WO2017050938A1 - Compounds and methods for inhibiting jak - Google Patents
Compounds and methods for inhibiting jak Download PDFInfo
- Publication number
- WO2017050938A1 WO2017050938A1 PCT/EP2016/072616 EP2016072616W WO2017050938A1 WO 2017050938 A1 WO2017050938 A1 WO 2017050938A1 EP 2016072616 W EP2016072616 W EP 2016072616W WO 2017050938 A1 WO2017050938 A1 WO 2017050938A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- methyl
- methoxy
- pyrazol
- indol
- amino
- Prior art date
Links
- BQZWKEHNKAZASP-JOCHJYFZSA-N CC[C@H](C(Nc(cccc12)c1[nH]cc2-c1nc(Nc2c[n](C)nc2OC)ncc1C)=O)N1CCN(C)CC1 Chemical compound CC[C@H](C(Nc(cccc12)c1[nH]cc2-c1nc(Nc2c[n](C)nc2OC)ncc1C)=O)N1CCN(C)CC1 BQZWKEHNKAZASP-JOCHJYFZSA-N 0.000 description 1
- OSQOEGUZLYZJGA-UHFFFAOYSA-N Cc1cnc(Nc2c[n](C)nc2OC)nc1-c(c1ccc2)c[nH]c1c2NC(CN1CCN(C)CC1)=O Chemical compound Cc1cnc(Nc2c[n](C)nc2OC)nc1-c(c1ccc2)c[nH]c1c2NC(CN1CCN(C)CC1)=O OSQOEGUZLYZJGA-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/13—Crystalline forms, e.g. polymorphs
Definitions
- JAK Japanese-associated kinase
- JAK1 non-receptor tyrosine kinases
- JAK2 JAK3
- Tyk2 non-receptor tyrosine kinases
- Cytokine and/or growth factor binding to cell-surface receptors facilitates activation of receptor- associated JAK kinases by autophosphorylation.
- Activated JAKs directly phosphorylate members of the STAT (signal transducers and activators of transcription) family of transcription factors (STAT1 , 2, 3, 4, 5a, 5b and 6) promoting their translocation to the nucleus and the transcriptional activation of target genes.
- STAT signal transducers and activators of transcription
- STAT3 Constitutive activation (i.e., tyrosine phosphorylation) of members of the STAT family, in particular STAT3, has been documented in a wide range of cancers and hyperproliferative disorders, and associated with poor prognosis in several cancers (Yu H, Jove R., Nat. Rev. Cancer 2004;4:97-105). Persistently activated STAT3 has been shown to be oncogenic (Bromberg JF, et al. Cell 1999;98:295-303) and to drive the expression of cellular proteins contributing to central processes in cancer progression (survival, proliferation, invasion, angiogenesis) (Yu and Jove, 2004, supra).
- cytokines typically members of the interleukin-6 (IL-6) cytokine family (Grivennikov, S. and Karin, M.
- JAK1 the key JAK kinase responsible for STAT3 activation
- Kim SM the key JAK kinase responsible for STAT3 activation
- JAK1/STAT3 signaling in multiple human tumors, the pathway has also been shown to be activated as a feedback resistance mechanism in response to inhibition of driver oncogenic pathways in cancer cells, such as the mutated epidermal growth factor receptor (EGFR) in non-small cell lung cancer (NSCLC), or the MAPK pathway in KRAS mutant tumors (Lee et al., Cancer Cell 2014;26;207-221 .; VanSchaeybroeck et al., Cell reports 2014;7; 1940-1955).
- driver oncogenic pathways in cancer cells, such as the mutated epidermal growth factor receptor (EGFR) in non-small cell lung cancer (NSCLC), or the MAPK pathway in KRAS mutant tumors.
- EGFR epidermal growth factor receptor
- NSCLC non-small cell lung cancer
- MAPK pathway in KRAS mutant tumors
- cancer cachexia is a significant contributor to increased mortality and poor response to chemotherapy in patients with advanced cancer.
- Elevated levels of inflammatory cytokines, such as IL-6, which signal through the JAK/STAT pathway have been shown to play a causal role, indicating the potential benefit of JAK1 inhibition in ameliorating cancer cachexia.
- JAK1 inhibition may be useful in treating a number of immune disorders, such as bone marrow disorders, rheumatoid arthritis, psoriasis, Crohn's disease, lupus and multiple sclerosis.
- JAK inhibitors Collectively, the observations of JAKs critical role in proliferative and immune disorders highlight broad potential for JAK inhibition as a therapeutic modality in a number of diseases and disorders. Accordingly, disclosed are compounds that are JAK inhibitors.
- R 1 is methyl or ethyl
- R 2 is selected from methyl, ethyl, methoxy and ethoxy
- R 3 is selected from hydrogen, chlorine, fluorine, bromine and methyl
- R 4 is selected from methyl, ethyl and -CH2OCH3;
- R 5 and R 6 are each individually methyl or hydrogen
- R 7 is selected from methyl, ethyl, -(Chb ⁇ OH and -(Ch ⁇ OCHs, or a pharmaceutically acceptable salt thereof.
- pharmaceutical compositions comprising a compound of Formula (I), or a pharmaceutically acceptable salt or solid form thereof, and a
- a JAK-related disorder in a subject in need thereof comprising administering to the subject an effective amount of a compound of Formula (I), or a pharmaceutically acceptable salt or solid form thereof.
- a compound of Formula (I), or a pharmaceutically acceptable salt or solid form thereof for use in treating a JAK-related disorder.
- compositions comprising a compound of Formula (I), or a pharmaceutically acceptable salt or solid form thereof, for use in treating a JAK-related disorder.
- a compound of Formula (I), or a pharmaceutically acceptable salt or solid form thereof in the manufacture of a medicament for treating a JAK-related disorder.
- a compound as of Formula (I), or a pharmaceutically acceptable salt or solid form thereof in combination with an anti-cancer therapeutic agent, or a pharmaceutically acceptable salt thereof.
- compositions comprising a compound of Formula (I), or a pharmaceutically acceptable salt or solid form thereof, in combination with an anti-cancer therapeutic agent, or a pharmaceutically acceptable salt thereof, for use in treating cancer.
- a compound of Formula (I), or a pharmaceutically acceptable salt or solid form thereof in combination with an anti-cancer therapeutic agent, or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for treating cancer.
- disclosed are methods of treating cancer cachexia in a subject in need thereof comprising administering to the subject an effective amount of a compound of Formula (I), or a pharmaceutically acceptable salt or solid form thereof.
- a compound of Formula (I), or a pharmaceutically acceptable salt or solid form thereof for use in treating cancer cachexia.
- pharmaceutical compositions comprising a compound of Formula (I), or a pharmaceutically acceptable salt or solid form thereof, for use in treating cancer cachexia.
- a compound of Formula (I), or a pharmaceutically acceptable salt or solid form thereof in the manufacture of a medicament for treating cancer cachexia.
- a method of treating an immune disorder in a subject in need thereof comprising administering to the subject an effective amount of a compound of Formula (I), or a pharmaceutically acceptable salt or solid form thereof.
- a compound of Formula (I), or a pharmaceutically acceptable salt or solid form thereof for use in treating an immune disorder.
- compositions comprising a compound of Formula (I), or a pharmaceutically acceptable salt or solid form thereof, for use in treating an immune disorder.
- a compound of Formula (I), or a pharmaceutically acceptable salt or solid form thereof in the manufacture of a medicament for treating an immune disorder.
- a method of inhibiting JAK in a subject in need thereof comprising administering to the subject an effective amount of a compound of Formula (I), or a pharmaceutically acceptable salt or solid form thereof.
- a compound of Formula (I), or a pharmaceutically acceptable salt or solid form thereof, for use in inhibiting JAK is disclosed.
- compositions comprising a compound of Formula (I), or a pharmaceutically acceptable salt or solid form thereof, for use in inhibiting JAK.
- a compound of Formula (I), or a pharmaceutically acceptable salt or solid form thereof in the manufacture of a medicament for inhibiting JAK.
- Figure 1 illustrates the powder X-ray diffraction diagram of Form A (2R)-N-(3- ⁇ 2-[(3- methoxy-1 -methyl-1 H-pyrazol-4-yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin- yl)propanamide.
- Figure 2 illustrates the differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) traces of Form A (2R)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 H-pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide.
- DSC differential scanning calorimetry
- TGA thermogravimetric analysis
- Figure 3 illustrates the powder X-ray diffraction diagram of Form B (2R)-N-(3- ⁇ 2-[(3- methoxy-1 -methyl-1 H-pyrazol-4-yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1 yl)propanamide.
- Figure 4 illustrates the differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) traces of Form B (2R)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 H-pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide.
- DSC differential scanning calorimetry
- TGA thermogravimetric analysis
- Figure 5 illustrates the powder X-ray diffraction diagram of Form C (2R)-N-(3- ⁇ 2-[(3- methoxy-1 -methyl-1 H-pyrazol-4-yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1 yl)propanamide.
- Figure 6 illustrates the differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) traces of Form C (2R)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 H-pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide.
- DSC differential scanning calorimetry
- TGA thermogravimetric analysis
- Figure 7 illustrates the powder X-ray diffraction diagram of Form D (2R)-N-(3- ⁇ 2-[(3- methoxy-1 -methyl-1 H-pyrazol-4-yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1 yl)propanamide.
- Figure 8 illustrates the differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) traces of Form D (2R)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 H-pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide.
- DSC differential scanning calorimetry
- TGA thermogravimetric analysis
- Figure 9 illustrates the powder X-ray diffraction diagram of Form A (2R)-N-(3- ⁇ 2-[(3- methoxy-1 -methyl-1 H-pyrazol-4-yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1 yl)propanamide saccharine salt.
- Figure 10 illustrates the differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) traces of Form A (2R)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 H-pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide saccharine salt.
- DSC differential scanning calorimetry
- TGA thermogravimetric analysis
- Figure 11 illustrates the powder X-ray diffraction diagram of Form B (2R)-N-(3- ⁇ 2-[(3- methoxy-1 -methyl-1 H-pyrazol-4-yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1 yl)propanamide saccharine salt.
- Figure 12 illustrates the differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) traces of Form B (2R)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 H-pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide saccharine salt.
- DSC differential scanning calorimetry
- TGA thermogravimetric analysis
- Figure 13 illustrates the powder X-ray diffraction diagram of Form C (2R)-N-(3- ⁇ 2-[(3- methoxy-1 -methyl-1 H-pyrazol-4-yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1 yl)propanamide saccharine salt.
- Figure 14 illustrates the powder X-ray diffraction diagram of Form D (2R)-N-(3- ⁇ 2-[(3- methoxy-1 -methyl-1 H-pyrazol-4-yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1 - yl)propanamide saccharine salt.
- Figure 15 illustrates the powder X-ray diffraction diagram of Form E (2R)-N-(3- ⁇ 2-[(3- methoxy-1 -methyl-1 H-pyrazol-4-yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1 - yl)propanamide saccharine salt.
- Figure 16 illustrates the powder X-ray diffraction diagram of (2R)-N-(3- ⁇ 2-[(3-methoxy-1- methyl-1 H-pyrazol-4-yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1- yl)propanamide saccharine hydrochloride salt.
- Figure 17 illustrates the powder X-ray diffraction diagram of (2R)-N-(3- ⁇ 2-[(3-methoxy-1- methyl-1 H-pyrazol-4-yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1- yl)propanamide napadisylic salt.
- Figure 18 illustrates the powder X-ray diffraction diagram of (2R)-N-(3- ⁇ 2-[(3-methoxy-1- methyl-1 H-pyrazol-4-yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1- yl)propanamide trimesic salt.
- Figure 19 illustrates NCI-H1975 tumor volumes after treatment with vehicle, osimertinib (2.5 mg/kg QD) administered as a single agent, Example 32 (12.5 mg/kg BID, 25 m/kg BID or 50 mg/kg BID) as a single agent, and osimertinib (2.5 mg/kg QD) in combination with Example 32 (12.5 mg/kg BID, 25 mg/kg BID or 50 mg/kg BID).
- ⁇ represents vehicle;
- ⁇ represents osimertinib administered as a single agent; represents 50 mg/kg BID Example 32
- ⁇ represents 25 mg/kg BID Example 32 administered as a single agent
- ⁇ represents 12.5 mg/kg BID Example 32 administered as a single agent
- ⁇ represents osimertinib administered in combination with 50 mg/kg BID Example 32
- ⁇ represents osimertinib administered in combination with 25 mg/kg BID Example 32
- osimertinib administered in combination with 12.5 mg/kg BID Example 32 represents osimertinib administered in combination with 12.5 mg/kg BID Example 32.
- Figure 20 illustrates body weights after treatment with vehicle, osimertinib (2.5 mg/kg QD) as a single agent, Example 32 (12.5 mg/kg BID, 25 mg/kg BID or 50 mg/kg BID) as a single agent, and osimertinib (2.5 mg/kg QD) in combination with Example 32 (12.5 mg/kg BID, 25 mg/kg BID or 50 mg/kg BID).
- ⁇ represents vehicle; represents osimertinib administered as a single agent; ⁇ & represents 50 mg/kg BID Example 32 administered as a single agent; J l - represents 25 mg/kg BID Example 32 administered as a single agent; - ⁇ - represents 12.5 mg/kg BID Example 32 administered as a single agent; "B * represents 50 mg/kg BID Example 32 administered in combination with osimertinib; ⁇ " represents 25 mg/kg BID Example 32 administered in combination with osimertinib; and " ⁇ "represents 12.5 mg/kg BID Example 32 administered in combination with osimertinib.
- Figure 21 illustrates knockdown of pSTAT3 in NCI-H1975 tumors after treatment with vehicle, AZD1480 as a single agent, osimertinib (2.5 mg/kg QD) as a single agent, Example 32 (12.5 mg/kg BID, 25 mg/kg BID or 50 mg/kg BID) as a single agent, and osimertinib (2.5 mg/kg QD) administered in combination with Example 32 (12.5 mg/kg BID, 25 mg/kg BID or 50 mg/kg
- ⁇ represents pSTAT3 and bars represent plasma levels of Example 32.
- Figure 22 illustrates PC-9 tumor volumes after treatment with vehicle, gefitinib (IRESSA ® , 6.25 mg/kg QD) administered as a single agent and gefitinib (IRESSA ® , 6.25 mg/kg QD) administered in combination with Example 32 (12.5 mg/kg BID, 50 mg/kg BID and 50 mg/kg BID dosed 2 days on/5 days off/wk).
- IRESSA ® gefitinib
- Example 32 (12.5 mg/kg BID, 50 mg/kg BID and 50 mg/kg BID dosed 2 days on/5 days off/wk).
- ⁇ ⁇ ⁇ represents vehicle; ⁇ represents gefitinib (IRESSA®) administered as a single agent; "A" represents gefinitib (IRESSA ® ) administered in combination with 50 mg/kg BID Example 32; ⁇ " represents gefitinib (IRESSA ® ) administered in combination with 12.5 mg/kg BID Example 32; and ⁇ " represents gefinitib (IRESSA ® ) administered in combination with 50 mg/kg BID with Example 32 dosed 2 days on/5 days off/wk.
- IRESSA® gefitinib administered as a single agent
- A represents gefinitib (IRESSA ® ) administered in combination with 50 mg/kg BID Example 32
- ⁇ represents gefitinib (IRESSA ® ) administered in combination with 12.5 mg/kg BID Example 32
- ⁇ " represents gefinitib (IRESSA ® ) administered in combination with 50 mg/kg BID with Example 32 dosed 2 days on/5 days off/wk.
- Figure 23 illustrates body weights after treatment with vehicle, gefitinib (IRESSA ® , 6.25 mg/kg QD) administered as a single agent and gefitinib (IRESSA ® , 6.25 mg/kg QD)
- Example 32 (12.5 mg/kg BID, 50 mg/kg BID and 50 mg/kg 2 dosed days on/5 days off/wk).
- - ⁇ represents vehicle;
- ⁇ * represents gefitinib (IRESSA ® ) administered as a single agent;
- ⁇ represents gefinitib (IRESSA ® ) administered in combination with 50 mg/kg BID Example 32;
- ⁇ represents gefinitib (IRESSA ® ) administered in combination with 50 mg/kg BID Example 32 dosed 2 days on/5 days off/wk.
- Figure 24 illustrates knockdown of pSTAT3 in PC-9 tumors after treatment with vehicle, gefitinib (IRESSA ® , 6.25 mg/kg QD) administered as a single agent and gefitinib (IRESSA ® , 6.25 mg/kg QD) administered in combination with Example 32 (12.5 mg/kg BID and 50 mg/kg BID).
- Figure 25 illustrates NCI-H1650 tumor volumes after treatment with vehicle, gefitinib (IRESSA ® , 6.25 mg/kg QD) administed as a single agent, Example 32 (25 mg/kg BID or 50 mg/kg BID) administered as a single agent and gefitinib (IRESSA ® , 6.25 mg/kg QD)
- Example 32 administered in combination with Example 32 (25 mg/kg BID or 50 mg/kg BID). represents vehicle; ⁇ - represents gefitinib (IRESSA ® ) administered as a single agent; ⁇ " represents 25 mg/kg BI D Example 32 administered as a single agent; “ ⁇ " represents 50 mg/kg BI D Example 32 administered as a single agent; ⁇ " represents gefinitib (I RESSA ® ) administered in combination with 25 mg/kg BID Example 32; and " ⁇ " represents gefinitib (I RESSA ® ) administered in combination with 50 mg/kg BI D Example 32.
- IRESSA ® gefitinib
- ⁇ " represents 25 mg/kg BI D Example 32 administered as a single agent
- ⁇ represents 50 mg/kg BI D Example 32 administered as a single agent
- ⁇ represents gefinitib (I RESSA ® ) administered in combination with 25 mg/kg BID Example 32
- ⁇ represents gefinitib (I RESSA ® ) administered
- Figure 26 illustrates body weights after treatment with vehicle, gefitinib (I RESSA ® , 6.25 mg/kg QD) administered as a single agent, Example 32 (25 mg/kg BI D or 50 mg/kg BI D) administered as a single agent and gefitinib (I RESSA ® , 6.25 mg/kg QD) administered in combination with Example 32 (25 mg/kg BI D or 50 mg/kg BI D), "•" represents vehicle;
- gefitinib (I RESSA ® ) administered as a single agent represents gefitinib (I RESSA ® ) administered as a single agent; ⁇ £r represents 25 mg/kg BID Example 32 administered as a single agent; represents 50 mg/kg BID Example 32 administered as a single agent; " ⁇ " represents gefinitib (I RESSA ® ) administered in combination with 25 mg/kg BI D Example 32; and " ⁇ ” represents gefinitib (I RESSA ® ) administered in combination with 50 mg/kg BI D Example 32.
