WO2017106064A1 - Biaryl monobactam compounds and methods of use thereof for the treatment of bacterial infections - Google Patents
Biaryl monobactam compounds and methods of use thereof for the treatment of bacterial infections Download PDFInfo
- Publication number
- WO2017106064A1 WO2017106064A1 PCT/US2016/066064 US2016066064W WO2017106064A1 WO 2017106064 A1 WO2017106064 A1 WO 2017106064A1 US 2016066064 W US2016066064 W US 2016066064W WO 2017106064 A1 WO2017106064 A1 WO 2017106064A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- tert
- mmol
- amino
- optionally substituted
- Prior art date
Links
- 238000000034 method Methods 0.000 title claims abstract description 155
- -1 Biaryl monobactam compounds Chemical class 0.000 title claims abstract description 98
- 208000035143 Bacterial infection Diseases 0.000 title claims abstract description 25
- 208000022362 bacterial infectious disease Diseases 0.000 title claims abstract description 25
- 238000011282 treatment Methods 0.000 title claims description 21
- 150000001875 compounds Chemical class 0.000 claims abstract description 519
- 150000003839 salts Chemical class 0.000 claims abstract description 194
- 239000003937 drug carrier Substances 0.000 claims abstract description 7
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical class OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 claims description 89
- 229910052757 nitrogen Inorganic materials 0.000 claims description 86
- 125000000217 alkyl group Chemical group 0.000 claims description 72
- 229910052717 sulfur Inorganic materials 0.000 claims description 60
- 229910052760 oxygen Inorganic materials 0.000 claims description 58
- 125000002950 monocyclic group Chemical group 0.000 claims description 57
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 51
- 229910052739 hydrogen Inorganic materials 0.000 claims description 50
- 239000001257 hydrogen Substances 0.000 claims description 48
- 229920006395 saturated elastomer Polymers 0.000 claims description 43
- 229910052736 halogen Inorganic materials 0.000 claims description 42
- 150000002367 halogens Chemical class 0.000 claims description 42
- 125000006413 ring segment Chemical group 0.000 claims description 36
- 125000003118 aryl group Chemical group 0.000 claims description 33
- 125000001424 substituent group Chemical group 0.000 claims description 28
- 125000005842 heteroatom Chemical group 0.000 claims description 26
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 25
- 125000002947 alkylene group Chemical group 0.000 claims description 24
- 125000000592 heterocycloalkyl group Chemical group 0.000 claims description 23
- 239000003781 beta lactamase inhibitor Substances 0.000 claims description 22
- 229940126813 beta-lactamase inhibitor Drugs 0.000 claims description 22
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 21
- 229940126085 β‑Lactamase Inhibitor Drugs 0.000 claims description 21
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 19
- 239000003814 drug Substances 0.000 claims description 17
- 150000002431 hydrogen Chemical group 0.000 claims description 17
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 16
- 235000019000 fluorine Nutrition 0.000 claims description 15
- 239000008194 pharmaceutical composition Substances 0.000 claims description 15
- 125000001153 fluoro group Chemical group F* 0.000 claims description 14
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 13
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 claims description 13
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 13
- 229910052799 carbon Inorganic materials 0.000 claims description 12
- 125000003831 tetrazolyl group Chemical group 0.000 claims description 11
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 9
- HZZVJAQRINQKSD-UHFFFAOYSA-N Clavulanic acid Natural products OC(=O)C1C(=CCO)OC2CC(=O)N21 HZZVJAQRINQKSD-UHFFFAOYSA-N 0.000 claims description 7
- HZZVJAQRINQKSD-PBFISZAISA-N clavulanic acid Chemical compound OC(=O)[C@H]1C(=C/CO)/O[C@@H]2CC(=O)N21 HZZVJAQRINQKSD-PBFISZAISA-N 0.000 claims description 7
- SMOBCLHAZXOKDQ-ZJUUUORDSA-N [(2s,5r)-7-oxo-2-(piperidin-4-ylcarbamoyl)-1,6-diazabicyclo[3.2.1]octan-6-yl] hydrogen sulfate Chemical compound O=C([C@H]1N2C[C@@](CC1)(N(C2=O)OS(O)(=O)=O)[H])NC1CCNCC1 SMOBCLHAZXOKDQ-ZJUUUORDSA-N 0.000 claims description 6
- 125000004429 atom Chemical group 0.000 claims description 6
- 229960002379 avibactam Drugs 0.000 claims description 6
- NDCUAPJVLWFHHB-UHNVWZDZSA-N avibactam Chemical compound C1N2[C@H](C(N)=O)CC[C@@]1([H])N(OS(O)(=O)=O)C2=O NDCUAPJVLWFHHB-UHNVWZDZSA-N 0.000 claims description 6
- 229960003324 clavulanic acid Drugs 0.000 claims description 6
- 229950011310 relebactam Drugs 0.000 claims description 6
- FKENQMMABCRJMK-RITPCOANSA-N sulbactam Chemical compound O=S1(=O)C(C)(C)[C@H](C(O)=O)N2C(=O)C[C@H]21 FKENQMMABCRJMK-RITPCOANSA-N 0.000 claims description 6
- 229960005256 sulbactam Drugs 0.000 claims description 6
- LPQZKKCYTLCDGQ-WEDXCCLWSA-N tazobactam Chemical compound C([C@]1(C)S([C@H]2N(C(C2)=O)[C@H]1C(O)=O)(=O)=O)N1C=CN=N1 LPQZKKCYTLCDGQ-WEDXCCLWSA-N 0.000 claims description 6
- 229960003865 tazobactam Drugs 0.000 claims description 6
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 claims description 5
- 238000004519 manufacturing process Methods 0.000 claims description 5
- 125000003342 alkenyl group Chemical group 0.000 claims description 4
- 241000589516 Pseudomonas Species 0.000 claims description 3
- 241000588748 Klebsiella Species 0.000 claims description 2
- 241000588923 Citrobacter Species 0.000 claims 1
- 241000588914 Enterobacter Species 0.000 claims 1
- 241000588722 Escherichia Species 0.000 claims 1
- 241000588771 Morganella <proteobacterium> Species 0.000 claims 1
- 241000607720 Serratia Species 0.000 claims 1
- 239000000203 mixture Substances 0.000 abstract description 224
- 229960003644 aztreonam Drugs 0.000 abstract description 8
- 239000003782 beta lactam antibiotic agent Substances 0.000 abstract description 7
- 239000002132 β-lactam antibiotic Substances 0.000 abstract description 7
- 229940124586 β-lactam antibiotics Drugs 0.000 abstract description 7
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 387
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 302
- 239000000243 solution Substances 0.000 description 230
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical group CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 207
- 235000019439 ethyl acetate Nutrition 0.000 description 186
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 168
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 138
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 125
- 229910001868 water Inorganic materials 0.000 description 125
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 115
- 238000006243 chemical reaction Methods 0.000 description 111
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 110
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 110
- 239000011541 reaction mixture Substances 0.000 description 100
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 88
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 87
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 81
- XBDQKXXYIPTUBI-UHFFFAOYSA-N Propionic acid Chemical compound CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 81
- 239000000741 silica gel Substances 0.000 description 81
- 229910002027 silica gel Inorganic materials 0.000 description 81
- 229960001866 silicon dioxide Drugs 0.000 description 81
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 75
- 239000000543 intermediate Substances 0.000 description 68
- 239000012044 organic layer Substances 0.000 description 67
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 64
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 61
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 description 60
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 58
- 239000012267 brine Substances 0.000 description 56
- 239000002904 solvent Substances 0.000 description 55
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 54
- 239000011734 sodium Substances 0.000 description 51
- 239000000706 filtrate Substances 0.000 description 50
- 238000004440 column chromatography Methods 0.000 description 49
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 43
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 38
- 238000005160 1H NMR spectroscopy Methods 0.000 description 33
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 33
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 31
- 239000010410 layer Substances 0.000 description 29
- 239000012071 phase Substances 0.000 description 29
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 29
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 29
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 28
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 26
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 26
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 26
- 239000003153 chemical reaction reagent Substances 0.000 description 25
- 239000003480 eluent Substances 0.000 description 25
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 25
- 229910052737 gold Inorganic materials 0.000 description 25
- 239000010931 gold Substances 0.000 description 25
- DUWWHGPELOTTOE-UHFFFAOYSA-N n-(5-chloro-2,4-dimethoxyphenyl)-3-oxobutanamide Chemical compound COC1=CC(OC)=C(NC(=O)CC(C)=O)C=C1Cl DUWWHGPELOTTOE-UHFFFAOYSA-N 0.000 description 25
- 235000019260 propionic acid Nutrition 0.000 description 25
- 239000007787 solid Substances 0.000 description 25
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 24
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 23
- AFABGHUZZDYHJO-UHFFFAOYSA-N 2-Methylpentane Chemical compound CCCC(C)C AFABGHUZZDYHJO-UHFFFAOYSA-N 0.000 description 22
- 238000000746 purification Methods 0.000 description 22
- 230000002441 reversible effect Effects 0.000 description 22
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 21
- 230000002829 reductive effect Effects 0.000 description 21
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 20
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 19
- 102000006635 beta-lactamase Human genes 0.000 description 19
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 18
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 18
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 18
- 235000019253 formic acid Nutrition 0.000 description 18
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 16
- 241000894006 Bacteria Species 0.000 description 15
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 15
- CDGJTKZKKHLQQN-GSVOUGTGSA-N [(3s)-3-amino-2,2-dimethyl-4-oxoazetidin-1-yl] hydrogen sulfate Chemical compound CC1(C)[C@H](N)C(=O)N1OS(O)(=O)=O CDGJTKZKKHLQQN-GSVOUGTGSA-N 0.000 description 15
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 15
- HIXDQWDOVZUNNA-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-hydroxy-7-methoxychromen-4-one Chemical compound C=1C(OC)=CC(O)=C(C(C=2)=O)C=1OC=2C1=CC=C(OC)C(OC)=C1 HIXDQWDOVZUNNA-UHFFFAOYSA-N 0.000 description 14
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 14
- 230000001580 bacterial effect Effects 0.000 description 14
- RTZKZFJDLAIYFH-UHFFFAOYSA-N ether Substances CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 14
- 125000001072 heteroaryl group Chemical group 0.000 description 14
- 238000003756 stirring Methods 0.000 description 14
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 13
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 13
- 239000003242 anti bacterial agent Substances 0.000 description 13
- 238000001914 filtration Methods 0.000 description 13
- 235000017557 sodium bicarbonate Nutrition 0.000 description 13
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 13
- DPZMUWXOAMOYDT-UHFFFAOYSA-N tert-butyl oxirane-2-carboxylate Chemical compound CC(C)(C)OC(=O)C1CO1 DPZMUWXOAMOYDT-UHFFFAOYSA-N 0.000 description 13
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 12
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 12
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 12
- 208000015181 infectious disease Diseases 0.000 description 12
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 12
- 150000003952 β-lactams Chemical class 0.000 description 12
- 239000007864 aqueous solution Substances 0.000 description 11
- 125000004567 azetidin-3-yl group Chemical group N1CC(C1)* 0.000 description 11
- 125000002619 bicyclic group Chemical group 0.000 description 11
- 229910000024 caesium carbonate Inorganic materials 0.000 description 11
- 125000004122 cyclic group Chemical group 0.000 description 11
- KTWOOEGAPBSYNW-UHFFFAOYSA-N ferrocene Chemical compound [Fe+2].C=1C=C[CH-]C=1.C=1C=C[CH-]C=1 KTWOOEGAPBSYNW-UHFFFAOYSA-N 0.000 description 11
- 125000001841 imino group Chemical group [H]N=* 0.000 description 11
- 229910000027 potassium carbonate Inorganic materials 0.000 description 11
- 239000000047 product Substances 0.000 description 11
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 description 10
- 108020004256 Beta-lactamase Proteins 0.000 description 10
- 229910002666 PdCl2 Inorganic materials 0.000 description 10
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 10
- 229940088710 antibiotic agent Drugs 0.000 description 10
- 230000003115 biocidal effect Effects 0.000 description 10
- 239000003795 chemical substances by application Substances 0.000 description 10
- 229910000160 potassium phosphate Inorganic materials 0.000 description 10
- 235000011009 potassium phosphates Nutrition 0.000 description 10
- 125000004495 thiazol-4-yl group Chemical group S1C=NC(=C1)* 0.000 description 10
- 229940086542 triethylamine Drugs 0.000 description 10
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 9
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 9
- 108090000204 Dipeptidase 1 Proteins 0.000 description 9
- WZPBZJONDBGPKJ-VEHQQRBSSA-N aztreonam Chemical compound O=C1N(S([O-])(=O)=O)[C@@H](C)[C@@H]1NC(=O)C(=N/OC(C)(C)C(O)=O)\C1=CSC([NH3+])=N1 WZPBZJONDBGPKJ-VEHQQRBSSA-N 0.000 description 9
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 9
- 239000003426 co-catalyst Substances 0.000 description 9
- 230000002401 inhibitory effect Effects 0.000 description 9
- 238000002953 preparative HPLC Methods 0.000 description 9
- 238000004007 reversed phase HPLC Methods 0.000 description 9
- 238000010898 silica gel chromatography Methods 0.000 description 9
- 239000000725 suspension Substances 0.000 description 9
- IOKGWQZQCNXXLD-UHFFFAOYSA-N tert-butyl n-(3-bromopropyl)carbamate Chemical compound CC(C)(C)OC(=O)NCCCBr IOKGWQZQCNXXLD-UHFFFAOYSA-N 0.000 description 9
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 9
- GZFGOTFRPZRKDS-UHFFFAOYSA-N 4-bromophenol Chemical compound OC1=CC=C(Br)C=C1 GZFGOTFRPZRKDS-UHFFFAOYSA-N 0.000 description 8
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 8
- XLYOFNOQVPJJNP-ZSJDYOACSA-N Heavy water Chemical compound [2H]O[2H] XLYOFNOQVPJJNP-ZSJDYOACSA-N 0.000 description 8
- 238000003820 Medium-pressure liquid chromatography Methods 0.000 description 8
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 8
- 125000004432 carbon atom Chemical group C* 0.000 description 8
- 229940079593 drug Drugs 0.000 description 8
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 8
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 8
- 239000012074 organic phase Substances 0.000 description 8
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 8
- 238000000926 separation method Methods 0.000 description 8
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 8
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 7
- LDTMNHIZDFDFFY-UHFFFAOYSA-N 2-[2-[(2-methylpropan-2-yl)oxycarbonylamino]-1,3-thiazol-4-yl]-2-oxoacetic acid Chemical compound CC(C)(C)OC(=O)NC1=NC(C(=O)C(O)=O)=CS1 LDTMNHIZDFDFFY-UHFFFAOYSA-N 0.000 description 7
- YEDUAINPPJYDJZ-UHFFFAOYSA-N 2-hydroxybenzothiazole Chemical compound C1=CC=C2SC(O)=NC2=C1 YEDUAINPPJYDJZ-UHFFFAOYSA-N 0.000 description 7
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 7
- 125000005336 allyloxy group Chemical group 0.000 description 7
- 230000015572 biosynthetic process Effects 0.000 description 7
- 239000012043 crude product Substances 0.000 description 7
- 238000004128 high performance liquid chromatography Methods 0.000 description 7
- 239000002808 molecular sieve Substances 0.000 description 7
- 239000000376 reactant Substances 0.000 description 7
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 7
- 239000012258 stirred mixture Substances 0.000 description 7
- 239000000126 substance Substances 0.000 description 7
- 238000003786 synthesis reaction Methods 0.000 description 7
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 6
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 6
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 6
- JRNVZBWKYDBUCA-UHFFFAOYSA-N N-chlorosuccinimide Chemical compound ClN1C(=O)CCC1=O JRNVZBWKYDBUCA-UHFFFAOYSA-N 0.000 description 6
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 6
- 239000002253 acid Substances 0.000 description 6
- 239000013543 active substance Substances 0.000 description 6
- 150000001412 amines Chemical group 0.000 description 6
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 6
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 6
- 238000004587 chromatography analysis Methods 0.000 description 6
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical compound CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 6
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 6
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 6
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 6
- 229910000029 sodium carbonate Inorganic materials 0.000 description 6
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 description 6
- 239000012453 solvate Substances 0.000 description 6
- QGXFDKHOSNJQKE-UHFFFAOYSA-N sulfuric acid;2,2,2-trifluoroacetic acid Chemical compound OS(O)(=O)=O.OC(=O)C(F)(F)F QGXFDKHOSNJQKE-UHFFFAOYSA-N 0.000 description 6
- WJKHJLXJJJATHN-UHFFFAOYSA-N triflic anhydride Chemical compound FC(F)(F)S(=O)(=O)OS(=O)(=O)C(F)(F)F WJKHJLXJJJATHN-UHFFFAOYSA-N 0.000 description 6
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 5
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 5
- 239000004480 active ingredient Substances 0.000 description 5
- IPWKHHSGDUIRAH-UHFFFAOYSA-N bis(pinacolato)diboron Chemical compound O1C(C)(C)C(C)(C)OB1B1OC(C)(C)C(C)(C)O1 IPWKHHSGDUIRAH-UHFFFAOYSA-N 0.000 description 5
- FAMRKDQNMBBFBR-BQYQJAHWSA-N diethyl azodicarboxylate Substances CCOC(=O)\N=N\C(=O)OCC FAMRKDQNMBBFBR-BQYQJAHWSA-N 0.000 description 5
- 239000000284 extract Substances 0.000 description 5
- 102000020235 metallo-beta-lactamase Human genes 0.000 description 5
- 108060004734 metallo-beta-lactamase Proteins 0.000 description 5
- 229940041009 monobactams Drugs 0.000 description 5
- 239000002244 precipitate Substances 0.000 description 5
- VVWRJUBEIPHGQF-MDZDMXLPSA-N propan-2-yl (ne)-n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)\N=N\C(=O)OC(C)C VVWRJUBEIPHGQF-MDZDMXLPSA-N 0.000 description 5
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 5
- 238000004366 reverse phase liquid chromatography Methods 0.000 description 5
- 239000007858 starting material Substances 0.000 description 5
- 238000004808 supercritical fluid chromatography Methods 0.000 description 5
- 238000004809 thin layer chromatography Methods 0.000 description 5
- XSRJWRWWEDIFOY-UHFFFAOYSA-N (2,2-dimethyl-4-oxoazetidin-1-yl) hydrogen sulfate Chemical compound CC1(C)CC(=O)N1OS(O)(=O)=O XSRJWRWWEDIFOY-UHFFFAOYSA-N 0.000 description 4
- SVKDKHOABDSHJE-UHFFFAOYSA-N 3-(4-bromophenoxy)-2-hydroxypropanoic acid Chemical compound BrC1=CC=C(OCC(C(=O)O)O)C=C1 SVKDKHOABDSHJE-UHFFFAOYSA-N 0.000 description 4
- 102000004190 Enzymes Human genes 0.000 description 4
- 108090000790 Enzymes Proteins 0.000 description 4
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 4
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 4
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 4
- WKDDRNSBRWANNC-UHFFFAOYSA-N Thienamycin Natural products C1C(SCCN)=C(C(O)=O)N2C(=O)C(C(O)C)C21 WKDDRNSBRWANNC-UHFFFAOYSA-N 0.000 description 4
- 239000008346 aqueous phase Substances 0.000 description 4
- 239000002585 base Substances 0.000 description 4
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 4
- 230000000694 effects Effects 0.000 description 4
- 125000000623 heterocyclic group Chemical group 0.000 description 4
- ZSKVGTPCRGIANV-ZXFLCMHBSA-N imipenem Chemical compound C1C(SCC\N=C\N)=C(C(O)=O)N2C(=O)[C@H]([C@H](O)C)[C@H]21 ZSKVGTPCRGIANV-ZXFLCMHBSA-N 0.000 description 4
- 229960002182 imipenem Drugs 0.000 description 4
- 239000003112 inhibitor Substances 0.000 description 4
- 230000005764 inhibitory process Effects 0.000 description 4
- IUYHWZFSGMZEOG-UHFFFAOYSA-M magnesium;propane;chloride Chemical compound [Mg+2].[Cl-].C[CH-]C IUYHWZFSGMZEOG-UHFFFAOYSA-M 0.000 description 4
- LXCFILQKKLGQFO-UHFFFAOYSA-N methylparaben Chemical compound COC(=O)C1=CC=C(O)C=C1 LXCFILQKKLGQFO-UHFFFAOYSA-N 0.000 description 4
- 239000003208 petroleum Substances 0.000 description 4
- 235000011056 potassium acetate Nutrition 0.000 description 4
- 238000002360 preparation method Methods 0.000 description 4
- 229940002612 prodrug Drugs 0.000 description 4
- 239000000651 prodrug Substances 0.000 description 4
- 239000002002 slurry Substances 0.000 description 4
- 229940124597 therapeutic agent Drugs 0.000 description 4
- 230000001225 therapeutic effect Effects 0.000 description 4
- CSRZQMIRAZTJOY-UHFFFAOYSA-N trimethylsilyl iodide Chemical compound C[Si](C)(C)I CSRZQMIRAZTJOY-UHFFFAOYSA-N 0.000 description 4
- 229910000404 tripotassium phosphate Inorganic materials 0.000 description 4
- 235000019798 tripotassium phosphate Nutrition 0.000 description 4
- MGBFMCQDMMADTF-UHFFFAOYSA-N 3-azidopropyl trifluoromethanesulfonate Chemical compound FC(F)(F)S(=O)(=O)OCCCN=[N+]=[N-] MGBFMCQDMMADTF-UHFFFAOYSA-N 0.000 description 3
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 3
- PTEFNEALEPSHLC-UHFFFAOYSA-N 6-bromopyridin-3-ol Chemical compound OC1=CC=C(Br)N=C1 PTEFNEALEPSHLC-UHFFFAOYSA-N 0.000 description 3
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 3
- JDZZHVYFAYPHEP-UHFFFAOYSA-N BrC1=CC=C(C=N1)OCC(C(=O)OC(C)(C)C)O[Si](C)(C)C(C)(C)C Chemical compound BrC1=CC=C(C=N1)OCC(C(=O)OC(C)(C)C)O[Si](C)(C)C(C)(C)C JDZZHVYFAYPHEP-UHFFFAOYSA-N 0.000 description 3
- PCBZRNYXXCIELG-WYFCWLEVSA-N COC1=CC=C(C[C@H](NC(=O)OC2CCCC3(C2)OOC2(O3)C3CC4CC(C3)CC2C4)C(=O)N[C@@H]2[C@@H](CO)O[C@H]([C@@H]2O)N2C=NC3=C2N=CN=C3N(C)C)C=C1 Chemical compound COC1=CC=C(C[C@H](NC(=O)OC2CCCC3(C2)OOC2(O3)C3CC4CC(C3)CC2C4)C(=O)N[C@@H]2[C@@H](CO)O[C@H]([C@@H]2O)N2C=NC3=C2N=CN=C3N(C)C)C=C1 PCBZRNYXXCIELG-WYFCWLEVSA-N 0.000 description 3
- 229930186147 Cephalosporin Natural products 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 3
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- 150000001204 N-oxides Chemical class 0.000 description 3
- 229930182555 Penicillin Natural products 0.000 description 3
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 3
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 3
- 238000004458 analytical method Methods 0.000 description 3
- 230000000845 anti-microbial effect Effects 0.000 description 3
- 239000004599 antimicrobial Substances 0.000 description 3
- JILPQYGWASXNCQ-UHFFFAOYSA-N benzhydryl 4-(4-bromophenyl)-2-hydroxybutanoate Chemical compound BrC1=CC=C(C=C1)CCC(C(=O)OC(C1=CC=CC=C1)C1=CC=CC=C1)O JILPQYGWASXNCQ-UHFFFAOYSA-N 0.000 description 3
- 229940041011 carbapenems Drugs 0.000 description 3
- 125000002837 carbocyclic group Chemical group 0.000 description 3
- 238000004113 cell culture Methods 0.000 description 3
- 229940124587 cephalosporin Drugs 0.000 description 3
- 150000001780 cephalosporins Chemical class 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 125000004177 diethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 3
- ZZVUWRFHKOJYTH-UHFFFAOYSA-N diphenhydramine Chemical group C=1C=CC=CC=1C(OCCN(C)C)C1=CC=CC=C1 ZZVUWRFHKOJYTH-UHFFFAOYSA-N 0.000 description 3
- 201000010099 disease Diseases 0.000 description 3
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 3
- BLYSELSMKSKVBQ-SNVBAGLBSA-N ethyl (2R)-3-(4-bromophenoxy)-2-hydroxypropanoate Chemical compound BrC1=CC=C(OC[C@H](C(=O)OCC)O)C=C1 BLYSELSMKSKVBQ-SNVBAGLBSA-N 0.000 description 3
- LSGWSXRILNPXKJ-UHFFFAOYSA-N ethyl oxirane-2-carboxylate Chemical group CCOC(=O)C1CO1 LSGWSXRILNPXKJ-UHFFFAOYSA-N 0.000 description 3
- 239000012458 free base Substances 0.000 description 3
- 150000004677 hydrates Chemical class 0.000 description 3
- 239000012535 impurity Substances 0.000 description 3
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 3
- 230000000155 isotopic effect Effects 0.000 description 3
- 230000014759 maintenance of location Effects 0.000 description 3
- 238000004949 mass spectrometry Methods 0.000 description 3
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 3
- GDOPTJXRTPNYNR-UHFFFAOYSA-N methyl-cyclopentane Natural products CC1CCCC1 GDOPTJXRTPNYNR-UHFFFAOYSA-N 0.000 description 3
- 125000002911 monocyclic heterocycle group Chemical group 0.000 description 3
- 231100000252 nontoxic Toxicity 0.000 description 3
- 230000003000 nontoxic effect Effects 0.000 description 3
- 150000002960 penicillins Chemical class 0.000 description 3
- 239000012041 precatalyst Substances 0.000 description 3
- 238000011321 prophylaxis Methods 0.000 description 3
- ZDYVRSLAEXCVBX-UHFFFAOYSA-N pyridinium p-toluenesulfonate Chemical compound C1=CC=[NH+]C=C1.CC1=CC=C(S([O-])(=O)=O)C=C1 ZDYVRSLAEXCVBX-UHFFFAOYSA-N 0.000 description 3
- 239000011369 resultant mixture Substances 0.000 description 3
- 239000000377 silicon dioxide Substances 0.000 description 3
- 229910052708 sodium Inorganic materials 0.000 description 3
- 229910000104 sodium hydride Inorganic materials 0.000 description 3
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical class [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 3
- POZFCYRQBIGCNN-LLVKDONJSA-N tert-butyl (2R)-3-(4-bromo-3-chlorophenoxy)-2-hydroxypropanoate Chemical compound BrC1=C(C=C(OC[C@H](C(=O)OC(C)(C)C)O)C=C1)Cl POZFCYRQBIGCNN-LLVKDONJSA-N 0.000 description 3
- ALLGJVKOLKRAED-LLVKDONJSA-N tert-butyl (2R)-3-(4-bromophenoxy)-2-hydroxypropanoate Chemical compound BrC1=CC=C(OC[C@H](C(=O)OC(C)(C)C)O)C=C1 ALLGJVKOLKRAED-LLVKDONJSA-N 0.000 description 3
- SLBRSWXIDHFOHV-UHFFFAOYSA-N tert-butyl 3-(6-bromopyridin-3-yl)oxy-2-hydroxypropanoate Chemical compound BrC1=CC=C(C=N1)OCC(C(=O)OC(C)(C)C)O SLBRSWXIDHFOHV-UHFFFAOYSA-N 0.000 description 3
- 125000001981 tert-butyldimethylsilyl group Chemical group [H]C([H])([H])[Si]([H])(C([H])([H])[H])[*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- DPKBAXPHAYBPRL-UHFFFAOYSA-M tetrabutylazanium;iodide Chemical compound [I-].CCCC[N+](CCCC)(CCCC)CCCC DPKBAXPHAYBPRL-UHFFFAOYSA-M 0.000 description 3
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 3
- 229910052725 zinc Inorganic materials 0.000 description 3
- 239000011701 zinc Substances 0.000 description 3
- BXMXMTJFBNXCTN-SSDOTTSWSA-N (2R)-1-(4-bromophenoxy)propan-2-ol Chemical compound BrC1=CC=C(OC[C@@H](C)O)C=C1 BXMXMTJFBNXCTN-SSDOTTSWSA-N 0.000 description 2
- FEJCUMQZLAVWKG-MRVPVSSYSA-N (2R)-3-(4-bromophenoxy)-2-hydroxypropanamide Chemical compound BrC1=CC=C(OC[C@H](C(=O)N)O)C=C1 FEJCUMQZLAVWKG-MRVPVSSYSA-N 0.000 description 2
- FCJLGOVVTAGJKG-ZCFIWIBFSA-N (2R)-3-(6-bromopyridin-3-yl)oxy-2-hydroxypropanoic acid Chemical compound BrC1=CC=C(C=N1)OC[C@H](C(=O)O)O FCJLGOVVTAGJKG-ZCFIWIBFSA-N 0.000 description 2
- ISUIVWNWEDIHJD-GBXIJSLDSA-N (2r,3s)-3-azaniumyl-2-methyl-4-oxoazetidine-1-sulfonate Chemical compound C[C@@H]1[C@H](N)C(=O)N1S(O)(=O)=O ISUIVWNWEDIHJD-GBXIJSLDSA-N 0.000 description 2
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 description 2
- 125000006570 (C5-C6) heteroaryl group Chemical group 0.000 description 2
- YZUPZGFPHUVJKC-UHFFFAOYSA-N 1-bromo-2-methoxyethane Chemical compound COCCBr YZUPZGFPHUVJKC-UHFFFAOYSA-N 0.000 description 2
- ZHXUWDPHUQHFOV-UHFFFAOYSA-N 2,5-dibromopyridine Chemical compound BrC1=CC=C(Br)N=C1 ZHXUWDPHUQHFOV-UHFFFAOYSA-N 0.000 description 2
- MPXSERZVGZXMNE-UHFFFAOYSA-N 2-(4-bromophenoxy)acetaldehyde Chemical compound BrC1=CC=C(OCC=O)C=C1 MPXSERZVGZXMNE-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- PXRFNCNIHPRBOT-UHFFFAOYSA-N 2-[5-chloro-2-[(2-methylpropan-2-yl)oxycarbonylamino]-1,3-thiazol-4-yl]-2-oxoacetic acid Chemical compound CC(C)(C)OC(=O)NC1=NC(C(=O)C(O)=O)=C(Cl)S1 PXRFNCNIHPRBOT-UHFFFAOYSA-N 0.000 description 2
- KENPFZUYYWVXNW-UHFFFAOYSA-N 2-bromoethyl trifluoromethanesulfonate Chemical compound FC(F)(F)S(=O)(=O)OCCBr KENPFZUYYWVXNW-UHFFFAOYSA-N 0.000 description 2
- CFMZSMGAMPBRBE-UHFFFAOYSA-N 2-hydroxyisoindole-1,3-dione Chemical compound C1=CC=C2C(=O)N(O)C(=O)C2=C1 CFMZSMGAMPBRBE-UHFFFAOYSA-N 0.000 description 2
- YZEUHQHUFTYLPH-UHFFFAOYSA-N 2-nitroimidazole Chemical compound [O-][N+](=O)C1=NC=CN1 YZEUHQHUFTYLPH-UHFFFAOYSA-N 0.000 description 2
- DVLFYONBTKHTER-UHFFFAOYSA-N 3-(N-morpholino)propanesulfonic acid Chemical compound OS(=O)(=O)CCCN1CCOCC1 DVLFYONBTKHTER-UHFFFAOYSA-N 0.000 description 2
- WHVSIWLMCCGHFW-UHFFFAOYSA-N 3-azidopropan-1-ol Chemical group OCCCN=[N+]=[N-] WHVSIWLMCCGHFW-UHFFFAOYSA-N 0.000 description 2
- WVGCPEDBFHEHEZ-UHFFFAOYSA-N 4-bromo-1h-pyrazole Chemical compound BrC=1C=NNC=1 WVGCPEDBFHEHEZ-UHFFFAOYSA-N 0.000 description 2
- MRQYTJXVULSNIS-UHFFFAOYSA-N 4-bromo-3-fluorophenol Chemical compound OC1=CC=C(Br)C(F)=C1 MRQYTJXVULSNIS-UHFFFAOYSA-N 0.000 description 2
- RGHHSNMVTDWUBI-UHFFFAOYSA-N 4-hydroxybenzaldehyde Chemical compound OC1=CC=C(C=O)C=C1 RGHHSNMVTDWUBI-UHFFFAOYSA-N 0.000 description 2
- FHZALEJIENDROK-UHFFFAOYSA-N 5-bromo-1h-imidazole Chemical compound BrC1=CN=CN1 FHZALEJIENDROK-UHFFFAOYSA-N 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- 241000589291 Acinetobacter Species 0.000 description 2
- 241000588626 Acinetobacter baumannii Species 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 2
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 2
- WZPBZJONDBGPKJ-UHFFFAOYSA-N Antibiotic SQ 26917 Natural products O=C1N(S(O)(=O)=O)C(C)C1NC(=O)C(=NOC(C)(C)C(O)=O)C1=CSC(N)=N1 WZPBZJONDBGPKJ-UHFFFAOYSA-N 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 2
- IFKOEVQXCXGVEW-LLVKDONJSA-N BrC1=C(C=C(OC[C@H](C(=O)OC(C)(C)C)O)C=C1)F Chemical compound BrC1=C(C=C(OC[C@H](C(=O)OC(C)(C)C)O)C=C1)F IFKOEVQXCXGVEW-LLVKDONJSA-N 0.000 description 2
- SLBRSWXIDHFOHV-SECBINFHSA-N BrC1=CC=C(C=N1)OC[C@H](C(=O)OC(C)(C)C)O Chemical compound BrC1=CC=C(C=N1)OC[C@H](C(=O)OC(C)(C)C)O SLBRSWXIDHFOHV-SECBINFHSA-N 0.000 description 2
- JDZZHVYFAYPHEP-CQSZACIVSA-N BrC1=CC=C(C=N1)OC[C@H](C(=O)OC(C)(C)C)O[Si](C)(C)C(C)(C)C Chemical compound BrC1=CC=C(C=N1)OC[C@H](C(=O)OC(C)(C)C)O[Si](C)(C)C(C)(C)C JDZZHVYFAYPHEP-CQSZACIVSA-N 0.000 description 2
- PWLFLNZLWWGREZ-AWEZNQCLSA-N BrC=1C=NN(C=1)C[C@H](CNC(OC(C)(C)C)=O)O[Si](C)(C)C(C)(C)C Chemical compound BrC=1C=NN(C=1)C[C@H](CNC(OC(C)(C)C)=O)O[Si](C)(C)C(C)(C)C PWLFLNZLWWGREZ-AWEZNQCLSA-N 0.000 description 2
- ZUGPZIFDTUPTSQ-UHFFFAOYSA-N BrC=1N(C(N(C=1)CC(CNC(OC(C)(C)C)=O)O[Si](C)(C)C(C)(C)C)=NC(=O)OC(C)(C)C)C Chemical compound BrC=1N(C(N(C=1)CC(CNC(OC(C)(C)C)=O)O[Si](C)(C)C(C)(C)C)=NC(=O)OC(C)(C)C)C ZUGPZIFDTUPTSQ-UHFFFAOYSA-N 0.000 description 2
- OWBVFPHZPYOUJC-UHFFFAOYSA-N BrC=1N=C(N(C=1)CCCNC(OC(C)(C)C)=O)[N+](=O)[O-] Chemical compound BrC=1N=C(N(C=1)CCCNC(OC(C)(C)C)=O)[N+](=O)[O-] OWBVFPHZPYOUJC-UHFFFAOYSA-N 0.000 description 2
- LGEGTMADVIXAJA-UHFFFAOYSA-N C(C)(C)(C)OC(=O)N(C1=CC=C(C=N1)OCC(C(=O)OC(C)(C)C)O[Si](C)(C)C(C)(C)C)C1=CC=NC=C1 Chemical compound C(C)(C)(C)OC(=O)N(C1=CC=C(C=N1)OCC(C(=O)OC(C)(C)C)O[Si](C)(C)C(C)(C)C)C1=CC=NC=C1 LGEGTMADVIXAJA-UHFFFAOYSA-N 0.000 description 2
- PDSNBNODLCWGEE-HXUWFJFHSA-N C(C)(C)(C)OC(=O)NCCCN1N=CC(=C1)C1=C(C=C(OC[C@H](C(=O)OC(C)(C)C)O)C=C1)F Chemical compound C(C)(C)(C)OC(=O)NCCCN1N=CC(=C1)C1=C(C=C(OC[C@H](C(=O)OC(C)(C)C)O)C=C1)F PDSNBNODLCWGEE-HXUWFJFHSA-N 0.000 description 2
- KQVWFUZNZAKYTM-HXUWFJFHSA-N C(C)(C)(C)OC(=O)NCCNC1=NC=CC(=C1)C1=CC=C(OC[C@H](C(=O)OC(C)(C)C)O)C=C1 Chemical compound C(C)(C)(C)OC(=O)NCCNC1=NC=CC(=C1)C1=CC=C(OC[C@H](C(=O)OC(C)(C)C)O)C=C1 KQVWFUZNZAKYTM-HXUWFJFHSA-N 0.000 description 2
- HFIPANSGKJBXGA-QGZVFWFLSA-N C(C)(C)(C)OC([C@@H](COC1=CC=C(C(=O)OC)C=C1)O[Si](C)(C)C(C)(C)C)=O Chemical compound C(C)(C)(C)OC([C@@H](COC1=CC=C(C(=O)OC)C=C1)O[Si](C)(C)C(C)(C)C)=O HFIPANSGKJBXGA-QGZVFWFLSA-N 0.000 description 2
- JPBPKZXVYOTUSB-UHFFFAOYSA-L C([O-])([O-])=O.[Cs+].[N+](=O)([O-])C=1N(C=CN1)CCCNC(OC(C)(C)C)=O.[Cs+] Chemical compound C([O-])([O-])=O.[Cs+].[N+](=O)([O-])C=1N(C=CN1)CCCNC(OC(C)(C)C)=O.[Cs+] JPBPKZXVYOTUSB-UHFFFAOYSA-L 0.000 description 2
- BVFPFLSOATXUFU-XCWUCEQJSA-N CC(C)([C@@H](C1=O)NC(/C(\C2=CSC(N)=N2)=N\O[C@@H](COC(C=C2)=CC=C2C2=CN(CCCN)C=[N+]2C)C(O)=O)=O)N1OS([O-])(=O)=O Chemical compound CC(C)([C@@H](C1=O)NC(/C(\C2=CSC(N)=N2)=N\O[C@@H](COC(C=C2)=CC=C2C2=CN(CCCN)C=[N+]2C)C(O)=O)=O)N1OS([O-])(=O)=O BVFPFLSOATXUFU-XCWUCEQJSA-N 0.000 description 2
- KCBAMQOKOLXLOX-BSZYMOERSA-N CC1=C(SC=N1)C2=CC=C(C=C2)[C@H](C)NC(=O)[C@@H]3C[C@H](CN3C(=O)[C@H](C(C)(C)C)NC(=O)CCCCCCCCCCNCCCONC(=O)C4=C(C(=C(C=C4)F)F)NC5=C(C=C(C=C5)I)F)O Chemical compound CC1=C(SC=N1)C2=CC=C(C=C2)[C@H](C)NC(=O)[C@@H]3C[C@H](CN3C(=O)[C@H](C(C)(C)C)NC(=O)CCCCCCCCCCNCCCONC(=O)C4=C(C(=C(C=C4)F)F)NC5=C(C=C(C=C5)I)F)O KCBAMQOKOLXLOX-BSZYMOERSA-N 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- RGSFGYAAUTVSQA-UHFFFAOYSA-N Cyclopentane Chemical compound C1CCCC1 RGSFGYAAUTVSQA-UHFFFAOYSA-N 0.000 description 2
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 2
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 2
- 241000588724 Escherichia coli Species 0.000 description 2
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 2
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 2
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 2
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 2
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 2
- IBGPUQBHQHNERZ-GFCCVEGCSA-N N1(C=NC=C1)C1=CC=C(C=N1)OC[C@H](C(=O)OC(C)(C)C)O Chemical compound N1(C=NC=C1)C1=CC=C(C=N1)OC[C@H](C(=O)OC(C)(C)C)O IBGPUQBHQHNERZ-GFCCVEGCSA-N 0.000 description 2
- 238000005481 NMR spectroscopy Methods 0.000 description 2
- 101100272976 Panax ginseng CYP716A53v2 gene Proteins 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- YZCKVEUIGOORGS-IGMARMGPSA-N Protium Chemical compound [1H] YZCKVEUIGOORGS-IGMARMGPSA-N 0.000 description 2
- 241000720974 Protium Species 0.000 description 2
- 241000589517 Pseudomonas aeruginosa Species 0.000 description 2
- 241000607715 Serratia marcescens Species 0.000 description 2
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 2
- 239000005708 Sodium hypochlorite Substances 0.000 description 2
- PLVITEWUBLSZIF-UHFFFAOYSA-N [3-[(2-methylpropan-2-yl)oxycarbonylamino]-2-[[(2-methylpropan-2-yl)oxycarbonylamino]methyl]propyl] methanesulfonate Chemical compound CS(=O)(=O)OCC(CNC(=O)OC(C)(C)C)CNC(=O)OC(C)(C)C PLVITEWUBLSZIF-UHFFFAOYSA-N 0.000 description 2
- FZKHVWYNJCXNKV-UHFFFAOYSA-N [Si](C)(C)(C(C)(C)C)OC(C(=O)OC(C)(C)C)COC=1C=NC(=CC=1)NC1=CC=NC=C1 Chemical compound [Si](C)(C)(C(C)(C)C)OC(C(=O)OC(C)(C)C)COC=1C=NC(=CC=1)NC1=CC=NC=C1 FZKHVWYNJCXNKV-UHFFFAOYSA-N 0.000 description 2
