WO2017007701A1 - Antiviral phosphodiamide compounds - Google Patents
Antiviral phosphodiamide compounds Download PDFInfo
- Publication number
- WO2017007701A1 WO2017007701A1 PCT/US2016/040606 US2016040606W WO2017007701A1 WO 2017007701 A1 WO2017007701 A1 WO 2017007701A1 US 2016040606 W US2016040606 W US 2016040606W WO 2017007701 A1 WO2017007701 A1 WO 2017007701A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- 3alkyl
- phenyl
- independently selected
- substituted
- unsubstituted
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- DSUAJFIEKRKPEE-GSVOUGTGSA-N C#C[C@H](C(O)=O)N Chemical compound C#C[C@H](C(O)=O)N DSUAJFIEKRKPEE-GSVOUGTGSA-N 0.000 description 1
- 0 C[C@@](C[n]1c2ncnc(N)c2nc1)OCP(*)(*)=O Chemical compound C[C@@](C[n]1c2ncnc(N)c2nc1)OCP(*)(*)=O 0.000 description 1
- SGOIRFVFHAKUTI-ZCFIWIBFSA-N C[C@H](C[n]1c(ncnc2N)c2nc1)OCP(O)(O)=O Chemical compound C[C@H](C[n]1c(ncnc2N)c2nc1)OCP(O)(O)=O SGOIRFVFHAKUTI-ZCFIWIBFSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6561—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing systems of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring or ring system, with or without other non-condensed hetero rings
- C07F9/65616—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing systems of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring or ring system, with or without other non-condensed hetero rings containing the ring system having three or more than three double bonds between ring members or between ring members and non-ring members, e.g. purine or analogs
Definitions
- HIV human immunodeficiency virus
- HIV-1 HIV type-1
- type-2 HIV-2
- HIV seropositive individuals are initially asymptomatic but typically develop AIDS related complex (ARC) followed by AIDS.
- Affected individuals exhibit severe immunosuppression which makes them highly susceptible to debilitating and ultimately fatal opportunistic infections.
- Replication of HIV by a host cell requires integration of the viral genome into the host cell's DNA.
- HIV replication cycle requires transcription of the viral RNA genome into DNA via an enzyme known as reverse transcriptase (RT).
- RT reverse transcriptase
- Reverse transcriptase has three known enzymatic functions: The enzyme acts as an RNA-dependent DNA polymerase, as a ribonuclease, and as a DNA-dependent DNA polymerase.
- RT transcribes a single-stranded DNA copy of the viral RNA.
- a ribonuclease RT destroys the original viral RNA and frees the DNA just produced from the original RNA.
- RT's ribonuclease activity is required for removing RNA and leaving the polypurine tract preserved for initiation of DNA-dependent polymerization.
- a DNA-dependent DNA polymerase RT makes a second,
- the two strands form double-stranded DNA, which is integrated into the host cell's genome by HIV integrase.
- NRTIs non-nucleoside active site competitive RT inhibitors
- EVF efavirenz
- NNP nevirapine
- ETR etravirine
- RPV rilpivirine
- NRTIs nucleos(t)ide reverse transcriptase inhibitors
- ZT 3'-azido- 3'-deoxythymidine
- ddl 2',3'-dideoxyinosine
- ddC dideoxycytidine
- d4T 3TC
- abacavir emtricitabine
- tenofovir TFV, also known as PMPA, 9-(2- phosphonyl-methoxypropy
- TFV belongs to a class of HIV anti-retro viral (ARV) agents known as nucleotide analog reverse transcriptase inhibitors (NRTIs).
- ARV HIV anti-retro viral
- NRTIs nucleotide analog reverse transcriptase inhibitors
- TFV After being taken up by cells, TFV is first converted to tenofovir-monophosphate (TFV-P) by adenosine monophosphate kinase and then to the active antiviral tenofovir-diphosphate (TFV-DP) by 5 '-nucleoside diphosphate kinase.
- TFV-P tenofovir-monophosphate
- TFV-DP active antiviral tenofovir-diphosphate
- TFV-DP inhibits HIV DNA synthesis by competing with the natural substrate, deoxy adenosine triphosphate, for incorporation into the complementary DNA strand by HIV reverse transcriptase; following incorporation, TFV acts as a chain terminator due to lack of a 3'-hydroxyl group that is required for addition of the next nucleotide. TFV has poor cellular permeability and thus has limited bioavailability. Tenofovir disoproxil fumarate (TDF) is approved for treating HIV infection and is marketed by Gilead under the trade name
- VIREADTM The disoproxil prodrug improves cell permeability and absorption after oral dosing, with the pro-moiety being cleaved rapidly after absorption to yield the parent TFV. As a result, the circulating level of TFV is much higher than that of TDF.
- Tenofovir alafenamide fumarate (TAF) is currently under review for treating HIV infection with a reduced level of circulating TFV. While each of the foregoing drugs is effective in treating HIV infection and
- AIDS there remains a need to develop additional HIV antiviral drugs including additional RT inhibitors.
- a particular problem is the development of mutant HIV strains that are resistant to the known inhibitors.
- the use of RT inhibitors to treat AIDS often leads to viruses that are less sensitive to the inhibitors. This resistance is typically the result of mutations that occur in the reverse transcriptase segment of the pol gene.
- the continued use of antiviral compounds to prevent HIV infection will inevitably result in the emergence of new resistant strains of HIV. Accordingly, there is a particular need for new RT inhibitors that are effective against mutant HIV strains.
- the present invention is directed to phosphodiamide prodrugs of tenofovir and their use in the inhibition of nucleotide reverse transcriptase.
- the invention is also directed to the use of said compounds for prophylaxis of infection by HIV, the treatment of infection by HIV, and the prophylaxis, treatment, and/or delay in the onset or progression of AIDS and/or ARC.
- the present invention is directed to compounds of structural Formula I:
- RA is a D-amino acid ester residue of formula (i) or a D-proline ester residue of formula (ii)
- heteroaryl unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5a -0-Ci-3alkyl, -SH, -NR6R7 or -Ci- 3alkyl, or
- RB is an L-amino acid ester residue of formula (iii) or an L-proline ester residue of formula
- heteroaryl unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5b -0-Ci-3alkyl, -SH, - NR8R9 or -Ci- 3alkyl, or
- R5a and R ⁇ b are each independently selected from -H or -C3-6cycloalkyl;
- R6 and R7 are each independently selected from -H, -Ci-3alkyl or -C3-6cycloalkyl;
- R8 and R9 are each independently selected from -H, -Ci-3alkyl or -C3-6cycloalkyl.
- Embodiment 1 of this invention are compounds of Formula I or the pharmaceutically acceptable salts thereof, wherein RA is a D-amino acid ester residue of formula (i) or a D-proline ester residue of formula (ii), and Rl is -Ci-4alkyl (particularly -
- Rl is— CH3, -CH(CH3)2, - CH(CH 3 )CH 2 CH 3 ,
- Rl is -CH3 or -CH2-phenyl
- Embodiment 2 of this invention are compounds of Formula I, Embodiment l,or Embodiment la, or a sub-class thereof, or the pharmaceutically acceptable salts thereof, wherein RB is an L-amino acid ester residue of formula (iii) or an L-proline ester residue of formula (iv) or a glycine ester residue of formula (v), and R3 is -Ci-4alkyl (particularly -
- R3 is -CFI3, -CH(CH3)2, - CH(CH 3 )CH 2 CH 3 ,
- R 3 is -CH3 or -CH2-phenyl.
- Embodiment 3 of this invention are compounds of Formula I, Embodiment 1, Embodiment la, Embodiment 2 or Embodiment 2a, each sub-class thereof, or the pharmaceutically acceptable salts thereof, wherein RA is a D-amino acid ester residue of formula (i) or a D-proline ester residue of formula (ii), and R2 is:
- R2 is -Ci-8alkyl, -CH2-phenyl, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl or phenyl, and in a sub-class thereof, R2 is -C3-7alkyl, cyclobutyl or cyclopentyl.
- Embodiment 4 of this invention are compounds of Formula I, Embodiment 1, Embodiment la, Embodiment 2, Embodiment 2a, Embodiment 3, or Embodiment 3a, each sub-class thereof, or the pharmaceutically acceptable salts thereof, wherein RB is an L-amino acid ester residue of formula (iii), or an L-proline ester residue of formula (iv) or a glycine ester residue of formula (v), and R4 is:
- R4 is -Ci-8alkyl, -CH2-phenyl, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl or phenyl, and in a sub-class thereof, R4 is -C3-7alkyl, cyclobutyl or cyclopentyl.
- Embodiment 5 of this invention are compounds of Formula I and each
- Embodiment 6 of this invention are compounds of Formula I and each Embodiment, class or sub-class thereof, or the pharmaceutically acceptable salts thereof, wherein of R6, R7, R8 and R9 are each -H or -Ci-3alkyl, and particularly each is H.
- Embodiment 7of this invention are compounds of Formula I or a pharmaceutically acceptable salt thereof, wherein:
- RA is a D-amino acid ester residue of formula (i) or a D-proline ester residue of formula (ii);
- RB is an L-amino acid ester residue of formula (iii), an L-proline ester residue of formula (iv) or a glycine ester residue of formula (v);
- pyridyl unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5b -0-Ci-3alkyl, -SH, -NR8R9 0 r -Ci-3alkyl, or (g) piperidinyl, pyrrolidinyl, tetrahydrofuranyl, or tetrahydropyranyl, each unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5b -0-Cl -3alkyl, -SH, -NR8R9-N or -Cl -3alkyl;
- R5a and R ⁇ b are each -H
- R6, R7, R8 and R9 are each independently selected from -H or Ci-3alkyl.
- Embodiment 8 of this invention are compounds of Formula I or a pharmaceutically acceptable salt thereof, wherein:
- RA is a D-amino acid ester residue of formula (i) or a D-proline ester residue of formula (ii);
- Rl is -CH3, -CH(CH3)2, -CH(CH3)CH2CH3, -CH2CH(CH3)2 . or -CH2-phenyl, or more particularly Rl is -CH3 or -CH2-phenyl;
- R2 is -Ci-8alkyl, -CH2-phenyl, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl or phenyl, or more particularly it is -C3-7alkyl, cyclobutyl or cyclopentyl;
- RB is an L-amino acid ester residue of formula (iii), an L-proline ester residue of formula
- R3 is -CH3, -CH(CH3)2, -CH(CH3)CH2CH3, -CH2CH(CH3)2 or -CH2-phenyl, or more particularly R3 is -CH3 or -CH2 -phenyl;
- R4 is -Ci-8alkyl, -CH2-phenyl, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl or phenyl, or more particularly it is -C3-7alkyl, cyclobutyl or cyclopentyl;
- R5a and R5b are each -H;
- R6, R7, R8 and R9 are each independently selected from -H or Ci-3alkyl.
- references to the compounds of Formula I herein encompasses the compounds of Formula I and all embodiments, classes and sub-classes thereof.
- the compounds of the invention encompass compounds of structural Formula I, embodiments, classes and subclasses thereof and salts thereof when such salts are possible, including the pharmaceutically acceptable salts of said compounds.
- alkyl refers to both branched- and straight-chain saturated aliphatic hydrocarbon groups having the specified number of carbon atoms in a specified range.
- Ci-4alkyl has 1, 2, 3 or 4 carbon atoms, and includes each of n-, iso-, sec- and fert-butyl, n- and z-propyl, ethyl and methyl.
- Cycloalkyl refers to a cyclized alkyl ring having the indicated number of carbon atoms in a specified range.
- C3-8 cycloalkyl encompasses each of cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cyloheptyl and cyclooctyl.
- C3-6cycloalkyl encompasses each of cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl.
- cycloalkyl is a substituent on an alkyl group in a compound of Formula I
- the cycloalkyl substituent can be bonded to any available carbon in the alkyl group.
- the following are illustrations of -C3-6cycloalkyl substituents wherein the substituent is cyclopropyl in bold:
- a "spiro-C3-6cycloalkyl" substituent refers to a cycloalkyl group bonded to an alkyl group via a a single, non-terminal carbon atom which is common to both the the cycloalkyl group and the alkyl group.
- Spiro-C3-6cycloalkyl encompasses each of spiro- cyclopropyl, spiro-cyclobutyl, spiro-cyclopentyl and spiro-cyclohexyl.
- the following is an illustration of a spiro-C3-6cycloalkyl substituent wherein the substituent is spiro-cyclopropyl in bold:
- Heteroalkyl refers to both branched- and straight-chain alkyl groups in which one non-terminal carbon atom is replaced with a heteroatom selected from N, O or S.
- the heteroalkyl group has the indicated total number of carbons and heteroatoms in a specifed range, e.g., 2-10 atom heteroalkyl encompasses each such group having one heteroatom plus 1, 2, 3, 4, 5, 6, 7, 8 or 9 carbon atom(s).
- heteroalkyl is -Ci-4alkyl-X-Ci-5alkyl wherein X is O, S or NH.
- Examples of heteroalkyl groups include, but are not limited to,
- Aryl refers to (i) phenyl, (ii) 9- or 10-membered bicyclic, fused carbocylic ring systems in which at least one ring is aromatic, and (iii) 11- to 14-membered tricyclic, fused carbocyclic ring systems in which at least one ring is aromatic.
- Suitable aryls include, for example, substituted and unsubstituted phenyl and substituted and unsubstituted naphthyl. An aryl of particular interest is unsubstituted or substituted phenyl.
- Heteroaryl refers to (i) a 5- or 6-membered heteroaromatic ring containing from 1 to 4 heteroatoms independently selected from N, O and S, wherein each N is optionally in the form of an oxide, and (ii) a 9- or 10-membered bicyclic fused ring system, wherein the fused ring system of (ii) contains from 1 to 6 heteroatoms independently selected from N, O and S, wherein each ring in the fused ring system contains zero, one or more than one heteroatom, at least one ring is aromatic, each N is optionally in the form of an oxide, and each S in a ring which is not aromatic is optionally S(O) or S(0)2- Suitable 5- and 6- membered heteroaromatic rings include, for example, pyridyl, 3-fluroropyridyl, 4- fluoropyridyl, 3-methoxypyridyl, 4-methoxypyridyl, pyrrolyl, pyrazinyl,
- Suitable 9- and 10-membered heterobi cyclic, fused ring systems include, for example, benzofuranyl, indolyl, indazolyl, naphthyridinyl, isobenzofuranyl, benzopiperidinyl, benzisoxazolyl, benzoxazolyl, chromenyl, quinolinyl, isoquinolinyl, isoindolyl, benzopiperidinyl, benzofuranyl, imidazo[l,2-a]pyridinyl, benzotriazolyl, indazolyl, indolinyl, and isoindolinyl.
- a class of heteroaryls includes unsubstituted or substituted pyridyl or pyrimidyl, and particularly unsubstituted or substituted pyridyl.
- heterocyclic ring refers to (i) a saturated 4- to 7-membered cyclized ring and (ii) an unsaturated, non-aromatic 4 to 7-membered cyclized ring comprised of carbon atoms and 1- 4 heteratoms independently selected from O, N and S.
- Heterocyclic rings within the scope of this invention include, for example, azetidinyl, piperidinyl, morpholinyl, thiomorpholinyl, thiazolidinyl, isothiazolidinyl, oxazolidinyl, isoxazolidinyl, pyrrolidinyl, imidazolidinyl, piperazinyl, tetrahydrofuranyl, tetrahydrothienyl, pyrazolidinyl, hexahydropyrimidinyl, thiazinanyl, thiazepanyl, azepanyl, diazepanyl, tetrahydropyranyl, tetrahydrothiopyranyl, and dioxanyl.
- Examples of 4- to 7-membered, unsaturated, non- aromatic heterocyclic rings within the scope of this invention include mono-unsaturated heterocyclic rings corresponding to the saturated heterocyclic rings listed in the preceding sentence in which a single bond is replaced with a double bond (e.g., a carbon-carbon single bond is replaced with a carbon-carbon double bond).
- a class of heterocyclic rings includes piperidinyl, pyrrolidinyl, tetrahydrofuranyl, or tetrahydropyranyl.
- a “stable” compound is a compound which can be prepared and isolated and whose structure and properties remain or can be caused to remain essentially unchanged for a period of time sufficient to allow use of the compound for the purposes described herein (e.g., therapeutic or prophylactic administration to a subject).
- the compounds of the present invention are limited to stable compounds embraced by Formula I and its embodiments.
- certain moieties as defined in Formula I may be unsubstituted or substituted, and the latter is intended to encompass substitution patterns (i.e., number and kind of substituents) that are chemically possible for the moiety and that result in a stable compound.
- Each compound of Formula I is comprised of a phosphodiamide amino acid ester, containing one D-amino acid ester residue and one achiral- or L-amino acid ester residue.
- Each compound also has a defined (R) chiral center in the alkyl-ether linking group connecting the nucleobase to the phosphodiamide.
- each compound of Formula I has mutliple chiral centers (also referred to as asymmetric or stereogenic centers), for which the spatial orientation of certain chiral centers are specifically defined.
- Each compound of Formula I also has an assymetric phosphorus center. This invention encompasses compounds having either the (R) or (S) stereoconfiguration at the phosphorus assymetric center, or mixtures thereof.
- This invention includes individual diastereomers, particularly epimers, i.e., compounds having the same chemical formula but which differ in the spatial arrangement around a single atom.
- This invention also includes mixtures of diastereomers, particularly mixtures of epimers, in all ratios.
- Embodiments of this invention also include a mixture of epimers enriched with 51 % or more of one of the epimers, including for example 60% or more, 70% or more, 80% or more, or 90% or more of one epimer.
- a single epimer is preferred.
- An individual or single epimer refers to an epimer obtained by chiral synthesis and/or using generally known separation and purification techniques, and which may be 100% of one epimer or may contain small amounts (e.g., 10% or less) of the opposite epimer.
- individual diasteromers are a subject of the invention in pure form, both as levorotatory and as dextrorotatory antipodes, in the form of racemates and in the form of mixtures of the two diasteromers in all ratios.
- the invention includes both the cis form and the trans form as well as mixtures of these forms in all ratios.
- the preparation of individual stereoisomers can be carried out, if desired, by separation of a mixture by customary methods, for example by chromatography or crystallization, by the use of stereochemically uniform starting materials for the synthesis or by stereoselective synthesis.
- a derivatization can be carried out before a separation of stereoisomers.
- the separation of a mixture of stereoisomers can be carried out at an intermediate step during the synthesis of a compound of Formula I or it can be done on a final racemic product.
- Absolute stereochemistry may be determined by X-ray crystallography of crystalline products or crystalline intermediates which are derivatized, if necessary, with a reagent containing a stereogenic center of known configuration.
- absolute stereochemistry may be determined by Vibrational Circular Dichroism (VCD) spectroscopy analysis.
- VCD Vibrational Circular Dichroism
- the present invention includes all such isomers, as well as salts, solvates (which includes hydrates) and solvated salts of such racemates, enantiomers, diastereomers and tautomers and mixtures thereof.
- the atoms in a compound of Formula I may exhibit their natural isotopic abundances, or one or more of the atoms may be artificially enriched in a particular isotope having the same atomic number, but an atomic mass or mass number different from the atomic mass or mass number predominantly found in nature.
- the present invention is meant to include all suitable isotopic variations of the compounds of Formula I, for example, different isotopic forms of hydrogen (H) include protium (lH) and deuterium (3 ⁇ 4T). Protium is the
- Isotopically-enriched compounds of Formula I can be prepared without undue
- the compounds can be administered in the form of pharmaceutically acceptable salts.
- pharmaceutically acceptable salt refers to a salt which is not biologically or otherwise undesirable (e.g., is neither toxic nor otherwise deleterious to the recipient thereof).
- the invention includes the corresponding pharmaceutically acceptable salts.
- the compounds of Formula I contain one or more acidic groups, the invention also includes the corresponding pharmaceutically acceptable salts.
- the compounds of Formula I that contain acidic groups e.g., -COOH
- salts include but are not limited to sodium salts, potassium salts, calcium salts, magnesium salts or salts with ammonia or organic amines such as, for example, ethylamine, ethanolamine, triethanolamine or amino acids.
- Compounds of Formula I which contain one or more basic groups, i.e.
- the invention also includes, in addition to the salt forms mentioned, inner salts or betaines (zwitterions). Salts can be obtained from the compounds of Formula I by customary methods which are known to the person skilled in the art, for example by combination with an organic or inorganic acid or base in a solvent or dispersant, or by anion exchange or cation exchange from other salts.
- the present invention also includes all salts of the compounds of Formula I which, owing to low physiological compatibility, are not directly suitable for use in pharmaceuticals but which can be used, for example, as intermediates for chemical reactions or for the preparation of pharmaceutically acceptable salts.
- compounds of the present invention may exist in amorphous form and/or one or more crystalline forms, and as such all amorphous and crystalline forms and mixtures thereof of the compounds of Formula I are intended to be included within the scope of the present invention.
- some of the compounds of the instant invention may form solvates with water (i.e., a hydrate) or common organic solvents.
- Such solvates and hydrates, particularly the pharmaceutically acceptable solvates and hydrates, of the compounds of of this invention are likewise encompassed within the scope of the compounds defined by Formula I and the pharmaceutically acceptable salts thereof, along with un- solvated and anhydrous forms of such compounds.
- the compounds of Formula I, embodiments thereof and specific compounds described and claimed herein encompass all possible pharmaceutically acceptable salts, stereoisomers, tautomers, physical forms (e.g., amorphous and crystalline forms), solvate and hydrate forms and any combination of the foregoing forms where such forms are possible.
- Compounds of Formula I are prodrug modifications of tenofovir, which is a mono- phosphonate.
- the compounds of Formula I may be converted intracellularly/z ? vivo by one or more mechanisms (e.g., enzyme-catalyzed chemical reactions) to the corresponding monophosphate or diphosphate of tenofovir .
- tenofovir diphosphate is generally understood to be responsible for inhibiting the HIV RT enzyme and for the resulting antiviral activity after administration of the compound of Formula I to a subject.
- prodrugs The compounds of Formula I described herein are prodrugs.
- a discussion of prodrugs is provided in (a) Stella, V. J.; Borchardt, R. T.; Hageman, M. J.; Oliyai, R.; Maag, H. et al. Prodrugs: Challenges and Rewards Part 1 and Part 2; Springer, p. 726: New York, NY, USA, 2007, (b) Rautio, I; Kumpulainen, H.; Heimbach, T.; Oliyai, R.; Oh, D. et al.
- compounds of Formula I are prodrug modifications of tenofovir, which is a mono- phosphonate.
- the compounds of Formula I may be converted intracellularly ⁇ in vivo or in vitro) to the corresponding monophosphate or diphosphate of tenofovir.
- the conversion may occur by one or more mechanisms, e.g., an enzyme-catalyzed chemical reaction, a metabolic chemical reaction, and/or a spontaneous chemical reaction (e.g., solvolysis), such as, for example, through hydrolysis in blood.
- tenofovir diphosphate is generally understood to be responsible for inhibiting the HIV RT enzyme and for the resulting antiviral activity after administration of the compound of Formula I to a subject.
- substantially pure means suitably at least about 60 wt.%, typically at least about 70 wt.%, preferably at least about 80 wt.%, more preferably at least about 90 wt.% (e.g., from about 90 wt.% to about 99 wt.%), even more preferably at least about 95 wt.% (e.g., from about 95 wt.% to about 99 wt.%, or from about 98 wt. % to 100 wt.
- a product containing a compound of Formula I or its salt e.g., the product isolated from a reaction mixture affording the compound or salt
- the level of purity of the compounds and salts can be determined using a standard method of analysis such as, high performance liquid chromatography, and/or mass spectrometry or NMR techniques. If more than one method of analysis is employed and the methods provide experimentally significant differences in the level of purity determined, then the method providing the highest purity level governs.
- a compound or salt of 100% purity is one which is free of detectable impurities as determined by a standard method of analysis.
- a substantially pure compound can be either a substantially pure mixture of the stereoisomers or a substantially pure individual
- the compounds of Formula I herein, and pharmaceutically acceptable salts thereof, are HIV reverse transcriptase inhibitors.
- the compounds are useful for inhibiting HIV reverse transcriptase and for inhibiting HIV replication in vitro and in vivo. More particularly, the compounds of Formula I inhibit the polymerase function of HIV- 1 reverse
- transcriptase The testing of compounds of the Examples of this invention in the Viking assay set forth in Example 10 below, illustrate the ability of compounds of the invention to inhibit the RNA-dependent DNA polymerase activity of HIV- 1 reverse transcriptase.
- the compounds of Formula I may also be useful agents against HIV-2.
- the compounds of Examples 1-9 of the present invention also exhibit activity against drug resistant forms of HIV (e.g., N RTI-associated mutant strains K103N and/or Y181C; NRTI-associated mutant strains Ml 84V and Ml 841 mutants).
- This invention also encompasses methods for the treatment or prophylaxis of infection by HIV, for the inhibition of HIV reverse transcriptase, for the treatment, prophylaxis, or delay in the onset of AIDS in a subject in need thereof, which comprise administering to the subject an effective amount of a compound of the invention or a pharmaceutically acceptable salt thereof.
- the invention further encompasses methods for the treatment or prophylaxis of infection by HIV, for the inhibition of HIV reverse transcriptase, for the treatment, prophylaxis, or delay in the onset of AIDS in a subject in need thereof, which comprise administering to the subject an effective amount of a compound of the invention or a pharmaceutically acceptable salt thereof in combination with an effective amount of one or more additional anti-HIV agents selected from the group consisting of HIV antiviral agents, immunomodulators, and anti-infective agents.
- the anti-HIV agent is an antiviral selected from the group consisting of HIV protease inhibitors, HIV reverse transcriptase inhibitors, HIV integrase inhibitors, HIV fusion inhibitors, HIV entry inhibitors, and HIV maturation inhibitors
- the invention encompasses a pharmaceutical composition comprising an effective amount of a compound of the invention, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
- the invention also encompasses a pharmaceutical composition comprising an effective amount of a compound of the invention, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier further comprising an effective amount of one or more additional anti-HIV agents selected from the group consisting of HIV antiviral agents, immunomodulators, and anti-infective agents.
- the anti-HIV agent is an antiviral selected from the group consisting of HIV protease inhibitors, HIV reverse transcriptase inhibitors, HIV integrase inhibitors, HIV fusion inhibitors, HIV entry inhibitors, and HIV maturation inhibitors.
- this invention could also be useful for inhibition of HBV reverse transcriptase. Accordingly, this invention also encompasses methods for the treatment of chronic hepatitis B which comprise administering to the subject an effective amount of a compound of the invention or a pharmaceutically acceptable salt thereof.
- the invention also encompasses a compound of the invention, or a pharmaceutically acceptable salt thereof, for use in the preparation of a medicament for the treatment or prophylaxis of infection by HIV, for the inhibition of HIV reverse transcriptase, or for the treatment, prophylaxis, or delay in the onset of AIDS in a subject in need thereof.
- composition comprising an effective amount of a compound of Formula I or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
- composition which comprises the product prepared by combining (e.g., mixing) an effective amount of a compound of Formula I or a
- anti-HIV agent is selected from one or more of an antiviral selected from the group consisting of HIV protease inhibitors, nucleoside HIV reverse transcriptase inhibitors, non-nucleoside HIV reverse transcriptase inhibitors, HIV integrase inhibitors, HIV fusion inhibitors, HIV entry inhibitors and HIV maturation inhibitors.
- a combination which is (i) a compound of Formula I or a pharmaceutically acceptable salt thereof and (ii) an anti-HIV agent selected from the group consisting of HIV antiviral agents, immunomodulators, and anti-infective agents; wherein the compound and the anti-HIV agent are each employed in an amount that renders the combination effective for inhibition of HIV reverse transcriptase, for treatment or prophylaxis of infection by HIV, or for treatment, prophylaxis of, or delay in the onset or progression of AIDS.
- anti-HIV agent is an antiviral selected from the group consisting of HIV protease inhibitors, nucleoside HIV reverse transcriptase inhibitors, non-nucleoside HIV reverse transcriptase inhibitors, HIV integrase inhibitors, HIV fusion inhibitors, HIV entry inhibitors and HIV maturation inhibitors.
- a method for the prophylaxis or treatment of infection by HIV e.g., HIV-1 in a subject in need thereof which comprises administering to the subject an effective amount of a compound of Formula I or pharmaceutically acceptable salt thereof.
- (j) A method for the prophylaxis, treatment or delay in the onset or progression of AIDS in a subject in need thereof which comprises administering to the subject an effective amount of a compound of Formula I or pharmaceutically acceptable salt thereof.
