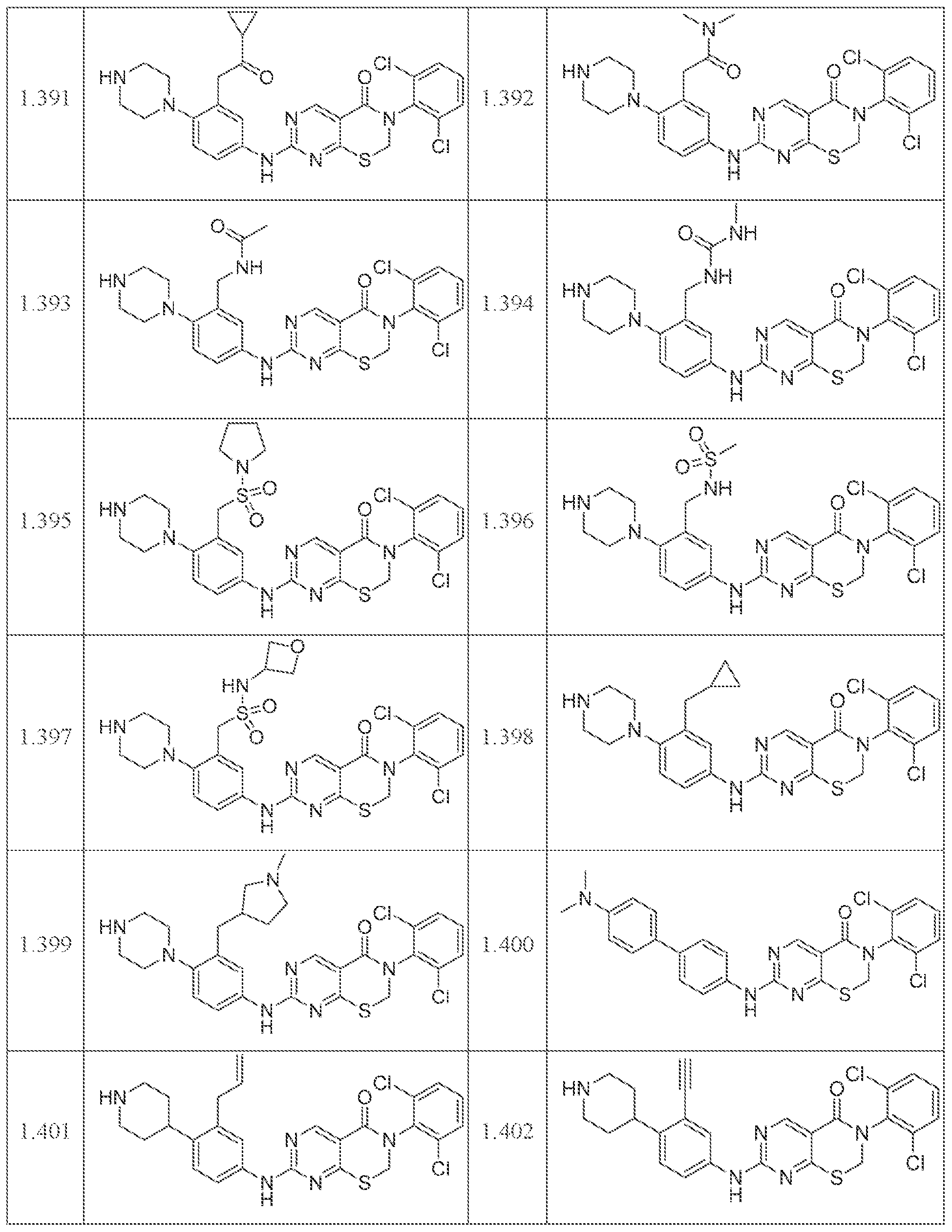
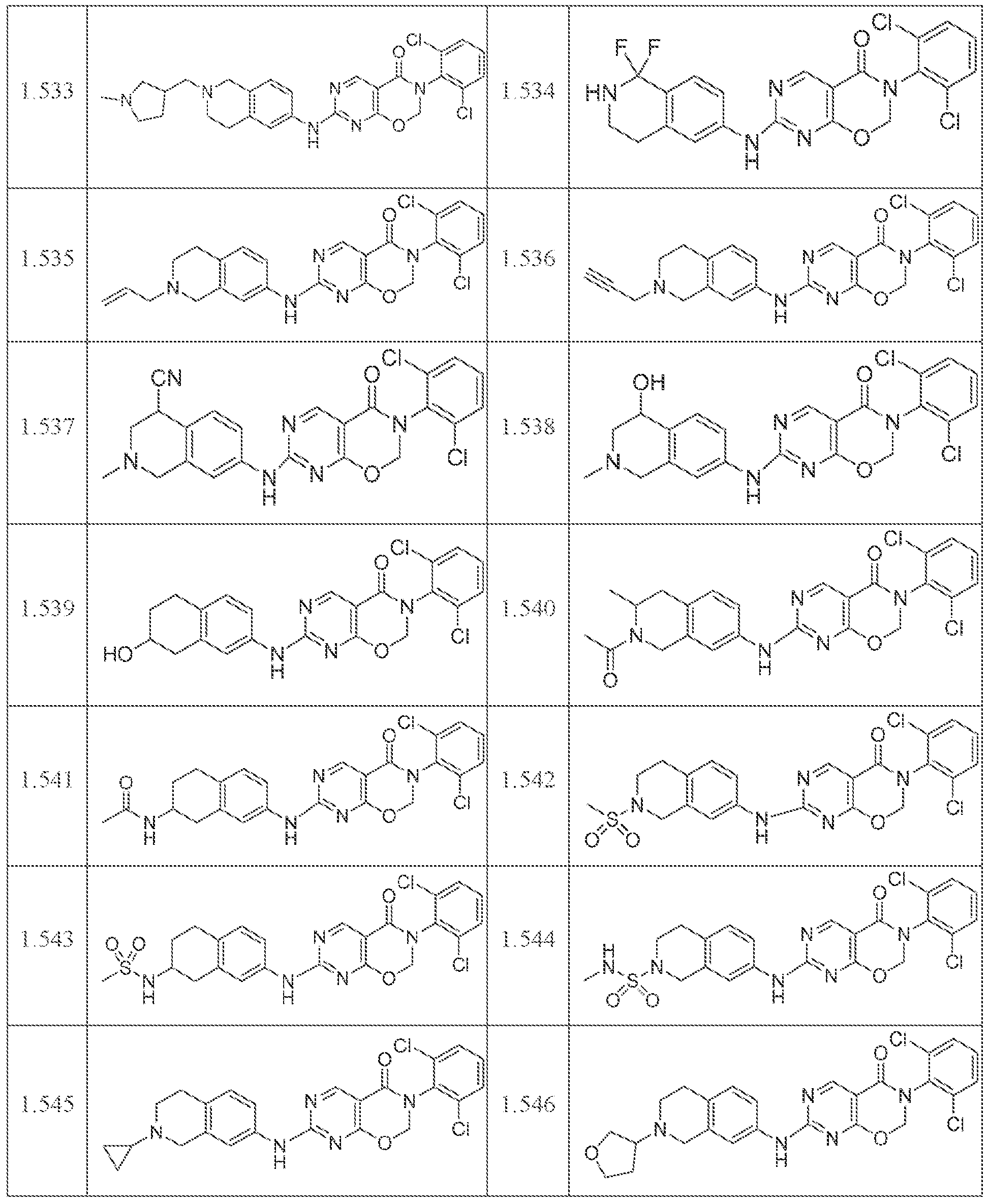
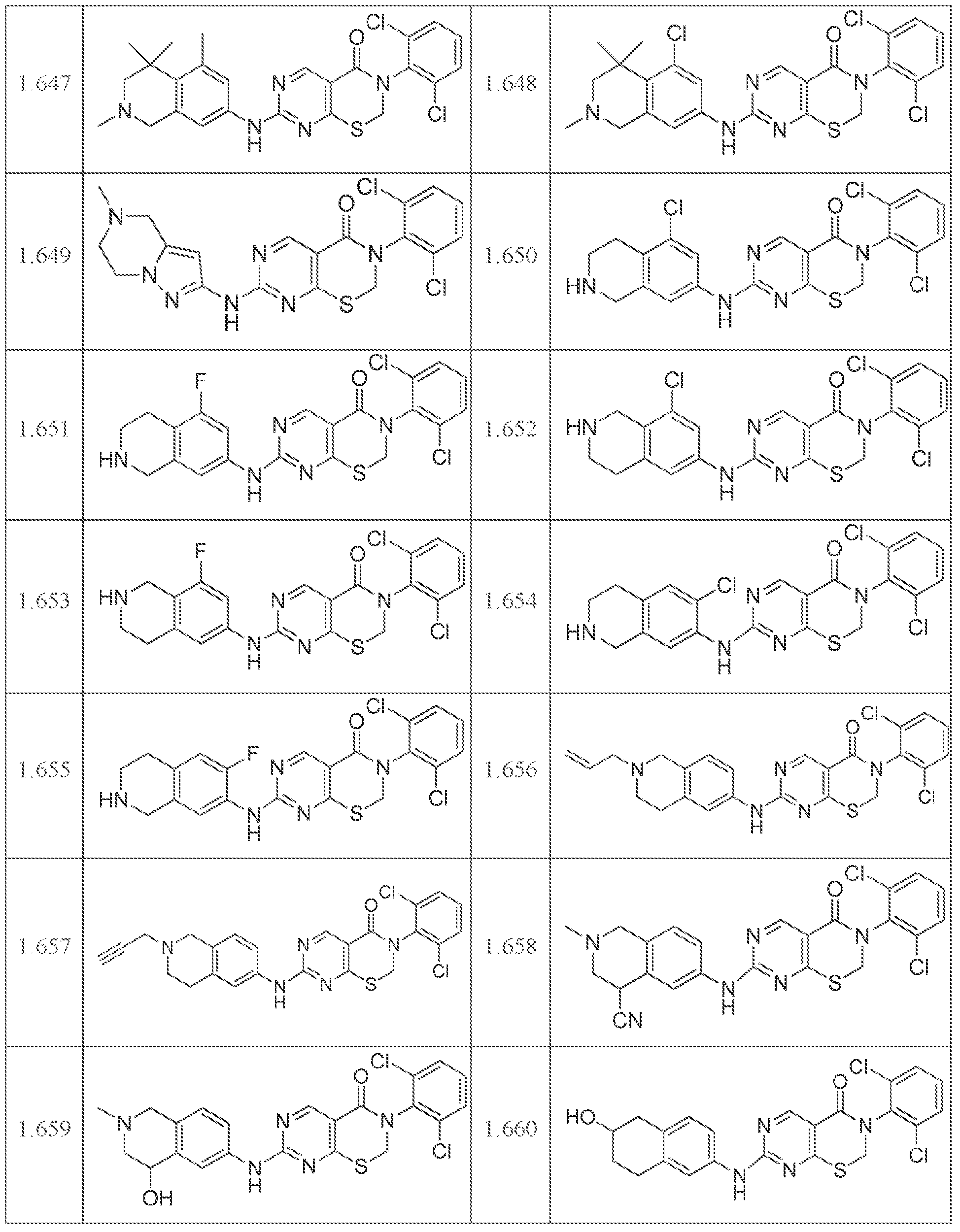
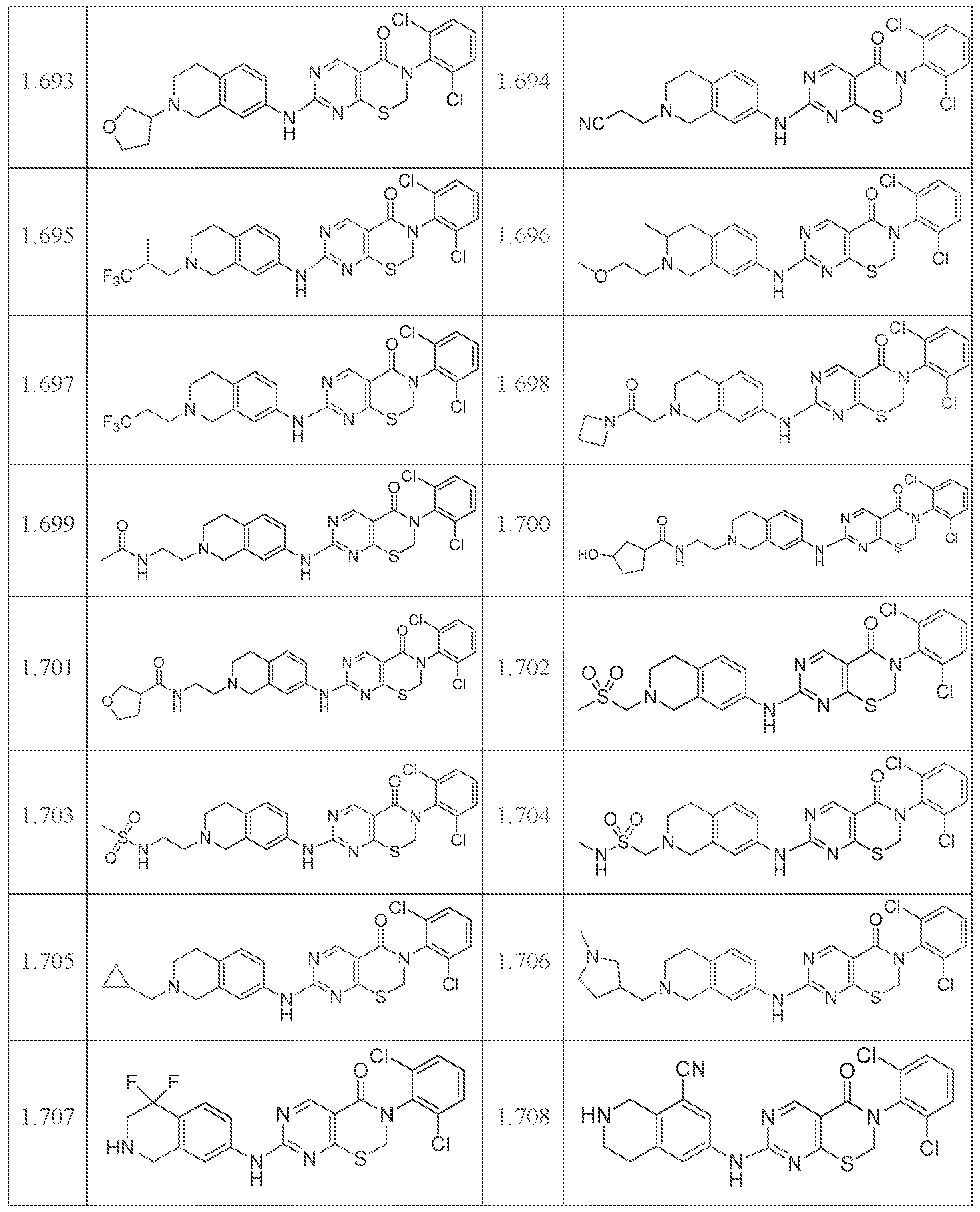
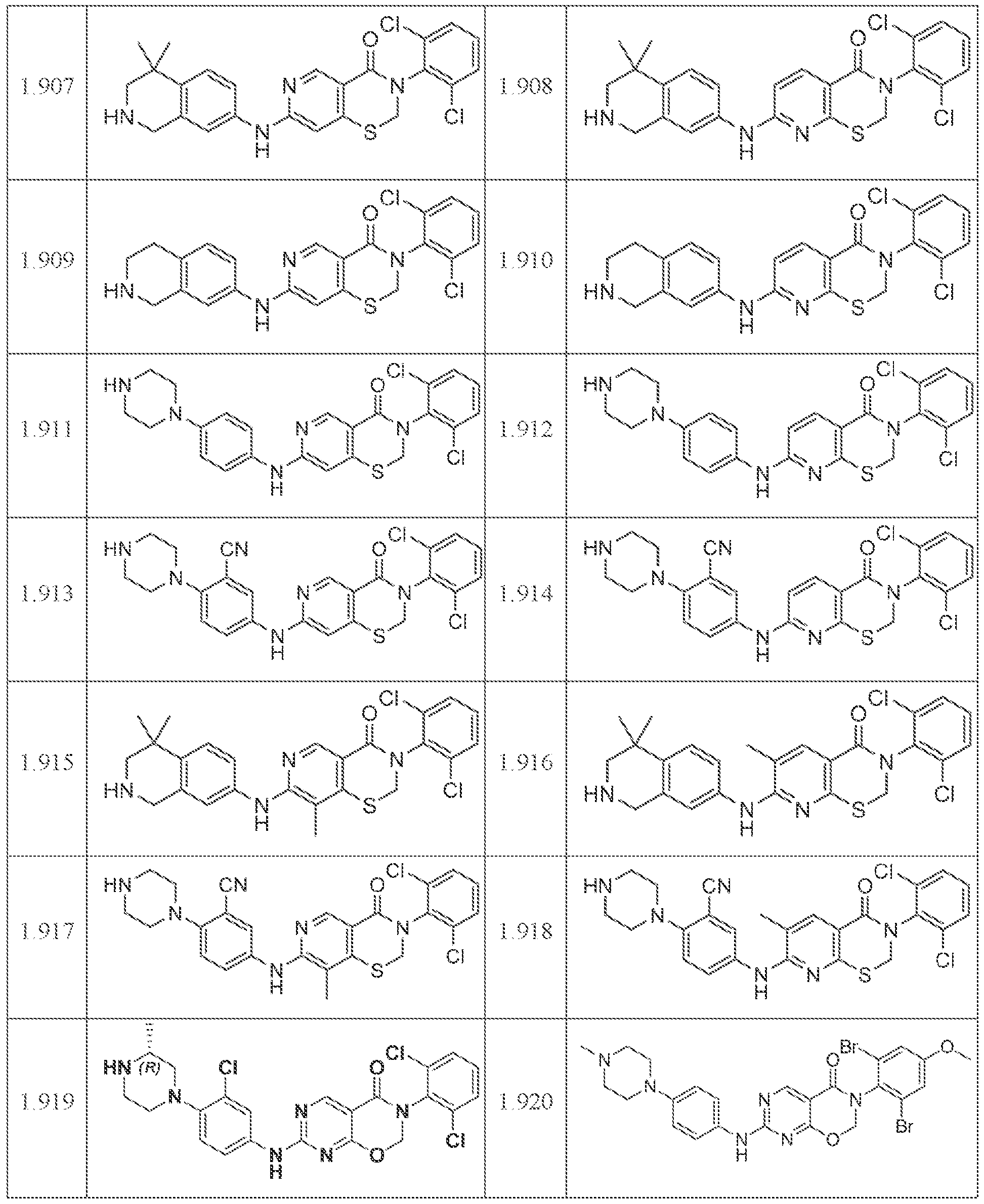
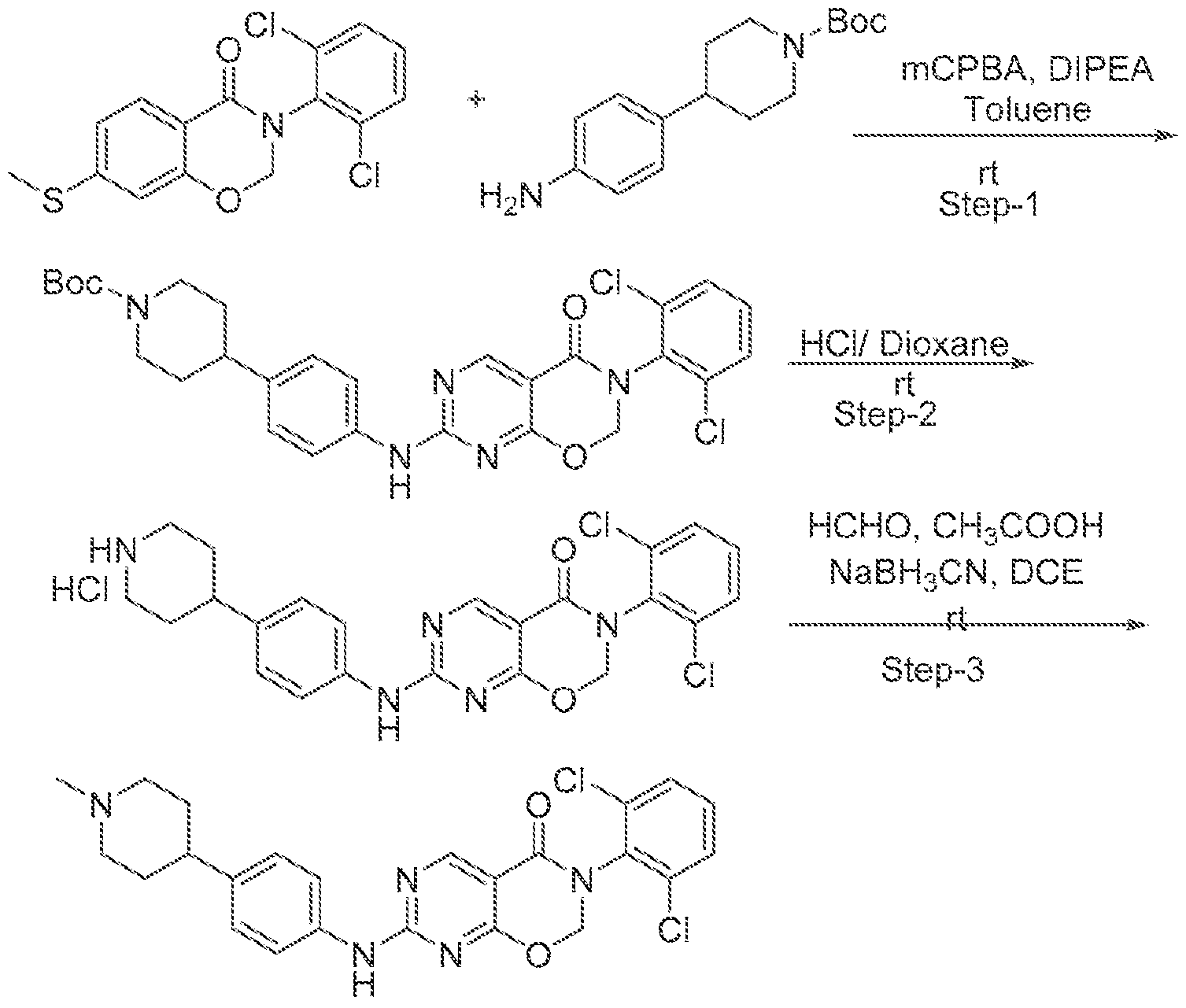
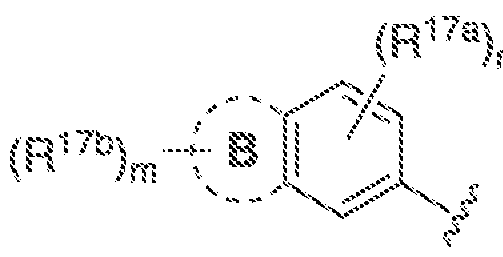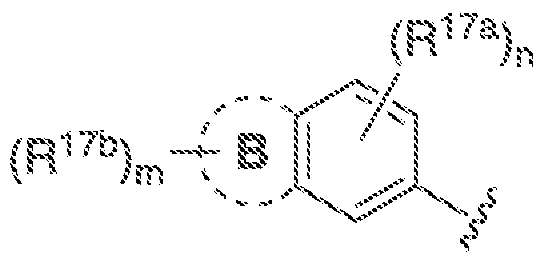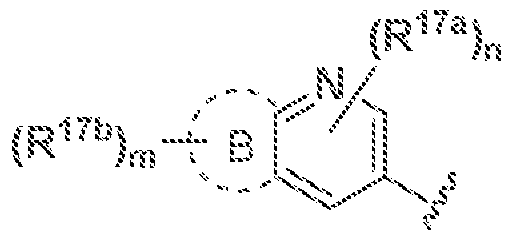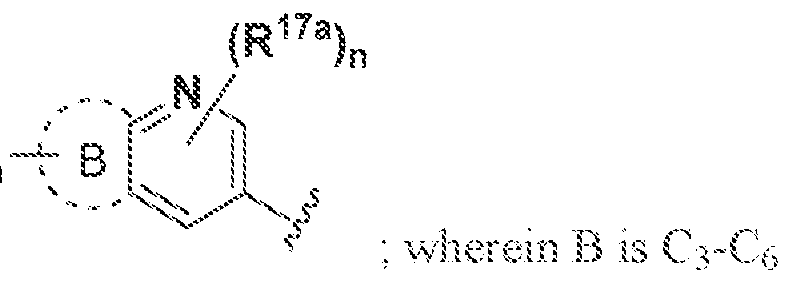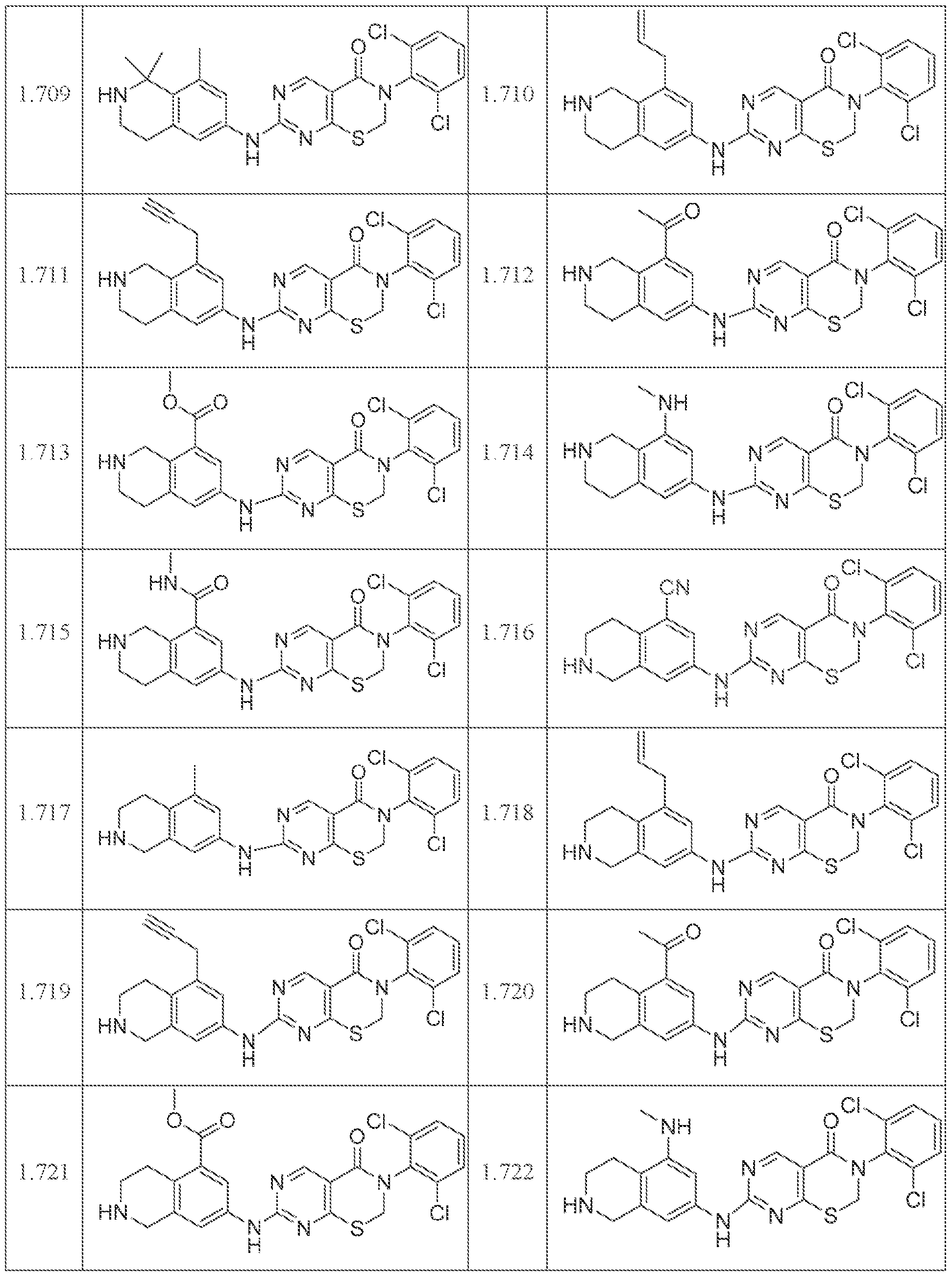HETEROCYCLIC COMPOUNDS AND USES THEREOF
CROSS- REFERENCE TO RELATED APPLICATIONS 001 I This application claims priority to U.S. Provisional Application No. 62/570,054, filed October 9, 2017, fee contents of winch are hereby incorporated by reference in their entirety.
FIELD OF THE INVENTION
[0002] Tins disclosure relates generally to therapeutics engaged n inhibition of the DNA damage checkpoint kinase, Weel , which potentiates genotoxic chemotherapies by abrogating cell-cycle arrest and proper DNA repair. The invention also provides pharmaceutically acceptable compositions comprising compounds of the present invention and methods of using said compositions in the treatment of diseases associated with this pathway.
BACKGROUND OF THE IN VENTION
[0003] Weel is a tyrosine kinase that phosphorylates and inactivates Cdc2 and is involved in G checkpoint Signaling. More particularly. Wee ; is involved in G2-M checkpoint Signaling. Because p53 is a key regulator in the G checkpoint, p53 -deficient tumors rely only on the G checkpoint after DNA damage. More particularly, because p53 is a key regulator in the (.1 · Λ checkpoint, p53--deficieni tumors rely only on the G :- \ i checkpoint after DNA damage. Hence, such tumors are selectively sensitized to DNA-damagmg agents by Weel inhibitio
[0004] Weel belongs to a family of protein kinases involved in the terminal phosphorylation and. inactivauon of cyclin-dependent kinase 1 -bound cyclm B, resulting in G cell cycle arrest in response to DNA damage. Weel was first identified in fission yeast, where Weel deficiency resulted in premature mitotic entry and replication of smaller-sized yeast. It is the major kinase responsible for the inhibitory phosphorylation of the tyrosine.
[0005] Before cells undergo mitosis, they progress through a tightly controlled, cascade of Gj -S, mtra-S, and G2-M checkpoints. Weel kinase has emerged as a key G .-M checkpoint regulator. This tyrosine kinase negatively regulates entry into mitosis by catalyzing an inhibitory phosphorylation of Cdc2 (the human homolog of cy dm- dependent kinase 1 (CDK1 ) on tyrosme- 1 5 (Y! 5). This results in mactrvation of the Cdc2/'cychn B complex, which arrests cells in G .- M
allowing for DNA repair. Such inhibition also occurs through Chkl -mediated inhibition of Cdc25 phosphatases, which remove the inhibitory phosphorylation on Cdc2. Thus, entry into mitosis rests on a balance between the opposing activities of Wee! and Chkl/Cdc25. Weel inhibition is thus expected to abrogate G2~M arrest and propel cells into premature mitosis, a hypothesis confirmed by studies documenting that Weel inhibition by either small molecule inhibitors or small interference 3R A leads to premature entry into mitosis and consequent cell death through mitotic catastrophe or apoptosis. (S. Miller, J. Clinical Oncology, 201 5).
[0006] Recenily, a few classes of Weel inhibitors have been disclosed. Among them is a selective inhibitor, AZD-1775 (L 2-allyl-l -(6-(2-hydroxypropan-2-yl)pyridin-2-yl)-6-((4- (4rnediylpi eraziml~yl)phenylSanun^ AZD-1 775 exhibited antitumor activity in various preclinical studies in potentiating cherno- and
radiotherapy and is currently in phase LIT clinical trials.
[0007] Weel is highly expressed in several cancer types, including hepatocellular carcinoma, breast cancers, cervical cancers, lung cancers, squamous cell carcinoma, diffuse intrinsic pontine glioma (DIPG), glioblastoma, medulloblastorna. leukemia, melanoma, and ovarian cancers. (P. Reigan et l, Trends in Pharmacol Sc , 2016).
[0008] There are few Weel inhibitors in clinical development. There is scope to improve Weel inhibitor selectivity and the properties of the inhibitors to permit targeting of specific cancer types.
BRIEF SUMMAR Y OF THE INVENTION
In one aspect, provided is a compound of Formula (I):
or a tautomer or isomer thereof, or a pharmaceutically acceptable salt of any of the foregoing. wherein U, W, X, Y, Z, R/", R '. and R" are as detailed herein.
[0010] In some embodiments, provided is a compound of Formula (la):
(la), or a salt thereof, wherein W, X Y, Z. R
~,<!, R
". and R
' are as detailed herein.
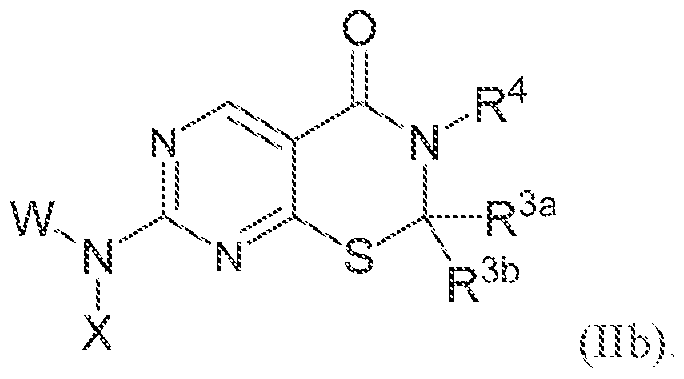
[0011] in some embodiments, the compound of Formula (I), or a tautomer or isomer thereof, or a. pharmaceutically accepiable salt of any of the foregoing, is of Formula (la ), (lb), i l ia ?. (lib), (Ilia), (fflb), (i:Va) or (IVb), as detailed herein.
[0012] In another aspect, provided is a. method of treating cancer in an individual in need thereof comprising administering to the individual a therapeutically effective amount of a compound detailed herein, such as a compound of Formula (I), or a pharmaceutically acceptable salt thereof in some embodiments, provided is a method of treating cancer in an individual in need thereof comprising administering to the individual a therapeutically effective amount of a compound detailed herein, such as a compound of Formula (!)„ or a tautomer or isomer thereof; or a pharmaceutically acceptable salt of any of the foregoing. Also provided is a method of inhibiting Wee! in a cell, comprising administering a compound detailed herein, or a salt thereof, to the cell. Also provided is a method of inhibiting Weel in a cell, comprising administering a compound detailed herein, or a tautomer or isomer thereof, or a pharmaceutically acceptable salt of any of the foregoing, to the cell.
[0013] in another aspect, provided are pharmaceutical compositions comprising a compound detailed herein and a pharmaceuticaily acceptable carrier or excipient. Pharmaceutical compositions comprising a compound detailed herein, or a tautomer or isomer thereof, or a pharmaceuticaily acceptable salt of any of the foregoing, and a pharmaceutically acceptable earner or excipientKits comprising a compound detailed herein or a pharmaceutically acceptable salt thereof are also provided. Kits comprising a compound detailed herein, or a tautomer or isomer thereof, or a pharmaceutically acceptable salt of any of the foregoing, are also provided. Compounds as detailed herein or a pharmaceutically acceptable salt thereof are also provided for the manufacture of a medicament for the treatment of cancer. Compounds as detailed herein, or a
iautomer or isomer thereof, or a pharmaceutically acceptable salt of any of the foregoing, are also provided for the manufacture of a medicament for the treatment of cancer.
DETAILED DESCRIPTIO OF THE INVENTION
[0014] " Alkyl" refers to and includes saturated linear and branched univalent hydrocarbon structures and combination thereof having the number of carbon atoms designated (i.e. , 0 · ·( ;: i means one to ten carbons). Particular alkyl groups are those havmg ! to 20 carbon atoms (a "(' · · (¾ alkyl"). More particular alkyl groups are those having 1 to 8 carbon atoms (a "0 ; ·ί > alkyl"), 3 to 8 carbon atoms (a alkyl")., 1 to 6 carbon atoms (a "€] -( , alkyl')}, 1 to 5 carbon atoms
(a "C j -Cs alkyl"), or 1 to 4 carbon atoms (a "C i --Cd alkyl"). Examples of alkyl include, but are not Umited to, groups such as methyl, ethyl, mpropyl, isopropyl, n-butyl, t-buryl, isobutyk sec- butyl homologs and isomers of, for example, n-pentyl, n-hexy!, n-heptyl, n-octyl, and die like.
[0015] "Alkenyl" as used herein refers to an unsaturated linear or branched univalent hydrocarbon chain or combination thereof, having at least one site of olefmic unsaturation (i. e. , having at least one moiety of the formula (>::C) and having the number of carbon atoms designated (i.e. , ( Cio means two to ten carbon atoms). The alkenyl group may be in "as" or "trans" configurations, or alternatively i n "E" or "2" configurations. Particular alkenyl groups are those having 2 to 20 carbon atoms (a "C2-C20 alkenyl"), having 2 to 8 carbon atoms (a "C?- C N alkenyl"). having 2 to 6 carbon atoms (a 'Τ¾ -(¾ aikenyl"), or having 2 to 4 carbon atoms (a "C2-C4 alkenyl"). Examples of alkenyl include, but are not limited to, groups such as ethenyl (or inyl), pfop-l -enyj, prop--2--enyi (or allyl). 2-metlwlprop-l -en l, but- l -enyl, but-2-enyl, but-3- enyl. buta- l ,3-dienyl, 2-methylbuta- 1 ,3-dienyL homologs and isomers thereof, and the like.
[0016] "Alkylene" as used herein refers to the same residues as alkyl, but having bivalency. Particular alkylene groups are those having 1 to 6 carbon atoms (a ' Ch -C ·: alkylene"), 1 to 5 carbon atoms (a ' CYC : alkylene"), 1 to 4 carbon atoms (a "Cj -Ci alkylene") or 1 to 3 carbon atoms (a "C 1-C3 alkylene"). Examples of alkylene include, but are not limited to, groups such as
methylene (-(¾-), ethylene i-CH2CH2~l propylene (~C¾C¾CH2~); buiylene
{••C! kO Π ;(Ί I .- ;, and the like.
[0017] "AlkynyP
" as used herein refers to an unsaturated linear or branched univalent hydrocarbon chain or combination thereof, having at least one site of acetyiemc unsaturation (i.e. , having at least one moiety of the formula C≡C) and having the number of carbon atoms designated (i.e., (¾rCio means two to ten carbon atoms). Particular alkynyi groups are those ha ving 2 to 20 carbon atoms (a "C
2-C2o alkynyi"). ha ving 2 ιο 8 carbon atoms (a "C
2-C§ alkynyi"). having 2 to 6 carbon atoms (a
alkynyi"), or having 2 to 4 carbon atoms (a "C
2- C.| alkynyi'
''). Examples of alkynyi include, but are not limited to, groups such as ethyny! (or acetylenyi), prop~l ~ynyl, prop-2-ynyl (or propargyl), bitt- l -ynyl, but-2-ynyl, but-3~ynyl, homologs and isomers thereof, and the like
[0018] "AryP refers to and includes polyunsaturated aromatic hydrocarbon groups. Aryl may contain additional fused rings (e.g., from 1 to 3 rings), including additionally fused aryl, heteroaryl, cycloalkyl, and/or heterocyclyl rings. In one variation, the aryl group contains from 6 to 14 annular carbon atoms. Examples of aryl groups include, but are not limited to, phenyl, naphthyl, biphenyl, and the like.
[0019] "Carbony!" refers to the group CO.
[0020] "Cycloalkyl" refers to and. includes cyclic univalent hydrocarbon structures, which may be fully saturated, mono- or polyunsaturated, but which are non-aromatic, having the number of carbon atoms designated (e.g., < ; --<' ·., means one to ten carbons). Cycloalkyl can consist of one ring, such as cyclohexyl, or multiple rings, such as adamantly, but excludes aryl groups. A cycloalkyl comprising more than one ring may be fused, spiro or bridged, or combinations thereof A preferred cycloalkyl is a cyclic hydrocarbon having from 3 to 13 annular carbon atoms. A more preferred cycloalkyl is a cyclic Iiydrocarbon having from 3 to 8 annular carbon atoms (a " Γ ;··(" Ν cycloalkyl"). Examples of cycloalkyl include, but are not limited to, oyclopropyl, cyclobutyl, cyclopeniyi, cyclohexyl, 1 -cyclohexe ryl, 3-cyclohexenyl, cycloheptyl, norbornyl, and the like.
[0021 ] 'ΉαΚν' or "halogen5 ' refers to elements of the Group 17 series having atomic number 9 to 85. Preferred halo groups include fluoro. chloro, bromo and iodo. Where a residue rs substituted with more than one halogen, ii may be referred to by using a prefix corresponding to the number of halogen moieties attached, e.g., dihaloaryi, dihaloalkyl, trihaioaryl etc. refer to aryl and alky! substituted with two {"di") or three O'tri") halo groups, which may be but are not necessarily the same halo; thus 4~chloro-3-£luorophenyl is within the scope of dihaloaryi. An alkyl group in which each hydrogen is replaced with a halo group is referred to as a
"perhaloalkyl." A preferred perhaloalkyl group is trifluoroalkyi (-CF3). Similarly,
"perknoa li- : :\v" refers to an alkoxy group in which a halogen takes the place of each H in the hydrocarbon making up the alkyl moiety of the alkoxy group. An example of a perhaloalkoxy group is tnfluoromeihoxy ; ··()( V -. ·.
[0022] "Heteroaryl" refers to and includes unsaturated aromatic cyclic groups having from 1 to 10 annular carbon atoms and at least one annular heteroatom , including but not limited to heteroatoms such as nitrogen, oxygen and sulfur, wherein the nitrogen and sulfur atoms are optionally oxidized, and the nitrogen atom(s) are optionally quaternized, A heteroaryl group can be attached to the remainder of the molecule at an annular carbon or at an annular heteroatom. Heteroaryl may contain additional fused, rings (e.g., from 1 to 3 rings), including additionally fused aryl, heteroaryl, cycloalkyl, and/or heierocyclyl rings. Examples of heteroaryl groups include, but are not limited to, pyrtdyt, pynmidyl, thiophenyl, furanyi, thiazolyl, and the like
[0023] "Heterocycle" or "heteroeyclyr refers to a. saturated or an unsaturated non-aromatic group having from 1 to 10 annular carbon atoms and from I to 4 annular heteroatoms, such as nitrogen, sulfur or oxygen, and the like, wherein the nitrogen and sulfur atoms are optionally oxidized, and the nitrogen atom(s) are optionally quaternized. A heierocyclyl group may have a. single ring or multiple condensed rings, but excludes heteroaryl groups. A heterocycle comprising more than one ring may be fused, spiro or bridged, or any combination thereof in fused ring systems, one or more of the fused rings can be aryl or heteroaryl. Examples of heierocyclyl groups include, but are not limited to. tetrahydropyranyl, dihydropyranyl, piperidinyl, piperazinyl, pyrrolidiuyl, tluazolmyl, thrazolidinyl. tettah drofuranyl,
teirahydrothiophenyl, 2,3- ihydrobenzo[b]thiophen-2-yl, 4-amiiio-2-oxopyrimidin-l(2H)- l, and the like.
[0024] · 0 ,ο' refers ΐο the moiety 0
[0025] "Optionally substituted" unless otherwise specified means that a group may be ims instituted or subsiiiirted by one or more (e.g., 1, 2, 3, 4 or 5) of the substituents listed for that group in which the subsiituents may be the same of different, in one embodiment an optionally substituted group has one substituent. In another embodiment, an optionally substituted group has two subsiituents. In another embodiment, an optionally substituted group has three subsiituents. in another embodiment, an optionally substituted group has four substituents. In some embodiments, an optionally substituted group has 1 to 2, 2 to 5, 3 to 5, 2 to 3, 2 to 4, 3 to 4, 1 to 3, 1 to 4 or 1 to 5 substituents.
[0026] A "pharmaceutically acceptable earner" refers to an ingredient in a pharmaceutical formulation, other than an active ingredient, which is nontoxic to a subject. A pharmaceutically acceptable carrier includes, but is not limited to, a buffer, excrpient stabil izer, or preservative.
[0027] As used herein, "treatment" or "treating" is an approach for obtaining beneficial or desired results including clinical results. For example, beneficial or desired results include, but are not limited to, one or more of the following: decreasing symptoms resulting from the disease, increasing the quality of life of those suffering from the disease, decreasing the dose of other medications required to treat the disease, delaying the progression of the disease, and/or prolonging survival of individuals. In reference to cancers or other unwanted cell proliferation, beneficial or desired results include shrinking a tumor (reducing tumor size); decreasing the growth rate of the tumor (such as to suppress tumor growth), reducing the number of cancer cells: inhibiting, retarding or slowing to some extent and preferably stopping cancer ceil infiltration into peripheral organs; inhibiting (slowing to some extent and preferably stopping) tumor metastasis; inhibiting tumor growth, preventing or delaying occurrence and/or recurrence of tumor, and/or relieving to some extent one or more of the symptoms associated with the cancer, in some embodiments, beneficial or desired results include preventing or delaying occurrence and/or recurrence, such as of unwanted cell proliferation,
[0028] As used herein, "delaying development of a disease" means to defer, hinder, slow, retard, stabilize, and/or postpone development of the disease (such as cancer). This delay can be of varying lengths of time, depending on the history of the disease and/or individual being treated. As is evident to one skilled m the art, a sufficient or significant delay can, in effect.
encompass prevention, in that the indi idual does not develop the disease. For example, a late stage cancer, such as development of metastasis, may be delayed.
[0029] As used herein, an "effective dosage" or "effective amount" of compound or salt thereof or pharmaceutical composition is an amount sufficient to effect beneficial or desired results. For prophylactic use, beneficial or desired results include results such as eliminating or reducing the risk, lessening the seventy of, or delaying the onset of the disease, including biochemical, histological and/or behavioral symptoms of the disease, its complications and intermediate pathological phenotypes presenting during development of the disease. For therapeutic use, beneficial or desired results include ameliorating, palliating, lessening, delaying or decreasing one or more symptoms resulting from the disease, increasing the qual ity of life of those suffering from the disease, decreasing the dose of other medications required to treat the disease, enhancing effect of another medication such as via targeting, delaying the progression of the disease, and/or prolonging survival In reference to cancers or other unwanted cell proliferation, an effective amount composes an amount sufficient to cause a tumor to shrink and/or to decrease the growth rate of the tumor (such as to suppress umor growth) or to prevent or delay other unwarned cell proliferation. In some embodiments, an effective amount is an amount sufficient to delay development In some embodiments, an effective amount is an amount sufficient to prevent or delay occurrence arid/or recurrence. An effective amount can be administered in one or more administrations, in the case of cancer, the effective amount of the drug or composition may: (i) reduce the number of cancer cells; (ii) reduce tumor size; (Hi) inhibit, retard, slow to some extent and preferably stop cancer cell infiltration into peripheral organs, (iv) inhibit (i.e., slow to some extent and preferably stop) tumor m tastasi , (v) inhibit tumor growth; (vi) prevent or delay occurrence and/or recurrence of tumor; and/or (vii) relieve to some exten one or more of the symptoms associated with the cancer. An effective dosage can be administered in one or more administrations. For purposes of this disclosure, an effective dosage of compound or a salt thereof, or pharmaceutical composition is an amount sufficient to accomplish prophylactic or therapeutic treatment either directly or indirectly. It is intended and understood that an effective dosage of a compound or salt thereofi or pharmaceutical
composition may or may not be achieved in conjunction with another drug, compound, or pharmaceutical composition. Thus, an "effective dosage" may be considered m the context of administering one or more therapeutic agents, and a single agent may be considered to be given
in an effective amount if, in conjunction with one or more other agents, a desirable result may be or is achieved.
[0030] As used herein, the term "individual''' is a mammal, including humans. An individual includes, but is not limited to. human, bovine, horse, feline, canine, rodent, or primate, in some embodiments, the individual is human. The individual (such as a human) may have advanced disease or lesser extent of disease, such as low tumor burden. In some embodiments, the individual is at an early stage of a proliferative disease (such as cancer). In some embodiments, the individual is at an advanced stage of a proliferative disease (such as an advanced cancer).
[0031] Reference to "about" a value or parameter herein includes (and describes) embodiments thai are directed to that value or parameter per se. For example, description referring to "about X" includes description of "X".
[0032] it is understood that aspects and variations described herein also include "consisting" and/or "consisting essentially of' aspects and variations.
Compounds
[0033] in one aspect, provided is a compound of Formula (I):
or a tautomer or isomer thereof, or a pharmaceutically acceptable salt of any of the foregoing, wherein:
U is O or S;
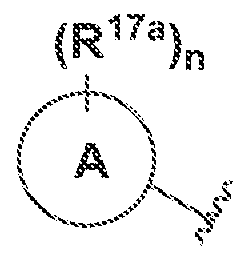
W is A or Ail wherein A and B are fused together;
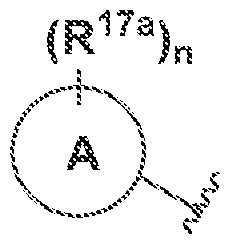
A is phenyl or 5·- to 6-membered heteroar l, each of which is optionally substituted with
R
! l>, wherein A and '
l> together are
and n is 0, 1, 2, 3, or 4:
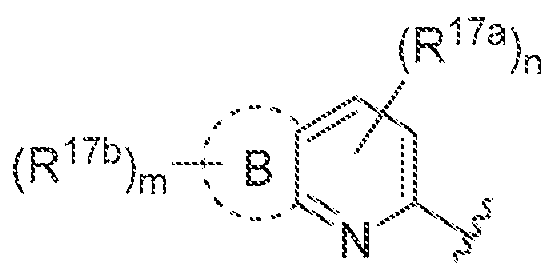
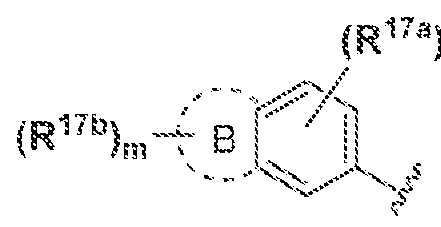
B is C-Co cycloalkyl.3-· ΐο 7~membered heterocyclyl, 5- to 7-mernbered heteroaryl, or (\. aryl, each of which is optionally substituted withR
"'\ wherein A, B, R
!"
!, andR"
"' together
(Ri7a)„ are "~ and m and n are independently ϋ, i.2, 3, or 4:
X is hydrogen or (Γχ~ί¾ alkyl:
Y is Nor C ;
Z is N or CR2;
R" and ." are independently hydrogen or 1·' ' ".
R" and IT ' are independently hydrogen or R*':>, or
R and are taken together wth fee carbon to winch they are attached to f nn a <V ('· . cycloalkyl,
each ' is independently oxo or R:'"1, or any two R! ' !' groups, when bound to the same carbon atom or two different carbon atoms, are taken together with the carbon or carbons to winch they are attached to form a (V
i o
('·. cycloalkyl or 3- to 7-memhered heterocyclyl, each is optionally substituted by R' '; each is independently (";-C1: alkyl. ( h--( alkenyl, *—C.. alkynyl, halogen, -CM, -OR10, -SR10, ~NE/1]K12, -C(())R10, -C(0)OR10, -Si(C C6 alkyl)3, -C(0)NRnR12,
-OC(0)NR]iR12, -NR10C(O)R]1, -NRi0C(O)NRuR12, ~S(0)2R10, -NR10S(O)RI!,
-S(i))2NRJ lRi2 3 C3--C6 cycloalkyl, 3- to 12-membered heterocyclyl, 5- to 10-membered heteroaryl, ( .- ' a aryl, -(CrC alkyiene)CN, · γ.(\ alkylene)ORIG, -if -Ch alkylene)SRi0, •n <h alkylene)MRUR12. ··(( ( alkylene)CF3, -f<* (\ alkylene)C(0>R10, -{C-r
(¾ alkylene)C(0)N R"", ·;(Υ·ί , alkylene)NR C(G)R'\ ·ΚΥ·
C3 alkylene)MR10C(OsNRUR12, -(C:-C3 alkylene)S(O)2Ri , -(C C3 alkylene)NRU,S(0)2RU, ·((·; ·( , alkylene)S(C))2NRijRi2, -(CrC3 alkyiene)(C C6 cycloalkyl), -iC C3 alkylene)(3- to 12-menibered heterocyclyl), -:C; -f . a!kylene}(5- to 10-menibered heteroaryl) or
··<;(' ikylene)(C6~ |4 aryl), wherem each R ' is independently optionally substituted by halogen, oxo, -OR13, -NR13R , -C(0)R13, -C(0)NR13R14, -NR13C(0)R14, ~SCO)2R13,
-NR13S(0)2R14, -S(0)2NR1 R14, ·{( Yb aikylene}C(0)NR13R14, ·{( ,·
C3 alkylene)NR C(O)R14, ··(( ;·( alkylene)S(0)2R1 , -(C]-C3 alkylene)NR1 S(O)2R14, -(C
C3 alkylene)S(0)2NR!3Ri 5 -(CrC alkyiene)(C3-C6 cycloaikyi), ··;( ( ; alkylene)(3- to 12- rnembered heterocyciyl), -Si(C C6 alkyl)3, -CN,■■(€;■■(¾ alkylene)GR13, -(Ci-
C3 alkylene)NRI3RU, -(CrC3 a1kylene)C(0)R13, C3-C3 cycloalkyl, or (Vi ,; alkyl optionally substituted by oxo, -OH or halogen,
R4 is hydrogen, Cj-Q alkyl C2-C6 alkenyl, C2-C6 alkynyl, C3-C6 cycloalkyk 3- to 12- rnenibered heierocyclyi, 5- to 10-memhered heteroaryl Qy-Cu aryl , ·(( ;-<b aikvjeneKC ;, cycloalkyl), ··{< ('·. alkylene)(3- to 12-membered heterocyciyl), ··{(';·( · alkylene}(5- to 10- mernbered heteroaryl) or ~(Cj-C alkylene)iLVC:4 aryl), wherem R" is independently optionally substituted by halogen, oxo, -OR33, -NR13Ri4, -C(Q)Ri3, -CN, C3-C8 cycloalkyl, or C C6 alkyl optionally substituted by oxo, -OH or halogen;
K " is independently hydrogen, C\~C(, alkyl, C2-C6 alkenyl alkynyl, 0 ~1¾ cycloalkyk C C14 aryl, 5-· to 6-membered heteroaryl or 3- to 6-membered heterocyciyl, wherein the < YC .··: alkyl, C2-C0 alkenyl, ( rC;, alkynyl, <YC .··. cycloalkyl, y-Cu aryl, 5- to 6-membered heteroaryl and 3- to 6-membered heterocyciyl are independently optionally substituted by halogen, oxo, -CN, -OR/'', - '''R!l, or C]-C6 alkyl optionally substituted by halogen, -OH or oxo,
R" and R'" are each independently hydrogen, Cj-Cg alkyl, C3~C6 cycloalkyl, C C14 aryl, 5- to 6-membered heteroaryl or 3- to 6-membered heterocyciyl, wherein the Cj-C6 alkyl, (',·( ·: cycloalkyl, C CM aryl, 5- to 6-membered heteroaryl and 3- to 6-membered heterocyciyl ofR." andR" " are independently optionally substituted by halogen, oxo, -CN, -OR15, - R15R j6 or (' ; ·( :·. alkyl optionally substituted by halogen, --Oil or oxo,
or R1 : and R' " are taken together with the atom to which they attached to form a 3- to 6- membered heterocycly! optionally substituted by halogen, oxo, or C1-C0 alkyl optionally substituted by halogen:
n 1 -i
R and R" ' are each independently hydrogen, ] - 6 alkyl, CVi .·. cycloaikyi, or 3·- to 6- membered heterocyclyl, herei the C C& alk l, C3--Q cycloaikyi, or 3- to 6-mern bared lieterocyciyl of R° and R '" are optionally substituted by halogen, -CN, -OR " . -\'R " R ' '. or oxo, or k : and " f are taken together with the atom to which they attached to form a 3·· 10 6- membered heterocyclyl optionally substituted by halogen, oxo or C C alkyl optionally substituted by halogen or oxo; and
R ' and K " ' are each independently hydrogen, C1-C6 alkyl optionally substituted by halogen or oxo, ry-C '.:. alkenyl optionally substituted by halogen or oxo, or ( : ·( .·. alkynyl optionally substituted by halogen or oxo, or ' are taken together with the atom to winch they attached to form a 3-· to 6- membered heterocyclyl optionally substituted by halogen, oxo or C i¾ alkyl optionally substituted by oxo or halogen,
[0034] In some embodiments of a compound of Formula (I), at least one of Y and Z is N [0035] in some embodiments, provided is a compound of Formula (la)'
or a salt thereof! wherein:
W is A or AB, wherein A and B are fused together;
A is phenyl or 6-membered heieroar l, each of which is optionally substituted with R' '.
wherein A and R
' together are
n is 0, 1, 2, 3, or 4;
B is cycloalkyl 3·· ιο 7-membered heterocyclyl 5- to 7-membered heteroaryl, or
C aryl, each of which is optionally substituted with R
; wherein A, B. R
J''!>, and R
J together
and ni and n are independently 0, 1 , 2, 3, or 4;
X is hydrogen or (YC .· alkyl;
Y is Nor CR ;
Z is N or CR2; and R." are independently hydrogen or R. ;
R " and R ' ' are independently hydrogen or R";>, or
R " and R are taken together with the carbon to which they are attached to form a ( ' : · ('· . cycloalkyl,
each R ' is independently oxo or R:'"1, or any two R! ' !' groups, when bound to the same carbon atom, are taken together with the carbon to which they are attached io form a (¾-C6 cycloalkyl; each R"'"" is independently i r< alkyl alkenyl, C'yi .··. alkynyl, halogen, -UN,
-OR10, -SR10, -NR;:Ri :. -nO- ^ -C(G)NR R12, -OC(0)NR1;R -NRi C(())Rn,
-NRi C(O)NRljRi2 5 -S(0}2R10, - R10S(O)2R , Α(();;ΝΙ ; :. C3-C6 cycloalkyl 3- to 12- niembered heterocyclyl, 5- to 10-niembered heteroaryl Q-Cu aryl, ···(';·( · aikylene}CN, -(Cj- C3 alkylene)OR1 , -(&-<¾ alkylene)SR10, -(C C alkylene)NRnR12, -iCrC3 alkylene)CF3, -(C C3 alkylene)C(O)R10 ; -(C C3 aikylene)C(0}NR] LR12 S -(€:■-(¾ alkylene)NRIGC(OjRri ;
-(C C aikyjene)NR]0C(O)NRnR12, -(C C aikylene)S(0)2R10, ·ΚΥ
C3 alkylene)NR1 SiO)2RU, -(C C3 nk viene;S(0)A : 1 R;\ -«V<\ alkylene)(C3-C6
cycloalkyl), -iC■.
■(': alkylene}(3- io 12-rnembered heterocyclyl), -(C C alkylene}(5- io 10- membered heteroaryl) or
ts independently optionally substituted by halogen, oxo,
-CN, -(CrC3 aikylene)OR , ·ίί>·<\ alkylene)NR RU ••;C:-f ; alkylenelQOsR13, C3-C¾ cycloalkyl, or <'···('>. alkyl optionally substituted by oxo, ~OH or halogen:
R
*; is hydrogen, ( :·'
',, alkyl, C
2-C
6 alkeny!, C
2-Q alkynyl, C
3-C
6 cycloalkyl, 3- io 12- membered heterocyclyl, 5- to 10~mernbered heteroaryl, Q5-C14 aryl . ·((
';··( , alkylene)(C
3-C6 cycloalkyl), -(Ci--C
3 alkylene)(3- to 12-membered heterocyclyl), -(C
rC
3 alkylene)(5- to 10· membered heteroaryl) or ·;(
';··(
" ,
aryl), wherein R
+ is independently optionally substituted by halogen, oxo, -OR
13, -blR
1 V\ ·( (0}R
: . -CN, C
3~C
8 cycloalkyl, or C
3-C alkyl optionally substituted by oxo, -OH or halogen;
R
:" is independently hydrogen, Cj-Cg alkyl,
cycloalkyl, aryl, 5- to (?· membered heteroaryl or 3-· to 6-mernbered heterocyclyl, wherem the
r ···(
'.;. alkyl, CT-Q, alkenyl, < ;-< alkynyl, CYC
!: cycloalkyl, C^- u aryl, 5~ to 6-membered heteroaiyl and 3·· 106- membered heterocyclyl are independently optionally substituted by halogen, oxo, -CN, -OR
J"', -NR
!" l , or C C
ft alkyl optionally substituted by halogen, -OH or oxo,
R" and R'" are each independently hydrogen, C C alkyl, C -C6 cycloalkyl, Y ·.· aryl, 5- 106-membered heteroaryl or 3- to 6-mernbered beierocyclyl, wherein the YC .··. alkyl, C Y cycloalkyl, C6-C14 aryl, 5- to 6-mernbered heteroaryl and 3- to 6-membered heterocyclyl oC R ' ' and K" ' are iridependently optionally substituted by halogen, oxo, -CN, -OR15, -NRV6 or C ;·(';, alkyl optionally substituted by halogen, -OH or oxo, or R" a:-d R' " are taken together with the atom to winch they attached to form a 3- to 6- membered heterocyclyl optionally substituted by halogen, oxo, or Cj-Cg alkyl optionally substituted by halogen;
R : and Λ" are each independently hydrogen or C| --C6 alkyl, wherein the CYC .·. a!kyl of are optionally substituted by halogen, -OR'",••NRi "R'v', or oxo., or R and R " ' are taken together with the atom to which ihey attached to form a 3-- to 6- mernbered beierocyclyl optionally substituted by halogen, oxo or Cj-Ce alkyl optionally substituted by halogen or oxo; and
'FT" and R " ' are each independently hydrogen, C C alkyl optionally substituted by halogen or oxo, !: alkenyl optionally substituted by halogen or oxo, or C2-C6 alkynyl optionally substituted by halogen or oxo, or R and R'" are taken together with the atom to which they attached to form a 3-- to 6- membered beierocyclyl optionally substituted by halogen, oxo or CVC alkyl optionally substituted by oxo or halogen.
[0036] in some embodiments, provided is a compound of Formula (la), or a tautomer or isomer thereof, or a pharmaceutically acceptable salt of any of the foregoing.
[0037 in some embodiments, the compound of formula (I) is of Formula (lb):
or a tautomer or isomer thereof, or a pharmaceutically acceptable salt of any of the foregoing, wherein W, X, Y, . '.. R ". RT', and ΙΓ are as detailed herein
[0038] In some embodiments,, provided is a. compound of Formula (11a):
or a iauiornef or isomer thereof, or a pharmaceutically acceptable salt of any of the foregoing, wherein W, X, R K and K
" are as derailed herein.
[0039] In some embodiments, the compound of Formula (lib):
or a tautoroer or isomer thereof, or a pharmaceutically acceptable salt of any of the foregoing, wherein W, X, R">d, K "' . and R'* are as detailed herein.
[0040| in some embodiments, provided is a compound of Formula (ilia):
or a iauiomer or isomer thereof, or a pharmaceutically acceptable salt of any of the foregoing, wherein W, X, !· R and R are as detailed herein.
[0041] in some embodiments, the compound of Formula (Hib):
or a iauiomer or isomer thereof, or a pharmaceutically acceptable salt of any of the foregoing, wherein W, X, R a, R and R'v are as detailed herein,
[0042] In some embodiments, provided is a. compound of Formula. (iVa):
or a tautomer or isomer thereof, or a pharmaceutically acceptable salt of any of the foregoing, wherein W, X, R ". !< ' . and !<" are as detai led herein.
|'0043) In some embodiments, tbe compound of Formula (!Vb):
or a tautomer or isomer thereof, or a pharmaceutically acceptable salt of any of the foregoing, wherein W, X. k " R and iV are as detailed herein.
[0044] In some embodiments of a compound of Formula (I ), U is (). In some embodiments of a compound of Formula (Ϊ), U is S.
[0045] In some embodiments of a compound of Formula (I ), (la), (lb), (Ila), (lib), (Ilia), (Iiib), (IVa) or (IVb), X is hydrogen, in some embodiments of a compound of Formula (I), (ia), (lb), (Ha), (lib), (Ilia), (Iiib), (IVa) or (IVb), X ·> C C6 alky!. In some embodiments of a compound of Formula (I), (la), (lb), (Ila), (lib), (Ilia), (Iiib), (IVa) or (IVb), X is methyl.
[0046! ^ some embodiments of a compound of Formula (I), Y is N. ! n some embodiments of a compound of Formula (I), Y is CR\ In some embodiments of a compound of Formula (I), Y is CFi.
[0047 In some embodiments of a compound of Formula (I), Z is N. In some embodiments of a compound of Formula (I), Z is CR \. In some embodiments of a compound of Formula (I), Z is ce.
|Ό048) in some embodiments of a compound of Formula (Ϊ), Y is N and Z is N. in some embodiments of a compound of Formula (I), Y is N and Z is CR \ in some embodiments of a compound of Formula (I)., Y is CR' and Z is N. in some embodiments of a compound of Formula (I), Y is N and Z is CH. In some embodiments of a compound of Formula (I), Y is CH and Z is N. j"G049| in some embodiments of a compound of Formula (I), X is !iydrogen; Y is N; and Z is N. In some embodiments of a compound of Formula (I), X is C|--(Y alkyl; Y is N; and Z is N. In some embodiments of a compound of Formula (I), X i hydrogen, Y is CH; and Z is N. in some embodiments of a compound of Formula (I), X is hydrogen; Y i N; and Z is CH.
[0050] In some embodiments of a compound of Formula (I), (la), (lb), (Ila), (lib), (Ilia), (Illb), (iVa) or (IVb), R " and R are mdependentiy hydrogen, ( : · ' ',, alkyl r ,..<\ . alkenyl, C2- C6 alkynyl, halogen, -CN, -OR10, -SR10, -NR1 !R12, -C(0)R10, -C(0)NR1 !R12; or R¾ and R¾ are taken together with the carbon to winch they are attached to form a <\ ·Γ,··. cycloalkyl. In some embodi ments of a compound of Formula (I), (la), (lb), (Ila), (lib), (Ilia), (Illb), (IVa) or (IVb), R and IF ' are independently hydrogen or C] -C6 alkyl, or R"~ and IF are taken together with the carbon to which they are attached to form a (¾»0> cycloalkyl. In some embodiments of a compound of Formula (I), (la), (lb), (Ila), (lib), (Ilia), (Illb), (IVa) or (IVb), R a is hydrogen and R '° is C C6 alky !, In some embodiments of a compound of Formula (I), (la), (lb), (Ila), (lib), (Ilia), ί I l b), (iVa) or (IVb), K ' is hydrogen and !< ' is hydrogen. In some embodiments of a compound, of Formula (I), (la), (lb), (Ma), (lib), (Ilia), (Illb), (IVa) or (IVb), R3a is C C6 alkyl and I¾V'' is CY-CY. alkyl. In some embodiments of a compound of Formula (I ), ( a), (lb), (I a), (lib, (Ilia), (Illb), (IVa) or (IVb), K ' is methyl and R ' is hydrogen. In some embodiments of a compound of Formula (I ), (la), Ob), (Ila), (lib, (Ola), Olib), (IVa) or (IVb), R"'a is methyl and R" !'' is methyl In some embodiments, R and R are taken together with the carbon to which they are attached to form a (¾■(¾ cycloalkyl OOSij In some embodiments of a compound of Formula (I), (la), or (lb), X is hydrogen; Y is N; Z is N; R~,<! and ! * " ' are independently hydrogen, C ; ·( ... alkyl, ί :- C;, alkenyl Cj-Q, alkynyl,
halogen, ~CN, -OR10, -SR.30, -NR.3 ¼12, -C(0)R10, -C(0)NR]V2; or R i! and R h are taken together with the carbon to which they are attached to form a ( Q cycloalkyl. in some embodiments of a compound of Formula (I), (la), or (lb), X is r · ··( '.;. alkyl; Y is N; Z ·> N; R" <! and " are independently hydrogen, ( ' : -<'.··. alkyl ( Cf. alkenyl C2-C6 alkynyl, halogen, --CN, -OR10, -SRi , -NRi ]R j 2, -C(O)Ri0, -C(0)NRUR12; or R3a and R¾ are taken together with the carbon to which they are attached to form a ( C^ cyc!oalky!. In some embodiments of a compound of Formula (I), (la), or (lb), X is hydrogen; Y is CH; Z is N; R and RV are independently hydrogen, C -< alkyl,€2-(¾ alkenyl, (V-CV alkynyl, halogen, -CN, -OR ' .
-SR10, -NRi !R j 2, -C(C))R? 0, -C(0)NR?V2; or R3a and R¾ are taken together with the carbon to winch they are attached to form a Cfo-Q, cycloalkyl. in some embodiments of a compound of Formula (!.}, (la), or (lb), X is hydrogen; Y J S N, Z IS CH; R"~ and . are independently hydrogen, C i -C6 alkyl, C2~C6 alkenyl ί alkynyl, halogen, -CN, -OR : " ~SRIG, ·Ν) ; ; !Γ:. ~C(0)R1 , -C(0)NRi !R12; or Ria and k ' are taken together with the carbon to which they are attached to form a < VC.:-. cycloalkyl
[0052] in some embodiments of a compound of Formula (I), (la), or (lb), X is hydrogen; Y is N; Z is N; R.Ja arid R" ' are independently hydrogen or < ' CY alkyl, or " and L' are taken together with the carbon to which they are attached to form a ( O, cycloalkyl. In some embodiments of a compound of Formula (I), (la), or (lb), X is hydrogen, Y is N; Z is N; R and R " are independently hydrogen or (' ; ·( .··. alkyl; or R ' ' and R '° are taken together with the carbon to which they are attached to form a ; :··(".. cycloalkyl. in some embodiments of a compound of Formula (i), (la), or (lb), X is hydrogen; Y is N; Z is N; R' and k " are independently hydrogen or C :■( ';.. alkyl. In some embodiments of a compound of Formula (I), (la), or (lb), X is liydrogen; Y is N; Z is N; R and R : ' are both liydrogen.
[0053] In some embodiments of a compound of Formula (I ), 0a), (lb), Ola), (lib), (Ilia), (iiib), (iVa) or (IVb), R a and R¾ are independently hydrogen, ( : · ' ',, a!kyi, C2--C6 alkenyl, Cfo- C6 alkynyl halogen, -CN, -ORi0\ -SR10, -NRUR12, -C(0)R10, -C(0)NR UR1 2. In some embodiments of a compound of Formula (I), (la), (lb), (Ha), (lib), (Ilia), (Mb), (IVa) or (IVb),
K and R are takers together with the carbon to which they are attached to form a (V
(\. cycloaikyl. in some embodiments of a compound of Formula (i), (la), (lb), (lis.), (lib), (Ills.),
(nib), (IVa) or (IVb), RJ<! and R. " ' are independently hydrogen or C1-G5 alkyl.
[0054] In some embodiments of a compound of Formula (I), (la), (lb), Ola). (lib), (Ilia). (Iiib), (IVa) or (IVb). κ " and Rih are independently hydrogen, C C6 alkyl, r-< aikenyl, C2- C6 aikynyl, halogen, -CN, -OR10, -SR10, -NR1 ¾R12, ~C(0)RIG, -C(0)NR] 1R12 and R4 is a 5- to 10-membered heieroaryl ( > alkyl -(Cs -<¾ aikylene)(C3-C6 cycloaikyl), -(C]-C3 alkylene)(3- to 12-membered heterocyclyi), -(C Q? alkylene)(5- to 10-memb red
heieroaryl), -(€¾-(¾ alkyiene C C aryl), or Qi-C u aryl each of which is optionally substituted by halogen, -CN, V \ -C(0)R i 3, -OR.13, or C C6 alkyl optionally substituted by -OH. In one such embodiment, K*'*' and R"'* are independently hydrogen or ( : · <*' :·. alkyl. In another such embedment, R" is a 5- to 10-membered heieroaryl or '. '·,··(." ;.: aryl, each of which is optionally substituted by halogen, -CN, ~NRI RU ·( (0}R : . -OR 5 " or C C6 alkyl optionally substituted by -OH. in some embodiments of a compound of Formula (I), (la), fib), (iia), (fib), (Ilia), (iiib), (IVa) or (IVb), R " and R are taken together with the carbon to which they are attached to form a ( VCV. cycloaikyl and k" is 5- to 1 0-membered heteroaryi, ί alkyl, ■{ '■■■ C3 alkyiene)(C3-C6 cycloaikyl), --(¾-(¾ alkylene)(3- ιο 12-membered heterocyclyi), ··((' ; · C3 aikylene)(5- to 10-membered heteroaryi), -(C1-C3 alkylene) C6~Ci4 aryl), or C Qd aryl, each of which is optionally substituted by halogen, -CN, -NR13RW, -C(0)R , -OR;J, or Cs -C alkyl optionaliy substituted by -OH. In one such embodiment R+ is a 5- to 1 0~membered beteroaiyl or (¾- ]4 aryl, each of which is optionally substituted by halogen, -CN, -NR nRM
-C(0)R i 3, -OR ; 3, or C] -C6 alkyl optionally substituted by -OH.