- Figure 27 illustrates knockdown of pSTAT3 in NCI-H 1650 tumors after treatment with vehicle, AZD1408 administered as a single agent, gefitinib (IRESSA ® , 6.25 mg/kg QD) administered as a single agent, Example 32 (25 mg/kg BID or 50 mg/kg BID) administered as a single agent and gefitinib (I RESSA ® , 6.25 mg/kg QD) administered in combination with Example
- Figure 28 illustrates LG1049 tumor volumes after treatment with vehicle ( ⁇ " ); osimertinib
- Example 32 (25 mg/kg QD) administered as a single agent for 28 days ( ⁇ ) ;
- Example 32 25 mg/kg BI D) administered as a single agent for 18 days ( "*");
- osimertinib 25 mg/kg QD administered in combination with Example 32 (25 mg/kg BID) dosed 7 days, then 3 days on/4 days off/wk until day 28 ( ⁇ " ).
- " ⁇ " represents mice treated with the combination for 28 days, then with Example 32 (25 mg/kg BI D) alone for 3 days on/4 days off/wk until the end of the study.
- Figure 29 illustrates body weights after treatment LG1049 tumor volumes after treatment with vehicle ( ⁇ ⁇ ), osimertinib (25 mg/kg QD) administered as a single agent for 28 days ( ⁇ ) i
- Example 32 25 mg/kg BI D
- Example 32 25 mg/kg BI D
- Example 32 25 mg/kg BID
- mice treated with the combination for 28 days, then with Example 32 (25 mg/kg BI D) alone for 3 days on/4 days off/wk until the end of the study
- Figure 30 illustrates knockdown of pSTAT3 and pEGFR in LG1049 tumors after five days of treatment with vehicle, osimertinib (25 mg/kg QD) administered as a single agent, Example 32 (25 mg/kg BID) administered as a single agent and osimertinib (25 mg/kg QD) administered in combination with Example 32 (25 mg/kg BID).
- Figure 31 illustrates NCI-H1975 tumor volumes after treatment with vehicle, Example 32 administered as a single agent, osimertinib administered as a single agent, and Example 32 administered in combination with osimertinib.
- Figure 31A illustrates tumor volume over time after continuous dosing with vehicle for 19 days (" ⁇ "); 50 mg/kg BID Example 32 administered as a single agent for 19 days ( * A-); 2.5 mg/kg QD osimertinib administered as a single agent for 26 days( ⁇ ); 2.5 mg/kg QD osimertinib administered in combination with 12.5 mg/kg BID Example 32 dosed for 26 days ( " ⁇ "' ); and 2.5 mg kg QD osimertinib administered in
- Figure 31 B illustrates tumor volume over time after dosing with vehicle (" ⁇ " ) for 19 days; 50 mg kg BID Example 32 administered as a single agent for 19 days ( "*-); 2.5 mg/kg QD osimertinib administered as a single agent for 26 days ( ⁇ ) ; 2.5 mg/kg QD osimertinib administered for 26 days in
- FIG. 31 C illustrates tumor volume over time after dosing with vehicle for 19 days (" ⁇ " ); 50 mg/kg BID Example 32 as a single agent for 19 days ( ⁇ *"); 2.5 mg/kg QD osimertinib administered as a single agent for 26 days ( " B " ); 2.5 mg/kg QD osimertinib administered for 29 days in combination with 25 mg/kg BID Example 32 dosed 7 days on/7 days off/2wk ( ⁇ " ); and 2.5 mg/kg QD osimertinib administered for 29 days in combination with 50 mg/kg BID Example 32 dosed 7 days on/7 days off/2wk ("V").
- Figure 31 D illustrates tumor volume over time after dosing with vehicle for 19 days ( * ⁇ " ); 50 mg/kg BID Example 32 administered as a single agent for 19 days ( "A-); 2.5 mg/kg QD osimertinib administered as a single agent for 26 days ( ⁇ ) ; 2.5 mg/kg QD osimertinib administered for 29 days in combination with 25 mg/kg BID Example 32 dosed 4 days on/3 days off/wk ( ⁇ ) ; and 2.5 mg/kg QD osimertinib administered for 29 days in combination with 50 mg/kg BID Example 32 dosed for 4 days on/3 days off/wk ("V").
- Figure 31 E illustrates tumor volume over time after dosing with vehicle for 19 days (" ⁇ "); 50 mg/kg BID Example 32 for 19 days ( -A-); 2.5 mg/kg QD osimertinib dosed for 26 days ( ⁇ " ); 2.5 mg/kg QD osimertinib dosed for 29 days in combination with 25 mg/kg BID Example 32 dosed for 2 days on/5 days off/wk ( * ⁇ *" ); and 2.5 mg/kg QD osimertinib dosed for 29 days in combination with 50 mg/kg BID Example 32 dosed 2 days on/5 days off/wk ("V").
- Figure 32 illustrates body weight over time after treatment with vehicle, Example 32 as a single agent, osimertinib as a single agent, and Example 32 in combination with osimertinib.
- * ⁇ represents vehicle dosed for 19 days; represents 2.5 mg/kg QD osimertinib dosed for 26 days; " ⁇ "represents 50 mg/kg BID Example 32 dosed for 19 days; ⁇ - represents 2.5 mg/kg QD osimertinib administered in combination with 12.5 mg/kg BID Example 32 dosed for 26 days; "•”represents 2.5 mg/kg QD osimertinib administered in combination with 50 mg/kg BID Example 32 dosed for 26 days; ⁇ - represents 2.5 mg/kg QD osimertinib administered for 26 days in combination with 25 mg/kg BID Example 32 dosed for 7 days; "O" represents 2.5 mg/kg QD osimertinib administered for 26 days in combination with 50 mg/
- R 1 is methyl or ethyl
- R 2 is selected from methyl, ethyl, methoxy and ethoxy
- R 3 is selected from hydrogen, chlorine, fluorine, bromine and methyl
- R 4 is selected from methyl, ethyl and -CH2OCH3;
- R 5 and R 6 are each individually methyl or hydrogen
- R 7 is selected from methyl, ethyl, -(Chb ⁇ OH and -(Ch ⁇ OCHs, or a pharmaceutically acceptable salt thereof.
- R 1 is methyl;
- R 2 is methoxy, ethoxy, methyl or ethyl;
- R 3 is hydrogen, fluorine methyl, chlorine or bromine;
- R 4 is methyl, ethyl or -CH 2 OCH 3 ;
- R 5 is hydrogen or methyl;
- R 6 is hydrogen or methyl;
- R 7 is methyl, -(CH 2 )20CH 3 , ethyl or -(CH 2 )20H.
- R 1 is ethyl
- R 2 is methoxy or ethoxy
- R 3 is methyl
- R 4 is methyl
- R 5 is hydrogen
- R 6 is hydrogen and R 7 is methyl.
- R 2 is methoxy;
- R 1 is methyl or ethyl;
- R 3 is hydrogen, fluorine, methyl, chlorine or bromine;
- R 4 is methyl, ethyl or -ChbOCHs;
- R 5 is hydrogen or methyl;
- R 6 is hydrogen or methyl;
- R 7 is methyl, ethyl, -(CH 2 )20CH 3 or -(CH 2 )20H.
- R 2 is ethoxy;
- R 1 is methyl or ethyl;
- R 3 is fluorine, methyl or chlorine;
- R 4 is methyl, ethyl or -ChbOCHs;
- R 5 is methyl or hydrogen;
- R 6 is methyl or hydrogen;
- R 7 is ethyl, methyl or -(CH 2 )20CH 3 .
- R 2 is methyl; R 1 is methyl; R 3 is hydrogen, methyl or fluorine; R 4 is methyl, ethyl or -ChbOCHs; R 5 is hydrogen; R 6 is hydrogen and R 7 is methyl.
- R 2 is ethyl; R 1 is methyl; R 3 is methyl; R 4 is methyl; R 5 is hydrogen; R 6 is hydrogen and R 7 is methyl.
- R 3 is hydrogen; R 1 is methyl; R 2 is methoxy or methyl; R 4 is methyl, ethyl or -CH2OCH3; R 5 is methyl or hydrogen; R 6 is hydrogen or methyl and R 7 is methyl, -(CH 2 ) 2 OCH 3 or ethyl.
- R 3 is fluorine; R 1 is methyl; R 2 is methoxy or ethoxy; R 4 is methyl, ethyl or -CH 2 OCH 3 ; R 5 is hydrogen or methyl; R 6 is hydrogen or methyl and R 7 is methyl, ethyl or -(CH 2 ) 2 OCH 3 .
- R 3 is methyl;
- R 1 is methyl or ethyl;
- R 2 is methoxy, ethoxy, methyl or ethyl;
- R 4 is methyl, ethyl or -ChbOCHs;
- R 5 is hydrogen or methyl;
- R 6 is hydrogen or methyl and
- R 7 is methyl, ethyl, -(CH 2 ) 2 OCH 3 or -(CH 2 ) 2 OH.
- R 3 is chlorine; R 1 is methyl; R 2 is methoxy or ethoxy; R 4 is methyl; R 5 is hydrogen; R 6 is hydrogen and R 7 is methyl.
- R 3 is bromine; R 1 is methyl; R 2 is methoxy; R 4 is methyl; R 5 is hydrogen; R 6 is hydrogen and R 7 is methyl.
- R 4 is methyl; R 1 is methyl or ethyl; R 2 is methoxy, ethoxy, methyl or ethyl; R 3 is hydrogen, fluorine, methyl, chlorine or bromine; R 5 is hydrogen or methyl; R 6 is hydrogen or methyl and R 7 is methyl, ethyl, -(CH 2 )20CH 3 or -(CH 2 )20H.
- R 4 is ethyl; R 1 is methyl; R 2 is methoxy, methyl or ethoxy; R 3 is methyl, hydrogen or fluorine; R 5 is hydrogen; R 6 is hydrogen and R 7 is methyl.
- R 4 is -CH 2 OCH 3 ;
- R 1 is methyl;
- R 2 is methoxy, methyl or ethoxy;
- R 3 is methyl, fluorine or hydrogen;
- R 5 is hydrogen;
- R 6 is hydrogen and R 7 is methyl.
- R 5 is hydrogen; R 1 is methyl or ethyl; R 2 is methoxy, ethoxy, methyl or ethyl; R 3 is hydrogen, fluorine, methyl, chlorine or bromine; R 4 is methyl, ethyl or -CH2OCH3; R 6 is hydrogen or methyl and R 7 is methyl, ethyl, -(CH 2 )20CH 3 or -(CH 2 ) 2 OH.
- R 5 is methyl; R 1 is methyl; R 2 is methoxy or ethoxy; R 3 is hydrogen, fluorine or methyl; R 4 is methyl; R 6 is hydrogen and R 7 is methyl.
- R 6 is hydrogen; R 1 is methyl or ethyl; R 2 is methoxy, ethoxy, methyl or ethyl; R 3 is hydrogen, fluorine, methyl, chlorine or bromine; R 4 is methyl, ethyl or -CH 2 OCH 3 ; R 5 is hydrogen or methyl and R 7 is methyl, ethyl, -(CH 2 ) 2 OCH 3 or -(CH 2 ) 2 OH.
- R 6 is methyl; R 1 is methyl; R 2 is methoxy or ethoxy; R 3 is fluorine, methyl or hydrogen; R 4 is methyl; R 5 is hydrogen and R 7 is methyl.
- R 7 is methyl;
- R 1 is methyl or ethyl;
- R 2 is methyl, ethyl, methoxy or ethoxy;
- R 3 is hydrogen, chlorine, fluorine, bromine or methyl;
- R 4 is methyl, ethyl or -CH 2 OCH 3 ;
- R 5 is hydrogen or methyl;
- R 6" is hydrogen or methyl.
- R 7 is ethyl; R 1 is methyl; R 2 is methoxy or ethoxy; R 3 is fluorine, methyl or hydrogen; R 4 is methyl; R 5 is hydrogen and R 6 is hydrogen.
- R 7 is -(CH 2 ) 2 OCH 3 ;
- R 1 is methyl;
- R 2 is methoxy or ethoxy;
- R 3 is fluorine, methyl or hydrogen;
- R 4 is methyl;
- R 5 is hydrogen and
- R 6 is hydrogen.
- R 7 is -(CH 2 ) 2 OH; R 1 is methyl, R 2 is methoxy; R 3 is methyl; R 4 is methyl; R 5 is hydrogen and R 6 is hydrogen.
- R 1 is methyl; R 2 is methoxy; R 3 is hydrogen; R 4 is methyl; R 5 is methyl; R 6 is hydrogen and R 7 is methyl.
- R 1 is methyl; R 2 is methoxy; R 3 is fluorine; R 4 is methyl; R 5 is hydrogen; R 6 is methyl and R 7 is methyl.
- R 1 is methyl; R 2 is methoxy; R 3 is fluorine; R 4 is methyl; R 5 is hydrogen; R 6 is hydrogen and R 7 is -(CH 2 ) 2 OCH 3 .
- R 1 is methyl; R 2 is methoxy; R 3 is fluorine; R 4 is methyl; R 5 is hydrogen; R 6 is hydrogen and R 7 is ethyl.
- R 1 is methyl; R 2 is methoxy; R 3 is fluorine; R 4 is methyl; R 5 is methyl; R 6 is hydrogen and R 7 is methyl.
- R 1 is methyl; R 2 is ethoxy; R 3 is fluorine; R 4 is methyl; R 5 is hydrogen; R 6 is hydrogen and R 7 is ethyl.
- R 1 is methyl; R 2 is ethoxy; R 3 is fluorine; R 4 is methyl; R 5 is hydrogen; R 6 is hydrogen and R 7 is -(CH 2 ) 2 0CH 3 .
- R 1 is methyl; R 2 is ethoxy; R 3 is fluorine; R 4 is methyl; R 5 is methyl; R 6 is hydrogen and R 7 is methyl.
- R 1 is methyl; R 2 is ethoxy; R 3 is fluorine; R 4 is methyl; R 5 is hydrogen; R 6 is methyl and R 7 is methyl.
- R 1 is methyl; R 2 is ethoxy; R 3 is fluorine; R 4 is methyl; R 5 is methyl; R 6 is hydrogen and R 7 is methyl.
- R 1 is methyl; R 2 is methoxy; R 3 is methyl; R 4 is methyl; R 5 is hydrogen; R 6 is methyl and R 7 is methyl.
- R 1 is methyl; R 2 is methoxy; R 3 is methyl; R 4 is methyl; R 5 is hydrogen; R 6 is hydrogen and R 7 is -(Ch ⁇ OH.
- R 1 is methyl; R 2 is methoxy; R 3 is methyl; R 4 is methyl; R 5 is hydrogen; R 6 is hydrogen and R 7 is ethyl.
- R 1 is methyl; R 2 is methoxy; R 3 is methyl; R 4 is methyl; R 5 is hydrogen; R 6 is hydrogen and R 7 is -(CH 2 )20CH 3 .
- R 1 is methyl; R 2 is methoxy; R 3 is methyl; R 4 is methyl; R 5 is methyl; R 6 is hydrogen and R 7 is methyl.
- R 1 is methyl; R 2 is ethoxy; R 3 is methyl; R 4 is methyl; R 5 is hydrogen; R 6 is hydrogen and R 7 is ethyl.
- R 1 is methyl; R 2 is ethoxy; R 3 is methyl; R 4 is methyl; R 5 is hydrogen; R 6 is hydrogen and R 7 is -(CH 2 ) 2 0CH 3 .
- R 1 is methyl; R 2 is ethoxy; R 3 is methyl; R 4 is methyl; R 5 is methyl; R 6 is hydrogen and R 7 is methyl.
- R 1 is methyl; R 2 is ethoxy; R 3 is methyl; R 4 is methyl; R 5 is hydrogen; R 6 is methyl and R 7 is methyl.
- R 1 is methyl; R 2 is methoxy; R 3 is fluorine; R 4 is methyl; R 5 is hydrogen; R 6 is hydrogen and R 7 is methyl.
- R 1 is methyl; R 2 is methoxy; R 3 is hydrogen; R 4 is methyl; R 5 is hydrogen; R 6 is methyl and R 7 is methyl.
- R 1 is methyl; R 2 is methoxy; R 3 is hydrogen; R 4 is methyl; R 5 is hydrogen; R 6 is hydrogen and R 7 is -(CH2)20CH3.
- R 1 is methyl; R 2 is methoxy; R 3 is hydrogen; R 4 is methyl; R 5 is hydrogen; R 6 is hydrogen and R 7 is ethyl.
- R 1 is methyl; R 2 is methoxy; R 3 is hydrogen; R 4 is methyl; R 5 is methyl; R 6 is hydrogen and R 7 is methyl.
- R 1 is methyl; R 2 is methoxy; R 3 is hydrogen; R 4 is methyl; R 5 is hydrogen; R 6 is methyl and R 7 is methyl.
- R 1 is methyl; R 2 is ethoxy; R 3 is methyl; R 4 is methyl; R 5 is hydrogen; R 6 is methyl and R 7 is methyl.
- R 1 is methyl; R 2 is methoxy; R 3 is hydrogen; R 4 is methyl; R 5 is hydrogen; R 6 is hydrogen and R 7 is methyl.
- R 1 is methyl; R 2 is methoxy; R 3 is methyl; R 4 is methyl; R 5 is hydrogen; R 6 is hydrogen and R 7 is methyl.
- R 1 is methyl; R 2 is ethoxy; R 3 is methyl; R 4 is methyl; R 5 is hydrogen; R 6 is hydrogen and R 7 is methyl.
- R 1 is methyl; R 2 is methoxy; R 3 is chlorine; R 4 is methyl; R 5 is hydrogen; R 6 is hydrogen and R 7 is methyl.
- R 1 is methyl; R 2 is methoxy; R 3 is bromine; R 4 is methyl; R 5 is hydrogen; R 6 is hydrogen and R 7 is methyl.
- R 1 is methyl; R 2 is ethoxy; R 3 is fluorine; R 4 is methyl; R 5 is hydrogen; R 6 is hydrogen and R 7 is methyl.
