WO2019173523A1 - Anti-tissue factor antibody-drug conjugates and their use in the treatment of cancer - Google Patents
Anti-tissue factor antibody-drug conjugates and their use in the treatment of cancer Download PDFInfo
- Publication number
- WO2019173523A1 WO2019173523A1 PCT/US2019/021024 US2019021024W WO2019173523A1 WO 2019173523 A1 WO2019173523 A1 WO 2019173523A1 US 2019021024 W US2019021024 W US 2019021024W WO 2019173523 A1 WO2019173523 A1 WO 2019173523A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cancer
- antibody
- subject
- drug conjugate
- months
- Prior art date
Links
- 229940049595 antibody-drug conjugate Drugs 0.000 title claims abstract description 419
- 239000000611 antibody drug conjugate Substances 0.000 title claims abstract description 409
- 206010028980 Neoplasm Diseases 0.000 title claims abstract description 375
- 201000011510 cancer Diseases 0.000 title claims abstract description 222
- 238000011282 treatment Methods 0.000 title claims description 145
- 238000000034 method Methods 0.000 claims abstract description 284
- 108010000499 Thromboplastin Proteins 0.000 claims abstract description 109
- 102000002262 Thromboplastin Human genes 0.000 claims abstract description 108
- 208000002154 non-small cell lung carcinoma Diseases 0.000 claims abstract description 86
- 208000029729 tumor suppressor gene on chromosome 11 Diseases 0.000 claims abstract description 86
- 206010061902 Pancreatic neoplasm Diseases 0.000 claims abstract description 79
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 claims abstract description 79
- 201000002528 pancreatic cancer Diseases 0.000 claims abstract description 79
- 208000008443 pancreatic carcinoma Diseases 0.000 claims abstract description 79
- 206010005003 Bladder cancer Diseases 0.000 claims abstract description 66
- 206010060862 Prostate cancer Diseases 0.000 claims abstract description 66
- 208000000236 Prostatic Neoplasms Diseases 0.000 claims abstract description 66
- 206010014759 Endometrial neoplasm Diseases 0.000 claims abstract description 65
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 claims abstract description 64
- 201000005112 urinary bladder cancer Diseases 0.000 claims abstract description 64
- 206010014733 Endometrial cancer Diseases 0.000 claims abstract description 61
- 208000001333 Colorectal Neoplasms Diseases 0.000 claims abstract description 60
- 206010009944 Colon cancer Diseases 0.000 claims abstract description 59
- 208000000461 Esophageal Neoplasms Diseases 0.000 claims abstract description 55
- 206010030155 Oesophageal carcinoma Diseases 0.000 claims abstract description 55
- 201000004101 esophageal cancer Diseases 0.000 claims abstract description 55
- 201000010536 head and neck cancer Diseases 0.000 claims abstract description 51
- 208000014829 head and neck neoplasm Diseases 0.000 claims abstract description 51
- 238000004519 manufacturing process Methods 0.000 claims abstract description 17
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 claims description 166
- 238000009121 systemic therapy Methods 0.000 claims description 154
- 239000000427 antigen Substances 0.000 claims description 126
- 108091007433 antigens Proteins 0.000 claims description 125
- 102000036639 antigens Human genes 0.000 claims description 125
- 239000003814 drug Substances 0.000 claims description 117
- 125000003275 alpha amino acid group Chemical group 0.000 claims description 111
- 239000012634 fragment Substances 0.000 claims description 107
- 238000002560 therapeutic procedure Methods 0.000 claims description 107
- 230000004044 response Effects 0.000 claims description 92
- 229910052697 platinum Inorganic materials 0.000 claims description 83
- 239000003795 chemical substances by application Substances 0.000 claims description 76
- 229940076838 Immune checkpoint inhibitor Drugs 0.000 claims description 75
- 239000012274 immune-checkpoint protein inhibitor Substances 0.000 claims description 75
- 229940124597 therapeutic agent Drugs 0.000 claims description 74
- 230000002411 adverse Effects 0.000 claims description 73
- 230000004083 survival effect Effects 0.000 claims description 66
- 229950004269 tisotumab vedotin Drugs 0.000 claims description 63
- 206010061818 Disease progression Diseases 0.000 claims description 49
- 230000005750 disease progression Effects 0.000 claims description 49
- 238000001959 radiotherapy Methods 0.000 claims description 38
- 208000037842 advanced-stage tumor Diseases 0.000 claims description 36
- 230000002829 reductive effect Effects 0.000 claims description 33
- 230000001225 therapeutic effect Effects 0.000 claims description 32
- 238000001356 surgical procedure Methods 0.000 claims description 30
- 108010044540 auristatin Proteins 0.000 claims description 27
- 239000003889 eye drop Substances 0.000 claims description 26
- 208000009956 adenocarcinoma Diseases 0.000 claims description 25
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 25
- IEDXPSOJFSVCKU-HOKPPMCLSA-N [4-[[(2S)-5-(carbamoylamino)-2-[[(2S)-2-[6-(2,5-dioxopyrrolidin-1-yl)hexanoylamino]-3-methylbutanoyl]amino]pentanoyl]amino]phenyl]methyl N-[(2S)-1-[[(2S)-1-[[(3R,4S,5S)-1-[(2S)-2-[(1R,2R)-3-[[(1S,2R)-1-hydroxy-1-phenylpropan-2-yl]amino]-1-methoxy-2-methyl-3-oxopropyl]pyrrolidin-1-yl]-3-methoxy-5-methyl-1-oxoheptan-4-yl]-methylamino]-3-methyl-1-oxobutan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]-N-methylcarbamate Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C)[C@@H](O)c1ccccc1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)OCc1ccc(NC(=O)[C@H](CCCNC(N)=O)NC(=O)[C@@H](NC(=O)CCCCCN2C(=O)CCC2=O)C(C)C)cc1)C(C)C IEDXPSOJFSVCKU-HOKPPMCLSA-N 0.000 claims description 24
- 229960002949 fluorouracil Drugs 0.000 claims description 24
- 108010093470 monomethyl auristatin E Proteins 0.000 claims description 24
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 claims description 23
- 206010041823 squamous cell carcinoma Diseases 0.000 claims description 21
- 229960001756 oxaliplatin Drugs 0.000 claims description 19
- DWAFYCQODLXJNR-BNTLRKBRSA-L oxaliplatin Chemical compound O1C(=O)C(=O)O[Pt]11N[C@@H]2CCCC[C@H]2N1 DWAFYCQODLXJNR-BNTLRKBRSA-L 0.000 claims description 19
- 108090000765 processed proteins & peptides Proteins 0.000 claims description 19
- 206010010741 Conjunctivitis Diseases 0.000 claims description 18
- 108010072866 Prostate-Specific Antigen Proteins 0.000 claims description 18
- 230000009467 reduction Effects 0.000 claims description 18
- 229960004768 irinotecan Drugs 0.000 claims description 17
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 claims description 17
- 108700012941 GNRH1 Proteins 0.000 claims description 15
- 239000000579 Gonadotropin-Releasing Hormone Substances 0.000 claims description 15
- 239000000556 agonist Substances 0.000 claims description 15
- 208000037819 metastatic cancer Diseases 0.000 claims description 15
- 208000011575 metastatic malignant neoplasm Diseases 0.000 claims description 15
- 229960001592 paclitaxel Drugs 0.000 claims description 15
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 claims description 15
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 claims description 14
- 229960003668 docetaxel Drugs 0.000 claims description 14
- 125000003396 thiol group Chemical group [H]S* 0.000 claims description 14
- 206010038111 Recurrent cancer Diseases 0.000 claims description 13
- 238000001990 intravenous administration Methods 0.000 claims description 13
- 206010023332 keratitis Diseases 0.000 claims description 13
- 230000001050 lubricating effect Effects 0.000 claims description 13
- 150000003431 steroids Chemical class 0.000 claims description 13
- 239000005526 vasoconstrictor agent Substances 0.000 claims description 13
- 230000002280 anti-androgenic effect Effects 0.000 claims description 12
- 239000000051 antiandrogen Substances 0.000 claims description 12
- 229960005277 gemcitabine Drugs 0.000 claims description 12
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 claims description 12
- 229930012538 Paclitaxel Natural products 0.000 claims description 11
- 238000001794 hormone therapy Methods 0.000 claims description 11
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 claims description 10
- 238000009167 androgen deprivation therapy Methods 0.000 claims description 10
- 210000004369 blood Anatomy 0.000 claims description 10
- 239000008280 blood Substances 0.000 claims description 10
- 229960005395 cetuximab Drugs 0.000 claims description 10
- 230000036961 partial effect Effects 0.000 claims description 10
- 229950000154 tisotumab Drugs 0.000 claims description 10
- 229960000397 bevacizumab Drugs 0.000 claims description 9
- WAVYAFBQOXCGSZ-UHFFFAOYSA-N 2-fluoropyrimidine Chemical compound FC1=NC=CC=N1 WAVYAFBQOXCGSZ-UHFFFAOYSA-N 0.000 claims description 7
- 229940124766 Cyp17 inhibitor Drugs 0.000 claims description 7
- 108010016626 Dipeptides Proteins 0.000 claims description 7
- 102000009465 Growth Factor Receptors Human genes 0.000 claims description 7
- 108010009202 Growth Factor Receptors Proteins 0.000 claims description 7
- 229960002173 citrulline Drugs 0.000 claims description 7
- 239000002474 gonadorelin antagonist Substances 0.000 claims description 7
- GAGWJHPBXLXJQN-UORFTKCHSA-N Capecitabine Chemical compound C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1[C@H]1[C@H](O)[C@H](O)[C@@H](C)O1 GAGWJHPBXLXJQN-UORFTKCHSA-N 0.000 claims description 6
- GAGWJHPBXLXJQN-UHFFFAOYSA-N Capecitabine Natural products C1=C(F)C(NC(=O)OCCCCC)=NC(=O)N1C1C(O)C(O)C(C)O1 GAGWJHPBXLXJQN-UHFFFAOYSA-N 0.000 claims description 6
- BMQGVNUXMIRLCK-OAGWZNDDSA-N cabazitaxel Chemical compound O([C@H]1[C@@H]2[C@]3(OC(C)=O)CO[C@@H]3C[C@@H]([C@]2(C(=O)[C@H](OC)C2=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=3C=CC=CC=3)C[C@]1(O)C2(C)C)C)OC)C(=O)C1=CC=CC=C1 BMQGVNUXMIRLCK-OAGWZNDDSA-N 0.000 claims description 6
- 229960001573 cabazitaxel Drugs 0.000 claims description 6
- 229960004117 capecitabine Drugs 0.000 claims description 6
- 238000012326 endoscopic mucosal resection Methods 0.000 claims description 6
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 claims description 6
- 229960004618 prednisone Drugs 0.000 claims description 6
- 229960002633 ramucirumab Drugs 0.000 claims description 6
- 229960000575 trastuzumab Drugs 0.000 claims description 6
- 208000004998 Abdominal Pain Diseases 0.000 claims description 5
- 206010052747 Adenocarcinoma pancreas Diseases 0.000 claims description 5
- 201000004384 Alopecia Diseases 0.000 claims description 5
- 206010010774 Constipation Diseases 0.000 claims description 5
- 206010012735 Diarrhoea Diseases 0.000 claims description 5
- 206010049438 General physical health deterioration Diseases 0.000 claims description 5
- 208000019025 Hypokalemia Diseases 0.000 claims description 5
- 206010021036 Hyponatraemia Diseases 0.000 claims description 5
- 206010027452 Metastases to bone Diseases 0.000 claims description 5
- 206010028813 Nausea Diseases 0.000 claims description 5
- 206010047700 Vomiting Diseases 0.000 claims description 5
- 231100000360 alopecia Toxicity 0.000 claims description 5
- 208000007502 anemia Diseases 0.000 claims description 5
- 206010061428 decreased appetite Diseases 0.000 claims description 5
- 229960004679 doxorubicin Drugs 0.000 claims description 5
- 208000001780 epistaxis Diseases 0.000 claims description 5
- 206010016256 fatigue Diseases 0.000 claims description 5
- 230000001976 improved effect Effects 0.000 claims description 5
- 230000008693 nausea Effects 0.000 claims description 5
- 201000002094 pancreatic adenocarcinoma Diseases 0.000 claims description 5
- 208000033808 peripheral neuropathy Diseases 0.000 claims description 5
- 208000024896 potassium deficiency disease Diseases 0.000 claims description 5
- 238000009097 single-agent therapy Methods 0.000 claims description 5
- 230000008673 vomiting Effects 0.000 claims description 5
- 239000008194 pharmaceutical composition Substances 0.000 claims description 4
- 102000007066 Prostate-Specific Antigen Human genes 0.000 claims 4
- 239000000203 mixture Substances 0.000 abstract description 42
- 210000004027 cell Anatomy 0.000 description 84
- 201000010099 disease Diseases 0.000 description 44
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 44
- 125000005647 linker group Chemical group 0.000 description 44
- -1 BSA Proteins 0.000 description 39
- 238000002591 computed tomography Methods 0.000 description 32
- 239000003112 inhibitor Substances 0.000 description 27
- 229940079593 drug Drugs 0.000 description 25
- 229960003301 nivolumab Drugs 0.000 description 25
- 229960002621 pembrolizumab Drugs 0.000 description 25
- 108010074708 B7-H1 Antigen Proteins 0.000 description 23
- 102100024216 Programmed cell death 1 ligand 1 Human genes 0.000 description 23
- 238000009472 formulation Methods 0.000 description 22
- 108010047041 Complementarity Determining Regions Proteins 0.000 description 21
- 108060003951 Immunoglobulin Proteins 0.000 description 21
- 102000018358 immunoglobulin Human genes 0.000 description 21
- 108090000623 proteins and genes Proteins 0.000 description 21
- 102100035360 Cerebellar degeneration-related antigen 1 Human genes 0.000 description 19
- 241000699670 Mus sp. Species 0.000 description 18
- 238000006722 reduction reaction Methods 0.000 description 17
- 101710117290 Aldo-keto reductase family 1 member C4 Proteins 0.000 description 16
- 238000002965 ELISA Methods 0.000 description 16
- 238000003364 immunohistochemistry Methods 0.000 description 16
- 238000002600 positron emission tomography Methods 0.000 description 16
- 108010021064 CTLA-4 Antigen Proteins 0.000 description 15
- 229940045513 CTLA4 antagonist Drugs 0.000 description 15
- 102100039498 Cytotoxic T-lymphocyte protein 4 Human genes 0.000 description 15
- 206010061289 metastatic neoplasm Diseases 0.000 description 15
- 102100038358 Prostate-specific antigen Human genes 0.000 description 14
- 229960004316 cisplatin Drugs 0.000 description 14
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 14
- 230000034994 death Effects 0.000 description 14
- 231100000517 death Toxicity 0.000 description 14
- 108700019146 Transgenes Proteins 0.000 description 13
- 230000000259 anti-tumor effect Effects 0.000 description 13
- 241000699666 Mus <mouse, genus> Species 0.000 description 12
- 238000002595 magnetic resonance imaging Methods 0.000 description 12
- 208000024891 symptom Diseases 0.000 description 12
- 238000002604 ultrasonography Methods 0.000 description 12
- 101000635804 Homo sapiens Tissue factor Proteins 0.000 description 11
- 238000002512 chemotherapy Methods 0.000 description 11
- 230000001394 metastastic effect Effects 0.000 description 11
- 235000018102 proteins Nutrition 0.000 description 11
- 102000004169 proteins and genes Human genes 0.000 description 11
- 102100038078 CD276 antigen Human genes 0.000 description 10
- 102100038929 V-set domain-containing T-cell activation inhibitor 1 Human genes 0.000 description 10
- 235000001014 amino acid Nutrition 0.000 description 10
- 229940024606 amino acid Drugs 0.000 description 10
- 239000003242 anti bacterial agent Substances 0.000 description 10
- 229950002916 avelumab Drugs 0.000 description 10
- KLNFSAOEKUDMFA-UHFFFAOYSA-N azanide;2-hydroxyacetic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OCC(O)=O KLNFSAOEKUDMFA-UHFFFAOYSA-N 0.000 description 10
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 10
- 230000003115 biocidal effect Effects 0.000 description 10
- 229960004562 carboplatin Drugs 0.000 description 10
- 238000011161 development Methods 0.000 description 10
- 230000018109 developmental process Effects 0.000 description 10
- 230000035772 mutation Effects 0.000 description 10
- 229950007221 nedaplatin Drugs 0.000 description 10
- 229950005566 picoplatin Drugs 0.000 description 10
- IIMIOEBMYPRQGU-UHFFFAOYSA-L picoplatin Chemical compound N.[Cl-].[Cl-].[Pt+2].CC1=CC=CC=N1 IIMIOEBMYPRQGU-UHFFFAOYSA-L 0.000 description 10
- 229960005399 satraplatin Drugs 0.000 description 10
- 190014017285 satraplatin Chemical compound 0.000 description 10
- 241000894007 species Species 0.000 description 10
- 190014017283 triplatin tetranitrate Chemical compound 0.000 description 10
- 229950002860 triplatin tetranitrate Drugs 0.000 description 10
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 9
- 150000001413 amino acids Chemical class 0.000 description 9
- 238000001802 infusion Methods 0.000 description 9
- 238000002347 injection Methods 0.000 description 9
- 239000007924 injection Substances 0.000 description 9
- 238000005259 measurement Methods 0.000 description 9
- 230000000306 recurrent effect Effects 0.000 description 9
- 229940011871 estrogen Drugs 0.000 description 8
- 239000000262 estrogen Substances 0.000 description 8
- 238000000684 flow cytometry Methods 0.000 description 8
- 150000003839 salts Chemical class 0.000 description 8
- 238000006467 substitution reaction Methods 0.000 description 8
- 210000001519 tissue Anatomy 0.000 description 8
- MFRNYXJJRJQHNW-DEMKXPNLSA-N (2s)-2-[[(2r,3r)-3-methoxy-3-[(2s)-1-[(3r,4s,5s)-3-methoxy-5-methyl-4-[methyl-[(2s)-3-methyl-2-[[(2s)-3-methyl-2-(methylamino)butanoyl]amino]butanoyl]amino]heptanoyl]pyrrolidin-2-yl]-2-methylpropanoyl]amino]-3-phenylpropanoic acid Chemical compound CN[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N(C)[C@@H]([C@@H](C)CC)[C@H](OC)CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 MFRNYXJJRJQHNW-DEMKXPNLSA-N 0.000 description 7
- BQQGAGGSEMLWRS-UHFFFAOYSA-N (4-aminophenyl)methyl carbamate Chemical compound NC(=O)OCC1=CC=C(N)C=C1 BQQGAGGSEMLWRS-UHFFFAOYSA-N 0.000 description 7
- 208000032818 Microsatellite Instability Diseases 0.000 description 7
- 229930006000 Sucrose Natural products 0.000 description 7
- 125000000539 amino acid group Chemical group 0.000 description 7
- 108010059074 monomethylauristatin F Proteins 0.000 description 7
- 108020004707 nucleic acids Proteins 0.000 description 7
- 102000039446 nucleic acids Human genes 0.000 description 7
- 150000007523 nucleic acids Chemical class 0.000 description 7
- 239000005720 sucrose Substances 0.000 description 7
- 206010010736 Conjunctival ulcer Diseases 0.000 description 6
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 6
- 239000000306 component Substances 0.000 description 6
- 230000001419 dependent effect Effects 0.000 description 6
- 230000014509 gene expression Effects 0.000 description 6
- 210000004602 germ cell Anatomy 0.000 description 6
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 6
- 238000009169 immunotherapy Methods 0.000 description 6
- 239000003381 stabilizer Substances 0.000 description 6
- 229940066453 tecentriq Drugs 0.000 description 6
- 238000011830 transgenic mouse model Methods 0.000 description 6
- 229940122815 Aromatase inhibitor Drugs 0.000 description 5
- 102100029822 B- and T-lymphocyte attenuator Human genes 0.000 description 5
- 102100022005 B-lymphocyte antigen CD20 Human genes 0.000 description 5
- 102100024217 CAMPATH-1 antigen Human genes 0.000 description 5
- 102100027207 CD27 antigen Human genes 0.000 description 5
- 101710185679 CD276 antigen Proteins 0.000 description 5
- 101150013553 CD40 gene Proteins 0.000 description 5
- 108010065524 CD52 Antigen Proteins 0.000 description 5
- 102100025221 CD70 antigen Human genes 0.000 description 5
- 206010052358 Colorectal cancer metastatic Diseases 0.000 description 5
- 102100031351 Galectin-9 Human genes 0.000 description 5
- 101100229077 Gallus gallus GAL9 gene Proteins 0.000 description 5
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 5
- 101150046249 Havcr2 gene Proteins 0.000 description 5
- 102100034458 Hepatitis A virus cellular receptor 2 Human genes 0.000 description 5
- 101000864344 Homo sapiens B- and T-lymphocyte attenuator Proteins 0.000 description 5
- 101000897405 Homo sapiens B-lymphocyte antigen CD20 Proteins 0.000 description 5
- 101000914511 Homo sapiens CD27 antigen Proteins 0.000 description 5
- 101000934356 Homo sapiens CD70 antigen Proteins 0.000 description 5
- 101001137987 Homo sapiens Lymphocyte activation gene 3 protein Proteins 0.000 description 5
- 101000934338 Homo sapiens Myeloid cell surface antigen CD33 Proteins 0.000 description 5
- 101000831007 Homo sapiens T-cell immunoreceptor with Ig and ITIM domains Proteins 0.000 description 5
- 101000934346 Homo sapiens T-cell surface antigen CD2 Proteins 0.000 description 5
- 101000914514 Homo sapiens T-cell-specific surface glycoprotein CD28 Proteins 0.000 description 5
- 101000914484 Homo sapiens T-lymphocyte activation antigen CD80 Proteins 0.000 description 5
- 101000801234 Homo sapiens Tumor necrosis factor receptor superfamily member 18 Proteins 0.000 description 5
- 101000851376 Homo sapiens Tumor necrosis factor receptor superfamily member 8 Proteins 0.000 description 5
- 101000955999 Homo sapiens V-set domain-containing T-cell activation inhibitor 1 Proteins 0.000 description 5
- 101000666896 Homo sapiens V-type immunoglobulin domain-containing suppressor of T-cell activation Proteins 0.000 description 5
- 102000017578 LAG3 Human genes 0.000 description 5
- 108010061593 Member 14 Tumor Necrosis Factor Receptors Proteins 0.000 description 5
- 241000699660 Mus musculus Species 0.000 description 5
- 101100407308 Mus musculus Pdcd1lg2 gene Proteins 0.000 description 5
- 102100025243 Myeloid cell surface antigen CD33 Human genes 0.000 description 5
- 238000011789 NOD SCID mouse Methods 0.000 description 5
- 108700030875 Programmed Cell Death 1 Ligand 2 Proteins 0.000 description 5
- 102100024213 Programmed cell death 1 ligand 2 Human genes 0.000 description 5
- 102000008115 Signaling Lymphocytic Activation Molecule Family Member 1 Human genes 0.000 description 5
- 108010074687 Signaling Lymphocytic Activation Molecule Family Member 1 Proteins 0.000 description 5
- 102100024834 T-cell immunoreceptor with Ig and ITIM domains Human genes 0.000 description 5
- 102100025237 T-cell surface antigen CD2 Human genes 0.000 description 5
- 102100027213 T-cell-specific surface glycoprotein CD28 Human genes 0.000 description 5
- 102100027222 T-lymphocyte activation antigen CD80 Human genes 0.000 description 5
- 102100028785 Tumor necrosis factor receptor superfamily member 14 Human genes 0.000 description 5
- 102100033728 Tumor necrosis factor receptor superfamily member 18 Human genes 0.000 description 5
- 102100040245 Tumor necrosis factor receptor superfamily member 5 Human genes 0.000 description 5
- 102100036857 Tumor necrosis factor receptor superfamily member 8 Human genes 0.000 description 5
- 108010079206 V-Set Domain-Containing T-Cell Activation Inhibitor 1 Proteins 0.000 description 5
- 102100038282 V-type immunoglobulin domain-containing suppressor of T-cell activation Human genes 0.000 description 5
- 239000003886 aromatase inhibitor Substances 0.000 description 5
- 229960003852 atezolizumab Drugs 0.000 description 5
- 230000008901 benefit Effects 0.000 description 5
- 239000000872 buffer Substances 0.000 description 5
- 229940121420 cemiplimab Drugs 0.000 description 5
- 150000001875 compounds Chemical class 0.000 description 5
- 239000000824 cytostatic agent Substances 0.000 description 5
- 230000001085 cytostatic effect Effects 0.000 description 5
- 238000003745 diagnosis Methods 0.000 description 5
- 229950009791 durvalumab Drugs 0.000 description 5
- 230000000694 effects Effects 0.000 description 5
- IJJVMEJXYNJXOJ-UHFFFAOYSA-N fluquinconazole Chemical compound C=1C=C(Cl)C=C(Cl)C=1N1C(=O)C2=CC(F)=CC=C2N=C1N1C=NC=N1 IJJVMEJXYNJXOJ-UHFFFAOYSA-N 0.000 description 5
- 210000004408 hybridoma Anatomy 0.000 description 5
- 230000006872 improvement Effects 0.000 description 5
- 229960005386 ipilimumab Drugs 0.000 description 5
- 208000020816 lung neoplasm Diseases 0.000 description 5
- 235000010355 mannitol Nutrition 0.000 description 5
- 230000033607 mismatch repair Effects 0.000 description 5
- 239000002736 nonionic surfactant Substances 0.000 description 5
- 210000000056 organ Anatomy 0.000 description 5
- 229950010773 pidilizumab Drugs 0.000 description 5
- 238000007920 subcutaneous administration Methods 0.000 description 5
- 230000009261 transgenic effect Effects 0.000 description 5
- 229950007217 tremelimumab Drugs 0.000 description 5
- 230000004614 tumor growth Effects 0.000 description 5
- 108010058566 130-nm albumin-bound paclitaxel Proteins 0.000 description 4
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 4
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 4
- 201000009030 Carcinoma Diseases 0.000 description 4
- SRBFZHDQGSBBOR-IOVATXLUSA-N D-xylopyranose Chemical compound O[C@@H]1COC(O)[C@H](O)[C@H]1O SRBFZHDQGSBBOR-IOVATXLUSA-N 0.000 description 4
- 101001012157 Homo sapiens Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 description 4
- 206010061534 Oesophageal squamous cell carcinoma Diseases 0.000 description 4
- RJKFOVLPORLFTN-LEKSSAKUSA-N Progesterone Chemical class C1CC2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H](C(=O)C)[C@@]1(C)CC2 RJKFOVLPORLFTN-LEKSSAKUSA-N 0.000 description 4
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 description 4
- 208000036765 Squamous cell carcinoma of the esophagus Diseases 0.000 description 4
- NKANXQFJJICGDU-QPLCGJKRSA-N Tamoxifen Chemical compound C=1C=CC=CC=1C(/CC)=C(C=1C=CC(OCCN(C)C)=CC=1)/C1=CC=CC=C1 NKANXQFJJICGDU-QPLCGJKRSA-N 0.000 description 4
- 239000002246 antineoplastic agent Substances 0.000 description 4
- 238000013459 approach Methods 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- 210000004204 blood vessel Anatomy 0.000 description 4
- 231100000504 carcinogenesis Toxicity 0.000 description 4
- 230000001413 cellular effect Effects 0.000 description 4
- 238000009096 combination chemotherapy Methods 0.000 description 4
- 229940127089 cytotoxic agent Drugs 0.000 description 4
- 239000002254 cytotoxic agent Substances 0.000 description 4
- 230000007423 decrease Effects 0.000 description 4
- 238000012217 deletion Methods 0.000 description 4
- 230000037430 deletion Effects 0.000 description 4
- 208000007276 esophageal squamous cell carcinoma Diseases 0.000 description 4
- 230000002209 hydrophobic effect Effects 0.000 description 4
- 238000004191 hydrophobic interaction chromatography Methods 0.000 description 4
- 230000002401 inhibitory effect Effects 0.000 description 4
- 238000003780 insertion Methods 0.000 description 4
- 230000037431 insertion Effects 0.000 description 4
- 238000007918 intramuscular administration Methods 0.000 description 4
- 238000007912 intraperitoneal administration Methods 0.000 description 4
- 150000002500 ions Chemical class 0.000 description 4
- 230000036210 malignancy Effects 0.000 description 4
- 238000010172 mouse model Methods 0.000 description 4
- 238000002638 palliative care Methods 0.000 description 4
- 238000002823 phage display Methods 0.000 description 4
- 239000000546 pharmaceutical excipient Substances 0.000 description 4
- 230000008569 process Effects 0.000 description 4
- 239000000583 progesterone congener Substances 0.000 description 4
- 238000002271 resection Methods 0.000 description 4
- 238000004007 reversed phase HPLC Methods 0.000 description 4
- 150000005846 sugar alcohols Chemical class 0.000 description 4
- 230000009885 systemic effect Effects 0.000 description 4
- 238000002626 targeted therapy Methods 0.000 description 4
- 230000001988 toxicity Effects 0.000 description 4
- 231100000419 toxicity Toxicity 0.000 description 4
- 238000005303 weighing Methods 0.000 description 4
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 3
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 3
- 108010069236 Goserelin Proteins 0.000 description 3
- BLCLNMBMMGCOAS-URPVMXJPSA-N Goserelin Chemical group C([C@@H](C(=O)N[C@H](COC(C)(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N1[C@@H](CCC1)C(=O)NNC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H]1NC(=O)CC1)C1=CC=C(O)C=C1 BLCLNMBMMGCOAS-URPVMXJPSA-N 0.000 description 3
- 241000282412 Homo Species 0.000 description 3
- 238000012450 HuMAb Mouse Methods 0.000 description 3
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 3
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 3
- 239000005551 L01XE03 - Erlotinib Substances 0.000 description 3
- 108010000817 Leuprolide Proteins 0.000 description 3
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 3
- 241000124008 Mammalia Species 0.000 description 3
- 206010027476 Metastases Diseases 0.000 description 3
- 241001529936 Murinae Species 0.000 description 3
- 229910019142 PO4 Inorganic materials 0.000 description 3
- 239000004365 Protease Substances 0.000 description 3
- 241000700159 Rattus Species 0.000 description 3
- 208000000102 Squamous Cell Carcinoma of Head and Neck Diseases 0.000 description 3
- 102100030859 Tissue factor Human genes 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- 239000013543 active substance Substances 0.000 description 3
- 230000033115 angiogenesis Effects 0.000 description 3
- 238000011717 athymic nude mouse Methods 0.000 description 3
- 238000012512 characterization method Methods 0.000 description 3
- 238000009104 chemotherapy regimen Methods 0.000 description 3
- 229940001468 citrate Drugs 0.000 description 3
- 239000000562 conjugate Substances 0.000 description 3
- 230000021615 conjugation Effects 0.000 description 3
- 230000003247 decreasing effect Effects 0.000 description 3
- 230000002950 deficient Effects 0.000 description 3
- 238000009110 definitive therapy Methods 0.000 description 3
- 239000003599 detergent Substances 0.000 description 3
- 238000005516 engineering process Methods 0.000 description 3
- 229960001433 erlotinib Drugs 0.000 description 3
- AAKJLRGGTJKAMG-UHFFFAOYSA-N erlotinib Chemical compound C=12C=C(OCCOC)C(OCCOC)=CC2=NC=NC=1NC1=CC=CC(C#C)=C1 AAKJLRGGTJKAMG-UHFFFAOYSA-N 0.000 description 3
- 229960002913 goserelin Drugs 0.000 description 3
- 230000012010 growth Effects 0.000 description 3
- 239000003102 growth factor Substances 0.000 description 3
- 201000000459 head and neck squamous cell carcinoma Diseases 0.000 description 3
- 229940127121 immunoconjugate Drugs 0.000 description 3
- 238000001727 in vivo Methods 0.000 description 3
- 239000004615 ingredient Substances 0.000 description 3
- 238000001361 intraarterial administration Methods 0.000 description 3
- 230000003902 lesion Effects 0.000 description 3
- GFIJNRVAKGFPGQ-LIJARHBVSA-N leuprolide Chemical group CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)CC1=CC=C(O)C=C1 GFIJNRVAKGFPGQ-LIJARHBVSA-N 0.000 description 3
- 229960004338 leuprorelin Drugs 0.000 description 3
- 201000005202 lung cancer Diseases 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 229960004296 megestrol acetate Drugs 0.000 description 3
- RQZAXGRLVPAYTJ-GQFGMJRRSA-N megestrol acetate Chemical compound C1=C(C)C2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(C)=O)(OC(=O)C)[C@@]1(C)CC2 RQZAXGRLVPAYTJ-GQFGMJRRSA-N 0.000 description 3
- HEBKCHPVOIAQTA-UHFFFAOYSA-N meso ribitol Natural products OCC(O)C(O)C(O)CO HEBKCHPVOIAQTA-UHFFFAOYSA-N 0.000 description 3
- 230000009401 metastasis Effects 0.000 description 3
- 238000011580 nude mouse model Methods 0.000 description 3
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 3
- 239000010452 phosphate Substances 0.000 description 3
- 229920001184 polypeptide Polymers 0.000 description 3
- 229920000136 polysorbate Polymers 0.000 description 3
- 230000004481 post-translational protein modification Effects 0.000 description 3
- 239000003755 preservative agent Substances 0.000 description 3
- 102000004196 processed proteins & peptides Human genes 0.000 description 3
- 229910052708 sodium Inorganic materials 0.000 description 3
- 239000011734 sodium Substances 0.000 description 3
- 229940083542 sodium Drugs 0.000 description 3
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 3
- 230000002459 sustained effect Effects 0.000 description 3
- 238000011200 topical administration Methods 0.000 description 3
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 3
- YXTKHLHCVFUPPT-YYFJYKOTSA-N (2s)-2-[[4-[(2-amino-5-formyl-4-oxo-1,6,7,8-tetrahydropteridin-6-yl)methylamino]benzoyl]amino]pentanedioic acid;(1r,2r)-1,2-dimethanidylcyclohexane;5-fluoro-1h-pyrimidine-2,4-dione;oxalic acid;platinum(2+) Chemical compound [Pt+2].OC(=O)C(O)=O.[CH2-][C@@H]1CCCC[C@H]1[CH2-].FC1=CNC(=O)NC1=O.C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 YXTKHLHCVFUPPT-YYFJYKOTSA-N 0.000 description 2
- LKJPYSCBVHEWIU-KRWDZBQOSA-N (R)-bicalutamide Chemical compound C([C@@](O)(C)C(=O)NC=1C=C(C(C#N)=CC=1)C(F)(F)F)S(=O)(=O)C1=CC=C(F)C=C1 LKJPYSCBVHEWIU-KRWDZBQOSA-N 0.000 description 2
- GVJHHUAWPYXKBD-UHFFFAOYSA-N (±)-α-Tocopherol Chemical compound OC1=C(C)C(C)=C2OC(CCCC(C)CCCC(C)CCCC(C)C)(C)CCC2=C1C GVJHHUAWPYXKBD-UHFFFAOYSA-N 0.000 description 2
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- VVIAGPKUTFNRDU-UHFFFAOYSA-N 6S-folinic acid Natural products C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)NC(CCC(O)=O)C(O)=O)C=C1 VVIAGPKUTFNRDU-UHFFFAOYSA-N 0.000 description 2
- 206010069754 Acquired gene mutation Diseases 0.000 description 2
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 2
- 241000282472 Canis lupus familiaris Species 0.000 description 2
- 241000283707 Capra Species 0.000 description 2
- 102100025064 Cellular tumor antigen p53 Human genes 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 2
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 2
- WQZGKKKJIJFFOK-QTVWNMPRSA-N D-mannopyranose Chemical compound OC[C@H]1OC(O)[C@@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-QTVWNMPRSA-N 0.000 description 2
- 108020004414 DNA Proteins 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- 208000013452 Fallopian tube neoplasm Diseases 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 101710113436 GTPase KRas Proteins 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- 108010017080 Granulocyte Colony-Stimulating Factor Proteins 0.000 description 2
- 102100039619 Granulocyte colony-stimulating factor Human genes 0.000 description 2
- 229940121710 HMGCoA reductase inhibitor Drugs 0.000 description 2
- 101100297421 Homarus americanus phc-2 gene Proteins 0.000 description 2
- 241000701806 Human papillomavirus Species 0.000 description 2
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 2
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 2
- 206010051792 Infusion related reaction Diseases 0.000 description 2
- 238000012449 Kunming mouse Methods 0.000 description 2
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- 208000001894 Nasopharyngeal Neoplasms Diseases 0.000 description 2
- 206010061535 Ovarian neoplasm Diseases 0.000 description 2
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 2
- 239000012270 PD-1 inhibitor Substances 0.000 description 2
- 239000012668 PD-1-inhibitor Substances 0.000 description 2
- 108010011536 PTEN Phosphohydrolase Proteins 0.000 description 2
- 241001494479 Pecora Species 0.000 description 2
- 102000035195 Peptidases Human genes 0.000 description 2
- 108091005804 Peptidases Proteins 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Natural products OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- 102100032543 Phosphatidylinositol 3,4,5-trisphosphate 3-phosphatase and dual-specificity protein phosphatase PTEN Human genes 0.000 description 2
- 208000026149 Primary peritoneal carcinoma Diseases 0.000 description 2
- 241000283984 Rodentia Species 0.000 description 2
- 102000012479 Serine Proteases Human genes 0.000 description 2
- 108010022999 Serine Proteases Proteins 0.000 description 2
- GUGOEEXESWIERI-UHFFFAOYSA-N Terfenadine Chemical group C1=CC(C(C)(C)C)=CC=C1C(O)CCCN1CCC(C(O)(C=2C=CC=CC=2)C=2C=CC=CC=2)CC1 GUGOEEXESWIERI-UHFFFAOYSA-N 0.000 description 2
- 241000011102 Thera Species 0.000 description 2
- 108090000190 Thrombin Proteins 0.000 description 2
- 108010050144 Triptorelin Pamoate Proteins 0.000 description 2
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 2
- 230000002159 abnormal effect Effects 0.000 description 2
- 230000021736 acetylation Effects 0.000 description 2
- 238000006640 acetylation reaction Methods 0.000 description 2
- 239000003708 ampul Substances 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 208000036878 aneuploidy Diseases 0.000 description 2
- 231100001075 aneuploidy Toxicity 0.000 description 2
- 230000001387 anti-histamine Effects 0.000 description 2
- 230000009830 antibody antigen interaction Effects 0.000 description 2
- 229940125644 antibody drug Drugs 0.000 description 2
- 239000000739 antihistaminic agent Substances 0.000 description 2
- HJBWBFZLDZWPHF-UHFFFAOYSA-N apalutamide Chemical compound C1=C(F)C(C(=O)NC)=CC=C1N1C2(CCC2)C(=O)N(C=2C=C(C(C#N)=NC=2)C(F)(F)F)C1=S HJBWBFZLDZWPHF-UHFFFAOYSA-N 0.000 description 2
- 229950007511 apalutamide Drugs 0.000 description 2
- PYMYPHUHKUWMLA-UHFFFAOYSA-N arabinose Natural products OCC(O)C(O)C(O)C=O PYMYPHUHKUWMLA-UHFFFAOYSA-N 0.000 description 2
- 235000010323 ascorbic acid Nutrition 0.000 description 2
- 239000011668 ascorbic acid Substances 0.000 description 2
- 210000003719 b-lymphocyte Anatomy 0.000 description 2
- 229960001950 benzethonium chloride Drugs 0.000 description 2
