WO2018170138A1 - Catalyst system for multi-block copolymer formation - Google Patents
Catalyst system for multi-block copolymer formation Download PDFInfo
- Publication number
- WO2018170138A1 WO2018170138A1 PCT/US2018/022454 US2018022454W WO2018170138A1 WO 2018170138 A1 WO2018170138 A1 WO 2018170138A1 US 2018022454 W US2018022454 W US 2018022454W WO 2018170138 A1 WO2018170138 A1 WO 2018170138A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- independently
- group
- atoms
- hydrocarbyl
- substituted
- Prior art date
Links
- 0 CCCC1C(*C)C(C)=CC1 Chemical compound CCCC1C(*C)C(C)=CC1 0.000 description 5
- STNDIJSPKXCAPQ-UHFFFAOYSA-N Bc1cc(-c2c(C)cc(C)cc2C)cc(-c2c(C)cc(C)cc2C)n1 Chemical compound Bc1cc(-c2c(C)cc(C)cc2C)cc(-c2c(C)cc(C)cc2C)n1 STNDIJSPKXCAPQ-UHFFFAOYSA-N 0.000 description 1
- UJOBWOGCFQCDNV-UHFFFAOYSA-N c1ccc2[nH]c3ccccc3c2c1 Chemical compound c1ccc2[nH]c3ccccc3c2c1 UJOBWOGCFQCDNV-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F297/00—Macromolecular compounds obtained by successively polymerising different monomer systems using a catalyst of the ionic or coordination type without deactivating the intermediate polymer
- C08F297/06—Macromolecular compounds obtained by successively polymerising different monomer systems using a catalyst of the ionic or coordination type without deactivating the intermediate polymer using a catalyst of the coordination type
- C08F297/08—Macromolecular compounds obtained by successively polymerising different monomer systems using a catalyst of the ionic or coordination type without deactivating the intermediate polymer using a catalyst of the coordination type polymerising mono-olefins
- C08F297/083—Macromolecular compounds obtained by successively polymerising different monomer systems using a catalyst of the ionic or coordination type without deactivating the intermediate polymer using a catalyst of the coordination type polymerising mono-olefins the monomers being ethylene or propylene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F210/00—Copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
- C08F210/16—Copolymers of ethene with alpha-alkenes, e.g. EP rubbers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F4/00—Polymerisation catalysts
- C08F4/42—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors
- C08F4/44—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides
- C08F4/60—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides together with refractory metals, iron group metals, platinum group metals, manganese, rhenium technetium or compounds thereof
- C08F4/62—Refractory metals or compounds thereof
- C08F4/64—Titanium, zirconium, hafnium or compounds thereof
- C08F4/64003—Titanium, zirconium, hafnium or compounds thereof the metallic compound containing a multidentate ligand, i.e. a ligand capable of donating two or more pairs of electrons to form a coordinate or ionic bond
- C08F4/64168—Tetra- or multi-dentate ligand
- C08F4/64172—Neutral ligand
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F210/00—Copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
- C08F210/14—Monomers containing five or more carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F297/00—Macromolecular compounds obtained by successively polymerising different monomer systems using a catalyst of the ionic or coordination type without deactivating the intermediate polymer
- C08F297/06—Macromolecular compounds obtained by successively polymerising different monomer systems using a catalyst of the ionic or coordination type without deactivating the intermediate polymer using a catalyst of the coordination type
- C08F297/08—Macromolecular compounds obtained by successively polymerising different monomer systems using a catalyst of the ionic or coordination type without deactivating the intermediate polymer using a catalyst of the coordination type polymerising mono-olefins
- C08F297/083—Macromolecular compounds obtained by successively polymerising different monomer systems using a catalyst of the ionic or coordination type without deactivating the intermediate polymer using a catalyst of the coordination type polymerising mono-olefins the monomers being ethylene or propylene
- C08F297/086—Macromolecular compounds obtained by successively polymerising different monomer systems using a catalyst of the ionic or coordination type without deactivating the intermediate polymer using a catalyst of the coordination type polymerising mono-olefins the monomers being ethylene or propylene the block polymer contains at least three blocks
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F4/00—Polymerisation catalysts
- C08F4/42—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors
- C08F4/44—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides
- C08F4/60—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides together with refractory metals, iron group metals, platinum group metals, manganese, rhenium technetium or compounds thereof
- C08F4/62—Refractory metals or compounds thereof
- C08F4/64—Titanium, zirconium, hafnium or compounds thereof
- C08F4/64003—Titanium, zirconium, hafnium or compounds thereof the metallic compound containing a multidentate ligand, i.e. a ligand capable of donating two or more pairs of electrons to form a coordinate or ionic bond
- C08F4/64006—Bidentate ligand
- C08F4/64041—Monoanionic ligand
- C08F4/64044—NN
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F4/00—Polymerisation catalysts
- C08F4/42—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors
- C08F4/44—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides
- C08F4/60—Metals; Metal hydrides; Metallo-organic compounds; Use thereof as catalyst precursors selected from light metals, zinc, cadmium, mercury, copper, silver, gold, boron, gallium, indium, thallium, rare earths or actinides together with refractory metals, iron group metals, platinum group metals, manganese, rhenium technetium or compounds thereof
- C08F4/62—Refractory metals or compounds thereof
- C08F4/64—Titanium, zirconium, hafnium or compounds thereof
- C08F4/64003—Titanium, zirconium, hafnium or compounds thereof the metallic compound containing a multidentate ligand, i.e. a ligand capable of donating two or more pairs of electrons to form a coordinate or ionic bond
- C08F4/64168—Tetra- or multi-dentate ligand
- C08F4/64186—Dianionic ligand
- C08F4/64193—OOOO
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2410/00—Features related to the catalyst preparation, the catalyst use or to the deactivation of the catalyst
- C08F2410/01—Additive used together with the catalyst, excluding compounds containing Al or B
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2410/00—Features related to the catalyst preparation, the catalyst use or to the deactivation of the catalyst
- C08F2410/04—Dual catalyst, i.e. use of two different catalysts, where none of the catalysts is a metallocene
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2500/00—Characteristics or properties of obtained polyolefins; Use thereof
- C08F2500/08—Low density, i.e. < 0.91 g/cm3
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2500/00—Characteristics or properties of obtained polyolefins; Use thereof
- C08F2500/12—Melt flow index or melt flow ratio
Definitions
- Embodiments relate to olefin polymerization catalysts, their manufacture, and the production of polyolefins using specific catalyst compositions, including the use of chain shuttling agents in the olefin polymerization process.
- polystyrene resin The properties and applications of polyolefins depend to varying degrees upon the specific features of the catalysts used in their preparation. Specific catalyst compositions, activation conditions, steric and electronic features, and the like all can factor into the characteristics of the resulting polymer product. Indeed, a multitude of polymer features, such as co-monomer incorporation, molecular weight, polydispersity, long-chain branching, and the related physical properties (e.g., density, modulus, melt properties, tensile features, and optical properties), can all be affected by catalyst design.
- block copolymers can be prepared from a common monomer environment by using a mixture of catalysts of different selectivities, such as different stereoselectivities or monomer selectivities. Under the right conditions, efficient chain shuttling can produce a multi -block copolymer that features a random distribution of hard and soft blocks of random length.
- One improvement to said processes would be to elevate reactor temperatures, which would increase production rate and decrease energy consumption while still producing desirable olefin block copolymer architecture with commercially acceptable catalyst efficiency and process control. Such an improvement has not been demonstrated in the state of the art.
- the present disclosure relates to a composition
- a composition comprising an admixture or reaction product resulting from combining:
- the second olefin polymerization procatalyst comprises a metal- ligand complex of Formula I):
- M is titanium, zirconium, or hafnium
- each Zl is independently a monodentate or polydentate ligand that is neutral, monoanionic, or dianionic, wherein nn is an integer, and wherein Zl and nn are chosen in such a way that the metal-ligand complex of Formula (I) is overall neutral;
- each Q 1 and Q 10 independently is selected from the group consisting of (C6-C4o)aryl, substituted (C6-C4o)aryl, (C 3 -C4o)heteroaryl, and substituted (C 3 - C4o)heteroaryl; wherein each Q 2 , Q 3 , Q 4 , Q 7 , Q 8 , and Q 9 independently is selected from a group consisting of hydrogen, (Ci-C4o)hydrocarbyl, substituted (Ci-C4o)hydrocarbyl, (Ci-C4o)heterohydrocarbyl, substituted (Ci-C4o)heterohydrocarbyl, halogen, and nitro (N0 2 );
- each Q 5 and Q 6 independently is selected from the group consisting of a (Ci-C4o)alkyl, substituted (Ci-C4o)alkyl, and [(Si)i-(C+Si)4o] substituted organosilyl;
- each N independently is nitrogen
- two or more of the Q 1"5 groups can combine together to form a ring structure, with such ring structure having from 5 to 16 atoms in the ring excluding any hydrogen atoms;
- two or more of the Q 6"10 groups can combine together to form a ring structure, with such ring structure having from 5 to 16 atoms in the ring excluding any hydrogen atoms.
- the present disclosure relates to a composition for use in the polymerization of at least one addition polymerizable monomer to form a multi-block (segmented) copolymer, said copolymer containing therein two or more blocks or segments differing in one or more chemical or physical properties, the composition comprising an admixture or reaction product resulting from combining:
- the second olefin polymerization procatalyst comprises a metal- ligand complex of Formula (I).
- the present disclosure relates to a composition for use in the polymerization of ethylene and at least one copolymerizable comonomer other than ethylene to form a multi-block (segmented) copolymer, said copolymer containing therein two or more blocks or segments differing in one or more chemical or physical properties, the composition comprising an admixture or reaction product resulting from combining:
- the second olefin polymerization procatalyst comprises a metal- ligand complex of Formula (I).
- the present disclosure relates to an olefin polymerization catalyst system comprising:
- the second olefin polymerization procatalyst comprises a metal- ligand complex of Formula (I).
- the present disclosure relates to a process for preparing a multi -block (segmented) copolymer, said process comprising contacting one or more addition polymerizable monomers under addition polymerizable conditions with a composition comprising an admixture or reaction product resulting from combining:
- the second olefin polymerization procatalyst comprises a metal- ligand complex of Formula (I).
- the present disclosure relates to a process for preparing a multi-block (segmented) copolymer comprising ethylene and at least one copolymerizable comonomer other than ethylene, said process comprising contacting ethylene and one or more addition polymerizable monomers other than ethylene under addition polymerizable conditions with a composition comprising an admixture or reaction product resulting from combining:
- the second olefin polymerization procatalyst comprises a metal- ligand complex of Formula (I).
- the present disclosure relates to a process for preparing a multi -block (segmented) copolymer, said process comprising contacting one or more addition polymerizable monomers under addition polymerizable conditions with an olefin polymerization catalyst system comprising:
- the second olefin polymerization procatalyst comprises a metal- ligand complex of Formula (I).
- the present disclosure relates to a process for preparing a multi -block (segmented) copolymer comprising ethylene and at least one copolymerizable comonomer other than ethylene, said process comprising contacting ethylene and one or more addition polymerizable monomers other than ethylene under addition polymerizable conditions with an olefin polymerization catalyst system comprising:
- the second olefin polymerization procatalyst comprises a metal- ligand complex of Formula (I).
- the foregoing processes take the form of continuous solution processes for forming block copolymers, such as multi-block copolymers (preferably linear multi -block copolymers of two or more monomers, especially ethylene and a C3-20 olefin or cycloolefin, and most especially ethylene and a C3-20 a- olefin), using multiple catalysts that are incapable of interconversion. That is, the catalysts are chemically distinct.
- the process is ideally suited for polymerization of mixtures of monomers at high monomer conversions. Under these polymerization conditions, shuttling from the chain shuttling agent to the catalyst becomes advantaged compared to chain growth, and multi-block copolymers, especially linear multi-block copolymers according to the present disclosure, are formed in high efficiency.
- segmented copolymer especially a copolymer comprising ethylene in polymerized form, said copolymer containing therein two or more
- the copolymer preferably possesses a molecular weight distribution, Mw/Mn, of equal to or less than 10.0 (e.g., equal to or less than 9.0, equal to or less than 8.0, equal to or less than 7.0, equal to or less than 6.0, equal to or less than 5.0, equal to or less than 4.0, equal to or less than 3.0, equal to or less than 2.8, etc.).
- Mw/Mn molecular weight distribution
- the polymers of the present disclosure are ethylene multi-block copolymers.
- FIG. 1 exemplifies the chain shuttling process that occurs in the
- FIGS. 2-4 provide CEF graphs demontrasting that olefin block copolymers are made in the examples of the present disclosure.
- FIG. 5 provides incorporation ratios for exemplary procatalysts of the present disclosure. Detailed Description
- Number ranges in this disclosure and as they relate to the compositions disclosed herein are approximate, and thus may include values outside of the range unless otherwise indicated. Number ranges include all values from and including the lower and the upper values and include fractional numbers or decimals.
- shuttling agent and “chain transfer agent” refer to those known to one of ordinary skill in the art. Specifically, the term “shuttling agent” or “chain shuttling agent” refers to a compound or mixture of compounds that is capable of causing polymeryl transfer between various active catalyst sites under conditions of polymerization. That is, transfer of a polymer fragment occurs both to and from an active catalyst site in a facile and reversible manner.
- an agent that acts merely as a "chain transfer agent,” such as some main-group alkyl compounds, may exchange, for example, an alkyl group on the chain transfer agent with the growing polymer chain on the catalyst, which generally results in termination of the polymer chain growth.
- the main-group center may act as a repository for a dead polymer chain, rather than engaging in reversible transfer with a catalyst site in the manner in which a chain shuttling agent does.
- the intermediate formed between the chain shuttling agent and the polymeryl chain is not sufficiently stable relative to exchange between this intermediate and any other growing polymeryl chain, such that chain termination is relatively rare.
- catalyst or “catalyst precursor” used herein refers to a transition metal species that, once combined with an activator co-catalyst, is capable of polymerization of unsaturated monomers.
- Cp2Zr(CH 3 )2 is a catalyst precursor, which, when combined with an activating cocatalyst, becomes the active catalyst species "Cp2Zr(CH 3 ) + " which is capable of polymerization of unsaturated monomers.
- Catalysts include those known in the art and those disclosed in WO 2005/090426, WO 2005/090427, WO 2007/035485, WO 2009/012215, WO 2014/105411, U.S. Patent Publication Nos. 2006/0199930, 2007/0167578,
- Co-catalyst or “activator” refer to those known in the art, e.g., those disclosed in WO 2005/090427 and U.S. Patent No. 8,501,885 B2, that can activate a procatalyst by combining with or contacting the procatalyst to form an active catalyst composition.
- BINAP 2,2'-bis(diphenylphosphino)-l,l '-binaphthyl; Acac: acetylacetonate; Mg(OH) 2 : magnesium hydroxide; NaO'Bii : sodium tert- butoxide; K3PO4 : potassium phosphate tribasic; brine : saturated aqueous sodium chloride; n-BuLi : «-butyllithium; MeMgBr: methylmagnesium bromide; HfC : hafnium(IV) chloride; HfBi : hafnium(IV) tetrabenzyl; ZrC : zirconium(IV) chloride; ZrBi : zirconium(IV) tetrabenzyl; Pd(OAc) 2 : palladium (II) acetate;
- Pd 2 dba3 Tris(dibenzylideneacetone)dipalladium(0); Ni(Acac) 2 : nickel (II)
- NiBr 2 (DME) nickel(II) bromide ethylene glycol dimethyl ether complex
- DEZ diethylzinc
- MMAO, MMAO-3A modified methylaluminoxane
- BHT butylated hydroxytoluene
- GPC gel permeation chromatography
- HT-GPC high-temperature gel permeation chromatography
- PDI polydispersity index
- GC MS gas chromatography mass spectrometry
- NMR nuclear magnetic resonance
- MHz megahertz
- g grams; mg: milligrams; mmol : millimoles; mL : milliliters; min : minutes; h : hours; d: days; RT: room temperature.
- Polymer refers to a compound prepared by polymerizing monomers, whether of the same or a different type.
- the generic term polymer thus embraces the term homopolymer, usually employed to refer to polymers prepared from only one type of monomer, and the term interpolymer as defined below. It also embraces all forms of interpolymers, e.g., random, block, homogeneous, heterogeneous, etc.
- Interpolymer and copolymer refer to a polymer prepared by the polymerization of at least two different types of monomers.
- polyethylene includes homopolymers of ethylene and copolymers of ethylene and one or more C3-8 a-olefins in which ethylene comprises at least 50 mole percent.
- crystalline if employed, refers to a polymer that possesses a first order transition or crystalline melting point (Tm) as determined by differential scanning calorimetry (DSC) or equivalent technique.
- DSC differential scanning calorimetry
- amorphous refers to a polymer lacking a crystalline melting point as determined by differential scanning calorimetry (DSC) or equivalent technique.
- OBC olefin block copolymer
- block copolymer multi- block copolymer
- shegmented copolymer refer to a polymer comprising two or more chemically distinct regions or segments (referred to as “blocks") preferably joined in a linear manner, that is, a polymer comprising chemically differentiated units which are joined (covalently bonded) end-to-end with respect to polymerized functionality, rather than in pendent or grafted fashion.
- the blocks may differ in the amount or type of comonomer incorporated therein, the density, the amount of crystallinity, the type of crystallinity (e.g., polyethylene versus polypropylene), the crystallite size attributable to a polymer of such composition, the type or degree of tacticity (isotactic or
- the olefin block copolymer may contain "hard blocks” (semicrystalline or high glass transition temperature) having lower comonomer content and "soft blocks” (low crystallinity or amorphous with low glass transition temperature) having higher comonomer content.
- block copolymers of the present disclosure are characterized by unique distributions of polymer polydispersity (PDI or Mw/Mn), block length distribution, and/or block number distribution, due, in a preferred embodiment, to the effect of the shuttling agent(s) in combination with catalysts.
- PDI polymer polydispersity
- Mw/Mn block length distribution
- block number distribution due, in a preferred embodiment, to the effect of the shuttling agent(s) in combination with catalysts.
- the block copolymers when produced in a continuous process, desirably possess PDI from 1.0 to 10.0 (e.g., from 1.0 to 9.0, from 1.0 to 8.0, from 1.0 to 7.0, from 1.0 to 6.0, from 1.0 to 5.0, from 1.0 to 4.0, from 1.0 to 3.5, from 1.0 to 3.0, from 1.7 to 2.9, from 1.8 to 2.5, from 1.8 to 2.2, and/or from 1.8 to 2.1).
- PDI 1.0 to 10.0 (e.g., from 1.0 to 9.0, from 1.0 to 8.0, from 1.0 to 7.0, from 1.0 to 6.0, from 1.0 to 5.0, from 1.0 to 4.0, from 1.0 to 3.5, from 1.0 to 3.0, from 1.7 to 2.9, from 1.8 to 2.5, from 1.8 to 2.2, and/or from 1.8 to 2.1).
- the block polymers When produced in a batch or semi-batch process, the block polymers desirably possess PDI from 1.0 to 10.0 (e.g., from 1.0 to 9.0, from 1.0 to 8.0, from 1.0 to 7.0, from 1.0 to 6.0, from 1.0 to 5.0, from 1.0 to 4.0, from 1.0 to 3.5, from 1.0 to 3.0, from 1.7 to 2.9, from 1.8 to 2.5, from 1.8 to 2.2, and/or from 1.8 to 2.1).
- PDI 1.0 to 10.0 (e.g., from 1.0 to 9.0, from 1.0 to 8.0, from 1.0 to 7.0, from 1.0 to 6.0, from 1.0 to 5.0, from 1.0 to 4.0, from 1.0 to 3.5, from 1.0 to 3.0, from 1.7 to 2.9, from 1.8 to 2.5, from 1.8 to 2.2, and/or from 1.8 to 2.1).
- ethylene multi-block copolymer means a multi-block copolymer comprising ethylene and one or more copolymerizable comonomers, wherein ethylene comprises a plurality of the polymerized monomer units of at least one block or segment in the polymer, preferably at least 90 mole percent, more preferably at least 95 mole percent, and most preferably at least 98 mole percent of said block.
- the ethylene multi -block copolymers of the present disclosure preferably have an ethylene content from 25 to 97 weight percent, more preferably from 40 to 96 weight percent, even more preferably from 55 to 95 percent weight, and most preferably from 65 to 85 weight percent.
- the polymer cannot be completely fractionated using standard selective extraction techniques. For example, polymers containing regions that are relatively crystalline (high density segments) and regions that are relatively amorphous (lower density segments) cannot be selectively extracted or fractionated using differing solvents.
- the quantity of extractable polymer using either a dialkyl ether- or an alkane- solvent is less than 10 percent, preferably less than 7 percent, more preferably less than 5 percent and most preferably less than 2 percent of the total polymer weight.
- the multi-block copolymers of the present disclosure desirably possess a PDI fitting a Schulz-Flory distribution rather than a Poisson distribution.
- the use of the present polymerization process results in a product having both a polydisperse block distribution as well as a polydisperse distribution of block sizes. This results in the formation of polymer products having improved and distinguishable physical properties.
- the theoretical benefits of a polydisperse block distribution have been previously modeled and discussed in Potemkin, Physical Review E (1998) 57(6), pp. 6902-6912, and Dobrynin. J. Chem. Phvs. (1997) 107(21), pp. 9234-9238.
- the polymers of the present disclosure possess a most probable distribution of block lengths.
- Exemplary copolymers according to the present disclosure are multi -block copolymers containing 4 or more blocks or segments including terminal blocks.
- the following mathematical treatment of the resulting polymers is based on theoretically derived parameters that are believed to apply to the presently disclosed polymers and demonstrate that, especially in a steady-state, continuous, well-mixed reactor, the block lengths of the resulting polymer prepared using 2 or more catalysts will each conform to a most probable distribution, derived in the following manner, wherein pi is the probability of propagation with respect to block sequences from catalyst i.
- the theoretical treatment is based on standard assumptions and methods known in the art and used in predicting the effects of polymerization kinetics on molecular architecture, including the use of mass action reaction rate expressions that are not affected by chain or block lengths. Such methods have been previously disclosed in W. H. Ray, J. Macromol. ScL Rev.
- Each catalyst has a probability of propagation (p and forms a polymer segment having a unique average block length and distribution.
- the probability of propagation is defined as:
- Rp[i] Rate of monomer consumption by catalyst i, (moles/L),
- Dormant polymer chains refers to polymer chains that are attached to a CSA.
- the overall monomer consumption or polymer propagation rate, Rp[i], is defined using an apparent rate constant, k pi , multiplied by a total monomer
- the total chain transfer rate is given below including values for chain transfer to hydrogen (H 2 ), beta hydride elimination, and chain transfer to chain shuttling agent (CSA).
- the reactor residence time is given by ⁇ and each subscripted k value is a rate constant.
- hydrocarbyl refers to univalent substituents containing only hydrogen and carbon atoms, including branched or unbranched, saturated or
- unsaturated, cyclic, polycyclic or noncyclic species examples include alkyl-, cycloalkyl-, alkenyl-, alkadienyl-, cycloalkenyl-, cycloalkadienyl-, aryl-, and alkynyl- groups.
- Substituted hydrocarbyl refers to a hydrocarbyl group that is substituted with one or more nonhydrocarbyl substituent groups.
- heteroatom containing hydrocarbyl or “heterohydrocarbyl” refer to univalent groups in which at least one atom other than hydrogen or carbon is present along with one or more carbon atom and one or more hydrogen atoms.
- heterocarbyl refers to groups containing one or more carbon atoms and one or more heteroatoms and no hydrogen atoms.
- the bond between the carbon atom and any heteroatom, as well as the bonds between any two heteroatoms, may be a single or multiple covalent bond or a coordinating or other donative bond.
- an alkyl group substituted with a heterocycloalkyl-, aryl- substituted heterocycloalkyl-, heteroaryl-, alkyl- substituted heteroaryl-, alkoxy-, aryloxy-, dihydrocarbylboryl-, dihydrocarbylphosphino-, dihydrocarbylamino-, trihydrocarbylsilyl-, hydrocarbylthio-, or hydrocarbyl seleno- group is within the scope of the term heteroalkyl.
- suitable heteroalkyl groups include cyanom ethyl-, benzoylmethyl-, (2- pyridyl)methyl-, and trifluoromethyl- groups.
- aromatic refers to a polyatomic, cyclic, conjugated ring system containing (4 ⁇ +2) ⁇ -electrons, wherein ⁇ is an integer greater than or equal to 1.
- fused as used herein with respect to a ring system containing two or more polyatomic, cyclic rings means that with respect to at least two rings thereof, at least one pair of adjacent atoms is included in both rings.
- aryl refers to a monovalent aromatic substituent which may be a single aromatic ring or multiple aromatic rings which are fused together, linked covalently, or linked to a common group such as a methylene or ethylene moiety. Examples of aromatic ring(s) include phenyl, naphthyl, anthracenyl, and biphenyl, among others.
