WO2015200648A1 - Use of peptides that block metadherin-snd1 interaction as treatment for cancer - Google Patents
Use of peptides that block metadherin-snd1 interaction as treatment for cancer Download PDFInfo
- Publication number
- WO2015200648A1 WO2015200648A1 PCT/US2015/037708 US2015037708W WO2015200648A1 WO 2015200648 A1 WO2015200648 A1 WO 2015200648A1 US 2015037708 W US2015037708 W US 2015037708W WO 2015200648 A1 WO2015200648 A1 WO 2015200648A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- mtdh
- peptide
- cancer
- sndl
- tumor
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/1703—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates
- A61K38/1709—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/08—Peptides having 5 to 11 amino acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/10—Peptides having 12 to 20 amino acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/46—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates
- C07K14/47—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals
- C07K14/4701—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans from vertebrates from mammals not used
- C07K14/4748—Tumour specific antigens; Tumour rejection antigen precursors [TRAP], e.g. MAGE
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/82—Translation products from oncogenes
Definitions
- the present disclosure relates to methods of treating cancer and reducing tumor growth, recurrence and metastasis comprising administering an inhibitor that blocks the interaction of metadherin (MTDH) and Staphylococcal nuclease domain-containing 1 (SNDl) and inhibits the function of the MTDH-SND1 complex.
- MTDH metadherin
- SNDl Staphylococcal nuclease domain-containing 1
- the inhibitors include peptides or fragments of MTDH that bind SNDl or peptides or fragments derived from SNDl that bind metadherin.
- Metadherin also called AEG1, LYRIC
- MTDH has previously been identified as a pro- metastasis gene that resides in 8q22, a frequently amplified genomic locus linked to poor relapse-free survival of breast cancer (Hu et al., 2009).
- Overexpression of MTDH is observed in more than 40% of primary tumors and is an independent poor-prognosis factor (Hu et al., 2009). What drives the strong selection of MTDH in primary breast tumors is unclear and the functional significance of MTDH in normal development and tumorigenesis remains poorly understood.
- MTDH transmembrane protein that mediates the adhesion of cancer cells to the lung endothelium
- MTDH has been linked to multiple oncogenic pathways such as PI3K/AKT and NF- ⁇ (Emdad et al., 2013). How MTDH regulates these pathways remains elusive.
- evolutionarily conserved in higher vertebrates MTDH contains no recognizable functional domain, rendering the understanding of its biological function challenging.
- Multiple groups have identified several MTDH-binding partners, including PLZF, BCCIPa and Staphylococcal nuclease domain-containing 1 (SND1) (Wan and Kang, 2013). However, whether and how the interactions with these proteins mediate the function of MTDH is largely unknown.
- Breast cancer is a heterogeneous disease that can be broadly classified into luminal and basal-like subtypes based on gene expression profiles (Perou et al., 2000). It has been speculated that different oncogenic signaling may target different cells of origin, thus leading to the formation of different subtypes of breast cancer. However, the origin, identity and regulation of tumor-initiating cells (TICs) in different oncogene-induced mammary tumors remain poorly characterized. Autochthonous tumorigenesis in mice offers great models for tracking the early changes during tumor initiation and for investigating the role of a gene of interest in mediating the transformation and expansion of TICs.
- TICs tumor-initiating cells
- MTDH metadherin
- TICs luminal and basal breast tumor initiating cells
- prostate tumors and underscores the selection pressure to overexpress MTDH in diverse tumor subtypes.
- the functional dependency of MTDH on its conserved interaction with SNDl suggests that targeting the MTDH-SND1 complex may offer an opportunity to control tumor initiation, recurrence and metastasis by preventing the expansion of TICs, with minimal impact on normal tissues.
- the disclosure provides a method for preventing or reducing the recurrence or expansion of tumor initiating cells in a subject comprising administering an agent that interferes with the interaction of MTDH and SNDl.
- the disclosure also provides a method for treating cancer in a subject having cancer comprising administering to the subject an inhibitor that disrupts the interaction between MTDH and SNDl.
- the inhibitor binds to a region of human MTDH within residues 364-582 of MTDH (such as amino acids 364-407 of MTDH) set forth in SEQ ID NO: 1. In some embodiments, the inhibitor binds SNDl SN1/2 domains. In some embodiments, the inhibitor binds to a region of human SNDl within residues 16-339 of SNDl (such as amino acids 39-43 of SNDl) set forth in SEQ ID NO: 2.
- the disclosure provides a method for reducing or decreasing the expansion of tumor initiating cells in a subject comprising administering an inhibitor of the interaction between metadherin (MTDH) and Staphylococcal nuclease domain-containing 1 (SNDl) wherein the inhibitor inhibits SNDl protein interaction within residues 364 to 407 of metadherin (SEQ ID NO: 1).
- MTDH metadherin
- SNDl Staphylococcal nuclease domain-containing 1
- the subject has cancer.
- cancers contemplated herein include, but are not limited to breast cancer, prostate cancer, liver cancer, lung cancer, colon cancer, colorectal cancer, non-small cell lung carcinoma, squamous cell carcinoma, cervical cancer, bladder cancer, as well those listed in the Detailed Description.
- the cancer is selected from the group consisting of breast cancer and prostate cancer.
- the cancer is liver cancer.
- the MTDH/SND1 inhibitor is a peptide of MTDH, a peptide of SNDl, a peptide mimetic having similar structure as a peptide comprising residues 393-403 of MTDH, a small molecule compound that has similar structure or part of the structure as a peptide comprising residues 393-403 of MTDH, or a nanoparticle conjugate containing a peptide or peptide mimetic described herein.
- the inhibitor is a peptide.
- the peptide comprises the consensus sequence XWXXXXXWXX (SEQ ID NO: 3), where X is any amino acid.
- the peptide comprises the consensus sequence XWXXXXXWXX (SEQ ID NO: 3) from residues 393-403 of MTDH (SEQ ID NO: 2), where X is any amino acid.
- the inhibitor is a peptide comprising the consensus sequence XWXXXXXWXX (SEQ ID NO: 4), where X is any amino acid.
- the inhibitor is a peptide of MTDH selected from i) a peptide within residues 364- 582 of MTDH, ii) a peptide within residues 364-407 of MTDH, iii) a peptide comprising residues 386-407 of MTDH, or iv) a peptide comprising residues 393-403 of MTDH.
- the inhibitor is a peptide of MTDH selected from i) a peptide consisting essentially of residues 364-582 of SEQ ID NO: 1, ii) a peptide having residues 364-407 of MTDH, iii) a peptide consisting essentially of residues 386-407 of SEQ ID NO: 1, or iv) a peptide consisting essentially of residues 393-403 of SEQ ID NO: 1.
- the peptide is a variant of any of the wild type MTDH peptides described herein, wherein the variant peptide retains W394 and W401, wherein the position is based on the amino acid sequence of wild type MTDH (i.e., SEQ ID NO: 1).
- the peptide comprises the sequence of DWNAPAEEWGN (SEQ ID NO: 5), or a variant thereof, wherein the variant peptide comprises at least one (such as any of 2, 3, 4, 5, 6, 7, 8, or 9) mutations at any position except for the W residues.
- the peptide is about 11 to about 22 amino acids long (such as about 15 to about 22 amino acids long).
- the peptide comprises about any of 1, 2, 3, 4, 5, 6, or 7 amino acids N-terminal to the sequence of DWNAPAEEWGN (SEQ ID NO: 5), or a variant thereof.
- the peptide comprises about any of 1, 2, 3, or 4 amino acids C- terminal to the sequence of DWNAPAEEWGN (SEQ ID NO: 5), or a variant thereof.
- the peptide comprises the sequence of DWNAPEEWGN (SEQ ID NO: 20), or a variant thereof, wherein the variant peptide comprises at least one (such as any of 2, 3, 4, 5, 6, 7, 8, or 9) mutations at any position except for the W residues.
- the peptide is about 10 to about 21 amino acids long (such as about 14 to about 21 amino acids long).
- the peptide comprises about any of 1, 2, 3, 4, 5, 6, or 7 amino acids N-terminal to the sequence of DWNAPEEWGN (SEQ ID NO: 20), or a variant thereof. In some embodiments, the peptide comprises about any of 1, 2, 3, or 4 amino acids C-terminal to the sequence of DWNAPEEWGN (SEQ ID NO: 20), or a variant thereof.
- the peptide comprises a mutation in MTDH at one or more amino acid residues selected from the group consisting of D389, A392, D393, N395, E399, E400, W404, D406 and E407, wherein the amino acid position is based on the wild type MTDH sequence (i.e., SEQ ID NO: 1).
- the peptide comprises a mutation in MTDH at one or more amino acid residues selected from the group consisting of D389R, D393R, E399R, E400R, W404D, D406R and E407R, wherein the position is based on the wild type MTDH sequence (i.e., SEQ ID NO: 1).
- the mutation is selected from the group consisting of D389R, D393R, E399R, E400R, W404D, D406R and E407R of SEQ ID NO: 1.
- the inhibitor is a peptide of SND1 within residues 16 to 339 of SND1 (i.e., SEQ ID NO: 2).
- the SND1 peptide comprises a mutation at one or more residues selected from the group consisting of F250, R324, R255, R327, R324, P39, P43, E247, L256, H279, 1284, L287, R259 and N281, wherein the position is based on the wild type SND1 sequence (i.e., SEQ ID NO: 2).
- the mutation in SND1 is selected from the group consisting of F250A R324E and R255E of SEQ ID NO: 2.
- the inhibitor peptide is part of a fusion protein.
- the peptide is fused to an FC domain.
- compositions comprising an MTDH/SND1 inhibitor described herein and a pharmaceutically acceptable carrier.
- isolated peptides inhibitors such as any one of the peptides described herein.
- present application further provides methods of making and using any one of the peptide inhibitors (as well as compositions comprising such peptide inhibitors) described herein.
- a method of disrupting the interaction between MTDH and SND1 in a subject comprising administering to the subject any one of the peptide inhibitors (or pharmaceutical compositions comprising such peptide inhibitors) described above.
- a method of inhibiting SND1 -dependent expression of prosurvival genes in a subject comprising administering to the subject any one of the peptide inhibitors (or pharmaceutical compositions comprising such peptide inhibitors) described above.
- crystal structure of the binding between MTDH and SND1 which is useful for identifying inhibitors of the interaction between MTDH and SND1.
- FIG. 1A Schematic representation of WT and mutant Mtdh allele. Green boxes represent exons 1-12. Primers (F, forward; R, reverse) used for genotyping are indicated above the corresponding genomic sequences.
- FIG. IB LacZ expression in WT and KO embryo at day 10.5, visualized by X-gal staining.
- FIG. 1C MTDH protein immunobloting in MECs freshly dissociated from 8-week-old female mice with indicated Mtdh genotype.
- FIG. IE Percentage of tumor-free mammary glands at indicated ages in the same cohort of mice as in (FIG. ID).
- FIG. IF Total tumor burden of PyMT;Mtdh+/+, PyMT;Mtdh+/- and PyMT;Mtdh-/- cohorts evaluated at indicated age.
- FIG. 1H Percentage of mice from same cohorts as in (FIG. 1H) bearing indicated number of tumors at 300 days of age.
- FIG. IK Number of metastatic lesions per lung section in the same cohorts of mice from (FIG. 1J). Error bars represent 5-95 percentiles.
- FIG. 1L Kinetics of mammary tumor onset in MMTV-Wnt mice of the indicated genotypes.
- FIG. 1M Percentage of mice from same cohorts as in (L) bearing indicated number of tumors at 300 days of age.
- FIG. 10 Percentage of mice from same cohorts as in (FIG.
- FIG. 2A Flow cytometry of
- FIG. 2E Mammary tumor incidence (left) and size (right) 3 months after orthotopic transplantations of unsorted MECs dissociated from preneoplastic glands of PyMT;Mtdh+/+ and PyMT;Mtdh-/- mice.
- FIG. 2F Mammary tumor incidence (left) and size (right) 8 weeks after orthotopic transplantations of indicated sorted CD24+CD291ow luminal or CD24+CD29high basal MECs from preneoplastic glands of PyMT;Mtdh+/+ mice.
- FIG. 2G, FIG. 2H Mammary tumor incidence (FIG. 2G) and volumes (FIG.
- FIG. 3A-L also shows that MTDH is required for the activities of oncogene-induced luminal and basal TICs but not MaSCs.
- FIG. 3E Mammosphere forming assays of CD61+ or CD61- cells from indicated cell populations dissociated from Wnt hyperplastic glands. Data represent mean + SEM.
- FIG. 3F FIG.
- FIG. 3G Mammary tumor incidence (FIG. 3F) and tumor volumes (FIG. 3G) 2 months after orthotopic transplantation of 10000 cells of the indicated sorted luminal and basal populations from 7-month tumor-free mammary glands into NSG mice.
- FIG. 3H H&E staining of spontaneous ErbB2-induced tumors and tumors generated by indicated transplanted cells. Scale bars: 200 ⁇ (top) and 100 ⁇ (bottom).
- FIG. 31 Schematic of mammary gland reconstitution assays used in (FIG. 3J-K).
- FIG. 3 J, FIG. 3K Tables showing transplantation of limiting numbers of Lin- MECs (FIG. 3J) or Lin- CD24+CD29hi MECs (FIG. 3K) dissociated from mammary glands of WT or KO female virgin mice into cleared mammary fat pads of WT recipients.
- FIG. 3L Quantification (% of fat pad area filled with outgrowth) of reconstituted mammary outgrowths in (FIG. 3K). None of the experimental groups was significant by Mann- Whitney test. Statistics: (FIG. 3D, FIG. 3E) Student's t-test. (FIG. 3G) Mann- Whitney test. (FIG. 3A, FIG. 3F) Fisher's exact test. (FIG. 3B) Chi-square test. (FIG. 3J, FIG. 3K) Limiting dilution analysis. ***p ⁇ 0.001, **p ⁇ 0.01, *p ⁇ 0.05.
- FIG. 4A Schematic diagram of MMTV-Mtdh transgene construct and breeding scheme used to generate PyMT;Mtdh-/- mice with (Mtdh-/- - ⁇ -Tg) or without the MMTV-Mtdh transgene.
- FIG. 4B MTDH protein levels in PyMT-induced tumors from Mtdh+/+, Mtdh-/-, or Mtdh-/- -i-Tg mice.
- FIG. 4 Average number of tumor-free mammary glands at indicated ages (left column: Mtdh-/- mice; right column: Mtdh -/- + Tg mice) in the same cohort of mice as in (FIG. 4D).
- FIG. 4F Tumor burden of same cohorts of mice as in (FIG. 4D).
- FIG. 4G, FIG. 4H MTDH was knocked down by two independent shRNA (KD1 and KD2) in freshly dissociated PyMT;Mtdh+/+ pMECs and in vitro mammosphere (FIG.
- FIG. 4G, n 5, each in triplicates; left column: Control; middle column: KD1; right column: KD2) and in vivo tumor formation assays were performed (FIG. 4H, incidence at 3 months). FC, fold changes.
- FIG. 4K Schematic diagram of experiments in L-O.
- FIG. 4L Mammosphere formation of ALDH-positive or ALDH-negative tumor cells from PyMT;Mtdh+/+ tumors.
- FIG. 4M MTDH was knocked down in sorted ALDH+ cells from PyMT;Mtdh+/+ tumors and mammosphere assays were performed.
- FIG. 4N Mammosphere formation of Lin-CD24+CD61+ or Lin- CD24+CD61- tumor cells from Wnt;Mtdh+/+ tumors.
- FIG. 40 MTDH was knocked down in sorted Lin-CD24+CD61+ cells from Wnt;Mtdh+/+ tumors and mammosphere assays were performed.
- FIG. 4G, FIG. 41, FIG. 4L-0 Student's t-test.
- FIG. 4D Log rank test.
- FIG. 4E Chi-square test.
- FIG. 4F Mann-Whitney test.
- FIG. 4H, FIG. 4J Limiting dilution analysis. ***p ⁇ 0.001, **p ⁇ 0.01, *p ⁇ 0.05. Data represent mean + SEM.
- FIG. 5A Schematic diagram of MMTV-Mtdh transgene construct including the MMTV-LTR (mouse mammary tumor virus long terminal repeat;) promoter, mouse Mtdh coding sequence and the SV40 polyadenylation sequences. Hindlll and EcoRI restriction sites were used to clone Mtdh; Sail and Spel restriction sites were used to linearize the fragment for microinjection into mouse zygotes.
- FIG. 5B Genomic DNA purified from transgenic mice tail snips were subjected to southern blot.
- FIG. 5C Mtdh mRNA levels in organs from 8-week-old transgene-positive or negative female littermates (left column: 9077 MMTV-Mtdh-/- ; middle column: 9078 MMTV-Mtdh-/- ; right column 9079 MMTV-Mtdh-/-). All mice harbor wild-type endogenous Mtdh allele.
- FIG. 5D H&E staining of spontaneous PyMT-induced tumors from PyMT;Mtdh-/- mice with or without MMTV-Mtdh transgene. Scale bars: 200 ⁇ (top) and 100 ⁇ (bottom).
- FIG. 5G, FIG. 5H Mammary tumor incidence after orthotopic transplantation of pMECs (from two independent PyMT;Mtdh-/- mice) transduced with either vector control or Mtdh-expressing lenti viruses.
- FIG. 51 Representative flow cytometry analysis of ALDH activity in cells derived from a PyMT;Mtdh+/+ tumor. DEAB inhibitor-treated samples served as gating control.
- FIG. 6 A Combination of MTDH re-expression and SNDl knockdown in PyMT;Mtdh-/- tumor cells. The efficiency of SNDl KD and MTDH re-expression was assessed by western blotting.
- FIG. 6B, FIG. 6C In vitro mammosphere (FIG. 6B) and in vivo tumor formation (FIG. 6C, 6 weeks) assays were performed with cells generated in (FIG. 6A). +/- indicate whether the denoted protein is present (+) or absent (-) based on western blotting in (FIG. 6A).
- FIG. 6D SNDl was knocked down in PyMT;Mtdh+/+ or Wnt;Mtdh+/+ pMECs cells and mammosphere assays were performed in triplicates.
- FIG. 6E, FIG. 6F Tumor incidence (FIG. 6E) and volume (FIG. 6F) after orthotopic transplantations of control or SND1-KD PyMT;Mtdh+/+ pMECs.
- Statistics: (FIG. 6B, FIG. 6D) Student's t-test.
- FIG. 6C, FIG. 6E Limiting dilution analysis.
- FIG. 6F Mann- Whitney test. ***p ⁇ 0.001, **p ⁇ 0.01, *p ⁇ 0.05. Data represent mean + SEM..
- Figures 7A-E represents determination of key regions and residues mediating the
- FIG. 7 A Schematics of MTDH fragments and mutants with indicated SNDl -binding capability. + indicates binding and - indicates no binding based on results shown below.
