WO2016057924A1 - Pyrrolidine amide compounds as histone demethylase inhibitors - Google Patents
Pyrrolidine amide compounds as histone demethylase inhibitors Download PDFInfo
- Publication number
- WO2016057924A1 WO2016057924A1 PCT/US2015/054949 US2015054949W WO2016057924A1 WO 2016057924 A1 WO2016057924 A1 WO 2016057924A1 US 2015054949 W US2015054949 W US 2015054949W WO 2016057924 A1 WO2016057924 A1 WO 2016057924A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- membered
- compound
- mmol
- cancer
- carbocyclyl
- Prior art date
Links
- 0 CC(*CC1)*1=C Chemical compound CC(*CC1)*1=C 0.000 description 13
- XCPMJJPLYOTMBF-UHFFFAOYSA-N CC(C(NC)O)O Chemical compound CC(C(NC)O)O XCPMJJPLYOTMBF-UHFFFAOYSA-N 0.000 description 1
- XTJBPFYSBZVQDD-UHFFFAOYSA-N CC(C)(C(F)(F)F)C(NC)=O Chemical compound CC(C)(C(F)(F)F)C(NC)=O XTJBPFYSBZVQDD-UHFFFAOYSA-N 0.000 description 1
- QMKKJBRRKIKWFK-UHFFFAOYSA-N CC(C)(C)C(NC)=O Chemical compound CC(C)(C)C(NC)=O QMKKJBRRKIKWFK-UHFFFAOYSA-N 0.000 description 1
- DZUBTKLPIBEHSR-SNVBAGLBSA-N CC(C)(C)OC(N(CC1)C[C@@H]1NC(C1CC1)=O)=O Chemical compound CC(C)(C)OC(N(CC1)C[C@@H]1NC(C1CC1)=O)=O DZUBTKLPIBEHSR-SNVBAGLBSA-N 0.000 description 1
- HRMNYZNGIGQSHN-UHFFFAOYSA-N CC(C)(c1n[nH]c(C(N(CC2)CC2NC(C2CC2)=O)=O)c1)O Chemical compound CC(C)(c1n[nH]c(C(N(CC2)CC2NC(C2CC2)=O)=O)c1)O HRMNYZNGIGQSHN-UHFFFAOYSA-N 0.000 description 1
- XFCGFENUIALWFJ-UHFFFAOYSA-N CC(C)[n]1cnc(C(O)=O)c1 Chemical compound CC(C)[n]1cnc(C(O)=O)c1 XFCGFENUIALWFJ-UHFFFAOYSA-N 0.000 description 1
- CDHLQTQHNTUZIB-UMDHDWCXSA-N CC(C)c1n[nH]c(C(N(CC2)CC2c2nccc(CC(C)N(C)/N=C\C)c2)=O)c1 Chemical compound CC(C)c1n[nH]c(C(N(CC2)CC2c2nccc(CC(C)N(C)/N=C\C)c2)=O)c1 CDHLQTQHNTUZIB-UMDHDWCXSA-N 0.000 description 1
- GBCHGKJHHHFXHR-UHFFFAOYSA-N CC(CO)C(NC)=O Chemical compound CC(CO)C(NC)=O GBCHGKJHHHFXHR-UHFFFAOYSA-N 0.000 description 1
- MGUMFMQACXTQMO-UHFFFAOYSA-N CC(COC)c1n[nH]c(C(N(CC2)CC2NC(C2CC2)=O)=O)c1 Chemical compound CC(COC)c1n[nH]c(C(N(CC2)CC2NC(C2CC2)=O)=O)c1 MGUMFMQACXTQMO-UHFFFAOYSA-N 0.000 description 1
- WRAGCBBWIYQMRF-UHFFFAOYSA-N CC(NC1CCCCC1)=O Chemical compound CC(NC1CCCCC1)=O WRAGCBBWIYQMRF-UHFFFAOYSA-N 0.000 description 1
- FZERHIULMFGESH-UHFFFAOYSA-N CC(Nc1ccccc1)=O Chemical compound CC(Nc1ccccc1)=O FZERHIULMFGESH-UHFFFAOYSA-N 0.000 description 1
- POHCKHGBDOTACV-UHFFFAOYSA-N CC(c(c(F)ccc1)c1OC)=O Chemical compound CC(c(c(F)ccc1)c1OC)=O POHCKHGBDOTACV-UHFFFAOYSA-N 0.000 description 1
- SBEROSHQCHPJIB-UHFFFAOYSA-N CC1(CNC)COC1 Chemical compound CC1(CNC)COC1 SBEROSHQCHPJIB-UHFFFAOYSA-N 0.000 description 1
- REBGBCACIZAEOO-UHFFFAOYSA-N CC1(CNCC1)NC(C1CC1)=O Chemical compound CC1(CNCC1)NC(C1CC1)=O REBGBCACIZAEOO-UHFFFAOYSA-N 0.000 description 1
- SAVFTDLAFBLSMP-UHFFFAOYSA-N CCCOc1ccccc1C(NC)=O Chemical compound CCCOc1ccccc1C(NC)=O SAVFTDLAFBLSMP-UHFFFAOYSA-N 0.000 description 1
- YCGOCBWNHSDQMZ-UHFFFAOYSA-N CCN(CC1)N=C1C(NC)=O Chemical compound CCN(CC1)N=C1C(NC)=O YCGOCBWNHSDQMZ-UHFFFAOYSA-N 0.000 description 1
- ZCANBNVEMHJJPC-UHFFFAOYSA-N CNC(C(C1)CC1N)=O Chemical compound CNC(C(C1)CC1N)=O ZCANBNVEMHJJPC-UHFFFAOYSA-N 0.000 description 1
- SBEGZUOLDKCLKP-UHFFFAOYSA-N CNC(C(C=C1)Nc2c1[nH]cc2)=O Chemical compound CNC(C(C=C1)Nc2c1[nH]cc2)=O SBEGZUOLDKCLKP-UHFFFAOYSA-N 0.000 description 1
- HABTUABYBLLAFT-UHFFFAOYSA-N CNC(C1(CC1)C(F)(F)F)=O Chemical compound CNC(C1(CC1)C(F)(F)F)=O HABTUABYBLLAFT-UHFFFAOYSA-N 0.000 description 1
- OEBHHRORUJDBNT-UHFFFAOYSA-N CNC(C1(CC1)Oc1ccccc1)=O Chemical compound CNC(C1(CC1)Oc1ccccc1)=O OEBHHRORUJDBNT-UHFFFAOYSA-N 0.000 description 1
- DAKVRMFQASVEFF-UHFFFAOYSA-N CNC(C1(CC1)c1ccncc1)=O Chemical compound CNC(C1(CC1)c1ccncc1)=O DAKVRMFQASVEFF-UHFFFAOYSA-N 0.000 description 1
- ZVQPSVUIMCRLCY-UHFFFAOYSA-N CNC(CC(F)(F)F)=O Chemical compound CNC(CC(F)(F)F)=O ZVQPSVUIMCRLCY-UHFFFAOYSA-N 0.000 description 1
- KLGVWITWFZDHIX-UHFFFAOYSA-N CNC(CC1CC1)=O Chemical compound CNC(CC1CC1)=O KLGVWITWFZDHIX-UHFFFAOYSA-N 0.000 description 1
- SIXSDXBPHBHLPS-UHFFFAOYSA-N CNC(CCOC)=O Chemical compound CNC(CCOC)=O SIXSDXBPHBHLPS-UHFFFAOYSA-N 0.000 description 1
- HBEOLDIEIBRCNI-UHFFFAOYSA-N CNC(COc1ccccc1)=O Chemical compound CNC(COc1ccccc1)=O HBEOLDIEIBRCNI-UHFFFAOYSA-N 0.000 description 1
- QNMWSLMZPATZTD-UHFFFAOYSA-N CNC(NCc1ccccc1)=O Chemical compound CNC(NCc1ccccc1)=O QNMWSLMZPATZTD-UHFFFAOYSA-N 0.000 description 1
- VOHNTXJBTPUGNG-DMTCNVIQSA-N CNC([C@H](C1)[C@H]1F)=O Chemical compound CNC([C@H](C1)[C@H]1F)=O VOHNTXJBTPUGNG-DMTCNVIQSA-N 0.000 description 1
- PIHJUDHVYWCZLS-UHFFFAOYSA-N CNC(c(cc1)ccc1F)=O Chemical compound CNC(c(cc1)ccc1F)=O PIHJUDHVYWCZLS-UHFFFAOYSA-N 0.000 description 1
- SIOAPROKUUGJAQ-UHFFFAOYSA-N CNC(c(cc1)ccc1OC)=O Chemical compound CNC(c(cc1)ccc1OC)=O SIOAPROKUUGJAQ-UHFFFAOYSA-N 0.000 description 1
- JKUNAQSMHWIGCE-UHFFFAOYSA-N CNC(c(cc1)n[n]1-c1c[nH]nc1)=O Chemical compound CNC(c(cc1)n[n]1-c1c[nH]nc1)=O JKUNAQSMHWIGCE-UHFFFAOYSA-N 0.000 description 1
- NAGFMACWWJYORB-UHFFFAOYSA-N CNC(c(cccc1)c1F)=O Chemical compound CNC(c(cccc1)c1F)=O NAGFMACWWJYORB-UHFFFAOYSA-N 0.000 description 1
- NXVAEVHJWOBXKG-UHFFFAOYSA-N CNC(c(cccc1)c1OC)=O Chemical compound CNC(c(cccc1)c1OC)=O NXVAEVHJWOBXKG-UHFFFAOYSA-N 0.000 description 1
- WYOJMCQPETZNFB-UHFFFAOYSA-N CNC(c(cccc1)c1OCCO)=O Chemical compound CNC(c(cccc1)c1OCCO)=O WYOJMCQPETZNFB-UHFFFAOYSA-N 0.000 description 1
- ICXYWUGPBQZWJP-UHFFFAOYSA-N CNC(c(cccc1)c1Oc1ccccc1)=O Chemical compound CNC(c(cccc1)c1Oc1ccccc1)=O ICXYWUGPBQZWJP-UHFFFAOYSA-N 0.000 description 1
- AOSZITJMCUMUCD-UHFFFAOYSA-N CNC(c1cc(F)ccc1)=O Chemical compound CNC(c1cc(F)ccc1)=O AOSZITJMCUMUCD-UHFFFAOYSA-N 0.000 description 1
- QCUZSRZQGYWQHC-UHFFFAOYSA-N CNC(c1cc(OC)ccc1)=O Chemical compound CNC(c1cc(OC)ccc1)=O QCUZSRZQGYWQHC-UHFFFAOYSA-N 0.000 description 1
- SDYBIYUDMDJQCS-UHFFFAOYSA-N CNC(c1ccc2[n](C)ccc2c1)=O Chemical compound CNC(c1ccc2[n](C)ccc2c1)=O SDYBIYUDMDJQCS-UHFFFAOYSA-N 0.000 description 1
- HXDVDJASCLYPOK-UHFFFAOYSA-N Cc1c[n](C2CNCC2)nn1 Chemical compound Cc1c[n](C2CNCC2)nn1 HXDVDJASCLYPOK-UHFFFAOYSA-N 0.000 description 1
- GYLHKTDJFVODCT-ZETCQYMHSA-N O=C(C1CC1)N[C@@H]1CNCC1 Chemical compound O=C(C1CC1)N[C@@H]1CNCC1 GYLHKTDJFVODCT-ZETCQYMHSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/4025—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil not condensed and containing further heterocyclic rings, e.g. cromakalim
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D237/00—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings
- C07D237/02—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings
- C07D237/04—Heterocyclic compounds containing 1,2-diazine or hydrogenated 1,2-diazine rings not condensed with other rings having less than three double bonds between ring members or between ring members and non-ring members
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
Definitions
- KDM5 histone demethylases
- KDM5/JARID1 family of demethylases in humans contains four members, KDM5A, KDM5B, KDM5C and KDM5D.
- KDM5 family members contain five conserved domains: JmjN, ARID, JmjC, PHD and a C 5 HC 2 zinc finger.
- KDM5A, KDM5B, KDM5C and KDM5D are known and are publicly available, e.g., see UniProtK B/Swiss-Prot (see e.g., KDM5A (e.g., P29375-1 and P29375-2), KDM5B (e.g., Q9UGL1-1 and Q9UGL1-2), KDM5C (e.g., P41229-1, P41229-2, P41229-3 and P41229-4) and KDM5D (e.g., Q9BY66-1, Q9BY66-2 and Q9BY66-3).
- KDM5A e.g., P29375-1 and P29375-2
- KDM5B e.g., Q9UGL1-1 and Q9UGL1-2
- KDM5C e.g., P41229-1, P41229-2, P41229-3 and P41229-4
- KDM5D e.g
- A is selected from the group consisting of: R 1 is halo, -N(R X )2, C 1-6 alkyl, C 2-6 alkenyl, C 2-6 alkynyl, 3-8 membered carbocyclyl, C 1 _
- R 2 is 5-10 membered carbocyclyl, 5-10 membered heterocyclyl, 5-10 membered aryl, 5-10 membered heteroaryl, -OR a , -C(O)N(R a ) 2 , or NR a R b , wherein each 5-10 membered carbocyclyl, 5- 10 membered heterocyclyl, 5-10 membered aryl, and 5-10 membered heteroaryl is optionally substituted with one or more groups R d ;
- R a and R b are each independently selected from the group consisting of H, C 1-6 alkyl, C 2- ealkenyl, C 2-6 alkynyl, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 5-10 membered aryl, 5-10 membered heteroaryl -C(O)R c , -CO 2 R c , -C(O)N(R c ) 2 , -C(O)SR c , and -C(O)C(O)R c , wherein each C 1- ⁇ alkyl, C 2-6 alkenyl, C 2-6 alkynyl, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 5- 10 membered aryl, and 5-10 membered heteroaryl is optionally substituted with one or more groups independently selected from the group consisting of halo, nitro, cyano, oxo, Ci ⁇ alkyl, C
- each R c is independently selected from the group consisting of H, C 1-6 alkyl, C 2 _ 6 alkenyl, C 2 . 6 alkynyl, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 5-10 membered aryl and 5-10 membered heteroaryl, wherein each C 1-6 alkyl, C 2-6 alkenyl, C 2-6 alkynyl, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 5-10 membered aryl and 5-10 membered heteToaryl is optionally substituted with one or more groups R h :
- each R d is independently selected from the group consisting of halo, nitro, cyano, oxo, C 1- 6 alkyl, C 2-6 alkenyl, C 2-6 alkynyl, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 5-10 membered aryl, 5- 10 membered heteroaryl, ,-OR*, -SR e , -N(R e ) 2 , -C(O)R e , -CO 2 R E , -C(O)N(R e ) 2 , -C(O)SR e , -C(O)C(O)R e , -S(O)R e , -SO 2 R e , -SO 2 N(R c ) 2 , -N(R e )C(O)R e , -N(R e )C(O)N(R e ) 2 ,
- carbocyclyl, 3-8 membered heterocyclyl, 5-10 membered aryl and 5-10 membered heteroaryl is optionally substituted with one or more groups independently selected from R ;
- each R e is independently selected from the group consisting of H, C 1-6 alkyl, C 2-6 alkenyl, C 2- 6 alkyny], 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 5-10 membered aryl and 5-10 membered heteroaryl, wherein each C 1-6 alkyl, C 2-6 alkenyl, C 2-6 alkynyl, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 5-10 membered aryl and 5-10 membered heteroaryl is optionally substituted with one or more groups independently selected from the group consisting of Ci ⁇ alkyl, C 2-4 alkenyl, C 2-4 alkynyl, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 5-10 membered aryl and 5-10 membered heteroaryl;
- each R is independently selected from the group consisting of halo, nitro, cyano, oxo, Ci_ 4 alkyl 5 C 2-4 alkenyl 5 C 2-4 alkynyl, 3-8 membered carbocyclyl ,-OR g , -SR S , -N(R g ) 2 , -C(O)R g , -CO 2 R g , -C(O)N(R s ) 2 , -C(O)SR g , -C(O)C(O)R g , -S(O)R s , -SO 2 R g , -SO 2 N(R g ) 2 , -N(R B )C(O)R g ,
- each R g is independently selected from the group consisting of H, C 1-6 alkyl, C 2 . 6 alkenyl, C 2 . 6 alkynyl, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 5-10 membered aryl and 5-10 membered heteroaryl, wherein each C 1-6 alkyl, C 2-6 alkenyl, C 2 .
- 6 alkynyl, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 5-10 membered aryl and 5-10 membered heteroaryl is optionally substituted with one or more groups independently selected from the group consisting of C 1-4 alkyl, C 2-4 alkenyl, C 2-4 alkynyl, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 5-10 membered aryl and 5-10 membered heteroaryl; and
- each R h is independently selected from the group consisting of halo, nitro. cyano, oxo, C 1- 4 aJkyl, C 2-4 alkenyl, C 2-4 alkynyl, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 5-10 membered aryl, 5-10 membered heteroaryl, -N(R k )2, and ,-OR k , wherein each C 1-4 alkyl, C 2- 4alkenyl, C 2-4 alkynyl, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 5-10 membered aryl and 5-10 membered heteroaryl is optionally substituted with one or more groups independently selected from the group consisting of halo, hydroxy, C 1-4 alkoxy, cyano, oxo, C 1-4 alkyl, C 2-4 alkenyl, C 2-4 alkynyl,3-8 membered carbocyclyl,
- each R k is independently selected from the group consisting of H, C 1-4 alkyl, C 2-4 alkenyl, C 2- 4 alkynyl, 3-8 membered carbocyclyl, and 5-10 membered aryl wherein any C 1-4 alkyl, C 2-4 alkenyl, C 2-4 alkynyl, 3-8 membered carbocyclyl, and 5-10 membered aryl carbocyclyl is optionally substituted with one or more groups independently selected from the group consisting of halo, cyano, oxo hydroxy, and 3-8 membered carbocyclyl; and
- R 3 is H or C,.6alkyl
- R 4 is H, C 1-6 alkyl, C 2-6 alkenyl, C 2-6 alkynyl, or 3-8 membered carbocyclyl, 5-10 membered aryl, 5-10 membered heteroaryl wherein each C 1-6 alkyl, C 2-6 alkenyl, C 2-6 alkynyl, and 3-8 membered carbocyclyl, 5-10 membered aryl, 5-10 membered heteroaryl is optionally substituted with one or more groups independently selected from halo, hydroxy, cyano, C 1-6 alkoxy, and 3-8 membered carbocyclyl; and
- R 5 is H, halo, or C 1-6 alkyl
- R 6 is H, C 1-6 alkyl, C 2-6 alkenyl, C 2-6 alkynyl, halo, hydroxy, or 3-8 membered carbocyclyl, wherein each C 1-6 alkyl, C 2-6 alkenyl, C 2-6 alkynyl, and 3-8 membered carbocyclyl, is optionally substituted with one or more groups independently selected from halo, hydroxy, cyano, C 1-6 alkoxy, and 3-8 membered carbocyclyl; or R 5 and R 6 taken together with the atom to which they are attached form a 3-8 membered carbocyclyl or a 3-8 membered heterocyclyl, which 3-8 membered carbocyclyl and 3-8 membered heterocyclyl are optionally substitutes with one or more groups independently selected from halo, hydroxy, cyano, C 1-6 alkoxy, and 3-8 membered carbocyclyl
- Another aspect includes a composition, comprising a compound of formula (I), or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable adjuvant, carrier, or vehicle.
- Another aspect includes a method of treating a disease associated with KDM5 activity, comprising administering an therapeutically effective amount of a compound of formula (I), or a pharmaceutically acceptable salt thereof, to a patient in need thereof.
- Another aspect includes the use of a compound of formula (I), or a pharmaceutically acceptable salt thereof, in therapy.
- Another aspect includes the use of a compound of formula (I), or a pharmaceutically acceptable salt thereof, in treating a disease associated with KDM5 activity.
- Another aspect includes the use of a compound of formula (I), or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for the treatment of a disease associated with KDM5 activity.
- Another aspect includes a method of increasing the efficacy of a cancer treatment comprising a cancer therapy agent, comprising administering to a patient (a) an effective amount of a compound of formula (I), or a pharmaceutically acceptable salt thereof, and (b) an effective amount of the cancer therapy agent.
- Another aspect includes a method of treating an individual with cancer who has an increased likelihood of developing resistance to a cancer therapy agent comprising administering to the individual (a) an effective amount of a compound of formula (I), or a pharmaceutically acceptable salt thereof, and (b) an effective amount of the cancer therapy agent.
- Another aspect includes a processes and synthetic intermediates that are useful for preparing a compound of formula (I), or a salt thereof.
- Another aspect includes compounds for the study of histone demethylases, such as KDM5, the study of intracellular signal transduction pathways mediated by such histone demethylases, and the comparative evaluation of modulators of these demethylases.
- KDM5 histone demethylases
- structures depicted herein are also meant to include all isomeric (e.g., enantiomeric, diastereomeric, and geometric (or conformational)) forms of the structure; for example, the R and S configurations for each asymmetric center, Z and E double bond isomers, and Z and E conformational isomers. Therefore, single stereochemical isomers as well as enantiomeric, diastereomeric, and geometric (or conformational) mixtures of the present compounds are included.
- isomeric e.g., enantiomeric, diastereomeric, and geometric (or conformational)
- structures depicted herein are also meant to include compounds that differ only in the presence of one or more isotopically enriched atoms.
- compounds having the present structures including the replacement of hydrogen by deuterium or tritium, or the replacement of a carbon by a 13 C- or 14 C-enriched carbon are included.
- Such compounds are useful, for example, as analytical tools, as probes in biological assays, or as therapeutic agents.
- a particular enantiomer may, in some embodiments be provided substantially free of the corresponding enantiomer, and may also be referred to as "optically enriched.”
- “Optically-enriched,” as used herein, means that the compound is made up of a significantly greater proportion of one enantiomer. In certain embodiments the compound is made up of at least about 90% by weight of a preferred enantiomer. In other embodiments the compound is made up of at least about 95%, 98%, or 99% by weight of a preferred enantiomer.
- Preferred enantiomers may be isolated from racemic mixtures by any method known to those skilled in the art, including chiral high pressure liquid chromatography (HPLC) and the formation and
- heteroatom means one or more of oxygen, sulfur, nitrogen, phosphorus, or silicon (including, any oxidized form of nitrogen, sulfur, phosphorus, or silicon; the quaternized form of any basic nitrogen or; a substitutable nitrogen of a heterocyclic ring, for example N (as in 3,4-dihydro-2H-pyrrolyl), NH (as in pyrrolidinyl) or NR + (as in N-substituted pyrrolidinyl)).
- a "direct bond” or “covalent bond” refers to a single bond.
- halo and "halogen” as used herein refer to an atom selected from fluorine (fluoro, -F), chlorine (chloro, -CI), bromine (bromo, -Br), and iodine (iodo, -I).
- aliphatic or "aliphatic group”, as used herein, denotes a hydrocarbon moiety that may be straight-chain (i.e., unbranched), branched, or cyclic (including fused, bridging, and spiro- fused polycyclic) and may be completely saturated or may contain one or more units of
- aliphatic groups contain 1-6 carbon atoms. In some embodiments, aliphatic groups contain 1—4 carbon atoms, and in yet other embodiments aliphatic groups contain 1-3 carbon atoms. Suitable aliphatic groups include, but are not limited to, linear or branched, alky], alkenyl, and alkynyl groups, and hybrids thereof such as (cycloalkyl)alkyl, (cycloalkenyl)alkyl or (cycloalkyl)alkenyl.
- unsaturated means that a moiety has one or more units of unsaturation.
- cycloaliphatic refers to a saturated or partially unsaturated cyclic aliphatic monocyclic, bicyclic, or spior ring systems, as described herein, having from 3 to 10 members, wherein the aliphatic ring system is optionally substituted as defined above and described herein.
- Cycloaliphatic groups include, without limitation, cyclopropyl, cyclobutyl, cyclopentyl,
- the cycloalkyl has 3-6 carbons.
- cycloaliphatic also include aliphatic rings that are fused to one or more aromatic or nonaromatic rings, such as decahydronaphthyl, tetrahydronaphthyl, decalin, or bicyclo[2.2.2]octane, where the radical or point of attachment is on an aliphatic ring.
- cycloalkylene refers to a bivalent cycloalkyl group.
- a cycloalkylene group is a 1,1 -cycloalkylene group (i.e., a spiro-fused ring).
- Exemplary 1,1 -cycloalkylene groups include .
- a cycloalkylene group is a 1 ,2-cycloalkylene group or a 1,3-cycloalkylene group.
- alkyl refers to a monovalent saturated, straight- or branched- chain hydrocarbon radical derived from an aliphatic moiety containing between one and six carbon atoms by removal of a single hydrogen atom. In some embodiments, alkyl contains 1-5 carbon atoms. In another embodiment, alkyl contains ⁇ -4 carbon atoms. In still other embodiments, alkyl contains 1-3 carbon atoms. In yet another embodiment, alkyl contains 1-2 carbons.
- alkyl radicals include, but are not limited to, methyl, ethyl, n-propyl, isopropyl, n-butyl, iso-butyl, sec-butyl, sec-p entyl, iso-p entyl, tert-butyl, n-pentyl, neopentyl, n-hexyl, sec-hexyl, n-heptyl, n- octyl, n-decyl, n-undecyl, dodecyl, and the like.
- alkenyl denotes a monovalent group derived from a straight- or branched-chain aliphatic moiety having at least one carbon-carbon double bond by the removal of a single hydrogen atom. In certain embodiments, alkenyl contains 2-6 carbon atoms. In certain embodiments, alkenyl contains 2-5 carbon atoms. In some embodiments, alkenyl contains 2—4 carbon atoms. In another embodiment, alkenyl contains 2-3 carbon atoms. Alkenyl groups include, for example, ethenyl ("vinyl”), propenyl ("allyl”), butenyl, l-methyl-2-buten-l-yl, and the like.
- alkynyl refers to a monovalent group derived from a straight- or branched-chain aliphatic moiety having at least one carbon-carbon triple bond by the removal of a single hydrogen atom.
- alkynyl contains 2-6 carbon atoms.
- alkynyl contains 2—5 carbon atoms.
- alkynyl contains 2-4 carbon atoms.
- alkynyl contains 2-3 carbon atoms.
- Representative alkynyl groups include, but are not limited to, ethynyl, 2-propynyl ("propargyl"), 1-propynyl, and the like.
- aryl used alone or as part of a larger moiety as in “aralkyl”, “aralkoxy”, or “aryloxyalkyi”, refers to monocyclic and bicyclic ring systems having a total of five to 10 ring members, wherein at least one ring in the system is aromatic and wherein each ring in the system contains three to seven ring members.
- aryl may be used interchangeably with the term “aryl ring”.
- aryl refers to an aromatic ring system which includes, but not limited to, phenyl, biphenyl, naphthyl, anthracyl and the like, which may bear one or more substituents.
- aryl is a group in which an aromatic ring is fused to one or more non-aromatic rings, such as indanyl, phthalimidyl, naphthimidyl, phenantriidinyl, or tetrahydronaphthyl, and the like.
- heteroaryl and “heteroar-”, used alone or as part of a larger moiety refer to groups having 5 to 10 ring atoms, preferably 5, 6, or 9 ring atoms; having 6, 10, or 14 ⁇ electrons shared in a cyclic array; and having, in addition to carbon atoms, from one to five heteroatoms.
- heteroatom refers to nitrogen, oxygen, or sulfur, and includes any oxidized form of nitrogen or sulfur, and any quaternized form of a basic nitrogen.
- Heteroaryl groups include, without limitation, thienyl, furanyl, pyrrolyl, imidazolyl, pyrazolyl, triazolyl, tetrazolyl, oxazolyl, isoxazolyl, oxadiazolyl, thiazolyl, isothiazolyl, thiadiazolyl, pyridyl, pyridazinyl, pyrimidinyl, pyrazinyl, indolizinyl, purinyl, naphthyridinyl, and pteridinyl.
- heteroaryl and “heteroar-”, as used herein, also include groups in which a heteroaromatic ring is fused to one or more aryl, cycloaliphatic, or heterocyclyl rings, where the radical or point of attachment is on the heteroaromatic ring.
- Nonlimiting examples include indolyl, isoindolyl, benzothienyl, benzofuranyl, dibenzofuranyl, indazolyl, benzimidazolyl, benzthiazolyl, quinolyl, isoquinolyl, cinnolinyl, phthalazinyl, quinazolinyl, quinoxalinyl, 4H-quinolizinyl, carbazolyl, acridinyl, phenazinyl, phenothiazinyl, phenoxazinyl, tetrahydroquinolinyl,
- heteroaryl group may be mono- or bicyclic.
- heteroaryl may be used interchangeably with the terms “heteroaryl ring”, “heteroaryl group”, or “heteroaromatic”, any of which terms include rings that are optionally substituted.
- heterooaralkyl refers to an alkyl group substituted by a heteroaryl, wherein the alkyl and heteroaryl portions independently are optionally substituted.
- heterocycle As used herein, the terms “heterocycle”, “heterocyclyl”, “heterocyclic radical”, and
- heterocyclic ring are used interchangeably and refer to a stable 4- to 7-membered monocyclic or 7-10-membered bicyclic heterocyclic moiety that is either saturated or partially unsaturated, and having, in addition to carbon atoms, one or more, preferably one to four, heteroatoms, as defined above.
- nitrogen includes a substituted nitrogen.
- the nitrogen may be N (as in 3,4-dihydro-2H- pyrrolyl), NH (as in pyrrolidinyl), or + NR (as in N-substituted pyrrolidinyl).
- a heterocyclic ring can be attached to its pendant group at any heteroatom or carbon atom that results in a stable structure and any of the ring atoms can be optionally substituted.
- saturated or partially unsaturated heterocyclic radicals include, without limitation, tetrahydrofuranyl, tetrahydrothienyl, pyrrolidinyl, pyrrolidonyl, piperidinyl, pyrrolinyl,
- heterocycle used interchangeably herein, and also include groups in which a heterocyclyl ring is fused to one or more aryl, heteroaryl, or cycloaliphatic rings, such as indolinyl, 3H-indolyl, chromanyl, phenanthridinyl, 2-azabicyclo[2.2.1]heptanyl, octahydroindolyl, or tetrahydroquinolinyl, where the radical or point of attachment is on the heterocyclyl ring.
- heterocyclyl group may be mono- or bicyclic.
- heterocyclylalkyl refers to an alkyl group substituted by a heterocyclyl, wherein the alkyl and heterocyclyl portions independently are optionally substituted.
- partially unsaturated refers to a ring moiety that includes at least one double or triple bond between ring atoms but is not aromatic.
- partially unsaturated is intended to encompass rings having multiple sites of unsaturation, but is not intended to include aryl or heteroaryl moieties, as herein defined.
- alkylene refers to a bivalent alkyl group.
- An “alkylene chain” is a
- a substituted alkylene chain is a polymethylene group in which one or more methylene hydrogen atoms are replaced with a substituent. Suitable substituents include those described below for a substituted aliphatic group.
- an inhibitor refers to a compound that binds to and inhibits a KDM5 enzyme with measurable affinity and activity.
- an inhibitor has an IC 50 and/or binding constant of less about 50 ⁇ , less than about 1 ⁇ , less than about 500 nM, less man about 100 nM, or less than about 10 nM.
- measurable affinity and “measurably inhibit,” as used herein, refer to a measurable reduction in activity of a KDM5 enzyme between: (i) a sample comprising a compound a compound as described herein and such KDM5 enzyme, and (ii) an equivalent sample comprising such KDM5 enzyme, in the absence of said compound.
- “Pharmaceutically acceptable salts” include both acid and base addition salts.
- “Pharmaceutically acceptable acid addition salt” refers to those salts which retain the biological effectiveness and properties of the free bases and which are not biologically or otherwise undesirable, formed with inorganic acids such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, carbonic acid, phosphoric acid and the like, and organic acids may be selected from aliphatic, cycloaliphatic, aromatic, araliphatic, heterocyclic, carboxylic, and sulfonic classes of organic acids such as formic acid, acetic acid, propionic acid, glycolic acid, gluconic acid, lactic acid, pyruvic acid, oxalic acid, malic acid, maleic acid, maloneic acid, succinic acid, fumaric acid, tartaric acid, citric acid, aspartic acid, ascorbic acid, glutamic acid, anthranilic acid, benzoic acid, cinnamic acid, mandelic acid, embonic acid, phenylacetic acid, methanes
- “Pharmaceutically acceptable base addition salts” include those derived from inorganic bases such as sodium, potassium, lithium, ammonium, calcium, magnesium, iron, zinc, copper, manganese, aluminum salts and the like. Particularly base addition salts are the ammonium, potassium, sodium, calcium and magnesium salts. Salts derived from pharmaceutically acceptable organic nontoxic bases includes salts of primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines and basic ion exchange resins, such as isopropylamine, trimethylamine, diethylamine, triethylamine, tripropylamine, ethanolamine, 2- diethylaminoethanol.
- tromethamine dicyclohexylamine, lysine, arginine, histidine, caffeine, procaine, hydrabamine, choline, betaine, ethylenediamine, glucosamine, methylglucamine, theobromine, purines, piperizine, piperidine, N-ethylpiperidine, polyamine resins and the like.
- Particularly organic non-toxic bases are isopropylamine, diethylamine, ethanolamine,
- tromethamine dicyclohexylamine, choline, and caffeine.
- tautomer or “tautomeric form” refers to structural isomers of different energies which are interconvertible via a low energy barrier.
- proton tautomers also known as prototropic tautomers
- Valence tautomers include interconversions by reorganization of some of the bonding electrons.
- a “solvate” refers to an association or complex of one or more solvent molecules and a compound or pharmaceutically acceptable salt thereof as described herein.
- solvents include water, isopropanol, ethanol, methanol, DMSO, ethyl acetate, acetic acid and ethanolamine.
- hydrate refers to the complex where the solvent molecule is water.
- “Therapeutically effective amount” refers to an amount of a a compound or
- the therapeutically effective amount of the drug may reduce the number of cancer cells; reduce the tumor size; inhibit (i.e., slow to some extent and preferably stop) cancer cell infiltration into peripheral organs; inhibit (i.e., slow to some extent and preferably stop) tumor metastasis; inhibit, to some extent, tumor growth; and/or relieve to some extent one or more of the symptoms associated with the cancer.
- efficacy can, for example, be measured by assessing the time to disease progression (TTP) and/or determining the response rate (RR).
- TTP time to disease progression
- RR response rate
- the therapeutic effective amount is an amount sufficient to decrease or alleviate an allergic disorder, the symptoms of an autoimmune and/or inflammatory disease, or the symptoms of an acute inflammatory reaction (e.g. asthma).
- a therapeutically effective amount is an amount of a chemical entity described herein sufficient to significantly decrease the activity or number of drug tolerant or drug tolerant persisting cancer cells.
- Treatment refers to clinical intervention in an attempt to alter the natural course of the individual or cell being treated, and can be performed either for prophylaxis or during the course of clinical pathology. Desirable effects of treatment include one or more of preventing occurrence or recurrence of disease, alleviation of symptoms, diminishment of any direct or indirect pathological consequences of the disease, stabilized (i.e., not worsening) state of disease, preventing metastasis, decreasing the rate of disease progression, amelioration or palliation of the disease state, prolonging survival as compared to expected survival if not receiving treatment and remission or improved prognosis.
- a compound as described herein is used to delay development of a disease or disorder or to slow the progression of a disease or disorder.
- Those individuals in need of treatment include those already with the condition or disorder as well as those prone to have the condition or disorder, (for example, through a genetic mutation or abberent expression of a gene or protein) or those in which the condition or disorder is to be prevented.
- the compound is a compound of formula (la):
- R 1 is C 1-6 alkyl, wherein said C 1-6 alkyl is optionally substituted with one or more groups independently selected from the group consisting of oxo, hydroxy, halo, C 1- 3 alkoxy, 3-8 membered carbocyclyl, 5-10 membered aryl, -N(R a ) 2 , -N(R a )C ⁇ 0)R a , and C 1-3 alkyl.
- R 1 is halo, -N(R a ) 2 , 3-8 membered carbocyclyl, C 1-6 alkoxy, 5-10 membered aryl, wherein said 3-8 membered carbocyclyl and 5-10 membered aryl are optionally substituted with one or more groups independently selected from the group consisting of oxo, hydroxy, halo, C 1-3 alkoxy, 3-8 membered carbocyclyl, 5-10 membered aryl, -N(R a ) 2 , -N(R a )C(O)R a , and C 1-3 alky!.
- R 1 is bromo, cyclohexyl, isopropyl, isobutyl, cyclopentyl, 1 - methoxyethyl, cyclopropyl, cyclobutyl, amino, 4-phenylbut-2-yl, butyl, phenethyl, cyclopentyl, 1- (acetylamino)ethyl, or l-(hydroxyrnethylcarbonylarnino)ethyl.
- R 1 is isopropyl
- the compound is a compound of formula (lb):
- R 4 is H, methyl, or isopropyl.
- R 2 is 5-10 membered carbocyclyl that is optionally substituted with one or more groups R d .
- R is 5-10 membered heterocyclyl, that is optionally substituted with one or more groups R d .
- R 2 is selected from the group consisting of:
- R 2 is 5-10 membered aryl that is optionally substituted with one or more groups R d
- R is 5-10 membered heteroaryl that is optionally substituted with one or more groups R d .
- R 2 is selected from the group consisting of:
- R 2 is -OR a .
- R 2 is:
- R 2 is NR a R b .
- R 2 is selected from the group consisting of:
- R 2 is -C(O)N(R a ) 2 .
- R 2 is selected from the group consisting of:
- R 3 is H. In one embodiment R 3 is methyl.
- R 5 is H
- R 6 is C 1-6 alkyl or hydroxy.
- R 6 is methyl or hydroxy.
- R 6 is H.
- R 5 and R 6 taken together with the atom to which they are attached form a 3-8 membered carbocyclyl or a 3-8 membered heterocyclyl, which 3-8 membered carbocyclyl and 3-8 membered heterocyclyl are optionally substitutes with one or more groups independently selected from halo, hydroxy, cyano, C 1-6 alkoxy, and 3-8 membered carbocyclyl.
- R 5 and R 6 taken together with the atom to which they are attached form a cyclopropyl ring.
- compositions are provided.
- Another aspect includes a pharmaceutical composition
- a pharmaceutical composition comprising a a compound as described herein or a pharmaceutically acceptable salt thereof.
- the pharmaceutical composition comprising a a compound as described herein or a pharmaceutically acceptable salt thereof.
- composition further comprises a pharmaceutically acceptable carrier, adjuvant, or vehicle.
- composition further comprises an amount of the compound effective to measurably inhibit KDM5.
- the composition is formulated for
- patient refers to an animal, such as a mammal, such as a human. In one embodiment, patient or individual refers to a human.
- compositions of this invention refers to a non-toxic carrier, adjuvant, or vehicle that does not destroy the pharmacological activity of the compound with which it is formulated.
- Pharmaceutically acceptable carriers, adjuvants or vehicles that may be used in the compositions of this invention include, but are not limited to, ion exchangers, alumina, aluminum stearate, lecithin, serum proteins, such as human serum albumin, buffer substances such as phosphates, glycine, sorbic acid, potassium sorbate, partial glyceride mixtures of saturated vegetable fatty acids, water, salts or electrolytes, such as protamine sulfate, disodium hydrogen phosphate, potassium hydrogen phosphate, sodium chloride, zinc salts, colloidal silica, magnesium trisilicate, polyvinyl pyrrolidone, cellulose-based substances, polyethylene glycol, sodium carboxymethylcellulose, polyacrylates, waxes, polyethylene-polyoxypropylene-block
- compositions comprising a compound as described herein may be administered orally, parenterally, by inhalation spray, topically, transdermally, rectally, nasally, buccally, sublingually, vaginally, intraperitoneal, intrapulmonary, intradermal, epidural or via an implanted reservoir.
- parenteral as used herein includes subcutaneous, intravenous, intramuscular, intra-articular, intra-synovial, intrasternal, intrathecal, intrahepatic, intralesional and intracranial injection or infusion techniques.
- composition comprising a compound as described herein is formulated as a solid dosage form for oral administration.
- the solid oral dosage form comprising a compound as described herein further comprises one or more of (i) an inert, pharmaceutically acceptable excipient or carrier, such as sodium citrate or dicalcium phosphate, and (ii) filler or extender such as starches, lactose, sucrose, glucose, mannitol, or silicic acid, (iii) binders such as carboxymethylcellulose, alginates, gelatin, polyvinylpyrrolidinone, sucrose or acacia, (iv) humectants such as glycerol, (v) disintegrating agent such as agar, calcium carbonate, potato or tapioca starch, alginic acid, certain silicates or sodium carbonate, (vi) solution retarding agents such as paraffin, (vii) absorption accelerators such as quaternary ammonium salts, (viii) a wetting agent such as cetyl alcohol
- the solid oral dosage form is formulated as capsules, tablets or pills.
- the solid oral dosage form further comprises buffering agents.
- such compositions for solid oral dosage forms may be formulated as fillers in soft and hard-filled gelatin capsules comprising one or more excipients such as lactose or milk sugar, polyethylene glycols and the like.
- tablets, dragees, capsules, pills and granules of the compositions comprising a compound as described herein optionally comprise coatings or shells such as enteric coatings. They may optionally comprise opacifying agents and can also be of a composition that they release the active ingredients) only, or preferentially, in a certain part of the intestinal tract, optionally, in a delayed manner.
- embedding compositions include polymeric substances and waxes, which may also be employed as fillers in soft and hard-filled gelatin capsules using such excipients as lactose or milk sugar as well as high molecular weight polethylene glycols and the like.
- a composition comprises micro-encapsulated compound as described herein, and optionally, further comprises one or more excipients.
- compositions comprise liquid dosage formulations comprising a compound as described herein for oral administration, and optionally further comprise one or more of pharmaceutically acceptable emulsions, microemulsions, solutions, suspensions, syrups and elixirs.
- the liquid dosage form optionally, further comprise one or more of an inert diluent such as water or other solvent, a solubilizing agent, and an emulsifier such as ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate, propylene glycol, 1 ,3-butylene glycol, dimethylformamide, oils (in particular, cottonseed, groundnut, corn, germ, olive, castor, and sesame oils), glycerol, tetrahydrofurfuryl alcohol, polyethylene glycols or fatty acid esters of sorbitan, and mixtures thereof.
- liquid oral compositions optionally, further comprise one or
- sterile injectable aqueous or oleaginous suspensions may be formulated according to the known art using suitable dispersing or wetting agents and suspending agents.
- the sterile injectable preparation may also be a sterile injectable solution, suspension or emulsion in a nontoxic parenterally acceptable diluent or solvent, for example, as a solution in 1,3-butanediol.
- acceptable vehicles and solvents that may be employed are water, Ringer's solution, U.S.P. and isotonic sodium chloride solution.
- sterile, fixed oils are conventionally employed as a solvent or suspending medium.
- any bland fixed oil can be employed including synthetic mono- or diglycerides.
- fatty acids such as oleic acid are used in the preparation of injectables.
- Injectable formulations can be sterilized, for example, by filtration through a bacterial- retaining filter, or by incorporating sterilizing agents in the form of sterile solid compositions which can be dissolved or dispersed in sterile water or other sterile injectable medium prior to use.
- the rate of compound release can be controlled.
- biodegradable polymers include poly(orthoesters) and poly(anhydrides).
- Depot injectable formulations are also prepared by entrapping the compound in liposomes or microemulsions that are compatible with body tissues.
- the composition for rectal or vaginal administration are formulated as suppositories which can be prepared by mixing a compound as described herein with suitable non-irritating excipients or carriers such as cocoa butter, polyethylene glycol or a suppository wax, for example those which are solid at ambient temperature but liquid at body temperature and therefore melt in the rectum or vaginal cavity and release the compound.
- suitable non-irritating excipients or carriers such as cocoa butter, polyethylene glycol or a suppository wax, for example those which are solid at ambient temperature but liquid at body temperature and therefore melt in the rectum or vaginal cavity and release the compound.
- Example dosage forms for topical or transdermal administration of a compound as described herein include ointments, pastes, creams, lotions, gels, powders, solutions, sprays, inhalants or patches.
- the compound as described herein is admixed under sterile conditions with a pharmaceutically acceptable carrier, and optionally preservatives or buffers. Additional
- Transdermal dosage forms can be made by dissolving or dispensing the compound as described herein in medium, for example ethanol or dimethylsulfoxide.
- Absorption enhancers can also be used to increase the flux of the compound across the skin. The rate can be controlled by either providing a rate controlling membrane or by dispersing the compound in a polymer matrix or gel.
- Nasal aerosol or inhalation formulations of a compound as described herein may be prepared as solutions in saline, employing benzyl alcohol or other suitable preservatives, absorption promoters to enhance bioavailability, fluorocarbons, and/or other conventional solubilizing or dispersing agents.
- compositions may be administered with or without food. In certain embodiments, pharmaceutically acceptable compositions are administered without food. In certain embodiments, pharmaceutically acceptable compositions of this invention are administered with food.
- Specific dosage and treatment regimen for any particular patient will depend upon a variety of factors, including age, body weight, general health, sex, diet, time of administration, rate of excretion, drug combination, the judgment of the treating physician, and the severity of the particular disease being treated.
- the amount of a compound as described herein in the composition will also depend upon the particular compound in the composition.
- the therapeutically effective amount of the compound of the invention administered parenterally per dose will be in the range of about 0.01-100 mg/kg, alternatively about
- oral unit dosage forms such as tablets and capsules, contain from about 5 to about 100 mg of the compound of the invention.
- An example tablet oral dosage form comprises about 2 mg, 5 mg, 25 mg, 50 mg, 100 mg, 250 mg or 500 mg of a compound as described herein, and further comprises about 95-30 mg anhydrous lactose, about 5-40 mg sodium croscarmellose, about 5-30 mg polyvinylpyrrolidone (PVP) K30 and about 1-10 mg magnesium stearate.
- the process of formulating the tablet comprises mixing the powdered ingredients together and further mixing with a solution of the PVP.
- the resulting composition can be dried, granulated, mixed with the magnesium stearate and compressed to tablet form using conventional equipment.
- An example of an aerosol formulation can be prepared by dissolving about 2-500 mg of a compound as described herein, in a suitable buffer solution, e.g. a phosphate buffer, and adding a tonicifier, e.g. a salt such sodium chloride, if desired.
- the solution may be filtered, e.g. using a 0.2 micron filter, to remove im
- Another aspect includes the use of a compound as described herein for the inhibition of KDM5.
- Ccompounds as described herein may also be used to inhibit the removal of methyl marks on histone lysine residues, including inhibiting the removal of methyl marks from mono-, di- or tri- methylation of histones HI, H2A, H2B, H3 and H4, such as H3K4 (including for example the KDM5 substrate H3K4me3), thereby altering interactions of these histone proteins with DNA and/or other proteins, and altering certain subsequent genetic or protein expression.
- Compounds as described herein may also be used to inhibit KDM5 and reduce drug-tolerant cells, thereby treating or preventing drug-resistant diseases, such as drug-resistant cancer.
- the disease can be treated using a compound as described herein to prevent resistance from forming, for example before targets of chemotherapies become mutated to confer resistance to such
- the binding or inhibition activity of a compound as described herein may be determined by running a competition experiment where the is incubated with the KDM5 enzyme bound to known radioligands.
- Detailed conditions for assaying a compound as an inhibitor of KDM5 or a mutant thereof are set forth in the Examples below.
- detection of KDM5 activity is achieved with in vitro assays, which can be either direct binding (non-catalytic) or enzymatic (catalytic) asssays.
- Types of substrates that are used in such assays may include: short synthetic peptides corresponding to a number of residues from the N-terminus of histone sequences comprising the target lysine residue, single recombinant histone polypeptides, histone octamers reconstituted with recombinant histone proteins, and reconstituted nucleosomes (using reconstituted octamers and specific recombinant DNA fragments).
- the reconstituted nucleosomes may be mononucleosomes or oligonucleosomes.
- Another aspect includes a method of treating or preventing a disease responsive to the inhibition of KDM5 activity in a patient.
- the method includes administering a therapeutically effective amount of a compound as described hereinto a patient in need thereof.
- Another aspect includes the use of a compound as described herein, in therapy.
- Another aspect includes the use of a pharmaceutical composition comprising a compound as described herein, in therapy.
- Another aspect includes the use of a compound as described herein, in treating a disease associated with KDM5 activity.
- Another aspect includes the use of a pharmaceutical composition comprising a compound as described herein, in treating a disease associated with KDM5 activity.
- Another aspect includes the use of a compound as described herein, in the manufacture of a medicament for the treatment of a disease associated with KDM5 activity.
- Another aspect includes the use of a pharmaceutical composition comprising a compound as described herein, in the manufacture of a medicament for the treatment of a disease associated with KDM5 activity.
- the disease or condition is a hyperproliferative disease, cancer, stroke, diabetes, hepatomegaly, cardiovascular disease, multiple sclerosis, Alzheimer's disease, cystic fibrosis, viral disease, autoimmune diseases, atherosclerosis, restenosis, psoriasis, rheumatoid arthritis, inflammatory bowel disease, asthma, allergic disorders, inflammation, neurological disorders, a hormone-related disease, conditions associated with organ transplantation, immunodeficiency disorders, destructive bone disorders, proliferative disorders, infectious diseases, conditions associated with cell death, thrombin-induced platelet aggregation, liver disease, pathologic immune conditions involving T cell activation, CNS disorders or a myeloproliferative disorder.
- treatment may be administered after one or more symptoms have developed. In other embodiments, treatment may be administered in the absence of symptoms. For example, treatment may be administered to a susceptible individual prior to the onset of symptoms (e.g., in light of a history of symptoms and/or in light of genetic or other susceptibility factors). Treatment may also be continued after symptoms have resolved, for example to prevent or delay their recurrence.
- Another aspect includes a method for treating, ameliorating or preventing cancer, drug- resistant cancer or another proliferative disorder by administration of an effective amount of a compound as described herein to a mammal, for example a human, in need of such treatment.
- the disease to be treated is cancer or drug resistant cancer.
- cancers that may be treated using the compounds and methods described herein include, but are not limited to, adrenal cancer, acinic cell carcinoma, acoustic neuroma, acral lentigious melanoma, acrospiroma, acute eosinophilic leukemia, acute erythroid leukemia, acute lymphoblastic leukemia, acute megakaryoblastic leukemia, acute monocytic leukemia, acute promyelocyte leukemia, adenocarcinoma, adenoid cystic carcinoma, adenoma, adenomatoid odontogenic tumor, adenosquamous carcinoma, adipose tissue neoplasm, adrenocortical carcinoma, adult T-cell leukemia/lymphoma, aggressive NK-cell leukemia, AlDS-related lymphoma, alveolar rhabdomyosarcoma, alveolar soft part sarcoma, ameloblastic a
- dysembryoplastic neuroepithelial tumor dysgerminoma, embryonal carcinoma, endocrine gland neoplasm, endodermal sinus tumor, enteropathy-associated T-cell lymphoma, esophageal cancer, fetus in fetu, fibroma, fibrosarcoma, follicular lymphoma, follicular thyroid cancer,
- ganglioneuroma gastrointestinal cancer, germ cell tumor, gestational choriocarcinoma, giant cell fibroblastoma, giant cell tumor of the bone, glial tumor, glioblastoma multiforme, glioma, gliomatosis cerebri, glucagonoma, gonadoblastoma, granulosa cell tumor, gynandroblastoma, gallbladder cancer, gastric cancer, hemangioblastoma, head and neck cancer, hemangiopericytoma, hematological malignancy, hepatoblastoma, hepatosplenic T-cell lymphoma, Hodgkin's
- lymphoma non-Hodgkin's lymphoma, invasive lobular carcinoma, intestinal cancer, kidney cancer, laryngeal cancer, lentigo maligna, leukemia, leydig cell tumor, liposarcoma, lung cancer, lymphangioma, lymphangiosarcoma, lymphoepithelioma, lymphoma, acute lymphocytic leukemia, acute myelogeous leukemia, chronic lymphocytic leukemia, liver cancer, small cell lung cancer, non-small cell lung cancer, MALT lymphoma, malignant fibrous histiocytoma, malignant peripheral nerve sheath tumor, malignant triton tumor, mantle cell lymphoma, marginal zone B-cell lymphoma, mast cell leukemia, mediastinal germ cell tumor, medullary carcinoma of the breast, medullary thyroid cancer, medulloblastoma, melanoma, meningioma, merkel cell cancer, mesot
- Another embodiment includes a method for the treatment of benign proliferative disorders.
- benign proliferative disorders include, but are not limited to, benign soft tissue rumors, bone tumors, brain and spinal tumors, eyelid and orbital tumors, granuloma, lipoma, meningioma, multiple endocrine neoplasia, nasal polyps, pituitary tumors, prolactinoma, pseudotumor cerebri, seborrheic keratoses, stomach polyps, thyroid nodules, cystic neoplasms of the pancreas, hemangiomas, vocal cord nodules, polyps, and cysts, Castleman disease, chronic pilonidal disease, dermatofibroma, pilar cyst, pyogenic granuloma and juvenile polyposis syndrome.
- Another embodiment includes a therapeutic method useful for modulating protein methylation, gene expression, cell proliferation, cell differentiation and/or apoptosis in vivo in diseases mentioned above, in particular cancer, comprising administering to a patient in need of such therapy a pharmacologically active and therapeutically effective amount of one or more of the compounds as described herein.
- Another embodiment includes a method for regulating endogenous or heterologous promotor activity by contacting a cell with a compound as described herein.
- Another embodiment includes the use of a compound as described herein for the production of pharmaceutical compositions which are employed for the treatment and/or prophylaxis and/or amelioration of the diseases, disorders, illnesses and/or conditions as mentioned herein.
- Another embodiment includes the use of a compound as described herein for the production of pharmaceutical compositions which are employed for the treatment and/or prophylaxis of diseases and/or disorders responsive or sensitive to the inhibition of histone demethylases, particularly those diseases mentioned above, such as e.g. cancer.
- Compounds as described herein may be administered using any amount and any route of administration effective for treating or lessening the severity of the disorder.
- the exact amount required will vary from patient to patient, depending on the species, age, and general condition of the patient, for example the severity of the disorder, the particular compound, its mode of administration, and the like.
- the total daily usage of a compound as described herein by a given patient will be decided by the attending physician within the scope of sound medical judgment.
- the specific effective dose level for any particular patient will depend upon a variety of factors including the disorder being treated and the severity of the disorder; the activity of the specific compound employed; the specific composition employed; the age, body weight, general health, sex and diet of the patient; the time of administration, route of administration, and rate of excretion of the specific compound employed; the duration of the treatment; drugs used in combination or coincidental with the specific compound employed, and like factors well known in the medical arts.
- Another embodiment includes a method of inhibiting KDM5 activity in a biological sample comprising contacting said biological sample with a compound as described herein.
- biological sample includes, without limitation, a cell, cell cultures or extracts thereof; biopsied material obtained from a mammal or extracts thereof; and blood, saliva, urine, feces, semen, tears, or other body fluids or extracts thereof.
- the compound as described herein may be employed alone or in combination with other agents for treatment.
- the second agent of the pharmaceutical combination may be employed alone or in combination with other agents for treatment.
- the second agent of the pharmaceutical combination may be employed alone or in combination with other agents for treatment.
- formulation or dosing regimen may have complementary activities to the compound as described herein such that they do not adversely affect each other.
- the compounds may be administered together in a unitary pharmaceutical composition or separately.
- a compound or a pharmaceutically acceptable salt can be co-administered with a cytotoxic agent to treat proliferative diseases and cancer.
- co-administering refers to either simultaneous admuiistration, or any manner of separate sequential administration, of a compound as described herein, and a further active pharmaceutical ingredient or ingredients, including cytotoxic agents and radiation treatment. If the admimstration is not simultaneous, the compounds are administered in a close time proximity to each other. Furthermore, it does not matter if the compounds are administered in the same dosage form, e.g. one compound may be administered topically and another compound may be
- any agent that has activity against a disease or condition being treated may be co- administered.
- agents can be found in Cancer Principles and Practice of
- the treatment method includes the co-administration of a compound as described herein and at least one cytotoxic agent.
- cytotoxic agent refers to a substance that inhibits or prevents a cellular function and/or causes cell death or destruction.
- Cytotoxic agents include, but are not limited to, radioactive isotopes (e.g., At 211 , 1 131 , 1 125 , Y 90 , Re 186 , Re 188 , Sm 153 , Bi 212 , P 32 , Pb 212 and radioactive isotopes of Lu); chemotherapeutic agents; growth inhibitory agents; enzymes and fragments thereof such as nucleolytic enzymes; and toxins such as small molecule toxins or enzymatically active toxins of bacterial, fungal, plant or animal origin, including fragments and/or variants thereof.
- radioactive isotopes e.g., At 211 , 1 131 , 1 125 , Y 90 , Re 186 , Re 188 , Sm 153 , Bi 212 , P 32 , Pb 212 and radioactive isotopes of Lu
- chemotherapeutic agents e.g., At 211 , 1 131 , 1 125
- Exemplary cytotoxic agents can be selected from anti-microtubule agents, platinum coordination complexes, alkylating agents, antibiotic agents, topoisomerase II inhibitors, antimetabolites, topoisomerase I inhibitors, hormones and hormonal analogues, signal transduction pathway inhibitors, non-receptor tyrosine kinase angiogenesis inhibitors, immunotherapeutic agents, proapoptotic agents, nhibitors of LDH-A; inhibitors of fatty acid biosynthesis; cell cycle signaling inhibitors; HDAC inhibitors, proteasome inhibitors; and inhibitors of cancer metabolism.
- “Chemotherapeutic agent” includes chemical compounds useful in the treatment of cancer.
- chemotherapeutic agents include erlotinib (TARCEVA ® , Genentech/OSI Pharm.), bortezomib (VELCADE ® , Millennium Pharm.), disulfiram , epigallocatechin gallate , salinosporamide A, carfilzomib, 17- AAG(geldanamycin), radicicol, lactate dehydrogenase A (LDH-A), fulvestrant (FASLODEX ® , AstraZeneca), sunitib (SUTENT ® , Pfizer/Sugen), letrozole (FEMARA ® , Novartis), imatinib mesylate (GLEEVEC ® ., Novartis), finasunate (VATALANIB ® , Novartis), oxaliplatin (ELOXATIN ® , Sanofi), 5-FU (5-
- Lonafamib (SCH 66336), sorafenib (NEXAVAR ® , Bayer Labs), gefitinib (IRESSA ® ,
- alkylating agents such as thiotepa and CYTOXAN ® cyclosphosphamide
- alkyl sulfonates such as busulfan, improsulfan and piposulfan
- aziridines such as benzodopa, carboquone, meturedopa, and uredopa
- ethylenimines and methylamelamines including altretamine, triethylenemelamine, triemylenephosphoramide, triethylenethiophosphoramide and
- nitrogen mustards such as chlorambucil, chlomaphazine, chlorophosphamide, estramustine, ifosfamide, mechlorethamine, mechlorethamine oxide hydrochloride, melphalan, novembichin, phenesterine, prednimustine, trofosfamide, uracil mustard; nitrosoureas such as carmustine, chlorozotocin, fotemustine, iomustine, nimustine, and ranimnustine; antibiotics such as the enediyne antibiotics (e.g., calicheamicin, especially calicheamicin ⁇ I and calicheamicin ⁇ 11 (Angew Chem. Intl. Ed. Engl. 1994 33:183-186); dynemicin, including dynemicin A;
- bisphosphonates such as clodronate; an esperamicin; as well as neocarzinostatin chromophore and related chromoprotein enediyne antibiotic chromophores), aclacinomysins, actinomycin, authramycin, azaserine, bleomycins, cactinomycin, carabicin, caminomycin, carzinophilin, chromomycinis, dactinomycin, daunorubicin, detorubicin, 6-diazo-5-oxo-L-norleucine,
- ADRIAMYCIN ® (doxorubicin), morpholino-doxorubicin. cyanomorpholino-doxorubicin, 2- pyrrolino-doxorubicin and deoxydoxorubicin), epirubicin, esorubicin, idarubicin, marcellomycin, mitomycins such as mitomycin C, mycophenolic acid, nogalamycin, olivomycins, peplomycin, porfiromycin, puromycin, quelamycin, rodorubicin, streptonigrin, streptozocin, tubercidin, ubenimex, zinostatin, zorubicin; anti-metabolites such as methotrexate and 5-fluorouracil (5-FU); folic acid analogs such as denopterin, methotrexate, pteropterin, trimetrexate; purine analogs such as fludarabine, 6-mercaptopurine, thiamiprine, thioguanine;
- TAXOTERE ® docetaxel, doxetaxel; Sanofi-Aventis); chloranmbucil; GEMZAR ® (gemcitabine); 6-thioguanine; mercaptopurine; methotrexate; platinum analogs such as cisplatin and carboplatin; vinblastine; etoposide (VP-16); ifosfamide; mitoxantrone; vincristine; NAVELBINE ®
- XELODA ® ibandronate
- CPT-11 topoisomerase inhibitor
- RFS 2000 difluoromemylornithine
- retinoids such as retinoic acid
- Chemotherapeutic agent also includes (i) anti-hormonal agents that act to regulate or inhibit hormone action on tumors such as anti-estrogens and selective estrogen receptor modulators (SERMs), including, for example, tamoxifen (including NOLVADEX ® ; tamoxifen citrate), raloxifene, droloxifene, iodoxyfene , 4-hydroxytamoxifen, trioxifene, keoxifene, LY1 17018, onapristone, and FARESTON ® (toremifine citrate); (ii) aromatase inhibitors that inhibit the enzyme aromatase, which regulates estrogen production in the adrenal glands, such as, for example, 4(5)- irnidazoles, aminoglutethimide, MEGASE ® (megestrol acetate), AROMASIN ® (exemestane;
- SERMs selective estrogen receptor modulators
- Pfizer formestanie, fadrozole, RTVISOR ® (vorozole), FEMARA ® (Ietrozole; Novartis), and ARIMIDEX ® (anastrozole; AstraZeneca);
- anti-androgens such as flutamide, nilutamide, bicalutamide, leuprolide and goserelin; buserelin, tripterelin, medroxyprogesterone acetate, diethylstilbestrol, premarin, fluoxymesterone, all transretionic acid, fenretinide, as well as troxacitabine (a 1,3-dioxolane nucleoside cytosine analog);
- protein kinase inhibitors v
- antisense oligonucleotides particularly those which inhibit expression of genes in signaling pathways implicated in aberrant cell proliferation, such as, for example, P
- Additional humanized monoclonal antibodies with therapeutic potential as agents in combination with the compounds of the invention include: apolizumab, aselizumab, atlizumab, bapineuzumab, bivatuzumab mertansine, cantuzumab mertansine, cedelizumab, certolizumab pegol, cidfusituzumab, cidtuzumab, daclizumab, eculizumab, efalizumab, epratuzumab, erlizumab, felvizumab, fontolizumab, gemtuzumab ozogamicin, inotuzumab ozogamicin, ipilimumab, labetuzumab, lintuzumab, matuzumab, mepolizumab, motavizumab, motovizumab, natalizumab, nimotuzumab, nolovizum
- Chemotherapeutic agent also includes "EGFR inhibitors,” which refers to compounds that bind to or otherwise interact directly with EGFR and prevent or reduce its signaling activity, and is alternatively referred to as an "EGFR antagonist.”
- EGFR inhibitors refers to compounds that bind to or otherwise interact directly with EGFR and prevent or reduce its signaling activity
- Examples of such agents include antibodies and small molecules that bind to EGFR.
- antibodies which bind to EGFR include MAb 579 (ATCC CRL HB 8506), MAb 455 (ATCC CRL HB8507), MAb 225 (ATCC CRL 8508), MAb 528 (ATCC CRL 8509) (see, US Patent No.
- EMD 55900 Stragliotto et al. Eur. J. Cancer 32A:636-640 (1996)
- EMD7200 (matuzumab) a humanized EGFR antibody directed against EGFR that competes with both EGF and TGF-alpha for EGFR binding (EMD/Merck); human EGFR antibody, HuMax-EGFR (GenMab); fully human antibodies known as E 1.1 , E2.4, E2.5, E6.2, E6.4, E2.l l, E6. 3 and E7.6. 3 and described in US 6,235,883; MDX-447 (Medarex Inc); and mAb 806 or humanized mAb 806 (Johns et al., J. Biol.
- the anti-EGFR antibody may be conjugated with a cytotoxic agent, thus generating an immunoconjugate (see, e.g., EP659.439A2, Merck Patent GmbH).
- EGFR antagonists include small molecules such as compounds described in US Patent Nos: 5,616,582, 5,457,105, 5,475,001, 5,654,307, 5,679,683, 6,084,095, 6,265,410, 6,455,534, 6,521,620, 6,596,726, 6,713,484, 5,770,599, 6,140,332, 5,866,572, 6,399,602,
- EGFR antagonists include OSI-774 (CP-358774, erlotinib, TARCEV A ® Genentech/OSI Pharmaceuticals); PD 183805 (CI 1033, 2-propenamide, N-[4-[(3-chloro-4- fluorophenyl)arnino]-7-[3-(4-morpholinyl)propoxy]-6-quirlazolinyl]-, dihydrochloride, Pfizer Inc.); ZD1839, gefitinib (IRESSA®) 4-(3'-Chloro-4'-fluoroanilino)-7-methoxy-6-(3-morpholinopropoxy)quinazoline, AstraZeneca); ZM 105180 ((6-amino-4-(3-methylphenyl-amino)- quinazoline, Zeneca); BIBX-1382 (N8K3-chloro-4-fluoro-phenyl)-N2-(l-methyI
- Chemotherapeutic agents also include "tyrosine kinase inhibitors" including the EGFR- targeted drugs noted in the preceding paragraph; small molecule HER2 tyrosine kinase inhibitor such as TAK165 available from Takeda; CP-724,714, an oral selective inhibitor of the ErbB2 receptor tyrosine kinase (Pfizer and OSI); dual-HER inhibitors such as EKB-569 (available from Wyeth) which preferentially binds EGFR but inhibits both HER2 and EGFR-overexpressing cells; lapatinib (GSK572016; available from Glaxo-SmithKIine), an oral HER2 and EGFR tyrosine kinase inhibitor; PKI-166 (available from Novartis); pan-HER inhibitors such as canertinib (CI- 1033; Pharmacia); Raf-1 inhibitors such as antisense agent ISIS-5132 available from ISIS
- non-HER targeted TK inhibitors such as imatinib mesylate (GLEEVEC®, available from Glaxo SmithKline); multi-targeted tyrosine kinase inhibitors such as sunitinib (SUTENT®, available from Pfizer); VEGF receptor tyrosine kinase inhibitors such as vatalanib (PTK787/ZK222584, available from Novartis/Schering AG); MAPK extracellular regulated kinase I inhibitor CI- 1040 (available from Pharmacia); quinazolines, such as PD 153035,4-(3-chloroanilino) quinazoline; pyridopyrimidines; pyrimidopyrimidines;
- pyrrolopyrimidines such as CGP 59326, CGP 60261 and CGP 62706; pyrazolopyrimidines, 4- (phenylamino)-7H-pyrrolo[2,3-d] pyrimidhies; curcumin (diferuloyl methane, 4,5-bis (4- fluoroanilino)phthalimide); tyrphostines containing nitrothiophene moieties; PD-0183805 (Warner- Lamber); antisense molecules (e.g. those that bind to HER-encoding nucleic acid); quinoxalines (US Patent No. 5,804,396); tryphostins (US Patent No.
- Cbemotherapeutic agents also include dexamethasone, interferons, colchicine, metoprine, cyclosporine, amphotericin, metronidazole, alemtuzumab, alitretinoin, allopurinol, amifostine, arsenic trioxide, asparaginase, BCG live, bevacuzimab, bexarotene, cladribine, clofarabine, darbepoetin alfa, denileukin, dexrazoxane, epoetin alfa, elotinib, filgrastim, histrelin acetate, ibritumomab, interferon alfa-2a, interferon alfa-2b, lenalidomide, levamisole, mesna, methoxsalen, nandrolone, nelarabine, nofetumomab, oprelvekin
- Chemotherapeutic agents also include hydrocortisone, hydrocortisone acetate, cortisone acetate, tixocortol pivalate, triamcinolone acetonide, triamcinolone alcohol, mometasone, amcinonide, budesonide, desonide, fluocinonide, fluocinolone acetonide, betamethasone, betamethasone sodium phosphate, dexamethasone, dexamethasone sodium phosphate,
- fluocortolone hydrocortisone- 17-butyrate, hydrocortisone- 17- valerate, aclometasone dipropionate, betamethasone valerate, betamethasone dipropionate, prednicarbate, clobetasone- 17-butyrate, clobetasol-17-propionate, fluocortolone caproate, fluocortolone pivalate and fluprednidene acetate; immune selective anti-inflammatory peptides (ImSAIDs) such as phenylalanine-glutamine-glycine (FEG) and its D-isomeric form (feG) (IMULAN BioTherapeutics, LLC); anti-rheumatic drugs such as azathioprine, cyclosporin (cyclosporine A), D-penicillamine, gold salts, hydroxychloroquine, leflunomideminocycline, sulfasalazine, tumor necros
- costimulation blockers such as abatacept (Orencia), Interleukin 6 (IL-6) blockers such as tocilizumab (ACTEMERA®); Interleukin 13 (IL-13) blockers such as lebrikizumab; Interferon alpha (IFN) blockers such as Rontalizumab; Beta 7 integrin blockers such as rhuMAb Beta7; IgE pathway blockers such as Anti-Mi prime; Secreted homotrimeric LTa3 and membrane bound heterotrimer LTal/p2 blockers such as Anti-lymphotoxin alpha (LTa); radioactive isotopes (e.g., At 211 , 1 13 ', I 125 , Y 90 , Re 186 , Re 188 , Sm 153 , Bi 212 , P 32 , Pb 212 and radioactive isotopes of Lu);
- radioactive isotopes e.g., At 211 , 1 13 ', I 125 ,
- miscellaneous investigational agents such as thioplatin, PS-341, phenylbutyrate, ET-18- OCH 3 , or famesyl transferase inhibitors (L-739749, L-744832); polyphenols such as quercetin, resveratrol, piceatannol, epigallocatechine gallate, theaflavins, flavanols, procyanidins, betulinic acid and derivatives thereof; autophagy inhibitors such as chloroquine; delta-9-tetrahydrocannabinol (dronabinol, MARTNOL®); beta-lapachone; lapachol; colchicines; betulinic acid;
- UTORAL® bexarotene
- TARGRETIN® bisphosphonates such as clodronate (for example, BONEFOS® or OSTAC®), etidronate (DIDROCAL®), NE-58095, zoledronic acid/zoledronate (ZOMETA®), alendronate (FOSAMAX®), pamidronate (AREDIA®), tiludronate (SKELID®), or risedronate (ACTONEL®); and epidermal growth factor receptor (EGF-R); vaccines such as THERATOPE® vaccine; perifosine, COX-2 inhibitor (e.g. celecoxib or etoricoxib), proteosome inhibitor (e.g.
- COX-2 inhibitor e.g. celecoxib or etoricoxib
- proteosome inhibitor e.g.
- PS341) CCI-779; tipifarnib (R11577); orafenib, ABT510; Bcl-2 inhibitor such as oblimersen sodium (GENASENSE®); pixantrone; farnesyltransferase inhibitors such as lonafarnib
- SCH 6636 SARASAR TM
- pharmaceutically acceptable salts, acids or derivatives of any of the above as well as combinations of two or more of the above such as CHOP, an abbreviation for a combined therapy of cyclophosphamide, doxorubicin, vincristine, and prednisolone; and
- FOLFOX an abbreviation for a treatment regimen with oxaliplatin (ELOXATINTM) combined with 5-FU and leucovorin.
- Chemotherapeutic agents also include non-steroidal anti-inflammatory drugswith analgesic, antipyretic and anti-inflammatory effects.
- NSAIDs include non-selective inhibitors of the enzyme cyclooxygenase.
- Specific examples of NSAIDs include aspirin, propionic acid derivatives such as ibuprofen, fenoprofen, ketoprofen, flurbiprofen, oxaprozin and naproxen, acetic acid derivatives such as indomethacin, sulindac, etodolac, diclofenac, enolic acid derivatives such as piroxicam, meloxicam, tenoxicam, droxicam, lornoxicam and isoxicam, fenamic acid derivatives such as mefenamic acid, meclofenamic acid, flufenamic acid, tolfenamic acid, and COX-2 inhibitors such as celecoxib, etoricoxib, lumirac
- NSAIDs can be indicated for the symptomatic relief of conditions such as rheumatoid arthritis, osteoarthritis, inflammatory arthropathies, ankylosing spondylitis, psoriatic arthritis, Reiter's syndrome, acute gout, dysmenorrhoea, metastatic bone pain, headache and migraine, postoperative pain, mild-to- moderate pain due to inflammation and tissue injury, pyrexia, ileus, and renal colic.
- conditions such as rheumatoid arthritis, osteoarthritis, inflammatory arthropathies, ankylosing spondylitis, psoriatic arthritis, Reiter's syndrome, acute gout, dysmenorrhoea, metastatic bone pain, headache and migraine, postoperative pain, mild-to- moderate pain due to inflammation and tissue injury, pyrexia, ileus, and renal colic.
- Chemotherapeutic agents also include treatments for Alzheimer's Disease such as donepezil hydrochloride and rivastigmine; treatments for Parkinson's Disease such as L-
- DOPA/carbidopa DOPA/carbidopa, entacapone, ropinrole, pramipexole, bromocriptine, pergolide, trihexephendyl, and amantadine
- agents for treating multiple sclerosis (MS) such as beta interferon (e.g., Avonex and Rebif ® ), glatiramer acetate, and mitoxantrone
- treatments for asthma such as albuterol and montelukast sodium
- agents for treating schizophrenia such as zyprexa, risperdal, seroquel, and haloperidol
- anti-inflammatory agents such as corticosteroids, TNF blockers, IL-1 RA,
- immunomodulatory and immunosuppressive agents such as cyclosporin, tacrolimus, rapamycin, mycophenolate mofetil, interferons,
- corticosteroids corticosteroids, cyclophophamide, azathioprine, and sulfasalazine
- neurotrophic factors such as acetylcholinesterase inhibitors, MAO inhibitors, interferons, anti-convulsants, ion channel blockers, riluzole, and anti-Parkinsonian agents
- agents for treating cardiovascular disease such as beta- blockers, ACE inhibitors, diuretics, nitrates, calcium channel blockers, and statins
- agents for treating liver disease such as corticosteroids, cholestyramine, interferons, and anti-viral agents
- agents for treating blood disorders such as corticosteroids, anti-leukemic agents, and growth factors
- immunodeficiency disorders such as gamma globulin.
- chemotherapeutic agents include pharmaceutically acceptable salts, acids or derivatives of any of chemotherapeutic agents, described herein, as well as combinations of two or more of them.
- compositions of this invention are formulated such that a dosage of between 0.01 - 100 mg/kg body weight/day of an inventive can be administered.
- the additional therapeutic agent and the compound as described herein may act synergistically. Therefore, the amount of additional therapeutic agent in such compositions may be less than that required in a monotherapy utilizing only that therapeutic agent, or there may be fewer side effects for the patient given that a lower dose is used.
- a dosage of between 0.01 - 1,000 ⁇ g/kg body weight/day of the additional therapeutic agent can be administered.
- Another aspect includes treating or preventing drug resistance in a patient using a compound as described herein.
- a method of treating or preventing drug resistant cancer in a patient comprises administering a therapeutically effective amount of a compound as described herein to the patient alone or in combination with a cytotoxic agent.
- the individual is selected for treatment with a cytotoxic agent (e.g., targeted therapies, chemotherapies, and/or radiotherapies).
- a cytotoxic agent e.g., targeted therapies, chemotherapies, and/or radiotherapies.
- the individual starts treatment comprising administration of a compound as described herein prior to treatment with the cytotoxic agent
- the individual concurrently receives treatment comprising the compound as described herein and the cytotoxic agent.
- the compound as described herein increases the period of cancer sensitivity and/or delays development of cancer resistance.
- a compound as described herein comprising administering to the individual (a) a compound as described herein and (b) a cytotoxic agent (e.g., targeted therapy, chemotherapy, and/or radiotherapy).
- a cytotoxic agent e.g., targeted therapy, chemotherapy, and/or radiotherapy.
- the respective amounts of the compound as described herein and the cytotoxic agent are effective to increase the period of cancer sensitivity and/or delay the development of cancer cell resistance to the cancer therapy agent.
- the respective amounts of the compound as described herein and the cytotoxic agent are effective to increase efficacy of a cancer treatment comprising the cancer therapy agent.
- the respective amounts of the compound as described herein and the cytotoxic agent are effective to increase efficacy compared to a treatment (e.g., standard of care treatment) (e.g., standard of care treatment) comprising administering an effective amount of the cancer therapy agent without (in the absence of) the compound as described herein.
- a treatment e.g., standard of care treatment
- the respective amounts of the compound as described herein and cytotoxic agent agent are effective to increase response (e.g., complete response) compared to a treatment (e.g., standard of care treatment) comprising administering an effective amount of cytotoxic agent without (in the absence of) the compound as described herein.
- methods of increasing efficacy of a cancer treatment comprising a cytotoxic agent in an individual comprising administering to the individual (a) an effective amount of a compound as described herein and (b) an effective amount of the cytotoxic agent.
- cancer treatment comprising administering to the individual (a) an effective amount of a compound as described herein and (b) an effective amount of a cytotoxic agent, wherein the cancer treatment has increased efficacy compared to a treatment (e.g., standard of care treatment) comprising administering an effective amount of cytotoxic agent without (in the absence of) the compound as described herein.
- a treatment e.g., standard of care treatment
- kits for delaying and/or preventing development of cancer resistant to a cancer therapy agent in an individual comprising administering to the individual (a) an effective amount of a compound as described herein and (b) an effective amount of the cytotoxic agent.
- kits for treating an individual with cancer who has an increased likelihood of developing resistance to a cancer therapy agent comprising administering to the individual (a) an effective amount of a compound as described herein and (b) an effective amount of the cytotoxic agent.
- kits for increasing sensitivity to a cancer therapy agent in an individual with cancer comprising administering to the individual (a) an effective amount of a compound as described herein and (b) an effective amount of the cytotoxic agent.
- kits for extending the period of a cancer therapy agent sensitivity in an individual with cancer comprising administering to the individual (a) an effective amount of a compound as described herein and (b) an effective amount of the cytotoxic agent.
- the cytotoxic agent is a targeted therapy.
- the targeted therapy is one or more of an EGFR antagonist, RAF inhibitor, and/or PI3K inhibitor.
- the targeted therapy is an EGFR antagonist.
- the EGFR antagonist is N-(3-ethynylphenyl)-6,7- bis(2-methoxyethoxy)-4-quinazolinamine and/or a pharmaceutical acceptable salt thereof.
- the EGFR antagonist is N-(3-ethynylphenyl)-6,7-bis(2-methoxyethoxy)-4- quinazolinamine.
- the EGFR antagonist is N-(4-(3-fluorobenzyloxy)-3- chlorophenyl)-6-(5-((2-(methylsulfony])ethylam ino)methyl)furan-2-yl)quinazoliin-4-amine,di4- methylbenzenesulfonate or a pharmaceutically acceptable salt thereof (e.g. , lapatinib).
- targeted therapy is a RAF inhibitor.
- the RAF inhibitor is a BRAF inhibitor.
- the RAF inhibitor is a CRAF inhibitor.
- the BRAF inhibitor is vemurafenib.
- the RAF inhibitor is 3-(2-cyanopropan-2-yl)-N-(4-methyl-3-(3-methyl-4- oxo-3,4-dihydroquinazolin-6-ylamino)phenyl)benzamide or a pharmaceutically acceptable salt thereof (e.g., AZ628 (CAS# 878739-06-1)).
- the targeted therapy is a PI3K inhibitor.
- the cytotoxic agent is chemotherapy.
- the chemotherapy is a taxane.
- the taxane is paclitaxel. in certain embodiments, the taxane is docetaxel.
- the cytotoxic agent is a platinum agent. In certain embodiments, the platinum agent is carboplatin. In certain embodiments, the platinum agent is cisplatin. In certain embodiments of any of the methods, the cytotoxic agent is a taxane and a platinum agent. In certain embodiments, the taxane is paclitaxel. In certain embodiments, the taxane is docetaxel. In certain embodiments, the platinum agent is carboplatin. In certain embodiments, the platinum agent is cisplatin.
- the cytotoxic agent is a vinca alkyloid. In certain embodiments, the vinca alkyloid is vinorelbine. In certain embodiments of any of the methods, the chemotherapy is a nucleoside analog. In certain embodiments, the nucleoside analog is gemcitabine.
- the cytotoxic agent is radiotherapy.
- the compound as described herein is concomitantly administered with the cytotoxic agent (e.g., targeted therapy, chemotherapy, and/or radiotherapy). In certain embodiments, the compound as described herein is administered prior to and/or concurrently with the cytotoxic agent (e.g., targeted therapy, chemotherapy, and/or radiotherapy).
- the cytotoxic agent e.g., targeted therapy, chemotherapy, and/or radiotherapy.
- the cancer is lung cancer, breast cancer, pancreatic cancer, colorectal cancer, and/or melanoma.
- the cancer is lung.
- the lung cancer is NSCLC.
- the cancer is breast cancer.
- the cancer is melanoma. EXEMPLIFICATION
- compounds are prepared according to the following general procedures. It will be appreciated that, although the general methods depict the synthesis of certain compounds, the following general methods, and other methods known to one of ordinary skill in the art, can typically be applied to all compounds and subclasses and species of each of these compounds, as described herein.
- Solvent A was water containing 0.038% TFA
- solvent B was acetonitrile containing 0.02% TFA.
- a gradient was run: starting with 95% A and 5% B, going to 5% A and 95% B over the next 0.7 min. This solvent ratio was maintained for 0.4 min before returning to 95% A and 5% B over the next 0.4 min. Total run time was 1.5 min.
- Trifluoromethane sulfonic anhydride 38 mL, 1.07 eqiv., 0.134 mol was added drop wise to a mixture of ferf-butyl 3-(4-hydroxy-6-methylpyridin-2-yl)pyrrolidine-l-carboxylate (35 g, 1.0 eqiv., 0.127 mol) and triethylamine (21 mL,1.2 eqiv., 0.151 mol) in CH 2 C1 2 (350 mL) cooled with an ice bath.
- Example 1 1H NMR (400 MHz, CDC1 3 ) ⁇ 7.84 (s, 1H), 7.77 - 7.75 (m, 1H), 7.13 - 7.11 (m, 2H), 6.50, 6.48 (2s, 1H), 4.33 - 4.29 (m, 0.5H), 4.22 - 4.14 (m, 1H), 4.03 - 4.92 (m, 1H), 3.97 (s, 3H), 3.78 - 3.70 (m, 0.5H), 3.64 - 3.55 (m, 1H), 3.07 - 3.01 (m, 1H), 2.55 (s, 3H), 2.40 - 2.25 (m, 2H), 1.33 - 1.28 (m, 6H).
- DMSO-iie 58.55, 8.50 (2s, 1H), 8.22, 8.19 (2s, 1H), 7.99, 7.84 (2s, 1H), 7.64 (s, 1H), 6.91, 6.87 (2s, 1H), 6.51, 6.50 (2s, 1H), 5.06 (m, 1H), 4.90 (m, 1H), 4.68 (m, 1H), 4.54 (m, 1H), 3.93, 3.89 (2s, 3H), 3.01 - 2.97 (m, 1H), 1.27 - 1.24 (m, 6H).
- EtONa (8.2 mL, 8.2 mmol) in EtOH was added into a mixture of l-phenylpropan-2-one (1.0 g, 7.5 mmol) and diethyl oxalate (1.3 g, 9.0 mmol) in EtOH (10 mL) at room temperature.
- the reaction mixture was stirred for 16 hrs and concentrated.
- the organic phase was combined and dried over anhydrous Na 2 SO 4 .
- the mixture was filtered and concentrated to dryness to give the desired crude product (0.9 g, 51% yield) as yellow oil.
- N-(pyiTolidin-3-yl)cyclopropanecarboxamide hydrochloride (97 mg, 0.51 mmol) in DMF (2 mL) was added HATU (290 mg, 0.77 mmol) and DIPEA (198 mg, .53 mmol). The mixture was stirred at room temperature for 16 hrs. The mixture was purified by preparative HPLC to give the title compound (26 mg, 17% yield) as a white solid.
- Biotin-H3K4me3 peptide was purchased from Mew England Biolabs.
- HTRF reagents containing Eu-labeled H3K4mel-2 antibody, and streptavidin-XL665 were purchased from Cis-Bio International. Plates were read on an Envision multi-label plate reader (Perkin Elmer).
- the HTRF assay mixture contained 2 nM full length KDM5A enzyme, 100 nM H3K4Me3 peptide substrate, 1 uM 2-OG, 100 uM Fe 2+ , 500 uM ascorbate. 50 mM HEPES pH 7.0 buffer, 0.01% Triton-X 100, 2 mM DTT, 0.25 % DMSO at a final volume of 10 uL.
- the enzyme reaction was carried out at room temperature in black Proxiplate 384-Plus plate (Corning, Costar) within 30 minutes, in the presence of varying concentration of a test compound.
- PC9 cells were seeded in a 384 well plate (2000 cells/well) with a test compound and incubated for 120 hours at 37 °C.
- H3K4Me3 mark level was assessed using HTRF reagents from CisBio. Briefly, media was removed and cells were lysed in 20 pL of Epigeneous Cell Histone lysis buffer C for 45 min at 26 °C, shaking. 10 pL of 4 ⁇ g/mL acceptor antibody (H3K4me3-d2) and 10 ⁇ . of 1.2pg/mL donor antibody (Total H3-K) in Cell Histone detection buffer were added and the mixture was incubated at 26 °C for 1 hour. Assay plate was read subsequently on Envision (Perkin Elmer). Each compound was run in duplicate. Data were normalized to DMSO treated wells as the low response and EC 5 o's were calculate using a four- parameter fit.
Abstract
The present invention relates to compounds of formula (I) useful as inhibitors of one or more histone demethylses, such as KDM5. The invention also provides pharmaceutically acceptable compositions comprising compounds of the present invention and the compounds for use in methods for the treatment of various disorders. Formula (I): or a salt thereof, wherein: A is selected from the group consisting of:
Description
PYRROLIDINE AMIDE COMPOUNDS AS HISTONE DEMETHYLASE INHIBITORS
This application claims the benefit of prioirity to International Application No.
PCT/CN2014/088309 which is hereby incorporated by reference in its entirety.
TECHNICAL FIELD
Compounds useful as inhibitors of histone demethylases, such as KDM5 are provided.
BACKGROUND
Packaging the 3 billion nucleotides of the human genome into the nucleus of a cell requires tremendous compaction. To accomplish this feat, DNA in our chromosomes is wrapped around spools of proteins called histones to form dense repeating protein/DNA polymers known as chromatin. Far from serving as mere packaging modules, chromatin templates form the basis of a newly appreciated and fundamentally important set of gene control mechanisms termed epigenetic regulation. By conferring a wide range of specific chemical modifications to histones and DNA, epigenetic regulators modulate the structure, function, and accessibility of our genome, thereby exerting a tremendous impact on gene expression. Hundreds of epigenetic effectors have recently been identified, many of which are chromatin-binding or chromatin-modifying enzymes.
Significantly, an increasing number of these enzymes have been associated with a variety of disorders such as cancer. Thus, therapeutic agents directed against this emerging class of gene regulatory enzymes promise new approaches to the treatment of human diseases.
Additionally, the relatively rapid acquisition of resistance to cancer drugs remains a key obstacle to successful cancer therapy. Substantial efforts to elucidate the molecular basis for such drug resistance have revealed a variety of mechanisms, including drug efflux, acquisition of drug binding-deficient mutants of the target, engagement of alternative survival pathways and epigenetic alterations. Rare, stochastic, resistance-conferring genetic alterations have been found within a tumor cell population selected during drug treatment. See Sharma et al, Cell 141(l):69-80 (2010). The KDM5/JARID1 family of histone demethylases was found to play a role in cancer resistance. The KDM5/JARID1 family of demethylases in humans contains four members, KDM5A, KDM5B, KDM5C and KDM5D. KDM5 family members contain five conserved domains: JmjN, ARID, JmjC, PHD and a C5HC2 zinc finger. Amino acid sequences of KDM5A, KDM5B, KDM5C and KDM5D are known and are publicly available, e.g., see UniProtK B/Swiss-Prot (see e.g., KDM5A (e.g., P29375-1 and P29375-2), KDM5B (e.g., Q9UGL1-1 and Q9UGL1-2), KDM5C (e.g., P41229-1, P41229-2, P41229-3 and P41229-4) and KDM5D (e.g., Q9BY66-1, Q9BY66-2 and Q9BY66-3). There is currently a need for compounds that inhibit of KDM5 demethylases for treating hyperproliferative diseases, preventing drug resistance, and/or for improving the efficacy of other cancer treatments (e.g. targeted therapies, chemotherapies, and radiotherapies.
SUMMARY OF THE INVENTION
One aspect provides a compound of formula (I):

