WO2015077520A1 - Methods of treating abnormal muscular activity - Google Patents
Methods of treating abnormal muscular activity Download PDFInfo
- Publication number
- WO2015077520A1 WO2015077520A1 PCT/US2014/066740 US2014066740W WO2015077520A1 WO 2015077520 A1 WO2015077520 A1 WO 2015077520A1 US 2014066740 W US2014066740 W US 2014066740W WO 2015077520 A1 WO2015077520 A1 WO 2015077520A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- deuterium
- compound
- less
- deuterium enrichment
- recited
- Prior art date
Links
- 238000000034 method Methods 0.000 title claims abstract description 119
- 230000000694 effects Effects 0.000 title claims abstract description 91
- 230000003387 muscular Effects 0.000 title claims abstract description 64
- 230000002159 abnormal effect Effects 0.000 title claims abstract description 44
- 229910052805 deuterium Inorganic materials 0.000 claims description 230
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 claims description 212
- 150000001875 compounds Chemical class 0.000 claims description 167
- 239000000203 mixture Substances 0.000 claims description 106
- 239000003814 drug Substances 0.000 claims description 49
- 229960005333 tetrabenazine Drugs 0.000 claims description 32
- MKJIEFSOBYUXJB-HOCLYGCPSA-N (3S,11bS)-9,10-dimethoxy-3-isobutyl-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-one Chemical group C1CN2C[C@H](CC(C)C)C(=O)C[C@H]2C2=C1C=C(OC)C(OC)=C2 MKJIEFSOBYUXJB-HOCLYGCPSA-N 0.000 claims description 30
- 239000003112 inhibitor Substances 0.000 claims description 27
- 150000003839 salts Chemical class 0.000 claims description 25
- 239000001257 hydrogen Substances 0.000 claims description 24
- 229910052739 hydrogen Inorganic materials 0.000 claims description 24
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 20
- 229940124597 therapeutic agent Drugs 0.000 claims description 15
- VYFYYTLLBUKUHU-UHFFFAOYSA-N dopamine Chemical compound NCCC1=CC=C(O)C(O)=C1 VYFYYTLLBUKUHU-UHFFFAOYSA-N 0.000 claims description 14
- 239000003795 chemical substances by application Substances 0.000 claims description 13
- WTDRDQBEARUVNC-LURJTMIESA-N L-DOPA Chemical group OC(=O)[C@@H](N)CC1=CC=C(O)C(O)=C1 WTDRDQBEARUVNC-LURJTMIESA-N 0.000 claims description 11
- 208000023105 Huntington disease Diseases 0.000 claims description 9
- 102000006378 Catechol O-methyltransferase Human genes 0.000 claims description 8
- 108020002739 Catechol O-methyltransferase Proteins 0.000 claims description 8
- 208000012661 Dyskinesia Diseases 0.000 claims description 8
- 229960003638 dopamine Drugs 0.000 claims description 7
- 238000012545 processing Methods 0.000 claims description 7
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 claims description 6
- 208000006083 Hypokinesia Diseases 0.000 claims description 6
- 206010006100 Bradykinesia Diseases 0.000 claims description 5
- 206010020651 Hyperkinesia Diseases 0.000 claims description 5
- 208000000269 Hyperkinesis Diseases 0.000 claims description 5
- 230000000561 anti-psychotic effect Effects 0.000 claims description 5
- 239000002243 precursor Substances 0.000 claims description 5
- 239000005557 antagonist Substances 0.000 claims description 4
- 239000000534 dopa decarboxylase inhibitor Substances 0.000 claims description 4
- 239000003136 dopamine receptor stimulating agent Substances 0.000 claims description 4
- 239000004090 neuroprotective agent Substances 0.000 claims description 4
- 239000000164 antipsychotic agent Substances 0.000 claims description 3
- 229940005529 antipsychotics Drugs 0.000 claims description 3
- 229960004170 clozapine Drugs 0.000 claims description 3
- QZUDBNBUXVUHMW-UHFFFAOYSA-N clozapine Chemical compound C1CN(C)CCN1C1=NC2=CC(Cl)=CC=C2NC2=CC=CC=C12 QZUDBNBUXVUHMW-UHFFFAOYSA-N 0.000 claims description 3
- MEZLKOACVSPNER-GFCCVEGCSA-N selegiline Chemical compound C#CCN(C)[C@H](C)CC1=CC=CC=C1 MEZLKOACVSPNER-GFCCVEGCSA-N 0.000 claims description 3
- 229940081615 DOPA decarboxylase inhibitor Drugs 0.000 claims description 2
- 229940098778 Dopamine receptor agonist Drugs 0.000 claims description 2
- WTDRDQBEARUVNC-UHFFFAOYSA-N L-Dopa Natural products OC(=O)C(N)CC1=CC=C(O)C(O)=C1 WTDRDQBEARUVNC-UHFFFAOYSA-N 0.000 claims description 2
- HOKKHZGPKSLGJE-GSVOUGTGSA-N N-Methyl-D-aspartic acid Chemical compound CN[C@@H](C(O)=O)CC(O)=O HOKKHZGPKSLGJE-GSVOUGTGSA-N 0.000 claims description 2
- FTALBRSUTCGOEG-UHFFFAOYSA-N Riluzole Chemical compound C1=C(OC(F)(F)F)C=C2SC(N)=NC2=C1 FTALBRSUTCGOEG-UHFFFAOYSA-N 0.000 claims description 2
- DKNWSYNQZKUICI-UHFFFAOYSA-N amantadine Chemical group C1C(C2)CC3CC2CC1(N)C3 DKNWSYNQZKUICI-UHFFFAOYSA-N 0.000 claims description 2
- 229960003805 amantadine Drugs 0.000 claims description 2
- 229960004046 apomorphine Drugs 0.000 claims description 2
- VMWNQDUVQKEIOC-CYBMUJFWSA-N apomorphine Chemical compound C([C@H]1N(C)CC2)C3=CC=C(O)C(O)=C3C3=C1C2=CC=C3 VMWNQDUVQKEIOC-CYBMUJFWSA-N 0.000 claims description 2
- 229960002802 bromocriptine Drugs 0.000 claims description 2
- OZVBMTJYIDMWIL-AYFBDAFISA-N bromocriptine Chemical compound C1=CC(C=2[C@H](N(C)C[C@@H](C=2)C(=O)N[C@]2(C(=O)N3[C@H](C(N4CCC[C@H]4[C@]3(O)O2)=O)CC(C)C)C(C)C)C2)=C3C2=C(Br)NC3=C1 OZVBMTJYIDMWIL-AYFBDAFISA-N 0.000 claims description 2
- 229960004205 carbidopa Drugs 0.000 claims description 2
- TZFNLOMSOLWIDK-JTQLQIEISA-N carbidopa (anhydrous) Chemical group NN[C@@](C(O)=O)(C)CC1=CC=C(O)C(O)=C1 TZFNLOMSOLWIDK-JTQLQIEISA-N 0.000 claims description 2
- 229960003337 entacapone Drugs 0.000 claims description 2
- JRURYQJSLYLRLN-BJMVGYQFSA-N entacapone Chemical group CCN(CC)C(=O)C(\C#N)=C\C1=CC(O)=C(O)C([N+]([O-])=O)=C1 JRURYQJSLYLRLN-BJMVGYQFSA-N 0.000 claims description 2
- 229960004502 levodopa Drugs 0.000 claims description 2
- 239000003703 n methyl dextro aspartic acid receptor blocking agent Substances 0.000 claims description 2
- FASDKYOPVNHBLU-ZETCQYMHSA-N pramipexole Chemical compound C1[C@@H](NCCC)CCC2=C1SC(N)=N2 FASDKYOPVNHBLU-ZETCQYMHSA-N 0.000 claims description 2
- 229960003089 pramipexole Drugs 0.000 claims description 2
- 229960004181 riluzole Drugs 0.000 claims description 2
- 229960001879 ropinirole Drugs 0.000 claims description 2
- UHSKFQJFRQCDBE-UHFFFAOYSA-N ropinirole Chemical compound CCCN(CCC)CCC1=CC=CC2=C1CC(=O)N2 UHSKFQJFRQCDBE-UHFFFAOYSA-N 0.000 claims description 2
- 229960004603 tolcapone Drugs 0.000 claims description 2
- MIQPIUSUKVNLNT-UHFFFAOYSA-N tolcapone Chemical compound C1=CC(C)=CC=C1C(=O)C1=CC(O)=C(O)C([N+]([O-])=O)=C1 MIQPIUSUKVNLNT-UHFFFAOYSA-N 0.000 claims description 2
- 241000011102 Thera Species 0.000 claims 1
- 238000012544 monitoring process Methods 0.000 abstract description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 96
- 239000000243 solution Substances 0.000 description 77
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 60
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 60
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 57
- 239000007787 solid Substances 0.000 description 57
- 239000002904 solvent Substances 0.000 description 55
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 51
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 48
- 229910001868 water Inorganic materials 0.000 description 48
- 230000033001 locomotion Effects 0.000 description 43
- -1 valine ester Chemical class 0.000 description 43
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 39
- 102000009659 Vesicular Monoamine Transport Proteins Human genes 0.000 description 37
- 108010020033 Vesicular Monoamine Transport Proteins Proteins 0.000 description 37
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 36
- 208000035475 disorder Diseases 0.000 description 35
- 239000011541 reaction mixture Substances 0.000 description 35
- 238000006467 substitution reaction Methods 0.000 description 32
- 229940079593 drug Drugs 0.000 description 30
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 24
- 239000007788 liquid Substances 0.000 description 24
- 238000006243 chemical reaction Methods 0.000 description 23
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 22
- 239000002207 metabolite Substances 0.000 description 20
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 20
- 238000011282 treatment Methods 0.000 description 20
- GEJDGVNQKABXKG-CFKGEZKQSA-N [(2r,3r,11br)-9,10-dimethoxy-3-(2-methylpropyl)-2,3,4,6,7,11b-hexahydro-1h-benzo[a]quinolizin-2-yl] (2s)-2-amino-3-methylbutanoate Chemical compound C1CN2C[C@@H](CC(C)C)[C@H](OC(=O)[C@@H](N)C(C)C)C[C@@H]2C2=C1C=C(OC)C(OC)=C2 GEJDGVNQKABXKG-CFKGEZKQSA-N 0.000 description 18
- 229950006411 valbenazine Drugs 0.000 description 18
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 17
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 16
- 239000008194 pharmaceutical composition Substances 0.000 description 16
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 16
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 15
- 239000000543 intermediate Substances 0.000 description 15
- 239000010410 layer Substances 0.000 description 15
- 239000000047 product Substances 0.000 description 15
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 14
- 238000005481 NMR spectroscopy Methods 0.000 description 14
- 239000004480 active ingredient Substances 0.000 description 13
- 238000009472 formulation Methods 0.000 description 13
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 12
- 229910052938 sodium sulfate Inorganic materials 0.000 description 12
- 235000011152 sodium sulphate Nutrition 0.000 description 12
- WEQLWGNDNRARGE-DJIMGWMZSA-N (2R,3R,11bR)-9,10-dimethoxy-3-(2-methylpropyl)-2,3,4,6,7,11b-hexahydro-1H-benzo[a]quinolizin-2-ol Chemical compound C1CN2C[C@@H](CC(C)C)[C@H](O)C[C@@H]2C2=C1C=C(OC)C(OC)=C2 WEQLWGNDNRARGE-DJIMGWMZSA-N 0.000 description 11
- 239000002552 dosage form Substances 0.000 description 11
- 239000012044 organic layer Substances 0.000 description 11
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 10
- 241001465754 Metazoa Species 0.000 description 10
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 10
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 10
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 10
- 238000001914 filtration Methods 0.000 description 10
- 238000005259 measurement Methods 0.000 description 10
- 230000001404 mediated effect Effects 0.000 description 10
- 239000003921 oil Substances 0.000 description 10
- 239000000651 prodrug Substances 0.000 description 10
- 229940002612 prodrug Drugs 0.000 description 10
- 239000000725 suspension Substances 0.000 description 10
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 9
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 9
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 9
- 239000002253 acid Substances 0.000 description 9
- 238000012377 drug delivery Methods 0.000 description 9
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 9
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 9
- 229910052757 nitrogen Inorganic materials 0.000 description 9
- 235000019198 oils Nutrition 0.000 description 9
- 229910000027 potassium carbonate Inorganic materials 0.000 description 9
- 239000000843 powder Substances 0.000 description 9
- 238000002360 preparation method Methods 0.000 description 9
- 208000024891 symptom Diseases 0.000 description 9
- UNILWMWFPHPYOR-KXEYIPSPSA-M 1-[6-[2-[3-[3-[3-[2-[2-[3-[[2-[2-[[(2r)-1-[[2-[[(2r)-1-[3-[2-[2-[3-[[2-(2-amino-2-oxoethoxy)acetyl]amino]propoxy]ethoxy]ethoxy]propylamino]-3-hydroxy-1-oxopropan-2-yl]amino]-2-oxoethyl]amino]-3-[(2r)-2,3-di(hexadecanoyloxy)propyl]sulfanyl-1-oxopropan-2-yl Chemical compound O=C1C(SCCC(=O)NCCCOCCOCCOCCCNC(=O)COCC(=O)N[C@@H](CSC[C@@H](COC(=O)CCCCCCCCCCCCCCC)OC(=O)CCCCCCCCCCCCCCC)C(=O)NCC(=O)N[C@H](CO)C(=O)NCCCOCCOCCOCCCNC(=O)COCC(N)=O)CC(=O)N1CCNC(=O)CCCCCN\1C2=CC=C(S([O-])(=O)=O)C=C2CC/1=C/C=C/C=C/C1=[N+](CC)C2=CC=C(S([O-])(=O)=O)C=C2C1 UNILWMWFPHPYOR-KXEYIPSPSA-M 0.000 description 8
- XLYOFNOQVPJJNP-ZSJDYOACSA-N Heavy water Chemical compound [2H]O[2H] XLYOFNOQVPJJNP-ZSJDYOACSA-N 0.000 description 8
- 241000720974 Protium Species 0.000 description 8
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 8
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 8
- 239000007864 aqueous solution Substances 0.000 description 8
- 238000013265 extended release Methods 0.000 description 8
- 238000010348 incorporation Methods 0.000 description 8
- 238000002347 injection Methods 0.000 description 8
- 239000007924 injection Substances 0.000 description 8
- AWJUIBRHMBBTKR-UHFFFAOYSA-N isoquinoline Chemical compound C1=NC=CC2=CC=CC=C21 AWJUIBRHMBBTKR-UHFFFAOYSA-N 0.000 description 8
- 229920000642 polymer Polymers 0.000 description 8
- 239000003826 tablet Substances 0.000 description 8
- 102000004190 Enzymes Human genes 0.000 description 7
- 108090000790 Enzymes Proteins 0.000 description 7
- 208000016285 Movement disease Diseases 0.000 description 7
- 239000002775 capsule Substances 0.000 description 7
- 239000003153 chemical reaction reagent Substances 0.000 description 7
- 239000003638 chemical reducing agent Substances 0.000 description 7
- 238000004587 chromatography analysis Methods 0.000 description 7
- 229940088598 enzyme Drugs 0.000 description 7
- 239000000706 filtrate Substances 0.000 description 7
- 125000004551 isoquinolin-3-yl group Chemical group C1=NC(=CC2=CC=CC=C12)* 0.000 description 7
- 235000011181 potassium carbonates Nutrition 0.000 description 7
- 230000002829 reductive effect Effects 0.000 description 7
- 238000013268 sustained release Methods 0.000 description 7
- 239000012730 sustained-release form Substances 0.000 description 7
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 6
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 6
- 241000282414 Homo sapiens Species 0.000 description 6
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 6
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 6
- YZCKVEUIGOORGS-IGMARMGPSA-N Protium Chemical compound [1H] YZCKVEUIGOORGS-IGMARMGPSA-N 0.000 description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 6
- 230000008901 benefit Effects 0.000 description 6
- 239000012267 brine Substances 0.000 description 6
- 239000003054 catalyst Substances 0.000 description 6
- 229940125797 compound 12 Drugs 0.000 description 6
- 239000012043 crude product Substances 0.000 description 6
- 230000005445 isotope effect Effects 0.000 description 6
- 239000008101 lactose Substances 0.000 description 6
- 238000004519 manufacturing process Methods 0.000 description 6
- 230000002503 metabolic effect Effects 0.000 description 6
- 238000012986 modification Methods 0.000 description 6
- 230000004048 modification Effects 0.000 description 6
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 6
- 238000000746 purification Methods 0.000 description 6
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- 230000001225 therapeutic effect Effects 0.000 description 6
- 231100000419 toxicity Toxicity 0.000 description 6
- 230000001988 toxicity Effects 0.000 description 6
- 230000007704 transition Effects 0.000 description 6
- 229910052722 tritium Inorganic materials 0.000 description 6
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 5
- QCDJYGZCMGEQNF-UHFFFAOYSA-N 3-[(dimethylamino)methyl]-5-methylhexan-2-one Chemical compound CC(C)CC(C(C)=O)CN(C)C QCDJYGZCMGEQNF-UHFFFAOYSA-N 0.000 description 5
- 206010010904 Convulsion Diseases 0.000 description 5
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 5
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 5
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 5
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 5
- 206010044565 Tremor Diseases 0.000 description 5
- 239000012298 atmosphere Substances 0.000 description 5
- 125000004429 atom Chemical group 0.000 description 5
- 229940126543 compound 14 Drugs 0.000 description 5
- 239000003085 diluting agent Substances 0.000 description 5
- 230000005764 inhibitory process Effects 0.000 description 5
- 229910052744 lithium Inorganic materials 0.000 description 5
- 239000012022 methylating agents Substances 0.000 description 5
- 239000012074 organic phase Substances 0.000 description 5
- 239000000546 pharmaceutical excipient Substances 0.000 description 5
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 5
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 5
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 5
- 239000011591 potassium Substances 0.000 description 5
- 229910052700 potassium Inorganic materials 0.000 description 5
- 238000001953 recrystallisation Methods 0.000 description 5
- 239000003381 stabilizer Substances 0.000 description 5
- 238000003756 stirring Methods 0.000 description 5
- QCTKXTXHDRLVAT-UHFFFAOYSA-M (2-acetyl-4-methylpentyl)-trimethylazanium;iodide Chemical compound [I-].CC(C)CC(C(C)=O)C[N+](C)(C)C QCTKXTXHDRLVAT-UHFFFAOYSA-M 0.000 description 4
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 4
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 4
- 241000282412 Homo Species 0.000 description 4
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 4
- 229930195725 Mannitol Natural products 0.000 description 4
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 4
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 4
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 4
- 206010043118 Tardive Dyskinesia Diseases 0.000 description 4
- 230000004913 activation Effects 0.000 description 4
- 238000013459 approach Methods 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- 210000000476 body water Anatomy 0.000 description 4
- 239000000969 carrier Substances 0.000 description 4
- 229940127573 compound 38 Drugs 0.000 description 4
- 230000003247 decreasing effect Effects 0.000 description 4
- 238000009826 distribution Methods 0.000 description 4
- 239000000839 emulsion Substances 0.000 description 4
- 238000005516 engineering process Methods 0.000 description 4
- 150000002148 esters Chemical class 0.000 description 4
- 230000000670 limiting effect Effects 0.000 description 4
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 4
