WO2016100633A1 - A process for preparing halogenated azaindole compounds using boroxine - Google Patents
A process for preparing halogenated azaindole compounds using boroxine Download PDFInfo
- Publication number
- WO2016100633A1 WO2016100633A1 PCT/US2015/066311 US2015066311W WO2016100633A1 WO 2016100633 A1 WO2016100633 A1 WO 2016100633A1 US 2015066311 W US2015066311 W US 2015066311W WO 2016100633 A1 WO2016100633 A1 WO 2016100633A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- reaction
- formula
- yield
- diaminocyclohexane
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 COc(c1c2[n](*)cc1)cnc2Br Chemical compound COc(c1c2[n](*)cc1)cnc2Br 0.000 description 2
- PWSWAPPSYGUHST-UHFFFAOYSA-N CC(C(N(CC1)CCN1C(c1ccccc1)=O)=O)=O Chemical compound CC(C(N(CC1)CCN1C(c1ccccc1)=O)=O)=O PWSWAPPSYGUHST-UHFFFAOYSA-N 0.000 description 1
- UPDSOLUKSUHRDC-UHFFFAOYSA-N COc1c(cc[nH]2)c2cnc1 Chemical compound COc1c(cc[nH]2)c2cnc1 UPDSOLUKSUHRDC-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6561—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing systems of two or more relevant hetero rings condensed among themselves or condensed with a common carbocyclic ring or ring system, with or without other non-condensed hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C17/00—Preparation of halogenated hydrocarbons
- C07C17/013—Preparation of halogenated hydrocarbons by addition of halogens
- C07C17/02—Preparation of halogenated hydrocarbons by addition of halogens to unsaturated hydrocarbons
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D249/00—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms
- C07D249/02—Heterocyclic compounds containing five-membered rings having three nitrogen atoms as the only ring hetero atoms not condensed with other rings
- C07D249/08—1,2,4-Triazoles; Hydrogenated 1,2,4-triazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F5/00—Compounds containing elements of Groups 3 or 13 of the Periodic Table
- C07F5/02—Boron compounds
- C07F5/022—Boron compounds without C-boron linkages
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/582—Recycling of unreacted starting or intermediate materials
Definitions
- the present invention relates to a process for preparing halogenated azaindole compounds which are used in obtaining HIV attachment inhibitor compounds useful as antivirals.
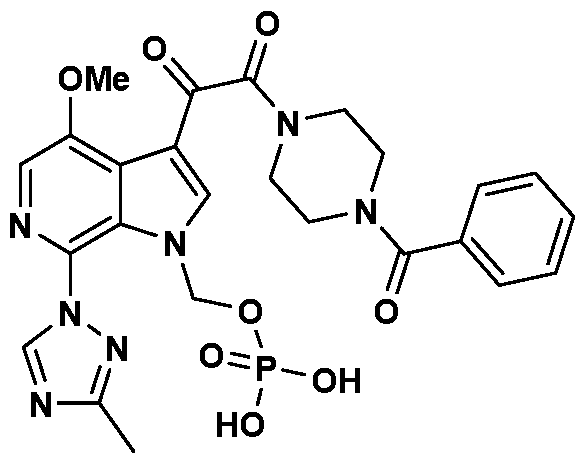
- the invention provides methods of making the piperazine prodrug compound identified as l-benzoyl-4-[2-[4-methoxy-7-(3-methyl-lH-l,2,4-triazol-l-yl)-l- [(phosphonooxy)methyl]-lH-pyrrolo[2,3-c]pyridin-3-yl]-l,2-dioxoethyl]-piperazine, as well as certain intermediates thereof.
- the invention also relates to the compounds produced by the processes herein.
- HIV-1 human immunodeficiency virus -1
- HIV-1 human immunodeficiency virus -1
- AIDS Abreliable Immuno Deficiency Syndrome
- the approved therapies to treat HIV infection fall into 4 general classes: (1) reverse-transcriptase inhibitors, (2) protease inhibitors, (3) integrase inhibitors and (4) entry inhibitors.
