WO2018005662A1 - Boronic acid derivatives and therapeutic uses thereof - Google Patents
Boronic acid derivatives and therapeutic uses thereof Download PDFInfo
- Publication number
- WO2018005662A1 WO2018005662A1 PCT/US2017/039787 US2017039787W WO2018005662A1 WO 2018005662 A1 WO2018005662 A1 WO 2018005662A1 US 2017039787 W US2017039787 W US 2017039787W WO 2018005662 A1 WO2018005662 A1 WO 2018005662A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- optionally substituted
- alkyl
- group
- carbocyclyl
- compound
- Prior art date
Links
- 0 *c([n]1)nnc1O Chemical compound *c([n]1)nnc1O 0.000 description 7
- SGPIGSHLDNBRQB-WAYWQWQTSA-N CC(C)(Oc1c2c(F)ccc1/C=C\B(O)O)OC2=O Chemical compound CC(C)(Oc1c2c(F)ccc1/C=C\B(O)O)OC2=O SGPIGSHLDNBRQB-WAYWQWQTSA-N 0.000 description 1
- BDVMMHJHDXNXPR-SREVYHEPSA-N CC(C)(Oc1c2c(F)ccc1/C=C\[Br]=C)OC2=O Chemical compound CC(C)(Oc1c2c(F)ccc1/C=C\[Br]=C)OC2=O BDVMMHJHDXNXPR-SREVYHEPSA-N 0.000 description 1
- CEXILKJKXFZEJD-FIMMUYGNSA-N CC(C)(Oc1c2c(F)ccc1C1C(B(O[C@H]3C(N(C)C)=O)O[C@H]3C(N(C)C)=O)C1)OC2=O Chemical compound CC(C)(Oc1c2c(F)ccc1C1C(B(O[C@H]3C(N(C)C)=O)O[C@H]3C(N(C)C)=O)C1)OC2=O CEXILKJKXFZEJD-FIMMUYGNSA-N 0.000 description 1
- CJDWRXRYXXZOFA-UHFFFAOYSA-N CC(C)C(C(N1)=O)SC1=O Chemical compound CC(C)C(C(N1)=O)SC1=O CJDWRXRYXXZOFA-UHFFFAOYSA-N 0.000 description 1
- OFELREQXHIMXKP-UHFFFAOYSA-N CC(C)C(N1)=NOC1=S Chemical compound CC(C)C(N1)=NOC1=S OFELREQXHIMXKP-UHFFFAOYSA-N 0.000 description 1
- WORFMGRCFYNZOZ-UHFFFAOYSA-N CC(C)N(C(N1)=O)OC1=O Chemical compound CC(C)N(C(N1)=O)OC1=O WORFMGRCFYNZOZ-UHFFFAOYSA-N 0.000 description 1
- JZDDUNJFAMFXMB-UHFFFAOYSA-N CC(C)c(nn[nH]1)c1F Chemical compound CC(C)c(nn[nH]1)c1F JZDDUNJFAMFXMB-UHFFFAOYSA-N 0.000 description 1
- FOTIBFXYPVRLQJ-UHFFFAOYSA-N CC(C)c1ncn[o]1 Chemical compound CC(C)c1ncn[o]1 FOTIBFXYPVRLQJ-UHFFFAOYSA-N 0.000 description 1
- RFFXUEDBNNOGDO-UHFFFAOYSA-N CC(C)c1nnn[nH]1 Chemical compound CC(C)c1nnn[nH]1 RFFXUEDBNNOGDO-UHFFFAOYSA-N 0.000 description 1
- WIEGQXSJSQWJCK-UHFFFAOYSA-N CC(O1)=C(CN)OC1=O Chemical compound CC(O1)=C(CN)OC1=O WIEGQXSJSQWJCK-UHFFFAOYSA-N 0.000 description 1
- CHHGQELXPZVXNA-GHMZBOCLSA-N CC(OCCOC(OCOC(c(c(F)ccc1[C@@H]2[C@H]3C2)c1OB3O)=O)=O)=O Chemical compound CC(OCCOC(OCOC(c(c(F)ccc1[C@@H]2[C@H]3C2)c1OB3O)=O)=O)=O CHHGQELXPZVXNA-GHMZBOCLSA-N 0.000 description 1
- IPWKHHSGDUIRAH-UHFFFAOYSA-N CC1(C)OB(B2OC(C)(C)C(C)(C)O2)OC1(C)C Chemical compound CC1(C)OB(B2OC(C)(C)C(C)(C)O2)OC1(C)C IPWKHHSGDUIRAH-UHFFFAOYSA-N 0.000 description 1
- ILILMGLCVSOUCG-UHFFFAOYSA-N CCOC(COC(OCCl)=O)=O Chemical compound CCOC(COC(OCCl)=O)=O ILILMGLCVSOUCG-UHFFFAOYSA-N 0.000 description 1
- JLQLTELAOKOFBV-UHFFFAOYSA-N CC[n]1nnnc1S Chemical compound CC[n]1nnnc1S JLQLTELAOKOFBV-UHFFFAOYSA-N 0.000 description 1
- MODVMQGLMDBFRZ-UHFFFAOYSA-N CCc(nn[nH]1)c1S Chemical compound CCc(nn[nH]1)c1S MODVMQGLMDBFRZ-UHFFFAOYSA-N 0.000 description 1
- PCYDYHRBODKVEL-PHDIDXHHSA-N CN(C)C([C@@H]([C@H](C(N(C)C)=O)O)O)=O Chemical compound CN(C)C([C@@H]([C@H](C(N(C)C)=O)O)O)=O PCYDYHRBODKVEL-PHDIDXHHSA-N 0.000 description 1
- MOEDEYVUTCDHEC-YGPZHTELSA-N COCC(OCOC(c(c(F)ccc1C2[C@H]3C2)c1OB3O)=O)=O Chemical compound COCC(OCOC(c(c(F)ccc1C2[C@H]3C2)c1OB3O)=O)=O MOEDEYVUTCDHEC-YGPZHTELSA-N 0.000 description 1
- ZSBCVGHLJGEASR-MPDSMELSSA-N C[C@@]12OB(/C=C\C)OC3C1C14C2CC1CC34 Chemical compound C[C@@]12OB(/C=C\C)OC3C1C14C2CC1CC34 ZSBCVGHLJGEASR-MPDSMELSSA-N 0.000 description 1
- YPWKAZSHYHFTIW-ZCFIWIBFSA-N C[C@H](CCC1)N1C(C)=O Chemical compound C[C@H](CCC1)N1C(C)=O YPWKAZSHYHFTIW-ZCFIWIBFSA-N 0.000 description 1
- VCFOEWURSYAGKP-UHFFFAOYSA-N Cc1c(C(O)=O)[nH]nn1 Chemical compound Cc1c(C(O)=O)[nH]nn1 VCFOEWURSYAGKP-UHFFFAOYSA-N 0.000 description 1
- PZKFSRWSQOQYNR-UHFFFAOYSA-N Cc1nnc[nH]1 Chemical compound Cc1nnc[nH]1 PZKFSRWSQOQYNR-UHFFFAOYSA-N 0.000 description 1
- XDWRMBIQUNYCCU-QXMHVHEDSA-N O=C(c(c(OCc1ccccc1)c(/C=C\Br)cc1F)c1F)OCc1ccccc1 Chemical compound O=C(c(c(OCc1ccccc1)c(/C=C\Br)cc1F)c1F)OCc1ccccc1 XDWRMBIQUNYCCU-QXMHVHEDSA-N 0.000 description 1
- MHZBCLGBMWAGMK-NXEZZACHSA-N OB1Oc2c(C(OCOC(C3CC3)=O)=O)c(F)ccc2[C@@H]2[C@H]1C2 Chemical compound OB1Oc2c(C(OCOC(C3CC3)=O)=O)c(F)ccc2[C@@H]2[C@H]1C2 MHZBCLGBMWAGMK-NXEZZACHSA-N 0.000 description 1
- FKOFJDOUUGNHTF-UHFFFAOYSA-N OC(C(C(c(c(OCc1ccccc1)c1C(OCc2ccccc2)=O)cc(F)c1F)Br)Br)=O Chemical compound OC(C(C(c(c(OCc1ccccc1)c1C(OCc2ccccc2)=O)cc(F)c1F)Br)Br)=O FKOFJDOUUGNHTF-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F5/00—Compounds containing elements of Groups 3 or 13 of the Periodic System
- C07F5/02—Boron compounds
- C07F5/05—Cyclic compounds having at least one ring containing boron but no carbon in the ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F5/00—Compounds containing elements of Groups 3 or 13 of the Periodic System
- C07F5/02—Boron compounds
- C07F5/025—Boronic and borinic acid compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/407—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with other heterocyclic ring systems, e.g. ketorolac, physostigmine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4196—1,2,4-Triazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
- A61K31/426—1,3-Thiazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/69—Boron compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Definitions
- the present application relates to the fields of chemistry and medicine. More particularly, the present application relates to boronic acid antimicrobial compounds, compositions, their preparation, and their use as therapeutic agents.
- Antibiotics have been effective tools in the treatment of infectious diseases during the last half-century. From the development of antibiotic therapy to the late 1980s there was almost complete control over bacterial infections in developed countries. However, in response to the pressure of antibiotic usage, multiple resistance mechanisms have become widespread and are threatening the clinical utility of anti-bacterial therapy.
- the increase in antibiotic resistant strains has been particularly common in major hospitals and care centers. The consequences of the increase in resistant strains include higher morbidity and mortality, longer patient hospitalization, and an increase in treatment costs.
- ⁇ -lactamases can be grouped into 4 classes based on their amino acid sequences, namely, Ambler classes A, B, C, and D. Enzymes in classes A, C, and D include active-site serine ⁇ -lactamases, and class B enzymes, which are encountered less frequently, are Zn-dependent. These enzymes catalyze the chemical degradation of ⁇ -lactam antibiotics, rendering them inactive. Some ⁇ -lactamases can be transferred within and between various bacterial strains and species. The rapid spread of bacterial resistance and the evolution of multi-resistant strains severely limits ⁇ -lactam treatment options available.
- class D ⁇ -lactamase-expressing bacterium strains such as Acinetobacter baumannii
- A. baumannii strains express A, C, and D class ⁇ -lactamases.
- the class D ⁇ -lactamases such as the OXA families are particularly effective at destroying carbapenem type ⁇ -lactam antibiotics, e.g., imipenem, the active carbapenems component of Merck's Primaxin® (Montefour, K. et al., Crit. Care Nurse 2008, 28, 15; Perez, F. et al, Expert Rev. Anti Infect. Ther.
- New ⁇ -lactamases have recently evolved that hydrolyze the carbapenem class of antimicrobials, including imipenem, biapenem, doripenem, meropenem, and ertapenem, as well as other ⁇ -lactam antibiotics.
- carbapenemases belong to molecular classes A, B, and D.
- Class A carbapenemases of the KPC-type predominantly in Klebsiella pneumoniae but now also reported in other Enterobacteriaceae, Pseudomonas aeruginosa and Acinetobacter baumannii.
- the KPC carbapenemase was first described in 1996 in North Carolina, but since then has disseminated widely in the US.
- the zinc-dependent class B metallo ⁇ -lactamases are represented mainly by the VIM, IMP, and NDM types.
- IMP and VIM-producing K. pneumonia were first observed in 1990s in Japan and 2001 in Southern Europe, respectively.
- IMP-positive strains remain frequent in Japan and have also caused hospital outbreaks in China and Australia.
- dissemination of IMP-producing Enterobacteriaceae in the rest of the word appears to be somewhat limited.
- VIM-producing enterobacteria can be frequently isolated in Mediterranean countries, reaching epidemic proportions in Greece. Isolation of VIM-producing strains remains low in Northern Europe and in the United States.
- a characteristic of NDM- producing K. pneumonia isolates has been their rapid dissemination from their epicenter, the Indian subcontinent, to Western Europe, North America, Australia and Far East.
- NDM genes have spread rapidly to various species other than K. pneumonia.
- the plasmid-expressed class D carbapenemases belong to OXA-48 type.
- OXA-48 producing K. pneumonia was first detected in Turkey, in 2001.
- the Middle East and North Africa remain the main centers of infection.
- recent isolation of OXA-48-type producing organisms in India, Senegal and Argentina suggest the possibility of a global expansion.
- Isolation of OXA-48 in bacteria other than K. pneumonia underlines the spreading potential of OXA-48.
- Treatment of strains producing any of these carbapenemases with carbapenems can be associated with poor outcomes.
- Another mechanism of ⁇ -lactamase mediated resistance to carbapenems involves combination of permeability or efflux mechanisms combined with hyper production of beta-lactamases.
- One example is the loss of a porin combined in hyperproduction of ampC beta- lactamase results in resistance to imipenem in Pseudomonas aeruginosa.
- Efflux pump over expression combined with hyperproduction of the ampC ⁇ -lactamase can also result in resistance to a carbapenem such as meropenem.
- Y 1 is N or CR 4 ;
- n is an integer of 0 or 1 ;
- R and R together with the atoms to which they are attached form a fused ring or ring system selected from the group consisting of C3_ 7 carbocyclyl, 3-10 membered heterocyclyl, Ce-w aryl, and 5-10 membered heteroaryl, each optionally substituted with one or more R 5 , and
- each of R 1 , R 4 , R a , and R b is independently selected from the group consisting of H, amino, halogen, cyano, hydroxy, optionally substituted Ci_ 6 alkyl, optionally substituted Ci_ 6 haloalkyl, optionally substituted Ci_ 6 alkoxy, optionally substituted Ci_ 6 haloalkoxy, optionally substituted (Ci_ 6 alkoxy)Ci_ 6 alkyl, optionally substituted C 2 - 10 alkenyl, optionally substituted C 2 -ioalkynyl, optionally substituted carbocyclyl, optionally substituted 3-10 membered heterocyclyl, optionally substituted C6-ioaryl, optionally substituted 5-10 membered heteroaryl, optionally substituted (C3- 7carbocyclyl)Ci_ 6 alkyl, optionally substituted (3-10 membered heterocyclyl)Ci_ 6 alkyl, optionally substituted (C6-ioaryl)Ci
- R and R together with the atoms to which they are attached form a spirocyclic ring or ring system selected from the group consisting of C3_ 7 carbocyclyl and 3-10 membered heterocyclyl, each optionally substituted with one or more R 5 , and
- each of R 1 , R 2 , R a , and R b is independently selected from the group consisting of H, amino, halogen, cyano, hydroxy, optionally substituted Ci_ 6 alkyl, optionally substituted Ci_ 6 haloalkyl, optionally substituted Ci_ 6 alkoxy, optionally substituted Ci_ 6 haloalkoxy, optionally substituted (Ci_ 6 alkoxy)Ci_ 6 alkyl, optionally substituted C 2 _ 10 alkenyl, optionally substituted C 2 _ 1 oalkynyl, optionally substituted carbocyclyl, optionally substituted 3-10 membered heterocyclyl, optionally substituted C6-ioaryl, optionally substituted 5-10 membered heteroaryl, optionally substituted (C 3 _ 7 carbocyclyl)Ci_ 6 alkyl, optionally substituted (3-10 membered heterocyclyl)Ci_ 6 alkyl, optionally substituted (C6-ioaryl)C
- R 1 and R 2 together with the atoms to which they are attached form a spirocyclic ring or ring system selected from the group consisting of C 3 _ 7 carbocyclyl and 3-10 membered heterocyclyl, each optionally substituted with one or more R 5 , and
- each of R 3 , R 4 , R a , and R b is independently selected from the group consisting of H, amino, halogen, cyano, hydroxy, optionally substituted Ci_ 6 alkyl, optionally substituted Ci_ 6 haloalkyl, optionally substituted Ci_ 6 alkoxy, optionally substituted Ci_ 6 haloalkoxy, optionally substituted (Ci_ 6 alkoxy)Ci_ 6 alkyl, optionally substituted C 2 _ 10 alkenyl, optionally substituted C 2 _ 1 oalkynyl, optionally substituted C 3 _ 7 carbocyclyl, optionally substituted 3-10 membered heterocyclyl, optionally substituted C6-ioaryl, optionally substituted 5-10 membered heteroaryl, optionally substituted (C 3 _ 7 carbocyclyl)Ci_ 6 alkyl, optionally substituted (3-10 membered heterocyclyl)Ci_ 6 alkyl, optionally substituted (C6-i
- R a and R together with the atoms to which they are attached form a spirocyclic ring or ring system selected from the group consisting of C 3 _7 carbocyclyl and 3-10 membered heterocyclyl, each optionally substituted with one or more R 5 , and
- each of R 1 , R2 , R 3 , and R 4 is independently selected from the group consisting of H, amino, halogen, cyano, hydroxy, optionally substituted Ci_ 6 alkyl, optionally substituted Ci_ 6 haloalkyl, optionally substituted Ci_ 6 alkoxy, optionally substituted Ci_ 6 haloalkoxy, optionally substituted (Ci_ 6 alkoxy)Ci_ 6 alkyl, optionally substituted C 2 _ joalkenyl, optionally substituted C 2 _ioalkynyl, optionally substituted C 3 _7 carbocyclyl, optionally substituted 3-10 membered heterocyclyl, optionally substituted C6-ioaryl, optionally substituted 5-10 membered heteroaryl, optionally substituted (C 3 .
- R a and R together with the atoms to which they are attached form a fused ring or ring system selected from the group consisting of C 3 _7 carbocyclyl, 3-10 membered heterocyclyl, C 6- io aryl, and 5-10 membered heteroaryl, each optionally substituted with one or more R 5 , and
- each of R 1 , R 2 , R 3 , and R b is independently selected from the group consisting of H, amino, halogen, cyano, hydroxy, optionally substituted Ci_ 6 alkyl, optionally substituted Ci_ 6 haloalkyl, optionally substituted Ci_ 6 alkoxy, optionally substituted Ci_ 6 haloalkoxy, optionally substituted (Ci_ 6 alkoxy)Ci_ 6 alkyl, optionally substituted C 2 _ 10 alkenyl, optionally substituted C 2 _ 1 oalkynyl, optionally substituted C 3 _ 7 carbocyclyl, optionally substituted 3-10 membered heterocyclyl, optionally substituted C6-ioaryl, optionally substituted 5-10 membered heteroaryl, optionally substituted (C 3 _ 7 carbocyclyl)Ci_ 6 alkyl, optionally substituted (3-10 membered heterocyclyl)Ci_ 6 alkyl, optionally substituted (C6-io
- R 5 is -Y 5 -(CH 2 ) t -G;
- t is an integer of 0 or 1 ;
- G is selected from the group consisting of H, amino, halogen, cyano, hydroxy, optionally substituted Q_ 6 alkyl, optionally substituted Q_ 6 haloalkyl, optionally substituted Ci- 6 alkoxy, optionally substituted Ci- 6 haloalkoxy, optionally substituted (Ci- 6 alkoxy)Ci_ 6 alkyl, optionally substituted C 2 _ioalkenyl, optionally substituted C 2 _ l oalkynyl, optionally substituted C 3 _7 carbocyclyl, optionally substituted 3-10 membered heterocyclyl, optionally substituted C6-ioaryl, optionally substituted 5-10 membered heteroaryl, optionally substituted (C 3 _ 7 carbocyclyl)Ci_ 6 alkyl, optionally substituted (3-10 membered heterocyclyl)C !
- A is selected from the group consisting of C 3 _7 carbocyclyl, 3-10 membered heterocyclyl, C6-ioaryl, and 5-10 membered heteroaryl, each optionally substituted by one or more R 12 ;
- R 6 is selected from the group consisting of H, halogen, optionally substituted Ci_ 6 alkyl, OH, -C(0)OR, optionally substituted d_ 6 alkoxy, amino, -N(OR 8 )R 9 , optionally substituted Q_ 6 alkylthiol, C-amido, S-sulfonamido, CN, sulfinyl, sulfonyl, and a carboxylic acid isostere;
- R is selected from the group consisting of H, d_ 9 alkyl, -CR 10 R 11 OC(O)C 1 _ 9 alkyl, -CR 10 R n OC(O)C 3 _ 7 carbocyclyl, -CR 10 R n OC(O)(3 to 7 membered heterocyclyl), -CR 10 R n OC(O)C 2 _ 8 alkoxyalkyl, -CR 10 R 11 OC(O)OC 1 _
- R 7 is selected from the group consisting of -OH, optionally substituted Ci_ 6 alkoxy, amino, and -N(OR 8 )R 9 ;
- each R 8 and R 9 is independently selected from the group consisting of H, halogen, optionally substituted Ci-4alkyl, optionally substituted C 3 _7 carbocyclyl, optionally substituted 3-10 membered heterocyclyl, optionally substituted C6-ioaryl, and optionally substituted 5-10 membered heteroaryl;
- each R 10 and R 11 is independently selected from the group consisting of H, optionally substituted optionally substituted C 3 _ 7 carbocyclyl, optionally substituted 3-10 membered heterocyclyl, optionally substituted C6-ioaryl, and optionally substituted 5-10 membered heteroaryl;
- R 12 is selected from the group consisting of hydrogen, amino, halogen, cyano, hydroxy, optionally substituted Ci_ 6 alkyl, optionally substituted Ci_ 6 haloalkyl, optionally substituted Ci- 6 alkoxy, optionally substituted Ci- 6 haloalkoxy, optionally substituted (Ci_ 6 alkoxy)Ci_ 6 alkyl, optionally substituted C 2 _ioalkenyl, optionally substituted C 2 _ioalkynyl, optionally substituted C 3 _7 carbocyclyl, optionally substituted 3- 10 membered heterocyclyl, optionally substituted C6-ioaryl, optionally substituted 5-10 membered heteroaryl, optionally substituted (C3 -7 carbocyclyl)Ci.