- LQDFFWAIHFYWPB-UHFFFAOYSA-N [Si](C)(C)(C(C)(C)C)OC(C(=O)OC(C)(C)C)COC=1C=NC(=CC=1)OC1=CC=NC=C1 Chemical compound [Si](C)(C)(C(C)(C)C)OC(C(=O)OC(C)(C)C)COC=1C=NC(=CC=1)OC1=CC=NC=C1 LQDFFWAIHFYWPB-UHFFFAOYSA-N 0.000 description 2
- UCDNRKQFCWVTGQ-INIZCTEOSA-N [Si](C)(C)(C(C)(C)C)O[C@@H](CNC(OC(C)(C)C)=O)CO[Si](C)(C)C(C)(C)C Chemical compound [Si](C)(C)(C(C)(C)C)O[C@@H](CNC(OC(C)(C)C)=O)CO[Si](C)(C)C(C)(C)C UCDNRKQFCWVTGQ-INIZCTEOSA-N 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 125000000304 alkynyl group Chemical group 0.000 description 2
- CHKQALUEEULCPZ-UHFFFAOYSA-N amino 2,4,6-trimethylbenzenesulfonate Chemical compound CC1=CC(C)=C(S(=O)(=O)ON)C(C)=C1 CHKQALUEEULCPZ-UHFFFAOYSA-N 0.000 description 2
- 235000011114 ammonium hydroxide Nutrition 0.000 description 2
- 150000008064 anhydrides Chemical class 0.000 description 2
- 239000008228 bacteriostatic water for injection Substances 0.000 description 2
- 150000007514 bases Chemical class 0.000 description 2
- 229940077388 benzenesulfonate Drugs 0.000 description 2
- FZNZKRHEWXUQLP-HXUWFJFHSA-N benzhydryl (2R)-3-(4-bromo-3-fluorophenoxy)-2-hydroxypropanoate Chemical compound BrC1=C(C=C(OC[C@H](C(=O)OC(C2=CC=CC=C2)C2=CC=CC=C2)O)C=C1)F FZNZKRHEWXUQLP-HXUWFJFHSA-N 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- IOJUPLGTWVMSFF-UHFFFAOYSA-N benzothiazole Chemical compound C1=CC=C2SC=NC2=C1 IOJUPLGTWVMSFF-UHFFFAOYSA-N 0.000 description 2
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 2
- 150000001649 bromium compounds Chemical class 0.000 description 2
- GZUXJHMPEANEGY-UHFFFAOYSA-N bromomethane Chemical compound BrC GZUXJHMPEANEGY-UHFFFAOYSA-N 0.000 description 2
- 230000005587 bubbling Effects 0.000 description 2
- RYYVLZVUVIJVGH-UHFFFAOYSA-N caffeine Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2C RYYVLZVUVIJVGH-UHFFFAOYSA-N 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 2
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 2
- 239000003054 catalyst Substances 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- 238000002512 chemotherapy Methods 0.000 description 2
- 239000000460 chlorine Substances 0.000 description 2
- 229910052801 chlorine Inorganic materials 0.000 description 2
- 229940125833 compound 23 Drugs 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 125000000392 cycloalkenyl group Chemical group 0.000 description 2
- 230000002939 deleterious effect Effects 0.000 description 2
- 229910052805 deuterium Inorganic materials 0.000 description 2
- 229940043279 diisopropylamine Drugs 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 description 2
- 229910001873 dinitrogen Inorganic materials 0.000 description 2
- 239000006185 dispersion Substances 0.000 description 2
- LZWLLMFYVGUUAL-UHFFFAOYSA-L ditert-butyl(cyclopenta-1,3-dien-1-yl)phosphane;dichloropalladium;iron(2+) Chemical compound [Fe+2].Cl[Pd]Cl.CC(C)(C)P(C(C)(C)C)C1=CC=C[CH-]1.CC(C)(C)P(C(C)(C)C)C1=CC=C[CH-]1 LZWLLMFYVGUUAL-UHFFFAOYSA-L 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 150000002118 epoxides Chemical class 0.000 description 2
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 2
- 239000011737 fluorine Substances 0.000 description 2
- 229910052731 fluorine Inorganic materials 0.000 description 2
- 238000009472 formulation Methods 0.000 description 2
- 238000004108 freeze drying Methods 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- KWIUHFFTVRNATP-UHFFFAOYSA-N glycine betaine Chemical compound C[N+](C)(C)CC([O-])=O KWIUHFFTVRNATP-UHFFFAOYSA-N 0.000 description 2
- 208000027096 gram-negative bacterial infections Diseases 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- XGIHQYAWBCFNPY-AZOCGYLKSA-N hydrabamine Chemical compound C([C@@H]12)CC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC[C@@]1(C)CNCCNC[C@@]1(C)[C@@H]2CCC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC1 XGIHQYAWBCFNPY-AZOCGYLKSA-N 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 239000004615 ingredient Substances 0.000 description 2
- 150000007529 inorganic bases Chemical class 0.000 description 2
- 150000004694 iodide salts Chemical class 0.000 description 2
- 239000011630 iodine Substances 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- MCVFFRWZNYZUIJ-UHFFFAOYSA-M lithium;trifluoromethanesulfonate Chemical compound [Li+].[O-]S(=O)(=O)C(F)(F)F MCVFFRWZNYZUIJ-UHFFFAOYSA-M 0.000 description 2
- 239000011777 magnesium Substances 0.000 description 2
- 229910052749 magnesium Inorganic materials 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 125000005394 methallyl group Chemical group 0.000 description 2
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Natural products C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 2
- OCVJWIIHKKCWBC-UHFFFAOYSA-N methyl 2-(4-bromophenoxy)-3-hydroxypropanoate Chemical compound BrC1=CC=C(OC(C(=O)OC)CO)C=C1 OCVJWIIHKKCWBC-UHFFFAOYSA-N 0.000 description 2
- HIVDYKZQIRSLRV-UHFFFAOYSA-N methyl 2-(4-bromophenoxy)-3-methoxypropanoate Chemical compound BrC1=CC=C(OC(C(=O)OC)COC)C=C1 HIVDYKZQIRSLRV-UHFFFAOYSA-N 0.000 description 2
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 2
- 244000005700 microbiome Species 0.000 description 2
- 239000002480 mineral oil Substances 0.000 description 2
- 235000010446 mineral oil Nutrition 0.000 description 2
- 150000007522 mineralic acids Chemical class 0.000 description 2
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 2
- SFMJNHNUOVADRW-UHFFFAOYSA-N n-[5-[9-[4-(methanesulfonamido)phenyl]-2-oxobenzo[h][1,6]naphthyridin-1-yl]-2-methylphenyl]prop-2-enamide Chemical compound C1=C(NC(=O)C=C)C(C)=CC=C1N1C(=O)C=CC2=C1C1=CC(C=3C=CC(NS(C)(=O)=O)=CC=3)=CC=C1N=C2 SFMJNHNUOVADRW-UHFFFAOYSA-N 0.000 description 2
- XBXCNNQPRYLIDE-UHFFFAOYSA-M n-tert-butylcarbamate Chemical compound CC(C)(C)NC([O-])=O XBXCNNQPRYLIDE-UHFFFAOYSA-M 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 125000004430 oxygen atom Chemical group O* 0.000 description 2
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 2
- 239000000546 pharmaceutical excipient Substances 0.000 description 2
- 238000005191 phase separation Methods 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- 230000003389 potentiating effect Effects 0.000 description 2
- 239000002243 precursor Substances 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 230000035755 proliferation Effects 0.000 description 2
- CAEWJEXPFKNBQL-UHFFFAOYSA-N prop-2-enyl carbonochloridate Chemical compound ClC(=O)OCC=C CAEWJEXPFKNBQL-UHFFFAOYSA-N 0.000 description 2
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 2
- 125000002206 pyridazin-3-yl group Chemical group [H]C1=C([H])C([H])=C(*)N=N1 0.000 description 2
- TUEVQPUBMYVDSI-UHFFFAOYSA-N pyridin-1-ium 2,2,2-trifluoroacetate Chemical compound C1=CC=[NH+]C=C1.C1=CC=[NH+]C=C1.[O-]C(=O)C(F)(F)F.[O-]C(=O)C(F)(F)F TUEVQPUBMYVDSI-UHFFFAOYSA-N 0.000 description 2
- 125000004076 pyridyl group Chemical group 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- SUKJFIGYRHOWBL-UHFFFAOYSA-N sodium hypochlorite Chemical compound [Na+].Cl[O-] SUKJFIGYRHOWBL-UHFFFAOYSA-N 0.000 description 2
- 235000009518 sodium iodide Nutrition 0.000 description 2
- JQWHASGSAFIOCM-UHFFFAOYSA-M sodium periodate Chemical compound [Na+].[O-]I(=O)(=O)=O JQWHASGSAFIOCM-UHFFFAOYSA-M 0.000 description 2
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 2
- 238000010561 standard procedure Methods 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 125000004434 sulfur atom Chemical group 0.000 description 2
- 208000024891 symptom Diseases 0.000 description 2
- OUJSJVLKCZFVQH-JOCHJYFZSA-N tert-butyl (2R)-2-hydroxy-3-[4-[6-[3-[(2-methylpropan-2-yl)oxycarbonylamino]prop-1-ynyl]pyridin-3-yl]phenoxy]propanoate Chemical compound C(C)(C)(C)OC(=O)NCC#CC1=CC=C(C=N1)C1=CC=C(OC[C@H](C(=O)OC(C)(C)C)O)C=C1 OUJSJVLKCZFVQH-JOCHJYFZSA-N 0.000 description 2
- RHONMXZMNSIHBT-CYBMUJFWSA-N tert-butyl (2R)-3-(4-ethenylphenoxy)-2-hydroxypropanoate Chemical compound O[C@@H](C(=O)OC(C)(C)C)COC1=CC=C(C=C1)C=C RHONMXZMNSIHBT-CYBMUJFWSA-N 0.000 description 2
- SVLNBADGWSNZOW-HXUWFJFHSA-N tert-butyl (2R)-3-[3-chloro-4-[1-[3-[(2-methylpropan-2-yl)oxycarbonylamino]propyl]pyrazol-4-yl]phenoxy]-2-hydroxypropanoate Chemical compound C(C)(C)(C)OC(=O)NCCCN1N=CC(=C1)C1=C(C=C(OC[C@H](C(=O)OC(C)(C)C)O)C=C1)Cl SVLNBADGWSNZOW-HXUWFJFHSA-N 0.000 description 2
- TVRGXJBRUFXXCQ-UMSFTDKQSA-N tert-butyl (2S)-2-(1,3-dioxoisoindol-2-yl)oxy-2-methyl-3-[4-[6-[2-[(2-methylpropan-2-yl)oxycarbonylamino]ethylamino]pyridin-3-yl]phenoxy]propanoate Chemical compound C(C)(C)(C)OC(=O)NCCNC1=CC=C(C=N1)C1=CC=C(OC[C@](C(=O)OC(C)(C)C)(C)ON2C(C3=CC=CC=C3C2=O)=O)C=C1 TVRGXJBRUFXXCQ-UMSFTDKQSA-N 0.000 description 2
- UWXKTIFNMXEQPM-SANMLTNESA-N tert-butyl (2S)-2-(1,3-dioxoisoindol-2-yl)oxy-3-[4-[1-[3-[(2-methylpropan-2-yl)oxycarbonylamino]propyl]pyrazol-4-yl]phenoxy]propanoate Chemical compound C(C)(C)(C)OC(=O)NCCCN1N=CC(=C1)C1=CC=C(OC[C@@H](C(=O)OC(C)(C)C)ON2C(C3=CC=CC=C3C2=O)=O)C=C1 UWXKTIFNMXEQPM-SANMLTNESA-N 0.000 description 2
- HNBUWVCFOYYHIF-SANMLTNESA-N tert-butyl (2S)-2-aminooxy-2-methyl-3-[4-[6-[2-[(2-methylpropan-2-yl)oxycarbonylamino]ethylamino]pyridin-3-yl]phenoxy]propanoate Chemical compound NO[C@](C(=O)OC(C)(C)C)(COC1=CC=C(C=C1)C=1C=NC(=CC=1)NCCNC(=O)OC(C)(C)C)C HNBUWVCFOYYHIF-SANMLTNESA-N 0.000 description 2
- QUSSBBJODDOWTD-AWEZNQCLSA-N tert-butyl (2S)-2-aminooxy-3-(4-bromophenoxy)-2-methylpropanoate Chemical compound NO[C@](C(=O)OC(C)(C)C)(COC1=CC=C(C=C1)Br)C QUSSBBJODDOWTD-AWEZNQCLSA-N 0.000 description 2
- RFOIQKPPXITQFO-QFIPXVFZSA-O tert-butyl (2S)-2-aminooxy-3-[4-[1-(2-azidoethyl)-2-[3-[(2-methylpropan-2-yl)oxycarbonylamino]propyl]pyrazol-1-ium-4-yl]phenoxy]propanoate Chemical compound NO[C@@H](COC1=CC=C(C=C1)C=1C=[N+](N(C=1)CCCNC(=O)OC(C)(C)C)CCN=[N+]=[N-])C(=O)OC(C)(C)C RFOIQKPPXITQFO-QFIPXVFZSA-O 0.000 description 2
- SGCBZXYEVPJNLZ-SFHVURJKSA-O tert-butyl (2S)-2-aminooxy-3-[6-[3-[3-[(2-methylpropan-2-yl)oxycarbonylamino]propyl]imidazol-3-ium-1-yl]pyridin-3-yl]oxypropanoate Chemical compound NO[C@@H](COC=1C=CC(=NC=1)N1C=[N+](C=C1)CCCNC(=O)OC(C)(C)C)C(=O)OC(C)(C)C SGCBZXYEVPJNLZ-SFHVURJKSA-O 0.000 description 2
- IFKWIFYTNIBYDT-IBGZPJMESA-N tert-butyl (2S)-3-(4-bromophenoxy)-2-methyl-2-[(2-methylpropan-2-yl)oxycarbonylamino]oxypropanoate Chemical compound BrC1=CC=C(OC[C@](C(=O)OC(C)(C)C)(C)ONC(=O)OC(C)(C)C)C=C1 IFKWIFYTNIBYDT-IBGZPJMESA-N 0.000 description 2
- TYSLPPILACEUDT-NRFANRHFSA-N tert-butyl (2S)-3-[6-[3-(3-aminopropyl)imidazol-3-ium-1-yl]pyridin-3-yl]oxy-2-(1,3-dioxoisoindol-2-yl)oxypropanoate Chemical compound NCCC[N+]1=CN(C=C1)C1=NC=C(C=C1)OC[C@@H](C(=O)OC(C)(C)C)ON1C(C2=CC=CC=C2C1=O)=O TYSLPPILACEUDT-NRFANRHFSA-N 0.000 description 2
- UJLDFDMDAGJZSV-SFYZADRCSA-N tert-butyl (3s,4r)-3,4-diaminopiperidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CC[C@@H](N)[C@@H](N)C1 UJLDFDMDAGJZSV-SFYZADRCSA-N 0.000 description 2
- NYOGNFGAOMCBNB-CABCVRRESA-N tert-butyl (3s,4r)-3-azido-4-(phenylmethoxycarbonylamino)piperidine-1-carboxylate Chemical compound [N-]=[N+]=N[C@H]1CN(C(=O)OC(C)(C)C)CC[C@H]1NC(=O)OCC1=CC=CC=C1 NYOGNFGAOMCBNB-CABCVRRESA-N 0.000 description 2
- GDUWCNDISUJOPX-UHFFFAOYSA-N tert-butyl 2-(1,3-dioxoisoindol-2-yl)oxy-2-methyl-3-[4-[1-[3-[(2-methylpropan-2-yl)oxycarbonylamino]propyl]pyrazol-4-yl]phenoxy]propanoate Chemical compound C(C)(C)(C)OC(=O)NCCCN1N=CC(=C1)C1=CC=C(OCC(C(=O)OC(C)(C)C)(C)ON2C(C3=CC=CC=C3C2=O)=O)C=C1 GDUWCNDISUJOPX-UHFFFAOYSA-N 0.000 description 2
- YEWVGSGIUGHBPA-UHFFFAOYSA-N tert-butyl 2-aminooxy-2-methyl-3-[4-[1-[3-[(2-methylpropan-2-yl)oxycarbonylamino]propyl]pyrazol-4-yl]phenoxy]propanoate Chemical compound NOC(C(=O)OC(C)(C)C)(COC1=CC=C(C=C1)C=1C=NN(C=1)CCCNC(=O)OC(C)(C)C)C YEWVGSGIUGHBPA-UHFFFAOYSA-N 0.000 description 2
- QUSSBBJODDOWTD-UHFFFAOYSA-N tert-butyl 2-aminooxy-3-(4-bromophenoxy)-2-methylpropanoate Chemical compound NOC(C(=O)OC(C)(C)C)(COC1=CC=C(C=C1)Br)C QUSSBBJODDOWTD-UHFFFAOYSA-N 0.000 description 2
- KTJRJTMYZHECEB-UHFFFAOYSA-N tert-butyl 2-hydroxy-3-(6-pyridin-4-yloxypyridin-3-yl)oxypropanoate Chemical compound OC(C(=O)OC(C)(C)C)COC=1C=NC(=CC=1)OC1=CC=NC=C1 KTJRJTMYZHECEB-UHFFFAOYSA-N 0.000 description 2
- HMAGZHPJACHPDG-UHFFFAOYSA-N tert-butyl 2-methyloxirane-2-carboxylate Chemical compound CC(C)(C)OC(=O)C1(C)CO1 HMAGZHPJACHPDG-UHFFFAOYSA-N 0.000 description 2
- YYGMRDIVZOVVTO-UHFFFAOYSA-N tert-butyl 3-(4-bromo-N-methylanilino)-2-hydroxypropanoate Chemical compound BrC1=CC=C(C=C1)N(CC(C(=O)OC(C)(C)C)O)C YYGMRDIVZOVVTO-UHFFFAOYSA-N 0.000 description 2
- MBCZNBURDIVTDV-UHFFFAOYSA-N tert-butyl 3-(4-bromoanilino)-2-hydroxypropanoate Chemical compound BrC1=CC=C(C=C1)NCC(C(=O)OC(C)(C)C)O MBCZNBURDIVTDV-UHFFFAOYSA-N 0.000 description 2
- HXRDRJKAEYHOBB-UHFFFAOYSA-N tert-butyl 3-(hydroxymethyl)azetidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CC(CO)C1 HXRDRJKAEYHOBB-UHFFFAOYSA-N 0.000 description 2
- LXJWAAYEIOZSNE-XIFFEERXSA-O tert-butyl 3-[[4-[4-[(2S)-2-(1,3-dioxoisoindol-2-yl)oxy-3-[(2-methylpropan-2-yl)oxy]-3-oxopropoxy]phenyl]-2-[3-[(2-methylpropan-2-yl)oxycarbonylamino]propyl]pyrazol-1-ium-1-yl]methyl]azetidine-1-carboxylate Chemical compound C(C)(C)(C)OC([C@H](COC1=CC=C(C=C1)C=1C=[N+](N(C=1)CCCNC(=O)OC(C)(C)C)CC1CN(C1)C(=O)OC(C)(C)C)ON1C(C2=CC=CC=C2C1=O)=O)=O LXJWAAYEIOZSNE-XIFFEERXSA-O 0.000 description 2
- BONSKTIHNARTGJ-UHFFFAOYSA-N tert-butyl 3-fluoro-3-(hydroxymethyl)azetidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CC(F)(CO)C1 BONSKTIHNARTGJ-UHFFFAOYSA-N 0.000 description 2
- FDLQADPNBWVVMY-UHFFFAOYSA-N tert-butyl N-[2-[(4-bromopyridin-2-yl)amino]ethyl]carbamate Chemical compound BrC1=CC(=NC=C1)NCCNC(OC(C)(C)C)=O FDLQADPNBWVVMY-UHFFFAOYSA-N 0.000 description 2
- HMRJEURUHXVKJW-UHFFFAOYSA-N tert-butyl N-[3-(2-aminoimidazol-1-yl)propyl]carbamate Chemical compound NC=1N(C=CN=1)CCCNC(OC(C)(C)C)=O HMRJEURUHXVKJW-UHFFFAOYSA-N 0.000 description 2
- DADZBYOXRZHEGG-UHFFFAOYSA-N tert-butyl N-[3-(2-nitroimidazol-1-yl)propyl]carbamate Chemical compound [N+](=O)([O-])C=1N(C=CN=1)CCCNC(OC(C)(C)C)=O DADZBYOXRZHEGG-UHFFFAOYSA-N 0.000 description 2
- QYISOPOXUWTEOS-UHFFFAOYSA-N tert-butyl N-[3-(4-bromopyrazol-1-yl)propyl]carbamate Chemical compound BrC=1C=NN(C=1)CCCNC(OC(C)(C)C)=O QYISOPOXUWTEOS-UHFFFAOYSA-N 0.000 description 2
- SOLZPXYDFWIYFX-UHFFFAOYSA-N tert-butyl N-[3-[(6-chloropyridazin-3-yl)amino]propyl]carbamate Chemical compound CC(C)(C)OC(=O)NCCCNC1=CC=C(Cl)N=N1 SOLZPXYDFWIYFX-UHFFFAOYSA-N 0.000 description 2
- XKSFMPBVQNXEPD-SECBINFHSA-N tert-butyl N-[4-[2-[[(3S)-2,2-dimethyl-4-oxo-1-sulfooxyazetidin-3-yl]amino]-2-oxoacetyl]-1,3-thiazol-2-yl]carbamate Chemical compound S(=O)(=O)(ON1C([C@@H](C1=O)NC(C(=O)C=1N=C(SC=1)NC(=O)OC(C)(C)C)=O)(C)C)O XKSFMPBVQNXEPD-SECBINFHSA-N 0.000 description 2
- IXZDIALLLMRYOU-UHFFFAOYSA-N tert-butyl hypochlorite Chemical compound CC(C)(C)OCl IXZDIALLLMRYOU-UHFFFAOYSA-N 0.000 description 2
- ACNRTYKOPZDRCO-UHFFFAOYSA-N tert-butyl n-(2-oxoethyl)carbamate Chemical compound CC(C)(C)OC(=O)NCC=O ACNRTYKOPZDRCO-UHFFFAOYSA-N 0.000 description 2
- WHIGRQBQVQDNDU-UHFFFAOYSA-N tert-butyl n-[2-[(5-bromopyridin-2-yl)amino]ethyl]carbamate Chemical compound CC(C)(C)OC(=O)NCCNC1=CC=C(Br)C=N1 WHIGRQBQVQDNDU-UHFFFAOYSA-N 0.000 description 2
- ISXSCDLOGDJUNJ-UHFFFAOYSA-N tert-butyl prop-2-enoate Chemical compound CC(C)(C)OC(=O)C=C ISXSCDLOGDJUNJ-UHFFFAOYSA-N 0.000 description 2
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 2
- YAPQBXQYLJRXSA-UHFFFAOYSA-N theobromine Chemical compound CN1C(=O)NC(=O)C2=C1N=CN2C YAPQBXQYLJRXSA-UHFFFAOYSA-N 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 231100000331 toxic Toxicity 0.000 description 2
- 230000002588 toxic effect Effects 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- SEDZOYHHAIAQIW-UHFFFAOYSA-N trimethylsilyl azide Chemical compound C[Si](C)(C)N=[N+]=[N-] SEDZOYHHAIAQIW-UHFFFAOYSA-N 0.000 description 2
- 238000001665 trituration Methods 0.000 description 2
- 238000000825 ultraviolet detection Methods 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- 239000003643 water by type Substances 0.000 description 2
- LSPHULWDVZXLIL-UHFFFAOYSA-N (+/-)-Camphoric acid Chemical compound CC1(C)C(C(O)=O)CCC1(C)C(O)=O LSPHULWDVZXLIL-UHFFFAOYSA-N 0.000 description 1
- UUPQXPOQKUEZPC-UHFFFAOYSA-N (2-oxoazetidin-1-yl) hydrogen sulfate Chemical compound OS(=O)(=O)ON1CCC1=O UUPQXPOQKUEZPC-UHFFFAOYSA-N 0.000 description 1
- OEDBYQLGYUBXJT-AREMUKBSSA-N (2R)-1-(4-bromophenoxy)-3-trityloxypropan-2-ol Chemical compound BrC1=CC=C(OC[C@H](COC(C2=CC=CC=C2)(C2=CC=CC=C2)C2=CC=CC=C2)O)C=C1 OEDBYQLGYUBXJT-AREMUKBSSA-N 0.000 description 1
- AJXYLTNPXYZSMP-CQSZACIVSA-N (2R)-2-[tert-butyl(dimethyl)silyl]oxy-3-(6-imidazol-1-ylpyridin-3-yl)oxypropanoic acid Chemical compound CC(C)(C)[Si](C)(C)O[C@H](COC1=CN=C(C=C1)N2C=CN=C2)C(=O)O AJXYLTNPXYZSMP-CQSZACIVSA-N 0.000 description 1
- PFNVNNUCTNTEPI-MRVPVSSYSA-N (2R)-3-(4-bromo-3-fluorophenoxy)-2-hydroxypropanoic acid Chemical compound BrC1=C(C=C(OC[C@H](C(=O)O)O)C=C1)F PFNVNNUCTNTEPI-MRVPVSSYSA-N 0.000 description 1
- HJUCAUCQKUELKW-LLVKDONJSA-N (2R)-3-(6-bromopyridin-3-yl)oxy-2-[tert-butyl(dimethyl)silyl]oxypropanoic acid Chemical compound CC(C)(C)[Si](C)(C)O[C@H](COC1=CN=C(C=C1)Br)C(=O)O HJUCAUCQKUELKW-LLVKDONJSA-N 0.000 description 1
- YYTSGNJTASLUOY-GSVOUGTGSA-N (2r)-1-chloropropan-2-ol Chemical compound C[C@@H](O)CCl YYTSGNJTASLUOY-GSVOUGTGSA-N 0.000 description 1
- ISUIVWNWEDIHJD-HRFVKAFMSA-N (2s,3s)-3-azaniumyl-2-methyl-4-oxoazetidine-1-sulfonate Chemical compound C[C@H]1[C@H](N)C(=O)N1S(O)(=O)=O ISUIVWNWEDIHJD-HRFVKAFMSA-N 0.000 description 1
- GHOKWGTUZJEAQD-ZETCQYMHSA-N (D)-(+)-Pantothenic acid Chemical compound OCC(C)(C)[C@@H](O)C(=O)NCCC(O)=O GHOKWGTUZJEAQD-ZETCQYMHSA-N 0.000 description 1
- XZNOAVNRSFURIR-UHFFFAOYSA-N 1,1,1,3,3,3-hexafluoro-2-(trifluoromethyl)propan-2-ol Chemical compound FC(F)(F)C(O)(C(F)(F)F)C(F)(F)F XZNOAVNRSFURIR-UHFFFAOYSA-N 0.000 description 1
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical group CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 1
- KTZQTRPPVKQPFO-UHFFFAOYSA-N 1,2-benzoxazole Chemical compound C1=CC=C2C=NOC2=C1 KTZQTRPPVKQPFO-UHFFFAOYSA-N 0.000 description 1
- PAAZPARNPHGIKF-UHFFFAOYSA-N 1,2-dibromoethane Chemical compound BrCCBr PAAZPARNPHGIKF-UHFFFAOYSA-N 0.000 description 1
- BCMCBBGGLRIHSE-UHFFFAOYSA-N 1,3-benzoxazole Chemical compound C1=CC=C2OC=NC2=C1 BCMCBBGGLRIHSE-UHFFFAOYSA-N 0.000 description 1
- WORJRXHJTUTINR-UHFFFAOYSA-N 1,4-dioxane;hydron;chloride Chemical compound Cl.C1COCCO1 WORJRXHJTUTINR-UHFFFAOYSA-N 0.000 description 1
- 125000005940 1,4-dioxanyl group Chemical group 0.000 description 1
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical group C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 1
- VFWCMGCRMGJXDK-UHFFFAOYSA-N 1-chlorobutane Chemical compound CCCCCl VFWCMGCRMGJXDK-UHFFFAOYSA-N 0.000 description 1
- VUQPJRPDRDVQMN-UHFFFAOYSA-N 1-chlorooctadecane Chemical class CCCCCCCCCCCCCCCCCCCl VUQPJRPDRDVQMN-UHFFFAOYSA-N 0.000 description 1
- LUQUSMOTPHBKHT-UHFFFAOYSA-N 1-hydroxypyrrolidine-2,5-dione;hydrate Chemical compound O.ON1C(=O)CCC1=O LUQUSMOTPHBKHT-UHFFFAOYSA-N 0.000 description 1
- 125000006017 1-propenyl group Chemical group 0.000 description 1
- HYZJCKYKOHLVJF-UHFFFAOYSA-N 1H-benzimidazole Chemical compound C1=CC=C2NC=NC2=C1 HYZJCKYKOHLVJF-UHFFFAOYSA-N 0.000 description 1
- KDSPLKNONIUZSZ-UHFFFAOYSA-N 2,4,5,6-tetrahydroxyhexanal Chemical compound OCC(O)C(O)CC(O)C=O KDSPLKNONIUZSZ-UHFFFAOYSA-N 0.000 description 1
- SURCGQGDUADKBL-UHFFFAOYSA-N 2-(2-hydroxyethylamino)-5-nitrobenzo[de]isoquinoline-1,3-dione Chemical compound [O-][N+](=O)C1=CC(C(N(NCCO)C2=O)=O)=C3C2=CC=CC3=C1 SURCGQGDUADKBL-UHFFFAOYSA-N 0.000 description 1
- PAWQVTBBRAZDMG-UHFFFAOYSA-N 2-(3-bromo-2-fluorophenyl)acetic acid Chemical compound OC(=O)CC1=CC=CC(Br)=C1F PAWQVTBBRAZDMG-UHFFFAOYSA-N 0.000 description 1
- WNSJUFSJUZSGSL-UHFFFAOYSA-N 2-(6-bromopyridin-3-yl)oxyethanol Chemical compound OCCOC1=CC=C(Br)N=C1 WNSJUFSJUZSGSL-UHFFFAOYSA-N 0.000 description 1
- VLRZNFJVVSSIAA-UHFFFAOYSA-N 2-(difluoromethyl)-6-[1-(trifluoromethyl)cyclopropyl]benzoic acid Chemical compound FC(C1=C(C(=O)O)C(=CC=C1)C1(CC1)C(F)(F)F)F VLRZNFJVVSSIAA-UHFFFAOYSA-N 0.000 description 1
- MSWZFWKMSRAUBD-IVMDWMLBSA-N 2-amino-2-deoxy-D-glucopyranose Chemical compound N[C@H]1C(O)O[C@H](CO)[C@@H](O)[C@@H]1O MSWZFWKMSRAUBD-IVMDWMLBSA-N 0.000 description 1
- LDLCZOVUSADOIV-UHFFFAOYSA-N 2-bromoethanol Chemical compound OCCBr LDLCZOVUSADOIV-UHFFFAOYSA-N 0.000 description 1
- 125000004974 2-butenyl group Chemical group C(C=CC)* 0.000 description 1
- SZIFAVKTNFCBPC-UHFFFAOYSA-N 2-chloroethanol Chemical compound OCCCl SZIFAVKTNFCBPC-UHFFFAOYSA-N 0.000 description 1
- BFSVOASYOCHEOV-UHFFFAOYSA-N 2-diethylaminoethanol Chemical compound CCN(CC)CCO BFSVOASYOCHEOV-UHFFFAOYSA-N 0.000 description 1
- 229940013085 2-diethylaminoethanol Drugs 0.000 description 1
- OPLCYGTZYZLOKI-UHFFFAOYSA-N 2-hydroxy-1,3-dihydroisoindole Chemical compound C1=CC=C2CN(O)CC2=C1 OPLCYGTZYZLOKI-UHFFFAOYSA-N 0.000 description 1
- 125000006029 2-methyl-2-butenyl group Chemical group 0.000 description 1
- 229940080296 2-naphthalenesulfonate Drugs 0.000 description 1
- WMPPDTMATNBGJN-UHFFFAOYSA-N 2-phenylethylbromide Chemical class BrCCC1=CC=CC=C1 WMPPDTMATNBGJN-UHFFFAOYSA-N 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- 125000001494 2-propynyl group Chemical group [H]C#CC([H])([H])* 0.000 description 1
- YSIXZOWGRJZVLY-UHFFFAOYSA-N 3,3-diphenyldiazirine Chemical compound C1=CC=CC=C1C1(C=2C=CC=CC=2)N=N1 YSIXZOWGRJZVLY-UHFFFAOYSA-N 0.000 description 1
- GUSWJGOYDXFJSI-UHFFFAOYSA-N 3,6-dichloropyridazine Chemical compound ClC1=CC=C(Cl)N=N1 GUSWJGOYDXFJSI-UHFFFAOYSA-N 0.000 description 1
- PQVLJKLTNMOGBT-UHFFFAOYSA-N 3-(1-tritylimidazol-4-yl)propan-1-amine Chemical compound C1=NC(CCCN)=CN1C(C=1C=CC=CC=1)(C=1C=CC=CC=1)C1=CC=CC=C1 PQVLJKLTNMOGBT-UHFFFAOYSA-N 0.000 description 1
- JEVPOYFZJLLZIA-UHFFFAOYSA-N 3-(1-tritylimidazol-4-yl)propan-1-ol Chemical compound C1=NC(CCCO)=CN1C(C=1C=CC=CC=1)(C=1C=CC=CC=1)C1=CC=CC=C1 JEVPOYFZJLLZIA-UHFFFAOYSA-N 0.000 description 1
- PDXBIRJSELAZBA-UHFFFAOYSA-N 3-(4-bromo-2-imino-3-methylimidazol-1-yl)propan-1-amine Chemical compound BrC=1N(C(N(C=1)CCCN)=N)C PDXBIRJSELAZBA-UHFFFAOYSA-N 0.000 description 1
- XLAKVMDAMKGUET-UHFFFAOYSA-N 3-bromo-1h-pyridazin-6-one Chemical group BrC=1C=CC(=O)NN=1 XLAKVMDAMKGUET-UHFFFAOYSA-N 0.000 description 1
- USEGQJLHQSTGHW-UHFFFAOYSA-N 3-bromo-2-methylprop-1-ene Chemical compound CC(=C)CBr USEGQJLHQSTGHW-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-M 3-carboxy-2,3-dihydroxypropanoate Chemical compound OC(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-M 0.000 description 1
- ALKYHXVLJMQRLQ-UHFFFAOYSA-M 3-carboxynaphthalen-2-olate Chemical compound C1=CC=C2C=C(C([O-])=O)C(O)=CC2=C1 ALKYHXVLJMQRLQ-UHFFFAOYSA-M 0.000 description 1
- 125000004575 3-pyrrolidinyl group Chemical group [H]N1C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- XKTYXVDYIKIYJP-UHFFFAOYSA-N 3h-dioxole Chemical compound C1OOC=C1 XKTYXVDYIKIYJP-UHFFFAOYSA-N 0.000 description 1
- UGNSMKDDFAUGFT-UHFFFAOYSA-N 4,4-dimethyl-2-phenyl-5h-1,3-oxazole Chemical compound CC1(C)COC(C=2C=CC=CC=2)=N1 UGNSMKDDFAUGFT-UHFFFAOYSA-N 0.000 description 1
- NSPJBMPRABAKOZ-UHFFFAOYSA-N 4-(1,3-dinitropropan-2-yl)phenol Chemical compound OC1=CC=C(C(C[N+]([O-])=O)C[N+]([O-])=O)C=C1 NSPJBMPRABAKOZ-UHFFFAOYSA-N 0.000 description 1
- ZFKXJCOPCYEQFO-UHFFFAOYSA-N 4-(4-bromophenyl)-2-hydroxybutanoic acid Chemical compound OC(=O)C(O)CCC1=CC=C(Br)C=C1 ZFKXJCOPCYEQFO-UHFFFAOYSA-N 0.000 description 1
- NUKYPUAOHBNCPY-UHFFFAOYSA-N 4-aminopyridine Chemical compound NC1=CC=NC=C1 NUKYPUAOHBNCPY-UHFFFAOYSA-N 0.000 description 1
- FQEYHIPPYOSPLF-UHFFFAOYSA-N 4-bromo-3-chlorophenol Chemical compound OC1=CC=C(Br)C(Cl)=C1 FQEYHIPPYOSPLF-UHFFFAOYSA-N 0.000 description 1
- AYVPVDWQZAAZCM-UHFFFAOYSA-N 4-bromo-n-methylaniline Chemical compound CNC1=CC=C(Br)C=C1 AYVPVDWQZAAZCM-UHFFFAOYSA-N 0.000 description 1
- WDFQBORIUYODSI-UHFFFAOYSA-N 4-bromoaniline Chemical compound NC1=CC=C(Br)C=C1 WDFQBORIUYODSI-UHFFFAOYSA-N 0.000 description 1
- BAQKUNMKVAPWGU-UHFFFAOYSA-N 4-bromopyridin-2-amine Chemical compound NC1=CC(Br)=CC=N1 BAQKUNMKVAPWGU-UHFFFAOYSA-N 0.000 description 1
- HVCNXQOWACZAFN-UHFFFAOYSA-N 4-ethylmorpholine Chemical compound CCN1CCOCC1 HVCNXQOWACZAFN-UHFFFAOYSA-N 0.000 description 1
- GCNTZFIIOFTKIY-UHFFFAOYSA-N 4-hydroxypyridine Chemical compound OC1=CC=NC=C1 GCNTZFIIOFTKIY-UHFFFAOYSA-N 0.000 description 1
- ZVTWZSXLLMNMQC-UHFFFAOYSA-N 4-phenylmethoxybenzaldehyde Chemical compound C1=CC(C=O)=CC=C1OCC1=CC=CC=C1 ZVTWZSXLLMNMQC-UHFFFAOYSA-N 0.000 description 1
- KHZPYJZDNWQLHF-AZKKKJBWSA-N 5-[4-[(2S)-2-(1,3-dioxoisoindol-2-yl)oxy-3-[(2-methylpropan-2-yl)oxy]-3-oxopropoxy]phenyl]-2-[(2-methylpropan-2-yl)oxycarbonyl-[2-[(2-methylpropan-2-yl)oxycarbonylamino]ethyl]amino]-5,6-dihydro-4H-pyrimidine-1-carboxylic acid Chemical compound CC(C)(C)OC([C@H](COC1=CC=C(C(C2)CN=C(N(CCNC(OC(C)(C)C)=O)C(OC(C)(C)C)=O)N2C(O)=O)C=C1)ON(C(C1=CC=CC=C11)=O)C1=O)=O KHZPYJZDNWQLHF-AZKKKJBWSA-N 0.000 description 1
- YNQUXLMARMDZRC-UHFFFAOYSA-N 6-bromopyridin-3-ol 2-(6-bromopyridin-3-yl)oxyethanol Chemical compound Oc1ccc(Br)nc1.OCCOc1ccc(Br)nc1 YNQUXLMARMDZRC-UHFFFAOYSA-N 0.000 description 1
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 1
- DLFVBJFMPXGRIB-UHFFFAOYSA-N Acetamide Chemical compound CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 1
- 239000005695 Ammonium acetate Substances 0.000 description 1
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonium chloride Substances [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 1
- AKBNVJDEJOAPCY-UHFFFAOYSA-N BrC1=CC=C(C=C1)N(CC(C(=O)OC(C)(C)C)O[Si](C)(C)C(C)(C)C)C Chemical compound BrC1=CC=C(C=C1)N(CC(C(=O)OC(C)(C)C)O[Si](C)(C)C(C)(C)C)C AKBNVJDEJOAPCY-UHFFFAOYSA-N 0.000 description 1
- ZQUBNVIQHPAXHE-UHFFFAOYSA-N BrC1=CC=C(C=C1)NCC(C(=O)OC(C)(C)C)O[Si](C)(C)C(C)(C)C Chemical compound BrC1=CC=C(C=C1)NCC(C(=O)OC(C)(C)C)O[Si](C)(C)C(C)(C)C ZQUBNVIQHPAXHE-UHFFFAOYSA-N 0.000 description 1
- IYLXUVAKVBKSSL-MRVPVSSYSA-N BrC1=CC=C(C=N1)OC[C@H](C(=O)OCC)O Chemical compound BrC1=CC=C(C=N1)OC[C@H](C(=O)OCC)O IYLXUVAKVBKSSL-MRVPVSSYSA-N 0.000 description 1
- DRHLDSKZYRWPGI-SSDOTTSWSA-N BrC1=CC=C(N=N1)OC[C@H](C(=O)OC(C)(C)C)O Chemical compound BrC1=CC=C(N=N1)OC[C@H](C(=O)OC(C)(C)C)O DRHLDSKZYRWPGI-SSDOTTSWSA-N 0.000 description 1
- VCAMRCTWHWISLT-CYBMUJFWSA-N BrC1=CC=C(OC[C@H](C(=O)N)O[Si](C)(C)C(C)(C)C)C=C1 Chemical compound BrC1=CC=C(OC[C@H](C(=O)N)O[Si](C)(C)C(C)(C)C)C=C1 VCAMRCTWHWISLT-CYBMUJFWSA-N 0.000 description 1
- GFAILPQYBPLVCS-UHFFFAOYSA-N BrC1=NC=C(C=C1)OCCO[Si](C)(C)C(C)(C)C Chemical compound BrC1=NC=C(C=C1)OCCO[Si](C)(C)C(C)(C)C GFAILPQYBPLVCS-UHFFFAOYSA-N 0.000 description 1
- YATBNGYNUYXSHD-UHFFFAOYSA-N BrC1=NC=C(C=C1)OCCO[Si](C)(C)C(C)(C)C.[Si](C)(C)(C(C)(C)C)OCCOC=1C=CC(=NC1)N1C=NC=C1 Chemical compound BrC1=NC=C(C=C1)OCCO[Si](C)(C)C(C)(C)C.[Si](C)(C)(C(C)(C)C)OCCOC=1C=CC(=NC1)N1C=NC=C1 YATBNGYNUYXSHD-UHFFFAOYSA-N 0.000 description 1
- QFOGVPVJGQVGTF-UHFFFAOYSA-N BrC=1N(C(N(C=1)CCCNC(OC(C)(C)C)=O)=N)C Chemical compound BrC=1N(C(N(C=1)CCCNC(OC(C)(C)C)=O)=N)C QFOGVPVJGQVGTF-UHFFFAOYSA-N 0.000 description 1
- RCVISFJJVBCFKO-DEDYPNTBSA-N BrC=1N(\C(\N(C=1)CCCNC(OC(C)(C)C)=O)=N/C(=O)OC(C)(C)C)C Chemical compound BrC=1N(\C(\N(C=1)CCCNC(OC(C)(C)C)=O)=N/C(=O)OC(C)(C)C)C RCVISFJJVBCFKO-DEDYPNTBSA-N 0.000 description 1
- GVYBCZVYELPQDB-RCCKNPSSSA-N BrC=1N(\C(\N(C=1)CCCNC(OC(C)(C)C)=O)=N/CCNC(=O)OC(C)(C)C)C Chemical compound BrC=1N(\C(\N(C=1)CCCNC(OC(C)(C)C)=O)=N/CCNC(=O)OC(C)(C)C)C GVYBCZVYELPQDB-RCCKNPSSSA-N 0.000 description 1
- ACANYHIBQWFCEW-UHFFFAOYSA-N BrC=1N=CN(C=1)CC(CNC(OC(C)(C)C)=O)O[Si](C)(C)C(C)(C)C Chemical compound BrC=1N=CN(C=1)CC(CNC(OC(C)(C)C)=O)O[Si](C)(C)C(C)(C)C ACANYHIBQWFCEW-UHFFFAOYSA-N 0.000 description 1
- PRHRUYKKUPTDDW-UHFFFAOYSA-N BrCC(CNC(OC(C)(C)C)=O)O[Si](C)(C)C(C)(C)C Chemical compound BrCC(CNC(OC(C)(C)C)=O)O[Si](C)(C)C(C)(C)C PRHRUYKKUPTDDW-UHFFFAOYSA-N 0.000 description 1
- JHKBXTJHHHNYQM-UHFFFAOYSA-N BrCC1CN(C(O1)(C)C)C(=O)OC(C)(C)C Chemical compound BrCC1CN(C(O1)(C)C)C(=O)OC(C)(C)C JHKBXTJHHHNYQM-UHFFFAOYSA-N 0.000 description 1
- DFQQIPSCCBYHOW-UHFFFAOYSA-N BrCCCNC(OC(C)(C)C)=O.BrCCCNC(OC(C)(C)C)=O Chemical compound BrCCCNC(OC(C)(C)C)=O.BrCCCNC(OC(C)(C)C)=O DFQQIPSCCBYHOW-UHFFFAOYSA-N 0.000 description 1
- CIVRLRFAKHDGIA-UHFFFAOYSA-N BrCCOC1=CC=C(C=C1)C=1C=NN(C=1)CCCNC(OC(C)(C)C)=O Chemical compound BrCCOC1=CC=C(C=C1)C=1C=NN(C=1)CCCNC(OC(C)(C)C)=O CIVRLRFAKHDGIA-UHFFFAOYSA-N 0.000 description 1
- PRHRUYKKUPTDDW-LLVKDONJSA-N BrC[C@H](CNC(OC(C)(C)C)=O)O[Si](C)(C)C(C)(C)C Chemical compound BrC[C@H](CNC(OC(C)(C)C)=O)O[Si](C)(C)C(C)(C)C PRHRUYKKUPTDDW-LLVKDONJSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-M Butyrate Chemical compound CCCC([O-])=O FERIUCNNQQJTOY-UHFFFAOYSA-M 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 1
- BBKKFENHADHVQU-ROPPNANJSA-N C(C)(C)(C)OC(=O)NCC(CN1N=CC(=C1)C1=CC=C(OC[C@H](C(=O)OC(C)(C)C)O)C=C1)F Chemical compound C(C)(C)(C)OC(=O)NCC(CN1N=CC(=C1)C1=CC=C(OC[C@H](C(=O)OC(C)(C)C)O)C=C1)F BBKKFENHADHVQU-ROPPNANJSA-N 0.000 description 1
- CRYYBPJKXHFPRT-CFSSBGJSSA-N C(C)(C)(C)OC(=O)NCCCN1/C(/N(C(=C1)C1=CC=C(OC[C@@H](C(=O)OCC)ON2C(C3=CC=CC=C3C2=O)=O)C=C1)C)=N/C(=O)OC(C)(C)C Chemical compound C(C)(C)(C)OC(=O)NCCCN1/C(/N(C(=C1)C1=CC=C(OC[C@@H](C(=O)OCC)ON2C(C3=CC=CC=C3C2=O)=O)C=C1)C)=N/C(=O)OC(C)(C)C CRYYBPJKXHFPRT-CFSSBGJSSA-N 0.000 description 1
- GKJSUNKAZGTUBG-UHFFFAOYSA-N C(C)(C)(C)OC(=O)NCCCN1C(=NC=C1)NC1=CC=C(C=N1)OCC(C(=O)OC(C)(C)C)O[Si](C)(C)C(C)(C)C Chemical compound C(C)(C)(C)OC(=O)NCCCN1C(=NC=C1)NC1=CC=C(C=N1)OCC(C(=O)OC(C)(C)C)O[Si](C)(C)C(C)(C)C GKJSUNKAZGTUBG-UHFFFAOYSA-N 0.000 description 1
- IWQZXDFVIWYQRB-AREMUKBSSA-N C(C)(C)(C)OC(=O)NCCCN1N=C(C=C1C=O)C1=CC=C(OC[C@H](C(=O)OC(C)(C)C)O[Si](C)(C)C(C)(C)C)C=C1 Chemical compound C(C)(C)(C)OC(=O)NCCCN1N=C(C=C1C=O)C1=CC=C(OC[C@H](C(=O)OC(C)(C)C)O[Si](C)(C)C(C)(C)C)C=C1 IWQZXDFVIWYQRB-AREMUKBSSA-N 0.000 description 1
- CLWKUEPZKREKPB-HXUWFJFHSA-N C(C)(C)(C)OC(=O)NCCCNC1=CC=C(N=N1)C1=CC=C(OC[C@H](C(=O)OC(C)(C)C)O)C=C1 Chemical compound C(C)(C)(C)OC(=O)NCCCNC1=CC=C(N=N1)C1=CC=C(OC[C@H](C(=O)OC(C)(C)C)O)C=C1 CLWKUEPZKREKPB-HXUWFJFHSA-N 0.000 description 1
- LZAPJYGPYPQFMB-AREMUKBSSA-N C(C)(C)(C)OC(=O)NCCCNC1=CC=C(N=N1)C1=CC=C(OC[C@H](C(=O)OC(C)(C)C)ON2C(C3=CC=CC=C3C2=O)=O)C=C1 Chemical compound C(C)(C)(C)OC(=O)NCCCNC1=CC=C(N=N1)C1=CC=C(OC[C@H](C(=O)OC(C)(C)C)ON2C(C3=CC=CC=C3C2=O)=O)C=C1 LZAPJYGPYPQFMB-AREMUKBSSA-N 0.000 description 1
- WDFQBOVWMJEYTB-WJOKGBTCSA-N C(C)(C)(C)OC(=O)NCCNCC1=CC(=NN1CCCNC(=O)OC(C)(C)C)C1=CC=C(OC[C@H](C(=O)OC(C)(C)C)O[Si](C)(C)C(C)(C)C)C=C1 Chemical compound C(C)(C)(C)OC(=O)NCCNCC1=CC(=NN1CCCNC(=O)OC(C)(C)C)C1=CC=C(OC[C@H](C(=O)OC(C)(C)C)O[Si](C)(C)C(C)(C)C)C=C1 WDFQBOVWMJEYTB-WJOKGBTCSA-N 0.000 description 1
- WSHHKISDTIKKBP-SANMLTNESA-N C(C)(C)(C)OC([C@H](COC1=CC=C(C=C1)C=1C=NN(C=1)CC1(CN(C1)C(=O)OC(C)(C)C)F)ON1C(C2=CC=CC=C2C1=O)=O)=O Chemical compound C(C)(C)(C)OC([C@H](COC1=CC=C(C=C1)C=1C=NN(C=1)CC1(CN(C1)C(=O)OC(C)(C)C)F)ON1C(C2=CC=CC=C2C1=O)=O)=O WSHHKISDTIKKBP-SANMLTNESA-N 0.000 description 1
- MVWUAMQIKNQNCX-UHFFFAOYSA-N C(C1=CC=CC=C1)(C1=CC=CC=C1)(C1=CC=CC=C1)N1C=NC(=C1)CCCNC(OC(C)(C)C)=O Chemical compound C(C1=CC=CC=C1)(C1=CC=CC=C1)(C1=CC=CC=C1)N1C=NC(=C1)CCCNC(OC(C)(C)C)=O MVWUAMQIKNQNCX-UHFFFAOYSA-N 0.000 description 1
- RXYAKXAJNQLYDQ-UHFFFAOYSA-L C([O-])([O-])=O.[K+].BrC1=CC=C(C=N1)OCC(C(=O)OC(C)(C)C)O.[K+] Chemical compound C([O-])([O-])=O.[K+].BrC1=CC=C(C=N1)OCC(C(=O)OC(C)(C)C)O.[K+] RXYAKXAJNQLYDQ-UHFFFAOYSA-L 0.000 description 1
- XNEOWXQMXLIUAA-UHFFFAOYSA-N C1(=CC=CC=C1)OC.N1C=NC(=C1)CCCNC(OC(C)(C)C)=O Chemical compound C1(=CC=CC=C1)OC.N1C=NC(=C1)CCCNC(OC(C)(C)C)=O XNEOWXQMXLIUAA-UHFFFAOYSA-N 0.000 description 1
- KJZVAHZBZCYETN-VFOTVAIQSA-N CC(C)(C)OC(NCCCN1[N+](C)=CC(C(C=C2)=CC=C2OC[C@@H](C2=NNN=N2)O/N=C(\C([O-])=O)/C2=CSC(N)=N2)=C1)=O Chemical compound CC(C)(C)OC(NCCCN1[N+](C)=CC(C(C=C2)=CC=C2OC[C@@H](C2=NNN=N2)O/N=C(\C([O-])=O)/C2=CSC(N)=N2)=C1)=O KJZVAHZBZCYETN-VFOTVAIQSA-N 0.000 description 1
- IKWGYLVDNPDMBP-KQNHUGOFSA-N CC(C)(C)OC([C@H](COC(C=C1)=CN=C1N1C=[N+](CCCNC(OC(C)(C)C)=O)C=C1)O/N=C(\C([O-])=O)/C1=CSC(NC(OC(C)(C)C)=O)=N1)=O Chemical compound CC(C)(C)OC([C@H](COC(C=C1)=CN=C1N1C=[N+](CCCNC(OC(C)(C)C)=O)C=C1)O/N=C(\C([O-])=O)/C1=CSC(NC(OC(C)(C)C)=O)=N1)=O IKWGYLVDNPDMBP-KQNHUGOFSA-N 0.000 description 1
- WPFOFRHFZCMKMA-UHFFFAOYSA-N CC1=CC=C(C=C1)S(=O)(=O)O.C(C)(C)(C)OC(=O)NCCCN1N=CC(=C1)C1=CC=C(OCC(C(=O)OC(C)(C)C)(C)ON2C(C3=CC=CC=C3C2=O)=O)C=C1 Chemical compound CC1=CC=C(C=C1)S(=O)(=O)O.C(C)(C)(C)OC(=O)NCCCN1N=CC(=C1)C1=CC=C(OCC(C(=O)OC(C)(C)C)(C)ON2C(C3=CC=CC=C3C2=O)=O)C=C1 WPFOFRHFZCMKMA-UHFFFAOYSA-N 0.000 description 1
- KKVUFSJFQXUKHD-UHFFFAOYSA-N CN.C(C1=CC=CC=C1)(C1=CC=CC=C1)(C1=CC=CC=C1)N1C=NC(=C1)CCCN Chemical compound CN.C(C1=CC=CC=C1)(C1=CC=CC=C1)(C1=CC=CC=C1)N1C=NC(=C1)CCCN KKVUFSJFQXUKHD-UHFFFAOYSA-N 0.000 description 1
- RDDRBIUGBRSSGI-UHFFFAOYSA-N COP(=O)(OC)C(COC1=CC=C(C=C1)C=1C=NN(C=1)CCCNC(OC(C)(C)C)=O)O Chemical compound COP(=O)(OC)C(COC1=CC=C(C=C1)C=1C=NN(C=1)CCCNC(OC(C)(C)C)=O)O RDDRBIUGBRSSGI-UHFFFAOYSA-N 0.000 description 1
- KKTWYOQDABSVJF-UHFFFAOYSA-N COP(=O)(OC)C(COC1=CC=C(C=C1)C=1C=NN(C=1)CCCNC(OC(C)(C)C)=O)ON1C(C2=CC=CC=C2C1=O)=O Chemical compound COP(=O)(OC)C(COC1=CC=C(C=C1)C=1C=NN(C=1)CCCNC(OC(C)(C)C)=O)ON1C(C2=CC=CC=C2C1=O)=O KKTWYOQDABSVJF-UHFFFAOYSA-N 0.000 description 1
- YSNGGLCHXCALJV-SBSPUUFOSA-N C[Si](C)(C)C=[N+]=[N-].BrC1=CC=C(OC[C@H](C(=O)OC)O)C=C1 Chemical compound C[Si](C)(C)C=[N+]=[N-].BrC1=CC=C(OC[C@H](C(=O)OC)O)C=C1 YSNGGLCHXCALJV-SBSPUUFOSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 241000588919 Citrobacter freundii Species 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 1
- PMPVIKIVABFJJI-UHFFFAOYSA-N Cyclobutane Chemical compound C1CCC1 PMPVIKIVABFJJI-UHFFFAOYSA-N 0.000 description 1
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 1
- LVZWSLJZHVFIQJ-UHFFFAOYSA-N Cyclopropane Chemical compound C1CC1 LVZWSLJZHVFIQJ-UHFFFAOYSA-N 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- MNQZXJOMYWMBOU-VKHMYHEASA-N D-glyceraldehyde Chemical compound OC[C@@H](O)C=O MNQZXJOMYWMBOU-VKHMYHEASA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 1
- 206010059866 Drug resistance Diseases 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 241000881810 Enterobacter asburiae Species 0.000 description 1
- 241000588697 Enterobacter cloacae Species 0.000 description 1
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical compound C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 1
- CWYNVVGOOAEACU-UHFFFAOYSA-N Fe2+ Chemical compound [Fe+2] CWYNVVGOOAEACU-UHFFFAOYSA-N 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 1
- LPHGQDQBBGAPDZ-UHFFFAOYSA-N Isocaffeine Natural products CN1C(=O)N(C)C(=O)C2=C1N(C)C=N2 LPHGQDQBBGAPDZ-UHFFFAOYSA-N 0.000 description 1
- RRHGJUQNOFWUDK-UHFFFAOYSA-N Isoprene Chemical compound CC(=C)C=C RRHGJUQNOFWUDK-UHFFFAOYSA-N 0.000 description 1
- 239000005909 Kieselgur Substances 0.000 description 1
- 241000588915 Klebsiella aerogenes Species 0.000 description 1
- 241000588749 Klebsiella oxytoca Species 0.000 description 1
- 241000588747 Klebsiella pneumoniae Species 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- 229930182821 L-proline Natural products 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 239000007993 MOPS buffer Substances 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 102000010750 Metalloproteins Human genes 0.000 description 1
- 108010063312 Metalloproteins Proteins 0.000 description 1
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 1
- LGDSHSYDSCRFAB-UHFFFAOYSA-N Methyl isothiocyanate Chemical compound CN=C=S LGDSHSYDSCRFAB-UHFFFAOYSA-N 0.000 description 1
- 241000588772 Morganella morganii Species 0.000 description 1
