3-yl group Chemical group [H]C1=C([H])C([H])=C(*)N=N1 0.000 claims description 2
- 125000004527 pyrimidin-4-yl group Chemical group N1=CN=C(C=C1)* 0.000 claims description 2
- 125000004528 pyrimidin-5-yl group Chemical group N1=CN=CC(=C1)* 0.000 claims description 2
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 claims description 2
- QMNUDYFKZYBWQX-UHFFFAOYSA-N 1H-quinazolin-4-one Chemical compound C1=CC=C2C(=O)N=CNC2=C1 QMNUDYFKZYBWQX-UHFFFAOYSA-N 0.000 claims 2
- OHUOSTWHICFKIF-UHFFFAOYSA-N 1h-1,4-diazepine-2,5-dione Chemical compound O=C1C=CNC(=O)C=N1 OHUOSTWHICFKIF-UHFFFAOYSA-N 0.000 claims 2
- UAYZCDICIHGXOR-UHFFFAOYSA-N 3-[(1-acetylpiperidin-4-yl)amino]-6-[3-(3,4-dihydro-1H-isoquinolin-2-yl)-2-hydroxypropyl]-7,8-dihydro-1,6-naphthyridin-5-one Chemical compound C(C)(=O)N1CCC(CC1)NC=1C=NC=2CCN(C(C=2C=1)=O)CC(CN1CC2=CC=CC=C2CC1)O UAYZCDICIHGXOR-UHFFFAOYSA-N 0.000 claims 2
- ZGYGJQUHLDOYKY-AREMUKBSSA-N 7-[(1-acetylpiperidin-4-yl)amino]-2-[(2R)-3-(3,4-dihydro-1H-isoquinolin-2-yl)-2-hydroxypropyl]-3,4-dihydroisoquinolin-1-one Chemical compound C(C)(=O)N1CCC(CC1)NC1=CC=C2CCN(C(C2=C1)=O)C[C@@H](CN1CC2=CC=CC=C2CC1)O ZGYGJQUHLDOYKY-AREMUKBSSA-N 0.000 claims 2
- IRYXSELPLSAVII-AREMUKBSSA-N C(C)(=O)N1CCC(CC1)NC1=CC=C2C3(CN(C(C2=C1)=O)C[C@@H](CN1CC2=CC=CC=C2CC1)O)CC3 Chemical compound C(C)(=O)N1CCC(CC1)NC1=CC=C2C3(CN(C(C2=C1)=O)C[C@@H](CN1CC2=CC=CC=C2CC1)O)CC3 IRYXSELPLSAVII-AREMUKBSSA-N 0.000 claims 2
- CUCLRKONTPWLHJ-UHFFFAOYSA-N C1N(CCC2=CC=CC=C12)CC(CN1C(C=2C=C(C=NC=2CC1)NC=1C=NN(C=1)C)=O)O Chemical compound C1N(CCC2=CC=CC=C12)CC(CN1C(C=2C=C(C=NC=2CC1)NC=1C=NN(C=1)C)=O)O CUCLRKONTPWLHJ-UHFFFAOYSA-N 0.000 claims 2
- OBHBHIBVGGRKEL-QHCPKHFHSA-N C1N(CCC2=CC=CC=C12)C[C@@H](CN1C(C2=CC(=CC=C2CC1)NC1CN(C1)S(=O)(=O)C)=O)O Chemical compound C1N(CCC2=CC=CC=C12)C[C@@H](CN1C(C2=CC(=CC=C2CC1)NC1CN(C1)S(=O)(=O)C)=O)O OBHBHIBVGGRKEL-QHCPKHFHSA-N 0.000 claims 2
- 125000000218 acetic acid group Chemical group C(C)(=O)* 0.000 claims 2
- 241001024304 Mino Species 0.000 claims 1
- 102200082402 rs751610198 Human genes 0.000 claims 1
- 125000001475 halogen functional group Chemical group 0.000 abstract description 2
- 101710084427 Protein arginine N-methyltransferase 5 Proteins 0.000 abstract 2
- 150000002431 hydrogen Chemical class 0.000 abstract 2
- 208000027755 Blood autoimmune disease Diseases 0.000 abstract 1
- 208000032895 autoimmune disorder of blood Diseases 0.000 abstract 1
- 208000037979 autoimmune inflammatory disease Diseases 0.000 abstract 1
- 239000000243 solution Substances 0.000 description 66
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 64
- 229910001868 water Inorganic materials 0.000 description 64
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 60
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 60
- 210000004027 cell Anatomy 0.000 description 58
- 238000005160 1H NMR spectroscopy Methods 0.000 description 54
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 54
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 48
- 239000011541 reaction mixture Substances 0.000 description 47
- 239000011369 resultant mixture Substances 0.000 description 46
- 239000007858 starting material Substances 0.000 description 42
- 239000007787 solid Substances 0.000 description 41
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 29
- 238000004440 column chromatography Methods 0.000 description 28
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 27
- 239000012074 organic phase Substances 0.000 description 27
- 239000000203 mixture Substances 0.000 description 23
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 22
- 125000004429 atom Chemical group 0.000 description 20
- 241001465754 Metazoa Species 0.000 description 17
- 238000003556 assay Methods 0.000 description 17
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 17
- 238000010992 reflux Methods 0.000 description 17
- 239000012267 brine Substances 0.000 description 15
- 239000003921 oil Substances 0.000 description 15
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 15
- 229940125904 compound 1 Drugs 0.000 description 14
- 239000005457 ice water Substances 0.000 description 14
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 13
- 239000013058 crude material Substances 0.000 description 13
- 239000000741 silica gel Substances 0.000 description 13
- 229910002027 silica gel Inorganic materials 0.000 description 13
- 239000011734 sodium Substances 0.000 description 13
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 12
- 238000006243 chemical reaction Methods 0.000 description 12
- 125000001072 heteroaryl group Chemical group 0.000 description 12
- 125000002950 monocyclic group Chemical group 0.000 description 12
- 125000001424 substituent group Chemical group 0.000 description 12
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 11
- 239000000463 material Substances 0.000 description 11
- 239000000523 sample Substances 0.000 description 11
- 125000003118 aryl group Chemical group 0.000 description 10
- LUCGBEPEAUHERV-UHFFFAOYSA-N pyridazin-4-amine Chemical compound NC1=CC=NN=C1 LUCGBEPEAUHERV-UHFFFAOYSA-N 0.000 description 10
- 239000002904 solvent Substances 0.000 description 10
- CXNIUSPIQKWYAI-UHFFFAOYSA-N xantphos Chemical compound C=12OC3=C(P(C=4C=CC=CC=4)C=4C=CC=CC=4)C=CC=C3C(C)(C)C2=CC=CC=1P(C=1C=CC=CC=1)C1=CC=CC=C1 CXNIUSPIQKWYAI-UHFFFAOYSA-N 0.000 description 10
- 0 CC(N(CC1)CCC1N/C1=C/*C/C=C(/CCN(CC(CN(CC2)Cc3c2cccc3)N)C2=O)\C2=C1)=O Chemical compound CC(N(CC1)CCC1N/C1=C/*C/C=C(/CCN(CC(CN(CC2)Cc3c2cccc3)N)C2=O)\C2=C1)=O 0.000 description 9
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 9
- 108010033040 Histones Proteins 0.000 description 9
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 9
- 230000003833 cell viability Effects 0.000 description 9
- 108090000623 proteins and genes Proteins 0.000 description 9
- 238000012360 testing method Methods 0.000 description 9
- FAAHGTZEAKFSIZ-JOCHJYFZSA-N 4-[(2R)-3-(3,4-dihydro-1H-isoquinolin-2-yl)-2-hydroxypropyl]-7-(pyridazin-4-ylamino)-2,3-dihydro-1,4-benzoxazepin-5-one Chemical compound C1N(CCC2=CC=CC=C12)C[C@H](CN1CCOC2=C(C1=O)C=C(C=C2)NC1=CN=NC=C1)O FAAHGTZEAKFSIZ-JOCHJYFZSA-N 0.000 description 8
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 8
- 208000025205 Mantle-Cell Lymphoma Diseases 0.000 description 8
- 238000004458 analytical method Methods 0.000 description 8
- 230000003197 catalytic effect Effects 0.000 description 8
- 125000000592 heterocycloalkyl group Chemical group 0.000 description 8
- 238000011534 incubation Methods 0.000 description 8
- 229920006395 saturated elastomer Polymers 0.000 description 8
- 108010034457 5'-methylthioadenosine phosphorylase Proteins 0.000 description 7
- 241000282472 Canis lupus familiaris Species 0.000 description 7
- 238000012054 celltiter-glo Methods 0.000 description 7
- 238000004587 chromatography analysis Methods 0.000 description 7
- 239000003814 drug Substances 0.000 description 7
- 238000001727 in vivo Methods 0.000 description 7
- 239000007788 liquid Substances 0.000 description 7
- 239000012071 phase Substances 0.000 description 7
- 238000000926 separation method Methods 0.000 description 7
- 229910000104 sodium hydride Inorganic materials 0.000 description 7
- 208000024891 symptom Diseases 0.000 description 7
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 7
- UWYZHKAOTLEWKK-UHFFFAOYSA-N 1,2,3,4-tetrahydroisoquinoline Chemical compound C1=CC=C2CNCCC2=C1 UWYZHKAOTLEWKK-UHFFFAOYSA-N 0.000 description 6
- WKERQDDTBNIAHT-UHFFFAOYSA-N 7-bromo-2-[3-(3,4-dihydro-1H-isoquinolin-2-yl)-2-hydroxypropyl]-3,4-dihydroisoquinolin-1-one Chemical compound BrC1=CC=C2CCN(C(C2=C1)=O)CC(CN1CC2=CC=CC=C2CC1)O WKERQDDTBNIAHT-UHFFFAOYSA-N 0.000 description 6
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 6
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 6
- 102000003708 Protein arginine N-methyltransferase Human genes 0.000 description 6
- 108020000912 Protein arginine N-methyltransferase Proteins 0.000 description 6
- MEFKEPWMEQBLKI-AIRLBKTGSA-N S-adenosyl-L-methioninate Chemical compound O[C@@H]1[C@H](O)[C@@H](C[S+](CC[C@H](N)C([O-])=O)C)O[C@H]1N1C2=NC=NC(N)=C2N=C1 MEFKEPWMEQBLKI-AIRLBKTGSA-N 0.000 description 6
- 102100034187 S-methyl-5'-thioadenosine phosphorylase Human genes 0.000 description 6
- 239000002253 acid Substances 0.000 description 6
- 229960001570 ademetionine Drugs 0.000 description 6
- 239000006143 cell culture medium Substances 0.000 description 6
- 230000008859 change Effects 0.000 description 6
- 239000003153 chemical reaction reagent Substances 0.000 description 6
- 239000000460 chlorine Substances 0.000 description 6
- 230000005764 inhibitory process Effects 0.000 description 6
- 239000000543 intermediate Substances 0.000 description 6
- 230000004048 modification Effects 0.000 description 6
- 238000012986 modification Methods 0.000 description 6
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 6
- 239000002244 precipitate Substances 0.000 description 6
- 230000035755 proliferation Effects 0.000 description 6
- 239000000758 substrate Substances 0.000 description 6
- 238000011282 treatment Methods 0.000 description 6
- BDFKEHMYAOSLHV-UHFFFAOYSA-N 2-[3-(3,4-dihydro-1H-isoquinolin-2-yl)-2-hydroxypropyl]-3-methyl-7-(oxetan-3-ylamino)-3,4-dihydroisoquinolin-1-one Chemical compound C1N(CCC2=CC=CC=C12)CC(CN1C(C2=CC(=CC=C2CC1C)NC1COC1)=O)O BDFKEHMYAOSLHV-UHFFFAOYSA-N 0.000 description 5
- BRLQWZUYTZBJKN-UHFFFAOYSA-N Epichlorohydrin Chemical compound ClCC1CO1 BRLQWZUYTZBJKN-UHFFFAOYSA-N 0.000 description 5
- 241000699666 Mus <mouse, genus> Species 0.000 description 5
- 208000002903 Thalassemia Diseases 0.000 description 5
- 125000003342 alkenyl group Chemical group 0.000 description 5
- 125000003545 alkoxy group Chemical group 0.000 description 5
- 239000011324 bead Substances 0.000 description 5
- 210000004369 blood Anatomy 0.000 description 5
- 239000008280 blood Substances 0.000 description 5
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 5
- 238000004113 cell culture Methods 0.000 description 5
- 231100000673 dose–response relationship Toxicity 0.000 description 5
- 238000002347 injection Methods 0.000 description 5
- 239000007924 injection Substances 0.000 description 5
- 230000011987 methylation Effects 0.000 description 5
- 238000007069 methylation reaction Methods 0.000 description 5
- 125000003367 polycyclic group Chemical group 0.000 description 5
- 238000002360 preparation method Methods 0.000 description 5
- 102000004169 proteins and genes Human genes 0.000 description 5
- 238000000746 purification Methods 0.000 description 5
- 125000004076 pyridyl group Chemical group 0.000 description 5
- 239000000725 suspension Substances 0.000 description 5
- DABJLFKRJWUXMV-UHFFFAOYSA-N 2-(4-bromophenyl)-2-methylpropanenitrile Chemical compound N#CC(C)(C)C1=CC=C(Br)C=C1 DABJLFKRJWUXMV-UHFFFAOYSA-N 0.000 description 4
- WSRQQVFTQGYDIY-UHFFFAOYSA-N 4-[3-(3,4-dihydro-1H-isoquinolin-2-yl)-2-hydroxypropyl]-1-methyl-7-(pyridazin-4-ylamino)-2,3-dihydro-1,4-benzodiazepin-5-one Chemical compound C1N(CCC2=CC=CC=C12)CC(CN1CCN(C2=C(C1=O)C=C(C=C2)NC1=CN=NC=C1)C)O WSRQQVFTQGYDIY-UHFFFAOYSA-N 0.000 description 4
- WUUGFSXJNOTRMR-IOSLPCCCSA-N 5'-S-methyl-5'-thioadenosine Chemical compound O[C@@H]1[C@H](O)[C@@H](CSC)O[C@H]1N1C2=NC=NC(N)=C2N=C1 WUUGFSXJNOTRMR-IOSLPCCCSA-N 0.000 description 4
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 4
- 101100434927 Caenorhabditis elegans prmt-5 gene Proteins 0.000 description 4
- 230000007067 DNA methylation Effects 0.000 description 4
- 102000006947 Histones Human genes 0.000 description 4
- 101000582546 Homo sapiens Methylosome protein 50 Proteins 0.000 description 4
- 102100030528 Methylosome protein 50 Human genes 0.000 description 4
- 241000700159 Rattus Species 0.000 description 4
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 4
- SOZRMVNRGDOZBY-UHFFFAOYSA-N [1-(4-bromophenyl)cyclopropyl]methanamine Chemical compound C=1C=C(Br)C=CC=1C1(CN)CC1 SOZRMVNRGDOZBY-UHFFFAOYSA-N 0.000 description 4
- 238000010171 animal model Methods 0.000 description 4
- 239000012298 atmosphere Substances 0.000 description 4
- 125000002619 bicyclic group Chemical group 0.000 description 4
- 229910000024 caesium carbonate Inorganic materials 0.000 description 4
- 239000012230 colorless oil Substances 0.000 description 4
- 238000007796 conventional method Methods 0.000 description 4
- DCOPUUMXTXDBNB-UHFFFAOYSA-N diclofenac Chemical compound OC(=O)CC1=CC=CC=C1NC1=C(Cl)C=CC=C1Cl DCOPUUMXTXDBNB-UHFFFAOYSA-N 0.000 description 4
- 229960001259 diclofenac Drugs 0.000 description 4
- 239000012470 diluted sample Substances 0.000 description 4
- 229910001873 dinitrogen Inorganic materials 0.000 description 4
- 229940079593 drug Drugs 0.000 description 4
- 230000002255 enzymatic effect Effects 0.000 description 4
- 235000019439 ethyl acetate Nutrition 0.000 description 4
- 239000012091 fetal bovine serum Substances 0.000 description 4
- 238000001914 filtration Methods 0.000 description 4
- 230000006870 function Effects 0.000 description 4
- 238000001990 intravenous administration Methods 0.000 description 4
- IOMMMLWIABWRKL-WUTDNEBXSA-N nazartinib Chemical compound C1N(C(=O)/C=C/CN(C)C)CCCC[C@H]1N1C2=C(Cl)C=CC=C2N=C1NC(=O)C1=CC=NC(C)=C1 IOMMMLWIABWRKL-WUTDNEBXSA-N 0.000 description 4
- AQHHHDLHHXJYJD-UHFFFAOYSA-N propranolol Chemical compound C1=CC=C2C(OCC(O)CNC(C)C)=CC=CC2=C1 AQHHHDLHHXJYJD-UHFFFAOYSA-N 0.000 description 4
- 235000018102 proteins Nutrition 0.000 description 4
- 238000013207 serial dilution Methods 0.000 description 4
- 229960005322 streptomycin Drugs 0.000 description 4
- 239000000126 substance Substances 0.000 description 4
- 238000003786 synthesis reaction Methods 0.000 description 4
- ITMCEJHCFYSIIV-UHFFFAOYSA-N triflic acid Chemical compound OS(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-N 0.000 description 4
- 239000003643 water by type Substances 0.000 description 4
- GLGNXYJARSMNGJ-VKTIVEEGSA-N (1s,2s,3r,4r)-3-[[5-chloro-2-[(1-ethyl-6-methoxy-2-oxo-4,5-dihydro-3h-1-benzazepin-7-yl)amino]pyrimidin-4-yl]amino]bicyclo[2.2.1]hept-5-ene-2-carboxamide Chemical compound CCN1C(=O)CCCC2=C(OC)C(NC=3N=C(C(=CN=3)Cl)N[C@H]3[C@H]([C@@]4([H])C[C@@]3(C=C4)[H])C(N)=O)=CC=C21 GLGNXYJARSMNGJ-VKTIVEEGSA-N 0.000 description 3
- VIJSPAIQWVPKQZ-BLECARSGSA-N (2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-acetamido-5-(diaminomethylideneamino)pentanoyl]amino]-4-methylpentanoyl]amino]-4,4-dimethylpentanoyl]amino]-4-methylpentanoyl]amino]propanoyl]amino]-5-(diaminomethylideneamino)pentanoic acid Chemical compound NC(=N)NCCC[C@@H](C(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(C)(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(C)=O VIJSPAIQWVPKQZ-BLECARSGSA-N 0.000 description 3
- ONBQEOIKXPHGMB-VBSBHUPXSA-N 1-[2-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)propan-1-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 ONBQEOIKXPHGMB-VBSBHUPXSA-N 0.000 description 3
- YHCYQNXJXRRFFM-UHFFFAOYSA-N 1-bromo-4-(2-nitroprop-1-enyl)benzene Chemical compound [O-][N+](=O)C(C)=CC1=CC=C(Br)C=C1 YHCYQNXJXRRFFM-UHFFFAOYSA-N 0.000 description 3
- LBGSWBJURUFGLR-UHFFFAOYSA-N 1-methylpyrazol-4-amine Chemical compound CN1C=C(N)C=N1 LBGSWBJURUFGLR-UHFFFAOYSA-N 0.000 description 3
- VDIBQWNRSYUNMV-UHFFFAOYSA-N 2-(4-bromophenyl)-2-methylpropan-1-amine Chemical compound NCC(C)(C)C1=CC=C(Br)C=C1 VDIBQWNRSYUNMV-UHFFFAOYSA-N 0.000 description 3
- AXNPVVJPGIMOGK-GOTSBHOMSA-N 2-[(2S)-3-(3,4-dihydro-1H-isoquinolin-2-yl)-2-hydroxypropyl]-7-[[(3S)-oxolan-3-yl]amino]-3,4-dihydroisoquinolin-1-one Chemical compound C1N(CCC2=CC=CC=C12)C[C@@H](CN1C(C2=CC(=CC=C2CC1)N[C@@H]1COCC1)=O)O AXNPVVJPGIMOGK-GOTSBHOMSA-N 0.000 description 3
- UBLCRUHXSMDNQZ-UHFFFAOYSA-N 2-[(4-methoxyphenyl)methylamino]ethanol Chemical compound COC1=CC=C(CNCCO)C=C1 UBLCRUHXSMDNQZ-UHFFFAOYSA-N 0.000 description 3
- SEPIARSNMPOZRD-UHFFFAOYSA-N 2-[3-(3,4-dihydro-1H-isoquinolin-2-yl)-2-hydroxypropyl]-7-[(1-methyl-2-oxopyridin-4-yl)amino]-3,4-dihydroisoquinolin-1-one Chemical compound C1N(CCC2=CC=CC=C12)CC(CN1C(C2=CC(=CC=C2CC1)NC1=CC(N(C=C1)C)=O)=O)O SEPIARSNMPOZRD-UHFFFAOYSA-N 0.000 description 3
- WCUKHIOLFPGARE-UHFFFAOYSA-N 2-[3-(3,4-dihydro-1H-isoquinolin-2-yl)-2-hydroxypropyl]-7-[[1-(oxan-4-yl)pyrazol-4-yl]amino]-3,4-dihydroisoquinolin-1-one Chemical compound C1N(CCC2=CC=CC=C12)CC(CN1C(C2=CC(=CC=C2CC1)NC=1C=NN(C=1)C1CCOCC1)=O)O WCUKHIOLFPGARE-UHFFFAOYSA-N 0.000 description 3
- YWPMKTWUFVOFPL-UHFFFAOYSA-N 3,4-dihydro-2h-isoquinolin-1-one Chemical compound C1=CC=C2C(=O)NCCC2=C1 YWPMKTWUFVOFPL-UHFFFAOYSA-N 0.000 description 3
- JHUUPUMBZGWODW-UHFFFAOYSA-N 3,6-dihydro-1,2-dioxine Chemical compound C1OOCC=C1 JHUUPUMBZGWODW-UHFFFAOYSA-N 0.000 description 3
- ZMGGIIZSNPNIER-UHFFFAOYSA-N 3-[3-(3,4-dihydro-1H-isoquinolin-2-yl)-2-hydroxypropyl]-6-(pyridazin-4-ylamino)quinazolin-4-one Chemical compound C1N(CCC2=CC=CC=C12)CC(CN1C=NC2=CC=C(C=C2C1=O)NC1=CN=NC=C1)O ZMGGIIZSNPNIER-UHFFFAOYSA-N 0.000 description 3
- FAAHGTZEAKFSIZ-QFIPXVFZSA-N 4-[(2S)-3-(3,4-dihydro-1H-isoquinolin-2-yl)-2-hydroxypropyl]-7-(pyridazin-4-ylamino)-2,3-dihydro-1,4-benzoxazepin-5-one Chemical compound C1N(CCC2=CC=CC=C12)C[C@@H](CN1CCOC2=C(C1=O)C=C(C=C2)NC1=CN=NC=C1)O FAAHGTZEAKFSIZ-QFIPXVFZSA-N 0.000 description 3
- YKZDTNIWDJJWMK-UHFFFAOYSA-N 4-[3-(3,4-dihydro-1H-isoquinolin-2-yl)-2-hydroxypropyl]-1-methyl-7-(pyridazin-4-ylamino)-3H-1,4-benzodiazepine-2,5-dione Chemical compound C1N(CCC2=CC=CC=C12)CC(CN1CC(N(C2=C(C1=O)C=C(C=C2)NC1=CN=NC=C1)C)=O)O YKZDTNIWDJJWMK-UHFFFAOYSA-N 0.000 description 3
- XZFUFOLVHMSXFK-UHFFFAOYSA-N 4-[3-(3,4-dihydro-1H-isoquinolin-2-yl)-2-hydroxypropyl]-7-(pyridazin-4-ylamino)-2,3-dihydropyrido[2,3-f][1,4]oxazepin-5-one Chemical compound C1N(CCC2=CC=CC=C12)CC(CN1CCOC2=C(C1=O)N=C(C=C2)NC1=CN=NC=C1)O XZFUFOLVHMSXFK-UHFFFAOYSA-N 0.000 description 3
- HYIOLVRBDBXGGX-UHFFFAOYSA-N 5-bromo-2-fluoro-n-[2-(methylamino)ethyl]benzamide Chemical compound CNCCNC(=O)C1=CC(Br)=CC=C1F HYIOLVRBDBXGGX-UHFFFAOYSA-N 0.000 description 3
- HKYKCMHDQALMNQ-UHFFFAOYSA-N 7-[(1-acetylpiperidin-4-yl)amino]-4-[3-(3,4-dihydro-1H-isoquinolin-2-yl)-2-hydroxypropyl]-2,3-dihydro-1,4-benzoxazepin-5-one Chemical compound C(C)(=O)N1CCC(CC1)NC=1C=CC2=C(C(N(CCO2)CC(CN2CC3=CC=CC=C3CC2)O)=O)C=1 HKYKCMHDQALMNQ-UHFFFAOYSA-N 0.000 description 3
- 108050006400 Cyclin Proteins 0.000 description 3
- 102000016736 Cyclin Human genes 0.000 description 3
- 108020004414 DNA Proteins 0.000 description 3
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 3
- 108091005886 Hemoglobin subunit gamma Proteins 0.000 description 3
- 102100038617 Hemoglobin subunit gamma-2 Human genes 0.000 description 3
- 239000007760 Iscove's Modified Dulbecco's Medium Substances 0.000 description 3
- 101100288142 Mus musculus Klkb1 gene Proteins 0.000 description 3
- YDGMGEXADBMOMJ-LURJTMIESA-N N(g)-dimethylarginine Chemical compound CN(C)C(\N)=N\CCC[C@H](N)C(O)=O YDGMGEXADBMOMJ-LURJTMIESA-N 0.000 description 3
- HVPFXCBJHIIJGS-LURJTMIESA-N N(omega),N'(omega)-dimethyl-L-arginine Chemical compound CN\C(=N/C)NCCC[C@H](N)C(O)=O HVPFXCBJHIIJGS-LURJTMIESA-N 0.000 description 3
- 229940125897 PRMT5 inhibitor Drugs 0.000 description 3
- 229920002565 Polyethylene Glycol 400 Polymers 0.000 description 3
- 101100288143 Rattus norvegicus Klkb1 gene Proteins 0.000 description 3
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical class [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 230000004913 activation Effects 0.000 description 3
- 230000011759 adipose tissue development Effects 0.000 description 3
- 150000001338 aliphatic hydrocarbons Chemical class 0.000 description 3
- 125000000304 alkynyl group Chemical group 0.000 description 3
- 239000008346 aqueous phase Substances 0.000 description 3
- 235000009697 arginine Nutrition 0.000 description 3
- 239000012131 assay buffer Substances 0.000 description 3
- 238000003149 assay kit Methods 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- 230000015572 biosynthetic process Effects 0.000 description 3
- 239000000872 buffer Substances 0.000 description 3
- 238000011088 calibration curve Methods 0.000 description 3
- 238000010609 cell counting kit-8 assay Methods 0.000 description 3
- 229940125758 compound 15 Drugs 0.000 description 3
- 229940126142 compound 16 Drugs 0.000 description 3
- 125000004122 cyclic group Chemical group 0.000 description 3
- 238000010511 deprotection reaction Methods 0.000 description 3
- 238000001514 detection method Methods 0.000 description 3
- 235000013305 food Nutrition 0.000 description 3
- 230000030279 gene silencing Effects 0.000 description 3
- 230000036541 health Effects 0.000 description 3
- 238000004128 high performance liquid chromatography Methods 0.000 description 3
- 150000002430 hydrocarbons Chemical group 0.000 description 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 238000001294 liquid chromatography-tandem mass spectrometry Methods 0.000 description 3
- 238000004020 luminiscence type Methods 0.000 description 3
- 239000002207 metabolite Substances 0.000 description 3
- MOFLZFPEDQFDFD-UHFFFAOYSA-N methyl n-[1-(4-bromophenyl)propan-2-yl]carbamate Chemical compound COC(=O)NC(C)CC1=CC=C(Br)C=C1 MOFLZFPEDQFDFD-UHFFFAOYSA-N 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- 238000003305 oral gavage Methods 0.000 description 3
- 230000002018 overexpression Effects 0.000 description 3
- JLFNLZLINWHATN-UHFFFAOYSA-N pentaethylene glycol Chemical compound OCCOCCOCCOCCOCCO JLFNLZLINWHATN-UHFFFAOYSA-N 0.000 description 3
- 239000002243 precursor Substances 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- 108090000765 processed proteins & peptides Proteins 0.000 description 3
- 230000002285 radioactive effect Effects 0.000 description 3
- 238000001959 radiotherapy Methods 0.000 description 3
