WO2016149535A1 - Cd48 antibodies and conjugates thereof - Google Patents
Cd48 antibodies and conjugates thereof Download PDFInfo
- Publication number
- WO2016149535A1 WO2016149535A1 PCT/US2016/022943 US2016022943W WO2016149535A1 WO 2016149535 A1 WO2016149535 A1 WO 2016149535A1 US 2016022943 W US2016022943 W US 2016022943W WO 2016149535 A1 WO2016149535 A1 WO 2016149535A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- antibody
- drug
- drug conjugate
- linker
- seq
- Prior art date
Links
- 101150023971 Cd48 gene Proteins 0.000 title 1
- 101000716130 Homo sapiens CD48 antigen Proteins 0.000 claims abstract description 113
- 102100036008 CD48 antigen Human genes 0.000 claims abstract description 94
- 241001529936 Murinae Species 0.000 claims abstract description 13
- 229940049595 antibody-drug conjugate Drugs 0.000 claims description 173
- 239000000611 antibody drug conjugate Substances 0.000 claims description 142
- 210000004027 cell Anatomy 0.000 claims description 95
- 229920001223 polyethylene glycol Polymers 0.000 claims description 87
- 206010035226 Plasma cell myeloma Diseases 0.000 claims description 67
- 208000034578 Multiple myelomas Diseases 0.000 claims description 62
- 206010028980 Neoplasm Diseases 0.000 claims description 55
- 239000003814 drug Substances 0.000 claims description 49
- 150000003839 salts Chemical class 0.000 claims description 46
- 230000027455 binding Effects 0.000 claims description 41
- 229940079593 drug Drugs 0.000 claims description 39
- 201000011510 cancer Diseases 0.000 claims description 35
- 102100035360 Cerebellar degeneration-related antigen 1 Human genes 0.000 claims description 33
- 239000002202 Polyethylene glycol Substances 0.000 claims description 31
- 238000000034 method Methods 0.000 claims description 30
- 125000006239 protecting group Chemical group 0.000 claims description 25
- 150000001875 compounds Chemical class 0.000 claims description 23
- 125000003275 alpha amino acid group Chemical group 0.000 claims description 21
- 125000003827 glycol group Chemical group 0.000 claims description 19
- 102000046585 human CD48 Human genes 0.000 claims description 19
- 239000000203 mixture Substances 0.000 claims description 17
- 102000039446 nucleic acids Human genes 0.000 claims description 17
- 108020004707 nucleic acids Proteins 0.000 claims description 17
- 150000007523 nucleic acids Chemical class 0.000 claims description 17
- 229910052739 hydrogen Inorganic materials 0.000 claims description 15
- 239000001257 hydrogen Substances 0.000 claims description 15
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 15
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 claims description 10
- 239000002254 cytotoxic agent Substances 0.000 claims description 10
- 125000000524 functional group Chemical group 0.000 claims description 10
- 229940127089 cytotoxic agent Drugs 0.000 claims description 9
- 239000013598 vector Substances 0.000 claims description 9
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 8
- 238000004519 manufacturing process Methods 0.000 claims description 7
- 208000031261 Acute myeloid leukaemia Diseases 0.000 claims description 6
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 claims description 5
- 210000003719 b-lymphocyte Anatomy 0.000 claims description 5
- 239000008194 pharmaceutical composition Substances 0.000 claims description 5
- 230000001268 conjugating effect Effects 0.000 claims description 4
- 230000036210 malignancy Effects 0.000 claims description 3
- 238000012258 culturing Methods 0.000 claims 4
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 claims 1
- 238000002648 combination therapy Methods 0.000 description 118
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 75
- 229960003957 dexamethasone Drugs 0.000 description 74
- GOTYRUGSSMKFNF-UHFFFAOYSA-N lenalidomide Chemical compound C1C=2C(N)=CC=CC=2C(=O)N1C1CCC(=O)NC1=O GOTYRUGSSMKFNF-UHFFFAOYSA-N 0.000 description 49
- GXJABQQUPOEUTA-RDJZCZTQSA-N bortezomib Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)B(O)O)NC(=O)C=1N=CC=NC=1)C1=CC=CC=C1 GXJABQQUPOEUTA-RDJZCZTQSA-N 0.000 description 48
- 229960004942 lenalidomide Drugs 0.000 description 48
- 229960001467 bortezomib Drugs 0.000 description 47
- BLMPQMFVWMYDKT-NZTKNTHTSA-N carfilzomib Chemical group C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC(C)C)C(=O)[C@]1(C)OC1)NC(=O)CN1CCOCC1)CC1=CC=CC=C1 BLMPQMFVWMYDKT-NZTKNTHTSA-N 0.000 description 40
- 108010021331 carfilzomib Proteins 0.000 description 40
- 229960002438 carfilzomib Drugs 0.000 description 40
- 239000003795 chemical substances by application Substances 0.000 description 36
- 125000005647 linker group Chemical group 0.000 description 36
- 229960002204 daratumumab Drugs 0.000 description 25
- FPOHNWQLNRZRFC-ZHACJKMWSA-N panobinostat Chemical compound CC=1NC2=CC=CC=C2C=1CCNCC1=CC=C(\C=C\C(=O)NO)C=C1 FPOHNWQLNRZRFC-ZHACJKMWSA-N 0.000 description 25
- 238000011282 treatment Methods 0.000 description 25
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 24
- 229960005184 panobinostat Drugs 0.000 description 24
- UVSMNLNDYGZFPF-UHFFFAOYSA-N pomalidomide Chemical compound O=C1C=2C(N)=CC=CC=2C(=O)N1C1CCC(=O)NC1=O UVSMNLNDYGZFPF-UHFFFAOYSA-N 0.000 description 23
- 229960000688 pomalidomide Drugs 0.000 description 22
- 241000699670 Mus sp. Species 0.000 description 21
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 20
- 230000000694 effects Effects 0.000 description 19
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 18
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 18
- 238000006243 chemical reaction Methods 0.000 description 18
- 229960004137 elotuzumab Drugs 0.000 description 18
- 108091033319 polynucleotide Proteins 0.000 description 16
- 102000040430 polynucleotide Human genes 0.000 description 16
- 239000002157 polynucleotide Substances 0.000 description 16
- 239000012453 solvate Substances 0.000 description 16
- 230000021615 conjugation Effects 0.000 description 15
- 239000000562 conjugate Substances 0.000 description 14
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 14
- 210000004881 tumor cell Anatomy 0.000 description 14
- -1 CD 150 Proteins 0.000 description 13
- 235000001014 amino acid Nutrition 0.000 description 13
- MXAYKZJJDUDWDS-LBPRGKRZSA-N ixazomib Chemical group CC(C)C[C@@H](B(O)O)NC(=O)CNC(=O)C1=CC(Cl)=CC=C1Cl MXAYKZJJDUDWDS-LBPRGKRZSA-N 0.000 description 13
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 12
- 150000001413 amino acids Chemical class 0.000 description 12
- 230000010056 antibody-dependent cellular cytotoxicity Effects 0.000 description 12
- 229960003648 ixazomib Drugs 0.000 description 12
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 12
- NFGXHKASABOEEW-UHFFFAOYSA-N 1-methylethyl 11-methoxy-3,7,11-trimethyl-2,4-dodecadienoate Chemical compound COC(C)(C)CCCC(C)CC=CC(C)=CC(=O)OC(C)C NFGXHKASABOEEW-UHFFFAOYSA-N 0.000 description 11
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 11
- 201000010099 disease Diseases 0.000 description 11
- 238000006467 substitution reaction Methods 0.000 description 11
- 229960004397 cyclophosphamide Drugs 0.000 description 10
- 239000013604 expression vector Substances 0.000 description 10
- 210000004602 germ cell Anatomy 0.000 description 10
- 229910052757 nitrogen Inorganic materials 0.000 description 10
- 108090000623 proteins and genes Proteins 0.000 description 10
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 9
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 9
- 208000035475 disorder Diseases 0.000 description 9
- 238000001990 intravenous administration Methods 0.000 description 9
- 239000002904 solvent Substances 0.000 description 9
- 229940124597 therapeutic agent Drugs 0.000 description 9
- PSVXZQVXSXSQRO-UHFFFAOYSA-N undecaethylene glycol Chemical compound OCCOCCOCCOCCOCCOCCOCCOCCOCCOCCOCCO PSVXZQVXSXSQRO-UHFFFAOYSA-N 0.000 description 9
- UEJJHQNACJXSKW-UHFFFAOYSA-N 2-(2,6-dioxopiperidin-3-yl)-1H-isoindole-1,3(2H)-dione Chemical compound O=C1C2=CC=CC=C2C(=O)N1C1CCC(=O)NC1=O UEJJHQNACJXSKW-UHFFFAOYSA-N 0.000 description 8
- 210000001744 T-lymphocyte Anatomy 0.000 description 8
- 108010044540 auristatin Proteins 0.000 description 8
- 239000012091 fetal bovine serum Substances 0.000 description 8
- 239000000047 product Substances 0.000 description 8
- 235000018102 proteins Nutrition 0.000 description 8
- 102000004169 proteins and genes Human genes 0.000 description 8
- 238000000746 purification Methods 0.000 description 8
- 230000004044 response Effects 0.000 description 8
- 229960003433 thalidomide Drugs 0.000 description 8
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 8
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 7
- 241001465754 Metazoa Species 0.000 description 7
- 239000000427 antigen Substances 0.000 description 7
- 102000036639 antigens Human genes 0.000 description 7
- 108091007433 antigens Proteins 0.000 description 7
- 210000004369 blood Anatomy 0.000 description 7
- 239000008280 blood Substances 0.000 description 7
- 239000012636 effector Substances 0.000 description 7
- 235000019253 formic acid Nutrition 0.000 description 7
- 238000001802 infusion Methods 0.000 description 7
- 239000007924 injection Substances 0.000 description 7
- 238000002347 injection Methods 0.000 description 7
- 238000002953 preparative HPLC Methods 0.000 description 7
- 238000001946 ultra-performance liquid chromatography-mass spectrometry Methods 0.000 description 7
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 6
- 241000699666 Mus <mouse, genus> Species 0.000 description 6
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 6
- 235000005311 Pandanus odoratissimus Nutrition 0.000 description 6
- 240000002390 Pandanus odoratissimus Species 0.000 description 6
- 239000012980 RPMI-1640 medium Substances 0.000 description 6
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 6
- 238000001727 in vivo Methods 0.000 description 6
- 229960001924 melphalan Drugs 0.000 description 6
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical group OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 6
- 239000000243 solution Substances 0.000 description 6
- 238000007920 subcutaneous administration Methods 0.000 description 6
- 208000024891 symptom Diseases 0.000 description 6
- OMRPLUKQNWNZAV-CONSDPRKSA-N (6as)-3-[3-[[(6as)-2-methoxy-8-(4-methoxyphenyl)-11-oxo-6a,7-dihydropyrrolo[2,1-c][1,4]benzodiazepin-3-yl]oxy]propoxy]-8-(4-aminophenyl)-2-methoxy-6a,7-dihydropyrrolo[2,1-c][1,4]benzodiazepin-11-one Chemical compound C1=CC(OC)=CC=C1C1=CN2C(=O)C3=CC(OC)=C(OCCCOC=4C(=CC=5C(=O)N6C=C(C[C@H]6C=NC=5C=4)C=4C=CC(N)=CC=4)OC)C=C3N=C[C@@H]2C1 OMRPLUKQNWNZAV-CONSDPRKSA-N 0.000 description 5
- 108010038940 CD48 Antigen Proteins 0.000 description 5
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 5
- 239000004472 Lysine Substances 0.000 description 5
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 description 5
- 208000033559 Waldenström macroglobulinemia Diseases 0.000 description 5
- 239000002253 acid Substances 0.000 description 5
- 230000002378 acidificating effect Effects 0.000 description 5
- 230000004913 activation Effects 0.000 description 5
- 230000005888 antibody-dependent cellular phagocytosis Effects 0.000 description 5
- 230000006907 apoptotic process Effects 0.000 description 5
- 239000002775 capsule Substances 0.000 description 5
- 210000004978 chinese hamster ovary cell Anatomy 0.000 description 5
- 230000004540 complement-dependent cytotoxicity Effects 0.000 description 5
- 239000012634 fragment Substances 0.000 description 5
- 230000003834 intracellular effect Effects 0.000 description 5
- 239000007928 intraperitoneal injection Substances 0.000 description 5
- 239000006187 pill Substances 0.000 description 5
- KZNICNPSHKQLFF-UHFFFAOYSA-N succinimide Chemical class O=C1CCC(=O)N1 KZNICNPSHKQLFF-UHFFFAOYSA-N 0.000 description 5
- 239000003826 tablet Substances 0.000 description 5
- 238000012447 xenograft mouse model Methods 0.000 description 5
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 4
- 0 CCC(C)C(C(C1)*=C1N(CCC1)C1C(C(C)C(NC(C)C(c1ccccc1)O)=O)OC)N(C)C(C(C(C)C)NC(C(C(C)C)N(C)C(OCc(cc1)cc(NC(CCNC(C(CCCCNC(C(C)CCOCC)=O)N*)=O)=O)c1O[C@@](C[C@](C1)O)O[C@@]1C(O)=O)=O)=O)=O Chemical compound CCC(C)C(C(C1)*=C1N(CCC1)C1C(C(C)C(NC(C)C(c1ccccc1)O)=O)OC)N(C)C(C(C(C)C)NC(C(C(C)C)N(C)C(OCc(cc1)cc(NC(CCNC(C(CCCCNC(C(C)CCOCC)=O)N*)=O)=O)c1O[C@@](C[C@](C1)O)O[C@@]1C(O)=O)=O)=O)=O 0.000 description 4
- 102000015713 CD48 Antigen Human genes 0.000 description 4
- VYZAMTAEIAYCRO-BJUDXGSMSA-N Chromium-51 Chemical compound [51Cr] VYZAMTAEIAYCRO-BJUDXGSMSA-N 0.000 description 4
- 206010020631 Hypergammaglobulinaemia benign monoclonal Diseases 0.000 description 4
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 4
- 229940079156 Proteasome inhibitor Drugs 0.000 description 4
- 108010076504 Protein Sorting Signals Proteins 0.000 description 4
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 4
- IEDXPSOJFSVCKU-HOKPPMCLSA-N [4-[[(2S)-5-(carbamoylamino)-2-[[(2S)-2-[6-(2,5-dioxopyrrolidin-1-yl)hexanoylamino]-3-methylbutanoyl]amino]pentanoyl]amino]phenyl]methyl N-[(2S)-1-[[(2S)-1-[[(3R,4S,5S)-1-[(2S)-2-[(1R,2R)-3-[[(1S,2R)-1-hydroxy-1-phenylpropan-2-yl]amino]-1-methoxy-2-methyl-3-oxopropyl]pyrrolidin-1-yl]-3-methoxy-5-methyl-1-oxoheptan-4-yl]-methylamino]-3-methyl-1-oxobutan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]-N-methylcarbamate Chemical compound CC[C@H](C)[C@@H]([C@@H](CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C)[C@@H](O)c1ccccc1)OC)N(C)C(=O)[C@@H](NC(=O)[C@H](C(C)C)N(C)C(=O)OCc1ccc(NC(=O)[C@H](CCCNC(N)=O)NC(=O)[C@@H](NC(=O)CCCCCN2C(=O)CCC2=O)C(C)C)cc1)C(C)C IEDXPSOJFSVCKU-HOKPPMCLSA-N 0.000 description 4
- 125000000217 alkyl group Chemical group 0.000 description 4
- 229940049706 benzodiazepine Drugs 0.000 description 4
- 150000001557 benzodiazepines Chemical class 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 4
- 229960004316 cisplatin Drugs 0.000 description 4
- 239000000539 dimer Substances 0.000 description 4
- 238000010494 dissociation reaction Methods 0.000 description 4
- 230000005593 dissociations Effects 0.000 description 4
- 229960004679 doxorubicin Drugs 0.000 description 4
- 229960005420 etoposide Drugs 0.000 description 4
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 4
- PTCGDEVVHUXTMP-UHFFFAOYSA-N flutolanil Chemical compound CC(C)OC1=CC=CC(NC(=O)C=2C(=CC=CC=2)C(F)(F)F)=C1 PTCGDEVVHUXTMP-UHFFFAOYSA-N 0.000 description 4
- 201000003444 follicular lymphoma Diseases 0.000 description 4
- 238000004128 high performance liquid chromatography Methods 0.000 description 4
- 210000002540 macrophage Anatomy 0.000 description 4
- 201000005328 monoclonal gammopathy of uncertain significance Diseases 0.000 description 4
- 108090000765 processed proteins & peptides Proteins 0.000 description 4
- 239000003207 proteasome inhibitor Substances 0.000 description 4
- 230000000284 resting effect Effects 0.000 description 4
- 230000009870 specific binding Effects 0.000 description 4
- 238000002560 therapeutic procedure Methods 0.000 description 4
- 239000003643 water by type Substances 0.000 description 4
- VGGWNGWXGFWLRK-UHFFFAOYSA-N 8,9-dihydro-1H-[1,3]oxazolo[4,5-i][1,2]benzodiazepine Chemical class C1=CC=NNC2=C(OCN3)C3=CC=C21 VGGWNGWXGFWLRK-UHFFFAOYSA-N 0.000 description 3
- 208000023761 AL amyloidosis Diseases 0.000 description 3
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 description 3
- 108060003951 Immunoglobulin Proteins 0.000 description 3
- 208000005531 Immunoglobulin Light-chain Amyloidosis Diseases 0.000 description 3
- 241000282567 Macaca fascicularis Species 0.000 description 3
- 208000025205 Mantle-Cell Lymphoma Diseases 0.000 description 3
- 102000035195 Peptidases Human genes 0.000 description 3
- 108091005804 Peptidases Proteins 0.000 description 3
- 206010036673 Primary amyloidosis Diseases 0.000 description 3
- 125000004429 atom Chemical group 0.000 description 3
- 210000004899 c-terminal region Anatomy 0.000 description 3
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 3
- 239000000824 cytostatic agent Substances 0.000 description 3
- 231100000599 cytotoxic agent Toxicity 0.000 description 3
- 230000001472 cytotoxic effect Effects 0.000 description 3
- 230000006870 function Effects 0.000 description 3
- 208000010726 hind limb paralysis Diseases 0.000 description 3
- 102000018358 immunoglobulin Human genes 0.000 description 3
- 239000007943 implant Substances 0.000 description 3
- 239000003446 ligand Substances 0.000 description 3
- XMYQHJDBLRZMLW-UHFFFAOYSA-N methanolamine Chemical compound NCO XMYQHJDBLRZMLW-UHFFFAOYSA-N 0.000 description 3
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 3
- 208000022256 primary systemic amyloidosis Diseases 0.000 description 3
- 230000002441 reversible effect Effects 0.000 description 3
- 229960004641 rituximab Drugs 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 230000004083 survival effect Effects 0.000 description 3
- 230000008961 swelling Effects 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- 230000001225 therapeutic effect Effects 0.000 description 3
- 239000003981 vehicle Substances 0.000 description 3
- NNUFGAKJIDSIFG-VIFPVBQESA-N (2,5-dioxopyrrolidin-1-yl) (2S)-2-(2,5-dioxopyrrol-1-yl)-3-[(2-methylpropan-2-yl)oxycarbonylamino]propanoate Chemical compound CC(C)(C)OC(=O)NC[C@H](N1C(=O)C=CC1=O)C(=O)ON1C(=O)CCC1=O NNUFGAKJIDSIFG-VIFPVBQESA-N 0.000 description 2
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 2
- UZOVYGYOLBIAJR-UHFFFAOYSA-N 4-isocyanato-4'-methyldiphenylmethane Chemical compound C1=CC(C)=CC=C1CC1=CC=C(N=C=O)C=C1 UZOVYGYOLBIAJR-UHFFFAOYSA-N 0.000 description 2
- 102100031585 ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 Human genes 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 2
- 102000047934 Caspase-3/7 Human genes 0.000 description 2
- 108700037887 Caspase-3/7 Proteins 0.000 description 2
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 2
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 2
- 206010061818 Disease progression Diseases 0.000 description 2
- 108010043121 Green Fluorescent Proteins Proteins 0.000 description 2
- 102000003964 Histone deacetylase Human genes 0.000 description 2
- 108090000353 Histone deacetylase Proteins 0.000 description 2
- 208000017604 Hodgkin disease Diseases 0.000 description 2
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- 101000777636 Homo sapiens ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 Proteins 0.000 description 2
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 2
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 2
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 2
- 229910019142 PO4 Inorganic materials 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- 108020004511 Recombinant DNA Proteins 0.000 description 2
- 108010003723 Single-Domain Antibodies Proteins 0.000 description 2
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 2
- 108060008539 Transglutaminase Proteins 0.000 description 2
- 229920004890 Triton X-100 Polymers 0.000 description 2
- 239000013504 Triton X-100 Substances 0.000 description 2
- 229960000583 acetic acid Drugs 0.000 description 2
- 229960000548 alemtuzumab Drugs 0.000 description 2
- 229940100198 alkylating agent Drugs 0.000 description 2
- 239000002168 alkylating agent Substances 0.000 description 2
- 230000006023 anti-tumor response Effects 0.000 description 2
- 238000003556 assay Methods 0.000 description 2
- 229940112129 campath Drugs 0.000 description 2
- 235000014633 carbohydrates Nutrition 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- 230000015556 catabolic process Effects 0.000 description 2
- 230000003833 cell viability Effects 0.000 description 2
- 238000003776 cleavage reaction Methods 0.000 description 2
- 238000004440 column chromatography Methods 0.000 description 2
- 239000000356 contaminant Substances 0.000 description 2
- 235000018417 cysteine Nutrition 0.000 description 2
- 230000009089 cytolysis Effects 0.000 description 2
- 231100000433 cytotoxic Toxicity 0.000 description 2
- 230000003013 cytotoxicity Effects 0.000 description 2
- 231100000135 cytotoxicity Toxicity 0.000 description 2
- 238000002784 cytotoxicity assay Methods 0.000 description 2
- 231100000263 cytotoxicity test Toxicity 0.000 description 2
- 238000006731 degradation reaction Methods 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 230000004069 differentiation Effects 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 2
- 230000005750 disease progression Effects 0.000 description 2
- 150000002019 disulfides Chemical class 0.000 description 2
- 239000003623 enhancer Substances 0.000 description 2
- 150000002148 esters Chemical class 0.000 description 2
- 239000007850 fluorescent dye Substances 0.000 description 2
- 238000009472 formulation Methods 0.000 description 2
- 239000012362 glacial acetic acid Substances 0.000 description 2
- 229930182480 glucuronide Natural products 0.000 description 2
- 239000001963 growth medium Substances 0.000 description 2
- 230000036541 health Effects 0.000 description 2
- 125000005842 heteroatom Chemical group 0.000 description 2
- 150000004677 hydrates Chemical class 0.000 description 2
- 150000002466 imines Chemical group 0.000 description 2