- each R 13 and R 14 is independently selected from the group consisting of H, optionally substituted Ci- 6 alkyl, optionally substituted C 3 _7 carbocyclyl, optionally substituted 3-10 membered heterocyclyl, optionally substituted C6-ioaryl, and optionally substituted 5-10 membered heteroaryl;
- R 15 is optionally substituted Ci- 6 alkyl
- Y 2 is selected from the group consisting of -0-, -S-, and -NR 9 -;
- Y 3 is selected from the group consisting of -OH, -SH, and -NHR 9 ;
- Y 4 is selected from the group consisting of -OH, optionally substituted Ci_6
- Y 5 is selected from the group consisting of -S-, -S(O)-, -S(0) 2 -, -0-, -CR f R g -, and -NR g -, or Y 5 is absent;
- Y 6 is selected from the group consisting of -S-, -S(O)-, -S(0) 2 -, -0-, -CR f R g -, and
- each p and q is independently 0 or 1.
- each of R 2 and R 3 is independently selected from the group consisting of H, amino, halogen, cyano, hydroxy, optionally substituted Ci_6 alkyl, optionally substituted Ci_6 haloalkyl, optionally substituted Ci_6 alkoxy, optionally substituted Ci_6 haloalkoxy, optionally substituted (Ci_ 6 alkoxy)Ci_ 6 alkyl, optionally substituted C 2 _ioalkenyl, optionally substituted C 2 _ioalkynyl, optionally substituted C 3 _7 carbocyclyl, optionally substituted 3-10 membered heterocyclyl, optionally substituted C6-ioaryl, optionally substituted 5-10 membered heteroaryl, optionally substituted (C3_ 7 carbocyclyl)Ci_ 6 alkyl, optionally substituted (3-10 membered heterocyclyl)Ci_ 6 alkyl, optionally substituted (C 6- ioaryl)Ci_ 6 alkyl, (
- n is an integer of 0 or 1 ;
- each R a and R b is independently selected from the group consisting of H, amino, halogen, cyano, hydroxy, optionally substituted Ci_ 6 alkyl, optionally substituted Ci_ 6 haloalkyl, optionally substituted Ci_ 6 alkoxy, optionally substituted Ci_ 6 haloalkoxy, optionally substituted (Ci_ 6 alkoxy)Ci_ 6 alkyl, optionally substituted C 2 _ioalkenyl, optionally substituted C 2 _ioalkynyl, optionally substituted C 3 _7 carbocyclyl, optionally substituted 3-10 membered heterocyclyl, optionally substituted C6-ioaryl, optionally substituted 5-10 membered heteroaryl, optionally substituted (C3_ 7 carbocyclyl)Ci- 6 alkyl, optionally substituted (3-10 membered heterocyclyl)Ci_ 6 alkyl, optionally substituted (C 6- ioaryl)Ci_ 6 alkyl,
- R a and R b together with the atoms to which they are attached form a spiro ring or ring system selected from the group consisting of C 3 _ 7 carbocyclyl and 3-10 membered heterocyclyl, each optionally substituted with one or more R ; or
- m 1 ;
- R a and R 3 together with the atoms to which they are attached form a fused ring or ring system selected from the group consisting of C 3 _ 7 carbocyclyl and 3-10 membered heterocyclyl, each optionally substituted with one or more R 5 ; and each R 2 and R b is independently selected from the group consisting of H, amino, halogen, cyano, hydroxy, optionally substituted Ci_ 6 alkyl, optionally substituted Ci_ 6 haloalkyl, optionally substituted Ci- 6 alkoxy, optionally substituted Ci- 6 haloalkoxy, optionally substituted (Ci_ 6 alkoxy)Ci_ 6 alkyl, optionally substituted C 2 _ioalkenyl, optionally substituted C 2 _ioalkynyl, optionally substituted C 3 _7 carbocyclyl, optionally substituted 3-10 membered heterocyclyl, optionally substituted C6-ioaryl, optionally substituted 5-10 membered hetero
- R 5 is -Y 5 -(CH 2 ) t -G;
- t is an integer of 0 or 1 ;
- G is selected from the group consisting of H, amino, halogen, cyano, hydroxy, optionally substituted Ci_ 6 alkyl, optionally substituted Ci_ 6 haloalkyl, optionally substituted Ci_ 6 alkoxy, optionally substituted Ci_ 6 haloalkoxy, optionally substituted (Ci_ 6 alkoxy)Ci_ 6 alkyl, optionally substituted C 2 _ioalkenyl, optionally substituted C 2 _ l oalkynyl, optionally substituted C 3 _7 carbocyclyl, optionally substituted 3-10 membered heterocyclyl, optionally substituted C6-io ryl, optionally substituted 5-10 membered heteroaryl, optionally substituted (C 3 _ 7 carbocyclyl)Ci_ 6 alkyl, optionally substituted (3-10 membered heterocyclyl)Ci_ 6 alkyl, optionally substituted (C6-ioaryl)Ci_ 6 alkyl, (C 6- i
- A is a ring system selected from the group consisting of C 3 _7 carbocyclyl, 3-10 membered heterocyclyl, C6-ioaryl, and 5-10 membered heteroaryl, each optionally substituted with one or more R 12 ;
- R 6 is selected from the group consisting of H, halogen, optionally substituted Ci_ 6 alkyl, OH, -C(0)OR, optionally substituted d_ 6 alkoxy, amino, -N(OR 8 )R 9 , optionally substituted Ci_ 6 alkylthiol, C-amido, S-sulfonamido, CN, sulfinyl, sulfonyl, and a carboxylic acid isostere; is selected from the group consisting of H, Ci_ alkyl, -CR R OC(0)Ci_ -CR 10 R n OC(O)C3- 7 carbocyclyl, -CR 10 R n OC(O)(3 to 7 membered heterocyclyl), -CR lu R 1 OC(0)C 2 - 8 alkoxyalkyl, -CR' OC C OC
- R 7 is selected from the group consisting of -OH, optionally substituted Ci_ 6 alkoxy, amino, and -N(OR 8 )R 9 ;
- each R 8 and R 9 is independently selected from the group consisting of H, halogen, optionally substituted Ci-4alkyl, optionally substituted C 3 _7 carbocyclyl, optionally substituted 3-10 membered heterocyclyl, optionally substituted C6-ioaryl, and optionally substituted 5-10 membered heteroaryl;
- each R 10 and R 11 is independently selected from the group consisting of H, optionally substituted Ci-4alkyl, optionally substituted C 3 _7 carbocyclyl, optionally substituted 3-10 membered heterocyclyl, optionally substituted C6-ioaryl, and optionally substituted 5-10 membered heteroaryl;
- R 12 is selected from the group consisting of hydrogen, amino, halogen, cyano, hydroxy, optionally substituted Ci_ 6 alkyl, optionally substituted Ci_ 6 haloalkyl, optionally substituted Ci_ 6 alkoxy, optionally substituted Ci_ 6 haloalkoxy, optionally substituted Ci_ 6 alkylthiol, optionally substituted (Ci_ 6 alkoxy)Ci_ 6 alkyl, optionally substituted C 2 _ioalkenyl, optionally substituted C 2 _ioalkynyl, optionally substituted C 3 _7 carbocyclyl, optionally substituted 3-10 membered heterocyclyl, optionally substituted C6-ioaryl, optionally substituted 5-10 membered heteroaryl, optionally substituted (C 3 _ 7 carbocyclyl)Ci_ 6 alkyl, optionally substituted (3-10 membered heterocyclyl)Ci_ 6 alkyl, optionally substituted (C6-ioaryl)
- R 15 is optionally substituted Ci- 6 alkyl
- Y is selected from the group consisting of -0-, -S-, and -NR -;
- Y is selected from the group consisting of -OH, -SH, and -NHR ;
- Y 4 is selected from the group consisting of -OH, optionally substituted Cj_ 6
- Y 5 is selected from the group consisting of -S-, -S(O)-, -S(0) 2 -, -0-, -CR f R g -, and -NR g -, or Y 5 is absent;
- Y 6 is selected from the group consisting of -S-, -S(O)-, -S(0) 2 -, -0-, -CR f R g -, and
- each p and q is independently 0 or 1.
- Y 1 is N or CR 4 ;
- n is an integer of 0 or 1 ;
- r is an integer of 0 or 1 ;
- R and R together with the atoms to which they are attached form a fused ring or ring system selected from the group consisting of C3_ 7 carbocyclyl, 3-10 membered heterocyclyl, Ce-w aryl, and 5-10 membered heteroaryl, each optionally substituted with one or more R 5 , and
- each of R 1 , R 4 , R a , and R b is independently selected from the group consisting of H, amino, halogen, cyano, hydroxy, optionally substituted Ci_ 6 alkyl, optionally substituted Ci_ 6 haloalkyl, optionally substituted Ci_ 6 alkoxy, optionally substituted Ci_ 6 haloalkoxy, optionally substituted (Ci_ 6 alkoxy)Ci_ 6 alkyl, optionally substituted C 2 _ l oalkenyl, optionally substituted C2-ioalkynyl, optionally substituted C3-7 carbocyclyl, optionally substituted 3-10 membered heterocyclyl, optionally substituted C6-ioaryl, optionally substituted 5-10 membered heteroaryl, optionally substituted (3 ⁇ 4_ 7 carbocyclyl)Ci_ 6 alkyl, optionally substituted (3-10 membered heterocyclyl)Ci_ 6 alkyl, optionally substituted (C6-ioaryl
- R and R together with the atoms to which they are attached form a spirocyclic ring or ring system selected from the group consisting of C3_ 7 carbocyclyl and 3-10 membered heterocyclyl, each optionally substituted with one or more R 5 , and
- each of R 1 , R 2 , R a , and R b is independently selected from the group consisting of H, amino, halogen, cyano, hydroxy, optionally substituted Ci_ 6 alkyl, optionally substituted Ci_ 6 haloalkyl, optionally substituted Ci_ 6 alkoxy, optionally substituted Ci_ 6 haloalkoxy, optionally substituted (Ci-6 alkoxy)Ci-6 alkyl, optionally substituted C 2 - joalkenyl, optionally substituted C 2 _ioalkynyl, optionally substituted C 3 _7 carbocyclyl, optionally substituted 3-10 membered heterocyclyl, optionally substituted C6-ioaryl, optionally substituted 5-10 membered heteroaryl, optionally substituted (C 3- 7 carbocyclyl)Ci_ 6 alkyl, optionally substituted (3-10 membered heterocyclyl)Ci_ 6 alkyl, optionally substituted (C6-ioaryl)
- R 1 and R 2 together with the atoms to which they are attached form a spirocyclic ring or ring system selected from the group consisting of C 3 _ 7 carbocyclyl and 3-10 membered heterocyclyl, each optionally substituted with one or more R 5 , and
- each of R 3 , R 4 , R a , and R b is independently selected from the group consisting of H, amino, halogen, cyano, hydroxy, optionally substituted Ci- 6 alkyl, optionally substituted Ci_ 6 haloalkyl, optionally substituted Ci_ 6 alkoxy, optionally substituted Ci_ 6 haloalkoxy, optionally substituted (Ci_ 6 alkoxy)Ci_ 6 alkyl, optionally substituted C 2 _ joalkenyl, optionally substituted C 2 _ioalkynyl, optionally substituted C 3 _7 carbocyclyl, optionally substituted 3-10 membered heterocyclyl, optionally substituted C6-ioaryl, optionally substituted 5-10 membered heteroaryl, optionally substituted (C 3 _ 7 carbocyclyl)Ci- 6 alkyl, optionally substituted (3-10 membered heterocyclyl)Ci- 6 alkyl, optionally substituted (C6-i
- R a and R together with the atoms to which they are attached form a spirocyclic ring or ring system selected from the group consisting of C 3 _7 carbocyclyl and 3-10 membered heterocyclyl, each optionally substituted with one or more R 5 , and
- each of R 1 , R 2 , R 3 , and R 4 is independently selected from the group consisting of H, amino, halogen, cyano, hydroxy, optionally substituted Ci_ 6 alkyl, optionally substituted Ci_ 6 haloalkyl, optionally substituted Ci_ 6 alkoxy, optionally substituted Ci_ 6 haloalkoxy, optionally substituted (Ci_ 6 alkoxy)Ci_ 6 alkyl, optionally substituted C 2 _ joalkenyl, optionally substituted C 2 _ioalkynyl, optionally substituted C 3 _7 carbocyclyl, optionally substituted 3-10 membered heterocyclyl, optionally substituted C6-ioaryl, optionally substituted 5-10 membered heteroaryl, optionally substituted (C 3 _ 7 carbocyclyl)Ci- 6 alkyl, optionally substituted (3-10 membered heterocyclyl)Ci- 6 alkyl, optionally substituted (C6-ioary
- R a and R 4 together with the atoms to which they are attached form a fused ring or ring system selected from the group consisting of C 3 _7 carbocyclyl, 3-10 membered heterocyclyl, Ce-w aryl, and 5-10 membered heteroaryl, each optionally substituted with one or more R 5 , and
- each of R 1 , R 2 , R 3 , and R b is independently selected from the group consisting of H, amino, halogen, cyano, hydroxy, optionally substituted Ci- 6 alkyl, optionally substituted Ci_ 6 haloalkyl, optionally substituted Ci_ 6 alkoxy, optionally substituted Ci_ 6 haloalkoxy, optionally substituted (Ci_ 6 alkoxy)Ci_ 6 alkyl, optionally substituted C 2 _ joalkenyl, optionally substituted C 2 _ioalkynyl, optionally substituted C 3 _7 carbocyclyl, optionally substituted 3-10 membered heterocyclyl, optionally substituted C6-ioaryl, optionally substituted 5-10 membered heteroaryl, optionally substituted (C 3 _ 7 carbocyclyl)Ci- 6 alkyl, optionally substituted (3-10 membered heterocyclyl)Ci- 6 alkyl, optionally substituted (C6-io
- R 5 is -Y 5 -(CH 2 ) t -G;
- t is an integer of 0 or 1 ;
- G is selected from the group consisting of H, amino, halogen, cyano, hydroxy, optionally substituted Ci_ 6 alkyl, optionally substituted Ci_ 6 haloalkyl, optionally substituted Cj_ 6 alkoxy, optionally substituted Cj_ 6 haloalkoxy, optionally substituted (Cj_ 6 alkoxy)Ci_ 6 alkyl, optionally substituted C 2 _ioalkenyl, optionally substituted C 2 _ l oalkynyl, optionally substituted C 3 _7 carbocyclyl, optionally substituted 3- 10 membered heterocyclyl, optionally substituted C6-ioaryl, optionally substituted 5-10 membered heteroaryl, optionally substituted (C 3 _ 7 carbocyclyl)Ci_ 6 alkyl, optionally substituted (3-10 membered heterocyclyl)Ci_ 6 alkyl, optionally substituted (C6-ioaryl)Ci_ 6 alkyl, (C 6-
- R 6 is selected from the group consisting of optionally substituted - (CH 2 )nC(0)OR and a carboxylic acid isostere;
- n is an integer selected from 0 to 6;
- R is selected from the group consisting of H, d_ 9 alkyl, -CR 10 R 11 OC(O)C 1 _ 9 alkyl, -CR 10 R n OC(O)C 3 - 7 carbocyclyl, -CR 10 R n OC(O)(3 to 7 membered heterocyclyl), -CR 10 R n OC(O)C 2 _ 8 alkoxyalkyl, -CR 10 R 11 OC(O)OC 1 _
- R 7 is selected from the group consisting of -OH, optionally substituted Ci_ 6 alkoxy, amino, and -N(OR 8 )R 9 ;
- each R 8 and R 9 is independently selected from the group consisting of H, halogen, optionally substituted Ci-4alkyl, optionally substituted C 3 _7 carbocyclyl, optionally substituted 3-10 membered heterocyclyl, optionally substituted C6-ioaryl, and optionally substituted 5-10 membered heteroaryl;
- each R 10 and R 11 is independently selected from the group consisting of H, optionally substituted Ci-4alkyl, optionally substituted C 3 _7 carbocyclyl, optionally substituted 3-10 membered heterocyclyl, optionally substituted C6-ioaryl, and optionally substituted 5-10 membered heteroaryl;
- each R 13 and R 14 is independently selected from the group consisting of H, optionally substituted Ci- 6 alkyl, optionally substituted C 3 _7 carbocyclyl, optionally substituted 3-10 membered heterocyclyl, optionally substituted C6-ioaryl, and optionally substituted 5-10 membered heteroaryl;
- R 15 is optionally substituted Ci- 6 alkyl
- Y is selected from the group consisting of -0-, -S-, and -NR -;
- Y 5 is selected from the group consisting of -S-, -S(O)-, -S(0) 2 -, -0-, -CR f R g -, and -NR g -, or Y 5 is absent;
- each R c , R d , R e , R f , and R g are independently selected from the group consisting of H, halogen, optionally substituted Ci-4alkyl, optionally substituted carbocyclyl, optionally substituted 3-10 membered heterocyclyl, optionally substituted C6-ioaryl, and optionally substituted 5-10 membered heteroaryl; and
- each R h and R 1 is independently selected from the group consisting of H, halogen, cyano, amino, C-amido, N-amido, optionally substituted Ci-4alkyl, optionally substituted C3_7 carbocyclyl, optionally substituted 3-10 membered heterocyclyl, optionally substituted C6-ioaryl, and optionally substituted 5-10 membered heteroaryl; or R h and R 1 together with the atoms to which they are attached form a spirocyclic ring or ring system selected from the group consisting of carbocyclyl, 3-10 membered heterocyclyl, C - io aryl, and 5-10 membered heteroaryl, each optionally substituted with one or more R 5 .
- r is an integer of 0 or 1 ;
- each of R and R is independently selected from the group consisting of H, amino, halogen, cyano, hydroxy, optionally substituted Ci- 6 alkyl, optionally substituted Ci_ 6 haloalkyl, optionally substituted Ci_ 6 alkoxy, optionally substituted Ci_ 6 haloalkoxy, optionally substituted (Ci_ 6 alkoxy)Ci_ 6 alkyl, optionally substituted C2-ioalkenyl, optionally substituted C2-ioalkynyl, optionally substituted C3-7 carbocyclyl, optionally substituted 3-10 membered heterocyclyl, optionally substituted C6-ioaryl, optionally substituted 5-10 membered heteroaryl, optionally substituted (C3- 7 carbocyclyl)Ci- 6 alkyl, optionally substituted (3-10 membered heterocyclyl)Ci_ 6 alkyl, optionally substituted (C6-ioaryl)Ci_ 6 aikyl, (C 6- ioary
- n is an integer of 0 or 1 ;
- each R a and R b is independently selected from the group consisting of H, amino, halogen, cyano, hydroxy, optionally substituted Cj_ 6 alkyl, optionally substituted Ci- 6 haloalkyl, optionally substituted Ci- 6 alkoxy, optionally substituted Ci_ 6 haloalkoxy, optionally substituted (Ci_ 6 alkoxy)Ci_ 6 alkyl, optionally substituted C 2 _ioalkenyl, optionally substituted C 2 _ioalkynyl, optionally substituted carbocyclyl, optionally substituted 3-10 membered heterocyclyl, optionally substituted C6-ioaryl, optionally substituted 5-10 membered heteroaryl, optionally substituted (C 3 _ 7 carbocyclyl)C 1 _ 6 aikyl, optionally substituted (3-10 membered heterocyclyl)Ci- 6 alkyl, optionally substituted (C6-ioaryl)Ci- 6 aiky
- R a and R b together with the atoms to which they are attached form a ring or ring system selected from the group consisting of C 3 _ 7 carbocyclyl, and 3-10 membered heterocyclyl, each optionally substituted with one or more R 5 ; or
- m 1 ;
- R a and R 3 together with the atoms to which they are attached form a ring or ring system selected from the group consisting of C 3 _ 7 carbocyclyl, and 3-10 membered heterocyclyl, each optionally substituted with one or more R 5 ; and each R 2 and R b is independently selected from the group consisting of H, amino, halogen, cyano, hydroxy, optionally substituted Ci_ 6 alkyl, optionally substituted Q_ 6 haloalkyl, optionally substituted Q_ 6 alkoxy, optionally substituted Ci_ 6 haloalkoxy, optionally substituted (Ci_ 6 alkoxy)Ci_ 6 alkyl, optionally substituted C 2 _ioalkenyl, optionally substituted C 2 _ioalkynyl, optionally substituted C 3 _7 carbocyclyl, optionally substituted 3-10 membered heterocyclyl, optionally substituted C6-ioaryl, optionally substituted 5-10 membered heteroary
- R 5 is -Y 5 -(CH 2 ) t -G;
- t is an integer of 0 or 1 ;
- G is selected from the group consisting of H, amino, halogen, cyano, hydroxy, optionally substituted Ci_ 6 alkyl, optionally substituted Ci_ 6 haloalkyl, optionally substituted Ci_ 6 alkoxy, optionally substituted Ci_ 6 haloalkoxy, optionally substituted (Ci_ 6 alkoxy)Ci_ 6 alkyl, optionally substituted C 2 _ioalkenyl, optionally substituted C 2 _ joalkynyl, optionally substituted C 3 _ 7 carbocyclyl, optionally substituted 3-10 membered heterocyclyl, optionally substituted C6-ioaryl, optionally substituted 5-10 membered heteroaryl, optionally substituted (C 3 _ 7 carbocyclyl)Ci_ 6 alkyl, optionally substituted (3-10 membered heterocyclyl)Ci_ 6 alkyl, optionally substituted (C6-ioaryl)Ci_ 6 alkyl, (C 6- ioary
- R 6 is selected from the group consisting of optionally substituted - (CH 2 )nC(0)OR and a carboxylic acid isostere;
- n is an integer selected from 0 to 6;
- R is selected from the group consisting of H, d_ 9 alkyl, -CR 10 R 11 OC(O)C 1 _ 9 alkyl, -CR 10 R n OC(O)C 3 _ 7 carbocyclyl, -CR 10 R n OC(O)(3 to 7 membered heterocyclyl), -CR 10 R n OC(O)C 2 _ 8 alkoxyalkyl, -CR 10 R 11 OC(O)OC 1 _
- R 7 is selected from the group consisting of -OH, optionally substituted Ci_ 6
- each R and R is independently selected from the group consisting of H, halogen, optionally substituted Ci-4alkyl, optionally substituted carbocyclyl, optionally substituted 3-10 membered heterocyclyl, optionally substituted C6-ioaryl, and optionally substituted 5-10 membered heteroaryl;
- each R 10 and R 11 is independently selected from the group consisting of H, optionally substituted optionally substituted Cj. 7 carbocyclyl, optionally substituted 3-10 membered heterocyclyl, optionally substituted C6-ioaryl, and optionally substituted 5-10 membered heteroaryl;
- each R 13 and R 14 is independently selected from the group consisting of H, optionally substituted Ci- 6 alkyl, optionally substituted carbocyclyl, optionally substituted 3-10 membered heterocyclyl, optionally substituted C6-ioaryl, and optionally substituted 5-10 membered heteroaryl;
- R 15 is optionally substituted Ci- 6 alkyl
- Y is selected from the group consisting of -0-, -S-, and -NR -;
- Y 5 is selected from the group consisting of -S-, -S(O)-, -S(0) 2 -, -0-, -CR f R g -, and -NR g -, or Y 5 is absent;
- each R c , R d , R e , R f , and R g are independently selected from the group consisting of H, halogen, optionally substituted Ci-4alkyl, optionally substituted carbocyclyl, optionally substituted 3-10 membered heterocyclyl, optionally substituted C6-ioaryl, and optionally substituted 5-10 membered heteroaryl; and
- each R h and R 1 is independently selected from the group consisting of H, halogen, cyano, amino, C-amido, N-amido, optionally substituted Ci-4alkyl, optionally substituted C3_7 carbocyclyl, optionally substituted 3-10 membered heterocyclyl, optionally substituted C6-ioaryl, and optionally substituted 5-10 membered heteroaryl; or R h and R 1 together with the atoms to which they are attached form a spirocyclic ring or ring system selected from the group consisting of carbocyclyl, 3-10 membered heterocyclyl, C - io aryl, and 5-10 membered heteroaryl, each optionally substituted with one or more R 5 .