- Examples of available drugs for the treatment of HIV include nucleoside reverse transcriptase (RT) inhibitors or approved single pill combinations: zidovudine (or AZT or RETROVIR ® ), didanosine (or VIDEX ® ), stavudine (or ZERIT ® ), lamivudine (or 3TC or EPIVIR ® ), zalcitabine (or DDC or HIVID ® ), abacavir succinate (or ZIAGEN ® ), Tenofovir disoproxil fumarate salt (or VIREAD ® ), emtricitabine (or FTC or EMTRIVA ® ), COMBIVIR ® (contains -3TC plus AZT), TRIZIVIR ® (contains abacavir, la
- VIRAMUNE ® delavirdine (or RESCRIPTOR ® ) and efavirenz (or SUSTIVA ® ),
- ATRIPLA ® (TRUVADA ® + SUSTIVA ® ), and etravirine, and peptidomimetic protease inhibitors or approved formulations: saquinavir, indinavir, ritonavir, nelfinavir, amprenavir, lopinavir, KALETRA ® (lopinavir and Ritonavir), darunavir, atazanavir (REYATAZ ® ), and tipranavir (APTIVUS ® ), and integrase inhibitors such as raltegravir (ISENTRESS ® ), and entry inhibitors such as enfuvirtide (T-20) (FUZEON ® ) and maraviroc (SELZENTRY ® ).
- saquinavir indinavir, ritonavir, nelfinavir, amprenavir, lopinavir, KALETRA ® (lopinavir and Ritonavir), darunavir
- HIV attachment inhibitors a novel subclass of antiviral compounds that bind to the HIV surface glycoprotein gpl20, and interfere with the interaction between the surface protein gpl20 and the host cell receptor CD4. Thus, they prevent HIV from attaching to the human CD4 T-cell, and block HIV replication in the first stage of the HIV life cycle.
- the properties of HIV attachment inhibitors have been improved in an effort to obtain compounds with maximized utility and efficacy as antiviral agents.
- HIV attachment inhibitor compound in particular, has now shown considerable prowess against HIV.
- This compound is identified as l-(4-benzoyl- piperazin- 1 -y l)-2- [4-methoxy-7-(3-methy l-[ 1 ,2,4] triazol- 1 -y 1)- 1 H-py rralo [2,3 -c] pyridine-3-yl] -ethane- 1,2-di one, and is set forth and described in U.S. 7,354,924, which is incorporated herein in its entirety:
- the compound is represented by the formula below:
- the above compound is the parent compound of the prodrug known as 1-benzoyl- 4-[2- [4-methoxy-7-(3 -methyl- 1H- 1 ,2,4-triazol - 1 -y 1)- 1 - [(phosphonooxy )methy 1] - 1H- pyrrolo[2,3-c]pyridin-3-yl]-l,2-dioxoethyl]-piperazine. It is set forth and described in U.S. Patent No. 7,745,625, which is incorporated by reference herein it its entirety. The compound is represented by the formula below:
- Prodrug Compound and Intermediates also details various procedures for making the piperazine prodrug compound. These include a multi-step process which uses the
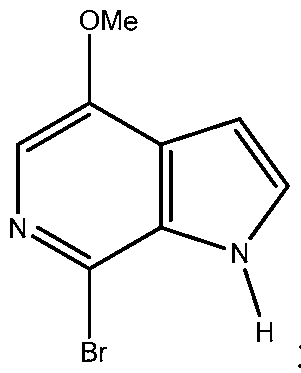
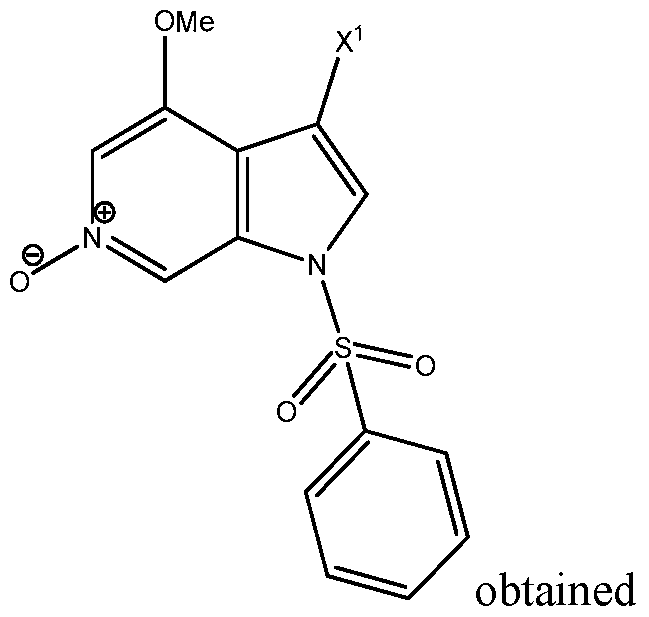
- the invention provides a process for preparing a compound of
- step (b) performing a bromination reaction in the presence of one or more boroxine compounds on the compound obtained in step (a) to obtain the compound
- step (c) performing a deprotection reaction on the compound obtained in step (b) to prepare the compound of formula I above;
- X 1 is selected from the group of H, ' H o and and Y is Br.