- a method for the inhibition of HIV reverse transcriptase in a subject in need thereof which comprises administering to the subject the pharmaceutical composition of (a), (b), (c) or (d) or the combination of (e) or (f).
- a method for the prophylaxis or treatment of infection by HIV in a subject in need thereof, which comprises administering to the subject the pharmaceutical composition of (a), (b), (c) or (d) or the combination of (e) or (f).
- HIV e.g., HIV-1
- a method for the prophylaxis, treatment, or delay in the onset or progression of AIDS in a subj ect in need thereof which comprises administering to the subj ect the pharmaceutical composition of (a), (b), (c) or (d) or the combination of (e) or (f).
- the present invention also includes a compound of Formula I or pharmaceutically acceptable salt thereof (i) for use in, (ii) for use as a medicament for, or (iii) for use in the preparation of a medicament for: (a) therapy (e.g., of the human body), (b) medicine, (c) inhibition of HIV reverse transcriptase, (d) treatment or prophylaxis of infection by HIV, or (e) treatment, prophylaxis of, or delay in the onset or progression of AIDS.
- the compounds of the present invention can optionally be employed in combination with one or more anti-HIV agents selected from HIV antiviral agents, anti-infective agents, and immunomodulators.
- Additional embodiments of the invention include the pharmaceutical compositions, combinations and methods set forth in (a)-(n) above and the uses (i)(a)-(e) through (iii)(a)-(e) set forth in the preceding paragraph, wherein the compound of the present invention employed therein is a compound of one of the embodiments, aspects, classes, sub-classes, or features described above. In all of these embodiments etc., the compound may optionally be used in the form of a pharmaceutically acceptable salt.
- Additional embodiments of the present invention include each of the pharmaceutical compositions, combinations, methods and uses set forth in the preceding paragraphs, wherein the compound of the present invention or its salt employed therein is substantially pure.
- Still additional embodiments of the present invention include the pharmaceutical compositions, combinations and methods set forth in (a)-(n) above and the uses (i)(a)-(e) through (iii)(a)-(e) set forth above, wherein the HIV of interest is HIV-1.
- the compound of Formula I is employed in an amount effective against HIV-1 and the anti-HIV agent is an HIV-1 antiviral selected from the group consisting of HIV-1 protease inhibitors, HIV-1 reverse transcriptase inhibitors, HIV-1 integrase inhibitors, HIV-1 fusion inhibitors, HIV-1 entry inhibitors and HIV-1 maturation inhibitors.
- the compounds of Formula I may also be useful agents against HIV-2.
- administration and variants thereof (e.g., “administering” a compound) in reference to a compound of Formula I means providing the compound to the individual in need of treatment or prophylaxis and includes both self-administration and administration to the patient by another person.
- a compound is provided in combination with one or more other active agents (e.g., antiviral agents useful for treating or prophylaxis of HIV infection or AIDS)
- administration and its variants are each understood to include provision of the compound and other agents at the same time or at different times.
- the agents of a combination are administered at the same time, they can be administered together in a single composition or they can be administered separately.
- composition is intended to encompass a product comprising the specified ingredients, as well as any product which results from combining the specified ingredients.
- Ingredients suitable for inclusion in a pharmaceutical composition are pharmaceutically acceptable ingredients, which means the ingredients must be compatible with each other and not deleterious to the recipient thereof.
- subject or "patient” as used herein refers to an animal, preferably a mammal, most preferably a human, who has been the object of treatment, observation or experiment.
- an effective amount means an amount of a compound sufficient to inhibit HIV reverse transcriptase, inhibit HIV replication, exert a prophylactic effect, and/or a exert a therapeutic effect after administration.
- One embodiment of "effective amount” is a “therapeutically effective amount” which is an amount of a compound that is effective for inhibiting HIV reverse transcriptase, inhibiting HIV replication (either of the foregoing which may also be referred to herein as an “inhibition effective amount”), treating HIV infection, treating AIDS, delaying the onset of AIDS, and/or slowing progression of AIDS in a patient.
- an effective amount is a “prophylactically effective amount” which is an amount of the compound that is effective for prophylaxis of HIV infection or prophylaxis of AIDS in a patient. It is understood that an effective amount can simultaneously be both a therapeutically effective amount, e.g., for treatment of HIV infection, and a prophylactically effective amount, e.g., for prevention or reduction of risk for developing AIDS.
- an effective amount can refer to each individual agent or to the combination as a whole, wherein the amounts of all agents administered in the combination are together effective, but wherein a component agent of the combination may or may not be present individually in an effective amount with reference to what is considered effective for that component agent if it were were were
- the compounds of this invention can be administered alone.
- the compounds of this invention can be administered by means that produces contact of the active agent with the agent's site of action. They can be administered by conventional means available for use in conjunction with pharmaceuticals, either as individual therapeutic agents or in a combination of therapeutic agents. They can be administered alone, but typically are administered with a pharmaceutical carrier selected on the basis of the chosen route of administration and standard pharmaceutical practice.
- the compounds of the invention can, for example, be administered orally (e.g., via tablet or capsule), parenterally (including subcutaneous injections, intravenous, intramuscular or intrastemal injection, or infusion techniques), by inhalation spray, or rectally, in the form of a unit dosage of a pharmaceutical composition containing an effective amount of the compound and conventional non-toxic pharmaceutically acceptable carriers, adjuvants and vehicles.
- the compound could also be administered via an implantable drug delivery device adapted to provide an effective amount of the compound or a pharmaceutical composition of the compound over an extended period of time.
- Liquid preparations suitable for oral administration can be prepared according to techniques known in the art and can employ any of the usual media such as water, glycols, oils, alcohols and the like.
- Solid preparations suitable for oral administration e.g., powders, pills, capsules and tablets
- Parenteral compositions can be prepared according to techniques known in the art and typically employ sterile water as a carrier and optionally other ingredients, such as a solubility aid.
- Injectable solutions can be prepared according to methods known in the art wherein the carrier comprises a saline solution, a glucose solution or a solution containing a mixture of saline and glucose.
- Implantable compositions can be prepared according to methods known in the art wherein the carrier comprises the active chemical ingredient with polymers as suitable excipients, or utilizing an implantable device for drug delivery. Further description of methods suitable for use in preparing pharmaceutical compositions for use in the present invention and of ingredients suitable for use in said compositions is provided in Remington's Pharmaceutical Sciences. 18th edition, edited by A. R. Gennaro, Mack Publishing Co., 1990 and in
- Formulations of compounds described by Formula I that result in drug supersaturation and/or rapid dissolution may be utilized to facilitate oral drug absorption.
- Formulation approaches to cause drug supersaturation and/or rapid dissolution include, but are not limited to, nanoparticulate systems, amorphous systems, solid solutions, solid dispersions, and lipid systems.
- Such formulation approaches and techniques for preparing them are well known in the art.
- solid dispersions can be prepared using excipients and processes as described in reviews (e.g., A.T.M. Serajuddin, J Pharm Sci, 88: 10, pp. 1058-1066 (1999)).
- Nanoparticulate systems based on both attrition and direct synthesis have also been described in reviews such as Wu et al (F. Kesisoglou, S. Panmai, Y. Wu, Advanced Drug Delivery Reviews, 59:7 pp. 631 -644 (2007)).
- the compounds of Formula I can be administered in a dosage range of 0.001 to 1000 mg/kg of mammal (e.g., human) body weight per day, or at longer time intervals on non- consecutive days as appropriate, in a single dose or in divided doses.
- a dosage range is 0.01 to 500 mg/kg body weight per day, or at other time intervals as appropriate, administered orally or via other routes of administration in a single dose or in divided doses.
- Another example of a dosage range is 0.1 to 100 mg/kg body weight per day, or at other time intervals as appropriate, administered orally or via other routes of administration in single or divided doses.
- Another example of a dosage range is 50 mg to 1 gram per day, in a single dose or divided doses.
- Daily or weekly adminstration can be via any suitable route of administration but is preferably via oral administration and can be in single or divided daily doses within each 24 hour period. Divided doses may be administered via mulitple dosage units at essentially the same time or at staggered times over the 24 hour period. For weekly or less frequent dosing regimens with longer time intervals on non-consecutive days, a parenteral route of adminstration may be employed.
- Examples of such dosing regimens with longer time intervals on non-consecutive days include but are not limited to administration once weekly, once bi-weekly (once every two weeks with leeway as to exact date of dosing), once monthly (e.g., once every 30 days, or the same calendar day each month with leeway as to exact date of dosing), once bimonthly (e.g., once every 60 days, or the same calendar day every two months with leeway as to exact date of dosing), once every 3 months (e.g., once every 90 days, or the same calendar day every three months with leeway as to exact date of dosing), or once every six months (e.g., once every 180 days, or the same calendar day every six months with leeway as to exact date of dosing), or once yearly (e.g., once every 12 months with leeway as to exact date of the annual dosing).
- the dosage units may contain 1.0 mg to l OOOmg of the active ingredient, for example but not limited to, 1 , 5, 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 350, 400, 450, 500, 600, 700, 800, 900 or 1000 milligrams of the active ingredient for the symptomatic adjustment of the dosage to the patient to be treated.
- the compound may be formulated in oral formulations for immediate or modified release such as extended or controlled release.
- the favorable pharmacokinetic profile of tested compounds of this invention may also render the the compounds suitable for less frequent dosing.
- the compounds of the invention could be administered orally, weekly or parenterally at longer time intervals as described above.
- the compositions can be administered, e.g., intravenously (IV) or intramuscularly (IM) via injection, or using other infusion techniques.
- IV intravenously
- IM intramuscularly
- One or more of such injections or infusions may be administered at each dosing time interval as needed to deliver the appropriate amount of active agent.
- the specific dose level and frequency of dosage for any particular patient may be varied and will depend upon a variety of factors including the activity of the specific compound employed, the metabolic stability and length of action of that compound, the age, body weight, general health, sex, diet, mode and time of administration, rate of excretion, drug combination, the severity of the particular condition, and the host undergoing therapy. In some cases, depending on the potency of the compound or the individual response, it may be necessary to deviate upwards or downwards from the given dose. The amount and frequency of administration will be regulated according to the judgment of the attending clinician considering such factors.
- an anti-HIV agent is any agent which is directly or indirectly effective in the inhibition of HIV, the treatment or prophylaxis of HIV infection, and/or the treatment, prophylaxis or delay in the onset or progression of AIDS. It is understood that an anti-HIV agent is effective in treating, preventing, or delaying the onset or progression of HIV infection or AIDS and/or diseases or conditions arising therefrom or associated therewith.
- the compounds of this invention may be effectively administered, whether at periods of pre-exposure and/or post-exposure, in combination with effective amounts of one or more anti-HIV agents selected from HIV antiviral agents, imunomodulators, antiinfectives, or vaccines useful for treating HIV infection or AIDS.
- Suitable HIV antivirals for use in combination with the compounds of the present invention include, for example, those listed in Table 1 as follows: Antiviral A ents for Treatin HIV infection or AIDS
- nevirapine NVP
- PPL- 100 also known as PL-462 (Ambrilia) PI
- Some of the drugs listed in the table are used in a salt form; e.g., abacavir sulfate, delavirdine mesylate, indinavir sulfate, atazanavir sulfate, nelfinavir mesylate, saquinavir mesylate.
- HIV antiviral agents and other agents will typically be employed in these combinations in their conventional dosage ranges and regimens as reported in the art, including, for example, the dosages described in the Physicians' Desk Reference, Thomson PDR, Thomson PDR, 57th edition (2003), the 58th edition (2004), or the 59th edition (2005) and the current Physicians' Desk Reference (68th ed.). (2014), Montvale, NJ: PDR Network.
- the dosage ranges for a compound of the invention in these combinations can be the same as those set forth above.
- the compounds of this invention are also useful in the preparation and execution of screening assays for antiviral compounds.
- the compounds of this invention are useful for isolating enzyme mutants, which are excellent screening tools for more powerful antiviral compounds.
- the compounds of this invention are useful in establishing or determining the binding site of other antivirals to HIV reverse transcriptase, e.g., by competitive inhibition.
- Compounds of Formula I can be prepared from (i?)-(((l-(6-amino-9H-purin-9- yl)propan-2-yl)oxy)methyl)phosphonic acid (1), also referred to herein as TFV, with D- amino esters under EDC coupling conditions.
- the reaction of phosphoamide (3) with the second L-amino esters under 2,2'-dipyridyldisulfide condensation conditions yields the products in formula (II).
- D or L-proline esters or glycine esters can likewise be employed.
- amino esters that are not commercially available they are generally prepared by condensation between corresponding amino acid with alcohols under thionyl chloride.
- compounds of Formula I can be prepared from (i?)-(((l-(6- amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonic acid (1), with L-amino esters under EDC coupling conditions to yield (5) first.
- the reaction of phosphoamide (5) with the D-amino esters under 2,2'-dipyridyldisulfide condensation conditions yields the products in formula (II).
- D or L-proline esters or glycine esters can likewise be employed.
- Compounds of Formula III can be prepared from (i?)-(((l-(6-amino-9H-purin- 9-yl)propan-2-yl)oxy)methyl)phosphonic acid (1), also referred to herein as TFV, by coupling (1) with an excess of an L-amino ester (4) under 2,2'-dipyridyldisulfide
- Amino esters that are not commercially available, can be prepared by condensation of the corresponding amino acid with an alcohol in the presence of thionyl chloride.
- L-proline esters or glycine esters can likewise be employed.
- reactions sensitive to moisture or air were performed under nitrogen or argon using anhydrous solvents and reagents.
- the progress of reactions was determined by either analytical thin layer chromatography (TLC) usually performed with E. Merck pre-coated TLC plates, silica gel 60F-254, layer thickness 0.25 mm or liquid chromatography-mass spectrometry (LC-MS).
- TLC analytical thin layer chromatography
- LC-MS liquid chromatography-mass spectrometry
- the analytical LC-MS system used consisted of a Waters ZQTM platform with electrospray ionization in positive ion detection mode with an Agilent 1100 series HPLC with autosampler.
- the column was usually a Water Xterra MS CI 8, 3.0 ⁇ 50 mm, 5 /mi.
- the flow rate was 1 mL/min, and the injection volume was 10 piL.
- UV detection was in the range 210-400 nm.
- the mobile phase consisted of solvent A (water plus 0.06% TFA) and solvent B (acetonitrile plus 0.05% TFA) with a gradient of 100% solvent A for 0.7 min changing to 100% solvent B over 3.75 min, maintained for 1.1 min, then reverting to 100% solvent A over 0.2 min.
- Preparative HPLC purifications were usually performed using a mass spectrometry directed system. Usually they were performed on a Waters Chromatography Workstation configured with LC-MS System Consisting of: Waters ZQ single quad MS system with Electrospray Ionization, Waters 2525 Gradient Pump, Waters 2767 Injecto /Collector, Waters 996 PDA Detector, the MS Conditions of: 150-750 amu, Positive
- the mobile phases consisted of mixtures of acetonitrile (10-100%) in water containing 0.1% TFA. Flow rates were maintained at 50 mL/min, the injection volume was 1800 //L, and the UV detection range was 210-400 nm. Mobile phase gradients were optimized for the individual compounds. Reactions performed using microwave irradiation were normally carried out using an Emrys Optimizer manufactured by Personal Chemistry, or an Initiator manufactured by Biotage. Concentration of solutions was carried out on a rotary evaporator under reduced pressure.
- Flash chromatography was usually performed using a Biotage® Flash Chromatography apparatus (Dyax Corp.) on silica gel (32-63 ⁇ , 60 A pore size) in pre-packed cartridges of the size noted.
- lH NMR spectra were acquired at 500 MHz spectrometers in CDCI3 solutions unless otherwise noted. Chemical shifts were reported in parts per million (ppm).
- Tetramethylsilane (TMS) was used as internal reference in CD3CI solutions, and residual CH3OH peak or TMS was used as internal reference in CD3OD solutions. Coupling constants (J) were reported in hertz (Hz). Chiral analytical
- CHIRALPAK® AS CHIRALPAK® AD
- CHIRALCEL® OD CHIRALCEL® IA
- CHIRALCEL® OJ CHIRALCEL® OJ columns
- CHIRALPAK AS Chiral preparative chromatography was conducted on one of of CHIRALPAK AS, of CHIRALPAK AD, CHIRALCEL® OD, CHIRALCEL ®IA, CHIRALCEL® OJ columns (20x250 mm) (Daicel Chemical Industries, Ltd.) with desired isocratic solvent systems identified on chiral analytical chromatography or by supercritical fluid (SFC) conditions.
- SFC supercritical fluid
- a chiral center in a compound may exist in the "S" or “R” stereoconfigurations, or as a mixture of both.
- the compounds in the examples herein contain a phosphorus chiral center, and additional compounds within the scope of Formula I may also contain a phosphorus chiral center.
- compounds having a chiral center were separated into stereoisomers, referred to as Isomer A (faster eluting isomer) and Isomer B (slower eluting isomer), based on their observed elution order resulting from the separation as performed. Elution order of separated isomers may differ if performed under conditions different than those employed herein.
- each of the A isomer and B isomer in each Example (e.g., 1A and IB) is not assigned to a particular chemical name.
- an asterisk (*) may be used in the associated chemical structure drawing that indicates the location of the unassigned chiral center.
- Step 1 CR)-cvclopropyl 2-((tert-butoxycarbonyl)amino)propanoate: To a suspension of cyclopropyl boronic acid (6.2 g, 72 mmol) in 10% aqueous NaOH (50 mL) was added a solution of aqueous H2O2 (30%, 20 mL, 200 mmol) dropwise with continuous stirring at 0 °C. After 1 hour at 0 °C, the reaction mixture was quenched with saturated aqueous Na2S203 (30 mL, exothermic) and neutralized with 6 N HCl. The resulting solution was extracted with DCM (3 x 50 mL).
- Step 1 (RVallyl 2-(tert-butoxycarbonylamino)propanoate: To a solution of (i?)-2-((tert- butoxycarbonyl)amino)propanoic acid (1.89 g, 9.99 mmol) and DBU (1.82 g, 11.97 mmol) in acetonitrile (70 mL) was added 3-bromoprop-l-ene (1.45 g, 11.99 mmol) at 0 °C.
- Example 2 The compounds in Table 2 were prepared in an analogous fashion to that described for Example 1 A and IB.
- the column having the heading INT provides the intermediate example compounds used to make each exemplified compound.
- the antiviral activity of the tenofovir prodrugs of the Examples herein was assessed in an assay that measures the rate of replication of HIV in cell culture, termed the Viking assay (Viral KINetics in Green cells) and performed as follows. HIV-1 replication was monitored using MT4-gag-GFP clone D3 (hereafter designated MT4-GFP), which are MT-4 cells modified to harbor a GFP reporter gene, the expression of which is dependent on the HIV-1 expressed proteins tat and rev. Productive infection of an MT4-GFP cell with HIV-1 results in GFP expression approximately 24 h post-infection.
- MT4-GFP MT4-gag-GFP clone D3
- MT4-GFP cells were maintained at 37°C/5% CO2/90% relative humidity in RPMI 1640 supplemented with 10% fetal bovine serum, 100 U/ml penicillin/streptomycin, and 400 ⁇ g/ml G418 to maintain the reporter gene.
- MT4-GFP cells were placed in the same medium lacking G418 and infected overnight with HIV-1 (H9/IIIB strain) virus at an approximate multiplicity of infection of 0.01 in the same incubation conditions. Cells were then washed and re- suspended in either RPMI 1640 supplemented with 10% or 50% normal human serum (NHS) at 1.6 x 105 cells/mL (10% NHS or 50% NHS, respectively).
- NHS normal human serum
- Compound plates were prepared by dispensing compounds dissolved in DMSO into wells of 384 well poly-D-lysine- coated plates (0.2 ⁇ /well) using an ECHO acoustic dispenser. Each compound was tested in a 10-point serial 3-fold dilution (typical final concentrations: 8.4 ⁇ - 0.42 nM). Controls included no inhibitor (DMSO only) and a combination of three antiviral agents (efavirenz, indinavir, an in-house integrase strand transfer inhibitor at final concentrations of 4 ⁇ each). Cells were added (50 ⁇ 11) to compound plates and the infected cells were maintained at 37°C/5% CO2/90% relative humidity.
- Infected cells were quantified at two time points, ⁇ 48h and- 72h postinfection, by counting the number of green cells in each well using an Acumen eX3 scanner. The increase in the number of green cells over ⁇ 24h period gives the reproductive ratio, R0, which is typically 5-15 and has been shown experimentally to be in logarithmic phase (data not shown). Inhibition of R0 is calculated for each well, and IC50S determined by non-linear 4-parameter curve fitting. Assay IC50 results are shown in Table 3.
- the following assay was employed to evaluate the stability of the prodrugs in simulated gastrointestinal tract conditions.
- Preparation of fasted state simulated intestinal fluid (FaSSIF) using Phares SIF Powder was carried out according to protocols from Phare Drug Delivery AG (Baselland, Switzerland).
- Phare Drug Delivery AG Baselland, Switzerland.
- stock solutions (10 mM) of prodrug substance in DMSO was added to 990 of 0.5 mg/mL Pancreatin solution (Fisher CAS#8049-47-6) in FaSSIF.
- Two samples were prepared for each compound at initial. If the sample was clear solution at the beginning, ran one sample directly as initial by HPLC; if the sample was not clear at starting, diluted the sample by 100% ACN.
- the flow rate was 1.8 mL/min, and the injection volume was 5 or 10 ⁇ .
- UV detection was in the range 210-400 nm.
- the mobile phase consisted of solvent A (water plus 10 mM tetrabutylammonium bromide) and solvent B (acetonitrile) with a gradient of 90% solvent A at 0 min changing to 95% solvent B over 6 min, maintained for 1.5 min, then reverting to 90% solvent A over 1.6 min.
- the area of the parent in prodrug at 5h time point was divided by the area of the parent in prodrug at 0 h time point, to generate the % claimed parent ratio, which are summarized in Table 3 for GI Tract stability.
- Prodrugs were administered to beagle dogs through intravenous (IV) and oral (P.O.) administrations in a non-crossover manner.
- the IV dose was prepared in 20% hydroxypropyl ⁇ -cyclodextrin (HPBCD) and was administered via cephalic or saphenous vein.
- the P.O. dose was prepared in 10% polysorbate 80 (Tween 80) and was administered via gavage.
- Blood samples were serially collected following dose administration for up to 48 hr and plasma was separated by centrifugation.
- the concentrations of prodrugs in dog plasma were determined by a LC-MS/MS assay following a protein precipitation step and addition of an appropriate internal standard (labetalol, imipramine or diclofenac). Quantification was done by determining peak area-ratios of the prodrugs and tenofovir to the internal standard. Additional blood sample(s) was collected following dose administration for up to 24 hr.
- PBMCs Peripheral blood mononuclear cells
- concentrations of tenofovir and/or its phosphate conjugate(s) in PBMCs were determined by an LC-MS/MS assay following a protein precipitation step and addition of an appropriate internal standard (labetalol, imipramine or diclofenac). Quantification was done by determining peak area-ratios of tenofovir and/or its phosphate conjugate(s) to the internal standard.
- Pharmacokinetic parameters were obtained using non-compartmental methods (Watson ® ).
- the area under the plasma concentration-time curve (AUCo- t ) was calculated from the first time point (0 min) up to the last time point with measurable drug concentration using the linear trapezoidal or linear/log-linear trapezoidal rule.
- the IV plasma clearance was calculated by dividing the dose by AUCo-inf.
- the terminal half-life of elimination was determined by unweighted linear regression analysis of the log-transformed data. The time points for determination of half-life were selected by visual inspection of the data.
- the volume of distribution at steady state (Vd ss ) was obtained from the product of plasma clearance and mean residence time (determined by dividing the area under the first moment curve by the area under the curve).
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Compounds of Formula (I): and their pharmaceutically acceptable salts are useful for the inhibition of HIV reverse transcriptase. The compounds may also be useful for the prophylaxis or treatment of infection by HIV and in the prophylaxis, delay in the onset or progression, and treatment of AIDS. The compounds and their salts can be employed as ingredients in pharmaceutical compositions, optionally in combination with other antiviral agents, immunomodulators, antibiotics or vaccines.
Description
TITLE OF THE INVENTION
ANTIVIRAL PHOSPHODIAMIDE COMPOUNDS
BACKGROUND OF THE INVENTION
The retrovirus designated human immunodeficiency virus (HIV), particularly the strains known as HIV type-1 (HIV-1) and type-2 (HIV-2), have been etiologically linked to the immunosuppressive disease known as acquired
immunodeficiency syndrome (AIDS). HIV seropositive individuals are initially asymptomatic but typically develop AIDS related complex (ARC) followed by AIDS. Affected individuals exhibit severe immunosuppression which makes them highly susceptible to debilitating and ultimately fatal opportunistic infections. Replication of HIV by a host cell requires integration of the viral genome into the host cell's DNA.
Since HIV is a retrovirus, the HIV replication cycle requires transcription of the viral RNA genome into DNA via an enzyme known as reverse transcriptase (RT).
Reverse transcriptase has three known enzymatic functions: The enzyme acts as an RNA-dependent DNA polymerase, as a ribonuclease, and as a DNA-dependent DNA polymerase. In its role as an RNA-dependent DNA polymerase, RT transcribes a single-stranded DNA copy of the viral RNA. As a ribonuclease, RT destroys the original viral RNA and frees the DNA just produced from the original RNA. During the viral RNA-dependent polymerization process, RT's ribonuclease activity is required for removing RNA and leaving the polypurine tract preserved for initiation of DNA-dependent polymerization. As a DNA-dependent DNA polymerase, RT makes a second,
complementary DNA strand using the first DNA strand as a template. The two strands form double-stranded DNA, which is integrated into the host cell's genome by HIV integrase.
Itis known that compounds that inhibit enzymatic functions of HIV RT will inhibit HIV replication in infected cells. These compounds are useful in the treatment of HIV infection in humans. There are two classes of RT inhibitors: one is non-nucleoside active site competitive RT inhibitors (NNRTIs), such as efavirenz (EFV), nevirapine (NVP), etravirine (ETR), and rilpivirine (RPV), and the other is nucleos(t)ide reverse transcriptase inhibitors (NRTIs) which are active site inhibitors, such as 3'-azido- 3'-deoxythymidine (AZT), 2',3'-dideoxyinosine (ddl), 2',3'- dideoxycytidine (ddC), d4T, 3TC, , abacavir,
emtricitabine, and tenofovir (TFV, also known as PMPA, 9-(2- phosphonyl-methoxypropyl)adenine).
TFV belongs to a class of HIV anti-retro viral (ARV) agents known as nucleotide analog reverse transcriptase inhibitors (NRTIs).