- Two putative nuclear localization signals are residues 432-451 for NLS2 and residues 561-580 for NLS3.
- W394D and W401D either completely or strongly reduced the binding, respectively.
- FIG. 7B Pulldown of His6-SND1AC by GST- tagged MTDH fragments with indicated boundaries. The bound proteins were examined by SDS-PAGE and visualized by Coomassie blue staining.
- FIG. 7C Pulldown of His6-
- FIG. 7C 1/10 of the His6-SND1AC input was shown, and GST alone was used as a negative control. Representative results of 3 independent experiments are shown.
- FIG. 7D, FIG. 7E Lysates from HEK293T cells expressing the indicated ectopic human SND1, AG02 or MTDH were immunoprecipitated with anti-Myc and immunoblotted with the indicated antibodies.
- FIG. 8A-H shows that SND1 -binding deficient MTDH fails to promote tumor- initiating potential of MECs.
- FIG. 8A, FIG. 8E Lysates from PyMT;Mtdh-/- MECs
- Mammosphere assays were performed with PyMT;Mtdh-/- pMECs reconstituted with indicated Mtdh constructs.
- FIG. 8C, FIG. 8D, FIG. 8G, FIG. 8H In vivo tumor formation (FIG. 8C, FIG. 8G for tumor incidence; FIG. 8D, FIG. 8H for tumor volumes) were performed at limiting numbers using PyMT;Mtdh-/- pMECs reconstituted with indicated WT or mutant MTDH.
- mouse W391D MTDH corresponds to human W394D MTDH; and mouse W398D MTDH corresponds to human W401D MTDH.
- FIG. 8C, FIG. 8G Limiting dilution analysis.
- FIG. 8D, FIG. 8H Mann- Whitney test. Data represent mean + SEM. ***p ⁇ 0.001, **p ⁇ 0.01, *p ⁇ 0.05.
- FIG. 9A Quantification of cleaved caspase
- FIG. 9D Protein levels of SND1 and ⁇ -actin (loading control) in control or MTDH-KD PyMT;Mtdh+/+ pMECs treated with CPT at indicated concentrations for
- FIG. 9G Ingenuity Pathway Analysis shows the top 5 molecular and cellular functions of SNDl-upregulated genes shown in (FIG. 9F) and the number of molecules/genes implicated in each category.
- FIG. 9H Effects of SNDl-upregulated genes in cell survival and cell death functions. Z scores were calculated based on gene expression changes and gene functions as specified by the ingenuity knowledge base. A given function is predicted to be significantly increased when z > 2 or decreased when z ⁇ -2.
- FIG. 10A-K shows MTDH and SND1 are important for in vitro sphere-forming and in vivo tumor-initiating activities of human breast cancer cells.
- FIG. 10A, FIG. 10B MTDH (FIG. 10A) or SND1 (FIG. 10B) was knocked down in HMLE-Neu cells and tumorsphere assays were performed in triplicates.
- FIG. IOC, FIG. 10D MTDH (FIG. IOC) or SND1 (FIG. 10D) was knocked down in the BCM-4013 patient-derived xenografted (PDX) tumor cells and tumorsphere assays were performed in triplicates.
- FIG. 10A, FIG. 10A MTDH (FIG. 10A) or SND1 (FIG. 10B) was knocked down in HMLE-Neu cells and tumorsphere assays were performed in triplicates.
- FIG. IOC, FIG. 10D MTDH (FIG. IOC) or SND1 (FI
- FIG. 10E MTDH or SND1 was knocked down in MDA-MB-231 cells, and the KD efficiency was measured by immunoblotting.
- FIG. 10F Tumorsphere assays of MDA-MB-231 cells were performed in triplicates.
- FIG. 10G, FIG. 10H Tumor incidence (FIG. 10G) and volumes (FIG. 10H) 5 weeks after injection of limiting numbers of MDA-MB-231 cells.
- FIG. 10J Bar graph presentation of (FIG. 101).
- FIG. 10K Schematic illustration depicting the essential role of MTDH in tumor initiation but not normal gland development. Under stress conditions during tumorigenesis, the MTDH-SND1 interaction protects SND1 from stress-induced degradation and supports the survival and activities of both basal and luminal TICs.
- FIG. 10G Limiting dilution analysis.
- FIG. 10H Mann- Whitney test.
- FIG. 101, FIG. 10J Chi- square test. Data represent mean + SEM. ***p ⁇ 0.001, **p ⁇ 0.01, *p ⁇ 0.05.
- FIG. 11A-M shows MTDH and SND1 positively correlate at protein levels in human breast cancers and cooperate in predicting poor prognosis.
- FIG. 11 A, FIG. 11B MTDH
- FIG. 11 A or SND1 (FIG. 11B) was knocked down in HMLE-Neu cells by 3 independent shRNAs, and the KD efficiency was measured by immunoblotting.
- FIG. 11C, FIG. 11D MTDH (FIG. 11C) or SND1 (FIG. 11D) was knocked down in BCM-4013 patient-derived xenografted (PDX) tumor cells (Zhang et al., 2013) by 2 independent shRNAs and the KD efficiency was measured by q-PCR. Data represent mean + SEM.
- FIG. HE, FIG. 11F
- FIG. HE Validation of the specificity of MTDH (FIG. HE) and SND1 (FIG. 11F) antibodies for immunohistochemical staining.
- the antibodies are reactive to both human and mouse proteins.
- FIG. HE Representative immunohistochemical staining of MTDH in PyMT;Mtdh+/+ and PyMT;Mtdh-/- mammary tumors.
- FIG. 11F Representative immunohistochemical staining of SND1 in PyMT;Mtdh+/+ control or SND1-KD tumors. Scale bars: 100 ⁇ .
- FIG. 11G The protein levels of MTDH and SND1 in human breast tumors were determined by
- FIG. 11H Bar graph presentation of (FIG. 11G).
- FIG. I ll Correlation between mRNA levels of MTDH and SND1 in the NKI295 dataset.
- FIG. 11 J, FIG. 11K Correlation between MTDH and SND1 mRNA levels with primary tumor size (FIG. 11 J) and differentiation (FIG. 11K) of breast tumors.
- FIG. 11L Distant metastasis free survival (DMFS) of breast cancer patients stratified by the mRNA levels of MTDH and SND1.
- DMFS Distant metastasis free survival
- FIG. 11J-L NKI295 human breast cancer dataset (van de Vijver et al., 2002) was used and samples were divided into four groups based on MTDH/SNDl mRNA levels using medium cutoff.
- FIG. 11M Correlation between MTDH mRNA levels with prognosis of different subtypes of human breast cancer. Kaplan-Meier plot of relapse-free survival of patients stratified by median MTDH expression in the KM Plotter breast cancer meta-analysis database (Gyorffy et al., 2010).
- FIG. 12A shows MTDH levels are associated with tumor progression and metastasis in human prostate cancer.
- FIG. 12A Two prostate tumor tissue microarrays were stained with an anti-MTDH antibody. MTDH levels were scored as negative (0), low (1), medium (2) or high (3). Representative images of MTDH immuno staining of prostate tissue of different stages and distant metastasis. Scale bar, 40 ⁇ .
- FIG. 12B Correlation of MTDH levels with prostate tumor Gleason scores. Top curve represents average MTDH score (mean + SEM) and bottom curve represents the percentage of samples with medium/high levels of MTDH within indicated groups.
- FIG. 12D Kaplan-Meier analysis of recurrence-free survival of prostate cancer patients based on MTDH expression in their tumors.
- FIG. 13 A shows MTDH genomic gain is associated with MTDH protein levels and clinical progression in prostate cancer.
- FIG. 13 A A prostate tumor tissue microarray was analyzed for MTDH genomic copy number by FISH. Shown are examples of tumors without (left) or with (right) MTDH genomic gains. SpectrumGreen (green) and SpectrumOrange (pale orange) probes detect chromosome 8 centromere and the 8q22 region, respectively. Scale bar, 1 ⁇ .
- FIG. 13D Kaplan-Meier analysis of recurrence-free survival of prostate cancer patients with and without MTDH genomic gain in their tumors. Cox proportional hazard ratio (HR) is shown.
- FIG. 14A shows generation and characterization of TRAMP mice with different Mtdh status.
- FIG. 14A Cross scheme for the generation of TRAMP mice in C57BL/6 background with different Mtdh status.
- FIG. 14B Genotyping of mice generated in (FIG. 14A). Top, the detected WT (602 bp) and gene-trapped mutant alleles (472 bp) of Mtdh. Bottom, the detected TRAMP transgene (600 bp) and internal genomic control (324 bp).
- FIG. 14C Mtdh mRNA in prostate tissues from mice with indicated genotypes analyzed by qPCR.
- Mtdh mRNA was undetectable in Mtdh 'A tissues, and elevated in TRAMP-positive prostate tissues as compared to normal glands. ** P ⁇ 0.01, *** P ⁇ 0.001 based on Mann-Whitney test.
- FIG. 14D Western blot analysis of Mtdh in prostates from WT non-transgenic mice and tumors from TRAMP/Mtdh +/+ mice. Arbitrary levels of Mtdh protein after normalized to ⁇ -actin are shown at the bottom.
- E-F Representative immunohistochemical staining of SV40 Tag oncoprotein in prostate glands from 8- (FIG. 14E) and 28-week-old (FIG. 14F) TRAMP/ ⁇ +/+ and TRAMP/Mtdh ' ⁇ mice. Scale bar, 50 ⁇ .
- FIG. 15A-E shows loss of Mtdh in mice inhibits tumor formation, reduces tumor burden and increases survival rate.
- FIG. 15A-B Wet weight (FIG. 15A) or relative weight as % of body weight (FIG. 15B) of lower urogenital tracts excised from TRAMP I Mtdh +/+ and
- TRAMPIMtdh +/ ⁇ mice (denoted as Mtdh + ) or TRAMP/Mtdh ' ⁇ (denoted as Mtdh ).
- Data represent mean + SEM. ** P ⁇ 0.01 based on Mann- Whitney test.
- FIG. 15C Representative images of lower urogenital tracts (B, bladder; P, prostate; SV, seminal vesicle) excised from 36-week-old male mice with indicated genotypes. Scale bar, 1 cm.
- FIG. 15D Tumor incidence scored by examining histological sections of prostate glands from cohorts of TRAMP mice with indicated Mtdh genotype at different ages.
- FIG. 16 A H&E-stained histologic sections of prostates dissected from TRAMP/ Mtdh + and TRAMP/Mtdh ' mice at indicated ages. Scale bar: 200 ⁇ .
- FIG. 16B Each prostate from mice with indicated genotypes and ages was assigned a single highest grade. '+' and '-' indicate TRAMP/ Mtdh + and TRAMP/Mtdh ' mice, respectively.
- Grade scores 1, normal; 2, low grade PIN; 3, high-grade PIN; 4, well differentiated adenocarcinoma and phyllode tumor; 5, moderately differentiated adenocarcinoma; 6, poorly differentiated adenocarcinoma and neuroendocrine tumors.
- the grading scheme followed standard protocol as previously described (Hurwitz et al., Current protocols in immunology / edited by John E Coligan [et al].
- FIG. 17A shows ablation of Mtdh reduces systemic metastasis of prostate cancer.
- FIG. 17A Incidence of lung, liver and lymph node metastasis in cohorts of one-year-old TRAMP mice with indicated Mtdh genotypes. P values based on Chi-square test.
- FIG. 17B Bar graph presentation (left column: Mtdh+; right column: Mtdh-) of (FIG. 17A).
- FIG. 18A-G shows silencing of Mtdh in TRAMP-C1 prostate cancer cells decreases proliferation in vitro and tumor formation in vivo.
- FIG. 18A-B Mtdh was knocked down by two independent shRNA, as quantified by qPCR (FIG. 18A) and western blot (FIG. 18B).
- FIG. 18C Proliferation rate of control and Mtdh-KD TRAMP-C1 cancer cells after 48 hours.
- FIG. 18E Tumor volumes 5 weeks post injection in (FIG. 18D). ***P ⁇ 0.001 based on Mann- Whitney test.
- FIG. 18F Images of tumors dissected 5 weeks post transplantation in (FIG. 18D). G, Mtdh mRNA levels in tumors formed in control and KD groups. Note the tumors that eventually grew in the KD groups expressed similar levels of Mtdh as the controls.
- FIG. 18 A, FIG. 18C, FIG. 18G Data represent mean + SEM and P values based on Student's t-test. ** P ⁇ 0.01, *** P ⁇ 0.001, n.s. not significant.
- Figure (19) shows mapping of SNDl -MTDH interaction and overall structure of their complex. Overall structure of MTDH-SND1 complex. Two perpendicular views are shown. The SNl and SN2 domains of SNDl and MTDH are colored cyan, magenta and yellow, respectively. SNDl is shown in ribbon (left) and surface (right). MTDH is shown in worm (backbone) and cylinder (side chain).
- FIG. 20A represents a structural and sequence comparison of SNl/2 with SN3/4 and SNase.
- FIG. 20A Overlay of the structures of SNl/2 (magenta, in the complex with MTDH), SN3/4 (blue, PDB code: 3BDL) and two models of SNase (yellow, PDB code: 2ENB) in stereo view. The difference in ⁇ 2- ⁇ 3 loop is emphasized by a dashed circle.
- FIG. 20B Sequence alignment of SNl/2 with SN3/4 from SNDl and SNase. Secondary structural elements are indicated above the sequences. conserveed residues are colored red. Residues that interact with MTDH are identified by green squares. The residues at the active site of SNase that contribute to the nuclease activity are indicated by red circles.
- FIG. 21 illustrates the MTDH-SND1 binding interface.
- a close-up stereo of MTDH- SND1 interface The ⁇ 2- ⁇ 3 loops from SN3 domain (light blue) and SNase (yellow) are shown for highlighting the unique structure of SNl ⁇ 2- ⁇ 3 loop required for MTDH-binding.
- Figures 22A-D shows the identification of MTDH and SND1 mutants deficient in binding.
- FIG. 22A In vitro pull-down of SND1 (16-339) by GST-tagged MTDH (364-582) harboring WT or mutant sequence. The proteins bound to GS4B were examined on SDS-PAGE and visualized by Coomassie blue staining.
- FIG. 22A In vitro pull-down of SND1 (16-339) by GST-tagged MTDH (364-582) harboring WT or mutant sequence.
- the proteins bound to GS4B were examined on SDS-PAGE and visualized by Coomassie blue staining.
- FIG. 22C HEK293T cells were transfected with human HA-SND1, WT Myc-MTDH or Myc-MTDH with indicated single point mutation. Lysates were immunoprecipitated with anti-Myc antibody and immunoblotted with the indicated antibodies.
- FIG. 22D HEK293T cells were transfected with human Myc-MTDH, WT HA-SND1 or mutant HA-SND1 with indicated single point mutations or deletions. Lysates were immunoprecipitated with anti-Myc and immunoblotted with the indicated antibodies.
- FIG. 23 A Lysates from PyMT;Mtdh-/- tumor cells
- FIG. 23C-E In vivo tumor formation (FIG. 23C for tumor incidence; FIG. 23D, FIG. 23E for tumor volumes) were performed at limiting numbers using PyMT;Mtdh-/- tumor cells reconstituted with indicated WT or mutant MTDH.
- FIG. 23C Limiting dilution analysis.
- FIG. 23D, FIG. 23E Mann- Whitney test. **p ⁇ 0.01, *p ⁇ 0.05.
- FIG. 24A shows that mutations in MTDH-binding pockets impair tumor- promoting function of SND1.
- FIG. 24A Lysates from SND1-KD PyMT;Mtdh+/+ tumor cells reconstituted with vector control, WT or mutant shRNA-resistant murine SND1 were immunoprecipitated with anti-MTDH antibody and immunoblotted for indicated proteins.
- FIG. 24B Mammosphere assays were performed with SND1-KD PyMT;Mtdh+/+ tumor cells reconstituted with vector control or indicated SND1 constructs.
- FIG. 24C, FIG. 24D Mammary tumor incidence (FIG. 24C) and tumor growth curve (FIG.
- Figures 25 shows MTDH interaction protects SND1 from heat shock stress-induced degradation.
- HEK293T cells were transfected with HA-SNDl together with either empty vector control or indicated WT or mutant Myc-MTDH constructs. Two days post infections, cells were treated under heat shock conditions and lysates were immunoblotted for indicated proteins, ⁇ - actin was used as loading control. Representative results of three independent experiments are shown.
- FIGS. 26A-D show wild type MTDH peptide significant block MTDH-SND1 interaction.
- 293T cells co-transfected with HA-SNDl and MYC-MTDH. After 48hr cells were collected and subjected to IP assay.
- FIG. 26A, FIG. 26B Cell lysate was incubated with anti- HA antibody or IgG overnight followed by 2hr incubation with Protein A/G beads to pull down HA-SNDl protein. The beads was washed with lysis buffer and split into 5 fractions. Each fraction was eluted with buffer or indicated peptides for 30min. The elution fractions and beads were collected for WB.
- FIG. 27A shows that the interaction interrupt effect was confirmed by endogenous immunoprecipitation of a breast cancer cell line.
- Breast cancer cells MDA-MB231 were collected and subjected to IP assay.
- FIG. 27 A Cell lysate was incubated with anti-SNDl antibody or IgG overnight followed by 2hr incubation with Protein A/G beads to pull down endogenous SND1 protein. The beads were washed with lysis buffer and split into 5 fractions. Each fraction was eluted with buffer or indicated peptides for 30min. The elution fractions and beads were collected for WB.
- FIG. 27 A Cell lysate was incubated with anti-SNDl antibody or IgG overnight followed by 2hr incubation with Protein A/G beads to pull down endogenous SND1 protein. The beads were washed with lysis buffer and split into 5 fractions. Each fraction was eluted with buffer or indicated peptides for 30min. The elution fractions and beads were collected for
- FIG. 27B Cell lyaste was split into 5 fractions, and incubated with anti-SNDl antibody plus either buffer or indicated peptides. After 24hr, the lysate was incubated with Protein A/G beads then the beads were washed in wash buffer.
- Figure 28 shows the identification of MTDH mutants bearing mutations in the SND1- binding motif that has no or little effect on SNDl -binding. In vitro pull-down of SNDl (amino acids 16-339 of SEQ ID NO: 2) by GST-tagged MTDH (amino acids 386-407 of SEQ ID NO: 1) harboring WT or mutant sequence. The proteins bound to GS4B were examined on SDS-PAGE and visualized by Coomassie blue staining.
- the present disclosure provides methods to reduce cancer growth and cancer metastasis by inhibiting the activity of the MTDH-SND1 complex using peptides or other compounds that inhibit the binding of SNDl with MTDH.
- the inventors discovered that MTDH deficiency inhibits diverse oncogene- or carcinogen-induced tumorigenesis. Results below show that MTDH is selectively required for TICs and MTDH-mediated stabilization of SNDl confers a survival advantage under stress. This is the first disclosure that MTDH interaction is required for SNDl -dependent expression of pro-survival genes.