- the present invention provides a process for preparing a compound of formula II
- step (b) performing a bromination reaction on the compound obtained in step (a) using one or more boroxine compounds along with a bromide source and an activating agent to
- step (c) performing a deprotection reaction on the compound obtained in step (b) using either substantially pure toluene or toluene in combination with a solvent, followed by crystallization, to prepare the compound of formula II or its salts thereof.
- the present invention provides a method of making a compound of formula III
- said process comprising the steps of: (a) performing an oxidation reaction on the compound Ph using H2O2, phthalic anhydride and dichloromethane to yield the compound
- step (b) performing a bromination reaction on the compound obtained in step (a) using one or more boroxine compounds along with a bromide source and an activating agent to
- step (c) performing a deprotection reaction on the compound obtained in step (b) using either substantially pure toluene or toluene in combination with a solvent, followed by
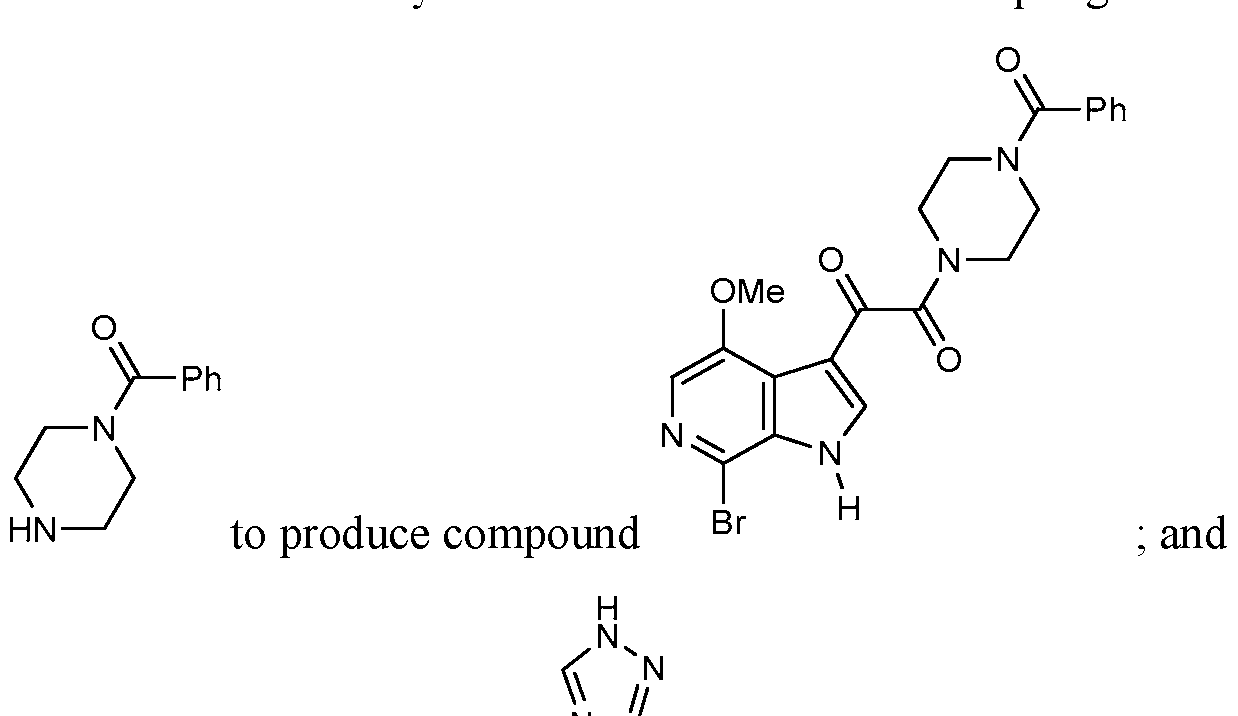
- step (d) reacting the compound obtained in step (c) to obtain the compound
- said ligand is selected from the group of 1,2-diaminocyclohexane, trans- 1,2-diaminocyclohexane, cz fraws-diaininocyclohexane, cis-N,N'- dimethyl-l,2-diaminocyclohexane, fra «s-N,N'-dimethyl- 1,2- diaminocyclohexane, cw-Zfrara-NN'-dimethyl-l ⁇ -diaminocyclohexane, 1 ,2-diaminoethane, N,N'-dimethyl-l,2-diaminoethane, 1,10- phenanthroline, 4,7-diphenyl-l,10-phenantroline, 5 -methyl- 1,10- phenanthroline, 5-chloro-l,10-phenantroline, and 5-nitro-l,10- phenanthroline; and (f) reacting the group
- step (g) the compound obtained in step (f) with an acid, such as acetic acid, to yield the compound of formula III above.