- 230000002519 immonomodulatory effect Effects 0.000 description 2
- 230000001900 immune effect Effects 0.000 description 2
- 229940127121 immunoconjugate Drugs 0.000 description 2
- 229940072221 immunoglobulins Drugs 0.000 description 2
- 239000004615 ingredient Substances 0.000 description 2
- 230000002452 interceptive effect Effects 0.000 description 2
- 210000004698 lymphocyte Anatomy 0.000 description 2
- 125000003588 lysine group Chemical group [H]N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 2
- 230000002132 lysosomal effect Effects 0.000 description 2
- 210000004962 mammalian cell Anatomy 0.000 description 2
- 239000003550 marker Substances 0.000 description 2
- 230000001404 mediated effect Effects 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 230000035772 mutation Effects 0.000 description 2
- 201000000050 myeloid neoplasm Diseases 0.000 description 2
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 2
- 230000009871 nonspecific binding Effects 0.000 description 2
- 239000000546 pharmaceutical excipient Substances 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- 239000002953 phosphate buffered saline Substances 0.000 description 2
- 230000003389 potentiating effect Effects 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 230000007017 scission Effects 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 238000009097 single-agent therapy Methods 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 239000003381 stabilizer Substances 0.000 description 2
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 2
- 229960002317 succinimide Drugs 0.000 description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 2
- 238000004809 thin layer chromatography Methods 0.000 description 2
- 125000003396 thiol group Chemical group [H]S* 0.000 description 2
- 150000003573 thiols Chemical class 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 102000003601 transglutaminase Human genes 0.000 description 2
- 230000003442 weekly effect Effects 0.000 description 2
- MFRNYXJJRJQHNW-DEMKXPNLSA-N (2s)-2-[[(2r,3r)-3-methoxy-3-[(2s)-1-[(3r,4s,5s)-3-methoxy-5-methyl-4-[methyl-[(2s)-3-methyl-2-[[(2s)-3-methyl-2-(methylamino)butanoyl]amino]butanoyl]amino]heptanoyl]pyrrolidin-2-yl]-2-methylpropanoyl]amino]-3-phenylpropanoic acid Chemical compound CN[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N(C)[C@@H]([C@@H](C)CC)[C@H](OC)CC(=O)N1CCC[C@H]1[C@H](OC)[C@@H](C)C(=O)N[C@H](C(O)=O)CC1=CC=CC=C1 MFRNYXJJRJQHNW-DEMKXPNLSA-N 0.000 description 1
- AGGWFDNPHKLBBV-YUMQZZPRSA-N (2s)-2-[[(2s)-2-amino-3-methylbutanoyl]amino]-5-(carbamoylamino)pentanoic acid Chemical compound CC(C)[C@H](N)C(=O)N[C@H](C(O)=O)CCCNC(N)=O AGGWFDNPHKLBBV-YUMQZZPRSA-N 0.000 description 1
- LJIOTBMDLVHTBO-CUYJMHBOSA-N (2s)-2-amino-n-[(1r,2r)-1-cyano-2-[4-[4-(4-methylpiperazin-1-yl)sulfonylphenyl]phenyl]cyclopropyl]butanamide Chemical compound CC[C@H](N)C(=O)N[C@]1(C#N)C[C@@H]1C1=CC=C(C=2C=CC(=CC=2)S(=O)(=O)N2CCN(C)CC2)C=C1 LJIOTBMDLVHTBO-CUYJMHBOSA-N 0.000 description 1
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 description 1
- GHOKWGTUZJEAQD-ZETCQYMHSA-N (D)-(+)-Pantothenic acid Chemical compound OCC(C)(C)[C@@H](O)C(=O)NCCC(O)=O GHOKWGTUZJEAQD-ZETCQYMHSA-N 0.000 description 1
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 1
- BDNKZNFMNDZQMI-UHFFFAOYSA-N 1,3-diisopropylcarbodiimide Chemical compound CC(C)N=C=NC(C)C BDNKZNFMNDZQMI-UHFFFAOYSA-N 0.000 description 1
- SVUOLADPCWQTTE-UHFFFAOYSA-N 1h-1,2-benzodiazepine Chemical compound N1N=CC=CC2=CC=CC=C12 SVUOLADPCWQTTE-UHFFFAOYSA-N 0.000 description 1
- WXTMDXOMEHJXQO-UHFFFAOYSA-N 2,5-dihydroxybenzoic acid Chemical compound OC(=O)C1=CC(O)=CC=C1O WXTMDXOMEHJXQO-UHFFFAOYSA-N 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-M 3-carboxy-2,3-dihydroxypropanoate Chemical compound OC(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-M 0.000 description 1
- UPMLOUAZCHDJJD-UHFFFAOYSA-N 4,4'-Diphenylmethane Diisocyanate Chemical compound C1=CC(N=C=O)=CC=C1CC1=CC=C(N=C=O)C=C1 UPMLOUAZCHDJJD-UHFFFAOYSA-N 0.000 description 1
- YXHLJMWYDTXDHS-IRFLANFNSA-N 7-aminoactinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=C(N)C=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 YXHLJMWYDTXDHS-IRFLANFNSA-N 0.000 description 1
- 108700012813 7-aminoactinomycin D Proteins 0.000 description 1
- 239000012114 Alexa Fluor 647 Substances 0.000 description 1
- 239000012099 Alexa Fluor family Substances 0.000 description 1
- 241000269858 Anarhichas lupus Species 0.000 description 1
- 108010032595 Antibody Binding Sites Proteins 0.000 description 1
- 241000701822 Bovine papillomavirus Species 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 1
- 238000003731 Caspase Glo 3/7 Assay Methods 0.000 description 1
- 102000004225 Cathepsin B Human genes 0.000 description 1
- 108090000712 Cathepsin B Proteins 0.000 description 1
- 102000005600 Cathepsins Human genes 0.000 description 1
- 108010084457 Cathepsins Proteins 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- 108010047041 Complementarity Determining Regions Proteins 0.000 description 1
- 108091035707 Consensus sequence Proteins 0.000 description 1
- 241000557626 Corvus corax Species 0.000 description 1
- 241000699802 Cricetulus griseus Species 0.000 description 1
- 241000701022 Cytomegalovirus Species 0.000 description 1
- DSLZVSRJTYRBFB-LLEIAEIESA-N D-glucaric acid Chemical compound OC(=O)[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O DSLZVSRJTYRBFB-LLEIAEIESA-N 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- AEMOLEFTQBMNLQ-AQKNRBDQSA-N D-glucopyranuronic acid Chemical compound OC1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-AQKNRBDQSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- BWGNESOTFCXPMA-UHFFFAOYSA-N Dihydrogen disulfide Chemical compound SS BWGNESOTFCXPMA-UHFFFAOYSA-N 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- IAYPIBMASNFSPL-UHFFFAOYSA-N Ethylene oxide Chemical group C1CO1 IAYPIBMASNFSPL-UHFFFAOYSA-N 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- 230000005526 G1 to G0 transition Effects 0.000 description 1
- 108700039691 Genetic Promoter Regions Proteins 0.000 description 1
- 108700007698 Genetic Terminator Regions Proteins 0.000 description 1
- 102000053187 Glucuronidase Human genes 0.000 description 1
- 108010060309 Glucuronidase Proteins 0.000 description 1
- 102000003886 Glycoproteins Human genes 0.000 description 1
- 108090000288 Glycoproteins Proteins 0.000 description 1
- 102000005744 Glycoside Hydrolases Human genes 0.000 description 1
- 108010031186 Glycoside Hydrolases Proteins 0.000 description 1
- 239000012981 Hank's balanced salt solution Substances 0.000 description 1
- 208000021519 Hodgkin lymphoma Diseases 0.000 description 1
- 101000633778 Homo sapiens SLAM family member 5 Proteins 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- 108010067060 Immunoglobulin Variable Region Proteins 0.000 description 1
- 102000017727 Immunoglobulin Variable Region Human genes 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 1
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 description 1
- 206010025323 Lymphomas Diseases 0.000 description 1
- 241000829100 Macaca mulatta polyomavirus 1 Species 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 206010060880 Monoclonal gammopathy Diseases 0.000 description 1
- NQTADLQHYWFPDB-UHFFFAOYSA-N N-Hydroxysuccinimide Chemical compound ON1C(=O)CCC1=O NQTADLQHYWFPDB-UHFFFAOYSA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- 206010028813 Nausea Diseases 0.000 description 1
- DFPAKSUCGFBDDF-UHFFFAOYSA-N Nicotinamide Chemical group NC(=O)C1=CC=CN=C1 DFPAKSUCGFBDDF-UHFFFAOYSA-N 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- 108091028043 Nucleic acid sequence Proteins 0.000 description 1
- 108091034117 Oligonucleotide Proteins 0.000 description 1
- 206010033128 Ovarian cancer Diseases 0.000 description 1
- 206010061535 Ovarian neoplasm Diseases 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 101150092610 PKH2 gene Proteins 0.000 description 1
- 208000002774 Paraproteinemias Diseases 0.000 description 1
- RFCVXVPWSPOMFJ-STQMWFEESA-N Phe-Leu Chemical compound CC(C)C[C@@H](C(O)=O)NC(=O)[C@@H](N)CC1=CC=CC=C1 RFCVXVPWSPOMFJ-STQMWFEESA-N 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 108020005067 RNA Splice Sites Proteins 0.000 description 1
- 102100029216 SLAM family member 5 Human genes 0.000 description 1
- 102100029215 Signaling lymphocytic activation molecule Human genes 0.000 description 1
- 101710163413 Signaling lymphocytic activation molecule Proteins 0.000 description 1
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 229920002253 Tannate Polymers 0.000 description 1
- 229940123237 Taxane Drugs 0.000 description 1
- 102000004243 Tubulin Human genes 0.000 description 1
- 108090000704 Tubulin Proteins 0.000 description 1
- 238000005411 Van der Waals force Methods 0.000 description 1
- 229940122803 Vinca alkaloid Drugs 0.000 description 1
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 1
- 230000003187 abdominal effect Effects 0.000 description 1
- 239000008351 acetate buffer Substances 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 230000009824 affinity maturation Effects 0.000 description 1
- 235000004279 alanine Nutrition 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- 230000007815 allergy Effects 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- 125000006242 amine protecting group Chemical group 0.000 description 1
- 125000000539 amino acid group Chemical group 0.000 description 1
- 206010002022 amyloidosis Diseases 0.000 description 1
- 230000001093 anti-cancer Effects 0.000 description 1
- 230000000259 anti-tumor effect Effects 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 229940072107 ascorbate Drugs 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 230000001580 bacterial effect Effects 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 125000003310 benzodiazepinyl group Chemical group N1N=C(C=CC2=C1C=CC=C2)* 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- PUJDIJCNWFYVJX-UHFFFAOYSA-N benzyl carbamate Chemical class NC(=O)OCC1=CC=CC=C1 PUJDIJCNWFYVJX-UHFFFAOYSA-N 0.000 description 1
- 230000001588 bifunctional effect Effects 0.000 description 1
- 210000003969 blast cell Anatomy 0.000 description 1
- 239000012267 brine Substances 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 229930195731 calicheamicin Natural products 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 238000012054 celltiter-glo Methods 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 238000007385 chemical modification Methods 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 208000032852 chronic lymphocytic leukemia Diseases 0.000 description 1
- 229940001468 citrate Drugs 0.000 description 1
- 230000000295 complement effect Effects 0.000 description 1
- 239000002131 composite material Substances 0.000 description 1
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 1
- 230000001085 cytostatic effect Effects 0.000 description 1
- 229940026692 decadron Drugs 0.000 description 1
- 230000001934 delay Effects 0.000 description 1
- 210000004443 dendritic cell Anatomy 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 230000006866 deterioration Effects 0.000 description 1
- 239000000032 diagnostic agent Substances 0.000 description 1
- 229940039227 diagnostic agent Drugs 0.000 description 1
- IJKVHSBPTUYDLN-UHFFFAOYSA-N dihydroxy(oxo)silane Chemical compound O[Si](O)=O IJKVHSBPTUYDLN-UHFFFAOYSA-N 0.000 description 1
- BGRWYRAHAFMIBJ-UHFFFAOYSA-N diisopropylcarbodiimide Natural products CC(C)NC(=O)NC(C)C BGRWYRAHAFMIBJ-UHFFFAOYSA-N 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 230000003292 diminished effect Effects 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 229960005501 duocarmycin Drugs 0.000 description 1
- 229930184221 duocarmycin Natural products 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 238000003821 enantio-separation Methods 0.000 description 1
- 210000001163 endosome Anatomy 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 229940088598 enzyme Drugs 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- PQVSTLUFSYVLTO-UHFFFAOYSA-N ethyl n-ethoxycarbonylcarbamate Chemical compound CCOC(=O)NC(=O)OCC PQVSTLUFSYVLTO-UHFFFAOYSA-N 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 238000003818 flash chromatography Methods 0.000 description 1
- 238000000684 flow cytometry Methods 0.000 description 1
- 229940050411 fumarate Drugs 0.000 description 1
- 238000001502 gel electrophoresis Methods 0.000 description 1
- 239000003862 glucocorticoid Substances 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 229940097042 glucuronate Drugs 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- 125000000487 histidyl group Chemical group [H]N([H])C(C(=O)O*)C([H])([H])C1=C([H])N([H])C([H])=N1 0.000 description 1
- 210000004408 hybridoma Anatomy 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 210000002865 immune cell Anatomy 0.000 description 1
- 238000003018 immunoassay Methods 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 230000008595 infiltration Effects 0.000 description 1
- 238000001764 infiltration Methods 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 230000001788 irregular Effects 0.000 description 1
- TWBYWOBDOCUKOW-UHFFFAOYSA-M isonicotinate Chemical compound [O-]C(=O)C1=CC=NC=C1 TWBYWOBDOCUKOW-UHFFFAOYSA-M 0.000 description 1
- 230000002147 killing effect Effects 0.000 description 1
- 229940000764 kyprolis Drugs 0.000 description 1
- 239000010410 layer Substances 0.000 description 1
- 208000032839 leukemia Diseases 0.000 description 1
- 238000012417 linear regression Methods 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 239000012669 liquid formulation Substances 0.000 description 1
- 229940040692 lithium hydroxide monohydrate Drugs 0.000 description 1
- GLXDVVHUTZTUQK-UHFFFAOYSA-M lithium hydroxide monohydrate Substances [Li+].O.[OH-] GLXDVVHUTZTUQK-UHFFFAOYSA-M 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 201000005202 lung cancer Diseases 0.000 description 1
- 208000020816 lung neoplasm Diseases 0.000 description 1
- 210000003712 lysosome Anatomy 0.000 description 1
- 230000001868 lysosomic effect Effects 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 125000005439 maleimidyl group Chemical group C1(C=CC(N1*)=O)=O 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 238000002483 medication Methods 0.000 description 1
- 230000009401 metastasis Effects 0.000 description 1
- 230000008880 microtubule cytoskeleton organization Effects 0.000 description 1
- 239000003607 modifier Substances 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- 210000004980 monocyte derived macrophage Anatomy 0.000 description 1
- 239000000178 monomer Substances 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 238000002703 mutagenesis Methods 0.000 description 1
- 231100000350 mutagenesis Toxicity 0.000 description 1
- YVFMFWAUSKFVGO-UHFFFAOYSA-N naphthalene-2-carboperoxoic acid Chemical class C1=CC=CC2=CC(C(=O)OO)=CC=C21 YVFMFWAUSKFVGO-UHFFFAOYSA-N 0.000 description 1
- 210000000822 natural killer cell Anatomy 0.000 description 1
- 230000008693 nausea Effects 0.000 description 1
- 239000013642 negative control Substances 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 239000002547 new drug Substances 0.000 description 1
- 229940030115 ninlaro Drugs 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 239000002773 nucleotide Substances 0.000 description 1
- 125000003729 nucleotide group Chemical group 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 238000011275 oncology therapy Methods 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 239000012044 organic layer Substances 0.000 description 1
- 230000000242 pagocytic effect Effects 0.000 description 1
- 229940014662 pantothenate Drugs 0.000 description 1
- 235000019161 pantothenic acid Nutrition 0.000 description 1
- 239000011713 pantothenic acid Substances 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 108010073101 phenylalanylleucine Proteins 0.000 description 1
- 230000035790 physiological processes and functions Effects 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- 229940012957 plasmin Drugs 0.000 description 1
- 230000008488 polyadenylation Effects 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 238000006116 polymerization reaction Methods 0.000 description 1
- 229920001184 polypeptide Polymers 0.000 description 1
- 229940008606 pomalyst Drugs 0.000 description 1
- 230000002028 premature Effects 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- 235000019833 protease Nutrition 0.000 description 1
- 235000019419 proteases Nutrition 0.000 description 1
- 238000001742 protein purification Methods 0.000 description 1
- 230000017854 proteolysis Effects 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 238000003259 recombinant expression Methods 0.000 description 1
- 230000010076 replication Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 229940120975 revlimid Drugs 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- 125000000467 secondary amino group Chemical group [H]N([*:1])[*:2] 0.000 description 1
- 230000003248 secreting effect Effects 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 238000007086 side reaction Methods 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 238000010898 silica gel chromatography Methods 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 1
- 125000006850 spacer group Chemical group 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 238000002660 stem cell treatment Methods 0.000 description 1
- 125000000547 substituted alkyl group Chemical group 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 125000004434 sulfur atom Chemical group 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 230000001839 systemic circulation Effects 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- RUPAXCPQAAOIPB-UHFFFAOYSA-N tert-butyl formate Chemical group CC(C)(C)OC=O RUPAXCPQAAOIPB-UHFFFAOYSA-N 0.000 description 1
- 238000004448 titration Methods 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 230000005030 transcription termination Effects 0.000 description 1
- 230000002103 transcriptional effect Effects 0.000 description 1
- 230000001131 transforming effect Effects 0.000 description 1
- 230000004614 tumor growth Effects 0.000 description 1
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 1
- 241000701161 unidentified adenovirus Species 0.000 description 1
- 229940099039 velcade Drugs 0.000 description 1
- 238000010626 work up procedure Methods 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
- C07K16/2803—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants against the immunoglobulin superfamily
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39533—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals
- A61K39/39566—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals against immunoglobulins, e.g. anti-idiotypic antibodies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/68031—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug being an auristatin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6801—Drug-antibody or immunoglobulin conjugates defined by the pharmacologically or therapeutically active agent
- A61K47/6803—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates
- A61K47/6807—Drugs conjugated to an antibody or immunoglobulin, e.g. cisplatin-antibody conjugates the drug or compound being a sugar, nucleoside, nucleotide, nucleic acid, e.g. RNA antisense
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6849—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting a receptor, a cell surface antigen or a cell surface determinant
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6883—Polymer-drug antibody conjugates, e.g. mitomycin-dextran-Ab; DNA-polylysine-antibody complex or conjugate used for therapy
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/02—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing at least one abnormal peptide link
- C07K5/0205—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing at least one abnormal peptide link containing the structure -NH-(X)3-C(=0)-, e.g. statine or derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/24—Immunoglobulins specific features characterized by taxonomic origin containing regions, domains or residues from different species, e.g. chimeric, humanized or veneered
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/56—Immunoglobulins specific features characterized by immunoglobulin fragments variable (Fv) region, i.e. VH and/or VL
- C07K2317/565—Complementarity determining region [CDR]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/73—Inducing cell death, e.g. apoptosis, necrosis or inhibition of cell proliferation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/90—Immunoglobulins specific features characterized by (pharmaco)kinetic aspects or by stability of the immunoglobulin
- C07K2317/92—Affinity (KD), association rate (Ka), dissociation rate (Kd) or EC50 value
Definitions
- the invention provides murine, chimeric, and humanized antibodies that specifically bind to CD48 and conjugates thereof.