- R 1 is methyl; R 2 is ethoxy; R 3 is chlorine; R 4 is methyl; R 5 is hydrogen; R 6 is hydrogen and R 7 is methyl.
- R 1 is methyl; R 2 is methyl; R 3 is hydrogen; R 4 is methyl; R 5 is hydrogen; R 6 is hydrogen and R 7 is methyl.
- R 1 is methyl; R 2 is methoxy; R 3 is methyl; R 4 is ethyl; R 5 is hydrogen; R 6 is hydrogen and R 7 is methyl.
- R 1 is methyl; R 2 is methoxy; R 3 is methyl; R 4 is -CH2OCH3; R 5 is hydrogen; R 6 is hydrogen and R 7 is methyl.
- R 1 is methyl; R 2 is methoxy; R 3 is hydrogen; R 4 is ethyl; R 5 is hydrogen; R 6 is hydrogen and R 7 is methyl.
- R 1 is methyl; R 2 is methyl; R 3 is fluorine; R 4 is ethyl; R 5 is hydrogen; R 6 is hydrogen and R 7 is methyl.
- R 1 is methyl; R 2 is methoxy; R 3 is fluorine; R 4 is ethyl; R 5 is hydrogen; R 6 is hydrogen and R 7 is methyl.
- R 1 is methyl; R 2 is methyl; R 3 is fluorine; R 4 is -CH 2 OCH 3 ; R 5 is hydrogen; R 6 is hydrogen and R 7 is methyl.
- R 1 is methyl; R 2 is ethyl; R 3 is methyl; R 4 is methyl; R 5 is hydrogen; R 6 is hydrogen and R 7 is methyl.
- R 1 is methyl; R 2 is methyl; R 3 is fluorine; R 4 is methyl; R 5 is hydrogen; R 6 is hydrogen and R 7 is methyl.
- R is methyl; R 2 is methyl; R 3 is hydrogen; R 4 is methyl; R 5 is hydrogen; R 6 is hydrogen and R 7 is methyl.
- R 1 is ethyl; R 2 is methoxy; R 3 is methyl; R 4 is methyl; R 5 is hydrogen; R 6 is hydrogen and R 7 is methyl.
- R 1 is ethyl; R 2 is ethoxy; R 3 is methyl; R 4 is methyl; R 5 is hydrogen;
- R 6 is hydrogen and R 7 is methyl.
- R 1 is methyl; R 2 is methoxy; R 3 is hydrogen; R 4 is -CH 2 OCH 3 ; R 5 is hydrogen; R 6 is hydrogen and R 7 is methyl.
- R 1 is methyl; R 2 is ethoxy; R 3 is methyl; R 4 is ethyl; R 5 is hydrogen; R 6 is hydrogen and R 7 is methyl.
- R 1 is methyl; R 2 is ethoxy; R 3 is methyl; R 4 is -CH 2 OCH 3 ; R 5 is hydrogen; R 6 is hydrogen and R 7 is methyl.
- the compounds of formula (I) are compounds of formula (la):
- R 1a is methyl or ethyl
- R 2a is selected from methyl, ethyl, methoxy and ethoxy
- R 3a is selected from hydrogen, chlorine, fluorine, bromine and methyl
- R 4a is selected from methyl, ethyl and -CH 2 OCH 3 ;
- R 5a and R 6a are each individually methyl or hydrogen
- R 7a is selected from methyl, ethyl, -(CH 2 ) 2 OH and -(CH 2 ) 2 OCH3, or a pharmaceutically acceptable salt thereof.
- R 1a is methyl;
- R 2a is methoxy, ethoxy, methyl or ethyl;
- R 3a is hydrogen, fluorine methyl, chlorine or bromine;
- R 4a is methyl, ethyl or -CH 2 OCH 3 ;
- R 5a is hydrogen or methyl;
- R 6a is hydrogen or methyl;
- R 7a is methyl, -(CH 2 )20CH 3 , ethyl or -(CH 2 ) 2 OH.
- R 1a is ethyl
- R 2a is methoxy or ethoxy
- R 3a is methyl
- R 4a is methyl
- R 5a is hydrogen
- R 6a is hydrogen and R 7a is methyl.
- R 2a is methoxy;
- R 1a is methyl or ethyl;
- R 3a is hydrogen, fluorine, methyl, chlorine or bromine;
- R 4a is methyl, ethyl or -CH 2 OCH3;
- R 5a is hydrogen or methyl;
- R 6a is hydrogen or methyl;
- R 7a is methyl, ethyl, -(CH 2 ) 2 OCH 3 or -(CH 2 ) 2 OH.
- R 2a is ethoxy;
- R 1a is methyl or ethyl;
- R 3a is fluorine, methyl or chlorine;
- R 4a is methyl, ethyl or -CH 2 OCH3;
- R 5a is methyl or hydrogen;
- R 6a is methyl or hydrogen;
- R 7a is ethyl, methyl or -(CH 2 ) 2 OCH3.
- R 2a is methyl; R 1a is methyl; R 3a is hydrogen, methyl or fluorine; R 4a is methyl, ethyl or -CH 2 OCH3; R 5a is hydrogen; R 6a is hydrogen and R 7a is methyl.
- R 2a is ethyl; R 1a is methyl; R 3a is methyl; R 4a is methyl; R 5a is hydrogen; R 6a is hydrogen and R 7a is methyl.
- R 3a is hydrogen; R 1a is methyl; R 2a is methoxy or methyl; R 4a is methyl, ethyl or -CH 2 OCH3; R 5a is methyl or hydrogen; R 6a is hydrogen or methyl and R 7a is methyl, -(CH 2 ) 2 OCH 3 or ethyl.
- R 3a is fluorine; R 1a is methyl; R 2a is methoxy or ethoxy; R 4a is methyl, ethyl or -CH 2 OCH 3 ; R 5a is hydrogen or methyl; R 6a is hydrogen or methyl and R 7a is methyl, ethyl or -(CH 2 ) 2 OCH 3 .
- R 3a is methyl;
- R 1a is methyl or ethyl;
- R 2a is methoxy, ethoxy, methyl or ethyl;
- R 4a is methyl, ethyl or -CH 2 OCH 3 ;
- R 5a is hydrogen or methyl;
- R 6a is hydrogen or methyl and
- R 7a is methyl, ethyl, -(CH 2 ) 2 OCH 3 or -(CH 2 ) 2 OH.
- R 3a is chlorine; R 1a is methyl; R 2a is methoxy or ethoxy; R 4a is methyl; R 5a is hydrogen; R 6a is hydrogen and R 7a is methyl.
- R 3a is bromine; R 1a is methyl; R 2a is methoxy; R 4a is methyl; R 5a is hydrogen; R 6a is hydrogen and R 7a is methyl.
- R 4a is methyl;
- R 1a is methyl or ethyl;
- R 2a is methoxy, ethoxy, methyl or ethyl;
- R 3a is hydrogen, fluorine, methyl, chlorine or bromine;
- R 5a is hydrogen or methyl;
- R 6a is hydrogen or methyl and
- R 7a is methyl, ethyl, -(CH 2 )20CH 3 or -(CH 2 ) 2 OH.
- R 4a is ethyl; R 1a is methyl; R 2a is methoxy, methyl or ethoxy; R 3a is methyl, hydrogen or fluorine; R 5a is hydrogen; R 6a is hydrogen and R 7a is methyl.
- R 4a is -CH 2 OCH 3 ;
- R 1a is methyl;
- R 2a is methoxy, methyl or ethoxy;
- R 3a is methyl, fluorine or hydrogen;
- R 5a is hydrogen;
- R 6a is hydrogen and
- R 7a is methyl.
- R 5a is hydrogen; R 1a is methyl or ethyl; R 2a is methoxy, ethoxy, methyl or ethyl; R 3a is hydrogen, fluorine, methyl, chlorine or bromine; R 4a is methyl, ethyl or -CH2OCH3; R 6a is hydrogen or methyl and R 7a is methyl, ethyl, -(CH 2 )20CH 3 or -(CH 2 ) 2 OH.
- R 5a is methyl; R 1a is methyl; R 2a is methoxy or ethoxy; R 3a is hydrogen, fluorine or methyl; R 4a is methyl; R 6a is hydrogen and R 7a is methyl.
- R 6a is hydrogen; R 1a is methyl or ethyl; R 2a is methoxy, ethoxy, methyl or ethyl; R 3a is hydrogen, fluorine, methyl, chlorine or bromine; R 4a is methyl, ethyl or -CH 2 OCH 3 ; R 5s is hydrogen or methyl and R 7a is methyl, ethyl, -(CH 2 ) 2 OCH 3 or -(CH 2 ) 2 OH.
- R 6a is methyl; R 1a is methyl; R 2a is methoxy or ethoxy; R 3a is fluorine, methyl or hydrogen; R 4a is methyl; R 5a is hydrogen and R 7a is methyl.
- R 7a is methyl;
- R 1a is methyl or ethyl;
- R 2a is methyl, ethyl, methoxy or ethoxy;
- R 3a is hydrogen, chlorine, fluorine, bromine or methyl;
- R 4a is methyl, ethyl or
- R 5a is hydrogen or methyl
- R 6a is hydrogen or methyl
- R 7a is ethyl; R 1a is methyl; R 2a is methoxy or ethoxy; R 3a is fluorine, methyl or hydrogen; R 4a is methyl; R 5a is hydrogen and R 6a is hydrogen.
- R 7a is -(CH 2 ) 2 OCH 3 ;
- R 1a is methyl;
- R 2a is methoxy or ethoxy;
- R 3a is fluorine, methyl or hydrogen;
- R 4a is methyl;
- R 5a is hydrogen and
- R 6a is hydrogen.
- R 7a is -(CH 2 ) 2 OH; R 1a is methyl, R 2a is methoxy; R 3a is methyl; R 4a is methyl; R 5a is hydrogen and R 6a is hydrogen.
- R 1a is methyl; R 2a is methoxy; R 3a is hydrogen; R 4a is methyl; R 5a is methyl; R 6a is hydrogen and R 7a is methyl.
- R 1a is methyl; R 2a is methoxy; R 3a is fluorine; R 4a is methyl; R 5a is hydrogen; R 6a is methyl and R 7a is methyl.
- R 1a is methyl; R 2a is methoxy; R 3a is fluorine; R 4a is methyl; R 5a is hydrogen; R 6a is hydrogen and R 7a is -(CH 2 ) 2 OCH 3 .
- R 1a is methyl; R 2a is methoxy; R 3a is fluorine; R 4a is methyl; R 5a is hydrogen; R 6a is hydrogen and R 7a is ethyl.
- R 1a is methyl; R 2a is methoxy; R 3a is fluorine; R 4a is methyl; R 5a is methyl; R 6a is hydrogen and R 7a is methyl.
- R 1a is methyl; R 2a is ethoxy; R 3a is fluorine; R 4a is methyl; R 5a is hydrogen; R 6a is hydrogen and R 7a is ethyl.
- R 1a is methyl; R 2a is ethoxy; R 3a is fluorine; R 4a is methyl; R 5a is hydrogen; R 6a is hydrogen and R 7a is -(CH 2 )20CH 3 .
- R 1a is methyl; R 2a is ethoxy; R 3a is fluorine; R 4a is methyl; R 5a is methyl; R 6a is hydrogen and R 7a is methyl.
- R 1a is methyl; R 2a is ethoxy; R 3a is fluorine; R 4a is methyl; R 5a is hydrogen; R 6a is methyl and R 7a is methyl.
- R 1a is methyl; R 2a is ethoxy; R 3a is fluorine; R 4a is methyl; R 5a is methyl; R 6a is hydrogen and R 7a is methyl.
- R 1a is methyl; R 2a is methoxy; R 3a is methyl; R 4a is methyl; R 5a is hydrogen; R 6a is methyl and R 7a is methyl.
- R 1a is methyl; R 2a is methoxy; R 3a is methyl; R 4a is methyl; R 5a is hydrogen; R 6a is hydrogen and R 7a is -(Chb ⁇ OH.
- R 1a is methyl; R 2a is methoxy; R 3a is methyl; R 4a is methyl; R 5a is hydrogen; R 6a is hydrogen and R 7a is ethyl.
- R 1a is methyl; R 2a is methoxy; R 3a is methyl; R 4a is methyl; R 5a is hydrogen; R 6a is hydrogen and R 7a is -(CH 2 )20CH 3 .
- R 1a is methyl; R 2a is methoxy; R 3a is methyl; R 4a is methyl; R 5a is methyl; R 6a is hydrogen and R 7a is methyl.
- R 1a is methyl; R 2a is ethoxy; R 3a is methyl; R 4a is methyl; R 5a is hydrogen; R 6a is hydrogen and R 7a is ethyl.
- R 1a is methyl; R 2a is ethoxy; R 3a is methyl; R 4a is methyl; R 5a is hydrogen; R 6a is hydrogen and R 7a is -(CH 2 )20CH 3 .
- R 1a is methyl; R 2a is ethoxy; R 3a is methyl; R 4a is methyl; R 5a is methyl; R 6a is hydrogen and R 7a is methyl.
- R 1a is methyl; R 2a is ethoxy; R 3a is methyl; R 4a is methyl; R 5a is hydrogen; R 6a is methyl and R 7a is methyl.
- R 1a is methyl; R 2a is methoxy; R 3a is fluorine; R 4a is methyl; R 5a is hydrogen; R 6a is hydrogen and R 7a is methyl.
- R 1a is methyl; R 2a is methoxy; R 3a is hydrogen; R 4a is methyl; R 5a is hydrogen; R 6a is methyl and R 7a is methyl.
- R 1a is methyl; R 2a is methoxy; R 3a is hydrogen; R 4a is methyl; R 5a is hydrogen; R 6a is hydrogen and R 7a is -(CH 2 )20CH 3 .
- R 1a is methyl; R 2a is methoxy; R 3a is hydrogen; R 4a is methyl; R 5a is hydrogen; R 6a is hydrogen and R 7a is ethyl.
- R 1a is methyl; R 2a is methoxy; R 3a is hydrogen; R 4a is methyl; R 5a is methyl; R 6a is hydrogen and R 7a is methyl.
- R 1a is methyl; R 2a is methoxy; R 3a is hydrogen; R 4a is methyl; R 5a is hydrogen; R 6a is methyl and R 7a is methyl.
- R 1a is methyl; R 2a is ethoxy; R 3a is methyl; R 4a is methyl; R 5a is hydrogen; R 6a is methyl and R 7a is methyl.
- R 1a is methyl; R 2a is methoxy; R 3a is hydrogen; R 4a is methyl; R 5a is hydrogen; R 6a is hydrogen and R 7a is methyl.
- R 1a is methyl; R 2a is methoxy; R 3a is methyl; R 4a is methyl; R 5a is hydrogen; R 6a is hydrogen and R 7a is methyl.
- R 1a is methyl; R 2a is ethoxy; R 3a is methyl; R 4a is methyl; R 5a is hydrogen; R 6a is hydrogen and R 7a is methyl.
- R 1a is methyl; R 2a is methoxy; R 3a is chlorine; R 4a is methyl; R 5a is hydrogen; R 6a is hydrogen and R 7a is methyl.
- R 1a is methyl; R 2a is methoxy; R 3a is bromine; R 4a is methyl; R 5a is hydrogen; R 6a is hydrogen and R 7a is methyl.
- R 1a is methyl; R 2a is ethoxy; R 3a is fluorine; R 4a is methyl; R 5a is hydrogen; R 6a is hydrogen and R 7a is methyl.
- R 1a is methyl; R 2a is ethoxy; R 3a is chlorine; R 4a is methyl; R 5a is hydrogen; R 6a is hydrogen and R 7a is methyl.
- R 1a is methyl; R 2a is methyl; R 3a is hydrogen; R 4a is methyl; R 5a is hydrogen; R 6a is hydrogen and R 7a is methyl.
- R 1a is methyl; R 2a is methoxy; R 3a is methyl; R 4a is ethyl; R 5a is hydrogen; R 6a is hydrogen and R 7a is methyl.
- R 1a is methyl; R 2a is methoxy; R 3a is methyl; R 4a is -CH2OCH3; R 5a is hydrogen; R 6a is hydrogen and R 7a is methyl.
- R 1a is methyl; R 2a is methoxy; R 3a is hydrogen; R 4a is ethyl; R 5a is hydrogen; R 6a is hydrogen and R 7a is methyl.
- R 1a is methyl; R 2a is methyl; R 3a is fluorine; R 4a is ethyl; R 5a is hydrogen; R 6a is hydrogen and R 7a is methyl.
- R 1a is methyl; R 2a is methoxy; R 3a is fluorine; R 4a is ethyl; R 5a is hydrogen; R 6a is hydrogen and R 7a is methyl.
- R 1a is methyl; R 2a is methyl; R 3a is fluorine; R 4a is -CH2OCH is hydrogen; R 6a is hydrogen and R 7a is methyl.
- R 1a is methyl; R 2a is ethyl; R 3a is methyl; R 4a is methyl; R 5a is hydrogen; R 6a is hydrogen and R 7a is methyl.
- R 1a is methyl; R 2a is methyl; R 3a is fluorine; R 4a is methyl; R 5a is hydrogen; R 6a is hydrogen and R 7a is methyl.
- R 1a is methyl; R 2a is methyl; R 3a is hydrogen; R 4a is methyl; R 5a is hydrogen; R 6a is hydrogen and R 7a is methyl.
- R 1a is ethyl; R 2a is methoxy; R 3a is methyl; R 4a is methyl; R 5a is hydrogen; R 6a is hydrogen and R 7a is methyl.
- R 1a is ethyl; R 2a is ethoxy; R 3a is methyl; R 4a is methyl; R 5a is hydrogen; R 6a is hydrogen and R 7a is methyl.
- R 1a is methyl; R 2a is methoxy; R 3a is hydrogen; R 4a is -CH2OCH3; R 5a is hydrogen; R 6a is hydrogen and R 7a is methyl.
- R 1a is methyl; R 2a is ethoxy; R 3a is methyl; R 4a is ethyl; R 5a is hydrogen; R 6a is hydrogen and R 7a is methyl.
- R 1a is methyl; R 2a is ethoxy; R 3a is methyl; R 4a is -CH2OCH3; R 5a is hydrogen; R 6a is hydrogen and R 7a is methyl.