After being taken up by cells, TFV is first converted to tenofovir-monophosphate (TFV-P) by adenosine monophosphate kinase and then to the active antiviral tenofovir-diphosphate (TFV-DP) by 5 '-nucleoside diphosphate kinase.
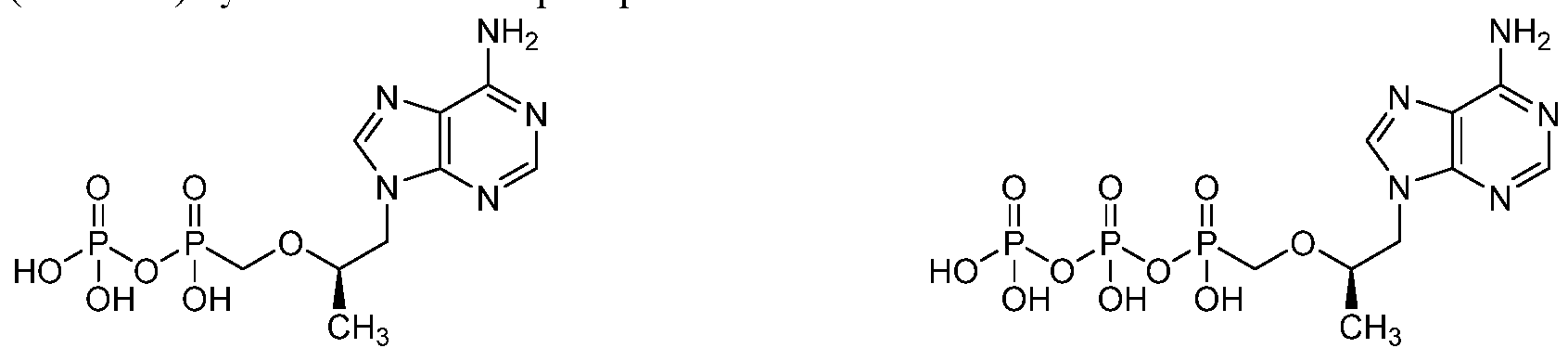
Tenofovir-monophosphate (TFV-P) Tenofovir-diphosphate (TFV-DP)
TFV-DP inhibits HIV DNA synthesis by competing with the natural substrate, deoxy adenosine triphosphate, for incorporation into the complementary DNA strand by HIV reverse transcriptase; following incorporation, TFV acts as a chain terminator due to lack of a 3'-hydroxyl group that is required for addition of the next nucleotide. TFV has poor cellular permeability and thus has limited bioavailability. Tenofovir disoproxil fumarate (TDF) is approved for treating HIV infection and is marketed by Gilead under the trade name
VIREAD™. The disoproxil prodrug improves cell permeability and absorption after oral dosing, with the pro-moiety being cleaved rapidly after absorption to yield the parent TFV. As a result, the circulating level of TFV is much higher than that of TDF. Tenofovir alafenamide fumarate (TAF) is currently under review for treating HIV infection with a reduced level of circulating TFV.
While each of the foregoing drugs is effective in treating HIV infection and
AIDS, there remains a need to develop additional HIV antiviral drugs including additional RT inhibitors. A particular problem is the development of mutant HIV strains that are resistant to the known inhibitors. The use of RT inhibitors to treat AIDS often leads to viruses that are less sensitive to the inhibitors. This resistance is typically the result of mutations that occur in the reverse transcriptase segment of the pol gene. The continued use of antiviral compounds to prevent HIV infection will inevitably result in the emergence of new resistant strains of HIV. Accordingly, there is a particular need for new RT inhibitors that are effective against mutant HIV strains.
SUMMARY OF THE INVENTION
The present invention is directed to phosphodiamide prodrugs of tenofovir and their use in the inhibition of nucleotide reverse transcriptase. In addition to the use of said compounds in the inhibition of HIV reverse transcriptase, the invention is also directed to the use of said compounds for prophylaxis of infection by HIV, the treatment of infection by HIV, and the prophylaxis, treatment, and/or delay in the onset or progression of AIDS and/or ARC.
DETAILED DESCRIPTION OF THE INVENTION
The present invention is directed to compounds of structural Formula I:

or pharmaceutically acceptable salt thereof, wherein:
RA is a D-amino acid ester residue of formula (i) or a D-proline ester residue of formula (ii)
 Rl is (a) -Cl -4alkyl, (b) -Cl -4alkyl substituted with-OH, -SH, -SCH3, -N¾ or -NH- C(=NH)-NH2,
Rl is (a) -Cl -4alkyl, (b) -Cl -4alkyl substituted with-OH, -SH, -SCH3, -N¾ or -NH- C(=NH)-NH2,
(c) -CH2-phenyl, (d) -CH2-phenol, (e) -(CH2)l-2-COOH, (f) -(CH2)l-2-CONH2, (g) - CH2-lH-indole, (h) -CH2-imidazole, (i) aryl (for example but not limited to phenyl or naphthyl) or (j) heteroaryl (for example but not limited to pyridine);
R2 is
(a) -Ci-ioalkyl unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5a5 -0-Ci-3alkyl, -SH, - NR6R75 -C3-6cycloalkyl or spiro-C3-6cycloalkyl,
(b) -CH2-phenyl unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5a -0-Ci-3alkyl, -SH, -NR6R7 0r -Ci- 3alkyl,
(c) -C3-8cycloalkyl unsubstituted or substituted with one to three substituents
independently selected from fluoro, chloro, bromo, -OR^ -0-Ci-3alkyl, -SH, -
NR6R7 or -C 1-3 alky 1,
(d) aryl unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5a -0-Ci-3alkyl, -SH, -NR6R7 0r -Ci-3alkyl,
(e) 2-10 atom heteroalkyl,
(f) heteroaryl unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5a -0-Ci-3alkyl, -SH, -NR6R7 or -Ci- 3alkyl, or
(g) a heterocyclic ring unsubstituted or substituted with one to three substituents
independently selected from fluoro, chloro, bromo, -OR^ -0-Ci-3alkyl, -SH, - NR6R7 or -Ci-3alkyl;
RB is an L-amino acid ester residue of formula (iii) or an L-proline ester residue of formula
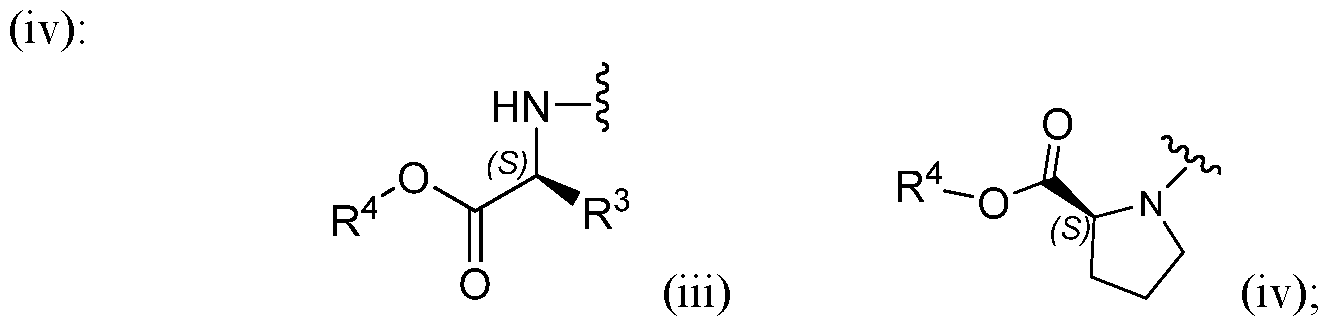

(c) -CH2-phenyl, (d) -CH2-phenol, (e) -(CH2)l-2-COOH, (f) -(CH2)l-2-CONH2, (g) - CH2-lH-indole, (h) -CH2-imidazole, (i) aryl (for example but not limited to phenyl or naphthyl) or (j) heteroaryl (for example but not limited to pyridine);
R4 is
(a) -Ci-ioalkyl unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5b5 -0-Ci-3alkyl, -SH, -
NR8R95 -C3-6cycloalkyl or spiro-C3-6cycloalkyl,
(b) -CH2-phenyl unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5b -0-Ci-3alkyl, -SH, - NR8R9 or -Ci-
3alkyl,
(c) -C3-8cycloalkyl unsubstituted or substituted with one to three substituents
independently selected from fluoro, chloro, bromo, -OR5b5 -0-Ci-3alkyl, -SH, - NR8R9 or -C 1-3 alky 1,
(d) aryl unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5b -0-Ci-3alkyl, -SH, - NR8R9 0r -Ci-3alkyl,
(e) 2-10 atom heteroalkyl,
(f) heteroaryl unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5b -0-Ci-3alkyl, -SH, - NR8R9 or -Ci- 3alkyl, or
(g) a heterocyclic ring unsubstituted or substituted with one to three substituents
independently selected from fluoro, chloro, bromo, -OR5b5 -SH, -NR8R9 or -Ci- 3alkyl;
R5a and R^b are each independently selected from -H or -C3-6cycloalkyl;.
R6 and R7 are each independently selected from -H, -Ci-3alkyl or -C3-6cycloalkyl; and
R8 and R9 are each independently selected from -H, -Ci-3alkyl or -C3-6cycloalkyl. In Embodiment 1 of this invention are compounds of Formula I or the pharmaceutically acceptable salts thereof, wherein RA is a D-amino acid ester residue of formula (i) or a D-proline ester residue of formula (ii), and Rl is -Ci-4alkyl (particularly -
CH3, -CH(CH3)2,
-CH(CH3)CH2CH3, -CH2CH(CH3)2), -CH2OH, -CH(OH)CH3, -CH2SH, -(CH2)2SCH3, - (CH2)4NH2, -(CH2)3NH-C(=NH)-NH2, -CH2-pheny 1, -CH2-phenol, -(CH2) 1 -2-COOH, -(CH2)l-2-CONH2, -CH2-lH-indole, -CH2-imidazole, phenyl, naphthyl or pyridyl. In a class of this embodiment referred to as Embodiment la, Rl is— CH3, -CH(CH3)2, - CH(CH3)CH2CH3,
-CH2CH(CH3)2 or -CH2-phenyl, and in a sub-class thereof Rl is -CH3 or -CH2-phenyl.
In Embodiment 2 of this invention are compounds of Formula I, Embodiment l,or Embodiment la, or a sub-class thereof, or the pharmaceutically acceptable salts thereof, wherein RB is an L-amino acid ester residue of formula (iii) or an L-proline ester residue of formula (iv) or a glycine ester residue of formula (v), and R3 is -Ci-4alkyl (particularly -
CH3, -CH(CH3)2,
-CH(CH3)CH2CH3, -CH2CH(CH3)2), -CH2OH, -CH(OH)CH3, -CH2SH, -(CH2)2SCH3, -(CH2)4NH2, -(CH2)3NH-C(=NH)-NH2, -CH2-phenyl, -CH2-phenol, -(CH2)l -2-COOH, - (CH2)l-2-CONH2, -CH2-lH-indole, -CH2-imidazole, phenyl, naphthyl or pyridyl. In a class of this embodiment referred to as Embodiment 2a, R3 is -CFI3, -CH(CH3)2, - CH(CH3)CH2CH3,
-CH2CH(CH3)2 or -CH2-phenyl, and in a sub-class thereof R3 is -CH3 or -CH2-phenyl.
In Embodiment 3 of this invention are compounds of Formula I, Embodiment 1, Embodiment la, Embodiment 2 or Embodiment 2a, each sub-class thereof, or the pharmaceutically acceptable salts thereof, wherein RA is a D-amino acid ester residue of formula (i) or a D-proline ester residue of formula (ii), and R2 is:
(a) -Ci -8alkyl, -CH2CH2OH -CH2CH2CH2OH -CH2CH2SH -CH2CH2CH2SH
-CH2CH2NH2, -CH2CH2CH2NH2,
(b) -CH2-phenyl, unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5a -0-Ci-3alkyl, -SH, -NR6R7 0r -Ci-3alkyl,
(c) -C3-6cycloalkyl, unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5^ -0-Ci-3alkyl, -SH, -NR6R7 0r -Ci-3alkyl, (d) phenyl or naphthyl, each unsubstituted or substituted with one to three substituents
independently selected from fluoro, chloro, bromo, -OR^ -0-Ci-3alkyl, -SH, -NR6R7 or -C 1-3 alky 1,
(e) -CH2CH20CH3, -CH2CH2CH20CH3, -CH2CH2SCH3, -CH2CH2CH2SCH3,
-CH2CH2NHCH3, -CH2CH2CH2NHCH3,
(f) pyridyl, unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5a -0-Ci-3alkyl, -SH, -NR6R7 0r -Ci-3alkyl, or
(g) piperidinyl, pyrrolidinyl, tetrahydrofuranyl, or tetrahydropyranyl, each unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo,
-OR5a -0-Ci-3alkyl, -SH, -NR6R7 0r -Ci-3alk l.
In a class of Embodiment 3, referred to as Embodiment 3a, R2 is -Ci-8alkyl, -CH2-phenyl, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl or phenyl, and in a sub-class thereof, R2 is -C3-7alkyl, cyclobutyl or cyclopentyl.
In Embodiment 4 of this invention are compounds of Formula I, Embodiment 1, Embodiment la, Embodiment 2, Embodiment 2a, Embodiment 3, or Embodiment 3a, each sub-class thereof, or the pharmaceutically acceptable salts thereof, wherein RB is an L-amino acid ester residue of formula (iii), or an L-proline ester residue of formula (iv) or a glycine ester residue of formula (v), and R4 is:
(a) -Ci-8alkyl, -CH2CH2OH -CH2CH2CH2OH -CH2CH2SH -CH2CH2CH2SH
-CH2CH2NH2, -CH2CH2CH2NH2,
(b) -CH2-phenyl, unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5b -0-Ci-3alkyl, -SH, -NR8R9 0r -Ci-3alkyl,
(c) -C3-6cycloalkyl, unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5b5 -0-Ci-3alkyl, -SH, -NR8R9 0r -Ci-3alkyl,
(d) phenyl or naphthyl, each unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5b5 -0-Ci-3alkyl, -SH, -NR8R9 or -C 1-3 alky 1,
(e) -CH2CH20CH3, -CH2CH2CH20CH3, -CH2CH2SCH3, -CH2CH2CH2SCH3,
-CH2CH2NHCH3, -CH2CH2CH2NHCH3,
(f) pyridyl, unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5b -0-Ci-3alkyl, -SH, -NR8R9 0r -Ci-3alkyl, or
(g) piperidinyl, pyrrolidinyl, tetrahydrofuranyl, or tetrahydropyranyl, each unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo,
-OR5b -0-Ci-3alkyl, -SH, -NR8R9 0r -Ci-3alkyl.
In a class of Embodiment 4, referred to as Embodiment 4a, R4 is -Ci-8alkyl, -CH2-phenyl, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl or phenyl, and in a sub-class thereof, R4 is -C3-7alkyl, cyclobutyl or cyclopentyl.
In Embodiment 5 of this invention are compounds of Formula I and each
Embodiment, class or sub-class thereof, or the pharmaceutically acceptable salts thereof, wherein R5a and R^b are each -H.
In Embodiment 6 of this invention are compounds of Formula I and each Embodiment, class or sub-class thereof, or the pharmaceutically acceptable salts thereof, wherein of R6, R7, R8 and R9 are each -H or -Ci-3alkyl, and particularly each is H.
In Embodiment 7of this invention are compounds of Formula I or a pharmaceutically acceptable salt thereof, wherein:
RA is a D-amino acid ester residue of formula (i) or a D-proline ester residue of formula (ii); Rl is -Ci-4alkyl (particularly -CH3, -CH(CH3)2, -CH(CH3)CH2CH3, or -CH2CH(CH3)2), -CH2OH, -CH(OH)CH3, -CH2SH, -(CH2)2SCH3, -(CH2)4NH2, -(CH2)3NH-C(=NH)-
NH2, -CH2-phenyl, -CH2-phenol, -(CH2)l-2-COOH, -(CH2)l-2-CONH2, -CH2-IH- indole, -CH2-imidazole, phenyl, naphthyl or pyridyl;
R2 is
(a) -Ci-8alkyl, -CH2CH2OH -CH2CH2CH2OH -CH2CH2SH -CH2CH2CH2SH
-CH2CH2NH2, -CH2CH2CH2NH2,
(b) -CH2-phenyl, unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5a -0-Ci-3alkyl, -SH, -NR6R7 0r -Ci-3alkyl,
(c) -C3-6cycloalkyl, unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5^ -0-Ci-3alkyl, -SH, -NR6R7 0r -Ci-3alkyl, (d) phenyl or naphthyl, each unsubstituted or substituted with one to three substituents
independently selected from fluoro, chloro, bromo, -OR^ -0-Ci-3alkyl, -SH, -NR6R7 or -C 1-3 alky 1,
(e) -CH2CH20CH3, -CH2CH2CH20CH3, -CH2CH2SCH3, -CH2CH2CH2SCH3,
-CH2CH2NHCH3, -CH2CH2CH2NHCH3,
(f) pyridyl, unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5a -0-Ci-3alkyl, -SH, -NR6R7 0r -Ci-3alkyl, or
(g) piperidinyl, pyrrolidinyl, tetrahydrofuranyl, or tetrahydropyranyl, each unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5a -0-Ci-3alkyl, -SH, -NR6R7 0r -Ci-3alkyl;
RB is an L-amino acid ester residue of formula (iii), an L-proline ester residue of formula (iv) or a glycine ester residue of formula (v);
R3 is -Ci-4alkyl (particularly -CH3, -CH(CH3)2, -CH(CH3)CH2CH3, or -CH2CH(CH3)2), -CH2OH, -CH(OH)CH3, -CH2SH, -(CH2)2SCH3, -(CH2)4NH2, -(CH2)3NH-C(=NH)- NH2, -CH2-phenyl, -CH2-phenol, -(CH2)l-2-COOH, -(CH2)l-2-CONH2, -CH2-IH- indole, -CH2-imidazole, phenyl, naphthyl or pyridyl;
R4 is
(a) -Ci-8alkyl, -CH2CH2OH -CH2CH2CH2OH -CH2CH2SH -CH2CH2CH2SH
-CH2CH2NH2, -CH2CH2CH2NH2,
(b) -CH2-phenyl, unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5b -0-Ci-3alkyl, -SH, -NR8R9 0r -Ci-3alkyl,
(c) -C3-6cycloalkyl, unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5b, -0-Ci-3alkyl, -SH, -NR8R9 0r -Ci-3alkyl,
(d) phenyl or naphthyl, each unsubstituted or substituted with one to three substituents
independently selected from fluoro, chloro, bromo, -OR5b5 -0-Ci-3alkyl, -SH, -NR8R9 or -C 1 -3 alky 1,
(e) -CH2CH20CH3, -CH2CH2CH20CH3, -CH2CH2SCH3, -CH2CH2CH2SCH3, -CH2CH2NHCH3, -CH2CH2CH2NHCH3,
(f) pyridyl, unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5b -0-Ci-3alkyl, -SH, -NR8R9 0r -Ci-3alkyl, or (g) piperidinyl, pyrrolidinyl, tetrahydrofuranyl, or tetrahydropyranyl, each unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5b -0-Cl -3alkyl, -SH, -NR8R9-N or -Cl -3alkyl;
R5a and R^b are each -H; and
R6, R7, R8 and R9 are each independently selected from -H or Ci-3alkyl.
In Embodiment 8 of this invention are compounds of Formula I or a pharmaceutically acceptable salt thereof, wherein:
RA is a D-amino acid ester residue of formula (i) or a D-proline ester residue of formula (ii); Rl is -CH3, -CH(CH3)2, -CH(CH3)CH2CH3, -CH2CH(CH3)2.or -CH2-phenyl, or more particularly Rl is -CH3 or -CH2-phenyl;
R2 is -Ci-8alkyl, -CH2-phenyl, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl or phenyl, or more particularly it is -C3-7alkyl, cyclobutyl or cyclopentyl;
RB is an L-amino acid ester residue of formula (iii), an L-proline ester residue of formula
(iv), or a glycine ester residue of formula (v);
R3 is -CH3, -CH(CH3)2, -CH(CH3)CH2CH3, -CH2CH(CH3)2 or -CH2-phenyl, or more particularly R3 is -CH3 or -CH2 -phenyl;
R4 is -Ci-8alkyl, -CH2-phenyl, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl or phenyl, or more particularly it is -C3-7alkyl, cyclobutyl or cyclopentyl;
R5a and R5b are each -H; and
R6, R7, R8 and R9 are each independently selected from -H or Ci-3alkyl.
Reference to the compounds of Formula I herein encompasses the compounds of Formula I and all embodiments, classes and sub-classes thereof. The compounds of the invention encompass compounds of structural Formula I, embodiments, classes and subclasses thereof and salts thereof when such salts are possible, including the pharmaceutically acceptable salts of said compounds.
As used herein, "alkyl" refers to both branched- and straight-chain saturated aliphatic hydrocarbon groups having the specified number of carbon atoms in a specified range. For example the term "Ci-io alkyl" means linear or branched chain alkyl groups, including all possible isomers, having 1, 2, 3, 4, 5, 7, 8, 9 or 10 carbon atoms, and includes each of the decyl, nonyl, octyl, heptyl, hexyl and pentyl isomers as well as n-, iso-, sec- and fert-butyl (butyl, s-butyl, /'-butyl, /-butyl; Bu = butyl), n- and z-propyl (Pr = propyl), ethyl (Et) and methyl (Me). "Ci-4alkyl" has 1, 2, 3 or 4 carbon atoms, and includes each of n-, iso-, sec- and fert-butyl, n- and z-propyl, ethyl and methyl.
"Cycloalkyl" refers to a cyclized alkyl ring having the indicated number of carbon atoms in a specified range. Thus, for example, "C3-8 cycloalkyl" encompasses each of cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cyloheptyl and cyclooctyl.
"C3-6cycloalkyl" encompasses each of cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl.
When cycloalkyl is a substituent on an alkyl group in a compound of Formula I, the cycloalkyl substituent can be bonded to any available carbon in the alkyl group. The following are illustrations of -C3-6cycloalkyl substituents wherein the substituent is cyclopropyl in bold:

A "spiro-C3-6cycloalkyl" substituent refers to a cycloalkyl group bonded to an alkyl group via a a single, non-terminal carbon atom which is common to both the the cycloalkyl group and the alkyl group. Spiro-C3-6cycloalkyl encompasses each of spiro- cyclopropyl, spiro-cyclobutyl, spiro-cyclopentyl and spiro-cyclohexyl. The following is an illustration of a spiro-C3-6cycloalkyl substituent wherein the substituent is spiro-cyclopropyl in bold:

"Heteroalkyl" refers to both branched- and straight-chain alkyl groups in which one non-terminal carbon atom is replaced with a heteroatom selected from N, O or S. The heteroalkyl group has the indicated total number of carbons and heteroatoms in a
specifed range, e.g., 2-10 atom heteroalkyl encompasses each such group having one heteroatom plus 1, 2, 3, 4, 5, 6, 7, 8 or 9 carbon atom(s). In an embodiment thereof, heteroalkyl is -Ci-4alkyl-X-Ci-5alkyl wherein X is O, S or NH. Examples of heteroalkyl groups include, but are not limited to,
-CH2CH20CH3, -CH2CH2CH2OCH3 ; -CH2CH2SCH3, -CH2CH2CH2SCH3, -
CH2CH2NHCH3 and -CH2CH2CH2NHCH3.
"Aryl" refers to (i) phenyl, (ii) 9- or 10-membered bicyclic, fused carbocylic ring systems in which at least one ring is aromatic, and (iii) 11- to 14-membered tricyclic, fused carbocyclic ring systems in which at least one ring is aromatic. Suitable aryls include, for example, substituted and unsubstituted phenyl and substituted and unsubstituted naphthyl. An aryl of particular interest is unsubstituted or substituted phenyl.
"Heteroaryl" refers to (i) a 5- or 6-membered heteroaromatic ring containing from 1 to 4 heteroatoms independently selected from N, O and S, wherein each N is optionally in the form of an oxide, and (ii) a 9- or 10-membered bicyclic fused ring system, wherein the fused ring system of (ii) contains from 1 to 6 heteroatoms independently selected from N, O and S, wherein each ring in the fused ring system contains zero, one or more than one heteroatom, at least one ring is aromatic, each N is optionally in the form of an oxide, and each S in a ring which is not aromatic is optionally S(O) or S(0)2- Suitable 5- and 6- membered heteroaromatic rings include, for example, pyridyl, 3-fluroropyridyl, 4- fluoropyridyl, 3-methoxypyridyl, 4-methoxypyridyl, pyrrolyl, pyrazinyl, pyrimidinyl, pyridazinyl, triazinyl, thienyl, furanyl, imidazolyl, pyrazolyl, triazolyl (i.e., 1,2,3-triazolyl or 1,2,4-triazolyl), tetrazolyl, oxazolyl, isooxazolyl, oxadiazolyl (i.e., the 1,2,3-, 1,2,4-, 1,2,5- (furazanyl), or 1,3,4-isomer), oxatriazolyl, thiazolyl, isothiazolyl, and thiadiazolyl. Suitable 9- and 10-membered heterobi cyclic, fused ring systems include, for example, benzofuranyl, indolyl, indazolyl, naphthyridinyl, isobenzofuranyl, benzopiperidinyl, benzisoxazolyl, benzoxazolyl, chromenyl, quinolinyl, isoquinolinyl, isoindolyl, benzopiperidinyl, benzofuranyl, imidazo[l,2-a]pyridinyl, benzotriazolyl, indazolyl, indolinyl, and isoindolinyl. A class of heteroaryls includes unsubstituted or substituted pyridyl or pyrimidyl, and particularly unsubstituted or substituted pyridyl.
The term "heterocyclic ring" refers to (i) a saturated 4- to 7-membered cyclized ring and (ii) an unsaturated, non-aromatic 4 to 7-membered cyclized ring comprised
of carbon atoms and 1- 4 heteratoms independently selected from O, N and S. Heterocyclic rings within the scope of this invention include, for example, azetidinyl, piperidinyl, morpholinyl, thiomorpholinyl, thiazolidinyl, isothiazolidinyl, oxazolidinyl, isoxazolidinyl, pyrrolidinyl, imidazolidinyl, piperazinyl, tetrahydrofuranyl, tetrahydrothienyl, pyrazolidinyl, hexahydropyrimidinyl, thiazinanyl, thiazepanyl, azepanyl, diazepanyl, tetrahydropyranyl, tetrahydrothiopyranyl, and dioxanyl. Examples of 4- to 7-membered, unsaturated, non- aromatic heterocyclic rings within the scope of this invention include mono-unsaturated heterocyclic rings corresponding to the saturated heterocyclic rings listed in the preceding sentence in which a single bond is replaced with a double bond (e.g., a carbon-carbon single bond is replaced with a carbon-carbon double bond). A class of heterocyclic rings includes piperidinyl, pyrrolidinyl, tetrahydrofuranyl, or tetrahydropyranyl.
It is understood that the specific rings and ring systems suitable for use in the present invention are not limited to those listed in the preceding paragraphs. These rings and ring systems are merely representative.
As would be recognized by one of ordinary skill in the art, certain compounds of the present invention maybe able to exist as tautomers. All tautomeric forms of these compounds, whether isolated individually or in mixtures, are within the scope of the present invention. For example, in instances where an -OH substituent is permitted on a
heteroaromatic ring and keto-enol tautomerism is possible, it is understood that the substituent might in fact be present, in whole or in part, in the oxo (=0) form.
A "stable" compound is a compound which can be prepared and isolated and whose structure and properties remain or can be caused to remain essentially unchanged for a period of time sufficient to allow use of the compound for the purposes described herein (e.g., therapeutic or prophylactic administration to a subject). The compounds of the present invention are limited to stable compounds embraced by Formula I and its embodiments. For example, certain moieties as defined in Formula I may be unsubstituted or substituted, and the latter is intended to encompass substitution patterns (i.e., number and kind of substituents) that are chemically possible for the moiety and that result in a stable compound.
Each compound of Formula I is comprised of a phosphodiamide amino acid ester, containing one D-amino acid ester residue and one achiral- or L-amino acid ester residue. Each compound also has a defined (R) chiral center in the alkyl-ether linking group
connecting the nucleobase to the phosphodiamide. Accordingly, each compound of Formula I has mutliple chiral centers (also referred to as asymmetric or stereogenic centers), for which the spatial orientation of certain chiral centers are specifically defined. Each compound of Formula I also has an assymetric phosphorus center. This invention encompasses compounds having either the (R) or (S) stereoconfiguration at the phosphorus assymetric center, or mixtures thereof.
This invention includes individual diastereomers, particularly epimers, i.e., compounds having the same chemical formula but which differ in the spatial arrangement around a single atom. This invention also includes mixtures of diastereomers, particularly mixtures of epimers, in all ratios. Embodiments of this invention also include a mixture of epimers enriched with 51 % or more of one of the epimers, including for example 60% or more, 70% or more, 80% or more, or 90% or more of one epimer. A single epimer is preferred. An individual or single epimer refers to an epimer obtained by chiral synthesis and/or using generally known separation and purification techniques, and which may be 100% of one epimer or may contain small amounts (e.g., 10% or less) of the opposite epimer. Thus, individual diasteromers are a subject of the invention in pure form, both as levorotatory and as dextrorotatory antipodes, in the form of racemates and in the form of mixtures of the two diasteromers in all ratios. In the case of a cis/trans isomerism the invention includes both the cis form and the trans form as well as mixtures of these forms in all ratios.
The preparation of individual stereoisomers can be carried out, if desired, by separation of a mixture by customary methods, for example by chromatography or crystallization, by the use of stereochemically uniform starting materials for the synthesis or by stereoselective synthesis. Optionally a derivatization can be carried out before a separation of stereoisomers. The separation of a mixture of stereoisomers can be carried out at an intermediate step during the synthesis of a compound of Formula I or it can be done on a final racemic product. Absolute stereochemistry may be determined by X-ray crystallography of crystalline products or crystalline intermediates which are derivatized, if necessary, with a reagent containing a stereogenic center of known configuration. Alternatively, absolute stereochemistry may be determined by Vibrational Circular Dichroism (VCD) spectroscopy analysis. The present invention includes all such isomers, as well as salts, solvates (which
includes hydrates) and solvated salts of such racemates, enantiomers, diastereomers and tautomers and mixtures thereof.
The atoms in a compound of Formula I may exhibit their natural isotopic abundances, or one or more of the atoms may be artificially enriched in a particular isotope having the same atomic number, but an atomic mass or mass number different from the atomic mass or mass number predominantly found in nature. The present invention is meant to include all suitable isotopic variations of the compounds of Formula I, for example, different isotopic forms of hydrogen (H) include protium (lH) and deuterium (¾T). Protium is the
predominant hydrogen isotope found in nature. Enriching for deuterium may afford certain therapeutic advantages, such as increasing in vivo half-life or reducing dosage requirements, or may provide a compound useful as a standard for characterization of biological samples. Isotopically-enriched compounds of Formula I can be prepared without undue
experimentation by conventional techniques well known to those skilled in the art or by processes analogous to those described in the Schemes and Examples herein using appropriate isotopically-enriched reagents and/or intermediates.
The compounds can be administered in the form of pharmaceutically acceptable salts. The term "pharmaceutically acceptable salt" refers to a salt which is not biologically or otherwise undesirable (e.g., is neither toxic nor otherwise deleterious to the recipient thereof).
Since the compounds of Formula I contain by definition at least one basic group, the invention includes the corresponding pharmaceutically acceptable salts. When the compounds of Formula I contain one or more acidic groups, the invention also includes the corresponding pharmaceutically acceptable salts. Thus, the compounds of Formula I that contain acidic groups (e.g., -COOH) can be used according to the invention as, for example but not limited to, alkali metal salts, alkaline earth metal salts or as ammonium salts.
Examples of such salts include but are not limited to sodium salts, potassium salts, calcium salts, magnesium salts or salts with ammonia or organic amines such as, for example, ethylamine, ethanolamine, triethanolamine or amino acids. Compounds of Formula I, which contain one or more basic groups, i.e. groups which can be protonated, can be used according to the invention in the form of their acid addition salts with inorganic or organic acids as, for example but not limited to, salts with hydrogen chloride, hydrogen bromide, phosphoric acid, sulfuric acid, nitric acid, benzenesulfonic acid, methanesulfonic acid, p-toluenesulfonic acid,
naphthalenedisulfonic acids, oxalic acid, acetic acid, trifluoroacetic acid, tartaric acid, lactic acid, salicylic acid, benzoic acid, formic acid, propionic acid, pivalic acid, diethylacetic acid, malonic acid, succinic acid, pimelic acid, fumaric acid, maleic acid, malic acid, sulfaminic acid, phenylpropionic acid, gluconic acid, ascorbic acid, isonicotinic acid, citric acid, adipic acid, etc. If the compounds of Formula I simultaneously contain acidic and basic groups in the molecule the invention also includes, in addition to the salt forms mentioned, inner salts or betaines (zwitterions). Salts can be obtained from the compounds of Formula I by customary methods which are known to the person skilled in the art, for example by combination with an organic or inorganic acid or base in a solvent or dispersant, or by anion exchange or cation exchange from other salts. The present invention also includes all salts of the compounds of Formula I which, owing to low physiological compatibility, are not directly suitable for use in pharmaceuticals but which can be used, for example, as intermediates for chemical reactions or for the preparation of pharmaceutically acceptable salts.
Furthermore, compounds of the present invention may exist in amorphous form and/or one or more crystalline forms, and as such all amorphous and crystalline forms and mixtures thereof of the compounds of Formula I are intended to be included within the scope of the present invention. In addition, some of the compounds of the instant invention may form solvates with water (i.e., a hydrate) or common organic solvents. Such solvates and hydrates, particularly the pharmaceutically acceptable solvates and hydrates, of the compounds of of this invention are likewise encompassed within the scope of the compounds defined by Formula I and the pharmaceutically acceptable salts thereof, along with un- solvated and anhydrous forms of such compounds.
Accordingly, the compounds of Formula I, embodiments thereof and specific compounds described and claimed herein encompass all possible pharmaceutically acceptable salts, stereoisomers, tautomers, physical forms (e.g., amorphous and crystalline forms), solvate and hydrate forms and any combination of the foregoing forms where such forms are possible.
Compounds of Formula I (or any embodiment thereof and pharmaceutically acceptable salts thereof) are prodrug modifications of tenofovir, which is a mono- phosphonate. The compounds of Formula I may be converted intracellularly/z ? vivo by one
or more mechanisms (e.g., enzyme-catalyzed chemical reactions) to the corresponding monophosphate or diphosphate of tenofovir . While not wishing to be bound by any particular theory, tenofovir diphosphate is generally understood to be responsible for inhibiting the HIV RT enzyme and for the resulting antiviral activity after administration of the compound of Formula I to a subject.
The compounds of Formula I described herein are prodrugs. A discussion of prodrugs is provided in (a) Stella, V. J.; Borchardt, R. T.; Hageman, M. J.; Oliyai, R.; Maag, H. et al. Prodrugs: Challenges and Rewards Part 1 and Part 2; Springer, p. 726: New York, NY, USA, 2007, (b) Rautio, I; Kumpulainen, H.; Heimbach, T.; Oliyai, R.; Oh, D. et al.
Prodrugs: design and clinical applications. Nat. Rev. Drug Discov. 2008, 7, 255, (c) T.
Higuchi and V. Stella, Pro-drugs as Novel Delivery Systems (1987) \4 of the A.C.S.
Symposium Series, and in (d) Bioreversible Carriers in Drug Design, (1987) Edward B. Roche, ed., American Pharmaceutical Association and Pergamon Press. More specifically, compounds of Formula I are prodrug modifications of tenofovir, which is a mono- phosphonate. The compounds of Formula I may be converted intracellularly {in vivo or in vitro) to the corresponding monophosphate or diphosphate of tenofovir. The conversion may occur by one or more mechanisms, e.g., an enzyme-catalyzed chemical reaction, a metabolic chemical reaction, and/or a spontaneous chemical reaction (e.g., solvolysis), such as, for example, through hydrolysis in blood. While not wishing to be bound by any particular theory, tenofovir diphosphate is generally understood to be responsible for inhibiting the HIV RT enzyme and for the resulting antiviral activity after administration of the compound of Formula I to a subject.
Another embodiment of the present invention is a compound of Formula I wherein the compound or its salt is in a substantially pure form. As used herein "substantially pure" means suitably at least about 60 wt.%, typically at least about 70 wt.%, preferably at least about 80 wt.%, more preferably at least about 90 wt.% (e.g., from about 90 wt.% to about 99 wt.%), even more preferably at least about 95 wt.% (e.g., from about 95 wt.% to about 99 wt.%, or from about 98 wt. % to 100 wt. %), and most preferably at least about 99 wt.% (e.g., 100 wt.%) of a product containing a compound of Formula I or its salt (e.g., the product isolated from a reaction mixture affording the compound or salt) consists of the compound or salt. The level of purity of the compounds and salts can be determined using a standard
method of analysis such as, high performance liquid chromatography, and/or mass spectrometry or NMR techniques. If more than one method of analysis is employed and the methods provide experimentally significant differences in the level of purity determined, then the method providing the highest purity level governs. A compound or salt of 100% purity is one which is free of detectable impurities as determined by a standard method of analysis. With respect to a compound of the invention which has one or more asymmetric centers and can occur as mixtures of stereoisomers, a substantially pure compound can be either a substantially pure mixture of the stereoisomers or a substantially pure individual
stereoisomer.
The compounds of Formula I herein, and pharmaceutically acceptable salts thereof, are HIV reverse transcriptase inhibitors. The compounds are useful for inhibiting HIV reverse transcriptase and for inhibiting HIV replication in vitro and in vivo. More particularly, the compounds of Formula I inhibit the polymerase function of HIV- 1 reverse
transcriptase. The testing of compounds of the Examples of this invention in the Viking assay set forth in Example 10 below, illustrate the ability of compounds of the invention to inhibit the RNA-dependent DNA polymerase activity of HIV- 1 reverse transcriptase. The compounds of Formula I may also be useful agents against HIV-2. The compounds of Examples 1-9 of the present invention also exhibit activity against drug resistant forms of HIV (e.g., N RTI-associated mutant strains K103N and/or Y181C; NRTI-associated mutant strains Ml 84V and Ml 841 mutants). This invention also encompasses methods for the treatment or prophylaxis of infection by HIV, for the inhibition of HIV reverse transcriptase, for the treatment, prophylaxis, or delay in the onset of AIDS in a subject in need thereof, which comprise administering to the subject an effective amount of a compound of the invention or a pharmaceutically acceptable salt thereof.
The invention further encompasses methods for the treatment or prophylaxis of infection by HIV, for the inhibition of HIV reverse transcriptase, for the treatment, prophylaxis, or delay in the onset of AIDS in a subject in need thereof, which comprise administering to the subject an effective amount of a compound of the invention or a pharmaceutically acceptable salt thereof in combination with an effective amount of one or more additional anti-HIV agents selected from the group consisting of HIV antiviral agents, immunomodulators, and anti-infective agents. Within this embodiment, the anti-HIV agent is
an antiviral selected from the group consisting of HIV protease inhibitors, HIV reverse transcriptase inhibitors, HIV integrase inhibitors, HIV fusion inhibitors, HIV entry inhibitors, and HIV maturation inhibitors
The invention encompasses a pharmaceutical composition comprising an effective amount of a compound of the invention, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier. The invention also encompasses a pharmaceutical composition comprising an effective amount of a compound of the invention, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier further comprising an effective amount of one or more additional anti-HIV agents selected from the group consisting of HIV antiviral agents, immunomodulators, and anti-infective agents.
Within this embodiment, the anti-HIV agent is an antiviral selected from the group consisting of HIV protease inhibitors, HIV reverse transcriptase inhibitors, HIV integrase inhibitors, HIV fusion inhibitors, HIV entry inhibitors, and HIV maturation inhibitors.
The compounds of this invention could also be useful for inhibition of HBV reverse transcriptase. Accordingly, this invention also encompasses methods for the treatment of chronic hepatitis B which comprise administering to the subject an effective amount of a compound of the invention or a pharmaceutically acceptable salt thereof.
The invention also encompasses a compound of the invention, or a pharmaceutically acceptable salt thereof, for use in the preparation of a medicament for the treatment or prophylaxis of infection by HIV, for the inhibition of HIV reverse transcriptase, or for the treatment, prophylaxis, or delay in the onset of AIDS in a subject in need thereof.
Other embodiments of the present invention include the following:
(a) A pharmaceutical composition comprising an effective amount of a compound of Formula I or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
(b) A pharmaceutical composition which comprises the product prepared by combining (e.g., mixing) an effective amount of a compound of Formula I or a
pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
(c) The pharmaceutical composition of (a) or (b), further comprising an effective amount of one or more an anti-HIV agents selected from the group consisting of HIV antiviral agents, immunomodulators, and anti-infective agents.
(d) The pharmaceutical composition of (c), wherein the anti-HIV agent is selected from one or more of an antiviral selected from the group consisting of HIV protease inhibitors, nucleoside HIV reverse transcriptase inhibitors, non-nucleoside HIV reverse transcriptase inhibitors, HIV integrase inhibitors, HIV fusion inhibitors, HIV entry inhibitors and HIV maturation inhibitors.
(e) A combination which is (i) a compound of Formula I or a pharmaceutically acceptable salt thereof and (ii) an anti-HIV agent selected from the group consisting of HIV antiviral agents, immunomodulators, and anti-infective agents; wherein the compound and the anti-HIV agent are each employed in an amount that renders the combination effective for inhibition of HIV reverse transcriptase, for treatment or prophylaxis of infection by HIV, or for treatment, prophylaxis of, or delay in the onset or progression of AIDS.
(f) The combination of (e), wherein the anti-HIV agent is an antiviral selected from the group consisting of HIV protease inhibitors, nucleoside HIV reverse transcriptase inhibitors, non-nucleoside HIV reverse transcriptase inhibitors, HIV integrase inhibitors, HIV fusion inhibitors, HIV entry inhibitors and HIV maturation inhibitors.
(g) A method for the inhibition of HIV reverse transcriptase in a subject in need thereof which comprises administering to the subject an effective amount of a compound of Formula I or pharmaceutically acceptable salt thereof.
(h) A method for the prophylaxis or treatment of infection by HIV (e.g., HIV-1) in a subject in need thereof which comprises administering to the subject an effective amount of a compound of Formula I or pharmaceutically acceptable salt thereof.
(i) The method of (h), wherein the compound of Formula I or a pharmaceutically acceptable salt thereof is administered in combination with an effective amount of at least one other HIV antiviral selected from the group consisting of HIV protease inhibitors, HIV integrase inhibitors, non-nucleoside HIV reverse transcriptase inhibitors, nucleoside HIV reverse transcriptase inhibitors, HIV fusion inhibitors, HIV entry inhibitors and HIV maturation inhibitors.
(j) A method for the prophylaxis, treatment or delay in the onset or progression of AIDS in a subject in need thereof which comprises administering to the subject an effective amount of a compound of Formula I or pharmaceutically acceptable salt thereof.
(k) The method of (j), wherein the compound is administered in combination with an effective amount of at least one other HIV antiviral selected from the group consisting of HIV protease inhibitors, HIV integrase inhibitors, non-nucleoside HIV reverse transcriptase inhibitors, nucleoside HIV reverse transcriptase inhibitors, HIV fusion inhibitors, HIV entry inhibitors and HIV maturation inhibitors.
(1) A method for the inhibition of HIV reverse transcriptase in a subject in need thereof which comprises administering to the subject the pharmaceutical composition of (a), (b), (c) or (d) or the combination of (e) or (f).
(m) A method for the prophylaxis or treatment of infection by HIV (e.g., HIV-1) in a subject in need thereof, which comprises administering to the subject the pharmaceutical composition of (a), (b), (c) or (d) or the combination of (e) or (f).
(n) A method for the prophylaxis, treatment, or delay in the onset or progression of AIDS in a subj ect in need thereof which comprises administering to the subj ect the pharmaceutical composition of (a), (b), (c) or (d) or the combination of (e) or (f).
The present invention also includes a compound of Formula I or pharmaceutically acceptable salt thereof (i) for use in, (ii) for use as a medicament for, or (iii) for use in the preparation of a medicament for: (a) therapy (e.g., of the human body), (b) medicine, (c) inhibition of HIV reverse transcriptase, (d) treatment or prophylaxis of infection by HIV, or (e) treatment, prophylaxis of, or delay in the onset or progression of AIDS. In these uses, the compounds of the present invention can optionally be employed in combination with one or more anti-HIV agents selected from HIV antiviral agents, anti-infective agents, and immunomodulators.
Additional embodiments of the invention include the pharmaceutical compositions, combinations and methods set forth in (a)-(n) above and the uses (i)(a)-(e) through (iii)(a)-(e) set forth in the preceding paragraph, wherein the compound of the present invention employed therein is a compound of one of the embodiments, aspects, classes, sub-classes, or features described above. In all of these embodiments etc., the compound may optionally be used in the form of a pharmaceutically acceptable salt.
Additional embodiments of the present invention include each of the pharmaceutical compositions, combinations, methods and uses set forth in the preceding paragraphs, wherein the compound of the present invention or its salt employed therein is substantially pure. With
respect to a pharmaceutical composition comprising a compound of Formula I or its salt and a pharmaceutically acceptable carrier and optionally one or more excipients, it is understood that the term "substantially pure" is in reference to a compound of Formula I or its salt per se.
Still additional embodiments of the present invention include the pharmaceutical compositions, combinations and methods set forth in (a)-(n) above and the uses (i)(a)-(e) through (iii)(a)-(e) set forth above, wherein the HIV of interest is HIV-1. Thus, for example, in the pharmaceutical composition (d), the compound of Formula I is employed in an amount effective against HIV-1 and the anti-HIV agent is an HIV-1 antiviral selected from the group consisting of HIV-1 protease inhibitors, HIV-1 reverse transcriptase inhibitors, HIV-1 integrase inhibitors, HIV-1 fusion inhibitors, HIV-1 entry inhibitors and HIV-1 maturation inhibitors. The compounds of Formula I may also be useful agents against HIV-2.
The term "administration" and variants thereof (e.g., "administering" a compound) in reference to a compound of Formula I means providing the compound to the individual in need of treatment or prophylaxis and includes both self-administration and administration to the patient by another person. When a compound is provided in combination with one or more other active agents (e.g., antiviral agents useful for treating or prophylaxis of HIV infection or AIDS), "administration" and its variants are each understood to include provision of the compound and other agents at the same time or at different times. When the agents of a combination are administered at the same time, they can be administered together in a single composition or they can be administered separately.
As used herein, the term "composition" is intended to encompass a product comprising the specified ingredients, as well as any product which results from combining the specified ingredients. Ingredients suitable for inclusion in a pharmaceutical composition are pharmaceutically acceptable ingredients, which means the ingredients must be compatible with each other and not deleterious to the recipient thereof.
The term "subject" or "patient" as used herein refers to an animal, preferably a mammal, most preferably a human, who has been the object of treatment, observation or experiment.
The term "effective amount" as used herein means an amount of a compound sufficient to inhibit HIV reverse transcriptase, inhibit HIV replication, exert a prophylactic effect, and/or a exert a therapeutic effect after administration. One embodiment of "effective
amount" is a "therapeutically effective amount" which is an amount of a compound that is effective for inhibiting HIV reverse transcriptase, inhibiting HIV replication (either of the foregoing which may also be referred to herein as an "inhibition effective amount"), treating HIV infection, treating AIDS, delaying the onset of AIDS, and/or slowing progression of AIDS in a patient.
Another embodiment of "effective amount" is a "prophylactically effective amount" which is an amount of the compound that is effective for prophylaxis of HIV infection or prophylaxis of AIDS in a patient. It is understood that an effective amount can simultaneously be both a therapeutically effective amount, e.g., for treatment of HIV infection, and a prophylactically effective amount, e.g., for prevention or reduction of risk for developing AIDS. The term "preventing," as used herein with respect to an HIV viral infection or AIDS, refers to reducing the likelihood or severity of HIV infection or AIDS.
When the compound of Formula I is administered as a salt, reference to an amount of the compound in milligrams or grams is based on the free form (i.e., the non-salt form) of the compound. In the combination therapies of the present invention, an effective amount can refer to each individual agent or to the combination as a whole, wherein the amounts of all agents administered in the combination are together effective, but wherein a component agent of the combination may or may not be present individually in an effective amount with reference to what is considered effective for that component agent if it were were
administered alone. In the method of the present invention (i.e., inhibiting HIV reverse transcriptase, treating or prophylaxis of HIV infection, inhibiting HIV replication, treating or prophylaxis of AIDS, delaying the onset of AIDS, or delaying or slowing progression of AIDS), the compounds of this invention, optionally in the form of a salt, can be administered by means that produces contact of the active agent with the agent's site of action. They can be administered by conventional means available for use in conjunction with pharmaceuticals, either as individual therapeutic agents or in a combination of therapeutic agents. They can be administered alone, but typically are administered with a pharmaceutical carrier selected on the basis of the chosen route of administration and standard pharmaceutical practice. The compounds of the invention can, for example, be administered orally (e.g., via tablet or capsule), parenterally (including subcutaneous injections, intravenous, intramuscular or intrastemal injection, or infusion techniques), by inhalation spray, or rectally, in the form of a
unit dosage of a pharmaceutical composition containing an effective amount of the compound and conventional non-toxic pharmaceutically acceptable carriers, adjuvants and vehicles. The compound could also be administered via an implantable drug delivery device adapted to provide an effective amount of the compound or a pharmaceutical composition of the compound over an extended period of time.
Liquid preparations suitable for oral administration (e.g., suspensions, syrups, elixirs and the like) can be prepared according to techniques known in the art and can employ any of the usual media such as water, glycols, oils, alcohols and the like. Solid preparations suitable for oral administration (e.g., powders, pills, capsules and tablets) can be prepared according to techniques known in the art and can employ such solid excipients as starches, sugars, kaolin, lubricants, binders, disintegrating agents and the like. Parenteral compositions can be prepared according to techniques known in the art and typically employ sterile water as a carrier and optionally other ingredients, such as a solubility aid. Injectable solutions can be prepared according to methods known in the art wherein the carrier comprises a saline solution, a glucose solution or a solution containing a mixture of saline and glucose.
Implantable compositions can be prepared according to methods known in the art wherein the carrier comprises the active chemical ingredient with polymers as suitable excipients, or utilizing an implantable device for drug delivery. Further description of methods suitable for use in preparing pharmaceutical compositions for use in the present invention and of ingredients suitable for use in said compositions is provided in Remington's Pharmaceutical Sciences. 18th edition, edited by A. R. Gennaro, Mack Publishing Co., 1990 and in
Remington - The Science and Practice of Pharmacy. 22nd Edition, published by
Pharmaceutical Press and Philadelphia College of Pharmacy at University of the Sciences, 2012, ISBN 978 0 85711-062-6 and prior editions.
Formulations of compounds described by Formula I that result in drug supersaturation and/or rapid dissolution may be utilized to facilitate oral drug absorption. Formulation approaches to cause drug supersaturation and/or rapid dissolution include, but are not limited to, nanoparticulate systems, amorphous systems, solid solutions, solid dispersions, and lipid systems. Such formulation approaches and techniques for preparing them are well known in the art. For example, solid dispersions can be prepared using excipients and processes as described in reviews (e.g., A.T.M. Serajuddin, J Pharm Sci,
88: 10, pp. 1058-1066 (1999)). Nanoparticulate systems based on both attrition and direct synthesis have also been described in reviews such as Wu et al (F. Kesisoglou, S. Panmai, Y. Wu, Advanced Drug Delivery Reviews, 59:7 pp. 631 -644 (2007)).
The compounds of Formula I can be administered in a dosage range of 0.001 to 1000 mg/kg of mammal (e.g., human) body weight per day, or at longer time intervals on non- consecutive days as appropriate, in a single dose or in divided doses. One example of a dosage range is 0.01 to 500 mg/kg body weight per day, or at other time intervals as appropriate, administered orally or via other routes of administration in a single dose or in divided doses. Another example of a dosage range is 0.1 to 100 mg/kg body weight per day, or at other time intervals as appropriate, administered orally or via other routes of administration in single or divided doses. Another example of a dosage range is 50 mg to 1 gram per day, in a single dose or divided doses.
Daily or weekly adminstration can be via any suitable route of administration but is preferably via oral administration and can be in single or divided daily doses within each 24 hour period. Divided doses may be administered via mulitple dosage units at essentially the same time or at staggered times over the 24 hour period. For weekly or less frequent dosing regimens with longer time intervals on non-consecutive days, a parenteral route of adminstration may be employed. Examples of such dosing regimens with longer time intervals on non-consecutive days include but are not limited to administration once weekly, once bi-weekly (once every two weeks with leeway as to exact date of dosing), once monthly (e.g., once every 30 days, or the same calendar day each month with leeway as to exact date of dosing), once bimonthly (e.g., once every 60 days, or the same calendar day every two months with leeway as to exact date of dosing), once every 3 months (e.g., once every 90 days, or the same calendar day every three months with leeway as to exact date of dosing), or once every six months (e.g., once every 180 days, or the same calendar day every six months with leeway as to exact date of dosing), or once yearly (e.g., once every 12 months with leeway as to exact date of the annual dosing). For oral (e.g., tablets or capsules) or other routes of administration, the dosage units may contain 1.0 mg to l OOOmg of the active ingredient, for example but not limited to, 1 , 5, 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 350, 400, 450, 500, 600, 700, 800, 900 or 1000 milligrams of the active ingredient for the symptomatic adjustment of the dosage to the patient to be treated. Furthermore, the
compound may be formulated in oral formulations for immediate or modified release such as extended or controlled release.
The favorable pharmacokinetic profile of tested compounds of this invention may also render the the compounds suitable for less frequent dosing. Thus, the compounds of the invention could be administered orally, weekly or parenterally at longer time intervals as described above. For parenteral administration, the compositions can be administered, e.g., intravenously (IV) or intramuscularly (IM) via injection, or using other infusion techniques. One or more of such injections or infusions may be administered at each dosing time interval as needed to deliver the appropriate amount of active agent.
The specific dose level and frequency of dosage for any particular patient may be varied and will depend upon a variety of factors including the activity of the specific compound employed, the metabolic stability and length of action of that compound, the age, body weight, general health, sex, diet, mode and time of administration, rate of excretion, drug combination, the severity of the particular condition, and the host undergoing therapy. In some cases, depending on the potency of the compound or the individual response, it may be necessary to deviate upwards or downwards from the given dose. The amount and frequency of administration will be regulated according to the judgment of the attending clinician considering such factors.
As noted above, the present invention is also directed to use of a compound of Formula I with one or more anti-HIV agents. An "anti-HIV agent" is any agent which is directly or indirectly effective in the inhibition of HIV, the treatment or prophylaxis of HIV infection, and/or the treatment, prophylaxis or delay in the onset or progression of AIDS. It is understood that an anti-HIV agent is effective in treating, preventing, or delaying the onset or progression of HIV infection or AIDS and/or diseases or conditions arising therefrom or associated therewith. For example, the compounds of this invention may be effectively administered, whether at periods of pre-exposure and/or post-exposure, in combination with effective amounts of one or more anti-HIV agents selected from HIV antiviral agents, imunomodulators, antiinfectives, or vaccines useful for treating HIV infection or AIDS. Suitable HIV antivirals for use in combination with the compounds of the present invention include, for example, those listed in Table 1 as follows:
Antiviral A ents for Treatin HIV infection or AIDS

lopinavir + ritonavir, Kaletra® PI
maraviroc, Selzentry® EI
nelfinavir, Viracept® PI
nevirapine, NVP, Viramune® nnRTI
PPL- 100 (also known as PL-462) (Ambrilia) PI
raltegravir, MK-0518, Isentress™ Inl
Rilpivirine nnRTI
ritonavir, Norvir® PI
saquinavir, Invirase®, Fortovase® PI
stavudine, d4T,didehydrodeoxythymidine, Zerit® nRTI
tipranavir, Aptivus® PI
vicriviroc EI
EI = entry inhibitor; FI = fusion inhibitor; Inl = integrase inhibitor; PI = protease inhibitor; nRTI = nucleoside reverse transcriptase inhibitor; nnRTI = non-nucleoside reverse transcriptase inhibitor. Some of the drugs listed in the table are used in a salt form; e.g., abacavir sulfate, delavirdine mesylate, indinavir sulfate, atazanavir sulfate, nelfinavir mesylate, saquinavir mesylate.
It is understood that the scope of combinations of the compounds of this invention with anti-HIV agents is not limited to the HIV antivirals listed in Table 1, but includes in principle any combination with any pharmaceutical composition useful for the treatment or prophylaxis of AIDS. The HIV antiviral agents and other agents will typically be employed in these combinations in their conventional dosage ranges and regimens as reported in the art, including, for example, the dosages described in the Physicians' Desk Reference, Thomson PDR, Thomson PDR, 57th edition (2003), the 58th edition (2004), or the 59th edition (2005) and the current Physicians' Desk Reference (68th ed.). (2014), Montvale, NJ: PDR Network. The dosage ranges for a compound of the invention in these combinations can be the same as those set forth above.
The compounds of this invention are also useful in the preparation and execution of screening assays for antiviral compounds. For example, the compounds of this invention are
useful for isolating enzyme mutants, which are excellent screening tools for more powerful antiviral compounds. Furthermore, the compounds of this invention are useful in establishing or determining the binding site of other antivirals to HIV reverse transcriptase, e.g., by competitive inhibition.
Abbreviations and acron ms em lo ed herein include the followin :


Several methods for preparing the compounds of this invention are described in the following Schemes and Examples. Starting materials and intermediates were purchased commercially from common catalog sources or were made using known procedures, or as otherwise illustrated. Some frequently applied routes to the compounds of Formula I are described in Schemes that follow. In some cases the order of carrying out the reaction steps in the Schemes and Examples may be varied to facilitate the reaction or to avoid unwanted reaction products.
SCHEME 1

Compounds of Formula I can be prepared from (i?)-(((l-(6-amino-9H-purin-9- yl)propan-2-yl)oxy)methyl)phosphonic acid (1), also referred to herein as TFV, with D- amino esters under EDC coupling conditions. The reaction of phosphoamide (3) with the second L-amino esters under 2,2'-dipyridyldisulfide condensation conditions yields the products in formula (II). D or L-proline esters or glycine esters can likewise be employed. For amino esters that are not commercially available, they are generally prepared by condensation between corresponding amino acid with alcohols under thionyl chloride.
SCHEME 2

Alternatively, compounds of Formula I can be prepared from (i?)-(((l-(6- amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonic acid (1), with L-amino esters under EDC coupling conditions to yield (5) first. The reaction of phosphoamide (5) with the D-amino esters under 2,2'-dipyridyldisulfide condensation conditions yields the products in formula (II). D or L-proline esters or glycine esters can likewise be employed.
SCHEME 3

(III)
Compounds of Formula III can be prepared from (i?)-(((l-(6-amino-9H-purin- 9-yl)propan-2-yl)oxy)methyl)phosphonic acid (1), also referred to herein as TFV, by coupling (1) with an excess of an L-amino ester (4) under 2,2'-dipyridyldisulfide
condensation conditions. Amino esters that are not commercially available, can be prepared by condensation of the corresponding amino acid with an alcohol in the presence of thionyl chloride. L-proline esters or glycine esters can likewise be employed.
Reactions sensitive to moisture or air were performed under nitrogen or argon using anhydrous solvents and reagents. The progress of reactions was determined by either analytical thin layer chromatography (TLC) usually performed with E. Merck pre-coated TLC plates, silica gel 60F-254, layer thickness 0.25 mm or liquid chromatography-mass spectrometry (LC-MS). Typically the analytical LC-MS system used consisted of a Waters ZQ™ platform with electrospray ionization in positive ion detection mode with an Agilent 1100 series HPLC with autosampler. The column was usually a Water Xterra MS CI 8, 3.0 χ 50 mm, 5 /mi. The flow rate was 1 mL/min, and the injection volume was 10 piL. UV detection was in the range 210-400 nm. The mobile phase consisted of solvent A (water plus 0.06% TFA) and solvent B (acetonitrile plus 0.05% TFA) with a gradient of 100% solvent A for 0.7 min changing to 100% solvent B over 3.75 min, maintained for 1.1 min, then reverting to 100% solvent A over 0.2 min.
Preparative HPLC purifications were usually performed using a mass spectrometry directed system. Usually they were performed on a Waters Chromatography
Workstation configured with LC-MS System Consisting of: Waters ZQ single quad MS system with Electrospray Ionization, Waters 2525 Gradient Pump, Waters 2767 Injecto /Collector, Waters 996 PDA Detector, the MS Conditions of: 150-750 amu, Positive
Electrospray, Collection Triggered by MS, and a Waters SUNFIRE® C-18 5 micron, 30 mm (id) x 100 mm column. The mobile phases consisted of mixtures of acetonitrile (10-100%) in water containing 0.1% TFA. Flow rates were maintained at 50 mL/min, the injection volume was 1800 //L, and the UV detection range was 210-400 nm. Mobile phase gradients were optimized for the individual compounds. Reactions performed using microwave irradiation were normally carried out using an Emrys Optimizer manufactured by Personal Chemistry, or an Initiator manufactured by Biotage. Concentration of solutions was carried out on a rotary evaporator under reduced pressure. Flash chromatography was usually performed using a Biotage® Flash Chromatography apparatus (Dyax Corp.) on silica gel (32-63 μΜ, 60 A pore size) in pre-packed cartridges of the size noted. lH NMR spectra were acquired at 500 MHz spectrometers in CDCI3 solutions unless otherwise noted. Chemical shifts were reported in parts per million (ppm). Tetramethylsilane (TMS) was used as internal reference in CD3CI solutions, and residual CH3OH peak or TMS was used as internal reference in CD3OD solutions. Coupling constants (J) were reported in hertz (Hz). Chiral analytical
chromatography was performed on one of CHIRALPAK® AS, CHIRALPAK® AD, CHIRALCEL® OD, CHIRALCEL® IA, or CHIRALCEL® OJ columns (250x4.6 mm) (Daicel Chemical Industries, Ltd.) with noted percentage of either ethanol in hexane
(%Et/Hex) or isopropanol in heptane (%IP A/Hep) as isocratic solvent systems. Chiral preparative chromatography was conducted on one of of CHIRALPAK AS, of CHIRALPAK AD, CHIRALCEL® OD, CHIRALCEL ®IA, CHIRALCEL® OJ columns (20x250 mm) (Daicel Chemical Industries, Ltd.) with desired isocratic solvent systems identified on chiral analytical chromatography or by supercritical fluid (SFC) conditions.
It is understood that a chiral center in a compound may exist in the "S" or "R" stereoconfigurations, or as a mixture of both. The compounds in the examples herein contain a phosphorus chiral center, and additional compounds within the scope of Formula I may also contain a phosphorus chiral center. In the following examples, compounds having a chiral center were separated into stereoisomers, referred to as Isomer A (faster eluting isomer) and Isomer B (slower eluting isomer), based on their observed elution order resulting from the
separation as performed. Elution order of separated isomers may differ if performed under conditions different than those employed herein. Except for the defined chiral centers in the parent stereoisomer mixture, absolute stereochemistry (R or S) of each of the "A" and "B" separated stereoisomers was not determined. Therefore, each of the A isomer and B isomer in each Example (e.g., 1A and IB) is not assigned to a particular chemical name. In examples where absolute stereochemistry of each of the separated isomers was not determined, an asterisk (*) may be used in the associated chemical structure drawing that indicates the location of the unassigned chiral center.
INTERMEDIATE A