- CD48 antigen (Cluster of Differentiation 48) is also known as B-lymphocyte activation marker (BLAST-1) or signaling lymphocytic activation molecule 2 (SLAMF2).
- BLAST-1 B-lymphocyte activation marker
- SLAMF2 signaling lymphocytic activation molecule 2
- CD48 is a member of the CD2 subfamily of the immunoglobulin superfamily (IgSF) which includes SLAM (signaling lymphocyte activation molecules) proteins, such as CD84, CD 150, CD229 and CD244.
- CD48 is found on the surface of lymphocytes and other immune cells, and dendritic cells, and participates in activation and differentiation pathways in these cells.
- CD48 is known to be expressed on multiple myeloma cells and other cancers of B cell origin, e.g., non-Hodgkins lymphoma (NHL), Chronic lymphocytic leukemia (CLL), Monoclonal Gammopathy of NHL.
- NHL non-Hodgkins lymphoma
- CLL Chronic lymphocytic leukemia
- MGUS Waldenstrom's Macroglobulinemia
- WM Primary/Systemic Amyloidosis patient tumor cells
- FL follicular lymphoma
- this disclosure provides a chimeric or humanized antibody that specifically binds to the human CD48 protein.
- the antibody includes heavy chain CDR sequences of SEQ ID NOs:3-5 and light chain CDR sequences of SEQ ID NOs:6-8.
- the antibody exhibits higher binding affinity to the human CD48 protein, as compared to a murine antibody that specifically binds to the human CD48 protein and also includes the heavy chain CDR sequences of SEQ ID NOs:3-5 and light chain CDR sequences of SEQ ID NOs:6-8.
- the chimeric or humanized antibody exhibits at least 2-fold higher binding affinity for the human CD48 protein, as compared to the murine antibody.
- the antibody is a humanized antibody.
- the antibody includes the heavy chain variable region of SEQ ID NO: l. In another embodiment, the antibody includes the light chain variable region of SEQ ID NO:2. In a further embodiment, the antibody includes the heavy chain variable region of SEQ ID NO: l and the light chain variable region of SEQ ID NO:2. In one embodiment, the antibody is conjugated to a cytotoxic drug attached to a linker.
- the drug-linker attached to the antibody has the formula:
- Z represents an organic moiety having a reactive site capable of reacting with a functional group on the antibody to form a covalent attachment thereto, n ranges from 8 to 36, R PR is hydrogen or a protecting group, R 21 is a capping unit for the polyethylene glycol moiety.
- this disclosure provides a humanized antibody that specifically binds to the human CD48 protein, that includes a heavy chain variable region of SEQ ID NO: l and a light chain variable region of SEQ ID NO:2.
- the antibody in one embodiment, is conjugated to a cytotoxic drug attached to a linker.
- An exemplary drug-linker has the formula:
- Z represents an organic moiety having a reactive site capable of reacting with a functional group on the antibody to form a covalent attachment thereto, n ranges from 8 to 36, R PR is hydrogen or a protecting group, R 21 is a capping unit for the polyethylene glycol moiety.
- Another exemplary drug-linker for conjugation to the disclosed antibodies has the formula:
- Z represents an organic moiety having a reactive site capable of reacting with a functional group on the antibody to form a covalent attachment thereto, n ranges from 8 to 36, R PR is hydrogen or a protecting group, R 21 is a capping unit for the polyethylene glycol moiety.
- Another exemplary drug-linker for conjugation to the disclosed antibodies has the formula
- n ranges from 8 to 36
- R is hydrogen or a protecting group
- R 21 is a capping unit for the polyethylene glycol moiety.
- Another exemplary drug-linker for conjugation to the disclosed antibodies has the formula
- n ranges from 8 to 36
- R is hydrogen or a protecting group
- R 21 is a capping unit for the polyethylene glycol moiety.
- the value n can range from 8 to 14.
- the value n ranges from 10 to 12. In a further embodiment of this disclosure, the value of n is 12. In another embodiment, R 21 is -C3 ⁇ 4 or -CH 2 CH 2 CO 2 H.
- this disclosure provides an anti-CD48 antibody-drug conjugate compound having the formula
- Z represents an organic moiety linking the antibody and the remainder of the drug-linker via covalent bonds
- n ranges from 8 to
- R 21 is a capping unit for the polyethylene glycol moiety, and p is from 1 to 16.
- the antibody can be any of the disclosed anti-CD48 antibodies.
- the antibody has the heavy chain variable region of SEQ ID NO: l and the light chain variable region of SEQ ID NO:2.
- Another exemplary drug-linker for conjugation to the disclosed antibodies has the formula:
- Another exemplary drug-linker for conjugation to the disclosed antibodies has the following properties:
- R is hydrogen or a protecting group.
- Another exemplary drug-linker for conjugation to the disclosed antibodies has the formula:
- Another exemplary drug-linker for conjugation to the disclosed antibodies has the following properties:
- the value n can range from 8 to 14. In other embodiment of this disclosure, the value n ranges from 10 to 12. In a further embodiment of this disclosure, the value of n is 12. In another embodiment, R 21 is -CH 3 or -CH 2 CH 2 CO 2 H. [0017] In another embodiment, any of the disclosed antibody-drug conjugates has a p value of 8. In another embodiment, the drug-linker is attached to the antibody via the cysteine residues of the interchain disulfide bonds of the antibody.
- the antibody-drug conjugate composition includes a population of anti-CD48 antibody-drug conjugate molecules with an average drug load of 8 and with the predominant drug load in the composition being 8.
- compositions [0019] In another aspect this disclosure provides pharmaceutical compositions and
- formulations that include the CD48 antibody-drug conjugate disclosed herein.
- the CD48 antibody-drug conjugates are used to treat patients with a cancer that expresses CD48.
- the CD48 expressing cancer in one embodiment, is multiple myeloma.
- the CD48 expressing cancer is a B cell malignancy, e.g., non- hodgkins lymphoma, follicular lymphoma, mantle cell lymphoma, Monoclonal Gammopathy of Unknown Significance (MGUS), Waldenstrom's Macroglobulinemia (WM), Primary/Systemic Amyloidosis patient tumor cells, and chronic lymphocytic leukemia.
- Another example of a CD48 expressing cancer that can be treated using the methods disclosed herein is acute myelogenous leukemia.
- the term "monoclonal antibody” as used herein refers to an antibody obtained from a population of substantially homogeneous antibodies, i.e., the individual antibodies comprising the population are identical except for possible naturally occurring mutations that may be present in minor amounts.
- the modifier "monoclonal” indicates the character of the antibody as being obtained from a substantially homogeneous population of antibodies, and is not to be construed as requiring production of the antibody by any particular method.
- the monoclonal antibodies to be used in accordance with the present invention may be made by the hybridoma method first described by Kohler et al. (1975) Nature 256:495, or may be made by recombinant DNA methods (see, for example, U.S. Patent No.
- the "monoclonal antibodies” may also be isolated from phage antibody libraries using the techniques described in Clackson et al. (1991) Nature, 352:624-628 and Marks et al. (1991) J. Mol. Biol, 222:581-597, for example or may be made by other methods.
- the antibodies described herein are monoclonal antibodies.
- Antibodies are typically provided in isolated form. This means that an antibody is typically at least 50% w/w pure of interfering proteins and other contaminants arising from its production or purification but does not exclude the possibility that the antibody is combined with an excess of pharmaceutical acceptable carrier(s) or other vehicle intended to facilitate its use. Sometimes antibodies are at least 60%, 70%, 80%, 90%, 95 or 99% w/w pure of interfering proteins and contaminants from production or purification.
- Antibodies, including isolated antibodies can be conjugated to cytotoxic agents and provided as antibody drug conjugates.
- An "isolated" polynucleotdie refers to a polynucleotide that has been identified and separated and/or recovered from components of its natural.
- Specific binding of a monoclonal antibody to its target antigen means an affinity of at least 10 6 , 10 7 , 10 s , 10 9 , or 10 10 M "1 . Specific binding is detectably higher in magnitude and distinguishable from non-specific binding occurring to at least one unrelated target. Specific binding can be the result of formation of bonds between particular functional groups or particular spatial fit (e.g., lock and key type) whereas nonspecific binding is usually the result of van der Waals forces.
- the CD48 directed antibody-drug conjugates and anti-CD48 antibodies specifically bind to CD48.
- the basic antibody structural unit is a tetramer of subunits.
- Each tetramer includes two identical pairs of polypeptide chains, each pair having one "light” (about 25 kDa) and one "heavy" chain (about 50-70 kDa).
- the amino-terminal portion of each chain includes a variable region of about 100 to 110 or more amino acids primarily responsible for antigen recognition. This variable region is initially expressed linked to a cleavable signal peptide.
- the variable region without the signal peptide is sometimes referred to as a mature variable region.
- a light chain mature variable region means a light chain variable region without the light chain signal peptide.
- Light chains are classified as either kappa or lambda.
- Heavy chains are classified as gamma, mu, alpha, delta, or epsilon, and define the antibody's isotype as IgG, IgM, IgA, IgD and IgE, respectively.
- the subscript and constant regions are joined by a "J" region of about 12 or more amino acids, with the heavy chain also including a "D” region of about 10 or more amino acids.
- variable regions of each light/heavy chain pair form the antibody binding site.
- the chains all exhibit the same general structure of relatively conserved framework regions (FR) joined by three hypervariable regions, also called complementarity determining regions or CDRs.
- the CDRs from the two chains of each pair are aligned by the framework regions, enabling binding to a specific epitope.
- FRl relatively conserved framework regions
- CDRs complementarity determining regions
- Kabat Kabat, Sequences of Proteins of Immunological Interest (National Institutes of Health, Bethesda, MD, 1987 and 1991), or Chothia & Lesk, J. Mol. Biol. 196:901-917 (1987); Chothia et al., Nature 342:878-883 (1989).
- Kabat also provides a widely used numbering convention (Kabat numbering system) in which corresponding residues between different heavy chain variable regions or between different light chain variable regions are assigned the same number. Numbering of the heavy chain constant region is via the EU index as set forth in Kabat (Kabat, Sequences of Proteins of Immunological Interest (National Institutes of Health, Bethesda, MD, 1987 and 1991).
- antibody includes intact antibodies and antigen binding fragments thereof.
- An "intact antibody” is one which comprises an antigen-binding variable region as well as a light chain constant domain (C L ) and heavy chain constant domains, C H I, C H 2, C H 3 and C H 4, as appropriate for the antibody class.
- the constant domains may be native sequence constant domains (e.g., human native sequence constant domains) or amino acid sequence variant thereof.
- Antibody fragments compete with the intact antibody from which they were derived for specific binding to the target including separate heavy chains, light chains Fab, Fab', F(ab') 2 , F(ab)c, diabodies, Dabs, nanobodies, and Fv. Fragments can be produced by recombinant DNA techniques, or by enzymatic or chemical separation of intact immunoglobulins.
- the term “intact antibody” is one which comprises an antigen-binding variable region as well as a light chain constant domain (C L ) and heavy chain constant domains, C H I
- antibody also includes a diabody (homodimeric Fv fragment) or a minibody (V L -V H -C H 3), a bispecific antibody or the like.
- a bispecific or bifunctional antibody is an artificial hybrid antibody having two different heavy/light chain pairs and two different binding sites (see, e.g., Songsivilai and Lachmann, Clin. Exp. Immunol., 79:315-321 (1990); Kostelny et al., J.
- the term "patient” includes human and other mammalian subjects that receive either prophylactic or therapeutic treatment.
- amino acids are grouped as follows: Group I (hydrophobic side chains): met, ala, val, leu, ile; Group II (neutral hydrophilic side chains): cys, ser, thr; Group III (acidic side chains): asp, glu; Group IV (basic side chains): asn, gin, his, lys, arg; Group V (residues influencing chain orientation): gly, pro; and Group VI (aromatic side chains): trp, tyr, phe. Conservative substitutions involve substitutions between amino acids in the same class. Non- conservative substitutions constitute exchanging a member of one of these classes for a member of another.
- Percentage sequence identities are determined with antibody sequences maximally aligned by the Kabat numbering convention. After alignment, if a subject antibody region (e.g., the entire mature variable region of a heavy or light chain) is being compared with the same region of a reference antibody, the percentage sequence identity between the subject and reference antibody regions is the number of positions occupied by the same amino acid in both the subject and reference antibody region divided by the total number of aligned positions of the two regions, with gaps not counted, multiplied by 100 to convert to percentage.
- a subject antibody region e.g., the entire mature variable region of a heavy or light chain
- compositions or methods "comprising" one or more recited elements may include other elements not specifically recited.
- a composition that comprises antibody may contain the antibody alone or in combination with other ingredients.
- a therapeutically effective amount of the conjugate refers to an amount of the antibody-drug conjugate that is effective to treat a disease or disorder in a mammal.
- a therapeutically effective amount of the conjugate may reduce the number of cancer cells; reduce the tumor size; inhibit (i.e., slow to some extent and preferably stop) cancer cell infiltration into peripheral organs; inhibit (i.e., slow to some extent and preferably stop) tumor metastasis; inhibit tumor growth; and/or relieve one or more of the symptoms associated with the cancer.
- efficacy can, for example, be measured by assessing the time to disease progression (TTP) and/or determining the response rate (RR).
- “effective regimen” refers to a combination of amount of the conjugate being administered and dosage frequency adequate to accomplish treatment of the disorder.
- treat refers to therapeutic treatment wherein the object is to inhibit or slow down (lessen) an undesired physiological change or disorder, such as the development or spread of cancer.
- beneficial or desired clinical results include, but are not limited to, alleviation of symptoms, diminishment of extent of disease, a stabilized (i.e., not worsening) state of disease, delay or slowing of disease progression, amelioration or palliation of the disease state, and remission (whether partial or complete), whether detectable or undetectable.
- Treatment can also mean prolonging survival as compared to expected survival if not receiving treatment.
- Those in need of treatment include those with detectable disease.
- Those in need of treatment can also include those with those with
- undetectable disease e.g., patients that have achieved a complete response after treatment for the CD48 expressing disorder but are in need of therapy in order to prevent relapse.
- pharmaceutically acceptable means approved or approvable by a regulatory agency of the Federal or a state government or listed in the U.S. Pharmacopeia or other generally recognized pharmacopeia for use in animals, and more particularly in humans.
- pharmaceutically compatible ingredient refers to a pharmaceutically acceptable diluent, adjuvant, excipient, or vehicle with which an anti-CD48 antibody or antibody-drug conjugate is administered to a subject.
- phrases "pharmaceutically acceptable salt,” refers to pharmaceutically acceptable organic or inorganic salts.
- Exemplary salts include sulfate, citrate, acetate, oxalate, chloride, bromide, iodide, nitrate, bisulfate, phosphate, acid phosphate, isonicotinate, lactate, salicylate, acid citrate, tartrate, oleate, tannate, pantothenate, bitartrate, ascorbate, succinate, maleate, gentisinate, fumarate, gluconate, glucuronate, saccharate, formate, benzoate, glutamate, methanesulfonate, ethanesulfonate, benzenesulfonate, p toluenesulfonate, and pamoate (i.e., 1,1 ' methylene bis -(2 hydroxy 3 naphthoate)) salts.