a salt thereof, wherein:
A is selected from the group consisting of:
 R1 is halo, -N(RX)2, C1-6alkyl, C2-6alkenyl, C2-6alkynyl, 3-8 membered carbocyclyl, C1_
R1 is halo, -N(RX)2, C1-6alkyl, C2-6alkenyl, C2-6alkynyl, 3-8 membered carbocyclyl, C1_
6alkoxy, 5-10 membered aryl, 5-10 membered heteroaryl, or 3-8 membered heterocyclyl, wherein said Chalky!, C1-6alkenyl, C1-6alkynyl, 3-8 membered carbocyclyl, C1-6alkoxy, 5-10 membered aryl, 5-10 membered heteroaryl, and 3-8 membered heterocyclyl are optionally substituted with one or more groups independently selected from the group consisting of oxo, hydroxy, halo, C1- 3alkoxy, 3-8 membered carbocyclyl, 5- 10 membered aryl, -N(RX)2, -N(Rx)C(O)Rx, and C1- 3alkyl; each Rx is independently selected from the group consisting of H and C1-6alkyl, that is optionally substituted with one or more groups independently selected from the group consisting of halo, hydroxy, and C1-_.alkoxy;
R2 is 5-10 membered carbocyclyl, 5-10 membered heterocyclyl, 5-10 membered aryl, 5-10 membered heteroaryl, -ORa, -C(O)N(Ra)2, or NRaRb, wherein each 5-10 membered carbocyclyl, 5- 10 membered heterocyclyl, 5-10 membered aryl, and 5-10 membered heteroaryl is optionally substituted with one or more groups Rd;
Ra and Rb are each independently selected from the group consisting of H, C1-6alkyl, C2- ealkenyl, C2-6alkynyl, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 5-10 membered aryl, 5-10 membered heteroaryl -C(O)Rc, -CO2Rc, -C(O)N(Rc)2, -C(O)SRc, and -C(O)C(O)Rc, wherein each C1-^alkyl, C2-6alkenyl, C2-6alkynyl, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 5- 10 membered aryl, and 5-10 membered heteroaryl is optionally substituted with one or more groups independently selected from the group consisting of halo, nitro, cyano, oxo, Ci^alkyl, C2-4alkenyl, C2-4alkynyl, 3-8 membered carbocyclyl,-ORc, -SRC, -N(RC)2, -C(O)Rc, -CO2Rc, -C(O)N(Rc)2, -C(O)SRc, -C(O)C(O)Rc, -S(O)Rc, -SO2Rc, -SO2N(Rc)2, -N(Rc)C(O)Rc, -Ν(Rc)(O)Ν(Rc)2, -
N(Rc)SO2 Rc, -N(Rc)SO2N(Rc)2, -N(RC)N(RC)2, -N(RC)C(=N(RC))N(RC)2, -C(=N)N(RC)2, -C=NORc, and -C(=N(Rc))N(Rc)2;
each Rc is independently selected from the group consisting of H, C1-6alkyl, C2_6alkenyl, C2. 6alkynyl, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 5-10 membered aryl and 5-10 membered heteroaryl, wherein each C1-6alkyl, C2-6alkenyl, C2-6alkynyl, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 5-10 membered aryl and 5-10 membered heteToaryl is optionally substituted with one or more groups Rh:
each Rd is independently selected from the group consisting of halo, nitro, cyano, oxo, C1- 6alkyl, C2-6alkenyl, C2-6alkynyl, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 5-10 membered aryl, 5- 10 membered heteroaryl, ,-OR*, -SRe, -N(Re)2, -C(O)Re, -CO2RE, -C(O)N(Re)2, -C(O)SRe, -C(O)C(O)Re, -S(O)Re, -SO2Re, -SO2N(Rc)2, -N(Re)C(O)Re, -N(Re)C(O)N(Re)2, -N(Re)SO2Re 5 -N(Re)SO2N(Rc)2, -N(Re)N(Re)2, -N(Re)C(=N(Re))N(Re)2, -C(=N)N(Re)2, -C=NORe, and -C(=N(Re))N(Re)2. wherein each C1-6alkyl, C2-6alkenyl, C2-6alkynyl, 3-8 membered
carbocyclyl, 3-8 membered heterocyclyl, 5-10 membered aryl and 5-10 membered heteroaryl is optionally substituted with one or more groups independently selected from R ;
each Re is independently selected from the group consisting of H, C1-6alkyl, C2-6alkenyl, C2- 6alkyny], 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 5-10 membered aryl and 5-10 membered heteroaryl, wherein each C1-6alkyl, C2-6alkenyl, C2-6alkynyl, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 5-10 membered aryl and 5-10 membered heteroaryl is optionally substituted with one or more groups independently selected from the group consisting of Ci^alkyl, C2-4alkenyl, C2-4alkynyl, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 5-10 membered aryl and 5-10 membered heteroaryl;
each R is independently selected from the group consisting of halo, nitro, cyano, oxo, Ci_ 4alkyl5 C2-4alkenyl5 C2-4alkynyl, 3-8 membered carbocyclyl ,-ORg, -SRS, -N(Rg)2, -C(O)Rg, -CO2Rg, -C(O)N(Rs)2, -C(O)SRg, -C(O)C(O)Rg, -S(O)Rs, -SO2Rg, -SO2N(Rg)2, -N(RB)C(O)Rg,
-N(RE)C(O)N(Rg)2, -N(Rg)SO2Rg, -N(Rg)SO2N(Rg)2, -N(Rs)N(Rg)2, -N(Rg)C(=N(Rg))N(Rg)2, - C(=N)N(Rg)2, and -C=NORg, -C(=N(Rg))N(Rg)2, wherein each C1-4alkyl, C2-4alkenyl, C^alkynyl, and 3-8 membered carbocyclyl is optionally substituted with one or more groups independently selected from the group consisting of halo, cyano, oxo, 3-8 membered carbocyclyl, -ORg, -N(Rg)2, -C(O)Rg, -CO2Rg, -C(O)N(Rg)2, -SO2Rg, -SO2N(Rg)2, and -N(Rg)C(O)Rg;
each Rg is independently selected from the group consisting of H, C1-6alkyl, C2.6alkenyl, C2. 6alkynyl, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 5-10 membered aryl and 5-10 membered heteroaryl, wherein each C1-6alkyl, C2-6alkenyl, C2.6alkynyl, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 5-10 membered aryl and 5-10 membered heteroaryl is optionally
substituted with one or more groups independently selected from the group consisting of C1-4alkyl, C2-4alkenyl, C2-4alkynyl, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 5-10 membered aryl and 5-10 membered heteroaryl; and
each Rh is independently selected from the group consisting of halo, nitro. cyano, oxo, C1- 4aJkyl, C2-4alkenyl, C2-4alkynyl, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 5-10 membered aryl, 5-10 membered heteroaryl, -N(Rk)2, and ,-ORk, wherein each C1-4alkyl, C2- 4alkenyl, C2-4alkynyl, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 5-10 membered aryl and 5-10 membered heteroaryl is optionally substituted with one or more groups independently selected from the group consisting of halo, hydroxy, C1-4alkoxy, cyano, oxo, C1-4alkyl, C2-4alkenyl, C2-4alkynyl,3-8 membered carbocyclyl, and 5-10 membered aryl;
each Rk is independently selected from the group consisting of H, C1-4alkyl, C2-4alkenyl, C2- 4alkynyl, 3-8 membered carbocyclyl, and 5-10 membered aryl wherein any C1-4alkyl, C2-4alkenyl, C2-4alkynyl, 3-8 membered carbocyclyl, and 5-10 membered aryl carbocyclyl is optionally substituted with one or more groups independently selected from the group consisting of halo, cyano, oxo hydroxy, and 3-8 membered carbocyclyl; and
R3 is H or C,.6alkyl;
R4 is H, C1-6alkyl, C2-6alkenyl, C2-6alkynyl, or 3-8 membered carbocyclyl, 5-10 membered aryl, 5-10 membered heteroaryl wherein each C1-6alkyl, C2-6alkenyl, C2-6alkynyl, and 3-8 membered carbocyclyl, 5-10 membered aryl, 5-10 membered heteroaryl is optionally substituted with one or more groups independently selected from halo, hydroxy, cyano, C1-6alkoxy, and 3-8 membered carbocyclyl; and
R5 is H, halo, or C1-6alkyl, and R6 is H, C1-6alkyl, C2-6alkenyl, C2-6alkynyl, halo, hydroxy, or 3-8 membered carbocyclyl, wherein each C1-6alkyl, C2-6alkenyl, C2-6alkynyl, and 3-8 membered carbocyclyl, is optionally substituted with one or more groups independently selected from halo, hydroxy, cyano, C1-6alkoxy, and 3-8 membered carbocyclyl; or R5 and R6 taken together with the atom to which they are attached form a 3-8 membered carbocyclyl or a 3-8 membered heterocyclyl, which 3-8 membered carbocyclyl and 3-8 membered heterocyclyl are optionally substitutes with one or more groups independently selected from halo, hydroxy, cyano, C1-6alkoxy, and 3-8 membered carbocyclyl.
Another aspect includes a composition, comprising a compound of formula (I), or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable adjuvant, carrier, or vehicle.
Another aspect includes a method of treating a disease associated with KDM5 activity, comprising administering an therapeutically effective amount of a compound of formula (I), or a pharmaceutically acceptable salt thereof, to a patient in need thereof.
Another aspect includes the use of a compound of formula (I), or a pharmaceutically acceptable salt thereof, in therapy.
Another aspect includes the use of a compound of formula (I), or a pharmaceutically acceptable salt thereof, in treating a disease associated with KDM5 activity.
Another aspect includes the use of a compound of formula (I), or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for the treatment of a disease associated with KDM5 activity.
Another aspect includes a method of increasing the efficacy of a cancer treatment comprising a cancer therapy agent, comprising administering to a patient (a) an effective amount of a compound of formula (I), or a pharmaceutically acceptable salt thereof, and (b) an effective amount of the cancer therapy agent.
Another aspect includes a method of treating an individual with cancer who has an increased likelihood of developing resistance to a cancer therapy agent comprising administering to the individual (a) an effective amount of a compound of formula (I), or a pharmaceutically acceptable salt thereof, and (b) an effective amount of the cancer therapy agent.
Another aspect includes a processes and synthetic intermediates that are useful for preparing a compound of formula (I), or a salt thereof.
Another aspect includes compounds for the study of histone demethylases, such as KDM5, the study of intracellular signal transduction pathways mediated by such histone demethylases, and the comparative evaluation of modulators of these demethylases.
DETAILED DESCRIPTION
Definitions
Definitions of specific functional groups and chemical terms are described in more detail below. Chemical elements are identified in accordance with the Periodic Table of the Elements, CAS version, Handbook of Chemistry and Physics, 75th Ed., inside cover, and specific functional groups are generally defined as described therein. Additionally, general principles of organic chemistry, as well as specific functional moieties and reactivity, are described in Organic
Chemistry, Thomas Sorrell, University Science Books, Sausalito, 1999; Smith and March March 's Advanced Organic Chemistry, 5th Edition, John Wiley & Sons, Inc., New York, 2001; Larock, Comprehensive Organic Transformations, VCH Publishers, Inc., New York, 1989; Carruthers,
Some Modern Methods of Organic Synthesis, 3rd Edition, Cambridge University Press, Cambridge, 1987; the entire contents of each of which are incorporated herein by reference.
Unless otherwise stated, structures depicted herein are also meant to include all isomeric (e.g., enantiomeric, diastereomeric, and geometric (or conformational)) forms of the structure; for example, the R and S configurations for each asymmetric center, Z and E double bond isomers, and Z and E conformational isomers. Therefore, single stereochemical isomers as well as enantiomeric, diastereomeric, and geometric (or conformational) mixtures of the present compounds are included.
Unless otherwise stated, all tautomeric forms of the compounds are included. For example, when the group A includes a pyrazole ring, it can be either tautomeric form shown below:

Additionally, unless otherwise stated, structures depicted herein are also meant to include compounds that differ only in the presence of one or more isotopically enriched atoms. For example, compounds having the present structures including the replacement of hydrogen by deuterium or tritium, or the replacement of a carbon by a 13C- or 14 C-enriched carbon are included. Such compounds are useful, for example, as analytical tools, as probes in biological assays, or as therapeutic agents.
Where a particular enantiomer is preferred, it may, in some embodiments be provided substantially free of the corresponding enantiomer, and may also be referred to as "optically enriched." "Optically-enriched," as used herein, means that the compound is made up of a significantly greater proportion of one enantiomer. In certain embodiments the compound is made up of at least about 90% by weight of a preferred enantiomer. In other embodiments the compound is made up of at least about 95%, 98%, or 99% by weight of a preferred enantiomer. Preferred enantiomers may be isolated from racemic mixtures by any method known to those skilled in the art, including chiral high pressure liquid chromatography (HPLC) and the formation and
crystallization of chiral salts or prepared by asymmetric syntheses. See, for example, Jacques et al., Enantiomers, Racemates and Resolutions (Wiley Interscience, New York, 1981); Wilen, et al., Tetrahedron 33:2725 (1977); Eliel, E.L. Stereochemistry of Carbon Compounds (McGraw-Hill, NY, 1962); Wilen, S.H. Tables of Resolving Agents and Optical Resolutions p. 268 (E.L. Eliel, Ed., Univ. of Notre Dame Press, Notre Dame, IN 1972).
The term "a compound as described herein" includes the compounds described in Examples
1-432 and salts thereof, as well as compounds of formula (1) and salts thereof.
The term "heteroatom" means one or more of oxygen, sulfur, nitrogen, phosphorus, or silicon (including, any oxidized form of nitrogen, sulfur, phosphorus, or silicon; the quaternized form of any basic nitrogen or; a substitutable nitrogen of a heterocyclic ring, for example N (as in 3,4-dihydro-2H-pyrrolyl), NH (as in pyrrolidinyl) or NR+ (as in N-substituted pyrrolidinyl)).
As used herein a "direct bond" or "covalent bond" refers to a single, double or triple bond.
Iη certain embodiments, a "direct bond" or "covalent bond" refers to a single bond.
The terms "halo" and "halogen" as used herein refer to an atom selected from fluorine (fluoro, -F), chlorine (chloro, -CI), bromine (bromo, -Br), and iodine (iodo, -I).
The term "aliphatic" or "aliphatic group", as used herein, denotes a hydrocarbon moiety that may be straight-chain (i.e., unbranched), branched, or cyclic (including fused, bridging, and spiro- fused polycyclic) and may be completely saturated or may contain one or more units of
unsaturation, but which is not aromatic. Unless otherwise specified, aliphatic groups contain 1-6 carbon atoms. In some embodiments, aliphatic groups contain 1—4 carbon atoms, and in yet other embodiments aliphatic groups contain 1-3 carbon atoms. Suitable aliphatic groups include, but are not limited to, linear or branched, alky], alkenyl, and alkynyl groups, and hybrids thereof such as (cycloalkyl)alkyl, (cycloalkenyl)alkyl or (cycloalkyl)alkenyl.
The term "unsaturated", as used herein, means that a moiety has one or more units of unsaturation.
The terms "cycloaliphatic", "carbocycle", "carbocyclyl", "carbocyclo", or "carbocyclic", used alone or as part of a larger moiety, refer to a saturated or partially unsaturated cyclic aliphatic monocyclic, bicyclic, or spior ring systems, as described herein, having from 3 to 10 members, wherein the aliphatic ring system is optionally substituted as defined above and described herein. Cycloaliphatic groups include, without limitation, cyclopropyl, cyclobutyl, cyclopentyl,
cyclopentenyl, cyclohexyl, cyclohexenyl, cycloheptyl, cycloheptenyl, cyclooctyl, cyclooctenyl, and cyclooctadienyl. In some embodiments, the cycloalkyl has 3-6 carbons. The terms
"cycloaliphatic", "carbocycle", "carbocyclyl", "carbocyclo", or "carbocyclic" also include aliphatic rings that are fused to one or more aromatic or nonaromatic rings, such as decahydronaphthyl, tetrahydronaphthyl, decalin, or bicyclo[2.2.2]octane, where the radical or point of attachment is on an aliphatic ring.
As used herein, the term "cycloalkylene" refers to a bivalent cycloalkyl group. In certain embodiments, a cycloalkylene group is a 1,1 -cycloalkylene group (i.e., a spiro-fused ring).
Exemplary 1,1 -cycloalkylene groups include
 . In other embodiments, a cycloalkylene
group is a 1 ,2-cycloalkylene group or a 1,3-cycloalkylene group. Exemplary 1,2-cycloalkylene
. In other embodiments, a cycloalkylene
group is a 1 ,2-cycloalkylene group or a 1,3-cycloalkylene group. Exemplary 1,2-cycloalkylene

The term "alkyl," as used herein, refers to a monovalent saturated, straight- or branched- chain hydrocarbon radical derived from an aliphatic moiety containing between one and six carbon atoms by removal of a single hydrogen atom. In some embodiments, alkyl contains 1-5 carbon atoms. In another embodiment, alkyl contains \-4 carbon atoms. In still other embodiments, alkyl contains 1-3 carbon atoms. In yet another embodiment, alkyl contains 1-2 carbons. Examples of alkyl radicals include, but are not limited to, methyl, ethyl, n-propyl, isopropyl, n-butyl, iso-butyl, sec-butyl, sec-p entyl, iso-p entyl, tert-butyl, n-pentyl, neopentyl, n-hexyl, sec-hexyl, n-heptyl, n- octyl, n-decyl, n-undecyl, dodecyl, and the like.
The term "alkenyl," as used herein, denotes a monovalent group derived from a straight- or branched-chain aliphatic moiety having at least one carbon-carbon double bond by the removal of a single hydrogen atom. In certain embodiments, alkenyl contains 2-6 carbon atoms. In certain embodiments, alkenyl contains 2-5 carbon atoms. In some embodiments, alkenyl contains 2—4 carbon atoms. In another embodiment, alkenyl contains 2-3 carbon atoms. Alkenyl groups include, for example, ethenyl ("vinyl"), propenyl ("allyl"), butenyl, l-methyl-2-buten-l-yl, and the like.
The term "alkynyl," as used herein, refers to a monovalent group derived from a straight- or branched-chain aliphatic moiety having at least one carbon-carbon triple bond by the removal of a single hydrogen atom. In certain embodiments, alkynyl contains 2-6 carbon atoms. In certain embodiments, alkynyl contains 2—5 carbon atoms. In some embodiments, alkynyl contains 2-4 carbon atoms. In another embodiment, alkynyl contains 2-3 carbon atoms. Representative alkynyl groups include, but are not limited to, ethynyl, 2-propynyl ("propargyl"), 1-propynyl, and the like.
The term "aryl" used alone or as part of a larger moiety as in "aralkyl", "aralkoxy", or "aryloxyalkyi", refers to monocyclic and bicyclic ring systems having a total of five to 10 ring members, wherein at least one ring in the system is aromatic and wherein each ring in the system contains three to seven ring members. The term "aryl" may be used interchangeably with the term "aryl ring". In certain embodiments, "aryl" refers to an aromatic ring system which includes, but not limited to, phenyl, biphenyl, naphthyl, anthracyl and the like, which may bear one or more substituents. Also included within the scope of the term aryl", as it is used herein, is a group in which an aromatic ring is fused to one or more non-aromatic rings, such as indanyl, phthalimidyl, naphthimidyl, phenantriidinyl, or tetrahydronaphthyl, and the like.
The terms "heteroaryl" and "heteroar-", used alone or as part of a larger moiety, e.g., "heteroaralkyl", or "heteroaralkoxy", refer to groups having 5 to 10 ring atoms, preferably 5, 6, or 9 ring atoms; having 6, 10, or 14 π electrons shared in a cyclic array; and having, in addition to carbon atoms, from one to five heteroatoms. The term "heteroatom" refers to nitrogen, oxygen, or sulfur, and includes any oxidized form of nitrogen or sulfur, and any quaternized form of a basic nitrogen. Heteroaryl groups include, without limitation, thienyl, furanyl, pyrrolyl, imidazolyl, pyrazolyl, triazolyl, tetrazolyl, oxazolyl, isoxazolyl, oxadiazolyl, thiazolyl, isothiazolyl, thiadiazolyl, pyridyl, pyridazinyl, pyrimidinyl, pyrazinyl, indolizinyl, purinyl, naphthyridinyl, and pteridinyl. The terms "heteroaryl" and "heteroar-", as used herein, also include groups in which a heteroaromatic ring is fused to one or more aryl, cycloaliphatic, or heterocyclyl rings, where the radical or point of attachment is on the heteroaromatic ring. Nonlimiting examples include indolyl, isoindolyl, benzothienyl, benzofuranyl, dibenzofuranyl, indazolyl, benzimidazolyl, benzthiazolyl, quinolyl, isoquinolyl, cinnolinyl, phthalazinyl, quinazolinyl, quinoxalinyl, 4H-quinolizinyl, carbazolyl, acridinyl, phenazinyl, phenothiazinyl, phenoxazinyl, tetrahydroquinolinyl,
tetrahydroisoquinolinyl, and pyrido[2,3-b]-1,4-oxazin-3(4H)-one. A heteroaryl group may be mono- or bicyclic. The term "heteroaryl" may be used interchangeably with the terms "heteroaryl ring", "heteroaryl group", or "heteroaromatic", any of which terms include rings that are optionally substituted. The term "heteroaralkyl" refers to an alkyl group substituted by a heteroaryl, wherein the alkyl and heteroaryl portions independently are optionally substituted.
As used herein, the terms "heterocycle", "heterocyclyl", "heterocyclic radical", and
"heterocyclic ring" are used interchangeably and refer to a stable 4- to 7-membered monocyclic or 7-10-membered bicyclic heterocyclic moiety that is either saturated or partially unsaturated, and having, in addition to carbon atoms, one or more, preferably one to four, heteroatoms, as defined above. When used in reference to a ring atom of a heterocycle, the term "nitrogen" includes a substituted nitrogen. As an example, in a saturated or partially unsaturated ring having 0-3 heteroatoms selected from oxygen, sulfur or nitrogen, the nitrogen may be N (as in 3,4-dihydro-2H- pyrrolyl), NH (as in pyrrolidinyl), or +NR (as in N-substituted pyrrolidinyl).
A heterocyclic ring can be attached to its pendant group at any heteroatom or carbon atom that results in a stable structure and any of the ring atoms can be optionally substituted. Examples of such saturated or partially unsaturated heterocyclic radicals include, without limitation, tetrahydrofuranyl, tetrahydrothienyl, pyrrolidinyl, pyrrolidonyl, piperidinyl, pyrrolinyl,
tetrahydroquinolinyl, tetrahydroisoquinolinyl, decahydroquinolinyl, oxazolidinyl, piperazinyl, dioxanyl, dioxolanyl, diazepinyl, oxazepinyl, thiazepinyl, morpholinyl, and quinuclidinyl. The terms "heterocycle", "heterocyclyl", "heterocyclyl ring", "heterocyclic group", "heterocyclic
moiety", and "heterocyclic radical", are used interchangeably herein, and also include groups in which a heterocyclyl ring is fused to one or more aryl, heteroaryl, or cycloaliphatic rings, such as indolinyl, 3H-indolyl, chromanyl, phenanthridinyl, 2-azabicyclo[2.2.1]heptanyl, octahydroindolyl, or tetrahydroquinolinyl, where the radical or point of attachment is on the heterocyclyl ring. A heterocyclyl group may be mono- or bicyclic. The term "heterocyclylalkyl" refers to an alkyl group substituted by a heterocyclyl, wherein the alkyl and heterocyclyl portions independently are optionally substituted.
As used herein, the term "partially unsaturated" refers to a ring moiety that includes at least one double or triple bond between ring atoms but is not aromatic. The term "partially unsaturated" is intended to encompass rings having multiple sites of unsaturation, but is not intended to include aryl or heteroaryl moieties, as herein defined.
The term "alkylene" refers to a bivalent alkyl group. An "alkylene chain" is a
polymethylene group, i.e., -(CH2)η-, wherein n is a positive integer, preferably from 1 to 6, from 1 to 4, from 1 to 3, from 1 to 2, or from 2 to 3. A substituted alkylene chain is a polymethylene group in which one or more methylene hydrogen atoms are replaced with a substituent. Suitable substituents include those described below for a substituted aliphatic group.
As used herein, the term "inhibitor" refers to a compound that binds to and inhibits a KDM5 enzyme with measurable affinity and activity. In certain embodiments, an inhibitor has an IC50 and/or binding constant of less about 50 μΜ, less than about 1 μΜ, less than about 500 nM, less man about 100 nM, or less than about 10 nM.
The terms "measurable affinity" and "measurably inhibit," as used herein, refer to a measurable reduction in activity of a KDM5 enzyme between: (i) a sample comprising a compound a compound as described herein and such KDM5 enzyme, and (ii) an equivalent sample comprising such KDM5 enzyme, in the absence of said compound.
"Pharmaceutically acceptable salts" include both acid and base addition salts.
"Pharmaceutically acceptable acid addition salt" refers to those salts which retain the biological effectiveness and properties of the free bases and which are not biologically or otherwise undesirable, formed with inorganic acids such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, carbonic acid, phosphoric acid and the like, and organic acids may be selected from aliphatic, cycloaliphatic, aromatic, araliphatic, heterocyclic, carboxylic, and sulfonic classes of organic acids such as formic acid, acetic acid, propionic acid, glycolic acid, gluconic acid, lactic acid, pyruvic acid, oxalic acid, malic acid, maleic acid, maloneic acid, succinic acid, fumaric acid, tartaric acid, citric acid, aspartic acid, ascorbic acid, glutamic acid, anthranilic acid, benzoic acid,
cinnamic acid, mandelic acid, embonic acid, phenylacetic acid, methanesulfonic acid, ethanesulfonic acid, benzenesulfonic acid, p-toluenesulfonic acid, salicyclic acid and the like.
"Pharmaceutically acceptable base addition salts" include those derived from inorganic bases such as sodium, potassium, lithium, ammonium, calcium, magnesium, iron, zinc, copper, manganese, aluminum salts and the like. Particularly base addition salts are the ammonium, potassium, sodium, calcium and magnesium salts. Salts derived from pharmaceutically acceptable organic nontoxic bases includes salts of primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines and basic ion exchange resins, such as isopropylamine, trimethylamine, diethylamine, triethylamine, tripropylamine, ethanolamine, 2- diethylaminoethanol. tromethamine, dicyclohexylamine, lysine, arginine, histidine, caffeine, procaine, hydrabamine, choline, betaine, ethylenediamine, glucosamine, methylglucamine, theobromine, purines, piperizine, piperidine, N-ethylpiperidine, polyamine resins and the like. Particularly organic non-toxic bases are isopropylamine, diethylamine, ethanolamine,
tromethamine, dicyclohexylamine, choline, and caffeine.
The term "tautomer" or "tautomeric form" refers to structural isomers of different energies which are interconvertible via a low energy barrier. For example, proton tautomers (also known as prototropic tautomers) include interconversions via migration of a proton, such as keto-enol and imine-enamine isomerizations. Valence tautomers include interconversions by reorganization of some of the bonding electrons.
A "solvate" refers to an association or complex of one or more solvent molecules and a compound or pharmaceutically acceptable salt thereof as described herein. Examples of solvents include water, isopropanol, ethanol, methanol, DMSO, ethyl acetate, acetic acid and ethanolamine. The term "hydrate" refers to the complex where the solvent molecule is water.
"Therapeutically effective amount" refers to an amount of a a compound or
pharmaceutically acceptable salt thereof as described herein that (i) treats the particular disease, condition or disorder, (ii) attenuates, ameliorates or eliminates one or more symptoms of the particular disease, condition, or disorder, or (iii) prevents or delays the onset of one or more symptoms of the particular disease, condition or disorder described herein. In the case of cancer, the therapeutically effective amount of the drug may reduce the number of cancer cells; reduce the tumor size; inhibit (i.e., slow to some extent and preferably stop) cancer cell infiltration into peripheral organs; inhibit (i.e., slow to some extent and preferably stop) tumor metastasis; inhibit, to some extent, tumor growth; and/or relieve to some extent one or more of the symptoms associated with the cancer. For cancer therapy, efficacy can, for example, be measured by assessing the time to disease progression (TTP) and/or determining the response rate (RR). In the
case of immunological disorders, the therapeutic effective amount is an amount sufficient to decrease or alleviate an allergic disorder, the symptoms of an autoimmune and/or inflammatory disease, or the symptoms of an acute inflammatory reaction (e.g. asthma). In some embodiments, a therapeutically effective amount is an amount of a chemical entity described herein sufficient to significantly decrease the activity or number of drug tolerant or drug tolerant persisting cancer cells.
"Treatment" (and variations such as "treat" or "treating") refers to clinical intervention in an attempt to alter the natural course of the individual or cell being treated, and can be performed either for prophylaxis or during the course of clinical pathology. Desirable effects of treatment include one or more of preventing occurrence or recurrence of disease, alleviation of symptoms, diminishment of any direct or indirect pathological consequences of the disease, stabilized (i.e., not worsening) state of disease, preventing metastasis, decreasing the rate of disease progression, amelioration or palliation of the disease state, prolonging survival as compared to expected survival if not receiving treatment and remission or improved prognosis. In certain embodiments, a compound as described herein is used to delay development of a disease or disorder or to slow the progression of a disease or disorder. Those individuals in need of treatment include those already with the condition or disorder as well as those prone to have the condition or disorder, (for example, through a genetic mutation or abberent expression of a gene or protein) or those in which the condition or disorder is to be prevented.
Examplary Values
In one embodiment the compound is a compound of formula (la):

or a salt thereof.
In one embodiment R1 is C1-6alkyl, wherein said C1-6alkyl is optionally substituted with one or more groups independently selected from the group consisting of oxo, hydroxy, halo, C1- 3alkoxy, 3-8 membered carbocyclyl, 5-10 membered aryl, -N(Ra)2, -N(Ra)C{0)Ra, and C1-3alkyl.
In one embodiment R1 is halo, -N(Ra)2, 3-8 membered carbocyclyl, C1-6alkoxy, 5-10 membered aryl, wherein said 3-8 membered carbocyclyl and 5-10 membered aryl are optionally substituted with one or more groups independently selected from the group consisting of oxo,
hydroxy, halo, C1-3alkoxy, 3-8 membered carbocyclyl, 5-10 membered aryl, -N(Ra)2, -N(Ra)C(O)Ra, and C1-3alky!.
In one embodiment R1 is bromo, cyclohexyl, isopropyl, isobutyl, cyclopentyl, 1 - methoxyethyl, cyclopropyl, cyclobutyl, amino, 4-phenylbut-2-yl, butyl, phenethyl, cyclopentyl, 1- (acetylamino)ethyl, or l-(hydroxyrnethylcarbonylarnino)ethyl.
In one embodiment R1 is isopropyl.
In one embodiment the compound is a compound of formula (lb):

or a salt thereof.
In one embodiment R4 is H, methyl, or isopropyl.
In one embodiment R is selected from the group consisting of:






In one embodiment R2 is 5-10 membered carbocyclyl that is optionally substituted with one or more groups Rd.
In one embodiment R is 5-10 membered heterocyclyl, that is optionally substituted with one or more groups Rd.
In one embodiment R2 is selected from the group consisting of:

In one embodiment R2 is 5-10 membered aryl that is optionally substituted with one or more groups Rd

In one embodiment R is 5-10 membered heteroaryl that is optionally substituted with one or more groups Rd.
In one embodiment R2 is selected from the group consisting of:




In one embodiment R2 is -ORa.
In one embodiment R2 is:

In one embodiment R2 is NRaRb.
In one embodiment R2 is selected from the group consisting of:



In one embodiment R2 is -C(O)N(Ra)2.
In one embodiment R2 is selected from the group consisting of:

In one embodiment R3 is H.
In one embodiment R3 is methyl.
In one embodiment R5 is H
In one embodiment R6 is C1-6alkyl or hydroxy.
In one embodiment R6 is methyl or hydroxy.
In one embodiment R6 is H.
In one embodiment R5 and R6 taken together with the atom to which they are attached form a 3-8 membered carbocyclyl or a 3-8 membered heterocyclyl, which 3-8 membered carbocyclyl and 3-8 membered heterocyclyl are optionally substitutes with one or more groups independently selected from halo, hydroxy, cyano, C1-6alkoxy, and 3-8 membered carbocyclyl.
In one embodiment R5 and R6 taken together with the atom to which they are attached form a cyclopropyl ring.
In another embodiment the compound is selected from the group consisting of:














and salts thereof.
In one embodiment the compound is not:

or a salt thereof.
Uses. Formulation and Administration
Pharmaceutically acceptable compositions
Another aspect includes a pharmaceutical composition comprising a a compound as described herein or a pharmaceutically acceptable salt thereof. In one embodiment, the
composition further comprises a pharmaceutically acceptable carrier, adjuvant, or vehicle. In another embodiment, the composition further comprises an amount of the compound effective to measurably inhibit KDM5. In certain embodiments, the composition is formulated for
administration to a patient in need thereofcertain embodiments.
The term "patient" or "individual" as used herein, refers to an animal, such as a mammal, such as a human. In one embodiment, patient or individual refers to a human.
The term "pharmaceutically acceptable carrier, adjuvant, or vehicle" refers to a non-toxic carrier, adjuvant, or vehicle that does not destroy the pharmacological activity of the compound with which it is formulated. Pharmaceutically acceptable carriers, adjuvants or vehicles that may be used in the compositions of this invention include, but are not limited to, ion exchangers, alumina, aluminum stearate, lecithin, serum proteins, such as human serum albumin, buffer substances such as phosphates, glycine, sorbic acid, potassium sorbate, partial glyceride mixtures of saturated vegetable fatty acids, water, salts or electrolytes, such as protamine sulfate, disodium hydrogen phosphate, potassium hydrogen phosphate, sodium chloride, zinc salts, colloidal silica,
magnesium trisilicate, polyvinyl pyrrolidone, cellulose-based substances, polyethylene glycol, sodium carboxymethylcellulose, polyacrylates, waxes, polyethylene-polyoxypropylene-block polymers, polyethylene glycol and wool fat.
Compositions comprising a compound as described herein may be administered orally, parenterally, by inhalation spray, topically, transdermally, rectally, nasally, buccally, sublingually, vaginally, intraperitoneal, intrapulmonary, intradermal, epidural or via an implanted reservoir. The term "parenteral" as used herein includes subcutaneous, intravenous, intramuscular, intra-articular, intra-synovial, intrasternal, intrathecal, intrahepatic, intralesional and intracranial injection or infusion techniques.
In one embodiment, the composition comprising a compound as described herein is formulated as a solid dosage form for oral administration. Solid dosage forms for oral
administration include capsules, tablets, pills, powders, and granules. In certain embodiments, the solid oral dosage form comprising a compound as described herein further comprises one or more of (i) an inert, pharmaceutically acceptable excipient or carrier, such as sodium citrate or dicalcium phosphate, and (ii) filler or extender such as starches, lactose, sucrose, glucose, mannitol, or silicic acid, (iii) binders such as carboxymethylcellulose, alginates, gelatin, polyvinylpyrrolidinone, sucrose or acacia, (iv) humectants such as glycerol, (v) disintegrating agent such as agar, calcium carbonate, potato or tapioca starch, alginic acid, certain silicates or sodium carbonate, (vi) solution retarding agents such as paraffin, (vii) absorption accelerators such as quaternary ammonium salts, (viii) a wetting agent such as cetyl alcohol or glycerol monostearate, (ix) absorbent such as kaolin or bentonite clay, and (x) lubricant such as talc, calcium stearate, magnesium stearate, polyethylene glycols or sodium lauryl sulfate. In certain embodiments, the solid oral dosage form is formulated as capsules, tablets or pills. In certain embodiments, the solid oral dosage form further comprises buffering agents. In certain embodiments, such compositions for solid oral dosage forms may be formulated as fillers in soft and hard-filled gelatin capsules comprising one or more excipients such as lactose or milk sugar, polyethylene glycols and the like.
In certain embodiments, tablets, dragees, capsules, pills and granules of the compositions comprising a compound as described herein optionally comprise coatings or shells such as enteric coatings. They may optionally comprise opacifying agents and can also be of a composition that they release the active ingredients) only, or preferentially, in a certain part of the intestinal tract, optionally, in a delayed manner. Examples of embedding compositions include polymeric substances and waxes, which may also be employed as fillers in soft and hard-filled gelatin capsules using such excipients as lactose or milk sugar as well as high molecular weight polethylene glycols and the like.
In another embodiment, a composition comprises micro-encapsulated compound as described herein, and optionally, further comprises one or more excipients.
In another embodiment, compositions comprise liquid dosage formulations comprising a compound as described herein for oral administration, and optionally further comprise one or more of pharmaceutically acceptable emulsions, microemulsions, solutions, suspensions, syrups and elixirs. In certain embodiments, the liquid dosage form optionally, further comprise one or more of an inert diluent such as water or other solvent, a solubilizing agent, and an emulsifier such as ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate, propylene glycol, 1 ,3-butylene glycol, dimethylformamide, oils (in particular, cottonseed, groundnut, corn, germ, olive, castor, and sesame oils), glycerol, tetrahydrofurfuryl alcohol, polyethylene glycols or fatty acid esters of sorbitan, and mixtures thereof. In certain embodiments, liquid oral compositions optionally further comprise one or more adjuvant, such as a wetting agent, a suspending agent, a sweetening agent, a flavoring agent and a perfuming agent.
Injectable preparations, for example, sterile injectable aqueous or oleaginous suspensions may be formulated according to the known art using suitable dispersing or wetting agents and suspending agents. The sterile injectable preparation may also be a sterile injectable solution, suspension or emulsion in a nontoxic parenterally acceptable diluent or solvent, for example, as a solution in 1,3-butanediol. Among the acceptable vehicles and solvents that may be employed are water, Ringer's solution, U.S.P. and isotonic sodium chloride solution. In addition, sterile, fixed oils are conventionally employed as a solvent or suspending medium. For this purpose any bland fixed oil can be employed including synthetic mono- or diglycerides. In addition, fatty acids such as oleic acid are used in the preparation of injectables.
Injectable formulations can be sterilized, for example, by filtration through a bacterial- retaining filter, or by incorporating sterilizing agents in the form of sterile solid compositions which can be dissolved or dispersed in sterile water or other sterile injectable medium prior to use.
In order to prolong the effect of a compound as described herein, it may be desirable to slow the absorption of the compound from subcutaneous or intramuscular injection. This may be accomplished by the use of a liquid suspension of crystalline or amorphous material with poor water solubility. The rate of absorption of the compound then depends upon its rate of dissolution that, in turn, may depend upon crystal size and crystalline form. Alternatively, delayed absorption of a parenterally administered compound form is accomplished by dissolving or suspending the compound in an oil vehicle. Injectable depot forms are made by forming microencapsule matrices of the compound in biodegradable polymers such as polylactide-polyglycolide. Depending upon the ratio of compound to polymer and the nature of the particular polymer employed, the rate of
compound release can be controlled. Examples of other biodegradable polymers include poly(orthoesters) and poly(anhydrides). Depot injectable formulations are also prepared by entrapping the compound in liposomes or microemulsions that are compatible with body tissues.
In certain embodiments, the composition for rectal or vaginal administration are formulated as suppositories which can be prepared by mixing a compound as described herein with suitable non-irritating excipients or carriers such as cocoa butter, polyethylene glycol or a suppository wax, for example those which are solid at ambient temperature but liquid at body temperature and therefore melt in the rectum or vaginal cavity and release the compound.
Example dosage forms for topical or transdermal administration of a compound as described herein include ointments, pastes, creams, lotions, gels, powders, solutions, sprays, inhalants or patches. The compound as described herein is admixed under sterile conditions with a pharmaceutically acceptable carrier, and optionally preservatives or buffers. Additional
formulation examples include an ophthalmic formulation, ear drops, eye drops,, transdermal patches. Transdermal dosage forms can be made by dissolving or dispensing the compound as described herein in medium, for example ethanol or dimethylsulfoxide. Absorption enhancers can also be used to increase the flux of the compound across the skin. The rate can be controlled by either providing a rate controlling membrane or by dispersing the compound in a polymer matrix or gel.
Nasal aerosol or inhalation formulations of a compound as described herein may be prepared as solutions in saline, employing benzyl alcohol or other suitable preservatives, absorption promoters to enhance bioavailability, fluorocarbons, and/or other conventional solubilizing or dispersing agents.
In certain embodiments, pharmaceutical compositions may be administered with or without food. In certain embodiments, pharmaceutically acceptable compositions are administered without food. In certain embodiments, pharmaceutically acceptable compositions of this invention are administered with food.
Specific dosage and treatment regimen for any particular patient will depend upon a variety of factors, including age, body weight, general health, sex, diet, time of administration, rate of excretion, drug combination, the judgment of the treating physician, and the severity of the particular disease being treated. The amount of a compound as described herein in the composition will also depend upon the particular compound in the composition.
In one embodiment, the therapeutically effective amount of the compound of the invention administered parenterally per dose will be in the range of about 0.01-100 mg/kg, alternatively about
0.1 to 20 mg/kg of patient body weight per day, with the typical initial range of compound used
being 0.3 to 15 mg/kg/day. In another embodiment, oral unit dosage forms, such as tablets and capsules, contain from about 5 to about 100 mg of the compound of the invention.
An example tablet oral dosage form comprises about 2 mg, 5 mg, 25 mg, 50 mg, 100 mg, 250 mg or 500 mg of a compound as described herein, and further comprises about 95-30 mg anhydrous lactose, about 5-40 mg sodium croscarmellose, about 5-30 mg polyvinylpyrrolidone (PVP) K30 and about 1-10 mg magnesium stearate. The process of formulating the tablet comprises mixing the powdered ingredients together and further mixing with a solution of the PVP. The resulting composition can be dried, granulated, mixed with the magnesium stearate and compressed to tablet form using conventional equipment. An example of an aerosol formulation can be prepared by dissolving about 2-500 mg of a compound as described herein, in a suitable buffer solution, e.g. a phosphate buffer, and adding a tonicifier, e.g. a salt such sodium chloride, if desired. The solution may be filtered, e.g. using a 0.2 micron filter, to remove impurities and contaminants.
Uses of Compounds and Pharmaceutically Acceptable Compositions
Another aspect includes the use of a compound as described herein for the inhibition of KDM5. Ccompounds as described herein may also be used to inhibit the removal of methyl marks on histone lysine residues, including inhibiting the removal of methyl marks from mono-, di- or tri- methylation of histones HI, H2A, H2B, H3 and H4, such as H3K4 (including for example the KDM5 substrate H3K4me3), thereby altering interactions of these histone proteins with DNA and/or other proteins, and altering certain subsequent genetic or protein expression. Compounds as described herein may also be used to inhibit KDM5 and reduce drug-tolerant cells, thereby treating or preventing drug-resistant diseases, such as drug-resistant cancer. In certain embodiments, the disease can be treated using a compound as described herein to prevent resistance from forming, for example before targets of chemotherapies become mutated to confer resistance to such
chemotherapies.
In certain embodiments, the binding or inhibition activity of a compound as described herein may be determined by running a competition experiment where the is incubated with the KDM5 enzyme bound to known radioligands. Detailed conditions for assaying a compound as an inhibitor of KDM5 or a mutant thereof are set forth in the Examples below.
In certain embodiments, detection of KDM5 activity is achieved with in vitro assays, which can be either direct binding (non-catalytic) or enzymatic (catalytic) asssays. Types of substrates that are used in such assays may include: short synthetic peptides corresponding to a number of residues from the N-terminus of histone sequences comprising the target lysine residue, single recombinant histone polypeptides, histone octamers reconstituted with recombinant histone proteins, and
reconstituted nucleosomes (using reconstituted octamers and specific recombinant DNA fragments). The reconstituted nucleosomes may be mononucleosomes or oligonucleosomes.
Another aspect includes a method of treating or preventing a disease responsive to the inhibition of KDM5 activity in a patient. The method includes administering a therapeutically effective amount of a compound as described hereinto a patient in need thereof.
Another aspect includes the use of a compound as described herein, in therapy. Another aspect includes the use of a pharmaceutical composition comprising a compound as described herein, in therapy.
Another aspect includes the use of a compound as described herein, in treating a disease associated with KDM5 activity. Another aspect includes the use of a pharmaceutical composition comprising a compound as described herein, in treating a disease associated with KDM5 activity.
Another aspect includes the use of a compound as described herein, in the manufacture of a medicament for the treatment of a disease associated with KDM5 activity. Another aspect includes the use of a pharmaceutical composition comprising a compound as described herein, in the manufacture of a medicament for the treatment of a disease associated with KDM5 activity.
In certain embodiments, the disease or condition is a hyperproliferative disease, cancer, stroke, diabetes, hepatomegaly, cardiovascular disease, multiple sclerosis, Alzheimer's disease, cystic fibrosis, viral disease, autoimmune diseases, atherosclerosis, restenosis, psoriasis, rheumatoid arthritis, inflammatory bowel disease, asthma, allergic disorders, inflammation, neurological disorders, a hormone-related disease, conditions associated with organ transplantation, immunodeficiency disorders, destructive bone disorders, proliferative disorders, infectious diseases, conditions associated with cell death, thrombin-induced platelet aggregation, liver disease, pathologic immune conditions involving T cell activation, CNS disorders or a myeloproliferative disorder.
In certain embodiments, treatment may be administered after one or more symptoms have developed. In other embodiments, treatment may be administered in the absence of symptoms. For example, treatment may be administered to a susceptible individual prior to the onset of symptoms (e.g., in light of a history of symptoms and/or in light of genetic or other susceptibility factors). Treatment may also be continued after symptoms have resolved, for example to prevent or delay their recurrence.
Another aspect includes a method for treating, ameliorating or preventing cancer, drug- resistant cancer or another proliferative disorder by administration of an effective amount of a compound as described herein to a mammal, for example a human, in need of such treatment. In certain embodiments, the disease to be treated is cancer or drug resistant cancer.
Examples of cancers that may be treated using the compounds and methods described herein include, but are not limited to, adrenal cancer, acinic cell carcinoma, acoustic neuroma, acral lentigious melanoma, acrospiroma, acute eosinophilic leukemia, acute erythroid leukemia, acute lymphoblastic leukemia, acute megakaryoblastic leukemia, acute monocytic leukemia, acute promyelocyte leukemia, adenocarcinoma, adenoid cystic carcinoma, adenoma, adenomatoid odontogenic tumor, adenosquamous carcinoma, adipose tissue neoplasm, adrenocortical carcinoma, adult T-cell leukemia/lymphoma, aggressive NK-cell leukemia, AlDS-related lymphoma, alveolar rhabdomyosarcoma, alveolar soft part sarcoma, ameloblastic fibroma, anaplastic large cell lymphoma, anaplastic thyroid cancer, androgen dependent cancer, angioimmunoblastic T-cell lymphoma, angiomyolipoma, angiosarcoma, astrocytoma, atypical teratoid rhabdoid tumor, B-cell chronic lymphocytic leukemia, B-cell lymphoma, basal cell carcinoma, biliary tract cancer, bladder cancer, blastoma, bone cancer, Brenner tumor, Brown tumor, Burkitt's lymphoma, breast cancer, brain cancer, carcinoma, carcinoma in situ, carcinosarcoma, cartilage tumor, cementoma, myeloid sarcoma, chondroma, chordoma, choriocarcinoma, choroid plexus papilloma, clear-cell sarcoma of the kidney, craniopharyngioma, cutaneous T-cell lymphoma, cervical cancer, colorectal cancer, Degos disease, desmoplastic small round cell tumor, diffuse large B-cell lymphoma,
dysembryoplastic neuroepithelial tumor, dysgerminoma, embryonal carcinoma, endocrine gland neoplasm, endodermal sinus tumor, enteropathy-associated T-cell lymphoma, esophageal cancer, fetus in fetu, fibroma, fibrosarcoma, follicular lymphoma, follicular thyroid cancer,
ganglioneuroma, gastrointestinal cancer, germ cell tumor, gestational choriocarcinoma, giant cell fibroblastoma, giant cell tumor of the bone, glial tumor, glioblastoma multiforme, glioma, gliomatosis cerebri, glucagonoma, gonadoblastoma, granulosa cell tumor, gynandroblastoma, gallbladder cancer, gastric cancer, hemangioblastoma, head and neck cancer, hemangiopericytoma, hematological malignancy, hepatoblastoma, hepatosplenic T-cell lymphoma, Hodgkin's
lymphoma, non-Hodgkin's lymphoma, invasive lobular carcinoma, intestinal cancer, kidney cancer, laryngeal cancer, lentigo maligna, leukemia, leydig cell tumor, liposarcoma, lung cancer, lymphangioma, lymphangiosarcoma, lymphoepithelioma, lymphoma, acute lymphocytic leukemia, acute myelogeous leukemia, chronic lymphocytic leukemia, liver cancer, small cell lung cancer, non-small cell lung cancer, MALT lymphoma, malignant fibrous histiocytoma, malignant peripheral nerve sheath tumor, malignant triton tumor, mantle cell lymphoma, marginal zone B-cell lymphoma, mast cell leukemia, mediastinal germ cell tumor, medullary carcinoma of the breast, medullary thyroid cancer, medulloblastoma, melanoma, meningioma, merkel cell cancer, mesothelioma, metastatic urothelial carcinoma, mixed Mullerian tumor, mucinous tumor, multiple myeloma, muscle tissue neoplasm, mycosis fungoides, myxoid liposarcoma, myxoma,
myxosarcoma, nasopharyngeal carcinoma, neurinoma, neuroblastoma, neurofibroma, neuroma, nodular melanoma, ocular cancer, oligoastrocytoma, oligodendroglioma, oncocytoma, optic nerve sheath meningioma, optic nerve tumor, oral cancer, osteosarcoma, ovarian cancer, Pancoast tumor, papillary thyroid cancer, paraganglioma, pinealoblastoma, pineocytoma, pituicytoma, pituitary adenoma, pituitary tumor, plasmacytoma, polyembryoma, precursor T-lymphoblastic lymphoma, primary central nervous system lymphoma, primary effusion lymphoma, preimary peritoneal cancer, prostate cancer, pancreatic cancer, pharyngeal cancer, pseudomyxoma periotonei, renal cell carcinoma, renal medullary carcinoma, retinoblastoma, rhabdomyoma, rhabdomyosarcoma, Richter's transformation, rectal cancer, sarcoma, Schwannomatosis, seminoma, Sertoli cell tumor, sex cord-gonadal stromal tumor, signet ring cell carcinoma, skin cancer, small blue round cell tumors, small cell carcinoma, soft tissue sarcoma, somatostatinoma, soot wart, spinal tumor, splenic marginal zone lymphoma, squamous cell carcinoma, synovial sarcoma, Sezary's disease, small intestine cancer, stomach cancer, T-cell lymphoma, testicular cancer, thecoma, thyroid cancer, transitional cell carcinoma, throat cancer, urachal cancer, urogenital cancer, urothelial carcinoma, uveal melanoma, uterine cancer, verrucous carcinoma, visual pathway glioma, vulvar cancer, vaginal cancer, Waldenstrom's macroglobulinemia, Warthin's tumor and Wilms' tumor.
Another embodiment includes a method for the treatment of benign proliferative disorders. Examples of benign proliferative disorders include, but are not limited to, benign soft tissue rumors, bone tumors, brain and spinal tumors, eyelid and orbital tumors, granuloma, lipoma, meningioma, multiple endocrine neoplasia, nasal polyps, pituitary tumors, prolactinoma, pseudotumor cerebri, seborrheic keratoses, stomach polyps, thyroid nodules, cystic neoplasms of the pancreas, hemangiomas, vocal cord nodules, polyps, and cysts, Castleman disease, chronic pilonidal disease, dermatofibroma, pilar cyst, pyogenic granuloma and juvenile polyposis syndrome.
Another embodiment includes a therapeutic method useful for modulating protein methylation, gene expression, cell proliferation, cell differentiation and/or apoptosis in vivo in diseases mentioned above, in particular cancer, comprising administering to a patient in need of such therapy a pharmacologically active and therapeutically effective amount of one or more of the compounds as described herein.
Another embodiment includes a method for regulating endogenous or heterologous promotor activity by contacting a cell with a compound as described herein.
Another embodiment includes the use of a compound as described herein for the production of pharmaceutical compositions which are employed for the treatment and/or prophylaxis and/or amelioration of the diseases, disorders, illnesses and/or conditions as mentioned herein.
Another embodiment includes the use of a compound as described herein for the production of pharmaceutical compositions which are employed for the treatment and/or prophylaxis of diseases and/or disorders responsive or sensitive to the inhibition of histone demethylases, particularly those diseases mentioned above, such as e.g. cancer.
Compounds as described herein may be administered using any amount and any route of administration effective for treating or lessening the severity of the disorder. The exact amount required will vary from patient to patient, depending on the species, age, and general condition of the patient, for example the severity of the disorder, the particular compound, its mode of administration, and the like. The total daily usage of a compound as described herein by a given patient will be decided by the attending physician within the scope of sound medical judgment. The specific effective dose level for any particular patient will depend upon a variety of factors including the disorder being treated and the severity of the disorder; the activity of the specific compound employed; the specific composition employed; the age, body weight, general health, sex and diet of the patient; the time of administration, route of administration, and rate of excretion of the specific compound employed; the duration of the treatment; drugs used in combination or coincidental with the specific compound employed, and like factors well known in the medical arts.
Another embodiment includes a method of inhibiting KDM5 activity in a biological sample comprising contacting said biological sample with a compound as described herein.
The term "biological sample", as used herein, includes, without limitation, a cell, cell cultures or extracts thereof; biopsied material obtained from a mammal or extracts thereof; and blood, saliva, urine, feces, semen, tears, or other body fluids or extracts thereof.
Co-Administration of Compounds and Other Agents
The compound as described herein may be employed alone or in combination with other agents for treatment. For example, the second agent of the pharmaceutical combination
formulation or dosing regimen may have complementary activities to the compound as described herein such that they do not adversely affect each other. The compounds may be administered together in a unitary pharmaceutical composition or separately. In one embodiment a compound or a pharmaceutically acceptable salt can be co-administered with a cytotoxic agent to treat proliferative diseases and cancer.
The term "co-administering" refers to either simultaneous admuiistration, or any manner of separate sequential administration, of a compound as described herein, and a further active pharmaceutical ingredient or ingredients, including cytotoxic agents and radiation treatment. If the admimstration is not simultaneous, the compounds are administered in a close time proximity to
each other. Furthermore, it does not matter if the compounds are administered in the same dosage form, e.g. one compound may be administered topically and another compound may be
administered orally.
Typically, any agent that has activity against a disease or condition being treated may be co- administered. Examples of such agents can be found in Cancer Principles and Practice of
Oncology by V.T. Devita and S. Hellman (editors), 6th edition (February 15, 2001), Lippincott Williams & Wilkins Publishers. A person of ordinary skill in the art would be able to discern which combinations of agents would be useful based on the particular characteristics of the drugs and the disease involved.
In one embodiment, the treatment method includes the co-administration of a compound as described herein and at least one cytotoxic agent. The term "cytotoxic agent" as used herein refers to a substance that inhibits or prevents a cellular function and/or causes cell death or destruction. Cytotoxic agents include, but are not limited to, radioactive isotopes (e.g., At211, 1131, 1125, Y90, Re186, Re188, Sm153, Bi212, P32, Pb212 and radioactive isotopes of Lu); chemotherapeutic agents; growth inhibitory agents; enzymes and fragments thereof such as nucleolytic enzymes; and toxins such as small molecule toxins or enzymatically active toxins of bacterial, fungal, plant or animal origin, including fragments and/or variants thereof.
Exemplary cytotoxic agents can be selected from anti-microtubule agents, platinum coordination complexes, alkylating agents, antibiotic agents, topoisomerase II inhibitors, antimetabolites, topoisomerase I inhibitors, hormones and hormonal analogues, signal transduction pathway inhibitors, non-receptor tyrosine kinase angiogenesis inhibitors, immunotherapeutic agents, proapoptotic agents, nhibitors of LDH-A; inhibitors of fatty acid biosynthesis; cell cycle signaling inhibitors; HDAC inhibitors, proteasome inhibitors; and inhibitors of cancer metabolism.
"Chemotherapeutic agent" includes chemical compounds useful in the treatment of cancer. Examples of chemotherapeutic agents include erlotinib (TARCEVA®, Genentech/OSI Pharm.), bortezomib (VELCADE®, Millennium Pharm.), disulfiram , epigallocatechin gallate , salinosporamide A, carfilzomib, 17- AAG(geldanamycin), radicicol, lactate dehydrogenase A (LDH-A), fulvestrant (FASLODEX®, AstraZeneca), sunitib (SUTENT®, Pfizer/Sugen), letrozole (FEMARA®, Novartis), imatinib mesylate (GLEEVEC®., Novartis), finasunate (VATALANIB®, Novartis), oxaliplatin (ELOXATIN®, Sanofi), 5-FU (5-fluorouracil), leucovorin, Rapamycin
(Sirolimus, RAPAMUNE®, Wyeth), Lapatinib (TYKERB®, GSK572016, Glaxo Smith Kline),
Lonafamib (SCH 66336), sorafenib (NEXAVAR®, Bayer Labs), gefitinib (IRESSA®,
AstraZeneca), AG1478, alkylating agents such as thiotepa and CYTOXAN® cyclosphosphamide; alkyl sulfonates such as busulfan, improsulfan and piposulfan; aziridines such as benzodopa,
carboquone, meturedopa, and uredopa; ethylenimines and methylamelamines including altretamine, triethylenemelamine, triemylenephosphoramide, triethylenethiophosphoramide and
trimethylomelamine; acetogenins (especially bullatacin and bullatacinone); a camptothecin
(including topotecan and irinotecan); bryostatin; callystatin; CC-1065 (including its adozelesin, carzelesin and bizelesin synthetic analogs); cryptophycins (particularly cryptophycin 1 and cryptophycin 8); adrenocorticosteroids (including prednisone and prednisolone); cyproterone acetate; 5cc-reductases including finasteride and dutasteride); vorinostat, romidepsin, panobinostat, valproic acid, mocetinostat dolastatin; aldesleukin, talc duocarmycin (including the synthetic analogs, KW-2189 and CB1-TM1); eleutherobin; pancratistatin; a sarcodictyin; spongistatin;
nitrogen mustards such as chlorambucil, chlomaphazine, chlorophosphamide, estramustine, ifosfamide, mechlorethamine, mechlorethamine oxide hydrochloride, melphalan, novembichin, phenesterine, prednimustine, trofosfamide, uracil mustard; nitrosoureas such as carmustine, chlorozotocin, fotemustine, iomustine, nimustine, and ranimnustine; antibiotics such as the enediyne antibiotics (e.g., calicheamicin, especially calicheamicin γΐ I and calicheamicin ω11 (Angew Chem. Intl. Ed. Engl. 1994 33:183-186); dynemicin, including dynemicin A;
bisphosphonates, such as clodronate; an esperamicin; as well as neocarzinostatin chromophore and related chromoprotein enediyne antibiotic chromophores), aclacinomysins, actinomycin, authramycin, azaserine, bleomycins, cactinomycin, carabicin, caminomycin, carzinophilin, chromomycinis, dactinomycin, daunorubicin, detorubicin, 6-diazo-5-oxo-L-norleucine,
ADRIAMYCIN® (doxorubicin), morpholino-doxorubicin. cyanomorpholino-doxorubicin, 2- pyrrolino-doxorubicin and deoxydoxorubicin), epirubicin, esorubicin, idarubicin, marcellomycin, mitomycins such as mitomycin C, mycophenolic acid, nogalamycin, olivomycins, peplomycin, porfiromycin, puromycin, quelamycin, rodorubicin, streptonigrin, streptozocin, tubercidin, ubenimex, zinostatin, zorubicin; anti-metabolites such as methotrexate and 5-fluorouracil (5-FU); folic acid analogs such as denopterin, methotrexate, pteropterin, trimetrexate; purine analogs such as fludarabine, 6-mercaptopurine, thiamiprine, thioguanine; pyrimidine analogs such as ancitabine, azacitidine, 6-azauridine, carmofur, cytarabine, dideoxyuridine, doxifluridine, enocitabine, floxuridine; androgens such as calusterone, dromostanolone propionate, epitiostanol, mepitiostane, testolactone; anti-adrenals such as aminoglutethimide, mitotane, trilostane; folic acid replenisher such as frolinic acid; aceglatone; aldophosphamide glycoside; aminolevulinic acid; eniluracil;
amsacrine; bestrabucil; bisantrene; edatraxate; defofamine; demecolcine; diaziquone; elfomitbine; elliptinium acetate; an epothilone; etoglucid; gallium nitrate; hydroxyurea; lentinan; lonidainine; maytansinoids such as maytanstne and ansamitocins; mitoguazone; mitoxantrone; mopidamnol;
nitraerine; pentostatin; phenamet; pirarubicin; losoxantrone; podophyllinic acid; 2-ethylhydrazide; procarbazine; PSK® polysaccharide complex (JHS Natural Products, Eugene, Oreg.); razoxane; rhizoxin; sizoftiran; spirogermanium; tenuazonic acid; triaziquone; 2,2,,2"-trichlorotriethylamine; trichothecenes (especially T-2 toxin, verracurin A, roridin A and anguidine); urethan; vindesine; dacarbazine; mannomustine; mitobronitol; mitolactol; pipobroman; gacytosine; arabinoside ("Ara- C"); cyclophosphamide; thiotepa; taxoids, e.g., TAXOL (paclitaxel; Bristol-Myers Squibb
Oncology, Princeton, N.J.), ABRAXANE® (Cremophor-rree), albumin-engineered nanoparticle formulations of paclitaxel (American Pharmaceutical Partners, Schaumberg, 111.), and
TAXOTERE® (docetaxel, doxetaxel; Sanofi-Aventis); chloranmbucil; GEMZAR® (gemcitabine); 6-thioguanine; mercaptopurine; methotrexate; platinum analogs such as cisplatin and carboplatin; vinblastine; etoposide (VP-16); ifosfamide; mitoxantrone; vincristine; NAVELBINE®
(vinorelbine); novantrone; teniposide; edatrexate; daunomycin; aminopterin; capecitabine
(XELODA®); ibandronate; CPT-11; topoisomerase inhibitor RFS 2000; difluoromemylornithine (DMFO); retinoids such as retinoic acid; and pharmaceutically acceptable salts, acids and derivatives of any of the above.
Chemotherapeutic agent also includes (i) anti-hormonal agents that act to regulate or inhibit hormone action on tumors such as anti-estrogens and selective estrogen receptor modulators (SERMs), including, for example, tamoxifen (including NOLVADEX®; tamoxifen citrate), raloxifene, droloxifene, iodoxyfene , 4-hydroxytamoxifen, trioxifene, keoxifene, LY1 17018, onapristone, and FARESTON® (toremifine citrate); (ii) aromatase inhibitors that inhibit the enzyme aromatase, which regulates estrogen production in the adrenal glands, such as, for example, 4(5)- irnidazoles, aminoglutethimide, MEGASE® (megestrol acetate), AROMASIN® (exemestane;
Pfizer), formestanie, fadrozole, RTVISOR® (vorozole), FEMARA® (Ietrozole; Novartis), and ARIMIDEX® (anastrozole; AstraZeneca); (iii) anti-androgens such as flutamide, nilutamide, bicalutamide, leuprolide and goserelin; buserelin, tripterelin, medroxyprogesterone acetate, diethylstilbestrol, premarin, fluoxymesterone, all transretionic acid, fenretinide, as well as troxacitabine (a 1,3-dioxolane nucleoside cytosine analog); (iv) protein kinase inhibitors; (v) lipid kinase inhibitors; (vi) antisense oligonucleotides, particularly those which inhibit expression of genes in signaling pathways implicated in aberrant cell proliferation, such as, for example, PKC- alpha, Ralf and H-Ras; (vii) ribozymes such as VEGF expression inhibitors (e.g., ANGIOZYME®) and HER2 expression inhibitors; (viii) vaccines such as gene therapy vaccines, for example, ALLOVECTIN®, LEUVECTIN®, and VAXID®; PROLEUKIN®, rIL-2; a topoisomerase 1 inhibitor such as LURTOTECAN®; ABARELIX® rmRH; and (ix) pharmaceutically acceptable salts, acids and derivatives of any of the above.
Chemotherapeutic agent also includes antibodies such as alemtuzumab (Campath), bevacizumab (AVASTIN®, Genentech); cetuximab (ERBITUX®, Imclone); panitumumab
(VECTIBIX®, Amgen), rituximab (RITUXAN®, Genentech/Biogen Idee), pertuzumab
(OMNITARG®, 2C4, Genentech), trastuzumab (HERCEPTIN®, Genentech), tositumoraab (Bexxar, Corixia), and the antibody drug conjugate, gemtuzumab ozogamicin (MYLOTARG®, Wyeth). Additional humanized monoclonal antibodies with therapeutic potential as agents in combination with the compounds of the invention include: apolizumab, aselizumab, atlizumab, bapineuzumab, bivatuzumab mertansine, cantuzumab mertansine, cedelizumab, certolizumab pegol, cidfusituzumab, cidtuzumab, daclizumab, eculizumab, efalizumab, epratuzumab, erlizumab, felvizumab, fontolizumab, gemtuzumab ozogamicin, inotuzumab ozogamicin, ipilimumab, labetuzumab, lintuzumab, matuzumab, mepolizumab, motavizumab, motovizumab, natalizumab, nimotuzumab, nolovizumab, numavizumab, ocrelizumab, omalizumab, palivizumab, pascolizumab, pecfusituzumab, pectuzumab, pexelizumab, ralivizumab, ranibizumab, reslivizumab, reslizumab, resyvizumab, rovelizumab, ruplizumab, sibrotuzumab, siplizumab, sontuzumab, tacatuzumab tetraxetan, tadocizumab, talizumab, tefibazumab, tocilizumab, toralizumab, tucotuzumab celmoleukin, tucusituzumab, umavizumab, urtoxazumab, ustekinumab, visilizumab, and the anti- interleukin-12 (ABT-874/J695, Wyeth Research and Abbott Laboratories) which is a recombinant exclusively human-sequence, full-length Igd λ antibody genetically modified to recognize interleukin-12 p40 protein.
Chemotherapeutic agent also includes "EGFR inhibitors," which refers to compounds that bind to or otherwise interact directly with EGFR and prevent or reduce its signaling activity, and is alternatively referred to as an "EGFR antagonist." Examples of such agents include antibodies and small molecules that bind to EGFR. Examples of antibodies which bind to EGFR include MAb 579 (ATCC CRL HB 8506), MAb 455 (ATCC CRL HB8507), MAb 225 (ATCC CRL 8508), MAb 528 (ATCC CRL 8509) (see, US Patent No. 4,943, 533, Mendelsohn et al.) and variants thereof, such as chimerized 225 (C225 or Cetuximab; ERBUTIX®) and reshaped human 225 (H225) (see, WO 96/40210, Imclone Systems Inc.); IMC-11F8, a fully human, EGFR-targeted antibody
(Imclone); antibodies that bind type II mutant EGFR (US Patent No. 5,212,290); humanized and chimeric antibodies that bind EGFR as described in US Patent No. 5,891,996; and human antibodies that bind EGFR, such as ABX-EGF or Panitumumab (see WO98/50433,
Abgenix/Amgen); EMD 55900 (Stragliotto et al. Eur. J. Cancer 32A:636-640 (1996)); EMD7200 (matuzumab) a humanized EGFR antibody directed against EGFR that competes with both EGF and TGF-alpha for EGFR binding (EMD/Merck); human EGFR antibody, HuMax-EGFR
(GenMab); fully human antibodies known as E 1.1 , E2.4, E2.5, E6.2, E6.4, E2.l l, E6. 3 and E7.6. 3 and described in US 6,235,883; MDX-447 (Medarex Inc); and mAb 806 or humanized mAb 806 (Johns et al., J. Biol. Chem. 279(29):30375-30384 (2004)). The anti-EGFR antibody may be conjugated with a cytotoxic agent, thus generating an immunoconjugate (see, e.g., EP659.439A2, Merck Patent GmbH). EGFR antagonists include small molecules such as compounds described in US Patent Nos: 5,616,582, 5,457,105, 5,475,001, 5,654,307, 5,679,683, 6,084,095, 6,265,410, 6,455,534, 6,521,620, 6,596,726, 6,713,484, 5,770,599, 6,140,332, 5,866,572, 6,399,602,
6,344,459, 6,602,863, 6,391,874, 6,344,455, 5,760,041, 6,002,008, and 5,747,498, as well as the following PCT publications: W098/14451, WO98/50038, WO99/09016, and WO99/24037.
Particular small molecule EGFR antagonists include OSI-774 (CP-358774, erlotinib, TARCEV A® Genentech/OSI Pharmaceuticals); PD 183805 (CI 1033, 2-propenamide, N-[4-[(3-chloro-4- fluorophenyl)arnino]-7-[3-(4-morpholinyl)propoxy]-6-quirlazolinyl]-, dihydrochloride, Pfizer Inc.); ZD1839, gefitinib (IRESSA®) 4-(3'-Chloro-4'-fluoroanilino)-7-methoxy-6-(3-morpholinopropoxy)quinazoline, AstraZeneca); ZM 105180 ((6-amino-4-(3-methylphenyl-amino)- quinazoline, Zeneca); BIBX-1382 (N8K3-chloro-4-fluoro-phenyl)-N2-(l-methyI-piperidin-4-yl)- pyrimido[5,4-d]pyrimidine-2,8-diamine, Boehringer Ingelheim); PKI-166 ((R)-4-[4-[(l- phenylethyl)amino]- 1 H-pyrrolo[2,3-d]pyrimidin-6-yl]-phenol); (R)-6-(4-hydroxyphenyI)-4-[(l- phenylethyl)amino]-7H-pyiTolo[2,3-d]pyrimidine); CL-387785 (N-[4-[(3-bromophenyl)amino]-6- qumazolinyl]-2-butynamide); EKB-569 (N-[4-[(3-chloro-4-fluorophenyl)amino]-3-cyano-7- emoxy-6-qmnolmyl]-4-(dimethylamino)-2-butenamide) (Wyeth); AG1478 (Pfizer); AG1571 (SU 5271; Pfizer); dual EGFR/HER2 tyrosine kinase inhibitors such as lapatinib (TYKERB®,
GSK572016 or N-[3-chloro-4-[(3 fluorophenyl)methoxy]phenyl]- 6[5[[[2memylsulfonyl)emy]]amino]memyl]-2-mranyl]-4-quinazolinamine).
Chemotherapeutic agents also include "tyrosine kinase inhibitors" including the EGFR- targeted drugs noted in the preceding paragraph; small molecule HER2 tyrosine kinase inhibitor such as TAK165 available from Takeda; CP-724,714, an oral selective inhibitor of the ErbB2 receptor tyrosine kinase (Pfizer and OSI); dual-HER inhibitors such as EKB-569 (available from Wyeth) which preferentially binds EGFR but inhibits both HER2 and EGFR-overexpressing cells; lapatinib (GSK572016; available from Glaxo-SmithKIine), an oral HER2 and EGFR tyrosine kinase inhibitor; PKI-166 (available from Novartis); pan-HER inhibitors such as canertinib (CI- 1033; Pharmacia); Raf-1 inhibitors such as antisense agent ISIS-5132 available from ISIS
Pharmaceuticals which inhibit Raf-1 signaling; non-HER targeted TK inhibitors such as imatinib mesylate (GLEEVEC®, available from Glaxo SmithKline); multi-targeted tyrosine kinase
inhibitors such as sunitinib (SUTENT®, available from Pfizer); VEGF receptor tyrosine kinase inhibitors such as vatalanib (PTK787/ZK222584, available from Novartis/Schering AG); MAPK extracellular regulated kinase I inhibitor CI- 1040 (available from Pharmacia); quinazolines, such as PD 153035,4-(3-chloroanilino) quinazoline; pyridopyrimidines; pyrimidopyrimidines;
pyrrolopyrimidines, such as CGP 59326, CGP 60261 and CGP 62706; pyrazolopyrimidines, 4- (phenylamino)-7H-pyrrolo[2,3-d] pyrimidhies; curcumin (diferuloyl methane, 4,5-bis (4- fluoroanilino)phthalimide); tyrphostines containing nitrothiophene moieties; PD-0183805 (Warner- Lamber); antisense molecules (e.g. those that bind to HER-encoding nucleic acid); quinoxalines (US Patent No. 5,804,396); tryphostins (US Patent No. 5,804,396); ZD6474 (Astra Zeneca); PTK- 787 (Novartis/Schering AG); pan-HER inhibitors such as CI- 1033 (Pfizer); Affinitac (ISIS 3521; Isis/Lilly); imatinib mesylate (GLEEVEC®); PKI 166 (Novartis); GW2016 (Glaxo SmithKline); CI- 1033 (Pfizer); EKB-569 (Wyeth); Semaxinib (Pfizer); ZD6474 (AstraZeneca); PTK-787 (Novartis/Schering AG); INC-lCl 1 (Imclone), rapamycin (sirolimus, RAPAMUNE®); or as described in any of the following patent publications: US Patent No. 5,804,396; WO 1999/09016 (American Cyanamid); WO 1998/43960 (American Cyanamid); WO 1997/38983 (Warner
Lambert); WO 1999/06378 (Warner Lambert); WO 1999/06396 (Warner Lambert); WO
1996/30347 (Pfizer, Inc); WO 1996/33978 (Zeneca); WO 1996/3397 (Zeneca) and WO
1996/33980 (Zeneca).
Cbemotherapeutic agents also include dexamethasone, interferons, colchicine, metoprine, cyclosporine, amphotericin, metronidazole, alemtuzumab, alitretinoin, allopurinol, amifostine, arsenic trioxide, asparaginase, BCG live, bevacuzimab, bexarotene, cladribine, clofarabine, darbepoetin alfa, denileukin, dexrazoxane, epoetin alfa, elotinib, filgrastim, histrelin acetate, ibritumomab, interferon alfa-2a, interferon alfa-2b, lenalidomide, levamisole, mesna, methoxsalen, nandrolone, nelarabine, nofetumomab, oprelvekin, palifermin, pamidronate, pegademase, pegaspargase, pegfilgrastim, pemetrexed disodium, plicamycin, porfimer sodium, quinacrine, rasburicase, sargramostim, temozolomide, VM-26, 6-TG, toremifene, tretinoin, ATRA, valrubicin, zoledronate, and zoledronic acid, and pharmaceutically acceptable salts thereof.
Chemotherapeutic agents also include hydrocortisone, hydrocortisone acetate, cortisone acetate, tixocortol pivalate, triamcinolone acetonide, triamcinolone alcohol, mometasone, amcinonide, budesonide, desonide, fluocinonide, fluocinolone acetonide, betamethasone, betamethasone sodium phosphate, dexamethasone, dexamethasone sodium phosphate,
fluocortolone, hydrocortisone- 17-butyrate, hydrocortisone- 17- valerate, aclometasone dipropionate, betamethasone valerate, betamethasone dipropionate, prednicarbate, clobetasone- 17-butyrate, clobetasol-17-propionate, fluocortolone caproate, fluocortolone pivalate and fluprednidene acetate;
immune selective anti-inflammatory peptides (ImSAIDs) such as phenylalanine-glutamine-glycine (FEG) and its D-isomeric form (feG) (IMULAN BioTherapeutics, LLC); anti-rheumatic drugs such as azathioprine, cyclosporin (cyclosporine A), D-penicillamine, gold salts, hydroxychloroquine, leflunomideminocycline, sulfasalazine, tumor necrosis factor alpha (TNFa) blockers such as etanercept (Enbrel), infliximab (Remicade), adalimumab (Humira), certolizumab pegol (Cimzia), golimumab (Simponi), Interleukin 1 (IL-1) blockers such as anakinra (Kineret), T cell
costimulation blockers such as abatacept (Orencia), Interleukin 6 (IL-6) blockers such as tocilizumab (ACTEMERA®); Interleukin 13 (IL-13) blockers such as lebrikizumab; Interferon alpha (IFN) blockers such as Rontalizumab; Beta 7 integrin blockers such as rhuMAb Beta7; IgE pathway blockers such as Anti-Mi prime; Secreted homotrimeric LTa3 and membrane bound heterotrimer LTal/p2 blockers such as Anti-lymphotoxin alpha (LTa); radioactive isotopes (e.g., At211, 113', I125, Y90, Re186, Re188, Sm153, Bi212, P32, Pb212 and radioactive isotopes of Lu);
miscellaneous investigational agents such as thioplatin, PS-341, phenylbutyrate, ET-18- OCH3, or famesyl transferase inhibitors (L-739749, L-744832); polyphenols such as quercetin, resveratrol, piceatannol, epigallocatechine gallate, theaflavins, flavanols, procyanidins, betulinic acid and derivatives thereof; autophagy inhibitors such as chloroquine; delta-9-tetrahydrocannabinol (dronabinol, MARTNOL®); beta-lapachone; lapachol; colchicines; betulinic acid;
acetylcamptothecin, scopolectin, and 9-aminocamptothecin); podophyllotoxin; tegafur
(UFTORAL®); bexarotene (TARGRETIN®); bisphosphonates such as clodronate (for example, BONEFOS® or OSTAC®), etidronate (DIDROCAL®), NE-58095, zoledronic acid/zoledronate (ZOMETA®), alendronate (FOSAMAX®), pamidronate (AREDIA®), tiludronate (SKELID®), or risedronate (ACTONEL®); and epidermal growth factor receptor (EGF-R); vaccines such as THERATOPE® vaccine; perifosine, COX-2 inhibitor (e.g. celecoxib or etoricoxib), proteosome inhibitor (e.g. PS341); CCI-779; tipifarnib (R11577); orafenib, ABT510; Bcl-2 inhibitor such as oblimersen sodium (GENASENSE®); pixantrone; farnesyltransferase inhibitors such as lonafarnib
(SCH 6636, SARASARTM ); and pharmaceutically acceptable salts, acids or derivatives of any of the above; as well as combinations of two or more of the above such as CHOP, an abbreviation for a combined therapy of cyclophosphamide, doxorubicin, vincristine, and prednisolone; and
FOLFOX, an abbreviation for a treatment regimen with oxaliplatin (ELOXATIN™) combined with 5-FU and leucovorin.
Chemotherapeutic agents also include non-steroidal anti-inflammatory drugswith analgesic, antipyretic and anti-inflammatory effects. NSAIDs include non-selective inhibitors of the enzyme cyclooxygenase. Specific examples of NSAIDs include aspirin, propionic acid derivatives such as ibuprofen, fenoprofen, ketoprofen, flurbiprofen, oxaprozin and naproxen, acetic acid derivatives
such as indomethacin, sulindac, etodolac, diclofenac, enolic acid derivatives such as piroxicam, meloxicam, tenoxicam, droxicam, lornoxicam and isoxicam, fenamic acid derivatives such as mefenamic acid, meclofenamic acid, flufenamic acid, tolfenamic acid, and COX-2 inhibitors such as celecoxib, etoricoxib, lumiracoxib, parecoxib, rofecoxib, rofecoxib, and valdecoxib. NSAIDs can be indicated for the symptomatic relief of conditions such as rheumatoid arthritis, osteoarthritis, inflammatory arthropathies, ankylosing spondylitis, psoriatic arthritis, Reiter's syndrome, acute gout, dysmenorrhoea, metastatic bone pain, headache and migraine, postoperative pain, mild-to- moderate pain due to inflammation and tissue injury, pyrexia, ileus, and renal colic.
Chemotherapeutic agents also include treatments for Alzheimer's Disease such as donepezil hydrochloride and rivastigmine; treatments for Parkinson's Disease such as L-
DOPA/carbidopa, entacapone, ropinrole, pramipexole, bromocriptine, pergolide, trihexephendyl, and amantadine; agents for treating multiple sclerosis (MS) such as beta interferon (e.g., Avonex and Rebif®), glatiramer acetate, and mitoxantrone; treatments for asthma such as albuterol and montelukast sodium; agents for treating schizophrenia such as zyprexa, risperdal, seroquel, and haloperidol; anti-inflammatory agents such as corticosteroids, TNF blockers, IL-1 RA,
azathioprine, cyclophosphamide, and sulfasalazine; immunomodulatory and immunosuppressive agents such as cyclosporin, tacrolimus, rapamycin, mycophenolate mofetil, interferons,
corticosteroids, cyclophophamide, azathioprine, and sulfasalazine; neurotrophic factors such as acetylcholinesterase inhibitors, MAO inhibitors, interferons, anti-convulsants, ion channel blockers, riluzole, and anti-Parkinsonian agents; agents for treating cardiovascular disease such as beta- blockers, ACE inhibitors, diuretics, nitrates, calcium channel blockers, and statins; agents for treating liver disease such as corticosteroids, cholestyramine, interferons, and anti-viral agents; agents for treating blood disorders such as corticosteroids, anti-leukemic agents, and growth factors; and agents for treating immunodeficiency disorders such as gamma globulin.
Additionally, chemotherapeutic agents include pharmaceutically acceptable salts, acids or derivatives of any of chemotherapeutic agents, described herein, as well as combinations of two or more of them.
The amount of both the compound as described herein and additional agent (in those compositions which comprise an additional therapeutic agent as described above) that may be combined with the carrier materials to produce a single dosage form will vary depending upon the host treated and the particular mode of administration. In certain embodiments, compositions of this invention are formulated such that a dosage of between 0.01 - 100 mg/kg body weight/day of an inventive can be administered.
The additional therapeutic agent and the compound as described herein may act synergistically. Therefore, the amount of additional therapeutic agent in such compositions may be less than that required in a monotherapy utilizing only that therapeutic agent, or there may be fewer side effects for the patient given that a lower dose is used. In certain embodiments, in such compositions a dosage of between 0.01 - 1,000 μg/kg body weight/day of the additional therapeutic agent can be administered.
Another aspect includes treating or preventing drug resistance in a patient using a compound as described herein. For example, a method of treating or preventing drug resistant cancer in a patient comprises administering a therapeutically effective amount of a compound as described herein to the patient alone or in combination with a cytotoxic agent. In certain
embodiments, the individual is selected for treatment with a cytotoxic agent (e.g., targeted therapies, chemotherapies, and/or radiotherapies). In certain embodiments, the individual starts treatment comprising administration of a compound as described herein prior to treatment with the cytotoxic agent In certain embodiments, the individual concurrently receives treatment comprising the compound as described herein and the cytotoxic agent. In certain embodiments, the compound as described herein increases the period of cancer sensitivity and/or delays development of cancer resistance.
In particular, provided herein are methods of treating cancer in an individual comprising administering to the individual (a) a compound as described herein and (b) a cytotoxic agent (e.g., targeted therapy, chemotherapy, and/or radiotherapy). In certain embodiments, the respective amounts of the compound as described herein and the cytotoxic agent are effective to increase the period of cancer sensitivity and/or delay the development of cancer cell resistance to the cancer therapy agent. In certain embodiments, the respective amounts of the compound as described herein and the cytotoxic agent are effective to increase efficacy of a cancer treatment comprising the cancer therapy agent. For example, in certain embodiments, the respective amounts of the compound as described herein and the cytotoxic agent are effective to increase efficacy compared to a treatment (e.g., standard of care treatment) (e.g., standard of care treatment) comprising administering an effective amount of the cancer therapy agent without (in the absence of) the compound as described herein. In certain embodiments, the respective amounts of the compound as described herein and cytotoxic agent agent are effective to increase response (e.g., complete response) compared to a treatment (e.g., standard of care treatment) comprising administering an effective amount of cytotoxic agent without (in the absence of) the compound as described herein.
Also provided herein are methods of increasing efficacy of a cancer treatment comprising a cytotoxic agent in an individual comprising administering to the individual (a) an effective amount of a compound as described herein and (b) an effective amount of the cytotoxic agent.
Provided herein are methods of treating cancer in an individual wherein cancer treatment comprising administering to the individual (a) an effective amount of a compound as described herein and (b) an effective amount of a cytotoxic agent, wherein the cancer treatment has increased efficacy compared to a treatment (e.g., standard of care treatment) comprising administering an effective amount of cytotoxic agent without (in the absence of) the compound as described herein.
In addition, provided herein are methods of delaying and/or preventing development of cancer resistant to a cancer therapy agent in an individual, comprising administering to the individual (a) an effective amount of a compound as described herein and (b) an effective amount of the cytotoxic agent.
Provided herein are methods of treating an individual with cancer who has an increased likelihood of developing resistance to a cancer therapy agent comprising administering to the individual (a) an effective amount of a compound as described herein and (b) an effective amount of the cytotoxic agent.
Further provided herein are methods of increasing sensitivity to a cancer therapy agent in an individual with cancer comprising administering to the individual (a) an effective amount of a compound as described herein and (b) an effective amount of the cytotoxic agent.
Provided herein are also methods of extending the period of a cancer therapy agent sensitivity in an individual with cancer comprising administering to the individual (a) an effective amount of a compound as described herein and (b) an effective amount of the cytotoxic agent.
Provided herein are methods of extending the duration of response to a cytotoxic agent in an individual with cancer comprising administering to the individual (a) an effective amount of a compound as described herein and (b) an effective amount of the cytotoxic agent.
Γη certain embodiments of any of the methods, the cytotoxic agent is a targeted therapy. In certain embodiments, the targeted therapy is one or more of an EGFR antagonist, RAF inhibitor, and/or PI3K inhibitor.
In certain embodiments of any of the methods, the targeted therapy is an EGFR antagonist. In certain embodiments of any of the methods, the EGFR antagonist is N-(3-ethynylphenyl)-6,7- bis(2-methoxyethoxy)-4-quinazolinamine and/or a pharmaceutical acceptable salt thereof. In certain embodiments, the EGFR antagonist is N-(3-ethynylphenyl)-6,7-bis(2-methoxyethoxy)-4- quinazolinamine. In certain embodiments, the EGFR antagonist is N-(4-(3-fluorobenzyloxy)-3-
chlorophenyl)-6-(5-((2-(methylsulfony])ethylam ino)methyl)furan-2-yl)quinazoliin-4-amine,di4- methylbenzenesulfonate or a pharmaceutically acceptable salt thereof (e.g. , lapatinib).
In certain embodiments of any of the methods, targeted therapy is a RAF inhibitor. In certain embodiments, the RAF inhibitor is a BRAF inhibitor. In certain embodiments, the RAF inhibitor is a CRAF inhibitor. In certain embodiments, the BRAF inhibitor is vemurafenib. In certain embodiments, the RAF inhibitor is 3-(2-cyanopropan-2-yl)-N-(4-methyl-3-(3-methyl-4- oxo-3,4-dihydroquinazolin-6-ylamino)phenyl)benzamide or a pharmaceutically acceptable salt thereof (e.g., AZ628 (CAS# 878739-06-1)).
In certain embodiments of any of the methods, the targeted therapy is a PI3K inhibitor. In certam embodiments of any of the methods, the cytotoxic agent is chemotherapy. Γη certain embodiments of any of the methods, the chemotherapy is a taxane. In certain embodiments, the taxane is paclitaxel. in certain embodiments, the taxane is docetaxel.
In certain embodiments of any of the methods, the cytotoxic agent is a platinum agent. In certain embodiments, the platinum agent is carboplatin. In certain embodiments, the platinum agent is cisplatin. In certain embodiments of any of the methods, the cytotoxic agent is a taxane and a platinum agent. In certain embodiments, the taxane is paclitaxel. In certain embodiments, the taxane is docetaxel. In certain embodiments, the platinum agent is carboplatin. In certain embodiments, the platinum agent is cisplatin.
In certain embodiments of any of the methods, the cytotoxic agent is a vinca alkyloid. In certain embodiments, the vinca alkyloid is vinorelbine. In certain embodiments of any of the methods, the chemotherapy is a nucleoside analog. In certain embodiments, the nucleoside analog is gemcitabine.
In certain embodiments of any of the methods, the cytotoxic agent is radiotherapy.
In certain embodiments of any of the methods, the compound as described herein is concomitantly administered with the cytotoxic agent (e.g., targeted therapy, chemotherapy, and/or radiotherapy). In certain embodiments, the compound as described herein is administered prior to and/or concurrently with the cytotoxic agent (e.g., targeted therapy, chemotherapy, and/or radiotherapy).
In certain embodiments of any of the methods, the cancer is lung cancer, breast cancer, pancreatic cancer, colorectal cancer, and/or melanoma. In certain embodiments, the cancer is lung. In certain embodiments, the lung cancer is NSCLC. In certain embodiments, the cancer is breast cancer. In certain embodiments, the cancer is melanoma.
EXEMPLIFICATION
As depicted in the Examples below, in certain exemplary embodiments, compounds are prepared according to the following general procedures. It will be appreciated that, although the general methods depict the synthesis of certain compounds, the following general methods, and other methods known to one of ordinary skill in the art, can typically be applied to all compounds and subclasses and species of each of these compounds, as described herein.
The general synthetic methods illustrateds in Schemes 1 -4 and the general LCMS isolations procedures identified as LCMS Method A - LCMS Method F were used to prepare the compounds of Examples 1 -247 as detailed below.
Scheme 1 (Method A)

The general synthesis of compounds 8 is illustrated in Scheme 1. Organolithium addition to a pyrrolidine Weinreb amide 1 provided ketone 2, which was transformed to pyridine 3 upon hydrogenation. Treatment of compound 3 with trilflic anhydride gave intermediate 4. Suzuki coupling of triflate 4 with a boronic acid or boronate ester 5 gave compound 6. Deprotection of Boc group, followed by amide coupling with carboxylic acid 7 resulted in pyrrolidine amide 8.
Alternatively, removal of Boc group in intermediate 4, followed by amide coupling with a carboxylic acid 7 gave intermediate 9. Subsequently, Suzuki coupling of intermediate 9 with a boronic acid or boronate ester also led to compound 8. Besides Suzuki coupling with a boronate ester or boronic acid, the triflate 9 could also be transformed into compound 8, through Stille coupling reaction, or carbonylation, or Buchwald coupling reaction, etc.
Scheme 2 (Method B)

An alternative synthesis of compound 15 is illustrated in Scheme 2. Suzuki coupling of an aryl bromide 10 with a boronate ester 11 gave 2,5-dihydropyrrole 12. This was followed by hydrogenation to provide pyrrolidine intermediate 13, which was subsequently deprotected with hydrochloric acid to give pyrrolidine 14. Amide coupling with a carboxylic acid 7 then led to compound 15.
Scheme 3 (Method C)


A general synthesis of compound 20 is described in Scheme 3. 3-amino pyrrlidine 16 reacted with an acid chloride 17 or a carboxylic acid 18 to give amide 19. This was followed by deprotection of Boc group, and subsequent amide formation with a carboxylic acid 7 gave pyrrolidine amide 20. Alternatively, pyrrolidine 21 could be coupled with a carboxylic acid 7 to give amide 22.
Subsequent removal of Boc group, and coupling with an acid chloride, or a carboxylic acid, produced compound 20. In addition, the amino group in compound 22 could react with other reagents. For example, reaction of 22 with an isocyanate gave rise to a urea. Compund 22 could also undergo SNAr reaction with an aryl fluoride, to form a C-N bond. Alternatively, it could also form an heterocycle.
Scheme 4 (Method D)

A general synthesis of compound 27 is described in Scheme 4. 3-hydroxypyrrolidine 24 was converted to its mesylate, which upon treatment with a heterocyclic compound 25 provided compound 26. Alternatively, coupling of 3-hydroxypyrrolidine 24 and the heterocycle 25 could
also be achieved by a Mitsunobu reaction. After de-protection of Boc group in 26, amide coupling with a carboxylic acid 7 then afforded compound 27.
LCMS Method A (Agilent 10-80 AB, ELSD, 2 min)
Experiments were performed on an Agilent 1200 HPLC (with a PDA detector) with Agilent 61 10 MSD mass spectrometer using ESI as ionization source using an Xtimate CI 8, 3 um, 30 x 2.1 mm and a 1.2 mL/min flow -rate. Solvent A was water containing 0.038% TFA, and solvent B was acetonitrile containing 0.02% TFA. A gradient was run: starting with 10% A and 90% B, going to 20% A and 80% B within 0.9 min, then holding at 20% A and 80% B for 0.6 min. Total run time was 2 min.
LCMS Method B (Agilent 0-30 AB, ELSD, 2 min)
Experiments were performed on an Agilent 1200 HPLC (with a PDA detector) with Agilent 6110 MSD mass spectrometer using ESI as ionization source using an Xtimate CI 8, 3 um, 30 x 2.1 mm and a 1.2 mL/min flow rate. Solvent A was water containing 0.038% TFA, and solvent B was acetonitrile containing 0.02% TFA. A gradient was run: starting at 100% A, going to 30% A and 70% B within 0.9 min, then holding at 30% A and 70% B for 0.6 min. Total run time was 2 min.
LCMS Method C (Agilent 0-60 AB, ELSD, 2 min)
Experiments were performed on an Agilent 1200 HPLC (with a PDA detector) with Agilent 6110 MSD mass spectrometer using ESI as ionization source using an Xtimate CI 8, 3 um, 30 x 2.1 mm and a 1.2 mL/min flow rate. Solvent A was water containing 0.038% TFA, and solvent B was acetonitrile containing 0.02% TFA. A gradient was run: starting with 100% A and going to 40% A and 60% B within 0.9 min, then holding at 40% A and 60% B for 0.6 min. Total run time was 2 min.
LCMS Method D (Agilent 30-90 AB, ELSD, 2 min)
Experiments were performed on an Agilent 1200 HPLC (with a PDA detector) with Agilent 6110 MSD mass spectrometer using ESI as ionization source using an Xtimate CI 8, 3 um, 30 x 2.1 mm and a 1.2 mL/min flow rate. Solvent A was water containing 0.038% TFA, and solvent B was acetonitrile containing 0.02% TFA. A gradient was run: starting with 30% A and 70% B, going to 10% A and 90% B within 0.9 min, then holding at 10% A and 90% B for 0.6 min. Total run time was 2 min.
LCMS Method E (SHIMADZU 5-95 AB, ELSD, 1.5 min)
Experiments were performed on a SHIMADZU 20A HPLC (with a PDA detector) with
SHIMADZU 2010EV MSD mass spectrometer using ESI as ionization source using an Merk RP- 18e 2 x 25mm column and a 1.5mL/min flow rate. Solvent A was water containing 0.038% TFA, and solvent B was acetonitrile containing 0.02% TFA. A gradient was run: starting with 95% A and 5% B, going to 5% A and 95% B over the next 0.7 min. This solvent ratio was maintained for 0.4 min before returning to 95% A and 5% B over the next 0.4 min. Total run time was 1.5 min.
LCMS Method F (Agilent 5-95 AB, ELSD, 10 min)
Experiments were performed on an Agilent 6140 quadrupole LC/MS system linked to a HPLC Agilent 1200 system with a UV detector monitoring at 254 nm, and mass spectrometry scanning 90-1300 amu in ESI+ ionization mode. This system uses an Agilent SB C18 (1.8 um 30 x 2.1 mm) column, maintained at 25 °C and a 0.4 mL/min flow rate. Solvent A was water containmg 0.05% TFA, and solvent B was acetonitrile containing 0.05% TFA. A gradient was run: starting with 95% A and 5% B for the first 0.3 min, going to 5% A and 95% B over the next 6.5 min. This solvent ratio was maintained for 1.5 min before returning to 95% A and 5% B over the next 0.1 min. Total run time was 10 min.
Examples 1 and 2
S)-(3-Isopropyl-1H -pyrazol-5-yl)(3-(6-methyl-4-(l-methyl-1H -pyrazol-4-yl)pyridin-2- yl)pyrrolidin-l-yl)methanone; and (R)-(3-Isopropyl-1H -pyrazol-5-yI)(3-(6-methyl-4-(l- methyl-l/jT-pyrazol-4-yl)pyridin-2-yl)pyrrolidin-l-yI)methanone
Step 1

terf-butyl 3-(2-(3-methyIisoxazol-5-yl)acetyl)pyrrolidine-l-carboxylate
To a cooled (-78 °C) rigorously stirred solution of 3,5-dimethylisoxazole (31.4 mL, 1.1 eqiv., 0.319 mol) in THF (175 mL), a 2.6 M o-BuLi solution (123 mL, 1.1 eqiv., 0.319 mol) in hexane was added under argon. The reaction mixture was stirred at -78 °C for 60 min and a solution of tert- butyl 3-(methoxy(methyl)carbamoyl)pyrrolidine-l -carboxylate (75 g, 1.0 eqiv., 0.2906 mol) in 300 mL of THF was added at this temperature to the reaction mixture. The mixture was kept at -78 °C for another 60 min, then warmed to room temperature overnight. The mixture was quenched with
solution of NH4C1 (2 M, 500 mL). The organic layer was separated, dried over sodium sulfate, and evaporated. Crude product was purified by flash column chromatography on silica gel eluting with hexane/EtOAc to give the desired product (50 g, 58.5 % yield) as an oil. Step 2

tert-butyl 3-(4-hydroxy-6-methylpyridin-2-yl)pyiTolidine-l-carboxylate
To a solution of tert-butyl 3-(2-(3-methylisoxazol-5-yl)acetyl)pyrrolidine-l-carboxylate (52 g, 1.0 eqiv.) in methanol (1400 mL) was added Pd/C (10%) (5.2 g) and the mixture was hydrogenated at 30 atm at 90 °C for 20 hrs. The reaction mixture was passed through Celite plug and the filtrate was evaporated on rotavap to yield (50 g) of the desired product which was used in the next step without further purification.
Step 3

/erf-butyl 3-(6-methyl-4-(((trifluoromethyl)sulfonyl)oxy)pyridin-2-yl)pyrroIidine-l- carboxylate
Trifluoromethane sulfonic anhydride (38 mL, 1.07 eqiv., 0.134 mol) was added drop wise to a mixture of ferf-butyl 3-(4-hydroxy-6-methylpyridin-2-yl)pyrrolidine-l-carboxylate (35 g, 1.0 eqiv., 0.127 mol) and triethylamine (21 mL,1.2 eqiv., 0.151 mol) in CH2C12 (350 mL) cooled with an ice bath. The mixture was allowed to warm to 10 °C, then it was diluted with CH2C12 (100 mL), washed with saturated NaHCC>3 (3 x 100 mL), dried Na2SO4 and then concentrated. Crude product was purified by flash column chromatography on silica gel eluting with hexane-EtOAc to give the desired product (30 g, 58 % yield) as solid.
Step 4

tert-Buty\ 3-(6-methyl-4-(l-methyl-1H -pyrazol-4-yl)pyridiii-2-yl)pyrroIidine-l-carboxyIate
To a solution of tert-butyl 4-(6-methyl-4-(((trifluoromethyl)sulfonyl)oxy)pyridin-2-yl)piperidine carboxylate (500 mg, 1.22 mmol), l-methyl-4-(4,4,5,5-tetramethyl-l ,3,2-dioxaborolan-2-yl)-lH- pyrazole (127 mg, 0.61 mmol) and Na2CO3 (388 mg, 3.66 mmol) in DME: H2O (5: 1 , 10 mL) was added Pd{dppf)Cl2 (9 mg, 12.2 μmol). The reaction mixture was purged with nitrogen and then heated at 100 °C in a microwave reactor for 30 min. After cooling to room temperature, the reaction mixture was concentrated and the residue was dissolved in EtOAc (180 mL) and washed with H2O (150 mL x 2). The organic phase was dried over anhydrous Na2SO4, filtered and the filtrate was concentrated to dryness. The residue was purified by column chromatography eluting with 0-7% MeOH in DCM to give the desired product (355 mg, 85% yield) as yellow oil which was used directly for the next step without further purification. Step 5