- the invention in further embodiments is also directed to each of the compounds of formulas I, II and III herein which are produced by the processes herein set forth.
- the present invention is directed to these, as well as other important ends, hereinafter described.
- alkyl group refers to a saturated aliphatic hydrocarbon including straight chain and branched chain groups.
- the alkyl group has 1 to 20 carbon atoms (whenever a numerical range; e.g., "1-20", is stated herein, it means that the group, in this case the alkyl group may contain 1 carbon atom, 2 carbon atoms, 3 carbon atoms, etc. up to and including 20 carbon atoms). More preferably, it is a medium size alkyl having 1 to 10 carbon atoms. Most preferably, it is a lower alkyl having 1 to 4 carbon atoms.
- the alkyl group may be substituted or unsubstituted.
- Ci-6 alkyl as used herein and in the claims means straight or branched chain alkyl groups with up to and including 6 carbon atoms, such as methyl, ethyl, propyl, isopropyl, butyl, isobutyl, t-butyl, amyl, hexyl and the like.
- An "aryl” “Aryl” or “Ar” group refers to an all carbon monocyclic or fused-ring poly cyclic (i.e., rings which share adj acent pairs of carbon atoms) groups having a completely conjugated pi-electron system. Examples, without limitation, of aryl groups are phenyl, napthalenyl and anthracenyl. The aryl group may be substituted or unsubstituted.
- Boroxines General term to refer to cyclic trimeric boronic acid anhydrides; these will include the trialkylboroxines such as trimethylboroxine, and also the triarylboroxines such as triphenylboroxine
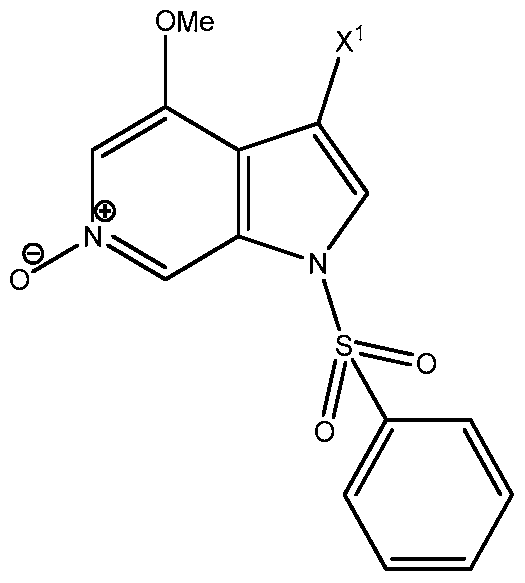
- the present invention provides a process for preparing a compo ,
- step (c) performing a deprotection reaction on the compound obtained in step (b) to obtain the compound of formula I above; O OH o wherein X 1 is selected from the group of H, ' ' oo a ann ⁇ dd H ' o
- the oxidation reaction is carried out using oxidizing agents selected from the group of catalytic methyltrioxorhenium (MTO) and hydrogen peroxide urea complex (UHP), m-CPBA (m-chloroperoxybenzoic acid), a mixture of AC2O and H2O2, and a mixture of phthalic anhydride and H2O2.
- MTO catalytic methyltrioxorhenium
- UHP hydrogen peroxide urea complex
- m-CPBA m-chloroperoxybenzoic acid
- AC2O and H2O2 a mixture of AC2O and H2O2
- step (a) obtained in step (a) is treated with aqueous Na2S03 followed by the addition of aqueous K3PO4.
- step (a) is a crystalline solid with about 85 % yield and > about 99 % purity.
- the compound obtained in step (a) is a crystalline solid which is not isolated.