- UREZNYTWGJKWBI-UHFFFAOYSA-M benzethonium chloride Chemical compound [Cl-].C1=CC(C(C)(C)CC(C)(C)C)=CC=C1OCCOCC[N+](C)(C)CC1=CC=CC=C1 UREZNYTWGJKWBI-UHFFFAOYSA-M 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- SRBFZHDQGSBBOR-UHFFFAOYSA-N beta-D-Pyranose-Lyxose Natural products OC1COC(O)C(O)C1O SRBFZHDQGSBBOR-UHFFFAOYSA-N 0.000 description 2
- 229960000997 bicalutamide Drugs 0.000 description 2
- 238000012575 bio-layer interferometry Methods 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- 230000005773 cancer-related death Effects 0.000 description 2
- YCIMNLLNPGFGHC-UHFFFAOYSA-N catechol Chemical compound OC1=CC=CC=C1O YCIMNLLNPGFGHC-UHFFFAOYSA-N 0.000 description 2
- 230000000973 chemotherapeutic effect Effects 0.000 description 2
- 210000000349 chromosome Anatomy 0.000 description 2
- 230000015271 coagulation Effects 0.000 description 2
- 238000005345 coagulation Methods 0.000 description 2
- 230000000295 complement effect Effects 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 239000003246 corticosteroid Substances 0.000 description 2
- 231100000433 cytotoxic Toxicity 0.000 description 2
- 230000001472 cytotoxic effect Effects 0.000 description 2
- 230000007547 defect Effects 0.000 description 2
- 230000006735 deficit Effects 0.000 description 2
- 238000004925 denaturation Methods 0.000 description 2
- 230000036425 denaturation Effects 0.000 description 2
- 238000001514 detection method Methods 0.000 description 2
- 230000006866 deterioration Effects 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- 238000010494 dissociation reaction Methods 0.000 description 2
- 230000005593 dissociations Effects 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- 231100000673 dose–response relationship Toxicity 0.000 description 2
- 239000003937 drug carrier Substances 0.000 description 2
- 230000009977 dual effect Effects 0.000 description 2
- 239000012636 effector Substances 0.000 description 2
- 230000002357 endometrial effect Effects 0.000 description 2
- 229960004671 enzalutamide Drugs 0.000 description 2
- WXCXUHSOUPDCQV-UHFFFAOYSA-N enzalutamide Chemical compound C1=C(F)C(C(=O)NC)=CC=C1N1C(C)(C)C(=O)N(C=2C=C(C(C#N)=CC=2)C(F)(F)F)C1=S WXCXUHSOUPDCQV-UHFFFAOYSA-N 0.000 description 2
- 239000013604 expression vector Substances 0.000 description 2
- MKXKFYHWDHIYRV-UHFFFAOYSA-N flutamide Chemical compound CC(C)C(=O)NC1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 MKXKFYHWDHIYRV-UHFFFAOYSA-N 0.000 description 2
- 229960002074 flutamide Drugs 0.000 description 2
- PJZDLZXMGBOJRF-CXOZILEQSA-L folfirinox Chemical compound [Pt+4].[O-]C(=O)C([O-])=O.[NH-][C@H]1CCCC[C@@H]1[NH-].FC1=CNC(=O)NC1=O.C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1.C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 PJZDLZXMGBOJRF-CXOZILEQSA-L 0.000 description 2
- VVIAGPKUTFNRDU-ABLWVSNPSA-N folinic acid Chemical compound C1NC=2NC(N)=NC(=O)C=2N(C=O)C1CNC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 VVIAGPKUTFNRDU-ABLWVSNPSA-N 0.000 description 2
- 235000008191 folinic acid Nutrition 0.000 description 2
- 239000011672 folinic acid Substances 0.000 description 2
- 238000005194 fractionation Methods 0.000 description 2
- 229940050411 fumarate Drugs 0.000 description 2
- 229940050410 gluconate Drugs 0.000 description 2
- RWSXRVCMGQZWBV-WDSKDSINSA-N glutathione Chemical compound OC(=O)[C@@H](N)CCC(=O)N[C@@H](CS)C(=O)NCC(O)=O RWSXRVCMGQZWBV-WDSKDSINSA-N 0.000 description 2
- 235000011187 glycerol Nutrition 0.000 description 2
- 230000036541 health Effects 0.000 description 2
- 230000002962 histologic effect Effects 0.000 description 2
- 108700020746 histrelin Proteins 0.000 description 2
- HHXHVIJIIXKSOE-QILQGKCVSA-N histrelin Chemical compound CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)CC(N=C1)=CN1CC1=CC=CC=C1 HHXHVIJIIXKSOE-QILQGKCVSA-N 0.000 description 2
- 229960002193 histrelin Drugs 0.000 description 2
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 2
- 230000001900 immune effect Effects 0.000 description 2
- 210000000987 immune system Anatomy 0.000 description 2
- 229940072221 immunoglobulins Drugs 0.000 description 2
- 230000000977 initiatory effect Effects 0.000 description 2
- 230000010354 integration Effects 0.000 description 2
- 238000007913 intrathecal administration Methods 0.000 description 2
- KLEAIHJJLUAXIQ-JDRGBKBRSA-N irinotecan hydrochloride hydrate Chemical compound O.O.O.Cl.C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 KLEAIHJJLUAXIQ-JDRGBKBRSA-N 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 229960001691 leucovorin Drugs 0.000 description 2
- 239000003446 ligand Substances 0.000 description 2
- 239000008176 lyophilized powder Substances 0.000 description 2
- RLSSMJSEOOYNOY-UHFFFAOYSA-N m-cresol Chemical compound CC1=CC=CC(O)=C1 RLSSMJSEOOYNOY-UHFFFAOYSA-N 0.000 description 2
- 230000003211 malignant effect Effects 0.000 description 2
- 230000001404 mediated effect Effects 0.000 description 2
- 208000010658 metastatic prostate carcinoma Diseases 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 229960000513 necitumumab Drugs 0.000 description 2
- 208000004235 neutropenia Diseases 0.000 description 2
- XWXYUMMDTVBTOU-UHFFFAOYSA-N nilutamide Chemical compound O=C1C(C)(C)NC(=O)N1C1=CC=C([N+]([O-])=O)C(C(F)(F)F)=C1 XWXYUMMDTVBTOU-UHFFFAOYSA-N 0.000 description 2
- 229960002653 nilutamide Drugs 0.000 description 2
- 238000001543 one-way ANOVA Methods 0.000 description 2
- 229940048191 onivyde Drugs 0.000 description 2
- 230000002611 ovarian Effects 0.000 description 2
- 230000002018 overexpression Effects 0.000 description 2
- 210000003101 oviduct Anatomy 0.000 description 2
- 229960005489 paracetamol Drugs 0.000 description 2
- 238000007911 parenteral administration Methods 0.000 description 2
- 229940121655 pd-1 inhibitor Drugs 0.000 description 2
- QOFFJEBXNKRSPX-ZDUSSCGKSA-N pemetrexed Chemical compound C1=N[C]2NC(N)=NC(=O)C2=C1CCC1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 QOFFJEBXNKRSPX-ZDUSSCGKSA-N 0.000 description 2
- 229960005079 pemetrexed Drugs 0.000 description 2
- AQIXEPGDORPWBJ-UHFFFAOYSA-N pentan-3-ol Chemical compound CCC(O)CC AQIXEPGDORPWBJ-UHFFFAOYSA-N 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- 238000011518 platinum-based chemotherapy Methods 0.000 description 2
- 238000010837 poor prognosis Methods 0.000 description 2
- 238000002360 preparation method Methods 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- 230000002250 progressing effect Effects 0.000 description 2
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 2
- 235000019419 proteases Nutrition 0.000 description 2
- 230000005180 public health Effects 0.000 description 2
- 230000005855 radiation Effects 0.000 description 2
- 238000011127 radiochemotherapy Methods 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- GHMLBKRAJCXXBS-UHFFFAOYSA-N resorcinol Chemical compound OC1=CC=CC(O)=C1 GHMLBKRAJCXXBS-UHFFFAOYSA-N 0.000 description 2
- 210000003705 ribosome Anatomy 0.000 description 2
- 238000012216 screening Methods 0.000 description 2
- 238000011519 second-line treatment Methods 0.000 description 2
- 229940126586 small molecule drug Drugs 0.000 description 2
- 230000037439 somatic mutation Effects 0.000 description 2
- 238000010186 staining Methods 0.000 description 2
- 238000011272 standard treatment Methods 0.000 description 2
- 230000003319 supportive effect Effects 0.000 description 2
- 239000004094 surface-active agent Substances 0.000 description 2
- 229960001603 tamoxifen Drugs 0.000 description 2
- 229940095064 tartrate Drugs 0.000 description 2
- 229960004072 thrombin Drugs 0.000 description 2
- 229960004824 triptorelin Drugs 0.000 description 2
- VXKHXGOKWPXYNA-PGBVPBMZSA-N triptorelin Chemical compound C([C@@H](C(=O)N[C@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)NCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)C1=CC=C(O)C=C1 VXKHXGOKWPXYNA-PGBVPBMZSA-N 0.000 description 2
- 210000004881 tumor cell Anatomy 0.000 description 2
- 210000004291 uterus Anatomy 0.000 description 2
- 239000013598 vector Substances 0.000 description 2
- HDTRYLNUVZCQOY-UHFFFAOYSA-N α-D-glucopyranosyl-α-D-glucopyranoside Natural products OC1C(O)C(O)C(CO)OC1OC1C(O)C(O)C(O)C(CO)O1 HDTRYLNUVZCQOY-UHFFFAOYSA-N 0.000 description 1
- PGOHTUIFYSHAQG-LJSDBVFPSA-N (2S)-6-amino-2-[[(2S)-5-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-4-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-5-amino-2-[[(2S)-5-amino-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S,3R)-2-[[(2S)-5-amino-2-[[(2S)-2-[[(2S)-2-[[(2S,3R)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-5-amino-2-[[(2S)-1-[(2S,3R)-2-[[(2S)-2-[[(2S)-2-[[(2R)-2-[[(2S)-2-[[(2S)-2-[[2-[[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-1-[(2S)-2-[[(2S)-2-[[(2S)-2-[[(2S)-2-amino-4-methylsulfanylbutanoyl]amino]-3-(1H-indol-3-yl)propanoyl]amino]-5-carbamimidamidopentanoyl]amino]propanoyl]pyrrolidine-2-carbonyl]amino]-3-methylbutanoyl]amino]-4-methylpentanoyl]amino]-4-methylpentanoyl]amino]acetyl]amino]-3-hydroxypropanoyl]amino]-4-methylpentanoyl]amino]-3-sulfanylpropanoyl]amino]-4-methylsulfanylbutanoyl]amino]-5-carbamimidamidopentanoyl]amino]-3-hydroxybutanoyl]pyrrolidine-2-carbonyl]amino]-5-oxopentanoyl]amino]-3-hydroxypropanoyl]amino]-3-hydroxypropanoyl]amino]-3-(1H-imidazol-5-yl)propanoyl]amino]-4-methylpentanoyl]amino]-3-hydroxybutanoyl]amino]-3-(1H-indol-3-yl)propanoyl]amino]-5-carbamimidamidopentanoyl]amino]-5-oxopentanoyl]amino]-3-hydroxybutanoyl]amino]-3-hydroxypropanoyl]amino]-3-carboxypropanoyl]amino]-3-hydroxypropanoyl]amino]-5-oxopentanoyl]amino]-5-oxopentanoyl]amino]-3-phenylpropanoyl]amino]-5-carbamimidamidopentanoyl]amino]-3-methylbutanoyl]amino]-4-methylpentanoyl]amino]-4-oxobutanoyl]amino]-5-carbamimidamidopentanoyl]amino]-3-(1H-indol-3-yl)propanoyl]amino]-4-carboxybutanoyl]amino]-5-oxopentanoyl]amino]hexanoic acid Chemical compound CSCC[C@H](N)C(=O)N[C@@H](Cc1c[nH]c2ccccc12)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N1CCC[C@H]1C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1CCC[C@H]1C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@@H](Cc1cnc[nH]1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](Cc1c[nH]c2ccccc12)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](Cc1ccccc1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](Cc1c[nH]c2ccccc12)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCCN)C(O)=O PGOHTUIFYSHAQG-LJSDBVFPSA-N 0.000 description 1
- ORFNVPGICPYLJV-YTVPMEHESA-N (2s)-2-[[(2r,3r)-3-[(2s)-1-[(3r,4s,5s)-4-[[(2s)-2-[[(2s)-2-[6-(2,5-dioxopyrrol-1-yl)hexanoyl-methylamino]-3-methylbutanoyl]amino]-3-methylbutanoyl]-methylamino]-3-methoxy-5-methylheptanoyl]pyrrolidin-2-yl]-3-methoxy-2-methylpropanoyl]amino]-3-phenylpropan Chemical compound C([C@H](NC(=O)[C@H](C)[C@@H](OC)[C@@H]1CCCN1C(=O)C[C@H]([C@H]([C@@H](C)CC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)CCCCCN1C(C=CC1=O)=O)C(C)C)OC)C(O)=O)C1=CC=CC=C1 ORFNVPGICPYLJV-YTVPMEHESA-N 0.000 description 1
- WOWDZACBATWTAU-FEFUEGSOSA-N (2s)-2-[[(2s)-2-(dimethylamino)-3-methylbutanoyl]amino]-n-[(3r,4s,5s)-1-[(2s)-2-[(1r,2r)-3-[[(1s,2r)-1-hydroxy-1-phenylpropan-2-yl]amino]-1-methoxy-2-methyl-3-oxopropyl]pyrrolidin-1-yl]-3-methoxy-5-methyl-1-oxoheptan-4-yl]-n,3-dimethylbutanamide Chemical compound CC(C)[C@H](N(C)C)C(=O)N[C@@H](C(C)C)C(=O)N(C)[C@@H]([C@@H](C)CC)[C@H](OC)CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C)[C@@H](O)C1=CC=CC=C1 WOWDZACBATWTAU-FEFUEGSOSA-N 0.000 description 1
- AGGWFDNPHKLBBV-YUMQZZPRSA-N (2s)-2-[[(2s)-2-amino-3-methylbutanoyl]amino]-5-(carbamoylamino)pentanoic acid Chemical compound CC(C)[C@H](N)C(=O)N[C@H](C(O)=O)CCCNC(N)=O AGGWFDNPHKLBBV-YUMQZZPRSA-N 0.000 description 1
- GHOKWGTUZJEAQD-ZETCQYMHSA-N (D)-(+)-Pantothenic acid Chemical compound OCC(C)(C)[C@@H](O)C(=O)NCCC(O)=O GHOKWGTUZJEAQD-ZETCQYMHSA-N 0.000 description 1
- AGBQKNBQESQNJD-SSDOTTSWSA-N (R)-lipoic acid Chemical compound OC(=O)CCCC[C@@H]1CCSS1 AGBQKNBQESQNJD-SSDOTTSWSA-N 0.000 description 1
- OWEGMIWEEQEYGQ-UHFFFAOYSA-N 100676-05-9 Natural products OC1C(O)C(O)C(CO)OC1OCC1C(O)C(O)C(O)C(OC2C(OC(O)C(O)C2O)CO)O1 OWEGMIWEEQEYGQ-UHFFFAOYSA-N 0.000 description 1
- HTCSFFGLRQDZDE-UHFFFAOYSA-N 2-azaniumyl-2-phenylpropanoate Chemical compound OC(=O)C(N)(C)C1=CC=CC=C1 HTCSFFGLRQDZDE-UHFFFAOYSA-N 0.000 description 1
- PBSPQZHJEDTHTL-UHFFFAOYSA-N 2-benzoylpentanoic acid Chemical compound CCCC(C(O)=O)C(=O)C1=CC=CC=C1 PBSPQZHJEDTHTL-UHFFFAOYSA-N 0.000 description 1
- ALKYHXVLJMQRLQ-UHFFFAOYSA-N 3-Hydroxy-2-naphthoate Chemical class C1=CC=C2C=C(O)C(C(=O)O)=CC2=C1 ALKYHXVLJMQRLQ-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-M 3-carboxy-2,3-dihydroxypropanoate Chemical compound OC(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-M 0.000 description 1
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 1
- AILRADAXUVEEIR-UHFFFAOYSA-N 5-chloro-4-n-(2-dimethylphosphorylphenyl)-2-n-[2-methoxy-4-[4-(4-methylpiperazin-1-yl)piperidin-1-yl]phenyl]pyrimidine-2,4-diamine Chemical compound COC1=CC(N2CCC(CC2)N2CCN(C)CC2)=CC=C1NC(N=1)=NC=C(Cl)C=1NC1=CC=CC=C1P(C)(C)=O AILRADAXUVEEIR-UHFFFAOYSA-N 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- BFYIZQONLCFLEV-DAELLWKTSA-N Aromasine Chemical compound O=C1C=C[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CC(=C)C2=C1 BFYIZQONLCFLEV-DAELLWKTSA-N 0.000 description 1
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 102000015081 Blood Coagulation Factors Human genes 0.000 description 1
- 108010039209 Blood Coagulation Factors Proteins 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 241000700198 Cavia Species 0.000 description 1
- 241000700199 Cavia porcellus Species 0.000 description 1
- 102000000844 Cell Surface Receptors Human genes 0.000 description 1
- 108010001857 Cell Surface Receptors Proteins 0.000 description 1
- 208000032170 Congenital Abnormalities Diseases 0.000 description 1
- 206010010356 Congenital anomaly Diseases 0.000 description 1
- 241000557626 Corvus corax Species 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- HEBKCHPVOIAQTA-QWWZWVQMSA-N D-arabinitol Chemical compound OC[C@@H](O)C(O)[C@H](O)CO HEBKCHPVOIAQTA-QWWZWVQMSA-N 0.000 description 1
- DSLZVSRJTYRBFB-LLEIAEIESA-N D-glucaric acid Chemical compound OC(=O)[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O DSLZVSRJTYRBFB-LLEIAEIESA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- AEMOLEFTQBMNLQ-AQKNRBDQSA-N D-glucopyranuronic acid Chemical compound OC1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-AQKNRBDQSA-N 0.000 description 1
- HMFHBZSHGGEWLO-SOOFDHNKSA-N D-ribofuranose Chemical compound OC[C@H]1OC(O)[C@H](O)[C@@H]1O HMFHBZSHGGEWLO-SOOFDHNKSA-N 0.000 description 1
- 230000006820 DNA synthesis Effects 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- 239000004375 Dextrin Substances 0.000 description 1
- 229920001353 Dextrin Polymers 0.000 description 1
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- 241000283074 Equus asinus Species 0.000 description 1
- 239000004386 Erythritol Substances 0.000 description 1
- UNXHWFMMPAWVPI-UHFFFAOYSA-N Erythritol Natural products OCC(O)C(O)CO UNXHWFMMPAWVPI-UHFFFAOYSA-N 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- 229930091371 Fructose Natural products 0.000 description 1
- RFSUNEUAIZKAJO-ARQDHWQXSA-N Fructose Chemical compound OC[C@H]1O[C@](O)(CO)[C@@H](O)[C@@H]1O RFSUNEUAIZKAJO-ARQDHWQXSA-N 0.000 description 1
- 239000005715 Fructose Substances 0.000 description 1
- 241000287828 Gallus gallus Species 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 108010024636 Glutathione Proteins 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 102000003886 Glycoproteins Human genes 0.000 description 1
- 108090000288 Glycoproteins Proteins 0.000 description 1
- SQUHHTBVTRBESD-UHFFFAOYSA-N Hexa-Ac-myo-Inositol Natural products CC(=O)OC1C(OC(C)=O)C(OC(C)=O)C(OC(C)=O)C(OC(C)=O)C1OC(C)=O SQUHHTBVTRBESD-UHFFFAOYSA-N 0.000 description 1
- 101000690301 Homo sapiens Aldo-keto reductase family 1 member C4 Proteins 0.000 description 1
- 101000878605 Homo sapiens Low affinity immunoglobulin epsilon Fc receptor Proteins 0.000 description 1
- 101001116548 Homo sapiens Protein CBFA2T1 Proteins 0.000 description 1
- 108091006905 Human Serum Albumin Proteins 0.000 description 1
- 102000008100 Human Serum Albumin Human genes 0.000 description 1
- 108010054477 Immunoglobulin Fab Fragments Proteins 0.000 description 1
- 102000001706 Immunoglobulin Fab Fragments Human genes 0.000 description 1
- 108700005091 Immunoglobulin Genes Proteins 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- LKDRXBCSQODPBY-AMVSKUEXSA-N L-(-)-Sorbose Chemical compound OCC1(O)OC[C@H](O)[C@@H](O)[C@@H]1O LKDRXBCSQODPBY-AMVSKUEXSA-N 0.000 description 1
- AHLPHDHHMVZTML-BYPYZUCNSA-N L-Ornithine Chemical compound NCCC[C@H](N)C(O)=O AHLPHDHHMVZTML-BYPYZUCNSA-N 0.000 description 1
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 1
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 1
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 1
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 1
- 239000005411 L01XE02 - Gefitinib Substances 0.000 description 1
- 239000002136 L01XE07 - Lapatinib Substances 0.000 description 1
- 239000002138 L01XE21 - Regorafenib Substances 0.000 description 1
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 1
- 102100038007 Low affinity immunoglobulin epsilon Fc receptor Human genes 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- GUBGYTABKSRVRQ-PICCSMPSSA-N Maltose Natural products O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@@H](CO)OC(O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-PICCSMPSSA-N 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 206010063569 Metastatic squamous cell carcinoma Diseases 0.000 description 1
- 208000003445 Mouth Neoplasms Diseases 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- 206010061309 Neoplasm progression Diseases 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- AHLPHDHHMVZTML-UHFFFAOYSA-N Orn-delta-NH2 Natural products NCCCC(N)C(O)=O AHLPHDHHMVZTML-UHFFFAOYSA-N 0.000 description 1
- UTJLXEIPEHZYQJ-UHFFFAOYSA-N Ornithine Natural products OC(=O)C(C)CCCN UTJLXEIPEHZYQJ-UHFFFAOYSA-N 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 208000002193 Pain Diseases 0.000 description 1
- 108090000526 Papain Proteins 0.000 description 1
- 102000057297 Pepsin A Human genes 0.000 description 1
- 108090000284 Pepsin A Proteins 0.000 description 1
- 229920001363 Polidocanol Polymers 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 229920002701 Polyoxyl 40 Stearate Polymers 0.000 description 1
- 229920001214 Polysorbate 60 Polymers 0.000 description 1
- 208000006994 Precancerous Conditions Diseases 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- 108010029485 Protein Isoforms Proteins 0.000 description 1
- 102000001708 Protein Isoforms Human genes 0.000 description 1
- 108010094028 Prothrombin Proteins 0.000 description 1
- 108020004511 Recombinant DNA Proteins 0.000 description 1
- JVWLUVNSQYXYBE-UHFFFAOYSA-N Ribitol Natural products OCC(C)C(O)C(O)CO JVWLUVNSQYXYBE-UHFFFAOYSA-N 0.000 description 1
- PYMYPHUHKUWMLA-LMVFSUKVSA-N Ribose Natural products OC[C@@H](O)[C@@H](O)[C@@H](O)C=O PYMYPHUHKUWMLA-LMVFSUKVSA-N 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- UQZIYBXSHAGNOE-USOSMYMVSA-N Stachyose Natural products O(C[C@H]1[C@@H](O)[C@H](O)[C@H](O)[C@@H](O[C@@]2(CO)[C@H](O)[C@@H](O)[C@@H](CO)O2)O1)[C@@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@H](CO[C@@H]2[C@@H](O)[C@@H](O)[C@@H](O)[C@H](CO)O2)O1 UQZIYBXSHAGNOE-USOSMYMVSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- 108091008874 T cell receptors Proteins 0.000 description 1
- 102000016266 T-Cell Antigen Receptors Human genes 0.000 description 1
- 229920002253 Tannate Polymers 0.000 description 1
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 1
- 239000004473 Threonine Substances 0.000 description 1
- 208000007536 Thrombosis Diseases 0.000 description 1
- HDTRYLNUVZCQOY-WSWWMNSNSA-N Trehalose Natural products O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-WSWWMNSNSA-N 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- YJQCOFNZVFGCAF-UHFFFAOYSA-N Tunicamycin II Natural products O1C(CC(O)C2C(C(O)C(O2)N2C(NC(=O)C=C2)=O)O)C(O)C(O)C(NC(=O)C=CCCCCCCCCC(C)C)C1OC1OC(CO)C(O)C(O)C1NC(C)=O YJQCOFNZVFGCAF-UHFFFAOYSA-N 0.000 description 1
- 241000251539 Vertebrata <Metazoa> Species 0.000 description 1
- 229930003427 Vitamin E Natural products 0.000 description 1
- TVXBFESIOXBWNM-UHFFFAOYSA-N Xylitol Natural products OCCC(O)C(O)C(O)CCO TVXBFESIOXBWNM-UHFFFAOYSA-N 0.000 description 1
- NLMBVBUNULOTNS-HOKPPMCLSA-N [4-[[(2s)-5-(carbamoylamino)-2-[[(2s)-2-[6-(2,5-dioxopyrrol-1-yl)hexanoylamino]-3-methylbutanoyl]amino]pentanoyl]amino]phenyl]methyl n-[(2s)-1-[[(2s)-1-[[(3r,4s,5s)-1-[(2s)-2-[(1r,2r)-3-[[(1s,2r)-1-hydroxy-1-phenylpropan-2-yl]amino]-1-methoxy-2-methyl-3-o Chemical compound C1([C@H](O)[C@@H](C)NC(=O)[C@H](C)[C@@H](OC)[C@@H]2CCCN2C(=O)C[C@H]([C@H]([C@@H](C)CC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)OCC=2C=CC(NC(=O)[C@H](CCCNC(N)=O)NC(=O)[C@@H](NC(=O)CCCCCN3C(C=CC3=O)=O)C(C)C)=CC=2)C(C)C)OC)=CC=CC=C1 NLMBVBUNULOTNS-HOKPPMCLSA-N 0.000 description 1
- 229960000853 abiraterone Drugs 0.000 description 1
- GZOSMCIZMLWJML-VJLLXTKPSA-N abiraterone Chemical group C([C@H]1[C@H]2[C@@H]([C@]3(CC[C@H](O)CC3=CC2)C)CC[C@@]11C)C=C1C1=CC=CN=C1 GZOSMCIZMLWJML-VJLLXTKPSA-N 0.000 description 1
- 229940022663 acetate Drugs 0.000 description 1
- 108010052004 acetyl-2-naphthylalanyl-3-chlorophenylalanyl-1-oxohexadecyl-seryl-4-aminophenylalanyl(hydroorotyl)-4-aminophenylalanyl(carbamoyl)-leucyl-ILys-prolyl-alaninamide Proteins 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 229960001686 afatinib Drugs 0.000 description 1
- ULXXDDBFHOBEHA-CWDCEQMOSA-N afatinib Chemical compound N1=CN=C2C=C(O[C@@H]3COCC3)C(NC(=O)/C=C/CN(C)C)=CC2=C1NC1=CC=C(F)C(Cl)=C1 ULXXDDBFHOBEHA-CWDCEQMOSA-N 0.000 description 1
- 230000009824 affinity maturation Effects 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 238000013019 agitation Methods 0.000 description 1
- 235000004279 alanine Nutrition 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 125000000217 alkyl group Chemical group 0.000 description 1
- 230000002152 alkylating effect Effects 0.000 description 1
- HDTRYLNUVZCQOY-LIZSDCNHSA-N alpha,alpha-trehalose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-LIZSDCNHSA-N 0.000 description 1
- HMFHBZSHGGEWLO-UHFFFAOYSA-N alpha-D-Furanose-Ribose Natural products OCC1OC(O)C(O)C1O HMFHBZSHGGEWLO-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-PHYPRBDBSA-N alpha-D-galactose Chemical compound OC[C@H]1O[C@H](O)[C@H](O)[C@@H](O)[C@H]1O WQZGKKKJIJFFOK-PHYPRBDBSA-N 0.000 description 1
- AGBQKNBQESQNJD-UHFFFAOYSA-N alpha-Lipoic acid Natural products OC(=O)CCCCC1CCSS1 AGBQKNBQESQNJD-UHFFFAOYSA-N 0.000 description 1
- 230000009435 amidation Effects 0.000 description 1
- 238000007112 amidation reaction Methods 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 239000012491 analyte Substances 0.000 description 1
- 229960002932 anastrozole Drugs 0.000 description 1
- YBBLVLTVTVSKRW-UHFFFAOYSA-N anastrozole Chemical compound N#CC(C)(C)C1=CC(C(C)(C#N)C)=CC(CN2N=CN=C2)=C1 YBBLVLTVTVSKRW-UHFFFAOYSA-N 0.000 description 1
- 230000002491 angiogenic effect Effects 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 230000001093 anti-cancer Effects 0.000 description 1
- 230000000843 anti-fungal effect Effects 0.000 description 1
- 229940121363 anti-inflammatory agent Drugs 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 238000009175 antibody therapy Methods 0.000 description 1
- 230000000890 antigenic effect Effects 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 210000001106 artificial yeast chromosome Anatomy 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 235000009582 asparagine Nutrition 0.000 description 1
- 229960001230 asparagine Drugs 0.000 description 1
- 229960001716 benzalkonium Drugs 0.000 description 1
- 229960000686 benzalkonium chloride Drugs 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 235000019445 benzyl alcohol Nutrition 0.000 description 1
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QUYVBRFLSA-N beta-maltose Chemical compound OC[C@H]1O[C@H](O[C@H]2[C@H](O)[C@@H](O)[C@H](O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@@H]1O GUBGYTABKSRVRQ-QUYVBRFLSA-N 0.000 description 1
- 230000007698 birth defect Effects 0.000 description 1
- HUTDDBSSHVOYJR-UHFFFAOYSA-H bis[(2-oxo-1,3,2$l^{5},4$l^{2}-dioxaphosphaplumbetan-2-yl)oxy]lead Chemical compound [Pb+2].[Pb+2].[Pb+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O HUTDDBSSHVOYJR-UHFFFAOYSA-H 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 230000023555 blood coagulation Effects 0.000 description 1
- 239000003114 blood coagulation factor Substances 0.000 description 1
- 210000001772 blood platelet Anatomy 0.000 description 1
- 210000000988 bone and bone Anatomy 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- 229950004272 brigatinib Drugs 0.000 description 1
- 239000006172 buffering agent Substances 0.000 description 1
- 239000004067 bulking agent Substances 0.000 description 1
- LRHPLDYGYMQRHN-UHFFFAOYSA-N butyl alcohol Substances CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 210000004899 c-terminal region Anatomy 0.000 description 1
- 239000004202 carbamide Substances 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 239000005018 casein Substances 0.000 description 1
- BECPQYXYKAMYBN-UHFFFAOYSA-N casein, tech. Chemical compound NCCCCC(C(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(CC(C)C)N=C(O)C(CCC(O)=O)N=C(O)C(CC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(C(C)O)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=N)N=C(O)C(CCC(O)=O)N=C(O)C(CCC(O)=O)N=C(O)C(COP(O)(O)=O)N=C(O)C(CCC(O)=N)N=C(O)C(N)CC1=CC=CC=C1 BECPQYXYKAMYBN-UHFFFAOYSA-N 0.000 description 1
- 235000021240 caseins Nutrition 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 235000019438 castor oil Nutrition 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 125000002091 cationic group Chemical group 0.000 description 1
- 230000032823 cell division Effects 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 239000002738 chelating agent Substances 0.000 description 1
- 229910052729 chemical element Inorganic materials 0.000 description 1
- 238000007385 chemical modification Methods 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 229940044683 chemotherapy drug Drugs 0.000 description 1
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 1
- 229920003211 cis-1,4-polyisoprene Polymers 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 238000011284 combination treatment Methods 0.000 description 1
- 230000024203 complement activation Effects 0.000 description 1
- 230000004154 complement system Effects 0.000 description 1
- 238000011443 conventional therapy Methods 0.000 description 1
- 150000004696 coordination complex Chemical class 0.000 description 1
- 238000004132 cross linking Methods 0.000 description 1
- 230000009260 cross reactivity Effects 0.000 description 1
- 150000003999 cyclitols Chemical class 0.000 description 1
- HPXRVTGHNJAIIH-UHFFFAOYSA-N cyclohexanol Chemical compound OC1CCCCC1 HPXRVTGHNJAIIH-UHFFFAOYSA-N 0.000 description 1
- 231100000599 cytotoxic agent Toxicity 0.000 description 1
- 229960002272 degarelix Drugs 0.000 description 1
- MEUCPCLKGZSHTA-XYAYPHGZSA-N degarelix Chemical group C([C@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCNC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N[C@H](C)C(N)=O)NC(=O)[C@H](CC=1C=CC(NC(=O)[C@H]2NC(=O)NC(=O)C2)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@@H](CC=1C=NC=CC=1)NC(=O)[C@@H](CC=1C=CC(Cl)=CC=1)NC(=O)[C@@H](CC=1C=C2C=CC=CC2=CC=1)NC(C)=O)C1=CC=C(NC(N)=O)C=C1 MEUCPCLKGZSHTA-XYAYPHGZSA-N 0.000 description 1
- 238000001212 derivatisation Methods 0.000 description 1
- 235000019425 dextrin Nutrition 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 230000029087 digestion Effects 0.000 description 1
- 150000002016 disaccharides Chemical class 0.000 description 1
- 230000008034 disappearance Effects 0.000 description 1
- JMGZBMRVDHKMKB-UHFFFAOYSA-L disodium;2-sulfobutanedioate Chemical compound [Na+].[Na+].OS(=O)(=O)C(C([O-])=O)CC([O-])=O JMGZBMRVDHKMKB-UHFFFAOYSA-L 0.000 description 1
- 238000004520 electroporation Methods 0.000 description 1
- 229940088598 enzyme Drugs 0.000 description 1
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 description 1
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 description 1
- 230000007705 epithelial mesenchymal transition Effects 0.000 description 1
- UNXHWFMMPAWVPI-ZXZARUISSA-N erythritol Chemical compound OC[C@H](O)[C@H](O)CO UNXHWFMMPAWVPI-ZXZARUISSA-N 0.000 description 1
- 235000019414 erythritol Nutrition 0.000 description 1
- 229940009714 erythritol Drugs 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- SFNALCNOMXIBKG-UHFFFAOYSA-N ethylene glycol monododecyl ether Chemical compound CCCCCCCCCCCCOCCO SFNALCNOMXIBKG-UHFFFAOYSA-N 0.000 description 1
- 229960000255 exemestane Drugs 0.000 description 1
- 238000013265 extended release Methods 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 238000011354 first-line chemotherapy Methods 0.000 description 1
- 230000022244 formylation Effects 0.000 description 1
- 238000006170 formylation reaction Methods 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- FBPFZTCFMRRESA-GUCUJZIJSA-N galactitol Chemical compound OC[C@H](O)[C@@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-GUCUJZIJSA-N 0.000 description 1
- 229930182830 galactose Natural products 0.000 description 1
- WIGCFUFOHFEKBI-UHFFFAOYSA-N gamma-tocopherol Natural products CC(C)CCCC(C)CCCC(C)CCCC1CCC2C(C)C(O)C(C)C(C)C2O1 WIGCFUFOHFEKBI-UHFFFAOYSA-N 0.000 description 1
- 229960002584 gefitinib Drugs 0.000 description 1
- XGALLCVXEZPNRQ-UHFFFAOYSA-N gefitinib Chemical compound C=12C=C(OCCCN3CCOCC3)C(OC)=CC2=NC=NC=1NC1=CC=C(F)C(Cl)=C1 XGALLCVXEZPNRQ-UHFFFAOYSA-N 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 229940097042 glucuronate Drugs 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 1
- 229960003180 glutathione Drugs 0.000 description 1
- 150000004676 glycans Chemical class 0.000 description 1
- YQEMORVAKMFKLG-UHFFFAOYSA-N glycerine monostearate Natural products CCCCCCCCCCCCCCCCCC(=O)OC(CO)CO YQEMORVAKMFKLG-UHFFFAOYSA-N 0.000 description 1
- SVUQHVRAGMNPLW-UHFFFAOYSA-N glycerol monostearate Natural products CCCCCCCCCCCCCCCCC(=O)OCC(O)CO SVUQHVRAGMNPLW-UHFFFAOYSA-N 0.000 description 1
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 1
- 230000013595 glycosylation Effects 0.000 description 1
- 238000006206 glycosylation reaction Methods 0.000 description 1
- 102000054751 human RUNX1T1 Human genes 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 229920001477 hydrophilic polymer Polymers 0.000 description 1
- 230000003463 hyperproliferative effect Effects 0.000 description 1
- 229950007440 icotinib Drugs 0.000 description 1
- QQLKULDARVNMAL-UHFFFAOYSA-N icotinib Chemical compound C#CC1=CC=CC(NC=2C3=CC=4OCCOCCOCCOC=4C=C3N=CN=2)=C1 QQLKULDARVNMAL-UHFFFAOYSA-N 0.000 description 1
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 1
- 229960001101 ifosfamide Drugs 0.000 description 1
- 238000002649 immunization Methods 0.000 description 1
- 230000003053 immunization Effects 0.000 description 1
- 230000005847 immunogenicity Effects 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000000099 in vitro assay Methods 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 238000011221 initial treatment Methods 0.000 description 1
- 239000003999 initiator Substances 0.000 description 1
- 229960000367 inositol Drugs 0.000 description 1
- CDAISMWEOUEBRE-GPIVLXJGSA-N inositol Chemical compound O[C@H]1[C@H](O)[C@@H](O)[C@H](O)[C@H](O)[C@@H]1O CDAISMWEOUEBRE-GPIVLXJGSA-N 0.000 description 1
- 230000009878 intermolecular interaction Effects 0.000 description 1
- 238000007917 intracranial administration Methods 0.000 description 1
- 230000008863 intramolecular interaction Effects 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- TWBYWOBDOCUKOW-UHFFFAOYSA-M isonicotinate Chemical compound [O-]C(=O)C1=CC=NC=C1 TWBYWOBDOCUKOW-UHFFFAOYSA-M 0.000 description 1
- 229940001447 lactate Drugs 0.000 description 1
- 239000000832 lactitol Substances 0.000 description 1
- 235000010448 lactitol Nutrition 0.000 description 1
- VQHSOMBJVWLPSR-JVCRWLNRSA-N lactitol Chemical compound OC[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O VQHSOMBJVWLPSR-JVCRWLNRSA-N 0.000 description 1
- 229960003451 lactitol Drugs 0.000 description 1
- 229960004891 lapatinib Drugs 0.000 description 1
- BCFGMOOMADDAQU-UHFFFAOYSA-N lapatinib Chemical compound O1C(CNCCS(=O)(=O)C)=CC=C1C1=CC=C(N=CN=C2NC=3C=C(Cl)C(OCC=4C=C(F)C=CC=4)=CC=3)C2=C1 BCFGMOOMADDAQU-UHFFFAOYSA-N 0.000 description 1
- 229950006462 lauromacrogol 400 Drugs 0.000 description 1
- 229960003881 letrozole Drugs 0.000 description 1
- HPJKCIUCZWXJDR-UHFFFAOYSA-N letrozole Chemical group C1=CC(C#N)=CC=C1C(N1N=CN=C1)C1=CC=C(C#N)C=C1 HPJKCIUCZWXJDR-UHFFFAOYSA-N 0.000 description 1
- 210000000265 leukocyte Anatomy 0.000 description 1
- 235000019136 lipoic acid Nutrition 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 201000005243 lung squamous cell carcinoma Diseases 0.000 description 1
- 210000004324 lymphatic system Anatomy 0.000 description 1
- 150000002680 magnesium Chemical class 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- 206010025482 malaise Diseases 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 230000035800 maturation Effects 0.000 description 1
- 229950008001 matuzumab Drugs 0.000 description 1
- 230000010534 mechanism of action Effects 0.000 description 1
- 229960002985 medroxyprogesterone acetate Drugs 0.000 description 1
- PSGAAPLEWMOORI-PEINSRQWSA-N medroxyprogesterone acetate Chemical compound C([C@@]12C)CC(=O)C=C1[C@@H](C)C[C@@H]1[C@@H]2CC[C@]2(C)[C@@](OC(C)=O)(C(C)=O)CC[C@H]21 PSGAAPLEWMOORI-PEINSRQWSA-N 0.000 description 1
- 201000001441 melanoma Diseases 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 229930182817 methionine Natural products 0.000 description 1
- 229960004452 methionine Drugs 0.000 description 1
- 235000006109 methionine Nutrition 0.000 description 1
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 1
- 239000004292 methyl p-hydroxybenzoate Substances 0.000 description 1
- 229960002216 methylparaben Drugs 0.000 description 1
- 230000000813 microbial effect Effects 0.000 description 1
- 230000008880 microtubule cytoskeleton organization Effects 0.000 description 1
- 235000013336 milk Nutrition 0.000 description 1
- 239000008267 milk Substances 0.000 description 1
- 210000004080 milk Anatomy 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 238000010369 molecular cloning Methods 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- DASWEROEPLKSEI-UIJRFTGLSA-N monomethyl auristatin e Chemical compound CN[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N(C)[C@@H]([C@@H](C)CC)[C@H](OC)CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C)[C@@H](O)C1=CC=CC=C1 DASWEROEPLKSEI-UIJRFTGLSA-N 0.000 description 1
- MFRNYXJJRJQHNW-NARUGQRUSA-N monomethyl auristatin f Chemical compound CN[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N(C)C([C@@H](C)CC)[C@H](OC)CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 MFRNYXJJRJQHNW-NARUGQRUSA-N 0.000 description 1
- 150000002772 monosaccharides Chemical class 0.000 description 1
- PJUIMOJAAPLTRJ-UHFFFAOYSA-N monothioglycerol Chemical compound OCC(O)CS PJUIMOJAAPLTRJ-UHFFFAOYSA-N 0.000 description 1
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 description 1
- 230000017074 necrotic cell death Effects 0.000 description 1
- 229950010203 nimotuzumab Drugs 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 229960003104 ornithine Drugs 0.000 description 1
- 229960003278 osimertinib Drugs 0.000 description 1
- DUYJMQONPNNFPI-UHFFFAOYSA-N osimertinib Chemical compound COC1=CC(N(C)CCN(C)C)=C(NC(=O)C=C)C=C1NC1=NC=CC(C=2C3=CC=CC=C3N(C)C=2)=N1 DUYJMQONPNNFPI-UHFFFAOYSA-N 0.000 description 1
- 229940043515 other immunoglobulins in atc Drugs 0.000 description 1
- 229940039748 oxalate Drugs 0.000 description 1
- LXCFILQKKLGQFO-UHFFFAOYSA-N p-hydroxybenzoic acid methyl ester Natural products COC(=O)C1=CC=C(O)C=C1 LXCFILQKKLGQFO-UHFFFAOYSA-N 0.000 description 1
- 230000036407 pain Effects 0.000 description 1
- 229960001972 panitumumab Drugs 0.000 description 1
- 229940014662 pantothenate Drugs 0.000 description 1
- 235000019161 pantothenic acid Nutrition 0.000 description 1
- 239000011713 pantothenic acid Substances 0.000 description 1
- 229940055729 papain Drugs 0.000 description 1
- 235000019834 papain Nutrition 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 230000006320 pegylation Effects 0.000 description 1
- 229940111202 pepsin Drugs 0.000 description 1
- 230000002085 persistent effect Effects 0.000 description 1
- 239000005426 pharmaceutical component Substances 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 230000001766 physiological effect Effects 0.000 description 1