- the compounds of formula (I) are compounds of formula (lb):
- R 2b is selected from methyl, ethyl, methoxy and ethoxy
- R 3b is selected from hydrogen, chlorine, fluorine, bromine and methyl; and R 7b is selected from methyl, ethyl, -(Chb ⁇ OH and -(CH2)20CH3, or a pharmaceutically acceptable salt thereof.
- R 2b is methoxy
- R 3b is hydrogen, fluorine, methyl, chlorine or bromine
- R 7b is methyl, ethyl, -(CH 2 )20CH 3 or -(CH 2 ) 2 OH.
- R 2b is ethoxy
- R 3b is fluorine, methyl or chlorine
- R 7b is ethyl, methyl or -(CH 2 ) 2 OCH 3 .
- R 2b is methyl; R 3b is hydrogen, methyl or fluorine; and R 7b is methyl.
- R 2b is ethyl; R 3b is methyl; and R 7b is methyl.
- R 3b is hydrogen; R 2b is methoxy or methyl; and R 7b is methyl, -(CH 2 ) 2 OCH 3 or ethyl.
- R 3b is fluorine; R 2b is methoxy or ethoxy; R 7b is methyl, ethyl or -(CH 2 ) 2 OCH 3 .
- R 3b is methyl; R 2b is methoxy, ethoxy, methyl or ethyl; and R 7b is methyl, ethyl, -(CH 2 ) 2 OCH 3 or -(CH 2 ) 2 OH.
- R 3b is chlorine; R 2b is methoxy or ethoxy; and R 7b is methyl.
- R 3b is bromine; R 2b is methoxy; and R 7b is methyl.
- R 7b is methyl;
- R 2b is methyl, ethyl, methoxy or ethoxy; and
- R 3b is hydrogen, chlorine, fluorine, bromine or methyl.
- R 7b is ethyl
- R 2b is methoxy or ethoxy
- R 3b is fluorine, methyl or hydrogen.
- R 7b is -(CH2)20CH 3 ;
- R 2b is methoxy or ethoxy; and
- R 3b is fluorine, methyl or hydrogen.
- R 7b is -(Chb ⁇ OH; R 2b is methoxy; and R 3a is methyl.
- R 2b is methoxy; R 3b is fluorine; and R 7b is -(CH2)20CH 3 .
- R 2b is methoxy; R 3b is fluorine; and R 7a is ethyl.
- R 2b is ethoxy
- R 3b is fluorine
- R 7a is ethyl
- R 2b is ethoxy; R 3b is fluorine; and R 7b is -(CH 2 )20CH 3 .
- R 2b is methoxy; R 3b is methyl; and R 7b is -(CH 2 )20H.
- R 2b is methoxy; R 3b is methyl; and R 7b is ethyl.
- R 2b is methoxy; R 3b is methyl; and R 7b is -(CH2)20CH 3 .
- R 2b is ethoxy; R 3b is methyl; and R 7b is ethyl. In some embodiments, R 2b is ethoxy; R 3b is methyl; and R 7b is ethyl. In some embodiments, R 2b is ethoxy; R 3b is methyl; and R 7b is ethyl.
- R 2b is ethoxy; R 3b is methyl; and R 7b is -(CH 2 )20CH 3 .
- R 2b is methoxy; R 3b is fluorine; and R 7b is methyl. In some embodiments, R 2b is methoxy; R 3b is hydrogen; and R 7b is -(CH 2 )20CH 3 .
- R 2b is methoxy; R 3b is hydrogen; and R 7b is methyl.
- R 2b is methoxy; R 3b is methyl; and R 7b is methyl.
- R 2b is ethoxy; R 3b is methyl; and R 7b is methyl.
- R 2b is methoxy; R 3b is chlorine; and R 7b is methyl.
- R 2b is methoxy; R 3b is bromine; and R 7b is methyl.
- R 2b is ethoxy; R 3b is fluorine; and R 7b is methyl.
- R 2b is ethoxy; R 3b is chlorine; and R 7b is methyl.
- R 2b is methyl; R 3b is hydrogen; and R 7b is methyl.
- R 2b is ethyl; R 3b is methyl; and R 7b is methyl.
- R 2b is methyl; R 3b is fluorine; and R 7b is methyl.
- R 2b is methyl; R 3b is hydrogen; and R 7b is methyl.
- the disclosed compounds are obtainable by any process described in the Examples. In one embodiment, the disclosed are the intermediate compounds described in the Examples.
- pharmaceutically acceptable salt includes acid addition or base salts that retain the biological effectiveness and properties of the compounds of Formula (I), (la), (lb) and Table 1 and, which typically are not biologically or otherwise undesirable.
- Formula (I), (la), (lb) and Table 1 are capable of forming acid and/or base salts by virtue of the presence of amino and/or carboxyl groups or groups similar thereto.
- Pharmaceutically acceptable acid addition salts can be formed with inorganic acids and organic acids, e.g., acetate, aspartate, benzoate, besylate, bromide/hydrobromide,
- chlortheophyllonate citrate, ethandisulfonate, fumarate, gluceptate, gluconate, glucuronate, hippurate, hydroiodide/iodide, isethionate, lactate, lactobionate, laurylsulfate, malate, maleate, malonate, mandelate, mesylate, methylsulphate, napadisylate, naphthoate, napsylate, nicotinate, nitrate, octadecanoate, oleate, oxalate, palmitate, palmoate, phosphate/hydrogen phosphate/dihydrogen phosphate, polygalacturonate, propionate, stearate, succinate, subsalicylate, tartrate, tosylate, trimesate and trifluoroacetate salts.
- Inorganic acids from which salts can be derived include, for example, hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, and the like.
- Organic acids from which salts can be derived include, for example, acetic acid, propionic acid, glycolic acid, oxalic acid, maleic acid, malonic acid, succinic acid, fumaric acid, tartaric acid, trimesic acid, citric acid, benzoic acid, mandelic acid, methanesulfonic acid, napadisylic acid, ethanesulfonic acid, toluenesulfonic acid, trifluoroacetic acid, sulfosalicylic acid, and the like.
- Pharmaceutically acceptable base addition salts can be formed with inorganic and organic bases.
- Inorganic bases from which salts can be derived include, for example, ammonium salts and metals from columns I to XII of the periodic table.
- the salts are derived from sodium, potassium, ammonium, calcium, magnesium, iron, silver, zinc, and copper; particularly suitable salts include ammonium, potassium, sodium, calcium and magnesium salts.
- Organic bases from which salts can be derived include, for example, primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines, basic ion exchange resins, and the like.
- Certain organic amines include isopropylamine, benzathine, cholinate, diethanolamine, diethylamine, lysine, meglumine, piperazine and tromethamine.
- the pharmaceutically acceptable salts of the compounds Formula (I), (la), (lb) and Table 1 can be synthesized from a basic or acidic moiety, by conventional chemical methods.
- such salts can be prepared by reacting free acid forms of these compounds with a stoichiometric amount of the appropriate base (such as Na + , Ca 2+ , Mg 2+ , or K + hydroxide, carbonate, bicarbonate or the like), or by reacting free base forms of these compounds with a stoichiometric amount of the appropriate acid.
- a stoichiometric amount of the appropriate base such as Na + , Ca 2+ , Mg 2+ , or K + hydroxide, carbonate, bicarbonate or the like
- Such reactions are typically carried out in water or in an organic solvent, or in a mixture of the two.
- use of non-aqueous media like ether, ethyl acetate, ethanol, isopropanol, or acetonitrile is desirable, where practicable.
- any formula given herein is also intended to represent unlabeled forms as well as isotopically labeled forms for the compounds of Formula (I), (la), (lb) and Table 1.
- Isotopically labeled compounds have structures depicted by the formulas given herein except that one or more atoms are replaced by an atom having a selected atomic mass or mass number.
- isotopes that can be incorporated into the compounds of Formula (I), (la), (lb) and Table 1 include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorous, fluorine and chlorine, such as 2 H, 3 H, 11 C, 13 C, 14 C, 15 N, 16 F, 31 P, 32 P, 35 S, 36 CI and 125 l.
- the compounds of Formula (I), (la), (lb) and Table 1 may include various isotopically labeled compounds into which radioactive isotopes, such as 2 H, 3 H, 13 C and 14 C, are present.
- Isotopically labeled compounds of formula (I), (la) and (lb) can generally be prepared by convention techniques known to those skilled in the art or by processes analogous to those described in the accompanying Examples using appropriate isotopically labeled reagents in place of the non-labeled reagents previously employed.
- the compounds of formula (I), (la), (lb) and Table 1 may have different isomeric forms.
- the language "optical isomer” or “stereoisomer” refers to any of the various stereoisomeric configurations which may exist for a given compound of formula (I), (la), (lb) and Table 1. It is understood that a substituent may be attached at a chiral center of a carbon atom and, therefore, the disclosed compounds include enantiomers, diastereomers and racemates.
- enantiomer includes pairs of stereoisomers that are non-superimposable mirror images of each other. A 1 :1 mixture of a pair of enantiomers is a racemic mixture. The term is used to designate a racemic mixture where appropriate.
- stereoisomers include stereoisomers that have at least two asymmetric atoms, but which are not mirror images of each other.
- the absolute stereochemistry is specified according to the Cahn-lngold-Prelog R-S system. When a compound is a pure enantiomer, the stereochemistry at each chiral center may be specified by either R or S.
- Resolved compounds whose absolute configuration is unknown can be designated (+) or (-) depending on the direction (dextro- or levorotatory) which they rotate plane polarized light at the wavelength of the sodium D line.
- Certain of the compounds of Formula (I), (la), (lb) and Table 1 contain one or more asymmetric centers or axes and may thus give rise to enantiomers, diastereomers or other stereoisomeric forms that may be defined, in terms of absolute stereochemistry, as (R)- or (S)-.
- the present disclosure is meant to include all such possible isomers, including racemic mixtures, optically pure forms and intermediate mixtures.
- Optically active (R)- and (S)-isomers may be prepared using chiral synthons or chiral reagents, or resolved using conventional techniques well known in the art, such as chiral HPLC.
- solid forms of the compounds of Formula (I), (la) and (lb), or a pharmaceutically acceptable salt thereof include polymorphs, crystalline salts, solvates, hydrates and amorphous forms of the compounds of Formula (I), (la) and (lb).
- polymorph includes crystalline materials that have the same chemical composition but different molecular packing.
- crystalline salt includes crystalline structures with the same chemical materials, but incorporating acid or base addition salts within the molecular packing of the crystalline structure.
- solvate includes crystalline structures of the same chemical material, but incorporating molecules of solvent within the molecular packing of the crystalline structure.
- hydrates includes crystalline structures of the same chemical material, but incorporating molecules of water within the molecular packing of the crystalline structure.
- amorphous form includes compounds of the same molecular material but without the molecular order of a crystalline structure ⁇ e.g., polymorph, crystalline salt, solvate or hydrate) of the same molecular material.
- solid materials may be characterized using conventional techniques such as X-Ray Powder Diffraction (XRPD), Differential Scanning Calorimetry (DSC), Thermal Gravimetric Analysis (TGA), Diffuse Reflectance Infrared Fourier Transform (DRIFT) spectroscopy, Near Infrared (NIR) spectroscopy, solution and/or solid state nuclear magnetic resonance spectroscopy.
- XRPD X-Ray Powder Diffraction
- DSC Differential Scanning Calorimetry
- TGA Thermal Gravimetric Analysis
- DRIFT Diffuse Reflectance Infrared Fourier Transform
- NIR Near Infrared
- solution and/or solid state nuclear magnetic resonance spectroscopy solution and/or solid state nuclear magnetic resonance spectroscopy.
- the water content of such solid materials may be determined by Karl Fischer analysis.
- the solid forms described herein provide XRPD patterns substantially the same as the XRPD patterns shown in the Figures, and have the various 2-theta (2 ⁇ ) values as shown in the Tables included herein.
- an XRPD pattern or diffractogram may be obtained which has one or more measurement errors depending on the recording conditions, such as the equipment or machine used.
- intensities in an XRPD pattern may fluctuate depending on measurement conditions or sample preparation as a result of preferred orientation.
- the relative intensity of peaks can also be affected by, for example, grains above 30 ⁇ in size and non-unitary aspect ratios.
- the skilled person understands that the position of reflections can be affected by the precise height at which the sample sits in the diffractometer, and also the zero calibration of the diffractometer.
- the surface planarity of the sample may also have a small effect.
- any solid forms providing XRPD patterns substantially the same as those shown in the Figures fall within the scope of the corresponding embodiment.
- a person skilled in the art of XRPD is able to judge the substantial identity of XRPD patterns.
- a measurement error of a diffraction angle in an XRPD is approximately 2 ⁇ ( ⁇ 0.2°), and such degree of a measurement error should be taken into account when considering the X-ray powder diffraction pattern in the Figures and when reading data contained in the Tables included herein.
- Form A (2f?)-N-(3- ⁇ 2-[(3-methoxy-1-methyl-1 H- pyrazol-4-yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide.
- Form A (2f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 7-pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has an XRPD pattern comprising at least one peak expressed as 2 ⁇ ( ⁇ 0.2°) selected from the peaks listed in Table 17.
- Form A (2f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has an XRPD pattern substantially similar to Figure 1.
- Form A (2f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has a DSC thermogram comprising an endotherm with a desolvation onset at about 1 10 °C and a peak at about 1 13 °C.
- Form A (2f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has a DSC thermogram substantially similar to Figure 2.
- Form A (2f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 7-pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has a TGA thermogram exhibiting a mass loss of about 7.8% upon heating from about 25 °C to about 150 °C.
- Form A 2f?)-N-(3- ⁇ 2-[(3-methoxy-1-methyl-1 7-pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has a TGA thermogram substantially similar to Figure 2.
- Form B (2f?)-N-(3- ⁇ 2-[(3-methoxy-1-methyl-1 H- pyrazol-4-yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide.
- Form B (2f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 /-/-pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 /-/-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has an XRPD pattern comprising at least one peak expressed as 2 ⁇ ( ⁇ 0.2°) selected from the peaks listed in Table 18.
- Form B (2f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has an XRPD pattern substantially similar to Figure 3.
- Form B (2f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has a DSC thermogram comprising an endotherm with a desolvation onset at about 1 12 °C and a peak at about 1 17 °C.
- Form B (2f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has a DSC thermogram substantially similar to Figure 4.
- Form B (2f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has a TGA thermogram exhibiting a mass loss of about 10.0% upon heating from about 25 °C to about 200 °C.
- Form B (2f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has a TGA thermogram substantially similar to Figure 4.
- Form C (2f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 H- pyrazol-4-yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide.
- Form C (2f?)-N-(3- ⁇ 2-[(3-methoxy-1-methyl-1 /-/-pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has an XRPD pattern comprising at least one peak expressed as 2 ⁇ ( ⁇ 0.2°) selected from the peaks listed in Table 19.
- Form C (2 fi)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has an XRPD pattern substantially similar to Figure 5.
- Form C (2 fi)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has a DSC thermogram comprising an endotherm with a desolvation onset at about 1 12 °C and a peak at about 1 14 °C.
- Form C (2 fi)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has a DSC thermogram substantially similar to Figure 6.
- Form C (2 fi)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has a TGA thermogram exhibiting a mass loss of about 9.2% upon heating from about 25 °C to about 175 °C.
- Form C (2 fi)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has a TGA thermogram substantially similar to Figure 6.
- Form D (2f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 H- pyrazol-4-yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide.
- Form D (2 fi)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has an XRPD pattern comprising at least one peak expressed as 2 ⁇ ( ⁇ 0.2°) at about 21.8°.
- Form D (2 fi)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has an XRPD pattern comprising at least one peak expressed as 2 ⁇ ( ⁇ 0.2°) at about 6.4°.
- Form D (2 fi)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has an XRPD pattern comprising at least one peak expressed as 2 ⁇ ( ⁇ 0.2°) at about 16.6°.
- Form D (2 fi)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has an XRPD pattern comprising at least one peak expressed as 2 ⁇ ( ⁇ 0.2°) at about 8.9°.
- Form D (2 fi)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has an XRPD pattern comprising at least one peak expressed as 2 ⁇ ( ⁇ 0.2°) at about 8.1 °.
- Form D (2 f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has an XRPD pattern comprising at least one peak expressed as 2 ⁇ ( ⁇ 0.2°) selected from about 21.8° and 6.4°.
- Form D (2 fi)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has an XRPD pattern comprising at least one peak expressed as 2 ⁇ ( ⁇ 0.2°) selected from about 21.8° and 16.6°.
- Form D (2 fi)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has an XRPD pattern comprising at least one peak expressed as 2 ⁇ ( ⁇ 0.2°) selected from about 21.8° and 8.9°.
- Form D (2 fi)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has an XRPD pattern comprising at least one peak expressed as 2 ⁇ ( ⁇ 0.2°) selected from about 21.8° and 8.1 °.
- Form D (2 fi)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has an XRPD pattern comprising at least one peak expressed as 2 ⁇ ( ⁇ 0.2°) selected from about 6.4° and 16.6°.
- Form D (2 f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has an XRPD pattern comprising at least one peak expressed as 2 ⁇ ( ⁇ 0.2°) selected from about 6.4° and 8.9°.
- Form D (2 fi)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has an XRPD pattern comprising at least one peak expressed as 2 ⁇ ( ⁇ 0.2°) selected from about 6.4° and 8.1 °.
- Form D (2 fi)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has an XRPD pattern comprising at least one peak expressed as 2 ⁇ ( ⁇ 0.2°) selected from about 16.6° and 8.9°.
- Form D (2 fi)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has an XRPD pattern comprising at least one peak expressed as 2 ⁇ ( ⁇ 0.2°) selected from about 16.6° and 8.1 °.
- Form D (2 fi)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has an XRPD pattern comprising at least one peak expressed as 2 ⁇ ( ⁇ 0.2°) selected from about 8.1 ° and 8.9°.
- Form D (2 fi)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has an XRPD pattern comprising at least one peak expressed as 2 ⁇ ( ⁇ 0.2°) selected from about 21.8°, 6.4° and 16.6°.
- Form D (2 fi)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has an XRPD pattern comprising at least one peak expressed as 2 ⁇ ( ⁇ 0.2°) selected from about 21.8°, 6.4° and 8.9°.
- Form D (2 fi)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has an XRPD pattern comprising at least one peak expressed as 2 ⁇ ( ⁇ 0.2°) selected from about 21.8°, 6.4° and 8.1 °.