- the bromination is carried out in the presence of a bromide source such as tetraoctyl ammonium bromide, an activating agent such as methanesulfonic anhydride, and a boroxine compound such as triphenylboroxine.
- a bromide source such as tetraoctyl ammonium bromide
- an activating agent such as methanesulfonic anhydride
- boroxine compound such as triphenylboroxine.
- the deprotection reaction is carried out using toluene in conjunction with isopropyl alcohol (IP A).
- the present invention provides a process for preparing a compound of formula II
- step (b) performing a bromination reaction on the compound obtained in step (a) using one or more boroxine compounds, along with a bromide source and an activating agent to
- step (c) performing a deprotection reaction on the compound obtained in step (b) using either pure toluene or toluene in combination with a solvent, followed by crystallization, to prepare the compound of formula II or its salts thereof.
- the present invention provides a method of making a compound of formula III
- said process comprising the steps of: (a) performing an oxidation reaction on the compound Ph using H2O2, phthalic anhydride and dichloromethane to yield the compound
- step (b) performing a bromination reaction on the compound obtained in step (a) using one or more boroxine compounds, along with a bromide source and an activating agent,
- step (c) performing a deprotection reaction on the compound obtained in step (b) using either pure toluene or toluene in combination with a solvent, followed by
- step (d) reacting the compound obtained in step (c) to obtain the compound
- said ligand is selected from the group of 1,2-diaminocyclohexane, trans- 1 ,2-diaminocyclohexane, cz ' s-/fra «s-diarulnocyclohexane, cw-NN'-dimethyl-l ,2- diaminocyclohexane, frafts-NN'-dimethyl- 1,2-diaminocyclohexane, cis-ltrans- NN'-dimethyl- 1 ,2-diaminocyclohexane, 1 ,2-diaminoethane, NN'-dimethyl- 1 ,2- diaminoethane, 1,10-phenanthroline, 4,7-diphenyl-l,10-phenantroline, 5-methyl- 1,10-phenanthroline, 5-chloro-l,10-phenantroline, and 5-nitro-l,10- phenanthroline; and reacting
- step (f) reacting compound obtained in step (f) with an acid, such as acetic acid, to yield the compound of formula III above.
- the compounds of the present invention may be prepared using the reactions and techniques described in this section, as well as, other synthetic methods which may be available to those of ordinary skill in the art.
- the reactions are performed in solvents appropriate to the reagents and materials employed and suitable for the transformation being affected.
- all proposed reaction conditions including choice of solvents, reaction temperature, duration of the experiment and workup procedures, are chosen to be the conditions standard for that reaction, which should be readily recognized by one skilled in the art. It is understood by one skilled in the art of organic synthesis that the functionality present on various portions of the molecule must be compatible with the reagents and reactions proposed. Such restrictions to the substituents which are compatible with the reaction conditions will be readily apparent to one skilled in the art and alternate methods must then be used.
- the mixture was cooled to 10 °C.
- the reaction was quenched by controlled addition of a solution of sodium sulfite (85.5 kg, 1 equiv) in water (1330 kg) such that the internal temperature remained below 20 °C.
- the resulting biphasic mixture was stirred vigorously at 20 °C for 2 hours to ensure complete reduction of any residual oxidant.
- a solution of K3PO4 (353 kg) in water (1330 kg) was then added to the quenched reaction mixture and the biphasic mixture stirred at 20 °C for 2 hours.
- the top aqueous phase was discarded and the lower product-rich organic phase was washed with water (1330 kg).
- the bottom product-rich organic phase was transferred to a clean 8000 L reactor.
- Toluene (1900 L) was added, and the batch was concentrated at ⁇ 0.075 MPa while maintaining the jacket temperature below 40 °C to a final volume of 3000 L.
- Toluene was added (1900 L) two more times with similar concentrations to volume batch volume of 3000 L in order to meet the specifications for KF ( ⁇ 200 ppm) and DCM (dichloromethane) ( ⁇ 1 wt%).
- the batch was cooled to 20 °C and toluene (1900 L) was added.
- Tetraoctylammonium bromide 450.4 kg, 1.25 equiv
- triphenylboroxine (267 kg, 1.3 equiv) were added, and the mixture was agitated for 1 h.
- Methanesulfonic anhydride 275.5 kg, 2.4 equiv
- toluene (413 kg) were then added and the mixture agitated for 30 min.