- 230000002829 reductive effect Effects 0.000 description 3
- 230000001718 repressive effect Effects 0.000 description 3
- 238000012216 screening Methods 0.000 description 3
- 210000002966 serum Anatomy 0.000 description 3
- 239000012258 stirred mixture Substances 0.000 description 3
- 229910052717 sulfur Inorganic materials 0.000 description 3
- 239000006228 supernatant Substances 0.000 description 3
- 230000001225 therapeutic effect Effects 0.000 description 3
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 2
- YQOLEILXOBUDMU-KRWDZBQOSA-N (4R)-5-[(6-bromo-3-methyl-2-pyrrolidin-1-ylquinoline-4-carbonyl)amino]-4-(2-chlorophenyl)pentanoic acid Chemical compound CC1=C(C2=C(C=CC(=C2)Br)N=C1N3CCCC3)C(=O)NC[C@H](CCC(=O)O)C4=CC=CC=C4Cl YQOLEILXOBUDMU-KRWDZBQOSA-N 0.000 description 2
- NLHBHVGPMMXWIM-UHFFFAOYSA-N 1-(4-aminopiperidin-1-yl)ethanone Chemical compound CC(=O)N1CCC(N)CC1 NLHBHVGPMMXWIM-UHFFFAOYSA-N 0.000 description 2
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 2
- OZAIFHULBGXAKX-UHFFFAOYSA-N 2-(2-cyanopropan-2-yldiazenyl)-2-methylpropanenitrile Chemical compound N#CC(C)(C)N=NC(C)(C)C#N OZAIFHULBGXAKX-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 2
- QHXILXDHCYOAQA-UHFFFAOYSA-N 2h-1,4-oxazepin-5-one Chemical compound O=C1C=COCC=N1 QHXILXDHCYOAQA-UHFFFAOYSA-N 0.000 description 2
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 2
- OLANYRNTAZEXRD-UHFFFAOYSA-N 3-bromo-6-[3-(3,4-dihydro-1H-isoquinolin-2-yl)-2-hydroxypropyl]-7,8-dihydro-1,6-naphthyridin-5-one Chemical compound BrC=1C=NC=2CCN(C(C=2C=1)=O)CC(CN1CC2=CC=CC=C2CC1)O OLANYRNTAZEXRD-UHFFFAOYSA-N 0.000 description 2
- KCUUWMHKSOQWKY-UHFFFAOYSA-N 7-bromo-1-methyl-3,4-dihydro-2H-1,4-benzodiazepin-5-one Chemical compound CN1CCNC(=O)c2cc(Br)ccc12 KCUUWMHKSOQWKY-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 2
- 238000012815 AlphaLISA Methods 0.000 description 2
- 239000004475 Arginine Substances 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- 108090000266 Cyclin-dependent kinases Proteins 0.000 description 2
- 102000003903 Cyclin-dependent kinases Human genes 0.000 description 2
- 102100024812 DNA (cytosine-5)-methyltransferase 3A Human genes 0.000 description 2
- 108010024491 DNA Methyltransferase 3A Proteins 0.000 description 2
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- 206010061218 Inflammation Diseases 0.000 description 2
- PIWKPBJCKXDKJR-UHFFFAOYSA-N Isoflurane Chemical compound FC(F)OC(Cl)C(F)(F)F PIWKPBJCKXDKJR-UHFFFAOYSA-N 0.000 description 2
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 2
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 2
- 241000699670 Mus sp. Species 0.000 description 2
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 2
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 2
- 108091007960 PI3Ks Proteins 0.000 description 2
- 108090000430 Phosphatidylinositol 3-kinases Proteins 0.000 description 2
- 102000003993 Phosphatidylinositol 3-kinases Human genes 0.000 description 2
- 229920001213 Polysorbate 20 Polymers 0.000 description 2
- 102000055027 Protein Methyltransferases Human genes 0.000 description 2
- 108700040121 Protein Methyltransferases Proteins 0.000 description 2
- 208000018020 Sickle cell-beta-thalassemia disease syndrome Diseases 0.000 description 2
- 108010090804 Streptavidin Proteins 0.000 description 2
- 206010043391 Thalassaemia beta Diseases 0.000 description 2
- 239000007983 Tris buffer Substances 0.000 description 2
- 102000044209 Tumor Suppressor Genes Human genes 0.000 description 2
- 108700025716 Tumor Suppressor Genes Proteins 0.000 description 2
- 206010047141 Vasodilatation Diseases 0.000 description 2
- 238000002441 X-ray diffraction Methods 0.000 description 2
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 230000002293 adipogenic effect Effects 0.000 description 2
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 2
- YDGMGEXADBMOMJ-UHFFFAOYSA-N asymmetrical dimethylarginine Natural products CN(C)C(N)=NCCCC(N)C(O)=O YDGMGEXADBMOMJ-UHFFFAOYSA-N 0.000 description 2
- YNHIGQDRGKUECZ-UHFFFAOYSA-L bis(triphenylphosphine)palladium(ii) dichloride Chemical compound [Cl-].[Cl-].[Pd+2].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 YNHIGQDRGKUECZ-UHFFFAOYSA-L 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 2
- 230000004663 cell proliferation Effects 0.000 description 2
- 230000033077 cellular process Effects 0.000 description 2
- 238000010568 chiral column chromatography Methods 0.000 description 2
- 125000001309 chloro group Chemical group Cl* 0.000 description 2
- 229940125844 compound 46 Drugs 0.000 description 2
- 229940126545 compound 53 Drugs 0.000 description 2
- 125000004093 cyano group Chemical group *C#N 0.000 description 2
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 2
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- 230000006718 epigenetic regulation Effects 0.000 description 2
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 2
- 230000001747 exhibiting effect Effects 0.000 description 2
- 230000001605 fetal effect Effects 0.000 description 2
- 238000001640 fractional crystallisation Methods 0.000 description 2
- 238000012226 gene silencing method Methods 0.000 description 2
- 108060003196 globin Proteins 0.000 description 2
- 239000001963 growth medium Substances 0.000 description 2
- 125000004438 haloalkoxy group Chemical group 0.000 description 2
- 125000001188 haloalkyl group Chemical group 0.000 description 2
- 208000020451 hereditary persistence of fetal hemoglobin Diseases 0.000 description 2
- 150000004677 hydrates Chemical class 0.000 description 2
- 125000002768 hydroxyalkyl group Chemical group 0.000 description 2
- 230000004054 inflammatory process Effects 0.000 description 2
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 2
- 229960002725 isoflurane Drugs 0.000 description 2
- 108010082117 matrigel Proteins 0.000 description 2
- 238000013160 medical therapy Methods 0.000 description 2
- 108020004999 messenger RNA Proteins 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- GWPIITVNOVIAMF-UHFFFAOYSA-N methyl 2-[[1-(4-bromophenyl)cyclopropyl]methylcarbamoyloxy]benzoate Chemical compound BrC1=CC=C(C=C1)C1(CC1)CNC(=O)OC1=C(C(=O)OC)C=CC=C1 GWPIITVNOVIAMF-UHFFFAOYSA-N 0.000 description 2
- SVGWVPZUKDEXRT-UHFFFAOYSA-N methyl N-[2-(4-bromophenyl)-2-methylpropyl]carbamate Chemical compound COC(=O)NCC(C)(C)c1ccc(Br)cc1 SVGWVPZUKDEXRT-UHFFFAOYSA-N 0.000 description 2
- XMJHPCRAQCTCFT-UHFFFAOYSA-N methyl chloroformate Chemical compound COC(Cl)=O XMJHPCRAQCTCFT-UHFFFAOYSA-N 0.000 description 2
- 239000012046 mixed solvent Substances 0.000 description 2
- 230000035772 mutation Effects 0.000 description 2
- YCJZWBZJSYLMPB-UHFFFAOYSA-N n-(2-chloropyrimidin-4-yl)-2,5-dimethyl-1-phenylimidazole-4-carboxamide Chemical compound CC=1N(C=2C=CC=CC=2)C(C)=NC=1C(=O)NC1=CC=NC(Cl)=N1 YCJZWBZJSYLMPB-UHFFFAOYSA-N 0.000 description 2
- 239000012299 nitrogen atmosphere Substances 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- OJEOJUQOECNDND-UHFFFAOYSA-N oxetan-3-amine Chemical compound NC1COC1 OJEOJUQOECNDND-UHFFFAOYSA-N 0.000 description 2
- ZRSNZINYAWTAHE-UHFFFAOYSA-N p-methoxybenzaldehyde Chemical compound COC1=CC=C(C=O)C=C1 ZRSNZINYAWTAHE-UHFFFAOYSA-N 0.000 description 2
- 229910052698 phosphorus Inorganic materials 0.000 description 2
- 125000004574 piperidin-2-yl group Chemical group N1C(CCCC1)* 0.000 description 2
- 125000004483 piperidin-3-yl group Chemical group N1CC(CCC1)* 0.000 description 2
- 125000004482 piperidin-4-yl group Chemical group N1CCC(CC1)* 0.000 description 2
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 2
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 2
- 238000012877 positron emission topography Methods 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 2
- 239000000047 product Substances 0.000 description 2
- CDQXHVDVGLVACE-UHFFFAOYSA-N propan-2-amine Chemical compound [CH2]C(C)N CDQXHVDVGLVACE-UHFFFAOYSA-N 0.000 description 2
- 229960003712 propranolol Drugs 0.000 description 2
- 125000003373 pyrazinyl group Chemical group 0.000 description 2
- VTGOHKSTWXHQJK-UHFFFAOYSA-N pyrimidin-2-ol Chemical compound OC1=NC=CC=N1 VTGOHKSTWXHQJK-UHFFFAOYSA-N 0.000 description 2
- 150000003254 radicals Chemical class 0.000 description 2
- 108020003175 receptors Proteins 0.000 description 2
- 102000005962 receptors Human genes 0.000 description 2
- 230000008672 reprogramming Effects 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- 238000005070 sampling Methods 0.000 description 2
- 230000019491 signal transduction Effects 0.000 description 2
- 150000003384 small molecules Chemical class 0.000 description 2
- DAEPDZWVDSPTHF-UHFFFAOYSA-M sodium pyruvate Chemical compound [Na+].CC(=O)C([O-])=O DAEPDZWVDSPTHF-UHFFFAOYSA-M 0.000 description 2
- 210000001082 somatic cell Anatomy 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- GSUTUOLZLSQMDB-UHFFFAOYSA-N tert-butyl N-[2-[(5-bromo-2-fluorobenzoyl)amino]ethyl]-N-methylcarbamate Chemical compound BrC=1C=CC(=C(C(=O)NCCN(C(OC(C)(C)C)=O)C)C=1)F GSUTUOLZLSQMDB-UHFFFAOYSA-N 0.000 description 2
- 238000004809 thin layer chromatography Methods 0.000 description 2
- 230000002103 transcriptional effect Effects 0.000 description 2
- 230000014616 translation Effects 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 2
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 2
- 238000001946 ultra-performance liquid chromatography-mass spectrometry Methods 0.000 description 2
- 230000024883 vasodilation Effects 0.000 description 2
- 210000003462 vein Anatomy 0.000 description 2
- ASGMFNBUXDJWJJ-JLCFBVMHSA-N (1R,3R)-3-[[3-bromo-1-[4-(5-methyl-1,3,4-thiadiazol-2-yl)phenyl]pyrazolo[3,4-d]pyrimidin-6-yl]amino]-N,1-dimethylcyclopentane-1-carboxamide Chemical compound BrC1=NN(C2=NC(=NC=C21)N[C@H]1C[C@@](CC1)(C(=O)NC)C)C1=CC=C(C=C1)C=1SC(=NN=1)C ASGMFNBUXDJWJJ-JLCFBVMHSA-N 0.000 description 1
- ABJSOROVZZKJGI-OCYUSGCXSA-N (1r,2r,4r)-2-(4-bromophenyl)-n-[(4-chlorophenyl)-(2-fluoropyridin-4-yl)methyl]-4-morpholin-4-ylcyclohexane-1-carboxamide Chemical compound C1=NC(F)=CC(C(NC(=O)[C@H]2[C@@H](C[C@@H](CC2)N2CCOCC2)C=2C=CC(Br)=CC=2)C=2C=CC(Cl)=CC=2)=C1 ABJSOROVZZKJGI-OCYUSGCXSA-N 0.000 description 1
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 1
- IUSARDYWEPUTPN-OZBXUNDUSA-N (2r)-n-[(2s,3r)-4-[[(4s)-6-(2,2-dimethylpropyl)spiro[3,4-dihydropyrano[2,3-b]pyridine-2,1'-cyclobutane]-4-yl]amino]-3-hydroxy-1-[3-(1,3-thiazol-2-yl)phenyl]butan-2-yl]-2-methoxypropanamide Chemical compound C([C@H](NC(=O)[C@@H](C)OC)[C@H](O)CN[C@@H]1C2=CC(CC(C)(C)C)=CN=C2OC2(CCC2)C1)C(C=1)=CC=CC=1C1=NC=CS1 IUSARDYWEPUTPN-OZBXUNDUSA-N 0.000 description 1
- YJLIKUSWRSEPSM-WGQQHEPDSA-N (2r,3r,4s,5r)-2-[6-amino-8-[(4-phenylphenyl)methylamino]purin-9-yl]-5-(hydroxymethyl)oxolane-3,4-diol Chemical compound C=1C=C(C=2C=CC=CC=2)C=CC=1CNC1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O YJLIKUSWRSEPSM-WGQQHEPDSA-N 0.000 description 1
- NWZSZGALRFJKBT-KNIFDHDWSA-N (2s)-2,6-diaminohexanoic acid;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.NCCCC[C@H](N)C(O)=O NWZSZGALRFJKBT-KNIFDHDWSA-N 0.000 description 1
- WWTBZEKOSBFBEM-SPWPXUSOSA-N (2s)-2-[[2-benzyl-3-[hydroxy-[(1r)-2-phenyl-1-(phenylmethoxycarbonylamino)ethyl]phosphoryl]propanoyl]amino]-3-(1h-indol-3-yl)propanoic acid Chemical compound N([C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)O)C(=O)C(CP(O)(=O)[C@H](CC=1C=CC=CC=1)NC(=O)OCC=1C=CC=CC=1)CC1=CC=CC=C1 WWTBZEKOSBFBEM-SPWPXUSOSA-N 0.000 description 1
- STBLNCCBQMHSRC-BATDWUPUSA-N (2s)-n-[(3s,4s)-5-acetyl-7-cyano-4-methyl-1-[(2-methylnaphthalen-1-yl)methyl]-2-oxo-3,4-dihydro-1,5-benzodiazepin-3-yl]-2-(methylamino)propanamide Chemical compound O=C1[C@@H](NC(=O)[C@H](C)NC)[C@H](C)N(C(C)=O)C2=CC(C#N)=CC=C2N1CC1=C(C)C=CC2=CC=CC=C12 STBLNCCBQMHSRC-BATDWUPUSA-N 0.000 description 1
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 description 1
- IWZSHWBGHQBIML-ZGGLMWTQSA-N (3S,8S,10R,13S,14S,17S)-17-isoquinolin-7-yl-N,N,10,13-tetramethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-amine Chemical compound CN(C)[C@H]1CC[C@]2(C)C3CC[C@@]4(C)[C@@H](CC[C@@H]4c4ccc5ccncc5c4)[C@@H]3CC=C2C1 IWZSHWBGHQBIML-ZGGLMWTQSA-N 0.000 description 1
- UDQTXCHQKHIQMH-KYGLGHNPSA-N (3ar,5s,6s,7r,7ar)-5-(difluoromethyl)-2-(ethylamino)-5,6,7,7a-tetrahydro-3ah-pyrano[3,2-d][1,3]thiazole-6,7-diol Chemical compound S1C(NCC)=N[C@H]2[C@@H]1O[C@H](C(F)F)[C@@H](O)[C@@H]2O UDQTXCHQKHIQMH-KYGLGHNPSA-N 0.000 description 1
- MIPHRQMEIYLZFZ-SCSAIBSYSA-N (3r)-oxolan-3-amine Chemical compound N[C@@H]1CCOC1 MIPHRQMEIYLZFZ-SCSAIBSYSA-N 0.000 description 1
- HUWSZNZAROKDRZ-RRLWZMAJSA-N (3r,4r)-3-azaniumyl-5-[[(2s,3r)-1-[(2s)-2,3-dicarboxypyrrolidin-1-yl]-3-methyl-1-oxopentan-2-yl]amino]-5-oxo-4-sulfanylpentane-1-sulfonate Chemical compound OS(=O)(=O)CC[C@@H](N)[C@@H](S)C(=O)N[C@@H]([C@H](C)CC)C(=O)N1CCC(C(O)=O)[C@H]1C(O)=O HUWSZNZAROKDRZ-RRLWZMAJSA-N 0.000 description 1
- MPDDTAJMJCESGV-CTUHWIOQSA-M (3r,5r)-7-[2-(4-fluorophenyl)-5-[methyl-[(1r)-1-phenylethyl]carbamoyl]-4-propan-2-ylpyrazol-3-yl]-3,5-dihydroxyheptanoate Chemical compound C1([C@@H](C)N(C)C(=O)C2=NN(C(CC[C@@H](O)C[C@@H](O)CC([O-])=O)=C2C(C)C)C=2C=CC(F)=CC=2)=CC=CC=C1 MPDDTAJMJCESGV-CTUHWIOQSA-M 0.000 description 1
- MIPHRQMEIYLZFZ-BYPYZUCNSA-N (3s)-oxolan-3-amine Chemical compound N[C@H]1CCOC1 MIPHRQMEIYLZFZ-BYPYZUCNSA-N 0.000 description 1
- 125000006645 (C3-C4) cycloalkyl group Chemical group 0.000 description 1
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 1
- AICIYIDUYNFPRY-UHFFFAOYSA-N 1,3-dihydro-2H-imidazol-2-one Chemical compound O=C1NC=CN1 AICIYIDUYNFPRY-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- IBXMKLPFLZYRQZ-UHFFFAOYSA-N 1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1C=CC(=O)C=CC1=CC=CC=C1 IBXMKLPFLZYRQZ-UHFFFAOYSA-N 0.000 description 1
- AESBEDHSBQUPGR-UHFFFAOYSA-N 1-(3,4-dihydro-1H-isoquinolin-2-yl)-3-[7-(pyridazin-4-ylamino)-1H-phthalazin-2-yl]propan-2-ol Chemical compound C1N(CCC2=CC=CC=C12)CC(CN1CC2=CC(=CC=C2C=N1)NC1=CN=NC=C1)O AESBEDHSBQUPGR-UHFFFAOYSA-N 0.000 description 1
- YUJHTWYBKVAQEN-UHFFFAOYSA-N 1-(3-aminoazetidin-1-yl)ethanone Chemical compound CC(=O)N1CC(N)C1 YUJHTWYBKVAQEN-UHFFFAOYSA-N 0.000 description 1
- PHMLYBPPMKYBKY-UHFFFAOYSA-N 1-(6-amino-2-azaspiro[3.3]heptan-2-yl)ethanone Chemical compound CC(=O)N1CC2(CC(N)C2)C1 PHMLYBPPMKYBKY-UHFFFAOYSA-N 0.000 description 1
- GZZNBQLBGSVOOZ-UHFFFAOYSA-N 1-(oxan-4-yl)pyrazol-4-amine Chemical compound C1=C(N)C=NN1C1CCOCC1 GZZNBQLBGSVOOZ-UHFFFAOYSA-N 0.000 description 1
- KQZLRWGGWXJPOS-NLFPWZOASA-N 1-[(1R)-1-(2,4-dichlorophenyl)ethyl]-6-[(4S,5R)-4-[(2S)-2-(hydroxymethyl)pyrrolidin-1-yl]-5-methylcyclohexen-1-yl]pyrazolo[3,4-b]pyrazine-3-carbonitrile Chemical compound ClC1=C(C=CC(=C1)Cl)[C@@H](C)N1N=C(C=2C1=NC(=CN=2)C1=CC[C@@H]([C@@H](C1)C)N1[C@@H](CCC1)CO)C#N KQZLRWGGWXJPOS-NLFPWZOASA-N 0.000 description 1
- MOGQNVSKBCVIPW-UHFFFAOYSA-N 1-methylpyrazol-3-amine Chemical compound CN1C=CC(N)=N1 MOGQNVSKBCVIPW-UHFFFAOYSA-N 0.000 description 1
- AUJHZIGJKOITDS-UHFFFAOYSA-N 1-methylsulfonylazetidin-3-amine Chemical compound CS(=O)(=O)N1CC(N)C1 AUJHZIGJKOITDS-UHFFFAOYSA-N 0.000 description 1
- RQEUFEKYXDPUSK-UHFFFAOYSA-N 1-phenylethylamine Chemical compound CC(N)C1=CC=CC=C1 RQEUFEKYXDPUSK-UHFFFAOYSA-N 0.000 description 1
- HUTNOYOBQPAKIA-UHFFFAOYSA-N 1h-pyrazin-2-one Chemical compound OC1=CN=CC=N1 HUTNOYOBQPAKIA-UHFFFAOYSA-N 0.000 description 1
- JLZVIWSFUPLSOR-UHFFFAOYSA-N 2,3-difluorobenzoic acid Chemical compound OC(=O)C1=CC=CC(F)=C1F JLZVIWSFUPLSOR-UHFFFAOYSA-N 0.000 description 1
- FQMZXMVHHKXGTM-UHFFFAOYSA-N 2-(1-adamantyl)-n-[2-[2-(2-hydroxyethylamino)ethylamino]quinolin-5-yl]acetamide Chemical compound C1C(C2)CC(C3)CC2CC13CC(=O)NC1=CC=CC2=NC(NCCNCCO)=CC=C21 FQMZXMVHHKXGTM-UHFFFAOYSA-N 0.000 description 1
- MFHFWRBXPQDZSA-UHFFFAOYSA-N 2-(4-bromophenyl)acetonitrile Chemical compound BrC1=CC=C(CC#N)C=C1 MFHFWRBXPQDZSA-UHFFFAOYSA-N 0.000 description 1
- WGABOZPQOOZAOI-UHFFFAOYSA-N 2-[4-[[(3,5-dimethoxy-4-methylbenzoyl)-(3-phenylpropyl)amino]methyl]phenyl]acetic acid Chemical compound COC1=C(C)C(OC)=CC(C(=O)N(CCCC=2C=CC=CC=2)CC=2C=CC(CC(O)=O)=CC=2)=C1 WGABOZPQOOZAOI-UHFFFAOYSA-N 0.000 description 1
- PYRKKGOKRMZEIT-UHFFFAOYSA-N 2-[6-(2-cyclopropylethoxy)-9-(2-hydroxy-2-methylpropyl)-1h-phenanthro[9,10-d]imidazol-2-yl]-5-fluorobenzene-1,3-dicarbonitrile Chemical compound C1=C2C3=CC(CC(C)(O)C)=CC=C3C=3NC(C=4C(=CC(F)=CC=4C#N)C#N)=NC=3C2=CC=C1OCCC1CC1 PYRKKGOKRMZEIT-UHFFFAOYSA-N 0.000 description 1
- CUKXRHLWPSBCTI-UHFFFAOYSA-N 2-amino-5-bromobenzoic acid Chemical compound NC1=CC=C(Br)C=C1C(O)=O CUKXRHLWPSBCTI-UHFFFAOYSA-N 0.000 description 1
- YSUIQYOGTINQIN-UZFYAQMZSA-N 2-amino-9-[(1S,6R,8R,9S,10R,15R,17R,18R)-8-(6-aminopurin-9-yl)-9,18-difluoro-3,12-dihydroxy-3,12-bis(sulfanylidene)-2,4,7,11,13,16-hexaoxa-3lambda5,12lambda5-diphosphatricyclo[13.2.1.06,10]octadecan-17-yl]-1H-purin-6-one Chemical compound NC1=NC2=C(N=CN2[C@@H]2O[C@@H]3COP(S)(=O)O[C@@H]4[C@@H](COP(S)(=O)O[C@@H]2[C@@H]3F)O[C@H]([C@H]4F)N2C=NC3=C2N=CN=C3N)C(=O)N1 YSUIQYOGTINQIN-UZFYAQMZSA-N 0.000 description 1
- TVTJUIAKQFIXCE-HUKYDQBMSA-N 2-amino-9-[(2R,3S,4S,5R)-4-fluoro-3-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-7-prop-2-ynyl-1H-purine-6,8-dione Chemical compound NC=1NC(C=2N(C(N(C=2N=1)[C@@H]1O[C@@H]([C@H]([C@H]1O)F)CO)=O)CC#C)=O TVTJUIAKQFIXCE-HUKYDQBMSA-N 0.000 description 1
- NTNWOCRCBQPEKQ-UHFFFAOYSA-N 2-azaniumyl-5-[(n'-methylcarbamimidoyl)amino]pentanoate Chemical compound CN=C(N)NCCCC(N)C(O)=O NTNWOCRCBQPEKQ-UHFFFAOYSA-N 0.000 description 1
- VRHKBNFQGHNLIJ-UHFFFAOYSA-N 2-bromo-5-fluoropyridine-4-carboxylic acid Chemical compound OC(=O)C1=CC(Br)=NC=C1F VRHKBNFQGHNLIJ-UHFFFAOYSA-N 0.000 description 1
- 125000004974 2-butenyl group Chemical group C(C=CC)* 0.000 description 1
- LFOIDLOIBZFWDO-UHFFFAOYSA-N 2-methoxy-6-[6-methoxy-4-[(3-phenylmethoxyphenyl)methoxy]-1-benzofuran-2-yl]imidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(C=1)=CC=CC=1OCC1=CC=CC=C1 LFOIDLOIBZFWDO-UHFFFAOYSA-N 0.000 description 1
- JESRNIJXVIFVOV-UHFFFAOYSA-N 2-methylpyrazol-3-amine Chemical compound CN1N=CC=C1N JESRNIJXVIFVOV-UHFFFAOYSA-N 0.000 description 1
- 125000004637 2-oxopiperidinyl group Chemical group O=C1N(CCCC1)* 0.000 description 1
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 125000004485 2-pyrrolidinyl group Chemical group [H]N1C([H])([H])C([H])([H])C([H])([H])C1([H])* 0.000 description 1
- QBWKPGNFQQJGFY-QLFBSQMISA-N 3-[(1r)-1-[(2r,6s)-2,6-dimethylmorpholin-4-yl]ethyl]-n-[6-methyl-3-(1h-pyrazol-4-yl)imidazo[1,2-a]pyrazin-8-yl]-1,2-thiazol-5-amine Chemical compound N1([C@H](C)C2=NSC(NC=3C4=NC=C(N4C=C(C)N=3)C3=CNN=C3)=C2)C[C@H](C)O[C@H](C)C1 QBWKPGNFQQJGFY-QLFBSQMISA-N 0.000 description 1
- MAFVYPWCNOGKEE-UHFFFAOYSA-N 3-bromo-7,8-dihydro-6h-1,6-naphthyridin-5-one Chemical compound C1CNC(=O)C2=CC(Br)=CN=C21 MAFVYPWCNOGKEE-UHFFFAOYSA-N 0.000 description 1
- 125000003682 3-furyl group Chemical group O1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 1
- 125000004575 3-pyrrolidinyl group Chemical group [H]N1C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- WYFCZWSWFGJODV-MIANJLSGSA-N 4-[[(1s)-2-[(e)-3-[3-chloro-2-fluoro-6-(tetrazol-1-yl)phenyl]prop-2-enoyl]-5-(4-methyl-2-oxopiperazin-1-yl)-3,4-dihydro-1h-isoquinoline-1-carbonyl]amino]benzoic acid Chemical compound O=C1CN(C)CCN1C1=CC=CC2=C1CCN(C(=O)\C=C\C=1C(=CC=C(Cl)C=1F)N1N=NN=C1)[C@@H]2C(=O)NC1=CC=C(C(O)=O)C=C1 WYFCZWSWFGJODV-MIANJLSGSA-N 0.000 description 1
- CRGNLDFNDMPVBN-UHFFFAOYSA-N 4-amino-1-methylpyridin-2-one Chemical compound CN1C=CC(N)=CC1=O CRGNLDFNDMPVBN-UHFFFAOYSA-N 0.000 description 1
- WBYJZPAHGAGMQX-UHFFFAOYSA-N 4-amino-1-methylpyrrolidin-2-one Chemical compound CN1CC(N)CC1=O WBYJZPAHGAGMQX-UHFFFAOYSA-N 0.000 description 1
- ZRYZBQLXDKPBDU-UHFFFAOYSA-N 4-bromobenzaldehyde Chemical compound BrC1=CC=C(C=O)C=C1 ZRYZBQLXDKPBDU-UHFFFAOYSA-N 0.000 description 1
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 1
- MVXAKOGJWVQPKC-UHFFFAOYSA-N 5-(3-ethynyl-5-fluorophenyl)-2-pyridin-2-yl-4,6,7,8-tetrahydro-[1,3]oxazolo[4,5-c]azepine Chemical compound FC1=CC(C#C)=CC(N2CC=3N=C(OC=3CCC2)C=2N=CC=CC=2)=C1 MVXAKOGJWVQPKC-UHFFFAOYSA-N 0.000 description 1
- JXLRQFLREJBDHU-UHFFFAOYSA-N 5-[(1-acetylpiperidin-4-yl)amino]-9-[3-(3,4-dihydro-1H-isoquinolin-2-yl)-2-hydroxypropyl]-9-azatricyclo[8.1.1.02,7]dodeca-2(7),3,5-trien-8-one Chemical compound C(C)(=O)N1CCC(CC1)NC=1C=CC2=C(C(N(C3CC2C3)CC(CN2CC3=CC=CC=C3CC2)O)=O)C=1 JXLRQFLREJBDHU-UHFFFAOYSA-N 0.000 description 1
- KUZSBKJSGSKPJH-VXGBXAGGSA-N 5-[(9R)-6-[(3R)-3-methylmorpholin-4-yl]-11-oxa-1,3,5-triazatricyclo[7.4.0.02,7]trideca-2,4,6-trien-4-yl]pyrazin-2-amine Chemical compound C[C@@H]1COCCN1c1nc(nc2N3CCOC[C@H]3Cc12)-c1cnc(N)cn1 KUZSBKJSGSKPJH-VXGBXAGGSA-N 0.000 description 1
- BLSMDXUAEFVYID-UHFFFAOYSA-N 5-bromo-2,4-difluorobenzoic acid Chemical compound OC(=O)C1=CC(Br)=C(F)C=C1F BLSMDXUAEFVYID-UHFFFAOYSA-N 0.000 description 1
- GCYMJHNRPKUCQH-UHFFFAOYSA-N 5-bromo-2-fluoro-N-(2-hydroxyethyl)-N-[(4-methoxyphenyl)methyl]pyridine-3-carboxamide Chemical compound BrC=1C=NC(=C(C(=O)N(CC2=CC=C(C=C2)OC)CCO)C=1)F GCYMJHNRPKUCQH-UHFFFAOYSA-N 0.000 description 1
- PEXAZYDITWXYNJ-UHFFFAOYSA-N 5-bromo-2-fluorobenzoic acid Chemical compound OC(=O)C1=CC(Br)=CC=C1F PEXAZYDITWXYNJ-UHFFFAOYSA-N 0.000 description 1
- YDQAWSYIQGVXML-UHFFFAOYSA-N 5-bromo-9-aza-tricyclo[8.1.1.0[2,7]]dodeca-2,4,6-trien-8-one Chemical compound N1C(=O)C2=CC(Br)=CC=C2C2CC1C2 YDQAWSYIQGVXML-UHFFFAOYSA-N 0.000 description 1
- 125000006163 5-membered heteroaryl group Chemical group 0.000 description 1
- XPXWYVCQCNFIIJ-UHFFFAOYSA-N 5-methyl-1,3,4-oxadiazol-2-amine Chemical compound CC1=NN=C(N)O1 XPXWYVCQCNFIIJ-UHFFFAOYSA-N 0.000 description 1