- 239000000594 mannitol Substances 0.000 description 4
- 235000010355 mannitol Nutrition 0.000 description 4
- 238000002483 medication Methods 0.000 description 4
- 230000004060 metabolic process Effects 0.000 description 4
- PIDFDZJZLOTZTM-KHVQSSSXSA-N ombitasvir Chemical compound COC(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@H]1C(=O)NC1=CC=C([C@H]2N([C@@H](CC2)C=2C=CC(NC(=O)[C@H]3N(CCC3)C(=O)[C@@H](NC(=O)OC)C(C)C)=CC=2)C=2C=CC(=CC=2)C(C)(C)C)C=C1 PIDFDZJZLOTZTM-KHVQSSSXSA-N 0.000 description 4
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 4
- 238000007254 oxidation reaction Methods 0.000 description 4
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 4
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 4
- 230000008569 process Effects 0.000 description 4
- 238000011160 research Methods 0.000 description 4
- 239000007858 starting material Substances 0.000 description 4
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 4
- JRMUNVKIHCOMHV-UHFFFAOYSA-M tetrabutylammonium bromide Chemical compound [Br-].CCCC[N+](CCCC)(CCCC)CCCC JRMUNVKIHCOMHV-UHFFFAOYSA-M 0.000 description 4
- 231100000027 toxicology Toxicity 0.000 description 4
- 230000009466 transformation Effects 0.000 description 4
- 238000000844 transformation Methods 0.000 description 4
- CSRZQMIRAZTJOY-UHFFFAOYSA-N trimethylsilyl iodide Chemical compound C[Si](C)(C)I CSRZQMIRAZTJOY-UHFFFAOYSA-N 0.000 description 4
- WEQLWGNDNRARGE-OIISXLGYSA-N (2s,3r,11br)-9,10-dimethoxy-3-(2-methylpropyl)-2,3,4,6,7,11b-hexahydro-1h-benzo[a]quinolizin-2-ol Chemical compound C1CN2C[C@@H](CC(C)C)[C@@H](O)C[C@@H]2C2=C1C=C(OC)C(OC)=C2 WEQLWGNDNRARGE-OIISXLGYSA-N 0.000 description 3
- HUWSZNZAROKDRZ-RRLWZMAJSA-N (3r,4r)-3-azaniumyl-5-[[(2s,3r)-1-[(2s)-2,3-dicarboxypyrrolidin-1-yl]-3-methyl-1-oxopentan-2-yl]amino]-5-oxo-4-sulfanylpentane-1-sulfonate Chemical compound OS(=O)(=O)CC[C@@H](N)[C@@H](S)C(=O)N[C@@H]([C@H](C)CC)C(=O)N1CCC(C(O)=O)[C@H]1C(O)=O HUWSZNZAROKDRZ-RRLWZMAJSA-N 0.000 description 3
- XEZNGIUYQVAUSS-UHFFFAOYSA-N 18-crown-6 Chemical compound C1COCCOCCOCCOCCOCCO1 XEZNGIUYQVAUSS-UHFFFAOYSA-N 0.000 description 3
- 238000005160 1H NMR spectroscopy Methods 0.000 description 3
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 3
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonium chloride Substances [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 3
- 0 CC(*)(*)C(*)(C(*)(*)*)C(*)(*)C(*)(C(*)(*)N(C(*)(*)C1(*)*)C(*)(C2(*)*)c(c(*)c3OC(*)(*)*)c1c(*)c3OC(*)(*)*)C2=O Chemical compound CC(*)(*)C(*)(C(*)(*)*)C(*)(*)C(*)(C(*)(*)N(C(*)(*)C1(*)*)C(*)(C2(*)*)c(c(*)c3OC(*)(*)*)c1c(*)c3OC(*)(*)*)C2=O 0.000 description 3
- HSVWPRINCAYYOG-UHFFFAOYSA-N CCOC(=O)C(C)(CCC(C)=O)C(O)=O Chemical compound CCOC(=O)C(C)(CCC(C)=O)C(O)=O HSVWPRINCAYYOG-UHFFFAOYSA-N 0.000 description 3
- VAEYOVFWKMQGGK-UHFFFAOYSA-N CN(C)CC(C(C)=O)CC(C)=C Chemical compound CN(C)CC(C(C)=O)CC(C)=C VAEYOVFWKMQGGK-UHFFFAOYSA-N 0.000 description 3
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 3
- 208000014094 Dystonic disease Diseases 0.000 description 3
- 108010010803 Gelatin Proteins 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 239000003810 Jones reagent Substances 0.000 description 3
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 3
- 208000001089 Multiple system atrophy Diseases 0.000 description 3
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 3
- 229920002125 Sokalan® Polymers 0.000 description 3
- QJJXYPPXXYFBGM-LFZNUXCKSA-N Tacrolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1\C=C(/C)[C@@H]1[C@H](C)[C@@H](O)CC(=O)[C@H](CC=C)/C=C(C)/C[C@H](C)C[C@H](OC)[C@H]([C@H](C[C@H]2C)OC)O[C@@]2(O)C(=O)C(=O)N2CCCC[C@H]2C(=O)O1 QJJXYPPXXYFBGM-LFZNUXCKSA-N 0.000 description 3
- 108090000373 Tissue Plasminogen Activator Proteins 0.000 description 3
- 102000003978 Tissue Plasminogen Activator Human genes 0.000 description 3
- QCBIDIAWUUDGKI-UHFFFAOYSA-M [I-].CC(=C)CC(C[N+](C)(C)C)C(C)=O Chemical compound [I-].CC(=C)CC(C[N+](C)(C)C)C(C)=O QCBIDIAWUUDGKI-UHFFFAOYSA-M 0.000 description 3
- FOHLKPGFQSXYMS-UHFFFAOYSA-M [I-].CCOC(=O)C(C)CC(C[N+](C)(C)C)C(C)=O Chemical compound [I-].CCOC(=O)C(C)CC(C[N+](C)(C)C)C(C)=O FOHLKPGFQSXYMS-UHFFFAOYSA-M 0.000 description 3
- 239000002671 adjuvant Substances 0.000 description 3
- 239000002246 antineoplastic agent Substances 0.000 description 3
- WEQLWGNDNRARGE-UHFFFAOYSA-N b-Dihydrotetrabenazine Chemical compound C1CN2CC(CC(C)C)C(O)CC2C2=C1C=C(OC)C(OC)=C2 WEQLWGNDNRARGE-UHFFFAOYSA-N 0.000 description 3
- 239000011230 binding agent Substances 0.000 description 3
- 230000037396 body weight Effects 0.000 description 3
- FUSUHKVFWTUUBE-UHFFFAOYSA-N buten-2-one Chemical compound CC(=O)C=C FUSUHKVFWTUUBE-UHFFFAOYSA-N 0.000 description 3
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 3
- 210000004027 cell Anatomy 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 229940125773 compound 10 Drugs 0.000 description 3
- 239000012351 deprotecting agent Substances 0.000 description 3
- 238000013461 design Methods 0.000 description 3
- KSWCYAJKDRMLKY-UHFFFAOYSA-N diethyl 2-methyl-2-(3-oxobutyl)propanedioate Chemical compound CCOC(=O)C(C)(CCC(C)=O)C(=O)OCC KSWCYAJKDRMLKY-UHFFFAOYSA-N 0.000 description 3
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical compound CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 3
- 239000000221 dopamine uptake inhibitor Substances 0.000 description 3
- 208000010118 dystonia Diseases 0.000 description 3
- VVDUZZGYBOWDSQ-UHFFFAOYSA-M eschenmoser's salt Chemical compound [I-].C[N+](C)=C VVDUZZGYBOWDSQ-UHFFFAOYSA-M 0.000 description 3
- DVIFNSDZOSQUGX-UHFFFAOYSA-N ethyl 2-acetyl-4-methylpent-4-enoate Chemical compound CCOC(=O)C(C(C)=O)CC(C)=C DVIFNSDZOSQUGX-UHFFFAOYSA-N 0.000 description 3
- ZJWLLXQQYYTJSW-UHFFFAOYSA-N ethyl 2-methyl-5-oxohexanoate Chemical compound CCOC(=O)C(C)CCC(C)=O ZJWLLXQQYYTJSW-UHFFFAOYSA-N 0.000 description 3
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 3
- 239000000499 gel Substances 0.000 description 3
- 239000008273 gelatin Substances 0.000 description 3
- 229920000159 gelatin Polymers 0.000 description 3
- 235000019322 gelatine Nutrition 0.000 description 3
- 235000011852 gelatine desserts Nutrition 0.000 description 3
- 239000008187 granular material Substances 0.000 description 3
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 3
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 3
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 3
- 230000007062 hydrolysis Effects 0.000 description 3
- 238000006460 hydrolysis reaction Methods 0.000 description 3
- 239000004615 ingredient Substances 0.000 description 3
- 238000011835 investigation Methods 0.000 description 3
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 3
- 230000000155 isotopic effect Effects 0.000 description 3
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 3
- 239000000314 lubricant Substances 0.000 description 3
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 3
- 239000008108 microcrystalline cellulose Substances 0.000 description 3
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 3
- 229940016286 microcrystalline cellulose Drugs 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- 230000003647 oxidation Effects 0.000 description 3
- 239000003208 petroleum Substances 0.000 description 3
- 230000003285 pharmacodynamic effect Effects 0.000 description 3
- 239000012071 phase Substances 0.000 description 3
- 229920001223 polyethylene glycol Polymers 0.000 description 3
- 230000002265 prevention Effects 0.000 description 3
- 239000003223 protective agent Substances 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- 239000012279 sodium borohydride Substances 0.000 description 3
- 229910000033 sodium borohydride Inorganic materials 0.000 description 3
- 239000012453 solvate Substances 0.000 description 3
- 230000003068 static effect Effects 0.000 description 3
- 239000004094 surface-active agent Substances 0.000 description 3
- 238000002560 therapeutic procedure Methods 0.000 description 3
- 210000001519 tissue Anatomy 0.000 description 3
- 229960000187 tissue plasminogen activator Drugs 0.000 description 3
- BQJOIGVCRGWWGH-UHFFFAOYSA-N trimethyl(5-methylhex-2-en-2-yloxy)silane Chemical compound CC(C)CC=C(C)O[Si](C)(C)C BQJOIGVCRGWWGH-UHFFFAOYSA-N 0.000 description 3
- SFLSHLFXELFNJZ-QMMMGPOBSA-N (-)-norepinephrine Chemical compound NC[C@H](O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-QMMMGPOBSA-N 0.000 description 2
- JJYKJUXBWFATTE-SECBINFHSA-N (2r)-3,3,3-trifluoro-2-methoxy-2-phenylpropanoic acid Chemical compound CO[C@](C(O)=O)(C(F)(F)F)C1=CC=CC=C1 JJYKJUXBWFATTE-SECBINFHSA-N 0.000 description 2
- IUSARDYWEPUTPN-OZBXUNDUSA-N (2r)-n-[(2s,3r)-4-[[(4s)-6-(2,2-dimethylpropyl)spiro[3,4-dihydropyrano[2,3-b]pyridine-2,1'-cyclobutane]-4-yl]amino]-3-hydroxy-1-[3-(1,3-thiazol-2-yl)phenyl]butan-2-yl]-2-methoxypropanamide Chemical compound C([C@H](NC(=O)[C@@H](C)OC)[C@H](O)CN[C@@H]1C2=CC(CC(C)(C)C)=CN=C2OC2(CCC2)C1)C(C=1)=CC=CC=1C1=NC=CS1 IUSARDYWEPUTPN-OZBXUNDUSA-N 0.000 description 2
- VIJSPAIQWVPKQZ-BLECARSGSA-N (2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-[[(2s)-2-acetamido-5-(diaminomethylideneamino)pentanoyl]amino]-4-methylpentanoyl]amino]-4,4-dimethylpentanoyl]amino]-4-methylpentanoyl]amino]propanoyl]amino]-5-(diaminomethylideneamino)pentanoic acid Chemical compound NC(=N)NCCC[C@@H](C(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(C)(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(C)=O VIJSPAIQWVPKQZ-BLECARSGSA-N 0.000 description 2
- STBLNCCBQMHSRC-BATDWUPUSA-N (2s)-n-[(3s,4s)-5-acetyl-7-cyano-4-methyl-1-[(2-methylnaphthalen-1-yl)methyl]-2-oxo-3,4-dihydro-1,5-benzodiazepin-3-yl]-2-(methylamino)propanamide Chemical compound O=C1[C@@H](NC(=O)[C@H](C)NC)[C@H](C)N(C(C)=O)C2=CC(C#N)=CC=C2N1CC1=C(C)C=CC2=CC=CC=C12 STBLNCCBQMHSRC-BATDWUPUSA-N 0.000 description 2
- WSSKRNHJTRPOTQ-WXIDZIMUSA-N (3R,11bR)-3-(2-hydroxy-2-methylpropyl)-9,10-bis(trideuteriomethoxy)-1,3,4,6,7,11b-hexahydrobenzo[a]quinolizin-2-one Chemical compound [2H]C([2H])([2H])Oc1cc2CCN3C[C@@H](CC(C)(C)O)C(=O)C[C@@H]3c2cc1OC([2H])([2H])[2H] WSSKRNHJTRPOTQ-WXIDZIMUSA-N 0.000 description 2
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 description 2
- UDQTXCHQKHIQMH-KYGLGHNPSA-N (3ar,5s,6s,7r,7ar)-5-(difluoromethyl)-2-(ethylamino)-5,6,7,7a-tetrahydro-3ah-pyrano[3,2-d][1,3]thiazole-6,7-diol Chemical compound S1C(NCC)=N[C@H]2[C@@H]1O[C@H](C(F)F)[C@@H](O)[C@@H]2O UDQTXCHQKHIQMH-KYGLGHNPSA-N 0.000 description 2
- OOKAZRDERJMRCJ-KOUAFAAESA-N (3r)-7-[(1s,2s,4ar,6s,8s)-2,6-dimethyl-8-[(2s)-2-methylbutanoyl]oxy-1,2,4a,5,6,7,8,8a-octahydronaphthalen-1-yl]-3-hydroxy-5-oxoheptanoic acid Chemical compound C1=C[C@H](C)[C@H](CCC(=O)C[C@@H](O)CC(O)=O)C2[C@@H](OC(=O)[C@@H](C)CC)C[C@@H](C)C[C@@H]21 OOKAZRDERJMRCJ-KOUAFAAESA-N 0.000 description 2
- MPDDTAJMJCESGV-CTUHWIOQSA-M (3r,5r)-7-[2-(4-fluorophenyl)-5-[methyl-[(1r)-1-phenylethyl]carbamoyl]-4-propan-2-ylpyrazol-3-yl]-3,5-dihydroxyheptanoate Chemical compound C1([C@@H](C)N(C)C(=O)C2=NN(C(CC[C@@H](O)C[C@@H](O)CC([O-])=O)=C2C(C)C)C=2C=CC(F)=CC=2)=CC=CC=C1 MPDDTAJMJCESGV-CTUHWIOQSA-M 0.000 description 2
- IOQORVDNYPOZPL-VQTJNVASSA-N (5S,6R)-5-(4-chlorophenyl)-6-cyclopropyl-3-[6-methoxy-5-(4-methylimidazol-1-yl)pyridin-2-yl]-5,6-dihydro-2H-1,2,4-oxadiazine Chemical compound ClC1=CC=C(C=C1)[C@@H]1NC(=NO[C@@H]1C1CC1)C1=NC(=C(C=C1)N1C=NC(=C1)C)OC IOQORVDNYPOZPL-VQTJNVASSA-N 0.000 description 2
- VUEGYUOUAAVYAS-JGGQBBKZSA-N (6ar,9s,10ar)-9-(dimethylsulfamoylamino)-7-methyl-6,6a,8,9,10,10a-hexahydro-4h-indolo[4,3-fg]quinoline Chemical compound C1=CC([C@H]2C[C@@H](CN(C)[C@@H]2C2)NS(=O)(=O)N(C)C)=C3C2=CNC3=C1 VUEGYUOUAAVYAS-JGGQBBKZSA-N 0.000 description 2
- BGRJTUBHPOOWDU-NSHDSACASA-N (S)-(-)-sulpiride Chemical compound CCN1CCC[C@H]1CNC(=O)C1=CC(S(N)(=O)=O)=CC=C1OC BGRJTUBHPOOWDU-NSHDSACASA-N 0.000 description 2
- ZYZCALPXKGUGJI-DDVDASKDSA-M (e,3r,5s)-7-[3-(4-fluorophenyl)-2-phenyl-5-propan-2-ylimidazol-4-yl]-3,5-dihydroxyhept-6-enoate Chemical compound C=1C=C(F)C=CC=1N1C(\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O)=C(C(C)C)N=C1C1=CC=CC=C1 ZYZCALPXKGUGJI-DDVDASKDSA-M 0.000 description 2
- DEVSOMFAQLZNKR-RJRFIUFISA-N (z)-3-[3-[3,5-bis(trifluoromethyl)phenyl]-1,2,4-triazol-1-yl]-n'-pyrazin-2-ylprop-2-enehydrazide Chemical compound FC(F)(F)C1=CC(C(F)(F)F)=CC(C2=NN(\C=C/C(=O)NNC=3N=CC=NC=3)C=N2)=C1 DEVSOMFAQLZNKR-RJRFIUFISA-N 0.000 description 2
- KKHFRAFPESRGGD-UHFFFAOYSA-N 1,3-dimethyl-7-[3-(n-methylanilino)propyl]purine-2,6-dione Chemical compound C1=NC=2N(C)C(=O)N(C)C(=O)C=2N1CCCN(C)C1=CC=CC=C1 KKHFRAFPESRGGD-UHFFFAOYSA-N 0.000 description 2
- KQZLRWGGWXJPOS-NLFPWZOASA-N 1-[(1R)-1-(2,4-dichlorophenyl)ethyl]-6-[(4S,5R)-4-[(2S)-2-(hydroxymethyl)pyrrolidin-1-yl]-5-methylcyclohexen-1-yl]pyrazolo[3,4-b]pyrazine-3-carbonitrile Chemical compound ClC1=C(C=CC(=C1)Cl)[C@@H](C)N1N=C(C=2C1=NC(=CN=2)C1=CC[C@@H]([C@@H](C1)C)N1[C@@H](CCC1)CO)C#N KQZLRWGGWXJPOS-NLFPWZOASA-N 0.000 description 2
- WZZBNLYBHUDSHF-DHLKQENFSA-N 1-[(3s,4s)-4-[8-(2-chloro-4-pyrimidin-2-yloxyphenyl)-7-fluoro-2-methylimidazo[4,5-c]quinolin-1-yl]-3-fluoropiperidin-1-yl]-2-hydroxyethanone Chemical compound CC1=NC2=CN=C3C=C(F)C(C=4C(=CC(OC=5N=CC=CN=5)=CC=4)Cl)=CC3=C2N1[C@H]1CCN(C(=O)CO)C[C@@H]1F WZZBNLYBHUDSHF-DHLKQENFSA-N 0.000 description 2
- QXOGPTXQGKQSJT-UHFFFAOYSA-N 1-amino-4-[4-(3,4-dimethylphenyl)sulfanylanilino]-9,10-dioxoanthracene-2-sulfonic acid Chemical compound Cc1ccc(Sc2ccc(Nc3cc(c(N)c4C(=O)c5ccccc5C(=O)c34)S(O)(=O)=O)cc2)cc1C QXOGPTXQGKQSJT-UHFFFAOYSA-N 0.000 description 2
- WXTMDXOMEHJXQO-UHFFFAOYSA-N 2,5-dihydroxybenzoic acid Chemical compound OC(=O)C1=CC(O)=CC=C1O WXTMDXOMEHJXQO-UHFFFAOYSA-N 0.000 description 2
- WGFNXGPBPIJYLI-UHFFFAOYSA-N 2,6-difluoro-3-[(3-fluorophenyl)sulfonylamino]-n-(3-methoxy-1h-pyrazolo[3,4-b]pyridin-5-yl)benzamide Chemical compound C1=C2C(OC)=NNC2=NC=C1NC(=O)C(C=1F)=C(F)C=CC=1NS(=O)(=O)C1=CC=CC(F)=C1 WGFNXGPBPIJYLI-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- LBLYYCQCTBFVLH-UHFFFAOYSA-N 2-Methylbenzenesulfonic acid Chemical compound CC1=CC=CC=C1S(O)(=O)=O LBLYYCQCTBFVLH-UHFFFAOYSA-N 0.000 description 2
- HVAUUPRFYPCOCA-AREMUKBSSA-N 2-O-acetyl-1-O-hexadecyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCCOC[C@@H](OC(C)=O)COP([O-])(=O)OCC[N+](C)(C)C HVAUUPRFYPCOCA-AREMUKBSSA-N 0.000 description 2
- PYRKKGOKRMZEIT-UHFFFAOYSA-N 2-[6-(2-cyclopropylethoxy)-9-(2-hydroxy-2-methylpropyl)-1h-phenanthro[9,10-d]imidazol-2-yl]-5-fluorobenzene-1,3-dicarbonitrile Chemical compound C1=C2C3=CC(CC(C)(O)C)=CC=C3C=3NC(C=4C(=CC(F)=CC=4C#N)C#N)=NC=3C2=CC=C1OCCC1CC1 PYRKKGOKRMZEIT-UHFFFAOYSA-N 0.000 description 2
- FMKGJQHNYMWDFJ-CVEARBPZSA-N 2-[[4-(2,2-difluoropropoxy)pyrimidin-5-yl]methylamino]-4-[[(1R,4S)-4-hydroxy-3,3-dimethylcyclohexyl]amino]pyrimidine-5-carbonitrile Chemical compound FC(COC1=NC=NC=C1CNC1=NC=C(C(=N1)N[C@H]1CC([C@H](CC1)O)(C)C)C#N)(C)F FMKGJQHNYMWDFJ-CVEARBPZSA-N 0.000 description 2
- VVCMGAUPZIKYTH-VGHSCWAPSA-N 2-acetyloxybenzoic acid;[(2s,3r)-4-(dimethylamino)-3-methyl-1,2-diphenylbutan-2-yl] propanoate;1,3,7-trimethylpurine-2,6-dione Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O.CN1C(=O)N(C)C(=O)C2=C1N=CN2C.C([C@](OC(=O)CC)([C@H](C)CN(C)C)C=1C=CC=CC=1)C1=CC=CC=C1 VVCMGAUPZIKYTH-VGHSCWAPSA-N 0.000 description 2
- YSUIQYOGTINQIN-UZFYAQMZSA-N 2-amino-9-[(1S,6R,8R,9S,10R,15R,17R,18R)-8-(6-aminopurin-9-yl)-9,18-difluoro-3,12-dihydroxy-3,12-bis(sulfanylidene)-2,4,7,11,13,16-hexaoxa-3lambda5,12lambda5-diphosphatricyclo[13.2.1.06,10]octadecan-17-yl]-1H-purin-6-one Chemical compound NC1=NC2=C(N=CN2[C@@H]2O[C@@H]3COP(S)(=O)O[C@@H]4[C@@H](COP(S)(=O)O[C@@H]2[C@@H]3F)O[C@H]([C@H]4F)N2C=NC3=C2N=CN=C3N)C(=O)N1 YSUIQYOGTINQIN-UZFYAQMZSA-N 0.000 description 2
- TVTJUIAKQFIXCE-HUKYDQBMSA-N 2-amino-9-[(2R,3S,4S,5R)-4-fluoro-3-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-7-prop-2-ynyl-1H-purine-6,8-dione Chemical compound NC=1NC(C=2N(C(N(C=2N=1)[C@@H]1O[C@@H]([C@H]([C@H]1O)F)CO)=O)CC#C)=O TVTJUIAKQFIXCE-HUKYDQBMSA-N 0.000 description 2
- NPRYCHLHHVWLQZ-TURQNECASA-N 2-amino-9-[(2R,3S,4S,5R)-4-fluoro-3-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-7-prop-2-ynylpurin-8-one Chemical compound NC1=NC=C2N(C(N(C2=N1)[C@@H]1O[C@@H]([C@H]([C@H]1O)F)CO)=O)CC#C NPRYCHLHHVWLQZ-TURQNECASA-N 0.000 description 2
- LFOIDLOIBZFWDO-UHFFFAOYSA-N 2-methoxy-6-[6-methoxy-4-[(3-phenylmethoxyphenyl)methoxy]-1-benzofuran-2-yl]imidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(C=1)=CC=CC=1OCC1=CC=CC=C1 LFOIDLOIBZFWDO-UHFFFAOYSA-N 0.000 description 2
- ZUXNHFFVQWADJL-UHFFFAOYSA-N 3,4,5-trimethoxy-n-(2-methoxyethyl)-n-(4-phenyl-1,3-thiazol-2-yl)benzamide Chemical compound N=1C(C=2C=CC=CC=2)=CSC=1N(CCOC)C(=O)C1=CC(OC)=C(OC)C(OC)=C1 ZUXNHFFVQWADJL-UHFFFAOYSA-N 0.000 description 2
- WYFCZWSWFGJODV-MIANJLSGSA-N 4-[[(1s)-2-[(e)-3-[3-chloro-2-fluoro-6-(tetrazol-1-yl)phenyl]prop-2-enoyl]-5-(4-methyl-2-oxopiperazin-1-yl)-3,4-dihydro-1h-isoquinoline-1-carbonyl]amino]benzoic acid Chemical compound O=C1CN(C)CCN1C1=CC=CC2=C1CCN(C(=O)\C=C\C=1C(=CC=C(Cl)C=1F)N1N=NN=C1)[C@@H]2C(=O)NC1=CC=C(C(O)=O)C=C1 WYFCZWSWFGJODV-MIANJLSGSA-N 0.000 description 2
- QCXJEYYXVJIFCE-UHFFFAOYSA-N 4-acetamidobenzoic acid Chemical compound CC(=O)NC1=CC=C(C(O)=O)C=C1 QCXJEYYXVJIFCE-UHFFFAOYSA-N 0.000 description 2
- DQAZPZIYEOGZAF-UHFFFAOYSA-N 4-ethyl-n-[4-(3-ethynylanilino)-7-methoxyquinazolin-6-yl]piperazine-1-carboxamide Chemical compound C1CN(CC)CCN1C(=O)NC(C(=CC1=NC=N2)OC)=CC1=C2NC1=CC=CC(C#C)=C1 DQAZPZIYEOGZAF-UHFFFAOYSA-N 0.000 description 2
- FFWSICBKRCICMR-UHFFFAOYSA-N 5-methyl-2-hexanone Chemical compound CC(C)CCC(C)=O FFWSICBKRCICMR-UHFFFAOYSA-N 0.000 description 2
- RSIWALKZYXPAGW-NSHDSACASA-N 6-(3-fluorophenyl)-3-methyl-7-[(1s)-1-(7h-purin-6-ylamino)ethyl]-[1,3]thiazolo[3,2-a]pyrimidin-5-one Chemical compound C=1([C@@H](NC=2C=3N=CNC=3N=CN=2)C)N=C2SC=C(C)N2C(=O)C=1C1=CC=CC(F)=C1 RSIWALKZYXPAGW-NSHDSACASA-N 0.000 description 2
- HCCNBKFJYUWLEX-UHFFFAOYSA-N 7-(6-methoxypyridin-3-yl)-1-(2-propoxyethyl)-3-(pyrazin-2-ylmethylamino)pyrido[3,4-b]pyrazin-2-one Chemical compound O=C1N(CCOCCC)C2=CC(C=3C=NC(OC)=CC=3)=NC=C2N=C1NCC1=CN=CC=N1 HCCNBKFJYUWLEX-UHFFFAOYSA-N 0.000 description 2