[0055] In some embodiments of a compound of Formula (I ), (la), ( b), (Ila), ( ib), (Ilia), (Iiib), (IVa) or (IVb), R4 is 5- to 10-membered heieroaryl, Γ ; ..(\, alkyl ··{(· ; ·( · a!kyiene)(C3-C6 cycloaikyl), -(C C3 aikylene)(3- to 12-membered heterocyclyi), -(C C aikylene)(5- to 10- membered heteroaryi), -(Cj-C3 alkyle«e)(Cr,-C j aryl), or < ( · .; aryl, each of winch is optionally substituted by halogen, -CN, -NRi 3R 14, ~C(0)Ri3, -ORr\ or CrC6 alkyl optionally
substituted by -OH. in some embodiments of a compound of Formula (Is. (la), (lb), (Ha), (lib),
(iiia) , (Nib). (IVa) or (IVb), IT' is 5- io lO-rnembered heteroaiyl or C-v-i ·.; aryl, each of which is unsubstituted. In some embodiments of a compound of Formula (I), (la), (lb), (Ha), (lib), (Ilia),
(iiib) , (IVa) or (iVb), K: is 5- io 10-membered heteroaiyl or C(,-Cu aryl, ach of which is optionally substituted by halogen, oxo, ·Οκ;\ ·\Κ! V"'.• 0 }Ri\ -CN, C3-C8 cyc!oalkyl, or Cj-Cg alkyl optionally substituted by oxo, -OH or halogen, in some embodiments of a compound of Formula (I), (la), (lb), (Iia), (lib), (Ilia), (111!··. (IVa) or (IVb), R4 is 5- to 10-membered heteroaryl or ( Ci*. aryl, each of which is optionally substituted by halogen, -CN, -NR"'"'¾! , -C(0)R" -OR."", or C]-C6 alkyl optionally substituted by -OH. in some embodiments of a compound of Formula (i), (ia), (lb), (iia), (lib), (Ilia), (Iiib), (IVa) or (I Vb), R4 is 5- to 10- membered heteroaiyl optionally substituted by halogen, -CN, ·ΛΚ' : '. -C(0)R'", -OR'"', or Ci- C alkyl optionally substituted by -OH.ln some embodiments of a compound of Formula (I), a), (lb), (Ha), (lib), (iiia). (Iiib), (IVa) or (IVb), R4 is ( <:~r;i aryl optionally substituted by halogen, -CN, -NR°R!* -C(0)Ri3, -OR13, or Ci-C6 aikyl optionally substituted by -OH. in some
4
embodiments of a compound of Formula (1), R. is phenyl optionally substituted by halogen, ·■ CN, -NRI3R 14, -C(0)Ri 5 -OR1 , or C C6 alkyl optionally substituted by -OH. In some embodiments of a compound of Formula (I), (ia), (lb), (Ha), (lib). (Ilia), (Iib), (IVa) or (IVb), R' is phenyl optionally substituted by halogen or 0:·Ο··. alkyl optionally substituted by -OH. In some embodiments of a compound of Formula (I), (la), (lb), (Iia), (lib), (Ilia), (Iiib), (IVa) or (IVb), R" is phenyl optionally substituted by halogen
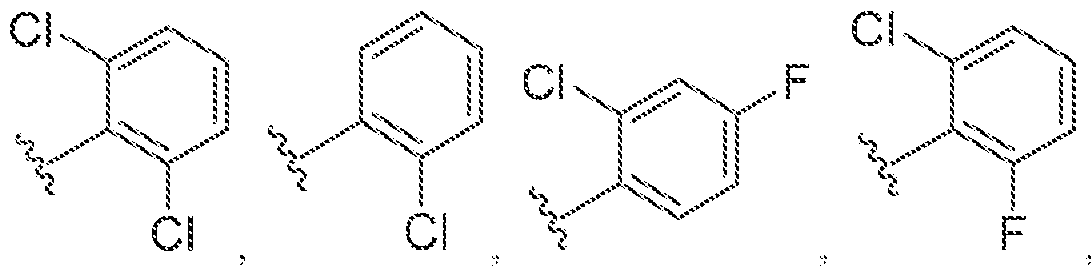
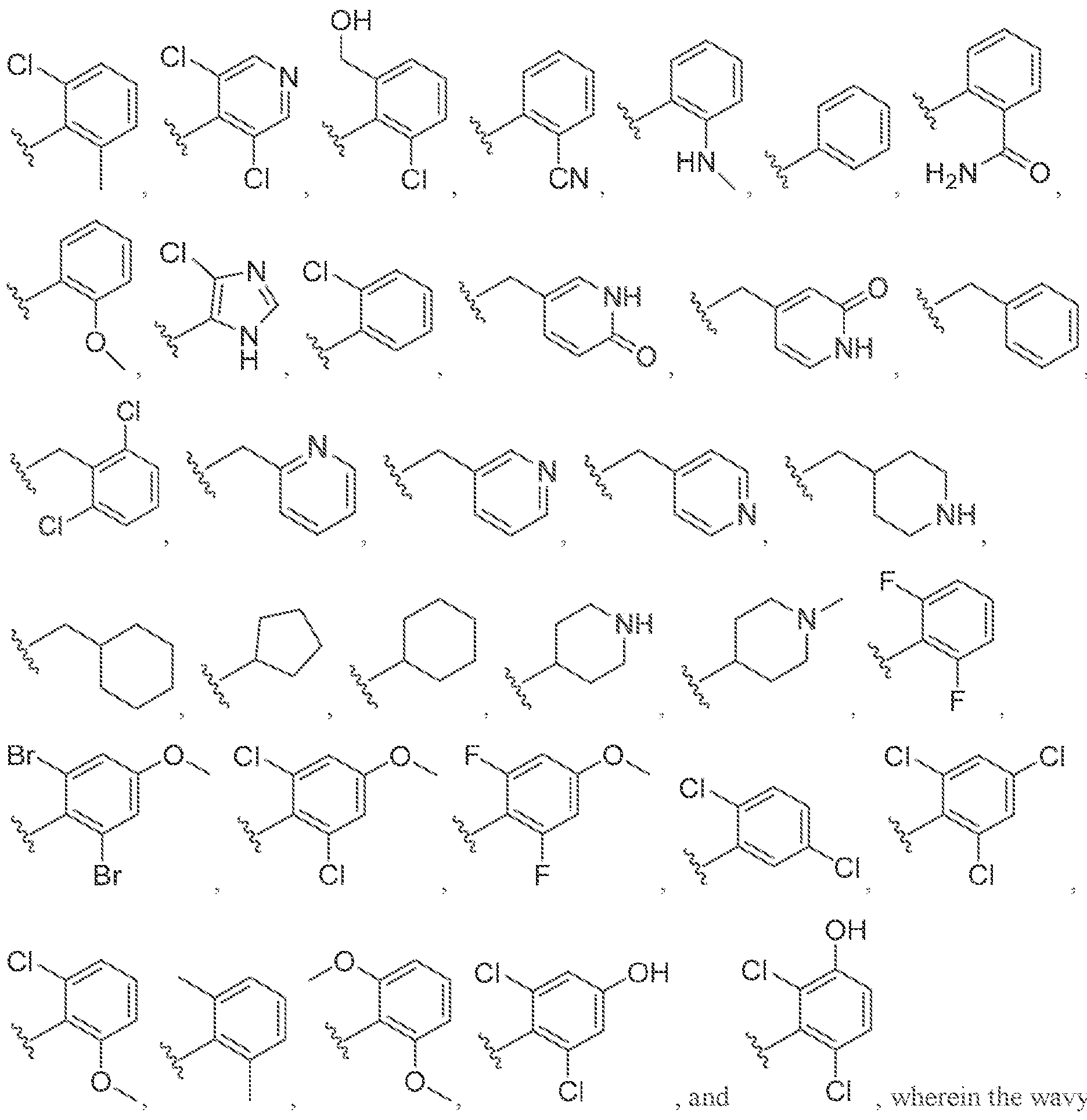
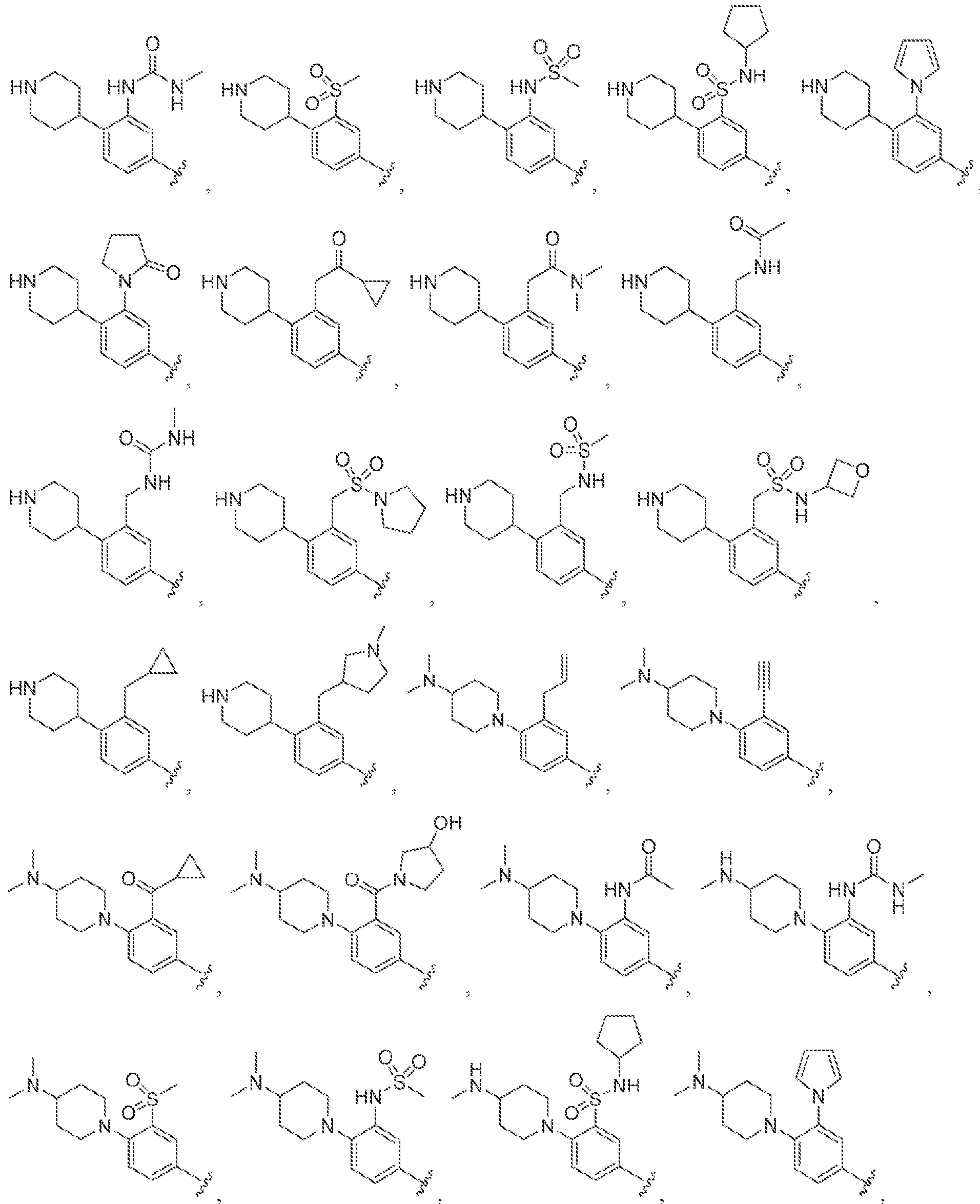
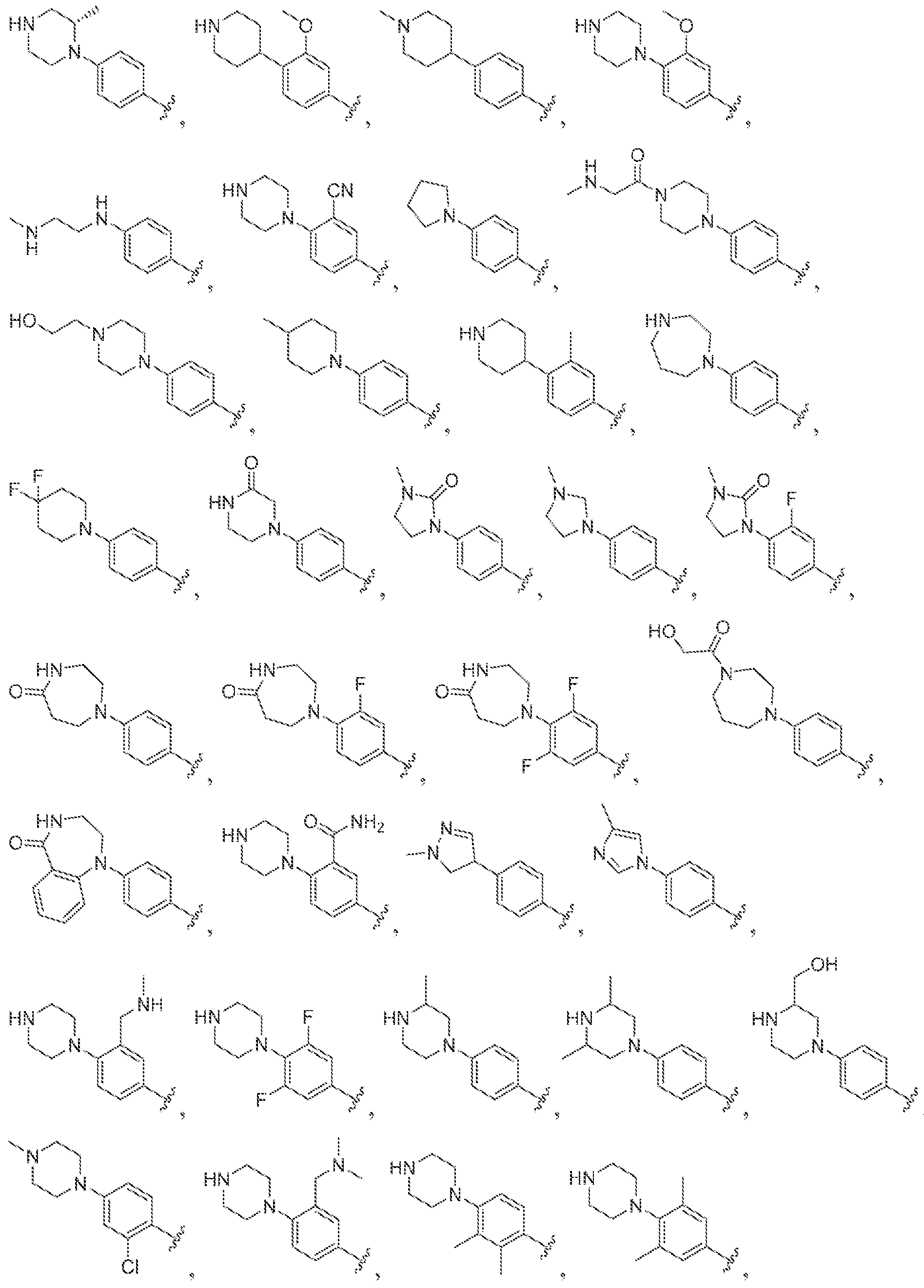
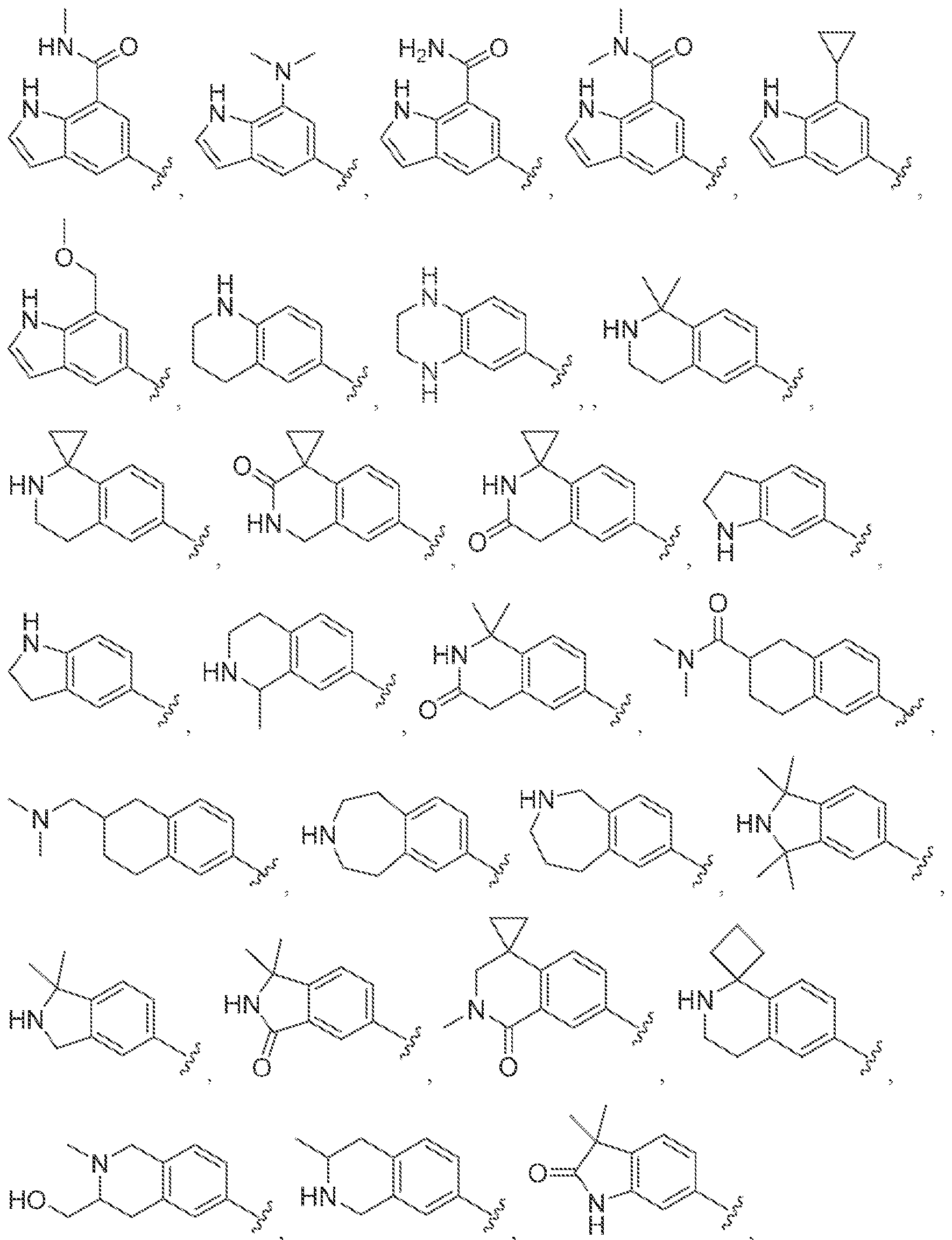
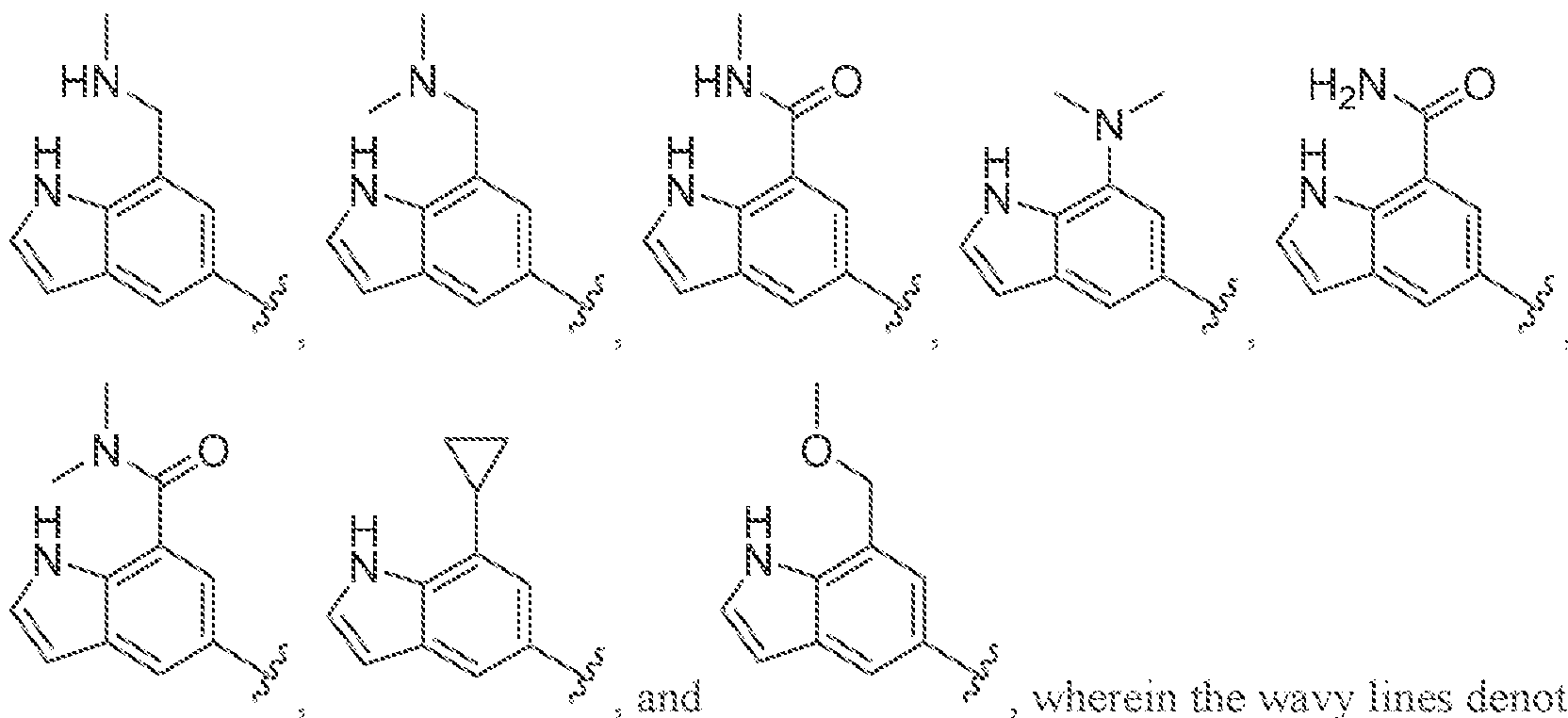
[0056] In some embodiments of a compound of Formula (I), (la), (lb), (Iia), (lib), (Ilia), (IHb), (IVa) or (IVb), IV is selected from the group consisting of:
methyl, ethyl, isopropyl, cyclopropyi.
s i
'iiV
methyl, ethyl, isopropyl, cyclopropyl.
lines denote attachment points to the parent molecule. In some embodiments, is
.
[0058] In some embodiments of a compound of Formula (I), (la), or (lb), X is hydrogen; Y is N; Z is N; Ef " and !<
"' are independently hydi
'ogen, (
" : - ·
' aJkyh t
.;·-<
*.·> aikenyl, ·'
" alkynyl, naiogeu, - iN, -υκ. , - r , -ΙΝΛ , --y.yU}«. , - ioj t , or is. ano Λ aie iaken together with the carbon to which they are attached ιο form a C3-C cycloalkyl; K
'; is 5- to 10· membered heteroaryl, Ci-Cr, a!kyl, ~(C]-C¾
cycloalkyl), -; C ; -i ; alkylene)(3- to 12-membered heterocyciyl). ·
( :··(
" : aikylene)(5- to 10-membered
heteroaryl), ~(C]-0* alkyiene)(C(?-Cj 4 aryl), or C i · · aryi, each of which is optionally substituted by halogen, -CN, -\K ' ' ". -O0)R' -OK"', or Ci-Ce alkyi optionally substituted
by -OH. In some embodiments of a compound of Formula (I), a), or (ib), X is Cj-C^ alkyl; Y is N; Z is N; Ef " and !< "' are independently hydrogen, (":-·' alkyl. ί ;-·<), alkenyl, ='* * --Ο. alkynyl, halogen, -CN, -ORiG, -SR10, -NR1 V2, ·( ( }R::'. -C(Q)NRnRi2; or R¾ and R are taken together with the carbon to which they are attached 10 form a. CVC6 cycloalkyl; K' is 5- to 10- membered heteroaryl or ,~C]4 aryl, each of which is optionally substituted by halogen,■- CN, -NR13RI ( ·( (i))R; . -ORL', or CrC6 a!kyl optionally substituted by -OH. in some embodiments of a compound of Formula (I), (ia). or (lb), X is hydrogen: Y is CH: Z is N; R ' ' and "" are independently hydrogen, Cj-Q, alkyl, (¾-ΐ¾ alkenyl, C-2-Q alkynyl, halogen, -CN, -OR10, -SR10, ~C(O)Ri , -Πϋ '^κ". or R3a and R¾ are taken together with the carbon to which they are attached io form a ( C^ cyc!oalkyl; R' is 5 - to 10-membered heteroaryl or Cv ;.: aryl, each of which is optionally substituted by halogen, -CN, ·ΛΚ''Ί<: '. -C(0)R' ', -OR"", or 1- 6 alkyl optionally substituted by -OH. in some embodiments of a compound of Formula (I), (ia), or (lb), X is hydrogen; Y is N; Z is ( Π.1< "' and k are independently hydrogen, C1-G5 alkyl. (' ···(>. alkenyl. {" . ···(>. alkynyl, halogen, -CN, -OR:".
-SRK!, -NRURi 3 -QO)R10, -C(0)NRSiR12; or R a and R3 are taken together with the carbon to which they are attached to form a C3-C6 cycloalkyl; R' is 5- to 10-membered heteroaryl or Q,- Ci aryl, each of which is optionally substituted by halogen, -CN, -NR; RM -C(G)R13, -OR13, or ('; ·( ... alkyl optionally substituted by -OH.
[0059] in some embodiments of a. compound of Formula (I), (ia), or (Ib), X is hydrogen; Y is N, Zts N; Rf" and R"1' ' are independently hydrogen or Cj-Cf, alkyl, or ' and K ' are taken together with the carbon to which they are attached to form a C3-Q cycloalkyl; R" is 5- to 10- nie beted heteroaryl, Ci-Cr, alkyl, -(C1-C3 alkylene)((¾~C6 cycloalkyl), ··;< V(" , alkyiene)(3- to 12-membered heterocyclyl), -(C1-C3 aikylene)(5- to 10-membered.
heteroaryl), -(C1-C3 alkylene)(Q,-'Ci4 aryl), or aryl. each of winch is optionally substituted by halogen, -CN, -NRi R.14, ~C(0)R13, -OR13, or C C6 alkyl optionally substituted by -OH. in some embodiments of a compound of Formula (i), (ia), or (Ib), X is hy drogen; Y is N; Z is N; R"" and R"1' ' are independently hydrogen or C1-G5 alkyl; or R and R are taken
optionally substituted by halogen, -CN, -NR R -C{0}RS'\ -OR13, or CrC6 a!ky! optionally hydrogen; Y is ; Z is N; R"'! and in eoenc asicys; i
AT H?A'>T?'- ifWO OP' alky! optional !y substituted by -OH.
ivdrogen; Y is N: ' N; and Κ ύ are both hydrogen; R+ is selected from the group
(IVa) or (IVb), X is hydrogen: R
J<! and R
' ' are both hydrogen; and il
" is a member of the preceding group described for Formula (I), (la) or (lb) In some embodiments of a compound of Formula (1), (la) or (lb), X is hydrogen, Y is X, Z is N: R
"" and
" ""' are both hydrogen: R is phenyl optionally substituted by halogen. In some embodiments of a compound of Formula (Ila), (lib), (Mia), (lllb), (IVa) or (IVb), X ·> hydrogen; R
¾ and R
¾ are both hydrogen; and R
4 is phenyl optionally substituted by halogen.
[0060] In some embodiments of a compound of Formula (I), (la), (lb), (Ila), (lib), (Ilia), (fflb), (IVa.) or ( IVb)., W i A, wherein A i phenyl or 5- ιο 6-membered heteroaryl, each of winch is optionally substituted with R! "!. In some embodiments of a compound of Formula (I ), (la), (lb), (Ila), (lib), (Ilia), (IHb), (IVa) or (IVb), W is AB, wherem A and B are fused together; A. is phenyl or 5~ to 6-membered heteroaryl, each of which is optionally substituted with R and B is C * -<V cycloalkyl, 3-· to 7-membered heterocyclyl, 5- to 7-membered heteroaryl, or ( aryl, each of which is optionally substituted with R" '. In some embodiments of a compound of Formula (I), (la), (lb), (Ila), (Rb), (Ilia), (Rib), (IVa) or (IVb), W is A, wherem A is phenyl or 5- to 6-mernbered heteroar l, each of which is optionally substituted with R' "". and wherem A and
Formula (I), (la), (lb), (Ha), (lib), (Ilia), (fflb), (IVa) or (IVb), W is AB, wherein A and B are fused together: A is phenyl or 5- ιο 6-membered heteroaryl, each of which is optionally
V"'!
substituted with R V; B is C AR cycloalkyl, 3- to ?-membered heterocyciyl, 5- to 7-membered heteroaiyl, or Q, aryl, each of which is optionally substituted with R ' ί ; and wherem A, B, R' ' ,
and R
! together are
and m and n are independently 0, 1 , 2, 3, or 4.
[0061] In some embodiments of a compound of Formula (I j, (la), (lb), (Ila), (lib), (ilia), (fflb), (IVa) or (IVb), W is A, wherein A is phenyl or 6-membered heteroaryl, each of which is optionally substituted with R "J In some embodiments of a compound of Formula (I), (la), (lb),
(Iia), ( lib), (I a), (nib), (IVa) or (IVb), W is AB, where n A and B are fused together; A is phenyl or 6-membered heteroaryl, each of which is optionally substituted with R" "": and B is Cy C( cycloalkyl, 3- to 7-membered heierocyeiyl, 5- to 7-membered heteroaryl, or (V, aryl, each of which is optionally substituted with R' In some embodiments of a compound of Formula (I), (la), (lb). (I ia), (Mb), (iiia), (Bib). (IVa) or (IVb), W is A. wherein A is pbenyl or 6-membered
: 7·¾ , 1
beteroar b each of which is optionally substituted with R ' ' and wherein A and k ' ' " together
are
and n is 0, 1 , 2, 3., or 4. in some embodiments of a compound of Formula (I), ( a),
(lb), (Ba), (Bb), (Bla), (Bib), (IVa) or (IVb), W is AB, wherein A and B are fused together, A is phenyl or 6-membered heteroaryl each of which is optionally substituted with R" "; B is (V (\. cycloalkyl, 3- to 7-membered heterocyclyl, 5 - to 7-membered heteroaryh or CA, aryl. each of which is optionally substituted with R ' ' '; and wherem A, B, R ' ". and R" ° together are
[0062] In some embodiments of a compound of Formula (I ), (la), (lb), i lia), (Bb), (Ilia), (Illb), (IVa) or (IVb), W is A, wherem A is phenyl or 5- to 6-membered beteroaryb each of which is optionally substituted with R ' In some embodiments of a compound of Formula (I), (la), (lb), (iia), (lib), (Ilia), (Illb), (IVa) or (IVb), W is A, wherem A is phenyl or 5- to 6- membered heteroar l, each of winch is optionally substituted with ':. wherein A and R! "!
together are
and n is 0, 1 , 2, 3, or 4. In some embodiments of a compound of Formula
(I), (la), (lb), (Iia), (lib), (Ilia), (Illb), (IVa) or (I Vb), n is 0. In some embodiments of a compound of Formula (I), (la), (lb), (Iia), (lib), (Ilia), (Illb), (IVa) or (IVb), n is 1. In some embodiments of a compound of Formula (I), (la), (lb), (Iia), ( ib), (Ilia), (Illb), (IVa) or (IVb), n is 2 In some embodiments of a compound of Formula (I), (la), (lb), (Iia), (lib), (Ilia), (Illb),
(IVa) or (IVb), n is 3. In some embodiments of a compound of Formula (J), (la), Ob), (!la), (Jib), (ffla), (Illb), (IVa) or (IVb), R1/a rs independently 3- to 12-membered heterocyclyl, ~(C
C
3 alky!enejOR
10. C C
6 alky! optionally substituted by halogen or -OH, -C(0) R
UR
", --(Cj- (¾ ikxk:ue)NR
;; : :. -CN, halogen, -NR
]iR
12, C
3-C
6 cycloalkyl, or -OR
i0, wherein the 3- to 12-membered heterocyclyl of R
1 "
! is optionally substituted with ( V-O, alkyl optionally substituted by halogen or -OH, -C(0)R , · >(\ alkyleneOR
1', -S(0)
2R
?A C
3-C
8 cycloalkyl, oxo, halogen, or -OR
1 w herein the 3- to 12-membered heterocyclyl of f¾f
"A is optionally fused wi th 5- to IO-mernbered heteroaryl or
aryl. In some embodiments of a compound of Formula (I), (ia), (lb), (iia), (lib), (Ilia), (Illb), (iVa) or (IVb), R
Va is independently Cj-Q alkyl optionally substituted by halogen or -OH, halogen, -CN, or 3- to 12-memhered heterocyclyl optionally substituted with Cj-Cg alkyl optionally substituted by halogen or -OH, In some embodiments of a compound of Formula (I), (la), (lb), (Iia), (Jib), (Ilia), (Illb), (IVa) or (IVb), W is A, wherem A is phenyl optionally substituted with R
' wherein R rs independently
"·· to 12-membered heterocyclyl,
■■{(.
' i ·( , a.lkylene)OR
' , Cj-CV, alkyl optionally substituted by halogen or -OH, ·
( (0;\R
; :R
: ' ~(C C
3 alkylene)NR
nR
i2, -CN, halogen, -NR
iSR
i2, C
3- O. cycloalkyl, or -O "", wherein the 3- to 12-membered heterocyclyl of R
' is optionally substituted with Cj-Cy alkyl optionally substituted by halogen or -
OH, -C(0)R13, -(Ci--C3 alkylene)()R , -S(0)2R C¾-Cg cycloalkyl, oxo, halogen, or -OR;3, wherem the 3- to 12-membered heterocyclyl of K is optionally fused with 5- to 10-mernbered heteroaryl or ( CM aryl. In some embodiments of a compound of Formula (I), ( a), (lb), (Iia), (lib), (Ilia), (Illb), (IVa) or (IVb), W is A wherein A is phenyl optionally substituted with R.! "!, wherem iV"~ is independently Cj-Q, alkyl optionally substituted by halogen or -Oi , halogen, -CN, or 3- to 12-membered heterocyclyl optionally substituted with Cj-CV, alkyl optionally substituted by halogen or -OH.
[0063
'| In some embodiments of a compound of Formula (I), W is A, wherein A is phenyl or 6-niembered heteroaryl, each of which rs optionally substituted wdh R
" ' '". In some embodiments of a compound of Formula (I), W is A. wherein A is phenyl or 6-memhered heteroaryl, each of
winch is optionally substituted with R
!
is 0, 1, 2, 3, or 4. in some embodiments of a compound of Formula (ϊ), n is 0, In some embodiments of a compound of Formula (I), n is I . In some embodiments of a compound of Formula (I), n is 2. In some embodiments of a compound of Formula (I), n is 3. In some embodiments of a compound of Formula (I), R
! ' is independently 3- to 12-membered heierocyclyl, -{Ci~
C3 alkyiene)OR10, ( r alkyl optionally substituted by halogen or -OH . ·('(() ;\ K 1 ; !\ "'. -(C C3 alkylene)NR.UR 12, -CN, halogen, -NR! jR12, C3-C6 cycloalkyl, or -ORl u, wherein the 3- to 12-membered heierocyclyl of R" " is optionally substituted with Γ ; ·Γ:·. alkyl optionally substituted by halogen or -OH, -CiO)R; 3, -(C C alkylene)OR.13, · ί Η ; \ C3-C8 cycloalkyl, oxo, halogen, or -ORJ"\ wherei the 3- to 12-membered heterocyclyl of R' is optionally fused with 5- to 10-membered heteroaryl or 0. d · · aryi. In some embodiments of a compound of Formula (I), R ' is independently j -C6 alkyl optionally substituted by halogen or -OH, halogen, -CN, or 3- to 12-membered heterocyclyl optionally substituted with Cj -Cf, alkyl optionally substituted by halogen or -OH. In some embodiments of a compound of Formula (I ), W is A, wherein A is phenyl optionally substituted with R ' wherein R is independently 3- to 12-membered heterocyclyl, ·-{ <* ; ·( , a.lkylene)OR' , < ' (b. alkyl optionally substituted by halogen or -OI L ·( ( ;\ R : ; : ? ~(C C3 alkyiene)NRi !R ; 2, -CN, halogen, -NRi ?Ri2, C3- (\. cycloalkyl, or - R ' ". wherein the 3- to 12-membered heterocyclyl of R ' is optionally substituted with (b ·( ... alkyl optionally substituted by halogen or -
OH, -C(G)R13, -<C i -C3 alkylene)OR , -SiO)2R , C3-C8 cycloalkyl, oxo, halogen, or -OR 3, wherein the 3- to 12~niembered heterocyclyl of R" is optionally fused with 5- to ICbniernbered heteroaryl or <VC ·.; aryl. In some embodiments of a compound of Formula (I), W is A wherein A is phenyl optionally substituted wrth R wherein R is independently 0}-Οή alkyl optionally substituted by halogen or -OH, halogen, -CN, or 3- to 12-membered heterocyclyl optionally substituted with C] -C6 alkyl optionally substituted by halogen or -OH.
[0064] In some embodiments of a compound of Formula (1), (la), (lb), (iia), (lib), (Ilia),
(111b), (IVa) or (IVb). W is A, wherein A is pyndmyi optionally substituted with R In some embodiments of a coinpound of Formula (I), (la), (lb), (Ila), (lib), (Ilia), (Illb), (IVa) or (IVb), W is A, wberem A is pyndmyi optionaily substituted with R" \ wherein R." is independently 3- to 12-membered heterocyclyi, -(C1 -C3 alkyjene)OR' Ci-Q, alky! optionally substituted by halogen or -OH, -C(0)NRnR; 2, -(Ci-lA, aikylene)NRuRi2, -CN, halogen, ~NRnR; \ <V C(5 cycloalkyl, or · Κ :". wherein the 3- to 12-membered heterocyclyi 0; R ' is optionally substituted with C Qi alky 1 optionally substituted by halogen or■·
OH, -CfO) 13, · ((· ; ·( , alkyle;ie)ORi3, · : (()) : : C3-C8 cycioalkyl, oxo, halogen, or -OR 13, wherein the 3- to 12-membered heterocyclyi ot R ' is optionally fused with 5- to 10-membered heteroaryi or C(,~C aryl. In some embodiments of a compound of Formula (I), (la), (lb), (Ha). (Rb, (iiia), (Rib), (iVa) or (IVb), W is A, wherein A is pyridinyl optionally substituted with R" "", wherein R' "' is independently ] -C6 alkyi optionally substituted by halogen or -OR, halogen, -CN, or 3- to 12-membered heterocyclyi optionally substituted with Cj-Cf, alkyl optionaily substituted by halogen or -OH.
[0065] In some embodiments of a compound of Formula (I ), (la), (lb), (Ha), (lib, (Ilia), (Illb), (IVa) or (IVb), W is selected from the group consisting of:
lines denote atta some ernboci s of a compound of Formula CI), (la),
Ilia), illlb i
'lVa) or (TVbVW is
, wne; m tee wavy une denotes attaomnei
0066] in some embodiments of a compound of Formula (I), (la), (lb), (Ma), (Mb), (MI nib), (iVa) or (IVb), W is selected from the group consisting of:
and , wherein the wavy lines denote attachment points to the parent molecule, in some embodiments of a compound of Formula (I), (ia), (Ib), (lia), (lib), (liia), (iilb), (IVa) or
(TVb), W is
, wherein the wavy !me denotes attachment point to the parent
17a
molecule and W is optionally substitu ted by K ' In some embodim nts of a compound of
Formula ( ! }. (ia), fib), (Ha). (Tib), (Ola), (nib), (IVa) or (IVb), W is
, wherein the wavy line denotes attachment point to the parent molecule.
[0067] it is understood that each description of W may be combined with each description of X, Y, Z, K '. R\ R !< "'. and !<" For example, in one aspect, ii is understood that each description of W may be combined in one aspect with a variation in which X is hydrogen, Y is N, Z is N, R '; is hydrogen, R ' is hydrogen, and R+ is 2,6-dichlorophenyl.
[0068] In some embodiments of a compound of Formula (I), (la) or (Ib), X is hydrogen, Y is N; Z is N; Ef " and !<
"' are independently hydrogen or C Ce alkyl; or k
" and R are taken together with the carbon to which they are attached to form a C
3-C
6 cycloalkyl; R" is 5- to 10- membered heieroaryl,
alkyl, -(€
¾-(¾ alkylenejiC Q cycloalkyl), -(C1-C3 alkylene)(3- to
12-membered heterocyclyl), AC; -(A alkylene)(5~ to 1 0-mernbered
heteroaryl), ~i ( ' i alkylene)(C¾- j aryl), or Cg-Cu aiyl, ach of which is optionally substituted by halogen, -CN, -NR/¼14, -C(G)R i 3, -QR i 3, or CrC6 alkyi optionally substituted
11- . "■ 7
by -OH; W is A , wherem A is phenyl optional iy substituted with R" ' \ wherein R" is independently 3-- to 12-membered heterocyclyl, -(C : -AA alkyl ene)ORf ', Ci -CA alky! optionally substituted by halogen or -OH, -C(G)NRnRi ?', -(C 1 -C3 alkylene)NRURi2, -CN, halogen,■■ NR l 'R'", C -Q cycloalkyl, or -OR"', wherein the 3- to 12-membered heterocyclyl of R is optionally substituted with Cj-(A alkyl optionally substituted by halogen or■■
OH, -C(0)R
13, -(C C
3 aikylene)OR
? 3, -S(0)
?R CA-Cg cycloalkyl oxo, halogen, or ~OR wherem the 3- to 12-membered heterocyclyl of
s optionally fused wife 5- to 10-rnembered heteroaryl or C Cu aryl. In some embodiments of a compound of Formula (I), (la) or (lb), X is hydrogen; Y is N; Z is N; K and R are independently hydrogen or
{" : ··(>. alkyl; or R
J<! and R
" 4 are taken together with the carbon to winch they are attached to form a CA-iA, cycloalkyl; R. is C ~C
14 aryl optionally substituted by halogen, -CN, -NR
i 3R
14, -C(0)R
i 3, -OR
i 3, or C C
6 alky! optionally substituted by -OR; W is A, wherein A is phenyl optionally substituted with Ι
' ", wherem R
' is independently Q-i-A alkyl optionally substituted by halogen or -OH, halogen, -CN, or 3- to 12-rnenibered heterocyclyl optionally substituted with Cj-(A alkyl optionally substituted by halogen or -OH. in some embodiments of a compound of Formula (I), (fa) or (lb), X is hydrogen; Y is N; Z is N; and k
" are independently hydrogen or (A · C
5 alkyl; R
*; is phenyl optionally substituted by halogen, -CN, · \ !<
; V
4. ·ί (0 }R
; . AAA
* or ( alkyl optionally substituted by -OH; W is A, wherein A is phenyl optionally substituted with R , wherein R
" " is independently Ci-CA alkyi optionally substituted by halogen or -OH, halogen, -CN, or 3 - to 12-membered heterocyclyl optionally substituted with ( : · (
';, alkyl optionally substituted by halogen or -OH. in some embodiments of a compound of Formula (Ϊ), (la) or (lb), X is hydrogen; Y is N; Z is N; R and k are both hydrogen; R
" is selected from
45
In some embodiments of a compound of Formula (I),
(la) or (lb). X is hydrogen; Y is N; " are both hydrogen: R'+ is phenyl
optionally substituted by nalog n;
optionally substituted by R. . in some embodiments of a compound of Formula (I), (la) or(Ib), X is hydrogen; Y is N; Z is N; K
" and
R
" 1' are both hydrogen; R ' is phenyl optionally substituted by halogen; W is
[0069] In some embodiments of a compound of Formula (I), (la) or (lb), X is hydrogen, Y is yZ is N; K
' and K
"' ' are independently hydrogen or C| -C6 alkyi; or R and R are taken together with the carbon to which they are attached to form a C:,-C
6 cycloalkyl; R
" is 5- to 10- membered heteroaryl,
alkyi, -(€
¾-(¾ alkylene)(C cycloalkyl), -(C
1 -C3 alkyiene)(3- to 12~mernbered heterocyclyl), · ; (
' · ··(
' < alkylene)(5- to 10~membered
heteroaryl), -(€
¾-(¾ alkylene)(C6-€i aryl), or Q-,-C } aryl, each of which is optionally substituted by halogen, -CN, -NR
i3R
i 4, -C(0)R
i3, -OR.
i3, or C C
6 alkyi optionally substituted by -OH; W is A , wherein A is phenyl optionally substituted with R wherein R
" is independently 3- to 12-membered heterocyclyl, -((
" ;·■(¾ alkylene)OR°
', 1 < R alkyi optionally substituted by halogen or -Oi l. -C(0)NR
UR
i 2, -{C
1-C3 alkylene}NR
i ]R
i2, -CN, halogen, - R
* "R
^' :1 C3-C6 cycloalkyl, or -OR
"', wherein ihe 3- to 12 -membered heterocyclyl of R
i !! is optionally substituted with Ci~C7, alkyi optionally substituted by halogen or - OIL -C(G)R
13, -(C C
3 alkylene)OR , -S(0)
2R
B, (¾-C
8 cycloalkyl, oxo, halogen, or -OR
13, wberem the ' · to 12-membered heterocyclyl of R is optionally fused with 5- to 10~niernbered heteroaryl or ( Ci *. aryl. In some embodiments of a compound of Formula (Ϊ), (la) or (lb), X is hydrogen; Y is N; Z is N; R and R
: ' are independently hydrogen or (
' ; ··0
;·. alkyi, or R
"" and R
"
are taken together with the carbon to which they are attached to form a C-^-C^ cycloaikyl; K
" is C
6-Ci4 &ryl optionally substituted by halogen, -CN, -NR
l 3R
14, ~C(0)R
3, -OR
1'3, or C C
6 aikyl optionally substituted by -OH; W is A, wherein A is phenyl optionally substituted with R
" ' '", wherein 1·'
' is independently Cj -Cg aikyl optionally substituted by halogen or -OB, halogen, -CM, or 3-· to 1 2-mernbered heterocyclyl optionally substituted with
aikyl optionally substituted by halogen or -OH. In some embodiments of a compound of Formula (I), (la) or (lb), X is hydrogen, Y is M , Z is N; R
"" and R
" ' are independently hydrogen or
¾ - C
6 aikyl: R
4 is phenyl optionally substituted by halogen, -CN, -NR^R
8 , -C(0)R
? 3, -OR
1"', or (
' : ·
( ':·: aikyl optionally substituted by -OH: W is A, wherein A is phenyl optionally substituted with R
" " wherein R
' is independently Ci- ^ aikyl optionally substituted by halogen or -OH. halogen, -CN, or 3·- to 12-menibered heterocyclyl optionally substituted with Ci~Cr, aikyl optionally substitu ted by halogen or -OH. in some embodiments of a compound of Formula (I), (la) or (lb). X is hydrogen; Y is N; Z is N; R
'"1 and R
" are both h drogen; II
" is selected from
the group consisting of methyl, ethyl, isopropyl, oyclopropyl,
, In some em Dodimenis or a compound or om ul U), λ J S nydrogen: is
N; Z is ; R: eotn is ohenyl ootionally substituted by haiogen; W is
Formula (! }, X is hydrogen: Y is N: Z is N; E " and RJl> are both hydrogen; R ' is phenyl
H '""^ CN
[0070] in some embodiments of a compound of Formula (I), (la), O b), (Ila), (Hb), (ilia), (ITTbl, (IVa) or (TVb), W is ΛΒ wherein A and B are fused together, A is phenyl or 5~ to 6- membered heteroaryl, each of which is optionally substituted with E! "!; .8 is Cs-C^ eycloalk] to 7-membered heterocyclyi, 5- to 7-membered heteroaryl, or {' aryi, each of which is o tionally substituted with R * ' ; and herein A, B, Ri , a, and R ' together are
m ict n are indepenaentiy 0, s , 2, , or 4
[0071 ] in some embodiments of a compound of Formula (1), (la), (lb), (iia), (lib), (iiia), (Iiib), (IVa) or (IVb). W is AB, wherein A and B are fused together; A is phenyl or 6-membered heteroaryi, each of which is optionally substituted with R
" ' "; B is - 6 cycloalkyl, 3·- to 7- membered heterocyclyl, 5- to 7-membered heieroaryl, or C¾ aryl, each of winch is o tionally
substi uted with R
1 ' L'; and wherein A, B, R
1 ' and R
1 ' together are
and m and n are independently 0, 1 , 2, 3, or 4.
[0072] In some embodiments of a compound of Formula (I), (la , (lb), (11a), (lib), (ilia),
(Iiib), (IVa) or (IVb), , . R
J" and R
1 ' 0 together are
, wherein B is
(h -C .··. cycloalkyl, ' · to 7-membered heterocyclyl 5~ to 7-membered heteroaryi, or (7. aryl and rn and n are independently 0, ; . 2. or 3. in some embodiments of a compound of Formula (I), (la), (lb), (Iia), (O b), (Ilia), (I I lb), (IVa) or (IVb), m is 0. In some embodiments of a compound of Formula (I), (la), (lb), ΐ f fa), i i i b} i l l la) (ffibj, (IVa) or (IVb), m is 1 , In some embodiments of a compound of Formula (I), rn is 2. In some embodiments of a compound of Formula (Ϊ), (la), (lb), (Iia), (lib), (Ilia), (Iiib), (IVa) or (IVb). m is 3. In some embodiments of a compound of Formula (I), (la), (lb), (Iia), (lib), (Ilia), ( Iiib), (IVa) or (IV b), m is 4. In some embodiments of a compound of Formula (I), n is 0. In some embodiments of a compound of Formula (I), (la). (Ib), (Iia), ( lib), (Ilia), (iiib), (IVa) or (IV b), n is 1. In some embodiments of a compound of Formula (I), n is 2. In some embodiments of a compound of Formula (I), (la). (Ib), (Iia). (lib), (Ilia), (131b), (IVa) or (IVb), n is 3. in some embodiments of a compound of Formula (I), (la), (Ib), (Iia), (lib), (Ilia), (ffib), (IVa) or (IVb), n is 4. In some embodiments, A, B, R
! ¾ and R
? VS' n together are
; wherein B is V < -C ... cycloalkyl, 3- to 7-membered heterocyclyl, 5- to 7-membered heteroaryi, or CY, aryl ; each R
' is independently -(C|-
(¾ alkylene)OR
10 s Ci-C
6 alky! optionally substituted by halogen, -C(0)NR
! 2,
■(<
'■
■■ C
3 alkylenelNRb ", -CN, halogen, -NR
!iR
12, C
3-C
6 cycloalkyl, or -OR
10, each R
i7b is independently oxo, ·;(Υ·ί ,
alkyl optionally substituted by
halogen, -C(0)NR
nR
1?', ···(>(
.. alkylene)NR R
i2, -CN, halogen, -NR.
;½
2, C C
6 cycloaJkyl, or -OR'
"; or any two groups
1 ' ", when bound to the same carbon atom or two different carbon atoms, are taken together wth fee carbon or carbons to which they are attached to form a (
',· (
'·, cycloalkyl or 3- to 7-memhered. heterocyclyl, each is optionally substituted by R
'" and m and n are inde endently 0, 1,2, or 3. In some embodiments, A, B, R
' and R
' together are
wherem B is CVQ, cycloalkyl, · to 7-membered heterocyclyl, 5- to
7-membered heteroaryk or (¾ aryl; each R" " is independently -(C C aikylenejOR'"', C
C6 alkyl optionally substituted by halogen,■-C(0)MRIiR12, ·{( : alkylene)NR R12, -CN, halogen, -NR^' ", C3-C0 cycloalkyl, or -OR.'"', each "° is independently oxo, -(Ci~
(¾ alkylene)OR10 s Ci-C6 alkyl optionally substituted by halogen, -C(0)NR R12 ■(<'■■■
Cb alkylene)NR7'R"", -CN, halogen, - R'T" cycloalkyl, or -OR ' '. or any two groups ""', when bound, to the same carbon atom, are takers together with the carbon to which they are attached to form a CYC !: cycloalkyl: and rn and n are independently 0, 1 , 2, or 3, In some
embodiments. A, B, R
' and R
' ' together are
; wherein B is (¾- (
y cycloalkyl, 3- to 7~membered heterocyclyl, 5- to 7-membered heteroaryk or Q aryl; each R '
:Λ is independently (h ·( .·. alkyl; each R
l ' is independently 0|~ί¾ alkyl optionally substituted by halogen, -€{0)R
"", or oxo; and each m and n are independently 0, 1,2, or 3. In some
embodiments, A, B,
; wherein B is (¾
■■ j cycloalkyl, 3- to 7~memhered heterocyclyl, 5- to 7-membered heteroaryl or (V, aryl; each R"
J is independently CV( .·. alkyl; two groups
1 are taken together with the carbon to which they are attached to form a C¾-C-6 cycloalkyl, and each m and n are i dependently 0, 1 , 2, or 3.
[0073] In some embodiments of a compound of Formula (I ), (ia), (lb), (iia), (lib), (iiia),
(Illb), (IVa.) or (I Vb), A, B, R
i7a and R
1? together are
; wherein 13 is
r !··(
'.. cycloalkyl,
"· · to 7-membered heterocyclyl, 5~ to 7-membered heteroaryl, or€5 aryl; and m and n are independently 0, 1 , 2, or 3. In some embodiments of a compound of Formula (I), m is 0. in some embodiments of a compound of Formula (I), (Ia), (lb), (Ha), (lib), (Ilia), (Illb), (IVa) or (IVb), ra is 1. In some embodiments of a compound of Formula (I), (Ia), (lb), (Ha), (lib), (Ilia), (Illb), (IVa.) or ( IVb), m is 2, In some embodiments of a compound of Formula (I), ( a), (lb), ( Iia), (lib), ( Ilia), (Illb), ( IVa) or (IVb), m is 3 In some embodiments of a compound of Formula (I), m is 4, In some embodiments of a compound of Formula ( I), (la), (lb), (Iia), (lib), (Ilia), (Illb), (IVa) or (IVb), n is 0. In some embodiments of a. compound of Formula (I), (la), (lb), (Iia), (lib), (Ilia), (Illb), (IVa) or ( IVb), n is 1 . In some embodiments of a compound of Formula (I), (la), (lb), (Iia), ( lib), (Ilia), (Illb), (IVa) or (IVb), n is 2. In some embodiments of a compound of Formula (I), (la), ( b), (Iia), ( ib), (Ilia), (I b), (IVa) or (I Vb), n is 3. In some embodiments of a compound of Formula (I), (la), (lb), (Iia), (lib ( Ilia), (Illb), (IVa) or (IVb), n
is 4. In some embodiments. A, .8, R '
<! and R7
' " together are
wherein B is C -C6 cycloalkyl, 3- io 7-membered heterocyclyl, 5- to 7-membered heteroaryl, or (\. aiyl; each K
' is independently -: <VC , alkylene)OR
' , Cb -CV, alkyl optionally substituted by
halogen, ·Π }\Κ
; !!
; '. -(€;-(¾ alkylene)NR
X ¾
37 -CN, halogen, -NR.
1 lR
U, C
3-C
6 cycloalkyi or -OR
" '7; each k
' is independently oxo, ~(C:--C
3 a!kylene)Oir 7 C
rC
6 alky] optionally substituted by halogen, -C(0)NR
! 2, · 7··(\ aikylene)NR
nR
i2, -CN, halogen, -NR
1]R
12, C
3- j cycloalkyi, or -OR
1"; or any two groups R.
' ' 7 when bound to the same carbon atom or two different carbon atoms, are taken together with the carbon or carbons to which they are attached to form a
!'7
•••(7. cycloalkyi or 3~ to 7-membered heteroeyclyl, each is optionally substituted by R
J"', and m and n are inde endently 0, 1, 2, or 3. in some embodiments, A, B, R
' and K
' )
n together are
; wherein B is C3--C
6 cycloalkyi, 3- to 7-membered heterocyclyL 5- to 7-mernbered heteroaryl, or 7 aryi, each K
' is independently -(Cj- (¾ alkylene)OR
10, Ci-C
6 alky! optionally substituted by halogen, -C(0)NR R
12 ■(<
'■
■■ (7 alkyiene)NR
11R
i5, -CN, halogen, -NR
!iR
12, C
3-C
6 cycloalkyi, or -OR.
10, each R
i7b is independently oxo, -;(7 ·( , alkylene)OR
' , C
\~C(, alkyl optionally substituted by
halogen, -C(0)NRnRi2, -{( { . alkylene)NR Ri2, -CN, halogen, -NR.;½ 2, C -C6 cycloalkyi, or -OR.'°; or any two groups R""Y when bound to the same carbon atony are taken together with the carbon to which they are attached to form a (¾~CY, c cloalkyi; and ni and n are independently
0, 1 , 2, or 3. In some embodiments. A, B,
wherein B is (¾-(¾ cycloalkyi, 3- to 7-membered heteroeyclyl, 5- 107-membered heteroaryl, or €5 aryl; each R' is independently C|-C6 alkyl; each RJ"~' is independently Γ···(7, alkyl optionally substituted by halogen, ~C(0)R'7 or oxo; and each in and n are independently 0, 1, 2,
or 3 In some embodiments, A, B,
"" and R
' "J together are
; wherein
B is CVi .·. cycloalkyi, 3- to 7~mernbered heteroeyclyl, 5- to 7-membered heteroaryl, or (7. aryl;
each R" is independently C] -C6 alkyl; two groups "" are taken together with the carbon to winch they are attached to lorn- a ( Q cycloalkyl; and each m and n are independently 0, 1. 2, or 3.
[0074] in some embodiments of a compound of Formula 1), (ia), (lb), (iia), (lib), (iiia).
(lllb), (IVa.) or (I Vb), A, B, R
17a and Κ
Γ,¾ together are
ί ,·Γ;, cycloalkyl, 3-· to 7-membered heierocyclyi, 5- to 7-membered heteroaryl. or C¾ aryl; and m and n are independently 0, ! . 2. or 3. In some embodiments of a compound of Formula (I), (ia), (lb), (Iia)., (lib), (Ilia), (lllb), (IVa.) or (IVb), m is 0. in some embodiments of a compound of Formula (I), m is 1. In some embodiments of a compound of Formula (I), (ia), (lb), (Iia), (lib), (Ilia), (i i i h). (IVa) or (IVb), m is 2. in some embodiments of a compound of Formula (I), (ia), (lb), (iia), (lib), (iiia), (Hib), (IVa) or (IVb), m is 3. In some embodiments of a compound of Formula (I), m is 4. In some embodiments of a compound of Formula (I), (la), (lb). (Iia), (lib), (ilia), (lllb), (IVa) or (IVb), n is 0. in some embodiments of a compound of Formula (I), (la), (lb), (Iia). (lib), (Ilia), (lllb), (IVa) or (IVb). n is 1. n some embodiments of a compound of Formula (I), (ia), (lb), (iia), (lib), (iiia), (liib), (iVa) or (IV b), n is 2. in some embodiments of a compound of Formula (I), (la), (lb), (Iia), (lib), (Ilia), (lllb), (IVa) or (I Vb), n is 3. In some embodiments of a compound of Formula (I), (la), (lb), (Iia), Va) or (IVb), n
is 4, In some embodiments, A, B, K
* and R
! together are
; wherein
B is CVi .·. cycloalkyl, 3- to 7-membered heierocyclyi, 5- to 7-membered heteroaryl, or€5 aryl; each K' is independently -: <VC , alkylene)OR' , C j-CV, alkyl optionally substituted by halogen, -C(0)NRl 3R12, -(€;-(¾ aikylene)NRI lR] 2, -CN, halogen, -NR.1 lRU, C -C6 cycloalkyl, or -OR"'"'; each k ' is independently 0x0,•n <\ alkylene)Oi ', C C0 alkyl optionally substituted by halogen, -C(0}NR; V2, -(C i -C3 aikylene)NRi !R? 2, -CN, halogen, -NR; iRi2, C - j cycloalkyl, or - kV ; or any two groups R' ' when bound to the same carbon atom or two
different carbon atoms, are taken together with the carbon or carbons to which they are attached to form a C --C6 cycloalkyi or 3·- to 7-membered heterocyc!yi, each is optionally substituted by R and m and n are inde endently 0, 1, 2, or 3. in some embodiments, A, B, R.J " and K '
together are
·ί V
: cycloalkyi 3- to 7-membered heterocyclyl, 5~ to 7-membered heteroaryl, or < aryi, each
' rs independently ~{C] - (¾ alkylene)GR
10, C
rC
6 alkyl optionally substituted by halogen, ·Γ(0}\ Κ
; < 7
■(
( ■
■■ C
3 alkyiene)NR
l iR
i ", -CN, halogen, -N
! lR
12, C
3-C
6 cycloalkyi or -OR
10, each R
i? is independently oxo, -; CVi alkylene)OR
"\ Cj -C6 alkyl optionally substituted by
halogen, -C(0)NRnR12, ~(C C alkylene)NRS iR12, -CN, halogen, -NRnR°, C C6 cycloalkyi, or -OR ' '; or any two groups RJ when bound to the same carbon atom, are taken together with the carbon to which they are attached to form a (' ,·( !: cycloalkyi; and rn and n are independently
0, 1 , 2, or 3. in some embodiments, A, B,
' and R
" together are
wherein B is '. ' :··(/<·. cycloalkyi, 3- to 7-membered. heterocyclyl, 5~ to 7-membered heteroaryl ('.. aryi; each K ' is independently Cj -Q, alkyl, each K ' is independently C ; ·Γ;·. alkyl
Ί 3
optionally substituted by halogen, -€(0)R"\ or oxo; and each rn and n are independently 0, 1
or 3. i.n some embodiments. A, B, R and R together are
; wherein
B is C3 -C6 cycloalkyi, 3- to 7-membered heterocyclyl 5- to 7-membered heteroaryl or Q, aryi; each R ' ' " is independently C G, alkyl: two groups R ' ' L' are taken together with the carbon to which they are attached to form a C3-C6 cycloalkyi; and each m and n are independently 0, 1 , 2, or .
[0075] in some embodiments of a compound of Formula (1), (la), (lb), (Ila), (lib), (Ilia),
r ;··('.;. cycloalkyl, 3-- to 7-membered heterocyclyl, 5- to 7-membered heteroaryl, or€5 aryi; and m and n are independently 0, L 2, or 3, In some embodiments of a compound of Formula (I), (la), Ob), (Ha). (Mb), (Ilia), (ttSb), (IVa) or (IVb), m is 0. In some embodiments of a compound of Formula (I), (la), (lb), (Ila), (lib), (Ilia), (Illb), (IVa) or (IVb), m is 1 . In some embodiments of a compound of Formula (I), (la), (lb). (Ila), (lib), (Il ia), (Illb), (IVa) or (IVb), m is 2. In some embodiments of a compound of Formula (I), (la), (lb), (Ila), (lib), (Ilia), (Illb), (IVa) or (IVb), ra is 3 In some embodiments of a compound of Formula (I), (la), (lb), (Ila), (lib), (Ilia), (Illb), (IVa) or (IVb), m is 4, In some embodiments of a compound of Formula (I), (la), (lb), (Ila), (lib), (Ilia), (Mb), (IVa) or (IVb), n is 0. In some embodiments of a compound of Formula (I), (la), (lb), (I a), (lib), (Ilia), (Illb), (I Va) or (IVb), n is 1. In some embodiments of a compound of Formula (I), (la), (lb), (Ha), (lib), (ilia), (Illb), (IVa) or (IV b), n is 2. In some embodiments of a compound of Formula (i), (la), (ib), (Ila), (Kb), (Ilia), (Illb), (IVa) or (I Vb), n is 3. In some embodiments of a compound of Formula (I), (la), (lb), (Ila), (Jib (Ilia), (Illb), (IVa) or (IVb), n
is 4. i.n some embodiments. A, B, K and R togetner are
;
wherein B is (¾-C6 cycloalkyl, 3- to 7-membered heterocyclyl, 5- to 7-membered heteroaryl or ('·, aryl; eacb R ' is independently ·;' <' ;.-(\ alkylene)OR"v', C]-C6 alkyl optionally substituted by halogen, -C(0)NRnR1 ?', ··; ( ; ·( · alkylene)NR Ri2, -CN, halogen, -NirV2, C3-( , cycloalkyl or -OR"'; each R"" is independently oxo, · ((· ; ·( .. alkylene)OR"\ CrC6 alkyl optionally substituted by halogen, -C(0)NRnRi 2, -(CVC : alky!enej R^R12, -CN, halogen, ~NRnRi 2, CV (\. cycloalkyl, or -OR""; or any ; wo groups R' ' " , when bound to the same carbon atom or two different carbon atoms, are taken together with the carbon or carbons to winch they are attached to form a CVC .·. cycloalkyl or 3- to 7-membered heterocyclyl each is optionally substituted by
Rl°; and m and n are independently 0, 1 , 2, or 3. In some embodiments, A, B, R. "! and R
ttooggeetthheerr aarree
;;;; wwwwhhhheeeerrrreeeeiiiinnnn BBBB ----ssss CC CC ,,,,····(((( ccyyccllooaallkkyyll,, 33-- ttoo 77--mmeemmbbeerreedd ii..77aa
heterocyclyi, 5- to 7-membered heteroaryl or (¾ aryl eacb R is independently --{(7 ·
C-3 aikylene)OR1 , CVi alky! optionally substituted by halogen, -C(0)NRnRi 2, ··{(
C3 alkylenejNR^R12, -CN, halogen, -N ^R12, C3-G5 cycloalkyl or -OR10; each Ri7 is independently oxo, -(Ci -CN aikylene)OR!^, (7 -f alky! optionally substituted by
halogen, -C(0)NRUR12, ·(( ;-<7 aiky]ene)NRnR12, -CN, halogen, -NR1 ]R12, (¾-C0 cycloalkyl. or -OR"''; or any two groups Rl ' when bound to the same carbon atom, are taken together with the carbon to which they are attached to form a C3-Q cycloalkyl, and m and n are inde endently
0, 1, 2, or 3. in some embodiments, A, B, R
' and R"° together are
; wherem B is Γ ;··(7, cycloalkyl, 3-- to 7-membered heterocyclyi, 5- to 7-membered heteroaryl, O. aryl; each R' is independently C <7. alkyl, each "° is independently < 7 -C !: alkyl optionally substituted by halogen, -C(0)R'7 or oxo; and each m and n are independently 0, 1 ,
or 3. Ri some embodiments, A, B, R
wherem B is (7-C/. cycloalkyl, 3- to 7-membered heterocyclyi. to 7-membered heteroaryl or C(5 aryl; each R. "! is independently€χ~ί¾ alkyl two groups R. ' 1' are taken together with the carbon to which they are attached to form a C3-C73 cycloalkyl; and each rn and n are
independently 0, 1 , 2, or 3
[0076] in some embodiments of a compound of Formula 1), (la), (lb), (Ila), (lib), (Ilia),
OTib), (IVa) or (IVb), A, B, R
i ;a and R
J'7i> together are

; wherein B is C -C6 cycloalkyi, 3- to 7-membered heierocyclyl, 5- o 7-membered heteroaryl. or C¾ aryl; and m and n are independently 0, L 2, or 3, In some embodiments of a compound of Formula (I), (la), (lb), (ila), (Mb), (Mia,), (Tiib), (IVa) or (IVb), m is 0. In some embodiments of a compound of Formula (I), (la), (lb), (Ila), (lib), (Ilia), (Illb), (IVa) or (IVb), m is 1 . In some embodiments of a compound of Formula (I), (la), (lb). (Ila), (lib), (Ilia.), (Illb), ( IVa) or (IVb), m is 2, In some embodiments of a compound of Formula (I), (la), (lb), (Ila), (lib), (Ilia), (Illb), (IVa) or (IVb), ra is 3 In some embodiments of a compound of Formula (I), ( a), (lb), (Ila), (lib), (Ilia), (Illb), (IVa) or (IVb), m is 4, n some embodiments of a compound of Formula (I), (la), (lb), (Ila), (Kb), (Ilia), (nib), (IVa) or (IVb), n is 0. in some embodiments of a compound of Formula (I), (la), (lb), (Ha), (lib), (Ilia), (I b), (I Va) or (IVb), n is 1. In some embodiments of a compound of Formula (Ϊ), n is 2. In some embodiments of a compound of Formula (I), (la), (lb), (Ila), (Kb), (Ilia), (Illb), (IVa) or (IVb), n is 3. In some embodiments of a compound of Formula (I), (la), (lb), (Ka k (lib), (iiia , (Illb), (IVa) or (IV b), n -s 4. in some embodiments, A, B, R
17a and R
1 ?b together are
, wherein B is
; vO. cycloalkyi, 3- to 7-membered beterocyclyl, 5~ to 7-membered heteroaryl, or aryl, each
' JS independently ~{C] - (¾ alkylene)GR
10, C
rC
6 alkyl optionally substituted by halogen, -C(()) R
; 1R
i2,
■(
( ■
■■ C3 alkylene)NRl iRi ", -CN, halogen, -NRi LR12, C3-C6 cycloalkyi, or -OK10, each Ri? is independently oxo, -■(€;■-(¾ alkylene)(¾¾V, C : --(V alkyl optionally substituted by
halogen, -C(0)NRnR12, -{ ( { .. alkylene)NRS iR12, -CN, halogen, -NRUR°, <VC cycloalkyi, or -CXRV'; or any two groups RJ when bound to the same carbon atom or two different carbon atoms, are taken together with the carbon or carbons to winch they are attached to form a€ 3-
€5 cycloalkyi or 3·- to 7-membered heierocyclyl, each is optionally substituted by R ' ": and m and
n are independently 0, 1, 2, or 3. In some embodiments, A. B, RJ and R1 together are
wherein B is CVQ, cycloalkyi, · to 7-membered heierocyclyl, 5- to 7-membered heteroaryl, or ¾ aryl; each
""" is independently ·((
r< aikylene)OR
"'
', C
\~
C6 alkyl optionally substituted by halogen, .·ΓίΟ)ΝΚ: ". ·{( : aikylene)NR R12, -CN, halogen, -NR^' ", C3-C0 cycloalkyi, or -OR'1', each is independently oxo, -(Ci-
(¾ alkyiene)OR10, Ci-C6 alkyl optionally substituted by halogen, -C(0)NR! 2,■(<'■■■
C\ alkylene)NR7'R!7 -CN, halogen, - R'^R"", cycloalkyi or -OR"1', or any two groups
R
"L', when bound to the same carbon atom, are takers together with the carbon to which they are attached to form a CYC
!: cycloalkyi; and rn and n are independentl 0, 1 , 2, or 3, In some )n embodiments. A, B, R
i,a and R"
J together are
; wherein B is Ci-
C
f} cycloalkyi, 3- to 7-membered heterocyclyl, 5-· to 7-membered heteroaryl, or€5 aryl; each K
' is independently Ci-Cs alkyl, each
' is independently < ·<
'.··. alkyl optionally substituted by halogen, -C(0)R
l", or oxo: and each m and. n are inde endently 0, L 2, or 3, In some )n embodiments. A, B, R
";" and R"
!' together are
; wherem B is C
3-
Cfj cycloalkyi, 3- to 7-memhefed heterocyclyl, 5- to 7-membered heteroaryl, or C6 aryl; each R ! :Λ is independently C; ·( ... alkyl; two groups R! l'are taken together with the carbon to which they are attached to form a Cy-Cg cycloalkyi, and each m and n are independently 0, 1 , 2, or 3.
[0077] in some embodiments of a compound of Formula if }. (ia), (lb), (iia), (Mb), (ilia), (I3Ib), (IVa) or (I.Vb), W is AB , wherein A and B are fused together and AB is selected from the group consisting of:
and each is optionally substituted bv R' '
In some embodiments of a compound of Formula (I), W is AB s wherein A and B ;
wherem the wavy line denotes attachment point to the parent molecule and AB is optionally substituted by R
" " and R
" ' ' In some embodiments, AB ts
, wherem tlie
ί 00791
100801 In some em odimenis oi a com ound ot onmua {!), jll ΠΤ ¾ ί
'ίίί·-Illb, IVa or IVb , W is selected from the rou consistin of:
. and , wherein the wavy lines denote attachment points to the parent molecule.
[0081] It is understood that each description of AB may be combined with each description of X, Y, Z, R . R' . k ' R/n and R'\ For example, in one aspect it is understood that each description of AB may be combined in one aspect with a variation in which X is hydrogen, Y is N, Z is N, R is hydrogen, R" " is hydrogen, and i 2,6-dichlorophenyh
[0082] in some embodiments of a compound of Formula (I), (la) or (lb), X is !iydrogen; Y is N; Z is N; R"'' and ' ' are independently hydrogen or C1-G5 a!kyl; or "',x and R are taken together with the carbon to which they are attached to form a (" Q cycloalkyl, R"* is 5- to 1 0- membered heteroaiyl or Q,~C] ary!, each of which is optionally substituted by halogen, ·· CN, -NR13RI ( -C(0)R , -O ; . or Cr-C6 a!kyl optionally substituted by -OH: W is
wherein B is <YC cycloaJkyl, 3~ to 7-membered heierocyelyl,
7-membered heteroaryl, or C,¾ aryl; each R : is independently --(C C3 alkylene)OR ' , Cr
C6 alkyl optionally substituted by halogen, -C(0)NRnR1 2, ··(( ( .. alkyleneS R^ R12, -CN, halogen, -NR. ' : R ' . ( Q, cycloalkyh or -OR '"; eacli R" "'' is independently oxo, -€■-
C\ alkyiene)OR1 , ( rO, alkyl optionally substituted by halogen, ·( (Ol M< i ; R ; :. ·(( r
C3 alkyl enelNR 'R", -CN, halogen, -NR' ' Γ'. ί , ·{" :, cyc!oalkyi, or -OK ' or any two groups
RJ when bound to the same carbon atom or two different carbon atoms, are taken together
with the carbon or carbons io which they are attached to form a {' 7··(7. cycloaikyl or 3-- to 7- rnembered heterocyclyl, each is optionally substituted by K " : and m and n are independently 0, 1, 2, or 3. in some embodiments of a compound of Formula (I), (la) or (lb), X is hydrogen; ¥' is
' are mdependentiy hydrogen or C]-C6 alkyi; or and RX' are taken together with die carbon io winch they are attached to form a C C6 cycloaikyl; ; is 5- to 10- membered heteroaryl or OCX: aryl, each of which is optionally substituted by halogen, - CN, -NRi3R , -C(0)Ri 3, -OR1 3, or C X-C . alkyl optionally substituted by -OH; W is
; wherem B is ( -1¾ cycloaikyl, 3- to /-membered heterocyclyl, >·· to 7--rnembered heteroaryl, or C¾ aryl; each R
' is independently -(C1-C3 aikylene)OR
J '', C|- C
6 alkyi optionally substituted by halogen, -€(0)Ν
! ίΚ
12, ·(( XX alkyiene)NR
11R
i -CN, halogen, ·Λ Κ
: ' R X «
' 7..(\ cycloaikyl, or -OR
" each R
: is independently oxo, ·(( : ·
C3 aJkylenejOR10. <XC alkyl optionally substituted by halogen, -C(0)NRnRi2, -·ΧΧ
(X aikyiene^R
1 !R
12, -CN, halogen, -NR
S ½
12, C
3-C
6 cycloaikyl, or -QR
' or any two groups R
' ', when bound to die same carbon atom, are taken together with the carbon to which diey are attached to form a ί XX, cycloaikyl; and m and n are independently 0, 1 , 2, or 3. In some embodiments of a compound of Formula (I), (la) or (lb), X is hydrogen: Y is N; Z is N; K
" and R are mdependentiy hydrogen or (
r C
;, alkyi; or 1'
': and R
Jl' are taken together with the carbon to which they are attached to form a (
'>. cycloaikyl; k
": is (¾
■■€; aryl optionally substituted by haiogen, -CN, -NR
; 3R
!4, -C(0)R
i 3, -OR
i 3, or C
rC
6 alkyi optionally substituted n by -OH; W is
wherein B is cycloaikyl. 3- to 7-membered heterocyclyl, 5- to 7-membered heteroaryl, or Q, aryl: each R.
: Λ is independently (7 ·( .·.. alkyl: each R
" i independently C X ·
: alkyl optionally substituted by haiogen,•·<70)
' . or oxo; and
each m and n axe independently 0, I, 2, or 3, In some embodiments of a compound of Formula (I), (la) or (lb), X is hydrogen; ¥
' is N; Z is ; R
'; and R
J" are independently hydrogen or C
\~ C
6 a!kyl; R
4 is phenyl optionally substituted by halogen, -CN, ~NR R
14, -C(C»R
13, ~OR , or
( { ;'
: alky! optionally substituted by -OH; W is
; wherein B is CV
€5 cycloalkyl, 3·- to 7-membered heterocyclyl, 5·- to 7-membered heteroaryl, or C<}- aryl; each R"" is independently C ; -·' alkyl; two groups R ' are taken together with the carbon to winch they are attached to form a (VCV. cycloalkyl; and each m and n are independently 0, 1 , 2, or 3.
[0083] in some embodiments of a compound of Formula (1), (la) or (lb), X is hydrogen; Y is N; Z is N; R.Ja arid ' are independently hydrogen or <' :·-0.··. alkyl, or R"" and R'1"' are taken together with the carbon to which they are attached to form a ( O, cycloalkyl; R' is 5- to 10- niembered heteroaryl or ( C^ aryl, each of winch is optionally substituted by halogen,■■ CM, -NRi3R14, -C(0) j3, -OR13, or C C6 alkyl optionally substituted by -OH; W is
cycloalkyl, 3- to 7-membered heterocyclyl, 5- to
7-membered heteroaryl. or ('.. aryl: each R"" is independently --iC3-C¾ alkylene)OR'\ Cj-
C6 alkyl optionally substituted by halogen, -C(0)NR R° ·(('; ·( , alkylene)NR!iR12, -CN, halogen, -ΝΙ 'Κ'", (¾'■(¾ cycloalkyl. or -OR'"; each R ' is independently oxo,■■(€;■■
C3 alkyiene)OR1', C C6 alkyl optionally substituted by halogen, -C(C)) Ri!R12, »(C
C3 ah- \-)e:-C \R " l : ' . -CN, halogen, -NR' : R '". ί ,·{" ;, cycloalkyl, or -O "; or any two groups
W ''. when bound to the same carbon atom or two different, carbon atoms, are taken together with the carbon or carbons to which they are attached to form a {'h--( V cycloalkyl or 3-- to 7- membered heterocyclyl, each is optionally substituted by R' , and m and n are independently 0, I, 2, or 3. in some embodiments of a compound of Formula (I), (la) or (lb), X is hydrogen; ¥' is
' are independently hydrogen or C]-C6 alkyi; or R and R"' are taken together with the carbon to which they are attached to form a ( Q cycloaikyl; R is 5- 1010- menibered heteroaryl or r ;,-CY; aryl, each of which is optionally substituted by halogen, · CN, -NR13RU, -C(0)Ri3, -OR13, or C C6 alkyl optionally substituted by Oil: W is
; wherein B is ί YY, cycloaikyl, 3- to 7-membered heterocyclyl, 5- to 7-mernbered heteroaryl. or (
'.. aryl; each R"" is independently dC
3-C¾ alkylenejOR Y Cj- C
6 alkyl optionally substituted by halogen, -C(0)NR R° ·((
'; ·( , alkylene)NR
!iR
12, -CN, halogen, -\
'R
' Y
' . CYY. cycloaikyl, or -OR
" ; each R
' is independently oxo, -(CY- C
3 alkylene)OR
i , C
rC
6 alkyl optionally substituted by halogen, -C(0)NR
i!R
?2, ·
;(
r
C3 alkylene)NRUR12, -CN, halogen, -N ^iCy CY-C6 cycloaikyl, or -ORi0; or any two groups K' :'. when bound to the same carbon atom, are taken together with the carbon to winch they are attached to form a <Y-C.:-. cycloaikyl; and m and n are independently 0, I, 2, or 3. In some embodiments of a compound of Formula (I), (la) or (lb), X is hydrogen; Y is N; Z is N; !< "' and RJ" are independently hydrogen or Y ··( .;. alkyl; or R and R'L' are taken together with the carbon io which they are attached to form a t :--(.,. cycloaikyl, R" is (';,·('·.: aryl optionally substituted by halo en, -CN, -NR: R;'\ -C(0)R33, -OR33, or CrCY alkyl optionally substituted
by -OH; Wis
; wherein B is C -C
6 cycloaikyl, 3- to 7-membered
. 17·:>
heterocyclyl, 5- to 7-menabered heteroaryl, or C
¾ aryl, each R
"" is independently C]-
> alkyl, each R
' is independently (
'· ·( .··. alkyl optionally substituted by halogen, -C(0)R
!\ or oxo: and each m and n are independently 0, I, 2, or 3, In some embodiments of a compound of Formula (I), (la) or (lb), X is hydrogen, Y is N, Z is N; R and k
'"' are independently hydrogen or CY C
6 alkyl; R
4 is phenyl optionally substituted by halogen, -CN, -NR
13R
i4, -C(0)R
13, -OR
13, or
C i --C
6 alky! optionally substituted by -OH; A, B. R
1 a and R
i 7 are
wherein B is < V(7. cycioalkyl, 3- to 7-membered heterocyclyl, 5- to 7-membered heteroaryl, or C¾ aryl; each R.: "! is ndependently 0 ; ·( .·. alkyl: two groups R : are taken together with the carbon to which they are attached to form a (^-C¾ cycioalkyl; and each m and n are
independently 0. 1, 2, or 3.
[0084] in some embodiments of a compound of Formula (1), (ia) or (lb), X is hydrogen; Y is N; Z is N; R"" and R ' ' are independently hydrogen or C1-G5 alkyl; or R " and K are taken together with the carbon to which they are attached to form a C3-C c cioalk l, R"* is 5- to 1 0- membered heteroatyl or QyCu ary!, each of which is optionally substituted by halogen, ·· CN, -NR13RI ( ·( (0}R ; . -OR"13, or CrC6 a!kyl optionally substituted by -OH: W ss
; wherein B is C3-C6 cycioalkyl, 3- to 7-membered heterocyclyl, 5- to 7-mernbered heteroaryl, or C
¾ aryl; each " is independently n
' r · ··(
' , a!kylene)OR
' . C]- C . alkyl optionally substituted by halogen, ~C(0) R
! i"R
12, ·π <b aikylene)NR
nR
i2, -CN, halogen, -NR.'