- JZSCYXRZCJCLLX-UHFFFAOYSA-N 6-[3-(3,4-dihydro-1H-isoquinolin-2-yl)-2-hydroxypropyl]-3-(oxetan-3-ylamino)-7,8-dihydro-1,6-naphthyridin-5-one Chemical compound C1N(CCC2=CC=CC=C12)CC(CN1C(C=2C=C(C=NC=2CC1)NC1COC1)=O)O JZSCYXRZCJCLLX-UHFFFAOYSA-N 0.000 description 1
- DXSMYDSFWCOSFM-UHFFFAOYSA-N 6-bromo-1h-3,1-benzoxazine-2,4-dione Chemical compound N1C(=O)OC(=O)C2=CC(Br)=CC=C21 DXSMYDSFWCOSFM-UHFFFAOYSA-N 0.000 description 1
- OVEISJPVPHWEHR-UHFFFAOYSA-N 6-bromo-1h-quinazolin-4-one Chemical compound N1C=NC(=O)C2=CC(Br)=CC=C21 OVEISJPVPHWEHR-UHFFFAOYSA-N 0.000 description 1
- BELKVKMBIAENSA-UHFFFAOYSA-N 6-bromo-3h-2-benzofuran-1-one Chemical compound BrC1=CC=C2COC(=O)C2=C1 BELKVKMBIAENSA-UHFFFAOYSA-N 0.000 description 1
- YGDRQLYJJGEHCC-UHFFFAOYSA-N 6-chloro-3-fluoropyridine-2-carboxylic acid Chemical compound OC(=O)C1=NC(Cl)=CC=C1F YGDRQLYJJGEHCC-UHFFFAOYSA-N 0.000 description 1
- WKERQDDTBNIAHT-IBGZPJMESA-N 7-bromo-2-[(2S)-3-(3,4-dihydro-1H-isoquinolin-2-yl)-2-hydroxypropyl]-3,4-dihydroisoquinolin-1-one Chemical compound BrC1=CC=C2CCN(C(C2=C1)=O)C[C@H](CN1CC2=CC=CC=C2CC1)O WKERQDDTBNIAHT-IBGZPJMESA-N 0.000 description 1
- KQCHCWYEVUFPDE-UHFFFAOYSA-N 7-bromo-2-[3-(3,4-dihydro-1H-isoquinolin-2-yl)-2-hydroxypropyl]-4,4-dimethyl-3H-isoquinolin-1-one Chemical compound BrC1=CC=C2C(CN(C(C2=C1)=O)CC(CN1CC2=CC=CC=C2CC1)O)(C)C KQCHCWYEVUFPDE-UHFFFAOYSA-N 0.000 description 1
- QZOFFZNKHKTVAB-UHFFFAOYSA-N 7-bromo-2-[3-(3,4-dihydro-1H-isoquinolin-2-yl)-2-hydroxypropyl]phthalazin-1-one Chemical compound BrC1=CC=C2C=NN(C(C2=C1)=O)CC(CN1CC2=CC=CC=C2CC1)O QZOFFZNKHKTVAB-UHFFFAOYSA-N 0.000 description 1
- UGOWZEJIWPQYDK-UHFFFAOYSA-N 7-bromo-4,4-dimethyl-2,3-dihydroisoquinolin-1-one Chemical compound BrC1=CC=C2C(C)(C)CNC(=O)C2=C1 UGOWZEJIWPQYDK-UHFFFAOYSA-N 0.000 description 1
- AEXMDEPFMMTBAY-GOSISDBHSA-N 7-bromo-4-[(2R)-3-(3,4-dihydro-1H-isoquinolin-2-yl)-2-hydroxypropyl]-2,3-dihydro-1,4-benzoxazepin-5-one Chemical compound BrC=1C=CC2=C(C(N(CCO2)C[C@@H](CN2CC3=CC=CC=C3CC2)O)=O)C=1 AEXMDEPFMMTBAY-GOSISDBHSA-N 0.000 description 1
- NGFFINMFCQKWGV-UHFFFAOYSA-N 7-bromo-4-[(4-methoxyphenyl)methyl]-2,3-dihydropyrido[3,2-f][1,4]oxazepin-5-one Chemical compound BrC1=CC=2C(N(CCOC=2N=C1)CC1=CC=C(C=C1)OC)=O NGFFINMFCQKWGV-UHFFFAOYSA-N 0.000 description 1
- REUJASKNFVPCDT-UHFFFAOYSA-N 7-bromospiro[2,3-dihydroisoquinoline-4,1'-cyclopropane]-1-one Chemical compound C=1C(Br)=CC=C2C=1C(=O)NCC21CC1 REUJASKNFVPCDT-UHFFFAOYSA-N 0.000 description 1
- MITGKKFYIJJQGL-UHFFFAOYSA-N 9-(4-chlorobenzoyl)-6-methylsulfonyl-2,3-dihydro-1H-carbazol-4-one Chemical compound ClC1=CC=C(C(=O)N2C3=CC=C(C=C3C=3C(CCCC2=3)=O)S(=O)(=O)C)C=C1 MITGKKFYIJJQGL-UHFFFAOYSA-N 0.000 description 1
- OZAIFHULBGXAKX-VAWYXSNFSA-N AIBN Substances N#CC(C)(C)\N=N\C(C)(C)C#N OZAIFHULBGXAKX-VAWYXSNFSA-N 0.000 description 1
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 1
- YCIPQJTZJGUXND-UHFFFAOYSA-N Aglaia odorata Alkaloid Natural products C1=CC(OC)=CC=C1C1(C(C=2C(=O)N3CCCC3=NC=22)C=3C=CC=CC=3)C2(O)C2=C(OC)C=C(OC)C=C2O1 YCIPQJTZJGUXND-UHFFFAOYSA-N 0.000 description 1
- 230000007730 Akt signaling Effects 0.000 description 1
- 206010002091 Anaesthesia Diseases 0.000 description 1
- 101100168911 Arabidopsis thaliana CUL4 gene Proteins 0.000 description 1
- 229930091051 Arenine Natural products 0.000 description 1
- 206010060999 Benign neoplasm Diseases 0.000 description 1
- 208000014644 Brain disease Diseases 0.000 description 1
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 description 1
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 description 1
- OPSIAXILNYHKPB-UHFFFAOYSA-N CC(CC1)C(CC(C)=O)CCC1Nc(cc12)ccc1OCCN(CCCN(CC1)Cc3c1cccc3)C2=O Chemical compound CC(CC1)C(CC(C)=O)CCC1Nc(cc12)ccc1OCCN(CCCN(CC1)Cc3c1cccc3)C2=O OPSIAXILNYHKPB-UHFFFAOYSA-N 0.000 description 1
- CLEGOPRKXGRLCN-UHFFFAOYSA-N CC(CCC(CCC1)CCC1Nc(cc(c(OCCN1CCCN(CC2)Cc3c2cccc3)c2)C1=[U])c2F)O Chemical compound CC(CCC(CCC1)CCC1Nc(cc(c(OCCN1CCCN(CC2)Cc3c2cccc3)c2)C1=[U])c2F)O CLEGOPRKXGRLCN-UHFFFAOYSA-N 0.000 description 1
- JGUWADHWNHKQLG-UHFFFAOYSA-N CCC1N=CC(N)=CC=C1 Chemical compound CCC1N=CC(N)=CC=C1 JGUWADHWNHKQLG-UHFFFAOYSA-N 0.000 description 1
- BQXUPNKLZNSUMC-YUQWMIPFSA-N CCN(CCCCCOCC(=O)N[C@H](C(=O)N1C[C@H](O)C[C@H]1C(=O)N[C@@H](C)c1ccc(cc1)-c1scnc1C)C(C)(C)C)CCOc1ccc(cc1)C(=O)c1c(sc2cc(O)ccc12)-c1ccc(O)cc1 Chemical compound CCN(CCCCCOCC(=O)N[C@H](C(=O)N1C[C@H](O)C[C@H]1C(=O)N[C@@H](C)c1ccc(cc1)-c1scnc1C)C(C)(C)C)CCOc1ccc(cc1)C(=O)c1c(sc2cc(O)ccc12)-c1ccc(O)cc1 BQXUPNKLZNSUMC-YUQWMIPFSA-N 0.000 description 1
- 101150041968 CDC13 gene Proteins 0.000 description 1
- WUZBOJXXYMKMMF-UHFFFAOYSA-N COC1=CC2=NC=3N(C(N(C(C=3N2C=C1)=O)CCC)=O)CCCCNC(=O)C1=CC=C(C=C1)S(=O)(=O)F Chemical compound COC1=CC2=NC=3N(C(N(C(C=3N2C=C1)=O)CCC)=O)CCCCNC(=O)C1=CC=C(C=C1)S(=O)(=O)F WUZBOJXXYMKMMF-UHFFFAOYSA-N 0.000 description 1
- BCDOUWOBZFQAGZ-PWKHNDMNSA-N C[C@H](CN(CC1)Cc2c1cccc2)CN(CCOc(c1c2)cc(F)c2NC2CCC(CCC(C)=O)CCC2)C1=O Chemical compound C[C@H](CN(CC1)Cc2c1cccc2)CN(CCOc(c1c2)cc(F)c2NC2CCC(CCC(C)=O)CCC2)C1=O BCDOUWOBZFQAGZ-PWKHNDMNSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- OKTJSMMVPCPJKN-NJFSPNSNSA-N Carbon-14 Chemical compound [14C] OKTJSMMVPCPJKN-NJFSPNSNSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 108010077544 Chromatin Proteins 0.000 description 1
- 108010060434 Co-Repressor Proteins Proteins 0.000 description 1
- 102000008169 Co-Repressor Proteins Human genes 0.000 description 1
- 229940126639 Compound 33 Drugs 0.000 description 1
- 229940127007 Compound 39 Drugs 0.000 description 1
- 102100041019 Coordinator of PRMT5 and differentiation stimulator Human genes 0.000 description 1
- XFXPMWWXUTWYJX-UHFFFAOYSA-N Cyanide Chemical compound N#[C-] XFXPMWWXUTWYJX-UHFFFAOYSA-N 0.000 description 1
- 108010025468 Cyclin-Dependent Kinase 6 Proteins 0.000 description 1
- 102000013698 Cyclin-Dependent Kinase 6 Human genes 0.000 description 1
- 108010009392 Cyclin-Dependent Kinase Inhibitor p16 Proteins 0.000 description 1
- 102100024458 Cyclin-dependent kinase inhibitor 2A Human genes 0.000 description 1
- 230000033616 DNA repair Effects 0.000 description 1
- 102100025450 DNA replication factor Cdt1 Human genes 0.000 description 1
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 101100168913 Dictyostelium discoideum culD gene Proteins 0.000 description 1
- OTMSDBZUPAUEDD-UHFFFAOYSA-N Ethane Chemical compound CC OTMSDBZUPAUEDD-UHFFFAOYSA-N 0.000 description 1
- RRSNDVCODIMOFX-MPKOGUQCSA-N Fc1c(Cl)cccc1[C@H]1[C@@H](NC2(CCCCC2)[C@@]11C(=O)Nc2cc(Cl)ccc12)C(=O)Nc1ccc(cc1)C(=O)NCCCCCc1cccc2C(=O)N(Cc12)C1CCC(=O)NC1=O Chemical compound Fc1c(Cl)cccc1[C@H]1[C@@H](NC2(CCCCC2)[C@@]11C(=O)Nc2cc(Cl)ccc12)C(=O)Nc1ccc(cc1)C(=O)NCCCCCc1cccc2C(=O)N(Cc12)C1CCC(=O)NC1=O RRSNDVCODIMOFX-MPKOGUQCSA-N 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 241000237858 Gastropoda Species 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 208000032843 Hemorrhage Diseases 0.000 description 1
- 241000238631 Hexapoda Species 0.000 description 1
- 101000748895 Homo sapiens Coordinator of PRMT5 and differentiation stimulator Proteins 0.000 description 1
- 101000914265 Homo sapiens DNA replication factor Cdt1 Proteins 0.000 description 1
- 101000702559 Homo sapiens Probable global transcription activator SNF2L2 Proteins 0.000 description 1
- 101000757216 Homo sapiens Protein arginine N-methyltransferase 1 Proteins 0.000 description 1
- 101100369992 Homo sapiens TNFSF10 gene Proteins 0.000 description 1
- 101000702545 Homo sapiens Transcription activator BRG1 Proteins 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 1
- 238000004566 IR spectroscopy Methods 0.000 description 1
- 102100023915 Insulin Human genes 0.000 description 1
- 108090001061 Insulin Proteins 0.000 description 1
- 102100025169 Max-binding protein MNT Human genes 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 102000016397 Methyltransferase Human genes 0.000 description 1
- 108060004795 Methyltransferase Proteins 0.000 description 1
- 101000687343 Mus musculus PR domain zinc finger protein 1 Proteins 0.000 description 1
- NTWVQPHTOUKMDI-YFKPBYRVSA-N N-Methyl-arginine Chemical compound CN[C@H](C(O)=O)CCCN=C(N)N NTWVQPHTOUKMDI-YFKPBYRVSA-N 0.000 description 1
- 229910017906 NH3H2O Inorganic materials 0.000 description 1
- 238000005481 NMR spectroscopy Methods 0.000 description 1
- 229910020889 NaBH3 Inorganic materials 0.000 description 1
- GEYURAWQNSPGES-UHFFFAOYSA-N OC(CCC(CC1)CCc2c1cccc2)CCC(CCOc(cc1)c2cc1NC1COC1)CC2=O Chemical compound OC(CCC(CC1)CCc2c1cccc2)CCC(CCOc(cc1)c2cc1NC1COC1)CC2=O GEYURAWQNSPGES-UHFFFAOYSA-N 0.000 description 1
- ZAPUSMSDYONTNT-UHFFFAOYSA-N OC(CN(CC1)Cc2c1cccc2)CN(CCc(c1c2)ccc2NI)C1=O Chemical compound OC(CN(CC1)Cc2c1cccc2)CN(CCc(c1c2)ccc2NI)C1=O ZAPUSMSDYONTNT-UHFFFAOYSA-N 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- 108091000080 Phosphotransferase Proteins 0.000 description 1
- 101150096028 Prmt7 gene Proteins 0.000 description 1
- 102100031021 Probable global transcription activator SNF2L2 Human genes 0.000 description 1
- 102100022985 Protein arginine N-methyltransferase 1 Human genes 0.000 description 1
- 229910019891 RuCl3 Inorganic materials 0.000 description 1
- 238000011579 SCID mouse model Methods 0.000 description 1
- 229910006069 SO3H Inorganic materials 0.000 description 1
- 101100094489 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) MRP21 gene Proteins 0.000 description 1
- 101100168914 Schizosaccharomyces pombe (strain 972 / ATCC 24843) pcu4 gene Proteins 0.000 description 1
- PNUZDKCDAWUEGK-CYZMBNFOSA-N Sitafloxacin Chemical compound C([C@H]1N)N(C=2C(=C3C(C(C(C(O)=O)=CN3[C@H]3[C@H](C3)F)=O)=CC=2F)Cl)CC11CC1 PNUZDKCDAWUEGK-CYZMBNFOSA-N 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 239000005864 Sulphur Substances 0.000 description 1
- 102000002259 TNF-Related Apoptosis-Inducing Ligand Receptors Human genes 0.000 description 1
- 108010000449 TNF-Related Apoptosis-Inducing Ligand Receptors Proteins 0.000 description 1
- 108091007178 TNFRSF10A Proteins 0.000 description 1
- 108700012411 TNFSF10 Proteins 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 102100031027 Transcription activator BRG1 Human genes 0.000 description 1
- 108091023040 Transcription factor Proteins 0.000 description 1
- 102000040945 Transcription factor Human genes 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- 102100024598 Tumor necrosis factor ligand superfamily member 10 Human genes 0.000 description 1
- 102100040113 Tumor necrosis factor receptor superfamily member 10A Human genes 0.000 description 1
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 description 1
- 102000006275 Ubiquitin-Protein Ligases Human genes 0.000 description 1
- 108010083111 Ubiquitin-Protein Ligases Proteins 0.000 description 1
- 208000025865 Ulcer Diseases 0.000 description 1
- SPXSEZMVRJLHQG-XMMPIXPASA-N [(2R)-1-[[4-[(3-phenylmethoxyphenoxy)methyl]phenyl]methyl]pyrrolidin-2-yl]methanol Chemical compound C(C1=CC=CC=C1)OC=1C=C(OCC2=CC=C(CN3[C@H](CCC3)CO)C=C2)C=CC=1 SPXSEZMVRJLHQG-XMMPIXPASA-N 0.000 description 1
- LNUFLCYMSVYYNW-ZPJMAFJPSA-N [(2r,3r,4s,5r,6r)-2-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[[(3s,5s,8r,9s,10s,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-3-yl]oxy]-4,5-disulfo Chemical compound O([C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1C[C@@H]2CC[C@H]3[C@@H]4CC[C@@H]([C@]4(CC[C@@H]3[C@@]2(C)CC1)C)[C@H](C)CCCC(C)C)[C@H]1O[C@H](COS(O)(=O)=O)[C@@H](OS(O)(=O)=O)[C@H](OS(O)(=O)=O)[C@H]1OS(O)(=O)=O LNUFLCYMSVYYNW-ZPJMAFJPSA-N 0.000 description 1
- PSLUFJFHTBIXMW-WYEYVKMPSA-N [(3r,4ar,5s,6s,6as,10s,10ar,10bs)-3-ethenyl-10,10b-dihydroxy-3,4a,7,7,10a-pentamethyl-1-oxo-6-(2-pyridin-2-ylethylcarbamoyloxy)-5,6,6a,8,9,10-hexahydro-2h-benzo[f]chromen-5-yl] acetate Chemical compound O([C@@H]1[C@@H]([C@]2(O[C@](C)(CC(=O)[C@]2(O)[C@@]2(C)[C@@H](O)CCC(C)(C)[C@@H]21)C=C)C)OC(=O)C)C(=O)NCCC1=CC=CC=N1 PSLUFJFHTBIXMW-WYEYVKMPSA-N 0.000 description 1
- SMNRFWMNPDABKZ-WVALLCKVSA-N [[(2R,3S,4R,5S)-5-(2,6-dioxo-3H-pyridin-3-yl)-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl] [[[(2R,3S,4S,5R,6R)-4-fluoro-3,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-hydroxyphosphoryl]oxy-hydroxyphosphoryl] hydrogen phosphate Chemical compound OC[C@H]1O[C@H](OP(O)(=O)OP(O)(=O)OP(O)(=O)OP(O)(=O)OC[C@H]2O[C@H]([C@H](O)[C@@H]2O)C2C=CC(=O)NC2=O)[C@H](O)[C@@H](F)[C@@H]1O SMNRFWMNPDABKZ-WVALLCKVSA-N 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- YLEIFZAVNWDOBM-ZTNXSLBXSA-N ac1l9hc7 Chemical compound C([C@H]12)C[C@@H](C([C@@H](O)CC3)(C)C)[C@@]43C[C@@]14CC[C@@]1(C)[C@@]2(C)C[C@@H]2O[C@]3(O)[C@H](O)C(C)(C)O[C@@H]3[C@@H](C)[C@H]12 YLEIFZAVNWDOBM-ZTNXSLBXSA-N 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 239000000853 adhesive Substances 0.000 description 1
- 230000001070 adhesive effect Effects 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 238000009098 adjuvant therapy Methods 0.000 description 1
- 125000004450 alkenylene group Chemical group 0.000 description 1
- 125000005083 alkoxyalkoxy group Chemical group 0.000 description 1
- 150000003973 alkyl amines Chemical class 0.000 description 1
- SRVFFFJZQVENJC-IHRRRGAJSA-N aloxistatin Chemical compound CCOC(=O)[C@H]1O[C@@H]1C(=O)N[C@@H](CC(C)C)C(=O)NCCC(C)C SRVFFFJZQVENJC-IHRRRGAJSA-N 0.000 description 1
- 125000003368 amide group Chemical group 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 239000003708 ampul Substances 0.000 description 1
- 230000037005 anaesthesia Effects 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 150000001484 arginines Chemical class 0.000 description 1
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 1
- 230000003190 augmentative effect Effects 0.000 description 1
- 125000004618 benzofuryl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000004622 benzoxazinyl group Chemical group O1NC(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- KGNDCEVUMONOKF-UGPLYTSKSA-N benzyl n-[(2r)-1-[(2s,4r)-2-[[(2s)-6-amino-1-(1,3-benzoxazol-2-yl)-1,1-dihydroxyhexan-2-yl]carbamoyl]-4-[(4-methylphenyl)methoxy]pyrrolidin-1-yl]-1-oxo-4-phenylbutan-2-yl]carbamate Chemical compound C1=CC(C)=CC=C1CO[C@H]1CN(C(=O)[C@@H](CCC=2C=CC=CC=2)NC(=O)OCC=2C=CC=CC=2)[C@H](C(=O)N[C@@H](CCCCN)C(O)(O)C=2OC3=CC=CC=C3N=2)C1 KGNDCEVUMONOKF-UGPLYTSKSA-N 0.000 description 1
- 150000001602 bicycloalkyls Chemical group 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000031018 biological processes and functions Effects 0.000 description 1
- 229960002685 biotin Drugs 0.000 description 1
- 235000020958 biotin Nutrition 0.000 description 1
- 239000011616 biotin Substances 0.000 description 1
- 230000000740 bleeding effect Effects 0.000 description 1
- 230000017531 blood circulation Effects 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- 210000001185 bone marrow Anatomy 0.000 description 1
- 229910052796 boron Inorganic materials 0.000 description 1
- UWTDFICHZKXYAC-UHFFFAOYSA-N boron;oxolane Chemical compound [B].C1CCOC1 UWTDFICHZKXYAC-UHFFFAOYSA-N 0.000 description 1
- 238000002725 brachytherapy Methods 0.000 description 1
- 210000000481 breast Anatomy 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 1
- 238000012754 cardiac puncture Methods 0.000 description 1
- 230000006369 cell cycle progression Effects 0.000 description 1
- 230000011712 cell development Effects 0.000 description 1
- 230000024245 cell differentiation Effects 0.000 description 1
- 230000033026 cell fate determination Effects 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 230000030570 cellular localization Effects 0.000 description 1
- 230000005754 cellular signaling Effects 0.000 description 1
- 125000003636 chemical group Chemical group 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- 125000003016 chromanyl group Chemical group O1C(CCC2=CC=CC=C12)* 0.000 description 1
- 210000003483 chromatin Anatomy 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 229940125773 compound 10 Drugs 0.000 description 1
- 229940126543 compound 14 Drugs 0.000 description 1
- 229940126212 compound 17a Drugs 0.000 description 1
- 229940125810 compound 20 Drugs 0.000 description 1
- 229940126208 compound 22 Drugs 0.000 description 1
- 229940125833 compound 23 Drugs 0.000 description 1
- 229940125846 compound 25 Drugs 0.000 description 1
- 229940125851 compound 27 Drugs 0.000 description 1
- 229940126214 compound 3 Drugs 0.000 description 1
- 229940125877 compound 31 Drugs 0.000 description 1
- 229940125878 compound 36 Drugs 0.000 description 1
- 229940125807 compound 37 Drugs 0.000 description 1
- 229940127573 compound 38 Drugs 0.000 description 1
- 229940126540 compound 41 Drugs 0.000 description 1
- 229940125936 compound 42 Drugs 0.000 description 1
- 229940127271 compound 49 Drugs 0.000 description 1
- 229940125898 compound 5 Drugs 0.000 description 1
- 230000008876 conformational transition Effects 0.000 description 1
- 239000012059 conventional drug carrier Substances 0.000 description 1
- 238000012937 correction Methods 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- 230000000875 corresponding effect Effects 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 230000001186 cumulative effect Effects 0.000 description 1
- 125000004966 cyanoalkyl group Chemical group 0.000 description 1
- KZZKOVLJUKWSKX-UHFFFAOYSA-N cyclobutanamine Chemical compound NC1CCC1 KZZKOVLJUKWSKX-UHFFFAOYSA-N 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000006547 cyclononyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000005508 decahydronaphthalenyl group Chemical group 0.000 description 1
- 230000006735 deficit Effects 0.000 description 1
- 238000012217 deletion Methods 0.000 description 1
- 230000037430 deletion Effects 0.000 description 1
- 230000017858 demethylation Effects 0.000 description 1
- 238000010520 demethylation reaction Methods 0.000 description 1
- 229910052805 deuterium Inorganic materials 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- UBHZUDXTHNMNLD-UHFFFAOYSA-N dimethylsilane Chemical compound C[SiH2]C UBHZUDXTHNMNLD-UHFFFAOYSA-N 0.000 description 1
- QLBHNVFOQLIYTH-UHFFFAOYSA-L dipotassium;2-[2-[bis(carboxymethyl)amino]ethyl-(carboxylatomethyl)amino]acetate Chemical compound [K+].[K+].OC(=O)CN(CC([O-])=O)CCN(CC(O)=O)CC([O-])=O QLBHNVFOQLIYTH-UHFFFAOYSA-L 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 230000035620 dolor Effects 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 238000010894 electron beam technology Methods 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 230000001973 epigenetic effect Effects 0.000 description 1
- 210000000267 erythroid cell Anatomy 0.000 description 1
- 230000006203 ethylation Effects 0.000 description 1
- 238000006200 ethylation reaction Methods 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 208000021045 exocrine pancreatic carcinoma Diseases 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 125000004428 fluoroalkoxy group Chemical group 0.000 description 1
- 125000003709 fluoroalkyl group Chemical group 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 230000008014 freezing Effects 0.000 description 1
- 238000007710 freezing Methods 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 101150034785 gamma gene Proteins 0.000 description 1
- 230000004077 genetic alteration Effects 0.000 description 1
- 231100000118 genetic alteration Toxicity 0.000 description 1
- 230000037442 genomic alteration Effects 0.000 description 1
- 210000004602 germ cell Anatomy 0.000 description 1
- 230000009477 glass transition Effects 0.000 description 1
- 208000005017 glioblastoma Diseases 0.000 description 1
- 102000018146 globin Human genes 0.000 description 1
- JAXFJECJQZDFJS-XHEPKHHKSA-N gtpl8555 Chemical compound OC(=O)C[C@H](N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@@H]1C(=O)N[C@H](B1O[C@@]2(C)[C@H]3C[C@H](C3(C)C)C[C@H]2O1)CCC1=CC=C(F)C=C1 JAXFJECJQZDFJS-XHEPKHHKSA-N 0.000 description 1
- 238000010438 heat treatment Methods 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine monohydrate Substances O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- 230000003301 hydrolyzing effect Effects 0.000 description 1
- 206010020718 hyperplasia Diseases 0.000 description 1
- 230000002390 hyperplastic effect Effects 0.000 description 1
- 230000001969 hypertrophic effect Effects 0.000 description 1
- 125000004282 imidazolidin-2-yl group Chemical group [H]N1C([H])([H])C([H])([H])N([H])C1([H])* 0.000 description 1
- 125000002632 imidazolidinyl group Chemical group 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 238000000099 in vitro assay Methods 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 239000003701 inert diluent Substances 0.000 description 1
- 230000002757 inflammatory effect Effects 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 238000011081 inoculation Methods 0.000 description 1
- 229940125396 insulin Drugs 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000003384 isochromanyl group Chemical group C1(OCCC2=CC=CC=C12)* 0.000 description 1
- 125000004594 isoindolinyl group Chemical group C1(NCC2=CC=CC=C12)* 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000000555 isopropenyl group Chemical group [H]\C([H])=C(\*)C([H])([H])[H] 0.000 description 1
- 125000003253 isopropoxy group Chemical group [H]C([H])([H])C([H])(O*)C([H])([H])[H] 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000005956 isoquinolyl group Chemical group 0.000 description 1
- 125000004628 isothiazolidinyl group Chemical group S1N(CCC1)* 0.000 description 1
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 1
- 239000004973 liquid crystal related substance Substances 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- RENRQMCACQEWFC-UGKGYDQZSA-N lnp023 Chemical compound C1([C@H]2N(CC=3C=4C=CNC=4C(C)=CC=3OC)CC[C@@H](C2)OCC)=CC=C(C(O)=O)C=C1 RENRQMCACQEWFC-UGKGYDQZSA-N 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- LVWZTYCIRDMTEY-UHFFFAOYSA-N metamizole Chemical compound O=C1C(N(CS(O)(=O)=O)C)=C(C)N(C)N1C1=CC=CC=C1 LVWZTYCIRDMTEY-UHFFFAOYSA-N 0.000 description 1