- 108010058207 Anistreplase Proteins 0.000 description 2
- 241001550224 Apha Species 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- JQUCWIWWWKZNCS-LESHARBVSA-N C(C1=CC=CC=C1)(=O)NC=1SC[C@H]2[C@@](N1)(CO[C@H](C2)C)C=2SC=C(N2)NC(=O)C2=NC=C(C=C2)OC(F)F Chemical compound C(C1=CC=CC=C1)(=O)NC=1SC[C@H]2[C@@](N1)(CO[C@H](C2)C)C=2SC=C(N2)NC(=O)C2=NC=C(C=C2)OC(F)F JQUCWIWWWKZNCS-LESHARBVSA-N 0.000 description 2
- OJRUSAPKCPIVBY-KQYNXXCUSA-N C1=NC2=C(N=C(N=C2N1[C@H]3[C@@H]([C@@H]([C@H](O3)COP(=O)(CP(=O)(O)O)O)O)O)I)N Chemical compound C1=NC2=C(N=C(N=C2N1[C@H]3[C@@H]([C@@H]([C@H](O3)COP(=O)(CP(=O)(O)O)O)O)O)I)N OJRUSAPKCPIVBY-KQYNXXCUSA-N 0.000 description 2
- AJAKXRTZHSPVHO-UHFFFAOYSA-N CCOC(=O)C(C)CC(CN(C)C)C(C)=O Chemical compound CCOC(=O)C(C)CC(CN(C)C)C(C)=O AJAKXRTZHSPVHO-UHFFFAOYSA-N 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- JZUFKLXOESDKRF-UHFFFAOYSA-N Chlorothiazide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC2=C1NCNS2(=O)=O JZUFKLXOESDKRF-UHFFFAOYSA-N 0.000 description 2
- 108010061846 Cholesterol Ester Transfer Proteins Proteins 0.000 description 2
- 102000012336 Cholesterol Ester Transfer Proteins Human genes 0.000 description 2
- 206010008748 Chorea Diseases 0.000 description 2
- 229940126639 Compound 33 Drugs 0.000 description 2
- 108010037462 Cyclooxygenase 2 Proteins 0.000 description 2
- 102000002004 Cytochrome P-450 Enzyme System Human genes 0.000 description 2
- 108010015742 Cytochrome P-450 Enzyme System Proteins 0.000 description 2
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 2
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 2
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 2
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 2
- 108030001679 Endothelin-converting enzyme 1 Proteins 0.000 description 2
- 102000048186 Endothelin-converting enzyme 1 Human genes 0.000 description 2
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- LVDRREOUMKACNJ-BKMJKUGQSA-N N-[(2R,3S)-2-(4-chlorophenyl)-1-(1,4-dimethyl-2-oxoquinolin-7-yl)-6-oxopiperidin-3-yl]-2-methylpropane-1-sulfonamide Chemical compound CC(C)CS(=O)(=O)N[C@H]1CCC(=O)N([C@@H]1c1ccc(Cl)cc1)c1ccc2c(C)cc(=O)n(C)c2c1 LVDRREOUMKACNJ-BKMJKUGQSA-N 0.000 description 2
- QIAFMBKCNZACKA-UHFFFAOYSA-N N-benzoylglycine Chemical compound OC(=O)CNC(=O)C1=CC=CC=C1 QIAFMBKCNZACKA-UHFFFAOYSA-N 0.000 description 2
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 2
- QOVYHDHLFPKQQG-NDEPHWFRSA-N N[C@@H](CCC(=O)N1CCC(CC1)NC1=C2C=CC=CC2=NC(NCC2=CN(CCCNCCCNC3CCCCC3)N=N2)=N1)C(O)=O Chemical compound N[C@@H](CCC(=O)N1CCC(CC1)NC1=C2C=CC=CC2=NC(NCC2=CN(CCCNCCCNC3CCCCC3)N=N2)=N1)C(O)=O QOVYHDHLFPKQQG-NDEPHWFRSA-N 0.000 description 2
- 102000003729 Neprilysin Human genes 0.000 description 2
- 108090000028 Neprilysin Proteins 0.000 description 2
- 102000004316 Oxidoreductases Human genes 0.000 description 2
- 108090000854 Oxidoreductases Proteins 0.000 description 2
- 229930040373 Paraformaldehyde Natural products 0.000 description 2
- 208000018737 Parkinson disease Diseases 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- 108010003541 Platelet Activating Factor Proteins 0.000 description 2
- 239000002202 Polyethylene glycol Substances 0.000 description 2
- 102100038280 Prostaglandin G/H synthase 2 Human genes 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- 241000283984 Rodentia Species 0.000 description 2
- YASAKCUCGLMORW-UHFFFAOYSA-N Rosiglitazone Chemical compound C=1C=CC=NC=1N(C)CCOC(C=C1)=CC=C1CC1SC(=O)NC1=O YASAKCUCGLMORW-UHFFFAOYSA-N 0.000 description 2
- 229940121991 Serotonin and norepinephrine reuptake inhibitor Drugs 0.000 description 2
- PNUZDKCDAWUEGK-CYZMBNFOSA-N Sitafloxacin Chemical compound C([C@H]1N)N(C=2C(=C3C(C(C(C(O)=O)=CN3[C@H]3[C@H](C3)F)=O)=CC=2F)Cl)CC11CC1 PNUZDKCDAWUEGK-CYZMBNFOSA-N 0.000 description 2
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 2
- 206010067672 Spasmodic dysphonia Diseases 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- 241000282898 Sus scrofa Species 0.000 description 2
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 2
- 206010044074 Torticollis Diseases 0.000 description 2
- 208000000323 Tourette Syndrome Diseases 0.000 description 2
- 208000016620 Tourette disease Diseases 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- LJOOWESTVASNOG-UFJKPHDISA-N [(1s,3r,4ar,7s,8s,8as)-3-hydroxy-8-[2-[(4r)-4-hydroxy-6-oxooxan-2-yl]ethyl]-7-methyl-1,2,3,4,4a,7,8,8a-octahydronaphthalen-1-yl] (2s)-2-methylbutanoate Chemical compound C([C@H]1[C@@H](C)C=C[C@H]2C[C@@H](O)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)CC1C[C@@H](O)CC(=O)O1 LJOOWESTVASNOG-UFJKPHDISA-N 0.000 description 2
- SPXSEZMVRJLHQG-XMMPIXPASA-N [(2R)-1-[[4-[(3-phenylmethoxyphenoxy)methyl]phenyl]methyl]pyrrolidin-2-yl]methanol Chemical compound C(C1=CC=CC=C1)OC=1C=C(OCC2=CC=C(CN3[C@H](CCC3)CO)C=C2)C=CC=1 SPXSEZMVRJLHQG-XMMPIXPASA-N 0.000 description 2
- PSLUFJFHTBIXMW-WYEYVKMPSA-N [(3r,4ar,5s,6s,6as,10s,10ar,10bs)-3-ethenyl-10,10b-dihydroxy-3,4a,7,7,10a-pentamethyl-1-oxo-6-(2-pyridin-2-ylethylcarbamoyloxy)-5,6,6a,8,9,10-hexahydro-2h-benzo[f]chromen-5-yl] acetate Chemical compound O([C@@H]1[C@@H]([C@]2(O[C@](C)(CC(=O)[C@]2(O)[C@@]2(C)[C@@H](O)CCC(C)(C)[C@@H]21)C=C)C)OC(=O)C)C(=O)NCCC1=CC=CC=N1 PSLUFJFHTBIXMW-WYEYVKMPSA-N 0.000 description 2
- RJURFGZVJUQBHK-UHFFFAOYSA-N actinomycin D Natural products CC1OC(=O)C(C(C)C)N(C)C(=O)CN(C)C(=O)C2CCCN2C(=O)C(C(C)C)NC(=O)C1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)NC4C(=O)NC(C(N5CCCC5C(=O)N(C)CC(=O)N(C)C(C(C)C)C(=O)OC4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-UHFFFAOYSA-N 0.000 description 2
- 239000013543 active substance Substances 0.000 description 2
- WNLRTRBMVRJNCN-UHFFFAOYSA-N adipic acid Chemical compound OC(=O)CCCCC(O)=O WNLRTRBMVRJNCN-UHFFFAOYSA-N 0.000 description 2
- 230000002411 adverse Effects 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- 239000003242 anti bacterial agent Substances 0.000 description 2
- 238000013528 artificial neural network Methods 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 2
- XGIUDIMNNMKGDE-UHFFFAOYSA-N bis(trimethylsilyl)azanide Chemical compound C[Si](C)(C)[N-][Si](C)(C)C XGIUDIMNNMKGDE-UHFFFAOYSA-N 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- MCQRPQCQMGVWIQ-UHFFFAOYSA-N boron;methylsulfanylmethane Chemical compound [B].CSC MCQRPQCQMGVWIQ-UHFFFAOYSA-N 0.000 description 2
- UWTDFICHZKXYAC-UHFFFAOYSA-N boron;oxolane Chemical compound [B].C1CCOC1 UWTDFICHZKXYAC-UHFFFAOYSA-N 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- CREMABGTGYGIQB-UHFFFAOYSA-N carbon carbon Chemical compound C.C CREMABGTGYGIQB-UHFFFAOYSA-N 0.000 description 2
- 229910002092 carbon dioxide Inorganic materials 0.000 description 2
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 2
- 239000012320 chlorinating reagent Substances 0.000 description 2
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 2
- WGLPBDUCMAPZCE-UHFFFAOYSA-N chromium trioxide Inorganic materials O=[Cr](=O)=O WGLPBDUCMAPZCE-UHFFFAOYSA-N 0.000 description 2
- 238000000576 coating method Methods 0.000 description 2
- 239000012230 colorless oil Substances 0.000 description 2
- 238000002648 combination therapy Methods 0.000 description 2
- 229940125904 compound 1 Drugs 0.000 description 2
- 229940125758 compound 15 Drugs 0.000 description 2
- 229940125782 compound 2 Drugs 0.000 description 2
- 229940125851 compound 27 Drugs 0.000 description 2
- 229940127204 compound 29 Drugs 0.000 description 2
- 229940126214 compound 3 Drugs 0.000 description 2
- 229940125877 compound 31 Drugs 0.000 description 2
- 229940125878 compound 36 Drugs 0.000 description 2
- 229940125807 compound 37 Drugs 0.000 description 2
- 229940126540 compound 41 Drugs 0.000 description 2
- 229940125936 compound 42 Drugs 0.000 description 2
- 229940127271 compound 49 Drugs 0.000 description 2
- 229940125898 compound 5 Drugs 0.000 description 2
- 229940126545 compound 53 Drugs 0.000 description 2
- 229940127113 compound 57 Drugs 0.000 description 2
- 238000007906 compression Methods 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- 229940127089 cytotoxic agent Drugs 0.000 description 2
- 230000002354 daily effect Effects 0.000 description 2
- GHVNFZFCNZKVNT-UHFFFAOYSA-N decanoic acid Chemical compound CCCCCCCCCC(O)=O GHVNFZFCNZKVNT-UHFFFAOYSA-N 0.000 description 2
- 230000002939 deleterious effect Effects 0.000 description 2
- JXTHNDFMNIQAHM-UHFFFAOYSA-N dichloroacetic acid Chemical compound OC(=O)C(Cl)Cl JXTHNDFMNIQAHM-UHFFFAOYSA-N 0.000 description 2
- PXBRQCKWGAHEHS-UHFFFAOYSA-N dichlorodifluoromethane Chemical compound FC(F)(Cl)Cl PXBRQCKWGAHEHS-UHFFFAOYSA-N 0.000 description 2
- 229960001760 dimethyl sulfoxide Drugs 0.000 description 2
- XHFGWHUWQXTGAT-UHFFFAOYSA-N dimethylamine hydrochloride Natural products CNC(C)C XHFGWHUWQXTGAT-UHFFFAOYSA-N 0.000 description 2
- IQDGSYLLQPDQDV-UHFFFAOYSA-N dimethylazanium;chloride Chemical compound Cl.CNC IQDGSYLLQPDQDV-UHFFFAOYSA-N 0.000 description 2
- 239000002270 dispersing agent Substances 0.000 description 2
- POULHZVOKOAJMA-UHFFFAOYSA-N dodecanoic acid Chemical compound CCCCCCCCCCCC(O)=O POULHZVOKOAJMA-UHFFFAOYSA-N 0.000 description 2
- 239000008298 dragée Substances 0.000 description 2
- 239000003937 drug carrier Substances 0.000 description 2
- JXVQSYFGPAPTFR-LUAWRHEFSA-N ethyl (Z)-2-methyl-5-trimethylsilyloxyhex-4-enoate Chemical compound CCOC(=O)C(C)C\C=C(\C)O[Si](C)(C)C JXVQSYFGPAPTFR-LUAWRHEFSA-N 0.000 description 2
- 230000003090 exacerbative effect Effects 0.000 description 2
- 230000029142 excretion Effects 0.000 description 2
- 239000000284 extract Substances 0.000 description 2
- 239000010685 fatty oil Substances 0.000 description 2
- 230000005021 gait Effects 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- 239000003193 general anesthetic agent Substances 0.000 description 2
- 229940005494 general anesthetics Drugs 0.000 description 2
- 239000003862 glucocorticoid Substances 0.000 description 2
- 238000005469 granulation Methods 0.000 description 2
- 230000003179 granulation Effects 0.000 description 2
- LNEPOXFFQSENCJ-UHFFFAOYSA-N haloperidol Chemical compound C1CC(O)(C=2C=CC(Cl)=CC=2)CCN1CCCC(=O)C1=CC=C(F)C=C1 LNEPOXFFQSENCJ-UHFFFAOYSA-N 0.000 description 2
- 230000036541 health Effects 0.000 description 2
- IPCSVZSSVZVIGE-UHFFFAOYSA-N hexadecanoic acid Chemical compound CCCCCCCCCCCCCCCC(O)=O IPCSVZSSVZVIGE-UHFFFAOYSA-N 0.000 description 2
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 2
- JYGXADMDTFJGBT-VWUMJDOOSA-N hydrocortisone Chemical compound O=C1CC[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 JYGXADMDTFJGBT-VWUMJDOOSA-N 0.000 description 2
- 230000003483 hypokinetic effect Effects 0.000 description 2
- 230000002267 hypothalamic effect Effects 0.000 description 2
- 239000012729 immediate-release (IR) formulation Substances 0.000 description 2
- 230000006872 improvement Effects 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 238000007912 intraperitoneal administration Methods 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 2
- 239000003589 local anesthetic agent Substances 0.000 description 2
- 229960005015 local anesthetics Drugs 0.000 description 2
- 230000007774 longterm Effects 0.000 description 2
- 235000019359 magnesium stearate Nutrition 0.000 description 2
- 230000014759 maintenance of location Effects 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 238000006241 metabolic reaction Methods 0.000 description 2
- CSJDCSCTVDEHRN-UHFFFAOYSA-N methane;molecular oxygen Chemical compound C.O=O CSJDCSCTVDEHRN-UHFFFAOYSA-N 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 238000000465 moulding Methods 0.000 description 2
- 201000006417 multiple sclerosis Diseases 0.000 description 2
- 239000000472 muscarinic agonist Substances 0.000 description 2
- XVDBWWRIXBMVJV-UHFFFAOYSA-N n-[bis(dimethylamino)phosphanyl]-n-methylmethanamine Chemical compound CN(C)P(N(C)C)N(C)C XVDBWWRIXBMVJV-UHFFFAOYSA-N 0.000 description 2
- XTEGVFVZDVNBPF-UHFFFAOYSA-N naphthalene-1,5-disulfonic acid Chemical compound C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1S(O)(=O)=O XTEGVFVZDVNBPF-UHFFFAOYSA-N 0.000 description 2
- 229960003512 nicotinic acid Drugs 0.000 description 2
- 235000001968 nicotinic acid Nutrition 0.000 description 2
- 239000011664 nicotinic acid Substances 0.000 description 2
- 239000012299 nitrogen atmosphere Substances 0.000 description 2
- 239000000041 non-steroidal anti-inflammatory agent Substances 0.000 description 2
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 description 2
- 239000002767 noradrenalin uptake inhibitor Substances 0.000 description 2
- 229940127221 norepinephrine reuptake inhibitor Drugs 0.000 description 2
- WWZKQHOCKIZLMA-UHFFFAOYSA-N octanoic acid Chemical compound CCCCCCCC(O)=O WWZKQHOCKIZLMA-UHFFFAOYSA-N 0.000 description 2
- KVWDHTXUZHCGIO-UHFFFAOYSA-N olanzapine Chemical compound C1CN(C)CCN1C1=NC2=CC=CC=C2NC2=C1C=C(C)S2 KVWDHTXUZHCGIO-UHFFFAOYSA-N 0.000 description 2
- 229960005017 olanzapine Drugs 0.000 description 2
- PXQPEWDEAKTCGB-UHFFFAOYSA-N orotic acid Chemical compound OC(=O)C1=CC(=O)NC(=O)N1 PXQPEWDEAKTCGB-UHFFFAOYSA-N 0.000 description 2
- 229920002866 paraformaldehyde Polymers 0.000 description 2
- 238000007911 parenteral administration Methods 0.000 description 2
- 239000006072 paste Substances 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- 239000003444 phase transfer catalyst Substances 0.000 description 2
- 150000003904 phospholipids Chemical class 0.000 description 2
- 230000037081 physical activity Effects 0.000 description 2
- 230000004962 physiological condition Effects 0.000 description 2
- 229960003634 pimozide Drugs 0.000 description 2
- YVUQSNJEYSNKRX-UHFFFAOYSA-N pimozide Chemical compound C1=CC(F)=CC=C1C(C=1C=CC(F)=CC=1)CCCN1CCC(N2C(NC3=CC=CC=C32)=O)CC1 YVUQSNJEYSNKRX-UHFFFAOYSA-N 0.000 description 2
- HYAFETHFCAUJAY-UHFFFAOYSA-N pioglitazone Chemical compound N1=CC(CC)=CC=C1CCOC(C=C1)=CC=C1CC1C(=O)NC(=O)S1 HYAFETHFCAUJAY-UHFFFAOYSA-N 0.000 description 2
- 229960002797 pitavastatin Drugs 0.000 description 2
- RHGYHLPFVJEAOC-FFNUKLMVSA-L pitavastatin calcium Chemical compound [Ca+2].[O-]C(=O)C[C@H](O)C[C@H](O)\C=C\C1=C(C2CC2)N=C2C=CC=CC2=C1C1=CC=C(F)C=C1.[O-]C(=O)C[C@H](O)C[C@H](O)\C=C\C1=C(C2CC2)N=C2C=CC=CC2=C1C1=CC=C(F)C=C1 RHGYHLPFVJEAOC-FFNUKLMVSA-L 0.000 description 2
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Chemical compound [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 description 2
- 229920000191 poly(N-vinyl pyrrolidone) Polymers 0.000 description 2
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 2
- 229920000053 polysorbate 80 Polymers 0.000 description 2
- 238000002953 preparative HPLC Methods 0.000 description 2
- 238000004237 preparative chromatography Methods 0.000 description 2
- WGYKZJWCGVVSQN-UHFFFAOYSA-N propylamine Chemical compound CCCN WGYKZJWCGVVSQN-UHFFFAOYSA-N 0.000 description 2
- 238000011002 quantification Methods 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- 239000011369 resultant mixture Substances 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- 102220310434 rs764401457 Human genes 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- CXMXRPHRNRROMY-UHFFFAOYSA-N sebacic acid Chemical compound OC(=O)CCCCCCCCC(O)=O CXMXRPHRNRROMY-UHFFFAOYSA-N 0.000 description 2
- QZAYGJVTTNCVMB-UHFFFAOYSA-N serotonin Chemical compound C1=C(O)C=C2C(CCN)=CNC2=C1 QZAYGJVTTNCVMB-UHFFFAOYSA-N 0.000 description 2
- 239000003775 serotonin noradrenalin reuptake inhibitor Substances 0.000 description 2
- BNRNXUUZRGQAQC-UHFFFAOYSA-N sildenafil Chemical compound CCCC1=NN(C)C(C(N2)=O)=C1N=C2C(C(=CC=1)OCC)=CC=1S(=O)(=O)N1CCN(C)CC1 BNRNXUUZRGQAQC-UHFFFAOYSA-N 0.000 description 2
- 239000012354 sodium borodeuteride Substances 0.000 description 2
- 229910000029 sodium carbonate Inorganic materials 0.000 description 2
- 239000011780 sodium chloride Substances 0.000 description 2
- 239000012312 sodium hydride Substances 0.000 description 2
- 229910000104 sodium hydride Inorganic materials 0.000 description 2
- 239000007909 solid dosage form Substances 0.000 description 2
- 239000000600 sorbitol Substances 0.000 description 2
- 201000002849 spasmodic dystonia Diseases 0.000 description 2
- 229960002256 spironolactone Drugs 0.000 description 2
- LXMSZDCAJNLERA-ZHYRCANASA-N spironolactone Chemical compound C([C@@H]1[C@]2(C)CC[C@@H]3[C@@]4(C)CCC(=O)C=C4C[C@H]([C@@H]13)SC(=O)C)C[C@@]21CCC(=O)O1 LXMSZDCAJNLERA-ZHYRCANASA-N 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 125000001424 substituent group Chemical group 0.000 description 2
- 239000000375 suspending agent Substances 0.000 description 2
- 238000007910 systemic administration Methods 0.000 description 2
- 229940037128 systemic glucocorticoids Drugs 0.000 description 2
- QJJXYPPXXYFBGM-SHYZHZOCSA-N tacrolimus Natural products CO[C@H]1C[C@H](CC[C@@H]1O)C=C(C)[C@H]2OC(=O)[C@H]3CCCCN3C(=O)C(=O)[C@@]4(O)O[C@@H]([C@H](C[C@H]4C)OC)[C@@H](C[C@H](C)CC(=C[C@@H](CC=C)C(=O)C[C@H](O)[C@H]2C)C)OC QJJXYPPXXYFBGM-SHYZHZOCSA-N 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- LMBFAGIMSUYTBN-MPZNNTNKSA-N teixobactin Chemical compound C([C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H](CCC(N)=O)C(=O)N[C@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CO)C(=O)N[C@H]1C(N[C@@H](C)C(=O)N[C@@H](C[C@@H]2NC(=N)NC2)C(=O)N[C@H](C(=O)O[C@H]1C)[C@@H](C)CC)=O)NC)C1=CC=CC=C1 LMBFAGIMSUYTBN-MPZNNTNKSA-N 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- ZMZDMBWJUHKJPS-UHFFFAOYSA-N thiocyanic acid Chemical compound SC#N ZMZDMBWJUHKJPS-UHFFFAOYSA-N 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- 230000002110 toxicologic effect Effects 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 2
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical compound CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 2
- WFPIAZLQTJBIFN-DVZOWYKESA-N zuclopenthixol Chemical compound C1CN(CCO)CCN1CC\C=C\1C2=CC(Cl)=CC=C2SC2=CC=CC=C2/1 WFPIAZLQTJBIFN-DVZOWYKESA-N 0.000 description 2