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 1
- UEEJHVSXFDXPFK-UHFFFAOYSA-N N-dimethylaminoethanol Chemical compound CN(C)CCO UEEJHVSXFDXPFK-UHFFFAOYSA-N 0.000 description 1
- HTLZVHNRZJPSMI-UHFFFAOYSA-N N-ethylpiperidine Chemical compound CCN1CCCCC1 HTLZVHNRZJPSMI-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- YDJMPFPDXAFDMQ-SFHVURJKSA-N N1(C=NC=C1)C1=CC=C(C=N1)OC[C@@H](C(=O)OC(C)(C)C)ON1C(C2=CC=CC=C2C1=O)=O Chemical compound N1(C=NC=C1)C1=CC=C(C=N1)OC[C@@H](C(=O)OC(C)(C)C)ON1C(C2=CC=CC=C2C1=O)=O YDJMPFPDXAFDMQ-SFHVURJKSA-N 0.000 description 1
- 229910017906 NH3H2O Inorganic materials 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- YNHCZQHYXFWIIG-DDWIOCJRSA-N O1C(C1)C(=O)OCC.BrC1=CC=C(C=N1)OC[C@H](C(=O)OCC)O Chemical compound O1C(C1)C(=O)OCC.BrC1=CC=C(C=N1)OC[C@H](C(=O)OCC)O YNHCZQHYXFWIIG-DDWIOCJRSA-N 0.000 description 1
- QSUAWFGWTVBQPT-UHFFFAOYSA-N O=C1N(C(C2=CC=CC=C12)=O)OCCOC1=CC=C(C=C1)C=1C=NN(C=1)CCCNC(OC(C)(C)C)=O Chemical compound O=C1N(C(C2=CC=CC=C12)=O)OCCOC1=CC=C(C=C1)C=1C=NN(C=1)CCCNC(OC(C)(C)C)=O QSUAWFGWTVBQPT-UHFFFAOYSA-N 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 102220472129 Protein Wnt-2_C23A_mutation Human genes 0.000 description 1
- 102220472231 Protein Wnt-2_C37A_mutation Human genes 0.000 description 1
- 241000588770 Proteus mirabilis Species 0.000 description 1
- 241000588777 Providencia rettgeri Species 0.000 description 1
- 241000589776 Pseudomonas putida Species 0.000 description 1
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- 239000000589 Siderophore Substances 0.000 description 1
- 241000122973 Stenotrophomonas maltophilia Species 0.000 description 1
- 229920002253 Tannate Polymers 0.000 description 1
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 1
- ZMZDMBWJUHKJPS-UHFFFAOYSA-M Thiocyanate anion Chemical compound [S-]C#N ZMZDMBWJUHKJPS-UHFFFAOYSA-M 0.000 description 1
- RHQDFWAXVIIEBN-UHFFFAOYSA-N Trifluoroethanol Chemical compound OCC(F)(F)F RHQDFWAXVIIEBN-UHFFFAOYSA-N 0.000 description 1
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 1
- RVZRLFUMFFBCSC-GJOBRYSDSA-N [(3S)-3-[[(2E)-2-[(1S)-2-[4-[1-(3-aminopropyl)-2-methylpyrazol-2-ium-4-yl]phenoxy]-1-carboxyethoxy]imino-2-(2-amino-1,3-thiazol-4-yl)acetyl]amino]-2,2-dimethyl-4-oxoazetidin-1-yl] sulfate Chemical compound S(=O)(=O)(ON1C([C@@H](C1=O)NC(/C(/C=1N=C(SC=1)N)=N/O[C@@H](COC1=CC=C(C=C1)C=1C=[N+](N(C=1)CCCN)C)C(=O)O)=O)(C)C)[O-] RVZRLFUMFFBCSC-GJOBRYSDSA-N 0.000 description 1
- SILRCLUGLHJTDD-QPXSJBHFSA-N [(3S)-3-[[(2Z)-2-(2-amino-1,3-thiazol-4-yl)-2-[(1S)-1-carboxy-2-[4-[1-[(2R)-2,3-diaminopropyl]-2-methylpyrazol-2-ium-4-yl]-3-fluorophenoxy]ethoxy]iminoacetyl]amino]-2,2-dimethyl-4-oxoazetidin-1-yl] sulfate Chemical compound S(=O)(=O)(ON1C([C@@H](C1=O)NC(\C(=N/O[C@@H](COC1=CC(=C(C=C1)C=1C=[N+](N(C=1)C[C@@H](CN)N)C)F)C(=O)O)\C=1N=C(SC=1)N)=O)(C)C)[O-] SILRCLUGLHJTDD-QPXSJBHFSA-N 0.000 description 1
- CLRVMSJQWAEVQT-NQYCYQSHSA-N [(3S)-3-[[(2Z)-2-[(1S)-2-[4-[1-(3-aminopropyl)-2-methylpyrazol-2-ium-4-yl]-3-fluorophenoxy]-1-carboxyethoxy]imino-2-(2-amino-1,3-thiazol-4-yl)acetyl]amino]-2,2-dimethyl-4-oxoazetidin-1-yl] sulfate Chemical compound S(=O)(=O)(ON1C([C@@H](C1=O)NC(\C(\C=1N=C(SC=1)N)=N/O[C@@H](COC1=CC(=C(C=C1)C=1C=[N+](N(C=1)CCCN)C)F)C(=O)O)=O)(C)C)[O-] CLRVMSJQWAEVQT-NQYCYQSHSA-N 0.000 description 1
- FSIQEWQYYRNGRM-BCBFWPAESA-N [(3S)-3-[[(2Z)-2-[1-[4-[1-(3-aminopropyl)-2-methylpyrazol-2-ium-4-yl]phenoxy]-2-carboxypropan-2-yl]oxyimino-2-(2-amino-1,3-thiazol-4-yl)acetyl]amino]-2,2-dimethyl-4-oxoazetidin-1-yl] sulfate Chemical compound C[n+]1cc(cn1CCCN)-c1ccc(OCC(C)(O\N=C(/C(=O)N[C@@H]2C(=O)N(OS([O-])(=O)=O)C2(C)C)c2csc(N)n2)C(O)=O)cc1 FSIQEWQYYRNGRM-BCBFWPAESA-N 0.000 description 1
- VXIYDYFOEDTARB-TWCQISOYSA-N [(3S)-3-[[(2Z)-2-[2-[4-[1-(3-aminopropyl)pyrazol-4-yl]phenoxy]ethoxyimino]-2-(2-amino-1,3-thiazol-4-yl)acetyl]amino]-2,2-dimethyl-4-oxoazetidin-1-yl] hydrogen sulfate Chemical compound CC1(C)[C@H](NC(=O)C(=N/OCCOc2ccc(cc2)-c2cnn(CCCN)c2)\c2csc(N)n2)C(=O)N1OS(O)(=O)=O VXIYDYFOEDTARB-TWCQISOYSA-N 0.000 description 1
- QFZLMCJENPYQBG-LVLGTLCVSA-N [(3S)-3-[[(2Z)-2-[2-[6-[3-(3-aminopropyl)imidazol-3-ium-1-yl]pyridin-3-yl]oxyethoxyimino]-2-(2-amino-1,3-thiazol-4-yl)acetyl]amino]-2,2-dimethyl-4-oxoazetidin-1-yl] sulfate 2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.CC1(C)[C@H](NC(=O)C(=N/OCCOc2ccc(nc2)-n2cc[n+](CCCN)c2)\c2csc(N)n2)C(=O)N1OS([O-])(=O)=O QFZLMCJENPYQBG-LVLGTLCVSA-N 0.000 description 1
- AMXAQVKNGGFRFE-QRWMCTBCSA-N [Si](C)(C)(C(C)(C)C)O[C@@H](C(=O)OC(C)(C)C)COC1=CC=C(C=C1)C(CN=[N+]=[N-])N=[N+]=[N-] Chemical compound [Si](C)(C)(C(C)(C)C)O[C@@H](C(=O)OC(C)(C)C)COC1=CC=C(C=C1)C(CN=[N+]=[N-])N=[N+]=[N-] AMXAQVKNGGFRFE-QRWMCTBCSA-N 0.000 description 1
- GLFODEYBGFXDKO-GOSISDBHSA-N [Si](C)(C)(C(C)(C)C)O[C@@H](C(=O)OC(C)(C)C)COC1=CC=C(C=C1)C(N(C)OC)=O Chemical compound [Si](C)(C)(C(C)(C)C)O[C@@H](C(=O)OC(C)(C)C)COC1=CC=C(C=C1)C(N(C)OC)=O GLFODEYBGFXDKO-GOSISDBHSA-N 0.000 description 1
- OXGCZSIEEJDGFC-GOSISDBHSA-N [Si](C)(C)(C(C)(C)C)O[C@@H](C(=O)OC(C)(C)C)COC1=CC=C(C=C1)C=C Chemical compound [Si](C)(C)(C(C)(C)C)O[C@@H](C(=O)OC(C)(C)C)COC1=CC=C(C=C1)C=C OXGCZSIEEJDGFC-GOSISDBHSA-N 0.000 description 1
- VFDHDMONRHQELN-NSHDSACASA-N [Si](C)(C)(C(C)(C)C)O[C@@H](CNC(OC(C)(C)C)=O)CO Chemical compound [Si](C)(C)(C(C)(C)C)O[C@@H](CNC(OC(C)(C)C)=O)CO VFDHDMONRHQELN-NSHDSACASA-N 0.000 description 1
- ZVQOOHYFBIDMTQ-UHFFFAOYSA-N [methyl(oxido){1-[6-(trifluoromethyl)pyridin-3-yl]ethyl}-lambda(6)-sulfanylidene]cyanamide Chemical compound N#CN=S(C)(=O)C(C)C1=CC=C(C(F)(F)F)N=C1 ZVQOOHYFBIDMTQ-UHFFFAOYSA-N 0.000 description 1
- WLLIXJBWWFGEHT-UHFFFAOYSA-N [tert-butyl(dimethyl)silyl] trifluoromethanesulfonate Chemical compound CC(C)(C)[Si](C)(C)OS(=O)(=O)C(F)(F)F WLLIXJBWWFGEHT-UHFFFAOYSA-N 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- WNLRTRBMVRJNCN-UHFFFAOYSA-L adipate(2-) Chemical compound [O-]C(=O)CCCCC([O-])=O WNLRTRBMVRJNCN-UHFFFAOYSA-L 0.000 description 1
- 229940072056 alginate Drugs 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 125000003545 alkoxy group Chemical group 0.000 description 1
- 150000001350 alkyl halides Chemical class 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- AWUCVROLDVIAJX-UHFFFAOYSA-N alpha-glycerophosphate Natural products OCC(O)COP(O)(O)=O AWUCVROLDVIAJX-UHFFFAOYSA-N 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 150000001409 amidines Chemical group 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 235000019257 ammonium acetate Nutrition 0.000 description 1
- 229940043376 ammonium acetate Drugs 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 229960003121 arginine Drugs 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- OISFUZRUIGGTSD-LJTMIZJLSA-N azane;(2r,3r,4r,5s)-6-(methylamino)hexane-1,2,3,4,5-pentol Chemical compound N.CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO OISFUZRUIGGTSD-LJTMIZJLSA-N 0.000 description 1
- KVVMXWRFYAGASO-UHFFFAOYSA-N azetidine-1-carboxylic acid Chemical compound OC(=O)N1CCC1 KVVMXWRFYAGASO-UHFFFAOYSA-N 0.000 description 1
- 125000005604 azodicarboxylate group Chemical group 0.000 description 1
- 239000003899 bactericide agent Substances 0.000 description 1
- 239000000022 bacteriostatic agent Substances 0.000 description 1
- 230000003385 bacteriostatic effect Effects 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000004603 benzisoxazolyl group Chemical group O1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000003354 benzotriazolyl group Chemical group N1N=NC2=C1C=CC=C2* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 235000019445 benzyl alcohol Nutrition 0.000 description 1
- MSWZFWKMSRAUBD-UHFFFAOYSA-N beta-D-galactosamine Natural products NC1C(O)OC(CO)C(O)C1O MSWZFWKMSRAUBD-UHFFFAOYSA-N 0.000 description 1
- 125000003460 beta-lactamyl group Chemical group 0.000 description 1
- 229960003237 betaine Drugs 0.000 description 1
- 125000002618 bicyclic heterocycle group Chemical group 0.000 description 1
- 239000012472 biological sample Substances 0.000 description 1
- XGIUDIMNNMKGDE-UHFFFAOYSA-N bis(trimethylsilyl)azanide Chemical compound C[Si](C)(C)[N-][Si](C)(C)C XGIUDIMNNMKGDE-UHFFFAOYSA-N 0.000 description 1
- 230000036765 blood level Effects 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- OQROAIRCEOBYJA-UHFFFAOYSA-N bromodiphenylmethane Chemical compound C=1C=CC=CC=1C(Br)C1=CC=CC=C1 OQROAIRCEOBYJA-UHFFFAOYSA-N 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229960001948 caffeine Drugs 0.000 description 1
- VJEONQKOZGKCAK-UHFFFAOYSA-N caffeine Natural products CN1C(=O)N(C)C(=O)C2=C1C=CN2C VJEONQKOZGKCAK-UHFFFAOYSA-N 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 125000001589 carboacyl group Chemical group 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- 235000011089 carbon dioxide Nutrition 0.000 description 1
- 230000030833 cell death Effects 0.000 description 1
- 238000004296 chiral HPLC Methods 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- 229940090805 clavulanate Drugs 0.000 description 1
- 229910017052 cobalt Inorganic materials 0.000 description 1
- 239000010941 cobalt Substances 0.000 description 1
- 229940125898 compound 5 Drugs 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 125000001047 cyclobutenyl group Chemical group C1(=CCC1)* 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001162 cycloheptenyl group Chemical group C1(=CCCCCC1)* 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- BALGDZWGNCXXES-UHFFFAOYSA-N cyclopentane;propanoic acid Chemical compound CCC(O)=O.C1CCCC1 BALGDZWGNCXXES-UHFFFAOYSA-N 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000298 cyclopropenyl group Chemical group [H]C1=C([H])C1([H])* 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 125000002704 decyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000000422 delta-lactone group Chemical group 0.000 description 1
- XUWHAWMETYGRKB-UHFFFAOYSA-N delta-valerolactam Natural products O=C1CCCCN1 XUWHAWMETYGRKB-UHFFFAOYSA-N 0.000 description 1
- 238000001212 derivatisation Methods 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 150000008050 dialkyl sulfates Chemical class 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- YLFBFPXKTIQSSY-UHFFFAOYSA-N dimethoxy(oxo)phosphanium Chemical compound CO[P+](=O)OC YLFBFPXKTIQSSY-UHFFFAOYSA-N 0.000 description 1
- SPCNPOWOBZQWJK-UHFFFAOYSA-N dimethoxy-(2-propan-2-ylsulfanylethylsulfanyl)-sulfanylidene-$l^{5}-phosphane Chemical compound COP(=S)(OC)SCCSC(C)C SPCNPOWOBZQWJK-UHFFFAOYSA-N 0.000 description 1
- GAFRWLVTHPVQGK-UHFFFAOYSA-N dipentyl sulfate Chemical class CCCCCOS(=O)(=O)OCCCCC GAFRWLVTHPVQGK-UHFFFAOYSA-N 0.000 description 1
- 239000001177 diphosphate Substances 0.000 description 1
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical compound [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 1
- 235000011180 diphosphates Nutrition 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- CPGLZCPYWQALMI-UHFFFAOYSA-N dodecane-1-sulfinic acid Chemical compound CCCCCCCCCCCCS(O)=O CPGLZCPYWQALMI-UHFFFAOYSA-N 0.000 description 1
- POULHZVOKOAJMA-UHFFFAOYSA-M dodecanoate Chemical compound CCCCCCCCCCCC([O-])=O POULHZVOKOAJMA-UHFFFAOYSA-M 0.000 description 1
- 125000003438 dodecyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 229940009662 edetate Drugs 0.000 description 1
- 229950005627 embonate Drugs 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 229940092559 enterobacter aerogenes Drugs 0.000 description 1
- 229950000206 estolate Drugs 0.000 description 1
- KQCBSWBQAXTILK-WYMLVPIESA-N ethyl (1e)-n-(2,4,6-trimethylphenyl)sulfonyloxyethanimidate Chemical compound CCO\C(C)=N\OS(=O)(=O)C1=C(C)C=C(C)C=C1C KQCBSWBQAXTILK-WYMLVPIESA-N 0.000 description 1
- XNVRKLCQBZTGNA-UHFFFAOYSA-N ethyl 2-(2-amino-1,3-thiazol-4-yl)-2-oxoacetate Chemical compound CCOC(=O)C(=O)C1=CSC(N)=N1 XNVRKLCQBZTGNA-UHFFFAOYSA-N 0.000 description 1
- YLWNLYIMPYOOFI-UHFFFAOYSA-N ethyl 2-[2-[(2-methylpropan-2-yl)oxycarbonylamino]-1,3-thiazol-4-yl]-2-oxoacetate Chemical compound CCOC(=O)C(=O)C1=CSC(NC(=O)OC(C)(C)C)=N1 YLWNLYIMPYOOFI-UHFFFAOYSA-N 0.000 description 1
- WMCSPGOMDNVCSW-UHFFFAOYSA-N ethyl 2-[2-[(2-methylpropan-2-yl)oxycarbonylamino]-1,3-thiazol-4-yl]-2-oxoacetate 2-[2-[(2-methylpropan-2-yl)oxycarbonylamino]-1,3-thiazol-4-yl]-2-oxoacetic acid Chemical compound CC(C)(C)OC(=O)Nc1nc(cs1)C(=O)C(O)=O.CCOC(=O)C(=O)c1csc(NC(=O)OC(C)(C)C)n1 WMCSPGOMDNVCSW-UHFFFAOYSA-N 0.000 description 1
- 125000000816 ethylene group Chemical group [H]C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 229960004979 fampridine Drugs 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- 238000003818 flash chromatography Methods 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 239000000499 gel Substances 0.000 description 1
- 238000001502 gel electrophoresis Methods 0.000 description 1
- 229960001731 gluceptate Drugs 0.000 description 1
- KWMLJOLKUYYJFJ-VFUOTHLCSA-N glucoheptonic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)[C@@H](O)C(O)=O KWMLJOLKUYYJFJ-VFUOTHLCSA-N 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 229960002442 glucosamine Drugs 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 125000001188 haloalkyl group Chemical group 0.000 description 1
- 230000035876 healing Effects 0.000 description 1
- DMEGYFMYUHOHGS-UHFFFAOYSA-N heptamethylene Natural products C1CCCCCC1 DMEGYFMYUHOHGS-UHFFFAOYSA-N 0.000 description 1
- MNWFXJYAOYHMED-UHFFFAOYSA-N heptanoic acid Chemical compound CCCCCCC(O)=O MNWFXJYAOYHMED-UHFFFAOYSA-N 0.000 description 1
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004366 heterocycloalkenyl group Chemical group 0.000 description 1
- IPCSVZSSVZVIGE-UHFFFAOYSA-M hexadecanoate Chemical compound CCCCCCCCCCCCCCCC([O-])=O IPCSVZSSVZVIGE-UHFFFAOYSA-M 0.000 description 1
- FFUAGWLWBBFQJT-UHFFFAOYSA-N hexamethyldisilazane Chemical compound C[Si](C)(C)N[Si](C)(C)C FFUAGWLWBBFQJT-UHFFFAOYSA-N 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- 125000006038 hexenyl group Chemical group 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 1
- XLVPCRSPMUNYAD-UHFFFAOYSA-N hydrazine;hydroiodide Chemical compound I.NN XLVPCRSPMUNYAD-UHFFFAOYSA-N 0.000 description 1
- ZMZDMBWJUHKJPS-UHFFFAOYSA-N hydrogen thiocyanate Natural products SC#N ZMZDMBWJUHKJPS-UHFFFAOYSA-N 0.000 description 1
- 230000003301 hydrolyzing effect Effects 0.000 description 1
- RCCPEORTSYDPMB-UHFFFAOYSA-N hydroxy benzenecarboximidothioate Chemical compound OSC(=N)C1=CC=CC=C1 RCCPEORTSYDPMB-UHFFFAOYSA-N 0.000 description 1
- 125000002768 hydroxyalkyl group Chemical group 0.000 description 1
- GCDSPLNVAMRZAF-UHFFFAOYSA-N hydroxymethyl azetidine-1-carboxylate Chemical compound OCOC(=O)N1CCC1 GCDSPLNVAMRZAF-UHFFFAOYSA-N 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 230000000415 inactivating effect Effects 0.000 description 1
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 1
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropanol acetate Natural products CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 description 1
- 125000000555 isopropenyl group Chemical group [H]\C([H])=C(\*)C([H])([H])[H] 0.000 description 1
- 229940011051 isopropyl acetate Drugs 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 1
- 125000005956 isoquinolyl group Chemical group 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- GWYFCOCPABKNJV-UHFFFAOYSA-N isovaleric acid Chemical compound CC(C)CC(O)=O GWYFCOCPABKNJV-UHFFFAOYSA-N 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 229940001447 lactate Drugs 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 229940099584 lactobionate Drugs 0.000 description 1
- JYTUSYBCFIZPBE-AMTLMPIISA-N lactobionic acid Chemical compound OC(=O)[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O JYTUSYBCFIZPBE-AMTLMPIISA-N 0.000 description 1
- 229940070765 laurate Drugs 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 235000019341 magnesium sulphate Nutrition 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-M mandelate Chemical compound [O-]C(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-M 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- OKKJLVBELUTLKV-VMNATFBRSA-N methanol-d1 Chemical compound [2H]OC OKKJLVBELUTLKV-VMNATFBRSA-N 0.000 description 1
- NVBLRKGCQVZDMQ-UHFFFAOYSA-N methyl 2-bromo-3-methoxypropanoate Chemical compound COCC(Br)C(=O)OC NVBLRKGCQVZDMQ-UHFFFAOYSA-N 0.000 description 1
- FSUWVSSCCGVYLS-UHFFFAOYSA-N methyl 3-(1-tritylimidazol-4-yl)propanoate Chemical compound C1=NC(CCC(=O)OC)=CN1C(C=1C=CC=CC=1)(C=1C=CC=CC=1)C1=CC=CC=C1 FSUWVSSCCGVYLS-UHFFFAOYSA-N 0.000 description 1
- LWTKERPWUWBNRS-GFCCVEGCSA-N methyl 4-[(2R)-2-hydroxy-3-[(2-methylpropan-2-yl)oxy]-3-oxopropoxy]benzoate Chemical compound C(C)(C)(C)OC([C@@H](COC1=CC=C(C(=O)OC)C=C1)O)=O LWTKERPWUWBNRS-GFCCVEGCSA-N 0.000 description 1
- 229940102396 methyl bromide Drugs 0.000 description 1
- LRMHVVPPGGOAJQ-UHFFFAOYSA-N methyl nitrate Chemical compound CO[N+]([O-])=O LRMHVVPPGGOAJQ-UHFFFAOYSA-N 0.000 description 1
- 239000004292 methyl p-hydroxybenzoate Substances 0.000 description 1
- JZMJDSHXVKJFKW-UHFFFAOYSA-M methyl sulfate(1-) Chemical compound COS([O-])(=O)=O JZMJDSHXVKJFKW-UHFFFAOYSA-M 0.000 description 1
- 229960002216 methylparaben Drugs 0.000 description 1
- 230000000813 microbial effect Effects 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 125000006578 monocyclic heterocycloalkyl group Chemical group 0.000 description 1
- 229940076266 morganella morganii Drugs 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- 230000036457 multidrug resistance Effects 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- 125000001421 myristyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- PSHKMPUSSFXUIA-UHFFFAOYSA-N n,n-dimethylpyridin-2-amine Chemical compound CN(C)C1=CC=CC=N1 PSHKMPUSSFXUIA-UHFFFAOYSA-N 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-M naphthalene-2-sulfonate Chemical compound C1=CC=CC2=CC(S(=O)(=O)[O-])=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-M 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- QEKXARSPUFVXIX-UHFFFAOYSA-L nickel(2+);triphenylphosphane;dibromide Chemical compound [Ni+2].[Br-].[Br-].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 QEKXARSPUFVXIX-UHFFFAOYSA-L 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 125000001400 nonyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 229910052762 osmium Inorganic materials 0.000 description 1
- SYQBFIAQOQZEGI-UHFFFAOYSA-N osmium atom Chemical compound [Os] SYQBFIAQOQZEGI-UHFFFAOYSA-N 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 125000003566 oxetanyl group Chemical group 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- YYROPELSRYBVMQ-UHFFFAOYSA-N p-toluenesulfonyl chloride Substances CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 1
- 229940014662 pantothenate Drugs 0.000 description 1
- 235000019161 pantothenic acid Nutrition 0.000 description 1
- 239000011713 pantothenic acid Substances 0.000 description 1
- VEDDBHYQWFOITD-UHFFFAOYSA-N para-bromobenzyl alcohol Chemical compound OCC1=CC=C(Br)C=C1 VEDDBHYQWFOITD-UHFFFAOYSA-N 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 230000007170 pathology Effects 0.000 description 1
- 150000002961 penems Chemical class 0.000 description 1
- 125000002255 pentenyl group Chemical group C(=CCCC)* 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- JRKICGRDRMAZLK-UHFFFAOYSA-L peroxydisulfate Chemical compound [O-]S(=O)(=O)OOS([O-])(=O)=O JRKICGRDRMAZLK-UHFFFAOYSA-L 0.000 description 1
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 1
- PARWUHTVGZSQPD-UHFFFAOYSA-N phenylsilane Chemical compound [SiH3]C1=CC=CC=C1 PARWUHTVGZSQPD-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- XKJCHHZQLQNZHY-UHFFFAOYSA-N phthalimide Chemical compound C1=CC=C2C(=O)NC(=O)C2=C1 XKJCHHZQLQNZHY-UHFFFAOYSA-N 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000005936 piperidyl group Chemical group 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Substances [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 1
- 229920000768 polyamine Polymers 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- 229960002429 proline Drugs 0.000 description 1
- OGHBATFHNDZKSO-UHFFFAOYSA-N propan-2-olate Chemical compound CC(C)[O-] OGHBATFHNDZKSO-UHFFFAOYSA-N 0.000 description 1
- SWWHCQCMVCPLEQ-UHFFFAOYSA-N propan-2-yl methanesulfonate Chemical compound CC(C)OS(C)(=O)=O SWWHCQCMVCPLEQ-UHFFFAOYSA-N 0.000 description 1
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- 239000004405 propyl p-hydroxybenzoate Substances 0.000 description 1
- 229960003415 propylparaben Drugs 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- 150000003212 purines Chemical class 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 125000000246 pyrimidin-2-yl group Chemical group [H]C1=NC(*)=NC([H])=C1[H] 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- 238000005070 sampling Methods 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 150000003335 secondary amines Chemical class 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- XGVXKJKTISMIOW-ZDUSSCGKSA-N simurosertib Chemical compound N1N=CC(C=2SC=3C(=O)NC(=NC=3C=2)[C@H]2N3CCC(CC3)C2)=C1C XGVXKJKTISMIOW-ZDUSSCGKSA-N 0.000 description 1
- AWUCVROLDVIAJX-GSVOUGTGSA-N sn-glycerol 3-phosphate Chemical compound OC[C@@H](O)COP(O)(O)=O AWUCVROLDVIAJX-GSVOUGTGSA-N 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 239000008354 sodium chloride injection Substances 0.000 description 1
- UKLNMMHNWFDKNT-UHFFFAOYSA-M sodium chlorite Chemical compound [Na+].[O-]Cl=O UKLNMMHNWFDKNT-UHFFFAOYSA-M 0.000 description 1
- 229960002218 sodium chlorite Drugs 0.000 description 1
- 229910000162 sodium phosphate Inorganic materials 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- 235000019345 sodium thiosulphate Nutrition 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 230000000707 stereoselective effect Effects 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 239000008227 sterile water for injection Substances 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 238000003419 tautomerization reaction Methods 0.000 description 1
- 229950002757 teoclate Drugs 0.000 description 1
- 125000004213 tert-butoxy group Chemical group [H]C([H])([H])C(O*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- UYWJIKGUTRZXPG-LJQANCHMSA-N tert-butyl (2R)-2-hydroxy-3-[4-[1-[3-[(2-methylpropan-2-yl)oxycarbonylamino]propyl]-2-nitroimidazol-4-yl]phenoxy]propanoate Chemical compound C(C)(C)(C)OC(=O)NCCCN1C(=NC(=C1)C1=CC=C(OC[C@H](C(=O)OC(C)(C)C)O)C=C1)[N+](=O)[O-] UYWJIKGUTRZXPG-LJQANCHMSA-N 0.000 description 1
- UCBHHTYHMQNBHU-HXUWFJFHSA-N tert-butyl (2R)-2-hydroxy-3-[4-[1-[3-[(2-methylpropan-2-yl)oxycarbonylamino]propyl]pyrazol-4-yl]phenoxy]propanoate Chemical compound C(C)(C)(C)OC(=O)NCCCN1N=CC(=C1)C1=CC=C(OC[C@H](C(=O)OC(C)(C)C)O)C=C1 UCBHHTYHMQNBHU-HXUWFJFHSA-N 0.000 description 1
- AGPQRRSDAVXYLQ-JOCHJYFZSA-N tert-butyl (2R)-2-hydroxy-3-[4-[6-[3-[(2-methylpropan-2-yl)oxycarbonylamino]propyl]pyridin-3-yl]phenoxy]propanoate Chemical compound C(C)(C)(C)OC(=O)NCCCC1=CC=C(C=N1)C1=CC=C(OC[C@H](C(=O)OC(C)(C)C)O)C=C1 AGPQRRSDAVXYLQ-JOCHJYFZSA-N 0.000 description 1
- KTAFACQBTYXJNN-CQSZACIVSA-N tert-butyl (2R)-3-[4-(1,3-dinitropropan-2-yl)phenoxy]-2-hydroxypropanoate Chemical compound [N+](=O)([O-])CC(C[N+](=O)[O-])C1=CC=C(OC[C@H](C(=O)OC(C)(C)C)O)C=C1 KTAFACQBTYXJNN-CQSZACIVSA-N 0.000 description 1
- AZVOPOGHLKRAMA-DAXIOTQJSA-N tert-butyl (2S)-2-(1,3-dioxoisoindol-2-yl)oxy-3-[4-[(2E)-3-methyl-1-[3-[(2-methylpropan-2-yl)oxycarbonylamino]propyl]-2-[(2-methylpropan-2-yl)oxycarbonylimino]imidazol-4-yl]phenoxy]propanoate Chemical compound C(C)(C)(C)OC(=O)NCCCN1\C(\N(C(=C1)C1=CC=C(OC[C@@H](C(=O)OC(C)(C)C)ON2C(C3=CC=CC=C3C2=O)=O)C=C1)C)=N/C(=O)OC(C)(C)C AZVOPOGHLKRAMA-DAXIOTQJSA-N 0.000 description 1
- FYZFVVUEVJPIFU-DEOSSOPVSA-O tert-butyl (2S)-2-(1,3-dioxoisoindol-2-yl)oxy-3-[6-[3-[3-[(2-methylpropan-2-yl)oxycarbonylamino]propyl]imidazol-3-ium-1-yl]pyridin-3-yl]oxypropanoate Chemical compound C(C)(C)(C)OC([C@H](COC=1C=CC(=NC=1)N1C=[N+](C=C1)CCCNC(=O)OC(C)(C)C)ON1C(C2=CC=CC=C2C1=O)=O)=O FYZFVVUEVJPIFU-DEOSSOPVSA-O 0.000 description 1
- PTHGTYANKKMAGD-QQJIGTMBSA-N tert-butyl (2S)-2-aminooxy-3-[4-[(2E)-3-methyl-1-[3-[(2-methylpropan-2-yl)oxycarbonylamino]propyl]-2-[(2-methylpropan-2-yl)oxycarbonylimino]imidazol-4-yl]phenoxy]propanoate Chemical compound NO[C@H](C(=O)OC(C)(C)C)COC1=CC=C(C=C1)C=1N(/C(/N(C=1)CCCNC(=O)OC(C)(C)C)=N/C(=O)OC(C)(C)C)C PTHGTYANKKMAGD-QQJIGTMBSA-N 0.000 description 1
- QMCRCDZMFIWKBO-QHCPKHFHSA-O tert-butyl (2S)-2-aminooxy-3-[4-[1-(3-azidopropyl)-2-[3-[(2-methylpropan-2-yl)oxycarbonylamino]propyl]pyrazol-2-ium-4-yl]phenoxy]propanoate Chemical compound NO[C@@H](COC1=CC=C(C=C1)C=1C=[N+](N(C=1)CCCNC(=O)OC(C)(C)C)CCCN=[N+]=[N-])C(=O)OC(C)(C)C QMCRCDZMFIWKBO-QHCPKHFHSA-O 0.000 description 1
- XCZZZYJNRYNTJG-PMERELPUSA-N tert-butyl (2S)-2-methyl-3-[4-[2-[2-[(2-methylpropan-2-yl)oxycarbonylamino]ethylamino]pyrimidin-5-yl]phenoxy]-2-[(2-methylpropan-2-yl)oxycarbonylamino]oxypropanoate Chemical compound C(C)(C)(C)OC(=O)NCCNC1=NC=C(C=N1)C1=CC=C(OC[C@](C(=O)OC(C)(C)C)(C)ONC(=O)OC(C)(C)C)C=C1 XCZZZYJNRYNTJG-PMERELPUSA-N 0.000 description 1
- XTQDECNYFGCFAA-NDEPHWFRSA-O tert-butyl (2S)-3-[4-[1-(2-bromoethyl)-2-[3-[(2-methylpropan-2-yl)oxycarbonylamino]propyl]pyrazol-2-ium-4-yl]phenoxy]-2-(1,3-dioxoisoindol-2-yl)oxypropanoate Chemical compound BrCC[N+]=1N(C=C(C=1)C1=CC=C(C=C1)OC[C@@H](C(=O)OC(C)(C)C)ON1C(C2=CC=CC=C2C1=O)=O)CCCNC(=O)OC(C)(C)C XTQDECNYFGCFAA-NDEPHWFRSA-O 0.000 description 1
- VYTHHZQTVIKASP-LJAQVGFWSA-O tert-butyl (2S)-3-[4-[1-(3-azidopropyl)-2-[3-[(2-methylpropan-2-yl)oxycarbonylamino]propyl]pyrazol-2-ium-4-yl]phenoxy]-2-(1,3-dioxoisoindol-2-yl)oxypropanoate Chemical compound N(=[N+]=[N-])CCC[N+]=1N(C=C(C=1)C1=CC=C(C=C1)OC[C@@H](C(=O)OC(C)(C)C)ON1C(C2=CC=CC=C2C1=O)=O)CCCNC(=O)OC(C)(C)C VYTHHZQTVIKASP-LJAQVGFWSA-O 0.000 description 1
- INRGNBQNTWTYHK-HKBQPEDESA-N tert-butyl (2S)-3-[4-[2-[bis[(2-methylpropan-2-yl)oxycarbonyl]amino]-1-[3-[(2-methylpropan-2-yl)oxycarbonylamino]propyl]imidazol-4-yl]phenoxy]-2-(1,3-dioxoisoindol-2-yl)oxypropanoate Chemical compound C(C)(C)(C)OC(=O)N(C=1N(C=C(N=1)C1=CC=C(OC[C@@H](C(=O)OC(C)(C)C)ON2C(C3=CC=CC=C3C2=O)=O)C=C1)CCCNC(=O)OC(C)(C)C)C(=O)OC(C)(C)C INRGNBQNTWTYHK-HKBQPEDESA-N 0.000 description 1
- XMNCGYHYYHDHEK-MRVPVSSYSA-N tert-butyl (3R)-3-(trifluoromethylsulfonyloxymethyl)pyrrolidine-1-carboxylate Chemical compound FC(S(=O)(=O)OC[C@H]1CN(CC1)C(=O)OC(C)(C)C)(F)F XMNCGYHYYHDHEK-MRVPVSSYSA-N 0.000 description 1
- HKIGXXRMJFUUKV-MRVPVSSYSA-N tert-butyl (3r)-3-(hydroxymethyl)pyrrolidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CC[C@@H](CO)C1 HKIGXXRMJFUUKV-MRVPVSSYSA-N 0.000 description 1
- GANFVPYUFFNOKQ-HUUCEWRRSA-N tert-butyl (3r,4r)-3-hydroxy-4-(phenylmethoxycarbonylamino)piperidine-1-carboxylate Chemical compound O[C@@H]1CN(C(=O)OC(C)(C)C)CC[C@H]1NC(=O)OCC1=CC=CC=C1 GANFVPYUFFNOKQ-HUUCEWRRSA-N 0.000 description 1
- ZBNWFTRJPQWGBI-CLJLJLNGSA-N tert-butyl (4S)-4-[4-[(2R)-2-[tert-butyl(dimethyl)silyl]oxy-3-[(2-methylpropan-2-yl)oxy]-3-oxopropoxy]phenyl]-2-[2-[(2-methylpropan-2-yl)oxycarbonylamino]ethylimino]imidazolidine-1-carboxylate Chemical compound C(C)(C)(C)OC([C@@H](COC1=CC=C(C=C1)[C@@H]1N=C(N(C1)C(=O)OC(C)(C)C)NCCNC(=O)OC(C)(C)C)O[Si](C)(C)C(C)(C)C)=O ZBNWFTRJPQWGBI-CLJLJLNGSA-N 0.000 description 1
- UBOHTDIRPPWKSV-UHFFFAOYSA-O tert-butyl 2-(1,3-dioxoisoindol-2-yl)oxy-2-methyl-3-[4-[1-methyl-2-[3-[(2-methylpropan-2-yl)oxycarbonylamino]propyl]pyrazol-1-ium-4-yl]phenoxy]propanoate Chemical compound C(C)(C)(C)OC(C(COC1=CC=C(C=C1)C=1C=[N+](N(C=1)CCCNC(=O)OC(C)(C)C)C)(C)ON1C(C2=CC=CC=C2C1=O)=O)=O UBOHTDIRPPWKSV-UHFFFAOYSA-O 0.000 description 1
- XRQYPUWAXLZUJD-UHFFFAOYSA-N tert-butyl 2-aminooxy-3-(4-bromophenoxy)-2-methylpropanoate tert-butyl 3-(4-bromophenoxy)-2-hydroxy-2-methylpropanoate Chemical compound CC(C)(C)OC(=O)C(C)(O)COc1ccc(Br)cc1.CC(C)(C)OC(=O)C(C)(COc1ccc(Br)cc1)ON XRQYPUWAXLZUJD-UHFFFAOYSA-N 0.000 description 1
- XZBGUMFZFGTDFF-UHFFFAOYSA-N tert-butyl 2-hydroxy-3-[6-[(2-methylpropan-2-yl)oxycarbonyl-pyridin-4-ylamino]pyridin-3-yl]oxypropanoate Chemical compound C(C)(C)(C)OC(=O)N(C1=CC=C(C=N1)OCC(C(=O)OC(C)(C)C)O)C1=CC=NC=C1 XZBGUMFZFGTDFF-UHFFFAOYSA-N 0.000 description 1
- SJMYWORNLPSJQO-UHFFFAOYSA-N tert-butyl 2-methylprop-2-enoate Chemical compound CC(=C)C(=O)OC(C)(C)C SJMYWORNLPSJQO-UHFFFAOYSA-N 0.000 description 1
- HYXNPAJRDQZIQH-UHFFFAOYSA-N tert-butyl 3-(4-bromophenoxy)-2-hydroxy-2-methylpropanoate Chemical compound BrC1=CC=C(OCC(C(=O)OC(C)(C)C)(C)O)C=C1 HYXNPAJRDQZIQH-UHFFFAOYSA-N 0.000 description 1
- HPURZGPZYRVDTD-UHFFFAOYSA-N tert-butyl 3-(trifluoromethylsulfonyloxymethyl)azetidine-1-carboxylate Chemical compound FC(S(=O)(=O)OCC1CN(C1)C(=O)OC(C)(C)C)(F)F HPURZGPZYRVDTD-UHFFFAOYSA-N 0.000 description 1
- QHCKOPQPUKHMSX-UHFFFAOYSA-N tert-butyl 3-fluoro-3-[(4-methylphenyl)sulfonyloxymethyl]azetidine-1-carboxylate Chemical compound FC1(CN(C1)C(=O)OC(C)(C)C)COS(=O)(=O)C1=CC=C(C)C=C1 QHCKOPQPUKHMSX-UHFFFAOYSA-N 0.000 description 1
- SXMSBPYZGBPFML-BTMGADRYSA-N tert-butyl 5-[[4-[4-[(2S)-2-(1,3-dioxoisoindol-2-yl)oxy-3-[(2-methylpropan-2-yl)oxy]-3-oxopropoxy]phenyl]-2-methylpyrazol-2-ium-1-yl]methyl]-2,2-dimethyl-1,3-oxazolidine-3-carboxylate Chemical compound C(C)(C)(C)OC([C@H](COC1=CC=C(C=C1)C=1C=[N+](N(C=1)CC1CN(C(O1)(C)C)C(=O)OC(C)(C)C)C)ON1C(C2=CC=CC=C2C1=O)=O)=O SXMSBPYZGBPFML-BTMGADRYSA-N 0.000 description 1
- BQXNHJFJZGXYHR-UHFFFAOYSA-O tert-butyl N-[2-[(4-bromo-3-methylimidazol-1-ium-1-yl)methyl]-3-[(2-methylpropan-2-yl)oxycarbonylamino]propyl]carbamate Chemical compound BrC=1[N+](=CN(C=1)CC(CNC(=O)OC(C)(C)C)CNC(=O)OC(C)(C)C)C BQXNHJFJZGXYHR-UHFFFAOYSA-O 0.000 description 1
- ISZMVNUOIWSNEO-UHFFFAOYSA-N tert-butyl N-[3-(1H-imidazol-5-yl)propyl]carbamate Chemical compound N1C=NC(=C1)CCCNC(OC(C)(C)C)=O ISZMVNUOIWSNEO-UHFFFAOYSA-N 0.000 description 1
- OPDRNUVLAYCEPW-UHFFFAOYSA-O tert-butyl N-[3-(4-bromo-3-methylimidazol-1-ium-1-yl)-2-[tert-butyl(dimethyl)silyl]oxypropyl]carbamate Chemical compound BrC=1[N+](=CN(C=1)CC(CNC(=O)OC(C)(C)C)O[Si](C)(C)C(C)(C)C)C OPDRNUVLAYCEPW-UHFFFAOYSA-O 0.000 description 1
- PWLFLNZLWWGREZ-UHFFFAOYSA-N tert-butyl N-[3-(4-bromopyrazol-1-yl)-2-[tert-butyl(dimethyl)silyl]oxypropyl]carbamate Chemical compound BrC=1C=NN(C=1)CC(CNC(OC(C)(C)C)=O)O[Si](C)(C)C(C)(C)C PWLFLNZLWWGREZ-UHFFFAOYSA-N 0.000 description 1
- TZRQZPMQUXEZMC-UHFFFAOYSA-N tert-butyl n-(2-bromoethyl)carbamate Chemical compound CC(C)(C)OC(=O)NCCBr TZRQZPMQUXEZMC-UHFFFAOYSA-N 0.000 description 1
- MXXNEFMIRPHICR-UHFFFAOYSA-N tert-butyl n-(2-fluoro-3-hydroxypropyl)carbamate Chemical group CC(C)(C)OC(=O)NCC(F)CO MXXNEFMIRPHICR-UHFFFAOYSA-N 0.000 description 1
- POHWAQLZBIMPRN-UHFFFAOYSA-N tert-butyl n-(3-aminopropyl)carbamate Chemical compound CC(C)(C)OC(=O)NCCCN POHWAQLZBIMPRN-UHFFFAOYSA-N 0.000 description 1
- OWAMQHJPVUGZSB-LURJTMIESA-N tert-butyl n-[(2s)-2,3-dihydroxypropyl]carbamate Chemical compound CC(C)(C)OC(=O)NC[C@H](O)CO OWAMQHJPVUGZSB-LURJTMIESA-N 0.000 description 1
- SOBBHYDDNWGZCN-UHFFFAOYSA-N tert-butyl n-[2-[(5-bromopyrimidin-2-yl)amino]ethyl]carbamate Chemical compound CC(C)(C)OC(=O)NCCNC1=NC=C(Br)C=N1 SOBBHYDDNWGZCN-UHFFFAOYSA-N 0.000 description 1
- PZSPXAMRSRUYNR-UHFFFAOYSA-N tert-butyl n-[3-(5-bromopyridin-2-yl)prop-2-ynyl]carbamate Chemical compound CC(C)(C)OC(=O)NCC#CC1=CC=C(Br)C=N1 PZSPXAMRSRUYNR-UHFFFAOYSA-N 0.000 description 1
- DSPYCWLYGXGJNJ-UHFFFAOYSA-N tert-butyl n-prop-2-ynylcarbamate Chemical compound CC(C)(C)OC(=O)NCC#C DSPYCWLYGXGJNJ-UHFFFAOYSA-N 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 229960004559 theobromine Drugs 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000001984 thiazolidinyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- 125000000165 tricyclic carbocycle group Chemical group 0.000 description 1
- IMNIMPAHZVJRPE-UHFFFAOYSA-N triethylenediamine Chemical compound C1CN2CCN1CC2 IMNIMPAHZVJRPE-UHFFFAOYSA-N 0.000 description 1
- 125000000026 trimethylsilyl group Chemical group [H]C([H])([H])[Si]([*])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- JBWKIWSBJXDJDT-UHFFFAOYSA-N triphenylmethyl chloride Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(Cl)C1=CC=CC=C1 JBWKIWSBJXDJDT-UHFFFAOYSA-N 0.000 description 1
- YFTHZRPMJXBUME-UHFFFAOYSA-N tripropylamine Chemical compound CCCN(CCC)CCC YFTHZRPMJXBUME-UHFFFAOYSA-N 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 229960000281 trometamol Drugs 0.000 description 1
- 238000003828 vacuum filtration Methods 0.000 description 1
- 229940070710 valerate Drugs 0.000 description 1
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical compound CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 1
- 229910052720 vanadium Inorganic materials 0.000 description 1
- 230000035899 viability Effects 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 239000003039 volatile agent Substances 0.000 description 1
- 230000036642 wellbeing Effects 0.000 description 1
- 238000010626 work up procedure Methods 0.000 description 1
- 238000002424 x-ray crystallography Methods 0.000 description 1
- 150000003954 δ-lactams Chemical class 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/397—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having four-membered rings, e.g. azetidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
- A61K31/427—Thiazoles not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/433—Thidiazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/444—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring heteroatom, e.g. amrinone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C53/00—Saturated compounds having only one carboxyl group bound to an acyclic carbon atom or hydrogen
- C07C53/15—Saturated compounds having only one carboxyl group bound to an acyclic carbon atom or hydrogen containing halogen
- C07C53/16—Halogenated acetic acids
- C07C53/18—Halogenated acetic acids containing fluorine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6558—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6558—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system
- C07F9/65583—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system each of the hetero rings containing nitrogen as ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2300/00—Mixtures or combinations of active ingredients, wherein at least one active ingredient is fully defined in groups A61K31/00 - A61K41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/42—Oxazoles
- A61K31/424—Oxazoles condensed with heterocyclic ring systems, e.g. clavulanic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
- A61K31/429—Thiazoles condensed with heterocyclic ring systems
- A61K31/43—Compounds containing 4-thia-1-azabicyclo [3.2.0] heptane ring systems, i.e. compounds containing a ring system of the formula, e.g. penicillins, penems
- A61K31/431—Compounds containing 4-thia-1-azabicyclo [3.2.0] heptane ring systems, i.e. compounds containing a ring system of the formula, e.g. penicillins, penems containing further heterocyclic rings, e.g. ticarcillin, azlocillin, oxacillin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/551—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having two nitrogen atoms, e.g. dilazep
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Definitions
- This invention relates to novel biaryl monobactam compounds, processes for their preparation and their use as therapeutic agents. More particularly, the invention relates to biaryl monobactam compounds and their use as antibiotic agents for the treatment of bacterial infections.