2-Methyl-4-(l-methy]-1H -pyrazol-4-y])-6-(pyrrolidin-3-yI)pyridine
To a solution of tert-butyl 3-(6-methyl-4-(l-methyl-l H-pyrazol-4-yl)pyridin-2-yl)pyrrolidine-l- carboxylate (710 mg, 2.07 mmol) in EtOAc (2 mL) was added dropwise HCl/EtOAc (5 mL) and stirred at room temperature for 2 hrs. The reaction mixture was concentrated to dryness to give the desired product (510 mg, 88% yield) as yellow solid which was used directly for the next step without further purification. 1H NMR (400 MHz, CD3OD) δ 8.63 (s, 1 H), 8.32 (s, 1H), 8.14 (s, 1 H), 7.97 (s, 1H), 4.00 (s, 3H), 3.96 - 3.86 (m, 2H), 3.72 - 3.65 (m, 2H), 3.52 - 3.45 (m, 1H), 2.77 (s, 3H), 2.72 - 2.64 (m, 1H), 2.46 - 2.36 (m, 1H).
Step 6

Example 1:
(S)-(3-IsopropyI-lJT-pyrazol-5-yl)(3-(6-methyl-4-(l-methyl-lH-pyrazol-4-y])pyridin-2- yl)pyrrolidin-l-yl)methanone

Example 2:
(R)-(3-Isopropyl-l-ff-pyrazol-5-yl)(3-(6-methyl-4-(l-methyl-1H -pyrazol-4-yl)pyridiii-2- yl)pyrrolidin-l-yl)methanone
To a stirred solution of 3-isopropyl-1H -pyrazole-5-carboxylic acid (133 mg, 0.86 mmol) in DMF (5 mL) was added DIPEA (372 mg, 2.88 mmol) and HATU (395 mg, 1.04 mmol) at room
temperature. The mixture was stirred at room temperature for 10 min, then 2-methyl-4-(l-methyl- 1H -pyrazol-4-yl)-6-(pyrrolidin-3-yl)pyridine hydrochloride (200 mg, 0.72 mmol) was added and stirred at room temperature for 2 hrs. The mixture was diluted with EtOAc (200 mL), washed with brine (90 mL x 3), dried over anhydrous Na2SO4, filtered, the filtrate was concentrated to give a residue which was purified by preparative TLC (DCM/ MeOH = 10: 1) to give a mixture of enantiomers (130 mg, 48% yield) as white solid, which was purified by chiral SFC to give two enantiomers Example 1 (with arbitrarily assigned stereochemistry) (44.7 mg, 16% yield) and Example 2 (with arbitrarily assigned stereochemistry) (34.5 mg, 13% yield).
Example 1: 1H NMR (400 MHz, CDC13) δ 7.84 (s, 1H), 7.77 - 7.75 (m, 1H), 7.13 - 7.11 (m, 2H), 6.50, 6.48 (2s, 1H), 4.33 - 4.29 (m, 0.5H), 4.22 - 4.14 (m, 1H), 4.03 - 4.92 (m, 1H), 3.97 (s, 3H), 3.78 - 3.70 (m, 0.5H), 3.64 - 3.55 (m, 1H), 3.07 - 3.01 (m, 1H), 2.55 (s, 3H), 2.40 - 2.25 (m, 2H), 1.33 - 1.28 (m, 6H). LCMS (ESI) m/z: 378.9 [M+H]+, RT = 0.673 min (LCMS method E).
Example 2: 1H NMR (400 MHz, CDC13) δ 7.84 (s, 1H)5 7.74 (s5 1H), 7.12 - 7.11 (m, 1H), 7.10 (s, 1H), 6.50, 6.48 (2s, 1H), 4.33 - 4.29 (m5 0.5H), 4.21 - 4.1 1 (m, 1H), 4.07 - 3.90 (m, 2H), 4.01 (s, 3H), 3.78 - 3.71 (m, 0.5H), 3.65 - 3.54 (m, 1H), 3.09 - 3.00 (m, 1H), 2.54 (s, 3H), 2.46 - 2.28 (m, 2H), 1.33 - 1.28 (m, 6H). LCMS (ESI) m/z: 379.0 [M+H]+, RT = 0.673 min (LCMS method E).
Example 3
Step l

2-Methyl-6-(pyrrolidin-3-yl)pyridin-4-yl trifluoromethanesulfonate
To a solution of 2-(l-(tert-butoxycarbonyl)pyrrolidin-3-yl)-6-methylpyridin-4-yl trifluoro methanesulfonate (1.5 g, 3.6 mmol) in DCM (60 mL) was added TFA (9 mL), the mixture was stirred at room temperature for 1.5 hrs. The mixture was poured into aqueous saturated NaHCOa (200 mL), extracted with EtOAc (100 mL x 3), dried over anhydrous Na2SO4, filtered and concentrated to give the desired product (1.0 g, 90% yield) as a pale brown oil. LCMS (ESI) m/z: 310.9 [M+H]+.
Step 2

2-{l -(3-Isopropyl-1H -py razole-5-carbonyl)pyrrolidin-3-yI)-6-methylpy ridin-4-yl trifluoro methanesulfonate
To a solution of 2-methyl-6-(pyrrolidin-3-yl)pyridin-4-yI trifluoromethanesulfonate (1.0 g, 3.2 mmol) and 3-isopropyl-1H -pyrazole-5-carboxylic acid (600 mg, 3,9 mmol), TEA (1.0 g, 9.9 mmol) in DCM (150 mL) was added PyBrOP (1.8 g, 3.9 mmol). The mixture was stirred at room temperature for 30 min, then diluted with DCM (100 mL), washed with brine (200 mL x 3), dried over anhydrous Na2SO4, filtered and concentrated to give the crude product which
was purified by flash column chromatography on silica gel eluting with 0-5% MeOH in DCM to give the desired product (1.1 g, 78% yield) as a pale browm solid. LCMS (ESI) m/z: 446.9 [M+H]+.
Step 3

(3-Isopropyl-1H -pyrazol-5-yl)(3-(6-methyl-4-(p-tolyl)pyridin-2-yl)pyrrolidin-l-yl) methanone
A mixture of 2-(l-(3-isopropyl-1H -pyrazole-5-carbonyl)pyrrolidin-3-yl)-6-memylpyridin-4-yl trifluoro methanesulfonate (50 mg, 0.11 mmol) and /?-tolylboronic acid (23 mg, 0.17 mmol), K2CO3 (46 mg, 0.33 rnrnole), Pd(dppf)Cl2 (10 mg, 0.01 mmol) in dioxane (2 mL) and H2O (0.5 mL) was heated at 110 °C in a microwave reactor for 30 min. The mixture was filtered through a silica gel pad, washed with EtOAc, concentrated to give the crude product which was purified by preparative TLC (DCM: MeOH = 20: 1) to give the desired product (16 mg, 37% yield) as a brown solid. 1H NMR (400 MHz, CDC13) 87.49 - 7.17 (m, 2H), 7.26 (m, 4H), 6.46 (s, 1H), 4.30 - 3.54 (m, 5H), 3.04 (m, 1H), 2.57 (2s, 3H), 2.47 - 2.25 (m, 5H), 1.28 (m, 6H). LCMS (ESI) m/z: 388.9 [M+H]+, RT - 0.743 min (LCMS Method E).
Example 4

Methyl 2-(1-(3-isopn)pyl-1H -pyrazole-5-carbonyl)pyrroIidin-3-yl)-6-methylisonicotinat€ To a solution of 2-(l-(3-isopropyl-1H -pyrazole-5-carbonyl)pyrrolidin-3-yl)-6-me1hylpyridin-4-yl trifluoromethanesulfonate (100 mg, 0.2 mmol) and TEA (80 mg, 0.8 mmol) in DMF (5 mL) and MeOH (2.5 mL) were added Pd(OAc)2 (10 mg, 0.08 mmole) and dppp (20 mg, 0.04 mmole). The mixture was stirred at 70 °C for 16 hrs under an atmosphere of carbon monoxide. After cooling to room temperature, the reaction mixture was diluted with EtOAc (100 mL), washed with brine (30 mL x 3), dried over anhydrous Na2SO4, filtered and concentrated to give a residue which was purified by preparative TLC (DCM: MeOH = 15: 1) to give the desired product (45 mg, 63% yield) as a white solid. 1H NMR (400 MHz, CD3OD) δ 8.25 - 8.28 (m, 2H), 6.67 (s, 1H), 4.59 - 4.54 (m,
0.5H), 4.34 - 4.17 (m, 2H), 4.14 - 3.84 (m, 4.5H), 4.02 (s, 3H), 3.12 - 3.05 (m, 1 H), 2.89 (2s, 3H), 1.32 (m, 6H); LCMS (ESI) m/z: 357.0 [M+H]+, RT = 0.933 min (LCMS Method A).
Example 5

2-(l-(3-Isopropyl-l-ff-pyra2H>le-5-carbonyl)pyrrolidin-3-yl)-6-methylisonicotinic acid
To a solution of methyl 2-(l-(3-isopropyl-1H -pyrazole-5-carbonyl)pyrrolidin-3-yl)-6- methylisonicotinate (33 mg, 0.1 mmol) in H2O (5 mL) and MeOH (5 mL) were added LiOH (40 mg, 1.0 mmol). The mixture was stirred at room temperature for 2 hrs. The mixture was acidified to pH = 2.0 with diluted aqueous HC1, extracted with DCM/MeOH = 10/1 (20 mL x 3), concentrated to give crude desired product (28 mg, 90% yield) as a white solid.
Step 2

2-(1-(3-Isopπ)pyl·l//-pyrazole-5-carbonyl)pyrrolidin-3-yl)-N,N,6-trimethylisonicotmamide
To a solution of 2-(l-(3-isopropyl-1H ^yrazole-5-carbonyl)pyiTolidin-3-yl)-6-methylisonicotinic acid (28 mg, 0.8 mmol), dimethylamine hydrochloride (26 mg, 0.32 mmol), TEA (50 mg, 0.5 mmol) in DCM (10 mL) was added PyBrOP (46 mg, 0.1 mmol), the mixture was stirred at room temperature for 1 hr. The mixture was concentrated to give the crude product which was purified by preparative TLC (DCM/MeOH = 10: 1) to give the desired product (13 mg, 43% yield) as a white solid. 1H NMR (400 MHz, CD3OD) δ 7.90 (s, 1 H), 7.83 (d, J=1.6 Hz, 1H), 6.61 (s, 1H), 4.56 (mz, 0.5H ), 4.32 - 4.12 (m, 1.5H), 4.09 - 3.68 (m, 3H), 3.12 (s, 3H), 3.09 - 2.99 (m, 1H), 2.95
(s, 3H), 2.83 (2s, 3H), 2.64 - 2.26 (m, 2H), 1.32 (m, 6H). LCMS (ESI) m/z: 370.1 [M+H]+, RT = 0.71 1 min (LCMS Method A).
Example 6

A mixture of 2-(l -(3-isopropyl- 1 H-pyrazole-5- :arbonyl)pyrrolidin-3-yl)-6-methylpyridin-4-yl trifluoromethanesulfonate (50 mg, 0.11 mmol) and 2-(tributylstannyl)pyridine (66 mg, 0.18 mmol), LiCl (7 mg, 0.15 mmol), Pd(PPh3)Cl2 (10 mg, 0.01 mmol) in DMF (2 mL) and H2O (0.5 mL) was heated at 120 °C in a microwave reactor for 30 min. After cooling to room temperature, the mixture was filtered through a silica-gel pad, washed with EtOAc and concentrated to give a residue which was purified by preparative TLC (DCM/MeOH = 10: 1) to give the desired product (8.8 mg, 21% yield) as a white solid. 1H NMR (400 MHz, CD3OD) δ 8.67 (d, J= 4.4 Hz, lH), 8.07 - 7.89 (m, 2H), 7.83 - 7.69 (m, 2H), 7.51 - 7.42 (m, 1H), 6.53 (2s, 1 H), 4.43 (m, 0.5H), 4.28 - 3.82 (m, 3H), 3.76 - 3.55 (m, 1.5H), 3.12 - 2.96 (m, 1H), 2.60 (2s, 3H), 2.52 - 2.24 (m, 2H), 1.33 - 1.25 (m, 6H). LCMS (ESI) m/z: 376.1 [M+H]+, RT = 0.829 min (LCMS method A).
Example 7

(3-IsopropyI-lJy-pyi^ol-5-yI)(3-(6-methyl-4-phenoxypyridin-2-yl)pyiTolidiii-l-yI) methanone
A mixture of (3-(4-bromo-6-methylpyridin-2-yl)pyrrolidin-l-yl)(3-isopropyl-1H -pyrazol-5- yl)methanone (40 mg, 0.10 mmol), phenol (56.4 mg, 0.6 mmol), Cul (57.2 mg, 0.3 mmol), K3PO4 (63.6 mg, 0.3 mmol), picolinic acid (37 mg, 0.3 mmol) in DMSO (4 mL) was heated at 120 °C in microwave reactor for 30 min. The reaction mixture was washed with water (20 mL), extracted with EtOAc (20 mL). The organic phase was dried over anhydrous Na2SO4, concentrated and the
residue was purified with preparative TLC (5% MeOH in DCM) to give the crude product which was further purified by preparative HPLC to give the desired product (2.0 mg, 2% yield). Ή NMR (400 MHz, CD3OD) δ 7.49 - 7.44 (m, 2H), 7.30 - 7.27 (m, 1H), 7.05-7.15 (m, 2H), 6.72-6.70 (m 1H), 6.65-6.55 (m, 1H)5 6.49 (s, 1H), 4.42 - 4.12 (m, 1H), 4.08 - 3.90 (m, 1.5H), 3.88 - 3.63 (m, 1.5H), 3.55 - 3.45 (m, 1H), 3.12 - 2,96 (m, 1H), 2.44, 2.42 (2s, 3H), 2.40 - 2.12 (m, 2H), 1.31 - 1.28 (m, 6H). LCMS (ESI) m/z: 391.1 [M+H]+, RT = 0.736 min (LCMS Method E).
Example 8

(3-Isopi >pyl-lJy-pyrazol-5-yl)(3-(4-methoxy-6-methylpyridin-2-yl)pyiToUdiii-l-yl) methanone
A mixture of (3-(4-bromo-6-methylpyridin-2-yl)pyrrolidin-l-yl)(3-isopropyl-1H -pyrazol-5-yl) methanone (40 mg, 0.11 mmol) and MeONa (2 mL, 4 mmole) in DMSO (2 mL) was heated at 90 °C for 16 h. The mixture was diluted with EtOAc (20 mL), washed with brine (20 mL x 3), concentrated to give a residue which was purified by preparative TLC (DCM/MeOH = 10: 1) to give the desired product (23.1 mg, 72% yield) as a white solid. 1H NMR (400 MHz, CD3OD) δ 6.70 - 6.80 (m, 2H), 6.49 (2s, 1 H), 4.44 - 4. 1 (m, 1H), 4.09 - 3.87 (m, 2H), 3.84 (m, 3H), 3.78 - 3.61 (m, 1H), 3.59 - 3.45 (m, 1H), 3.02 (m, 1H)5 2.46 (2s, 3H), 2.40 - 2.13 (m, 2H), 1.28 (m, 6H); LCMS (ESI) m/z: 376.1 [M+H]+ 5 RT = 0.829 min (LCMS method A). Example 9

(3-(4-Isopropoxy-6-methylpyridin-2-yl)pyrroIidin-l-yI)(3-isopropyl-1H r-pyraz»l-5- yl)methanone
Sodium metal (25 mg, 1.06 mmol) was added into z-PrOH (3 mL) at room temperature. The mixture was stirred at 60 °C for 2 hrs. (3-(4-bromo-6-memylpyridm-2-yl)pyrrolidin-l-yl)(3- isopropyl-1H -pyrazol- 5-yl)methanone (100 mg. 0.27 mmol) was added into the reaction mixture and stirred at 60 °C for 16 hrs. After cooling to room temperature, the reaction mixture was diluted
with DCM (15 mL) and washed with ¾0 (10 mL), brine (10 mL). The organic phase was dried over anhydrous Na2SO , concentrated and the residue was purified by preparative TLC (9% MeOH in DCM) to give the desired product (4.0 mg, 4% yield) as a white solid. Ή NMR (400 MHz, CDCI3) δ 12.92 (s, 1H), 6.77 (s, 2H), 6.40 (s, 1H), 4.60 - 4.80 (m, 1H), 4.31 - 4.14 (m, 1H), 3.94 - 3.48 (m, 4H), 3.05 - 2.85 (m, 1H), 2.41 (s, 3H), 2.35 - 1.97 (m5 2H), 1.29 - 1.21 (m, 12H). LCMS (ESI) m/z: 356.9 [M+H]+, RT = 0.730 min (LCMS Method E).
Example 10

(3-Isopropyl-l T-pyrazol-5-yl)(3-(6-methyI-4-morpholinopyridiii-2-yl)pyrroIidiii-l- yl)methanone
A mixture of 2-( 1 -(3-isopropyl- 1 H^yr.izole-5 arbonyl)pyrrolidin-3-yl)-6-methylpyridin-4-yl trifluoromethanesulfonate (50 mg, 0.11 mmol) and morpholine (1 mL) was heated at 120 °C in microwave reactor for 30 min. The mixture concentrated to give a residue which was purified by preparative TLC (DCM MeOH = 10: 1) to give the desired product (17.7 mg, 41% yield) as a white solid. 1H NMR (400 MHz, CDC13) δ 6.52 - 6.29 (m, 3H), 4.24 - 4.14 (m, 0.5H), 4.09 - 3.94 (m, 1H), 3.89 - 3.71 (m, 5.5H), 3.68 - 3.48 (m, 2H), 3.26 (m, 4H), 3.02 - 2.86 (m, 1H), 2.51 (s, 3H), 2.30 - 2.26 (m, 2H), 1.22 (m, 6H). LCMS (ESI) m/z: 384.1 [M+H]+, RT = 0.771 min (LCMS Method A). Example 11

(3-(4-(Benzylamino)^-methylpyridin-2-yl)pyrroHdin-l-yl)(3-isopropyl-l^ -pyrazol-5- yl)methanone
A solution of 2-(l -(3-isopropyl-l H-pyi^ole-5-carbonyI)pyrTolidin-3-yl)-6-methylpyridin-4-yl trifluoromethanesulfonate (80 mg, 179.2 umol) in benzyl amine (96.0 mg, 896.0 umol) was heated
in microware reactor at 120 °C for 30 min. After cooling to room temperature, the mixture was concentrated and the residue was purified by preparative HPLC (Base) to give the desired product (2.1 mg, 3% yield) as yellow solid. Ή NMR (400 MHz, CDC13) 6 7.36 - 7.31 (m, 5H), 6.46, 6.45 (2s, 1H), 6.27 - 6.25 (m, 2H), 4.36 (m, 2H), 4.23 - 4.19 (m, lH), 4.10 - 4.03 (m, 1 H), 3.96 - 3.81 (m, 2H), 3.72 - 3.65 (m, lH), 3.53 - 3.44 (m, 1H), 3.06 - 2.98 (m, 1H), 2.41 (s, 3H), 2.32 - 2.18 (m, 2H), 1.32 - 1.28 (m, 6 H). LCMS (ESI) m/z: 404.2 [M+H]+, RT = 1.087 min (LCMS method C).
Method B
Example 12
Step l

/eif-Butyl 3-(4-amino-6-chloropyridin-2-yl)-2^-dihydro-lJi-r-pyiTole-l-carboxylate
To a solution of ter/-butyl 3-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)-2,5-dihydro-1H -pyrrole- 1 -carboxylate (10 g, 34 mmol), 2,6-dichloropyridin-4-amine (8.3 g, 51 mmol) and Na2C<¼ (11 g, 0.10 mol) in Dioxane/H2O (5: 1, 240 mL) was added Pd(dppf)Cl2 (1.2 g, 1.7 mmol) at room temperature. The mixture was heated at 70 °C for 16 hrs under a N2 atomsphere. After cooling to room temperature, the reaction mixture was filtered, concentrated and the residue was diluted with EtOAc (500 mL) and washed with ¾0 (400 mL x 2), brine (400 mL). The organic phase was dried over anhydrous a2SO4, filtered and concentrated to dryness. The residue was purified by flash column chromatography eluting with 0-50% EtOAc in hexanes) to give the desired product (5 g, 50% yield) as white solid.
Step 2

/ert-Butyl 3-(4-bromo-6→:hloropyridiii-2-yl)-2,5-dihydro-l-H-pyrrole-l-carboxylate
To a solution of ferf-butyl 3-(4-amino-6-chloropyridin-2-yl)-2,5-dihydro-1H -pyrrole-l-carboxylate (4 g, 14 mmol), t-BuONO (1.8 g, 18 mmol) in C¾CN (150 mL) was added CuBr2 (4.5 g, 20 mmol ) at 0 °C and stirred at 0 °C for 1 hr. Then the reaction mixture was warmed up to room
temperature for 2 hrs under N2 atmosphere. The reaction mixture was concentrated and the residue was diluted with EtOAc (300 mL). washed with H2O (250 mL), NH3 ¾0 (18%, 250 mL x3), brine (250 mL). The organic phase was dried over anhydrous Na2SO4, filtered and the filtrate was concentrated to dryness. The residue was purified by silica gel column chromatography eluting with 0-9% MeOH in DCM) to give the desired product (2.4 g, 49% yield) as yellow solid.
Step 3

To a solution of tert-butyl 3-(4-bromo-6-chloropyridin-2-y])-2,5-dihydro-1H -pyrrole-l-carboxylate (3 g, 8.3 mmol), l-methyl-4-(4,4,5,5-tetramethyl-l,3,2-dioxaboroIan-2-yl)-1H -pyrazole (2.6 g, 13 mmol) and Na2CO3 (2.6 g, 25 mmol) in dioxane/ H2O (5:1, 150 mL) was added Pd(dppf)Cl2 (305 mg, 0.42 mmol) at room temperature. The mixture was heated at 80 °C for 3 hrs under N2 atomsphere. After cooling to room temperature, the reaction mixture was filtered, concentrated and the residue was diluted with EtOAc (200 mL) and washed with H2O (150 mL), brine (150 mL). The organic phase was dried over anhydrous Na2SO , filtered and the filtrate was concentrated to dryness. The residue was purified by silica gel column chromatography eluting with 0-9% MeOH in DCM) to give the desired product (2.5 g, 83% yield) as yellow solid. 1H NMR (400 MHz,
CDCI3)G δ 7.86, 7.83 (2s, 1H), 7.78, 7.74 (2s, 1H), 7.28 (s, lH), 7.11 (s, 1H), 6.7, 6.6 (2s, 1H), 4.60 - 4.50 (m, 2H), 4.43 - 4.30 (m, 2H), 3.99 (s., 3H), 1.53, 1.52 (2s, 9H).
Step 4

2-Chloro-6-(2,5^ihydro-lJ.T-pyrrol-3-yl)-4^1-methyI-1H -pyrazoM-yl)pyridine
hydrochloride
To a stirred solution of ferf-butyl 3-(6-chloro-4-(l-methyl-1H -pyrazx)l-4-yl)pyridin-2-yl)-2J5- dihydro-1H -pyrroIe-l-carboxylate (2.5 g, 6.9 mmol) in EtOAc (2 mL) was added dropwise
HCl EtOAc (5 mL, 20 mmol, 4 M) and stirred at room temperature for 1 hr. The reaction mixture was concentrated and the solid was washed with EtOAc (50 mL x 2) to give the crude desired product (1.8 g) as yellow solid. 1H NMR (400 MHz, CD3OD) 58.35 (s, 1H), 8.16 (s, 1H), 7.89 (s, 1H), 7.60 (s, 1H), 6.81 (s, 1H), 4.53 (s, 2H), 4.36 (s, 2H), 4.00, 3.98 (2s, 3H). Step 5

(3-(6-Chloro-4-(l-methyl-l//-pyrazol-4-y^
isopropyl-1H -pyrazol-5-yl)methanone
To a stirred solution of 3-isopropyl-1H -pyraz»le-5-carboxylic acid (1.1 g, 7.3 mmol) in DMF (20 mL) was added DIEA (3.1 g, 24 mmol) and HATU (3.2 g, 8.5 mmol) and stirred at room
temperature for 10 min before the addition of 2-chloro-6-(2,5-dihydro-1H -pyrrol-3-yl)-4-(l- methyl-1H -pyrazol-4-yl)pyridine hydrochloride (1.8 g, 6.1 mmol). The mixture was stirred at room temperature for 2 hrs under a N2 atomsphere. The mixture was diluted with EtOAc (250 mL), washed with ¾0 (240 mL x 3). The organic phase was dried over anhydrous Na2SO4, filtered and the filtrate was concentrated and purified by silica gel column chromatography eluting with 0-10%
MeOH in DCM to give the desired product (1.8 g, 75% yield) as yellow solid. 1H NMR (400 MHz,
DMSO-iie) 58.55, 8.50 (2s, 1H), 8.22, 8.19 (2s, 1H), 7.99, 7.84 (2s, 1H), 7.64 (s, 1H), 6.91, 6.87
(2s, 1H), 6.51, 6.50 (2s, 1H), 5.06 (m, 1H), 4.90 (m, 1H), 4.68 (m, 1H), 4.54 (m, 1H), 3.93, 3.89 (2s, 3H), 3.01 - 2.97 (m, 1H), 1.27 - 1.24 (m, 6H).
Step 6

(3-(6-Chloro-4-(l-methyl-1H -pyrazol-4-yl)pyridin-2-yl)pyrroIidin-l-yl)(3-isopropyl-lfl- pyrazol-5-yl)methanone
To a stirred solution of (3-(6-Chloro-4-(l-me1iiyl-1H -pyrazol-4-yl)pyridin-2-yl)-2,5-dihydro-1H - pyrrol-l-yl)(3-isopropyl-1H -pyrazol-5-yl)methanone (1.4 g, 3.5 mmol) in THF (150 mL) was added PtO2 (500 mg, 2.2 mmol) and stirred under hydrogen at room temperature for one week. The reaction mixture was filtered and the filtrate was concentrated give the desired product (1.3 g, 92% yield) as yellow solid. 1H NMR (400 MHz, CDC13) 67.84 (s, 1H), 7.80, 7.78 (2s, 1H), 7.28 (s, 1H), 7.22, 7.21 (2s, lH), 6.46, 6.45 (2s, 1H), 4.32 - 4.26 (m, 0.5H), 4.20 - 4.10 (m, 1 H), 4.08 - 3.99 (m, 0.5H), 3.98, 3.97 (2s, 3H), 3.96 - 3.87 (m, 1H), 3.75 - 3.2 (m, 1H), 3.62 - 3.54 (m, 1H), 3.19 - 3.18 (m, 1H), 3.04 - 3.02 (m, 1H), 2.47 - 2.29 (m, 2H), 1.32 - 1.28 (m, 6H).
Step 7

(3-Isopropyl-l f-pyrazol-5-yl)(3-(4-(l-methyl-l -r-pyrazol-4-yl) pyridin-2-yl) pyrrolidin-l-yl) methanone
A solution of (3-(6-chloro-4-(l -methyl- 1H -pyrazol-4-yl) pyridin-2-yl)pyrrolidin-l -yl)(3-isoprop yl- 1H -pyrazol-5-yl)methanone (50 mg, 0.13 mmol), Pd C (10 mg) in MeOH (10 mL) was stirred under 1 arm H2 for 16 hrs and filtered through a pad of Celite. The filtrate was concentrated and the residue was purified by preparative HPLC to give the desired product (10 mg, 22% yield) as a white solid. 1H NMR (400 MHz, CD3OD) δ 8.41 (s, 1H), 8.21 (s, 1H), 8.01 (s, 1H), 7.58 (s, 1H), 7.47 (s, 1H), 6.52 (s, 1H), 4.50 - 4.40 (m, 0.5H), 4.30 - 4.20 (m, 0.5H), 4.19 - 3.90 (m, 5H), 3.80 -
3.50 (m, 2H), 3.10 - 2.90 (m, 1H), 2.50 - 2.10 (m, 2H), 1.31 - 1.28 (m, 6H). LCMS (ESI) m/z: 365.2 [M+H]+, RT = 0.665 min(LCMS method E).
Example 13

(3-Isopropyl-ljtf-pyrazol-5-yl)(3-(6-methoxy-4-(l -methyl- l -pyraz»l-4-yI)pyridiii-2-yl) pyrrolidin-l-yl)methanone
Na (57.6 mg) was added into CH3OH (8 mL) at room temperature and the mixture was stirred for 1 hr. (3-(6-chloro-4-(l -methyl- 1 H-pyrazol-4-yl)pyrjdin-2-yl)pyrrolidin- 1 -yl)(3-isopropyl- 1 H-pyra zol-5-yl) methanone (100 mg, 0.25 mmol) was added to this mixture. The mixture was stirred at 70 °C for 16 hrs. The reaction mixture was concentrated.and the residue was purified by preparative HPLC to give the desired product (4 mg, 4% yield) as a white solid. Ή NMR (400 MHz, DMSO- d6) δ 7.79 (s, 1H), 7.70 (m, 1H), 6.88 (m, 1H), 6.66 (m, 1H), 6.45, 6.42 (2s, 1H), 4.1 1 - 3.87 (m, 9H), 3.56 - 3.46 (m, 1H), 3.12 - 3.00 (m, 1H), 2.43 - 2.27 (m, 2H), 1.31 - 1.27 (m, 6H). LCMS (ESI) m/z: 398.4 [M+H]+, RT = 0.776 min (LCMS method E).
Example 14
Step

tert- utyl 3>(2-chloropyrimidiii-4-yl)-2^-dihydro-l r-pyrrole-l-carboxylate
A mixture of 2,4-dichloropyrimidine (1.0 g, 6.7 mmol), tert-butyl 3-(4,4,5,5-tetramethyl-l,3,2- dioxaborolan-2-yl)-2,5-dihydro-1H -pyrrole-l-carboxylate (1.6 g, 5.4 mmol), Pd(dppf)Cl2 (0.5 g, 0.34 mmol), Na2CO3 (1.4 g, 13 mmol) in Dioxane/H2O (12 mL, 5: 1) was stirred at 80 °C for 2 hrs. After cooling to room temperature, the reaction mixture was washed with H2O (10 mL), and extracted with EtOAc (1 mL). The organic phase was dried with anhydrous Na2SC>4, filtered and concentrated. The residue was purified with flash column choromatograpy on silica gel eluting with 20% EtOAc in hexanes to give the desired product (0.8 g, 42% yield) as a white solid. 1H
NMR (400 MHz, CDC13) δ 8.60 - 8.57 (m, 1H), 7.27, 7.10 (2s, 1H), 6.94, 6.87 (2s5 1H), 4.51 (s, 2H), 4.36 - 4.20 (m, 2H), 1.51 - 1.49 (m, 9H).
Step 2

terf-Butyl 3-(2-ch!oropyrimidiii-4-yl) pyrrolidine-l-carboxylate
A mixture of tert-butyl 3-(2-cUoropyr-midin-4-yl)-2,5-dihydro-1H -pyrrole-l-carboxylate (800 mg, 2.8 mmol), PtO2 (129 mg, 0.57 mmol) in THF (10 mL) was stirred under 1 atm of H2 at room temperature for 12 hrs. The suspension was filtered through a pad of celite. The filtrate was concentrated and the residue was purified with column chromatograph on silica gel eluting with 20% EtOAc in hexanes to give the desired product (0.28 g, 35% yield) as a white solid. 1H NMR (400 MHz, CDCI3) δ 8.55 (d, J= 4.8 Hz, 1H), 7.16 (d, J= 4.4 Hz, 1H), 3.88 - 3.70 (m, 1H), 3.64 - 3.44 (m, 4H), 2.30 - 2.18 (m, 2H), 1.47 (s, 9H). Step 3

ferf-Butyl 3-(2-oxo-l,2-dihydropyrimidin-4-yl)pyrrolidine-l-carboxylate
A mixture of tert-butyl 3-(2-chloropyrimidin-4-yl) pyrrolidine-l-carboxylate (1.2 g, 4.1 mmol), l,4-diazabicyclo[2.2.2]octane (0.23 gt 2.1 mmol), K2CO3 (1.0 g, 7.4 mmol) in Dioxane/H2O (10 mL/10mL) was stirred at room temperature for 12 hrs. The reaction mixture was concentrated and extracted with DCM (15 mL). The organic phase was dried over Na2SO4, filtered and
concentrated. The residue was purified with column chromatograph on silica gel eluting with 9% MeOH in DCM to give desired product (0.6 g, 64% yield) as a white solid. Step 4

teif-Butyl 3-(l-isopropyl-2- xo-l,2-dihydropyrimidin-4-yl)pyrrolidiiie-l-carboxylate
A mixture of teri-butyl 3-(2-oxo-l ,2-dihydropyrimidin-4-yl)pyTrolidme-l-carboxylate (300 mg, 1.1 mmol), 2-iodopropane (211 mg, 1.2 mmol), CS2CO3 (737 mg, 2.3 mmol) in DMF (8 mL) was stirred at 60 °C for 5 hrs. After cooling down to room temperature, the reaction mixture was washed with H2O (10 mL), and extracted with DCM (10 mL x 3). The reaction mixture was concentrated to give the crude product (200 mg, 57% yield) as yellow oil, contaminated with O- isopropyl pyrimidine
Step S

Isopropyl-4-(pyrrolidin-3-yl)pyrimidin-2(l )-one hydrochloride
To a mixture of iert-butyl 3-(l-isopropyl-2-oxo-l,2-dihydropyrimidin-4-yl)pyrrolidine-l-carbo- xylate and Ο-ίΡτ pyrimidine (200 mg, 0.65 mmol) in EtOAc (4 mL) was added 4M HCl EtOAc (4 mL) at room temperature. The mixture was stirred at room temperature for 2 hrs and then concentrated to dryness to give the crude product (150 mg, 95% yield) as yellow solid, which was directly taken to the next step.
Step 6

l-Isopropyl-4-(l-(3-isopropyI-lfl-pyrazole-5-carbonyl)pyrrolidin-3-yI)pyrimidin-2(1H )- one
A mixture of 3-isopropyl-1H -pyrazole-5-carboxylic acid (114 mg, 0.74 mmol), HATU (393 mg, 1.0 mmol), N-emyl-N-isopropylpropan-2-amine (382 mg, 3.0 mmol) in DMF (5 mL) was stirred at room temperature for 10 min. A solution of the mixture of l-isopropyl-4-(pyrrolidin-3- yl)pyrimidin-2(1H )-one hydrochloride and 2-isopropoxy-4-(pym>lidin-3-yl)pyrimidine hydro chloride (180 mg, 0.74 mmol) in DMF (4 mL) was added into the mixture. The reaction mixture was stirred at room temperature for 2 hrs and then washed with H2O (15 mL), extracted with EtOAc (15 mL). The organic phase was washed with ¾0 (15mLx3), then washed with brine (10 mL), dried over anhydrous Na2SO4. The mixture was filtered. The filtrate was concentrated via rotavap to give a residue which was purified with preparative HPLC to give the desired product (50
mg, 20% yield) as white solid. Ή NMR (400 MHz, CD3OD) δ 8.17 - 8.14 (m, 1H), 6.63 - 6.59 (m, 1H), 6.49 (s, 1H), 4.44 - 4.30 (m, 0.5H), 4.27 - 3.93 (m, 2H), 3.87-3.82 (m, 1H), 3.76 - 3.63 (m, 0.5H), 3.59 - 3.42 (m, 1H), 3.12 - 2.94 (m, 1H), 2.51 - 2.11 (m, 2H), 1.43 - 1.29 (m, 12H); LCMS (ESI) m/z: 344.1 [M+H]+, RT = 1.033 min (LCMS method C).
Method C
Example 15
Step 1

tert-B tyl 3-(cyclopropanecarboxamido)-3-methylpyrrolidine-l-carboxylate
To a solution of /f?r/-butyl 3-amino-3-methylpyrrolidbie-l-carboxylate (560 mg, 2.80 mmol) in DCM (5 mL) was added DIEA (0.93 mL, 5.59 mmol), then cyclopropanecarbonyl chloride (300 mg, 2,80 mmol) was added dropwise at 0 °C. The resulting mixture was stirred at room temperature for 1 hour. The reaction was quenched with water, and then extracted with DCM (50 mL x 3). Combined organic layers were washed with ¾0 (50 mL), dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified by column chromatograph on silica gel eluted with 0- 30% EtOAc in hexanes to give the desired product (680 mg, 91% yield) as light yellow oil. Ή NMR (400 MHz, CDC13) δ 5.56 (s, 1H), 3.60 - 3.32 ( m, 4H), 1.85 - 1.82 (m, 1H), 1.29 - 1.26 (m, 2H), 0.93 - 0.85 (m, 4H).
Step 2

iV-(3-methylpyrroIidin-3-yl)cyclopropanecarboxaiiiide hydrochloride To a solution of teri-butyl 3-(cyclopropanecarboxamido)-3-methylpyrrolidine-l-carboxylate (680 mg, 2.53 mmol) in EtOAc (3 mL) was added dropwise HCl/EtOAc (5 mL). The mixture was stirred at room temperature for 1 hr. Then the reaction mixture was concentrated and the crude product was used in the next step directly.
Step 3

-V-(l-(3-isopropyl-liff-pyrazole-5-carbonyl)-3-methy]pyrrolidiii-3- yl)cyclopropanecarboxamide
To a solution of 3-isopropyl-1H -pyrazole-5-carboxylic acid (431 mg, 2.79 mmol) in DMF (6 mL) were added HATU (1.38 g, 3.63 mmol) and DIEA (1.85 mL, 1 1.18 mmol) and then stirred at room temperature for 5 minutes before the addition of N-(3-methylpyrrolidin-3-yl)cyclopropane carboxamide hydrochloride (572 mg, 2.79 mmol) was added. The resulting mixture was stirred at room temperature for 16 hrs. The reaction mixture was filtered and the crude product was purified by preparative HPLC (Gemini C18, 150 x 25 mm xlO um, 26-56% MeCN/H2O) to give the desired product (210 mg, 25% yield) as white solid. Ή NMR (400 MHz, DMSO-flfe) δ 8.09 (s, 1H), 6.41 (s, 1H), 4.17 - 4.14 (m, 0.5H), 3.88 - 3.72 (m, 2H), 3.55 - 3.52 (m, 1H), 3.38 - 3.35 (m, 0.5H), 2.96 - 2.93 (m, 1H), 2.31 - 2.19 (m, 1H), 1.88 - 1.72 (m, 1H), 1.56 - 1.53 (m, 1H), 1.38 (s, 3H), 1.22 (d, J= 7.2 Hz, 6H), 0.63 - 0.57 (m, 4H). LCMS (ESI) m/z: 305.1 [M+H]+, RT = 0.904 min (LCMS Method E).
Example 16
Step 1

/er/-Butyl 3-(cycIopropanecarboxamido)pyrrolidine-l-carboxyIate
To a stirred solution of /erf-butyl 3-aminopyrrolidine-l -carboxylate (15.0 g, 80.5 mmol) in DCM (45 mL) was added TEA (24.5 g, 241.6 mmol) and cooled to 0 °C in an ice bath. Then
cyclopropanecarbonyl chloride (10.1 g, 96.6 mmol) was added dropwise into the mixture and stirred at room temperature for 2 hrs. The mixture was diluted with DCM (200 mL) and washed with ¾0 (180 mL x 3). The organic phase was dried over anhydrous Na2SO , filtered and the filtrate was concentrated to give the desired product (18.0 g, 88% yield) as brown oil. Ή NMR (400 MHz, CDC13)D δ 6.00-5.96 (m, 1H), 4.51 - 4.43 (m, 1H), 3.65 - 3.56 (m, 1H), 3.46 - 3.35 (m,
2H), 3.30 - 3.1 1 (m, 1H), 2.20 - 2.08 (m, 1H), 1.83 (br. s., 1H), 1.38 - 1.31 (m, 1H), 1.01 - 0.92 (m, 2H), 0.79 - 0.69 (m, 2H).
Step 2

7V-(PyrroIidin-3-yl)cyclopropanecarboxamide hydrochloride
To a stirred solution of terf-butyl 3-(cyclopropanecarboxamido)pyrrolidine-l-carboxylate (7 g, 27.5 mmol) in EtOAc (10 mL) was added dropwise HCl/EtOAc (35 mL, 140 mmol, 4 M) and stirred at room temperature for 1 hr. The reaction mixture was concentrated and the solid was washed with EtOAc (50 mL x 3) to give the desired product (5.0 g, 96% yield) as yellow solid. 1H NMR (400 MHz, CD3OD) δ 4.45 - 4.37 (m, 1H), 3.55 - 3.45 (m, 2H), 3.40 - 3.33 (m, 1H), 3.25 - 3.20 (m, 1H), 2.38 - 2.26 (m, 1H), 2.36 - 2.27 (m, 1H), 1.70 - 1.58 (m, 1H), 0.90 - 0.83 (m, 2H), 0.83 - 0.75 (m, 2H). Step 3

Ethyl 3-acetyl-l -T-pyrazo]e-5-carboxy]ate
To a solution of 3-butyn-2-one (5 g, 73 mmol) in H2O (80 mL) was added ethyl diazoacetate (12.5 g, 110 mmol). The mixture was stirred at room temperature for 4 hrs, and then filtered to give the desired product (10 g, 74% yield) as a white solid. 1HNMR (400 MHz, CD3OD) δ 7.38, 7.21 (m, 1H), 4.43 - 4.34 (m, 2H), 2.55, 2.52 (2s, 3H),1.39 - 1.32 (m, 3H).
Step 4

3-acetyl-l -f-pyrazole-5-carboxylic acid
To a solution of ethyl 3-acetyl-1H -pyrazole-5-carboxylate (1 g, 5.5 mmol) in a mixture of MeOH
(5 mL) and H2O (5 mL) was added NaOH (1.1 g, 27 mmol). The mixture was stirred at room temperature for 2 hrs before being acidified by cone. HC1 to pH 2.0, and then filtered to give the desired product (800 mg, 94% yield) as a white solid. Step 5

-V-(l-(3-acetyl-lJ¥-pyi^ole-5-carbonyl)pyrrolidin-3-yl)cyclopTOpanecarboxamide
To a solution of 3-acetyl-l//-pyrazole-5-carboxyIic acid (800 mg, 5 mmol) in DMF (15 mL) was added N-pyrrolidin-3-ylcyclopropanecarboxamide (1.2 g, 8 mmol), HATU (3 g, 8 mmol) and DIE A (2.8 mL, 16 mmol). The mixture was stirred at room temperature for 15 hrs and then concentrated to dryness under reduced pressure. The residue was partitioned between water (8 mL) and EtOAc (30 mL). The aqueous layer was extracted with EtOAc (20 mL x 2) and combined organic layers were dried over anhydrous Na2SO , filterd, concentrated, and purified by silica gel column chromatography eluting with 0-5% MeOH in DCM to give the desired product (1.8 g, 53% yield) as a white solid. LCMS (ESI) m/z: 290.9 [M+H]+, RT = 0.99 min (LCMS Method C).
Step 6

Ar-(l-(3-(l-hydroxyethyl)-li/-pyrazole-5-carbonyl)pyrrolidin-3-yl) cyclopropanecarboxamide To a solution of N-(\ -(3-acetyl-1H -pyrazole-5-carbonyl)pyrrolidin-3-yl)cyclopropanecarbox amide (500 mg, 0.9 mmol) in MeOH (8 mL) was added NaBiLt (93 mg, 2.4 mmol) at 0 °C under nitrogen atmosphere. The mixture was warmed to room temperature and stirred for 2 hrs before being concentrated under reduced pressure. The residue was treated with water (3 mL) and extracted with EtOAc (20 mL x 2). The combined organic layers were dried over anhydrous Na2SO4, filterd, concentrated, and purified by preparative HPLC (Gemini CI 8 150 x 25 mm x 10 um, 12-42% MeCN/H2O) to give the desired product (33.3 mg, 13% yield) as a white solid. 1H NMR (400
MHz, CD3OD) δ 6.61 (s, 1H), 4.93 - 4.91 (m, 1H), 4.44 - 3.68 (m, 5H), 2.21 - 2.18 (m, 1H), 2.03 -
1.92 (m, 1H), 1.58 - 1,52 (m, 1H), 1.51 - 1.48 (m, 3H), 0.85 - 0.72 (m, 4H). LCMS (ESI) m/z: 293.1 [M+H]+, RT = 0.85 min (LCMS Method C).
Example 17

^-(l-(3-(l-fIuoroethyl)-l-f.r-pyrazole-5-carbonyl)pyrrolidin-3-yl)cyc]opropanecarboxamide
To a suspension of JV-(l-(3-(l-hydroxyethyl)-1H -pyrazole-5-carbonyl)pyrrolidin-3- yi)cyclopropanecarboxamide (120 mg, 0.4 mmol) in DCM (5 mL) was added dropwise DAST (198 mg, 1 mmol) at -78 °C under a nitrogen atmosphere. After addition, the reaction was warmed to room temperature and stirred for 30 min before being quenched with saturated NaHCC>3 solution, then extracted with EtO Ac (20 mL x 3). The combined organic layers were dried over anhydrous a2SC>4, filtered, concentrated and purified by preparative HPLC (Gemini CI 8 150 x 25 mm x 10 μπι, 20-50% MeCN H2O) to give the title compound (30.8 mg, 25% yield) as a white solid. 1H NMR (400 MHz, CD3OD) δ 6.78, 6.75 (2s, lH), 5.80 - 5.63 (m, 1H), 4.41 - 4.37 (m, 1H), 4.12 - 3.68 (m, 4H), 2.24 - 2.18 (m, 1H), 2.07 - 1.96 (m, 1H); 1.72 - 1.62 (m, 3H), 1.59 - 1.50 (m, 1H), 0.85 - 0J3 (m, 4H). LCMS (ESI) m/z: 295.2 [M+H]+, RT = 0.95 min (LCMS Method C).
Example 18

-V^l-(3-(2-hydroxypropan-2-yl)-l-tf-pyra-tole-5-carbonyl)pyrrolidiii-3- yl)cyclopropanecarboxamide
To a solution of N-[l-(3-acetyl-1H -pyrazole-5-carrx>nyl)pyrrolidin-3-yl]cyclopropane carboxamide (100 mg, 0.3 mmol) in THF (3 mL) was added MeMgBr (0.7 mL, 3 M in THF) at 0 °C under a nitrogen atmosphere. The mixture was warmed to room temperature and stirred for 5 hrs before being quenched with saturated NH4CI solution, and extracted with EtOAc (20 mL x 3). The combined organic layers were dried over anhydrous Na2SC>4, filterd, concentrated, and purified by preparative HPLC to give the desired product (8.2 mg, 8% yield) as a white solid. 1H NMR (400 MHz, CD3OD) δ 6.61 (s, 1H), 4.46 - 4.40 (m, 1 H), 4.18 - 3.70 (m, 3H), 2.29 - 2.19 (m, 1H), 2.06 -
1.94 (m, 1H), 1.59 (m, 7H), 0.90 - 0.73 (m, 4H). LCMS (ESI) m/z: 307.1 [M+H]+, RT (LCMS Method C).
Example 19
Step 1