- GJJFMKBJSRMPLA-HIFRSBDPSA-N (1R,2S)-2-(aminomethyl)-N,N-diethyl-1-phenyl-1-cyclopropanecarboxamide Chemical compound C=1C=CC=CC=1[C@@]1(C(=O)N(CC)CC)C[C@@H]1CN GJJFMKBJSRMPLA-HIFRSBDPSA-N 0.000 description 1
- ASGMFNBUXDJWJJ-JLCFBVMHSA-N (1R,3R)-3-[[3-bromo-1-[4-(5-methyl-1,3,4-thiadiazol-2-yl)phenyl]pyrazolo[3,4-d]pyrimidin-6-yl]amino]-N,1-dimethylcyclopentane-1-carboxamide Chemical compound BrC1=NN(C2=NC(=NC=C21)N[C@H]1C[C@@](CC1)(C(=O)NC)C)C1=CC=C(C=C1)C=1SC(=NN=1)C ASGMFNBUXDJWJJ-JLCFBVMHSA-N 0.000 description 1
- UAOUIVVJBYDFKD-XKCDOFEDSA-N (1R,9R,10S,11R,12R,15S,18S,21R)-10,11,21-trihydroxy-8,8-dimethyl-14-methylidene-4-(prop-2-enylamino)-20-oxa-5-thia-3-azahexacyclo[9.7.2.112,15.01,9.02,6.012,18]henicosa-2(6),3-dien-13-one Chemical compound C([C@@H]1[C@@H](O)[C@@]23C(C1=C)=O)C[C@H]2[C@]12C(N=C(NCC=C)S4)=C4CC(C)(C)[C@H]1[C@H](O)[C@]3(O)OC2 UAOUIVVJBYDFKD-XKCDOFEDSA-N 0.000 description 1
- XUFXOAAUWZOOIT-SXARVLRPSA-N (2R,3R,4R,5S,6R)-5-[[(2R,3R,4R,5S,6R)-5-[[(2R,3R,4S,5S,6R)-3,4-dihydroxy-6-methyl-5-[[(1S,4R,5S,6S)-4,5,6-trihydroxy-3-(hydroxymethyl)-1-cyclohex-2-enyl]amino]-2-oxanyl]oxy]-3,4-dihydroxy-6-(hydroxymethyl)-2-oxanyl]oxy]-6-(hydroxymethyl)oxane-2,3,4-triol Chemical compound O([C@H]1O[C@H](CO)[C@H]([C@@H]([C@H]1O)O)O[C@H]1O[C@@H]([C@H]([C@H](O)[C@H]1O)N[C@@H]1[C@@H]([C@@H](O)[C@H](O)C(CO)=C1)O)C)[C@@H]1[C@@H](CO)O[C@@H](O)[C@H](O)[C@H]1O XUFXOAAUWZOOIT-SXARVLRPSA-N 0.000 description 1
- WEQLWGNDNRARGE-XIRDDKMYSA-N (2S,3S,11bS)-9,10-dimethoxy-3-(2-methylpropyl)-2,3,4,6,7,11b-hexahydro-1H-benzo[a]quinolizin-2-ol Chemical compound C1CN2C[C@H](CC(C)C)[C@@H](O)C[C@H]2C2=C1C=C(OC)C(OC)=C2 WEQLWGNDNRARGE-XIRDDKMYSA-N 0.000 description 1
- YJLIKUSWRSEPSM-WGQQHEPDSA-N (2r,3r,4s,5r)-2-[6-amino-8-[(4-phenylphenyl)methylamino]purin-9-yl]-5-(hydroxymethyl)oxolane-3,4-diol Chemical compound C=1C=C(C=2C=CC=CC=2)C=CC=1CNC1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O YJLIKUSWRSEPSM-WGQQHEPDSA-N 0.000 description 1
- WEQLWGNDNRARGE-BHYGNILZSA-N (2r,3s,11bs)-9,10-dimethoxy-3-(2-methylpropyl)-2,3,4,6,7,11b-hexahydro-1h-benzo[a]quinolizin-2-ol Chemical compound C1CN2C[C@H](CC(C)C)[C@H](O)C[C@H]2C2=C1C=C(OC)C(OC)=C2 WEQLWGNDNRARGE-BHYGNILZSA-N 0.000 description 1
- DIWRORZWFLOCLC-HNNXBMFYSA-N (3s)-7-chloro-5-(2-chlorophenyl)-3-hydroxy-1,3-dihydro-1,4-benzodiazepin-2-one Chemical compound N([C@H](C(NC1=CC=C(Cl)C=C11)=O)O)=C1C1=CC=CC=C1Cl DIWRORZWFLOCLC-HNNXBMFYSA-N 0.000 description 1
- YQOLEILXOBUDMU-KRWDZBQOSA-N (4R)-5-[(6-bromo-3-methyl-2-pyrrolidin-1-ylquinoline-4-carbonyl)amino]-4-(2-chlorophenyl)pentanoic acid Chemical compound CC1=C(C2=C(C=CC(=C2)Br)N=C1N3CCCC3)C(=O)NC[C@H](CCC(=O)O)C4=CC=CC=C4Cl YQOLEILXOBUDMU-KRWDZBQOSA-N 0.000 description 1
- DEQANNDTNATYII-OULOTJBUSA-N (4r,7s,10s,13r,16s,19r)-10-(4-aminobutyl)-19-[[(2r)-2-amino-3-phenylpropanoyl]amino]-16-benzyl-n-[(2r,3r)-1,3-dihydroxybutan-2-yl]-7-[(1r)-1-hydroxyethyl]-13-(1h-indol-3-ylmethyl)-6,9,12,15,18-pentaoxo-1,2-dithia-5,8,11,14,17-pentazacycloicosane-4-carboxa Chemical compound C([C@@H](N)C(=O)N[C@H]1CSSC[C@H](NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCCCN)NC(=O)[C@@H](CC=2C3=CC=CC=C3NC=2)NC(=O)[C@H](CC=2C=CC=CC=2)NC1=O)C(=O)N[C@H](CO)[C@H](O)C)C1=CC=CC=C1 DEQANNDTNATYII-OULOTJBUSA-N 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- MPIPASJGOJYODL-SFHVURJKSA-N (R)-isoconazole Chemical compound ClC1=CC(Cl)=CC=C1[C@@H](OCC=1C(=CC=CC=1Cl)Cl)CN1C=NC=C1 MPIPASJGOJYODL-SFHVURJKSA-N 0.000 description 1
- TVYLLZQTGLZFBW-ZBFHGGJFSA-N (R,R)-tramadol Chemical compound COC1=CC=CC([C@]2(O)[C@H](CCCC2)CN(C)C)=C1 TVYLLZQTGLZFBW-ZBFHGGJFSA-N 0.000 description 1
- MIOPJNTWMNEORI-GMSGAONNSA-N (S)-camphorsulfonic acid Chemical compound C1C[C@@]2(CS(O)(=O)=O)C(=O)C[C@@H]1C2(C)C MIOPJNTWMNEORI-GMSGAONNSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- AHOJOGBEEUGNQI-CLFYSBASSA-N (Z)-2-methyl-5-trimethylsilyloxyhex-4-enoic acid Chemical compound CC(C\C=C(\C)O[Si](C)(C)C)C(O)=O AHOJOGBEEUGNQI-CLFYSBASSA-N 0.000 description 1
- WSPOMRSOLSGNFJ-AUWJEWJLSA-N (Z)-chlorprothixene Chemical compound C1=C(Cl)C=C2C(=C/CCN(C)C)\C3=CC=CC=C3SC2=C1 WSPOMRSOLSGNFJ-AUWJEWJLSA-N 0.000 description 1
- WBYWAXJHAXSJNI-VOTSOKGWSA-M .beta-Phenylacrylic acid Natural products [O-]C(=O)\C=C\C1=CC=CC=C1 WBYWAXJHAXSJNI-VOTSOKGWSA-M 0.000 description 1
- PXUIZULXJVRBPC-UHFFFAOYSA-N 1'-[3-(3-chloro-10,11-dihydro-5H-dibenzo[b,f]azepin-5-yl)propyl]hexahydro-2H-spiro[imidazo[1,2-a]pyridine-3,4'-piperidin]-2-one Chemical compound C12=CC(Cl)=CC=C2CCC2=CC=CC=C2N1CCCN1CCC2(C(NC3CCCCN32)=O)CC1 PXUIZULXJVRBPC-UHFFFAOYSA-N 0.000 description 1
- DDMOUSALMHHKOS-UHFFFAOYSA-N 1,2-dichloro-1,1,2,2-tetrafluoroethane Chemical compound FC(F)(Cl)C(F)(F)Cl DDMOUSALMHHKOS-UHFFFAOYSA-N 0.000 description 1
- NAOLWIGVYRIGTP-UHFFFAOYSA-N 1,3,5-trihydroxyanthracene-9,10-dione Chemical compound C1=CC(O)=C2C(=O)C3=CC(O)=CC(O)=C3C(=O)C2=C1 NAOLWIGVYRIGTP-UHFFFAOYSA-N 0.000 description 1
- DKMFBWQBDIGMHM-UHFFFAOYSA-N 1-(4-fluorophenyl)-4-(4-methyl-1-piperidinyl)-1-butanone Chemical compound C1CC(C)CCN1CCCC(=O)C1=CC=C(F)C=C1 DKMFBWQBDIGMHM-UHFFFAOYSA-N 0.000 description 1
- TUSDEZXZIZRFGC-UHFFFAOYSA-N 1-O-galloyl-3,6-(R)-HHDP-beta-D-glucose Natural products OC1C(O2)COC(=O)C3=CC(O)=C(O)C(O)=C3C3=C(O)C(O)=C(O)C=C3C(=O)OC1C(O)C2OC(=O)C1=CC(O)=C(O)C(O)=C1 TUSDEZXZIZRFGC-UHFFFAOYSA-N 0.000 description 1
- ONBQEOIKXPHGMB-VBSBHUPXSA-N 1-[2-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)propan-1-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 ONBQEOIKXPHGMB-VBSBHUPXSA-N 0.000 description 1
- MDLAAYDRRZXJIF-UHFFFAOYSA-N 1-[4,4-bis(4-fluorophenyl)butyl]-4-[4-chloro-3-(trifluoromethyl)phenyl]-4-piperidinol Chemical compound C1CC(O)(C=2C=C(C(Cl)=CC=2)C(F)(F)F)CCN1CCCC(C=1C=CC(F)=CC=1)C1=CC=C(F)C=C1 MDLAAYDRRZXJIF-UHFFFAOYSA-N 0.000 description 1
- 102100025573 1-alkyl-2-acetylglycerophosphocholine esterase Human genes 0.000 description 1
- SQEPZQFOYGZCJQ-UHFFFAOYSA-N 1-methoxy-3,4-dihydroisoquinoline Chemical compound C1=CC=C2C(OC)=NCCC2=C1 SQEPZQFOYGZCJQ-UHFFFAOYSA-N 0.000 description 1
- VSNHCAURESNICA-NJFSPNSNSA-N 1-oxidanylurea Chemical compound N[14C](=O)NO VSNHCAURESNICA-NJFSPNSNSA-N 0.000 description 1
- FRPZMMHWLSIFAZ-UHFFFAOYSA-N 10-undecenoic acid Chemical compound OC(=O)CCCCCCCCC=C FRPZMMHWLSIFAZ-UHFFFAOYSA-N 0.000 description 1
- FUFLCEKSBBHCMO-UHFFFAOYSA-N 11-dehydrocorticosterone Natural products O=C1CCC2(C)C3C(=O)CC(C)(C(CC4)C(=O)CO)C4C3CCC2=C1 FUFLCEKSBBHCMO-UHFFFAOYSA-N 0.000 description 1
- KKFDCBRMNNSAAW-UHFFFAOYSA-N 2-(morpholin-4-yl)ethanol Chemical compound OCCN1CCOCC1 KKFDCBRMNNSAAW-UHFFFAOYSA-N 0.000 description 1
- JIVPVXMEBJLZRO-CQSZACIVSA-N 2-chloro-5-[(1r)-1-hydroxy-3-oxo-2h-isoindol-1-yl]benzenesulfonamide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC([C@@]2(O)C3=CC=CC=C3C(=O)N2)=C1 JIVPVXMEBJLZRO-CQSZACIVSA-N 0.000 description 1
- BFSVOASYOCHEOV-UHFFFAOYSA-N 2-diethylaminoethanol Chemical compound CCN(CC)CCO BFSVOASYOCHEOV-UHFFFAOYSA-N 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- OHXAOPZTJOUYKM-UHFFFAOYSA-N 3-Chloro-2-methylpropene Chemical compound CC(=C)CCl OHXAOPZTJOUYKM-UHFFFAOYSA-N 0.000 description 1
- FEBOTPHFXYHVPL-UHFFFAOYSA-N 3-[1-[4-(4-fluorophenyl)-4-oxobutyl]-4-piperidinyl]-1H-benzimidazol-2-one Chemical compound C1=CC(F)=CC=C1C(=O)CCCN1CCC(N2C(NC3=CC=CC=C32)=O)CC1 FEBOTPHFXYHVPL-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- GUJRSXAPGDDABA-NSHDSACASA-N 3-bromo-N-[[(2S)-1-ethyl-2-pyrrolidinyl]methyl]-2,6-dimethoxybenzamide Chemical compound CCN1CCC[C@H]1CNC(=O)C1=C(OC)C=CC(Br)=C1OC GUJRSXAPGDDABA-NSHDSACASA-N 0.000 description 1
- UOQHWNPVNXSDDO-UHFFFAOYSA-N 3-bromoimidazo[1,2-a]pyridine-6-carbonitrile Chemical compound C1=CC(C#N)=CN2C(Br)=CN=C21 UOQHWNPVNXSDDO-UHFFFAOYSA-N 0.000 description 1
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 1
- PMXMIIMHBWHSKN-UHFFFAOYSA-N 3-{2-[4-(6-fluoro-1,2-benzoxazol-3-yl)piperidin-1-yl]ethyl}-9-hydroxy-2-methyl-6,7,8,9-tetrahydropyrido[1,2-a]pyrimidin-4-one Chemical compound FC1=CC=C2C(C3CCN(CC3)CCC=3C(=O)N4CCCC(O)C4=NC=3C)=NOC2=C1 PMXMIIMHBWHSKN-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- SWLAMJPTOQZTAE-UHFFFAOYSA-N 4-[2-[(5-chloro-2-methoxybenzoyl)amino]ethyl]benzoic acid Chemical class COC1=CC=C(Cl)C=C1C(=O)NCCC1=CC=C(C(O)=O)C=C1 SWLAMJPTOQZTAE-UHFFFAOYSA-N 0.000 description 1
- MINMDCMSHDBHKG-UHFFFAOYSA-N 4-[4-[[6-methoxy-2-(2-methoxyimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-1-benzofuran-4-yl]oxymethyl]-5-methyl-1,3-thiazol-2-yl]morpholine Chemical compound N1=C2SC(OC)=NN2C=C1C(OC1=CC(OC)=C2)=CC1=C2OCC(=C(S1)C)N=C1N1CCOCC1 MINMDCMSHDBHKG-UHFFFAOYSA-N 0.000 description 1
- WUBBRNOQWQTFEX-UHFFFAOYSA-N 4-aminosalicylic acid Chemical compound NC1=CC=C(C(O)=O)C(O)=C1 WUBBRNOQWQTFEX-UHFFFAOYSA-N 0.000 description 1
- WRFYIYOXJWKONR-UHFFFAOYSA-N 4-bromo-2-methoxyaniline Chemical compound COC1=CC(Br)=CC=C1N WRFYIYOXJWKONR-UHFFFAOYSA-N 0.000 description 1
- 125000004203 4-hydroxyphenyl group Chemical group [H]OC1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- XFJBGINZIMNZBW-CRAIPNDOSA-N 5-chloro-2-[4-[(1r,2s)-2-[2-(5-methylsulfonylpyridin-2-yl)oxyethyl]cyclopropyl]piperidin-1-yl]pyrimidine Chemical compound N1=CC(S(=O)(=O)C)=CC=C1OCC[C@H]1[C@@H](C2CCN(CC2)C=2N=CC(Cl)=CN=2)C1 XFJBGINZIMNZBW-CRAIPNDOSA-N 0.000 description 1
- ODHCTXKNWHHXJC-VKHMYHEASA-N 5-oxo-L-proline Chemical compound OC(=O)[C@@H]1CCC(=O)N1 ODHCTXKNWHHXJC-VKHMYHEASA-N 0.000 description 1
- QOYHHIBFXOOADH-UHFFFAOYSA-N 8-[4,4-bis(4-fluorophenyl)butyl]-1-phenyl-1,3,8-triazaspiro[4.5]decan-4-one Chemical compound C1=CC(F)=CC=C1C(C=1C=CC(F)=CC=1)CCCN1CCC2(C(NCN2C=2C=CC=CC=2)=O)CC1 QOYHHIBFXOOADH-UHFFFAOYSA-N 0.000 description 1
- MKJIEFSOBYUXJB-UHFFFAOYSA-N 9,10-dimethoxy-3-isobutyl-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-one Chemical compound C1CN2CC(CC(C)C)C(=O)CC2C2=C1C=C(OC)C(OC)=C2 MKJIEFSOBYUXJB-UHFFFAOYSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- LRFVTYWOQMYALW-UHFFFAOYSA-N 9H-xanthine Chemical class O=C1NC(=O)NC2=C1NC=N2 LRFVTYWOQMYALW-UHFFFAOYSA-N 0.000 description 1
- 239000005541 ACE inhibitor Substances 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- 206010001497 Agitation Diseases 0.000 description 1
- 206010001541 Akinesia Diseases 0.000 description 1
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 1
- 239000005695 Ammonium acetate Substances 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- 208000019901 Anxiety disease Diseases 0.000 description 1
- CEUORZQYGODEFX-UHFFFAOYSA-N Aripirazole Chemical compound ClC1=CC=CC(N2CCN(CCCCOC=3C=C4NC(=O)CCC4=CC=3)CC2)=C1Cl CEUORZQYGODEFX-UHFFFAOYSA-N 0.000 description 1
- 108010024976 Asparaginase Proteins 0.000 description 1
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 206010003591 Ataxia Diseases 0.000 description 1
- XUKUURHRXDUEBC-KAYWLYCHSA-N Atorvastatin Chemical compound C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CC[C@@H](O)C[C@@H](O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-KAYWLYCHSA-N 0.000 description 1
- XUKUURHRXDUEBC-UHFFFAOYSA-N Atorvastatin Natural products C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CCC(O)CC(O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-UHFFFAOYSA-N 0.000 description 1
- NOWKCMXCCJGMRR-UHFFFAOYSA-N Aziridine Chemical class C1CN1 NOWKCMXCCJGMRR-UHFFFAOYSA-N 0.000 description 1
- 239000005552 B01AC04 - Clopidogrel Substances 0.000 description 1
- 239000005528 B01AC05 - Ticlopidine Substances 0.000 description 1
- 206010058504 Ballismus Diseases 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 229940123208 Biguanide Drugs 0.000 description 1
- 108010006654 Bleomycin Proteins 0.000 description 1
- VMIYHDSEFNYJSL-UHFFFAOYSA-N Bromazepam Chemical compound C12=CC(Br)=CC=C2NC(=O)CN=C1C1=CC=CC=N1 VMIYHDSEFNYJSL-UHFFFAOYSA-N 0.000 description 1
- RKLNONIVDFXQRX-UHFFFAOYSA-N Bromperidol Chemical compound C1CC(O)(C=2C=CC(Br)=CC=2)CCN1CCCC(=O)C1=CC=C(F)C=C1 RKLNONIVDFXQRX-UHFFFAOYSA-N 0.000 description 1
- KFVNVMJIPOXHPC-LUAWRHEFSA-N CCOC(C(C)C/C=C(/C)\O[N](C)(C)C)=O Chemical compound CCOC(C(C)C/C=C(/C)\O[N](C)(C)C)=O KFVNVMJIPOXHPC-LUAWRHEFSA-N 0.000 description 1
- PKMUHQIDVVOXHQ-HXUWFJFHSA-N C[C@H](C1=CC(C2=CC=C(CNC3CCCC3)S2)=CC=C1)NC(C1=C(C)C=CC(NC2CNC2)=C1)=O Chemical compound C[C@H](C1=CC(C2=CC=C(CNC3CCCC3)S2)=CC=C1)NC(C1=C(C)C=CC(NC2CNC2)=C1)=O PKMUHQIDVVOXHQ-HXUWFJFHSA-N 0.000 description 1
- 229940127291 Calcium channel antagonist Drugs 0.000 description 1
- 241000282465 Canis Species 0.000 description 1
- 239000005632 Capric acid (CAS 334-48-5) Substances 0.000 description 1
- 239000005635 Caprylic acid (CAS 124-07-2) Substances 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- JWBOIMRXGHLCPP-UHFFFAOYSA-N Chloditan Chemical compound C=1C=CC=C(Cl)C=1C(C(Cl)Cl)C1=CC=C(Cl)C=C1 JWBOIMRXGHLCPP-UHFFFAOYSA-N 0.000 description 1
- 229920001268 Cholestyramine Polymers 0.000 description 1
- WBYWAXJHAXSJNI-SREVYHEPSA-N Cinnamic acid Chemical compound OC(=O)\C=C/C1=CC=CC=C1 WBYWAXJHAXSJNI-SREVYHEPSA-N 0.000 description 1
- KAAZGXDPUNNEFN-UHFFFAOYSA-N Clotiapine Chemical compound C1CN(C)CCN1C1=NC2=CC=CC=C2SC2=CC=C(Cl)C=C12 KAAZGXDPUNNEFN-UHFFFAOYSA-N 0.000 description 1
- CHBRHODLKOZEPZ-UHFFFAOYSA-N Clotiazepam Chemical compound S1C(CC)=CC2=C1N(C)C(=O)CN=C2C1=CC=CC=C1Cl CHBRHODLKOZEPZ-UHFFFAOYSA-N 0.000 description 1
- 206010010071 Coma Diseases 0.000 description 1
- 229940127007 Compound 39 Drugs 0.000 description 1
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 1
- 208000011990 Corticobasal Degeneration Diseases 0.000 description 1
- MFYSYFVPBJMHGN-ZPOLXVRWSA-N Cortisone Chemical compound O=C1CC[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 MFYSYFVPBJMHGN-ZPOLXVRWSA-N 0.000 description 1
- MFYSYFVPBJMHGN-UHFFFAOYSA-N Cortisone Natural products O=C1CCC2(C)C3C(=O)CC(C)(C(CC4)(O)C(=O)CO)C4C3CCC2=C1 MFYSYFVPBJMHGN-UHFFFAOYSA-N 0.000 description 1
- 241000699800 Cricetinae Species 0.000 description 1
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 1
- 108010036941 Cyclosporins Proteins 0.000 description 1
- 208000000130 Cytochrome P-450 CYP3A Inducers Diseases 0.000 description 1
- 208000009011 Cytochrome P-450 CYP3A Inhibitors Diseases 0.000 description 1
- CKLJMWTZIZZHCS-UHFFFAOYSA-N D-OH-Asp Natural products OC(=O)C(N)CC(O)=O CKLJMWTZIZZHCS-UHFFFAOYSA-N 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-N D-gluconic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O RGHNJXZEOKUKBD-SQOUGZDYSA-N 0.000 description 1
- RGHNJXZEOKUKBD-UHFFFAOYSA-N D-gluconic acid Natural products OCC(O)C(O)C(O)C(O)C(O)=O RGHNJXZEOKUKBD-UHFFFAOYSA-N 0.000 description 1
- 108010092160 Dactinomycin Proteins 0.000 description 1
- 108020005199 Dehydrogenases Proteins 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- 239000004338 Dichlorodifluoromethane Substances 0.000 description 1
- 229940094659 Dopamine reuptake inhibitor Drugs 0.000 description 1
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 241000792859 Enema Species 0.000 description 1
- 108010056764 Eptifibatide Proteins 0.000 description 1
- 241000283073 Equus caballus Species 0.000 description 1
- 108090000371 Esterases Proteins 0.000 description 1
- 108010008165 Etanercept Proteins 0.000 description 1
- CUCHJCMWNFEYOM-UHFFFAOYSA-N Ethyl loflazepate Chemical compound C12=CC(Cl)=CC=C2NC(=O)C(C(=O)OCC)N=C1C1=CC=CC=C1F CUCHJCMWNFEYOM-UHFFFAOYSA-N 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 239000001263 FEMA 3042 Substances 0.000 description 1
- 229940123583 Factor Xa inhibitor Drugs 0.000 description 1
- 241000282324 Felis Species 0.000 description 1
- PLDUPXSUYLZYBN-UHFFFAOYSA-N Fluphenazine Chemical compound C1CN(CCO)CCN1CCCN1C2=CC(C(F)(F)F)=CC=C2SC2=CC=CC=C21 PLDUPXSUYLZYBN-UHFFFAOYSA-N 0.000 description 1
- 229940123414 Folate antagonist Drugs 0.000 description 1
- 108700012941 GNRH1 Proteins 0.000 description 1
- DSLZVSRJTYRBFB-UHFFFAOYSA-N Galactaric acid Natural products OC(=O)C(O)C(O)C(O)C(O)C(O)=O DSLZVSRJTYRBFB-UHFFFAOYSA-N 0.000 description 1
- IAJILQKETJEXLJ-UHFFFAOYSA-N Galacturonsaeure Natural products O=CC(O)C(O)C(O)C(O)C(O)=O IAJILQKETJEXLJ-UHFFFAOYSA-N 0.000 description 1
- 241000699694 Gerbillinae Species 0.000 description 1
- FAEKWTJYAYMJKF-QHCPKHFHSA-N GlucoNorm Chemical compound C1=C(C(O)=O)C(OCC)=CC(CC(=O)N[C@@H](CC(C)C)C=2C(=CC=CC=2)N2CCCCC2)=C1 FAEKWTJYAYMJKF-QHCPKHFHSA-N 0.000 description 1
- 102000004366 Glucosidases Human genes 0.000 description 1
- 108010056771 Glucosidases Proteins 0.000 description 1
- 239000000579 Gonadotropin-Releasing Hormone Substances 0.000 description 1
- 241000282575 Gorilla Species 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- WYCLKVQLVUQKNZ-UHFFFAOYSA-N Halazepam Chemical compound N=1CC(=O)N(CC(F)(F)F)C2=CC=C(Cl)C=C2C=1C1=CC=CC=C1 WYCLKVQLVUQKNZ-UHFFFAOYSA-N 0.000 description 1
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 1
- 208000002972 Hepatolenticular Degeneration Diseases 0.000 description 1
- 206010019851 Hepatotoxicity Diseases 0.000 description 1
- 102000007625 Hirudins Human genes 0.000 description 1
- 108010007267 Hirudins Proteins 0.000 description 1
- 101001120086 Homo sapiens P2Y purinoceptor 12 Proteins 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 102000004877 Insulin Human genes 0.000 description 1
- 108090001061 Insulin Proteins 0.000 description 1
- 102100025306 Integrin alpha-IIb Human genes 0.000 description 1
- 101710149643 Integrin alpha-IIb Proteins 0.000 description 1
- YQEZLKZALYSWHR-UHFFFAOYSA-N Ketamine Chemical compound C=1C=CC=C(Cl)C=1C1(NC)CCCCC1=O YQEZLKZALYSWHR-UHFFFAOYSA-N 0.000 description 1
- CKLJMWTZIZZHCS-UWTATZPHSA-N L-Aspartic acid Natural products OC(=O)[C@H](N)CC(O)=O CKLJMWTZIZZHCS-UWTATZPHSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-N L-arginine Chemical compound OC(=O)[C@@H](N)CCCN=C(N)N ODKSFYDXXFIFQN-BYPYZUCNSA-N 0.000 description 1
- 229930064664 L-arginine Natural products 0.000 description 1
- 235000014852 L-arginine Nutrition 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 1
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 1