- Substituted aryl refers to an aryl group in which one or more hydrogen atoms bound to any carbon is replaced by one or more functional groups such as alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, heterocycloalkyl, substituted heterocycloalkyl, halogen, alkylhalos (e.g., CF 3 ), hydroxy, amino, phosphido, alkoxy, amino, thio, nitro, and both saturated and unsaturated cyclic hydrocarbons which are fused to the aromatic ring(s), linked covalently or linked to a common group such as a methylene or ethylene moiety.
- the common linking group may also be a carbonyl as in benzophenone or oxygen as in diphenylether or nitrogen in diphenylamine.
- the individual rates of polymerization of comonomer and monomer are typically complex functions of temperature, catalyst, and monomer/comonomer concentrations. In the limit as comonomer concentration in the reaction media drops to zero, R P 2 drops to zero, F2 becomes zero and the polymer consists of pure monomer. In the limiting case of no monomer in the reactor, R p i becomes zero and F2 is one
- the ratio of comonomer to monomer in the reactor largely determines polymer composition as determined according to either the Terminal Copolymerization Model or the Penultimate Copolymerization Model.
- a simplified equation for comonomer composition can be derived as disclosed in George Odian, Principles of Polymerization, Second Edition, John Wiley and Sons 1970, as follows: [0050] From this equation, the mole fraction of comonomer in the polymer is solely dependent on the mole fraction of comonomer in the reaction media and two temperature dependent reactivity ratios defined in terms of the insertion rate constants as:
- the comonomer content can be calculated (again as disclosed in George Odian, Supra.) as:
- the polymer composition is a function only of temperature dependent reactivity ratios and comonomer mole fraction in the reactor. The same is also true when reverse comonomer or monomer insertion may occur or in the case of the interpolymerization of more than two monomers.
- Reactivity ratios for use in the foregoing models may be predicted using well known theoretical techniques or empirically derived from actual polymerization data. Suitable theoretical techniques are disclosed, for example, in B. G. Kyle, Chemical and Process Thermodynamics, Third Addition, Prentice-Hall, 1999 and in Redlich-Kwong-Soave (RKS) Equation of State, Chemical Engineering Science, 1972, pp. 1197-1203.
- RKS Redlich-Kwong-Soave
- the present disclosure may alternatively be related to a composition or catalyst system for use in the polymerization of two or more addition polymerizable monomers, especially ethylene and at least one copolymerizable comonomer, to form a high molecular weight, segmented copolymer (multi-block copolymer), said copolymer containing therein two or more (preferably three or more) segments or blocks differing in one or more chemical or physical properties as further disclosed herein, the catalyst system or composition comprising the admixture or reaction product resulting from combining:
- the ratio of the reactivity ratios ( ⁇ / rm) under the polymerization conditions is 0.5 or less (e.g., 0.25 or less, 0.125 or less, 0.08 or less,
- a process for use in the polymerization of two or more addition polymerizable monomers (especially ethylene and at least one copolymerizable comonomer) to form a high molecular weight, segmented copolymer (multi-block copolymer), said copolymer containing therein two or more (preferably three or more) segments or blocks differing in one or more chemical or physical properties as further disclosed herein, the process comprising the steps of combining two or more addition polymerizable monomers (especially ethylene and at least one copolymerizable comonomer) under polymerization conditions with the catalyst system or composition comprising the admixture or reaction product resulting from combining:
- the ratio of the reactivity ratios ( ⁇ / rm) under the polymerization conditions is 0.5 or less (e.g., 0.25 or less, 0.125 or less, 0.08 or less,
- composition or catalyst system for use in the polymerization of two or more addition polymerizable monomers (referred to as monomer and comonomer(s) respectively), especially ethylene and at least one copolymerizable comonomer, to form a high molecular weight, segmented copolymer (multi-block copolymer), said copolymer containing therein two or more (preferably three or more) segments or blocks differing in one or more chemical or physical properties as further disclosed herein, the catalyst system or composition comprising the admixture or reaction product resulting from combining:
- the ratio (Fi / F 2 ) under the polymerization conditions is 2 or more (e.g., 4 or more, 10 or more, 15 or more, and 20 or more).
- a process for use in the polymerization of two or more addition polymerizable monomers (referred to as monomer and comonomer(s) respectively), especially ethylene and at least one copolymerizable comonomer, to form a high molecular weight, segmented copolymer (multi-block copolymer), said copolymer containing therein two or more (preferably three or more) segments or blocks differing in one or more chemical or physical properties as further disclosed herein, the process comprising the steps of combining under polymerization conditions:
- A a first olefin polymerization procatalyst
- B a second olefin polymerization procatalyst capable of preparing polymers differing in chemical or physical properties from the polymer prepared by the first olefin polymerization procatalyst (A) under equivalent polymerization conditions
- Suitable monomers for use in preparing the olefin block copolymers or multi-block copolymers of the present disclosure include ethylene and one or more addition polymerizable monomers (i.e., comonomers) other than ethylene.
- suitable comonomers include straight-chain or branched a-olefins of 3 to 30, preferably
- carbon atoms such as propylene, 1-butene, 1-pentene, 3 -methyl- 1-butene, 1- hexene, 4-methyl-l- pentene, 3 -methyl- 1-pentene, 1-octene, 1-decene, 1-dodecene, 1- tetradecene, 1 -hexadecene, 1- octadecene and 1-eicosene; cycloolefins of 3 to 30, preferably 3 to 20 carbon atoms, such as cyclopentene, cycloheptene, norbornene, 5- methyl-2-norbornene, tetracyclododecene, and 2-methyl- 1,4,5, 8-dimethano- l,2,3,4,4a,5,8,8a-octahydronaphthalene; di- and poly-olefins, such as butadiene, isoprene, 4-methyl- 1,3-p
- shuttling agent refers to a compound or mixture of compounds employed in the composition/catalyst system/process of the present disclosure that is capable of causing polymeryl exchange between at least two active catalyst sites of the catalysts included in the composition/catalyst system/process under the conditions of the polymerization. That is, transfer of a polymer fragment occurs both to and from one or more of the active catalyst sites.
- a "chain transfer agent” causes termination of polymer chain growth and amounts to a one-time transfer of growing polymer from the catalyst to the transfer agent.
- the shuttling agent has an activity ratio RA-B/RB-A of from 0.01 and 100, more preferably from 0.1 to 10, most preferably from 0.5 to 2.0, and most highly preferably from 0.8 to 1.2, wherein RA-B is the rate of polymeryl transfer from catalyst A active site to catalyst B active site via the shuttling agent, and RB-A is the rate of reverse polymeryl transfer, i.e., the rate of exchange starting from the catalyst B active site to catalyst A active site via the shuttling agent.
- the intermediate formed between the shuttling agent and the polymeryl chain is sufficiently stable such that chain termination is relatively rare.
- the rate of chain shuttling (defined by the time required to transfer a polymer chain from a catalyst site to the chain shuttling agent and then back to a catalyst site) is similar to or faster than the rate of polymer termination, even up to 10 or even 100 times faster than the rate of polymer termination. This permits polymer block formation on the same time scale as polymer propagation.
- Suitable chain shuttling agents for use herein include Group 1, 2, 12 or 13 metal compounds or complexes containing at least one Ci-20 hydrocarbyl group, preferably hydrocarbyl substituted magnesium, aluminum, gallium or zinc compounds containing from 1 to 12 carbons in each hydrocarbyl group, and reaction products thereof with a proton source.
- Preferred hydrocarbyl groups are alkyl groups, preferably linear or branched, C2-8 alkyl groups.
- shuttling agents for use in the present invention are trialkyl aluminum and dialkyl zinc compounds, especially triethylaluminum, tri(i-propyl) aluminum, tri(i-butyl)aluminum, tri(n-hexyl)aluminum, tri(n-octyl)alummum, triethylgallium, or diethylzinc.
- Additional suitable shuttling agents include the reaction product or mixture formed by combining the foregoing organometal compounds, preferably a tri(Ci- 8 ) alkyl aluminum or di(Ci- 8 ) alkyl zinc compound, especially triethylaluminum, tri(i-propyl) aluminum, tri(i-butyl)aluminum, tri(n-hexyl)aluminum, tri(n-octyl)aluminum, or diethylzinc, with less than a stoichiometric quantity (relative to the number of hydrocarbyl groups) of a secondary amine or a hydroxyl compound, especially bis(trimethylsilyl)amine, t- butyl(dimethyl)siloxane, 2- hydroxymethylpyridine, di(n-pentyl)amine, 2,6-di(t- butyl)phenol, ethyl(l-naphthyl)amine, bis(2,3,6,7-di
- shuttling agents are n- octylaluminum di(bis(trimethylsilyl)amide), i-propylaluminum bis(dimethyl(t- butyl)siloxide), and n-octyl aluminum di(pyridinyl-2-methoxide), i-butylaluminum bis(dimethyl(t-butyl)siloxane), i-butylaluminum bis(di(trimethylsilyl)amide), n- octylaluminum di(pyridine-2-methoxide), i-butylaluminum bis(di(n-pentyl)amide), n- octylaluminum bis(2,6-di-t- butylphenoxide), n-octylaluminum di(2,6-di-t- butylphenoxide), n-octylaluminum bis(2,6-di-t- buty
- suitable chain shuttling agents include metal alkyls containing a divalent metal (e.g., Zn), a trivalent metal (e.g., Al), or a mixture of divalent metal and trivalent metal.
- the chain shuttling agent is a divalent metal alkyl, such as dialkylzinc.
- the chain shuttling agent is a trivalent metal alkyl, such as
- the organometallic compound is a mixture of divalent metal alkyl (e.g., dialkylzinc) and trivalent metal alkyl (e.g.,
- the chain shuttling agent is a mixture of trivalent metal and divalent metal at a trivalent/divalent metal ratio from 99: 1 to 1 :99 (e.g., from 95:5 to 50:50, from 90: 10 to 80:20, from 90: 10 to 70:30, etc.).
- the chain shuttling agent is a metal alkyl containing a mixture of aluminum and zinc metals at an aluminum/zinc ratio from 99: 1 to 1 :99 (e.g., from 95:5 to 50:50, from 90: 10 to 80:20, from 90: 10 to 70:30, etc.).
- shuttling agent for one catalyst or catalyst combination may not necessarily be as good or even satisfactory for use with a different catalyst or catalyst combination. Some potential shuttling agents may adversely affect the performance of one or more catalysts and may be undesirable for use for that reason as well. Accordingly, the activity of the chain shuttling agent desirably is balanced with the catalytic activity of the catalysts to achieve the desired polymer properties. In some embodiments of the present disclosure, best results may be obtained by use of shuttling agents having a chain shuttling activity (as measured by a rate of chain transfer) that is less than the maximum possible rate.
- shuttling agents possess the highest rates of polymer transfer as well as the highest transfer efficiencies (reduced incidences of chain termination). Such shuttling agents may be used in reduced concentrations and still achieve the desired degree of shuttling. In addition, such shuttling agents result in production of the shortest possible polymer block lengths.
- chain shuttling agents with a single exchange site are employed due to the fact that the effective molecular weight of the polymer in the reactor is lowered, thereby reducing viscosity of the reaction mixture and consequently reducing operating costs.
- Suitable procatalysts that would fall within the scope of the first olefin polymerization procatalyst (A) of the present disclosure include the
- the first olefin polymerization procatalyst (A) is the soft block/segment catalyst (i.e., high comonomer incorporator) of the olefin block copolymers of the present disclosure.
- heterogeneous catalysts examples include the well known Ziegler-Natta
- compositions especially Group 4 metal halides supported on Group 2 metal halides or mixed halides and alkoxides and the well known chromium or vanadium based catalysts.
- the catalysts for use herein are homogeneous catalysts comprising a relatively pure organometallic compound or metal complex, especially compounds or complexes based on metals selected from Groups 3-15 or the Lanthanide series of the Periodic Table of the Elements.
- Metal complexes for use herein may be selected from Groups 3 to 15 of the Periodic Table of the Elements containing one or more delocalized, ⁇ -bonded ligands or polyvalent Lewis base ligands. Examples include metallocene, half-metallocene, constrained geometry, and polyvalent pyridylamine, or other polychelating base complexes.
- the complexes are generically depicted by the formula: MKkX x Z z , or a dimer thereof, wherein
- M is a metal selected from Groups 3-15, preferably 3-10, more preferably 4-10, and most preferably Group 4 of the Periodic Table of the Elements;
- K independently at each occurrence is a group containing delocalized ⁇ - electrons or one or more electron pairs through which K is bound to M, said K group containing up to 50 atoms not counting hydrogen atoms, optionally two or more K groups may be joined together forming a bridged structure, and further optionally one or more K groups may be bound to Z, to X or to both Z and X;
- X independently at each occurrence is a monovalent, anionic moiety having up to 40 non- hydrogen atoms, optionally one or more X groups may be bonded together thereby forming a divalent or polyvalent anionic group, and, further optionally, one or more X groups and one or more Z groups may be bonded together thereby forming a moiety that is both covalently bound to M and coordinated thereto; or two X groups together form a divalent anionic ligand group of up to 40 non-hydrogen atoms or together are a conjugated diene having from 4 to 30 non-hydrogen atoms bound by means of delocalized ⁇ -electrons to M, whereupon M is in the +2 formal oxidation state, and
- Z independently at each occurrence is a neutral, Lewis base donor ligand of up to 50 non- hydrogen atoms containing at least one unshared electron pair through which Z is coordinated to M;
- Suitable metal complexes include those containing from 1 to 3 ⁇ -bonded anionic or neutral ligand groups, which may be cyclic or non-cyclic delocalized ⁇ - bonded anionic ligand groups. Exemplary of such ⁇ -bonded groups are conjugated or nonconjugated, cyclic or non-cyclic diene and dienyl groups, allyl groups,
- boratabenzene groups phosphole, and arene groups.
- ⁇ -bonded is meant that the ligand group is bonded to the transition metal by a sharing of electrons from a partially delocalized ⁇ -bond.
- Each atom in the delocalized ⁇ -bonded group may independently be substituted with a radical selected from the group consisting of hydrogen, halogen, hydrocarbyl, halohydrocarbyl, hydrocarbyl -substituted heteroatoms wherein the heteroatom is selected from Group 14-16 of the Periodic Table of the Elements, and such hydrocarbyl- substituted heteroatom radicals further substituted with a Group 15 or 16 hetero atom containing moiety.
- two or more such radicals may together form a fused ring system, including partially or fully hydrogenated fused ring systems, or they may form a metallocycle with the metal.
- hydrocarbyl is Ci-20 straight, branched and cyclic alkyl radicals, C 6 -2o aromatic radicals, C7-20 alkyl-substituted aromatic radicals, and C7-20 aryl-substituted alkyl radicals.
- Suitable hydrocarbyl -substituted heteroatom radicals include mono-, di- and tri-substituted radicals of boron, silicon, germanium, nitrogen, phosphorus or oxygen wherein each of the hydrocarbyl groups contains from 1 to 20 carbon atoms.
- Examples include ⁇ , ⁇ -dimethylamino, pyrrolidinyl, trimethylsilyl, tri ethyl silyl, t- butyldimethylsilyl, methyldi(t-butyl)silyl, triphenylgermyl, and trimethylgermyl groups.
- Examples of Group 15 or 16 hetero atom containing moieties include amino, phosphino, alkoxy, or alkylthio moieties or divalent derivatives thereof, for example, amide, phosphide, alkyleneoxy or alkylenethio groups bonded to the transition metal or Lanthanide metal, and bonded to the hydrocarbyl group, ⁇ -bonded group, or hydrocarbyl- substituted heteroatom.
- Suitable anionic, delocalized ⁇ -bonded groups include cyclopentadienyl, indenyl, fluorenyl, tetrahydroindenyl, tetrahydrofluorenyl, octahydrofluorenyl, pentadienyl, cyclohexadienyl, dihydroanthracenyl,
- hexahydroanthracenyl decahydroanthracenyl groups, phosphole, and boratabenzyl groups, as well as inertly substituted derivatives thereof, especially Ci-10 hydrocarbyl- substituted or tris(Ci-io hydrocarbyl)silyl- substituted derivatives thereof.
- Preferred anionic delocalized ⁇ -bonded groups are cyclopentadienyl, pentamethylcyclopentadienyl, tetramethylcyclopentadienyl,
- the boratabenzenyl ligands are anionic ligands which are boron containing analogues to benzene. They are previously known in the art having been described by G. Herberich, et al., in Organometallics. 14,1, 471-480 (1995). Preferred
- boratabenzenyl ligands correspond to the formula:
- R 1 is an inert substituent, preferably selected from the group consisting of hydrogen, hydrocarbyl, silyl, halo or germyl, said R 1 having up to 20 atoms not counting hydrogen, and optionally two adjacent R 1 groups may be joined together.
- R 1 is an inert substituent, preferably selected from the group consisting of hydrogen, hydrocarbyl, silyl, halo or germyl, said R 1 having up to 20 atoms not counting hydrogen, and optionally two adjacent R 1 groups may be joined together.
- R 1 is an inert substituent, preferably selected from the group consisting of hydrogen, hydrocarbyl, silyl, halo or germyl, said R 1 having up to 20 atoms not counting hydrogen, and optionally two adjacent R 1 groups may be joined together.
- Phospholes are anionic ligands that are phosphorus containing analogues to a cyclopentadienyl group. They are previously known in the art having been described by WO 98/50392, and elsewhere. Preferred phosphole ligands correspond to the formula:
- transition metal complexes for use herein correspond to the formula: MKkXxZz, or a dimer thereof, wherein:
- M is a Group 4 metal
- K is a group containing delocalized ⁇ -electrons through which K is bound to M, said K group containing up to 50 atoms not counting hydrogen atoms, optionally two K groups may be joined together forming a bridged structure, and further optionally one K may be bound to X or Z;
- X at each occurrence is a monovalent, anionic moiety having up to 40 non- hydrogen atoms, optionally one or more X and one or more K groups are bonded together to form a metallocycle, and further optionally one or more X and one or more Z groups are bonded together thereby forming a moiety that is both covalently bound to M and coordinated thereto;
- Z independently at each occurrence is a neutral, Lewis base donor ligand of up to 50 non- hydrogen atoms containing at least one unshared electron pair through which Z is coordinated to M;
- Suitable complexes include those containing either one or two K groups.
- the latter complexes include those containing a bridging group linking the two K groups.
- Suitable bridging groups are those corresponding to the formula
- (ER'2) e wherein E is silicon, germanium, tin, or carbon, R' independently at each occurrence is hydrogen or a group selected from silyl, hydrocarbyl, hydrocarbyloxy and combinations thereof, said R' having up to 30 carbon or silicon atoms, and e is 1 to 8.
- R' independently at each occurrence is methyl, ethyl, propyl, benzyl, tert- butyl, phenyl, methoxy, ethoxy or phenoxy.
- Examples of the complexes containing two K groups are compounds corresponding to the formula:
- M is titanium, zirconium or hafnium, preferably zirconium or hafnium, in the +2 or +4 formal oxidation state;
- R 3 at each occurrence independently is selected from the group consisting of hydrogen, hydrocarbyl, silyl, germyl, cyano, halo and combinations thereof, said R 3 having up to 20 non- hydrogen atoms, or adjacent
- R 3 groups together form a divalent derivative (that is, a hydrocarbadiyl, siladiyl or germadiyl group) thereby forming a fused ring system, and
- X independently at each occurrence is an anionic ligand group of up to 40 non-hydrogen atoms, or two X" groups together form a divalent anionic ligand group of up to 40 non-hydrogen atoms or together are a conjugated diene having from 4 to 30 non-hydrogen atoms bound by means of delocalized ⁇ -electrons to M, whereupon M is in the +2 formal oxidation state, and
- R', E and e are as previously defined.
- dimethylbis(cyclopentadienyl)silane dimethylbis(tetramethylcyclopentadienyl)silane, dimethylbis(2-ethylcyclopentadien-l-yl)silane, dimethylbis(2-t-butylcyclopentadien-l- yl)silane, 2,2-bis(tetramethylcyclopentadienyl)propane, dimethylbis(inden-l-yl)silane, dimethylbis(tetrahydroinden-l-yl)silane, dimethylbis(fluoren-l-yl)silane,
- dimethylbis(tetrahydrofluoren-l-yl)silane dimethylbis(2-methyl-4-phenylinden-l-yl)- silane, dimethylbis(2-methylinden-l -yl)silane, dimethyl(cyclopentadienyl)(fluoren-l- yl)silane, dimethyl(cyclopentadienyl)(octahydrofluoren-l-yl)silane,
- Suitable X" groups are selected from hydride, hydrocarbyl, silyl, germyl, halohydrocarbyl, halosilyl, silylhydrocarbyl and aminohydrocarbyl groups, or two X" groups together form a divalent derivative of a conjugated diene or else together they form a neutral, ⁇ -bonded, conjugated diene.
- Exemplary X" groups are CI -20 hydrocarbyl groups.
- metal complexes of the foregoing formula suitable for use in the present disclosure include:
- a further class of metal complexes utilized in the present disclosure corresponds to the preceding formula: MKZ Z X X , or a dimer thereof, wherein M, K, X, x and z are as previously defined, and Z is a substituent of up to 50 non-hydrogen atoms that together with K forms a metallocycle with M.
- Suitable Z substituents include groups containing up to 30 non-hydrogen atoms comprising at least one atom that is oxygen, sulfur, boron or a member of Group 14 of the Periodic Table of the Elements directly attached to K, and a different atom, selected from the group consisting of nitrogen, phosphorus, oxygen or sulfur that is covalently bonded to M.
- this class of Group 4 metal complexes used according to the present invention includes "constrained geometry catalysts" corresponding to the formula:
- M is titanium or zirconium, preferably titanium in the +2, +3, or +4 formal oxidation state
- K 1 is a delocalized, ⁇ -bonded ligand group optionally substituted with from 1 to 5 R 2 groups,
- R 2 at each occurrence independently is selected from the group consisting of hydrogen, hydrocarbyl, silyl, germyl, cyano, halo and combinations thereof, said R 2 having up to 20 non- hydrogen atoms, or adjacent R 2 groups together form a divalent derivative (that is, a hydrocarbadiyl, siladiyl or germadiyl group) thereby forming a fused ring system,
- each X is a halo, hydrocarbyl, heterohydrocarbyl, hydrocarbyloxy or silyl group, said group having up to 20 non-hydrogen atoms, or two X groups together form a neutral C5-30 conjugated diene or a divalent derivative thereof; x is 1 or 2;
- Y is -O-, -S-, - R'-, -PR'-;
- R' independently at each occurrence is hydrogen or a group selected from silyl, hydrocarbyl, hydrocarbyloxy and combinations thereof, said R' having up to 30 carbon or silicon atoms.
- Ar is an aryl group of from 6 to 30 atoms not counting hydrogen
- R 4 independently at each occurrence is hydrogen, Ar, or a group other than Ar selected from hydrocarbyl, trihydrocarbylsilyl, trihydrocarbylgermyl, halide, hydrocarbyloxy, trihydrocarbylsiloxy, bis(trihydrocarbylsilyl)amino,
- R group having up to 40 atoms not counting hydrogen atoms, and optionally two adjacent R 4 groups may be joined together forming a polycyclic fused ring group;
- M is titanium;
- Y is -0-, -S-, - R 5 -, -PR 5 -; - R 5 2 , or -PR 5 2 ;
- R 5 independently at each occurrence is hydrocarbyl, trihydrocarbylsilyl, or trihydrocarbylsilylhydrocarbyl, said R 5 having up to 20 atoms other than hydrogen, and optionally two R 5 groups or R 5 together with Y or Z form a ring system;
- R 6 independently at each occurrence, is hydrogen, or a member selected from hydrocarbyl, hydrocarbyl oxy, silyl, halogenated alkyl, halogenated aryl, - R 5 2 , and combinations thereof, said R 6 having up to 20 non-hydrogen atoms, and optionally, two R 6 groups or R 6 together with Z forms a ring system;
- Z is a neutral diene or a monodentate or polydentate Lewis base optionally bonded to R 5 , R 6 , or X;
- X is hydrogen, a monovalent anionic ligand group having up to 60 atoms not counting hydrogen, or two X groups are joined together thereby forming a divalent ligand group;
- x is 1 or 2;
- z 0, 1 or 2.
- Suitable examples of the foregoing metal complexes are substituted at both the 3- and 4- positions of a cyclopentadienyl or indenyl group with an Ar group.