To a solution of methyl 2-isocyanoacetate (1 g, 10 mmol) in EtOH (10 mL) was added DMF-DMA (2.4 g, 20 mmol) at 0 °C under a nitrogen atmosphere and the mixture was stirred at room temperature for 24 hrs before being concentrated under reduced pressure. The residue was purified by neutral alumina column eluting with 0-20% EtOAc in hexanes to give the desired product (1 g, 64% yield) as a yellow oil. Step 2

Methyl 1 -isopropyl-1H -imidazole-4-carboxylate
A mixture of (Z)-methyl 3-(dimethylamino)-2-isocyanoacrylate (400 mg, 2 mmol) and
isopropylamine (0.43 mL, 25 mmol) in an autoclave was stirred at 70 °C for 2 hrs. After cooling to room temperature, the reaction mixture was treated with water (3 mL) and EtOAc (20 ml x 3). The combined organic layers were dried over anhydrous Na2SO4, filtered, concentrated to dryness to give the desired product (400 mg, 92% yield) as a yellow oil. 1H NMR (400 MHz, CD3OD): δ 7.93 (d, J= 0.8 Hz, 1H), 7.84 (s, 1H), 4.58 - 4.48 (m, 1H), 3.86 (s, 3H), 1.52 (d, J= 6.8 Hz, 6H).
Step 3

l-Isopropyl-1H -imidazole-4-carboxyIic acid
To a solution of methyl l-isopropyl-1H -imidazole-4-carboxylate (400 mg, 2 mmol) in a mixture of MeOH (2 mL) and H2O (1 mL) was added LiOH»H2O (300 mg, 7 mmol). The mixture was stirred at room temperature for 2 hrs, and then concentrated under reduced pressure to dryness to give the desired crude product (750 mg) as a yellow solid.
Step 4

N^l-(l- opropyI-liy-imidazole-4-carbonyl)pyrrolidin-3-yI)cyclopropanecarboxamide
To a solution of l-isopropyl-1H -imidazole-4-carboxylic acid (200 mg, 1.3 mmol) in DMF (3 mL) was added N-pyrrolidin-3-ylcyclopropanecarboxamide (260 mg, 1.7 mmol), HATU (739 mg, 2 mmol) and DIEA (0.7 mL, 4 mmol). The mixture was stirred at room temperatue for 3 hrs, filtered, concentrated, and purified by preparative HPLC (Gemini 150 x 25 mm x 10 μπι, 0-30%
MeCN/H2O) to give the desired product (93.7 mg, 24% yield) as a white solid. 1H NMR (400 MHz, DMSO-i¾): δ 8.31 - 8.29 (m, 1H), 7.77 - 7.74 (m, 1H), 4.44 - 4.41 (m, 1H), 4.26 - 4.19 (m, 1H), 4.08 - 3.76 (m, 2H), 3.58 - 3.48 (m, 2H), 2.07 - 1.98 (m, 1H), 1.82 - 1.70 (m, lH), 1.58 - 1.52 (m, 1H), 1.40 - 1.38 (m, 6H), 0.64 - 0.60 (m, 4H). LCMS (ESI) m/z: 291.1 [M+H]+, RT = 0.79 min (LCMS Method C).
Example 20
Ste l
 Dess-Martin reagent (9.3 g, 22.0 mmol) was added into a solution of 1 -phenylpropan-2-ol (2.0 g, 15 mmol) in DCM (20 mL) at room temperature. The mixture was stirred at room temperature for 12 hrs and washed with Na2S2O3 aq. solution (20 mL), NaHCO3 aq. solution (20 mL). The organic phase was dried over anhydrous Na2SO4 and then filtered. The filtrate was concentrated and the residue was purified by flash column chromatography on silica gel eluting with 20% EtOAc in hexanes to give the desired product (1.5 g, 76% yield) as colorless oil. H NMR (400 MHz, CDC13) δ 7.35 -7.32 (m, 2H), 7.29 - 7.27 (m, 1H), 7.25 - 7.19 (m, 2H), 3.69 (s, 2H), 2.15 (s, 3H).
Dess-Martin reagent (9.3 g, 22.0 mmol) was added into a solution of 1 -phenylpropan-2-ol (2.0 g, 15 mmol) in DCM (20 mL) at room temperature. The mixture was stirred at room temperature for 12 hrs and washed with Na2S2O3 aq. solution (20 mL), NaHCO3 aq. solution (20 mL). The organic phase was dried over anhydrous Na2SO4 and then filtered. The filtrate was concentrated and the residue was purified by flash column chromatography on silica gel eluting with 20% EtOAc in hexanes to give the desired product (1.5 g, 76% yield) as colorless oil. H NMR (400 MHz, CDC13) δ 7.35 -7.32 (m, 2H), 7.29 - 7.27 (m, 1H), 7.25 - 7.19 (m, 2H), 3.69 (s, 2H), 2.15 (s, 3H).
Step 2

EtONa (8.2 mL, 8.2 mmol) in EtOH was added into a mixture of l-phenylpropan-2-one (1.0 g, 7.5 mmol) and diethyl oxalate (1.3 g, 9.0 mmol) in EtOH (10 mL) at room temperature. The reaction mixture was stirred for 16 hrs and concentrated. The residue was dissolved in ¾0 (10 mL), HC1 (1 N) was added to adjust to pH = 2.0, then extracted with DCM (15 mL x 2). The organic phase was combined and dried over anhydrous Na2SO4. The mixture was filtered and concentrated to dryness to give the desired crude product (0.9 g, 51% yield) as yellow oil.
Step 3

A mixture of ethyl 2,4-dioxo-5-phenylpentanoate (0.9 g, 3.8 mmol), hydrazine hydrate (0.37 g, 5.8 mmol), AcOH (0.35 g, 5.8 mmol) in EtOH (10 mL) was stirred at room temperature for 12 hrs. The reaction mixture was concentrated and the residue was dissolved in DCM (10 mL) and washed with NaHCO3 aqueous solution (10 mL). The organic phase was washed with brine (10 mL) and dried over anhydrous Na2SO4. The mixture was filtered and evaporated to dryness to give the crude product (0.5 g, 57% yield) as a yellow oil.
Step 4

LiOH (0.26 g, 11 mmol) was added into a solution of ethyl 3-benzyl-1H -pyrazole-5-carboxylate (0.5 g, 2.2 mmol) in THF/H2O (5 mU 5mL) at room temperature. The mixture was stirred for 12 hrs. To the reaction mixture was added concentrated HC1 (1 N) to adjust to pH = 2.0. The mixture was filtered and dried to give the desired product (0.4 g, 91% yield) as a white solid.
Step 5

A mixture of 3-benzyl-1H -pyrazole-5-carboxylic acid (100 mg, 0.50 mmol), N-(pyrrolidin-3-yl) cyclopropanecarboxamide hydrochloride (94 mmg, 0.5 mmol), T3P (472 mg, 0.74 mmol), Et3N (200 mg, 2.0 mmol) in DMF (5 mL) was stirred at room temperature for 2 hrs. The reaction mixture was purified by preparative HPLC to give the desired product (39 mg, 23%
yield) as a white solid. 1H NMR (400MHz, DMSO-dLs) δ 13.07 (2s, 1H), 8.33, 8.31 (2s, 1H), 7.38 - 7.16 (m, 5H), 6.38, 6.37 (2s, 1H), 4.30 - 4.20 (m, 1H), 4.03 - 3.89 (m, 3H), 3.75 - 3.45 (m, 3H), 2.04 - 1.99 (m, 1H), 1.87 - 1.72 (m, 1H), 1.55 - 1.45 (m, 1H), 0.75 - 0.54 (m, 4H). LCMS (ESI) m/z: 339.1 [M+H]+, RT = 1.149 min.(Method C).
Example 21
Step 1

Step 2

Eth l 3-(l -phenylethyl)-1H -pyrazole-5-carboxy late
To a solution of (Z)-ethyl 3-(l-(2-tosylhydrazono)ethyl)-1H -pyrazole-5-carboxylate (300 mg, 0.9 mmol) in 1,4-dioxane (5 mL) was added phenylboronic acid (163 mg, 1.3 mmol) and 2CO3 (370 mg, 2.7 mmol) and the mixture was heated at 110 °C for 5 hrs. After cooling to room temperature, the reaction mixture was concentrated and the residue was partitioned with water (5 mL) and EtOAc (20 mL x 3). The combined organic layers were dried over anhydrous Na SO4, filterd, concentrated, and purified by silica gel column chromatography
eluting with 0-5% MeOH in DCM to give the desired product (250 mg, 43% yield) as a white solid. LCMS (ESI) m/z: 244.9 [M+H]+. Step 3

3-(l-phenylethyl)-l//-pyrazole-5-carboxylic acid
To a solution of ethyl 3-(l-phenylethyl)-l /-pyrazole-5-carboxylate (250 mg, 1 mmol) in a mixture of MeOH (1 mL) and ¾0 (1 mL) was added LiOH H2O (137 mg, 3 mmol). The mixture was stirred at room temperature for 2 hrs, then was concentrated under reduced pressure to dryness to give the crude desired product (300 mg) as a white solid. LCMS (ESI) m z: 217.2 [M+H]+.
Step 4

iV-(l-(3^1-phenylethyl)-l/f-pyrazole-5-carbonyI)pyrro^
To a solution of 3-(l-phenylemyl)-1H -pyrazole-5-carboxylic acid (300 mg, 1.4 mmol) in DMF (3 mL) was added JV-pyrrolidin-3-ylcyclopropanecarboxamide (214 mg, 1.4 mmol), HATU (527 mg, 1.4 mmol) and D1EA (0.7 mL, 4.2 mmol). The mixture was stirred at room temperature for 3 hrs before being purified by preparative HPLC (Gemini CI 8 150 x 25 mm x 10 urn, 22-52%
MeCN/H2O) to give the title compound (35.7 mg, 7% yield) as a white solid. 1H NMR (400 MHz, DMSO-£¾): δ 8.31, 8.29 (2s, 1H), 7.29 - 7.15 (m, 5H), 6.44, 6.42 (2s, 1H), 4.26 - 4.15 (m, 2H)5 3.98 - 3.81 (m, 2H), 3.63 - 3.48 (m, 2H), 2.10 - 1.94 (m, IH); 1.83 - 1.70 (m, 1H), 1.54 (d, J= 7.2 Hz, 3H), 1.48 - 1.41 (m, 1H), 0.69 - 0.59 (m, 4H). LCMS (ESI) m/z: 353.2 [M+H]+, RT = 1.16 min (LCMS Method C).
Example 22

Methyl 2-methyl-5-phenylpentanoate
To a stirred solution of methyl 5-phenylpentanoate (10 g, 52.02 mmol) in THF (100 mL) was added LDA (28.7 mL, 57.32 mmol) dropwise in an ice bath. After stirring at this temperature for 30 min, Mel (8.74 mL, 140.44 mmol) was added. The reaction mixture was stirred at 20 °C for 12 hrs. The reaction was quenched with sat NH4CI (100 mL), washed with EtOAc (100 mL x 2). The combined organic layers were dried over anhydrous jSC^, filtered and concentrated to give the desired product (9.6 g, 90% yield) as a yellow oil. Ή NMR (400 MHz, CDC13) δ: 7.28 - 7.15 (m, 5H), 3.65 (s, 3H), 2.62 - 2.58 (m, 2H), 2.48 - 2.43 (m, 1H), 1.67 - 1.47 (m, 4H), 1.13 (d, J= 7.2 Hz, 3H).
Step 2

2-Methyl-5-phenylpentanoic acid
A mixture of methyl 2-methyl-5-phenyl-pentanoate (9.6 g, 46.54 mmol) and LiOH (3.34 g, 139.62 mmol) in methanol (70 mL) and water (70 mL) was stirred at room temperature for 12 hrs. The organic solvent was removed, the mixture was diluted in water (30 mL), washed with EtOAc (100 mL). The aqueous layer was acidified by 2 N HC1 to pH 6.0. The mixture was extracted EtOAc (100 mL x 2). Combined organic layers were dried over anhydrous Na2SO , filtered and concentrated to give the desired product (8.5 g, 95% yield) as a colorless oil. 1H NMR (400 MHz, CDC13) 5: 7.30 - 7.17 (m, 5H), 2.64 - 2.61 (m, 2H), 2.52 - 2.46 (m, 1H), 1.75 - 1.65 (m, 3H), 1.50 - 1.48 (m, 1H), 1.18 (d, J= 7.2 Hz, 3H)
Step 3

N-methoxy-N,2-dimethyl-5-phenylpentanamide
A mixture of 2-methyl-5-phenyl-pentanoic acid (8.5 g, 44.21 mmol), Ν,Ο-dimethyl hydroxyamine hydrochloride (5.18 g, 53.06 mmol), HATU (20.16 g, 53.06 mmol) and DIEA (17.1 lg, 132.64 mmol) in DCM (85 mL) was stirred at room temperature for 2 hrs. The reaction mixture was diluted in DCM (50 mL), washed with 1 N HC1 (150 mL), sat NaHCO3 (150 mL) and brine (150 mL). The organic layer was dried over anhydrous aaSC^, filtered and concentrated. The crude residue was purified by silica gel column chromatography (petroleum ether/EtOAc = 30/1) to give the desired product (7.8 g, 75% yield) as colorless oil. 1H NMR (400 MHz, CDC13) δ 7.30 - 7.16 (m, 5H), 3.66 (s, 3H), 3.18 (s, 3H), 2.90 - 2.88 (m, 1H), 2.65 - 2.56 (m, 2H), 1.77 - 1.73 (m, 1H), 1.64 - 1.58 (m, 2H), 1.45 - 1.41 (m, 1H), 1.1 1 (d, J= 6.8 Hz, 3H)
Step 4

To a stirred soluion of N-methoxy-N,2-dimethyl-5-phenyl-pentanamide (4 g, 17 mmol) in THF (40 mL) was added MeMgBr (6.8 mL, 20.4 mmol) in an ice bath. The reaction was stirred at room temperature for 2 hrs. The reaction was quenched with sat NH4C1 (50 mL), washed with EtOAc (50 mL x 2). The combined organic layers were dried over anhydrous Na2SC>4, filtered and concentrated to give the desired product (2.9 g, 90% yield) as a light yellow oil. 1H NMR (400 MHz, CDC ) 5: 7.28 - 7.14 (m, 5H)5 2.62 - 2.57 (m, 2H), 2.52 - 2.50 (m, 1H), 2.09 (s, 3H), 1.60 - 1.57 (m, 3H), 1.40 - 1.38 (m, 1 H), 1.06 (d, J= 6.8 Hz, 3H).
Step S

To a stirred solution of 3-methyl-6-phenylhexan-2-one (2.9 g, 15.24 mmol) in ethanol (50 mL) was added NaOEt (1.14 g, 16.76 mmol). After stirring at room temperature for 30 min, diethyl oxalate (2.28 mL, 16.76 mmol) was added and then stirred at room temperature for 4 hrs. The solvent was removed, the mixture was partitioned with water (40 mL) and EtOAc (40 mL x 2). The combined organic layers were dried over anhydrous Na2SC>4, filtered and concentrated. The crude residue was purified by silica gel column chromatography (petroleum ether/EtO Ac = 20/1 ) to give the desired product (2.1 g, 47% yield) as yellow oil.
Step 6

Ethyl 5-(5-phenyIpentan-2-yl)-l//-pyrazole-3-carboxylate
A mixture of ethyl 5-methyl-2,4-dioxo-8-phenyl-octanoate (2.1g, 7.23mmol) and hydrazine monohydrate (0.51 mL, 8.68 mmol) in ethanol (30 mL) was stirred at room temperature for 2 hrs. The solvent was removed, the crude residue was purified by silica gel column chromatography (petroleum ether EtOAc = 5/1) to give the desired product (0.6 g, 29% yield) as a yellow oil-
Step 7

5-(5-phenylpentan-2-yl)-1H -pyrazoIe-3-carboxylic acid
A mixture of ethyl 5-(l-methyl-4-phenyl-butyI)-1H -pyrazole-3-carboxylate (600 mg, 2.1 mmol) and LiOH (251 mg, 10.48 mmol) in methanol (6 mL) and water (6 mL) was stirred at room temperature for 3 hrs. The organic solvent was removed, the mixture was diluted in water (40 mL), washed with EtOAc (40mL x 2). The aqueous layer was acidified by 2 N HC1 to pH = 5.0. The mixture was washed with EtOAc (40 mL x 2). The combined layers were dried over anhydrous Na2SO45 filtered and concentrated to give the crude desired product (400 mg, 74% yield) as a yellow solid. LCMS M/Z (M+H) = 258.9.
Step 8

O
-V-il-iS-iS-phenylpentan- -yli-l T-pyrazole-S-carbony^pyrrolidiii-S-yl) cyclopropanecarboxamide
A mixture of 5-(l-methyl-4-phenyl-butyl)-1H -pyrazole-3-carboxylic acid (150 mg, 0.58 mmol), N- pyrrolidin-3-ylcyclopropanecarboxamide hydrochloride (133 mg, 0.70 mmol), HATU (265 mg, 0.70 mmol) and DIEA (225 mg, 1.74 mmol) in DMF (3 mL) was stirred at room temperature for 12 hrs. The reaction mixture was diluted in water (20 mL), and extracted with EtOAc (20 mL x 2). Combined organic layers were dried over anhydrous Na2SC>4, filtered and concentrated. The crude residue was purified by preparative HPLC to give the desired product (25 mg, 11% yield) as a white solid. Ή NMR (400 MHz, DMSO-rf6) δ 7.23 - 7.09 (m, 5H), 6.43 (s, 1H), 4.39 - 4.37 (m, 1H), 4.08 - 3.68 (m, 4H), 2.91 - 2.89 (m, 1H), 2.59 - 2.55 (m, 2H), 2.20 - 2.18 (m, 1H), 1.95 - 1.93 (m, 1H), 1.62 - 1.56 (m, 5H), 1.24 (d, J = 7.2 Hz, 3H), 0.84- 0.71 (m, 4H). LCMS (ESI), M/Z (M+H) = 395.0, RT - 0.722 min (LCMS method E).
Example 23
Step 1

Ethyl 3-(but-3-en-2-yI)-1H -pyrazole-5-carboxylate
To a solution of ethyl 3-(l-(2-tosylhydTazono)ethyl)-1H -pyrazole-5-carboxylate (1.0 g, 2.85 mmol) and vinylboronic anhydride pyridine complex (1.03 g, 4.28 mmol) in dioxane (10 mL) was added K2CO3 (1.18 g, 8.56 mmol). The resulting mixture was heated at 110 °C for 16 hrs. After cooling to room temperature, the reaction mixture was filtered and concentrated. The residue was purified by flash column chromatography on silica gel (eluting with 0-2% MeOH in DCM) to give the crude title compound (400 mg) as a yellow oil. LCMS (ESI) m/z: 194.9 [M+H]+.
Step 2

Ethyl 3-(sec-buty])-1H -pyrazole-5-carboxylate
To a solution of ethyl 3-(but-3-en-2-yl)-1H -pyrazole-5-carboxylate (400 mg crude, 2.06 mmol) in MeOH (10 mL) was added Pd/C (10% wt., 100 mg). The mixture was stirred under H2 (15 psi) at room temperature for 4 hrs. The mixture was filtered and concentrated to give desired crude product (370 mg) as a pale yellow oil. lH NMR (400 MHz, CD3OD): δ 6.55 (s, 1H), 4.34 (q, J= 6.8 Hz, 2H), 2.83 - 2.79 (m, 1H), 1.69 - 1.61 (m, 2H), 1.37 (t, J= 6.8 Hz, 3H), 1.28 (d, J= 6.8 Hz, 3H), 0.87 (t, J= 6.8 Hz, 3H).
Step 3

Lithium 3-(sec-buryl)-1H -pyrazole-5-carboxylate
To a solution of ethyl 3 -(.sec-butyl)- 1 //-pyrazole-5-carboxylate (100 mg, 0.51 mmol) in MeOH (2 mL) and ¾0 (1 mL) was added lithium hydroxide monohydrate (64 mg, 1.53 mmol). The resulting mixture was stirred at room temperature for 2 hrs and concentrated to give the crude title compound (150 mg) as a white solid. LCMS (ESI) m/z: 169.1 [M-Li+H]+. Step 4

N-il^-isec-butyli-Ltf-pyrazole-S-carbony pyrrolidin-S-ylJcyclopropanecarboxainide
To a solution of lithium 3-(sec-butyl)-1H -pyrazole-5-carboxylate (150 mg crude, 0.51 mmol) and
N-(pyiTolidin-3-yl)cyclopropanecarboxamide hydrochloride (97 mg, 0.51 mmol) in DMF (2 mL) was added HATU (290 mg, 0.77 mmol) and DIPEA (198 mg, .53 mmol). The mixture was stirred at room temperature for 16 hrs. The mixture was purified by preparative HPLC to give the title
compound (26 mg, 17% yield) as a white solid. Ή NMR (400 MHz, DMSO-c¾) 5 8.35, 8.33 (s, 1H total), 6.41, 6.39 (s, 1H total), 4.26 - 4.24 (m, 1H), 3.99 - 3.93 (m, 2H), 3.67 - 3.35 (m, 2H), 2.78 - 2.72 (m, 1H), 2.04 -2.01 (m, 1H), 1.78 -1.73 (m, 1H), 1.60 - 1.53 (m, 3H), 1.20 (d, J= 6.8 Hz, 3H), 0.80 (t, J= 7.2 Hz, 3H), 0.68 -0.62 (m, 4H). LCMS (ESI) m/z: 305.0 [M+H]+, Rt = 1.081 min (LCMS Method C),
Example 24
Step 1

N-(l-(3-bromo-lH-pyrazole-5-carbonyl)pyrrolidin-3-yI)cyclopropanecarboxamide
To a solution of 3-bromo-1H -pyrazole-5-carboxylic acid (1.0 g, 5.24 mmol) and N-(pyrrolidin-3- y])cyclopropanecarboxamide hydrochloride (1.0 g, 5.24 mmol) in DMF (20 mL) was added HATU (2.39 g, 6.29 mmol) and DIPEA (2.03 g, 15.72 mmol). The mixture was stirred at room
temperature for 16 hrs and concentrated. The residue was dissloved in EtO Ac (50 mL) and washed with ¾0 (10 mL). The organic phase was dried over anhydrous Na2SC> , filtered and concentrated. T he residue was purified by flash column chromatography on silica gel (0-3% MeOH in DCM) to give the desired product (900 mg, 53% yield) as a white solid. Ή NMR (400 MHz, DMSO-i/6) δ 13.87 (s, 1H), 8.36 (s, 1H), 6.89, 6.84 (2s, 1H), 4.31 - 4.26 (m, 1H), 3.89 - 3.34 (m, 4H), 2.06 - 2.01 (m, 1H), 1.89 - 1.72 (m, 1H), 1.53 - 1.50 (m, 1H), 0.67 - 0.64 (m, 4H). LCMS (ESI) m z: 329.0 [M+H]+, RT = 1.007 min (LCMS Method C).
Step 2

jV-{l-(3-bromo-l-((2-(trimethylsiIyl)etho
yI)cyclopropanecarboxamide
To a solution of N-(l-(3-bromo-1H -pyrazole-5-carbonyl)pyrrolidin-3-yl)cyclopropane carboxamide (500 mg, 1.53 mmol) in DMF (5 mL) was added NaH (60% in mineral oil, 153 mg, 3.82 mmol)
portionwise at 0 °C under a nitrogen atmosphere. After stirring at 0 °C for 15 min, a solution of (2- (chloromethoxy)ethyl)trimethylsilane (280 mg, 1.68 mmol) in DMF (2 mL) was added dropwise. cAfter addition, the resulting mixture was stirred at room temperature for 16 hrs. The reaction was quenched with saturated NH4CI (20 mL) and extracted with EtOAc (20 mL). The organic phase was dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified by flash column chromatography on silica gel (eluting with 0-50% EtOAc in hexanes) to give the desired product (450 mg, 64% yield) as a colorless oil. LCMS (ESI) m/z: 457.1 [M+H]+ .
Step 3

N-(l-(3^3 -trifluoroprop-l-en-2-yl)-l-((2-^
carbonyl)pyrrolidin-3-yl)cyclopropanecarboxamide
To a solution of iV-(l-(3-bromo-l-((2-(trimethylsilyl)ethoxy)methyl)-1H -pyrazole-5- carbonyl)pyrrolidin-3-yl)cyclopropanecarboxamide (300 mg, 0.656 mmol) and 4,4,6-trimethyl-2- (3,3,3-trifluoroprop-l-en-2-yl)-l,3,2-dioxaborinane (291 mg. 1.31 mmol) in dioxane (6 mL) and H2O (3 mL) was added Cs2CO3 (642 mg, 1.97 mmol) and Pd(PPh3)4 (76 mg, 0.066 mmol). The resulting mixture was purged with N2 for 1 min and then heated at 110 °C for 30 min in a microwave reactor. The reaction mixture was colled to room temperature, extracted with EtOAc (10 mL). The organic phase was dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified by flash column chromatography on silica gel (eluteing with 0-50% EtOAc in hexanes) to give the desired product (180 mg, 62% yield) as a colorless oil. LCMS (ESI) m/z: 473.2 [ +H]+.
Step 4

7V-(l-(3-(1 5l-trifluoropropan-2-yl)-l-((2-(trim
carbonyl)pyrrolidin-3-yl)cyclopropanecarboxamide
To a solution of N-(l-(3-(3,33-trifluoroprop-l-en-2-yl)-l-((2-(trimethylsilyl)ethoxy)methyl)-1H - pyrazole-5-carbonyl)pyrrolidin-3-yl)cyclopropanecarboxamide (150 mg, 0.317 mmol) in MeOH (5 mL) was added Pd/C (10% wt., 50 mg). The resulting mixture was stirred at room temperature under H2 for 2 hrs. The reaction mixture was filtered and concentrated to give the crude desired product (150 mg) as colorless oil. LCMS (ESI) m/z: 475.2 [M+H]+.
Step 5

-(l-(3-(1 -frifl»oropropan-2-yl)-l/f-pyrazole-5-carbonyl)pyrrolidiii-3-yl)
cyc.opropanecarboxam.de
To a solution of N-(1^3-(l,l,l-trifluoropropan-2-yl)-l-((2-(trimethylsiIyl)ethoxy)memy pyrazole-5-carbonyl)pyrrolidin-3-yl)cyclopropanecarboxamide (150 mg, 0.316 mmol) in DC (3 mL) was added TFA(1.5 mL) dropwise. The resulting mixture was stirred at room temperature for 2 hrs. The reaction mixture was concentrated and the residue was dissolved in EtOAc (10 mL) and adjusted the pH = 8 ~ 9 using saturated NaHCO3 (1 mL). The mixture was dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified by preparative HPLC to give the desired product (26 mg, 24% yield) as a white solid. 1H NMR (400 MHz, CD3OD) δ 6.76, 6.72 (s, 1H total), 4.46 - 4.40 (m, 1H), 4.35 - 3.58 (m, 5H), 2.29 - 1.96 (m, 2H), 1.65 - 1.52 (m, 4H), 0.89- 0.75 (m, 4H). LCMS (ESI) m/z: 345.1[M+H]+, RT = 1.070 min (LCMS Method C).
Example 25
Step 1

Ethyl 3-acetyl-l-((2-(trimethyIsilyl)ethoxy)methyl)-1H-pyraz le-5-carboxyIate
To a stirred solution of ethyl 3-acetyl-1H -pyrazole-5-carboxylate (7.5g, 41.17mmol) in DMF (75mL) was added NaH (1.98 g, 49.4 mmol) in an ice bath. After stirred at this temperature for 30 min5 the reaction was added SEMC1 (8.24 g, 49.4 mmol) dropwise. The reaction mixture was stirred at room temperature for 12 his. The mixture was diluted in EtOAc (100 mL), washed with brine (100 mL x 3). The organic layer was dried over anhydrous Na2SO4, filtered and
concentrated. The residue was purified by silica gel flash column chromatography (petroleum ether: EtOAc = 50: 1) to give the desired product (4.8 g, 37% yield) as a colorless oil. Ή NMR (400 MHz, CDC13) δ 7.37 (s, 1H), 5.90 (s, 2H), 4.40 - 4.35 (m, 2H), 3.63 (t, J= 8.0 Hz, 2H), 2.61 (s, 3H), 1.38 (t, J= 6.8 Hz, 3H), 0.91 (t, J= 8.0 Hz, 2H), 0.03 (s, 9H).
Step 2
(is)-ethyl S^l-methoxyprop-l-en^-yl^l-^-^rimethylsilylJethoxyimethylJ-lJi-pyrazole-S- carboxylate
A mixture of methoxy methyl(triphenyl)phosphonium chloride (15.8 g, 46.09 mmol) and /-BuOK (5.17 g, 46.09 mmol) in THF (90 mL) was stirred at 20 °C for 30 min. To the mixture was added ethyl 5-aceryl-2-(2-trimethylsilylethoxymethyl)pyrazole-3-carboxylate (4.8 g, 15.36 mmol) in an ice bath under N2 atmosphere. The mixture was stirred at 20 °C for 12 hrs. The reeaction was quenched by sat NH4CI (100 mL), and extracted with EtOAc (100 mL x 2). The combined organic layers were dried over anhydrous Na2S(¾, filtered and concentrated. The crude residue was purified by silica gel flash column chromatography (petroleum ether: EtOAc = 50: 1) to give the desired product (1 g, 19% yield) as a colorless oil. LCMS (ESI) M/Z (M+H) = 340.9.
Step 3

(lT)-3^1-methoxyprop-l-en-2-yl)-l^(2-(trim
carboxylic acid
A mixture of ethyl 5-[(£)-2-methoxy-l -methyl-vinyl]-2-(2-trimethylsilylet oxymethyl) pyrazole-3- carboxylate (300 mg, 0.88 mmol) and TBAF (1.1 g, 4.41 mmol) in THF (6 mL) was stirred at 20 °C for 12 hrs. The mixture was diluted in EtOAc (30 mL), washed with brine (30 mL x 3). The organic layer was dried over anhydrous Na2SO_i, filtered and concentrated to give the desired product (200 mg, 73% yield) as a yellow oil. LC S M/Z (M+H) = 312.9.
Step 4

3-(l-Methoxypropan-2-yl)-l-((2-(trimethylsiIyl)ethoxy)methyl)-1H -pyrazole-5-carboxylic acid
A mixture of 5-[(E)-2-methoxy-l -methyl-vinyl] -2-(2-trimethylsilylethoxymethyl)pyrazole-3- carboxylic acid (200 mg, 0.64 mmol) and Pd/C (100 mg) in ethyl acetate (4 mL) was stirred at room temperature for 12 hrs, under 1 atm of ¾. The reaction mixture was filtered over a short of Celite pad. The filtrate was concentrated to give the crude desired product (200 mg, 99% yield) as a yellow solid. LCMS M/Z (M+H) = 314.9. Step 5

A^l-(3-(l-methoxypropan-2-yl)-l-((2-(trimethylsi^^^
carbonyl)pyrrolidhi-3-yl)cyclopropanecarboxamide
A mixture of 5-(2-memoxy-l-memyl-emyl)-2-(2-trimemylsilylethoxymemyl)pyrazole-3-carboxyH acid (200 mg, 0.64mmol), N-pyrrolidin-3-ylcyclopropanecarboxamide hydrochloride (146 mg, 0.76 mmol), HATU (290 mg, 0.76 mmol) and DIE A (246 mg, 1.91 mmol) in DMF (5 mL) was stirred at
room temperature for 12 hrs. The mixture was diluted in EtOAc (30 mL), washed with brine (30 mL x 2). The organic layer was dried over anhydrous Na2SO4, filtered and concentrated. The crude residue was purified by preparative TLC (DCM: MeOH = 20: 1) to give the desired product (200 mg, 70% yield) as a yellow solid. LCMS M/Z (M+Na) = 473.0.
Step 6

iV-(l-(3-(l-niethoxypropan-2-yl)-l^r-pyra-io]e-5-carbonyl)pyiTolidin-3-yl) cyclopropanecarboxamide
A solution of N-[l-[5-(2-methoxy-l -methyl-ethyl)-2-(2-trimethylsilylethoxymethyl) pyrazole-3- carbonyl]pyiTolidin-3-yl]cyclopropanecarboxamide (350 mg, 0.78 mmol) in DCM (4 mL) and TFA (4 mL) was stirred at room temperature for 2 hrs. T he solvent was removed, the crude residue was purified by preparative HPLC to give the desired product (38 mg, 15% yield) as a white solid. Ή NMR (400 MHz, DMSO-i¾) δ 6.54 (s, 1H), 4.41 - 4.39 (m, 1H), 4.14 - 3.82 (m, 4H), 3.39 (d, J= 6.4 Hz, 2H), 3.34 (s, 3H), 3.17 - 3.15 (m, 1H), 2.20 - 1.98 (m, 1H), 1.97 - 1.95 (m, 1H), 1.60 - 1.58 (m, 1 H), 1.30 (d, J= 7.2 Hz, 3H), 0.87 - 0.73 (m, 4H). LCMS (ESI) M/Z (M+H) = 321.2, RT = 0.656 min (LCMS method E).
Example 26
Step 1

Methyl 3-isopropoxy-1H -pyrazoIe-5-carboxylate
To a mixture of methyl 3-hydroxy-1H -pyrazole-5-carboxylate (300 mg, 2.11 mmol) and K2CO3 (438 mg, 3.17 mmol) in DMF (3 mL) was added 2-iodopropane (4 1 mg, 2.53 mmol). The resulting mixture was stirred at room temperature for 16 hrs. The reaction was quenched with water and the mixture was extracted with EtOAc (30 mL x 3). The combined organic layers were washed with water and brine, dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified by flash column chromatograph on silica gel eluting with 0-10% EtOAc in hexanes to
give the desired product (100 mg, 26% yield) as a light yellow solid. Ή NMR (400 MHz, CDC13) δ 6.19 (s, 1H), 4.78 - 4.69 (m, 1H), 3.90 (s, 3H), 1.35 (d, J= 6.0 Hz, 6H).
Step 2

3-Isopropoxy-l//-pyrazole-5-carboxyIic acid
To a solution of methyl 3-isopropoxy-1H -pyrazole-5-carboxylate (100 mg, 0.543 mmol) in
MeOH H2O (3/2, 3 mL) was added LiOH H O (91 mg, 2.17 mmol). The resulting mixture was stirred at room temperature for 1 hrs. After removal of MeOH under reduced pressure, the mixture was acidized with 3 N HC1 to pH = 1.0 and then extracted with EtOAc (50 mL). The organic layer was washed with brine, dried over anhydrous Na2SC>4, filtered and concentrated to give the desired product (80 mg, 87% yield) as a light yellow solid.
Step 3

A^l-(3-isopropoxy-ljtf-pyi^oIe-5-carbonyI)pyrrolidin-3-yl)cyclopropanecarboxamide
To a solution of 3-isopropoxy-1H -pyrazole-5-carboxylic acid (70 mg, 0.411 mmol) in DMF (1.5 mL) were added HATU (235 mg, 0.617 mmol) and DIEA (0.27 mL, 1.65 mmol). After the mixture stirred for 5 min, N-(pyrrolidin-3-yl)cyclopropanecarboxamide hydrochloride (78 mg, 0.411 mmol) was added. The resulting mixture was stirred at room temperature for 16 hrs. Filtered, the mixture was purified by preparative HPLC (Gemini CI 8, 150 x 25 mm x 10 μιη, 14-44% MeCN H2O) to give the desired product (35 mg, 28% yield) as a white solid. Ή NMR (400 MHz, CD3OD) δ 6.14, 6.09 (2s, 1H), 4.66 - 4.62 (m, 1H), 4.44 - 4.40 (m, 1H), 4.05 - 3.50 (m, 4H), 2.28 - 2.12 (m, 1H), 2.10 - 1.90 (m, 1H), 1.33 (d, J= 6.0 Hz, 6H), 0.87 - 0.84 (m, 2H), 0.77 - 0.74 (m, 2H). LCMS (ESI) mix: 307.2 [M+H]+, RT = 1.036 min (LCMS method C).
Example 27
Step 1

tert-Buty 1 (l-(3-isopropyl- l i-py razolc-5-carbony l)py rrolidin-3-yl)carbamate
To a solution of tert-butyl N-pyirolidin-3-ylcarbamate (1 g, 5.4 mmol) in DMF (15 ml) was added 3-isopropyl-1H -pyrazole-5-carboxyhc acid (0.9 g, 5.9 mmol), DIEA (1.9 ml, 10.7 mmol) and HATU (2.6 g, 7.0 mmol). The mixture was stirred at room temperature for 15 hrs, then diluted with H2O (20 ml), and extracted with EtOAc (60 ml x 2). The combined organic layers were dried over anhydrous Na2SO4, filtered, concentrated and the residue was purified by silica gel flash chromatography eluted with 0-10% MeOH in DCM to give the desired product (1.3 g, 75% yield) as a white solid. LCMS (ESI) m/z: 323.0 [M+H]+, RT = 1.1 1 min (LCMS Method A).
Step 2

(3-AmiiiopyrroIidin-l-yl)(3-isopropyl-1H -pyra2oI-5-yl)methanone
To a suspension of r/-butyl (l-(3-isopropyl-1H -pyrazole-5-carbonyl)pyrrolidin-3-yl)carbamate (1.3 g, 4.0 mmol) in EtOAc (7 ml) was added HC1 (7 ml, 4 M in EtOAc). The mixture was stirred at room temperature for 30 min, then solvent was removed under reduced pressure to give the desired crude product (1.1 g) as a white solid. LCMS (ESI) m/z: 223.2 [M+H]+, RT = 0.74 min (LCMS Method C).
Step 3

-V-il^-isopropyl-l/T-pyrazole-S-carbonylJpyrrolidiii-S-y acetaniide
To a solution of (3-aminopyirolidin-l-yl)(3-isopropyl-1H -pyrazol-5-yl)methanone (200 mg, 0.9 ramol) in DMF (4 ml) was added acetyl acetate (1 19 mg, 1.2 mmol) and DIEA (0.5 ml, 2.7 mmol). The mixture was stirred at room temperature for 2 hrs, filtered and purified by preparative HPLC (Gemini C18 150 x 25 mm x 10 μιη, 11-41% MeCN/H2O) to give the desired product (62.3 mg, 26% yield) as a white solid. Ή NMR (400 MHz, DMSO-£¾) δ 12.91 (s, lH), 7.78 (m, 1H), 6.39 (s, 1H), 4.22 - 4.20 (m, lH), 3.97 - 3.90 (m, 2H), 3.64 - 3.50 (m, 2H), 2.97 - 2.94 (m, 1H), 2.02 - 1.99 (m, 1H), 1.81 - 1.74 (m, 4H), 1.22 (d, J= 7.2 Hz, 6H). LCMS (ESI) m/z: 265.1 [M+H]+, RT = 0.94 min (LCMS Method C). Examples 28
Step 1

(S)- r/-buryl 3-(cycIopropanecarboxamido)pyrrolidine-l-carboxylate
To a stirred solution of (5)-iert-butyl 3-aminopyrrolidine-l-carboxylate (1.0 g, 5.37 mmol) in DCM (5 mL) was added TEA (1.63 g, 16.11 mmol) and cooled to 0 °C. Then cyclopropanecarbonyl chloride (673.5 mg, 6.44 mmol) was added drop wise into the mixture and the reaction mixture was warned to room temperature over 2 hrs. The mixture was diluted with DCM (150 mL) and washed with ¾0 (120 mL x 3). The organic phase was dried over anhydrous Na2SO-i, filtered and concentrated to afford the desired product (1.2 g, 86% yield) as yellow solid which was used to the next step directly without further purification.
Step 2

(5)- V-(pyrrolidin-3-yl)cyclopropanecarboxamide hydrochloride
To a stirred solution of (S)-fert-butyl 3-(cyclopropanecarboxamido) pyrrolidine- 1 -carboxylate (1.2 g, 4.7 mmol) in EtOAc (2 mL) was added dropwise HCl/EtOAc (4 N, 3 mL, 12 mmol) at room temperature and stirred for 1 hr. The reaction mixture was concentrated and washed with EtOAc
(30 mL x 2) to afford the crude desired product (700 mg, 78% yield) as yellow solid.
Step 3

(5)-JV-(1^3-isopropyl-1H -pyrazole-5-carbon I)pyrrolidin-3-yl)cyclopropanecarboxamide To a stirred solution of 3-isopropyl-1H -pyrazole-5-carboxylic acid (221 mg, 1.16 mmol) in DMF (3 mL) was added DIEA(500 mg, 3.87 mmol) and HATU (539.2 mg, 1.42 mmol) at room
temperature. The mixture was stirred for 10 min before (5)-N-(pyrrolidin-3- yl)cyclopropanecarboxamide hydrochloride (200 mg, 1.29 mmol) was added. The mixture was stirred at room temperature for another 2 hrs before being purified by preparative HPLC to afford the desired product (40 mg, 1 1 % yield) as white solid. lK NMR (400 MHz, CD3OD) δ 6.48 (s, 1 H), 4.47 - 4.34 (m, 1H), 4.17 - 3.97 (m, 1.5 H), 3.85 - 3.80 (m, 1 H), 3.78 - 3.64 (m, 1 H), 3.55 - 3.52 (m, 0.5 H), 3.07 - 3.00 (m, 1 H), 2.27 - 2.12 (m, 1 H), 2.02 - 1.90 (m, 1 H), 1.65 - 1.52 (m, 1 H), 1.30 (<LJ = 7.2 Hz, 6 H), 0.90 - 0.79 (m, 2 H), 0.79 - 0.69 (m, 2 H). LCMS (ESI) m/z: 291.1 [M+H]+, RT = 1.023 min (LCMS method C).
Examples 29
Step 1

(R)-/er/-butyl 3-(cycIopropanecarboxamido)pyrroIidine-l-carboxylate
To a solution of (K)-ferf-butyl 3-aminopyrrolidine-l-carboxylate (1.0 g, 5.37 mmol) in DCM (10 mL) was added Et3 (1.09 g, 10.74 mmol), followed by the addition of cyclopropanecarbonyl chloride (0.6 mL, 0.64 mmol) dropwise at 0 °C. The mixture was stirred for 1 min, then quenched with H2O (25 mL), and extracted with DCM (20 mL x 3). Combined organic layers were dried over anhydrous Na2SO4 and concentrated to give desired product (1.30 g, crude) as a yellow solid.
Step 2

(R)-N-(pyrroUdin-3-yl)cyclopropanecarboxamide hydrochloride
To a solution of (R)-tert-butyl 3 cyclopropanecarboxaniido)pyrrolidine-l-carboxylate (1.3 g, 5.11 mmol) in EtOAc (10 mL) was added HCl/EtOAc (10 mL). The mixture was stirred at room temperature for 12 hrs. Then the mixture was evaporated under reduced pressure to give the crude desired product (800 mg) as a dark solid.
Step 3

(Jf)-A^-(l-(3-isopropyl-lfl-pyra2ole-5-carboiiyl) pyrrolidin-3-yl)cyclopropane carboxamide
To a solution of 3-isopropyl-1H -pyrazole-5-carboxylic acid (220 mg, 1.43 mmol) in DMF (5 mL) was added HATU (690 mg, 1.82 mmol) and DIPEA (0.7 mL, 0.39 mmol) at room temperature. The reaction mixture was stirred for 30 min and then (i?)-N-(pyrrolidin-3-yl) cyclopropanecarboxamide hydrochloride (200 mg, 1.30 mmol) was added and the mixture was stirred for another 2 hrs. Then the mixture was filtered and purified by preparative HPLC to give desired product (136.8 mg, 36% yield). 1HNMR (400 MHz, CD3OD) δ 6.48 (s, 1H), 4.41 - 4.38 (m, 1H), 4.03 - 3.75 (m, 4H), 3.05 - 3.02 (m, 1H), 2.20 - 2.15 (m, 1H), 1.97 - 1.96 (m, 1H), 1.59 - 1.57 (m, 1H), 0.86 - 0.82 (m, 2H), 0.75 - 0.73 (m, 2H). LCMS (ESI) m/z: 291.1 [M+H]+, RT = 0.687 min (LCMS Method E).
Example 30
Step l


2-(l-(3-IsopropyHH-pyra-!;ole-5-carbonyl)pyrrolidiii-3-yl)isoindolin-l-one
To a solution of (3-aminopyrrolidin-l-yl)(3-isopropyl-l /-pyrazol-5-yl)methanone hydrochloride (100 mg, 0.386 mmol) and DIPEA (150 mg, 1.16 mmol) in MeCN (2 mL) was added methyl 2- (bromomethyl)benzoate (89 mg, 0.386 mmol). The mixture was stirred at room temperature for 16 hrs. The mixture was concentrated and the residue was purified by preparative HPLC (Gemini C|g 150 x25 mm x 10 urn, 30-60-16% MeCN condition) to give the desired product (19 mg, 15% yield) as a white solid. 1H NMR (400 MHz, CD3OD): δ 7.80 - 7.76 (m, 1H), 7.64 - 7.49 (m, 3H), 6.53 (s, 1H), 5.01 - 4.96 (m, 1H), 4.60, 4.58 (s, 2H total), 4.26-4.22 (m, 1H), 4.28 - 3.71 (m, 4H), 3.05 - 3.02 (m, 1H), 2.39 - 2.31 (m, 2H), 1.82-1.75 (m, 1H), 1.31, 1.29 (d, J= 6.8 Hz, 6H total). LCMS (LCMS Method C): Rt = 1.145 min, m/z: 339.2 [M+H]+.
Example 31

Methyl (l-(3-isopropyl-lJi-pyrazole-5-carbonyl)pyrrolidiii-3-yl)carbamate
A solution of methyl carbonochloridate (120 mg, 1.27 mmol) in DCM (1 mL) was added into a solution of (3-aminopyrrolidin-l-yl)(3-isopropyl-1H -pyrazol-5-yl) methanone hydrochloride (40 mg, 1.85 mmol), DIPEA (600 mg, 4.64 mmol) in DCM (5 mL) at 0 °C under a N2 atmosphere. The
reaction mixture was stirred at room temperature for 12 hrs. The reaction mixture was concentrated and the residue was purified by preparative HPLC to give the title product (67 mg, 18% yield) as a white solid. Ή NMR (400 MHz, methanol-^) δ 6.48 (s, 1H), 4.25-4.16 (m, 1H), 4.15-4.07 (m, 0.5H), 4.05-3.95 (m, 1H), 3.84-3.80 (m, 0.5H), 3.75-3.47 (m, 5H), 3.05-3.02 (m, 1H), 2.25-2.12 (m, 1H), 2.00-1.88 (m, 1H), 1.30 (d, J= 8.0 Hz, 6H). LCMS (ESI) m/z: 280.8 [M+H]+, RT = 0.690 min.
Example 32