- 230000006461 physiological response Effects 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 150000003058 platinum compounds Chemical class 0.000 description 1
- 229920001983 poloxamer Polymers 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 229940099429 polyoxyl 40 stearate Drugs 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 229940068965 polysorbates Drugs 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 150000003109 potassium Chemical class 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 229940002612 prodrug Drugs 0.000 description 1
- 239000000651 prodrug Substances 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- 239000004405 propyl p-hydroxybenzoate Substances 0.000 description 1
- 229960003415 propylparaben Drugs 0.000 description 1
- 230000017854 proteolysis Effects 0.000 description 1
- 230000006337 proteolytic cleavage Effects 0.000 description 1
- 238000002708 random mutagenesis Methods 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 238000010188 recombinant method Methods 0.000 description 1
- 229960004836 regorafenib Drugs 0.000 description 1
- FNHKPVJBJVTLMP-UHFFFAOYSA-N regorafenib Chemical compound C1=NC(C(=O)NC)=CC(OC=2C=C(F)C(NC(=O)NC=3C=C(C(Cl)=CC=3)C(F)(F)F)=CC=2)=C1 FNHKPVJBJVTLMP-UHFFFAOYSA-N 0.000 description 1
- 230000001177 retroviral effect Effects 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- HEBKCHPVOIAQTA-ZXFHETKHSA-N ribitol Chemical compound OC[C@H](O)[C@H](O)[C@H](O)CO HEBKCHPVOIAQTA-ZXFHETKHSA-N 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- CDAISMWEOUEBRE-UHFFFAOYSA-N scyllo-inosotol Natural products OC1C(O)C(O)C(O)C(O)C1O CDAISMWEOUEBRE-UHFFFAOYSA-N 0.000 description 1
- 238000011333 second-line chemotherapy Methods 0.000 description 1
- 230000003248 secreting effect Effects 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 230000009131 signaling function Effects 0.000 description 1
- 238000002741 site-directed mutagenesis Methods 0.000 description 1
- 238000005549 size reduction Methods 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- HRZFUMHJMZEROT-UHFFFAOYSA-L sodium disulfite Chemical compound [Na+].[Na+].[O-]S(=O)S([O-])(=O)=O HRZFUMHJMZEROT-UHFFFAOYSA-L 0.000 description 1
- APSBXTVYXVQYAB-UHFFFAOYSA-M sodium docusate Chemical group [Na+].CCCCC(CC)COC(=O)CC(S([O-])(=O)=O)C(=O)OCC(CC)CCCC APSBXTVYXVQYAB-UHFFFAOYSA-M 0.000 description 1
- 229940001584 sodium metabisulfite Drugs 0.000 description 1
- 235000010262 sodium metabisulphite Nutrition 0.000 description 1
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 1
- 229940001474 sodium thiosulfate Drugs 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- 239000008137 solubility enhancer Substances 0.000 description 1
- 239000000243 solution Substances 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 235000010356 sorbitol Nutrition 0.000 description 1
- 125000006850 spacer group Chemical group 0.000 description 1
- 230000003393 splenic effect Effects 0.000 description 1
- 210000004988 splenocyte Anatomy 0.000 description 1
- UQZIYBXSHAGNOE-XNSRJBNMSA-N stachyose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO[C@@H]2[C@@H]([C@@H](O)[C@@H](O)[C@@H](CO[C@@H]3[C@@H]([C@@H](O)[C@@H](O)[C@@H](CO)O3)O)O2)O)O1 UQZIYBXSHAGNOE-XNSRJBNMSA-N 0.000 description 1
- SFVFIFLLYFPGHH-UHFFFAOYSA-M stearalkonium chloride Chemical compound [Cl-].CCCCCCCCCCCCCCCCCC[N+](C)(C)CC1=CC=CC=C1 SFVFIFLLYFPGHH-UHFFFAOYSA-M 0.000 description 1
- 238000011146 sterile filtration Methods 0.000 description 1
- 239000008227 sterile water for injection Substances 0.000 description 1
- 230000004936 stimulating effect Effects 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 229940086735 succinate Drugs 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 238000011477 surgical intervention Methods 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- RTKIYNMVFMVABJ-UHFFFAOYSA-L thimerosal Chemical compound [Na+].CC[Hg]SC1=CC=CC=C1C([O-])=O RTKIYNMVFMVABJ-UHFFFAOYSA-L 0.000 description 1
- 229940033663 thimerosal Drugs 0.000 description 1
- 229960002663 thioctic acid Drugs 0.000 description 1
- 229940035024 thioglycerol Drugs 0.000 description 1
- 229940071127 thioglycolate Drugs 0.000 description 1
- CWERGRDVMFNCDR-UHFFFAOYSA-M thioglycolate(1-) Chemical compound [O-]C(=O)CS CWERGRDVMFNCDR-UHFFFAOYSA-M 0.000 description 1
- 229960002952 tipiracil Drugs 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 238000012876 topography Methods 0.000 description 1
- 238000012448 transchromosomic mouse model Methods 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
- 238000011269 treatment regimen Methods 0.000 description 1
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical class CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 150000004043 trisaccharides Chemical class 0.000 description 1
- GPRLSGONYQIRFK-MNYXATJNSA-N triton Chemical compound [3H+] GPRLSGONYQIRFK-MNYXATJNSA-N 0.000 description 1
- 230000005751 tumor progression Effects 0.000 description 1
- ZHSGGJXRNHWHRS-VIDYELAYSA-N tunicamycin Chemical compound O([C@H]1[C@@H]([C@H]([C@@H](O)[C@@H](CC(O)[C@@H]2[C@H]([C@@H](O)[C@@H](O2)N2C(NC(=O)C=C2)=O)O)O1)O)NC(=O)/C=C/CC(C)C)[C@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1NC(C)=O ZHSGGJXRNHWHRS-VIDYELAYSA-N 0.000 description 1
- MEYZYGMYMLNUHJ-UHFFFAOYSA-N tunicamycin Natural products CC(C)CCCCCCCCCC=CC(=O)NC1C(O)C(O)C(CC(O)C2OC(C(O)C2O)N3C=CC(=O)NC3=O)OC1OC4OC(CO)C(O)C(O)C4NC(=O)C MEYZYGMYMLNUHJ-UHFFFAOYSA-N 0.000 description 1
- 230000004222 uncontrolled growth Effects 0.000 description 1
- 229940045136 urea Drugs 0.000 description 1
- 229960002066 vinorelbine Drugs 0.000 description 1
- GBABOYUKABKIAF-GHYRFKGUSA-N vinorelbine Chemical compound C1N(CC=2C3=CC=CC=C3NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC GBABOYUKABKIAF-GHYRFKGUSA-N 0.000 description 1
- 235000019165 vitamin E Nutrition 0.000 description 1
- 229940046009 vitamin E Drugs 0.000 description 1
- 239000011709 vitamin E Substances 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 1
- 230000004580 weight loss Effects 0.000 description 1
- 208000016261 weight loss Diseases 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 230000029663 wound healing Effects 0.000 description 1
- 238000012447 xenograft mouse model Methods 0.000 description 1
- 239000000811 xylitol Substances 0.000 description 1
- 235000010447 xylitol Nutrition 0.000 description 1
- HEBKCHPVOIAQTA-SCDXWVJYSA-N xylitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)CO HEBKCHPVOIAQTA-SCDXWVJYSA-N 0.000 description 1
- 229960002675 xylitol Drugs 0.000 description 1
- 229950008250 zalutumumab Drugs 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6851—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a determinant of a tumour cell
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/05—Dipeptides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/62—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid
- A61K47/65—Peptidic linkers, binders or spacers, e.g. peptidic enzyme-labile linkers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/68031—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug being an auristatin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6843—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a material from animals or humans
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6849—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a receptor, a cell surface antigen or a cell surface determinant
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6889—Conjugates wherein the antibody being the modifying agent and wherein the linker, binder or spacer confers particular properties to the conjugates, e.g. peptidic enzyme-labile linkers or acid-labile linkers, providing for an acid-labile immuno conjugate wherein the drug may be released from its antibody conjugated part in an acidic, e.g. tumoural or environment
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/36—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against blood coagulation factors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/545—Medicinal preparations containing antigens or antibodies characterised by the dose, timing or administration schedule
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/56—Immunoglobulins specific features characterized by immunoglobulin fragments variable (Fv) region, i.e. VH and/or VL
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/56—Immunoglobulins specific features characterized by immunoglobulin fragments variable (Fv) region, i.e. VH and/or VL
- C07K2317/565—Complementarity determining region [CDR]
Definitions
- the present invention relates to anti-tissue factor (TF) antibody-drug conjugates and methods of using the same to treat cancer, such as colorectal cancer, non-small cell lung cancer, pancreatic cancer, head and neck cancer, bladder cancer, endometrial cancer, esophageal cancer and prostate cancer.
- cancer such as colorectal cancer, non-small cell lung cancer, pancreatic cancer, head and neck cancer, bladder cancer, endometrial cancer, esophageal cancer and prostate cancer.
- Tissue factor also called thromboplastin, factor III or CD142 is a protein present in subendothelial tissue, platelets, and leukocytes necessary for the initiation of thrombin formation from the zymogen prothrombin. Thrombin formation ultimately leads to the coagulation of blood.
- TF enables cells to initiate the blood coagulation cascade, and it functions as the high-affinity receptor for the coagulation factor Vila (FVIIa), a serine protease.
- FVIIa coagulation factor Vila
- the resulting complex provides a catalytic event that is responsible for initiation of the coagulation protease cascades by specific limited proteolysis. Unlike the other cofactors of these protease cascades, which circulate as nonfunctional precursors, TF is a potent initiator that is fully functional when expressed on cell surfaces.
- TF is the cell surface receptor for the serine protease factor Vila (FVIIa). Binding of FVIIa to TF starts signaling processes inside the cell, said signaling function playing a role in angiogenesis.
- angiogenesis is a normal process in growth and development, as well as in wound healing, it is also a fundamental step in the transition of tumors from a dormant state to a malignant state.
- cancer cells gain the ability to produce proteins that participate in angiogenesis (i.e., angiogenic growth factors), these proteins are released by the tumor into nearby tissues, thereby stimulating new blood vessels to sprout from existing healthy blood vessels toward and into the tumor. Once new blood vessels enter the tumor, the tumor can rapidly expand its size and invade local tissue and organs. Through the new blood vessels, cancer cells may further escape into the circulation and lodge in other organs to form new tumors, also known as metastasis.
- TF expression is observed in many types of cancer, and is associated with more aggressive disease. Furthermore, human TF also exists in a soluble alternatively-spliced form, asHTF. It has been found that asHTF promotes tumor growth (Hobbs et al ., 2007, Thrombosis Res. 120(2): S13-S21).
- Systemic therapies for non-operable colorectal cancer include fluorouracil (5-FU), immunotherapy such as pembrolizumab and nivolumab, regorafenib, trifluridine-tipiracil doublet (TAS-102), and irinotecan or oxaliplatin in combination with 5-FU. More effective treatments for these later stage patients are urgently needed.
- NSCLC Non-small cell lung cancer
- SCC/NSCLC squamous cell carcinoma
- SCC/NSCLC have shown limited benefit and are primarily aimed at prolonging survival and maintaining the quality of life for as long as possible, while minimizing side effects due to treatment.
- First line treatment for patients with SCC/NSCLC whose tumors do not express high levels of PD-L1 include a platinum -based chemotherapy doublet that does not contain pemetrexed, anti-VEGF antibody, or an anti-EGFR antibody necitumumab in combination with gemcitabine and cisplatin.
- Patients with at least 50% tumor cell staining for PD-L1 are offered first-line treatment with the anti -PD- 1 inhibitor pembrolizumab.
- Patients who progress on an initial combination chemotherapy regimen may receive an anti -PD- 1 or PD- Ll antibody, and combination chemotherapy is considered for patients whose disease has progressed after receiving PD-1/L1 inhibitors.
- New classes of therapy are urgently needed that can provide meaningful benefit to SCC/NSCLC patients.
- Pancreatic cancer is considered a“silent killer” because patients often do not feel symptoms until their disease has advanced and spread— in the US, 52% of patients had metastatic disease at diagnosis in 2017. More than 53,000 cases are estimated to have been diagnosed in the US in 2017, with over 43,000 deaths. Five year survival for people with metastatic pancreatic cancer remains a dismal 8% in the US and may be as low as 4% worldwide. Most patients diagnosed with pancreatic cancer succumb to the disease within the first year. Surgical resection offers the only chance of cure. However, only 15% to 20% of patients have resectable disease at initial diagnosis; the majority have either locally advanced or metastatic cancer. Metastatic pancreatic cancer patients have very few effective treatment options and are often treated only with palliative care.
- First line combination treatments include FOLFIRINOX or nab-paclitaxel plus gemcitabine.
- Second line and later treatments offer limited efficacy with significant treatment-related toxicity.
- Preferred regimens in this group include liposomal irinotecan (Onivyde) with 5-FU/leucovorin, FOLFOX, and gemcitabine in combination with nab-paclitaxel, erlotinib, or bevacizumab. Enrollment in available clinical trials is a preferred option for patients with advanced exocrine pancreatic adenocarcinoma, if available, due to the significant unmet medical need in this disease.
- Head and neck cancers make up approximately 3% of cancers in the United States. Over 63,000 cases are estimated to have been diagnosed in 2017 and more than 13,000 patients died from this disease. Though human papilloma virus (HPV) infection also appears to contribute to head and neck cancers. More than 90-95% of oral and
- nasopharyngeal cancers are of squamous histology. Surgical resection, radiotherapy, and/or chemoradiation are frequently recommended for patients with early-stage or localized disease. Palliative chemotherapy, immunotherapy and/or supportive care are the most appropriate options for patients with locally recurrent or metastatic disease that are not amenable to definitive therapy. Platinum-based regimens are the preferred standard of care treatment for patients with recurrent or de novo metastatic squamous cell carcinoma of the head and neck (SCCHN). Cetuximab in combination with a platinum/5-FU regimen has demonstrated clinically meaningful benefits compared to platinum/5-FU alone.
- second line treatment is with single agent chemotherapy, targeted therapy, or a checkpoint inhibitor such as nivolumab or pembrolizumab.
- a checkpoint inhibitor such as nivolumab or pembrolizumab.
- Bladder cancer is the sixth most common cancer in the United States, with an estimated 76,960 new cases diagnosed in 2016. Of these patients, 16,390 deaths were estimated to have occurred, with men being more likely to be affected than women.
- the 5- year relative survival rate for all stages combined is 77%. However, survival rates depend on many factors, including the histology and stage of bladder cancer diagnosed. For patients with bladder cancer that is invasive but not yet spread outside the bladder, the 5-year survival rate is 70%. For patients with bladder cancer that extends through the bladder to the surrounding tissue and/or organs, the 5-year survival rate is 34%.
- a cisplatin-based chemotherapy regimen followed by surgical removal of the bladder or radiation therapy and concomitant chemotherapy is currently the standard treatment for patients with invasive bladder cancer. More effective treatments for bladder cancer, particularly for patients with advanced or metastatic bladder cancer, are urgently needed.
- Endometrial cancer is the most common gynecologic malignancy in the United States, accounting for 6% of cancers in women. In 2017, an estimated 61,380 women were diagnosed with endometrial cancer, and approximately 11,000 died from this disease. From 1987 to 2008, there was a 50% increase in the incidence of endometrial cancer, with an approximate 300% increase in the number of associated deaths. Endometrial
- adenocarcinomas can be classified into two histologic categories— type 1 or type 2.
- Type 1 endometrial carcinomas are of endometrioid histology, lower grade, and often confined to the uterus at diagnosis. These tumors are estrogen-mediated, and often, women diagnosed with type 1 endometrial carcinomas are obese, with excess endogenous estrogen production.
- Type 1 carcinomas (estrogen dependent) have high rates of K-ras and PTEN loss or mutation, as well as defects in mismatch repair genes, which lead to microsatellite instability (MSI).
- MSI microsatellite instability
- Type 2 (non estrogen dependent) carcinomas are higher-grade adenocarcinomas and are of non- endometrioid histology, occurring in older, leaner women, although an association with increasing body mass index (BMI) has been observed.
- BMI body mass index
- Type 2 cancers have p53 mutations, may have overexpression of human epidermal growth factor receptor 2 (HER-2/neu), and show aneuploidy.
- HER-2/neu human epidermal growth factor receptor 2
- chemotherapeutic and targeted therapy agents approved for ovarian, fallopian tube and primary peritoneal cancers since the 1971 approval of megestrol acetate for the palliative treatment of advanced endometrial cancer, only pembrolizumab has been Food and Drug Administration (FDA)-approved for high microsatellite instability (MSI-H) or mismatch repair deficient (dMMR) endometrial cancer; this highlights the need for new therapies to treat advanced, recurrent, metastatic endometrial cancer.
- FDA Food and Drug Administration
- MSI-H microsatellite instability
- dMMR mismatch repair deficient
- Esophageal cancer is the sixth leading cause of cancer-related mortality worldwide due to its overall poor prognosis.
- the global age-standardized incidence rate of esophageal squamous cell carcinoma (ESCC) is 1.4-13.6 per 100,000 people.
- Esophageal cancer is estimated to be responsible for 15,690 deaths and 16,940 new cases in the ETnited States in 2016. The majority of patients present with locally advanced or systemic disease and outcomes remain poor despite advances in treatment. More effective treatments for these patients with locally advanced or systemic disease are urgently needed.
- Prostate cancer is the most common non-cutaneous malignancy in males, with a projected 161,360 incident cases and 26,730 deaths estimated in the United States in 2017 alone.
- Curative modalities for localized prostate cancer include surgery and/or radiation therapy, with or without androgen deprivation therapy. While contemporary treatment methods, such as intensity-modulated radiotherapy, are used to deliver radiation with high accuracy, defining the position and the extent of the tumor is still quite challenging. Other issues in the treatment of the radiotherapy patient include the choice of the radiotherapy technique (hypo- or standard fractionation) and the use and length of androgen deprivation therapy. More effective treatments are needed, especially for patients with advanced and metastatic prostate cancer.
- the present invention meets the need for improved treatment of colorectal cancer, non-small cell lung cancer, pancreatic cancer, head and neck cancer, bladder cancer, endometrial cancer, esophageal cancer and prostate cancer by providing highly specific and effective anti-TF antibody-drug conjugates.
- TF tissue factor
- the antibody-drug conjugate comprises an anti-TF antibody or an antigen binding fragment thereof conjugated to a monomethyl auristatin or a functional analog thereof or a functional derivative thereof, wherein the antibody-drug conjugate is
- the antibody-drug conjugate is administered at a dose ranging from about 1.5 mg/kg to about 2.1 mg/kg, and wherein the cancer is selected from the group consisting of colorectal cancer, non-small cell lung cancer, pancreatic cancer, head and neck cancer, bladder cancer, endometrial cancer, esophageal cancer and prostate cancer.
- the antibody-drug conjugate is administered at a dose of about 2.0 mg/kg. In some embodiments, the antibody-drug conjugate is administered at a dose of 2.0 mg/kg. In some of any of the embodiments herein, the antibody-drug conjugate is administered once about every 1 week, 2 weeks, 3 weeks or 4 weeks. In some of any of the embodiments herein, the antibody-drug conjugate is administered once about every 3 weeks.
- the subject has been previously treated with one or more therapeutic agents and did not respond to the treatment, wherein the one or more therapeutic agents is not the antibody-drug conjugate. In some of any of the embodiments herein, the subject has been previously treated with one or more therapeutic agents and relapsed after the treatment , wherein the one or more therapeutic agents is not the antibody-drug conjugate. In some of any of the
- the subject has been previously treated with one or more therapeutic agents and has experienced disease progression during treatment, wherein the one or more therapeutic agents is not the antibody-drug conjugate.
- the cancer is colorectal cancer.
- the subject received prior systemic therapy and experienced disease progression on or after the systemic therapy.
- the subject received 1, 2 or 3 rounds of prior systemic therapy.
- the colorectal cancer is non-operable.
- the subject has been previously treated with one or more agents selected from the group consisting of fluoropyrimidine, oxaliplatin, irinotecan and bevacizumab. In some of any of the embodiments herein, the subject has been previously treated with one or more agents selected from the group consisting of cetuximab, panitumab and a checkpoint inhibitor.
- the cancer is non-small cell lung cancer. In some of any of the embodiments herein, the non-small cell lung cancer is squamous cell carcinoma. In some of any of the embodiments herein, the non-small cell lung cancer has predominant squamous histology.
- the non-small cell lung cancer is adenocarcinoma.
- the subject received prior systemic therapy and experienced disease progression on or after the systemic therapy.
- the subject received 1 or 2 rounds of prior systemic therapy.
- the subject has been previously treated with one or more agents selected from the group consisting of a platinum-based therapy and a checkpoint inhibitor.
- the cancer is pancreatic cancer.
- the pancreatic cancer is exocrine pancreatic adenocarcinoma. In some of any of the
- the pancreatic cancer has predominant adenocarcinoma histology. In some of any of the embodiments herein, greater than 85% of the pancreatic cancer cells have adenocarcinoma histology. In some of any of the embodiments herein, the subject received prior systemic therapy and experienced disease progression on or after the systemic therapy. In some of any of the embodiments herein, the subject received 1 round of prior systemic therapy. In some of any of the embodiments herein, the subject has been previously treated with one or more agents selected from the group consisting of gemcitabine and 5- fluorouracil. In some of any of the embodiments herein, the pancreatic cancer is not resectable.
- the cancer is head and neck cancer. In some of any of the embodiments herein, the head and neck cancer is squamous cell carcinoma. In some of any of the embodiments herein, the subject received prior systemic therapy and experienced disease progression on or after the systemic therapy. In some of any of the embodiments herein, the subject received 1 or 2 rounds of prior systemic therapy. In some of any of the embodiments herein, the subject has been previously treated with one or more agents selected from the group consisting of a platinum-based therapy and a checkpoint inhibitor. In some of any of the embodiments herein, the subject has been previously treated with an anti-epithelial growth factor receptor therapy. In some of any of the embodiments herein, the cancer is bladder cancer. In some of any of the embodiments herein, the subject received prior systemic therapy and experienced disease progression on or after the systemic therapy. In some of any of the embodiments herein, the subject received 1,
- the subject has been previously treated with a platinum -based therapy. In some of any of the embodiments herein, the subject has previously undergone surgery or radiation therapy for the bladder cancer. In some of any of the embodiments herein, the cancer is endometrial cancer. In some of any of the embodiments herein, the subject received prior systemic therapy and experienced disease progression on or after the systemic therapy. In some of any of the embodiments herein, the subject received 1, 2 or 3 rounds of prior systemic therapy. In some of any of the embodiments herein, the subject has been previously treated with one or more agents selected from the group consisting of a platinum-based therapy, hormone therapy, and a checkpoint inhibitor.
- the subject has previously been treated with doxorubicin. In some of any of the embodiments herein, the subject has previously been treated with paclitaxel. In some of any of the embodiments herein, the subject has previously undergone surgery or radiation therapy for the endometrial cancer. In some of any of the embodiments herein, the cancer is esophageal cancer. In some of any of the embodiments herein, the subject received prior systemic therapy and
- the subject received 1, 2 or 3 rounds of prior systemic therapy.
- the subject has been previously treated with one or more agents selected from the group consisting of a platinum-based therapy and a checkpoint inhibitor.
- the subject has been previously treated with one or more agents selected from the group consisting of ramucirumab, paclitaxel, 5- fluorouracil, docetaxel, irinotecan, capecitabine and trastuzumab.
- the subject has previously undergone surgery, radiation therapy or endoscopic mucosal resection for the esophageal cancer.
- the cancer is prostate cancer.
- the subject received prior systemic therapy and experienced disease progression on or after the systemic therapy.
- the subject received 1, 2 or 3 rounds of prior systemic therapy.
- the prostate cancer is castration-resistant prostate cancer.
- the subject experienced bone metastases.
- the subject has been previously treated with one or more agents selected from the group consisting of androgen deprivation therapy, a luteinizing hormone-releasing hormone agonist, a luteinizing hormone-releasing hormone antagonist, a CYP17 inhibitor, and an anti-androgen.
- the subject has been previously treated with one or more agents selected from the group consisting of docetaxel, prednisone and cabazitaxel. In some of any of the embodiments herein, the subject has previously undergone surgery or radiation therapy for the prostate cancer.
- the cancer is an advanced stage cancer. In some of any of the embodiments herein, the advanced stage cancer is a stage 3 or stage 4 cancer. In some of any of the embodiments herein, the advanced stage cancer is metastatic cancer. In some of any of the embodiments herein, the cancer is recurrent cancer. In some of any of the embodiments herein, the subject received prior treatment with standard of care therapy for the cancer and failed the prior treatment.
- the monomethyl auristatin is monomethyl auristatin E (MMAE).
- the anti-TF antibody or antigen binding fragment thereof of the antibody-drug conjugate is a monoclonal antibody or a monoclonal antigen-binding fragment thereof.
- the anti-TF antibody or antigen-binding fragment thereof of the antibody-drug conjugate comprises a heavy chain variable region and a light chain variable region, wherein the heavy chain variable region comprises:
- the anti-TF antibody or antigen-binding fragment thereof of the antibody-drug conjugate comprises a heavy chain variable region comprising an amino acid sequence at least 85% identical to the amino acid sequence of SEQ ID NO:7 and a light chain variable region comprising an amino acid sequence at least 85% identical to the amino acid sequence of SEQ ID NO: 8.
- the anti-TF antibody or antigen-binding fragment thereof of the antibody-drug conjugate comprises a heavy chain variable region comprising the amino acid sequence of SEQ ID NO:7 and a light chain variable region comprising the amino acid sequence of SEQ ID NO:8.
- the anti-TF antibody of the antibody-drug conjugate is tisotumab.
- the antibody-drug conjugate further comprises a linker between the anti-TF antibody or antigen-binding fragment thereof and the monomethyl auristatin.
- the linker is a cleavable peptide linker.
- the cleavable peptide linker has a formula: -MC-vc-RAB-, wherein:
- vc is the dipeptide valine-citrulline
- PAB is:
- the linker is attached to sulphydryl residues of the anti-TF antibody obtained by partial reduction or full reduction of the anti-TF antibody or antigen-binding fragment thereof.
- the linker is attached to monomethyl auristatin E (MMAE), wherein the antibody-drug conjugate has the following structure:
- Afc- MC-v3 ⁇ 4--?AU-MMAI wherein p denotes a number from 1 to 8, S represents a sulphydryl residue of the anti-TF antibody, and Ab designates the anti-TF antibody or antigen-binding fragment thereof.
- the average value of p in a population of the antibody-drug conjugates is about 4.
- the antibody- drug conjugate is tisotumab vedotin.
- the route of administration for the antibody-drug conjugate is intravenous.
- the one or more therapeutic effects in the subject is improved after administration of the antibody-drug conjugate relative to a baseline.
- the one or more therapeutic effects is selected from the group consisting of: size of a tumor derived from the cancer, objective response rate, duration of response, time to response, progression free survival, overall survival and prostate-specific antigen (PSA) level.
- PSA prostate-specific antigen
- the subject exhibits a reduction in PSA level in a blood sample from the subject by at least about 5%, at least about 10%, at least about 15%, at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least about 45%, at least about 50%, at least about 60%, at least about 70%, or at least about 80% relative to the PSA level in a blood sample obtained from the subject before administration of the antibody-drug conjugate.
- the size of a tumor derived from the cancer is reduced by at least about 10%, at least about 15%, at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least about 45%, at least about 50%, at least about 60%, at least about 70%, or at least about 80% relative to the size of the tumor derived from the cancer before administration of the antibody-drug conjugate.
- the objective response rate is at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least about 45%, at least about 50%, at least about 60%, at least about 70%, or at least about 80%.
- the subject exhibits progression-free survival of at least about 1 month, at least about 2 months, at least about 3 months, at least about 4 months, at least about 5 months, at least about 6 months, at least about 7 months, at least about 8 months, at least about 9 months, at least about 10 months, at least about 11 months, at least about 12 months, at least about eighteen months, at least about two years, at least about three years, at least about four years, or at least about five years after administration of the antibody-drug conjugate.
- the subject exhibits overall survival of at least about 1 month, at least about 2 months, at least about 3 months, at least about 4 months, at least about 5 months, at least about 6 months, at least about 7 months, at least about 8 months, at least about 9 months, at least about 10 months, at least about 11 months, at least about 12 months, at least about eighteen months, at least about two years, at least about three years, at least about four years, or at least about five years after administration of the antibody-drug conjugate.
- the duration of response to the antibody-drug conjugate is at least about 1 month, at least about 2 months, at least about 3 months, at least about 4 months, at least about 5 months, at least about 6 months, at least about 7 months, at least about 8 months, at least about 9 months, at least about 10 months, at least about 11 months, at least about 12 months, at least about eighteen months, at least about two years, at least about three years, at least about four years, or at least about five years after administration of the antibody-drug conjugate.
- the subject has one or more adverse events and is further administered an additional therapeutic agent to eliminate or reduce the severity of the one or more adverse events.
- the subject is at risk of developing one or more adverse events and is further administered an additional therapeutic agent to prevent or reduce the severity of the one or more adverse events.
- the one or more adverse events is anemia, abdominal pain, hypokalemia, hyponatremia, epistaxis, fatigue, nausea, alopecia, conjunctivitis, constipation, decreased appetite, diarrhea, vomiting, peripheral neuropathy, or general physical health deterioration.
- the one or more adverse events is a grade 3 or greater adverse event.
- the one or more adverse events is a serious adverse event.
- the one or more adverse events is conjunctivitis and/or keratitis and the additional agent is a preservative-free lubricating eye drop, an ocular vasoconstrictor and/or a steroid eye drop.
- the antibody-drug conjugate is administered as a monotherapy.
- the subject is a human.
- the antibody-drug conjugate is in a pharmaceutical composition comprising the antibody-drug conjugate and a pharmaceutical acceptable carrier.
- kits comprising:
- FIG. l is a diagram showing the mechanism of action (MO A) of the antibody- drug conjugate tisotumab vedotin.
- FIG. 2A-2B show dose-dependent anti-tumor effects of single-dose tisotumab vedotin treatment in a NCI-H441 cell line-derived (CDX) mouse xenograft model.
- FIG. 2 A shows tumor growth of the NCI-H441 xenografts after treatment with different doses of tisotumab vedotin, isotype control antibody (IgGl-bl2), or isotype control ADC (IgGl-bl2- vcMMAE).
- Mean and standard of error of the mean (SEM) of each group is shown at each time point.
- FIG. 2B shows mean tumor size in each mouse on day 47. Mean and SEM of each group are indicated. Differences among the groups were analyzed by one-way ANOVA. Statistically significant differences are indicated as follows: *: p ⁇ 0.05; **: p ⁇ 0.0l; ***: pO.OOl.
- FIG. 3 shows anti -tumor effects of tisotumab vedotin treatment in a squamous cell lung carcinoma patient-derived xenograft (PDX) mouse model LXFE 690.
- Mean and SEM of tumor size of the LXFE 690 xenografts at each time point in groups treated with two doses of tisotumab vedotin at 4 mg/kg, IgGl-bl2 or IgGlbl2-vcMMAE are shown.
- FIG. 4A-4B show dose-dependent anti-tumor effects of tisotumab vedotin treatment in a HPAF II CDX mouse model.
- FIG. 4 A shows tumor growth of the HPAF II xenografts after treatment with tisotumab vedotin, or IgGl-bl2. Mean and SEM of each group is shown at each time point.
- FIG. 4B shows mean tumor size in each mouse on day 25. Mean and SEM of each group are indicated. Differences among the groups were analyzed by one-way ANOVA. Statistically significant differences versus the IgGl-bl2 group are indicated as follows: *: p ⁇ 0.05; **: p ⁇ 0.0l; ***: p ⁇ 0.00l.
- FIG. 5 shows anti-tumor effects of tisotumab vedotin treatment in a pancreatic cancer PDX mouse model PAXF 1657. Mean and SEM of tumor size of the PAXF 1657 xenografts at each time point in groups treated with two doses of tisotumab vedotin at 4 mg/kg, IgGl-bl2 or IgGlbl2-vcMMAE are shown.
- FIG. 6 shows anti-tumor effects of tisotumab vedotin treatment in a SCCHN cancer CDX mouse model FaDu. Mean and SEM of tumor size of the FaDu xenografts at each time point in groups treated with three doses of tisotumab vedotin, PBS or IgGlbl2- vcMMAE are shown.
- FIG. 7 shows anti -tumor effects of tisotumab vedotin treatment in the BXF 1036 bladder cancer patient-derived xenograft model.
- Tumor size was assessed by caliper measurement. Error bars indicate standard error of the mean (S.E.M.).
- FIG. 8 shows anti -tumor effects of tisotumab vedotin treatment in the BXF 1036 bladder cancer patient-derived xenograft model.
- Tumor size was assessed by caliper measurement. Symbols represent individual mice, horizontal lines represent mean tumor size per treatment group and error bars represent standard error of the mean (S.E.M.)