- the slurry was heated to 75 °C for 10 h, then sampled and analyzed. During this time the reaction mixture transformed from a thick slurry to a homogenous solution. After completion of the bromination reaction, the batch was concentrated at ⁇ 0.075 MPa while maintaining the jacket temperature below 40 °C to a final volume of 3000 L. The resulting slurry was cooled to 25 °C and acetonitrile (1200 kg) was added and agitated for 2 h. The slurry was filtered and the solids were rinsed with acetonitrile (450 kg). The solids were triphenylboroxine, and can be dried (50 °C under vacuum) and re-used in subsequent bromination reactions.
- the organic stream was transferred to an 8000 L glass lined vessel and toluene (950 L) was added. The mixture was then concentrated (T ⁇ 50 °C, 40-90 mbar) to a final volume of 1300 L, at which point the water content of the toluene solution was ⁇ 1.0 wt.
- Isopropanol 450 kg was added and the batch was heated to 40 °C.
- Aqueous HC1 (162.5 kg, 35 w/w %, 2.5 equiv) was then added over 3 hours with high agitation. The resulting suspension was cooled to 20 °C over 1 hour and then stirred for 2 hours.
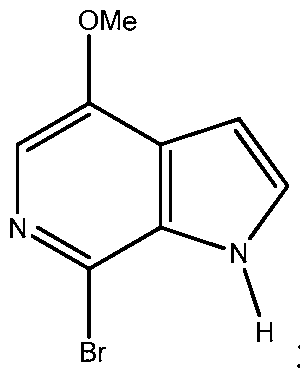
- the product was collected by centrifugation, washed with a mixture of toluene (400 L) and isopropanol (171 L), a mixture of toluene (752 L) and isopropanol (293 L), and toluene (570 L), and dried at 50 °C at ⁇ 0.1 MPa to afford the brominated azaindole Id as an off-white solid, 129.5 kg (99.64 AP, 99.79 wt%, 69.7% corrected yield).
- halogenated azaindole compounds and the reactions described above can be used in the production of the piperazine prodrug compound as shown further along in the scheme above.
- particularly le may be converted to li using the schemes described in PCT application number PCT/US2013/024880 filed February, 6, 2013, entitled “Methods for the Preparation of HIV Attachment Inhibitor Piperazine Prodrug Compound", and incorporated herein in its entirety.
- the effective preparation of the brominated intermediate Id is an important step for the effective synthesis of compound li.
- the bromination process uses readily available and stable reagents such as triphenylboroxine, Oct4NBr and Ms20and has several advantages. It provides high selectivity for the desired brominated azaindole and reduces the number of undesired impurities. This translates into higher yields and increased purity (average of 99 wt%). The ability to recycle and reuse the
- triphenylboroxine (-60 %) reagent results in a reduction of the cost of the overall process, and increases the sustainability of the manufacturing process. Initial cost estimations have shown that this process can be -20-35% less expensive than other preparations of compound le.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Virology (AREA)
- Epidemiology (AREA)
- Communicable Diseases (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Oncology (AREA)
- Tropical Medicine & Parasitology (AREA)
- AIDS & HIV (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description



































Claims

















Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA2971104A CA2971104A1 (en) | 2014-12-18 | 2015-12-17 | A process for preparing halogenated azaindole compounds using boroxine |
| CN201580076438.0A CN107406470A (en) | 2014-12-18 | 2015-12-17 | The method that halogenation azaindole compounds are prepared using boron oxygen hexatomic ring |
| RU2017123259A RU2017123259A (en) | 2014-12-18 | 2015-12-17 | METHOD FOR PRODUCING HALOGENED ASAINDOLE COMPOUNDS USING BOROXIN |
| KR1020177019923A KR20170096021A (en) | 2014-12-18 | 2015-12-17 | A process for preparing halogenated azaindole compounds using boroxine |
| US15/529,537 US9908882B2 (en) | 2014-12-18 | 2015-12-17 | Process for preparing halogenated azaindole compounds using boroxine |
| AU2015364600A AU2015364600B2 (en) | 2014-12-18 | 2015-12-17 | A process for preparing halogenated azaindole compounds using boroxine |