- ⁇ -lactams are the most widely used antibiotics for treatment of serious bacterial infections. These include carbapenems, cephalosporins, penicillins, and monobactams. As has been observed for other antibiotic classes, resistance to ⁇ -lactams has emerged. For most Gram-negative bacteria, this resistance is primarily driven by the expression of ⁇ -lactamases, enzymes that hydrolyze ⁇ -lactam compounds. There are 4 different classes of ⁇ -lactamases (A, B, C, and D) capable of hydrolyzing overlapping but distinct subsets of ⁇ -lactams (Drawz and Bonomo, Clin. Micro. Rev., 2010, 23 : 160-201).
- MBLs metallo ⁇ -lactamases
- Aztreonam a monobactam, was first approved in the U.S in 1986 for the treatment of aerobic Gram-negative bacterial infections and remains the only monobactam in use in the U.S. today.
- aztreonam has poor activity against Pseudomonas and Acinetobacter strains. Because monobactams are inherently resistant to hydrolysis by MBLs, several companies have begun developing novel monobactam compounds for the treatment of infections caused by Gram-negative bacteria. Monobactam compounds comprising a siderophore moiety are disclosed in WO 2007/065288, WO2012/073138, J. Medicinal Chemistry 56: 5541-5552 (2013), and Bioorganic and Medicinal Chemstry Letters 22:5989 (2012).
- U.S. Patent Application Publication No. US 2014/0275007 discloses oxamazin monobactams and their use as antibacterial agents
- U.S. Patent Application Publication No. US 2015/0266867 also discloses novel monobactam compounds for the use as antibacterial agents
- International Patent Application Publication No. WO 2013/110643 discloses novel amidine substituted monobactam derivatives and their use as antimicrobial reagents.
- the invention relates to the design and synthesis of a series of bi-aryl monobactam analogs, a novel class of highly potent antibiotics effective against a broad range of Gram- negative bacteria.
- These compounds and their pharmaceutically acceptable salts may be useful as therapeutic agents for clinical treatment of various infections caused by Gram- negative bacteria, including strains that are multidrug resistant.
- the compounds can be used alone or in combination with a suitable ⁇ -lactamase inhibitor. More particularly, the present invention includes compounds of Formula I:
- W is a bond or O
- R x and R z are independently hydrogen, -SCi-C 3 alkyl, C1-C3 alkyl, -(Ci- C 3 alkylene)nOCi-C 3 alkyl, or -(Ci-C 3 alkylene) n NCi-C 3 alkyl, wherein the -SCi-C 3 alkyl, the Ci-C 3 alkyl, the -(Ci-C 3 alkylene)nOCi-C 3 alkyl and the -(Ci-C 3 alkylene)nNCi-C 3 alkyl are optionally substituted with one to seven fluorines;
- R x and R z together with the carbon to which they are attached, form a monocyclic C4-C7 cycloalkyl or a monocyclic C4-C7 heterocycloalkyl with 1, 2, or 3 heteroatom ring atoms independently selected from N, O and S, wherein said C4-C7 cycloalkyl and said C4-C7 heterocycloalkyl are optionally substituted with one to three substituents independently selected from -F, -OH and -OCi-C 3 alkyl;
- X is N or CR 1 ;
- R 1 is hydrogen, Ci.C 3 alkyl, or halogen; wherein said C1-C 3 alkyl is optionally substituted with one to three R a ;
- each occurrence of R a is independently hydrogen, halogen, Ci-C 3 alkyl, - R c R d or -
- Z is C1-C 3 alkylene, optionally substituted with one to three R b ;
- each occurrence of R b is independently -Ci-C 8 alkyl, -C 3 -C7 cycloalkyl, -C(0)OR e , - C(0) R c R d , tetrazolyl, oxadiazolonyl, HetA, AryA, -S(0) m R e , -S(0) m R c R d , or "P(0)(R e ) p wherein said -Ci-C 8 alkyl and said -C 3 -C7 cycloalkyl are optionally substituted with one to three R a ;
- HetA is a 4- to 6-membered saturated or monounsaturated monocyclic ring with 1, 2, or 3 heteroatom ring atoms independently selected from N, N as a quaternary salt, O and S, optionally substituted with one to four R 4 ;
- AryA is a 5- to 6-membered monocyclic aromatic ring with 0, 1, 2, or 3 ring atoms independently selected from N, N as a quaternary salt, O and S, optionally substituted with one to four R 4 ;
- Y is a bond, O, R 2 , S, or CH 2 ;
- R 2 is hydrogen, -C1-C 3 alkyl, -C(0)R e , -C(0) R c R d , -S(0) m R e , or -S(0) m R c R d , wherein said -C1-C 3 alkyl is optionally substituted with one to three R a ;
- a 1 is AryA
- a 2 is -(CH 2 ) n N(R 3 ) 2 -, C 3 -C7 cycloalkyl, AryC or HetC, wherein said C 3 -C7 cycloalkyl is optionally substituted with one to four R 4 ;
- AryC is a 5- to 6-membered monocyclic aromatic ring with 0, 1, 2, or 3 ring atoms independently selected from N, N as a quaternary salt, O and S, optionally substituted with one to four R 4 ;
- HetC is a 4- to 7-membered saturated or monounsaturated monocyclic ring with 1, 2, or 3 heteroatom ring atoms independently selected from N, N as a quaternary salt, O and S, optionally substituted with one to four R 4 ;
- each occurrence of R 4 is independently: -Ci-C 8 alkyl, -C 2 -C 8 alkenyl, -C 2 -C 8 alkynyl, halogen, -OR e , -S(0) m R e , -S(0) m R c R d , -C(0)R e , -OC(0)R e , -C(0)OR e , -CN, -
- cycloheteroalkyl optionally containing one to two additional heteroatoms independently selected from O, S and - R g ;
- HetB is a 3- to 6-membered saturated or monounsaturated monocyclic ring with 1, 2, or 3 heteroatom ring atoms independently selected from N, N as a quaternary salt, O and S, optionally substituted with one to three R a ;
- Q is a bond, CH 2 , O, S, -(CH 2 ) n R 3 -, or - R 3 (CH 2 ) n -, wherein each CH 2 is unsubstituted or substituted with one to two substituents selected from halogen, -Ci-C 6 alkyl, OR e and -(CH 2 ) n R c R d ;
- R 3 is hydrogen or -Ci-C 3 alkyl, wherein said -Ci-C 3 alkyl is optionally substituted with one to three R a ;
- M is R 5 , - HR 5 , -N(R 5 ) 2 , -OR 5 , -(CH 2 ) n R 5 , -C(0)R 5 , -C( H)R 5 , or -S(0) m R 5 ;
- R 5 is H, C 2 -Cio alkyl, C 3 -C 7 cycloalkyl, Ci-C 6 alkyl-C 3 -C 7 cycloalkyl, HetB, AryB, or - H(Ci-C 6 alkyl), wherein said Ci-C 6 alkyl, said C 2 -Cio alkyl and said C 3 -C 7 cycloalkyl are optionally substituted with one to four R 6 ;
- AryB is a 5- to 6-membered monocyclic aromatic ring with 0, 1, 2, or 3 ring atoms independently selected from N, O and S, optionally substituted with one to four R 4 ;
- each occurrence of R 6 is independently selected from the group consisting of:
- each occurrence of R c and R d is independently: hydrogen, -Ci-Cio alkyl, -C 2 -C 10 alkenyl, -C 3 -C6 cycloalkyl, -C 1 -C 10 alkylene-C 3 -C 6 cycloalkyl, HetA, -Ci-Cioalkylene-HetB, AryB, -C 1 -C 10 alkylene-AryB, and - C
- each occurrence of R e is independently: hydrogen, -Ci-Cioalkyl, -C 2 -C 10 alkenyl, - OH, -OC1-C4 alkyl, -C 3 -C 6 cycloalkyl, -C1-C10 alkylene-C 3 -C 6 cycloalkyl, HetB, -C1-C10 alkylene-HetB, AryB, -C 1 -C 10 alkylene-AryB, or -C 1 -C 10 alkylene-HetB; wherein each R e is optionally substituted with one to three R h ;
- each occurrence of R is independently: halogen, -C 1 -C 10 alkyl, -OH, -OC 1 -C4 alkyl, - S(0) m Ci-C 4 alkyl, -CN, -CF 3 , -OCHF 2 , -OCF3, or H 2 , wherein said -C1-C10 alkyl is optionally substituted with one to three substituents independently selected from: -OH, halogen, cyano, and -S(0) 2 CH 3 ;
- each occurrence of R g is independently: hydrogen, -C(0)R e , and -C 1 -C 10 alkyl, wherein said -Ci-Cioalkyl is optionally substituted with one to five fluorines;
- each occurrence of R h is independently: halogen, -Ci-Cioalkyl, -OH, -OC 1 -C4 alkyl, - S(0) m Ci-C 4 alkyl, -CN, -CF 3 , -OCHF 2 , or -OCF 3 ; wherein said -C1-C10 alkyl is optionally substituted with one to three substituents independently selected from: -OH, halogen, cyano, or-S(0) 2 CH 3 ;
- each n is independently 0, 1, 2, 3 or 4;
- each m is independently 0, 1 or 2
- each p is independently 1 or 2.
- the present invention also includes compounds of Formula I:
- W is a bond or O;
- R x and R z are independently hydrogen, -SCi-C 3 alkyl, C 1 -C3 alkyl, -(Ci- C 3 alkylene)nOCi-C 3 alkyl, or -(Ci-C 3 alkylene) n NCi-C 3 alkyl, wherein the -SCi-C 3 alkyl, the Ci-C 3 alkyl, the -(Ci-C 3 alkylene) n OCi-C 3 alkyl and the -(Ci-C 3 alkylene) n NCi-C 3 alkyl are optionally substituted with one to seven fluorines;
- R x and R z together with the carbon to which they are attached, form a monocyclic C 4 -C 7 cycloalkyl or a monocyclic C 4 -C 7 heterocycloalkyl with 1, 2, or 3 heteroatom ring atoms independently selected from N, O and S, wherein said C 4 -C 7 cycloalkyl and said C 4 -C 7 heterocycloalkyl are optionally substituted with one to three substituents independently selected from -F, -OH and -OCi-C 3 alkyl;
- X is N or CR 1 ;
- R 1 is hydrogen, Ci.C 3 alkyl, or halogen; wherein said Ci-C 3 alkyl is optionally substituted with one to three R a ;
- each occurrence of R a is independently hydrogen, halogen, Ci-C 3 alkyl, - R c R d or -
- Z is Ci-C 3 alkylene, optionally substituted with one to three R b ;
- each occurrence of R b is independently -Ci-C 8 alkyl, -C 3 -C 7 cycloalkyl, -C(0)OR e , - C(0) R c R d , tetrazolyl, oxadiazolonyl, HetA, AryA, -S(0) m R e , -S(0) m R c R d , or "P(0)(R e ) p wherein said -Ci-C 8 alkyl and said -C 3 -C 7 cycloalkyl are optionally substituted with one to three R a ;
- HetA is a 4- to 6-membered saturated or monounsaturated monocyclic ring with 1, 2, or 3 heteroatom ring atoms independently selected from N, N as a quaternary salt, O and S, optionally substituted with one to four R 4 ;
- AryA is a 5- to 6-membered monocyclic aromatic ring with 0, 1, 2, or 3 ring atoms independently selected from N, N as a quaternary salt, O and S, optionally substituted with one to four R 4 ;
- Y is a bond, O, R 2 , S, or CH 2 ;
- R 2 is hydrogen, -Ci-C 3 alkyl, -C(0)R e , -C(0) R c R d , -S(0) m R e , or -S(0) m R c R d , wherein said -Ci-C 3 alkyl is optionally substituted with one to three R a ;
- a 1 is AryA
- a 2 is C 3 -C 7 cycloalkyl, AryC or HetC, wherein said C 3 -C 7 cycloalkyl is optionally substituted with one to four R 4 ;
- AryC is a 5- to 6-membered monocyclic aromatic ring with 0, 1, 2, or 3 ring atoms independently selected from N, N as a quaternary salt, O and S, optionally substituted with one to four R 4 ;
- HetC is a 4- to 7-membered saturated or monounsaturated monocyclic ring with 1, 2, or 3 heteroatom ring atoms independently selected from N, N as a quaternary salt, O and S, optionally substituted with one to four R 4 ;
- each occurrence of R 4 is independently: -Ci-C 8 alkyl, -C 2 -C 8 alkenyl, -C 2 -C 8 alkynyl, halogen, -OR e , -S(0) m R e , -S(0) m R c R d , -C(0)R e , -OC(0)R e , -C(0)OR e , -CN, -
- HetB is a 3- to 6-membered saturated or monounsaturated monocyclic ring with 1, 2, or 3 heteroatom ring atoms independently selected from N, N as a quaternary salt, O and S, optionally substituted with one to three R a ;
- Q is a bond, CH 2 , O, S, -(CH 2 ) n R 3 -, or - R 3 (CH 2 ) n -;
- R 3 is hydrogen or -Ci-C 3 alkyl, wherein said -Ci-C 3 alkyl is optionally substituted with one to three R a ;
- M is R 5 , - R 5 , -OR 5 , -(CH 2 ) n R 5 , -C(0)R 5 , -CH( H)R 5 , or -S(0) m R 5 ;
- R 5 is C 2 -Cio alkyl, C 3 -C 7 cycloalkyl, HetB, or AryB, wherein said C 2 -Cio alkyl and said C 3 -C 7 cycloalkyl are optionally substituted with one to four R 6 ;
- AryB is a 5- to 6-membered monocyclic aromatic ring with 0, 1, 2, or 3 ring atoms independently selected from N, O and S, optionally substituted with one to four R 4 ;
- each occurrence of R 6 is independently selected from the group consisting of:
- each occurrence of R c and R d is independently: hydrogen, -C 1 -C 10 alkyl, -C 2 -Cio alkenyl, -C 3 -C 6 cycloalkyl, -C 1 -C 10 alkylene-C 3 -C 6 cycloalkyl, HetA, -Ci-Cioalkylene-HetB, AryB, -C 1 -C 10 alkylene-AryB, and - C 1 -C 10 alkylene-HetB, or, alternatively, R c and R d together with the nitrogen atom to which they are attached, form a 4- to 7-membered cycloheteroalkyl optionally containing one to two additional heteroatoms independently selected from O, S and -NR g , and wherein each R c and R d is optionally substituted with one to three R f ;
- each occurrence of R e is independently: hydrogen, -Ci-Cioalkyl, -C 2 -C 10 alkenyl, - OH, -OC1-C4 alkyl, -C 3 -C 6 cycloalkyl, -C1-C10 alkylene-C 3 -C 6 cycloalkyl, HetB, -C1-C10 alkylene-HetB, AryB, -Ci-Cio alkylene-AryB, or -Ci-Cio alkylene-HetB; wherein each R e is optionally substituted with one to three R h ;
- each occurrence of R is independently: halogen, -Ci-Cio alkyl, -OH, -OC 1 -C4 alkyl, - S(0) m Ci-C 4 alkyl, -CN, -CF 3 , -OCHF 2 , or -OCF 3 ; wherein said -C 1 -C 10 alkyl is optionally substituted with one to three substituents independently selected from: -OH, halogen, cyano, and -S(0) 2 CH 3 ;
- each occurrence of R g is independently: hydrogen, -C(0)R e , and -C 1 -C 10 alkyl, wherein said -Ci-Cioalkyl is optionally substituted with one to five fluorines;
- each occurrence of R h is independently: halogen, -Ci-Cioalkyl, -OH, -OC 1 -C4 alkyl, - S(0) m Ci-C 4 alkyl, -CN, -CF 3 , -OCHF 2 , or -OCF 3 ; wherein said -C 1 -C 10 alkyl is optionally substituted with one to three substituents independently selected from: -OH, halogen, cyano, or-S(0) 2 CH 3 ;
- each n is independently 0, 1, 2, 3 or 4;
- each m is independently 0, 1 or 2
- each p is independently 1 or 2.
- the present invention also relates to a pharmaceutical composition for treating a bacterial infection in a subject, including infection with multidrug resistant Gram-negative bacterial strains, comprising a biaryl monobactam compound of the invention and a pharmaceutically acceptable carrier, diluent or excipient.
- the Compounds of Formula (I) (also referred to herein as the "biaryl monobactam compounds”) and pharmaceutically acceptable salts thereof can be useful, for example, for inhibiting the growth of Gram-negative bacterial strains, including but not limited to, Pseudomonas and Acinetobacter strains, and/or for treating or preventing the clinical maifestations thereof in a patient.
- the present invention is also directed to methods of treating Gram-negative bacterial infections in a subject in need of treatment thereof, comprising administering to the subject an effective amount of a biaryl monobactam compound of the invention.
- the method includes administration of a beta lactamase inhibitor compound.
- the invention relates to novel biaryl monobactam analogs, a class of highly potent antibiotics effective against a broad range of Gram-negative bacteria. These compounds have utility as therapeutic agents for clinical treatment of various infections caused by Gram- negative bacteria, including strains that are multidrug resistant, and for the treatment or prevention of the clinical pathologies associated therewith.
- each variable including those of Formulas I, II- 1, II-2, II-3, III- 1 , III-2, and III-3, and the various embodiments thereof, each variable is selected independently of the others unless otherwise indicated.
- the present invention encompasses all compounds of Formulas I, II- 1, II-2, II-3, III- 1, III-2, and III-3, and the various embodiments thereof, for example, any solvates, hydrates, stereoisomers, and tautomers of said compounds and of any pharmaceutically acceptable salts thereof.
- a first embodiment of the invention is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein X, Y, Z, A 1 , Q, A 2 , M, W, R x and R z are as defined in Formula I in the Summary of the Invention.
- a second embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is a bond, and all other variables are as defined in Embodiment El .
- a third embodiment is a compound of Formula I, or a
- a fourth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is N, and all other variables are as defined in Embodiment El .
- a fifth embodiment is a compound of Formula I, or a
- R 1 is hydrogen
- R 1 is halogen.
- R 1 is chlorine.
- R 1 is fluorine
- R 1 is Ci.C 3 alkyl optionally substituted with one to three R a , wherein each occurrence of R a is independently hydrogen, halogen, Ci-C 3 alkyl, - R c R d or - OR e .
- a sixth embodiment is a compound of Formula I, or a
- W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is C 1 -C3 alkylene optionally substituted with one to three R b , wherein each occurrence of R b is independently hydrogen, Ci-C 8 alkyl, C 3 -C 7 cycloalkyl, -C(0)OR e , -C(0) R c R d , tetrazolyl, oxadiazolonyl, HetA, AryA, -S(0) m R e , -S(0) m R c R d , or - P(0)(R e ) p , wherein said Ci-C 8 alkyl and said C 3 -C 7 cycloalkyl are optionally substituted with one to three R a ;and all other variables are as defined in Embodi
- Z is Ci-C 3 alkylene substituted with one occurrence of R b .
- Z is Ci-C 3 alkylene substituted with two occurrences of R b .
- Z is Ci-C 3 alkylene substituted with three occurrences of R b .
- a seventh embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is Ci-C 3 alkylene substituted with -C(0)OR e ; and all other variables are as defined in Embodiment
- An eighth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is Ci-C 3 alkylene substituted with -C(0)OH; and all other variables are as defined in Embodiment El .
- a ninth embodiment is a compound of Formula I, or a
- a tenth embodiment is a compound of Formula I, or a
- W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is C 1 -C 3 alkylene substituted with Ci-C 8 alkyl, optionally substituted with one to three R a , and all other variables are as defined in Embodiment El .
- An eleventh embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is C 1 -C 3 alkylene substituted with methyl; and all other variables are as defined in Embodiment El .
- a twelfth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is C 1 -C 3 alkylene substituted with methyl and -C(0)OH; and all other variables are as defined in Embodiment El .
- a thirteenth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is - CH(C(0)OH)CH 2 - and all other variables are as defined in Embodiment El .
- a fourteenth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is C 1 -C3 alkylene substituted with oxadiazolonyl, and all other variables are as defined in
- Embodiment E15 is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is a bond, and all other variables are as defined in
- Embodiment El A sixteenth embodiment (Embodiment E16) is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is O, and all other variables are as defined in Embodiment El .
- a seventeenth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is NR 2 , and all other variables are as defined in Embodiment El .
- R 2 is hydrogen. In another sub- embodiment of Embodiment E17, R 2 is C 1-C3 alkyl optionally substituted with one to three R a . In a further sub-embodiment of Embodiment El 7, R 2 is C(0)R e In yet another sub- embodiment of Embodiment El 7, R 2 is -C(0)NR c R d . In still another sub-embodiment of Embodiment El 7, R 2 is -S(0) m R e . In a further sub -embodiment of Embodiment El 7, R 2 is - S(0) m NR c R d .
- An eighteenth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is S, and all other variables are as defined in Embodiment El .
- An nineteenth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is CH 2 , and all other variables are as defined in Embodiment El .
- a twentieth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 a 5- to 6- membered monocyclic aromatic ring with 0, 1, 2, or 3 ring atoms independently selected from N, N as a quaternary salt, O and S, optionally substituted with one to four R 4 , and all other variables are as defined in Embodiment El .
- a twenty-first embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is a 5- membered monocyclic aromatic ring with 0, 1, 2, or 3 ring atoms independently selected from N, N as a quaternary salt, O and S, optionally substituted with one to four R 4 , and all other variables are as defined in Embodiment El .
- a twenty-second embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is a 6- membered monocyclic aromatic ring with 0, 1, 2, or 3 ring atoms independently selected from N, N as a quaternary salt, O and S, optionally substituted with one to four R 4 , and all other variables are as defined in Embodiment El .
- a 1 has 1 ring atoms independently selected from N, N as a quaternary salt, O and S.
- Embodiment E21 or E22 A 1 has 2 ring atoms independently selected from N, N as a quaternary salt, O and S. In another sub-embodiment of Embodiment E21 or E22, A 1 has 2 ring atoms independently selected from N, N as a quaternary salt, O and S. In yet another sub-embodiment of Embodiment E21 or E22, A 1 has 3 ring atoms independently selected from N, N as a quaternary salt, O and S.
- a twenty -third embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is an 6- membered monocyclic aromatic ring, optionally substituted with one to four R 4 , and all other variables are as defined in Embodiment El .
- a twenty-fourth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is an unsubstituted 6-membered monocyclic aromatic ring, and all other variables are as defined in Embodiment El .
- a twenty-fifth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is:
- a twenty-sixth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is a bond, and all other variables are as defined in
- a twenty-seventh embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is CH 2 , and all other variables are as defined in
- a twenty-eighth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is O, and all other variables are as defined in Embodiment El .
- a twenty-ninth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is S, and all other variables are as defined in Embodiment El .
- a thirtieth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is -(CH 2 ) n NR 3 -, and all other variables are as defined in Embodiment El .
- a thirty-first embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is - R 3 (CH 2 ) n -, and all other variables are as defined in Emb odi ment E 1.
- a thirty-second embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is H, and all other variables are as defined in
- a thirty-third embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is N(Ci-C 3 alkyl) optionally substituted with one to three R a , and all other variables are as defined in Embodiment El .
- a thirty-fourth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is C 3 -C 7 cycloalkyl optionally substituted with one to four R 4 ; and all other variables are as defined in Embodiment El .
- a thirty-fifth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is AryC; and all other variables are as defined in Embodiment El .
- a thirty-sixth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is a 5- membered monocyclic aromatic ring with 0, 1, 2, or 3 ring atoms independently selected from N, N as a quaternary salt, O and S, optionally substituted with one to four R 4 ; and all other variables are as defined in Embodiment El .
- a thirty-seventh embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is a 6- membered monocyclic aromatic ring with 0, 1, 2, or 3 ring atoms independently selected from N, N as a quaternary salt, O and S, optionally substituted with one to four R 4 , and all other variables are as defined in Embodiment El .
- a thirty-eighth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is HetC; and all other variables are as defined in Embodiment El .
- a thirty-ninth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is a 4- to 7- membered saturated or monounsaturated monocyclic ring with 1 heteroatom ring atom selected from N, N as a quaternary salt, O and S, optionally substituted with one to four R 4 ; and all other variables are as defined in Embodiment El .
- a fortieth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is a 4- to 7- membered saturated or monounsaturated monocyclic ring with 2 heteroatom ring atoms independently selected from N, N as a quaternary salt, O and S, optionally substituted with one to four R 4 ; and all other variables are as defined in Embodiment El .
- a forty-first embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is a 4- to 7- membered saturated or monounsaturated monocyclic ring with 3 heteroatom ring atoms independently selected from N, N as a quaternary salt, O and S, optionally substituted with one to four R 4 ; and all other variables are as defined in Embodiment El .
- a 2 is unsubstituted. In other sub- embodiments, A 2 is substituted with one R 4 . In further sub -embodiments A 2 is substituted with two R 4 . In additional sub-embodiments, A 2 is substituted with three R 4 . In still further sub-embodiments, A 2 is substituted with four R 4 .
- a forty-second embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in
- a forty -third embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is:
- a forty-fourth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is E26-E33, A 2 is:
- a forty -fifth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is of Embodiments E26-E33, A 2 is:
- a forty-sixth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is:
- a forty-seventh embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is E26-E33, A 2 is:
- a forty-eighth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is:
- a forty-ninth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is:
- a fiftieth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is:
- a fifty-first embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is:
- a fifty-second embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is:
- a fifty -third embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is:
- a fifty -fourth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is:
- a fifty -fifth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is:
- a fifty-sixth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is:
- a fifty-seventh embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is: N->
- a fifty-eighth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is E26-E33, A 2 is:
- a fifty-ninth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is defined in any of Embodiments E34-E58, M is R 5 , and all other variables are as defined in
- Embodiment E60 is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is defined in any of Embodiments E34-E58, M is C2-C10 alkyl optionally substituted with one to four R 6 , and all other variables are as defined in Embodiment El .
- a sixty-first embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is defined in any of Embodiments E34-E58, M is C3-C7 cycloalkyl optionally substituted with one to four R 6 , and all other variables are as defined in Embodiment El .
- a sixty-second embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is defined in any of Embodiments E34-E58, M is HetB, and all other variables are as defined in Embodiment El .
- a sixty -third embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is defined in any of Embodiments E34-E58, M is AryB, and all other variables are as defined in Embodiment El .
- a sixty-fourth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is defined in any of Embodiments E34-E58, M is - R 5 , and all other variables are as defined in Emb odi ment E 1.
- a sixty-fifth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is defined in any of Embodiments E34-E58, M is -OR 5 , and all other variables are as defined in Embodiment El .
- a sixty-sixth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is defined in any of Embodiments E34-E58, M is -(CH 2 ) n R 5 , and all other variables are as defined in Embodiment El .
- a sixty-seventh embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is defined in any of Embodiments E34-E58, M is -C(0)R 5 , and all other variables are as defined in Embodiment El .
- a sixty-eighth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is defined in any of Embodiments E34-E58, M is -CH( H)R 5 , and all other variables are as defined in Embodiment El .
- a sixty-ninth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is defined in any of Embodiments E34-E58, M is -S(0) m R 5 and all other variables are as defined in Emb odi ment E 1.
- a seventieth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is defined in any of Embodiments E34-E58, M is -(CH 2 )3 H 2 , and all other variables are as defined in Embodiment El .
- a seventy-first embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is defined in any of Embodiments E34-E58, M is -(CH 2 )i -2 HetB, and all other variables are as defined in Embodiment El .
- a seventy-second embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is defined in any of Embodiments E34-E58, M is -CH 2 AryB, and all other variables are as defined in Embodiment El .
- a seventy -third embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is defined in any of Embodiments E34-E58, M is defined in any of Embodiments E59- E72, R x and R z are methyl, and all other variables are as defined in Embodiment El .
- a seventy -fourth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is defined in any of Embodiments E34-E58, M is defined in any of Embodiments E59- E72, R x is hydrogen and R z is methyl, and all other variables are as defined in Embodiment El .
- a seventy -fifth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is defined in any of Embodiments E34-E58, M is defined in any of Embodiments E59- E72, one of R x and R z is SCH 3 , and all other variables are as defined in Embodiment El .
- a seventy-sixth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is defined in any of Embodiments E34-E58, M is defined in any of Embodiments E59- E72, one of R x and R z is SC 1 -C3 alkyl, optionally substituted with one to seven fluorines, and all other variables are as defined in Embodiment El .
- a seventy-seventh embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is defined in any of Embodiments E34-E58, M is defined in any of Embodiments E59- E72, one of R x and R z is SCH 3 , optionally substituted with one to three fluorines, and all other variables are as defined in Embodiment El .
- a seventy-eighth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is defined in any of Embodiments E34-E58, M is defined in any of Embodiments E59- E72, one of R x and R z is Ci-C 3 alkyl, optionally substituted with one to seven fluorines, and all other variables are as defined in Embodiment El .
- a seventy -ninth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is defined in any of Embodiments E34-E58, M is defined in any of Embodiments E59- E72, one of R x and R z is (Ci-C3alkylene) n OCi-C3alkyl, optionally substituted with one to seven fluorines, and all other variables are as defined in Embodiment El .
- An eightieth embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is defined in any of Embodiments E34-E58, M is defined in any of Embodiments E59- E72, one of R x and R z is (Ci-C3alkylene) n NCi-C3alkyl, optionally substituted with one to seven fluorines, and all other variables are as defined in Embodiment El .
- An eighty -first embodiment is a compound of Formula I, or a pharmaceutically acceptable salt thereof, wherein W is defined in Embodiment E2 or E3, X is defined in any of Embodiment E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E, Z is defined in any of Embodiments E6-E13, Y is defined in any of Embodiments E15-E19, A 1 is defined in any of Embodiments E20-E25, Q is defined in any of Embodiments E26-E33, A 2 is defined in any of Embodiments E34-E58, M is defined in any of Embodiments E59- E72, R x and R z , together with the carbon to which they are attached, come together to form a monocyclic C 4 - C 7 cycloalkyl or a monocyclic C 4 -C 7 heterocycloalkyl with 1, 2, or 3 heteroatom ring
- An eighty-second embodiment is a compound of Formula II- 1, II-
- X is defined in any of Embodiments El, E4, E5, E5-A, E5-B, E5-C, E5-D, or E5-E;
- a 1 is defined in any of Embodiments El, E20-E25;
- a 2 is defined in any of Embodiments El, E34-E58;
- M is defined in any of Embodiments El, E59- E72;
- R x and R z are defined in any of Embodiments El, E73-E81;
- R bl , R b2 , and R b3 are independently hydrogen, Ci-C 8 alkyl, C 3 -C 7 cycloalkyl, -C(0)OR e , - C(0) R c R d , tetrazolyl, oxadiazolonyl, HetA, AryA, -S(0) m R e , -S(0) m R c R d , or - P(O)
- An eighty -third embodiment is a compound of Formula II- 1, II-2, II-3, III- 1 , III-2, or III-3, or a pharmaceutically acceptable salt thereof, wherein R bl and R b2 are independently hydrogen, Ci-C 3 alkyl, tetrazolyl, oxadiazolonyl or -C(0)OR e ; and R b3 is hydrogen, and all other variables are as defined in Embodiment E82.
- An eighty-fourth embodiment is a compound having the structure:
- each occurrence of R 4 is independently: -Ci-C 8 alkyl C 2 -C 8 alkenyl, -C 2 -C 8 alkynyl, halogen, -OR e , -S(0) m R e , -S(0) m R c R d , -C(0)R e
- R 5 is C 2 -Cio alkyl, C 3 -C 7 cycloalkyl, Ci-C 6 alkyl-C 3 - C 7 cycloalkyl, HetB, AryB, or - H(Ci-C 6 alkyl), wherein said Ci-C 6 alkyl, said C 2 -Ci 0 alkyl and said C 3 -C 7 cycloalkyl are optionally substituted with one to four R 6 .
- Reference to different embodiments with respect to Formula I compounds specifically includes different embodiments of Formula II and Formula III such as Formula II- 1, II-2, II-3, III-l, ⁇ -2, and ⁇ -3, sub-embodiments of Formula I, II- 1, II-2, II-3, III-l, III- 2, and III-3, other embodiments provided herein, and individual compounds described herein.
- Formula II and Formula III such as Formula II- 1, II-2, II-3, III-l, ⁇ -2, and ⁇ -3, sub-embodiments of Formula I, II- 1, II-2, II-3, III-l, III- 2, and III-3, other embodiments provided herein, and individual compounds described herein.
- a pharmaceutical composition comprising an effective amount of a compound of Formula I, II- 1, II-2, II-3, III-l, III-2, or III-3, as defined herein, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
- composition comprising (i) a compound of Formula I, II- 1,
- a method for treating a bacterial infection in a subject which comprises administering to a subject in need of such treatment an effective amount of a compound of Formula I, II- 1, II-2, II-3, III-l, III-2, or III-3, or a pharmaceutically acceptable salt thereof.
- a method for treating a bacterial infection which comprises administering to a subject in need of such treatment a therapeutically effective amount of the composition of (a), (b), (c), (d), or (e).
- the present invention also includes a compound of Formula I, II-l, II-2, II-3, III- 1 , III-2, or III-3, or a pharmaceutically acceptable salt thereof, (i) for use in, (ii) for use as a medicament for, or (iii) for use in the preparation (or manufacture) of a medicament for, medicine or treating bacterial infection, including infection with a multidrug resistant bacterial strain.
- the compounds of the present invention can optionally be employed in combination with one or more second therapeutic agents including relebactam, tazobactam, clavulanic acid, sulbactam, and avibactam.
- Additional embodiments of the invention include the pharmaceutical compositions, combinations and methods set forth in (a)-(j) above and the uses set forth in the preceding paragraph, wherein the compound of the present invention employed therein is a compound of one of the embodiments, sub-embodiments, classes or sub-classes described above.
- the compound may optionally be used in the form of a pharmaceutically acceptable salt in these embodiments.
- each embodiment may be combined with one or more other embodiments, to the extent that such a combination provides a stable compound or salt and is consistent with the description of the embodiments. It is further to be understood that the embodiments of compositions and methods provided as (a) through (j) above are understood to include all embodiments of the compounds and/or salts, including such embodiments as result from combinations of embodiments.
- Additional embodiments of the present invention include each of the pharmaceutical compositions, combinations, methods and uses set forth in the preceding paragraphs, wherein the compound of the present invention or its salt employed therein is substantially pure.
- a pharmaceutical composition comprising a compound of Formula I, II-l, II- 2, II-3, III- 1 , III-2, or III-3 or its salt and a pharmaceutically acceptable carrier and optionally one or more excipients
- substantially pure is in reference to a compound of Formula I, II-l, II-2, II-3, III- 1 , III-2, or III-3 or its salt per se; i.e., the purity of this active ingredient in the composition.
- ⁇ -lactamase inhibitor refers to a compound which is capable of inhibiting enzyme activity from ⁇ -lactamases.
- inhibiting ⁇ -lactamase activity means inhibiting the activity of a class A, C, and/or D ⁇ -lactamase.
- inhibition at a 50% inhibitory concentration is preferably achieved at or below about 100 micrograms/mL, or at or below about 50 micrograms/mL, or at or below about 25 micrograms/mL.
- class A”, “class B”, “class C”, and “class D” ⁇ -lactamases are understood by those skilled in the art and are described in S. G. Waley, ⁇ -lactamase:
- metallo ⁇ -lactamase denotes a metalloprotein capable of inactivating a ⁇ -lactam antibiotic.
- the ⁇ -lactamase can be an enzyme which catalyzes the hydrolysis of the ⁇ -lactam ring of a ⁇ -lactam antibiotic.
- microbial metallo- ⁇ -lactamases can be, for example, a zinc metallo ⁇ -lactamase.
- ⁇ -Lactamases of interest include those disclosed in, e.g., S. G. Waley, ⁇ -lactamase:
- ⁇ -Lactamases of particular interest herein include metallo- ⁇ -lactamases of Escherichia coli (such as New Delhi Metallo-fi-lactamase, NDM), Serratia marcescens (such as IMP), and Klebsiella spp. (such as Verona integron-encoded metallo- ⁇ - lactamase, VIM).). Additional metallo ⁇ -lactamases of interest herein include SPM-, GIM-, SIM-, KHM-, AIM-, DIM-, SMB-, TMB-, and FIM-type enzymes.
- antibiotic refers to a compound or composition which decreases the viability of a microorganism, or which inhibits the growth or proliferation of a
- antibiotics include those which can increase the generation time by at least about 10-fold or more (e.g., at least about 100-fold or even indefinitely, as in total cell death).
- an antibiotic is further intended to include an antimicrobial, bacteriostatic, or bactericidal agent. Examples of antibiotics include penicillins, cephalosporins and carbapenems.
- ⁇ -lactam antibiotic refers to a compound with antibiotic properties that contains a ⁇ -lactam functionality.
- Non-limiting examples of ⁇ -lactam antibiotics include penicillins, cephalosporins, penems, carbapenems, and monobactams.
- the term "about”, when modifying the quantity (e.g., kg, L, or equivalents) of a substance or composition, or the value of a physical property, or the value of a parameter characterizing a process step (e.g., the temperature at which a process step is conducted), or the like refers to variation in the numerical quantity that can occur, for example, through typical measuring, handling and sampling procedures involved in the preparation, characterization and/or use of the substance or composition; through inadvertent error in these procedures; through differences in the manufacture, source, or purity of the ingredients employed to make or use the compositions or carry out the procedures; and the like.
- "about” can mean a variation of ⁇ 0.1, 0.2, 0.3, 0.4, 0.5, 1.0, 2.0, 3.0, 4.0, or 5.0 of the appropriate unit. In certain embodiments, "about” can mean a variation of ⁇ 1%, 2%, 3%, 4%, 5%, 10%, or 20%.