- FIG. 9 shows anti -tumor effects of tisotumab vedotin treatment in an esophageal cancer patient-derived xenograft model in nude mice.
- Tumor size was assessed by caliper measurement. Error bars indicate standard error of the mean (S.E.M.).
- FIG. 10 shows anti -tumor effects of tisotumab vedotin treatment in a PAXF1657 pancreatic cancer patient-derived xenograft model in nude mice.
- Tumor size was assessed by caliper measurement. Error bars indicate standard error of the mean (S.E.M. ).
- FIG. 11 shows anti -tumor effects of tisotumab vedotin treatment in a PA5415 pancreatic cancer patient-derived xenograft model in NOD-SCID mice.
- Tumor size was assessed by caliper measurement. Error bars indicate standard error of the mean (S.E.M.).
- FIG. 12 shows anti-tumor effects of tisotumab vedotin treatment in PA5415 pancreatic cancer patient-derived xenograft model in NOD-SCID mice.
- Tumor-free survival after treatment with tisotumab vedotin 0.5, 1 or 2 mg/kg
- an isotype control ADC IgGl- bl2-MMAE, 2 mg/kg
- an isotype control IgG IgGl-bl2, 2 mg/kg.
- Tumor size was assessed by caliper measurement. A tumor size of 500 mm 3 was used as a cut-off for tumor progression.
- FIG. 13 shows anti -tumor effects of tisotumab vedotin treatment in a diverse panel of colorectal cancer (CRC) patient-derived xenograft (PDX) models in NOD-SCID mice.
- Responding models (R) were defined as models showing DT/DO ⁇ 10% (tumor stasis or tumor regression), and non-responding models were defined as DT/DO > 70%.
- FIG. 14 shows anti-tumor effects of tisotumab vedotin treatment in a diverse panel of colorectal cancer (CRC) patient-derived xenograft (PDX) models in NOD-SCID mice.
- Responding models (R) were defined as models showing DT/DO ⁇ 10% (tumor stasis or tumor regression), and non-responding models were defined as DT/DO > 70%.
- FIG. 15 shows average TF mRNA expression levels in PDX models classified as responder, non-responder or intermediate.
- tissue factor tissue factor
- TF tissue factor
- CD142 tissue factor antigen
- TF antigen tissue factor antigen
- CD142 antigen tissue factor antigen
- tissue factor comprises the amino acid sequence found under Genbank accession NP_00l984.
- immunoglobulin refers to a class of structurally related glycoproteins consisting of two pairs of polypeptide chains, one pair of light (L) low molecular weight chains and one pair of heavy (H) chains, all four inter-connected by disulfide bonds.
- L light
- H heavy
- each heavy chain typically is comprised of a heavy chain variable region (abbreviated herein as V H or VH) and a heavy chain constant region (C H or CH).
- the heavy chain constant region typically is comprised of three domains, C H l, C H 2, and C H 3.
- the heavy chains are generally inter-connected via disulfide bonds in the so-called“hinge region.”
- Each light chain typically is comprised of a light chain variable region (abbreviated herein as V L or VL) and a light chain constant region (C L or CL).
- the light chain constant region typically is comprised of one domain, C L.
- the CL can be of k (kappa) or l (lambda) isotype.
- the terms“constant domain” and“constant region” are used interchangeably herein. Unless stated otherwise, the numbering of amino acid residues in the constant region is according to the EU-index as described in Kabat et al.
- immunoglobulin can derive from any of the commonly known isotypes, including but not limited to IgA, secretory IgA, IgG, and IgM.
- IgG subclasses are also well known to those in the art and include but are not limited to human IgGl, IgG2, IgG3 and IgG4.
- immunotype refers to the antibody class or subclass ( e.g ., IgM or IgGl) that is encoded by the heavy chain constant region genes.
- variable region or“variable domain” refers to the domain of an antibody heavy or light chain that is involved in binding the antibody to antigen.
- the variable regions of the heavy chain and light chain (V H and V L , respectively) of a native antibody may be further subdivided into regions of hypervariability (or hypervariable regions, which may be hypervariable in sequence and/or form of structurally defined loops), also termed complementarity-determining regions (CDRs), interspersed with regions that are more conserved, termed framework regions (FRs).
- CDRs complementarity-determining regions
- CDRs complementarity determining regions
- HVRs hypervariable regions
- CDR-H1, CDR-H2, CDR-H3 three CDRs in each heavy chain variable region
- CDR-L1, CDR-L2, CDR-L3 three CDRs in each light chain variable region
- “Framework regions” and“FR” are known in the art to refer to the non-CDR portions of the variable regions of the heavy and light chains.
- FR-H1, FR-H2, FR-H3, and FR-H4 there are four FRs in each full-length heavy chain variable region (FR-H1, FR-H2, FR-H3, and FR-H4), and four FRs in each full-length light chain variable region (FR-L1, FR-L2, FR-L3, and FR-L4).
- FR1, CDR1, FR2, CDR2, FR3, CDR3, FR4 See also Chothia and Lesk ./. Mot. Biol., 195, 901-917 (1987)).
- antibody in the context of the present invention refers to an immunoglobulin molecule, a fragment of an immunoglobulin molecule, or a derivative of either thereof, which has the ability to specifically bind to an antigen under typical physiological conditions with a half-life of significant periods of time, such as at least about 30 min, at least about 45 min, at least about one hour (h), at least about two hours, at least about four hours, at least about eight hours, at least about 12 hours (h), about 24 hours or more, about 48 hours or more, about three, four, five, six, seven or more days, etc., or any other relevant functionally-defined period (such as a time sufficient to induce, promote, enhance, and/or modulate a physiological response associated with antibody binding to the antigen and/or time sufficient for the antibody to recruit an effector activity).
- significant periods of time such as at least about 30 min, at least about 45 min, at least about one hour (h), at least about two hours, at least about four hours, at least about eight hours, at least about 12 hours (h), about
- variable regions of the heavy and light chains of the immunoglobulin molecule contain a binding domain that interacts with an antigen.
- the constant regions of the antibodies may mediate the binding of the immunoglobulin to host tissues or factors, including various cells of the immune system (such as effector cells) and components of the complement system such as Clq, the first component in the classical pathway of complement activation.
- An antibody may also be a bispecific antibody, diabody, multispecific antibody or similar molecule.
- the term "monoclonal antibody” as used herein refers to a preparation of antibody molecules that are recombinantly produced with a single primary amino acid sequence.
- a monoclonal antibody composition displays a single binding specificity and affinity for a particular epitope.
- the term “human monoclonal antibody” refers to antibodies displaying a single binding specificity which have variable and constant regions derived from human germline immunoglobulin sequences.
- the human monoclonal antibodies may be generated by a hybridoma which includes a B cell obtained from a transgenic or
- transchromosomal non-human animal such as a transgenic mouse, having a genome comprising a human heavy chain transgene and a light chain transgene, fused to an immortalized cell.
- an "isolated antibody” refers to an antibody that is substantially free of other antibodies having different antigenic specificities (e.g ., an isolated antibody that binds specifically to TF is substantially free of antibodies that bind specifically to antigens other than TF).
- An isolated antibody that binds specifically to TF can, however, have cross- reactivity to other antigens, such as TF molecules from different species.
- an isolated antibody can be substantially free of other cellular material and/or chemicals.
- an isolated antibody includes an antibody conjugate attached to another agent (e.g, small molecule drug).
- an isolated anti-TF antibody includes a conjugate of an anti-TF antibody with a small molecule drug (e.g, MMAE or MMAF).
- a “human antibody” refers to an antibody having variable regions in which both the FRs and CDRs are derived from human germline immunoglobulin sequences. Furthermore, if the antibody contains a constant region, the constant region also is derived from human germline immunoglobulin sequences.
- the human antibodies of the disclosure can include amino acid residues not encoded by human germline immunoglobulin sequences (e.g, mutations introduced by random or site-specific mutagenesis in vitro or by somatic mutation in vivo). However, the term "human antibody,” as used herein, is not intended to include antibodies in which CDR sequences derived from the germline of another
- human antibodies and “fully human antibodies” and are used synonymously.
- humanized antibody refers to a genetically engineered non-human antibody, which contains human antibody constant domains and non-human variable domains modified to contain a high level of sequence homology to human variable domains. This can be achieved by grafting of the six non-human antibody complementarity determining regions (CDRs), which together form the antigen binding site, onto a CDRs.
- CDRs complementarity determining regions
- FR human acceptor framework region
- substitution of framework residues from the parental antibody (i.e. the non-human antibody) into the human framework regions may be required.
- Structural homology modeling may help to identify the amino acid residues in the framework regions that are important for the binding properties of the antibody.
- a humanized antibody may comprise non-human CDR sequences, primarily human framework regions optionally comprising one or more amino acid back-mutations to the non-human amino acid sequence, and fully human constant regions.
- additional amino acid modifications which are not necessarily back-mutations, may be applied to obtain a humanized antibody with preferred characteristics, such as affinity and biochemical properties.
- chimeric antibody refers to an antibody wherein the variable region is derived from a non-human species (e.g. derived from rodents) and the constant region is derived from a different species, such as human.
- Chimeric antibodies may be generated by antibody engineering.“Antibody engineering” is a term used generic for different kinds of modifications of antibodies, and which is a well-known process for the skilled person.
- a chimeric antibody may be generated by using standard DNA techniques as described in Sambrook et al. , 1989, Molecular Cloning: A laboratory Manual, New York: Cold Spring Harbor Laboratory Press, Ch. 15.
- the chimeric antibody may be a genetically or an enzymatically engineered recombinant antibody.
- Chimeric monoclonal antibodies for therapeutic applications are developed to reduce antibody immunogenicity. They may typically contain non-human (e.g . murine) variable regions, which are specific for the antigen of interest, and human constant antibody heavy and light chain domains.
- an anti-antigen antibody refers to an antibody that binds to the antigen.
- an anti-TF antibody is an antibody that binds to the antigen TF.
- an "antigen-binding portion" or antigen-binding fragment” of an antibody refers to one or more fragments of an antibody that retain the ability to bind specifically to the antigen bound by the whole antibody.
- antibody fragments include but are not limited to Fv, Fab, Fab', Fab’-SH, F(ab') 2 ; diabodies; linear antibodies; single-chain antibody molecules (e.g. scFv); and multispecific antibodies formed from antibody fragments.
- Papain digestion of antibodies produces two identical antigen binding fragments, called“Fab” fragments, each with a single antigen-binding site, and a residual“Fc” fragment, whose name reflects its ability to crystallize readily. Pepsin treatment yields an F(ab’) 2 fragment that has two antigen-combining sites and is still capable of cross-linking antigen.
- Percent (%) sequence identity with respect to a reference polypeptide sequence is defined as the percentage of amino acid residues in a candidate sequence that are identical with the amino acid residues in the reference polypeptide sequence, after aligning the sequences and introducing gaps, if necessary, to achieve the maximum percent sequence identity, and not considering any conservative substitutions as part of the sequence identity. Alignment for purposes of determining percent amino acid sequence identity can be achieved in various ways that are within the skill in the art, for instance, using publicly available computer software such as BLAST, BLAST-2, ALIGN or Megalign (DNASTAR) software. Those skilled in the art can determine appropriate parameters for aligning sequences, including any algorithms needed to achieve maximal alignment over the full length of the sequences being compared.
- % sequence identity of a given amino acid sequence A to, with, or against a given amino acid sequence B is calculated as follows:
- binding typically is a binding with an affinity corresponding to a K D of about 10 6 M or less, e.g.
- the amount with which the K D of binding is lower is dependent on the K D of the antibody, so that when the K D of the antibody is very low, then the amount with which the K D of binding to the antigen is lower than the K D of binding to a non-specific antigen may be at least 10,000-fold (that is, the antibody is highly specific).
- K D (M) refers to the dissociation equilibrium constant of a particular antibody-antigen interaction. Affinity, as used herein, and K D are inversely related, that is that higher affinity is intended to refer to lower K D , and lower affinity is intended to refer to higher K D.
- ADC refers to an antibody-drug conjugate, which in the context of the present invention refers to an anti-TF antibody, which is coupled to a drug moiety (e.g ., MMAE or MMAF) as described in the present application.
- PAB refers to the self-immolative spacer
- the term“Ab-MC-vc-PAB-MMAE” refers to an antibody conjugated to the drug MMAE through a MC-vc-PAB linker.
- A“platinum-based therapy” refers to treatment with a platinum-based agent.
- a “platinum-based agent” refers to a molecule or a composition comprising a molecule containing a coordination complex comprising the chemical element platinum and useful as a chemotherapy drug. Platinum-based agents generally act by inhibiting DNA synthesis and some have alkylating activity. Platinum-based agents encompass those that are currently being used as part of a chemotherapy regimen, those that are currently in development, and those that may be developed in the future.
- a “cancer” refers to a broad group of various diseases characterized by the uncontrolled growth of abnormal cells in the body.
- a “cancer” or “cancer tissue” can include a tumor. ETnregulated cell division and growth results in the formation of malignant tumors that invade neighboring tissues and can also metastasize to distant parts of the body through the lymphatic system or bloodstream. Following metastasis, the distal tumors can be said to be "derived from” the pre-metastasis tumor.
- Treatment or “therapy” of a subject refers to any type of intervention or process performed on, or the administration of an active agent to, the subject with the objective of reversing, alleviating, ameliorating, inhibiting, slowing down, or preventing the onset, progression, development, severity, or recurrence of a symptom, complication, condition, or biochemical indicia associated with a disease.
- the disease is cancer.
- a “subject” includes any human or non-human animal.
- the term “non-human animal” includes, but is not limited to, vertebrates such as non-human primates, sheep, dogs, and rodents such as mice, rats, and guinea pigs. In some embodiments, the subject is a human.
- the terms “subject” and “patient” and“individual” are used interchangeably herein.
- an“effective amount” or “therapeutically effective amount” or “therapeutically effective dosage” of a drug or therapeutic agent is any amount of the drug that, when used alone or in combination with another therapeutic agent, protects a subject against the onset of a disease or promotes disease regression evidenced by a decrease in severity of disease symptoms, an increase in frequency and duration of disease symptom-free periods, or a prevention of impairment or disability due to the disease affliction.
- the ability of a therapeutic agent to promote disease regression can be evaluated using a variety of methods known to the skilled practitioner, such as in human subjects during clinical trials, in animal model systems predictive of efficacy in humans, or by assaying the activity of the agent in in vitro assays.
- a therapeutically effective amount of a drug includes a "prophylactically effective amount," which is any amount of the drug that, when administered alone or in combination with an anti -cancer agent to a subject at risk of developing a cancer (e.g., a subject having a pre-malignant condition) or of suffering a recurrence of cancer, inhibits the development or recurrence of the cancer.
- the prophylactically effective amount prevents the development or recurrence of the cancer entirely. “Inhibiting" the development or recurrence of a cancer means either lessening the likelihood of the cancer’s development or recurrence, or preventing the development or recurrence of the cancer entirely.
- subtherapeutic dose means a dose of a therapeutic compound (e.g, an anti-TF antibody-drug conjugate) that is lower than the usual or typical dose of the therapeutic compound when administered alone for the treatment of a hyperproliferative disease (e.g, cancer).
- a therapeutic compound e.g, an anti-TF antibody-drug conjugate
- an anti-cancer agent promotes cancer regression in a subject.
- a therapeutically effective amount of the drug promotes cancer regression to the point of eliminating the cancer.
- Promoting cancer regression means that administering an effective amount of the drug, alone or in combination with an anti cancer agent, results in a reduction in tumor growth or size, necrosis of the tumor, a decrease in severity of at least one disease symptom, an increase in frequency and duration of disease symptom-free periods, or a prevention of impairment or disability due to the disease affliction.
- the terms “effective” and “effectiveness” with regard to a treatment includes both pharmacological effectiveness and physiological safety.
- Pharmacological effectiveness refers to the ability of the drug to promote cancer regression in the patient.
- Physiological safety refers to the level of toxicity or other adverse physiological effects at the cellular, organ and/or organism level (adverse effects) resulting from administration of the drug.
- Sustained response refers to the sustained effect on reducing tumor growth after cessation of a treatment.
- the tumor size may remain to be the same or smaller as compared to the size at the beginning of the administration phase.
- the sustained response has a duration that is at least the same as the treatment duration, or at least 1.5, 2.0, 2.5, or 3 times longer than the treatment duration.
- complete response or “CR” refers to disappearance of all target lesions
- partial response or “PR” refers to at least a 30% decrease in the sum of the longest diameters (SLD) of target lesions, taking as reference the baseline SLD
- stable disease or “SD” refers to neither sufficient shrinkage of target lesions to qualify for PR, nor sufficient increase to qualify for PD, taking as reference the smallest SLD since the treatment started.
- progression free survival refers to the length of time during and after treatment during which the disease being treated (e.g, cancer) does not get worse. Progression-free survival may include the amount of time patients have experienced a complete response or a partial response, as well as the amount of time patients have experienced stable disease.
- ORR all response rate
- weight-based dose means that a dose administered to a patient is calculated based on the weight of the patient. For example, when a patient with 60 kg body weight requires 2 mg/kg of an anti-TF antibody-drug conjugate, one can calculate and use the appropriate amount of the anti-TF antibody-drug conjugate (i.e., 120 mg) for administration.
- flat dose means a dose that is administered to a patient without regard for the weight or body surface area (BSA) of the patient.
- the flat dose is therefore not provided as a mg/kg dose, but rather as an absolute amount of the agent (e.g ., the anti-TF antibody-drug conjugate).
- an antibody-drug conjugate e.g., 240 mg of an anti-TF antibody-drug conjugate.
- phrases "pharmaceutically acceptable” indicates that the substance or composition must be compatible chemically and/or toxicologically, with the other ingredients comprising a formulation, and/or the mammal being treated therewith.
- Exemplary salts include, but are not limited, to sulfate, citrate, acetate, oxalate, chloride, bromide, iodide, nitrate, bisulfate, phosphate, acid phosphate, isonicotinate, lactate, salicylate, acid citrate, tartrate, oleate, tannate, pantothenate, bitartrate, ascorbate, succinate, maleate, gents sinate, fumarate, gluconate, glucuronate, saccharate, formate, benzoate, glutamate, methanesulfonate "mesylate", ethanesulfonate, benzenesulfonate, p-toluenesuifonate, pamoate (i.e., 4,4’-methy!ene-bis ⁇ (2-hydroxy-3-naphthoate)) salts, alkali metal (e.g., sodium and potassium)
- a pharmaceutically acceptable salt may involve the inclusion of another molecule such as an acetate ion, a succinate ion or other counter ion.
- the counter ion may be any organic or inorganic moiety that stabilizes the charge on the parent compound.
- a pharmaceutically acceptable salt may have more than one charged atom in its structure.
- a pharmaceutically acceptable salt can have one or more charged atoms and/or one or more counter ion.
- administering refers to the physical introduction of a therapeutic agent to a subject, using any of the various methods and delivery systems known to those skilled in the art.
- exemplary routes of administration for the anti-TF antibody-drug conjugate include intravenous, intramuscular, subcutaneous, intraperitoneal, spinal or other parenteral routes of administration, for example by injection or infusion (e.g ., intravenous infusion).
- parenteral administration means modes of administration other than enteral and topical administration, usually by injection, and includes, without limitation, intravenous, intramuscular, intraarterial, intrathecal, intralymphatic, intralesional, intracapsular, intraorbital, intracardiac, intradermal, intraperitoneal, transtracheal, subcutaneous, subcuticular, intraarticular, subcapsular, subarachnoid, intraspinal, epidural and intrasternal injection and infusion, as well as in vivo electroporation.
- a therapeutic agent can be administered via a non-parenteral route, or orally.
- non-parenteral routes include a topical, epidermal or mucosal route of administration, for example, intranasally, vaginally, rectally, sublingually or topically.
- Administering can also be performed, for example, once, a plurality of times, and/or over one or more extended periods.
- baseline or “baseline value” used interchangeably herein can refer to a measurement or characterization of a symptom before the administration of the therapy (e.g., an antibody-drug conjugate as described herein) or at the beginning of administration of the therapy.
- the baseline value can be compared to a reference value in order to determine the reduction or improvement of a symptom of a TF-associated disease contemplated herein (e.g., colorectal cancer, non-small cell lung cancer, pancreatic cancer and head and neck cancer).
- reference or “reference value” used interchangeably herein can refer to a measurement or characterization of a symptom after administration of the therapy (e.g., an antibody-drug conjugate as described herein).
- the reference value can be measured one or more times during a dosage regimen or treatment cycle or at the completion of the dosage regimen or treatment cycle.
- a "reference value” can be an absolute value; a relative value; a value that has an upper and/or lower limit; a range of values; an average value; a median value: a mean value; or a value as compared to a baseline value.
- a “baseline value” can be an absolute value; a relative value; a value that has an upper and/or lower limit; a range of values; an average value; a median value; a mean value; or a value as compared to a reference value.
- the reference value and/or baseline value can be obtained from one individual, from two different individuals or from a group of individuals (e.g., a group of two, three, four, five or more individuals).
- the term“monotherapy” as used herein means that the antibody drug conjugate is the only anti-cancer agent administered to the subject during the treatment cycle.
- Other therapeutic agents can be administered to the subject.
- anti inflammatory agents or other agents administered to a subject with cancer to treat symptoms associated with cancer, but not the underlying cancer itself, including, for example inflammation, pain, weight loss, and general malaise, can be administered during the period of monotherapy.
- An "adverse event” as used herein is any unfavorable and generally unintended or undesirable sign (including an abnormal laboratory finding), symptom, or disease associated with the use of a medical treatment.
- a medical treatment can have one or more associated AEs and each AE can have the same or different level of severity.
- Reference to methods capable of "altering adverse events” means a treatment regime that decreases the incidence and/or severity of one or more AEs associated with the use of a different treatment regime.
- A“serious adverse event” or“SAE” as used herein is an adverse event that meets one of the following criteria:
- Requires inpatient hospitalization or prolongation of existing hospitalization excluding the following: 1) routine treatment or monitoring of the underlying disease, not associated with any deterioration in condition, 2) elective or pre-planned treatment for a pre-existing condition that is unrelated to the indication under study and has not worsened since signing the informed consent, and social reasons and respite care in the absence of any deterioration in the patient’s general condition.
- the terms "about” or “comprising essentially of refer to a value or composition that is within an acceptable error range for the particular value or composition as determined by one of ordinary skill in the art, which will depend in part on how the value or composition is measured or determined, i.e., the limitations of the measurement system. For example, “about” or “comprising essentially of can mean within 1 or more than 1 standard deviation per the practice in the art. Alternatively, “about” or “comprising essentially of can mean a range of up to 20%. Furthermore, particularly with respect to biological systems or processes, the terms can mean up to an order of magnitude or up to 5-fold of a value. When particular values or compositions are provided in the application and claims, unless otherwise stated, the meaning of "about” or “comprising essentially of should be assumed to be within an acceptable error range for that particular value or composition.
- the terms "once about every week,” “once about every two weeks,” or any other similar dosing interval terms as used herein mean approximate numbers. "Once about every week” can include every seven days ⁇ one day, i.e., every six days to every eight days. "Once about every two weeks” can include every fourteen days ⁇ two days, i.e., every twelve days to every sixteen days. "Once about every three weeks” can include every twenty-one days ⁇ three days, i.e., every eighteen days to every twenty-four days. Similar approximations apply, for example, to once about every four weeks, once about every five weeks, once about every six weeks, and once about every twelve weeks.
- a dosing interval of once about every six weeks or once about every twelve weeks means that the first dose can be administered any day in the first week, and then the next dose can be administered any day in the sixth or twelfth week, respectively.
- a dosing interval of once about every six weeks or once about every twelve weeks means that the first dose is administered on a particular day of the first week (e.g., Monday) and then the next dose is administered on the same day of the sixth or twelfth weeks (i.e., Monday), respectively.
- any concentration range, percentage range, ratio range, or integer range is to be understood to include the value of any integer within the recited range and, when appropriate, fractions thereof (such as one tenth and one hundredth of an integer), unless otherwise indicated. [0086] Various aspects of the disclosure are described in further detail in the following subsections.
- the present invention provides an anti-TF antibody-drug conjugate that binds to TF for use in the treatment of colorectal cancer, non-small cell lung cancer, pancreatic cancer, head and neck cancer, bladder cancer, endometrial cancer, esophageal cancer and prostate cancer in a subject, wherein the antibody-drug conjugate comprises an anti-TF antibody or an antigen-binding fragment thereof conjugated to a monomethyl auristatin or a functional analog thereof or a functional derivative thereof.
- the cancer is colorectal cancer.
- the cancer is non-small cell lung cancer.
- the cancer is pancreatic cancer.
- the cancer is head and neck cancer.
- the cancer is bladder cancer.
- the cancer is endometrial cancer.
- the cancer is esophageal cancer.
- the cancer is prostate cancer.
- the colorectal cancer, non-small cell lung cancer, pancreatic cancer, head and neck cancer, bladder cancer, endometrial cancer, esophageal cancer or prostate cancer is a metastatic cancer.
- the subject has relapsed, recurrent and/or metastatic colorectal cancer, non-small cell lung cancer, pancreatic cancer, head and neck cancer, bladder cancer, endometrial cancer, esophageal cancer or prostate cancer.
- anti-TF antibodies of the disclosure bind TF, e.g ., human TF, and exert cytostatic and cytotoxic effects on malignant cells, such as colorectal cancer, non-small cell lung cancer, pancreatic cancer, head and neck cancer, bladder cancer, endometrial cancer, esophageal cancer and prostate cancer cells.
- Anti-TF antibodies of the disclosure are preferably monoclonal, and may be multispecific, human, humanized or chimeric antibodies, single chain antibodies, Fab fragments, F(ab') fragments, fragments produced by a Fab expression library, and TF binding fragments of any of the above.
- the anti-TF antibodies of the disclosure specifically bind TF.
- the immunoglobulin molecules of the disclosure can be of any type (e.g, IgG, IgE, IgM, IgD, IgA and IgY), class (e.g, IgGl, IgG2, IgG3, IgG4, IgAl and IgA2) or subclass of immunoglobulin molecule.
- type e.g, IgG, IgE, IgM, IgD, IgA and IgY
- class e.g, IgGl, IgG2, IgG3, IgG4, IgAl and IgA2
- subclass of immunoglobulin molecule e.g, immunoglobulin molecule.
- the anti-TF antibodies are antigen binding fragments (e.g, human antigen-binding fragments) as described herein and include, but are not limited to, Fab, Fab' and F(ab') 2 , Fd, single-chain Fvs (scFv), single-chain antibodies, disulfide-linked Fvs (sdFv) and fragments comprising either a V L or V H domain.
- Antigen-binding fragments, including single-chain antibodies may comprise the variable region(s) alone or in combination with the entirety or a portion of the following: hinge region, CH1, CH2, CH3 and CL domains.
- antigen-binding fragments comprising any combination of variable region(s) with a hinge region, CH1, CH2, CH3 and CL domains.
- the anti-TF antibodies or antigen-binding fragments thereof are human, murine (e.g . , mouse and rat), donkey, sheep, rabbit, goat, guinea pig, camelid, horse, or chicken.
- the anti-TF antibodies of the present disclosure may be monospecific, bispecific, trispecific or of greater multi specificity. Multispecific antibodies may be specific for different epitopes of TF or may be specific for both TF as well as for a heterologous protein. See, e.g., PCT publications WO 93/17715; WO 92/08802; WO 91/00360; WO 92/05793;
- Anti-TF antibodies of the present disclosure may be described or specified in terms of the particular CDRs they comprise.
- the precise amino acid sequence boundaries of a given CDR or FR can be readily determined using any of a number of well-known schemes, including those described by Rabat et al. (1991),“Sequences of Proteins of Immunological Interest,” 5th Ed. Public Health Service, National Institutes of Health, Bethesda, MD
- a“CDR” or“complementary determining region,” or individual specified CDRs (e.g, CDR-H1, CDR-H2, CDR-H3), of a given antibody or region thereof (e.g, variable region thereof) should be understood to encompass a (or the specific) CDR as defined by any of the aforementioned schemes.
- a particular CDR e.g ., a CDR-H3
- contains the amino acid sequence of a corresponding CDR in a given V H or V L region amino acid sequence it is understood that such a CDR has a sequence of the corresponding CDR (e.g., CDR-H3) within the variable region, as defined by any of the aforementioned schemes.
- the scheme for identification of a particular CDR or CDRs may be specified, such as the CDR as defined by the Rabat, Chothia, AbM or IMGT method.
- CDR sequences provided herein are according to the IMGT numbering scheme as described in Lefranc, M. P. et al., Dev. Comp. Immunol., 2003, 27, 55-77.
- antibodies of the disclosure comprise one or more CDRs of the antibody 011. See WO 2011/157741 and WO 2010/066803.
- the disclosure encompasses an antibody or derivative thereof comprising a heavy or light chain variable domain, said variable domain comprising (a) a set of three CDRs, in which said set of CDRs are from monoclonal antibody 011, and (b) a set of four framework regions, in which said set of framework regions differs from the set of framework regions in monoclonal antibody 011, and in which said antibody or derivative thereof binds to TF.
- said antibody or derivative thereof specifically binds to TF.
- the anti-TF antibody is 011.
- the antibody 011 is also known as tisotumab.
- anti-TF antibodies that compete with tisotumab binding to TF are also provided herein.
- Anti-TF antibodies that bind to the same epitope as tisotumab are also provided herein.
- an anti-TF antibody comprising 1, 2, 3, 4, 5, or 6 of the CDR sequences of tisotumab.
- an anti-TF antibody comprising a heavy chain variable region and a light chain variable region
- the heavy chain variable region comprises (i) CDR-H1 comprising the amino acid sequence of SEQ ID NO: l, (ii) CDR-H2 comprising the amino acid sequence of SEQ ID NO:2, and (iii) CDR-H3 comprising the amino acid sequence of SEQ ID NO:3
- the light chain variable region comprises (i) CDR-L1 comprising the amino acid sequence of SEQ ID NO:4, (ii) CDR-L2 comprising the amino acid sequence of SEQ ID NO:5, and (iii) CDR-L3 comprising the amino acid sequence of SEQ ID NO:6.
- an anti-TF antibody described herein may comprise any suitable framework variable domain sequence, provided that the antibody retains the ability to bind TF (e.g ., human TF).
- heavy chain framework regions are designated "HC-FR1-FR4”
- light chain framework regions are designated "LC-FR1-FR4.”
- the anti-TF antibody comprises a heavy chain variable domain framework sequence of SEQ ID NO:9, 10, 11, and 12 (HC-FR1, HC-FR2, HC-FR3, and HC-FR4, respectively).
- the anti-TF antibody comprises a light chain variable domain framework sequence of SEQ ID NO: 13, 14, 15, and 16 (LC-FR1, LC-FR2, LC-FR3, and LC-FR4, respectively).
- the heavy chain variable domain comprises the amino acid sequence of
- the heavy chain CDR sequences comprise the following:
- the heavy chain FR sequences comprise the following: a) HC-FR1 (EVQLLESGGGLVQPGGSLRLSCAAS (SEQ ID NO:9));
- the light chain CDR sequences comprise the following: a) CDR-L1 (QGISSR (SEQ ID NO:4));
- the light chain FR sequences comprise the following:
- an anti-TF antibody that binds to TF (e.g ., human TF), wherein the antibody comprises a heavy chain variable region and a light chain variable region, wherein the antibody comprises:
- HC-FR1 comprising the amino acid sequence of SEQ ID NO:9;
- HC-FR4 comprising the amino acid sequence of SEQ ID NO: 12, and/or
- an LC-FR1 comprising the amino acid sequence of SEQ ID NO: 13;
- an anti-TF antibody comprising a heavy chain variable domain comprising the amino acid sequence of SEQ ID NO:7 or comprising a light chain variable domain comprising the amino acid sequence of SEQ ID NO:8. In one aspect, provided herein is an anti-TF antibody comprising a heavy chain variable domain comprising the amino acid sequence of SEQ ID NO:7 and comprising a light chain variable domain comprising the amino acid sequence of SEQ ID NO:8.
- an anti-TF antibody comprising a heavy chain variable domain comprising an amino acid sequence having at least 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% sequence identity to the amino acid sequence of SEQ ID NO:7.
- a heavy chain variable domain comprising an amino acid sequence having at least 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% sequence identity to the amino acid sequence of SEQ ID NO:7 contains substitutions (e.g, conservative substitutions), insertions, or deletions relative to the reference sequence and retains the ability to bind to a TF (e.g, human TF). In certain embodiments, a total of 1 to 10 amino acids have been substituted, inserted and/or deleted in SEQ ID NO:7.
- the anti-TF antibody comprises a heavy chain variable domain sequence of SEQ ID NO:7 including post-translational modifications of that sequence.
- the heavy chain variable domain comprises one, two or three CDRs selected from: (a) CDR-H1 comprising the amino acid sequence of SEQ ID NO: l, (b) CDR- H2 comprising the amino acid sequence of SEQ ID NO:2, and (c) CDR-H3 comprising the amino acid sequence of SEQ ID NO:3.
- an anti-TF antibody comprising a light chain variable domain comprising an amino acid sequence having at least 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% sequence identity to the amino acid sequence of SEQ ID NO:8.
- a light chain variable domain comprising an amino acid sequence having at least 85%, 86%, 87%, 88%, 89%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, or 99% sequence identity to the amino acid sequence of SEQ ID NO:8 contains substitutions (e.g, conservative substitutions), insertions, or deletions relative to the reference sequence and retains the ability to bind to a TF (e.g, human TF). In certain embodiments, a total of 1 to 10 amino acids have been substituted, inserted and/or deleted in SEQ ID NO:8.
- the anti-TF antibody comprises a light chain variable domain sequence of SEQ ID NO:8 including post-translational modifications of that sequence.
- the light chain variable domain comprises one, two or three CDRs selected from: (a) CDR-L1 comprising the amino acid sequence of SEQ ID NO:4, (b) CDR- L2 comprising the amino acid sequence of SEQ ID NO:5, and (c) CDR-L3 comprising the amino acid sequence of SEQ ID NO:6.
- the anti-TF antibody comprises a heavy chain variable domain as in any of the embodiments provided above, and a light chain variable domain as in any of the embodiments provided above.
- the antibody comprises the heavy chain variable domain sequence of SEQ ID NO: 7 and the light chain variable domain sequence of SEQ ID NO:8, including post-translational modifications of those sequences.
- the anti-TF antibody of the anti-TF antibody-drug conjugate comprises: i) a heavy chain CDR1 comprising the amino acid sequence of SEQ ID NO: 1, a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 2, a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 3; and ii) a light chain CDR1 comprising the amino acid sequence of SEQ ID NO: 4, a light chain CDR2 comprising the amino acid sequence of SEQ ID NO: 5, and a light chain CDR3 comprising the amino acid sequence of SEQ ID NO: 6.
- the anti-TF antibody of the anti-TF antibody-drug conjugate comprises: i) an amino acid sequence having at least 85% sequence identity to a heavy chain variable region comprising the amino acid sequence of SEQ ID NO: 7, and ii) an amino acid sequence having at least 85% sequence identity to a light chain variable region comprising the amino acid sequence of SEQ ID NO: 8.
- the anti-TF antibody of the anti-TF antibody-drug conjugate is a monoclonal antibody.
- the anti-TF antibody of the anti-TF antibody-drug conjugate is tisotumab, which is also known as antibody 011 as described in WO
- Anti-TF antibodies of the present invention may also be described or specified in terms of their binding affinity to TF (e.g ., human TF).
- Preferred binding affinities include those with a dissociation constant or Kd less than 5 xlO 2 M, 10 2 M, 5xl0 3 M, 10 3 M, 5xl0 4 M, 10 4 M, 5x10 5 M, 10 5 M, 5x1 O 6 M, 10 6 M, 5x1 O 7 M, 10 7 M, 5x1 O 8 M, lO 8 M, 5x1 O 9 M, 10 9 M, 5xlO 10 M, 10 10 M, 5xl0 u M, 10 11 M, 5xl0 12 M, 10 12 M, 5xlO 13 M, 10 13 M, 5x1 O 14 M, 10 14 M, 5x1 O 15 M, or 10 15 M.
- IgA immunoglobulins
- IgD immunoglobulins
- IgE immunoglobulins
- IgG immunoglobulins
- IgG immunoglobulins
- IgG2 immunoglobulins
- IgG3 immunoglobulins
- IgA2 immunoglobulins
- IgG3 immunoglobulins
- IgA2 immunoglobulins
- IgG3 immunoglobulins
- IgAl antibodies can exist in multiple polymorphic variants termed allotypes (reviewed in Jefferis and Lefranc 2009. mAbs Vol 1 Issue 4 1-7) any of which are suitable for use in some of the embodiments herein.
- the antibody may comprise a heavy chain Fc region comprising a human IgG Fc region.
- the human IgG Fc region comprises a human IgGl .
- the antibodies also include derivatives that are modified, i.e., by the covalent attachment of any type of molecule to the antibody such that covalent attachment does not prevent the antibody from binding to TF or from exerting a cytostatic or cytotoxic effect on HD cells.
- the antibody derivatives include antibodies that have been modified, e.g., by glycosylation, acetylation, PEGylation, phosphylation, amidation, derivatization by known protecting/blocking groups, proteolytic cleavage, linkage to a cellular ligand or other protein, etc. Any of numerous chemical modifications may be carried out by known techniques, including, but not limited to specific chemical cleavage, acetylation, formylation, metabolic synthesis of tunicamycin, etc.
- the derivative may contain one or more non-classical amino acids.
- the anti-TF antibody-drug conjugates described herein comprise a linker between an anti-TF antibody or antigen-binding fragment thereof as described herein and a cytostatic or cytotoxic drug.
- the linker is a non-cleavable linker. In some embodiments the linker is a cleavable linker.
- the linker is a cleavable peptide linker comprising maleimido caproyl (MC), the dipeptide valine-citrulline (vc) and p-aminobenzylcarbamate (PAB).
- MC maleimido caproyl
- vc dipeptide valine-citrulline
- PAB p-aminobenzylcarbamate
- the cleavable peptide linker has the formula: MC-vc-PAB-, wherein: a) MC is:
- vc is the dipeptide valine-citrulline
- PAB is:
- the linker is a cleavable peptide linker comprising maleimido caproyl (MC).
- MC maleimido caproyl
- the cleavable peptide linker has the formula: MC-, wherein:
- the linker is attached to sulphydryl residues of the anti-TF antibody or antigen-binding fragment thereof obtained by partial or full reduction of the anti- TF antibody or antigen-binding fragment thereof. In some embodiments, the linker is attached to sulphydryl residues of the anti-TF antibody or antigen-binding fragment thereof obtained by partial reduction of the anti-TF antibody or antigen-binding fragment thereof. In some embodiments, the linker is attached to sulphydryl residues of the anti-TF antibody or antigen-binding fragment thereof obtained by full reduction of the anti-TF antibody or antigen-binding fragment thereof.