' k
: " . C Cg cycioalkyl, or -OR
*'"; each R
' is independently oxo, ~(Ci - C
3 a!kyiene)GR
Ui, C
rC alkyl optionally substituted by halogen, -QO sNR
1 V
2, ··;<
1 : i l l ? f>
C3 alkylene)NR ' "R , -CN, halogen, - R", C3-C6 cycioalkyl, or -OR ' : or any two groups R" \ when bound to the same carbon atom or two different carbon atoms, are taken together with the carbon or carbons to which they are attached to form a C3-C6 cycioalkyl or 3- to 7- membered heterocyclyl, each is optionally substituted by R " and m and n are independently 0, 1, 2, or 3. in some embodiments of a compound, of Formula (I), (ia) or (lb), X is hydrogen, Y is N; Z is N; K ' and l< " ' are independently hydrogen or C Ce alkyl; or R " and R are taken together with the carbon to which they are attached to form a C3-C6 cycioalkyl; R" is 5- to 10- me bered heieroaiyl or (" : ( ' : .;. aryl, each of which is optionally subsiituied by halogen, -
(IN, -NR1 RW, ·( 0}R! -OR13, or C C6 alky! optionally substituted by OU: W is
; wherein B i C Ce cycloalkyl 3- to 7-mernbered heterocyclyL 5·· to 7-mernbered heteroaryl, or C
6 aryl; each
'l; is independently ·ί
Γ ···(
' .. a!kylene)OR
" . C]- (\. alkyl optionally substituted by halogen, -CCOjNR'
2, ·π
rC: aikylene)NR
nR
12, -CN, halogen, ·ΛΚ
: 'R
' 7
{'7··(7, cycloalkyl, or -OR
" each R
: ts independently oxo,■■{( :· C
3 a!kyiene)GR
Ui, C
r-C a!ky! optionally substituted by halogen, -CiOsNR
1 V
2, ··;(
1 : 5 i l l? i f>
C
3 a1kylene)NR
'-R , -CN, halogen, - R
", C
3 »C
6 cycloalkyl, or -OR
' : or any two groups K
' \ when bound io the same carbon atom, are taken together v- -Ah the carbon to which they are attached to form a C3-C6 cycloalkyl; and m and n are independently 0, 1, 2, or 3. In some embodiments of a compound of Formula (I), (la) or (lb), X is Iiydrogen, Y is , Z is N: R
"d and R are independently hydrogen or ί
r<7 alkyl; or 1'
': and R
JL' are taken together with the carbon to which they are attached to form a Cj-Q cycloalkyl;
" is CG-C ·
4 aryl optionally substituted by halo en, -CN, -NR
13R
i4,
0r C
3-C
6 alkyl optionally substituted
by -OH; Wis
wherein B is (77.·.. cycloalkyl, 3- to 7~mernbered heterocyclyl, 5- to 7-membered heteroaryl, or (V. aryl: each R
: '; is independently (7 ·( .·. alkyl; each R
' is independently C7d >. alky! optionally substituted by halogen, ~C(O)R
!7 or oxo; and each m and n are independently 0, 1, 2, or 3, In some embodiments of a compound of Formula (Ϊ), (la) or (lb), X is hydrogen; Y is N: Z is N; R and R
'"' are independently Iiydrogen or (7· C
6 alkyl;
; is phenyl optionally substituted by halogen, -CN, ~NR
15R
U, -C(C»R , -OR
13, or
(R17a)n
Cj-Q, alkyl optionally substituted by -Oil; rs (R1¾ BCL ··* wherem B is C3--
Cf} cycloalkyl, 3- to 7-niembered heterocyclyl, 5- to 7»mer«bered heteroaryl, or (7. aryl; each
R"J is independently C i .·. alkyl; two groups R1 are taken together with the carbon to which they are attached to form a C¾-C-6 cycloaikyl, and each m and n are independently 0, 1, 2, or 3.
[0085] In some embodiments of a compound of Formula (Ϊ), 0a) or (lb), X is hydrogen, Y is
' are independently hydrogen or C]-C6 alkyl; or R and R"' are taken together with the carbon to winch they are attached to form a ( Q cycloaikyl; R is 5- io 10- membered heteroaryl or {" :,-C;.; aryl, each of which is optionally substituted by halogen, · CN, -NRi3R , -CiO)R;3, -OR13, or i><\. alkyl optionally substituted by -OH; W is
(R17a)n
' * ; wheremB is Li-Cs cycloaikyl, 3 - to /-membereel neterocyciyl, ~ to 7-membered heteroaryl, or Q, aryl; each RJ''!> is independently ··<;( alkylene )OR1J,Ci~ C6 alkyl optionally substituted by halogen, -C(0)NRnR12, ·ίΟ;·( .. aiky!eneS R^R12, -CN, halogen, -N .' :R ' . ( Q, cycloaikyl. or -OR'"; eacliR""'' is independently oxo, -€■- C\ alkyl ene)OR1 , CrC6 alkyl optionally substituted by halogen, ·( (0;M<i;R; :. ·(( r
alkyj.ene)NRf "Rli, -CN, halogen, · N k ; ", cycloaikyl, or -OR ' '. or any two groups
RJ when bound to the same carbon atom or two different carbon atoms, are taken together with the carbon or carbons to which they are attached to form a <b ··('>. cycloaikyl or '· to 7~ membered heterocyclyl, each is optionally substituted by R" ; and ni and n are independently 0, 1, 2, or 3 In some embodiments of a compound of Formula (I), (la) or (lb), X is hydrogen, Y is N; Z is N; R.Ja and " ' are independently hydrogen or C]-C6 alkyl; or R '* and R " are taken together with the carbon to which they are attached to form a Cs-C cycloaikyl; R* is 5- to 10- membered heteroaryl or <>( ;.; aryl, each of winch is optionally substituted by halogen, - CN -NR13Ri4, -C(0)Ri3, -OR13, or Ci-C6 alkyl optionally substituted by OH. W is
; wherem B is -C6 cycioaikyi, 5- to '-membered. heterocyciyi,
ΐο 7-membered heteroaryl, or C
¾ aryl; each
'l; is independently ·ί
Γ ···(
' .. a!kylene)OR
" . C|- (\. alkyl optionally substituted by halogen, ^(Oj R^
12, -(C1-C3 aikylene)NR
nR
12, -CN, halogen, ·ΛΚ
: d
' 7
{'7··(7. cycloalkyl, or -OR
" each R
: is independently oxo,■■{( :· C
3 aJkylenesOR
1. <7-C alkyl optionally substituted by halogen, -QOJNR^R
12, - V
1 · 1 f P i? 1 f>
(¾ alkylene)NR
' R . -CN, halogen, -NR
" 'k 7 C
3-C
6 cycloalkyl, or -OR
' or any two groups R
" \ when bound io the same carbon atom, are taken together v- -Ah the carbon to which they are attached to form a C3-C6 cycloalkyl; and m and n are independently 0, 1, 2, or 3. In some embodiments of a compound of Formula (I), (la) or (lb), X is Iiydrogen, Y is , Z is N; R
"d and R are independently hydrogen or ί
r<7 alkyl; o R
': and R
Jl' are taken together with the carbon to which they are attached to form a ( ,·<
'>. cycloalkyl; k is i
' ;y- aryl optionally substituted by halo en, -CN, -NR
i3R
i4, -C(0)R
i3, -OR.
J'3, or C C
6 alkyl optionally substituted )n by -OH; W is
: wherein B is (77.·.. cycloalkyl, 3- to 7-mernbered heterocyclyi, 5- to 7-membered heteroaryl, or C\. aryl, each K
' is independently <7··(7. alkyl, each R7 is independently (7 ·( ..·. alkyl optionally substituted by halogen, -C(0)R'7 or oxo: and each m and n are independently 0, 1, 2, or 3, In some embodiments of a compound of Formula (I), (la) or (lb), X is hydrogen; Y is N: Z is N; R and k are independently Iiydrogen or (7· C
6 alkyl; R
4 is phenyl optionally substituted by halo en, -CN, ~NR
1 R
14, -C(C»R , ~OR , or )r t
r <7 alkyl optionally substituted by -OH; W is
, wherein B i
Cf} cycloalkyl, 3- to 7-membered heterocyclyL 5-· to 7~memberecl heteroaryl, or€5 aryl; each R ' " is independently C : ··(¾ alkyl, two groups R * ' ' are taken together with the carbon to wh¾cl they are attached to form a C3-C6 cycloalkyl; and each rn and n are independently 0, 1,2, or 3.
[0086] In some embodiments of a compound of Formula (Is. (ia) or (lb), X is hydrogen; Y is N; Z is N; R" and R ' ' are independently hydrogen or C1-G5 alkyl; or and K are taken together with the carbon to which they are attached 10 form a. CVC6 cycioalkyl; K" is 5- to 10- niembered heteroaryl or ,~Ci4 aryl, each of which is optionally substituted by halogen,■- CN, -NR13RI ( ·( (0}R; . -OR13, or CrC6 a!kyl optionally substituted by -OH: W is
; wherein B is C3-C6 cycioalkyl 3- to 7-membered heterocyclyi, 5- to 7~ lembered heteroaryl, or C
¾ aryl; each
'l; is independently ·ί
Γ ···(
' .. alkylene)OR
" . C]- C. alkyi optionally substituted by halogen, -C(Os R
iLR
12, ·π <h alkyiene)NR
UR
il -CN, halogen, ·ΛΚ
: 'R
' \
{'7··0
:·. cycioalkyl, or -OR
" each R
: is independently oxo,■-(€:-·
C3 alkylene)ORK!. C C6 alkyl optionally substituted by halogen, -C(0)NRnRi2, ···<>
1 : 5 i l l? 1 f>
C3 alkylene)NR'-R , -CN, halogen, -NR. R'", C3-C6 cycioalkyl, or -OR' : or any two groups R' \ when bound to the same carbon atom or two different carbon atoms, are taken togeiher with the carbon or carbons to which they are attached to form a ( Ck, cycioalkyl or 3- to 7- membered heterocyclyi, each is optionally substituted by RJ and m and n are independently 0, 1, 2, or 3. in some embodiments of a compound, of Formula (I), (ia) or (lb), X is hydrogen, Y is N; Z is N; " and R "' are independently hydrogen or Ci-C^ alkyl: or R " and R are taken together with the carbon to which they are attached to form a C3-C6 cycioalkyl; R" is 5- to 10- membered heteroaryl or ( : (':.;. aryl, each of which is optionally substituted by halogen, - CN -NR1 RW, ~C(0)Ri3, -OR13, or C C6 aikyl optionally substituted by -OH; W is
wherein B is CVC .·. cycioalkyl, 3-· to 7-membered heterocyclyi, 5- to 7-membered heteroaiyl, or (
' ;, aiyl; each
""" is independently u
( <\ alkyiene)OR"", Cj- C
6 alkyl optionally substituted by halogen,
■-C(0) R
IiR
12, -(C C
3 aikylene)NR. R
12, -CN, halogen, -NR
^ ^, C3-C cycioalkyl, or -OR'
1', each R
"° is independently oxo, --((V
(¾ alkylene)OR
10 s C i -C
6 alky! optionally substituted by halogen, -C(0)NR
! 2, Y Y- C\ a!kyierse)N
" 'R"\ -CN, halogen, - R' T
". cycloalkyi or -OR
' '. or any two groups when bound to the same carbon atom, are takers together with the carbon to which they are atttiched to form a CYC cycloalkyi; and rn and n are independently 0, 1 , 2, or 3 , In some embodiments of a compound of Formula (I), (la) or (lb), X is hydrogen; Y is N; Z is N; R and R
' " are independently liydrogen or C C alky I; or K and are taken together w th fee carbon to which they are attached to form a YC ... cycloalkyi: R
+ is C :
; aryl optionally substituted by halo en, -CN, -NR
i 3R
H, YiOji y -OR
1 \, or C C
6 alky! optionally substituted ), by ΟΠ . W is
. wherem B is CY-Q cycloalkyi, ·· · to 7-niembered heterocyclyl, 5~ to 7-membered heteroaryi, or aryi, each
' rs independently f C aikyl; each R
J is independently CY Y. alk l optionally substituted by halogen, -C(0}R
l '. or oxo, and each m and n are independently 0, L 2, or 3, in some embodiments of a compound of Formula (I), (la) or (lb), X is hydrogen; Y is N: Z is N; .
' ' and R are independently hydrogen or (■. · C alkyl; IT is phenyl optionally substituted by halogen, -CN, -NR R
14, -C(0)R
i 3, -OR
1 , or
(R17a)n
(>( ): alkyl optionally substituted by -OH; W is N -· wherem B is CY¬
CY. cycloalkyi, 3- to 7-membered heterocyclyl, 5~ to 7-membered. heteroaryi , or i¾ aryl; each ' ' i! is independently ( Y ' ;. alkyl ; two groups R * ' " are taken together with the carbon to which they are attached to form a CYC !: cycloalkyi; and each m and n are independently 0, 1 , 2, or 3,
[0087] in some embodiments of a compound of Formula (D, (la) or (ib), X is hydrogen; Y is N; Z. is N; R
'" and '
' are both hydrogen; R
" is selected from the group consisting of methyl,
ethyl, isopropyl, cyciopropy!.
:
is selected from the group consisting of:
wherei the wa !mes denote attachment points to the parent molecule.
[0088] In some embodiments of a compound of Formula (I), (la) or (lb), X is hydrogen, Y N; Z is N; Ef " and !< " ' are both hydrogen; R ' i selected from the group consisting of m thyl,
W
is selected from the group consisting of:
parent molecule.
[0089] In some embodiments of a compound of Formula (I ), (la) or (lb), W is A, wherein A
ί j
rs phenyl optionally substituted with " " and R is independent ly 3- to 12-membered heterocyclyl, -(CrC3 alkylenejOR10. C : -( .··. alkyl, -C(0)NR1 j'R12, ~(Cr
C-3 aikylene)NRUR12, -CN, halogen, - N ^C ". or -OR i 0, wherein the 3- to 12-membered heterocyclyl of R is optionally substituted with Ci - ,
alkyl, ~C(0)R -(C C3 alkylene)OR -SfO)2R13, C3-C8 cycloaikyi, oxo, halogen, or -OR1 ; X is hydrogen , Y is N, Z is N; R ': i hydrogen, R is hydrogen, and ' is 2,6~dichlorophenyl. in some embodimenis of a compound of Formula (I), (ia) or (ib), W is A, wherein A is phenyl optionally substituted with R and R' ' " is independently 3-· to 12-membered heterocyclyl or -CN, X is hydrogen; Y is N; Z is N, R is hydrogen; K ' is hydrogen; and R'* is 2,6-- dichiorophenyl.
In some embodiments of a compound of Formula (i), (ia) or (lb), W is
wherein B is 3·- to 7-membered heterocyc!yl; each R
' is
independently Ci-C6 alkyl; each II" L' is independently C|-C6 a kyl optionally substituted by halogen, ~C(0)R"". or oxo: X is hydrogen; Y is N; Z is N; R '; is liydrogen; K is !iydrogen; and R** is 2.6-dichloropherryl , i some embodiments of a compound of Formula (I), (la) or (lb), W is
wherein B is 6-membered heterocyclyl: each R
: "' is independently alkyl ; each R
' is independently Ci -Cs alkyl optionally substituted by halogen., ·( i ( ) ) R
' '. or oxo; X is hydrogen; V :> N; Z is N; k
*' is hydrogen; R
" * is hydrogen; and R
' is 2,6- dichlorophenyl In some embodiments of a compound of Formula (I), (la) or (lb), W is
wherem B is 6-membered heterocyc!yl; each
1 l! is independently < ;-< alkyl ; eaoh R/
" '"' is independently C i -Q alkyl , X is hydrogen; Y is N; Z is N, R
" is hydrogen; R is liydrogen; and R is 2,6-dichlorophenyl.
[0091] in some embodiments of a compound of Fommla (I), (la) or (lb), W is selected from the group consisting of:
] 7a lines denote aiiachment points to the parent molecule and each is optionally substituted with R and R ' X is hydrogen; Y is N; Z is N; R"'~ is hydrogen; R is hydrogen, and R. is 2,6- diehlorophenyl.
[0092] In some embodiments of a compound of Formula (I), (la) or ( l b), W is
substituted by -CN; X is hydrogen; Y is N, Z is N; R
' is hydrogen; R
"'° is hydrogen, and R
"1 is 2,6-dichlorophenyl. in some embodiments of a compound of Formula (1),
(la) or (lb), W is
substituted by C C
6 alkyl; X i hydrogen, Y is N, Z is N;
K " is hydrogen; R " * is hydrogen; and R ' is 2,6-dichlorophenyi.
[0093] Also provided are salts of compounds referred 10 herein, such as pharmaceutically acceptable salts. The invention also includes any or all of the stereochemical forms, including any enantiomeric or diastereomenc forms, and any tautomers or other forms of the compounds descri bed.
[0094] A compound as detailed herein may in one aspect be in a. purified form arid
compositions comprising a compound in purified forms are detailed herein. Compositions comprising a compound as detailed herein or a salt thereof are provided, such as compositions of substantially pure compounds. Compositions comprising a compound as deiailed herein, or a tautomer or isomer thereof, or a pharmaceutically acceptable salt of any of the foregoing, are provided, such as compositions of substantially pure compounds, in some embodiments, a composition containing a compound as detailed herein or a salt thereof is in substantially pure form. Unless otherwise stated, "substantially pure" intends a composition that contains no more than 35% impurity, w herein the impurity denotes a compound other than the compound comprising the majority of the composition or a salt thereof In some embodiments, a
composition of substantially pure compound or a salt thereof is provided wherein the
composition contains no more than 25%, 20%, 15%, 10%, or 5% impurity, in some
embodiments, a composition of substantially pure compound or a salt thereof ts provided wherein the composition contains or no more than 3%, 2%, 1 % or 0,5% impurity. 009S] Representative compounds are listed in Table 1.
Table 1
Com Com i poun Structure poun Structure 1 d No d No.
S .5
\,- A- ■ il ArV ά H
1 7
† ! .·'■" ->
[0096] In some embodiments, provided herein is a compound described in Table 1 , or a tautomer thereof, or a salt of a ny of the foregoing, and uses thereof In some embodiments, provided herein is a compound described in Table .1 , or a salt thereof In some embodiments, provided herein is a compound described in Table 1 , or a lautomer or isomer thereof, or a. pharmaceutically acceptable salt of any of the foregoing,
[0097] The embodiments and variations described herein are suitable for compounds of any formulae detailed herein, where applicable.
[0098] Representative examples of compounds detailed herein, including intermediates and final compounds according to the present disclosure are depicted herein. It is understood that in one aspect, any of the compounds may be nsed in the methods detailed herein, including, where applicable, intermediate compounds that may be isolated and administered to an individual.
[0099] The compounds depicted herein may be present as salts even if salts are not depicted and it is understood that the present disclosure embraces all salts and solvates of the compounds depicted here, as well as the non-salt and non-sol vate form of the compound, as is well understood by the skilled artisan. In some embodiments, the salts of the compounds provided herem are pharmaceutically acceptable salts. Where one or more tertiary amine moiety is present in the compound, the N-oxides are also provided and described.
[0100] Where tautomeric forms may be present for any of the compounds described herein, each and every tautomeric form is intended even though only one or some of the tautomeric forms may be explicitly depicted. The tautomeric forms specifically depicted may or may not be the predominant forms in solution or when used according to the methods described herei It is understood that tautomeric forms of a compound of the formulae described herein may be present, for example, when tautomeric forms of a substituent are present such as when a substituent embraces a keto-enol tautomer or the like.
[0101 ] The present disclosure also includes any or all of the stereochemical forms, including any enantiomeric or diastereomeric forms of the compounds described, such as compounds of Table 1 . The structure or name is intended to embrace all possible stereoisomers of a compound depicted, and each unique stereoisomer has a compound number bearing a suffix "a", "b", etc. All forms of the compounds are also embraced by the invention, such as crystalline or noncrystalline forms of the compound Compositions comprising a compound of the invention are also intended, such as a composition of substantially pure compound, including a specific stereochemical form thereof or a composition comprising mixtures of compounds of the invention m any ratio, including two or more stereochemical forms, such as in a racenuc or non- racemic mixture.
[0102] The invention also intends isotopieally-kbeled and/or isotopicaliy-enriched forms of compounds described herein. The compounds herein may contain unnatural proportions of atomic isotopes at one or more of the atoms that constitute such compounds. In some
embodiments, die compound is isotopieally-!abeled, such as an isotopicaily -labeied compound of Formula (I) or variations thereof described herein, where a fraction of one or more atoms are replaced by an isotope of the same element. Exemplary isotopes that can be incorporated into compounds of the invention include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorus, sulfur, chlorine, such as Ί Ι 11 1 ! CS : ί . ' ¾ ? 5(), 1 Ό, "p, 35S, l 8F, 36Cj. Certain isotope !abeled compounds (e.g. "H and " C) are useful compound or substrate tissue distribution studies. Incorporation of heavier isotopes such as deuterium ('Ή) can afford certain therapeutic advantages resulting from greater metabolic stability, for example, increased in vivo half-life, or reduced dosage requirements and, hence may be preferred in some instances.
[0103] Isotopicaily-labeied compounds of die present invention can generally be prepared by standard methods and techniques known to those skilled m the art or by procedures similar to those described in the accompanying Examples substituting appropriate isotopieally-labeled reagents in place of the corresponding non-labeled reagent.
[0104] The invention also includes any or ail metabolites of any of the compounds described. The metabolites may include any chemical species generated by a biotransformation of any of the compounds described, such as intermediates and products of metabolism of the compound, such as would be generated in vivo following administration to a human.
[0105] Articles of manufacture comprising a compound described herein, or a salt or solvate thereof, m a suitable container are provided. The container may be a vial, jar, ampoule, preloaded s inge, i.v. bag, and the like.
[0106] Preferably, the compounds detailed herein are orally bioavailable. However, the compounds may also be formulated for parenteral (e.g., intravenous) administration. OJ 07] One or several compounds described herein can be used in the preparation of a medicament by combining die compound or compounds as an active ingredient with a pharmacol gically acceptable carrier, which are known in the art. Depending on the therapeutic form of the medication, the carrier may be m various forms, in one variation, the manufacture of a medicament is for use in any of the methods disclosed herein, e.g., for the treatment of cancer.
General synthetic methods
[0108] The compounds of fee invention may be prepared by a number of processes as generally described below and more specifically in the Examples hereinafter (such as fee schemes provided in the Examples below), in the following process descriptions, the symbols when used in the formulae depicted are to be understood to represent those groups described above in relation to the formulae herein.
[0109] Where it is desired to obtain a particular enantiomer of a compound, this may be accomplished from a corresponding mixture of enantiomers using any suitable conventional procedure for separating or resolving enantiomers. Thus, for example, d astereomeric derivatives may be produced by reaction of a mixture of enantiomers, e.g., a racemate, and an appropriate chiral compound. The diastereomers may then be separated by any convenient means, for example by crystallization and the desired enantiomer recovered. In another resolution process, a racemate may be separated using chiral High Performance Liquid
Chromatography. Alternatively, if desired a particular enantiomer may be obtained by using an appropriate chiral intermediate in one of the processes described.
[01 10] Chromatography, recrystaliization and oilier conventional separation procedures may also be used with intermediates or final products where it is desired to obtain a particular isomer of a compound or to otherwise purify a product of a reaction.
[0111] Solvates and/or polymorphs of a compound provided herein or a pharmaceutically acceptable salt thereof are also contempl ted. Solvates contain either stoichiometric or non- stoichiometric amounts of a solvent, and are often formed during the process of crystallization. Hydrates are formed when the solvent is water, or alcoholates are formed when the solvent is alcohol. Polymorphs include the different crystal packing arrangements of the same elemental composition of a. compound. Polymorphs usually have different X-ray diffraction patterns, infrared spectra, melting points, density, hardness, crystal shape, optical and electrical properties, stability, and/or solubility Various ctors such as the recrystaliization solvent, rate of crystallization, and storage temperature may cause a single crystal form to dominate
[0 12] in some embodiments, compounds of Formula. (I) may be synthesized according to Schemes 1 -3 ,
Scheme I
SS
'^
S"Z^ OH Step-3 Ό
" \
R3b Siep-4
Scheme 3
wherein W, Y, /.. R ". R"", and IT are as defined for Formula (I). Particular examples are provided in the Example Section below. Here L i a leaving group like Br, G or ί etc.
Pharmaceutical Compositions and Formulations
[01 13] Pharmaceut cal compositions of any of the compounds detai led herein are embraced by this disclosure. Thus, the present disclosure includes pharmaceutical compositions comprising a compound as detailed herein or a pharmaceutically acceptable salt thereof and a pharmaceutically acceptable earner or excipient. The present disclosure also includes pharmaceutical compositions comprising a compound as detailed herein or a tautomer or isomer thereof, or a pharmaceutically acceptable salt of any of die foregoing and a pharmaceutically acceptable carrier or excipient. In one aspect the pharmaceutically acceptable salt is an acid addition salt, such as a salt formed with an inorganic or organic acid. Pharmaceutical compositions may take a form suitable for oral, buccal, parenteral, nasal, topical or rectal administration or a form suitable for administration by inhalation. jO f 14] A compound as detailed herein may m one aspect be in a purified form and compositions comprising a compound in purified forms are detailed herein. Compositions comprising a compound as detailed herein or a salt thereof are provided, such as compositions of
substantially pure compounds, in some embodiments, a composition containing a compound as detailed herein or a salt thereof is in substantially pure form.
[01 15] in one variation, the compounds herein are synthetic compounds prepared for administration to an individual. In another variation, compositions are provided containing a compound m substantially pure form. In another variation, the present disclosure embraces pharmaceutical compositions comprising a compound detailed herein and a pharmaceutically acceptable carrier. In another variation, methods of administering a compound are provided. The purified forms, pharmaceutical compositions and methods of administering the compounds are suitable for any compound or form thereof detailed herein.
(011 ] A compound detailed herein or salt thereof may be formulated for any available delivery route, including an oral, mucosal (e.g. , nasal, sublingual, vaginal, buccal or rectal), parenteral (e.g. , intramuscular, subcutaneous or intravenous), topical or transdermal delivery form. A compound or salt thereof may be formulated with suitable earners to provide delivery forms that include, but are not limited to, tablets, caplets, capsules (such as hard gelatin capsules or soft elastic gelatin capsules), cachets, troches, lozenges, gums, dispersions, suppositories, ointments, cataplasms (poultices), pastes, powders, dressings, creams, solutions, patches, aerosols (e.g. , nasal spray or inhalers), gels, suspensions (e.g., aqueous or non-aqueous liquid suspensions, oibm-water emulsions or water-m-oil liquid emulsions), solutions and elixirs,
[0117] One or several compounds described herein or a salt thereof can be used in the preparation of a formulation, such as a pharmaceutical formulation, by combining the compound or compounds, or a salt thereof, as an active ingredient with a pharmaceutically acceptable carrier, such as those mentioned above. Depending on the therapeutic form of the system (e.g; , transdermal patch vs. oral tablet), the carrier may be in various forms. In addition,
pharmaceutical formulations may contain preservatives, solubilizers, stabilizers, re-wetting agents, emulgators, sweeteners, dyes, adj usters, and salts for the adjustment of osmotic pressure, buffers, coating agents or antioxidants. Formulations comprising the compound may also contain other substances which have valuable therapeutic properties. Pharmaceutical formulations may be prepared by known pharmaceutical methods. Suitable formulations can be found, e.g. , in Remington 's Pharmaceutical Sciences, Mack Publishing Company, Philadelphia, PA, 20'" ed. (2000). which i incorporated herein by reference
[01 18] Coxnpounds as descr bed herein may be administered to individuals in a form of generally accepted oral compositions, such as tablets, coated tablets, and gel capsules in a hard or in soft shell, emulsions or suspensions. Examples of carriers, winch may be used for the preparation of such compositions, are lactose, corn starch or its derivatives, talc, stearate or its salts, t.-' c Acceptable earners for gel capsules with soft shell are, for instance, plant oils, wax, fats, semisolid and liquid poly-ols, and so on. In addition, pharmaceutical formulations may contain preservatives, solubilizers, stabilizers, re-wetting agents, eniulgaiors, sweeteners, dyes, adjusters, and salts for the adjustment of osmotic pressure, buffers, coating agents or
antioxidants.
(0119] Any of the compounds described herein can be formulated in a tablet in any dosage form described, for example, a compound as described herein or a pharmaceutically acceptable salt thereof can be formulated as a 10 mg tablet.
[0120] Compositions comprising a compound provided herein are also described, in one variation, the composition comprises a compound or salt thereof and a pharmaceutically acceptable carrier or excipient. In another variation, a composition of substantially pure compound is provided.
Adethods of Use
(0121] Compounds and compositions detailed herein, such as a pharmaceutical composition containing a compound of any formula provided herein or a salt thereof and a pharmaceutically acceptable earner or excipient, may be used in methods of administration and treatment as provided herei Compounds and compositions detailed herein, such as a pharmaceutical composi tion containing a compound of any formula provided herein, or a tautomer or isomer thereof, or a pharmaceutically acceptable salt of any of the foregoing, and a pharmaceutically acceptable earner or excipient, may be used in methods of administration and treatment as provided herein The compounds and compositions may also be used in in vitro methods, such as in vitro methods of administering a compound or composition to cells for screening purposes and/or for conducting quality control assays.
[01 2] Provided herein is a method of treating a disease in an individual comprising administering an effective amount of a compound of Formula (I), ( a), (lb), (Ila), (lib), (Il a),
(Illb), (IVa) or (IVb), or any embodiment, variation or aspect thereof (collectively, a compound of Formula (I), (la), (lb), (Ila), (lib), (Ilia), (illb), (IVa) or (IVb), or die present compounds or the compounds detailed or described herein) or a pharmaceutically acceptable salt thereof, to the individual. Further provided herein is a method of treating a proliferative disease m an individual, comprising administering an effective amonnt of the compoursd of Formula (1) , (la), (lb), (Ha), (lib), (I ia), (Illb), (IVa) or (IVb), or a pharmaceutically acceptable salt thereof, to the individual. Also provided herein is a method of treating cancer i an individual comprising administering an effective amonnt of the compound of Formula (I), (la). (Ib), (Ha). (lib), (Ilia), (131b), (IVa) or (IVb), or a pharmaceutically acceptable salt thereof, to the individual, in some embodiments, the compound is administered to the individual according to a dosage and/or method of administration described herein.
[0123] in some embodiments, the cancer in the individual has one or more TP53 gene mutations or expresses mutant p53. ZP53 is the human gene that encodes p53. in some embodiments, provided herein is a method of treating a cancer in an individual, comprising (a) selecting the individual for treatmen t based on (i) the presence of one or more mutations of the TP 53 gene in the cancer, or (ii) expression of mutant p53 m the cancer, and administering an effective amount of the compound of Formula (I), (la), (lb), ( a), (lib), (Ilia), (Illb), (IVa) or (IVb), or a pharmaceutically acceptable salt thereof, to the individual. In some embodiments, the cancer is assayed for the expression of mutant p53. In some embodiments, the 7P5J gene of the cancer is sequenced to detect the one or more mutations, in some embodiments, the TP 53 gene is sequenced by biopsying the cancer and sequencing the TP53 gene from the biopsied cancer, in some embodiments, the TP 53 gene is sequenced by sequencing circulating- tumor DNA (ctDNA) from the indi idual.
[01 4] in some embodiments, provided herein is a method of using a compound of Formula (I), (la), (ib), (ila), (iib), (Ilia), (Illb), (IVa) or (IVb), or any embodiment in the manufacture of a medicament for treatment of a disease In some embodiments, provided herein is a method of using a compound of Formula (i), (ia), (ib), (iia), (iib), (iiia), ( I l l b). (IVa) or (IVb), or any embodiment in the manufacture of a medicament for treatment of cancer.
[0125] In some embodiments, a compound of formula (I), (la), (ib), (Ila), (lib), (Ilia), (Iiib), (IVa) or (IVb), or a salt thereof is used to treat an individual having a proliferative disease, such
as cancer as described herein. In some embodiments, the individaal is at risk of developing a proliferative disease, such as cancer. In some of these embodiments, the individual is deiermmed to be at nsk of developing cancer based upon one or more nsk factors. In some of these embodiments, the nsk factor is a family history and/or gene associated with cancer.
[0126] The present compounds or salts thereof are believed to be effective for treating a variety of diseases and disorders. For example, in some embodiments, the present compositions may be used to treat a proliferative disease, such as cancer, in some embodiments the cancer is a solid umor. In some embodiments the cancer is any of adult and pediatric oncology, myxoid and round cell carcinoma, locally advanced tumors, metastatic cancer, human soft tissue sarcomas, including Ewmg's sarcoma, cancer metastases, including lymphatic metastases, squamous cell carcinoma, particularly of the head and neck, esophageal squamous cell carcinoma, oral carcinoma, blood cell malignancies, including multiple myeloma, leukemias, including acute lymphocytic leukemia, acute nonlymphocytic leukemia, chronic lymphocytic leukemia, chronic myelocytic leukemia, and hair cell leukemia, effusion lymphomas (body cavity based lymphomas), thymic lymphoma, cutaneous T cell lymphoma, Hodgkin's lymphoma, non- Hodgkm's lymphoma, cancer of the adrenal cortex, ACTH-producing tumors, lung cancer, including small cell carcinoma and nonsmall ceil cancers, breast cancer, including small cell carcinoma and ductal carcinoma, gastrointestinal cancers, including stomach cancer, colon cancer, colorectal cancer, polyps associated with colorectal neoplasia, pancreatic cancer, Hver cancer, urologies! cancers, including bladder cancer, including primary superficial bladder tumors, mvasive transitional cell carcinoma of the bladder, and muscle-invasive bladder cancer, prostate cancer, malignancies of the female genital tract, including ovarian carcinoma, prirnaiy peritoneal epithelial neoplasms, cervical carcinoma, uterine endometrial cancers, vaginal cancer, cancer of the vulva, uterine cancer and solid tumors in the ovarian follicle, malignancies of the male genital tract, including testicular cancer and penile cancer, kidney cancer, including renal cell carcinoma, brain cancer, including intrinsic brain tumors, neuroblastoma, astrocytic brain tumors, gliomas, metastatic tumor cell invasion m the central nervous system, bone cancers, including osteomas and osteosarcomas, skin cancers, including melanoma, tumor progression of human skin keratmocytes, squamous cell cancer, thyroid cancer, retinoblastoma, neuroblastoma, peritoneal effusion, malignant pleural effusion, mesothelioma, Wibns's tumors, gall bladder cancer, trophoblastic neoplasms, hemangiopericytoma, and Kaposi's sarcoma.
[0127] irs some embodiments, the compounds and compositions described herein suppress C¾-M checkpoint in a ceil (such as a. cancer cell). In some embodiments, the cancer cell is a cancer cell from any of the cancer types described herein. Suppression of the (¾-M D A damage checkpoint results in premature mitosis of the ceil, and consequently apoptosis. In some embodiments, provided herein is a method of suppressing the C½-M DNA damage checkpoint in a cell comprising administering an effective amount of the compound of Formula (I), (la), (lb), (Ha), (IlbS, (Ilia), ([[ b), (IVa) or (IVb), or a pharmaceutically acceptable salt thereof, to the cell. In some embodiments, the (¾-M DNA damage checkpoint is suppressed in about 40% or more, about 50% or more, about 60% or more, about 70% or more, about 80% or more, about 85% or more, about 90% or more, about 95% or more, about 96% or more, about 97% or more, about 98% or more, or about 99% or more of cells in a cell population. In some embodiments, the G.;-\ j DNA damage checkpoint is suppressed in up to about 99%, up to about 98%, up to about 97%, up io about 96%, up io about 95%, up io about 90%, up to about 85%, or up to about 80% of cells in the cell population.
[0128] In some embodiments, provided herein is a method of inducing premature mitosis in a cell comprising administering an effective amount of the compound of Formula (I), (la), (lb), (Ha), (lib), (Ilia), (Illb), (IVa) or (IVb), or a pharmaceutically acceptable salt thereof, to the cell. In some embodiments, premature mitosis is induced in about 40% or more, about 50% or more, about 60% or more, about 70% or more, about 80% or more, about 85% or more, about 90% or more, about 95% or more, about 96% or more, about 97% or more, about 98% or more, or about 99% or more of cells in a cell population. In some embodiments, premature mitosis is induced in up to about 99%, up to about 98%, up to about 97%, up to about 96%, up to about 95%, up to about 90%, up to about 85%, or up to about 80% of cells in the cell population.
[0129] In some embodiments, provided herein is a method of inducing apoptosis in a ceil comprising administering an effective amount of the compound of Formula (I), (la), (lb), (Ha), (lib), (Ilia), (Illb), (IVa) or (IVb), or a pharmaceutically acceptable salt thereof, to the cell. In some embodiments, apoptosis is induced in about 40% or more, about 50% or more, about 60% or more, about 70% or more, about 80% or more, about 85% or more, about 90% or more, about 95% or more, about 96% or more, about 97% or more, about 98% or more, or about 99% or more of cells in a cell population. In some embodiments, apoptosis is induced in up to about
99%, up io about 98%, up io about 97%, up io about 96%, up to about 95%, up to about 90%, u to about 85%, or u to about 80% of cells in the cell population.
[0130] in some embodiments, provided herein is a method of inhibiting Weel in a cell comprising administering an effective amount of the compound of Fomuila (Ϊ), (la), (lb), (Ha), (lib), (Ilia), (I3Ib), (IVa) or (IVb), or a pharmaceutically acceptable salt thereof) to the cell. In some embodiments, Weei is inhibited by about 10% or more, about 20% or more, about 30% or more, about 40% or more, about 50% or more, about 60% or more, about 70% or more, about 75% or more, about 80% or more, about 90% or more, about 95% or more, about 96% or more, about 97% or more, about 98% or more, or about 99% or more. In some embodiments, Weei is inhibited up to about 99%, up to about 98%, up to about 97%, up to about 96%, up to about 95%, up to about 90%, up to about 85%, up to about 80%, up to about 70%, or up to about 60%. In some embodiments, the activity of Weel is measured according to a kinase assay,
[0!3l| In some embodiments, provided herein is a method of inhibiting Weel composing contacting Weel with an effective amount of the compound of Formula (I), (la). (lb), (Tla). (lib), (Ilia), (Illb), ( Va ) or (IVb), or a pharmaceutically acceptable salt th reof In some
embodiments, the compound of Formula (I), (la), (lb), (Ila), (lib), (Ilia), (Illb), (IVa) or (I Vb), or a pharmaceutically acceptable salt thereof binds to Weel with an !C ;. of less than 1 μΜ, less than 900 nM, less than 800 nM, less than 700 nM, less than 600 nM, less than 500 nM, less than 400 nM, less than 300 nM, less than 200 nM, less than 100 nM, less than 50 nM, or less than 10 nM. In some embodiments, the compound of Formula (I) or a pharmaceutically acceptable salt thereof binds to Weel with an ICJO between 1 nM and 10 nM, between 10 nM and 50 nM, between 50 nM and 100 nM, between 100 nM and 200 nM . between 200 n : and 300 nM, between 300 nM: and 400 nM, between 400 nM and 500 nM, between 500 nM and 600 nM, between 600 nM and 700 nM, between 700 nM and 800 nM, between 800 nM: and 900 nM, or between 900 nM: and 1 μΜ. In some embodiments, the IC50 is measured according to a kinase assay. I some embodiments, the IC50 is measured according to a cell cytotoxicity assay.
[0132] In some embodiments, provided herein is a method of inhibiting the proliferation of a cell, comprising contacting the ceil with an effective amount of the compound of Formula (Ϊ), (la), (lb), (ila), (Kb), (Ilia), (Illb), (IVa) or (IVb), or a pharmaceutically acceptable salt thereof In some embodiments, the compound of Formula (I), (la), (lb), (Ila), (lib), (Ilia), (Illb), (IVa) or
(IVb), or a pharmaceutically accepiable salt thereof is effective in inhibiting the proliferation of the ceil with an Κ½ of less than 10 μΜ, less than 5 μΜ, or less than 2 μΜ, or less than 1 μΜ. Irs some embodiments, die compound of Formula (I)., (la), (lb), (Ila), (lib)., (Ilia), (Illb)., (IVa) or (IVb):1 or a pharmaceutically accepiable salt is effective in inhibiting the proliferation of the cell with n A. between 1 M and 2 Μ, between 2 Μ arid 5 μΜ, or between 2 μΜ and 10 μΜ. In some embodiments, the I C =..·.. is measured according to a cell proliferation assay.
Combination Therapy
[0133] As provided herein, the presently disclosed compounds or a salt thereof may activate the immune system, for example by inducing apoptosis or suppressing mitosis of cancer cells.
Accordingly, the present compounds or a. salt thereof may be used in combination with other anti-cancer agents to enhance tumor immunotherapy In some embodiments., provided herein is a method of treating a disease in an individual comprising administering an effective amount of a compound of Formula (I). (La), (lb), (Ha), (lib), (lite), (lift), (iVa) or (IVb), or a
pharmaceutically acceptable salt thereof and an additional therapeutic agent to the individual In some embodiments, the disease is a proliferative disease such as cancer.
[0134] In some embodiments., the additional therapeutic agent is a cancer immunotherapy agent In some embodiments, the additional therapeutic agent is a chemotherapeutic agent. In some embodiments, the additional therapeutic agent is an inmrunosumulatory agent. In some embodiments, the additional therapeutic agent targets a checkpoint protein (for example an immune checkpoint inhibitor). In some embodiments, the additional therapeutic agent is effective to stimulate, enhance or improve an immune response against a rumor. In some embodiments, the additional chemotherapeutic agent is a DNA alkylating agent, a platinum- based chemotherapeutic agent, a kmase inhibitor or a DNA damage repair (DDR) pathwa inhibitor. In some embodiments, the additional chemotherapeutic agent is a DNA alkylating agent In some embodiments, the additional chemotherapeutic agent is a platinum-based chemotherapeutic agent. In some embodiments, the additional chemotherapeutic agent is a kinase inhibitor. In some embodiments, the additional chemotherapeutic agent is a DNA damage repair (DDR) pathway inhibitor.
[0135] In another aspect, provided herein is a combination therapy for the treatment of a disease, such as cancer, in some embodiments, provided herem is a method of treating a disease in an individual comprising administering an effective amount of a compound of Formula (I), (la), H b). (Ha), (lib), (Mia), i ! ! ! b). (IVa) or (IVb), or a pharmaceutically acceptable salt thereof, in combination with a radiation therapy.
[0136] in some embodiments, provided herem is a method of treating a disease in an individual comprising (a) administering an effective amount of a compound of Formula (I), (ia), (lb), (Ma), (lib), (Mia), (ffib), (IVa) or (IVb), or a pharmaceutically acceptable salt thereof; and (b) administering an effective amount of an additional chemotherapeutic agent. In some embodiments, the chemotherapeutic agent is a kinase inhibitor or an agent that inhibits one or more DNA damage repair (DDR) pathways. In some embodiments, a compound of Formula (I), (la), (lb), (Ma), (lib), (M¾), (Illb), (IVa) or (IVb), or a pharmaceutically acceptable salt thereof is administered prior to, after, or simultaneously co-administered with the additional
chemotherapeutic agent. In some embodiments, a compound of Formula (I), (la), (lb), (Ha), (lib), (Ilia), (Illb), (IVa) or (IVb), or a pharmaceutically acceptable salt thereof is administered 1 or more hours (such as 2 or more hours, 4 or more hours, 8 or more hours. 12 or more hours, 24 or more hours, or 48 or more hours) prior to or after the additional chemotherapeutic agent.
[0137] Examples of chemotherapeutic agents that can be used in combination with a compound of Formula (I), (la), (lb), (Ila), (lib), (Ilia), (Illb), (IVa) or (IVb), or a
pharmaceutically acceptable salt thereof include DNA -targeted agents, a DNA alkylating agent (such as cyclophosphamide, mechlorethamine, chlorambucil, melphaian, dacarbaz e, or ni rosoureas), a topoisomerase inhi bitor (such as a Topoisomerase I inhibitor (e.g. , innotecan or topoteean) or a Topoisomerase H inhibitor (e.g. , etoposide or tenyposide)), an anthracycline (such as daunorubicin, doxorubicin, epirubicin, idarubicin, mi oxantrone, or valrubiein), a histone deacetylase inhibitor (such as vorinostat or romidepsin), a bromodomain inhibitor, other epigenetic inhibitors, a taxane (such as paclitaxel or dooetaxel), a kinase inhibitor (such as bortezonub, erlotinib, gefitinib, miatmib, vemurafenib, or vismodegib), an anti-angiogenie inhibitor, a nucleotide analog or precursor analog (such as azacitidine, azathioprine,
capecitabme, cytarabine, doxifluridine, 5-fluorouracil, gemcitabine, hydroxyurea,
mercaptopurine, methotrexate, or tioguanine), or a platinum-based chemotherapeutic agent (such as cisplatin, carboplatin, or oxaliplatin), pemetrexed, or a combination diereof.
[0138] in some embodiments, provided herein is a method of treating a disease in an individual comprising (a) administering an effective amount of a compound of Formula (Ϊ), (ia), (lb), (Ha), (lib), (Ilia), (Illb), (IVa) or (IVb), or a pharmaceutically acceptable salt thereof, and (b) administering an effective amount of a kinase inhibitor (such as bortezomib, erloimib, gefitimb, imatinib, vemurafenib, or vismodegib). In some embodiments, a compound of Formula (I), (la), (lb), (Ha), (lib), (Ilia), (1Mb), (IVa) or (IVb), or a pharmaceutically acceptable salt thereof is administered prior to, after, or simultaneously co-administered with the kinase inhibitor. In some embodiments, a compound of Formula (1), (ia), (lb), (iia), (lib), (ilia), (1Mb), (IVa) or (IVb), or a pharmaceutically acceptable salt thereof is administered 1 or more hours (such as 2 or more hours, 4 or more hours, 8 or more hours, 12 or more hours, 24 or more hours, or 48 or more hours) prior to or after the kinase inhibitor.
[0139] In some embodiments, provided herein is a method of treating a disease in an individual comprising (a) administering an effective amount of a compound of Formula (I), (la), ( b), (Ha), ( b), (Ilia), (Mb), (IVa) or (IVb), or a pharmaceutically acceptable salt thereof, and (b) administering an effective amount of a DNA damaging agent. In some embodiments, a compound of Formula (I), (la), (lb), (Iia), (lib), (Ilia), (Il b), (IVa) or (IVb), or a
pharmaceutically acceptable salt thereof is administered prior to, after, or simultaneously coadministered with the DNA damaging agent. In some embodiments, a compound of Formula (I), (la), (ib), (Iia), (iib), (Ilia), (Illb), (IVa) or (IVb), or a pharmaceutically acceptable salt thereof is administered 1 or more hours (such as 2 or more hours, 4 or more hours, 8 or more hours, 12 or more hours, 24 or more hours, or 48 or more hours) prior to o after the DNA damaging agent
[0140] In some embodiments, provided herein is a method of treating a disease in an individual coinprising (a) administering an effective amount of a. compound of Formula (I), (la), (ib), (iia). (iib), (Ilia), (Illb), (IVa) or (IVb). or a pharmaceutically acceptable salt thereof, and (b) administering an effective amount of a DNA alkylating agent (such as cyclophosphamide, meohloretharmne, chlorambucil, melphalan, dacarbazine, or nitrosoureas), in some
embodiments, a compound of Formula (I), (la), (Ib), (Iia), (lib), (Ilia), (Illb), (IVa) or (I Vb), or a pharmaceutically acceptable salt thereof is administered prior to, after, or simultaneously coadministered with the DNA alkylating agent. In some embodiments, a compound of Formula (I), (la), (l b), (Iia), (Ji b), (Ilia), ( Illb), (IVa) or (IVb), or a pharmaceutically acceptable salt thereof is
administered 1 or more hours (such as 2 or more hours, 4 or more hours, 8 or m re hours, 12 or more hours, 24 or more hours, or 48 or more hours) prior to or after the D A alkylating agent
[01 1 ] in some embodiments, provided herein is a method of treating a disease in an individual comprising (a) administering an effective amount of a compound of Formula (Ϊ), (ia), (lb), (Ha), (lib), (Ilia), (Illb), (IVa) or (IV b), or a pharmaceutically acceptable salt thereof, and (b) administering an effective amount of a topoisonierase inhibitor (such as a Topoisonierase I inhibitor (e.g., irmotecan or topoteean) or a 'Iopoisomerase II inhibitor (e.g., etopostde or ieniposide)). In some embodiments, a compound of Formula (I), (la), (lb), (Ila), (lib), (Ilia), (I3Ib), (IVa) or (IVb), or a pharmaceutically acceptable salt thereof is administered prior to, after, or simultaneously co-administered with the topoisonierase inhibitor. In some embodiments, a compound of f ormula (I), (la), fib), (Ila), (lib), (Ilia), (iiib), (IVa) or (IVb), or a
pharmaceutically acceptable salt thereof is administered 1 or more hours (such as 2 or more hours, 4 or more hours, 8 or more hours, 12 or more hours, 24 or more hours, or 48 or more hours) prior to or after the topoisonierase inhibitor.
[0142] In some embodiments, provided herein is a method of treating a disease in an individual composing (a) administering an effective amount of a compound of Formula (I), (la), (lb), (Ila), (lib), (Ilia), (Illb), (IVa) or (IVb), or a pharmaceutically acceptable salt thereof and (b) administering an effective amount of an anthracyclme (such as daunorubicin, doxorubicin, epirubicm, idarubicin, niitoxantrone, or valrubicin). In some embodiments, a compound of Formula (Ϊ), (la), (lb), (Ila), (lib), (Ilia), (Illb), (IVa) or (IVb), or a pharmaceutically acceptable salt thereof is administered prior to, after, or simultaneously co-administered with the anthracyclme In some embodiments, a compound of Formula (I), (la ), ( lb), (Ila ), (lib), (Ilia ), (Illb), (IVa.) or (IVb), or a. pharmaceutically acceptable salt thereof is administered 1 or more hours (such as 2 or more hours, 4 or more hours, 8 or more hours, 12 or more hours, 24 or more hours, or 48 or more hours) prior to or after the anthraeycJine.
[0143] In some embodiments, provided herein is a method of treating a disease in an individual comprising (a) administering an effective amount of a. compound of Formula (I), (la), (lb), (Ila ), ( lib), (Ilia), (Illb), (IVa) or (iVb), or a pharmaceutically acceptable salt thereof, and (b) administering an effective amount of a hisione deacetyiase inhibitor (such as vorinostat or romidepsin). In some embodiments, a compound of Formula (I), (la), (lb), (Ila), (lib), (Ilia),
(IHb), (IVa) or (IVb), or a pharmaceutically acceptable salt thereof is administered pnor to, after, or simultaneously co-administered with the histone deacetylase inhibitor, in some embodiments, a compound of Formula (I), (la), (lb), (Ila), (lib), (Ilia), (fflb), (IVa) or (IVb), or a
pharmaceutically acceptable salt thereof is administered 1 or more hours (such as 2 or more hotrrs, 4 or more hours, 8 or more hours, 12 or more hours, 24 or more hours, or 48 or more hours) prior to or after the histone deacetylase inhibitor.
[0144] In some embodiments, provided herein is a method of treating a disease m an individual comprising (a) administering an effective amount of a compound of Formula (I), (la), (lb), (Ila), (lib), (Ilia), (IHb), (IVa) or (IVb), or a pharmaceutically acceptable salt thereof, and (b) administering an effective amount of a taxane (such as pachtaxel or docetaxel). In some embodiments, a compound of Formula (I), (la), (lb), (Ila), (lib), (Ilia), (iilb), (IVa) or (IVb), or a pharmaceutically acceptable salt thereof is administered poor to, after, or simultaneously coadministered with the taxane. In some embodiments, a compound of Formula (I), (la), (lb), (Ila), (Tib), OTia), (nib), (IVa) or (IVb), or a pharmaceutically acceptable salt thereof is administered 1 or more hours (such as 2 or more hours, 4 or more hours, 8 or more hours, 12 or more hours, 24 or more hours, or 48 or more hours) prior to or after the taxane,
[0145] In some embodiments, provided herei is a method of treating a disease in a individual comprising (a) administering an effective amount of a compound of Formula (I), (la), (lb), (Ila), (lib), (Ilia), (IHb), (IVa.) or (IVb), or a. pharmaceutically acceptable salt thereof, and (b) administering an effective amount of a nucleotide analog or precursor analog (such as azaeiiidine, azathiopnne, capeciiabine, cyiarabine, doxifluridine, 5~fluorouracil, gemciiabme, hydrox urea., mercaptopurme, methoirexate, or tioguamne). In some embodiments, a. compound of Formula. (I) or a pharmaceutically acceptable salt thereof is administered prior ιο, after, or simultaneously co-administered with the nucleotide analog or precursor analog. In some embodiments, a compound of Formula (I), (la), (lb), (Ha), (lib),. (Ilia), (iilb), (IVa) o (IVb), or a pharmaceutically acceptable salt thereof is administered 1 or more hours (such as 2 or more hours, 4 or more hours, 8 or more hours, 12 or more hours, 24 or more hours, or 48 or more hours) prior to or after the nucleotide analog or precursor analog.
[0146] In some embodiments, provided herein is a method of treating a disease in an individual comprising (a) administering an effective amount of a compound of Formula (I), (la),
(lb), ( I la h (lib), (Ilia), (!!Ib), (IVa) or (IV b), or a pharmaceutically acceptable salt thereof and (b) administering an effective amount of a platinum-based chemotherapeuuc agent (such as cisplaim, carboplaim, or oxalrplatm). In some embodiments, a compound of Formula (I), (la), (lb), (Ma), (Kb), (Mia), (ffib), (IVa) or (IVb), or a pharmaceutically acceptable salt thereof is administered prior to, after, or simultaneously co-administered with the platinum-based chemoiherapeutic agent. In some embodiments, a compound of Formula (ij, (la), (Ibj, (ila), (lib), (Ilia), (I3Ib), (IVa) or (IVb), or a pharmaceutically acceptable salt thereof is administered 1 or more hours (such as 2 or more hours, 4 or more hours, 8 or more hours, 12 or more hours, 24 or more hours, or 48 or more hours) prior to or after the platinum-based chemotherapeuttc agent.
[0147] in some embodiments, provided herein is a method of treating a disease in an individual comprising (a) administering an effective amount of a compound of Formula (1), (la), (lb), (Ha), (lib), (ffla), (1Mb), (IVa) or (IVb), or a pharmaceutically acceptable salt thereof, and (b) administering an effective amount of pernetrexed. In some embodiments, a compound of Formula (I), (la), (lb), (Ha), (lib), (Mia), (IHb), (IVa) or (IVb), or a pharmaceutically acceptable salt thereof is administered prior to, after, or simultaneously co-admi istered with the pernetrexed. In some embodimen ts, a compound of Formula (I), (la), (lb), (Ila), (lib), (Ilia), (IT bl, (IVa) or (IVb), or a pharmaceutically acceptable salt thereof is administered i or more hours (such as 2 or more hours, 4 or more hours, 8 or more hours, 12 or more hours, 24 or more hours, or 48 or more hours) prior to or after the pernetrexed,
[0148] In some embodiments, provided herein is a method of treating a disease in an individual comprising (a) administering an effective amount of a. compound of Formula (I), (la), (lb), (Ila). ( lib), (Ilia), (IHb), (IVa) or (IVb). or a pharmaceutically acceptable salt thereof, and (b) administering an effective amount of a DDR pathway inhibitor. In some embodiments, a compound of Formula (I). ( la), (lb), (Ila), (Mb). (I lia), (Bib). ( IVa) or (IVb), or a
pharmaceutically acceptable salt thereof is administered prior to, after, or simultaneously coadministered with the DDR pathway inhibitor. In some embodiments, a compound of Formula (I), (la), (lb), (Ha), (lib), (Ilia), (ffib), (IVa) or (IVb). or a pharmaceutically acceptable salt thereof is administered 1 or more hours (such as 2 or more hours, 4 or more hours, 8 or more hours, 12 or more hours, 24 or more hours, or 48 or more hours) prior to or after the DDR pathway inhibitor. Examples of inhibitors of the DDR pathway include polyi ADF-ribose) polymerase (PA]¾P) inhibitors (such as olaparib, rucaparib, niraparib, or talazopanb), ataxia
telangiectasia mutated (ATM) protein inhibitors, ataxia telangiectasia and RacB -related (ATR) protein inhibitors, checkpoint kinase I (Chk l ) inhibitors, or combinations thereof
[0149] in some embodiments, provided herein is a method of treating a disease in an individual comprising (a) administering an effective amount of a compound of Formula (i), (ia), (lb), (Ha), (lib), (Ilia), (Illb), (IVa) or (IVb), or a pharmaceutically acceptable salt thereof, and (b) administering an effective amount of a PARP inhibitor (such as olapanb, rucaparib, mrapanb, or taiazoparib). In some embodiments, a compound of Formula (I), (ia), (lb), (iia), (lib), (Ilia), (IMb), (IVa) or (IVb), or a pharmaceutically acceptable salt thereof is administered prior to, after, or simultaneously co-administered with the PA R P inhibitor. In some embodiments, a compound of Formula (I), (ia), (lb), (Iia), (lib), (Ilia), (IHb), (IVa) or (IVb), or a pharmaceutically acceptable salt thereof is administered I or more hours (such as 2 or more hours, 4 or more hours, 8 or more hours, 12 or more hours, 24 or more hours, or 48 or more hours) poor to or after the PARP nhibitor.
[0150] in some embodiments, provided herein is a method of treating a disease in an individual comprising (a) administering an effective amount of a compound of Formula (I), (ia), fib), (iia), (Mb), (ffla), fUib), (IVa) or (IVb), or a pharmaceutically acceptable salt thereof, and (b) administering an effective amount of an ATM protein inhibitor. In some embodiments, a compound of Formula (I), (¾), (lb), (Iia), (lib), (Ilia), (Il b), (IVa) or (IVb), or a
pharmaceutically acceptable salt thereof is administered prior to, after, or simultaneously coadmini tered with the ATM protein inhibitor. In some embodiments, a. compound of Formula (I), ( a), (lb), (Iia), (lib), (Ilia), (Illb), (IVa) or (IVb), or a pharmaceutically acceptable salt thereof is administered 1 or more hours (such as 2 or more hours, 4 or more hours, 8 or more hours, 12 or more hours, 24 or more hours, or 48 or more hours) prior ιο or after the ATM protein inhibito
[0151] In some embodiments, provided herein is a method of treating a disease in an individual comprising (a) administering an effective amount of a compound of Formula (I), (la), (lb), (I a), (lib), (Il a), (Illb), (IVa) or ( IVb), or a pharmaceutically acceptable salt thereof, and (b) administering an effective amount of an ATR protein inhibitor, in some embodiments, a compound of Formula (I), (la), (lb), (Iia), (lib), (Ilia), (Illb), (IVa) or (I Vb), or a
pharmaceutically acceptable salt thereof is administered prior to, after, or simultaneously co-
administered with the ATR protein inhibitor. In some embodiments, a compound of Formula (I), (la), (lb), (Ha), (lib), (Ilia), (Illb), (IVa) or (IVb), or a pharmaceuiically acceptable salt thereof is administered 1 or more hours (such as 2 or more hours, 4 or more hours, 8 or more hours, 12 or more hours, 24 or more hours, or 48 or more hours) prior to or after the ATR protein inhibitor. 01S2] in some embodiments, provided herein is a method of treating a disease m an individual comprising (a) administering an effective amount of a compound of (I), (la), (lb), (iia), (lib), (Ilia), (Illb), (IVa) or (IVb), or a pharmaceutically acceptable salt thereof, and (bj administering an effective amount of an Chkl inhibitor. In some embodiments, a compound of Formula (I), (la), (lb), (Ila), (lib), (Ilia), (Illb), (IVa) or (IVb), or a pharmaceutically acceptable salt thereof is administered prior to, after, or simultaneously co-administered with the Chkl inhibitor. In some embodiments, a compound of Formula (I), (la), (lb), (Ila), (lib), (Ilia), (Illb), (IVa) or (IVb), or a pharmaceutically acceptable salt thereof is administered 1 or more hours (such as 2 or more hours, 4 or more hours, 8 or more hours, 12 or more hours, 24 or more hours, or 48 or more hours) prior to or after the Chk l inhibitor.
[0153] In another aspect, provided herein is a combination therapy in winch a compound of Formula ( ! }. (ia), (lb), (iia), (lib), (Ilia), (Illb), (IVa) or (IVb), is coadministered (which may be separately or simultaneously) with one or more additional agents that are effective in stimulating immune responses to thereby further enhance, stimulate or upregulate immune responses in a subject. For example, provided is a. method for stimulating an immune response in a. subject comprising administering to the subject a compound of Formula (I), (Ia), (lb), ffia), (lib), (ffia), (Illb), (IVa.) or ( IVb), or a. salt thereof and one or more irnmunostimulatory antibodies, such as an anti-PD-1 antibody, an anti-PD-Ll antibody and/or an anti-CTLA~4 antibody, such thai an immune response is stimulated in the subject, for example to inhi bit tumor growth. In one embodiment, the subject, is administered a compound of Formula (I), (la), (lb), (Ila), (lib), (ffia), (Illb), (IVa.) or (IVb),or a sail thereof and an anti-PD-1 antibody. In another embodiment, the subject is administered a compound of Formula (I), (la), (lb), (Ila), (lib), (Ilia), (Illb), (IVa) or (IVb), or a salt thereof and an anti-PD-Ll antibody. In yet another embodiment, the subject is administered a compound of Formula (I), ( la), (lb), ( Ila), ( lib), ( Ilia), ( Illb), ( IVa) or (IVb), or a salt thereof and an anti-CTLA- antibody. In another embodiment, the irnmunostimulatory antibody (e.g., anti-PD-1, anti-PD-Ll and/or anti-CTLA-4 antibody) is a human antibody.
Alternatively, the inummostimu!atory antibody can be, for example, a chimeric or humanized antibody (e.g., prepared from a mouse anti-PD-1 , anti-PD-Ll and/or am; ·ί Ί ! Λ··4 antibody). 0J 54] in one embodiment die present disclosure provides a method for treating a proliferative disease (e.g., cancer), comprising administering a compound of Formula (I ), (la), (lb), (Ha), (lib), (Ilia), (ffib), (IVa) or (IVb),or a salt thereof and an anti-PD-1 antibody to a subject, in further embodiments, a compound of Formula (I), (la), (lb), (Ma), (lib), (Mia), (Mib), (IVa) or (IVb), or a salt thereof is administered at a subtherapeutic dose, the anti-PD-1 antibody is administered at a subtherapeutic dose, or both are administered at a subtherapeutic dose. In another embodiment, the present disclosure provides a method for altering an adverse event associated with treatment of a hyperprohferative disease with an immunostimu!atory agent, comprising administering a compound of Formula (I), (la), (lb), (iia), (lib), (Ilia), (Illb), (IVa) or (IVb), or a salt thereof and a subtherapeutic dose of anti-PD-1 antibody to a subject. In certain embodiments, the subject is human. In certain embodiments, the anti-PD-1 antibody is a human sequence monoclonal antibody.
[0155] In one embodiment the present invention provides a method for treating a
liyperproliferative disease (e.g , cancer), comprising administering a compound of Formula 0), (la), ( l b -. (Iia), (lib), (I ia), (illb), (IVa) or (IVb), or a salt thereof and an anti-PD-Ll antibody to a subject. In further embodiments, a compound of Formula (I), (la), (lb), (Iia), (Mb). (Ilia), (Illb), (IVa) or (IVb), or a salt thereof is administered at a subtherapeutic dose, the anti-PD-Ll antibody is administered at a subtherapeutic dose, or both are administered at a subtherapeutic dose In another embodimen the present invention provides a method for altering an adverse event associated with treatment of a liyperproliferative disease with an immunostimulatory agent, comprising administering a compound of Formula (I), (la), (lb), (Iia), (lib), (Ilia), (Illb), (IVa) or (IVb), or a salt thereof and a subtherapeutic dose of anti-PD-Ll anti body to a subject. In certain embodiments, the subject is human. In certain embodiments, the anti-PD-Ll antibody is a. human sequence monoclonal antibody.
[0156] In certain embodiments, the combination of therapeutic agents discussed herein can be administered concurrently as a single composition in a. pharmaceutically acceptable carrier, or concurrently as separate compositions each m a pharmaceutically acceptable carrier, in another embodiment, the combination of therapeutic agents can be administered sequentially. For
example, an an -CI i . A -i antibody and a compound of Formula (Ϊ), (la). (Ib), (lla), (lib), (Ilia), (Illb), (IVa) or (IVb). or a salt thereof can be administered sequentially, such as and -CTL A -4 antibody being administered first and a compound of Formula (I). (la), (ib), (Ma), (lib). (Mia), (lilb), (IVa) or (IVb), or a salt thereof second, or a compound of Formula ø), (la), (ib), (iia), (lib), (Ilia), (Illb), (IVa) or (IVb), or a salt thereof being administered first and anti-CTLA-4 antibody second. Additionally or alternatively, an anti-PD- antibody and a compound of Formula (I), (la), (Ib), (lla), (lib), (Ilia), (Illb), (IVa) or (IVb), or a salt thereof can be administered sequentially, such as anti-PD-1 antibody being administered first and a compound of Formula (I), (ia), (Lb), (iia), (lib), (ilia), (IMbj, (IVa) or (IVb), or a salt thereof second, or a compound of Formula (I), (la), (Ib), (lla), (lib), (Mia), (Illb), (IVa) or (IVb), or a salt thereof being administered first and anti-PD-l antibody second. Additionally or alternatively, an anti- PD-L1 antibody and a compound of Formula (I), (la), (ib), (lla), (lib), (Ilia), (illb), (IVa) or (IVb), or a salt thereof can be administered sequentially, such as anti-PD-Ll antibody being administered first and a compound of Formula (I), (la), (Ib), (lla), (lib), (Ilia), (Illb), (IVa) or (IVb), or a salt thereof second, or a compound of Formula (I), (la), (Ib), (iia), (lib), (ffla), (Mb), (IVa) or (IVb), or a salt thereof being administered first and anti-PD~Ll antibody second,
[0157] Furthermore, if more than one dose of the combination therapy is administered sequentially, the order of the sequential administration can be reversed or kept in the same order at each time point of administration, sequential administrations can be combined with concurrent- administrations, or any combination thereof
[0158] Optionally, the combination of a compound of Formula (I), (la), (Ib), (lla), (lib), (Ilia), (nib), (IVa) or (IVb), or a salt thereof can be f rther combined with an immunogenic agent, such as cancerous cells, purified tumor antigens (including recombinant proteins, peptides, and carbohydrate molecules), cells, and cells transfected with genes encoding immune stimulating cytokine
[0159] A compound of Formula (I), (la), (Tb), (lla), (lib), (Ilia), ( I l l b;. (IVa) or (IVb), or a salt thereof can also be further combined with standard cancer treatments. For example, a compound of Formula (I), (la), Ob), (lla), (Jib), (I ia), Ollb), (IVa) or (IVb), or a salt thereof can be effectively combined with chemotherapeutic regimes. In these instances, it is possible to reduce the dose of other chemotherapeutic reagent administered with the combination of the
instant disclosure (Mokyr et al. (1 998) Cancer Research 58: 5301 -5304). Other combination therapies wit a compound of Formula (I), (la), (lb), (Ila), (Kb), (Ilia), (Illb), (IVa) or (IVb), or a salt thereof include radiation, surgery, or hormone deprivation. Angiogenesi inhibitors can also be combined with a compound of Formula (I), (la), (lb), (Ma), (lib), (Mia), (Illb), {IVa) or (IVb), or a salt thereof Inhibition of angiogenesis leads to tumor cell death, which can be a source of tumor antigen fed into host antigen presentation pathways.
[0160] in another example, a compound of Formula (I), (la), (lb), (Ha), (lib), (Ilia), (Illb), (IVa) or (IVb), or a salt thereof can be used in conjunction with anti -neoplastic antibodies. By way of example and not wishing to be bound by theory, treatment with an ana-cancer antibody or an anti-cancer antibody conjugated to a toxin can lead to cancer ceil death (e.g., tumor cells) which would potentiate an immune response mediated by CTLA-4, FD~I, PD-LI or a compound of Formula (I), (la), (Ib), (Ila), (lib), (Ilia), (Illb), (IVa) or (IVb), or a salt thereof In an exemplary embodiment, a treatment of a hyperproliferative disease (e.g., a cancer tumor) can include an anti-cancer antibody in combination with a compound of Formula (I), (la), (Ib), (Ila), (lib), (Ilia), (Illb), (IVa) or (IVb), or a salt thereof and anti-CTLA-4 and/or anti-PD-I and/or anti-PD-LI antibodies, concurrently or sequentially or any combination thereof which can potentiate anti-tumor immune responses by the host. Other antibodies that can be used to activate host immune responsiveness can be further used in combination with a compound of Formula (I), (la), (Ib), (Ila), (lib), (Ilia), (Illb), (IVa) or (IVb), or a salt thereof
[01 1] In some embodiments, a compound of Formula (I), (la), (Ib), (Ila), ( ib), (Ilia), (Illb), (IVa) or (IVb), or a salt thereof can be combined with an anti~CD73 therapy, such as an anti- CD73 antibody.
[0162] In yet further embodiments, the compound of Formula (I), (la), (i b), (Ha), (lib), (I ia), (I lb), (IVa) or (IVb), or a salt thereof is administered in combination with another Wee! inhibi or.
Oosiay. and Method of Administration
[0163] The dose of a. compound administered to an individual (such as a, human) may vary with the particular compound or salt thereof the method of administration, arid the particular disease, such as type and stage of cancer, being treated In some embodiments, the amount of the compound or salt thereof is a. therapeutically effective amount.
[0164] The effective amount of the compound may in one aspect be a dose of between about 0.01 and about 100 mg/kg. Effective amounts or doses of the compounds of the invention may be ascertained by routine methods, such as modeling, dose escalation, or clinical trials, taking into account routine factors, e.g., the mode or route of administration or drug delivery, the pharmacokinetics of the agent, the severity and course of the disease to be treated, the subject's health status, condition, and weight. An exemplary dose is in the range of about from about 0.7 mg to 7 g daily, or about 7 mg to 350 mg daily, or about 350 mg to 1.75 g daily, or about 1.75 to 7 g daily.
[0!6S Any of the methods provided herein may in one aspect comprise administering to an individual a pharmaceutical composition that contains an effective amount of a compound provided herein or a salt thereof and a pharmaceutically acceptable excipient.
(0166'| A compound or composition of the invention may be administered to an individual in accordance with an effective dosing regimen for a desired period of time or duration, such as at least about one month, at least about 2 months, at least about 3 months, at least about 6 .months, or at least about 12 months or longer, winch in some variations may be for the duration of the individual's life, in one variation, the compound is administered on a daily or intermittent schedule The compound can be administered to an individual continuously (for example, at least once daily) over a period of time. The dosing frequency can also be less than once daily, e.g., about a once weekly dosing. The dosing frequency can be more than once daily, e.g., twice or three times daily. The dosing frequency can also be intermittent, including a 'drug holiday' (e.g. , once daily dosing for 7 days followed by no doses tor 7 days, repeated for any 14 day time period, such as about 2 months, about 4 months, about 6 months or more). Any of the dosing frequencies can employ any of the compounds described herein together with any of the dosages described herein,
[0167] The compounds provided herein or a salt th reof may be administered to an individual via various routes, including, e.g. , intravenous, intramuscular, subcutaneous, oral and transdermal. A compound provided herein can be administered frequently at low doses, known as 'metronomic therapy,' or as part of a maintenance therapy using compound alone or in combination with one or more additional drugs. Metronomic therapy or maintenance therapy
car* comprise administration of a compound provided herein in cycles. eirononiic therapy or maintenance therapy can comprise intra- turn oral administration of a compound provided herein.
[0168] in one aspect, the invention provides a method of treating cancer in an individual by parenteral iy administering to the individual (e.g., a human) an effective amount of a compound or salt thereof in some embodiments, the route of administration is intravenous, intra-arte ial, intramuscular, or subcutaneous, in some embodiments, the route of administration is oral. In still other embodiments, the route of administration is transdermal.
[0169] The invention also provides compositions (including pharmaceutical compositions) as described herem for the use in treating, preventing, and/or delaying the onset and/or development of cancer and other methods described herein, in certain embodiments, the composition comprises a pharmaceutical formulation which is present m a unit dosage form.
[0170] Also provided are articles of manufacture comprising a compound of the disclosure or a salt thereof composition, and unit dosages described herein in suitable packaging for use m the methods described herem. Suitable packaging is known in the art and includes, for example, vials, vessels, ampules, bottles, jars, flexible packaging and the like. An article of manufacture may further be sterilized and/or sealed.
Kits
[0171] The present disclosure further provides kits for carrying out the methods of the invention, which comprises one or more compounds described herein or a composition comprising a compound described herein. I'he kits may employ any of the compounds disclosed herein. In one variation, the kit employs a compound described herein or a pharmaceutically acceptable salt thereof The kits may be used for any one or more of the uses described herein, and, accordingly, may contain instructions for the treatment of cancer,
[Oi 72] Kits generally comprise suitable packaging. The kits may comprise one or more containers comprising any compound described herein. Each component (if there is more than one component) can be packaged in separate containers or some components can be combined in one container where cross-reactivity and shelf life permit.
[Oi 73] The kits may be in unit dosage forms, bulk packages (e.g., multi-dose packages) or sub-unit doses. For example, kits may be provided that contain sufficient dosages of a
compound as disclosed herein and/or an additional pharmaceutical ly active compound useful for a disease detailed herein to provide effective treatment of an individual for an extended period, such as any of a week, 2 weeks, 3 weeks, 4 weeks. 6 weeks, 8 weeks, 3 months, 4 months, 5 months. 7 months, 8 months. 9 months, or more. Kits may also include multiple unit doses of the compounds and instructions for use and be packaged quantities sufficient for storage and use in pharmacies (e.g., hospital pharmacies and compounding pharmacies).
[0174] The kits may optionally include a set of instructions, generally written instructions, although electronic storage media (e.g. , magnetic diskette or optical disk) containing instructions are also acceptable, relating to the use of components) of the methods of the present invention. The instructions included with the kit generally include information as to the components and their administration to an individual.
[0175] The invention can be further understood by reference to the following examples, which are provided by way of illustration and are not meant to be limiting.
EXAMPLES
Synthetic Exannyies
Example SI. Synthesis of 5-/2, 6-dichlorophenyi)-7-((4-(4-methylpiperazm-l~yl)^
2, 3-dihydra-4H~pyrimido[5, 4-ej[L 3]oxazin-4-one
(Compound. No. 1.1}
[0176] Step- 1: Synthesis of 4-hydroxy-2-methyisaIfaiiyi-pyriiRidioe-5-carboxyik acid:
To a stnxecl solution of ethyl 4-chlofo-2-methylsiiifanyl-pyrimidine-5-carbox late ( 10 g, 43.1 mmol, 1.0 eq) in 1 50 niL of eihanol: water (2: 1 ) was added aOH (17.2 g, 431 rrrmo!, 10 eq). Reaction was heated at 110 °C for 2 h. Progress of reaction was monitored by LCMS. Upon the consumption of starting material, solvent was removed under reduced pressnre. Residue was diluted with i 00 mL of water and pH of mixture was adjusted up to 5 with 3 HCl solution. Precipitated compound was filtered off, washed with water (50 nil,) and dried ursder vacuum to obtain the desired product, 4--hydroxy"2"met!iylsulfanyl~pyriniidine--5--carboxyhc acid (6.0 g, 75%).
LCMS: 1 87 [M+I]
+
[0177] Step-2: Synthesis of N-(2,6-dichIorophenyl)-4-hydroxy-2-methyis«lfanyi- pyrii«idi«e-5-carboxamide: A stirred solution of -(2,6-dich !of ophen l)-4-h droxy-2-
(6.0 g, 32.2 rnmol, 1.0 eq) and 2.0-diehloroanilme (5.20 g, 32.2 rnmol, 1 .0 eq) in 200 mL of toluene was purged with nitrogen gas for 1 5 rmn. To the above solution PCC (30 ml,) was added. Reaction mixture was heated at 100 °C for 48 h. Progress of reaction was monitored by ! .CMS After completion of reaction, solvent was removed under reduced pressure, residue was diluted with 1 00 ml, of mixture of diethyl ether. MeOH (1 0' 1 stirred for 15 mm then filtered off Solid was suspended in MeOH (20 ml,), stirred for 5 nun, filtered off and washed with MeOH ( 10 mL), and then dried under vacuum to obtain
(6.0 g, 57%).
[0178] Step-3: Synthesis of 3-(2,6-dich!orophenyl)-7-methylsu!fanyI-2H-pyi-hn«do[5,4- e] l^]oxa«in-4-one: To a solution of N-(2,6~dichlorophenyl)~4~hydroxy-2-metliykulfarsyl~ pyrinudme~5~carboxarmde (4, 0 g, 12.1 rnmol, 1 .0 eq) in CfCCN (200 ml,) was added CH?!? (4.90 g, 18.5 mmoL 1 .5 eq) and CS2CO3 ( 12.0 g, 36.4 mmo!, 3.0 eq). Reaction mixture was heated a t 80 °C for 12 h. Progress of reaction was monitored by LCMS. After completion of the reaction, solvent was removed under reduced pressure, residue was diluted with 50 ml, of water and extracted with ethyl acetate ( 100 mL
x 3), The combined organic layer was washed with water (50 mL
x 3), dned over anhydrous N ^SC and concentrated under reduced pressure. Crude product obtained was purified by flash chromatography using ethyl acetate: hexane (3- 5%) as e!uenis to obtain 3-(2,6-dichlorophenyj)-7-methylsulfanyl~2H.-pyrimido[5,4- e] [l,3]oxazin~4~one (400 mg, 10%).
[0179] S ep~4: Synthesis of 3-(2,6-dichiorophenyl)-7-[4-(4-metliyipiperazin-i- yI)anilmo]-2H-pyrimido[5,4-e] [.1.^]oxazin-4-one: To a stirred solution of 3-(2,6- d ehlorophen !)-7-methy 1 sulfanyl-2H-pyrimido[5 ,4-e'j 1 ,3}oxazm-4-one (200 mg, 0.587 mrno!, 1.0 eq) in toluene (5 ml,) was added rn-CPRA (202 mg, 1. 17 rnmol, 2.0 eq) and allowed 10 stir at rt for 30 min, 4--(4--mediylpiperaziml~y!)amline (1 23 mg, 0.646 rnmol, 1 , 1 0 eq) and DIPEA
(378 rng. 2.94 mmoL 5.0 eq) were added and allowed to siir at it for 2 h. Progress of reaction was monitored by LCMS. After completion of reaction, precipitated compound was f ltered off and washed with toluene (5 mL) and dried under vacuum. Solid was triturated with MeOH (3 mL) then filtered off and dried under vacuum to afford 3~(2,6-clichlorophenyl)-?-[4-{4- methylpiperazin-dwl)anmno]~2H"pyriniido[5,4"e"] 1.3]oxazin~4~One (45 mg, 16%). This 45 nig compound was dissolved in 2 nil, of MeOH and 0.5 mL of 2M HQ m MeOH was then added to it. Solvent was removed under reduced pressure and thereafter compound was freeze dried to afford 3-(2,6-dichlorophenyl)-7-[4-(4-ineihyj i^^
e][},3]oxazm- -one (50 rng) as HQ salt.