- Examples of the foregoing metal complexes include:
- M is titanium in the +2, +3 or +4 formal oxidation state
- R 7 independently at each occurrence is hydride, hydrocarbyl, silyl, germyl, halide, hydrocarbyl oxy, hydrocarbylsiloxy, hydrocarbyl silyl amino,
- hydrocarbylene-phosphino hydrocarbylsulfido, halo-substituted hydrocarbyl, hydrocarbyloxy-substituted hydrocarbyl, silyl -substituted hydrocarbyl,
- R 7 group having up to 40 atoms not counting hydrogen, and optionally two or more of the foregoing groups may together form a divalent derivative
- R 8 is a divalent hydrocarbylene- or substituted hydrocarbyl ene group forming a fused system with the remainder of the metal complex, said R 8 containing from 1 to 30 atoms not counting hydrogen;
- X a is a divalent moiety, or a moiety comprising one ⁇ -bond and a neutral two electron pair able to form a coordinate-covalent bond to M, said X a comprising boron, or a member of Group 14 of the Periodic Table of the Elements, and also comprising nitrogen, phosphorus, sulfur or oxygen;
- X is a monovalent anionic ligand group having up to 60 atoms exclusive of the class of ligands that are cyclic, delocalized, ⁇ -bound ligand groups and optionally two X groups together form a divalent ligand group;
- Z independently at each occurrence is a neutral ligating compound having up to 20 atoms
- x 0, 1 or 2;
- z is zero or 1.
- Suitable examples of such complexes are 3-phenyl-substituted s-indecenyl complexes corresponding to the formula:
- Specific metal complexes include:
- M is titanium in the +2, +3 or +4 formal oxidation state
- T is - R 9 - or -0-;
- R 9 is hydrocarbyl, silyl, germyl, dihydrocarbylboryl, or halohydrocarbyl or up to 10 atoms not counting hydrogen;
- R 10 independently at each occurrence is hydrogen, hydrocarbyl
- X a is a divalent moiety lacking in delocalized ⁇ -electrons, or such a moiety comprising one ⁇ -bond and a neutral two electron pair able to form a coordinate- covalent bond to M, said X a comprising boron, or a member of Group 14 of the Periodic Table of the Elements, and also comprising nitrogen, phosphorus, sulfur or oxygen;
- X is a monovalent anionic ligand group having up to 60 atoms exclusive of the class of ligands that are cyclic ligand groups bound to M through delocalized ⁇ - electrons or two X groups together are a divalent anionic ligand group;
- Z independently at each occurrence is a neutral ligating compound having up to 20 atoms
- x 0, 1, 2, or 3;
- T N(CH 3 )
- X is halo or hydrocarbyl
- x is 2
- X a is dimethylsilane
- z is 0, and R 10 at each occurrence is hydrogen, a hydrocarbyl, hydrocarbyloxy, dihydrocarbylamino, hydrocarbyleneamino, dihydrocarbylamino- substituted hydrocarbyl group, or hydrocarbyleneamino- substituted hydrocarbyl group of up to 20 atoms not counting hydrogen, and optionally two R 10 groups may be joined together.
- Illustrative metal complexes of the foregoing formula that may be employed in the practice of the present invention further include the following compounds:
- Illustrative Group 4 metal complexes that may be employed in the practice of the present disclosure further include:
- T is a bridging group, preferably containing 2 or more atoms other than hydrogen
- X and Y are each independently selected from the group consisting of nitrogen, sulfur, oxygen and phosphorus; more preferably both X and Y are nitrogen,
- R and R ' independently each occurrence are hydrogen or Ci-50 hydrocarbyl groups optionally containing one or more heteroatoms or inertly substituted derivative thereof.
- suitable R and R ' groups include alkyl, alkenyl, aryl, aralkyl, (poly)alkylaryl and cycloalkyl groups, as well as nitrogen, phosphorus, oxygen and halogen substituted derivatives thereof.
- Rb and Rb' groups include methyl, ethyl, isopropyl, octyl, phenyl, 2,6-dimethylphenyl, 2,6-di(isopropyl)phenyl, 2,4,6-trimethylphenyl, pentafluorophenyl, 3,5- trifluoromethylphenyl, and benzyl;
- g and g' are each independently 0 or 1;
- M is a metallic element selected from Groups 3 to 15, or the Lanthanide series of the Periodic Table of the Elements.
- M is a Group 3-13 metal, more preferably M is a Group 4-10 metal;
- L is a monovalent, divalent, or trivalent anionic ligand containing from 1 to 50 atoms, not counting hydrogen.
- suitable L groups include halide; hydride; hydrocarbyl, hydrocarbyloxy; di(hydrocarbyl)amido, hydrocarbyleneamido, di(hydrocarbyl)phosphido; hydrocarbylsulfido; hydrocarbyloxy,
- L groups are CI -20 alkyl, C7-20 aralkyl, and chloride;
- h and h' are each independently an integer from 1 to 6, preferably from 1 to 4, more preferably from 1 to 3, and j is 1 or 2, with the value h x j selected to provide charge balance;
- Z is a neutral ligand group coordinated to M , and containing up to 50 atoms not counting hydrogen.
- Preferred Z groups include aliphatic and aromatic amines, phosphines, and ethers, alkenes, alkadienes, and inertly substituted derivatives thereof.
- Suitable inert substituents include halogen, alkoxy, aryloxy, alkoxycarbonyl, aryloxycarbonyl, di(hydrocarbyl)amine, tri(hydrocarbyl)silyl, and nitrile groups.
- Preferred Z groups include triphenylphosphine, tetrahydrofuran, pyridine, and 1,4- diphenylbutadiene;
- f is an integer from 1 to 3;
- T , R and R ' may be joined together to form a single or multiple ring structure
- h is an integer from 1 to 6, preferably from 1 to 4, more preferably from 1 to 3; indicates any form of electronic interaction, especially coordinate or covalent bonds, including multiple bonds, arrows signify coordinate bonds, and dotted lines indicate optional double bonds.
- R it is preferred that R have relatively low steric hindrance with respect to X .
- most preferred R groups are straight chain alkyl groups, straight chain alkenyl groups, branched chain alkyl groups wherein the closest branching point is at least 3 atoms removed from X , and halo,
- Highly preferred R groups in this embodiment are CI -8 straight chain alkyl groups.
- R ' preferably has relatively high steric hindrance with respect to Y .
- suitable R ' groups for this embodiment include alkyl or alkenyl groups containing one or more secondary or tertiary carbon centers, cycloalkyl, aryl, alkaryl, aliphatic or aromatic heterocyclic groups, organic or inorganic oligomeric, polymeric or cyclic groups, and halo, dihydrocarbylamino, alkoxy or trihydrocarbylsilyl substituted derivatives thereof.
- Preferred R ' groups in this embodiment contain from 3 to 40, more preferably from 3 to 30, and most preferably from 4 to 20 atoms not counting hydrogen and are branched or cyclic.
- Examples of preferred T groups are structures corresponding to the followin formulas:
- Each R d is Cl-10 hydrocarbyl group, preferably methyl, ethyl, n-propyl, i- propyl, t-butyl, phenyl, 2,6-dimethylphenyl, benzyl, or tolyl.
- Each R e is Cl-10 hydrocarbyl, preferably methyl, ethyl, n-propyl, i-propyl, t-butyl, phenyl, 2,6- dimethylphenyl, benzyl, or tolyl.
- two or more R d or R e groups, or mixtures of Rd and Re groups may together form a polyvalent derivative of a hydrocarbyl group, such as, 1,4-butylene, 1,5-pentylene, or a multicyclic, fused ring, polyvalent hydrocarbyl- or heterohydrocarbyl- group, such as naphthalene-l,8-diyl.
- a hydrocarbyl group such as, 1,4-butylene, 1,5-pentylene, or a multicyclic, fused ring
- polyvalent hydrocarbyl- or heterohydrocarbyl- group such as naphthalene-l,8-diyl.
- Suitable examples of the foregoing polyvalent Lewis base complexes include:
- R d at each occurrence is independently selected from the group consisting of hydrogen and CI -50 hydrocarbyl groups optionally containing one or more heteroatoms, or inertly substituted derivative thereof, or further optionally, two adjacent R d groups may together form a divalent bridging group; d' is 4;
- M is a Group 4 metal, preferably titanium or hafnium, or a Group 10 metal, preferably Ni or Pd;
- L ' is a monovalent ligand of up to 50 atoms not counting hydrogen, preferably halide or hydrocarbyl, or two L groups together are a divalent or neutral ligand group, preferably a C2-50 hydrocarbyl ene, hydrocarbadiyl or diene group.
- the polyvalent Lewis base complexes for use in the present invention especially include Group 4 metal derivatives, especially hafnium derivatives of hydrocarbylamine substituted heteroaryl compounds corresponding to the formula: wherein:
- R 11 is selected from alkyl, cycloalkyl, heteroalkyl, cycloheteroalkyl, aryl, and inertly substituted derivatives thereof containing from 1 to 30 atoms not counting hydrogen or a divalent derivative thereof;
- T 1 is a divalent bridging group of from 1 to 41 atoms other than hydrogen, preferably 1 to 20 atoms other than hydrogen, and most preferably a mono- or di- Cl- 20 hydrocarbyl substituted methylene or silane group;
- R 12 is a C5-20 heteroaryl group containing Lewis base functionality, especially a pyridin-2-yl- or substituted pyridin-2-yl group or a divalent derivative thereof;
- M 1 is a Group 4 metal, preferably hafnium
- X 1 is an anionic, neutral or dianionic ligand group
- x' is a number from 0 to 5 indicating the number of such X 1 groups; and bonds, optional bonds and electron donative interactions are represented by lines, dotted lines and arrows respectively.
- Suitable complexes are those wherein ligand formation results from hydrogen elimination from the amine group and optionally from the loss of one or more additional groups, especially from R 12 .
- electron donation from the Lewis base functionality preferably an electron pair, provides additional stability to the metal center.
- Suitable metal complexes correspond to the formula:
- R 13 , R 14 , R 15 and R 16 are hydrogen, halo, or an alkyl, cycloalkyl, heteroalkyl, heterocycloalkyl, aryl, or silyl group of up to 20 atoms not counting hydrogen, or adjacent R 13 , R 14 , R 15 or R 16 groups may be joined together thereby forming fused ring derivatives, and bonds, optional bonds and electron pair donative interactions are represented by lines, dotted lines and arrows respectively.
- Suitable examples of the foregoing metal com lexes correspond to the formula:
- R 13 , R 14 , R 15 and R 16 are as previously defined, preferably R 13 , R 14 , and R 15 are hydrogen, or CI -4 alkyl, and R 16 is C 6 -20 aryl, most preferably naphthalenyl;
- R a independently at each occurrence is Ci-4 alkyl, and a is 1-5, most preferably R a in two ortho- positions to the nitrogen is isopropyl or t-butyl;
- R 17 and R 18 independently at each occurrence are hydrogen, halogen, or a Ci-20 alkyl or aryl group, most preferably one of R 17 and R 18 is hydrogen and the other is a C6-20 aryl group, especially 2-isopropyl, phenyl or a fused polycyclic aryl group, most preferably an anthracenyl group, and bonds, optional bonds and electron pair donative interactions are represented by lines, dotted lines and arrows respectively.
- Exemplary metal complexes for use herein as catalysts correspond to the formula:
- X 1 at each occurrence is halide, ⁇ , ⁇ -dimethylamido, or C1-4 alkyl, and preferably at each occurrence X 1 is methyl;
- R f independently at each occurrence is hydrogen, halogen, CI -20 alkyl, or C6- 20 aryl, or two adj acent R f groups are j oined together thereby forming a ring, and f is 1 - 5;
- R c independently at each occurrence is hydrogen, halogen, Ci-20 alkyl, or C 6 -2o aryl, or two adjacent R c groups are joined together thereby forming a ring, and c is 1-5.
- Suitable examples of metal complexes for use as catalysts according to the present invention are complexes of the following formulas:
- R x is CI -4 alkyl or cycloalkyl, preferably methyl, isopropyl, t-butyl or cyclohexyl;
- X 1 at each occurrence is halide, ⁇ , ⁇ -dimethylamido, or CI -4 alkyl, preferably methyl.
- the hydrogen of the 2-position of the a-naphthalene group substituted at the 6-position of the pyridin-2-yl group is subject to elimination, thereby uniquely forming metal complexes wherein the metal is covalently bonded to both the resulting amide group and to the 2-position of the a- naphthalenyl group, as well as stabilized by coordination to the pyridinyl nitrogen atom through the electron pair of the nitrogen atom.
- Additional suitable metal complexes of polyvalent Lewis bases for use herein include compounds corresponding to the formula:
- R 20 is an aromatic or inertly substituted aromatic group containing from 5 to 20 atoms not counting hydrogen, or a polyvalent derivative thereof;
- T 3 is a hydrocarbylene or hydrocarbyl silane group having from 1 to 20 atoms not counting hydrogen, or an inertly substituted derivative thereof;
- M 3 is a Group 4 metal, preferably zirconium or hafnium;
- G is an anionic, neutral or dianionic ligand group; preferably a halide, hydrocarbyl, silane, trihydrocarbylsilylhydrocarbyl, trihydrocarbylsilyl, or
- dihydrocarbylamide group having up to 20 atoms not counting hydrogen
- g is a number from 1 to 5 indicating the number of such G groups; and bonds and electron donative interactions are represented by lines and arrows respectively.
- T 3 is a divalent bridging group of from 2 to 20 atoms not counting hydrogen, preferably a substituted or unsubstituted, C3-6 alkylene group;
- Ar 2 independently at each occurrence is an arylene or an alkyl- or aryl- substituted arylene group of from 6 to 20 atoms not counting hydrogen;
- M 3 is a Group 4 metal, preferably hafnium or zirconium; G independently at each occurrence is an anionic, neutral or dianionic ligand group;
- g is a number from 1 to 5 indicating the number of such X groups; and electron donative interactions are represented by arrows.
- Suitable examples of metal complexes of foregoing formula include the followin compounds
- M 3 is Hf or Zr
- Ar 4 is C 6 -2o aryl or inertly substituted derivatives thereof, especially 3,5- di(isopropyl)phenyl, 3,5-di(isobutyl)phenyl, dibenzo-lH-pyrrole-l-yl, or anthracen-5- yl, and
- T 4 independently at each occurrence comprises a C3-6 alkylene group, a C3-6 cycloalkylene group, or an inertly substituted derivative thereof;
- R 21 independently at each occurrence is hydrogen, halo, hydrocarbyl, trihydrocarbylsilyl, or trihydrocarbylsilylhydrocarbyl of up to 50 atoms not counting hydrogen;
- G independently at each occurrence is halo or a hydrocarbyl or
- trihydrocarbylsilyl group of up to 20 atoms not counting hydrogen, or 2 G groups together are a divalent derivative of the foregoing hydrocarbyl or trihydrocarbylsilyl groups.
- Suitable compounds are compounds of the formulas:
- Ar 4 is 3,5-di(isopropyl)phenyl, 3,5-di(isobutyl)phenyl, dibenzo-lH- pyrrole-l-yl, or anthracen-5-yl,
- R 21 is hydrogen, halo, or CI -4 alkyl, especially methyl
- T 4 is propan-l,3-diyl or butan-l,4-diyl
- G is chloro, methyl or benzyl.
- M is zirconium or hafnium
- R 20 independently at each occurrence is a divalent aromatic or inertly substituted aromatic group containing from 5 to 20 atoms not counting hydrogen;
- T 3 is a divalent hydrocarbon or silane group having from 3 to 20 atoms not counting hydrogen, or an inertly substituted derivative thereof;
- R D independently at each occurrence is a monovalent ligand group of from 1 to 20 atoms, not counting hydrogen, or two R D groups together are a divalent ligand group of from 1 to 20 atoms, not counting hydrogen.
- Ar 2 independently at each occurrence is an arylene or an alkyl-, aryl-, alkoxy- or amino- substituted arylene group of from 6 to 20 atoms not counting hydrogen or any atoms of any substituent;
- T 3 is a divalent hydrocarbon bridging group of from 3 to 20 atoms not counting hydrogen, preferably a divalent substituted or unsubstituted C3-6 aliphatic,
- cycloaliphatic or bis(alkylene)- substituted cycloaliphatic group having at least 3 carbon atoms separating oxygen atoms;
- R D independently at each occurrence is a monovalent ligand group of from 1 to 20 atoms, not counting hydrogen, or two R D groups together are a divalent ligand group of from 1 to 40 atoms, not counting hydrogen.
- Ar 4 independently at each occurrence is C 6 -2o aryl or inertly substituted derivatives thereof, especially 3,5-di(isopropyl)phenyl, 3,5-di(isobutyl)phenyl, dibenzo- lH-pyrrole-l-yl, naphthyl, anthracen-5-yl, 1,2,3,4,6,7,8, 9-octahydroanthracen-5-yl;
- T 4 independently at each occurrence is a propyl ene-1, 3 -diyl group, a
- R 21 independently at each occurrence is hydrogen, halo, hydrocarbyl, trihydrocarbylsilyl, trihydrocarbylsilylhydrocarbyl, alkoxy or amino of up to 50 atoms not counting hydrogen;
- R D independently at each occurrence is halo or a hydrocarbyl or
- trihydrocarbylsilyl group of up to 20 atoms not counting hydrogen, or 2 R D groups together are a divalent hydrocarbylene, hydrocarbadiyl or trihydrocarbylsilyl group of up to 40 atoms not counting hydrogen.
- Exemplary metal complexes are compounds of the formula:
- Ar 4 independently at each occurrence, is 3,5-di(isopropyl)phenyl, 3,5- di(isobutyl)phenyl, dibenzo-lH-pyrrole-l-yl, or anthracen-5-yl,
- R 21 independently at each occurrence is hydrogen, halo, hydrocarbyl, trihydrocarbylsilyl, trihydrocarbylsilylhydrocarbyl, alkoxy or amino of up to 50 atoms not counting hydrogen;
- T 4 is propan-l,3-diyl or bis(methylene)cyclohexan-l,2-diyl;
- R D independently at each occurrence is halo or a hydrocarbyl or
- trihydrocarbylsilyl group of up to 20 atoms not counting hydrogen, or 2 R D groups together are a hydrocarbylene, hydrocarbadiyl or hydrocarbylsilanediyl group of up to 40 atoms not counting hydrogen.
- Suitable metal complexes according to the present disclosure correspond to the formulas:
- suitable metal complexes are the following compounds: A) bis((2-oxoyl-3-(l,2,3,4,6,7,8,9-octahydroanthracen-5-yl)-5-(methyl)phenyl)-2- phenoxy)-l,3-propanediylhafnium (IV) dimethyl,
- the foregoing metal complexes may be conveniently prepared by standard metallation and ligand exchange procedures involving a source of the transition metal and a neutral polyfunctional ligand source.
- the techniques employed are the same as or analogous to those disclosed in USP 6,827,976 and US2004/0010103, and elsewhere.
- the metal complex is activated to form the active catalyst composition by combination with the cocatalyst.
- the activation may occur prior to addition of the catalyst composition to the reactor with or without the presence of other components of the reaction mixture, or in situ through separate addition of the metal complex and activating cocatalyst to the reactor.
- the foregoing polyvalent Lewis base complexes are conveniently prepared by standard metallation and ligand exchange procedures involving a source of the Group 4 metal and the neutral polyfunctional ligand source.
- the complexes may also be prepared by means of an amide elimination and hydrocarbylation process starting from the corresponding Group 4 metal tetraamide and a hydrocarbylating agent, such as trimethylaluminum. Other techniques may be used as well.
- These complexes are known from the disclosures of, among others, US patents 6,320,005, 6, 103,657, WO 02/38628, WO 03/40195, and US 04/0220050.
- Catalysts having high comonomer incorporation properties are also known to reincorporate in situ prepared long chain olefins resulting incidentally during the polymerization through ⁇ - hydride elimination and chain termination of growing polymer, or other process.
- concentration of such long chain olefins is particularly enhanced by use of continuous solution polymerization conditions at high conversions, especially ethylene conversions of 95 percent or greater, more preferably at ethylene conversions of 97 percent or greater. Under such conditions a small but detectable quantity of olefin terminated polymer may be reincorporated into a growing polymer chain, resulting in the formation of long chain branches, that is, branches of a carbon length greater than would result from other deliberately added comonomer.
- chains reflect the presence of other comonomers present in the reaction mixture. That is, the chains may include short chain or long chain branching as well, depending on the comonomer composition of the reaction mixture. Long chain branching of olefin polymers is further described in USP's 5,272,236, 5,278,272, and 5,665,800.
- branching including hyper-branching, may be induced in a particular segment of the present multi -block copolymers by the use of specific catalysts known to result in "chain-walking" in the resulting polymer.
- specific catalysts known to result in "chain-walking" in the resulting polymer.
- certain homogeneous bridged bis indenyl- or partially hydrogenated bis indenyl- zirconium catalysts disclosed by Kaminski, et al., J. Mol. Catal. A: Chemical 102 (1995) 59-65; Zambelli, et al., Macromolecules. 1988, 21, 617- 622; or Dias, et al., I Mol. Catal.
- A: Chemical 185 (2002) 57-64 may be used to prepare branched copolymers from single monomers, including ethylene.
- Higher transition metal catalysts, especially nickel and palladium catalysts are also known to lead to hyper- branched polymers (the branches of which are also branched) as disclosed in
- the presence of such branching (long chain branching, 1,3 -addition, or hyper-branching) in the polymers of the invention can be confined to only the blocks or segments resulting from activity of the first olefin polymerization procatalyst (A). Accordingly, in one embodiment of the disclosure a multi-block copolymer containing blocks or segments differing in the presence of such branching in combination with other segments or blocks substantially lacking such branching (especially high density or highly crystalline polymer blocks), can be produced from a single monomer containing reaction mixture, that is, without the addition of a deliberately added comonomer.
- a multi-block copolymer comprising alternating unbranched, ethylene homopolymer segments and branched polyethylene segments, especially ethylene/propylene copolymer segments, may be prepared from an initial reaction mixture consisting essentially of ethylene as the addition polymerizable monomer.
- the presence of such branching in the multi -block copolymers of the invention can be detected by certain physical properties of the resulting copolymers, such as reduced surface imperfections during melt extrusion (reduced melt fracture), reduced glass transition temperature, Tg, for the amorphous segments compared to a non-branched polymer segment, and/or the presence of 1,3 -addition sequences or hyper-branching as detected by MR techniques.
- procatalysts that fall within the scope of the first olefin polymerization procatalyst (A) of the present disclosure include but are not limited to Procatalysts (A1)-(A7), as listed below.
- Procatalyst (A2) (E)-((2,6-diisopropylphenyl)(2-methyl-3- (octylimino)butan-2-yl)amino)trimethyl hafnium prepared according to methods known in the art.
- Procatalyst (A3) [[2',2" '-[l,2-cyclohexanediylbis(methyleneoxy- KO)]bis[3-(9H-carbazol-9-yl)-5-methyl[l,l '-biphenyl]-2-olato-KO]](2-)]dimethyl hafnium prepared according to methods known in the art.
- Procatalyst (A4) [[6',6' "-[l,4-butanediylbis(oxy-KO)]bis[3-(9H-carbazol- 9-yl)-3'-fluoro-5-methyl-[l, -biphenyl]-2-olato-KO]](2-)]-dimethyl hafnium prepared according to methods known in the art.
- Procatalyst (A6) [[6',6" '-[l,4-butanediylbis(oxy-KO)]bis[3-(9H-carbazol- 9-yl)-3'-fluoro-5-(butyldimethylsilyl)-[l, l '-biphenyl]-2-olato-KO]](2-)]-dimethyl hafnium prepared according to methods known in the art.
- the second olefin polymerization procatalyst (B) of the present disclosure comprises a metal-li and complex of Formula (I):
- M is titanium, zirconium, or hafnium
- each Zl is independently a monodentate or polydentate ligand that is neutral, monoanionic, or dianionic, wherein nn is an integer, and wherein Zl and nn are chosen in such a way that the metal-ligand complex of Formula (I) is overall neutral; wherein each Q 1 and Q 10 independently is selected from the group consisting of (C6-C4o)aryl, substituted (C6-C4o)aryl, (C3-C4o)heteroaryl, and substituted (C 3 - C4o)heteroaryl;
- each Q 2 , Q 3 , Q 4 , Q 7 , Q 8 , and Q 9 independently is selected from a group consisting of hydrogen, (Ci-C4o)hydrocarbyl, substituted (Ci-C4o)hydrocarbyl, (Ci-C4o)heterohydrocarbyl, substituted (Ci-C4o)heterohydrocarbyl, halogen, and nitro (N0 2 );
- each Q 5 and Q 6 independently is selected from the group consisting of a (Ci-C4o)alkyl, substituted (Ci-C4o)alkyl, and [(Si)i-(C+Si)4o] substituted organosilyl; wherein each N independently is nitrogen;
- two or more of the Q 1"5 groups can combine together to form a ring structure, with such ring structure having from 5 to 16 atoms in the ring excluding any hydrogen atoms;
- two or more of the Q 6"10 groups can combine together to form a ring structure, with such ring structure having from 5 to 16 atoms in the ring excluding any hydrogen atoms.