- 241000283953 Lagomorpha Species 0.000 description 1
- 239000005639 Lauric acid Substances 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- YLCXGBZIZBEVPZ-UHFFFAOYSA-N Medazepam Chemical compound C12=CC(Cl)=CC=C2N(C)CCN=C1C1=CC=CC=C1 YLCXGBZIZBEVPZ-UHFFFAOYSA-N 0.000 description 1
- 102000018697 Membrane Proteins Human genes 0.000 description 1
- 108010052285 Membrane Proteins Proteins 0.000 description 1
- CESYKOGBSMNBPD-UHFFFAOYSA-N Methyclothiazide Chemical compound ClC1=C(S(N)(=O)=O)C=C2S(=O)(=O)N(C)C(CCl)NC2=C1 CESYKOGBSMNBPD-UHFFFAOYSA-N 0.000 description 1
- DUGOZIWVEXMGBE-UHFFFAOYSA-N Methylphenidate Chemical compound C=1C=CC=CC=1C(C(=O)OC)C1CCCCN1 DUGOZIWVEXMGBE-UHFFFAOYSA-N 0.000 description 1
- 229930192392 Mitomycin Natural products 0.000 description 1
- KLPWJLBORRMFGK-UHFFFAOYSA-N Molindone Chemical compound O=C1C=2C(CC)=C(C)NC=2CCC1CN1CCOCC1 KLPWJLBORRMFGK-UHFFFAOYSA-N 0.000 description 1
- PCZOHLXUXFIOCF-UHFFFAOYSA-N Monacolin X Natural products C12C(OC(=O)C(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 PCZOHLXUXFIOCF-UHFFFAOYSA-N 0.000 description 1
- 102000010909 Monoamine Oxidase Human genes 0.000 description 1
- 108010062431 Monoamine oxidase Proteins 0.000 description 1
- 229940123685 Monoamine oxidase inhibitor Drugs 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- 208000008238 Muscle Spasticity Diseases 0.000 description 1
- 208000021642 Muscular disease Diseases 0.000 description 1
- 241000282339 Mustela Species 0.000 description 1
- 208000002033 Myoclonus Diseases 0.000 description 1
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 1
- JTVPZMFULRWINT-UHFFFAOYSA-N N-[2-(diethylamino)ethyl]-2-methoxy-5-methylsulfonylbenzamide Chemical compound CCN(CC)CCNC(=O)C1=CC(S(C)(=O)=O)=CC=C1OC JTVPZMFULRWINT-UHFFFAOYSA-N 0.000 description 1
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 1
- UEEJHVSXFDXPFK-UHFFFAOYSA-N N-dimethylaminoethanol Chemical compound CN(C)CCO UEEJHVSXFDXPFK-UHFFFAOYSA-N 0.000 description 1
- CMWTZPSULFXXJA-UHFFFAOYSA-N Naproxen Natural products C1=C(C(C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-UHFFFAOYSA-N 0.000 description 1
- 206010028813 Nausea Diseases 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- 206010029216 Nervousness Diseases 0.000 description 1
- 201000005625 Neuroleptic malignant syndrome Diseases 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 108010016076 Octreotide Proteins 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- 235000019502 Orange oil Nutrition 0.000 description 1
- 206010031127 Orthostatic hypotension Diseases 0.000 description 1
- XCWPUUGSGHNIDZ-UHFFFAOYSA-N Oxypertine Chemical compound C1=2C=C(OC)C(OC)=CC=2NC(C)=C1CCN(CC1)CCN1C1=CC=CC=C1 XCWPUUGSGHNIDZ-UHFFFAOYSA-N 0.000 description 1
- 102100026171 P2Y purinoceptor 12 Human genes 0.000 description 1
- 108010016731 PPAR gamma Proteins 0.000 description 1
- 235000021314 Palmitic acid Nutrition 0.000 description 1
- 241000282577 Pan troglodytes Species 0.000 description 1
- 208000027089 Parkinsonian disease Diseases 0.000 description 1
- 206010034010 Parkinsonism Diseases 0.000 description 1
- LRBQNJMCXXYXIU-PPKXGCFTSA-N Penta-digallate-beta-D-glucose Natural products OC1=C(O)C(O)=CC(C(=O)OC=2C(=C(O)C=C(C=2)C(=O)OC[C@@H]2[C@H]([C@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)[C@@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)[C@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)O2)OC(=O)C=2C=C(OC(=O)C=3C=C(O)C(O)=C(O)C=3)C(O)=C(O)C=2)O)=C1 LRBQNJMCXXYXIU-PPKXGCFTSA-N 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 102100038825 Peroxisome proliferator-activated receptor gamma Human genes 0.000 description 1
- RGCVKNLCSQQDEP-UHFFFAOYSA-N Perphenazine Chemical compound C1CN(CCO)CCN1CCCN1C2=CC(Cl)=CC=C2SC2=CC=CC=C21 RGCVKNLCSQQDEP-UHFFFAOYSA-N 0.000 description 1
- YGYAWVDWMABLBF-UHFFFAOYSA-N Phosgene Chemical compound ClC(Cl)=O YGYAWVDWMABLBF-UHFFFAOYSA-N 0.000 description 1
- ZPHBZEQOLSRPAK-UHFFFAOYSA-N Phosphoramidon Natural products C=1NC2=CC=CC=C2C=1CC(C(O)=O)NC(=O)C(CC(C)C)NP(O)(=O)OC1OC(C)C(O)C(O)C1O ZPHBZEQOLSRPAK-UHFFFAOYSA-N 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- CYLWJCABXYDINA-UHFFFAOYSA-N Polythiazide Polymers ClC1=C(S(N)(=O)=O)C=C2S(=O)(=O)N(C)C(CSCC(F)(F)F)NC2=C1 CYLWJCABXYDINA-UHFFFAOYSA-N 0.000 description 1
- 229940127315 Potassium Channel Openers Drugs 0.000 description 1
- TUZYXOIXSAXUGO-UHFFFAOYSA-N Pravastatin Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(O)C=C21 TUZYXOIXSAXUGO-UHFFFAOYSA-N 0.000 description 1
- MWQCHHACWWAQLJ-UHFFFAOYSA-N Prazepam Chemical compound O=C1CN=C(C=2C=CC=CC=2)C2=CC(Cl)=CC=C2N1CC1CC1 MWQCHHACWWAQLJ-UHFFFAOYSA-N 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- ZGUGWUXLJSTTMA-UHFFFAOYSA-N Promazinum Chemical compound C1=CC=C2N(CCCN(C)C)C3=CC=CC=C3SC2=C1 ZGUGWUXLJSTTMA-UHFFFAOYSA-N 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 208000028017 Psychotic disease Diseases 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 108091007187 Reductases Proteins 0.000 description 1
- 208000005793 Restless legs syndrome Diseases 0.000 description 1
- 208000006289 Rett Syndrome Diseases 0.000 description 1
- PPTYJKAXVCCBDU-UHFFFAOYSA-N Rohypnol Chemical compound N=1CC(=O)N(C)C2=CC=C([N+]([O-])=O)C=C2C=1C1=CC=CC=C1F PPTYJKAXVCCBDU-UHFFFAOYSA-N 0.000 description 1
- RYMZZMVNJRMUDD-UHFFFAOYSA-N SJ000286063 Natural products C12C(OC(=O)C(C)(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 RYMZZMVNJRMUDD-UHFFFAOYSA-N 0.000 description 1
- 206010040047 Sepsis Diseases 0.000 description 1
- 208000009106 Shy-Drager Syndrome Diseases 0.000 description 1
- 208000013738 Sleep Initiation and Maintenance disease Diseases 0.000 description 1
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 1
- 206010041349 Somnolence Diseases 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- 108010023197 Streptokinase Proteins 0.000 description 1
- 208000006011 Stroke Diseases 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- 229930006000 Sucrose Natural products 0.000 description 1
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 1
- 229940100389 Sulfonylurea Drugs 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-ZSJDYOACSA-N Sulfuric acid-d2 Chemical compound [2H]OS(=O)(=O)O[2H] QAOWNCQODCNURD-ZSJDYOACSA-N 0.000 description 1
- UNRHXEPDKXPRTM-UHFFFAOYSA-N Sultopride Chemical compound CCN1CCCC1CNC(=O)C1=CC(S(=O)(=O)CC)=CC=C1OC UNRHXEPDKXPRTM-UHFFFAOYSA-N 0.000 description 1
- 208000027522 Sydenham chorea Diseases 0.000 description 1
- 229940123237 Taxane Drugs 0.000 description 1
- SEQDDYPDSLOBDC-UHFFFAOYSA-N Temazepam Chemical compound N=1C(O)C(=O)N(C)C2=CC=C(Cl)C=C2C=1C1=CC=CC=C1 SEQDDYPDSLOBDC-UHFFFAOYSA-N 0.000 description 1
- KLBQZWRITKRQQV-UHFFFAOYSA-N Thioridazine Chemical compound C12=CC(SC)=CC=C2SC2=CC=CC=C2N1CCC1CCCCN1C KLBQZWRITKRQQV-UHFFFAOYSA-N 0.000 description 1
- GFBKORZTTCHDGY-UWVJOHFNSA-N Thiothixene Chemical compound C12=CC(S(=O)(=O)N(C)C)=CC=C2SC2=CC=CC=C2\C1=C\CCN1CCN(C)CC1 GFBKORZTTCHDGY-UWVJOHFNSA-N 0.000 description 1
- 229940122388 Thrombin inhibitor Drugs 0.000 description 1
- 102000003938 Thromboxane Receptors Human genes 0.000 description 1
- 108090000300 Thromboxane Receptors Proteins 0.000 description 1
- RUJBDQSFYCKFAA-UHFFFAOYSA-N Tofisopam Chemical compound N=1N=C(C)C(CC)C2=CC(OC)=C(OC)C=C2C=1C1=CC=C(OC)C(OC)=C1 RUJBDQSFYCKFAA-UHFFFAOYSA-N 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- FNYLWPVRPXGIIP-UHFFFAOYSA-N Triamterene Chemical compound NC1=NC2=NC(N)=NC(N)=C2N=C1C1=CC=CC=C1 FNYLWPVRPXGIIP-UHFFFAOYSA-N 0.000 description 1
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 1
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 1
- 108060008683 Tumor Necrosis Factor Receptor Proteins 0.000 description 1
- 208000025865 Ulcer Diseases 0.000 description 1
- 108090000435 Urokinase-type plasminogen activator Proteins 0.000 description 1
- 102000003990 Urokinase-type plasminogen activator Human genes 0.000 description 1
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 1
- SECKRCOLJRRGGV-UHFFFAOYSA-N Vardenafil Chemical compound CCCC1=NC(C)=C(C(N=2)=O)N1NC=2C(C(=CC=1)OCC)=CC=1S(=O)(=O)N1CCN(CC)CC1 SECKRCOLJRRGGV-UHFFFAOYSA-N 0.000 description 1
- 229940122803 Vinca alkaloid Drugs 0.000 description 1
- 208000018839 Wilson disease Diseases 0.000 description 1
- 208000013142 Writer cramp Diseases 0.000 description 1
- SMNRFWMNPDABKZ-WVALLCKVSA-N [[(2R,3S,4R,5S)-5-(2,6-dioxo-3H-pyridin-3-yl)-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl] [[[(2R,3S,4S,5R,6R)-4-fluoro-3,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-hydroxyphosphoryl]oxy-hydroxyphosphoryl] hydrogen phosphate Chemical compound OC[C@H]1O[C@H](OP(O)(=O)OP(O)(=O)OP(O)(=O)OP(O)(=O)OC[C@H]2O[C@H]([C@H](O)[C@@H]2O)C2C=CC(=O)NC2=O)[C@H](O)[C@@H](F)[C@@H]1O SMNRFWMNPDABKZ-WVALLCKVSA-N 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- 229960002632 acarbose Drugs 0.000 description 1
- XUFXOAAUWZOOIT-UHFFFAOYSA-N acarviostatin I01 Natural products OC1C(O)C(NC2C(C(O)C(O)C(CO)=C2)O)C(C)OC1OC(C(C1O)O)C(CO)OC1OC1C(CO)OC(O)C(O)C1O XUFXOAAUWZOOIT-UHFFFAOYSA-N 0.000 description 1
- 230000001133 acceleration Effects 0.000 description 1
- NOSIYYJFMPDDSA-UHFFFAOYSA-N acepromazine Chemical compound C1=C(C(C)=O)C=C2N(CCCN(C)C)C3=CC=CC=C3SC2=C1 NOSIYYJFMPDDSA-UHFFFAOYSA-N 0.000 description 1
- 229960005054 acepromazine Drugs 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- WNTYBHLDCKXEOT-UHFFFAOYSA-N acetophenazine Chemical compound C12=CC(C(=O)C)=CC=C2SC2=CC=CC=C2N1CCCN1CCN(CCO)CC1 WNTYBHLDCKXEOT-UHFFFAOYSA-N 0.000 description 1
- 229960000276 acetophenazine Drugs 0.000 description 1
- 229960001138 acetylsalicylic acid Drugs 0.000 description 1
- ODHCTXKNWHHXJC-UHFFFAOYSA-N acide pyroglutamique Natural products OC(=O)C1CCC(=O)N1 ODHCTXKNWHHXJC-UHFFFAOYSA-N 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- RJURFGZVJUQBHK-IIXSONLDSA-N actinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-IIXSONLDSA-N 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 231100000403 acute toxicity Toxicity 0.000 description 1
- 229960003148 adinazolam Drugs 0.000 description 1
- GJSLOMWRLALDCT-UHFFFAOYSA-N adinazolam Chemical compound C12=CC(Cl)=CC=C2N2C(CN(C)C)=NN=C2CN=C1C1=CC=CC=C1 GJSLOMWRLALDCT-UHFFFAOYSA-N 0.000 description 1
- 239000001361 adipic acid Substances 0.000 description 1
- 235000011037 adipic acid Nutrition 0.000 description 1
- 229960000250 adipic acid Drugs 0.000 description 1
- 239000000048 adrenergic agonist Substances 0.000 description 1
- 238000013019 agitation Methods 0.000 description 1
- 239000000556 agonist Substances 0.000 description 1
- 229940083712 aldosterone antagonist Drugs 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 125000001931 aliphatic group Chemical group 0.000 description 1
- 125000000217 alkyl group Chemical group 0.000 description 1
- 229940045714 alkyl sulfonate alkylating agent Drugs 0.000 description 1
- 150000008052 alkyl sulfonates Chemical class 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- AEMOLEFTQBMNLQ-WAXACMCWSA-N alpha-D-glucuronic acid Chemical compound O[C@H]1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-WAXACMCWSA-N 0.000 description 1
- 229960004538 alprazolam Drugs 0.000 description 1
- VREFGVBLTWBCJP-UHFFFAOYSA-N alprazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NN=C2CN=C1C1=CC=CC=C1 VREFGVBLTWBCJP-UHFFFAOYSA-N 0.000 description 1
- 229960000473 altretamine Drugs 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 230000001668 ameliorated effect Effects 0.000 description 1
- XSDQTOBWRPYKKA-UHFFFAOYSA-N amiloride Chemical compound NC(=N)NC(=O)C1=NC(Cl)=C(N)N=C1N XSDQTOBWRPYKKA-UHFFFAOYSA-N 0.000 description 1
- 229960002576 amiloride Drugs 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 229940024606 amino acid Drugs 0.000 description 1
- 235000001014 amino acid Nutrition 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 229960004909 aminosalicylic acid Drugs 0.000 description 1
- 229960003036 amisulpride Drugs 0.000 description 1
- NTJOBXMMWNYJFB-UHFFFAOYSA-N amisulpride Chemical compound CCN1CCCC1CNC(=O)C1=CC(S(=O)(=O)CC)=C(N)C=C1OC NTJOBXMMWNYJFB-UHFFFAOYSA-N 0.000 description 1
- ZPBWCRDSRKPIDG-UHFFFAOYSA-N amlodipine benzenesulfonate Chemical compound OS(=O)(=O)C1=CC=CC=C1.CCOC(=O)C1=C(COCCN)NC(C)=C(C(=O)OC)C1C1=CC=CC=C1Cl ZPBWCRDSRKPIDG-UHFFFAOYSA-N 0.000 description 1
- 229960004005 amlodipine besylate Drugs 0.000 description 1
- 229940043376 ammonium acetate Drugs 0.000 description 1
- 235000019257 ammonium acetate Nutrition 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 235000011114 ammonium hydroxide Nutrition 0.000 description 1
- LSQZJLSUYDQPKJ-NJBDSQKTSA-N amoxicillin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)=CC=C(O)C=C1 LSQZJLSUYDQPKJ-NJBDSQKTSA-N 0.000 description 1
- 229960003022 amoxicillin Drugs 0.000 description 1
- MZZLGJHLQGUVPN-HAWMADMCSA-N anacetrapib Chemical compound COC1=CC(F)=C(C(C)C)C=C1C1=CC=C(C(F)(F)F)C=C1CN1C(=O)O[C@H](C=2C=C(C=C(C=2)C(F)(F)F)C(F)(F)F)[C@@H]1C MZZLGJHLQGUVPN-HAWMADMCSA-N 0.000 description 1
- 229950000285 anacetrapib Drugs 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 239000003098 androgen Substances 0.000 description 1
- 229940030486 androgens Drugs 0.000 description 1
- 229940044094 angiotensin-converting-enzyme inhibitor Drugs 0.000 description 1
- 210000003423 ankle Anatomy 0.000 description 1
- 229940045799 anthracyclines and related substance Drugs 0.000 description 1
- 230000002280 anti-androgenic effect Effects 0.000 description 1
- 230000000879 anti-atherosclerotic effect Effects 0.000 description 1
- 230000001078 anti-cholinergic effect Effects 0.000 description 1
- 229940046836 anti-estrogen Drugs 0.000 description 1
- 230000001833 anti-estrogenic effect Effects 0.000 description 1
- 229940121363 anti-inflammatory agent Drugs 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 230000000340 anti-metabolite Effects 0.000 description 1
- 230000001028 anti-proliverative effect Effects 0.000 description 1
- 239000000051 antiandrogen Substances 0.000 description 1
- 229940030495 antiandrogen sex hormone and modulator of the genital system Drugs 0.000 description 1
- 239000003416 antiarrhythmic agent Substances 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 229940065524 anticholinergics inhalants for obstructive airway diseases Drugs 0.000 description 1
- 239000003146 anticoagulant agent Substances 0.000 description 1
- 229940127219 anticoagulant drug Drugs 0.000 description 1
- 229940125708 antidiabetic agent Drugs 0.000 description 1
- 239000003472 antidiabetic agent Substances 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 229940100197 antimetabolite Drugs 0.000 description 1
- 239000002256 antimetabolite Substances 0.000 description 1
- 229940045686 antimetabolites antineoplastic purine analogs Drugs 0.000 description 1
- 229940034982 antineoplastic agent Drugs 0.000 description 1
- 229940045719 antineoplastic alkylating agent nitrosoureas Drugs 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 235000006708 antioxidants Nutrition 0.000 description 1
- 229940127218 antiplatelet drug Drugs 0.000 description 1
- 229940124522 antiretrovirals Drugs 0.000 description 1
- 239000003903 antiretrovirus agent Chemical class 0.000 description 1
- 230000036506 anxiety Effects 0.000 description 1
- 230000009118 appropriate response Effects 0.000 description 1
- 239000008135 aqueous vehicle Substances 0.000 description 1
- 229960004372 aripiprazole Drugs 0.000 description 1
- 150000004982 aromatic amines Chemical class 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 229960005261 aspartic acid Drugs 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 208000006673 asthma Diseases 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- VHGCDTVCOLNTBX-QGZVFWFLSA-N atomoxetine Chemical compound O([C@H](CCNC)C=1C=CC=CC=1)C1=CC=CC=C1C VHGCDTVCOLNTBX-QGZVFWFLSA-N 0.000 description 1
- 229960002430 atomoxetine Drugs 0.000 description 1
- 229960005370 atorvastatin Drugs 0.000 description 1
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 1
- 229960003515 bendroflumethiazide Drugs 0.000 description 1
- HDWIHXWEUNVBIY-UHFFFAOYSA-N bendroflumethiazidum Chemical compound C1=C(C(F)(F)F)C(S(=O)(=O)N)=CC(S(N2)(=O)=O)=C1NC2CC1=CC=CC=C1 HDWIHXWEUNVBIY-UHFFFAOYSA-N 0.000 description 1
- UPABQMWFWCMOFV-UHFFFAOYSA-N benethamine Chemical compound C=1C=CC=CC=1CNCCC1=CC=CC=C1 UPABQMWFWCMOFV-UHFFFAOYSA-N 0.000 description 1
- 229960002507 benperidol Drugs 0.000 description 1
- JUHORIMYRDESRB-UHFFFAOYSA-N benzathine Chemical compound C=1C=CC=CC=1CNCCNCC1=CC=CC=C1 JUHORIMYRDESRB-UHFFFAOYSA-N 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 229940049706 benzodiazepine Drugs 0.000 description 1
- 150000001557 benzodiazepines Chemical class 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 229960004365 benzoic acid Drugs 0.000 description 1
- 150000004283 biguanides Chemical class 0.000 description 1
- 229920000080 bile acid sequestrant Polymers 0.000 description 1
- 229940096699 bile acid sequestrants Drugs 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 229960000074 biopharmaceutical Drugs 0.000 description 1
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical class N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 1
- 206010005159 blepharospasm Diseases 0.000 description 1
- 230000000744 blepharospasm Effects 0.000 description 1
- KGBXLFKZBHKPEV-UHFFFAOYSA-N boric acid Chemical compound OB(O)O KGBXLFKZBHKPEV-UHFFFAOYSA-N 0.000 description 1
- 239000004327 boric acid Substances 0.000 description 1
- 208000029028 brain injury Diseases 0.000 description 1
- 229960002729 bromazepam Drugs 0.000 description 1
- 229960004037 bromperidol Drugs 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 229960004064 bumetanide Drugs 0.000 description 1
- MAEIEVLCKWDQJH-UHFFFAOYSA-N bumetanide Chemical compound CCCCNC1=CC(C(O)=O)=CC(S(N)(=O)=O)=C1OC1=CC=CC=C1 MAEIEVLCKWDQJH-UHFFFAOYSA-N 0.000 description 1