LCMS: 485 [M+l f i MR (400 MHz, DMSO-de): δ 10.41 (br s, 11:3), 10.30 (br s, I H), 8.80 (s, IH), 7.65 id, J - 8.33 Hz, 2H), 7,60 (d, J - 8,77 Hz, 2H), 7.50 (dd, J - 7.67, 8.55 U/. IH), 7.01 i d. J - 9.21 Hz, 2H), 5.72 (s, 2H), 3.77 (d, J - 13.59 Hz, 2H), 3.08■- 3,26 (m, 2H), 2.92 ~ 3.08 (m, 2H), 2,83 (d, J - 4.82 Hz, 3H)
Example S2. Synlhesis of3~(2, 6~dichk>rophenyl)~7~(4~morpholinoaniUno)-2H~pynnddo[S, 4~ ejj'l, 3 oxazin~4~one
(Compound No, 1.2)
[0180] Synthesis of 3-(2
¾6-dkhlorophenyl)-7-(4-i«orphoiiooaaiii«o)-2H-pyrh«ido[5,4^ e| 113]oxazin-4-one: To a stirred solution of 3-(2,6-dichlorophenyl)-7-meihylsi3lfanyl~2H- pyrimido[5,4-e] [l ,3]oxazm--4--one (130 nig, 0.381 nirnol, 1 .0 eq) in toluene (2 nil,) was added m- CPBA (164 nig, 0.953 nimol, 2.5 eq) and allowed to stir at ri for 30 nun. 4-Morpholmoaniline (75 mg, 0.420 ni nol, 1. 1 eq) and DIFEA (196 nig, 1.52 nirnol, 5.0 eq) were added and stirred at rt for 1 h. Progress of reaction was monitored by LCMS. After completion of reaction, precipitated compound was filtered off and washed with toluene (5 mL) and dried under vacuum to afford 3--(2,6-dichlorop!ieny!V7"(4-m
Hi MR (400 MHz, DMSO~d6): δ 10.28 (br s, IH), 8.79 (s. I H), 7.65 (d, J - 8 1 1 Hz, 21:3),. 7.56 (d, J - 7.45 Hz, 2H), 7.47 - 7,52 (ni, IH), 6.94 (d, J - 8.99 Hz, 2Hj, 5,71 (s, 2HI 3.71 -· 3.77 (m, 4H), 3.04 - 3. 10 (m, 4H).
Example S3. Synthesis of 3~(2 i~dichkiropheriyl)~ 7~((2~(2~hydroxyacetyl^
tetrahydroisoquinolm-7-yl)amino}~2,3~dihydro-4H^y
(Compound No, 1.3)
98
[01811 Siep-1: Synthesis of tert-butyl 7-|];3-(2,6-dichiorophenyl}-4-oxo-2H-pyrimido|S,4- eJ jl^Joxaa&ta-T- ljarainoJ-^HH^ To a stirred solution
; i l ;: · :■···! -one (400 nig, i .17 nirnoi, 1 .0 eq) in toluene (10 nil,) was added ?w~CPBA (504 mg, 2.94 mmo!. 2, 5 eq) and allowed to stir at rt for 30 min. Tert-43uty!- 7-aniino- ,4
»dinied^^^
earboxylate (323 mg, 1 .1 7 mmol, 1.0 eq) and DiPEA (606 mg, 4, 70 mmol , 4.0 eq) were added and. stirred, at rt for 1 h. After completion of reaction, mixture was diluted with water (50 mL) and extracted with ethyl acetate (50 mL 2), Hie combined organic extracts were wasbed with water (50 mL) and brine (50 mL), dried over χ·α -HO.; and concentrated under reduced pressure. Crude product obtained was purified by flash chromatography to afford ten--bir yi--7--[ [ 3--(2,6-
dichlorophenyl}-4-oxo-2H-pyfimido[5,4-e ] ,3 joxazin-7-yl jaminoJ-4,4-diinelliyl-l ,3- dihydroisoquinoline-2-carboxylate (300 mg, 45.1%).
LCMS: 570 [M÷l]÷
[0182] S ep~2: Synthesis of 3-(2,6-dichioropheiByl)-7 (4,4-dimethyI-2^-dihydro-lH- isoq molm-7-yi)aramo]-2H-pyriraido[5,4-e][.l^]oxa¾in-4-one: tert-Butyl - 3-(2,6~ dichlorophenyl)-4-oxo-2H^yrimM^
dihydroisoqiunoiine-2~earboxylaie (300 rng, 0,528 mmo!) was dissolved n 4M I 'i in dioxane (6 mL) at 0 °C, Reaction was sin red at ri for 2 h. Progress of reaction w monitored by LCMS After completion of reaction, preerpi ed compound was filtered oft, washed with dioxane (3 mL) and dried under reduced pressure to obtain 3-(2,6-dic-hiorophenyl)-7-[(4,4-diraeihyi-2,3- dihydro-lH-isoquino n-7-yi)amino]-2H-pyrimido['5,4-e"S l ,3]oxa.zm-4-one (200 mg, 80.6%) as eCl salt.
LCMS; 470 [ Hj+
[0J 83] Step-3 Synthesis of 3-(2,6-dichIorophenyl)-7-((2-(2-hydroxyacetyl)-4,4-dimethyI- l^,3,4~tetrahydroisoquinolin-7-yl)an^^
sse: To a stirred solution of giycotic acid (36 mg, 0.473 mmol.1.2 eq) in DMF (5 mL) was added HATU (225 mg, 0.592 mmol, 1.5 eq) at rt. The resulting mixture was stirred at rt for 5 min.3-(2,6-dich]orophenyl)-7-((4J4-dimethy]-1.253,4-tetrahydroisoqiu
di.hydro-4H-pyrimido[5,4-e][.l,3]oxazin-4-one hydrochloride (200 mg, 0.395 mmol, 1.0 eq) and
DIPEA (0.135 usL.0,790 mmol, 2.0 eq) were added and stirred at n for 1 h. Progress of reaction was monitored by LCMS, After completion of reaction, mixture was diluted with water (50 mL) and extracted with ethyl acetate (50 mL 2), Hie combined organic extracts were washed with water (50 mL) and brine (50 mL), dried over Na2S()4 and concentrated under reduced pressure. Crude residue was purified by flash chromatography to afford 3d . -dichioor>l: nx π·· ·((2·: · !·γu:o,;^ acet l }··4.;-d ;:■:·;· hx 1 · i . . \ -;e dn^\-Hi:-i; ··?·, n:::; :: r- , -d!b ;in,-4| ! · pyrimido[5,4-e][l,3]oxazin-4-one (50 mg, 24%).
LCMS: 528 [M÷lf
JH NMR (400 MBz, DMSO-de): δ 10.41 (d J 10.96 Hz, I H), 8.84 (s, ΪΗ), 7.66 (d, J ==== 8.1 1 Hz. 2H), 7.47 - 7.58 (rn, 3H), 7.36 (dd. J - 5.04, 8.99 Hz, 1 ! ! }. 5.74 (s, 7! ! }. 4.62 (d. J - 14.03 Hz, 3H), 4.21 idd. ./ - 5.48, 19.51 Hz, 2H), 3.50 (s, I H), 3.36 is, I H), 1.20 (s, 3H), 1.23 (s, 3H).
Example S4. Sy ihesis of 3-(2, 6~dichhrophenyl)~7~((2, 4,4~rrimethyI~.1, 2, 3,4- ietrahydroisoqumo]in-7-yl)ammo)~2,3-dihy
(Compound
[0184] Synthesis of 3~(2,6-die torophe y!)~7~((2,4,4~trImethyI-l92 tesrahydro lso idm4i½»7-^ To a stirred solution of 3-(2,6~dichloi phenyl)-7-((4,4-dime†hy
2
J3-dihydro-4H~pymnido[5,4~e] .l ,3 oxazin-4-one hydrochloride (200 nig, 0.394 mraol, 1.0 eq) and HCHO (0.096 mL, I A M mmol, 3.0 eq) m dichloroethane (1 0 ml,) acetic acid (0. 120 mL, 1 , 970 nimol, 5,0 eq) was added drop-wise at 0 °C. The respiting mixture was stirred at rt for 1 h followed by addition of NaCNBH^ (74 rng, 1. 1 82 nimol, 3.0 eq) at 0 °C. The resulting mixture was stirred at rt for 1 h. The progress of reaction was monitored by LCMS. After completion of reaction, solvent was removed under reduced pressure, residue was basified with saturated solution of NaHCO
.3 (50 rnL) and extracted with ethyl acetate (100 ml,
χ 2). The combined organic layer was washed with water ( 50 mL) and brine (50 mL), dried over anhydrous
and concentrated under reduced. Crude residue was purified by flash chromatography to afford
LCMS: 484 ΓΜ+l
λΗ MM (400 MB'z, DMSO-d6): δ 10.31 (br s, 1H), 882 (s,
_> IS.
ixatnvie a synthesis o
[0185] Step-
'J : Synthesis of tert-butyl 4-(4-aitropheayi)pipera2;i«e-l-carbox iate. To a stirred solution of 5-broxno-2-nitro-pyridine (4.0 g, 21.5 mmol) and tert-butyi pi erazine-l - carboxylate (3.30 g, 23.7 mmol, 1.0 eq) m 30 ml . of DMF was added
2C0
3 (3.30 g, 23.7 mmol 1.0 eq). Reaction m xtur was heated ai 80 °C for 12 h. Progress of d e reaction was rnomiored by LCMS. Upon the consumption of starting material, the reaction mixture was acidified up to pl:l 5 with IN HC1 solution and extracted with ethyl acetate (200 mL
x 2), Combined organic layer was washed with water (50 mL >
■ 3), dried over anhydrous Ν¾8ϋ¼ and concentrated under reduced pressure to obtain 6.5 g (89%) of tert-butyl 4-i4"niirophenyl)piperazine--] --carboxy!ate.
LCMS: 308 [M+l ]+
[0186] Step-2: Synthesis of tert-butyl 4-(4-gminopb yl)psperazkui-l--csrboxylste; To a stirred solution of tert-butyl 4-(4-nitrophenyl)piperazine-l -carboxylate (6.50 g, 21 17 mmol, 1.0 eq) in 100 mL of ethanol: water (1 : 1 ) mixture were added NH4C1 ( 1 1.40 g, 21 1 mmol, 10 eq) and Fe(0) (4.70 g, 84.68 mmol, 4.0 eq). Reaction mixture was heated at 80 °C for 4 h. Progress of reaction was monitored by LCMS. Upon the consumption of starting material, solvent was removed under reduced pressure. Aqueous layer was basified with saturated solution of sodium carbonate and extracted with ethyl acetate (200 mL χ 2). Combined organic layer was washed with 50 mL of brine solution, dned over anhydrous Na^SCL and concentrated under reduced pressure to obtain 4.0 g (68%) of tert-butyl 4-(4-aminophenyl)piperazine-l -carboxylate. L< MS: 278 [M+l ] +
[0187] Step-3: Synthesis of ter -b yl 4-[4-[[^(2,6-dkhIoropheiayl)-4-oxo-2H- pyrimido[5,4~e] [Ι \ oxazin-7-y I] amino] henyl] piperazine- i -carboxyla te; To a stirred solution of 3~(2>6-diclnorophenyl)-7-methylsulfanyl~2H-pyrinii (200 mg, 0.587 mmol, 1 .0 eq) in 3 mL of toluene was added ffi-CPBA (252 rug. 1 47 mmol, 2.5 eq) and allowed to stir at rt for 30 mm. Tert-butyl 4-(4-armnopheny !)piperazine- .1 -carboxylate (179 mg, 0.65 mmol, 1 .1 eq) and DIFEA (303 mg, 2.35 mmol, 4.0 eq) were added and stirred at rt for 1 h. Progress of reaction was monitored by LCMS, After consumption of starting material, precipitated compound was fi ltered off and washed with toluene (5 mL) and dried under vacuum to afford tert-butyl 4-[4-[[3 -(2,6-dichlorophenyl)-4-oxo-2F]-pyriinido[5,4-e][l ,3 ]oxazin-7- yljamino]phenyl]piperazine- l -carboxylate i 40 mg, 41 .8%).
LCMS: 571 [M÷Ij
[0188] Step-4: Syn esis of 3-(2,6-dichioropheHyl)-7-(4-piperazin-i-yianaiao)-2H- pyrimido [5,4-e] [ 1 ) oxazin-4-one; 2M MeOHic HCl solution (5 mL) was added in to a RB tlask containing iert~butyl 4-[4-[[3-(2,6-dichlorophenyl)-4-ox -2H-pyrimido[5J4-e][l,3]oxazin- 7yl]arnino] phenyl] piperazine-1 -earboxylaie (140 rng, 0.246 rnmol, 1.0 eq) at 0 °C, Mixture was stirred at ri for 12 h. Progress of reaction was monitored by LCMS. After consumption of starting material, precipitated compound was filtered off and washed with MeOB (3 mL) and dried under vacuum to afford 3-(2,6-dichlorophenyl)-7-(4-piperazin- 1 -y lam!ino)-2H- pyrimido 5,4-e [l ,3]oxazin-4-one (40 rng, 32%) as BCI sail.
LCMS: 471 [M+l f
¾. NMR (400 M¾ DMSO-d6): δ 10.31 (br s, IH), 9.08 (br s, 2H), 8.80 (s, IH), 7.66 (d, J - 7.89 Hz, 2I¾ 7.55 - 7.64 nu. J - 8.77 Hz, 2H), 7.42 - 7.55 (m, I H), 6.91 ·· 7 08 (m, J - 9 21 Hz, 2H), 5.72 (s, 2! ! }. 3.27 - 3.37 (m, 4B), 3 23 (br s, 4H).
Example S6. Synthesis oj'3-(2, 6-dicklorophenyl)-7-{{2-{2-kydm.^-2-meihyipropimoyi}-4, ·· -· dimeS.hyl-1, 2, 3, 4-le†rahydroisoquinolin~ - 7-yl)amino}-2, 3~dihydro~4H-pyrimido[5, -eJfl.3 joxazin- 4-one
(Compound No. 1.6)
[0J 89] Synthesis of 3-( 2,6-dichIorophenyi)-?-( ( 2-( 2-hydroxy-2-roethySpropanoyl)-4,4- d« ethyl-l,2,3,4-tetrahydroiso^
oxazin ~4~one: l'o a stirred solution of 2-hydroxy-2-meihylpropanoic acid (49 mg. 0.473 mmol, 1.2 eq) DMF (5 ml.) was added ! iAl i (225 mg, 0.591 mmol, 1.5 eq) at rt. The resulting mixture was stirred at rt for 5 rmn, followed by addition of 3-(2,6--didtiorophenyl)-7-((4,4» dimethyl-1 ,25i,4- etrahydroisoqiunolin»7--yl¼
4 one hydrochloride (200 mg, 0,395 mmol, 1.0 eq) and DIFEA (0.135 niL, 0.788 mmol, 2.0 eq), and stirred at rt for I h. Progress of reaction was monitored by LCMS. After consumption of starting material mixture was dilnted with water (50 rnL) and extracted with ethyl acetate (50 mi, x 2). The combined organic layer was washed with water (50 niL) and brine (50 m'L). dried over anhydrous NajSGi and concentrated under reduced. Crude residue was purified by SFC to afford 3 -{2,6--dichloropheny 1 )-7-((2-{2-h droxy~2~metiiylpfopanoyl)-4,4-diineiliyl- 1 ,2,3,4-- tetxahydfOiSoquinolm--7--yi)ammo}--2,3 ^ (35 mg,
16%).
LCMS; 556 [M-H f
{H NMR (400 M¾ DMSO-d6)t δ 10, 38 (br s, 1 H), 8.85 (s, !H), 7.66 (d, J - 8.31 Hz, 2H), 7.46 · 7.58 (m, 3H), 7.36 id, ./ === 8.80 Hz, 1H), 5.74 (s, 2H), 5.51 (br s, IH), 5.15 (br s, 2H), 3.54 (br s, 2H), 1.34 (s, 6H), 1 .20 (br s, 61 ) }
Example S7. Synthesis 3--6?,
isoqiiinoHn~7yl aminoj~2H^yrimido[5, 4~e [l, 3 ox zin"4~one
(Compound No. 1.7)
[0190] Synthesis of 3-(2,6-dichIoroph«myl)-7-[[2-(2-hydrox acetyl)-3,4-dihydro-1 H- -Soqumolin-7 yI]amino]-2H-pyrimido[5,4-ej [i ]ox∞in~4~one: To a stirred solution of glycolic acid ( 19 mg, 0.253 inmol, 1. 1 eq) In 3 mi, of DMF was added HATU (1 31 mg, 0.345 mmol, 1.5 eq) ai ri. The resulting mixture was stirred at rt for 5 min, followed by addition of 3- (2,6- ichlorophenyl)-7-Yl ,2,3,44etrahy
e][ l ,3joxazm~4-one hydrochloride (100 mg, 0.230 mmol, 1.0 eq) and DIPEA (59 mg, 0,460 mmol, 2.0 eq) and stirred at rt for 1 h. Reaction was monitored by LCMS. After completion of reaction, mixture vvas poured on cold water (20 mL). Precipitated compound was filtered off, washed with water (30 mL) and dried under reduced pressure. Crude residue was purified by flash chromatography using MeOH: CU Ch as eluents to afford 3-(2,6-dichlorophenyi)-7-[[2-(2- hydroxyacetyl)-3,4-dihydro-lH-^
JH NMR (400 MHz, DMSO-d6): δ 10.42 ( br s, I H), 8.85 (s, 10), 7.63 - 7.71 (m, J 8.33 Hz, 21:3), 7 59 (br s. 1 1 1 ). 7.48 ·· 7 55 (m, 2H), 7. 1 (d, 7 - 8.33 Hz, IH), 5.74 (s, 2IiI), 4.52 4.70 (m, 3H), 4.18 (d, J - 4.82 Hz, 21 1 ). 3.65 (br s, 21:3),. 2.75 (br s, 2H).
Example S8. Synthesis of3-{2, 6-dichloropheny])~7~[4-[4-(2~hydrox\'aceSy])piperazin-l- y]]anilinoj~2hI-pynmido[5, 4~e lil, 3 oxazin-4-one
(Compound No, 1.8)
F
[0191] Synthesis of 3~(2,6-dic tor phe y!)~7~[4~|4~(2-hydr xyi¾cetyI)pIperi¾z½-l~ yI]anilmo]-2H-pyrimido|
'5,4-e]
To a stirred solution of glyeoiie acid (23 rag, 0.304 mrrjol 1 . 1 eq) in 2 ml, of DMF was added HATU (160 mg, 0 420 rnmoL 2.0 eq ) at rt The resulting mixture was stirred at rt for 5 mm., followed by addition of 3-(2,6-dichlorophenyl)-7-(4- pi perazin - 1 -y lani no)-2H-pyrimido 5 ,4-e] 1 Jioxa.zm-4-one bydrochloiide (130 mg, 0 280 mraoi, 1 0 eq) and DIPEA ( 108 mg, 0.840 mmol, 3 0 eq). then stirred at rt for 1 hr. Reaction was monitored by ! .(
'MS After completion of reaction, mixture was poured on ice cold water (20 mL). Precipitated compound was filtered off, washed w th water (30 mL) and dried under reduced pressure to afford 3-(2,6-dichlorophenyl)-7-[4-[4-(2-hydroxyacetyl)piperazin- ] - yl]anilino]-2∑-I-pyrimido[5,4-e][l ,3 Joxazin-4-one (70 mg, 47.3%).
LCMS: 529 [M+1 ]
\H NMI (400 MB¾ DMSO-d6): δ 1 0.29 (br s, 1 H), 8.79 (s, 1 H1 7.62 - 7.72 (m, J - 8.33 Hz, 2H), 7.52■■ 7.62 (m, 2B), 7.47■■ 7.52 (m, I B), 6.85■■ 7.00 (m, J === 8.77 \ \v. 2H), 5.71 (s, 2H), 4.63 (1, J --= 5.48 Hz, 1H), 4.13 (d, J === 5.70 Bz, 2H), 3.62 (br s, 2B), 3.49 (br s, 2B), 3.1 0 (br s,
[0192 Step-1: Synthesis of -Cl-chloropheayl^'i-hydroxy-l-niethylsulfanyl-pyrimicIiae- S-carboxaimide. To a stirred suspension of 4-hydroxy-2-m i ylsulfanyI-pyrimidi!ie-5-carboxyiic acid (5, 0 g, 26.88 rnmol, 1 .Oeq) and 2-chloroaniIine (3.75 g, 29.56 mmol, 1 1 eq) in toluene (200
nil,) was added PC¾ (30 aiL) and heated at 100 °C for 12 h. Reaction was monitored by LCMS. After completion of reaction., solvent was removed under reduced pressure; residue wa cooled to 0 °C and basrfied by saturated NaHCCL solution. Ethyl acetate (50 mL) was added into it and extracted. Product was insoluble in this biphasic system which was filtered and dried to afford N-
(2.6 g. 29.7%) as solid product.
LCMS: 296 [M+l f
[0193] Siep-2: Synthesis of 3-(2-ehloropheny!)-7-methy!s ifanyl-2H-pyrimido[5,4- e] [l,3]oxa2:in-4-one: To a stirred suspension of N-(2-chloi
'ophenyl)-4-hydroxy-2- meti lsidi¼iybpyrmndme--5--carboxan'nde ( 1.0 g, 3.38 mrnol, 1 ,0eq) in CTI
3CN (30 mL) and DMSO (5 ml,) was added cesium carbonate (3.29 g, 10.14 nimol, 3.0eq) and stirred at rt for 5 r m. ( i bb (1.36 g, 5.08 mrnof l . Seq) was added and heated at 80 °C for 12 b. Reaction was monitored by LCMS. After completion of reaction, solvent was removed under reduced pressure; residue was diluted with ice-cold water and extracted with etbyl acetate (100 niL x 2). Combined organic layer was washed with brine solution (50 mL), dried over anhydrous -
'u -SO.; and concentrated under reduced pressure to give crude product which was purified by flash chromatography to afford
; i .3 ioxrcmv 4~one ( 150 nig, 14.4·%} of as white solid,
LCMS: 308 [M+]
[0194] Siep-3: Synthesis of tert- butyl 4-|;4-|; 3-(2-chiorophe«yl)-4-oxo-2H-pyrimido[5¾4- e j ( 1 ,3] oxa2ia-7-yl] amino] phen l ] piperaashi - 1-car boxy late: To a stirred solution of 3 -(2- chlorophenyl)-7-nieihyisiHfanyl-2H-pyriinido[ 5,4-e ] 1 ,3 ]oxazin-4-one (130 nig, 0.423 mrnol, 1 .0 eq ) in 5 mL of toluene was added /?J~CFBA (224 mg, 0.84 rnol, 2.0 eq) and stirred at rt for 30 mm. Tert- butyl 4~(4~-aniinophenyi)piperazine-d--carboxylate (117 nig, 0.423 nimol, 1 .0 eq) and DIPEA (163 nig, i .27 nimol, 3.0 eq) were added and allowed to stir at rt for 12 h. Reaction was momtored by LCMS. After completion of reaction, solvent was removed under reduced pressure. Residue was diluted with saturated NaHCCh solution and extracted with CH2CI2 (100 mL / 2) (Combined organic layer was washed with brine solution (50 mL). dried over anhydrous Na?S(>4
and concentrated under reduced pressure to give crude product winch was purified by flash chromatography to afford tert-butyl 4-[4-[[3-(2-chlofophenyl)-4-oxo-2∑-]-pyrimido[5,4- e][ l,3]osazin-?-yl]aniino]phenyl]piperazine-'l-carboxylaie (120 nig, 52.8%).
LCMS: 53? [M-H ]÷
[0195] Siep-4: Synthesis of 3~(2-chlorophenyI)-7-(4-piperazm~l-ylaniliao)-2H- pyrimido[5,4-e][.l^joxa¾in-4-one: To a stirred solution of tert-butyl 4-[4-[[3-(2-chlorophenyl)-
4~oxo-2H-pyrirmdo[5,4- ][I,3 oxazin-7^ (120 rng,
0.22 mrnol, 1 .0 eq) in CH2C12 (5 ml,) was added TFA (0.5 mL) at 0 °C and allowed to stir at rt for 5 h. After completion of reaction, solvent was removed under reduced pressure to dryness. Residue was recrystaiiized with e hanol . Precipitated product was filtered and dried io afford 3 · (2"Chlorophenyi}"7--(4--pi erazin-d-ylanilino)~2Ii-^yrimido[5,4--e][l ,3]oxazm--4»one (52 mg, TFA salt. 43%) as an off-white solid.
LCMS: 437 [M+] f
¾ MR (400 MHz, CD3OD): δ 8 82 (br s, i l l ). 7.62 (d J - 8.33 Hz, 3H), 7.45 (br s, 3H), 7.04 (d, J ------ 8.33 Hz, 2H), 5.65 (br s, 2HI 3.38 (br s, 8H).
Example SI 0. Synthesis of 3~(2, 6-dich!orophem -7-f(4, 4~dimethyi~2, 3~dihydr ~ IH-i soquinolin-
7-yI) minoj-2H~pyrimidof~5>4-sffI>3Jox zin~4~one
(Compound No, 1,10)
[0196] Synthesis of .H2,6-dichlorophc^
7-yl)araino]-2H-pyrimid [5,4-e][l,3] xazin-4- ne: tert-Butyl 7-[[3-(2,6-dichlorophenyl)-4- oxo--2H-^yrirmdo 5,4--e][I,3 oxazii - l]arnino'M
carboxylate (100 m g, 0.176 mmol) was dissolved n 4M HCl (2 mL) in dioxane solution at 0 °C. Reaction was stirred ai r t tor 2 h. Progress of reaction was monitored by 1.CMS After completion of reaction, precipitated compound was filtered off, washed with dioxane (3 mL) and dned under reduced pressure to obtain 3-(2!6-dichlorophenyl)-7-[{4,4-dimethyl-2,3-dihydiO-lH- isoquinoi in~7~yl)attiino]42H-pyri ido j'5,4-e"j \ 1 ,3 ]oxazin-4-one (40 mg, HCl salt).
¾ R (400 MEte, DMSO-d6): δ 10.48 (br s, IH), 9.24 (br s, 2B), 8.85 (s, IB), 7.66 (d, ./ === 7.83 Hz, 2H), 7.60 (d J 8.31 Hz, IH), 7.56 (s, !H), 7.52 (d, J - 7.83 Hz, IH), 7.46 (d, J - 8.31 Hz, IH), 5.75 (s, 2H), 4.26 (br s.2H), 3.22 (br s, 2H), 1.35 (s, 6H).
.Example Sll. Synthesis qf3~(2~chioro~4~iJiioro»phenyI)~7~f4-( - 2H-pyrimido[5, 4-e][L3]oxazm-4-one
(Compound No, 1.11)
[0197] Step-J : Synthesis of N-(2-chloro-4-fluoro-pheoyi)-4-hydroxy-2-i«ethylsuifanyl- pyrh«idiiie~S~carbosamkle: A stirred solution of 4-hydfoxy-2-ineihyjsulfanyl~pyrimidme-5- carboxylie acid (5.0 g, 26.9 mmol) and 2~chloro-4-fluoro-aniline (4.30 g, 29.5? mmo!) in 200 ml, of Toluene was purged with nitrogen gas for 15 mi To the above soluiion P(¾ (50 mL) was added. Reaction was heated at 100 °C for 48 h. Progress of reaction was monitored by LCMS. After completion of reaction, solvent was removed under reduced pressure, residue was basified with saturated solution of sodium bicarbonate. In basified layer, ethyl acetate (200 mL) was added, and then stirred for 10 ni in. Precipitated compound was filtered off and washed with 50 niL of water and dried under vacuum to obtain N-(2-chloro-4-fluoro-phenyi)-4-hydiOxy-2- methylsuifanyl-'pynmidine'-5'-carboxamide (2.60 g).
LCMS: 314 [M+l ]+
[0198] Step-2: S h sis of 3-( 2-c.hioro-4-fluoro-phenyl)-7-methylsu!fainyl-2H- pyrimido[5,4-ej .l^]oxa¾in-4-one: To a solution of N-(2-chlo.ro-4-fluoro-p.henyl)-4-hydroxy~2- meihylsulfanyl-pyrimidine-5-earboxarmde (2 40 g, 7,52 mmol) in mixture of Q¾CN (200 ml .) and DMSC) (10 niL) was added diiodomethane (3.02 g, 1 1.28 rnmo!) and cesium carbonate (7.30
·.·.. 22.53 mmoi). Reaction was heated at 80 °€ for 12 h. Progress of reaction was monitored by LCMS. After the completion of the reaction, solvent was removed under reduced pressure, residue was diluted with 50 mL of water and extracted with ethyl acetate (100 mL 3).
Combined organic layer was washed with water (50 mL >< 3), dried over anhydrous Na SC^ and concentrated under reduced pressure. Crude was purified by flash chromatography using ethyl acetate: hexane (3-5%) as eluents to obiam 3-(2-chloro-4-iluoi'o-pheny] )-7-rneihy]sulfanyl~2H- pynmido 5,4-e] l ,3]oxazm--4--one (300 rng).
LCMS: 326 [M÷l ]÷
[0199] Siep-3: Synthesis of 3-(2-cldoro-4-fl«oro-phenyi)-?-(4-(4-methylpipera2in-l- yl)aaH«ao]-2H-pyrimido{5,4-eI 1 l,3]oxazin-4-oiie: To a stirred solution of 3-(2-chloro-4-fluoro- phenyl)~7~meihylsulfanyl-2H-pyrimido 5.4-e][l ,3]o.x;azin-4-one (80 mg, 0.256 mmoi) in 3 mL of toluene was added JW-CPBA (110 mg, 0.64 mmoi) and allowed to stir at rt for 30 mm. 4-(4~ Methylpiperazin-l-yj) aniline (49 mg, 0.256 mmoi) and DIPEA (1 32 mg, 1.024 mmoi) were then added and allowed to stir at rt for 2 h. Progress of reaction was monitored by LCMS. After completion of reaction solvent was removed under reduced pressure, residue was diluted with water (20 ml,) and. extracted with ethyl acetate (50 ml, χ 3). Combined organic layer was washed with water (20 mL χ 3), dried over anhydrous Na?SOi and concentrated under reduced, pressure. Crude residue was purified by reversed phase chromatography to obtain 3-(2~chloro-4- †1uoro-phenyi)~7- 4-(4-nieihyl iperazm^ (25
LCMS: 469 [M÷l f
\H NMM (400 MI¾ DMSO-de): «5 10 20 (br s, IH), 8.76 (s, i l l }. 7.66 fdd, J - 2.85, 8.55 Hz, IH), 7.47 - 7,63 (ny, 3H), 7,35 (dt, ./ - 3.07, 8.55 Hz, IH), 6.92 (d, J - 9.21 Hz, 2H), 5.73 (br s, I H), 5.61 (br s, IH), 3.02 - 3.15 (m, 4H), 2.35 -· 2.47 (m, 4H), 2,22 (s, 3H),
Example SI 2. Synthesis of 3~(2 )-dichlorophenyi)-7-f4~(4~piperidyI) m
ejfl, 3 oxazin~4~one
(Compound No. 1.12)
[0200] Step-1 : Synthesis of tert-butyl -( -amjnopheRyl)-3,6-dihydr< -2H-pyridiRe-l- carboxylate: To a stirred solution of 4-bromoanii ine (1.0 g, 5.81 mmol) and tert-buty! 4-
(4,4,5,5-tetrauiethyl- ] 33,2- ioxaborolan"2"yl)-3,6'-dihydro--2H^yridme"l --carboxylate (2.1 5 g, 6.97 rmnol) in DMF (1 5 mL) was added sodium carbonate (1 ,23 g, 1 1.62 mmol, 2 aqueous solution). Nitrogen purging was done for 10 nun and then added Pd(PPh3)4 (335 mg, 0.29 mmol) into it. Reaction mixture was stirred at 90 °C for 3 h. After completion of reaction, reaction mixture was cooled to it, diluted with ice-cold water ( 10 mL) and extracted with ethyl acetate (50 ml, x 3 ). Combined organic layer was washed with bnne solution (1 0 mL x 5), dned over anhydrous Na^SC^ and concentrated under reduced pressure to afford crude product, which was purified by eombi-flash chromatography (eluent-0-30% ethyl acetate m hexane) to afford the desired product (1 .0 g).
LCMS: 275 [M÷l f
[0201] Step-2: Synthesis of tert-butyl 4-{4-aramophenyl)piperidine-l-carboxylate: To a stirred solution of tert-butyl 4-(4-aniinophenyl)-3,6-dihydro-2I i-pyridiiie-l -carboxylaie (274 nig.,
1.0 mmol) in MeOH (10 nil) was added Pd/C (50 nig) and stirred under hydrogen atmosphere. After completion of reaction, rmxiure was filtered through celite and filtrate was concentrated under reduced pressure to afford the desired product (250 mg) as viscous oil. LC .S: 221
[M+I f
[0202] Siep-3: synthesis of tert-butyl 4-[4-[[^i2,6-dich!oropheiayl)-4-oxo-2H- pyrksm!o[5,4-e] [l^joxaKin~7~yl]amin jphenyI]piperid»u l- arboxyiate: To a stirred solution of 3-(2,6-dichioropheny!)-7-m<shy!sutf (100 mg, 0.292 mmol) in toluene (4 ml.) was added m-CPBA (155 mg., 0, 584 mmol. 65% purity) and stirred at i for 30 mm. Tert-butyl 4-(4-aminophenyl)piperidine-l -carboxylaie (81 nig., 0.292 mmol) and DiPEA (1 13 mg, 0.876 mmol) were added and allowed to stir at rt for 3b, After completion of reaction, solvent was removed under reduced pressure, diluted with saturated NaHCCh solution (20 mL) and extracted with Ci¾Cl2 (20 mL x 3). (Combined organic layer was washed with brine solution, dried over anhydrous Na?SCh and concentrated under reduced pressure to give crude product which was purified by cornbi -flash chromatography to afford tert- butyl 4-[ -] -(2,6-dichlorophenyl)-4-oxo-2H-pyfimido[5,4-e] l ,3']oxazin-7- yi]annno]pheny!]piperidme-d --carboxy!ate (70 mg).
LCMS: 570 [M÷l f
[0203] S ep~4: Synthesis of 3-{2,6-dichIoropheny!)-7-[4-{4-piperidy!)anHino]-2H- pyrimido [5,4-e] [ 1 ) oxazin-4-one; To a stirred solution of tert-butyl 444 ·: i 2.0··
dichioropheny!)-4-oxo-2H-pyriraido[5^
carboxylate (70 mg, 0.22 mmol) in Q-FCb (3 mL) was added TFA (0.5 mL) at 0 °C and allowed to stir at rt for 3 h. After completion of reaction, solvent was removed under reduced pressure to dryness, basifled by using saturated NaHCCh solution (20 mL) and extracted with C3¾C¾ (20 mL x 3). Combined organic layer was washed with brine solution, dried over anhydrous ^ -SO.; and concentrated under reduced pressure to afford crude product winch was recrystall tzed by CH2CI2 and n-pentane to afford 3~(2,6-dichlorophenyl)-7-[4-(4~piperidyl)anilino]-2H- pyrimido 5,4-e][1 ,3joxazin-4-one ( 1 1 mg).
LCMS: 470 [M÷l ]÷
JH NMR (400 MH2, DMSO-d6): S 10.39 (br s, I H), 8.83 (s, 1H), 7.56 ■■ 7.83 (m, 4H), 7.41 ■· 7.54 (m, I H), 7.20 (d, J = 8.31 Hz, 21:1), 5.57 ·· 5.86 (m, 2H), 3.05 (d. J - 12.23 ϊ¾ 2H), 2.55 ·■ 2.74 (m, 2H), 1.69 (d, J - 12.23 Hz, 2H), 1.39 ·· 1.61 (m, 2H), 1.21 (s, IH).
Example SI 3. Synthesis of3-{2, 6~dichIoropheriyi)--7--f4~i2--fhydrox)methyi)p
y^]anilinoj~2hI-pynmido[5, 4~e]il, 3 oxazin-4-one
(Compound No, 1.13)
[0204] Step-1 : Synthesis of tert-butyl 3-(hydroxyraethyl)-4-(4-RttrophenyI)piperazme-l- carboxylate: To a stirred solution of l -fluoro~4- tro-benzene (500 mg, 3.54 mmol, 1.0 eq) and tert-butyl 3-(hydi
'oxymetbyl)piperazin -l-carboxylate (560 mg, 3.54 mmol, 1 .0 eq) in DMF (10 mL) vvas added K2CO3 (732 mg, 5.31 mmol 1 .5 eq). Reaction mixture vvas heated at 50 O lor 12 h. Progress of the reaction was monitored by ! CMS Upon the consumption of starting
material, the reaction mixture was diluted with water (100 rnL) and extracted wife ethyl acetate (200 rnL x 2). Combined organic layer was washed with water (50 rnL x 3 ), dried over anhydrous a2SOd and concentrated under reduced pressure io obtain the crude. The crude was purified by eombi-ilash (0-50% EtOAc-hexane) to obtain tert-butyl 3 -(bydroxymeihyl)-4-(4- mtrophenyl) piperazine-1
■- carboxylase (400 rng, 33%).
[0205] Step-2: Synthesis of tert-butyl 4-(4-amiaophenyl)-3-(hydroxyiRefhyI)piperaz«ne- 1-carhoxylate: To a stirred solution of tert-butyl 3-(hydroxymethyl)~4~(4- nitrophenyl)piperazine-l "Carboxylate (0.3 g, 0.890 mmol. 1.0 eq) in MeOH (10 mL) was added Pd/C (100 mg, 1 0 mol%). Reaction mixture was stirred at rt under hydrogen atmosphere for 1 h Progress of reaction was monitored by LCMS. Upon the consumption of starting material, the reaction mixture was filtered through celite bed and washed with MeOH (20 mL), solvent was removed under reduced pressure. The solid obtained was triturated with ether to obtain tert-butyi 4-(4-ami.nophenyl)-3-(hydroxy.metb.yl) piperazine-Lcarboxylate (200 mg, 73.26%),
LCMS: 308 [M+] f
[0206] Step-3: Synthesis tert-butyl 4-[4-[[3-(2,6-dkhloropheRyl)-4-oxo-2H- pyrimido[5 4~e] [.l^]oxazin-7-y^
carboxylate: To a stirred solution of 3-(2,6-dichiofopheiiyl)-7-methylsulfanyl-2H-pyriniido[5!4- e][ l ,3]oxazin-4-one (200 mg, 0,587 mmol, 1.0 eq) in toluene ( 10 mL) was added /ί/-€ΡΒΑ (252 mg, 1 ,47 mmol, 2.5 eq) and allowed to stir at rt tor 30 nnn. Tert-butyl 4--(4--ammopheny!)-3- (hydrox methyl) piperazine-l -carboxylate (197 mg. 0.65 mmol, 1.1 eq) and D1PEA (0.4 rnL. 2.35 mmol, 4.0 eq) were added and stirred at rt for 1 h. Progress of reaction was monitored by LCMS. After consumption of starting material, the reaction mixture was diluted with water (1 0 mL) and was extracted with EtOAc (10 ml, X 2). The organic layer was dried and purified by combi-flash using 040% MeOH-CH2Cl2 as e!uent to afford tert-butyl ..| ..Π ·; 2. ο- dich!oropheny!)- --oxO"2H~pynmido 5,4~^^
pipera.zine--I --carboxyla.te (200 mg, 51.28%).
LCMS; 601 [M-H/f
[0207] Step-4: Synthesis of 3-(2,6-dichIorophenyl)-7-|4-[2-(hydroxymethyl)pipera-5ia-l- yI]aoiHao)-2H-pyrimido[5,4-e] [1 | xaz«i-4-one: To the stirred solution of tert-buiyl 4-{4-}}3- (2,6-dichlorophenyj)-4-oxo-2H~pyrimido 5.4-e][l ,3]osazin-?-yj]amino]phen ]~3^
(hydroxyrneihyi) prperazine-I-carboxylaiee (200 mg, 0.33 rnmoi, 1 .0 eq) in Dioxane (2 mL) was added 4M Dioxane-HCi at rt Mixture was stirred at s ·■ for 1 h. Progress of reaction was morsitored by LCMS. After corssumption of starting materia! solvent was removed under vacuum and the HCl salt obtained was purified by Reverse phase column chromatography to afford 3-(2,6-dichiorophenyl)-7 4 2--i
e][ l ,3 joxazin-4-one (40 mg, 24%).
LCMS: 501 [M÷l f i NMR (400 MHz, »MSO-d6); δ I O.33 (br s. 1 H), 8.81 (s. 1 H), 8.38 (br s, 1 H), 7.65 (d, J - 8.31 Hz, 2H), 7.54 - 7.63 (m, J - 8.31 Hz, 2H), 7.42 · 7.54 (m, 1H), 6.86 · 7.02 (m, J - 8.80 Hz, 2H), 5.72 (s, 2H), 3.74 - 3.94 (m, 3H), 2.97 (br s, 3Hj, 2.84 ( br s, 3H).
Example SI 4, Synthesis of 3~(2~chloro~4~flu ro~phenyl)~ 7~[(4, 4~drmeihyl~2, 3-dihydro-lH~ isoquinolin- 7-yl)animo]-2H-pyrimida[S, 4-ej' fl, 3]oxazin~4~one
(Compound No, 1 , 14)
[0208] Synthesis of 3-(2-chioro-4-flaoro-pheayi)-7- (4,4-dimethyl-2 -dihydro-i H- isoquinolia-7-yi)amiao]-2H-pyrimido 5,4-e] [l^]oxazm-4-onei Tert-butyl 7-[[3-(2-ch!oxO-4- iluorO"phenyl)- -oxo--2H
»pynin^
dihydroisoquinoline-2-carboxylate (80 mg, 0.144 mmol, 1 .0 eq) was dissolved in C¾C (15 niL) was added triflouro acetic acid (0 4 mL) at 0 °C, Reaction was stirred at rt for 4 h. Progress of reaction was monitored by LCMS. After completion of reaction, solvent was removed under reduced pressure. Residue was triturated with diethyl ether; solid was filtered off, dried under vacuum and basified with NaHCC solution. Precipitated compound was filtered off and purified by reversed phase chromatography to obtain 3-(2-chlofo-4-fluofo-phenyl)~7-[(4,4-dimeti yb2.3- dihydro-1 H-isoquinolin-7-yi)atnino]-2H.-py.rimido[5,4~e][l ,3]oxa in-4-one (4 mg. 5%).
¾. NMR (400 M¾ DMSO-d6): δ 10.27 (br s, IH), 8.80 (s, IH), 7.66 (dd, J - 2.45, 8,31 Hz, IH), 7.58 (dd, J === 5.62, 8.56 Hz, I B), 7.47 (d, J === 8.80 Hz, I H), 7.21 ■· 7.39 (tn, 2H), 5.75 ( br s, I B), 5.62 (br s, IH), 3.85 (s, 2H), 2.66 - 2.77 (ni, 2H), 1 .91 (s, IH), 1 .21 (s, 6H).
Example SIS. SyrHhesis of3~(2.6-dichlorQphenyi)-7-i4~(4~eihylsulfonylpiperazin~ l~yl)aniUnoj~ 2H~pyriniidol5, 4~e]l ./, 3joxazin-4-one
(Compound. No. 1.15)
[0209] Synthesis of 3~(2, -dic tor h y!)~ 4~ 4~ * Is Ifo yI i r^i ~^ I)arsI!mo]~ 2H-pyriinido [5,4-e] [1 ,3] oxasrin-4-one: To a stirred solution of 3~(2,6-dichiorophenyl)--7-(4- p perazin - 1 -y lani imo)-2H-pyrimido[5 ,4-e] 1 ,3}oxazm-4-one (50 rag, 0.106 mrnol) in C¾C (4
niL) were added TEA (21 mg, 0.212 mrnol) and ethanes uifonyi chloride (13.5 mg, 0.106 r rsol) and stirred at rt tor 3 h. After completion of reaction, reaction mixture was di kited with water ( 10 mL) and extracted with C3¾(¾ (10 nil, χ 3). Combined organic layer was washed with brine solution, dned over anhydrous Na?SC>4 and concentrated under reduced pressure to afford crude product which was washed with, diethyl ether and dried to afford the desired product (29 mg).
\y NMM (400 MHz, CD<¾): δ 8.96 (s, IH), 7.51 (d, ./ - 8.77 Hz, 2H), 7.45 (d, J - 7.89 Hz, IH), 7.24 · 7.35 (m, 2HI 6,95 (d, J - 9.21 Hz, 2H), 5.54 (s, 2H), 3.35■- 3.54 (m, 5H), 3.09 · 3.34 (m, 4H), 3.01 (q, J - 7,45 Hz, 2H), 1 .41 (t, J - 7.45 Hz, 3H).
Example SI 6. Synthesis of 3~(2!6~dichlorophem! "7--[3~(h}^x)xymeihyi ~4^
anilino]~2H-pywmido[5, 4~e][L 3]axazin-4-one
(Compound No. 1,16}
[0210] Step-J : Synthesis of tert-butyi 4-(3-fori«yi-4-niiro-pheayl)pipera2«ae-
'l- carboxylate: To a stirred solution of 5-fluofo-2-nitro-benzaldehyde (500 mg, 2.95 mmol, 1.0 eqj and tert-butyi piperazine-1 --carboxylate (550 nig, 2.95 mmol, 1.0 eq) in DMF (10 mL) was added K2CO (611 nig, 4.42 mmol, 1 .5 eq). Reaction mixture was heated at 50 °C for 12 h. Progress of the reaction was monitored by i .C S. Upon the consumption of starting material, the reaction mixture was diluted with water (100 mL) and extracted with ethyl acetate (200 mL 2). Combined organic layer was washed w th water (50 mL > 3). dned over anhydrous Na
2S() and concentrated under reduced pressure to obtain the crude. The crude was purified by combi- f!ash (0-50% EtOAc-hexane) to obtain tert-butyi 4~(3-formyl-4-ni1xo-phenyT)piperazme-l- earboxylate (500 mg, 55%).
[0211] Step-2: Synthesis of tert-butyi 4-[2-(hydroxymethyI)-4-n«tr< -phenyi]piperazine-
J -carboxylate: To a stirred solution of tert-butyi 4-(2~i½Tnyl-4-niixo-phenyl)piperazine~l - carboxylase (0,7 g, 2.57 mmol, 1.0 eq) m Π Π (10 mL) was added NaBB
4 (97 mg, 2.57 mmol, 1,0 eq) at 0 °C Reaction mixture was stirred at 0 °C for 2 h. Progress of the reaction was monitored by LCMS, Upon the consumption of starting material, the reaction mixture was diluted with water (20 mL and extracted with C¾C1? (100 mL x 2). Combined organic layer was dried over anhydrous - SO.; and concentrated under reduced pressure to obtain tert-butyi 4-[2-(h droxymethyl)-4-nitro-phenyl]piperazme-l -carboxylate (0.5 g. 71 %).
[0212] Step-3: Synthesis of tert-butyi 4- [4-a mo-2-(hy droxymettryI)pheny i ] piperazi«e- l-carboxylate: To a stirred solution of tert-butyi 4- 2-(hydroxymetlryl)--4--nitro- phenyl]piperazme-l -carboxylate (0.5 g, 1.483 mmol. 1 .0 eq) in MeOH (10 niL) was added Pd/C (200 mg, 10 mol%). Reaction mixture was stirred at rt under hydrogen atmosphere for 1 fr Progress of reaction was monitored by LCMS. Upon the consumption of starting material, the reaction mixture was filtered through the celite bed and washed with MeOH (50 mL), solvent was removed under reduced pressure. The solid obtained was triturated with ether to obtain tert- butyi 4-i4-amino-2-(hydroxymethyi)pherryi]piperazine--l --carboxylate (400 mg. 87.91 %).
LCMS: 308 [M+l]
[0213] S ep~4: Synthesis u-ri-hm> i 4 4 P-(2, ~dkMoro te I 4~oxo»2B~
pyrimido [5,4-e] [ 1 ) oxazin-7-yl] amino]-2-(hydroxy methyl)phenyl] piperazine- 1 - carboxylate: To a. stirred solution of 3-(2>6-dic]ilorophenyl)-7-meihylsuli½iyl-2∑i-pyrimido 5,4- e][ l ,3]oxazm--4-one (300 mg, 0..877 mmol, 1.0 eq) in toluene ( 10 mL) was added /M-CPBA (377 mg, 2.1 92 mmol, 2.5 eq) and allowed to stir at rt for 30 min, Tert- butyl 4-[4-[[3-(2,6- dichk>rophenyl)-4-ox 2H-pyrinudo 5,4-e] [l,3]oxazin-7-yl]arnino]-2-
(hydroxyinethyl)phenyrjpiperazine-l -carboxylate (296 nig., 0.96 mraoi, 1.1 eq) and DiPEA (0,6 mL, 3.508 mmo!, 4.0 eq) were added and stirred at rt for 1 b. Progress of reaction was monitored by LCMS. After consumption of starting material, die reaction mixture was diluted with water (10 mL) and was extracted with EtOAc ( 10 mL X 2). The organic layer was dried and purified by combi-flash using 0-10% MeOBA:H2Cl2 as elueni to afford tert-buty! 4-'[4-{[3-(2,6- dichIoroph ny])-4-oxo-2H-pyrimido[5,4-e] [l ,3]oxazin-7-y]]amino]-2- (hydroxymethyl)pbenyl]piperazine~l -carboxylate (300 mg, 34.20%),
LCMS: 601 [M÷l f
[0214] Siep-5: Synthesis 3 2,6-dichI rophenyl)-7-[3 hydr xyniefhyI)-4-piperaz«n-l-yl- aniimo]-2H-pyrimido|S,4-e) [l,3]oxa«i«-4-one. To a stirred solution of tert-buJyl 4-(4-( [3-(2,6- dichlorophenyl)-4~oxO-2H-pyrimidoi'5,4-e] [ l^
piperazine- 1 -carboxylate (300 mg, 0.50 mmol, 1 .0 eq) in dioxane (2 niL) was added 4M dioxane-HCl ( 1 0 niL) and reaction mixture was stirred at it for 1 hr. Progress of reaction was monitored by LCMS. After consumption of starting material, solvent was removed under vacuum and die HCi salt obtained was purified by reversed phase column chromatography to afford 3-(2,6--dich!orophenyl)-7-[3--(hydro
e][L3]oxazrn-4~one (200 mg, 80%).
*H NMR (400 MHz, DMSO-d6): S 1 1 .63 (br s, 1H), .10.1.0 (s, 1H), 9.56 (s, 1H), 9.03 (br s, 1H), 8.95 (d, J - 7.89 Hz, 2H), 8.88 (d, J - 8,33 Hz, Hi), 8.72 - 8.82 (m, 1H), 8.33 (d, J - 8,77 Hz, I H), 7.02 (s, 2H), 6.42 (br s, 1H), 5.84 (s, 2H), 4.21 (br s, 4Hj, 4.09 (br s, 4H).
fj a p!e SI 7. Synthesis of 3~(2 )~dichlorophenyi)~2~methyi~ 7~(4~piperazin~l~ylam pyrimidofS, 4~e/fl, 3joxazin-4-one
(Compound No. 1. 17)
[0215] Step-1: Synthesis of 3~(2,6-dichlorophenyl)-2-methyl-7-methylsulfanyl-2H- pyriniidoj
'5,4-eJ
To a stirred solution of ~(2,6-clichlorophenyl)-4-hyclroxy- 2-rnethy]sulfa.nyl-pyrimidine-5-carboxamide (2 g, 6 060 mmoi, 1 eq) in CF CN (20 mL) was added cesium carbonate (5.9 g, 18.00 mmoi, 3.0 eq). The reaction mixture was degassed with nitrogen tor 10 min and CB?!? (2 g, 9.09 mmoi 1 .5 eq) was added dropwise. Tbe reaction mixture was heated at 80 °C for 12h. Progress of the reaction was monitored by LCMS, Upon the consumption of starting material, the reaction mixture was diluted, with water (200 mL) and extracted with ethyl acetate (200 ml, 2) Combined organic layer was washed with water (50 mL x 3), dried over anhydrous \ -SO : and concentrated under reduced pressure. The crude residue obtained was purified by combi-fiash 0-100% EtOAc-hexane to obtain (2,6-
(1 50 mg,
[0216] Step-2: Syn esis of tert-butyl 4~[4~[[3-(2,6-dkhiorophenyi)~2~methyi~4~oxo-2H~ pyr nido [5,4-e] [ 1 ,3] oxazin-7-yl] mino] phenyl] piperazin e- l-earboxylate: To a stirred solution of 3-(2,6~dichloi phenyl)-2-m thyl-7-m
one ( 100 mg, 0.290 mmol, 1 0 eq) in toluene (10 mL) was added m- PBA (100 mg, 0 58 mmol, 2.0 eq) and allowed to stir at rt for 30 ratn. Tert-butyl 4--{4--a.minophenyl) piperazine-1- earboxylaie (1 07 mg, 0.35 mmol, 1.10 eq) and DIPEA. (149 rng, 1 , 1 6 rnmoi, and 4.0 eq) were added and allowed io stir at ri for 1 h. Progress of reaction was monitored by LCMS. After completion of reaction, solvent was removed under reduced pressure ("rude residue was suspended in 20 mL of water, extracted with ethyl acetate (50 mL x 2), Combined organic layer was washed with water (20 mL), dried over anhydrous Na^SC^ and concentrated under reduced pressure Crude residue was purified by flash chromatography using ethyl acetate : hexane io obtain tert-butyl 4-[4-[[3-(2,6~dich!oropbenyl)-2~methyl- -oxo~2H-pyrirnido[5,4-e]['i,3]oxazin~ 7~yi]armno] phenyl]piperazme-l -carboxylate (100 mg, 59. 1%).
LCMS: 585 [M÷l f
[0217] Sfep-2: Synthesis of 3-(2,6-dkhlorophenyl)-2-methyl-7-(4-piperazin-J-ylanilino)-
2H-py rimtdo | S,4-e] [1,3] oxazio-4-one: To a stirred solution of tert-butyl 4-[4-[[3-(2-chloro-6- fliJoro-pbenyl)--4-oxo-2H"pyriniido| 5,4-e [ 1 ,3 joxazu -yl jamuio iphenyi ] piperazme-4 -
■ carboxylate ( 100 mg, 0. 171 mmol, 1.0 eq) in 4M 1 Ί in dioxaue (3 mL). The reaction mixture was stirred at ri for 1 hr. Progress of reaction was monitored by LCMS. After consumption of starting material, precipitated compound was filtered off and washed with dioxaue and dried under vacuum. Crude residue was purified by reversed phase chromatography to obtain 3--(2,6- diehlorophenyl)-2-methyl-7-(4-piperazin- (7 mg, 8.53%) as TFA salt.
*H MM (400 MBz, DMSO-d6): «5 10.30 (s, 1H), 8.78 (s, IH), 8.67 (br s, 2IT), 7.67 (dd, J 2.63. 8.33 Hz, 2H), 7.53 · 7.63 (m, 2H), 7,44 · 7.53 (m, I H), 7.00 (d, J - 9.21 Hz, 2H), 6.14 (d, J - 5.70 Hz, i l l ; 3,26 (br s, 5H), 3.16 (br s, J Pi), 1.38 (d, J - 5.70 Hz, 3H).
fj a p!e SI 8. Synthesis o/3-(2, 6~dichiorophen} )-7-f^~{^~eihyipiperazin~]~yl)amiinoj~2 - pyri MofS, 4~e/fl, 3]oxazin~4~one
(Compound No. 1.18)
[0218} Synthesis of 3-(2,6-dichloropheny!)-7-j4-(4-eihylpipera2in-l-yl)aaaino)-2H- pyrimido(5,4-eI 11 ]oxaaan-4-one: To a stirred solution of 3-(2.6-dichlorophenyl}-7-(4- pip ra m-l -y
j.aniIino)-2H-pyrimido[5
J4-e][ ]o fazin-4-one hydrochloride (80 nig, 0.169 mmol, 1.0 eq) and acetaldehyde (35%) (0.65 nil, 0.507 mmol, 3.0 eq) in CH
2C¾ (5 ml.) acetic acid (51 mg, 0.845 mmol, 5.0 eq) was added drop- wise at 0 °C. The resulting mixture was stirred at rt for 1 !:. followed by addition of NaCNB¾ (32 mg, 0.507 mmo!, 3.0 eqj at 0 °C. The resulting mixture was stirred at rt for 1 hr The progress of reaction was monitored by LCM5. After completion of reaction, solvent was removed under reduced pressure, residue was basificd with saturated solution of NaHC0
3 (50 mL). Precipitated compound was filtered off, dried under vacuum and purified by flash chromatography using MeOH: C¾(¾ as eluersis to afford 3-·(2,6~
*H NMR (400 MHz, CD3OD): «5 8,82 (s, 1H), 7.57 (d, J - 7.89 Hz, 3H), 7.39 -· 7,48 (m, Hi), 7.00 (d, J - 9 21 Hz, 2H), 5.66 (s, 2H), 3.26 (br s, 4H), 2 83 (br s, 4H), 2.66 (s, 2H), 1.21 (t, J 7 24 Hz, 3H).
fj a p!e SI 9. Synthesis o/3-(2.6~dichiorophenyi)~7~i 4~(4~isopropyipiperazin~l~yi)aniibioj~2H~ pyrimidofS, 4~e/fl, 3]oxazin~4~one
(Compound No. J.19)
[0219] Step-1 : Synthesis of l~(4-nitr phe»yI)pipera2:ine: To a stirred solution of l-fluoro- 4~mtro-- benzene (5, 0 g, 35.46 mmol) m ethanol (30 rnL) was added piperazme (7.62 g, 88,65 rrsrno!) and stirred at reflux tor 3 h. After completion of reaction, solvent was removed under reduced pressure. Residue obtained was basified by saturated NaHC(>3 solution (20 mL) and extracted with CH?C1? (20 ml, x 3), Cornbmed organic layer was washed with brine solution, dried over aniiydrous
and concentrated under reduced pressure to afford 1 -(4- iiitrophenyi jpiperazine (6.60 g) as a yellow solid.
LCMS: 208 [M+l
[0220] Step-2: Synthesis of l-isopropyI-4-(4-nitrophenyl)pipera¾inej To a stirred solution of 1 -(4-nitrophenyl)piperazine (500 nig, 2.41 nunc! ) and acetone (420 mg, 7.24 mmol) in MeOH (10 mL) was added glacial acetic acid (0.2 mL) and allowed to stir at rt 12 h. NaCNBH3 (302
nig, 4.82 n mol) was then added portion- wise and allowed to stir at n for 3 h. After completion of reaction, solvent was removed under reduced pressure, residue obtained was diluted wife saturated NaHCCh solution (20 mL) and extracted with C!¾C¾ (20 mL x 3). Combined organic layer was washed with brine solution, dried over anhydrous N 2S(>4 and concentrated under reduced pressure to afford l-isopropyl-4-{4-nitrophenyl)piperazine (450 mg). LCMS: 250
[M+l f
[0221] Step-3: Sy t es s of 4-{4-«sopropyIpiperazuj-l -yl)aniiine: To a stirred solution of ].~isopropyi~4~(4-mirophenyj)piperazme (450 mg, 1.80 mmol) in ethanol (15 mi..) and water (3 niL) were added iron powder (695 nig, 12.65 nimol) and N¾.C1 (670 nig, 12.65 nimol). The reaction mixture was stirred at 80 °C for 4h. After completion of reaction, solvent was removed under reduced pressure, diluted with water (20 mL) and extracted with CH?Ci? (20 ml, x 3), Combined organic layer was washed with bnne solution, dried over anhydrous Na2S(>4 and concentrated under reduced pressure to afford the desired product (300 mg),
LCMS: 220 [M+l f
[0222] Step-4: Synthesis of 3-{2,6-dichIorophenyl)-7-[4-(4-«sopropyIpipera¾in-J- yI)aoiHao)-2H-pyrimido[5,4-e][l |oxaz«i-4-one: To a stirred solution of 3-(2,6- dichlorophenyl}-7-niethylsulfaiiyl-2H-pyrimido 5,4-e"j ] ,3]oxazin'-4'-one (70 mg, 0.204 rrarsol) in toluene (3 mL) was added /w-CPBA (71 nig, 0.408 n mol) and stirred at rt for 30 mm. 4-(4- Isopropylpiperazin--l --yl }amlii e (44 mg, 0.204 mmoi) and DiPEA (78 mg, 0.614 mmoi) were then added and allowed to stir at rt for 12 h After completion of reaction, solvent was removed under reduced pressure, diluted with saturated NaHCOj solution (20 mL) and extracted with CI¾C¾ (20 mL x 3). (Combined organic layer was washed with bnne solution, dried over anhydrous f-h jSC and concentrated under reduced pressure to afford crude product which was purified by combi-flash chromatography to afford 3-(2,6-dichloropheny!)-7-[4-(4- isopropylpiperazin-l-yl)amlino]-2H-pyrimido[5>4-e][l ,3] oxazin-4-one (1 8 mg),
LCMS: 51 3 [M+lf
*11 NMR (400 MHz, DMSO-d6): δ 10.24 ( br s, I H), 8.78 (s, I B), 7.59 - 7.72 On. J === 8.33 Hz. 21:3), 7 39 7.59 (m, 3H), 6.79 - 7.01 (m, J - 8.77 Η¾ 2Π ;. 5.70 (s. 21:3), 3 08 (br s. 4H), 2 67 (br s. IH), 2.58 (br s, 4H), 1.01 (d, J - 6.14 Hz, 6H).
Example S20. Synthesis of3-{2, 6~dichioropheny])-7~[3-(hydroxyme†h) )~4~(4-meih^
yi}aniHno]~2H~pynmido[5, 4~ej[l , 3]oxazm~4~one
(Compound No, 1.20)
[0223] Synthesis of 3-(2,6-dich1orophenyl)-7-[3-(hydroxymethy1)-4-{4-methyipiperaziR~ i - i)aniii» ] -2H~pyrimido[5,4~e] [i ,3]oxa¾m-4-one To a stirred solution of 3-(2,6- dichlorophenyl)-7-methylsulfany]-2H-pynraido[5,4-e][l ,3]oxazin-4-one (300 mg, 0 879 mmol,
1.0 eq) in CH2C12 ( 15 mL) was added /J/-CPBA (454 nig, 2.64 mmoi, 2.5 eq) and allowed to stir at ft for 30 min. 5-amino-2- 4-meihylpiperazin-I-yl)phenyl]MeOH (1 94 mg, 0.879 mrnol, 1.0 eqj and DIPEA (567 mg. 12.9 mmol, 4.0 eq) were added and allowed to stir at rt for 12 h.
Progress of reaction was monitored by LCMS. After completion of reaction solvent was removed under reduced pressure, residue was diluted with water (20 mL) and extracted with ethyl acetate (50 mL x 3), Combined organic layer was washed with water (20 mL x 3), dried over anhydrous Na
2S0 and concentrated under reduced pressure. Crude was purified by reversed phase chromatography to obtain
yl)anilino]-2H"pyrimido 5,4-e][ ; ,3 ]oxazin-4-one (20 nig, 4.4%).
JH NMR (400 MHz, DMSO-de): δ 10.31 (br s, IH), 8.80 (s, IH), 8.28 (br s, 10), 7.74 (br s, HI), 7.65 ( J - 8.33 Hz, 2H), 7.57 (dd. J - 2.63, 8.33 Hz. i l l ?. 7.50 (dd, J - 7.67, 8.55 Hz, IH). 7.04 (d, J - 8.77 Hz, I H), 5.72 (s, 2H), 5.09 (br s. 1 H), 4 54 (s, 2H), 3.51 (s, HI), 2 82 (t, J - 4 38 Hz, 4H), 2.23 (s. 3 IT), 1 .24 (br s, 3H).
Example S21. Synthesis of 7-f 4~(4-cyclopropylpiperazm-l-yl) niiino -3~(2, 6-dichioropheny!)~ 2H~pynmido[S, 4~ej[l, 3]oxazin~4~one
(Compound No, 1.21)
[0224] Step- 1: Synthesis of l-cyciopropyi-4-(4-nitropheoyI)piperazine. To a stirred solution of l -fluoro-4-nitro-benzene (500 mg, 3.54 mnio!) and 1 --cyc!opropy!piperazme (536 mg, 4.25 mmol) in 10 mL of eihanoi was added DIPEA (913 mg, 7.08 mrao!) and stirred at reflux for 12 h. After completion of reaction, solvent was removed under reduced pressure, diluted with water (20 mL) and extracted with CH2CI2 (20 mL x 3). Combined organic layer w s washed with bnne solution, dried over anhydrous N'u :SO.: and concentrated under reduced pressure to afford
crude product which was washed wrih diethyl ether and dried io afford 1 -cyciopropyi-4--(4-- miropheriy!)piperazme (450 nig).
LCMS: 248 [M÷l f
[0225] Step- 2: Syn esis of 4-{4-cydopropyIpipera¾jn-i -yi)aniline: To a stirred solution of l -cyclopropyl-4-(4-nitrophenyl)piperazi:ie (450 rug, 1 , 82 mmol) ethanol (1 5 .ml,) and water (3 mL) were added iron powder (675 nig, 12.75 mmol} and ammonium chloride (701 mg, 12.75 mmol) The reaction mixture was stirred at 80 °C tor 4 h. After completion of reaction., solvent was removed under reduced pressure, diluted with water (20 mL) and extracted with CH2G2 (20 mL x 3). Combined organic layer was washed with brine solution, dried over anhydrous .SO.; and concentrated under reduced pressure to afford the desired product (300 nig).
LCMS; 218 [M÷l f
[0226] Siep-3: Synthesis of 7-[4-(4-cyclopropyipiperaztR-l-yI)anaino]-3-(2,6- dkhlorophenyl)~2H-pyriniidoj
'5,4-e {l,3]oxaziii-4~one. To a stirred solution of 3-(2,6- dichlorophenyl)-7-methylsulfanyl-2H^ Tiniido[5,4~e][3 ,3]oxazin-4-one (1 00 mg, 0.292 mmol) in toluene (4 mL) was added /w-CPBA (1 55 mg, 0.58 mmol) and stirred at ft for 30 niin. 4-(4- Cyclopropylpipefazin-l-yl)aniline (63 nig, 0.292 mmol) and D.IPEA (113 nig, 0.876 mmol) were then added and allowed to stir at ft for 12 h. After completion of reaction, solvent was removed under reduced pressure, diluted with saturated NaHCCh solution (20 mL) and extracted with Q!bCi? (20 niL x 3). Combined organic layer was washed with brine solution, dried over anhydrous V· ΗΟ.
; and concentrated under reduced pressure to afford crude product which was purified by flash chromatography to afford 7-[4--i4--cyciopropy!piperazin-4 -yl)anilino]-3 -(2,6- dichiorophenyl)-2H"pyrimido 5,4-eJ[
' l ,3 oxazin-4-one (30 mg).
\H NMR (400 MB¾ DMSO-d6): δ 10.24 (br s, 1H), 8 78 (s. 1 H), 7.65 (d, ./ - 7 89 Hz, 2H), 7.41 ■■ 7.60 (m, 3H), 6.92 (d, J === 8.77 Hz, 2H), 5.71 (s, 21 ) ·. 3.05 (br s, A W ) 2.67 (br s, A W ) 1.65 (br s, 1 H), 0.44 (d, J --= 3.95 Hz, 2H), 0.34 (br s, 2H).
fa-ample S22. Synthesis of 3~(2.6-dichioropheny -7-[(2- ihyi--4,4-dM
dihydroisoqiiiftolin-7-yl)amifto]-2H~pyrimido[5,4^
(Compound. No. 1.22
[0227] Synthesis of 3-(2,6-dichtorophenyl)-7-[(2-et^
dihydroisoquinolui-7-yi)a uio]-2'B-pyr^ To a stirred solution of 3--(2,6'-dichlorophenylV-7 (4,4--d^
pyrimido[5,4-e:| '1 ,3 joxazin-4-one hydrochloride (100 mg. 0,213 mmol, 1.0 eq) and acetaldehyde (35%) (0.8 mL, 0,638 mmol, 3.0 eq) in (¾(¾ (5 mL) acetic acid (64 mg, 1.10 nirnoi, 5.0 eq) was added dropwise ai 0 '"C The resulting mixture was stirred ai rt for 1 h, followed by addition of NaCNBLL (40 mg, 0.638 mmol, 3.0 eq) at 0 " 0. The resulting mixture was stirred at rt for I hr. The progress of reaction was monitored by LCMS, After completion of reaction, solvent was removed under reduced pressure, residue was basified with saturated solution of NaHCC (50 mL). Precipitated compound was filtered off, dried under vacuum and purified by reversed phase chromatography using to afford 3-(2,6-dic-hIorophenyl)~7~[(2-ethyI-4, ~dimethy}-l ,3- di.hydroisoquinolin-7-yi)aDiino]-2H.-pyrimido[5,4~e][l ,3]oxazin-4-one (25 mg, 23.6%)
LCMS: 498 [M+l
Hi NMR (400 MHz, DMSO-de): δ 10.31 (br s, 1H), 8.82 (s. I H). 7.66 (d, J - 8.31 Hz, 2H), 7.47 (d, J -- 8.80 Hz, IH), 7.51 (d, ./ - 7.83 Hz, 1H), 7 37 (br s, IH), 7.30 (d, ./ - 8.31 Hz, 2H), 5.73 (s, 2H), 3.49 (s, 2H), 2.39 - 2.47 (m, 2H), 2.28 - 2.39 (m, 2H), 1 .83 (s, 2H), 1.23 (s, 6H), 1.09 (t J ------- 7 09 Hz, 3H).
Example S23. Synthesis qf3-(2, 6-dichlorophenyiJ~ 7~((l~oxo~1, 2, 3.4-ietrahydroisoqidnolin- 7- yl)amino)~2, 3~dibydro~4H~pyrimido[5, 4~e]jl, 3 ]oxazin~4~one
(Compound No. 1.23)
mCPBA, Toluene
DIPEA.RT, 2b
[0228] Synthesis of 3-(2}6-dkhl rophenyl)-7-((l-oxo-l,23,4-fetrahydroi$ q«inolin-7- yl)amino)-23-d«hydro-4H-pyriraido[5,4-e] [i^]oxa¾in-4-one: To a stirred solution of 3-(2,6- dichloropheiiyl)-7-inethylsulfanyl-2H-pyfimido 5,4-eJ l ,3 oxazin-4-one (200 mg, 0.59 mmo!, 1.0 eq) m toluene (1 5 mL) was added >;.···(Ί ·' Λ (203 mg, 1.1 8 rnmol, 2.0 eq) and allowed to stir at rt for 30 rmn. 7~Amino-3,4-dihydro-2H-isoquinolin-l -one ( 100 mg. 0.590 rnmo!, 1.0 eq ) and DiPEA (300 nig, 2.36 rnmol, 4.0 eq) were added and allowed to stir at rt for 2 h. Progress of reaction was monitored by LCMS. After completion of reaction solvent was removed under reduced pressure, residue was diluted with water (20 mL) and extracted with ΟΠ 7 Ί · (50 ml, * 3). Combined organic layer was washed with water (20 ml, >< 3). dried over anhydrous a^SC^ and concentrated under reduced pressure. Crude residue was purified by reversed phase chromatography to obtain 3-(2.6-dichlorophenyl}-7-(( 1 -oxo~l,2,3,4-tetrahydroisoquinolin--7- yl)amino)-2,3-dihydro-4H.-pyrimido[5,4-e][] ,3]oxazm-4--one (13 mg, 4.8%).
LCMS: 456 [M+l]
*H NMR (400 MHz, DMSO-d6): δ 10.51 (br s, 1H), 8.86 (s, IH), 8.22 (br s, 1 H), 7.95 (br
IH), 7.83 (dd, J ------ 1.97, 8, 1 1 Hz, IH), 7,59 - 7.71 (m, 2H), 7.48 ~ 7.56 (m, Hi), 7.28 (d, J - 8.
Hz, Hi), 5.75 (s, 2H), 3.35 (d, J - 3.95 Hz, 2H), 2.86 (t, J - 6.58 Hz, 2H).
Example S24. Synthesis o/3~(2, 6-dichIorophenyiJ~ 7~(( 2, 3, 4-t.etrahydroi$oquinohn 2, 3~dihydro~4H-pyrimido[5, 4~e]j L 3joxazin-4-one hydrochloride
(Compound No. 1.24)
[0229] Step-Is Synthesis of tert-butyl 7-{{3-(2,6-d«chIorophenyl)-4-oxo-2H-pyrimiclo[5,4- e] [ly3)oxazia-7-yi]amin©)-3,4-d¾^ To a stirred solution of 3-(2,6-dicMorophenyl)-7Hmethy (200 g, 0.59 rnmol, 1 .0 eq) in toluene (3 ml,) was added /w-CPRA (250 mg, 0.147 mrnol, 2, 5 eq) a d allowed lo stir at rt for 30 min. Tert-butyl 7-amino-3,4-dihytiro-lI-i-i3oqumo]ine-2-carboxylai (1 50 mg, 0.590 mrnol, 1 .0 eq) and DIPEA (300 mg, 2,36 rnmol, 4.0 eq) were added and allowed to stir at :·;, for 1 h. Progress of reaction was monitored by LCfVlS After completion of reaction solvent was removed under reduced pressure, residue was diluted with water (20 niL) and extracted with ethyl acetate (50 mi., x 3). Combined organic layer was washed with water (20 ml., 3), dried over anhydrous

and concentrated under reduced pressure. Crude residue was purified by reversed phase chromatography to obtain test-butyl 7-[[3-(2,6~dich]orophe.n.yl)~4~oxo-2H- pynmido[5,4~e] [l ,3]oxazin-7-yj]amino]-3,4-dihydro-lH-isoqiiinoline-2-carboxylaie (150 .mg, 45.6%),
LCMS: 542 [M+l
[0230] Step-2: Synthesis of 3-{2,6-dichIorophenyl)-7-(i,2,3,4-tetrahydroisoquhH>ljn-7- yIai«ino)-2H-pyrimido[5,4-ej (l,3]oxa2ia-4-ooe hydrochloride: tert-Butyl 7-[[3-(2,6-
dichlorophenyl}-4-oxo-2H-pyfimido[5,4-e]
isoquinoline^-carboxylate (150 mg, 0.277 mrnol, 1 .0 eq) was dissolved in 4M FIG in dioxane (2 mL) at 0 °C. The reaction mixture was stirred at rt for 1 h. Progress of reaction was monitored by LCMS. After completion of reaction, precipitated compound was filtered off, washed with dioxane (3 mL) and dried under reduced pressure to obtain 3-(2,6-diehlorophenyl)-7-(l ,2,3,4- ietrahydroisoquinolin-7-ylamino)-2H-pyfimido[5,4~e][l ,31oxazni--4--one hydrochloride (30 mg, 24.5%) as HC! salt.
LCMS; 442 [Μ+l '
¾ N R (400 MHz, DMSO-d6)i δ 10.48 (br s, I H), 9.21 (br s, 2B), 8.85 (s, I B), 7.66 (d, J 8.3 1 Hz, 2H), 7.55 ~ 7.63 (m, 2H), 7.46■- 7.54 (m, Hi), 7.21 (d, J - 8.3 i Hz, Hi), 5.75 (s, 21i), 4.27 (br s, 2H), 3.57 (s, 2H), 3.36 (br s, 2H), 2.97 (t, J - 5.87 Hz, 2B).
Example S25, Synthesis of 3~(2~chloro~6~fluoro~phenyl)~ 7~f4-(4-methyipiperazin-l-yI) mimoj- 2H~pytimid [5, 4~e][L 3 ]axa∑in-4-one
(Compound No. 1.25)
[0231 ] Step-
'J : Synthesis of N-(2-chloro-6-fluoro-pheoyi)-4-hydroxy-2-i«ethylsuifanyl- pyrii«idi«e-5-carboxamide: A stirred solution of 4-hy koxy-2-niethyisulfaiiyl-pyriniidine-5- carboxy!ic acid (5.70 g, 30.65 mmol, 1.0 eq) and 2~chloro-6-£luoro-aniline (4.50 g, 30.65 mmo!, 1.0 eq) in toluene (200 mL) was purged with nitrogen gas for 15 mm. To the above solution PC 1 (30 mL) was added. The reaction was heated at 100 °C for 72 h. Progress of reaction was monitored by ! .(
'MS After completion of reaction, solvent was removed under reduced pressure, residue was diluted with a mixture of diethyl ether: MeOH (10: 1 ) (1 0 mL) stirred for 1 5 mm then f ltered off. Solid was suspended in MeOH (20 mL) stirred for 5 mm, filtered off and washed with MeOH (10 mL), and then dried under vacuum to obtain of N-(2-ch!oro-6-fluoro- phenyl)- "hydroxy--2--methylsulfanyl-'pynmidine'-5'-carboxamide (5.0 g, 52.8%).
LCMS: 314 [M+l
[0232] Step-2: Synthesis 3-(2-chloro-6-fluoro-phenyl)~7~methylsu!fanyl-2H- pyrimido(5,4-eI 11 ]oxaaan-4-one: To a stirred solution of !-(2"Chloro-6-flooro-phenyl)~4~ iwdroxy-2-methy]su]t nyl-pyrimidine-5-carboxamide (5.0 ;.·. 15.97 mrnol, 1 ,0 eq) in CH3CN: DMSO (200 mL: 10 ml) was added Π Ν . (6.40 g, 23.96 mo!, 1.5 eq) and Cs
2C()
3 (1 5.60 g, 47.91 mmol, 3.0 eq). The reaction mixture was heated at 80 °C for 36 h Progress of reaction was monitored by LCMS. After the completion of the reaction, solvent was removed under reduced pressure, residue was diluted with water (50 mL) and extracted with ethyl acetate (100 mL
χ 3), Combined organic layer was washed with water (50 mi ,
χ 3), dried over anhydrous
and concentrated under reduced pressure. Crude was purified by flash chromatography using ethyl acetate: hexane as eluents to obtain 3- 2-chloro~6-fli3oro--phenyl)-7-meihylsi3lfanyl-2H- pyrimido 5,4-e] [l ,3]oxazin-4-one (300 mg, 5.76%).
LCMS; 326 [M-H ]÷
[0233] Siep-3: Synthesis of 3-(2-chloro-6-{luoro-phenyl)-7-[4-(4-raethylpiperazin-.l- yl)anilino]-2H-pyrimido[5,4-e][1 ] >xa«»n-4-One To a stirred solution of 3-(2-chloro-6-†1uoro- phenyi)-7-raet)iyisuifanyl-2H-pyrirriido 5>4-e'][I,3]oxazin-4-one (60 mg, 0. 185 mraol, 1.0 eq) in toluene (2 ml.) was added i»-CPBA (79 mg, 0.452 mmol 2 5 eq) and allowed to stir at rt for 30 min. 4-(4-Methyipiperazm-Lyl) aniline (35 mg, 0.185 mmol, 1 .0 eq) and DIFEA (96 mg. 0.744 mmol, 4. 0 eq) were added and allowed to stir at rt for 1 h. Progress of reaction was monitored
by LCMS. After completion of reaction, precipitated compound was filtered off and washed with toluene (2 mL) and dried under vacuum. Solid was triturated with McOi ) (2 mL) then filtered off and dried under vacuum to afford 3-(2-chioro-6"fluoro-p3ieny3)-7-[4-(4-metliylpiperazin~ 1 - yl)anilmo]~2H-pyrimiclo[5,4-e] L3]oxazin~4--one (15 nig, 17.6%).
LCMS: 469 [M÷l f
XH MR (400 MHz, DMSO-d6): 3 10.25 (br s, 1 H), 8.78 (s, 1H), 7.45 - 7.60 (m, 4H), 7.38 - 7.45 (m, IH), 6.92 (ds J - 8, 80 Hz, 2HI 5,65 - 5.77 (m, 2H), 3.01 - 3.1 6 (m, 4H), 2.42 -· 2.48 (m, 4H)S 2.22 (s, 3H).