- the anti-TF antibody-drug conjugates described herein comprise a linker as described herein between an anti-TF antibody or antigen-binding fragment thereof as described herein and a cytostatic or cytotoxic drug.
- Auristatins have been shown to interfere with microtubule dynamics, GTP hydrolysis and nuclear and cellular division (see Woyke et al (2001 ) Antimicrob. Agents and Chemother. 45(12): 3580-3584) and have anti cancer ( See U.S. Patent Nos. 5663149) and antifungal activity (See Pettit et al. , (1998) Antimicrob. Agents and Chemother. 42: 2961-2965.
- auristatin E can be reacted with para-acetyl benzoic acid or benzoylvaleric acid to produce AEB and AEVB, respectively.
- Other typical auristatin derivatives include AFP, MMAF (monomethyl auristatin F), and MMAE (monomethyl auristatin E).
- Suitable auristatins and auristatin analogs, derivatives and prodrugs, as well as suitable linkers for conjugation of auristatins to Abs, are described in, e.g ., ET.S. Patent Nos. 5,635,483, 5,780,588 and 6,214,345 and in International patent application publications W002088172, W02004010957,
- the cytostatic or cytotoxic drug is an auristatin or a functional analog thereof (e.g, functional peptide thereof) or a functional derivative thereof.
- the auristatin is a monomethyl auristatin or a functional analog thereof (e.g, functional peptide thereof) or a functional derivative thereof.
- the auristatin is monomethyl auristatin E (MMAE):
- MMAE wherein the wavy line indicates the attachment site for the linker.
- the auristatin is monomethyl auristatin F (MMAF):
- MMAF wherein the wavy line indicates the attachment site for the linker.
- the cleavable peptide linker has the formula: MC-vc-PAB-, and is attached to MMAE.
- the resulting linker-auri statin, MC-vc-PAB -MMAE is also designated vcMMAE.
- the vcMMAE drug linker moiety and conjugation methods are disclosed in W02004010957, US7659241, US7829531 and US7851437.
- vcMMAE is attached to an anti-TF antibody or antigen-binding fragment thereof as described herein, the resulting structure is:
- p denotes a number from 1 to 8, e.g, 1, 2, 3, 4, 5, 6, 7 or 8, e.g., p may be from 3-5,
- S represents a sulphydryl residue of the anti-TF antibody and Ab designates an anti-TF antibody or antigen-binding fragment thereof as described herein.
- the average value of p in a population of antibody-drug conjugates is about 4.
- p is measured by hydrophobic interaction chromatography (HIC), for example by resolving drug-loaded species based on the increasing hydrophobicity with the least hydrophobic, unconjugated form eluting first and the most hydrophobic, 8-drug form eluting last with the area percentage of a peak representing the relative distribution of the particular drug-loaded antibody-drug conjugate species. See Ouyang, J., 2013, Antibody-Drug
- p is measured by reversed phase high-performance liquid chromatography (RP-HPLC), for example by first performing a reduction reaction to completely dissociate the heavy and light chains of the ADC, then separating the light and heavy chains and their corresponding drug- loaded forms on an RP column, where the percentage peak are from integration of the light chain and heavy chain peaks, combined with the assigned drug load for each peak, is used to calculate the weighted average drug to antibody ration.
- RP-HPLC reversed phase high-performance liquid chromatography
- the cleavable peptide linker has the formula: MC-vc-PAB-, and is attached to MMAF.
- the resulting linker-auri statin, MC-vc-PAB -MMAF is also designated vcMMAF.
- a non-cleavable linker MC is attached to MMAF.
- the resulting linker-auristatin MC-MMAF is also designated mcMMAF.
- Both the vcMMAF and mcMMAF drug linker moieties and conjugation methods are disclosed in W02005081711 and US7498298.
- p denotes a number from 1 to 8, e.g, 1, 2, 3, 4, 5, 6, 7 or 8, e.g, p may be from 3-5
- S represents a sulphydryl residue of the anti-TF antibody and Ab or mAb designates an anti- TF antibody or antigen-binding fragment thereof as described herein.
- the average value of p in a population of antibody-drug conjugates is about 4.
- p is measured by hydrophobic interaction chromatography (HIC), for example by resolving drug-loaded species based on the increasing hydrophobicity with the least hydrophobic, unconjugated form eluting first and the most hydrophobic, 8-drug form eluting last with the area percentage of a peak representing the relative distribution of the particular drug-loaded antibody-drug conjugate species.
- HIC hydrophobic interaction chromatography
- p is measured by reversed phase high-performance liquid chromatography (RP-HPLC), for example by first performing a reduction reaction to completely dissociate the heavy and light chains of the ADC, then separating the light and heavy chains and their corresponding drug- loaded forms on an RP column, where the percentage peak are from integration of the light chain and heavy chain peaks, combined with the assigned drug load for each peak, is used to calculate the weighted average drug to antibody ration.
- RP-HPLC reversed phase high-performance liquid chromatography
- the antibody-drug conjugate is tisotumab vedotin.
- nucleic acids encoding an anti-TF antibody or antigen-binding fragment thereof as described herein are nucleic acids encoding an anti-TF antibody or antigen-binding fragment thereof as described herein.
- vectors comprising the nucleic acids encoding an anti-TF antibody or antigen-binding fragment thereof as described herein.
- host cells expressing the nucleic acids encoding an anti-TF antibody or antigen-binding fragment thereof as described herein.
- host cells comprising the vectors comprising the nucleic acids encoding an anti-TF antibody or antigen-binding fragment thereof as described herein.
- the anti-TF antibodies described herein may be prepared by well-known recombinant techniques using well known expression vector systems and host cells.
- the antibodies are prepared in a CHO cell using the GS expression vector system as disclosed in De la Cruz Edmunds et al, 2006, Molecular Biotechnology 34; 179- 190, EP216846, U.S. Pat. No. 5,981,216, WO 87/04462, EP323997, U.S. Pat. No. 5,591,639, U.S. Pat. No. 5,658,759, EP338841, U.S. Pat. No. 5,879,936, and U.S. Pat. No. 5,891,693.
- Monoclonal anti-TF antibodies described herein may e.g. be produced by the hybridoma method first described by Kohler et al. , Nature , 256, 495 (1975), or may be produced by recombinant DNA methods. Monoclonal antibodies may also be isolated from phage antibody libraries using the techniques described in, for example, Clackson et al. , Nature , 352, 624-628 (1991) and Marks et al, JMol, Biol., 222(3):58l-597 (1991).
- Monoclonal antibodies may be obtained from any suitable source.
- monoclonal antibodies may be obtained from hybridomas prepared from murine splenic B cells obtained from mice immunized with an antigen of interest, for instance in form of cells expressing the antigen on the surface, or a nucleic acid encoding an antigen of interest.
- Monoclonal antibodies may also be obtained from hybridomas derived from antibody- expressing cells of immunized humans or non-human mammals such as rats, dogs, primates, etc.
- the antibody (e.g ., anti-TF antibody) of the invention is a human antibody.
- Human monoclonal antibodies directed against TF may be generated using transgenic or transchromosomal mice carrying parts of the human immune system rather than the mouse system.
- transgenic and transchromosomic mice include mice referred to herein as HuMAb mice and KM mice, respectively, and are collectively referred to herein as “transgenic mice”.
- the HuMAb mouse contains a human immunoglobulin gene minilocus that encodes unrearranged human heavy (m and g) and k light chain immunoglobulin sequences, together with targeted mutations that inactivate the endogenous m and k chain loci (Lonberg, N. et al., Nature , 368, 856-859 (1994)). Accordingly, the mice exhibit reduced expression of mouse IgM or k and in response to immunization, the introduced human heavy and light chain transgenes undergo class switching and somatic mutation to generate high affinity human IgG,K monoclonal antibodies (Lonberg, N. et al. (1994), supra; reviewed in Lonberg, N.
- the HCo7 mice have a JKD disruption in their endogenous light chain (kappa) genes (as described in Chen et al, EMBO J. 12:821-830 (1993)), a CMD disruption in their endogenous heavy chain genes (as described in Example 1 of WO 01/14424), a KCo5 human kappa light chain transgene (as described in Fishwild et al, Nature Biotechnology, 14:845- 851 (1996)), and a HCo7 human heavy chain transgene (as described in U.S. Pat. No.
- the HCol2 mice have a JKD disruption in their endogenous light chain (kappa) genes (as described in Chen et al. , EMBO J. 12:821-830 (1993)), a CMD disruption in their endogenous heavy chain genes (as described in Example 1 of WO 01/14424), a KCo5 human kappa light chain transgene (as described in Fishwild et al. , Nature Biotechnology, 14:845- 851 (1996)), and a HCol2 human heavy chain transgene (as described in Example 2 of WO 01/14424).
- kappa endogenous light chain
- CMD disruption in their endogenous heavy chain genes as described in Example 1 of WO 01/14424
- KCo5 human kappa light chain transgene as described in Fishwild et al. , Nature Biotechnology, 14:845- 851 (1996)
- HCol2 human heavy chain transgene as described in Example 2 of WO 01/14424.
- the HCol7 transgenic mouse strain (see also ETS 2010/0077497) was generated by coinjection of the 80 kb insert of pHC2 (Taylor et al. (1994) Int. Immunol ., 6:579-591), the Kb insert of pVX6, and a -460 kb yeast artificial chromosome fragment of the yIgH24 chromosome. This line was designated (HCol7) 25950. The (HCol7) 25950 line was then bred with mice comprising the CMD mutation (described in Example 1 of PCT Publication WO 01109187), the JKD mutation (Chen et al, (1993 ) EMBO J.
- mice express human immunoglobulin heavy and kappa light chain transgenes in a
- the HCo20 transgenic mouse strain is the result of a co-injection of minilocus 30 heavy chain transgene pHC2, the germline variable region (Vh)-containing YAC ylgHlO, and the minilocus construct pVx6 (described in W009097006).
- the (HCo20) line was then bred with mice comprising the CMD mutation (described in Example 1 of PCT Publication WO 01/09187), the JKD mutation (Chen et al. (1993) EMBO J. 12:811-820), and the (KC05) 9272 trans gene (Fishwild eta). (1996) Nature Biotechnology, 14:845-851).
- the resulting mice express human 10 immunoglobulin heavy and kappa light chain transgenes in a background homozygous for disruption of the endogenous mouse heavy and kappa light chain loci.
- HuMab mice were crossed with KCO05 [MIK] (Balb) mice which were generated by backcrossing the KC05 strain (as described in Fishwild et al. (1996) Nature Biotechnology , 14:845-851) to wild-type Balb/c mice to generate mice as described in W009097006. ETsing this crossing Balb/c hybrids were created for HCol2, HCol7, and HCo20 strains.
- the endogenous mouse kappa light chain gene has been homozygously disrupted as described in Chen et al ., EMBO J 12:811-820 (1993) and the endogenous mouse heavy chain gene has been homozygously disrupted as described in Example 1 of WO 01/09187
- This mouse strain carries a human kappa light chain transgene, KCo5, as described in Fishwild et al., Nature Biotechnology, 14:845-851 (1996).
- This mouse strain also carries a human heavy chain transchromosome composed of chromosome 14 fragment hCF (SC20) as described in WO 02/43478.
- Splenocytes from these transgenic mice may be used to generate hybridomas that secrete human monoclonal antibodies according to well-known techniques, Human monoclonal or polyclonal antibodies of the present invention, or antibodies of the present invention originating from other species may also be generated transgenically through the generation of another non-human mammal or plant that is transgenic for the immunoglobulin heavy and light chain sequences of interest and production of the antibody in a recoverable form therefrom.
- antibodies may be produced in, and recovered from, the milk of goats, cows, or other mammals. See for instance U.S. Pat. No. 5,827,690, U.S. Pat. No. 5,756,687, U.S. Pat. No. 5,750,172 and U.S. Pat. No. 5,741,957.
- human antibodies of the present invention or antibodies of the present invention from other species may be generated through display-type technologies, including, without limitation, phage display, retroviral display, ribosomal display, and other techniques, using techniques well known in the art and the resulting molecules may be subjected to additional maturation, such as affinity maturation, as such techniques are well known in the art (See for instance Hoogenboom et al, J. Mol, Biol.
- the invention provides methods for treating cancer in a subject with an anti-TF antibody-drug conjugate described herein, wherein the cancer is colorectal cancer, non-small cell lung cancer, pancreatic cancer, head and neck cancer, bladder cancer, endometrial cancer, esophageal cancer or prostate cancer.
- the antibody-drug conjugate is tisotumab vedotin.
- the subject is a human.
- the present invention provides an antibody-drug conjugate that binds to TF for use in the treatment of cancer
- the antibody-drug conjugate comprises an anti-TF antibody or an antigen-binding fragment thereof conjugated to a monomethyl auristatin or a functional analog thereof or a functional derivative thereof and wherein the cancer is colorectal cancer, non-small cell lung cancer, pancreatic cancer, head and neck cancer, bladder cancer, endometrial cancer, esophageal cancer or prostate cancer.
- the antibody-drug conjugate is tisotumab vedotin.
- the subject is a human.
- the subject has been previously treated for the colorectal cancer, non-small cell lung cancer, pancreatic cancer, head and neck cancer, bladder cancer, endometrial cancer, esophageal cancer or prostate cancer.
- the subject did not respond to the treatment (e.g ., the subject experienced disease progression during treatment).
- the subject relapsed after the treatment.
- the subject experienced disease progression after the treatment.
- the treatment previously administered to the subject was not an anti-TF antibody-drug conjugate as described herein.
- Colorectal cancer is the third leading cause of cancer-related deaths in men and women in the United States. Though colorectal cancer mortality rates have steadily declined in recent years (dropping an estimated 4% per year between 2008 and 2011) due in part to better screening rates for early detection, 5 year survival for patients with metastatic colorectal cancer is only 21%.
- the approach to subsequent therapy is variable and may include maintenance chemotherapy or a switch to a different regimen altogether because of disease progression or intolerance to the initial regimen.
- the model of distinct "lines" of chemotherapy in which regimens containing non-cross-resistant drugs are each used in succession until disease progression is being abandoned in favor of a
- the invention provides methods for treating colorectal cancer in a subject with an antibody-drug conjugate described herein.
- the antibody-drug conjugates described herein are for use in a method of treating colorectal cancer in a subject.
- the antibody-drug conjugate is tisotumab vedotin.
- the subject has not been previously treated for the colorectal cancer.
- the subject has received at least one previous treatment for the colorectal cancer.
- the subject received prior systemic therapy for the colorectal cancer.
- the subject experienced disease progression on or after the systemic therapy.
- the subject received no more than 3 rounds of prior systemic therapy.
- the subject received 1, 2 or 3 rounds of prior systemic therapy. In some embodiments, the subject received 1 round of prior systemic therapy. In some embodiments, the subject received 2 rounds of prior systemic therapy. In some embodiments, the subject received 3 rounds of prior systemic therapy. In some embodiments, the colorectal cancer is non-operable. In some embodiments, the subject has been previously treated with one or more agents selected from the group consisting of fluoropyrimidine, oxaliplatin, irinotecan, bevacizumab, cetuximab, panitumab and a checkpoint inhibitor. In some embodiments, the subject has been previously treated with one or more agents selected from the group consisting of fluoropyrimidine, oxaliplatin, irinotecan and bevacizumab. In some embodiments,
- the subject has been previously treated with fluoropyrimidine. In some embodiments, the subject has been previously treated with oxaliplatin. In some
- the subject has been previously treated with irinotecan. In some embodiments, the subject has been previously treated with bevacizumab. In some embodiments, the subject has been previously treated with one or more agents selected from the group consisting of cetuximab, panitumab and a checkpoint inhibitor. In some embodiments, the subject has been previously treated with cetuximab. In some embodiments, the subject has been previously treated with panitumab. In some embodiments, the subject has been previously treated with a checkpoint inhibitor.
- the checkpoint inhibitor is an inhibitor of PD-l, PD-L1, CTLA-4, PD-L2, LAG3, Tim3, 2B4, A2aR, ID02, B7-H3, B7-H4, BTLA, CD2, CD20, CD27, CD28, CD30, CD33, CD40, CD52, CD70, CD80, CD86, CD112, CD137, CD 160, CD226, CD276, DR3, OX-40, GAL9, GITR, ICOS, HVEM, IDOI, KIR, LAIR, LIGHT, MARCO, PS, SLAM, TIGIT, VISTA, and/or VTCN1.
- PD-l PD-L1, CTLA-4, PD-L2, LAG3, Tim3, 2B4, A2aR, ID02, B7-H3, B7-H4, BTLA, CD2, CD20, CD27, CD28, CD30, CD33, CD40, CD52, CD70, CD80, CD86, CD112, CD137, CD
- the checkpoint inhibitor is an inhibitor of PD-l, PD-L1 and/or CTLA-4. In some embodiments, the checkpoint inhibitor is an inhibitor of PD-L In some embodiments, the checkpoint inhibitor is selected from the group consisting of nivolumab (OPDIVO®, BMS-936558, MDX-1106 or MK-34775), pembrolizumab (KEYTRUDA®, MK-3475), pidilizumab (CT-011) and cemiplimab (REGN2810). In some embodiments, the checkpoint inhibitor is an inhibitor of PD-L1.
- the checkpoint inhibitor is selected from the group consisting of atezolizumab (TECENTRIQ®, MPDL3280A), avelumab (BAVENCIO®), durvalumab and BMS-936559.
- the checkpoint inhibitor is an inhibitor of CTLA-4.
- the checkpoint inhibitor is selected from the group consisting of ipilimumab and tremelimumab.
- the colorectal cancer is an advanced stage cancer.
- the advanced stage cancer is a stage 3 or 4 cancer.
- the advanced stage cancer is a metastatic cancer.
- the colorectal cancer is a recurrent cancer.
- the subject received prior treatment with standard of care therapy for the cancer and failed the prior treatment.
- the subject is a human.
- At least 0.1%, at least 1%, at least 2%, at least 3%, at least 4%, at least 5%, at least 6%, at least 7%, at least 8%, at least 9%, at least 10%, at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 60%, at least 70%, or at least 80% of the colorectal cancer cells from the subject express TF.
- the percentage of cells that express TF is determined using immunohistochemistry (IHC).
- the percentage of cells that express TF is determined using flow cytometry.
- the percentage of cells that express TF is determined using an enzyme- linked immunosorbent assay (ELISA).
- Lung cancer remains the leading cause of death from cancer in the United States. Treatments with curative intent for patients with early stage disease include surgery, chemotherapy, radiation therapy, or a combined modality approach. Lung cancer typically undergoes epithelial-mesenchymal transition with early metastatic spread. The symptoms are often difficult for patients to recognize in the early stages of disease. Because of these two factors, a majority of patients are diagnosed with advanced stage disease, which is usually incurable.
- NSCLC represents up to 80% of all lung cancers. Within the subtypes of NSCLC, squamous cell carcinoma (SCC/NSCLC) represents approximately 30% of NSCLC.
- SCC/NSCLC squamous cell carcinoma
- Systemic therapy can significantly prolong survival and help maintain quality of life in patients who present with stage IV squamous NSCLC or who develop advanced disease following their initial definitive therapy. Histology provides insight into the optimal agents to combine with a platinum compound and molecular characterization of the tumor. Patients with SCC/NSCLC should have tumor assessed for the expression of programmed death ligand- 1 (PD-L1). The choice of initial therapy is guided by this information.
- PD-L1 programmed death ligand- 1
- the preferred first line option is a platinum-based chemotherapy doublet that does not contain pemetrexed or anti-VEGF.
- platinum partners that may be used in initial therapy for SCC/NSCLC include necitumumab, a monoclonal antibody that targets EGFR, e.g. , in combination with gemcitabine and cisplatin.
- necitumumab a monoclonal antibody that targets EGFR, e.g. , in combination with gemcitabine and cisplatin.
- first-line treatment with the anti -PD- 1 inhibitor pembrolizumab should be offered. Pembrolizumab should be continued until progression or intolerable toxicity occurs.
- Systemic therapy trials for second line and later metastatic NSCLC include docetaxel, vinorelbine or ifosfamide, OPDIVO ® , docetaxel, KEYTRUDA ® , and TECENTRIQ ® .
- the most preferred treatment regimen for SCC/NSCLC patients who progress on an initial combination chemotherapy regimen is immunotherapy with an anti -PD- 1 or PD-L1 antibody. Combination chemotherapy should be considered for patients whose disease has progressed after receiving PD-1/L1 inhibitors.
- the invention provides methods for treating non-small cell lung cancer with an antibody-drug conjugate described herein.
- the antibody-drug conjugates described herein are for use in a method of treating non-small cell lung cancer in a subject.
- the antibody-drug conjugate is tisotumab vedotin.
- the subject has not been previously treated for the non-small cell lung cancer.
- the subject has received at least one previous treatment for the non-small cell lung cancer.
- the subject received prior systemic therapy for the non small cell lung cancer.
- the subject experienced disease progression on or after the systemic therapy.
- the subject received no more than 2 rounds of prior systemic therapy.
- the subject received 1 or 2 rounds of prior systemic therapy.
- the subject received 1 round of prior systemic therapy.
- the subject received 2 rounds of prior systemic therapy.
- the subject has been previously treated with one or more agents selected from the group consisting of a platinum-based therapy and a checkpoint inhibitor. In some embodiments, the subject has been previously treated with a platinum- based therapy. In some embodiments, the platinum-based therapy is selected from the group consisting of carboplatin, cisplatin, oxaliplatin, nedaplatin, triplatin tetranitrate,
- the platinum-based therapy is carboplatin. In some embodiments, the platinum-based therapy is cisplatin. In some embodiments, the platinum-based therapy is oxaliplatin. In some embodiments, the platinum-based therapy is nedaplatin. In some embodiments, the platinum-based therapy is triplatin tetranitrate. In some embodiments, the platinum-based therapy is phenanthriplatin. In some embodiments, the platinum-based therapy is picoplatin. In some embodiments, the platinum-based therapy is satraplatin. In some embodiments, the subject has been previously treated with a checkpoint inhibitor.
- the checkpoint inhibitor is an inhibitor of PD-l, PD-L1, CTLA-4, PD-L2, LAG3, Tim3, 2B4, A2aR, ID02, B7-H3, B7-H4, BTLA, CD2, CD20, CD27, CD28, CD30, CD33, CD40, CD52, CD70, CD80, CD86, CD112, CD137, CD 160, CD226, CD276, DR3, OX-40, GAL9, GITR, ICOS, HVEM, IDOI, KIR, LAIR, LIGHT, MARCO, PS, SLAM, TIGIT, VISTA, and/or VTCN1.
- the checkpoint inhibitor is an inhibitor of PD-l, PD-L1 and/or CTLA-4. In some embodiments, the checkpoint inhibitor is an inhibitor of PD-L In some embodiments, the checkpoint inhibitor is selected from the group consisting of nivolumab (OPDIVO®, BMS-936558, MDX-1106 or MK-34775), pembrolizumab (KEYTRUDA®, MK-3475), pidilizumab (CT-011) and cemiplimab (REGN2810). In some embodiments, the checkpoint inhibitor is an inhibitor of PD-L1.
- the checkpoint inhibitor is selected from the group consisting of atezolizumab (TECENTRIQ®, MPDL3280A), avelumab (BAVENCIO®), durvalumab and BMS-936559.
- the checkpoint inhibitor is an inhibitor of CTLA-4.
- the checkpoint inhibitor is selected from the group consisting of ipilimumab and tremelimumab.
- the non-small cell lung cancer is squamous cell carcinoma. In some embodiments, the non small cell lung cancer has predominant squamous histology.
- greater than 75%, greater than 80%, greater than 85%, greater than 90% or greater than 95% of the non-small cell lung cancer cells have squamous histology. In some embodiments greater than 75% of the non-small cell lung cancer cells have squamous histology. In some embodiments greater than 80% of the non-small cell lung cancer cells have squamous histology. In some embodiments greater than 85% of the non-small cell lung cancer cells have squamous histology. In some embodiments greater than 90% of the non-small cell lung cancer cells have squamous histology. In some embodiments greater than 95% of the non-small cell lung cancer cells have squamous histology.
- the non-small cell lung cancer is adenocarcinoma. In some embodiments, the non-small cell lung cancer is an advanced stage cancer. In some embodiments, the advanced stage cancer is a stage 3 or 4 cancer. In some embodiments, the advanced stage cancer is a metastatic cancer. In some embodiments, the non-small cell lung cancer is a recurrent cancer. In some embodiments, the subject received prior treatment with standard of care therapy for the cancer and failed the prior treatment. In a particular embodiment, the subject is a human.
- the percentage of cells that express TF is determined using immunohistochemistry (IHC).
- the percentage of cells that express TF is determined using flow cytometry.
- the percentage of cells that express TF is determined using an enzyme-linked immunosorbent assay (ELISA).
- Pancreatic cancer is the third leading cause of cancer-related death in the United States in 2016. Five year survival for people with metastatic pancreatic cancer remains a dismal 8% in the US and may be as low as 4% worldwide. Surgical resection offers the only chance of cure. However, only 15% to 20% of patients have resectable disease at initial diagnosis; the majority have either locally advanced or metastatic cancer. Metastatic pancreatic cancer patients have very few effective treatment options and are often treated only with palliative care. First line combination regimens including FOLFIRINOX or nab- paclitaxel plus gemcitabine are often options for patients with a reasonable performance status and have been shown to prolong OS by several months. Second line and later treatments offer limited efficacy with significant treatment-related toxicity.
- Preferred regimens in this group include liposomal irinotecan (ONIVYDE ® ) with 5-FU/leucovorin, FOLFOX, and gemcitabine in combination with nab-paclitaxel, erlotinib, or bevacizumab.
- the invention provides methods for treating pancreatic cancer with an antibody- drug conjugate described herein.
- the antibody-drug conjugates described herein are for use in a method of treating pancreatic cancer in a subject.
- the antibody-drug conjugate is tisotumab vedotin.
- the subject has not been previously treated for the pancreatic cancer.
- the subject has received at least one previous treatment for the pancreatic cancer.
- the subject received prior systemic therapy for the pancreatic cancer.
- the subject experienced disease progression on or after the systemic therapy.
- the subject received no more than 1 round of prior systemic therapy. In some embodiments, the subject received 1 round of prior systemic therapy. In some embodiments, the subject has been previously treated with one or more agents selected from the group consisting of gemcitabine and 5-fluorouracil (5-FU). In some embodiments, the subject has been previously treated with gemcitabine. In some embodiments, the subject has been previously treated with 5-fluorouracil. In some embodiments, the pancreatic cancer is not resectable. In some embodiments, the pancreatic cancer is exocrine pancreatic
- the pancreatic cancer has predominant
- adenocarcinoma histology In some embodiments greater than 75%, greater than 80%, greater than 85%, greater than 90% or greater than 95% of the pancreatic cancer cells have adenocarcinoma histology. In some embodiments greater than 75% of the pancreatic cancer cells have adenocarcinoma histology. In some embodiments greater than 80% of the pancreatic cancer cells have adenocarcinoma histology. In some embodiments greater than 85% of the pancreatic cancer cells have adenocarcinoma histology. In some embodiments greater than 90% of the pancreatic cancer cells have adenocarcinoma histology.
- the pancreatic cancer is an advanced stage cancer.
- the advanced stage cancer is a stage 3 or 4 cancer.
- the advanced stage cancer is a metastatic cancer.
- the pancreatic cancer is a recurrent cancer.
- the subject received prior treatment with standard of care therapy for the cancer and failed the prior treatment.
- the subject is a human.
- At least 0.1%, at least 1%, at least 2%, at least 3%, at least 4%, at least 5%, at least 6%, at least 7%, at least 8%, at least 9%, at least 10%, at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 60%, at least 70%, or at least 80% of the pancreatic cancer cells from the subject express TF.
- the percentage of cells that express TF is determined using immunohistochemistry (IHC).
- the percentage of cells that express TF is determined using flow cytometry.
- the percentage of cells that express TF is determined using an enzyme- linked immunosorbent assay (ELISA).
- Head and neck cancers account for approximately 4% of all cancers in the United States. More than 90-95% of oral and nasopharyngeal cancers are of squamous histology.
- Surgical resection, radiotherapy, and/or chemoradiation are frequently recommended for patients with early-stage or localized disease.
- Palliative chemotherapy, immunotherapy and/or supportive care are the most appropriate options for patients with locally recurrent or metastatic disease that are not amenable to definitive therapy.
- primary treatment is with systemic therapy.
- Platinum-based regimens are the preferred standard of care in this setting. Cetuximab in combination with a platinum-5-FU regimen has demonstrated clinically meaningful benefit with an improvement in median OS of 10.1 months compared to 7.4 months for platinum/5-FU alone.
- second line treatment is with single agent chemotherapy, targeted therapy, or a checkpoint inhibitor (CPI).
- CPI checkpoint inhibitor
- the invention provides methods for treating head and neck cancer with an antibody-drug conjugate described herein.
- the antibody-drug conjugates described herein are for use in a method of treating head and neck cancer in a subject.
- the antibody-drug conjugate is tisotumab vedotin.
- the subject has not been previously treated for the head and neck cancer.
- the head and neck cancer is squamous cell carcinoma.
- the subject has received at least one previous treatment for the head and neck cancer.
- the subject received prior systemic therapy for the head and neck cancer.
- the subject experienced disease progression on or after the systemic therapy.
- the subject received no more than 2 rounds of prior systemic therapy. In some embodiments, the subject received 1 or 2 rounds of prior systemic therapy. In some embodiments, the subject received 1 round of prior systemic therapy. In some embodiments, the subject received 2 rounds of prior systemic therapy. In some embodiments, the subject has been previously treated with one or more agents selected from the group consisting of a platinum-based therapy, a checkpoint inhibitor and an anti-epithelial growth factor receptor therapy. In some embodiments, the subject has been previously treated with one or more agents selected from the group consisting of a platinum-based therapy and a checkpoint inhibitor. In some embodiments, the subject has been previously treated with a platinum- based therapy. In some embodiments, the platinum-based therapy is selected from the group consisting of carboplatin, cisplatin, oxaliplatin, nedaplatin, triplatin tetranitrate,
- the platinum-based therapy is carboplatin. In some embodiments, the platinum-based therapy is cisplatin. In some embodiments, the platinum-based therapy is oxaliplatin. In some embodiments, the platinum-based therapy is nedaplatin. In some embodiments, the platinum-based therapy is triplatin tetranitrate. In some embodiments, the platinum-based therapy is phenanthriplatin. In some embodiments, the platinum-based therapy is picoplatin. In some embodiments, the platinum-based therapy is satraplatin. In some embodiments, the subject has been previously treated with a checkpoint inhibitor.
- the checkpoint inhibitor is an inhibitor of PD-l, PD-L1, CTLA-4, PD-L2, LAG3, Tim3, 2B4, A2aR, ID02, B7-H3, B7-H4, BTLA, CD2, CD20, CD27, CD28, CD30, CD33, CD40, CD52, CD70, CD80, CD86, CD112, CD137, CD 160, CD226, CD276, DR3, OX-40, GAL9, GITR, ICOS, HVEM, IDOI, KIR, LAIR, LIGHT, MARCO, PS, SLAM, TIGIT, VISTA, and/or VTCN1.
- PD-l PD-L1, CTLA-4, PD-L2, LAG3, Tim3, 2B4, A2aR, ID02, B7-H3, B7-H4, BTLA, CD2, CD20, CD27, CD28, CD30, CD33, CD40, CD52, CD70, CD80, CD86, CD112, CD137, CD
- the checkpoint inhibitor is an inhibitor of PD-l, PD-L1 and/or CTLA-4. In some embodiments, the checkpoint inhibitor is an inhibitor of PD-L In some embodiments, the checkpoint inhibitor is selected from the group consisting of nivolumab (OPDIVO®, BMS-936558, MDX-1106 or MK-34775), pembrolizumab (KEYTRUDA®, MK-3475), pidilizumab (CT-011) and cemiplimab (REGN2810). In some embodiments, the checkpoint inhibitor is an inhibitor of PD-L1.
- the checkpoint inhibitor is selected from the group consisting of atezolizumab (TECENTRIQ®, MPDL3280A), avelumab (BAVENCIO®), durvalumab and BMS-936559.
- the checkpoint inhibitor is an inhibitor of CTLA-4.
- the checkpoint inhibitor is selected from the group consisting of ipilimumab and tremelimumab.
- the subject has been previously treated with an anti-epithelial growth factor receptor therapy.
- the anti-epithelial growth factor receptor therapy is selected from the group consisting of gefitinib, erlotinib, afatinib, brigatinib, icotinib, lapatinib, osimertinib, cetuximab, panitumumab, zalutumumab, nimotuzumab and matuzumab.
- the head and neck cancer is an advanced stage cancer.
- the advanced stage cancer is a stage 3 or 4 cancer.
- the advanced stage cancer is a metastatic cancer.
- the head and neck cancer is a recurrent cancer.
- the subject received prior treatment with standard of care therapy for the cancer and failed the prior treatment.
- the subject is a human.
- At least 0.1%, at least 1%, at least 2%, at least 3%, at least 4%, at least 5%, at least 6%, at least 7%, at least 8%, at least 9%, at least 10%, at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 60%, at least 70%, or at least 80% of the head and neck cancer cells from the subject express TF.
- the percentage of cells that express TF is determined using immunohistochemistry (IHC).
- the percentage of cells that express TF is determined using flow cytometry.
- the percentage of cells that express TF is determined using an enzyme-linked immunosorbent assay (ELISA).
- Bladder cancer is the sixth most common cancer in the United States, with an estimated 76,960 new cases diagnosed in 2016. Of these patients, 16,390 deaths were estimated to have occurred, with men being more likely to be affected than women.
- the 5- year relative survival rate for all stages combined is 77%. However, survival rates depend on many factors, including the histology and stage of bladder cancer diagnosed. For patients with bladder cancer that is invasive but not yet spread outside the bladder, the 5-year survival rate is 70%. For patients with bladder cancer that extends through the bladder to the surrounding tissue and/or organs, the 5-year survival rate is 34%.
- a cisplatin-based chemotherapy regimen followed by surgical removal of the bladder or radiation therapy and concomitant chemotherapy is currently the standard treatment for patients with invasive bladder cancer.
- the invention provides methods for treating bladder cancer with an antibody-drug conjugate described herein.
- the antibody-drug conjugates described herein are for use in a method of treating bladder cancer in a subject.
- the antibody-drug conjugate is tisotumab vedotin.
- the subject has not been previously treated for the bladder cancer.
- the subject received at least one previous treatment for the bladder cancer.
- the subject received prior systemic therapy for the bladder cancer.
- the subject experienced disease progression on or after the systemic therapy.
- the subject received no more than 3 rounds of prior systemic therapy. In some embodiments, the subject received 1, 2 or 3 rounds of prior systemic therapy. In some embodiments, the subject received 1 round of prior systemic therapy. In some embodiments, the subject received 2 rounds of prior systemic therapy. In some embodiments, the subject received 3 rounds of prior systemic therapy. In some embodiments, the subject has been previously treated with a platinum-based therapy. In some embodiments, the platinum-based therapy is selected from the group consisting of carboplatin, cisplatin, oxaliplatin, nedaplatin, triplatin tetranitrate, phenanthriplatin, picoplatin and satraplatin. In some embodiments, the platinum-based therapy is carboplatin.
- the platinum-based therapy is cisplatin. In some embodiments, the platinum-based therapy is oxaliplatin. In some embodiments, the platinum-based therapy is nedaplatin. In some embodiments, the platinum-based therapy is triplatin tetranitrate. In some embodiments, the platinum-based therapy is phenanthriplatin. In some embodiments, the platinum-based therapy is picoplatin. In some embodiments, the platinum-based therapy is satraplatin. In some embodiments, the subject has previously undergone surgery or radiation therapy for the bladder cancer. In some embodiments, the subject has previously undergone surgery for the bladder cancer. In some embodiments, the subject has previously undergone radiation therapy for the bladder cancer.
- the bladder cancer is an advanced stage cancer.
- the advanced stage cancer is a stage 3 or 4 cancer.
- the advanced stage cancer is a metastatic cancer.
- the bladder cancer is a recurrent cancer.
- the subject received prior treatment with standard of care therapy for the cancer and failed the prior treatment.
- the subject is a human.
- the percentage of cells that express TF is determined using immunohistochemistry (IHC).
- the percentage of cells that express TF is determined using flow cytometry. In some embodiments, the percentage of cells that express TF is determined using an enzyme- linked immunosorbent assay (ELISA).
- ELISA enzyme- linked immunosorbent assay
- Endometrial cancer is the most common gynecologic malignancy in the ETnited States, accounting for 6% of cancers in women. In 2017, an estimated 61,380 women were diagnosed with endometrial cancer, and approximately 11,000 died from this disease. From 1987 to 2008, there was a 50% increase in the incidence of endometrial cancer, with an approximate 300% increase in the number of associated deaths. Endometrial
- adenocarcinomas can be classified into two histologic categories— type 1 or type 2.
- Type 1 endometrial carcinomas are of endometrioid histology, lower grade, and often confined to the uterus at diagnosis. These tumors are estrogen-mediated, and often, women diagnosed with type 1 endometrial carcinomas are obese, with excess endogenous estrogen production.
- Type 1 carcinomas (estrogen dependent) have high rates of K-ras and PTEN loss or mutation, as well as defects in mismatch repair genes, which lead to microsatellite instability (MSI).
- MSI microsatellite instability
- Type 2 (non estrogen dependent) carcinomas are higher-grade adenocarcinomas and are of non- endometrioid histology, occurring in older, leaner women, although an association with increasing body mass index (BMI) has been observed.
- BMI body mass index
- Type 2 cancers have p53 mutations, may have overexpression of human epidermal growth factor receptor 2 (HER-2/neu), and show aneuploidy.
- HER-2/neu human epidermal growth factor receptor 2
- chemotherapeutic and targeted therapy agents approved for ovarian, fallopian tube and primary peritoneal cancers since the 1971 approval of megestrol acetate for the palliative treatment of advanced endometrial cancer, only pembrolizumab has been Food and Drug Administration (FDA)-approved for high microsatellite instability (MSI-H) or mismatch repair deficient (dMMR) endometrial cancer; this highlights the need for new therapies to treat advanced, recurrent, metastatic endometrial cancer.
- FDA Food and Drug Administration
- MSI-H microsatellite instability
- dMMR mismatch repair deficient
- the invention provides methods for treating endometrial cancer with an antibody- drug conjugate described herein.
- the antibody-drug conjugates described herein are for use in a method of treating endometrial cancer in a subject.