TFV
P- { f( li?)-2-(6-amino-9H-purin-9-yl)-l -methylethoxylmethyl}-N-r(l^)-l -methyl-2-( 1 - methylethoxy)-2-oxoethyllphosphonamidic acid: To a solution of (i?)-(((l-(6-amino-9H- purin-9-yl)propan-2-yl)oxy)methyl)phosphonic acid (referred to herein as TFV, 3.0 g, 10.5 mmol) in water (50 mL) were added EDC (10.0 g, 52.2 mmol) and (<S)-isopropyl 2- aminopropanoate hydrochloride (8.8 g, 52.2 mmol) at ambient temperature. The pH value of the resulting solution was adjusted to 7.2-7.6 with TEA (5.3 g, 52.2 mmol). The resulting mixture was stirred at 40 °C for 16 h. The reaction was monitored by LC-MS. Upon reaction completion, the reaction was cooled down to ambient temperature and concentrated under reduced pressure. The residue was purified by flash chromatography (Column: C18, 330 g, 20-35 urn, 100 A; Mobile Phase A: Water plus 5 mM NH4HCO3, Mobile Phase B: ACN;
Gradient: 5-20% B in 25 min; Flow rate: 50 mL/min; Detector 254 nm, retention time: 18 min) to afford the title compound as a colorless solid: LC/MS: [(M+l)]+ = 401.2.
INTERMEDIATE B

TFV
P- ir(li?)-2-(6-arnino-9H-purin-9-yl)-l-methylethoxylmethyl}-N-r(li?)-l-methyl-2-(l- methylethoxy)-2-oxoethyllphosphonamidic acid: INTERMEDIATE B was prepared in a similar fashion to that described for the synthesis of INTERMEDIATE A starting from TFV with (i?)-isopropyl 2-aminopropanoate hydrochloride: LC/MS: [(M+l)]+ = 401.2
INTERMEDIATE C

Step 1
Step 2 HCI/dioxane/rt/2 h
O
NHpHCI
INTERMEDIATE C
(RVcvclopropyl 2-aminopropanoate hydrochloride
Step 1: CR)-cvclopropyl 2-((tert-butoxycarbonyl)amino)propanoate: To a suspension of cyclopropyl boronic acid (6.2 g, 72 mmol) in 10% aqueous NaOH (50 mL) was added a solution of aqueous H2O2 (30%, 20 mL, 200 mmol) dropwise with continuous stirring at 0 °C. After 1 hour at 0 °C, the reaction mixture was quenched with saturated aqueous Na2S203
(30 mL, exothermic) and neutralized with 6 N HCl. The resulting solution was extracted with DCM (3 x 50 mL). The organic layers were combined, dried over anhydrous sodium sulfate and filtered. To the filtration were added (i?)-2-((tert-butoxycarbonyl)amino)propanoic acid (6.6 g, 34.9 mmol), HATU (13.3 g, 34.9 mmol) and Et3N (3.8 g, 34.9 mmol). The resulting mixture was stirred for 16 h at ambient temperature. The reaction was quenched by water (10 mL) and washed with a solution of saturated aqueous solution of NaHC03 (3 x 200 mL).
The organic layer was dried with anhydrous sodium sulfate and filtered. The filtrate was concentrated under reduced pressure and the residue was purified by silica gel column chromatography, eluted with petroleum ether/EtOAc (50: 1) to provide the title compound: LC/MS: [(M+l)]+ = 230.0.
Step 2: (i?)-cvclopropyl 2-aminopropanoate hydrochloride: To a solution of (R)-cyclopropyl 2-((tert-butoxycarbonyl)amino)propanoate (1 g, 4.36 mmol) in 1,4-dioxane (10 mL) was added 12 N HCl (3 mL). The resulting solution was stirred for 3 h at ambient temperature. Upon reaction completion, the resulting solution was concentrated under reduced pressure to give the title compound: LC/MS: [(M+l)]+ = 130.0.
INTERMEDIATE D

INTERMEDIATE D
(i?)-cvclobutyl 2-aminopropanoate hydrochloride: Thionyl chloride (8 g, 67.3 mmol) was added dropwise to a solution of (i?)-2-aminopropanoic acid (2 g, 22.5 mmol) in cyclobutanol (30 mL) at -50 °C. The mixture was allowed to warm to ambient temperature, and then heated at 80 °C for 6 h. The reaction mixture was concentrated under reduced pressure to give a residue, which was triturated with ice-cold diethyl ether to provide the title compound: LC/MS: [(M+l)]+ = 144.0.
INTERMEDIATE E

INTERMEDIATE E
(i<?)-propyl 2-aminopropanoate hydrochloride: INTERMEDIATE E was prepared in a similar fashion to that described for the synthesis of INTERMEDIATE D starting from thionyl chloride (8.0 g, 67.3 mmol) and (i?)-2-aminopropanoic acid (2 g, 22.5 mmol) in propan-l-ol (50 mL): LC/MS: [(M+l)]+ = 132.0.
INTERMEDIATE F

INTERMEDIATE F
(i?)-butyl 2-aminopropanoate hydrochloride: INTERMEDIATE F was prepared in similar fashion to that described for the synthesis of INTERMEDIATE D starting from thionyl chloride (8.0 g, 67.3 mmol) and (i?)-2-aminopropanoic acid (2 g, 22.5 mmol) in butan-l-ol (50 mL): LC/MS: [(M +1)]+ = 146.2.
INTERMEDIATE G

INTERMEDIATE G
(i<?)-pentyl 2-aminopropanoate hydrochloride: INTERMEDIATE G was prepared in a similar fashion to that described for the synthesis of INTERMEDIATE D starting from
thionyl chloride (8.0 g, 67.3 mmol) and (i?)-2-aminopropanoic acid (2 g, 22.5 mmol) in pentan-l-ol (50 mL): LC/MS: [(M+l)]+ = 160.1.
INTERMEDIATE H

INTERMEDIATE H
(R)-hexyl 2-aminopropanoate hvdrochlorideTNTERMEDIATE H was prepared in a similar fashion to that described for the synthesis of INTERMEDIATE D starting from thionyl chloride (8.0 g, 67.3 mmol) and (K)-2-aminopropanoic acid (2 g. 22.5 mmol) in hexan-l-ol (50 mL): LC/MS: [(M+l )]+ = 174.1.

INTERMEDIATE I
(i?)-cvclopentyl 2-aminopropanoate hydrochloride: INTERMEDIATE I was prepared in a similar fashion to that described for the synthesis of INTERMEDIATE D starting from thionyl chloride (8.0 g, 67.3 mmol) and (i?)-2-aminopropanoic acid (2 g, 22.5 mmol) in cyclopentanol (50 mL): LC/MS: [(M+l)]+ = 157.9.

INTERMEDIATE J
(i<?)-isopropyl 2-amino-3-phenylpropanoate hydrochloride: INTERMEDIATE J was prepared in a similar fashion to that described for the synthesis of INTERMEDIATE D starting from thionyl chloride (4.3 g, 36.3 mmol) and (i?)-2-amino-3-phenylpropanoic acid (2 g, 12.11 mmol) in 2-propanol (50 mL): LC/MS: [(M+l)]+ = 208.0.
INTERMEDIATE K

Step 1 Step 2 INTERMEDIATE K
(i?)-allyl 2-aminopropanoate hydrochloride
Step 1: (RVallyl 2-(tert-butoxycarbonylamino)propanoate: To a solution of (i?)-2-((tert- butoxycarbonyl)amino)propanoic acid (1.89 g, 9.99 mmol) and DBU (1.82 g, 11.97 mmol) in acetonitrile (70 mL) was added 3-bromoprop-l-ene (1.45 g, 11.99 mmol) at 0 °C. After stirring for 16 h at ambient temperature, the resulting mixture was concentrated under reduced pressure and the residue was purified by silica gel column chromatography, eluted with petroleum ether/EtOAc (8 : 1) to provide the title compound: LC/MS: [(M+l)]+ = 230.1.
Step 2: (i<?)-cvclopropyl 2-aminopropanoate hydrochloride: To a solution of (R)-allyl 2-((tert- butoxycarbonyl)amino)propanoate (0.75 g, 3.27 mmol) in 1,4-dioxane (10 mL) was added 12 NHC1 (3 mL). The resulting solution was stirred for 3 h at ambient temperature. Upon reaction completion, the resulting solution was concentrated under reduced pressure to give the title compound: LC/MS: [(M+l)]+ = 129.9.
INTERMEDIATE L
INTERMEDIATE L
(i<?)-heptyl 2-aminopropanoate: To a suspension of heptan-l-ol (1 g, 8.61 mmol) and (i?)-2-aminopropanoic acid (1.15 g, 12.91 mmol) in benzene (15 mL) was add 4- methylbenzenesulfonic acid (2.52 g, 14.63 mmol). The resulting mixture was refluxed for 16 h under nitrogen atmosphere. Upon reaction completion, the resulting mixture was cooled down to ambient temperature and quenched by the addition of saturated aqueous solution of NaHC03 (50 mL), extracted with ethyl acetate (3 x 100 mL). The combined organic layers was dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated under reduced pressure to afford the title compound: LC/MS: [(M+l)]+ = 188.1.
INTERMEDIATE M

INTERMEDIATE M
(i?)-nonyl 2-aminopropanoate: INTERMEDIATE M was prepared in a similar fashion to that described for the synthesis of INTERMEDIATE L starting from (R)-2- aminopropanoic acid (0.93 g, 10.4 mmol), nonan-l-ol (1 g, 6.93 mmol) and 4- methylbenzenesulfonic acid (2.01 g, 11.78 mmol) in benzen (15 mL): LC/MS: [(M+l)]+ = 216.1.

INTERMEDIATE N
(7<?)-decyl 2-aminopropanoate: INTERMEDIATE N was prepared in a similar fashion to that described for the synthesis of INTERMEDIATE L starting from (R)-2- aminopropanoic acid (0.84 g, 9.48 mmol), decan- l -ol (1.0 g, 6.32 mmol) and 4- methylbenzenesulfonic acid (1.85 g, 10.74 mmol) in benzen (15 mL): LC/MS : [(M+l)]+ = 230.1.

INTERMEDIATE O
CSyisobutyl 2-amino-3-phenylpropanoate hydrochloride: INTERMEDIATE O was prepared in a similar fashion to that described for the synthesis of INTERMEDIATE D starting from thionyl chloride (4.3 g, 36.3 mmol) and (<S)-2-amino-3-phenylpropanoic acid (2 g, 12.1 1 mmol) in 2-methylpropan-l-ol (50 mL): LC/MS: [(M+l)]+ = 222. 1.
INTERMEDIATE P
(£)-isopropyl pyrrolidine-2-carboxylate hydrochloride: INTERMEDIATE P was prepared in a similar fashion to that described for the synthesis of INTERMEDIATE D
starting from thionyl chloride (6.2 g, 52.2 mmol) and (<S)-pyrrolidine-2-carboxylic acid (2 g, 17.4 mmol) in propan-2-ol (70 mL): LC/MS : [(M+l)]+ = 158.1.

INTERMEDIATE A 1A and 1B
(RVbutyl 2-(((S)-((((R)- 1 -(6-amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)(((^)- 1 - isopropoxy-1 -oxopropan-2-yl)amino)phosphoryl)arnino)propanoate and
(RVbutyl 2-(((i?)-((((i?)-l-(6-arnino-9H-purin-9-yl)propan-2-yl)oxy)methyl)(((^)-l- isopropoxy-1 -oxopropan-2-yl)amino)phosphoryl)arnino)propanoate : To a mixture of P- ((((i?)- 1 -(6-amino-9H-purin-9-y l)propan-2-y l)oxy )methy 1)-N-((S)- 1 -isopropoxy-1 - oxopropan-2-yl)phosphonamidic acid (INTERMEDIATE A, 0.3 g, 0.75 mmol), (R)-butyl 2-aminopropanoate hydrochloride (INTERMEDIATE F, 0.27 g, 1.50 mmol) and triethylamine (0.38 g, 3.75 mmol) in pyridine (5 mL) were added triphenylphosphine (0.59 g, 2.25 mmol) and 2,2'-dipyridyldisulfide (Aldrithiol-2, 0.49 g, 2.25 mmol) at ambient temperature. The resulting mixture was heated at 60 °C for 3 h under nitrogen atmosphere. After cooling down to ambient temperature, the resulting mixture was concentrated under reduced pressure and the residue was purified by silica gel column chromatography, eluted with CH2Cl2/MeOH (50/1-10/1) to afford the mixture of the two diastereoisomers. Then the two title compounds were separated by Prep-HPLC (Column: X Bridge C I 8, 19 x 150 mm, 5 urn; Mobile Phase: Water (contains 10 mmol NH4HCO3) and CH3CN, (25-41 % in 8.5 min);
Flow rate: 20 mL/min; Detector: 254 nm; to afford Isomer 1A (faster eluting, RT=6.8 min) as a colorless solid: lH NMR (400 MHz; CD3OD) δ 8.24 (s, 1H), 8.23 (s, 1H), 5.00 (q, J = 6.4 Hz, 1H), 4.40 (dd, J = 2.8; 14.4 Hz, 1H), 4.26 (dd, J = 6.0; 14.4 Hz, 1H), 4.18-4.09 (m, 2H),
3.98-3.92 (m, 3H), 3.81 (dd, J = 8.8; 13.2 Hz, 1H), 3.57 (dd, J = 10.0; 13.2 Hz, 1H), 1.64 (q, J= 6.8 Hz, 2H), 1.42 (q, J = 7.2 Hz, 2H), 1.34 (d, J = 7.2 Hz, 3H), 1.29 (d, J= 7.2 Hz, 3H), 1.27 (d, J= 6.4 Hz, 6H), 1.21 (d, J= 6.4 Hz, 3H), 0.96 (t, J = 7.2 Hz, 3H); 31p NMR (162 MHz; CD3OD) δ 23.58 (decoupled); LC/MS: [(M+l)]+ = 528.2; and Isomer IB (slower eluting, RT=7.3 min) as a colorless solid: lH NMR (400 MHz; CD3OD) δ 8.24 (s, 1H), 8.23 (s, 1H), 4.93 (q, J= 6.4 Hz, 1H), 4.43 (dd, J = 2.8; 14.4 Hz, 1H), 4.25 (dd, J = 7.2; 14.8 Hz, 1H), 4.18-4.10 (m, 2H), 4.00-3.93 (m, 1H), 3.92-3.81 (m, 3H), 3.62 (dd, J= 10.0; 13.2 Hz, 1H), 1.63 (q, J = 6.8 Hz, 2H), 1.42 (q, J = 7.6 Hz, 2H), 1.37 (d, J= 7.2 Hz, 3H), 1.31 (d, J = 7.2 Hz, 3H), 1.24-1.22 (m, 9H), 0.96 (t, J= 7.6 Hz, 3H); 31p NMR (162 MHz; CD3OD) δ 24.02 (decoupled); LC/MS: [(M+l)]+ = 528.2.

1-methylethyl N-r(i?)-ir(li?)-2-(6-amino-9H-purin-9-yl)-l-methylethoxyl methyl } (Γ(1^)-1- methyl-2-(l-methylethoxy)-2-oxoethyll amino Iphosphoryll-D-phenylalaninate and
1 -methylethyl N-fftS1)- ί Γ( li?)-2-(6-amino-9H-purin-9-yl)- 1 -methylethoxylmethyl} { \(\S)- 1 - methyl-2-(l-methylethoxy)-2-oxoethyll amino Iphosphoryll-D-phenylalaninate :
The two title compounds were prepared in the same way as described for example 1A and IB (from INTERMEDIATE A and INTERMEDIATE J) except the purification was by chiral- HPLC with the following conditions: Column: Chiralpak IC 2 x 25 cm, 5um; Mobile Phase: Hexane/EtOH (0-20% EtOH in 30 min); Flow rate: 20 mL/min; Detector: 254 nm; to afford Isomer 2A (faster eluting, RT=15.5 min) as a colorless solid: lH NMR (300 MHz; CD3OD) δ 8.20 (s, 1H), 8.19 (s, 1H), 7.14-7.12 (m, 3H), 7.03-6.98 (m, 2H), 5.05-4.93 (m, 2H), 4.30 (dd, J= 2.7; 10.8 Hz, 1H), 4.13 (d, J = 7.2 Hz, 1H), 4.10-4.06 (m, 1H), 3.91-3.86 (m, 1H), 3.65-3.62 (m, 1H), 3.42 (dd, J = 8.1; 12.9 Hz, 1H), 2.96-2.89 (m, 2H), 2.67 (dd, J= 8.4; 13.5
Hz, 1H), 1.30 (d, J= 6.9 Hz, 3H), 1.24-1.19 (m, 9H), 1.13 (d, J = 6.3 Hz, 3H), 1.08 (d, J = 6.3 Hz, 3H); 31p NMR (121 MHz; CD3OD) δ 23.52 (decoupled); LC/MS: [(M+l)]+ = 590.4; and
Isomer 2B (slower eluting, RT=24.5 min) as a colorless solid: lH NMR (300 MHz; CD3OD) δ 8.17 (s, 1H), 8.15 (s, 1H), 7.27-7.17 (m, 5H), 4.94-4.81 (m, 2H), 4.35 (dd, J = 3.0; 14.4 Hz, 1H), 4.18 (dd, J= 6.9; 14.7 Hz, 1H), 4.03-3.97 (m, 1H), 3.92-3.85 (m, 1H), 3.70 (dd, J= 8.4; 13.2 Hz, 1H), 3.53-3.45 (m, 2H), 3.04-3.00 (m, 1H), 2.83 (dd, J= 8.1 ; 16.5 Hz, 1H), 1.20- 1.11 (m, 18H); 31p NMR (121 MHz; CD3OD) δ 23.73 (decoupled); LC/MS: [(M+l)]+ =
590.4.
The compounds in Table 2 were prepared in an analogous fashion to that described for Example 1 A and IB. The column having the heading INT provides the intermediate example compounds used to make each exemplified compound. The
diastereoisomers were separated by one of the four listed methods: reverse phase HPLC, chiral HPLC, SFC, or prep TLC. Absolute sterochemistry of the "A" isomer (faster eluting) and "B" isomer (slower eluting ) of each example was not determined.
TABLE 2




9A 9A: 540.6
1 -methy lethy 1 l-[(R)-{ [(lR)-2-(6-ammo-9H-purin-9-y 1)- and B+P and
1 -methy lethoxy ] methy 1 } { [( \R)- 1 -methy l-2-( 1 - 9B 9B: 540.6 methylethoxy)-2-oxoethyl] amino} phosphoryl] -L- prolinate; and
1 -methy lethy 1 1 - [(S)- { [(lR)-2-(6-ammo-9H-purin-9-y 1)- 1 -methy lethoxy ] methy 1 } { [( \R)- 1 -methy l-2-( 1 - methylethoxy)-2-oxoethyl] amino} phosphoryl] -L- prolinate
TABLE 2A