- Another embodiment of the present invention is a compound of Formula I, II- 1, II-2, II-3, III- 1 , III-2, or III-3, or a pharmaceutically acceptable salt thereof, as originally defined or as defined in any of the foregoing embodiments, sub-embodiments, aspects, classes or sub-classes, wherein the compound or its salt is in a substantially pure form.
- substantially pure means suitably at least about 60 wt.%, typically at least about 70 wt.%, preferably at least about 80 wt.%, more preferably at least about 90 wt.% (e.g., from about 90 wt.%) to about 99 wt.%>), even more preferably at least about 95 wt.%> (e.g., from about 95 wt.%) to about 99 wt.%>, or from about 98 wt.%> to 100 wt.%>), and most preferably at least about 99 wt.%) (e.g., 100 wt.%>) of a product containing a compound of Formula I, II- 1, II-2, II-3, III- 1 , III-2, or III-3 or its salt (e.g., the product isolated from a reaction mixture affording the compound or salt) consists of the compound or salt.
- the level of purity of the compounds and salts can be determined using a standard method of analysis such as thin layer chromatography, gel electrophoresis, high performance liquid chromatography, and/or mass spectrometry. If more than one method of analysis is employed and the methods provide experimentally significant differences in the level of purity determined, then the method providing the highest level of purity governs.
- a compound or salt of 100% purity is one which is free of detectable impurities as determined by a standard method of analysis.
- a substantially pure compound can be either a substantially pure mixture of the stereoisomers or a substantially pure individual diastereomer or enantiomer unless expressly depicted otherwise.
- the present invention encompasses all stereoisomeric forms of the compounds of Formula I, II-l, II-2, II-3, III- 1 , III-2, and III-3. Unless a specific stereochemistry is indicated, the present invention is meant to comprehend all such isomeric forms of these compounds.
- Centers of asymmetry that are present in the compounds of Formula I, II-l, II-2, II-3, III- 1 , III-2, and III-3 can all independently of one another have (R) configuration or (S) configuration.
- bonds to the chiral carbon are depicted as straight lines in the structural Formulas of the invention, it is understood that both the (R) and (S) configurations of the chiral carbon, and hence both enantiomers and mixtures thereof, are embraced within the Formula.
- a compound name is recited without a chiral designation for a chiral carbon, it is understood that both the (R) and (S) configurations of the chiral carbon, and hence individual
- the invention includes all possible enantiomers and diastereomers and mixtures of two or more stereoisomers, for example mixtures of enantiomers and/or diastereomers, in all ratios.
- enantiomers are a subject of the invention in enantiomerically pure form, both as levorotatory and as dextrorotatory antipodes, in the form of racemates and in the form of mixtures of the two enantiomers in all ratios.
- the invention includes both the cis form and the trans form as well as mixtures of these forms in all ratios.
- the preparation of individual stereoisomers can be carried out, if desired, by separation of a mixture by customary methods, for example by chromatography or crystallization, by the use of stereochemically uniform starting materials for the synthesis or by stereoselective synthesis.
- a derivatization can be carried out before a separation of stereoisomers.
- the separation of a mixture of stereoisomers can be carried out at an intermediate step during the synthesis of a compound of Formula I, II-l, II-2, II-3, III- 1 , III-2, and III-3 or it can be done on a final racemic product.
- Absolute stereochemistry may be determined by X-ray crystallography of crystalline products or crystalline intermediates which are derivatized, if necessary, with a reagent containing a stereogenic center of known configuration. Where compounds of this invention are capable of tautomerization, all individual tautomers as well as mixtures thereof are included in the scope of this invention.
- the present invention includes all such isomers, as well as salts, solvates (including hydrates) and solvated salts of such racemates, enantiomers, diastereomers and tautomers and mixtures thereof.
- Alkyl means saturated carbon chains which may be linear or branched or combinations thereof, unless the carbon chain is defined otherwise.
- alkyl groups include methyl, ethyl, propyl, isopropyl, butyl, sec- and tert-butyl, pentyl, hexyl, heptyl, octyl, nonyl, and the like.
- Alkylene refers to an alkyl group, as defined above, wherein one of the alkyl group's hydrogen atoms has been replaced with a bond.
- alkylene groups include -CH 2 -, -CH 2 CH 2 -, -CH 2 CH 2 CH 2 -, -CH 2 CH 2 CH 2 CH 2 -, - CH(CH 3 )CH 2 CH 2 -, -CH(CH 3 )- and -CH 2 CH(CH 3 )CH 2 -.
- an alkylene group has from 1 to about 6 carbon atoms.
- an alkylene group has from 1 to about 3 carbon atoms.
- an alkylene group is branched.
- an alkylene group is linear.
- an alkylene group is -CH 2 -.
- the term "Ci-C 6 alkylene" refers to an alkylene group having from 1 to 6 carbon atoms.
- alkenyl means carbon chains which contain at least one carbon-carbon double bond, and which may be linear or branched, or combinations thereof, unless otherwise defined. Examples of alkenyl include vinyl, allyl, isopropenyl, pentenyl, hexenyl, heptenyl, 1-propenyl, 2-butenyl, 2-methyl-2-butenyl, and the like.
- Alkynyl means carbon chains which contain at least one carbon-carbon triple bond, and which may be linear or branched, or combinations thereof, unless otherwise defined. Examples of alkynyl include ethynyl, propargyl, 3 -methyl- 1-pentynyl, 2-heptynyl and the like.
- Aromatic ring system or “aromatic” in reference to a ring means monocyclic, bicyclic or tricyclic aromatic ring or ring system containing 5-14 ring atoms, wherein at least one of the rings is aromatic.
- the term may be used to describe a saturated or
- a 5-7-membered cycloalkyl can be fused through two adjacent ring atoms to a 5-6-membered heteroaryl containing 1, 2, or 3 heteroatom ring atoms selected from N, O, and S.
- a heteromonocyclic ring is fused through two ring atoms to a phenyl or 5-6-membered heteroaryl containing 1, 2, or 3 heteroatoms selected from N, O, and S.
- the N can be in the form of quaternary amine.
- an N ring atom can be in the form of an N-oxide.
- Aryl means a monocyclic, bicyclic or tricyclic carbocyclic aromatic ring or ring system containing 6-14 carbon atoms, wherein at least one of the rings is aromatic.
- aryl examples include phenyl and naphthyl. In one embodiment of the present invention, aryl is phenyl.
- Cycloalkyl means a saturated monocyclic, bicyclic or bridged carbocyclic ring, having a specified number of carbon atoms.
- Examples of cycloalkyl include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, indanyl, and the like.
- cycloalkyl is selected from: cyclopropane, cyclobutane,
- Cycloalkenyl means a nonaromatic monocyclic or bicyclic carbocylic ring containing at least one double bond.
- Examples of cycloalkenyl include cyclopropenyl, cyclobutenyl, cyclopentenyl, cyclohexenyl, cycloheptenyl, cyclooxtenyl and the like.
- heterocycloalkyl refers to a non-aromatic saturated monocyclic or multicyclic ring system comprising 3 to 11 ring atoms, wherein from 1 to 4 of the ring atoms are independently N, H, S (including SO and SO2) and O, and the remainder of the ring atoms are carbon atoms.
- a heterocycloalkyl group can be joined via a ring carbon or ring nitrogen atom (if present). Where the ring or ring system contains one or more N atoms, the N can be in the form of quaternary amine.
- a heterocycloalkyl group is monocyclic and has from about 3 to about 7 ring atoms.
- a heterocycloalkyl group is monocyclic has from about 4 to about 7 ring atoms. In another embodiment, a heterocycloalkyl group is bicyclic and has from about 7 to about 11 ring atoms. When a heterocycloalkyl contains two rings, the rings may be fused or spirocyclic. In still another embodiment, a heterocycloalkyl group is monocyclic and has 5 or 6 ring atoms. In one embodiment, a heterocycloalkyl group is monocyclic. In another embodiment, a heterocycloalkyl group is bicyclic. There are no adjacent oxygen and/or sulfur atoms present in the ring system. Any -NH group in a heterocycloalkyl ring may exist protected such as, for example, as an -N(BOC), -N(Cbz), -N(Tos) group and the like; such protected
- heterocycloalkyl groups are considered part of this invention.
- a heterocycloalkyl group can be optionally substituted by one or more "ring system substituents" which may be the same or different, and are as defined herein below.
- heterocycloalkyl (if present) can be optionally oxidized to the corresponding N-oxide, S- oxide or S,S-dioxide.
- monocyclic heterocycloalkyl rings include oxetanyl, piperidyl, pyrrolidinyl, piperazinyl, morpholinyl, thiomorpholinyl, thiazolidinyl, 1,4-dioxanyl, tetrahydrofuranyl, delta-lactam, delta-lactone, silacyclopentane, silapyrrolidine and the like, and all isomers thereof.
- “Drug resistant” means, in connection with a Gram-negative bacterial strain, a strain which is no longer susceptible to at least one previously effective drug; which has developed the ability to withstand antibiotic attack by at least one previously effective drug.
- “Multidrug resistant” means a strain that is no longer susceptible to two or more previously effective drugs; which has developed the ability to withstand antibiotic attack by two or more previously effective drugs.
- a drug resistant strain may relay that ability to withstand to its progeny. Said resistance may be due to random genetic mutations in the bacterial cell that alters its sensitivity to a single drug or to different drugs.
- Heterocycloalkenyl means a nonaromatic monocyclic, bicyclic or bridged carbocyclic ring or ring system containing at least one double bond and containing at least one heteroatom selected from N, H, S and O.
- Heteroaryl means monocyclic, bicyclic or tricyclic ring or ring system containing 5- 14 carbon atoms and containing at least one ring heteroatom selected from N, NH, S
- heteroaryl ring system where one or more of the rings are saturated and contain one or more N atoms, the N can be in the form of quaternary amine.
- a heteroaryl group has 5 to 10 ring atoms.
- a heteroaryl group is monocyclic and has 5 or 6 ring atoms.
- a heteroaryl group is bicyclic.
- a heteroaryl group can be optionally substituted by one or more ring system substituents which may be the same or different. Any nitrogen atom of a heteroaryl can be optionally oxidized to the corresponding N-oxide.
- heteroaryl also encompasses a heteroaryl group, as defined above, which is fused to a benzene ring.
- heteroaryl include pyrrolyl, isoxazolyl, isothiazolyl, pyrazolyl, pyridyl, oxazolyl, oxadiazolyl, thiadiazolyl, thiazolyl, imidazolyl, triazolyl, tetrazolyl, furanyl, triazinyl, thienyl, pyrimidyl, pyridazinyl, pyrazinyl, benzisoxazolyl, benzoxazolyl, benzothiazolyl, benzimidazolyl, benzopyrazolyl, benzofuranyl, benzothiophenyl (including S-oxide and dioxide),
- heteroaryl is selected from: pyridine, pyrimidine, thiazole, benzimidazole, benzthiazole, benzoxazole, and benzisoxazole.
- heteroaryl is pyridine.
- bicyclic rings include: "Heterocycle” means a monocyclic or bicyclic saturated, partially unsaturated, or unsaturated ring system containing 5-10 atoms and containing at least one ring heteroatom selected from N, S and O. In select embodiments, the ring system contains 1-4 heteroatoms selected from N, S and O. When a heterocycle contains two rings, the rings may be fused, bridged or spirocyclic. Examples of monocyclic heterocycle rings include piperazine, piped dine, and morpholine. Examples of bicyclic heterocycle rings include 1,4- diazabicyclo[2,2,2]octane and 2,6-diazaspiroheptane.
- Halogen includes fluorine, chlorine, bromine and iodine.
- any variable e.g., Rl, Ra, etc.
- its definition on each occurrence is independent of its definition at every other occurrence.
- combinations of substituents and/or variables are permissible only if such combinations result in stable compounds.
- a squiggly line across a bond in a substituent variable represents the point of attachment.
- a “stable” compound is a compound which can be prepared and isolated and whose structure and properties remain or can be caused to remain essentially unchanged for a period of time sufficient to allow use of the compound for the purposes described herein (e.g., therapeutic administration to a subject).
- the compounds of the present invention are limited to stable compounds embraced by Formula I, II- 1, II-2, II-3, III- 1 , III-2, and III-3.
- substituted shall be deemed to include multiple degrees of substitution by a named substitutent. Where multiple substituent moieties are disclosed or claimed, the substituted compound can be independently substituted by one or more of the disclosed or claimed substituent moieties, singly or plurally. By independently substituted, it is meant that the (two or more) substituents can be the same or different. When a group, e.g., Ci-C 8 alkyl, is indicated as being substituted, such substitutions can also occur where such group is part of a larger substituent, e.g., -Ci-C6alkyl-C3-C7Cycloalkyl and -Ci-C 8 alkyl-aryl.
- the atoms may exhibit their natural isotopic abundances, or one or more of the atoms may be artificially enriched in a particular isotope having the same atomic number, but an atomic mass or mass number different from the atomic mass or mass number predominantly found in nature.
- the present invention is meant to include all suitable isotopic variations of the compounds of Formula I, II- 1, II-2, II-3, III- 1 , III-2, and III-3.
- different isotopic forms of hydrogen (H) include protium (1H) and deuterium ( 2 H or D).
- Protium is the predominant hydrogen isotope found in nature. Enriching for deuterium may afford certain therapeutic advantages, such as increasing in vivo half-life or reducing dosage requirements, or may provide a compound useful as a standard for characterization of biological samples.
- Isotopically-enriched compounds within Formula I, II- 1, II-2, II-3, III- 1 , III-2, and III-3 can be prepared without undue experimentation by conventional techniques well known to those skilled in the art or by processes analogous to those described in the Schemes and Examples herein using appropriate isotopically-enriched reagents and/or intermediates.
- any of the various cyclic rings and ring systems described herein may be attached to the rest of the compound at any ring atom (i.e., any carbon atom or any heteroatom) provided that a stable compound results.
- a heteroaromatic ring described as containing from “ 1 to 4 heteroatoms” means the ring can contain 1, 2, 3 or 4 heteroatoms. It is also to be understood that any range cited herein includes within its scope all of the sub-ranges within that range.
- a heterocyclic ring described as containing from “ 1 to 4 heteroatoms” is intended to include as aspects thereof, heterocyclic rings containing 2 to 4 heteroatoms, 3 or 4 heteroatoms, 1 to 3 heteroatoms, 2 or 3 heteroatoms, 1 or 2 heteroatoms, 1 heteroatom, 2 heteroatoms, 3 heteroatoms, and 4 heteroatoms.
- Ci-C 6 when used with a chain means that the chain can contain 1, 2, 3, 4, 5 or 6 carbon atoms. It also includes all ranges contained therein including C1-C5, C 1 -C 4 , C1-C3, C 1 -C 2 , C2-C6, C3-C6, C4-C6, C 5 - C 6 , and all other possible combinations.
- the compounds of the present invention have at least one asymmetric center and can have one or more additional centers as a result of the presence of certain substituents and/or substituent patterns. Accordingly, compounds of the invention can occur as mixtures of stereoisomers, or as individual diastereomers, or enantiomers. All isomeric forms of these compounds, whether individually or in mixtures, are within the scope of the present invention.
- compound refers to the free compound and, to the extent they are stable, any hydrate or solvate thereof.
- a hydrate is the compound complexed with water
- a solvate is the compound complexed with an organic solvent.
- the compounds of the present invention can be employed in the form of pharmaceutically acceptable salts. It will be understood that, as used herein, the compounds of the instant invention can also include the pharmaceutically acceptable salts, and also salts that are not pharmaceutically acceptable when they are used as precursors to the free compounds or their pharmaceutically acceptable salts or in other synthetic manipulations.
- pharmaceutically acceptable salt refers to a salt which possesses the effectiveness of the parent compound and which is not biologically or otherwise undesirable (e.g., is neither toxic nor otherwise deleterious to the recipient thereof).
- pharmaceutically acceptable salt refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids including inorganic or organic bases and inorganic or organic acids. Salts of basic compounds encompassed within the term “pharmaceutically acceptable salt” refer to non-toxic salts of the compounds of this invention which are generally prepared by reacting the free base with a suitable organic or inorganic acid.
- Representative salts of basic compounds of the present invention include, but are not limited to, the following: acetate, ascorbate, adipate, alginate, aspirate, benzenesulfonate, benzoate, bicarbonate, bisulfate, bitartrate, borate, bromide, butyrate, camphorate, camphorsulfonate, camsylate, carbonate, chloride, clavulanate, citrate, cyclopentane propionate, diethyl acetic, digluconate, dihydrochloride, dodecylsulfanate, edetate, edisylate, estolate, esylate, ethanesulfonate, formic, fumarate, gluceptate, glucoheptanoate, gluconate, glutamate, glycerophosphate, glycollylarsanilate, hemisulfate, heptanoate, hexanoate, hex
- suitable pharmaceutically acceptable salts thereof include, but are not limited to, salts derived from inorganic bases including aluminum, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, manganic, mangamous, potassium, sodium, zinc, and the like. Particularly preferred are the ammonium, calcium, magnesium, potassium, and sodium salts.
- Salts derived from pharmaceutically acceptable organic non-toxic bases include salts of primary, secondary, and tertiary amines, cyclic amines, dicyclohexyl amines and basic ion-exchange resins, such as arginine, betaine, caffeine, choline, ⁇ , ⁇ -dibenzylethylenediamine, diethylamine, 2-diethylaminoethanol, 2- dimethylaminoethanol, ethanolamine, ethylamine, ethylenediamine, N-ethylmorpholine, N- ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine, and the like.
- the basic nitrogen-containing groups may be quaternized with such agents as lower alkyl halides, such as methyl, ethyl, propyl, and butyl chloride, bromides and iodides; dialkyl sulfates like dimethyl, diethyl, dibutyl; and diamyl sulfates, long chain halides such as decyl, lauryl, myristyl and stearyl chlorides, bromides and iodides, aralkyl halides like benzyl and phenethyl bromides and others.
- lower alkyl halides such as methyl, ethyl, propyl, and butyl chloride, bromides and iodides
- dialkyl sulfates like dimethyl, diethyl, dibutyl
- diamyl sulfates long chain halides
- salts can be obtained by known methods, for example, by mixing a compound of the present invention with an equivalent amount and a solution containing a desired acid, base, or the like, and then collecting the desired salt by filtering the salt or distilling off the solvent.
- the compounds of the present invention and salts thereof may form solvates with a solvent such as water, ethanol, or glycerol.
- the compounds of the present invention may form an acid addition salt and a salt with a base at the same time according to the type of substituent of the side chain.
- the compound of the invention can also be employed in the form of a prodrug. Any prodrug precursor known in the art can be used to form a prodrug of the invention.
- the hydrogen in -COOH in formula I can be replaced with any the following groups: Ci-6alkyl, C3-6cycloalkyl, -Ci-6alkylene-C3-6cycloalkyl, C3- 7cycloheteroalkyl,-C 1 -6alkylene-C3 -7cycloheteroalkyl, aryl, -C 1.1 oalkylene-aryl, heteroaryl, and -Ci-ioalkylene-heteroaryl.
- the Ci- 6alkyl, C3-6cycloalkyl, or C3-7cycloheteroalkyl can be substituted.
- each aryl and heteroaryl can be substituted.
- the present invention includes pharmaceutical compositions comprising a compound of Formula I of the present invention, optionally one other active components (e.g., a ⁇ -lactamase inhibitor), and a pharmaceutically acceptable carrier.
- active components e.g., a ⁇ -lactamase inhibitor
- pharmaceutically acceptable carrier is meant that the ingredients of the pharmaceutical composition must be compatible with each other, do not interfere with the effectiveness of the active ingredient(s), and are not deleterious (e.g., toxic) to the recipient thereof.
- compositions according to the invention may, in addition to the inhibitor, contain diluents, fillers, salts, buffers, stabilizers, solubilizers, and other materials well known in the art.
- the present invention includes a method for treating a bacterial infection which comprises administering to a subject in need of such treatment a therapeutically effective amount of a compound of Formula I, II- 1, II-2, II-3, III- 1 , III-2, and III-3, or a pharmaceutically acceptable salt thereof, optionally in combination with a ⁇ -lactamase inhibitor.
- subject or, alternatively, “patient” as used herein refers to an animal, preferably a mammal, most preferably a human, who has been the object of treatment, observation or experiment.
- administration and variants thereof (e.g., “administering” a compound) in reference to a compound of Formula I, II- 1, II-2, II-3, III- 1 , III-2, and III-3 mean providing the compound, or a pharmaceutically acceptable salt thereof, to the individual in need of treatment.
- a compound or a salt thereof is provided in combination with one or more other active agents (e.g., a ⁇ -lactamase inhibitor),
- administration and its variants are each understood to include provision of the compound or its salt and the other agents at the same time or at different times.
- agents of a combination are administered at the same time, they can be administered together in a single composition or they can be administered separately.
- a "combination" of active agents can be a single composition containing all of the active agents or multiple compositions each containing one or more of the active agents.
- a combination can be either a single composition comprising both agents or two separate compositions each comprising one of the agents; in the case of three active agents a combination can be either a single composition comprising all three agents, three separate compositions each comprising one of the agents, or two compositions one of which comprises two of the agents and the other comprises the third agent; and so forth.
- compositions and combinations of the present invention are suitably administered in effective amounts.
- effective amount means the amount of active compound sufficient to inhibit bacterial growth and thereby elicit the response being sought (i.e., an "inhibition effective amount" in a cell, tissue, system, animal or human.
- the effective amount is a "therapeutically effective amount” for the alleviation of the symptoms of the disease or condition being treated (e.g., the healing of conditions associated with bacterial infection, and/or bacterial drug resistance).
- the effective amount is a "prophy tactically effective amount” for prophylaxis of the symptoms of the disease or condition being prevented.
- compositions of the present invention are suitably parenteral, oral, sublingual, transdermal, topical, intranasal, intratracheal, intraocular, or intrarectal, wherein the composition is suitably formulated for administration by the selected route using formulation methods well known in the art, including, for example, the methods for preparing and administering formulations described in chapters 39, 41, 42, 44 and 45 in Remington - The Science and Practice of Pharmacy, 21 st edition, 2006.
- compounds of the invention are administered intravenously in a hospital setting.
- administration is oral in the form of a tablet or capsule or the like.
- a therapeutic composition is for example, suitably administered at a sufficient dosage to attain a blood level of inhibitor of at least about 1 microgram/mL, and in additional embodiment at least about 10 micrograms/mL, and at least about 25
- micrograms/mL For localized administration, much lower concentrations than this may be effective, and much higher concentrations may be tolerated.
- Intravenous administration of a compound of the invention can be conducted by reconstituting a powdered form of the compound with an acceptable solvent.
- suitable solvents include, for example, saline solutions (e.g., 0.9% Sodium Chloride Injection) and sterile water (e.g., Sterile Water for Injection, Bacteriostatic Water for Injection with methylparaben and propylparaben, or Bacteriostatic Water for Injection with 0.9% benzyl alcohol).
- the powdered form of the compound can be obtained by gamma-irradiation of the compound or by lyophilization of a solution of the compound, after which the powder can be stored (e.g., in a sealed vial) at or below room temperature until it is reconstituted.
- the concentration of the compound in the reconstituted IV solution can be, for example, in a range of from about 0.1 mg/mL to about 20 mg/mL.
- the present invention also includes a method for inhibiting bacterial growth which comprises administering to a bacterial cell culture, or to a bacterially infected cell culture, tissue, or organism, an inhibition effective amount of a compound of Formula I, II-l, II-2, II- 3, III- 1 , III-2, and III-3.
- Additional embodiments of the invention include the bacterial growth inhibiting method just described, wherein the compound of the present invention employed therein is a compound of one of the embodiments, sub-embodiments or classes described above. The compound may optionally be used in the form of a pharmaceutically acceptable salt in these embodiments.
- the method can involve administration of a compound of Formula I, II- 1, II-2, II-3, III- 1 , III-2, and III-3 to an experimental cell culture in vitro to prevent the growth of ⁇ -lactam resistant bacteria.
- the method can alternatively involve administration of a compound of Formula I to an animal, including a human, to prevent the growth of ⁇ -lactam resistant bacteria in vivo.
- the compound of Formula I, II- 1, II-2, II-3, III- 1 , III-2, and III-3 is typically co-administered with a ⁇ - lactamase inhibitor.
- the methods of the presently disclosed subject matter are useful for treating these conditions in that they inhibit the onset, growth, or spread of the condition, cause regression of the condition, cure the condition, or otherwise improve the general well-being of a subject afflicted with, or at risk of, contracting the condition.
- the terms “treat”, “treating”, and grammatical variations thereof, as well as the phrase “method of treating”, are meant to encompass any desired therapeutic intervention, including but not limited to a method for treating an existing infection in a subject, and a method for the prophylaxis (i.e., preventing) of infection, such as in a subject that has been exposed to a microbe as disclosed herein or that has an expectation of being exposed to a microbe as disclosed herein.
- Compounds of the invention can be employed for the treatment, prophylaxis or inhibition of bacterial growth or infections due to bacteria that are resistant to ⁇ -lactam antibiotics. More particularly, the bacteria can be metallo ⁇ -lactamase positive strains that are highly resistant to ⁇ -lactam antibiotics.
- the terms "slightly resistant” and “highly resistant” are well-understood by those of ordinary skill in the art (see, e.g., Payne et al., Antimicrobial Agents and Chemotherapy 38:767-772 (1994); Hanaki et al., Antimicrobial Agents and Chemotherapy 30: 11.20-11.26 (1995)).
- bacterial strains which are highly resistant to imipenem are those against which the MIC of imipenem is >16 ⁇ g/mL, and bacterial strains which are slightly resistant to imipenem are those against which the MIC of imipenem is >4 ⁇ g/mL.
- ⁇ -lactamase inhibitors can be used in combination with a ⁇ -lactamase inhibitor for the treatment of infections caused by ⁇ -lactamase producing strains, in addition to those infections which are subsumed within the antibacterial spectrum of the antibiotic agent.
- ⁇ -lactamase producing bacteria are Pseudomonas aeruginosa, Pseudomonas putida, Enterobacter cloacae, Klebsiella pneumoniae, Klebsiella oxytoca, Escherichia coli, Serratia marcescens, Enterobacter aerogenes, Enterobacter asburiae, Citrobacter freundii, Proteus mirabilis, Morganella morganii, Providencia rettgeri, Stenotrophomonas maltophilia and Acinetobacter baumannii.
- a compound of Formula I, II- 1, II-2, II-3, III- 1 , III- 2, and III-3 in admixture or conjunction with a ⁇ -lactamase inhibitor, or a prodrug thereof. It is advantageous to use a compound of Formula I in combination with a class A and C ⁇ - lactamase inhibitor because of the class B ⁇ -lactamase resistant properties of the compounds. It is also advantageous to use a compound of Formula I in combination with one or more Class A, C, or D ⁇ -lactamase inhibitors to further limit ⁇ -lactam susceptability. As already noted, the compound of Formula I and the ⁇ -lactamase inhibitor can be administered separately (at the same time or as different times) or in the form of a single composition containing both active ingredients.
- Relebactam, tazobactam, clavulanic acid, sulbactam, avibactam and other ⁇ -lactamase and metallo ⁇ -lactamase inhibitors suitable for use in the present invention include those known to show inhibitory activity to ⁇ -lactamases.
- DIAD diisopropyl azodicarboxylate
- DIPEA di-isopropylethylamine
- DMA dimethylacetamide
- DMAP 4-dimethylaminopyridine or N,N-dimethylamino- pyridine
- DMF ⁇ , ⁇ -dimethylformamide
- DMSO dimethyl sulfoxide
- EDC l-ethyl-3- (3-dimethylaminopropyl) carbodiimide; eq. or equiv.
- IPA isopropyl alcohol
- iPrMgCl isopropyl magnesium chloride
- IP Ac isopropyl acetate
- LDA lithium diisopropylamide
- m-CPBA m-chloroperoxybenzoic acid
- MOPS 3-(N-morpholino)propanesulfonic acid
- MPLC medium pressure liquid chromatography
- MTBE methyl tert-butyl ether
- BS N-bromosuccinimide
- NCS N-chloros
- TBS tert-butyldimethylsilyl
- TBS-C1 tert- butyldimethylsilyl chloride
- TBDMS-C1 tert-butyldimethylsilyl chloride
- t-BuOH tert- butanol
- TBSO tert-butyldimethylsilyl
- TEA triethylamine
- TEMPO is (2,2,6,6- tetramethylpiperidin-l-yl)oyl
- TFA trifluoroacetic acid
- THF tetrahydrofuran
- TLC thin layer chromatography
- TMS trimethylsilyl
- TMS-C1 trimethylsilyl chloride
- TMS-I trimethylsilyl
- the ⁇ -lactam intermediate can be either purchased from commercial sources or synthesized following the scheme below, which shows the synthesis of ⁇ -lactam analogs wherein Rx and Rz come together to form a 4-membered spirocyclic ring.
- the synthetic scheme has been discussed in detail in the literature. (See EP 0229012). This amine can be converted to the final monobactam compounds with a similar procedure demonstrated in the following Examples.
- Step A ethyl 2-(2-((tert-butoxycarbonyl)amino)thiazol-4-yl)-2-oxoacetate
- acetonitrile 250 ml
- BOC-anhydride 23.2 ml, 100 mmol
- ⁇ , ⁇ , ⁇ ', ⁇ '-tetramethyl- ethylenediamine 9.80 ml, 64.9 mmol
- Step B 2-(2-((tert-butoxycarbonyl)amino)thiazol-4-yl)-2-oxoacetic acid
- Ethyl 2-(2-((tert- butoxycarbonyl)amino)thiazol-4-yl)-2-oxoacetate (10.2g, 34.1 mmol) was dissolved in THF (140 ml)/MeOH (50 ml) and sodium hydroxide was added (68.3 ml, 68.3 mmol, 1M). The solution was stirred at room temperature for 4 hours. The reaction mixture was poured into water (1L) and extracted with EtOAc (3x200ml).
- Step A tert-butyl (3-methyl-L2,4-thiadiazol-5-yl)carbamate
- 3- methyl-l,2,4-thiadiazol-5-amine 167 g, 1.45 mol, 1.00 equiv
- 4-dimethylamino-pyridine 17.7 g, 144.88 mmol, 0.10 equiv
- di-tert-butyl dicarbonate 348 g, 1.59 mol, 1.10 equiv
- butan-l-ol (1670 mL).
- the resulting solution was stirred for 1 hour at 40°C.
- the resulting mixture was concentrated under vacuum.
- the residue was washed with Hexane. This resulted in the title compound.
- Step B 2-(5-((tert-butoxycarbonyl)amino)-L2,4-thiadiazol-3-yl)acetic acid
- the pH value of the solution was adjusted to 2 with HC1 (2M mol/L).
- the resulting solution was extracted with 2.5 L of ethyl acetate and the organic layers were combined.
- the resulting mixture was washed with 2000 mL of brine.
- the mixture was dried over anhydrous sodium sulfate and concentrated under vacuum, which resulted in the title compound.
- Step C 2-(5-((fert-Butoxycarbonyl)amino)-L2,4-thiadiazol-3-yl)-2-oxoacetic acid
- a solution of 2-(5-[[(tert-butoxy)carbonyl]amino]-l,2,4-thiadiazol-3- yl)acetic acid (76 g, 293.12 mmol, 1.00 equiv) in dioxane (1520 mL), Se0 2 (65.14 g, 587 mmol, 2.00 equiv).
- Step A tert-butyl 3-((6-bromopyridin-3-yl)oxy)-2-hydroxypropanoate Potassium carbonate
- Step B tert-butyl 3-((6-bromopyridin-3-yl)oxy)-2-((tert-butyldimethyl-silyl)oxy)propanoate
- tert-butyl 3-((6-bromopyridin-3-yl)oxy)-2-hydroxypropanoate 6700 mg, 21.1 mmol
- imidazole 3440 mg, 50.5 mmol
- TBDMS-C1 3800 mg, 25.3 mmol
- DMAP 257 mg, 2.11 mmol
- Step A tert-butyl 2-methyloxirane-2-carboxylate
- a solution of tert-butyl methacrylate (44 g, 309 mmol) in CH 2 CI 2 (1000 mL) was added portion wise w-CPBA (120 g, 557 mmol).
- the resulting solution was stirred at 30 °C for 20 hours, resulting large amount of precipitate.
- the slurry was filtered, to the filtrate was added in small portions of saturated sodium thiosulfate solution (about 100 mL, added in small portions until no more heat is generated).
- Step B tert-butyl 3-(4-bromophenoxy)-2-hydroxy-2-methylpropanoate
- a mixture of 4- bromophenol (15 g, 87 mmol), K 2 C0 3 (17.97 g, 130 mmol) and tert-butyl 2-methyloxirane- 2-carboxylate (20.6 g, 130 mmol) in DMF (200 mL) was heated and stirred at 80 °C for 16 hours. TLC indicated complete conversion.
- the mixture was diluted with water (100 mL) and extracted with CH 2 C1 2 (3x 100 mL), washed with brine (2x 40 mL). The organic layer was dried over Na 2 S0 4 , filtered and concentrated in vacuum.
- Step C Q-(mesitylsulfonyl)hydroxylamine
- (E)-ethyl N- (mesitylsulfonyl)oxyacetimidate (20 g, 70.1 mmol) in 1,4-dioxane (20 mL) was cooled to 0 °C.
- Perchloric acid (8.45 g, 84 mmol) was added dropwise (slowly). After stirring for 15 minutes, the mixture solidified. The contents of the reaction were transferred to 200 mL of ice water, and the flask rinsed with water (100 mL) and tert-butyl methyl ether (100 mL).
- Step D tert-butyl 2-(aminooxy)-3-(4-bromophenoxy)-2-methylpropanoate
- Tert-Butyl 3-(4- bromophenoxy)-2-hydroxy-2-methylpropanoate (9 g, 27.2 mmol) was dissolved in anhydrous THF (120 mL) under N 2 , cooled to 0 °C. NaH (1.96 g, 48.9 mmol, (60 % in paraffin) was added in 3 portions. The mixture was stirred at 0 °C for 30 min. Then O- (mesitylsulfonyl)hydroxylamine (6.43 g, 29.9 mmol) was added to this mixture.
- Step A l-bromo-2-fluoro-4-((2-methylallyl)oxy)benzene 3-Bromo-2-methylprop-l-ene (5.3 mL, 52 mmol) was added to a stirred suspension of 4-bromo-3-fluorophenol (10 g, 52 mmol) and K 2 C0 3 (8.7 g, 63 mmol) in DMF (105 mL). The reaction mixture was stirred at rt for 1.5 h, then partitioned between ether and water. The layers were separated, and the organic layer was washed with water and brine, dried (MgS0 4 ), filtered and concentrated in vacuo to afford the title compound, which was used without further purification.
- Step B (R)-3-(4-bromo-3-fluorophenoxy)-2-methylpropane-L2-diol.
- AD-mix a (12 g, 0.035 mmol of osmium) was added to a suspension of l-bromo-2-fluoro-4-((2-methyl- allyl)oxy)benzene (2.1 g, 8.5 mmol) in tert-butanol (41 mL) and water (41 mL).
- the reaction was stirred at rt for 4 h, then partitioned between EtOAc and water. The layers were separated, and the aqueous layer was extracted with EtOAc.
- Step C ( ) 5 -3-(4-bromo-3-fluorophenoxy)-2-hvdroxy-2-methylpropanoic acid.
- a mixture of NaH 2 P0 4 (2.43 g, 20.2 mmol) in water (41 mL) was added to a stirred solution of (R)-3-(4- bromo-3-fluorophenoxy)-2-methylpropane-l,2-diol (2.26 g, 8.10 mmol) in THF (41 mL).
- Step D fert-butyl ( ) 5 r )-3-(4-bromo-3-fluorophenoxy)-2-hydroxy-2-methylpropanoate.
- tert- Butyl-N,N'-diisopropylcarbammidate (1.87 mL, 8.0 mmol) was added to a stirred solution of ( ⁇ S)-3-(4-bromo-3-fluorophenoxy)-2-hydroxy-2-methylpropanoic acid (1.17 g, 4.0 mmol) in THF (20 mL). The resulting mixture was heated to 60 °C. After 3 h, the reaction was cooled to rt, filtered through a pad of CeliteTM and concentrated in vacuo.
- Step E fert-butyl ( ) 5 -2-(aminooxy)-3-(4-bromo-3-fluorophenoxy)-2-methylpropanoate .
- the title compound was prepared following a procedure similar to the procedure of
- Step A (R)-3-(4-bromophenoxy)-2-hydroxypropanoic acid TFA (850 ⁇ , 11.0 mmol) was added to a stirred solution of Intermediate 3 (350 mg, 1.10 mmol) in DCM (5.5 mL). The reaction mixture was stirred at rt. After 1.5 h, the reaction was concentrated in vacuo, and the resulting solid was carried on without further purification. LC-MS [M + Na]: m/z 283.2.
- Step B Methyl (R)-3-(4-bromophenoxy)-2-hydroxypropanoate Trimethylsilyldiazomethane (2.2 mL of a 2.0 M solution in hexanes, 4.41 mmol) was added to a (1 : 1) mixture of
- Step A fert-butyl 3-fluoro-3-((tosyloxy)methyl)azetidine-l-carboxylate ⁇ -Toluenesulfonyl chloride (1.02 g, 5.32 mmol) was added to a stirred solution of tert-butyl 3-fluoro-3-
- Step B fert-butyl 3-((4-bromo-lH-pyrazol-l-yl)methyl)-3-fluoroazetidine-l-carboxylate
- Sodium hydride 116 mg of a 60% dispersion in mineral oil, 2.89 mmol
- tert-butyl 3-fluoro-3-((tosyloxy)methyl)azetidine-l- carboxylate (1.04 g, 2.89 mmol)
- 4-bromo-lH-pyrazole (425 mg, 2.89 mmol) in DMF (13 mL) at 0 °C.
- the reaction mixture was allowed to warm to rt and stirred overnight.
- Step A N 1 -(5-bromopyridin-2-yl)ethane-L2-diamine
- Ethylenediamine (25.4 mL, 380 mmol) was added to a stirred solution of 2,5-dibromopyridine (5.00 g, 21.1 mmol).
- the reaction was heated to 100 °C for 16 h, then cooled to rt, and concentrated in vacuo.
- the resulting residue was diluted in DCM.
- the organic layers were washed with water, dried (MgS0 4 ), filtered and concentrated in vacuo to give the title compound, which was used in the next step without purification.
- Step B fert-butyl (2-((5-bromopyridin-2-yl)amino)ethyl)carbamate
- Di-tert-butyl di- carbonate (5.53 g, 25.3 mmol) was added to a stirred suspension of ⁇ -(S-bromopyridin ⁇ - yl)ethane-l,2-diamine (4.56 g, 21.1 mmol) and sodium carbonate (2.35 g, 22.2 mmol) in a (1 : 1) dioxane:water solution (64 mL total volume).
- the reaction mixture was stirred at rt overnight, then the reaction mixture was extracted with EtOAc.
- Step A tert-butyl (3-((6-chloropyridazin-3-yl)amino)propyl)carbamate
- DMSO 1,2,4-dichloropyridazine
- tert-butyl 3-aminopropyl)carbamate
- triethylamine 1.50 mL, 10.8 mmol
- Step A 3-((tert-butoxycarbonyl)amino)-2-fluoropropyl 4-methyl benzenesulfonate
- the title compound was prepared using a procedure similar to the procedure of Intermediate 16, Step A substituting tert-butyl (2-fluoro-3-hydroxypropyl)carbamate for tert-butyl 3-fluoro-3- (hydroxymethyl)azetidine-l-carboxylate.
- Step B tert-butyl (3-(4-bromo-lH-pyrazol-l-yl)-2-fluoropropy0carbamate
- the title compound was prepared according to the procedure of intermediate 16 Step B.
- Step A 3-(l-trityl-lH-imidazol-4-yl)propan-l-ol Lithium aluminum hydride (2.5 mL of a 1 M THF solution, 2.5 mmol) was added to a stirred solution of methyl 3-(l-trityl-lH- imidazol-4-yl)propanoate (500 mg, 1.26 mmol) in THF (4.0 mL) at 0 °C. The resulting mixture was warmed to 70 °C and allowed to stir at 70 °C for 2 h. Then the reaction was cooled to rt and quenched by the addition of water, followed by 2 N NaOH.
- Step B 2-(3-(l-trityl-lH-imidazol-4-yl)propyl)isoindoline-L3-dione Phthalimide (278 mg, 1.89 mmol) was added to a stirred solution of 3-(l-trityl-lH-imidazol-4-yl)propan-l-ol (465 mg, 1.26 mmol) and triphenylphosphine (496 mg, 1.89 mmol) in THF (12 mL). The reaction was cooled to 0 °C, then DIAL ) (0.37 mL, 1.89 mmol) was slowly added dropwise, and the resulting mixture was allowed to warm to rt. After stirring overnight, the reaction mixture was filtered, and the solids were washed with THF to give the title compound. LC-MS [M + H]: m/z 498.5.
- Step C 3-(l-trityl-lH-imidazol-4-yl)propan-l-amine Methylamine (11 mL of a 33% w/v EtOH solution, 88 mmol) was added to a stirred solution of 2-(3-(l-trityl-lH-imidazol-4- yl)propyl)isoindoline-l,3-dione (1.92 g, 3.86 mmol) in EtOH (40 mL) and water (3.5 mL).
- Step D fert-butyl (3-(l-trityl-lH-imidazol-4-yl)propyl)carbamate
- Di-tertbutyl di carbonate (1.69 g, 7.72 mmol) was added to a stirred solution of 3-(l-trityl-lH-imidazol-4-yl)propan-l- amine (1.42 g, 3.86 mmol) and triethylamine (1.08 mL, 7.72 mmol) in DCM (30 mL). The reaction mixture was stirred at rt.
- Step E tert-butyl (3-(lH-imidazol-4-yl)propyl)carbamate Anisole (377 mg, 3.49 mmol) was added to a stirred solution of tert-butyl (3-(l-trityl-lH-imidazol-4-yl)propyl)carbamate (1.63 g, 3.49 mmol) in acetic acid (5.0 mL), and the reaction mixture was heated to 60 °C. After 24 h, the reaction mixture was concentrated in vacuo and azeotroped successively with toluene and acetonitrile. The resulting residue was suspended in EtOAc and DCM, and the mixture was adsorbed onto CeliteTM.
- Step B tert-butyl (S)-(2,3-bis((tert-butyldimethylsilyl)oxy)propyl)carbamate Into a 500mL
- Step C tert-butyl (S)-(2-((tert-butyldimethylsilyl)oxy)-3-hydroxypropyl)carbamate Into a 5L
- Step D (S)-3-((tert-butoxycarbonyl)amino)-2-((tert-butyldimethylsilyl)oxy)propyl methanesulfonate
- tert-butyl N-[(2S)-2-[(tert-butyldimethylsilyl)oxy]-3- hydroxypropyl]carbamate 40 g, 131 mmol, 1 equiv
- dichloromethane 400 mL
- TEA 36.5 mL, 2 eq
- methanesulfonyl chloride (14.5 mL, 1.50 eq) was stirred for 2 h at room temperature.
- Step E tert-butyl (S)-(3-bromo-2-((tert-butyldimethylsilyl)oxy)propyl)carbamate
- reaction mixture was diluted with H 2 0 (500 mL), and the resulting solution was extracted with 500 mL of ethyl acetate. The combined organic layers were dried over anhydrous sodium sulfate and concentrated under vacuum. The resulting residue was purified on a silica gel column with ethyl
- Step F tert-butyl (S)-(3-(4-bromo-lH-pyrazol-l-yl)-2-((tertbu dimetfaylsilyl)oxy)- propyPcarbamate
- reaction mixture was diluted with 2 L of EA.
- the resulting mixture was washed with H 2 0 (2x4L).
- the mixture was dried over anhydrous magnesium sulfate and concentrated under vacuum.
- the resulting residue was applied onto a silica gel column with ethyl acetate/petroleum ether (1 : 10) to give the title compound.
- Step A fert-Butyl (3-(4-bromo-7H-pyrazol-l-yl)propyl)carbamate
- 4-bromo-7H- pyrazole (3 g, 20.4 mmol), tert-butyl (3-bromopropyl)carbamate (4.86 g, 20.4 mmol) and cesium carbonate (9.98 g, 30.6 mmol) in DMF (20 ml) was stirred at room temperature overnight. The mixture was diluted with water (30 ml) and extracted with EtOAc (3x10 ml). The extracts were combined, dried over MgS0 4 and concentrated to dryness.
- Step B fert-Butyl (R)-3-(4-(l-(3-((fert-butoxycarbonyl)amino)propyl)-7H-pyrazol-4- yl)phenoxy)-2-hydroxypropanoate
- (S)-tert-butyl 2-hydroxy-3-(4-(4,4,5,5- tetramethyl-l,3,2-dioxaborolan-2-yl)phenoxy)propanoate 5 g, 13.7 mmol
- tert-butyl (3-(4- bromo-7H-pyrazol-l-yl)propyl)carbamate (4.18 g, 13.7 mmol)
- l, l'-bis(di-tert- butylphosphino)ferrocene palladium dichloride 0.95 g, 1.37 mmol
- THF 150 ml
- Step C fert-Butyl (S)-3-(4-(l-(3-((fert-butoxycarbonyl)amino)propyl)-7H-pyrazol-4- yl)phenoxy)-2-(( 1 , 3 -dioxoi soindolin-2-yl)oxy)propanoate
- (S)-tert-butyl 3- (4-(l-(3-((tert-butoxycarbonyl)amino)propyl)-7H-pyrazol-4-yl)phenoxy)-2- hydroxypropanoate (2.2g, 4.77 mmol), 2-hydroxyisoindoline-l,3-dione (0.933 g, 5.72 mmol), and triphenylphosphine ( 1.50 g, 5.72 mmol) in DCM ( 100ml) was added DIAL ) (1.1 1 ml, 5.72 mmol).
- Step D (S)-4-(4-(3-(fert-Butoxy)-2-((L3-dioxoisoindolin-2-yl)oxy)-3-oxopropoxy)phenyl)- l-(3-((fert-butoxycarbonyl)amino)propyl)-2-methyl-7H-pyrazol-2-ium
- Step E (S)-4-(4-(2-( Aminooxy)-3 -(fert-butoxy)- 3 -oxopropoxy)phenyl)- 1 -(3 -((fert- butoxycarbonyl)amino)propyl)-2-methyl-7H-pyrazol-2-ium
- (S)-4-(4-(3- (ter/-butoxy)-2-(( 1 ,3 -dioxoi soindolin-2-yl)oxy)-3 -oxopropoxy)phenyl)- 1 -(3 -((tert- butoxycarbonyl)amino)propyl)-2-methyl-7H-pyrazol-2-ium iodide (2g, 2.67 mmol) in acetonitrile (20 ml) was added hydrazine (0.092 ml, 2.94 mmol).