- 230000001394 metastastic effect Effects 0.000 description 1
- 206010061289 metastatic neoplasm Diseases 0.000 description 1
- XZWYZXLIPXDOLR-UHFFFAOYSA-N metformin Chemical compound CN(C)C(=N)NC(N)=N XZWYZXLIPXDOLR-UHFFFAOYSA-N 0.000 description 1
- 229960003105 metformin Drugs 0.000 description 1
- QAWMIDIJFWHLAQ-UHFFFAOYSA-N methyl 2-(2-methoxycarbonylphenoxy)carbonyloxybenzoate Chemical compound COC(=O)C1=CC=CC=C1OC(=O)OC1=CC=CC=C1C(=O)OC QAWMIDIJFWHLAQ-UHFFFAOYSA-N 0.000 description 1
- UFEJKYYYVXYMMS-UHFFFAOYSA-N methylcarbamic acid Chemical compound CNC(O)=O UFEJKYYYVXYMMS-UHFFFAOYSA-N 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003506 n-propoxy group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])O* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 230000001338 necrotic effect Effects 0.000 description 1
- 238000009099 neoadjuvant therapy Methods 0.000 description 1
- 210000005036 nerve Anatomy 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- LYGJENNIWJXYER-UHFFFAOYSA-N nitromethane Chemical compound C[N+]([O-])=O LYGJENNIWJXYER-UHFFFAOYSA-N 0.000 description 1
- 230000001473 noxious effect Effects 0.000 description 1
- 238000010534 nucleophilic substitution reaction Methods 0.000 description 1
- 239000002773 nucleotide Substances 0.000 description 1
- 125000003729 nucleotide group Chemical group 0.000 description 1
- PIDFDZJZLOTZTM-KHVQSSSXSA-N ombitasvir Chemical compound COC(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@H]1C(=O)NC1=CC=C([C@H]2N([C@@H](CC2)C=2C=CC(NC(=O)[C@H]3N(CCC3)C(=O)[C@@H](NC(=O)OC)C(C)C)=CC=2)C=2C=CC(=CC=2)C(C)(C)C)C=C1 PIDFDZJZLOTZTM-KHVQSSSXSA-N 0.000 description 1
- 231100000590 oncogenic Toxicity 0.000 description 1
- 230000002246 oncogenic effect Effects 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- AHVQYHFYQWKUKB-UHFFFAOYSA-N oxan-4-amine Chemical compound NC1CCOCC1 AHVQYHFYQWKUKB-UHFFFAOYSA-N 0.000 description 1
- 125000000160 oxazolidinyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 230000036407 pain Effects 0.000 description 1
- 230000001717 pathogenic effect Effects 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 239000008177 pharmaceutical agent Substances 0.000 description 1
- 229940124531 pharmaceutical excipient Drugs 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 102000020233 phosphotransferase Human genes 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 230000036470 plasma concentration Effects 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 230000004481 post-translational protein modification Effects 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- 238000002661 proton therapy Methods 0.000 description 1
- XFTQRUTUGRCSGO-UHFFFAOYSA-N pyrazin-2-amine Chemical compound NC1=CN=CC=N1 XFTQRUTUGRCSGO-UHFFFAOYSA-N 0.000 description 1
- 125000004289 pyrazol-3-yl group Chemical group [H]N1N=C(*)C([H])=C1[H] 0.000 description 1
- 125000003072 pyrazolidinyl group Chemical group 0.000 description 1
- LETVJWLLIMJADE-UHFFFAOYSA-N pyridazin-3-amine Chemical compound NC1=CC=CN=N1 LETVJWLLIMJADE-UHFFFAOYSA-N 0.000 description 1
- OYRRZWATULMEPF-UHFFFAOYSA-N pyrimidin-4-amine Chemical compound NC1=CC=NC=N1 OYRRZWATULMEPF-UHFFFAOYSA-N 0.000 description 1
- FVLAYJRLBLHIPV-UHFFFAOYSA-N pyrimidin-5-amine Chemical compound NC1=CN=CN=C1 FVLAYJRLBLHIPV-UHFFFAOYSA-N 0.000 description 1
- HZFPPBMKGYINDF-UHFFFAOYSA-N pyrimidin-5-ylboronic acid Chemical compound OB(O)C1=CN=CN=C1 HZFPPBMKGYINDF-UHFFFAOYSA-N 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- 125000004621 quinuclidinyl group Chemical group N12C(CC(CC1)CC2)* 0.000 description 1
- 238000003044 randomized block design Methods 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 230000007420 reactivation Effects 0.000 description 1
- 230000007115 recruitment Effects 0.000 description 1
- 230000001172 regenerating effect Effects 0.000 description 1
- 230000022532 regulation of transcription, DNA-dependent Effects 0.000 description 1
- 230000000754 repressing effect Effects 0.000 description 1
- 230000001850 reproductive effect Effects 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 102200082946 rs33948578 Human genes 0.000 description 1
- 230000036185 rubor Effects 0.000 description 1
- YBCAZPLXEGKKFM-UHFFFAOYSA-K ruthenium(iii) chloride Chemical compound [Cl-].[Cl-].[Cl-].[Ru+3] YBCAZPLXEGKKFM-UHFFFAOYSA-K 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229940054269 sodium pyruvate Drugs 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 239000007790 solid phase Substances 0.000 description 1
- 238000002798 spectrophotometry method Methods 0.000 description 1
- 210000000130 stem cell Anatomy 0.000 description 1
- 230000000707 stereoselective effect Effects 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 230000008961 swelling Effects 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 238000002626 targeted therapy Methods 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 125000004213 tert-butoxy group Chemical group [H]C([H])([H])C(O*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 1
- JYRWUSXRTGACLY-UHFFFAOYSA-N tert-butyl 4-[[3-(4-methylsulfonylphenyl)-[1,2]oxazolo[4,5-d]pyrimidin-7-yl]oxy]piperidine-1-carboxylate Chemical compound C1CN(C(=O)OC(C)(C)C)CCC1OC1=NC=NC2=C1ON=C2C1=CC=C(S(C)(=O)=O)C=C1 JYRWUSXRTGACLY-UHFFFAOYSA-N 0.000 description 1
- QYJVBVKFXDHFPQ-UHFFFAOYSA-N tert-butyl n-(2-aminoethyl)-n-methylcarbamate Chemical compound NCCN(C)C(=O)OC(C)(C)C QYJVBVKFXDHFPQ-UHFFFAOYSA-N 0.000 description 1
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 1
- UQCWXKSHRQJGPH-UHFFFAOYSA-M tetrabutylazanium;fluoride;hydrate Chemical compound O.[F-].CCCC[N+](CCCC)(CCCC)CCCC UQCWXKSHRQJGPH-UHFFFAOYSA-M 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 125000004192 tetrahydrofuran-2-yl group Chemical group [H]C1([H])OC([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000001984 thiazolidinyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 108091006107 transcriptional repressors Proteins 0.000 description 1
- 238000006276 transfer reaction Methods 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 238000013519 translation Methods 0.000 description 1
- 230000000472 traumatic effect Effects 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- UCPYLLCMEDAXFR-UHFFFAOYSA-N triphosgene Chemical compound ClC(Cl)(Cl)OC(=O)OC(Cl)(Cl)Cl UCPYLLCMEDAXFR-UHFFFAOYSA-N 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- 210000004881 tumor cell Anatomy 0.000 description 1
- 230000005748 tumor development Effects 0.000 description 1
- 208000001072 type 2 diabetes mellitus Diseases 0.000 description 1
- 241000701447 unidentified baculovirus Species 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/472—Non-condensed isoquinolines, e.g. papaverine
- A61K31/4725—Non-condensed isoquinolines, e.g. papaverine containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/517—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with carbocyclic ring systems, e.g. quinazoline, perimidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/553—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having at least one nitrogen and one oxygen as ring hetero atoms, e.g. loxapine, staurosporine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/06—Antianaemics
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D217/00—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems
- C07D217/22—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to carbon atoms of the nitrogen-containing ring
- C07D217/24—Oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D451/00—Heterocyclic compounds containing 8-azabicyclo [3.2.1] octane, 9-azabicyclo [3.3.1] nonane, or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane or granatane alkaloids, scopolamine; Cyclic acetals thereof
- C07D451/02—Heterocyclic compounds containing 8-azabicyclo [3.2.1] octane, 9-azabicyclo [3.3.1] nonane, or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane or granatane alkaloids, scopolamine; Cyclic acetals thereof containing not further condensed 8-azabicyclo [3.2.1] octane or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane; Cyclic acetals thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/08—Bridged systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D498/04—Ortho-condensed systems
Definitions
- the present invention generally relates to inhibitors of protein arginine methyltransferase 5 (PRMT5), pharmaceutical products thereof, and methods thereof.
- PRMT5 protein arginine methyltransferase 5
- the present invention can also include application of the class of compounds and derivatives thereof in treating other disorders or diseases related to PRMT5, for example, prostate cancer, colon cancer, ovarian cancer, skin cancer, testicular cancer, uterine cancer, cervical cancer, esophageal cancer, bladder cancer, gastric cancer, epidermoid cancer, hematopoietic cancer, leukemia such as acute myeloid leukemia (AML), acute lymphocytic leukemia, chronic myeloid leukemia, hairy cell leukemia, myelodysplasia, myeloproliferative disorders, acute myelogenous leukemia
- AML acute myeloid leukemia
- AML acute lymphocytic leukemia
- chronic myeloid leukemia hairy cell leukemia
- Epigenetic regulation of gene expression is an important biological determinant of protein production and cellular differentiation, and it plays a significant pathogenic role in a number of human disorders and diseases.
- epigenetic regulation involves heritable modification of genetic material without changing its nucleotide sequence, and it is mediated by selective and reversible modification of DNA and proteins (e g., histones) that control the conformational transition between transcriptionally active and inactive states of chromatin.
- DNA and proteins e g., histones
- These covalent modifications can be controlled by enzymes, many of which are associated with specific genetic alterations that can cause human diseases.
- the methylation of protein arginine is catalyzed by the protein arginine methyl transferases (PRMTs) family of methyltransferases.
- PRMTs protein arginine methyl transferases
- methylation is a common post-translational modification that regulates numerous cellular processes, including gene transcription, mRNA splicing, DNA repair, protein cellular localization, cell fate determination, and signaling.
- PRMTs there are nine PRMTs annotated in the human genome. The majority of these enzymes are Type I enzymes (PRMT-l, -2, -3, -4, -6, -8) that are capable of mono- and asymmetric dimethylation of arginine, with S-adenosylmethionine (SAM) as the methyl donor.
- SAM S-adenosylmethionine
- PRMT-5, -7 and -9 are Type II enzymes that catalyze symmetric dimethylation of arginines.
- Each PRMT species harbors the characteristic motifs of seven beta strand methyltransferases, as well as additional“double E” and“THW” sequence motifs particular to the PRMT subfamily.
- PRMTS is a general transcriptional repressor that functions with numerous transcription factors and repressor complexes, including BRG1 and hBRM, Blimp 1, and Snail. This enzyme, once recruited to a promoter, symmetrically dimethylates H3R8 and H4R3. Importantly, the H4R3 site is a major target for PRMT1 methylation (ADMA) and is generally regarded as a transcriptional activating mark. Thus, both H4R3me2s (repressive; me2s indicates SDMA modification) and H4R3me2a (active; me2a indicates ADMA modification) marks are produced in vivo. The specifieity of PRMT5 for H3R8 and H4R3 can be altered by its interaction with COPR5 and this could perhaps play an important role in determining PRMT5 corepressor status.
- Chromatin-modifying enzymes may play a role in diseases such as proliferative disorders, metabolic disorders, and blood disorders.
- PRMTs play an important role in cancer. Aberrant expression of PRMTs has been identified in human cancers, and PRMTs are considered to be therapeutic targets. For example, global analysis of histone modifications in prostate cancer has shown that the dim ethylation of histone H4R3 is positively correlated with increasing grade, and these changes are predictive of clinical outcome.
- PRMT5 levels have been shown to be elevated in a panel of lymphoid cancer cell lines as well as mantle cell lymphoma clinical samples.
- PRMT5 interacts with a number of substrates that are involved in a variety of cellular processes, including RNA processing, signal transduction, and transcriptional regulation.
- PRMT5 can directly modify histone H3 and H4, resulting in the repression of gene expression.
- PRMT5 overexpression can stimulate cell growth and induce transformation by directly repressing tumor suppressor genes.
- the transcription factor MYC also safeguards proper pre-messenger-RNA splicing as an essential step in lymphomagenesis.
- MTAP methylthioadenosine phosphorylase
- PRMT5 also plays a role in hemoglobinopathies.
- the developmental switch in human globin gene subtype from fetal to adult that begins at birth heralds the onset of the hemoglobinopathies, b-thalassemia and sickle cell disease (SCD).
- SCD sickle cell disease
- DNA methylation Central to silencing of the gamma-genes is DNA methylation, which marks critical CpG dinucleotides flanking the gene transcriptional start site in adult bone marrow erythroid cells. It has been shown that these marks are established as a consequence of recruitment of the DNA methyl transferase, DNMT3A to the gamma-promoter by PRMT5.
- PRMT5 -mediated methylation of histone H4R3 recruits DNMT3A, coupling histone and DNA methylation in gene silencing.
- PRMT5 induces the repressive histone mark, H4R3me2s, which serves as a template for direct binding of DNMT3 A, and subsequent DNA methylation.
- PRMT5 binding or its enzymatic activity leads to demethylation of the CpG dinucleotides and gene activation.
- PRMT5 binding to the gamma-promoter, and its enzymatic activity are essential for assembly of a multi protein complex on the gamma-promoter, which induces a range of coordinated repressive epigenetic marks. Disruption of this complex leads to reactivation of gamma gene expression.
- [Oi l] One aspect of the present invention provides a compound represented by Formula (I), or a pharmaceutically acceptable salt, ester, prodrug, complex, solvate, isomer, or hydrate thereof, in any crystalline form or in amorphous form.
- Another aspect of the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of Formula (I), or a pharmaceutically acceptable salt, ester, prodrug, complex, solvate, hydrate, or isomer thereof, in any crystalline form or in amorphous form; and a pharmaceutically acceptable excipient.
- kits or packaged pharmaceutical comprising a compound of Formula (I), or a pharmaceutically acceptable salt, ester, prodrug, complex, solvate, hydrate or isomer thereof, in any crystalline form or in amorphous form; and instructions for use thereof.
- Still another aspect of the invention provides a method of inhibiting PRMT5 enzyme, comprising: contacting the PRMT5 enzyme with an effective amount of a compound of Formula (I), or a pharmaceutically acceptable salt, ester, prodrug, complex, solvate, hydrate or isomer thereof, in any crystalline form or in amorphous form.
- a compound of Formula (I) or a pharmaceutically acceptable salt, ester, prodrug, complex, solvate, hydrate or isomer thereof, in any crystalline form or in amorphous form.
- Another aspect of the invention provides a method of a method of treating a disorder or disease mediated by PRMT5 or associated with aberrant PRMT5 activity, comprising administering to a subject in need thereof a therapeutically effective amount of a compound of Formula (I), or a pharmaceutically acceptable salt, ester, prodrug, complex, solvate, hydrate or isomer thereof, in any crystalline form or in amorphous form; or a pharmaceutical composition thereof.
- the disorder or disease may be a proliferative disorder such as cancer, a metabolic disorder such as diabetes or obesity, or a blood disorder such as hemoglobinopathy, e.g. sickle cell anemia or beta-thalessemia.
- Figure 1 shows the tumor volume change after the treatment in mantle cell lymphoma (MCL) Z-138 xenograft efficacy studies using a compound of formula (I) in accordance with an exemplary embodiment of the present invention.
- MCL mantle cell lymphoma
- substituents of compounds of the invention are disclosed in groups or in ranges. It is specifically intended that the invention include each and every individual sub-combination of the members of such groups and ranges.
- the term“C l-6 alkyl” is specifically intended to include Ci alkyl (methyl), C 2 alkyl (ethyl), C 3 alkyl, C 4 alkyl, C 5 alkyl, and C 6 alkyl.
- a numerical range is disclosed herein, unless otherwise specified, such range is continuous, inclusive of both the minimum and maximum values of the range as well as every value between such minimum and maximum values.
- a range refers to integers, only the integers from the minimum value to and including the maximum value of such range are included.
- multiple ranges are provided to describe a feature or characteristic, such ranges can be combined.
- the present invention provides a compound of Formula (1), or a pharmaceutically acceptable salt, ester, prodrug, complex, solvate, or hydrate thereof.
- a bivalent spiro ring-forming group is defined as a group comprising two atoms (two valents) which can be bonded to a same ring member atom.
- R 3 is a bivalent spiro ring-forming group such as an ethylene spiro ring-forming group -CH 2 CH 2 -, in which two carbon atoms each can be bonded to a same ring member atom such as C.
- two specific compounds of the invention include such an ethylene spiro ring-forming group -CH 2 CH 2 -, i.e.
- a bivalent bridge-forming group is defined as a group that can be bonded to two ring member atoms.
- the two ring member atoms may be adjacent to each other (fused), or not adjacent to each other in the ring (bridged).
- R 3 is a bivalent bridge-forming group such as a methylene bridge-forming group -CH 2 - that can be bonded or attached to two ring-member atoms such as two carbon atoms in a compound of the invention, e.g.
- R is indepedntly of each other any suitable group e.g. alkyl group.
- -OR may be an alkoxy or alkyloxy group, i.e. an -O-alkyl group.
- C l-6 alkoxy/alkyloxy is an -0-(C l-6 alkyl) group.
- alkoxy include methoxy, ethoxy, propoxy (e.g , n-propoxy and isopropoxy), tert-butoxy, and the like.
- the alkoxy or alkyloxy group optionally can be substituted by 1 or more (e.g., 1 to 5) suitable substituents.
- Class (2) Alkyl group, i.e. saturated aliphatic hydrocarbon including straight chains and branched chains.
- the alkyl group has 1 to 20 carbon atoms, 1 to 10 carbon atoms, 1 to 6 carbon atoms, or 1 to 4 carbon atoms.
- the term“Ci -6 alkyl” refers to linear or branched radicals of 1 to 6 carbon atoms (e.g , methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl, n-pentyl, or n-hexyl).
- An alkyl group optionally can be substituted by one or more (e.g., 1 to 5) suitable substituents,
- Class (3) Alkenyl group, i.e. aliphatic hydrocarbon having at least one carbon-carbon double bond, including straight chains and branched chains having at least one carbon-carbon double bond.
- the alkenyl group has 2 to 20 carbon atoms, 2 to 10 carbon atoms, 2 to 6 carbon atoms, 3 to 6 carbon atoms, or 2 to 4 carbon atoms.
- C 2-6 alkenyl includes straight or branched chain unsaturated radicals (having at least one carbon-carbon double bond) of 2 to 6 carbon atoms, including, but not limited to, ethenyl.
- alkenyl group optionally can be substituted by one or more (e.g., 1 to 5) suitable substituents.
- the alkenyl group may exist as the pure E form, the pure Z form, or any mixture thereof.
- Cycloalkyl group may be saturated or unsaturated, non-aromatic, monocyclic or polycyclic (such as bicyclic) hydrocarbon rings (e.g., monocyclics such as cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, cyclononyl, or bicyclics including spiro, fused, or bridged systems (such as bicyclo[ 1.1.1 ]pentanyl, bicyclo[2.2.l]heptanyl, bicyclo[3.2.
- monocyclics such as cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, cyclononyl
- bicyclics including spiro, fused, or bridged systems (such as bicyclo[ 1.1.1 ]pentanyl, bicyclo[
- the cycloalkyl group has 3 to 15 carbon atoms. In some embodiments the cycloalkyl may optionally contain one, two or more non-cumulative non-aromatic double or triple bonds and/or one to three oxo groups. In some embodiments, the bicycloalkyl group has 6 to 14 carbon atoms.
- “C 3-l4 cycloalkyl” includes saturated or unsaturated, non-aromatic, monocyclic or polycyclic (such as bicyclic) hydrocarbon rings of 3 to 14 ring-forming carbon atoms (e.g., cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, bicyclo [l. l.l]pentanyl, or cyclodecanyl).
- the cycloalkyl group optionally can be substituted by 1 or more (e.g., 1 to 5) suitable substituents.
- Class (7) Heteroaryl group, i.e monocyclic or fused-ring polycyclic aromatic heterocyclic groups with one or more heteroatom ring members (ring-forming atoms) each independently selected from O, S and N in at least one ring.
- the heteroaryl group has 5 to 14 ring-forming atoms, including 1 to 13 carbon atoms, and 1 to 8 heteroatoms selected from O, S, and N.
- the heteroaryl group has 5 to 10 ring-forming atoms including one to four heteroatoms.
- the heteroaryl group has 5 to 8 ring-forming atoms including one, two or three heteroatoms.
- 5-membered heteroaryl group is a monocyclic heteroaryl group as defined above with 5 ring-forming atoms in the monocyclic heteroaryl ring