- compositions comprising a therapeutically effective amount of a compound having the structure of Formula I, II, III, IV, V or VI, as described herein, or pharmaceutically acceptable salts thereof, and a pharmaceutically acceptable excipient.
- the pharmaceutical composition may further comprise an additional medicament.
- Some additional embodiments described herein relate to methods of treating a bacterial infection comprising administering a compound having the structure of Formula I, II, III, IV, V or VI as described herein, or pharmaceutically acceptable salts thereof to a subject in need thereof.
- the method further comprises administering to the subject an additional medicament, for example, the additional medicament may be selected from an antibacterial agent, an antifungal agent, an antiviral agent, an anti-inflammatory agent, or an antiallergic agent.
- compounds that contain a boronic acid moiety that act as antimicrobial agents and/or as potentiators of antimicrobial agents.
- Various embodiments of these compounds include compounds having the structures of Formula I or II as described above or pharmaceutically acceptable salts thereof. In some embodiments of the
- the compounds of Formula I or II are also represented by the structure of Formula la or Ila, or pharmaceutically acceptable salts thereof:
- the compounds of Formula la or Ila are also represented by the structure of Formula lb or lib, or pharmaceutically acceptable salts thereof:
- m is 0 and the compounds of Formula lb or lib are also represented by the structure of Formula Ic or lie, or pharmaceutically acceptable salts thereof:
- the compounds of Formula Ic or lie are in various stereoisomeric form, including those represented by the structure of Formula Ic-1, Ic-2, IIc-1 or IIc-2,
- R 2 and R 3 together with the atoms to which they are attached form a ring or ring system selected from the group consisting of C3_ 7 carbocyclyl, 3-10 membered heterocyclyl, C6-10 aryl, and 5-10 membered heteroaryl, each optionally substituted with one or more R 5 .
- R and R together with the atoms to which they are attached form C3_ 7 carbocyclyl optionally substituted with one or more R 5 .
- R and R together with the atoms to which they are attached form cyclopropyl, bicyclo[2.2.1]heptyl, bicyclo[2.2.1]heptenyl, tetrahydrofuranyl, or dihydrofuranyl, each optionally substituted with one or more R 5 .
- the compound of Formula Ic or lie is also represented by the structure of Formula Id or lid, or pharmaceutically
- i is
- R 5 substituted with one R 5 .
- R 5 is substituted with two R 5 .
- the compounds of Formula Id or lid are in various stereoisomeric forms, including those represented by the structure of Formula Id-1 , Id-2, IId-1 or IId-2, or ph
- R 1 is hydrogen. In another embodiment, R 1 is an optionally substituted Ci- 6 alkyl, for example, Ci- 6 hydroxyalkyl.
- R 4 is hydrogen
- R 3 and R 4 together with the atoms to which they are attached form a spiro ring or ring system selected from the group consisting of C3_ 7 carbocyclyl, and 3-10 membered heterocyclyl, each optionally substituted with one or more R".
- R and R together with the atoms to which they are attached form C3_ 7 carbocyclyl optionally substituted with one or more R 5 .
- R 3 and R 4 together with the atoms to which they are attached form cyclopropyl optionally substituted with one or more R 5.
- R 3 and R 4 together with the atoms to which they are attached form 3-10 membered heterocyclyl optionally substituted with one or more R 5 , for example, 3, 4, 5, 6, or 7 membered heterocyclyl comprising one, two or three heteroatoms selected from the group consisting of oxygen, nitrogen or sulfur.
- R 1 is hydrogen.
- R 2 is hydrogen.
- R 6 is -C(0)OR.
- R is H or Q_9 alkyl.
- R is -CR 10 R n OC(O)Ci- 9 alkyl, -CR 10 R n OC(O)C 3 - 7 carbocyclyl, -CR 10 R n OC(O)(3 to 7 membered heterocyclyl), or - 10 R n OC
- the 3 to 7 membered heterocyclyl is
- R is -CR 10 R 11 OC(O)OC 1 _ 9 alkyl, -CR 10 R n OC(O)OC 3 - 7 carbocyclyl, -CR 10 R n OC(O)O(3 to 7 membered heterocyclyl), or -CR 10 R n OC(O)OC 2 - alkyl.
- heterocyclyl is 0 , , or .
- R is
- each of R 13 and R 14 is independently H or d_ 6 alkyl.
- R is -CR 10 R n OC(O)O(CH 2 )i- 3 C(0)NR 13 R 14 , -CR 10 R 11 OC(O)O(CH 2 )2-3OC(O)C 1 _4 alkyl, -CR 10 R 11 OC(O)(CH 2 ) 1 - 3 OC(O)C 1 alkyl, or -CR 10 R n OC(O)O(CH 2 )i-3C(O)OCi alkyl.
- each R 10 and R 11 is independently hydrogen or Ci_ 6 alkyl.
- R 7 is -OH.
- Y is -0-.
- Y 3 is -OH. In some embodiments, Y 4 is -OH.
- each J, L and M is CR 12.
- R 12 is hydrogen, halogen, Ci_6 alkoxy, or Ci_6 haloalkoxy.
- at least one of J, L and M of Formula lb, Ic, Id, lib, lie, or lid is N (nitrogen).
- M is nitrogen.
- the pharmaceutically acceptable salts are selected from alkaline metal salts or ammonium salts.
- the pharmaceutically acceptable salts are sodium salts with the structures selected from the group consisting of:
- compounds that contain a boronic acid moiety that act as antimicrobial agents and/or as potentiators of antimicrobial agents.
- Various embodiments of these compounds include compounds having the structures of Formula III or IV as described above or pharmaceutically acceptable salts thereof.
- R is selected from H, Ci -9 alkyl, -CR 10 R n OC( , -
- the compounds of Formula III or IV are also represented by the structure of Formula Ilia or IVa, or pharmaceutically acceptable salts thereof:
- M is independently selected from CR or N (nitrogen).
- m is 0 and the compounds of Formula Ilia or IVa are also represented by the structure of Formula Illb or IVb, or pharmaceutically acceptable salts thereof:
- R is selected from H, halogen, or Ci_ 6 alkyl.
- R is hydrogen
- R 2 and R 3 together with the atoms to which they are attached form a fused ring or ring system selected from the group consisting of C3_ 7 carbocyclyl, 3-10 membered heterocyclyl, C6- 10 aryl, and 5-10 membered heteroaryl, each optionally substituted with one or more R 5.
- R 2 and R 3 together with the atoms to which they are attached form C3- 7 carbocyclyl optionally substituted with one or more R 3 .
- R 2 and R 3 together with the atoms to which they are attached form cyclopropyl optionally substituted with one or more R 5 .
- R 6 is -C(0)OR.
- R is H or C 1 -9 alkyl.
- R is -CR lu R 1 OC(0)C 1 _ 9 alkyl, -CR lu R u OC(0)C 3-
- the 3 to 7 membered heterocyclyl is or some further embodiments, R is -CR 1U R 1 OC(0)0C 1
- R is
- each of R and R is independently H or d_ 6 alkyl.
- R is -CR 10 R n OC(O)O(CH 2 )i- 3 C(0)NR 13 R 14 , -CR 10 R 11 OC(O)O(CH 2 )2-3OC(O)C 1 _4 alkyl, -CR 10 R 11 OC(O)(CH 2 ) 1 - 3 OC(O)C 1 alkyl, or -CR 10 R 11 OC(O)O(CH 2 )i- 3 C(O)OC 1 alkyl.
- each R 10 and R 11 is independently hydrogen or Ci_ 6 alkyl.
- R is -OH.
- Y is -0-.
- Y 3 is -OH. In some embodiments, Y 4 is -OH.
- each J, L and M is CR 12.
- R 12 is selected from hydrogen, halogen or Ci_ 6 alkoxy.
- at least one of J, L and M is N (nitrogen).
- M is N.
- the pharmaceutically acceptable salts are selected from alkaline metal salts or ammonium salts. In one embodiment, the pharmaceutically acceptable salts are sodium salts.
- compounds that contain a boronic acid moiety that act as antimicrobial agents and/or as potentiators of antimicrobial agents.
- Various embodiments of these compounds include compounds having the structures of Formula V as described above or pharmaceutically acceptable salts thereof.
- R is selected from H, C 1 - 9 alkyl, -CR 10 R n OC(O -
- the compounds of Formula V are also represented by the structure of Formula Va, or pharmaceutically acceptable salts thereof:
- m is 1 and the compounds of Formula Va represented by the structure of Formula Vb, or pharmaceutically acceptable salts thereof:
- both R a and R b are H.
- R 2 and R 3 together with the atoms to which they are attached form a fused ring or ring system selected from the group consisting of carbocyclyl, 3-10 membered heterocyclyl, C6-10 aryl, and 5-10 membered heteroaryl, each optionally substituted with one or more R 5.
- R 2 and R 3 together with the atoms to which they are attached form C3_ 7 carbocyclyl optionally substituted with one or more R 5 .
- R and R together with the atoms to which they are attached form cyclopropyl optionally substituted with one or more R 5 .
- r is 1
- both R h and R 1 are H.
- R 6 is -(CH 2 )nC(0)OR and n is 0.
- the compound is also represented by the structure of Formula Vc or pharmaceutically acceptable salts thereof:
- the compounds of Formula Vc are in various stereoisomeric forms, including those represented by the structure of Formula Vc-1 or Vc-2, or pharmaceutically acceptable salts thereof:
- R 7 is -OH.
- Y is -0-.
- compounds that contain a boronic acid moiety that act as antimicrobial agents and/or as potentiators of antimicrobial agents.
- Various embodiments of these compounds include compounds having the structures of Formula VI as described above or pharmaceutically acceptable salts thereof.
- R is selected from H, d_ 9 alkyl, -CR 10 R 11 OC(O)C 1 _ 9 alkyl, -
- m is 1 and the compounds of Formula VI are also represented by the structure of Formula Via, or pharmaceutically acceptable salts thereof:
- both R a and R b are H.
- r is 1, and both R h and R l are H.
- R 6 is -(CH 2 )nC(0)OR and n is 0.
- the compound is also represented by the structure of Formula VIb or pharmaceutically acceptable salts thereof:
- R 7 is -OH.
- Y is -0-.
- the pharmaceutically acceptable salts are selected from alkaline metal salts or ammonium salts. In one embodiment, the pharmaceutically acceptable salts are sodium salts, including disodium salts.
- the compounds disclosed herein may exist as individual enantiomers and diastereomers or as mixtures of such isomers, including racemates. Separation of the individual isomers or selective synthesis of the individual isomers is accomplished by application of various methods which are well known to practitioners in the art. Unless otherwise indicated, all such isomers and mixtures thereof are included in the scope of the compounds disclosed herein. Furthermore, compounds disclosed herein may exist in one or more crystalline or amorphous forms. Unless otherwise indicated, all such forms are included in the scope of the compounds disclosed herein including any polymorphic forms. In addition, some of the compounds disclosed herein may form solvates with water (i.e., hydrates) or common organic solvents. Unless otherwise indicated, such solvates are included in the scope of the compounds disclosed herein.
- the compounds described herein may convert to or exist in equilibrium with alternate forms. Accordingly, in some embodiments, the compounds described herein may exist in combination with one or more of these forms. For example, as shown below, the compounds disclosed herein may exist in cyclic boronate monoesters with the structure of Formulae I, la, lb, Ic, and Id or in acyclic form as boronic acids with the structure of Formulae II, Ila, lib, lie, lid, or may exist as a mixture of the two forms depending on the medium.
- the compounds disclosed herein may exist in cyclic form as cyclic boronate monoesters with the structure of Formulae III, Ilia, and Illb or in acyclic form as boronic acids with the structure of Formulae IV, IVa and IVb, or may exist as a mixture of the two forms depending on the medium.
- the compounds described herein may exist in cyclic dimeric form, trimeric form or tetrameric form.
- the compound of Formula II may exist in dimeric form (II-A), trimeric form (II-B), or tetrameric form (II-C):
- C a to Q or “C a _ b " in which "a” and “b” are integers refer to the number of carbon atoms in the specified group. That is, the group can contain from “a” to "b", inclusive, carbon atoms.
- a “Ci to C4 alkyl” or “Ci_4 alkyl” group refers to all alkyl groups having from 1 to 4 carbons, that is, CH 3 -, CH 3 CH2-, CH 3 CH2CH2-, (CH 3 ) 2 CH-, CH 3 CH2CH2CH2-, CH 3 CH 2 CH(CH 3 )- and (CH 3 ) 3 C-.
- halogen or "halo,” as used herein, means any one of the radio- stable atoms of column 7 of the Periodic Table of the Elements, e.g., fluorine, chlorine, bromine, or iodine, with fluorine and chlorine being preferred.
- alkyl refers to a straight or branched hydrocarbon chain that is fully saturated (i.e., contains no double or triple bonds).
- the alkyl group may have 1 to 20 carbon atoms (whenever it appears herein, a numerical range such as “1 to 20” refers to each integer in the given range; e.g., "1 to 20 carbon atoms” means that the alkyl group may consist of 1 carbon atom, 2 carbon atoms, 3 carbon atoms, etc., up to and including 20 carbon atoms, although the present definition also covers the occurrence of the term "alkyl” where no numerical range is designated).
- the alkyl group may also be a medium size alkyl having 1 to 9 carbon atoms.
- the alkyl group could also be a lower alkyl having 1 to 4 carbon atoms.
- the alkyl group may be designated as "C1-4 alkyl” or similar designations.
- Q_4 alkyl indicates that there are one to four carbon atoms in the alkyl chain, i.e., the alkyl chain is selected from the group consisting of methyl, ethyl, propyl, iso-propyl, n-butyl, iso- butyl, sec-butyl, and t-butyl.
- Typical alkyl groups include, but are in no way limited to, methyl, ethyl, propyl, isopropyl, butyl, isobutyl, tertiary butyl, pentyl, hexyl, and the like.
- alkoxy refers to the formula -OR wherein R is an alkyl as is defined above, such as "Ci_ alkoxy”, including but not limited to methoxy, ethoxy, n- propoxy, 1-methylethoxy (isopropoxy), n-butoxy, iso-butoxy, sec-butoxy, and tert-butoxy, and the like.
- alkylthio refers to the formula -SR wherein R is an alkyl as is defined above, such as "Ci_ alkylthio” and the like, including but not limited to methylmercapto, ethylmercapto, n-propylmercapto, 1-methylethylmercapto (isopropylmercapto), n-butylmercapto, iso-butylmercapto, sec-butylmercapto, tert- butylmercapto, and the like.
- alkenyl refers to a straight or branched hydrocarbon chain containing one or more double bonds.
- the alkenyl group may have 2 to 20 carbon atoms, although the present definition also covers the occurrence of the term "alkenyl” where no numerical range is designated.
- the alkenyl group may also be a medium size alkenyl having 2 to 9 carbon atoms.
- the alkenyl group could also be a lower alkenyl having 2 to 4 carbon atoms.
- the alkenyl group may be designated as "C2-4 alkenyl" or similar designations.
- C2-4 alkenyl indicates that there are two to four carbon atoms in the alkenyl chain, i.e., the alkenyl chain is selected from the group consisting of ethenyl, propen-l-yl, propen-2-yl, propen-3-yl, buten-l-yl, buten-2-yl, buten-3-yl, buten-4-yl, 1-methyl-propen-l-yl, 2-methyl-propen-l-yl, 1-ethyl-ethen-l-yl, 2-methyl-propen-3-yl, buta-l,3-dienyl, buta-1,2,- dienyl, and buta-l,2-dien-4-yl.
- Typical alkenyl groups include, but are in no way limited to, ethenyl, propenyl, butenyl, pentenyl, and hexenyl, and the like.
- alkynyl refers to a straight or branched hydrocarbon chain containing one or more triple bonds.
- the alkynyl group may have 2 to 20 carbon atoms, although the present definition also covers the occurrence of the term "alkynyl” where no numerical range is designated.
- the alkynyl group may also be a medium size alkynyl having 2 to 9 carbon atoms.
- the alkynyl group could also be a lower alkynyl having 2 to 4 carbon atoms.
- the alkynyl group may be designated as "C2-4 alkynyl" or similar designations.
- C2-4 alkynyl indicates that there are two to four carbon atoms in the alkynyl chain, i.e., the alkynyl chain is selected from the group consisting of ethynyl, propyn-l-yl, propyn-2-yl, butyn-l-yl, butyn-3-yl, butyn-4-yl, and 2-butynyl.
- Typical alkynyl groups include, but are in no way limited to, ethynyl, propynyl, butynyl, pentynyl, and hexynyl, and the like.
- heteroalkyl refers to a straight or branched hydrocarbon chain containing one or more heteroatoms, that is, an element other than carbon, including but not limited to, nitrogen, oxygen and sulfur, in the chain backbone.
- the heteroalkyl group may have 1 to 20 carbon atom, although the present definition also covers the occurrence of the term "heteroalkyl” where no numerical range is designated.
- the heteroalkyl group may also be a medium size heteroalkyl having 1 to 9 carbon atoms.
- the heteroalkyl group could also be a lower heteroalkyl having 1 to 4 carbon atoms.
- the heteroalkyl group may be designated as "Q-4 heteroalkyl" or similar designations.
- the heteroalkyl group may contain one or more heteroatoms.
- C 1 -4 heteroalkyl indicates that there are one to four carbon atoms in the heteroalkyl chain and additionally one or more heteroatoms in the backbone of the chain.
- alkylene means a branched, or straight chain fully saturated di-radical chemical group containing only carbon and hydrogen that is attached to the rest of the molecule via two points of attachment (i.e., an alkanediyl).
- the alkylene group may have 1 to 20 carbon atoms, although the present definition also covers the occurrence of the term alkylene where no numerical range is designated.
- the alkylene group may also be a medium size alkylene having 1 to 9 carbon atoms.
- the alkylene group could also be a lower alkylene having 1 to 4 carbon atoms.
- the alkylene group may be designated as "Ci_4 alkylene" or similar designations.
- Q ⁇ alkylene indicates that there are one to four carbon atoms in the alkylene chain, i.e., the alkylene chain is selected from the group consisting of methylene, ethylene, ethan-l,l-diyl, propylene, propan-l,l-diyl, propan-2,2-diyl, 1-methyl- ethylene, butylene, butan-l,l-diyl, butan-2,2-diyl, 2-methyl-propan-l,l-diyl, 1-methyl- propylene, 2-methyl-propylene, 1,1-dimethyl-ethylene, 1 ,2-dimethyl-ethylene, and 1-ethyl- ethylene.
- alkenylene means a straight or branched chain di-radical chemical group containing only carbon and hydrogen and containing at least one carbon-carbon double bond that is attached to the rest of the molecule via two points of attachment.
- the alkenylene group may have 2 to 20 carbon atoms, although the present definition also covers the occurrence of the term alkenylene where no numerical range is designated.
- the alkenylene group may also be a medium size alkenylene having 2 to 9 carbon atoms.
- the alkenylene group could also be a lower alkenylene having 2 to 4 carbon atoms.
- the alkenylene group may be designated as "C2-4 alkenylene" or similar designations.