Phenyl (l-(3-isopropyI-l.H-pyrazole-5-carbonyl)p rrolidin-3-yl)carbamate
A solution of phenyl carbonochloridate (303 mg, 1.93 mmol) in DCM (1 mL), was added into a solution of (3-aminopyrrolidin-l-yl)(3-isopropyl-1H -pyrazol-5-yl) methanone hydrochloride (500 mg, 1.93 mmol), DIPEA (749 mg, 5.79 mmol) in DCM (5 mL) at 0 °C under a N2 atmosphere. T he reaction mixture was stirred at room temperature for 12 hrs. The reaction mixture was concentrated and the residue was purified by preparative HPLC to give the title product ( 10 mg„ 1.5% yield) as a white solid. 1H NMR (400 MHz, methanol-^) δ 7.44-7.31 (m, 2H), 7.25-7.15 (m, 1H), 7.12-7.08 (m, 2H), 6.49 (s, 1H), 4.31-4.25 (m, 0.5H), 4.20-4.02 (ra, 1.5H), 3.96-3.84 (m, 1H), 3.80-3.58 (m, 2H), 3.10-2.98 (m, 1H), 2.32-2.18 (m, 1H), 2.10-2.00 (m, 1H), 1.31 (d, J = 4.0 Hz, 6H). LCMS (ESI) m/z: 343.1 [M+H]+, RT = 0.786 min.
Example 33

l-C^clopropyl-S-il-iS-isopropyl-l/T-pyrazole-S-carbony^pyrrolidin-S-ylJurea
A solution of phenyl (l-(3-isopropyl-1H -pyrazole-5-carbonyl)pyrrolidin-3-yl)carbamate (150 mg,
0.44 mmol), cyclopropanamine (38 mg, 0.68 mmol), DIEA (170 mg, 1.31 mmol) was stirred at room temperature for 12 hrs. The reaction mixture was concentrated. The residue was purified by preparative HPLC to give the desired product (46.9 mg, 35% yield) as a white solid. Ή NMR (400
MHz, DMSO-4 δ 12.90 (s, 1H), 6.37 (s, 1H), 6.13 - 5.98 (m, 2H), 4.15 - 4.05 (m, 1H), 4.00 - 3.85
(m, 1.5H), 3.64 - 3.44 (m, 2.5H), 2.99-2.91 (m, 1H), 2.40 - 2.38(m, 1H), 2.02 - 1.97(m, 1H), 1.83- 1.71 (m, 1H), 1.22 (d, J= 6.8 Hz, 6H), 0.58- 0.49 (m, 2H), 0.34-0.25 (m, 2H). LCMS (ESI) m/z: 306.3 [M+H]+, RT = 0.686 min (LCMS method E). Example 34

l-(l-(3-Isopropyl-li -pyrazole-5-carboiiyl) pyrrolidin-3-yl)-3-methylurea
A solution of methanamine hydrochloride (30 mg, 0.43 mmol), phenyl (l-(3-isopropyl-1H - pyrazole-5-carbonyl)-pyrrolidin-3-yl)carbamate (100 mg, 0.29 mmol) in MeCN (3 mL) was stirred at 60 °C for 12 hrs. The reaction mixture was concentrated. The residue was purified by preparative HPLC to give the desired product (47.7 mg, 40% yield) as a white solid. Ή NMR (400 MHz, DMSC-rf6) δ 12.92 (s, 1H), 6.36 (s, 1H), 6.21 - 6.19 (m, 1H), 5.66 - 5.54 (m, 1H), 4.13 - 4.02 (m, 1 H), 4.00 - 3.85 (m, 1H), 3.69 - 3.42 (m, 2H), 3.30 - 3.22 (m, 1H), 3.00 - 2.89 (m, 1H), 2.54- 2.52 (m, 3H), 2.07 - 1.94 (m, 1H), 1.81 - 1.66 (m, 1H), 1.21 (d, J = 6.8 Hz, 6H). LCMS (ESI) m/z: 280.1 [M+H]+, RT = 0.569 min (LCMS method E).
Example 35

l-IsopropyI-3-(l-(3-isopropyl-lH-pyrazoIe-5-carbonyl)pyrrolidin-3-yl)urea A mixture of (3-aminopyiTolidm-l-yl)(3-isopropyl-1H -pyrazol-5-yl)methanone hydrochloride (200 mg, 0.77 mmol), 2-isocyanatopropane (66 mg, 0.77 mmol), DIEA (300 mg, 2.32 mmol) in C¾CN (5 mL) was stirred at room temperature for 12 hrs. The reaction mixture was diluted with ¾0 (10 mL) and extracted with EtOAc (10 mL). The organic phase was separated and then concentrated to give the crude product which was purified with preparative HPLC to give the desired product (3.6 mg, 1.5% yield) as a white solid. 1H NMR (400MHz, DMSO-rf6) δ 12.92 (s, 1 H), 6.37 (s, 1 H), 6.02-6.00 (m, 1H), 5.58 - 5.55 (m, 1H), 4.19 - 3.82 (m, 3H), 3.70 - 3.46 (m, 3H), 2.99 - 2.93 (m, 1H), 2.07 - 1.96 (m, 1H), 1.79 - 1.65 (m, 1H), 1.22 (d, J= 8.0 Hz, 6H), 1.10 - 0.90 (m, 6H). LCMS (ESI) m z: 308.3 [M+H]+, RT = 0.703 min (LCMS method E).
Example 36

(3-Isopropyl-liy-pyrazol-5-yl)(3-(pyridm-2-ylamino)pyrrolidin-l-yI)methanone To a solution of (3-aminopyrrolidin-l -yl)(3-isopropyl-1H -pyrazol-5-yl)methanone (200 mg, 0.9 mmol) in DMF (4 ml) was added 2-fluoropyridine (131 mg, 1.3 mmol) and CS2CO3 (879 mg, 2.7 mmol). Then the mixture was stirred at 150 °C for 15 hrs. After cooling to room temperature, the reaction mixture was filtered and purified by preparative HPLC (Gemini 150 x 25 mm x 5 μπι, 50% MeCN) to give the desired product (30.7 mg, 11% yield) as a white solid. Ή NMR (400 MHz, CD3OD) δ 8.53 (d, J= 4.0 Hz, 1H), 8.03 - 8.00 (m, 1H), 7.87 - 7.85 (m, 1H), 7.45 - 7.42 (m, 1H), 6.78 (s, 1H), 4.26 - 4.25 (m, 1H), 3.94 - 3.92 (m, 0.5H), 3.89 - 3.83 (m, 3H), 3.69 - 3.66 (m, 1H), 3.51 - 3.49 (m, 0.5H), 2.26 - 2.21 (m, 1H), 1.93 - 1.83 (m, 1 H), 1.28 (d, J= 6.8 Hz, 6H). LCMS (ESI) m/z: 300.1 [M+H]+, RT = 0.95 min (LCMS Method C). Example 37
Step 1

iV-(l,l-dichloropropan-2-ylidene)-4-methyIbenzenesuIfonohydrazide To a solution of 1 , 1 -dichloropropan-2-one ( 10 g, 78.76 mmol) in EtCOOH (60 mL) was added 4- methylbenzenesulfonohydrazide (13.4 g, 71.95 mmol) dropwise. The resulting mixture was stiired at room temperature for 16 hrs. The white solid was collected by Alteration and washed with cyclohexane (20 mL) to give the crude desired product (18.0 g) as a white solid. Ή NMR (400 MHz, DMSC s): δ 11.91 (brs, 1H), 7.80 (d, J= 8.0 Hz, 2H), 7.42 (d, J= 8.0 Hz, 2H), 2.38 (s, 3H), 1.84(s, 3H).
Step 2

ferf-Butyl 3-(4-methyl-1H -1^2^-triazol-l-yI)pyrroIidine-l-carboxylate
To a solution of tert-b\i y\ 3-aminopyrrolidine-l-carboxylate (2.0 g, 10.74 mmol) in EtOH (50 mL) was added DIPEA (8.33 g, 64.43 mmol) at 0 °C. After stirring at 0 °C for 10 min, a solution of (N- (l,l-dichloropropan-2-ylidene)-4-methylbenzenesulfonohydrazide (4.12 g, 13.96 mmol) in MeCN was added to the cooled solution. The resulting mixture was stirred at room temperature for 16 hrs. After reaction, the mixture was concentrated. The residue was redissloved in EtOAc (50 mL) and washed with ¾0 (20 mL). The organic phase was dried over anhydrous Na2SO , filtered and concentrated. The residue was purified by flash column chromatography on silica gel eluting with 0-50% EtOAc in hexanes to give the desired product (2.1 g, 77% yield) as a brown oil. lH NMR (400 MHz, CDC13): 6 7.28 (s, 1H)5 5.15 -5.12 (m, 1H), 3.91 - 3.86 (m, 1H), 3.79 - 3.58 (m, 3H), 2.45 - 2.39 (m, 2H), 2.36 (s, 3H), 1.48 (s, 9H). Step 3

4-Methyl-l-(pyrrolidin-3-yl)-l/M^^-triazole hydrochloride
To a solution of terf-butyl 3-(4-methyl-1H -l,2,3-triazol-l-yl)pyrrolidine-l-carboxylate (1.0 g, 3.96 mmol) in EtOAc (2 mL) was added HCl/EtOAc (10 mL). The resulting mixture was stirred at room temperature for 2 hrs and concentrated to give the crude desired product (700 mg) as a brown solid. 1H NMR (400 MHz, CD3OD): δ 8.62 (s, 1H), 5.80 -5.75 (m, 1H), 3.98 (d, J= 4.8 Hz, 2H)5 3.64 - 3.60 (m, 2H), 2.81 - 2.67 (m, 2H), 2.55 (s, 3H). Step 4

(5-IsopropyI-ll/-pyrazol-3-yl)(3-(4-methyI-l/W
To a solution of 5-isopropyl-lH-pyrazole-3-carboxylic acid (160 mg, 1.04 mmol) and 4-methyl-l- (pyrrolidin-3-yl)-lH-l,2,3-triazole hydrochloride (196 mg, 1.04 mmol) in DMF (4 mL) was added HATU (593 mg, 1.56 mmol) and DIPEA (402 mg, 3.11 mmol). The mixture was stirred at room temperature for 16 hrs. The mixture was purified by prep.-HPLC (Gemini Cjg 150*25 mm*10 urn, 15-45% MeCN condition) to give the desired product (108 mg, 36% yield) as a white solid. !H NMR (400 MHz, MeOD): 5 7.86, 7.83 (s, 1H total), 6.52, 6.50 (2s, 1H), 5.33 - 5.29 (m, 1H), 4.46 - 4.42 (m, lH), 4.17 - 4.11 (m, 2H), 3.85 - 3.82 (m, 1H), 3.05-3.00 (m, 1H), 2.58 - 2.49 (m, 2H), 2.32, 2.31 (2s, 1H), 1.29, 1.27 (d, J= 6.8 Hz, 6H total). LCMS (LCMS Method C): Rt = 1.017 min, m/z: 289.1 [M+H]+.
Method D
Example 38
Step l

tert- utyl 3-(4-nitro-lii-pyrazol-l-y])pyrroIidine-l-carboxylate
To a solution of 4-nitro-1H -pyrazole (2 g, 17.68 mmol), terf -butyl 3-hydroxypyrrolidine-l- carboxylate (3.32 g, 17.68 mmol) and PPI13 (5.56 g, 21.2 mmol) in THF (20 mL) under a nitrogen atmosphere at 10 °C was added DIAD (5.3 g, 23 mmol). The reaction mixture was stirred at 10 °C for 16 hrs. The reaction mixture was concentrated and purified by flash column chromatography on silica gel, eluting with 0-50% EtOAc in hexanes to afford the desired product (4 g, 80% yield) as a white solid. LCMS (ESI) m/z: 305.1 [M+Na]+, RT = 1.04 min (LCMS Method A).
Step 2

4-nitro-l-(pyrrolidin-3-yl)-lii-pyrazole
To the reaction mixture of tert-butyl 3-(4-nitro-1H -pyrazol-l-yl)pynOlidine-l-carboxylate (400 mg
1.42 mmol) in EtOAc (8 mL) was added HCl/EtOAc (8 mL, 4M). The reaction mixture was stirred at 10 °C for 30 min. Then the reaction mixture was concentrated to afford the desired product (100 mg, 39% yield) as an off-white solid.
Step 3

(3-isopropyl-liy-pyi^ol-5-yl)(3-(4-nitro-1H -pyrazol-l-yl)pyrroUdiii-l-yI)methanone To the reaction mixture of 3-isopropyl-1H -pyrazole-5-carboxylic acid (360 mg, 2.2 mmol), HATU (1.0 g, 4.4 mmol) in DMF (10 mL) was added DIEA (851 mg, 6.6 mmol). The reaction mixture was stirred at 10 °C for 30 min, then 4-nitro-l-(pyrrolidin-3-yl)-1H -pyrazole (400 mg, 2.2 mmol) was added. The reaction mixture was stirred at 10 °C for 16 hrs. The reaction mixture was concentrated and purified by flash column chromatography on silica gel, eluting with 0-2% MeOH in DCM to afford the desired product (200 mg, 40% yield) as a yellow oil. LCMS (ESI) m/z: 319.1 [M+Na]\ RT = 0.655 min (LCMS Method A).
Step 4

(3-(4-amino-1H -pyrazol-l-yl)pyrrolidin-l-yl)(3-isopropyl-1H -pyrazol-5-yl)methanone
To the reaction mixture of (3-isopropyl-1H -pyrazol-5-yl)(3-(4-nitro-1H -pyrazol-l-yl)pynOlidm-l- yl)methanone (200 mg, 0.63 mmol) was added Pd/C (20 mg). The reaction mixture was stirred at 10 °C under a ¾ atmosphere for 16 hrs. The reaction mixture was filtered and the filtrate was concentrated and purified by preparative HPLC to afford the desired product (5 mg, 3% yield) as a white solid. 1H NMR (400 MHz, DMSO-i¾ ) δ 7.09 (s, 1H), 6.98 (s, 1H), 6.39 (s, 1H), 4.81 (m, 1H), 4.25 - 3.65(m, 4H), 3.10 - 2.90 (m, 1 H), 2.33 - 2.30 (m, 2H), 1.24 (d, J= 8 Hz, 6H). LCMS (ESI) m/z: 289.1 [M+H]+, RT = 0.95 min (LCMS Method B).
Example 39
Stcp l

Benzyl 3-((metliylsulfonyl)oxy)pyrroIidine-l-carboxyIate
To a stirred solution of benzyl 3-hydroxypyrrolidtne-l-carboxylate (500 mg, 2.26 mmol) in DCM (10 mL) was added triethylamine (686 mg, 0.94 mL, 6.78 mmol) at room temperature. The resulting mixture was cooled to 0 °C. Then methanesulfonyl chloride (388 mg, 3.39 mmol) was added dropwise into the mixture and the mixture was slowly warmed to room temperature over 2 hrs under N2 atomsphere. The mixture was diluted with DCM (20 mL) and washed with H2O (10 mL x 2), hydrochloric acid (10 ml x 2, IN), saturated NaHCO3 (10 mL). The organic phase was dried over anhydrous Na2SO4, filtered and concentrated to give the crude desired product (600 mg) as a brown oil which was used in the next step without further purification. Step 2

Benzyl 3-(2-oxopyridin-l (2H)-yl)pyrrolidine-l-carboxylate
To a solution of benzyl 3-((methylsulfonyl)oxy)pyrrolidine-l-carboxylate (1.0 g, 3.34 mmol), 2CO3 (692.5 mg, 5.01 mmol) in DMF (5 mL) was added pyridin-2(1H )-one (381.2 mg, 4.01 mmol) at room temperature. The mixture was stirred at 100 °C for 16 hrs under a N2 atmosphere. After cooling to room temperature, the reaction mixture was diluted with EtOAc (120 mL), washed with H2O (80 mL x 2) and brine (80 mL). The organic phase was dried over anhydrous Na2SO4, filtered and the filtrate was concentrated to dryness. The residue obtained was purified by silica gel flash column chromatography (0-25% EtOAc in hexanes and 0-3% MeOH in DCM) to give the crude desired product (200 mg) as a colourless oil, which was used to the next step directly without further purification.
Step 3

l-(PyrTolidin-3-yl)pyridin-2(l//)-oiie
To a solution of benzyl 3-(2-oxopyrid-n-l(2H)-yl)pyiTolidine-l-carboxylate (200 mg, 0.67 mmol) in EtOH (2 mL) was added Pd/C (50 mg, wet, 10%) and the mixture was stirred at room
temperature for 16 hrs under a ¾ balloon. The mixture was filtered and the filtrate was concentrated to dryness under reduced pressure to afford a mixture of the crude desired product (98 mg) as a colourless oil which was used to the next step directly without furhter purification.
Step 4

l-(l-(3-IsopropyI-1H -pyraz le-5-carbonyl)pyrrolidin-3-yl)pyridhi-2(1H )-one
To a stirred solution of 3-Isopropyl-1H -pyrazole-5-carboxylic acid (100 mg, 0.65 mmol) in DMF (2 mL) was added DIE A (252 mg, 1.95 mmol) and HATU (271 mg, 0.78 mmol) at room temperature. The mixture was stirred at room temperature for 10 min before the addition of the mixture l-(Pynolidin-3-yl)pyridin-2(1H )-one and l-(Pyrrolidin-3-yl)piperidin-2-one (98 mg). The mixture was stierred at room temperature for 2 hrs and purified by preparative HPLC to afford the desired product (16 mg, 8% yield) as white solid. LH NMR (400 MHz, CD3OD) δ 7.64 (t, J= 5.2 Hz, 1H), 7.58 - 7.47 (m, 1H), 6.62 - 6.55 (m, 1H), 6.52 (s, 1H), 6.48 - 6.40 (m, 1H), 5.49 - 5.44 (m, 1H), 4.47 - 4.37 (m, 0.5H), 4.25 - 4.16 (m, 1H), 4.12 - 4.07 (m, 1H), 3.94 - 3.75 (m, 1.5H), 3.08 - 3.00 (m, 1H), 2.53 - 2.30 (m, 2H), 1.30 (t, J= 6.8 Hz, 6H). LCMS (ESI) m/z: 301.1 [M+H]+, RT = 0.682 min
(LCMS method E).
Example 40
Step 1

teff-but l 3-((metby]sulfonyl)oxy)pyrrolidine-l-carboxylate
To a stirred solution of /erf-butyl 3-hydroxypyrrolidine-l-carboxylate (5 g, 27 mmol) in DC (20 mL) was added triethylarnine (8.1 g, 80 mmol) at room temperature. The mixture was cooled to 0 °C and then methanesulfonyl chloride (3.9 g, 34 mmol) was added dropwise into the mixture. The resulting mixture was slowly warmed to room temperature over 2 hrs under N2 atomsphere. The mixture was diluted with DCM (80 mL) and washed with ¾0 (80 mL), hydrochloric acid (80 ml x 2, IN), saturated NaHCO3 (80 mL). The organic phase was dried over anhydrous Na2SO4, filtered and concentrated to give the crude desired product (3.8 g, 54% yield) as colourless oil which was used in the next step without further purification. Step 2

toi-Butyl 3-(3-methyl-2-oxopyridiii-l(2H)-yl)pyrrolidine-l-carboxylate
To a solution of terf-Butyl 3-((methylsulfonyl)oxy)pyrrolidine-l-carboxylate (1.0 g, 3.77 mmol), 2CO3 (781.3 mg, 5.70 mmol) in DMF (7 mL) was added 3-Methylpyridin-2(lH)-one (493.6 mg, 4.52 mmol) at room temperature. The mixture was stirred at 100 °C for 16 hrs under a N2 atmosphere. After cooling to room temperature, it was diluted with EtOAc (120 mL), washed with H2O (80 mL x 2) and brine (80 mL). The organic phase was dried over lSJa2SO4, filtered and concentrated to dryness to afford the crude title compounds (800 mg) as yellow oil which was used to the next step directly without further purification.
Step 3

3-Methyl-l-(pyrrolidin-3-yl)pyridin-2(l /)-one
To a solution of the mixture of /erf-butyl 3-(3-methyl-2-oxopyridin-l(2H)-yl)pyrrolidine-l- carboxylate and tert-butyl 3-((3-methylpyridin-2-yl)oxy)pyrrolidine-l-carboxylate (800 mg, 2.87 mmol) in EtOAc (2 mL) was added dropwise HCl/EtOAc (2 mL, 8 mmol) and stirred at room temperature for 1 nr. The reaction mixture was concentrated and the residue obtained was washed with EtOAc (30 mL x 2) to afford the crude desired product (600 mg) as a yellow oil which was used to the next step directly without further purification.
Step 4

l^l-(3-Isopropyl-l i-pyrazole-S-carbonyl)pyrrolidin-3-yl)-3-methylpyridin-2(l -one
To a solution of 3-Isopropyl-1H -pyrazole-5-carboxylic acid (474 mg, 3.07 mmol) in DMF (3 mL) was added DIEA (1.44 g, 11.18 mmol) and HATU (1.28 g, 3.35 mmol) at room temperature. The mixture was stirred at room temperature for 10 min before the addition of the 3-Methyl-l- (pyrrolidin-3-yl)pyridin-2(l /)-one. The mixture was stirred at room temperature for 2 hrs and purified by preparative HPLC to afford the desired product (30 mg, 3.1% yield) as white solid. 1H NMR (400 MHz, CD3OD) δ (m, 1H), 7.39 (m, 1H), 6.52, 6.51 (2s, 1H), 6.37 - 6.33 (m, 1H), 5.51 - 5.42 (m, 1H), 4.44 - 4.39 (m, 0.5H), 4.25 - 4.04 (m, 2H), 3.98 - 3.72 (m, 1.5H), 3.08 - 2.99 (m, 1H), 2.52 - 2.26 (m, 2H), 2.13, 2.11 (2s, 3H), 1.30 (t, J= 6.6 Hz, 6H). LCMS (ESI) m/z: 315.2 [M+H]+, RT = 0.719 min (LCMS method E).
Example 41 Step l

(S)-terf-butyl 3-((methyIsulfonyl)oxy)pyrrolidine-l-carboxylate
To a cooled solution of teri-butyl (3S)-3-hydroxypyrrolidine-l-carboxylate (1 g, 80.11 mmol) in
DCM (150 mL) was added TEA (22.3 mL, 160.22 mmol) and MsCl (7.44 mL, 96.13 mmol). The reaction mixture was stirred at 28 °C for 1 hr. The reaction mixture was diluted in DCM (100 mL), washed with 1 N HC1 (100 mL), saturated NaHCO3 (100 mL) and brine (100 mL). The organic layer was dried over anhydrous Na2SO4, filtered and concentrated to afford the desired product
(19.4 g, 91% yield) as a yellow oil. 1H NMR (400 MHz, CDC13) δ 5.30 - 5.26 (m, 1H), 3.68 - 3.43 (m, 4H), 3.05 (s, 3H), 2.27 - 2.13 (m, 2H), 1.46 (s, 9H).
Step 2

(R)-tert-buty\ 3-(3-bromo-2-oxopyridin-l (2 /)-yl)p rrolidine-l -carboxy late
A mixture of 3-bromo-1H -pyridin-2-one (9 g, 51.72 mmol), terr-butyl (3S)-3-methylsulfonyl oxypyrrolidine-l-carboxylate (16.47 g, 62.07 mmol) and K2CO3 (14.28 g, 103.45 mmol) in DMF (90 mL) was stirred at 80 °C for 12 hours. The reaction mixture was diluted in water (150 mL), washed with EtO Ac (150 mL x 2). The combined organic layers were dried over anhydrous Na2SO4, filtered and concentrated. The residue was purified by silica gel flash column
chromatography (petroleum ether EtOAc = 10/1) to afford the desired product (4.2 g, 24% yield) as a colorless oil. Step 3

(R)-3-bromo-l-(pyrrohdin-3-yl)pyridin-2(1H )-one
To a stirred solution of tert-butyl (3jf?)-3-(3-bromo-2-oxo-l -pyridyl)pyrrolidine-l-carboxylate (4.2 g, 12.24 mmol) in methanol (40 mL) was added HCl/MeOH (10 mL) in an ice bath. The reaction was stirred at 30 °C for 2 hrs. The solvent was removed under reduced pressure to give the crude product (3 g) which was used in next step without further purification.
Step 4

(^) -bromo-l-(l-(3-isopropyl-l-ff-pyrazole-5-car^
A mixture of (i?)-3-bromo-l-(pyrrolidin-3-yl)pyridin-2(l /)-one (3 g, 12.34 mmol), 3-isopropyl- lH-pyrazole-5-carboxylic acid (2.1 g, 13.58 mmol), HATU (5.6 g. 14.81mmol) and DIEA (6.58 mL, 37.02 mmol) in DMF (30 mL) was stirred at room temperature for 12 hrs. The mixture was
diluted with EtOAc (60 mL), washed with brine (60 mL x 2). The organic layer was dried over anhydrous Na2SO4, filtered and concentrated. The crude residue was purified by preparative TLC (DC : MeOH = 50: 1) to afford the desired product (1.6 g, 34% yield) as a light yellow solid. LCMS M/Z (M+Na) = 402.8.
Step 5

(Ji)-3-cyclopropyl-l-(l-(3-isopropyl-l//-pyrazole-^^
one
A mixture of 3-bromo- 1 -[(3Λ)- 1 -(3-isopropyl- 1H -pyrazole-5-carbonyl)pyrrolidin-3-yl]pyridin-2- one (200 mg, 0.53 rnmol), cyclopropylboronic acid (54 rag, 0.63 mmol), Pd(dppf)Cl2 (39 mg, 0.05 mmol) and Na2CC>3 (168 mg, 1.58 mmol) in 1,4-dioxane (5 mL) and water (1 mL) was stirred at 100 °C for 12 hrs under a N2 atmosphere. After cooling to room temperature, the reaction mixture was diluted in water (20 mL), washed with DCM (20 mL x 2). Combined organic layers were dried over anhydrous Na2SO4, filtered and concentrated. The crude residue was purified by preparative HPLC to afford the desired product (31 mg, 17% yield) as a white solid. 'Η NMR (400 MHz, DMSO-4 δ 7.46 - 7.44 (m, 1H); 7.10 - 7.08 (m, 1H), 6.53 (s, 1H), 6.37 - 6.32 (m, 1H), 5.51 - 5.46 (m, 1H), 4.44 - 3.77 (m, 4H), 3.06 - 3.01 (m, 1H), 2.50 - 2.32 (m, 2H), 2.06 - 2.01 (m, 1H), 1.30 (m, 6H), 0.95 - 0.90 (m, 2H), 0.63 - 0.58 (m, 2H). LCMS (ESI) M/Z (M+H) = 341.1, RT = 0.762 min (LCMS method E).
Example 42 Step 1

(Jf^l-il-iS-isoprop l-1H -p razole-S-carbon lJp iTolidin-S- ^-S-i rop-l-en- - l) pyridin-
2(l#)-one
A mixture of 3-bromo-l-[(3i2)-l-(3-isopropyl-1H -pyrazole-5-carbonyl)pyiTolidin-3-yl]pyri one (300 mg, 0.79 mmol), potassium trifluoro(isopropenyl)boranuide (140 mg, 0.95 mmol), Pd(OAc)2 (27 mg, 0.12 mmol), RuPhos (111 mg, 0.24 mmol) and K3P04 (504 mg, 2.37 mmol) in toluene (5 mL) and water (0.5 mL) was stirred at 110 °C for 12 hrs under a N2 atmosphere. After cooling to room temperature, the reaction mixture was diluted in water (20 mL), and then extracted with DCM (20 mL x 2). The combined organic layers were dried over anhydrous Na2SO4, filtered and concentrated. The crude residue was purified by silica gel flash column chromatography (DCM/MeOH = 30/1) to afford the desired product (90 mg, 25% yield) as a light yellow solid. LCMS M/Z (M+H) = 340.9.
Step 2

(Jt)-3-isopropyI-l-(l-(3-isopropyl-l-ff-pyrazole»5-carbonyI)pyiToUdin-3-yI)pyrid
A mixture of 3-isopropenyl-l-[(3i?)-l-(3-isopropyl-1H -pyrazole-5-carbonyl)pyrrolidin-3- yl]pyridin-2-one (90 mg, 0.26 mmol) and Pd/C (45 mg) in ethyl acetate (2 mL) was stirred at room temperature for 12 hrs under an atmposhere of H2 balloon. The reaction was filtered over a short of Celite pad. The filtrate was concentrated and the crude residue was purified by preparative HPLC to afford the desired product (38 mg, 32% yield) as a white solid. Ή NMR (400 MHz, DMSO-c/6) δ 7.49 - 7.47 (m, 1H), 7.40 - 7.37 (m, 1H), 6.51 (s, 1H), 6.42 - 6.39 (m, 1H), 5.50 - 5.46 (m, 1H), 4.44 - 4.07 (m, 4H), 3.16 - 3.03 (m, 2H), 2.47 - 2.31 (m, 2H), 1.30 (m, 6H), 1.18 (m, 6H). LCMS (ESI) M Z (M+H) = 343.1 , RT = 0.798 min (LCMS method E).
Example 43 Step 1

(if)-l-(l-(3-isopropyl-ljff-pyi^ole-5-carbonyl)pyrroIidin-3-yl)-3-vinylpyridin-2(li^
A mixture of 3-bromo-l -[(3Λ)-1 -(3-isopropyl-l H-pyrazole-5-carbonyl)pyrrolidin-3-yl]pyridin-2-
one (200 mg, 0.53 mmol), potassium trifluoro(vinyl)boranuide (141 mg, 1.05 mmol), Pd(dppf)Cl2 (39 mg, 0.05 mmol) and Na2CO3 (168 mg, 1.58 mmol) in 1,4-dioxane (5 mL) and water (1 mL) was stirred at 110 °C for 12 hrs, under a N2 atmosphere. After cooling to room temperature, the reaction mixture was diluted in water (20 mL), and extracted with DCM (20 mL x 2). The combined organic layers were dried over anhydrous Na2SO4, filtered and concentrated. The crude residue was purified by silica gel flash column chromatography (DCM/MeOH = 30/1) to afford the desired product (50 mg, 29% yield) as a light yellow solid.
Step 2

(J?)-3-ethyl-l-(l-(3-isopropyl-l^-pyi^oIe-5-carbonyl)pyrrolidin-3-yl)pyridin-2(lii)-one
A mixture of l-[(3 Z)-l-(3-isorropyl-1H ^yrazole-5-carbonyl)pyrrolidin-3-yl]-3-vinyl-pyridin-2- one (50 mg, 0.15 mmol), Pd/C (25 mg, 0.1500mmol) in ethyl acetate (2 mL) was stirred at room temperature for 12 hrs, under 1 arm of H2. The reaction mixture was filtered over a short Celite pad and then the filtrate was concentrated. The resulting residue was purified by preparative HPLC to afford the desired product (35 mg, 70% yield) as a white solid. Ή NMR (400 MHz, DMSO-ife) 6 7.49 - 7.46 (m, 1H), 7.36 - 7.35 (m, 1H), 6.49 (s, 1H), 6.38 - 6.35 (m, 1H), 5.48 - 5.43 (m, 1H), 4.17 - 3.78 (m, 4H), 3.03 - 3.01 (m, lH), 2.54 - 2.49 (m, 2H), 2.42 - 2.31 (m, 2H), 1.30 (m, 6H), 1.17 - 1.13 (m, 3H). LCMS (ESI) M/Z (M+H) = 329.0, RT = 0.626 min (LCMS method E).
Using the General Synthetic Method (Syn. Met.) and the General LCMS Method shown, the following compounds were also prepared.
Table 1

















1







2

4
5









4


6


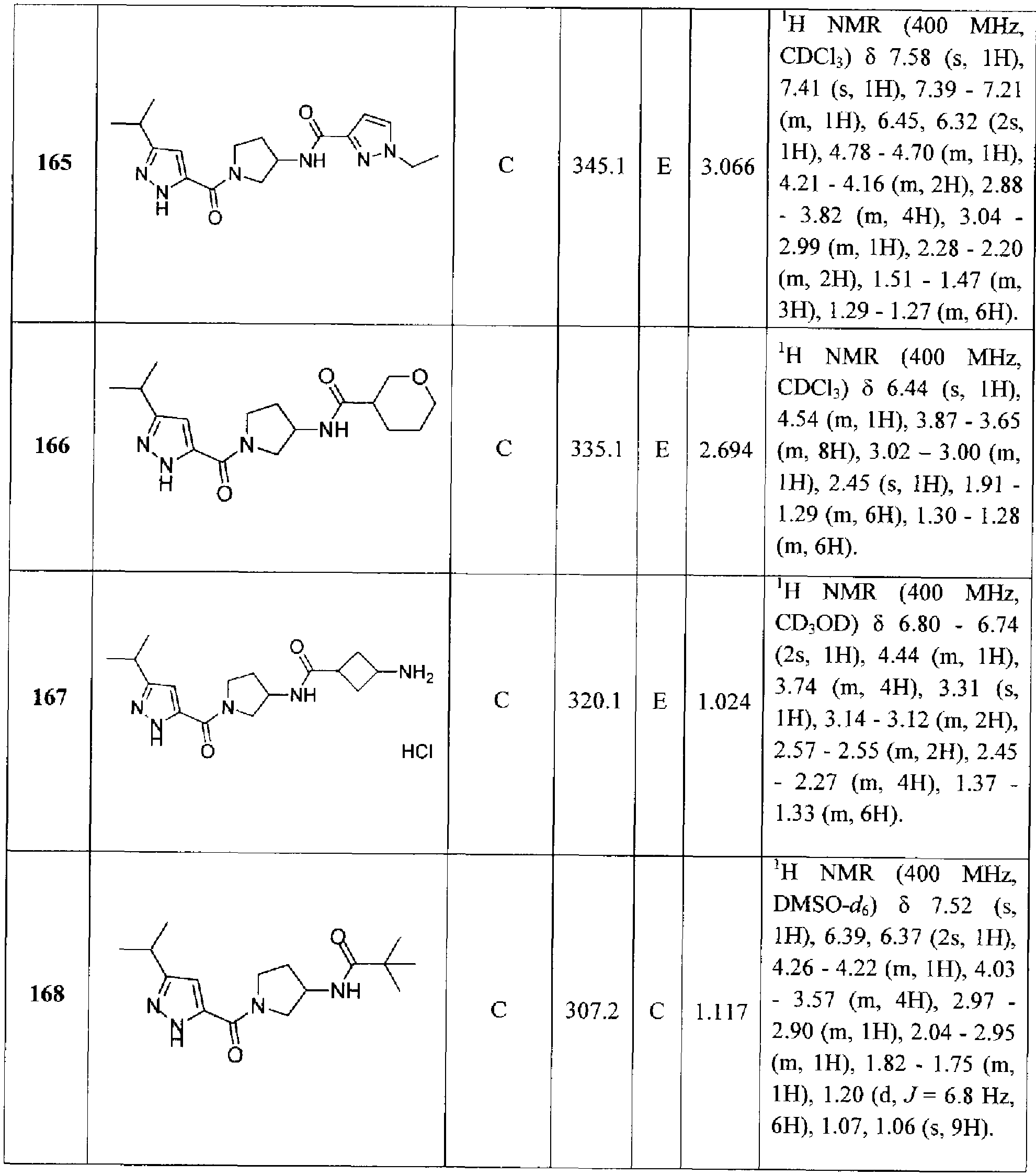



2





167





2











EXAMPLE 248
KDM5 Enzyme Assay Procedure
Full length KDM5A enzyme was expressed and purified inhouse. Biotin-H3K4me3 peptide was purchased from Mew England Biolabs. HTRF reagents (containing Eu-labeled H3K4mel-2
antibody, and streptavidin-XL665) were purchased from Cis-Bio International. Plates were read on an Envision multi-label plate reader (Perkin Elmer).
The HTRF assay mixture contained 2 nM full length KDM5A enzyme, 100 nM H3K4Me3 peptide substrate, 1 uM 2-OG, 100 uM Fe2+, 500 uM ascorbate. 50 mM HEPES pH 7.0 buffer, 0.01% Triton-X 100, 2 mM DTT, 0.25 % DMSO at a final volume of 10 uL. The enzyme reaction was carried out at room temperature in black Proxiplate 384-Plus plate (Corning, Costar) within 30 minutes, in the presence of varying concentration of a test compound. At the end of enzyme reaction, 5 uL of 1 mM EDTA were added to quench the reaction and then the detection reagents (5 uL) were added to give final concentrations of 0.5 nM Eu-labeled H3K4mel-2 antibody, and 50 nM streptavidin-XL665. The plates were incubated at room temperature for 60 minutes and then read in the Envision plate reader. The readouts were transformed into % inhibition, and IC50 value of a test compounds was generated by using four parameters curve fitting (Model 205 in XLFIT5, iDBS).
EXAMPLE 249
KDM5 Cell Assay Procedure:
PC9 cells were seeded in a 384 well plate (2000 cells/well) with a test compound and incubated for 120 hours at 37 °C. H3K4Me3 mark level was assessed using HTRF reagents from CisBio. Briefly, media was removed and cells were lysed in 20 pL of Epigeneous Cell Histone lysis buffer C for 45 min at 26 °C, shaking. 10 pL of 4μg/mL acceptor antibody (H3K4me3-d2) and 10 μΐ. of 1.2pg/mL donor antibody (Total H3-K) in Cell Histone detection buffer were added and the mixture was incubated at 26 °C for 1 hour. Assay plate was read subsequently on Envision (Perkin Elmer). Each compound was run in duplicate. Data were normalized to DMSO treated wells as the low response and EC5o's were calculate using a four- parameter fit.
Data for the compounds of Examples 1-247 from the assays described in Examples 248 and
249 is provided in the following table.
Table 2


1






192

1







2

2


2

2

2

2


2


210

2

2

2

2

2

216

2


2

2


222


2

225

While a number of embodiments have been described, these examples may be altered to provide other embodiments that utilize the compounds and methods described herein. Therefore, the scope of this invention is to be defined by the appended claims rather than by the specific embodiments that have been represented by way of example.
Claims
We claim:
1. A compound of formula (I):
,R3
*R2

O
I
or a salt thereof, wherein:
A is selected from the group consisting of:
R1


R1 is halo, -N(R )2, C1-6alkyl, C2-6alkenyl, C2-6alkynyl, 3-8 membered carbocyclyl, C1- 6alkoxy, 5-10 membered aryl, 5-10 membered heteroaryl, or 3-8 membered heterocyclyl, wherein said Chalky., C1-6alkenyl, C1-6alkynyl, 3-8 membered carbocyclyl, C1-6alkoxy, 5-10 membered aryl, 5-10 membered heteroaryl, and 3-8 membered heterocyclyl are optionally substituted with one or more groups independently selected from the group consisting of oxo, hydroxy, halo, C1- 3alkoxy, 3-8 membered carbocyclyl, 5-10 membered aryl, -N(R )2, -N(Rx)C(O)R , and C1- 3alkyl; each R is independently selected from the group consisting of H and C1-6alkyl, that is optionally substituted with one or more groups independently selected from the group consisting of halo, hydroxy, and C1-3alkoxy;
R2 is 5-10 membered carbocyclyl, 5-10 membered heterocyclyl, 5-10 membered aryl, 5-10 membered heteroaryl, -ORa, -C(O)N(Ra)2, or NRaRb, wherein each 5-10 membered carbocyclyl, 5- 10 membered heterocyclyl, 5-10 membered aryl, and 5-10 membered heteroaryl is optionally substituted with one or more groups Rd;
Ra and R are each independently selected from the group consisting of H, C1-6alkyl, C2. 6alkenyl, C2-6alkynyl, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 5-10 membered aryl, 5-10 membered heteroaryl -C(O)Rc, -CO2Rc, -C(O)N(Rc)2, -C(O)SRc, and -C(O)C(O)Rc, wherein each C1-6alkyl, C2-6alkenyl, C2-&alkynyl, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 5- 10 membered aryl, and 5-10 membered heteroaryl is optionally substituted with one or more groups independently selected from the group consisting of halo, nitro, cyano, oxo, CMalkyl, C2-4alkenyl, C2-4alkynyl, 3-8 membered carbocyclyl,-ORc, -SRC, -N(RC)2, -C(O)Rc, -CO2Rc, -C(O)N(Rc)2,
-C(O)SRc, -C(O)C(O)Rc, -S(O)Rc, -SO2Rc, -SO2N(RC)2, ~N(Rc)C(O)Rc, -NCR^C OiNCR^, - N(R°)SO2RC, -N(Rc)SO2N(Rc)2, -N(RC)N(RC)2, -N(RC)C(=N(RC))N(RC)2, -C(=N)N(RC)2, -C=NORc, and -C(=N(Rc))N(Rc)2;
each Rc is independently selected from the group consisting of H, C1-6alkyl, C2-6alkenyl, C2. ealkynyl, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 5-10 membered aryl and 5-10 membered heteroaryl, wherein each Cj-salkyl, C2-6alkenyl, C _6alkynyl, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 5-10 membered aryl and 5-10 membered heteroaryl is optionally substituted with one or more groups R ;
each Rd is independently selected from the group consisting of halo, nitro, cyano, oxo, Ci_ ealkyl, C2.6alkenyl, C2-6alkynyl, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 5-10 membered aryl, 5-10 membered heteroaryl, ,-ORe, -SRe, -N{Re)2, -C(O)Rc, -CO2Re, -C(O)N(Re)2, -C(O)SRc, -C(O)C(O)Re, -S(O)Re, -SO2Re, -SO2N(Rc)2, -N(Re)C(O)Re, -N(Re)C(O)N(Re)2, -N(Re)SO2Re, -N(Re)SO2N(Re)2, -N(Re)N(Re)2, -N(Re)C(=N(Re))N(Re)2, -C(=N)N(Re)2, -C=NORe, and -C(=N(Re))N(Re)2, wherein each C1-6aHcyl, C2-6alkenyl, C2-6alkynyl, 3-8 membered
carbocyclyl, 3-8 membered heterocyclyl, 5-10 membered aryl and 5-10 membered heteroaryl is optionally substituted with one or more groups independently selected from R ;
each Rc is independently selected from the group consisting of H, C1-6alkyl, C2-6alkenyl, C2- fialkynyl, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 5-10 membered aryl and 5-10 membered heteroaryl, wherein each C1-6alkyl, C2-6alkenyl, C2-6alkynyl, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 5-10 membered aryl and 5-10 membered heteroaryl is optionally substituted with one or more groups independently selected from the group consisting of Cj^alkyl, C2-4alkenyl, C2-4alkynyl, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 5-10 membered aryl and 5-10 membered heteroaryl;
each R is independently selected from the group consisting of halo, nitro, cyano, oxo, C[- 4alkyl, C2-4alkenyl, C^alkynyl, 3-8 membered carbocyclyl,-OR6, -SRg, -N(Rg)2, -C(O)Rg, -CO2Rg, -C(O)N(Rg)2, -C(O)SRg -C(O)C(O)Rg, -S(O)Rg, -SO2Rg, -SO2N(Rg)2, -N(Rg)C(O)Rg
-N(Rg)C(O)N(Rg)2, -N(Rg)SO2Rg, -N(Rg)SO2N(Rg)2, -N(Rg)N(R )2, -N(RE)C(=N(Rg))N(Rg)2, - CC^NtR^, and -C=NORg, -C(=N(Rg))N(R6)2, wherein each C alkyl, Qwalkenyl, C^alkynyl, and 3-8 membered carbocyclyl is optionally substituted with one or more groups independently selected from the group consisting of halo, cyano, oxo, 3-8 membered carbocyclyl,-ORg, -N(R )2, -C(O)R -CO2RB, -C(O)N(Rg)2, -SO2R -SO2N(Rg)2, and -N(Rg)C(O)Rg;
each Rg is independently selected from the group consisting of H, C1-6alkyl, C2-6alkenyl, C2- ealkynyl, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 5-10 membered aryl and 5-10 membered heteroaryl, wherein each C1-6alkyl, C2-6alkenyl, C2-6alkynyl, 3-8 membered carbocyclyl,
3-8 membered heterocyclyl, 5-10 membered aryl and 5-10 membered heteroaiyl is optionally substituted with one or more groups independently selected from the group consisting of C1-4alkyl, C2-4alkenyl, C2-4alkyn l, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 5-10 membered aryl and 5-10 membered heteroaryl; and
each Rh is independently selected from the group consisting of halo, nitro, cyano, oxo, d- 4alkyl, C2-4aIken l, C2-4alkynyl, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 5-10 membered aryl, 5-10 membered heteroaryl, -N(Rk)2, and ,-ORk, wherein each C1-4aHcyl, C2.
4alkenyl, C2-4alkynyl, 3-8 membered carbocyclyl, 3-8 membered heterocyclyl, 5-10 membered aryl and 5-10 membered heteroaryl is optionally substituted with one or more groups independently selected from the group consisting of halo, hydroxy, Ci^alkoxy, cyano, oxo, C alkyl, C2-4alkenyl, C2-4alkynyl,3-8 membered carbocyclyl, and 5-10 membered aryl;
each Rk is independently selected from the group consisting of H, C alkyl, C2-4alkenyl, C2- 4alkynyl, 3-8 membered carbocyclyl, and 5-10 membered aryl wherein any C alkyl, C2-6alkenyl, C2-4alkynyI, 3-8 membered carbocyclyl, and 5-10 membered aryl carbocyclyl is optionally substituted with one or more groups independently selected from the group consisting of halo, cyano, oxo hydroxy, and 3-8 membered carbocyclyl; and
R3 is H or C1-6alkyl;
R4 is H, CMalkyl, C2-6alkenyl, C2-6alk nyl, or 3-8 membered carbocyclyl, 5-10 membered aryl, 5-10 membered heteroaryl wherein each C1-6alkyl, C2-6alkenyl, C2-6alkynyl, and 3-8 membered carbocyclyl, 5-10 membered aryl, 5-10 membered heteroaryl is optionally substituted with one or more groups independently selected from halo, hydroxy, cyano, C1-6alkoxy, and 3-8 membered carbocyclyl; and
R5 is H, halo, or CMalkyl, and R6 is H, CMalkyl, C2-ealkenyl, Cj-salkynyl, halo, hydroxy, or 3-8 membered carbocyclyl, wherein each CMalkyl, C2-6alkenyl, C2-6alkynyl, and 3-8 membered carbocyclyl, is optionally substituted with one or more groups independently selected from halo, hydroxy, cyano, C1-6alkoxy, and 3-8 membered carbocyclyl; or R5 and R6 taken together with the atom to which they are attached form a 3-8 membered carbocyclyl or a 3-8 membered heterocyclyl, which 3-8 membered carbocyclyl and 3-8 membered heterocyclyl are optionally substitutes with one or more groups independently selected from halo, hydroxy, cyano, C1-6alkoxy, and 3-8 membered carbocyclyl
provided the compound is not:

O
2. The compound of claim 1 which is a compound of formula (la):

or a salt thereof.
3. The compound of claim 1 wherein R is Cj-ealkyl, wherein said C1-6alk l is optionally substituted with one or more groups independently selected from the group consisting of oxo, hydroxy, halo, C1- 3alkoxy, 3-8 membered carbocyclyl. 5-10 membered aryl, -N(Ra)2,
-N(Ra)C(O)Ra, and C1- 3alkyl.
4. The compound of claim 1 wherein R1 is halo, -N(Ra)2, 3-8 membered carbocyclyl, C1- ealkoxy, 5-10 membered aryl, wherein said 3-8 membered carbocyclyl and 5-10 membered aryl are optionally substituted with one or more groups independently selected from the group consisting of oxo, hydroxy, halo, C1- 3alkoxy, 3-8 membered carbocyclyl, 5-10 membered aryl, -N(Ra)2,
-N(Ra)C(O)Ra, and C1- 3alkyl.
5. The compound of claim 1 wherein R1 is bromo, cyclohexyl, isopropyl, isobutyl, cyclopentyl, 1-methoxyethyl, cyclopropyl, cyclobutyl, amino, 4-phenylbut-2-yl, butyl, phenethyl, cyclopentyl, 1 -(acetylamino)ethyl, or l-(hydroxymethylcarbonylamino)ethyl.
6. The compound of claim 1 wherein Rl is isopropyl.
7. The compound of claim 1 which is a compound of formula (lb):

compound of claim 7 wherein R is H, methyl, or isopropyl.