- a pharmaceutically acceptable salt may involve the inclusion of another molecule such as an acetate ion, a succinate ion or other counterion.
- the counterion may be any organic or inorganic moiety that stabilizes the charge on the parent compound.
- a pharmaceutically acceptable salt may have more than one charged atom in its structure. Instances where multiple charged atoms are part of the pharmaceutically acceptable salt can have multiple counter ions. Hence, a pharmaceutically acceptable salt can have one or more charged atoms and/or one or more counterion.
- Solvates in the context of the invention are those forms of the compounds of the invention that form a complex in the solid or liquid state through coordination with solvent molecules. Hydrates are one specific form of solvates, in which the coordination takes place with water. Preferred solvates in the context of the present invention are hydrates.
- Figure 1 shows the amino acid sequences for the heavy chain variable region of the murine MEM 102 antibody and the humanized vHA, vHB, and vHC heavy chain and selected human germline acceptor variable region sequences.
- Figure 2 shows the amino acid sequences for the light chain variable region of the murine MEM 102 antibody and the humanized vLA, vLB,and vLC light chain and selected human germline acceptor variable region sequences.
- Figure 3 shows saturation binding curves for MEM102 antibodies on human U-266 multiple myeloma cells.
- Figure 4 shows saturation binding curves for MEM102 antibodies on CHO cells transfected with cynomolgous monkey CD48.
- Figure 5 shows caspase 3/7 activation (apoptotic cell death) in human NCI-H929 multiple myeloma cells after treatment with MEM-102 ADCs.
- Figure 6 shows caspase 3/7 activation in human U-266 multiple myeloma cells after treatment with MEM-102 ADCs.
- Figure 7 shows in vivo activity of hMEM102 ADCs in a mouse xenograft model implanted with NCI-H929 cells. This is a disseminated model of multiple myeloma.
- Figure 8 shows in vivo activity of hMEM102 ADCs in a mouse xenograft model implanted with NCI-H929 cells. This is a subcutaneous model of multiple myeloma.
- Figure 9 shows in vivo activity of hMEM102 ADCs in a mouse xenograft model implanted with MM.1R cells. This is a disseminated model of multiple myeloma.
- Figure 10 shows in vivo activity of hMEM102 ADCs in a mouse xenograft model implanted with MM.1R cells. This is a subcutaneous model of multiple myeloma.
- Figure 11 shows in vivo activity of hMEM102 ADCs in a mouse xenograft model implanted with EJM cells. This is a disseminated model of multiple myeloma.
- the present invention is based, in part, on the discovery that antibody-drug conjugates, including pegylated-MMAE antibody-drug conjugates targeted to CD48 are particularly effective at killing CD48+ expressing cells.
- a high affinity MEM102 humanized antibody could be constructed using as the heavy chain variable region acceptor sequence the hlgG VH7-4-l/hIgG-JH5 heavy chain variable region human germline.
- a preferred acceptor sequence is the hIgG-VK6-21/hIgG-JK4 light chain variable region human germline.
- the high affinity MEM102 humanized antibody was constructed without the need for performing affinity maturation and while retaining the identity of the CDRs of the murine antibody.
- the high affinity MEM 102 humanized antibody was also effective at drug delivery as part of an antibody drug conjugate.
- the resultant hMEM102-MDpr- PEG(12)-gluc-MMAE 8-load conjugate was highly active against a panel of multiple myeloma cell lines.
- CD48 refers to human CD48.
- An exemplary human sequence is assigned GenBank accession number CAG33293.1.
- a humanized antibody is a genetically engineered antibody in which the CDRs from a non-human "donor” antibody are grafted into human "acceptor” antibody sequences (see, e.g., Queen, US 5,530,101 and 5,585,089; Winter, US 5,225,539; Carter, US 6,407,213; Adair, US 5,859,205; and Foote, US 6,881,557).
- the acceptor antibody sequences can be, for example, a mature human antibody sequence, a composite of such sequences, a consensus sequence of human antibody sequences, or a germline region sequence.
- a humanized antibody is an antibody having some or all CDRs entirely or substantially from a non-human donor antibody and variable region framework sequences and constant regions, if present, entirely or substantially from human antibody sequences.
- a humanized heavy chain has at least one, two and usually all three CDRs entirely or
- a humanized light chain has at least one, two and usually all three CDRs entirely or substantially from a donor antibody light chain, and a light chain variable region framework sequence and light chain constant region, if present, substantially from human light chain variable region framework and constant region sequences.
- a humanized antibody typically comprises a humanized heavy chain and a humanized light chain.
- a CDR in a humanized or human antibody is substantially from or substantially identical to a corresponding CDR in a non-human antibody when at least 60%, 85%, 90%, 95% or 100% of corresponding residues (as defined by Kabat) are identical between the respective CDRs.
- a CDR in a humanized antibody or human antibody is substantially from or substantially identical to a corresponding CDR in a non-human antibody when there are no more than 3 conservative amino acid substitutions in each CDR.
- the variable region framework sequences of an antibody chain or the constant region of an antibody chain are substantially from a human variable region framework sequence or human constant region respectively when at least 70%, 80%, 85%, 90%, 95% or 100% of corresponding residues defined by Kabat are identical.
- humanized antibodies often incorporate all six CDRs (preferably as defined by Kabat) from a mouse antibody, they can also be made with less than all CDRs (e.g., at least 3, 4, or 5) CDRs from a mouse antibody (e.g., Pascalis et al., J. Immunol. 169:3076, 2002; Vajdos et al., Journal of Molecular Biology, 320: 415-428, 2002; Iwahashi et al., Mol. Immunol.
- Certain amino acids from the human variable region framework residues can be selected for substitution based on their possible influence on CDR conformation and/or binding to antigen. Investigation of such possible influences is by modeling, examination of the characteristics of the amino acids at particular locations, or empirical observation of the effects of substitution or mutagenesis of particular amino acids.
- the invention provides antibodies directed against the CD48 antigen.
- Preferred antibodies are chimeric or humanized antibodies derived from the murine MEM102 antibody.
- a preferred acceptor sequence for the heavy chain variable region is the hlgG VH7-4-l/hIgG- JH5 heavy chain variable region human germline.
- a preferred acceptor sequence is the hIgG-VK6-21/hIgG-JK4 light chain variable region human germline.
- An exemplary anti-CD48 antibody is a humanized antibody that includes the heavy chain CDRs as set forth in SEQ ID NO: l and the light chain CDRs as set forth in SEQ ID NO:2 and additionally has a mature heavy chain variable region with at least 90%, 91%, 92%, 93%, 94% or 95% identity to SEQ ID NO: l and a mature light chain variable region with at least 90%, 91%, 92%, 93%, 94% or 95% identity to SEQ ID NO:2.
- the CDRs are as defined by Kabat.
- Humanized forms of the mouse MEM 102 antibody include three exemplified humanized heavy chain mature variable regions (HA-HC) and three exemplified humanized light chain mature variable regions (LA-LC).
- the permutations of these chains include HALA, HALB, HALC, HBLA, HBLB, HBLC, HCLA, HCLB, and HCLC. Of these permutations, HALA is preferred.
- HALA comprises the heavy chain set forth in SEQ ID NO: l and light chain set forth in SEQ ID NO:2. Any one of HALB, HALC, HBLA, HBLB, HBLC, HCLA, HCLB, and HCLC can be used, however, in place of HALA.
- the apparent dissociation constant (kd) of the humanized MEM 102 antibodies for human CD48 is preferably within a range of 0.1 nM to 10 nM, even more preferably within a range of 0.1 nM to 5 nM, even preferably within a range of 1 nM to 3 nM or 2 nM to about 3 nM.
- the antibodies of the present invention have an apparent dissociation constant within a range of 0.1 to 2.0 times, or even 0.5 to 4 times that of the apparent dissociation constant of the murine MEM102 antibody for human CD48.
- the apparent dissociation constant (kd) of the antibodies for human CD48 is about 5.0.
- Heavy and light chain variable regions of humanized MEM 102 antibodies can be linked to at least a portion of a human constant region.
- the choice of constant region can depend, in part, whether antibody-dependent cell-mediated cytotoxicity, antibody dependent cellular phagocytosis and/or complement dependent cytotoxicity are desired.
- human isotopes IgGl and IgG3 have strong complement-dependent cytotoxicity
- human isotype IgG2 has weak complement-dependent cytotoxicity
- human IgG4 lacks complement-dependent cytotoxicity.
- Human IgGl and IgG3 also induce stronger cell mediated effector functions than human IgG2 and IgG4.
- Light chain constant regions can be lambda or kappa.
- Antibodies can be expressed as tetramers containing two light and two heavy chains, as separate heavy chains, light chains, as Fab, Fab', F(ab')2, and Fv, or as single chain antibodies in which heavy and light chain subscript domains are linked through a spacer.
- Human constant regions show allotypic variation and isoallotypic variation between different individuals, that is, the constant regions can differ in different individuals at one or more polymorphic positions.
- Isoallotypes differ from allotypes in that sera recognizing an isoallotype binds to a non-polymorphic region of a one or more other isotypes.
- One or several amino acids at the amino or carboxy terminus of the light and/or heavy chain may be missing or derivatized in a proportion or all of the molecules. Substitutions can be made in the constant regions to reduce or increase effector function such as complement-mediated cytotoxicity or ADCC (see, e.g., Winter et al., US Patent No. 5,624,821; Tso et al., US Patent No. 5,834,597; and Lazar et al., Proc. Natl. Acad. Sci. USA 103:4005, 2006), or to prolong half-life in humans (see, e.g., Hinton et al., J. Biol.
- the constant region can be modified to allow for site specific conjugation of a drug- linker.
- Such techniques include the use of naturally occurring or engineered cysteine residues, disulfide bridges, poly-histidine sequences, glycoengineering tags, and transglutaminase recognition sequences.
- An exemplary substitution for site specific conjugation using bacterial transglutaminase is N297S or N297Q.
- An exemplary substitution for site specific conjugation using an engineered cysteine is S239C.
- Antibody fragments can also be modified for site- specific conjugation of a drug-linker, see for example, Kim et al., Mol Cancer Ther 2008;7(8).
- Humanized or chimeric MEM 102 antibodies can be produced by recombinant expression.
- Recombinant polynucleotide constructs typically include an expression control sequence operably linked to the coding sequences of antibody chains, including naturally- associated or heterologous promoter regions.
- the expression control sequences are eukaryotic promoter systems in vectors capable of transforming or transfecting eukaryotic host cells. Once the vector has been incorporated into the appropriate host, the host is maintained under conditions suitable for high level expression of the nucleotide sequences, and the collection and purification of the crossreacting antibodies.
- Mammalian cells are a preferred host for expressing nucleotide segments encoding immunoglobulins or fragments thereof. See Winnacker, From Genes to Clones, (VCH
- suitable host cell lines capable of secreting intact heterologous proteins include CHO cell lines (e.g., DG44), various COS cell lines, HeLa cells, HEK293 cells, L cells, and non- antibody-producing myelomas including Sp2/0 and NS0.
- the cells are nonhuman.
- Expression vectors for these cells can include expression control sequences, such as an origin of replication, a promoter, an enhancer (Queen et al., Immunol. Rev. 89:49 (1986)), and necessary processing information sites, such as ribosome binding sites, RNA splice sites, polyadenylation sites, and transcriptional terminator sequences.
- Preferred expression control sequences are promoters derived from endogenous genes, cytomegalovirus, SV40, adenovirus, bovine papillomavirus, and the like. See Co et al., J. Immunol. 148: 1149 (1992). [0064] Once expressed, antibodies can be purified according to standard procedures of the art, including HPLC purification, column chromatography, gel electrophoresis and the like (see generally, Scopes, Protein Purification (Springer- Verlag, NY, 1982)).
- the invention further provides nucleic acids encoding any of the humanized heavy and light chains described herein.
- the nucleic acids also encode a signal peptide fused to the mature heavy and light chain variable regions. Coding sequences on nucleic acids can be in operable linkage with regulatory sequences to ensure expression of the coding sequences, such as a promoter, enhancer, ribosome binding site, transcription termination signal and the like.
- the nucleic acids encoding heavy and light chains can occur in isolated form or can be cloned into one or more vectors.
- the nucleic acids can be synthesized by for example, solid state synthesis or PCR of overlapping oligonucleotides. Nucleic acids encoding heavy and light chains can be joined as one contiguous nucleic acid, e.g., within an expression vector, or can be separate, e.g., each cloned into its own expression vector.
- this disclosure provides an isolated polynucleotide encoding an antibody heavy chain variable region comprising the amino acid sequence as set forth in HA, HB, or HC.
- the isolated polynucleotide can encode an antibody heavy chain variable region comprising the amino acid sequence of SEQ ID NO: l.
- polynucleotide can further encode a human IgG heavy chain constant region.
- the isotype of the IgG constant region is, e.g., IgGl, IgG2, IgG3, or IgG4.
- the isotype of the IgG constant region is IgGl.
- the encoded IgGl constant region has an amino acid sequence comprising a substitution at residue 239, according to the EU index as set forth in Kabat system, i.e., S239C.
- the disclosure also provides an expression vector comprising the isolated polynucleotide encoding the antibody heavy chain variable region comprising the amino acid sequence as set forth in HA, HB, or HC (e.g., SEQ ID NO: l or variants thereof), and further, a host cell comprising that expression vector.
- the host cell is a mammalian host cell, e.g., a CHO cell.
- this disclosure provides an isolated polynucleotide encoding an antibody light chain variable region comprising the amino acid sequence as set forth in LA, LB or LC.
- an isolated polynucleotide encoding an antibody light chain variable region comprising the amino acid sequence of SEQ ID NO:2.
- This isolated polynucleotide can further encode a human IgG light chain constant region.
- the isotype of the IgG light chain constant region is, e.g., a kappa constant region.
- the disclosure also provides an expression vector comprising the isolated polynucleotide encoding the antibody light chain variable region comprising the amino acid sequence as set forth in LA or LB or LC (e.g., SEQ ID NO:2 or variants thereof), and further, a host cell comprising that expression vector.
- the host cell is a mammalian host cell, e.g., a CHO cell.
- this disclosure provides an isolated polynucleotide or polynucleotides encoding an antibody heavy chain variable region comprising the amino acid sequence of SEQ ID NO: l and an antibody light chain variable region comprising the amino acid sequence of SEQ ID NO:2, the heavy chain variable domain and the light chain variable domain forming an antibody or antigen binding fragment that specifically binds to human CD48.
- This disclosure also provides an expression vector comprising the isolated polynucleotide or polynucleotides the encode the antibody heavy chain variable region comprising the amino acid sequence of SEQ ID NO: l and the antibody light chain variable region comprising the amino acid sequence of SEQ ID NO:2 .
- a host cell comprising the expression vector or vectors is also provided.
- the host cell is preferably a mammalian cell, e.g., a CHO cell.
- this disclosure provides first and second vectors comprising a polynucleotide encoding an antibody heavy chain variable region comprising the amino acid sequence of SEQ ID NO: l and a polynucleotide encoding an antibody light chain variable region comprising the amino acid sequence of SEQ ID NO:2, the heavy chain variable domain and the light chain variable domain forming an antibody or antigen binding fragment that specifically binds to human CD48.
- Host cell comprising the vectors are provided, preferably mammalian host cells, such as a CHO cell.
- Anti-CD48 antibodies can be conjugated to therapeutic agents, diagnostic agents or stabilizing agents to form antibody conjugates.
- Anti-CD48 antibodies conjugated to therapeutic agents are referred to herein as antibody-drug conjugates (ADCs).
- Exemplary therapeutic agents have cytostatic or cytotoxic effect and can also be referred to as cytotoxic agents or cytostatic agents.
- cytotoxic agents include, for example, auristatins, camptothecins, calicheamicins, duocarmycins, etoposides, maytansinoids (e.g., DM1, DM2, DM3, DM4) , taxanes, benzodiazepines (e.g., pyrrolo[l,4]benzodiazepines, indolinobenzodiazepines, and oxazolidinobenzodiazepines including pyrrolo[l,4]benzodiazepine dimers,
- the therapeutic agent is conjugated to the antibody via a linker unit.
- the linker unit can be cleavable or non-cleavable.
- the therapeutic agent can be attached to the antibody with a cleavable linker that is sensitive to cleavage in the intracellular environment of the CD48-expressing cancer cell but is not substantially sensitive to the extracellular environment, such that the conjugate is cleaved from the antibody when it is internalized by the CD48-expressing cancer cell (e.g., in the endosomal, lysosomal environment, or in the caveolear environment).
- the therapeutic agent can be conjugated to the antibody via a non-cleavable linker and drug release is by total antibody degradation following internalization by the CD48-expressing cancer cell.
- the ADC will comprise a linker region between the cytotoxic or cytostatic agent and the anti-CD48 antibody.
- the linker can be cleavable under intracellular conditions, such that cleavage of the linker releases the therapeutic agent from the antibody in the intracellular environment (e.g., within a lysosome or endosome or caveolea).
- the linker can be, e.g., a peptidyl linker that is cleaved by an intracellular peptidase or protease enzyme, including a lysosomal or endosomal protease.
- Cleaving agents can include cathepsins B and D and plasmin (see, e.g., Dubowchik and Walker, Pharm. Therapeutics 83:67-123, 1999).
- Most typical are peptidyl linkers that are cleavable by enzymes that are present in CD48- expressing cells.
- a peptidyl linker that is cleavable by the thiol-dependent protease cathepsin-B, which is highly expressed in cancerous tissue can be used (e.g., a linker comprising a Phe-Leu or a Val-Cit peptide).
- the linker can also be a carbohydrate linker, including a sugar linker that is cleaved by an intracellular glycosidase (e.g., a glucuronide linker cleavable by a glucuronidase).
- an intracellular glycosidase e.g., a glucuronide linker cleavable by a glucuronidase
- the linker also can be a non-cleavable linker, such as an maleimido-alkylene- or maleimide-aryl linker that is directly attached to the therapeutic agent and released by proteolytic degradation of the antibody.
- a non-cleavable linker such as an maleimido-alkylene- or maleimide-aryl linker that is directly attached to the therapeutic agent and released by proteolytic degradation of the antibody.
- the anti-CD48 antibody can be conjugated to the linker via a heteroatom of the antibody. These heteroatoms can be present on the antibody in its natural state or can be introduced into the antibody. In some aspects, the anti-CD48 antibody will be conjugated to the linker via a nitrogen atom of a lysine residue. In other aspects, the anti-CD48 antibody will be conjugated to the linker via a sulfur atom of a cysteine residue. The cysteine residue can be naturally-occurring or one that is engineered into the antibody. Methods of conjugating linkers and drug-linkers to antibodies via lysine and cysteine residues are known in the art.
- Exemplary antibody-drug conjugates include auristatin based antibody-drug conjugates (i.e., the drug component is an auristatin drug).
- Auristatins bind tubulin, have been shown to interfere with microtubule dynamics and nuclear and cellular division, and have anticancer activity.
- the auristatin based antibody-drug conjugate comprises a linker between the auristatin drug and the anti-CD48 antibody.
- the linker can be, for example, a cleavable linker (e.g., a peptidyl linker, a carbohydrate linker) or a non-cleavable linker (e.g., linker released by degradation of the antibody).