- 6-membered heteroaryl is a monocyclic heteroaryl group as defined above with 6 ring-forming atoms in the monocyclic heteroaryl ring
- 5 ⁇ l0-membered heteroaryl is a monocyclic or bicyclic heteroaryl group as defined above with 5, 6, 7, 8, 9 or 10 ring-forming atoms in the monocyclic or bicyclic heteroaryl ring.
- a heteroaryl group optionally can be substituted by 1 or more (e.g., 1 to 5) suitable substituents.
- suitable substituents e.g. 1 to 5
- monocyclic heteroaryls include those with 5 ring-forming atoms including one to three heteroatoms or those with 6 ring-forming atoms including one, two or three nitrogen heteroatoms.
- fused bicyclic heteroaryls include two fused 5- and/or 6-membered monocyclic rings including one to four heteroatoms.
- pyrazolyl e g., pyrazol-l-yl, pyrazol-3-yl, pyrazol-4-yl
- tetrazolyl e.g., triazolyl
- Class (8) Heterocycloalkyl group, i.e. monocyclic or polycyclic (including 2 or more rings that are fused together, including spiro, fused, or bridged systems, for example, a bicyclic ring system), saturated or unsaturated, non-aromatic 4- to 15-membered ring system including 1 to 14 ring-forming carbon atoms and 1 to 10 ring-forming heteroatoms each independently selected from O, S, N, P and B.
- 4- to l2-membered heterocycloalkyl is a monocyclic or polycyclic, saturated or unsaturated, non-aromatic 4- to 12-membered ring system that comprises one or more ring-forming heteroatoms.
- heterocycloalkyl rings include azetidinyl, tetrahy dr ofuranyl , imidazoli dinyl, pyrrolidinyl, piperidinyl, piperazinyl, oxazolidinyl, thiazolidinyl, pyrazolidinyl, thiomorpholinyl, tetrahy drothi azinyl , tetrahydrothiadiazinyl, morpholinyl, oxetanyl, tetrahy drodi azinyl , oxazinyl, oxathi azinyl, quinuclidinyl, chromanyl, isochro
- heterocycloalkyl rings include tetrahydrofuran-2-yl, tetrahy drofuran-3 -yl, tetrahy dropyranyl (e.g., tetrahydro-2H-pyran-4-yl), imidazolidin-l-yl, imidazolidin-2-yl, imidazolidin-4-yl, pyrrolidin-l-yl, pyrrolidin-2-yl, pyrrolidin-3-yl, piperidin-l-yl, piperidin-2-yl, piperidin-3-yl, piperidin-4-yl, piperazin-l-yl, piperazin-2-yl, l,3-oxazolidin-3-yl, l,4-oxazepan-l-yl, isothiazoli dinyl, l,3-thiazolidin
- heterocycloalkyl group is optionally substituted by 1 or more (e.g., 1 to 5) suitable substituents.
- suitable substituents include 5- or 6-membered monocyclic rings and 9- or lO-membered fused bi cyclic rings.
- the teim“non-hydrogen monovalent group” may include a combination of any number of groups selected from the above 8 classes. By a combination of two groups, it means that one group (Gl) is substituted with another group (G2) to form a new group -G1-G2. By combination of three groups, it means that a first group (Gl) is substituted with a second group (G2) which is substituted with a third group (G3), forming a new group -G1-G2-G3.
- a group from Classes (2)-(8) may be substituted with a group from Class (1): (i) Flaloalkyl group such as fluoroalkyl, i.e.
- Cl-6 haloalkyl is a Cl-6 alkyl group having one or more halogen substituents (up to perhaloalkyl, i.e., every hydrogen atom of the alkyl group has been replaced by a halogen atom).
- Cl haloalkyl is a methyl group having one, two, or three halogen substituents (ii) Hydroxylalkyl or hydroxyalkyl, i.e.
- Cyanoalkyl group i.e an alkyl group having one or more (e.g., 1, 2, or 3) OF! substituents
- Cyanoalkyl group i.e an alkyl group having one or more (e.g., 1, 2, or 3) -CN substituents.
- a group from Class (1) may be substituted with another group from Class (1), e.g. haloalkoxy group such as fluoroalkoxy, i.e. an -O-haloalkyl group.
- Cl-6 haloalkoxy refers to an -0-(C 1 -6 haloalkyl) group.
- Formula (I) is Formula (Ia-l) or Formula (Id- 1 ):
- Ri in Formula (I), (Ia-l) and (Id-l) may be piperidinyl, acetylpiperidinyl, a C 3 -C l2 spiro-, fused-, or bridged- bicyclic group containing 0-2 hetero atoms, azaspiroheptanyl, acetylazaspiroheptanyl, azetidinyl, acetylazetidinyl, (methylsulfonyl)azetidinyl, azabi cyclooctanyl , acetyl azabi cyclooctanyl , azabicyclohexanyl, acetylazabicyclohexanyl, cycloalkyl, pyrrolidinyl, oxopyrrolidinyl, alkyloxopyrrolidinyl, methyl oxopyrrolidinyl
- pyridinyl can be 2-pyridinyl (or pyridin-2-yl), 3-pyiidinyl (or pyridin-3-yl), or 4-pyridinyl (or pyridin-4-yl).
- the point of attachment of the non-hydrogen monovalent group can be specified to indicate the position where the non-hydrogen monovalent group is attached to another moiety.
- “-Ci -2 alkyl-(C 3- 4 cycloalkyl)” means the point of attachment occurs at the“C l-2 alkyl” part.
- Table 1 A lists some representative examples of Formula (I) compound and their codes used through the present description. These compounds can effectively inhibit the enzymatic activity of PRMT5, as will be described in more details in the EXAMPLE section.
- compounds of Formula (I) include (R)-7-((l-acetylpiperidin-4-yl)amino)-2-(3-(3,4-dihydroisoquinolin-2(lH)-yl)-2-hydroxyprGpyl)- 3,4-dihydroisoquinolin-l (2H)-one (24a), and (R)-7-((l-acetylpiperidin-4-yl)amino)-4-(3-(3,4- dihydroisoquinolin-2(lH)-yl)-2-hydroxypropyl)-3,4-dihydrobenzo[f][l,4]oxazepin-5(2H)-one (52a).
- the present invention may include all pharmaceutically acceptable isotopically labelled compounds of Formula (I) or salts thereof, wherein one or more atoms are replaced by atoms having the same atomic number, but an atomic mass or mass number different from the atomic mass or mass number which predominates in nature.
- isotopes suitable for inclusion in the compounds of the invention include isotopes of hydrogen, such as 2 H and H, carbon, such as n C, 13 C and 14 C, chlorine, such as 36 Cl, fluorine, such as 18 F, iodine, such as 123 I and 125 I, nitrogen, such as N and N, oxygen, such as 0, 0 and O, phosphorus, such as P, and sulphur, such as 35 S.
- Certain isotopically labelled compounds of Formula (I), for example, those incorporating a radioactive isotope, are useful in drug and/or substrate tissue distribution studies.
- the radioactive isotopes tritium, i.e., 3 H, and carbon- 14, i.e., 14C, are particularly useful for this purpose in view of their ease of incorporation and detection.
- Substitution with heavier isotopes such as deuterium, i.e., 2 H may afford certain therapeutic advantages resulting from greater metabolic stability, for example, increased in vivo half-life or reduced dosage requirements, and hence may be preferred in some circumstances.
- Isotopically labeled compounds of Formula (I) can generally be prepared by conventional techniques known to those skilled in the art using an appropriate isotopically labeled reagent in place of the non-labeled reagent previously employed.
- Stereoisomers of Formula (I) include cis and trans isomers, optical isomers such as R and S enantiomers, diastereomers, geometric isomers, rotational isomers, atropisomers, and conformational isomers of the compounds of Formula (I), including compounds exhibiting more than one type of isomerism; and mixtures thereof (such as racemates and diastereomeric pairs)
- the compounds of Formula (I) may exist in the form of pharmaceutically acceptable salts such as acid addition salts and/or base salts thereof.
- Suitable acid addition salts are formed from acids which form non-toxic salts e.g. hydrochloride/chloride.
- Suitable base salts are formed from bases which form non-toxic salts such as calcium and sodium salts. Hemisalts of acids and bases may also be formed, for example, hemisulfate and hemi calcium salts.
- the compounds of Formula (I) or a pharmaceutically acceptable salt thereof include all forms of the compound of Formula (I) or pharmaceutically salt thereof, including hydrates, solvates, isomers (e.g. rotational stereoisomers), crystalline and non-crystalline forms, isomorphs, polymorphs, metabolites, and prodrugs thereof.
- Compounds of Formula (I) may exist in unsolvated and solvated forms. When the solvent or water is tightly bound, the complex will have a well-defined stoichiometry independent of humidity. When, however, the solvent or water is weakly bound, as in channel solvates and hygroscopic compounds, the water/solvent content will be dependent on humidity and drying conditions.
- the compounds of Formula (I) may exist in a continuum of solid states ranging from fully amorphous to fully crystalline.
- amorphous refers to a state in which the material lacks long-range order at the molecular level and, depending upon temperature, may exhibit the physical properties of a solid or a liquid. Typically such materials do not give distinctive X-ray diffraction patterns and, while exhibiting the properties of a solid, are more formally described as a liquid.
- a change from apparent solid to a material with liquid properties occurs, which is characterized by a change of state, typically second order (“glass transition”).
- the invention also relates to prodrugs of the compounds of Formula (I).
- Some compounds of Formula (I) may have little or no pharmacological activity themselves, but they can, when administered into or onto the body, be converted into compounds of Formula (1) having the desired activity, for example, by hydrolytic cleavage. Such derivatives are referred to as“prodrugs”.
- Prodrugs in accordance with the invention can, for example, be produced by replacing appropriate functionalities present in the compounds of Formula (I) with certain moieties known to those skilled in the art as“pro-moieties”.
- certain compounds of Formula (I) may themselves act as prodrugs of other compounds of Formula (I) Metabolites of compounds of Formula (I) formed in vivo upon administration of the drug are also included within the scope of the invention.
- Compounds of the invention can be prepared using known organic synthesis techniques and can be synthesized according to any of numerous possible synthetic routes.
- the reactions for preparing compounds of the invention can be carried out in suitable solvents, which can be readily selected by one of skill in the art of organic synthesis.
- suitable solvents can be substantially non-reactive with the starting materials (reactants), the intermediates, or products at the temperatures at which the reactions are carried out, e.g., temperatures that can range from the solvent’s freezing temperature to the solvent’s boiling temperature.
- a given reaction can be carried out in one solvent or a mixture of more than one solvent.
- suitable solvents for a particular reaction step can be selected by the skilled artisan.
- Preparation of compounds of the invention may involve the protection and deprotection of various chemical groups.
- the need for protection and deprotection, and the selection of appropriate protecting groups, can be readily determined by one skilled in the art.
- a -CN group can be hydrolyzed to afford an amide group;
- a carboxylic acid can be converted to an amide;
- a carboxylic acid can be converted to an ester, which in turn can be reduced to an alcohol, which in turn can be further modified.
- an OH group can be converted into a better leaving group such as a methanesulfonate, which in turn is suitable for nucleophilic substitution, such as by a cyanide ion.
- an unsaturated bond such as C-C double bond or C-C triple bond can be reduced to a saturated bond by hydrogenation.
- Functional (reactive) groups can be protected/deprotected in the course of the synthetic scheme, if appropriate and/or desired.
- an OH group can be protected by a benzyl, methyl, or acetyl group, which can be deprotected and converted back to the OH group in a later stage of the synthetic process.
- an NH 2 group can be protected by a benzyloxycarbonyl (Cbz) or BOC group; conversion back to the NH 2 group can be carried out at a later stage of the synthetic process via deprotection.
- the compounds may exist as stereoisomers, such as atropisomers, racemates, enantiomers, or diastereomers.
- Conventional techniques for the preparation/isolation of individual enantiomers include chiral synthesis from a suitable optically pure precursor or resolution of the racemate using, for example, chiral high-performance liquid chromatography (HPLC).
- HPLC high-performance liquid chromatography
- the racemate (or a racemic precursor) may be reacted with a suitable optically active compound, for example, an alcohol, or, in the case where the compound contains an acidic or basic moiety, an acid or base such as tartaric acid or 1 -phenyl ethyl amine.
- Stereoisomeric conglomerates may be separated by conventional techniques known to those skilled in the art. Suitable stereoselective techniques are well known to those of ordinary skill in the art.
- geometric cis/trans (or Z/E) isomers are possible.
- Cis/trans isomers may be separated by conventional techniques well known to those skilled in the art, for example, chromatography and fractional crystallization.
- the present invention also provides a pharmaceutical composition
- a pharmaceutical composition comprising a therapeutically effective amount of the compound of Formula (I), or a pharmaceutically acceptable salt, ester, prodrug, complex, solvate, isomer, or hydrate thereof, in any crystalline form or in amorphous form, a pharmaceutically acceptable carrier or excipient, and optionally comprising at least one additional medicinal or pharmaceutical agent.
- the pharmaceutically acceptable carrier or excipient may comprise any conventional pharmaceutical carrier or excipient. Suitable pharmaceutical carriers include inert diluents or fillers, water and various organic solvents such as hydrates and solvates. The pharmaceutical compositions may, if desired, contain additional ingredients such as flavorings, binders. excipients and the like
- a therapeutically effective amount refers to that amount of the compound (including a pharmaceutically acceptable salt thereof) being administered which will relieve to some extent one or more of the symptoms of the disorder being treated.
- a therapeutically effective amount refers to that amount which has the effect of relieving to some extent or eliminating one or more symptoms associated with the PRMT5-mediated disease or disorder.
- the term“treating”, as used herein, unless otherwise indicated, means reversing, alleviating, inhibiting the progress of, or preventing the disorder or condition to which such term applies, or one or more symptoms of such disorder or condition.
- the term “treating” also includes adjuvant and neo-adjuvant treatment of a subject.
- Administration of the compounds of Formula (I) may be effected by any method that enables delivery of the compounds to the site of action. These methods include, for example, enteral routes (e.g., oral routes, buccal routes, sublabial routes, and sublingual routes), oral routes, intranasal routes, inhaled routes, intraduodenal routes, parenteral injection (including intravenous, subcutaneous, intramuscular, intravascular or infusion), intrathecal routes, epidural routes, intracerebral routes, intracerbroventricular routes, topical, and rectal administration.
- enteral routes e.g., oral routes, buccal routes, sublabial routes, and sublingual routes
- oral routes e.g., intranasal routes, inhaled routes, intraduodenal routes, parenteral injection (including intravenous, subcutaneous, intramuscular, intravascular or infusion), intrathecal routes, epidural routes, intracerebral routes, intracerbroventricular routes, topical, and rectal administration.
- the compounds of Formula (I) may be admini stered/effected by parenteral injection routes (e.g., intravenous injection route). In one embodiment of the present invention, the compounds of Formula (I) may be administered or effected by oral routes.
- Dosage of the compounds of Formula (I) may be adjusted to provide the desired response. It is to be noted that dosage values may vary with the type and severity of the condition to be alleviated, and may include single or multiple doses.
- the present invention provides a kit or packaged pharmaceutical comprising a compound of Formula (I), or a pharmaceutically acceptable salt, ester, prodrug, complex, solvate, isomer, or hydrate thereof, in any crystalline form or in amorphous form, and instructions for use thereof
- kits may include a provided pharmaceutical composition or compound and a container (e.g., a vial, ampule, bottle, syringe, and/or dispenser package, or other suitable container).
- the kits may optionally further include a second container comprising a pharmaceutical excipient for dilution or suspension of the pharmaceutical composition or compound.
- a pharmaceutical composition or compound is provided in two containers, and when it is needed, the contents in the two containers are combined to form one unit dosage form.
- the present invention provides a method of inhibiting a protein arginine methyltransferase 5 (PRMT5) enzyme, comprising: contacting the PRMT5 enzyme with an effective amount of a compound of Formula (I), or a pharmaceutically acceptable salt, ester, prodrug, complex, solvate, isomer, or hydrate thereof, in any crystalline form or in amorphous form.
- PRMT5 protein arginine methyltransferase 5
- the step of inhibiting may be carried out in vitro or in vivo.
- “In vitro” refers to procedures performed in an artificial environment such as, e.g., without limitation, in a test tube or culture medium.
- “In vivo” refers to procedures performed within a living organism such as, without limitation, a human, a mouse, dog, rat or rabbit.
- IC50 refers to the half maximal inhibitory concentration of an inhibitor in inhibiting biological or biochemical function. This quantitative measure indicates how much of a particular inhibitor is needed to inhibit a given biological process (or component of a process, he. an enzyme, cell, cell receptor or microorganism) by half. In other words, it is the half maximal (50%) inhibitory concentration (IC) of a substance (50% IC, or IC50).
- IC50 refers to the plasma concentration required for obtaining 50%> of a maximum effect in vivo.
- the method of the invention utilizes the PRMT5 inhibitor of Formula (I) with an IC50 value of about or less than a predetermined value, as ascertained in an in vitro assay.
- the PRMT5 inhibitor inhibits PRMT5 a with an IC50 value of about 10 nM or less, 20 nM or less, 30 nM or less, 40 nM or less, 50 nM or less, 60 nM or less, 70 nM or less, 80 nM or less, 90 nM or less, 100 nM or less, 150 nM or less, 200 nM or less, 300 nM or less, 400 nM or less, 500 nM or less, 600 nM or less, 700 nM or less, 800 nM or less, 900 nM or less, 1000 nM or less, 1500 nM or less, 2000 nM or less, or 2500 nM or less (or a number in the range defined by and including
- a compound of Formula (I) is useful in somatic cell reprogramming, such as reprogramming somatic cells into stem cells.
- a compound of Formula (I) is useful in germ cell development, and are thus envisioned useful in the areas of reproductive technology and regenerative medicine.
- the present invention provides a method of treating a disorder or disease mediated by PRMT5 or associated with aberrant PRMT5 activity, comprising administering to a subject in need thereof a therapeutically effective amount of a compound of Formula (I), or a pharmaceutically acceptable salt, ester, prodrug, complex, solvate, isomer, or hydrate thereof, in any crystalline form or in amorphous form; or a pharmaceutical composition thereof.
- a compound of Formula (I) or a pharmaceutically acceptable salt, ester, prodrug, complex, solvate, isomer, or hydrate thereof, in any crystalline form or in amorphous form; or a pharmaceutical composition thereof.
- the term“PRMT 5 -mediated disorder” means any disease, disorder, or other pathological condition in which PRMT5 is known to play a role. Accordingly, in some embodiments, the present invention relates to treating or lessening the severity of one or more diseases in which PRMT 5 is known to play a role.
- the methods of the invention are useful for treating a disease condition associated with PRMT5. Any disease condition that results directly or indirectly from an abnormal activity or expression level of PRMT5 can be an intended disease condition. Different disease conditions associated with PRMT5 have been reported. PRMT5 has been implicated, for example, in a variety of human cancers as well as a number of hemoglobinopathies.
- the diabetes is Type 2 diabetes.
- a compound of Formula (I) is useful to delay the onset of, slow the progression of, or ameliorate the symptoms of, obesity.
- a compound of Formula (I) is useful to help a subject lose weight.
- a compound of Formula (I) could be used in combination with other compounds, drugs, or therapeutics, such as metformin and insulin, to treat diabetes and/or obesity.
- a compound of Formula (I) is useful in treating a blood disorder, e.g., a hemoglobinopathy, such as sickle cell disease or .beta.-thalassemia.
- a blood disorder e.g., a hemoglobinopathy, such as sickle cell disease or .beta.-thalassemia.
- PRMT5 is a known repressor of gamma-globin gene expression, and increased fetal gamma-globin (HbF) levels in adulthood are associated with symptomatic amelioration in sickle cell disease and P3 -thalassemia.
- HbF fetal gamma-globin
- the inhibition of PRMT5 by a compound Formula (I) is useful in treating a blood disorder, such as a hemoglobinopathy such as sickle cell disease or beta-thalassemia.
- a compound of Formula (I) is useful to delay the onset of, slow the progression of, or ameliorate the symptoms of, sickle cell disease. In some embodiments, a compound of Formula (I) is useful to delay the onset of, slow the progression of, or ameliorate the symptoms of, beta-thalassemia.