- Form D (2 fi)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has an XRPD pattern comprising at least one peak expressed as 2 ⁇ ( ⁇ 0.2°) selected from about 21.8°, 16.6° and 8.9°.
- Form D (2 f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has an XRPD pattern comprising at least one peak expressed as 2 ⁇ ( ⁇ 0.2°) selected from about 21.8°, 16.6° and 8.1 °.
- Form D (2 fi)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has an XRPD pattern comprising at least one peak expressed as 2 ⁇ ( ⁇ 0.2°) selected from about 21.8°, 8.9° and 8.1 °.
- Form D (2 fi)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has an XRPD pattern comprising at least one peak expressed as 2 ⁇ ( ⁇ 0.2°) selected from about 16.6°, 8.9° and 8.1 °.
- Form D (2 fi)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has an XRPD pattern comprising at least one peak expressed as 2 ⁇ ( ⁇ 0.2°) selected from about 21.8°, 6.4°, 16.6° and 8.9°.
- Form D (2 fi)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has an XRPD pattern comprising at least one peak expressed as 2 ⁇ ( ⁇ 0.2°) selected from about 21.8°, 6.4°, 16.6° and 8.1 °.
- Form D (2 fi)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has an XRPD pattern comprising at least one peak expressed as 2 ⁇ ( ⁇ 0.2°) selected from about 6.4°, 16.6°, 8.9° and 8.1 °.
- Form D (2 fi)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has an XRPD pattern comprising at least one peak expressed as 2 ⁇ ( ⁇ 0.2°) selected from about 21.8°, 6.4°, 16.6°, 8.9° and 8.1 °.
- Form D (2 f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has an XRPD pattern comprising at least one peak expressed as 2 ⁇ ( ⁇ 0.2°) selected from the peaks listed in Table 20.
- Form D (2 fi)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has an XRPD pattern substantially similar to Figure 7.
- Form D (2 fi)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has a DSC thermogram comprising an endotherm with a desolvation onset at about 1 16 °C and a peak at about 1 19 °C.
- Form D (2 fi)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has a DSC thermogram substantially similar to Figure 8.
- Form D (2 fi)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has a TGA thermogram exhibiting a mass loss of about 8.0% upon heating from about 25 °C to about 200 °C.
- Form D (2 fi)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide has a TGA thermogram substantially similar to Figure 8.
- Form A (2f?)-N-(3- ⁇ 2-[(3-methoxy-1-methyl-1 H- pyrazol-4-yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide saccharine salt.
- Form A (2f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 7-pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide saccharine salt has an XRPD pattern comprising at least one peak expressed as 2 ⁇ ( ⁇ 0.2°) selected from the peaks listed in Table 21 .
- Form A (2f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide saccharine salt has an XRPD pattern substantially similar to Figure 9.
- Form A (2f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide saccharine salt has a DSC thermogram comprising an endotherm with a melting point onset at about 163 °C and a peak at about 169 °C.
- Form A (2f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide saccharine salt has a DSC thermogram substantially similar to Figure 10.
- Form A (2f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide saccharine salt has a TGA thermogram exhibiting a mass loss of about 3.1 % upon heating from about 25 °C to about 150 °C.
- Form A (2f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide saccharine salt has a TGA thermogram substantially similar to Figure 10.
- Form B-Saccharine Salt
- Form B (2f?)-N-(3- ⁇ 2-[(3-methoxy-1-methyl-1 H- pyrazol-4-yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide saccharine salt.
- Form B (2f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide saccharine salt has an XRPD pattern comprising at least one peak expressed as 2 ⁇ ( ⁇ 0.2°) selected from the peaks listed in Table 22.
- Form B (2f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 7-pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide saccharine salt has an XRPD pattern substantially similar to Figure 11.
- Form B (2f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 7-pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide saccharine salt has a DSC thermogram comprising an endotherm with a broad desolvation peak at about 53 °C and two endotherm events with an onset at about 153 °C and a peak at 162 °C and an onset at about 176 °C and a peak at about 182 °C.
- Form B (2f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide saccharine salt has a DSC thermogram substantially similar to Figure 12.
- Form B (2f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide saccharine salt has a TGA thermogram exhibiting a mass loss of about 2.7% upon heating from about 25 °C to about 100 °C.
- Form B (2f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide saccharine salt has a TGA thermogram substantially similar to Figure 12.
- Form C (2f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 H- pyrazol-4-yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide saccharine salt.
- Form C (2 f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide saccharine salt has an XRPD pattern comprising at least one peak expressed as 2 ⁇ ( ⁇ 0.2°) selected from the peaks listed in Table 23.
- Form C (2 f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide saccharine salt has an XRPD pattern substantially similar to Figure 13.
- Form D (2f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 H- pyrazol-4-yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide saccharine salt.
- Form D (2 f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide saccharine salt has an XRPD pattern comprising at least one peak expressed as 2 ⁇ ( ⁇ 0.2°) selected from the peaks listed in Table 24.
- Form D (2 f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl- 1 /- -pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide saccharine salt has an XRPD pattern substantially similar to Figure 14.
- Form E (2f?)-N-(3- ⁇ 2-[(3-methoxy-1-methyl-1 H- pyrazol-4-yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide saccharine salt.
- Form E (2f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 7-pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide saccharine salt has an XRPD pattern comprising at least one peak expressed as 2 ⁇ ( ⁇ 0.2°) selected from the peaks listed in Table 25.
- Form E (2f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 7-pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide saccharine salt has an XRPD pattern substantially similar to Figure 15.
- (2f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 H-pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide hydrochloride saccharine salt has an XRPD pattern comprising at least one peak expressed as 2 ⁇ ( ⁇ 0.2°) selected from the peaks listed in Table 26.
- (2f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 H-pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide hydrochloride saccharine salt has an XRPD pattern substantially similar to Figure 16.
- (2f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 H-pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide hydrochloride napadisylic salt has an XRPD pattern comprising at least one peak expressed as 2 ⁇ ( ⁇ 0.2°) selected from the peaks listed in Table 27.
- (2f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 H-pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide napadisylic salt has an XRPD pattern substantially similar to Figure 17.
- (2f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 H-pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide hydrochloride trimesic salt has an XRPD pattern comprising at least one peak expressed as 2 ⁇ ( ⁇ 0.2°) selected from the peaks listed in Table 28.
- (2f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 H-pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1 -yl)propanamide trimesic salt has an XRPD pattern substantially similar to Figure 18.
- compositions comprising a compound of formula (I), (la), (lb) or of Table 1 , and a pharmaceutically acceptable excipient, carrier or diluent.
- pharmaceutically acceptable excipient, carrier or diluent includes compounds, materials, compositions, and/or dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, or other problem or complication, as ascertained by one of skill in the art.
- compositions may be in a form suitable for oral use (for example, as tablets, lozenges, hard or soft capsules, aqueous or oily suspensions, emulsions, dispersible powders or granules, syrups or elixirs), for topical use (for example, as creams, ointments, gels, or aqueous or oily solutions or suspensions), for administration by inhalation (for example, as a finely divided powder or a liquid aerosol), for administration by insufflation (for example, as a finely divided powder) or for parenteral administration (for example, as a sterile aqueous or oily solution for intravenous, subcutaneous, intramuscular or intramuscular dosing or as a suppository for rectal dosing).
- oral use for example, as tablets, lozenges, hard or soft capsules, aqueous or oily suspensions, emulsions, dispersible powders or granules, syrups or e
- compositions intended for oral use may contain, for example, one or more coloring, sweetening, flavoring and/or preservative agents.
- Suitable pharmaceutically acceptable excipients for a tablet formulation include, for example, inert diluents such as lactose, sodium carbonate, calcium phosphate or calcium carbonate; granulating and disintegrating agents such as corn starch or algenic acid; binding agents such as starch; lubricating agents such as magnesium stearate, stearic acid or talc; preservative agents such as ethyl or propyl p-hydroxybenzoate; and anti-oxidants, such as ascorbic acid. Tablet formulations may be uncoated or coated either to modify their
- Compositions for oral use may be in the form of hard gelatin capsules in which the active ingredient is mixed with an inert solid diluent, for example, calcium carbonate, calcium phosphate or kaolin, or as soft gelatin capsules in which the active ingredient is mixed with water or oil, such as peanut oil, liquid paraffin, or olive oil.
- an inert solid diluent for example, calcium carbonate, calcium phosphate or kaolin
- water or oil such as peanut oil, liquid paraffin, or olive oil.
- Aqueous suspensions generally contain the active ingredient in finely powdered form or in the form of nano or micronized particles together with one or more suspending agents, such as sodium carboxymethylcellulose, methylcellulose, hydroxypropylmethylcellulose, sodium alginate, polyvinyl-pyrrolidone, gum tragacanth and gum acacia; dispersing or wetting agents such as lecithin or condensation products of an alkylene oxide with fatty acids (for example polyoxethylene stearate), or condensation products of ethylene oxide with long chain aliphatic alcohols, for example heptadecaethyleneoxycetanol, or condensation products of ethylene oxide with partial esters derived from fatty acids and a hexitol such as polyoxyethylene sorbitol monooleate, or condensation products of ethylene oxide with long chain aliphatic alcohols, for example heptadecaethyleneoxycetanol, or condensation products of ethylene oxide with partial esters derived from fatty acids and a hexito
- the aqueous suspensions may also contain one or more preservatives such as ethyl or propyl p-hydroxybenzoate; anti-oxidants such as ascorbic acid; coloring agents; flavoring agents; and/or sweetening agents such as sucrose, saccharine or aspartame.
- preservatives such as ethyl or propyl p-hydroxybenzoate
- anti-oxidants such as ascorbic acid
- coloring agents such as ascorbic acid
- flavoring agents such as ascorbic acid
- sweetening agents such as sucrose, saccharine or aspartame.
- Oily suspensions may be formulated by suspending the active ingredient in a vegetable oil such as arachis oil, olive oil, sesame oil or coconut oil or in a mineral oil such as liquid paraffin.
- the oily suspensions may also contain a thickening agent such as beeswax, hard paraffin or cetyl alcohol. Sweetening agents such as those set out above, and flavoring agents may be added to provide a palatable oral preparation. These compositions may be preserved by the addition of an anti-oxidant such as ascorbic acid.
- Dispersible powders and granules suitable for preparation of an aqueous suspension by the addition of water generally contain the active ingredient together with a dispersing or wetting agent, suspending agent and one or more preservatives. Suitable dispersing or wetting agents and suspending agents are exemplified by those already mentioned above. Additional excipients such as sweetening, flavoring and coloring agents, may also be present.
- the pharmaceutical compositions may also be in the form of oil-in-water emulsions.
- the oily phase may be a vegetable oil, such as olive oil or arachis oil, or a mineral oil, such as for example liquid paraffin or a mixture of any of these.
- Suitable emulsifying agents may be, for example, naturally-occurring gums such as gum acacia or gum tragacanth, naturally-occurring phosphatides such as soya bean, lecithin, an esters or partial esters derived from fatty acids and hexitol anhydrides (for example sorbitan monooleate) and condensation products of the said partial esters with ethylene oxide such as polyoxyethylene sorbitan monooleate.
- the emulsions may also contain sweetening, flavoring and preservative agents.
- Syrups and elixirs may be formulated with sweetening agents such as glycerol, propylene glycol, sorbitol, aspartame or sucrose, and may also contain a demulcent, preservative, flavoring and/or coloring agent.
- the pharmaceutical compositions may also be in the form of a sterile injectable aqueous or oily suspension, which may be formulated according to known procedures using one or more of the appropriate dispersing or wetting agents and suspending agents, which have been mentioned above.
- a sterile injectable preparation may also be a sterile injectable solution or suspension in a non-toxic parenterally-acceptable diluent or solvent, for example a solution in 1 ,3-butanediol.
- Compositions for administration by inhalation may be in the form of a conventional pressurized aerosol arranged to dispense the active ingredient either as an aerosol containing finely divided solid or liquid droplets.
- Conventional aerosol propellants such as volatile fluorinated hydrocarbons or hydrocarbons may be used and the aerosol device is conveniently arranged to dispense a metered quantity of active ingredient.
- the amount of active ingredient that is combined with one or more excipients to produce a single dosage form will necessarily vary depending upon the host treated and the particular route of administration.
- Routes of Administration and Dosage Regimes the reader is referred to Chapter 25.3 in Volume 5 of Comprehensive Medicinal Chemistry (Corwin Hansch; Chairman of Editorial Board), Pergamon Press 1990.
- the compounds of Formula (I), (la), (lb) and Table 1 may be administered once, twice, three times a day or as many times in a 24 hour period as medically necessary.
- One of skill in the art would readily be able to determine the amount of each individual dose based on the subject.
- the compounds of Formula (I), (la), (lb) or Table 1 are administered in one dosage form.
- the compounds of formula (I), (la), (lb) or Table are administered in multiple dosage forms.
- a JAK-related disorder in a subject in need thereof, comprising administering to the subject an effective amount of a compound of Formula (I), (la), (lb) or Table 1 , or a pharmaceutically acceptable salt or solid form thereof.
- a compound of Formula (I), (la), (lb) or Table 1 or a pharmaceutically acceptable salt or solid form thereof, for use in treating a JAK-related disorder.
- a compound of Formula (I), (la), (lb) or Table 1 or a pharmaceutically acceptable salt or solid form thereof, in the manufacture of a medicament for treating a JAK-related disorder.
- compositions comprising a compound of Formula (I), (la), (lb) or Table 1 , or a pharmaceutically acceptable salt or solid form thereof, for use in treating a JAK-related disorder.
- JK-related disorder includes cancer, cancer cachexia and immune disorders.
- cancer includes cancers with: (i) an EGFR-related etiology such as non-small cell lung cancer (NSCLC), head and neck: squamous cell cancer (HNSCC) and colorectal cancer; (ii) and activating RAS family mutations such as NSCLC, pancreatic cancer, colorectal cancer, prostate cancer, melanoma, thyroid cancer, bladder cancer, cholangiocarcinoma, and leukemia; (iii) a HER2 amplification or mutation such as breast cancer, gastric cancer, lung cancer; (iv) an ALK gene activation such as lung cancer, breast cancer, colorectal cancer, diffuse large B-cell lymphoma, anaplastic large cell lymphoma; (v) a MET amplification or mutation such as NSCLC, gastric cancer, colorectal cancer, papillary renal cell carcinoma; and (vi) an FGFR-related etiology such as breast cancer, gastric cancer, endometrial cancer,
- the cancer is pancreatic cancer, gastrointestinal cancer, breast cancer, a gynecological cancer (e.g., ovarian cancer or cervical cancer), bladder cancer, SCHN, non-small cell lung cancer or small cell lung cancer. In some embodiments, the cancer has metastasized.
- a compound of Formula (I), (la), (lb), or Table 1 comprising administering to the subject an effective amount of a compound of Formula (I), (la), (lb), or Table 1 , or a pharmaceutically acceptable salt or solid form thereof in combination with an anti-cancer therapeutic agent, or a pharmaceutically acceptable salt thereof.
- a compound of Formula (I), (la), (lb) or Table 1 or a pharmaceutically acceptable salt or solid form thereof in combination with anti-cancer therapeutic agent, or a pharmaceutically acceptable salt thereof, for use in treating a cancer.
- a compound of Formula (I), (la), (lb) or Table 1 or a pharmaceutically acceptable salt or solid form thereof, in combination with an anti-cancer therapeutic agent, or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for treating cancer.
- compositions comprising a compound of Formula (I), (la), (lb) or Table 1 , or a pharmaceutically acceptable salt or solid form thereof, in combination with an anti-cancer therapeutic agent, or a pharmaceutically acceptable salt thereof, for use in treating cancer.
- the language "in combination with” includes administering the compound of Formula (I), (la), (lb) or Table 1 , or a pharmaceutically acceptable salt thereof, and the anti-cancer therapeutic agent, or pharmaceutically acceptable salt thereof, sequentially, separately or simultaneously.
- the compound of Formula (I), (la), (lb) or Table 1 , or a pharmaceutically acceptable salt thereof, and the anti-cancer therapeutic agent, or pharmaceutically acceptable salt thereof are administered in separate formulations, and are administered at substantially the same time, sequentially or separately.
- the compound of Formula (I), (la), (lb) or Table 1 , or pharmaceutically acceptable salt thereof is administered for one day, two days, three days, four days, five days, six days, seven days, eight days, nine days, ten days, 1 1 days, 12 days, 13 days, 14 days, three weeks or one month in a row.
- compound of Formula (I), (la), (lb) or Table 1 is administered intermittently, for example, for 7 days followed by a 7 day clearance period (e.g., 7 days on/7 days off), for 1 day followed by a 6 day clearance period (e.g., 1 day on/6 days off), for 2 days followed by a 5 day clearance period (2 days on/5 days off), for 3 days followed by a 4 day clearance period (e.g., 3 days on/4 days off), for 4 days followed by a 3 day clearance period (e.g., 4 days on/3 days off), for 5 days followed by a 2 day clearance period (5 days on/2 days off), or for 6 days followed by a 1 day clearance period (6 days on/1 day off).
- a 7 day clearance period e.g., 7 days on/7 days off
- a 6 day clearance period e.g., 1 day on/6 days off
- 2 days followed by a 5 day clearance period (2 days on/5 days off
- 3 days followed by a 4 day clearance period e.g., 3 days on
- anti-cancer therapeutic agent includes, for example, EGFR inhibitors, MAPK pathway inhibitors, Raf inhibitors, HER2 inhibitors, FGFR inhibitors, antimetabolites, alkylating agents and antimitotic agents, and pharmaceutically acceptable salts thereof.
- a compound of Formula (I), (la), (lb) or Table 1 is administered in combination with one or more EGFR inhibitors.
- EGFR inhbitors include EGFR antibodies, ABX-EGF, anti-EGFR immunoliposomes, EGF-vaccine, EMD-7200, ERBITUX® (cetuximab), HR3, IgA antibodies, IRESSA® (gefitinib), TARCEVA® (erlotinib or OSI-774), TP- 38, EGFR fusion protein, TYKERB® (lapatinib), TAGRISSOTM (osimertinib or AZD9291 ), GILOTRIF® (afatinib), CO-1686, WZ4002, PD153035, PF 00299804 and the like.
- a compound of Formula (I), (la), (lb) or Table 1 is administered in combination with osimertinib. In some embodiments, a compound of Formula (I), (la), (lb) or Table 1 is administered in combination with gefitinib.