- C2-4 alkenylene indicates that there are two to four carbon atoms in the alkenylene chain, i.e., the alkenylene chain is selected from the group consisting of ethenylene, ethen-l,l-diyl, propenylene, propen-l,l-diyl, prop-2-en-l,l-diyl, 1-methyl-ethenylene, but-l-enylene, but-2- enylene, but-l,3-dienylene, buten-l,l-diyl, but-l,3-dien-l,l-diyl, but-2-en-l,l-diyl, but-3-en- 1,1-diyl, l-methyl-prop-2-en-l,l-diyl, 2-methyl-prop-2-en-l,l-diyl, 1-ethyl-ethenylene, 1,2- dimethyl-ethenylene
- aromatic refers to a ring or ring system having a conjugated pi electron system and includes both carbocyclic aromatic (e.g., phenyl) and heterocyclic aromatic groups (e.g., pyridine).
- carbocyclic aromatic e.g., phenyl
- heterocyclic aromatic groups e.g., pyridine
- the term includes monocyclic or fused-ring polycyclic (i.e., rings which share adjacent pairs of atoms) groups provided that the entire ring system is aromatic.
- aryl refers to an aromatic ring or ring system (i.e., two or more fused rings that share two adjacent carbon atoms) containing only carbon in the ring backbone. When the aryl is a ring system, every ring in the system is aromatic.
- the aryl group may have 6 to 18 carbon atoms, although the present definition also covers the occurrence of the term "aryl” where no numerical range is designated. In some embodiments, the aryl group has 6 to 10 carbon atoms.
- the aryl group may be designated as "C6-io aryl,” “C6 or Cio aryl,” or similar designations. Examples of aryl groups include, but are not limited to, phenyl, naphthyl, azulenyl, and anthracenyl.
- aryloxy and arylthio refers to RO- and RS-, in which R is an aryl as is defined above, such as “C6-io aryloxy” or “C6-io arylthio” and the like, including but not limited to phenyloxy.
- an "aralkyl” or “arylalkyl” is an aryl group connected, as a substituent, via an alkylene group, such as "CT-U aralkyl” and the like, including but not limited to benzyl, 2- phenylethyl, 3-phenylpropyl, and naphthylalkyl.
- the alkylene group is a lower alkylene group (i.e., a Ci_4 alkylene group).
- heteroaryl refers to an aromatic ring or ring system (i.e., two or more fused rings that share two adjacent atoms) that contain(s) one or more heteroatoms, that is, an element other than carbon, including but not limited to, nitrogen, oxygen and sulfur, in the ring backbone.
- heteroaryl is a ring system, every ring in the system is aromatic.
- the heteroaryl group may have 5-18 ring members (i.e., the number of atoms making up the ring backbone, including carbon atoms and heteroatoms), although the present definition also covers the occurrence of the term "heteroaryl" where no numerical range is designated.
- the heteroaryl group has 5 to 10 ring members or 5 to 7 ring members.
- the heteroaryl group may be designated as "5-7 membered heteroaryl,” "5-10 membered heteroaryl,” or similar designations.
- heteroaryl rings include, but are not limited to, furyl, thienyl, phthalazinyl, pyrrolyl, oxazolyl, thiazolyl, imidazolyl, pyrazolyl, isoxazolyl, isothiazolyl, triazolyl, thiadiazolyl, pyridinyl, pyridazinyl, pyrimidinyl, pyrazinyl, triazinyl, quinolinyl, isoquinlinyl, benzimidazolyl, benzoxazolyl, benzothiazolyl, indolyl, isoindolyl, and benzothienyl.
- a “heteroaralkyl” or “heteroarylalkyl” is heteroaryl group connected, as a substituent, via an alkylene group. Examples include but are not limited to 2-thienylmethyl, 3- thienylmethyl, furylmethyl, thienylethyl, pyrrolylalkyl, pyridylalkyl, isoxazoUylalkyl, and imidazolylaikyl.
- the alkylene group is a lower alkylene group (i.e., a C 1 -4 alkylene group).
- carbocyclyl means a non-aromatic cyclic ring or ring system containing only carbon atoms in the ring system backbone.
- carbocyclyl is a ring system, two or more rings may be joined together in a fused, bridged or spiro-connected fashion.
- Carbocyclyls may have any degree of saturation provided that at least one ring in a ring system is not aromatic.
- carbocyclyls include cycloalkyls, cycloalkenyls, and cycloalkynyls.
- the carbocyclyl group may have 3 to 20 carbon atoms, although the present definition also covers the occurrence of the term "carbocyclyl” where no numerical range is designated.
- the carbocyclyl group may also be a medium size carbocyclyl having 3 to 10 carbon atoms.
- the carbocyclyl group could also be a carbocyclyl having 3 to 6 carbon atoms.
- the carbocyclyl group may be designated as "C3_6 carbocyclyl" or similar designations.
- carbocyclyl rings include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cyclohexenyl, 2,3-dihydro-indene, bicycle[2.2.2]octanyl, adamantyl, and spiro[4.4]nonanyl.
- a "(carbocyclyl)alkyl” is a carbocyclyl group connected, as a substituent, via an alkylene group, such as "CMO (carbocyclyl)alkyl” and the like, including but not limited to, cyclopropylmethyl, cyclobutylmethyl, cyclopropylethyl, cyclopropylbutyl, cyclobutylethyl, cyclopropylisopropyl, cyclopentylmethyl, cyclopentylethyl, cyclohexylmethyl, cyclohexylethyl, cycloheptylmethyl, and the like.
- the alkylene group is a lower alkylene group.
- cycloalkyl means a fully saturated carbocyclyl ring or ring system. Examples include cyclopropyl, cyclobutyl, cyclopentyl, and cyclohexyl.
- cycloalkenyl means a carbocyclyl ring or ring system having at least one double bond, wherein no ring in the ring system is aromatic.
- An example is cyclohexenyl.
- heterocyclyl means a non-aromatic cyclic ring or ring system containing at least one heteroatom in the ring backbone. Heterocyclyls may be joined together in a fused, bridged or spiro-connected fashion. Heterocyclyls may have any degree of saturation provided that at least one ring in the ring system is not aromatic. The heteroatom(s) may be present in either a non-aromatic or aromatic ring in the ring system.
- the heterocyclyl group may have 3 to 20 ring members (i.e., the number of atoms making up the ring backbone, including carbon atoms and heteroatoms), although the present definition also covers the occurrence of the term "heterocyclyl” where no numerical range is designated.
- the heterocyclyl group may also be a medium size heterocyclyl having 3 to 10 ring members.
- the heterocyclyl group could also be a heterocyclyl having 3 to 6 ring members.
- the heterocyclyl group may be designated as "3-6 membered heterocyclyl" or similar designations.
- the heteroatom(s) are selected from one up to three of O, N or S, and in preferred five membered monocyclic heterocyclyls, the heteroatom(s) are selected from one or two heteroatoms selected from O, N, or S.
- heterocyclyl rings include, but are not limited to, azepinyl, acridinyl, carbazolyl, cinnolinyl, dioxolanyl, imidazolinyl, imidazolidinyl, morpholinyl, oxiranyl, oxepanyl, thiepanyl, piperidinyl, piperazinyl, dioxopiperazinyl, pyrrolidinyl, pyrrolidonyl, pyrrolidionyl, 4-piperidonyl, pyrazolinyl, pyrazolidinyl, 1,3-dioxinyl, 1,3-dioxanyl, 1 ,4-dioxinyl, 1,4-dioxanyl, 1,3-oxathianyl, 1,4- oxathiinyl, 1,4-oxathianyl, 2H-l,2-oxazinyl, trioxanyl, a
- a "(heterocyclyl)alkyl” is a heterocyclyl group connected, as a substituent, via an alkylene group. Examples include, but are not limited to, imidazolinylmethyl and indolinylethyl.
- Non-limiting examples include formyl, acetyl, propanoyl, benzoyl, and acryl.
- a "cyano” group refers to a "-CN” group.
- a "cyanato” group refers to an "-OCN” group.
- An "isocyanato” group refers to a "-NCO” group.
- a "thiocyanato" group refers to a "-SCN” group.
- a “sulfonyl” group refers to an "-SO2 " group in which R is selected from hydrogen, optionally substituted Ci_6 alkyl, optionally substituted C2-6 alkenyl, optionally substituted C2-6 alkynyl, optionally substituted carbocyclyl, optionally substituted Ce-w aryl, optionally substituted 5- 10 membered heteroaryl, and optionally substituted 3- 10 membered heterocyclyl, as defined herein.
- S-sulfonamido refers to a "-S02NRA B" group in which RA and R B are each independently selected from hydrogen, halogen, optionally substituted Ci_6 alkyl, optionally substituted Ci_6 alkoxy, optionally substituted C2-6 alkenyl, optionally substituted C2-6 alkynyl, optionally substituted carbocyclyl, optionally substituted Ce-w aryl, optionally substituted 5- 10 membered heteroaryl, and optionally substituted 3- 10 membered heterocyclyl, as defined herein.
- RA and R B are each independently selected from hydrogen, halogen, optionally substituted Ci_6 alkyl, optionally substituted Ci_6 alkoxy, optionally substituted C2-6 alkenyl, optionally substituted C2-6 alkynyl, optionally substituted carbocyclyl, optionally substituted Ce-w aryl, optionally substituted 5- 10 membered heteroaryl, and optionally substituted 3- 10 membered heterocycly
- N-sulfonamido refers to a "-N(RA)S02RB” group in which RA and R B are each independently selected from hydrogen, halogen, optionally substituted Ci_6 alkyl, optionally substituted C2-6 alkenyl, optionally substituted C2-6 alkynyl, optionally substituted carbocyclyl, optionally substituted Ce-w aryl, optionally substituted 5- 10 membered heteroaryl, and optionally substituted 3- 10 membered heterocyclyl, as defined herein.
- An “amino” group refers to a "-NR A R B " group in which R A and R B are each independently selected from hydrogen, halogen, optionally substituted Cj_6 alkyl, optionally substituted C2-6 alkenyl, optionally substituted C2-6 alkynyl, optionally substituted C3-7 carbocyclyl, optionally substituted Ce-w aryl, optionally substituted 5- 10 membered heteroaryl, and optionally substituted 3- 10 membered heterocyclyl as defined herein.
- a non-limiting example includes free amino (i.e., -NH 2 ).
- aminoalkyl refers to an amino group connected via an alkylene group.
- alkoxyalkyl refers to an alkoxy group connected via an alkylene group, such as a “C2-8 alkoxyalkyl” and the like.
- a substituted group is derived from the unsubstituted parent group in which there has been an exchange of one or more hydrogen atoms for another atom or group. Unless otherwise indicated, when a group is deemed to be "substituted,” it is meant that the group is substituted with one or more substituents independently selected from C 1 -C6 alkyl, C 1 -C6 alkenyl, Q-C6 alkynyl, C 1 -C6 heteroalkyl, C3-C7 carbocyclyl (optionally substituted with halo, C 1 -C6 alkyl, C 1 -C6 alkoxy, C 1 -C6 haloalkyl, and C 1 -C6 haloalkoxy), C3-C7-carbocyclyl-Ci- C6-alkyl (optionally substituted with halo, Q-C6 alkyl, C 1 -C6 alkoxy, Q-C6 halo
- radical naming conventions can include either a mono-radical or a di-radical, depending on the context.
- a substituent requires two points of attachment to the rest of the molecule, it is understood that the substituent is a di-radical.
- a substituent identified as alkyl that requires two points of attachment includes di-radicals such as -CH 2 -, -CH 2 CH 2 -, -CH 2 CH(CH 3 )CH 2 -, and the like.
- Other radical naming conventions clearly indicate that the radical is a di-radical such as "alkylene” or "alkenylene.”
- R 1 and R2 are defined as selected from the group consisting of hydrogen and alkyl, or R 1 and R2 are defined as selected from the group consisting of hydrogen and alkyl, or R 1 and R2 are defined as selected from the group consisting of hydrogen and alkyl, or R 1 and R2 are defined as selected from the group consisting of hydrogen and alkyl, or R 1 and R2 are defined as selected from the group consisting of hydrogen and alkyl, or R 1 and R2 are defined as selected from the group consisting of hydrogen and alkyl, or R 1 and
- R 2 together with the nitrogen to which they are attached form a heterocyclyl, it is meant that R 1 and R can be selected from hydrogen or alkyl, or alternatively, the substructure has structure:
- ring A is a heteroaryl ring containing the depicted nitrogen.
- R 1 and R z are defined as selected from the group consisting of hydrogen and alkyl, or R 1 and R together with the atoms to which they are attached form an aryl or carbocylyl, it is meant that
- R and R" can be selected from hydrogen or alkyl, or alternatively, the substructure has structure:
- A is an aryl ring or a carbocylyl containing the depicted double bond.
- a substituent is depicted as a di-radical (i.e. , has two points of attachment to the rest of the molecule), it is to be understood that the substituent can be attached in any directional configuration unless otherwise indicated.
- a substituent depicted as -AE- or v 3 ⁇ 4 A ⁇ E A includes the substituent being oriented such that the A is attached at the leftmost attachment point of the molecule as well as the case in which A is attached at the rightmost attachment point of the molecule.
- isosteres of a chemical group are other chemical groups that exhibit the same or similar properties.
- tetrazole is an isostere of carboxylic acid because it mimics the properties of carboxylic acid even though they both have very different molecular formulae. Tetrazole is one of many possible isosteric replacements for carboxylic acid.
- carboxylic acid isosteres contemplated include -SO 3 H, -SO2HNR, -P(3 ⁇ 4(R)2, - P0 3 (R) 2 , -CONHNHSO2R, -COHNSO2R, and -CONRCN, where R is selected from hydrogen, Ci_6 alkyl, C2-6 alkenyl, C2-6 alkynyl, carbocyclyl, Ce-w aryl, 5-10 membered heteroaryl, and 3-10 membered heterocyclyl, as defined herein.
- carboxylic acid isosteres can include 5-7 membered carbocycles or heterocycles containing any combination of C3 ⁇ 4, O, S, or N in any chemically stable oxidation state, where any of the atoms of said ring structure are optionally substituted in one or more positions.
- the following structures are non-limiting examples of carbocyclic and heterocyclic isosteres contemplated.
- the atoms of said ring structure may be optionally substituted at one or more positions with R as defined above.
- the placement of one or more R substituents upon a carbocyclic or heterocyclic carboxylic acid isostere is not a substitution at one or more atom(s) that maintain(s) or is/are integral to the carboxylic acid isosteric properties of the compound, if such substituent(s) would destroy the carboxylic acid isosteric properties of the compound.
- Subject as used herein, means a human or a non-human mammal, e.g., a dog, a cat, a mouse, a rat, a cow, a sheep, a pig, a goat, a non-human primate or a bird, e.g., a chicken, as well as any other vertebrate or invertebrate.
- mammal is used in its usual biological sense. Thus, it specifically includes, but is not limited to, primates, including simians (chimpanzees, apes, monkeys) and humans, cattle, horses, sheep, goats, swine, rabbits, dogs, cats, rodents, rats, mice guinea pigs, or the like.
- primates including simians (chimpanzees, apes, monkeys) and humans, cattle, horses, sheep, goats, swine, rabbits, dogs, cats, rodents, rats, mice guinea pigs, or the like.
- pharmaceutically acceptable carrier or “pharmaceutically acceptable excipient” includes any and all solvents, dispersion media, coatings, antibacterial and antifungal agents, isotonic and absorption delaying agents and the like.
- the use of such media and agents for pharmaceutically active substances is well known in the art. Except insofar as any conventional media or agent is incompatible with the active ingredient, its use in the therapeutic compositions is contemplated.
- various adjuvants such as are commonly used in the art may be included. Considerations for the inclusion of various components in pharmaceutical compositions are described, e.g., in Gilman et al. (Eds.) (1990); Goodman and Gilman's: The Pharmacological Basis of Therapeutics, 8th Ed., Pergamon Press.
- a therapeutic effect relieves, to some extent, one or more of the symptoms of a disease or condition, and includes curing a disease or condition. "Curing” means that the symptoms of a disease or condition are eliminated; however, certain long-term or permanent effects may exist even after a cure is obtained (such as extensive tissue damage).
- Treatment refers to administering a compound or pharmaceutical composition to a subject for prophylactic and/or therapeutic purposes.
- prophylactic treatment refers to treating a subject who does not yet exhibit symptoms of a disease or condition, but who is susceptible to, or otherwise at risk of, a particular disease or condition, whereby the treatment reduces the likelihood that the patient will develop the disease or condition.
- therapeutic treatment refers to administering treatment to a subject already suffering from a disease or condition.
- the compounds disclosed herein may exist as individual enantiomers and diastereomers or as mixtures of such isomers, including racemates. Separation of the individual isomers or selective synthesis of the individual isomers is accomplished by application of various methods which are well known to practitioners in the art. Unless otherwise indicated, all such isomers and mixtures thereof are included in the scope of the compounds disclosed herein. Furthermore, compounds disclosed herein may exist in one or more crystalline or amorphous forms. Unless otherwise indicated, all such forms are included in the scope of the compounds disclosed herein including any polymorphic forms. In addition, some of the compounds disclosed herein may form solvates with water (i.e., hydrates) or common organic solvents. Unless otherwise indicated, such solvates are included in the scope of the compounds disclosed herein.
- Isotopes may be present in the compounds described. Each chemical element as represented in a compound structure may include any isotope of said element.
- a hydrogen atom may be explicitly disclosed or understood to be present in the compound.
- the hydrogen atom can be any isotope of hydrogen, including but not limited to hydrogen- 1 (protium) and hydrogen-2 (deuterium).
- reference herein to a compound encompasses all potential isotopic forms unless the context clearly dictates otherwise.
- a "prodrug” refers to an agent that is converted into the parent drug in vivo. Prodrugs are often useful because, in some situations, they may be easier to administer than the parent drug. They may, for instance, be bioavailable by oral administration whereas the parent is not. The prodrug may also have improved solubility in pharmaceutical compositions over the parent drug.
- An example, without limitation, of a prodrug would be a compound which is administered as an ester (the "prodrug") to facilitate transmittal across a cell membrane where water solubility is detrimental to mobility but which then is metabolically hydrolyzed to the carboxylic acid, the active entity, once inside the cell where water- solubility is beneficial.
- a further example of a prodrug might be a short peptide (polyaminoacid) bonded to an acid group where the peptide is metabolized to reveal the active moiety.
- a prodrug derivative Conventional procedures for the selection and preparation of suitable prodrug derivatives are described, for example, in Design of Prodrugs, (ed. H. Bundgaard, Elsevier, 1985), which is hereby incorporated herein by reference in its entirety.
- pro-drug ester refers to derivatives of the compounds disclosed herein formed by the addition of any of several ester-forming groups that are hydrolyzed under physiological conditions.
- pro-drug ester groups include pivoyloxymethyl, acetoxymethyl, phthalidyl, indanyl and methoxymethyl, as well as other such groups known in the art, including a (5-R-2-oxo-l,3-dioxolen-4-yl)methyl group.
- Other examples of pro-drug ester groups can be found in, for example, T. Higuchi and V. Stella, in "Pro-drugs as Novel Delivery Systems", Vol. 14, A.C.S.
- Methodabolites of the compounds disclosed herein include active species that are produced upon introduction of the compounds into the biological milieu.
- Solvate refers to the compound formed by the interaction of a solvent and a compound described herein, a metabolite, or salt thereof. Suitable solvates are pharmaceutically acceptable solvates including hydrates.
- pharmaceutically acceptable salt refers to salts that retain the biological effectiveness and properties of a compound, which are not biologically or otherwise undesirable for use in a pharmaceutical.
- the compounds herein are capable of forming acid and/or base salts by virtue of the presence of amino and/or carboxyl groups or groups similar thereto.
- Pharmaceutically acceptable acid addition salts can be formed with inorganic acids and organic acids. Inorganic acids from which salts can be derived include, for example, hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, and the like.
- Organic acids from which salts can be derived include, for example, acetic acid, propionic acid, glycolic acid, pyruvic acid, oxalic acid, maleic acid, malonic acid, succinic acid, fumaric acid, tartaric acid, citric acid, benzoic acid, cinnamic acid, mandelic acid, methanesulfonic acid, ethanesulfonic acid, p-toluenesulfonic acid, salicylic acid, and the like.
- Pharmaceutically acceptable base addition salts can be formed with inorganic and organic bases.
- Inorganic bases from which salts can be derived include, for example, sodium, potassium, lithium, ammonium, calcium, magnesium, iron, zinc, copper, manganese, aluminum, and the like; particularly preferred are the ammonium, potassium, sodium, calcium and magnesium salts.
- Organic bases from which salts can be derived include, for example, primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines, basic ion exchange resins, and the like, specifically such as isopropylamine, trimethylamine, diethylamine, triethylamine, tripropylamine, and ethanolamine.
- each of Z ® and R may be independently selected from an alkali metal cation or an ammodium cation (NH 4 + ).
- each of Z ® and R may be indpendently selected from an alkali metal cation or an ammodium cation (NH/ t + ).
- R may be indpendently an alkali metal cation or an ammodium cation (NH/ t + ).
- the compounds disclosed herein may be synthesized by methods described below, or by modification of these methods. Ways of modifying the methodology include, among others, temperature, solvent, reagents etc., known to those skilled in the art.
- Ways of modifying the methodology include, among others, temperature, solvent, reagents etc., known to those skilled in the art.
- it may be necessary and/or desirable to protect sensitive or reactive groups on any of the molecules concerned. This may be achieved by means of conventional protecting groups, such as those described in Protective Groups in Organic Chemistry (ed. J.F.W. McOmie, Plenum Press, 1973); and P.G.M. Green, T.W.