- Auristatins include MMAF, and MMAE.
- the synthesis and structure of exemplary auristatins are described in U.S. Publication Nos. 7,659,241, 7,498,298, 2009-0111756, 2009-0018086, and 7,968, 687 each of which is incorporated herein by reference in its entirety and for all purposes.
- exemplary antibody-drug conjugates include maytansinoid antibody-drug conjugates (i.e., the drug component is a maytansinoid drug), and benzodiazepine antibody drug conjugates (i.e., the drug component is a benzodiazepine (e.g., pyrrolo[l,4]benzodiazepine dimers (PBD dimer), indolinobenzodiazepine dimers, and oxazolidinobenzodiazepine dimers)).
- PBD dimer pyrrolo[l,4]benzodiazepine dimers
- indolinobenzodiazepine dimers e.g., indolinobenzodiazepine dimers
- oxazolidinobenzodiazepine dimers e.g., pyrrolo[l,4]benzodiazepine dimers (PBD dimer), indolinobenzodiazepine dimers, and oxazolidinobenzodiazepine dimers
- a preferred PBD dimer for use in the present invention is represented by formula I.
- the preferred stereochemistry of the PBD dimer is as shown in formula la:
- n 1 or 3.
- Solvates of formula (I) and (la) are typically formed from addition of water or alcoholic solvent across the imine functional group of one or both PBD monomers to form
- R 10 is H, and R 11 is OH or OR A , where R A is saturated C 1-4 alkyl (preferably methyl); or (b) R and R form a nitrogen-carbon double bond between the nitrogen and carbon atoms to which they are bound; or
- R 10 is H, and R 11 is OH or OR A , where R A is saturated C 1-4 alkyl (preferably methyl); and the other of R 10 and R 11 form a nitrogen-carbon double bond between the nitrogen and carbon atoms to which they are bound.
- the PBD dimer of formula I or la is typically linked to the antibody via a Linker Unit, LU.
- the Linker Unit acts to release the PBD dimer of formula I or la (or a pharmaceutically salt, solvate, or solvate of the salt thereof ) at the target site (e.g., inside the cancer cell) .
- a PBD drug-linker compound for use in the present invention is represented below by formula II (preferred stereochemistry as shown in Ila) wherein LU is a Linker Unit.
- the Linker Unit can be, for example, a cleavable peptide Linker Unit (e.g., a linker comprising the valine- alanine peptide) or a cleavable disulfide Linker Unit:
- n 1 or 3.
- a preferred PBD drug-linker compound for use in the present invention is represented by Formula III below:
- the PBD drug-linker is conjugated to an anti-CD48 antibody to produce a CD48 targeted antibody-drug conjugate.
- the antibody can be conjugated to a drug-linker of formula II or formula III.
- An exemplary CD48 targeted antibody-drug conjugate is shown below in formulas IV, IVa, and IVb:
- Exemplary drug-linkers include MMAE drug-linkers.
- the present inventors have found that the incorporation of a polyethylene glycol polymer as a side chain into a cleavable ⁇ - glucuronide MMAE drug-linker provides antibody drug-conjugates with descreased plasma clearance and increased antitumor activity in xenograft models as compared to a non-PEGylated control. Accordingly, particularly advantageous drug-linkers for attachment to the antibodies of the present invention are as follows:
- Z represents an organic moiety having a reactive site capable of reacting with a functional group on the antibody to form a covalent attachment thereto
- n ranges from 8 to 36 and most preferably ranges from 8 to 14 (most preferably 12)
- R 21 is a capping unit for the polyethylene glycol moiety, preferably- CH 3 or-CH 2 CH 2 C0 2 H.
- a preferred Z moiety is a maleimido-containing moiety. Particularly preferred Z moieities are shown in the drug-linkers below:
- n ranges from 8 to 36 and most preferably ranges from 8 to 14 (most preferably 12)
- R PR is hydrogen or a protecting group, e.g., acid labile protecting group, e.g., BOC
- R 21 is a capping unit for the polyethylene glycol moiety, preferably-CH 3 or -CH 2 CH 2 CO 2 H.
- R PR can be hydrogen or a protecting group.
- Protective groups as used herein refer to groups which selectively block, either temporarily or permanently, a reactive site in a multifunctional compound.
- a protecting group is a suitable protecting group when it is capable of preventing or avoiding unwanted side-reactions or premature loss of the protecting group under reaction conditions required to effect desired chemical transformation elsewhere in the molecule and during purification of the newly formed molecule when desired, and can be removed under conditions that do not adversely affect the structure or stereochemical integrity of that newly formed molecule.
- Suitable amine protecting groups include acid-labile nitrogen protecting groups, including those provided by Isidro-Llobel et al. "Amino acid-protecting groups" Chem. Rev. (2009) 109: 2455-2504.
- an acid-labile nitrogen-protecting group transforms a primary or secondary amino group to its corresponding carbamate and includes t- butyl, allyl, and benzyl carbamates.
- R 21 is a capping unit for the polyethylene glycol moiety.
- polyethylene glycol units can be terminally capped with a wide diversity of organic moieties, typically those that are relatively non-reactive.
- Alkyl and substituted alkyl groups are preferred, including, for example, -Ci_io alkyl, -C 2-1 o alkyl-C0 2 H, - C 2 _io alkyl-OH, -C 2 _i 0 alkyl-NH 2 , C 2 _i 0 alkyl-NH(Ci_ 3 alkyl), or C 2 _i 0 alkyl-N(Ci_ 3 alkyl) 2 .
- the subscript p represents the drug load and, depending on the context, can represent the number of molecules of drug-linker molecules attached to an individual antibody molecule and as such, is an integer value, or can represent an average drug load and, as such, can be an integer or non-integer value but is typically a non-integer value.
- An average drug load represents the average number of drug- linker molecules per antibody in a population. Often, but not always, when we refer to an antibody, e.g., a monoclonal antibody, we are referring to a population of antibody molecules.
- the average drug load is an important quality attribute as it determines the amount of drug that can be delivered to a target cell.
- the percentage of unconjugated antibody molecules in the composition is included in the average drug load value.
- the average drug load when referring to a composition comprising a population of antibody-drug conjugate compounds is from 1 to about 16, preferably about 2 to about 14, more preferably about 2 to about 10.
- a particularly preferred average drug load is about 2.
- the actual drug load for individual antibody molecules in the population of antibody-drug conjugate compounds is from 1 to 4, 1 to 3 or 1 to 2 with a predominant drug loading of 2.
- the average drug load of 2 is achieved via site specific conjugation techniques (e.g., engineered cysteines introduced to the antibody including at position 239, according to the EU Index numbering system).
- a particularly preferred average drug load is about 8.
- the drug-linkers are conjugated to the cysteine residues of the reduced inter-chain disulfides.
- the actual drug load for individual antibody molecules in the population of antibody-drug conjugate compounds is from 1 to 10 (or from 6 to 10 or from 6 to 8) with a predominant drug loading of 8.
- a higher drug load can be achieved, for example, if, in addition to the interchain disulfides, drug- linker is conjugated to introduced cysteine residues (such as a cysteine residue introduced at position 239, according to the EU index).
- Exemplary ADCs include the following:
- n ranges from 8 to 36 and most preferably ranges from 8 to 14 (most preferably 12)
- R PR is hydrogen or a protecting group, e.g., acid labile protecting group, e.g., BOC
- R is a capping unit for the polyethylene glycol moiety, preferably-CH 3 or -CH 2 CH 2 CO 2 H
- Ab represents an anti- CD48 antibody and p represents an integer ranging from 1 to 16, preferably 1 to 14, 6 to 12, 6 to 10, or 8 to 10 when referring to individual antibody molecules or to an average drug load of from about 4 or about 6 to about 14, preferably about 8 when referring to a population of antibody molecules.
- the PEG (polyethylene glycol) portion of the drug linker can range from 8 to 36, however, it has been found that a PEG of 12 ethylene oxide units is particularly preferably. It has been found that longer PEG chains can result in slower clearance whereas shorter PEG chains can result in diminished activity. Accordingly, the subscript n in all of the embodiments above is preferably 8 to 14, 8 to 12, 10 to 12 or 10 to 14 and is most preferably 12.
- Polydisperse PEGS, monodisperse PEGS and discrete PEGs can be used to make the PEGylated antibody drug conjugates of the present invention.
- Polydisperse PEGs are a heteregenous mixture of sizes and molecular weights whereas monodisperse PEGs are typically purified from heterogenous mxitures and are therefore provide a single chain length and molecular weight.
- Preferred PEG Units are discrete PEGs, compounds that are synthesized in step-wise fashion and not via a polymerization process.
- Discrete PEGs provide a single molecule with defined and specified chain length.
- the value for the subscript "n" can be an average number and can be an integer or non-integer number.
- covalent attachment of the antibody to the drug-linker is accomplished through a sulfhydryl functional group of the antibody interacting with a maleimide functional group of a drug linker to form a thio-substituted succinimide.
- the sulfhydryl functional group can be present on the Ligand Unit in the Ligand' s natural state, for example, in a naturally-occurring residue (inter-chain disulfide resides), or can be introduced into the Ligand via chemical modification or by biological engineering, or a combination of the two.
- an antibody-substituted succinimide may exist in hydrolyzed form(s).
- an ADC is comprised of a succinimide moiety that when bonded to the antibody is represented by the structure of
- the wavy line indicates linkage to the remainder of the drug-linker.
- the CD48 targeted antibody-drug conjugates described herein can be used to treat a CD48 expresssing disorder, such as CD48 expressing cancer.
- a CD48 expresssing disorder such as CD48 expressing cancer.
- cancers show detectable levels of CD48 measured at the protein (e.g., by immunoassay) or RNA level.
- Some such cancers show elevated levels of CD48 relative to noncancerous tissue of the same type, preferably from the same patient.
- a level of CD48 in a cancer is measured before performing treatment.
- cancers associated with CD48 expression include multiple myeloma, and other B cell malignancies, including Hodgkin' s disease, non-Hodgkin' s lymphoma, Follicular lymphoma, chronic lymphocytic leukemia (CLL), mantle cell lymphoma, Waldenstrom's Macroglobulinemia, Primary/Systemic Amyloidosis patient tumor cells, MGUS, and
- AML acute myeloid leukemia
- Methods of the present invention include treating a patient that has a cancer that expresses CD48 comprising administering to the patient an antibody-drug conjugate of the present invention.
- the cancer can be any CD48 expressing cancer, including, for example, multiple myeloma.
- CD48 directed antibody-drug conjugates are administered in an effective regimen meaning a dosage, route of administration and frequency of administration that delays the onset, reduces the severity, inhibits further deterioration, and/or ameliorates at least one sign or symptom of cancer.
- Exemplary dosages for CD48 directed pegylated-MMAE conjugates are generally from about 1.0 ⁇ g/kg to 10.0 mg/kg, or from about 0.1 mg/kg to 5.0 mg/kg or from about 0.5 mg/kg to 1.0, 2.0, or 4.0 ⁇ g/kg, although alternate dosages are contemplated.
- a preferred dose range is from about 0.3 mg/kg to about 2.0 mg/kg.
- Administration can be by a variety of administration routes.
- the conjugates are administered parenterally, such as intravenously, intramuscularly, or subcutaneously.
- the delivery can be into the systemic circulation by intravenous or subcutaneous administration.
- administration is via intravenous delivery.
- Intravenous administration can be, for example, by infusion over a period such as 30-90 minutes or by a single bolus injection.
- administration will be via slow IV push (i.e., over 30-60 seconds) in a peripherally inserted central catheter.
- the frequency of administration depends upon many different factors, including means of administration, target site, physiological state of the patient, whether the patient is human or an animal, and other medications administered.
- the frequency can be daily, weekly, monthly, quarterly, or at irregular intervals in response to changes in the patient's condition or progression of the cancer being treated.
- An exemplary frequency for intravenous administration is between twice a week and quarterly over a continuous course of treatment, although more or less frequent dosing is also possible.
- Other exemplary frequencies for intravenous administration are every three weeks or between once weekly or once monthly over a continuous course of treatment, although more or less frequent dosing is also possible.
- Another exemplary frequency is administration every six weeks.
- compositions for parenteral administration are preferably sterile and substantially isotonic and manufactured under GMP conditions.
- Pharmaceutical compositions can be provided in unit dosage form (i.e., the dosage for a single administration).
- compositions can be formulated using one or more physiologically acceptable carriers, diluents, excipients or auxiliaries.
- the formulation depends on the route of
- conjugates can be formulated in aqueous solutions, preferably in physiologically compatible buffers such as Hank's solution, Ringer's solution, or physiological saline or acetate buffer (to reduce discomfort at the site of injection).
- physiologically compatible buffers such as Hank's solution, Ringer's solution, or physiological saline or acetate buffer (to reduce discomfort at the site of injection).
- the solution can contain formulatory agents such as suspending, stabilizing and/or dispersing agents.
- antibodies can be in lyophilized form for constitution with a suitable vehicle, e.g., sterile pyrogen-free water, before use.
- a suitable vehicle e.g., sterile pyrogen-free water
- the concentration of conjugate in a liquid formulation can vary widely.
- the ADC is present at a concentration from about 0.5 mg/ml to about 30 mg/ml, from about 0.5 mg/ml to about 10 mg/ml, from about 1 mg/ml to about 10 mg/ml, from about 2 mg/ml to about 10 mg/ml, or from about 2 mg/ml to about 5 mg/ml.
- Treatment with conjugates of the invention can be combined with chemotherapy, radiation, stem cell treatment, surgery, and other treatments effective against the disorder being treated, including standard of care for the particular disorder being treated.
- the present invention encompasses methods of treating the disease and disorders described herein as a monotherapy or in combination therapy with, for example, standard of care or investigational drugs for treatment of such diseases and/or disorders.
- Methods for the treatment of cancer include administering to a patient in need thereof an effective amount of a CD48 directed antibody-drug conjugate of the present invention in combination with an additional anti-cancer agent or other agent to treat cancer.
- An exemplary agent for combination therapy is carfilzomib (e.g. KYPROLIS®), a proteasome inhibitor used to treat multiple myeloma (see Siegel DS et al. A phase 2 study of single-agent carfilzomib (PX-171-003-A1) in patients with relapsed and refractory multiple myeloma. Blood 2012; 120:2817-2825).
- Carfilzomib can be administered as an intravenous/IV infusion.
- carfilzomib is administered in a combination therapy with a CD48 directed antibody-drug conjugate of the present invention.
- carfilzomib is administered in a combination therapy with an hMEM102 antibody-drug conjugate of the present invention. In a further embodiment, carfilzomib is administered in a combination therapy with an hMEM102-MDpr-PEG(12)-gluc-MMAE 8-load of the present invention.
- Carfilzomib can also be administered in a combination therapy with a CD48 directed antibody-drug conjugate of the present invention and an additional agent.
- Carfilzomib has been combined with various additional agents to treat multiple myeloma.
- carfilzomib has been combined with lenalidomide and dexamethasone (see Stewart KA et al. Carfilzomib, lenalidomide, and dexamethasone for relapsed multiple myeloma. N Engl J Med. 2015; 372: 142- 152).
- carfilzomib is administered in a combination therapy with
- carfilzomib is administered in a combination therapy with lenalidomide, dexamethasone, and an hMEM102 antibody-drug conjugate of the present invention.
- carfilzomib is administered in a combination therapy with lenalidomide, dexamethasone, and an hMEM102-MDpr-PEG(12)-gluc-MMAE 8-load of the present invention.
- Carfilzomib has also been combined with dexamethasone (see Dimopoulos MD et al. Carfilzomib and dexamethasone versus bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma (ENDEAVOR): a randomised, phase 3, open-label, multicentre study. Lancet Oncology 2016; 17:27-38).
- carfilzomib is administered in a combination therapy with dexamethasone and a CD48 directed antibody-drug conjugate of the present invention.
- carfilzomib is administered in a combination therapy with dexamethasone and an hMEM102 antibody-drug conjugate of the present invention. In a further embodiment, carfilzomib is administered in a combination therapy with dexamethasone and an hMEM102-MDpr-PEG(12)-gluc-MMAE 8-load of the present invention.
- Carfilzomib has also been combined with panobinostat (see Berdeja JG et al. Phase I/II study of the combination of panobinostat and carfil omib in patients with relapsed/refractory multiple myeloma. Haematologica 2015; 100:670-676).
- carfilzomib is administered in a combination therapy with panobinostat and a CD48 directed antibody-drug conjugate of the present invention.
- carfilzomib is administered in a combination therapy with panobinostat and an hMEM102 antibody-drug conjugate of the present invention.
- carfilzomib is administered in a combination therapy with panobinostat and an hMEM102-MDpr-PEG(12)-gluc-MMAE 8-load of the present invention.
- Carfilzomib has also been combined with pomalidomide and dexamethasone (see Shah J et al. Carfilzomib, pomalidomide, and dexamethasone (CPD) in patients with relapsed and/or refractory multiple myeloma. Blood 2015; 126: 2284-2290).
- carfilzomib is administered in a combination therapy with pomalidomide, dexamethasone, and a CD48 directed antibody-drug conjugate of the present invention.
- carfilzomib is administered in a combination therapy with pomalidomide, dexamethasone, and an hMEM 102 antibody-drug conjugate of the present invention.
- carfilzomib is administered in a combination therapy with pomalidomide, dexamethasone, and an hMEM 102- MDpr-PEG(12)-gluc-MMAE 8-load of the present invention.
- daratumumab e.g. DARZALEXTM
- CD38 a glycoprotein highly expressed on multiple myeloma cells
- Daratumumab can be administered to patients by intravenous infusion to treat multiple myeloma (see Lokhorst HM et al. Targeting CD38 with daratumumab monotherapy in multiple myeloma. N Engl J Med 2015; 373: 1207-1219).
- daratumumab is administered in a combination therapy with a CD48 directed antibody-drug conjugate of the present invention.
- daratumumab is administered in a combination therapy with an hMEM 102 antibody-drug conjugate of the present invention. In a further embodiment, daratumumab is administered in a combination therapy with an hMEM102-MDpr- PEG(12)-gluc-MMAE 8-load of the present invention.
- Daratumumab can also be administered in a combination therapy with a CD48 directed antibody-drug conjugate of the present invention and an additional agent.
- Daratumumab has been combined with various additional agents to treat multiple myeloma.
- daratumumab has been combined with bortezomib and lenalidomide (see Phipps C et al.
- daratumumab and its potential in the treatment of multiple myeloma overview of the preclinical and clinical development. Ther Adv Hematol 2015; 6: 120-127).
- daratumumab is administered in a combination therapy with bortezomib, lenalidomide, and a CD48 directed antibody-drug conjugate of the present invention.
- daratumumab is administered in a combination therapy with bortezomib, lenalidomide, and an hMEM 102 antibody-drug conjugate of the present invention.
- daratumumab is administered in a combination therapy with bortezomib, lenalidomide, and an hMEM102-MDpr- PEG(12)-gluc-MMAE 8-load of the present invention.
- Daratumumab has also been combined with bortezomib and dexamethasone (see Phipps C et al.).
- daratumumab is administered in a combination therapy with bortezomib, dexamethasone, and a CD48 directed antibody-drug conjugate of the present invention.
- daratumumab is administered in a combination therapy with bortezomib, dexamethasone, and an h MEM 102 antibody-drug conjugate of the present invention.