| BR112017012643A BR112017012643A2 (en) | 2014-12-18 | 2015-12-17 | process for preparing a compound, and method for making a compound. |
| JP2017532065A JP6554542B2 (en) | 2014-12-18 | 2015-12-17 | Method for preparing halogenated azaindole compounds using boroxine |
| EP15820021.2A EP3233853B1 (en) | 2014-12-18 | 2015-12-17 | A process for preparing halogenated azaindole compounds using boroxine |
| ES15820021T ES2728089T3 (en) | 2014-12-18 | 2015-12-17 | Preparation procedure for halogenated azaindole compounds using boroxine |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201462093645P | 2014-12-18 | 2014-12-18 | |
| US62/093,645 | 2014-12-18 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016100633A1 true WO2016100633A1 (en) | 2016-06-23 |
Family
ID=55069171
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2015/066311 Ceased WO2016100633A1 (en) | 2014-12-18 | 2015-12-17 | A process for preparing halogenated azaindole compounds using boroxine |
| PCT/US2015/066299 Ceased WO2016100625A1 (en) | 2014-12-18 | 2015-12-17 | A process for preparing halogenated azaindole compounds using boroxine |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2015/066299 Ceased WO2016100625A1 (en) | 2014-12-18 | 2015-12-17 | A process for preparing halogenated azaindole compounds using boroxine |
Country Status (12)
| Country | Link |
|---|---|
| US (1) | US9908882B2 (en) |
| EP (1) | EP3233853B1 (en) |
| JP (1) | JP6554542B2 (en) |
| KR (1) | KR20170096021A (en) |
| CN (1) | CN107406470A (en) |
| AU (1) | AU2015364600B2 (en) |
| BR (1) | BR112017012643A2 (en) |
| CA (1) | CA2971104A1 (en) |
| ES (1) | ES2728089T3 (en) |
| PT (1) | PT3233853T (en) |
| RU (1) | RU2017123259A (en) |
| WO (2) | WO2016100633A1 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020148679A1 (en) | 2019-01-17 | 2020-07-23 | ViiV Healthcare UK (No.4) Limited | Process for preparing fostemsavir |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007127731A1 (en) * | 2006-04-25 | 2007-11-08 | Bristol-Myers Squibb Company | 4-squarylpiperazine derivatives as antiviral agents |
| US7354924B2 (en) | 2001-02-02 | 2008-04-08 | Bristol-Myers Squibb Company | Composition and antiviral activity of substituted azaindoleoxoacetic piperazine derivatives |
| WO2009158394A1 (en) * | 2008-06-25 | 2009-12-30 | Bristol-Myers Squibb Company | Diketo azolopiperidines and azolopiperazines as anti-hiv agents |
| US7745625B2 (en) | 2004-03-15 | 2010-06-29 | Bristol-Myers Squibb Company | Prodrugs of piperazine and substituted piperidine antiviral agents |
| US8436168B2 (en) | 2011-01-31 | 2013-05-07 | Bristol-Myers Squibb Company | Methods of making HIV attachment inhibitor prodrug compound and intermediates |
| WO2013119625A1 (en) * | 2012-02-08 | 2013-08-15 | Bristol-Myers Squibb Company | Methods for the preparation of hiv attachment inhibitor piperazine prodrug compound |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2013542966A (en) * | 2010-11-19 | 2013-11-28 | エフ.ホフマン−ラ ロシュ アーゲー | Pyrazolopyridines and their use as TYK2 inhibitors and their use |
-
2015
- 2015-12-17 EP EP15820021.2A patent/EP3233853B1/en active Active
- 2015-12-17 AU AU2015364600A patent/AU2015364600B2/en not_active Ceased
- 2015-12-17 WO PCT/US2015/066311 patent/WO2016100633A1/en not_active Ceased
- 2015-12-17 BR BR112017012643A patent/BR112017012643A2/en not_active Application Discontinuation
- 2015-12-17 CA CA2971104A patent/CA2971104A1/en not_active Abandoned
- 2015-12-17 WO PCT/US2015/066299 patent/WO2016100625A1/en not_active Ceased
- 2015-12-17 CN CN201580076438.0A patent/CN107406470A/en active Pending
- 2015-12-17 ES ES15820021T patent/ES2728089T3/en active Active
- 2015-12-17 KR KR1020177019923A patent/KR20170096021A/en not_active Withdrawn
- 2015-12-17 RU RU2017123259A patent/RU2017123259A/en not_active Application Discontinuation
- 2015-12-17 JP JP2017532065A patent/JP6554542B2/en active Active
- 2015-12-17 PT PT15820021T patent/PT3233853T/en unknown