- an inhibitor of the interaction between metadherin (MTDH) and Staphylococcal nuclease domain-containing 1 (SND1) or a “MTDH/SND1 inhibitor” refers to a compound or composition that inhibits the interaction of MTDH with SND1 at the binding sites of the two proteins.
- the inhibitor inhibits binding between the proteins at residues 393-403 of MTDH (i.e., SEQ ID NO: 1). Additional inhibitors are described in greater detail in the Detailed Description.
- a “therapeutically effective amount” or “effective amount” refers to that amount of a peptide or other inhibitor product described herein, sufficient to result in amelioration of symptoms, for example, treatment, healing, prevention or amelioration of the relevant medical condition, or an increase in rate of treatment, healing, prevention or
- a therapeutically effective dose refers to that ingredient alone.
- a therapeutically effective dose refers to combined amounts of the active ingredients that result in the therapeutic effect, whether administered in combination, including serially or simultaneously.
- a therapeutically effective amount of the peptide or other inhibitor product ameliorates symptoms associated with various cancers, including but not limited to, loss of appetite, oral pain, upper abdominal pain, fatigue, abdominal swelling, persistent aches, bone pain, nausea, vomiting, constipation, weight loss, headaches, rectal bleeding, night sweats, digestive discomfort, and painful urination.
- Treatment refers to administration of a compound or composition to a subject for therapeutic or prophylactic purposes.
- a “therapeutic” treatment is a treatment administered to a subject who exhibits signs or symptoms of pathology for the purpose of diminishing or eliminating those signs or symptoms.
- the signs or symptoms may be biochemical, cellular, histological, functional or physical, subjective or objective.
- a "prophylactic" treatment is a treatment administered to a subject who does not exhibit signs of a disease or exhibits only early signs of the disease, for the purpose of decreasing the risk of developing pathology.
- the compounds or compositions of the disclosure may be given as a prophylactic treatment to reduce the likelihood of developing a pathology or to minimize the severity of the pathology, if developed.
- composition refers to a composition suitable for pharmaceutical use in a subject animal, including humans and mammals.
- a pharmaceutical composition comprises a therapeutically effective amount of a peptide or other product described herein, optionally another biologically active agent, and optionally a pharmaceutically acceptable excipient, carrier or diluent.
- a pharmaceutical composition encompasses a composition comprising the active ingredient(s), and the inert ingredient(s) that make up the carrier, as well as any product that results, directly or indirectly, from combination, complexation or aggregation of any two or more of the ingredients, or from dissociation of one or more of the ingredients, or from other types of reactions or interactions of one or more of the ingredients.
- the pharmaceutical compositions of the present disclosure encompass any composition made by admixing a compound of the disclosure and a pharmaceutically acceptable excipient, carrier or diluent.
- “Pharmaceutically acceptable carrier” refers to any of the standard pharmaceutical carriers, buffers, and the like, such as a phosphate buffered saline solution, 5% aqueous solution of dextrose, and emulsions (e.g., an oil/water or water/oil emulsion).
- excipients include adjuvants, binders, fillers, diluents, disintegrants, emulsifying agents, wetting agents, lubricants, glidants, sweetening agents, flavoring agents, and coloring agents.
- Suitable pharmaceutical carriers, excipients and diluents are described in Remington's Pharmaceutical Sciences, 19th Ed.
- Preferred pharmaceutical carriers depend upon the intended mode of administration of the active agent. Typical modes of administration include enteral (e.g., oral) or parenteral (e.g., subcutaneous, intramuscular, intravenous or intraperitoneal injection; or topical, transdermal, or transmucosal administration).
- enteral e.g., oral
- parenteral e.g., subcutaneous, intramuscular, intravenous or intraperitoneal injection; or topical, transdermal, or transmucosal administration.
- pharmaceutically acceptable or “pharmacologically acceptable” salt, ester or other derivative of an active agent comprise, for example, salts, esters or other derivatives refers to a material that is not biologically or otherwise undesirable, i.e., the material may be administered to an individual without causing any undesirable biological effects or without interacting in a deleterious manner with any of the components of the composition in which it is contained or with any components present on or in the body of the individual.
- unit dosage form refers to physically discrete units suitable as unitary dosages for human and animal subjects, each unit containing a predetermined quantity of a compound of the disclosure calculated in an amount sufficient to produce the desired effect, optionally in association with a pharmaceutically acceptable excipient, diluent, carrier or vehicle.
- the specifications for the novel unit dosage forms of the present disclosure depend on the particular compound employed and the effect to be achieved, and the pharmacodynamics associated with each compound in the host.
- the term "subject” encompasses mammals.
- mammals include, but are not limited to, any member of the mammalian class: humans, non-human primates such as chimpanzees, and other apes and monkey species; farm animals such as cattle, horses, sheep, goats, swine; domestic animals such as rabbits, dogs, and cats; laboratory animals including rodents, such as rats, mice and guinea pigs, and the like.
- the term does not denote a particular age or gender.
- the subject is human.
- Metadherin Metadherin (MTDH; also known as AEG-1, 3D3/LYRIC) was identified as a pro-metastasis gene that resides in 8q22, a frequently amplified genomic locus linked to poor relapse-free survival of breast cancer (Hu et al., 2009).
- the amino acid sequence of human metadherin can be found in Genbank Accession No. AAH45642, herein incorporated by reference, and is also provided herein as SEQ ID NO: 1.
- elevated levels of MTDH have been reported in more than 20 cancer types (4), suggesting a potentially crucial and broad functionality of this gene in human cancer.
- MTDH pleiotropic tumor-promoting roles may stem from the complex nature of this protein, as revealed by its initial identification.
- MTDH was originally reported as an HIV- induced gene in astrocytes (Su et al., 2002), a cell-surface molecule mediating the homing of mammary tumor cells to the lung endothelium (Brown and Ruoslahti et al., 2004), a lysine -rich CEACAM1 co-isolated (LYRIC) protein associated with tight junctions in prostate epithelial cells (Britt et al., 2004), and as a novel transmembrane protein present in the different subcellular compartments (Sutherland et al., 2004).
- LYRIC lysine -rich CEACAM1 co-isolated
- the human MTDH encodes a 582-amino acid protein with no recognizable domains that could indicate its biological function, except for a putative transmembrane domain and three lysine-rich nuclear localization signals (Thirkettle et al., 2009).
- MTDH has recently been reported to interact with multiple proteins. In the nucleus, MTDH was shown to interact with PLZF (Thirkettle et al., 2009), BCCIPa (Ash et al., 2008) and NFKB subunit p65 (Emdad et al., 2006; Sarkar et al., 2008).
- MTDH was reported to interact with staphylococcal nuclease domain-containing protein 1 (SNDl) (Blanco et al, 2011; Yoo et al., 2011; Meng et al., 2012).
- SNDl staphylococcal nuclease domain-containing protein 1
- MTDH has also been linked to multiple classical oncogenic signaling pathways such as PI3K/AKT and Wnt signaling (Emdad et al., 2013) in a cancer cell-type dependent manner.
- PI3K/AKT PI3K/AKT
- Wnt signaling Emdad et al., 2013
- SNDl is a multifunctional protein harboring four tandem repeats of
- SN domain (TSN5 domain) at the C terminus (Callebaut and Mornon, 1997; Ponting, 1997). It belongs to the oligonucleotide/oligosaccharide binding-fold (OB-fold) superfamily consisting of proteins that primarily participate in DNA/RNA-binding via the typical ⁇ -barrel of the OB-fold
- SNDl was suggested to be an essential component of the RNA-induced silencing complex (RISC) and involved in miRNA-mediated silencing (Caudy et al., 2003). It was also shown to have a nuclease activity toward hyper-edited miRNA primary transcripts (Scadden, 2005).
- RISC RNA-induced silencing complex
- SNDl Structural and biochemical analysis of SNDl suggested that the N-terminal SN domains, particularly SN3/4, possess RNA-binding and nuclease activity (Li et al., 2008), and the C-terminal TSN domain interacts with methylated Lys/Arg ligands and small nuclear ribonucleoprotein (snRNP) complexes (Shaw et al., 2007).
- SNDl is among the very few members of the OB-fold superfamily that participate in interaction with diverse proteins.
- SNDl was identified as a binding partner of MTDH in multiple types of cancer, and has been shown to be important for cancer cell survival under oncogenic or chemotherapeutic stresses (Blanco et al., 2011; Meng et al., 2012; Wan et al., 2014; Yoo et al., 2011).
- SNDl is an interacting partner of MTDH that possesses tumor-promoting function similar to that of MTDH (Blanco et al., 2011; Meng et al., 2012; Wang et al., 2012; Yoo et al., 2011). It is shown herein that the biochemically identified MTDH mutants with compromised SNDl -binding exhibited a reduced activity in the expansion and survival of tumor- initiating cells in diverse subtypes of breast cancer (Wan et al., 2014). No structural insight has yet been available for understanding the interaction between MTDH and its binding partners and how these interactions affect its role in cancer. Whether the function of SNDl in cancer relies on MTDH-binding remains unclear. The range of identified SNDl -interacting proteins suggests that its SN domains have been evolved into protein-protein interaction domains; the mode of interaction, however, has been unclear until the present disclosure.
- the interaction of MTDH with SNDl plays an important role in the progression of tumor growth and metastasis.
- Administration of an inhibitor of this protein interaction is as a useful therapy for the treatment of cancer.
- residues 394 and 401 of wild type MTDH set forth in SEQ ID NO: 1 are required for interaction with SNDl.
- the inhibitor is a peptide of MTDH comprising (a) the consensus sequence XWXXXXXWXX (SEQ ID NO: 3), wherein X is any amino acid, or (b) XWXXXXXWXX (SEQ ID NO: 4), wherein X is any amino acid.
- the inhibitor is a peptide of MTDH comprising the consensus sequence XWXXXXXWXX (SEQ ID NO: 3) from residues 393-403 of MTDH, wherein X is any amino acid.
- the inhibitor is a peptide of MTDH comprising the consensus sequence
- the MTDH/SND1 inhibitor is a peptide of MTDH, a peptide of SND1, a peptide mimetic having similar structure as a peptide comprising residues 393-403 of SEQ ID NO: 1, a peptide mimetic having a similar structure as a peptide comprising residues 390-403 of SEQ ID NO: 1, a small molecule compound that has similar structure or part of the structure as a peptide comprising residues 393-403 of SEQ ID NO: 1, a small molecule compound that has similar structure or part of the structure as a peptide comprising residues 390- 403 of SEQ ID NO: 1, or a nanoparticle conjugate containing a peptide or peptide mimetic described herein.
- the inhibitor is a peptide of MTDH selected from i) a peptide within residues 364-582 of SEQ ID NO: 1, ii) a peptide within residues 364-407 of SEQ ID NO: 1, iii) a peptide comprising residues 386-407 of SEQ ID NO: 1, iv) a peptide comprising residues 393-403 of SEQ ID NO: 1, or v) a peptide comprising residues 390-403 of SEQ ID NO: 1; or vi) a peptide within residues 364-582, residues 364-407, residues 386-407, residues 393-403 or residues 390-403 of SEQ ID NO: 1 comprising the consensus sequence XWXXXXXWXX (SEQ ID NO: 3) at residues 393-403, wherein X is any amino acid.
- the peptide comprises the sequence of DWNAPAEEWGN (SEQ ID NO: 5), or a variant thereof, wherein the variant peptide comprises at least one (such as any of 2, 3, 4, 5, 6, 7, 8, or 9) mutations at any position except for the W residues.
- the peptide is about 11 to about 22 amino acids long (such as about 15 to about 22 amino acids long).
- the peptide comprises about any of 1, 2, 3, 4, 5, 6, or 7 amino acids N-terminal to the sequence of DWNAPAEEWGN (SEQ ID NO: 5), or a variant thereof.
- the peptide comprises about any of 1, 2, 3, or 4 amino acids C- terminal to the sequence of DWNAPAEEWGN (SEQ ID NO: 5), or a variant thereof.
- the peptide comprises the sequence of DWNAPEEWGN (SEQ ID NO: 20), or a variant thereof, wherein the variant peptide comprises at least one (such as any of 2, 3, 4, 5, 6, 7, 8, or 9) mutations at any position except for the W residues.
- the peptide is about 10 to about 21 amino acids long (such as about 14 to about 21 amino acids long).
- the peptide comprises about any of 1, 2, 3, 4, 5, 6, or 7 amino acids N-terminal to the sequence of DWNAPEEWGN (SEQ ID NO: 20), or a variant thereof. In some embodiments, the peptide comprises about any of 1, 2, 3, or 4 amino acids C- terminal to the sequence of DWNAPEEWGN (SEQ ID NO: 20), or a variant thereof.
- the peptide competes with a wild type MTDH peptide comprising the sequence of DWNAPAEEWGN (SEQ ID NO: 5) for binding to SNDl (e.g., a wildtype MTDH peptide having the same length).
- SNDl e.g., a wildtype MTDH peptide having the same length.
- the peptide binds to SNDl with an affinity that is at least about any of 50%, 60%, 70%, 80%, 90%, 95%, or 100% of the binding affinity of a wild type MTDH peptide comprising the sequence of
- DWNAPAEEWGN (SEQ ID NO: 5) for SNDl (e.g., a wildtype MTDH peptide having the same length).
- the peptide comprises a mutation at one or more amino acid residues in MTDH selected from the group consisting of D389, A392, D393, N395, E399, E400, W404, D406 and E407, wherein the position corresponds to the wild type MTDH (i.e., SEQ ID NO: 1).
- the peptide comprises a mutation at one or more (such as 2, 3, 4, 5, 6, or 7) amino acid residues selected from the group consisting of N395, A396, P397, A398, E399, E400, and N403, wherein the position corresponds to the wild type MTDH (i.e., SEQ ID NO: 1).
- the mutation in MTDH is selected from the group consisting of N395K, A396D, PAE(397-399)SQ, and E400D/N403L, wherein the position corresponds to the wild type MTDH (i.e., SEQ ID NO: 1).
- the mutation in MTDH (SEQ ID NO: 1) is selected from the group consisting of D389R, D393R, E399R, E400R, W404D, D406R and E407R.
- the inhibitor is a peptide of SNDl within residues 16 to 339 of SNDl (i.e., SEQ ID NO: 2).
- the SNDl peptide comprises a mutation at one or more residues selected from the group consisting of F250, R324, R255, R327, R324, P39, P43, E247, L256, H279, 1284, L287, R259 and N281 , wherein the position corresponds to wild type SND1 (i.e., SEQ ID NO: 2).
- the mutation in SND1 i.e., SEQ ID NO: 2 is selected from the group consisting of F250A R324E and R255E.
- the MTDH or SND1 peptide comprises (e.g., has) an amino acid sequence having 60 amino acids or less, 55 amino acids or less, 40 amino acids or less, 35 amino acids or less, or 30 amino acids or less.
- the peptide comprises (e.g., has) an amino acid sequence having 25 amino acids or less, 20 amino acids or less, 15 amino acids or less, or 10 amino acids or less.
- the peptide comprises (e.g., has) 10-35 amino acid residues (e.g., 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, or 35 amino acid residues).
- amino acids are removed from the peptide described herein from within the amino acid sequence, at the N-terminus, and/or at the C-terminus.
- Such peptide fragments can comprise 3-14 amino acid residues (e.g., 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, or 14 amino acid residues).
- a peptide described herein can be fused or complexed to a second peptide domain that, for example, binds another target or increases the half-life or stability of the peptide.
- the peptide further comprises one or more amino acids that facilitate synthesis, handling, or use of the peptide, including, but not limited to, one or two lysines at the N-terminus and/or C-terminus to increase solubility of the peptide.
- Suitable fusion proteins include, but are not limited to, proteins comprising a peptide described herein linked to one or more polypeptides, polypeptide fragments, or amino acids not generally recognized to be part of the protein sequence.
- a fusion peptide comprises the entire amino acid sequences of two or more peptides or, alternatively, comprises portions (fragments) of two or more peptides.
- a peptide described herein is operably linked to, for instance, one or more of the following: a marker protein, a peptide that facilitates purification, a peptide sequence that promotes formation of multimeric proteins, or a fragment of any of the foregoing.
- Suitable fusion partners include, but are not limited to, a His tag, a FLAG tag, a strep tag, and a myc tag.
- a peptide described herein is fused to one or more entities that enhance the half life of the peptide.
- Half life can be increased by, e.g., increasing the molecular weight of the peptide to avoid renal clearance and/or incorporating a ligand for the nFc receptor-mediated recycling pathway.
- the peptide is fused to or chemically conjugated to an albumin polypeptide or a fragment thereof (e.g., human serum albumin (HSA) or bovine serum albumin (BSA)).
- HSA human serum albumin
- BSA bovine serum albumin
- the peptide is fused to or complexed with an albumin binding domain or fatty acid that binds albumin when administered in vivo.
- an albumin binding domain is "albu-tag," a moiety derived from on 4-(p-iodophenyl)-butanoic acid (Dumelin et al., Angew Chem Int Ed Engl 47:3196-3201 (2008)).
- Other suitable fusion partners include, but are not limited to, a proline- alanine- serine multimer (PASylation) and an antibody or fragment thereof (e.g., an Fc portion of an antibody).
- Derivatives are contemplated and include peptides that have been chemically modified in some manner distinct from addition, deletion, or substitution of amino acids.
- a peptide provided herein is chemically bonded with polymers, lipids, other organic moieties, and/or inorganic moieties. Examples of peptide and protein modifications are given in
- the peptides described herein optionally comprise a functional group that facilitates conjugation to another moiety (e.g., a peptide moiety).
- exemplary functional groups include, but are not limited to, isothiocyanate, isocyanate, acyl azide, NHS ester, sulfonyl chloride, aldehyde, epoxide, oxirane, carbonate, arylating agent, imidoester, carbodiimide, anhydride, alkyl halide derivatives (e.g., haloacetyl derivatives), maleimide, aziridine, acryloyl derivatives, arylating agents, thiol-disulfide exchange reagents (e.g., pyridyl disulfides or TNB thiol), diazoalkane, carboyldiimadazole, ⁇ , ⁇ '- Disuccinyl carbonate
- isothiocyanate e.g.,
- Maleimide is useful, for example, for generating a Protein S-binding peptide that binds with albumin in vivo.
- the invention includes peptides described herein covalently modified to include one or more water soluble polymer attachments.
- a water soluble polymer (or other chemical moiety) is attached to any amino acid residue, although attachment to the N- or C- terminus is preferred in some embodiments.
- Useful polymers include, but are not limited to,
- PEG e.g., PEG approximately 40 kD, 30 kD, 20 kD, 10 kD, 5 kD, or 1 kD in size
- the peptide of the invention is a PEGylated peptide.