- protecting groups for oxygen atoms are selected for their compatibility with the requisite synthetic steps as well as compatibility of the introduction and deprotection steps with the overall synthetic schemes (P.G.M. Green, T.W. Wutts, Protecting Groups in Organic Synthesis (3rd ed.) Wiley, New York (1999)). Handling of protecting and/or sterodirecting groups specific to boronic acid derivatives is described in a recent review of chemistry of boronic acids: D.G. Hall (Ed.), Boronic Acids. Preparation and Application in Organic Synthesis and Medicine, Wiley VCH (2005) and in earlier reviews: Matteson, D. S. (1988). Asymmetric synthesis with boronic esters.
- Compounds of formula IX can be made starting from protected aryl or heteroaryl precursors of formula 1-1 via Z-vinyl boronate (1-3) followed by cyclopropanation and deprotection.
- the compounds of formula 1-3 may be attained from 1-2 (where X is halogen), which may be made by means of known methods of Z-haloalkene formation ⁇ Tetrahedron Lett., 2001, 42, 3893-3896) with conventional protecting groups for R', R", and R' ", such as those described in Protective Groups in Organic Chemistry (ed. J.F.W. McOmie, Plenum, 1973, which is incorporated herein by reference in its entirety); and Protecting Groups in Organic Synthesis P.G.M.
- Aryl compounds of formula 1-2 upon borylation by well-known available methods ⁇ Chem. Rev. 2010, 110, 890-931, which is incorporated herein by reference in its entirety) and boronate ester formation with desired chiral auxiliary give intermediates of formula 1-3.
- vinyl boronate derivative 1-3 can also be made via acetylene derivative of formula 1-5, which can be made from compounds of formula 1-1 by acetylene coupling such as in Sonogoshira reaction.
- Phenyl acetylene derivatives of formula 1-5 can be transformed into Z-vinylboronates (1-3) by ruthenium hydride pincer complex catalysed addition of pinacolborane to terminal alkynes. (/. Am. Chem. Soc , 2012, 134, 14349-14352).
- a Cu- catalysed Z-selective hydroboration of alkynes with 1,8-diaminonaphthaleneborane may also be utilized to make compounds of formula 1-3 from terminal alkynes (1-5) ⁇ Org. Lett , 2016, 18, 1390-1393).
- Terminal acetylenes of formula 1-5 can be selectively transformed under silver catalyzed hydroboration conditions to compounds of formula 1-6 (Tetrahedron, 2014, 70, 5815- 5819).
- Such alkynyl boronates of formula 1-6 can be reduced stereoselectively to the cw-alkenyl pinacolboronates (1-3) via hydroboration with dicyclohexylborane (/. Org. Chem. , 2008, 73, 6841-6844).
- Cyclopropanation of compounds of formula 1-3 to 1-4 may be attained by palladium or Zn mediated carbene additions (/. Am. Chem. Soc, 2015, 137, 13176-13182). Such transformations can also be done to give compounds of 1-4 in high enantioselectivity (Tetrahedron, 2008, 64, 7041-7095; Eur. J. Org. Chem. 2000, 2557-2562). Alternatively, dimethyloxosulfonium methylide also reacts with enones to undergo 1,4-addition followed by ring closure to give a cyclopropane derivatives (Tetrahedron Lett. , 2003, 44, 3629-3630).
- a phosphate carbenoid (RO) 2 P(0)OZnCH 2 I (/. Org. Chem. , 2010, 75, 1244-1250; Org. Process Res. Dev. , 2016, 20, 786-798) that can be stored may be utilized in such cyclopropanations from 1-3 to 1-4.
- Iodonium ylides derived from malonate methyl ester may also be utilized for higher reactivity in the Rh catalyzed cyclopropanation (Org. Lett. , 2012, 14, 317-3173).
- compounds of formula IX can be made via a convergent approach from intermediates II-3 and II-4 as shown in Scheme 2.
- Salicylic acid derivatives of formula II-4 where Y is a leaving group undergo coupling reaction with Reformatsky reagent of II-3 in Negishi conditions to give intermediates of formula II-5 (Tetrahedron, 2014, 1508-1515; /. Org. Chem. , 2013, 78, 8250-8266, each of which is incorporated herein by reference in its entirety).
- Intermediates of formula II-4 where Y is - B(OH)2 undergo palladium mediated Suzuki type cross-coupling with II-3 (/. Org. Chem.
- compounds of formula XI can be made via borylation followed by cyclopropanation from acetylene intermediate III-2 as shown in Scheme 3.
- Acetylene intermediates of formula III-2 can be made from oxidation of intermediates of III-l followed by Corey-Fuchs method (Org. Synth. 2005, 81, 1). Alternatively, aldehydes of III-l can also be transformed to III-2 by treating with dimethyl- 1- (l-diazo-2-oxopropyl)phosphonate (/. Am. Chem. Soc, 2003, 125, 3714-3715). Such acetylene intermediates of III-2 can be further converted to compounds of XI via borylation, cyclopropanation and deprotection sequence as described above in Scheme 1. tion
- Compounds of formula IV-2 (embodiments of compounds of Formula III), IV-4 (embodiments of compounds of Formula I), and IV-6 (embodiments of compounds of Formula I), may be prepared from appropriately protected vinyl boronate intermediates of formula IV-1 (prepared in Scheme 1) as shown in Scheme 4. Derivatives of formula IV-1 can be directly transformed to vinyl boronates of IV-2 by deprotection in the conditions described above in scheme 1. Intermediates of formula IV-1 may be treated with diazoacetates ⁇ Tetrahedron, 2008, 64, 7041-7095) to undergo cyclopropanation followed by selective ester deprotection to carboxylic acid intermediates of formula IV-3.
- Such carboxylic acids undergo amide formation followed by deprotection to give amide analogs of formula IV-4 (Org. Process Res. Dev. , 2016, 20, 140-177).
- the carboxylic acids of IV-3 may be converted to carbamates (IV-5) via Curtius rearrangement (Chem. Rev. 1988, 88, 297-368; Org. Lett., 2005, 4107-4110; Eur. J. Org. Chem. 2009, 5998 -6008 which is incorporated herein by reference in its entirety).
- Intermediates of IV-5 upon selective hydrolysis of carbamate followed by appropriate amide formation give compounds of formula IV-6.
- Compounds of formula IV-5 may also be transformed to compounds of formula IX where Y 5 is -NHC(0)-0- by hydrolysis.
- Intermediates of formula V-3 may be prepared as shown in Scheme 5.
- V-3 may be used in the preparation of compound of formula IX.
- Such intermediates of formula V-3 can be synthesized from V-2 where X' is a triflate or bromo or iodo group.
- Synthesis of boronates of V-3 may be achieved via Miyaura borylation reaction by cross-coupling of bis(pinacolato)diboron ( ⁇ 2 ⁇ 3 ⁇ 4) with aryl halides (/. Org. Chem. , 1995, 60, 7508-7510).
- prodrugs include but are not limited to substituted or non-substituted alkyl esters, (acyloxy)alkyl (Synthesis 2012, 44, 207, which is incorporated herein by reference in its entirety), [(alkoxycarbonyl)oxy]methyl esters (WO10097675, which is incorporated herein by reference in its entirety), or (oxodioxolyl)methyl esters (/. Med. Chem. 1996, 39, 323-338, which is incorporated herein by reference in its entirety).
- R H by treatment with acid or in neutral conditions (e.g., carbodiimide coupling) in the presence of alcohols (ROH) or via base promoted esterification with RX where X is a leaving group in the presence of an appropriate base.
- VI-1 Formula IX [0154]
- One exemplary but non-limiting general synthetic scheme for preparing prodrugs is shown in Scheme 6.
- the boronic acid of formula VI-1 where R is hydrogen can react with a chloro/bromo-substituted prodrug moiety to form a prodrug of formula IX where R is a prodrug moiety.
- Examples of the prodrug moiety R can be -C 1 _ 9 alkyl, -CR 10 R 11 OC(O)C 1 _
- boronate derivatives of formula VII-1 where Z is a boronate ester of pinacol or pinanediol or boramide of 1,8-diaminonaphthalene may be also utilized for introduction of prodrugs and convert them to final prodrugs as shown in Scheme 7.
- Such carboxylic acids (VII-1) can be made from compounds of formula 1-4 by selective deprotection of OR'.
- the prodrug group may also be introduced earlier in the sequence in compounds of formula 1-1 where R' is R. Such sequence where prodrug is introduced in earlier intermediates is only feasible when the ester is stable under the final deprotection conditions to remove the phenol protective group and boronate ester group.
- a daily dose may be from about 0.25 mg/kg to about 120 mg/kg or more of body weight, from about 0.5 mg/kg or less to about 70 mg/kg, from about 1.0 mg/kg to about 50 mg/kg of body weight, or from about 1.5 mg/kg to about 10 mg/kg of body weight.
- the dosage range would be from about 17 mg per day to about 8000 mg per day, from about 35 mg per day or less to about 7000 mg per day or more, from about 70 mg per day to about 6000 mg per day, from about 100 mg per day to about 5000 mg per day, or from about 200 mg to about 3000 mg per day.
- the amount of active compound administered will, of course, be dependent on the subject and disease state being treated, the severity of the affliction, the manner and schedule of administration and the judgment of the prescribing physician.
- Administration of the compounds disclosed herein or the pharmaceutically acceptable salts thereof can be via any of the accepted modes of administration for agents that serve similar utilities including, but not limited to, orally, subcutaneously, intravenously, intranasally, topically, transdermally, intraperitoneally, intramuscularly, intrapulmonarilly, vaginally, rectally, or intraocularly. Oral and parenteral administrations are customary in treating the indications that are the subject of the preferred embodiments.
- compositions comprising: (a) a safe and therapeutically effective amount of a compound described herein (including enantiomers, diastereoisomers, tautomers, polymorphs, and solvates thereof), or pharmaceutically acceptable salts thereof; and (b) a pharmaceutically acceptable carrier, diluent, excipient or combination thereof.
- compositions containing a pharmaceutically-acceptable carrier include compositions containing a pharmaceutically-acceptable carrier.
- pharmaceutically acceptable carrier or “pharmaceutically acceptable excipient” includes any and all solvents, dispersion media, coatings, antibacterial and antifungal agents, isotonic and absorption delaying agents and the like. The use of such media and agents for pharmaceutically active substances is well known in the art. Except insofar as any conventional media or agent is incompatible with the active ingredient, its use in the therapeutic compositions is contemplated. In addition, various adjuvants such as are commonly used in the art may be included. Considerations for the inclusion of various components in pharmaceutical compositions are described, e.g., in Gilman et al. (Eds.) (1990); Goodman and Gilman's: The Pharmacological Basis of Therapeutics, 8th Ed., Pergamon Press, which is incorporated herein by reference in its entirety.
- substances which can serve as pharmaceutically- acceptable carriers or components thereof, are sugars, such as lactose, glucose and sucrose; starches, such as corn starch and potato starch; cellulose and its derivatives, such as sodium carboxymethyl cellulose, ethyl cellulose, and methyl cellulose; powdered tragacanth; malt; gelatin; talc; solid lubricants, such as stearic acid and magnesium stearate; calcium sulfate; vegetable oils, such as peanut oil, cottonseed oil, sesame oil, olive oil, corn oil and oil of theobroma; polyols such as propylene glycol, glycerine, sorbitol, mannitol, and polyethylene glycol; alginic acid; emulsifiers, such as the TWEENS; wetting agents, such sodium lauryl sulfate; coloring agents; flavoring agents; tableting agents, stabilizers; antioxidants; preservatives; pyr
- a pharmaceutically-acceptable carrier to be used in conjunction with the subject compound is basically determined by the way the compound is to be administered.
- compositions described herein are preferably provided in unit dosage form.
- a "unit dosage form" is a composition containing an amount of a compound that is suitable for administration to an animal, preferably mammal subject, in a single dose, according to good medical practice.
- the preparation of a single or unit dosage form does not imply that the dosage form is administered once per day or once per course of therapy.
- Such dosage forms are contemplated to be administered once, twice, thrice or more per day and may be administered as infusion over a period of time (e.g., from about 30 minutes to about 2-6 hours), or administered as a continuous infusion, and may be given more than once during a course of therapy, though a single administration is not specifically excluded.
- the skilled artisan will recognize that the formulation does not specifically contemplate the entire course of therapy and such decisions are left for those skilled in the art of treatment rather than formulation.
- compositions useful as described above may be in any of a variety of suitable forms for a variety of routes for administration, for example, for oral, nasal, rectal, topical (including transdermal), ocular, intracerebral, intracranial, intrathecal, intra-arterial, intravenous, intramuscular, or other parental routes of administration.
- routes for administration for example, for oral, nasal, rectal, topical (including transdermal), ocular, intracerebral, intracranial, intrathecal, intra-arterial, intravenous, intramuscular, or other parental routes of administration.
- oral and nasal compositions comprise compositions that are administered by inhalation, and made using available methodologies.
- a variety of pharmaceutically-acceptable carriers well-known in the art may be used.
- Pharmaceutically-acceptable carriers include, for example, solid or liquid fillers, diluents, hydrotropies, surface-active agents, and encapsulating substances.
- Optional pharmaceutically-active materials may be included, which do not substantially interfere with the inhibitory activity of the compound.
- the amount of carrier employed in conjunction with the compound is sufficient to provide a practical quantity of material for administration per unit dose of the compound.
- Various oral dosage forms can be used, including such solid forms as tablets, capsules, granules and bulk powders. Tablets can be compressed, tablet triturates, enteric- coated, sugar-coated, film-coated, or multiple-compressed, containing suitable binders, lubricants, diluents, disintegrating agents, coloring agents, flavoring agents, flow-inducing agents, and melting agents.
- Liquid oral dosage forms include aqueous solutions, emulsions, suspensions, solutions and/or suspensions reconstituted from non-effervescent granules, and effervescent preparations reconstituted from effervescent granules, containing suitable solvents, preservatives, emulsifying agents, suspending agents, diluents, sweeteners, melting agents, coloring agents and flavoring agents.
- the pharmaceutically-acceptable carrier suitable for the preparation of unit dosage forms for peroral administration is well-known in the art.
- Tablets typically comprise conventional pharmaceutically-compatible adjuvants as inert diluents, such as calcium carbonate, sodium carbonate, mannitol, lactose and cellulose; binders such as starch, gelatin and sucrose; disintegrants such as starch, alginic acid and croscarmelose; lubricants such as magnesium stearate, stearic acid and talc.
- Glidants such as silicon dioxide can be used to improve flow characteristics of the powder mixture.
- Coloring agents such as the FD&C dyes, can be added for appearance.
- Sweeteners and flavoring agents such as aspartame, saccharin, menthol, peppermint, and fruit flavors, are useful adjuvants for chewable tablets.
- Capsules typically comprise one or more solid diluents disclosed above. The selection of carrier components depends on secondary considerations like taste, cost, and shelf stability, which are not critical, and can be readily made by a person skilled in the art.
- Peroral compositions also include liquid solutions, emulsions, suspensions, and the like.
- the pharmaceutically-acceptable carriers suitable for preparation of such compositions are well known in the art.
- Typical components of carriers for syrups, elixirs, emulsions and suspensions include ethanol, glycerol, propylene glycol, polyethylene glycol, liquid sucrose, sorbitol and water.
- typical suspending agents include methyl cellulose, sodium carboxymethyl cellulose, AVICEL RC-591, tragacanth and sodium alginate;
- typical wetting agents include lecithin and polysorbate 80; and typical preservatives include methyl paraben and sodium benzoate.
- Peroral liquid compositions may also contain one or more components such as sweeteners, flavoring agents and colorants disclosed above.
- compositions may also be coated by conventional methods, typically with pH or time-dependent coatings, such that the subject compound is released in the gastrointestinal tract in the vicinity of the desired topical application, or at various times to extend the desired action.
- dosage forms typically include, but are not limited to, one or more of cellulose acetate phthalate, polyvinylacetate phthalate, hydroxypropyl methyl cellulose phthalate, ethyl cellulose, Eudragit coatings, waxes and shellac.
- compositions described herein may optionally include other drug actives.
- compositions useful for attaining systemic delivery of the subject compounds include sublingual, buccal and nasal dosage forms.
- Such compositions typically comprise one or more of soluble filler substances such as sucrose, sorbitol and mannitol; and binders such as acacia, microcrystalline cellulose, carboxymethyl cellulose and hydroxypropyl methyl cellulose. Glidants, lubricants, sweeteners, colorants, antioxidants and flavoring agents disclosed above may also be included.
- a liquid composition which is formulated for topical ophthalmic use, is formulated such that it can be administered topically to the eye.
- the comfort should be maximized as much as possible, although sometimes formulation considerations (e.g. drug stability) may necessitate less than optimal comfort.
- the liquid should be formulated such that the liquid is tolerable to the patient for topical ophthalmic use.
- an ophthalmically acceptable liquid should either be packaged for single use, or contain a preservative to prevent contamination over multiple uses.
- solutions or medicaments are often prepared using a physiological saline solution as a major vehicle.
- Ophthalmic solutions should preferably be maintained at a comfortable pH with an appropriate buffer system.
- the formulations may also contain conventional, pharmaceutically acceptable preservatives, stabilizers and surfactants.
- Preservatives that may be used in the pharmaceutical compositions disclosed herein include, but are not limited to, benzalkonium chloride, PHMB, chlorobutanol, thimerosal, phenylmercuric, acetate and phenylmercuric nitrate.
- a useful surfactant is, for example, Tween 80.
- various useful vehicles may be used in the ophthalmic preparations disclosed herein. These vehicles include, but are not limited to, polyvinyl alcohol, povidone, hydroxypropyl methyl cellulose, poloxamers, carboxymethyl cellulose, hydroxyethyl cellulose and purified water.
- Tonicity adjustors may be added as needed or convenient. They include, but are not limited to, salts, particularly sodium chloride, potassium chloride, mannitol and glycerin, or any other suitable ophthalmically acceptable tonicity adjustor.
- buffers include acetate buffers, citrate buffers, phosphate buffers and borate buffers. Acids or bases may be used to adjust the pH of these formulations as needed.
- an ophthalmically acceptable antioxidant includes, but is not limited to, sodium metabisulfite, sodium thiosulfate, acetylcysteine, butylated hydroxyanisole and butylated hydroxytoluene.
- excipient components which may be included in the ophthalmic preparations, are chelating agents.
- a useful chelating agent is edetate disodium, although other chelating agents may also be used in place or in conjunction with it.
- Topical formulations may generally be comprised of a pharmaceutical carrier, co-solvent, emulsifier, penetration enhancer, preservative system, and emollient.
- the compounds and compositions described herein may be dissolved or dispersed in a pharmaceutically acceptable diluent, such as a saline or dextrose solution.
- a pharmaceutically acceptable diluent such as a saline or dextrose solution.
- Suitable excipients may be included to achieve the desired pH, including but not limited to NaOH, sodium carbonate, sodium acetate, HC1, and citric acid.
- the pH of the final composition ranges from 2 to 8, or preferably from 4 to 7.
- Antioxidant excipients may include sodium bisulfite, acetone sodium bisulfite, sodium formaldehyde, sulfoxylate, thiourea, and EDTA.
- excipients found in the final intravenous composition may include sodium or potassium phosphates, citric acid, tartaric acid, gelatin, and carbohydrates such as dextrose, mannitol, and dextran. Further acceptable excipients are described in Powell, et al., Compendium of Excipients for Parenteral Formulations, PDA J Pharm Sci and Tech 1998, 52 238-311 and Nema et al., Excipients and Their Role in Approved Injectable Products: Current Usage and Future Directions, PDA J Pharm Sci and Tech 2011, 65 287-332, both of which are incorporated herein by reference in their entirety.
- Antimicrobial agents may also be included to achieve a bacteriostatic or fungistatic solution, including but not limited to phenylmercuric nitrate, thimerosal, benzethonium chloride, benzalkonium chloride, phenol, cresol, and chlorobutanol.
- compositions for intravenous administration may be provided to caregivers in the form of one more solids that are reconstituted with a suitable diluent such as sterile water, saline or dextrose in water shortly prior to administration.
- a suitable diluent such as sterile water, saline or dextrose in water shortly prior to administration.
- the compositions are provided in solution ready to administer parenterally.
- the compositions are provided in a solution that is further diluted prior to administration.
- the combination may be provided to caregivers as a mixture, or the caregivers may mix the two agents prior to administration, or the two agents may be administered separately.
- the actual dose of the active compounds described herein depends on the specific compound, and on the condition to be treated; the selection of the appropriate dose is well within the knowledge of the skilled artisan.
- Some embodiments of the present invention include methods of treating bacterial infections with the compounds and compositions comprising the compounds described herein. Some methods include administering a compound, composition, pharmaceutical composition described herein to a subject in need thereof.
- a subject can be an animal, e.g., a mammal (including a human).
- the bacterial infection comprises a bacteria described herein.
- methods of treating a bacterial infection include methods for preventing bacterial infection in a subject at risk thereof.
- the subject is a human.
- Further embodiments include administering a combination of compounds to a subject in need thereof.
- a combination can include a compound, composition, pharmaceutical composition described herein with an additional medicament.
- Some embodiments include co-administering a compound, composition, and/or pharmaceutical composition described herein, with an additional medicament.
- coadministration it is meant that the two or more agents may be found in the patient's bloodstream at the same time, regardless of when or how they are actually administered.
- the agents are administered simultaneously.
- administration in combination is accomplished by combining the agents in a single dosage form.
- the agents are administered sequentially.
- the agents are administered through the same route, such as orally.
- the agents are administered through different routes, such as one being administered orally and another being administered intravenous (i.v.).
- Examples of additional medicaments include an antibacterial agent, antifungal agent, an antiviral agent, an anti-inflammatory agent and an anti-allergic agent.