- a compound of Formula (I), (la), (lb) or Table 1 is administered in combination with one or more MAPK pathway inhibitors.
- MAPK pathway inhibitors include MEK inhibitors such as Selumetinib, Mekinist® (trametinib), Cobimetinib, PD0325901 , Pimasertib, MEK162, Refametinib and the like; Raf and B-Raf inhibitors which include vemurafenib, dabrafenib, Encorafenib (LGX818) and the like.
- a compound of Formula (I), (la), (lb) or Table 1 is administered in combination with one or more HER2 inhibitors.
- HER2 inhibitors include CP-724-714, CI-1033 (canertinib), HERCEPTIN® (trastuzumab), TYKERB® (lapatinib), OMNITARG® (2C4, petuzumab), TAK-165, GW-572016 (ionafarnib), GW-282974, EKB-569, PI-166, dHER2 (HER2 vaccine), APC-8024 (HER-2 vaccine), anti-HER/2neu bispecific antibody, B7.her2lgG3, AS HER2 bifunctional bispecific antibodies, mAB AR-209, mAB 2B-1 and the like.
- a compound of Formula (I), (la), (lb) or Table 1 is administered in combination with one or more ALK inhibitors.
- ALK inhibitors include crizotinib, ceritinib, and the like.
- a compound of Formula (I), (la), (lb) or Table 1 is administered in combination with one or more FGFR inhibitors.
- FGFR inhibitors include AZD4547, BJG398, Dovitinib, Lucitanib, MGFR1877S, FP-1039 and the like.
- a compound of Formula (I), (la), (lb) or Table 1 is administered in combination with one or more MET inhibitors.
- MET inhibitors include Savolitinib, Onartuzumab, Rilotumumab, Cabozantinib, Tivantinib, LY2875358, Ficlatuzumab, Foretinib, Crizotinib, INC280, AMG337, MSC21561 19J and the like
- a compound of Formula (I), (la), (lb) or Table 1 is administered in combination with one or more antimetabolites.
- Antimetabolites include ALIMTA® (pemetrexed disodium, LY231514, MTA), 5-azacitidine, XELODA® (capecitabine), carmofur, LEUSTAT® (cladribine), clofarabine, cytarabine, cytarabine ocfosfate, cytosine arabinoside, decitabine, deferoxamine, doxifluridine, eflornithine, EICAR (5-ethynyl-1-3-D-ribofuranosylimidazole-4- carboxamide), enocitabine, ethnylcytidine, fludarabine, 5-fluorouracil alone or in combination with leucovorin, GEMZAR® (gemcitabine), hydroxyurea, ALKERAN® (melphalan),
- mercaptopurine 6-mercaptopurine riboside, methotrexate, mycophenolic acid, nelarabine, nolatrexed, ocfosfate, pelitrexol, pentostatin, pemextred, raltitrexed, Ribavirin, triapine, trimetrexate, S-1 , tiazofurin, tegafur, TS-1 , vidarabine, UFT and the like.
- a compound of Formula (I), (la), (lb) or Table 1 is administered in combination with one or more alkylating agents.
- Alkylating agents include altretamine, AMD- 473, AP-5280, apaziquone, bendamustine, brostallicin, busulfan, cisplatin, carboplatin, carboquone, carmustine (BCNU), chlorambucil, CLORETAZINE® (laromustine, VNP 40101 M), cyclophosphamide, decarbazine, estramustine, fotemustine, glufosfamide, ifosfamide, KW- 2170, lomustine (CCNU), mafosfamide, melphalan, mitobronitol, mitolactol, nimustine, nitrogen mustard N-oxide, nitrosoureas, oxaliplatin, ranimustine, temozolomide, thiotepa, TREANDA® (
- a compound of Formula (I), (la), (lb) or Table 1 is administered in combination with one or more antimitotic agents.
- Antimitotic agents include batabulin, epothilone D (KOS-862), N-(2-((4-hydroxyphenyl)amino)pyridin-3-yl)-4- methoxybenzenesulfonamide, ixabepilone (BMS 247550), paclitaxel, TAXOTERE® (docetaxel),
- PNU100940 (109881 ), patupilone, XRP-9881 (larotaxel), vinflunine, ZK-EPO (synthetic epothilone) and the like.
- cancer cachexia includes a syndrome with symptoms that includes host tissue wasting, anorexia, asthenia and abnormal host intermediary metabolism.
- the subject suffering from cancer cachexia has pancreatic cancer or an upper gastrointestinal cancer, for example, esophageal cancer, stomach cancer, gastric cancer, liver cancer, gall bladder cancer, neuroendocrine cancer or Barrett's esophagus.
- the subject suffering from cancer cachexia has terminal cancer.
- disclosed are methods for treating cancer cachexia in a subject in need thereof comprising administering to the subject an effective amount of a compound of Formula
- a compound of Formula (I), (la), (lb) or Table 1 or a pharmaceutically acceptable salt or solid form thereof, for use in treating cancer cachexia.
- a compound of Formula (I), (la), (lb) or Table 1 or a pharmaceutically acceptable salt or solid form thereof, in the manufacture of a medicament for treating cancer cachexia.
- compositions comprising a compound of
- Formula (I), (la), (lb) or Table 1 or a pharmaceutically acceptable salt or solid form thereof, for use in treating cancer cachexia.
- immune disorder includes, for example, bone marrow disorders ⁇ e.g., myelofibrosis and polycythemia vera), rheumatoid arthritis, psoriasis, irritable bowel disease (IBD), Crohn's disease, lupus, multiple sclerosis, asthma, autoimmune thyroid disorders (e.g.,
- Hashimoto's thyroiditis Hashimoto's thyroiditis, Graves' disease or post-partum thyroiditis), ulcerative colitis, Alopecia areata, vitiligo and myositis.
- a method for treating an immune disorder in a subject in need thereof comprising administering to the subject an effective amount of a compound of Formula (I), (la), (lb), or Table 1 , or a pharmaceutically acceptable salt or solid form thereof.
- a compound of Formula (I), (la), (lb) or Table 1 or a pharmaceutically acceptable salt or solid form thereof, for use in treating an immune disorder.
- a compound of Formula (I), (la), (lb) or Table 1 or a pharmaceutically acceptable salt or solid form thereof, in the manufacture of a medicament for treating an immune disorder.
- compositions comprising a compound of Formula (I), (la), (lb) or Table 1 , or a pharmaceutically acceptable salt or solid form thereof, for use in treating an immune disorder.
- a compound of Formula (I), (la), (lb) or Table 1 comprising administering to the subject an effective amount of a compound of Formula (I), (la), (lb) or Table 1 , or a pharmaceutically acceptable salt or solid form thereof.
- a compound of Formula (I), (la), (lb) or Table 1 or a pharmaceutically acceptable salt or solid form thereof, for use in inhibiting JAK.
- a compound of Formula (I), (la), (lb) or Table 1 or a pharmaceutically acceptable salt or solid form thereof, in the manufacture of a medicament for inhibiting JAK.
- compositions comprising a compound of Formula (I), (la), (lb) or Table 1 , or a pharmaceutically acceptable salt or solid form thereof, for use in inhibiting JAK.
- JAK includes a family of Janus kinases that are intracellular, nonreceptor tyrosine kinases that transduce cytokine-mediated signals via the JAK-STAT pathway.
- JAK includes JAK1 , JAK2 and JAK3.
- the compounds of Formula (I), (la) and (lb) are selective inhibitors of JAK1 , JAK2 and/or JAK3.
- selective inhibitor includes compounds that have a greater inhibitory effect (as demonstrated, for example, by a lower IC50) for one or two of the JAK family members over the other JAK family members.
- a JAK1 -selective inhibitor exhibits greater inhibitory effect on JAK1 over JAK2 and JAK3; a JAK2-selective inhibitor exhibits a greater inhibitory effect on JAK2 over JAK1 and JAK3; a JAK3-selective inhibitor exhibits a greater inhibitory effect on JAK3 over JAK1 and JAK2; a JAK1/2-selective inhibitor exhibits a greater inhibitory effect on JAK1 and JAK2 over JAK3; a JAK1/3-selective inhibitor exhibits a greater inhibitory effect on JAK1 and JAK3 over JAK2; and a JAK2/3-selective inhibitor exhibits a greater inhibitory effect on JAK2 and JAK3 over JAK1 .
- the compounds of Formula (I), (la) and (lb) are JAK1 -selective inhibitors. In some embodiments, the compounds of Formula (I), (la) and (lb) are JAK1/2-selective inhibitors.
- the language "effective amount” includes an amount of a compound of Formula (I), (la), (lb) or Table 1 that will elicit a biological or medical response in a subject, for example, the reduction or inhibition of enzyme or protein activity related to JAK, cancer or an immune disorder; amelioration of symptoms of cancer or an immune disorder; or the slowing or delaying of progression of cancer or an immune disorder.
- the language "effective amount” includes the amount of a compound of Formula (I), (la), (lb) or Table 1 , that when administered to a subject, is effective to at least partially alleviate, inhibit, and/or ameliorate cancer or an immune disorder or inhibit JAK, and/or reduce or inhibit the growth of a tumor or proliferation of cancerous cells in a subject.
- the term "subject” includes warm blooded mammals, for example, primates, dogs, cats, rabbits, rats, and mice.
- the subject is a primate, for example, a human.
- the subject is suffering from cancer or an immune disorder.
- the subject is in need of treatment ⁇ e.g., the subject would benefit biologically or medically from treatment).
- the subject is suffering from cancer cachexia.
- the subject is suffering from cancer.
- the subject is suffering from cancer cachexia.
- the subject is suffering from an immune disorder.
- the subject may have elevated blood levels of inflammatory biomarkers e.g., serum systemic C-reactive protein (CRP), IL-6, TNFa, IL-1 , procalcitonin and IL-8.
- the subject may be suffering from a high STAT3- positive tumor.
- the subject is suffering from a EGFR-M positive cancer (e.g., non-small cell lung cancer).
- the EGFR-M positive cancer has a predominately T790M-positive mutation.
- the EGFR-M positive cancer has a predominately T790M-negative mutation.
- the subject is suffering from a KRAS mutant cancer (e.g., KRAS mutated non-small cell lung cancer).
- the subject is suffering from metastatic pancreatic cancer, metastatic
- metastatic breast cancer metastatic breast cancer
- a metastatic gynecologic cancer e.g., metastatic ovarian cancer or metastatic cervical cancer
- metastatic bladder cancer metastatic squamous cell head and neck cancer (SCHN)
- metastatic non-small cell lung cancer metastatic haematological cancers ⁇ e.g., non-Hodgkin's lymphoma) or metastatic small cell lung cancer.
- the subject suffering from cancer may show evidence of immune inflammation, including, for example, the presence of PDL1 , interferon gamma, tumor-infiltrating leukocytes and gene expression signatures indicating increased type I or type II interferon signaling, abnormal levels tumor suppressive cells, such as regulatory T lymphocytes or myeloid-derived cells, abnormal levels of granulocytes or proteins indicating the presence of granulocytes.
- inhibitor includes a decrease in the baseline activity of a biological activity or process.
- the compounds of Formula (I), (la), (lb) or Table 1 inhibit JAK. .
- the language “treat,” “treating” and “treatment” includes the reduction or inhibition of enzyme or protein activity related to JAK, cancer or an immune disorder in a subject, amelioration of one or more symptoms of a cancer or an immune disorder in a subject, or the slowing or delaying of progression of cancer or an immune disorder in a subject.
- the language “treat,” “treating” and “treatment” also includes the reduction or inhibition of the growth of a tumor or proliferation of cancerous cells in a subject.
- MultiGram III SFC instrument with UV collection
- the column used was a Waters Acquity HSS T3 (1 .8 ⁇ , 2.1 x 50 mm), for basic analysis the column used was a Waters Acquity BEH C18 (1 .7 ⁇ 2.1x50 mm).
- HATU (1 -[Bis(dimethylamino)methylene]-1 H-1 ,2,3-triazolo[4,5-b]pyridinium 3- oxid hexafluorophosphate)
- the reaction mixture was diluted with DCM (2 L), and washed sequentially with water (500 mL x 2), 10% aqueous K 2 C0 3 (500 mL x 2), and 1 M HCI (500 mL x 2) and saturated NaCI (500 mL x 2).
- the organic layer was dried over Na2S0 4 , filtered and evaporated.
- 500 mL EA was added.
- the solvent was removed under reduced pressure.
- 1000 mL TBME was added.
- the reaction was allowed to warm up to -15°C and stirred for 16 hour.
- the reaction mixture was quenched with ice water (100 mL), extracted with ethyl acetate (3 x 200 mL), the organic layer was dried, filtered and evaporated to afford a tan solid.
- the crude product was purified by flash silica chromatography, elution gradient 100 to 0% petroleum ether in ethyl acetate.
- Trifluoromethanesulfonic anhydride (53.6 mL, 317 mmol) was added dropwise to (S)- methyl 2-hydroxypropanoate (30 g, 288 mmol) and 2,6-lutidine (37 mL, 317 mmol) in DCM (500 mL) at -78°C over a period of 1 hour. The resulting solution was stirred at -78°C for 0.5 hours. The solution was then warmed to room temperature for 1 hour. The organic phase was washed with 1 N HCI (aq.) (2 x 100 mL) and dried over sodium sulfate, then filtered and evaporated.
- methylpiperazin-1 -yl)propanamide (0.38 g, 0.7 mmol, Intermediate 58) was dissolved in DMSO (7 ml.) and cesium fluoride (0.32 g, 2.1 mmol) was added. The reaction mixture was then heated at 100°C and allowed to stir for 2 hours. The reaction mixture was diluted with ethyl acetate and water. The organic layer was separated and the aqueous layer was extracted with ethyl acetate.
- Stereochemical assignment of the enantiomers was made based on the major product formation of the reaction and validated by biological activity against JAK1 in the Enzyme Inhibition Studies, as shown in Example 66.
- Stereochemical assignment of the enantiomers was made based on the major product formation of the reaction and validated by biological activity against JAK1 in the Enzyme Inhibition Studies, as shown in Example 66.
- Stereochemical assignment of the enantiomers was made based on the major product formation of the reaction and validated by biological activity against JAK1 in the Enzyme Inhibition Studies, as shown in Example 66.
- Example 31b analytical chiral-HPLC: Chiralpak IA, 5 ⁇ silica, 0.46x25 cm column, hexanes (0.1 % TEA): EtOH (60:40) at
- the reaction mixture was diluted with ethyl acetate (100 mL), and washed with saturated aqueous Na2C03 (50 mL), water (50 mL) and brine (50 mL). The organic layer was dried, filtered and evaporated to afford crude product.
- the crude product was purified by preparative HPLC (XSelect CSH Prep C18 OBD column, 5 ⁇ , 19x150 mm ), employing a gradient of 30-70% acetonitrile in 0.03% aqueous ammonia as eluents.
- Stereochemical assignment of the enantiomers was made based on the biological activity against JAK1 in the Enzyme Inhibition Studies, as shown in Example 66.
- Examples 47 and 48 i2ff)-N-i3-f2-ri3-Methoxy-1 -methyl-1 H-pyrazol-4-yl)amino1pyrimidin- 4-ylM H-indol-7-yl)-2-i4-methylpiperazin-1 -yl)butanamide and (2S)-N-(3-(2-r(3-Methoxy-1 - methyl-1 H-pyrazol-4-yl)amino1pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1 - vDbutanamide
- Stereochemical assignment of the enantiomers was made based on the biological activity against JAK1 in the Enzyme Inhibition Studies, as shown in Example 66.
- Stereochemical assignment of the enantiomers was made based on the biological activity against JAK1 in
- Example 60 Example 61
- the crude product was purified by preparative HPLC (XBridge Prep C18 OBD column, 5 ⁇ silica, 19 mm diameter, 150 mm length), using decreasingly polar mixtures of water (containing 0.2% ammonia) and acetonitrile as eluents.
- the crude product was purified by preparative chiral-HPLC on a Lux Cellulose-4 column, eluting isocratically with 50% ethanol in isohexane (modified with 0.1 % triethylamine) as eluent.
- Stereochemical assignment of the enantiomers was made based on the biological activity against JAK1 in the Enzyme Inhibition Studies, as shown in Example 66.
- Stereochemical assignment of the enantiomers was made based on the biological activity against JAK1 in the Enzyme Inhibition Studies, as shown in Example 66.
- JAK2 amino acids 831 -1 132
- JAK3 amino acids 781 -1 1244
- buffer conditions 50 mM HEPES pH 7.3, 1 mM DTT, 0.01 % Tween® 20, 50 ⁇ g/mL BSA, and 10 mM MgC .
- JAK enzyme was expressed as an N-terminal GST fusion in insect cells and purified by glutathione-affinity and size-exclusion
- Enzymes were assayed both at their respective ATP Km (JAK1 : 55 ⁇ , JAK2: 15 ⁇ , JAK3: 3 ⁇ ) and the approximated high end of physiological ATP concentration of 5 mM, in the presence of inhibitor dosed at 30, 3, 0.3, 0.03, 0.003 and 0 ⁇ final test concentrations.
- JAK1 6 nM of enzyme (for Km ATP assay) or 4 nM enzyme (for high ATP assay) was incubated with 1.5 ⁇ peptide substrate (FITC-C6-KKHTDDGYMPMSPGVA-NH2 (SEQ ID NO:1 ), Intonation, Boston, MA).
- NCI-H1975 cells were plated onto Costar #3701 96 or 384 well tissue-culture treated plates at 5,000 cells/well in 30uL medium (RPMI, 10% FBS, supplemented with L-glutamine) and incubated overnight at 37 °C in 5% CO2.
- Phospho STAT3 signal was quantitated utilizing Cell Signaling Technology #7146B Pathscan 97hosphor STAT3 antibody pair kit, following manufacturer's instructions.
- Cells were dosed with compound and incubated at 37 °C in 5% CO2 for 2 hours, after which medium and compound were aspirated and cells lysed with 35uL cold 1x Cell Signaling Lysis buffer and chilled at 4°C for 1 -2 hours.
- Lysate was incubated on STAT3 capture plates at 4°C overnight, washed 3x with Tris-Buffered Saline with 0.05% Tween® 20 (TBST), then 98 phosphor STAT3 detection antibody was applied for 2 hours. Following washing with TBST (3x), HRP-secondary antibody was applied for 2 hours. After additional washing, signal was detected using TMB and Stop solution and read at 450 nm using Tecan Infinite M100.