Column: Chiralpak IA 20 * 250 mm, 5
A 23.38 (162 MHz; CD3OD) 13.43 um; Mobile Phase A: Hexane (plus
0.2% DEA, v/v); Mobile Phase B:
EtOH; Gradient: 30% B in 33 min;
B 23.71 (162 MHz; CD3OD) Flow rate: 20 mL/min; Detector: UV 27.21
254 nm.
Less polarA 24.00 (121 MHz; CD3OD) diastereomer
Preparative TLC with CH2Cl2/MeOH (Rf = 0.31)
(10/1, v/v) more polarB 23.58 (121 MHz; CD3OD) diastereomer
(Rf = 0.27)
Column: XBridge C18, 19 * 150 mm,
A 24.00 (162 MHz; CD3OD) 5 um; Mobile phase A: water (plus 10 7.51 mM NH4HCO3); Mobile phase B:
CH3CN; Gradient: 15%~75% B in 10
B 23.56 (162 MHz; CD3OD) min); Flow rate: 20 mL/min; Detector: 8.50
UV 254 nm.
Column: Sunfire C18, 19 * 150 mm, 5
A 23.21 (162 MHz; CD3OD) um; Mobile Phase A: Water (plus 10 8.30 mM NH4HCO3); Mobile Phase B:
CH3CN; Flow rate: 20 mL/min;
B 23.27 (162 MHz; CD3OD) Gradient: 28% B to 28% B in 13 min; 9.50
Detector: UV 254 nm.
EXAMPLE 10
Assessing antiviral potency in a Multiple Round HIV-1 Infection Assay (Viking Assay)
The antiviral activity of the tenofovir prodrugs of the Examples herein was assessed in an assay that measures the rate of replication of HIV in cell culture, termed the Viking assay (Viral KINetics in Green cells) and performed as follows. HIV-1 replication was monitored using MT4-gag-GFP clone D3 (hereafter designated MT4-GFP), which are MT-4 cells modified to harbor a GFP reporter gene, the expression of which is dependent on the HIV-1 expressed proteins tat and rev. Productive infection of an MT4-GFP cell with HIV-1 results in GFP expression approximately 24 h post-infection. MT4-GFP cells were maintained at 37°C/5% CO2/90% relative humidity in RPMI 1640 supplemented with 10% fetal bovine serum, 100 U/ml penicillin/streptomycin, and 400μg/ml G418 to maintain the reporter gene. For infections, MT4-GFP cells were placed in the same medium lacking G418 and infected overnight with HIV-1 (H9/IIIB strain) virus at an approximate multiplicity of infection of 0.01 in the same incubation conditions. Cells were then washed and re- suspended in either RPMI 1640 supplemented with 10% or 50% normal human serum (NHS) at 1.6 x 105 cells/mL (10% NHS or 50% NHS, respectively). Compound plates were prepared by dispensing compounds dissolved in DMSO into wells of 384 well poly-D-lysine- coated plates (0.2 μΐ/well) using an ECHO acoustic dispenser. Each compound was tested in a 10-point serial 3-fold dilution (typical final concentrations: 8.4 μΜ - 0.42 nM). Controls included no inhibitor (DMSO only) and a combination of three antiviral agents (efavirenz, indinavir, an in-house integrase strand transfer inhibitor at final concentrations of 4 μΜ each). Cells were added (50μΕΛνβ11) to compound plates and the infected cells were maintained at 37°C/5% CO2/90% relative humidity.
Infected cells were quantified at two time points, ~48h and- 72h postinfection, by counting the number of green cells in each well using an Acumen eX3 scanner. The increase in the number of green cells over ~24h period gives the reproductive ratio, R0, which is typically 5-15 and has been shown experimentally to be in logarithmic phase (data not shown). Inhibition of R0 is calculated for each well, and IC50S determined by non-linear 4-parameter curve fitting. Assay IC50 results are shown in Table 3.
EXAMPLE 11
Prodrug stability assay in bio-relevant media
The following assay was employed to evaluate the stability of the prodrugs in simulated gastrointestinal tract conditions. Preparation of fasted state simulated intestinal fluid (FaSSIF) using Phares SIF Powder was carried out according to protocols from Phare Drug Delivery AG (Baselland, Switzerland). For sample preparation, \0 μΐ. stock solutions (10 mM) of prodrug substance in DMSO was added to 990 of 0.5 mg/mL Pancreatin solution (Fisher CAS#8049-47-6) in FaSSIF. Two samples were prepared for each compound at initial. If the sample was clear solution at the beginning, ran one sample directly as initial by HPLC; if the sample was not clear at starting, diluted the sample by 100% ACN. Put the other sample under 37°C and observed the sample at 5h time-point. At 5 h time point, if the sample was clear solution then performed HPLC analysis directly; if it was not clear solution, diluted the sample by 100% ACN and assayed by HPLC. All the samples were vortexed for 3 min and observed before injection. For the diluted samples, the area will be multiplied by a factor when data analysis. The analysis was carried out with an Agilent 1100 series HPLC with autosampler. The column was usually a Poroshell 120 EC-C18, 4.6 χ 50mm, 2.7 μηι.
The flow rate was 1.8 mL/min, and the injection volume was 5 or 10 μί. UV detection was in the range 210-400 nm. The mobile phase consisted of solvent A (water plus 10 mM tetrabutylammonium bromide) and solvent B (acetonitrile) with a gradient of 90% solvent A at 0 min changing to 95% solvent B over 6 min, maintained for 1.5 min, then reverting to 90% solvent A over 1.6 min. The area of the parent in prodrug at 5h time point was divided by the area of the parent in prodrug at 0 h time point, to generate the % claimed parent ratio, which are summarized in Table 3 for GI Tract stability.
EXAMPLE 12
Pharmacokinetic Studies in Dogs - In Vivo Dog PK
Prodrugs were administered to beagle dogs through intravenous (IV) and oral (P.O.) administrations in a non-crossover manner. The IV dose was prepared in 20% hydroxypropyl β-cyclodextrin (HPBCD) and was administered via cephalic or saphenous vein. The P.O. dose was prepared in 10% polysorbate 80 (Tween 80) and was administered via gavage.
Blood samples were serially collected following dose administration for up to 48 hr and plasma was separated by centrifugation. The concentrations of prodrugs in dog plasma were determined by a LC-MS/MS assay following a protein precipitation step and addition of an appropriate internal standard (labetalol, imipramine or diclofenac). Quantification was done by determining peak area-ratios of the prodrugs and tenofovir to the internal standard. Additional blood sample(s) was collected following dose administration for up to 24 hr.
Peripheral blood mononuclear cells (PBMCs) were isolated by centrifugation, using tubes and reagents specified for such application. The concentrations of tenofovir and/or its phosphate conjugate(s) in PBMCs were determined by an LC-MS/MS assay following a protein precipitation step and addition of an appropriate internal standard (labetalol, imipramine or diclofenac). Quantification was done by determining peak area-ratios of tenofovir and/or its phosphate conjugate(s) to the internal standard.
Pharmacokinetic parameters were obtained using non-compartmental methods (Watson®). The area under the plasma concentration-time curve (AUCo-t) was calculated from the first time point (0 min) up to the last time point with measurable drug concentration using the linear trapezoidal or linear/log-linear trapezoidal rule. The IV plasma clearance was calculated by dividing the dose by AUCo-inf. The terminal half-life of elimination was determined by unweighted linear regression analysis of the log-transformed data. The time points for determination of half-life were selected by visual inspection of the data. The volume of distribution at steady state (Vdss) was obtained from the product of plasma clearance and mean residence time (determined by dividing the area under the first moment curve by the area under the curve). The maximum plasma concentration (Cmax) and the time at which maximum concentration occurred (Tmax) were obtained by inspection of the plasma concentration-time data. Absolute oral bioavailability (%F) was determined from dose- adjusted IV and P.O. AUC ratios of the prodrug. Table 3 shows in vivo dog PK data in the form of TFV-DP concentrations (μΜ) in dog PBMCs at 24 h following a 10 mg/kg P.O. dose of the indicated prodrug.
TABLE 3

Employing the assays described in Examples 11 and 12, GI Tract stability data and in vivo dog PK for compounds comprised of L,L amino acid residues are shown in Table

Claims
WHAT IS CLAIMED IS:
A compound of structural Formula I

I
or pharmaceutically acceptable salt thereof, wherein:
RA is a D-amino acid ester residue of formula (i) or a D-proline ester residue of formula (ii)
 Rl is (a) -Ci -4alkyl, (b) -Ci -4alkyl substituted with-OH, -SH, -SCH3, -N¾ or -NH- C(=NH)-NH2,
Rl is (a) -Ci -4alkyl, (b) -Ci -4alkyl substituted with-OH, -SH, -SCH3, -N¾ or -NH- C(=NH)-NH2,
(c) -CH2-phenyl, (d) -CH2-phenol, (e) -(CH2)l-2-COOH, (f) -(CH2)l-2-CONH2, (g) - CH2-lH-indole, (h) -CH2-imidazole, (i) aryl or (j) heteroaryl;
R2 is
(a) -Ci-ioalkyl unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR^a^ -0-Ci-3alkyl, -SH, - NR6R 5 -C3-6cycloalkyl or spiro-C3-6cycloalkyl,
(b) -CH2-phenyl unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5a -0-Ci-3alkyl, -SH, -NR6R7 0r -Ci- 3alkyl,
(c) -C3-8cycloalkyl unsubstituted or substituted with one to three substituents
independently selected from fluoro, chloro, bromo, -OR^a^ -0-Ci-3alkyl, -SH, - NR6R7 or -C 1-3 alky 1,
(d) aryl unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5a -0-Ci-3alkyl, -SH, -NR6R7 0r -Ci-3alkyl,
(e) 2-10 atom heteroalkyl,
(f) heteroaryl unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5a -0-Ci-3alkyl, -SH, -NR6R7 or -Ci- 3alkyl, or
(g) a heterocyclic ring unsubstituted or substituted with one to three substituents
independently selected from fluoro, chloro, bromo, -OR^ -0-Ci-3alkyl, -SH, - NR6R7 or -Ci-3alkyl;
RB is an L-amino acid ester residue of formula (iii) or an L-proline ester residue of formula