- the second olefin polymerization procatalyst (B) is the hard block/segment catalyst (i.e., low comonomer incorporator) of the olefin block copolymers of the present disclosure.
- embodiments thereof herein is intended to include every possible stereoisomer, including coordination isomers, thereof.
- the metal ligand complex of Formula (I) above provides for homoleptic as well as heteroleptic procatalyst components.
- each of the (C1-C40) hydrocarbyl and (C1-C40) heterohydrocarbyl of any one or more of Q 1 , Q 2 , Q 3 , Q 4 , Q 5 , Q 6 , Q 7 , Q 8 , Q 9 and Q 10 each independently is unsubstituted or substituted with one or more R s substituents, and wherein each R s independently is a halogen atom, polyfluoro substitution, perfluoro substitution, unsubstituted (Ci-Ci 8 )alkyl, (C6-Ci 8 )aryl, F 3 C, FCH2O, F2HCO, F 3 CO, (R cl ) 3 Si, (R cl ) 3 Ge, (R L1 )0, (R L1 )S, (R u )S(0), (R C1 )S(0) 2 , (R U ) 2 P, (R C1 ) 2 N, (
- Q 5 and Q 6 are each independently (C1-C40) primary or secondary alkyl groups with respect to their connection to the amine nitrogen of the parent ligand structure.
- the terms primary and secondary alkyl groups are given their usual and customary meaning herein; i.e., primary indicating that the carbon atom directly linked to the ligand nitrogen bears at least two hydrogen atoms and secondary indicates that the carbon atom directly linked to the ligand nitrogen bears only one hydrogen atom.
- two or more Q 1"5 groups or two or more Q 6"10 groups each independently can combine together to form ring structures, with such ring structures having from 5 to 16 atoms in the ring excluding any hydrogen atoms.
- Q 5 and Q 6 are each independently (C1-C40) primary or secondary alkyl groups and most preferably, Q 5 and Q 6 are each
- Q 1 and Q 10 of the olefin polymerization procatalyst of Formula I) are substituted phenyl groups; as shown in Formula (II),
- J 1 -! 10 are each independently selected from the group consisting of R s substituents and hydrogen; and wherein each R s independently is a halogen atom, polyfluoro substitution, perfluoro substitution, unsubstituted (Ci-Ci8)alkyl,
- R cl (R C1 )C(0)N(R C1 ), or (R C1 ) 2 NC(0), or two of the R s are taken together to form an unsubstituted (Ci-Ci8)alkylene where each R s independently is an
- J 1 , J 5 , J 6 and J 10 of Formula (II) are each independently selected from the group consisting of halogen atoms, (Ci-C 8 ) alkyl groups, and (Ci-C 8 ) alkoxyl groups.
- J 1 , J 5 , J 6 and J 10 of Formula (II) are each independently methyl; ethyl or isopropyl.
- the parenthetical expression (C1-C40) can be represented by the form "(Cx-Cy)," which means that the unsubstituted version of the chemical group comprises from a number x carbon atoms to a number y carbon atoms, wherein each x and y independently is an integer as described for the chemical group.
- the R s substituted version of the chemical group can contain more than y carbon atoms depending on nature of R s .
- the substituted (C x -C y ) chemical group may comprise more than y total carbon atoms; i.e., the total number of carbon atoms of the carbon atom-containing substituent(s)-substituted (C x -C y ) chemical group is equal to y plus the sum of the number of carbon atoms of each of the carbon atom-containing substituent(s).
- Any atom of a chemical group that is not specified herein is understood to be a hydrogen atom.
- each of the chemical groups (e.g., Q 1"10 ) of the metal- ligand complex of Formula (I) may be unsubstituted, that is, can be defined without use of a substituent R s , provided the above-mentioned conditions are satisfied.
- at least one of the chemical groups of the metal-ligand complex of Formula (I) independently contain one or more of the substituents R s .
- each R s independently is bonded to a same or different substituted chemical group.
- two or more R s are bonded to a same chemical group, they independently are bonded to a same or different carbon atom or heteroatom, as the case may be, in the same chemical group up to and including persubstitution of the chemical group.
- substitution means each hydrogen atom (H) bonded to a carbon atom or heteroatom of a corresponding unsubstituted compound or functional group, as the case may be, is replaced by a substituent (e.g., R s ).
- polysubstitution means each of at least two, but not all, hydrogen atoms (H) bonded to carbon atoms or heteroatoms of a corresponding unsubstituted compound or functional group, as the case may be, is replaced by a substituent (e.g., R s ).
- substituent e.g., R s
- the term “monosubstitution” means that only one hydrogen atom (H) bonded to a carbon atom or heteroatom of a corresponding unsubstituted compound or functional group, as the case may be, is replaced by a substituent (e.g., R s ).
- hydrocarbyl, heterohydrocarbyl, hydrocarbylene, heterohydrocarbylene, alkyl, alkylene, heteroalkyl, heteroalkylene, aryl, arylene, heteroaryl, heteroarylene, cycloalkyl, cycloalkylene, heterocycloalkyl, and heterocycloalkylene are intended to include every possible stereoisomer.
- (Ci-C4o)hydrocarbyl means a hydrocarbon radical of from 1 to 40 carbon atoms and the term "(Ci-C4o)hydrocarbylene” means a hydrocarbon diradical of from 1 to 40 carbon atoms, wherein each hydrocarbon radical and diradical independently is aromatic (6 carbon atoms or more) or non-aromatic, saturated or unsaturated, straight chain or branched chain, cyclic (including mono- and polycyclic, fused and non-fused polycyclic, including bicyclic; 3 carbon atoms or more) or acyclic, or a combination of two or more thereof; and each hydrocarbon radical and diradical independently is the same as or different from another
- a (Ci-C4o)hydrocarbyl independently is an unsubstituted or substituted (Ci-C4o)alkyl, (C3-C4o)cycloalkyl, (C3-C2o)cycloalkyl-(Ci-C2o)alkylene, (C 6 - C4o)aryl, or (C6-C2o)aryl-(Ci-C2o)alkylene.
- All individual values and subranges from 1 to 40 carbons in the (Ci-C4o)hydrocarbyl are included and disclosed herein; for example, the number of carbon atoms in the (Ci-C4o)hydrocarbyl may range from an upper limit of 40 carbon atoms, preferably 30 carbon atoms, more preferably 20 carbon atoms, more preferably 15 carbon atoms, more preferably 12 carbon atoms and most preferably 10 carbon atoms.
- the (Ci-C4o)hydrocarbyl includes (Ci- C4o)hydrocarbyl groups, (Ci-C 3 o)hydrocarbyl) groups, (Ci-C2o)hydrocarbyl) groups, (Ci-Ci5)hydrocarbyl) groups, (Ci-Ci2)hydrocarbyl) groups, (Ci-Cio)hydrocarbyl) groups,. (Cio-C 3 o)hydrocarbyl) groups, (Ci5-C4o)hydrocarbyl) groups, (C 5 - C2 5 )hydrocarbyl) groups, or (Ci5-C25)hydrocarbyl) groups.
- (Ci-C4o)alkyl means a saturated straight or branched
- unsubstituted (Ci-C4o)alkyl are unsubstituted (Ci- C2o)alkyl; unsubstituted (Ci-Cio)alkyl; unsubstituted (Ci-C5)alkyl; methyl; ethyl; 1- propyl; 2-propyl; 2,2-dimethylpropyl, 1 -butyl; 2-butyl; 2-methylpropyl; 1, 1- dimethyl ethyl; 1-pentyl; 1-hexyl; 2-ethylhexyl, 1-heptyl; 1-nonyl; and 1-decyl; 2,2,4- trimethylpentyl.
- substituted (Ci-C4o)alkyl examples include substituted (Ci-C2o)alkyl; substituted (Ci-Cio)alkyl; trifluorom ethyl; trimethylsilylmethyl; methoxymethyl;
- (C6-C4o)aryl means an unsubstituted or substituted (by one or more R s ) mono-, bi- or tricyclic aromatic hydrocarbon radical of from 6 to 40 carbon atoms, of which at least from 6 to 14 of the carbon atoms are aromatic ring carbon atoms, and the mono-, bi- or tricyclic radical comprises 1, 2 or 3 rings, respectively; wherein one ring is aromatic and the optional second and third rings independently are fused or non-fused and the second and third rings are each independently optionally aromatic.
- unsubstituted (C6-C4o)aryl examples include unsubstituted (C6-C2o)aryl; unsubstituted (C6-Ci8)aryl; phenyl; biphenyl; ort/zo-terphenyl; weto-terphenyl;
- dihydroindenyl dihydroindenyl; naphthyl; tetrahydronaphthyl; phenanthrenyl and triptycenyl.
- substituted (C6-C4o)aryl examples include substituted (C6-C2o)aryl; substituted (C 6 - Ci 8 )aryl; 2,6-bis[(Ci-C 2 o)alkyl]-phenyl; 2-(Ci-C 5 )alkyl-phenyl; 2,6-bis(Ci-C 5 )alkyl- phenyl; 2,4,6-tris(Ci-C5)alkyl-phenyl; polyfluorophenyl; pentafluorophenyl; 2,6- dimethylphenyl; 2,6-diisopropylphenyl; 2,4,6-triisopropylphenyl; 2,4,6- trimethylphenyl; 2-methyl-6-trimethylsilylphenyl; 2-methyl-4,6-diisopropylphenyl; 4- methoxyphenyl; and 4-methoxy-2,6-dimethylphenyl.
- (C3-C4o)cycloalkyl means a saturated cyclic hydrocarbon radical of from 3 to 40 carbon atoms that is unsubstituted or substituted by one or more R s .
- Other cycloalkyl groups e.g., (C3-Ci 2 )alkyl) are defined in an analogous manner.
- unsubstituted (C3-C4o)cycloalkyl examples include unsubstituted (C3-C 2 o)cycloalkyl; unsubstituted (C3-Cio)cycloalkyl; cyclopropyl; cyclobutyl; cyclopentyl; cyclohexyl; cycloheptyl; cyclooctyl; cyclononyl; cyclodecyl; cyclopentyl; cyclohexyl;
- substituted (C3-C4o)cycloalkyl are substituted (C3-C 2 o)cycloalkyl;
- Examples of (Ci-C4o)hydrocarbylene are unsubstituted or substituted (C3- C4o)hydrocarbylene; (C6-C4o)arylene, (C3-C4o)cycloalkylene, and (C3-C4o)alkylene (e.g., (C3-C20)alkylene).
- the diradicals are on the terminal atoms of the hydrocarbylene as in a 1,3-alpha, omega diradical (e.g., -CH 2 CH 2 CH 2 -) or a 1,5-alpha, omega diradical with internal substitution (e.g., - CH2CH 2 CH(CH3)CH 2 CH2-).
- the diradicals are on the nonterminal atoms of the hydrocarbylene as in a C7 2,6-diradical (e.g.,
- (Ci-C4o)heterohydrocarbyl and “(Ci-C4o)heterohydrocarbylene” mean a heterohydrocarbon radical or diradical, respectively, of from 1 to 40 carbon atoms, and each heterohydrocarbon independently has one or more heteroatoms or heteroatomic groups O; S; N; S(O); S(0) 2 ; S(0) 2 N; Si(R cl ) 2 ; Ge(R cl ) 2 ; P(R C1 );
- (Ci-C4o)heterohydrocarbylene independently is unsubstituted or substituted (by one or more R s ), aromatic or non-aromatic, saturated or unsaturated, straight chain or branched chain, cyclic (including mono- and poly-cyclic, fused and non-fused polycyclic) or acyclic, or a combination of two or more thereof; and each is
- (Ci-C4o)alkylene means a saturated or unsaturated straight chain or branched chain diradical of from 1 to 40 carbon atoms that is unsubstituted or substituted by one or more R s .
- unsubstituted (Ci-C4o)alkylene examples include unsubstituted (C3-C 2 o)alkylene, including unsubstituted l,3-(C3-Cio)alkylene; 1,4- (C 4 -Cio)alkylene; -(CH 2 ) 3 -; -(CH 2 ) 4 -; -(CH 2 ) 5 -; -(CH 2 ) 6 -; -(CH 2 ) 7 -; -(CH 2 ) 8 -;
- substituted (Ci-C4o)alkylene are substituted (C 3 - C 2 o)alkylene; -CF 2 CF 2 CF 2 -; and -(CH 2 )i4C(CH 3 ) 2 (CH 2 )5- (i.e., a 6,6-dimethyl substituted normal- 1,20-eicosylene).
- examples of substituted (Ci-C4o)alkylene also include l,2-bis(methylene)cyclopentane; l,2-bis(methylene)cyclohexane; 2,3- bis(methylene)-7,7-dimethyl-bicyclo[2.2.1]heptane; and 2,3- bis(methylene)bicyclo[2.2.2]octane.
- (C 3 -C4o)cycloalkylene means a cyclic diradical (i.e., the radicals are on ring atoms) of from 3 to 40 carbon atoms that is unsubstituted or substituted by one or more R s .
- Connection of the chelating substituents to a cycloalkylene Q 3 group of Formula (I) must also satisfy the requirement that there be at least three atoms in the shortest chain connecting the bridged N atoms of Formula (I).
- Examples of unsubstituted (C3-C4o)cycloalkylene are 1,3-cyclobutylene, 1,3-cyclopentylene, and 1,4-cyclohexylene.
- Examples of substituted (C3-C4o)cycloalkylene are 2-trimethylsilyl- 1,4-cyclohexylene and l,2-dimethyl-l,3-cyclohexylene.
- the (Ci-C4o)heterohydrocarbyl independently is unsubstituted or substituted (Ci-C4o)heteroalkyl, (Ci-C4o)hydrocarbyl-0-, (Ci-C4o)hydrocarbyl-S-, (Ci-C 4 o)hydrocarbyl-S(0)-, (Ci-C 4 o)hydrocarbyl-S(0) 2 -,
- each R C1 is hydrogen, unsubstituted (Ci-Ci 8 )hydrocarbyl or an unsubstituted (Ci-Ci 8 )heterohydrocarbyl, or absent (e.g., absent when N comprises
- (Ci-C4o)heteroaryl means an unsubstituted or substituted (by one or more R s ) mono-, bi- or tricyclic heteroaromatic hydrocarbon radical of from 1 to 40 total carbon atoms and from 1 to 6 heteroatoms, and the mono-, bi- or tricyclic radical comprises 1, 2 or 3 rings, respectively, wherein one ring is heteroaromatic and the optional second and third rings independently are fused or non-fused; and the second or third rings are each independently optionally heteroaromatic.
- Other heteroaryl groups e.g., (C3-Ci 2 )heteroaryl) are defined in an analogous manner.
- the monocyclic heteroaromatic hydrocarbon radical is a 5- membered or 6-membered ring.
- the 5-membered ring has from 1 to 4 carbon atoms and from 4 to 1 heteroatoms, respectively, each heteroatom being O, S, N, or P, and preferably O, S, or N.
- Examples of 5-membered ring heteroaromatic hydrocarbon radical are pyrrol-l-yl; pyrrol-2-yl; furan-3-yl; thiophen-2-yl; pyrazol-l-yl; isoxazol-2- yl; isothiazol-5-yl; imidazol-2-yl; oxazol-4-yl; thiazol-2-yl; 1,2,4-triazol-l-yl; 1,3,4- oxadiazol-2-yl; l,3,4-thiadiazol-2-yl; tetrazol-l-yl; tetrazol-2-yl; and tetrazol-5-yl.
- the 6-membered ring has 3 to 5 carbon atoms and 1 to 3 heteroatoms, the heteroatoms being N or P, and preferably N.
- 6-membered ring heteroaromatic hydrocarbon radical are pyridine-2-yl; pyrimidin-2-yl; and pyrazin-2-yl, triazinyl.
- the bicyclic heteroaromatic hydrocarbon radical preferably is a fused 5,6- or 6,6-ring system. Examples of the fused 5,6-ring system bicyclic heteroaromatic hydrocarbon radical are indol-l-yl; and benzimidazole-l-yl.
- Examples of the fused 6,6-ring system bicyclic heteroaromatic hydrocarbon radical are quinolin-2-yl; and isoquinolin-l-yl.
- the tricyclic heteroaromatic hydrocarbon radical preferably is a fused 5,6,5-; 5,6,6-; 6,5,6-; or 6,6,6-ring system.
- An example of the fused 5,6,5-ring system is 1,7- dihydropyrrolo[3,2- Jindol-l-yl.
- An example of the fused 5, 6,6-ring system is IH- benzo[ ]indol-l-yl.
- An example of the fused 6,5,6-ring system is 9H-carbazol-9-yl.
- An example of the fused 6,5,6-ring system is 9H-carbazol-9-yl.
- An example of the fused 6,6,6-ring system is acrydin-9-yl.
- [00152] The term "[(Si)i-(C+Si)4o] substituted organosilyl” means a substituted silyl radical with 1 to 40 silicon atoms and 0 to 39 carbon atoms such that the total number of carbon plus silicon atoms is between 1 and 40.
- Examples of [(Si)i-(C+Si)4o] substituted organosilyl include trimethyl silyl, triisopropylsilyl, dimethylphenyl silyl, diphenylmethylsilyl, triphenyl silyl, and triethylsilyl.
- the (C 3 -C4o)heteroaryl is 2, 7-di substituted carbazolyl or 3, 6-di substituted carbazolyl, more preferably wherein each R s independently is phenyl, methyl, ethyl, isopropyl, or tertiary-butyl, still more preferably 2,7-di(tertiary- butyl)-carbazolyl, 3,6-di(tertiary-butyl)-carbazolyl, 2,7-di(tertiary-octyl)-carbazolyl, 3,6-di(tertiary-octyl)-carbazolyl, 2,7-diphenylcarbazolyl, 3,6-diphenylcarbazolyl, 2,7- bis(2,4,6-trimethylphenyl)-carbazolyl or 3,6-bis(2,4,6-trimethylphenyl)-carbazolyl.
- a heteroalkyl group may optionally be cyclic, i.e. a heterocycloalkyl group.
- Examples of unsubstituted (C3-C4o)heterocycloalkyl are unsubstituted (C 3 - C2o)heterocycloalkyl, unsubstituted (C 3 -Cio)heterocycloalkyl, oxetan-2-yl,
- halogen atom means fluorine atom (F), chlorine atom (CI), bromine atom (Br), or iodine atom (I) radical.
- F fluorine atom
- CI chlorine atom
- Br bromine atom
- I iodine atom
- halide means fluoride (F “ ), chloride (CI “ ), bromide (Br “ ), or iodide ( ⁇ ) anion.
- O-O, S-S, or O-S bonds there are no O-O, S-S, or O-S bonds, other than O-S bonds in an S(O) or S(0)2 diradical functional group, in the metal -ligand complex of Formula (I).
- saturated means lacking carbon-carbon double bonds, carbon- carbon triple bonds, and (in heteroatom-containing groups) carbon-nitrogen, carbon- phosphorous, and carbon-silicon double bonds and carbon-nitrogen triple bonds. Where a saturated chemical group is substituted by one or more substituents R s , one or more double and/or triple bonds optionally may or may not be present in substituents R s .
- M is titanium, zirconium, or hafnium. In one embodiment, M is titanium. In another embodiment, M is zirconium. In another embodiment, M is hafnium. In some embodiments, M is in a formal oxidation state of +2, +3, or +4.
- Each Zl independently is a monodentate or polydentate ligand that is neutral, monoanionic, or dianionic. Zl and nn are chosen in such a way that the metal-ligand complex of Formula (I) is, overall, neutral. In some embodiments each Zl independently is the monodentate ligand. In one embodiment when there are two or more Zl monodentate ligands, each Zl is the same. In some embodiments the monodentate ligand is the monoanionic ligand. The monoanionic ligand has a net formal oxidation state of -1.
- monoanionic ligand may independently be hydride, (Ci-C4o)hydrocarbyl carbanion, (Ci-C4o)heterohydrocarbyl carbanion, halide, nitrate, carbonate, phosphate, borate, borohydride, sulfate, HC(0)0 “ , alkoxide or aryloxide (RO " ),
- At least one monodentate ligand of Zl independently is the neutral ligand.
- the neutral ligand is a neutral Lewis base group that is R X1 R K R L , R K OR L , R K SR L , or R X1 PR K R L , wherein each R X1
- R K and R L independently is as defined above.
- each Zl is a monodentate ligand that independently is a halogen atom, unsubstituted (Ci-C2o)hydrocarbyl, unsubstituted
- each monodentate ligand Zl is a chlorine atom, (Ci-Cio)hydrocarbyl (e.g., (Ci-C6)alkyl or benzyl), unsubstituted (Ci-Cio)hydrocarbylC(0)0-, or R K R L N- wherein each of R K and R L independently is an unsubstituted (Ci-Cio)hydrocarbyl.
- the bidentate ligand is a neutral bidentate ligand.
- the bidentate ligand is a monoanionic-mono(Lewis base) ligand.
- the bidentate ligand is a dianionic ligand.
- the dianionic ligand has a net formal oxidation state of -2.
- each dianionic ligand independently is carbonate, oxalate (i.e., " 0 2 CC(0)0 " ), (C 2 -C 4Q )hydrocarbylene dicarbanion, (C 1 -C 4Q )heterohydrocarbylene dicarbanion, phosphate, or sulfate.
- number and charge (neutral, monoanionic, dianionic) of Zl are selected depending on the formal oxidation state of M such that the metal-ligand complex of Formula (I) is, overall, neutral.
- each Zl is the same, wherein each Zl is methyl; isobutyl; neopentyl; neophyl; trimethylsilylmethyl; phenyl; benzyl; or chloro.
- nn is 2 and each Zl is the same.
- each Zl is a different one of methyl; isobutyl; neopentyl; neophyl;
- the metal-ligand complex of Formula (I) is a mononuclear metal complex.
- olefin polymerization catalyst systems of the present invention demonstrate reversible chain transfer indicative of chain shuttling behavior in the presence of appropriate chain shuttling agents. Such combination of attributes is particularly of interest in the preparation of olefin block copolymers. In general, the ability to tune alpha-olefin incorporation and thus short- chain branching distribution is critical to accessing materials with performance differentiation.
- the metal -ligand complex of Formula (I) is a metal - ligand complex of Formula II):
- J 1 -! 10 are each independently selected from the group consisting of R s substituents (as defined above) and hydrogen.
- J 1 , J 5 , J 6 and J 10 of Formula (II) are each independently selected from the group consisting of halogen atoms, (Ci-C 8 ) alkyl groups, and (Ci-C 8 ) alkoxyl groups.
- An activator is an additive which renders a procatalyst active with respect to olefin polymerization by contacting it to, or combining it with, the procatalyst.
- activators abstract a monoanionic ligand, typically an alkyl group, in some cases a benzyl or methyl group, to form a cationic metal-ligand complex of the procatalyst, which has a weakly coordinating or noncoordinating anion derived or present as a portion of the activating agent.
- activators of this type include: Bransted acids, such as [R 3 NH] + (ammonium) based activators, e.g.
- alumoxane alone is used as the activator, preferably the number of moles of the alumoxane that are employed is at least 100 times the number of moles of the metal-ligand complex. Lower loading of alumoxanes do not act as activators, rather they serve as scavenging agent.
- a scavenging agent sequesters impurities in the reactor prior to addition of the catalyst, and as such, does not constitute an activator.
- Suitable activating co-catalysts for use herein include alkyl aluminums; polymeric or oligomeric alumoxanes (also known as aluminoxanes); neutral Lewis acids; and non-polymeric, non-coordinating, ion-forming compounds (including the use of such compounds under oxidizing conditions).
- a suitable activating technique is bulk electrolysis. Combinations of one or more of the foregoing activating co-catalysts and techniques are also contemplated.
- alkyl aluminum means a monoalkyl aluminum dihydride or monoalkylaluminum dihalide, a dialkyl aluminum hydride or dialkyl aluminum halide, or a trialkylaluminum.
- Aluminoxanes and their preparations are known at, for example, United States Patent Number (USPN) US 6, 103,657.
- Exemplary Lewis acid activating co-catalysts are Group 13 metal compounds containing from 1 to 3 hydrocarbyl substituents as described herein. In some embodiments, exemplary Group 13 metal compounds are tri(hydrocarbyl)- substituted-aluminum or tri(hydrocarbyl)-boron compounds. In some other
- exemplary Group 13 metal compounds are tri((Ci-Cio)alkyl)aluminum or tri((C6-Ci8)aryl)boron compounds and halogenated (including perhalogenated) derivatives thereof.
- exemplary Group 13 metal compounds are tris(fluoro-substituted phenyl)boranes, in other embodiments, tris(pentafluorophenyl)borane.
- the activating co-catalyst is a tris((Ci-C2o)hydrocarbyl)methane borate (e.g., trityl tetrakis(pentafluorophenyl)borate) or a tri((Ci-C2o)hydrocarbyl)ammonium tetra((Ci-C2o)hydrocarbyl)borate (e.g., bis(octadecyl)methyl ammonium tetrakis(pentafluorophenyl)borate).