Example S26. Synthesis of3~(2, 6~dichJoraphenyl)~ 7-((4~{2~methylpiperazm~ l~yi)phenyl)a ino)~
4~ ] [ l, 3 oxa∑iri-4-one
[0234] Step-J : Synthesis of tert-butyi 3-methyI-4-(4-mtropheayi)pipera2;i«e-l- carboxylate: To a stirred solution of i -fkioro-4-nitrobenzene (5.0 g, 35.46 mrnol, 1 .0 eq) and tert-butyi 3
»methyipiperazine~l -carboxyiate (7.09 g, 35.46 rnrnoi, 1 .0 eqj in DMSO (40 mL) was added K
2C0
3 (9,78 g, 70.92 mrnol, 2.0 eq). The reaction mixture was heated at 120 °C for 16 h. Progress of the reaction was monitored by LCMS. Upon the consumption of starting material, mixture was diluted with water (100 mL-) and extracted with ethyl acetate (200 s in . * 2).
Combined organic layer was washed with water (50 mL χ 3), dried over anhydrous Na?S(>4 and concentrated under reduced pressure. Crude residue was purified by flash chromatography using ethyl acetate: hexane as eiuents to obtain tert-butyi 3-methyh4-(4-nitrophenyj)piperazme- 3 - earboxylate (5.5 g, 48.67%).
LCMS: 322 [M÷l f
[0235] Siep-2: Synthesis of tert-butyi 4~{4~am epbeRyI)-3-metbyIpiperazine-:! - carboxylate: To a stirred solution of tert-butyi 3 -methyl-4-(4-niirophenyl)piperazine-l " carboxylate (5 g, 15.57 mrnol. 1 .0 eq) in 100 mL of ethanol: water (1 : 1 ) mixture were added NH4CI (1 .65 g, 31.1 5 rnrnoi, 2 eq) and iron (2.75 g, 46.73 mrnol. 3.0 eq). The reaction mixture was heated at 90 °C for 2 h. Progress of reaction was monitored by 1 CMS Upon the consumption of starting material, solvent was removed under reduced pressure Aqueous layer was basified with saturated solution of sodium carbonate solution and extracted with ethyl acetate (200 mL x 2), Combined organic layer was washed with 50 mL of brine solution, dried over anhydrous a^SC^ and concentrated under reduced pressure. ( rude residue was purified by flash chromatography using MeOH: O CCC as eiuents to obtain tert-butyi 4-(4-aniinophenyi)-3- methylpiperazine-i -carboxylate (1.5 g, 33%).
LCMS: 291 [M÷lf
[0236] Siep-3: Synthesis of tert-butyi 4-(4-((3-(2,6-dichioropheayl)-4-oxo-3,4-dihydro- 2H-pyriaiido 15,4-e) [13
.1 oxaz«a-7-yl)ara«ao)phenyl)-3-ffiethylpiperaziiie-l -carboxylate: To a stirred solution of 3-(2.6-dichlorophenyl)-7-(methylthio)~2,3-clihydro-4H--pyrimido 5,4- e][ i ,3
"joxazin-4-one (250 mg, 0.733 mrnol, L0 eq) in toluene (15 mL) was added H-CFBA (315 mg, 1.83 mmoi, 2.5 eq) and allowed to stir at rt for 30 mia Tert-butyi 4-(4-aminophenyl)-3- niethylpiperazine-1 -carboxylate (256 mg, 0.87 rnmo!, 1 .2 eq) and DIPEA (37/8 mg, 2.93 mrnol,
4.0 eq) were added and stirred at ri for I h. Progress of reaction vvas monitored by LCMS. After consumption of starting material the reaction mixture was extracted with ethyl acetate (200 rni, x 2). Combined organic layer was washed with water (50 nil, x 3), dried over anhydrous a2S(>
4 ami concentrated under reduced pressure. Crude residue was purified by flash chromatography using MeOH: ( ! I ;
(.
') ; as eiuents to obtain teri-b tyi
ihydrG-2H-pyTimido[5,4~e][l ,3]oxazm^
[0237] Step-4: Synthesis of 3-{2,6-dichIorophenyl)-7-(i4-{2-raethylpiperaziR-l-yI)-ll4- pyridin-l-yl)amino)-2^dihydrc II-pyr^ A solution of tert- butyl 4-(4-((3-(2,6-dichiorophenyl)-4-oxo-3,4-dihydix>-2H-pyrimidoj ,4-e][l.,3]oxaan-7- yl)arnino)pheiiyl)-3-raethylpiperazine-l -carboxylate (100 mg, 0.170 nrnoi, 1 eq) in 4M dioxane HQ (2 niL) was stirred at rt for 1 h. Progress of reaction was monitored by LCMS. After consumption of starting material, precipitated compound was filtered off and washed with diethyl ether and dried under vacuum. Crude residue was purified by reversed phase
chromatography to obtain 3-(2,6-dichloiOpheiiyl)-7-((4-(2-methylpiperazin-l -yl)-l 14-pyridin-l - yl}ainino)-2,3-dihydfo-4H-pyriffiido]'5,4-e'| l,3 oxazin-4-one ( 15 nig, 18%).
LCMS; 485 [M+l f
¼ MR (400 MB¾ DMSO-d6): δ 8.78 (s, 1H), 7.65 (d, J 8.31 Hz, 2H), 7.42 - 7.56 (m, 3H), 6.88 i d. J === 8.80 Bz, 2H), 5.70 (s, 2B), 3.76 (br s, 1H), 3.07 id, J ==== 1 1.25 Hz, 2H), 2.94 (d, J === 8.31 Bz, 2H), 2.84 (d, J === 8.80 Hz, I B), 2.74 (d, J === 9.78 Bz, 2H), 1.90 (s, 2H), 0.95 (d, J 6.36 Bz, 3B).
Example S27. Synthesis of 3~(2~chloro~6~fltioro~phenyl)~7~(4^iperazin~ I ~yianilmo)~2 i~ pyrimidofS, 4~e/fl, 3]oxazin~4~one
(Compound No. 1, 27}
[0238] Step-1: tert-butyl 4-[4-[[3-(2-chIoro-6-fluoro-phenyl)-4-oxo-2H-pyrimido[5,4- e] [1 ]oxa«in-7-yi]aramo]phenyl]piperazine-l-carboxylatej To a stirred solution of 3~(2~ chloro-6-fluoro-phenyl)-7nni^ (1 50 rrsg, 0,462 rrsrno!, 1 .0 eq) in toluene (3 mL) was added TK-CPRA (199 mg, 1.16 mmol, 2, 5 eq) and allowed to stir at rt for 30 min, Tert-butyl 4-(4-aminophenyl) piperazine-l -earboxyiate (128 mg, 0.462 rrsrno!, 1 .0 eq) and DiPEA (238 rrsg, 1 ,85 siunoi. and 4,0 eq) were added and allowed ΐο sti at rt for 1 h Progress of reaction was monitored by LCMS, After completion of reaction, precipitated compound was fil ered off and washed with toluene (2 mL) and dried under vacuum to afford tert-butyl 4-[4-[ [3-{2-chlofo-6-fiuofo-phenyl)-4-oxo-2∑-]-pyrimido[5,4-e][l .3 ]oxazm-7- yl]amino]phenyl] piperazine- l -car boxy late (90 mg, 354 %). LCMS: 555 [ M+l j
[0239] Step-2: Synthesis of 3 2-chloro-6-iluoro-pheny!)-7-(4-pipera3Ein-l-ylaaaino)-2H~ pyrimido [5,4-e] [ 1 ) oxazm-4-one: To a stirred solution of tert-butyl 4-[4-[[3-(2-chlo.ro-6- fii3oro-phenyl)-4-oxo-2H-pyrimido[554-e][L3]oxazin-7-yl]am.ino]phenyI]piperazi
carboxylase (80 mg, 0 144 mmol, 1 .0 eq) in 4M HC1 in dioxane (2 mL). The reaction mixture was stirred at rt for 1 h. Progress of reaction was monitored by LCMS. After consumption of
siartmg material, precipitated compound vvas tillered off arid washed with dioxane and dried under vacuum. Oude residue vvas purified by reversed phase chromatography to obtain 3··ϊ 2·· cliloro-6-fluoro-phenyl)-7-(4~piperazi (30 mg, 46,8%).
iH NMR (400 MBz, DMSO-d
6): S 10.27 (br s, IH), 8.78 (s, IH), 7.48 - 7.73 (m, 4H), 7.40 - 7.45 (m, IH), 6.91 (d, J - 8.80 Hz, 2Hj, 5.67■- 5.75 (m, 2Hj, 2.97 - 3.03 (m, 4H), 2.83 (d, J - 4.89 Hz, 4H), 1.83 (s, IH).
Example S28. Synthesis of 3~(2~chioro~6fIuorophejryI)~7-(4<4~dffiie4hyl~] ,2.3,4- telrahydraisaquinoHn-7-y amina)~2H-pyrimido[5, ^
Compound No. L 28}
[0240] Step-I s Synthesis of tert-butyl 7-((3-(2-cWoro-6-iluoropheny!)-4-oxo-3,4- diliydro~2H~pyrimidoj
'5,4-e| [ ^
2(J H)-carboxyiaie: To a stirred solution
dih dro-4H-pyrimido|'5,4-e'| \ 1 ,3 ]oxazin-4-one (250 mg, 0.769 mmol, 1.0 eq) in toluene (5 m'L) was added #?-CFBA (330 mg, 1.923 mmol, 2.5 eq) and allowed to stir at ft for 30 mm. Tert-butyl 7-amino-4,4-dimet yl-3,4-dihydfoisoquinoline-2 lH)--cafboxylate (254 mg, 0.92 mmoi, 1.2 eq) and DIPEA (396 mg, 3.07 mmol, 4.0 eq) were added and stirred at ft for 1 h. Progress of reaction was monitored by LCMS. After consumption of starting material, the reaction mixture was extracted with ethyl acetate (200 mL χ 2). The combined organic layer was washed with water (50 mL x 3), dned over anhydrous \ .-Si) ; and concentrated under reduced pressure. Crude was purified by flash chromatography using eOH: f 1 LO ½ eluents to obtain tert-butyl 7-((3-(2- chloro-6-f1uorophenyl)-4-oxo-3,4-dihydro-2H-pyrimido[5,4-e] [] 53 ]oxa.zin-7-yl)a..mino)-4,4- diffietbyl-3J4-dihydroisoqmnoline-2(l H)carboxyj.ate (280 mg, 65%).
LCMS: 554 [M+l
[0241] 8ϋ¾ρ-2: Synthesis of 3-{2-chl ro-6-fluorophenyi)-7-((4,4-dHnethyl-l,2,3,4- tef rahydroisoquinoIin-7-yl)amino)-2 -dihydro-4H-pyrimido [5,4-e j 11 ,3] oxa2in-4-one: To a stirred solution of tert-butyl 7-{ -(2-chiofo-6-fiuofophenyl)-4-oxo-3!4-dihydro-ZH- pynmido[5,4-e: | 4 ,3 ]oxazm--7--yl}anuno)- ,4-dimetl l^,4-dihydroisoqin^olme-2(l H)- carboxylate (250 mg, 0.450 mmol, 1 eq) in 4M HQ m dioxane (2 mL). Mixture vvas stirred at rt for 1 hr. Progress of reaction was monitored by LCMS. After consumption of starting material, precipitated compound was filtered off and washed with dioxane and dned under vacuum to obtain " 4 - 0"d:;. h!o: o bo::yi ;..?..: : ..(2 -:; ;;:ihv ipi era/sn- 1 - ) - = \ -) - r:d:u- : "Y! ) n ·u;o ···2..^ dlhydro■ H-pyπmido 5,4--e]Π ,3]oxazm■-4■-one (200 mg, 90%).
LCMS; 453 [M+l
¾ NMR (400 MHz, DMSO-dc): δ 8 78 (s, 1H), 7.48 - 7.61 (m, 4H), 7.39 - 7 46 (rat ΓΗ), 6.91 (d, J - 8.80 Hz, 2H), 5.66 - 5.79 (m, 2H), 2,91 - 3.06 (m, 41 1 ). 2.83 (d, J - 4.89 Hz, 4Hj, 1.83 (s, 1H).
Example S29, Synthesis of3-(2, 6-dichIorophenyI)~7~(3~fluoro~4~piper zm~I~y^
pyrimidofS, 4~e][l, 3]oxazin~4~ ne
[0242] Step-1 : Synthesis of tert-butyl 4-[4-[[3-(2,6-dkhiorophenyI)-4-oxo-2H-
To a stirred solution of
one (250 mg, 0.733 mmoL 1.0 eq) in toluene (3 m'L) was added #¾-CPBA (31 5 rng. 1.83 nunol, 2.5 eq) and allowed to stir at rt for 30 min. Tert- butyl 4-(4-ammo-2-fluoro-phenyl }piperazine-l - carboxylaie (21 6 nig, 0.733 rnrnol, 1 .0 eq) and DIPEA (378 mg, 2.93 maioi, 4.0 eq) vvere added and allowed to stir at rt tor 1 hr. Progress of reaction was monitored by I .CMS After completion of reaction solvent was removed under reduced pressure, residue was diluted with water (20 mL) and extracted with ethyl acetate (50 mL χ 3). The combined organic layer was washed with water (20 mL x 3), dried over anhydrous Na^SO^ and concentrated under reduced pressure. Crude residue was purified by reversed phase chromatography to obtain tert- butyl 4-[4-[[3~(2,6- di chioropheny l)- --oxo--2 H~pynmi do 5 ,4 ^
1 -carboxylase (250 mg, 70%).
LCMS: 589 [M÷l ]÷
10243] Step-2: Synthesis of 3-(2,6-dichIorophenyl)-7-(3-fluoro-4-pipera2;i«-l-yI-aaaiao)- 2H-pyrimido[5,4-ej(l,3]oxa-5«a-4-ooe: tert-Butyl 4- 4- 3-(2,6-dichloropheny!)-4-oxo-2H- pyrimido[5,4-e] [l,3]oxazin-7-yl]amino]^ (250 mg,
0.425 rnmol, 1.0 eq) was dissolved in 4M HCl in dioxane (4 mL) at 0 °C. The reaction mixture was stirred at rt for i h. Progress of reaction was monitored by LCMS. After completion of reaction, precipitated compound was filtered off, washed with dioxane (3 mL) and dried under reduced pressure. Crude residue was purified by reversed phase chromatography to obtain 3- (2,6~diehlorophenyl)-7-(3~fluGro^
(50 mg, 24.15%).
LCMS: 489 [M+] f i NMR (400 MHz, DMSO-d«): δ 1.0.48 (br s, IH), 8.85 (s, IH), 8.26 (br s, I H), 7.60 - 7.74 (m, 3H), 7.46 - 7.56 (m, Hi), 7.40 (br s, IH), 7.03 (br s, IH), 5.74 (s, 2H), 3.17 (br. s., IH), 2.98 (br s, 7H).
Example S30, Synthesis of 3-(2-chloro-6-mei yiphenyl)~7~((4-(pipera
dihydro~4hI~pytimidoiS, 4~e][l, 3 oxazm-4-one
(Compound No. 1.30)
Step-1 Step-2
DIPEA, rt 1 h
Step-3
[0244] Step-1 : Synthesis of N-(2-chloro-6-methyipheoyI)-4-hydroxy-2- (methyithi )pyrimidiue~5~carboxainide: To a stirred solution of 4~hydroxy-2-
(meihykhio)pyfimidirse--5--carboxy!iC acid (5 g, 26.88 mmol, 1.0 eq) m toluene (100 ml,) were added 2-chloro~6»meihylanitine (3.8 g, 26.88 mmol, 1 eq) and P(¾ (25 mLj. The reaction mixture was allowed to stir at 100 °C for 72 h. Progress of reaction was monitored by LCMS. After consumption of starting material solvent was removed under reduced pressure, residue was diluted with diethyl ether (100 mL) and MeOH (10 ml.) stirred at rt for 10 min then filtered off and dried under vacuum to obtain N-(2-chloro-6-ni thylpheny{)-4-hydiOxy-2- (metlwlthio}pyrinudine-5-carboxamide (4 g, 48%).
LCMS: 310 [M+l
[0245] Step-2: Synthesis of 3-(2-chloro-6-methylphenyi)-7-(methyhhio)-2,3-dihydro-4H- pyrii«ido[5,4-e| | i ,3] oxa2in-4-one: To a stirred solution of N-(2-chloro-6-methylphenyl}-4- hydroxy~-2~-(methy!thio)pyrim irie~-5--carboxamide (5 g, 16.18 mmoi. 1.0 eq) n ( I I ;C\ (150 mL) were added ( s;CO . (1 5.75 g, 48 53 mmoi, 3 eq) and DMSO (10 rnL). The reaction mixture was purged with nitrogen for 10 min. CI ! ·'· · (6.5 g, 24.27 mmoi, 1.5 eq) was added and allowed lo stir a t 80 °C for 48 h. Progress of reaction was monitored by LCMS. After consumption of starting matenal, solvent was removed under reduced pressure; residue was diluted with water (200 mL) and extracted with ethyl acetate (400 mL χ 2). The c mbined orgamc layer was washed with water (50 mL >■ 3), dried over anhydrous Na^SC^ and concentrated under reduced pressure. Crude residue was purified by flash chromatography was using ethyl acetate: hexane as eluents to obtain 3^2~chloro-6-methylphenyl)~7~(meth
e][l,3]oxazin~4~one (800 mg, 1 5%).
LCMS: 322 [M+I]+
[0246] Step-3: Synthesis of tart-butyl 4 4 (3-(2~e Ioro-6-∞et yIpheny!)- -oxo-3,4~ dihydro-2H-pyrimid0[5,4-e] [l^]0xazin^^ To a stirred solution of 3-i2-ehloro~6~metnylphenyl )-7-(m^
e][ l ,3joxazin~4-one (200 mg, 0.643 mmoi 1 .0 eq) in toluene (5 niL) was added »?-CPBA (267 nig, 1.557 nimol, 2.5 eq) and allowed to stir at rt for 30 min. Teri-butyi 4-(4- aminophenyl)piperazine-l -carboxylate (207 mg, 0.747 mmoi, 1 .2 eq) and DIFEA (321 mg, 2.40 mmoi, 4.0 eq) were added and stirred at rt for 1 h. Progress of reaction was monitored by LCMS. After consumption of starting matenal, precipitated compound was fdtered off to obtain ten- butyl 4--(4»((3--(2--chioro--6-nieihy
y!)aniino)pheny!) piperazme- l -earbo.xylate (85 mg, 24.85%). LCMS: 551 [M÷l f
[0247] Step-4: Synthesis of 3-{2-chloro-6-methy!phenyl)-7-((4-(pipera¾in-l- yI)phenyl)araino)-2^-d«hydro-4H^yriraido[5,4-e][.l ]ox^«n-4-otH:5J To a stirred solution of tert-butyl 4-(4-((3-(2-cbloro-6-raetliylpheny{)-4-oxo-3,4-dihydro-2H-pyrimjdo['5,4- e)[ ] ,3 oxazm-7-yl Sammo)pheriyl) piperaz ne- 1. -carbox !ate (80 mg, 0, 145 mmoi, 1 eq) in 4M Cl in dioxane (3 mL). The reaction mixture . s stirred at rt for 1 h. Progress of reaction was
monitored by LCMS. After consumption of starting material, precipitated compound vvas filtered off and dned under reduced pressure, ("rude residue was purified by reversed phase
chromatography to obtain 3~(2-chlorG-6-methylphenyj)-7-((4-(piperazin- 1 -yl)phenyl)amino)-2,3- dihydro-4H-pyriinido[5,4-el[l,3]oxazin-4-one (30 mg, 46. 15%).
*H NMM (400 MHz, DMSO-d6): δ 8.77 (s, 1H), 7.53 (d, J - 8.33 Hz, 2H), 7 45 (d, ./ - 5.26 Hz, I H), 7.35 (d, J - 5.26 Hz, 2H), 6.90 i d. J - 9.21 Hz, 2H), 5.58 - 5.70 (m, 2H), 3.00 (br s, 4H), 2.84 (br s, 4H), 2.18 · 2.28 (m, 3H), 1.90 (s, IH).
Example S31. Synthesis of3~(2, 6~dichlorophe d)-7-(4-{4~methylpiperazin~l~\ )ph^
2H~pyrido[3, 2-eJfl, 3j xaz -4(3H)-one
Compound No. 1.31)
[0248] Step-!: Synthesis of 6-ehloro-2-raethoxynieotink acid: To a stirred solution of 2,6- dichloromcotinic acid (20.0 g, 104.16 mmol, 1 .0 eq) in Ή-IF (80 mL) and MeOH (40 mL) was added LiOH ¾0 (13, 1 1 g, 312.49 mmol, 3,0 eq) at r The resulting mixture was stirred at same
temperature for 12 h. Progress of reaction was monitored by LCMS. After completion of reaction, mixture vvas concentrated and diluted with water (1 00 mL), followed by dropwsse addition of I N HCl (50 mL), the formation of white precipitate was observed which was filtered and dried under vacuum to afford 6--chloro-2»methoxynicotinic acid as white sol id (19.0 g, 97.23%).
LCMS: 188 [M+l
[0249] Sfep-2: Synthesis of 6-chIoro-N-(2,6-dichlorophenyI)-2-methoxynic tinainide. A solution of 6-chloro-2-m thoxymcotimc acid ( 10.0 g, 53.310 mraoi, 1.0 eq) and 2,6-- dichloroamline (9,5 g, 58.641 mmo!, 1 . 1 eq) in toluene (100 mi..) was purged with nitrogen for 10 mm at n, followed by addition of P(¾ (50 mL). The resulting solution was stirred at 100 °C for 48 h. The progress of reaction was monitored by LCMS. The reaction mixture was concentrated, basified. with saturated solution of NaHCC (500 ml,), extracted, with EtOAc (100 mL x 2). The combined organic layers were washed with water (50 mL), with brine (50 mL), dried over a^SO,^, concentrated and purified by flash chromatography using ethyl
acetate:hexane to afford 6~chloro-N~(2,6-dichlorophesiyl)-2-methoxynicotniamide (6, 5 g, 36.77%) as off white solid.
[0250] Step-3: Synthesis of 6-chioro- -(2,6-dkhIorophenyi)-2-hydroxyakotinai«ide: To a stirred solution of 6-chloro-N-(2,6-dichlorophenyl )-2-niethoxynicotinaffiide (4.0 g, 12.06 mmol, 1 .0 eq), in CH2C¾ (50 mL) was drop- wise added 1 M solution of BBr3 (36.1 9 mL, 36, 19 rnmol, 3.0 eq) at 0 °C, The resulting mixture was stirred at the same temperature for 10 niin Progress of reaction was monitored by LCMS. After completion the reaction mixture was basified with saturated solution of NaHCCh ( 100 mL), extracted with CH2CI2 (100 mL χ 2). The combined organic layers were washed with water (50 mL), with brine (50 mL), dried over
a2S(>4. concentrated and purified by flash chromatography using ethylaeetate.'hexane to afford 6~chloi ~N~(2,6- ichlorophenyl)~2~hydroxynicotinamide as white solid (3.81 g, 99.47%).
LCMS; 317[M÷1]
÷
[0251 ] Step-4: Synthesis of 7-chioro-3-(2,6-dichioropke»yI)-2 - Iihydro-4II-pyrido[3,2- e) [l,3]oxazio-4-one: To a stirred solution of 6-chloro-N-(2
!6-dichlorophenyl}-2- hydroxynicotinamide (3.8 g, 1 1.96 mmol, 1.0 eq), in CH
3CN (50 siiL) was added Cs
2C0
3 (11.70 ;.·. 35.898 mmol 3, 0 eq) at rt. 1 he resulting mixture was stirred and pnrged with nitrogen for 10 min followed by addition of diiodomethane (6,41 g, 23.933 inrrsol., 2, 0 eq) at rt. The reaction mixture was heated at 90 °C for 48 h. The progress of reaction was monitored by 1 .CMS The reaction mixture was diluted with wate (100 mL), extracted with ethyl aeetate ( 100 rnL
x 2), The combined organic layers were washed with water (50 mL), with brine (50 rnL), dried over a2S(>4, concentrated and purified by flash chromatography using ethyl acetate : hexane to afford 7-chk>ro-3~(2
>6-dichk>rophe as off white solid (0.235 g, 5.95%,).
[0252] Step-5: Synthesis of 3-(2,6-dichIorophenyl)-7-((4-(4-raethyipiperazin-l- yI)phenyi}amino)-2^-dihydro-4H-pyrido[3
>2-ej |1 ]ox^in-4-oae: To a stirred solution of 7- cliloro-3- 2,6-diclilorophenyl)--2,3- ibydro- Il-^yrido|3,2-'e][l ,3 ]oxazm-4-one
(0.230 g, 0.697 mmol, 1.0 eq), and 4-{4-methy]piperazin -yl)aniiine (0.134 g, 0.697 mmol, 1 .0 eq) in DMSO (5.0 mL) was added K3PO
4 (0.296 g, 1.394 mmol, 2.0 eq) at ri. The resulting mixture was stirred and purged with nitrogen for 1 0 min. followed by addition of Cui (0.01 4 g, 0.0697 mmol, 0, 1 eq) and L-proline (0.016 g, 0 1394 mmol. 0.2 eq) and again purged with nitrogen. The reaction mixture was heated at 90 °C for 24h. The progress of reaction was monitored by LCMS. The reaction mixture was diluted with water (100 mL), extracted with EtOAc (1 00 ml, >< 2), The combined organic layer was washed with water (50 mL), with brine (50 ml,), dned over Na.?S()4, concentrated and purified by reversed phase chromatography to afford
1(4 -(4 ·:■:<.;· ! l; ; : ; H; /:: ;-
(7 mg, 2.08%,) as an off white solid.
LCMS: 484 [M+l
Hi mm (400 MHz, DMSO-d«): δ 9.61 (s, 1 H). 7.91 (d. J - 8.33 Hz, 1H), 7.64 (d, J - 8.33 Hz, 2HI 7.44 - 7.52 (m, 3H), 6.93 (d, J - 9.21 Hz, 2H), 6.54 (s, IH), 5.57 (s, 2Hj, 3.09 (i, 4H), 2.45 (t, 4HI 2,22 (s, 3H).
Example S32. Synthesis qf3-(2, 6HiichlorophenyiJ~7~(4~(4-methylpiperazin-i-yiJpheny!a ino)~ 2H-pyrido[3, ~e][L 3]oxazm-4(3H)-one
(Compound No. 1.52
10253] Step-l: Synthesis of 6-chloro-4-methoxyiiicotink acids To a stirred solution of methyl 4,6»dichloromcotinate (20.0 g, 97.09 mmoL 1.0 eq), in THF (80 mL) and MeOH (40 mL) was added LiOH.H?0 (12.22 g, 291,29 mmoL 3.0 eq) at rt. The resulting mixture was stirred at the same temperature for 12h. The reaction mixture was concentrated and diluted with water ( 100 mL), followed by dropwise addition of IN HQ (50 mL). white precipitate formed was collected by filtered and dried under vacuum to afford the desired compound, 6-ch!oro--4-- methoxynieotinie acid (10.0 g, 49.45%) as white solid.
LCMS: 188[M÷1]"'
[0254] Step-2: Synthesis of 6-chioro- -(2,6-dkhIorophenyi)-4-hydroxyakotinai«idei A solution of 6-chloro-4-niethoxynicotmic acid (9,0 g, 47.97 mmol, 1 .0 eq) and 2,6-dichloroamline (7.7? g, 47.97 mmoi, 1.0 eq) in toluene (270 ml) was purged with nitrogen for 10 niin at rt, followed by addition of PCh (45 ml,). I'he resulting solution was stirred at 100 °C for 48 h. The progress of reaction was monitored by LCMS, Tbe reaction mixture was concentrated., basrfied with saturated solution of NaHCC (500 mi,), extracted with EtOAc (2x 100 niL), The combined organic layers were washed with water (50 .ml,), with bone (50 ml,), dried over a:>SC> , concentrated and purified by combi- lasb [Silica gel 100-200 mesh, e!ution 0-30 EtOAc in hexane] to afford the desired compound, 6--chloro- --(2,6-dichloropheriyl)-4- bydroxyriicotinamide ( 3.5 g, 22.98%) as an off white solid.
LCMS: 317[M H f
[0255] Step-3: Synthesis of 7-chloro-3-(2,6-dichlorophenyI)-2^-dihyclro-4H~pyridoj;3,4- e| 113]oxazin-4~one: To a stirred solution of 6~chloro- ~(2,6-dichlorophenyi)-4- hydroxy nicotinamide (3.05 g, 9 60 mmol 1.0 eq), in CH3CN (120 mL) was added Cs2CG3 (9.38 g, 28.80 rnmoL 3.0 eq) at rt. The resulting mixture was stirred and purged with nitrogen tor 10 niin followed by addition of diiodomethane (5.14 g, 19.20 rnmoL 2.0 eq) at rt. The reaction mixture was heated at 90 °C tor 48 h. The progress of reaction was monitored by LCMS. The reaction mixture vvas diluted with water (1 00 mi,), extracted with EtOAc (2x 1 00 ml,). The combined organic layers were washed with water (50 mL), with brine (50 mL), dried over Na2SQ4. concentrated and purified with combi-flash [Silica gel 100-200 mesh; eluti.on 0-20 EtOAc in bexane] to afford tbe desired compound, 7-ch!oroLL(2,6-dieh!orophenyi}~2,3~dihydro~ 4H-pyrido 3,4-e [l ,3]oxazm-4-one (1 .71 g, 54 1 1 %) as light yellow solid.
LCMS; 329.1 [M÷ 1]÷
[0256] Step-4: Synthesis of 3-(2,6-dichIorophenyl)-7-(metkylthio)-2,3-dikydro-4H- p rido^^-e] [1 ]os:azia" ~on : A stirred solution of 7-chioro-3-(2,6-clichiorophenyi)-2,3- dihydro-4H-pyrido[3,4-e] [l,3]oxazin-4-one (0.500 g, 1.51 7 mmol, 1 .0 eq) and sodium- thiomethoxide (0.531 g, 7.585 mmol, 5.0 eq) in dioxane (10.0 mL) was stirred at rt for 4h. The progress of reaction was monitored by LCMS. The reaction mixture was diluted with water (1 00 mL). extracted with EtOAc (2x 100 mL). Tbe combined organic layers were washed with water
(50 iiiL), with brine (50 mL), dried over ^SO^ concentrated and purified by flash chromatography (elution 0-50 1 i Ac in hexane) to afford the desired compound, 3 ·-(
· '·
···'>-·
(0.292 g, 53.39%,) as an off white solid.
[0257] Step-5:- Synthesis of 3-(2,6-dkhIorophenyl)-7-(methyls«lfooyI)-2¾3-dihydro-4H- pyridoj3,4-e] [1 Joxaz«a-4-oae. To a stirred solution of 3-(2,6-dic3ilorophenyl)-7-(methylthio)-
23--dihydrO"4H-pyndo[3,4-e][L3]oxazin-4-one (0.290 g, 0.852 niiiiol, 1 .0 eq) in Acetone (6 niL) at 0 °C was added Oxone (1.94 g, 12.79 mmo! in 1 mL of water). The resulting mixture was stirred at rt for 3h. The progress of reaction was monitored by LCMS. The reaction mixture was concentrated, diluted with water (100 mL). extracted with EtOAc (2x100 mL). The combined organic layers were washed with water (50 nil,), with brine (50 ml.,), dried over Na?S(> concentrated and purified by flash chromatography (Elution: 0-60% EiOAc in hexane) to afford the desired compound, 3-(2,6-dichlorophenyl)-7-(ffiethylsulfonyl)-2,3-dihydfo-4H-pyrido 3,4- e][ l,3]oxazin-4-one (0.202 g, 63.68%,) as an off white solid.
LCMS; 373.2 [M÷l]÷ 025S] Step-6:- Synthesis of 3-(2,6-dic orophenyl)-7~((4~(4-methyIpiperazin-l- yl)phenyl }amino)-2v3-dihydro-4H-pyrido (3,4-eJ [1,3] oxa«in-4-one: To a stirred solution of 3- (2,6-dichk>rophenyI)-7-(me^ (0.150 g, 0.403 rarnol, 1.0 eq), and 4-(4-rnethylpiperazin- 1 -yi)aniline (0.084 g, 0,443 mnioh 1.1 eq) in DMSO (5.0 mL) was added tBuOK (0.090 g, 0.806 mmol, 2.0 eq) at rt. The resulting mixture was heated at 120 °C in microwave for 30 mm. The progress of reaction was monitored by LCMS. The reaction mixture was diluted with water (100 ml,), extracted with EtOAc (2x100 rnL). The combined organic layers were washed with water (50 mL), with brme (50 mi..), dried over Na^SQ^ concentrated and purified by reversed phase purification to afford the desired compound, 3-(2,6-dichiorophenyl)~7~((4-(4-methyl^
4H"pyrido[3,4-e][l ,3]oxazm-4-one (0.004 g, 2.05%j as an off white solid.
LCMS: 484.2 [M+l
*11 M (400 MHz, DMSO-d6): δ 9.37 (s, 1 H), 8.54 (s, 3H), 7.64 (d, ../ === 7.89 Hz, 4ΕΪ), 7.49 (d ./ - 8.33 Hz. 3B), 7 39 id, 7 8.33 Hz, 41:1), 6.92 (d, J ------- 8.77 Hz, 3H), 6.24 (s. 2H), 5.55 (s. 41:1),
4. 10 (d, J - 4 82 Hz, 4H), 3.16 (d / - 5.26 Hz 3H), 3.08 (br a, 4H), 2.22 (s, 3i-[).
Example S33. Synthesis of3-(2, 6 iichloropheny)~7-(3-me†hyi-4-{piperazin-l-yl)p
2H-pyri ido[5, 4-ejfI, 3]oxazin~4(3H)-one
(Compound No, 1.33)
[0259] Step-1: Synthesis of tert- butyl 4-|;4-|; 3-(2,6-dichioropheayl)-4-oxo-2H- pyriniido 5,4-e] [ί ] oxazin-7-yi | mino ] -2-raetfayi-pfaeny 1] piperazine- l~carboxy late: To a stirred solution of 3~(2.6-dichlorophenyl)-7-methy^
one (250 nig, 0.733 nirnol, 1.0 eq) in 3 mL of toluene was added m-CPBA (315 nig, 1.83 nirnol, 2.5 eq) and allowed to stir at rt for 30 mi Tert-butyl 4-(4-ammo-2-methyl-ph !iyl)piperazine-.l - earboxylate (21 3 rng, 0 733 rnmol, 1.0 eq) and DI.PE (378 mg, 2 93 mmol, 4.0 eq) were added and allowed to stir at n for 1 br. Progress of reaction was monitored by LCMS, After completion of reaction solvent was removed under reduced pressure, residue was diluted with 20 mL of water and. extracted with ethyl acetate (50 ml, χ 3). Combined organic layer was washed with water (20 mL x 3), dried over anhydrous Na2S04 and concentrated under reduced pressure. rud was purified by flash chromatography using eOITCiLCb as e!uents to obtam 180 mg of
tert-butyi 4-4-jp~(2,6-diehlorophenyl)-4-oxo-2i}-pyri^^
metiiyl-phenyl Jpiperazine- 1 -carboxyiaie as a reebase.
LCMS: 585 [M÷l]÷
[0260] Step-2: Syn esis of 3-{2,6-dichIoropheny!)-7-(3-raethyl-4-piperazin-l-yI- aniIino)-2H-pyriraido5,4-e][1 ]oxazm-4-one hydrochloride salt: Tert-butyl 4-[4-[[3-(2,6- dichlorophenyl)-4-oxo-2H^yrim^
1 -carboxyiaie (150 mg, 0,256 rsniol, 1.0 eq) was dissolved in 4 si , of 4 HQ m dioxane solution at 0 °C. Reaction wa stirred at rt. for 1 hr. Progress of reaction was monitored by LCMS. After completion of reaction., precipitated compound was filtered off, washed with 3 niL of dioxane and dried ursder reduced pressure to obtain 3-(2,6-dicbloi'ophenyl)-7-(3-methyl-4- φ ra^m-l- l- nili o)-Zί:ϊ-p riΐnid [5,4-eJ ,3] azin-4-one (90 mg., 67,7%) as an HQ salt
LCMS: 485 [M l
*H r E, (400 MHz, DMSO~d6): δ 10,31 (br s, 1H)S 8.97 (br. s., 2H), 8.82 (s, 1H), 7,66 (d, J 7 Hv Hz, 2H), 7.42 - 7,59 (m, 3H), 7,04 (d, J - 8.33 Hz, 2H), 5.72 (s, 2H), 3.23 (br s, 3H), 3.16 (br s.1I¾ 3,02 (br s., 4H), 2.26 (s, 3H).
Example S34. Synthesis (rf 3~(2.6~dichk)rophenyl)-7-(3~eiydrox inethyi)
yljphenyiam moJ-2fI~pynm idol 5, 4~e If 1,3 joxazin-4(3Hj-one
(Compound No.1.34)
[0261] Step-1 : Synthesis of (2-brorao-5-nitrophenyl) MeOH: To a stirred solution of 2- bromo-5-nitrobenzaldehy e (5 g, 21.73 mmoL 1.0 eq) m 100 ml, of THF: MeOH (1 : 1) was added L (1 .67 g:1 43 47 nvmoi, 2.eq) ai 0 °C and allowed to stir ai rt for 1 hr. Progress of reaction was monitored by TLC. After consumption of starting material, solvent was removed under reduced pressure: the residue was diluted with 100 mL of water and extracted with ethyl acetate (200 mL ··- 2). Combined organic layer was washed with water (50 mL χ 3), dried over anhydrous 2SOd and concentrated under reduced pressure to obtain 4.5 g (89.28 %) of (2- bromo- 5 - ni iroph eny 1 S MeO .
[0262] Step-2: Synthesis of tert-butyl 4-{2-{hydrox 'methyI)-4-nitropheny -3,6- dihydropyridine-l(2H)-carboxylate: To a stirred solution of (2-bromo-5-nitrophenyl)M C)H (1
g, 4.31 mmol. 1 .0 eq) in 5 mL of dioxane were added tert-butyl 4-(4
34,5,5 etfameihyi--l ,3- dioxolan-2-yl)-3,6-dihydropyridine-] (2H)-carboxylate ( 1.59 g, 5.17 mmol, 1 .2 eq) and solution of Na^CC (1 .35 g, 12.93 mmol, 3 eq) in 2 mL of water. The reaction mixture was purged with nitrogen for 1 5 mm. Ρά(¾ίάρρί) (0 351 g, 0,431 mmol 0. 1 eq) was added. The reaction mixture was stirred at 120 °C for 20 mm in microwave. Progress of reaction was monitored by LCMS. After consumption of starting material, solvent was removed under reduced pressure; residue was diluted with 50 mL of water and extracted with ethyl acetate (200 mL >< 2). The combined organic layer was washed with water (50 mL x 3), dried over anhydrous X
.-SC
) ; and concentrated under reduced pressure, ("rude was purified by flash chromatography using ethyl acetate: hexane as eluents to obtain 460 mg (46%) of tert- butyl 4-(2-(hydroxymemyl)-4- nilxopheny l)-3,6-dihydiOpyri dine- l (ZH)-carboxy late.
[0263] Step-3: Synthesis of tert-butyl 4-(4-ara«ao-2-(hydroayffiethyl)pheayl)piperidiiie-
1-carboxylate: To a stirred solution of tert-butyl 4-(2-(hydrox.y.methyl)-4-.nitropheny])-3.6- dihydropyridme~l (2H)-carboxylate (1.2 g, 3.58 nirnof 1 , 0 eq) in 50 mL of MeOH was added Pd/C (200 mg). The reaction mixture was stirred at rt under hydrogen environment for 2h, Progress of reaction is moni tored by LCMS After consumption of starting material, reaction mixture was filtered eff using celite bed. The filtrate was collected and concentrated under reduce pressure to obtain 0.891 g (97%) of tert-butyl 4-(4-ar no~2~
(hydroxymethyl)pheny!)piperidine~l -carboxylate.
[0264] Step-4: Synthesis of tert-butyl 4-(4-((3-(2,6-dicMorophenyI)-4-oxo-3,4-dihy 2M-pyrimido[5y4-e] (l,3]oxa:«½-7-y^^
carboxylate: To a stirred solution of 3~(2,6-dichlorophenyl)-7-{methylthio)--2,3-dihydro-4H-- pyrimido[5,4-el [l ,3]oxazm-4-one (300 mg, 0.879 mmol, 1 .0 eq) in 5 mL of toluene was added ni-CPBA (378 mg, 2.199 mmol, 2.5 eq) and allowed to stir at rt for 30 mm. Tert-butyl 4-(4~ amino-2-(!iydroxymethyl)phenyl)piperidine-l-carboxylate (323 mg, 1.05 mmol, 1 .2 eq) and DIPEA (453 mg, 3.51 mmol, 4.0 eq) were added and stirred at rt for 1 nr. Progress of reaction was monitored by LCMS. After consumption of starting material, solvent w¾s removed under
reduced pressure, residue was diluted with 50 mL of water and extracted with ethyl acetate (200 mL x 2). The combined organic layer was washed with water (50 niL x 3 ), dried over anhydrous Na2S04 and concentrated ursder reduced pressure. Crude was purified by flash chromatography usmg MeOH:CH2C12 as eluents to obtarn 180 mg (34%) tert-butyi 4. ; .|..π ··; 2. ·
dichlorophenyl)-4-Gxo-3,4-dihydro-2H-py^^
(hydroxymetliyi)pheny!) pipendme-1 -carboxylate.
[0265] Srep~S: Synthesis of 3-(2,6-dichioropheHyl)-7-((;^(hydrox 'meth I)- -(piperidin- 4-yI)phenyl)aramo)-2^-dihydro-4H-pyr n«do[5,4-e] [ly3]oxazin-4-ORe. To a stirred solution of tert-hutyl 4-(4-((3-(2,6~dichjorophenyl)~4~oxo-3,4-d^
yI)aroino)-2- (hydroxyraethyI)phenyl) piperidme- 1 -carboxylate (180 nig, 0.300 mraol, I eq) in 4 ; l( i in dioxane (3 mi..). Mixture was stirred at rt for 1 hr. Progress of reaction was monitored by LCMS. After consumption of starting material, precipitated compound vvas filtered off and washed with dioxane and dried under vacuum. Crude was purified by reversed phase
chromatography to obtain 30 nig (20%) of 3-(2,6-dichlorophenyi)-7-((3-(hydroxyniethyl}-4- (pipefidm-4-yl}phenyl)araiiio)-2,3-dihydro-4H-pyriinidof'5,4-e p ,3 Joxazin-4-one.
LCMS; 500 [M+l f
{H MR (400 MHz, »MSO-d6); δ 1 0.39 (br s, 1.H), 8.82 (s, 1.H), 8.43 (br s, 2H), 7.55 - 7.74 (m, 4ΕΪ), 7.47 - 7.54 (m, I B), 7.19 i d. ./ === 8.77 Hz, 1 H), 5.73 is, 2H), 4.55 (s, 2H), 3.1 9 (br s, 4B), 2.93 (br s, 2H), 2.80 (br s, 2H), 1.70 ( br s, 4H).
Example S35. Synthesis of 3-(2, 6 Uchlorophenyl)~7~(2~methyl- 1 ,2,3,4-tetrahyxlro ^
ylamino)~2H~pyrimido[5.4~e][L 3]oxazin~4(3H)-one
[0266] Synthesis of 3-(2,6-dichIorophCTiyl)-7^^
yl}a«i«no]-2H-pyrimido|S,4-e) [l,3]oxazio-4-oae: To a stirred solution of 3-(2,6- dichiorophei !)-7-{'L2,334 etrah^
one hydrochloride ( 150 mg, 0,340 mmol, 1.0 eq) and HCHO (0,096 mL, 1.02 mmol, 3,0 eq) in dichioroeihane (5 nil,) was added acetic acid (102 mg. 1 ,70 mmol, 5.0 eq) dropwise at 0"'J€, The resulting mixture was stirred at rt for 1 hr followed by addition of NaCNBiL (64 mg, 1 , 02 mmo!, 3.0 eq) at 0°C. The resulting mixture was stirred at rt for 30 min. The progress of reaction was monitored by LCMS After completion of reaction, mixture was basified with saturated solution of NaHCO-} (20 mL) and extracted with CH2Q2 (50 mL x 2) The combined organic layer was washed with water (50 ml,) ami bnne (50 mL), dried over anhydrous \·;· .·ΗΟ.· and concentrated under reduced, Crude was purified by reversed phase chromatography to afford 3 mg (1.58%) formate salt of 3~(2:16-dic!tioropheny!S--7-- (2-m
2 H-pymni do[5 ,4-e] 1 , 3 oxazi n-4-one.
LCMS: 456 [M+Ij
' i t NMR (400 M¾ DMSO-d6): δ 8.78 (s, H), 7.62 - 7.69 (m, J - 8,33 Hz, 2H), 7.50 (d, J 7.45 Hz, 2H), 7.54 (d, J - 7,89 Hz, I B), 6 87 6,96 (m, J - 8.77 Hz, 2H). 5.70 (s, 2H), 3.04 (d, J - 5.26 Hz, 4H), 2.89 (d, J - 4.82 Hz, 4H), 1.90 (s, i l l )
Example S36. Synthesis of3-{2, 6~dichhrophemiJ--7~(6~(piperazirs~!-yi)py
pyrimido/5, 4-e / I.3 Joxazin- 7(3/70 -one
(Compound No. 1.36)
H
[0267] Step-1 : Synthesis of tert-butyl 4-(5-nitropyridin-2-yI)piperazine-l-carboxyIatej
To a stirred solution of 2-bromo-5-niiropyridine (1.0 g, 4.926 mmoL 1 eq ) and tert-butyl piperazine- 1 -carboxyiaie (0.917 g, 4.926 rnmoL 1.0 eq) in 4 niL of ¾(> was added K CO . (1 .02 ;.·. 7.389 mrno!, 1 , 5 eq). The reaction mixture was heated at 150 °C tor 1 5 rrun in microwave. The progress of reaction was monitored by LCMS. Upon the consumption of starting material, the precipitated compound was filtered off and dried to obtain the desired product tert-butyl 4-(5- nitropyridin-2-yl)piperazine-l -carbox j.ate (1 , 283 g. 84.18%) as a yellow solid.
LCMS- 309 [M l
[0268] Step-2: Synthesis of tert-butyi 4-(5-anMoopyrid«i-2-yl}pipera2ine-l-carbox iate:
To a stirred solution of tert-butyl 4-(5-nitfopyfidm-2-yl)piperaziiie-l -carboxyiate (2.9 g, 9.405 nmso!, 1.0 eq) in 6 mL of ethanol: water (1 : 1 ) mixture were added ammonium chloride (4.04 g, 75.24 rnrno!, 4 eq) and Fe(0) (2.10 g, 37.6:2 mmol, 4.0 eq). Reaction mixture was heated at 50 °C for 30 mm. Progress of reaction was monitored by LCMS. Upon the consumption of starting materia!, the reaction mixture was filtered over ce!ite and filtrate was concentrated under reduced pressure. The crude obtained was diluted with 50 niL of water and extracted with ethyl acetate (200 mL x 2), Combined organic layer was washed with water and brine, dried over a^S0 and concentrated under reduced pressure. The crude vvas purified by flash chromatography using 0- 2% MeOH in C¾Cb as eluents to obtain the desired product, teri-butyl 4-(5-aminopyndin-2- yl)piperazine-l -carboxy!ate (1.072 g, 40%).
LCMS: 279 [M+I]+
[0269] S ep-3: Synthesis of tert-butyl 4-(5-((-^(2,6-dkhIorophenyI)-4-oxo-3,4-dihydro- 2B- rlm^ To a stirred solution of 3-(2,6-dichlorophenyl)-7-(methylthio)-2,3-dihydro-4∑J-pyriinidof'5,4- e][ L3 joxazin-4-one (300 nig, 0.876 mmol, 1 ,0 eq) m 3 mL of toluene was added mCPBA (376.9 mg, 2.19 mmol, 2.5 eq) and allowed to stir at rt for 30 in. Tert-butyl 4-(5-aminopyndin-2- yl)piperazine-l "Carbox late (244.03 mg, 0.876 mmol, 1 eq) and D!PEA (453 mg, 3.50 mmol, 4.0 eq ) were added and allowed to stirred at rt for 1 hr. Progress of reaction vvas monitored by LCMS. After consumption of starting material, the reaction mixture was extracted with ethyl acetate (50 niL 2). Combined organic layer was washed with water (20 mL χ 3), dried over anhydrous Na^SO^ and concentrated under reduced pressure. The crude was purified by flash chromatography using MeOH: CH2<¾ as eluents to obtain 105 mg (20%) of tert-butyl 4~(5-((3- (2,6-dichlorophenyj)-4-oxo-3,4~dihydro~2H-pyrim
yi)piperazme- 1 -car oxy!ate.
LCMS: 571 [M+l
[0270] Step-4: Synthesis of 3-(2?6-dle Ioropheny!)-7-((6-(psp r8zh5~l~yI)pyrid¾~3- yl)amino)-2 -d«hydro-4H-pyriniido[5,4-ej [i,3]oxa¾m-4-on ten-Butyl 4-(5-((3-(2,6- di chloropheny i)- --oxO"3 ,4- ihy di ~2H-py
yl)piperazine-l -cafboxy!ate (100 mg. 0.175 mmol, 1 eq) was dissolved in 4N F CI in droxane (3 mL). Reaction mixture vvas stirred at ft for 1 hr. Progress of reaction was monitored by LCMS. After consumption of starting matenal, precipitated compound was filtered off and washed with diethyl ether and dried under vacuum. The crude was purified by reversed phase to obtain 70 mg (84.86%) 3~(2;0-diehlorophenyl)-7-((0-(piperaz^
pyrimido[5,4-e] [l ,3]oxazm-4-one.
Hi MR (400 MHz, DMSO~d6): δ 9.24 (br s> 2H), 8.83 (s> IH), 8.47 (br. s., IH), 8.02 (d, J - 7.89 Hz, Hi), 7.66 (d, J - 7.89 Hz, 2H), 7.42 -· 7.54 (m, IH), 7. 14 i d. J - 9.21 Hz, IH), 5.73 (s, 2H), 3.73 ·· 3.84 (m, 3H), 3.70 (br s, Hi), 3,03 · 3.25 (m, 4H).
Example S37. Synthesis of (R)~3~ ,6HiichlorophenyI)-7-(4-(2-(hydroxym
yi. phenylam inoj~2H~pyrimido[5, 4~e] [1 , 3>oxazin~4(3H)~one
(Compound No. L 37}
[0271] Synthesis of (R)-3-(2^dkhlorophenyl)-7-(4-(2-(hydroxymethyI)piperazin-l- y!)phenyIamino)-2H-pyrimido 5,4-e][1 ]oxaziin-4(3H)-one: (R)-3-(2,6-dichlo.rophenyl)-7-(4-
(2-(hyd.rox.ymethyl)piperazin-l ~yl)phe!iykniino)-2H~pynmido[5,4-e][l ,3]oxaziri-4(3H)~one is separated from die product synthesized in the scheme described in Example S I 3.
Example S38. Sytithesfs /'('¾~5~f¾ 6-i¾c/?/oi
yi}phenylamino)~2H~pyrimido[5, 4~e][i , 3 oxazin-4 (3H)~one
(Compound No. 1.38)
[0272] Synthesis of (S)-3-(2,6-dkh!orophenyi)-7-(4-(2-(hydroxyraethyl)piperazin-l- yl)phenylaraino)-2H-pyrimid [5,4-e] [l,3] xazin-4(3H)-one. (S)-3-(2,6-dichlorophenyl)-7-(4- (2-(hydroxymethyl)piperazin-l-yl)^ is separated trom the product synthesized in the scheme described in Example S13.
fj a p!e S39. Synthesis o/3-(2, 6~difhiorophenyi)~ 7~{4~(piperazbi~l~yi)phenylamino)~2H~ pyrimidofS, 4~e/fl, 3]oxazin-4(3H)-one
(Com ound. No. 1.39)
Siep~3
[0273] Step-1: Synthesis of IS;-(2,6~dif!uoropheayl)~4~hydroxy-2-{methyIihio)pyrimidhi&- 5-carboxamide: To a stirred solution of 4-hydrox ,,-2-(m thylthio)pyrimidine-5-carboxy]ic add (5 g, 26 88 mmol, 1.0 eq) in 100 mL of Toluene was added 2,6-difIuoroaniline (3,81 g, 29.55 mraoi, L I eq) and P(¾ (25 mL). Reaction was allowed to stir ai 100 °C for 72 h. Progress of reaction was monitored by LCMS. After consumption of starting material, solvent was removed under reduced pressure, residue was diluted with diethyl ether (100 mL) and 10 mL of MeOPi. Solid was filtered out and dried under vacuum to obtain 7 g (88%) of N-{2,6-difluorophenyl)-4- hydroxy-2-(methylthio)pyrimidine-5-cafboxaniide.
LCMS: 298 [M+l]
[0274] Siop-2: Synthesis of 3-{2,6-difloorophenyi)-7-(methyhhio)-2^-dihydro-4H- pyr do [5,4-e] [ 1 ) oxazin-4-one; To a stirred solution of N~(2,6-difl uoropheny l)-4-hy droxy-2-
(rneihy]ihio)pyrimidine-5-carboxamide (7 g, 23.56 rnmoi, 1.0 eq) m 200 nU , o! <Ί I f \ were added Cs2C03 (22.97 g, 70.70 mrnoi, 3 eq) and DMSO (15 mi,). Reaction mixture was purged with nitrogen for 10 mm. Diiodomethane (9.47 g, 35, 35 mmo!, 1.5 eq) was added and reaction was allowed to stir at 80 °C for 48h. Progress of reaction was monitored by LCMS. After consumption of starting material solvent was removed under reduced pressure; residue was diluted with 100 mL of water and extracted with ethyl acetate (400 mL 2). Combined organic layer was washed with water (1 00 rnL >< 3), dried over anhydrous Na2S(>4 and concentrated under reduced pressure. Crude was purified with flash chromatography usmg EtoAc: Hexane as eluents to obtain 400 mg (5%) of 3-(2!6-difluorophenyl)-7-(ffietb.ylthio)-2,3-dihydro-4H- pyrimido[5,4-e] [.l ,3]oxazm-4-one.
LCMS: 310 [M+l f
[0275] Sfep-3: Synthesis of tert-butyl 4-(4-(i3
»i2,6~dsfi¾sor p ssyI}-4-oxo-3,4
»di ydr ~ 2II-pyri idoj5,4-ej [l^]oxazm-7-yl)a ^^ To a stirred solution of
L3 ji rxa in-i- c (220 mg, 0.7 ; 1 maioi, 1 .0 eq) in 5 mL of toluene was added mCPBA (306 nig, 1.779 rnmoi, 2.5 eq) and allowed to stir at i for 30 nun. Tert-buiyl 4~(4-aminophenyl)piperazine-l - carboxylate (238 mg, 0.854 mmo!, 1.2 eq) and DJPEA (367 mg, 2.84 mmoL 4.0 eq) were added and stirred at rt for 1 hr. Progress of reaction was monitored by LCMS. After consumption of starting material, solvent was removed under reduced pressure; residue was diluted with water (20 mL) and extracted with et!iyi acetate (100 mL x 2). Combined organic layer was washed with water (50 mL 3), dried over anhydrous Na
2S0
4 and concentrated under reduced pressure. To obtain 100 mg (26.10%) of ert-butyl -M 4-ii.v.{.?..6~dn l uo; ophe yi r- f- ^ . :κΗπ> Jro-2i b pyrimido[5,4-e][l ,3]oxazm^ flash chromatography was used using MeOH. CH2C12.
LCMS; 539 [M+l f
[0276] Step-4: Synthesis of 3-(2,6-diflaorophenyl)-7-((4-(piperazm-'l-yl}phe«yI)aiRi«o)- 2^-dihydro-4H-pyrii«ido{5,4-e] [1 ,3]oxa2in-4-one: To a stirred solution of tert-butyl 4-(4-((3-
(2,6-difli3orophenyl)-4-Gxo-3,4-dihydro-^
piperazine-l -earbo.xylate (100 mg, 0.185 mmoi, I eq) in dioxane HCi (4 ml,). Reaction mixture was stirred at rt for 1 hr. Progress of reaction was monitored by LCMS. After consumption of starting material, precipitated compound was filtered off. Crude was purified by reversed phase to obtain 6 mg (7%) of 3~(2,6-clifiuorophenyl)-7-((4-(piperazin-l-yj)pheny )amino)-2.3-dihycliO- 4H-pyrirnido[5,4--e1iI ,3 oxazin- --one.
LCMS: 439 [M+l
Hi NMR (400 MHz, »MSO-d6) δ 8.83 is, i l l ?. 7.58 (d, J - 8.77 Hz, 3H), 7.34 (br s, 2H), 6.97 (d, ·/ - 9.65 Hz, 2H), 5. Si (br s, 2Hj, 3.23 (d, J - 4.82 Hz, 2H), 3.06 (br s, 2HI 2,89 (br s, 2H).
Example S40, Synthesis of 7~(3~chloro~4~(piperazin~l~yl)phenyla.mino)~3~(2, 6-dichloroph nyl)~ 2H~pytim id [5 ', 4~e ][1 , 3 ]oxa∑in-4(3H)~one
(Compound No. 1.40)
[0277] Step-1: Syn es s of tert-butyl 4-(2-chloro-4-(3-(2
>6-dich!orophenyl)-4-oxo-3
>4-
stirred solution of 3-(2,6-dichiorophenyl)-7-methylsul.fany}-2H~py.rimido[5,4-e][l ,3]oxa in-4-
one (250 mg, 0.733 mmoi, 1 .0 eq) in 3 mL of toluene was added mCPBA (31 5 nig, 1.83 maioi, 2.5 eq) and allowed to stir at n for 30 mm. Tert-butyl 4-(4-amino-2-chlorophenyl)pipefazine- 1 - carboxylate (227 mg, 0.733 mmoi, 1.0 eq) and DIPEA (378 mg, 2.93 mmoi, 4.0 eq) were added and allowed to stir at rt for i h. Progress of reaction was monitored by LCMS. After completion of reaction solvent was removed under reduced pressure, residue was diluted with 20 mL of water and extracted with ethyl acetate (50 rnl, x 3). Combined organic layer was washed with water (20 ml, χ 3), dried over anhydrous NajSGi and concentrated under reduced pressure. Crude was purified by reversed phase chromatography to obtain 140 mg (45.5%) of tert-butyl ··';· f - ohlo.: <>4 -( C( .0--d:i:hi : o i -e;:\ i }···;··; :χο··5.4 - d - i dro- 3; H?y; b -doj '7·ί··:: |! i ? io ;iz- ii- '';.
ylamino)phenyi)piperazine-l -earbox late.
LCMS; 605 [M-H j+
[0278] Step-2: Synthesis of 7~(3-chIoro-4-(piper¾Ein-l-yl)phenylami«o)~3~(2,6- dichlorophenyl)-2H-pyrimido[5,4-eI | ]oxazin-4(3H)-one: Teri-bnty! 4-(2~chioro-4-{3~(2,6- dichlorophenyl)-4~oxo~354-dihydro-2H~pynmido[5,4~e [l ,3 ]oxazin-7- ylamino)phenyl)piperazme -carboxyiate (200 mg, 0.33 mmoi, 1 ,0 eq) was dissolved m 5 mL of 4N H.C1 in dioxane solution at 0 °C. Reaction was stirred at rt for 30 mm. Progress of reaction was monitored by LCMS. After completion of reaction, precipitated compound was filtered off, washed with 2 ml, of dioxane and dried under reduced pressure. Crude was purified by reversed phase chromatography to obtain 20 mg (11 , 05%) formate salt of 7-(3-chloro-4-(piperazin-.l - yl)phenylammo)- --(2,6- iohlorop
LCMS; 505 [M-MX
¾ MR (400 MEte, DMSG-d6) <5 8, 86 (s, I H), 8, 25 (s, I H), 7, 55 ■■ 7.70 (ni, 3H), 7.51 (d, J 7 Hv Hz, IH), 7.15 (d, J - 8.77 Hz, IH), 5.75 (s, 2H), 2.91 (s, 8H).
Example S41. Synthesis of3-(2, 6-dichIorophenyIJ~7-f4-f2-oxopi/m^azin~]-yl)phenyiamim)J-2H- pyrimidofS, 4-ejfl.3 ]oxazin-4(3hI)~one
(Compound No, 1.41)
[0279] Step-1: Synthesis of tert-butyl 4~(4~((3-(2,6-dichloropheayI)~4~oxo-3,4-dihydro-
2Il
» yrimldoj5,y4"e] To a stirred solution
e][ l ,3]oxazm-4-one (300 nig, 0.934 rnmol, 1 .0 eq) in 4 mL of toluene was added niCPBA (401.86 mg, 2.336 mmol, 2.5 eq) and allowed to stir at rt for 30 nun. Tert-butyl 4-(4- aminopbenyl)-3-oxopiperazine-l-cai
'boxylate (326.3 mg, 1. 121 mmol, 1.2 eq) and DiPEA (482.24 mg, 3.78 mmol 4.0 eq) were added and stirred at rt for 1 hr, Progress of reaction was monitored by LCMS. After consumption of starting material, solvent was evaporated under reduced pressure. Residue was diluted with 30 mL of water and extracted with ethyl acetate (200 mL x 2). Combined organic layer was washed with water (50 mL x 3)., dried over anhydrous Na2S(>4 and concentrated under reduced pressure. Oude vvas purified by flash chromatography usnig MeOH : CH
2C¾ as eiuents to obtain 200 mg (38%)
LCMS: 585 [M÷l ]÷
[0280| Step-5: Synthesis of 3~(2,6-dichiorophenyI)-7-((4-(2-oxopiperazin-l- yl }phenyi)arajno)-2^-dihydro-4H-pyriraido [5,4-e] [ 1-3] ©xay.m-4-one: To the tert-butyl 4-(4-
((3-(2,6~dichloropbenyl)-4~oxo-3,4-dihydro-2H.-pyrimido[5,4~e][l ,3]oxa in-7-yi)amino)phenyl)-
3~oxopiperazine-.l -carboxy.late (200 nig, 0.340 mmol, I eq) was added 4N HC1 in dioxane (4
HiL). Mixture was stirred ai rt for 1 hr. Progress of reaction was monitored by LCMS. After consumption of starting material, precipitated compound was filtered off and washed with diethyl ether and dried under vacuum. Crude was purified by reversed phase chromatography to obtain 20 mg (12%) of 3-(2.6-dichlorophenyl)-7-((4-(2~oxopiperazin-3 -yl)phenyl)amino)-2,3-- diliydrO"4H-pyrimido[5,4~-e][] ,3 oxazin-4"One.
LCMS; 485 [M÷l f
¾ NMM (400 MHz, »MSO-d6); «5 10.50 (br s> 1.H), 8.86 (s, 1.H), 8.34 (br s, I H), 7.70 - 7.78 (m, J - 8.77 Hz, 2H), 7.66 (d, J - 8,33 Hz, 2H)S 7.49 (d, J - 7.89 Hz, Hi), 7.24 ~ 7.32 (m, J - 9,21 Hz, 2H), 5.75 (s, 2H), 3.58 (t, J - 5.04 Hz, 2H), 3.37 (s, 2H), 3.01 (d, J - 5.26 Hz, 2H).
Example S42, Synthesis of 3~(2, 6~dichlorophenylj~7~(3~(hydroxymeihyJj
yl)phenylamino)-2H~pyri ido[5, 4~e [l,3]oxa in~4(3E)-one
Compound No. 1.42)
[0281] Synthesis of 3-(2¾6-dichlorophenyl)-7-(3-(hydroxymethyl)-4-(i-methyipiperidin- 4-yl)pheaylain«uo)-2H-pyrimido{5,4-eI | l,3]oxa¾n-4(3H)~oae: To a stirred solution of 3 -(2,6- dichiorophenylV7- 3--(hydroxy
e][l,3]osazin-4 3H)~one (50 nig, 0.1 mmol, 1 .0 eq) in 3 mL of dichioroethane were added HCHO (0 025 mL, 0.3 mmol, 3 eq) and CH3COQH. (30 mg, 0.5 mmol 5eq) at 0 °C and allowed to stir at rt for 45 min. NaCNBIl? (18.9 mg, 0.3 mmol 3.0 eq) was added to the reaction mixture at 0 °C and stirred at rt for 20 min. Progress of reaction was monitored by LCMS. After consumption of starting material, the reaction mixture was diluted with NallCC , extracted, with
ί Π (Ί (20 nil, χ 2). The combined organic layer was washed with water (10 rnL χ 3), dried over aniiydrous a^SC^ and concentrated under reduced pressure. ( rude residue was purified by reversed phase column chromatography to obtain 20 mg (39.2%) of 3-(2,6~dichlorophenyl)~7~(3- (hydroxymeihy!V4-(l-methy
4(3H)-one as an off white solid.
*H NMI (400 MB'z, DMSO-d6): δ 10.33 (s, IH), 8.81 (s, IH), 7.55 - 7.82 (m, ' ! !}. 7.44 - 7.53 (m, IH), 7,22 (d, J - 8.33 Hz, I H ;. 6,56 (s, 2H), 5,65■- 5.82 (m, 2H), 5.13 (s, Hi), 4.52 i d. J - 4.38 Hz, M l ) 4.13 (s, I H), 3.16 (d, J - 4.38 Hz, 2H)S 2.86 (d, J - 1 1 .40 Hz, 2H)S 2.67 (s, 2H)S 2.19 (br s, 3H).
Example S43. Synthesis of 3~(3,5~dichIoropyiidin~4~yi)-7-(4~(4~me4 y piperazin~l~
yi}pheny amino)~2H-pyrimido[5, 4-e][l>3joxasin~4(3H)~one
(Compound No. L 43}
[0282] Step-1: Synthesis of 4-raethoxy-2-(methyItlu )pyriinidme-5-carboxylic add: To a stirred solution of etby! 4-cbloro-2-(niethyUhio)pyrimidine-5-carbox late (3, 0 g, 12,893 rnmol, 1.0 eq ), n MeOH (15 mi..) added .1 M soluiiors of NaOH (20 mL) at rt. The resulting mixture was stnred at the same temperature tor 3h, The reaction mixture wa concentrated and acidified with I N HQ solution ( 10 mL) to adjusted the pH 4-5, the ίοπ πΡ η of white precipitate was observed which was filtered and dried under vacuum to afford the desired oompound 4~nietboxy- 2-(rneihyltbio)pymnidine-5-carboxylic acid (1.71 g, 66.25%,) as white solid.
LCMS: 201.1 [M- l f
[0283] Step-2: Synthesis of -(3 "dk lor pyrld½"4"yi)-4-me oxy-2~
(methyithi )pyrimidiue~5~carhoxainide: To a stirred solution of 4~methoxy-2-
(methylthio)pyrimidine~5~carboxyhc acid (1 .7 g, 8,491 mmol. 1.0 eq), in C¾C¾ (50 ml.,) was added (C()C1)2 (1.45 mL, 16.982 mrno!, 2.0 eq) and DMF (0.01 mL) at 0 °C. The resulting
mixture was stirred at rt for 5h. I'he reaction mixture was concentrated, dissolved m€¾(¾ (5.0 rnL) and added to a stirred solution of 3,5-dichloropyridin-4-a.mine (1 .385 g, 8 491 mmol, 1.0 eq S and Nail (680 mg, 16.982 mmol, 2 0 eq) in DMF (30 ml,) at 0 °C. The resulting mixture was stirred a t ri for 2h, The progress of reaction was monitored by LCMS. The reaction mixture was poured in ice cold water (50 mL), extracted with EtOAc (2x 00 mL), the combined organic layers were washed with water (50 rnL), with brine (50 mL), dried over \ - SO.; concentrated and purified by flash chromatography [silica gel 1 00-200 mesh elution 0-30% EtOAc in hexane] to afford the desired compound N~(3,5-clichloropyridin-4-yj)-4-i'neihoxy-2- (methy!thiojpyrimidine-S-carboxamide (2.2 g, 75.06%) as off white solid.
LCMS: 34 9[M+l f
[0284] Step-3: Synthesis of -(3,5-dichloropyridin-4-yl)-4-hydroxy-2- (methyIthio)pyrimidiRe-5-carboxamidej To a stirred solution of N-(3,5-dichloropyridin-4-yl)- 4--meUioxy'-2-{niethylihio}pyrimidine -'Carboxamide (2.2 g, 6.373 mmol, 1.0 eq ), in ( 1 LOL (50 rnL) was added 1M BBr3 (19.119 mL, 19. 1 19 .mmol, 3.0 eq) at 0 °C. The resulting mixture was stirred at rt for 1 2h. The reaction mixture was basified with saturated sol tion of NaHCCL ( 100 mL). extracted with CILCL (2x50 mL), the combined organic layers were washed with water (50 rnL), with brine (50 mL), dried over N 2SC>4, concentrated and purified by flash
chromatography [Silica gel 100-200 mesh, e!ution 0-20% EtOAc in Hexane] to afford the desired compound N-(3 , 5-dichloropyri din -4-y{)-4-hydroxy-2-(meth ! thio)p rim idme-5- carboxamide (2,0 g, 94.78%) off* white solid.
[0285] Siep-4: Synthesis of 3-(3,5-dkhloropyridin-4-y!)-7-(raethy!thio)-2 dihydro-4H- pyrimido[5,4-e] [.l^joxa¾in-4-one: To a stirred solution of N-(3,5-dichioropyridin-4-yl)-4- hydroxy-2-(rnethylthio)pyi
'irnidme-5-cai
'boxamide (2,0 g, 6.039 mmol, 1 , 0 eq), m Q-BC (50 mL) was added ί >
.-CO . (5.9 g, 18. 1 17 mmol, 3.0 eq) at rt. The resulting mixture was stirred and purged with nitrogen for 10 mm. followed by addition of dhodomethane (0 97 mL, 12,078 mmol, 2.0 eq) at ri. The reaction mixture was heated at 90 °C for 48 h. The progress of reaction was monitored by LCMS. The reaction mixture was diluted with water ( 100 mL), extracted with EtOAc (2x 100 mL). The combined organic layers were washed with water (50 mL), with brine
(50 mL), dried over
concentrated and purified with flash chromatography [Silica gel 100-200 mesh; elution 0-20% EtOAc in hexane] to afford the desired compound -{.·!. · dichloropyridm-4~yl)-7-(meihyk^^ (0, 300 g,
14.47%,) as off
* white solid.
[0286] Step-5: Synthesis of 3-(3,5-dkhIoropyridin-4-yI)-7-((4-(4-methyipiperazin-i - yl)phenyl)amino)-2 -dihydro-4H-pyrimido [5,4-e] ( 1 ,3) oxazin-4-one: To a stirred solution of 3-(3,5--dichloropyndi n-4-y V7- met^^
(0.1 00 g, 0.291 mmol, 1 .0 eq), in toluene (5.0 mL) was added mCPBA (0. 100 g, 0,582 mmol, 2.0 eqj at rt. The resulting mixture was stirred at rt for 10 mm. followed by addition of and 4-(4- methy!piperazin-1 -yljanilme (0.061 g, 0.320 mmol, 1.1 eq) and DIPEA (0.200 mi . 1 , 164 mmol, 4.0 eqj. The reaction mixture was stirred at rt for 30 mm. The progress of reaction was monitored by LCMS. The reaction mixture was diluted with water (100 mL), extracted with EtOAc (2 χ 100 mL). The combined organic layers were washed with water (50 mL), with brine (50 mL), dried over NaiSOd concentrated and purified by reversed phase chromatography to afford the desired compound 3-(3,5-dichlofopyridm-4-yl)-7-((4-(4-methylpipefazin-l-yl)phenyl)aniino)-2!3- dihydro-4H-pyfimido[5,4-ej [l ,3]oxazin-4-one (0.0026 g, 18.44%,} as a yellow solid.
LCMS; 486.2[M- l j+'
¼ NMR (400 MI¾ DMSO-d6): δ 10 35 (br s, 1H), 8.78 - 8.87 (m, 3H), 7.55 u!. J 6.58 Hz, 2H), 6.95 (d, J === 8.77 Hz, 2H), 5.79 (s, 2H), 3,22-3.10 (m, 4H), 2.75-2.65 (m, 4H), 2,39 (br s, 3H).
2H~pyrimido[5, 4~ej[L 3joxazm-4(3B)-one
(Compound No. 1.44)
[0287] Step- 1 : Synthesis of tert-butyl 6~((3~(2,6-dic orophenyI)-4~oxo-3,4-dihydro-2H- pyrimido[5,4-e][1 ]oxazm-7-yl)amino)-3,4-dihydroisoquinoIme-2( To a stirred solution of · θ · ·ϋ:^ η !θ: ίηί i } -" -(n:;;th> i; i:io}- 3-dd:vd; o-4j | - s nnud i 5.4 · e][ l ,3]oxazm-4-one (250 mg, 0.730 rnmol, 1 .0 eq) in 4 mL of toluene was added mCPBA (314 mg, 1.827 rnmol, 2.5 eq) and allowed to stir at rt for 30 mm, Tert-butyl 6-a.rmno-3.4-- dihydroisoqtmioline~2(lHS--carboxylate (217 mg, 0.877 mmol, 1.2 eq) and DIPEA (377 mg, 2.923 mmol, 4.0 eq) were added and stirred at rt for 1 hr, Progress of reaction was monitored by LCMS. After consumption of starting material solvent was removed under reduced pressure. Residue was diluted with 20 mL of water and extracted with ethyl acetate (200 mL >< 2).
Combined organic layer was washed with water (50 mL
χ 3), dried over anhydrous Na?S(>4 and concentrated under reduced pressure. Crude was purified by flash chromatography using MeOH: CH
2C!
2 as eluents to obtain 180 mg (37.87%) tert-butyl
dihydro~2H-pyrinudo]'5,4-e]ri,3 oxazin-7~yl)am
LCMS: 543 [M÷l]÷
[0288] Step-2: Synthesis of 3-(2,6-dkh!orophenyl)-7-((l,2^, -tetrahydroisoquinolin-6~ yl }amino)-2;3-dihydro-4H-pyriitnido[5,4-e] [ 1 ,3]oxazin-4-one; To the tert-butyl 6-((3~(2,6- dicldoropheriylV4-oxo-3,4~dihydro~2H^
dihydroisoquinoline~2(lH)-carboxylate (150 nig, 0.276 mmol, 1 eq) was added 4 N HC1 in dioxane (3 ml,) Reaction mixture was stirred, at rt for 1 hr. Progress of reaction was monitored.
by LCMS. After consumption of starting material, precipitated compound was filtered off and washed with diethyl ether and dried under vacuum to obtain 35 mg (28%) of ? ·( 2 ··
d:-.b!o: o he:p π·· ··{{ i
e] [ 1,3 ] oxazm-4»one.
LCMS: 443 [M÷l]÷
\H NMR (400 MHz» DMSO-d6): δ 10.49 (br s, 1H), 9.10 (br s, 2H), 8.87 (s, !H), 7.54 - 7.81 (mt 4H), 7.46 - 7.54 (m, 1 H)S 7.20 (d, J ------ 8.33 Hz, IHj, 5.75 (s, 21 ! ;. 4.23 (br s, 2H), 3.33 (br s, 2H),
3.00 (br s, 2H).
Example S45. Synthesis o/3~(2, 6~dichIorophem!0-7-(4' -(4- ^droxypipe?id^
2H~pyrimido[5, 4-ejfI, 3]οχ ήη~4(3Η)-οηβ
(Compound No. 1, 45)
STEP-3
[0289] §tep-l: Synthesis of l-(4-mtropheayi)piperidin-4-o!: To a stirred solution of 1 - fJuoro-4-nitrobenzene (3 g, 21, 27 mrnol . 1 eq) and piperidin-4-o! (3.2 g, 31,91 mmoL i .5eq) in DMF (20 ml,) was added
.:( <) ·. (3 8 g, 27.65 rnmof 1 .3 eq). Reaction mixture was stirred at 85
°C for 1 hr. Progress of reaction was monitored by LCMS. After consumption of starting material, reaction mixture was poured on ice cold water and precipitated compound was filtered off and dried under vacuum. Solid was washed with diethyl ether and pentane to obtain 4 g (84.74%) of l-(4-nitrophenyj)piperidin-4-ol.
[0290] Step-2: Synthesis of l~{4~ammephenyl)pipi>ridin~4~ i: To a stirred solution of 1 ~(4~ mtropheny{)piperidin~4~ol (3 g., 13, 51 mmo!, 1 eq) in EtOH (50 txiL) was added Fe(0) (4,45 g, 81.08 m ole, 6eq ) and I4C1 (5.72 g. 108. 10 m mole, 8eq) solution m water (50 mL) Reaction mixture was stirred at 90 °C for 1 r. Progress of reaction was monitored by LCMS. After consumption of starting material, the reaction mixture was filtered through cehte bed. Filtrate was concentrated under reduced pressure. Residue was diluted with 50 mL of water and extracted with ethyl acetate (200 mi, χ 2). Combined organic layer was washed with water (50 mL v- 3), dried over anhydrous Na2SC> and concentrated under reduced pressure to obtain 2 g (80%) of I--(4--anunophenyi)piperidin- --ol.
LCMS: 193 [M+l f
[0291] S ep-3: Synthesis of 3-(2,6-dkhIorophenyl)-7-((4-(4-hyclroxyp«peridin-l- yl)phenyl)mniuo)-2 "d«hydro-4H-pyriffiido(5,4~e]II^]ox^in-4-one: To a stirred solution of 3-(2,6~dich!orophenyl)-7~(met ylthio)-2,3-dihydro-4H-pyrimido[5,4 (250 nig, 0.730 nimol, 1 0 eq) in 5 mL of toluene was added mCPBA (314.3 mg. 1.827 rnmol, 2.5 eq) and allowed to stir at rt for 30 min. 1 -(4-aminopheny})piperidin-4-oI (168 mg. 0.877 rnmol, 1 .2 eq) and DIPLA. (377 mg, 2.92 mmol, 4.0 eq) were added and stirred at rt for 1 h. Progress of reaction was monitored by LCMS, After consumption of starting material, solvent was removed under reduced pressure, 20 mL of water was added and extracted with ethyl acetate (200 ml, >< 2). Combined organic layer was washed with water (50 ml, >< 3), dried over anhydrous a^SC^ and concentrated under reduced pressure. Crude was purified by reversed phase chromatography to obtain 50 mg ( 14.08%) of 3-(2,6-dichlorophenyl)-7-((4-'(4-'hydroxypiperidin-l - yl)phenyl)amino)-2,3-dihydfo-4H-pyrimido 5.4-e][l ,3]o.x:azin-4-one.