- a compound of Formula (I) is useful in treating inflammatory and autoimmune disease.
- PRMT5 is reported to activate NFkB signaling pathway through the methylation of p65.
- PRMT5 is reported to interact with Death receptor 4 and Death receptor 5 contributing to TRAIL-induced activation of inhibitor or kB kinase (IKK) and nuclear factor-kB (NF-kB).
- IKK inhibitor or kB kinase
- NF-kB nuclear factor-kB
- inflammatory disease refers to those diseases, disorders or conditions that are characterized by signs of pain (dolor, from the generation of noxious substances and the stimulation of nerves), heat (cal or, from vasodilatation), redness (rubor, from vasodilatation and increased blood flow), swelling (tumor, from excessive inflow or restricted outflow of fluid), and/or loss of function.
- Inflammation takes on many forms and includes, but is not limited to, acute, adhesive, atrophic, catarrhal, chronic, cirrhotic, diffuse, disseminated, exudative, fibrinous, fibrosing, focal, granulomatous, hyperplastic, hypertrophic, interstitial, metastatic, necrotic, obliterative, parenchymatous, plastic, productive, proliferous, pseudomembranous, purulent, sclerosing, seroplastic, serous, simple, specific, subacute, suppurative, toxic, traumatic, and/or ulcerative inflammation.
- PRMT5 has also been implicated in accelerating cell cycle progression through Gl phase and modulating regulators of Gl ; for example, PRMT5 may upregulate cyclin-dependent kinase (CDK) 4, CDK6, and cyclins Dl, D2 and El. Moreover, PRMT5 may activate phosphoinositide 3-kinase (PI3K)/AKT signaling.
- CDK cyclin-dependent kinase
- PI3K phosphoinositide 3-kinase
- the disorder or disease includes breast cancer, lung cancer, pancreatic cancer, prostate cancer, colon cancer, ovarian cancer, skin cancer, testicular cancer, uterine cancer, cervical cancer, esophageal cancer, bladder cancer, gastric cancer, liver cancer, epidermoid cancer, brain cancer, hematopoietic cancer, leukemia such as acute myeloid leukemia (AML), acute lymphocytic leukemia, chronic myeloid leukemia, hairy cell leukemia, myelodysplasia, myeloproliferative disorders, acute myelogenous leukemia (AML), chronic myelogenous leukemia(CML), mastocytosis, chronic lymphocytic leukemia (CLL), multiple myeloma (MM), myelodysplastic syndrome (MDS), hemoglobinopathies such as beta-thalassemia and sickle cell disease (SCD), lymphoma, medulloblastoma, rectum adenocarcinoma
- the disorder or disease comprises lymphoma representable by cell lines such as Raji, SU-DHL4, and Z138; or glioma representable by cell lines such as U87MG, U251 and T98G.
- MIAPaCa2, Aspcl, A6L, SKPC1 and Panc-l MIAPaCa2, Aspcl, A6L, SKPC1 and Panc-l.
- the disorder or disease includes breast cancer representable by cell lines such as
- Example 1 2-(3-(3,4-dihydroisoquinolin-2(lH)-yl)-2-hydroxypropyl)-7-(oxetan-3-ylamino)- 3,4-dihydroisoquinolin-l(2H)-one (1)
- Step 1 l-chloro-3-(3,4-dihydroisoquinolin-2(lH)-yl)propan-2-ol
- Step 2 7 -bromo-2-(3-(3, 4-dihydroi soquinolin-2(lH)-yl)-2-hydroxypropyl)-3, 4- dihydroi soquinolin- 1 (2H)-one (I- 1)
- Step 3 2-(3 -(3 ,4-dihydroi soquinolin-2( 1 H)-yl)-2-hydroxypropyl)-7-(oxetan-3 -yl amino)-
- the reaction mixture was treated with water, and was extracted with EA. Organic phase was washed with brine, dried over Na 2 S0 4 , and was concentrated to dryness.
- Step 1 2-(4-bromophenyl)-2-methylpropanenitrile
- Step 2 2-(4-bromophenyl)-2-methylpropan-l -amine
- Step 3 methyl (2-(4-bromophenyl)-2-methylpropyl)carbamate
- Step 5 7-bromo-4,4-dimethyl-2-(oxiran-2-ylmethyl)-3,4-dihydroisoquinolin-l(2H)-one
- Step 7 2-(3-(3,4-dihydroisoquinolin-2(lH)-yl)-2-hydroxypropyl)-4,4-dimethyl- 7-(oxetan-3-ylamino) -3,4-dihydroisoquinolin-l (2H)-one (15)
- the resultant mixture was heated to reflux under nitrogen atmosphere for 6 hours.
- the reaction mixture was treated with water and EA, and organic phase was separated. Organic phase was washed with water, dried over Na 2 S0 4 , and was concentrated to dryness.
- Step 1 tert-butyl 3-bromo-7,8-dihydro-l,6-naphthyridine-6(5H)-carboxylate
- Step 2 tert-butyl 3-bromo-5-oxo-7, 8-dihydro- l,6-naphthyridine-6(5H)-carboxylate
- Step 3 3-bromo-7,8-dihydro-l,6-naphthyridin-5(6H)-one [0111] To a solution of tert-butyl 3-bromo-5-oxo-7,8-dihydro-l,6-naphthyridine- 6(5H)-carboxylate (4.40 g, 13.4 mmol, 1.0 eq.) in dichloromethane (60 ml) was added
- Trifluoroacetic acid (TFA, 20 ml), and the resultant mixture was stirred at ambient temperature for 3 hours.
- the reaction mixture was concentrated to dryness, and was treated with water.
- the obtained solid was dried to afford 3 -bromo-7,8-dihydro- 1 , 6-naphthyridin-5 (6H)-one (2.60 g, 86% yield) as a light yellow solid.
- Step 4 3-bromo-6-(oxiran-2-ylmethyl)-7,8-dihydro-l,6-naphthyridin-5(6H)-one
- Step 5 3-bromo-6-(3-(3,4-dihydroisoquinolin-2(lH)-yl)-2-hydroxypropyl)- 7,8-dihydro-i,6- naphthyridin-5(6H)-one (1-2)
- Step 6 6-(3-(3,4-dihydroisoquinolin-2(lH)-yl)-2-hydroxypropyl)-3-(oxetan-3-ylamino)- 7, 8-dihydro- l,6-naphthyridin-5(6H)-one (16)
- Step 1 1 -bromo-4-(2-nitroprop- 1 -en- 1 -yl)benzene
- Step 2 l-(4-bromophenyl)propan-2-amine
- Step 3 methyl ( 1 -(4-bromophenyl)propan-2-yl)carbamate
- Step 4 7-bromo-3-methyl-3,4-dihydroisoquinolin-l(2H)-one
- Example 6 (R)-7-((l-acetylpiperidin-4-yl)amino)-2-(3-(3,4-dihydroisoquinolin- 2(1 H)-yl)-2-hydroxy propyl)-3,4-dihydroisoquinolin-l(2H)-one (24a) and (S)-7-((l-acetyl piperidin-4-yl)amino)-2-(3-(3,4-dihydroisoquinolin-2(lH)-yl)-2-hydroxypropyl)-3,4-dihydroisoq uinolin- 1 (2H)-one (24b)
- Step 1 7-bromo-2-(2-((tert-butyldimethylsilyl)oxy)-3-(3,4-dihydroisoquinolin- 2(lH)-yl)propyl)-3,4- dihydroisoquinolin-l (2H)-one
- Step 2 7-((l-acetylpiperidin-4-yl)amino)-2-(2-((tert-butyldimethylsilyl)oxy)- 3 -(3, 4-dihydro isoquinolin-2(lH)-yl)propyl)-3,4-dihydroisoquinolin-l(2H)-one
- Step 3 (R)-7-((l-acetylpiperidin-4-yl)amino)-2-(3-(3,4-dihydroisoquinolin- 2(lH)-yl)-2-hydroxy propyl)-3 ,4-dihydroi soquinolin- 1 (2H)-one (24a) and (S)-7-((l -acetyl piperidin-4-yl)amino)-2-(3-(3,4-dihydroisoquinolin-2(lEl)-yl)-2-hydroxypropyl)-3,4-dihydroisoq uinolin-l(2H)-one (24b) [0133] To a stirred solution of 7-((l-acetylpiperidin-4-yl)amino)-2-(2-((tert-butyldimethyl silyl)oxy)-3-(3,4-dihydroisoquinolin-2(lH)-
- reaction mixture was concentrated to dryness, and the obtained residue was purified by column chromatography to afford 7-((l-acetylpiperidin-4-yl)amino)- 2-(3-(3,4-dihydroisoquinolin-2(lH)-yl)-2-hydroxypropyl)-3,4-dihydroisoquinolin-l(2H)-one.
- Step 1 7-bromo-4-(3-(3,4-dihydroisoquinolin-2(lH)-yl)-2-hydroxypropyl)- 3,4-dihydrobenzo[f][l,4] oxazepin-5(2H)-one
- Step 2 (R)-4-(3 -(3 ,4-dihydroisoquinolin-2( lH)-yl)-2-hydroxypropyl)-7-(pyridazin- 4-ylamino)-3,4- dihydrobenzo[f] [ 1 ,4]oxazepin-5(2H)-one (30a) and (S)-4-(3-(3, 4-dihydro isoquinolin-2(lH)-yl)-2-hydroxypropyl)-7-(pyridazin-4-ylamino)-3,4-dihydrobenzo[f][l,4]oxaze pin-5(2H)-one (30b)
- Example 10 6-chloro-2-(3-(3,4-dihydroisoquinolin-2(lH)-yl)-2-hydroxypropyl)-7-((l-methyl- lH-pyrazol-4-yl)amino)-3,4-dihydroisoquinolin-l(2H)-one (26)
- Compound 26 was synthesized in a method similar to that for compound 1 in Example 1, using 7-bromo-6-chloro-l,2,3,4-tetrahydroisoquinolin-l-one and 1 -methyl- 1 H-py razol-4-amine as starting material.
- 1 HNMR 400 MHz, DMSO- ⁇ 3 ⁇ 4 6(pprn) 7.66 (s, 1H), 7.35 (s, 1H), 7.25 (s, 2H), 7.02-7.09 (m, 5H), 4.79 (brs, 1H), 4.00-4.03 (m, 1H), 3.84 (s, 3H), 3.72-3.76 (m, 1H),
- Example 11 (R)-2-(3-(3,4-dihydroisoquinolin-2(lH)-yl)-2-hydroxypropyl)-7-((l-methyl-lH- pyrazol-4-yl)amino)-3,4-dihydroisoquinolin-l(2H)-one (I2a) and (S)-2-(3-(3,4-dihydro isoquinolin-2(lH)-yl)-2-hydroxypropyl)-7-((l-methyl-lH-pyrazol-4-yl)amino)-3,4-dihydroisoqu inolin-l(2H)-one (l2b)
- Compound 12a and 12b was synthesized in a method similar to that for compound 30a and 30b in Example 7, using 7-bromo-2-(3-(3,4-dihydroisoquinolin-2(lH)-yl)-2-hydroxypropyl)- 3,4-dihydroisoquinolin-l(2H)-one (1-1) and 1 -methyl- lH-pyrazol -4-amine as starting
- Example 12 (R)-2-(3-(3,4-dihydroisoquinolin-2(lH)-yl)-2-hydroxypropyl)- 7-(pyridazin-4-ylamino)-3,4-dihydroisoquinolin-l(2H)-one (28a) and (S)-2-(3-(3 ,4-dihydro isoquinolin-2(lH)-yl)-2-hydroxypropyl)-7-(pyridazin-4-ylamino)-3,4-dihydroisoquinolin-l(2H)- one (28b)
- Compound 28a and 28b was synthesized in a method similar to that for compound 30a and 30b in Example 7, using 7-bromo-2-(3-(3,4-dihydroisoquinolin-2(lH)-yl)-2-hydroxypropyl)- 3 ,4-dihydroisoquinolin- 1 (2H)-one (1-1) and pyridazin-4-amine as starting material. !
- Example 13 (R)-4-(3-(3,4-dihydroisoquinolin-2(lH)-yl)-2-hydroxypropyl)- 7-((l-methyl-lH-pyrazol-4-yl)amino)-3,4-dihydrobenzo[f][l,4]oxazepin-5(2H)-one (29a) and (S)-4-(3-(3,4-dihydroisoquinolin-2(lH)-yl)-2-hydroxypropyl)-7-((l-methyl-lH-pyrazol-4-yl)ami no)-3,4-dih
- Example 14 (R)-7-bromo-4-(3-(3,4-dihydroisoquinolin-2(lH)-yl)-2-hydroxypropyl)- 3,4-dihydrobenzo[f][l,4]oxazepin-5(2H)-one and (S)-7-bromo-4-(3-(3,4-dihydroisoquinolin- 2(lH)-yl)-2-hydroxypropyl)-3,4-dihydrobenzo[f [l ,4]oxazepin-5(2H)-one
- Analytical separation method Instrument: waters UPLC; Column: Daicel Chiralcel IC, 2. l x l50mm ID., 3pm; Mobile phase: A for C0 2 and B for ethanol (0. l%DEA); Gradient: B 30%; Flow rate: lmL/min; Back pressure: l500psi; Column temperature: 40°C; Wavelength: 220nm.
- Preparative separation method Instrument: waters SFC200; Column: Daicel Chiralcel IC, 250x30mm ID., 5 pm; Mobile phase: A for C0 2 and B for ethanol(0.l%NH 3 H 2 0); Gradient: B 35%; Flow rate: 70mL /min; Back pressure: lOObar; Column temperature: 38°C; Wavelength: 2l0nm; Cycle time: l4min.
- Example 15 7-((l-acetylazetidin-3-yl)amino)-2-(3-(3,4-dihydroisoquinolin-2(lH)-yl)- 2-hydroxypropyl)-3,4-dihydroisoquinolin-l(2H)-one (6)
- Example 16 7-(cyclobutylamino)-2-(3-(3,4-dihydroisOquinolin-2(lH)-yl)-2-hydroxypropyl)- 3,4-dihydroisoquinolin-l (2H)-one (7)
- Compound 10 was synthesized in a method similar to that for compound 1 in Example 1, using (3S)-oxolan-3-amine and (S)-7-bromo-2-(3-(3,4-dihydroisoquinolin-2(lH)-yl)- 2-hydroxypropyl)-3,4-dihydroisoquinolin-l(2H)-one as starting material.
- Compound 11 was synthesized in a method similar to that for compound 1 in Example 1, using (3R)-oxolan-3-amine and (S)-7-bromo-2-(3-(3,4-dihydroisoquinolin-2(lH)-yl)- 2-hydroxypropyl)-3,4-dihydroisoquinolin-l(2H)-one as starting material.
- Example 23 2-(3-(3,4-dihydroisoquinolin-2(lH)-yl)-2-hydroxypropyl)-7-((l-(tetrahydro- 2H-pyran-4-yl)-lH-pyrazol-4-yl)amino)-3,4-dihydroisoquinolin- 1 (2H)-one (25)
- Example 28 4-(3-(3,4-dihydroisoquinolin-2(lH)-yl)-2-hydroxypropyl)-7-(oxetan-3-ylamino)- 3,4-dihydrobenzo[f][l,4]oxazepin-5(2H)-one (5)
- Compound 13 was synthesized in a method similar to that for compound 14 in Example 2, using 1 -methyl -4-(4, 4, 5 , 5 -tetramethyl - 1 , 3 , 2-di oxab orol an-2-yl )- 1 H-pyrazol e and
- Step 4 7-bromo-2-(3-(3,4-dihydroisoquinolin-2(lH)-yl)-2-hydroxypropyl)
- Step 5 2-(3-(3,4-dihydroisoquinolin-2(lH)-yl)-2-hydroxypropyl)-7-(pyridazin- 4-ylamino)phthalazin -l(2H)-one (33)
- Compound 33 was synthesized in a method similar to that for compound 1 in Example 1, using pyridazin-4-amine and 7-bromo-2-(3-(3,4-dihydroisoquinolin-2(lH)-yl)-2-hydroxypropyl) phthalazin- 1 (2H)-one as starting material.
- Example 33 2-(3-(3,4-dihydroisoquinolin-2(lH)-yl)-2-hydroxypropyl)-4,4-dimethyl- 7-(pyridazin-4-ylamino)-3,4-dihydroisoquinolin-l(2H)-one (34)
- Example 34 2'-(3-(3,4-dihydroisoquinolin-2(lH)-yl)-2-hydroxypropyl)-7'-(pyridazin- 4-ylamino)-2',3 '-dihydro- 1 'H-spiro[cyclopropane- 1 ,4'-isoquinolin]- l'-one (35)
- Step 1 ( 1 -(4-bromophenyl)cyclopropyl)methanamine [0178] To a stirred solution of 1 -(4-bromophenyl)eyclopropane- 1 -carbonitrile (1.00 g, 4.50 mmol, 1.0 eq) in toluene (10 ml) was added solution of BH 3 in THF (1 N, 22.5 mmol, 5.0 eq) at ice-water bath temperature, and the resultant mixture was heated to reflux for 4 hours. The reaction mixture was concentrated to dryness, and was treated with methanol. The resultant mixture was heated to reflux for additional 0.5 hour. The reaction mixture was concentrated to dryness, and was treated with DCM and water.
- Step 2 methyl 2-((((l-(4-bromophenyl)cyclopropyl)methyl)carbamoyl)oxy)benzoate
- Step 3 7'-bromo-2', 3 '-dihydro- 1 'H-spiro[cyclopropane- 1 ,4'-isoquinolin]- 1 '-one
- Step 4 2'-(3-(3,4-dihydroisoquinolin-2(lH)-yl)-2-hydiOxypropyl)-7'-(pyridazin- 4-ylamino)-2',3'- dihydro- rH-spiro[cyclopropane-l,4'-isoquinolin]-r-one (35)
- Compound 36 was synthesized in a method similar to that for compound 30a in Example 7, using pyrimidin-5-amine and (R)-7-bromo-4-(3-(3,4-dihydroisoquinolin-2(lH)-yl)- 2-hydroxypropyl)-3,4-dihydrobenzo[f][l,4]oxazepin-5(2H)-one as starting material.
- 1 HNMR 1 HNMR
- Example 7 using pyrazin-2-amine and (R)-7-bromo-4-(3 -(3 ,4-dihydroi soquinolin-2( 1 H)-yl)- 2-hydroxypropyl)-3,4-dihydrobenzo[f][l,4]oxazepin-5(2H)-one as starting material.
- Example 7 using pyridazin-3 -amine and (R)-7-bromo-4-(3 -(3 ,4-dihydroi soquinolin-2( 1 H)-yl)- 2-hydroxypropyl)-3,4-dihydrobenzo[f][l,4]oxazepin-5(2H)-one as starting material.
- Example 7 using 1 -methyl- lH-pyrazol-5-amine and (R)-7-bromo-4-(3-(3,4-dihydroisoquinolin- 2(lH)-yl)-2-hydroxypropyl)-3,4-dihydrobenzo[f][l,4] oxazepin-5(2H)-one as starting
- Compound 43 was synthesized in a method similar to that for compound 30a in Example 7, using 4-amino- l-methylpyrrolidin-2-one and (R)-7-bromo-4-(3-(3, 4-dihydro isoquinolin-2(lH)-yl)-2-hydroxypropyl)-3,4-dihydrobenzo[f][l,4]oxazepin-5(2H)-one as starting material.
- Example 43 4-(3-(3,4-dihydroisoquinolin-2(lH)-yl)-2-hydroxypropyl)-l-methyl-7-(pyridazin- 4-ylamino)-3,4-dihydro-lH-benzo[e][l,4]diazepine-2,5-dione (44)
- Step 1 6-bromo-2H-benzo[d][ 1 ,3]oxazine-2,4( 1 H)-dione
- Step 2 6-bromo-l-methyl-2H-benzo[d][l,3]oxazine-2,4(lH)-dione
- Step 3 7-bromo-l -methyl-3, 4-dihydro-lH-benzo[e][l,4]diazepine-2,5-di one
- Step 4 4-(3-(3,4-dihydroisoquinolin-2(lH)-yl)-2-hydroxypropyl)-l-methyl- 7-(pyridazin-4- ylamino) -3, 4-dihydro- lH-benzo[e] [ 1 ,4]diazepine-2,5-dione (44)
- Example 44 4-(3-(3,4-dihydroisoquinolin-2(lH)-yl)-2-hydroxypropyl)-l-methyl- 7-(pyridazin-4- ylamino)-l,2,3,4-tetrahydro-5H-benzo[e][l,4]diazepin-5-one (45)
- Step 1 tert-butyl (2-(5-bromo-2-fluorobenzamido)ethyl)(methyl)carbamate Boc
- reaction mixture was washed with water followed by 0.1 N HC1 solution, washed with brine, dried over MgS0 4 , and was concentrated to dryness.
- Step 3 7-bromo-l-methyl-l,2,3,4-tetrahydro-5H-benzo[e][l,4]diazepin-5-one
- Step 4 4-(3-(3,4-dihydroisoquinolin-2(lH)-yl)-2-hydroxypropyl)- l-methyl-7-(pyridazin-4- ylamino) - 1 ,2,3 ,4-tetrahydro-5H-benzo[e] [ 1 ,4]diazepin-5-one (45)
- Example 45 4-(3-(3,4-dihydroisoquinolin-2(lH)-yl)-2-hydroxypropyl)-7-(pyridazin- 4-ylamino)-3,4- dihydropyrido[3,2-f][l,4]oxazepin-5(2H)-one (47)
- Step 1 2-((4-methoxybenzyl)amino)ethan- 1 -ol
- Step 2 5-bromo-2-fluoro-N-(2-hydrQxyethyl)-N-(4-methoxybenzyl)nicotinamide
- Step 3 7-bromo-4-(4-methoxybenzyl)-3,4-dihydropyrido[3,2-f] [ 1 ,4]oxazepin- 5(2H)-one
- Step 4 7-bromo-3,4-dihydropyrido[3,2-f][l,4]oxazepin-5(2H)-one
- Step 5 4-(3-(3,4-dihydroisoquinolin-2(lH)-yl)-2-hydroxypropyl)- 7-(pyridazin-4-ylamino)-3,4- dihydropyrido[3,2-f [l,4]oxazepin-5(2H)-one (47)
- Example 46 4-(3-(3,4-dihydroisoquinolin-2(lH)-yl)-2-hydroxypropyl)-7-(pyridazin-4-yl amino)-3,4-dihydropyrido[4,3-f][l,4]oxazepin-5(2H)-one (48)
- Example 47 4-(3-(3,4-dihydroisoquinolin-2(lH)-yl)-2-hydroxypropyl)-7-(pyridazin-4-yl amino)-3,4-dihydropyrido[2,3-f] [ 1 ,4]oxazepin-5(2H)-one (49)
- Example 48 4-(3-(3,4-dihydroisoquinolin-2(lH)-yl)-2-hydroxypropyl)-8-fluoro- 7-(pyridazin-4-ylamino)-3,4-dihydrobenzo[f][l,4]oxazepin-5(2H)-one (50)
- Example 49 4-(3-(3,4-dihydroisoquinolin-2(lH)-yl)-2-hydroxypropyl)-9-fluoro-7-(pyridazin- 4-ylamino)-3,4-dihydrobenzo[f][l,4]oxazepin-5(2H)-one (51)
- Step 3 4-(3-(3,4-dihydroisoquinolin-2(lH)-yl)-2-hydroxypropyl)-9-fluoro- 7-(pyridazin-4-ylamino)- 3,4-dihydrobenzo[f][l,4]oxazepin-5(2H)-one (51)
- Example 50 7-((l-acetylpiperidin-4-yl)amino)-4-(3-(3,4-dihydroisoquinolin-2(lH)-yl)- 2-hydroxy propyl)-3,4-dihydrobenzo[f] [1 ,4]oxazepin-5(2H)-one (52)
- Compound 52 was synthesized in a method similar to that for compound 24a/24b in Example 6, using 7-bromo-4-(3-(3,4-dihydroisoquinolin-2(lH)-yl)-2-hydroxypropyl)- 3,4-dihydrobenzo[f][l,4]oxazepin-5(2H)-one (example 7) as intermediate.
- Example 51 7'-((l-acetylpiperidin-4-yl)amino)-2'-(3-(3,4-dihydroisoquinolin-2(lH)-yl)- 2-hydroxypropyl)-2',3'-dihydro-rH-spiro[cyclopropane-l,4'-isoquinolin]-r-one (53)
- Compound 53 was synthesized in a method similar to that for compound 24a/24b in Example 6, using 7'-bromo-2',3 '-dihydro- 1 H-spiro[cyclopropane-l ,4'-isoquinolin]- 1 '-one (example 34) as intermediate.
- Example 54 7-((l-acetylpiperidin-4-yl)amino)-4-(3-(3,4-dihydroisoqumolin-2(lH)-yl)- 2-hydroxy propyl)-6-fluoro-3 ,4-dihydrobenzo[f] [ 1 ,4]oxazepin-5(2H)-one (46)
- Example 55 7-((2-acetyl-2-azaspiro[3.3]heptan-6-yl)amino)-4-(3-(3,4-dihydroisoquinolin- 2(lH)-yl)-2-hydroxypropyl)-3,4-dihydrobenzo[f][l,4]oxazepin-5(2H)-one (54)
- Example 56 7-((8-acetyl-8-azabicyclo[3.2. l]octan-3-yl)amino)-4-(3-(3,4-dihydroisoquinolin- 2(lH)-yl)-2-hydroxypropyl)-3,4-dihydrobenzo[f][l,4]oxazepin-5(2H)-one (55)
- Compound 55 was synthesized in a method similar to that for compound 24a/24b in Example 6, using l-(3-amino-8-azabicyclo[3.2. l]octan-8-yl)ethan-l-one as the starting material.
- Example 57 7-((3 -acetyl -3 -azabi cy cl o [3.1.0]hexan-6-yl)amino)-4-(3-(3,4-dihydroisoquinolin- 2(lH)-yl)-2-hydroxypropyl)-3,4-dihydrobenzo[f][l,4]oxazepin-5(2H)-one (56)
- Compound 56 was synthesized in a method similar to that for compound 24a/24b in Example 6, using l-(6-amino-3-azabicyclo[3. l .O]hexan-3-yl)ethan-l-one as the starting material.
- Example 58 (R)-8-((l-acetylpiperidin-4-yl)amino)-2-(3-(3,4-dihydroisoquinolin-2(lH)-yl)-2- hy droxypropyl )-2, 3 ,4,5 -tetrahydro- GH-3 , 5 -m ethanobenzo [e] azepin- 1 -one (57a) and
- SAM S-adenosylmethionine
- Tween20 Tween20
- DMSO dimethyl sulfoxide
- Tris buffer Tris buffer
- Methyl transfer reaction was initiated by adding 2.5 pL of Biotin-H4 (1-21) peptide substrate solution in the assay buffer. Final concentrations of PRMT 5/MEP50, SAM, Biotin-H4 peptide substrate, and DMSO were 25 nM, 10 mM, 30 nM, and 1%, respectively. The reaction was allowed to perform for 120 minutes in dark with gentle shaking at room
- Example 60 Cell Proliferation Inhibition Assay:
- CMOS fetal bovine serum
- FBS heat inactivated fetal bovine serum
- horse serum was purchased from Therm oFisher, Waltham, MA, USA.
- Corning 96- and 384-well cell culture plates were purchased from ThermoFisher, Waltham, MA, USA.
- Cell-Titer Glo® was purchased from Promega Corporation, Madison, WI, USA.
- Luminescence was read after 10 minute of incubation with the Envision multiple plate reader from PerkinElmer (Waltham, MA, USA). The concentrations of compounds inhibiting cell viability by 50% (IC50 values) were determined using a sigmoidal dose-response model
- cell viability was measured using the CellTiter-Glo assay kit from Promega (Madison, WI, USA) by quantitating the ATP present in the cell cultures. 20 pL of CellTiter-Glo reagent was added to each well of the cell plates.
- Luminescence was read after 10 minute of incubation with the Envision multiple plate reader from PerkinElmer (Waltham, MA, USA). The concentrations of compounds inhibiting cell viability by 50% (IC50 values) were determined using a sigmoidal dose-response model
- Example 61 Cell Proliferation Inhibitory Activity Screening on Breast, Liver and Lung Cancer Cell Line Panels
- cell viability was measured using the CellTiter-Glo assay kit from Promega (Madison, WI, USA) by quantitating the ATP present in the cell cultures. 20 pL of CellTiter-Glo reagent was added to each well of the cell plates. Luminescence was read after 10 minute of incubation with the Envision multiple plate reader from PerkinElmer (Waltham, MA, USA). The concentrations of compounds inhibiting cell viability by 50% (IC50 values) were determined using a sigmoidal dose-response model (variable slopes, four parameters) in Prism 7 (La Jolla, CA, USA).
- Example 62 Pharmacokinetic study in mouse, rat and dog
- Standard calibration curves were constructed by analyzing a series of control plasma aliquots containing Diclofenac (100 ng/mL) as an internal standard and 1.0-3,000 ng/mL testing compounds. Compound concentrations were determined with UPLC -MS/MS-22 (Triple QuadTM 6500).
- V ss (L/kg) 2. l7 ⁇ 0.0643 7.l5 ⁇ 2.86 5.02 ⁇ 0.389
- V ss (L/kg) l .32 ⁇ 0. l44 l .807 ⁇ 0.l30 l .564 ⁇ 0.Q4l
- Example 63 Mantle cell lymphoma (MCL) Z-138 xenograft efficacy studies
- CB-17 female SCID mice (19-21 g; 10-12 weeks) were purchased from Shanghai SLAC laboratory animal CO. LTD. Each mouse was inoculated subcutaneously at the right flank with Z-138 tumor cells (5 c 10 6 cells/mouse, 50% Matrigel) in 0.2 mL of a mixture of base media and Matrigel (RPMIMatrigel, 1 : 1) for tumor development. The treatments were started when the mean tumor size reached 121.55 mm 3 , which was 11 days after inoculation. Mice were assigned into groups using a randomized block design. Testing compounds or vehicle (5%DMSO+45%PEG400 ⁇ 50%H 2 O) was administered orally BID for 21 days.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Diabetes (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Epidemiology (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Oncology (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Emergency Medicine (AREA)
- Endocrinology (AREA)
- Immunology (AREA)
- Child & Adolescent Psychology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Description