- PEG moieties are available in different shapes, e.g., linear or branched.
- moieties useful for improving peptide half life or stability include, for instance, albumin (optionally modified to allow conjugation to the inventive peptide), fatty acid chains (e.g., C12-C18 fatty acid, such as a C14 fatty acid, or dicarboxylic acids, such as octadecane dicarboxylic acid (oddc)), an antibody or fragment thereof (e.g., an Fc portion of an antibody), and proline- alanine- serine multimers.
- albumin optionally modified to allow conjugation to the inventive peptide
- fatty acid chains e.g., C12-C18 fatty acid, such as a C14 fatty acid, or dicarboxylic acids, such as octadecane dicarboxylic acid (oddc)
- oddc octadecane dicarboxylic acid
- an antibody or fragment thereof e.g., an Fc portion of an antibody
- a peptide derivative in another aspect, includes a targeting moiety specific for a particular cell type, tissue, and/or organ.
- the peptide is linked to one or more chemical moieties that facilitate purification, detection, multimerization, binding with an interaction partner, and characterization of peptide activity.
- An exemplary chemical moiety is biotin.
- Other moieties suitable for conjugation to the peptide of the invention include, but are not limited to, a photosensitizer, a dye, a fluorescence dye, a radionuclide, a radionuclide-containing complex, an enzyme, a toxin, and a cytotoxic agent.
- Photosensitizers include, e.g., Photofrin, Visudyne, Levulan, Foscan, Metvix, Hexvix®, CysviewTM, Laserphyrin, Antrin, Photochlor, Photosens, Photrex, Lumacan, Cevira, Visonac, BF-200 ALA, and Amphinex. If desired, a His tag, a FLAG tag, a strep tag, or a myc tag is conjugated to the peptide.
- the peptides of the invention are acylated at the N-terminal amino acid of the peptide. In another aspect, the peptides of the invention are amidated at the C- terminal amino acid of the peptide. In a still further aspect, the peptides of the invention are acylated at the N-terminal amino acid of the peptide and are amidated at the C-terminal amino acid of the peptide.
- Derivatives also include peptides comprising modified or non-proteinogenic amino acids or a modified linker group (see, e.g., Grant, Synthetic Peptides: A User's Guide, Oxford University Press (1992)).
- Modified amino acids include, for example, amino acids wherein the amino and/or carboxyl group is replaced by another group.
- Non-limiting examples include modified amino acids incorporating thioamides, ureas, thioureas, acylhydrazides, esters, olefines, sulfonamides, phosphoric acid amides, ketones, alcohols, boronic acid amides, benzodiazepines and other aromatic or non-aromatic heterocycles (see Estiarte et al., Burgers Medicinal Chemistry, 6 edition, Volume 1, Part 4, John Wiley & Sons, New York (2002)).
- Non- proteinogenic amino acids include, but are not limited, to ⁇ -alanine (Bal), norvaline (Nva), norleucine (Nle), 4-aminobutyric acid ( ⁇ -Abu), 2-aminoisobutyric acid (Aib), 6-aminohexanoic acid ( ⁇ -Ahx), ornithine (Orn), hydroxyproline (Hyp), taurine, sarcosine, citrulline (Cit), cysteic acid (Coh), cyclohexylalanine (Cha), methioninesulfoxide (Meo), methioninesulfone (Moo), homoserinemethylester (Hsm), propargylglycine (Eag), 5-fluorotryptophan (5Fw), 6- fluorotryptophan (6Fw), 3',4'-dimethoxyphenyl-alanine (Ear), 3',4'-difluorophenylalanine (Dff), 4'
- modified linkers include, but are not limited to, the flexible linker 4,7,10-trioxa-l,13-tridecanediamine (Ttds), glycine, 6- aminohexanoic acid, beta-alanine (Bal), pentynoic acid (Pyn), and combinations of Ttds, glycine, 6-aminohexanoic acid and Bal.
- Homologs of the amino acids constituting the peptides of the invention may be as set forth in Table 1. In any embodiment, one or more amino acids of the peptide of the invention are substituted with an amino acid or building block set forth in Table 1.
- Methylphenylalanine Nitrophenylalanine, Y, Naphtylalanine, 1,2,3,4-L- tetrahydroisoquinolinecarboxylic acid (Tic), ⁇ -Homo tyro sine (Bhy)
- the peptide (CO-NH) linkages joining amino acids within the peptide of the invention are reversed to create a "retro-modified" peptide, i.e., a peptide comprising amino acid residues assembled in the opposite direction (NH-CO bonds) compared to the reference peptide.
- the retro-modified peptide comprises the same amino acid chirality as the reference peptide.
- An "inverso-modified" peptide is a peptide of the invention comprising amino acid residues assembled in the same direction as a reference peptide, but the chirality of the amino acids is inverted.
- the "inverso-modified” peptide comprises D-amino acids, and vice versa.
- Inverso-modified peptides comprise CO-NH peptide bonds.
- a "retro-inverso modified” peptide refers to a peptide comprising amino acid residues assembled in the opposite direction and which have inverted chirality.
- a retro-inverso analogue has reversed termini and reversed direction of peptide bonds (i.e., NH-CO), while approximately maintaining the side chain topology found in the reference peptide.
- Retro-inverso peptidomimetics are made using standard methods, including the methods described in Meziere et al, J. Immunol. , 159, 3230-3237 (1997), incorporated herein by reference. Partial retro-inverso peptides are peptides in which only part of the amino acid sequence is reversed and replaced with enantiomeric amino acid residues.
- a peptide inhibitor described herein is conjugated to a carrier peptide or protein, such as a cargo protein or peptide, a transport peptide or peptide, or a cell penetrating protein or peptide (CPP) to improve pharmacokinetic behavior.
- a carrier peptide or protein such as a cargo protein or peptide, a transport peptide or peptide, or a cell penetrating protein or peptide (CPP) to improve pharmacokinetic behavior.
- CPP cell penetrating protein or peptide
- a peptide inhibitor described herein is conjugated with a transport peptide or cell penetrating peptide selected from the group consisting of the HIV TAT peptide, TATm, PTD, PTR (also referred to as PTD), pVEC, SynB, R9, R9-TAT, MTS, PreS2-TLM, HTLV-II REX, MAP, TP, PEP, and PrP.
- a transport peptide or cell penetrating peptide selected from the group consisting of the HIV TAT peptide, TATm, PTD, PTR (also referred to as PTD), pVEC, SynB, R9, R9-TAT, MTS, PreS2-TLM, HTLV-II REX, MAP, TP, PEP, and PrP.
- a peptide inhibitor described herein is admixed with a carrier peptide or protein, such as a cargo protein or peptide, a transport peptide or peptide, or a cell penetrating protein or peptide (CPP) to improve pharmacokinetic behavior.
- a carrier peptide or protein such as a cargo protein or peptide, a transport peptide or peptide, or a cell penetrating protein or peptide (CPP) to improve pharmacokinetic behavior.
- Suitable molar ratio between the peptide inhibitor and the carrier peptide or protein can be, for example, about 1: 100 to about 100:1, including for example about 1:50 to about 50:1, about 1:20 to about 20:1, about 1:10 to about 10:1, about 1:5 to about 5:1, or about 1:1.
- the molar ratio between the peptide inhibitor and the carrier peptide or protein about any of about 1:1 to about 20:1, about 1:1 to about 10:1, about 1:1 to about 1:5:1, about 1:1 to about 2:1, about 1:1, about 1:1 to about 1:2, about 1:1 to about 1:5, about 1:1 to about 1:10, or about 1:1 to about 1:20.
- Intracellular delivery of the peptides described herein for prophylaxis or treatment of a cancer as described herein can be achieved utilizing a "facilitator moiety" for facilitating passage or translocation of the peptide or nucleic acid across the outer cell/plasma membrane into the cytoplasm and/or nucleus of cells, such as a carrier peptide.
- a "facilitator moiety" for facilitating passage or translocation of the peptide or nucleic acid across the outer cell/plasma membrane into the cytoplasm and/or nucleus of cells, such as a carrier peptide.
- a facilitator moiety as described herein may facilitate the entry of a peptide, agent or nucleic acid embodied by the invention into a cancer cell in any of a number of ways and the invention is not limited to any particular mechanism (e.g., direct penetration into the cell (e.g., via enhanced cell membrane solubility or formation of a transient pore in the cell membrane), endocytosis-mediated cell entry (e.g., via interaction with cell a surface expressed receptor, or macropinocytosis), and cell entry via formation of a transitory structure on the cell membrane.
- direct penetration into the cell e.g., via enhanced cell membrane solubility or formation of a transient pore in the cell membrane
- endocytosis-mediated cell entry e.g., via interaction with cell a surface expressed receptor, or macropinocytosis
- cell entry via formation of a transitory structure on the cell membrane e.g., direct penetration into the cell (e.g., via enhanced cell membrane solubility or formation of
- the facilitator moiety can be a lipid moiety or other non-peptide moiety (e.g., a carbohydrate moiety) which enhances cell membrane solubility of an anti-cancer peptide in accordance with the invention for passage across the outer cell membrane of the target cell or whereby entry of the peptide into the cell is facilitated.
- the lipid moiety can for instance be selected from triglycerides, including mixed triglycerides. Fatty acids and particularly, C16-C20 fatty acids can also be used. Typically, the fatty acid will be a saturated fatty acid and most usually, stearic acid.
- a peptide described herein is conjugated with a conjugation agent for forming a complex with a label, signalling, or other molecule (e.g., a contrast agent, imaging agent, biotin, streptavidin, radioisotope, fluorescent dye,
- a peptide embodied by the invention can be coupled to a facilitator moiety for facility passage of the peptide into a target cell and a conjugation agent complexed with, or for being complexed to, a label, signaling molecule, radioisotope or the like for detection of the peptide within the cell utilising a suitable imaging technique (e.g., magnetic resonance imaging (MRI)).
- MRI magnetic resonance imaging
- DOTA (1,4,7, 10-tetraazacyclodecane-l,4,7,10-tetraacetic acid
- a conjugation agent that may be used and can be complexed to a range of compounds for use in cancer therapy and diagnosis such as monoclonal antibodies, radioisotopes, and metal cations (e.g., calcium and gadolinium).
- a peptide described herein is conjugated to DOTA complexed with gadolinium (Sturzu A et al, 2008) as a contrast agent for imaging of target cells.
- the peptides described herein are formulated into liposomes, microparticles, or nanoparticles, for example for targeted delivery and/or sustained release.
- Liposomes, microparticles, and nanoparticles suitable for peptide delivery are known in the art. See, e.g., Tan et al., Recent
- Nanoparticle conjugates comprising one ore more of the peptides described herein are also contemplated.
- nanoparticle refers to means a particle whose size is measured in the nanometers range (i.e., less than ⁇ ). In some embodiments, the
- nanoparticle has a total diameter in the range of approximately 2-500 nm, including for example about 2 to about 200 nm, about 10 to about 200 nm, about 50 to about 100 nm.
- the nanoparticle core material can be a metal or semiconductor and may be formed of more than one type of atom.
- the core material is a metal selected from Au, Fe or Cu.
- Nanoparticle cores may also be formed from alloys including Au/Fe, Au/Cu, Au/Gd, Au/Fe/Cu, Au/Fe/Gd and Au/Fe/Cu/Gd, and may be used in the present invention.
- Preferred core materials are Au and Fe, with the most preferred material being Au.
- the cores of the nanoparticles preferably comprise between about 100 and 500 atoms (e.g. gold atoms) to provide core diameters in the nanometer range.
- NMR active atoms include Mn +2 , Gd +3 , Eu +2 , Cu +2 , V +2 , Co +2 , Ni +2 , Fe +2 , Fe +3 and lanthanides "1"3 , or quantum dots.
- Nanoparticle cores comprising semiconductor atoms can be detected as nanometre scale semiconductor crystals are capable of acting as quantum dots, that is they can absorb light thereby exciting electrons in the materials to higher energy levels, subsequently releasing photons of light at frequencies characteristic of the material.
- An example of a semiconductor core material is cadmium selenide, cadmium sulphide, cadmium tellurium.
- the zinc compounds such as zinc sulphide.
- the nanoparticle conjugate comprises a detectable label.
- the label may be an element of the core of the nanoparticle or the ligand.
- the label may be detectable because of an intrinsic property of that element of the nanoparticle or by being linked, conjugated or associated with a further moiety that is detectable.
- Preferred examples of labels include a label which is a fluorescent group, a radionuclide, a magnetic label or a dye.
- Fluorescent groups include fluorescein, rhodamine or tetramethyl rhodamine, Texas-Red, Cy3, Cy5, etc., and may be detected by excitation of the fluorescent label and detection of the emitted light using Raman scattering spectroscopy (Y. C. Cao, R. Jin, C. A. Mirkin, Science 2002, 297: 1536-1539).
- the nanoparticle conjugate comprise a radionuclide for use in detecting the nanoparticle using the radioactivity emitted by the radionuclide, e.g. by using PET, SPECT, or for therapy, i.e. for killing target cells.
- radionuclides commonly used in the art that could be readily adapted for use in the present invention include "mTc, which exists
- radionuclides as labels and tracers is well known in the art and could readily be adapted by the skilled person for use in the aspects of the present invention.
- the radionuclides may be employed most easily by doping the cores of the nanoparticles or including them as labels present as part of ligands immobilised on the nanoparticles.
- the nanoparticles conjugates can be detected using a number of techniques well known in the art using a label associated with the nanoparticle as indicated above or by employing a property of them. These methods of detecting nanoparticles can range from detecting the aggregation that results when the nanoparticles bind to another species, e.g. by simple visual inspection or by using light scattering (transmittance of a solution containing the nanoparticles), to using sophisticated techniques such as transmission electron microscopy (TEM) or atomic force microscopy (AFM) to visualize the nanoparticles.
- TEM transmission electron microscopy
- a further method of detecting metal particles is to employ plasmon resonance that is the excitation of electrons at the surface of a metal, usually caused by optical radiation.
- SPR surface plasmon resonance
- a metal such as Ag or Au
- a dielectric material such as air or water.
- SPR surface plasmon resonance
- the nanoparticles include or are doped with atoms which are NMR active, then this technique can be used to detect the particles, both in vitro or in vivo, using techniques well known in the art.
- Nanoparticles can also be detected using a system based on quantitative signal amplification using the nanoparticle-promoted reduction of silver (I). Fluorescence spectroscopy can be used if the nanoparticles include ligands as fluorescent probes. Also, isotopic labelling of the carbohydrate can be used to facilitate their detection.
- a peptide described herein is conjugated to a gold nanoparticle for imaging or for assisted cell death of the target cells through laser irradiation of branched gold particles. Gold nanoparticle transfer across plasma and nuclear membranes has been reported previously (de la Fuente J. M. and Berry C. C, 2005).
- the peptides described herein can be useful for many purposes. For example, in some embodiments, there is provided a method of disrupting the interaction between MTDH and SND1 in a subject, comprising administering to the subject any one of the peptide inhibitors (or pharmaceutical compositions comprising such peptide inhibitors) described herein.
- a method of inhibiting SND1 -dependent expression of prosurvival genes in a subject comprising administering to the subject any one of the peptide inhibitors (or pharmaceutical compositions comprising such peptide inhibitors) described herein.
- a method of treating cancer in a subject comprising administering to the subject any one of the peptide inhibitors (or pharmaceutical compositions comprising such peptide inhibitors) described herein.
- a method of inhibiting tumor metastasis in a subject having cancer comprising administering to the subject any one of the peptide inhibitors (or pharmaceutical compositions comprising such peptide inhibitors) described herein.
- exemplary cancers include, but are not limited to, adrenocortical carcinoma, AIDS- related cancers, AIDS-related lymphoma, anal cancer, anorectal cancer, cancer of the anal canal, appendix cancer, childhood cerebellar astrocytoma, childhood cerebral astrocytoma, basal cell carcinoma, skin cancer (non-melanoma), biliary cancer, extrahepatic bile duct cancer, intrahepatic bile duct cancer, bladder cancer, urinary bladder cancer, bone and joint cancer, osteosarcoma and malignant fibrous histiocytoma, brain cancer, brain tumor, brain stem glioma, cerebellar astrocytoma, cerebral astrocytoma/malignant glioma, epen
- hypopharyngeal cancer intraocular melanoma, ocular cancer, islet cell tumors (endocrine pancreas), Kaposi Sarcoma, kidney cancer, renal cancer, kidney cancer, laryngeal cancer, acute lymphoblastic leukemia, acute myeloid leukemia, chronic lymphocytic leukemia, chronic myelogenous leukemia, hairy cell leukemia, lip and oral cavity cancer, liver cancer, lung cancer, non-small cell lung cancer, small cell lung cancer, AIDS-related lymphoma, non-Hodgkin lymphoma, primary central nervous system lymphoma, Waldenstram macroglobulinemia, meduUoblastoma, melanoma, intraocular (eye) melanoma, merkel cell carcinoma, mesothelioma malignant, mesothelioma, metastatic squamous neck cancer, mouth cancer, cancer of the tongue, multiple endocrine neoplasia syndrome, mycosis fun
- rhabdomyosarcoma salivary gland cancer, ewing family of sarcoma tumors, Kaposi Sarcoma, soft tissue sarcoma, uterine cancer, uterine sarcoma, skin cancer (non-melanoma), skin cancer (melanoma), merkel cell skin carcinoma, small intestine cancer, soft tissue sarcoma, squamous cell carcinoma, stomach (gastric) cancer, supratentorial primitive neuroectodermal tumors, testicular cancer, throat cancer, thymoma, thymoma and thymic carcinoma, thyroid cancer, transitional cell cancer of the renal pelvis and ureter and other urinary organs, gestational trophoblastic tumor, urethral cancer, endometrial uterine cancer, uterine sarcoma, uterine corpus cancer, vaginal cancer, vulvar cancer, and Wilm's Tumor.
- the cancer is selected from the group consisting of breast cancer, liver cancer, colon cancer, lung cancer and prostate cancer.
- the cancer is prostate cancer.
- the cancer is breast cancer.
- Prostate Cancer is the most common type of cancer among men in the United States. It accounts for 28% of the total cancer incidences and 10% of the total cancer deaths of American men (1). Prostate cancer-related mortality is mainly caused by advanced cancers that have disseminated and metastasized to distant organs such as the bone, lung and liver (2). Once cancer cells establish secondary tumors in these vital organs, treatment usually becomes more complicated with worse outcome. Defining and understanding novel molecular targets that drive prostate cancer progression and metastasis is important for developing effective therapeutic approaches for prostate cancer.
- MTDH was shown to be overexpressed in prostate tumor tissues and tumorigenic cell lines compared to benign hyperplastic tissues and normal epithelial cells (Thirkettle et al., 2009; Kikuno et al., 2007). Knockdown of MTDH in cultured prostate cancer cells enhanced apoptosis and inhibited Matrigel invasion.