- the antibody-drug conjugate is tisotumab vedotin.
- the subject has not been previously treated for the endometrial cancer.
- the subject received at least one previous treatment for the endometrial cancer.
- the subject received prior systemic therapy for the endometrial cancer.
- the subject experienced disease progression on or after the systemic therapy.
- the subject received no more than 3 rounds of prior systemic therapy. In some embodiments, the subject received 1, 2 or 3 rounds of prior systemic therapy. In some embodiments, the subject received 1 round of prior systemic therapy. In some embodiments, the subject received 2 rounds of prior systemic therapy. In some
- the subject received 3 rounds of prior systemic therapy. In some embodiments, the subject received 3 rounds of prior systemic therapy.
- the subject has been previously treated with one or more agents selected from the group consisting of a platinum-based therapy, hormone therapy, and a checkpoint inhibitor. In some embodiments, the subject has been previously treated with a platinum- based therapy. In some embodiments, the platinum-based therapy is selected from the group consisting of carboplatin, cisplatin, oxaliplatin, nedaplatin, triplatin tetranitrate,
- the platinum-based therapy is carboplatin. In some embodiments, the platinum-based therapy is cisplatin. In some embodiments, the platinum-based therapy is oxaliplatin. In some embodiments, the platinum-based therapy is nedaplatin. In some embodiments, the platinum-based therapy is triplatin tetranitrate. In some embodiments, the platinum-based therapy is phenanthriplatin. In some embodiments, the platinum-based therapy is picoplatin. In some embodiments, the platinum-based therapy is satraplatin. In some embodiments, the subject has been previously treated with a hormone therapy.
- the hormone therapy is selected from the group consisting of a progestin, tamoxifen, a luteinizing hormone-releasing hormone agonist, and an aromatase inhibitor.
- the hormone therapy is a progestin.
- the progestin is medroxyprogesterone acetate.
- the progestin is megestrol acetate.
- the hormone therapy is tamoxifen.
- the hormone therapy is a luteinizing hormone-releasing hormone agonist.
- the luteinizing hormone-releasing hormone agonist is goserelin.
- the luteinizing hormone-releasing hormone agonist is leuprolide.
- the hormone therapy is an aromatase inhibitor.
- the aromatase inhibitor is letrozole.
- the aromatase inhibitor is anastrozole.
- the aromatase inhibitor is exemestane.
- the subject has been previously treated with a checkpoint inhibitor.
- the checkpoint inhibitor is an inhibitor of PD-l, PD-L1, CTLA-4, PD-L2, LAG3, Tim3, 2B4, A2aR, ID02, B7-H3, B7-H4, BTLA, CD2, CD20, CD27, CD28, CD30, CD33, CD40, CD52, CD70, CD80, CD86, CD112, CD137, CD 160, CD226, CD276, DR3, OX-40, GAL9, GITR, ICOS, HVEM, IDOI, KIR, LAIR, LIGHT, MARCO, PS, SLAM, TIGIT, VISTA, and/or VTCN1.
- the checkpoint inhibitor is an inhibitor of PD-l, PD-L1 and/or CTLA-4. In some embodiments, the checkpoint inhibitor is an inhibitor of PD-L In some embodiments, the checkpoint inhibitor is selected from the group consisting of nivolumab (OPDIVO®, BMS-936558, MDX-1106 or MK-34775), pembrolizumab (KEYTRUDA®, MK-3475), pidilizumab (CT-011) and cemiplimab
- the checkpoint inhibitor is an inhibitor of PD-L1.
- the checkpoint inhibitor is selected from the group consisting of atezolizumab (TECENTRIQ®, MPDL3280A), avelumab (BAVENCIO®), durvalumab and BMS-936559.
- the checkpoint inhibitor is an inhibitor of CTLA-4.
- the checkpoint inhibitor is selected from the group consisting of ipilimumab and tremelimumab.
- the subject has been previously treated with doxorubicin.
- the subject has been previously treated with paclitaxel.
- the subject has previously undergone surgery or radiation therapy for the endometrial cancer. In some embodiments, the subject has previously undergone surgery for the endometrial cancer. In some embodiments, the subject has previously undergone radiation therapy for the endometrial cancer. In some embodiments, the endometrial cancer is an advanced stage cancer. In some embodiments, the advanced stage cancer is a stage 3 or 4 cancer. In some embodiments, the advanced stage cancer is a metastatic cancer. In some embodiments, the endometrial cancer is a recurrent cancer. In some embodiments, the subject received prior treatment with standard of care therapy for the cancer and failed the prior treatment. In a particular embodiment, the subject is a human.
- At least 0.1%, at least 1%, at least 2%, at least 3%, at least 4%, at least 5%, at least 6%, at least 7%, at least 8%, at least 9%, at least 10%, at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 60%, at least 70%, or at least 80% of the endometrial cancer cells from the subject express TF.
- the percentage of cells that express TF is determined using immunohistochemistry (IHC).
- the percentage of cells that express TF is determined using flow cytometry.
- the percentage of cells that express TF is determined using an enzyme-linked immunosorbent assay (ELISA).
- Esophageal cancer is the sixth leading cause of cancer-related mortality worldwide due to its overall poor prognosis.
- the global age-standardized incidence rate of esophageal squamous cell carcinoma (ESCC) is 1.4-13.6 per 100,000 people.
- Esophageal cancer is estimated to be responsible for 15,690 deaths and 16,940 new cases in the Einited States in 2016. The majority of patients present with locally advanced or systemic disease and outcomes remain poor despite advances in treatment. More effective treatments for these patients with locally advanced or systemic disease are urgently needed.
- the invention provides methods for treating esophageal cancer with an antibody- drug conjugate described herein.
- the antibody-drug conjugates described herein are for use in a method of treating esophageal cancer in a subject.
- the antibody-drug conjugate is tisotumab vedotin.
- the subject has not been previously treated for the esophageal cancer.
- the subject received at least one previous treatment for the esophageal cancer.
- the subject received prior systemic therapy for the esophageal cancer.
- the subject experienced disease progression on or after the systemic therapy.
- the subject received no more than 3 rounds of prior systemic therapy. In some embodiments, the subject received 1, 2 or 3 rounds of prior systemic therapy. In some embodiments, the subject received 1 round of prior systemic therapy. In some embodiments, the subject received 2 rounds of prior systemic therapy. In some embodiments, the subject received 3 rounds of prior systemic therapy. In some embodiments, the subject has been previously treated with one or more agents selected from the group consisting of a platinum- based therapy and a checkpoint inhibitor. In some embodiments, the subject has been previously treated with a platinum-based therapy.
- the platinum-based therapy is selected from the group consisting of carboplatin, cisplatin, oxaliplatin, nedaplatin, triplatin tetranitrate, phenanthriplatin, picoplatin and satraplatin.
- the platinum-based therapy is carboplatin.
- the platinum-based therapy is cisplatin.
- the platinum-based therapy is oxaliplatin.
- the platinum-based therapy is nedaplatin.
- the platinum-based therapy is triplatin tetranitrate.
- the platinum-based therapy is phenanthriplatin.
- the platinum-based therapy is picoplatin. In some embodiments, the platinum-based therapy is satraplatin. In some embodiments, the subject has been previously treated with a checkpoint inhibitor. In some embodiments, the checkpoint inhibitor is an inhibitor of PD-l, PD-L1, CTLA-4, PD-L2, LAG3, Tim3, 2B4, A2aR, ID02, B7-H3, B7-H4, BTLA, CD2, CD20, CD27, CD28, CD30, CD33, CD40, CD52, CD70, CD80, CD86, CD112, CD137, CD 160, CD226, CD276, DR3, OX-40, GAL9, GITR, ICOS, HVEM, IDOI, KIR, LAIR, LIGHT, MARCO, PS, SLAM, TIGIT, VISTA, and/or VTCN1.
- the checkpoint inhibitor is an inhibitor of PD-l, PD-L1, CTLA-4, PD-L2, LAG3, Tim3, 2B4, A2
- the checkpoint inhibitor is an inhibitor of PD-l, PD-L1 and/or CTLA-4. In some embodiments, the checkpoint inhibitor is an inhibitor of PD-L In some embodiments, the checkpoint inhibitor is selected from the group consisting of nivolumab (OPDIVO®, BMS-936558, MDX-1106 or MK-34775), pembrolizumab
- the checkpoint inhibitor is an inhibitor of PD-L1.
- the checkpoint inhibitor is selected from the group consisting of atezolizumab (TECENTRIQ®, MPDL3280A), avelumab (BAVENCIO®), durvalumab and BMS-936559.
- the checkpoint inhibitor is an inhibitor of CTLA-4.
- the checkpoint inhibitor is selected from the group consisting of ipilimumab and tremelimumab.
- the subject has been previously treated with one or more agents selected from the group consisting of ramucirumab, paclitaxel, 5-fluorouracil, docetaxel, irinotecan, capecitabine and trastuzumab.
- the subject has been previously treated with ramucirumab.
- the subject has been previously treated with paclitaxel.
- the subject has been previously treated with 5- fluorouracil.
- the subject has been previously treated with docetaxel.
- the subject has been previously treated with irinotecan. In some embodiments, the subject has been previously treated with capecitabine. In some embodiments, the subject has been previously treated with trastuzumab. In some
- the subject has previously undergone surgery, radiation therapy, or endoscopic mucosal resection for the esophageal cancer. In some embodiments, the subject has previously undergone surgery for the esophageal cancer. In some embodiments, the subject has previously undergone radiation therapy for the esophageal cancer. In some embodiments, the subject has previously undergone endoscopic mucosal resection for the esophageal cancer. In some embodiments, the esophageal cancer is an advanced stage cancer. In some embodiments, the advanced stage cancer is a stage 3 or 4 cancer. In some embodiments, the advanced stage cancer is a metastatic cancer. In some embodiments, the esophageal cancer is a recurrent cancer. In some embodiments, the subject received prior treatment with standard of care therapy for the cancer and failed the prior treatment. In a particular embodiment, the subject is a human.
- the percentage of cells that express TF is determined using immunohistochemistry (IHC).
- the percentage of cells that express TF is determined using flow cytometry.
- the percentage of cells that express TF is determined using an enzyme-linked immunosorbent assay (ELISA).
- Prostate cancer is the most common non-cutaneous malignancy in males, with a projected 161,360 incident cases and 26,730 deaths estimated in the United States in 2017 alone.
- Curative modalities for localized prostate cancer include surgery and/or radiation therapy, with or without androgen deprivation therapy. While contemporary treatment methods, such as intensity-modulated radiotherapy, are used to deliver radiation with high accuracy, defining the position and the extent of the tumor is still quite challenging. Other issues in the treatment of the radiotherapy patient include the choice of the radiotherapy technique (hypo- or standard fractionation) and the use and length of androgen deprivation therapy. More effective treatments are needed, especially for patients with advanced and metastatic prostate cancer.
- the invention provides methods for treating prostate cancer with an antibody-drug conjugate described herein.
- the antibody-drug conjugates described herein are for use in a method of treating prostate cancer in a subject.
- the antibody-drug conjugate is tisotumab vedotin.
- the subject has not been previously treated for the prostate cancer.
- the subject received at least one previous treatment for the prostate cancer.
- the subject received prior systemic therapy for the prostate cancer.
- the subject experienced disease progression on or after the systemic therapy.
- the subject received no more than 3 rounds of prior systemic therapy.
- the subject received 1, 2 or 3 rounds of prior systemic therapy.
- the subject received 1 round of prior systemic therapy. In some embodiments, the subject received 2 rounds of prior systemic therapy. In some embodiments, the subject received 3 rounds of prior systemic therapy.
- the prostate cancer is castration-resistant prostate cancer. In some embodiments, the subject has experienced bone metastases. In some embodiments, the prostate cancer has metastasized to a bone. In some embodiments, the subject has been previously treated with one or more agents selected from the group consisting of androgen deprivation therapy, a luteinizing hormone-releasing hormone agonist, a luteinizing hormone-releasing hormone antagonist, a CYP17 inhibitor, and an anti androgen. In some embodiments, the subject has been previously treated with androgen deprivation therapy.
- the subject has been previously treated with a luteinizing hormone-releasing hormone agonist.
- the luteinizing hormone-releasing hormone agonist is selected from the group consisting of leuprolide, goserelin, triptorelin and histrelin.
- the luteinizing hormone-releasing hormone agonist is leuprolide.
- the luteinizing hormone-releasing hormone agonist is goserelin.
- the luteinizing hormone-releasing hormone agonist is triptorelin.
- the luteinizing hormone-releasing hormone agonist is histrelin.
- the subject has been previously treated with a luteinizing hormone-releasing hormone antagonist.
- the luteinizing hormone-releasing hormone antagonist is degarelix.
- the subject has been previously treated with a CYP17 inhibitor.
- the CYP17 inhibitor is abiraterone.
- the subject has been previously treated with an anti -androgen.
- the anti-androgen is selected from the group consisting of flutamide, bicalutamide, nilutamide, enzalutamide and apalutamide.
- the anti -androgen is flutamide.
- the anti-androgen is bicalutamide.
- the anti-androgen is nilutamide.
- the anti-androgen is enzalutamide. In some embodiments, the anti -androgen is apalutamide. In some embodiments, the subject has been previously treated with one or more agents selected from the group consisting of docetaxel, prednisone and cabazitaxel. In some embodiments, the subject has been previously treated with docetaxel. In some embodiments, the subject has been previously treated with prednisone. In some embodiments, the subject has been previously treated with cabazitaxel. In some embodiments, the subject has previously undergone surgery or radiation therapy for the prostate cancer. In some embodiments, the subject has previously undergone surgery for the prostate cancer. In some embodiments, the subject has previously undergone radiation therapy for the prostate cancer. In some embodiments, the prostate cancer is an advanced stage cancer. In some
- the advanced stage cancer is a stage 3 or 4 cancer. In some embodiments, the advanced stage cancer is a metastatic cancer. In some embodiments, the prostate cancer is a recurrent cancer. In some embodiments, the subject received prior treatment with standard of care therapy for the cancer and failed the prior treatment. In a particular embodiment, the subject is a human.
- At least 0.1%, at least 1%, at least 2%, at least 3%, at least 4%, at least 5%, at least 6%, at least 7%, at least 8%, at least 9%, at least 10%, at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 60%, at least 70%, or at least 80% of the prostate cancer cells from the subject express TF.
- the percentage of cells that express TF is determined using immunohistochemistry (IHC).
- the percentage of cells that express TF is determined using flow cytometry.
- the percentage of cells that express TF is determined using an enzyme- linked immunosorbent assay (ELISA).
- An anti-TF antibody-drug conjugate or antigen-binding fragment thereof described herein can be administered by any suitable route and mode. Suitable routes of administering antibody-drug conjugate of the present invention are well known in the art and may be selected by those of ordinary skill in the art. In one embodiment, the antibody-drug conjugate is administered parenterally.
- Parenteral administration refers to modes of administration other than enteral and topical administration, usually by injection, and include epidermal, intravenous, intramuscular, intraarterial, intrathecal, intracapsular, intraorbital, intracardiac, intradermal, intraperitoneal, intratendinous, transtracheal, subcutaneous, subcuticular, intraarticular, subcapsular, subarachnoid, intraspinal, intracranial, intrathoracic, epidural and intrastemal injection and infusion.
- the route of administration of an anti-TF antibody-drug conjugate or antigen-binding fragment described herein is intravenous injection or infusion.
- the route of administration of an anti-TF antibody-drug conjugate or antigen-binding fragment described herein is intravenous infusion.
- the present invention provides for methods of treating a subject with colorectal cancer, non-small cell lung cancer, pancreatic cancer, head and neck cancer, bladder cancer, endometrial cancer, esophageal cancer or prostate cancer as described herein with a particular dose of an anti-TF antibody-drug conjugate or antigen-binding fragment thereof as described herein, wherein the subject is administered the antibody-drug conjugate or antigen-binding fragment thereof as described herein with a particular frequency.
- an anti-TF antibody-drug conjugate or antigen-binding fragment thereof as described herein is administered to the subject at a dose ranging from about 0.9 mg/kg to about 2.1 mg/kg of the subject’s body weight.
- the dose is about 0.9 mg/kg, about 1.0 mg/kg, about 1.1 mg/kg, about 1.2 mg/kg, about 1.3 mg/kg, about l.4mg/kg, about 1.5 mg/kg, about 1.6 mg/kg, about 1.7 mg/kg, about 1.8 mg/kg, about 1.9 mg/kg, about 2.0 mg/kg or about 2.1 mg/kg.
- the dose is about 2.0 mg/kg.
- the dose is 0.9 mg/kg, 1.0 mg/kg, 1.1 mg/kg, 1.2 mg/kg, 1.3 mg/kg, l.4mg/kg, 1.5 mg/kg, 1.6 mg/kg, 1.7 mg/kg, 1.8 mg/kg, 1.9 mg/kg, 2.0 mg/kg or 2.1 mg/kg.
- the dose is 2.0 mg/kg.
- the dose is 2.0 mg/kg and the anti-TF antibody-drug conjugate is tisotumab vedotin.
- the dose of the anti-TF antibody-drug conjugate administered is the amount that would be administered if the subject weighed 100 kg.
- the dose of the anti-TF antibody-drug conjugate administered is 200 mg.
- an anti-TF antibody-drug conjugate or antigen-binding fragment thereof as described herein is administered to the subject once about every 1 to 4 weeks. In certain embodiments, an anti- TF antibody-drug conjugate or antigen-binding fragment thereof as described herein is administered once about every 1 week, once about every 2 weeks, once about every 3 weeks or once about every 4 weeks. In one embodiment, an anti-TF antibody-drug conjugate or antigen-binding fragment thereof as described herein is administered once about every 3 weeks. In one embodiment, an anti-TF antibody-drug conjugate or antigen-binding fragment thereof as described herein is administered once every 3 weeks.
- the dose is about 0.9 mg/kg and is administered once about every 1 week. In some embodiments, the dose is about 0.9 mg/kg and is administered once about every 2 weeks. In some embodiments, the dose is about 0.9 mg/kg and is administered once about every 3 weeks. In some embodiments, the dose is about 0.9 mg/kg and is administered once about every 4 weeks. In some embodiments, the dose is about 1.0 mg/kg and is administered once about every 1 week. In some embodiments, the dose is about 1.0 mg/kg and is administered once about every 2 weeks. In some embodiments, the dose is about 1.0 mg/kg and is administered once about every 3 weeks. In some embodiments, the dose is about 1.0 mg/kg and is administered once about every 4 weeks.
- the dose is about 1.1 mg/kg and is administered once about every 1 week. In some embodiments, the dose is about 1.1 mg/kg and is administered once about every 2 weeks. In some embodiments, the dose is about 1.1 mg/kg and is administered once about every 3 weeks. In some embodiments, the dose is about 1.1 mg/kg and is administered once about every 4 weeks. In some
- the dose is about 1.2 mg/kg and is administered once about every 1 week. In some embodiments, the dose is about 1.2 mg/kg and is administered once about every 2 weeks. In some embodiments, the dose is about 1.2 mg/kg and is administered once about every 3 weeks. In some embodiments, the dose is about 1.2 mg/kg and is administered once about every 4 weeks. In some embodiments, the dose is about 1.3 mg/kg and is administered once about every 1 week. In some embodiments, the dose is about 1.3 mg/kg and is administered once about every 2 weeks. In some embodiments, the dose is about 1.3 mg/kg and is administered once about every 3 weeks. In some embodiments, the dose is about 1.3 mg/kg and is administered once about every 4 weeks. In some embodiments, the dose is about 1.4 mg/kg and is administered once about every 1 week. In some embodiments, the dose is about 1.4 mg/kg and is administered once about every 2 weeks. In some embodiments, the dose is about 1.4 mg/kg and is administered once about every 2 weeks. In some embodiments, the
- the dose is about 1.4 mg/kg and is administered once about every 3 weeks. In some embodiments, the dose is about 1.4 mg/kg and is administered once about every 4 weeks. In some embodiments, the dose is about 1.5 mg/kg and is administered once about every 1 week. In some embodiments, the dose is about 1.5 mg/kg and is administered once about every 2 weeks. In some embodiments, the dose is about 1.5 mg/kg and is administered once about every 3 weeks. In some embodiments, the dose is about 1.5 mg/kg and is administered once about every 4 weeks. In some embodiments, the dose is about 1.6 mg/kg and is administered once about every 1 week. In some embodiments, the dose is about 1.6 mg/kg and is administered once about every 2 weeks. In some embodiments, the dose is about 1.6 mg/kg and is administered once about every 3 weeks. In some embodiments, the dose is about 1.6 mg/kg and is administered once about every 4 weeks. In some embodiments, the dose is about 1.6 mg/kg and is administered once about every 1 week. In some embodiments, the dose is about
- the dose is about 1.7 mg/kg and is administered once about every 1 week. In some embodiments, the dose is about 1.7 mg/kg and is administered once about every 2 weeks. In some embodiments, the dose is about 1.7 mg/kg and is administered once about every 3 weeks. In some embodiments, the dose is about 1.7 mg/kg and is administered once about every 4 weeks. In some embodiments, the dose is about 1.8 mg/kg and is administered once about every 1 week. In some embodiments, the dose is about 1.8 mg/kg and is administered once about every 2 weeks. In some embodiments, the dose is about 1.8 mg/kg and is administered once about every 3 weeks. In some embodiments, the dose is about 1.8 mg/kg and is administered once about every 4 weeks. In some embodiments, the dose is about 1.9 mg/kg and is administered once about every 1 week. In some embodiments, the dose is about 1.9 mg/kg and is administered once about every 2 weeks. In some embodiments, the dose is about 1.9 mg/kg and is administered once about every 2 weeks. In some embodiments, the
- the dose is about 1.9 mg/kg and is administered once about every 3 weeks. In some embodiments, the dose is about 1.9 mg/kg and is administered once about every 4 weeks. In some embodiments, the dose is about 2.0 mg/kg and is administered once about every 1 week. In some embodiments, the dose is about 2.0 mg/kg and is administered once about every 2 weeks. In some embodiments, the dose is about 2.0 mg/kg and is administered once about every 3 weeks. In some embodiments, the dose is about 2.0 mg/kg and is administered once about every 4 weeks. In some embodiments, the dose is about 2.1 mg/kg and is administered once about every 1 week. In some embodiments, the dose is about 2.1 mg/kg and is administered once about every 2 weeks. In some embodiments, the dose is about 2.1 mg/kg and is administered once about every 3 weeks. In some embodiments, the dose is about 2.1 mg/kg and is administered once about every 4 weeks. In some embodiments, the dose is about 1.9 mg/kg and is administered once about every 1 week. In some embodiments, the dose is about
- the dose is 0.9 mg/kg and is administered once about every 1 week. In some embodiments, the dose is 0.9 mg/kg and is administered once about every 2 weeks. In some embodiments, the dose is 0.9 mg/kg and is administered once about every 3 weeks. In some embodiments, the dose is 0.9 mg/kg and is administered once about every 4 weeks. In some embodiments, the dose is 1.0 mg/kg and is administered once about every 1 week. In some embodiments, the dose is 1.0 mg/kg and is administered once about every 2 weeks. In some embodiments, the dose is 1.0 mg/kg and is administered once about every 3 weeks. In some embodiments, the dose is 1.0 mg/kg and is administered once about every 4 weeks.
- the dose is 1.1 mg/kg and is administered once about every 1 week. In some embodiments, the dose is 1.1 mg/kg and is administered once about every 2 weeks. In some embodiments, the dose is 1.1 mg/kg and is administered once about every 3 weeks. In some embodiments, the dose is 1.1 mg/kg and is administered once about every 4 weeks. In some embodiments, the dose is 1.2 mg/kg and is administered once about every 1 week. In some embodiments, the dose is 1.2 mg/kg and is administered once about every 2 weeks. In some embodiments, the dose is 1.2 mg/kg and is administered once about every 3 weeks. In some embodiments, the dose is 1.2 mg/kg and is administered once about every 4 weeks.
- the dose is 1.3 mg/kg and is administered once about every 1 week. In some embodiments, the dose is 1.3 mg/kg and is administered once about every 2 weeks. In some embodiments, the dose is 1.3 mg/kg and is administered once about every 3 weeks. In some embodiments, the dose is 1.3 mg/kg and is administered once about every 4 weeks. In some embodiments, the dose is 1.4 mg/kg and is administered once about every 1 week. In some embodiments, the dose is 1.4 mg/kg and is administered once about every 2 weeks. In some embodiments, the dose is 1.4 mg/kg and is administered once about every 3 weeks. In some embodiments, the dose is 1.4 mg/kg and is administered once about every 4 weeks.
- the dose is 1.5 mg/kg and is administered once about every 1 week. In some embodiments, the dose is 1.5 mg/kg and is administered once about every 2 weeks. In some embodiments, the dose is 1.5 mg/kg and is administered once about every 3 weeks. In some embodiments, the dose is 1.5 mg/kg and is administered once about every 4 weeks. In some embodiments, the dose is 1.6 mg/kg and is administered once about every 1 week. In some embodiments, the dose is 1.6 mg/kg and is administered once about every 2 weeks. In some embodiments, the dose is 1.6 mg/kg and is administered once about every 3 weeks. In some embodiments, the dose is 1.6 mg/kg and is administered once about every 4 weeks.
- the dose is 1.7 mg/kg and is administered once about every 1 week. In some embodiments, the dose is 1.7 mg/kg and is administered once about every 2 weeks. In some embodiments, the dose is 1.7 mg/kg and is administered once about every 3 weeks. In some embodiments, the dose is 1.7 mg/kg and is administered once about every 4 weeks. In some embodiments, the dose is 1.8 mg/kg and is administered once about every 1 week. In some embodiments, the dose is 1.8 mg/kg and is administered once about every 2 weeks. In some embodiments, the dose is 1.8 mg/kg and is administered once about every 3 weeks. In some embodiments, the dose is 1.8 mg/kg and is administered once about every 4 weeks.
- the dose is 1.9 mg/kg and is administered once about every 1 week. In some embodiments, the dose is 1.9 mg/kg and is administered once about every 2 weeks. In some embodiments, the dose is 1.9 mg/kg and is administered once about every 3 weeks. In some embodiments, the dose is 1.9 mg/kg and is administered once about every 4 weeks. In some embodiments, the dose is 2.0 mg/kg and is administered once about every 1 week. In some embodiments, the dose is 2.0 mg/kg and is administered once about every 2 weeks. In some embodiments, the dose is 2.0 mg/kg and is administered once about every 3 weeks. In some embodiments, the dose is 2.0 mg/kg and is administered once about every 4 weeks.
- the dose is 2.1 mg/kg and is administered once about every 1 week. In some embodiments, the dose is 2.1 mg/kg and is administered once about every 2 weeks. In some embodiments, the dose is 2.1 mg/kg and is administered once about every 3 weeks. In some embodiments, the dose is 2.1 mg/kg and is administered once about every 4 weeks. In some embodiments, the dose is 2.0 mg/kg and is administered once about every 3 weeks ( e.g ., ⁇ 3 days). In some embodiments, the dose is 2.0 mg/kg and is administered once every 3 weeks. In some embodiments, the dose is 2.0 mg/kg and is administered once every 3 weeks and the antibody-drug conjugate is tisotumab vedotin.
- the dose is 2.0 mg/kg and is administered once every 3 weeks and the antibody-drug conjugate is tisotumab vedotin and the dose is decreased to 1.3 mg/kg if one or more adverse events occur. In some embodiments, the dose is 1.3 mg/kg and is administered once every 3 weeks and the antibody-drug conjugate is tisotumab vedotin and the dose is decreased to 0.9 mg/kg if one or more adverse events occur. In some embodiments, for a subject weighing more than 100 kg, the dose of the anti-TF antibody-drug conjugate administered is the amount that would be administered if the subject weighed 100 kg. In some embodiments, for a subject weighing more than 100 kg, the dose of the anti-TF antibody-drug conjugate administered is 200 mg.
- an anti-TF antibody-drug conjugate or antigen-binding fragment thereof as described herein is administered to the subject at a flat dose ranging from about 50 mg to about 200 mg such as at a flat dose of about 50 mg or a flat dose of about 60 mg or a flat dose of about 70 mg or a flat dose of about 80 mg or a flat dose of about 90 mg or a flat dose of about 100 mg or a flat dose of about 110 mg or a flat dose of about 120 mg or a flat dose of about 130 mg or a flat dose of about 140 mg or a flat dose of about 150 mg or a flat dose of about 160 mg or a flat dose of about 170 mg or a flat dose of about 180 mg or a flat dose of about 190 mg or a flat dose of about 200 mg.
- the flat dose is administered to the subject once about every 1 to 4 weeks. In certain embodiments, the flat dose is administered to the subject once about every 1 week, once about every 2 weeks, once about every 3 weeks or once about every 4 weeks. In some embodiments, the flat dose is administered to the subject once about every 3 weeks ( e.g ., ⁇ 3 days). In some embodiments, the flat dose is
- the flat dose is administered to the subject once every 3 weeks and the antibody-drug conjugate is tisotumab vedotin.
- an anti-TF antibody-drug conjugate or antigen-binding fragment thereof as described herein is administered to the subject at a flat dose ranging from 50 mg to 200 mg such as at a flat dose of 50 mg or a flat dose of 60 mg or a flat dose of 70 mg or a flat dose of 80 mg or a flat dose of 90 mg or a flat dose of 100 mg or a flat dose of 110 mg or a flat dose of 120 mg or a flat dose of 130 mg or a flat dose of 140 mg or a flat dose of 150 mg or a flat dose of 160 mg or a flat dose of 170 mg or a flat dose of 180 mg or a flat dose of 190 mg or a flat dose of 200 mg.
- the flat dose is administered to the subject once about every 1 to 4 weeks. In certain embodiments, the flat dose is administered to the subject once about every 1 week, once about every 2 weeks, once about every 3 weeks or once about every 4 weeks. In some embodiments, the flat dose is administered to the subject once about every 3 weeks (e.g., ⁇ 3 days). In some embodiments, the flat dose is administered to the subject once every 3 weeks. In some embodiments, the flat dose is administered to the subject once every 3 weeks and the antibody-drug conjugate is tisotumab vedotin.
- a method of treatment or use or product for use described herein further comprises the administration of one or more additional therapeutic agents.
- the one or more additional therapeutic agents are administered simultaneously with an anti-TF antibody-drug conjugate or antigen-binding fragment thereof as described herein, such as tisotumab vedotin.
- the one or more additional therapeutic agents and an anti-TF antibody-drug conjugate or antigen-binding fragment thereof as described herein are administered sequentially.
- simultaneous means that the anti-TF antibody-drug conjugate and the one or more additional therapeutic agents are administered to the subject less than one hour apart, such as less than about 30 minutes apart, less than about 15 minutes apart, less than about 10 minutes apart or less than about 5 minutes apart.
- sequential administration means that the anti-TF antibody-drug conjugate and the one or more additional therapeutic agents are administered a least 1 hour apart, at least 2 hours apart, at least 3 hours apart, at least 4 hours apart, at least 5 hours apart, at least 6 hours apart, at least 7 hours apart, at least 8 hours apart, at least 9 hours apart, at least 10 hours apart, at least 11 hours apart, at least 12 hours apart, at least 13 hours apart, at least 14 hours apart, at least 15 hours apart, at least 16 hours apart, at least 17 hours apart, at least 18 hours apart, at least 19 hours apart, at least 20 hours apart, at least 21 hours apart, at least 22 hours apart, at least 23 hours apart, at least 24 hours apart, at least 2 days apart, at least 3 days apart, at least 4 days apart, at least 5 days apart, at least 5 days apart, at least 7 days apart, at least 2 weeks apart, at least 3 weeks apart or at least 4 weeks apart.
- a method of treating colorectal cancer, non-small cell lung cancer, pancreatic cancer, head and neck cancer, bladder cancer, endometrial cancer, esophageal cancer or prostate cancer with an anti-TF antibody-drug conjugate or antigen-binding fragment thereof as described herein, such as e.g. , tisotumab vedotin, results in an anti-TF antibody-drug conjugate or antigen-binding fragment thereof as described herein, such as e.g. , tisotumab vedotin, results in an anti-TF antibody-drug conjugate or antigen-binding fragment thereof as described herein, such as e.g. , tisotumab vedotin, results in an anti-TF antibody-drug conjugate or antigen-binding fragment thereof as described herein, such as e.g. , tisotumab vedotin, results in an anti-TF antibody-drug conjugate
- the one or more therapeutic effects is the size of the tumor derived from the cancer (e.g, colorectal cancer, non-small cell lung cancer, pancreatic cancer, head and neck cancer, bladder cancer, endometrial cancer, esophageal cancer or prostate cancer), the objective response rate, the duration of response, the time to response, progression free survival, overall survival, or any combination thereof.
- the one or more therapeutic effects is the size of the tumor derived from the cancer.
- the one or more therapeutic effects is decreased tumor size.
- the one or more therapeutic effects is stable disease.
- the one or more therapeutic effects is partial response.
- the one or more therapeutic effects is complete response. In one embodiment, the one or more therapeutic effects is the objective response rate. In one embodiment, the one or more therapeutic effects is the duration of response. In one embodiment, the one or more therapeutic effects is the time to response. In one embodiment, the one or more therapeutic effects is progression free survival. In one embodiment, the one or more therapeutic effects is overall survival. In one embodiment, the one or more therapeutic effects is cancer regression. In one embodiment, the one or more therapeutic effects is a reduction in prostate specific antigen level.
- response to treatment with an anti-TF antibody-drug conjugate or antigen-binding fragment thereof as described herein may include the following criteria (RECIST Criteria 1.1):
- the effectiveness of treatment with an anti-TF antibody-drug conjugate or antigen-binding fragment thereof described herein, such as e.g ., tisotumab vedotin is assessed by measuring the objective response rate.
- the objective response rate is the proportion of patients with tumor size reduction of a predefined amount and for a minimum period of time.
- the objective response rate is based upon RECIST vl .1.
- the objective response rate is at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least about 45%, at least about 50%, at least about 60%, at least about 70%, or at least about 80%.
- the objective response rate is at least about 20%-80%. In one embodiment, the objective response rate is at least about 30%-80%. In one embodiment, the objective response rate is at least about 40%-80%. In one embodiment, the objective response rate is at least about 50%-80%. In one embodiment, the objective response rate is at least about 60%-80%.
- the objective response rate is at least about 70%-80%. In one embodiment, the objective response rate is at least about 70%-80%. In one
- the objective response rate is at least about 80%. In one embodiment, the objective response rate is at least about 85%. In one embodiment, the objective response rate is at least about 90%. In one embodiment, the objective response rate is at least about 95%. In one embodiment, the objective response rate is at least about 98%. In one embodiment, the objective response rate is at least about 99%. In one embodiment, the objective response rate is at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 60%, at least 70%, or at least 80%. In one embodiment, the objective response rate is at least 20%-80%. In one embodiment, the objective response rate is at least 30%- 80%. In one embodiment, the objective response rate is at least 40%-80%. In one
- the objective response rate is at least 50%-80%. In one embodiment, the objective response rate is at least 60%-80%. In one embodiment, the objective response rate is at least 70%-80%. In one embodiment, the objective response rate is at least 80%. In one embodiment, the objective response rate is at least 85%. In one embodiment, the objective response rate is at least 90%. In one embodiment, the objective response rate is at least 95%. In one embodiment, the objective response rate is at least 98%. In one embodiment, the objective response rate is at least 99%. In one embodiment, the objective response rate is 100%.
- response to treatment with an anti-TF antibody-drug conjugate or antigen-binding fragment thereof described herein, such as e.g ., tisotumab vedotin is assessed by measuring the size of a tumor derived from the cancer (e.g, colorectal cancer, non-small cell lung cancer, pancreatic cancer, head and neck cancer, bladder cancer, endometrial cancer, esophageal cancer or prostate cancer).
- a tumor derived from the cancer e.g, colorectal cancer, non-small cell lung cancer, pancreatic cancer, head and neck cancer, bladder cancer, endometrial cancer, esophageal cancer or prostate cancer.
- the size of a tumor derived from the cancer is reduced by at least about 10%, at least about 15%, at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least about 45%, at least about 50%, at least about 60%, at least about 70%, or at least about 80% relative to the size of the tumor derived from the cancer before administration of the anti-TF antibody-drug conjugate.
- the size of a tumor derived from the cancer is reduced by at least aboutl0%-80%. In one embodiment, the size of a tumor derived from the cancer is reduced by at least about 20%-80%. In one embodiment, the size of a tumor derived from the cancer is reduced by at least about 30%-80%.
- the size of a tumor derived from the cancer is reduced by at least about 40%-80%. In one embodiment, the size of a tumor derived from the cancer is reduced by at least about 50%-80%. In one embodiment, the size of a tumor derived from the cancer is reduced by at least about 60%-80%. In one
- the size of a tumor derived from the cancer is reduced by at least about 70%- 80%. In one embodiment, the size of a tumor derived from the cancer is reduced by at least about 80%. In one embodiment, the size of a tumor derived from the cancer is reduced by at least about 85%. In one embodiment, the size of a tumor derived from the cancer is reduced by at least about 90%. In one embodiment, the size of a tumor derived from the cancer is reduced by at least about 95%. In one embodiment, the size of a tumor derived from the cancer is reduced by at least about 98%. In one embodiment, the size of a tumor derived from the cancer is reduced by at least about 99%.
- the size of a tumor derived from the cancer is reduced by at least 10%, at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 60%, at least 70%, or at least 80% relative to the size of the tumor derived from the cancer before administration of the anti-TF antibody-drug conjugate.
- the size of a tumor derived from the cancer is reduced by at least l0%-80%. In one embodiment, the size of a tumor derived from the cancer is reduced by at least 20%-80%. In one embodiment, the size of a tumor derived from the cancer is reduced by at least 30%-80%.
- the size of a tumor derived from the cancer is reduced by at least 40%-80%. In one embodiment, the size of a tumor derived from the cancer is reduced by at least 50%-80%. In one embodiment, the size of a tumor derived from the cancer is reduced by at least 60%-80%. In one embodiment, the size of a tumor derived from the cancer is reduced by at least 70%-80%. In one embodiment, the size of a tumor derived from the cancer is reduced by at least 80%. In one embodiment, the size of a tumor derived from the cancer is reduced by at least 85%. In one embodiment, the size of a tumor derived from the cancer is reduced by at least 90%. In one embodiment, the size of a tumor derived from the cancer is reduced by at least 95%.
- the size of a tumor derived from the cancer is reduced by at least 98%. In one embodiment, the size of a tumor derived from the cancer is reduced by at least 99%. In one embodiment, the size of a tumor derived from the cancer is reduced by 100%. In one embodiment, the size of a tumor derived from the cancer is measured by magnetic resonance imaging (MRI). In one embodiment, the size of a tumor derived from the cancer is measured by computed tomography (CT). In one embodiment, the size of a tumor derived from the cancer is measured by positron emission tomography (PET). In one embodiment, the size of a tumor derived from the cancer is measured by ultrasound.