- I C50 values (the concentration that causes 50% inhibition) were calculated by plotting percent inhibition of the phosphor-signal relative to untreated sample (maximum signal) and positive control treated sample (maximum inhibition/minimum signal), using Xlfit4 version 4.2.2 for Microsoft Excel.
- Example 68 Solid Forms of i2ff)-A -i3-f2-ri3-methoxy-1 -methyl-1 H-pyrazol-4- yl)amino1pyrimidin-4-yl>-1 H-indol-7-yl)-2-i4-methylpiperazin-1 -yl)propanamide
- XRPD analysis was performed using a Bruker D4 (or D8) diffractometer, which is commercially available from Bruker AXS IncTM (Madison, Wisconsin).
- the XRPD spectra were obtained by mounting a sample (approximately 20 mg) of the material for analysis on a single silicon crystal wafer mount (e.g., a Bruker silicon zero background X-ray diffraction sample holder) and spreading out the sample into a thin layer with the aid of a microscope slide.
- the sample was spun at 30 revolutions per minute (to improve counting statistics) and irradiated with X-rays generated by a copper long-fine focus tube operated at 40 kV and 40 mA with a wavelength of 1.5406 angstroms (i.e., about 1.54 angstroms).
- the sample was exposed for 1 second per 0.02 degree 2-theta increment (continuous scan mode) over the range 5 degrees (or 2 degrees) to 40 degrees 2-theta in theta-theta mode.
- the running time was -17 min for D4 and -15 min for D8.
- XRPD 2 ⁇ values may vary with a reasonable range, e.g., in the range ⁇ 0.2° and that XRPD intensities may vary when measured for essentially the same crystalline form for a variety of reasons including, for example, preferred orientation.
- Principles of XRPD are described in publications, such as, for example, Giacovazzo, C. et al. (1995), Fundamentals of
- DSC analysis was performed on samples prepared according to standard methods using a Q SERIESTM Q1000 DSC calorimeter available from TA INSTRUMENTS® (New Castle, Delaware). A sample (approximately 2 mg) was weighed into an aluminum sample pan and transferred to the DSC. The instrument was purged with nitrogen at 50 mL/min and data collected between 22 °C and 300 °C, using a dynamic heating rate of 10 °C/minute. Thermal data was analyzed using standard software, e.g., Universal v.4.5A from TA INSTRUMENTS®. Thermoqravimetry Analysis (TGA)
- TGA TGA was performed on samples prepared according to standard methods using a Q SERIESTM Q5000 thermogravimetry analyzer available from TA Instruments INSTRUMENTS® (New Castle, Delaware). A sample (approximately 5 mg) was placed into an aluminum sample pan and transferred to the TGA furnace. The instrument was purged with nitrogen at 50 mL/min and data collected between 25°C and 300 °C, using a dynamic heating rate of 10 °C/minute. Thermal data was analyzed using standard software, e.g., Universal v.4.5A from TA
- Example 68A Form A i2R)-N-i3-f2-[i3-methoxy- 1-methyl-1H ⁇ yrazol-4-yl)aminolpyrimidin-4-yl ⁇ - 1H-indol-7-yl)-2-(4-methylpiperazin- 1 -vDpropanamide
- Method 1 50 mg of off-white amorphous (2f?)-N-(3- ⁇ 2-[(3-methoxy-1 -methyl-1 H-pyrazol-4- yl)amino]pyrimidin-4-yl ⁇ -1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide was dissolved in 0.4 ml of TBME in a 4 mL vial. The solid precipitated out from the solution after 30 minutes. The slurry was stirred under ambient conditions overnight. The resulting white solid material was identified as Form A by XRPD analysis.
- crystalline seeds obtained from Method 1 were mixed in a 20 mL vial.
- 5 mL of TBME was added to form a slurry.
- the slurry was stirred under ambient conditions overnight and a homogenous slurry formed.
- the slurry was filtered, and the resulting solid was washed with TBME and dried in air. 498 mg of a white crystalline solid was obtained and identified as Form A by XRPD analysis.
- Form A (Method 2) was analyzed by XRPD and the results are tabulated below (Table 17) and shown in Figure 1 .
- Form A (Method 2) was analyzed by thermal techniques. DSC analysis indicated that Form A has an endotherm event of desolvation with an onset at 1 10 °C and a peak at 1 13 °C. TGA indicated that Form A exhibits a mass loss of about 7.8 % upon heating from about 25 °C to about 150 °C. A representative DSC/TGA thermogram of Form A is shown in Figure 2.
- Example 68B Form B (2R)-N-(3-f2-f(3-methoxy- 1-methyl-1H-pyrazol-4-yl)aminolpyrimid 1H-indol-7-yl)-2-(4-methylpiperazin- 1 -yQpropanamide
- Form B was analyzed by XRPD and the results are tabulated below (Table 18) and shown in Figure 3.
- Form B was analyzed by thermal techniques. DSC analysis indicated that Form B has an endotherm event of desolvation with an onset at 1 12 °C and a peak at 1 17 °C. TGA indicated that Form B exhibits a mass loss of about 10.0 % upon heating from about 25 °C to about 200 °C. A representative DSC/TGA thermogram of Form B is shown in Figure 4.
- Example 68C Form C ⁇ 2R)-N- ⁇ 3- ⁇ 2-[ ⁇ 3-methoxy- 1-methyl-1H ⁇ yrazol-4-yl)amino]pyrimidin-4-yl ⁇ -
- a white crystalline solid was obtained and was identified as Form C by XRPD.
- Form C was analyzed by thermal techniques. DSC analysis indicated that Form C has an endotherm event of desolvation with an onset at 1 12 °C and a peak at 1 14 °C. TGA indicated that Form C exhibits a mass loss of about 9.2 % upon heating from about 25 °C to about 175 °C. A representative DSC/TGA thermogram of Form C is shown in Figure 6.
- Example 68D Form D (2R ⁇ -N-(3- ⁇ 2-f(3-methoxy- 1-methyl-1 H-pyrazol-4-yl)amino]pyrimidin-4- yl ⁇ - 1 H-indol- 7- yl) -2- (4-meth ylpiperazin - 1 - vDpropanamide
- Method 1 Approximately 100 mg of light yellow amorphous (2f?)-N-(3- ⁇ 2-[(3-methoxy-1- methyl-1 H-pyrazol-4-yl)amino]pyrimidin-4-yl ⁇ -1 /- -indol-7-yl)-2-(4-methylpiperazin-1- yl)propanamide was dissolved in 1 ml of EtOAC to get a clear solution. The solution was placed in the freezer overnight and solid was precipitated out. The slurry was stirred at room
- Form D (Method 2) was analyzed by XRPD and the results are tabulated below (Table 20) and shown in Figure 7.
- Form D was analyzed by thermal techniques. DSC analysis indicated that Form D has an endotherm event of desolvation with an onset at 1 16 °C and a peak at 1 19 °C. TGA indicated that Form D exhibits a mass loss of about 8.0 % upon heating from about 25 °C to about 200 °C. A representative DSC/TGA thermogram of Form D is shown in Figure 8.
- Example 68E Form A (2R ⁇ -N-(3- ⁇ 2-f(3-methoxy- 1-methyl-1 H-pyrazol-4-yl)amino]pyrimidin-4- yl ⁇ - 1 H-indol-7-yl)-2-(4-methylpiperazin- 1 -vDpropanamide Saccharine Salt
- Form A of the saccharin salt was analyzed by thermal techniques. DSC analysis indicated that Form A has an endotherm event of melting point with an onset at 163 °C and a peak at 169 °C. TGA indicated that Form A exhibits a mass loss of about 3.1 % upon heating from about 25 °C to about 150 °C. A representative DSC/TGA thermogram of Form D is shown in Figure 10.
- a yellow crystal material was obtained and identified as Form B (2f?)-N-(3- ⁇ 2-[(3-methoxy-1-methyl-1 H-pyrazol-4-yl)amino]pyrimidin-4-yl ⁇ - 1 H-indol-7-yl)-2-(4-methylpiperazin-1-yl)propanamide saccharine salt by XRPD.
- Form B of the saccharin salt was analyzed by XRPD and the results are tabulated below (Table 22) and shown in Figure 1 1 .
- Form B of the saccharin salt was analyzed by thermal techniques.
- DSC analysis indicated that Form B has a broad endotherm event of desolvation with a peak at 53 °C, followed by two endotherm events, one with an onset at 153 °C and a peak at 162 °C and the other with an onset at 176 °C and a peak at 182 °C.
- TGA indicated that Form B exhibits a mass loss of about 2.7 % upon heating from about 25 °C to about 100 °C.
- a representative DSC/TGA thermogram of Form B is shown in Figure 12.
- Example 68G Form C i2R) ⁇ -i3-f2-[i3-methoxy-1-methyl-1H-pyrazol-4-yl)amino]pyrimidin-4- ylj- 1 H-indol-7-yl)-2-(4-methylpiperazin- 1 -vDpropanamide Saccharine Salt
- a yellow crystal material was obtained and identified as Form C (2f?)-N-(3- ⁇ 2-[(3- methoxy-1 -methyl-1 H-pyrazol-4-yl)amino]pyrimidin-4-yl ⁇ -1 /- -indol-7-yl)-2-(4-methylpiperazin-1 - yl)propanamide saccharine salt by XRPD.
- Form C of the saccharin salt was analyzed by XRPD and the results are tabulated below (Table 23) and shown in Figure 13.
- Example 68H Form D (2R)-N-(3- ⁇ 2-f(3-methoxy- 1-methyl-1 H-pyrazol-4-yl)amino]pyrimidin-4- yl ⁇ - 1 H-indol-7-yl)-2-(4-methylpiperazin- 1 -yPpropanamide Saccharine Salt
- Form D of the saccharin salt was analyzed by XRPD and the results are tabulated below
- Example 681 Form E i2R)-N-i3- ⁇ 2-[i3-methoxy- 1-methyl-1H ⁇ yrazol-4-yl)amino]pyrimidin-4-yl ⁇ -
- Form E of the saccharin salt was analyzed by XRPD and the results are tabulated below
- Example 68J (2R)-N-(3- ⁇ 2-f(3-methoxy- 1 -methyl- 1 H-pyrazol-4-yl)amino]pyrimidin-4-yl)- 1 H- indol-7-yl ⁇ -2-(4-methylpiperazin- 1-yl ⁇ propanamide Saccharine Hydrochloride Salt
- the counter ion solution was added to the solution of (2f?)-N-(3- ⁇ 2-[(3- methoxy-1 -methyl-1 H-pyrazol-4-yl)amino]pyrimidin-4-yl ⁇ -1 /- -indol-7-yl)-2-(4-methylpiperazin-1 - yljpropanamide dropwise.
- the resulting solution was subjected to slow evaporation and isolated by centrifugation.
- trimesic acid salt was analyzed by XRPD and the results are tabulated below (Table 28) and shown in Figure 18.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Immunology (AREA)
- Epidemiology (AREA)
- Physical Education & Sports Medicine (AREA)
- Vascular Medicine (AREA)
- Rheumatology (AREA)
- Dermatology (AREA)
- Urology & Nephrology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Description




































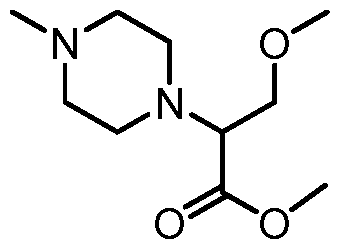












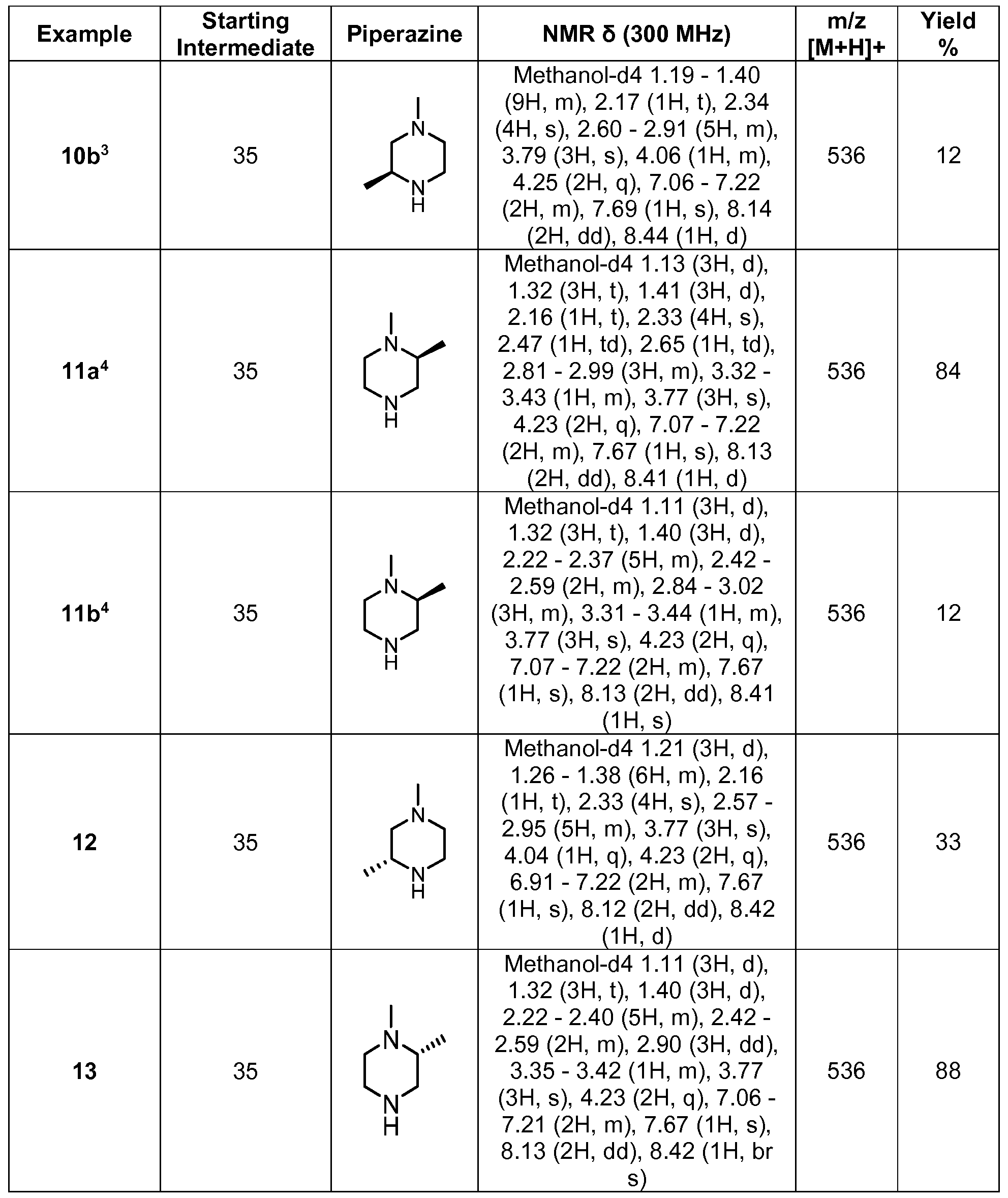































Claims



Priority Applications (16)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP16770031.9A EP3353168B1 (en) | 2015-09-25 | 2016-09-22 | Compounds and methods for inhibiting jak |
| CA2995430A CA2995430C (en) | 2015-09-25 | 2016-09-22 | Compounds and methods for inhibiting jak |
| CN201680055491.7A CN108368091B (en) | 2015-09-25 | 2016-09-22 | Compounds and methods for inhibiting JAK |
| MX2018003590A MX375724B (en) | 2015-09-25 | 2016-09-22 | COMPOUNDS AND METHODS FOR JAK INHIBITION. |
| AU2016328764A AU2016328764B2 (en) | 2015-09-25 | 2016-09-22 | Compounds and methods for inhibiting JAK |
| ES16770031T ES2956642T3 (en) | 2015-09-25 | 2016-09-22 | Compounds and methods for JAK inhibition |
| DK16770031.9T DK3353168T3 (en) | 2015-09-25 | 2016-09-22 | COMPOUNDS AND METHODS FOR INHIBITING JAK |
| EP23150992.8A EP4219482A1 (en) | 2015-09-25 | 2016-09-22 | Compounds and methods for inhibiting jak |
| KR1020257006096A KR20250035597A (en) | 2015-09-25 | 2016-09-22 | Compounds and methods for inhibiting jak |
| JP2018535238A JP6767491B2 (en) | 2015-09-25 | 2016-09-22 | Compounds and methods for inhibiting JAK |
| BR112018005833-5A BR112018005833B1 (en) | 2015-09-25 | 2016-09-22 | COMPOUND, PHARMACEUTICALLY ACCEPTABLE SALT, PHARMACEUTICAL COMPOSITION AND USE OF THE COMPOUND |
| CN202010626114.2A CN111848586B (en) | 2015-09-25 | 2016-09-22 | Compounds and methods for inhibiting JAK |
| RU2018112993A RU2760359C2 (en) | 2015-09-25 | 2016-09-22 | Compounds and methods for inhibiting jak |
| KR1020187008186A KR102774784B1 (en) | 2015-09-25 | 2016-09-22 | Compounds and methods for inhibiting JAK |
| HK19101824.9A HK1259422B (en) | 2015-09-25 | 2016-09-22 | Compounds and methods for inhibiting jak |
| ZA2018/00782A ZA201800782B (en) | 2015-09-25 | 2018-02-06 | Compounds and methods for inhibiting jak |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562232629P | 2015-09-25 | 2015-09-25 | |
| US62/232,629 | 2015-09-25 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017050938A1 true WO2017050938A1 (en) | 2017-03-30 |
Family
ID=56979589
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2016/072616 WO2017050938A1 (en) | 2015-09-25 | 2016-09-22 | Compounds and methods for inhibiting jak |
Country Status (17)
| Country | Link |
|---|---|
| US (5) | US9714236B2 (en) |
| EP (2) | EP3353168B1 (en) |
| JP (1) | JP6767491B2 (en) |
| KR (2) | KR102774784B1 (en) |
| CN (4) | CN111646980B (en) |
| AR (1) | AR106138A1 (en) |
| AU (1) | AU2016328764B2 (en) |
| BR (1) | BR112018005833B1 (en) |
| CA (1) | CA2995430C (en) |
| DK (1) | DK3353168T3 (en) |
| ES (1) | ES2956642T3 (en) |
| MX (1) | MX375724B (en) |
| PT (1) | PT3353168T (en) |
| RU (1) | RU2760359C2 (en) |
| TW (1) | TWI740843B (en) |
| WO (1) | WO2017050938A1 (en) |
| ZA (1) | ZA201800782B (en) |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2018134213A1 (en) * | 2017-01-17 | 2018-07-26 | Astrazeneca Ab | Jak1 selective inhibitors |
| WO2020016302A1 (en) * | 2018-07-18 | 2020-01-23 | Astrazeneca Ab | A xinafoate salt of a jak inhibiting compound |
| WO2020057669A1 (en) * | 2018-09-21 | 2020-03-26 | 上海轶诺药业有限公司 | Aromatic heterocyclic compound with kinase inhibitory activity |
| WO2020211839A1 (en) | 2019-04-19 | 2020-10-22 | Dizal (Jiangsu) Pharmaceutical Co., Ltd. | Jak1 selective kinase inhibitor |
| CN111961037A (en) * | 2020-09-17 | 2020-11-20 | 嘉兴特科罗生物科技有限公司 | Pharmaceutical compound as JAK kinase inhibitor |