- daratumumab is administered in a combination therapy with bortezomib, dexamethasone, and an hMEM102-MDpr-PEG(12)-gluc-MMAE 8-load of the present invention.
- elotuzumab e.g. EMPLICITITM
- SLAMF7 signaling lymphocytic activation molecule F7
- Elotuzumab can be administered to patients by intravenous infusion to treat multiple myeloma (see Zonder JA et al. A phase 1, multicenter, open-label, dose escalation study of elotuzumab in patients with advanced multiple myeloma. Blood 2012; 120: 552-559).
- elotuzumab is administered in a combination therapy with a CD48 directed antibody-drug conjugate of the present invention. In a further embodiment, elotuzumab is administered in a combination therapy with an hMEM102 antibody-drug conjugate of the present invention. In a further embodiment, elotuzumab is administered in a combination therapy with an hMEM102-MDpr-PEG(12)-gluc-MMAE 8-load of the present invention.
- Elotuzumab can also be administered in a combination therapy with a CD48 directed antibody-drug conjugate of the present invention and an additional agent.
- Elotuzumab has been combined with various additional agents to treat multiple myeloma.
- elotuzumab has been combined with lenalidomide and dexamethasone (see Lonial S et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med 2015; 373:621-631 ;).
- elotuzumab is administered in a combination therapy with lenalidomide, dexamethasone, and a CD48 directed antibody-drug conjugate of the present invention.
- elotuzumab is administered in a combination therapy with lenalidomide, dexamethasone, and an hMEM102 antibody-drug conjugate of the present invention.
- elotuzumab is administered in a combination therapy with lenalidomide, dexamethasone, and an hMEM102-MDpr-PEG(12)-gluc-MMAE 8-load of the present invention.
- lenalidomide e.g. REVLIMID®
- an immunomodulatory agent given to patients to treat multiple myeloma
- Lenalidomide can be packaged as a capsule, pill, or tablet for oral administration.
- lenalidomide is administered in a combination therapy with a CD48 directed antibody-drug conjugate of the present invention.
- lenalidomide is administered in a combination therapy with an hMEM102 antibody-drug conjugate of the present invention.
- lenalidomide is administered in a combination therapy with an hMEM102-MDpr-PEG(12)-gluc-MMAE 8-load of the present invention.
- Lenalidomide can also be administered in a combination therapy with a CD48 directed antibody-drug conjugate of the present invention and an additional agent.
- Lenalidomide has been combined with various additional agents that treat multiple myeloma.
- lenalidomide has been combined with bortezomib and dexamethasone (see Richardson PG et al.
- lenalidomide is administered in a combination therapy with bortezomib, dexamethasone, and a CD48 directed antibody-drug conjugate of the present invention.
- lenalidomide is administered in a combination therapy with bortezomib, dexamethasone, and an hMEM102 antibody-drug conjugate of the present invention.
- lenalidomide is administered in a combination therapy with bortezomib, dexamethasone, and an hMEM102- MDpr-PEG(12)-gluc-MMAE 8-load of the present invention.
- Lenalidomide has also been combined with carfilzomib and dexamethasone (see Stewart KA et al.).
- lenalidomide is administered in a combination therapy with carfilzomib, dexamethasone, and a CD48 directed antibody-drug conjugate of the present invention.
- lenalidomide is administered in a combination therapy with carfilzomib, dexamethasone, and an hMEM102 antibody-drug conjugate of the present invention.
- lenalidomide is administered in a combination therapy with carfilzomib, dexamethasone, and an hMEM102-MDpr-PEG(12)-gluc-MMAE 8-load of the present invention.
- Lenalidomide has also been combined with daratumumab and bortezomib (see Phipps C et al.). In an embodiment, lenalidomide is administered in a combination therapy with
- lenalidomide is administered in a combination therapy with daratumumab, bortezomib, and an hMEM102 antibody-drug conjugate of the present invention.
- lenalidomide is administered in a combination therapy with
- daratumumab daratumumab, bortezomib, and an hMEM102-MDpr-PEG(12)-gluc-MMAE 8-load of the present invention.
- Lenalidomide has also been combined with elotuzumab and dexamethasone (see Lonial S et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med 2015; 373:621-631).
- lenalidomide is administered in a combination therapy with elotuzumab, dexamethasone, and a CD48 directed antibody-drug conjugate of the present invention.
- lenalidomide is administered in a combination therapy with elotuzumab, dexamethasone, and an hMEM102 antibody-drug conjugate of the present invention.
- lenalidomide is administered in a combination therapy with elotuzumab, dexamethasone, and an hMEM102-MDpr-PEG(12)-gluc-MMAE 8-load of the present invention.
- bortezomib e.g. VELCADE®
- proteasome inhibitor given to patients to treat multiple myeloma and mantle cell lymphoma
- Bortezomib can be administered to patients via intravenous injection.
- bortezomib is administered in a combination therapy with a CD48 directed antibody-drug conjugate of the present invention.
- bortezomib is administered in a combination therapy with an hMEM102 antibody-drug conjugate of the present invention. In a further embodiment, bortezomib is administered in a combination therapy with an hMEM102-MDpr-PEG(12)-gluc-MMAE 8-load of the present invention.
- Bortezomib can also be administered in a combination therapy with a CD48 directed antibody-drug conjugate of the present invention and an additional agent.
- Bortezomib has been combined with various additional agents to treat multiple myeloma.
- bortezomib has been combined with thalidomide and dexamethasone (see Kapoor P et al. Bortezomib combination therapy in multiple myeloma. Semin Hematol 2012; 3:228-242).
- bortezomib is administered in a combination therapy with thalidomide, dexamethasone, and a CD48 directed antibody-drug conjugate of the present invention.
- bortezomib is administered in a combination therapy with thalidomide, dexamethasone, and an hMEM102 antibody-drug conjugate of the present invention.
- bortezomib is administered in a combination therapy with thalidomide, dexamethasone, and an hMEM102-MDpr-PEG(12)-gluc-MMAE 8-load of the present invention.
- Bortezomib has also been combined with dexamethasone, thalidomide, cisplatin, doxorubicin, cyclophosphamide, and etoposide (see Kapoor P et al.).
- bortezomib is administered in a combination therapy with dexamethasone, thalidomide, cisplatin, doxorubicin, cyclophosphamide, etoposide, and a CD48 directed antibody-drug conjugate of the present invention.
- bortezomib is administered in a combination therapy with dexamethasone, thalidomide, cisplatin, doxorubicin, cyclophosphamide, etoposide, and an hMEM102 antibody-drug conjugate of the present invention.
- bortezomib is administered in a combination therapy with dexamethasone, thalidomide, cisplatin, doxorubicin, cyclophosphamide, etoposide, and an hMEM102-MDpr-PEG(12)-gluc-MMAE 8- load of the present invention.
- Bortezomib has also been combined with daratumumab and lenalidomide (see Phipps C et al.).
- bortezomib is administered in a combination therapy with
- bortezomib is administered in a combination therapy with daratumumab, lenalidomide, and an hMEM102 antibody-drug conjugate of the present invention.
- bortezomib is administered in a combination therapy with daratumumab, lenalidomide, and an hMEM102-MDpr-PEG(12)-gluc-MMAE 8-load of the present invention.
- bortezomib is administered in a combination therapy with lenalidomide, dexamethasone, and a CD48 directed antibody-drug conjugate of the present invention.
- bortezomib is administered in a combination therapy with lenalidomide, dexamethasone, and an hMEM102 antibody-drug conjugate of the present invention.
- bortezomib is administered in a combination therapy with lenalidomide, dexamethasone, and an hMEM102-MDpr-PEG(12)-gluc-MMAE 8-load of the present invention.
- bortezomib is administered in a combination therapy with panobinostat, dexamethasone, and a CD48 directed antibody-drug conjugate of the present invention.
- bortezomib is administered in a combination therapy with panobinostat, dexamethasone, and an hMEM102 antibody-drug conjugate of the present invention.
- bortezomib is administered in a combination therapy with panobinostat, dexamethasone, and an hMEM102-MDpr-PEG(12)-gluc-MMAE 8-load of the present invention.
- dexamethasone e.g.
- DECADRON® a glucocorticosteroid used to treat cancer (including multiple myeloma, leukemia, and lymphoma), inflammation, allergies, and nausea.
- Dexamethasone can be administered as a tablet, pill, or capsule for oral administration, or by intravenous infusion.
- dexamethasone is administered in a combination therapy with a CD48 directed antibody-drug conjugate of the present invention.
- dexamethasone is administered in a combination therapy with an hMEM102 antibody-drug conjugate of the present invention.
- dexamethasone is administered in a combination therapy with an hMEM102-MDpr-PEG(12)-gluc-MMAE 8-load of the present invention.
- Dexamethasone can also be administered in a combination therapy with a CD48 directed antibody-drug conjugate of the present invention and an additional agent.
- Another exemplary agent for combination therapy is cyclophosphamide (e.g.
- Cyclophosphamide can be administered by injection, infusion, as a tablet, pill, or capsule for oral administration, or by injection into a muscle, into the abdominal lining, or into lung lining. In an embodiment, cyclophosphamide is administered in a combination therapy with a CD48 directed antibody-drug conjugate of the present invention.
- cyclophosphamide is administered in a combination therapy with an hMEM102 antibody-drug conjugate of the present invention.
- cyclophosphamide is administered in a combination therapy with an hMEM102-MDpr-PEG(12)-gluc-MMAE 8-load of the present invention.
- Cyclophosphamide can also be administered in a combination therapy with a CD48 directed antibody-drug conjugate of the present invention and an additional agent.
- melphalan an alkylating agent used to treat cancer (including multiple myeloma and ovarian cancer).
- Melphalan can be administered orally, as an injection or infusion.
- melphalan is administered in a combination therapy with a CD48 directed antibody-drug conjugate of the present invention.
- melphalan is administered in a combination therapy with an hMEM102 antibody- drug conjugate of the present invention.
- melphalan is administered in a combination therapy with an hMEM102-MDpr-PEG(12)-gluc-MMAE 8-load of the present invention.
- Melphalan can also be administered in a combination therapy with a CD48 directed antibody-drug conjugate of the present invention and an additional agent.
- pomalidomide e.g.
- Pomalidomide can be administered as a capsule, pill, or tablet for oral administration.
- pomalidomide is administered in a combination therapy with a CD48 directed antibody-drug conjugate of the present invention.
- pomalidomide is administered in a combination therapy with an hMEM102 antibody-drug conjugate of the present invention.
- pomalidomide is administered in a combination therapy with an hMEM102- MDpr-PEG(12)-gluc-MMAE 8-load of the present invention.
- Pomalidomide can also be administered in a combination therapy with a CD48 directed antibody-drug conjugate of the present invention and an additional agent.
- Pomalidomide has been combined with various additional agents to treat multiple myeloma.
- Pomalidomide has been combined with dexamethasone (see Richardson P et al. Pomalidomide alone or in combination with low-dose dexamethasone in relapsed and refractory multiple myeloma: a randomized phase 2 study. Blood 2014; 123: 1826- 1832).
- pomalidomide is administered in a combination therapy with dexamethasone and a CD48 directed antibody-drug conjugate of the present invention.
- pomalidomide is administered in a combination therapy with dexamethasone and an hMEM102 antibody-drug conjugate of the present invention. In a further embodiment, pomalidomide is administered in a combination therapy with dexamethasone and an hMEM102-MDpr-PEG(12)-gluc-MMAE 8-load of the present invention.
- Pomalidomide has also been combined with carfilzomib and dexamethasone (see Shah J et al. Carfilzomib, pomalidomide, and dexamethasone (CPD) in patients with relapsed and/or refractory multiple myeloma. Blood 2015; 126: 2284-2290).
- pomalidomide is administered in a combination therapy with carfilzomib, dexamethasone, and a CD48 directed antibody-drug conjugate of the present invention.
- pomalidomide is administered in a combination therapy with carfilzomib, dexamethasone, and an hMEM102 antibody-drug conjugate of the present invention.
- pomalidomide is administered in a combination therapy with carfilzomib, dexamethasone, and an hMEM102- MDpr-PEG(12)-gluc-MMAE 8-load of the present invention.
- panobinostat e.g. FARYDAK®
- HDAC histone deacetylase
- panobinostat can be administered as a pill, capsule, or tablet for oral administration.
- panobinostat is administered in a combination therapy with a CD48 directed antibody-drug conjugate of the present invention.
- panobinostat is administered in a combination therapy with an hMEM102 antibody-drug conjugate of the present invention. In a further embodiment, panobinostat is administered in a combination therapy with an hMEM102-MDpr- PEG(12)-gluc-MMAE 8-load of the present invention.
- Panobinostat can also be administered in a combination therapy with a CD48 directed antibody-drug conjugate of the present invention and an additional agent.
- Panobinostat has been combined with various additional agents to treat multiple myeloma.
- panobinostat has been combined with carfilzomib (see Berdeja JG et al.).
- panobinostat is administered in a combination therapy with carfilzomib and a CD48 directed antibody-drug conjugate of the present invention.
- panobinostat is administered in a combination therapy with carfilzomib and an hMEM102 antibody-drug conjugate of the present invention.
- panobinostat is administered in a combination therapy with carfilzomib and an hMEM102-MDpr-PEG(12)-gluc-MMAE 8-load of the present invention.
- Panobinostat has also been combined with bortezomib and dexamethasone (see Richardson P et al. 2013).
- panobinostat is administered in a combination therapy with bortezomib, dexamethasone, and a CD48 directed antibody-drug conjugate of the present invention.
- panobinostat is administered in a combination therapy with bortezomib, dexamethasone, and an h MEM 102 antibody-drug conjugate of the present invention.
- panobinostat is administered in a combination therapy with bortezomib, dexamethasone, and an hMEM102-MDpr-PEG(12)-gluc-MMAE 8-load of the present invention.
- ixazomib (NINLARO®), a proteasome inhibitor used to treat cancer (including multiple myeloma).
- Ixazomib can be administered orally.
- ixazomib is administered in a combination therapy with a CD48 directed antibody-drug conjugate of the present invention.
- ixazomib is administered in a combination therapy with an hMEM102 antibody-drug conjugate of the present invention.
- ixazomib is administered in a combination therapy with an hMEM102-MDpr-PEG(12)-gluc-MMAE 8-load of the present invention.
- Ixazomib can also be administered in a combination therapy with a CD48 directed antibody-drug conjugate of the present invention and an additional agent.
- Ixazomib has been combined with various additional agents to treat multiple myeloma.
- ixazomib has been combined with lenalidomide and dexamethasone (see Moreau P et al. Ixazomib, an investigational oral proteasome inhibitor, in combination with lenalidomide and dexamethasone, significantly extends progression-free survival for patients with relapsed and/or refractory multiple myeloma: the phase 3 tourmaline-MMl study. ASH Annual Meeting Abstracts, 2015).
- ixazomib is administered in a combination therapy with lenalidomide, dexamethasone, and a CD48 directed antibody-drug conjugate of the present invention.
- ixazomib is administered in a combination therapy with lenalidomide, dexamethasone, and an hMEM102 antibody-drug conjugate of the present invention.
- ixazomib is administered in a combination therapy with lenalidomide, dexamethasone, and an hMEM102- MDpr-PEG(12)-gluc-MMAE 8-load of the present invention.
- Example 1 Antibody selection and humanization
- the murine MEM102 antibody binds to the human CD48 protein and was first disclosed in Bazil et al., Folia Biologica 35:289-297 (1989). Nucleic acids encoding the murine MEM 102 antibody were sequenced and encoded heavy and light chain CDR sequences were identified, i.e., SEQ ID NO:3-8.
- Several humanized MEM102 antibodies were constructed using the hlgG VH7-4-l/hIgG-JH5 heavy chain variable region human germline and the hIgG-VK6- 21/hIgG-JK4 light chain variable region human germline as the human acceptor sequences.
- the antibodies differed in the selection of amino acid residues to be mutated back to the mouse antibody or mouse germline sequence.
- the antibody designated HALA dashed chain as set forth in SEQ ID NO: l (vHA) and the light chain as set forth in SEQ I D NO:2 (vLA) was selected as the lead humanized MEM 102 antibody on the basis of its (i) binding characteristics, (ii) ability to deliver drug and (iii) number of back mutations as compared to the other variants.
- Humanized MEM102 antibody is also referred to as hMEM102.
- Antibodies designated HCLA (antibody having the heavy chain variable region designated vHC and the light chain variable region designated vLA), HALB (antibody having the heavy chain variable region designated vHA and the light chain variable region designated vLB), HBLA (antibody having the heavy chain variable region designated vHB and the light chain variable region designated vLA), HBLB (antibody having the heavy chain variable region designated vHB and the light chain variable region designated vLB), HCLB (antibody having the heavy chain variable region designated vHC and the light chain variable region designated vLB) can be used in the present invention in place of the HALA antibody.
- Results Table 1 and Figures 3 and 4 show that hMEM 102-H ALA antibody has similar binding affinity compared to cMEM102 in U-266 cells, as indicated by the low Kd values. Binding affinity of hMEM 102-H ALA and cMEM102 to cynomolgus monkey CD48 in stably transfected CHO-DG44 clonal cells was also comparable. Both hMEM102-HALA and cMEM102 have greater than 4-fold lower Kd compared to mMEM102 in both cell lines. The anti-CD48 antibodies had approximately 6-fold stronger binding affinity to human CD48 compared to cynomolgus monkey CD48.
- LC-MS system 1 consisted of a ZMD Micromass mass spectrometer interfaced to an HP Agilent 1100 HPLC instrument equipped with a C12 Phenomenex Synergi 2.0 x 150 mm, 4 ⁇ , 80 A reverse phase column.
- LC-MS system 2 consisted of a Waters Xevo G2 Tof mass spectrometer interfaced to a Waters 2695 Separations Module with a Waters 2996 Photodiode Array Detector; the column, mobile phases, gradient, and flow rate were same as for LC-MS system 1.
- UPLC-MS was carried out on a Waters SQ mass detector interfaced to an Acquity Ultra Performance LC equipped with an Acquity UPLC BEH C18 2.1 x 50 mm, 1.7 ⁇ reverse phase column.
- Preparative HPLC was carried out on a Varian ProStar 210 solvent delivery system configured with a Varian ProStar 330 PDA detector.
- glucuronide-MMAE intermediate 2 (40 mg, 26.8 ⁇ ) was added 0.9 mL methanol and 0.9 mL tetrahydrofuran. The solution was then cooled in an ice bath and lithium hydroxide monohydrate (6.8 mg, 161 ⁇ ) was added drop wise in as a solution in 0.9 mL water. The reaction was then stirred on ice for 1.5 h, at which time LC/MS revealed complete conversion to product. Glacial acetic acid (9.2 ⁇ , 161 ⁇ ) was then added and the reaction was concentrated to dryness.
- N-hydroxysuccinimide (178 mg, 1.5 mmol) was added, followed by l-ethyl-3-(3-dimethylaminopropyl) carbodiimide (298 mg, 1.5 mmol).
- the reaction was stirred at room temperature under nitrogen for 3 hours.
- Aqueous workup was achieved through dilution into 120 mL water; the aqueous layer was then extracted three times with 60 mL ethyl acetate.
- the combined organic layer was then washed with brine, dried over sodium sulfate, and concentrated to dryness.