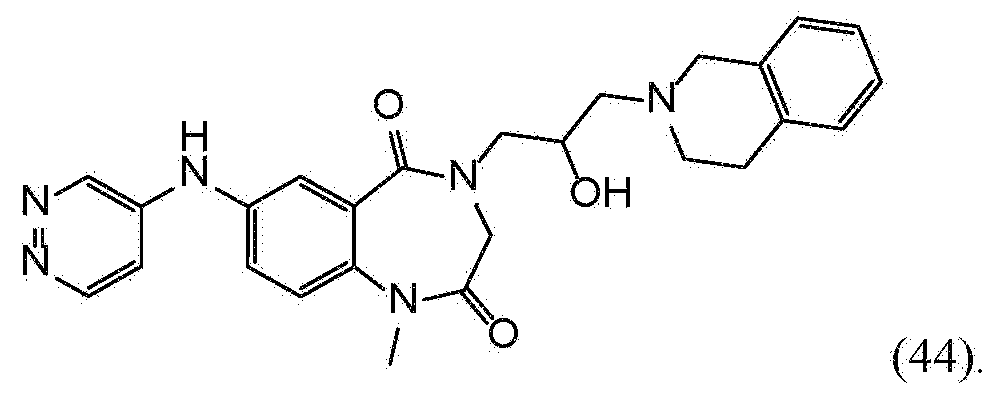

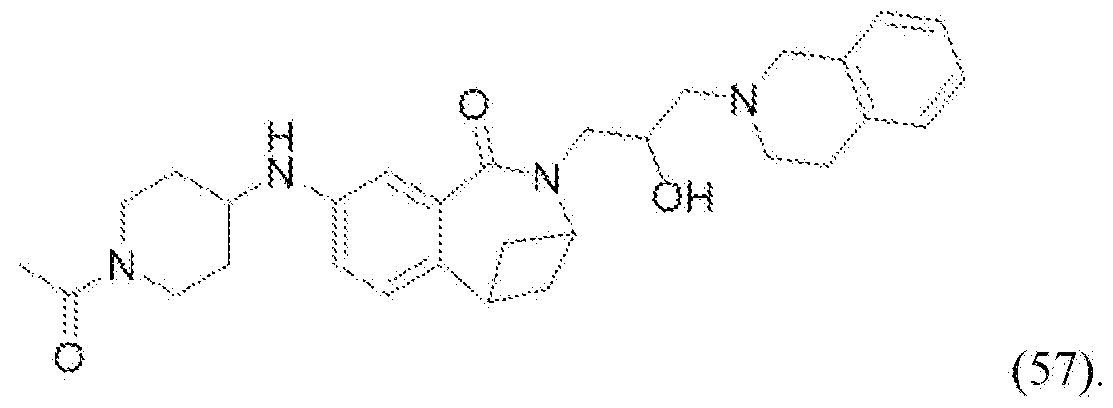
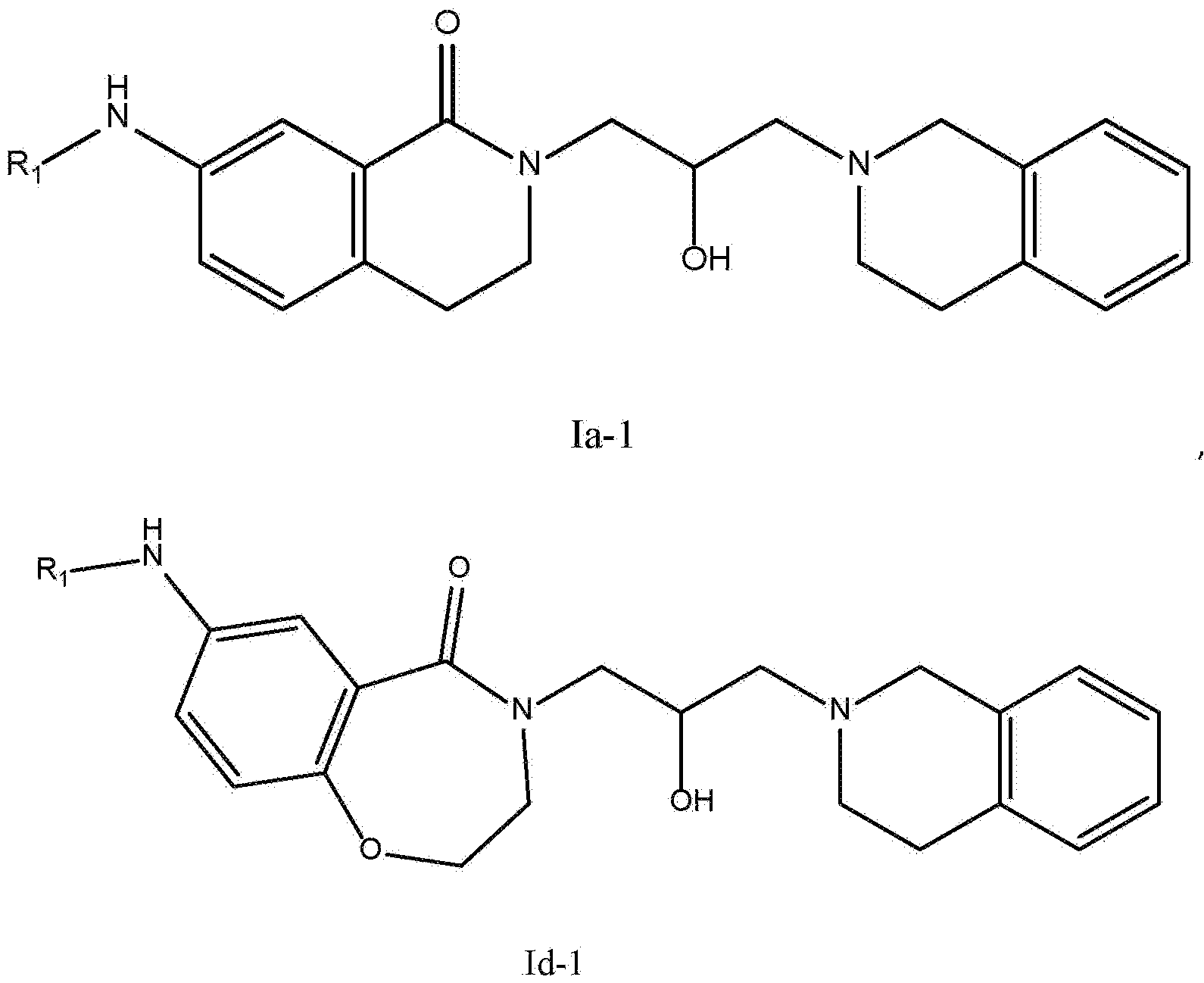






