- ammonium means a nitrogen cation that is a ((Ci-C2o)hydrocarbyl)4N + , a
- each (Ci-C2o)hydrocarbyl may be the same or different.
- Exemplary combinations of neutral Lewis acid activating co-catalysts include mixtures comprising a combination of a tri((Ci-C4)alkyl)aluminum and a halogenated tri((C6-Ci8)aryl)boron compound, especially a
- tris(pentafluorophenyl)borane tris(pentafluorophenyl)borane.
- Other exemplary embodiments are combinations of such neutral Lewis acid mixtures with a polymeric or oligomeric alumoxane, and
- ratios of numbers of moles of (metal-ligand complex):(tris(pentafluoro-phenylborane): (alumoxane) are from 1 : 1 : 1 to 1 : 10:30, other exemplary embodiments are from 1 : 1 : 1.5 to 1 :5: 10.
- Examples of suitable salts of a cationic oxidizing agent and a non-coordinating, compatible anion as activating co-catalysts for addition polymerization catalysts are disclosed in US 5,321,106.
- Examples of suitable carbenium salts as activating co-catalysts for addition polymerization catalysts are disclosed in US 5,350,723.
- Examples of suitable silylium salts as activating co-catalysts for addition polymerization catalysts are disclosed in US 5,625,087.
- Examples of suitable complexes of alcohols, mercaptans, silanols, and oximes with tris(pentafluorophenyl)borane are disclosed in US 5,296,433.
- the procatalysts of the present disclosure may be activated to form an active catalyst composition by combination with one or more cocatalyst such as a cation forming cocatalyst, a strong Lewis acid, or a combination thereof.
- cocatalysts for use include polymeric or oligomeric aluminoxanes, especially methyl aluminoxane, as well as inert, compatible, noncoordinating, ion forming compounds.
- Exemplary suitable cocatalysts include, but are not limited to modified methyl aluminoxane (MMAO); bis(hydrogenated tallow
- alkyl methylammonium tetrakis(pentafluorophenyl)borate; triethyl aluminum (TEA); and any combinations thereof.
- one or more of the foregoing activating co-catalysts are used in combination with each other.
- An especially preferred combination is a mixture of a tri((C 1 -C 4 )hydrocarbyl)aluminum, tri((C 1 -C 4 )hydrocarbyl)borane, or an ammonium borate with an oligomeric or polymeric alumoxane compound.
- the co-catalyst is [(C16-18H33-37)- 2CH3 H] tetrakis(pentafluorophenyl)borate salt.
- the ratio of total number of moles of one or more catalysts to total number of moles of one or more of the activating co-catalysts is from 1 : 10,000 to 100: 1. In some embodiments, the ratio is at least 1 :5000, in some other embodiments, at least 1 : 1000; and 10: 1 or less, and in some other embodiments, 1 : 1 or less.
- the number of moles of the alumoxane that are employed is at least 100 times the number of moles of the catalysts.
- tris(pentafluorophenyl)borane alone is used as the activating co- catalyst, in some other embodiments, the number of moles of the
- tris(pentafluorophenyl)borane that are employed to the total number of moles of one or more catalysts form 1 :0.5 to 1 : 10, in some other embodiments, from 1 : 1 to 1 :6, in some other embodiments, from 1 : 1 to 1 :5.
- the remaining activating co-catalysts are generally employed in approximately mole quantities equal to the total mole quantities of one or more catalysts.
- Any conventional polymerization processes may be employed to produce the block copolymers of the present disclosure.
- Such conventional polymerization processes include, but are not limited to, solution polymerization processes, gas phase polymerization processes, slurry, or particle forming polymerization processes, and combinations thereof using one or more conventional reactors, e.g., loop reactors, isothermal reactors, fluidized bed reactors, stirred tank reactors, batch reactors in parallel, series, and/or any combinations thereof.
- multi-block copolymers are prepared via a solution polymerization process employing a first olefin polymerization procatalyst (A), a second olefin polymerization procatalyst (B), one or more cocatalysts, and a chain shuttling agent (C).
- A first olefin polymerization procatalyst
- B second olefin polymerization procatalyst
- C chain shuttling agent
- the polymerization processes of the disclosure employing a first olefin polymerization procatalyst A, a second olefin polymerization procatalyst B, one or more cocatalysts, and chain shuttling agent C may be further elucidated by reference to Figure 1, where there are illustrated activated catalyst site A, 10, which under polymerization conditions forms a polymer chain, 13, attached to the active catalyst site, 12. Similarly, active catalyst site B, 20, produces a differentiated polymer chain, 23, attached to the active catalyst site, 22. A chain shuttling agent C I, attached to a polymer chain produced by active catalyst B, 14, exchanges its polymer chain, 23, for the polymer chain, 13, attached to catalyst site A.
- Additional chain growth under polymerization conditions causes formation of a multi -block copolymer, 18, attached to active catalyst site A.
- chain shuttling agent C2 attached to a polymer chain produced by active catalyst site A, 24, exchanges its polymer chain, 13, for the polymer chain, 23, attached to catalyst site B.
- Additional chain growth under polymerization conditions causes formation of a multi-block copolymer, 28, attached to active catalyst site B.
- the growing multi -block copolymers are repeatedly exchanged between active catalyst A and active catalyst B by means of shuttling agent C resulting in formation of a block or segment of differing properties whenever exchange to the opposite active catalyst site occurs.
- the growing polymer chains may be recovered while attached to a chain shuttling agent and functionalized if desired. Alternatively, the resulting polymer may be recovered by scission from the active catalyst site or the shuttling agent, through use of a proton source or other killing agent.
- composition of the respective segments or blocks, and especially of the end segments of the polymer chains may be influenced through selection of process conditions or other process variables.
- the nature of the end segments is determined by the relative rates of chain transfer or termination for the respective catalysts as well as by the relative rates of chain shuttling.
- Possible chain termination mechanisms include, but are not limited to, ⁇ -hydrogen elimination, ⁇ -hydrogen transfer to monomer, ⁇ -methyl elimination, and chain transfer to hydrogen or other chain- terminating reagent such as an organosilane or chain functionalizing agent.
- the ratio of the shuttling rate to the propagation rate for the three catalysts follows the order 1>2>3, then the average block length for the three block types will follow the order 3>2>1, and there will be fewer instances of 2-type blocks adjacent to 3 -type blocks than 1-type blocks adjacent to 2-type blocks.
- a method exists for controlling the block length distribution of the various block types. For example, by selecting catalysts 1, 2, and 3 (wherein 2 and 3 produce substantially the same polymer block type), and a chain shuttling agent, and the shuttling rate follows the order 1 > 2 > 3, the resulting polymer will have a bimodal distribution of block lengths made from the 2 and 3 catalysts.
- the reaction mixture comprising one or more monomers is contacted with the activated catalyst composition according to any suitable polymerization conditions.
- the process is characterized by use of elevated temperatures and pressures. Hydrogen may be employed as a chain transfer agent for molecular weight control according to known techniques if desired.
- the monomers and solvents employed be of sufficiently high purity that catalyst deactivation does not occur. Any suitable technique for monomer purification such as devolatilization at reduced pressure, contacting with molecular sieves or high surface area alumina, or a combination of the foregoing processes may be employed.
- Suitable supports include solid, particulated, high surface area, metal oxides, metalloid oxides, or mixtures thereof (interchangeably referred to herein as an inorganic oxide). Examples include: talc, silica, alumina, magnesia, titania, zirconia, Sn 2 0 3 , aluminosilicates, borosilicates, clays, and mixtures thereof.
- Suitable supports preferably have a surface area as determined by nitrogen porosimetry using the B.E.T. method from 10 to 1000 m /g, and preferably from 100 to 600 m 2 /g.
- the average particle size typically is from 0.1 to 500 ⁇ , preferably from 1 to 200 ⁇ , more preferably 10 to 100 ⁇ .
- the present catalyst composition and optional support may be spray dried or otherwise recovered in solid, particulated form to provide a composition that is readily transported and handled.
- Suitable methods for spray diying a liquid containing slurry are well known in the art and usefully employed herein. Preferred techniques for spray diying catalyst compositions for use herein are described in US-A's-5 ,648,310 and 5,672,669.
- the polymerization is desirably carried out as a continuous polymerization, preferably a continuous, solution polymerization, in which catalyst components, shuttling agent(s), monomers, and optionally solvent, adjuvants, scavengers, and polymerization aids are continuously supplied to the reaction zone and polymer product continuously removed there from.
- a continuous polymerization preferably a continuous, solution polymerization, in which catalyst components, shuttling agent(s), monomers, and optionally solvent, adjuvants, scavengers, and polymerization aids are continuously supplied to the reaction zone and polymer product continuously removed there from.
- the catalyst compositions can be advantageously employed in a high pressure, solution, slurry, or gas phase polymerization process.
- a solution polymerization process it is desirable to employ homogeneous dispersions of the catalyst components in a liquid diluent in which the polymer is soluble under the polymerization conditions employed.
- One such process utilizing an extremely fine silica or similar dispersing agent to produce such a homogeneous catalyst dispersion where either the metal complex or the cocatalyst is only poorly soluble is disclosed in US-A-5,783,512.
- a solution process to prepare the novel polymers of the present invention is preferably carried out at a temperature between 80°C and 250°C, more preferably between 100°C and 210°C, and most preferably between 110°C and 210°C.
- a high pressure process is usually carried out at temperatures from 100°C to 400°C and at pressures above 500 bar (50 MPa).
- a slurry process typically uses an inert hydrocarbon diluent and temperatures of from 0°C up to a temperature just below the temperature at which the resulting polymer becomes substantially soluble in the inert polymerization medium. Preferred temperatures in a slurry polymerization are from 30°C, preferably from 60°C up to 115°C, preferably up to 100°C.
- Pressures typically range from atmospheric (100 kPa) to 500 psi (3.4 MPa).
- continuous or substantially continuous polymerization conditions are preferably employed.
- the use of such polymerization conditions, especially continuous, solution polymerization processes employing two or more active polymerization catalyst species, allows the use of elevated reactor temperatures which results in the economical production of multi-block or segmented copolymers in high yields and efficiencies.
- Both homogeneous and plug-flow type reaction conditions may be employed. The latter conditions are preferred where tapering of the block composition is desired.
- Both catalyst compositions (A) and (B) may be prepared as a homogeneous composition by addition of the requisite metal complexes to a solvent in which the polymerization will be conducted or in a diluent compatible with the ultimate reaction mixture.
- the desired cocatalyst or activator and the shuttling agent may be combined with the catalyst composition either prior to, simultaneously with, or after combination with the monomers to be polymerized and any additional reaction diluent.
- the individual ingredients as well as any active catalyst composition must be protected from oxygen and moisture. Therefore, the catalyst components, shuttling agent and activated catalysts must be prepared and stored in an oxygen and moisture free atmosphere, preferably a dry, inert gas such as nitrogen.
- one means for carrying out such a polymerization process is as follows.
- the monomers to be polymerized are introduced continuously together with any solvent or diluent.
- the reactor contains a liquid phase composed substantially of monomers together with any solvent or diluent and dissolved polymer.
- Preferred solvents include C4-10 hydrocarbons or mixtures thereof, especially alkanes such as hexane or mixtures of alkanes, as well as one or more of the monomers employed in the polymerization.
- Procatalysts along with cocatalyst and chain shuttling agent are continuously or intermittently introduced in the reactor liquid phase or any recycled portion thereof.
- the reactor temperature and pressure may be controlled by adjusting the
- the polymerization rate is controlled by the rate of catalyst addition.
- the ethylene content of the polymer product is determined by the ratio of ethylene to comonomer in the reactor, which is controlled by manipulating the respective feed rates of these components to the reactor.
- the polymer product molecular weight is controlled, optionally, by controlling other polymerization variables such as the temperature, monomer concentration, or by the previously mentioned chain transfer agent, as is well known in the art.
- a catalyst kill agent such as water, steam or an alcohol.
- the polymer solution is optionally heated, and the polymer product is recovered by flashing off gaseous monomers as well as residual solvent or diluent at reduced pressure, and, if necessary, conducting further devolatilization in equipment such as a devolatilizing extruder.
- the mean residence time of the catalyst and polymer in the reactor generally is from 5 minutes to 8 hours, and preferably from 10 minutes to 6 hours.
- the foregoing polymerization may be carried out in a continuous loop reactor with or without a monomer, catalyst or shuttling agent gradient established between differing regions thereof, optionally accompanied by separated addition of catalysts and/or chain transfer agent, and operating under adiabatic or non- adiabatic solution polymerization conditions or combinations of the foregoing reactor conditions.
- suitable loop reactors and a variety of suitable operating conditions for use therewith are found in USP's 5,977,251, 6,319,989 and 6,683, 149.
- the catalyst composition may also be prepared and employed as a heterogeneous catalyst by adsorbing the requisite components on an inert inorganic or organic particulated solid, as previously disclosed.
- a heterogeneous catalyst is prepared by co-precipitating the metal complex and the reaction product of an inert inorganic compound and an active hydrogen containing activator, especially the reaction product of a tri (CI .4 alkyl) aluminum compound and an ammonium salt of a
- hydroxyaryltris(pentafluorophenyl)borate such as an ammonium salt of (4-hydroxy- 3,5-ditertiarybutylphenyl)tris(pentafluorophenyl)borate.
- the catalyst composition may be employed in a slurry or a gas phase polymerization.
- slurry polymerization takes place in liquid diluents in which the polymer product is substantially insoluble.
- the diluent for slurry polymerization is one or more hydrocarbons with less than 5 carbon atoms.
- saturated hydrocarbons such as ethane, propane or butane may be used in whole or part as the diluent.
- the ⁇ -olefin comonomer or a mixture of different a- olefin monomers may be used in whole or part as the diluent.
- Most preferably at least a major part of the diluent comprises the a-olefin monomer or monomers to be polymerized.
- the support material and resulting catalyst has a median particle diameter from 20 to 200 ⁇ , more preferably from 30 ⁇ to 150 ⁇ , and most preferably from 50 ⁇ to 100 ⁇ .
- the support has a median particle diameter from 1 ⁇ to 200 ⁇ , more preferably from 5 ⁇ to 100 ⁇ , and most preferably from 10 ⁇ to 80 ⁇ .
- Suitable gas phase polymerization process for use herein are substantially similar to known processes used commercially on a large scale for the manufacture of polypropylene, ethylene/ a- olefin copolymers, and other olefin polymers.
- the gas phase process employed can be, for example, of the type which employs a
- the gas employed to fluidize the bed comprises the monomer or monomers to be polymerized, and also serves as a heat exchange medium to remove the heat of reaction from the bed.
- the hot gases emerge from the top of the reactor, normally via a tranquilization zone, also known as a velocity reduction zone, having a wider diameter than the fluidized bed and wherein fine particles entrained in the gas stream have an opportunity to gravitate back into the bed. It can also be advantageous to use a cyclone to remove ultra-fine particles from the hot gas stream.
- the gas is then normally recycled to the bed by means of a blower or compressor and one or more heat exchangers to strip the gas of the heat of polymerization.
- a preferred method of cooling of the bed is to feed a volatile liquid to the bed to provide an evaporative cooling effect, often referred to as operation in the condensing mode.
- the volatile liquid employed in this case can be, for example, a volatile inert liquid, for example, a saturated hydrocarbon having 3 to 8, preferably 4 to 6, carbon atoms.
- the monomer or comonomer itself is a volatile liquid, or can be condensed to provide such a liquid, this can suitably be fed to the bed to provide an evaporative cooling effect.
- the volatile liquid evaporates in the hot fluidized bed to form gas which mixes with the fluidizing gas.
- the volatile liquid is a monomer or comonomer, it will undergo some polymerization in the bed.
- the evaporated liquid then emerges from the reactor as part of the hot recycle gas, and enters the compression/heat exchange part of the recycle loop.
- the recycle gas is cooled in the heat exchanger and, if the temperature to which the gas is cooled is below the dew point, liquid will precipitate from the gas.
- This liquid is desirably recycled continuously to the fluidized bed. It is possible to recycle the precipitated liquid to the bed as liquid droplets carried in the recycle gas stream. This type of process is described, for example in EP-89691; U.S. 4,543,399; WO-94/ 25495 and U.S. 5,352,749.
- a particularly preferred method of recycling the liquid to the bed is to separate the liquid from the recycle gas stream and to reinject this liquid directly into the bed, preferably using a method which generates fine droplets of the liquid within the bed.
- This type of process is described in WO-94/28032.
- the polymerization reaction occurring in the gas fluidized bed is catalyzed by the continuous or semi -continuous addition of catalyst composition according to the invention.
- the catalyst composition may be subjected to a prepolymerization step, for example, by polymerizing a small quantity of olefin monomer in a liquid inert diluent, to provide a catalyst composite comprising supported catalyst particles embedded in olefin polymer particles as well.
- the polymer is produced directly in the fluidized bed by polymerization of the monomer or mixture of monomers on the fluidized particles of catalyst composition, supported catalyst composition or prepolymerized catalyst composition within the bed. Start-up of the polymerization reaction is achieved using a bed of preformed polymer particles, which are preferably similar to the desired polymer, and conditioning the bed by drying with inert gas or nitrogen prior to introducing the catalyst composition, the monomers and any other gases which it is desired to have in the recycle gas stream, such as a diluent gas, hydrogen chain transfer agent, or an inert condensable gas when operating in gas phase condensing mode.
- the produced polymer is discharged continuously or semi-continuously from the fluidized bed as desired.
- the gas phase processes most suitable for the practice of this invention are continuous processes which provide for the continuous supply of reactants to the reaction zone of the reactor and the removal of products from the reaction zone of the reactor, thereby providing a steady-state environment on the macro scale in the reaction zone of the reactor. Products are readily recovered by exposure to reduced pressure and optionally elevated temperatures (devolatilization) according to known techniques.
- the fluidized bed of the gas phase process is operated at temperatures greater than 50°C, preferably from 60°C to 110°C, more preferably from 70°C to 110°C.
- metallated polymers wherein the metal is the remnant of the catalyst or chain shuttling agent employed, as well as further derivatives thereof, for example, the reaction product of a metallated polymer with an oxygen source and then with water to form a hydroxyl terminated polymer.
- sufficient air or other quench agent is added to cleave some or all of the shuttling agent-polymer bonds thereby converting at least a portion of the polymer to a hydroxyl terminated polymer.
- Additional examples include olefin terminated polymers formed by ⁇ -hydride elimination and ethylenic unsaturation in the resulting polymer.
- the multi-block copolymer may be functionalized by maleation (reaction with maleic anhydride or its equivalent), metallation (such as with an alkyl lithium reagent, optionally in the presence of a Lewis base, especially an amine, such as tetramethylethylenediamine), or by incorporation of a diene or masked olefin in a copolymerization process.
- the masking group for example a trihydrocarbylsilane, may be removed thereby exposing a more readily functionalized remnant.
- metallated polymer species can be utilized in well known chemical reactions such as those suitable for other alkyl- aluminum, alkyl-gallium, alkyl-zinc, or alkyl-Group 1 compounds to form amine-, hydroxy-, epoxy-, ketone, ester, nitrile, and other functionalized terminated polymer products. Examples of suitable reaction techniques that are adaptable for use here in are described in Negishi, "Organometallics in Organic Synthesis", Vol. 1 and 2, (1980), and other standard texts in organometallic and organic synthesis.
- multi-block copolymers i.e., olefin block copolymers or OBCs
- OBCs olefin block copolymers
- (A) Mw/Mn from 1.0 to 10.0 e.g., from 1.0 to 9.0, from 1.0 to 8.0, from 1.0 to 7.0, from 1.0 to 6.0, from 1.0 to 5.0, from 1.5 to 5.0, from 1.5 to 4.0, from 1.7 to 3.5, etc.
- Tm melting point
- d density
- the CRYSTAF peak is determined using at least 5 percent of the cumulative polymer, and if less than 5 percent of the polymer has an identifiable CRYSTAF peak, then the CRYSTAF temperature is 30° C; and/or
- the olefin block copolymer may have one, some, all, or any combination of properties (A)-(G).
- Block Index can be determined as described in detail in U.S. Pat. No. 7,608,668 herein incorporated by reference for that purpose. Analytical methods for determining properties (A) through (G) are disclosed in, for example, U.S. Pat. No 7,608,668, Col. 31, line 26 through Col. 35, line 44, which is herein incorporated by reference for that purpose.
- the olefin block copolymers prepared by the compositions/catalyst systems/processes of the present disclosure have a density of from 0.820 g/cc to 0.925 g/cc (e.g., from 0.860 g/cc to 0.890 g/cc).
- the olefin block copolymers prepared by the compositions/catalyst systems/processes of the present disclosure have a melt index (MI) from 0.1 g/10 min to 1000 g/10 min (e.g., from 0.1 g/10 min to 500 g/10 min, from 0.1 g/10 min to 100 g/10 min, from 0.1 g/10 min to 50 g/10 min, from 0.1 g/10 min to 35 g/10 min, from 0.1 g/10 min to 30 g/10, from 0.1 g/10 min to 20 g/10 min, and/or from 0.1 g/10 min to 15 g/10 min), as measured by ASTM D 1238 (190° C/2.16 kg).
- MI melt index
- the olefin block copolymers prepared by the compositions/catalyst systems/processes of the present disclosure have a molecular weight of 10,000 to 250,000 g/mole (e.g., from 10,000 to 200,000 g/mole and/or from 20,000 to 175,000 g/mole).
- the olefin block copolymers prepared by the compositions/catalyst systems/processes of the present disclosure have a residual zinc content from 50 ppm to 1000 ppm (e.g., from 50 ppm to 750 ppm, from 50 ppm to 500 ppm, and/or from 75 ppm to 400 ppm).
- the olefin block copolymers of the present disclosure have a molecular weight distribuction (MWD or PDI) of less than 5.0 (e.g., less than 4.0, less than 3.5, less than 3.0, less than 2.9, less than 2.8, etc.). In certain embodiments, the olefin block copolymers of the present disclosure have a thermo-mechanical resistance (TMA) of greater than 100 °C. Examples
- Combined Catalyst Efficiency The combined catalyst efficiency is calculated by dividing the mass (e.g., the number of kilograms (kg po iymer)) of the olefin block copolymer prepared by the mass (e.g., the total number of milligrams or grams (gmetai)) of metal from both procatalysts.
- 3D-GPC A high temperature Triple Detector Gel Permeation
- the concentration detector is an Infra-red concentration detector (IR4 from Polymer Char, Valencia, Spain), which was used to determine the molecular weight and molecular weight distribution.
- the other two detectors are a Precision Detectors (Agilent) 2-angle laser light scattering detector, Model 2040, and a 4-capillary differential viscometer detector, Model 150R, from Malvern (Houston, TX). The 15° angle of the light scattering detector was used for calculation purposes.
- B has a value of 1.0, and the experimentally determined value of A is around 0.39.
- a fifth order polynomial was used to fit the respective polyethylene- equivalent calibration points obtained from equation (1) to their observed elution volumes.
- the actual polynomial fit was obtained so as to relate the logarithm of polyethylene equivalent molecular weights to the observed elution volumes (and associated powers) for each polystyrene standard.
- Wf is the weight fraction of the z ' -th component and M, is the molecular weight of the z ' -th component.
- the MWD was expressed as the ratio of the weight average molecular weight (Mw) to the number average molecular weight (Mn).
- KLS LS-MW calibration constant.
- the response factor, KLS, of the laser detector was determined using the certificated value for the weight average molecular weight of NIST 1475 (52,000 g/mol).
- DSC Differential Scanning Calorimetry
- Density measurements are conducted according to ASTM D792.
- Melt index: h and I 10 are measured in accordance with ASTM D-1238 (190°C; 2.16 kg and 10 kg).
- IR-4 or IR-5 detector is used.
- ODCB Eight hundred milligrams of BHT and five grams of the silica gel are added to two liters of ODCB.
- ODCB can be also dried by passing through a column or columns packed with silica gel.
- Silica gel 40 is packed into two 300 x 7.5 mm GPC size stainless steel columns and the Silica gel 40 columns are installed at the inlet of the pump of the CEF instrument to dry ODCB; and no BHT is added to the mobile phase.
- This "ODCB containing BHT and silica gel" or ODCB dried with silica gel 40 is now referred to as "ODCB.”
- This ODBC is sparged with dried nitrogen (N2) for one hour before use.
- Dried nitrogen is such that is obtained by passing nitrogen at ⁇ 90 psig over CaC0 3 and 5A molecular sieves.
- the resulting nitrogen should have a dew point of approximately -73°C.
- Sample preparation is done with autosampler at 4 mg/ml (unless otherwise specified) under shaking at 160°C for 2 hours.
- the injection volume is 300 ⁇ 1.