- Compound 11 was conjugated via its interchain thiols to the anti-CD48 antibody at an average drug loading of 8 drugs per antibody using methods known in the art (see, for example, U.S. Patent No 7,659,241).
- Example 4 Cytotoxicity of hMEM-102 ADCs on Multiple Myeloma cancer cell lines.
- Anti-CD48 auristatin antibody drug conjugates were serially diluted 3-fold in media to produce 10 point dose curves (1,000 ng/mL - 0.05081 ng/mL) and applied to multiple myeloma cells cultured in 96-well assay plates (10,000 to 15,000 cells per well in 200 ⁇ ⁇ media). Cells were incubated with ADCs for 96 hours total at 37°C, 5% C02. Cell viability was assayed using the Cell Titer Glo luminescent cytotoxicity assay (Promega), and data collected using an EnVision plate reader (PerkinElmer). All cytotoxicity assays were performed with quadruplicate data points and the mean IC50 values from 2-3 independent experiments are reported.
- hMEM 102 antibody was conjugated to vcMMAE(4-load), mcMMAF(4-load), and MDpr-PEG(12)-gluc-MMAE, as an eight load, also referred to as hMEM102-5088(8). Similar conjugations were performed on a control antibody, a non-CD48 binding antibody. hMEM102-5088(8) exhibited improved cytotoxic activity as compared to the same antibody conjugated to vcMMAE(4) and mcMMAF(4). As a negative control, a cell line that does not express CD48, LP-1, was also included.
- the hMEM102- MDpr-PEG(12)-gluc-MMAE (8) conjugate is represented by filled squares, while the control antibody- MDpr-PEG(12)-gluc- MMAE (8) conjugate is represented by open squares.
- mice Female NSG (NOD scid gamma; NOO.Cg-Prkdc scid Il2rg tmlw ISzi) mice were implanted with 2.5 million NCI-H929 cells, per animal intravenously to generate a disseminated model of multiple myeloma.
- NOD scid gamma NOO.Cg-Prkdc scid Il2rg tmlw ISzi mice were implanted with 2.5 million NCI-H929 cells, per animal intravenously to generate a disseminated model of multiple myeloma.
- ADC dose levels examined were 0.33 mg/kg and 1.0 mg/kg. Mice with advanced tumor burden were sacrificed upon showing symptoms of hind limb paralysis, cranial swelling, and/or moribundity. As shown in Figure 7, both ADCs produced durable complete responses in 8/8 mice at all dose levels (single dose), while non-binding control ADC dosed mice were all sacrificed due to disease by day-60 of the study.
- mice Female NSG (NOD scid gamma; NOO.Cg-Prkdc scid Il2rg tmlw ISzi) mice were implanted with 1 million NCI-H929 multiple myeloma cells per animal subcutaneously.
- mice per treatment group were given a single intraperitoneal injection of hMEM102-MDpr-PEG(12)-gluc-MMAE (5088) ADC or non-binding control hIgG-MDpr-PEG(12)-gluc-MMAE (5088) ADC or hMEM102-Auristatin T (4830) or non-binding control hlgG-Auristatin T (4830), or hMEM 102- vcMM AE (1006) ADC or non- binding control hlgG-vcMMAE (1006). ADC dose levels examined were 0.33 mg/kg and 1.0 mg/kg.
- mice were sacrificed when subcutaneous NCI-H929 tumor volume reached 1,000 mm 3 .
- the of hMEM102-MDpr-PEG(12)-gluc-MMAE (5088) ADC produced durable complete responses in all mice at the 1.0 mg/kg dose levels.
- the lower dose level of 0.33 mg/kg hMEM102-MDpr-PEG(12)-gluc-MMAE (5088) produced a tumor delay.
- the vcMMAE and Auristatin T ADCs induced tumor delay only at the highest doses.
- mice Female NSG (NOD scid gamma; NOO.Cg-Prkdc scid Il2rg tmlw ISzi) mice were implanted with 1 million MM.1R cells, per animal intravenously to generate a disseminated model of multiple myeloma.
- NOD scid gamma NOO.Cg-Prkdc scid Il2rg tmlw ISzi mice were implanted with 1 million MM.1R cells, per animal intravenously to generate a disseminated model of multiple myeloma.
- ADC dose levels examined were 1.0 mg/kg and 3.0 mg/kg. Mice with advanced tumor burden were sacrificed upon showing symptoms of hind limb paralysis, cranial swelling, and/or moribundity. As shown in Figure 9, both ADCs produced durable complete responses in mice at all dose levels tested (single dose), while non-binding control ADC dosed mice were all sacrificed due to disease by day-60 of the study.
- mice Female NSG (NOD scid gamma; NOO.Cg-Prkdc scid Il2rg tmlw ISzi) mice were implanted with 5 million MM.1R multiple myeloma cells per animal subcutaneously.
- mice per treatment group were given a single intraperitoneal injection of hMEM102-MDpr-PEG(12)-gluc-MMAE (5088) ADC or non-binding control hIgG-MDpr-PEG(12)-gluc-MMAE (5088) ADC or hMEM102-Auristatin T (4830) or non-binding control hlgG-Auristatin T (4830), or hMEM 102- vcMM AE (1006) ADC or non- binding control hlgG-vcMMAE (1006). ADC dose levels examined were 0.33 mg/kg and 1.0 mg/kg.
- mice were sacrificed when subcutaneous MM.1R tumor volume reached 1,000 mm 3 .
- the of hMEM102-MDpr-PEG(12)-gluc-MMAE (5088) ADC produced the most potent antitumor response and the longest tumor delay.
- mice Female NSG (NOD scid gamma; NOO.Cg-Prkdc scid Il2rg tmlw ISzi) mice were implanted with 5 million EJM cells, per animal intravenously to generate a disseminated model of multiple myeloma.
- NOD scid gamma NOO.Cg-Prkdc scid Il2rg tmlw ISzi mice were implanted with 5 million EJM cells, per animal intravenously to generate a disseminated model of multiple myeloma.
- ADCC antibody dependent cellular cytotoxicity
- NK natural killer cells
- Methods for WIL2-S target cells antibody dependent cellular cytotoxicity (ADCC) was measured through chromium-51 release using purified natural killer (NK) cells combined with antibody coated CD48-positive target cells.
- WIL2-S tumor cells were labeled with chromium-51 and pre-incubated for 30 minutes with antibody (0.1 ng/niL - 10 ⁇ g/mL).
- Target cells were then combined with NK effector cells (effector/target ratio 10: 1) and incubated an additional 4 hours at 37°C, 5% C02. Chromium-51 in the supernatant was then quantified on a Perkin Elmer TopCount plate reader.
- ADCC activity is measured as a percentage of maximum lysis relative to 1% Triton X-100 treated control target cells.
- ADCC was assayed for normal human resting T cells (All Cells) using the same NK cell effector ratio and anti-CD48 antibody or ADC titration range described above; however, the PKH2 green fluorescent cell linker kit (Sigma) was used to label the cell membrane (not chromium-51). 7-AAD dye was used to measure T cell viability by flow cytometry, using the LSRII flow cytometer (Becton Dickinson).
- Complement dependent cytotoxicity was measured by incubating normal human T cells or WIL2-S tumor cells with serially diluted antibody (0.02 - 50 ⁇ g/mL) in RPMI 1640 culture media containing 10% heat-inactivated human AB serum and 5 ⁇ Sytox Green fluorescent dye. Cells were then incubated for 2 hours at 37°C, 5% C0 2 . Fluorescence from lysed cells was measured on an Envision plate reader. Maximum specific lysis of target cells was calculated as a percentage of 1% Triton X-100 treated control cells.
- Antibody dependent cellular phagocytosis was measured using monocyte derived macrophages as effector cells and PKH26 red fluorescent dye labeled normal human T cells (All Cells), WIL2-S, or Raji tumor cells.
- Target cells were pre-incubated with serially diluted antibody (0.2 ng/mL - 2 ⁇ g/mL) for 30 minutes, and then washed twice with phosphate buffered saline.
- Macrophages were added to target cells at a 4: 1 ratio in RPMI 1640 culture media containing 10% low IgG fetal bovine serum and incubated for 2 hours at 37°C, 5% C0 2 .
- Macrophages were then labeled with Alexa Fluor®-488 conjugated mouse anti-human CD l ib antibody. Tumor cell positive macrophages were detected as events showing green and red dual fluorescence on a FACSCalibur flow cytometer. Maximum specific phagocytic activity is presented as the percentage of tumor positive macrophages after subtraction of non-binding isotype control background activity.
- CDC activity results are shown in Table 4. Neither the unconjugated hMEM102 antibody nor the hMEM102-MDpr-PEG(12)-gluc-MMAE (5088) ADC exhibited CDC activity against normal resting T cells and WIL2-S tumor cells. Both alemtuzumab and rituximab exhibited substantial CDC activity against the target cells.
- ADCP results are shown in Table 5. Both the unconjugated hMEM102 antibody and the hMEM102-MDpr-PEG(12)-gluc-MMAE (5088) ADC exhibited moderate ADCP activity against normal resting T cells or WIL2-S and Raji tumor cell lines, consistent with levels observed with Campath and Rituximab.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- Immunology (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Organic Chemistry (AREA)
- Epidemiology (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Genetics & Genomics (AREA)
- Biophysics (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Cell Biology (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Microbiology (AREA)
- Mycology (AREA)
- Crystallography & Structural Chemistry (AREA)
- Biotechnology (AREA)
- Hematology (AREA)
- Oncology (AREA)
- Peptides Or Proteins (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
- Medicinal Preparation (AREA)
Abstract
Description





















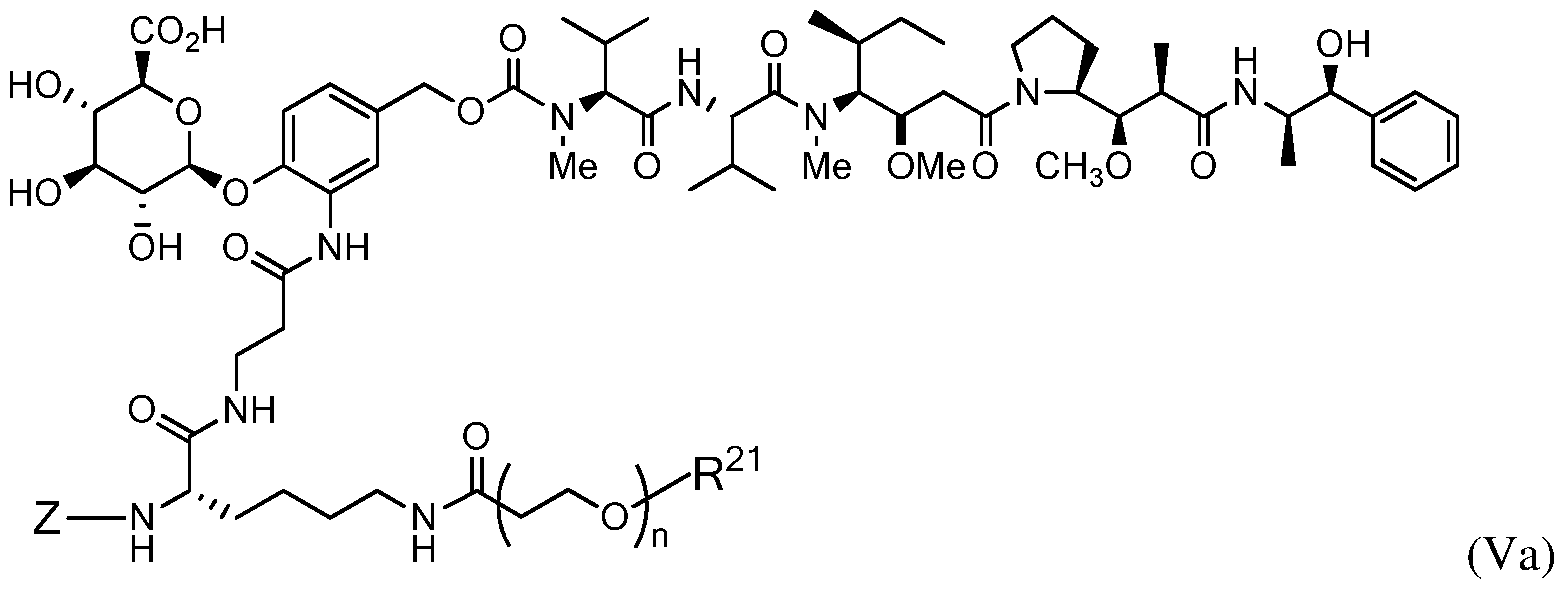



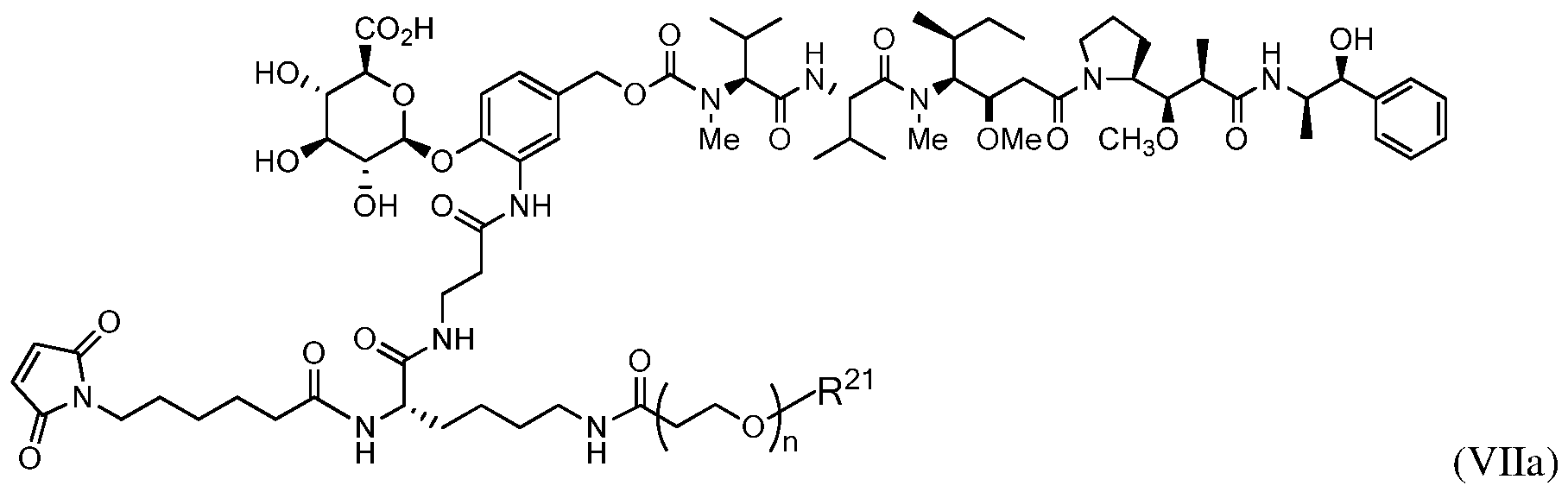





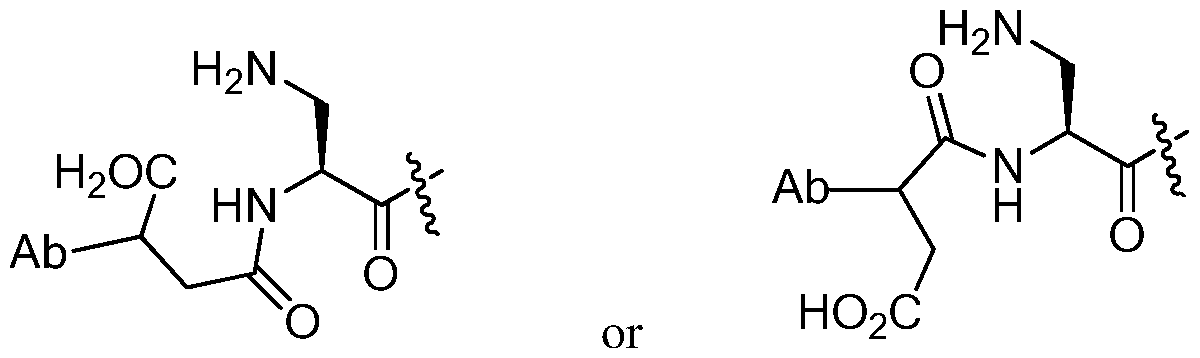
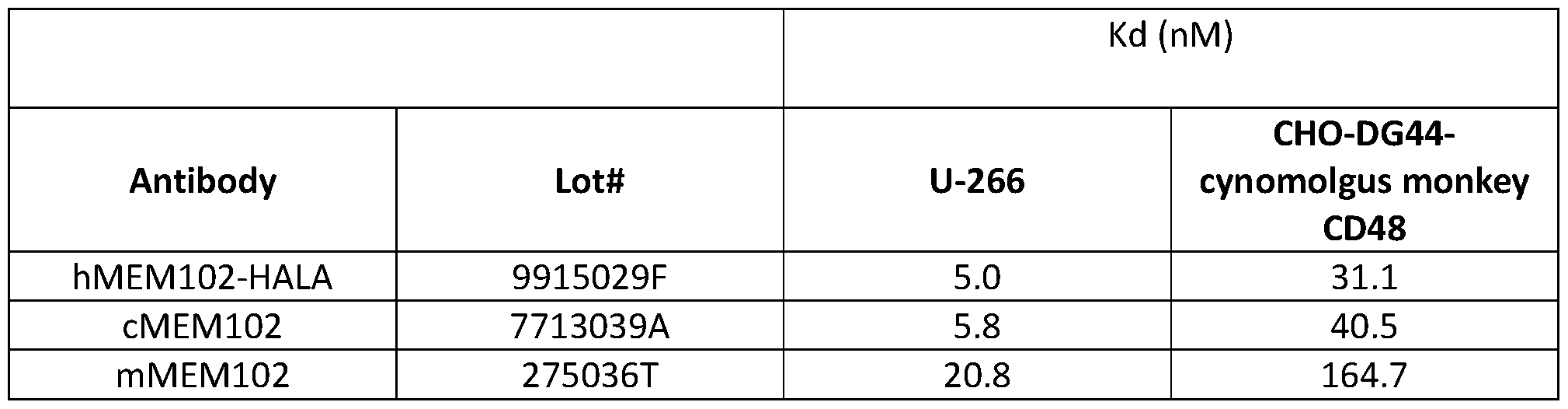
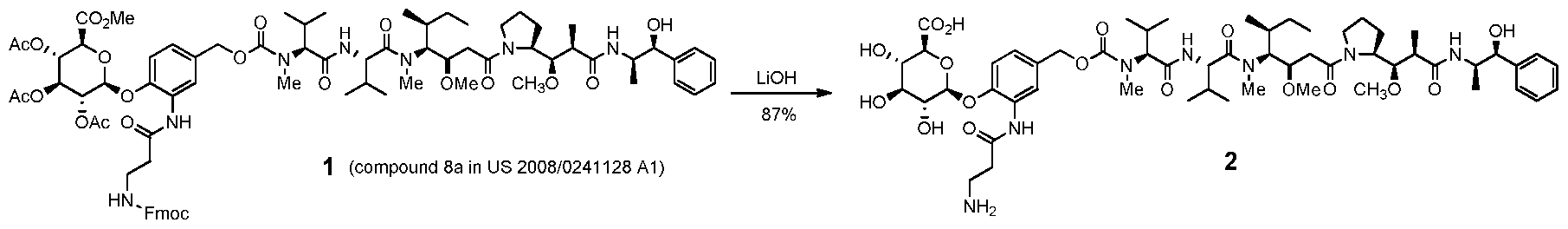






Claims













Priority Applications (18)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2017548953A JP6892826B2 (en) | 2015-03-18 | 2016-03-17 | CD48 antibody and its complex |
| EP19219476.9A EP3662933A1 (en) | 2015-03-18 | 2016-03-17 | Cd48 antibodies and conjugates thereof |
| CN201680013161.1A CN107530422B (en) | 2015-03-18 | 2016-03-17 | CD48 antibodies and conjugates thereof |
| BR112017019617-4A BR112017019617A2 (en) | 2015-03-18 | 2016-03-17 | ANTIBODY, ANTIBODY-PHARMACEUTICAL CONJUGATE COMPOUND, COMPOSITION OF ANTIBODY-PHARMACEUTICAL CONJUGATE, METHODS FOR TREATING A PATIENT WITH A CANCER THAT EXPRESSES CD48, TO PRODUCE ANTI-CD48 ANTIBODY, AND TO PRODUCE AN ANTIBODY, ISOLATED NUCLEIC, ISOLATED VECTOR, AND, ISOLATED HOST CELL |
| MX2017011432A MX2017011432A (en) | 2015-03-18 | 2016-03-17 | Cd48 antibodies and conjugates thereof. |
| US15/557,910 US10722592B2 (en) | 2015-03-18 | 2016-03-17 | CD48 antibodies and conjugates thereof |
| DK16765773.3T DK3270965T3 (en) | 2015-03-18 | 2016-03-17 | CD48 ANTIBODIES AND CONJUGATES THEREOF |
| CA2976740A CA2976740A1 (en) | 2015-03-18 | 2016-03-17 | Cd48 antibodies and conjugates thereof |
| EP16765773.3A EP3270965B1 (en) | 2015-03-18 | 2016-03-17 | Cd48 antibodies and conjugates thereof |
| SG11201706786UA SG11201706786UA (en) | 2015-03-18 | 2016-03-17 | Cd48 antibodies and conjugates thereof |
| EA201792055A EA035374B1 (en) | 2015-03-18 | 2016-03-17 | Cd48 antibodies and conjugates thereof |
| KR1020177024226A KR20170128256A (en) | 2015-03-18 | 2016-03-17 | CD48 antibody and its conjugate |
| ES16765773T ES2795818T3 (en) | 2015-03-18 | 2016-03-17 | CD48 antibodies and conjugates thereof |
| AU2016232839A AU2016232839B2 (en) | 2015-03-18 | 2016-03-17 | CD48 antibodies and conjugates thereof |
| IL254027A IL254027B (en) | 2015-03-18 | 2017-08-16 | Cd48 antibodies and conjugates thereof |
| ZA2017/05935A ZA201705935B (en) | 2015-03-18 | 2017-08-31 | Cd48 antibodies and conjugates thereof |
| HK18108175.0A HK1248538A1 (en) | 2015-03-18 | 2018-06-26 | Cd48 antibodies and conjugates thereof |
| US16/892,529 US20200289661A1 (en) | 2015-03-18 | 2020-06-04 | Cd48 antibodies and conjugates thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201562134981P | 2015-03-18 | 2015-03-18 | |
| US62/134,981 | 2015-03-18 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US15/557,910 A-371-Of-International US10722592B2 (en) | 2015-03-18 | 2016-03-17 | CD48 antibodies and conjugates thereof |
| US16/892,529 Continuation US20200289661A1 (en) | 2015-03-18 | 2020-06-04 | Cd48 antibodies and conjugates thereof |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016149535A1 true WO2016149535A1 (en) | 2016-09-22 |
Family
ID=56919489
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2016/022943 WO2016149535A1 (en) | 2015-03-18 | 2016-03-17 | Cd48 antibodies and conjugates thereof |
Country Status (17)
| Country | Link |
|---|---|
| US (2) | US10722592B2 (en) |
| EP (2) | EP3662933A1 (en) |
| JP (2) | JP6892826B2 (en) |
| KR (1) | KR20170128256A (en) |
| CN (1) | CN107530422B (en) |
| AU (1) | AU2016232839B2 (en) |
| BR (1) | BR112017019617A2 (en) |
| CA (1) | CA2976740A1 (en) |