| WO2022189496A1 (en) * | 2021-03-11 | 2022-09-15 | Janssen Pharmaceutica Nv | Lorpucitinib for use in the treatment of jak mediated disorders |
| US11524968B2 (en) | 2017-10-18 | 2022-12-13 | Hk Inno.N Corporation | Heterocyclic compound as a protein kinase inhibitor |
| RU2818002C2 (en) * | 2019-04-19 | 2024-04-23 | Дизал (Цзянсу) Фармасьютикал Ко., Лтд. | Selective kinase jak1 inhibitor |
| US12180185B2 (en) | 2018-11-15 | 2024-12-31 | Hk Inno.N Corporation | Compound as protein kinase inhibitor, and pharmaceutical composition comprising thereof |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2760359C2 (en) * | 2015-09-25 | 2021-11-24 | Дизал (Цзянсу) Фармасьютикалз Ко., Лимитед | Compounds and methods for inhibiting jak |
| CN109311858B (en) | 2016-05-26 | 2021-12-03 | 里科瑞尔姆Ip控股有限责任公司 | EGFR inhibitor compounds |
| CN110627775A (en) * | 2019-10-24 | 2019-12-31 | 嘉兴特科罗生物科技有限公司 | Small molecule compound |
| JP7613778B2 (en) | 2020-11-26 | 2025-01-15 | 科輝智薬(深▲セン▼)新薬研究中心有限公司 | Amide compounds, pharmaceutical compositions and uses thereof |
| CN115260128B (en) * | 2022-09-21 | 2022-12-09 | 苏州凯瑞医药科技有限公司 | Preparation method of novel JAK inhibitor key intermediate |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20080287475A1 (en) | 2005-10-28 | 2008-11-20 | Astrazeneca Ab | 4-(3-Aminopyrazole) Pyrimidine Derivatives for Use as Tyrosine Kinase Inhibitors in the Treatment of Cancer |
| WO2012030910A1 (en) * | 2010-09-01 | 2012-03-08 | Ambit Biosciences Corporation | 2-cycloquinazoline derivatives and methods of use thereof |
| WO2012116247A1 (en) * | 2011-02-25 | 2012-08-30 | Synta Pharmaceuticals Corp. | Hsp90 inhibitory compounds in treating jak/stat signaling-mediated cancers |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE602005027825D1 (en) | 2004-03-30 | 2011-06-16 | Vertex Pharma | AS AZHIBITORS OF JAK AND OTHER PROTEIN KINASES SUITABLE AZAINDOLE |
| GB0500492D0 (en) | 2005-01-11 | 2005-02-16 | Cyclacel Ltd | Compound |
| AU2007209928B2 (en) * | 2006-01-30 | 2013-03-28 | Exelixis, Inc. | 4-aryl-2-amino-pyrimidines or 4-aryl-2-aminoalkyl-pyrimidines as JAK-2 modulators and pharmaceutical compositions containing them |
| BRPI0720264B1 (en) * | 2006-12-08 | 2022-03-03 | Novartis Ag | COMPOUNDS AND COMPOSITIONS AS PROTEIN KINASE INHIBITORS |
| EP2440559B1 (en) * | 2009-05-05 | 2018-01-10 | Dana-Farber Cancer Institute, Inc. | Egfr inhibitors and methods of treating disorders |
| US8716303B2 (en) | 2009-05-22 | 2014-05-06 | Incyte Corporation | N-(hetero)aryl-pyrrolidine derivatives of pyrazol-4-yl-pyrrolo[2,3-d]pyrimidines and pyrrol-3-yl-pyrrolo[2,3-d]pyrimidines as janus kinase inhibitors |
| TWI466885B (en) * | 2009-07-31 | 2015-01-01 | Japan Tobacco Inc | Nitrogen-containing spiro compound and its medical use |
| US9198911B2 (en) | 2010-11-02 | 2015-12-01 | The Trustees Of Columbia University In The City Of New York | Methods for treating hair loss disorders |
| CA2815330A1 (en) * | 2010-11-09 | 2012-05-18 | Cellzome Limited | Pyridine compounds and aza analogues thereof as tyk2 inhibitors |
| EA026201B1 (en) * | 2010-11-19 | 2017-03-31 | Инсайт Холдингс Корпорейшн | Cyclobutyl substituted pyrrolopyridine and pyrrolopyrimidine derivatives as jak inhibitors |
| AU2012273164B2 (en) * | 2011-06-20 | 2015-05-28 | Incyte Holdings Corporation | Azetidinyl phenyl, pyridyl or pyrazinyl carboxamide derivatives as JAK inhibitors |
| EP3459565A1 (en) | 2012-03-29 | 2019-03-27 | The Trustees of Columbia University in the City of New York | Methods for treating hair loss disorders |
| RU2760359C2 (en) * | 2015-09-25 | 2021-11-24 | Дизал (Цзянсу) Фармасьютикалз Ко., Лимитед | Compounds and methods for inhibiting jak |
-
2016
- 2016-09-22 RU RU2018112993A patent/RU2760359C2/en active
- 2016-09-22 WO PCT/EP2016/072616 patent/WO2017050938A1/en active Application Filing
- 2016-09-22 AU AU2016328764A patent/AU2016328764B2/en active Active
- 2016-09-22 KR KR1020187008186A patent/KR102774784B1/en active Active
- 2016-09-22 CA CA2995430A patent/CA2995430C/en active Active
- 2016-09-22 CN CN202010629286.5A patent/CN111646980B/en active Active
- 2016-09-22 KR KR1020257006096A patent/KR20250035597A/en active Pending
- 2016-09-22 EP EP16770031.9A patent/EP3353168B1/en active Active
- 2016-09-22 MX MX2018003590A patent/MX375724B/en active IP Right Grant
- 2016-09-22 BR BR112018005833-5A patent/BR112018005833B1/en active IP Right Grant
- 2016-09-22 CN CN202010629290.1A patent/CN111606893B/en active Active
- 2016-09-22 JP JP2018535238A patent/JP6767491B2/en active Active
- 2016-09-22 CN CN202010626114.2A patent/CN111848586B/en active Active
- 2016-09-22 PT PT167700319T patent/PT3353168T/en unknown
- 2016-09-22 US US15/272,554 patent/US9714236B2/en active Active
- 2016-09-22 DK DK16770031.9T patent/DK3353168T3/en active
- 2016-09-22 CN CN201680055491.7A patent/CN108368091B/en active Active
- 2016-09-22 ES ES16770031T patent/ES2956642T3/en active Active
- 2016-09-22 EP EP23150992.8A patent/EP4219482A1/en active Pending
- 2016-09-23 AR ARP160102919A patent/AR106138A1/en active IP Right Grant
- 2016-09-23 TW TW105130891A patent/TWI740843B/en active
-
2017
- 2017-05-22 US US15/601,324 patent/US10167276B2/en active Active
-
2018
- 2018-02-06 ZA ZA2018/00782A patent/ZA201800782B/en unknown
- 2018-11-20 US US16/196,038 patent/US10654835B2/en active Active
-
2020
- 2020-04-28 US US16/860,169 patent/US11247983B2/en active Active
-
2021
- 2021-12-28 US US17/563,913 patent/US12319670B2/en active Active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20080287475A1 (en) | 2005-10-28 | 2008-11-20 | Astrazeneca Ab | 4-(3-Aminopyrazole) Pyrimidine Derivatives for Use as Tyrosine Kinase Inhibitors in the Treatment of Cancer |
| WO2012030910A1 (en) * | 2010-09-01 | 2012-03-08 | Ambit Biosciences Corporation | 2-cycloquinazoline derivatives and methods of use thereof |
| WO2012116247A1 (en) * | 2011-02-25 | 2012-08-30 | Synta Pharmaceuticals Corp. | Hsp90 inhibitory compounds in treating jak/stat signaling-mediated cancers |
Non-Patent Citations (23)
| Title |
|---|
| "Comprehensive Medicinal Chemistry", vol. 5, 1990, PERGAMON PRESS |
| "Remington's Pharmaceutical Sciences", 1985, MACK PUBLISHING COMPANY |
| BRANTLEY ET AL., CLIN. CANCER RES., vol. 14, 2008, pages 4694 - 4704 |
| BROMBERG J.; WANG TC., CANCER CELL, vol. 15, 2009, pages 79 - 80 |
| BROMBERG JF ET AL., CELL, vol. 98, 1999, pages 295 - 303 |
| BUNN, C.W.: "Chemical Crystallography", 1948, CLARENDON PRESS |
| GIACOVAZZO, C. ET AL.: "Fundamentals of Crystallography", 1995, OXFORD UNIVERSITY PRESS |
| GRIVENNIKOV, S.; KARIN, M., CANCER CELL, vol. 13, 2008, pages 7 - 9 |
| GUSCHIN ET AL., EMBO J, vol. 14, 1995, pages 1421 - 1429 |
| JENKINS, R.; SNYDER, R. L.: "Introduction to X-Ray Powder Diffractometry", 1996, JOHN WILEY & SONS |
| JENKINS, R; SNYDER, R.L.: "Introduction to X-Ray Powder Diffractometry", 1996, JOHN WILEY & SONS |
| KIM SM ET AL., MOL. CANCER THER, vol. 11, 2012, pages 2254 - 2264 |
| KLUG, H. P.; ALEXANDER, L. E., X-RAY DIFFRACTION PROCEDURES, 1974 |
| KLUG, H. P.; ALEXANDER, L. E.: "X-ray Diffraction Procedures", 1974, JOHN WILEY AND SONS |
| LEE ET AL., CANCER CELL, vol. 26, 2014, pages 207 - 221 |
| LEE ET AL., MOL. CANCER THER., vol. 5, 2006, pages 8 - 19 |
| MOTTOK ET AL., BLOOD, vol. 110, 2007, pages 3387 - 90 |
| OGATA ET AL., GASTROENTEROLOGY, vol. 131, 2006, pages 179 - 193 |
| SCHINDLER C; DARNELL JE JR., ANNU. REV. BIOCHEM., vol. 64, 1995, pages 621 - 651 |
| SONG, MOL. CANCER THER.., vol. 10, 2011, pages 481 - 494 |
| STAHL; WERMUTH: "Handbook of Pharmaceutical Salts: Properties, Selection, and Use", 2002, WILEY-VCH |
| VANSCHAEYBROECK ET AL., CELL REPORTS, vol. 7, 2014, pages 1940 - 1955 |
| YU H; JOVE R., NAT. REV. CANCER, vol. 4, 2004, pages 97 - 105 |
Cited By (29)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20190104215A (en) * | 2017-01-17 | 2019-09-06 | 아스트라제네카 아베 | JAK1 selective inhibitor |
| KR102659213B1 (en) | 2017-01-17 | 2024-04-18 | 아스트라제네카 아베 | Jak1 selective inhibitors |
| US11897869B2 (en) | 2017-01-17 | 2024-02-13 | Astazeneca Ab | JAK1 selective inhibitors |
| WO2018134213A1 (en) * | 2017-01-17 | 2018-07-26 | Astrazeneca Ab | Jak1 selective inhibitors |
| KR20230141938A (en) * | 2017-01-17 | 2023-10-10 | 아스트라제네카 아베 | Jak1 selective inhibitors |
| EA037067B1 (en) * | 2017-01-17 | 2021-02-02 | Астразенека Аб | Jak1 selective inhibitors |
| KR102585048B1 (en) | 2017-01-17 | 2023-10-05 | 아스트라제네카 아베 | JAK1 selective inhibitor |
| US10961228B2 (en) | 2017-01-17 | 2021-03-30 | Astrazeneca Ab | JAK1 selective inhibitors |
| US11524968B2 (en) | 2017-10-18 | 2022-12-13 | Hk Inno.N Corporation | Heterocyclic compound as a protein kinase inhibitor |
| JP7422732B2 (en) | 2018-07-18 | 2024-01-26 | アストラゼネカ・アクチエボラーグ | JAK inhibitor compound xinafoate |
| CN112424187A (en) * | 2018-07-18 | 2021-02-26 | 阿斯利康(瑞典)有限公司 | Xinafoate salts of JAK-inhibiting compounds |
| US11149029B2 (en) | 2018-07-18 | 2021-10-19 | Astrazeneca Ab | Salts of IJAK inhibitors and methods of using the same |
| JP2021530526A (en) * | 2018-07-18 | 2021-11-11 | アストラゼネカ・アクチエボラーグAstrazeneca Aktiebolag | Xinafoate of JAK inhibitor compound |
| JP7644190B2 (en) | 2018-07-18 | 2025-03-11 | アストラゼネカ・アクチエボラーグ | Xinafoate salt of JAK inhibitor compound |
| WO2020016302A1 (en) * | 2018-07-18 | 2020-01-23 | Astrazeneca Ab | A xinafoate salt of a jak inhibiting compound |
| JP7349750B2 (en) | 2018-09-21 | 2023-09-25 | シャンハイ エンノババイオ ファーマシューティカルズ カンパニー リミテッド | Aromatic heterocyclic compounds with kinase inhibitory activity |
| KR102669660B1 (en) * | 2018-09-21 | 2024-05-27 | 상하이 엔노바바이오 파마슈티컬스 씨오 엘티디. | Aromatic heterocyclic compound with kinase inhibitory activity |
| JP2022502484A (en) * | 2018-09-21 | 2022-01-11 | シャンハイ エンノババイオ ファーマシューティカルズ カンパニー リミテッドShanghai Ennovabio Pharmaceuticals Co., Ltd. | Aromatic heterocyclic compound with kinase inhibitory activity |
| KR20210095621A (en) * | 2018-09-21 | 2021-08-02 | 상하이 엔노바바이오 파마슈티컬스 씨오 엘티디. | Aromatic heterocyclic compounds with kinase inhibitory activity |
| US12234232B2 (en) | 2018-09-21 | 2025-02-25 | Shanghai Ennovabio Pharmaceuticals Co., Ltd. | Aromatic heterocyclic compound with kinase inhibitory activity |
| CN112823159A (en) * | 2018-09-21 | 2021-05-18 | 上海轶诺药业有限公司 | Heteroaromatic compound with kinase inhibition activity |
| AU2019344878B2 (en) * | 2018-09-21 | 2022-08-11 | Shanghai Ennovabio Pharmaceuticals Co., Ltd. | Aromatic heterocyclic compound with kinase inhibitory activity |
| WO2020057669A1 (en) * | 2018-09-21 | 2020-03-26 | 上海轶诺药业有限公司 | Aromatic heterocyclic compound with kinase inhibitory activity |
| US12180185B2 (en) | 2018-11-15 | 2024-12-31 | Hk Inno.N Corporation | Compound as protein kinase inhibitor, and pharmaceutical composition comprising thereof |
| WO2020211839A1 (en) | 2019-04-19 | 2020-10-22 | Dizal (Jiangsu) Pharmaceutical Co., Ltd. | Jak1 selective kinase inhibitor |
| RU2818002C2 (en) * | 2019-04-19 | 2024-04-23 | Дизал (Цзянсу) Фармасьютикал Ко., Лтд. | Selective kinase jak1 inhibitor |
| EP3956322A4 (en) * | 2019-04-19 | 2023-05-03 | Dizal (Jiangsu) Pharmaceutical Co., Ltd. | SELECTIVE KINASE JAK1 INHIBITOR |
| CN111961037A (en) * | 2020-09-17 | 2020-11-20 | 嘉兴特科罗生物科技有限公司 | Pharmaceutical compound as JAK kinase inhibitor |
| WO2022189496A1 (en) * | 2021-03-11 | 2022-09-15 | Janssen Pharmaceutica Nv | Lorpucitinib for use in the treatment of jak mediated disorders |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US12319670B2 (en) | Compounds and methods for inhibiting JAK | |
| BR112020023068A2 (en) | triazolopyrimidine compounds and their use in cancer treatment | |
| WO2021003417A1 (en) | Tyrosine kinase non-receptor 1 (tnk1) inhibitors and uses thereof | |
| KR20200104336A (en) | Heterocyclic compounds as Tyro3, Axl and Mertk (TAM) families of receptor tyrosine kinase inhibitors | |
| HK40035295A (en) | Compounds and methods for inhibiting jak | |
| HK40026896B (en) | Compounds and methods for inhibiting jak | |
| HK40026896A (en) | Compounds and methods for inhibiting jak | |
| HK40026768B (en) | Compounds and methods for inhibiting jak | |
| HK40026768A (en) | Compounds and methods for inhibiting jak | |
| HK1259422B (en) | Compounds and methods for inhibiting jak | |
| HK40067698A (en) | Tyrosine kinase non-receptor 1 (tnk1) inhibitors and uses thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16770031 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2995430 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2016328764 Country of ref document: AU Date of ref document: 20160922 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20187008186 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2018/003590 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2018535238 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112018005833 Country of ref document: BR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2018112993 Country of ref document: RU Ref document number: 2016770031 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 112018005833 Country of ref document: BR Kind code of ref document: A2 Effective date: 20180323 |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020257006096 Country of ref document: KR |