- the temperature profile of CEF is:
- the flow rate during crystallization is 0.052 ml/min.
- the flow rate during cooling step is 0.052mL/min.
- the flow rate during elution is 0.50 ml/min.
- the data is collected at one data point/second.
- the CEF column is packed with glass beads at 125 ⁇ 6% (MO-SCI Specialty Products) with 1/8 inch stainless tubing according to U. S. Patent No. 8,372,931.
- the column outside diameter (OD) is 1/8 inch.
- the critical parameters needed to duplicate the method include the column internal diameter (ID), and column length (L).
- ID column internal diameter
- L column length
- ID and L must be such that when packed with the 125 ⁇ diameter glass beads, the liquid internal volume is 2.1 to 2.3 mL. If L is 152 cm, then ID must be 0.206 cm and the wall thickness must be 0.056cm. Different values for L and ID can be used, as long as the glass bead diameter is 125 ⁇ and the internal liquid volume is between 2.1 and 2.3 mL.
- Column temperature calibration is performed by using a mixture of NIST Standard Reference Material Linear polyethylene 1475a (l .Omg/ml) and Eicosane (2mg/ml) in ODCB.
- CEF temperature calibration consists of four steps: (1) Calculating the delay volume defined as the temperature offset between the measured peak elution temperature of Eicosane minus 30.00° C; (2) Subtracting the temperature offset of the elution temperature from CEF raw temperature data.
- this temperature offset is a function of experimental conditions, such as elution temperature, elution flow rate, etc.; (3) Creating a linear calibration line transforming the elution temperature across a range of 30.00° C and 140.00° C so that NIST linear polyethylene 1475a has a peak temperature at 101.0°C, and Eicosane has a peak temperature of 30.0° C; (4) For the soluble fraction measured isothermally at 30° C, the elution temperature is extrapolated linearly by using the elution heating rate of 3° C/min. The reported elution peak temperatures are obtained such that the observed comonomer content calibration curve agrees with those previously reported in U.S. Patent No. 8,372,931.
- Residual zinc content may be measured by standard industry procedure, such as mass balance or an x-ray fluorescence (XRF) method.
- TMA The temperature resistance of the OBC resins was evaluated using Therm omechanical Analysis. TMA was conducted on a TA Instruments Q400. A small puck about 8 mm in diameter was cut from a 3.2 mm thick compression molded plaque. The probe was 1.1 mm in diameter with a rounded tip. Samples were tested under ambient conditions with a temperature ramp rate of 5 °C/min. The temperature at which the probe indented 1 mm into the surface under a constant force of 1 N was recorded.
- Reactivity Ratios Reactivity ratios of the olefin polymerization procatalysts may be determined by the discussion and mathematical formulas above.
- procatalysts falling within the scope of the first olefin polymerization procatalyst (A) of the present disclosure (Procatalysts (A4), shown below:
- Exemplary, non-limiting procatalysts falling within the scope of the second olefin polymerization procatalyst (B) of the present disclosure have the following structures:
- Procatalysts of the comparative CSA and dual catalyst combination representative of the state of the art have the following structures:
- Step 1 Synthesis of 2-mesityl-6-bromopyridine. Inside the glove box, a 200 mL jar is charged with 2,6-dibromopyridine (8.00 g, 33.77 mmol), 2,6- diisopropylphenylimidazolium chloride (0.287 g, 0.68 mmol), nickel
- Step 2 Synthesis of 2-mesityl-6-neopentylaminopyridine.
- the crude product from the previous reaction is added to a 250 mL round bottom flask.
- Neopentylamine (4.42 g, 50.66 mmol), NaOtBu (7.14 g, 74.30 mmol), Pd 2 dba 3 (0.309 g, 0.34 mmol), and BINAP (0.420 g, 0.68 mmol) are all added to the flask, followed by anhydrous toluene (120 mL).
- the reaction is heated overnight at 100 °C under a nitrogen purge.
- the reaction is cooled to room temperature, then all volatiles are removed.
- Procatalyst (B 10) is prepared following the General Procedure for metallation of 2-aminopyridine ligands using HfCl 4 . Yield: 86%.
- 3 ⁇ 4 MR (400 MHz, C 6 D 6 ) ⁇ 6.96 - 6.86 (m, 2H), 6.70 (s, 4H), 5.88 (d, J 8.7 Hz, 2H), 5.85 - 5.81 (m, 2H), 2.41 (s, 4H), 2.18 (s, 6H), 2.10 - 1.94 (m, 6H), 1.71 - 1.51 (m, 6H), 0.95 (s, 18H), 0.47 (s, 6H).
- Proatalyst (B 14) is then prepared following the General Procedure for metallation of 2-aminopyridine ligands described above using HfCU. Yield: 87%. 3 ⁇ 4
- Procatalyst (Al) is synthesized in accordance with the procedures described in U.S. Patent No. 6,953,764 B2, which is incorporated herein by reference in its entirety.
- a 5-L three-neck round bottom flask is fitted with an overhead stirrer in a drybox under N 2 atmosphere and charged with ligand (393 g, 1.19 mol), zirconium tetrachloride (139 g, 0.597 mmol) and dry toluene (3.5 L).
- ligand 393 g, 1.19 mol
- zirconium tetrachloride 393 g, 0.597 mmol
- dry toluene 3.5 L.
- 3.0 M methylmagnesium bromide in ether 800 mL, 2400 mmol
- Temperature and methane evolution are monitored carefully during addition of the first two equivalents of the Grignard reagent. After stirring for an additional hour, the ether and toluene are removed under vacuum.
- the solids are slurried with toluene (4 L) and the magnesium salts are removed on a fritted funnel.
- the salts are washed with an additional 4 L toluene and the filtrates are combined in a B-5 cylinder.
- the cylinder is sampled for gravimetric analysis and batch reactor evaluation. Typical concentrations are near 5.4 wt. %, equivalent to -370 g complex (80% yield).
- the I 2 (25.0 g, 98.3 mmol, 1.35 equiv.) was dissolved in 58mL THF.
- the reaction flask was cooled to -78°C and the I 2 solution was added dropwise via syringe.
- the mixture was stirred overnight, gradually warming to room temperature in the process. Quenched with lOOmL sat. aq. Na2S 2 03 and lOOmL H 2 0. Stirred 30min. Diluted with lOOmL Et 2 0, separated organic layer. Extracted aqueous phase with additional lOOmL Et 2 0. Dried combined organic fractions over MgS0 4 , filtered and concentrated.
- the crude material was filtered through a short silica plug, eluting with hexane.
- the crude purity was excellent and the recovery was quantitative.
- the silane-derived impurity present from the preceding step was still present.
- the impurity could be removed by reverse phase chromatography, eluting with 0— >30% THF/MeCN gradient.
- THF/MeCN Collected product in two fractions.
- the later eluting fraction contained significant amounts of an unidentified impurity and was discarded.
- the remaining fraction was resubjected to column chromatography under identical conditions.
- Step 1 Preparation of 2-methyldecene: A 2 L 3 -neck flask equipped with a mechanical stirrer, thermocouple, and a nitrogen pad, was charged with
- methyltriphenylphosphonium bromide (297.18 g, 831.9 mmol, 1.3 equiv.) and 850 mL tetrahydrofuran (THF, anhydrous, 1 mL/mmol, via cannula).
- THF tetrahydrofuran
- the white suspension was cooled to 0 °C in an ice bath and NaOtBu (79.95 g, 831.9 mmol, 1.3 equiv.) was added (slight exotherm observed, ⁇ 2 °C increase upon addition).
- the resulting reaction mixture (yellow slurry) was stirred at 0 °C for 45 minutes.
- Step 2 Preparation of 4-(2-methyldecan-2-vQphenol: A 2 L 4-neck flask equipped with a magnetic stirrer, thermocouple, water condenser, and nitrogen pad was charged with phenol (133.5 g, 1418 mmol, 2 equiv.), 2-methyldecene (109.4 g, 709 mmol, 1 equiv.), and 709 mL 1,2-dichloroethane (DCE). The reaction mixture was sparged with nitrogen for 30 minutes.
- phenol 133.5 g, 1418 mmol, 2 equiv.
- 2-methyldecene 109.4 g, 709 mmol, 1 equiv.
- DCE 1,2-dichloroethane
- the organic layer was washed with 2 N NaOH (300 mL x 2), water (300 mL), brine (300 mL), and passed through a plug of silica gel topped with Na 2 S04.
- the plug was rinsed with hexanes and the filtrate was concentrated via rotary evaporation to give the crude product as a light brown oil.
- the crude product was dissolved in hexanes and concentrated (repeated 3 times) to remove residual EtOAc.
- the crude product was obtained as a light brown oil (175 g, 95.8% purity by GC analysis, 95% isolated yield).
- Step 3 Preparation of 2-iodo-4-(2-methyldecan-2-vnphenol: A 3 L 4-neck flask equipped with a mechanical stirrer, a thermocouple, and nitrogen pad was charged with 4-(2-methyldecan-2-yl)phenol (176 g, 678 mmol, 1 equiv.), p- toluenesulfonic acid monohydrate (pTSA H 2 0, 12.9 g, 67.4 mmol, 0.1 equiv.), and 1000 mL acetonitrile. The resulting reaction mixture (pink solution) was stirred at ambient temperature for 30 minutes.
- NIS N-iodosuccinimide
- the reaction mixture was stirred for 1 hour at 5 °C, then the ice bath was removed, the flask was covered in foil, and the reaction mixture (orange solution) was stirred at ambient temperature.
- a sample of the reaction mixture was quenched with Na 2 S03 (20% aq) and analyzed by GC. The GC trace showed full consumption of the starting material.
- the reaction mixture was quenched with Na 2 S0 3 (20% aq., -150 mL), diluted with water (1500 mL), and stirred at ambient temperature for 1 h. A brown oil settled to the bottom of the flask after stirring was ceased. The solution was decanted away from the oil and additional water (1500 mL) was added. The resulting mixture was stirred at ambient temperature for 1 h and the solution was decanted away from the oil (repeated one more time).
- the crude product was dissolved in 1 L of hexanes, washed with water (500 mL x 2), brine (500 mL), and passed through a plug of silica gel topped with Na 2 SC"4. The filtrate was concentrated via rotary evaporation and dried in a vacuum oven at 40 °C (20 Torr) to give the crude product as a brown oil (254 g, -96% purity by
- Step 4 Preparation of 2-iodo-l-(methoxymethoxy)-4-(2-methyldecan-2- yPbenzene: A I L 3 -neck flask equipped with a mechanical stirrer, reflux condenser, and nitrogen pad was charged with dimethoxymethane (59 mL, 664.0 mmol, 1.3 equiv. relative to ArOH), anhydrous toluene (250 mL), and Zn(OAc) 2 (20 mg, 0.02 mol%). Acetyl chloride (47 mL, 664.0 mmol, 1.3 equiv.) was added via addition funnel over 45 minutes.
- reaction mixture was stirred at ambient temperature for 1 hour then heated to 60 °C overnight (15 hours). After the reaction was deemed complete by GC analysis, the reaction mixture was allowed to cool to ambient temperature then cooled in an ice-water bath. Saturated NH4CI solution (500 mL) was added at such a rate to maintain the temperature below 30 °C (an exothermic reaction was observed, used an ice water bath to control the temperature). The biphasic reaction mixture was stirred for 2 hours then diluted with toluene (500 mL) and water (500 mL). The two phases were separated and the aqueous layer was washed with toluene (500 mL x 2).
- Step 5 A 2 L 3 -neck flask equipped with a mechanical stirrer, reflux condenser, and nitrogen pad was charged with 2-iodo-l-(methoxymethoxy)-4-(2- methyldecan-2-yl)benzene (323.0 g, 710.3 mmol, 1 equiv.) and anhydrous toluene (890 mL). The solution was sparged with nitrogen for 30 minutes.
- Step 6 A 3 -neck 1 L round bottom flask equipped with a magnetic stirrer, thermowell with therm oprobe, and nitrogen purge was charged with DOC-61128 top group (81.0 g, 177 mmol, 1 equiv.) and anhydrous THF (366 g, via cannula, 18.1 wt% solution) under a nitrogen atmosphere. The mixture was cooled to -20 to -15°C in a dry-ice acetonitrile bath.
- the reaction mixture was re-cooled to between -20 and - 15 °C, and 2-isopropoxy-4,4,5,5,-tetramethyl-l,3,2-dioxaborolane (iPrOBPin, 49 mL, 239 mmol, 1.35 equiv.) was added via syringe pump at 1 mL/min. After the addition was complete, the reaction mixture was stirred for 40 minutes and allowed to warm to 0 °C. A sample was taken, quenched into saturated H4CI, extracted with hexanes, diluted with THF, and analyzed by HPLC. The HPLC trace showed 95.6 % BPin + boronic acid and 1.5% protodeborylated byproduct.
- iPrOBPin 2-isopropoxy-4,4,5,5,-tetramethyl-l,3,2-dioxaborolane
- reaction mixture had warmed to 4 °C.
- the light brown slurry reaction mixture was re-cooled in the dry-ice bath to 0 - 5 °C, diluted with hexanes (200 mL), and ice water (115 mL) was slowly added keeping the temperature below 5 °C.
- the mixture was transferred to a 1 L separately funnel and the bottom aqueous layer was removed.
- the aqueous layer was extracted with hexanes (60 mL x 2).
- the combined organics were washed with saturated NH4CI (115 mL), water (115 mL), and saturated NaCl (60 mL), and vacuum filtered through a pad of anhydrous MgS04.
- Step 7 Preparation of 2-iodo-4-fluorophenol: This step is performed as seen in the synthesis of procatalyst (A4).
- Step 8 This step is perfomed as seen in the synthesis of procatalyst (A4).
- Step 9 A I L 4-neck flask equipped with an overhead stirrer, thermocouple, water condenser, and a nitrogen pad was charged with DOC-61128 iodo-bottom group
- BPin (20.7 g, 35.8 wt% soln, 0.2 equiv., degassed) and Pd(PPh 3 ) 4 (0.7 g, 1 mol%) were added and the reaction mixture was stirred at 50 °C overnight. After stirring overnight (15 h), a sample of the reaction mixture (brown solution) was taken and analyzed by HPLC. The HPLC trace showed full consumption of the monocoupled intermediate.
- the reaction mixture was cooled to ambient temperature and the layers were separated. The aqueous layer was extracted with THF (160 mL x 2). The combined organic layers were passed through a filter then washed with brine (500 mL total, 5 portions) and placed into a 2 L 4-neck flask.
- Deprotection step A 2 L 4-neck flask equipped with a magnetic stirrer, water condenser, thermocouple, and nitrogen pad was charged with the crude protected product in THF. Methanol (MeOH, 370 mL) and concentrated HC1 (31 mL, 362 mmol, 6 equiv.) were added and the reaction mixture (clear-red solution) was stirred at 60 °C. After stirring for 5 h, a sample of the reaction mixture was taken and analyzed by 19 F NMR spectroscopy. The 19 F NMR spectrum showed that the deprotection was almost complete.
- Step 10 In a glove box, a glass reactor was charged with HfCU (39.428 g, 123.10 mmol) and 5.4 L toluene (anhydrous). The mixture was cooled to -20°C and then MeMgBr (172 mL, 517.01 mmol) was added with stirring. After stirring for 8 minutes, the DOC-61128 ligand (135.59 g, 123.10 mmol) was added, and the resulting mixture was allowed to warm to room temperature over 20 minutes. Once at room temperature, the reaction mixture was stirred for 4.5 hours. When the reaction completion was confirmed by NMR, the crude mixture was filtered, and the solids were rinsed with additional toluene (0.4 L).
- the filtrate was dried under vacuum to obtain a brown solid.
- the solids were taken up in anhydrous methyl cycl oh exane (4.5 L), stirred for 2 hours and then filtered through a pad of Celite. The solids were rinsed with additional methyl cycl ohexane (0.5 L) and the filtrate was dried under vacuum to obtain 145.666 g, 90.5 % yield of the desired metal complex.
- Step 2 Preparation of N-isobutyl-6-mesitylpyridin-2-amine: A 500 mL round bottom flask was charged with 2-bromo-6-mesitylpyridine (20.0 g, 72.4 mmol), NaOtBu (20.9 g, 217.3 mmol), Pd 2 dba 3 (0.33 g, 0.36 mmol), rac-BINAP (0.45 g, 0.72 mmol), isobutylamine (10.8 mL, 109 mmol), and toluene (200 mL, anhydrous). The reaction mixture was heated to 100 °C for 15 h, then all volatiles were removed. The crude product was washed with water and extracted with EtOAc.
- Step 3 Inside a glovebox a 200 mL jar was charged with HfCl 4 (8.11 g, 25.3 mmol) and CH 2 C1 2 (100 mL). The suspension was cooled to -25 °C then MeMgBr (3 M in ether, 38.0 mL, 114.0 mmol) was added. The solution was allowed to stir for 2 min then a cold CH2CI2 (50 mL) solution of 2-mesityl-N-neopentylpyridin-2-amine (13.6 g, 50.7 mmol) was added. The solution quickly changed to a yellow color and was allowed to stir at room temperature for 3 h.
- the exemplary, non-limiting procatalysts described above are used to polymerize olefin block copolymers in continuous mixed polymerizations.
- the continuous mixed polymerizations are carried out in either a 5 L or a 3.8 L
- CSTR continuously stirred-tank reactor
- 114 L loop reactor a 114 L loop reactor in accordance with the following, non-limiting procedures and with the process conditions of Tables 1-3.
- the properties of the olefin block copolymers polymerized with the exemplary, non- limiting procatalysts of the present disclosure are presented in Table 4.
- Catl refers to a first olefin polymerization procatalyst (A)
- Cat2 refers to a second olefin polymerization procatalyst (B)
- CoCat 1 refers to [HNMe(C18H37)2][B(C6F5)4], an exemplary cocatalyst
- CoCat2 refers to TEA
- CSA refers to DEZ, an exemplary chain shuttling agent.
- Raw materials ethylene, 1-octene, butene
- process solvents SBP100/140, commercially available from Shell, or a narrow boiling range high-purity isoparaffinic solvent trademarked as Isopar-E available from Exxon Mobil Corp.
- Isopar-E isoparaffinic solvent trademarked as Exxon Mobil Corp.
- Triethylaluminum commercially available from Akzo Nobel, is used as an impurity scavenger.
- Diethylzinc (DEZ) commercially available from Akzo Nobel, is used as a chain shuttling agent.
- the individual catalyst components are manually batch diluted to specified component concentrations with purified solvent and are added to the reactor under a positive nitrogen pressure.
- the cocatalyst is
- the combined solvent, monomer, comonomer and hydrogen are fed to the reactor at a controlled temperatures between 5° C and 30° C.
- the reactor runs liquid full with the reactor feed (ethylene, 1-octene hydrogen and process solvent) entering the reactor from the bottom and exiting at the top.
- the reactor is heated with hot oil and normal process pressure is 28 bar.
- the catalyst is fed to the reactor to reach a specified conversion of ethylene.
- the cocatalyst is fed separately based on a calculated specified molar ratio (1.2 molar equivalents) to the catalyst component.
- the TEA and DEZ share the same line as the cocatalyst.
- TEA flow is based on either an Al concentration in the reactor or a specified molar ratio to the catalyst component.
- DEZ flow is based on a Zn concentration in the polymer.
- polymerization reactor (containing solvent, monomer, comonomer, hydrogen, catalyst components, and molten polymer) exits the reactor and water is injected into the stream to terminate polymerization.
- various stabilizers or additives such as antioxidants are added at this point.
- the stream then goes through a static mixer to evenly disperse the catalyst kill and additives.
- the molten polymer is preheated before it enters the first devolatilizer, where majority of the solvent and non-converted feeds are removed via a flash.
- the solvent rich polymer is transported through a second preheater in a vacuum devolatilizer where the remaining solvent is removed.
- the final polymer is pumped through a static mixer to a pelletizer.