- Step F (S.Z)-4-(4-(3-(fert-Butoxy)-2-((((2-((fert-butoxycarbonvnamino)- thiazol- 4- yl)(carboxy)methylene)amino)oxy)-3-oxopropoxy)phenyl)-l-(3-((fert- butoxycarbonyl)amino)propyl)-2-methyl-7H-pyrazol-2-ium
- Step G (S)-3-((Z)-2-((((S)-l-(fert-Butoxy)-3-(4-(l-(3-((fer/-butoxy-carbonvnamino)propyn- 2-methyl-77J-pyrazol-2-ium-4-yl)phenoxy)-l-oxopropan-2-yl)oxy)imino)-2-(2-((fert- butoxycarbonyl)amino)thiazol-4-yl)acetamido)-2,2-dimethyl-4-oxoazetidin-l-yl sulfate
- Step H Mono(4-(4-((S)-2-((((Z)- 1 -(2-aminothiazol-4-yl)-2-(((S)-2.2-dimethyl-4-oxo- 1 - (sulfooxy)azetidin-3-yl)amino)-2-oxoethylidene)amino)oxy)-2-carboxyethoxy)phenyl)-l-(3- ammoniopropyl)-2-methyl-lH-pyrazol-2-ium) mono(2,2,2-trifluoroacetate)
- Step A fert-Butyl (3-(4-bromo-7H-imidazol-l-yl)propyl)carbamate.
- Step B fert-Butyl (R)-3-(4-(l-(3-((fert-butoxycarbonyl)amino)propyl)-7H-imidazol-4- yl)phenoxy)-2-hydroxypropanoate
- the procedure was the same as step B in Example 1 with the reagent of tert-butyl (3-(4-bromo-7H-imidazol-l-yl)propyl)carbamate, (R)-tert-butyl 2- hydroxy-3-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenoxy)propanoate (300 mg, 0.824 mmol), tert-butyl (3-(4-bromo-7H-imidazol-l-yl)propyl)carbamate (276 mg, 0.906 mmol)( intermediate 4), l,l'-bis(di-tert-butylphosphino
- Step C fert-Butyl (S)-3-(4-(l-(3-((fert-butoxycarbonyl)amino)propyl)-7H-imidazol-4- yl)phenoxy)-2-(( 1 , 3 -dioxoi soindolin-2-yl)oxy)propanoate
- the procedure was the same as Step C in Example 1 with the reagent of tert-butyl (R)-3-(4-(l-(3-((tert-butoxycarbonyl)- amino)propyl)-7H-imidazol-4-yl)phenoxy)-2-hydroxypropanoate (0.25g, 0.542 mmol), 2- hydroxyisoindoline-l,3-dione (0.097 g, 0.596 mmol), triphenylphosphine (0.170 g, 0.650 mmol), DIAD (0.126 ml, 0.650 mmol) and THF (2 ml).
- Step D (S)-4-(4-(3-(fert-Butoxy)-2-((L3-dioxoisoindolin-2-yl)oxy)-3-oxopropoxy)phenyl)- l-(3-((fert-butoxycarbonyl)amino)propyl)-3-methyl-7H-imidazol-3-ium iodide
- the procedure was the same as Step D in Example 1 with the reagents tert-butyl (S)-3-(4-(l-(3- ((tert-butoxycarbonyl)amino)propyl)-77J-imidazol-4-yl)phenoxy)-2-((l,3-dioxoisoindolin-2- yl)oxy)propanoate (0.17g, 0.280 mmol), Mel (0.105 ml, 1.681 mmol) in MeCN (2 ml).
- LC- MS [M] + m/z 6
- Step E (S)-4-(4-(2-( Aminooxy)-3 -(fert-butoxy)- 3 -oxopropoxy)phenyl)- 1 -(3 -((fert- butoxycarbonyl)amino)propyl)-3-methyl-77J-imidazol-3-ium iodide
- the procedure was the same as step E in Example 1 with the reagents (S)-4-(4-(3-(tert-butoxy)-2-((l,3- dioxoisoindolin-2-yl)oxy)-3-oxopropoxy)phenyl)-l-(3-((tert-butoxycarbonyl)amino)propyl)- 3-methyl-777-imidazol-3-ium iodide (0.210 g, 0.28mmol), hydrazine (8.79 ⁇ , 0.280 mmol) in EtOH (4 ml).
- Step F (S.Z)-4-(4-(3-(fert-butoxy)-2-((((2-((fert-butoxycarbonvnamino)-thiazol-4- yl)(carboxy)methylene)amino)oxy)-3-oxopropoxy)phenyl)-l-(3-((fert-butoxy- carbonyl)amino)propyl)-3-methyl-77J-imidazol-3-ium iodide
- the procedure was the same as step Fin Example 1 with the reagents (S)-4-(4-(2-(aminooxy)-3-(tert-butoxy)-3- oxopropoxy)phenyl)-l-(3-((tert-butoxycarbonyl)amino)propyl)-3-methyl-77J-imidazol-3- ium(0.173 g, 0.28 mmol), 2-(2-((tert-butoxycarbonyl)amino)thiazol-4-
- Step G (S)-3-((Z)-2-((((S)-l-(fert-Butoxy)-3-(4-(l-(3-((fert-butoxycarbonvnamino)propyn- 3-methyl-77J-imidazol-3-ium-4-yl)phenoxy)-l-oxopropan-2-yl)oxy)imino)-2-(2-((fert- butoxycarbonyl)amino)thiazol-4-yl)acetamido)-2,2-dimethyl-4-oxoazetidin-l-yl sulfate
- the procedure was same as step G in Example 1 with the reagent (S,Z)-4-(4-(3-(tert-butoxy)-2- ((((2-((tert-butoxycarbonyl)amino)thiazol-4-yl)(carboxy)methylene)amino)oxy)-3- oxopropoxy)phenyl)-l-(
- Step H Mono(4-(4-((S)-2-((((Z)- 1 -(2-aminothiazol-4-yl)-2-(((S)-2.2-dimethyl-4-oxo- 1 - (sulfooxy)azetidin-3-yl)amino)-2-oxoethylidene)amino)oxy)-2-carboxyethoxy)phenyl)-l-(3- ammoniopropyl)-3-methyl-lH-imidazol-3-ium) mono(2,2,2-trifluoroacetate)
- Step A fert-Butyl (3-(4-(4.4.5.5-tetramethyl-1.3.2-dioxaborolan-2-ylVlH-pyrazol-l- yl)propyl)carbamate
- tert-butyl (3-bromopropyl)carbamate (2.45 g, 10.3 mmol)
- cesium carbonate (3.36 g, 10.3 mmol) in DMF (20 ml) was stirred at 60°C overnight.
- Step B l-Bromo-4-(2-bromoethoxy)benzene 1,2-Dibromoethane (4.26 mL, 49.4 mmol) and cesium carbonate (6.83 g, 49.4 mmol) were added to a stirred solution of 4-bromophenol (1.40 g, 8.09 mmol) in acetone (10 mL). The reaction mixture was heated to 80°C for 8 hours, at which time, the reaction was cooled to room temperature and filtered. The organic filtrate was concentrated, and the resulting crude residue was purified by column
- Step C fert-Butyl (3-(4-(4-(2-bromoethoxy)phenyl)-lH-pyrazol-l-yl)propyl)carbamate 2.0 M aqueous sodium carbonate (1.60 mL, 3.20 mmol) was added to a microwave vial containing a stirred solution of l-bromo-4-(2-bromoethoxy)benzene (300 mg, 1.07 mmol), tert-butyl (3-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazol-l-yl)propyl)- carbamate (565 mg, 1.61 mmol) and l, l'-bis(di-tert-butylphosphino)ferrocene palladium dichloride (78 mg, 0.1 1 mmol) in 1,4-dioxane (4.3 mL).
- Step D fert-Butyl (3-(4-(4-(2-((1.3-dioxoisoindolin-2-vnoxy)ethoxy)phenvn-lH-pyrazol-l- yl)propyl)carbamate
- N-Hydroxyphthalimide 185 mg, 1.13 mmol
- tert-butyl 3-(4-(4-(2-bromoethoxy)phenyl)-lH-pyrazol-l-yl)propyl)carbamate
- DBU 138 mg, 0.905 mmol
- Compound 8 was prepared following procedures similar to those described above for Example 1, by omitting step D and substituting (2R,3,S)-3-amino-2-methyl-4-oxoazetidine-l- sulfonic acid for (,S)-3-amino-2,2-dimethyl-4-oxoazetidin-l-yl hydrogen sulfate where appropriate.
- Step A fert-Butyl (S)-2-(aminooxy)-3-(4-(l-(3-((fert-butoxycarbonyl)amino)propyl)-7H- imidazol-4-yl)phenoxy)propanoate
- the procedure was the same as step E in Example 1 with the reagents (S)-tert-butyl 3-(4-(l -(3-((tert-butoxycarbonyl)amino)propyl)-77J-imidazol-4- yl)phenoxy)-2-((l,3-dioxoisoindolin-2-yl)oxy)propanoate (80 mg, 0.109 mmol) (from step C in Example 2), hydrazine (3.42 ⁇ , 0.109 mmol) in EtOH (4 ml).
- LC-MS [M+H] + m/z 477.26.
- Step B (S.Z)-2-(((l-(fert-Butoxy)-3-(4-(l-(3-((fert-butoxycarbonyl)amino)propyl)-7H- imidazol-4-yl)phenoxy)-l-oxopropan-2-yl)oxy)imino)-2-(2-((fert-butoxycarbonyl)- amino)thiazol-4-yl)acetic acid
- the procedure was the same as step Fin Example 1 with the reagents 2-(2-((tert-butoxycarbonyl)amino)thiazol-4-yl)-2-oxoacetic acid (42.0 mg, 0.154 mmol) (intermediate 6) and (S)-tert-butyl 2-(aminooxy)-3-(4-(l-(3-((tert- butoxycarbonyl)amino)propyl)-7H-imidazol-4-yl)phenoxy)propanoate (70 mg, 0.
- Step C fert-Butyl (S)-3-(4-(l-(3-((fert-butoxycarbonyl)amino)propyl)-7H-imidazol-4- yl)phenoxy)-2-( ((( ⁇ )-!-( 2-( ( fert-butoxycarbonvnamino)thiazol-4-vn-2-( ( ( S)-2.2-dimethyl-4- oxo-l-(sulfooxy)azetidin-3-yl)amino)-2-oxoethylidene)amino)oxy)propanoate
- the procedure was the same as step G in Example 1 with the reagents (S,Z)-2-(((l-(tert-butoxy)- 3-(4-(l-(3-((tert-butoxycarbonyl)amino)propyl)-7H-imidazol-4-yl)phenoxy)-l-oxopropan-2- yl)
- Step D (S)-3 -(4-(l -(3 -AminopropylV7H-imidazol-4-vnphenoxyy2-((Y(Z)- 1 -(2- aminothiazol-4-yl)-2-(((S)-2,2-dimethyl-4-oxo-l-(sulfooxy)azetidin-3-yl)amino)-2- oxoethylidene)amino)oxy)propanoic acid
- the procedure was the same as step H in Example 2 with the reagents (S)-tert-butyl 3-(4-(l-(3-((tert-butoxycarbonyl)amino)propyl)-77J- imidazol-4-yl)phenoxy)-2-((Z)-(l-(2-((tert-butoxycarbonyl)amino)thiazol-4-yl)-2-(((S)-2,2- dimethyl-4-oxo
- Step A (S.Z)-4-(4-(3-(fert-Butoxy)-2-((((2-((fert-butoxycarbonvnamino)-5-chlorothiazol-4- yl)(carboxy)methylene)amino)oxy)-3-oxopropoxy)phenyl)-l-(3-((fert-butoxycarbonyl)- amino)propyl)-2-methyl-77J-pyrazol-2-ium iodide
- the procedure was the same as step F in Example 1 with the reagents 2-(2-((tert-butoxycarbonyl)amino)-5-chlorothiazol-4-yl)-2- oxoacetic acid (0.052 g, 0.170 mmol, Intermediate 6) and (S)-4-(4-(2-(aminooxy)-3-(tert- butoxy)-3-oxopropoxy)phenyl)-l-(3-((tert-butoxycarbonyl)
- Step B (S)-3-((Z)-2-((((S)-l-(fert-Butoxy)-3-(4-(l-(3-((fert-butoxycarbonvnamino)propyn- 2-methyl-7H-pyrazol-2-ium-4-yl)phenoxy)-l-oxopropan-2-yl)oxy)imino)-2-(2-((fert- butoxycarbonyl)amino)-5-chlorothiazol-4-yl)acetamido)-2,2-dimethyl-4-oxoazetidin-l-yl sulfate
- the procedure was the same as step G in Example 2 with the reagents (S,Z)-4-(4-(3- (tert-butoxy)-2-((((2-((tert-butoxycarbonyl)amino)-5-chlorothiazol-4-yl)(carboxy)- methylene)amino)oxy)-3-oxo
- Step C (S)-3-((Z)-2-(2-Amino-5-chlorothiazol-4-vn-2-(((S)-2-(4-(l-(3-aminopropyn-2- methyl-77J-pyrazol-2-ium-4-yl)phenoxy)-l-carboxyethoxy)imino)acetamido)-2,2-dimethyl- 4-oxoazetidin-l-yl sulfate
- the procedure was the same as step H in Example 2 with the reagents 4-(4-((S)-3-(tert-butoxy)-2-(((Z)-(l-(2-((tert-butoxycarbonyl)amino)-5-chloro- thiazol-4-yl)-2-(((S)-2,2-dimethyl-4-oxo-l-(sulfooxy)azetidin-3-yl)amino)-2-oxo- ethylid
- Step A ( S.Z>4-(4-(3-( fert-ButoxyV2-(((( 5-( ( fert-butoxycarbonvnamino)-1.2.4-thiadiazol-3- yl)(carboxy)methylene)amino)oxy)-3-oxopropoxy)phenyl)-l-(3-((fert-butoxycarbonyl)- amino)propyl)-2-methyl-7H-pyrazol-2-ium iodide
- the procedure was the same as step Fin Example 1 with the reagents 2-(5-((tert-butoxycarbonyl)amino)-l,2,4-thiadiazol-3-yl)-2- oxoacetic acid (0.046 g, 0.170 mmol) (Intermediate 7) and (S)-4-(4-(2-(aminooxy)-3-(fert- butoxy)-3-oxopropoxy)phenyl)-l-(3-((tert-butoxycarbon
- Step B (S)-3-(YZ)-2-(Y(YS)-l-( fert-Butoxy)-3 -(4-( 1 -(3 -(( fert-butoxycarbonvnamino)propyn- 2-methyl-7H-pyrazol-2-ium-4-yl)phenoxy)-l-oxopropan-2-yl)oxy)imino)-2-(5-((fert- butoxycarbonvDamino)- 1 ,2,4-thiadiazol-3 -yl)acetamido)-2,2-dimethyl-4-oxoazetidin- 1 -yl sulfate
- the procedure was the same as step G in Example 2 with the reagents (S,Z)-4-(4-(3- (tert-butoxy)-2-((((5-((tert-butoxycarbonyl)amino)-l,2,4-thiadiazol-3-yl)(carboxy)- methylene)
- Step C (SV3-((Z)-2-(5-Amino-1.2.4-thiadiazol-3-vn-2-(((S)-2-(4-(l-(3-aminopropyn-2- methyl-7H-pyrazol-2-ium-4-yl)phenoxy)-l-carboxyethoxy
- Example 13 was synthesized and characterized by LC/MS.
- Step A fert-butyl ( ⁇ )-2-1 ⁇ -3-(4-(1-(2- ⁇ 1 ⁇ 1 ⁇ 6 ⁇ 1 ⁇ -1 ⁇ - ⁇ 1-4- yl)phenoxy)propanoate
- Intermediate 3 (R)-tert-butyl 3-(4-bromophenoxy)- 2-hydroxypropanoate 500 mg, 1.58 mmol
- 4-(2-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan- 2-yl)-lH-pyrazol-l-yl)ethyl)morpholine 726 mg, 2.36 mmol
- l,l'-bis(di-tert- butylphosphino)ferrocene palladium dichloride 103 mg, 0.158 mmol
- THF 10 ml
- Step B teit-butyl (S)-2-((1.3-dioxoisoindolin-2-vnoxy)-3-(4-(l-(2-morpholinoethvn-lH- pyrazol-4-yl)phenoxy)propanoate
- the procedure was same as step C in Example 1 with the reagent of: tert-butyl (R)-2-hydroxy-3-(4-(l-(2-mo ⁇ holinoethyl)-lH-pyrazol-4- yl)phenoxy)propanoate (0.480 g, 1.15 mmol), 2-hydroxyisoindoline-l,3-dione (0.225 g, 1.38 mmol), triphenylphosphine (0.362 g, 1.38 mmol), DIAD (0.272 ml, 1.380 mmol) and THF (4 ml).
- LC-MS [M + H] + m/z 563.47.
- Step C (S)-4-(2-(4-(4-(3-(tert-butoxy)-2-((1.3-dioxoisoindolin-2-vnoxy)-3-oxopropoxy)- phenyl -lH-pyrazol-l-yl ethyl -4-methylmo ⁇ holin-4-ium iodide
- (S)-tert- butyl 2-(( 1 , 3 -dioxoi soindolin-2-yl)oxy)-3 -(4-( 1 -(2- ⁇ 1 ⁇ 6 ⁇ 1)- 1 H-pyrazol-4-yl)- phenoxy)propanoate 250 mg, 0.444 mmol
- acetonitrile (4 ml) was added iodomethane (0.167 ml, 2.67 mmol)
- Step D (S)-4-(2-(4-(4-(2-(aminooxy)-3-(tert-butoxy)-3-oxopropoxy)phenyl)-lH-pyrazol-l- yl)ethyl)-4-methylmorpholin-4-ium iodide
- the procedure is the same as step E in Example 1 with the reagent (S)-4-(2-(4-(4-(3-(tert-butoxy)-2-((l,3-dioxoisoindolin-2-yl)oxy)-3- oxopropoxy)phenyl)-lH-pyrazol-l-yl)ethyl)-4-methylmo holin-4-ium iodide (310 mg, 0.440 mmol), hydrazine (14.0 ⁇ , 0.440 mmol) in EtOH (4 ml).
- Step E (S.Z)-4-(2-(4-(4-(3-(tert-butoxy)-2-((((2-((tert-butoxycarbonvnamino)thiazol-4- yl)(carboxy)methylene)amino)oxy)-3-oxopropoxy)phenyl)-lH-pyrazol-l-yl)ethyl)-4- methylmorpholin-4-ium iodide
- the procedure was the same as step F in Example 1 with the reagents 2-(5-((tert-butoxycarbonyl)amino)-l,2,4-thiadiazol-3-yl)-2-oxoacetic acid (0.118 g, 0.435 mmol) (Intermediate 7) and (S)-4-(2-(4-(4-(2-(aminooxy)-3-(tert-butoxy)-3- oxopropoxy)phenyl)
- Step F 4-(2-(4-(4-((S)-3-(tert-butoxy)-2-((((Z)-l-(2-((tert-butoxycarbonvnamino)thiazol-4- vn-2-(((S)-2.2-dimethyl-4-oxo-l-(sulfooxy)azetidin-3-vnamino)-2- oxoethylidene)amino)oxy)-3-oxopropoxy)phenyl)-lH-pyrazol-l-yl)ethyl)-4- methylmo ⁇ holin-4-ium iodide
- the procedure was the same as step Fin Example 2 with the reagents (S,Z)-4-(2-(4-(4-(3-(tert-butoxy)-2-((((2-((tert-butoxycarbonyl)amino)thiazol-4- yl)(carboxy)methylene)amino)oxy)-3-
- Step G (S)-3-((Z)-2-(2-aminothiazol-4-vn-2-(((S)-l-carboxy-2-(4-(l-(2-(4-methyl- morpholino-4-ium)ethyl)-lH-pyrazol-4-yl)phenoxy)ethoxy)imino)acetamido)-2,2-dimethyl- 4-oxoazetidin-l-yl sulfate
- the procedure was the same as step H in Example 2 with the reagents 4-(2-(4-(4-((S)-3-(tert-butoxy)-2-((((Z)-l-(2-((tert-butoxycarbonyl)amino)thiazol-4- yl)-2-(((S)-2,2-dimethyl-4-oxo-l-(sulfooxy)azetidin-3-yl)amino)-2-oxoethylidene)-
- Step A teit-butyl (3-(4-(4.4.5.5-tetramethyl-1.3.2-dioxaborolan-2-yl)-lH-pyrazol-l- yl)propyl)carbamate
- Cesium carbonate (12.2 g, 37.5 mmol) was added to a room
- Step B tert-butyl 3-(4-(l-(3-((tert-butoxycarbonyl)amino)propyl)-lH-pyrazol-4- yl)phenoxy)-2-(( 1 , 3 -dioxoi soindolin-2-yl)oxy)-2-methylpropanoate
- tert- butyl 2-(aminooxy)-3-(4-bromophenoxy)-2-methylpropanoate 100 mg, 0.289 mmol
- tert- butyl 3-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazol-l-yl)propyl)carbamate (101 mg, 0.289 mmol)
- 1, 1 '-bis (di-tert-butylphosphino)ferrocenepalladium dichloride (18.82 mg, 0.029 mmol) in diox
- Step C tert-butyl 3-(4-(l-(3-((tert-butoxycarbonyl)amino)propyl)-lH-pyrazol-4- yl)phenoxy)-2-(( 1 , 3 -dioxoi soindolin-2-yl)oxy)- 2-methylpropanoate
- 4-methylbenzene- sulfonic acid (12% in acetic acid) (63.2 mg, 0.044 mmol) was added to a room temperature mixture of tert-butyl 2-(aminooxy)-3-(4-(l-(3-((tert-butoxycarbonyl)amino)propyl)-lH- pyrazol-4-yl)phenoxy)-2-methylpropanoate (540 mg, 1.10 mmol), molecular seives (200 mg) in toluene and the mixture was stirred at 100 °C for 4 hours.
- Step D 4-(4-(3-(tert-butoxy)-2-((L3-dioxoisoindolin-2-yl)oxy)-2-methyl-3-oxopropoxy)- phenyl)-l-(3-((tert-butoxycarbonyl)amino)propyl)-2-methyl-lH-pyrazol-2-ium iodide
- the procedure was the same as step D in Example 1 with the reagent tertbutyl 3-(4-(l-(3-((tert- butoxycarbonyl)amino)propyl)-lH-pyrazol-4-yl)phenoxy)-2-((l,3-dioxoisoindolin-2-yl)oxy)- 2-methylpropanoate (0.495g, 0.797 mmol), Mel (0.399 ml, 6.38 mmol) in MeCN (3 ml).
- Step E (Z)-4-(4-(3-(tert-butoxy)-2-((((2-((tert-butoxycarbonvnamino)thiazol-4- yl)(carboxy)methylene)amino)oxy)-2-methyl-3-oxopropoxy)phenyl)-l-(3-((tert- butoxycarbonyl)amino)propyl)-2-methyl-lH-pyrazol-2-ium iodide Hydrazine (0.024 ml, 0.763 mmol) in 0.3 mL of DCM was added to a stirred, room temperature mixture of 4-(4- (3-(tert-butoxy)-2-((l,3-dioxoisoindolin-2-yl)oxy)-2-methyl-3-oxopropoxy)phenyl)-l-(3- ((tert-butoxycarbonyl)amino)propyl)-2-methyl-lH-pyrazol-2-ium (485 mg
- Step F 4-(4-(3-(tert-butoxy)-2-((((Z)-l-(2-((tert-butoxycarbonvnamino)thiazol-4-vn-2- (((2R,3 S)-2-methyl-4-oxo-l-sulfoazetidin-3-yl)amino)-2-oxoethylidene)amino)oxy)-2- methyl-3-oxopropoxy)phenyl)-l-(3-((tert-butoxycarbonyl)amino)propyl)-2-methyl-lH- pyrazol-2-ium To a solution of (Z)-4-(4-(3-(tert-butoxy)-2-((((2-((tert- butoxycarbonyl)amino)thiazol-4-yl)(carboxy)methylene)amino)oxy)-2-methyl-3- oxopropoxy)phenyl)-l-(3-((ter
- Step G (S)-3 -((Z)-2-((((S)- 1 -(4-( 1 -(3 -aminopropyn-2-methyl- lH-pyrazol-2-ium-4- yl)phenoxy)-2-carboxypropan-2-yl)oxy)imino)-2-(2-aminothiazol-4-yl)acetamido)-2,2- dimethyl-4-oxoazetidin-l-yl sulfate compound with 2,2,2-trifluoroacetic acid (1 : 1) To a solution of 4-(4-(3-(tert-butoxy)-2-((((Z)-l-(2-((tert-butoxycarbonyl)amino)thiazol-4-yl)-2- (((S)-2,2-dimethyl-4-oxo-l-(sulfooxy)azetidin-3-yl)amino)-2-oxoethylidene)a
- Step A tert-butyl (R)-3-(4-bromo-3-fluorophenoxy)-2-hydroxypropanoate
- the procedure was the same as Intermediate 3 with the reagent of tert-butyl oxirane-2-carboxylate (4.19 g, 29.1 mmol), (R,R)-Co catalyst (0.595g, 0.709 mmol), 4-bromo-3-fluorophenol (2.71 g, 14.19 mmol).
- LC-MS [M + Na] + m/z 358.92.
- Step B tert-butyl (R)-3-(4-(l-(3-((tert-butoxycarbonyl)amino)propyl)-lH-pyrazol-4-yl)-3- fluorophenoxy)-2-hydroxypropanoate
- the procedure was the same as Step B in Example 1 with the reagent of tert-butyl (R)-3-(4-bromo-3-fluorophenoxy)-2-hydroxypropanoate (470 mg, 1.40 mmol), tert-butyl (3-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazol-l- yl)propyl)carbamate (512 mg, 1.458 mmol), l,l'-bis(di-tert-butylphosphino)ferrocene palladium dichloride (91 mg, 0.140 mmol), 1,4-dioxane (4
- Step C tert-butyl (S)-3-(4-(l-(3-((tert-butoxycarbonyl)amino)propyl)-lH-pyrazol-4-yl)-3- fluorophenoxy)-2-((L3-dioxoisoindolin-2-yl)oxy)propanoate
- the procedure was same as Step C in Example 1 with the reagent of tert-butyl (R)-3-(4-(l-(3-((tert-butoxycarbonyl)- amino)propyl)-lH-pyrazol-4-yl)-3-fluorophenoxy)-2-hydroxypropanoate (0.45 lg, 0.94 mmol), 2-hydroxyisoindoline-l,3-dione (0.153 g, 0.94 mmol), triphenylphosphine (0.271 g, 1.04 mmol), DEAD (0.164 ml, 1.04 mmol) and THF (4
- Step D (S)-4-(4-(3-(tert-butoxy)-2-((L3-dioxoisoindolin-2-yl)oxy)-3-oxopropoxy)-2- fluorophenyl)-l-(3-((tert-butoxycarbonyl)amino)propyl)-2-methyl-lH-pyrazol-2-ium iodide
- the procedure was same as step D in Example 1 with the reagent tert-butyl (S)-3-(4-(l-(3- ((tert-butoxycarbony l)amino)propyl)- 1 H-pyrazol-4-yl)-3 -fluorophenoxy)-2-(( 1,3- dioxoisoindolin-2-yl)oxy)propanoate (0.576g, 0.922 mmol), Mel (0.461 ml, 7.38 mmol)
- Step E (S.Z)-4-(4-(3-(tert-butoxy)-2-((((2-((tert-butoxycarbonvnamino)thiazol-4-vn- (carboxy)methylene)amino)oxy)-3-oxopropoxy)-2-fluorophenyl)-l-(3-((tert-butoxy- carbonyl)amino)propyl)-2-methyl-lH-pyrazol-2-ium iodide
- the procedure was the same as step E in Example 1 with the reagents hydrazine (0.028 ml, 0.89 mmol), (S)-4-(4-(3-(tert- butoxy)-2-(( 1 , 3 -dioxoi soindolin-2-yl)oxy)-3 -oxopropoxy)-2-fluorophenyl)- 1 -(3 -((tert- butoxycarbonyl)amino)propyl)-2-
- Step F 4-(4-((S)-3-(tert-butoxy)-2-((((Z)-l-(2-((tert-butoxycarbonvnamino)thiazol-4-vn-2- (((S)-2,2-dimethyl-4-oxo-l-(sulfooxy)azetidin-3-yl)amino)-2-oxoethylidene)amino)oxy)-3- oxopropoxy)-2-fluorophenyl)-l-(3-((tert-butoxycarbonyl)amino)propyl)-2-methyl-lH- pyrazol-2-ium
- the procedure was the same as step F in Example 1 with the reagents (S,Z)- 4-(4-(3-(tert-butoxy)-2-((((2-((tert-butoxycarbonyl)amino)thiazol-4-yl)(carboxy)methylene)- amino)oxy)-3-o
- Step G (S)-3-((Z)-2-(((S)-2-(4-(l-(3-aminopropyn-2-methyl-lH-pyrazol-2-ium-4-vn-3- fluorophenoxy)-l-carboxyethoxy)imino)-2-(2-aminothiazol-4-yl)acetamido)-2,2-dimethyl-4- oxoazetidin-l-yl sulfate compound with formic acid (1 : 1)
- the procedure was the same as step G in Example 1 with the reagents 4-(4-((S)-3-(tert-butoxy)-2-((((Z)-l-(2-((tert- butoxycarbonyl)amino)thiazol-4-yl)-2-(((S)-2,2-dimethyl-4-oxo-l-(sulfooxy)azetidin-3- yl)amino)-2-oxoethy
- Step A 4-(4-(3-( tert-butoxy V2-( (((Z)-l-(2-(( tert-butoxycarbonvnamino)thiazol-4-vn-2- (((2R,3 S)-2-methyl-4-oxo-l-sulfoazetidin-3-yl)amino)-2-oxoethylidene)amino)oxy)-2- methyl-3-oxopropoxy)phenyl)-l-(3-((tert-butoxycarbonyl)amino)propyl)-2-methyl-lH- pyrazol-2-ium
- the procedure was the same as step F in Example 1 with the reagents (Z)-4- (4-(3-(tert-butoxy)-2-((((2-((tert-butoxycarbonyl)amino)thiazol-4- yl)(carboxy)methylene)amino)oxy)-2-methyl-3-oxopropoxy
- Step B (2R.3 SV3 -((Z)-2-((((S)- 1 -(4-( 1 -(3 -aminopropyn-2-methyl- lH-pyrazol-2-ium-4- yl)phenoxy)-2-carboxypropan-2-yl)oxy)imino)-2-(2-aminothiazol-4-yl)acetamido)-2-methyl- 4-oxoazetidine-l -sulfonate compound with 2,2,2-trifluoroacetic acid (1 : 1)
- the procedure was the same as step G in Example 1 with the reagents 4-(4-(3-(tert-butoxy)-2-((((Z)-l-(2- ((tert-butoxycarbonyl)amino)thiazol-4-yl)-2-(((2R,3 S)-2-methyl-4-oxo-l-sulfoazetidin-3- yl)amino)-2
- Step A tert-butyl 3-((6-bromopyridin-3-yl)oxy)-2-hydroxypropanoate K 2 C0 3 (6.35 g, 46.0 mmol) was added to a stirred, room temperature mixture of tert-butyl oxirane-2-carboxylate (6.63 g, 46.0 mmol), 6-bromopyridin-3-ol (4 g, 22.99 mmol) in acetonitrile and the mixture was stirred at 90 °C for 3 hours, cooled to room temperature and filtered. The filtrate was concentrated down. The residue was purified by column chromatography on silica gel, eluting with EtOAc/isohexane (0-50%) to give the title compound.
- Example 26 (S)-3-((Z)-2-(((S)-2-((6-(l-(3-aminopropyl)-lH-pyrazol-4-yl)-l-methylpyridin- l-ium-3-yl)oxy)-l-carboxyethoxy)imino)-2-(2-aminothiazol-4-yl)acetamido)-2,2-dimethyl- 4-oxoazetidin-l-yl sulfate compound with 2,2,2-trifluoroacetic acid (1 : 1) was prepared from tert-butyl 3-((6-bromopyridin-3-yl)oxy)-2-hydroxypropanoate by 6 steps. The procedures of the six steps were same as in step B, C, D, E, F, G of Example 2. LC-MS [M] + : m/z 682.20
- Step A fert-Butyl (3-(3-amino-4-bromo-7H-pyrazol-l-yl)propyl)carbamate
- the procedure was the same as step A in Example 1 with the reagents 4-bromo-/H-pyrazol-3 -amine and tert-butyl (3 -bromopropyl)carbamate.
- Step B fert-Butyl (S)-3-(4-(3-amino-l-(3-((fert-butoxycarbonyl)amino)-propyl)-7H-pyrazol- 4-yl)phenoxy)-2-((fert-butyldimethylsilyl)oxy)propanoate
- the procedure was the same as step B in Example 1 with the reagents tert-butyl 2-((tert-butyldimethylsilyl)oxy)-3-(4- (4,4,5, 5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenoxy)-propanoate (0.21g, 0.439 mmol) (Intermediate 8), tert-butyl (3-(3-amino-4-bromo-7H-pyrazol-l-yl)propyl)carbamate (0.17g, 0.533 mmol), andl,l'-bis(di-tert-buty
- Step C fert-Butyl 3-(4-(3-(((allyloxy)carbonyl)amino)-l-(3-((fert-butoxycarbonyl)amino) propyl)- 7H-pyrazol-4-yl)phenoxy)-2-((fert-butyldimethylsilyl)oxy)propanoate
- tert-butyl 3-(4-(3-amino-l-(3-((tert-butoxycarbonyl)amino)propyl)-7H-pyrazol-4- yl)phenoxy)-2-((tert-butyldimethylsilyl)oxy)propanoate (0.54g, 0.914 mmol) in CH 2 CI 2 (10 ml) was added DIPEA (0.319 ml, 1.83 mmol) followed by allyl carbonochloridate (0.194 ml, 1.828 mmol) at room temperature.
- Step D fert-Butyl 3-(4-(3-(((allyloxy)carbonyl)amino)-l-(3-((fert-butoxycarbonyl)- amino)propyl)-7H-pyrazol-4-yl)phenoxy)-2-hydroxypropanoate To a solution of fert-butyl
- Step E fert-Butyl 3-(4-(3-(((allyloxy)carbonyl)amino)-l-(3-((fert-butoxycarbonyl)- amino)propyl)-7H-pyrazol-4-yl)phenoxy)-2-((L3-dioxoisoindolin-2-yl)oxy)propanoate
- the procedure was the same as step C in Example 1 with the reagents tert-butyl 3-(4-(3- (((allyloxy)carbonyl)amino)-l-(3-((tert-butoxycarbonyl)amino)propyl)-77J-pyrazol-4- yl)phenoxy)-2-hydroxypropanoate (0.3g, 0.535 mmol), 2-hydroxyisoindoline-l,3-dione (0.096 g, 0.589 mmol), triphenylphosphine (0.168 g, 0.642 mmol),
- Step F fert-Butyl 3-(4-(3-(((allyloxy)carbonyl)amino)-l-(3-((fert-butoxycarbonyl)- amino)propyl)-77J-pyrazol-4-yl)phenoxy)-2-(aminooxy)propanoate
- the procedure was the same as step E in Example 1 with the reagents tert-butyl 3-(4-(3-(((allyloxy)carbonyl)- amino)-l-(3-((tert-butoxycarbonyl)amino)propyl)-77J-pyrazol-4-yl)phenoxy)-2-((l,3- dioxoisoindolin-2-yl)oxy)propanoate (35 mg, 0.050 mmol) and hydrazine (3.1 1 ⁇ , 0.099 mmol) in THF (2mL).
- LC-MS [M+H] + m/z 576.47
- Step G (Z)-2-(((3-(4-(3-(((Allyloxy)carbonyl)amino)-l-(3-((fert-butoxycarbonyl)amino)- propyl)-77J-pyrazol-4-yl)phenoxy)- 1 -(fert-butoxy)- 1 -oxopropan-2-yl)oxy)imino)-2-(2-((fert- butoxycarbonyl)amino)thiazol-4-yl)acetic acid
- the procedure was the same as step Fin Example 1 with the reagents 2-(2-((tert-butoxycarbonyl)amino)thiazol-4-yl)-2-oxoacetic acid (0.028 g, 0.101 mmol) and fert-butyl 3-(4-(3-(((allylox )carbonyl)amino)-l-(3-((tert- butoxycarbonyl)amino)propyl
- Step H fert-Butyl 3-(4-(3-(((allyloxy)carbonyl)amino)-l-(3-((fert-butoxy
- Step J (SV3-(4-(3 - Amino- 1 -(3 -aminopropylV 7H-pyrazol-4-vnphenoxy)-2-(Y(YZ 1 -(2- aminothiazol-4-yl)-2-(((S)-2,2-dimethyl-4-oxo-l-(sulfooxy)azetidin-3-yl)amino)-2- oxoethylidene)amino)oxy)propanoic acid
- the procedure was the same as step H in Example 2 with the reagents tert-butyl 3-(4-(3-amino-l-(3-((tert-butoxycarbonyl)amino)propyl)-7H- pyrazol-4-yl)phenoxy)-2-(((Z)-(l-(2-((tert-butoxycarbonyl)amino)thiazol-4-yl)-2-(((S)-2,2- di
- Step A (S)-3-(((Allyloxy)carbonvnamino)-4-(4-(3-(fert-butoxy)-2-((1.3-dioxoisoindolin-2- yl)oxy)-3-oxopropoxy)phenyl)-l-(3-((fert-butoxycarbonyl)amino)propyl)-2-methyl-7H- pyrazol-2-ium iodide
- the procedure was the same as step D in Example 1 with the reagents (S)-tert-butyl 3-(4-(3-(((allyloxy)carbonyl)amino)-l-(3-((tert-butoxycarbonyl)amino)propyl)- 7H-pyrazol-4-yl)phenoxy)-2-((l,3-dioxoisoindolin-2-yl)oxy)propanoate (110 mg, 0.156 mmol), Mel (0.078 ml,
- Step B-F The procedures of Step B-F were the same as step F-J in Example 28 to give the title compound.
- LC-MS [M+H] + m/z 696.41.
- Step A (R)-ethyl 3-(4-bromophenoxy)-2-hydroxypropanoate To a mixture of 4- bromophenol (2 g, 11.6 mmol), R,R-Co Catalyst(III) (0.485 g, 0.578 mmol) (J. Am. Chem. Soc, 1999, 121, 6086-6087), molecular sieves 4A (4 g), ethyl oxirane-2-carboxylate (2.95 g, 25.4 mmol) was added tert-butyl methyl ether (6 ml) under N 2 , the resulting suspension was stirred at room temperature under N 2 over the weekend. The filtrate was concentrated after filtration and the residue was purified on silica gel column using EtOAc/hexane as eluting solvents to give the title compound. LC/MS: [M+H] + : 288.9, 290.9.
- Step B (R)-3-(4-bromophenoxy)-2-hydroxypropanoic acid To the solution of (R)-ethyl 3-(4- bromophenoxy)-2-hydroxypropanoate (3.13 g, 10.8 mmol) in THF (60 ml) and MeOH (20 ml) at 0 °C was added NaOH (21.6 ml, 21.6 mmol), and the resulting solution was stirred at 0 °C for 4 hours.
- Step C (R)-3-(4-bromophenoxy)-2-hydroxypropanamide
- DCM DCM
- 1- hydroxy-2,5-pyrrolidinedione monohydrate 1.50 g, 11.3 mmol
- EDC 3-hydroxy-2,5-pyrrolidinedione monohydrate
- the resulting mixture was stirred at room temperature for 2 hours.
- the mixture was patitioned between water and DCM, the organic phase was washed with water, brine, dried over Na 2 S0 4 , concentrated and the residue was dissolved in dioxane (80 mL) and cooled to 0 °C.
- Step D (R)-3-(4-bromophenoxy)-2-((tert-butyldimethylsilyl)oxy)-propanamide
- imidazole (2.36 g, 34.6 mmol
- TBS-C1 (2.61 g, 17.3 mmol)
- DMAP 0.085 g, 0.692 mmol
- the reaction mixture was partitioned between EtOAc and sat. NaHC0 3 , the aqueous phase was extracted with EtOAc three times, the combined organic phase was dried over Na 2 S0 4 , concentrated and the residue was purified on silica gel column using
- Step F (S)-5-(2-(4-bromophenoxy)- 1 -((tert-butyldimethylsilyl)oxy)ethyl)-2H-tetrazole
- dibutyltin oxide 0.381 g, 1.53 mmol
- TMS- N 3 2.03 ml, 15.3 mmol
- Step G (S)-5-(2-(4-bromophenoxy)-l-((tert-butyldimethylsilvnoxy)ethvn-2-(4- methoxybenzyl)-2H-tetrazole
- K 2 C0 3 (1.13 g, 8.19 mmol)
- Step H (SVteit-butyl (3-(4-(4-(2-((tert-butyldimethylsilvnoxy)-2-(2-(4-methoxybenzvn-2H- tetrazol-5-yl)ethoxy)phenyl)- lH-pyrazol- 1 -yl)propyl)carbamate
- (S)-5-(2- (4-bromophenoxy)-l-((tert-butyldimethylsilyl)oxy)ethyl)-2-(4-methoxybenzyl)-2H-tetrazole (0.57 g, 1.10 mmol)
- tert-butyl (3-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH- pyrazol-l-yl)propyl)carbamate (0.424 g, 1.21 mmol) in dioxane (20 ml)
- Step J (R)-tert-butyl (3-(4-(4-(2-((1.3-dioxoisoindolin-2-vnoxy)-2-(2-(4-methoxybenzvn- 2H-tetrazol-5-yl)ethoxy)phenyl)-lH-pyrazol-l-yl)propyl)carbamate
- (S)- tert-butyl (3-(4-(4-(2-hydroxy-2-(2-(4-methoxybenzyl)-2H-tetrazol-5-yl)ethoxy)phenyl)-lH- pyrazol-l-yl)propyl)carbamate (0.19 g, 0.346 mmol) in THF (10 ml) was added 2- hydroxyisoindoline-l,3-dione (0.068 g, 0.415 mmol), triphenylphosphine (0.136 g, 0.519 mmol), and DEAD
- Step K (R)- 1 -(3 -((tert-butoxy carbonyl)amino)propyl)-4-(4-(2-(( 1 , 3 -dioxoi soindolin-2- yl)oxy)-2-(2-(4-methoxybenzyl)-2H-tetrazol-5-yl)ethoxy)phenyl)-2-methyl-lH-pyrazol-2- ium iodide To the mixture of (R)-tert-butyl (3-(4-(4-(2-((l,3-dioxoisoindolin-2-yl)oxy)-2- (2-(4-methoxybenzyl)-2H-tetrazol-5-yl)ethoxy)phenyl)-lH-pyr
- Step L (R)-4-(4-(2-(aminooxy)-2-(2-(4-methoxybenzvn-2H-tetrazol-5-vnethoxy)phenvn-l- (3-((tert-butoxycarbonyl)amino)propyl)-2-methyl-lH-pyrazol-2-ium
- Step M (R.Z)-l-(3-((tert-butoxycarbonvnamino)propyn-4-(4-(2-((((2-((tert-butoxy- carbonyl)amino)thiazol-4-yl)(carboxy)methylene)amino)oxy)-2-(2-(4-methoxybenzyl)-2H- tetrazol-5-yl)ethoxy)phenyl)-2-methyl-lH-pyrazol-2-ium
- Step N (R.Z)-4-(4-(2-((((2-aminothiazol-4-vn(carboxy)methylene)-amino)oxy)-2-(2H- tetrazol-5-yl)ethoxy)phenyl)-l-(3-((tert-butoxycarbonyl)amino)propyl)-2-methyl-lH- pyrazol-2-ium
- Step O (S)-3-((Z)-2-(2-aminothiazol-4-vn-2-(((R)-2-(4-(l-(3-((tert-butoxycarbonvn- amino)propyl)-2-methyl-lH-pyrazol-2-ium-4-yl)phenoxy)-l-(2H-tetrazol-5-yl)ethoxy)- imino)acetamido)-2,2-dimethyl-4-oxoazetidin-l-yl sulfate
- Step P l-(3-aminopropyn-4-(4-((R)-2-((((Z)-l-(2-aminothiazol-4-vn-2-(((S)-2.2-dimethyl- 4-oxo-l-(sulfooxy)azetidin-3-yl)amino)-2-oxoethylidene)amino)oxy)-2-(2H-tetrazol-5- yl)ethoxy)phenyl)-2-methyl-lH-pyrazol-2-ium 2,2,2-trifluoroacetate
- Step A 4-(4-((R)-2-(((Z)-(l-(2-aminothiazol-4-vn-2-(((2S.3S)-2-methyl-4-oxo-l- sulfoazetidin-3-yl)amino)-2-oxoethylidene)amino)oxy)-2-(2H-tetrazol-5-yl)ethoxy)phenyl)- l-(3-((tert-butoxycarbonyl)amino)propyl)-2-methyl-lH-pyrazol-2-ium To the solution of (R,Z)-4-(4-(2-((((2-aminothiazol-4-yl)(carboxy)methylene)amino)oxy)-2-(2H-tetrazol-5- yl)ethoxy)phenyl)-l-(3-((tert-butoxycarbonyl)amino)propyl)-2-methyl-l
- Step B l-(3-aminopropyn-4-(4-((R)-2-((((Z)-l-(2-aminothiazol-4-vn-2-(((2S.3S)-2-methyl- 4-oxo-l-sulfoazetidin-3-yl)amino)-2-oxoethylidene)amino)oxy)-2-(2H-tetrazol-5- yl)ethoxy)phenyl)-2-methyl-lH-pyrazol-2-ium 2,2,2-trifluoroacetate To the solution of 4-(4- ((R)-2-(((Z)-(l-(2-aminothiazol-4-yl)-2-(((2S,3S)-2-methyl-4-oxo-l-sulfoazetidin-3- yl)amino)-2-oxoethylidene)amino)oxy)-2-(2H-tetra
- Step A tert-butyl (3-(2-nitro-lH-imidazol-l-yl)propyl)carbamate
- Cesium carbonate (2240 mg, 6.88 mmol) was added to a stirred mixture of 3-(boc-amino)propyl bromide (901 mg, 3.78 mmol) and 2-nitro-lH-imidazole (389 mg, 3.44 mmol) in DMF (20 ml) and the mixture was stirred at 60 °C overnight.