- the disclosure provides MTDH/SND1 inhibitors useful in the treatment of cancer (e.g., to inhibit or suppress growth of or metastasis of a tumor).
- a composition comprising one or more pharmaceutically acceptable carriers.
- Pharmaceutically or pharmacologically acceptable carriers or vehicles refer to molecular entities and compositions that do not produce allergic, or other adverse reactions when administered using routes well-known in the art, as described below, or are approved by the U.S. Food and Drug Administration or a counterpart foreign regulatory authority as an acceptable additive to orally or parenterally administered pharmaceuticals.
- Pharmaceutically acceptable carriers include any and all clinically useful solvents, dispersion media, coatings, antibacterial and antifungal agents, isotonic and absorption delaying agents and the like.
- Pharmaceutical carriers include pharmaceutically acceptable salts, particularly where a basic or acidic group is present in a compound.
- an acidic substituent such as— COOH
- the ammonium, sodium, potassium, calcium and the like salts are contemplated for administration.
- pharmaceutically acceptable esters of the compound ⁇ e.g., methyl, tert-butyl, pivaloyloxymethyl, succinyl, and the like
- esters being known in the art for modifying solubility and/or hydrolysis characteristics for use as sustained release or prodrug formulations.
- an acidic salt such as hydrochloride, hydrobromide, acetate, maleate, pamoate, phosphate, methanesulfonate, p-toluenesulfonate, and the like, is contemplated as a form for administration.
- compounds may form solvates with water or common organic solvents. Such solvates are contemplated as well.
- the inhibitors herein may be administered orally, parenterally, transocularly, intranasally, transdermally, transmucosally, by inhalation spray, vaginally, rectally, or by intracranial injection.
- parenteral as used herein includes subcutaneous injections, intravenous, intramuscular, intracisternal injection, or infusion techniques. Administration by intravenous, intradermal, intramusclar, intramammary, intraperitoneal, intrathecal, retrobulbar, intrapulmonary injection and or surgical implantation at a particular site is contemplated as well.
- compositions for administration by any of the above methods are essentially free of pyrogens, as well as other impurities that could be harmful to the recipient. Further,
- compositions for administration parenterally are sterile.
- the MTDH/SND1 inhibitor is administered in a therapeutically effective amount; typically, the composition is in unit dosage form.
- the amount of inhibitor administered is, of course, dependent on the age, weight, and general condition of the patient, the severity of the condition being treated, and the judgment of the prescribing- physician. Suitable therapeutic amounts will be known to those skilled in the art and/or are described in the pertinent reference texts and literature.
- the dose is administered either one time per day or multiple times per day.
- the MTDH/SND1 inhibitor may be administered one, two, three or four times per day.
- an effective dosage of inhibitor may be within the range of 0.01 mg to 1000 mg per kg (mg/kg) of body weight per day.
- the inhibitor is administered at a daily dose ranging from about 10 mg/kg to about 250 mg/kg, or from about 100 mg/kg to about 250 mg/kg, or from about 60 mg/kg to about 100 mg/kg or from about 50 mg/kg to about 90 mg/kg, or from about 30 mg/kg to about 80 mg/kg, or from about 20 mg/kg to about 60 mg/kg, or from about 10 mg/kg to about 50 mg/kg.
- the effective dose may be 0.5 mg/kg, 1 mg/kg, 5 mg/kg, 10 mg/kg, 15 mg/kg, 20 mg/kg/ 25 mg/kg, 30 mg/kg, 35 mg/kg, 40 mg/kg, 45 mg/kg, 50 mg/kg, 55 mg/kg, 60 mg/kg, 70 mg/kg, 75 mg/kg, 80 mg/kg, 90 mg/kg, 100 mg/kg, 125 mg/kg, 150 mg/kg, 175 mg/kg, 200 mg/kg, 250 mg/kg, 300 mg/kg, 350 mg/kg, 400 mg/kg, 450 mg/kg, 500 mg/kg, and may increase by 25 mg/kg increments up to 1000 mg/kg, or may range between any two of the foregoing values.
- Administration may continue for at least 3 months, 6 months, 9 months, 1 year, 2 years, or more.
- compositions described herein can be administered in therapeutically effective dosages alone or in combination with adjunct cancer therapy such as surgery, chemotherapy, radiotherapy, immunotherapy, thermotherapy, and laser therapy, and may provide a beneficial effect, e.g. reducing tumor size, slowing rate of tumor growth, inhibiting metastasis, sensitizing tumors to cancer treatments, or otherwise improving overall clinical condition, without necessarily eradicating the cancer.
- adjunct cancer therapy such as surgery, chemotherapy, radiotherapy, immunotherapy, thermotherapy, and laser therapy
- a beneficial effect e.g. reducing tumor size, slowing rate of tumor growth, inhibiting metastasis, sensitizing tumors to cancer treatments, or otherwise improving overall clinical condition, without necessarily eradicating the cancer.
- Cytostatic and cytotoxic agents that target the cancer cells are specifically contemplated for combination therapy.
- agents that target angiogenesis or lymphangiogenesis, or immune therapies targeting checkpoint pathways are specifically contemplated for combination therapy.
- chemotherapeutic agent is a chemical compound useful in the treatment of cancer.
- chemotherapeutic agents include: alkylating agents such as thiotepa and CYTOXAN® cyclosphosphamide; alkyl sulfonates such as busulfan, improsulfan and piposulfan; aziridines such as benzodopa, carboquone, meturedopa, and uredopa;
- ethylenimines and methylamelamines including altretamine, triethylenemelamine,
- trietylenephosphoramide, triethiylenethiophosphoramide and tiimethylolomelamine acetogenins (especially bullatacin and bullatacinone); a camptothecin (including the synthetic analogue topotecan); bryostatin; callystatin; CC-1065 (including its adozelesin, carzelesin and bizelesin synthetic analogues); cryptophycins (particularly cryptophycin 1 and cryptophycin 8); dolastatin; duocarmycin (including the synthetic analogues, KW-2189 and CB1-TM1); eleutherobin;
- pancratistatin a sarcodictyin; spongistatin; nitrogen mustards such as chlorambucil,
- chlornaphazine cholophosphamide, estramustine, ifosfamide, mechlorethamine, mechlorethamine oxide hydrochloride, melphalan, novembichin, phenesterine, prednimustine, trofosfamide, uracil mustard; nitrosureas such as carmustine, chlorozotocin, fotemustine, lomustine, nimustine, and ranimnustine; vinca alkaloids; epipodophyllotoxins; antibiotics such as the enediyne antibiotics (e.g., calicheamicin, especially calicheamicin gammall and
- calicheamicin omegall calicheamicin omegall
- L-asparaginase anthracenedione substituted urea
- methyl hydrazine derivatives dynemicin, including dynemicin A
- bisphosphonates such as clodronate
- an esperamicin as well as neocarzino statin chromophore and related chromoprotein enediyne antiobiotic chromophore s
- aclacinomysins actinomycin, authramycin, azaserine, bleomycins, cactinomycin, carabicin, carminomycin, carzinophilin, chromomycinis, dactinomycin, daunorubicin, detorubicin, 6-diazo-5-oxo-L-norleucine, ADRIAMYCIN® doxorubicin
- morpholino-doxorubicin including morpholino-doxorubicin, cyanomorpholino-doxorubicin, 2-pyrrolino-doxorubicin and deoxydoxorubicin
- epirubicin including morpholino-doxorubicin, cyanomorpholino-doxorubicin, 2-pyrrolino-doxorubicin and deoxydoxorubicin
- epirubicin including esorubicin, idarubicin, marcellomycin, mitomycins such as mitomycin C, mycophenolic acid, nogalamycin, olivomycins, peplomycin, potfiromycin, puromycin, quelamycin, rodorubicin, streptonigrin, streptozocin, tubercidin, ubenimex, zinostatin, zorubicin
- anti-metabolites such as methotrexate and 5-fluorouracil (5-FU); folic acid analogs such as denopterin, methot
- diaziquone diaziquone; elfornithine; elliptinium acetate; an epothilone; etoglucid; gallium nitrate;
- hydroxyurea lentinan; lonidainine; maytansinoids such as maytansine and ansamitocins;
- mitoguazone mitoxantrone; mopidanmol; nitiaerine; pentostatin; phenamet; pirarubicin;
- losoxantione podophyllinic acid; 2-ethylhydrazide; procarbazine; PSK® polysaccharide complex (JHS Natural Products, Eugene, OR); razoxane; rhizoxin; sizofiran; spirogermanium; tenuazonic acid; triaziquone; 2,2 2"-trichlorotiiethylamine; trichothecenes (especially T-2 toxin, verracurin A, roridin A and anguidine); urethan; vindesine; dacarbazine; mannomustine;
- DFMO difluoromethylornithine
- retinoids such as retinoic acid
- capecitabine leucovorin (LV)
- irenotecan adrenocortical suppressant
- adrenocortico steroids progestins; estrogens; androgens; gonadotropin-releasing hormone analogs; and pharmaceutically acceptable salts, acids or derivatives of any of the above.
- anti-hormonal agents that act to regulate or inhibit hormone action on tumors
- SERMs selective estrogen receptor modulators
- tamoxifen including NOLVADEX® tamoxifen
- raloxifene including NOLVADEX® tamoxifen
- droloxifene 4-hydroxytamoxifen
- trioxifene keoxifene
- LY117018 onapristone
- aromatase inhibitors that inhibit the enzyme aromatase, which regulates estrogen production in the adrenal glands, such as, for example, 4(5)- imidazoles, aminoglutethimide, MEGASE® megestrol acetate, AROMASL® exemestane, formestanie, fadrozole, RIVISOR® vorozole, FEMARA® letrozole, and ARTMIDEX® anastrozole
- anti-androgens such as flutamide, n
- the peptides described herein are administered in conjunction with any number of immune checkpoint inhibitors.
- Immune checkpoint inhibitors include antibodies, or antigen binding fragments thereof, that bind to and block or inhibit the activity of one or more of CTLA-4, PDL1, PDL2, PD1, B7-H3, B7-H4, BTLA, HVEM, TIM3, and GAL9.
- Exemplary immune checkpoint inhibitors include, but are not limited to, Tremelimumab (CTLA- 4 blocking antibody), anti-OX40, PD-L1 monoclonal Antibody (Anti-B7-Hl; MEDI4736), ipilimumab, MK-3475 (PD-1 blocker) and Nivolumamb (anti-PDl antibody).
- the treatment methods described herein optionally include monitoring the effect of the therapeutic composition on the tumor.
- the size of the tumor can be determined, as can the presence of metastases.
- measurement of the degree of metastasis e.g., by measuring the number of metastatic modules or by measurement of ascites associated with metastasis.
- the MTDH/SND1 inhibitor and other drugs/therapies can be administered in combination either simultaneously in a single composition or in separate compositions.
- the administration is sequential. Simultaneous administration is achieved by administering a single composition or pharmacological protein formulation that includes both the inhibitor and other therapeutic agent(s). Alternatively, the other therapeutic agent(s) are taken separately at about the same time as a pharmacological formulation (e.g., tablet, injection or drink) of the inhibitor.
- a pharmacological formulation e.g., tablet, injection or drink
- kits for carrying out the methods of the disclosure.
- the kit contains, e.g., bottles, vials, ampoules, tubes, cartridges and/or syringes that comprise a liquid (e.g., sterile injectable) formulation or a solid (e.g., lyophilized) formulation.
- a liquid e.g., sterile injectable
- a solid e.g., lyophilized
- kits can also contain pharmaceutically acceptable vehicles or carriers (e.g., solvents, solutions and/or buffers) for reconstituting a solid (e.g., lyophilized) formulation into a solution or suspension for administration (e.g., by injection), including without limitation reconstituting a lyophilized formulation in a syringe for injection or for diluting concentrate to a lower concentration.
- pharmaceutically acceptable vehicles or carriers e.g., solvents, solutions and/or buffers
- extemporaneous injection solutions and suspensions can be prepared from, e.g., sterile powder, granules, or tablets comprising a composition comprising an inhibitor as described herein.
- kits can also include dispensing devices, such as aerosol or injection dispensing devices, pen injectors, autoinjectors, needleless injectors, syringes, and/or needles.
- the kit also provides an oral dosage form, e.g., a tablet or capsule or other oral formulation described herein, of the inhibitor for use in the method.
- the kit also provides instructions for use.
- mice All experimental protocols involving mice were approved by Institutional Animal Care and Use Committee (IACUC) of Princeton University. Mtdh-KO mice were generated by injecting ES cell line XB780 (BayGenomics) into the C57BL/6 blastocysts followed by confirmation of germline transmission. KO mice were then backcrossed to FVB background for >6 generations before breeding with MMTV-PyMT, MMTV-ErbB2, MMTV- Wnt transgenic mice (Jackson Laboratory) in FVB background.
- IACUC Institutional Animal Care and Use Committee
- mouse Mtdh coding sequence was inserted into pMMTV-SV40 vector, then the expression cassette was linearized and microinjected into the pronuclei of zygotes from FVB mice.
- female mice carrying the specific oncogenes were examined weekly for mammary tumors. Tumors were considered established when they became palpable for two consecutive weeks, and tumors were measured by calipers for calculation of tumor volumes ( ⁇ x length x width 16). Lung nodules were counted directly after fixation (MMTV-PyMT models) or after sectioning and staining of the lungs (MMTV-ErbB2 model).
- CTGCAAAACAAGCACCAGAG -3' (located in the second exon of Mtdh) (SEQ ID NO: 6) and reverse primer 5'-GTTTTCCCAGTCACGACGTT -3' (located in the targeting vector pGTOpfs) ID NO: 7) were used to amplify the fragment covering the insertion site using the Expand 20 kb PUS PCR System (Roche).
- the PCR product was sequenced, and the insertion site was determined to be at the 3041 bp of the second intron of Mtdh. Based on the insertion site, three genotyping primers were designed to distinguish Mtdh , Mtdh " and MtdK ⁇ mice as indicated below.
- Wild-type allele corresponds to a 602 bp PCR fragment
- gene trap allele corresponds to a 472 bp PCR fragment.
- the PCR primers for genotyping the wild-type and gene- trapped Mtdh alleles are in Table 1 :
- Genotyping of MMTV-Mtdh transgene Southern blot genotyping was performed by the Transgenic & Knockout Shared Resource at the Rutgers Cancer Institute of New Jersey to confirm transgenic lines.
- the primers used to identify MMTV-Mtdh transgenic progeny were designed to amplify the linking region between Mtdh and SV40, while primers targeting other genomic DNA region served as internal control.
- the PCR primers for genotyping the MMTV-Mtdh transgene are in Table 2.
- Mammary carcinogenesis Mammary carcinogenesis experiments were performed using the protocol previously described. Briefly, 6-week-old female FVB/N mice with indicated Mtdh genotypes were injected subcutaneously with 15 mg MPA (Depo-Provera, NY, Pfizer) one week before receiving 1 mg DMBA (Sigma- Aldrich Chemical Co, St. Louis, MO) in 0.1 ml cottonseed oil by gavage weekly for four consecutive weeks. Mice were examined weekly for tumor formations.
- MPA Depo-Provera, NY, Pfizer
- DMBA Sigma- Aldrich Chemical Co, St. Louis, MO
- HRP was developed in 3, 3-diaminobenzidein according to manufacturer's instructions (Invitrogen). Tissue sections were counterstained with hemotoxylin, dehydrated and mounted. To quantify cleaved-caspase 3-postive cells in Figure 9A, more than 20 random images (> 2000 cells) were taken under microscope and the positive cells were manually counted.
- Tissue array immunostaining Two breast cancer tissue microarrays were used and stained with antibodies against MTDH (Invitrogen, catalogue # 40-6500) and SND1 (Sigma, HPA002632).
- One TMA was obtained from Biomax (BR1921a) and composed of 160 primary tumors, 5 normal tissue and 27 normal adjacent tissues. Out of these 192 samples, 154 samples were stained successfully for both MTDH and SND1.
- a second TMA was obtained from the Cancer Institute of New Jersey (CINJ YMTA_201) and composed of 399 primary tumors. Out of these 399 samples, 270 samples were stained successfully for both MTDH and SND1. Each sample was scored as negative (0), weak (1), moderate (2), or strong (3) according to staining intensities.
- IP immunoprecipitation
- IP bead preparation 30 ⁇ of protein A/G beads were incubated with 5 ⁇ g of antibodies for 2 h at 4 °C. IPs were carried out for overnight at 4 °C, and beads were centrifuged, washed and subsequently boiled for 5 min in SDS protein loading buffer to elute bound protein. IP lysates were subjected to western blotting with indicated antibodies.
- Antibodies against MTDH (invitrogen, catalogue # 40-6500), SND1 (Santa cruz, catalogue # sc-271590), ERBB2 (Cell Signaling, catalogue # 2165), p- ERBB2 (Cell Signaling, catalogue # 2243), EGFR (Cell Signaling, catalogue # 4267), anti-Myc (Santa Cruz, catalogue # sc-40), and anti-HA (Santa Cruz, catalogue # sc-7392) were diluted 1:1000.
- ALDEFLUOR assay and separation of the ALDH-positive population by FACS The ALDEFLUOR kit (StemCell Technologies, Durham, NC, USA) was used to isolate the population with a high ALDH enzymatic activity according to the manufacturer's instructions. Briefly, tumor cells freshly dissociated from spontaneous or transplanted PyMT tumors were suspended in ALDEFLUOR assay buffer containing ALDH substrate (1 ml buffer containing 1 ⁇ /L BAAA per 1 x 10 6 cells) and incubated for 45 min at 37 °C. For each sample of cells, an aliquot was treated with 50 mM ALDH inhibitor diethylaminoben-zaldehyde (DEAB) right before adding the substrate. The sorting gates were established using the cells stained with PI only (for viability) and the ALDEFLUOR- stained cells treated with DEAB as negative controls.
- ALDEFLUOR assay and separation of the ALDH-positive population by FACS The ALDEFLUOR kit (StemCell Technologies, Durham, NC
- Human breast cancer TMAs Two human breast TMAs were used in the study to examine the correlation of MTDH and SND1 protein levels. One TMA was purchased from US Biomax (BR 192 la) and a second TMA was obtained from the Cancer Institute of New Jersey (YMTA_201). Both set of TMAs used de-identified tumor samples and were considered exempt by the Institutional Review Boards of Princeton University and the Rutgers New Jersey Medical School. [0105] Harvesting mammary epithelial cells and flow cytometry: Single cell suspensions of mammary glands or tumors were prepared as previously described (Shackleton et al., 2006).
- tissues were dissected, minced into small pieces and digested for 1 h at 37 °C in culture medium (1:1 DMEM : Ham's F-12 medium containing 5% FBS, 10 ng/ml EGF, 500 ng/ml hydrocortisone, 5 ⁇ g/ml Insulin, 20 ng/ml cholera toxin and 1% Pen/Strep) supplemented with 300 U/ml type 1A collagenase (Sigma) and 100 U/ml hyaluronidase (Sigma).