- 2015-12-17 US US15/529,537 patent/US9908882B2/en active Active
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7354924B2 (en) | 2001-02-02 | 2008-04-08 | Bristol-Myers Squibb Company | Composition and antiviral activity of substituted azaindoleoxoacetic piperazine derivatives |
| US7745625B2 (en) | 2004-03-15 | 2010-06-29 | Bristol-Myers Squibb Company | Prodrugs of piperazine and substituted piperidine antiviral agents |
| WO2007127731A1 (en) * | 2006-04-25 | 2007-11-08 | Bristol-Myers Squibb Company | 4-squarylpiperazine derivatives as antiviral agents |
| WO2009158394A1 (en) * | 2008-06-25 | 2009-12-30 | Bristol-Myers Squibb Company | Diketo azolopiperidines and azolopiperazines as anti-hiv agents |
| US8436168B2 (en) | 2011-01-31 | 2013-05-07 | Bristol-Myers Squibb Company | Methods of making HIV attachment inhibitor prodrug compound and intermediates |
| WO2013119625A1 (en) * | 2012-02-08 | 2013-08-15 | Bristol-Myers Squibb Company | Methods for the preparation of hiv attachment inhibitor piperazine prodrug compound |
| US8889869B2 (en) | 2012-02-08 | 2014-11-18 | Bristol-Myers Squibb Company | Methods for the preparation of HIV attachment inhibitor piperazine prodrug compound |
Non-Patent Citations (2)
| Title |
|---|
| KE CHEN ET AL: "Synthesis of the 6-Azaindole Containing HIV-1 Attachment Inhibitor Pro-Drug, BMS-663068", THE JOURNAL OF ORGANIC CHEMISTRY, vol. 79, no. 18, 19 September 2014 (2014-09-19), US, pages 8757 - 8767, XP055251332, ISSN: 0022-3263, DOI: 10.1021/jo5016008 * |
| SARAH E. WENGRYNIUK ET AL: "Regioselective Bromination of Fused Heterocyclic N -Oxides", ORGANIC LETTERS, vol. 15, no. 4, 15 February 2013 (2013-02-15), US, pages 792 - 795, XP055251397, ISSN: 1523-7060, DOI: 10.1021/ol3034675 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2020148679A1 (en) | 2019-01-17 | 2020-07-23 | ViiV Healthcare UK (No.4) Limited | Process for preparing fostemsavir |
Also Published As
| Publication number | Publication date |
|---|---|
| BR112017012643A2 (en) | 2018-01-09 |
| RU2017123259A3 (en) | 2019-04-22 |
| ES2728089T3 (en) | 2019-10-22 |
| AU2015364600B2 (en) | 2018-05-10 |
| US9908882B2 (en) | 2018-03-06 |
| WO2016100625A1 (en) | 2016-06-23 |
| AU2015364600A1 (en) | 2017-07-13 |
| JP2018504377A (en) | 2018-02-15 |
| CA2971104A1 (en) | 2016-06-23 |
| PT3233853T (en) | 2019-05-27 |
| JP6554542B2 (en) | 2019-07-31 |
| CN107406470A (en) | 2017-11-28 |
| US20170298064A1 (en) | 2017-10-19 |
| KR20170096021A (en) | 2017-08-23 |
| EP3233853A1 (en) | 2017-10-25 |
| EP3233853B1 (en) | 2019-03-06 |
| RU2017123259A (en) | 2019-01-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2020131895A1 (en) | Substituted heterocycle fused gamma-carbolines synthesis | |
| US9394322B2 (en) | Methods for the preparation of HIV attachment inhibitor piperazine prodrug compound | |
| WO2021209900A1 (en) | Inhibitors of human immunodeficiency virus replication | |
| EP3233853B1 (en) | A process for preparing halogenated azaindole compounds using boroxine | |
| EP3233856A1 (en) | A process for preparing halogenated azaindole compounds using pybrop |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15820021 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15529537 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 2971104 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2017532065 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112017012643 Country of ref document: BR |
|
| REEP | Request for entry into the european phase |
Ref document number: 2015820021 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2015364600 Country of ref document: AU Date of ref document: 20151217 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20177019923 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2017123259 Country of ref document: RU Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 112017012643 Country of ref document: BR Kind code of ref document: A2 Effective date: 20170613 |