- Preferred embodiments include combinations of a compound, composition or pharmaceutical composition described herein with an antibacterial agent such as a ⁇ -lactam.
- ⁇ -lactams include Amoxicillin, Ampicillin (e.g., Pivampicillin, Hetacillin, Bacampicillin, Metampicillin, Talampicillin), Epicillin, Carbenicillin (Carindacillin), Ticarcillin, Temocillin, Azlocillin, Piperacillin, Mezlocillin, Mecillinam (Pivmecillinam), Sulbenicillin, Benzylpenicillin (G), Clometocillin, Benzathine benzylpenicillin, Procaine benzylpenicillin, Azidocillin, Penamecillin, Phenoxymethylpenicillin (V), Propicillin, Benzathine phenoxymethylpenicillin, Pheneticillin, Cloxacillin (e.g., Dicloxacillin, Flucloxacillin), Ox
- Preferred embodiments include ⁇ -lactams such as Ceftazidime, Biapenem, Doripenem, Ertapenem, Imipenem, Meropenem, Tebipenem, Tebipenem pivoxil, Apapenem, and Panipenem.
- ⁇ -lactams such as Ceftazidime, Biapenem, Doripenem, Ertapenem, Imipenem, Meropenem, Tebipenem, Tebipenem pivoxil, Apapenem, and Panipenem.
- Additional preferred embodiments include ⁇ -lactams such as Aztreonam, Tigemonam, and Carumonam.
- Some embodiments include a combination of the compounds, compositions and/or pharmaceutical compositions described herein with an additional agent, wherein the additional agent comprises a monobactam.
- additional agent comprises a monobactam.
- monobactams include aztreonam, tigemonam, nocardicin A, carumonam, and tabtoxin.
- the compound, composition and/or pharmaceutical composition comprises a class A, C, or D beta- lactamase inhibitor.
- Some embodiments include co-administering the compound, composition or pharmaceutical composition described herein with one or more additional agents.
- Some embodiments include a combination of the compounds, compositions and/or pharmaceutical compositions described herein with an additional agent, wherein the additional agent comprises a class B beta lactamase inhibitor.
- a class B beta lactamase inhibitor includes ME1071 (Yoshikazu Ishii et al, "In Vitro Potentiation of Carbapenems with ME1071, a Novel Metallo-6-Lactamase Inhibitor, against Metallo- ⁇ - lactamase Producing Pseudomonas aeruginosa Clinical Isolates.” Antimicrob. Agents Chemother. doi:10.1128/AAC.01397-09 (July 2010)).
- Some embodiments include coadministering the compound, composition or pharmaceutical composition described herein with one or more additional agents.
- Some embodiments include a combination of the compounds, compositions and/or pharmaceutical compositions described herein with an additional agent, wherein the additional agent comprises one or more agents that include a class A, B, C, or D beta lactamase inhibitor. Some embodiments include co-administering the compound, composition or pharmaceutical composition described herein with the one or more additional agents.
- Bacterial infections that can be treated with the compounds, compositions and methods described herein can comprise a wide spectrum of bacteria.
- Example organisms include gram-positive bacteria, gram-negative bacteria, aerobic and anaerobic bacteria, such as Staphylococcus, Lactobacillus, Streptococcus, Sarcina, Escherichia, Enterobacter, Klebsiella, Pseudomonas, Acinetobacter, Mycobacterium, Proteus, Campylobacter, Citrobacter, Nisseria, Baccillus, Bacteroides, Peptococcus, Clostridium, Salmonella, Shigella, Serratia, Haemophilus, Brucella and other organisms.
- More examples of bacterial infections include Pseudomonas aeruginosa, Pseudomonas fluorescens, Pseudomonas acidovorans, Pseudomonas alcaligenes, Pseudomonas putida, Stenotrophomonas maltophilia, Burkholderia cepacia, Aeromonas hydrophilia, Escherichia coli, Citrobacter freundii, Salmonella typhimurium, Salmonella typhi, Salmonella paratyphi, Salmonella enteritidis, Shigella dysenteriae, Shigella flexneri, Shigella sonnei, Enterobacter cloacae, Enterobacter aerogenes, Klebsiella pneumoniae, Klebsiella oxytoca, Serratia marcescens, Francisella tularensis, Morganella morganii, Proteus mirabilis, Prote
- a 10-mL syringe filled with trifluoroacetic anhydride (11.25 mL, 81 mmol, 2 eq) and a 20-mL syringe filled with acetone (17 mL, 232 mmol, 5.7 eq) were added simultaneously via syringe pump over 24 hours to a clear solution of IB (10 g, 40 mmol) in TFA (10 mL) at 70°C. After 1 hour, the starting material began to crystallize out. TFA (5 mL) was added, affording a clear solution. After another hour at 70°C the solution became slightly heterogeneous. Upon completion of the addition, HPLC showed 89: 11 product to starting material.
- Compound 4A was prepared from Boc-t-Butyl ester intermediate (previously disclosed in WO 2015/179308) by TFA deprotection followed by isopropylidene protection as described in step 2 of Example 1.
- the biphasic orange reaction mixture was cooled to rt and filtered over Celite 545 (2 g). The flask and pad were rinsed with ethyl acetate (2 x 10 mL). The filtrate was partitioned. The organic layer was washed with water (60 mL), then concentrated to dryness. The orange solid was taken up in 3/7 1 -propanol/water (80 mL) and heated at 90°C. A biphasic solution was obtained. Propanol (6 mL) was added to get a homogeneous solution. Upon cooling, at 60°C, a biphasic mixture was obtained. Seeds were added and the mixture was allowed to cool to 50°C; a heterogeneous mixture was obtained.
- Step 1 Synthesis of 12A and 12B
- Step 1 Synthesis of compounds 16A and 16B
- Step 8 Synthesis of compound 201
- the reaction mixture was purified by prep-HPLC (C18, 250 x 21 mm, 0.1% formic acid in both acetonitrile and water ) to give the free acid compound 20 (34 mg, 94%).
- the acid product (34 mg, 0.16 mmol) in acetonitrile/water (1 :2, 5 mL) was treated with 0.1 N NaOH (3.5 mL), and stirred for 4h, and lyophilized to give sodium salt compound 20' (50.6 mg) as an off-white solid.
- a 10 mL-flask was flame-dried under vacuum, back-filled with nitrogen and cooled to rt.
- the flask was charged with compound 13 (100 mg, 0.45 mmol, 1 eq.), potassium carbonate (186 mg, 1.35 mmol, 3 eq.), and potassium iodide (224 mg, 1.35 mmol, 3 eq.).
- the reaction flask was placed under vacuum and back-filled with nitrogen three times.
- Anhydrous DMF (2 mL, 0.25 M) followed by freshly prepared chloride (0.90 mmol, 2 eq.) were added via syringe under nitrogen. The resulting mixture was stirred at 50 °C for 12 hrs under a nitrogen balloon.
- ⁇ -lactamase inhibitors were determined by assessing their aztreonam potentiation activity in a dose titration potentiation assay using strains of various bacteria that are resistant to aztreonam due to expression of various ⁇ - lactamases.
- Aztreonam is a monobactam antibiotic and is hydrolyzed by the majority of beta- lactamases that belong to class A or C (but not class B or D).
- the potentiation effect was observed as the ability of BLI compounds to inhibit growth in the presence of sub-inhibitory concentration of aztreonam.
- MICs of test strains varied from 64 ⁇ g/mL to > 128 ⁇ g/mL.
- Aztreonam was present in the test medium at 4 ⁇ g/mL. Compounds were tested at concentrations up to 40 ⁇ g/mL. In this assay, potency of compounds was reported as the minimum concentration of BLI required to inhibit growth of bacteria in the presence of 4 ⁇ g/mL of aztreonam (MPC@/ t ). Table 2 summarizes the BLI potency of aztreonam potentiation (MPC@/ t ) for various strains overexpressing class A (ESBL and KPC), and class C beta- lactamases. Aztreonam MIC for each strain is also shown. Table 2. Activity of BLIs to potentiate aztreonam against strains expressing class A and class C enzymes.
- Selected ⁇ -lactamase inhibitors were also tested for their ability to potentiate the monobactam tigemonam.
- the potentiation effect was observed as the ability of BLI compounds to inhibit growth in the presence of sub-inhibitory concentration of tigemonam.
- MICs of test strains varied from 16 ⁇ g/mL to > 64 ⁇ g/mL.
- Tigemonam was present in the test medium at 4 ⁇ g/mL.
- Compounds were tested at concentrations up to 40 ⁇ g/mL.
- potency of compounds was reported as the minimum concentration of BLI required to inhibit growth of bacteria in the presence of 4 ⁇ g/mL of aztreonam (MPC@ 4 ).
- Table 3 summarizes the BLI potency of tigemonam potentiation (MPC@ 4 ) for various strains overexpressing class A (ESBL) and class C beta-lactamases. Tigemonam MIC for each strain is also shown.
Abstract
Description
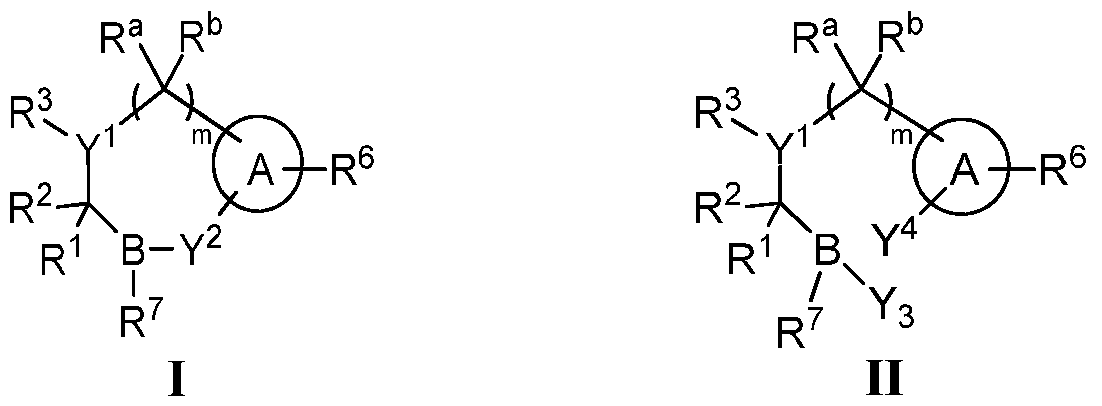














































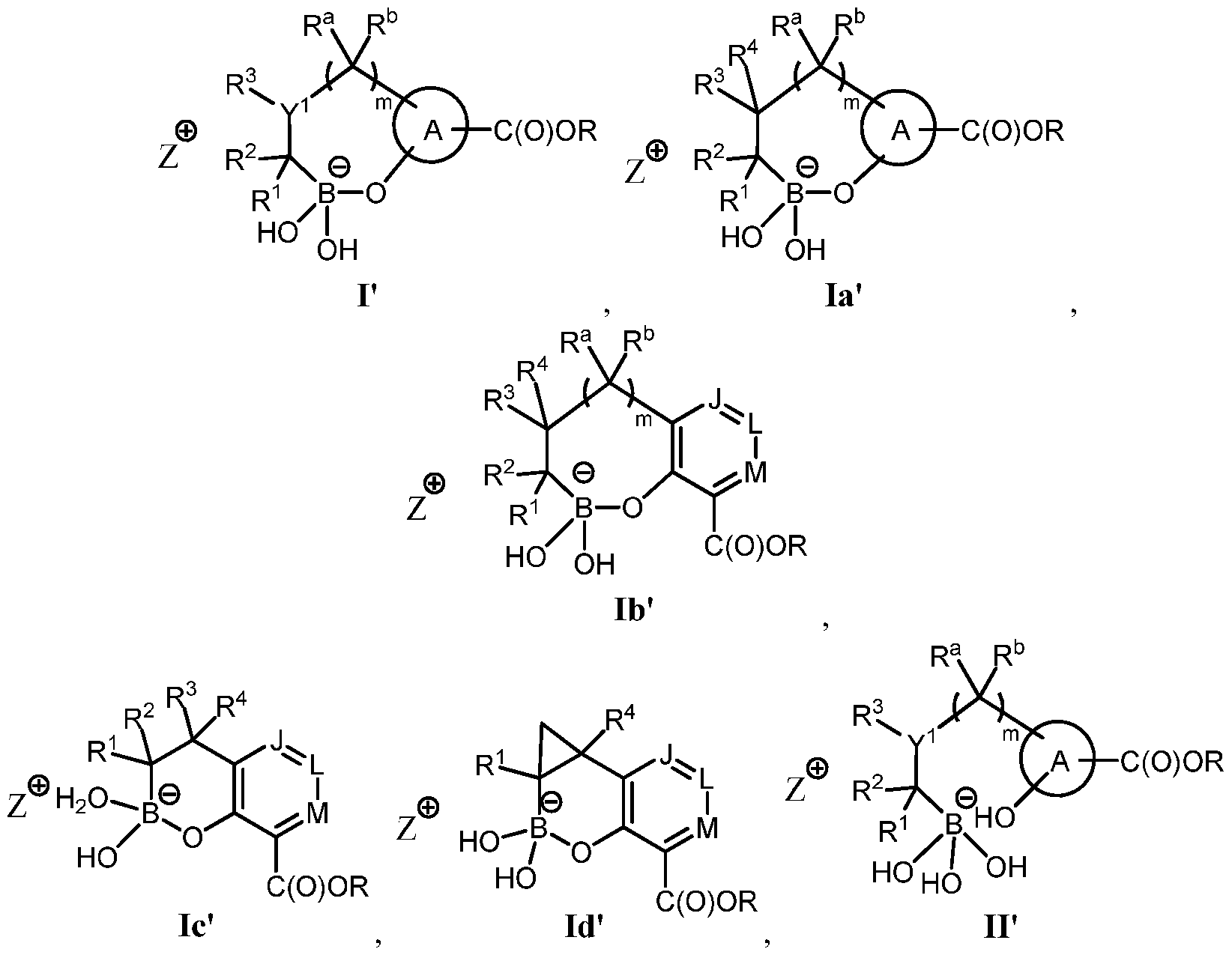























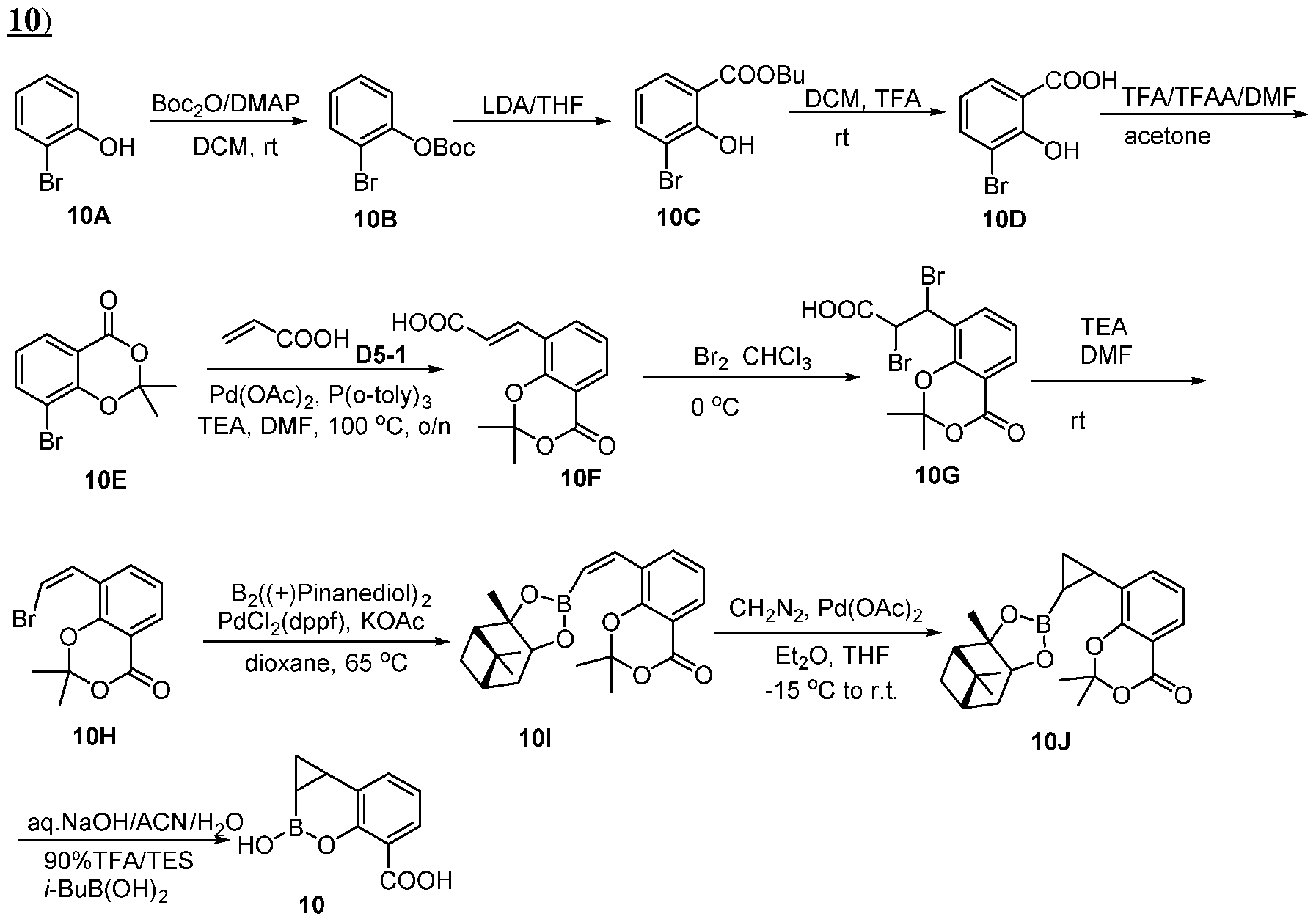



















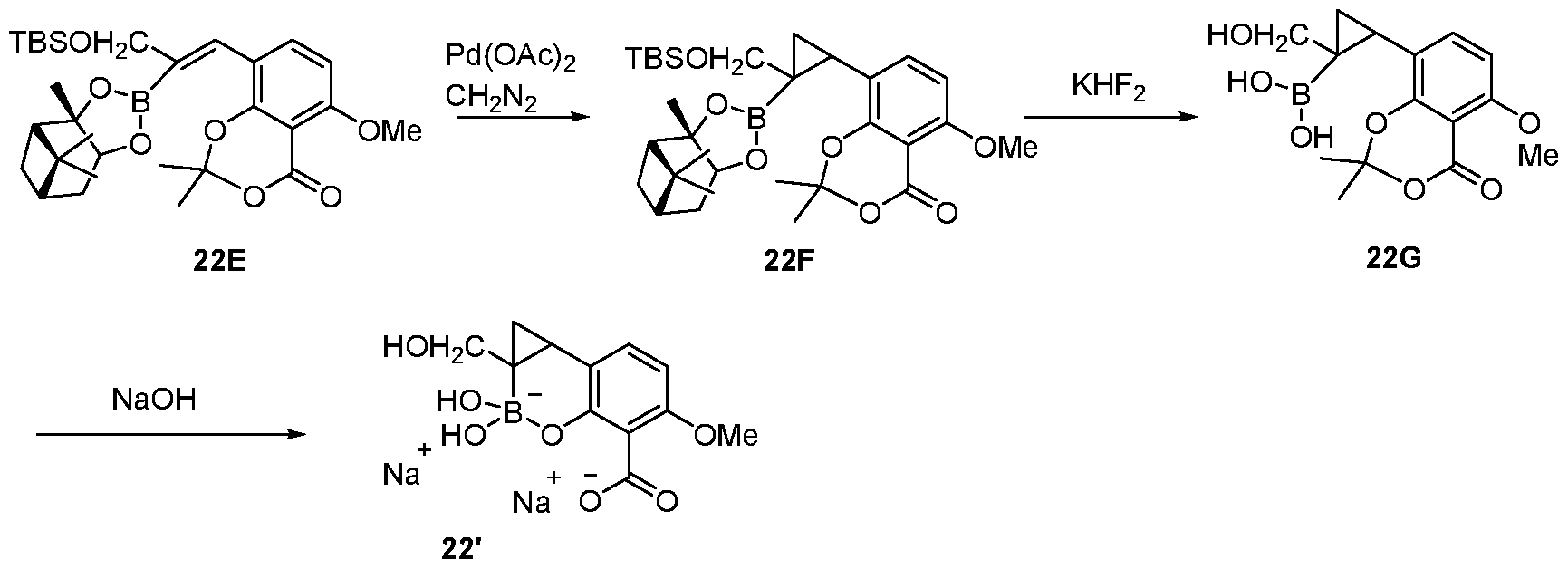






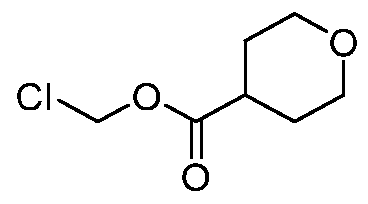




























Claims





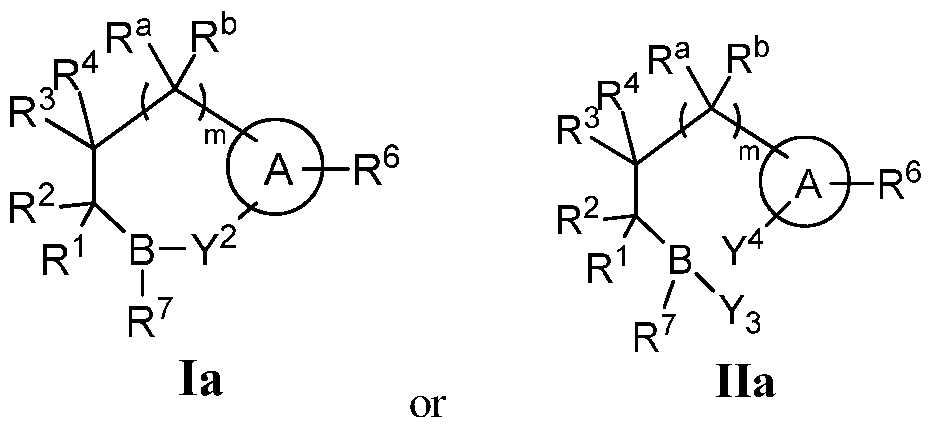










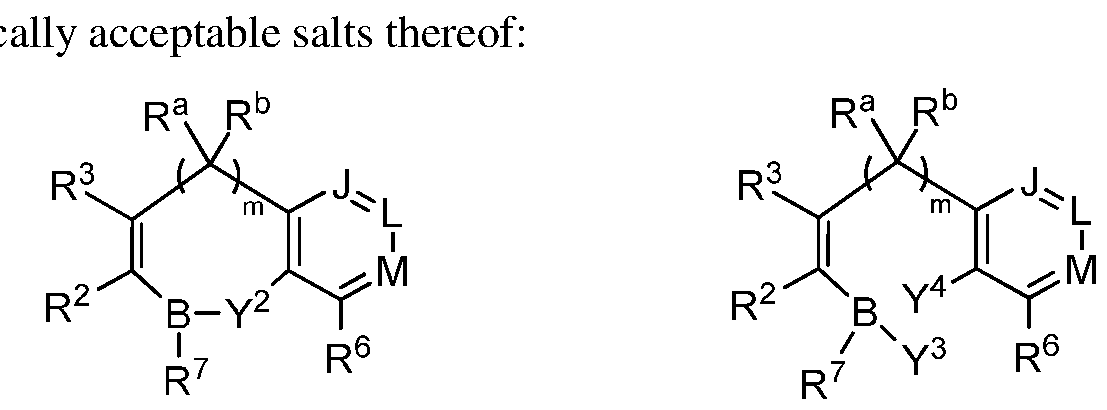










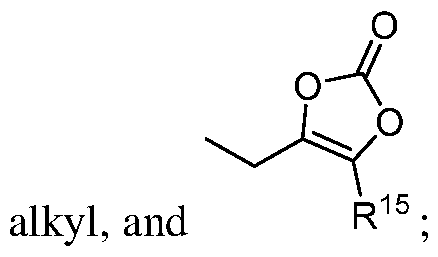



Priority Applications (20)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP17821161.1A EP3478693B1 (en) | 2016-06-30 | 2017-06-28 | Boronic acid derivatives and therapeutic uses thereof |
| KR1020197003015A KR102403296B1 (en) | 2016-06-30 | 2017-06-28 | Boronic acid derivatives and therapeutic uses thereof |
| CA3026582A CA3026582A1 (en) | 2016-06-30 | 2017-06-28 | Boronic acid derivatives and therapeutic uses thereof |
| BR112018077015-9A BR112018077015B1 (en) | 2016-06-30 | 2017-06-28 | Boronic acid derivatives and therapeutic uses thereof |
| MX2018015892A MX2018015892A (en) | 2016-06-30 | 2017-06-28 | Boronic acid derivatives and therapeutic uses thereof. |
| SG11201811549UA SG11201811549UA (en) | 2016-06-30 | 2017-06-28 | Boronic acid derivatives and therapeutic uses thereof |
| PL17821161T PL3478693T3 (en) | 2016-06-30 | 2017-06-28 | Boronic acid derivatives and therapeutic uses thereof |
| DK17821161.1T DK3478693T3 (en) | 2016-06-30 | 2017-06-28 | BORONIC ACID DERIVATIVES AND THERAPEUTIC USES THEREOF |
| JP2018565257A JP7060245B2 (en) | 2016-06-30 | 2017-06-28 | Boronic acid derivatives and their therapeutic use |
| AU2017290060A AU2017290060B2 (en) | 2016-06-30 | 2017-06-28 | Boronic acid derivatives and therapeutic uses thereof |
| ES17821161T ES2894251T3 (en) | 2016-06-30 | 2017-06-28 | Boronic acid derivatives and therapeutic uses thereof |
| RU2018143900A RU2773346C2 (en) | 2016-06-30 | 2017-06-28 | Boronic acid derivatives and their therapeutic use |
| UAA201812390A UA124464C2 (en) | 2016-06-30 | 2017-06-28 | Boronic acid derivatives and therapeutic uses thereof |
| MYPI2018002676A MY196909A (en) | 2016-06-30 | 2017-06-28 | Boronic acid derivatives and therapeutic uses thereof |
| CN201780041247.XA CN109415386B (en) | 2016-06-30 | 2017-06-28 | Boronic acid derivatives and their therapeutic use |
| IL263450A IL263450B (en) | 2016-06-30 | 2018-12-03 | Boronic acid derivatives and therapeutic uses thereof |
| ZA2018/08327A ZA201808327B (en) | 2016-06-30 | 2018-12-10 | Boronic acid derivatives and therapeutic uses thereof |
| CONC2018/0013978A CO2018013978A2 (en) | 2016-06-30 | 2018-12-21 | Boronic acid derivatives and therapeutic uses thereof. |
| PH12018502747A PH12018502747A1 (en) | 2016-06-30 | 2018-12-21 | Boronic acid derivatives and therapeutic uses thereof |
| SA518400742A SA518400742B1 (en) | 2016-06-30 | 2018-12-25 | Boronic acid derivatives and therapeutic uses thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201662357165P | 2016-06-30 | 2016-06-30 | |
| US62/357,165 | 2016-06-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2018005662A1 true WO2018005662A1 (en) | 2018-01-04 |
Family
ID=60785544
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2017/039787 WO2018005662A1 (en) | 2016-06-30 | 2017-06-28 | Boronic acid derivatives and therapeutic uses thereof |
Country Status (23)
| Country | Link |
|---|---|
| US (4) | US10294249B2 (en) |
| EP (1) | EP3478693B1 (en) |
| JP (1) | JP7060245B2 (en) |
| KR (1) | KR102403296B1 (en) |
| CN (1) | CN109415386B (en) |
| AU (1) | AU2017290060B2 (en) |
| BR (1) | BR112018077015B1 (en) |
| CA (1) | CA3026582A1 (en) |
| CL (1) | CL2018003681A1 (en) |
| CO (1) | CO2018013978A2 (en) |
| DK (1) | DK3478693T3 (en) |
| ES (1) | ES2894251T3 (en) |
| IL (1) | IL263450B (en) |
| MX (1) | MX2018015892A (en) |
| MY (1) | MY196909A (en) |
| PH (1) | PH12018502747A1 (en) |
| PL (1) | PL3478693T3 (en) |
| PT (1) | PT3478693T (en) |
| SA (1) | SA518400742B1 (en) |
| SG (1) | SG11201811549UA (en) |
| UA (1) | UA124464C2 (en) |
| WO (1) | WO2018005662A1 (en) |
| ZA (1) | ZA201808327B (en) |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020257306A1 (en) | 2019-06-19 | 2020-12-24 | Qpex Biopharma, Inc. | Boronic acid derivatives and therapeutic uses thereof |
| WO2021049600A1 (en) * | 2019-09-13 | 2021-03-18 | 塩野義製薬株式会社 | Pharmaceutical composition having antibacterial activity |
| JP2021522167A (en) * | 2018-04-20 | 2021-08-30 | キューペックス バイオファーマ, インコーポレイテッド | Boronic acid derivatives and their therapeutic use |
| WO2021188700A1 (en) * | 2020-03-18 | 2021-09-23 | Qpex Biopharma, Inc. | Boronic acid derivatives and therapeutic uses thereof |
| WO2021226114A1 (en) * | 2020-05-05 | 2021-11-11 | Qpex Biopharma, Inc. | Boronic acid derivatives and synthesis, polymorphic forms, and therapeutic uses thereof |
| WO2022037680A1 (en) | 2020-08-20 | 2022-02-24 | 辉诺生物医药科技(杭州)有限公司 | Boracic acid compound |
| US11286270B2 (en) | 2017-10-11 | 2022-03-29 | Qpex Biopharma, Inc. | Boronic acid derivatives and synthesis thereof |