- Organoids were sequentially suspended with 0.25% trypsin-EDTA for 1.5 min, 5 mg/ml Dispase (Invitrogen) and 0.1 mg/ml DNase (Sigma) for 5 min, and 0.64% ammonium chloride for 5 min at 37 °C before filtration through a 40 ⁇ nylon cell strainer and antibody staining.
- Mammary epithelial cells were incubated with an antibody cocktail containing CD31, CD45, TER119, CD24, CD29 and CD61 for 30 min followed by secondary antibody staining for 20 min before FACS analysis or sorting.
- Mammosphere/tumorsphere assays Single cells were plated in ultralow attachment plates (Corning, Tewksbury, MA) with sphere media (1:1 DMEM: Ham's 12 supplemented with B27 (Invitrogen), 20 ng/mL EGF (Novoprotein), 20 ng/mL bFGF, and 4 ⁇ g/mL heparin).
- Lentiviral infection For knockdown studies, pLKO plasmids containing shRNA sequences that target murine Mtdh (KD1, TRCN0000125816; KD2, TRCN0000125818), murine Sndl (TRCN0000054742), human MTDH (KD1, TRCN0000151467; KD2, TRCN0000322872; KD3, TRCN0000322949) and Human SND1 (KD1, TRCN0000245140; KD2,
- TRCN0000245141 KD3, TRCN0000245144 were purchased from Sigma- Aldrich (St Louis, MO, USA). All lentivirus construct plasmids were packaged into virus using HEK293-T cells as packaging cell lines together with helper plasmids VSVG (envelope) and p8.91 (gag-pol, pCMV- dR8.91) following standard protocols. Primary cells were either spin infected with concentrated virus-containing media supplemented with 8 ⁇ g/mL Polybrene for 2 hr at 1000 g at 4°C and then transplanted, or cultured in plates following infections and sorted before being used in experiments.
- GSEA Gene set enrichment analysis: Normalized microarray expression data were rank-ordered by expression using the provided ratio of classes (i.e. fold change) metric. Multiple probe matches for the same gene were collapsed into one value, with the highest probe reading being used in each case.
- SND1_CPT_UP signature contains 504 genes that are specifically upregulated by SND1 by > 2 fold (p ⁇ 0.05) under CPT treatment.
- SND1_CPT_UP signature was tested for enrichment in rank ordered lists via GSEA using the classic enrichment statistic and compared to enrichment results from 1000 random permutations of the gene set to obtain p values.
- Raw enrichment scores were converted to normalized enrichment scores using default GSEA parameters. Enrichment cores are defined as the members of the gene set that lie before or at the running sum peak (i.e. the enrichment score) of the ranked gene list.
- Mtdh-/- mice were viable, fertile, and displayed no obvious abnormalities when monitored for up to two years.
- MTDH was also detected in normal mammary epithelial cells (MECs) and the expression levels correlated with Mtdh genetic status (Figure 1C).
- MECs normal mammary epithelial cells
- Figure 1C To assess the influence of MTDH deficiency in postnatal mammary gland development, whole mounts of inguinal mammary fat pads from WT and KO virgin mice were examined. Except for a transient delay in ductal outgrowth of mammary glands from 3- and 5-week-old KO mice as compared to WT littermates, significant difference in branching morphogenesis was not observed at later time points or during pregnancy and lactation. The largely comparable mammary epithelium in WT and Mtdh-/- mice starting at puberty therefore allows us to use Mtdh-/- mice to examine the necessity of MTDH for mammary tumor formation.
- Mtdh KO inhibits tumor formation and metastasis in luminal mammary tumors: To dissect the roles of MTDH during autochthonous mammary tumor progression, the MMTV- PyMT and MMTV-ErbB2 transgenic models were first used, both of which develop luminal adenocarcinoma with high incidence of lung metastasis. In the aggressive MMTV-PyMT model, mammary tumors occurred as early as 42 days of age, and by day 63, 50% of Mtdh+/+ mice developed tumors ( Figure ID). In contrast, the first palpable tumor was detected in the Mtdh-/- group at day 50, and 50% of these mice developed tumors only after 80 days.
- MTDH protein levels were elevated in PyMT and ErbB2-driven tumors as compared to age-matched normal controls, suggesting that high levels of MTDH may confer growth advantage to MECs during tumorigenesis.
- Mtdh KO restrains the formation of basal-like and mixed subtypes of mammary tumors:
- the investigation of MTDH in tumor formation was expanded to the MMTV-Wnt model, which develops tumors that exhibit mammary stem cell (MaSC)-like gene expression profiles and resemble the basal subtype of human breast cancer (Herschkowitz et al., 2007). While virtually all Wnt;Mtdh+/+ mice succumbed to cancer at 300 days of age, no tumors were detected in 35% of Wnt;Mtdh+/- and 62% of Wnt;Mtdh-/- mice (Figure 1L). The multiplicity of tumors was also highly dependent on the gene dosage of Mtdh (Figure 1M).
- Mtdh KO impairs the expansion and activities of onco ene-induced basal and luminal TICs: The dramatic effect of Mtdh deletion on mammary tumor formation prompted investigatation of early events during tumorigenesis. To this end, we examined whole mounts (Figure 2A, top panels) and haematoxylin/eo sin- stained sections ( Figure 2A, bottom panels) of mammary glands from different tumor models at preneoplastic stages were examined. Both the PyMT and Wnt oncogenes induced extensive hyperplasia as early as four weeks in Mtdh+/+ mice; however, Mtdh-/- glands exhibited significantly fewer and smaller hyperplasia foci mingled with normal ductal structures.
- MMTV-ErbB2 mice have the longest tumor latency, and this corresponds to significantly delayed and the least severe hyperplasia.
- Whole mount analysis of mammary glands from 6-month-old tumor-free MMTV-ErbB2 females revealed close to 100% incidence of hyperplasia in Mtdh-positive mice, while only 20% of those from
- CD29 ⁇ integrin
- CD61 ⁇ 3 integrin
- Lin-CD24+CD291ow luminal subset (CD24+CD291ow) ( Figures 2B and 2C), consistent with previous reports of "luminal-like" gene expression profiles (Herschkowitz et al., 2007).
- Lin-CD24+CD29high CD24+CD29hi basal population, which enriches for MaSCs, was markedly increased in preneoplastic tissues from Wnt mammary glands
- Mtdh-/- preneoplastic glands indeed contain fewer TICs
- pMECs primary MECs
- MTDH is largely dispensable for adult MaSCs activities, as indicated by similar in vivo mammary gland reconstitution (Figure 31) efficiency of either unfractionated Lin- MECs (Figure 3J) or MaSCs- enriched basal cells ( Figures 3K and 3L) from WT and KO mice.
- MEC-intrinsic role of MTDH in promoting mammary tumor-initiating capacities As MTDH is widely expressed in mice, the tumorigenesis defects in whole-organism KO mice could result from either loss of MTDH in MECs or other cell/tissue types. To distinguish between these two possibilities, mouse MTDH was re-introduced specifically in MECs of Mtdh- /- mice in vivo and it was tested whether this would rescue the tumorigenic defects.
- MMTV-Mtdh transgenic mouse line was created ( Figures 4A, 5A and 5B) and expression of the Mtdh transgene was observed specifically in the mammary gland, and, to a lesser extent, the salivary gland ( Figure 5C).
- these MMTV-Mtdh mice were crossed with PyMT;Mtdh-/- mice to generate PyMT;Mtdh-/- mice with or without exogenous Mtdh transgene ( Figure 4A).
- transgene (Tg)-rescued PyMT;Mtdh-/- tumors expressed similar levels of MTDH as that of PyMT;Mtdh+/+ tumors ( Figure 4B).
- MTDH KD compromised the sphere-forming activities in CD61+ cells from MMTV-Wnt tumors (Figure 40). These results suggest that MTDH is continuously required for the full functionality of TICs in MTDH-positive tumors.
- Pro-tumorigenic role of MTDH requires its interacting partner SNDl: SNDl has been previously identified as the major binding partner of MTDH in human breast cancer cells and it had metastasis-promoting functions similar to MTDH (Blanco et al., 2011). In this study, it was found that the interaction between MTDH and SNDl was well conserved in human and murine breast cancer cells.
- SNDl contains four N-terminal Staphylococcal nuclease (SN) repeats and a C-terminal Vietnamese-SN hybrid domain.
- SN Staphylococcal nuclease
- TPM triple- point mutants
- TPM1 comprises mutations D389R, D393R and W394D.
- TPM2 comprises mutations E399R, E400R and W401D.
- TPM3 comprises mutations W404D, D406R and E407R.
- In vitro binding assays showed that both TPM1 and TPM2 could not bind SNDl AC whereas TPM3 bound SNDl AC as effectively as the WT MTDH ( Figure 7C).
- the mutations are also set out in Table 3.
- MTDH-mediated stabilization of SNDl confers MECs survival advantage under stress conditions: SNDl has been reported as a survival factor under various stress conditions (Gao et al., 2010; Sundstrom et al., 2009; Weissbach and Scadden, 2012). The more prominent role of MTDH in tumor initiation but not normal physiology of mammary glands led to a hypothesis that MTDH, through its interaction with SNDl, confers MECs advantages under stress conditions during tumorigenesis.
- MTDH and SND1 are important for tumor- initiating activities of human breast cancer cells: To demonstrate the important roles of both MTDH and SND1 in tumor- initiating activities in human breast cancer, MTDH or SND1 were silenced in multiple human breast cancer models, including: (1) HER2/Neu-transformed human breast epithelial cells (HMLE-N) (Mani et al., 2008) ( Figures 10A, 10B, 11A and 11B), (2) human primary patient-derived xenografts (DeRose et al., 2011; Zhang et al., 2013b) ( Figures IOC, 10D, 11C and 11D), and (3) the MDA-MB-231 human breast cancer cell line ( Figures 10E-10H).
- HMLE-N Human HER2/Neu-transformed human breast epithelial cells
- Figures 10A, 10B, 11A and 11B Human primary patient-derived xenografts
- Figures IOC, 10D, 11C and 11D Figures IOC
- metastasis-promoting genes have been shown to promote primary tumor growth in xenograft models (Vanharanta and Massague, 2013; Wan et al., 2013).
- the recurrent amplification/overexpression of MTDH in human primary breast tumors may therefore implicate MTDH in tumorigenesis in addition to its reported role in promoting breast cancer metastasis.
- TICs from MTDH-positive established mammary tumors such as ALDH+ cells and CD61+ cells from PyMT and Wnt- induced tumors, respectively, remain sensitive to MTDH inhibition, suggesting that MTDH-dependent mechanisms are at play in established tumors to maintain the optimal functionality of TICs, and therefore blocking MTDH and its regulated pathways will be beneficial to cancer patients with aberrant expression of MTDH.
- tumorigenesis by virtue of their abilities to mediate the conversion between differentiated cells and more primitive stem/progenitor cells in both normal and malignant contexts.
- SNDl As a major binding partner of MTDH (Blanco et al., 2011; Meng et al., 2012; Yoo et al., 2011).
- SNDl a major binding partner of MTDH
- two non- overlapping regions of MTDH namely amino acid 364-470 (Blanco et al., 2011) and 101-205 (Yoo et al., 2011)
- Our current study further determines a minimal fragment of MTDH (386-407) sufficient for the interaction and identifies two key residues within this fragment critical for the interaction.
- Tumorigenesis is accompanied by diverse stresses that tumor cells have to overcome, including oncogene-induced DNA replication/damage stress. It was demonstrated herein that SNDl is required for the expression of a cohort of pro-survival genes in cells under stress conditions, and silencing of SNDl sensitizes transformed MECs to replication stress-induced apoptosis. These results are consistent with previous reports that established SNDl as a pro- survival protein under various stress conditions (Gao et al., 2010; Sundstrom et al., 2009;
- SNDl is a multifunctional protein that has been reported to be involved in several gene regulatory processes, including transcriptional control, mRNA splicing, RNA stress granule formation, and RNA-induced silencing complex (RISC) machinery (reviewed in Wan and Kang, 2013). Future studies are warranted to investigate how SNDl modulates gene expression in response to stress conditions to promote cellular survival.
- RISC RNA-induced silencing complex
- MTDH knockout mice were crossed with transgenic adenomcarcnoma of mouse prostate (TRAMP) mice in order to generate animals that have heterogenous expression of MTDH and TRAMP.
- TRAMP transgenic adenomcarcnoma of mouse prostate
- the TRAMP model results in animals in which the expression of the SV40 T antigen is controlled by the rat Probasin promoter and induced during sex maturity of the mice, leading to prostate cancer development that resembles the clinical progression of human prostate cancer (Greenberg et al., 1995; Gingrich et al., 1999).
- mice All experimental protocols involving mice were approved by the Institutional Animal Care and Use Committee (IACUC) of Princeton University.
- Mtdh-/- mice were generated by injecting ES cell line XB780 (Bay Genomics) containing a gene-trapped allele of Mtdh into C57BL/6 blastocysts followed by confirmation of germline transmission by PCR. All Mtdh-/- mice were backcrossed to C57BL/6 background for > 6 generations before breeding with C57BL/6 TRAMP transgenic mice (Jackson Laboratory) (Greenberg, 2005).
- Female TRAMP mice were bred to Mtdh-/- male mice to obtain female TRAMP/Mtdh+/- and male Mtdh+/- founder mice. Subsequently, these founder mice were bred to generate
- TRAMP/Mtdh+/+, TRAMP/Mtdh+/-, and TRAMP/Mtdh-/- male mice were obtained from the tail biopsy and PCR was performed for genotypic analysis using the following primers: (1) common forward primer 5 '-G AG AGG AGGTTTTGGGG A AG- 3 ' (SEQ ID NO: 8); (2) reverse primer for WT allele 5'- CCCATGTCTAAAAAGCCAATC -3' (SEQ ID NO: 9); (3) reverse primer for mutant allele 5'- GTTCATATGGTGCCGTGCAG -3' (SEQ ID NO: 11).
- athymic nude male mice were injected subcutaneously with 5 x 10 5 TRAMP-C1 cells, and tumors were measured by calipers twice a week for calculation of tumor volumes ( ⁇ x length x width 16).
- TRAMP-C1 cell line was obtained from ATCC and authenticated using Short Tandem Repeat (STR) profiling method. The cells were maintained in Dulbecco's modified Eagle's medium supplemented with 0.005 mg/ml bovine insulin, 10 nM
- Tissue examination and tumor grading At the time of euthanization, the lower genitourinary tract, including the bladder (emptied), seminal vesicles, and the prostate was removed and weighed. Periaortic lymph nodes, lungs and livers were removed and inspected for evidence of visible metastases. Tissues were fixed in 10% phosphate-buffered formalin, embedded in paraffin, and sectioned at 5 ⁇ thickness.
- the H&E- stained dorsal-lateral lobe sections were each blindly scored on a scale of 1-6 (with T representing normal prostate and '6' representing poorly differentiated prostate adenocarcinomas or neuroendocrine tumors) (Gingrich, 1999; Kaplan-Lefko, 2003; Hurwitz, 2001).
- MTDH is associated with tumor progression and metastasis in human prostate cancer: Initially, the clinical relevance of MTDH in human prostate cancer samples was investigated. Two prostate tissue microarrays, consisting samples of normal prostates (NP), benign prostatic hyperplasia (BPH), prostatic intraepithelial neoplasia (PIN), prostate primary tumors and distant metastasis were analyzed by immunohistochemistry, and the MTDH protein levels were compared between samples of different clinical stages ( Figure 12A). MTDH protein was undetectable or expressed at very low levels in all normal or BPH prostate tissues.
- MTDH levels were also correlated with Gleason scores of primary tumors, as evidenced by higher average staining intensity of MTDH ( Figure 12B, top curve) or a greater percentage of samples with at least medium levels of MTDH ( Figure 12B, bottom curve) in late-stage tumors.
- PSA Prostate-Specific Antigen
- MTDH genomic gain is associated with MTDH overexpression and clinical progression in prostate cancer.
- a prostate tissue microarray was analyzed by interphase fluorescent in situ hybridization (FISH) using probes against either the MTDH genomic locus or the chromosome 8 centromere region (as control). FISH analysis revealed a significant portion of prostate cancer samples harbored extra genomic copies of the MTDH gene ( Figure 13A).
- MTDH DNA gain was significantly associated with higher levels of protein in the cells ( Figure 13B), indicating that DNA gain is a mechanism for MTDH overexpression in prostate cancer.
- Mtdh-deleted TRAMP mice To investigate the roles of Mtdh in normal development and cancer, whole-organism Mtdh-knockout (KO) mice were created using ESC line XB780 from Bay Genomics gene trap database (Stryke et al., 2003). The mutant allele contains a LacZ transgene insertion into the second intron of Mtdh, which results in premature termination of transcription. Mtdh-KO (Mtdh-/- ) mice were viable and fertile, and no overt abnormality in any organs were seen at autopsy.
- the gross morphology and relative weights of the lower genitourinary tract that includes both the prostate and seminal vesicles were comparable between wild-type (WT, Mtdh+/+), heterozygous (Mtdh+/-) and Mtdh-/- male mice. When examined at microscopic levels, there was no morphological abnormality in the prostate epithelium of Mtdh-/- mice.
- TRAMP/Mtdh+/+ and TRAMP/Mtdh+/- mice compared to matched control mice that do not express SV40 T antigen (Figure 14C). Consistently, a significant increase in protein levels of Mtdh was detected in TRAMP/Mtdh+/+ prostates compared to normal controls ( Figure 14D). These data indicate that Mtdh is upregulated during prostate tumorigenesis in mice and suggest that higher levels of Mtdh may confer a competitive advantage for tumor cells.
- TRAMP mice (n > 100) with different Mtdh status were studied for their prostate tumor development and cancer-related mortality.
- genitourinary weight is a reliable indicator of tumor burden in TRAMP mice (Kaplan-Lefko et al., 2003), the urogenital apparatus from
- TRAMP mice at different ages was dissected and measured to monitor tumor formation at a gross level. Since Mtdh expression was comparable between Mtdh+/+ and Mtdh+/- prostate tissues (Figure 14C), these mice were grouped as Mtdh-positive group (Mtdh+) versus Mtdh- negative group (Mtdh-).
- the SV40 T antigens induced a drastic expansion of the urogenital mass in TRAMP/Mtdh+ mice as a function of age ( Figure 15 A, diamond), such that many of these mice displayed severely enlarged abdomen at 36 weeks of age.
- prostate tissues from TRAMP mice were sectioned and hematoxylin and eosin staining was done.
- About 50% of 23 or 28-week-old TRAMP/Mtdh+ mice developed prostate cancer lesions, and by 36 weeks of age, almost all the mice in this group developed multifocal prostate tumors (Figure 15D).