LCMS: 486 [M÷l f
*11 MR (400 MHz, DMSQ-d6): δ 10.24 ( br s, ΓΗ), 8.78 (s, 1H), 7.65 (d, J ==== 7.89 Hz, 2B), 7.51 (d, J - 7.89 Hz, 3H), 6.92 (d, J - 8 77 Hz, 2H), 5.71 (s, 2H), 4.67 id. J - 4.38 Hz, HI), 3.61 (dt, J = 4.38, 8.55 H/.. I K), 3.48 (d, J - 12 72 H/.. 2H), 2.79 (i, ../ - 10.09 Hz, 21:1), 1.81 (d, ,/ = 9.21 H/.. 2H), 1 37 - 1 .55 (m, 2H)
Example S46. Synthesis of 7~(4~(4~(2~aminoacetyl)piperazin-l~yl)phenylamino)-3-(2, 6- dichlorophenyl)-2hI~pynmido[5, 4~e [ L3 oxazin~4(3H)~one
(Compound No. 1.46)
[0292] Siep-l: Synthesis of tart-butyl 2-{4-{4-(3-(2,6-dkhlor phenyi)-4-oxo-3,4-dihydro- 2H-pyrimidoj5,4-e] [l^]oxaz-«-7-ylami«©)phe«y ^ To a stirred solution of 2-(tert-butoxycarbonylai'nmo)acetic acid (104 mg, 0.5940 mmol, 1 .0 eq) in 4 rnL of DMF was added BATIJ (338.7 mg, 0.8910 mrnoL 1.5 eq) and allowed to stir ai rt for 30 min. A,
(0.25 rnL, 1 .485 mmol, 2.5 eq) and
(4-(piperazin-l-yl)pheny½mino)-2H-pyrimido[5,4-e] L3]oxazin~4(3H)-one (280 mg, 0.5940 mmol 1.0 eq) were added to the reaction mixture and stirred ai ri for 5 h . Progress of reaction
was monitored by LCMS. After consumption of starting material the reaction mixture vvas poured into ice cold water. The precipitate obtained was filtered and dried to afford tert- butyl 2-
y!amino)phenyi)piperazm"I"yl)--2"Oxoeihylcarbamaie (273 nig, 73.19 %) as a greenish solid. LCMS: 628 [M÷l f
[0293] Step-2: Syn esis of 7-{4-i4-i2-aminoaeety!)piperazm-l-yi)pheny!amino)-3-{2,6- dkhIorophenyl)~2H~pyrimido[5,4-e] [ i ] oxa¾in-4(3H)-one. To a stirred solution of ten-butyl 2"(4-(4-(3-(2.,6--diehiorophenyiM^
ylarniiio)phenyl)pipera2;in-l -yi)-2-oxoethy]cai'barnat (100 g ) in 1 mL of 1 ,4-dioxane was added the solution of 4N HQ in 1,4-dioxane (2 mL) drop-wise and allowed to stir at rt for 30 min. Progress of reaction was monitored by LCMS. After consumption of starting material, the precipitate obtained was filtered and washed with diethyl ether to obtain the crude. Crude was puniled by reversed phase column chromatography to obtain the desired compound. 7-(4-(4-(2- anmioaceiyl)piperazin- f-yi)phenyiamino^
oxazin- (3H) -one (25 mg, 29.7%) as an off white solid.
LCMS: 528 [M÷l ]÷
1Ά m (400 MHz, »MSO-d6): «5 10.25 (br s, IH), 8.79 (s, 1 H), 7.65 (d, J = 8.33 Hz, 2H), 7.53■■ 7.61 (m, J === 8.33 Hz, 2H), 7.45■■ 7.53 (m, ill), 6.91■■ 7.06 (m, J === 8.77 Hz, 2H), 5.71 (s, 2H), 3.62 (br s, 2H), 3.51 (br s, 2H), 3.4 1 (s, 2H), 3.09 ( br s, 4H), 1 .90 (s, 2H).
Example S47. SyrHhesis o/'J-f 6~dichiorophe 7yi)~7~{4~({2R,5S)~2.5~di eihyipipe zin~I~ yl)phenyiammo)~2 i~pyrimido[5, 4-e fl, 3f' oxazin-4(3H)~one hydrochloride
(Compound No. 1.47)
[0294] Step-1: Synthesis of tert-butyl (2R>5S)-2>5~climethy!-4~(4-nitiOpheayl)piperazine- 1-carhoxylate: To a stirred solution of I~fli3oro~4~mtrobenzene (1 g, 7.08 mmol, 1 eq) and piperidin-4-ol (1.56 ;·;!..7.08 mmol, 1.0 eq) in DMSO (12 rnL) was added K2C03 (1.95 g, 14.16 rmnol, 2.0 eq). Reaction mixture was stirred at 85 °C for 6h. Progress of reaction vvas monitored by LC7MS. After consumption of starting material, ice cold water was poured to the reaction mixture. The formation of precipitate was observed which was filtered to afford the desired compound, tert-butyl · 2k.5SrC. · duuoih. l-4-(4-:}ijr;:ph::r:> ! ; :; ο:·>π:ο·· I ··:;;:: bo.-o laic (840 mg, 35.44%) as a yellow solid.
LCMS: 336.2[M÷1]÷
[0295] Step-2: Synthesis of tert-butyl (2 ,5S)-4-(4-aminophenyi)-2,5- dimethylpiperaxine-'l-carboxylate: To a stirred solution of tert-butyl (2R,5S)-2,5-dimethyl-4-
(4-nitrophen T)piperazine- 1 -carboxylate (840 nig, 2.50 mmol, 1 eq) in EtOH and water (1 : 1 , 20
· :■! . · was added Fe(0) (420 mg, 7.51 mmol, 3 eq) and NLLCl (267 nig, 5.00 musol, 2 eq). The reaction mixture was stirred at 90 °C for 2h. Progress of reaction was monitored by LCMS, After consumption of starting material the reaction mixture was filtered through ceiite bed. The reaction mixture was concentrated, diluted with water, basified with NaHCCL solution and was extracted with EtOAc, The organic layers were dried over a?S04, concentrated and purified by flash chromatography (elutiom 0-2% MeO'H in C!¾C } io afford the desired product tert-butyl f2R,5S)-4-(4-arninophenyj.)-2,5-dimethylpiperazine-l.-carboxylate (472 mg, 61.70%) as an off white solid.
LCMS: 306.4 [M+l]+
[0296] Siep-3: Synthesis of tert-butyl (2S,5 H-(4-((3-(2,6-dkhiorophenyI)-4-oxt -3,4- dihydro~2H~pyrhnido [5,4-e] [ 1 ,3] oxazin-7-yI)ainino)pheny l)-2,5-diinethyIpiperazme-l - carboxylate: To a stirred solution of 3-(2,6-dichioiOphenyl)-7-(methylthio)-2,3-dihydro-4H- pyrimidoj
"5,4-e1 [ l,3
"joxazin-4-one (481 nig, 1 .404 mmol, 1.0 eq) in 5 mL of toluene was added m-CPBA (485 mg, 2.808 mmol, 2.0 eq) and allowed to stir at i for 30 mm. T rn h I (2R,5S)- 4-(4-an nophenyl )--2,5-'dimethylpiperazine-'l -'Carboxylate (472 mg, 1.545 mmol, 1 . 1 eq) and DIPEA (0.97 mL, 5.616 mmol, 4.0 eq ) were added and stirred at rt for lOh. The formation of precipitates was observed which was filtered to afford the desired compound, tert-butyl - 2S 5R -- 4-(4-((3"{2,6-dichlorophenyl)-4-oxo-3,4-dihydro-2H-pyrimido[5,4-e][l ,3 oxazin-7- yijamino)phenyl)-2,5~dimethyipiperazine--Lcarboxylate (21 5 mg. 25.51%) as an off white solid.
[0297] Step-4: Synthesis of 3-(2,6-dkhloropheRyl)-7-((4-((2R,5S)-2,5-dimethyIpiperaz«n- i-yl)plH¾nyI)anu»H>)-2 -dihydro-4H-pyriniido[5,4-e] [1 ^]o.xaziR-4-one hydrochloride: Tert- butyl (2S,5R)~4-(4-((3-(2,6-dichloropheny
7-yj)amino)phenyl)-2,5-dimethy]pipei'azine-l -cai"boxylate (50 mg, 0, 136 mmol, 1 ,0 eq) was dissolved in (1 mL) of dioxane and added 4M dioxane-HC! (2 mL) and allowed to stir at rt for 1 hr. After completion of reaction, the reaction mixture was filtered and dried under reduced
pressure io afford the desired compound, 1 ·(.'.:/:··· lieh! ; pi -cnv! }■■"'■■{ i -M · 2K.5H ··2.5· dimethylpipefazin-l -yi)phenyl)ammo)-2!3-dihydro-4H-pyfimido 5,4-eJ
hydrochloride (33 mg, 73.69%) as an off white solid.
\H NMR (400 MHz, DMSO-d6): δ 10.43 (b.r s, 1 H), 9.12 (br s, 1H), 8.84 (s, 2H), 7.66 (d, J - 8.33 Hz, 2B), 7.69 (d, J --= 8.77 Hz, I B), 7.50 (i, J ==== 7.89 Hz, I B), 7.1 3 (d, J === 8.33 Hz, 2H),
5.74 (s, 2H), 3.20 (d, J ------ 11.84 Hz, 2H), 2.85 (br s, 2H), 1 .23 (d, J - 6.58 Hz, 3H), 0.91 (d, J -
6.14 Hz, 3H).
Example S48. Synthesis qf3~(2!6~dichIorophem4)-7-(4-(2~(meihyhmm
2H~pyrimido[5, 4-ejfI, 3 oxazin~4(3H)-one
(Compound No. 1, 48)
[0298] Step-J : Synthesis of tert-butyi (2-hydr0xyethy.)(methy.)carbamate: To a stirred solution of 2--(methylarnino)ethan--Lol ( 1.0 g, 1 3.313 mmol, 1.0 eq), in CI C
ri ; (50 mL) was added di-tert-butyi dicarbonate (3.36 mL, 14.645 mmol, 1 . 1 eq) ai 0 °C The resulting mixture was stirred at rt for I hr. The progress of reaction was monitored by 'HNMR. The reaction mixture was diluted with C¾(¾ (50 mL) and washed with water (50 mL) and bri ne (50 ml) dned over a^SO,^, fil ered and concentrated to afford the desired compound, tert-butyi (2- hydroxyetliyl)(methy!)carbamate (2.30 g, 98.71%) as colorless liquid.
*H NMR (400 MHz, CiK. ;: δ 3.75 (d, J - 4.82 Hz, 2Hj, 3.40 (br s, 2H), 2.92 (s, 3H), 2.82 (br s. M l ). 1 .46 (s. 9i;i).
[0299] S ep-2: Synthesis of tert-butyi methyl(2-(4-nitrophenoxy)ethyl)carbaraate: To a stirred solution of 4-nitropb.enol ( 1.0 g . 7.188 mmol, 1 .0 eq), tert-butyi (2- hydroxyethyl)(meihyl)carbama.te ( 1.51 g . 8.626 mmol, 1 .2 eq) and triphenyl pbosphine (2.26 g, 8.626 mmol, 1.2 eq) in THF (100 mL) was dropwise added a solution of DIAL) (1.69 mL, 8.626 mmol, 1 .2 eq) in THF ( 1.0 mL) at 0 °C. The resulting mixture was stirred ai rt for 12h. The progress of reaction was moniiored by LCMS. The reaction mixture was concentrated and purified by combiflash [silic gel 1 00-200 mesh, elution 0-10% EtOAc in hexane] io afford the desired compound ten-butyl methyl (2-(4-nitrophenoxy) ethyl) carbamate (0.510 g, 23.94%) as yellow viscous.
LCMS; 297.2 ΓΜ÷1]÷
[0300] Step-3: Synthesis of tert-butyi (2-{4-arainophenoxy) ethyl) (methyl) carbamate:
To a stirred solution of tert-butyi methyl(2-(4~nitrophenoxy)ethyi}carbamate (0.500 g, 1 687 mmol, 1 0 eq) in EtOH (25 mL) was added Fe(0) (0.753, 13.498 mmol, 8.0 eq) and a solution of NH4CI (0.90 g, 16 87 mmol, 10.0 eq ) ai it. The resulting mixture was heated at 90 °C for 60 min The progress of reaction was monitored by LCMS. The reaction mixture was filtered through celite the residue was washed with EtOH (50 mL) the filtrate was concentrated and the residue was dissolved in EtOAc (50 mL), washed with water (2 50 mL), dried over
and concentrated to afford the desired compound tert-butyi (2-(4-a.nunophenoxy)etbyl) (methyl) carbamate (0, 270 g, 60.1 3%) as yellow viscous oil.
[0301] S ep~4: Syn esis of tert-butyl (2-(4-((3-(2,6-dkhlorophenyI)-4-oxo-3,4-dihydro-
2Il»p .rhiildoj5,y4"e] L^^ To a stirred solution of 3-(2,6~dichloi pbenyl)-7-(methyl^^^
one (280 mg, 0.818 mmoL 1 0 eq) in (10 ml,) of toluene was added m-CPBA (400 nig, 1 .636 mmol, 2,0 eq) and allowed to stir at rt. or 30 mm. Tet -butyi (2-(4-aminophenoxy jethyl)
(methyl) carbamate (261 mg, 0.981 mmol, 1.2 eq) ami DIPEA (0.570 mL, 3 272 rnmol, 4.0 eq) were added and allowed to stir at rt 12h. The reaction mixture was diluted with water (50 ml,) and extracted with EtOAc (2 >< 50 mL), The combined organic layers were washed with water (50 mL) and brine (50 mL), dried over Na.2S(>
4, filtered, concentrated and purified by flash chromatography[silica gel 100-200 mesh: elution 0-30% EtOAc in hexane] to afford the desired compound tert-butyl (2-(4-((3-(2,6-dichlorophenyl)-4-oxo-3,4-dihydro-2H-pyrimido[5,4- e][l,3]oxazin~7~yl)amino)phenoxy)etliyl)(meihyl)carbamaie (250 mg, 54,58%) as brown solid.
[0302] Step-5: Synthesis of 3-(2,6~dkhIorophenyl)-7-({4-(2-
(methyiattM«o)ethoxy)pheoyi)amioo)-2 -dihydro-4H-pyrimido|S,4-e] [1 ]οχ¾ε.«-4-οηβ:
Tert-butyl ( ·(4 ·(( L O-ndnoio bem I ;.·4·» ,\<> ?yi-dih dro--2i Ι- ^ : i!- doj "\4-- .:ji j ,:-/ : 7. yl)amino)phenoxy) ethyl) (methyl) carbamate (250 mg, 0.444 mmol, 1.0 eq) was dissolved m 4M HQ in dioxane (4.0 mL) and allowed to stir at rt for 1 h. After completion of reaction, the reaction mixture was concentrated under reduced pressure and purified by reversed phase purification to afford the desired compound 3-(2,6-dichJorophenyl)-7-((4-(2-
nig, 48.78%) as white solid.
LCMS; 460.1 ΓΜ÷1]÷ i NMR (400 MHz, DMSO-d«): δ 10.31 (br s, IH), 8.81 (s, IH), 8.31 (br s, I H), 7.57 ·· 7.69 (m, 4H)S 7.45■- 7,55 (m, IH), 6.96 (d, J - 8,77 Hz, 2H), 5,72 (s, 2H), 4.08 (br s, 3H), 3.00 (br s, 2H)S 2.44 (br s, 3H).
Example S49. Symthesis of(R)~3~(2, 6~dichiorophenyl)-7~(4~(2~methyipiperazbi~l~ yiphenyiam in oj- 2H-pyrim Mof 5,4~e/f 1 ,3/'ox zin~4(3H)~one
(Compound No.1.49)
[0303] Synthesis of (R)-3-(2,6-dichlorophenyl)-7-(4-(2-j(nethylpiperaz-n-i-
(R)-3-(2,6-dichlorophenyl)-7-(4- (2·:·;·;ϋ·ν φ::Η;Γ;· !·:· 1
•• Π
ηηοο h: · o i : · ; · ! I · | >\ Γ- π · ;· I· ί i "
'·. -i · c i i i 3 lox;: ·:· --ΙΠ Π }··· · ;·; was separated from the product synthesized in the scheme described m Example 826.
Example S50, Synthesis / 3-43.6-dicMorophenyl)~7~(3~methoxy-4~(pipendin~4~yl)pheny 21i~py nido[5,4~e] [ L3]oxa∑in-4(3H)~one
(Compound No.1.50)

[0304] Step-ί : Synthesis of tert-butyl 4-(2-methoxy-4-aitrophenyl)-3,6-dihydropyridioe- l(2H)-carboxylate: To a stirred solution of 1 romo~2-metnoxy-4-nitrobenzene ( 1.0 g, 4.310 nmso!, 1 .0 eq) and tert-butyl 4-(4,4,5,5-tetrainethyj-l,3,2-dioxaborolan-2-yl)-3,6- dihydropyridine-l(2HVcarboxylate (1.46 g, 4.740 mmoi, 1.1 eq) in dioxane (30 ml,) was added a 2M solution of Na^CC (913 nig, 8.620 mmoi, 2.0 eq) at ri. The resulting mixture was purged w th nitrogen for 10 min, followed by addition of Pd(dppf)(¾. ( I LCL (352 mg, 0.431 mmoi, 0.1 eq). The resulting mixture was heated at 90 °C for 12 h. The progress of reaction was monitored by 'HNMR. The reaction mixture was filtered through ceiite the residue was washed w th EtOAc (50 mL) the filtrate wash concentrated and purified by flash chromatography [silica gel 100-200 mesh, elution 0-10% EtOAc in hexane] to afford the desired compound tert-butyl h (2-ra thoxy-4-nitrophenyi)-3,6-dihydropyridine-] (2H}-carboxylate (1.12 g, 77.77%) as brown iSCOUS.
*11 MR (400 MHz, OX¾): δ 7.82 (dd, J ==== 2.19, 8.33 Hz, I B), 7.72 (d, J === 2.19 Bz, I B), 7.28 (d, ./ - 8.33 Hz, 1H), 5 88 (br s, 1H), 4.07 (br s, 2H), 3.61 (t, ./ - 5.26 Hz, 2H), 2 49 (br s, 2H), 1.45 - 1 53 (m, 9H).
[0305] Step- 2: Synthesis of tert-butyl 4-(4-amino-2-n»ethoxyphenyl)piperidine-l- carboxylate: To a stirred solution of tert-butyl 4-(2-methoxy-4-ni†rop.h.enyl)-3,6- dihydropyridine-1 (2H)-carboxylate (1 1 g, 3.289 mmoi, 1.0 eq) m MeOH (30 niL) was added 10% Pd/C ( 100 mg) at rt. The resulting mixture was purged with hydrogen for 60 min. The progress of reaction was monitored by LCMS. The reaction mixture was filtered through ceiite ihe residue was washed with MeOH (50 mL) ihe filtrate was concentrated and purified by pentane to afford the desired compound tert-butyl 4-(4-amino--2--methoxyphenyl)piperidine~l - carboxylase (0.760 g, 82.96%) as brown solid.
LCMS: 307.3 [M- l f
[0306] Step-3: Synthesis of tert-butyl 4-(4-((3-(2,6-dic orophenyI)-4-oxo-3,4-dihydro- 2H-pyrimido [ 5,4-e) [13.1 oxaz«a-7-yl)ara«ao)-2-niethoxypheny!)piper«d«ae- 1-car boxylate: To a stirred solution of 3-(2,6~d.ichlorophenyl)-7-(methylthio)-2,3-d.ihydro-4H-pyrimido[5,4- e~j[] ,3]oxazin-4-one (200 mg, 0.584 mmoi, 1 .0 eq) in ( 10 mL) of toluene was added mCPBA (201 mg, 1.168 mmoi, 2.0 eq) and allowed to stir at rt for 30 min, Tert-butyl 4-(4-amino-2- methoxyphenyl)piperidme-l -carboxylase ( 197 mg, 0.642 mmoi, 1 1 eq) and DIPEA (0.406 ml,,
2.336 nimo!, 4.0 eq) were added and allowed to stir at rt tor 12 h. The reaction mixture was diluted with water (50 mL) and extracted with EtOAc (2 x 50 mL), the combined organic layers were washed with water (50 mL) with brine (50 mL) dried over Na^SO^, filtered and
concentrated and purified by flash chromatography [silica gel 100-200 mesh; elution 0-30% EtOAc in hexane to afford the desired compound tert-butyl 4-(4-((3--(2,6-diohlorophenyl)-4-oxo- 3,4-dihydi ¾-2IT-pyrimido[5J4-e][l,3]oxazin-7-yl)arnino)-2-methoxy phenyl ) piperidine-1 - earboxylaie (1 52 mg, 43,42%) as off white solid.
LCMS: 600.2[M÷1]÷
[0307] Step-4: Synthesis of 3-(2¾6-dichlorophe«yl)-7-((3-methoxy-4-(piperid«i-4- yl)phenyl)affiino)-2 -d«hydro-4H-pyriffiido[5,4-e|II^]ox^in-4-one: tert-butyl 4-(4-((3-(2,6- dichioropheny!)--4--oxO"3,4~diliydro--2Ii"pyrimido[5,4--e][l ,3]oxazin-7-yl)amino)-2~methoxy phenyl) piperidine-l-carboxylate (152 mg, 0.249 mniol. 1.0 eq) was dissolved in 4.0 M-HC1 in dioxane (20.0 mL) and allowed to stir at rt for 1 hr. After completion of reaction, the reaction mixture was concentrated under reduced pressure and purified by reversed phase
chromatography to afford the desired compound 3-(2,6-dichlorophenyl)-7-((3-methoxy-4- (piperidin-4-y})phenyj)ammo)-2,3~dihyd (32 mg,
*11 MR (400 MHz, DMSO-de): δ 10.40 ( br s, Hi), 8.85 (s, 1H), 7.66 (d, J ==== 7.89 Hz, 2H), 7.51 (t, J - 8.33 Hz, 1H), 7.45 (br s, I I I }. 7.35 d, ./ - 8.33 Hz, i l l) 7.09 (d, ./ - 7.89 Hz, 1H), 5.75 (s, 2H), 3.79 (s, 2H), 3.08 (br s. 2H), 2 96 ft, J - 1 1.18 Hz, 2H), 1 .79 - 1 .88 (m, 2H), 1.65 - 1.79 (m, 2H).
.Example Λ Synthesis of 3~(2,f>-di hlorophenyT)~7-(2',3'-dihydro-i 'H~spirofcyc!oprop f7e~ 1 ,4 - isoqwmiUne -7'~yIa ino}-2H~pynmido[5, 4-e/fl, 3]oxazm~4(3H)-one
[0308] Step-1 : Synthesis of tert-butyl 7'-(3-(2,6-dkhioropheayi)-4-oxo-3,4-dihydro-2H- pyrtmido[5,4-e] | ί ^]oxa2m-7-ylammo)-l'H-spiro cydopropaae-'l ,4'-isoquinoime]-2'(3'Il)- carboxylate: To a stirred solution of 3~(2,6-dichlorophenyl)-7-{meihylthio)-2H-pyrimiclo[5,4-' e|[l,3]oxazin-4(3H)"One (300 mg, 0,879 mmol, i .O eq) in 5 rnL of toluene was added mCPBA (378 mg, 2,20 mmol, 2.5 eq) and allowed to stir at rt for 30 mm. Tert-butyl 7--ammo- lTi- spifo[cyclopropane-l ,4'~isoquinoline]'-2,(3'H)-cafbo.x;ylate (241 mg, 0.879 mmol, 1.0 eq) and DIPEA (453 nig, 3.52 mmol, 4.0 eq) were added and allowed to stir at rt for 1 h. Progress of reaction was monitored by LCMS After completion of reaction solvent was removed under reduced pressure, residue was diluted with 20 ml, of water and extracted with etliyl acetate (50 ml, v- 3). Combined organic layer was washed with water (20 ml, χ 3), dried over anhydrous a.2S(>4 arid concentrated under reduced pressure Crude was purified by flash chromatography to obtain 200 mg (40.1 %) of tert-butyl 7^ ;H2.6"d:chi; :ropi: nv! )- :0 o >-- , ! -d : ο - 2\ ) ·· pyfimido[5,4~e]n,3 ]oxazin-7-ylammo)-l ^
carboxylate.
LCMS; 568 [M+l f
[0309] Step-2: Synthesis of 3-(2,6-dkhlorophenyl)-7-(2*3'-dihydro-i Ή- spirofoycIopropane- '-isoquiaoHne]^
ylaminoH I-spiro [cyclopropane- l ,4'-isoquinolm ]-2!(3' )-carboxylate (200 mg, 0,353 mmol,
1 .0 eqj was dissolved in 5 mL of 4N HCl in dioxane solution at 0 C. Reaction was stirred at rt for 30 mi Progress of reaction was rnomtored by LCMS. After completion of reaction, precipitated compound was filtered off washed with 2 mL of dioxane and dried under reduced pressure. Crude was purified by reversed phase chromatography to obtain 50 mg (36 40%) of 3~ (2J6--dich!orophenyi)--7--(2i :"dihydro^
pyrimido[5,4-e] [I,3 Qxazin~4(3H)-one.
LCMS; 468 [MH j+
¾H M (400 MHz, DMSO-de): S 10.30 (br s, IH), 8.81 (s, I H), 7.65 (d, J ==== 8.33 Hz, 2H), 7.46 - 7.60 (in, 2H), 7.43 (d, J - 8.33 Hz, IH), 7.36 (br s, IH). 6.70 (d, J - 8.33 Hz, I H), 5.72 (s, 2H), 3.91 (s, 2H), 2.74 (s, 2H), 1.88 (s, 2H), 0.89 (br s, 2H), 0.79 ( br s. 21:3).
Example S52. Synthesis of3-{2, 6~dichhrophe i)~7~(4~(]~methyljnp
2H-pyri ido[5, 4-ejfI, 3]oxazin-4(3H)-one
(Compound No, 1.52)
[0310] Step-ί : Synthesis of tert-butyl 4-(4-((3-(2,6-dkhiorophenyI)-4-oxo-3,4-dihydro- 2J7~py rimido [5,4-ej ( 1 ,3] oxa«in-7-yi )amino)pheny i)piperidine~ 1-car boxylate: To a stirred solution of 3--(2,6-dichiorophenyly7-(mefo^
(700 mg, 2.04 mmol, 1 ,0 eq) in 6 mL toluene. «--CFBA (879 nig, 5.11 mmol, 2.5 eq) was added under stirring and resulting mixture was allowed to stir at rt for 30 mm. Further, tert-butyl 4-(4- aniinophenyl)piperidine-3 -earbo.x:ylate (565 mg, 2.04 mmoi, I eq) and DIPEA (1.075 g. 8.16 mmol, 4 eq) were added and the reaction was allowed to stir at rt for 1 2 h. The reaction was monitored by LCMS. After completion reaction was quenched with water and extracted with ethyl acetate (1 0 ml, x 3). The combined organic layer was washed with brme solution (20 mL), dried over anhydrous Na2SC> and concentrated under reduced pressure to afford crude product which was purified by flash chromatography (elut n: 0-10% MeOH in f 1 hO ; to afford the desired compound, tert-butyl 4~(4~((3~(2,6-dichlorophenyl}-4-oxo-3y -dihydro-2f7-pyrimido[5,4- e] [I,3]oxazin~7~yl)amino)phenyl)pipendine-Lcarboxylate (300 mg, 26%) as a yellow solid,
[0311] Step-2: Synthesis of 3-{2,6-dichIorophenyl)-7-(i4-{piperidin-4-yl)phenyI)aniino)- 2^dihydro-4J
E/-pyrii do[5,4-e] |Ί .3 jo a2hi-4-one hydrochloride: 20% HC1 in dioxane (2 mL) was added to a stirred solution of tert-but l 4-(4-((3 -(2,6-dichlorophenyl)-4-oxo-3,4-
(300 mg, 0.52 mmol, 1 .0 eq) at 0 °C and the resulting solution was allowed to stir at rt for 3 h. After completion of reaction, solvent was removed under reduced pressure, and the resulting solid was filtered and washed with diethylether and dried m vacuo to yield, 3-i 2,6~dichlorophenyl)-7-((4~ (piperidin- -yl)phenyl)ammoy2,3-di hydrochloride (1 50 mg, 56%) as a white solid.
LCMS; 470.1 ΓΜ÷1]÷
[0312] Step-3: Synthesis of 3-(2,6-dkhlorophenyl)-7-((4-(i-methyIpiperidin-4- yl)plH¾nyI)aniino)-2y3-dihydro-4Jf-pyrinudo[5,4-ej[J3]oxazin-4-onej To a solution of 3-(2,6- di iloropheiw!)-7-((4~(piperidin~4~y!)ph
t'] I ,3]oxazin~4~one hydrochloride (90 mg, 0.17 mmol, 1 .0 eq) in 5 mL DCE at 0°C HCHO (37%) (16.06 mg, 0.53 mmol 3 eq) and acetic acid (53.5 mg. 0.85 mmol, 5 eq) were added under
stirring under inert atmosphere. The resulting solution was stirred at rt for 1 hr. The mass was again cooled to 0 °C, followed by the addition of NaCNB¾ (33.6 nig, 0.51 mmol, 3 eq) under stirring. The reaction was stirred at rt for 15-20 nun and monitored by LCMS. After completion, the reaction was quenched will
's ice cold water followed by extraction using ethyl acetate. The combined organic layer was washed with brme (50 ml.) and concentrated under reduced pressure. Th residue was purif ed by flash chromatography (elution 0-10% MeO
'H in (Ί Ob · to afford
pyrimido 5,4-g][l,3]oxazin-4-one (15 mg, 17 %) as white solid.
¾ M (400 UX, DMSO- ): δ 10.39 (br s. IHh 883 (s, 1H), 767 - 7.62 (m, 4H), 7.58- 7.52 (m, IH), 7,24■- 7.20 (m, 2Hj, 5.73 (s, 2H) 2.87 - 2.85 (ny 2H), 2.18 (ss 3HI 1.984.91 (m, 2H), 1.75 - 1.62 (m, .M r
Example S53. Synthesis qf3-(2; 6HiichIorophenyI)~7~(3~methoxy-4-{piperazin-I-yI)ph
2H~pytimid [5,4~ ] [ l,3 oxa∑iri-4(3H)~on cHhydrochlori.de
(Compound No.1.53)
(0313] Step~l: Syn esis of test- but l 4-(2-methoxy-4-aitrophenyl)pipera¾ine-l- carboxylate: To a stirred solution of i--fiuoi ~2--methoxy--4--niirobenzene ( 1 ,0 g, 5.843 mmol, 1,0 eq) and teri-hulyl 4-brorno-3,6-dihydropyridine~l (2H)--carboxylate ( 1.2 g, 6.427 mmol, 1 . 1 eq) in DMSO (20 mL) was added
2CC (2.43 g, 17,53 mmol, 3.0 eq) ai rt. The resulting mixture was stirred at 90 °C for 12b The progress of reaction was monitored by LCM5. The reaction mixture was poured into ice cold water (100 mL), stirred for 5 mm, formation of precipitates was observed, which was filtered and dried under vacuum to afford the desired compound ten-butyl 4-(2-meihoxy~4~nitrophenyl)piperazine-l-carboxylate (1.75 g, 88,83%) as a yellow solid,
[0314] Step-2: Synthesis of tert-butyl 4-(4-ainino-2-methoxyphenyl)p«pera2:me-i- carboxylate: To a stirred solution of tert- butyl 4-i2-methoxy-4-nitxophenyl)piperazine-l- carboxylate (1.0 g, 2.964 mmol 1.0 eq) m EtOH (25 mL) was added Fe(0) (1.32 g, 23.712
ninio!, 8.0 eq) and a solution of H
4CI (1 .58 g, 29.64 mmol, 10.0 eq) t ri. The resulting mixture was heated at 90 °C for 1 h. The progress of reaction was rnomtored by LCMS. The reaction mixture was filtered through oeliie the residue was washed Witt's EtOH (50 mL) the filtrate was concentrated and the residue was dissolved in EtOAe ( 100 mL), washed Witt's water (2 100 mL), dried over
and concentrated to afford the desired compound tert-butyl 4~(4-amino- 2miethoxyphenyl)piperazine
»l -carboxylate (0.750 g, 82.22%) as brown viscous.
LCMS; 308.4 ΓΜ÷1]÷
[0315] Siep-3: Synthesis of ter -b tyl 4-{4-{{M2,6-dkhIorophenyl)-4-o^^
2II-pyrimido[5,4~e] [l^]o.xaz-n~7~y^^ To a stirred solution of 3-(2
J6-dicbk>rophenyl)-7-(methyltbio)-2
J3-dihydro-4 -pymnido[5,4- e][L3]oxazin~4-one (300 nig, 0.868 mmol, 1 ,0 eq) in (5.0 mL) of toluene was added m-CPRA (428 mg, 1 ,736 mmol, 2,0 eq) and allowed to stir at rt for 30 min. Ten-butyl 4-(4~amino-2- niethoxyphenyl)piperazine-l -carboxylate (293 mg, 0.955 ismol. 1 .1 eq) and DIFEA (0.606 mL, 3.472 mmol, 4.0 eq ) were added and allowed to stir at rt for 12h. The reaction mixture was diluted with water (50 mL) and extracted with EtOAc (2 >
■ 50 mL), the combined organic layers were washed with water (50 mL) with brine (50 mL), dried over \ - SO.; filtered and concentrated and. purified by flash chromatography (elution 0-35% EtOAc in Hexane) to afford the desired compound, test-butyl 4-(4-((3-(2,6-dichlorophenyl)-4-oxo-3,4-dihydro-2H.- pyriraido[5,4-e}[.l
>3]oxazm-7-y])an-imo)-2-raet
'hoxy phenyl) piperazine-l -carboxylate (1 50 rng, 28.79%) as an off white solid.
[0316] Step-4: Synthesis of 3-(2,6-dkhIorophenyl)-7-((3-methoxy-4-(piperazm-l- yI)phenyi)amino)-2^-dihydro-4H-pyrimido 5,4-e] [l^]oxazm-4-one dihydrochloride: Text- butyl 4-(4~((3~(2,0-dichlorophenyl)-4-oxG-3,4^
yl)amino)-2-methoxy phenyl) piperazine- 1 -carboxylate (150 mg, 0.249 mmol, 1.0 eq) was dissolved m 4.0M HCl in dioxane (3 mL) and allowed to stir at rt for 1 hr. After completion of reaction, the reaction mixture was filtered and dried under reduced pressure and purified by diethyl ether to afford the desired compound, 3-T2,6-dichlorophenyl)--7--((3--niethoxy~-4-
ll y o TOG i o
\H mm (400 MI¾ DMSO-d
6): δ 10.34 (br
S> 1H), 9.00 (br s, 2H), 8.83 is, 1H), 7.66 id, J Hz, 1 H), 5.73 is, 2H), 3.79 is, 3H), 3.22 (br s, 4H), 3. 15 (br s, 4H) .
"' NH2 S" Ό'" Step-3
[0317] Step-1 : Synthesis of tert-butyl i«ethyl(2-((4-aitrophenyl)amino)ethyi)carbamate:
To a stirred solution of l -fiuofo-4-nitrobenzene ( 1.0 g, 7.09 mmol, 1 .0 eq) and tert-butyl (2- aminoethy!)(mem.y!)carbamate (1.36 g, 7.79 mmo!, 1.1 eq) in DMF (20 mL) was added K ^CCL (1.96 g, 14. 18 mmol, 2 0 eq) at t The resulting mi ture was stirred at 90 °C for 12h. The progress of reaction was monitored by 1 .CMS The reaction mixture was poured into ice cold water (100 rnL), stirred for 5 min, formation of precipitates was observed., which was filtered and dried under vacuum to afford the desired compound, tert-butyl roethyl(2-((4- riitrophenyi)a.ffiino)ethy!)oa.rbamate (1 .72 g, 82.29%) as a yellow solid
LCMS: 296.1 [M÷ l]÷
[0318} Step-2: Synthes s of ieri~b¾iy! (2-((4~
ami»opheuyl)ainino)ethyi)(inethyi)carbamate. To a stirred solution of tert-butyl raethyi(2-((4- nitrophenyl)amino)etliy!)carbamate (0.50 g, 1.692 mmol, 1 .0 eq) m EtOH (25 mL) was added FefO) (756 g, 1 3.543 mmoL 8.0 eq) and a solution of NH
4CI (0 905 ;.·. 16.920 rarnol, 10.0 eq) at rt. The resulting mixture was beaied at 90 °C for 1 br. The progress of reaction was monitored by LCMS. The reaction mixture was tillered through ceiiie the residue was washed with EtOH (50 mL), the filtrate was concentrated and the residue was dissolved in EtOAc (50 mL), washed with water (2
x 50 mL), dried over N 2SC>4, and concentrated to afford the desired compound, tert- butyl (2-((4-aminophenyj.)amino)ethyl)(methyl)carbamate (0 30 g. 66.81%) as brown viscous.
[0319] Step-3s Synthesis of tert-butyl (2-((4-((3-(2,6-dichlorophe$*yi)-4-ox
2H-pyrimido[5,4-e] (l,3]oxa-5m-7-yi)ammo)phenyl)ammo)ethyi)(methyl)ca To a stirred solution of 3-(2,6-dichiorophenyl)~-7~-(methylthio}-2,3~-dihydro~4H-pyrimido[5,4- e][l,3]oxazm-4»one (300 mg, 0.868 mmol. 1.0 eq) in ( 10.0 mL) of toluene was added m-CFBA (432 mg. 1.753 mmoi, 2.0 eq) and allowed to stir at rt for 30 min. Pert-butyl (2-((4- aminophenyl)amino)ethyi)(met!iyl)carbamate (256 mg. 0.964 mmol, 1.1 eq) and DIPEA (0.611 mL. 3.504 mmol, 4.0 eqj were added and allowed to stir at rt for 12h. The reaction mixture was diluted with water (50 mL) and extracted with EtOAc (2 χ 50 mL). The combined organic layers were washed with water (50 mL) with brine (50 mL) dried over \ .-SO ·. filtered and
concentrated and purified by flash chromatography (eluiion 0-35% EtOAc m Hexane) to afford
the desired compound tert-butyl ( 2· Π · π 3· ; 2 -dich !orop e:} ; rd -oxo- 74--di hvdr: :--2! i - pyfimido[5,4-e] [l ,3 joxazin-7-yl)ai'nino)phenyl) ammo) ethyl )(meihyl)carbarnaie (250 mg, 51 .12%) as dark brown solid.
LCMS: 559.3 ΓΜ÷1]÷
[0320] Siep-4: Synthesis of 3-(2,6-dkhlorophenyl}-7-((4-((2-
{methyiam<no)ethyi)ami«> )phenyi) amino)-2v3-dihydro-4H-pyriitnicIo[5»4-e] [1 ,3]oxazin~4~ ne hydrochlorides Tert-butyl (2-((4^(3 2,6-drch rophersyl)-4-oxo-334~dihydro~2e- pyrimido[5J4-e] [l,3]oxazin-7-yl)arnino)pbenyl) ammo)efchyl)(roethyl)carbarriate (220 rrsg, 0,393 mraoi, 1.0 eq) was dissolved m 4M HO in dioxane (2 ml,) arsd allowed to stir at ri for 1 hr. After completion of reaction, ibe ead ion mixture was filtered and dried under reduced pressure and purified by diethyl ether to afford the desired compound, 3-(2>6-di chlorophen l)-7-( (4-((2- (methy]ammo)eAyl)amino)phenyi) araino)-2,3-dihydro-4H-pyrimido[5,4-e][l ,3}oxazm-4-one hydrochloride (161 mg, 89.44%) as brown solid.
LCMS: 459.2 [M- l f
*H NMR (400 MHz, DMSO-d6): δ 10. 17 (br s, IH), 8.67 · 8,97 (m, 3H), 7.61 ■- 7.70 (m, J - 8.33 Hz, 2H), 7.50 (d J - 8.33 Hz, I H), 7.41 - 7.48 (m, 2H), 6.62 -■ 6.74 (m, J - 8 77 Hz, 2H), 5.70 (s, 2H), 3.35 (i, J === 6.14 Hz, 2B), 3.07 (br s, 2H), 2.58 (br s, 3H). fj a p!e S55. Symthesis qf5~(3~(2, 6~dichiorophenyi)~4~oxo~3, 4~dihydro~2H~pynmido(5, 4- e][l, 3]axiizin-7-ylamim)~2~(piperazin~I~yl)benzonitrile
(Compound No. 1.55)
Step-4
[0321] Step-1 : tert-butyl 4-(2-cyano-4-a5trophenyl}pipera2ine-l-carboxyiate: To a stirred solution of 2-finoro~5-mtrobenzomtnle (4.0 g. 21.47 mmol, 1.0 eq) and tert-butyl pipendme-4 carboxylaie (3.92 g, 23,62 mmol 1.1 eq) in DMF (70 asL) was added K
2C0
3 (5.935 g, 42.95 mmol., 2, 0 eq) at rt. The resulting inixture was stirred at 90 °C for I2h, The progress of reaction was monitored by i .C S. The reaction mixture was poured into ice cold water (200 rrsL)., stirred for 5 nnn, formation of precipitates was observed, which was filtered and dried under vacuum to afford the desired compound, tert-butyl 4~(2~cya«o-4--niiroplieriynpipei
'azine~ l ~ carboxylate (6.0 g, 84.26% ) as a yellow solid.
[0322] Step-2: Synthesis of tert-butyl 4-(4-ara io-2-cyanophenyl)piperaane-l- carboxylate: To a stirred solution of ten-butyl 4-(2~cyano-4-nitrophenyl}piperazirse-1 ~ earboxylate (2.0 g, 6,021 mmol 1.0 eq) m EtOH (30 ml,) was added Fe(0) (1 ,0 g, 18.064 mmol, 3 eq) and a solution of NH4CI (644 mg, 12.042 mmol, 2 eq) in water (30 ml.,) at rt. The resulting
mixture was heated ai 90 °C for 1 hr. The progress of reaction was monitored by LCMS. The reaction mixture was filtered through celhe the residue was washed wife EtOH ( 50 mL). The filtrate was concentrated and the residue was dissolved in EtOAc (100 mL), washed with water (2 x 100 mL), dried over NajSO-i, and concentrated to afford the desired compound, teri-butyl 4~ (4-amino-2-cyanopheny])piperazine- l -carboxylate (1 .77 g, 88, 5%).
LCMS; 303.2 [MH j+
[0323] Step-3: Syathesis of tert- butyl 4-(2-cyaHo-4-((3-(2,6-dichIorophenyl)-4-oxo-3,4- dihydro-2H-pyr«mido[5,4-eI [ l,3]ox¾Ein-7-yl)amino)pheayl)piperazine- 1-carboxy!ate: To a stirred solution of 3-(2.6-dichlorophenyl)-7-(methylthio)~2 ,3-clihydro-4H~pyrimido 5.4-e][l ,3] oxazin~4~one (300 mg, 0.8767 mmol, 1.0 eq) m (1 0.0 mL) of toluene was added m-CPBA (376 mg, 2. 1917 mmol, 2.5 eq) and allowed to stir at rt for 30 mm followed by addition of tert- butyl 4--(4~amino~2-cyanophenyl)piperazine--l --carboxylate (265 mg, 0.8767 mmol, 1 eq) and DIPEA (452 mg, 3.506 mmol, 4.0 eq) and allowed to stir at rt for 12 h. The reaction mixture was diluted with water (50 mL) and extracted with EtOAc (2 χ 50 mL). The combined organic layers were washed with water (50 mL), brine (50 mL), dried over Na?S(>4., concentrated and purified by flash chromatography (elution 0-35% EtOAc in Hexane) to afford the desired compound, tert- butyl 4-(2-cyano^-((3-(2,6-dichlorophen^^
7-yl)amino)phenyl)piperazine-l -carboxylaie (1 50 mg, 28.68%) as an off white solid.
[0324] Srep~4: Syathesis of 5-((3-(2,6-dichIoropheHyl)-4-oxo-3^ -dihydro-2H- p^imido[5, -e] [l^]oxazin-7^ tert-butyl 4-(2-cyano-
4-((3-(2,6-dichIorophenyl)~4~oxo-3.4-dihydro-2H.-pyrimido[5,4~e][l ,3]oxazin-7- yl)armno)phenyl) piperazine- 1 -carboxykte (150 mg, 0.252 mmol, 1 0 eq) was dissolved m 4M BCI in dioxane (2 mL) and allowed to stir ai rt for 1 h. After completion of reaction, the reaction mixture was filtered and dried under reduced pressure. Solid was triturated with diethyl ether, filtered off and dried under vacuum to afford the desired compound, 5-((3-(2,6-dichlorophenyl)- 4-0X0-3, 4-dihydro-2∑i-pyrimido[5,4-e] l ,3 Joxa.zin-7-yl)an^mo)-2-(piperazin-l-yl)benzonitrile (36 mg, 26.39%) as white solid.
LCMS; 496.2[M- l j+
JH NMR (400 MHz, DMSO-d6): δ 10.54 (bf s, IH), 8.87 (s, IH), 8.32 is, 2H), 8.09 (br s, 1 H), 7.85 id J - 8.33 Hz, 2H), 7.66 (d J - 8 33 ; I/. 1 H)5 7 51 id. ./ - 8.33 Hz, I H), 7.18 (d, J - 9.65 Hz5 IH), 5.75 (s, 2H), 3.02 (br a, 41 n. 2.87 (br s. 4H).
Example S56. Synthesis of3-(2, 6 iichioropheny])-7~(4~(pyrroiidw-l-yi)phenylam
pyrimidofS, 4-ejfl.3 ]oxazin-4(3hI)~one
(Compound No, 1.56)
[0325] §tep-l: Synthesis of l-(4-nltropheayi)pyrroIidine. To a stirred solution of i-fluoro-
4-nitrobenzene (0.50 g, 3.543 mmol, 1 ,0 eq) and pyrrolidine iO.277 g, 3.897 mmol, 1 , 1 eq) in DMF (10 ml,) was added
.:( < ) ·. (0 980 g, 7.086 mrnol 2.0 eq) at rt. The resulting mixture was stirred ai 90 °C for 12 h. The progress of reaction vvas monitored by LCTMS. The reaction mixture was poured into ice cold, water (100 mL), stirred for 5 mm, formation of precipitates was obseived, which was filtered and dried under vacuum to afford the desired compound, 1~(4~ nitrophenyj)pyrrolidme (0.605 g, 88.83%) as a yellow solid.
[0326] Step-2: Synthesis of 4-{pyrroIidin-l-yI)anilin. To a stirred solution of l-(4- nitrophenyl)pyrrolidine (0.60 g. 3. 121 mmol, 1 0 eq) in EiOB (20 mL) was added FeiO) (1 ,395 g,
24.971 mmoL 8,0 eq) and a solution of NH
4CI (1.67 g, 31.210 mmoL 10.0 eq) at ri. The resulting mixture was heated at 90 °C for 60 min. The progress of reaction was monitored by LCMS. The reaction mixture was filtered through oehte the residue was washed wilt's EtOH (50 mL) the filtrate was concentrated and the residue was dissolved in EtOAe (50 mL), washed with water (2
x 50 raL). dried over a
2SQ
4, and concentrated to afford 4-ipyrrolidm-.l-yl}amline (0.30 g, 59.28%) as brown viscous.
[0327] Siep-3: Synthesis of 3-(2,6-dkhlorophmyl)-7-((4-(pyrroHdm-l-yl)phenyI)amino)- 23-d«hydro-4H-pyrimido[5,4-e][.l^]oxa¾in-4-one: To a stirred solution of 3 -(2,6- dichlorophenyl)-7-(methylthio)-2,3-dihydiO-4H-pyrimido[
'5
>4-e
"S ] ,3]oxazm-4-one (200 mg, 0.584 rnmol, 1.0 eq) in (5.0 mL) of toluene was added mCPBA (288 mg, 1.168 mmol, 2.0 eq) and allowed to stir at rt for 30 min.4--(Pyrro diml- l)anihne (105 mg, 0,642 mmo!, 1.1 eq) and DIPEA (0.408 mL, 2.336 mmol, 4.0 eq) were added and allowed to stir at rt for 12 h. Th reaction mixture was diluted with water (50 mL) and extracted with EiOAc (2 >
■ 50 mL), the combmed organic layers were washed with water (50 mL) with brme (50 mL) dried over a2S(>
4, filtered and concentrated and purified by reversed phase chromatography to afford the desired compound Τί To-U:d:!oropho:i i ι.
" .
;; ·( rro rlin- i · π ίηί . ί};π:ΐ:η· ··· . · i;h dr · ·;Π · pyrimido[5,4-e][l,3] oxazin-4-one (39 mg, 1466%) as a yellow solid
*11 MR (400 MHz, DMSO-d6): 610.13 (br s, 1H), 8.75 (s, 1H), 7.65 (d, J ==== 7.89 Hz, 2H), 7.50 (d, J - 7.89 Hz, 2H), 7.46 (d, ./ - 7.45 Hz, 1H), 6.53 (d, J - 8.77 Hz, 2H), 5.69 (s, 2H), 3.01 - 3.28 (m, 4H), 1.95 (t, J 6.36 Hz, 4H).
Example S57. Synlhesis qf3~(2, 6~dichlorophenyl)~ 7~(4~(4~(2~(methylamme)iicetyl)pipera ri~ 1~ yijphenylamino)~2B~pyr ido[5, 4-e [l, 3 oxazin~4(3H)~one hydrochloride
(Compound No, 1.57)
[0328] Step-1: Synthesis of/etf-butyl 4~(4~((3-(2,6-dich!oropheayI)~4-oxo-3,4-dihydro- 217-p rimido \S,4~e] [1 ]oxazia-7-yi)araiao)phenyl)piperazine-l-carboxyiatei To a stirred solution of 3~(2,0-dichJorophenyl)-7-(methy^
(0.5 g, 1 46 mmol, 1.0 eq) m 5 mL toluene /wCPRA (0.628 g, 3 65 mmol, 2.5 eq) was added under stirring and resulting mixture was allowed to stir at rt for 30 niin. Further, ie f-buty! 4-(4- aro.inophenyl)piperazine~l~carboxylate (0.405 g, 1 46 mmol, 1 eq) and. DIFEA (0 75 g. 5.85 mmol, 4 eq) were added and the reaction was allowed to stir at rt for 12h. The reaction was monitored by i .C S. After completion reaction was quenched with water and extracted with ethyl aceMe (10 rnL x 3). The combined organic layer was washed with brine solution (20 mL),
dried over anhydrous .SO.. and concentrated under reduced pressure to afford crude product winch was purified by flash chromatography (MeOH: (ΙΗ?(¾ 1 -1 0%) to afford teri- butyl 4-(4- ((3- 2,6~diclilorophenyl)-4~oxo~3,4-dihy^
piperazme- 1 -carboxyiaie (0.270 g, 33%) as white solid.
LCMS: 570 [Μ-Η ]+'
[0329] Step-2: Synthesis of 3-(2,6-dkhlorophmyl)-7-((4-(pipera7Jn-l-yl)phenyI)amino)- 23-d«hydro-4 /-pyrifnido[5,4- ][.l 3]oxa¾in-4-one hydrochloride: 20% HC1 in dioxane (2 mL) was added to a stirred solution of fc'/t-butyl
di h dr ··2 · i^ ruvsidol 5.4-;· Ij 1 . θ¾^^η:·· i )a: ^ i: ; hi;r: i } : cr nc-- i ~,:a; box laic (0.270 g, 0,47 mmo!, 1.0 eq) at 0 °C and the resulting solution was allowed to stir at rt for 3 h. After completion of reaction, solvent was removed under reduced pressure, and the resulting solid was filtered, washed with dieihylether, and dried to afford 3 -(2,6-dichlorophen l)-7-(( 4-(piperazin~ 1. - yl }pheiiyl)araino)-2,3-dihydro-4H-pyrimido[5,4-e ]'l ,3 joxazin-4-one hydrochloride (0.155 g, 65%) as a white solid.
LCMS: 506 [M÷l ]÷
[0330] Step-3: Synthesis of tert-butyl (2~(4~(4-((3-(2>6-dich!orophenyl)-4-oxo-3>4- dihydro-2ii-pyr«mklo{5,4-eI|l^]oxazia~7-y!)amiao)pheayl)pi^
oxoethyi)(methyi)carban»ate: To a solution of 3-(2,6-dichlorophenyl)-7-((4-(piperazin~l.~ yl)pheny])amino)-2,3~dihydro~4H-^^^ hydrochloride (0.150 g,
0.29 mmol, 1 .0 eq) in 5 mL DMF, HATU (0.165 g, 0.43 mmol) was added under stirring at rt under inert atmosphere. The resulting solution was stirred for 5 mm. Further N~ tsrt- butox.ycarhonyl)-N-methylg]ycine (0,061 mg, 0, 33 mmol) and DiPEA (0.131 g, 1 ,01 mmol) was added. The resulting mixture was stirred at rt for 1 --2 h and monitored by LCMS. After completion, the reaction was quenched with water and the resulting solid was filtered and dried to afford tert- butyl
e] I ,3]oxazim7wl)animo)pheiwl)piperazin ^ (0.145 g, 76%) as white solid,
LCMS: 641 [M÷l ]÷
[0331] Step-4: Synthesis of 3-(2,6-didUoropheHyl)-7-((4-(4-(methyiglycyl)piperazin-l- yI)phenyi)amino)-2^-dihydro-4//-pyrimidof5,4-e]{l^)oxazin-4-o«e hydrochloride: 20%
HQ in dioxane (2 niL) was added to a stirred solution of iert-buiyi (2-(4-(4-((3-(2,6- dichloropheiiylV4-oxo-3,4»dilw^
p^erazm- "yl)--2"Oxoeihylj(niethyl)carbaniate (0.14 g.0.21 rnmol, 1.0 eq) at 0 °C and the resulting solution was allowed to stir at rt for 3 h. After completion of reaction, solvent was removed under reduced pressure, and the resu!tmg solid was filtered and washed with
diethylether, and dried to afford 3·ί . ?-d; !:ior b :-x 1•• -((4-(pis¾ra/:-rd · !}ι;;·;ην η:;!·:·ηο)· .3· dihydro-4H-pyrimido[5,4~e][l ,3]oxazm-4-one hydrochloride (0.155 g, 1%) as a white solid.
¾ MR (400 MHz, DMSO-<f6): δ 102 (br s, 1H), 8.79 (s, !iu.7.65 (d. J - 7.89 \W.2H), 7,59- 7.54 (m, 2H), 7.52■- 7.46 (m, 1H), 6.96 (d, J - 8.77 Hz, 2H), 5.71 (s, 2H) 3.64-3.59 (m, 2H), 3.57-3.53 (m, 2H), 3.38-3.46 (m, 2H), 3.14-3.1 (m, 2H), 3.04-3.09 (m, 2H).
Example S58. Synthesis of 3~(2 )~dichlorophenyi)~7~(4~{4~{24^droxyethyi)pipe
yl)phenyiaminoj~2H-pyrimido[5,4-e
(Compound No, 1.58)
[0332] Step-
'J : Synthesis of 2-(4-(4-niiropheoyl)pipera-5ia-l-yi)eiha«-l-oI: To a stirred solution of 2~(piperazin-3 -yl)ethan-3 -ol (1.014 g, 7.79 mmol, 1.1 eq) in 5 mL DMF, K . >(
'(
) ·. (1.95 g, 14.1 6 mmol, 2 eq) was added under stirring and resulting mixture was allowed to stir at rt for 5-10 mm. Further, 1 -tl uoro-4-ni robenzen e ( i g, 7, 79 mmol, 1 eq) was added and the reaction was heated at 80 °C 12 h. The reaction was monitored by LCMS. After completion, reaction was cooled to rt and quenched with cold water. The resulting yellow precipitate was filtered and dried., washed with water ιο afford 2-(4-(4-nitrophenyl)piperazin- 1 - !)eihan- 1 -o! (0.905 g, 51 %) as a yellow solid
LCMS; 252 [M-H j+
[0333] Step-2: Synthesis of 2-(4~(4-aminopheny!)pipeimin-l-yl)ethaii-l~o 2-(4-(4- nitrophenyj)piperazin~] ~yi)ethan
»l -oi (0.9 g, 3.58 mmol) was dissolved in 1 0 mL EiOH, further 10 ml, water was added under stirring. Iron powder (0.6 g, 10, 75 mmol) and N f Id
'.
'! (0.383 g, 7.16 mmol) were then added. The resulting mixture was heated at 90 °C for 1-2 h The reaction was monitored by LCMS. After completion of reaction, mixture was filtered through cetite bed, solvent form die filtrate was removed under reduced pressure. Water was added to the residue and extracted using MeOH: Ci¾(¾ ( 10%), dried over
The solvent was removed under reduced pressure to afford 2~( -(4-ammopheny 1) piperazin-
'i-yl) ethan-1 -oi (0, 550 g, 70%) as a brown solid
[0334] Step-3: Synthesis of 3-(2,6-dichIorophenyl}-7-((4-(4-(2 iydroxyethyi)pipera2.in-l- yI)phenyi}amino)-2^-dihydro-4//-pyrimido[5,4-e] {l^)oxazin-4-onei To a stirred solution of 3--(2,6"dichlorophenyl)-7tiniethylthio^ (0.2 g,
0.58 mmol, 1.0 eq) in 5 mL toluene M-CPBA (0.251 g, 1.46 mmol, 2.5 eq ) was added under stirring and resulting mixture was allowed o stir at rt for 30 min. Further, 2-(4"(4·- aminophenyl)piperazin--i --yi)eihan--i --oi (0. 129 g, 0.58 mmol, 1.0 eq) and DIFEA (0.302 g, 2.33 mmol, 4 eq) were added and the reaction was allowed to stir at rt for 12 h. The reaction was monitored by LCMS. After completion reaction was quenched with water and extracted with ethyl acetate (5 niL x 3). The combined organic layer was washed with bnne solution (f 0 ml,), dried over anhydrous Na2SC> and concentrated under reduced pressure to afford crude product
which was purified by flash chromaiograpliy (MeOH:€¾(¾ 1-10%) ΐο afford 3--(2,6- dkh!oropherry IV 7--( ( 4--(4--^
pyrimido[5,4~£ l,3]oxazm-4-one (0.008 g, 10%) as white solid.
¾ R (400 MHz, DMSO-d6): S 10.18 (s, IB), 8.78 (s, IB), 8.20 (s, IH), 7.65 id, J === 8.33 Hz, 21:3), 7,51 (d, 7 - 7.45 Hz.2Π;.6.92 (d, J - 8.33 Hz, 7!!}. 5.71 (br s.2H), 4.43 (br s, 2H), 3.53 (d../ O.N H 2H), 3.17 (d J - 5.26 Hz, !H).3.09 (br s, ill).1.23 (br s, IH), ;.!6(d. / 10.09 Hz, IH).
Example S59. Synthesis of3-{2, 6~dichhrophe i)~7~(4~(4~methyljnp
2H-pyri ido[5, 4-ejfI, 3]oxazin~4(3H)-one
(Compound No, 1.59)
[0335] Step-1: Synthesis of 4-methyl-l-(4-nhr phe«y!)piperidme: To a stirred solution of l~iluoro-4~mtrobenzene (1.0 g, 7087 rrsmoi, 1.0 eqS and 4-me†hylpiperidine (0.773 g, 7.795
mmol, L i eq) in DMF (20 m.L) was added K2C03 (1 .95 g, 14. 17 mmol, 2.0 eq) at ri. The resulting mixture was stirred at 90 °C for 12 h. The progress of reaction was monitored by LCMS. The reaction mixture was poured into ice cold water (150 ml,), stirred for 5 mm, formation of precipitates was observed, which was filtered and dried under vacuum to afford the desired compound, 4-meAyl- l -(4~m†.rophenyl)piperidine (1.5 g 96.09% ) as a yellow solid LCMS: 221 [M÷l ]÷
[0336] Step-2: Synthesis of 4-(4-metfaylpiperidin- l-yl)ani!ine. To a stirred solution of 4- methyl- 3 -(4-nitrophenyl)piperidine (1.5 g, 6.809 mmol, 1.0 eq) in EtOH (12 rnL) was added Fe(O) (1 .14 g, 20.42 mmol, 3 eq) and a solution of NH4C1 (0 728 g, 1 .619 mmol, 2 eq) in water (12 mL) at ri The resulting mixture wa,s heated at 90 °C for 1 hr. The progress of reaction was monitored by LCMS. The reaction mixture was filtered through celiie the residue was washed with EtOH (50 mL). The filtrate was concentrated and the residue was dissolved in EtOAc (1 00 mL), washed with water (2 χ 100 mL), dried over Na?S(>4, and concentrated to afford the desired compound, 4-(4-methy Ipiperi din- 1. ~yl)ani line (0,900 g, 69.52%),
LCMS: 1 91 [M+l
[0337] Step-3: Synthesis of 3-{2,6-dichIorophenyl)-7-((4-{4-raethylpiperidin-i- yI)phenyi)amino)-2^-dihydro-4H-pyrimido[5,4-e] [l^]oxazin-4-o«ei To a stirred solution of 3--(2,6"dichlorophenyl)-7-(methylm^ (0.200 g, 0.5844 mmol, 1.0 eq) m (5.0 ml,) of toluene was added m-CPBA (0.201 g, 1 . 168 mmol, 2 eq) and allowed to stir at rt for 30 mm followed by addition of 4-(4-methylpiperidin-l -yl)ani!ine (0.122 g, 0,6428 mmol, 1.1 eq) and DJPEA (0.301 g, 2.337 mmol, 4.0 eq) and allowed to stir at rt for 12h. The reaction mixture was diluted with water (50 mL) and extracted with EtOAc (2 x 50 mL). The combined organic layers were washed with water (50 mL), brine (50 mL), dried over Na jSC , concentrated and purified by co nbi-flash [silica gel- 100-200 mesh; eiution 0-35% EtOAc in hexane] to afford the desired compound, 3--(2,6-dichlorophenyl)-7-((4-(4~
methylpiperidm-l -y!)pheny!)amino)-2J (0.1 1 0 g,
39.00%) as an off white solid.
LCMS; 484 [M-H j+
*11 NMR (400 MHz, DMSQ-d6): δ 10.21 (br s, IH), 8.78 (s, 1 H), 7.65 (d, J ==== 8.33 Hz, 2H), 7.35 - 7.57 (in, 31:1), 6.91 (d, J - 9.21 Hz, 2H), 5.70 (s. 2H), 3.61 (d, ,/ - 13 15 Hz, 2H), 2.52 -■ 2.78 (in, 2H), 1.69 (d, J - 12.72 Hz, 2H), 1.48 (br s, IH), 1.18 - 1.31 (m, 2H), 0.94 (d, J - 6.14 Hz, 3H).
Example S60. Synthesis oj'3~(2.6-dichlorophenyl)-7-(3-rneihyl-4-(piperidm~4~yi)phenykim 2H-pytimido[5, 4-e][ 3]oxazin-4(3H)-one
(Compound No. 1.60)
[0338] Step-1 : Synthesis of tert-butyl 4-(2-methyI- -nitrophenyl)-3,6-dihydropyridini'- l(2H)~carboxyiate: To a stirred solution of l.~hromo~2~methyl~4~ †robenzene (1 .5 g, 6 94
mmol, 1 .0 eq) and tert- butyl 4--(4,4,5,5"teiramethyl-.l33,2- ioxaborolari"2--yl)"3,6- dihydropyndine-l (2H)-carboxylate (3.0 g, 9.72 mmol, 1.4 eq) in dioxane (1 5 mi . ) was added a 2M solution of Na^CC (2.20 g. 20.82 mmol, 3.0 eq) at rt. The resulting mixture was purged with nitrogen for 10 min, followed by addition of Pdidppf )(¾.€¾(¾ (283 mg, 0.347 mmol, 0.05 eq). The resulting mixture was heated at 90 °C for 12 h. The progress of reaction was monitored by 'HNMR. The reaction mixture was filtered through cebie the residue was washed with EiOAc (50 mL) the filtrate wash concentrated and purified by ilash chromatography (elution 0- 10% EiOAc in hexane) to afford the desired compound, tert-butyl 4-(2-methyl-4- nitrophenyl)- ,6- ihydropyridine" l (2H)-carboxylaie (2.0 g. 90.49%) as white solid.
LCMS: 319.2 [M÷ l]÷
[0339] Step-2: Synthesis of tert-butyl 4-(4-ain«uo-2-aiethy!phenyl)p«pericIiae-l~ carboxylate: To a stirred solution of tert-butyl 4-(2-me hyl-4-mirophenyl)-3,6-dihydropyridme- I (2H)-carboxylaie (2.0 g, 6.28 mmol, 1 .0 eq) in MeOH (24 mL) was added 10% Pd/
'C (200 mg) at rt. The resulting mixture was purged with hydrogen for 60 min. The progress of reaction was monitored by LCMS. The reaction mixture was filtered through ceiite the residue was washed with MeOH (50 mL) the filtrate was concentrated and purified by n-pentane to afford the desired compound, tert-butyl 4~(4~amino~2-methylphenyi)pipendine- i-carboxylate (0.900 ;.·. 49.45%) as gray solid.
[0340] Step-3: Synthesis of tert-butyl 4-(4-((3-(2,6-dichloropheoyI)-4-oxo-3,4-dihydro- 2H-pyrimido [5,4-ej ( 1 ,3] oxa2«a-7- l)amiao)-2-i«ethylphen l)piperidioe- 1-car boxylate: To a stirred solution of 3-(2,6-dichloropheiiyl)-7-(methylthio)-2,3-dihydro-4∑J-pyriinidof'5,4- ejj i ,3 ]oxazm-4-one (500 mg, 1.46 mmol, 1 .0 eq) in toluene (6 mL) was added m-CPBA (504 mg, 2.92 mmol, 2.0 eq) and allowed to stir at rt for 30 min. Tert-butyl 4-(4~amino-2- rnethy!phenyl)piperidme-i -carboxylate (509 mg, 1 .75 mmol, 1.2 eq) and Dl'FEA (1.01 mL, 5.84 mmol, 4.0 eq) were added and allowed to stir at rt for 12 h. The reaction mixture was diluted with water (50 mL) and extracted with EiOAc (2 χ 50 mL), the combined organic layers were washed with water (50 mL) with bone (50 ml,) dried over Na2S04, filtered and concentrated and purified by flash chromatography (elution 0-30% EtOAc m hexane) io afford the desired
compound, tert- butyl 4-{ -((3-(2,6-dichlorophenyi)-4-oxo-3,4-dihydro-2∑-]-pyrimido[5,4- eij !. '¾;'ϋ·-7·; I - muKH ·2 ·:^ι^ Ip c^vilpme; i ; :=e- i -anlHrwi i (400 mg, 46.83%) as off white soncl.
LCMS: 584.2 ΓΜ÷1]÷
[0341] Siep-4: Synthesis of 3~(2,6-dic.h1oropheRyl}-7-((3~methyI-4~(piperidin~4~
yl)phenyl)ainino) ^ tert-butyJ 4-(4-((3-(2,6- dt ii!oro heu l)-4-o o-3.4--d;i:vdr
»2i l-p nimd l 5.4-ei
phenyl) pipendine- 1 -carboxyiate (100 rag, 0.171 ramol, 1.0 eq) was dissolved in 4MHC1 in dioxane (2,0 nU,) and allowed to stir at rt for 1 h. After completion of reaciiors, the reaction mixture was concentrated under reduced pressure and purified by reversed phase purification to afford the desired compound, 3-(2,6-dichloiOpbenyl)-7-((3-methyl-4-(piperidm-4- yl)phenyl)arnino)-2J3-dihydro-4H-pyrimido[5,4-e][.l,3]oxazin-4-one (24 mg, 28,96%) as white solid.
LCMS: 484.2 [M- lf
*H NMR (400 MH¾ DMSO-d6): δ 10.33 (br. 1H), 8.83 (s, 1 H), 7,66 (d, J = 7.89 Hz, 2 H), 7.57 (d. J - 833 Hz, 1 !!}.7.52 - 7.47 ( n, 2H), 7.13 (d, J = 8.77 Hz, I H), 5.73 (s, 2 !!}.3.32- 3.29 (m 2H), 3.14 (br. s.„ 1 H), 2.99-2.94 (m, 2H), 2.28 (m, 3H), 1.82 - 1.71 (ni, 4∑-]). fja p!e S61. Symthesis of 7~(4~(1, 4~diazepan~l~y!)pheriylamino)-3-{2, 6-dichiorophenyi)~2H~ pyrimido[5, 4~e/fl, 3]oxazin~4(3H forte
(Compound. No.1.61)
[0342] Step-1: Synthesis of tert-butyl 4~(4~nitiOpheayl)-l,4-dia8epane-l-carhoxylate: To a stirred solution of 1 -fluoro-4-nitrobenzene ( 1.0 g, 7.08? mrnol. 1.0 eq) and tert-butyl 1,4- diazepane-Lcarboxylate (1.561 g, 7.795 mrnoi, 1.1 eq) in DMF (20 mL) was added K
2C0i (1 96 g, 14.174 mmoi, 2.0 eq) at rt The resulting mixture was stirred at 90 °C tor 12 h. The progress of reaction was momtored by LCMS. The reaction mixture was poured into ice cold water (100 mL), stirred for 5 min, formation of precipitate was observed, which was filtered and dried under vacuum to afford the desired compound, tert-butyl 4-(4-nitrophenyl)-l ,4-diazepane- l - carboxylase ( 1.9 g, 83.44%) as a yellow solid.
[0343] Step- 2: Synthesis of tert-butyl 4-(4-aminophenyl}-l,4-dia«epaae-l-carboxylate:
To a stirred solution of tert-butyl 4-(4-nitropheny
j.)-.l ,4-diazepane-.l-carbox
,late (1 ,0 g, 3, 1 1 1
mmol, 1 .0 eq) in EtOH (25 mL) was added Fe(0) ( 1.39 g, 24.892 mmol, 8.0 eq) and a solution of NH4CI (1.66 g, 31 . 1 1 mnicsl. 10.0 eq) in water (25 mL) at rt. The resulting mixture was heated at 90 °C for 60 min. The progress of reaction was monitored by LCMS. The reactiors mixture was filtered through eelite the residue was w a hed with EtOB (50 mL). The filtrate was concentrated and the residue was dissolved in EtOAe (50 mL), washed with water (2 50 mi..), dned over 8
2S0
4, and concentrated to yield tert-butyl 4-{4-atninophenyl)- 1 ,4-diazepane- 1 -carboxyiate (0.95 g, 95.28%) as brown viscous.
[0344] Step-3: Synthesis of tert-butyl 4-(4-((-^(2,6-dkhiorophenyI)-4-oxo-3,4-dihydro- 2H-pyrimido[5,4-e] [ly3] xazm-7-yi)aramo)phenyl)-l,4-dia2:epane-.l: To a stirred solution of 3-(2,6-dich]orophenyl)-7-(meihyltlM^ (300 mg, 0.879 mmol, 1.0 eq) in (1 0.0 mL) of toluene was added m-CPRA (432 nig . 1.753 mmol, 2.0 eq ) and allowed to stir at rt for 30 min. Tert- butyl 4--(4--anunophen l H ,4-diazepane- I-- carboxylaie (281 nig, 0.964 mmol, 1 . 1 eq) and DIPEA (0.61 1 ml 3.506 mmol, 4,0 eq) were added and allowed to stir at rt for 12b. The reaction mixture was diluted with water (50 mL) and extracted with EtOAc (2 ··- 50 mL), the combined organic layers were washed wife water (50 mL) with brine (50 mL) dried over .·:>(}.·. filtered and concentrated and purified by combi flash (elution 0-35% EtOAc in hexane) to afford the desired compound, tert-butyl 4-(4-((3-(2J6- dichlorophenyl)-4~oxO~3,4-dihydro-2H~pyrim ^^
diazepane-L carboxyiate (195 nig, 38.16%) as brown solid.
[0345] Step-4: Synthesis of 7-((4-(l ,4-diai¾epan-l-yl)pheoyI)aiRi«o)-3-(2,6- dkhIorophenyl)-2 -dihydro-4H-pyrimido|S,4-e| [l^]oxa¾i«-4-one: Tert-butyl ^ ^iXj ' dichlorophenyl)-4-oxo-3,4-dihydro-2H-pyrimido 5.4-e][ l ,3]oxazin-7-yl)amino)p
diazepane- -carboxyiate ( 90 mg, 0.325 mmol, 1.0 eq) was dissolved in 4M HQ in dioxane (2 mL) and allowed to stir at rt for 1 h. After completion of reaction, the reaction mixture was filtered and dried under reduced pressure and purified by diethyl ether to afford the desired compound, 7-{{4-(l
pyriniido[5,4-e] [L3]oxazm-4-one formate (155 mg, 91.17%) as a yellow solid.
\H N L (400 MHz, DMSO-d6): S 10.1 5 (br s, ! ! § ;. 8.75 (s, 1 H), 8.42 (s, 1H), 7.65 (d, J 7.89 Hz, 2H), 7.31■■ 7.57 (m, 3H), 6.69 id, J === 8.33 Hz, 2H), 5.69 (br s, 2H), 3.39 - 3.53 (m, 6H), 2.89 (br s, 2H), 2.68 uL ./ === 5.26 Hz, 2H), 1.83 (br s, 2H).
Example S62. Synthesis qf3-(2; 6HiichIorophenyIJ~7~(3~(hydroxymet iyIJ~ ~(piper zifi~J~ yI)pheny!amin )~2~meihyl~2H^) imido[5,4~e fI,3 oxazin~4{3Hj~ fie
(Com ound No. 1.62)
[0346] Step-l : Synthesis of tert-butyl 4-(4-((-^(2,6-dkhIorophenyI)-2-methyI-4-oxo-3,4- dihydro-2H-pyr«mido [5,4-e] [l^]oxazin-7-yl)amino)-2- (hydroxyraethyi) phenyl) piperazme-l-carboxyiate: To a stirred solution of 3-(2,6-dichiofophenyl)-7-(methylthio)-2,3- dihyd.ro-4H-pyrinudo]
'5,4-e
"j l,3 oxazin-4-one ( 140 nig, 0.393 mnioL 1.0 eq) in (3.0 mL} of toluene was added /w-CPBA (200 mg, 0.786 nimoi, 2.0 eq) and allowed to stir at rt for 30 min. Tert-butyl 4-(2-(hydroxyni thyl)-4-nilxophenyl)pipefazine-l -carboxylaie (133 nig, 0.432 nimoi, 1.1 eqj and DiPEA (0.275 mL, 1.572 nimoi, 4.0 eq) were added and allowed to stir at rt for 12b, The reaction mixture was diluted with water (50 mL) and extracted with EtOAc (2 >
■ 50 mL), the combined organic layers were washed with water (50 mL) with bri e (50 mL) dried over Na
2S0 , filtered and concentrated and purified by flash chromatography (eiution 0-35% EtOAc
in hexane) to afford the desired compound, tert-buyl 4~(4-((3-(2,6-dichiofopheiiyl)-2-methyl-4- oxo-3,4-dihydro-2I3-pyrirnido [5,4-e] [1,3 !oxazm-7-yi)amtiio)-2- (hydrox methyl) phenyl) pmerazme-l-carboxyiate (115 nig, 47.71%) as off white solid.
[0347] Step-2: Synthesis of 3-(2,6-diehIor phe Yl)-7-((3-(hydroxymehyi)~4-{piper8¾¾~ i-yl)plH¾iyI)am. >)~2~methyl~2 -d^ tert-butyl 4-
animo)-2-(hydt'oxymetbyl)phenyl)pipera2;ine-l -oarboxylate (110 mg, 0.178 mmo!, 1.0 eqS was dissolved in 4M HQ in dioxane (2 mL) and allowed to stir at rt for 1 h. After completion of reaction, the reaction mixture was filtered and dried under reduced pressure and purified by reversed phase purification to afford the desired compound, 3-(2,6-dic-hlorophenyl)-7-((3- (hydroxymethyl)-4-(piperazm-l-y!)pheny
e][1 ,3joxazm--4-one (formate salt) (39 mg, 42.39%) as brown solid. LCMS: 515.2 [M- lf
*H NMR (400 MHz, I)MSO-d6): «510,29 (br s., Hi), 8.78 (s, 1H), 8.28 (br s, IH), 7,63■- 7.75 (m, 2ί-ί), 7.58 (d, IH), 7.52 (t, J - 8.11 > IH), 7.04 (d, J - 9.21 Hz, IH).6.14 (d, J - 570 Hz, IH), 5,03 (br s, 2H), 4.55 (s, 2H), 2.91 (br s, Hi}.2.79 (br s, 4H), 1 ,38 (d, J --= 5.70 Hz, 3H). fja p!e S63. Symthesis of 3~(2 )~dichlorophenyi)~7~{4~(4,4~difltioropiperidin~ 1 - yiphenyiam in oj- 2H ~^yrimido[ 5,4-e] [ 1 ,3 ox zin-4(3H)~one
(Compound No.1.63)
[0348] Step-1: Synthesis of 4,4~difl or ~i~(4~nitr pheayi)piperi line: To a stirred solution of 1 -fluoro-4-nitrobenzene (0.5 g, 3.54 mmoL 1.0 eq) and 4,4-diiluQropiperidme (0.6.15 g, 3,897 mmo!, 1.1 eq) m DMT (10 mL) was added
2C0
3 (0.978 g. 7.08 mmoL 2 0 eq) at rt The resulting mixture was stirred at 90 °C for 12h. The progress of reaction was monitored by LCMS. The reaction mixture was ponred into ice cold water (100 mL), stirred for 5 mm, formation of precipitate was observed, which was filtered and dried under vacuum to afford the desired compound, 4,4-difluoro-l-(4~nitrophenyl)piperidine (0.635 g, 74.0%) as a yellow solid,
[0349] Sfep-2: Synthesis of 4-(4
>4-(lifliioropiperid«n-i -yl)aaiiioe: To a stirred solution of 4,4-difluoro- 1 -(4~«itrophenyi)piperidi«e (0.6 g, 2.476 mmol, 1.0 eq) in EtOH (20 mL) was added Fe(0) (1 .1 g, 19.815 mrnol, 8.0 eq) and a solution of NH4CI ( 1.33 g. 24.76 mmoL 10.0 eq) in water (20 niL) at rt. The resulting mixture was heated at 90 °C for 60 ruin. The progress of reaction was monitored by LCMS, The reaction mixture was filtered, through celite the residue was washed with EtOH (50 rnE) the filtrate was concentrated and. the residue was dissolved in EiOAc (50 rnE)., washed with water (2 x 50 mL), dried over \a -SO.; , and conce trated to yield 4-(4,4-difluoropiperidin- 1 -yi)anilme (0.95 g, 95.28%) as brown viscous.