- SNPPWIUOZRMYNY-UHFFFAOYSA-N bupropion Chemical compound CC(C)(C)NC(C)C(=O)C1=CC=CC(Cl)=C1 SNPPWIUOZRMYNY-UHFFFAOYSA-N 0.000 description 1
- 229960001058 bupropion Drugs 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- DVLBYTMYSMAKHP-UHFFFAOYSA-N butaperazine Chemical compound C12=CC(C(=O)CCC)=CC=C2SC2=CC=CC=C2N1CCCN1CCN(C)CC1 DVLBYTMYSMAKHP-UHFFFAOYSA-N 0.000 description 1
- 229960000608 butaperazine Drugs 0.000 description 1
- 239000000480 calcium channel blocker Substances 0.000 description 1
- AXCZMVOFGPJBDE-UHFFFAOYSA-L calcium dihydroxide Chemical compound [OH-].[OH-].[Ca+2] AXCZMVOFGPJBDE-UHFFFAOYSA-L 0.000 description 1
- 239000000920 calcium hydroxide Substances 0.000 description 1
- 229910001861 calcium hydroxide Inorganic materials 0.000 description 1
- 229960000926 camazepam Drugs 0.000 description 1
- PXBVEXGRHZFEOF-UHFFFAOYSA-N camazepam Chemical compound C12=CC(Cl)=CC=C2N(C)C(=O)C(OC(=O)N(C)C)N=C1C1=CC=CC=C1 PXBVEXGRHZFEOF-UHFFFAOYSA-N 0.000 description 1
- LSPHULWDVZXLIL-QUBYGPBYSA-N camphoric acid Chemical compound CC1(C)[C@H](C(O)=O)CC[C@]1(C)C(O)=O LSPHULWDVZXLIL-QUBYGPBYSA-N 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 230000005907 cancer growth Effects 0.000 description 1
- KHAVLLBUVKBTBG-UHFFFAOYSA-N caproleic acid Natural products OC(=O)CCCCCCCC=C KHAVLLBUVKBTBG-UHFFFAOYSA-N 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 229960004424 carbon dioxide Drugs 0.000 description 1
- 229960004562 carboplatin Drugs 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 229940082638 cardiac stimulant phosphodiesterase inhibitors Drugs 0.000 description 1
- 239000012876 carrier material Substances 0.000 description 1
- NPAKNKYSJIDKMW-UHFFFAOYSA-N carvedilol Chemical compound COC1=CC=CC=C1OCCNCC(O)COC1=CC=CC2=NC3=CC=C[CH]C3=C12 NPAKNKYSJIDKMW-UHFFFAOYSA-N 0.000 description 1
- 229960004195 carvedilol Drugs 0.000 description 1
- 229960000590 celecoxib Drugs 0.000 description 1
- RZEKVGVHFLEQIL-UHFFFAOYSA-N celecoxib Chemical compound C1=CC(C)=CC=C1C1=CC(C(F)(F)F)=NN1C1=CC=C(S(N)(=O)=O)C=C1 RZEKVGVHFLEQIL-UHFFFAOYSA-N 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 206010008129 cerebral palsy Diseases 0.000 description 1
- 201000002866 cervical dystonia Diseases 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 229960004782 chlordiazepoxide Drugs 0.000 description 1
- ANTSCNMPPGJYLG-UHFFFAOYSA-N chlordiazepoxide Chemical compound O=N=1CC(NC)=NC2=CC=C(Cl)C=C2C=1C1=CC=CC=C1 ANTSCNMPPGJYLG-UHFFFAOYSA-N 0.000 description 1
- 229960002155 chlorothiazide Drugs 0.000 description 1
- DBOUGBAQLIXZLV-UHFFFAOYSA-N chlorproethazine Chemical compound C1=C(Cl)C=C2N(CCCN(CC)CC)C3=CC=CC=C3SC2=C1 DBOUGBAQLIXZLV-UHFFFAOYSA-N 0.000 description 1
- 229960003030 chlorproethazine Drugs 0.000 description 1
- ZPEIMTDSQAKGNT-UHFFFAOYSA-N chlorpromazine Chemical compound C1=C(Cl)C=C2N(CCCN(C)C)C3=CC=CC=C3SC2=C1 ZPEIMTDSQAKGNT-UHFFFAOYSA-N 0.000 description 1
- 229960001076 chlorpromazine Drugs 0.000 description 1
- 229960001552 chlorprothixene Drugs 0.000 description 1
- 229960001523 chlortalidone Drugs 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- 239000000812 cholinergic antagonist Substances 0.000 description 1
- 229940117975 chromium trioxide Drugs 0.000 description 1
- GAMDZJFZMJECOS-UHFFFAOYSA-N chromium(6+);oxygen(2-) Chemical compound [O-2].[O-2].[O-2].[Cr+6] GAMDZJFZMJECOS-UHFFFAOYSA-N 0.000 description 1
- 229960004588 cilostazol Drugs 0.000 description 1
- RRGUKTPIGVIEKM-UHFFFAOYSA-N cilostazol Chemical compound C=1C=C2NC(=O)CCC2=CC=1OCCCCC1=NN=NN1C1CCCCC1 RRGUKTPIGVIEKM-UHFFFAOYSA-N 0.000 description 1
- 229930016911 cinnamic acid Natural products 0.000 description 1
- 235000013985 cinnamic acid Nutrition 0.000 description 1
- NJMYODHXAKYRHW-DVZOWYKESA-N cis-flupenthixol Chemical compound C1CN(CCO)CCN1CC\C=C\1C2=CC(C(F)(F)F)=CC=C2SC2=CC=CC=C2/1 NJMYODHXAKYRHW-DVZOWYKESA-N 0.000 description 1
- 229960004316 cisplatin Drugs 0.000 description 1
- DQLATGHUWYMOKM-UHFFFAOYSA-L cisplatin Chemical compound N[Pt](N)(Cl)Cl DQLATGHUWYMOKM-UHFFFAOYSA-L 0.000 description 1
- 229960001403 clobazam Drugs 0.000 description 1
- CXOXHMZGEKVPMT-UHFFFAOYSA-N clobazam Chemical compound O=C1CC(=O)N(C)C2=CC=C(Cl)C=C2N1C1=CC=CC=C1 CXOXHMZGEKVPMT-UHFFFAOYSA-N 0.000 description 1
- 229960003120 clonazepam Drugs 0.000 description 1
- DGBIGWXXNGSACT-UHFFFAOYSA-N clonazepam Chemical compound C12=CC([N+](=O)[O-])=CC=C2NC(=O)CN=C1C1=CC=CC=C1Cl DGBIGWXXNGSACT-UHFFFAOYSA-N 0.000 description 1
- 229960001184 clopenthixol Drugs 0.000 description 1
- 229960003009 clopidogrel Drugs 0.000 description 1
- GKTWGGQPFAXNFI-HNNXBMFYSA-N clopidogrel Chemical compound C1([C@H](N2CC=3C=CSC=3CC2)C(=O)OC)=CC=CC=C1Cl GKTWGGQPFAXNFI-HNNXBMFYSA-N 0.000 description 1
- ULEUKTXFAJZAAV-UHFFFAOYSA-M clorazepate monopotassium Chemical compound [K+].C12=CC(Cl)=CC=C2NC(=O)C(C(=O)[O-])N=C1C1=CC=CC=C1 ULEUKTXFAJZAAV-UHFFFAOYSA-M 0.000 description 1
- 229960003864 clotiapine Drugs 0.000 description 1
- 229960003622 clotiazepam Drugs 0.000 description 1
- 229960003932 cloxazolam Drugs 0.000 description 1
- ZIXNZOBDFKSQTC-UHFFFAOYSA-N cloxazolam Chemical compound C12=CC(Cl)=CC=C2NC(=O)CN2CCOC21C1=CC=CC=C1Cl ZIXNZOBDFKSQTC-UHFFFAOYSA-N 0.000 description 1
- 238000011260 co-administration Methods 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 230000001149 cognitive effect Effects 0.000 description 1
- 229940126142 compound 16 Drugs 0.000 description 1
- 229940125844 compound 46 Drugs 0.000 description 1
- 229940126179 compound 72 Drugs 0.000 description 1
- 239000007891 compressed tablet Substances 0.000 description 1
- 230000006835 compression Effects 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 239000000599 controlled substance Substances 0.000 description 1
- 230000036461 convulsion Effects 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 1
- 229960004544 cortisone Drugs 0.000 description 1
- 239000006184 cosolvent Substances 0.000 description 1
- 239000007822 coupling agent Substances 0.000 description 1
- 239000006071 cream Substances 0.000 description 1
- SLFGIOIONGJGRT-UHFFFAOYSA-N cyamemazine Chemical compound C1=C(C#N)C=C2N(CC(CN(C)C)C)C3=CC=CC=C3SC2=C1 SLFGIOIONGJGRT-UHFFFAOYSA-N 0.000 description 1
- 229960004278 cyamemazine Drugs 0.000 description 1
- 239000000625 cyclamic acid and its Na and Ca salt Substances 0.000 description 1
- HCAJEUSONLESMK-UHFFFAOYSA-N cyclohexylsulfamic acid Chemical compound OS(=O)(=O)NC1CCCCC1 HCAJEUSONLESMK-UHFFFAOYSA-N 0.000 description 1
- 229960004397 cyclophosphamide Drugs 0.000 description 1
- 229930182912 cyclosporin Natural products 0.000 description 1
- 210000000172 cytosol Anatomy 0.000 description 1
- 231100000433 cytotoxic Toxicity 0.000 description 1
- 239000002254 cytotoxic agent Substances 0.000 description 1
- 231100000599 cytotoxic agent Toxicity 0.000 description 1
- 230000001472 cytotoxic effect Effects 0.000 description 1
- 230000003013 cytotoxicity Effects 0.000 description 1
- 231100000135 cytotoxicity Toxicity 0.000 description 1
- 229960000640 dactinomycin Drugs 0.000 description 1
- 238000013481 data capture Methods 0.000 description 1
- 229960002887 deanol Drugs 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 239000012024 dehydrating agents Substances 0.000 description 1
- 239000003405 delayed action preparation Substances 0.000 description 1
- 238000010520 demethylation reaction Methods 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 229960003957 dexamethasone Drugs 0.000 description 1
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- 229960003529 diazepam Drugs 0.000 description 1
- AAOVKJBEBIDNHE-UHFFFAOYSA-N diazepam Chemical compound N=1CC(=O)N(C)C2=CC=C(Cl)C=C2C=1C1=CC=CC=C1 AAOVKJBEBIDNHE-UHFFFAOYSA-N 0.000 description 1
- 235000019404 dichlorodifluoromethane Nutrition 0.000 description 1
- 229940042935 dichlorodifluoromethane Drugs 0.000 description 1
- YMWUJEATGCHHMB-DICFDUPASA-N dichloromethane-d2 Chemical compound [2H]C([2H])(Cl)Cl YMWUJEATGCHHMB-DICFDUPASA-N 0.000 description 1
- 229940087091 dichlorotetrafluoroethane Drugs 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- 229940043237 diethanolamine Drugs 0.000 description 1
- UPQZOUHVTJNGFK-UHFFFAOYSA-N diethyl 2-methylpropanedioate Chemical compound CCOC(=O)C(C)C(=O)OCC UPQZOUHVTJNGFK-UHFFFAOYSA-N 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- 229940043279 diisopropylamine Drugs 0.000 description 1
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 1
- WEHWNAOGRSTTBQ-UHFFFAOYSA-N dipropylamine Chemical compound CCCNCCC WEHWNAOGRSTTBQ-UHFFFAOYSA-N 0.000 description 1
- 229940042399 direct acting antivirals protease inhibitors Drugs 0.000 description 1
- 201000010099 disease Diseases 0.000 description 1
- 239000002934 diuretic Substances 0.000 description 1
- 229940030606 diuretics Drugs 0.000 description 1
- MSYUMPGNGDNTIQ-UHFFFAOYSA-N dixyrazine Chemical compound C12=CC=CC=C2SC2=CC=CC=C2N1CC(C)CN1CCN(CCOCCO)CC1 MSYUMPGNGDNTIQ-UHFFFAOYSA-N 0.000 description 1
- 229960005146 dixyrazine Drugs 0.000 description 1
- 208000002173 dizziness Diseases 0.000 description 1
- 239000003534 dna topoisomerase inhibitor Substances 0.000 description 1
- 229960003668 docetaxel Drugs 0.000 description 1
- MOTZDAYCYVMXPC-UHFFFAOYSA-N dodecyl hydrogen sulfate Chemical compound CCCCCCCCCCCCOS(O)(=O)=O MOTZDAYCYVMXPC-UHFFFAOYSA-N 0.000 description 1
- 230000035622 drinking Effects 0.000 description 1
- 239000006196 drop Substances 0.000 description 1
- RMEDXOLNCUSCGS-UHFFFAOYSA-N droperidol Chemical compound C1=CC(F)=CC=C1C(=O)CCCN1CC=C(N2C(NC3=CC=CC=C32)=O)CC1 RMEDXOLNCUSCGS-UHFFFAOYSA-N 0.000 description 1
- 229960000394 droperidol Drugs 0.000 description 1
- 239000000890 drug combination Substances 0.000 description 1
- 238000009510 drug design Methods 0.000 description 1
- 238000007876 drug discovery Methods 0.000 description 1
- 230000036267 drug metabolism Effects 0.000 description 1
- 230000008406 drug-drug interaction Effects 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 230000002500 effect on skin Effects 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 239000007920 enema Substances 0.000 description 1
- 229940079360 enema for constipation Drugs 0.000 description 1
- 229960000610 enoxaparin Drugs 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 210000002615 epidermis Anatomy 0.000 description 1
- 229960001208 eplerenone Drugs 0.000 description 1
- JUKPWJGBANNWMW-VWBFHTRKSA-N eplerenone Chemical compound C([C@@H]1[C@]2(C)C[C@H]3O[C@]33[C@@]4(C)CCC(=O)C=C4C[C@H]([C@@H]13)C(=O)OC)C[C@@]21CCC(=O)O1 JUKPWJGBANNWMW-VWBFHTRKSA-N 0.000 description 1
- 229930013356 epothilone Natural products 0.000 description 1
- HESCAJZNRMSMJG-KKQRBIROSA-N epothilone A Chemical class C/C([C@@H]1C[C@@H]2O[C@@H]2CCC[C@@H]([C@@H]([C@@H](C)C(=O)C(C)(C)[C@@H](O)CC(=O)O1)O)C)=C\C1=CSC(C)=N1 HESCAJZNRMSMJG-KKQRBIROSA-N 0.000 description 1
- 229960004468 eptifibatide Drugs 0.000 description 1
- GLGOPUHVAZCPRB-LROMGURASA-N eptifibatide Chemical compound N1C(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@H](CCCCNC(=N)N)NC(=O)CCSSC[C@@H](C(N)=O)NC(=O)[C@@H]2CCCN2C(=O)[C@@H]1CC1=CN=C2[C]1C=CC=C2 GLGOPUHVAZCPRB-LROMGURASA-N 0.000 description 1
- 201000006517 essential tremor Diseases 0.000 description 1
- 229940011871 estrogen Drugs 0.000 description 1
- 239000000262 estrogen Substances 0.000 description 1
- 239000000328 estrogen antagonist Substances 0.000 description 1
- AVOLMBLBETYQHX-UHFFFAOYSA-N etacrynic acid Chemical compound CCC(=C)C(=O)C1=CC=C(OCC(O)=O)C(Cl)=C1Cl AVOLMBLBETYQHX-UHFFFAOYSA-N 0.000 description 1
- 229960003199 etacrynic acid Drugs 0.000 description 1
- 229960000403 etanercept Drugs 0.000 description 1
- AFAXGSQYZLGZPG-UHFFFAOYSA-N ethanedisulfonic acid Chemical compound OS(=O)(=O)CCS(O)(=O)=O AFAXGSQYZLGZPG-UHFFFAOYSA-N 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- XYIBRDXRRQCHLP-UHFFFAOYSA-N ethyl acetoacetate Chemical compound CCOC(=O)CC(C)=O XYIBRDXRRQCHLP-UHFFFAOYSA-N 0.000 description 1
- 229960004759 ethyl loflazepate Drugs 0.000 description 1
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 230000003203 everyday effect Effects 0.000 description 1
- 210000003414 extremity Anatomy 0.000 description 1
- 206010016256 fatigue Diseases 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 229940125753 fibrate Drugs 0.000 description 1
- 239000003527 fibrinolytic agent Substances 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- IRYFCWPNDIUQOW-UHFFFAOYSA-N fluanisone Chemical compound COC1=CC=CC=C1N1CCN(CCCC(=O)C=2C=CC(F)=CC=2)CC1 IRYFCWPNDIUQOW-UHFFFAOYSA-N 0.000 description 1
- 229960005220 fluanisone Drugs 0.000 description 1
- 229960004930 fludiazepam Drugs 0.000 description 1
- ROYOYTLGDLIGBX-UHFFFAOYSA-N fludiazepam Chemical compound N=1CC(=O)N(C)C2=CC=C(Cl)C=C2C=1C1=CC=CC=C1F ROYOYTLGDLIGBX-UHFFFAOYSA-N 0.000 description 1
- RGUQWGXAYZNLMI-UHFFFAOYSA-N flumethiazide Chemical compound C1=C(C(F)(F)F)C(S(=O)(=O)N)=CC2=C1NC=NS2(=O)=O RGUQWGXAYZNLMI-UHFFFAOYSA-N 0.000 description 1
- 229960003028 flumethiazide Drugs 0.000 description 1
- 229960002200 flunitrazepam Drugs 0.000 description 1
- 229960002419 flupentixol Drugs 0.000 description 1
- 229960002690 fluphenazine Drugs 0.000 description 1
- 229960003532 fluspirilene Drugs 0.000 description 1
- 201000002865 focal hand dystonia Diseases 0.000 description 1
- 239000008098 formaldehyde solution Substances 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- WBJINCZRORDGAQ-UHFFFAOYSA-N formic acid ethyl ester Natural products CCOC=O WBJINCZRORDGAQ-UHFFFAOYSA-N 0.000 description 1
- 238000007710 freezing Methods 0.000 description 1
- 230000008014 freezing Effects 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- DSLZVSRJTYRBFB-DUHBMQHGSA-N galactaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)[C@@H](O)[C@H](O)C(O)=O DSLZVSRJTYRBFB-DUHBMQHGSA-N 0.000 description 1
- LRBQNJMCXXYXIU-QWKBTXIPSA-N gallotannic acid Chemical compound OC1=C(O)C(O)=CC(C(=O)OC=2C(=C(O)C=C(C=2)C(=O)OC[C@H]2[C@@H]([C@@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)[C@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)[C@@H](OC(=O)C=3C=C(OC(=O)C=4C=C(O)C(O)=C(O)C=4)C(O)=C(O)C=3)O2)OC(=O)C=2C=C(OC(=O)C=3C=C(O)C(O)=C(O)C=3)C(O)=C(O)C=2)O)=C1 LRBQNJMCXXYXIU-QWKBTXIPSA-N 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- YRSVDSQRGBYVIY-GJZGRUSLSA-N gemopatrilat Chemical compound O=C1N(CC(O)=O)C(C)(C)CCC[C@@H]1NC(=O)[C@@H](S)CC1=CC=CC=C1 YRSVDSQRGBYVIY-GJZGRUSLSA-N 0.000 description 1
- 229950006480 gemopatrilat Drugs 0.000 description 1
- 229960005219 gentisic acid Drugs 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 229960004580 glibenclamide Drugs 0.000 description 1
- WIGIZIANZCJQQY-RUCARUNLSA-N glimepiride Chemical compound O=C1C(CC)=C(C)CN1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)N[C@@H]2CC[C@@H](C)CC2)C=C1 WIGIZIANZCJQQY-RUCARUNLSA-N 0.000 description 1
- 229960004346 glimepiride Drugs 0.000 description 1
- 229960001381 glipizide Drugs 0.000 description 1
- ZJJXGWJIGJFDTL-UHFFFAOYSA-N glipizide Chemical compound C1=NC(C)=CN=C1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)NC2CCCCC2)C=C1 ZJJXGWJIGJFDTL-UHFFFAOYSA-N 0.000 description 1
- 235000012208 gluconic acid Nutrition 0.000 description 1
- 229950006191 gluconic acid Drugs 0.000 description 1
- 229960002989 glutamic acid Drugs 0.000 description 1
- ZNNLBTZKUZBEKO-UHFFFAOYSA-N glyburide Chemical compound COC1=CC=C(Cl)C=C1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)NC2CCCCC2)C=C1 ZNNLBTZKUZBEKO-UHFFFAOYSA-N 0.000 description 1
- 125000005456 glyceride group Chemical group 0.000 description 1
- 150000002344 gold compounds Chemical class 0.000 description 1
- 230000005283 ground state Effects 0.000 description 1
- 239000003102 growth factor Substances 0.000 description 1
- 239000003324 growth hormone secretagogue Substances 0.000 description 1
- 229960002158 halazepam Drugs 0.000 description 1
- 229960003878 haloperidol Drugs 0.000 description 1
- BCQZXOMGPXTTIC-UHFFFAOYSA-N halothane Chemical compound FC(F)(F)C(Cl)Br BCQZXOMGPXTTIC-UHFFFAOYSA-N 0.000 description 1
- 229960003132 halothane Drugs 0.000 description 1
- 210000004247 hand Anatomy 0.000 description 1
- 230000007686 hepatotoxicity Effects 0.000 description 1
- 231100000304 hepatotoxicity Toxicity 0.000 description 1
- 208000008675 hereditary spastic paraplegia Diseases 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- UUVWYPNAQBNQJQ-UHFFFAOYSA-N hexamethylmelamine Chemical compound CN(C)C1=NC(N(C)C)=NC(N(C)C)=N1 UUVWYPNAQBNQJQ-UHFFFAOYSA-N 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 229940006607 hirudin Drugs 0.000 description 1
- WQPDUTSPKFMPDP-OUMQNGNKSA-N hirudin Chemical compound C([C@@H](C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC(OS(O)(=O)=O)=CC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(O)=O)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCCN)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@@H](NC(=O)[C@@H](NC(=O)[C@H]1NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)CNC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)CC)NC(=O)[C@@H]2CSSC[C@@H](C(=O)N[C@@H](CCC(O)=O)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@H](C(=O)N[C@H](C(NCC(=O)N[C@@H](CCC(N)=O)C(=O)NCC(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCCN)C(=O)N2)=O)CSSC1)C(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]1NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=2C=CC(O)=CC=2)NC(=O)[C@@H](NC(=O)[C@@H](N)C(C)C)C(C)C)[C@@H](C)O)CSSC1)C(C)C)[C@@H](C)O)[C@@H](C)O)C1=CC=CC=C1 WQPDUTSPKFMPDP-OUMQNGNKSA-N 0.000 description 1
- 210000003630 histaminocyte Anatomy 0.000 description 1
- 229940125697 hormonal agent Drugs 0.000 description 1
- XGIHQYAWBCFNPY-AZOCGYLKSA-N hydrabamine Chemical compound C([C@@H]12)CC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC[C@@]1(C)CNCCNC[C@@]1(C)[C@@H]2CCC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC1 XGIHQYAWBCFNPY-AZOCGYLKSA-N 0.000 description 1
- 229960002003 hydrochlorothiazide Drugs 0.000 description 1