- MRI magnetic resonance imaging
- CT computed tomography
- PET positron emission tomography
- the size of a tumor derived from a colorectal cancer is measured by computed tomography (CT), positron emission tomography (PET) or magnetic resonance imaging (MRI). See Goh et al., 2014, Br. J. Radiol. 87(l034):20l308l 1.
- CT computed tomography
- PET positron emission tomography
- MRI magnetic resonance imaging
- CT computed tomography
- PET positron emission tomography
- MRI magnetic resonance imaging
- the size of a tumor derived from a pancreatic cancer is measured by computed tomography (CT), magnetic resonance imaging (MRI), ultrasound or positron emission tomography (PET). See Wolfgang et al., 2013, CA Cancer J. Clin. 63(5)318-348.
- CT computed tomography
- MRI magnetic resonance imaging
- PET positron emission tomography
- the size of a tumor derived from a bladder cancer is measured by positron emission tomography (PET).
- the size of a tumor derived from an endometrial cancer is measured by ultrasound, magnetic resonance imaging (MRI) or computed tomography (CT). See Nyen et al., 2018, Int. J. Mol. Sci. l9(8):2348. In some embodiments, the size of a tumor derived from an esophageal cancer is measured by ultrasound, computed tomography (CT) or positron emission tomography (PET). See Park and Kim, 2018, Ann. Transl. Med. 6(4):82.
- CT computed tomography
- PET positron emission tomography
- the size of a tumor derived from a prostate cancer is measured by ultrasound, magnetic resonance imaging (MRI), computed tomography (CT) or positron emission tomography (PET). See Das et al., 2018, Indian J. Tirol., 34(3): 172-179.
- response to treatment with an antibody-drug conjugate or antigen-binding fragment thereof described herein, such as e.g ., tisotumab vedotin promotes regression of a tumor derived from the cancer (e.g, colorectal cancer, non-small cell lung cancer, pancreatic cancer, head and neck cancer, bladder cancer, endometrial cancer, esophageal cancer or prostate cancer).
- a tumor derived from the cancer e.g, colorectal cancer, non-small cell lung cancer, pancreatic cancer, head and neck cancer, bladder cancer, endometrial cancer, esophageal cancer or prostate cancer.
- a tumor derived from the cancer regresses by at least about 10%, at least about 15%, at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least about 45%, at least about 50%, at least about 60%, at least about 70%, or at least about 80% relative to the size of the tumor derived from the cancer before administration of the anti-TF antibody-drug conjugate.
- a tumor derived from the cancer regresses by at least about 10% to about 80%.
- a tumor derived from the cancer regresses by at least about 20% to about 80%.
- a tumor derived from the cancer regresses by at least about 30% to about 80%.
- a tumor derived from the cancer regresses by at least about 40% to about 80%. In one embodiment, a tumor derived from the cancer regresses by at least about 50% to about 80%. In one embodiment, a tumor derived from the cancer regresses by at least about 60% to about 80%. In one embodiment, a tumor derived from the cancer regresses by at least about 70% to about 80%. In one embodiment, a tumor derived from the cancer regresses by at least about 80%. In one embodiment, a tumor derived from the cancer regresses by at least about 85%. In one embodiment, a tumor derived from the cancer regresses by at least about 90%.
- a tumor derived from the cancer regresses by at least about 95%. In one embodiment, a tumor derived from the cancer regresses by at least about 98%. In one embodiment, a tumor derived from the cancer regresses by at least about 99%. In one embodiment, a tumor derived from the cancer regresses by at least 10%, at least 15%, at least 20%, at least 25%, at least 30%, at least 35%, at least 40%, at least 45%, at least 50%, at least 60%, at least 70%, or at least 80% relative to the size of the tumor derived from the cancer before administration of the anti-TF antibody- drug conjugate. In one embodiment, a tumor derived from the cancer regresses by at least 10% to 80%.
- a tumor derived from the cancer regresses by at least 20% to 80%. In one embodiment, a tumor derived from the cancer regresses by at least 30% to 80%. In one embodiment, a tumor derived from the cancer regresses by at least 40% to 80%. In one embodiment, a tumor derived from the cancer regresses by at least 50% to 80%. In one embodiment, a tumor derived from the cancer regresses by at least 60% to 80%. In one embodiment, a tumor derived from the cancer regresses by at least 70% to 80%. In one embodiment, a tumor derived from the cancer regresses by at least 80%.
- a tumor derived from the cancer regresses by at least 85%. In one embodiment, a tumor derived from the cancer regresses by at least 90%. In one embodiment, a tumor derived from the cancer regresses by at least 95%. In one embodiment, a tumor derived from the cancer regresses by at least 98%. In one embodiment, a tumor derived from the cancer regresses by at least 99%. In one embodiment, a tumor derived from the cancer regresses by 100%. In one embodiment, regression of a tumor is determined by measuring the size of the tumor by magnetic resonance imaging (MRI). In one embodiment, regression of a tumor is determined by measuring the size of the tumor by computed tomography (CT).
- MRI magnetic resonance imaging
- CT computed tomography
- regression of a tumor is determined by measuring the size of the tumor by positron emission tomography (PET). In one embodiment, regression of a tumor is determined by measuring the size of the tumor by ultrasound. In some embodiments, regression of a tumor derived from a colorectal cancer is measured by computed tomography (CT), positron emission tomography (PET) or magnetic resonance imaging (MRI). See Goh et al., 2014, Br. J.
- regression of a tumor derived from a non-small cell lung cancer is measured by computed tomography (CT) or positron emission tomography (PET). See Aydin et al., 2013, Diagn. Interv. Radiol. l9(4):27l-8.
- regression of a tumor derived from a pancreatic cancer is measured by computed tomography (CT), magnetic resonance imaging (MRI), ultrasound or positron emission tomography (PET). See Wolfgang et al., 2013, CA Cancer J. Clin. 63(5)318-348.
- regression of a tumor derived from a head and neck cancer is measured by computed tomography (CT), magnetic resonance imaging (MRI), ultrasound or positron emission tomography (PET). See Nooij et al., 2018, Curr. Radiol. Rep. 6(l):2.
- regression of a tumor derived from a bladder cancer is measured by positron emission tomography (PET). See Vlachostergios et al., 2018, Bladder Cancer 4(3):247-259.
- regression of a tumor derived from an endometrial cancer is measured by ultrasound, magnetic resonance imaging (MRI) or computed tomography (CT). See Nyen et al., 2018, Int. J. Mol. Sci.
- regression of a tumor derived from an esophageal cancer is measured by ultrasound, computed tomography (CT) or positron emission tomography (PET). See Park and Kim, 2018, Ann. Transl. Med. 6(4):82.
- regression of a tumor derived from a prostate cancer is measured by ultrasound, magnetic resonance imaging (MRI), computed tomography (CT) or positron emission tomography (PET).
- MRI magnetic resonance imaging
- CT computed tomography
- PET positron emission tomography
- response to treatment with an anti-TF antibody-drug conjugate or antigen-binding fragment thereof described herein, such as e.g ., tisotumab vedotin, is assessed by measuring the time of progression free survival after administration of the anti-TF antibody-drug conjugate.
- the subject exhibits progression-free survival of at least about 1 month, at least about 2 months, at least about 3 months, at least about 4 months, at least about 5 months, at least about 6 months, at least about 7 months, at least about 8 months, at least about 9 months, at least about 10 months, at least about 11 months, at least about 12 months, at least about eighteen months, at least about two years, at least about three years, at least about four years, or at least about five years after administration of the anti-TF antibody-drug conjugate. In some embodiments, the subject exhibits progression-free survival of at least about 6 months after administration of the anti-TF antibody-drug conjugate.
- the subject exhibits progression-free survival of at least about one year after administration of the anti-TF antibody-drug conjugate. In some embodiments, the subject exhibits progression-free survival of at least about two years after administration of the anti- TF antibody-drug conjugate. In some embodiments, the subject exhibits progression-free survival of at least about three years after administration of the anti-TF antibody-drug conjugate. In some embodiments, the subject exhibits progression-free survival of at least about four years after administration of the anti-TF antibody-drug conjugate. In some embodiments, the subject exhibits progression-free survival of at least about five years after administration of the anti-TF antibody-drug conjugate.
- the subject exhibits progression-free survival of at least 1 month, at least 2 months, at least 3 months, at least 4 months, at least 5 months, at least 6 months, at least 7 months, at least 8 months, at least 9 months, at least 10 months, at least 11 months, at least 12 months, at least eighteen months, at least two years, at least three years, at least four years, or at least five years after administration of the anti-TF antibody-drug conjugate.
- the subject exhibits progression-free survival of at least 6 months after administration of the anti-TF antibody-drug conjugate.
- the subject exhibits progression-free survival of at least one year after administration of the anti-TF antibody-drug conjugate.
- the subject exhibits progression-free survival of at least two years after administration of the anti-TF antibody-drug conjugate. In some embodiments, the subject exhibits progression-free survival of at least three years after administration of the anti-TF antibody-drug conjugate. In some embodiments, the subject exhibits progression-free survival of at least four years after administration of the anti-TF antibody-drug conjugate. In some embodiments, the subject exhibits progression-free survival of at least five years after administration of the anti-TF antibody-drug conjugate.
- response to treatment with an anti-TF antibody-drug conjugate or antigen-binding fragment thereof described herein, such as e.g ., tisotumab vedotin, is assessed by measuring the time of overall survival after administration of the anti-TF antibody-drug conjugate.
- the subject exhibits overall survival of at least about 1 month, at least about 2 months, at least about 3 months, at least about 4 months, at least about 5 months, at least about 6 months, at least about 7 months, at least about 8 months, at least about 9 months, at least about 10 months, at least about 11 months, at least about 12 months, at least about eighteen months, at least about two years, at least about three years, at least about four years, or at least about five years after administration of the anti-TF antibody-drug conjugate. In some embodiments, the subject exhibits overall survival of at least about 6 months after administration of the anti-TF antibody-drug conjugate. In some embodiments, the subject exhibits overall survival of at least about one year after administration of the anti-TF antibody-drug conjugate.
- the subject exhibits overall survival of at least about two years after administration of the anti-TF antibody-drug conjugate. In some embodiments, the subject exhibits overall survival of at least about three years after administration of the anti-TF antibody-drug conjugate. In some embodiments, the subject exhibits overall survival of at least about four years after administration of the anti-TF antibody-drug conjugate. In some embodiments, the subject exhibits overall survival of at least about five years after administration of the anti-TF antibody-drug conjugate.
- the subject exhibits overall survival of at least 1 month, at least 2 months, at least 3 months, at least 4 months, at least 5 months, at least 6 months, at least 7 months, at least 8 months, at least 9 months, at least 10 months, at least 11 months, at least 12 months, at least eighteen months, at least two years, at least three years, at least four years, or at least five years after administration of the anti-TF antibody-drug conjugate.
- the subject exhibits overall survival of at least 6 months after administration of the anti-TF antibody-drug conjugate.
- the subject exhibits overall survival of at least one year after administration of the anti-TF antibody-drug conjugate.
- the subject exhibits overall survival of at least two years after
- the subject exhibits overall survival of at least three years after administration of the anti-TF antibody- drug conjugate. In some embodiments, the subject exhibits overall survival of at least four years after administration of the anti-TF antibody-drug conjugate. In some embodiments, the subject exhibits overall survival of at least five years after administration of the anti-TF antibody-drug conjugate.
- response to treatment with an anti-TF antibody-drug conjugate or antigen-binding fragment thereof described herein, such as e.g ., tisotumab vedotin is assessed by measuring the duration of response to the anti-TF antibody-drug conjugate after administration of the anti- TF antibody-drug conjugate.
- the duration of response to the anti-TF antibody-drug conjugate is at least about 1 month, at least about 2 months, at least about 3 months, at least about 4 months, at least about 5 months, at least about 6 months, at least about 7 months, at least about 8 months, at least about 9 months, at least about 10 months, at least about 11 months, at least about 12 months, at least about eighteen months, at least about two years, at least about three years, at least about four years, or at least about five years after administration of the anti-TF antibody-drug conjugate. In some embodiments, the duration of response to the anti-TF antibody-drug conjugate is at least about 6 months after
- the duration of response to the anti-TF antibody-drug conjugate is at least about one year after administration of the antibody-drug conjugate. In some embodiments, the duration of response to the anti- TF antibody-drug conjugate is at least about two years after administration of the antibody- drug conjugate. In some embodiments, the duration of response to the anti-TF antibody-drug conjugate is at least about three years after administration of the antibody-drug conjugate. In some embodiments, the duration of response to the anti-TF antibody-drug conjugate is at least about four years after administration of the antibody-drug conjugate.
- the duration of response to the anti-TF antibody-drug conjugate is at least about five years after administration of the antibody-drug conjugate. In some embodiments, the duration of response to the anti-TF antibody-drug conjugate is at least 1 month, at least 2 months, at least 3 months, at least 4 months, at least 5 months, at least 6 months, at least 7 months, at least 8 months, at least 9 months, at least 10 months, at least 11 months, at least 12 months, at least eighteen months, at least two years, at least three years, at least four years, or at least five years after administration of the anti-TF antibody-drug conjugate.
- the duration of response to the anti-TF antibody-drug conjugate is at least 6 months after administration of the antibody-drug conjugate. In some embodiments, the duration of response to the anti-TF antibody-drug conjugate is at least one year after administration of the antibody-drug conjugate. In some embodiments, the duration of response to the anti-TF antibody-drug conjugate is at least two years after administration of the antibody-drug conjugate. In some embodiments, the duration of response to the anti-TF antibody-drug conjugate is at least three years after administration of the antibody-drug conjugate. In some embodiments, the duration of response to the anti-TF antibody-drug conjugate is at least four years after administration of the antibody-drug conjugate. In some embodiments, the duration of response to the anti-TF antibody-drug conjugate is at least five years after administration of the antibody-drug conjugate.
- response to treatment of prostate cancer with an anti-TF antibody-drug conjugate or antigen binding fragment thereof described herein, such as e.g ., tisotumab vedotin is assessed by measuring prostate specific antigen (PSA) level in a blood sample from the subject.
- PSA prostate specific antigen
- the PSA levels are assessed based on the Prostate Cancer Clinical Trials Working Group Guidelines (PCWG2). See Scher et al., 2008, J. Clin. Oncol. 26(7): 1148-59.
- the subject exhibits a reduction in PSA level in a blood sample from the subject by at least about 5%, at least about 10%, at least about 15%, at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least about 45%, at least about 50%, at least about 60%, at least about 70%, or at least about 80% relative to the PSA level in a blood sample obtained from the subject before administration of the antibody-drug conjugate.
- a method of treating cancer e.g., colorectal cancer, non-small cell lung cancer, pancreatic cancer, head and neck cancer, bladder cancer, endometrial cancer, esophageal cancer or prostate cancer
- an anti-TF antibody-drug conjugates or antigen binding fragments thereof described herein such as e.g., tisotumab vedotin
- the subject is administered an additional therapeutic agent to eliminate or reduce the severity of the adverse event.
- the one or more adverse events the subject develops is anemia, abdominal pain, hypokalemia, hyponatremia, epistaxis, fatigue, nausea, alopecia,
- the one or more adverse events is a grade 1 or greater adverse event. In some embodiments, the one or more adverse events is a grade 2 or greater adverse event. In some embodiments, the one or more adverse events is a grade 3 or greater adverse event. In some embodiments, the one or more adverse events is a grade 1 adverse event. In some embodiments, the one or more adverse events is a grade 2 adverse event. In some embodiments, the one or more adverse events is a grade 3 adverse event. In some embodiments, the one or more adverse events is a grade 4 adverse event. In some embodiments, the one or more adverse events is a serious adverse event. In some embodiments, the one or more adverse events is
- conjunctivitis, conjunctival ulceration, and/or keratitis and the additional therapeutic agent is a preservative-free lubricating eye drop, an ocular vasoconstrictor, an antibiotic, a steroid eye drop, or any combination thereof.
- the one or more adverse events is conjunctivitis, conjunctival ulceration, and keratitis and the additional therapeutic agent is a preservative-free lubricating eye drop, an ocular vasoconstrictor, an antibiotic, a steroid eye drop, or any combination thereof.
- the one or more adverse events is conjunctivitis and keratitis and the additional therapeutic agent is a preservative-free lubricating eye drop, an ocular vasoconstrictor, an antibiotic, a steroid eye drop, or any combination thereof.
- the one or more adverse events is conjunctivitis and the additional therapeutic agent is a preservative-free lubricating eye drop, an ocular vasoconstrictor, an antibiotic, a steroid eye drop, or any combination thereof.
- the one or more adverse events is keratitis and the additional therapeutic agent is a preservative-free lubricating eye drop, an ocular vasoconstrictor, an antibiotic, a steroid eye drop, or any combination thereof.
- the subject is administered a treatment with the additional therapeutic agent to eliminate or reduce the severity of the adverse event (e.g ., conjunctivitis, conjunctival ulceration, and/or keratitis).
- the treatment is eye cooling pads (e.g. THERA PEARL Eye Mask or similar).
- the one or more adverse events is a recurrent infusion related reaction and the additional therapeutic agent is an antihistamine, acetaminophen and/or a corticosteroid.
- the one or more adverse events is neutropenia and the additional therapeutic agent is growth factor support (G-CSF).
- the subject treated with an anti-TF antibody-drug conjugates or antigen-binding fragments thereof described herein, such as e.g ., tisotumab vedotin is at risk of developing one or more adverse events.
- the subject is administered an additional therapeutic agent to prevent the development of the adverse event or to reduce the severity of the adverse event.
- the one or more adverse events the subject is at risk of developing is anemia, abdominal pain, hypokalemia, hyponatremia, epistaxis, fatigue, nausea, alopecia, conjunctivitis, constipation, decreased appetite, diarrhea, vomiting, peripheral neuropathy, general physical health deterioration, or any combination thereof.
- the one or more adverse events is a grade 1 or greater adverse event. In some embodiments, the one or more adverse events is a grade 2 or greater adverse event. In some embodiments, the one or more adverse events is a grade 3 or greater adverse event. In some embodiments, the one or more adverse events is a grade 1 adverse event. In some embodiments, the one or more adverse events is a grade 2 adverse event. In some embodiments, the one or more adverse events is a grade 3 adverse event. In some
- the one or more adverse events is a grade 4 adverse event. In some embodiments, the one or more adverse events is a grade 4 adverse event.
- the one or more adverse events is a serious adverse event.
- the one or more adverse events is conjunctivitis, conjunctival ulceration, and/or keratitis and the additional therapeutic agent is a preservative-free lubricating eye drop, an ocular vasoconstrictor, an antibiotic, a steroid eye drop, or any combination thereof.
- the one or more adverse events is conjunctivitis, conjunctival ulceration, and keratitis and the additional therapeutic agent is a preservative-free lubricating eye drop, an ocular vasoconstrictor, an antibiotic, a steroid eye drop, or any combination thereof.
- the one or more adverse events is conjunctivitis and keratitis and the additional therapeutic agent is a preservative-free lubricating eye drop, an ocular vasoconstrictor, an antibiotic, a steroid eye drop, or any combination thereof.
- the one or more adverse events is conjunctivitis and the additional therapeutic agent is a preservative-free lubricating eye drop, an ocular vasoconstrictor, an antibiotic, a steroid eye drop, or any combination thereof.
- the one or more adverse events is keratitis and the additional therapeutic agent is a preservative-free lubricating eye drop, an ocular vasoconstrictor, an antibiotic, a steroid eye drop, or any combination thereof.
- the subject is administered a treatment with the additional therapeutic agent to prevent the development of the adverse event or to reduce the severity of the adverse event (e.g ., conjunctivitis, conjunctival ulceration, and/or keratitis).
- the treatment is eye cooling pads (e.g. THERA PEARL Eye Mask or similar).
- the one or more adverse events is a recurrent infusion related reaction and the additional therapeutic agent is an antihistamine, acetaminophen and/or a corticosteroid.
- the one or more adverse events is neutropenia and the additional therapeutic agent is growth factor support (G-CSF).
- compositions e.g, pharmaceutical compositions and therapeutic formulations
- Therapeutic formulations are prepared for storage by mixing the active ingredient having the desired degree of purity with optional pharmaceutically acceptable carriers, excipients or stabilizers (Remington: The Science and Practice of Pharmacy, 20th Ed., Lippincott Williams & Wiklins, Pub., Gennaro Ed., Philadelphia, Pa. 2000).
- Acceptable carriers, excipients, or stabilizers are nontoxic to recipients at the dosages and concentrations employed, and include buffers, antioxidants including ascorbic acid, methionine, Vitamin E, sodium metabi sulfite; preservatives, isotonicifiers, stabilizers, metal complexes (e.g. Zn-protein complexes); chelating agents such as EDTA and/or non ionic surfactants.
- Buffers can be used to control the pH in a range which optimizes the therapeutic effectiveness, especially if stability is pH dependent. Buffers can be present at concentrations ranging from about 50 mM to about 250 mM.
- Suitable buffering agents for use with the present invention include both organic and inorganic acids and salts thereof. For example, citrate, phosphate, succinate, tartrate, fumarate, gluconate, oxalate, lactate, acetate.
- buffers may be comprised of histidine and trimethylamine salts such as Tris.
- Preservatives can be added to prevent microbial growth, and are typically present in a range from about 0.2%- 1.0% (w/v).
- Suitable preservatives for use with the present invention include octadecyldimethylbenzyl ammonium chloride; hexamethonium chloride; benzalkonium halides (e.g, chloride, bromide, iodide), benzethonium chloride; thimerosal, phenol, butyl or benzyl alcohol; alkyl parabens such as methyl or propyl paraben; catechol; resorcinol; cyclohexanol, 3-pentanol, and m-cresol.
- octadecyldimethylbenzyl ammonium chloride hexamethonium chloride
- benzalkonium halides e.g, chloride, bromide, iodide
- Tonicity agents can be present to adjust or maintain the tonicity of liquid in a composition.
- stabilizers When used with large, charged biomolecules such as proteins and antibodies, they are often termed “stabilizers” because they can interact with the charged groups of the amino acid side chains, thereby lessening the potential for inter and intramolecular interactions.
- Tonicity agents can be present in any amount between about 0.1% to about 25% by weight or between about 1% to about 5% by weight, taking into account the relative amounts of the other ingredients.
- tonicity agents include polyhydric sugar alcohols, trihydric or higher sugar alcohols, such as glycerin, erythritol, arabitol, xylitol, sorbitol and mannitol.
- Additional excipients include agents which can serve as one or more of the following: (1) bulking agents, (2) solubility enhancers, (3) stabilizers and (4) and agents preventing denaturation or adherence to the container wall.
- Such excipients include:
- polyhydric sugar alcohols (enumerated above); amino acids such as alanine, glycine, glutamine, asparagine, histidine, arginine, lysine, ornithine, leucine, 2-phenylalanine, glutamic acid, threonine, etc.; organic sugars or sugar alcohols such as sucrose, lactose, lactitol, trehalose, stachyose, mannose, sorbose, xylose, ribose, ribitol, myoinisitose, myoinisitol, galactose, galactitol, glycerol, cyclitols ( e.g ., inositol), polyethylene glycol; sulfur containing reducing agents, such as urea, glutathione, thioctic acid, sodium
- thioglycolate, thioglycerol, a-monothioglycerol and sodium thio sulfate low molecular weight proteins such as human serum albumin, bovine serum albumin, gelatin or other immunoglobulins
- hydrophilic polymers such as polyvinylpyrrolidone; monosaccharides (e.g., xylose, mannose, fructose, glucose; disaccharides (e.g, lactose, maltose, sucrose); trisaccharides such as raffmose; and polysaccharides such as dextrin or dextran.
- Non-ionic surfactants or detergents can be present to help solubilize the therapeutic agent as well as to protect the therapeutic protein against agitation-induced aggregation, which also permits the formulation to be exposed to shear surface stress without causing denaturation of the active therapeutic protein or antibody.
- Non-ionic surfactants are present in a range of about 0.05 mg/ml to about 1.0 mg/ml or about 0.07 mg/ml to about 0.2 mg/ml. In some embodiments, non-ionic surfactants are present in a range of about 0.001% to about 0.1% w/v or about 0.01% to about 0.1% w/v or about 0.01% to about 0.025% w/v.
- Suitable non-ionic surfactants include polysorbates (20, 40, 60, 65, 80, etc.), polyoxamers (184, 188, etc.), PLURONIC® polyols, TRITON®, polyoxyethylene sorbitan monoethers (TWEEN®-20, TWEEN®-80, etc.), lauromacrogol 400, polyoxyl 40 stearate, polyoxyethylene hydrogenated castor oil 10, 50 and 60, glycerol monostearate, sucrose fatty acid ester, methyl celluose and carboxymethyl cellulose.
- Anionic detergents that can be used include sodium lauryl sulfate, dioctyle sodium sulfosuccinate and dioctyl sodium sulfonate.
- Cationic detergents include benzalkonium chloride or benzethonium chloride.
- an anti-TF antibody-drug conjugate described herein for use in methods of treatment provided herein are described in W02015/075201.
- an anti-TF antibody-drug conjugate described herein is in a formulation comprising the anti-TF antibody drug conjugate, histidine, sucrose, and D-mannitol, wherein the formulation has a pH of about 6.0.
- an anti-TF antibody-drug conjugate described herein is in a formulation comprising the anti-TF antibody drug conjugate at a concentration of about 10 mg/ml, histidine at a concentration of about 30 mM, sucrose at a concentration of about 88 mM, D-mannitol at a concentration of about 165 mM, wherein the formulation has a pH of about 6.0.
- an anti-TF antibody- drug conjugate described herein is in a formulation comprising the anti-TF antibody drug conjugate at a concentration of 10 mg/ml, histidine at a concentration of 30 mM, sucrose at a concentration of 88 mM, D-mannitol at a concentration of 165 mM, wherein the formulation has a pH of 6.0.
- the formulation comprises tisotumab vedotin at a concentration of 10 mg/ml, histidine at a concentration of 30 mM, sucrose at a concentration of 88 mM, D-mannitol at a concentration of 165 mM, wherein the formulation has a pH of 6 0
- a formulation comprising the anti-TF antibody-conjugate described herein does not comprise a surfactant (z.e., is free of surfactant).
- the formulations In order for the formulations to be used for in vivo administration, they must be sterile.
- the formulation may be rendered sterile by filtration through sterile filtration membranes.
- the therapeutic compositions herein generally are placed into a container having a sterile access port, for example, an intravenous solution bag or vial having a stopper pierceable by a hypodermic injection needle.
- the route of administration is in accordance with known and accepted methods, such as by single or multiple bolus or infusion over a long period of time in a suitable manner, e.g, injection or infusion by subcutaneous, intravenous, intraperitoneal, intramuscular, intraarterial, intralesional or intraarticular routes, topical administration, inhalation or by sustained release or extended-release means.
- the formulation herein may also contain more than one active compound as necessary for the particular indication being treated, preferably those with complementary activities that do not adversely affect each other.
- the formulation herein may also contain more than one active compound as necessary for the particular indication being treated, preferably those with complementary activities that do not adversely affect each other.
- composition may comprise a cytotoxic agent, cytokine or growth inhibitory agent.
- cytotoxic agent cytokine or growth inhibitory agent.
- Such molecules are suitably present in combination in amounts that are effective for the purpose intended.
- compositions comprising a population of anti-TF antibody-drug conjugates or antigen-binding fragments thereof as described herein for use in a method of treating colorectal cancer, non-small cell lung cancer, pancreatic cancer, head and neck cancer bladder cancer, endometrial cancer, esophageal cancer or prostate cancer as described herein.
- compositions comprising a population of antibody-drug conjugates, wherein the antibody-drug conjugates comprise a linker attached to MMAE, wherein the antibody-drug conjugate has the following structure:
- p denotes a number from 1 to 8, e.g, 1, 2, 3, 4, 5, 6, 7 or 8
- S represents a sulphydryl residue of the anti-TF antibody or antigen-binding fragment thereof
- Ab designates the anti-TF antibody or antigen-binding fragment thereof as described herein, such as tisotumab.
- p denotes a number from 3 to 5.
- the average value of p in the composition is about 4.
- the population is a mixed population of antibody-drug conjugates in which p varies from 1 to 8 for each antibody-drug conjugate.
- the population is a homogenous population of antibody-drug conjugates with each antibody-drug conjugate having the same value for p.
- a composition comprising an anti-TF antibody-drug conjugate as described herein, such as e.g ., tisotumab vedotin, is coadministered with one or more additional therapeutic agents. In some embodiments the coadministration is
- simultaneous means that the anti-TF antibody-drug conjugate as described herein is administered simultaneously with the one or more additional therapeutic agents.
- simultaneous means that the anti-TF antibody-drug conjugate and the one or more additional therapeutic agents are administered to the subject less than about one hour apart, such as less than about 30 minutes apart, less than about 15 minutes apart, less than about 10 minutes apart or less than about 5 minutes apart.
- simultaneous means that the anti-TF antibody-drug conjugate and the one or more additional therapeutic agents are administered to the subject less than one hour apart, such as less than 30 minutes apart, less than 15 minutes apart, less than 10 minutes apart or less than 5 minutes apart.
- the anti-TF antibody-drug conjugate is administered sequentially with the one or more additional therapeutic agents.
- sequential administration means that the anti-TF antibody-drug conjugate and the one or more additional therapeutic agents are administered a least 1 hour apart, at least 2 hours apart, at least 3 hours apart, at least 4 hours apart, at least 5 hours apart, at least 6 hours apart, at least 7 hours apart, at least 8 hours apart, at least 9 hours apart, at least 10 hours apart, at least 11 hours apart, at least 12 hours apart, at least 13 hours apart, at least 14 hours apart, at least 15 hours apart, at least 16 hours apart, at least 17 hours apart, at least 18 hours apart, at least 19 hours apart, at least 20 hours apart, at least 21 hours apart, at least 22 hours apart, at least 23 hours apart, at least 24 hours apart, at least 2 days apart, at least 3 days apart, at least 4 days apart, at least 5 days apart, at least 5 days apart, at least 7 days apart, at least 2 weeks apart, at least 3 weeks apart or at least 4 weeks apart
- composition comprising an anti-TF antibody-drug conjugate as described herein, such as e.g. , tisotumab vedotin, is coadministered with one or more therapeutic agents to eliminate or reduce the severity of one or more adverse events.
- the coadministration is simultaneous or sequential.
- the anti-TF antibody-drug conjugate is administered simultaneously with the one or more therapeutic agents to eliminate or reduce the severity of one or more adverse events.
- simultaneous means that the anti-TF antibody-drug conjugate and the one or more therapeutic agents to eliminate or reduce the severity of one or more adverse events are administered to the subject less than about one hour apart, such as less than about 30 minutes apart, less than about 15 minutes apart, less than about 10 minutes apart or less than about 5 minutes apart.
- simultaneous means that the anti-TF antibody-drug conjugate and the one or more therapeutic agents to eliminate or reduce the severity of one or more adverse events are administered to the subject less than one hour apart, such as less than 30 minutes apart, less than 15 minutes apart, less than 10 minutes apart or less than 5 minutes apart.
- the anti-TF antibody-drug conjugate is administered sequentially with the one or more therapeutic agents to eliminate or reduce the severity of one or more adverse events.
- sequential administration means that the anti-TF antibody-drug conjugate and the one or more therapeutic agents are administered a least 1 hour apart, at least 2 hours apart, at least 3 hours apart, , at least 4 hours apart, at least 5 hours apart, at least 6 hours apart, at least 7 hours apart, at least 8 hours apart, at least 9 hours apart, at least 10 hours apart, at least 11 hours apart, at least 12 hours apart, at least 13 hours apart, at least 14 hours apart, at least 15 hours apart, at least 16 hours apart, at least 17 hours apart, at least 18 hours apart, at least 19 hours apart, at least 20 hours apart, at least 21 hours apart, at least 22 hours apart, at least 23 hours apart, at least 24 hours apart, at least 2 days apart, at least 3 days apart, at least 4 days apart, at least 5 days apart, at least 5 days apart, at least 7 days apart, at least 2 weeks apart
- the anti-TF antibody-drug conjugate is administered prior to the one or more therapeutic agents to eliminate or reduce the severity of one or more adverse events. In some embodiments, the one or more therapeutic agents to eliminate or reduce the severity of one or more adverse events is administered prior to the anti-TF antibody-drug conjugate.
- an article of manufacture or kit which comprises an anti-TF antibody-drug conjugate described herein, such as e.g ., tisotumab vedotin.
- the article of manufacture or kit may further comprise instructions for use of the anti-TF antibody-drug conjugate in the methods of the invention.
- the article of manufacture or kit comprises instructions for the use of an anti-TF antibody-drug conjugate in methods for treating cancer (e.g, colorectal cancer, non-small cell lung cancer, pancreatic cancer, head and neck cancer, bladder cancer, endometrial cancer, esophageal cancer or prostate cancer) in a subject comprising administering to the subject an effective amount of an anti-TF antibody-drug conjugate.
- cancer e.g, colorectal cancer, non-small cell lung cancer, pancreatic cancer, head and neck cancer, bladder cancer, endometrial cancer, esophageal cancer or prostate cancer
- the cancer is colorectal cancer as described herein.
- the cancer is non-small cell lung cancer as described herein.
- the cancer is pancreatic cancer as described herein.
- the cancer is head and neck cancer as described herein. In some embodiments, the cancer is bladder cancer as described herein. In some embodiments, the cancer is endometrial cancer as described herein. In some embodiments, the cancer is esophageal cancer as described herein. In some embodiments, the cancer is prostate cancer as described herein. In some embodiments, the subject is a human.
- the article of manufacture or kit may further comprise a container.
- Suitable containers include, for example, bottles, vials (e.g ., dual chamber vials), syringes (such as single or dual chamber syringes) and test tubes.
- the container is a vial.
- the container may be formed from a variety of materials such as glass or plastic. The container holds the formulation.
- the article of manufacture or kit may further comprise a label or a package insert, which is on or associated with the container, may indicate directions for reconstitution and/or use of the formulation.
- the label or package insert may further indicate that the formulation is useful or intended for subcutaneous, intravenous (e.g., intravenous infusion), or other modes of administration for treating colorectal cancer, non-small cell lung cancer, pancreatic cancer, head and neck cancer, bladder cancer, endometrial cancer, esophageal cancer or prostate cancer as described herein in a subject.
- the container holding the formulation may be a single-use vial or a multi-use vial, which allows for repeat administrations of the reconstituted formulation.
- the article of manufacture or kit may further comprise a second container comprising a suitable diluent.
- the article of manufacture or kit may further include other materials desirable from a commercial, therapeutic, and user standpoint, including other buffers, diluents, filters, needles, syringes, and package inserts with instructions for use.
- the article of manufacture or kit herein optionally further comprises a container comprising a second medicament, wherein the anti-TF antibody-drug conjugate is a first medicament, and which article or kit further comprises instructions on the label or package insert for treating the subject with the second medicament, in an effective amount.
- the label or package insert indicates that the first and second medicaments are to be administered sequentially or simultaneously, as described herein.
- the label or package insert indicates that the first medicament is to be administered prior to the administration of the second medicament.
- the label or package insert indicates that second medicament is to be administered prior to the first medicament.
- the article of manufacture or kit herein optionally further comprises a container comprising a second medicament, wherein the second medicament is for eliminating or reducing the severity of one or more adverse events, wherein the anti-TF antibody-drug conjugate is a first medicament, and which article or kit further comprises instructions on the label or package insert for treating the subject with the second medicament, in an effective amount.
- the label or package insert indicates that the first and second medicaments are to be administered sequentially or simultaneously, as described herein.
- the label or package insert indicates that the first medicament is to be administered prior to the administration of the second medicament.
- the label or package insert indicates that second medicament is to be administered prior to the first medicament.
- the anti-TF antibody-drug conjugate is present in the container as a lyophilized powder.
- the lyophilized powder is in a hermetically sealed container, such as a vial, an ampoule or sachette, indicating the quantity of the active agent.
- an ampoule of sterile water for injection or saline can be, for example, provided, optionally as part of the kit, so that the ingredients can be mixed prior to administration.
- kits can further include, if desired, one or more of various conventional pharmaceutical components, such as, for example, containers with one or more pharmaceutically acceptable carriers, additional containers, etc., as will be readily apparent to those skilled in the art.
- Printed instructions either as inserts or as labels, indicating quantities of the components to be administered, guidelines for administration, and/or guidelines for mixing the components can also be included in the kit.
- a method of treating cancer in a subject comprising administering to the subject an antibody-drug conjugate that binds to tissue factor (TF), wherein the antibody-drug conjugate comprises an anti-TF antibody or an antigen-binding fragment thereof conjugated to a monomethyl auristatin or a functional analog thereof or a functional derivative thereof, wherein the antibody-drug conjugate is administered at a dose ranging from about 1.5 mg/kg to about 2.1 mg/kg, and wherein the cancer is selected from the group consisting of colorectal cancer, non-small cell lung cancer, pancreatic cancer, head and neck cancer, bladder cancer, endometrial cancer, esophageal cancer and prostate cancer.
- TF tissue factor
- pancreatic cancer is exocrine pancreatic adenocarcinoma.
- pancreatic cancer has predominant adenocarcinoma histology.
- the anti-TF antibody or antigen binding fragment thereof of the antibody-drug conjugate comprises a heavy chain variable region and a light chain variable region, wherein the heavy chain variable region comprises:
- the anti-TF antibody or antigen binding fragment thereof of the antibody-drug conjugate comprises a heavy chain variable region comprising an amino acid sequence at least 85% identical to the amino acid sequence of SEQ ID NO:7 and a light chain variable region comprising an amino acid sequence at least 85% identical to the amino acid sequence of SEQ ID NO:8.
- the anti-TF antibody or antigen binding fragment thereof of the antibody-drug conjugate comprises a heavy chain variable region comprising the amino acid sequence of SEQ ID NO:7 and a light chain variable region comprising the amino acid sequence of SEQ ID NO:8.
- the antibody-drug conjugate further comprises a linker between the anti-TF antibody or antigen-binding fragment thereof and the monomethyl auristatin.
- vc is the dipeptide valine-citrulline
- PAB is:
- the one or more therapeutic effects is selected from the group consisting of: size of a tumor derived from the cancer, objective response rate, duration of response, time to response, progression free survival, overall survival and prostate specific antigen (PSA) level.