R3 is (a) -Ci -4alkyl, (b) -Ci -4alkyl substituted with-OH, -SH, -SCH3, -N¾ or -NH- C(=NH)-NH2,
(c) -CH2-phenyl, (d) -CH2-phenol, (e) -(CH2)l-2-COOH, (f) -(CH2)l-2-CONH2, (g) - CH2-lH-indole, (h) -CH2-imidazole, (i) aryl or (j) heteroaryl;
R4 is
(a) -Ci-ioalkyl unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5b5 -0-Ci-3alkyl, -SH, - NR8R95 -C3-6cycloalkyl or spiro-C3-6cycloalkyl,
(b) -CH2-phenyl unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5b -O-C 1-3 alky 1, -SH, - NR8R9 0r -C\- 3alkyl,
(c) -C3-8cycloalkyl unsubstituted or substituted with one to three substituents
independently selected from fluoro, chloro, bromo, -OR5b5 -O-C i -3 alky 1, -SH, -
NR8R9 or -C 1-3 alky 1,
(d) aryl unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5b -0-Ci-3alkyl, -SH, - NR8R9 0r -Ci-3alkyl,
(e) 2-10 atom heteroalkyl,
(f) heteroaryl unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5b -0-Ci-3alkyl, -SH, - NR8R9 or -Ci- 3alkyl, or
(g) a heterocyclic ring unsubstituted or substituted with one to three substituents
independently selected from fluoro, chloro, bromo, -OR5b5 -SH, -NR8R9 or -Ci- 3alkyl;
R5a and R^b are each independently selected from -H or -C3-6cycloalkyl;.
R6 and R7 are each independently selected from -H, -Ci-3alkyl or -C3-6cycloalkyl; and
R8 and R9 are each independently selected from -H, -Ci-3alkyl or -C3-6cycloalkyl.
2. The compound of claim 1 or a pharmaceutically acceptable salt thereof wherein:
Rl is -Ci-4alkyl, -CH2OH, -CH(OH)CH3, -CH2SH, -(CH2)2SCH3, -(CH2)4NH2, -(CH2)3NH-C(=NH)-NH2, -CH2-phenyl, -CH2-phenol, -(CH2)l-2-COOH, -(CH2)l-2- CONH2,
-CH2-lH-indole, -CH2-imidazole, phenyl, naphthyl or pyridyl.
3. The compound of claim 1 or a pharmaceutically acceptable salt thereof wherein
Rl is -CH3, -CH(CH3)2, -CH(CH3)CH2CH3, -CH2CH(CH3)2 or -CH2-phenyl.
4. The compound of claim 1 or a pharmaceutically acceptable salt thereof wherein
R3 is -Ci -4alkyl, -CH2OH, -CH(OH)CH3, -CH2SH, -(CH2)2SCH3, -(CH2)4NH2, -(CH2)3NH-C(=NH)-NH2, -CH2-phenyl, -CH2-phenol, -(CH2)l-2-COOH, -(CH2)l-2- CONH2,
-CH2-lH-indole, -CH2-imidazole, phenyl, naphthyl or pyridyl.
5. The compound of claim 1 or a pharmaceutically acceptable salt thereof wherein
R3 is -CH3, -CH(CH3)2, -CH(CH3)CH2CH3, -CH2CH(CH3)2 or -CH2-phenyl.
6. The compound of claim 1 or a pharmaceutically acceptable salt thereof wherein
Rl is -Ci -4alkyl, -CH2OH, -CH(OH)CH3, -CH2SH, -(CH2)2SCH3, -(CH2)4NH2, -(CH2)3NH-C(=NH)-NH2, -CH2-phenyl, -CH2-phenol, -(CH2)l-2-COOH, -(CH2)l-2- CONH2,
-CH2-lH-indole, -CH2-imidazole, phenyl, naphthyl or pyridyl; and
R3 is -Cl -4alkyl, -CH2OH, -CH(OH)CH3, -CH2SH, -(CH2)2SCH3, -(CH2)4NH2,
-(CH2)3NH-C(=NH)-NH2, -CH2-phenyl, -CH2-phenol, -(CH2)l-2-COOH, -(CH2)l-2-
CONH2,
-CH2-lH-indole, -CH2-imidazole, phenyl, naphthyl or pyridyl.
7. The compound of claim 1 or a pharmaceutically acceptable salt thereof wherein Rl is -CH3, -CH(CH3)2, -CH(CH3)CH2CH3, -CH2CH(CH3)2 or -CH2-phenyl; and
R3 is -CH3, -CH(CH3)2, -CH(CH3)CH2CH3, -CH2CH(CH3)2 or -CH2-phenyl.
8. The compound of claim 1 or a pharmaceutically acceptable salt thereof wherein
R2 1S
(a) -Ci -8alkyl, -CH2CH2OH -CH2CH2CH2OH -CH2CH2SH -CH2CH2CH2SH
-CH2CH2NH2, -CH2CH2CH2NH2,
(b) -CH2-phenyl, unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5a -0-Ci-3alkyl, -SH, -NR6R7 0r -Ci-3alkyl,
(c) -C3-6cycloalkyl, unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5^ -0-Ci-3alkyl, -SH, -NR6R7 0r -Ci-3alkyl, (d) phenyl or naphthyl, each unsubstituted or substituted with one to three substituents
independently selected from fluoro, chloro, bromo, -OR^ -0-Ci-3alkyl, -SH, -NR6R7 or -C 1-3 alky 1,
(e) -CH2CH20CH3, -CH2CH2CH20CH3, -CH2CH2SCH3, -CH2CH2CH2SCH3,
-CH2CH2NHCH3, -CH2CH2CH2NHCH3,
(f) pyridyl, unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5a -0-Ci-3alkyl, -SH, -NR6R7 0r -Ci-3alkyl, or
(g) piperidinyl, pyrrolidinyl, tetrahydrofuranyl, or tetrahydropyranyl, each unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo,
-OR5a -0-Ci-3alkyl, -SH, -NR6R7 0r -Ci-3alk l.
9. The compound of claim 1 or a pharmaceutically acceptable salt thereof wherein
R2 is -Ci-8alkyl, -CH2-phenyl, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl or phenyl.
10. The compound of any one of claims 1 to 9 or a pharmaceutically acceptable salt thereof wherein
R is:
(a) -Ci-8alkyl, -CH2CH2OH -CH2CH2CH2OH -CH2CH2SH -CH2CH2CH2SH
-CH2CH2NH2, -CH2CH2CH2NH2,
(b) -CH2-phenyl, unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5b -0-Ci-3alkyl, -SH, -NR8R9 0r -Ci-3alkyl,
(c) -C3-6cycloalkyl, unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5b5 -0-Ci-3alkyl, -SH, -NR8R9 0r -Ci-3alkyl,
(d) phenyl or naphthyl, each unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5b5 -0-Ci-3alkyl, -SH, -NR8R9 or -C 1-3 alky 1,
(e) -CH2CH20CH3, -CH2CH2CH20CH3, -CH2CH2SCH3, -CH2CH2CH2SCH3,
-CH2CH2NHCH3, -CH2CH2CH2NHCH3,
(f) pyridyl, unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5b -0-Ci-3alkyl, -SH, -NR8R9 0r -Ci-3alkyl, or
(g) piperidinyl, pyrrolidinyl, tetrahydrofuranyl, or tetrahydropyranyl, each unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo,
-OR5b -0-Cl -3alkyl, -SH, -NR8R9 0r -Ci-3alkyl.
11. The compound of any one of claims 1 to 9 or a pharmaceutically acceptable salt thereof wherein
R4 is -Ci-8alkyl, -CH2-phenyl, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl or phenyl.
12. The compound of claim 1 or a pharmaceutically acceptable salt thereof wherein:
RA is a D-amino acid ester residue of formula (i) or a D-proline ester residue of formula (ii);
Rl is -Ci-4alkyl, -CH2OH, -CH(OH)CH3, -CH2SH, -(CH2)2SCH3, -(CH2)4NH2, - (CH2)3NH-C(=NH)-NH2, -CH2-phenyl, -CH2-phenol, -(CH2)l-2-COOH, -(CH2)l-2- CONH2, -CH2-lH-indole, -CH2-imidazole, phenyl, naphthyl or pyridyl;
R2 is:
(a) -Ci -8alkyl, -CH2CH2OH -CH2CH2CH2OH -CH2CH2SH -CH2CH2CH2SH
-CH2CH2NH2, -CH2CH2CH2NH2,
(b) -CH2-phenyl, unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5a -0-Ci-3alkyl, -SH, -NR6R7 0r -Ci-3alkyl,
(c) -C3-6cycloalkyl, unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5^ -0-Ci-3alkyl, -SH, -NR6R7 0r -Ci-3alkyl,
(d) phenyl or naphthyl, each unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5a5 -0-Ci-3alkyl, -SH, -NR6R7 or -C 1-3 alky 1,
(e) -CH2CH20CH3, -CH2CH2CH20CH3, -CH2CH2SCH3, -CH2CH2CH2SCH3,
-CH2CH2NHCH3, -CH2CH2CH2NHCH3,
(f) pyridyl, unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5a -0-Ci-3alkyl, -SH, -NR6R7 0r -Ci-3alkyl, or
(g) piperidinyl, pyrrolidinyl, tetrahydrofuranyl, or tetrahydropyranyl, each unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5a -0-Ci-3alkyl, -SH, -NR6R7 0r -Ci-3alkyl;
RB is an L-amino acid ester residue of formula (iii), an L-proline ester residue of formula (iv) or a glycine ester residue of formula (v);
R3 is -Ci-4alkyl, -CH2OH, -CH(OH)CH3, -CH2SH, -(CH2)2SCH3, -(CH2)4NH2, -
(CH2)3NH-C(=NH)-NH2, -CH2-phenyl, -CH2-phenol, -(CH2)l-2-COOH, -(0¾)ΐ-2- CONH2, -CH2-lH-indole, -CH2-imidazole, phenyl, naphthyl or pyridyl;
R4 is
(a) -Ci-8alkyl, -CH2CH2OH -CH2CH2CH2OH -CH2CH2SH -CH2CH2CH2SH
-CH2CH2NH2, -CH2CH2CH2NH2,
(b) -CH2-phenyl, unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5b -0-Ci-3alkyl, -SH, -NR8R9 0r -Ci-3alkyl,
(c) -C3-6cycloalkyl, unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5b, -0-Ci-3alkyl, -SH, -NR8R9 0r -Ci-3alkyl,
(d) phenyl or naphthyl, each unsubstituted or substituted with one to three substituents
independently selected from fluoro, chloro, bromo, -OR5b5 -0-Ci-3alkyl, -SH, -NR8R9 or -C 1-3 alky 1,
(e) -CH2CH20CH3, -CH2CH2CH20CH3, -CH2CH2SCH3, -CH2CH2CH2SCH3,
-CH2CH2NHCH3, -CH2CH2CH2NHCH3,
(f) pyridyl, unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro, bromo, -OR5b -0-Ci-3alkyl, -SH, -NR8R9 0r -Ci-3alkyl, or (g) piperidinyl, pyrrolidinyl, tetrahydrofuranyl, or tetrahydropyranyl, each unsubstituted or substituted with one to three substituents independently selected from fluoro, chloro,
bromo,
-OR5b -0-Ci -3alkyl, -SH, -NR8R9-N or -Ci-3alkyl;
R5a and R5b are each -H; and
R6, R7, R8 and R9 are each independently selected from -H or Ci-3alkyl.
13. The compound of claim 1 or a pharmaceutically acceptable salt thereof wherein:
RA is a D-amino acid ester residue of formula (i) or a D-proline ester residue of formula (ii); Rl is -CH3, -CH(CH3)2, -CH(CH3)CH2CH3, -CH2CH(CH3)2.or -CH2-phenyl;
R2 is -Ci-8alkyl, -CH2-phenyl, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl or phenyl; RB is an L-amino acid ester residue of formula (iii), an L-proline ester residue of formula
(iv), or a glycine ester residue of formula (v);
R3 is -CH3, -CH(CH3)2, -CH(CH3)CH2CH3, -CH2CH(CH3)2 or -CH2-phenyl;
R4 is -Ci-8alkyl, -CH2-phenyl, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl or phenyl; R5a and R5b are each -H; and
R6, R7, R8 and R9 are each independently selected from -H or Ci-3alkyl.
14. The compound of claim 1 that is:
(R)-butyl 2-(((5)-((((R)-l -(6-amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)(((S)-l - isopropoxy-l-oxopropan-2-yl)amino)phosphoryl)arnino)propanoate ;
(R)-buty 1 2-(((R)-((((R)- 1 -(6-amino-9H-purin-9-y l)propan-2-y l)oxy )methy 1)(((S) - 1 - isopropoxy-l-oxopropan-2-yl)amino)phosphoryl)arnino)propanoate;
1 -methylethyl N-[(R)- { [(lR)-2-(6-amino-9H-purin-9-yl)-l -methylethoxy] methyl} { [(\S)-\- methy l-2-( 1 -methylethoxy )-2-oxoethy 1] amino} phosphory 1] -D-pheny lalaninate ;
1 -methylethyl N-[(S)- { [(li?)-2-(6-amino-9H-purin-9-yl)-l -methylethoxy] methyl} { [(1S)-1- methy l-2-( 1 -methylethoxy )-2-oxoethy 1] amino} phosphory 1] -D-pheny lalaninate;
cyclobutyl (2R,4R,6S)-4- { [(li?)-2-(6-amino-9H-purin-9-yl)-l -methylethoxy] methyl} -2,6,9- trimethyl-7-oxo-8-oxa-3,5-diaza-4-phosphadecan-l-oate 4-oxide;
cyclobutyl (2i?,45',65)-4-{[(li?)-2-(6-amino-9H-purin-9-yl)-l-methylethoxy]methyl}-2,6,9- trimethyl-7-oxo-8-oxa-3,5-diaza-4-phosphadecan-l-oate 4-oxide;
1 -methylethyl (25',45',6i?)-4-{[(li?)-2-(6-amino-9H-purin-9-yl)-l-methylethoxy]methyl}-
2,6-dimethyl-7-oxo-8-oxa-3,5-diaza-4-phosphaundecan-l-oate 4-oxide;
1 -methy lethy 1 (2S,4R,6R)-4- { [( li?)-2-(6-amino-9H-purin-9-y 1)- 1 -methy lethoxy ]methy 1 } -
2,6-dimethyl-7-oxo-8-oxa-3,5-diaza-4-phosphaundecan-l-oate 4-oxide;
1 -methy lethy 1 (2R,4R,6S)-4- { [( li?)-2-(6-amino-9H-purin-9-y 1)- 1 -methy lethoxy ]methy 1 } -
2,6,9-trimethyl-7-oxo-8-oxa-3,5-diaza-4-phosphadecan-l-oate 4-oxide;
1- methylethyl (2i?,45',65)-4-{[(li?)-2-(6-amino-9H-purin-9-yl)-l-methylethoxy]methyl}- 2,6,9-trimethyl-7-oxo-8-oxa-3,5-diaza-4-phosphadecan-l-oate 4-oxide;
2- methylpropyl N-[(i?)-{[(li?)-2-(6-amino-9H-purin-9-yl)-l -methy lethoxy] methyl} {[(IR)- 1 -methy l-2-( 1 -methy lethoxy )-2-oxoethy 1] amino} phosphory 1] -L-pheny lalaninate;
2-methylpropyl N-[(5)-{[(li?)-2-(6-amino-9H-purin-9-yl)-l-methylethoxy]methyl} {[(IR)-
1 -methy l-2-(l -methy lethoxy )-2-oxoethyl] amino} phosphory 1] -L-pheny lalaninate;
1-methylethyl (25',45',6i?)-4-{[(li?)-2-(6-amino-9H-purin-9-yl)-l-methylethoxy]methyl}-
2,6-dimethyl-7-oxo-8-oxa-3,5-diaza-4-phosphapentadecan-l-oate 4-oxide;
1 -methy lethy 1 (2S,4R,6R)-4- { [( li?)-2-(6-amino-9H-purin-9-y 1)- 1 -methy lethoxy ]methy 1 } -
2,6-dimethyl-7-oxo-8-oxa-3,5-diaza-4-phosphapentadecan-l-oate 4-oxide;
cyclopentyl (2i?,4i?,65)-4-{[(li?)-2-(6-amino-9H-purin-9-yl)-l-methylethoxy]methyl}-
2,6,9-trimethyl-7-oxo-8-oxa-3,5-diaza-4-phosphadecan-l-oate 4-oxide;
cyclopentyl (2i?,45',65 -4-{[(li?)-2-(6-amino-9H-purin-9-yl)-l-methylethoxy]methyl}-
2,6,9-trimethyl-7-oxo-8-oxa-3,5-diaza-4-phosphadecan-l-oate 4-oxide;
1 -methylethyl 1 -[(R)- { [(lR)-2-(6-ammo-9H-purin-9-yl)-l -methylethoxy]methyl} { [(\R)- 1 - methy l-2-( 1 -methy lethoxy )-2-oxoethy 1] amino} phosphory 1] -L-prolinate; or
1 -methylethyl 1 -[($)- {[(lR)-2-(6-ammo-9H-purin-9-yl)-l -methy lethoxy] methyl} { [(1R)-1 - methy l-2-( 1 -methy lethoxy )-2-oxoethy 1] amino} phosphory 1] -L-prolinate;
or a pharmaceutically acceptable salt thereof.
15. A pharmaceutical composition comprising an effective amount of the compound claim 1, or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable carrier.
16. The pharmaceutical composition of claim 15 further comprising an effective amount of one or more additional HIV antiviral agent selected from HIV protease inhibitors,
HIV integrase inhibitors, non-nucleoside HIV reverse transcriptase inhibitors, nucleoside HIV reverse transcriptase inhibitors, HIV fusion inhibitors and HIV entry inhibitors.
17. A method for the prophylaxis or treatment of infection by HIV or for the prophylaxis, treatment, or delay in the onset of AIDS in a subject in need thereof which comprises administering to the subject an effective amount of the compound of claim 1 or a pharmaceutically acceptable salt thereof.
18. The method of claim 17 further comprising administering to the subject an effective amount of one or more additional HIV antiviral agent selected from HIV protease inhibitors, HIV integrase inhibitors, non-nucleoside HIV reverse transcriptase inhibitors, nucleoside HIV reverse transcriptase inhibitors, HIV fusion inhibitors and HIV entry inhibitors.
19. A compound of claim 1 or a pharmaceutically acceptable salt thereof, for use in the preparation of a medicament for the prophylaxis or treatment of infection by HIV or for the prophylaxis, treatment, or delay in the onset of AIDS in a subject in need thereof.
20. Use of a compound of claim 1 in the manufacture of a medicament for the prophylaxis or treatment of infection by HIV or for the prophylaxis, treatment, or delay in the onset of AIDS in a subject in need thereof.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562189578P | 2015-07-07 | 2015-07-07 | |
| US62/189,578 | 2015-07-07 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2017007701A1 true WO2017007701A1 (en) | 2017-01-12 |
Family
ID=57686008
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2016/040606 Ceased WO2017007701A1 (en) | 2015-07-07 | 2016-07-01 | Antiviral phosphodiamide compounds |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2017007701A1 (en) |
Cited By (49)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9822138B2 (en) | 2015-08-10 | 2017-11-21 | Merck Sharp & Dohme Corp. | Antiviral beta-amino acid ester phosphodiamide compounds |
| CN108101942A (en) * | 2017-12-26 | 2018-06-01 | 深圳科兴生物工程有限公司 | Half fumaric acid tenofovir Chinese mugwort draws the synthetic method of potential impurity in the production of phenol amine |
| CN108623632A (en) * | 2018-08-17 | 2018-10-09 | 上海麦步医药科技有限公司 | A kind of tenofovir Chinese mugwort draws the preparation method of phenol amine |
| WO2019165374A1 (en) | 2018-02-26 | 2019-08-29 | Gilead Sciences, Inc. | Substituted pyrrolizine compounds as hbv replication inhibitors |
| WO2019195181A1 (en) | 2018-04-05 | 2019-10-10 | Gilead Sciences, Inc. | Antibodies and fragments thereof that bind hepatitis b virus protein x |
| WO2019193543A1 (en) | 2018-04-06 | 2019-10-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3'3'-cyclic dinucleotides |
| WO2019193533A1 (en) | 2018-04-06 | 2019-10-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'2'-cyclic dinucleotides |
| WO2019193542A1 (en) | 2018-04-06 | 2019-10-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'3'-cyclic dinucleotides |
| WO2019200247A1 (en) | 2018-04-12 | 2019-10-17 | Precision Biosciences, Inc. | Optimized engineered meganucleases having specificity for a recognition sequence in the hepatitis b virus genome |
| US10450335B2 (en) | 2015-12-15 | 2019-10-22 | Merck Sharp & Dohme Corp. | Antiviral oxime phosphoramide compounds |
| US10449208B2 (en) | 2016-08-25 | 2019-10-22 | Merck Sharp & Dohme Corp. | Antiviral prodrugs of tenofovir |
| WO2019211799A1 (en) | 2018-05-03 | 2019-11-07 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'3'-cyclic dinucleotide analogue comprising a cyclopentanyl modified nucleotide |
| US10519159B2 (en) | 2016-12-22 | 2019-12-31 | Merck Sharp & Dohme Corp. | Antiviral aliphatic ester prodrugs of tenofovir |
| WO2020028097A1 (en) | 2018-08-01 | 2020-02-06 | Gilead Sciences, Inc. | Solid forms of (r)-11-(methoxymethyl)-12-(3-methoxypropoxy)-3,3-dimethyl-8-0x0-2,3,8,13b-tetrahydro-1h-pyrido[2,1-a]pyrrolo[1,2-c] phthalazine-7-c arboxylic acid |
| WO2020092528A1 (en) | 2018-10-31 | 2020-05-07 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds having hpk1 inhibitory activity |
| WO2020092621A1 (en) | 2018-10-31 | 2020-05-07 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds as hpk1 inhibitors |
| US10662416B2 (en) | 2016-10-14 | 2020-05-26 | Precision Biosciences, Inc. | Engineered meganucleases specific for recognition sequences in the hepatitis B virus genome |
| US10736908B2 (en) | 2016-10-26 | 2020-08-11 | Merck Sharp & Dohme Corp. | Antiviral aryl-amide phosphodiamide compounds |
| US10745428B2 (en) | 2015-12-10 | 2020-08-18 | Idenix Pharmaceuticals Llc | Antiviral phosphodiamide prodrugs of tenofovir |
| WO2020178770A1 (en) | 2019-03-07 | 2020-09-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3'3'-cyclic dinucleotides and prodrugs thereof |
| WO2020178768A1 (en) | 2019-03-07 | 2020-09-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3'3'-cyclic dinucleotide analogue comprising a cyclopentanyl modified nucleotide as sting modulator |
| WO2020178769A1 (en) | 2019-03-07 | 2020-09-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'3'-cyclic dinucleotides and prodrugs thereof |
| WO2020214652A1 (en) | 2019-04-17 | 2020-10-22 | Gilead Sciences, Inc. | Solid forms of a toll-like receptor modulator |
| WO2020214663A1 (en) | 2019-04-17 | 2020-10-22 | Gilead Sciences, Inc. | Solid forms of a toll-like receptor modulator |
| CN111909204A (en) * | 2020-07-03 | 2020-11-10 | 佛山科学技术学院 | Tenofovir disopropiolate-based phosphoramidate compound and pharmaceutical composition and application thereof |
| CN111909205A (en) * | 2020-07-03 | 2020-11-10 | 佛山科学技术学院 | A kind of tenofovir dipropionate phosphoramidate compound and its pharmaceutical composition and use |
| WO2020237025A1 (en) | 2019-05-23 | 2020-11-26 | Gilead Sciences, Inc. | Substituted exo-methylene-oxindoles which are hpk1/map4k1 inhibitors |
| WO2020263830A1 (en) | 2019-06-25 | 2020-12-30 | Gilead Sciences, Inc. | Flt3l-fc fusion proteins and methods of use |
| WO2021034804A1 (en) | 2019-08-19 | 2021-02-25 | Gilead Sciences, Inc. | Pharmaceutical formulations of tenofovir alafenamide |
| US10966999B2 (en) | 2017-12-20 | 2021-04-06 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3′3′ cyclic dinucleotides with phosphonate bond activating the sting adaptor protein |
| WO2021067181A1 (en) | 2019-09-30 | 2021-04-08 | Gilead Sciences, Inc. | Hbv vaccines and methods treating hbv |
| WO2021113765A1 (en) | 2019-12-06 | 2021-06-10 | Precision Biosciences, Inc. | Optimized engineered meganucleases having specificity for a recognition sequence in the hepatitis b virus genome |
| US11123355B2 (en) | 2016-12-22 | 2021-09-21 | Idenix Pharmaceuticals Llc | Antiviral benzyl-amine phosphodiamide compounds |
| WO2021188959A1 (en) | 2020-03-20 | 2021-09-23 | Gilead Sciences, Inc. | Prodrugs of 4'-c-substituted-2-halo-2'-deoxyadenosine nucleosides and methods of making and using the same |
| US11203610B2 (en) | 2017-12-20 | 2021-12-21 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2′3′ cyclic dinucleotides with phosphonate bond activating the sting adaptor protein |
| WO2022031894A1 (en) | 2020-08-07 | 2022-02-10 | Gilead Sciences, Inc. | Prodrugs of phosphonamide nucleotide analogues and their pharmaceutical use |
| WO2022087149A2 (en) | 2020-10-22 | 2022-04-28 | Gilead Sciences, Inc. | Interleukin-2-fc fusion proteins and methods of use |
| CN114409705A (en) * | 2022-01-28 | 2022-04-29 | 四川科伦药业股份有限公司邛崃分公司 | Preparation method of prophenoltenofovir polyphenyl-removed impurity |
| WO2022241134A1 (en) | 2021-05-13 | 2022-11-17 | Gilead Sciences, Inc. | COMBINATION OF A TLR8 MODULATING COMPOUND AND ANTI-HBV siRNA THERAPEUTICS |
| WO2022271677A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271684A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271659A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271650A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| US11826375B2 (en) | 2018-07-19 | 2023-11-28 | Merck Sharp & Dohme Llc | Phosphinic amide prodrugs of tenofovir |
| CN119080837A (en) * | 2024-11-07 | 2024-12-06 | 上海柯君医药科技有限公司 | Antiviral drug and use thereof |
| WO2025240242A1 (en) | 2024-05-13 | 2025-11-20 | Gilead Sciences, Inc. | Combination therapies with ribavirin |
| WO2025240246A1 (en) | 2024-05-13 | 2025-11-20 | Gilead Sciences, Inc. | Combination therapies with ribavirin |
| WO2025240243A1 (en) | 2024-05-13 | 2025-11-20 | Gilead Sciences, Inc. | Combination therapies with bulevirtide and an inhibitory nucleic acid targeting hepatitis b virus |
| WO2025240244A1 (en) | 2024-05-13 | 2025-11-20 | Gilead Sciences, Inc. | Combination therapies comprising bulevirtide and lonafarnib for use in the treatment of hepatitis d virus infection |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8088754B2 (en) * | 2003-12-30 | 2012-01-03 | Gilead Sciences, Inc. | Anti-proliferative compounds, compositions, and methods of use thereof |
| US8754065B2 (en) * | 2011-08-16 | 2014-06-17 | Gilead Sciences, Inc. | Tenofovir alafenamide hemifumarate |
-
2016
- 2016-07-01 WO PCT/US2016/040606 patent/WO2017007701A1/en not_active Ceased
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8088754B2 (en) * | 2003-12-30 | 2012-01-03 | Gilead Sciences, Inc. | Anti-proliferative compounds, compositions, and methods of use thereof |
| US8754065B2 (en) * | 2011-08-16 | 2014-06-17 | Gilead Sciences, Inc. | Tenofovir alafenamide hemifumarate |
Non-Patent Citations (2)
| Title |
|---|
| JANSA ET AL.: "A novel and efficient one-pot synthesis of symmetrical diamide (bis-amidate) prodrugs of acyclic nucleoside phosphonates and evaluation of their biological activities", EUROPEAN JOURNAL OF MEDICINAL CHEMISTRY, vol. 46, 23 May 2011 (2011-05-23), pages 3748 - 3754, XP028278271 * |
| PERTUSATI ET AL.: "PMPA and PMEA prodrugs for the treatment of HIV infections and human papillomavirus (HPV) associated neoplasia and cancer", EUROPEAN JOURNAL OF MEDICINAL CHEMISTRY, vol. 78, 17 March 2014 (2014-03-17), pages 259 - 268, XP028847859 * |
Cited By (67)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9822138B2 (en) | 2015-08-10 | 2017-11-21 | Merck Sharp & Dohme Corp. | Antiviral beta-amino acid ester phosphodiamide compounds |
| US10745428B2 (en) | 2015-12-10 | 2020-08-18 | Idenix Pharmaceuticals Llc | Antiviral phosphodiamide prodrugs of tenofovir |
| US10450335B2 (en) | 2015-12-15 | 2019-10-22 | Merck Sharp & Dohme Corp. | Antiviral oxime phosphoramide compounds |
| US10449208B2 (en) | 2016-08-25 | 2019-10-22 | Merck Sharp & Dohme Corp. | Antiviral prodrugs of tenofovir |
| US11274285B2 (en) | 2016-10-14 | 2022-03-15 | Precision Biosciences, Inc. | Engineered meganucleases specific for recognition sequences in the Hepatitis B virus genome |
| US10662416B2 (en) | 2016-10-14 | 2020-05-26 | Precision Biosciences, Inc. | Engineered meganucleases specific for recognition sequences in the hepatitis B virus genome |
| US10736908B2 (en) | 2016-10-26 | 2020-08-11 | Merck Sharp & Dohme Corp. | Antiviral aryl-amide phosphodiamide compounds |
| US10519159B2 (en) | 2016-12-22 | 2019-12-31 | Merck Sharp & Dohme Corp. | Antiviral aliphatic ester prodrugs of tenofovir |
| US11123355B2 (en) | 2016-12-22 | 2021-09-21 | Idenix Pharmaceuticals Llc | Antiviral benzyl-amine phosphodiamide compounds |
| US10966999B2 (en) | 2017-12-20 | 2021-04-06 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3′3′ cyclic dinucleotides with phosphonate bond activating the sting adaptor protein |
| US11203610B2 (en) | 2017-12-20 | 2021-12-21 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2′3′ cyclic dinucleotides with phosphonate bond activating the sting adaptor protein |
| CN108101942B (en) * | 2017-12-26 | 2020-12-04 | 深圳科兴药业有限公司 | Method for synthesizing potential impurities in production of tenofovir alafenamide hemifumarate |
| CN108101942A (en) * | 2017-12-26 | 2018-06-01 | 深圳科兴生物工程有限公司 | Half fumaric acid tenofovir Chinese mugwort draws the synthetic method of potential impurity in the production of phenol amine |
| WO2019165374A1 (en) | 2018-02-26 | 2019-08-29 | Gilead Sciences, Inc. | Substituted pyrrolizine compounds as hbv replication inhibitors |
| WO2019195181A1 (en) | 2018-04-05 | 2019-10-10 | Gilead Sciences, Inc. | Antibodies and fragments thereof that bind hepatitis b virus protein x |
| US11292812B2 (en) | 2018-04-06 | 2022-04-05 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3′3′-cyclic dinucleotides |
| WO2019193543A1 (en) | 2018-04-06 | 2019-10-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3'3'-cyclic dinucleotides |
| US11149052B2 (en) | 2018-04-06 | 2021-10-19 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2′3′-cyclic dinucleotides |
| WO2019193533A1 (en) | 2018-04-06 | 2019-10-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'2'-cyclic dinucleotides |
| WO2019193542A1 (en) | 2018-04-06 | 2019-10-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'3'-cyclic dinucleotides |
| US11142750B2 (en) | 2018-04-12 | 2021-10-12 | Precision Biosciences, Inc. | Optimized engineered meganucleases having specificity for a recognition sequence in the Hepatitis B virus genome |
| WO2019200247A1 (en) | 2018-04-12 | 2019-10-17 | Precision Biosciences, Inc. | Optimized engineered meganucleases having specificity for a recognition sequence in the hepatitis b virus genome |
| US11788077B2 (en) | 2018-04-12 | 2023-10-17 | Precision Biosciences, Inc. | Polynucleotides encoding optimized engineered meganucleases having specificity for a recognition sequence in the Hepatitis B virus genome |
| WO2019211799A1 (en) | 2018-05-03 | 2019-11-07 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'3'-cyclic dinucleotide analogue comprising a cyclopentanyl modified nucleotide |
| US11826375B2 (en) | 2018-07-19 | 2023-11-28 | Merck Sharp & Dohme Llc | Phosphinic amide prodrugs of tenofovir |
| WO2020028097A1 (en) | 2018-08-01 | 2020-02-06 | Gilead Sciences, Inc. | Solid forms of (r)-11-(methoxymethyl)-12-(3-methoxypropoxy)-3,3-dimethyl-8-0x0-2,3,8,13b-tetrahydro-1h-pyrido[2,1-a]pyrrolo[1,2-c] phthalazine-7-c arboxylic acid |
| CN108623632A (en) * | 2018-08-17 | 2018-10-09 | 上海麦步医药科技有限公司 | A kind of tenofovir Chinese mugwort draws the preparation method of phenol amine |
| EP4371987A1 (en) | 2018-10-31 | 2024-05-22 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds as hpk1 inhibitors |
| EP4671244A1 (en) | 2018-10-31 | 2025-12-31 | Gilead Sciences, Inc. | SUBSTITUTED 6-AZABENZIMIDAZOLE COMPOUNDS AS HPK1 HIBITORS |
| WO2020092621A1 (en) | 2018-10-31 | 2020-05-07 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds as hpk1 inhibitors |
| WO2020092528A1 (en) | 2018-10-31 | 2020-05-07 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds having hpk1 inhibitory activity |
| US11766447B2 (en) | 2019-03-07 | 2023-09-26 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3′3′-cyclic dinucleotide analogue comprising a cyclopentanyl modified nucleotide as sting modulator |
| US12318403B2 (en) | 2019-03-07 | 2025-06-03 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2′3′-cyclic dinucleotides and prodrugs thereof |
| WO2020178769A1 (en) | 2019-03-07 | 2020-09-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'3'-cyclic dinucleotides and prodrugs thereof |
| WO2020178770A1 (en) | 2019-03-07 | 2020-09-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3'3'-cyclic dinucleotides and prodrugs thereof |
| WO2020178768A1 (en) | 2019-03-07 | 2020-09-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3'3'-cyclic dinucleotide analogue comprising a cyclopentanyl modified nucleotide as sting modulator |
| WO2020214652A1 (en) | 2019-04-17 | 2020-10-22 | Gilead Sciences, Inc. | Solid forms of a toll-like receptor modulator |
| WO2020214663A1 (en) | 2019-04-17 | 2020-10-22 | Gilead Sciences, Inc. | Solid forms of a toll-like receptor modulator |
| EP4458416A2 (en) | 2019-04-17 | 2024-11-06 | Gilead Sciences, Inc. | Solid forms of a toll-like receptor modulator |
| WO2020237025A1 (en) | 2019-05-23 | 2020-11-26 | Gilead Sciences, Inc. | Substituted exo-methylene-oxindoles which are hpk1/map4k1 inhibitors |
| WO2020263830A1 (en) | 2019-06-25 | 2020-12-30 | Gilead Sciences, Inc. | Flt3l-fc fusion proteins and methods of use |
| WO2021034804A1 (en) | 2019-08-19 | 2021-02-25 | Gilead Sciences, Inc. | Pharmaceutical formulations of tenofovir alafenamide |
| EP4458975A2 (en) | 2019-09-30 | 2024-11-06 | Gilead Sciences, Inc. | Hbv vaccines and methods treating hbv |
| WO2021067181A1 (en) | 2019-09-30 | 2021-04-08 | Gilead Sciences, Inc. | Hbv vaccines and methods treating hbv |
| EP4567109A2 (en) | 2019-12-06 | 2025-06-11 | Precision Biosciences, Inc. | Optimized engineered meganucleases having specificity for a recognition sequence in the hepatitis b virus genome |
| WO2021113765A1 (en) | 2019-12-06 | 2021-06-10 | Precision Biosciences, Inc. | Optimized engineered meganucleases having specificity for a recognition sequence in the hepatitis b virus genome |
| US12410418B2 (en) | 2019-12-06 | 2025-09-09 | Precision Biosciences, Inc. | Optimized engineered meganucleases having specificity for a recognition sequence in the Hepatitis B virus genome |
| WO2021188959A1 (en) | 2020-03-20 | 2021-09-23 | Gilead Sciences, Inc. | Prodrugs of 4'-c-substituted-2-halo-2'-deoxyadenosine nucleosides and methods of making and using the same |
| CN111909204A (en) * | 2020-07-03 | 2020-11-10 | 佛山科学技术学院 | Tenofovir disopropiolate-based phosphoramidate compound and pharmaceutical composition and application thereof |
| CN111909205A (en) * | 2020-07-03 | 2020-11-10 | 佛山科学技术学院 | A kind of tenofovir dipropionate phosphoramidate compound and its pharmaceutical composition and use |
| EP4667477A2 (en) | 2020-08-07 | 2025-12-24 | Gilead Sciences, Inc. | Prodrugs of phosphonamide nucleotide analogues and their pharmaceutical use |
| US12110305B2 (en) | 2020-08-07 | 2024-10-08 | Gilead Sciences, Inc. | Prodrugs of phosphonamide nucleotide analogues and their pharmaceutical use |
| WO2022031894A1 (en) | 2020-08-07 | 2022-02-10 | Gilead Sciences, Inc. | Prodrugs of phosphonamide nucleotide analogues and their pharmaceutical use |
| EP4667499A2 (en) | 2020-10-22 | 2025-12-24 | Gilead Sciences, Inc. | Interleukin-2-fc fusion proteins and methods of use |
| WO2022087149A2 (en) | 2020-10-22 | 2022-04-28 | Gilead Sciences, Inc. | Interleukin-2-fc fusion proteins and methods of use |
| WO2022241134A1 (en) | 2021-05-13 | 2022-11-17 | Gilead Sciences, Inc. | COMBINATION OF A TLR8 MODULATING COMPOUND AND ANTI-HBV siRNA THERAPEUTICS |
| WO2022271659A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271684A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271677A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271650A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| CN114409705A (en) * | 2022-01-28 | 2022-04-29 | 四川科伦药业股份有限公司邛崃分公司 | Preparation method of prophenoltenofovir polyphenyl-removed impurity |
| WO2025240246A1 (en) | 2024-05-13 | 2025-11-20 | Gilead Sciences, Inc. | Combination therapies with ribavirin |
| WO2025240243A1 (en) | 2024-05-13 | 2025-11-20 | Gilead Sciences, Inc. | Combination therapies with bulevirtide and an inhibitory nucleic acid targeting hepatitis b virus |
| WO2025240244A1 (en) | 2024-05-13 | 2025-11-20 | Gilead Sciences, Inc. | Combination therapies comprising bulevirtide and lonafarnib for use in the treatment of hepatitis d virus infection |
| WO2025240242A1 (en) | 2024-05-13 | 2025-11-20 | Gilead Sciences, Inc. | Combination therapies with ribavirin |
| CN119080837B (en) * | 2024-11-07 | 2025-03-11 | 上海柯君医药科技有限公司 | Antiviral medicine and application thereof |
| CN119080837A (en) * | 2024-11-07 | 2024-12-06 | 上海柯君医药科技有限公司 | Antiviral drug and use thereof |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2017007701A1 (en) | Antiviral phosphodiamide compounds | |
| AU2016305941B2 (en) | Antiviral beta-amino acid ester phosphodiamide compounds | |
| EP3386512B1 (en) | Antiviral phosphodiamide prodrugs of tenofovir | |
| US10519159B2 (en) | Antiviral aliphatic ester prodrugs of tenofovir | |
| EP3558322B1 (en) | Antiviral benzyl-amine phosphodiamide compounds | |
| EP3532069A1 (en) | Antiviral aryl-amide phosphodiamide compounds | |
| EP3390413B1 (en) | Antiviral oxime phosphoramide compounds | |
| EP3503895B1 (en) | Antiviral prodrugs of tenofovir |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16821847 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 16821847 Country of ref document: EP Kind code of ref document: A1 |