Abstract
Description













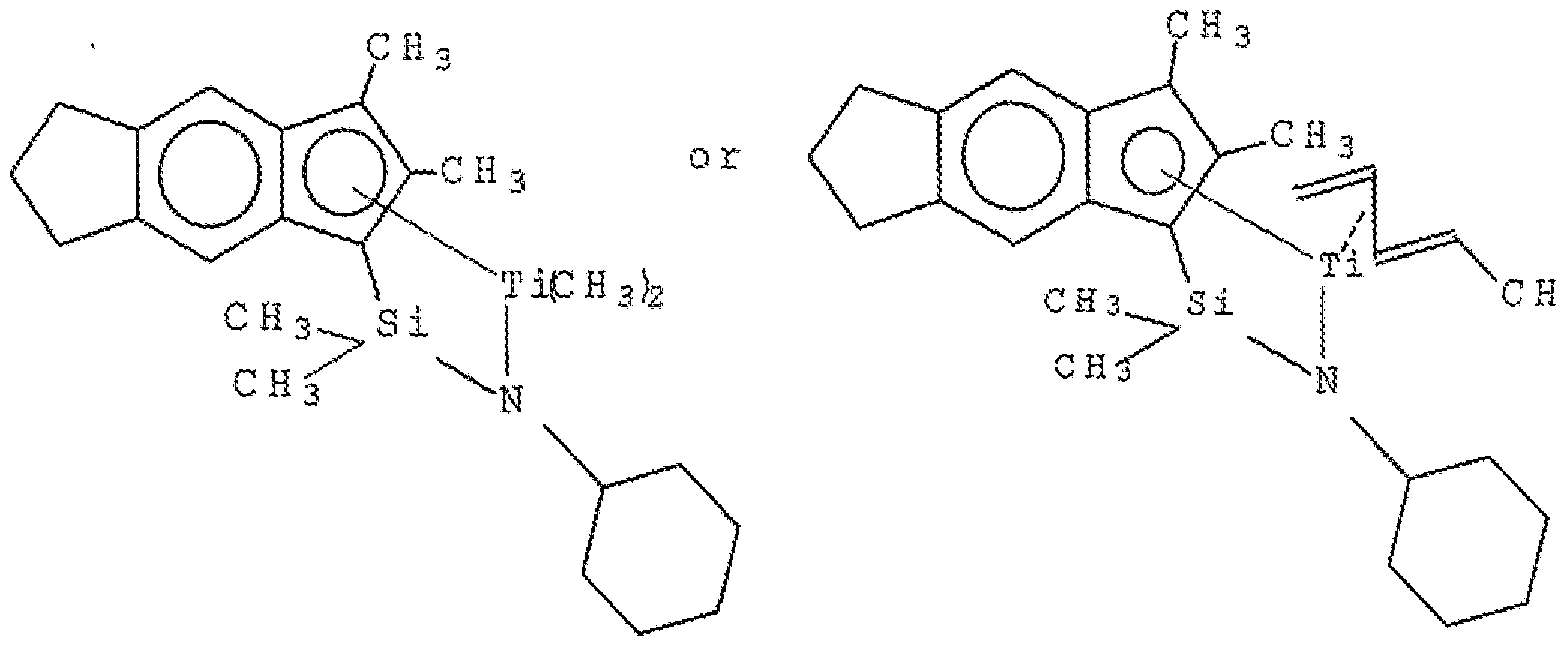

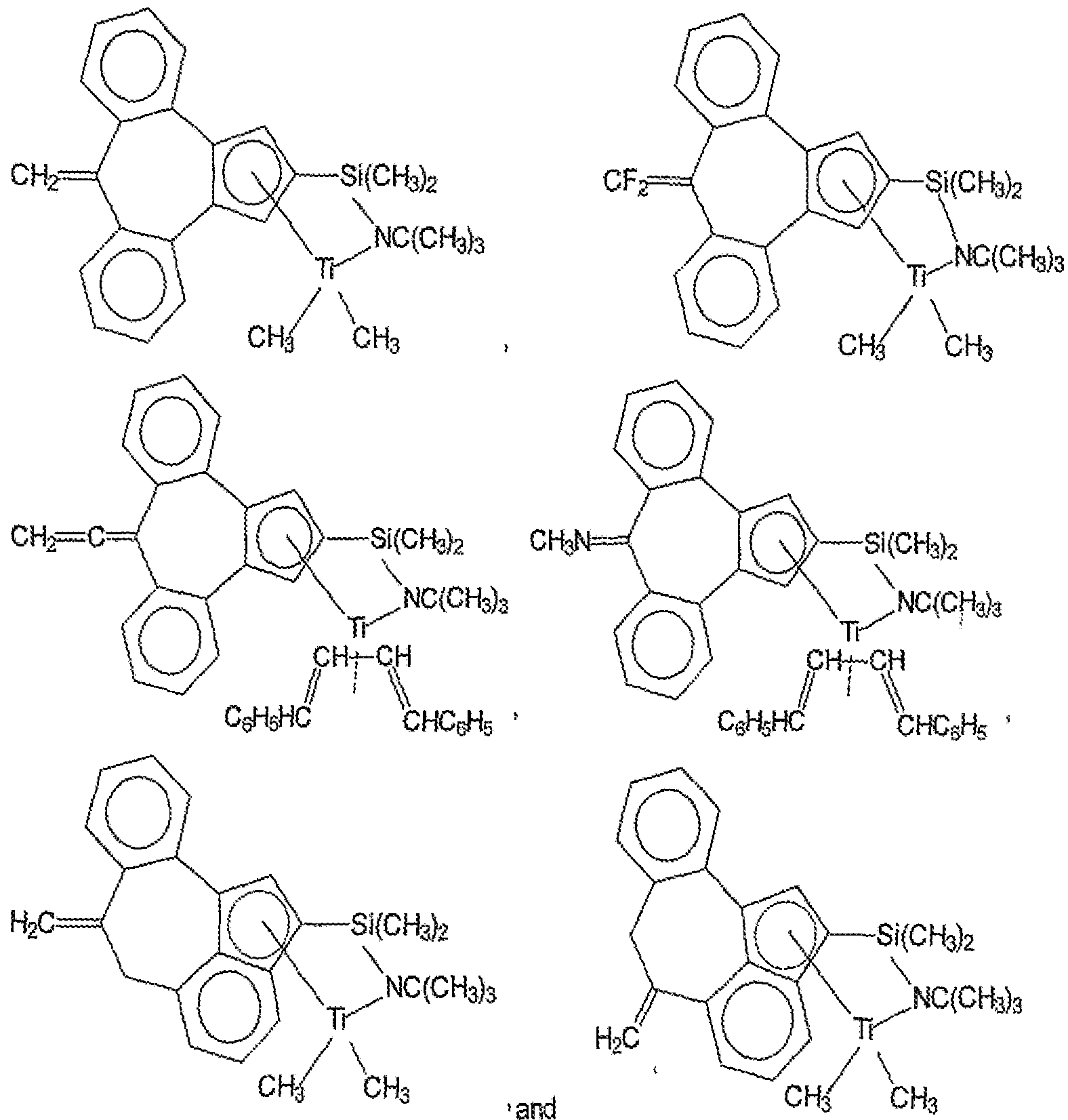












































































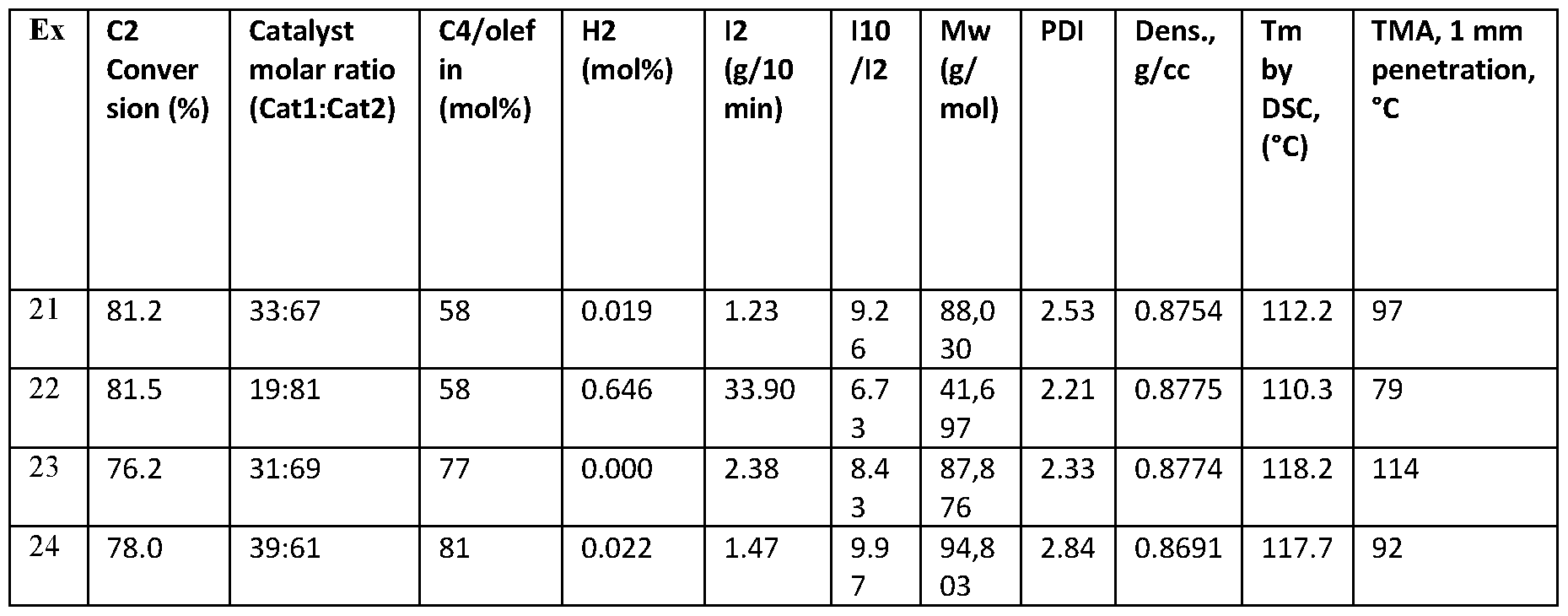






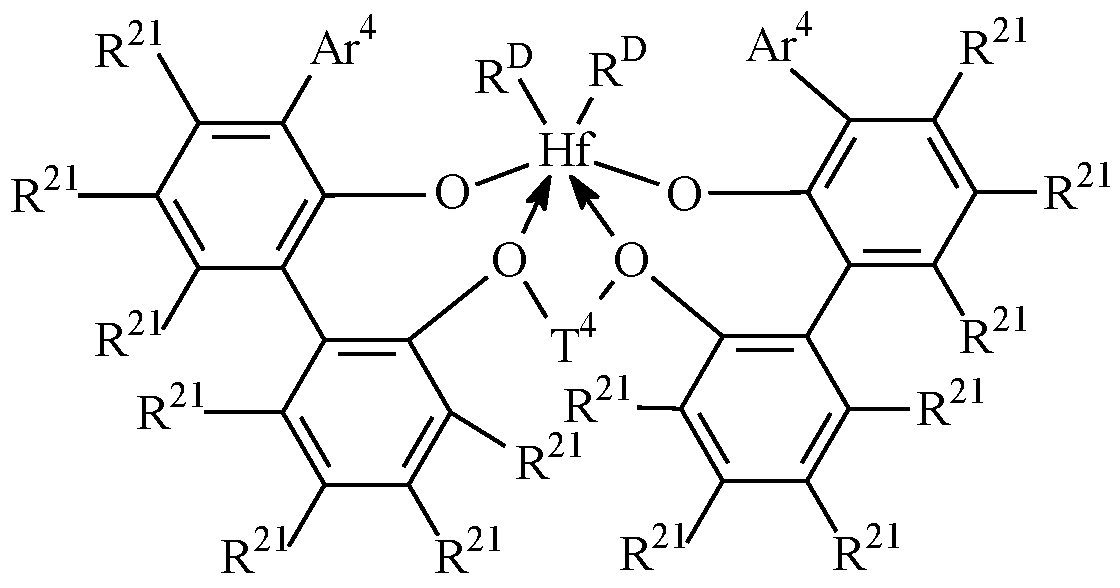


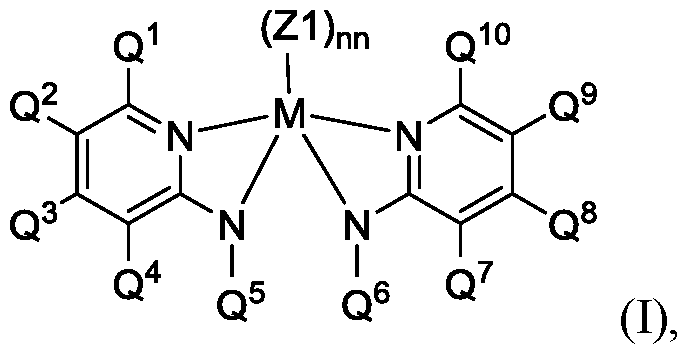








Claims







Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201880027869.1A CN110582518B (en) | 2017-03-15 | 2018-03-14 | Catalyst system for forming multi-block copolymers |
| BR112019019130-5A BR112019019130B1 (en) | 2017-03-15 | 2018-03-14 | OLEFIN POLYMERIZATION CATALYST SYSTEM AND PROCESS FOR PREPARING A MULTI-BLOCK COPOLYMER |
| CN202210873641.2A CN115260366A (en) | 2017-03-15 | 2018-03-14 | Catalyst system for forming multi-block copolymers |
| KR1020197030261A KR102653652B1 (en) | 2017-03-15 | 2018-03-14 | Catalyst system for forming multi-block copolymers |
| US16/494,085 US11459409B2 (en) | 2017-03-15 | 2018-03-14 | Catalyst system for multi-block copolymer formation |
| SG11201908414Y SG11201908414YA (en) | 2017-03-15 | 2018-03-14 | Catalyst system for multi-block copolymer formation |
| EP18715859.7A EP3596141B1 (en) | 2017-03-15 | 2018-03-14 | Catalyst system for multi-block copolymer formation |
| JP2019550232A JP7287895B2 (en) | 2017-03-15 | 2018-03-14 | Catalyst system for forming multi-block copolymers |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201762471534P | 2017-03-15 | 2017-03-15 | |
| US62/471,534 | 2017-03-15 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2018170138A1 true WO2018170138A1 (en) | 2018-09-20 |
Family
ID=61899371
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2018/022454 WO2018170138A1 (en) | 2017-03-15 | 2018-03-14 | Catalyst system for multi-block copolymer formation |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US11459409B2 (en) |
| EP (1) | EP3596141B1 (en) |
| JP (1) | JP7287895B2 (en) |
| KR (1) | KR102653652B1 (en) |
| CN (2) | CN110582518B (en) |
| BR (1) | BR112019019130B1 (en) |
| SG (1) | SG11201908414YA (en) |
| TW (1) | TW201840604A (en) |
| WO (1) | WO2018170138A1 (en) |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019182983A1 (en) | 2018-03-19 | 2019-09-26 | Dow Global Technologies Llc | Silicon-terminated organo-metal compounds and processes for preparing the same |
| WO2019182993A1 (en) | 2018-03-19 | 2019-09-26 | Dow Global Technologies Llc | Silicon-terminated telechelic polyolefin compositions and processes for preparing the same |
| WO2019182988A1 (en) | 2018-03-19 | 2019-09-26 | Dow Global Technologies Llc | Silicon-terminated organo-metal compounds and processes for preparing the same |
| WO2019182986A1 (en) | 2018-03-19 | 2019-09-26 | Dow Global Technologies Llc | Process for functionalization of organo-metal compounds with silyl-based functionalization agents and silyl-functionalized compounds prepared thereby |
| WO2020140067A1 (en) | 2018-12-28 | 2020-07-02 | Dow Global Technologies Llc | Curable compositions comprising unsaturated polyolefins |
| WO2020140064A1 (en) | 2018-12-28 | 2020-07-02 | Dow Global Technologies Llc | Organometallic chain transfer agents |
| WO2020140061A1 (en) | 2018-12-28 | 2020-07-02 | Dow Global Technologies Llc | Curable compositions comprising telechelic polyolefins |
| WO2020140058A1 (en) | 2018-12-28 | 2020-07-02 | Dow Global Technologies Llc | Telechelic polyolefins and processes for preparing the same |
| WO2021048030A1 (en) | 2019-09-10 | 2021-03-18 | Sabic Global Technologies B.V. | Compounds for use in catalyst compositions for the production of polyolefins |
| WO2021133925A1 (en) * | 2019-12-27 | 2021-07-01 | Dow Global Technologies Llc | Ethylene/butene multi-block copolymer and process for producing same |
| WO2021133945A1 (en) * | 2019-12-27 | 2021-07-01 | Dow Global Technologies Llc | Ethylene/octene multi-block copolymer and process for producing same |
| WO2021155168A1 (en) | 2020-01-31 | 2021-08-05 | Dow Global Technologies Llc | Polymerization processes that include group iii and lanthanide bis-phenyl-phenoxy metal-ligand complexes and chain transfer agents |
| WO2021155158A1 (en) | 2020-01-31 | 2021-08-05 | Dow Global Technologies Llc | Group iii and lanthanide bis-phenyl-phenoxy metal-ligand complexes |
| EP4234594A1 (en) | 2018-12-28 | 2023-08-30 | Dow Global Technologies LLC | Curable compositions comprising unsaturated polyolefins |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR102100142B1 (en) * | 2017-12-21 | 2020-04-14 | 사빅 에스케이 넥슬렌 컴퍼니 피티이 엘티디 | Metal-ligand complexes, catalyst composition for ethylene-based polymerization containing the same, and production methods of ethylene-based polymers using the same |
| CA3180371A1 (en) * | 2020-05-29 | 2021-12-02 | Dow Global Technologies Llc | Attenuated post-metallocene catalysts |
| CN115515991A (en) * | 2020-05-29 | 2022-12-23 | 陶氏环球技术有限责任公司 | Attenuated post metallocene catalyst |
| CA3180283A1 (en) * | 2020-05-29 | 2021-12-02 | Rhett A. BAILLIE | Chemically converted catalysts |
| CN116023537A (en) * | 2021-10-26 | 2023-04-28 | 中国石油化工股份有限公司 | Catalyst composition for olefin polymerization and preparation method and application thereof |
| CN116162185A (en) * | 2023-01-04 | 2023-05-26 | 万华化学集团股份有限公司 | Metallocene catalyst, preparation method and application thereof in olefin polymerization |
Citations (75)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0089691A2 (en) | 1982-03-24 | 1983-09-28 | Union Carbide Corporation | Continuous process for the production of polymer in a fluidized bed reactor |
| US4543399A (en) | 1982-03-24 | 1985-09-24 | Union Carbide Corporation | Fluidized bed reaction systems |
| US4588790A (en) | 1982-03-24 | 1986-05-13 | Union Carbide Corporation | Method for fluidized bed polymerization |
| US5028670A (en) | 1988-07-15 | 1991-07-02 | Bp Chemicals Limited | Process for the gas-phase polymerization of olefins in a fluidized-bed reactor |
| US5032562A (en) | 1989-12-27 | 1991-07-16 | Mobil Oil Corporation | Catalyst composition and process for polymerizing polymers having multimodal molecular weight distribution |
| US5064802A (en) | 1989-09-14 | 1991-11-12 | The Dow Chemical Company | Metal complex compounds |
| US5106804A (en) | 1989-12-22 | 1992-04-21 | Bp Chemicals Limited | Catalyst and prepolymer used for the preparation of polyolefins |
| US5153157A (en) | 1987-01-30 | 1992-10-06 | Exxon Chemical Patents Inc. | Catalyst system of enhanced productivity |
| US5272236A (en) | 1991-10-15 | 1993-12-21 | The Dow Chemical Company | Elastic substantially linear olefin polymers |
| US5278272A (en) | 1991-10-15 | 1994-01-11 | The Dow Chemical Company | Elastic substantialy linear olefin polymers |
| US5296433A (en) | 1992-04-14 | 1994-03-22 | Minnesota Mining And Manufacturing Company | Tris(pentafluorophenyl)borane complexes and catalysts derived therefrom |
| US5321106A (en) | 1990-07-03 | 1994-06-14 | The Dow Chemical Company | Addition polymerization catalyst with oxidative activation |
| US5350723A (en) | 1992-05-15 | 1994-09-27 | The Dow Chemical Company | Process for preparation of monocyclopentadienyl metal complex compounds and method of use |
| US5352749A (en) | 1992-03-19 | 1994-10-04 | Exxon Chemical Patents, Inc. | Process for polymerizing monomers in fluidized beds |
| WO1994025495A1 (en) | 1993-05-20 | 1994-11-10 | Exxon Chemical Patents Inc. | Process for polymerizing monomers in fluidized beds |
| WO1994028032A1 (en) | 1993-05-20 | 1994-12-08 | Bp Chemicals Limited | Polymerisation process |
| US5425872A (en) | 1993-06-24 | 1995-06-20 | The Dow Chemical Company | Electrochemical preparation of addition polymerization catalysts |
| US5436304A (en) | 1992-03-19 | 1995-07-25 | Exxon Chemical Patents Inc. | Process for polymerizing monomers in fluidized beds |
| US5453471A (en) | 1994-08-02 | 1995-09-26 | Union Carbide Chemicals & Plastics Technology Corporation | Gas phase polymerization process |
| US5461123A (en) | 1994-07-14 | 1995-10-24 | Union Carbide Chemicals & Plastics Technology Corporation | Gas phase fluidized bed polyolefin polymerization process using sound waves |
| US5462999A (en) | 1993-04-26 | 1995-10-31 | Exxon Chemical Patents Inc. | Process for polymerizing monomers in fluidized beds |
| US5470993A (en) | 1993-06-24 | 1995-11-28 | The Dow Chemical Company | Titanium(II) or zirconium(II) complexes and addition polymerization catalysts therefrom |
| US5473028A (en) | 1992-12-28 | 1995-12-05 | Mobil Oil Corporation | Process and a catalyst for preventing reactor fouling |
| US5543458A (en) | 1995-01-31 | 1996-08-06 | Shell Oil Company | Process for making graft block copolymers by growing anionic polymer chains from functionalized polyolefin backbones |
| US5556238A (en) | 1993-05-19 | 1996-09-17 | Bp Chemicals Limited | Process and apparatus for introducing a solid into a reactor |
| US5608019A (en) | 1992-12-28 | 1997-03-04 | Mobil Oil Corporation | Temperature control of MW in olefin polymerization using supported metallocene catalyst |
| US5616661A (en) | 1995-03-31 | 1997-04-01 | Union Carbide Chemicals & Plastics Technology Corporation | Process for controlling particle growth during production of sticky polymers |
| US5625087A (en) | 1994-09-12 | 1997-04-29 | The Dow Chemical Company | Silylium cationic polymerization activators for metallocene complexes |
| US5665800A (en) | 1991-10-15 | 1997-09-09 | The Dow Chemical Company | Elastic substantially linear olefin polymers |
| WO1997045434A1 (en) * | 1996-05-31 | 1997-12-04 | Borealis A/S | Novel transition metal complexes and method for preparing them |
| US5721185A (en) | 1991-06-24 | 1998-02-24 | The Dow Chemical Company | Homogeneous olefin polymerization catalyst by abstraction with lewis acids |
| US5783512A (en) | 1996-12-18 | 1998-07-21 | The Dow Chemical Company | Catalyst component dispersion comprising an ionic compound and solid addition polymerization catalysts containing the same |
| WO1998050392A1 (en) | 1997-05-08 | 1998-11-12 | Nova Chemicals (International) S.A. | Process to prepare bridged phosphole-cyclopentadienyl compounds |
| US5866704A (en) | 1996-12-19 | 1999-02-02 | The Dow Chemical Company | 3-aryl substituted indenyl containing metal complexes and polymerization process |
| US5883204A (en) | 1996-03-27 | 1999-03-16 | The Dow Chemical Company | Solution polymerization process with dispersed catalyst activator |
| US5919983A (en) | 1996-03-27 | 1999-07-06 | The Dow Chemical Company | Highly soluble olefin polymerization catalyst activator |
| US5977251A (en) | 1996-04-01 | 1999-11-02 | The Dow Chemical Company | Non-adiabatic olefin solution polymerization |
| US6015868A (en) | 1996-10-03 | 2000-01-18 | The Dow Chemical Company | Substituted indenyl containing metal complexes and olefin polymerization process |
| US6034022A (en) | 1996-12-19 | 2000-03-07 | The Dow Chemical Company | Fused ring substituted indenyl metal complexes and polymerization |
| US6103657A (en) | 1997-07-02 | 2000-08-15 | Union Carbide Chemicals & Plastics Technology Corporation | Catalyst for the production of olefin polymers |
| US6150297A (en) | 1997-09-15 | 2000-11-21 | The Dow Chemical Company | Cyclopentaphenanthrenyl metal complexes and polymerization process |
| US6268444B1 (en) | 1996-08-08 | 2001-07-31 | Dow Chemical Company | 3-heteroatom substituted cyclopentadienyl-containing metal complexes and olefin polymerization process |
| US6319989B1 (en) | 1997-07-21 | 2001-11-20 | The Dow Chemical Company | Broad MWD, compositionally uniform ethylene interpolymer compositions, process for making the same and article made therefrom |
| WO2002002577A1 (en) | 2000-06-30 | 2002-01-10 | Dow Global Technology Inc. | Polycyclic, fused ring compounds, metal complexes and polymerization process |
| WO2002038628A2 (en) | 2000-11-07 | 2002-05-16 | Symyx Technologies, Inc. | Substituted pyridyl amine ligands, complexes and catalysts therefrom; processes for producing polyolefins therewith |
| WO2002092610A1 (en) | 2001-05-14 | 2002-11-21 | Dow Global Technologies Inc. | 3-aryl-substituted cyclopentadienyl metal complexes and polymerization process |
| US20030004286A1 (en) | 1999-12-10 | 2003-01-02 | Jerzy Klosin | Alkaryl-substituted Group 4 metal complexes, catalysts and olefin polymerization process |
| US6515155B1 (en) | 1999-12-10 | 2003-02-04 | Dow Global Technologies Inc. | Substituted group 4 metal complexes, catalysts and olefin polymerization process |
| US6555634B1 (en) | 1999-05-13 | 2003-04-29 | The Dow Chemical Company | Di- and tri-heteroatom substituted indenyl metal complexes |
| WO2003040195A1 (en) | 2001-11-06 | 2003-05-15 | Dow Global Technologies Inc. | Supported catalysts for manufacture of polymers |
| WO2003078483A1 (en) | 2002-03-14 | 2003-09-25 | Dow Global Technologies Inc. | Substituted indenyl metal complexes and polymerization process |
| WO2003078480A2 (en) | 2002-03-14 | 2003-09-25 | Dow Global Technologies Inc. | Polycyclic, fused heteroring compounds, metal complexes and polymerization process |
| US20040010103A1 (en) | 2002-04-24 | 2004-01-15 | Symyx Technologies, Inc. | Bridged bi-aromatic ligands, catalysts, processes for polymerizing and polymers therefrom |
| US6683149B2 (en) | 1997-09-19 | 2004-01-27 | Dow Global Technologies Inc. | Narrow MWD, compositionally optimized ethylene interpolymer composition, process for making the same and article made therefrom |
| US6696379B1 (en) | 1997-09-19 | 2004-02-24 | The Dow Chemical Company | Supported modified alumoxane catalyst activator |
| WO2004024740A1 (en) | 2002-09-12 | 2004-03-25 | Dow Global Technologies Inc. | Preparation of metal complexes |
| US20040220050A1 (en) | 2003-05-02 | 2004-11-04 | Frazier Kevin A. | High activity olefin polymerization catalyst and process |
| US6827976B2 (en) | 1998-04-29 | 2004-12-07 | Unaxis Trading Ag | Method to increase wear resistance of a tool or other machine component |
| WO2005090426A1 (en) | 2004-03-17 | 2005-09-29 | Dow Global Technologies Inc. | Catalyst composition comprising shuttling agent for higher olefin multi-block copolymer formation |
| WO2005090427A2 (en) | 2004-03-17 | 2005-09-29 | Dow Global Technologies Inc. | Catalyst composition comprising shuttling agent for ethylene multi-block copolymer formation |
| US20060199930A1 (en) | 2004-03-17 | 2006-09-07 | Dow Global Technologies Inc. | Ethylene/alpha-olefins block interpolymers |
| WO2006101966A1 (en) | 2005-03-17 | 2006-09-28 | Dow Global Technologies, Inc. | ETHYLENE/α-OLEFINS BLOCK INTERPOLYMERS |
| US7163907B1 (en) | 1987-01-30 | 2007-01-16 | Exxonmobil Chemical Patents Inc. | Aluminum-free monocyclopentadienyl metallocene catalysts for olefin polymerization |
| WO2007035485A1 (en) | 2005-09-15 | 2007-03-29 | Dow Global Technologies Inc. | Catalytic olefin block copolymers with controlled block sequence distribution |
| US7355089B2 (en) | 2004-03-17 | 2008-04-08 | Dow Global Technologies Inc. | Compositions of ethylene/α-olefin multi-block interpolymer for elastic films and laminates |
| WO2009012215A1 (en) | 2007-07-13 | 2009-01-22 | Dow Global Technologies Inc. | Ethylene/a-olefin interpolymers containing low crystallinity hard blocks |
| WO2010022228A2 (en) | 2008-08-21 | 2010-02-25 | Dow Global Technologies, Inc. | Metal-ligand complexes and catalysts |
| US8058373B2 (en) | 2006-05-17 | 2011-11-15 | Dow Global Technologies Llc | Polypropylene solution polymerization process |
| EP2436703A1 (en) * | 2010-09-30 | 2012-04-04 | Dow Global Technologies LLC | Comb architecture olefin block copolymers |
| US8372931B2 (en) | 2009-07-01 | 2013-02-12 | Dow Global Technologies Llc | Ethylene-based polymer compositions |
| US8501885B2 (en) | 2009-07-29 | 2013-08-06 | Dow Global Technologies, Llc | Dual- or multi-headed chain shuttling agents and their use for preparation of block copolymers |
| WO2014105411A1 (en) | 2012-12-27 | 2014-07-03 | Dow Global Technologies Llc | Catalyst systems for olefin polymerization |
| US8785554B2 (en) | 2010-06-21 | 2014-07-22 | Dow Global Technologies Llc | Crystalline block composites as compatibilizers |
| WO2017173080A1 (en) * | 2016-03-31 | 2017-10-05 | Dow Global Technologies Llc | Olefin polymerization catalyst systems and methods of use thereof |
| WO2017173074A1 (en) * | 2016-03-31 | 2017-10-05 | Dow Global Technologies Llc | An olefin polymerization catalyst |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SG151301A1 (en) * | 2004-03-17 | 2009-04-30 | Dow Global Technologies Inc | Catalyst composition comprising shuttling agent for ethylene multi- block copolymer formation |
| AU2005224258B2 (en) | 2004-03-17 | 2010-09-02 | Dow Global Technologies Inc. | Catalyst composition comprising shuttling agent for ethylene copolymer formation |
| US7863379B2 (en) * | 2004-03-17 | 2011-01-04 | Dow Global Technologies Inc. | Impact modification of thermoplastics with ethylene/alpha-olefin interpolymers |
| ITMI20070877A1 (en) * | 2007-05-02 | 2008-11-03 | Dow Global Technologies Inc | PROCESS FOR THE PRODUCTION OF MULTI-BLOCKED COPOLYMERS WITH THE USE OF POLAR SOLVENTS |
| SG11201505073SA (en) * | 2012-12-27 | 2015-07-30 | Dow Global Technologies Llc | A polymerization process for producing ethylene based polymers |
| US20150337062A1 (en) * | 2012-12-27 | 2015-11-26 | Dow Global Technologies Llc | Ethylene Based Polymer |
| CN107406546B (en) * | 2015-03-17 | 2021-02-19 | Jxtg能源株式会社 | Process for producing oligomer and catalyst |
| CN108884196B (en) * | 2016-03-31 | 2021-06-18 | 陶氏环球技术有限责任公司 | Olefin polymerization catalyst system and method of using same |
-
2018
- 2018-03-14 CN CN201880027869.1A patent/CN110582518B/en active Active
- 2018-03-14 BR BR112019019130-5A patent/BR112019019130B1/en active IP Right Grant
- 2018-03-14 CN CN202210873641.2A patent/CN115260366A/en active Pending
- 2018-03-14 US US16/494,085 patent/US11459409B2/en active Active
- 2018-03-14 EP EP18715859.7A patent/EP3596141B1/en active Active
- 2018-03-14 WO PCT/US2018/022454 patent/WO2018170138A1/en unknown
- 2018-03-14 SG SG11201908414Y patent/SG11201908414YA/en unknown
- 2018-03-14 KR KR1020197030261A patent/KR102653652B1/en active IP Right Grant
- 2018-03-14 JP JP2019550232A patent/JP7287895B2/en active Active
- 2018-03-15 TW TW107108879A patent/TW201840604A/en unknown
Patent Citations (83)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4543399A (en) | 1982-03-24 | 1985-09-24 | Union Carbide Corporation | Fluidized bed reaction systems |