| US11352373B2 (en) | 2017-02-01 | 2022-06-07 | Melinta Subsidiary Corp. | Apparatus and continuous flow process for production of boronic acid derivative |
| KR20220088736A (en) | 2019-10-25 | 2022-06-28 | 스미토모 파마 가부시키가이샤 | Novel substituted condensed cyclic compound |
| WO2022217199A1 (en) * | 2021-04-05 | 2022-10-13 | Qpex Biopharma, Inc. | Ceftibuten dosing regimens |
| WO2022218328A1 (en) | 2021-04-13 | 2022-10-20 | 上海拓界生物医药科技有限公司 | BORIC ACID DERIVATIVE ACTING AS β-LACTAMASE INHIBITOR |
Families Citing this family (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8680136B2 (en) | 2010-08-10 | 2014-03-25 | Rempex Pharmaceuticals, Inc. | Cyclic boronic acid ester derivatives and therapeutic uses thereof |
| EP2943204B1 (en) | 2013-01-10 | 2019-03-13 | Venatorx Pharmaceuticals Inc | Beta-lactamase inhibitors |
| PL3154989T3 (en) | 2014-06-11 | 2021-10-18 | VenatoRx Pharmaceuticals, Inc. | Beta-lactamase inhibitors |
| WO2016081297A1 (en) | 2014-11-18 | 2016-05-26 | Rempex Pharmaceuticals, Inc. | Cyclic boronic acid ester derivatives and therapeutic uses thereof |
| US20180051041A1 (en) | 2015-03-17 | 2018-02-22 | Rempex Pharmaceuticals, Inc. | Boronic acid derivatives and therapeutic uses thereof |
| EP3347008B1 (en) | 2015-09-11 | 2022-03-09 | Venatorx Pharmaceuticals, Inc. | Beta-lactamase inhibitors |
| SG11201811549UA (en) * | 2016-06-30 | 2019-01-30 | Qpex Biopharma Inc | Boronic acid derivatives and therapeutic uses thereof |
| CN110678186A (en) | 2017-03-06 | 2020-01-10 | 维纳拓尔斯制药公司 | Solid forms and combination compositions comprising beta-lactamase inhibitors and uses thereof |
| CN110959008A (en) | 2017-05-26 | 2020-04-03 | 维纳拓尔斯制药公司 | Penicillin binding protein inhibitors |
| EP3630783A4 (en) | 2017-05-26 | 2021-03-03 | Venatorx Pharmaceuticals, Inc. | Penicillin-binding protein inhibitors |
| WO2020112542A1 (en) * | 2018-11-29 | 2020-06-04 | VenatoRx Pharmaceuticals, Inc. | Combination compositions comprising a beta-lactamase inhibitor and uses thereof |
| CN113150022B (en) * | 2021-04-23 | 2022-12-06 | 四川大学 | 3-substituted five-membered cyclic borate derivative, and pharmaceutical composition and pharmaceutical application thereof |
| TW202317085A (en) * | 2021-10-19 | 2023-05-01 | 大陸商上海濟煜醫藥科技有限公司 | Tricyclic boronic acid derivatives and preparation method and application thereof |
| WO2023155869A1 (en) * | 2022-02-18 | 2023-08-24 | 辉诺生物医药科技(杭州)有限公司 | Crystalline form of new boric acid-based compound and preparation method therefor |
| CN114716465A (en) * | 2022-05-06 | 2022-07-08 | 上海中医药大学 | Preparation method and application of bicyclic borate compound |
Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3686398A (en) * | 1970-07-20 | 1972-08-22 | Chevron Res | 10,9-boroxarophenanthrene as fungicides |
| US4353807A (en) * | 1981-03-24 | 1982-10-12 | Mobil Oil Corporation | Lubricants and fuels containing boroxarophenanthrene compounds |
| WO1987005297A1 (en) | 1986-03-03 | 1987-09-11 | The University Of Chicago | Cephalosporin derivatives |
| US7271186B1 (en) | 2002-12-09 | 2007-09-18 | Northwestern University | Nanomolar β-lactamase inhibitors |
| WO2009064413A1 (en) | 2007-11-13 | 2009-05-22 | Protez Pharmaceuticals, Inc. | Beta-lactamase inhibitors |
| WO2010097675A1 (en) | 2009-02-27 | 2010-09-02 | Dhanuka Laboratories Ltd. | An improved preparation process for cefpodoxime proxetil |
| WO2010130708A1 (en) * | 2009-05-12 | 2010-11-18 | Novartis International Pharmaceutical Ltd. | Beta-lactamase inhibitors |
| WO2011154953A1 (en) | 2010-06-10 | 2011-12-15 | Technion R&D Foundation Ltd. | Process for the preparation of iodides |
| WO2012021455A1 (en) * | 2010-08-10 | 2012-02-16 | Rempex Pharmaceuticals, Inc. | Cyclic boronic acid ester derivatives and therapeutic uses thereof |
| WO2012106995A1 (en) | 2011-02-11 | 2012-08-16 | Merck Sharp & Dohme Corp. | Rorgammat inhibitors |
| WO2016003929A1 (en) * | 2014-07-01 | 2016-01-07 | Rempex Pharmaceuticals, Inc. | Boronic acid derivatives and therapeutic uses thereof |
Family Cites Families (91)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4194047A (en) | 1975-11-21 | 1980-03-18 | Merck & Co., Inc. | Substituted N-methylene derivatives of thienamycin |
| US4260543A (en) | 1978-07-03 | 1981-04-07 | Merck & Co., Inc. | Crystalline N-formimidoyl thienamycin |
| US4409214A (en) | 1979-11-19 | 1983-10-11 | Fujisawa Pharmaceutical, Co., Ltd. | 7-Acylamino-3-vinylcephalosporanic acid derivatives and processes for the preparation thereof |
| FR2573070B1 (en) | 1984-11-13 | 1987-01-30 | Rhone Poulenc Sante | PROCESS FOR THE PREPARATION OF CARBONYL COMPOUNDS |
| CA1283404C (en) | 1986-07-01 | 1991-04-23 | Shigeru Sanai | Cephalosporin compounds, processes for their preparation and antibacterial agents |
| ZA893284B (en) | 1988-05-04 | 1990-03-28 | Igen Inc | Peptide analogs and their use as haptens to elicit catalytic antibodies |
| US5442100A (en) | 1992-08-14 | 1995-08-15 | The Procter & Gamble Company | β-aminoalkyl and β-N-peptidylaminoalkyl boronic acids |
| US5888998A (en) | 1997-04-24 | 1999-03-30 | Synphar Laboratories, Inc. | 2-oxo-1-azetidine sulfonic acid derivatives as potent β-lactamase inhibitors |
| US6184363B1 (en) | 1997-06-13 | 2001-02-06 | Northwestern University | Inhibitors of β-lactamases and uses therefor |
| WO1998056392A1 (en) | 1997-06-13 | 1998-12-17 | Northwestern University | INHIBITORS OF β-LACTAMASES AND USES THEREFOR |
| AU2190400A (en) | 1998-12-16 | 2000-07-03 | Northwestern University | Inhibitors of beta-lactamases and uses therefor |
| AU3124300A (en) | 1998-12-16 | 2000-07-03 | Northwestern University | Inhibitors of beta-lactamases and uses therefor |
| CN1190432C (en) | 1999-09-25 | 2005-02-23 | 史密丝克莱恩比彻姆有限公司 | Piperazing derivatives as 5-HT1B antagonists |
| JP2003513890A (en) | 1999-10-28 | 2003-04-15 | メルク エンド カムパニー インコーポレーテッド | Novel succinic metallo-beta-lactamase inhibitors and their use in treating bacterial infections |
| AU2001292732A1 (en) | 2000-09-12 | 2002-03-26 | Larry C. Blasczcak | Beta-lactam analogs and uses therefor |
| CN1189469C (en) | 2000-09-14 | 2005-02-16 | 潘斯瑞克有限公司 | 3-(heteroaryl acetamido)-2-oxo-azetidine-1-sulfonic acid derivatives as anti-bacterial agents |
| US6586615B1 (en) | 2001-01-10 | 2003-07-01 | Bristol-Myers Squibb Company | α-aminoboronic acids prepared by novel synthetic methods |
| DE10118698A1 (en) | 2001-04-17 | 2002-11-07 | Jerini Ag | Immobilization method and arrangement of connections produced therewith on a planar surface |
| AU2002360732A1 (en) * | 2001-12-26 | 2003-07-24 | Guilford Pharmaceuticals | Change inhibitors of dipeptidyl peptidase iv |
| FR2835186B1 (en) | 2002-01-28 | 2006-10-20 | Aventis Pharma Sa | NOVEL HETEROCYCLIC COMPOUNDS ACTIVE AS BETA-LACTAMASES INHIBITORS |
| JP2003229277A (en) | 2002-02-04 | 2003-08-15 | Matsushita Electric Ind Co Ltd | Material for light emitting element, light emitting element and light emitting device using the same |
| AUPS065102A0 (en) | 2002-02-20 | 2002-03-14 | Unisearch Limited | Fluorous acetalation |
| WO2004024697A1 (en) | 2002-09-11 | 2004-03-25 | Kureha Chemical Industry Company, Limited | Amine compounds and use thereof |
| AU2003268752A1 (en) | 2002-10-30 | 2004-05-25 | Sumitomo Chemical Company, Limited | High-molecular compounds and polymerer light emitting devices made by using the same |
| US7439253B2 (en) | 2002-12-06 | 2008-10-21 | Novexel | Heterocyclic compounds, their preparation and their use as medicaments, in particular as antibacterials and beta-lactamase inhibitors |
| BR0317613A (en) | 2002-12-20 | 2005-11-29 | Migenix Corp | Nucleotide translocase (ant) adenine binders and related compositions and methods |
| TW200418791A (en) | 2003-01-23 | 2004-10-01 | Bristol Myers Squibb Co | Pharmaceutical compositions for inhibiting proteasome |
| JP4233365B2 (en) | 2003-03-25 | 2009-03-04 | 三井化学株式会社 | Azadiol complex compound and optical recording medium using the compound |
| WO2005033090A1 (en) | 2003-10-06 | 2005-04-14 | Sumitomo Chemical Company, Limited | Aromatic compound |
| ES2288270T3 (en) | 2003-10-10 | 2008-01-01 | Pfizer Products Incorporated | 2H- (1,2,4) TRIAZOLO (4,3-A) REPLACED PIRAZINS AS INHIBITORS OF GSK-3. |
| NZ547752A (en) * | 2003-11-12 | 2009-12-24 | Phenomix Corp | Heterocyclic boronic acid compounds for inhibiting dipeptidyl peptidase-IV |
| TW200600494A (en) | 2004-03-08 | 2006-01-01 | Chugai Pharmaceutical Co Ltd | Bisphenyl compounds useful as vitamin d3 receptor agonists |
| US20060019116A1 (en) | 2004-07-22 | 2006-01-26 | Eastman Kodak Company | White electroluminescent device with anthracene derivative host |
| TW200618820A (en) | 2004-11-05 | 2006-06-16 | Alza Corp | Liposome formulations of boronic acid compounds |
| US20060178357A1 (en) | 2005-02-10 | 2006-08-10 | Buynak John D | Chphalosporin-derived mercaptans as inhibitors of serine and metallo-beta-lactamases |
| HUE040060T2 (en) | 2005-02-16 | 2019-02-28 | Anacor Pharmaceuticals Inc | Halogen-substituted boronophthalides for the treatment of infections |
| PL1851206T3 (en) | 2005-02-22 | 2013-01-31 | Teva Pharma | Rosuvastatin and salts thereof free of rosuvastatin alkylether and a process for the preparation thereof |
| US9184428B2 (en) | 2005-03-15 | 2015-11-10 | Uchicago Argonne Llc | Non-aqueous electrolytes for lithium ion batteries |
| US7825139B2 (en) * | 2005-05-25 | 2010-11-02 | Forest Laboratories Holdings Limited (BM) | Compounds and methods for selective inhibition of dipeptidyl peptidase-IV |
| TW200734311A (en) | 2005-11-21 | 2007-09-16 | Astrazeneca Ab | New compounds |
| CN101410111B (en) | 2005-12-07 | 2012-12-12 | 巴斯利尔药物股份公司 | Useful combinations of monobactam antibiotics with beta-lactamase inhibitors |
| US8168614B2 (en) | 2006-02-16 | 2012-05-01 | Anacor Pharmaceuticals, Inc. | Boron-containing small molecules as anti-inflammatory agents |
| US20100009957A1 (en) | 2006-09-27 | 2010-01-14 | Blizzard Timothy A | Novel inhibitors of beta-lactamase |
| CN101641095B (en) | 2007-03-23 | 2012-10-31 | 巴斯利尔药物股份公司 | Combination medicaments for treating bacterial infections |
| CA2694068C (en) * | 2007-07-23 | 2016-08-30 | The Board Of Trustees Of The University Of Illinois | System for controlling the reactivity of boronic acids |
| MX2010002608A (en) * | 2007-09-06 | 2010-06-01 | Merck Sharp & Dohme | Soluble guanylate cyclase activators. |
| GB0719366D0 (en) | 2007-10-03 | 2007-11-14 | Smithkline Beecham Corp | Compounds |
| US20100120715A1 (en) | 2007-11-13 | 2010-05-13 | Burns Christopher J | Beta-lactamase inhibitors |
| PT2231667E (en) | 2008-01-18 | 2013-12-13 | Merck Sharp & Dohme | Beta-lactamase inhibitors |
| US8129398B2 (en) | 2008-03-19 | 2012-03-06 | Bristol-Myers Squibb Company | HIV integrase inhibitors |
| EP2285384A4 (en) | 2008-05-12 | 2012-04-25 | Anacor Pharmaceuticals Inc | Boron-containing small molecules |
| WO2009139834A1 (en) | 2008-05-13 | 2009-11-19 | Poniard Pharmaceuticals, Inc. | Bioactive compounds for treatment of cancer and neurodegenerative diseases |
| WO2010075286A1 (en) | 2008-12-24 | 2010-07-01 | University Of Washington | MOLECULAR ACTIVATORS OF THE Wnt/β-CATENIN PATHWAY |
| US8278316B2 (en) * | 2009-03-09 | 2012-10-02 | Bristol-Myers Squibb Company | Aza pyridone analogs useful as melanin concentrating hormone receptor-1 antagonists |
| AU2010259002B2 (en) | 2009-06-08 | 2014-03-20 | Nantbio, Inc. | Triazine derivatives and their therapeutical applications |
| WO2011017125A1 (en) | 2009-07-28 | 2011-02-10 | Anacor Pharmaceuticals, Inc. | Trisubstituted boron-containing molecules |
| WO2011103686A1 (en) | 2010-02-26 | 2011-09-01 | Viswanatha , Sundaramma | CEPHALOSPORIN DERIVATIVES USEFUL AS β-LACTAMASE INHIBITORS AND COMPOSITIONS AND METHODS OF USE THEREOF |
| NZ602622A (en) | 2010-03-31 | 2015-01-30 | Millennium Pharm Inc | Derivatives of 1-amino-2-cyclopropylethylboronic acid |
| US20110288063A1 (en) | 2010-05-19 | 2011-11-24 | Naeja Pharmaceutical Inc. | Novel fused bridged bicyclic heteroaryl substituted 6-alkylidene penems as potent beta-lactamase inhibitors |
| DK2632927T3 (en) | 2010-10-26 | 2016-04-11 | Mars Inc | Boronates AS ARGINSASEINHIBITORER |
| TW201221518A (en) | 2010-11-18 | 2012-06-01 | Glaxo Group Ltd | Compounds |
| EP2508506A1 (en) | 2011-04-08 | 2012-10-10 | LEK Pharmaceuticals d.d. | Preparation of sitagliptin intermediates |
| CN102320960A (en) | 2011-08-02 | 2012-01-18 | 安徽东健化工科技有限公司 | Preparation method of 6-fluoro salicylic acid |
| US9012491B2 (en) | 2011-08-31 | 2015-04-21 | Rempex Pharmaceuticals, Inc. | Heterocyclic boronic acid ester derivatives and therapeutic uses thereof |
| WO2013053372A1 (en) | 2011-10-13 | 2013-04-18 | Therabor Pharmaceuticals | Boronic acid inhibitors of beta-lactamases |
| WO2013056163A1 (en) | 2011-10-14 | 2013-04-18 | The Regents Of The University Of California | Beta-lactamase inhibitors |
| AU2012356890B2 (en) | 2011-12-22 | 2017-06-08 | Ares Trading S.A. | Alpha-amino boronic acid derivatives, selective immunoproteasome inhibitors |
| CA2862710A1 (en) | 2012-01-09 | 2013-07-18 | University Of Tromso | Therapeutic boron-containing compounds |
| EP2615080A1 (en) | 2012-01-12 | 2013-07-17 | LEK Pharmaceuticals d.d. | Preparation of Optically Pure ß-Amino Acid Type Active Pharmaceutical Ingredients and Intermediates thereof |
| WO2013122888A2 (en) | 2012-02-15 | 2013-08-22 | Rempex Pharmaceuticals, Inc. | Methods of treating bacterial infections |
| US9156858B2 (en) | 2012-05-23 | 2015-10-13 | Rempex Pharmaceuticals, Inc. | Boronic acid derivatives and therapeutic uses thereof |