- 229960000890 hydrocortisone Drugs 0.000 description 1
- DMDGGSIALPNSEE-UHFFFAOYSA-N hydroflumethiazide Chemical compound C1=C(C(F)(F)F)C(S(=O)(=O)N)=CC2=C1NCNS2(=O)=O DMDGGSIALPNSEE-UHFFFAOYSA-N 0.000 description 1
- 229960003313 hydroflumethiazide Drugs 0.000 description 1
- 150000002431 hydrogen Chemical class 0.000 description 1
- 229940071870 hydroiodic acid Drugs 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 230000033444 hydroxylation Effects 0.000 description 1
- 238000005805 hydroxylation reaction Methods 0.000 description 1
- 230000001660 hyperkinetic effect Effects 0.000 description 1
- BBPRUNPUJIUXSE-DXKRWKNPSA-N ifetroban Chemical compound CCCCCNC(=O)C1=COC([C@H]2[C@H]([C@@H]3CC[C@H]2O3)CC=2C(=CC=CC=2)CCC(O)=O)=N1 BBPRUNPUJIUXSE-DXKRWKNPSA-N 0.000 description 1
- 229950004274 ifetroban Drugs 0.000 description 1
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Substances C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 229960003444 immunosuppressant agent Drugs 0.000 description 1
- 239000003018 immunosuppressive agent Substances 0.000 description 1
- 238000002513 implantation Methods 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 239000003701 inert diluent Substances 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 206010022437 insomnia Diseases 0.000 description 1
- 239000004026 insulin derivative Substances 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 230000002427 irreversible effect Effects 0.000 description 1
- 229960004849 isoconazole Drugs 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- FZWBNHMXJMCXLU-BLAUPYHCSA-N isomaltotriose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1OC[C@@H]1[C@@H](O)[C@H](O)[C@@H](O)[C@@H](OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C=O)O1 FZWBNHMXJMCXLU-BLAUPYHCSA-N 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 1
- 238000012804 iterative process Methods 0.000 description 1
- 229960003299 ketamine Drugs 0.000 description 1
- 229960004423 ketazolam Drugs 0.000 description 1
- PWAJCNITSBZRBL-UHFFFAOYSA-N ketazolam Chemical compound O1C(C)=CC(=O)N2CC(=O)N(C)C3=CC=C(Cl)C=C3C21C1=CC=CC=C1 PWAJCNITSBZRBL-UHFFFAOYSA-N 0.000 description 1
- 239000004922 lacquer Substances 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 1
- 229940099563 lactobionic acid Drugs 0.000 description 1
- 229940033355 lauric acid Drugs 0.000 description 1
- 239000003199 leukotriene receptor blocking agent Substances 0.000 description 1
- 229960004145 levosulpiride Drugs 0.000 description 1
- 239000000865 liniment Substances 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 229940057995 liquid paraffin Drugs 0.000 description 1
- 229960001078 lithium Drugs 0.000 description 1
- 239000012280 lithium aluminium hydride Substances 0.000 description 1
- 231100001252 long-term toxicity Toxicity 0.000 description 1
- 229960004391 lorazepam Drugs 0.000 description 1
- 239000006210 lotion Substances 0.000 description 1
- 229960004844 lovastatin Drugs 0.000 description 1
- PCZOHLXUXFIOCF-BXMDZJJMSA-N lovastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 PCZOHLXUXFIOCF-BXMDZJJMSA-N 0.000 description 1
- QLJODMDSTUBWDW-UHFFFAOYSA-N lovastatin hydroxy acid Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(C)C=C21 QLJODMDSTUBWDW-UHFFFAOYSA-N 0.000 description 1
- 229940127215 low-molecular weight heparin Drugs 0.000 description 1
- 229960000423 loxapine Drugs 0.000 description 1
- XJGVXQDUIWGIRW-UHFFFAOYSA-N loxapine Chemical compound C1CN(C)CCN1C1=NC2=CC=CC=C2OC2=CC=C(Cl)C=C12 XJGVXQDUIWGIRW-UHFFFAOYSA-N 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 230000001050 lubricating effect Effects 0.000 description 1
- VTHJTEIRLNZDEV-UHFFFAOYSA-L magnesium dihydroxide Chemical compound [OH-].[OH-].[Mg+2] VTHJTEIRLNZDEV-UHFFFAOYSA-L 0.000 description 1
- 239000000347 magnesium hydroxide Substances 0.000 description 1
- 229910001862 magnesium hydroxide Inorganic materials 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 229940098895 maleic acid Drugs 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N mandelic acid Chemical compound OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- 231100000682 maximum tolerated dose Toxicity 0.000 description 1
- 229960002225 medazepam Drugs 0.000 description 1
- 229950004994 meglitinide Drugs 0.000 description 1
- 229960001861 melperone Drugs 0.000 description 1
- SLVMESMUVMCQIY-UHFFFAOYSA-N mesoridazine Chemical compound CN1CCCCC1CCN1C2=CC(S(C)=O)=CC=C2SC2=CC=CC=C21 SLVMESMUVMCQIY-UHFFFAOYSA-N 0.000 description 1
- 229960000300 mesoridazine Drugs 0.000 description 1
- 230000037323 metabolic rate Effects 0.000 description 1
- XZWYZXLIPXDOLR-UHFFFAOYSA-N metformin Chemical compound CN(C)C(=N)NC(N)=N XZWYZXLIPXDOLR-UHFFFAOYSA-N 0.000 description 1
- 229960003105 metformin Drugs 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- GBMDVOWEEQVZKZ-UHFFFAOYSA-N methanol;hydrate Chemical compound O.OC GBMDVOWEEQVZKZ-UHFFFAOYSA-N 0.000 description 1
- 229960000485 methotrexate Drugs 0.000 description 1
- VRQVVMDWGGWHTJ-CQSZACIVSA-N methotrimeprazine Chemical compound C1=CC=C2N(C[C@H](C)CN(C)C)C3=CC(OC)=CC=C3SC2=C1 VRQVVMDWGGWHTJ-CQSZACIVSA-N 0.000 description 1
- 229940042053 methotrimeprazine Drugs 0.000 description 1
- WBYWAXJHAXSJNI-UHFFFAOYSA-N methyl p-hydroxycinnamate Natural products OC(=O)C=CC1=CC=CC=C1 WBYWAXJHAXSJNI-UHFFFAOYSA-N 0.000 description 1
- 229960001344 methylphenidate Drugs 0.000 description 1
- IUBSYMUCCVWXPE-UHFFFAOYSA-N metoprolol Chemical compound COCCC1=CC=C(OCC(O)CNC(C)C)C=C1 IUBSYMUCCVWXPE-UHFFFAOYSA-N 0.000 description 1
- 229960002237 metoprolol Drugs 0.000 description 1
- 229960000600 milnacipran Drugs 0.000 description 1
- 239000002394 mineralocorticoid antagonist Substances 0.000 description 1
- 244000309715 mini pig Species 0.000 description 1
- CFCUWKMKBJTWLW-BKHRDMLASA-N mithramycin Chemical compound O([C@@H]1C[C@@H](O[C@H](C)[C@H]1O)OC=1C=C2C=C3C[C@H]([C@@H](C(=O)C3=C(O)C2=C(O)C=1C)O[C@@H]1O[C@H](C)[C@@H](O)[C@H](O[C@@H]2O[C@H](C)[C@H](O)[C@H](O[C@@H]3O[C@H](C)[C@@H](O)[C@@](C)(O)C3)C2)C1)[C@H](OC)C(=O)[C@@H](O)[C@@H](C)O)[C@H]1C[C@@H](O)[C@H](O)[C@@H](C)O1 CFCUWKMKBJTWLW-BKHRDMLASA-N 0.000 description 1
- 229960004857 mitomycin Drugs 0.000 description 1
- 229960000350 mitotane Drugs 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 239000007932 molded tablet Substances 0.000 description 1
- 229960004938 molindone Drugs 0.000 description 1
- 239000002899 monoamine oxidase inhibitor Substances 0.000 description 1
- AGAHNABIDCTLHW-UHFFFAOYSA-N moperone Chemical compound C1=CC(C)=CC=C1C1(O)CCN(CCCC(=O)C=2C=CC(F)=CC=2)CC1 AGAHNABIDCTLHW-UHFFFAOYSA-N 0.000 description 1
- 229960000758 moperone Drugs 0.000 description 1
- 229960003894 mosapramine Drugs 0.000 description 1
- RTGDFNSFWBGLEC-SYZQJQIISA-N mycophenolate mofetil Chemical compound COC1=C(C)C=2COC(=O)C=2C(O)=C1C\C=C(/C)CCC(=O)OCCN1CCOCC1 RTGDFNSFWBGLEC-SYZQJQIISA-N 0.000 description 1
- 229960004866 mycophenolate mofetil Drugs 0.000 description 1
- WQEPLUUGTLDZJY-UHFFFAOYSA-N n-Pentadecanoic acid Natural products CCCCCCCCCCCCCCC(O)=O WQEPLUUGTLDZJY-UHFFFAOYSA-N 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-N naphthalene-2-sulfonic acid Chemical compound C1=CC=CC2=CC(S(=O)(=O)O)=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-N 0.000 description 1
- CMWTZPSULFXXJA-VIFPVBQESA-N naproxen Chemical compound C1=C([C@H](C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-VIFPVBQESA-N 0.000 description 1
- 229960002009 naproxen Drugs 0.000 description 1
- 239000006225 natural substrate Substances 0.000 description 1
- 230000008693 nausea Effects 0.000 description 1
- IOMMMLWIABWRKL-WUTDNEBXSA-N nazartinib Chemical compound C1N(C(=O)/C=C/CN(C)C)CCCC[C@H]1N1C2=C(Cl)C=CC=C2N=C1NC(=O)C1=CC=NC(C)=C1 IOMMMLWIABWRKL-WUTDNEBXSA-N 0.000 description 1
- 239000006199 nebulizer Substances 0.000 description 1
- 230000017074 necrotic cell death Effects 0.000 description 1
- 230000000926 neurological effect Effects 0.000 description 1
- 239000002858 neurotransmitter agent Substances 0.000 description 1
- 229960001454 nitrazepam Drugs 0.000 description 1
- KJONHKAYOJNZEC-UHFFFAOYSA-N nitrazepam Chemical compound C12=CC([N+](=O)[O-])=CC=C2NC(=O)CN=C1C1=CC=CC=C1 KJONHKAYOJNZEC-UHFFFAOYSA-N 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- LYGJENNIWJXYER-UHFFFAOYSA-N nitromethane Chemical compound C[N+]([O-])=O LYGJENNIWJXYER-UHFFFAOYSA-N 0.000 description 1
- 229960002640 nordazepam Drugs 0.000 description 1
- AKPLHCDWDRPJGD-UHFFFAOYSA-N nordazepam Chemical compound C12=CC(Cl)=CC=C2NC(=O)CN=C1C1=CC=CC=C1 AKPLHCDWDRPJGD-UHFFFAOYSA-N 0.000 description 1
- 229960002748 norepinephrine Drugs 0.000 description 1
- SFLSHLFXELFNJZ-UHFFFAOYSA-N norepinephrine Natural products NCC(O)C1=CC=C(O)C(O)=C1 SFLSHLFXELFNJZ-UHFFFAOYSA-N 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 229960002446 octanoic acid Drugs 0.000 description 1
- 229960001494 octreotide acetate Drugs 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- LVRLSYPNFFBYCZ-VGWMRTNUSA-N omapatrilat Chemical compound C([C@H](S)C(=O)N[C@H]1CCS[C@H]2CCC[C@H](N2C1=O)C(=O)O)C1=CC=CC=C1 LVRLSYPNFFBYCZ-VGWMRTNUSA-N 0.000 description 1
- 229950000973 omapatrilat Drugs 0.000 description 1
- 229940005483 opioid analgesics Drugs 0.000 description 1
- 239000010502 orange oil Substances 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 239000003791 organic solvent mixture Substances 0.000 description 1
- 201000002851 oromandibular dystonia Diseases 0.000 description 1
- 229960005010 orotic acid Drugs 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 229960004535 oxazepam Drugs 0.000 description 1
- ADIMAYPTOBDMTL-UHFFFAOYSA-N oxazepam Chemical compound C12=CC(Cl)=CC=C2NC(=O)C(O)N=C1C1=CC=CC=C1 ADIMAYPTOBDMTL-UHFFFAOYSA-N 0.000 description 1
- 239000007800 oxidant agent Substances 0.000 description 1
- 230000004783 oxidative metabolism Effects 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 229960002841 oxypertine Drugs 0.000 description 1
- LSQZJLSUYDQPKJ-UHFFFAOYSA-N p-Hydroxyampicillin Natural products O=C1N2C(C(O)=O)C(C)(C)SC2C1NC(=O)C(N)C1=CC=C(O)C=C1 LSQZJLSUYDQPKJ-UHFFFAOYSA-N 0.000 description 1
- 229960001057 paliperidone Drugs 0.000 description 1
- WLJNZVDCPSBLRP-UHFFFAOYSA-N pamoic acid Chemical compound C1=CC=C2C(CC=3C4=CC=CC=C4C=C(C=3O)C(=O)O)=C(O)C(C(O)=O)=CC2=C1 WLJNZVDCPSBLRP-UHFFFAOYSA-N 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 230000001314 paroxysmal effect Effects 0.000 description 1
- 235000010603 pastilles Nutrition 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 230000035515 penetration Effects 0.000 description 1
- 229960004505 penfluridol Drugs 0.000 description 1
- 235000019371 penicillin G benzathine Nutrition 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- WEYVCQFUGFRXOM-UHFFFAOYSA-N perazine Chemical compound C1CN(C)CCN1CCCN1C2=CC=CC=C2SC2=CC=CC=C21 WEYVCQFUGFRXOM-UHFFFAOYSA-N 0.000 description 1
- 229960002195 perazine Drugs 0.000 description 1
- LUALIOATIOESLM-UHFFFAOYSA-N periciazine Chemical compound C1CC(O)CCN1CCCN1C2=CC(C#N)=CC=C2SC2=CC=CC=C21 LUALIOATIOESLM-UHFFFAOYSA-N 0.000 description 1
- 229960000769 periciazine Drugs 0.000 description 1
- 229960000762 perphenazine Drugs 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 239000002570 phosphodiesterase III inhibitor Substances 0.000 description 1
- 239000002590 phosphodiesterase V inhibitor Substances 0.000 description 1
- 239000002571 phosphodiesterase inhibitor Substances 0.000 description 1
- 108010072906 phosphoramidon Proteins 0.000 description 1
- BWSDNRQVTFZQQD-AYVHNPTNSA-N phosphoramidon Chemical compound O([P@@](O)(=O)N[C@H](CC(C)C)C(=O)N[C@H](CC=1[C]2C=CC=CC2=NC=1)C(O)=O)[C@H]1O[C@@H](C)[C@H](O)[C@@H](O)[C@@H]1O BWSDNRQVTFZQQD-AYVHNPTNSA-N 0.000 description 1
- UHZYTMXLRWXGPK-UHFFFAOYSA-N phosphorus pentachloride Chemical compound ClP(Cl)(Cl)(Cl)Cl UHZYTMXLRWXGPK-UHFFFAOYSA-N 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 239000000049 pigment Substances 0.000 description 1
- 229960002034 pinazepam Drugs 0.000 description 1
- MFZOSKPPVCIFMT-UHFFFAOYSA-N pinazepam Chemical compound C12=CC(Cl)=CC=C2N(CC#C)C(=O)CN=C1C1=CC=CC=C1 MFZOSKPPVCIFMT-UHFFFAOYSA-N 0.000 description 1
- 229960005095 pioglitazone Drugs 0.000 description 1
- AXKPFOAXAHJUAG-UHFFFAOYSA-N pipamperone Chemical compound C1CC(C(=O)N)(N2CCCCC2)CCN1CCCC(=O)C1=CC=C(F)C=C1 AXKPFOAXAHJUAG-UHFFFAOYSA-N 0.000 description 1
- 229960002776 pipamperone Drugs 0.000 description 1
- JOMHSQGEWSNUKU-UHFFFAOYSA-N pipotiazine Chemical compound C12=CC(S(=O)(=O)N(C)C)=CC=C2SC2=CC=CC=C2N1CCCN1CCC(CCO)CC1 JOMHSQGEWSNUKU-UHFFFAOYSA-N 0.000 description 1
- 229960003252 pipotiazine Drugs 0.000 description 1
- VGYFMXBACGZSIL-MCBHFWOFSA-N pitavastatin Chemical compound OC(=O)C[C@H](O)C[C@H](O)\C=C\C1=C(C2CC2)N=C2C=CC=CC2=C1C1=CC=C(F)C=C1 VGYFMXBACGZSIL-MCBHFWOFSA-N 0.000 description 1
- 230000036470 plasma concentration Effects 0.000 description 1
- 239000004014 plasticizer Substances 0.000 description 1
- 239000000106 platelet aggregation inhibitor Substances 0.000 description 1
- 229910052697 platinum Inorganic materials 0.000 description 1
- 229960003171 plicamycin Drugs 0.000 description 1
- 229920000136 polysorbate Polymers 0.000 description 1
- 229950008882 polysorbate Drugs 0.000 description 1
- 229960005483 polythiazide Drugs 0.000 description 1
- 229920000046 polythiazide Polymers 0.000 description 1
- 230000001144 postural effect Effects 0.000 description 1
- 235000015320 potassium carbonate Nutrition 0.000 description 1
- 239000004036 potassium channel stimulating agent Substances 0.000 description 1
- 229940028868 potassium clorazepate Drugs 0.000 description 1
- WSHYKIAQCMIPTB-UHFFFAOYSA-M potassium;2-oxo-3-(3-oxo-1-phenylbutyl)chromen-4-olate Chemical compound [K+].[O-]C=1C2=CC=CC=C2OC(=O)C=1C(CC(=O)C)C1=CC=CC=C1 WSHYKIAQCMIPTB-UHFFFAOYSA-M 0.000 description 1
- DTGLZDAWLRGWQN-UHFFFAOYSA-N prasugrel Chemical compound C1CC=2SC(OC(=O)C)=CC=2CN1C(C=1C(=CC=CC=1)F)C(=O)C1CC1 DTGLZDAWLRGWQN-UHFFFAOYSA-N 0.000 description 1
- 229960004197 prasugrel Drugs 0.000 description 1
- 229960002965 pravastatin Drugs 0.000 description 1
- TUZYXOIXSAXUGO-PZAWKZKUSA-N pravastatin Chemical compound C1=C[C@H](C)[C@H](CC[C@@H](O)C[C@@H](O)CC(O)=O)[C@H]2[C@@H](OC(=O)[C@@H](C)CC)C[C@H](O)C=C21 TUZYXOIXSAXUGO-PZAWKZKUSA-N 0.000 description 1
- 229960004856 prazepam Drugs 0.000 description 1
- 229960004618 prednisone Drugs 0.000 description 1
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 description 1
- 125000001844 prenyl group Chemical group [H]C([*])([H])C([H])=C(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- CPTBDICYNRMXFX-UHFFFAOYSA-N procarbazine Chemical compound CNNCC1=CC=C(C(=O)NC(C)C)C=C1 CPTBDICYNRMXFX-UHFFFAOYSA-N 0.000 description 1
- 229960000624 procarbazine Drugs 0.000 description 1
- WIKYUJGCLQQFNW-UHFFFAOYSA-N prochlorperazine Chemical compound C1CN(C)CCN1CCCN1C2=CC(Cl)=CC=C2SC2=CC=CC=C21 WIKYUJGCLQQFNW-UHFFFAOYSA-N 0.000 description 1
- 229960003111 prochlorperazine Drugs 0.000 description 1
- 239000000583 progesterone congener Substances 0.000 description 1
- 229940072288 prograf Drugs 0.000 description 1
- 201000002212 progressive supranuclear palsy Diseases 0.000 description 1
- 229960003598 promazine Drugs 0.000 description 1
- 239000003380 propellant Substances 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- 239000003528 protein farnesyltransferase inhibitor Substances 0.000 description 1
- JTTAUPUMOLRVRA-UHFFFAOYSA-N prothipendyl Chemical group C1=CN=C2N(CCCN(C)C)C3=CC=CC=C3SC2=C1 JTTAUPUMOLRVRA-UHFFFAOYSA-N 0.000 description 1
- 229960000957 prothipendyl Drugs 0.000 description 1
- 150000003212 purines Chemical class 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 229940073095 questran Drugs 0.000 description 1
- 229960004431 quetiapine Drugs 0.000 description 1
- URKOMYMAXPYINW-UHFFFAOYSA-N quetiapine Chemical compound C1CN(CCOCCO)CCN1C1=NC2=CC=CC=C2SC2=CC=CC=C12 URKOMYMAXPYINW-UHFFFAOYSA-N 0.000 description 1
- SBYHFKPVCBCYGV-UHFFFAOYSA-N quinuclidine Chemical compound C1CC2CCN1CC2 SBYHFKPVCBCYGV-UHFFFAOYSA-N 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 1
- 238000009790 rate-determining step (RDS) Methods 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 238000010223 real-time analysis Methods 0.000 description 1
- 229940044551 receptor antagonist Drugs 0.000 description 1
- 239000002464 receptor antagonist Substances 0.000 description 1
- 229960003448 remoxipride Drugs 0.000 description 1
- 239000002461 renin inhibitor Substances 0.000 description 1
- 229940086526 renin-inhibitors Drugs 0.000 description 1
- 229960002354 repaglinide Drugs 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 206010039073 rheumatoid arthritis Diseases 0.000 description 1
- 229960001534 risperidone Drugs 0.000 description 1
- RAPZEAPATHNIPO-UHFFFAOYSA-N risperidone Chemical compound FC1=CC=C2C(C3CCN(CC3)CCC=3C(=O)N4CCCCC4=NC=3C)=NOC2=C1 RAPZEAPATHNIPO-UHFFFAOYSA-N 0.000 description 1
- 229960000371 rofecoxib Drugs 0.000 description 1
- RZJQGNCSTQAWON-UHFFFAOYSA-N rofecoxib Chemical compound C1=CC(S(=O)(=O)C)=CC=C1C1=C(C=2C=CC=CC=2)C(=O)OC1 RZJQGNCSTQAWON-UHFFFAOYSA-N 0.000 description 1
- 229960004586 rosiglitazone Drugs 0.000 description 1
- 229960000672 rosuvastatin Drugs 0.000 description 1
- BPRHUIZQVSMCRT-VEUZHWNKSA-N rosuvastatin Chemical compound CC(C)C1=NC(N(C)S(C)(=O)=O)=NC(C=2C=CC(F)=CC=2)=C1\C=C\[C@@H](O)C[C@@H](O)CC(O)=O BPRHUIZQVSMCRT-VEUZHWNKSA-N 0.000 description 1
- 238000010079 rubber tapping Methods 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 108010073863 saruplase Proteins 0.000 description 1
- 229960005399 satraplatin Drugs 0.000 description 1
- 190014017285 satraplatin Chemical compound 0.000 description 1
- 229940116353 sebacic acid Drugs 0.000 description 1
- 150000003335 secondary amines Chemical class 0.000 description 1
- 229940125723 sedative agent Drugs 0.000 description 1
- 239000000932 sedative agent Substances 0.000 description 1