- the mixture was diluted with ethyl acetate, washed with water three times and brine, dried over NaS0 4 , filtered and the solvent was evaporated under reduced pressure to give crude product.
- Step B tert-butyl (3-(4-bromo-2-nitro-lH-imidazol-l-yl)propyl)carbamate
- BS 0.847 g, 4.76 mmol
- Step C (R)-tert-butyl 3-(4-(l-(3-((tert-butoxycarbonyl)amino)propyl)-2-nitro-lH-imidazol- 4-yl)phenoxy)-2-hydroxypropanoate
- tert-butyl (3-(4-bromo-2-nitro-lH- imidazol-l-yl)propyl)carbamate (0.82 g, 2.348 mmol) in dioxane (20 ml) was added (R)-tert- butyl 2-hydroxy-3-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenoxy)propanoate (1.112 g, 3.05 mmol) and Na 2 C0 3 (3.52 ml, 7.05 mmol).
- Step D (R)-tert-butyl 3-(4-(l-(3-((tert-butoxycarbonyl)amino)propyl)-2-nitro-lH-imidazol- 4-yl)phenoxy)-2-((tert-butyldimethylsilyl)oxy)propanoate
- To the solution of (R)-tert-butyl 3-(4-(l-(3-((tert-butoxycarbonyl)amino)propyl)-2-nitro-lH-imidazol-4-yl)phenoxy)-2- hydroxypropanoate (2460 mg, 4.86 mmol) in DMF (20 ml) was added imidazole (1653 mg, 24.28 mmol), TBS-C1 (1464 mg, 9.71 mmol) and DMAP (59.3 mg, 0.486 mmol).
- Step E (R)-tert-butyl 3-(4-(2-amino-l-(3-((tert-butoxycarbonyl)amino)propyl)-lH-imidazol- 4-yl)phenoxy)-2-((tert-butyldimethylsilyl)oxy)propanoate
- Step F (R)-tert-butyl 3-(4-(2-(bis(tert-butoxycarbonyl)amino)-l-(3-((tert-butoxycarbonyl)- amino)propyl)-lH-imidazol-4-yl)phenoxy)-2-((tert-butyldimethylsilyl)oxy
- To the solution of (R)-tert-butyl 3-(4-(2-amino-l-(3-((tert-butoxycarbonyl)amino)propyl)-lH- imidazol-4-yl)phenoxy)-2-((tert-butyldimethylsilyl)oxy)propanoate (2.53 g, 4.28 mmol) in CH 2 C1 2 (40 ml) was added BOC 2 0 (2.98 ml, 12.85 mmol), TEA (1.194 ml, 8.56 mmol), and DMAP (0.052 g, 0.428
- Step G (R)-tert-butyl 3-(4-(2-(bis(tert-butoxycarbonyl)amino)-l-(3-((tert-butoxy- carbonyl)amino)propyl)-lH-imidazol-4-yl)phenoxy)-2-hydroxypropanoate
- Step H (S)-tert-butyl 3-(4-(2-(bis(tert-butoxycarbonyl)amino)-l-(3-((tert-butoxy- carbonyl)amino)propyl)-lH-imidazol-4-yl)phenoxy)-2-((L3-dioxoisoindolin-2-yl)- oxy)propanoate
- To the solution of (R)-tert-butyl 3-(4-(2-(bis(tert-butoxycarbonyl)amino)-l- (3-((tert-butoxycarbonyl)amino)propyl)-lH-imidazol-4-yl)phenoxy)-2-hydroxypropanoate (1.39 g, 2.05 mmol) in THF (20 ml) was added 2-hydroxyisoindoline-l,3-dione (0.402 g, 2.46 mmol), triphenylphosphine (0.808 g
- Step I (S)-tert-butyl 2-(aminooxy)-3-(4-(2-(bis(tert-butoxycarbonyl)amino)-l-(3-((tert- butoxycarbonyl)amino)propyl)-lH-imidazol-4-yl)phenoxy)propanoate
- Step J (S,Z)-2-(((3-(4-(2-(bis(tert-butoxycarbonyl)amino)-l-(3-((tert-butoxycarbonyl)- amino)propyl)-lH-imidazol-4-yl)phenoxy)-l-(tert-butoxy)-l-oxopropan-2-yl)oxy)imino)- (2-((tert-butoxycarbonyl)amino)thiazol-4-yl)acetic acid To the solution of (S)-tert-butyl-2- (aminooxy)-3-(4-(2-(bis(tert-butoxycarbonyl)amino)-l-(3-((tert-butoxycarbonyl)-amino)- propyl)-lH-imidazol-4-yl)phenoxy)propanoate (0.61 g, 0.882 mmol) in ethanol (8 ml) and DCM (8mL) was
- Step K (S)-tert-butyl 3-(4-(2-(bis(tert-butoxycarbonvnamino)-l-(3-((tert- butoxycarbonyl)amino)propyl)-lH-imidazol-4-yl)phenoxy)-2-(((Z)-(l-(2-((tert- butoxycarbonyl)amino)thiazol-4-yl)-2-(((S)-2,2-dimethyl-4-oxo-l-(sulfooxy)azetidin-3- yl)amino)-2-oxoethylidene)amino)oxy)propanoate To the solution of (S,Z)-2-(((3-(4-(2- (bis(tert-butoxycarbonyl)amino)-l-
- Step L 3-(2-amino-4-(4-((S)-2-((((Z)-l-(2-aminothiazol-4-vn-2-(((S)-2.2-dimethyl-4-oxo-l- (sulfooxy)azetidin-3-yl)amino)-2-oxoethylidene)amino)oxy)-2-carboxyethoxy)phenyl)-lH- imidazol-l-yl)propan-l-aminium 2,2,2-trifluoroacetate
- Step A (S,E)-tert-butyl 3-(4-(l-(3-((tert-butoxycarbonyl)amino)propyl)-2-((tert- butoxycarbonyl)imino)-3-methyl-2 -dihvdro-lH-imidazol-4-yl)phenoxy)-2-((L3- dioxoisoindolin-2-yl)oxy)propanoate
- Step B (S,E)-tert-butyl 2-(aminooxy)-3-(4-(l-(3-((tert-butoxycarbonyl)amino)propyl)-2- ((tert-butoxycarbonyl)imino)-3-methyl-2,3-dihvdro-lH-imidazol-4-yl)phenoxy)propanoate
- Step C (Z)-2-((((S)- 1 -(tert-butoxy V3 -(4-((E 1 -(3 -(( tert-butoxycarbonvnamino)propyn-2- ((tert-butoxycarbonyl)imino)-3-methyl-2,3-dihvdro-lH-imidazol-4-yl)phenoxy)-l-oxo- propan-2-yl)oxy)imino)-2-(2-((tert-butoxycarbonyl)amino)thiazol-4-yl)acetic acid To the solution of (S,E)-tert-butyl 2-(aminooxy)-3-(4-(l -(3 -((tert-butoxy carbonyl)amino)propyl)-2- ((tert-butoxycarbonyl)imino)-3-methyl-2,3-dihydro-lH-imidazol-4-yl)phenoxy)
- Step D (S)-tert-butyl 3-(4-((E)-l-(3-((tert-butoxycarbonvnamino)propyn-2-((tert- butoxycarbonyl)imino)-3-methyl-2,3-dihydro-lH-imidazol-4-yl)phenoxy)-2-(((Z)-(l-(2- ((tert-butoxycarbonyl)amino)thiazol-4-yl)-2-(((S)-2,2-dimethyl-4-oxo-l-(sulfooxy)azetidin- 3-yl)amino)-2-oxoethylidene)amino)oxy)propanoate To the solution of (Z)-2-((((S)-l-(tert-butoxy)-3-(4-((E)-l-(3-((tert-butoxycarbonyl)amino)propyl)-2-((tert-butoxy
- Step E 3 -(4-(4-((S)-2-((((Z)- 1 -(2-aminothiazol-4-yl)-2-(((S)-2.2-dimethyl-4-oxo- 1 - (sulfooxy)azetidin-3-yl)amino)-2-oxoethylidene)amino)oxy)-2-carboxyethoxy)phenyl)-2- imino-3-methyl-2,3-dihydro-lH-imidazol-l-yl)propan-l-aminium 2,2,2-trifluoroacetate To the solution of (S)-tert-butyl 3-(4-((E)-l-(3-((tert-butoxycarbonyl)amino)propyl)-2-((tert- butoxycarbonyl)imino)-3-methyl-2,3-dihydro-lH-imidazol-4-yl)phenoxy)-2-
- Step A tert-butyl 2-((tert-butyldimethylsilyl)oxy)-3-((6-(pyridin-4-ylamino)pyridin-3- yl)oxy)propanoate
- the mixture of J009 pre-catalyst G3 (Aldrich # 88755, 250 mg,
- Step B tert-butyl 3-((6-((tert-butoxycarbonyl)(pyridin-4-yl)amino)pyridin-3-yl)oxy)-2-((tert- butyldimethylsilyl)oxy)propanoate
- tert-butyl 2-((tert-butyldimethyl- silyl)oxy)-3-((6-(pyridin-4-ylamino)pyridin-3-yl)oxy)propanoate (0.61 g, 1.37 mmol) in CH 2 C1 2 (25 ml) was added TEA (0.382 ml, 2.74 mmol), BOC 2 0 (0.381 ml, 1.64 mmol), and DMAP (0.167 g, 1.37 mmol). The resulting solution was stirred at room temperature overnight. After concentration, the residue was purified on silica gel column using
- Step C tert-butyl 3-((6-((tert-butoxycarbonyl)(pyridin-4-yl)amino)pyridin-3-yl)oxy)-2- hydroxypropanoate
- TBAF 0.513 ml, 0.513 mmol
- Step D tert-butyl 3-((6-((tert-butoxycarbonyl)(pyridin-4-yl)amino)pyridin-3-yl)oxy)-2-((L3- dioxoisoindolin-2-yl)oxy)propanoate
- tert-butyl 3-((6-((tert- butoxycarbonyl)(pyridin-4-yl)amino)pyridin-3-yl)oxy)-2-hydroxypropanoate (0.23 g, 0.533 mmol) in THF (6 ml) was added 2-hydroxyisoindoline-l,3-dione (0.104 g, 0.640 mmol), triphenylphosphine (0.210 g, 0.80 mmol), and DEAD (0.127 ml, 0.800 mmol).
- Step E 4-((5-(3-(tert-butoxy)-2-((L3-dioxoisoindolin-2-yl)oxy)-3-oxopropoxy)pyridin-2- yl)(tert-butoxycarbonyl)amino)- 1 -(3 -((tert-butoxycarbonyOamino)propyOpyridin- 1 -ium
- tert-butyl 3-((6-((tert-butoxycarbonyl)(pyridin-4-yl)amino)pyridin-3-yl)oxy)- 2-((l,3-dioxoisoindolin-2-yl)oxy)propanoate (0.25 g, 0.434 mmol) in acetonitrile (4 ml) was added tert-butyl (3-iodopropyl)carbamate (0.148 g, 0.520 mmol).
- Step F 4-((5-(2-(aminooxy)-3-(tert-butoxy)-3-oxopropoxy)pyridin-2-yl)(tert-butoxy- carbonyl)amino)-l-(3-((tert-butoxycarbonyl)amino)propyl)pyridin-l-ium
- Step G (Z)-4-((5-(3-(tert-butoxy)-2-((((2-((tert-butoxycarbonvnamino)thiazol-4-vn- (carboxy)methylene)amino)oxy)-3-oxopropoxy)pyridin-2-yl)(tert-butoxycarbonyl)amino)-l- (3-((tert-butoxycarbonyl)amino)propyl)pyridin-l-ium To the solution of 4-((5-(2- (aminooxy)-3-(tert-butoxy)-3-oxopropoxy)pyridin-2-yl)(tert-butoxycarbonyl)amino)-l-(3- ((tert-butoxycarbonyl)amino)propyl)pyridin-l-ium (100 mg, 0.165 mmol) in ethanol (4 ml) and CH 2 C1 2 (4mL) was added 2-(2-((tert-butoxycarbony
- Step H 4-((5-(3-(tert-butoxy)-2-(((Z)-(l-(2-((tert-butoxycarbonvnamino)thiazol-4-vn-2-)
- Step A tert-butyl 2-((tert-butyldimethylsilyl)oxy)-3-((6-(pyridin-4-yloxy)pyridin-3- yl)oxy)propanoate
- Step B tert-butyl 2-hydroxy-3-((6-(pyridin-4-yloxy)pyridin-3-yl)oxy)propanoate
- tert-butyl 2-((tert-butyldimethylsilyl)oxy)-3-((6-(pyridin-4-yloxy)pyridin-3- yl)oxy)propanoate (220 mg, 0.493 mmol) in THF (6 ml) at 0 °C was added TBAF (0.493 ml, 0.493 mmol).
- TBAF 0.493 ml, 0.493 mmol
- Step C tert-butyl 2-((L3-dioxoisoindolin-2-yl)oxy)-3-((6-(pyridin-4-yloxy)pyridin-3- yl)oxy)propanoate
- tert-butyl 2-hydroxy-3-((6-(pyridin-4-yloxy)pyridin-3- yl)oxy)propanoate (331 mg, 0.996 mmol) in THF (6 ml) was added 2-hydroxyisoindoline-
- Step D 4-((5-(3-(tert-butoxy)-2-((L3-dioxoisoindolin-2-yl)oxy)-3-oxopropoxy)pyridin-2- yl)oxy)- 1 -(3 -((tert-butoxycarbonyOamino)propyOpyridin- 1 -ium
- tert-butyl 2-((l,3-dioxoisoindolin-2-yl)oxy)-3-((6-(pyridin-4-yloxy)pyridin-3-yl)oxy)propanoate (0.81 g, 1.696 mmol) in acetonitrile (10 ml) was added tert-butyl (3-iodopropyl)carbamate (1.451 g, 5.09 mmol), the resulting solution was heated at 60 °C overnight.
- Step E (Z)-4-((5-(3-(tert-butoxy)-2-((((2-((tert-butoxycarbonvnamino)thiazol-4-vn- (carboxy)methylene)amino)oxy)-3-oxopropoxy)pyridin-2-yl)oxy)-l-(3-((tert-butoxy- carbonyl)amino)propyl)pyridin-l-ium To the solution of 4-((5-(3-(tert-butoxy)-2-(( 1,3 - dioxoisoindolin-2-yl)oxy)-3-oxopropoxy)pyridin-2-yl)oxy)-l-(3-((tert-butoxycarbonyl)- amino)
- Step F 4-((5-(3-(tert-butoxy)-2-(((Z)-(l-(2-((tert-butoxycarbonvnamino)thiazol-4-vn-2- (((S)-2,2-dimethyl-4-oxo-l-(sulfooxy)azetidin-3-yl)amino)-2-oxoethylidene)amino)oxy)-3- oxopropoxy)pyridin-2-yl)oxy)-l-(3-((tert-butoxycarbonyl)amino)propyl)pyridin-l-ium To the solution of (Z)-4-((5-(3-(tert-butoxy)-2-((((2-((tert-butoxycarbonyl)amino)thiazol-4- yl)(carboxy)methylene)amino)oxy)-3-oxopropoxy)pyridin-2-yl)oxy)-l-(3-(
- Step G l-(3-aminopropyn-4-((5-((S)-2-((((Z)-l-(2-aminothiazol-4-vn-2-(((S)-2.2-dimethyl- 4-oxo-l-(sulfooxy)azetidin-3-yl)amino)-2-oxoethylidene)amino)oxy)-2-carboxyethoxy)- pyridin-2-yl)oxy)pyridin- 1 -ium 2,2,2-trifluoroacetate
- Step B 2-bromo-5-(2-((tert-butyldimethylsilyl)oxy)ethoxy)pyridine
- 2-((6- bromopyridin-3-yl)oxy)ethan-l-ol 315 mg, 1.445 mmol
- imidazole 492 mg, 7.22 mmol
- acetonitrile 9 mL
- DMAP 17.65 mg, 0.144 mmol
- Step C 5-(2-((tert-butyldimethylsilyl)oxy)ethoxy)-2-(lH-imidazol-l-yl)pyridine 2-Bromo- 5-(2-((tert-butyldimethylsilyl)oxy)ethoxy)pyridine (480 mg, 1.44 mmol), imidazole (148 mg, 2.17 mmol), L-Proline (27 mg, 0.231 mmol), K 2 C0 3 (399 mg, 2.89 mmol) and Cul (13.8 mg, 0.072 mmol) were combined and suspended in DMSO (3 mL). A stream of N 2 gas was bubbled through a septum into the solution for 10 minutes and the resultant solution was heated at 90 °C overnight.
- Step D 2-((6-(lH-imidazol-l-yl)pyridin-3-yl)oxy)ethan-l-ol
- 5-(2-((tert- butyldimethylsilyl)oxy)ethoxy)-2-(lH-imidazol-l-yl)pyridine 140 mg, 0.438 mmol
- TBAF TBAF
- Step E 2-(2-((6-(lH-imidazol-l-yl)pyridin-3-yl)oxy)ethoxy)isoindoline-L3-dione To a solution of 2-((6-(lH-imidazol-l-yl)pyridin-3-yl)oxy)ethan-l-ol (80 mg, 0.390 mmol) in
- Step F 3 -(3 -((tert-butoxy carbonyl)amino)propyl)- 1 -(5 -(2-(( 1 ,3 -dioxoi soindolin-2- yl)oxy)ethoxy)pyridin-2-yl)-lH-imidazol-3-ium
- 2-(2-((6-(lH-imidazol-l- yl)pyridin-3-yl)oxy)ethoxy)isoindoline-l,3-dione 131 mg, 0.374 mmol
- 3-(boc-amino)propyl bromide 214 mg, 0.89 mmol
- DMAP 5 mg, 0.041 mmol
- Step G l-(5-(2-(aminooxy)ethoxy)pyridin-2-yl)-3-(3-((tert-butoxycarbonyl)amino)propyl)- lH-imidazol-3-ium 3-(3-((tert-butoxycarbonyl)amino)propyl)-l-(5-(2-((l,3-dioxoisoindolin- 2-yl)oxy)ethoxy)pyridin-2-yl)-lH-imidazol-3-ium (0.19 g, 0.374 mmol) was suspended in MeOH (3 mL) and a 1M solution of hydrazine in MeOH (0.41 mL, 0.41 mmol) was added.
- Step H (Z)-3 -(3 -((tert-butoxycarbonyl)amino)propyl)- 1 - (5 - (2-((((2-((tert- butoxycarbonyl)amino)thiazol-4-yl)(carboxy)methylene)amino)oxy)ethoxy)pyridin-2-yn lH-imidazol-3-ium
- EtOH 2-(aminooxy)ethoxy)pyridin-2-yl)-3-(3-((tert- butoxycarbonyl)amino)propyl)-lH-imidazol-3-ium
- Step I (S.Z)-3-(3-((tert-butoxycarbonvnamino)propyn-l-(5-(2-(((l-(2-((tert-butoxy- carbonyl)amino)thiazol-4-yl)-2-((2,2-dimethyl-4-oxo-l-(sulfooxy)azetidin-3-yl)amino)-2- oxoethylidene)amino)oxy)ethoxy)pyridin-2-yl)-lH-imidazol-3-ium
- Step J (S,Z)-3 -(2-((2-((6-(3 -(3 -aminopropyl)- lH-imidazol-3 -ium- 1 -yl)pyridin-3 - yl)oxy)ethoxy)imino)-2-(2-aminothiazol-4-yl)acetamido)-2,2-dimethyl-4-oxoazetidin-l-yl sulfate 2,2,2-trifluoroacetate
- Step A Ethyl (R)-3-((6-bromopyridin-3-yl)oxy)-2-hydroxypropanoate
- Ethyl oxirane-2- carboxylate (1.47 g, 12.6 mmol) was suspended in MTBE (25 mL) and 6-bromopyridin-3-ol (1.0 g, 5.75 mmol) and 4A molecular sieves (2 g) were added.
- Intermediate 2 (0.482 g, 0.575 mmol) was added and the resultant mixture stirred at ambient temperature for 3 days.
- Step B (R)-3-((6-bromopyridin-3-yl)oxy)-2-hydroxypropanoic acid
- ethyl (R)-3-((6-bromopyridin-3-yl)oxy)-2-hydroxypropanoate 513 mg, 1.77 mmol
- 1M NaOH 3.54 ml, 3.54 mmol
- the pH was adjusted to 3 with IN HCl and the mixture partitioned between EtOAc and water.
- Step C tert-butyl (R)-3-((6-bromopyridin-3-yl)oxy)-2-hydroxypropanoate
- (R)-3-((6-bromopyridin-3-yl)oxy)-2-hydroxypropanoic acid 464 mg, 1.77 mmol
- 2-tert-butyl-l,3-diisopropylisourea 886 mg, 4.43 mmol
- Step D tert-butyl (R)-3-((6-bromopyridin-3-yl)oxy)-2-((tert-butyldimethylsilyl)oxy)- propanoate
- tert-butyl (R)-3-((6-bromopyridin-3-yl)oxy)-2-hydroxy- propanoate (265 mg, 0.833 mmol) in CH 2 C1 2 (6 mL) at 0 °C was added tri ethyl amine (0.348 ml, 2.50 mmol).
- Step E tert-butyl (R)-3-((6-(lH-imidazol-l-vnpyridin-3-vnoxy)-2-((tert- butyldimethylsilyl)oxy)propanoate
- the procedure was the same as described for Step C, Example 35 using the reactants (R)-3-((6-bromopyridin-3-yl)oxy)-2-((tert- butyldimethylsilyl)oxy)propanoate (318 mg, 0.735 mmol), imidazole (90 mg, 1.32 mmol), L- Proline (51 mg, 0.44 mmol), K 2 C0 3 (102 mg, 0.735 mmol) and Cul (42 mg, 0.22 mmol) in DMSO (3 mL).
- Step F tert-butyl (R)-3-((6-(lH-imidazol-l-yl)pyridin-3-yl)oxy)-2-hydroxypropanoate
- the procedure is the same as described for Step D in Example 35 using reactants (R)-3-((6-(lH- imidazol-l-yl)pyridin-3-yl)oxy)-2-((tert-butyldimethylsilyl)oxy)propanoate (80 mg, 0.191 mmol) in THF (4 mL) and 1M TBAF in THF (0.191 mL, 0.191 mmol).
- LC-MS [M + H] + m/z 306.27.
- Step G tert-butyl (S)-3-((6-(lH-imidazol-l-yl)pyridin-3-yl)oxy)-2-((L3-dioxoisoindolin-2- yl)oxy)propanoate
- the procedure was the same as described for Step E in Example 35 using reactants tert-butyl (R)-3-((6-(lH-imidazol-l-yl)pyridin-3-yl)oxy)-2-hydroxypropanoate (76 mg, 0.249 mmol), N-hydroxyphthalimide (48.7 mg, 0.299 mmol), triphenylphosphine (78 mg, 0.299 mmol), and DIAD (0.058 ml, 0.299 mmol) in THF (3 mL).
- Step H (S)-3 -(3 -aminopropyl)- 1 -(5 -(3 -(tert-butoxy)-2-(( 1 , 3 -dioxoi soindolin-2-yl)oxy)-3 - oxopropoxy)pyridin-2-yl)-lH-imidazol-3-ium
- To a solution of tert-butyl (S)-3-((6-(lH- imidazol-l-yl)pyridin-3-yl)oxy)-2-((l,3-dioxoisoindolin-2-yl)oxy)propanoate (85 mg, 0.189 mmol) in dioxane (2 mL) was added 3-(boc-amino)propyl bromide (67.4 mg, 0.283 mmol) and the mixture was stirred at 60 °C overnight.
- Step I (S)-l-(5-(3-(tert-butoxy)-2-((L3-dioxoisoindolin-2-yl)oxy)-3-oxopropoxy)pyridin-2- yl)-3-(3-((tert-butoxycarbonyl)amino)propyl)-lH-imidazol-3-ium
- (S)-3-(3- aminopropyl)- 1 -(5 -(3 -(tert-butoxy)-2-(( 1 ,3 -dioxoi soindolin-2-yl)oxy)-3 -oxopropoxy)pyridin- 2-yl)-lH-imidazol-3-ium 46 mg, 0.090 mmol) in CH 2 C1 2 (3 mL) was added BOC-anhydride (29.6 mg, 0.136 mmol) and ⁇ , ⁇ , ⁇ ', ⁇ '-tetramethylethylenediamine (15 ⁇
- Step J (S)-l-(5-(2-(aminooxy)-3-(tert-butoxy)-3-oxopropoxy)pyridin-2-yl)-3-(3-((tert- butoxycarbonyl)amino)propyl)-lH-imidazol-3-ium
- the procedure was the same as described for Step F of Example 35 using reactants (S)-l-(5-(3-(tert-butoxy)-2-((l,3- dioxoisoindolin-2-yl)oxy)-3-oxopropoxy)pyridin-2-yl)-3-(3-((tert-butoxycarbonyl)- amino)propyl)-lH-imidazol-3-ium (26 mg, 0.043 mmol) 1M hydrazine in MeOH (47 ⁇ ,
- Step K (S.Z)-l-(5-(3-(tert-butoxy)-2-((((2-((tert-butoxycarbonvnamino)thiazol-4- yl)(carboxy)methylene)amino)oxy)-3-oxopropoxy)pyridin-2-yl)-3-(3-((tert-butoxy- carbonyl)amino)propyl)-lH-imidazol-3-ium
- the procedure was the same as described for Step H of Example 35 using reactants (S)-l-(5-(2-(aminooxy)-3-(tert-butoxy)-3- oxopropoxy)pyridin-2-yl)-3-(3-((tert-butoxycarbonyl)amino)propyl)-lH-imidazol-3-ium (21 mg, 0.044 mmol) and Intermediate 6 (12.6 mg, 0.046 mmol) in Ethanol (0.5 mL) and C1CH
- Step L l-(5-((S)-3-(tert-butoxy)-2-((((Z)-l-(2-((tert-butoxycarbonvnamino)thiazol-4-vn-2- (((S)-2,2-dimethyl-4-oxo-l-(sulfooxy)azetidin-3-yl)amino)-2-oxoethylidene)amino)oxy)-3- oxopropoxy)pyridin-2-yl)-3-(3-((tert-butoxycarbonyl)amino)propyl)-lH-imidazol-3-ium
- the procedure was the same as described for Step I of Example 35 using reactants (S,Z)-1- (5-(3-(tert-butoxy)-2-((((2-((tert-butoxycarbonyl)amino)thiazol-4-yl)(carboxy)methylene)- amino)oxy)-3-oxopropoxy)
- Step M (SV 3 -(YZ)-2-((YS)-2-(Y6-(3 -(3 -aminopropyl)- lH-imidazol-3 -ium- 1 -yl)pyridin-3 - yl)oxy)-l-carboxyethoxy)imino)-2-(2-aminothiazol-4-yl)acetamido)-2,2-dimethyl-4- oxoazetidin-l-yl sulfate compound with 2,2,2-trifluoroacetic acid (1 : 1)
- the procedure was the same as described for Step J of Example 35 using reactants l-(5-((S)-3-(tert-butoxy)-2- ((((Z)-l-(2-((tert-butoxycarbonyl)amino)thiazol-4-yl)-2-(((S)-2,2-dimethyl-4-oxo-l-
- Step A tert-butyl (3-(2-nitro-lH-imidazol-l-yl)propyl)carbamate
- Cesium carbonate (576 mg, 1.77 mmol) was added to a stirred mixture of 3-(Boc-amino)propyl bromide (232 mg, 0.973 mmol) and 2-nitro-lH-imidazole (100 mg, 0.884 mmol) in DMF (2 ml) and the mixture was stirred at room temperature for 1 hour before it was stirred overnight at 60 °C.
- the mixture was diluted with ethyl acetate, washed with water (3x) and brine, dried
- Step B tert-butyl (3-(2-amino-lH-imidazol-l-yl)propyl)carbamate
- tert-butyl (3-(2-nitro-lH-imidazol-l-yl)propyl)carbamate 500 mg, 1.85 mmol
- 3%Pt/0.6%V/C 100 mg, 0.015 mmol
- the reaction mixture was diluted with EtOAc, filtered through Celite®. The filtrate was concentrated under reduced pressure to provide the title compound, which was used as-is for the next step.
- LC-MS [M + H] m/z 241.6.
- Step C tert-butyl 3-((6-((l-(3-((tert-butoxycarbonyl)amino)propyl)-lH-imidazol-2- yl)amino)pyridin-3-yl)oxy)-2-((tert-butyldimethylsilyl)oxy)propanoate
- tert-butyl (3-(2-amino-lH-imidazol-l-yl)propyl)carbamate 90 mg, 0.375 mmol
- palladium G3 precatalyst J009 and cesium carbonate (366 mg, 1.12 mmol) in dioxane (3.5 ml) was vacuum/N 2 exchanged 3 times before the mixture
- Step D tert-butyl 3-((6-((l-(3-(bis(tert-butoxycarbonyl)amino)propyl)-lH-imidazol-2- yl)(tert-butoxycarbonyl)amino)pyridin-3-yl)oxy)-2-((tert-butyldimethylsilyl)oxy)propanoate
- Step E tert-butyl 3-((6-((l-(3-(bis(tert-butoxycarbonyl)amino)propyl)-lH-imidazol-2- yl)(tert-butoxycarbonyl)amino)pyridin-3-yl)oxy)-2-hydroxypropanoate TBAF (1.0 M in THF, 0.606 ml, 0.606 mmol) was added to a stirred mixture of tert-butyl 3-((6-((l-(3- (bis(tert-butoxycarbonyl)amino)propyl)-lH-imidazol-2-yl)(tert-butoxycarbonyl)amino)- pyridin-3-yl)oxy)-2-((tert-butyldimethylsilyl)oxy)propanoate (320 mg, 0.404 mmol) in THF (3 ml) and the mixture was stirred at room temperature for 1 hour. Solvent was
- step E The product from step E (tert-butyl 3-((6-((l-(3-(bis(tert-butoxycarbonyl)amino)propyl)-lH- imidazol-2-yl)(tert-butoxycarbonyl)amino)pyridin-3-yl)oxy)-2-hydroxypropanoate) was converted to C37A and C37B as TFA salts following a similar procedure from step C, step E to step H in Example 1.
- Compound 37A 3-(2-((5-((S)-2-((((Z)-l-(2-aminothiazol-4-yl)-2-(((S)-2,2-dimethyl-4-oxo- l-(sulfooxy)azetidin-3-yl)amino)-2-oxoethylidene)amino)oxy)-2-carboxyethoxy)pyridin-2- yl)amino)-lH-imidazol-l-yl)propan-l-aminium 2,2,2-trifluoroacetate.
- Step A tert-butyl (R)-3-(4-bromo-3-chlorophenoxy)-2-hydroxypropanoate (R,R)-Co catalyst (0.24 g, 0.29 mmol) was added to a stirred, room temperature mixture of tert-butyl oxirane-2-carboxylate (1.7 g, 12 mmol), and 4-bromo-3-chlorophenol (1.2 g, 5.8 mmol) in t- butylmethyl ether. Then molecular sieves (2.3 g) were added, and the mixture was stirred at room temperature overnight, and filtered. The filtrate was concentrated, and the resulting residue was purified on silica gel column (80g), eluting with EtOAc/Hexane (5-40%) to give the title compound.
- Step B tert-butyl (R)-3-(4-(l-(3-((tert-butoxycarbonyl)amino)propyl)-lH-pyrazol-4-yl)-3- chlorophenoxy)-2-hydroxypropanoate
- (R)-tert-butyl 3-(4-bromo-3- chlorophenoxy)-2-hydroxypropanoate (0.42 g, 1.2 mmol)
- tert-butyl (R)-3-(4-bromo-3- chlorophenoxy)-2-hydroxypropanoate (0.42 g, 1.2 mmol)
- l,l'-bis(di-tert- butylphosphino)ferrocene palladium dichloride 0.078 g, 0.12 mmol)
- Step C tert-butyl (SV3-(4-(l-(3-((tert-butoxycarbonvnanuno propyn-lH-pyrazol-4-ylV3- chlorophenoxy)-2-((L3-dioxoisoindolin-2-yl)oxy)propanoate (E)-di ethyl diazene-1,2- dicarboxylate (0.20 ml, 1.3 mmol) was added to a 0 °C mixture of (R)-tert-butyl 3-(4-(l-(3- ((tert-butoxycarbonyl)amino)propyl)-lH-pyrazol-4-yl)-3-chlorophenoxy)-2- hydroxypropanoate (580 mg, 1.2 mmol), 2-hydroxyisoindoline-l,3-dione (190 mg, 1.2 mmol), triphenylphosphine (340 mg, 1.3 mmol)
- Step D (S)-4-(4-(3-(tert-butoxy)-2-((L3-dioxoisoindolin-2-yl)oxy)-3-oxopropoxy)-2- chlorophenyl)-l-(3-((tert-butoxycarbonyl)amino)propyl)-2-methyl-lH-pyrazol-2-ium iodide Mel (610 ⁇ , 9.8 mmol) was added to a mixture of (S)-tert-butyl 3-(4-(l-(3-((tert- butoxycarbonyl)amino)propyl)-lH-pyrazol-4-yl)-3-chlorophenoxy)-2-((l,3-dioxoisoindolin- 2-yl)oxy)propanoate (625 mg, 0.98 mmol) in CH 3 CN. The reaction mixture was stirred at 74°C overnight, and then cooled to RT. The reaction mixture was concentrated to give
- Step E ( S.Z)-4-( 4-( 3 -(tert-butoxyV2-( (((2-(( tert-butoxycarbonvnamino)thiazol-4-vn- (carboxy)methylene)amino)oxy)-3-oxopropoxy)-2-chlorophenyl)-l-(3-((tert-butoxy- carbonyl)amino)propyl)-2-methyl-lH-pyrazol-2-ium iodide Hydrazine (28 mg, 0.86 mmol) was added to a mixture of (S)-4-(4-(3-(tert-butoxy)-2-((l,3-dioxoisoindolin-2-yl)oxy)-3- oxopropoxy)-2-chlorophenyl)-l-(3-((tert-butoxycarbonyl)amino)propyl)-2-methyl-lH- pyrazol-2-ium iodide (610 mg
- Step F 4-( 4-( ( SV 3 -(tert-butoxyV2-( ((( ⁇ )-!-( 2-((tert-butoxycarbonvnamino)thiazol-4-vn-2- (((S)-2,2-dimethyl-4-oxo-l-(sulfooxy)azetidin-3-yl)amino)-2-oxoethylidene)amino)oxy)-3- oxopropoxy)-2-chlorophenyl)-l-(3-((tert-butoxycarbonyl)amino)propyl)-2-methyl-lH- pyrazol-2-ium To the solution of (S,Z)-4-(4-(3-(tert-butoxy)-2-((((2-((tert-butoxycarbonyl)- amino)thiazol-4-yl)(carboxy)methylene)amino)oxy)-3-oxopropoxy)-2-chlorophenyl)-
- Step G (S)-3-((Z)-2-(((S)-2-(4-(l-(3-aminopropyn-2-methyl-lH-pyrazol-2-ium-4-vn-3- chlorophenoxy)-l-carboxyethoxy)imino)-2-(2-aminothiazol-4-yl)acetamido)-2,2-dimethyl-4- oxoazetidin-l-yl sulfate TFA (2 ml, 26 mmol) was added to a stirred, room temperature mixture of 4-(4-((S)-3-(tert-butoxy)-2-(((Z)-(l-(2-((tert-butoxycarbonyl)amino)thiazol-4-yl)- 2-(((S)-2,2-dimethyl-4-oxo-l-(sulfooxy)azetidin-3-yl)amino)-2-oxoethylidene)amin
- Step A teit-butyl (3-(4-(4.4.5.5-tetramethyl-1.3.2-dioxaborolan-2-vn-lH-pyrazol-l- yl)propyl)carbamate
- Cesium carbonate (12 g, 38 mmol) was added to a room temperature mixture of 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (4.8 g, 25 mmol), tert-butyl (3-bromopropyl)carbamate (6.0 g, 25 mmol) in DMF (40 mL).
- Step B tert-butyl 3-(4-(l-(3-((tert-butoxycarbonyl)amino)propyl)-lH-pyrazol-4-yl)- phenoxy)-2-((L3-dioxoisoindolin-2-yl)oxy)-2-methylpropanoate
- tert-butyl 2-(aminooxy)-3-(4-bromophenoxy)-2-methylpropanoate 100 mg, 0.29 mmol)
- tert-butyl (3- (4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazol-l-yl)propyl)carbamate 100 mg, 0.29 mmol)
- 1, 1-bis (di-tert-butylphosphino) ferrocenepalladium dichloride (19 mg, 0.029
- the vial was sealed, degassed, refilled with N 2 , and stirred at 70 °C for 1 h. Then the reaction was diluted with water, extracted with twice with ethyl acetate. The combined organic layers were dried over MgS0 4 , filtered and concentrated. The resulting residue was purified by column chromatography on silica gel, eluting with
- Step C tert-butyl 3-(4-(l-(3-((tert-butoxycarbonyl)amino)propyl)-lH-pyrazol-4- yl)phenoxy)-2-((L3-dioxoisoindolin-2-yl)oxy)-2-methylpropanoate 4-Methyl- benzenesulfonic acid (12% in acetic acid, 63 mg, 0.044 mmol) was added to a room temperature mixture of tert-butyl 2-(aminooxy)-3-(4-(l-(3-((tert-butoxycarbonyl)- amino)propyl)-lH-pyrazol-4-yl)phenoxy)-2-methylpropanoate (540 mg, 1.1 mmol), and molecular seives (200 mg) in toluene. The reaction mixture was stirred at 100 °C for 4 h, then filtered and concentrated. The resulting residue was purified by column
- Step D 4-(4-(3-( teit-butoxy)-2-( (1.3 -dioxoi soindolin-2-vnoxy)-2-methyl-3 - oxopropoxy)phenyl)-l-(3-((tert-butoxycarbonyl)amino)propyl)-2-methyl-lH-pyrazol-2-ium iodide
- the title compound was prepared according to the procedure of Example 1 Step D using tert-butyl 3-(4-(l-(3-((tert-butoxycarbonyl)amino)propyl)-lH-pyrazol-4-yl)phenoxy)- 2-((l,3-dioxoisoindolin-2-yl)oxy)-2-methylpropanoate (0.50 g, 0.80 mmol), and Mel (0.40 ml, 6.4 mmol) in MeCN (3 ml).
- Step E (Z)-4-(4-(3-(tert-butoxy)-2-((((2-((tert-butoxycarbonvnamino)thiazol-4- yl)(carboxy)methylene)amino)oxy)-2-methyl-3-oxopropoxy)phenyl)-l-(3-((tert- butoxycarbonyl)amino)propyl)-2-methyl-lH-pyrazol-2-ium iodide Hydrazine (0.024 ml, 0.76 mmol) in 0.3 mL of DCM was added to a mixture of 4-(4-(3-(tert-butoxy)-2-((l,3- dioxoisoindolin-2-yl)oxy)-2-methyl-3-oxopropoxy)phenyl)-l-(3-((tert-butoxy-carbonyl)- amino)propyl)-2-methyl-lH-pyrazol-2-ium (480 mg, 0.76 mmol
- Step F 4-(4-(3-(tert-butoxy)-2-((((Z)-l-(2-((tert-butoxycarbonvnamino)thiazol-4-vn-2-(((S)- 2,2-dimethyl-4-oxo-l-sulfoazetidin-3-yl)amino)-2-oxoethylidene)amino)oxy)-2-methyl-3- oxopropoxy)phenyl)-l-(3-((tert-butoxycarbonyl)amino)propyl)-2-methyl-lH-pyrazol-2-ium To a solution of (Z)-4-(4-(3-(tert-butoxy)-2-((((2-((tert-butoxycarbonyl)amino)thiazol-4- yl)(carboxy)methylene)amino)oxy)-2-methyl-3-oxopropoxy)phenyl)-l-(3-((tert-
- Step G (S)-3 -((Z)-2-((((S)- 1 -(4-( 1 -(3 -aminopropyn-2-methyl- lH-pyrazol-2-ium-4- yl)phenoxy)-2-carboxypropan-2-yl)oxy)imino)-2-(2-aminothiazol-4-yl)acetamido)-2,2- dimethyl-4-oxoazetidin- 1 -yl sulfate
- Step A l-bromo-4-(2,2-diethoxyethoxy)benzene Potassium carbonate (1 1 g, 78 mmol) was added to a stirred, room temperature mixture of 4-bromophenol (5.4 g, 31 mmol) and 2- bromo-l, l-diethoxy ethane (1.8 ml, 12 mmol) in DMF. The reaction mixture was stirred at 80 °C overnight, then cooled to room temperature and diluted with EtOAc. The mixture was washed with water and brine. The organic layer was separated, dried over MgS0 4 , filtered and concentrated. The resulting residue was purified by column chromatography on silica gel, eluting with EtOAc/isohexane (0-40%) to give the title compound.
- Step B 2-(4-bromophenoxy)acetaldehyde HC1 (3.7 ml, 22 mmol) was added to a stirred, room temperature mixture of l-bromo-4-(2,2-diethoxyethoxy)benzene (2.6 g, 9.0 mmol) in THF. The reaction mixture was stirred at room temperature overnight, then the mixture was extracted with ether. The organic layer was separated, washed with water and brine, dried over MgS0 4 , filtered and concentrated to give the title compound.
- Step C dimethyl (2-(4-bromophenoxy)-l -hydroxy ethyPphosphonate
- Dimethyl phosphonate (1.20 ml, 13 mmol) was added dropwise to a stirred 0 °C mixture of 2-(4-bromophenoxy)- acetaldehyde (1.2 g, 5.3 mmol), triethylamine (1.8 ml, 13 mmol) in THF.
- the reaction mixture was stirred at 0 °C for 1 h, and then at RT for lh. Then the reaction mixture was concentrated.
- Step D tert-butyl (3-(4-(4-(2-(dimethoxyphosphoryl)-2-hydroxyethoxy)phenyl)-lH-pyrazol- 1 -yl)propyl)carbamate
- the title compound was prepared according to the procedure of Example 38, Step B.
- Step E tert-butyl (3-(4-(4-(2-(dimethoxyphosphoryl)-2-((L3-dioxoisoindolin-2-yl)oxy)- ethoxy)phenyl)-lH-pyrazol-l-yl)propyl)carbamate
- the title compound was prepared according to the procedure of Example 38, Step C.
- Step F l-(3-((tert-butoxycarbonyl)amino)propyl)-4-(4-(2-(dimethoxyphosphoryl)-2-((L3- dioxoisoindolin-2-yl)oxy)ethoxy)phenyl)-2-methyl-lH-pyrazol-2-ium trifluoromethanesulfonate Methyl trifluoromethanesulfonate (0.069 ml, 0.63 mmol) was added to a stirred, cooled 0 °C mixture of tert-butyl (3-(4-(4-(2-(dimethoxyphosphoryl)-2-((l,3- dioxoisoindolin-2-yl)oxy)ethoxy)phenyl)-lH-pyrazol-l-yl)propyl)carbamate (370 mg, 0.60 mmol) in CH 3 CN.
- Step G 1 -(3 -aminopropyl)-4-(4-(2-(( 1, 3 -di oxoi soindolin-2-yl)oxy)-2-phosphonoethoxy)- phenyl)-2 -methyl- lH-pyrazol-2-ium trifluoromethanesulfonate
- Bromotrimethylsilane (0.77 ml, 5.9 mmol) was added to a stirred, room temperature mixture of l-(3-((tert-butoxy- carbonyl)amino)propyl)-4-(4-(2-(dimethoxyphosphoryl)-2-((l,3-dioxoisoindolin-2-yl)oxy)- ethoxy)phenyl)-2-methyl-lH-pyrazol-2-ium trifluoromethanesulfonate (570 mg, 0.59 mmol) in DCM. The reaction mixture was stirred at room temperature overnight, and then concentrated to give the
- Step H l-(3-((tert-butoxycarbonyl)amino)propyl)-4-(4-(2-((L3-dioxoisoindolin-2-yl)oxy)-2- phosphonoethoxy)phenyl)-2-methyl-lH-pyrazol-2-ium
- Triethylamine (0.31 ml, 2.2 mmol) was added to a stirred, cooled room temperature mixture of l-(3-aminopropyl)-4-(4-(2-((l,3- dioxoisoindolin-2-yl)oxy)-2-phosphonoethoxy)phenyl)-2-methyl-lH-pyrazol-2-ium trifluoromethanesulfonate (0.28 g, 0.56 mmol), and di-tert-butyl dicarbonate (0.24 g, 1.12 mmol) in DMF.
- Step I l-(3-((tert-butoxycarbonyl)amino)propyl)-4-(4-(2-((((Z)-l-(2-((tert-butoxycarbonyl)- amino)thiazol-4-yl)-2-(((S)-2,2-dimethyl-4-oxo-l-(sulfooxy)azetidin-3-yl)amino)-2-oxo- ethylidene)amino)oxy)-2-phosphonoethoxy)phenyl)-2-methyl-lH-pyrazol-2-ium Hydrazine (10 ⁇ , 0.33 mmol) was added to a stirred, room temperature mixture of l-(3-((tert- butoxycarbonyl)amino)propyl)-4-(4-(2-((l,3-dioxoisoindolin-2-yl)oxy)-2-phosphonoethoxy)- phenyl)-2-methyl
- Step J (3 SV3 -(YZV 2-(Y2-(4-( 1 -(3 -aminopropyn-2-methyl- 1 H-pyrazol-2-ium-4-vnphenoxy)- l-phosphonoethoxy)imino)-2-(2-aminothiazol-4-yl)acetamido)-2,2-dimethyl-4-oxoazetidin-
- Step A l-(3-((tert-butoxycarbonyl)amino)propyl)-4-(4-(2-((L3-dioxoisoindolin-2-yl)oxy)- 2-(hydroxy(methoxy)phosphoryl)ethoxy)phenyl)-2-methyl-lH-pyrazol-2-ium iodide Mel (0.145 ml, 2.327 mmol) was added to a stirred, room temperature mixture of tert-butyl (3-(4- (4-(2-(dimethoxyphosphoryl)-2-((l,3-dioxoisoindolin-2-yl)oxy)ethoxy)phenyl)-lH-pyrazol- l-yl)propyl)carbamate (from Example 40 Step E, 143 mg, 0.23 mmol) in CH 3 CN. The reaction mixture was stirred at 72°C overnight, then concentrated to give the title compound. LC-MS [M] + : m
- Step B 1 -(3 -( (tert-butoxycarbonyl)amino)propyl)-4-( 4-(Y2R)-2-(Y(YZ)-(2-(Y tert- butoxycarbonyl)amino)thiazol-4-yl)(carboxy)methylene)amino)oxy)-2-(hvdroxyl- (methoxy)phosphoryl)ethoxy)phenyl)-2-methyl-lH-pyrazol-2-ium
- the title compound was prepared according to the procedure of Example 38 Step E.