- PSA prostate specific antigen
- any one of embodiments 55-62 wherein the subject exhibits a reduction in PSA level in a blood sample from the subject by at least about 5%, at least about 10%, at least about 15%, at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least about 45%, at least about 50%, at least about 60%, at least about 70%, or at least about 80% relative to the PSA level in a blood sample obtained from the subject before administration of the antibody-drug conjugate.
- 86 The method of any one of embodiments 1-85, wherein the size of a tumor derived from the cancer is reduced by at least about 10%, at least about 15%, at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least about 45%, at least about 50%, at least about 60%, at least about 70%, or at least about 80% relative to the size of the tumor derived from the cancer before administration of the antibody-drug conjugate.
- the objective response rate is at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least about 45%, at least about 50%, at least about 60%, at least about 70%, or at least about 80%.
- the duration of response to the antibody-drug conjugate is at least about 1 month, at least about 2 months, at least about 3 months, at least about 4 months, at least about 5 months, at least about 6 months, at least about 7 months, at least about 8 months, at least about 9 months, at least about 10 months, at least about 11 months, at least about 12 months, at least about eighteen months, at least about two years, at least about three years, at least about four years, or at least about five years after administration of the antibody-drug conjugate.
- a kit comprising:
- an antibody-drug conjugate that binds to TF for use in the treatment of cancer in a subject wherein the antibody-drug conjugate comprises an anti-TF antibody or an antigen binding fragment thereof conjugated to a monomethyl auristatin or a functional analog thereof or a functional derivative thereof, wherein the antibody-drug conjugate is administered to the subject at a dose ranging from about 0.9 mg/kg to about 2.1 mg/kg, and wherein the cancer is selected from the group consisting of colorectal cancer, non-small cell lung cancer, pancreatic cancer, head and neck cancer, bladder cancer, endometrial cancer, esophageal cancer and prostate cancer.
- the antibody-drug conjugate for use of any one of embodiments 155-159, wherein the subject has been previously treated with one or more agents selected from the group consisting of androgen deprivation therapy, a luteinizing hormone-releasing hormone agonist, a luteinizing hormone-releasing hormone antagonist, a CYP17 inhibitor, and an anti androgen.
- agents selected from the group consisting of androgen deprivation therapy, a luteinizing hormone-releasing hormone agonist, a luteinizing hormone-releasing hormone antagonist, a CYP17 inhibitor, and an anti androgen selected from the group consisting of androgen deprivation therapy, a luteinizing hormone-releasing hormone agonist, a luteinizing hormone-releasing hormone antagonist, a CYP17 inhibitor, and an anti androgen.
- MMAE monomethyl auri statin E
- the antibody-drug conjugate for use of any one of embodiments 101-170, wherein the anti-TF antibody or antigen-binding fragment thereof of the antibody-drug conjugate comprises a heavy chain variable region comprising an amino acid sequence at least 85% identical to the amino acid sequence of SEQ ID NO:7 and a light chain variable region comprising an amino acid sequence at least 85% identical to the amino acid sequence of SEQ ID NO:8.
- the antibody-drug conjugate for use of any one of embodiments 101-173, wherein the antibody-drug conjugate further comprises a linker between the anti-TF antibody or antigen binding fragment thereof and the monomethyl auristatin.
- vc is the dipeptide valine-citrulline
- PAB is:
- MMAE monomethyl auristatin E
- S represents a sulphydryl residue of the anti-TF antibody
- Ab designates the anti-TF antibody or antigen-binding fragment thereof.
- the antibody-drug conjugate for use of any one of embodiments 101-181, wherein at least about 0.1%, at least about 1%, at least about 2%, at least about 3%, at least about 4%, at least about 5%, at least about 6%, at least about 7%, at least about 8%, at least about 9%, at least about 10%, at least about 15%, at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least about 45%, at least about 50%, at least about 60%, at least about 70%, or at least about 80% of the cancer cells express TF.
- the antibody-drug conjugate for use of embodiment 183, wherein the one or more therapeutic effects is selected from the group consisting of: size of a tumor derived from the cancer, objective response rate, duration of response, time to response, progression free survival, overall survival and prostate specific antigen (PSA) level.
- PSA prostate specific antigen
- the antibody-drug conjugate for use of any one of embodiments 155-162, wherein the subject exhibits a reduction in PSA level in a blood sample from the subject by at least about 5%, at least about 10%, at least about 15%, at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least about 45%, at least about 50%, at least about 60%, at least about 70%, or at least about 80% relative to the PSA level in a blood sample obtained from the subject before administration of the antibody-drug conjugate.
- the antibody-drug conjugate for use of any one of embodiments 101-187, wherein the subject exhibits progression-free survival of at least about 1 month, at least about 2 months, at least about 3 months, at least about 4 months, at least about 5 months, at least about 6 months, at least about 7 months, at least about 8 months, at least about 9 months, at least about 10 months, at least about 11 months, at least about 12 months, at least about eighteen months, at least about two years, at least about three years, at least about four years, or at least about five years after administration of the antibody-drug conjugate.
- the antibody-drug conjugate for use of any one of embodiments 101-189, wherein the duration of response to the antibody-drug conjugate is at least about 1 month, at least about 2 months, at least about 3 months, at least about 4 months, at least about 5 months, at least about 6 months, at least about 7 months, at least about 8 months, at least about 9 months, at least about 10 months, at least about 11 months, at least about 12 months, at least about eighteen months, at least about two years, at least about three years, at least about four years, or at least about five years after administration of the antibody-drug conjugate.
- the antibody-drug conjugate for use of any one of embodiments 101-190, wherein the subject has one or more adverse events and is further administered an additional therapeutic agent to eliminate or reduce the severity of the one or more adverse events.
- the antibody-drug conjugate for use of embodiment 191 or embodiment 192, wherein the one or more adverse events is anemia, abdominal pain, hypokalemia, hyponatremia, epistaxis, fatigue, nausea, alopecia, conjunctivitis, constipation, decreased appetite, diarrhea, vomiting, peripheral neuropathy, or general physical health deterioration.
- an antibody-drug conjugate that binds to tissue factor (TF) for the manufacture of a medicament for treating cancer in a subject, wherein the antibody-drug conjugate comprises an anti-TF antibody or an antigen-binding fragment thereof conjugated to a monomethyl auristatin or a functional analog thereof or a functional derivative thereof, wherein the antibody-drug conjugate is administered at a dose ranging from about 0.9 mg/kg to about 2.1 mg/kg, and wherein the cancer is selected from the group consisting colorectal cancer, non-small cell lung cancer, pancreatic cancer, head and neck cancer, bladder cancer, endometrial cancer, esophageal cancer and prostate cancer.
- TF tissue factor
- embodiment 208 wherein the subject received prior systemic therapy and experienced disease progression on or after the systemic therapy.
- embodiment 216 wherein greater than 85% of the non-small cell lung cancer cells have squamous histology.
- pancreatic cancer is exocrine pancreatic adenocarcinoma.
- embodiment 224 wherein greater than 85% of the pancreatic cancer have adenocarcinoma histology.
- embodiment 230 wherein the head and neck cancer is squamous cell carcinoma.
- embodiment 230 or embodiment 231 wherein the subject received prior systemic therapy and experienced disease progression on or after the systemic therapy.
- any one of embodiments 200-268, wherein the anti-TF antibody or antigen binding fragment thereof of the antibody-drug conjugate comprises a heavy chain variable region and a light chain variable region, wherein the heavy chain variable region comprises:
- any one of embodiments 200-269, wherein the anti-TF antibody or antigen binding fragment thereof of the antibody-drug conjugate comprises a heavy chain variable region comprising an amino acid sequence at least 85% identical to the amino acid sequence of SEQ ID NO:7 and a light chain variable region comprising an amino acid sequence at least 85% identical to the amino acid sequence of SEQ ID NO:8.
- any one of embodiments 200-270, wherein the anti-TF antibody or antigen binding fragment thereof of the antibody-drug conjugate comprises a heavy chain variable region comprising the amino acid sequence of SEQ ID NO:7 and a light chain variable region comprising the amino acid sequence of SEQ ID NO:8.
- vc is the dipeptide valine-citrulline
- PAB is:
- S represents a sulphydryl residue of the anti-TF antibody
- Ab designates the anti-TF antibody or antigen-binding fragment thereof.
- any one of embodiments 200-280 wherein at least about 0.1%, at least about 1%, at least about 2%, at least about 3%, at least about 4%, at least about 5%, at least about 6%, at least about 7%, at least about 8%, at least about 9%, at least about 10%, at least about 15%, at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least about 45%, at least about 50%, at least about 60%, at least about 70%, or at least about 80% of the cancer cells express TF.
- embodiment 282 wherein the one or more therapeutic effects is selected from the group consisting of: size of a tumor derived from the cancer, objective response rate, duration of response, time to response, progression free survival, overall survival and prostate specific antigen (PSA) level.
- PSA prostate specific antigen
- any one of embodiments 254-261 wherein the subject exhibits a reduction in PSA level in a blood sample from the subject by at least about 5%, at least about 10%, at least about 15%, at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least about 45%, at least about 50%, at least about 60%, at least about 70%, or at least about 80% relative to the PSA level in a blood sample obtained from the subject before administration of the antibody-drug conjugate.
- any one of embodiments 200-284 wherein the size of a tumor derived from the cancer is reduced by at least about 10%, at least about 15%, at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least about 45%, at least about 50%, at least about 60%, at least about 70%, or at least about 80% relative to the size of the tumor derived from the cancer before administration of the antibody-drug conjugate.
- any one of embodiments 200-285, wherein the objective response rate is at least about 20%, at least about 25%, at least about 30%, at least about 35%, at least about 40%, at least about 45%, at least about 50%, at least about 60%, at least about 70%, or at least about 80%.
- any one of embodiments 200-287 wherein the subject exhibits overall survival of at least about 1 month, at least about 2 months, at least about 3 months, at least about 4 months, at least about 5 months, at least about 6 months, at least about 7 months, at least about 8 months, at least about 9 months, at least about 10 months, at least about 11 months, at least about 12 months, at least about eighteen months, at least about two years, at least about three years, at least about four years, or at least about five years after
- any one of embodiments 200-288 wherein the duration of response to the antibody-drug conjugate is at least about 1 month, at least about 2 months, at least about 3 months, at least about 4 months, at least about 5 months, at least about 6 months, at least about 7 months, at least about 8 months, at least about 9 months, at least about 10 months, at least about 11 months, at least about 12 months, at least about eighteen months, at least about two years, at least about three years, at least about four years, or at least about five years after administration of the antibody-drug conjugate.
- embodiment 290 or embodiment 291 wherein the one or more adverse events is anemia, abdominal pain, hypokalemia, hyponatremia, epistaxis, fatigue, nausea, alopecia, conjunctivitis, constipation, decreased appetite, diarrhea, vomiting, peripheral neuropathy, or general physical health deterioration.
- Example 1 A Phase study of tisotumab vedotin in subjects with locally advanced or
- Tisotumab vedotin is an antibody-drug conjugate comprising a TF-targeted human monoclonal immunoglobulin Gl (subtype K) conjugated via a protease-cleavable valine citrulline linker to the drug monomethyl auristatin E (MMAE), a dolastatin 10 analog.
- MMAE monomethyl auristatin E
- High, differential levels of TF have been observed on the membranes of neoplastic cells as well as on tumor-associated endothelium in multiple cancers, including SCCHN, NSCLC, colorectal cancer, and pancreatic cancer.
- Tisotumab vedotin selectively targets TF to deliver a clinically validated toxic payload to tumor cells (FIG. 1). See Breij EC et al. Cancer Res.
- Dolastatins and auristatins belong to a class of chemotherapies that act as microtubule disrupting agents.
- squamous NSCLC exocrine pancreatic adenocarcinoma, squamous cell carcinoma of the head and neck (SCCHN), bladder cancer, endometrial cancer, esophageal cancer or prostate cancer.
- second and third line therapeutic options are available for the patient populations in this study, response rates are low (ORRs of 15% or lower) and long term survival is poor.
- Patients with locally-advanced or metastatic colorectal or pancreatic cancer, squamous NSCLC, SCCHN, bladder cancer, endometrial cancer, esophageal cancer or prostate cancer whose disease has progressed after first and subsequent lines of treatment have significant unmet medical need for therapies that can meaningfully improve their prognosis.
- This global, open label, multicenter trial is designed to assess the safety, tolerability, and activity of tisotumab vedotin for the treatment of selected solid tumors.
- Eligible patients are at least 18 years of age with inoperable, locally advanced or metastatic cancer. Patients are enrolled into one of 8 cohorts based on tumor type, including colorectal cancer, non-small cell lung cancer with squamous cell histology only (squamous NSCLC), exocrine pancreatic adenocarcinoma, squamous cell carcinoma of the head and neck
- SCCHN bladder cancer
- endometrial cancer esophageal cancer
- prostate cancer esophageal cancer
- tisotumab vedotin is administered at a dose of 2.0 mg/kg as a 30 minutes IV infusion on Day 1 of each 21 -day cycle (Q3W). For patients weighing more than 100 kg, dosing is capped at 200 mg per infusion. An individual’s dose may be modified based upon treatment-related adverse events. Response is assessed every 6 weeks for the first 6 months, every 12 weeks for the next 6 months, and then every 6 months after that. RECIST vl .1 is used by the investigator to score responses for primary and secondary endpoints as well as progression. Objective responses are confirmed with repeat scans 4-6 weeks after the first documentation of response.
- Patients with endometrial cancer must have experienced disease progression on or after their most recent systemic therapy for locally advanced or metastatic disease. Patients should have received no more than 3 systemic regimens in the recurrent/metastatic setting.
- Patients with esophageal cancer must have experienced disease progression on or after their most recent systemic therapy for locally advanced or metastatic disease. Patients should have received no more than 3 systemic regimens in the recurrent/metastatic setting.
- Patients with prostate cancer must have experienced disease progression on or after their most recent systemic therapy for locally advanced or metastatic disease. Patients should have received no more than 3 systemic regimens in the recurrent/metastatic setting.
- ECOG Eastern Cooperative Oncology Group
- ANC absolute neutrophil count
- ALT alanine aminotransferase
- AST aspartate aminotransferase
- ASA prophylactic acetylsalicylic acid
- malignancies History of another malignancy within 3 years before the first dose of study drug, or any evidence of residual disease from a previously diagnosed malignancy. Exceptions are malignancies with a negligible risk of metastasis or death (e.g ., 5-year overall survival >90%), such as adequately treated carcinoma in situ of the cervix, non-melanoma skin carcinoma, localized prostate cancer, ductal carcinoma in situ, or Stage I uterine cancer.
- Inflammatory bowel disease including Crohn’s disease and colitis ulcerosa. Ongoing, acute or chronic inflammatory skin disease.
- Inflammatory lung disease including moderate and severe asthma and chronic obstructive pulmonary disease, requiring chronic medical therapy Medications or treatment regimens:
- ASA e.g., aspirin
- Surgery/procedures Major surgical procedure (defined as a surgery requiring inpatient hospitalization of at least 48 hours) within 4 weeks or excisional biopsy within 7 days prior to the first study drug administration. Patients who have planned major surgery during the treatment period must be excluded from the trial.
- live vaccines include, but are not limited to, the following: measles, mumps, rubella, varicella/zoster (chicken pox), yellow fever, rabies, Bacillus Calmette-Guerin, and typhoid vaccine.
- Seasonal influenza vaccines for injection are generally killed virus vaccines and are allowed; however, intranasal influenza vaccines ( e.g ., FLUMIST ® ) are live attenuated vaccines and are not allowed.
- At least 42 days must have elapsed from the last administration of chemo-radi otherapy .
- Lyophilized vials containing 40 mg of tisotumab vedotin are stored in a refrigerator at 2°C to 8°C.
- Tisotumab vedotin is reconstituted in 4 ml of water leading to a reconstituted solution comprising 10 mg/mL tisotumab vedotin.
- the reconstituted tisotumab vedotin is diluted into a 0.9% NaCl 100 mL infusion bag according to the dose calculated for the patient to receive 2.0 mg/kg tisotumab vedotin.
- Intravenous infusion is completed within 24 hours after the tisotumab vedotin vial has been reconstituted.
- a 0.2 pm in-line filter is used for the intravenous infusion.
- the entire 100 mL volume from the prepared infusion bag is administered. No dead volume is provided.
- dose reductions are permitted in order to allow the patient to continue treatment with tisotumab vedotin (Table 2).
- the confirmed objective response rate is defined as the proportion of patients who achieve a confirmed CR or PR according to RECIST vl .1 as assessed by the investigator.
- the confirmed ORR of each cohort and its exact 2-sided 95% Cl using the Clopper-Pearson method is calculated.
- Confirmed and unconfirmed ORR is defined as the proportion of patients who achieve a CR or PR according to RECIST vl.l as assessed by the investigator. These include patients with confirmed responses as well as those whose responses were not confirmed or had not yet been assessed for confirmation.
- DCR is defined as the proportion of patients who achieve a CR or PR according to RECIST vl .1 as assessed by the investigator, or meet the SD criteria at least once after start of study treatment at a minimum interval of 12 weeks. The confirmed and unconfirmed ORR and the DCR are estimated for each cohort and the 95%
- CIs are calculated using the Clopper-Pearson method.
- DOR is defined as the time from the first documentation of objective response (CR or PR that is subsequently confirmed) to the first documentation of PD or death due to any cause, whichever comes first.
- TTR is defined as the time from the start of study treatment to the first documentation of objective response (CR or PR that is subsequently confirmed).
- PFS is defined as the time from the start of study treatment to the first documentation of PD or death due to any cause, whichever comes first.
- OS is defined as the time from the start of study treatment to date of death due to any cause. In the absence of death, survival time will be censored at the last date the patient is known to be alive (i.e., date of last contact).
- the DOR, TTR, PFS, and OS are estimated for each cohort using the Kaplan-Meier methodology, and the medians and associated 95% CIs are calculated. Kaplan-Meier plots are provided as appropriate.
- the 3- and 6-month PFS rates, as well as the 6- and l2-month OS rates, are summarized.
- the TTR of patients who achieve an objective response is summarized.
- TTR time to RECIST vl .
- PFS progression-free survival
- Tissue Factor expression-response TF expression-response relationship relationship following treatment with tisotumab vedotin Assess biomarkers of biological activity and Relationship between biomarkers in blood resistance and predictive biomarkers of and tumor tissue to efficacy, safety, or other response biomarker endpoints following treatment with tisotumab vedotin
- ocular pre- medication guidelines are followed: (1) Administration of local ocular vasoconstrictor before infusion (brimonidine tartrate 0.2% eye drops or similar, 3 drops in each eye immediately prior to start of infusion; otherwise to be used in accordance with the product prescribing information). If the patient does not tolerate ocular vasoconstrictors due to adverse reactions, continued treatment with these may be stopped at the discretion of the investigator and following discussion with the sponsor’s medical monitor. (2) Use of refrigerator-based eye cooling pads during infusion, e.g ., THERA PEARL ® Eye Mask or similar. To be applied immediately before infusion in accordance with the instructions provided with the eye cooling pads.
- Steroid eye drops (dexamethasone 0.1% eye drops or equivalent) during the first 3 days of each treatment cycle (i.e., first drop to be given prior to start of infusion; continue treatment for 72 hours thereafter).
- Steroid eye drops should be administered as 1 drop in each eye, 3 times daily, for 3 days, or used in accordance with the product prescribing information.
- Use of preservative-free lubricating eye drops during the whole treatment phase of the trial i.e., from first dose of study drug until 30 days after the last dose of study drug).
- Lubricating eye drops should be administered according to the product prescribing information.
- Tisotumab vedotin may cause Infusion-Related Reactions including severe hypersensitivity or anaphylaxis. Signs and symptoms usually develop during or shortly after drug infusion. In case any clinical significant IRR is observed during or after the first infusion of tisotumab vedotin or at subsequent treatment cycles, the patient should be observed for 2 hours after the end of tisotumab vedotin administration for all subsequent infusions. At all times during infusion, immediate emergency treatment of an anaphylactic reaction according to institutional standards must be assured.
- Example 2 Anti-tumor activity of tisotumab vedotin in cell line-derived and patient- derived xenograft mouse models of non-small-cell lung carcinoma
- NSCLC non-small-cell lung carcinoma
- SCC squamous cell carcinoma
- AC adenocarcinoma
- mice were treated by intraperitoneal injection of tisotumab vedotin at various doses (0.5, 1.5, or 4.5 mg/kg) once on day 27 to evaluate dose-dependent anti-tumor effect of tisotumab vedotin.
- mice were treated with isotype control antibody IgGl-bl2 at 4 mg/kg, or with isotype ADC control IgGl-bl2-vcMMAE at 0.5, 1.5, or 4.5 mg/kg.
- treatment with tisotumab vedotin at 4.5 mg/kg showed superior efficacy in comparison with the other treatment groups in the NCI-H441 CDX model.
- Treatment with tisotumab vedotin at 1.5 mg/kg and particularly at 4.5 mg/kg significantly inhibited tumor development on day 47 compared to treatment with IgGl-bl2- vcMMAE at corresponding doses (FIG. 2B).
- mice were randomized into different groups. Mice received intravenous administrations of tisotumab vedotin, IgGl-bl2 control, or IgGl-bl2-vcMMAE control at 4 mg/kg on days 0 and 7 respectively. Tumor growth was calculated by measuring the tumor volumes every 3-4 days.
- LXFA 1674 (subtype AC) and LUO 395 (subtype SCC) respectively.
- FIG. 3 shows exemplary efficacy results of tisotumab vedotin in a squamous cell lung carcinoma model LXFE 690.
- LXFE 690 a squamous cell lung carcinoma model
- Tisotumab vedotin also showed anti -tumor activity in the LXFE 772, LXFA 289, LXFA 1041, LXFA 1674 and LUO 395 NSCLC xenograft models.
- Example 3 Anti-tumor activity of tisotumab vedotin in cell line-derived and patient- derived xenograft mouse models of pancreatic cancer
- a CDX model using HPAF-II cells (pancreatic adenocarcinoma, ATCC, cat. No. CRL-1997) was induced by injecting subcutaneously in the flank of SCID mice with 200 pL tumor cell suspension containing 2xl0 6 cells on day 0. On days 10, 13, 17 and 20, the mice received intraperitoneal administration of tisotumab vedotin at a dose of 0.3 mg/kg or 1 mg/kg or IgGl-bl2 control at 3 mg/kg.
- FIG. 5 shows exemplary efficacy results of tisotumab vedotin in the PAXF 1657 model.
- Example 4 Anti-tumor activity of tisotumab vedotin in cell line-derived xenograft mouse models of head and neck cancer
- Squamous cell carcinoma of the head and neck (SCCHN) cell lines FaDu ATCC cat. No. HTB-43
- VU-SCC-040 VU-SCC-OE
- VU-SCC-OE Hermsen et al (1996).
- Chromosomes. Cancer 15: 1-9) were used to produce CDX mice models of SCCHN. FaDu and VU-SCC-040 cell lines and xenograft tumors both had abundant TF expression. In comparison, the VU-SCC-OE cell line and xenograft tumors had significantly less, but detectable levels of TF expression. [0236] Cells from these SCCHN cell lines were subcutaneously injected in both flanks of nude mice at approximately 2 x 10 6 cells/per flank. When tumors reached an average size of 100 mm 3 (range 40-180 mm 3 ; day 0), intraperitoneal treatment of the mice with tisotumab vedotin was started.
- FIG. 6 shows the effect of tisotumab vedotin treatment in the FaDu CDX model.
- PBS or IgGl-bl2- vcMMAE treated mice from the control groups tumor outgrowth was rapid and the majority of mice had to be sacrificed on day 7.
- mice treated with tisotumab vedotin at 2 mg/kg tumor growth was significantly inhibited, and tumor regression was observed after 3 doses. However, tumors started to re-grow by day 30.
- mice treated with tisotumab vedotin at 4 mg/kg significant tumor regression was observed after the first dose. Moreover, complete tumor regression was observed by day 30 in all mice without recurrence of tumors until the end of the experiment (z.e., day 76).
- Example 5 Anti-tumor activity of tisotumab vedotin in a bladder cancer patient- derived xenograft model
- mice were randomized and treated intravenously with a single dose of 0.5, 1, 2 or 4 mg/kg tisotumab vedotin, the isotype control ADC IgGl-bl2-MMAE (4 mg/kg) or the unconjugated isotype control antibody IgGl-bl2 (4 mg/kg) diluted in PBS. The day of randomization and treatment was designated day 0.
- Example 6 Anti-tumor activity of tisotumab vedotin in an esophageal cancer PDX model
- mice were randomized into treatment groups according to their tumor sizes (8 mice per group).
- animals were treated intravenously with 4 mg/kg tisotumab vedotin, the isotype control ADC IgGl-bl2-MMAE or the unconjugated isotype control antibody IgGl-bl2 diluted in PBS.
- the day of randomization and first treatment was designated day 0.
- a second treatment was administered at day 7.
- mice were randomized into groups of 8 mice with an equal tumor size distribution, and treated intravenously with 4 mg/kg tisotumab vedotin, the isotype control ADC IgGl-bl2- MMAE or the unconjugated isotype control antibody IgGl-bl2 diluted in PBS. The day of randomization and first treatment was designated day 0. A second treatment was used.
- PA5415 pancreatic cancer patient-derived xenograft model was performed at Crown Bio (San Diego, U.S.).
- Patient-derived tumor cell suspensions (PA5415) were thawed, washed PBS and resuspended in cold PBS at concentrations of 74,000 viable cells/lOO m ⁇ .
- Cell suspensions were mixed with an equal volume of Cultrex® extracellular matrix (ECM) and kept on ice.
- ECM extracellular matrix
- NCID-SCID mice Female non-obese diabetic severe combined immunodeficient mice were injected subcutaneously with 200 m ⁇ of the cell suspension in ECM, under isoflurane anesthesia (day -37).
- mice were randomized into groups of 8 mice with comparable tumor size distribution.
- mice were treated intravenously with tisotumab vedotin (0.5, 1 or 2 mg/kg), the isotype control ADC IgGl-bl2-MMAE (2 mg/kg) or the unconjugated isotype control antibody IgGl-bl2 (2 mg/kg) diluted in PBS.
- the day of randomization and first treatment was designated day 0.
- a second treatment was administered at day 7. Tumor growth was assessed every 3-4 days.
- tisotumab vedotin induced inhibition of tumor growth in the PA5415 pancreatic cancer xenograft model (FIG. 11). Moreover, tisotumab vedotin prolonged tumor-free survival (using a tumor size of 500 mm 3 as a cut-off for tumor progression; FIG. 12).
- Example 8 Anti-tumor activity of tisotumab vedotin in a colorectal cancer PDX mouse model
- tisotumab vedotin was evaluated in a diverse panel of colorectal cancer (CRC) patient-derived xenograft (PDX) models in NOD-SCID mice in a“mouse clinical trial” (MCT).
- CRC colorectal cancer
- PDX patient-derived xenograft
- Xenografts were derived from frozen tumor cells from cancer patients. Establishment and characterization of the PDX models was performed following subcutaneous injection of 100 pl of the PDX tumor cell suspension into the rear flank of the mouse.
- mice were randomized into 2 groups per PDX model: either the tisotumab vedotin-treated group or the PBS control group (one mouse in each arm, n l). Mice were administered the following treatments by intravenous injections: 1) tisotumab vedotin alone at dose level 2 mg/kg (dose volume 10 ml/kg) weekly for two weeks (QWx2); 2) PBS control (dose volume 10 ml/kg) weekly for two weeks (QWx2).
- Models were excluded from the final analysis if the control tumor did not show at least doubling in tumor volume compared to day 0.
- Responding models (R) were defined as models showing AT/AC ⁇ 10% (tumor stasis or tumor regression), and non-responding models were defined as AT/AC > 70%.
- FIG. 13 and FIG. 14 tisotumab vedotin induced potent anti -tumor activity (tumor stasis or tumor regression) in 5/33 of the PDX models and no response in 16/33 of the models. 12/33 of the PDX models were classified as intermediate.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Immunology (AREA)
- Epidemiology (AREA)
- Organic Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Genetics & Genomics (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Hematology (AREA)
- Biophysics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Cell Biology (AREA)
- Dermatology (AREA)
- Zoology (AREA)
- Gastroenterology & Hepatology (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Peptides Or Proteins (AREA)
Abstract
Description

















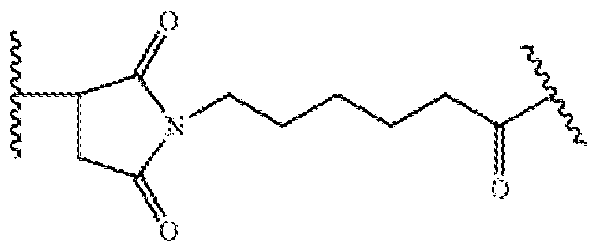













Claims
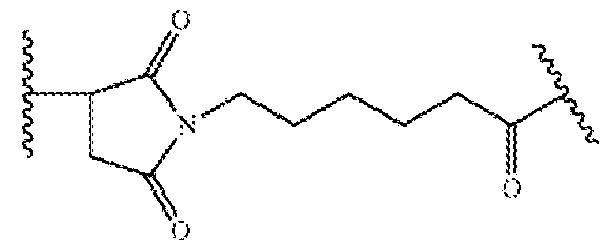


Priority Applications (13)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| SG11202007865VA SG11202007865VA (en) | 2018-03-07 | 2019-03-06 | Anti-tissue factor antibody-drug conjugates and their use in the treatment of cancer |
| BR112020018092-0A BR112020018092A2 (en) | 2018-03-07 | 2019-03-06 | METHOD FOR TREATING CANCER IN A SUBJECT, CASE, USE OF AN ANTIBODY-PHARMACEUTICAL CONJUGATE THAT LINKS TO THE TISSUE FACTOR, AND, ANTIBODY-PHARMACEUTICAL CONJUGATE. |
| CN201980031334.6A CN112105388A (en) | 2018-03-07 | 2019-03-06 | Anti-tissue factor antibody-drug conjugates and their use in cancer therapy |
| CA3091217A CA3091217A1 (en) | 2018-03-07 | 2019-03-06 | Anti-tissue factor antibody-drug conjugates and their use in the treatment of cancer |
| JP2020546443A JP7471227B2 (en) | 2018-03-07 | 2019-03-06 | Anti-tissue factor antibody-drug conjugates and their use in the treatment of cancer - Patents.com |
| EA202091688A EA202091688A1 (en) | 2018-09-25 | 2019-03-06 | ANTIBODY CONJUGATES TO TISSUE FACTOR WITH MEDICINES AND THEIR APPLICATION IN TREATMENT OF CANCER |
| US16/978,539 US20210030888A1 (en) | 2018-03-07 | 2019-03-06 | Anti-tissue factor antibody-drug conjugates and their use in the treatment of cancer |
| KR1020207028401A KR20200130356A (en) | 2018-03-07 | 2019-03-06 | Anti-tissue factor antibody-drug conjugates and their use in cancer treatment |
| EP19764722.5A EP3762032A4 (en) | 2018-03-07 | 2019-03-06 | Anti-tissue factor antibody-drug conjugates and their use in the treatment of cancer |
| MX2020008613A MX2020008613A (en) | 2018-03-07 | 2019-03-06 | Anti-tissue factor antibody-drug conjugates and their use in the treatment of cancer. |
| AU2019231696A AU2019231696A1 (en) | 2018-03-07 | 2019-03-06 | Anti-tissue factor antibody-drug conjugates and their use in the treatment of cancer |
| IL276768A IL276768A (en) | 2018-03-07 | 2020-08-17 | Anti-tissue factor antibody-drug conjugates and their use in the treatment of cancer |
| JP2024002970A JP2024045219A (en) | 2018-03-07 | 2024-01-12 | Anti-tissue factor antibody-drug conjugates and use thereof in treatment of cancer |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201862639891P | 2018-03-07 | 2018-03-07 | |
| US62/639,891 | 2018-03-07 | ||
| US201862736343P | 2018-09-25 | 2018-09-25 | |
| US62/736,343 | 2018-09-25 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2019173523A1 true WO2019173523A1 (en) | 2019-09-12 |
Family
ID=67845742
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2019/021024 WO2019173523A1 (en) | 2018-03-07 | 2019-03-06 | Anti-tissue factor antibody-drug conjugates and their use in the treatment of cancer |
Country Status (12)
| Country | Link |
|---|---|
| US (1) | US20210030888A1 (en) |
| EP (1) | EP3762032A4 (en) |
| JP (2) | JP7471227B2 (en) |
| KR (1) | KR20200130356A (en) |
| CN (1) | CN112105388A (en) |
| AU (1) | AU2019231696A1 (en) |
| BR (1) | BR112020018092A2 (en) |
| CA (1) | CA3091217A1 (en) |
| IL (1) | IL276768A (en) |
| MX (1) | MX2020008613A (en) |
| SG (1) | SG11202007865VA (en) |
| WO (1) | WO2019173523A1 (en) |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022111618A1 (en) * | 2020-11-26 | 2022-06-02 | 正大天晴药业集团股份有限公司 | Combined pharmaceutical composition of anti-pd-l1 antibody and c-met kinase inhibitor for treating lung cancer |
| WO2023213960A1 (en) * | 2022-05-06 | 2023-11-09 | Genmab A/S | Methods of treating cancer with anti-tissue factor antibody-drug conjugates |
| US12036286B2 (en) | 2021-03-18 | 2024-07-16 | Seagen Inc. | Selective drug release from internalized conjugates of biologically active compounds |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN114569734B (en) * | 2022-01-17 | 2023-08-29 | 北京化工大学 | Preparation method of nanoparticle for delivering cationic platinum drug based on reduction-sensitive polymer |
| WO2024067864A1 (en) * | 2022-09-30 | 2024-04-04 | Shanghai Junshi Biosciences Co., Ltd. | Anti-lair1 antibodies and their uses |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017042352A1 (en) * | 2015-09-11 | 2017-03-16 | Genmab A/S | Dosing regimens for anti-tf-antibody drug-conjugates |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2582728B1 (en) * | 2010-06-15 | 2017-08-23 | Genmab A/S | Human antibody drug conjugates against tissue factor |
-
2019
- 2019-03-06 US US16/978,539 patent/US20210030888A1/en active Pending
- 2019-03-06 WO PCT/US2019/021024 patent/WO2019173523A1/en unknown
- 2019-03-06 BR BR112020018092-0A patent/BR112020018092A2/en unknown
- 2019-03-06 JP JP2020546443A patent/JP7471227B2/en active Active
- 2019-03-06 EP EP19764722.5A patent/EP3762032A4/en active Pending
- 2019-03-06 MX MX2020008613A patent/MX2020008613A/en unknown
- 2019-03-06 SG SG11202007865VA patent/SG11202007865VA/en unknown
- 2019-03-06 KR KR1020207028401A patent/KR20200130356A/en unknown
- 2019-03-06 CA CA3091217A patent/CA3091217A1/en active Pending
- 2019-03-06 CN CN201980031334.6A patent/CN112105388A/en active Pending
- 2019-03-06 AU AU2019231696A patent/AU2019231696A1/en active Pending
-
2020
- 2020-08-17 IL IL276768A patent/IL276768A/en unknown
-
2024
- 2024-01-12 JP JP2024002970A patent/JP2024045219A/en active Pending
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017042352A1 (en) * | 2015-09-11 | 2017-03-16 | Genmab A/S | Dosing regimens for anti-tf-antibody drug-conjugates |
Non-Patent Citations (2)
| Title |
|---|
| I VERGOTE ET AL.: "A PHASE IIA STUDY OF TISOTUMAB VEDOTIN (HUMAX®-TF-ADC) IN PATIENTS WITH RELAPSED, RECURRENT AND/OR METASTATIC CERVICAL CANCER", ANNALS OF ONCOLOGY, vol. 28, no. Suppl 5, 8 September 2017 (2017-09-08), pages 1 - 16, XP055727581 * |
| See also references of EP3762032A4 * |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2022111618A1 (en) * | 2020-11-26 | 2022-06-02 | 正大天晴药业集团股份有限公司 | Combined pharmaceutical composition of anti-pd-l1 antibody and c-met kinase inhibitor for treating lung cancer |
| US12036286B2 (en) | 2021-03-18 | 2024-07-16 | Seagen Inc. | Selective drug release from internalized conjugates of biologically active compounds |
| WO2023213960A1 (en) * | 2022-05-06 | 2023-11-09 | Genmab A/S | Methods of treating cancer with anti-tissue factor antibody-drug conjugates |
Also Published As
| Publication number | Publication date |
|---|---|
| MX2020008613A (en) | 2020-09-21 |
| AU2019231696A1 (en) | 2020-09-03 |
| IL276768A (en) | 2020-10-29 |
| EP3762032A1 (en) | 2021-01-13 |
| CN112105388A (en) | 2020-12-18 |
| JP7471227B2 (en) | 2024-04-19 |
| US20210030888A1 (en) | 2021-02-04 |
| EP3762032A4 (en) | 2021-12-22 |
| JP2021515017A (en) | 2021-06-17 |
| JP2024045219A (en) | 2024-04-02 |
| KR20200130356A (en) | 2020-11-18 |
| BR112020018092A2 (en) | 2020-12-22 |
| SG11202007865VA (en) | 2020-09-29 |
| CA3091217A1 (en) | 2019-09-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7471227B2 (en) | Anti-tissue factor antibody-drug conjugates and their use in the treatment of cancer - Patents.com | |
| JP7460608B2 (en) | Methods for treating cancer using a combination of anti-PD-1 antibody and anti-tissue factor antibody-drug conjugate | |
| US20210015939A1 (en) | Methods of treating cancer with a combination of a platinum-based agent and an anti-tissue factor antibody-drug conjugate | |
| JP2024009998A (en) | Anti-tissue factor antibody-drug conjugates and their use in treatment of cancer | |
| EP3790584A1 (en) | Methods of treating cancer with a combination of an anti-pd-1 antibody and an anti-tissue factor antibody-drug conjugate | |
| AU2019321442A1 (en) | Anti-tissue factor antibody-drug conjugates and their use in the treatment of cancer | |
| JP2024129143A (en) | Methods of treating cancer using a combination of anti-VEGF antibody and anti-tissue factor antibody-drug conjugate | |
| CN114650846A (en) | Methods of treating cancer with combinations of platinum-based agents and anti-tissue factor antibody-drug conjugates |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 19764722 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 3091217 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2019231696 Country of ref document: AU Date of ref document: 20190306 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2020546443 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20207028401 Country of ref document: KR Kind code of ref document: A |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112020018092 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 2019764722 Country of ref document: EP Effective date: 20201007 |
|
| ENP | Entry into the national phase |
Ref document number: 112020018092 Country of ref document: BR Kind code of ref document: A2 Effective date: 20200904 |