[0350] S ep~3: Synthesis of 3-(2,6- lichIoropheny!)-7-({4-{4,4- lifl«oropiperidin-i- yl }phenyi)arajno)-2^-dihydro-4H-pyriraido [5,4-e] [ 1-3] ©xazm-4-one: To a stirred solution of 3-(2,6-dicii!orophenyl)-7-(methyUlijo)-2,3-dihydix>4H-pyrimido[5,4-e][l,3]oxazin-4-o (300 mg, 0.876 mmol, i 0 eq) in (5.0 mL) of toluene was added mCPBA (432 mg, 1.753 mmol, 2.0 eq) and allowed to stir at rt tor 30 rain. 4-(4,4-Difluoropiperidm-l -yi)aniljne (205 mg, 0, 963 mmol, 1.1 eq) and DIPEA (0,61 1 m l .. 3, 506 mmol, 4,0 eq) were added and allowed to stir at rt for 12 h. The reaction mixture was diluted with water (50 mL) and extracted with EtOAc (2 x 50 mL), the combined organic layers were washed with water (50 mL) with bnne (50 mL) dried over \
':t S(
}.; filtered and concentrated and purified by Bash chromatography (e!uxion 0-35% EtOAc m hexane) to afford the desired compound, LC .(;·· !:;. b !o: opi -os ·\ 0··?··; ·; ··( . ·ί · difl.uoropiperidin-l-y!)phenyl)amino)~2,3-dihydro-4H~py.rimido[5.4 (139 mg, 1 .30%) as an off white solid.
\li NMR (400 MHz, DMSO-de): δ 10.27 (br s, ! i u. 8.79 (s. ! i i ). 7.65 (d, J - 7.89 \ \ Y. 21:3), 7.56 d, J - 8.33 Hz, 2H), 7.45 - 7.54 (m, IH), 7.00 (d, J - 9.21 Hz, 2H), 5.71 (s, 2H), 3.26 - 3.30 (m, 4H), 1.96 - 2. 14 (m, 4H).
ejfl, 3joxazin~4(3H)~one
(Compound No, 1.64)
DIPEA, rt, 2h
[0351] Synthesis of 3-(2,6-dichIorophenyi)-?^isoqaiooii«-6-yIai«ino)-2H-pyrimido[5,4- e] [L3]oxazin-4i3fl)-0ne: To a stirred solution of 3-i2,6~dich!orophenyl)--7--(meihykhio)--2H-- pyrimido 5,4-e][l, ]oxazin-4(3H)-one (250 mg, 0.733 mmol, 1 .0 eq) in toluene (10 nil,) was added /B-CPBA (315 mg, i .83 mmol, 2.5 eq) and allowed to stir at rt for 30 mm. isoqinnolin-6- amme (105 mg, 0.733 mmol, 1.0 eq) and DIPEA (378 mg, 2.93 mmol, 4.0 eq) were added and stirred at ri for 2 h. Progress of reaction was monitored by LCMS. After consumption of starting material, precipitated compound was filtered off, dried and. purified by reversed phase chromatography to afford 3-(2,6~dicblorophenyl)~7~(isoq inoiin-6-ykmino)-2H-pynmido[5,4- e][l ,3]oxazm-4i3H)~one (6 mg, 1.86%).
LCMS: -438.2 [M+I]+ hi MR (400 MHz, CD30»)J «5 10.16 (s. l id), 9.40 is, 1 H). 9.20 (d, J - 6. 14 Hz, l id), 8.25 id. J 9.21 Hz, 1H), 7.75 (d, J - 7.45 Hz, HI), 7.62 (d, J - 8.33 Hz, 2H), 7.44 · 7.55 (m, 1 1 1 } . 7.34 id, J - 9.21 Hz, i l l) 7 02 (s, 1 1 1 ) 5 96 (s, 2B)
tetrahydreisequmolin~7~ylamine)-2H^yrimido[5,4~e]f
(Compound No. 1.65)
[0352] Siep-1: Synthesis of/erf-butyl 7-((3-(2,6-dichiorophenyl)-4-oxo-3,4-dihydro-2//- pyrimido5,4-e]{l^]oxazui-7-y^ To a stirred solution of 3-(2.6-dichlorophenyl)-7-{meihylthio)~23-dihydro-4H~benzo[?lj'l,3]oxazin~4~ one (200 nig, 0.58 nimol, 1.0 eq) in 5 mL toluene ?H~CPBA (251 mg, 1.48 nimol, 2,5 eq) was added under stirring and resulting mixture was allowed to stir at rt for 30 min. Further, ierf-butyl 7~ammo~3,4~dmydroisoqumohne~2(]/: -carboxylaie (145 nig, 0.58 nirnol, 1 eq) andDTPEA (0.4 ml,, 2,33 mrnol, 4 eq) were added and the reaction was allowed to stir at rt for 12 h. The progress of the reaction was monitored by 1 CMS After completion reaction was quencbed with water and. extracted with ethyl acetate (10 mL x 3), The combined organic layers were washed with brine solution (20 mL), dried over anhydrous a^SC^ and concentrated under reduced pressure io afford crude product which was purified by flash chromatography (1-10% MeOH: CH2C12) to afford, tert-butyl ί 3·ί . -dich] p!:o I i -i ·ο ;ο· > -i-^U i\^ ;··2//·ί rnnid i 5.-i · e][l,3]oxazm-7-y])ammo)-3,4-dihydro^ (225 mg, 71%) as white solid.
LCMS: 542 [Μ÷1 ] '
[0353] Siop-2: Synthesis of 3-{2,6- lichIoropheny!)-7-((l^,4-tetrahy lroisoquinolin-7- yl)ammo)-2^-dihydro- BT-pyriitnicIo [5,4-ej ( 1 ,3] oxazin-4-one hydrochloride:: 20% HC1 in dioxane (2 txiL) was added to a stirred solution of teri-butyl 7-((3-(2,6-dichlorophenyi)-4-oxo- 3J4-dihydro-2i -pymnido[5,4- ] l ,3 oxazin-7-yj)amino)-3,4-dihydiOis
carboxylase (220 mg, 0.40 mraol, 1.0 eq) in 1 mL dioxane maintained at 0 °C and the resulting solution was allowed to stir at rt for 3h, The progress of the reaction was monitored by LCMS, After completion of reaction, solvent was removed under reduced pressure, and the resulting solid was filtered and washed with ether., dried to afford 3 -(2,6-dich! orophenyl)-7-((4-(piperi din- 4-y])phenyl)araino)-2,3-dihydro-4H-pyrimido[5,4-t>][l ,3]oxazin-4-one hydrochloride (.160 mg, 82%) as a pale yellow solid.
LCMS: 441 [M÷l ]÷
[0354] Siep~3: Synthesis of 3^2,6-dichiorophenyI)-7-((2 2 ^
tetrafaydroisoquHiolHi-7-yl)amino^ [ 1,3] oxazia-4-oae: To a solution of 3--(2,6"dichlorophenyl}--7--((4"(pipen^
o[5,4-e][ 1.3]oxazin-4-one hydrochloride (100 nig, 0.21 rarnol, 1.0 eq) in 2 mL dry DTvlF at K2CO3 (86.6 rrsg, 0.63 rnmol, 3 eq) was added under stirring under inert atmosphere. Further, 2 · bromo- 1 , 1 -dif!uoroethane (61 mg, 0.42 mmoi, 1.0 eq) was added and the resulting solution was heated at 80 °C 12 h. The progress of the reaction was monitored by LCMS. After completion, the reaction was cooled to rt and quenched wife ice cold water followed by extraction using ethyl acetate (5 rnL x 3). The combined organic layer was washed with brine, dried over anhydrous Na2SC> and concentrated under reduced pressure. The residue was purified by flash
chromatography to afford 3-(2J6-dichloi'ophenyl)-7-((2-(2,2-diiluoi'oethyl)-l ,2,3,4- retra.hydroisoquinohm7w )a (14 mg, 13
%) as white solid formate salt,
LCMS: 505 [M-HX
JH NMM (400 MB*, DMSO-<l6): δ 10.36 (br s, Ϊ Η), 8.83 (s, 1H), 8.41 (br., 1 H), 7.66 (d, ../ === 7.89 Η¾ 2H), 7 35 - 7.55 (m, 3H), 7.08 (d, J - 8 33 Hz, 2H), 6.18 - 6 30 (rn, 1 H), 5.73 (s, 21:3), 3.71 (s, 2! !}. 2 87 2.97 (m, 2B), 2 79 (dd. ./ - 4.82, 13.59 Hz, 4H).
Example S66. Synthesis of3-(2,6 iichIoropheny)~7-((4-(3-{hydroxymeihy)piperazin~l- yijpkenyl)amino)-2, 3~dihydro-4H-pyrimido[5, 4-eJfl, 3 oxazin-4-one
(Compound No, 1.96)
[0355] Step-1: fer/-butyl 2~(hydroxymethyl)-4-(4-nitrophenyl)piperaziiie~l~carboxylate:
To a stirred solution of l-fluoro-4-nitrobenzene (1.0 g, 4.623 mrno!, 1.0 eq) and eri-butyl 2- (hy roxymethy )piperazine-l-carboxylate (0.717 g, 5.085 mmol, 1 .1 eq) in DMF (20 mL) was
added K -CO . (3.27 g, 9.247 mmol, 2.0 eq) at rt The resulting mixture was stirred ai 90°C for overnight. The progress of reaction was monitored by LCMS. The reaction mixture was poured into ice cold water (100 mL), stirred for 5 min, formation of precipitates was observed, which was filtered and dried under vacuum to afford the desired compound, tert-butyl 2- (hydroxymeihyl)-4-(4-nilTOpheny])pipera2;ine-l -caTboxylaie (600 mg. 32%) as yellow solid.
LCMS; 338 [M÷l] +
[0356] Sfep-2: Synthesis ofte/f- butyl 4-(4-amiaophenyl)-2-(hydroxyiRethyI)piperaz«ne-
J -carboxylate: To a stirred solution of tert-butyl 4-(4~aminophe«yl)-2- (hydroxymeihyl)piperazine-l -carboxylaie (0.5 g, 1.482 mmol, 1.0 eq) in EtOH (6 mL) was added Fe powder (0,248 g, 4.446 mmol, 3 eq) and a solution of NH C1 (0, 1 58 g, 2.964 mmol, 2 eq) in water (6 ml) at rt The resulting mixture was heated at 90°C for I h. The pi'ogress of reaction was monitored by LCMS. The reaction mixture was filtered through celite the residue was washed with EtOH (30 mL). The filtrate was concentrated and the residue was dissolved in EtOAc (30 mL), washed with water (2 >■ 30 mL), dried over Na2S(>4. and concentrated to afford the desired compound, tert-butyl 4"(4-aminophenylj"2"(liydroxymeihyl)piperazine--Lcarboxylaie (0.5 g, 96%).
LCMS: 308 [M+l
[0357] Step-3: Synthesis of tert-butyl 4-(4-((-^(2,6-dkhiorophenyI)-4-oxo-3,4-dihydro- 2H-pyriraido[5,4-e] [ly3]oxazm-7-yl)aramo)phenyl)-2-(hydroxynH¾thyI)piperazin
carboxylate: To a stirred solution of 3-(2,6-dicWorophenyl)-7-(methylthio)-2>3-dihydro-4H- pyrimido[5,4-e: | 1 ,3 joxazin-4-one (0.5 g 1.461 mmol. 1 .0 eq) in (10.0 mL) of toluene was added ffi-CPBA (0.502 g, 2.922 mmol. 2 eq) and allowed to stir at rt for 30 min, followed by addition of tert-butyl 4-(4-aminophenyi)-2-(hydroxymeth I )piperazme- 1 -carboxylate (0.494 g, 1.607 mmol, 1.1 eq) and DIPEA (0.753 g 5.844 mmol, 4.0 eq) and allowed to stir at rt for overnight. The reaction mixture was diluted with water (50 mL) and extracted with EtOAc (2 >■ 50 mL). The combined organic layers were washed with water (50 mL), brine (50 mL), dried over \ ·ΗΟ.· . concentrated and purified by combi flash [silica gel- .100-200 mesh; elation 0-35% EtOAc m hexane] to afford the desired compound, tert-butyl 4-(4-((3-(2,6-dichlorophenyl)-4-oxo-3,4-
dihydro--2H-pyrmiido[5,4-e] [l J jo^
carboxylase (0,230 g, 26%) as solid.
[0358J Siep~4: Synthesis of 3-(2,6-dichioropheHyl)-7-((4-(3-(hydroxymeth I)piperazia-l- yI) I)ami )- ^dlh dro-4Il- yrimld 554~ Mi ] x^i5i-4- sie: tert-butyl 4-(4-((3-(2,6- dichlorophenyl)-4-oxo-3,4-di^
(hydroxymeihyOpiperaziiie-l --carboxylaie (0.230 mg. 0.3823 mmol, 1 .0 eq) was dissolved in 4.0M-HC1 in dioxane (2 mL) and allowed to stir ai rt. for 1 b After completion of reaction, the reaction mixture was filtered arsd dried under reduced pressure and purified by diethyl etber to afford the desired compound, 3-(2,6-dicWorophenyl)-7-((4-(3-(hydroxyraethyl)piperazin-l- yl)pheny])amino)-2,3-dihydiO-4H-pyrinudo 5,4-e][l,3]oxazin-4-one (36 mg, 79%) as white solid.
LCMS ( \ l · M .·· 501 liPLC @ 254 ran = 95.36% and @ 220 ns = 97.11%.
*H NM (400 MH¾ DMSO-d*): 5 10.26 (br s> H), 8.79 (s, Hi), 7.65 (d, J - 8.33 Hz, 2H), 7.43 - 7.58 (m, 3B), 6 92 (d. 7 8.77 Hz, 2H), 5.71 (s, 2H), 4.76 (brs, IH), 3.55 (d, J --- 10.96 Hz, Hi!), 3.48 (d, / 10.96 Hz, I H), 3.40 (br s, 2Π . 3.03 (d, / 12.28 Hz, I H), 2 83 (br s, 2H), 2.56 - 2.72 (m, 21:1), 2.23 - 2.36 (m, 21 !)
Example S67. Synthesis of(R}-3-(2 )~dicPdorophenyl)-7-((4-(3~methylpiperimn-l- yijphenyI)amino)-2, 3~dihydro-4H-pyrimido[5, 4-eJfl, 3 oxazin-4-one
(Compound No, 1.173}
[0359] Step-1: Synthesis of tert-butyl 2-ffiethyl-4-(4-nitropheayl)piperazine~ 1- carboxylate: To a stirred solution of l-fiuoro- -nitrobenzene (1.0 g, 7.087 mrnoL 1 .0 eq) and tert-butyl 2~meihylpiperazine- i-carboxylate (1 .56 ;.·. 7.795 mmol, 1 , 1 eq) in DMT
' (20 m
'L) was added IsTCC (1.96 g, 14.174 mmol 2,0 eq) at rt. The resulting mixture was stirred at 90°C for overnight. The progress of reaction was monitored by LCMS. The reaction mixture was poured into ice cold vvater (50 niL), extracted with EtOAc (2 >< 50 niL) the combined organic layers were washed with water (50 aiL) with brine (50 ml) dned over
filtered and concentrated and, purified by com hi flash [silica gel 100-200 mesh: elution 0-5% EtOAc m hexane] to afford the desired compound, tert-butyi 2-methyl-4-(4-m†rophenyl)pip razine-I - earboxykie ( 1 ,4 g, 62%) as yellow solid.
[0360] Step-2: Synthesis of tert-butyi 4-(4-a mophe«yI)-2-ineihyIpiperazuie-l- carboxylafe: To a stirred solution of ieri-buiyl 2-methyl-4-(4-nitrophenyl )piperadne-l - carboxylate (1.10 g, 3.422 mmol, 1.0 eq) in EiOH (25 mi,) was added Fe (1.53 g, 27.381 musol, 8.0 eq) and a solution of NH C! (1,83 g, 34.22 mmol, 10.0 eq) in water (25 mL) at rt. The resulting mixture was heated at 90°C for 60 min The progress of reaction was monitored by LCMS. The reaction mixture was filtered through eehte the residue was w a hed with EtOB (50 mL) the filtrate was concentrated and the residue was dissolved in EtOAc (50 mL), w ashed with water (2 >
■ 50 mL), dried over N 2S0 , and concentrated to afford the desired compound, iert- butyl 4"(4"aminophenyl)--2"methylpiperazine"I"Carboxylaie (0.95 g, 95%) as brown viscous oil
[0361] Step-3: Synthesis of tart-butyl 4-(4-((-^(2,6-dkhiorophenyI)-4-oxo-3,4-dihydro- 2H-pyrimido[5,4-e][ly3]oxazm-7-yi)aramo)phenyl)-2-raethylpipera¾m To a stirred solution of 3-(2,6-dichloropheiiyl)-7-(methylthio)-2,3-dihydro-4∑J-pyriinidof'5,4- ejj' i ,3 joxazm--4-one (500 mg, 1.461 mmol, ; .0 eq) in (20.0 mL) of toluene was added /w-CFBA (720 mg. 2.922 mmol, 2.0 eq) and allowed to stir at rt for 30 min. iert-Butyl 4-(4-aminopheny !)- 2-methylpiperazine-I-carboxvlate (468 g, 1.607 mmol 1.1 eq) and DiPEA (1.02 mL, 5.844 mmol, 4.0 eq) were added and allowed to stir at rt for overnight. The reaction mixture was diluted with water (50 mL) and extracted with EtOAc (2 χ 50 mL), the combined organic layers were washed with water (50 ml,) with brine (50 mL) dried over Na^SO^, filtered and concentrated and purified by combi flash [silica gel 100-200 mesh, elution 0-35% EtOAc in hexane] to afford the desired compound, tert-butyl 4-(4-((3-(2,6-dichlorophenyl)-4-oxo-3,4- dihydro-2H~pymnido[5,4-e][l ,3]oxazm^
(0.413 g, 48%) as grey solid.
LCMS; 585.3 [MH j+
[0362] Step-4: Synthesis of 3-(2,6-dichIorophenyl)-7-((4-(3-methylpiperazm-j - yl)phenyl)mnino)-2 -difaydr0-4H-p}™ ieri-buiyl 4-(4-((3-(2,6- dichiorophenylV4-oxo-3,4»dihydro~2H^
niethylpiperazine-l -carboxylaie (400 mg, 0.683 mmol, 1.0 eq) was dissolved in 4.0 M-HC1 m dioxane (5 mL) and allowed to stir at ri for 1 h. After completion of reaction, the reaction
mixture was filtered and dried under reduced pressure and purified by diethyl ether to afford fee desired compound, 3-(2
!6-dichlorophenyl}-7-({ -(3-m thylpipefazin-l -yl)phenyl)an no)-2,3- dihydrO"4H-pyrimido[5,4~-e][] ,3 oxazm--4"One hydrochloride (160 mg, 48%) as yellow solid,
UPLC @ 254 nm - 97.98% and @ 220 nm = 98.24%
1Ή. MR (400 MBz, DMSO-d6): δ 10.24 (brs, HI), 8.78 (s, 1H), 7.59 - 7.71 (m, J= 8.33 Hz, 2H), 7.42■■ 7.58 (m, 3H), 6.80 - 6.98 (ni, J 8.77 Hz, 2B), 5.71 (s, 2B), 3.47 (t, J ------- 9.87 Hz,
2H), 2.96 (d, J-------- 1 1.84 Hz, I B), 2.73■■ 2.86 (m, 2H), 2.26 (brs, 1H), 2.17 (t ·/- 10.74 Bz, I B),
1.03 (d, J= 6.58 Hz, 3H).
Example S68. SyrHhesis of{R)-3-(2, 6-dichlorophenyl)~7-((3-fhioro-4-(3-methylpiperazin-l- yi phenyij amino)- 2, 3~dihydro~4H-pyrimido [5,4-ejiI, 3]oxazin~4-one
(Compound No. 1.174)
[0363] Step-1: Synthesis of tert-butyl 4-(2-fluoro-4-nitrophenyl)-2~methylpiper^ine~l~ carboxylate: To a stirred solution of 1 ,2-difluoro-4-nitrobenzene (1 .0 g, 6, 285 mmoL 1.0 eq) and test-bury! 2-methylpiperazrne-l -carboxy!ate (1.38 g, 6.914 mmol, 1 1 eq) in DMF (20 m.L) was added K2CQ3 (3,47 ;.·. 25.14 mmoL 4 0 eq) at rt. The resulting mixture was stirred ai 90°C for overnight. The progress of reaction was monitored by LCM.S. The reaction mixture was poured into ice cold water (100 mL), stirred for 5 mm, formation of precipitates was observed, which was filtered and dried under vacuum to afford the desired compound, tert-butyl 4-(2- fluorO"4--nitropheny!)-2-niethylpiperazine~l~-carboxyiate (1.92 g. 89%) as yellow solid.
LCMS: 340.1 ΓΜ÷1]÷
[0364] Siep-2: Synthesis of tert-butyl 4-{4-araiao-2-fi orophenyl)-2-methylpipera2:ine-i- carhoxylate: To a stirred solution of tert-butyl 4-(2dluoro-4-iutrophenyl)-2~met ylpiperazme--i -- carboxylase (1.0 g, 2.946 mrnol, 1.0 eq) in EtOH (20 mL) was added Fe ( 1.32 g, 23.573 mmol,
8.0 eqj and a solution of NH C! ( 1.56 g, 29.46 mmol, 10.0 eq) in water (20 mL) at rt. The resulting mixture w heated at 90°C for 60 nun. The progress of reaction was monitored by LCMS. The reaction mixture was filtered through cebte the residue was washed with EiOH (50 ml.) the filtrate was concentrated and the residue was dissolved in EtOAo (50 mL), washed with water (2
χ 50 mL), dried over \
'a ;HO.
; . and concentrated to afford the desired compound, tert- butyl 4-(4~amino-2-fluorophenyl)-2-methylpiperazine- ] -carboxylate (0.81 g, 89%) as brown viscous.
[0365] Step-3: Synthesis of tert-butyl 4-(4-((3-(2,6-dkhiorophe^^
2H-pyrimido[5,4-e][ly3] xazm-7-yi)aramo)-2-fiuorophenyl)-2-methylp«pera
carboxylate: To a stirred solution of 3-(2,6-dicWorophenyl)-7-(methylthio)-2,3-dihydro-4H- pyrimido[5,4-e] .l ,3 oxazin-4-one (350 mg, 1.461 mmol, 1.0 eq) in ( 10,0 mL) of toluene was added /JJ-CFBA (720 mg, 2.922 mmol, 2.0 eq) and al lowed ΐο stir at rt for 30 rain. tert-Buiyl 4- (4-aminO"2"fl tiorophenyl)'-2-metiiylpiperazine- l -carboxylate (497 mg, 1 .607 mmol, 1.1 eq) and DIPEA (1 .02 mL, 5.844 mmol. 4.0 eq) were added and al lowed to stir at rt for overnight. The reaction mixture was diluted with water (50 mL) and extracted with EtOAc (2 >■ 50 mL), the combined organic layers were washed with water (50 mL) with brine (50 mL) dried over Na2S(>4, iiliered and concentrated and purified by combi flash [silica gel 100-200 mesh; eiution 0-35% EtOAc m hexane] to afford the desired compound, tert-butyl ·ί ·(4·((5 ·(7.ό··
-2-metbylpiperazine- 1 --carboxylate (360 mg, 41%) as dark brown solid.
[0366] Step-4: Synthesis of 3-(2,6-dichIorophenyl)-7-((3-fluoro-4-(3-methyIpiperaziR- J - yl)phenyl) amino)~2 -dihydro-4H~pyrimido [5,4-e] l,3]oxazin-4-one: tert-butyl 4-(4-((3- (2,6~dichlorophenyl)-4-oxo~3x4"dihydrO"2H~pyriniidoi5,4~e] [ l ,3 oxazm -7-yl) amino)~-2~- fluorophenyl) -2-nietlwlpiperazine-l-carboxylate (350 mg, 0.579 mmol, 1.0 eqj was dissolved m 4.0 M-HC1 in dioxane (3 mL) and allowed to stir at rt for 1 h. After completion of reaction, the reaction mixture was filtered and dried under reduced pressure and purified by diethyl ether to afford the desired compound, 3"(2,6~dichlorophenyi)~7~((3~i uoro~4~(3~memylpiperazin-- l "
y!)phenyl)araixio)-2,3-dihydro-4H-pyrimido}5,4-e]n,3 joxazin-4-one hydrochloride (45 mg, 15%) as light yellow solid.
LCMS [M+l 504.2
UFLC 254 MI = 97.62% and (¾ 220 nm = 98.34%
{H NMM (400 M¾ DMSO-d6): δ 10.46 (brs, 1.H), 8.85 (s, 1.H), 7.61 - 7.75 (m, 2H), 7.46 - 7.56 (m, \ \ \ ). 7,40 (d, J:::: 7.02 Hz, 1 H), 7.03 it, J ------ 9.21 Hz, I B), 5.74 (s, 2H), 3.20 (d, J - 1 1.40
Hz, 2H), 2.99 · 3.08 (rn, 1H), 2.95 i d. J ------ 9.21 Hz, 2B), 2.59■■ 2.73 (m, 2H), 2.23■■ 2.40 (m, .?.! ) }
1.06 (d, J - 6, 58 Hz, 3H).
Example S69. SyrHhesis of 7~(lH-indol~5~yiamino)-3-(2, 6~diehiorophenyl)~2H-pyrimidof5.4- ejfl, 3]oxiizin-4(3H)~one
(Compound No. 1. 175)
[0367] Synthesis of 7-(lH-indoI-5-yl^^
e] l,3joxazin-4(3ii)-one: To a stirred solution of 3-(2,6-dicblorophenyl)-7-(methyltbio)-2I-i- pyrimido[5,4-e][I,3]oxazin~4(3H)-one (250 rrsg, 0.733 rnmol, 1 .0 eq) in 10 mL of toluene was added /JZ-CPBA (31 5 mg, 1.83 rnmol 2.5 eq) and allowed to stir ai rt for 30 min. l H-indol-5- amme (95 rng, 0.733 rnmol, 1 .0 eq) and DiPEA (378 mg, 2.93 rnmol, 4.0 eq) were added and stirred at rt for 2 h. Progress of reaction was monitored by LCMS. After consumption of starling material, precipitated compound was filtered off dned and purified by reverse phase
.CMS: 426
H MR (400 MHz, DMSO--4): δ 1 1.07 (br s, H), 8.80 (s, I H), 7.93 (br s, I H), 7.65 (d,
.89 .39 (ni.
[0368] Step-J : Synthesis of tert-butyi 4-(2
¾6-(limethyi-4-nitropheoyi)pipera¾ine-l- carboxylate: To a stirred solution of 2"fiuoro ,3 -dimeihyl--5-mitrobenzene ( 1.43 g, 8.45 mmoi, l eqj and tert-butyl piperazine- 1 -carboxylate (1 .73 mL, 9.29 mmoi, 1 .1 eq) ui DMSO (20 nil,) was added K2CO (3.5 g, 25.36 mmoi, 3.0 eq). Reaction mixture was stirred at 150°C for 1 6 h. Progress of reaction was monitored by LCMS. After consumption of starting material, ice cold water poured to the reaction mixture. The formation of precipitates was observed which was filtered to afford the desired compound, tert-butyl 4-(2,6-dimethyl-4-nitrophenyl)piperazine-I - carboxylase (370 mg, 13%) as yellow solid.
LCMS; 336.2 [M÷ l]÷
[0369] Step-2: Synthesis of (5~amino-2-(4-(tert-butoxj'carbony!)piper¾Ein- l~yl)-3- raethyl phenyl) methyiium: To a stirred solution of tert-butyl 4-(2,6-diroethyi-4- mh"ophenyl)piperazine-l-carboxyiaie (365 nig, i .08 mmoi, 1 eq) in EiOH and water (1 : 1, 10 mL) was added Fe (182.3 mg, 3.26 mmoi, 2 eq) and N¾Ci (1 16.4 mg, 2. 17 mmoi, 2 eq). The reaction mixture was stirred at 90 °€ for 2h. Progress of reaction was monitored by LC S. After consumption of starting material the reaction mixture was filtered through celite bed. The reaction mixture was concentrated, diluted with water, basifsed with NaHCC solution and was extracted with EtOAc, The organic layers were dried over a?S0 , concentrated and purified by column chromatography (Combiflash. elution: 0-2% MeOH in ( i Ι ΤΊ } to afford the desired product (5--aminO"2"(4"(tert-butoxycait!on (180 mg, 54%) as an off white solid.
[0370] Step-3: Synthesis of tert-butyl 4-(4-((3-(2,6-dichiorophenyl)-4-oxo-3,4-dihydro- 2II-pyri idoj5,4-ej [l^]oxazm-7-yl)a m^
To a stirred solution of (5--amino--2--(4~(tert~-butoxycarbonyl)piperazin--] ~yl)-3- methylphenyl)methyliimi (168 mg, 0.491 mmoi, 1 .0 eq) in toluene (5 mL) was added ??f~CPBA (212 mg, 1.22 mmoi, 2.5 eq) and allowed to stir at rt for 30 mm. iert-Butyl (2R,5S)-4-(4- am inophenyl}"2,5-dimet ylpiperazine~ l~carboxyiate (180 mg, 0.589 mmoi, 1 .2 eq) and DiPEA (0.33 mL, 1.96 mmoi. 4.0 eq) were added and stirred at rt for 10 h. The formation of precipitates was observed which was filtered to afford the desired compound, tert-butyl 4-(4-((3-(2,6
»
dimethylphenyl)piperazine-l -cafboxylate ( 100 mg, 30%) as an off white solid.
[0371] Step~4: Synthesis of 3-(2,6-dichioropheHyl)-7-((3,S-dimethyI- -(piperaziini-l- yl)phenyi) amino)-2 -dihydro~4H-pyriitnido[5,4-e] [1 ,3]oxazin-4-one hydrochloride:: tert- Butyl 4-(4-((3-(2,0-dichiorQphenyl)-4-oxo-3,4~d^
yl)arniiK))-2J6-dimei.hylpbenyl)pip razine-l -carboxy]ate (100 mg, 0.136 ramol, 1 .0 eq) was dissolved in dioxane (1 ml,) arsd added 4M dioxane-HCl (2 ηη . and allowed to stir ai rt for 1 h. After completion of reaction, the reaction mixture was filtered and dried under reduced pressure to afford the desired compound, 3-(2>6-dichioropheny!)-7-((3,5-diraethyi-4-(pipera2,.jn-l- yl)pheny])amino)-2,3-dihydiO-4H-pyrinudo 5>4-e] [l,3]oxazin-4-one hydrochloride (40 mg, 74%) as an off white solid,
LC S: 499.3 [M+l ]+ liPLC: @ 254 nni = 86.75% and @ 220 nm = 89.17%
*H NM (400 MH¾ DMSO- , HO salt): δ 10.32 (br s, 1H), 8.88 -· 8.8:2 (m, 2HI 7,70 (d,
./ >: .vi HZ, 2H), 7.56 - 7.52 (m, 1 I¾ 7.40 (br s, 2H), 5.77 (br s, 2H), 3.23 ·· 3.20 (m, 8H), 2.32 (s,
6H).
Example S7L Synthesis of 7-{lH-mdazol-5-yIamino}-3-(2, 6~dich!orophenyij~2H~pytimidoi5, 4- e [l, 3joxazin~4(3H)-one
(Compound No, 1.87)
[0372] To a stirred solution of 3-(2,6-dichlorophenyl)-7-(niethylthio)-2H-pyfimido[5,4- e][ l ,3"joxazin-4(3H)-one (250 mg, 0.733mmol, 1.0 eq) in 10 mL of toluene was added m-CPBA (315 mg, 1.83 rnmol, 2.5 eq) arid allowed io stir at rt for 30 nun. 1 H-indazol-5-amine (98 mg, 0.733 rnmo!, 1 ,0 eq) and DIPEA (378 mg, 2.93 mmol, 4.0 eq) were added and stirred at rt for 2h. Progress of reaction was monitored by LCMS. After consumption of starting material, precipitated compound was filtered off, dried arsd purified by reverse phase chromatography to afford (2 mg, 0.64%) of 7~(lH-indol~5~ylainino)~3~(2,6-dichlorophenyl)-2H~pyrimido 5.4- e] [ 1 ,3]oxazi n-4(3H)~one.
LCMS: 427 [M+l ]+
¾ MR (400 MHz, DMSO-d6): δ 13 02 (brs, I B), 8.84 is, I B), 8.07 is, I B), 7.66 (d, J 8.33 Hz, 3H), 7.48■- 7.59 (m, 4H), 5.73 (s, 2H).
Example S72. Synthesis qf3"(2; 6--dichIorOpherwl)-- 7-- methyI(4- f^
2, 3~dihydro~4H"pyrimido[5, 4~e]j L 3loxazin-4-one hydrochloride
(Compound No.1.104)
[0373] Ste -1: Synthesis of Jert-butyl 4-(4-((3-(2,6-didilorophenyi)-4-oxo-3,4-dihydro- 2/J-pyrimido [5,4-e] [l,3] xazm-7-yi)aramo)phenyl)pipera¾me-l-carboxylate To a stirred solution of 3--(2,6"diehlorophenyl)~7"(mei ^ ]oxazin-4-one
(300 mg, 0.87 rnmoL 1.0 eq) in toluene (4 niL); m-CPBA (376.9 nig, 2.19 nirnoL 2.5 eq) was added under stirring and resulting mixture was allowed to stir at rt for 30 minutes. Jen-butyl 4- (4-aminophenyl)ptperazine-i "Carboxylate (243. 1 nig, 0.87 rnmo!. 1 eq) and DIPEA (0.61 mL, 3.5 rnmol, 4 eq) were then added and the reaction was allowed to stir at rt for 16 h. The progress of the reaction was monitored by LCMS. After completion reaction vvas quenched with water and extracted with ethyl acetate (10 mL x 3). The combined organic layer was washed with brme solution (20 mL), dried over anliydrous a2S(> and concentrated under reduced pressure to afford crude product which was purified by flash chromatography (10-70% EtOAc/pet ether) to afford /m-hutyl 4-(4-((3-(2,6-dichlorophenyl)-4-oxo-3,4-dihydi-o-2H- pyriinido[5,4- ][.l J3]oxazin-7-yj)aniino)phenyl)piperaziiie-l -cai'boxylaie (205 mg, 41%) as yellow solid.
LCMS; 571 [M-H j
+
[0374] Step-2: Synthesis of tert-lmt l 4-(4-((3-(2,6-dichloropheny!)-4-oxo-3,4-dih¥dro- 2/i-pyrimido[5y4-e](l,3]oxa«n-7-y^ To a suspension of aH (21 mg, 0,52 nirnol, 1.5 eq) in dry DMF (2 mL), a solution of tert-buty] 4-(4- u 3· C -di i r ; Ο·4·ο;ο·34-|!h\ 4··ί··2//·! r:::;ido;5.4-!.'jj i .3 j x ui-^-
-earbo.xylate (200 mg.0.34 mmol, ieq) in dry DMF (2 mL) was added dropwise at 0 °C under inert atmosphere. The resulting mixture was stirred for 1-2 h at rt; followed by addition of Ol (0,05 ml.,, 0.69 mmol, 2 eq) The resulting solution was stirred at same temperature for 16 h. The progress of the reaction was monitored by LCMS. After completion of water (10 nfL), was added followed by extraction usmg MeOH/DCM (10 mL x 3). The combined organic layer was washed with bnne solution (10 mL x 2), dried over anhydrous Na
2S(>4 and concentrated under reduced pressure to afford crude product which was purified by fiash chromatography (MeOHDCM 1-10%) to afford ½7 ~butyi 4-(4-((3-(2,6-dich1orophenyl)-4- oxo-3, -dihydro~2H-pyrimido[5,4-e^
carboxylase (70 mg, 34%) as white solid.
LCMS: 585 [M l
[0375] Step-3: Synthesis of 3-(2,6-dichIorophenyi)-?-(methyl(4-(pipera2in-l- yl)phenyl)affiino)-2 "dihydro-4i-p unido(5,4-e]jI!3'|oxazin-4-one hydrochloride: 20% HCi m dioxane (2 mL) was added to a stirred solution of fert-butyl ·1··;.Ι··:;3··{2.0· dichlorophenyl)-4-oxo-3,4-dihydro-2H-pyrimido 5,4-g][l,3]oxazin-7- yl)(methyl)amino)phenyl)piperaziiie~l --carboxylate (65 mg, 0.11 mmoi, 1.0 eq) in dioxane (1 mL) at 0 °C under inert atmosphere and the resulting solution was allowed to stir at rt for 3 h. After completion, solvent was removed under reduced pressure and the resulting solid was filtered and washed with ether, dried to afford 3-(2!6~dichlorophenyl)-7~((4~(piperidin~4~ yl)pheiwl)a:iMno)~2,3-dihydro-4f/~pynmido[5,4~t, [l,3 oxazin-4-one hydrochloride (7 mg, 12%) as a yellow solid.
LCMS; 485 [M÷lf
¾ MR (400 MEte, DMSO- , FB): δ 8.71 (br s. Ills.7.64 (d. J-8.3 Hz, 2H) 7.51-7.46 (m, 1H) 7.21-7.18 (m, 2H), 7-6.98 (m, 2H), 5.65 (br s, 2H), 3.46 (br s, 3H), 3.19-3.17 (m, 4H), 2.99-2.97 (m, 4H).
FTxa ple S "3. Synthesis of 3 -(2, 6~dichiorophenyi)~ 7~{{3~((dimethyiamino)meihyi)~lH~indoi~6~ yl)amino)~2, 3~dihydro~4H~pyrimido 5.4-ef' fi, 3]oxazin~4~one
(Compound No. 1.107)
[0376] Step-l : Synthesis of 3-((dHnethylamino)raethyi)-J I3rindol~6-amine: To a. stirred solution of N,N-dim¾hyl-l-(6-niiro-i∑i-indol-3-yl)rneihananim (500 mg, 2,28 mraol, 1.0 eq) in (20.0 mL) of methanol was added 10% pd/C (50 mg) and purged with H2 at RT foe ih. s he reaction mixture was filtered through cel ite and residne was washed with methanol . The filtrate was concentrated to afford the desired compound 3~((diniet ylamino)niethyl)--] Ii"indoi--6~amine (400 mg, 92, 80%) as brown viscous.
[0377] Step-2: Synthesis of 3-(2,6-dichiorophenyl}-7-((3-((dimethyiamiao)methyl)-lH- i«dol-6-yl)aniioo)-2y3-dihydro-4H-pyrimido|S,4-e] [1 ]ο
χ82ΐ-«-4-ο
ηβ: 3-(2
s6-dich!orophenyf)-
( 180 mg, 0.526 mmoL 1.0 eq) in (3.0 mL) of toluene was added m-CPBA (260 nig. 1 .052 nimol, 2.0 eq) and allowed to stir at rt for 30 minutes. 3-((diineihylamino)meihyl)-lH-inclol-6-amine (1 10 mg, 0.578 nimol. 1.1 eq) and DIPEA. (0.37 mL, 2. 104 nimol, 4.0 eq) were added and allowed to stir at rt for overnight. The reaction mixture was concentrated, diluted with EtOAc (50 mL), washed with water (50 mi,), with brme (50 mL), dried over
'IN^SQ^ and concentrated, purified by reverse phase purification 3-(2,6-dichlorophenyl)-7-((3-((dirnethy!ammo)rnethy! )-lH-indo!-6-yl)amino)- pyrimido 5,4-e] [l,3]oxazin-4-one (4,0 mg, 1 ,57%%) as off white solid.
LCMS: 483.2 [M+l ; ! PLC @ 254 ran = 95.79% and @ 220 ns = 97.02%.
XH MR (400 M¾ DMSO-d6) δ 1 1.03 (brs, IH), 10.42 (s, 1.H), 8.82 is, IH), 8.20 (brs, IH), 8.02 (s, I H), 7.66 (d, J = 7.89 Hz, 2Hj, 7.33■- 7.61 (m, 2H), 7.1 0 - 7.33 (ni, 2H), 5.74 (s, 2H), 3.68 (brs, 21 k 2.26 (s, 6H). }, 1.95 (t, J = 6.36 ! ! /. 4H).
Example S74. Synthesis qf3-(2; 6 Uchlorophenyl)~7~((4~(4~(dimethylamino)piperidin~l~ yl)phenyl)aniino)~2, 3~dihydro~4H-pyrimidof5, 4-e]fI, 3 oxazin-4-one
(Compound No. 1.223)
[0378] Step-1 : Synthesis of AyV-dimethyl-l-(4-nitrophenyl)piperidin-4-amme: To a stirred solution of l-- 1uofo-4--niirobenzene (1 g, 7.08 rnmol, 1.0 eq) n DMSO ( 10 mL); N,N~ dimethylpiperidin-4-amine (1.56 g. 7.7 mniol, 2.5 eq) and DIPEA (4.93 niL. 28.3 rnmol) were added under stirring and resulting mixture was heated at 100 °C. The progress of the reaction was monitored by LCMS. After completion reaction was cooled and quenched with ice cold water.
The resulting solid precipitate was filtered, dried to afford AW-dimethyl-d --(4-- niirophenyi)piperidin-4-amine (1.6 g, 90%)as yellow solid.
LCMS: 250 [M÷l]÷
Step-2: Synthesis of ^-(^amin hm l^AyV-dinHif l i er dm-^ani ie. To a solution of
Ayv-dirneihyl-l-(4-nitrophenyl)piperidin-4-amme (1 .5 g, 6.01 mmo!, 1 eq) in EtO (15 mL), Fe powder (1 .GG8 g, 1 8,04 mmol, 3 eq) fol lowed by NB4C1 (0.643 g, 12.03 mmol, 2 eq) in water (15 mL) were added at rt under stirring. The resulting mixture was heated at 90 °C for 3-4 h. The progress of the reaction was monitored by LCMS. After completion reaction was cooled and the mixture was filtered through celite. The filtrate was concentrated, washed with water (5 mL x 2), followed by extraction using MeOH/DCM (10 ml, x 3). The combined organic layer was dried over anhydrous NajSGi and concentrated under reduced pressure to afford 1 -(4-aminophenyl)- Ayv-dimethylpiperidin-4-ai'nine (0.8 g, 37%) as brown solid.
[0379] Step-3: Synthesis of 3-(2,6-dkhlorophenyI)-7-((4-(4-(d^
yl}phenyi)ammo)-2^-dihydro-4£T-pyri^ To a stirred solution of
3-(2,6-dicli]oroph nyl)-7-(meihyM^ (200 nig, 0.58 mmol, 1.0 eq) in toluene (5 mL), m-CPBA (251 mg, 1 .46 mmol, 2.5 eq) was added under stirring and resulting mixture was allowed to stir at rt for 30 minutes. Further. ] ~(4~aminophenyl)-Av\- dunethylpiperidm--4-amine ( 128.1 mg, 0 58 mmol, 1 eq) and DIPEA (0 4 mL., 2,33 mmol., 4 eq) were added and the reaction was heated at 80 °C for 1 6 h The progress of the reaction was monitored by LCMS. After completion reaction was quenched with water and extracted with MeOII DCM (10 mL x 3). The combined organic layer was washed with brine solution (20 niL), dned over anhydrous x-a ·ΗΟ.· and concentrated under reduced pressure to afford crude product which was purified by flash chromatography (1- 10% MeOH/DCM) to afford · : ΤΟ- dichloropheny])-7-((4-(4-(dimethylam
pyrimido[5,4~t,)[l ,3 ]oxazin-4-one (1 00 mg, 33%) as white solid.
¾ MR (400 MHz, DMSO- ): □ 10.23 (s, IH), 8.78 (s, I H), 8.27 (brs, IH), 7.65 i d. ./ 8.33 Hz, 2H), 7.40 - 7.58 (m, 3H), 6 92 (d. ./ 8.77 Hz, 2H), 5.70 (s, 2H), 3.80 (s, 2H), 2.55 -■ 2.70 (m, 2H), 2.20 (s, 6i;i), 1.83 (d, J 1 1.84 Hz, 2H), 1.29 -■ 1.57 (m, 21 U
fj a p!e S75. Symthesis of 3~(2 )~dichiorophenyi)~7~{l,2,3,4-teirahydroquinoUn-7^ pyrimidofS, 4~e/fl, 3]oxazin-4(3H)-one
(Compound No. 1.121)
[0380] Step-1 : Synthesis of tert-butyl 7-(3-{2,6- lichIoropheny!)-4-o.xo-3,4-<i«hydro-2H- pyrimido [5,4-e] [ i ] oxaxm-T-ylainino^S^dihydroquinoIine- i (2II)-carboxyIa te: To a stirred solution of
(200 mg, 0.586 ismoi, 1.0 eq. ) m 3 mL of Toluene was added m-CFBA (352 mg, 1 .46 rranol, 2.5 eq.) and allowed to stir at RT for 30 minutes, tert-butyl 7-ammo-3,4~dihydroqiunoline~l (:2I j- earboxykie (144 mg, 0.586 mrnol, 1.0 eq. ) and DIPEA. (302 mg, 2.30 mmol, 4.0 eq. ) were added and allowed ιο stir at RT for I h. Progress of reaction was monitored by LCMS. After completion of reaction solvent was removed under reduced pressure, residue was diluted with 20ml of water and extracted with ethyl acetate (50 m'L-"3). Combined organic layer was washed with water (20mlx3), dried over anhydrous sodium sulfate and concentrated under reduced pressure. Crude was purified by flash chromatography to obtain 100 mg (31 .5%) of tert-butyl ?·· (3-(2,6-dichlofophenyl)-4-oxo-3,4-dihydro-2H-pyfirnido 5,4-eJf l ,3 oxazin-7-ylamino}-3,4~ dihydroquinohne-l(2H)-car boxy late.
LCMS: 542 [M+l ]+
[0381] Step-2; Synthesis of 3-( 2,6-dichIorophenyl)-7-{ i ^,3,4-tetrahydroqninoIin-7- ylamino)-2H-pyrimid [5,4-e] [l,3] .xazin-4(3H)-one. tert-butyl tert-butyl 7-(3-(2,6-
j 3 ίο.·;
;·/;ο· 7· ;;· : ·
·: - no) - 4 · dihydroqu oline~] (2H)-earboxylate ( l OOmg, 0.1 84 mrnol, 1 .0 eq. } was dissolved in 3 rnL of 4N HQ in dioxarse solution at Cf'C. Reaction was stirred at RT tor In. Progress of reaction was monitored by LCMS. Alter completion of reaction, precipitated compound was filtered oft, wasbed with 2 mL of dioxane and dned under reduced pressure, (
"rude was purified by reverse phase chromatography to obtain 12 mg (14.8%) tree base of 3-(2,6-dicblorophenyl)-7-(.l ,2,3,4- teti
"ahydi )qiimolin-7-ylamino)-2H-pyrirnido[5
J4-e][ l ,3 oxazin-4(3H)-one.
LCMS: 442 [M+l f
ill NMR (400 MB'z, DMSO-d6) δ 10.09 (br. 1H), 8.78 (s, 1H), 7.65 fd, J = 8.33 Hz, 2H), 7.43 ■■ 7.58 (m, 1H), 6.85 (br. s., 1H), 6.76 (br. s., 2H), 5.71 (s, 2H), 3.15 (br. s. , 3ΕΪ), 2.67 (br. s„ I B), 2.61 (d, J - 6.58 Hz, 2H), 1 .77 (br. s., 2H).
Example S76. Synthesis of 3"(2;6--dichIoropherwiJ--7--(i , 3,4- etrahydroqui
pyrimido/5, 4~e][L 3 oxazm~4(3 i) -one
(Compound No, 1.224)
[0382] Step-1: Synthesis of tert-butyl 6-(3-(2,6-dkh!orophenyl)-4-oxo-3,4-d«hydro-2H- pyrimido 5,4-e]
To a stirred
solution of 3--i2,6-dichiorophenyl)w
;-(mem^^
(200 mg, 0.586 mmol 1.0 eq. ) m 3 mL of Toluene was added m-€PBA (352 mg, 1 .46 mmol, 2.5 eq.) and allowed to stir at RT tor 30 minutes, tert-buty! 6-amino-3,4-dihydfoquinoline-l(2H)- carboxylate (144 mg, 0.586 mmol. 1.0 eq.) and DiPEA (302 mg, 2.30 mmol. 4.0 eq.) were added and allowed to stir at RT for 1 h. Progress of reaction was monitored by LCMS. After completion of reaction solvent was removed under reduced pressure, residue was diluted with 20ml of water and extracted with ethyl acetate (50 niLx3). Combined organic layer was washed with water (20mlx3), dried over anhydrous sodium sulfate and concentrated under reduced pressure. Crude was purified by flash chromatography to obtain 1 10 mg (34.7%) of tert-butyl 6- (3-(2,6~dich!orophenyl)- ~oxo-3,4-dihydro-2H.-pyrimido[5,4~e][l ,3]oxazm~7~ylanuno)~3,4- dihydroquinoline-l (2H)-carboxyla†e.
LCMS: 542 [M+l f
[0383] Siep-2: Synthesis of 3-(2,6-dichIor phenyl)-7-(j,2,3, -tetra ydroquhH>Im-6- yla«imo)-2H-pyrimido|S,4-e) [l,3;|oxazio-4(3H)-one: tert-butyl 6-(3-(2,6-dichloropheny I )-4- ox "3,4'-diliydro-2B"pyrimido| 5,4-e |Ί ,3 Joxazin-7-ylamino)-3,4-dihydi quinoline-l(2H)- carboxylate (90mg, 0. 166 mmol, 1 .0 eq. } was dissolved in 3 mL of 4N HQ in dioxane solution at 0'''C. Reaction was stirred at RT for Ih. Progress of reaction was monitored by LCMS. After completion of reaction, precipitated compound was filtered off, washed with 2 mL of dioxane and dried under reduced pressure. Crude was purified by reverse phase chromatography ιο obtain 50 mg (68.5%) HC1 of 3~(2,6--dic!tiofophenylV7- } ,23,4 e^
pyrimido 5>4-e] ['l,3]oxazin-4(3H)-one.
¾ N R (400 MHz, DMSO-d6): 6 1 0.84 (br. s., IH), 10.54 (br. s,, I H), 8.87 (s, Hi), 7.66 (d, J
7.89 Hz, 3H), 7.41 -■ 7.53 (m, I H), 7.22 (d, J 8.77 Hz, IH), 5.76 (s, 2H), 3.57 (s, 4H), 3.35 (br. s., 2H), 2.83 (L 7 6.36 Hz, 2H), 2.00 (br. s. , 2H).
Example S77. Synthesis oj'3~(2.6~dich!orophenyl)~7~((4~(2-(die†hylamm^
2, 3-dihydro-4H-pyrimido[5, 4~ej[l, 3]oxazin~4-one
(Compound No. 1.225)
|Ό384) To a stirred solution of 3-(2,6-dichloropheny!)-7-(methy!ihio)-2,3-dihydxO-4H- pynmido[5,4-e] [l,3]oxazin-4-one (150 mg, 0.438 mmol, 1 .0 eq) in (2.0 ■
■■)
'■.■ of toluene was added m-CPBA. (189 mg, 1 .095 mmol, 2.0 eq) and allowed to stir at rt for 30 minutes. 4-(2- (diethylamino)ethoxy)amlme (110 mg, 0.526 mmol 1.2 eqj and DIPEA (0.30 mL, 1.752 mmol, 4.0 eqj were added and allowed to stir at rt for overnight. The formation of precipitates was observed which was filtered to afford the desired compound 3-C2,6-dichiorophenyl)~7~((4-(2-
(18 mg,
8.1 7%) as white solid.
LOIS:: 502.2 [M÷l ; UPLC @ 220 nm - 90.72%.
JH NMR (400 MHz, DMSO~d6) d 1 0.31 (br. s., IH), 8.81 (s, IH), 7.67 (s, IH), 7.58 - 7.62 (m, J = 7.89 Hz, 2H), 7 48 (s, I H), 6 89 - 6.95 (m, . / 8.33 Hz, 2H), 5.72 is. 2H), 2.75 (br. s., 2H), 1.23 (br s., 2H), 1.00 (br. s, 9H)
Example S78, Synthesis qf3~(2, 6~ ichiorophenylj~ 7~((l,2, 3, 4~tetrahydroqidnox Iin~6~yl)csmijio)- 2, 3-djhydro-4H-pyrimidof5t 4-ejfl, 5]oxazin~4~one dihydrochloride
(Compound No. 1.226)
HCi
[0385] Step-I s Synthesis of di~tert-butyl 6-aitro-23-dihycIroquiiioxaHae- 1,4- dicarboxylate: To a stirred solution of 6-nit.ro- .2,3, -t trahydroquinoxaline (0.5 g, 2.79 mrnol, 1.0 eq) n DCM (20 ml,) was added tnethylarmne (1.1 7 mL, 8.37 mrnol, 3 0 eq) and DMAP (68 rng, 0, 558 mrnol, 0.2 eq) at rt. The resulting mixture was allowed to cool to 0°C followed by addition of li-tert-buiyl diearbonate (1 ,41 mi,, 6, 139 rnmol, 2, 2 eq), the reaction mixture was stirred at RT for overnight. The progress of reaction was monitored by LCMS. The reaction
mixture was diluted with DCM (50 mLj, and washed with water (2 x 50 nil,) dried over N
2S04. filtered and concentrated and purified by combi flash [silica gel 100-200 mesh; elution 0-35% EtOAe in Hexanej ιο afford the desired compound di-tert-butyl 6- tro-2,3-dihydroquinoxalme~ 1 ,4-dicarboxylat (0.65 g., 61 ,43%) as yellow viscous.
[0386] Step-2: Synthesis of di-tert-batyl 6-attM«o-2,3-dihydroqaiooxaii«e-l¾4- dicarboxylate: To a stirred solution of di-teri-butyl 6-nitro-2,3~dihydroquinGxalme- l ,4- dicarboxylate (0.65 g, 1 .713 mmo!, 1.0 eq) m EtOH (25 mL) was added Fe (765 g, 13.705 mmoL 8.0 eq) and a solution of N¾C1 (0,916 g, 17. 13 mmol, 10,0 eq) in water (25 mi . ) at rt. The resulting mixture was heated at 90°C tor 60 mm. The progress of reaction was monitored by LCMS. The reaction mixture was filtered through celite the residue was washed with EtOH (50 mL) the filtrate was concentrated and the residue was dissolved in EtOAc (50 mL), washed with water (2 x 50 mL), dried over Na^SCL, and concentrated to a fiord the desired di-tert-butyl 6- amino-2 -dihydroquinoxaiine~E4-dicarboxyiate (0,45 g, 76.79%) as brown viscous.
[0387] Step-3: Synthesis di-tert-butyl 6-((3-(2,6-dichIorophenyi)-4-oxo-3,4-dihydro-2H- ri.mk!o[5,4»e] ]o.x^ To a stirred solution of 3-(2,6-dichloropheny!)-7-(methy!ihio)-2,3-dihydxO-4H-pyrinudoi"5,4- ejj i ,3 joxazin--4-one (200 mg, 0.584 mmol. 1.0 eq) in (3.0 mL) of toluene was added m-CPBA (270 mg, 1.096 mmol, 2.0 eq) and allowed to stir at rt for 30 mmutes. di-tert-butyl 6»arnino~2,3- dihydroquinoxaiine-l ,4--dicarboxyiate (245 mg, 0.701 mmol, 1.2 eq) and DIPEA (0.40 mL, 2.336 mmol, 4.0 eq) were added and allowed to stir at rt for overnight. The reaction mixture was diluted with water (50 nil,) and extracted with EtOAc (2 / 50 mLj, the combined organic layers were washed with water (50 mL) with brine (50 mL) dried over Na?SOi, filtered and concentrated and purified by combi flash [silica gel 100-200 mesh; elution 0-50% EtOAc in Hexane to afford the desired compound di-tert-butyl (>■{ { ( 2/ ··d:c n : i■cπ^3 }·· !··· :\· :··3.1 · dihydro-2H~pyrimido[5,4-e][l ,3]oxazin-7-yl)^
(100 mg, 26.59%) as off white solid.
LCMS: 643.3 [M+lf
[0388] Step-4: Synthesis of 3-(2,6-dichioropke»yI)-7-((l,2 ,4-tetra ydroquiRoxaIin-6- yI)ai«ino)-2^-dihydro-4H-pyrimido{5,4-e] [1 ,3]oxa2in-4-one dlhydrochloride: di-tert-butyl 6- ((3--(2,6--dichlorophenyi)- --oxo-^,4^
dihydroquinoxaime-l ,4--di carboxylase (100 mg, 0,155 mmoL 1.0 eq) was dissolved in 4.0 M- HCi n dioxane (2 mL) and allowed to stir at rt for ih. After completion of reaction, the reaction mixture was filtered and dried under reduced pressure to afford the desired compound 3··;2.ίν
U:i. biO:O hO:: U" -(( i .2.3.4· · C:■ ! : dn ?i : ί ; ί Π i > - ¾; S : : ;■·'■· \ ' I )i; : : : :$ >}··2, - d : *· d:0-4l |··;.;\7! : : · id j 5.4· e][L3]oxazm-4»one dihydrochlori.de (60 mg, 71.42%) as solid.
LOIS:: 443.2 [M÷l ; UPLC @ 254 nm - 88.06% and @ 220 nm = 94.15%.
\li M (400 UX, DMSO~d6) δ 1038 (brs, IH), 8.83 (s, IH), 7.66 (d, / 8.33 Hz, 2H), 7.46
- 7.58 ( , IH), 7.24 - 7.38 (m, IH), 7.03 (brs, 2H), 5.95 (brs, 2H), 5.75 (s, 2H), 3.37 (d, ./ 8.33
Hz, 4H).
Example S?9, Synthesis of 3-(2,6-dichloropkenyI)~?~((I. ~dimethyl~l, 2,3,4- tetr hydroisoqidnolin~6~yl)amino)-2,3~dihydra~4H-^
(Compound No.1.227)
[0389] Step-1: Synthesis tert-bulyl 6-((3-(2,6-dichIorophenyi)-4-oxo-3,4-dihyd p riinidoi5,4-e|j ]©xaayhfi-7^
carboxylase: To a stirred solution of 3-(2,6-dichlorophenyi)-7-(methy!ihio)-2,3-dihydfo-4H- pyfimido[5,4-e] ]
'l ,3 joxazm-4-one ( 137 rng, 0.4 mraol 1.0 eq) in (2.0 rnL) of toluene was added m-CPBA (121 mg, 0.44 mmol, 1.1 eq) and allowed ΐο sin at rt for 30 minutes, tert- butyl 6- aniino-1 , l-dimethyl-3,4-dihydfoisoquinoline-2(lH)-cafboxylaie (121 nig, 0.44 mmol, 3 .1 eq) and DIPEA (0.28 mL, 0.28 mmol, 4.0 eq) were added and allowed to stir at rt for overnight. After completion of reaction, the reaction mixture was diluted with water and extracted with EtOAc (50 mL x 2). I
'he combined organic layer was washed with water (50 ml,), brme solution (50 mL), dried over anhydrous sodium sulfate and concentrated under reduced pressure to afford crude product, which was purified by flash chromatography [silica gel 100-200 mesh; elution 0- 50% EtOAc in hexane] to afford the desired compound, test-butyl 6-((3-(2,6-dichlorophenyl)-4- oxo-3, 4-dihydro-2H~pyrimido[5,4~e][l ,3]oxazirw~yi)ammo)- 1 ,1 -diniethyl~3,4- dihyd.roisoquino!me-2(!H)~carboxyiate (50mg, 21 .9 %) as sticky solid.
[0390] Step-2: Synthesis of 3-(2,6-dkhlorophenyI)-7-((l J -dsm t yl- 1 ,2,3.4- tef rahydroisoquinoIin-6-yl)amino)-2 -dihydro-4H-pyrimido [5,4-e j j 1 ,3] oxa2in-4-one: tert- butyl 6"ii3"i2,6-dichlorophenyl)'-4'-oxo-3,4"dihydrO"2H-pynmido[5,4'-e
l, l "dimeihyl"3,4'-dihydroisoqiunoline-2(] i
:i -carboxylate (40mg, 0.07 mmol) was dissolved in dioxane (0.4 mL), followed by dropwise addition of 4.0 M-HCi (0.4 mL) and allowed to stir at rt for 2h. After completion of reaction, the reaction mixture was filtered and dried to give 3··· 2. · d:·. h!o: o bo: · π·· ··{{ i
pyrimido[5,4-el [l ,3]oxazm-4-one (10 mg, 30.3%) as off white solid.
LCMS: 470.2 [M+l ; UPLC @ 254 ran = 90.49% and @ 220 nm = 91.73%.
; i l N R (400 MHz, DMSO-d6): δ 10.47 (brs., I ll), 9.12 (brs. 2H), 8.86 is, i l l ). 7.66 (d, J 780 Hz, 2H), 7.53 -· 7.61 (m, 21 k 7,50 (d, J = 7.45 Hz, 1H), 7.40 (d, J = 8.33 Hz, 1H), 5.75 (s, 2H), 3.44 (brs, 2H), 3.01 (brs, 2H), 1.64 (s, 6Hj.
Example SS0. Synthesis of 7-((3-chh ro-4-(3-methylpiperazm-l~yl)phenyi)am 6~ dichlorophenyi)"2, 3~dihydro~4H-pyrimido[5, -eIfl, 3 oxazin~4~one
[0391] Step-1: tert-butyl 4-(2-chloro-4-((3-(2,6-dkh!orophenyl)-4-oxo-3,4-d»hyclro-2H- pyrimido[5,4-e][1 ]oxazm-7-yl)amm^^ To a stirred solution of 3-(2,6~dichloropheny!)~7~(m
e][ l ,3]oxazm-4-one (1.70mg, 0, 5 mrnol . 1.0 eq) in of toluene (5.0 ml,) was added m-CPBA (247 rrrg, 1 ,0 mrnol, 2,0 eq) and allowed, to stir at rt for 30 minutes, tert-butyl 4-( -ami.n.o-2- chioro henyl)-
■2-
■πleίhyl φe .zine·
■l·
■carb xylaίe (1 80 mg, 0.55 mmol, 1 . 1 eq) and DIPEA (0.44 m l .. 2.5 mmol, 5 0 eq) were added nd allowed to stir at ri for overnight After completion of reaction, the reaction mixture was diluted with water and extracted with EtOAo (50 mL x 2), The combined organic layer was washed with water (50 mL), brine solution (50 mL), dried over anhydrous sodium sulfate arid concentrated under reduced pressnre to afford crude product, winch was purified by flash chromatography [silica gel 1 00-200 mesh, elution 0-50% EtOAc in hcxane] ιο afford the desired tert-butyl 4-(2-ch!oro-4-((3-(2,6-dichlorophenyl)-4-oxo-3,4- dihydro-2H~pymnido[5,4-e][l ,3]oxazin-7-yl)ainino)phenyi)-2-nieihyipiperazine-l-carboxylate (40mg, 1 2.8%) as light brown sticky liquid.
[0392] Step-2: Synthesis of 7 (3-chloro-4-(3-metfaylpiperazm^
(2, S
»dkMoro f^ tert-butyl 4~(2~
.
"! U i rs -
7· y})arnino)phenyI)-2-methyIpiperazine- 1 -carboxylate (40rng, 0.064mmol, 1 eq) was dissolved in
dioxane (0.4 mL), followed by dropwise addition of 4.0 M-HCi (0.4 mL) and allowed io stir at rt for Hi. After completion of reaction, the reaction mixture was filtered and dned io give s7-((3- chlofo-4- 3-ineihyj iperazm~l ~yl)phenyj)amino)-3-{2,6-dichloi phenyl)-2,3-dihydro-4H- pynmido[5,4-e][l,3]oxazin-4-one (10 mg, 30.3%) as off white solid.
LCMS: 519.2 [M+l ; UPLC @ 254 ran = 85.93 % and @ 220 rrm = 87.94 %.
; i l NMR (400 MHz, DMSO-d6). δ 10.54 (bra, 1 H). 8.87 (s, i l l ). 8.72 (bra, ! HS. 7.96 (brs, i ll), 7.60■- 7.72 (m, 3H)S 7.45 - 7.56 (m, 2H), 7.23 (d, 8.77 Hz, 1 H)S 5.76 (ss 2H), 3, 16 (brs, 3H), 2.94 (brs, 2H), 2.81 (d, . = 10.96 Hz, 2HI 1,28 (d, 6.14 Hz, 3H).
Example SSI. Synthesis qf(S)- 7~{{3~chioro~4~(3~meihyIpiperazin~l~yi)phenyi)amino)~3~{2, 6- dichlorophenyi)"2, 3~dihydro~4H-pyrimido[5, 4-eIfl, 3 oxazin~4~one
(Compound No.1.229)
[0393] Step-l: Synthesis of tert-butyl iS)-4-(2-chIorii-4-((3-(2,6-dkhiorophenyl)-4-oxo-
3,4-dihy d r -2H-pyrim id [5,4-e] [ .1,3 ] «xa¾in-7-yl)amin )phei¾yI)-2-KiethyIpipera¾me-
'i- earfooxylate: To a stirred solution of
pyrimido 5,4-e][l,3]oxazin-4-one (170 mg, 0,5 mmol, 1.0 eq) in of toluene (5.0 mL) was added m-CPRA (247 nig, 1 0 rnmol 2,0 eq) and allowed io stir a ri for 30 minute teit-buiyl (S}-4-{4-
anunO'-2'-chloropheny!)-2-nieihylpiperazine"i "Carboxylate ( 180 mg, 0.55 mtnol, 1. 1 eq) and DIPEA (0.44 mL, 2.5 mmol, 5.0 eq) were added and allowed to stir at rt for overnight After completion of reaction, the reaction mixture was diluted with water and extracted with EtOAc (50 ml, x 2). The combined organic layer was washed with water (50 mL), brine solution (50 mL). dried over anhydrous sodium sulfate and concentrated under reduced pressure to afford crude product, which was purified by flash chromatography [silica gel 100-200 mesh; elutton 0- 50% EtOAc m hexane] to afford the desrred tert-butyj (5)-4-(2-chloro~4~((3-(2s6~ dichiorophenyl)~4-oxo-3,4~dihydro~2H-py
methylpiperazme»l -carboxylate (60mg, 19.3%) as l ight brown sticky liquid. LCMS: 619.3 [M+l
[0394] Step-2: Synt esis of (S)-7-((3~c toro-4-i3-methYipiperaz¾~l~y!)phe yi)am½ )-3~ (2,6~dkhIorophenyl)-2 dihydro-4II^ tert-butyl ίό>4--(2~ ciiloro-4-((3-(2,6-dicMorophenyl)-4-oxo-3,^^
yl)aminoSphenyl)~2--niethylpiperazine-l -cai'boxykte (60mg, 0.096mmoi, 1 eq) was dissolved in dioxane (0.4 mL), followed by dropwise addition of 4.0 if i (0.4 mL) and allowed to stir at rt for lb. After completion of reaction, the reaction mixture was filtered and purified by preparative chromatography to give (6)-7-{ -chiofo-4-(3-niethyipiperazin-l -yl)phenyi)amino)-3-(2,6- dichlorophenyl}-2,3-dihydro-4H-pyrimido[5,4-e"j ] ,3 !oxazin-4-one (10 mg, 30.3%) as off white solid.
LCMS: 519.08 [M+l j+; I P! . @ 254 tim - 90.09 % and @ 220 nm - 93.75%.
*H MR (400 M¾ DMSO-d6): δ 10.47 (brs, IH), 8,85 (s, IH), 7.89 (brs, IH), 7.66 (d, J - 7.89 Hz. 2H), 7.58 7 62 (m, I H), 7.47 - 7.53 (rn, l id), 7.12 (d, J :::: 8.77 Hz, IH), 5.74 is, 2H), 3.06 (d, J 1 0.96 Hz, 3H), 2.79 - 2.93 (m, 4H), 1 .02 (d, / 6.14 Hz, 31:3).
Example S82. Synthesis of3-{2, 6~dichhrophe i)~7~(4~(4~methyljnp
2H-p rimido[5, - - jj j , 3]thiazin-4(3H)-one
(Compound No, 1,230}
1 395] Step-1: Synthesis of 4-hydr xy-2-(inethyIthio)pyrinii line-5-carboxyiic acid; To a sol ution of eiii l 4-chlofo-2-imethyl thiG)pyriniidine-5-earboxylate (30 g, 130.4 mmol, 1 eq) in EtOH (270 :)·! . ;. a solution of NaOH (52, 1 g, 1.3 rnol, 10 eq) in water (180 ml) was added under stirring. The resulting solution vvas heated at 100 °C for 2 h The progress of die reaction WHS monitored by LCMS. After completion of the reaction, mass was cooled to rt and acidified with cone. HQ (55 niL). The resulting precipitate was filtered, washed with water and dried io give 4- liydroxy'-2'-(inediylihio)pyriniidiiie -'Carboxylic acid (16.5 g, 69%) as a wh te solid.
[0396] LCMS: 187 [M÷ i f
[0397] Step-2: Synthesis of 4-c oro~2-(methylthio)pyrimicline-5-carbonyl chloride: To ethyl 4-c.b.loro-2-(met.bylthio)pyri.midine-5-carboxyj.ate ( 16.5 g, 88 7 mmol, 1 eq) in 500 niL round bottom flask, SO(¾ (1 50 ml,) followed by DMF (2.0 nil,) were added under stirring and the resulting solution was warmed io reflux for 2 h. Further, the solution was al lowed to cool io
ft and was concentfaied m vacuo io give 4-chloro-2-(meihyltluo)pyrifnidine-5-carbonyl chloride (12.5 g, 63%) as a white so d.
LCMS: 223 [M÷l ]÷
[0398] Step-3: Synthesis of 4-ch!oro-A-(2,6-dichloropheny!)-2-(methyhhio)pyriraidine- S-carboxaimide. To a stirred solution of 2,6-dichloroanilme (9.07 g, 56 mmol, leq)> in dry DCM
(60 mL) Et]N (23.43 ml.,, 1 68, 1 mmof 3 eq) was added followed by addition of 4-chloro-2- (memylthiojpyrinudme -catbonyl chloride (12.5 g, 56 mmol, 1 eq) in DCM (60 mL) under stirring under inert atmosphere. The resulting solution was stirred at rt for 2 h. The progress of the reaction was monitored by LCMS. After complete consumption of starting material, the reaction mass was diluted with water (50 niL) and extracted with DOM (50 mL x 3). The combined organic layer was dried over NasSO^ concentrated to afford 4~chioro--A
/--(2,6- dichloropheiwl)'-2-(methylthio)pyrimidine-5-carboxa.mide (12, 0 g, 61%) which was nsed without further purification.
[0399] Step-4: Synthesis of N-(2,6-dichl rophenyl)-4-mercapto-2-
(methyithio)pyriinidi«e-5-carhoxamide: To a stirring solution of 4-chloro-N-(2,6- dicbloropheiiyl}"2--(meiiiyluuo)pyrimidine--5--carboxamide (3.5 g, 10. 1 mnioL 1 eq ) in DMF (20 mL), was added sodium sulfide (1.60 g, 20. 17 mmol, 2.0 eq). The reaction was stirred at rt for 1 h. The progress of the reaction was monitored by LCMS. After completion of reaction, mixture was poured on ice cold water under stirring condition, precipitated compound was filtered off, washed with water and dried under vacuum to obtam 1 .35g (38.5%) o; Xd 2.6-d ic i r p! :enx h - 4"rnercapio~2~-(methylthio)pyrimidme~-5--carboxamide.
LCMS: 346 [M÷l]÷
[0400] S ep~S: S h sis of 3-(2,6-dk orophenyI)-7-(methyIthio)-2H-pyrimido[5,4- ej [l^]thia«in-4(3H)-one: N-(2,6-dichj.oroph nyl)-4-mercapto-2-(methylthio)py.rimidine-5- carboxamide (1 .35 g, 3.9 mmol. 1 eq) was suspended in acetonitrile (30 mL) and C iC L (3.80 g, 1 1.70 mmol, 4 eq) was added under stirring at rt. Dibromoniethane ( 1 .02 g, 5.85 mmol, 1.5 eq)
was then added and the resulting solution was stirred at HO 0 for 12 h. The progress of the reaction was monitored by LCMS. After completion of the reaction, fee nnxinre was diluted with water (20 nil,) and then extracted with EtOAc (20 ml, x 3). The combined organic extracts were washed with brine then dried over Na^SC^, filtered and concentrated to afford SOOrng (57.30%)
LCMS; 358 [M÷l f
[0401] Siep-6: Synthesis of 3-(2,6-dichl rophenyl)-7-(4-(4-raethylpiperazin-.l- yl)pheoyIattM«o)-2H-pyrimido[5>4-ej(1 ]th»^in-4(3H)-o«e: To a stirred solution of 3-(2,6- d :Ci : k>r :::':> - 7- i i) :C- i-:> i i h : }"2 Π · ί : : · ^ dol 5 4 I j ! . j I ί : ii> / :> :··4{ II V'- V- (200 ITig, 0.56 nimol,
1 eq) in toluene (7 niL), /w-CPBA (385 mg, 2.24 rnniol, 4.0 eq) was added under stirring and resulting mixture was allowed to stir at RT for In. Further, 4-(4--methyl iperazin
» l
»y!)anmne (1 1 8 mg, 0.616 rnmol, 1.1 eq) and DIPEA (434 nig, 3.36 nimol, 6.0 eq) were added and the reaction was stirred at rt for 1 h. The progress of the reaction was monitored by LCMS. After completion reaction, toluene was removed nnder reduced pressure; the mixture was diluted with water (20 mL) and then extracted with EtOAc (20 niL x 3). The combined organic extracts were washed with brine then dried over
filtered and concentrated. Crude was purified by reverse phase chromatography to afford 70nig (25.0%) formate
(4-(4--methylpiperazm'-h-yl)phen^
LCMS: 501 [M÷l]÷
*H M1 (400 MHz, BMSO-CI6) δ 10, 19 (s, 1 HI 8,76 (s, 1 H)S 7,64 (d, J - 7.89 Hz, 2H), 7.54 (d, J === 8.77 Hz, 2H), 7.38 - 7.50 (rn, 1H), 6.94 (d. J === 8.77 l . 2H), 5.22 (s, 2H), 3.16 (br. s., 4i-[), 2.66 (br. s , 4H1 2.1 5 -■ 2.43 (m, 31 Π
Example S83. Synlhesis qf3~(2, 6~dichIorophemyl)- 7-(4, 4-dimetkyi-l,2>3,4~tetrahy
7-ylamino)~2H~pyrimidof5.4-e/ ' [.!, 3j†hiazin-4(3H)-one
(Compound No, 1 , 231)
[0402] Step-I s Synthesis of tert-butyl 7-(3~(2,6-dichloropheny!)-4-oxo~3,4-dihydro-2H~ pyrimido[5,4-e] [.l^]thia;i^
carboxylate: To a stirred solution of 3-(2,6-dicWorophenyl)~7~(methylthio)-2H-pyriraido[5.4- e][I ,3]thiaziri-4(3i;i)~one (200 mg, 0.56 mmol. 1 eq) In toluene (5 mL), m-CPBA (385 mg, 2, 24 mmol, 4.0 eq) was added under stirring and resulting mixture was allowed to stir at RT for 1 h. Further, tert-butyl 7-axiimo-4>4-dimethyl-3>4-dihydroisoquinoline-2(l H)-carboxylate (1 55 mg, 0, 56 mraol, I eq) and DiPEA (433 mg, 3.36 mmol, 6 eq) were added and the reaction was stirred at ri for 12 h. The progress of the reaction was monitored by LCMS. After completion of reaction solvent was removed nnder reduced pressure, residue was diluted with 20ml of water and extracted with ethyl acetate (50 mL*3). Combined organic layer was washed with water (20mlx3). dried over anhydrous sodium sulfate and concentrated under reduced pressure. Crude was purified by flash chromatography using methario dichloromethane as eluents to obtain 220 mg ( 50% pure by LCMS) of tert-buiyl '' --; .i ihd:k r4 ;n π ·4 ·ο ,ο· ·> d-dihvdro- i ! · pyrirnidoj"5,4--e1 [ i,3 thiazir -yiamino}-4^
carboxylate.
LCMS: 586 [M÷l]
[0403] Step-2: Sy« thesis of 3-(2,6-dkhloropheayl)-7-(4,4-dii«ethyl-l,2 »4- teirahydro.soquinolin-7^ 4N OC! in dioxane (4 mL) was added to a stirred solution of tert-butyl 7-(3-(2,6~dichlorophenyl)~4~oxo~3.4- dihydro-2H-pyriinido[5,4-e] [l,3]iM^
2(l H)-car boxy late (200 mg, 0.34 nimol, 1 eq) in dioxane (1 mL) at 0 CC and the resulting solution was allowed to stir at rt for 1 h under inert atmosphere. The progress of the reaction was monitored by LCMS. After completion of reaction, resulting sol id was filtered off and washed with ether and dried. Crude was purified by reverse phase chromatography to afford 25 mg (30.5%) formate salt of 3--(2,6--dichlorophenyl)--7-(4,4»dimed
ylamino)-2H-pyriro.ido[5.,4-e][i ,3]thiazin-4(3H)-one.
LCMS: 486 [M+I]+
Hi N R (400 MHz, D.\ SO-d6: 610.27 (s, 1 H), 8.79 (s, 1 1 U. 7.65 (d, J --- 7.89 Π/. 2H), 7.40 - 7.57 (m, 2H), 7. 18 - 7.38 (m, 2H)S 5.24 (s, 2H), 3.84 (br. s., 3H), 2.61 - 2.84 (m, 2H), 1.21 (s, 6H)
Example S84. SyrHhesis of 3-(2.6-dichiorophexyi)-7~(4-(4-( imeihy
yi)phenyiam oE2H^ynmidoi5 -eli
(Compound No. 1.232)
|
'0404) To a stirred solution of 3-(2,6-dichioiOphenyl)-7-(methylthio)-2I-i-pyrimidoi
'5
!4- e][ l ,3 ]ti azuv-4(3H)--one (200 mg, 0.56 mmol, 1 eq) m toluene (5 ml), /w-CPBA (240 mg, 1.40 nmso!, 2.5 eq) was added under stirring and resulting mixture was allowed to stir at RT for Ih. Further, 1.~(4-aminophenyl)~N.N-diniethylpiperidin-4-ainine (147 mg, 0.672 mmol, 1.0 eq) and DIFEA (290 mg, 2.24 mrao!, 4.0 eq) were added and the reaction was stirred at rt for 12 h. The progress of the reaction was monitored by LCMS. After completion reaction, toluene was removed under reduced pressure; the mixture was diluted with water (20 ml,) and then extracted with EtOAc (20 niL x 3). The combined organic extracts were washed with brine then dried over Na
2S04, filtered and concentrated. Crude was purified by reverse phase chromatography to afford 5 nig free base of 3-(2,6-dichlorophenyl )-7-(4-(4-(diniethylai'nino)pipefidin-l - yl)phenylaxnino)-2H-pyrimido}5,4-e]n ,3 ]tluazin-4(3H)-one.