| DK (1) | DK3270965T3 (en) |
| EA (2) | EA035374B1 (en) |
| ES (1) | ES2795818T3 (en) |
| HK (1) | HK1248538A1 (en) |
| IL (1) | IL254027B (en) |
| MX (1) | MX2017011432A (en) |
| SG (1) | SG11201706786UA (en) |
| WO (1) | WO2016149535A1 (en) |
| ZA (1) | ZA201705935B (en) |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2021067861A1 (en) | 2019-10-04 | 2021-04-08 | Seagen Inc. | Camptothecin peptide conjugates |
| WO2021067820A1 (en) | 2019-10-04 | 2021-04-08 | Seagen Inc. | Formulation of antibody-drug conjugate |
| US11084801B2 (en) | 2018-05-29 | 2021-08-10 | Intocell, Inc. | Benzodiazepine derivatives and uses thereof |
| WO2021207701A1 (en) | 2020-04-10 | 2021-10-14 | Seagen Inc. | Charge variant linkers |
| WO2022112942A3 (en) * | 2020-11-24 | 2022-07-14 | Novartis Ag | Anti-cd48 antibodies, antibody drug conjugates, and uses thereof |
| WO2022251850A1 (en) | 2021-05-28 | 2022-12-01 | Seagen Inc. | Anthracycline antibody conjugates |
| US11730822B2 (en) | 2017-03-24 | 2023-08-22 | Seagen Inc. | Process for the preparation of glucuronide drug-linkers and intermediates thereof |
| WO2023178289A2 (en) | 2022-03-17 | 2023-09-21 | Seagen Inc. | Camptothecin conjugates |
| US11844839B2 (en) | 2016-03-25 | 2023-12-19 | Seagen Inc. | Process for the preparation of pegylated drug-linkers and intermediates thereof |
| WO2024129756A1 (en) | 2022-12-13 | 2024-06-20 | Seagen Inc. | Site-specific engineered cysteine antibody drug conjugates |
| US12036286B2 (en) | 2021-03-18 | 2024-07-16 | Seagen Inc. | Selective drug release from internalized conjugates of biologically active compounds |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2022519273A (en) | 2019-02-05 | 2022-03-22 | シージェン インコーポレイテッド | Anti-CD228 antibody and antibody drug conjugate |
| WO2021067776A2 (en) | 2019-10-04 | 2021-04-08 | Seagen Inc. | Anti-pd-l1 antibodies and antibody-drug conjugates |
| WO2022115451A1 (en) * | 2020-11-24 | 2022-06-02 | Novartis Ag | Mcl-1 inhibitor antibody-drug conjugates and methods of use |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20050260213A1 (en) * | 2004-04-16 | 2005-11-24 | Scott Koenig | Fcgamma-RIIB-specific antibodies and methods of use thereof |
| US20090280116A1 (en) * | 2007-11-13 | 2009-11-12 | Cogenesys, Inc. | Humanized antibodies against tl1a |
| US20120076790A1 (en) * | 2010-09-27 | 2012-03-29 | Regeneron Pharmaceuticals, Inc. | Anti-cd48 antibodies and uses thereof |
| US20130266579A1 (en) * | 2012-03-08 | 2013-10-10 | Ge Wei | Conditionally active anti-epidermal growth factor receptor antibodies and methods of use thereof |
| US20130333061A1 (en) * | 2008-02-05 | 2013-12-12 | Wei Wu | Isolated novel nucleic acid and protein molecules from soy and methods of using those molecules to generate transgenic plants with enhanced agronomic traits |
Family Cites Families (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4816567A (en) | 1983-04-08 | 1989-03-28 | Genentech, Inc. | Recombinant immunoglobin preparations |
| US5225539A (en) | 1986-03-27 | 1993-07-06 | Medical Research Council | Recombinant altered antibodies and methods of making altered antibodies |
| WO1988007089A1 (en) | 1987-03-18 | 1988-09-22 | Medical Research Council | Altered antibodies |
| US5530101A (en) | 1988-12-28 | 1996-06-25 | Protein Design Labs, Inc. | Humanized immunoglobulins |
| US5859205A (en) | 1989-12-21 | 1999-01-12 | Celltech Limited | Humanised antibodies |
| WO1992022653A1 (en) | 1991-06-14 | 1992-12-23 | Genentech, Inc. | Method for making humanized antibodies |
| CA2249998A1 (en) | 1996-03-27 | 1997-10-02 | Crc For Biopharmaceutical Research Pty. Ltd. | The use of antibodies against cd48 for the treatment of t and b cell lymphomas and leukemias |
| US5834597A (en) | 1996-05-20 | 1998-11-10 | Protein Design Labs, Inc. | Mutated nonactivating IgG2 domains and anti CD3 antibodies incorporating the same |
| JP2005538706A (en) | 2001-07-12 | 2005-12-22 | ジェファーソン フーテ, | Super humanized antibody |
| US7659241B2 (en) | 2002-07-31 | 2010-02-09 | Seattle Genetics, Inc. | Drug conjugates and their use for treating cancer, an autoimmune disease or an infectious disease |
| BR122018071808B8 (en) | 2003-11-06 | 2020-06-30 | Seattle Genetics Inc | conjugate |
| WO2005094886A1 (en) * | 2004-03-31 | 2005-10-13 | Kirin Beer Kabushiki Kaisha | Methods of inducing differentiation of regulatory t-cells and proliferating the same with gpi anchor protein agonist and medicinal composition therefor |
| WO2007008848A2 (en) | 2005-07-07 | 2007-01-18 | Seattle Genetics, Inc. | Monomethylvaline compounds having phenylalanine carboxy modifications at the c-terminus |
| CA2614436C (en) | 2005-07-07 | 2016-05-17 | Seattle Genetics, Inc. | Monomethylvaline compounds having phenylalanine side-chain modifications at the c-terminus |
| PT3248613T (en) * | 2005-07-18 | 2022-03-16 | Seagen Inc | Beta-glucuronide drug linker conjugates |
| ES2526079T3 (en) * | 2005-12-20 | 2015-01-05 | Sbi Biotech Co., Ltd. | Anti-ILT7 antibody |
| LT2211904T (en) | 2007-10-19 | 2016-11-25 | Seattle Genetics, Inc. | Cd19 binding agents and uses thereof |
| EP2418222B1 (en) | 2009-04-10 | 2016-11-02 | Osaka University | Therapeutic agent for treating diseases in which neoplastic proliferation of plasma cells occurs |
| EP2287197A1 (en) * | 2009-08-21 | 2011-02-23 | Pierre Fabre Medicament | Anti-cMET antibody and its use for the detection and the diagnosis of cancer |
| SG194099A1 (en) * | 2011-04-15 | 2013-11-29 | Compugen Ltd | Polypeptides and polynucleotides, and uses thereof for treatment of immune related disorders and cancer |
| EA037203B1 (en) * | 2012-05-15 | 2021-02-18 | Сиэтл Джинетикс, Инк. | Antibody-drug conjugates with self-stabilizing linkers |
| EA201690780A1 (en) * | 2013-10-15 | 2016-08-31 | Сиэтл Дженетикс, Инк. | PEDIATED MEDICINE-LINKS FOR IMPROVED PHARMACOKINETICS OF LJAND-MEDICINE CONJUGATES |
-
2016
- 2016-03-17 ES ES16765773T patent/ES2795818T3/en active Active
- 2016-03-17 CA CA2976740A patent/CA2976740A1/en not_active Abandoned
- 2016-03-17 JP JP2017548953A patent/JP6892826B2/en active Active
- 2016-03-17 KR KR1020177024226A patent/KR20170128256A/en not_active Application Discontinuation
- 2016-03-17 WO PCT/US2016/022943 patent/WO2016149535A1/en active Application Filing
- 2016-03-17 SG SG11201706786UA patent/SG11201706786UA/en unknown
- 2016-03-17 DK DK16765773.3T patent/DK3270965T3/en active
- 2016-03-17 MX MX2017011432A patent/MX2017011432A/en unknown
- 2016-03-17 EP EP19219476.9A patent/EP3662933A1/en not_active Withdrawn
- 2016-03-17 BR BR112017019617-4A patent/BR112017019617A2/en not_active IP Right Cessation
- 2016-03-17 EP EP16765773.3A patent/EP3270965B1/en active Active
- 2016-03-17 AU AU2016232839A patent/AU2016232839B2/en not_active Ceased
- 2016-03-17 CN CN201680013161.1A patent/CN107530422B/en active Active
- 2016-03-17 US US15/557,910 patent/US10722592B2/en active Active
- 2016-03-17 EA EA201792055A patent/EA035374B1/en unknown
- 2016-03-17 EA EA201992756A patent/EA201992756A3/en unknown
-
2017
- 2017-08-16 IL IL254027A patent/IL254027B/en active IP Right Grant
- 2017-08-31 ZA ZA2017/05935A patent/ZA201705935B/en unknown
-
2018
- 2018-06-26 HK HK18108175.0A patent/HK1248538A1/en unknown
-
2020
- 2020-05-26 JP JP2020091335A patent/JP2020141700A/en active Pending
- 2020-06-04 US US16/892,529 patent/US20200289661A1/en not_active Abandoned
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20050260213A1 (en) * | 2004-04-16 | 2005-11-24 | Scott Koenig | Fcgamma-RIIB-specific antibodies and methods of use thereof |
| US20090280116A1 (en) * | 2007-11-13 | 2009-11-12 | Cogenesys, Inc. | Humanized antibodies against tl1a |
| US20130333061A1 (en) * | 2008-02-05 | 2013-12-12 | Wei Wu | Isolated novel nucleic acid and protein molecules from soy and methods of using those molecules to generate transgenic plants with enhanced agronomic traits |
| US20120076790A1 (en) * | 2010-09-27 | 2012-03-29 | Regeneron Pharmaceuticals, Inc. | Anti-cd48 antibodies and uses thereof |
| US20130266579A1 (en) * | 2012-03-08 | 2013-10-10 | Ge Wei | Conditionally active anti-epidermal growth factor receptor antibodies and methods of use thereof |
Non-Patent Citations (3)
| Title |
|---|
| CHOTHIA ET AL., NATURE, vol. 342, 1989, pages 878 - 883 |
| CHOTHIALESK, J. MOL. BIOL., vol. 196, 1987, pages 901 - 917 |
| See also references of EP3270965A4 |
Cited By (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11844839B2 (en) | 2016-03-25 | 2023-12-19 | Seagen Inc. | Process for the preparation of pegylated drug-linkers and intermediates thereof |
| US11730822B2 (en) | 2017-03-24 | 2023-08-22 | Seagen Inc. | Process for the preparation of glucuronide drug-linkers and intermediates thereof |
| US11084801B2 (en) | 2018-05-29 | 2021-08-10 | Intocell, Inc. | Benzodiazepine derivatives and uses thereof |
| US11807628B2 (en) | 2018-05-29 | 2023-11-07 | Intocell, Inc. | Benzodiazepine derivatives and uses thereof |
| WO2021067861A1 (en) | 2019-10-04 | 2021-04-08 | Seagen Inc. | Camptothecin peptide conjugates |
| WO2021067820A1 (en) | 2019-10-04 | 2021-04-08 | Seagen Inc. | Formulation of antibody-drug conjugate |
| WO2021207701A1 (en) | 2020-04-10 | 2021-10-14 | Seagen Inc. | Charge variant linkers |
| WO2022112942A3 (en) * | 2020-11-24 | 2022-07-14 | Novartis Ag | Anti-cd48 antibodies, antibody drug conjugates, and uses thereof |
| US11999786B2 (en) | 2020-11-24 | 2024-06-04 | Novartis Ag | Anti-CD48 antibodies, antibody drug conjugates, and uses thereof |
| US12036286B2 (en) | 2021-03-18 | 2024-07-16 | Seagen Inc. | Selective drug release from internalized conjugates of biologically active compounds |
| WO2022251850A1 (en) | 2021-05-28 | 2022-12-01 | Seagen Inc. | Anthracycline antibody conjugates |
| WO2023178289A2 (en) | 2022-03-17 | 2023-09-21 | Seagen Inc. | Camptothecin conjugates |
| WO2024129756A1 (en) | 2022-12-13 | 2024-06-20 | Seagen Inc. | Site-specific engineered cysteine antibody drug conjugates |
Also Published As
| Publication number | Publication date |
|---|---|
| US20200289661A1 (en) | 2020-09-17 |
| KR20170128256A (en) | 2017-11-22 |
| US20180092984A1 (en) | 2018-04-05 |
| JP6892826B2 (en) | 2021-06-23 |
| IL254027B (en) | 2021-05-31 |
| EP3270965A4 (en) | 2018-09-05 |
| SG11201706786UA (en) | 2017-09-28 |
| CN107530422A (en) | 2018-01-02 |
| EP3270965A1 (en) | 2018-01-24 |
| IL254027A0 (en) | 2017-10-31 |
| EA201992756A2 (en) | 2020-03-31 |
| DK3270965T3 (en) | 2020-06-08 |
| JP2020141700A (en) | 2020-09-10 |
| AU2016232839A1 (en) | 2017-08-31 |
| US10722592B2 (en) | 2020-07-28 |
| CA2976740A1 (en) | 2016-09-22 |
| AU2016232839B2 (en) | 2021-02-25 |
| BR112017019617A2 (en) | 2018-05-22 |
| MX2017011432A (en) | 2017-11-10 |
| EA201792055A1 (en) | 2018-01-31 |
| EP3270965B1 (en) | 2020-05-06 |
| ZA201705935B (en) | 2021-01-27 |
| EA035374B1 (en) | 2020-06-03 |
| CN107530422B (en) | 2021-09-21 |
| JP2018509908A (en) | 2018-04-12 |
| HK1248538A1 (en) | 2018-10-19 |
| EP3662933A1 (en) | 2020-06-10 |
| ES2795818T3 (en) | 2020-11-24 |
| EA201992756A3 (en) | 2020-06-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20200289661A1 (en) | Cd48 antibodies and conjugates thereof | |
| US20210128742A1 (en) | CD123 Antibodies and Conjugates Thereof | |
| US11819553B2 (en) | Glypican 3 antibodies and conjugates thereof | |
| US20230173093A1 (en) | Charge variant linkers | |
| CA3221398A1 (en) | Anthracycline antibody conjugates |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16765773 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2976740 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 254027 Country of ref document: IL |
|
| ENP | Entry into the national phase |
Ref document number: 20177024226 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2016232839 Country of ref document: AU Date of ref document: 20160317 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11201706786U Country of ref document: SG |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2017/011432 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 2017548953 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112017019617 Country of ref document: BR |
|
| REEP | Request for entry into the european phase |
Ref document number: 2016765773 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 201792055 Country of ref document: EA |
|
| ENP | Entry into the national phase |
Ref document number: 112017019617 Country of ref document: BR Kind code of ref document: A2 Effective date: 20170914 |