- no prostate cancer was detected in 28-week-old TRAMP/Mtdh-/- mice, and prostate cancer lesions were detected in only 50% of 36-week-old TRAMP/Mtdh-/- mice at very limited regions of the prostate epithelium.
- TRAMP/Mtdh-/- mice did not display visible signs for prostate tumor formations by 36 weeks of age.
- tumors formed in TRAMP mice exhibit diverse histological features that are characteristic of adenocarcinomas, phyllode-like tumors, and neuroendocrine tumors, and it has been suggested that these morphologically distinct tumors may originate from different cell lineages within the prostate or develop independently from common early progenitors (Chiaverotti et al., 2008).
- TRAMP/Mtdh-/- mice showed a delay in the occurrence of all these different tumor subtypes, often appearing around 28 or 36 weeks, suggesting a possible lineage-independent role of Mtdh in prostate cancer.
- Prostate cancer progression consists of multiple stages starting from premalignant lesions such as PIN to well, moderately, and poorly differentiated carcinoma.
- Mtdh loss affects the initiation and progression of prostate cancer.
- histological examination was performed on dorsal and lateral lobes of each prostate gland, the most frequent and severe sites subjected to tumorigenesis in TRAMP mice (21) ( Figure 16A).
- each prostate was assigned a highest score ranging from a score of 1 for normal prostate and 6 for poorly differentiated carcinoma including neuroendocrine tumors.
- TRAMP/Mtdh+ mice developed well or moderately differentiated adenocarcinoma with invasive lesions at 23 weeks and 28 weeks, respectively, and by 36 weeks, 84% of TRAMP/Mtdh+ mice displayed moderately to poorly differentiated adenocarcinoma characteristic of analplastic sheets of tumor cells.
- TRAMP/Mtdh- mice only developed PIN or early- stage, well-differentiated adenocarcinomas or phyllode-like tumors by 36 weeks.
- TRAMP/Mtdh-/- mice examined developed visible metastases in the liver.
- multiple metastatic deposits in the lung were found in 39% of TRAMP/Mtdh+ mice, but only 2 out of 17 TRAMP/Mtdh-/- mice had one or two metastatic lesions detected in their lungs.
- Enlargement of lymph node were frequently identified in TRAMP/Mtdh+ mice (9/20) and this incidence decreased to 30% in TRAMP/Mtdh-/- mice.
- immunohistochemical analysis was performed on serial sections of the liver, lung and lymph node to detect the T antigen oncoprotein.
- the TRAMP model closely resembles human prostate cancer as it spontaneously develops progressive prostate cancer that can metastasize to multiple different organs.
- Mtdh levels are elevated in both human and SV40-driven murine prostate tumors compared to normal prostate tissues.
- Mtdh was recently shown to be overexpressed through genomic amplification in a p53/Pten-deficient metastatic prostate cancer mouse model in which telomerase activity was reactivated following telomere dysfunction (Ding, 2012), suggesting that Mtdh can be activated in mice through genomic amplification mechanism similar to what we found in human prostate cancer. Future studies are needed to investigate the molecular basis of Mtdh overexpresison in SV40-transformed tumors.
- TRAMP-C1 cancer cells prolonged tumor free- survival and reduced tumor growth in vivo lend further support for a prostate tumor cell-intrinsic role of Mtdh in prostate cancer.
- tumors eventually formed by TRAMP-C1 cancer cells that presumably carried Mtdh-targeting shRNA regained the expression of Mtdh, reinforcing the notion that Mtdh is essential for prostate cancer formation in vivo.
- a blockage of prostate cancer progression was observed at benign or well-differentiated stages in TRAMP/Mtdh-/- mice, a notion that is further supported by constitutively high levels of epithelial marker E-cadherin in TRAMP/Mtdh-/- prostate tissues.
- TRAMP/Mtdh+ mice displayed enlarged prostate glands and obstructed seminal vesicles, a phenomenon often resulted from invasive and malignant growth of prostate tumor cells into the urethra and seminal vesicles, urogenital track from TRAMP/Mtdh-/- mice appeared largely normal.
- MTDH exhibits a progression-correlated expression pattern in human prostate cancer.
- TRAMP/Mtdh-/- mice still developed invasive prostate cancer after long latency and died from the disease (1 out of 14) suggests that some TRAMP/Mtdh-/- tumors have overcome the deficiency of Mtdh and utilized Mtdh-independent pathways to support their malignant growth and progression. Indeed, the induction of SV40 oncogene occurs uniformly in prostate epithelial cells as early as puberty; however, the development of invasive prostate cancer and metastasis occurs much later and appears to be highly heterogeneous, suggesting that individual tumor may acquire different genetic and epigenetic alterations to fully transform SV40 antigen-positive cells.
- Mtdh While inactivation of Mtdh inhibits Mtdh-dependent signaling and prostate cancer progression as observed in the majority of TRAMP/Mtdh-/- mice, it would not block tumor growth that is supported by Mtdh-independent mechanisms. This is also in line with the observation that MTDH is highly expressed in a large percentage but not all human prostate cancer. Nevertheless, cancer cells that have depended on high levels of MTDH for their progression to a malignant stage would be sensitive to MTDH- targeting therapeutics, as supported by the finding that inhibition of Mtdh in Mtdh-positive TRAMP-C1 cells significantly impairs the tumorigenic potential of these cells.
- telomerase reactivation following telomere dysfunction in a prostate-cancer prone mouse model driven by Pten and p53 loss facilitates the selection of copy number alterations at cancer- relevant loci
- Mtdh was found to be at the top of a selective list of genes whose genetic amplification is strongly associated with cancer progression and metastasis in this mouse model (Ding et al., 2012). This suggests a possibly important role of Mtdh in prostate cancer driven by genetic events in addition to SV40 oncogenes.
- metastasis- promoting function of MTDH is likely to exist in other cancer types, beyond breast and prostate cancers.
- MTDH is overexpressed in both mouse and human prostate cancer and this elevated level of MTDH is critical for spontaneous prostate cancer progression and metastasis in vivo.
- the tumor-promoting roles of MTDH may not be restricted to the models used in the study. Indeed, ongoing studies have also found that deletion of Mtdh drastically inhibits spontaneous mammary tumor formation and metastasis. Future studies are needed to understand the underlying mechanisms and signaling pathways that responsible for the tumor-promoting function of MTDH and to facilitate the development of MTDH-targeting therapeutics to control human cancer.
- MTDH Metadherin
- SNDl Staphylococcal nuclease domain containing 1
- MTDH and SNDl interact with diverse cellular machineries and signaling proteins and are implicated in multiple cancer-related cellular processes and signaling pathways, it is possible that MTDH and SNDl enhance malignant features by coordinating tumor-promoting signaling activities via their multiple interaction domains/motifs.
- Protein Preparation All constructs and point mutations were generated using a standard PCR-based cloning strategy. Briefly, SND1 and MTDH with different boundaries and mutants were cloned in pQlink vector (Addgene) harboring an N-terminal His8-tag and a GST- tag, respectively, with a TEV cleavage site after the affinity tag. The proteins were
- the soluble fraction of lysate was purified over Ni-NTA resin (Qiagen) with lysis buffer containing 2 mM ⁇ - ⁇ , 0.5 M NaCl, 5 mM immidazole and 25 mM Tris PH8. His-SNDl proteins were eluted by buffer containing 0.2 M NaCl, 250 mM
- SND1 (16-339) was fused to the MTDH peptide (386-407) via a flexible linker with 21 residues (L21).
- L21 21 residues
- the protein was purified by Ni-NTA and Heparin column as described above before cleaved by TEV protease in the presence of 10 mM DTT.
- the sample was adjust to 1.4 M (NH4)2S04 and spun to remove pellet before further purified by Hydrophobic Interaction Chromatography (HiTrap Phenyl HP, GE Healthcare) to remove impurities, including TEV and the dimer of fusion protein at later peak.
- the monomer of fusion protein was then purified by gel filtration chromatography (Superdex 200, GE Healthcare) at final step to change the buffer to 10 mM Tris PH8, 150 mM NaCl and 10 mM DTT.
- the sample was concentrated to 15 mg/ml for crystallization.
- Crystals of SND1 and MTDH fusion protein were grown at 23° C by the sitting-drop vapor-diffusion method by mixing 250 nl of 15 mg/ml SeMet-SNDl(16-339)-L21-MTDH (386-407) with 250 nl of well buffer (21.6% pEG3350, 0.1M Sodium Citrate, PH8.0, 0.1M CsCl ), plus 50 nl of micro seeds. Single crystals grew in 4 days and matured after 7 days. Crystals were gradually changed to well buffer with 0 to 25% glycerol before flash frozen in liquid nitrogen.
- GST-Mediated Pull-Down Assay -20 ⁇ g of GST-MTDH (different truncation forms and site mutants) was bound to 10 ⁇ of glutathione resin via GST tag. The resin was washed with 200 ⁇ assay buffer (25 mM Tris, PH8.0, 150 mM NaCl, 3 mM DTT) three times to remove the excess unbound protein. 15 ⁇ g of His-SNDl (16-339) was added to the resin in a 100 ⁇ volume suspended in the assay buffer with 2 mg/ml of BSA. The mixture was washed with 200 ⁇ assay buffer twice before examined by SDS-PAGE, and visualized by Coomassie blue staining.
- IP immunoprecipitation and western blot: For in vivo immunoprecipitation (IP) experiments, cultured cells were washed in cold PBS and lysed in lysis buffer (20 mM Tris pH 7.4, 0.15 M NaCl, 1 mM EDTA, 1 mM EGTA, 1% Tx-100, 0.0025 M Na2P207, ImM ⁇ - glycerolphosphate, 1 mM Na3V04, and 1 mM NaF) with an EDTA-free protease inhibitor mixture (Roche Applied Science) and PMSF.
- IP in vivo immunoprecipitation
- IP lysates were subjected to western blotting with indicated antibodies.
- Tumorsphere assays Single cells were plated in ultra low attachment plates (Corning, Tewksbury, MA) with sphere media (1:1 DMEM: Ham's 12 supplemented with B27
- Tumongenesis assays All procedures involving mice and all experimental protocols were approved by Institutional Animal Care and Use Committee (IACUC) of Princeton
- tumorigenesis assays indicated numbers of PyMT tumor cells were transplanted into mammary fat of FVB recipient mice and tumor formations were monitored twice every week. Tumors were considered established when they became palpable for two consecutive weeks, and tumor size was measured by calipers for calculation of tumor volumes ( ⁇ x length x width 2/6).
- FRET Assay For FRET assay, CFP-MTDH fusion protein and a tetracysteine peptide fused to (TC) SND1 (16-339) were cloned into pQlink vector (Addgene) harboring an N- terminal GST- tag with a TEV cleavage site after the affinity tag. The proteins were
- E. coli strain DH5a overexpressed at 23° C in E. coli strain DH5a.
- the soluble fraction of the E. coli cell lysate was purified over GS4B resin (Qiagen), followed by TEV cleavage to remove the GST-tag.
- the untagged proteins were further fractionated by anion exchange chromatography (Source 15Q, GE Healthcare) and gel filtration chromatography (Superdex 200, GE Healthcare).
- the FlAsH-EDC2 compound (Invitrogen) was added to TC-SND1 (16-339) at 1:1.1 molar ratio to create the highly fluorescent TC-FLASH-SND1 that serves as the acceptor for CFP in the FRET assay.
- the donor fluorescent signal of CFP of 0.2 ⁇ CFP-MTDH was measured in the presence and absence of increasing concentrations of TC-FLASH-SND1 (0.25, 0.5, 1, 2, 4, 8 ⁇ ) using a Victor X5 Multilabel Plate Reader (Perkin Elmer) with excitation at 450 nm and emission at 490 nm.
- the TC- FLASH-SND1 concentration-dependent FRET efficiency was fitted in GraphPad Prism
- Biolayer interferometry BLI sensors immobilized by anti-GST antibody were activated by incubation with 150 nM GST-MTDH (386-407), followed by incubation with lmg/ml BSA and wash by binding buffer containing 25 mM Tris pH 8.0, 100 mM NaCl, and 3 mM DTT, with three minutes each step. Seven sensors activated by GST-MTDH were simultaneously dipped into seven wells containing the binding buffer control and increasing concentrations of His8-SND1 (16-339) (0.1, 0.2, 0.4, 0.8, 1.6, 3.2 ⁇ ) to measure the on rate of SNDl-binding. After three minutes of binding, the sensors were dipped into binding buffer to measure the off rate. Data collection and analysis were performed using ForteBio Octet RED96 (Pall Life Science).
- Each SN domain contains a ⁇ -barrel ( ⁇ 1- ⁇ 2- ⁇ 3- ⁇ 7- ⁇ 5) capped by a three-helix bundle ( ⁇ 1- ⁇ 2- ⁇ 3) and a short ⁇ -hairpin ( ⁇ 4- ⁇ 8) ( Figure 16).
- the MTDH peptide (D393WNAPAEEWGN403 of SEQ ID NO: 1) occupies the shallow groove between SNl and SN2 domains, with the two tryptophan residues, W394 and W401 of SEQ ID NO: 1, making extensive hydrophobic contacts with two well-defined hydrophobic pockets in SNDl.
- SNDl possesses three extended protruding structural elements (the ⁇ 6- ⁇ 7 hairpin in SNl and the extended ⁇ 4- 1 loop in both SNl and SN2), resulting in a spiky surface capable of diverse binding modes.
- the SN1/2 domains were previously suggested to participate in DNA/RNA- binding, and contribute to the nuclease activity of SNDl (Li et al., 2008). It remains to be determined whether the hilly surface of SNDl opposite to the MTDH-SND1 interface might participate in binding with DNA/RNA or other proteins and contribute to the nuclease activity or signaling mediated by SNDl.
- the elongated ⁇ 2- ⁇ 3 loop in SNl is crucial for mediating MTDH binding.
- the Lp4- l loops in SNl and SN3 are significant longer than that in SNase, and they adopt distinctly different conformations, which likely define different specificity for the two SN domains.
- Two out of six residues at the SNase active site are retained in SN3, only one of these residues remains the same or similar in SNl and SN4, but none of them is retained in SN2 ( Figure 20B).
- MTDH-SND1 interaction interface The fact that MTDH occupies an extended groove between SNl and SN2 on the back of the hilly surface of SNDl is consistent with the notion that MTDH might serve as a scaffold signaling protein. This architecture may allow MTDH to bridge SNDl and other MTDH-associated signaling complexes without interfering with major binding surfaces of SNDl.
- the MTDH-SND1 interface thus provides an important basis for understanding diverse downstream signaling and their function in cancer.
- the interface is dominated by hydrophobic van der Waals contacts of W394 and W401 in MTDH with two separate, well-defined hydrophobic pockets in SNDl, which are buttressed by hydrogen bond (H-bond) and salt bridge interactions at the periphery of the pockets ( Figure 21).
- the hydrophobic pocket for W394 is formed by residues P39, P43, and P44 in the SNl ⁇ 2- ⁇ 3 loop and the side chains of E247 and F250 on the SN2 al helix.
- the pocket for W401 is about 15 A away and located between the al and a2 helices from SN2, and contoured by hydrophobic residues L256, H279, 1284, and L287 and the carbon chain region of residues R255, R259, and N281.
- R327 and R324 in SNDl form several H-bond and salt bridge interactions with D393 and N395 in MTDH and its backbone carbonyl group at 392 in two of the five complexes in the
- the surface between the two hydrophobic pockets in SN1/2 is basic, which in part favors the electrostatic interaction with E400, but is not ideal for interaction with nonpolar residues (A396PA398) between W394 and W401. This likely explains the relatively weak interaction between SNDl and MTDH and the fast off-rate of this interaction.
- the protein groove between SN3 and SN4 is largely basic, underlying another structural feature of SN3/4 that disfavours MTDH-binding.
- the proline residues in the SN1 ⁇ 2- ⁇ 3 loop that are essential for lining the pocket for W394 are all absent in SN3 or SNase ( Figure 20B, Figure 21), further defining the binding specificity of SN1/2 to MTDH.
- the R324E mutation significantly weakened the MTDH-binding, likely by introducing a repulsive charge-charge contact with D393 in MTDH.
- a different mutation to this residue, R324A which was expected to perturb its H-bond interactions, barely affected MTDH-binding, similar to the negative control, R316E, an SNDl mutation located outside the interface.
- MTDH mutation W401D significantly reduced the binding ( Figure 22C, in blue), whereas other mutations, including the negative control D389R, a mutation located outside the MTDH-SND1 interface, did not affect the interaction ( Figure 22C).
- the SNDl mutations that affected MTDH-binding in vitro also affected the binding of full-length proteins in vivo to similar levels.
- Both WT HA-SND1 and the negative control mutant, HA-SND1 R316E bound readily with Myc-MTDH, whereas other mutations, ⁇ 39-43, F250A, or R255E, nearly completely abolished MTDH-binding, and R324E significantly reduced the binding (Figure 22D).
- MTDH mutants deficient in SNDl -binding had reduced pro-tumorigenic activities: As demonstrated above MTDH plays a role in regulating mammary tumorigenesis.
- genetic deletion of Mtdh in mice impairs the tumor-initiating potential of mammary epithelial cells transformed by diverse oncogenes (PyMT, Wnt, ErbB2) or carcinogen stimuli, and this defect can be readily rescued by reintroducing MTDH into Mtdh-knockout (Mtdh-/-) tumor cells by lentiviral transduction.
- the murine form of WT or mutant MTDH (W394A or W401A, corresponding mutations in mouse are W391A, W398A) was stably expressed in mammary tumor cells from PyMT;Mtdh-/- mice.
- the MTDH mutants W394A or W401A completely lost the ability to interact with SNDl ( Figure 23 A), suggesting that the SNDl -interacting residues of MTDH are well conserved between mouse and human.
- SNDl mutants deficient in MTDH-binding were inactive in tumor promotion:
- the well-defined pockets in SNDl for MTDH-binding and the role of this interaction in tumor initiation suggests that the protein pockets in SNDl represent a novel therapeutic target for cancer.
- knockdown (KD) of SNDl impairs the tumor initiating activities of PyMT/Mtdh+/+ tumor cells, supporting a tumor-promoting role of SNDl (Wan et al, 2014).
- MTDH has gained increasing interest in recent years given its broad implication in diverse cancer types, and SNDl has been documented as a MTDH-binding protein that possesses tumor-promoting functions similar to MTDH (Emdad et al., 2013; Wan and Kang, 2013).
- the MTDH-SND1 interface characterized in this study provides key insights into the molecular basis of their interaction.
- the essential SNDl -binding motif was previously mapped to two different regions of MTDH, residues 364-470 of SEQ ID NO: 1 (Blanco et al., 2011) and residues 101-205 of SEQ ID NO: 1 (Yoo et al., 2011).