- LC-MS [M] + m/z 739.46.
- Step C 1 -(3 -(( tert-butoxycarbonyl)amino)propyl)-4-( 4-(2-((((Z)- 1 -(2-(( tert- butoxycarbonyl)amino)thiazol-4-yl)-2-(((S)-2,2-dimethyl-4-oxo-l-(sulfooxy)azetidin-3- yl)amino)-2-oxoethylidene)amino)oxy)-2-(hydroxy(methoxy)phosphoryl)ethoxy)phenyl)-2- methyl-lH-pyrazol-2-ium
- the title compound was prepared according to the procedure of Example 38 Step F.
- LC-MS [M] + m/z 931.38.
- Step D (3 S)-3-((Z)-2-(((lR)-2-(4-(l-(3-aminopropyn-2-methyl-lH-pyrazol-2-ium-4- yl)phenoxy)-l-(hydroxy(methoxy)phosphoryl)ethoxy)imino)-2-(2-aminothiazol-4- yl)acetamido)-2,2-dimethyl-4-oxoazetidin-l-yl sulfate compound with 2,2,2-trifluoroacetic acid (1 : 1)
- the title compound was prepared according to the procedure of Example 38 Step G.
- EXAMPLE 42 (3 S)-3-((Z)-2-(((lR)-2-(4-(l-(3-aminopropyn-2-methyl-lH-pyrazol-2-ium-4- yl)phenoxy)-l-(hydroxy(methoxy
- Step A teit-butyl 2.2-dimethyl-5-((4-(4.4.5.5-tetramethyl-1.3.2-dioxaborolan-2-vn-lH- pyrazol-l-yl)methyl)oxazolidine-3-carboxylate
- Cesium carbonate (3.3 g, 10.2 mmol) was added to a room temperature mixture of 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH- pyrazole (1.3 g, 6.8 mmol)), and tert-butyl 5-(bromomethyl)-2,2-dimethyloxazolidine-3- carboxylate (2 g, 6.8 mmol)) in DMF (10 mL).
- Step B tert-butyl 5-((4-(4-((SV3-(tert-butoxyV2-((1.3-dioxoisoindolin-2-vnoxyV3- oxopropoxy)phenyl)-lH-pyrazol-l-yl)methyl)-2,2-dimethyloxazolidine-3-carboxylate
- the title compound was prepared according to the procedure of Example 38 Steps A-C.
- Step C 4-(4-((S)-3 -(tert-butoxy)-2-(( 1 ,3 -dioxoi soindolin-2-yl)oxy)-3 -oxopropoxy)phenyl)- 1 - ((3-(tert-butoxycarbonyl)-2,2-dimethyloxazolidin-5-yl)methyl)-2-methyl-lH-pyrazol-2-ium trifluoromethanesulfonate
- the title compound was prepared according to the procedure of Example 40 Step D.
- Step D l-(3-amino-2-hvdroxypropyl)-4-(4-((S)-3-(tert-butoxy)-2-((L3-dioxoisoindolin-2- yl)oxy)-3-oxopropoxy)phenyl)-2-methyl-lH-pyrazol-2-ium trifluoromethanesulfonate
- DL- 10-camphorsulfonic acid 0.028 g, 0.122 mmol
- Step E 4-(4-((S)-3-(tert-butoxy)-2-((L3-dioxoisoindolin-2-yl)oxy)-3-oxopropoxy)phenyl)-l- (3-((tert-butoxycarbonyl)amino)-2-hvdroxypropyl)-2-methyl-lH-pyrazol-2-ium 2,2,2- trifluoroacetate Et 3 N (170 ⁇ , 1.2 mmol) was added to a stirred, room temperature mixture of l-(3-amino-2-hydroxypropyl)-4-(4-((S)-3-(tert-butoxy)-2-((l,3-dioxoisoindolin-2-yl)oxy)-3- oxopropoxy)phenyl)-2-methyl-lH-pyrazol-2-ium trifluoromethanesulfonate (830 mg, 1.2 mmol), and BOC-Anhydride (225 ⁇ , 0.
- Step E (3SV3-((ZV2-(T(1 S)-2-(4-(l-(3-amino-2-hvdroxypropyn-2-methyl-lH-pyrazol-2- ium-4-yl)phenoxy)-l-carboxyethoxy)imino)-2-(2-aminothiazol-4-yl)acetamido)-2,2- dimethyl-4-oxoazetidin-l-yl sulfate
- the title compound was prepared according to the procedure of Example 38 Steps E-G.
- Step A (R)-benzhydryl 3-(4-bromo-3-fluorophenoxy)-2-hydroxypropanoate
- (R)-3-(4-bromo-3-fluorophenoxy)-2-hydroxypropanoic acid 6 g, 22 mmol
- Et 3 N 4.5 ml, 32 mmol
- acetonitrile 40 ml
- (bromomethylene)dibenzene 6.9 g, 28 mmol)
- Step B benzhydryl (R)-3-(3-fluoro-4-(4.4.5.5-tetramethyl-1.3.2-dioxaborolan-2- yl)phenoxy)-2-hydroxypropanoate Potassium acetate (3.0 g, 31 mmol) was added to a stirred, room temperature mixture of (R)-benzhydryl 3-(4-bromo-3-fluorophenoxy)-2- hydroxypropanoate (4.6 g, 9.4 mmol), 4,4,4',4',5,5,5',5'-octamethyl-2,2'-bi(l,3,2- dioxaborolane) (3.2 g, 12 mmol), and di-t-BuDPPF-PdCl 2 (0.34 g, 0.52 mmol) in dioxane.
- Step C benzhydryl (2R)-3-(4-(l-(3-((tert-butoxycarbonyl)amino)-2-((tert-butyldimethyl- silyl)oxy)propyl)-lH-pyrazol-4-yl)-3-fluorophenoxy)-2-hvdroxypropanoate
- Aqueous potassium phosphate (2.6 ml, 2.6 mmol) was added to a room temperature mixture of (R)- benzhydryl 3-(3-fluoro-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenoxy)-2- hydroxypropanoate (480 mg, 0.97 mmol),
- S -tert-butyl (3-(4-bromo-lH-pyrazol-l-yl)-2- ((tert-butyldimethylsilyl)oxy)propyl)carbamate (380 mg, 0.88 mmol)
- Step D (Z)-2-((((2S)-l-(benzhvdryloxy)-3-(4-(l-(3-((tert-butoxycarbonvnamino)-2-((tert- butyldimethylsilyl)oxy)propyl)-lH-pyrazol-4-yl)-3-fluorophenoxy)-l-oxopropan-2- yl)oxy)imino)-2-(2-((tert-butoxycarbonyl)amino)thiazol-4-yl) acetic acid DEAD (0.12 ml, 0.78 mmol) was added to a stirred, cooled 0 °C mixture of benzhydryl (2R)-3-(4-(l-(3-((tert- butoxycarbonyl)amino)-2-((tert-butyldimethylsilyl)oxy)propyl)-lH-pyrazol-4-yl)-3- fluorophenoxy)-2-
- Step F 4-( 4-( ( SV 3 -(benzhvdryloxy)-2-( ( ((Z)-( 2-((tert-butoxycarbonvnamino)thiazol-4- yl)(carboxy)methylene)amino)oxy)-3-oxopropoxy)-2-fluorophenyl)-l-(3-((tert- butoxycarbonyl)amino)-2-hydroxypropyl)-2-methyl-lH-pyrazol-2-ium TBAF (0.28 g, 1.06 mmol) was added to a stirred, cooled 0 °C mixture of (Z)-2-((((S)-l-(benzhydryloxy)-3-(4- (l-((S)-3-((tert-butoxycarbonyl)amino)-2-((tert-butyldimethyl-silyl)oxy)propyl)-2-methyl- lH-214-pyrazol-4-yl)-3-fluor
- Step G 4-(4-( ( S)-3-(benzhvdryloxy)-2-( (((Z)- 1 -(2-((tert-butoxycarbonvnamino)thiazol-4- yl)-2-(((S)-2,2-dimethyl-4-oxo-l-(sulfooxy)azetidin-3-yl)amino)-2-oxoethylidene)- amino)oxy)-3-oxopropoxy)-2-fluorophenyl)-l-((S)-3-((tert-butoxycarbonyl)amino)-2- hydroxypropyl)-2-methyl-lH-pyrazol-2-ium To a 4-(4-((S)-3-(benzhydryloxy)-2-((((Z)-(2- ((tert-butoxycarbonyl)amino)thiazol-4-yl)(carboxy)methylene)amino)oxy)-3-oxopropoxy)-2
- Nl-((ethylimino)methylene)-N3,N3-dimethylpropane-l,3-diamine hydrochloride (102 mg, 0.53 mmol) was added. The reaction was stirred at -10-0 °C for 1 h, then concentrated. The resulting residue was purified by preparative reverse phase (C-18), eluting with acetonitrile/water + 0.1% TFA to give the title compound.
- Step H ((3 SV3-((ZV2-(((l S)-2-(4-(l-(3-amino-2-hvdroxypropylV2-methyl-lH-pyrazol-2- ium-4-yl)-3-fluorophenoxy)-l-carboxyethoxy)imino)-2-(2-aminothiazol-4-yl)acetamido)- 2,2-dimethyl-4-oxoazetidin-l-yl sulfate compound with formic acid (1 : 1) TFA (2ml) was added to a stirred, room temperature mixture of 4-(4-((S)-3-(benzhydryloxy)-2-((((Z)-l-(2- ((tert-butoxycarbonyl)amino)thiazol-4-yl)-2-(((S)-2,2-dimethyl-4-oxo-l-(sulfooxy)azetidin- 3-yl)amino
- Step A tert-butyl (2-((tert-butyldimethylsilvnoxy)-3-(4-(4.4.5.5-tetramethyl-1.3.2- dioxaborolan-2-vD-lH-pyrazol-l-vDpropyDcarbamate Potassium acetate (3.48 g, 36 mmol) was added to a stirred, room temperature mixture of 4,4,4',4',5,5,5',5'-octamethyl-2,2'- bi(l,3,2-dioxaborolane) (3.6 g, 14 mmol), tert-butyl (3-(4-bromo-lH-pyrazol-l-yl)-2-((tert- butyldimethylsilyl)oxy)propyl)carbamate (5.1 g, 12 mmol) and di-t-BuDPPF PdCl 2 (0.38 g, 0.591 mmol) in dioxane.
- reaction mixture was degassed with N 2 twice and stirred at 70°C for 4 h. Then the reaction mixture was filtered and the filtrate was concentrated. The resulting residue was purified by column chromatography on silica gel 120 g column, eluting with EtOAc/isohexane (0-50%) to give the title compound.
- Step B tert-butyl (2-hvdroxy-3-(4-(4.4.5.5-tetramethyl-1.3.2-dioxaborolan-2-vn-lH-pyrazol- 1 -yl)propyl)carbamate TBAF (0.61 g, 2.3 mmol) was added to a stirred, 0 °C mixture of tert- butyl (2-((tert-butyldimethylsilyl)oxy)-3-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)- lH-pyrazol-l-yl)propyl)carbamate (0.93 g, 1.9 mmol) in THF.
- Step C l-((tert-butoxycarbonyl)amino)-3-(4-(4,4,5,5-tetramethyl-L3,2-dioxaborolan-2-yl)- lH-pyrazol-l-yl)propan-2-yl methanesulfonate MsCl (0.104 ml, 1.33 mmol) was added in one portion to a stirred solution of tert-butyl (2-hydroxy-3-(4-(4,4,5,5-tetramethyl-l,3,2- dioxaborolan-2-yl)-lH-pyrazol-l-yl)propyl)carbamate (480 mg, 1.30 mmol) and Et 3 N (0.24 ml, 1.7 mmol) in dichloromethane (5 mL) at 0 °C under N 2 .
- reaction mixture was stirred at 0 °C for 1 h, then concentrated in vacuo and partitioned between water (50 mL) and ethyl acetate (40 mL). The aqueous layer was separated, and extracted with ethyl acetate (2 x 40 mL). The combined organic layers were washed with brine (40 mL), dried (magnesium sulfate) and concentrated in vacuo to give the title compound.
- Step D tert-butyl ( " 2-azido-3-( " 4-( " 4.4.5.5-tetramethyl-1.3.2-dioxaborolan-2-vn-lH-pyrazol-l- yl)propyl)carbamate Sodium azide (173 mg, 2.7 mmol) was added in one portion to a stirred solution of l-((tert-butoxycarbonyl)amino)-3-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2- yl)-lH-pyrazol-l-yl)propan-2-yl methanesulfonate (590 mg, 1.33 mmol)) in dimethyl formamide (5 mL).
- Step E tert-butyl (2-amino-3-(4-(4,4,5,5-tetramethyl-L3,2-dioxaborolan-2-yl)-lH-pyrazol-l- yl)propyl)carbamate Pd-C (280 mg, 0.27 mmol) was added to a room temperature mixture of tert-butyl (2-azido-3-(4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazol-l- yl)propyl)carbamate (525 mg, 1.34 mmol) in MeOH. The reaction mixture was stirred at room temperature under H 2 overnight. Then the mixture was filtered and the filtrate was concentrated to dryness to give the title compound. LC-MS [M + H] + : 367.24.
- Step F di-teit-butyl (3-(4-(4.4.5.5-tetramethyl-1.3.2-dioxaborolan-2-vn-lH-pyrazol-l- yl)propane-L2-diyl)dicarbamate BOC-Anhydride (0.34 ml, 1.5 mmol) was added to a stirred, room temperature mixture of tert-butyl (2-amino-3-(4-(4,4,5,5-tetramethyl-l,3,2- dioxaborolan-2-yl)-lH-pyrazol-l-yl)propyl)carbamate (570 mg, 1.3 mmol) in THF. The mixture was stirred at room temperature for 1 h, and then concentrated to give the title compound. LC-MS [M + H] + : 467.36.
- Step G benzhydryl (2R)-3-(4-(l-(2,3-bis((tert-butoxycarbonyl)amino)propyl)-lH-pyrazol-4- yl)-3-fluorophenoxy)-2-hydroxypropanoate
- (R)-benzhydryl 3-(4-bromo-3- fluorophenoxy)-2-hydroxypropanoate 590 mg, 1.3 mmol
- di-tert-butyl 3-(4-(4-(4,4,5,5- tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- lH-pyrazol- 1 -yl)propane- 1 ,2-diyl)dicarbamate (630 mg, 1.3 mmol) and di-t-BuDPPF PdCl 2 (52 mg, 0.079 mmol) in dioxane.
- Example 45 was prepared from the faster eluting isomer according to the procedure of Example 1 Steps B-H.
- Example 46 was prepared from the slower eluting isomer according to the procedure of Example 1 Steps B-H. LC-MS [M + H] + : m/z 714.36.
- (R)-tert- butyl 2-((tert-butyldimethylsilyl)oxy)-3-(3-fluoro-4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan- 2-yl)phenoxy)propanoate 2.6 g, 5.24 mmol
- tert-butyl (3-(3-amino-4-bromo-7H-pyrazol-l- yl)propyl)carbamate 2.0 g, 6.3 mmol
- Step B fert-Butyl (R)-3-(4-(3-(((allyloxy)carbonvnamino l-(3-((fert- butoxycarbonvDamino) propyl)-7H-pyrazol-4-yl)-3-fluorophenoxy)-2-((fert- butyldimethylsilyl)oxy)propanoate
- Step C fert-Butyl (R)-3-(4-(3-(((allyloxy)carbonyl)amino)-l-(3-((fert-butoxycarbonyl)- amino)propyl)-7H-pyrazol-4-yl)-3-fluorophenoxy)-2-hydroxypropanoate
- Step D fert-Butyl (S)-3-(4-(3-(((allyloxy)carbonyl)amino)-l-(3-((fert-butoxycarbonyl) amino)propyl)-7H-pyrazol-4-yl)-3-fluorophenoxy)-2-((L3-dioxoisoindolin-2-yl)oxy)- propanoate
- (R)-tert-butyl 3-(4-(3-(((allyloxy)carbonyl)amino)-l-(3-((tert- butoxycarbonyl) amino)propyl)-7H-pyrazol-4-yl)-3-fluorophenoxy)-2-hydroxypropanoate (1.13g, 2.0 mmol) in THF (5 ml) was added 2-hydroxyisoindoline-l,3-dione (0.38 g, 2.3 mmol), and triphenylphosphine (0.62 g
- Step E (S)-3-(((Allyloxy)carbonyl)amino)-4-(4-(2-(aminooxy)-3-(fert-butoxy)-3- oxopropoxy)-2-fluorophenyl)-l-(3-((fert-butoxycarbonyl)amino)propyl)-2-methyl-7H- pyrazol-2-ium triflate
- Step F (S.Z)-3-(((Allyloxy)carbonvnamino)-4-(4-(3-(fert-butoxy)-2-((((2-((fert-butoxy- carbonyl)amino)thiazol-4-yl)(carboxy)methylene)amino)oxy)-3-oxopropoxy)-2-fluoro- phenyl)- 1 -(3 -((fert-butoxycarbonyl)amino)propyl)-2-methyl-7H-pyrazol-2-ium triflate
- 2-(2-((tert-butoxycarbonyl)amino)thiazol-4-yl)-2-oxoacetic acid (0.16 g, 0.60 mmol)
- Step G (S)-3-((Z)-2-((((S)-3-(4-(3-(((Allyloxy)carbonvnamino)-l-(3-((fert-butoxy- carbonyl)amino)propyl)-2-methyl-7H-pyrazol-2-ium-4-yl)-3-fluorophenoxy)-l-(fert-butoxy)- l-oxopropan-2-yl)oxy)imino)-2-(2-((fert-butoxycarbonyl)amino)thiazol-4-yl)acetamido)-2,2- dimethyl-4-oxoazetidin-l-yl sulfate
- Step I (S)-3-((Z)-2-(((S)-2-(4-(3-Amino-l-(3-aminopropyn-2-methyl-7H-pyrazol-2-ium-4- yl)-3 -fluorophenoxy)- l-carboxyethoxy)imino)-2-(2-aminothiazol-4-yl)acetamido)-2,2- dimethyl-4-oxoazetidin- 1 -yl sulfate
- Example 51 A LC-MS [M+l]: m/z 727.36.
- 1 HNMR 500 MHz, D 2 0) ⁇ ⁇ 8.49 (IH, s), 8.43 (IH, s), 7.49 (2H, d), 7.02 (2H, s), 4.57-4.53 (3H, m), 4.48 (IH, d), 4.40 (IH, d), 4.11 (3H, s), 3.12(2H, t), 2.32 (2H, m), 1.60 (3H, s), 1.38 (3H, s), 1.17 (3H, s).
- Example 51B LC-MS [M+l]: m/z 727.36.
- Step A fert-Butyl (R)-3-(4-(l-((S)-3-((fert-butoxycarbonvnamino)-2-((fert-butyl- dimethyl silyl) oxy)propyl)-7H-pyrazol-4-yl)phenoxy)-2-((fert-butyldimethylsilyl)- oxy)propanoate
- tert-butyl- 2-hydroxy-3-(4- (4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenoxy)propanoate (1 g, 2.8 mmol)
- Step B fert-Butyl (S)-3-(4-(l-((S)-3-((fert-butoxycarbonvnamino)-2-((fert-butyldimethyl- silyl) oxy)propyl)-7H-pyrazol-4-yl)phenoxy)-2-((L3-dioxoisoindolin-2-yl)oxy)propanoate
- (2R)-tert-butyl 3-(4-(l-(3-((tert-butoxycarbonyl)amino)-2-((tert-butyl- dimethyl silyl) oxy) ropyl)-77J-pyrazol-4-yl)phenoxy)-2-hydroxypropanoate (1.12 g, 1.9 mmol) in THF (10 ml) was added 2-hydroxyisoindoline-l,3-dione (0.37 g, 2.3 mmol), triphenylphosphine (0.60
- Step C 4-(4-((S)-3-(fert-Butoxy)-2-((L3-dioxoisoindolin-2-yl)oxy)-3-oxopropoxy)phenyl)- l-((S)-3-((fert-butoxycarbonyl)amino)-2-((fert-butyldimethylsilyl)oxy)propyl)-2-((l-(fert- butoxycarbonyl)azetidin-3-yl)methyl)-7H-pyrazol-2-ium, trifluoromethanesulfonate
- a solution of tert-butyl-3-(hydroxymethyl)azetidine-l-carboxylate (510 mg, 2.7 mmol) in CH 2 CI 2 (5 ml) was cooled to -78 °C, and trifluoromethanesulfonic anhydride (0.56 ml, 3.4 mmol) and Hunig's base (0.95 ml,
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Oncology (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Communicable Diseases (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description

































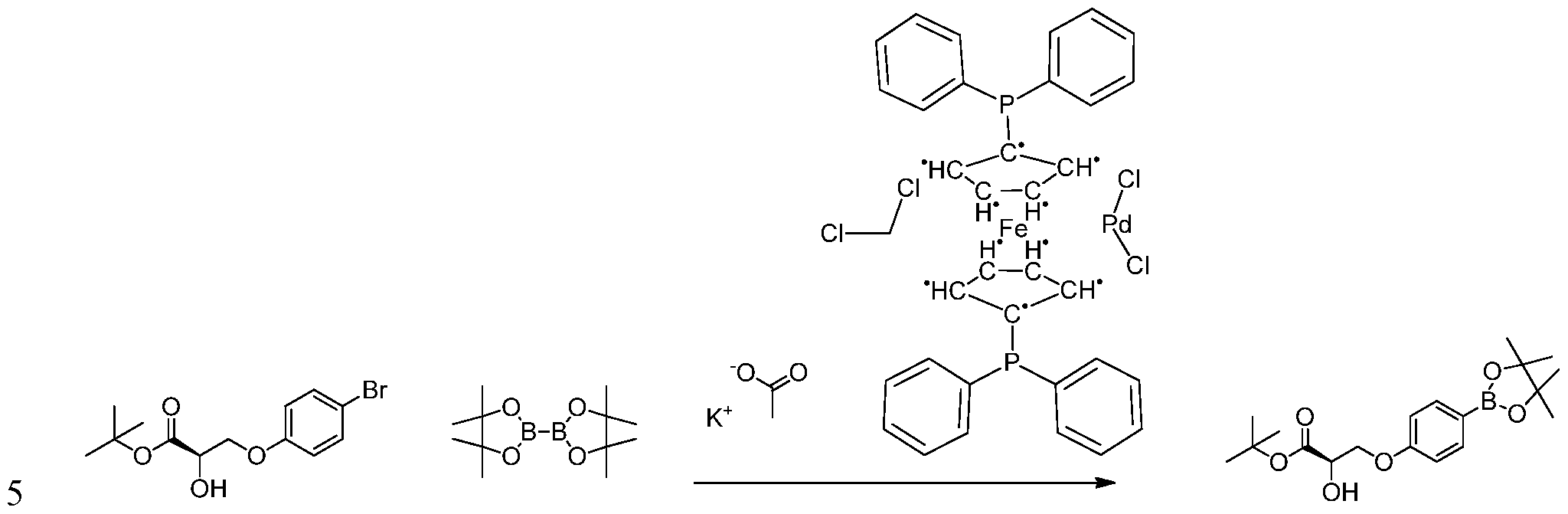
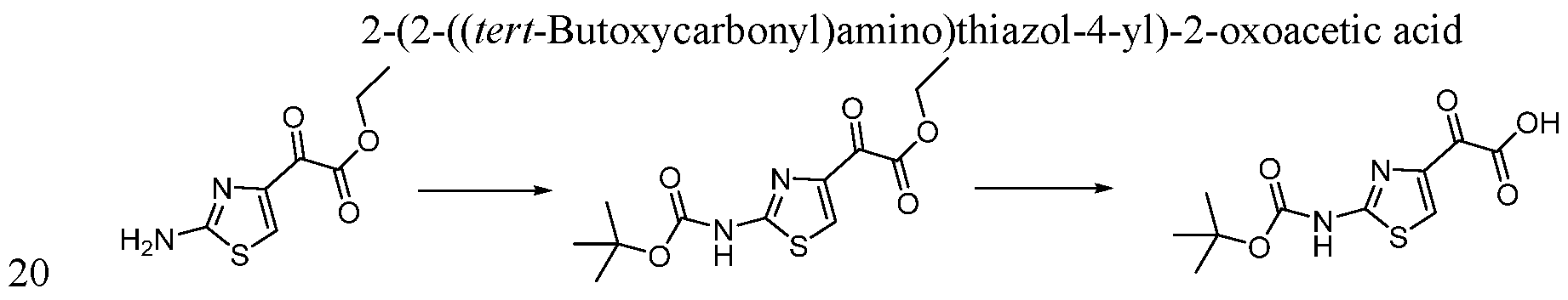




















































































































Claims













Priority Applications (12)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2016371600A AU2016371600B2 (en) | 2015-12-15 | 2016-12-12 | Biaryl monobactam compounds and methods of use thereof for the treatment of bacterial infections |
| US15/775,487 US20180339983A1 (en) | 2015-12-15 | 2016-12-12 | Biaryl monobactam compounds and methods of use thereof for the treatment of bacterial infections |
| BR112018010962A BR112018010962A8 (en) | 2015-12-15 | 2016-12-12 | COMPOUND, TRIFLUOROACETIC ACID SALT, PHARMACEUTICAL COMPOSITION, METHOD FOR TREATING A BACTERIAL INFECTION, AND, USE OF A COMPOUND |
| RU2018123484A RU2746129C2 (en) | 2015-12-15 | 2016-12-12 | Biarile monobactam compounds and methods of their application for treatment of bacterial infections |
| JP2018530835A JP7184646B2 (en) | 2015-12-15 | 2016-12-12 | Biaryl monobactam compounds and methods of their use for treating bacterial infections |
| KR1020187016482A KR20180093925A (en) | 2015-12-15 | 2016-12-12 | Biaryl monobactam compounds for the treatment of bacterial infections and methods for their use |
| MX2018005382A MX2018005382A (en) | 2015-12-15 | 2016-12-12 | Biaryl monobactam compounds and methods of use thereof for the treatment of bacterial infections. |
| CN201680074091.0A CN108368040A (en) | 2015-12-15 | 2016-12-12 | Diaryl monocycle beta-lactam compound and its method for treating bacterium infection |
| EP21204101.6A EP3978472A1 (en) | 2015-12-15 | 2016-12-12 | Biaryl monabactam compounds and methods of use thereof for the treatment of bacterial infections |
| CA3008006A CA3008006C (en) | 2015-12-15 | 2016-12-12 | Biaryl monobactam compounds and methods of use thereof for the treatment of bacterial infections |
| EP16876436.3A EP3390357B1 (en) | 2015-12-15 | 2016-12-12 | Biaryl monobactam compounds for the treatment of bacterial infections |
| US16/912,146 US11230543B2 (en) | 2015-12-15 | 2020-06-25 | Biaryl monobactam compounds and methods of use thereof for the treatment of bacterial infections |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562267855P | 2015-12-15 | 2015-12-15 | |
| US62/267,855 | 2015-12-15 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US15/775,487 A-371-Of-International US20180339983A1 (en) | 2015-12-15 | 2016-12-12 | Biaryl monobactam compounds and methods of use thereof for the treatment of bacterial infections |
| US16/912,146 Continuation US11230543B2 (en) | 2015-12-15 | 2020-06-25 | Biaryl monobactam compounds and methods of use thereof for the treatment of bacterial infections |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017106064A1 true WO2017106064A1 (en) | 2017-06-22 |
Family
ID=59057420
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2016/066064 WO2017106064A1 (en) | 2015-12-15 | 2016-12-12 | Biaryl monobactam compounds and methods of use thereof for the treatment of bacterial infections |
Country Status (12)
| Country | Link |
|---|---|
| US (2) | US20180339983A1 (en) |
| EP (2) | EP3390357B1 (en) |
| JP (1) | JP7184646B2 (en) |
| KR (1) | KR20180093925A (en) |
| CN (1) | CN108368040A (en) |
| AU (1) | AU2016371600B2 (en) |
| BR (1) | BR112018010962A8 (en) |
| CA (1) | CA3008006C (en) |
| MA (1) | MA44083A (en) |
| MX (1) | MX2018005382A (en) |
| RU (1) | RU2746129C2 (en) |
| WO (1) | WO2017106064A1 (en) |
Cited By (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN108362789A (en) * | 2018-01-19 | 2018-08-03 | 珠海优润医药科技有限公司 | A kind of high-efficiency liquid chromatography method for detecting of AVM hereinafter Batan sodium optical isomer |
| WO2019070492A1 (en) | 2017-10-02 | 2019-04-11 | Merck Sharp & Dohme Corp. | Chromane monobactam compounds for the treatment of bacterial infections |
| WO2019144969A1 (en) | 2018-01-29 | 2019-08-01 | 南京明德新药研发股份有限公司 | Monocyclic β-lactam compound for treating bacterial infection |
| US10385028B2 (en) | 2017-12-14 | 2019-08-20 | Nmd Pharma A/S | Compounds for the treatment of neuromuscular disorders |
| WO2020048828A1 (en) | 2018-09-03 | 2020-03-12 | Bayer Pharma Aktiengesellschaft | 5-heteroaryl-3,9-diazaspiro[5.5]undecane compounds |
| WO2020048827A1 (en) | 2018-09-03 | 2020-03-12 | Bayer Aktiengesellschaft | 1, 3, 9-triazaspiro[5.5] undecan-2-one compounds |
| CN111171014A (en) * | 2018-11-13 | 2020-05-19 | 南京圣和药业股份有限公司 | Monocyclic lactam compounds and uses thereof |
| US10934244B2 (en) | 2015-06-15 | 2021-03-02 | Nmd Pharma A/S | Compounds for use in treating neuromuscular disorders |
| US11034669B2 (en) | 2018-11-30 | 2021-06-15 | Nuvation Bio Inc. | Pyrrole and pyrazole compounds and methods of use thereof |
| WO2021121387A1 (en) * | 2019-12-19 | 2021-06-24 | 南京明德新药研发有限公司 | Application of compound in drug preparation |
| US11147788B2 (en) | 2017-12-14 | 2021-10-19 | Nmd Pharma A/S | Compounds for the treatment of neuromuscular disorders |
| US11230543B2 (en) | 2015-12-15 | 2022-01-25 | Merck Sharp & Dohme Corp. | Biaryl monobactam compounds and methods of use thereof for the treatment of bacterial infections |
| US11591284B2 (en) | 2017-12-14 | 2023-02-28 | Nmd Pharma A/S | Compounds for the treatment of neuromuscular disorders |
| WO2023091438A1 (en) | 2021-11-18 | 2023-05-25 | Merck Sharp & Dohme Llc | Chromane amidine monobactam antibiotics |
| US11730714B2 (en) | 2017-12-14 | 2023-08-22 | Nmd Pharma A/S | Compounds for the treatment of neuromuscular disorders |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013075068A1 (en) | 2011-11-18 | 2013-05-23 | Regeneron Pharmaceuticals, Inc. | Polymer protein microparticles |
| CN111303144B (en) * | 2019-12-13 | 2020-11-27 | 苏州信诺维医药科技有限公司 | Compound for treating bacterial infection |
| WO2022027439A1 (en) * | 2020-08-06 | 2022-02-10 | Ningxia Academy Of Agriculture And Forestry Sciences | β-LACTAM COMPOUNDS, THEIR PREPARATION AND USE AS ANTIBACTERIAL AGENTS |
| CN112661667B (en) * | 2020-12-28 | 2023-02-03 | 浦拉司科技(上海)有限责任公司 | Preparation method of trifluoroacetamidine |
| CN115463219A (en) * | 2021-11-09 | 2022-12-13 | 中国医学科学院医药生物技术研究所 | Pharmaceutical composition containing beta-lactam compound and application thereof |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4537886A (en) * | 1982-03-31 | 1985-08-27 | Beecham Group P.L.C. | β-lactam antibacterial agents and compositions containing them |
| EP0229012A1 (en) | 1986-01-06 | 1987-07-15 | E.R. Squibb & Sons, Inc. | Monosulfactams |
| WO2007065288A2 (en) | 2005-12-07 | 2007-06-14 | Basilea Pharmaceutica Ag | Useful combinations of monobactam antibiotics with beta-lactamase inhibitors |
| WO2012073138A1 (en) | 2010-11-29 | 2012-06-07 | Pfizer Inc. | Monobactams |
| WO2013110643A1 (en) | 2012-01-24 | 2013-08-01 | Aicuris Gmbh & Co. Kg | Amidine substituted beta - lactam compounds, their preparation and use as antibacterial agents |
| US20130296555A1 (en) * | 2012-03-30 | 2013-11-07 | Cubist Pharmaceuticals, Inc. | Beta-lactamase inhibitors |
| US20140275007A1 (en) | 2013-03-14 | 2014-09-18 | Rempex Pharmaceuticals, Inc. | Oxamazin antibiotics |
| US20150266867A1 (en) | 2014-03-24 | 2015-09-24 | Novartis Ag | Monobactam organic compounds for the treatment of bacterial infections |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20160326157A1 (en) | 2014-01-06 | 2016-11-10 | President And Fellows Of Harvard College | Monobactams and methods of their synthesis and use |
| KR20180093925A (en) | 2015-12-15 | 2018-08-22 | 머크 샤프 앤드 돔 코포레이션 | Biaryl monobactam compounds for the treatment of bacterial infections and methods for their use |
-
2016
- 2016-12-12 KR KR1020187016482A patent/KR20180093925A/en not_active Application Discontinuation
- 2016-12-12 US US15/775,487 patent/US20180339983A1/en not_active Abandoned
- 2016-12-12 JP JP2018530835A patent/JP7184646B2/en active Active
- 2016-12-12 EP EP16876436.3A patent/EP3390357B1/en active Active
- 2016-12-12 RU RU2018123484A patent/RU2746129C2/en active
- 2016-12-12 BR BR112018010962A patent/BR112018010962A8/en active Search and Examination
- 2016-12-12 EP EP21204101.6A patent/EP3978472A1/en active Pending
- 2016-12-12 CA CA3008006A patent/CA3008006C/en active Active
- 2016-12-12 MA MA044083A patent/MA44083A/en unknown
- 2016-12-12 WO PCT/US2016/066064 patent/WO2017106064A1/en active Application Filing
- 2016-12-12 AU AU2016371600A patent/AU2016371600B2/en active Active
- 2016-12-12 CN CN201680074091.0A patent/CN108368040A/en active Pending
- 2016-12-12 MX MX2018005382A patent/MX2018005382A/en unknown
-
2020
- 2020-06-25 US US16/912,146 patent/US11230543B2/en active Active
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4537886A (en) * | 1982-03-31 | 1985-08-27 | Beecham Group P.L.C. | β-lactam antibacterial agents and compositions containing them |
| EP0229012A1 (en) | 1986-01-06 | 1987-07-15 | E.R. Squibb & Sons, Inc. | Monosulfactams |
| WO2007065288A2 (en) | 2005-12-07 | 2007-06-14 | Basilea Pharmaceutica Ag | Useful combinations of monobactam antibiotics with beta-lactamase inhibitors |
| WO2012073138A1 (en) | 2010-11-29 | 2012-06-07 | Pfizer Inc. | Monobactams |
| WO2013110643A1 (en) | 2012-01-24 | 2013-08-01 | Aicuris Gmbh & Co. Kg | Amidine substituted beta - lactam compounds, their preparation and use as antibacterial agents |
| US20150045340A1 (en) | 2012-01-24 | 2015-02-12 | Aicuris Gmbh & Co. Kg | Amidine substituted beta-lactam compounds, their preparation and use as antibacterial agents |
| US20130296555A1 (en) * | 2012-03-30 | 2013-11-07 | Cubist Pharmaceuticals, Inc. | Beta-lactamase inhibitors |
| US20140275007A1 (en) | 2013-03-14 | 2014-09-18 | Rempex Pharmaceuticals, Inc. | Oxamazin antibiotics |
| US20150266867A1 (en) | 2014-03-24 | 2015-09-24 | Novartis Ag | Monobactam organic compounds for the treatment of bacterial infections |
Non-Patent Citations (9)
| Title |
|---|
| "Remington - The Science and Practice of Pharmacy", 2006 |
| BIOORGANIC AND MEDICINAL CHEMSTRY LETTERS, vol. 22, 2012, pages 5989 |
| DRAWZBONOMO, CLIN. MICRO. REV., vol. 23, 2010, pages 160 - 201 |
| HANAKI ET AL., ANTIMICROBIAL AGENTS AND CHEMOTHERAPY, vol. 30, 1995, pages 20 - 26 |
| J. AM. CHEM. SOC., vol. 121, 1999, pages 6086 - 6087 |
| J. MEDICINAL CHEMISTRY, vol. 56, 2013, pages 5541 - 5552 |
| PAYNE ET AL., ANTIMICROBIAL AGENTS AND CHEMOTHERAPY, vol. 38, 1994, pages 767 - 772 |
| S. G. WALEY: "P-lactamase: mechanisms of action, in The Chemistry of β-Lactams", 1992, CHAPMAN AND HALL, pages: 198 - 228 |
| See also references of EP3390357A4 |
Cited By (36)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10934244B2 (en) | 2015-06-15 | 2021-03-02 | Nmd Pharma A/S | Compounds for use in treating neuromuscular disorders |
| US11230543B2 (en) | 2015-12-15 | 2022-01-25 | Merck Sharp & Dohme Corp. | Biaryl monobactam compounds and methods of use thereof for the treatment of bacterial infections |
| CN111201021A (en) * | 2017-10-02 | 2020-05-26 | 默沙东公司 | Chroman monocyclic β -lactams for use in the treatment of bacterial infections |
| JP2020536082A (en) * | 2017-10-02 | 2020-12-10 | メルク・シャープ・アンド・ドーム・コーポレーションMerck Sharp & Dohme Corp. | Chroman monobactam compounds for the treatment of bacterial infections |
| JP7252948B2 (en) | 2017-10-02 | 2023-04-05 | メルク・シャープ・アンド・ドーム・エルエルシー | Chromanmonobactam compounds for treating bacterial infections |
| EP3691639A4 (en) * | 2017-10-02 | 2021-03-31 | Merck Sharp & Dohme Corp. | Chromane monobactam compounds for the treatment of bacterial infections |
| WO2019070492A1 (en) | 2017-10-02 | 2019-04-11 | Merck Sharp & Dohme Corp. | Chromane monobactam compounds for the treatment of bacterial infections |
| US11433055B2 (en) | 2017-10-02 | 2022-09-06 | Merck Sharp & Dohme Llc | Chromane monobactam compounds for the treatment of bacterial infections |
| KR102425959B1 (en) | 2017-10-02 | 2022-07-27 | 머크 샤프 앤드 돔 코포레이션 | Chroman Monobactam Compound for Treatment of Bacterial Infections |
| KR20200058520A (en) * | 2017-10-02 | 2020-05-27 | 머크 샤프 앤드 돔 코포레이션 | Chroman monobactam compounds for treatment of bacterial infections |
| US11147788B2 (en) | 2017-12-14 | 2021-10-19 | Nmd Pharma A/S | Compounds for the treatment of neuromuscular disorders |
| US10385028B2 (en) | 2017-12-14 | 2019-08-20 | Nmd Pharma A/S | Compounds for the treatment of neuromuscular disorders |
| US11730714B2 (en) | 2017-12-14 | 2023-08-22 | Nmd Pharma A/S | Compounds for the treatment of neuromuscular disorders |
| US11591284B2 (en) | 2017-12-14 | 2023-02-28 | Nmd Pharma A/S | Compounds for the treatment of neuromuscular disorders |
| CN108362789B (en) * | 2018-01-19 | 2020-09-01 | 珠海优润医药科技有限公司 | High performance liquid chromatography detection method for abamectin sodium optical isomer |
| CN108362789A (en) * | 2018-01-19 | 2018-08-03 | 珠海优润医药科技有限公司 | A kind of high-efficiency liquid chromatography method for detecting of AVM hereinafter Batan sodium optical isomer |
| US11459323B2 (en) | 2018-01-29 | 2022-10-04 | Medshine Discovery Inc. | Monocyclic β-lactam compound for treating bacterial infection |
| CN111511737B (en) * | 2018-01-29 | 2022-10-18 | 南京明德新药研发有限公司 | Monocyclic beta-lactam compounds for the treatment of bacterial infections |
| WO2019144969A1 (en) | 2018-01-29 | 2019-08-01 | 南京明德新药研发股份有限公司 | Monocyclic β-lactam compound for treating bacterial infection |
| JP2021512866A (en) * | 2018-01-29 | 2021-05-20 | メッドシャイン ディスカバリー インコーポレイテッド | Monocyclic β-lactam compounds for treating bacterial infections |
| JP7280273B2 (en) | 2018-01-29 | 2023-05-23 | メッドシャイン ディスカバリー インコーポレイテッド | Monocyclic β-Lactam Compounds for Treating Bacterial Infections |
| EP3747883A4 (en) * | 2018-01-29 | 2020-12-16 | Medshine Discovery Inc. | Monocyclic b-lactam compound for treating bacterial infection |
| CN111511737A (en) * | 2018-01-29 | 2020-08-07 | 南京明德新药研发有限公司 | Monocyclic β -lactam compounds for the treatment of bacterial infections |
| WO2020048828A1 (en) | 2018-09-03 | 2020-03-12 | Bayer Pharma Aktiengesellschaft | 5-heteroaryl-3,9-diazaspiro[5.5]undecane compounds |
| WO2020048827A1 (en) | 2018-09-03 | 2020-03-12 | Bayer Aktiengesellschaft | 1, 3, 9-triazaspiro[5.5] undecan-2-one compounds |
| US20220002288A1 (en) * | 2018-11-13 | 2022-01-06 | Nanjing Sanhome Pharmaceutical Co., Ltd. | Monobactam compounds and use therefor |
| WO2020098635A1 (en) | 2018-11-13 | 2020-05-22 | 南京圣和药业股份有限公司 | Monobactam compounds and use therefor |
| CN111171014A (en) * | 2018-11-13 | 2020-05-19 | 南京圣和药业股份有限公司 | Monocyclic lactam compounds and uses thereof |
| JP2022507470A (en) * | 2018-11-13 | 2022-01-18 | 南京聖和薬業股▲ふん▼有限公司 | Monobactam compounds and their use |
| EP3882246A4 (en) * | 2018-11-13 | 2021-12-29 | Nanjing Sanhome Pharmaceutical Co., Ltd. | Monobactam compounds and use therefor |
| CN111171014B (en) * | 2018-11-13 | 2023-11-07 | 南京圣和药业股份有限公司 | Monocyclolactam compounds and uses thereof |
| US11034669B2 (en) | 2018-11-30 | 2021-06-15 | Nuvation Bio Inc. | Pyrrole and pyrazole compounds and methods of use thereof |
| CN114828850A (en) * | 2019-12-19 | 2022-07-29 | 南京明德新药研发有限公司 | Application of compound in pharmacy |
| WO2021121387A1 (en) * | 2019-12-19 | 2021-06-24 | 南京明德新药研发有限公司 | Application of compound in drug preparation |
| WO2023091438A1 (en) | 2021-11-18 | 2023-05-25 | Merck Sharp & Dohme Llc | Chromane amidine monobactam antibiotics |
| US11932637B2 (en) | 2021-11-18 | 2024-03-19 | Merck Sharp & Dohme Llc | Chromane amidine monobactam compounds for the treatment of bacterial infections |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3978472A1 (en) | 2022-04-06 |
| AU2016371600A1 (en) | 2018-05-10 |
| KR20180093925A (en) | 2018-08-22 |
| EP3390357B1 (en) | 2021-11-10 |
| EP3390357A1 (en) | 2018-10-24 |
| RU2018123484A (en) | 2020-01-17 |
| BR112018010962A8 (en) | 2023-04-11 |
| CN108368040A (en) | 2018-08-03 |
| EP3390357A4 (en) | 2019-06-12 |
| JP2018537483A (en) | 2018-12-20 |
| CA3008006A1 (en) | 2017-06-22 |
| AU2016371600B2 (en) | 2021-02-18 |
| RU2746129C2 (en) | 2021-04-07 |
| MX2018005382A (en) | 2018-08-16 |
| US20180339983A1 (en) | 2018-11-29 |
| JP7184646B2 (en) | 2022-12-06 |
| RU2018123484A3 (en) | 2020-03-26 |
| US20200361928A1 (en) | 2020-11-19 |
| CA3008006C (en) | 2024-04-09 |
| US11230543B2 (en) | 2022-01-25 |
| MA44083A (en) | 2021-06-02 |
| BR112018010962A2 (en) | 2018-12-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2016371600B2 (en) | Biaryl monobactam compounds and methods of use thereof for the treatment of bacterial infections | |
| AU2017228870B2 (en) | Bicyclic aryl monobactam compounds and methods of use thereof for the treatment of bacterial infections | |
| AU2018345523B2 (en) | Chromane monobactam compounds for the treatment of bacterial infections | |
| JP2018138609A (en) | Naphthyridinedione derivatives |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16876436 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2018/005382 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 2016371600 Country of ref document: AU Date of ref document: 20161212 Kind code of ref document: A |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112018010962 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 3008006 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 20187016482 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2018530835 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2018123484 Country of ref document: RU Ref document number: 2016876436 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2016876436 Country of ref document: EP Effective date: 20180716 |
|
| ENP | Entry into the national phase |
Ref document number: 112018010962 Country of ref document: BR Kind code of ref document: A2 Effective date: 20180529 |