| US10561675B2 (en) | 2012-06-06 | 2020-02-18 | Rempex Pharmaceuticals, Inc. | Cyclic boronic acid ester derivatives and therapeutic uses thereof |
| RU2654692C2 (en) | 2012-12-07 | 2018-05-22 | Венаторкс Фармасьютикалс, Инк. | Beta-lactamase inhibitors |
| EP2941247A4 (en) | 2013-01-04 | 2017-02-08 | Rempex Pharmaceuticals, Inc. | Boronic acid derivatives and therapeutic uses thereof |
| US9101638B2 (en) | 2013-01-04 | 2015-08-11 | Rempex Pharmaceuticals, Inc. | Boronic acid derivatives and therapeutic uses thereof |
| EA201591003A1 (en) | 2013-01-04 | 2015-12-30 | Ремпекс Фармасьютикалз, Инк. | DERIVATIVES OF BORONIC ACID AND THEIR THERAPEUTIC APPLICATION |
| US9241947B2 (en) | 2013-01-04 | 2016-01-26 | Rempex Pharmaceuticals, Inc. | Boronic acid derivatives and therapeutic uses thereof |
| EP2943204B1 (en) | 2013-01-10 | 2019-03-13 | Venatorx Pharmaceuticals Inc | Beta-lactamase inhibitors |
| EP2970340B1 (en) | 2013-03-14 | 2020-02-12 | Venatorx Pharmaceuticals, Inc. | Beta-lactamase inhibitors |
| WO2014144380A1 (en) | 2013-03-15 | 2014-09-18 | Intrexon Corporation | Boron-containing diacylhydrazines |
| RS59488B1 (en) | 2014-05-05 | 2019-12-31 | Rempex Pharmaceuticals Inc | Synthesis of boronate salts and uses thereof |
| US9687497B1 (en) | 2014-05-05 | 2017-06-27 | Rempex Pharmaceuticals, Inc. | Salts and polymorphs of cyclic boronic acid ester derivatives and therapeutic uses thereof |
| US9963467B2 (en) | 2014-05-19 | 2018-05-08 | Rempex Pharmaceuticals, Inc. | Boronic acid derivatives and therapeutic uses thereof |
| PL3154989T3 (en) | 2014-06-11 | 2021-10-18 | VenatoRx Pharmaceuticals, Inc. | Beta-lactamase inhibitors |
| US9511142B2 (en) | 2014-06-11 | 2016-12-06 | VenatoRx Pharmaceuticals, Inc. | Beta-lactamase inhibitors |
| WO2016065282A1 (en) | 2014-10-24 | 2016-04-28 | The Regents Of The University Of Michigan | Nasal formulation, nasal kit, and method for enhancing nasal nitric oxide (no) levels |
| WO2016081297A1 (en) | 2014-11-18 | 2016-05-26 | Rempex Pharmaceuticals, Inc. | Cyclic boronic acid ester derivatives and therapeutic uses thereof |
| US20180051041A1 (en) | 2015-03-17 | 2018-02-22 | Rempex Pharmaceuticals, Inc. | Boronic acid derivatives and therapeutic uses thereof |
| CA2982911C (en) | 2015-04-24 | 2023-10-03 | Rempex Pharmaceuticals, Inc. | Methods of treating bacterial infections |
| SG11201811549UA (en) * | 2016-06-30 | 2019-01-30 | Qpex Biopharma Inc | Boronic acid derivatives and therapeutic uses thereof |
| EP3694864A4 (en) | 2017-10-11 | 2021-10-13 | Qpex Biopharma, Inc. | Boronic acid derivatives and synthesis thereof |
-
2017
- 2017-06-28 SG SG11201811549UA patent/SG11201811549UA/en unknown
- 2017-06-28 EP EP17821161.1A patent/EP3478693B1/en active Active
- 2017-06-28 JP JP2018565257A patent/JP7060245B2/en active Active
- 2017-06-28 AU AU2017290060A patent/AU2017290060B2/en active Active
- 2017-06-28 ES ES17821161T patent/ES2894251T3/en active Active
- 2017-06-28 UA UAA201812390A patent/UA124464C2/en unknown
- 2017-06-28 PL PL17821161T patent/PL3478693T3/en unknown
- 2017-06-28 CN CN201780041247.XA patent/CN109415386B/en active Active
- 2017-06-28 CA CA3026582A patent/CA3026582A1/en active Pending
- 2017-06-28 US US15/636,424 patent/US10294249B2/en active Active
- 2017-06-28 MY MYPI2018002676A patent/MY196909A/en unknown
- 2017-06-28 MX MX2018015892A patent/MX2018015892A/en unknown
- 2017-06-28 DK DK17821161.1T patent/DK3478693T3/en active
- 2017-06-28 KR KR1020197003015A patent/KR102403296B1/en active IP Right Grant
- 2017-06-28 BR BR112018077015-9A patent/BR112018077015B1/en active IP Right Grant
- 2017-06-28 WO PCT/US2017/039787 patent/WO2018005662A1/en unknown
- 2017-06-28 PT PT17821161T patent/PT3478693T/en unknown
-
2018
- 2018-12-03 IL IL263450A patent/IL263450B/en unknown
- 2018-12-10 ZA ZA2018/08327A patent/ZA201808327B/en unknown
- 2018-12-19 CL CL2018003681A patent/CL2018003681A1/en unknown
- 2018-12-21 CO CONC2018/0013978A patent/CO2018013978A2/en unknown
- 2018-12-21 PH PH12018502747A patent/PH12018502747A1/en unknown
- 2018-12-25 SA SA518400742A patent/SA518400742B1/en unknown
-
2019
- 2019-04-08 US US16/378,323 patent/US10570159B2/en active Active
-
2020
- 2020-02-18 US US16/793,965 patent/US11180512B2/en active Active
-
2021
- 2021-11-05 US US17/520,365 patent/US20220056055A1/en active Pending
Patent Citations (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3686398A (en) * | 1970-07-20 | 1972-08-22 | Chevron Res | 10,9-boroxarophenanthrene as fungicides |
| US4353807A (en) * | 1981-03-24 | 1982-10-12 | Mobil Oil Corporation | Lubricants and fuels containing boroxarophenanthrene compounds |
| WO1987005297A1 (en) | 1986-03-03 | 1987-09-11 | The University Of Chicago | Cephalosporin derivatives |
| US7271186B1 (en) | 2002-12-09 | 2007-09-18 | Northwestern University | Nanomolar β-lactamase inhibitors |
| WO2009064413A1 (en) | 2007-11-13 | 2009-05-22 | Protez Pharmaceuticals, Inc. | Beta-lactamase inhibitors |
| WO2009064414A1 (en) | 2007-11-13 | 2009-05-22 | Protez Pharmaceuticals, Inc. | Beta-lactamase inhibitors |
| WO2010097675A1 (en) | 2009-02-27 | 2010-09-02 | Dhanuka Laboratories Ltd. | An improved preparation process for cefpodoxime proxetil |
| WO2010130708A1 (en) * | 2009-05-12 | 2010-11-18 | Novartis International Pharmaceutical Ltd. | Beta-lactamase inhibitors |
| WO2011154953A1 (en) | 2010-06-10 | 2011-12-15 | Technion R&D Foundation Ltd. | Process for the preparation of iodides |
| WO2012021455A1 (en) * | 2010-08-10 | 2012-02-16 | Rempex Pharmaceuticals, Inc. | Cyclic boronic acid ester derivatives and therapeutic uses thereof |
| WO2012106995A1 (en) | 2011-02-11 | 2012-08-16 | Merck Sharp & Dohme Corp. | Rorgammat inhibitors |
| WO2016003929A1 (en) * | 2014-07-01 | 2016-01-07 | Rempex Pharmaceuticals, Inc. | Boronic acid derivatives and therapeutic uses thereof |
Non-Patent Citations (54)
| Title |
|---|
| "Bioreversible Carriers in Drug Design: Theory and Application", 1987, PERGAMON PRESS, pages: 14 - 21 |
| "Design of Prodrugs", 1985, ELSEVIER |
| "Goodman and Gilman's: The Pharmacological Basis of Therapeutics", 1990, PERGAMON PRESS |
| "Modern Pharmaceutics", 2002 |
| "Organic Synthesis, Medicine and Materials", 2011, WILEY-VCH, article "Boronic Acids: Preparations and Applications" |
| "Preparation and Application in Organic Synthesis and Medicine", 2005, WILEY VCH, article "Boronic Acids" |
| "Prodrugs: Challenges and Rewards,", 2007, SPRINGER |
| "Protective Groups in Organic Chemistry", 1973, PLENUM PRESS |
| "Remington's The Science and Practice of Pharmacy", 2005, LIPPINCOTT WILLIAMS & WILKINS |
| ANGEW. CHEM. INT. ED., vol. 55, 2016, pages 785 - 789 |
| ANTIMICROB AGENTS CHEMOTHER, vol. 50, 2006, pages 2280 |
| BOU, G. ET AL., J. ANTIMICROB. AGENTS CHEMOTHER., vol. 44, 2000, pages 1556 |
| BOU, G.MARTINEZ-BELTRAN, J., ANTIMICROB. AGENTS CHEMOTHER., vol. 40, 2000, pages 428 |
| CAREYSUNDBERG, ADVANCED ORGANIC CHEMISTRY |
| CHEM. REV., vol. 108, 2008, pages 288 - 325 |
| CHEM. REV., vol. 110, 2010, pages 890 - 931 |
| CHEM. REV., vol. 88, 1988, pages 297 - 368 |
| CHEM. SOC. REV., vol. 40, 2011, pages 5084 - 5121 |
| COUTTS, S. J. ET AL., TETRAHEDRON LETTERS, vol. 35, no. 29, 1994, pages 5109 - 5112 |
| DATABASE REGISTRY 15 February 2005 (2005-02-15), ANONYMOUS, XP055587452, retrieved from STN Database accession no. 831209-98-4 * |
| DATABASE REGISTRY 15 February 2005 (2005-02-15), ANONYMOUS, XP055587454, retrieved from STN Database accession no. 831210-03-8 * |
| EUR. J. ORG. CHEM., 2000, pages 2557 - 2562 |
| EUR. J. ORG. CHEM., 2009, pages 5998 - 6008 |
| J. AM. CHEM. SOC., vol. 129, 2007, pages 758 - 759 |
| J. AM. CHEM. SOC., vol. 134, 2012, pages 14349 - 14352 |
| J. AM. CHEM. SOC., vol. 137, 2015, pages 13176 - 13182 |
| J. MED. CHEM., vol. 39, 1996, pages 323 - 338 |
| J. MED. CHEM., vol. 46, 2003, pages 3437 - 3440 |
| J. ORG. CHEM., vol. 60, 1995, pages 7508 - 7510 |
| J. ORG. CHEM., vol. 61, 1996, pages 8718 - 8719 |
| J. ORG. CHEM., vol. 73, 2008, pages 6841 - 6844 |
| J. ORG. CHEM., vol. 75, 2010, pages 1244 - 1250 |
| J. ORG. CHEM., vol. 76, 2011, pages 3571 - 3575 |
| J. ORG. CHEM., vol. 78, 2013, pages 8250 - 8266 |
| MATTESON, D. S., TETRAHEDRON, vol. 45, no. 7, 1989, pages 1859 - 1885 |
| MATTESON, D. S.: "Asymmetric synthesis with boronic esters", ACCOUNTS OF CHEMICAL RESEARCH, vol. 21, no. 8, 1988, pages 294 - 300 |
| MONTEFOUR, K. ET AL., CRIT. CARE NURSE, vol. 28, 2008, pages 15 |
| NEMA ET AL.: "Excipients and Their Role in Approved Injectable Products: Current Usage and Future Directions", PDA J PHARM SCI AND TECH, vol. 65, 2011, pages 287 - 332, XP009166667, DOI: 10.5731/pdajpst.2011.00634 |
| ORG. LETT., 2005, pages 4107 - 4110 |
| ORG. LETT., vol. 14, 2012, pages 317 - 3173 |
| ORG. LETT., vol. 18, 2016, pages 1390 - 1393 |
| ORG. PROCESS RES. DEV., vol. 20, 2016, pages 140 - 177 |
| P.G.M. WUTTST.W. GREEN: "Protecting Groups in Organic Synthesis", 2007, JOHN WILEY & SONS |
| PEREZ, F. ET AL., EXPERT REV. ANTI INFECT. THER., vol. 6, 2008, pages 269 |
| POWELL ET AL.: "Compendium of Excipients for Parenteral Formulations", PDA J PHARM SCI AND TECH, vol. 52, 1998, pages 238 - 311, XP009119027 |
| SUMIDA, Y. ET AL.: "Boron-Selective Biaryl Coupling Approach to Versatile Dibenzoxaborins and Application to Concise Synthesis of Defucogilvocarcin M", ORGANIC LETTERS, vol. 16, no. 23, 2014, pages 6240 - 6243, XP055309267 * |
| SYNTHESIS, vol. 44, 2012, pages 207 |
| T. HIGUCHIV. STELLA: "A.C.S. Symposium Series", vol. 14, 1975, AMERICAN CHEMICAL SOCIETY, article "Pro-drugs as Novel Delivery Systems" |
| TETRAHEDRON LETT., vol. 42, 2001, pages 3893 - 3896 |
| TETRAHEDRON LETT., vol. 44, 2003, pages 3629 - 3630 |
| TETRAHEDRON, vol. 64, 2008, pages 7041 - 7095 |
| TETRAHEDRON, vol. 70, 2014, pages 1508 - 1515 |
| YOSHIKAZU ISHII ET AL.: "In Vitro Potentiation of Carbapenems with ME1071, a Novel Metallo-B-Lactamase Inhibitor, against Metallo-B-lactamase Producing Pseudomonas aeruginosa Clinical Isolates", ANTIMICROB. AGENTS CHEMOTHER., July 2010 (2010-07-01) |
| YUEN, A. K. L.HUTTON, C. A., TETRAHEDRON LETTERS, vol. 46, no. 46, 2005, pages 7899 - 7903 |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11352373B2 (en) | 2017-02-01 | 2022-06-07 | Melinta Subsidiary Corp. | Apparatus and continuous flow process for production of boronic acid derivative |
| JP7377545B2 (en) | 2017-10-11 | 2023-11-10 | キューペックス バイオファーマ, インコーポレイテッド | Boronic acid derivatives and their synthesis |
| US11286270B2 (en) | 2017-10-11 | 2022-03-29 | Qpex Biopharma, Inc. | Boronic acid derivatives and synthesis thereof |
| JP7329260B2 (en) | 2018-04-20 | 2023-08-18 | キューペックス バイオファーマ, インコーポレイテッド | Boronic acid derivatives and their therapeutic use |
| JP2021522167A (en) * | 2018-04-20 | 2021-08-30 | キューペックス バイオファーマ, インコーポレイテッド | Boronic acid derivatives and their therapeutic use |
| WO2020257306A1 (en) | 2019-06-19 | 2020-12-24 | Qpex Biopharma, Inc. | Boronic acid derivatives and therapeutic uses thereof |
| WO2021049600A1 (en) * | 2019-09-13 | 2021-03-18 | 塩野義製薬株式会社 | Pharmaceutical composition having antibacterial activity |
| EP4049680A4 (en) * | 2019-10-25 | 2024-01-10 | Sumitomo Pharma Co Ltd | Novel substituted condensed ring compound |
| KR20220088736A (en) | 2019-10-25 | 2022-06-28 | 스미토모 파마 가부시키가이샤 | Novel substituted condensed cyclic compound |
| WO2021188700A1 (en) * | 2020-03-18 | 2021-09-23 | Qpex Biopharma, Inc. | Boronic acid derivatives and therapeutic uses thereof |
| WO2021226114A1 (en) * | 2020-05-05 | 2021-11-11 | Qpex Biopharma, Inc. | Boronic acid derivatives and synthesis, polymorphic forms, and therapeutic uses thereof |
| WO2022037680A1 (en) | 2020-08-20 | 2022-02-24 | 辉诺生物医药科技(杭州)有限公司 | Boracic acid compound |
| WO2022217199A1 (en) * | 2021-04-05 | 2022-10-13 | Qpex Biopharma, Inc. | Ceftibuten dosing regimens |
| WO2022218328A1 (en) | 2021-04-13 | 2022-10-20 | 上海拓界生物医药科技有限公司 | BORIC ACID DERIVATIVE ACTING AS β-LACTAMASE INHIBITOR |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2017290060B2 (en) | Boronic acid derivatives and therapeutic uses thereof | |
| US10618918B2 (en) | Substituted boronic acids as antimicrobials | |
| EP3145936B1 (en) | Boronic acid derivatives and therapeutic uses thereof | |
| US9241947B2 (en) | Boronic acid derivatives and therapeutic uses thereof | |
| EP2941246A1 (en) | Boronic acid derivatives and therapeutic uses thereof | |
| AU2019256351B2 (en) | Boronic acid derivatives and therapeutic uses thereof | |
| US10662205B2 (en) | Cyclic boronic acid ester derivatives and therapeutic uses thereof | |
| RU2773346C2 (en) | Boronic acid derivatives and their therapeutic use | |
| NZ749864A (en) | Boronic acid derivatives and therapeutic uses thereof | |
| BR112020021356B1 (en) | BORONIC ACID DERIVATIVES AND THERAPEUTIC USES THEREOF |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 17821161 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 3026582 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2018565257 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2017290060 Country of ref document: AU Date of ref document: 20170628 Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112018077015 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 20197003015 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2017821161 Country of ref document: EP Effective date: 20190130 |
|
| ENP | Entry into the national phase |
Ref document number: 112018077015 Country of ref document: BR Kind code of ref document: A2 Effective date: 20181221 |