- the study here defined a short 11 -residue peptide motif (residues 393-403 of SEQ ID NO: 1) of MTDH as the primary SNDl -binding motif, which is located within the fragment identified by Blanco et al (Blanco et al., 2011).
- the structure of the MTDH-SND1 interface provides an important platform for understanding the cellular signaling coordinated by this interaction.
- SN3/4 does not possess the unique pockets and surface feature required for MTDH-binding.
- the hilly surfaces harboring protruding structures in SN1/2 and SN3/4 are distinctly different and are expected to confer different binding specificity.
- the domains in SNDl fragment containing SN3/4-TSN5 domains were shown to be arranged in a linear orientation with a crescent shape (Li et al., 2008).
- Example 4 Wild type MTDH peptides significantly block MTDH-SNDl interaction.
- IP endogenous immunoprecipitation
- Figure 27B Cell lyaste was split into 5 fractions, and incubated with anti-SNDl antibody plus either buffer or indicated peptides. After 24hr, the lysate was incubated with Protein A/G beads then the beads was washed followed by western blot.
- Staphylococcal nuclease domain containing 1 SNDl
- MTDH and SNDl mutants at this interface deficient in binding have reduced activity in cancer promotion, which establishes the significance of this interface as a novel target for developing cancer therapeutics.
- the peptide motifs at this interface and their derivatives are potential inhibitors for blocking this interaction.
- structure-guided mutational analysis was performed for residues other than W394 and W401, the two residues that bind to two well-defined pockets in SNDl. None of the mutations have an obvious effect on MTDH-SND1 binding. This provides a large flexibility in designing peptide and peptide mimic inhibitors and allow flexible chemistry in making such inhibitors.
- Protein Preparation All constructs and point mutations were generated using a standard PCR-based cloning strategy. Briefly, SND1 and MTDH with different boundaries and mutants were cloned in pQlink vector (Addgene) harboring an N-terminal His8-tag and a GST- tag, respectively, with a TEV cleavage site after the affinity tag. The proteins were
- GST-Mediated Titration Pull-Down Assay -20 ⁇ g of GST-MTDH (386-407, wild type and different mutants) was bound to 10 ⁇ of glutathione resin via GST tag. The resin was washed with 200 ⁇ assay buffer (25 mM Tris, PH8.0, 150 mM NaCl, 3 mM DTT) three times to remove the excess unbound protein. 15 ⁇ g of His-SNDl (16-339) with titrated concentrations (10, 3, 1, 0.5, 0.25, 0.12 ⁇ ) was added to the resin in a 500 ⁇ volume suspended in the assay buffer with 2 mg/ml of BSA. The mixture was washed with 500 ⁇ assay buffer twice before examined by SDS-PAGE, and visualized by Coomassie blue staining.
- Amino acid sequence variations of MTDH that retain SND1 -binding The degree of sequence variations in the peptide motif represents the level of flexibility in the chemistry of developing peptide mimic inhibitors.
- the degree of sequence variations in the peptide motif represents the level of flexibility in the chemistry of developing peptide mimic inhibitors.
- Gata-3 is an essential regulator of mammary-gland morphogenesis and luminal-cell differentiation. Nature cell biology 9, 201-209.
- ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome.
- GATA-3 links tumor differentiation and dissemination in a luminal breast cancer model. Cancer Cell 13, 141-152.
- Cytoplasmic Metadherin provides survival advantage under conditions of stress by acting as RNA-binding protein. J Biol Chem 287, 4485-4491. [0243] Perou, C. M., Sorlie, T., Eisen, M. B., van de Rijn, M., Jeffrey, S. S., Rees, C. A., Pollack, J. R., Ross, D. T., Johnsen, H., Akslen, L. A., et al. (2000). Molecular portraits of human breast tumours. Nature 406, 747-752.
- a NIK-IKKalpha module expands ErbB2-induced tumor-initiating cells by stimulating nuclear export of p27/Kipl. Cancer Cell 23, 647-659.
- LYRIC/AEG-1 is targeted to different subcellular compartments by ubiquitinylation and intrinsic nuclear localization signals. Clin Cancer Res. 2009;15:3003-13.
- BayGenomics a resource of insertional mutations in mouse embryonic stem cells. Nucleic Acids Res. 2003;31:278-81. [0282] Su ZZ, Kang DC, Chen Y, Pekarskaya O, Chao W, Volsky DJ, et al. Identification and cloning of human astrocyte genes displaying elevated expression after infection with HIV-1 or exposure to HIV-1 envelope glycoprotein by rapid subtraction hybridization, RaSH. Oncogene. 2002;21:3592-602.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Gastroenterology & Hepatology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Organic Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Immunology (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Epidemiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Zoology (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Marine Sciences & Fisheries (AREA)
- Oncology (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Toxicology (AREA)
- Peptides Or Proteins (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Medicinal Preparation (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
- Enzymes And Modification Thereof (AREA)
Abstract
Description



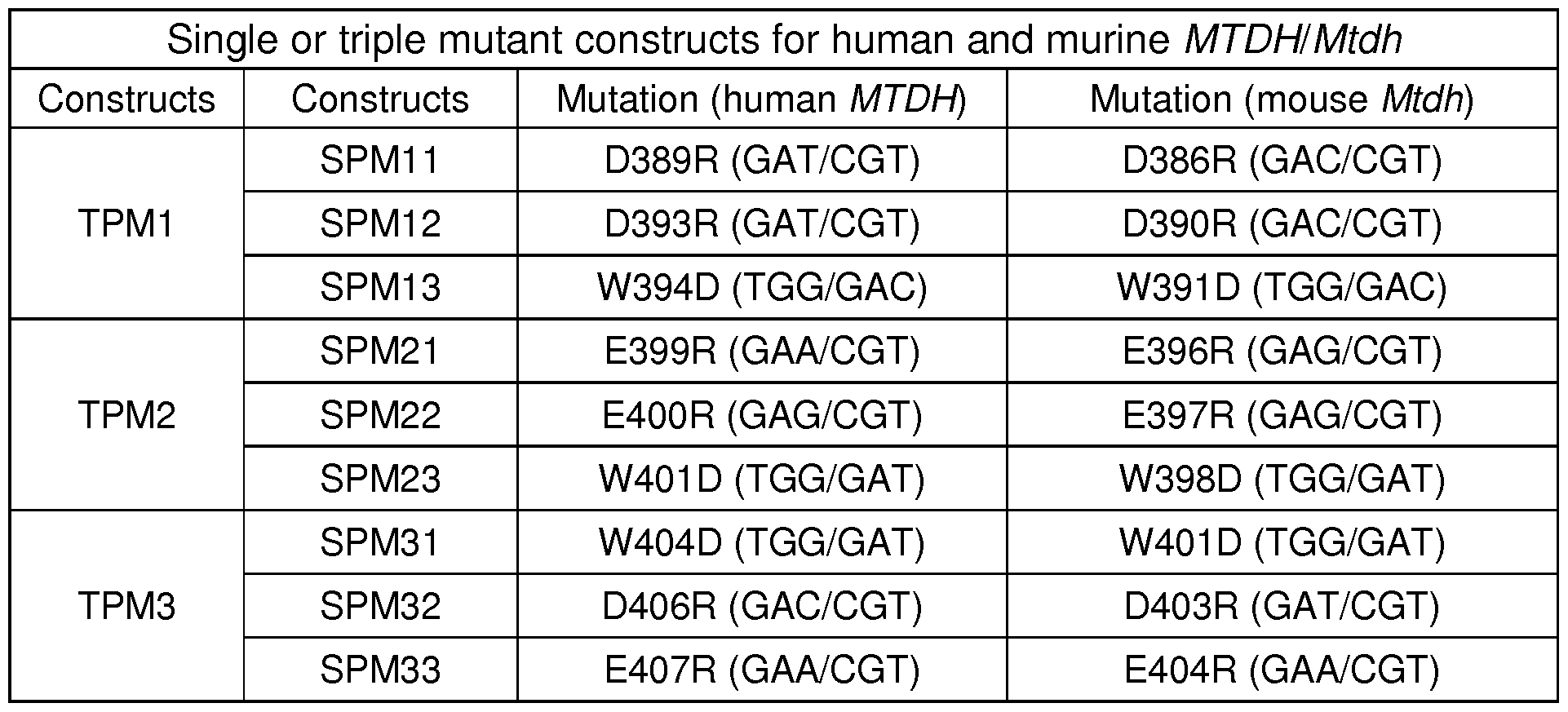
Claims
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US15/321,337 US10357539B2 (en) | 2014-06-25 | 2015-06-25 | Use of peptides that block metadherin-SND1 interaction as treatment for cancer |
| JP2016575208A JP6797027B2 (en) | 2014-06-25 | 2015-06-25 | Use of peptides that block the metadoherin-SND1 interaction as a treatment for cancer |
| CN201580041336.5A CN106794216B (en) | 2014-06-25 | 2015-06-25 | Use of peptides blocking the hetero-mucin-SND 1 interaction as cancer treatment |
| EP15812501.3A EP3177308B1 (en) | 2014-06-25 | 2015-06-25 | Use of peptides that block metadherin-snd1 interaction as treatment for cancer |
| AU2015279834A AU2015279834A1 (en) | 2014-06-25 | 2015-06-25 | Use of peptides that block metadherin-SND1 interaction as treatment for cancer |
| US16/406,837 US10786545B2 (en) | 2014-06-25 | 2019-05-08 | Use of peptides that block metadherin-SND1 interaction as treatment for cancer |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201462016914P | 2014-06-25 | 2014-06-25 | |
| US62/016,914 | 2014-06-25 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US15/321,337 A-371-Of-International US10357539B2 (en) | 2014-06-25 | 2015-06-25 | Use of peptides that block metadherin-SND1 interaction as treatment for cancer |
| US16/406,837 Division US10786545B2 (en) | 2014-06-25 | 2019-05-08 | Use of peptides that block metadherin-SND1 interaction as treatment for cancer |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2015200648A1 true WO2015200648A1 (en) | 2015-12-30 |
| WO2015200648A8 WO2015200648A8 (en) | 2016-12-29 |
Family
ID=54938807
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2015/037708 WO2015200648A1 (en) | 2014-06-25 | 2015-06-25 | Use of peptides that block metadherin-snd1 interaction as treatment for cancer |
Country Status (6)
| Country | Link |
|---|---|
| US (2) | US10357539B2 (en) |
| EP (1) | EP3177308B1 (en) |
| JP (2) | JP6797027B2 (en) |
| CN (1) | CN106794216B (en) |
| AU (1) | AU2015279834A1 (en) |
| WO (1) | WO2015200648A1 (en) |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015200648A1 (en) * | 2014-06-25 | 2015-12-30 | The Trustees Princeton University | Use of peptides that block metadherin-snd1 interaction as treatment for cancer |
| US11331019B2 (en) | 2017-08-07 | 2022-05-17 | The Research Foundation For The State University Of New York | Nanoparticle sensor having a nanofibrous membrane scaffold |
| CN112326961B (en) * | 2020-10-30 | 2021-08-06 | 福州迈新生物技术开发有限公司 | Analysis method and storage device for proportion of PD-L1 positive tumor cells in non-small cell lung cancer |
| CN112898383A (en) * | 2021-01-29 | 2021-06-04 | 深圳海创生物科技有限公司 | Oligopeptide, active peptide composition and application of oligopeptide and active peptide composition in preparation of product with anti-inflammatory effect |
| CN112933212B (en) * | 2021-04-07 | 2022-05-13 | 华中科技大学同济医学院附属协和医院 | Application of SND1 in preparation of medicine for preventing and treating vascular injury ischemia |
| WO2023064669A2 (en) | 2021-09-21 | 2023-04-20 | The Trustees Of Princeton University | Inhibitors of the mtdh-snd1 protein complex for cancer therapy |
| CN117865886B (en) * | 2024-03-07 | 2024-05-10 | 中国药科大学 | N- (quinoline-8-yl) quinoline-8-sulfonamide compound and application thereof |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005050198A2 (en) | 2003-11-13 | 2005-06-02 | The Burnham Institute | Metadherin polypeptides, encoding nucleic acids and methods of use |
| JP2006335659A (en) | 2005-05-31 | 2006-12-14 | Japan Health Science Foundation | Cancer cell proliferation inhibitor |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20060205934A1 (en) * | 2002-12-05 | 2006-09-14 | Macina Roberto A | Compositions, splice variants and methods relating to breast specific genes and proteins |
| CN101993935A (en) * | 2009-08-10 | 2011-03-30 | 芮屈生物技术(上海)有限公司 | In-situ hybridization assay kit and detection method for MTDH gene and application of kit |
| WO2015200648A1 (en) * | 2014-06-25 | 2015-12-30 | The Trustees Princeton University | Use of peptides that block metadherin-snd1 interaction as treatment for cancer |
-
2015
- 2015-06-25 WO PCT/US2015/037708 patent/WO2015200648A1/en active Application Filing
- 2015-06-25 EP EP15812501.3A patent/EP3177308B1/en active Active
- 2015-06-25 JP JP2016575208A patent/JP6797027B2/en active Active
- 2015-06-25 US US15/321,337 patent/US10357539B2/en active Active
- 2015-06-25 CN CN201580041336.5A patent/CN106794216B/en active Active
- 2015-06-25 AU AU2015279834A patent/AU2015279834A1/en not_active Abandoned
-
2019
- 2019-05-08 US US16/406,837 patent/US10786545B2/en active Active
-
2020
- 2020-01-30 JP JP2020013947A patent/JP2020097598A/en active Pending
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005050198A2 (en) | 2003-11-13 | 2005-06-02 | The Burnham Institute | Metadherin polypeptides, encoding nucleic acids and methods of use |
| JP2006335659A (en) | 2005-05-31 | 2006-12-14 | Japan Health Science Foundation | Cancer cell proliferation inhibitor |
Non-Patent Citations (11)
| Title |
|---|
| "Genbank", Database accession no. AAH45642 |
| "Sciences", 1995, MACK PUBLISHING CO. |
| "THE CAMBRIDGE DICTIONARY OF SCIENCE AND TECHNOLOGY", 1988 |
| BLANCO ET AL.: "Identification of Staphylococcal Nuclease Domain-containing 1 (SND1) as a Metadherin-interacting Protein with Metastasis-promoting Functions", THE JOUMAL OF BIOLOGICAL CHEMISTRY, vol. 286, no. 22, 3 June 2011 (2011-06-03), pages 19982 - 19992, XP055247921 * |
| GUO ET AL.: "Structural Insights into the Tumor-Promoting Function of the MTDH-SND1 Complex", CELL REPORTS, vol. 8, 25 September 2014 (2014-09-25), pages 1704 - 1713, XP055247925 * |
| HALEMARHAM, THE HARPER COLLINS DICTIONARY OF BIOLOGY, 1991 |
| MEZIERE ET AL., J. IMMUNOL., vol. 159, 1997, pages 3230 - 3237 |
| SINGLETON ET AL.: "DICTIONARY OF MICROBIOLOGY AND MOLECULAR BIOLOG", 1994 |
| WAN ET AL.: "MTDH-SND1 Interaction Is Crucial for Expansion and Activity of Tumor-Initiating Cells in Diverse Oncogene -andCarcinogen-InducedMammaryTumors", CANCER CELL, vol. 26, 14 July 2014 (2014-07-14), pages 92 - 105, XP029034082 * |
| WANG ET AL.: "Prognostic impact of Metadherin-SND1 interaction in colon cancer", MOLECULAR BIOLOGY REPORTS, vol. 39, no. Iss. 12, 14 October 2012 (2012-10-14), pages 10497 - 10504, XP035132995 * |
| XING ET AL.: "Inhibiting Metadherin/SND1 Interaction to Treat Cancer", WARF:P140424 US 01, 9 March 2015 (2015-03-09), pages 1 - 2, XP055247929, Retrieved from the Internet <URL:www .warf.org/documents/ technology-summary/P140424 US 01.pdf> [retrieved on 20150917] * |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3177308A4 (en) | 2018-04-04 |
| US10786545B2 (en) | 2020-09-29 |
| US20190336574A1 (en) | 2019-11-07 |
| WO2015200648A8 (en) | 2016-12-29 |
| EP3177308A1 (en) | 2017-06-14 |
| JP2020097598A (en) | 2020-06-25 |
| JP2017520574A (en) | 2017-07-27 |
| AU2015279834A1 (en) | 2016-12-22 |
| CN106794216A (en) | 2017-05-31 |
| CN106794216B (en) | 2022-03-11 |
| EP3177308B1 (en) | 2020-11-11 |
| JP6797027B2 (en) | 2020-12-09 |
| AU2015279834A8 (en) | 2019-08-08 |
| US10357539B2 (en) | 2019-07-23 |
| US20170189481A1 (en) | 2017-07-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US10786545B2 (en) | Use of peptides that block metadherin-SND1 interaction as treatment for cancer | |
| JP6661645B2 (en) | RNA aptamer for transferrin receptor (TFR) | |
| JP2019531699A (en) | Diagnosis and treatment method of cancer by expression state and mutation state of NRF2 and downstream target gene of the gene | |
| JP7101118B2 (en) | Stabilized BCL9 peptide for the treatment of abnormal WNT signaling | |
| US20150080315A1 (en) | SALL4 And Uses Thereof | |
| AU2013307383A1 (en) | Aminoheteroaryl compounds as MTH1 inhibitors | |
| JP2017526670A (en) | Inhibition of tumor metastasis by inhibition of necroptosis | |
| WO2020168110A1 (en) | Ephb4-ephrin b2 receptor ligand pair as a novel marker for the treatment of prostate cancer | |
| US20230227823A1 (en) | Fmrp and cancer treatment | |
| WO2019028055A1 (en) | Compounds, compositionals, and methods for treating t-cell acute lymphoblastic leukemia | |
| KR20200068647A (en) | RNA aptamer for transferrin receptor (TfR) | |
| WO2023064669A2 (en) | Inhibitors of the mtdh-snd1 protein complex for cancer therapy | |
| CN107106697B (en) | PDGFR RNA aptamer | |
| Pinheiro et al. | Targeting metabolic reprogramming as an anti-cancer strategy: aiming at monocarboxylate transporters | |
| JP7254020B2 (en) | Methods of treating cancers containing fusion genes | |
| JP7538798B2 (en) | Phosphorylated Dicer Antibodies and Methods of Use Thereof | |
| US20220144950A1 (en) | Targeting regulatory b cells and their regulators for cancer immunotherapy | |
| WO2014023869A2 (en) | Therapeutic use of cd44 inhibitor agents against human acute lymphoblastic leukaemia (all) | |
| WO2023137447A1 (en) | Alk gene fusions and uses thereof | |
| CA2765770A1 (en) | Agents and uses thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 15812501 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2016575208 Country of ref document: JP Kind code of ref document: A Ref document number: 2015279834 Country of ref document: AU Date of ref document: 20150625 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15321337 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2015812501 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2015812501 Country of ref document: EP |