LCMS: 529.3 [M- l j+; UPLC @ 254 nm - 93.92% and @ 220 nm - 91.04%.
*H NMR (400 MHz, DMSO-d6): 610.17 (br, s., IH), 8.76 (s, IH), 7.64 (d, J = 8.33 Hz, 2H), 7.41 - 7.57 (m, 3H), 6 93 id, J === 9.21 Hz, 2H), 5.22 (s, 2H), 3.66 (d, J === 10.52 Hz, 2H), 2.65 (d, J - 19.73 Hz, 2H), 2.19 (s, 6H), 1.82 (d, J ===: 10.52 Hz. 2H), 1.64 (br. s.. 2H), 1 .38 - 1.53 (m, 3H).
Example S85. Synthesis of '3-(2,6-dichIoropheny])~7-((} ,2,3, -ie†rahydroisoquinohn-7-yI)amino}- 2, 3-dihydro-4H-pyrimido[5, 4-ejfl.3 ] thiazin-4-one hydrochloride ipoundNo. 1,233)
[0405] Step-1 : Synthesis of tert- butyl ?-((3-(2,6-dichIorophenyl)-4-oxo-3,4-dihydro-2H- pyrii«ido{5,4-e] [i^]thia2in-7-yI)ai«ino)-3,4-dihydroisoq«iooIine-2(l To a stirred solution of 3--(2,6-dichlorophenyl)-7-(metl^
e][l,3]thiazin-4-one (150 mg, 0.418 mmol, 1.0 eq) in (2.0 mL) of toluene was added rn-CPB (206 mg, 0.836 mmol, 2.0 eq) and allowed to stir at rt for 30 minutes, tert-butyl 7»amino~3,4- dihydroisoquinoline-2(lH)-carboxylate (115 mg, 0.46 mmol, 1 .1 eq) and DIPEA (0.29 mL, 1 .672 mmol. 4.0 eq) were added and allowed to stir at rt for overnight. After completion of reaction, the reaction mixture was diluted with water and extracted with EtOAc (50 mL x 2). Combined organic layer was washed wrth water (50 mL) bnne solution (50 ml,), dned over anhydrous sodium sulfate and concentrated under reduced pressure to afford crude product, which was purified by flash chromatography [silica gel 100-200 mesh: elution 0-50% EtOAc in hexane] to afford the desired tert-butyl 7-((3-(2,6-dichiorophenyl)-4-oxo-3,4-dihydro-2H- pyrnnido[5,4~e] [l ,3]thiazm~7~yi)amn^ (130 mg.
[0406] Step-2: Synthesis of ^2,6-di iIoro heByl)-7-^
yl)amino)-2 -dihydro-4H-pyriniido[5,4-ej[1 ] *«a?in- -one hydrochloride; tert-butyl 7-((3-
(2,6"dichlorophenyl)-4-' x "3,4'-diiiydrO'-2B"pyrimido| 5,4-'e"||" ; .3 ]thiazin-7-yl)amino)-3,4- dihydroisoquinoiine-2(lH)--carboxylate (130 mg, 0.233 mmol, 1.0 eq) was dissolved in 4.0 M~
HCl (2 mL) and allowed to stir at rt for ; h After completion of reaction, the reaction mixture was filtered and dned under reduced pressure io afford the desired compound 3--{2,6- dichlorophenyl)-7-({l ,2,3,4-tetrahyck isoqiiinolm-7-yl }amino)-2
e] J ,3 ]thiazin~4~one hydrochloride (105 mg, 91.30%) as light yellow solid.
LCMS: 458. 1 [M+l ; UPLC @ 254 ran = 98.16% and @ 220 nm = 97.43%.
\ NMR (400 MBz, BMSO-d6) δ 10.44 (s, Hi), 9.27 (brs, 2H), 8.82 (ss ΓΗ), 7,65 (d, J = 7.89 Hz, 2H), 7.58 (brs, 21 ) } 7.42■■ 7,52 (m, I H), 7.20 (d, J === 8.77 Hz, ΗΪ), 5.26 (s, 2H), 4.26 (brs, 3H), 3,36 (brs, 21 1 } 2.96 it, ./ 6.36 Hz, 2H).
fj a p!e S86. Symthesis of 3~(2 )-dich}orophenyi)-7-{{ 1 ,2,3,4-teirahydroisoqid
2, 5--i:Mr¾ k>- ?f 4~e][l, 3]thiazin~4~one hydrochloride
(Compound No. 1.234)
[0407] Step-1 : Synthesis of tert- butyl 6-((3-(2,6-dichIorophenyl)-4-oxo-3,4-dihydro-2H- pyrimido(5,4~eJ
To a stirred solution of 3--(2,6--dichloropheriyl)--7-- ^
e][L3]thiazin-4-one (150 mg, 0.418 mmol, 1.0 eq) in (2.0 mL) of toluene was added m-CPBA. (206 mg, 0.836 mmol, 2.0 eq) and allowed to stir at rt for 30 minutes, iert-buryl 6»amino~3.4- dihydroiSoquinotine-2(lH)-carboxy!ate (115 mg, 0.46 mmol, 1 .1 eq) and DIPEA (0.29 mL, 1 ,672 mmol . 4.0 eq) were added and allowed to stir at rt for overnight After completion of reaction, the reaction mixture was diluted, with water (20 ml..,) and extracted with EtOAc (20 mL x 2). Combined organic layer was washed with water (50 ml,) brine solution (50 mL), dned over anhydrous sodium sulfate and concentrated under reduced pressure to afford crude product, which was purified by flash chromatography [silica gel 100-200 mesh: elation 0-50% EtOAc in hexane] to afford the desired tert-butyl 6-((3-(2,6-dichioropheriyl)~4-ox.o-3.4-dihydro-2H- pyrimido[5,4-e] [ 1.3]thiazm-7-yl)ammo) -3 ,4~dihydroisoquinoline~2(lH}-carboxylate ( 130 mg, 56.41%) as brown solid
LC S; 558 3 [M÷ l]
[0408] Step-2: Synthesis of 3-(2,6-dichIorophenyl)-7-((l,2 ,4-tetra ydroisGquinoIin-6- yl)ai«ino)-2 dihydro-4H-pyriiRidoi5,4-e|| 1 ] thia2in-4-one hydrochloride: tert- butyl 6-((3■■ (2,6-dichloi"Gphenyj)-4-oxo-3,4~dihydro~2H- pynrmdo[5,4--e1 [ i ,3 thiazm--7yyl)ammo) -3,4- dilwdroiSoquirsoime-:2(lH)--carboxy!ate (130 nig, 0.233 mnio!, 1.0 eq) was dissolved in 4.0 M- HCi (2 mL) arsd allowed to stir at rt for lh. After completion of reaction, the reaction mixture was filtered arsd dried under reduced pressure to afford the desired compound 3-(2,6- <.!:·. b!o:o he:: n- ··(( 1.2.3.4··; e; nil : ;in. ·\· ·!·:>ί ϋ :··(; Ϊ• ::-;!:0 -2.3-d-bvdro--i; : ;- -doj :\ - e][l,3]thiazin- -oiie hydrochloride (70 mg, 60.86%) as yellow solid.
LOIS:: 458.1 [M÷l ; PLC @ 254 nm - 97.98% and @ 220 nm = 97.33%
1Ή. MR (400 MHz, DMSO-d6) δ 10.43 (s, IH), 9.34 (brs, 2H), 8.83 (s, IH), 7.55 - 7.70 (m, 4H), 7.45 - 7,53 (ni, IH), 7.19 (d, J = 8.33 Hz, 1H), 5.26 (s, 2H), 4,21 (brs, 2H), 3.35 (brs, 2H), 3.00 (t, J= 5.92 Hz, 2Hj.
Example SS7. Synthesis qf3-(2; 6 UchloropheMyl)~7~((3~methyl-4-(pipemzin-l-yl)phenyl)^ 2, 3~dihydro~4H-pyrimido[5, 4-ef 'j , 3]thiazin-4-one hydrochloride
(Compound No.1.235)
3 7
[0409] Step-l : Synthesis of tert-butyl 4-(4-((3-(2,6-dichloropheoyi)-4-oxo-3,4-dihydro- 2H~pyrimido[5,4Hc] (l,3]tht^ To a stirred solution of 3-(2,6-dichlorophenyl)-7-(methylm^
e][l,3]thiazin- -one (150 mg, 0.418 mmol, 1.0 eq) in (2.0 mL) of toluene was added m-CPBA. (206 mg, 0.836 mmol, 2.0 eq) and allowed to stir at rt for 30 minutes, tert- butyl 4- 4-amino-2- methylphenyl)piperazine~T~-carboxylate (134 mg, 0.459 mmol, 1.1 eq) and DIPEA (0.29 mL, 1 .672 mmol. 4.0 eq) were added and allowed to stir at rt for overnight. After completion of reaction, the reaction mixture was diluted with water (20 mL) and extracted with EtOAc (20 mL x 2). Combined organic layer was washed with water (50 mL) brine solution (50 mL), dned over anhydrous sodium sulfate ami concentrated under reduced pressure to afford crude product, which was purified by flash chromatography [silica gel 100-200 mesh: elution 0-50% EtOAc in hexane] to afford the desired tert-butyl 4-(4-((3-(2,6-dichlorophenyl)~4-oxo-3,4-dihydro-2H- pyrrmido[5,4~e] [l ,3]thiazin-7-yl)amino)-2-methylphe!iy])piperazine-l -carboxylate (1 32 mg, 52.38%) as brown solid.
[0410] Step-2: Synt esis of 3-(2,6-dkhlorophenyi)-7-((3-methyl-4-(pipera¾in-l- yI)phenyl)araino)-2^-d«hydro-4H-pyriraido[5,4-e][.l ]thiazin-4-cme hydrochloride: tert- butyl
; I . i- i ::a U :--?-- yl)amino)-'2-methylphenyl)piperazine- 1 -carboxylate (1 30 mg, 0.2 ; 6 mmol, 1 .0 eq) vvas dissolved in 4.0 M-HC! (2 mL) and allowed to stir at rt for I h. After completion of reaction, the reaction mixture was filtered and dned under reduced pressure to afford the desired compound 3··· . · d::: io: o l1c : · }··?··· · ··: ·Hm^ ϊ··4··{ pfoera/m- - vi e^ i -;mm io . -ui lw dr -
;H i - . ;m -dol 5 4·· e][ ; ,3 ]thiazi«~4~one hydrochloride (98 nig, 85.21 %) as yellow solid.
LCMS: 501.2 [M+l ; UPLC @ 254 ran = 98.40% and @ 220 ns = 97.30%.
NMR (400 MBz, BMSO-d6) δ 10.28 (s, Hi), 9.10 (brs, 2H)S 8.79 (s, IH), 7,65 (d, J = 7.89 Hz, 2H), 7.57 (d, J - 7.45 Hz, I l l s. 7.42■■ 7.51 (m, 2H), 7.04 (d, J - 8.77 Hz, IH), 5.24 (s, 2H), 3.22 (brs, 4H), 3.02 (brs, 4H), 2.26 (s, 3H).
fj a p!e S88. Synthesis of 7~((3~ehloro~4~(piperazin~ !~yl)phenyl)amin )-3-(2, 6-dichiorophenyi)- 2, 3-dihydro-4H~pyrimido[5, 4~e][l, 3]thiazin~4~one 2, 2, 2-trifluoroaeeta†e
(Compound No. 1.236)
[0411] Step-1 : Synthesis of tert-butyl 4-(2-chI ro-4-((3-(2,6-dkhl rophenyl)-4- xa-3,4- dihydro-2H-pyrimid0|5,4-e] [l^]th^ To a stirred solution of 3-(2,6-dichloropheny!)-7-(methy!ihio)-2,3-dihydxO-4H-pyrinudoi"5,4- e][ l ,3jtInazuv-4-one (1 50 mg, 0.41 8 mmol, 1 .0 eq) in (2.0 mL) of toluene was added rn-CPBA (206 mg, 0.836 mmol, 2.0 eq) and allowed io stir at it for 30 minutes, tert-butyl 4-(4-amino-2- chlorophenyl)piperazme--l --carboxylate (144 nig, 0.460 mmol, 1.1 eq) and D1PEA (0.29 mL, 1.672 mmol, 4.0 eq) were added and allowed to stir at rt for overnight. After completion of reaction, the reaction mixture was diluted with water (20 mL) and extracted with EiOAc (20 mL x 2). Combined organic layer was washed with water (50 mL) brine solution (50 mL), dried over anhydrous sodium sulfate and concentrated under reduced pressure to afford crude product, which was purified by flash chromatography [silica gel 100-200 mesh: elution 0-50% EtOAc in hexane] to afford the desired tert-butyl 4-(2-chloro-4-((3~(2,6-dichlorophenyl)-4-oxo-3,4-
dihydro-2H~pyfimido[5,4-e][l,3]thi (80 rng.
52.38%) as yellow solid.
LCMS: 621.3 [M- lf
[0412] S ep-2: Syn ss of 7-((3-dilor&-4~(piperazia-l-yi)pheMyl)amino)-3-(2,6- dkhlorophenyl^l^dihydro-^-pyrimicIoiS^-e] [ 1 ) thiazin-4-one 2,2,2-trifluoroacetate: teri-butyl 4-(2~chloro-4-((3-(2,6-dichlorophenyl)-4-oxo-3,4~dihydro-2H-pyrirnido[5,4^ e][l,3]thiazin-7-yl)arnino)phenyj.)pipera2:ine-l-mrboxylate (75 mg. 0,120 mmol, 1.0 eq) was dissolved in 4.0 M-HC1 (2 ml.) and allowed to stir at rt for 1h. After completion of reaction, the reaction mixture was filtered and dried under reduced pressure and purified by reverse phase purification to afford the desired compound 7-((3-chloro-4-(piperazin-l-yl)phenyl)ainjno)-3- (2J6-dichk>u)phenyl}-2,3-dihydt)-4H-pyrinudo 5J4-e][l,3]ihiazin-4-one 2,2,2-irifluoroacetaie (25 mg, 32.89%) as yellow solid.
LCMS: 5211 [M÷lf ; IJPLC @ 254 ran === 99.01% and @ 220 nm == 97.34%.
JH NM (400 MHz, DMSO-d6) δ 1.0.49 (s, 1H), 8.84 is, 1H), 8.73 (brs.2H), 7.89 (d, J- 2.19 Hz, 1H), 7.6! - 7.72 (m, 311) 7.44 - 7.52 ( , 1H), 7.23 (d, J= 8.77 Hz, 1H), 5.26 (s, 3H), 3.26 (brs, 4H), 3.06 - 3.21 (m, 4H).
Example S89, Synthesis of 3~(2,6-dkMorophenyl)~7~((3~meth xy-4~(piperazin~l~
yI)phenyi) mino)-2.3~dihydro~4N-pyrmidof5, 4-ej'fl, 3]thiazin-4-one hydrochloride
(Compound No.1.237)
[0413] Siep-1: Synthesis of tert-butyl 4-(4-((3-(2,6-dkhioropheayi)-4-oxo-3,4-dihydro- 2H-pyrinndoj5,4-e] [1 .Ithiazhi-7-yl)arahio)-2-metfaox}^ To a stirred solution of 3·ί ^. -di i r honx π· ·(: ·;·ϊ i;hi )- .3 ·ϋ;)·ν4κ···4Π·ρ nsmdoi 5.·;· e][l,3]thiazin-4-one (120 mg, 0.335 mmol, 1.0 eq) in (2.0 ml,) of toluene was added m-CPBA (165 mg, 0.670 mmol, 2.0 eq) and allowed to stir at rt for 30 minutes, tert-butyl 4»(4»ammo-2- methoxyphenylspiperazine-l -carboxylate (114 mg, 0.368 mmol.1.1 eq) and DIPEA (0.233 mL. 1.34 mmol, 40 eq) were added and allowed to stir at rt for overnight. After completion of reaction, the reaction mixture was diluted with water (20 mL) and extracted with EtOAc (20 ml, x 2), Combined organic layer was washed with water (50 mL) bone solution (50 mL). dried over anhydrous sodium sulfate and concentrated under reduced pressure to afford crude product, which was purified by flash chromatography [silica gel 100-200 mesh, elutiors 0-50% EtOAc in hexane] to afford the desired tert-butyl 4~(4-((3-(2,6~dichlorophenyl)-4-oxo~3,4-dihydro-2H~ pyrimido[5,4-e][l,3]thiazin-7-yl)am (110 mg,
53.39%) as off white solid.
LCMS: 617.4 [MHj
[0414] Step-2: Synthesis of 3-(2,0-dkMorophen i)-?-((3-methoxy-^ yI)pheny.)amino)-2^-dihydro-4H-^^ hydrochloride:; tert- bntyi 4~(4~((3-<2,6-dich!orophenylM--oxo^4--d
(110 nig, 0.1 78 -nmoL 1.0 eq) was dissolved m 4.0 M-HCi (1.5 rnL) and allowed to stir at rt for I . After completion of reaction, the reaction mixture was filtered and dried under reduced pressure to afford the desired compound 3
»(2,6--dichlorophenyl}--7--((3-rnem^
4H-pyrimi o[5,4-el[l ,3]ihiazin-4-one hydrochloride (98 mg, 99.21%) as yellow solid. LCMS: 517.2 [M÷l ; UPLC @ 254 nm - 95. 1% and @ 220 nm = 93.36%
1Ή. m (400 MB¾ DMSO-d6) δ 10.3.1 (s, 1H), 9.00 (brs, 2H), 8.81 (s, I H1 7.65 (d, J - 8.33 Hz, 2H), 7.48 (t, J = 7.89 Hz, 2H), 7.27 (d, 7 9.21 Hz, 1 H), 6.93 (d, J = 8.33 Hz, IH), 5.24 (s, 2H), 3.79 (s, 3H), 3.22 (brs, 4H), 3.1 6 (brs, 4H).
Example S90. Synthesis of 3~(2~chIoro~6~fl orophenyi)~ 7-((4,4~dimethyl~!, 2,3,4- ietrahydroisoqainolin- 7-y])amino)-2, 3-dihydro~4H~pyrimidof5, 4~e]jl, 3f' ihiazin~4~one
(Compound No. 1.238)
[0415] Sfep-l: Synthesis of tert-butyl 7-((3-(2-chl ro-6-i1 rophenyl)-4-o.x -3,4- d.Llr dro-7¾T-§¾TO
2( lH)-carboxyiate: To a stirred solution of 3 -(2-chloro--6--fluorophenyl)-7-(metlwlihio}"2,3-
dihydro-4H-pyfirnido[5,4-e] l ,3']thiazm-4'-oiie (170 mg, 0.5 mrnol, 1.0 eq) in (5.0 mL) of toluene was added m-CPBA (173 mg, 1 nmol, 2.0 eq) and allowed to stir at ri for 30 minutes, tert-buty! ·;:· ·:·ο· ·ί 4·· !;·:Η : \ l .4 -ddr. h :H q ;! :i;ne- ( I Π ;···:. a: bo ^ kao (152 mg, 0.55 mrnoi, 1.1 eq) and DIPEA (0.36 mL, 2 mmo!, 4.0 eq) were added and allowed to stir at ri for overnight After completion of reaction, the reaction mixture was diluted with water and extracted with EtOAc (50 ml, x 2). The combined organic layer was washed with water (50 mL), brine solution (50 mL). dried over anhydrous sodium sulfate and concentrated under reduced pressure to afford crude product, which was purified by flash chromatography [silica gel 100-200 mesh; elution 0- 50% EtOAc in hexane] to afford the desired compound, tert-buty! 7-((3-(2-ch!oro--6- f1uo.ropheny!)-4~oxo~3,4-dihydro-2H~pyri
dihydroisoquinoj.ine-2(l.H)-carboxylate (80 mg, 28%) as an off white solid. LCMS: 570.4 [M+l
[0416] Siep-2: Synthesis of 3-(2-ch!oro-6-flaorophenyi)-7-((4, -dimethy!-l,2 ,4- tetrahydro«soquinolin-7-yl)amin^^ tert- butyl 7-((3-(2-chloro-6-†1uorophenyl)-4^^
yl)ainino)-4,4-diin thyl-3J4-dihytiroisoquinoline-2(li:i)-carboxylat (80 mg, 0. 14 mraoi, 1 .0 eq) was dissolved in dioxane (1 mL), followed by dropwise addition of 4.0 M-HC1 (0.5 mL) and allowed to stir at rt for I h. After completion of reaction, the reaction mixture was filtered and dned under reduced pressure to afford the crude product and the desired compound, 3--(2--ch!oro- 6--i!uorophenyi)-7-((4,4'-dm½t!^
pyrimido[5,4-e][i ,3jtinazim4-one is obtained by prep 1 1 Pi .C (12 mg, 30.16%) as light brown solid.
LCMS; 470.2 [fVHlf : UFLC @ 254 ran - 93.49% and @ 220 nm - 89.67%.
!H NMR (400 MHz, DMSO-d6): d 10.30 br. s., IH), 8.80 (s, IH), 8.17 (br. s, I H), 7.50 (br. s. , 2i;i), 7.42 (br s., IH), 7.34 (d, J --- 7 45 Hz, 2H), 5.30 (d, / 13.1 5 Hz, I H), 5.22 (d, J :::: 12.72 Hz, IH), 3.93 (br. s., 21 ) }. 2.82 (br. s„ 2H), 1.24 (s, 6H).
Example S9L Synthesis qf5~((3-(2,6-di hlorophenyi}-4-oxo-3,4Hlihydro~2H-pynmM
e [ 1 ,3]ih}az -7-yl}arnino)-2-(4~(dimethylamino)piperidin^
(Compound No. 1.239)
[0417] To a sdrred solution of 3·ί 2 O-did' oi'ophem i ;·- /·-( -ν^ϋν. It h icy 13 -d; I :vdro--4! 1- pyrimido[5,4-e] [l,3]thiazin-4-one (1 50 mg, 0.418 mmoL 1.0 eq) in (2,0 ml.) of toluene was added m-CPBA (206 rrsg, 0.836 mmol, 2.0 eq) and allowed to stir at rt tor 30 minutes. 5-amino- 2-(4-(dimeth laxninojp endin- 1 -y Obexizonitrile (123 mg, 0.502 mmol, 1.1 eq) and DIPEA (0.29 mL, 1 ,672 mmol, 4,0 eq) were added and allowed to stir at ri for overnight After completion of reaction, the reaction mixture was diluted with water (20 niL) and extracted with EtOAc (20 mL x 2). Combined organic layer was washed with water (50 ml.) brine solution (50 niL). dned over anhydrous sodium sulfate and concentrated under reduced pressure to afford crude product, which was purified by reverse phase chromatography to afford the desired 5··; ; 3··; 22ν diehlorophenyl)-4-Gxo-3,4-dihydro-2H-pyri
(dimeihyiamino)piperidin~-l~-yi)benzonitn!e (5.0 mg, 2.1 5%) as off white solid. LCMS: 554.2 [M+l ; UPLC @ 254 ran = 99.03% and @ 220 nm = 97.82%.
\H NMR (400 MHz, D SO- ) δ 10.54 (brs, HI), 9.62 (brs, 1H), 8.85 (brs, H I ). 8.06 (brs, 1H), 7.88 (brs, 1 H), 7.66 (d, ./ 7.89 Hz, 2H), 7.49 (d, ./ 8.33 Hz, 1H), 7.24 (d, J - 9.65 Hz, I l l s. 5.27 (brs, 2H), 4.34 (s. I l l s. 2.79-2.67 (rn, 6H), 2. 12 (brs, 4H), 1 .77 i d J 12.28 Hz, 4H).
Example S92. Synthesis of 7-(3-ch]oro-4-(4-(dimethylamino)piperidin-l-yr)phenyla 6- dichiorophenyl)~2H~pyrimido[5A~s j [Ί ,3 thiazin~4(3 i)~one
(Compound No. 1.240)
[0418] Step-1 : Synthesis of 4-hydroxy-2-(methyIthlo)pyriitnicIine-5-earboxyik dd:: To a stirred solution of ethyl 4~chioro-2-imethyit io)pynmidine-5-carboxylate (5 ;.·. 21 425 mrnol, 1 eq) in EtOH (25 rnL) was added a solution of NaOH (8.38 g, 214 25 moL 1 0 eq) in water (25 mL) and the resultant mixture was stirred at RT for 16 h The progress of the reaction was monitored by TLC, After completion, the reaction mixture was acidified using 2N-HC1 (5 mi,) to obtain a precipitate was filtered, washed with water and dried to give 4-hydi
'oxy-2- (methy!thio)pynraidine-5-carboxylic acid (2.7 g, 67.5 %) as a white solid.
[0419] Step-2: Synthesis of 4-chloro-2-(methyhhio)pyrimidiae-5-carboayl chloride : To a stirred solution of 44 droxy--2--(meiiiylihio)pyri;Tiidme--5--carboxylic acid (2.5 g, 13.44 mrnol, 1
eqj in DMF (2.5 mL) was added SOC¾ (10 ml,} and the resultant mixture was heated at 100 °C tor 2 h. After completion, the mixture was cooled to RT was then concentrated in vacuo to give 4~cliloro~2~(meil ylthio)pyninidine~5'-carbonyl chlonde (2,65 g, 83.59 %) as an off-white solid.
[0420] Sfep-3: Synthesis of 4--chI r '-A^ 2, --dl€ld r he yI)~2~(meftyUhs ) lraW ¾e'- 5-carboxamide: To a stirred solution of 2,6-dichloroaniline (1.92 g, 1 1.8 mniol, l eq) m DCM (20 mL) was added E13N (4.96 ml,, 35.6 rnmol, 3 eq) followed by fee addition of 4--chloro-2- (methylthio)pyrimidine-5-carbonyl chloride (2,65 g, 11 8 mniol, 1 eq) in DCM (20 mi.-}. The resultant mixture was stirred at RT for 5 h. The progress of the reaction was monitored by TLC After completion, the reaction mixture was diluted with water (50 mL) and extracted with DCM (50 mL >< 3). The combined organic layers were dried over ^SO,^ filtered and concentrated to afford 4-chloro~A/:- (2,6--dichlorophenyl)"2-4meihylthio)pyrimidine--5--carboxamide (1.9 g. 47 %) which was used in the next step without further purification.
LCMS; 349 [M÷! ]÷
[0421] Step-4: Synthesis of N-(2,6~dichIorophenyl)-4-mercapto~2- iraethyithio)pyriraidine-5-carbo.xamidei To a stirred solution of 4~chioro-.A?-(2,6- dichlorophenyl)-2~(methylthio)pyrimidine~5~carboxamide (1.3 g, 5.72 mniol, 1 eq) sn DMF (20 mL) ·■ water (1 0 ml,) was added Na2S (0.582 g, 4.47 mmoh 1, 2 eq) ai RT and the resultant mixture was stirred at RT for 1 h. The progress of the reaction was monitored by TLC. After completion, the mixture was diluted with water (50 ml,) and extracted with DCM (100 ml, x 2). The combined organic layers were dried over Na?S(>4., filtered and concentrated to obtain a crude residue which was purified by column chromatography compound eluting at 50% EtOAc/ hexane to afford N-(2,6-dicMorophenyl)-4-mercapto^
(0,35 g, 27.13 %) as an off- white solid.
LCMS: 347 [M÷l'f
[0422] Step-5: Synthesis of 3-(2,6-dkhloropheoyI)-7-(methyitlMo)-2H-pyrimido[5¾4- e) [l,3]thiazia-4(3'H)-one: To a stirred solution N-(2,6-dichlofopheny!)-4-mercapto-2-
(methylthio)pyrimidine-5-carboxamicle (0.35 g, 1.03 3 mmoi, i eq) in acetoniirile (5 mL) and added ί s.-CO (1.3 g, 4.043 mmoi, 4 eq) at T and the mixture was stirred for 10 mm. Dibromomethane (0.263 g, 1 .516 mmoi, 3 eq) was then added and the rmxtute and the mixture was stirred at 50 °C for 3 h. The progress of the reaction was monitored by ! I i After complet on, the mixture was diluted with water (20 mL) and then extracted with EtOAc (20 mL x 3). The combined organic layers were washed with brme (25 mL), dried over a^SO,^, filtered and concentrated to afford 3~(2,6- ichlorophenyi}"?"(methyithio)--2H"pyriniido[5,4-- e] [ 1 ,3]thiazin-4(3H)-one (0.2 g, 55.24 %) as an off-white solid which was taken to next step without further purification,
LCMS: 359 [M+l
[0423] Step-6: Synthesis of 7-(3-ch!oro-4-( -(dimethy!amino)piperidin-l- yl)phenylaraino)-3-(2,6~dkhlorophmy^^^ a stirred solution of 3-(2,6-dicMorophenyl)-7-(methylth^
one (100 mg, 0.279 mmoi, 1 eq) in toluene (5 mL) was added ???~CFBA (96.33 mg, 0.558 mmoi, 2 eq) at 0 °C and mixture was stirred at RT for 1 h. DIPEA (0.25 mL, 1.395 mmoi, 5 eq) and !■■ (4-'aminO"2"Chlorophenyi)--N(N'-dimeihylpiperidin--4--amine (77.7 nig, 0.334 mmoi, 1 eq) were then successively added to the mixture and the mixture was heated at 60 "(' for 3 h. The progress of the reaction was monitored by TLC. After completion, the mixture was diluted ife water (20 niL) and the extracted with EtOAc (20 mL >■ 3). The combined organic layers were washed with brine (25 mL), dried over a2S(>4. filtered and concentrated to afford crude which was purified by reversed-phase HPLC to afford 7-(3-chloiO-4-(4-(dimethylamino)piperidin-l - yl)phenylarmno)~3~(2,6-dichlorophenyl)-2H~py (1 mg, 0.46
%) as an off white solid.
LCMS- 564 [M l
[0424] Step-1: Synthesis of tert-butyl 7-((3-{2,6-<JjchIorophenyl)-4-o.xo-3,4-<Jjhydro-2H- pyrimidoi5,4-e][1 ]thjazm-7-yl)ammo)-4,4-diraethyi-3,4-dihy
carbo n>; - o a stir red jiution of )-(2,6 -dich! orophens l)-7-(methylthio)- i,3~dihydro~4H- do[: -i-e. ! JU i ia one (300 n Oi.} iO ^.ώ.',' Oil., j oi t iueoe wat> aodoo Γ ::»Χ .i?/ Λv (413 ffi¾ 1.67 nd allowed to rruies, tert-butyl
7-ami. 10-4 ,4-d nethyl-3, 4-dih 'droisoqtun ^Ή^ΓΪΟ~ (lH)-cai ate (278 mg.1.00 SrnmoL 1.2 eq) and Γ (0 ! I 8 mrrsoL 4 were a ieo < Hid allow !°<"> +V
; ed to st Ί
ύ 8.1 f!f
i I : O
>vl
overnight. After completion of reaction, the reaction mixture was diluted with water (20 mL) and extracted with EtOAc (20 nL x 2). Combined organic layer was washed with water (50 rnL) brme solution (50 mL), dried over anhydrous sodium sulfate and concentrated under reduced pressure to afford crude product, which was purified by flash chromatography [silica gel 1 00-200 mesh; elution 0-50% EtOAc in hexane] to afford the desired tert-bntyl 7-((3-(2,6- dichioropheny!)-4-oxo-3,4-diliydro-2Ii-pyrimido[5,4-e][l ,3]thiazin-7-yl)amino)-4,4-dimethyi- 3,4-dihydroisoquinolme-2(IH)-carboxylate (260 mg. 52.95%) as brown solid.
[0425] Siep-2: Synt esis of 3-(2,6-dich!orophenyi)-7-((4^ -dimethyl-L2y3,4~ tetrahydroisoqulnoHn-7-yl)amino)-2 -dihydro-4H-pyrjmi
hydrochloride; tert-butyl 7-((3-(2,6-dichlorophenyl)-4-oxo-3,4-dihydro-2H-pyrimido[5,4- ][l ,3]ihiazm-7-yj)ammo)-4,4~dim ^ (260 mg,
0,443 rnmol, 1 .0 eq) was dissolved in 4.0 M--HC! (2, 0 mL) and allowed to stir at rt for 1 h. After completion of reaction, the reaction mixture was filtered and dried under reduced pressure to afford the desired compound 3-(2,6-dichlorophenyl)-7-((4,4-dimethy 1-1 ,2,3, 4- teira.hydroisoquinotin-7-y^
hydrochloride (220 mg, 95, 23%) as brown solid.
LCMS; 486.2 [MH j+
[0426] Step-3:- Synthesis of 3-(2,6-dichlorophenyl)-7-((2,4,4-trimethyl-l,2^ tetrafaydroisoquinolia-7-yl)amin^^ a stirred solution of 3-(2,6-dichlorophenyl)-7-((4,4-dime hyl- ,2.3,4-tetrahydroisoquinolin -7- yljammo)- 2,3-dihydro-4H- pyrimido [5,4-e
"j [1 ,3] thiazin-4-one hydrochloride (220 mg, 0.42 rnmol, 1.0 eq) and Hi U (0.063 mL, 0.841 rnmol, 2.0 eq) in 1,2-dich!oroethane (10 mL) was dropwise added acetic acid (0 025 mL, 0.42 mmol, 1.0 eq) at 0"C. The resulting mixture was stirred at rt for lb followed by addition of NaBH(CH
3CO)
3 (98 mg, 0.462 mmol, 1 . 1 eq) at 0°C. The resulting mixture was stirred at rt for Ih. The progress of reaction was monitored by LCMS. The resection mixture was concentrated, basified with saturated solution of NaHCCL (50 mL) extracted with EtOAc (2.x 50 niL). The combined organic extracts were washed with water (50
: · ;! . ). with brine (50 rnL) dried over Na2SOd arsd concentrated ursder reduced and purified by combi flash chromatography io afford the desired compound 3'-(2.6-diclnoropheriy0-7-((2,4.,4-
e] [ 1 ,3]thiazi n-4-one (35 rng, 16.66%) as white solid.
LCMS: 500 2 [M÷ lf ; IJPLC @ 254 ran === 96.66% and @ 220 nrn == 96.84%.
*11 M:R (400 MH2, DMSO-de) δ 10.28 (brs, 1H), 8.79 (s, IH), 7.65 (d, ·/ 8.33 Hz, 2H), 7.41 - 7.54 (m, 2H), 7.26 - 7.36 (m, 2H), 5.24 (s5 2H), 3.43 (brs, 2H), 2.32 (brs, 5H), 1 .23 (s, 6H).
Example S94. Synthesis of 3~(2,6~dichhrop- heml)-7-(4-(4~(dimei yIam
methoxyphenyIam o}~2H~pyr ido[5, 4-e j [1 ,3]thiazin-4(3H)~one 2,2, 2~ir(fiuoroaceuc acid
(Compound No. 1,242)
[0427] Step-1 : Synthesis of 1^2-methoxy-4-«itropheayi)-N^i-dimethylpiperid«i-4- amine: To a stirred solution of l -fluoro-2-methoxy-4-nitrobenzene ( 1.0 g. 5.843 mmol, 1 .0 eq) and N, ~diniethylpipendni--4--amine dihydrochlonde (1.40 g, 7.012 mnio!, 1 .2 eq) in DMF ( 10 mL) was added potassium carbonate (2.42 g, 17.529 mmo!, 2.0 eq) at rt. The resulting mixture was stirred at 90°C for overnight. The progress of reaction was monitored by LCMS. The reaction mixture was poured into ice cold water (1 00 mL), extracted with EtOAc (2x50 mL). The combined organic extracts were washed with water (50 mL), with brine (50 mL) dried over Na?SC>4 and concentrated under reduced pressure to afford mixture of desired compound l-(2-
(1 .6 g. 98.15%) as yellow viscous.
LCMS: 280.2 [M- l f
[0428] Step-2: Sy« thesis of l-(4-ammo-2-methoxyphenyi)- ¾ -d5methyIpiperidm-4- amine: To a stirred solution of l-(2-meihoxy-4-nitrophenyl)~N,N-dimethylpiperidin-4--amine (1.6 g, 5.727 mmoi, 1.0 eq) in EtOH (25 mL) was added Fe (2.56 g, 45.622 mmol 8.0 eq) and a solution of H4CI (3.0 g, 57.27 mmol, 10.0 eq) in water (25 mL) at rt. Th resulting mixture was heated at 90°C for 60 mm. The progress of reaction was monitored by LCMS. The reaction mixture was filtered through cehte the residue was washed with EiOH (50 mL) the filtrate was concentrated and the residue WHS dissolved in EtOAc (50 mL), washed with water (2 x 50 mL), dried over Na^SCL, and concentrated to afford the desired l -(4-amino-2-metlioxyphenyl)- ,N- dimethyipiperidin-4-amine (1 , 10 g, 77 46%) as brown solid.
LCMS: 250.1 [M+l
[0429] Step-3: Synthesis of ^(2,6-dich!oropheiny -7-i4-(4-(dimethylammo)piperidin-l- yl)-3-methoxyphenylamin )-2ii-pyrimjdo[5,4-e] [i ,3] th«azin-4(3H)-one 2,2^2-trifiuoroacetk acid : To a stirred solution of 3-(2,6-dicblorophenyl)-7-(methylt io)-2,3-dihydro-4 - pyrimido 5J4-el [l,3"|thiazin-4-one (1 60 mg. 0.446 mmol. 1.0 eq) in (5.0 mL) of toluene was added mCPBA (220 mg. 0.892 mmol 2.0 eq) and allowed 10 stir at rt for 30 minutes. l -(4- aminO'-2'-methoxyphenyl)'-N,N-'dimethylpiperidin--4--annne ( 134 mg, 0.536 mmol, 1.2 eq) and DIPEA (0.31 mL, 1 .784 mmol, 4.0 eq) were added and allowed to stir at rt for overnight. The reaction mixture WHS diluted with water (50 mL) and extracted with EtOAc (2 x 50 mL), the
combined orgamc layers were washed with water (50 mL) wife brme (50 mL) dried over Na2S(>4, filtered and concentrated and purified by reverse phase chromatography to afford the desired compound 3-(2,6-dichlorophenyl)-7-((4-(4-(dimethylarnino)piperidin-l - yl)phenyl)arnino)-2,3-djhydro-4E}-pyriraido[5,4-e][.l >3]thiazin-4-one 2,2,2-ftifluoroacetic acid (32 mg, 11.1 1%) as off ye! low solid.
LCMS: 559.3 [Mt-l ; UPLC @ 254 nm == 98.95% and @ 220 nm === 98.80%. i NMR (400 ΜΗΪ, DMSO-de) δ 10.31 (s, I B), 9.99 (brs, 1H), 8.81 (s, 1H), 7.65 (d, J - 7.89 Hz, 2H), 7.48 (t, . 8. I I Hz, 2H), 7.26 (d, J= 8.77 Hz, IH), 6.98 (brs, IH), 5.25 i s. 2H), 3.80 i s. 3H), 3.48 (d, ./ Π .40 Hz, 2H), 3 27 (brs, I H), 2.78 (d, J = 4.82 Hz, 6H), 2.67 (brs, IH), 2.08 (d, ./ 11.40 Hz, 21:3),. 1 .83 (brs, 2H).
.Example S95. Synthesis of 7~((3~chloro~4~(2~methylpiperazin~ l~yi)pheny!)amiiKp~3~(2i 6- dichlorophenyl)-2, 3-dihydro~4H~pyrimido[5, 4-ejfl, 3]thiazin-4-one dihydrochloride
(Compound No. 1.243)
[0430] Step-1 : Sy« thesis of tert-butyi 4-(2-chIoro-4-((3-(2
¾6-dichlorophenyl)-4-oxo-3
¾4- dihydro-2H-pyrimido|5,4-e] [1 jthi^
carboxyiate: To a stirred solution of 3-(2,6-dichlorophenyl)-7- ineihyjihio)-2,3-dihydfo-4H- pynmido[5,4-e] [l,3]thiazin~4-one (200 mg. 0.558 mmol, 1.0 eq) in (3,0 niL) of toluene was added mCPBA (275 mg, 3 .116 mmol, 2.0 eq) and allowed to stir at rt for 30 minutes, tert-butyi 4-·(4"3^ι ο·-2·-ε!ι!θΓορ1ΐ6η 1)--3--ηΐ6ί1ιγ1 φ6Γ3ζί η6~Τ~-θ3Αοχνΐ3ίβ (200 mg, 0.61 5 mmol. 1 . 1 eq) and DIPEA (0.387 mL, 2.232 mrnol, 4.0 eq) were added and allowed to stir at ft for overnight. The reaction mixture was diluted with water (50 mL) and extracted with EtOAc (2 x 50 mL), the combined organic layers were washed with water (50 mL) with brine (50 mL) dried over Na?SC>
4, filtered and concentrated and purified by combi flash chromatography [silic gel 100- 200 mesh, elution 0-40% EtOAc in Hexane] to afford the desired compound tert-butyl 4-(2- chiofo-4-((3-(2,6-dichiofopheiiyl)-4-oxo-3
!4-dihydro-2iLpyfimido[5,4-e] 3
yl)aniino)phenyl)-3-methylpiperazine- 1 -carboxyiate (60 mg, 16.90%) as brown solid.
LCMS: 635.1 [M- l f
[0431] Step-2: Synthesis of 7 (3-chloro-4-(2-metfaylpiperazm-^
(2,6-didiiorophenyl)-2^~di^ dihydrochioride: tert-butyl 4-(2~chloro-4-{{3-{2,6-dichlorophenyl)-4-oso-3,4~dihydro-2H-pyrimido[5,4- e] [ 1 ,3]thiazin-7-yl)arnino)phenyI)-3-rnethylpiperazine- 1 -carboxyiate (60 mg, 0.095 mmol, 1 .0 eq) was dissolved in 4.0 M-HCi (2.0 mL) and allowed to stir at rt for 2h. After completion of reaction, the reaction mixture was filtered and dried under reduced pressure to afford the desired compound 7-CC3-chk)ro~4~(2~meth.ylpipera^
dihydro-4H.-pyrimido[5,4-e][l,3]thiazin~4-one dihydrochioride (22 mg, 38.58%) as brown solid, LCMS: 535.1 [M+l f ; UFLC @ 254 nm = 93.38% and @ 220 nm - 93.54%.
¾ NMR (400 »MSO-d6) <Π 0.55 (s, 1 H), 8.96 (brs, 2H), 8 86 (s, 1H), 7.93 (d. J --- 2.1 9
Hz, 1H), 7.71 (brs, 1H), 7.66 (d, J = 7 89 Hz, 2H), 7.49 (d, ./ 7.89 Hz, I I ! ) 7 30 (d, ./ 8.33 Hz, 1H), 5.27 (s, 2H), 3.36 (d, J = 15.79 Hz, 2H), 3.28 (brs, 2H), 3 12 (d, J = 10.52 Hz, 2H), 2.90 (brs, 2H), 2.82 (brs, 2H), 0.88 (d, J= 6.14 Hz, 3H)
Fixa ple S96. Symthesis of 7~((3~chhro~4~(3-methylpiperaz ~i-yiJphenyi)a ino)-3~(2s 6- dichlorophenyl)~2, 3-dihydro-4H-pyrimido[5, 4-ej[i, 3]thiazm-4~one dihydrochlori.de
(Compound No. 1.244)
[0432] Step-1 : Synthesis of tert-buty! 4-(2-chIoro-4-((3-(2,6-dich!orophenyl)-4-oxo-3,4- djhydro-2H-pyrimido(5,4~e) [l^]thia«^^
carhoxylate: To a stirred solution of 3-(2>6-dicii!orophenyl)-7-(mt^hyUiiio)-2,3-dihydi-o-4H- pyrimido[5,4~e] l ,3]thiazm-4~one (200 mg, 0.558 mmoi, 1 .0 eq) in (3.0 mL) of toluene was added mCPBA (275 mg, 1.116 mmol, 2.0 eq) and allowed to stir at it for 30 minutes. tert-bi yl 4-(4-amino-2-chlorophenyl)-2-rnethylpiperazine-l -carboxylate (200 mg, 0, 615 ramol, 1.1 eq) and 1)11 -1 .Λ (0 387 mi.., 2.232 mmol, 4.0 eq) were added and allowed to stir at ri for overnight. The reaction mixture was diluted with water (50 mL) and extracted with EiOAc (2 >< 50 mL), the combined organic layers were washed with water (50 mL) w fe brine (50 mL) dried over a2S(>4, filtered and concentrated and purified by combi flash chromatography [silic gel ; 00- 200 mesh; ekraon 0-40% EtOAc in Hexane] to afford the desired compound ten-butyl 4-(2-
chlofo-4-((3"(2,6'-dtchloropheiiyi)'-4'-oxo-3.4--dih^
yl)amino)phenyl)-2-methylpipefazine- 1 -carboxyiaie (120 mg, 33.80%) as brown solid. LCMS: 635.2 [M- l f
[0433] Step-2: Synthesis of 7 (3-chloro-4-(3-metfaylpiperazm^
(2,6-didiiorophenyl)-2^~diliy dihydroc oride): iert-butyl 4-(2~chloro-4-((3-(2,6-dichk>rophe^^
e] [ 1 ,3]thiasin-7-y})arnino)phenyI)-2-rnethyIpiperazine- 1 -carboxyiate (120 mg„ 0, 199 mmoL 1.0 eq) was dissolved m 4.0 M-HCl (2.0 rrd .} and allowed to stir ,u rt for 2h. After completion of reaction, the reaction mixture was filtered and dried under reduced pressure to afford the desired compound 7-((3-chloro~4-(3-melhylpiperaz ^^
dihydro-4H-pyrimido 5,4-e] [ 1. ,3 ]thiazin-4-one dihydrocblonde (59 n g, 51 , 75%) as yellow solid. LCMS; 535.1 [Mt-l ; UPLC @ 254 nm == 94.22% and @ 220 nm === 92.83%. l H NMR (400 ife, »MSO-d6) δ 10.49 (s, ! M ). 9 23 (brs, IH), 8.84 is, IH), 7.90 (d, J --- 2.63 Hz, IH), 7.65 (d, ./ 7.89 Hz, 3H), 7.45 - 7.54 (n , IH), 7 23 (d, 8.77 ! !/. IH), 5.26 (s, 2H), 3.38 (d, ./ 11.40 Hz, 3H), 3.30 (d, d 12.28 Hz, 2H), 3.15 (d, J = 10.52 Hz, 2H), 2.90 - 3.00 (m, IH), 2.80 (d, J= 1 1 .84 Hz, IH), 1.29 (d, J = 6.58 Hz, 3H).
Example S97, Synthesis 7-((3-chlaro-4-(4-(dimethylamino)piperidin~
chk)ro~6~fluorophenyI)~2, 3-dihydro-4H^ hnddof5i4-e]{l,3]thiazm~4~orie
(Compound No. 1.245)
H
[0434] To a stirred solution of 3-(2~chloro-6-t1uoropheny!)~7~(methytdiio)-2 ~o!ihydro~ H- pyriraido[5,4-e][l ,3]thiazin-4-one (170 mg, 0.5 mrno , 1.0 eq) in (2,0 mL) of toluene was added rn~CFBA (123 mg, 1 mmoL 2.0 eq) and allowed to stir at rt for 30 minutes. l~(4~amirso-2- chj.oroph n.yl)-N,N-dimet.bylpip ridin-4-amine (147 mg, 0.6 mrnol, 1 2 eq) and DIPEA (0.44 ml.,, 2.5 mmoi, 5,0 eq) were added, and allowed to stir at n for overnight. After completion of reaction, the reaction mixture was diluted with water and extracted with EtOAc (50 mL x 2). The combined organic layer was washed with water (50 ml,), brine solution (50 mL), dried over anhydrous sodium sulfate and concentrated under reduced pressure to afford crude product, which was purified by preparative chromatography to afford the desired compound, 7-((3- ciiloro-4-(4-(dirnethy!am o)pipen^
dihydro-4H-pynraido[5,4-e][l,3]thiazin-4-one (15mg, 6.7%) as an off white solid (formate sail). LC 8; 547 3 [M÷lf ; LPI .C @ 254 ran ===89.38% and @ 220 ran === 90,42%.
4 ) NMR (400 MHz, DMSO-d6) d 10.43 (brs, 1H), 8.82 (s, I B), 8.37 (brs, 2H), 7.84 (d, ./ 2,63 Hz, 1H), 7.61 (d, J = 7.02 Hz, IH), 7.46 - 7.55 (m, 2! if 7 39 - 7.46 (m, 1H), 7.15 (d, J = 8.77 Hz, 1H), 5.16 - 5.35 (m, 2H), 3.26 (d, ./ 1 1 .40 Hz, 3H), 2.59 - 2.69 (m, 2H), 2.22 (s, 6H), 1 .84 (d, ./ ==== 1 1.40 Hz, 2H), 1.55 (d. ./ 8.33 Hz, 2H)
Example S9S. Synthesis ofS-Q^whloro^e ylj-y-tfl -dir iethyl-l ^- tetrahydroisoquiriolm-6-yl}amirio}~2, 3~dihydro-4H~pyrimido[5, 4~e?[l , 3]ϋϊίαήη~4~οηβ
(Compound No. 1.246)
[0435] Step-1: S h sis tert-buty! 6-((3-(2,6-dkhlorophenyl)-4-oxo-3,4-dihy lro-2H- pyrimido[5,4~e] [.l^]thiazin-7^^^
carboxylases To a stirred solution of
pyrimidof 5,4-e] f 1 ,31thiazm-4-one (143mg, 0.4 rnmol, 1.0 eq) in of toluene (2.0 mL) was added m-CFBA (108 rng, 0,44 rnmol, .1 .1 eq) and allowed ιο stir at rt for 30 minutes, tert-butyi 6- animo-.l >I-dimethyl-3,4-dihydroisoqujno ne-2(l E})-carboxy!ate (121 mg, 0.44 mraol, 1 . 1 eq) and DIPEA (0.28 mL, 0.28 mmoh 4.0 eq) were added and allowed to stir at rt for overnight. After completion of reaction, the reaction mixture was diluted with water and extracted with EtOAe (50 mL x 2), The combined organic layer was washed with water (50 mL), brine solution (50 mL), dned over anhydrous sodium sulfate and concentrated under reduced pressure to afford crude product, which was purified by Bash chromatography [silica gel 100-200 mesh: efution 0- 50% EtOAc in hexane] to afford the desired tert buiyl i n ; .( d :cMo:T>phe; I ! ·4 ·ο.. ο· \4- dihy dro-2H-pyrimido|'5,4-e"i \ 1 ,3 ]thiazm-7-y !)ammo)- 1 , 1 -diraethy 1-3,4-dilrydroisoquinoline- 2(1 H) -car boxy late (40mg, 1 7. 1 %) as an off white solid.
LCMS: 486.2 [M÷ l]
[0436] Step-2: Synthesis of 3-(2,6-dichlorophenyl)-7-((l,l-dim^ tetrahydroisoquinoIin-6-yl)amino)-2 -dihydro-4H-pyrimido [5,4-e j 11 ,3] thia2iii-4-one: tert- biUyl 6~((3-(2,6-diehlorophenyl)-4-Gxo-3,4-dihyd^
lJ -dimethyl-S^-dihydroisoquinoline^ilHi-carbosylate (40r«g, 0.068mr«oi, 1 eq) was dissolved in dioxane (0.4 niL), followed by dropwise addition of 4.0 M-HCi (0.4 mL) and allowed to stir at rt for Ih. After completion of reaction, the reaction mixture was filtered and dried to give 3-(2,6-dichlorophenyf)-7-( l ,1. -dimethyl- l ,2.3,4-tetrahydroisoquinolin-6-yl)amino)- 2,3-dihydro-4H-pyrimido[5,4~e] [ 1 ,3 jthiaz in-4-one (10 mg, 30.3%) as off white solid.
LOIS:: 486.2 [M÷l ; UPLC @ 254 nm - 92.57 % and @ 220 nm - 95.39%.
! H NMR (400 MHz, DMSO-d6): d I 0.44 (s, 1 H), 9. 12 (brs, 2H), 8 83 is, I H), 7.65 (d. ./ 8.33 Hz, 21 k 7.44 - 7.58 (m, 2H), 7,40 (d, J = 8.33 ! ! . I H), 5.26 (s, 2H), 3.48 (s, 2H), 3.01 (brs, 2H), 1 .64 (s, 6H).
yl }phenykmino)-'2II"pyrimido|"5,4"el [ l,3"|oxazin-4(3II)--one ' '; · ///
(Compound No. 1.172)
[0437] Syn s s of {S)~^(2,&-dichlorophenyl)~7~(4~(2~methylpiperazin~l~
(Peak-II): (S}-3-(2,6- dichlorop.heny.l)-7-(4-(2-me†hylp^
4(3H)~one was separated from the product synthesized in the scheme described in Example S26.
Example SI 00, Synthesis of (R)~7~((3-chloro~4-(3-methyipiperasin~l~yl)phenyi)amino)-3 dichlorophenyl)-2, 3-dihydro~4N-pyrimidofS, 4-ej' fl, 3Joxazin~4~one
(Compound No.1.919)
[0438] Step-1: tert-buty! (R)-4~(2-chJoro-4-((3-(2,6-dkhlorophe^
2M-pyrimido[5,4-e][ly3]oxazm-7-yi)ara To a stirred solution of 3-(2,6-dich]orophenyl)-7-(metiiyU^
e][l,3]oxazm--4-one (170 mg, 0.5 mrnol, 1.0 eq) in of toluene (5.0 rrsL) was added m-CPBA (247 mg, 1.0 mnio!.2.0 eq) and allowed to stir at it for 30 minutes, tert-butyi (R)-4-(4-ainiriO"2-- chlorophenyl)-2-methylpiperazine-l-cafboxylate (180 mg, 0.55 rnmoi, 1.1 eq) and DIPEA (0.44 rnE.2.5 rnmoi, 5.0 eq) were added and allowed to stir at it for overnight. After completion of reaction, the reaction mixture was diluted with water and extracted wife EtOAc (50 mL x 2). The combined organic layer was washed with water (50 mL), brine solution (50 mL), dried over anhydrous sodium sulfate and concentrated under reduced pressure to afford crude product, wiuch was purified by flash chromatography [silica gel 100-200 mesh, elution 0-50% EtOAc m hexane] to afford the desired tert-butyi (K ;··;·: 3-ci-U-! - -((34 .i-"d! hl : :;!K:-:\-i}-- --;,\ i--3.-;- ihyclrG-2H-pynmido[5,4~e][1
(60 mg, 19.3%) as light brown sticky liquid.
LCMS: 619.3 [M+l
[0439] Step-2: Synthesis of (R)-7-((^chloro-4-(3-me^
(2,0-dkMor©plmiyI)-2 -d»hydro-4H-py^ tert-butyl (R)-4-(2- chloro- -T(3-(2,6~dichlorophenyiV4~^^
yl)amino)phenyl)~2~nieihylpipefazine-I -carboxylaie (60 mg, 0.096 mmoi, 1 eq) was dissolved in dioxane (0.4 nil,), followed by dropwise addition of 4.0 M-HCi (0.4 ml,) and allowed to stir at rt for Ih. After completion of reaction, the reaction mixture was filtered and purified by preparative chromatography to give
(7 mg, 21.21%) as an off white solid.
LCMS: 519.3/521.3 [M÷l.]+, UPLC @ 254 nm = 95.97% and @ 220 ran = 97.56%.
je NMR (400 MHZ, DMSG-D6): 510.44 (S, 1 H) 8.85 (BR. S„ 1 H) 7.89 (BR. S. , 1 H) 7.59■■ 7.70 (M, 1 H) 7.59 (BR. S., I H) 7.52 (BR. S.. 1 H) 7.12 ( ! ) ./ H M HZ, 1 H) 5.74 (BR S., 2 H) 4.11 (S, 1 H) 3.16 (BR. S , 3 H) 3 06 (D, J=9.29 HZ, 2 H) 2.89 (BR. S.. 2 H) 2.84 (BR. S , 2 H) 2.26 - 2.34 (M, 2 H) .
[0440] Compounds 1 .66- 1.95, 1.97-1 .171 and 1 .177-1 .222 can be prepared according to the experimental details exemplified in Examples SI-S70 and Scheme I, using the appropriate starting materials and reagents.
[0441] Other compounds can be prepared according to the experimental details exemplified in Examples S i -SI.00 and Scheme 1 to Scheme 3, using the appropriate starting materials and reagents.
Biological Examples
Example B 1. WEE I IC^o Determination
[0442] iC : values of compounds against WEE1 kinase enzyme were determined by
L-anthaScreen™ Terbium Labeled TR-FRET assay. Kinase assays were performed in I X kinase buffer (#PV6135,Invitrogen, Life Technologies Grand Island, NY) where total reaction volume was 1 ΟμΙ in low-volume 384-well plates (#451 1 , Corning) Serially diluted compounds (3-fold) were incubated with WEEi Enzyme ( In ) (#PR7373A, Invitrogen, Life Technologies Grand
Island, NY) for 10 min, following winch a mixture of ATP (lOuM) (#A1852, Sigma, St-Loms, MO) and
(200nM) (#PV3610, Invitrogem Lift; Technologies Grand Island, NY) was added and mcubaied m dark at room temperature for t h. After t h, Ι ΟμΙ stop solution containing Terbium labeled antibody (4nM) (#PV3529, invitrogen, Life
Technologies Grand Island, NY) and EDTA (#E5134, Sigma, Si-Louis, MO) (20mM) in TR- FRET dilution buffer (# PV3574, Invitrogen, Life Technologies Grand Island, NY) was added. Readings were taken in a Synergy Neo Plate reader (BioTek ,Wmooski) at single excitation of 340 nm and Dnal emission at 495nm and 520nm respectively.
[0443] The % activity of test samples was calculated as (Sample - Min)*100/(Max - Mm). [Max: DMSO control, complete reaction with enzyme & DMSO and Min: No enzyme & DMSO], Percent inhibition (100 -% activity) was fitted to the "four-parameter logistic model" in XLfii for determination of Ι(¾ο values. The results are shown in Table 2,
Table 2
Sy tiiesis Compound Enzyme Activity
Example No. Weel K:so (μ )
No.
S-i 1.1 0.056
S-2 1.2 3.26
S-3 1.3 0.04
S-4 1.4 0.009
S-5 1.5 0.069
S-6 1.6 0.279
S-7 1.7 0.074
S-8 1.8 0.162
S -9 1.9 0. 193
S-10 1. 0 0.01 1
S-I I 1.1 1 0.837
S-12 1.12 0.0735
S-I 3 1.13 0. 1437
S-14 1.14 0.173
$-15 1.15 1.558
S-16 1.16 0.049
S-17 1.17 0.156
S-I8 1.18 0.192
S-I9 1.19 0.206
S-20 1.20 0.048
S-21 1.21 1.389
S-22 1.22 0.03
S~23 1.23 0.607
S-24 1.24 0.035
S-25 1.25 0.068
S-26 1.26 0.088
S-27 1.27 0.052
S-28 1.28 0.035
S-29 1.29 0.212
S-30 1.30 0.325
S-31 1.31 30
S-32 1.32 0.366
S-33 1.33 0.05
S-34 1.34 0.014
S-35 1.35 0.035
S-36 1.36 0.104
S~37 1.37 0.113
S-¾ 1.38 0.176
S-39 1.39 0.54
S-40 1.40 0.02
S-4I 1.41 0.791
S-42 1.42 0.025
S-43 1.43 0.479
S-44 1.44 0.023
S-45 1.45 0.288
S-46 1.46 0.053
S-47 1.47 0.07
S-48 1.48 0.103
S-49 1.49 0.04
S-50 1.50 0.028
S-51 1.51 0.011
8-52 1.52 0.0505
S~53 1.53 0.033
S-54 1.54 0.2
S-55 1.55 0.106
S-56 1.56 5
S-57 1.57 0.101
S-58 1.58 0.234
S-59 1.59 >5
S-60 1.60 0.0435
8-61 1.61 0.1 1
S-62 1.62 0. 123
S-63 1.63 >5
S-64 1.64 >5 s - - 1.65 1.332
S-66 1.96 0.047
S-67 1.173 0.087
S -68 1.174 0.032
S-69 1.175 0.066
S-70 1.176 0.105
S-7I 1.87 0.777
S-72 1.104 • 5
S-73 1.107 0. 1 10
S-7-; 1.223 0.032
S-75 1.121 0.501
S-76 1.224 0.232
S~77 1.225 0.282
S-78 1.226 0. 188
S-79 1 ??7 0.021
S-80 1.228 0.127
8-81 1.229 0.185
S-82 1.230 0.561
S-83 1.231 0.142
S-84 1.232 0.273
S~85 1.233 0.165
S-86 1.234 0.202
S-87 1.235 0.111
S -88 1.236 0.335
S-87 1.237 ND
S-90 1.238 0.045
S-91 1.239 0.508
S-92 1.240 0.303
S-93 1.241 0.095
S-94 1.242 0.331
S-95 1.243 0.948
8-96 1.244 0.351
S-97 1.245 0.297
S-98 1.246 0.061
S-99 1. 172 0.066
S-100 1.919 0.340
ND: K oi Determined
Example 82. Determination of potency of compounds in cytotoxicity assay in A427, A 172, A549, Panc~10, 05, MDAMB231 and s~Pc-l ceU lines
[0444] A549 (CCL- 1 85; ATCCS and A427 (HTB--53, ATCC), both king epithelial cell lines were seeded in their respective medium (DMEM/MEM, 1 0569044/41090101 ; Gibco) at a cell count of 1500 cells per 100 iiL per well in a 96 well edge plate ( 167425; ThermoFisherj. Cells were allowed to grow at 37 °C for 24 hr in 5% ( () · environment (culture conditions) in a Nuaire incubator (humidified). Serially diluted test compounds (100 μΐ,) within the desired testing concentration ranges were added to the culture plate were further incubated in culture conditions for 72 br and 96 hr tor A.427 and A549 respectively. The experiment was terminated at the designated incubation time by replacing the medium with 100 LIL of 1 mM of resazunn (R70 7; Sigma) prepared in respective culture medium, and the plates were further incubated m culture conditions for 4-6 hr. Fluorescence was recorded using a multimodal plate reader (Biotek Synergy Neo) at an excitation wavelength of 535 run and emission wavelength of 590 nm to obtain relative fluorescence units. Data analysis was done by subtracting the background fluorescence (only medium blank) value from each reading and then normalizing with the vehicle control (DMSO treated cells) to obtain percent survival /proliferati n. Percent survival was then subtracted by 100 to get the percent inhibition of proliferation which was used to calculate IC5 values. Potency of compounds in other cell lines (As-Pc-1 , Panc-10.05,
DAMB231 , A172) was determined in an analogous manner. The results are shown in Table 3.
Table 3
Synthesi Comp Cell Cell Cell Cell Cell Cell s ound Viability Viability Viability Viability Viability Viability
Example No. A.427 IC5 A549 A 172 Pane 10.05 MDAMB AsPC-1
No. ίμ ) ICs Ao ICso (μΜ) 231 KAo ICio ίμ,Μ) (μΜ) (μΜ) (μΜ)
S-1 h i 4.47 9 7 ND ND ND ND
S-2 1 .2 ND ND NT) ND ND ND
S-3 1 .3 4.52 2.99 NT) ND ND ND
S-4 1 4 ND 5.74 ND ND ND ND
So 1.5 3,91 3.78 ND ND ND ND
S»6 1.6 ND ND ND ND ND ND
S-7 1.7 ND ND ND ND ND ND
S-8 1.8 ND ND ND ND ND ND
S-9 1.9 ND ND ND ND ND ND
S-10 1.10 1.99 2.6 ND ND ND ND
S-11 111 ND NT) ND ND ND ND
S- ! 1.12 3.5825 47025 7.135 3.17 9.045 7.875
S-13 1.13 ND ND ND ND ND ND
S-14 1.14 ND ND ND ND ND ND
S-15 1.15 ND ND ND ND ND ND
S-16 1.16 2.13 3.98 ND ND ND ND
S-17 1.17 ND ND ND ND ND ND
S-18 1.18 ND ND ND ND ND ND
S-19 1.19 ND ND ND ND ND ND
S-20 1.20 5.5 6.02 ND ND ND ND
S-2 1.21 ND ND ND ND ND ND
S-22 1.22 ND ND ND ND ND ND
S-23 1.23 ND ND ND ND ND ND
S-24 1.24 2.96 6.45 ND ND ND ND
S-25 1.25 ND ND ND ND ND ND
S-26 1.26 ND ND ND ND ND ND
S~2'7 127 6.97 4.39 ND ND ND ND
S-2K 1.28 ND ND ND ND ND ND
S-29 1.29 ND 8.88 ND ND ND ND
S-30 1. 0 ND 8.65 NT.) ND ND ND
S-31 1.31 ND ND ND ND ND ND
S-32 1.32 ND 7.43 ND ND ND ND
S-33 1.33 2.82 2.69 ND ND ND ND
S-34 1.34 4.81 21.02 ND ND ND ND
S-35 135 ND 111 ND ND ND ND
S-36 1.36 10.4 8.12 ND ND ND ND
S--37 1.37 ND 9.82 ND ND ND ND
S-38 1.38 ND 9.15 ND ND ND ND
S-39 1 .39 ND 10.45 ND ND ND ND
S-40 1 .40 2.65 2.73 ND NT) ND ND
S-41 1 41 ND >30 ND ND ND ND
S-42 1.42 3 2 5.6 ND ND ND ND
S-43 1.43 ND 22.09 ND ND ND ND
S-44 1.44 ND 5.79 ND ND ND ND
S-45 1.45 ND >30 ND ND ND ND
S--46 1 ,46 ND 9.86 ND ND ND ND
S-47 1 ,47 7.1 4.1 ND ND ND ND
S-48 1 .48 1 1.06 8.38 ND ND ND ND
S-49 1 .49 5.42 4.39 ND ND ND ND
S-50 1 50 3.48 2.79 ND ND ND ND
Sol 1.51 3.0425 6 4525 7.995 3.34 8.76 3.21
S-52 1.52 3.345 4.72 ND ND ND ND
S-53 1.53 3.6125 4.7225 9.675 9 335 10.47 2.41
S--54 1.54 10.48 9.575 ND ND ND ND
S--55 1.55 2.32 1.59 ND ND ND ND
S»56 1 56 10.465 1.86 ND ND ND ND
S-57 1.57 4.855 6.23 ND ND ND ND
S-58 1.58 7.115 12.35 ND ND ND ND
S-59 1 .59 13.99 ND ND ND ND ND
S-60 1 60 2.5825 8.335 4.8 2 85 5.34 2 905
S-6 1.61 4.12 ND ND ND ND ND
S-62 1.62 2.80 ND NT.) ND ND ND
S-63 1 .63 ND ND ND ND ND ND
S-64 1.64 ND ND ND ND ND ND
S-65 1.65 ND ND ND ND ND ND
S-66 1 .96 5.89 ND ND ND ND ND
S-67 1. 1 73 3.32 ND ND ND ND ND
S-68 1.174 3.4 ND ND ND ND ND
S-69 1. 175 ND ND ND ND ND
S-70 1 176 3.25 ND ND ND ND ND
S-71 1 .87 25.43 ND ND ND ND ND
S-72 1.104 27.75 ND ND ND ND ND
S-73 1. 107 7.70 ND ND ND ND ND
S-74 1 223 1.66 ND ND ND ND ND
S-75 1.121 7.33 ND ND ND ND ND
S-76 1 .224 12. 10 ND NT) ND ND ND
S-77 1.225 2. 16 ND ND ND ND ND
S-78 1.226 5.36 ND ND ND ND ND
S»79 1.227 2.1975 9.035 6.615 4 93 8.78 2 935
S-80 1 .228 1.46 ND NT) ND ND ND
S-81 1.229 2.25 ND ND ND ND ND
S-82 1.230 4.94 ND ND ND ND ND
S-83 1.231 ND ND ND ND ND
S-84 1 .232 1.12 ND NT) ND ND ND
S--85 1 233 7.21 ND ND ND ND ND
S-86 1.234 4.84 ND ND ND ND ND
S-87 1.235 2.90 ND ND ND ND ND
S-88 1.236 1 .77 ND ND ND ND ND
S-89 1 237 ND ND ND ND ND ND
S-90 1.238 5.29 ND ND ND ND ND
S-91 1.239 0.765 ND ND ND ND ND
S-92 1.240 1.64 ND ND ND ND ND
S-93 1.241 3.49 ND ND ND ND ND
S-94 1.242 1.62 ND ND ND ND ND
S-95 1 .243 0.625 ND NT) ND ND ND
S-96 1.244 0.510 ND ND ND ND ND
S-97 1.245 1.14 ND ND ND ND ND
S-98 1.246 2.44 ND ND ND ND ND
S-99 1. 172 4.90 ND ND ND ND ND
ND: Not Determined
.Example B3. Determination of potency of compounds in ceil proliferation assay in selecied cancer cell lines and cellular PD effects,
[0445] The effects of test compounds were studied in five cell lines with various histotypes. The cancer cells (Table 4) were harvested during the logarithmic growth period and counted. Adjust cell concentrations to the appropriated number with respective medium, ami add 90 s.iL cell suspensions to 96-well plates. After cells were seeded, the plates were shaken gently to distribute cells evenly and incubated at 37 °C, 5% CO2 on day 1.
Table 4: Cell Culture Conditions
No. Cell Line Histopathology
MEM÷10%FBS+NEAA+Sodium
1 A427 Lung adenocarcinoma
Pyruvate
LoVo Colorectal adenocarcinoma Ham's F12K-H 0%FBS
3 NCI-H460 Large-cell lung carcinoma RP I1 640 H 0%FBS
4 HCT-1 16 Colorectal carcinoma McCoy's 5a÷10%FBS
5 A2780 Ovarian cancer RPMI.1640÷10%FBS
[0446] Cells were treated with test compounds at 9 concentrations within a desired concentration range (e.g. 1 .5 nM - 10 μ.Μ) on day 2 by series diluting the test compound stock solution (10 mM in DMSO) with culture medium. Cell viability was assessed by Cell Titer-Glo® as recommended by Prornega (Cat. No, : G7572, Promega) typically 72 hrs post- treatment.
[0447] Cell viability data were plotted using GraphPad Prism (version 5, GraphPad
Software. Inc. , San Diego, CA). In addition, a nonlinear regression model with a sigmoidal dose response and variable slope within GraphPad Prism was used to calculate the Ι(¾ο value of individual test compounds.
ICso (μΜ)
Compound
LOVO HCT1 16 NCDH460 A427 A2780
No.
1 .3 8.263 8.1 21 >1 0 5.983 NDa
1.5 ND ND ND 4.291 ND
1. 10 ND ND ND 1 .665 3.847
1.12 ND ND ND 3.425 ND
1. 16 ND ND ND 3.038 ND
1.20 ND ND 3.639 ND
1 .24 ND ND ND 2.845 ND
1.27 ND ND \ ) > 7.748 ND
1.55 ND ND ND ND 6.805
ND means Not Determined
[0448] Additional test compounds will be studied in the same and/or other cancer cell lines with various sensitivities to reported Weel compounds using similar proliferation method, with possible variables, such as cell seeding densities and/or incubation durations.
Example B-i Detennmation of potency of compounds by assay pfcelhdarPD effects.
[0449] pCDC2 and - Π2ΛΧ are two clinical relevant biomarkers associated with Weel inhibition. CDC2Y15 phosphorylation in cells vvas reported to be abolished by Wee ; inhibitors (reference: Gavory G et. a!., Almae Discovery, AACR poster, 2016). γ-Η2ΑΧ, a DNA doublestrand break marker, was upregulated by Weel treatment in Weel senstive cell lines (Chiertin AD et a!.. Molecular Cancer Therapeutics, 2013), The effects of selected test compounds on pCDC2 and Y-H2AX will be assessed in selected cancer cell lines post 24 or 48 hr
treatment using Western blotting methods with selective antibodies (Guertin AD et al..
Molecular Cancer Therapeutics, 201 3).
[0450] Changes in the levels of phospho--CDC2 following treatment of cells with test compounds are assessed by enzyme-linked immunosorbent assay (ELBA) or Western blotting. A.427 cells (or other suitable cell line) are plated in 6-well plates and cultured for 24 hr to approximately 80-90% confluency. Medium is then replaced, and the cells are treated with test compound at several different concentrations as well as vehicle control. After incubation of treated ceils in cell culture conditions for a specified time (e.g., 24 hr), cells are rinsed with ice- cold PBS and lysed in I X cell lysis buffer containing protease inhibitors and phosphatase inhibitors. The cells are scraped from the plate with a cell scraper after a brief incubation on ice and transferred to a centrifuge tube, and then subjected to three freeze-thaw cycles in liquid nitrogen and a 37°C water bath for further lysis. The lysaies are centrifuged to pellet cell debns (using, for example, a 10 mm centnfugation of 2000 X g at 4°C) and the supernatants transferred to fresh tubes on ice. The proteui concentrations of the samples are estimated by the Bradford method or equivalent. The ELISA is carried out with the PathSean® Phospho-CDC2 (Tyrl S) Sandwich ELISA Kit (Cat. #7176, Cell Signaling Technology, Danvers, MA.) or similar product according to the manufacturer's instructions Changes in the levels of phospho-CDC2 may alternatively or additionally be analyzed by Western blotting of the samples using a primary antibody to phospho-CDC2 such as phospho~CDC2 (Tyrl S ) (10A1 1 ) rabbit mAb (Cat. #4539, Cell Signaling Technology) or rabbit polyclonal anti-CDKI (phospho Y15) antibody (Cat. #ab47594, Abeam, Cambridge, United Kingdom).
Example B5. Determination of activity of compounds in cancer cells in combination with various DNA-datnaging agents
[0451] The activity of test compounds in combination with eisplatin in A427 ceils was determined. Cells were seeded in a 96 well plate at 2000 cells/well. The next day, cells were treated with 2.5 μΜ cispiatm or vehicle ( I X PBS) and incubated in culture conditions (37AC, 5% CO O for 24 hr. Following the incubation, culture medium was replaced with medium containing test compound and cisplatin/vehicie, and cells were further incubated in culture conditions for another 72 hr. By this procedure, cispiatm was either continued or discontinued after an initial 24-hr incubation. Addition of test compound was in concentrations needed to obtain an 8-point
close response curve, with concentrations prepared by 3-fold dilution. The assay was terminated upon addition of resazurin, incubation for 4 hr m culture conditions and measurement of fluorescence at excitation and emission wavelengths of 535 and 590 nm, respectively. Assay results are shown in Table 6. 04S2] k was reported that M - 1775 (aka AZD- 1775), a potent and selective small molecule inhibitor of Weel , in combination with gemcitabtne, carboplatin, or cisplatin abolished tire phosphorylation of CDC2 at Tyrl 5 residue and abrogate the DNA deniage checkpoint, leading to apoptosis (Hirai H et al. Mo! Cancer Ther 2009; 8: 2992-3000, Small-molecule inhibition of Weel kinase by M -1 775 selectively sensitizes p53 -deficient tumor cells to DNA-damagnig agents).
[0453] Vanous cancer cell lines with p53 mutation will be studied by eo-incubation of test compound and one of the various DNA- -damaging agents, such as pemetrexed, doxorubicin, camptothecin, mitomycin C, gemcitabine. and 5-1 17 etc. The anti-proliferation effects of the DNA-damaging agents will be evaluated in the presence or absence of individual test compound using CTG assays described in Example B3. The concentrations of test compounds in -he combination studies will be selected based on die anti-cell proliferation effects of test compounds in cancer cell tines as monotherapy using CTG assays. Incubation time will be optimized prior to the combination treatment for individual test compound, in vitro mechanism based studies using histology staining and/or flow cytometry methods as described by Hirai H. et al. (Hirai H 201 0. MK-1 775 enhances antitumor efficacy 5--FU) may be used.
[0454] In addition, the sensitization of test compounds in drug-induced resistance cell lines (e.g., A2780cis) will be studied in vitro as combination therapy (e.g , cisplatin - Weel inhibitor) with the an ti --proliferation assays, histology staining and/or flow cytometry methods mentioned therebefore
Table 6. Ceil viability IC50 (μΜ) m A427 cells with 2 5 μΜ cisplatin
24 hr cisplatin then
Compound 24 hr cisplatm then
compound +
No. compound
cisplatin
1 . 10 0.36 <0.01 4
1.16 0.18 <0.014
1 .24 0.12 0.06
1.55 0.53 <0.014
.Example B6. Determination of synergistic activity of compounds combined with
chemotherapeutics in cancer celts
[0455] Determination of synergistic activity of compounds in combination with a chemotherapeutic drug on cell viability is determined by a combination matrix method.
Chemotherapeutics that may be used in the combination include but are not limited to a platinum-based chemotherapeutic agent, a DNA alkylating agent, a topoisomera.se inhibitor, an anthracycline, a hi stone deaeetylase inhibitor, a bromodomain inhibitor, a kinase inhibitor, a mTOR inhibitor, a PARP inhibitor, an ATM inhibitor, an ATR inhibitor, a Weel inhibitor, a proteasome inhibitor, and a nucleotide analog or precursor analog. Cancer cell lines that may be used in the assay include but are not limited to lung cancer, leukemia, lymphoma, multiple myeloma, ovarian cancer, breast cancer, pancreatic cancer, stomach cancer, colon cancer, liver cancer, bead and neck cancer, kidney cancer, skin cancer and brain cancer cell lines, ("ells are seeded in a 96-well plate and incubated at 37 C in ceil culture condi ions for 24 hr. Drugs are added and then the cells are incubated further at 37"'C in cell culture conditions for 72 hr. Cells are treated with single agents to obtain a dose response curve for each agent, ("ells are also treated with combinations of the drugs, based on a matrix generated by combining the two drugs at all different combinations of the doses used in the dose response curves. The assay is terminated by addition of Resazurin. incubation for 4 hr at 37°C, 5% COY Measurement of fluorescence at an excitation and emission wavelength of 535 and 590 nm respectively. Synergy is evaluated with the combination index (CD value using the Chou-Talaiay method in which additive effect (CI :::: 1), synergism (CI < 1 ), and antagonism (CI > 1) in drug combinations is determined (Chou TC. Cancer Res 2010;70:440-6.). A fixed drug ratio dilution method in which
drugs are combined in a fi ed ratio which is diluted to 5 or more dilutions, may also be used in place of die combination matrix method.
[0456] Although the foregoing invention has been described in some detail by way of illustration and exanip{e for purposes of clarity of understanding, it is apparent to those skilled in the art that certain minor changes and modifications will be practiced in light of the above teaching. Therefore, the description and examples should not be construed as l imiting the scope of the invention.