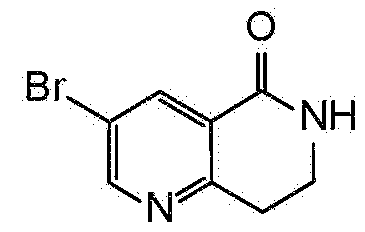




















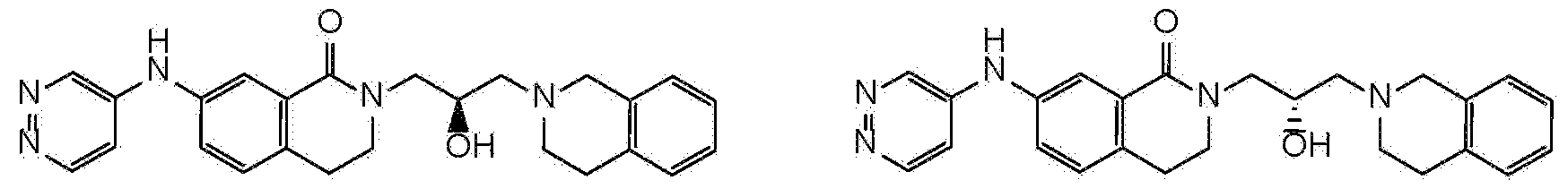




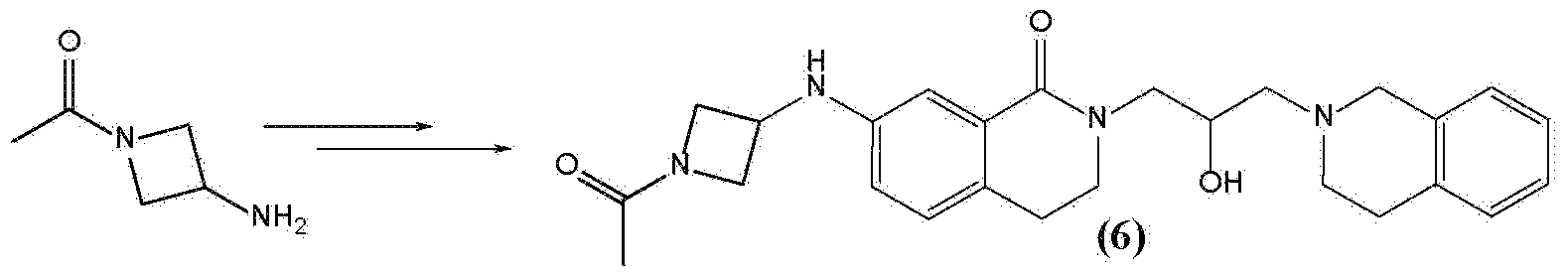











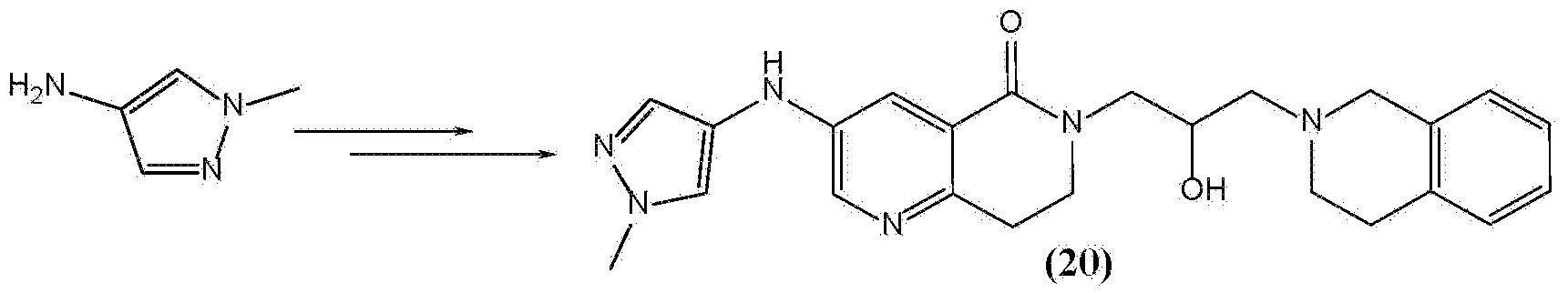











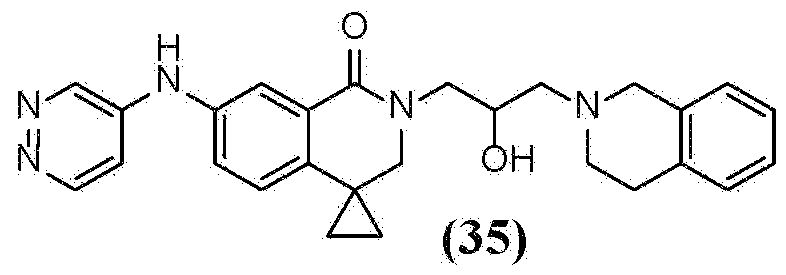






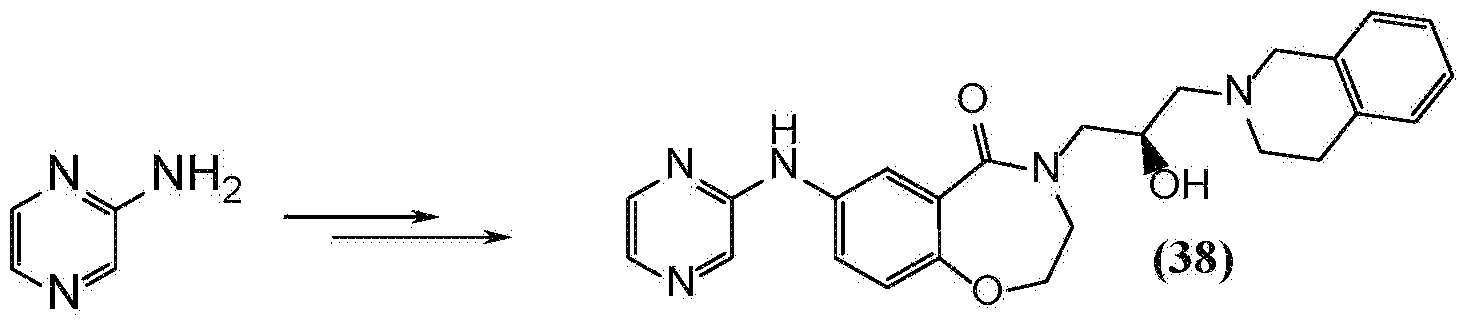












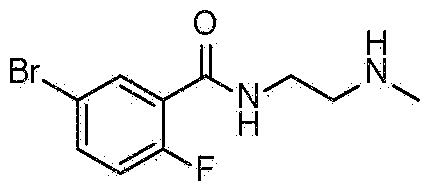


























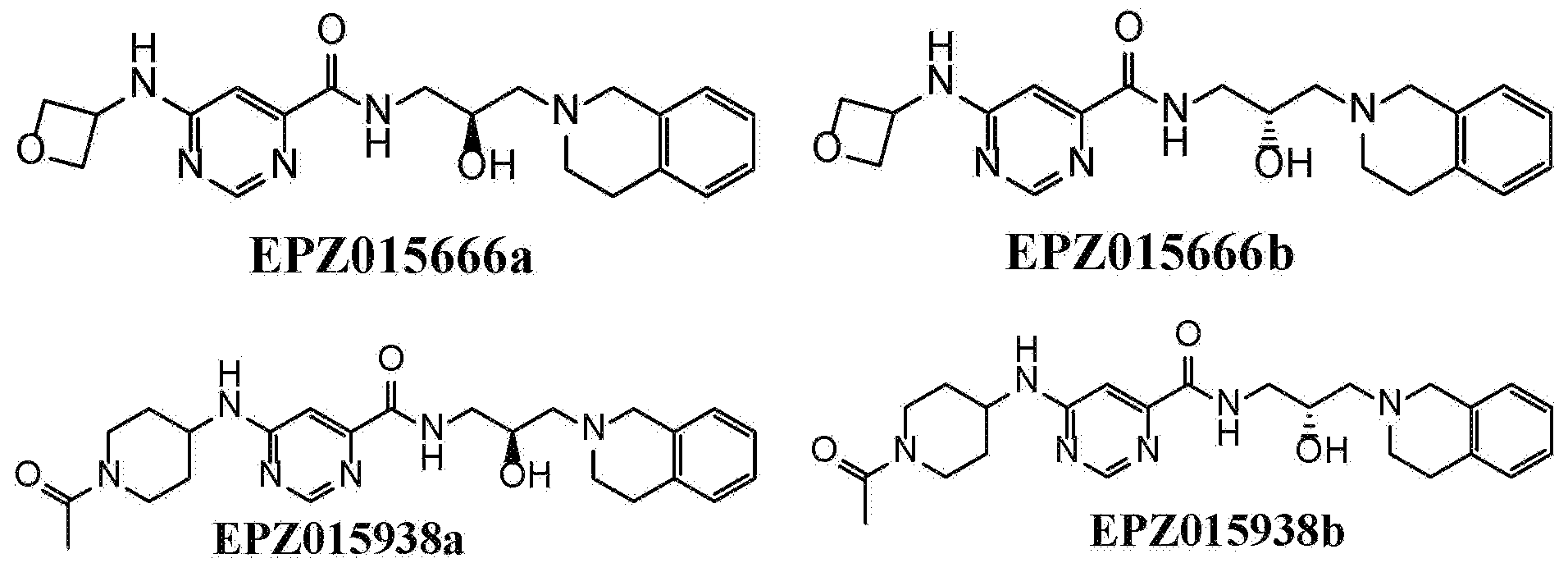





Claims


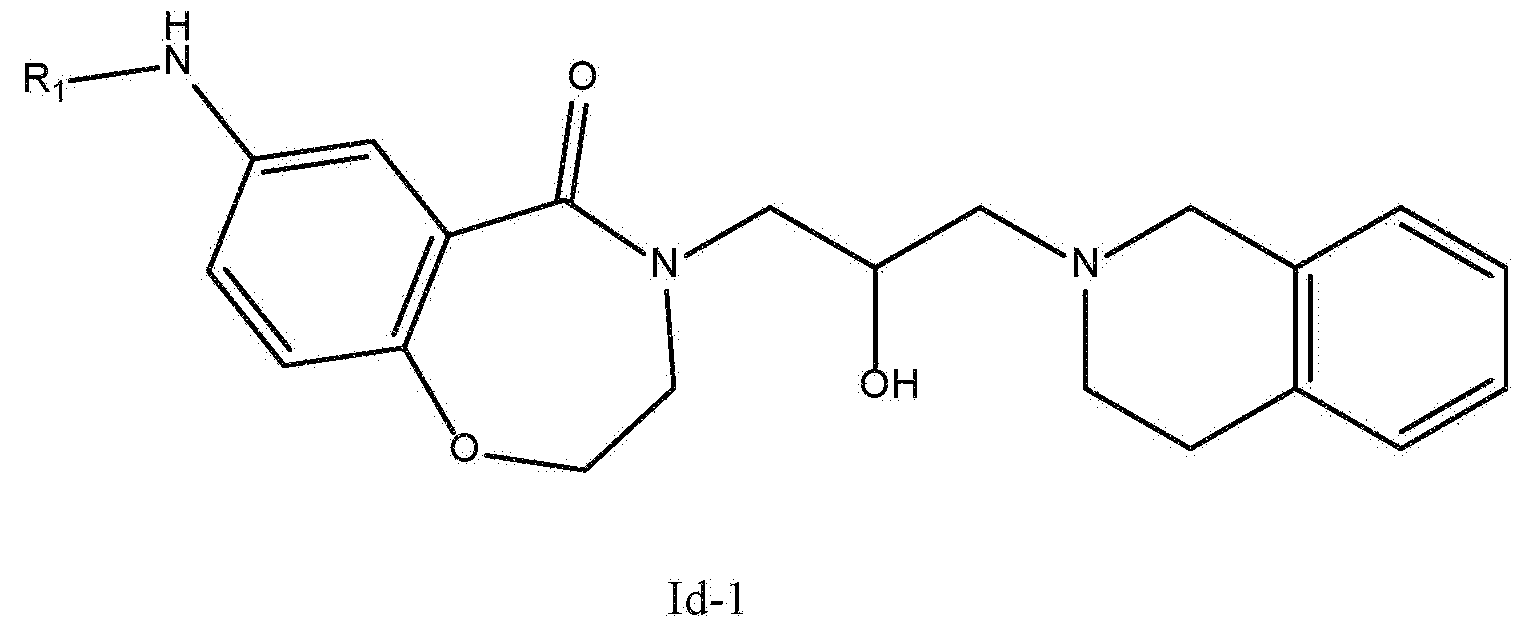




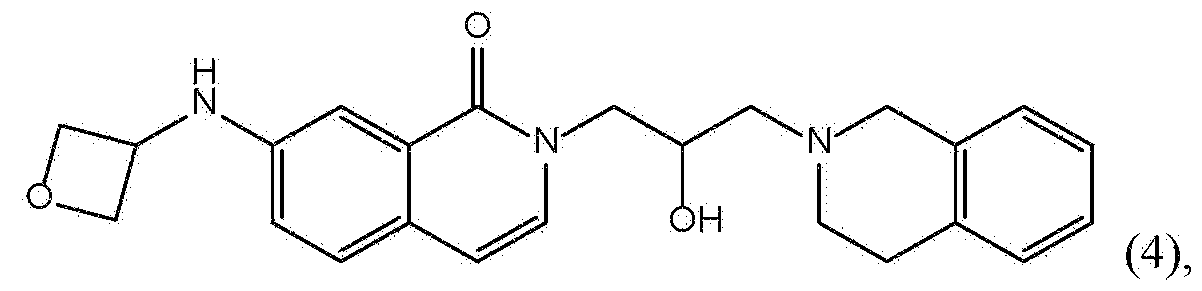















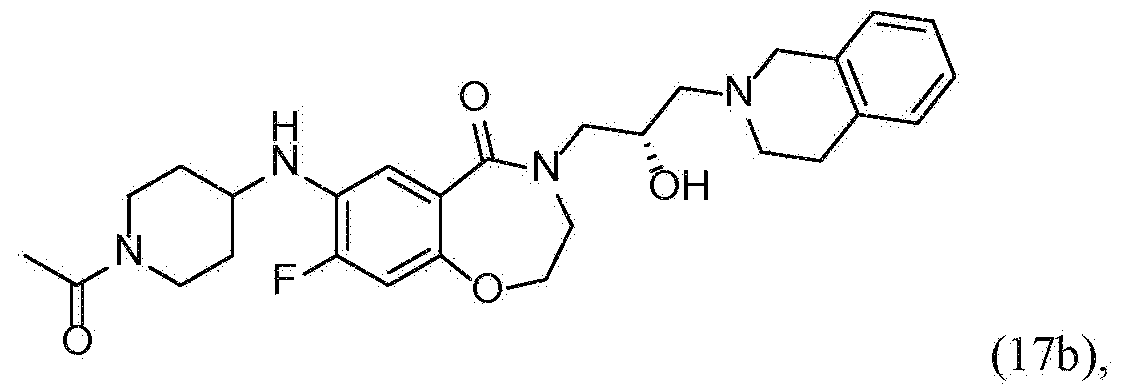




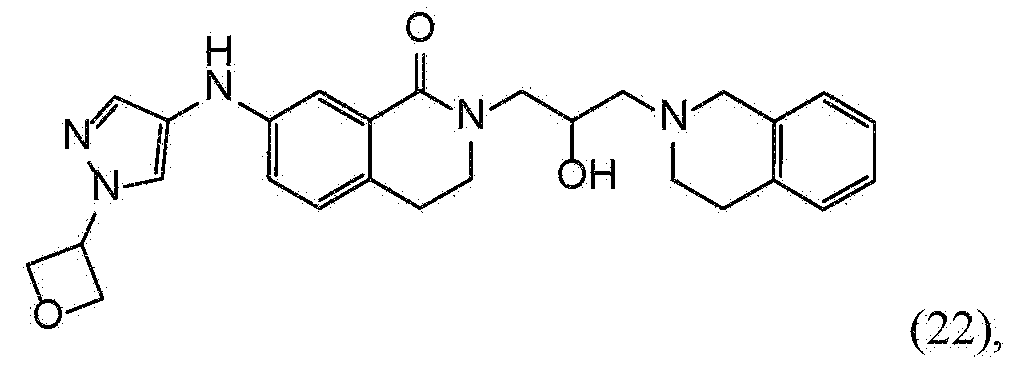

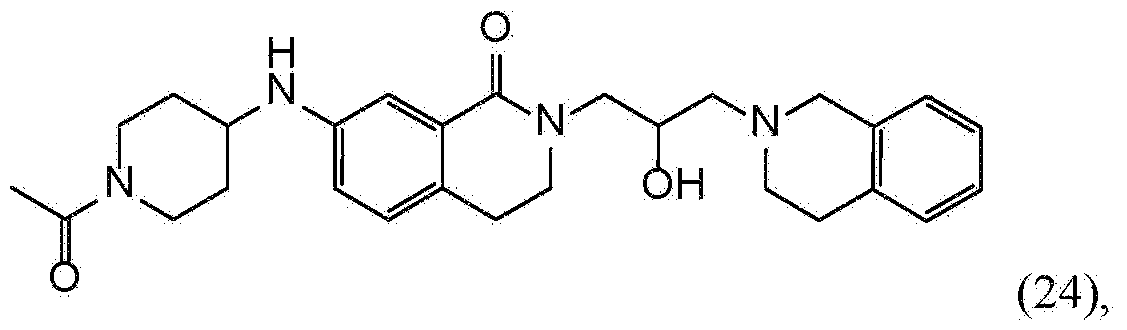























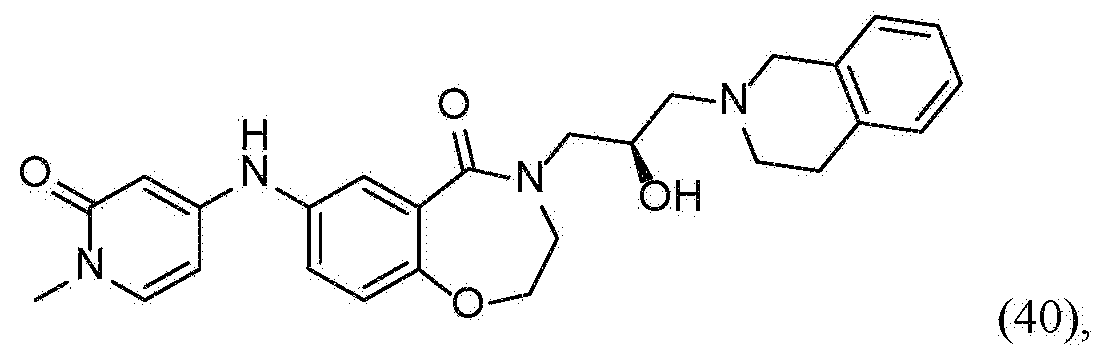


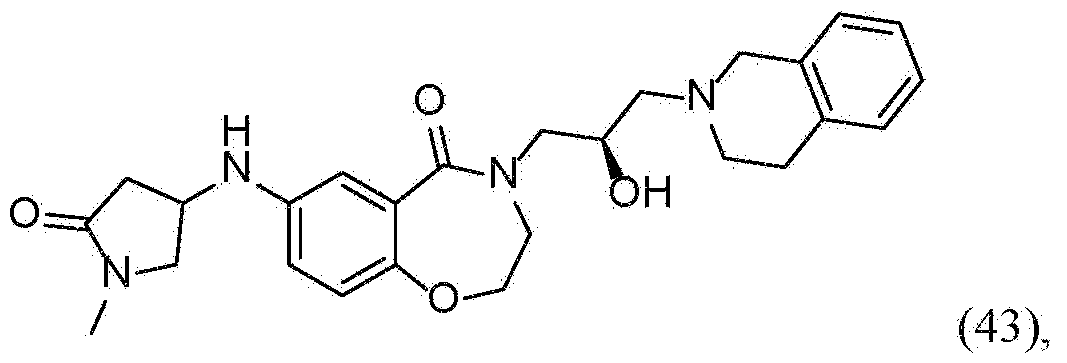










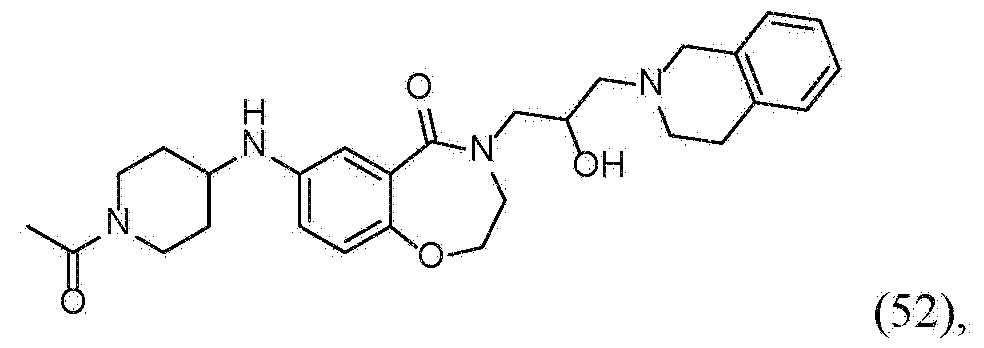









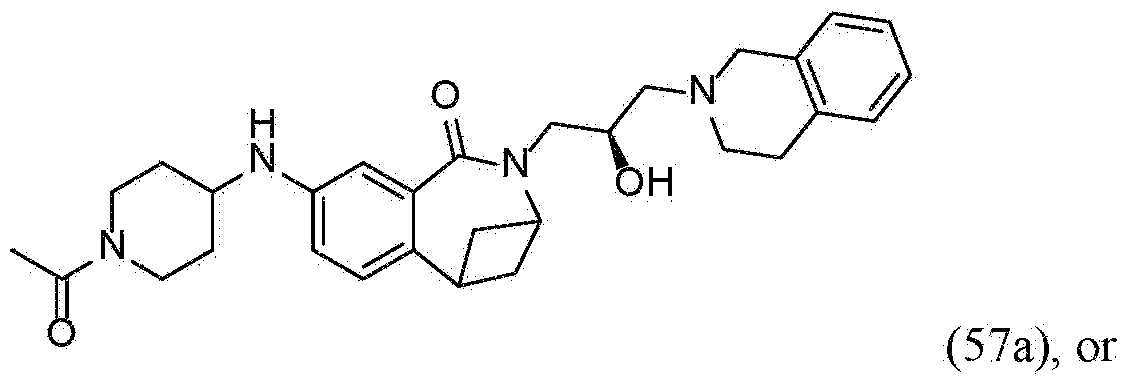



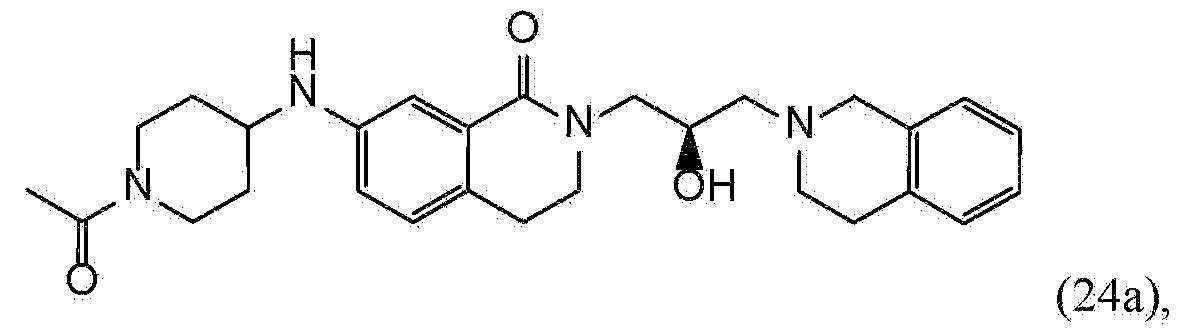


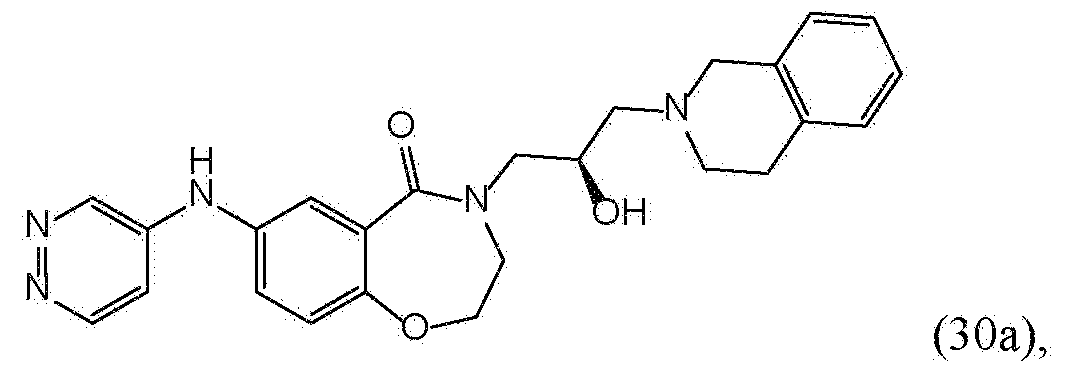




Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US16/969,652 US11591326B2 (en) | 2018-03-09 | 2019-03-08 | Inhibitors of protein arginine methyltransferase 5 (PRMT5), pharmaceutical products thereof, and methods thereof |
| JP2020566553A JP7092895B2 (en) | 2018-03-09 | 2019-03-08 | Inhibitors of protein arginine methyltransferase 5 (PRMT5), pharmaceuticals thereof and methods thereof |
| CA3090836A CA3090836C (en) | 2018-03-09 | 2019-03-08 | 3,4-dihydroisoquinolin-2(1h)-yl derivates useful as inhibitors of protein arginine methyltransferase 5 (prmt5), and pharmaceutical products thereof |
| EP19763499.1A EP3761978A4 (en) | 2018-03-09 | 2019-03-08 | Inhibitors of protein arginine methyltransferase 5 (prmt5), pharmaceutical products thereof, and methods thereof |
| KR1020207026248A KR102522856B1 (en) | 2018-03-09 | 2019-03-08 | Inhibitors of protein arginine methyltransferase 5 (PRMT 5), pharmaceutical products thereof and methods thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201862641241P | 2018-03-09 | 2018-03-09 | |
| US62/641,241 | 2018-03-09 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2019173804A1 true WO2019173804A1 (en) | 2019-09-12 |
Family
ID=67846363
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2019/021497 WO2019173804A1 (en) | 2018-03-09 | 2019-03-08 | Inhibitors of protein arginine methyltransferase 5 (prmt5), pharmaceutical products thereof, and methods thereof |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US11591326B2 (en) |
| EP (1) | EP3761978A4 (en) |
| JP (1) | JP7092895B2 (en) |
| KR (1) | KR102522856B1 (en) |
| CA (1) | CA3090836C (en) |
| WO (1) | WO2019173804A1 (en) |
Cited By (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110950841A (en) * | 2019-11-22 | 2020-04-03 | 济南大学 | Synthesis and application of novel triazole compounds |
| CN112125886A (en) * | 2019-06-24 | 2020-12-25 | 南京圣和药物研发有限公司 | Tricyclic compound serving as PRMT5 inhibitor and application thereof |
| WO2021066578A1 (en) * | 2019-10-02 | 2021-04-08 | 에스케이바이오팜 주식회사 | Bicyclic compound and use thereof |
| CN112645946A (en) * | 2019-10-12 | 2021-04-13 | 南京圣和药业股份有限公司 | Substituted tricyclic compound as PRMT5 inhibitor and application thereof |
| WO2021080359A1 (en) * | 2019-10-23 | 2021-04-29 | 에스케이바이오팜 주식회사 | Bicyclic compound and use thereof |
| US11077101B1 (en) | 2018-07-18 | 2021-08-03 | Tango Therapeutics, Inc. | Compounds and methods of use |
| US11111237B2 (en) | 2019-10-02 | 2021-09-07 | Sk Biopharmaceuticals Co., Ltd. | Bicyclic compound and use thereof |
| WO2021244542A1 (en) * | 2020-06-02 | 2021-12-09 | 石药集团中奇制药技术(石家庄)有限公司 | 3,4-dihydroisoquinoline compound and use thereof |
| WO2022002142A1 (en) * | 2020-06-30 | 2022-01-06 | 江苏先声药业有限公司 | Tetrahydroisoquinoline compounds and use thereof |
| WO2022048631A1 (en) | 2020-09-04 | 2022-03-10 | 上海翊石医药科技有限公司 | Compound having antitumor activity and use thereof |
| WO2022206964A1 (en) * | 2021-04-02 | 2022-10-06 | 海思科医药集团股份有限公司 | Prmt5 inhibitor and use thereof |
| US11492350B2 (en) | 2020-07-31 | 2022-11-08 | Tango Therapeutics, Inc. | Compounds and methods of use |
| WO2022237858A1 (en) | 2021-05-13 | 2022-11-17 | 上海翊石医药科技有限公司 | Compound having anti-tumor activity and use thereof |
| WO2023125947A1 (en) * | 2021-12-30 | 2023-07-06 | 江苏先声药业有限公司 | Pharmaceutically acceptable salt of tetrahydroisoquinoline compound, and crystal form and use thereof |
| WO2023131305A1 (en) * | 2022-01-06 | 2023-07-13 | 江苏先声药业有限公司 | Combination of prmt5 inhibitor and anti-cancer therapeutic agent |
| WO2023223099A3 (en) * | 2022-05-20 | 2023-12-28 | Hangzhou Unogen Biotech, Ltd. | Prmt5 inhibitors and methods of treatment |
| RU2825312C1 (en) * | 2020-06-02 | 2024-08-23 | СиЭсПиСи ЧЖУНЦИ ФАРМАСЬЮТИКАЛ ТЕКНОЛОДЖИ (ШИЦЗЯЧЖУАН) КО., ЛТД | 3,4-dihydroisoquinoline compound and use thereof |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP7298992B2 (en) | 2018-02-05 | 2023-06-27 | 横河電機株式会社 | Driving evaluation device, driving evaluation method, and driving evaluation program |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013120980A1 (en) | 2012-02-17 | 2013-08-22 | F. Hoffmann-La Roche Ag | Tricyclic compounds and methods of use therefor |
| WO2014100716A1 (en) | 2012-12-21 | 2014-06-26 | Epizyme, Inc. | Prmt5 inhibitors and uses thereof |
| WO2014100719A2 (en) | 2012-12-21 | 2014-06-26 | Epizyme, Inc. | Prmt5 inhibitors and uses thereof |
| US9777008B2 (en) * | 2012-12-21 | 2017-10-03 | Epizyme, Inc. | PRMT5 inhibitors and uses thereof |
-
2019
- 2019-03-08 US US16/969,652 patent/US11591326B2/en active Active
- 2019-03-08 KR KR1020207026248A patent/KR102522856B1/en active IP Right Grant
- 2019-03-08 EP EP19763499.1A patent/EP3761978A4/en active Pending
- 2019-03-08 JP JP2020566553A patent/JP7092895B2/en active Active
- 2019-03-08 CA CA3090836A patent/CA3090836C/en active Active
- 2019-03-08 WO PCT/US2019/021497 patent/WO2019173804A1/en active Application Filing
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013120980A1 (en) | 2012-02-17 | 2013-08-22 | F. Hoffmann-La Roche Ag | Tricyclic compounds and methods of use therefor |
| WO2014100716A1 (en) | 2012-12-21 | 2014-06-26 | Epizyme, Inc. | Prmt5 inhibitors and uses thereof |
| WO2014100719A2 (en) | 2012-12-21 | 2014-06-26 | Epizyme, Inc. | Prmt5 inhibitors and uses thereof |
| US9611257B2 (en) * | 2012-12-21 | 2017-04-04 | Epizyme, Inc. | PRMT5 inhibitors and uses thereof |
| US9777008B2 (en) * | 2012-12-21 | 2017-10-03 | Epizyme, Inc. | PRMT5 inhibitors and uses thereof |
Non-Patent Citations (2)
| Title |
|---|
| DATABASE Pubchem Compound 20 January 2016 (2016-01-20), "3-[3-(3,4-Dihydro-1H-isoquinolin-2-yl)-2-hydroxypropyl]-8-methylquinazolin-4-one", XP055637376, retrieved from NCBI Database accession no. 111149296 * |
| See also references of EP3761978A4 |
Cited By (37)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11986471B2 (en) | 2018-07-18 | 2024-05-21 | Tango Therapeutics, Inc. | Compounds and methods of use |
| US11077101B1 (en) | 2018-07-18 | 2021-08-03 | Tango Therapeutics, Inc. | Compounds and methods of use |
| CN112125886A (en) * | 2019-06-24 | 2020-12-25 | 南京圣和药物研发有限公司 | Tricyclic compound serving as PRMT5 inhibitor and application thereof |
| WO2020259478A1 (en) * | 2019-06-24 | 2020-12-30 | 南京圣和药物研发有限公司 | Tricyclic compound as prmt5 inhibitor and application thereof |
| CN112125886B (en) * | 2019-06-24 | 2023-06-23 | 南京圣和药物研发有限公司 | Tricyclic compounds as PRMT5 inhibitors and application thereof |
| CN114929691A (en) * | 2019-10-02 | 2022-08-19 | 爱思开生物制药株式会社 | Bicyclic compounds and uses thereof |
| WO2021066578A1 (en) * | 2019-10-02 | 2021-04-08 | 에스케이바이오팜 주식회사 | Bicyclic compound and use thereof |
| US11725001B2 (en) | 2019-10-02 | 2023-08-15 | Sk Biopharmaceuticals Co., Ltd. | Bicyclic compound and use thereof |
| EP4039686A4 (en) * | 2019-10-02 | 2023-08-09 | SK Biopharmaceuticals Co., Ltd. | Bicyclic compound and use thereof |
| US11111237B2 (en) | 2019-10-02 | 2021-09-07 | Sk Biopharmaceuticals Co., Ltd. | Bicyclic compound and use thereof |
| JP2022551947A (en) * | 2019-10-12 | 2022-12-14 | 南京聖和薬業股▲ふん▼有限公司 | Substituted tricyclic compounds as PRMT5 inhibitors and their applications |
| EP4043459A4 (en) * | 2019-10-12 | 2022-12-21 | Nanjing Sanhome Pharmaceutical Co., Ltd. | Substituted tricyclic compound as prmt5 inhibitor and use thereof |
| CN114466846A (en) * | 2019-10-12 | 2022-05-10 | 南京圣和药业股份有限公司 | Substituted tricyclic compound as PRMT5 inhibitor and application thereof |
| CN112645946A (en) * | 2019-10-12 | 2021-04-13 | 南京圣和药业股份有限公司 | Substituted tricyclic compound as PRMT5 inhibitor and application thereof |
| WO2021068953A1 (en) * | 2019-10-12 | 2021-04-15 | 南京圣和药业股份有限公司 | Substituted tricyclic compound as prmt5 inhibitor and use thereof |
| JP7301222B2 (en) | 2019-10-12 | 2023-06-30 | 南京聖和薬業股▲ふん▼有限公司 | Substituted tricyclic compounds as PRMT5 inhibitors and their applications |
| CN112645946B (en) * | 2019-10-12 | 2024-03-12 | 南京圣和药业股份有限公司 | Substituted tricyclic compounds as PRMT5 inhibitors and application thereof |
| CN114466846B (en) * | 2019-10-12 | 2024-08-20 | 南京圣和药业股份有限公司 | Substituted tricyclic compounds as PRMT5 inhibitors and application thereof |
| AU2020363144B2 (en) * | 2019-10-12 | 2023-08-24 | Nanjing Sanhome Pharmaceutical Co., Ltd. | Substituted tricyclic compound as PRMT5 inhibitor and use thereof |
| CN114945562A (en) * | 2019-10-23 | 2022-08-26 | 爱思开生物制药株式会社 | Bicyclic compounds and uses thereof |
| WO2021080359A1 (en) * | 2019-10-23 | 2021-04-29 | 에스케이바이오팜 주식회사 | Bicyclic compound and use thereof |
| CN110950841A (en) * | 2019-11-22 | 2020-04-03 | 济南大学 | Synthesis and application of novel triazole compounds |
| CN115697988A (en) * | 2020-06-02 | 2023-02-03 | 石药集团中奇制药技术(石家庄)有限公司 | 3, 4-dihydroisoquinoline compound and application thereof |
| WO2021244542A1 (en) * | 2020-06-02 | 2021-12-09 | 石药集团中奇制药技术(石家庄)有限公司 | 3,4-dihydroisoquinoline compound and use thereof |
| CN115697988B (en) * | 2020-06-02 | 2024-04-23 | 石药集团中奇制药技术(石家庄)有限公司 | 3, 4-Dihydro isoquinoline compound and application thereof |
| RU2825312C1 (en) * | 2020-06-02 | 2024-08-23 | СиЭсПиСи ЧЖУНЦИ ФАРМАСЬЮТИКАЛ ТЕКНОЛОДЖИ (ШИЦЗЯЧЖУАН) КО., ЛТД | 3,4-dihydroisoquinoline compound and use thereof |
| CN115768769A (en) * | 2020-06-30 | 2023-03-07 | 江苏先声药业有限公司 | Tetrahydroisoquinoline compound and application thereof |
| CN115768769B (en) * | 2020-06-30 | 2024-10-22 | 南京再明医药有限公司 | Tetrahydroisoquinoline compound and application thereof |
| WO2022002142A1 (en) * | 2020-06-30 | 2022-01-06 | 江苏先声药业有限公司 | Tetrahydroisoquinoline compounds and use thereof |
| US11999727B2 (en) | 2020-07-31 | 2024-06-04 | Tango Therapeutics, Inc. | Compounds and methods of use |
| US11492350B2 (en) | 2020-07-31 | 2022-11-08 | Tango Therapeutics, Inc. | Compounds and methods of use |
| WO2022048631A1 (en) | 2020-09-04 | 2022-03-10 | 上海翊石医药科技有限公司 | Compound having antitumor activity and use thereof |
| WO2022206964A1 (en) * | 2021-04-02 | 2022-10-06 | 海思科医药集团股份有限公司 | Prmt5 inhibitor and use thereof |
| WO2022237858A1 (en) | 2021-05-13 | 2022-11-17 | 上海翊石医药科技有限公司 | Compound having anti-tumor activity and use thereof |
| WO2023125947A1 (en) * | 2021-12-30 | 2023-07-06 | 江苏先声药业有限公司 | Pharmaceutically acceptable salt of tetrahydroisoquinoline compound, and crystal form and use thereof |
| WO2023131305A1 (en) * | 2022-01-06 | 2023-07-13 | 江苏先声药业有限公司 | Combination of prmt5 inhibitor and anti-cancer therapeutic agent |
| WO2023223099A3 (en) * | 2022-05-20 | 2023-12-28 | Hangzhou Unogen Biotech, Ltd. | Prmt5 inhibitors and methods of treatment |
Also Published As
| Publication number | Publication date |
|---|---|
| US11591326B2 (en) | 2023-02-28 |
| EP3761978A1 (en) | 2021-01-13 |
| JP7092895B2 (en) | 2022-06-28 |
| KR20200119859A (en) | 2020-10-20 |
| EP3761978A4 (en) | 2021-10-27 |
| KR102522856B1 (en) | 2023-04-19 |
| CA3090836A1 (en) | 2019-09-12 |
| CA3090836C (en) | 2024-04-16 |
| US20200399261A1 (en) | 2020-12-24 |
| JP2021514401A (en) | 2021-06-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11591326B2 (en) | Inhibitors of protein arginine methyltransferase 5 (PRMT5), pharmaceutical products thereof, and methods thereof | |
| US10597405B2 (en) | Chemical compounds | |
| KR102495687B1 (en) | fused ring compounds | |
| CN113166103B (en) | EGFR inhibitor and application thereof | |
| KR20210135539A (en) | Fused tricyclic compounds useful as anticancer agents | |
| KR20230167386A (en) | Oxazepine compounds and their use in cancer treatment | |
| US20230065740A1 (en) | Cyclin-dependent kinase inhibitors | |
| EA028890B1 (en) | Heterocyclylamines as pi3k inhibitors | |
| JP7307729B2 (en) | Exo-Azaspiro Inhibitors of the Menin-MLL Interaction | |
| EP3053923B1 (en) | Triazolopyrazine derivatives as tyrosin kinase inhibitors | |
| CN111825656B (en) | Inhibitors of protein arginine methyltransferase 5 (PRMT 5), pharmaceutical products thereof, and methods thereof | |
| JP2021515767A (en) | Identification and use of ERK5 inhibitors | |
| JP2018526393A (en) | Tricyclic PI3K inhibitor compounds and methods of use | |
| WO2020207991A1 (en) | Hexahydro-1h-pyrazino[1,2-a]pyrazine compounds for the treatment of autoimmune disease | |
| JP2024502301A (en) | Aromatic heterocyclic compounds, pharmaceutical compositions, and uses thereof | |
| CN106188038A (en) | One class has the compound of kinase inhibiting activity, preparation method and purposes | |
| KR20140022229A (en) | Novel triazolopyrazine derivatives and use thereof | |
| CN109790160B (en) | Pyrido five-membered aromatic ring compound, preparation method and application thereof | |
| CN116981673A (en) | Novel thiadiazolopyrimidinone derivatives |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 19763499 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 3090836 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2020566553 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20207026248 Country of ref document: KR Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2019763499 Country of ref document: EP |