231





10. The compound of any one of claims 1-8 wherein R2 is 5- 10 membered carbocyclyl that is optionally substituted with one or more groups Rd.
11. The compound of any one of claims 1 -8 wherein R2 is 5-10 membered heterocyclyl, that is optionally substituted with one or more groups Rd.
The compound of any one of claims 1-8 wherein R2 is selected from the group consisting

14. The compound of any one of claims 1 -8 wherein R2 is:

15. The compound of any one of claims 1 -8 wherein R is 5- 10 membered heteroaryl that is optionally substituted with one or more groups Rd.
16. The compound of any one of claims 1-8 wherein R2 is selected from the group consisting of:

2

238

239

17. The compound of any one of claims 1-8 wherein R2 is -ORa.
18. The compound of any one of claims 1 -8 wherein R2 is:

i-O
The compound of any one of claims 1-8 wherein R2 is NRaRb.
The compound of any one of claims 1-8 wherein R2 is selected from the group consisting

2


242
The compound of any one of claims 1-8 wherein R2 is -C(O)N(Ra)2.
The compound of any one of claims 1-5 wherein R2 is selected from the group consisting

23. The compound of any one of claims 1-22 wherein R is H.
24. The compound of any one of claims 1-22 wherein R is methyl.
25. The compound of any one of claims 1-24 wherein R5 is H
26. The compound of any one of claims 1 -25 wherein R is Ci_6alkyl or hydroxy.
27. The compound of any one of claims 1 -25 wherein R6 is methyl or hydroxy.
28. The compound of any one of claims 1-25 wherein R6 is H.
29. The compound of any one of claims 1 -24 wherein R5 and R6 taken together with the atom to which they are attached form a 3-8 membered carbocyclyl or a 3-8 membered heterocyclyl, which 3-8 membered carbocyclyl and 3-8 membered heterocyclyl are optionally substitutes with one or more groups independently selected from halo, hydroxy, cyano, C|.6alkoxy, and 3-8 membered carbocyclyl.
30. The compound of any one of claims 1 -24 wherein R5 and R6 taken together with the atom to which they are attached form a cyclopropyl ring.
The compound of claim 1 which is selected from the group consisting of:

244


246



249

20

251




O
and salts thereof.
32. A composition comprising a compound as described in any one of claims 1-31 or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable adjuvant, carrier, or vehicle.
33. The composition according to claim 32, in combination with an additional therapeutic agent.
34. The composition according to claim 33, wherein the additional therapeutic agent is a chemotherapeutic agent.
35. A method of increasing efficacy of a cancer treatment comprising a cytotoxic agent in an individual comprising administering to the individual (a) an effective amount of a compound as described in any one of claims 1-31 or a pharmaceutically acceptable salt thereof, and (b) an effective amount of the cytotoxic agent.
36. A method of treating an individual with cancer who has an increased likelihood of developing resistance to a cytotoxic agent comprising administering to the individual (a) an effective amount of a compound as described in any one of claims 1-31 or a pharmaceutically acceptable salt thereof, and (b) an effective amount of the cytotoxic agent.
37. A method of treating cancer in an individual comprising administering to the individual (a) a a compound as described in any one of claims 1-31 or a pharmaceutically acceptable salt thereof, and (b) cytotoxic agent.
38. The method of claim 37, wherein the respective amounts of the compound as described in any one of claims 1-31 or a pharmaceutically acceptable salt thereof and the cytotoxic agent are effective to increase the period of cancer sensitivity and/or delay the development of cell resistance to the cytotoxic agent.
255
3 . A method of increasing efficacy of a cancer treatment comprising a cytotoxic agent in an individual comprising administering to the individual an effective amount of a compound as described in any one of claims 1-31 or a pharmaceutically acceptable salt thereof.
40. A method of treating cancer in an individual wherein cancer treatment comprises administering to the individual (a) an effective amount of a compound as described in any one of claims 1-31 or a pharmaceutically acceptable salt thereof and (b) a cytotoxic agent, wherein the cancer treatment has increased efficacy compared to a standard treatment comprising administering an effective amount of the cytotoxic agent without (in the absence of) the compound as described in any one of claims 1-31 or the pharmaceutically acceptable salt thereof,
41. A method of delaying and/or preventing development of cancer resistant to a cytotoxic agent in an individual, comprising administering to the individual an effective amount of a compound as described in any one of claims 1-31 or a pharmaceutically acceptable salt thereof.
42. A method of treating an individual with cancer who has increased likelihood of developing resistance to a cytotoxic agent comprising administering to the individual (a) an effective amount of a compound as described in any one of claims 1-31 or a pharmaceutically acceptable salt thereof and (b) an effective amount of the cytotoxic agent.
43. A method of increasing sensitivity to a cytotoxic agent in an individual with cancer comprising administering to the individual an effective amount of a compound as described in any one of claims 1-31 or a pharmaceutically acceptable salt thereof.
44. A method of extending the period of a cancer therapy agent sensitivity in an individual with cancer comprising administering to the individual an effective amount of a compound as described in any one of claims 1-31 or a pharmaceutically acceptable salt thereof.
45. A method of extending the duration of response to a cancer therapy in an individual with cancer comprising administering to the individual an effective amount of a compound as described in any one of claims 1-31 or a pharmaceutically acceptable salt thereof.
46. The method of any one of claims 38, 39, 41 , and 43 wherein the method further comprises (b) administering to the individual an effective amount of the cytotoxic agent.
47. The method of any one of claims 35-46, wherein the cytotoxic agent is a chemotherapeutic agent.
48. The method of claim 47 wherein the chemotherapeutic agent is a taxane.
49. The method of claim 48, wherein the taxane is paclitaxel or docetaxel.
50. The method of claim 47 wherein the chemotherapeutic agent is a platinum agent.
51. The method of any one of claims 35-46, wherein the cytotoxic agent is selected from anti- microtubule agents, platinum coordination complexes, alkylating agents, antibiotic agents, topoisomerase II inhibitors, antimetabolites, topoisomerase I inhibitors, hormones and hormonal analogues, signal transduction pathway inhibitors, non-receptor tyrosine kinase angiogenesis inhibitors, irnmunotherapeutic agents, proapoptotic agents, ; inhibitors of LDH-A; inhibitors of fatty acid biosynthesis; cell cycle signaling inhibitors; HDAC inhibitors, proteasome inhibitors; and inhibitors of cancer metabolism.
52. The method of claim 47 wherein the chemotherapeutic agentis an antagonist of EGFR.
53. The method of claim 52, wherein the antagonist of EGFR is N-(3-ethynylphenyl)-6,7-bis(2- methoxyethoxy)quinazolin-4-amine or a pharmaceutically acceptable salt thereof.
54. The method of claim 47, wherein the chemotherapeutic agentis a RAF inhibitor.
55. The method of claim 54, wherein the RAF inhibitor is a BRAF and/or CRAF inhibitor.
56. The method of claim 54, wherein the RAF inhibitor is vemurafenib.
57. The method of claim 47 wherein the chemotherapeutic agent is a PI3 inhibitor.
58. A method of treating a proliferative disorder in an individual comprising administering to the individual an effective amount of a compound as described in any one of claims 1-31 or a pharmaceutically acceptable salt thereof.
59. The method of claim 58 wherein the proliferative disorder is lung cancer, melanoma, colorectal cancer, pancreatic cancer, and/or breast cancer.
60. A compound as described in any one of claims 1 -31 , or a pharmaceutically acceptable salt thereof, for use in medical therapy.
61. A compound as described in any one of claims 1 -31 , or a pharmaceutically acceptable salt thereof, for the prophylactic or therapeutic treatment of a proliferative disorder.
62. The use of a compound as described in any one of claims 1 -31 , or a pharmaceutically acceptable salt thereof, to prepare a medicament useful for treating a proliferative disorder.
63. The compound or use of any one of claims 61-62 wherein the proliferative disorder is lung cancer, melanoma, colorectal cancer, pancreatic cancer, and/or breast cancer.
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP15784257.6A EP3204379B1 (en) | 2014-10-10 | 2015-10-09 | Pyrrolidine amide compounds as histone demethylase inhibitors |
| JP2017518802A JP6783230B2 (en) | 2014-10-10 | 2015-10-09 | Pyrrolidone amide compounds as inhibitors of histone demethylase |
| CN201580066844.9A CN107912040B (en) | 2014-10-10 | 2015-10-09 | Pyrrolidine amide compounds as histone demethylase inhibitors |
| US15/482,584 US10022354B2 (en) | 2014-10-10 | 2017-04-07 | Pyrrolidine amide compounds as histone demethylase inhibitors |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN2014088309 | 2014-10-10 | ||
| CNPCT/CN2014/088309 | 2014-10-10 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US15/482,584 Continuation US10022354B2 (en) | 2014-10-10 | 2017-04-07 | Pyrrolidine amide compounds as histone demethylase inhibitors |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016057924A1 true WO2016057924A1 (en) | 2016-04-14 |
Family
ID=54337935
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2015/054949 WO2016057924A1 (en) | 2014-10-10 | 2015-10-09 | Pyrrolidine amide compounds as histone demethylase inhibitors |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US10022354B2 (en) |
| EP (1) | EP3204379B1 (en) |
| JP (1) | JP6783230B2 (en) |
| CN (1) | CN107912040B (en) |
| HK (1) | HK1253559A1 (en) |
| WO (1) | WO2016057924A1 (en) |
Cited By (50)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017205115A1 (en) | 2016-05-27 | 2017-11-30 | Gilead Sciences, Inc. | Compounds for the treatment of hepatitis b virus infection |
| WO2017205078A1 (en) | 2016-05-27 | 2017-11-30 | Gilead Sciences, Inc. | Methods for treating hepatitis b virus infections using ns5a, ns5b or ns3 inhibitors |
| WO2018039531A1 (en) | 2016-08-26 | 2018-03-01 | Gilead Sciences, Inc. | Substituted pyrrolizine compounds and uses thereof |
| WO2018045144A1 (en) | 2016-09-02 | 2018-03-08 | Gilead Sciences, Inc. | Toll like receptor modulator compounds |
| WO2018045150A1 (en) | 2016-09-02 | 2018-03-08 | Gilead Sciences, Inc. | 4,6-diamino-pyrido[3,2-d]pyrimidine derivaties as toll like receptor modulators |
| WO2018083288A1 (en) | 2016-11-07 | 2018-05-11 | Bayer Aktiengesellschaft | Substituted sulfonyl amides for controlling animal pests |
| WO2018144605A1 (en) | 2017-02-02 | 2018-08-09 | Gilead Sciences, Inc. | Compounds for the treatment of hepatitis b virus infection |
| WO2018144390A1 (en) | 2017-01-31 | 2018-08-09 | Gilead Sciences, Inc. | Crystalline forms of tenofovir alafenamide |
| WO2018195321A1 (en) | 2017-04-20 | 2018-10-25 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| WO2019160882A1 (en) | 2018-02-13 | 2019-08-22 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| WO2019165374A1 (en) | 2018-02-26 | 2019-08-29 | Gilead Sciences, Inc. | Substituted pyrrolizine compounds as hbv replication inhibitors |
| WO2019195181A1 (en) | 2018-04-05 | 2019-10-10 | Gilead Sciences, Inc. | Antibodies and fragments thereof that bind hepatitis b virus protein x |
| WO2019193543A1 (en) | 2018-04-06 | 2019-10-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3'3'-cyclic dinucleotides |
| WO2019193533A1 (en) | 2018-04-06 | 2019-10-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'2'-cyclic dinucleotides |
| WO2019193542A1 (en) | 2018-04-06 | 2019-10-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'3'-cyclic dinucleotides |
| WO2019200247A1 (en) | 2018-04-12 | 2019-10-17 | Precision Biosciences, Inc. | Optimized engineered meganucleases having specificity for a recognition sequence in the hepatitis b virus genome |
| WO2019204609A1 (en) | 2018-04-19 | 2019-10-24 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| WO2019211799A1 (en) | 2018-05-03 | 2019-11-07 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'3'-cyclic dinucleotide analogue comprising a cyclopentanyl modified nucleotide |
| WO2020014643A1 (en) | 2018-07-13 | 2020-01-16 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| WO2020028097A1 (en) | 2018-08-01 | 2020-02-06 | Gilead Sciences, Inc. | Solid forms of (r)-11-(methoxymethyl)-12-(3-methoxypropoxy)-3,3-dimethyl-8-0x0-2,3,8,13b-tetrahydro-1h-pyrido[2,1-a]pyrrolo[1,2-c] phthalazine-7-c arboxylic acid |
| WO2020086556A1 (en) | 2018-10-24 | 2020-04-30 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| WO2020092621A1 (en) | 2018-10-31 | 2020-05-07 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds as hpk1 inhibitors |
| WO2020092528A1 (en) | 2018-10-31 | 2020-05-07 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds having hpk1 inhibitory activity |
| US10662416B2 (en) | 2016-10-14 | 2020-05-26 | Precision Biosciences, Inc. | Engineered meganucleases specific for recognition sequences in the hepatitis B virus genome |
| WO2020178769A1 (en) | 2019-03-07 | 2020-09-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'3'-cyclic dinucleotides and prodrugs thereof |
| WO2020178770A1 (en) | 2019-03-07 | 2020-09-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3'3'-cyclic dinucleotides and prodrugs thereof |
| WO2020178768A1 (en) | 2019-03-07 | 2020-09-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3'3'-cyclic dinucleotide analogue comprising a cyclopentanyl modified nucleotide as sting modulator |
| WO2020214652A1 (en) | 2019-04-17 | 2020-10-22 | Gilead Sciences, Inc. | Solid forms of a toll-like receptor modulator |
| WO2020214663A1 (en) | 2019-04-17 | 2020-10-22 | Gilead Sciences, Inc. | Solid forms of a toll-like receptor modulator |
| WO2020237025A1 (en) | 2019-05-23 | 2020-11-26 | Gilead Sciences, Inc. | Substituted exo-methylene-oxindoles which are hpk1/map4k1 inhibitors |
| WO2020263830A1 (en) | 2019-06-25 | 2020-12-30 | Gilead Sciences, Inc. | Flt3l-fc fusion proteins and methods of use |
| WO2021011891A1 (en) | 2019-07-18 | 2021-01-21 | Gilead Sciences, Inc. | Long-acting formulations of tenofovir alafenamide |
| WO2021034804A1 (en) | 2019-08-19 | 2021-02-25 | Gilead Sciences, Inc. | Pharmaceutical formulations of tenofovir alafenamide |
| CN112424596A (en) * | 2018-09-10 | 2021-02-26 | 株式会社岛津制作所 | Method for separating phosphoinositide from biological membrane |
| US10966999B2 (en) | 2017-12-20 | 2021-04-06 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3′3′ cyclic dinucleotides with phosphonate bond activating the sting adaptor protein |
| WO2021067181A1 (en) | 2019-09-30 | 2021-04-08 | Gilead Sciences, Inc. | Hbv vaccines and methods treating hbv |
| WO2021113765A1 (en) | 2019-12-06 | 2021-06-10 | Precision Biosciences, Inc. | Optimized engineered meganucleases having specificity for a recognition sequence in the hepatitis b virus genome |
| US11040027B2 (en) | 2017-01-17 | 2021-06-22 | Heparegenix Gmbh | Protein kinase inhibitors for promoting liver regeneration or reducing or preventing hepatocyte death |
| WO2021188959A1 (en) | 2020-03-20 | 2021-09-23 | Gilead Sciences, Inc. | Prodrugs of 4'-c-substituted-2-halo-2'-deoxyadenosine nucleosides and methods of making and using the same |
| WO2021223699A1 (en) | 2020-05-07 | 2021-11-11 | Ono Pharmaceutical Co., Ltd. | 3-azabicyclo(3.1.0)hexane derivatives having kdm5 inhibitory activity and use thereof |
| US11203610B2 (en) | 2017-12-20 | 2021-12-21 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2′3′ cyclic dinucleotides with phosphonate bond activating the sting adaptor protein |
| WO2022031894A1 (en) | 2020-08-07 | 2022-02-10 | Gilead Sciences, Inc. | Prodrugs of phosphonamide nucleotide analogues and their pharmaceutical use |
| WO2022087149A2 (en) | 2020-10-22 | 2022-04-28 | Gilead Sciences, Inc. | Interleukin-2-fc fusion proteins and methods of use |
| WO2022087422A1 (en) * | 2020-10-22 | 2022-04-28 | Chulalongkorn University | Pyrrolidine-3-carboxamide derivatives and related uses |
| WO2022241134A1 (en) | 2021-05-13 | 2022-11-17 | Gilead Sciences, Inc. | COMBINATION OF A TLR8 MODULATING COMPOUND AND ANTI-HBV siRNA THERAPEUTICS |
| WO2022271659A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271677A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271650A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271684A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| EP3999183A4 (en) * | 2019-07-17 | 2023-11-15 | Ono Pharmaceutical Co., Ltd. | Compound having kdm5 inhibitory activity and pharmaceutical use thereof |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20190063745A (en) * | 2017-11-30 | 2019-06-10 | (주)인핸스드바이오 | NEW COMPOUNDS FOR INHIBITING BINDING BETWEEN p34 PROTEIN AND NEDD4-1 PROTEIN AND USE THEREOF |
| CN109852662B (en) * | 2018-12-25 | 2021-02-19 | 华南农业大学 | Application of histone methylated H3K4me3 in porcine ovarian granulosa cells |
| UY38934A (en) * | 2019-10-29 | 2021-05-31 | Biogen Ma Inc | O-GLUCOPROTEIN-2-ACETAMIDE-2-DEOXY-3-D-GLUCOPYRANOSIDASE INHIBITORS SPIROCYCLIC |
| CA3168103A1 (en) * | 2020-01-29 | 2021-08-05 | Kamari Pharma Ltd. | Compounds and compositions for use in treating skin disorders |
| CN111138289B (en) * | 2020-02-27 | 2022-08-30 | 奥锐特药业(天津)有限公司 | Compound and process for synthesizing 5-acetyl-1H-pyrazole-3-carboxylic acid by using same |
| CN116284202A (en) * | 2023-03-28 | 2023-06-23 | 华侨大学 | ProTACs compound of betulinic acid, preparation method and application thereof |
Citations (34)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4943533A (en) | 1984-03-01 | 1990-07-24 | The Regents Of The University Of California | Hybrid cell lines that produce monoclonal antibodies to epidermal growth factor receptor |
| US5212290A (en) | 1989-09-08 | 1993-05-18 | The Johns Hopkins University | Antibodies specific for type II mutant EGTR |
| EP0659439A2 (en) | 1993-12-24 | 1995-06-28 | MERCK PATENT GmbH | Immunoconjugates |
| US5457105A (en) | 1992-01-20 | 1995-10-10 | Zeneca Limited | Quinazoline derivatives useful for treatment of neoplastic disease |
| US5475001A (en) | 1993-07-19 | 1995-12-12 | Zeneca Limited | Quinazoline derivatives |
| WO1996003397A1 (en) | 1994-07-21 | 1996-02-08 | Akzo Nobel N.V. | Cyclic ketone peroxide formulations |
| WO1996030347A1 (en) | 1995-03-30 | 1996-10-03 | Pfizer Inc. | Quinazoline derivatives |
| WO1996033978A1 (en) | 1995-04-27 | 1996-10-31 | Zeneca Limited | Quinazoline derivative |
| WO1996033980A1 (en) | 1995-04-27 | 1996-10-31 | Zeneca Limited | Quinazoline derivatives |
| WO1996040210A1 (en) | 1995-06-07 | 1996-12-19 | Imclone Systems Incorporated | Antibody and antibody fragments for inhibiting the growth of tumors |
| US5654307A (en) | 1994-01-25 | 1997-08-05 | Warner-Lambert Company | Bicyclic compounds capable of inhibiting tyrosine kinases of the epidermal growth factor receptor family |
| WO1997038983A1 (en) | 1996-04-12 | 1997-10-23 | Warner-Lambert Company | Irreversible inhibitors of tyrosine kinases |
| WO1998014451A1 (en) | 1996-10-02 | 1998-04-09 | Novartis Ag | Fused pyrazole derivative and process for its preparation |
| US5747498A (en) | 1996-05-28 | 1998-05-05 | Pfizer Inc. | Alkynyl and azido-substituted 4-anilinoquinazolines |
| US5760041A (en) | 1996-02-05 | 1998-06-02 | American Cyanamid Company | 4-aminoquinazoline EGFR Inhibitors |
| US5804396A (en) | 1994-10-12 | 1998-09-08 | Sugen, Inc. | Assay for agents active in proliferative disorders |
| WO1998043960A1 (en) | 1997-04-03 | 1998-10-08 | American Cyanamid Company | Substituted 3-cyano quinolines |
| WO1998050038A1 (en) | 1997-05-06 | 1998-11-12 | American Cyanamid Company | Use of quinazoline compounds for the treatment of polycystic kidney disease |
| WO1998050433A2 (en) | 1997-05-05 | 1998-11-12 | Abgenix, Inc. | Human monoclonal antibodies to epidermal growth factor receptor |
| US5866572A (en) | 1996-02-14 | 1999-02-02 | Zeneca Limited | Quinazoline derivatives |
| WO1999006378A1 (en) | 1997-07-29 | 1999-02-11 | Warner-Lambert Company | Irreversible inhibitors of tyrosine kinases |
| WO1999006396A1 (en) | 1997-07-29 | 1999-02-11 | Warner-Lambert Company | Irreversible bicyclic inhibitors of tyrosine kinases |
| WO1999009016A1 (en) | 1997-08-01 | 1999-02-25 | American Cyanamid Company | Substituted quinazoline derivatives and their use as tyrosine kinase inhibitors |
| US5891996A (en) | 1972-09-17 | 1999-04-06 | Centro De Inmunologia Molecular | Humanized and chimeric monoclonal antibodies that recognize epidermal growth factor receptor (EGF-R); diagnostic and therapeutic use |
| WO1999024037A1 (en) | 1997-11-06 | 1999-05-20 | American Cyanamid Company | Use of quinazoline derivatives as tyrosine kinase inhibitors for treating colonic polyps |
| US6002008A (en) | 1997-04-03 | 1999-12-14 | American Cyanamid Company | Substituted 3-cyano quinolines |
| US6084095A (en) | 1994-01-25 | 2000-07-04 | Warner-Lambert Company | Substituted pyrido[3,2-d]pyrimidines capable of inhibiting tyrosine kinases of the epidermal growth factor receptor family |
| US6140332A (en) | 1995-07-06 | 2000-10-31 | Novartis Ag | Pyrrolopyrimidines and processes for the preparation thereof |
| US6344455B1 (en) | 1998-11-19 | 2002-02-05 | Warner-Lambert Company | N-[4-(3-chloro-4-fluoro-phenylamino)-7-(3-morpholin-4-yl-propoxy)-quinazolin-6-yl]-acrylamide, and irreversible inhibitor of tyrosine kinases |
| US6391874B1 (en) | 1996-07-13 | 2002-05-21 | Smithkline Beecham Corporation | Fused heterocyclic compounds as protein tyrosine kinase inhibitors |
| US6596726B1 (en) | 1994-01-25 | 2003-07-22 | Warner Lambert Company | Tricyclic compounds capable of inhibiting tyrosine kinases of the epidermal growth factor receptor family |
| US20100261701A1 (en) * | 2008-10-02 | 2010-10-14 | Asahi Kasei Pharma Corporation | 8-Substituted isoquinoline derivative and the use thereof |
| US20140275092A1 (en) * | 2013-03-13 | 2014-09-18 | Constellation Pharmaceuticals, Inc. | Pyrazolo compounds and uses thereof |
| WO2014164708A1 (en) * | 2013-03-12 | 2014-10-09 | Quanticel Pharmaceuticals, Inc. | Histone dementhylase inhibitors |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2835216A1 (en) * | 2011-05-31 | 2012-12-06 | Theravance, Inc. | Neprilysin inhibitors |
| EP2852584B1 (en) * | 2012-05-22 | 2018-02-28 | F. Hoffmann-La Roche AG | Substituted dipyridylamines and uses thereof |
| CN104918939B (en) * | 2012-11-16 | 2018-08-28 | 默沙东公司 | The purine inhibitors of human phosphatidyl-inositol 3-kinase delta |
-
2015
- 2015-10-09 EP EP15784257.6A patent/EP3204379B1/en active Active
- 2015-10-09 WO PCT/US2015/054949 patent/WO2016057924A1/en active Application Filing
- 2015-10-09 CN CN201580066844.9A patent/CN107912040B/en active Active
- 2015-10-09 JP JP2017518802A patent/JP6783230B2/en active Active
-
2017
- 2017-04-07 US US15/482,584 patent/US10022354B2/en active Active
-
2018
- 2018-10-08 HK HK18112763.0A patent/HK1253559A1/en unknown
Patent Citations (45)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5891996A (en) | 1972-09-17 | 1999-04-06 | Centro De Inmunologia Molecular | Humanized and chimeric monoclonal antibodies that recognize epidermal growth factor receptor (EGF-R); diagnostic and therapeutic use |
| US4943533A (en) | 1984-03-01 | 1990-07-24 | The Regents Of The University Of California | Hybrid cell lines that produce monoclonal antibodies to epidermal growth factor receptor |
| US5212290A (en) | 1989-09-08 | 1993-05-18 | The Johns Hopkins University | Antibodies specific for type II mutant EGTR |
| US5457105A (en) | 1992-01-20 | 1995-10-10 | Zeneca Limited | Quinazoline derivatives useful for treatment of neoplastic disease |
| US5616582A (en) | 1992-01-20 | 1997-04-01 | Zeneca Limited | Quinazoline derivatives as anti-proliferative agents |
| US5475001A (en) | 1993-07-19 | 1995-12-12 | Zeneca Limited | Quinazoline derivatives |
| EP0659439A2 (en) | 1993-12-24 | 1995-06-28 | MERCK PATENT GmbH | Immunoconjugates |
| US6265410B1 (en) | 1994-01-25 | 2001-07-24 | Warner-Lambert Company | Bicyclic compounds capable of inhibiting tyrosine kinases of the epidermal growth factor receptor family |
| US6084095A (en) | 1994-01-25 | 2000-07-04 | Warner-Lambert Company | Substituted pyrido[3,2-d]pyrimidines capable of inhibiting tyrosine kinases of the epidermal growth factor receptor family |
| US6455534B2 (en) | 1994-01-25 | 2002-09-24 | Warner-Lambert Company | Bicyclic compounds capable of inhibiting tyrosine kinases of the epidermal growth factor receptor family |
| US5654307A (en) | 1994-01-25 | 1997-08-05 | Warner-Lambert Company | Bicyclic compounds capable of inhibiting tyrosine kinases of the epidermal growth factor receptor family |
| US5679683A (en) | 1994-01-25 | 1997-10-21 | Warner-Lambert Company | Tricyclic compounds capable of inhibiting tyrosine kinases of the epidermal growth factor receptor family |
| US6713484B2 (en) | 1994-01-25 | 2004-03-30 | Warner-Lambert Company | Bicyclic compounds capable of inhibiting tyrosine kinases of the epidermal growth factor receptor family |
| US6596726B1 (en) | 1994-01-25 | 2003-07-22 | Warner Lambert Company | Tricyclic compounds capable of inhibiting tyrosine kinases of the epidermal growth factor receptor family |
| US6521620B1 (en) | 1994-01-25 | 2003-02-18 | Warner-Lambert Company | Bicyclic compounds capable of inhibiting tyrosine kinases of the epidermal growth factor receptor family |
| WO1996003397A1 (en) | 1994-07-21 | 1996-02-08 | Akzo Nobel N.V. | Cyclic ketone peroxide formulations |
| US5804396A (en) | 1994-10-12 | 1998-09-08 | Sugen, Inc. | Assay for agents active in proliferative disorders |
| WO1996030347A1 (en) | 1995-03-30 | 1996-10-03 | Pfizer Inc. | Quinazoline derivatives |
| US5770599A (en) | 1995-04-27 | 1998-06-23 | Zeneca Limited | Quinazoline derivatives |
| WO1996033980A1 (en) | 1995-04-27 | 1996-10-31 | Zeneca Limited | Quinazoline derivatives |
| WO1996033978A1 (en) | 1995-04-27 | 1996-10-31 | Zeneca Limited | Quinazoline derivative |
| WO1996040210A1 (en) | 1995-06-07 | 1996-12-19 | Imclone Systems Incorporated | Antibody and antibody fragments for inhibiting the growth of tumors |
| US6140332A (en) | 1995-07-06 | 2000-10-31 | Novartis Ag | Pyrrolopyrimidines and processes for the preparation thereof |
| US5760041A (en) | 1996-02-05 | 1998-06-02 | American Cyanamid Company | 4-aminoquinazoline EGFR Inhibitors |
| US5866572A (en) | 1996-02-14 | 1999-02-02 | Zeneca Limited | Quinazoline derivatives |
| US6399602B1 (en) | 1996-02-14 | 2002-06-04 | Zeneca Limited | Quinazoline derivatives |
| US6344459B1 (en) | 1996-04-12 | 2002-02-05 | Warner-Lambert Company | Irreversible inhibitors of tyrosine kinases |
| WO1997038983A1 (en) | 1996-04-12 | 1997-10-23 | Warner-Lambert Company | Irreversible inhibitors of tyrosine kinases |
| US6602863B1 (en) | 1996-04-12 | 2003-08-05 | Warner-Lambert Company | Irreversible inhibitors of tyrosine kinases |
| US5747498A (en) | 1996-05-28 | 1998-05-05 | Pfizer Inc. | Alkynyl and azido-substituted 4-anilinoquinazolines |
| US6391874B1 (en) | 1996-07-13 | 2002-05-21 | Smithkline Beecham Corporation | Fused heterocyclic compounds as protein tyrosine kinase inhibitors |
| WO1998014451A1 (en) | 1996-10-02 | 1998-04-09 | Novartis Ag | Fused pyrazole derivative and process for its preparation |
| WO1998043960A1 (en) | 1997-04-03 | 1998-10-08 | American Cyanamid Company | Substituted 3-cyano quinolines |
| US6002008A (en) | 1997-04-03 | 1999-12-14 | American Cyanamid Company | Substituted 3-cyano quinolines |
| US6235883B1 (en) | 1997-05-05 | 2001-05-22 | Abgenix, Inc. | Human monoclonal antibodies to epidermal growth factor receptor |
| WO1998050433A2 (en) | 1997-05-05 | 1998-11-12 | Abgenix, Inc. | Human monoclonal antibodies to epidermal growth factor receptor |
| WO1998050038A1 (en) | 1997-05-06 | 1998-11-12 | American Cyanamid Company | Use of quinazoline compounds for the treatment of polycystic kidney disease |
| WO1999006396A1 (en) | 1997-07-29 | 1999-02-11 | Warner-Lambert Company | Irreversible bicyclic inhibitors of tyrosine kinases |
| WO1999006378A1 (en) | 1997-07-29 | 1999-02-11 | Warner-Lambert Company | Irreversible inhibitors of tyrosine kinases |
| WO1999009016A1 (en) | 1997-08-01 | 1999-02-25 | American Cyanamid Company | Substituted quinazoline derivatives and their use as tyrosine kinase inhibitors |
| WO1999024037A1 (en) | 1997-11-06 | 1999-05-20 | American Cyanamid Company | Use of quinazoline derivatives as tyrosine kinase inhibitors for treating colonic polyps |
| US6344455B1 (en) | 1998-11-19 | 2002-02-05 | Warner-Lambert Company | N-[4-(3-chloro-4-fluoro-phenylamino)-7-(3-morpholin-4-yl-propoxy)-quinazolin-6-yl]-acrylamide, and irreversible inhibitor of tyrosine kinases |
| US20100261701A1 (en) * | 2008-10-02 | 2010-10-14 | Asahi Kasei Pharma Corporation | 8-Substituted isoquinoline derivative and the use thereof |
| WO2014164708A1 (en) * | 2013-03-12 | 2014-10-09 | Quanticel Pharmaceuticals, Inc. | Histone dementhylase inhibitors |
| US20140275092A1 (en) * | 2013-03-13 | 2014-09-18 | Constellation Pharmaceuticals, Inc. | Pyrazolo compounds and uses thereof |
Non-Patent Citations (16)
| Title |
|---|
| "Cancer Principles and Practice", 15 February 2001, LIPPINCOTT WILLIAMS & WILKINS PUBLISHERS |
| "Handbook of Chemistry and Physics" |
| ANGEW CHEM. INTL. ED. ENGL., vol. 33, 1994, pages 183 - 186 |
| CARRUTHERS: "Some Modern Methods of Organic Synthesis", 1987, CAMBRIDGE UNIVERSITY PRESS |
| DATABASE CA [online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; 2000, YU, GUIXUE ET AL: "Preparation of fused pyridopyridazine inhibitors of cGMP phosphodiesterase", XP002752101, retrieved from STN Database accession no. 2000:688225 * |
| ELIEL, E.L.: "Stereochemistry of Carbon Compounds", 1962, MCGRAW-HILL |
| JACQUES ET AL.: "Enantiomers, Racemates and Resolutions", 1981, WILEY INTERSCIENCE |
| JOHNS ET AL., ./ BIOL. CHEM., vol. 279, no. 29, 2004, pages 30375 - 30384 |
| LAROCK: "Comprehensive Organic Transformations", 1989, VCH PUBLISHERS, INC. |
| SHARMA ET AL., CELL, vol. 141, no. 1, 2010, pages 69 - 80 |
| SMITH; MARCH: "March's Advanced Organic Chemistry", 2001, JOHN WILEY & SONS, INC. |
| STRAGLIOTTO ET AL., EUR..1 CANCER, vol. 32A, 1996, pages 636 - 640 |
| THOMAS SORRELL: "Organic Chemistry", 1999 |
| WILEN ET AL., TETRAHEDRON, vol. 33, 1977, pages 2725 |
| WILEN, S.H.: "Tables of Resolving Agents and Optical Resolutions", 1972, UNIV. OF NOTRE DAME PRESS, pages: 268 |
| YU, GUIXUE ET AL: "Preparation of fused pyridopyridazine inhibitors of cGMP phosphodiesterase", PCT INT. APPL., 137 PP. CODEN: PIXXD2, 2000 * |
Cited By (66)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017205078A1 (en) | 2016-05-27 | 2017-11-30 | Gilead Sciences, Inc. | Methods for treating hepatitis b virus infections using ns5a, ns5b or ns3 inhibitors |
| WO2017205115A1 (en) | 2016-05-27 | 2017-11-30 | Gilead Sciences, Inc. | Compounds for the treatment of hepatitis b virus infection |
| WO2018039531A1 (en) | 2016-08-26 | 2018-03-01 | Gilead Sciences, Inc. | Substituted pyrrolizine compounds and uses thereof |
| EP3922634A1 (en) | 2016-08-26 | 2021-12-15 | Gilead Sciences, Inc. | Substituted pyrrolizine compounds and uses thereof |
| WO2018045144A1 (en) | 2016-09-02 | 2018-03-08 | Gilead Sciences, Inc. | Toll like receptor modulator compounds |
| WO2018045150A1 (en) | 2016-09-02 | 2018-03-08 | Gilead Sciences, Inc. | 4,6-diamino-pyrido[3,2-d]pyrimidine derivaties as toll like receptor modulators |
| US11274285B2 (en) | 2016-10-14 | 2022-03-15 | Precision Biosciences, Inc. | Engineered meganucleases specific for recognition sequences in the Hepatitis B virus genome |
| US10662416B2 (en) | 2016-10-14 | 2020-05-26 | Precision Biosciences, Inc. | Engineered meganucleases specific for recognition sequences in the hepatitis B virus genome |
| WO2018083288A1 (en) | 2016-11-07 | 2018-05-11 | Bayer Aktiengesellschaft | Substituted sulfonyl amides for controlling animal pests |
| US11040027B2 (en) | 2017-01-17 | 2021-06-22 | Heparegenix Gmbh | Protein kinase inhibitors for promoting liver regeneration or reducing or preventing hepatocyte death |
| WO2018144390A1 (en) | 2017-01-31 | 2018-08-09 | Gilead Sciences, Inc. | Crystalline forms of tenofovir alafenamide |
| US10442804B2 (en) | 2017-02-02 | 2019-10-15 | Gilead Sciences, Inc. | Compounds for the treatment of hepatitis B virus infection |
| WO2018144605A1 (en) | 2017-02-02 | 2018-08-09 | Gilead Sciences, Inc. | Compounds for the treatment of hepatitis b virus infection |
| WO2018195321A1 (en) | 2017-04-20 | 2018-10-25 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| EP4026835A2 (en) | 2017-04-20 | 2022-07-13 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| US11203610B2 (en) | 2017-12-20 | 2021-12-21 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2′3′ cyclic dinucleotides with phosphonate bond activating the sting adaptor protein |
| US10966999B2 (en) | 2017-12-20 | 2021-04-06 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3′3′ cyclic dinucleotides with phosphonate bond activating the sting adaptor protein |
| WO2019160882A1 (en) | 2018-02-13 | 2019-08-22 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| EP4227302A1 (en) | 2018-02-13 | 2023-08-16 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| WO2019165374A1 (en) | 2018-02-26 | 2019-08-29 | Gilead Sciences, Inc. | Substituted pyrrolizine compounds as hbv replication inhibitors |
| WO2019195181A1 (en) | 2018-04-05 | 2019-10-10 | Gilead Sciences, Inc. | Antibodies and fragments thereof that bind hepatitis b virus protein x |
| WO2019193533A1 (en) | 2018-04-06 | 2019-10-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'2'-cyclic dinucleotides |
| WO2019193542A1 (en) | 2018-04-06 | 2019-10-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'3'-cyclic dinucleotides |
| WO2019193543A1 (en) | 2018-04-06 | 2019-10-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3'3'-cyclic dinucleotides |
| US11292812B2 (en) | 2018-04-06 | 2022-04-05 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3′3′-cyclic dinucleotides |
| US11149052B2 (en) | 2018-04-06 | 2021-10-19 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2′3′-cyclic dinucleotides |
| US11788077B2 (en) | 2018-04-12 | 2023-10-17 | Precision Biosciences, Inc. | Polynucleotides encoding optimized engineered meganucleases having specificity for a recognition sequence in the Hepatitis B virus genome |
| WO2019200247A1 (en) | 2018-04-12 | 2019-10-17 | Precision Biosciences, Inc. | Optimized engineered meganucleases having specificity for a recognition sequence in the hepatitis b virus genome |
| US11142750B2 (en) | 2018-04-12 | 2021-10-12 | Precision Biosciences, Inc. | Optimized engineered meganucleases having specificity for a recognition sequence in the Hepatitis B virus genome |
| WO2019204609A1 (en) | 2018-04-19 | 2019-10-24 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| WO2019211799A1 (en) | 2018-05-03 | 2019-11-07 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'3'-cyclic dinucleotide analogue comprising a cyclopentanyl modified nucleotide |
| EP4234030A2 (en) | 2018-07-13 | 2023-08-30 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| WO2020014643A1 (en) | 2018-07-13 | 2020-01-16 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| WO2020028097A1 (en) | 2018-08-01 | 2020-02-06 | Gilead Sciences, Inc. | Solid forms of (r)-11-(methoxymethyl)-12-(3-methoxypropoxy)-3,3-dimethyl-8-0x0-2,3,8,13b-tetrahydro-1h-pyrido[2,1-a]pyrrolo[1,2-c] phthalazine-7-c arboxylic acid |
| CN112424596B (en) * | 2018-09-10 | 2023-07-18 | 株式会社岛津制作所 | Separation method of biomembrane inositol phosphate |
| CN112424596A (en) * | 2018-09-10 | 2021-02-26 | 株式会社岛津制作所 | Method for separating phosphoinositide from biological membrane |
| WO2020086556A1 (en) | 2018-10-24 | 2020-04-30 | Gilead Sciences, Inc. | Pd-1/pd-l1 inhibitors |
| WO2020092621A1 (en) | 2018-10-31 | 2020-05-07 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds as hpk1 inhibitors |
| WO2020092528A1 (en) | 2018-10-31 | 2020-05-07 | Gilead Sciences, Inc. | Substituted 6-azabenzimidazole compounds having hpk1 inhibitory activity |
| US11766447B2 (en) | 2019-03-07 | 2023-09-26 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3′3′-cyclic dinucleotide analogue comprising a cyclopentanyl modified nucleotide as sting modulator |
| WO2020178769A1 (en) | 2019-03-07 | 2020-09-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 2'3'-cyclic dinucleotides and prodrugs thereof |
| WO2020178768A1 (en) | 2019-03-07 | 2020-09-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3'3'-cyclic dinucleotide analogue comprising a cyclopentanyl modified nucleotide as sting modulator |
| WO2020178770A1 (en) | 2019-03-07 | 2020-09-10 | Institute Of Organic Chemistry And Biochemistry Ascr, V.V.I. | 3'3'-cyclic dinucleotides and prodrugs thereof |
| WO2020214663A1 (en) | 2019-04-17 | 2020-10-22 | Gilead Sciences, Inc. | Solid forms of a toll-like receptor modulator |
| WO2020214652A1 (en) | 2019-04-17 | 2020-10-22 | Gilead Sciences, Inc. | Solid forms of a toll-like receptor modulator |
| WO2020237025A1 (en) | 2019-05-23 | 2020-11-26 | Gilead Sciences, Inc. | Substituted exo-methylene-oxindoles which are hpk1/map4k1 inhibitors |
| WO2020263830A1 (en) | 2019-06-25 | 2020-12-30 | Gilead Sciences, Inc. | Flt3l-fc fusion proteins and methods of use |
| EP3999183A4 (en) * | 2019-07-17 | 2023-11-15 | Ono Pharmaceutical Co., Ltd. | Compound having kdm5 inhibitory activity and pharmaceutical use thereof |
| WO2021011891A1 (en) | 2019-07-18 | 2021-01-21 | Gilead Sciences, Inc. | Long-acting formulations of tenofovir alafenamide |
| WO2021034804A1 (en) | 2019-08-19 | 2021-02-25 | Gilead Sciences, Inc. | Pharmaceutical formulations of tenofovir alafenamide |
| WO2021067181A1 (en) | 2019-09-30 | 2021-04-08 | Gilead Sciences, Inc. | Hbv vaccines and methods treating hbv |
| WO2021113765A1 (en) | 2019-12-06 | 2021-06-10 | Precision Biosciences, Inc. | Optimized engineered meganucleases having specificity for a recognition sequence in the hepatitis b virus genome |
| WO2021188959A1 (en) | 2020-03-20 | 2021-09-23 | Gilead Sciences, Inc. | Prodrugs of 4'-c-substituted-2-halo-2'-deoxyadenosine nucleosides and methods of making and using the same |
| CN115461338A (en) * | 2020-05-07 | 2022-12-09 | 小野药品工业株式会社 | 3-azabicyclo (3.1.0) hexane derivatives having KDM5 inhibitory activity and uses thereof |
| CN115461338B (en) * | 2020-05-07 | 2024-02-20 | 小野药品工业株式会社 | 3-azabicyclo (3.1.0) hexane derivatives having KDM5 inhibitory activity and uses thereof |
| KR20230007352A (en) | 2020-05-07 | 2023-01-12 | 오노 야꾸힝 고교 가부시키가이샤 | Compounds with KDM5 inhibitory activity and pharmaceutical uses thereof |
| US11691968B2 (en) | 2020-05-07 | 2023-07-04 | Ono Pharmaceutical Co., Ltd. | Compound having KDM5 inhibitory activity and pharmaceutical use thereof |
| WO2021223699A1 (en) | 2020-05-07 | 2021-11-11 | Ono Pharmaceutical Co., Ltd. | 3-azabicyclo(3.1.0)hexane derivatives having kdm5 inhibitory activity and use thereof |
| WO2022031894A1 (en) | 2020-08-07 | 2022-02-10 | Gilead Sciences, Inc. | Prodrugs of phosphonamide nucleotide analogues and their pharmaceutical use |
| WO2022087422A1 (en) * | 2020-10-22 | 2022-04-28 | Chulalongkorn University | Pyrrolidine-3-carboxamide derivatives and related uses |
| WO2022087149A2 (en) | 2020-10-22 | 2022-04-28 | Gilead Sciences, Inc. | Interleukin-2-fc fusion proteins and methods of use |
| WO2022241134A1 (en) | 2021-05-13 | 2022-11-17 | Gilead Sciences, Inc. | COMBINATION OF A TLR8 MODULATING COMPOUND AND ANTI-HBV siRNA THERAPEUTICS |
| WO2022271684A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271650A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271677A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271659A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
Also Published As
| Publication number | Publication date |
|---|---|
| US20170312252A1 (en) | 2017-11-02 |
| CN107912040A (en) | 2018-04-13 |
| JP6783230B2 (en) | 2020-11-11 |
| EP3204379B1 (en) | 2019-03-06 |
| JP2017530984A (en) | 2017-10-19 |
| US10022354B2 (en) | 2018-07-17 |
| HK1253559A1 (en) | 2019-06-21 |
| CN107912040B (en) | 2021-04-06 |
| EP3204379A1 (en) | 2017-08-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10022354B2 (en) | Pyrrolidine amide compounds as histone demethylase inhibitors | |
| EP2970307B1 (en) | Pyrazolo compounds and uses thereof | |
| US10308614B2 (en) | Therapeutic compounds and uses thereof | |
| US10301253B2 (en) | Therapeutic compounds and uses thereof | |
| US10258603B2 (en) | Therapeutic compounds and uses thereof | |
| EP3041474B1 (en) | Antiproliferative compounds | |
| EP3464286A1 (en) | Pyrazolopyridine derivatives for the treatment of cancer | |
| EP3224258A1 (en) | 4,5,6,7-tetrahydro-1 h-pyrazolo[4,3-c]pyridin-3-amine compounds as cbp and/or ep300 inhibitors | |
| WO2022266206A1 (en) | Kras inhibitor conjugates | |
| WO2016123391A1 (en) | Therapeutic compounds and uses thereof | |
| WO2015135094A1 (en) | Therapeutic compounds and uses thereof | |
| US10280149B2 (en) | Therapeutic compounds and uses thereof | |
| US10202354B2 (en) | Therapeutic compounds and uses thereof | |
| WO2015049325A1 (en) | Therapeutic inhibitors of cdk8 and uses thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15784257 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2017518802 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2015784257 Country of ref document: EP |