| US4588790A (en) | 1982-03-24 | 1986-05-13 | Union Carbide Corporation | Method for fluidized bed polymerization |
| EP0089691A2 (en) | 1982-03-24 | 1983-09-28 | Union Carbide Corporation | Continuous process for the production of polymer in a fluidized bed reactor |
| US5153157A (en) | 1987-01-30 | 1992-10-06 | Exxon Chemical Patents Inc. | Catalyst system of enhanced productivity |
| US7163907B1 (en) | 1987-01-30 | 2007-01-16 | Exxonmobil Chemical Patents Inc. | Aluminum-free monocyclopentadienyl metallocene catalysts for olefin polymerization |
| US5028670A (en) | 1988-07-15 | 1991-07-02 | Bp Chemicals Limited | Process for the gas-phase polymerization of olefins in a fluidized-bed reactor |
| US5064802A (en) | 1989-09-14 | 1991-11-12 | The Dow Chemical Company | Metal complex compounds |
| US5106804A (en) | 1989-12-22 | 1992-04-21 | Bp Chemicals Limited | Catalyst and prepolymer used for the preparation of polyolefins |
| US5032562A (en) | 1989-12-27 | 1991-07-16 | Mobil Oil Corporation | Catalyst composition and process for polymerizing polymers having multimodal molecular weight distribution |
| US5321106A (en) | 1990-07-03 | 1994-06-14 | The Dow Chemical Company | Addition polymerization catalyst with oxidative activation |
| US5721185A (en) | 1991-06-24 | 1998-02-24 | The Dow Chemical Company | Homogeneous olefin polymerization catalyst by abstraction with lewis acids |
| US5272236A (en) | 1991-10-15 | 1993-12-21 | The Dow Chemical Company | Elastic substantially linear olefin polymers |
| US5278272A (en) | 1991-10-15 | 1994-01-11 | The Dow Chemical Company | Elastic substantialy linear olefin polymers |
| US5665800A (en) | 1991-10-15 | 1997-09-09 | The Dow Chemical Company | Elastic substantially linear olefin polymers |
| US5436304A (en) | 1992-03-19 | 1995-07-25 | Exxon Chemical Patents Inc. | Process for polymerizing monomers in fluidized beds |
| US5352749A (en) | 1992-03-19 | 1994-10-04 | Exxon Chemical Patents, Inc. | Process for polymerizing monomers in fluidized beds |
| US5296433A (en) | 1992-04-14 | 1994-03-22 | Minnesota Mining And Manufacturing Company | Tris(pentafluorophenyl)borane complexes and catalysts derived therefrom |
| US5350723A (en) | 1992-05-15 | 1994-09-27 | The Dow Chemical Company | Process for preparation of monocyclopentadienyl metal complex compounds and method of use |
| US5608019A (en) | 1992-12-28 | 1997-03-04 | Mobil Oil Corporation | Temperature control of MW in olefin polymerization using supported metallocene catalyst |
| US5473028A (en) | 1992-12-28 | 1995-12-05 | Mobil Oil Corporation | Process and a catalyst for preventing reactor fouling |
| US5462999A (en) | 1993-04-26 | 1995-10-31 | Exxon Chemical Patents Inc. | Process for polymerizing monomers in fluidized beds |
| US5405922A (en) | 1993-04-26 | 1995-04-11 | Exxon Chemical Patents Inc. | Process for polymerizing monomers in fluidized beds |
| US5556238A (en) | 1993-05-19 | 1996-09-17 | Bp Chemicals Limited | Process and apparatus for introducing a solid into a reactor |
| US5541270A (en) | 1993-05-20 | 1996-07-30 | Bp Chemicals Limited | Polymerization process |
| WO1994028032A1 (en) | 1993-05-20 | 1994-12-08 | Bp Chemicals Limited | Polymerisation process |
| WO1994025495A1 (en) | 1993-05-20 | 1994-11-10 | Exxon Chemical Patents Inc. | Process for polymerizing monomers in fluidized beds |
| US5425872A (en) | 1993-06-24 | 1995-06-20 | The Dow Chemical Company | Electrochemical preparation of addition polymerization catalysts |
| US5470993A (en) | 1993-06-24 | 1995-11-28 | The Dow Chemical Company | Titanium(II) or zirconium(II) complexes and addition polymerization catalysts therefrom |
| US5461123A (en) | 1994-07-14 | 1995-10-24 | Union Carbide Chemicals & Plastics Technology Corporation | Gas phase fluidized bed polyolefin polymerization process using sound waves |
| US5453471B1 (en) | 1994-08-02 | 1999-02-09 | Carbide Chemicals & Plastics T | Gas phase polymerization process |
| US5453471A (en) | 1994-08-02 | 1995-09-26 | Union Carbide Chemicals & Plastics Technology Corporation | Gas phase polymerization process |
| US5625087A (en) | 1994-09-12 | 1997-04-29 | The Dow Chemical Company | Silylium cationic polymerization activators for metallocene complexes |
| US5543458A (en) | 1995-01-31 | 1996-08-06 | Shell Oil Company | Process for making graft block copolymers by growing anionic polymer chains from functionalized polyolefin backbones |
| US5616661A (en) | 1995-03-31 | 1997-04-01 | Union Carbide Chemicals & Plastics Technology Corporation | Process for controlling particle growth during production of sticky polymers |
| US5883204A (en) | 1996-03-27 | 1999-03-16 | The Dow Chemical Company | Solution polymerization process with dispersed catalyst activator |
| US5919983A (en) | 1996-03-27 | 1999-07-06 | The Dow Chemical Company | Highly soluble olefin polymerization catalyst activator |
| US5977251A (en) | 1996-04-01 | 1999-11-02 | The Dow Chemical Company | Non-adiabatic olefin solution polymerization |
| WO1997045434A1 (en) * | 1996-05-31 | 1997-12-04 | Borealis A/S | Novel transition metal complexes and method for preparing them |
| US6268444B1 (en) | 1996-08-08 | 2001-07-31 | Dow Chemical Company | 3-heteroatom substituted cyclopentadienyl-containing metal complexes and olefin polymerization process |
| US6015868A (en) | 1996-10-03 | 2000-01-18 | The Dow Chemical Company | Substituted indenyl containing metal complexes and olefin polymerization process |
| US5783512A (en) | 1996-12-18 | 1998-07-21 | The Dow Chemical Company | Catalyst component dispersion comprising an ionic compound and solid addition polymerization catalysts containing the same |
| US6034022A (en) | 1996-12-19 | 2000-03-07 | The Dow Chemical Company | Fused ring substituted indenyl metal complexes and polymerization |
| US5866704A (en) | 1996-12-19 | 1999-02-02 | The Dow Chemical Company | 3-aryl substituted indenyl containing metal complexes and polymerization process |
| WO1998050392A1 (en) | 1997-05-08 | 1998-11-12 | Nova Chemicals (International) S.A. | Process to prepare bridged phosphole-cyclopentadienyl compounds |
| US6103657A (en) | 1997-07-02 | 2000-08-15 | Union Carbide Chemicals & Plastics Technology Corporation | Catalyst for the production of olefin polymers |
| US6320005B1 (en) | 1997-07-02 | 2001-11-20 | Union Carbide Chemicals & Plastics Technology Corporation | Catalyst for the production of olefin polymers |
| US6319989B1 (en) | 1997-07-21 | 2001-11-20 | The Dow Chemical Company | Broad MWD, compositionally uniform ethylene interpolymer compositions, process for making the same and article made therefrom |
| US6150297A (en) | 1997-09-15 | 2000-11-21 | The Dow Chemical Company | Cyclopentaphenanthrenyl metal complexes and polymerization process |
| US6696379B1 (en) | 1997-09-19 | 2004-02-24 | The Dow Chemical Company | Supported modified alumoxane catalyst activator |
| US6683149B2 (en) | 1997-09-19 | 2004-01-27 | Dow Global Technologies Inc. | Narrow MWD, compositionally optimized ethylene interpolymer composition, process for making the same and article made therefrom |
| US6827976B2 (en) | 1998-04-29 | 2004-12-07 | Unaxis Trading Ag | Method to increase wear resistance of a tool or other machine component |
| US6555634B1 (en) | 1999-05-13 | 2003-04-29 | The Dow Chemical Company | Di- and tri-heteroatom substituted indenyl metal complexes |
| US20030004286A1 (en) | 1999-12-10 | 2003-01-02 | Jerzy Klosin | Alkaryl-substituted Group 4 metal complexes, catalysts and olefin polymerization process |
| US6515155B1 (en) | 1999-12-10 | 2003-02-04 | Dow Global Technologies Inc. | Substituted group 4 metal complexes, catalysts and olefin polymerization process |
| WO2002002577A1 (en) | 2000-06-30 | 2002-01-10 | Dow Global Technology Inc. | Polycyclic, fused ring compounds, metal complexes and polymerization process |
| WO2002038628A2 (en) | 2000-11-07 | 2002-05-16 | Symyx Technologies, Inc. | Substituted pyridyl amine ligands, complexes and catalysts therefrom; processes for producing polyolefins therewith |
| WO2002092610A1 (en) | 2001-05-14 | 2002-11-21 | Dow Global Technologies Inc. | 3-aryl-substituted cyclopentadienyl metal complexes and polymerization process |
| WO2003040195A1 (en) | 2001-11-06 | 2003-05-15 | Dow Global Technologies Inc. | Supported catalysts for manufacture of polymers |
| WO2003078483A1 (en) | 2002-03-14 | 2003-09-25 | Dow Global Technologies Inc. | Substituted indenyl metal complexes and polymerization process |
| WO2003078480A2 (en) | 2002-03-14 | 2003-09-25 | Dow Global Technologies Inc. | Polycyclic, fused heteroring compounds, metal complexes and polymerization process |
| US20040010103A1 (en) | 2002-04-24 | 2004-01-15 | Symyx Technologies, Inc. | Bridged bi-aromatic ligands, catalysts, processes for polymerizing and polymers therefrom |
| WO2004024740A1 (en) | 2002-09-12 | 2004-03-25 | Dow Global Technologies Inc. | Preparation of metal complexes |
| US20040220050A1 (en) | 2003-05-02 | 2004-11-04 | Frazier Kevin A. | High activity olefin polymerization catalyst and process |
| US6953764B2 (en) | 2003-05-02 | 2005-10-11 | Dow Global Technologies Inc. | High activity olefin polymerization catalyst and process |
| US7608668B2 (en) | 2004-03-17 | 2009-10-27 | Dow Global Technologies Inc. | Ethylene/α-olefins block interpolymers |
| US20060199930A1 (en) | 2004-03-17 | 2006-09-07 | Dow Global Technologies Inc. | Ethylene/alpha-olefins block interpolymers |
| WO2005090426A1 (en) | 2004-03-17 | 2005-09-29 | Dow Global Technologies Inc. | Catalyst composition comprising shuttling agent for higher olefin multi-block copolymer formation |
| WO2005090427A2 (en) | 2004-03-17 | 2005-09-29 | Dow Global Technologies Inc. | Catalyst composition comprising shuttling agent for ethylene multi-block copolymer formation |
| US20070167578A1 (en) | 2004-03-17 | 2007-07-19 | Arriola Daniel J | Catalyst composition comprising shuttling agent for ethylene multi-block copolymer formation |
| US7355089B2 (en) | 2004-03-17 | 2008-04-08 | Dow Global Technologies Inc. | Compositions of ethylene/α-olefin multi-block interpolymer for elastic films and laminates |
| US20080311812A1 (en) | 2004-03-17 | 2008-12-18 | Arriola Daniel J | Catalyst Composition Comprising Shuttling Agent for Higher Olefin Multi-Block Copolymer Formation |
| WO2006101966A1 (en) | 2005-03-17 | 2006-09-28 | Dow Global Technologies, Inc. | ETHYLENE/α-OLEFINS BLOCK INTERPOLYMERS |
| WO2007035485A1 (en) | 2005-09-15 | 2007-03-29 | Dow Global Technologies Inc. | Catalytic olefin block copolymers with controlled block sequence distribution |
| US8058373B2 (en) | 2006-05-17 | 2011-11-15 | Dow Global Technologies Llc | Polypropylene solution polymerization process |
| WO2009012215A1 (en) | 2007-07-13 | 2009-01-22 | Dow Global Technologies Inc. | Ethylene/a-olefin interpolymers containing low crystallinity hard blocks |
| WO2010022228A2 (en) | 2008-08-21 | 2010-02-25 | Dow Global Technologies, Inc. | Metal-ligand complexes and catalysts |
| US8372931B2 (en) | 2009-07-01 | 2013-02-12 | Dow Global Technologies Llc | Ethylene-based polymer compositions |
| US8501885B2 (en) | 2009-07-29 | 2013-08-06 | Dow Global Technologies, Llc | Dual- or multi-headed chain shuttling agents and their use for preparation of block copolymers |
| US8785554B2 (en) | 2010-06-21 | 2014-07-22 | Dow Global Technologies Llc | Crystalline block composites as compatibilizers |
| EP2436703A1 (en) * | 2010-09-30 | 2012-04-04 | Dow Global Technologies LLC | Comb architecture olefin block copolymers |
| WO2014105411A1 (en) | 2012-12-27 | 2014-07-03 | Dow Global Technologies Llc | Catalyst systems for olefin polymerization |
| WO2017173080A1 (en) * | 2016-03-31 | 2017-10-05 | Dow Global Technologies Llc | Olefin polymerization catalyst systems and methods of use thereof |
| WO2017173074A1 (en) * | 2016-03-31 | 2017-10-05 | Dow Global Technologies Llc | An olefin polymerization catalyst |
Non-Patent Citations (18)
| Title |
|---|
| A. E. HAMIELEC; J. F. MACGREGOR: "Polymer Reaction Engineering", 1983, HANSER |
| B. G. KYLE: "Chemical and Process Thermodynamics", 1999, PRENTICE-HALL |
| BROOKHART ET AL., J. AM. CHEM. SOC., vol. 117, 1995, pages 64145 - 6415 |
| CHEMICAL ENGINEERING SCIENCE, 1972, pages 1197 - 1203 |
| DIAS ET AL., J. MOL. CATAL. A: CHEMICAL, vol. 185, 2002, pages 57 - 64 |
| DOBRYNIN, J. CHEM. PHVS., vol. 107, no. 21, 1997, pages 9234 - 9238 |
| G. HERBERICH ET AL., ORGANOMETALLICS, vol. 14, no. 1, 1995, pages 471 - 480 |
| GEORGE ODIAN: "Principles of Polymerization", 1970, JOHN WILEY AND SONS |
| IAN WESTMORELAND ET AL: "Chiral Titanium Bis(aminopyridinates) Based on a Biaryl Backbone", ORGANOMETALLICS, vol. 22, no. 14, 1 July 2003 (2003-07-01), US, pages 2972 - 2976, XP055383571, ISSN: 0276-7333, DOI: 10.1021/om030055k * |
| J. POLYM. SCI., POLYM. LET., vol. 6, 1968, pages 621 - 624 |
| KAMINSKI ET AL., J. MOL. CATAL. A: CHEMICAL, vol. 102, 1995, pages 59 - 65 |
| KRETSCHMER ET AL: "Highly active/selective and adjustable zirconium polymerization catalysts stabilized by aminopyridinato ligands", JOURNAL OF ORGANOMETALLIC CHEMISTRY, ELSEVIER-SEQUOIA S.A. LAUSANNE, CH, vol. 692, no. 21, 14 September 2007 (2007-09-14), pages 4569 - 4579, XP022248192, ISSN: 0022-328X, DOI: 10.1016/J.JORGANCHEM.2007.04.041 * |
| MONRABAL ET AL., MACROMOL. SYMP., vol. 257, 2007, pages 71 - 79 |
| NEGISHI, ORGANOMETALLICS IN ORGANIC SYNTHESIS, vol. 1 and 2, 1980 |
| POTEMKIN, PHYSICAL REVIEW, vol. 57, no. 6, 1998, pages 6902 - 6912 |
| TALJA ET AL: "Bis(alkylphenylaminopyridinato) titanium dichlorides as ethylene polymerization catalysts", JOURNAL OF MOLECULAR CATALYSIS A: CHEMICAL, ELSEVIER, AMSTERDAM, NL, vol. 280, no. 1-2, 4 November 2007 (2007-11-04), pages 102 - 105, XP022422895, ISSN: 1381-1169, DOI: 10.1016/J.MOLCATA.2007.10.029 * |
| W. H. RAY: "Rev. Macromol. Chem.", J. MACROMOL. SCI., vol. C8, 1972, pages 1 |
| ZAMBELLI ET AL., MACROMOLECULES, vol. 21, 1988, pages 617 - 622 |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019182993A1 (en) | 2018-03-19 | 2019-09-26 | Dow Global Technologies Llc | Silicon-terminated telechelic polyolefin compositions and processes for preparing the same |
| WO2019182988A1 (en) | 2018-03-19 | 2019-09-26 | Dow Global Technologies Llc | Silicon-terminated organo-metal compounds and processes for preparing the same |
| WO2019182986A1 (en) | 2018-03-19 | 2019-09-26 | Dow Global Technologies Llc | Process for functionalization of organo-metal compounds with silyl-based functionalization agents and silyl-functionalized compounds prepared thereby |
| WO2019182983A1 (en) | 2018-03-19 | 2019-09-26 | Dow Global Technologies Llc | Silicon-terminated organo-metal compounds and processes for preparing the same |
| EP4234594A1 (en) | 2018-12-28 | 2023-08-30 | Dow Global Technologies LLC | Curable compositions comprising unsaturated polyolefins |
| WO2020140067A1 (en) | 2018-12-28 | 2020-07-02 | Dow Global Technologies Llc | Curable compositions comprising unsaturated polyolefins |
| WO2020140064A1 (en) | 2018-12-28 | 2020-07-02 | Dow Global Technologies Llc | Organometallic chain transfer agents |
| WO2020140061A1 (en) | 2018-12-28 | 2020-07-02 | Dow Global Technologies Llc | Curable compositions comprising telechelic polyolefins |
| WO2020140058A1 (en) | 2018-12-28 | 2020-07-02 | Dow Global Technologies Llc | Telechelic polyolefins and processes for preparing the same |
| WO2021048030A1 (en) | 2019-09-10 | 2021-03-18 | Sabic Global Technologies B.V. | Compounds for use in catalyst compositions for the production of polyolefins |
| WO2021133945A1 (en) * | 2019-12-27 | 2021-07-01 | Dow Global Technologies Llc | Ethylene/octene multi-block copolymer and process for producing same |
| CN115427466A (en) * | 2019-12-27 | 2022-12-02 | 陶氏环球技术有限责任公司 | Ethylene/octene multi-block copolymer and preparation method thereof |
| WO2021133925A1 (en) * | 2019-12-27 | 2021-07-01 | Dow Global Technologies Llc | Ethylene/butene multi-block copolymer and process for producing same |
| WO2021155168A1 (en) | 2020-01-31 | 2021-08-05 | Dow Global Technologies Llc | Polymerization processes that include group iii and lanthanide bis-phenyl-phenoxy metal-ligand complexes and chain transfer agents |
| WO2021155158A1 (en) | 2020-01-31 | 2021-08-05 | Dow Global Technologies Llc | Group iii and lanthanide bis-phenyl-phenoxy metal-ligand complexes |
| EP4332133A2 (en) | 2020-01-31 | 2024-03-06 | Dow Global Technologies LLC | Group iii and lanthanide bis-phenyl-phenoxy metal-ligand complexes |
Also Published As
| Publication number | Publication date |
|---|---|
| CN110582518A (en) | 2019-12-17 |
| TW201840604A (en) | 2018-11-16 |
| BR112019019130B1 (en) | 2023-03-28 |
| JP2020514503A (en) | 2020-05-21 |
| BR112019019130A2 (en) | 2020-04-14 |
| KR20190123345A (en) | 2019-10-31 |
| EP3596141B1 (en) | 2021-07-28 |
| US20200131283A1 (en) | 2020-04-30 |
| JP7287895B2 (en) | 2023-06-06 |
| US11459409B2 (en) | 2022-10-04 |
| KR102653652B1 (en) | 2024-04-03 |
| EP3596141A1 (en) | 2020-01-22 |
| CN110582518B (en) | 2022-08-09 |
| CN115260366A (en) | 2022-11-01 |
| SG11201908414YA (en) | 2019-10-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP3596141B1 (en) | Catalyst system for multi-block copolymer formation | |
| CN109952324B (en) | Blocked multi-or double-headed compositions suitable for chain shuttling and methods of making the same | |
| EP3596138B1 (en) | Catalyst system for multi-block copolymer formation | |
| KR20200135420A (en) | Silicon-terminated organo-metal compound and method for preparing same | |
| US11208502B2 (en) | Catalyst system for multi-block coploymer formation | |
| US11897992B2 (en) | Catalyst system for multi-block copolymer formation | |
| KR102648625B1 (en) | Catalyst system for forming multi-block copolymers |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 18715859 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2019550232 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112019019130 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 20197030261 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2018715859 Country of ref document: EP Effective date: 20191015 |
|
| ENP | Entry into the national phase |
Ref document number: 112019019130 Country of ref document: BR Kind code of ref document: A2 Effective date: 20190914 |