- 229960003946 selegiline Drugs 0.000 description 1
- 229940076279 serotonin Drugs 0.000 description 1
- GZKLJWGUPQBVJQ-UHFFFAOYSA-N sertindole Chemical compound C1=CC(F)=CC=C1N1C2=CC=C(Cl)C=C2C(C2CCN(CCN3C(NCC3)=O)CC2)=C1 GZKLJWGUPQBVJQ-UHFFFAOYSA-N 0.000 description 1
- 229960000652 sertindole Drugs 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 238000007493 shaping process Methods 0.000 description 1
- 229960003310 sildenafil Drugs 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 229960002855 simvastatin Drugs 0.000 description 1
- RYMZZMVNJRMUDD-HGQWONQESA-N simvastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)C(C)(C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 RYMZZMVNJRMUDD-HGQWONQESA-N 0.000 description 1
- 229960002930 sirolimus Drugs 0.000 description 1
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 206010040882 skin lesion Diseases 0.000 description 1
- 231100000444 skin lesion Toxicity 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- 208000018198 spasticity Diseases 0.000 description 1
- 210000000278 spinal cord Anatomy 0.000 description 1
- 208000020431 spinal cord injury Diseases 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 239000004059 squalene synthase inhibitor Substances 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 229960004274 stearic acid Drugs 0.000 description 1
- 230000003637 steroidlike Effects 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 229960005202 streptokinase Drugs 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- WPLOVIFNBMNBPD-ATHMIXSHSA-N subtilin Chemical compound CC1SCC(NC2=O)C(=O)NC(CC(N)=O)C(=O)NC(C(=O)NC(CCCCN)C(=O)NC(C(C)CC)C(=O)NC(=C)C(=O)NC(CCCCN)C(O)=O)CSC(C)C2NC(=O)C(CC(C)C)NC(=O)C1NC(=O)C(CCC(N)=O)NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C1NC(=O)C(=C/C)/NC(=O)C(CCC(N)=O)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)CNC(=O)C(NC(=O)C(NC(=O)C2NC(=O)CNC(=O)C3CCCN3C(=O)C(NC(=O)C3NC(=O)C(CC(C)C)NC(=O)C(=C)NC(=O)C(CCC(O)=O)NC(=O)C(NC(=O)C(CCCCN)NC(=O)C(N)CC=4C5=CC=CC=C5NC=4)CSC3)C(C)SC2)C(C)C)C(C)SC1)CC1=CC=CC=C1 WPLOVIFNBMNBPD-ATHMIXSHSA-N 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 229910052717 sulfur Inorganic materials 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 229940032330 sulfuric acid Drugs 0.000 description 1
- 229960004940 sulpiride Drugs 0.000 description 1
- 229960004724 sultopride Drugs 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000002511 suppository base Substances 0.000 description 1
- 210000002504 synaptic vesicle Anatomy 0.000 description 1
- 230000001360 synchronised effect Effects 0.000 description 1
- 208000011580 syndromic disease Diseases 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 229940065721 systemic for obstructive airway disease xanthines Drugs 0.000 description 1
- 229960001967 tacrolimus Drugs 0.000 description 1
- 229960000835 tadalafil Drugs 0.000 description 1
- IEHKWSGCTWLXFU-IIBYNOLFSA-N tadalafil Chemical compound C1=C2OCOC2=CC([C@@H]2C3=C([C]4C=CC=CC4=N3)C[C@H]3N2C(=O)CN(C3=O)C)=C1 IEHKWSGCTWLXFU-IIBYNOLFSA-N 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 229940033123 tannic acid Drugs 0.000 description 1
- 235000015523 tannic acid Nutrition 0.000 description 1
- 229920002258 tannic acid Polymers 0.000 description 1
- 235000019640 taste Nutrition 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- 229960003188 temazepam Drugs 0.000 description 1
- LXIKEPCNDFVJKC-QXMHVHEDSA-N tenidap Chemical compound C12=CC(Cl)=CC=C2N(C(=O)N)C(=O)\C1=C(/O)C1=CC=CS1 LXIKEPCNDFVJKC-QXMHVHEDSA-N 0.000 description 1
- 229960003676 tenidap Drugs 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- AIUHRQHVWSUTGJ-UHFFFAOYSA-N thiopropazate Chemical compound C1CN(CCOC(=O)C)CCN1CCCN1C2=CC(Cl)=CC=C2SC2=CC=CC=C21 AIUHRQHVWSUTGJ-UHFFFAOYSA-N 0.000 description 1
- 229960004728 thiopropazate Drugs 0.000 description 1
- VZYCZNZBPPHOFY-UHFFFAOYSA-N thioproperazine Chemical compound C12=CC(S(=O)(=O)N(C)C)=CC=C2SC2=CC=CC=C2N1CCCN1CCN(C)CC1 VZYCZNZBPPHOFY-UHFFFAOYSA-N 0.000 description 1
- 229960003397 thioproperazine Drugs 0.000 description 1
- 229960002784 thioridazine Drugs 0.000 description 1
- 239000003868 thrombin inhibitor Substances 0.000 description 1
- 229960000103 thrombolytic agent Drugs 0.000 description 1
- 229960005344 tiapride Drugs 0.000 description 1
- 208000016686 tic disease Diseases 0.000 description 1
- 229960005001 ticlopidine Drugs 0.000 description 1
- PHWBOXQYWZNQIN-UHFFFAOYSA-N ticlopidine Chemical compound ClC1=CC=CC=C1CN1CC(C=CS2)=C2CC1 PHWBOXQYWZNQIN-UHFFFAOYSA-N 0.000 description 1
- 229960005013 tiotixene Drugs 0.000 description 1
- COKMIXFXJJXBQG-NRFANRHFSA-N tirofiban Chemical compound C1=CC(C[C@H](NS(=O)(=O)CCCC)C(O)=O)=CC=C1OCCCCC1CCNCC1 COKMIXFXJJXBQG-NRFANRHFSA-N 0.000 description 1
- 229960003425 tirofiban Drugs 0.000 description 1
- 239000010936 titanium Substances 0.000 description 1
- 239000004408 titanium dioxide Substances 0.000 description 1
- 229960002501 tofisopam Drugs 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 229940044693 topoisomerase inhibitor Drugs 0.000 description 1
- 231100000440 toxicity profile Toxicity 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- 229960004380 tramadol Drugs 0.000 description 1
- TVYLLZQTGLZFBW-GOEBONIOSA-N tramadol Natural products COC1=CC=CC([C@@]2(O)[C@@H](CCCC2)CN(C)C)=C1 TVYLLZQTGLZFBW-GOEBONIOSA-N 0.000 description 1
- 239000003204 tranquilizing agent Substances 0.000 description 1
- 230000002936 tranquilizing effect Effects 0.000 description 1
- 239000003558 transferase inhibitor Substances 0.000 description 1
- 238000011269 treatment regimen Methods 0.000 description 1
- 150000003626 triacylglycerols Chemical class 0.000 description 1
- 229960001288 triamterene Drugs 0.000 description 1
- 150000004654 triazenes Chemical class 0.000 description 1
- 229960003386 triazolam Drugs 0.000 description 1
- JOFWLTCLBGQGBO-UHFFFAOYSA-N triazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NN=C2CN=C1C1=CC=CC=C1Cl JOFWLTCLBGQGBO-UHFFFAOYSA-N 0.000 description 1
- CYRMSUTZVYGINF-UHFFFAOYSA-N trichlorofluoromethane Chemical compound FC(Cl)(Cl)Cl CYRMSUTZVYGINF-UHFFFAOYSA-N 0.000 description 1
- 229940029284 trichlorofluoromethane Drugs 0.000 description 1
- ZEWQUBUPAILYHI-UHFFFAOYSA-N trifluoperazine Chemical compound C1CN(C)CCN1CCCN1C2=CC(C(F)(F)F)=CC=C2SC2=CC=CC=C21 ZEWQUBUPAILYHI-UHFFFAOYSA-N 0.000 description 1
- 229960002324 trifluoperazine Drugs 0.000 description 1
- PNQBEPDZQUOCNY-UHFFFAOYSA-N trifluoroacetyl chloride Chemical compound FC(F)(F)C(Cl)=O PNQBEPDZQUOCNY-UHFFFAOYSA-N 0.000 description 1
- GPMXUUPHFNMNDH-UHFFFAOYSA-N trifluperidol Chemical compound C1CC(O)(C=2C=C(C=CC=2)C(F)(F)F)CCN1CCCC(=O)C1=CC=C(F)C=C1 GPMXUUPHFNMNDH-UHFFFAOYSA-N 0.000 description 1
- 229960002341 trifluperidol Drugs 0.000 description 1
- XSCGXQMFQXDFCW-UHFFFAOYSA-N triflupromazine Chemical compound C1=C(C(F)(F)F)C=C2N(CCCN(C)C)C3=CC=CC=C3SC2=C1 XSCGXQMFQXDFCW-UHFFFAOYSA-N 0.000 description 1
- 229960003904 triflupromazine Drugs 0.000 description 1
- UCPYLLCMEDAXFR-UHFFFAOYSA-N triphosgene Chemical compound ClC(Cl)(Cl)OC(=O)OC(Cl)(Cl)Cl UCPYLLCMEDAXFR-UHFFFAOYSA-N 0.000 description 1
- 239000003174 triple reuptake inhibitor Substances 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 229960001641 troglitazone Drugs 0.000 description 1
- GXPHKUHSUJUWKP-UHFFFAOYSA-N troglitazone Chemical compound C1CC=2C(C)=C(O)C(C)=C(C)C=2OC1(C)COC(C=C1)=CC=C1CC1SC(=O)NC1=O GXPHKUHSUJUWKP-UHFFFAOYSA-N 0.000 description 1
- GXPHKUHSUJUWKP-NTKDMRAZSA-N troglitazone Natural products C([C@@]1(OC=2C(C)=C(C(=C(C)C=2CC1)O)C)C)OC(C=C1)=CC=C1C[C@H]1SC(=O)NC1=O GXPHKUHSUJUWKP-NTKDMRAZSA-N 0.000 description 1
- 229960000281 trometamol Drugs 0.000 description 1
- 229940046728 tumor necrosis factor alpha inhibitor Drugs 0.000 description 1
- 239000002452 tumor necrosis factor alpha inhibitor Substances 0.000 description 1
- 102000003298 tumor necrosis factor receptor Human genes 0.000 description 1
- 239000005483 tyrosine kinase inhibitor Substances 0.000 description 1
- 231100000397 ulcer Toxicity 0.000 description 1
- 229960002703 undecylenic acid Drugs 0.000 description 1
- 229960005356 urokinase Drugs 0.000 description 1
- 229940005605 valeric acid Drugs 0.000 description 1
- 239000004474 valine Substances 0.000 description 1
- 229960002381 vardenafil Drugs 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 229960004688 venlafaxine Drugs 0.000 description 1
- PNVNVHUZROJLTJ-UHFFFAOYSA-N venlafaxine Chemical compound C1=CC(OC)=CC=C1C(CN(C)C)C1(O)CCCCC1 PNVNVHUZROJLTJ-UHFFFAOYSA-N 0.000 description 1
- RYJXBGGBZJGVQF-UHFFFAOYSA-N veralipride Chemical compound COC1=CC(S(N)(=O)=O)=CC(C(=O)NCC2N(CCC2)CC=C)=C1OC RYJXBGGBZJGVQF-UHFFFAOYSA-N 0.000 description 1
- 229960001968 veralipride Drugs 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 230000000007 visual effect Effects 0.000 description 1
- 229960005080 warfarin Drugs 0.000 description 1
- PJVWKTKQMONHTI-UHFFFAOYSA-N warfarin Chemical compound OC=1C2=CC=CC=C2OC(=O)C=1C(CC(=O)C)C1=CC=CC=C1 PJVWKTKQMONHTI-UHFFFAOYSA-N 0.000 description 1
- 239000003232 water-soluble binding agent Substances 0.000 description 1
- 239000002023 wood Substances 0.000 description 1
- 210000000707 wrist Anatomy 0.000 description 1
- 229940025158 xenazine Drugs 0.000 description 1
- UGZADUVQMDAIAO-UHFFFAOYSA-L zinc hydroxide Chemical compound [OH-].[OH-].[Zn+2] UGZADUVQMDAIAO-UHFFFAOYSA-L 0.000 description 1
- 229910021511 zinc hydroxide Inorganic materials 0.000 description 1
- 229940007718 zinc hydroxide Drugs 0.000 description 1
- MVWVFYHBGMAFLY-UHFFFAOYSA-N ziprasidone Chemical compound C1=CC=C2C(N3CCN(CC3)CCC3=CC=4CC(=O)NC=4C=C3Cl)=NSC2=C1 MVWVFYHBGMAFLY-UHFFFAOYSA-N 0.000 description 1
- 229960000607 ziprasidone Drugs 0.000 description 1
- 229960004496 zotepine Drugs 0.000 description 1
- HDOZVRUNCMBHFH-UHFFFAOYSA-N zotepine Chemical compound CN(C)CCOC1=CC2=CC=CC=C2SC2=CC=C(Cl)C=C12 HDOZVRUNCMBHFH-UHFFFAOYSA-N 0.000 description 1
- 229960004141 zuclopenthixol Drugs 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D455/00—Heterocyclic compounds containing quinolizine ring systems, e.g. emetine alkaloids, protoberberine; Alkylenedioxy derivatives of dibenzo [a, g] quinolizines, e.g. berberine
- C07D455/03—Heterocyclic compounds containing quinolizine ring systems, e.g. emetine alkaloids, protoberberine; Alkylenedioxy derivatives of dibenzo [a, g] quinolizines, e.g. berberine containing quinolizine ring systems directly condensed with at least one six-membered carbocyclic ring, e.g. protoberberine; Alkylenedioxy derivatives of dibenzo [a, g] quinolizines, e.g. berberine
- C07D455/04—Heterocyclic compounds containing quinolizine ring systems, e.g. emetine alkaloids, protoberberine; Alkylenedioxy derivatives of dibenzo [a, g] quinolizines, e.g. berberine containing quinolizine ring systems directly condensed with at least one six-membered carbocyclic ring, e.g. protoberberine; Alkylenedioxy derivatives of dibenzo [a, g] quinolizines, e.g. berberine containing a quinolizine ring system condensed with only one six-membered carbocyclic ring, e.g. julolidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4738—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4745—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems condensed with ring systems having nitrogen as a ring hetero atom, e.g. phenantrolines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/13—Amines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
- A61K31/19—Carboxylic acids, e.g. valproic acid
- A61K31/195—Carboxylic acids, e.g. valproic acid having an amino group
- A61K31/197—Carboxylic acids, e.g. valproic acid having an amino group the amino and the carboxyl groups being attached to the same acyclic carbon chain, e.g. gamma-aminobutyric acid [GABA], beta-alanine, epsilon-aminocaproic acid or pantothenic acid
- A61K31/198—Alpha-amino acids, e.g. alanine or edetic acid [EDTA]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4375—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a six-membered ring having nitrogen as a ring heteroatom, e.g. quinolizines, naphthyridines, berberine, vincamine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/551—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having two nitrogen atoms, e.g. dilazep
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/02—Muscle relaxants, e.g. for tetanus or cramps
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B59/00—Introduction of isotopes of elements into organic compounds ; Labelled organic compounds per se
- C07B59/002—Heterocyclic compounds
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07B—GENERAL METHODS OF ORGANIC CHEMISTRY; APPARATUS THEREFOR
- C07B2200/00—Indexing scheme relating to specific properties of organic compounds
- C07B2200/05—Isotopically modified compounds, e.g. labelled
Definitions
- the present disclosure relates to methods for treating abnormal muscular activity, more specifically to methods for treating abnormal muscular activity associated with at least one of bradykinesia, dyskinesia, and hyperkinesia.
- Movement disorders can be classified into two basic categories: those characterized by disordered or excessive movement (referred to as “hyperkinesia” or “dyskinesia”), and those that are characterized by slowness, or a lack of movement (referred to as “hypokinesia,”
- bradykinesia or "akinesia”
- An example of a “hyperkinetic” movement disorder is a tremor or a tic while Parkinson's disease can be classified as “hypokinetic,” because it is often
- Movement disorders include ataxia, corticobasal degeneration, dyskinesias
- dystonia generally, segmental, focal
- dystonia including blepharospasm, spasmodic torticollis (cervical dystonia), writer's cramp (limb dystonia), laryngeal dystonia (spasmodic dysphonia), and oromandibular dystonia, essential tremor, hereditary spastic paraplegia, Huntington' s Disease, multiple system atrophy (Shy Drager Syndrome), myoclonus, Parkinson's Disease, progressive supranuclear palsy, restless legs syndrome, Rett Syndrome, spasticity due to stroke, cerebral palsy, multiple sclerosis, spinal cord or brain injury, Sydenham's Chorea, tardive dyskinesia/dystonia, tics, Tourette's Syndrome, and Wilson's Disease.
- the inventors herein disclose new methods for assessing and treating abnormal muscular activity.
- the methods may be performed remotely and permit monitoring of a subject at home and in the community for un-biased, real-time analysis of a muscular activity disorder.
- step f treating the subject based upon the level of the subject's abnormal muscular activity as determined in step e.
- the term "and/or" when used in a list of two or more items, means that any one of the listed items can be employed by itself or in combination with any one or more of the listed items.
- the expression “A and/or B” is intended to mean either or both of A and B, i.e. A alone, B alone or A and B in combination.
- the expression “A, B and/or C” is intended to mean A alone, B alone, C alone, A and B in combination, A and C in combination, B and C in combination or A, B, and C in combination.
- abnormal refers to an activity or feature that differs from a normal activity or feature.
- abnormal muscular activity refers to muscular activity that differs from the muscular activity in a healthy subject.
- the abnormal activity may be decreased or increased in comparison to normal activity.
- An increase in muscular activity can result in excessive abnormal movements, excessive normal movements, or a combination of both.
- the term "accelerometer” is defined to include any electronics components that measure the three dimensional movement, including gyros and related products.
- processing refers to gathering, manipulating, storing, retrieving, and classifying the measured data. These steps may be performed by a microprocessor that includes one or more processing elements that are adapted to perform the recited operations.
- a processor may comprise all or part of one or more integrated circuits, firmware code, and/or software code that receive electrical signals from various sources and generate appropriate responses.
- all processing elements that comprise the processor are located together. In other embodiments, the elements of a processor may spread across multiple devices in multiple locations.
- the term "remote access unit” refers to a unit having a remote connection to the muscular activity measurement device.
- the unit may perform any of the steps of manipulating, storing, retrieving, and classifying the measured data. It may also communicate with the measurement device wirelessly.
- the remote access unit may feature a user interface to display raw or processed measured data.
- bond refers to a covalent linkage between two atoms, or two moieties when the atoms joined by the bond are considered part of larger substructure.
- a bond may be single, double, or triple unless otherwise specified.
- a dashed line between two atoms in a drawing of a molecule indicates that an additional bond may be present or absent at that position.
- disorder as used herein is intended to be generally synonymous, and is used interchangeably with, the terms “disease”, “syndrome”, and “condition” (as in medical condition), in that all reflect an abnormal condition of the human or animal body or of one of its parts that impairs normal functioning, is typically manifested by distinguishing signs and symptoms.
- treat are meant to include alleviating or abrogating a disorder or one or more of the symptoms associated with a disorder; or alleviating or eradicating the cause(s) of the disorder itself.
- treatment of a disorder is intended to include prevention.
- prevent refer to a method of delaying or precluding the onset of a disorder; and/or its attendant symptoms, barring a subject from acquiring a disorder or reducing a subject's risk of acquiring a disorder.
- terapéuticaally effective amount refers to the amount of a compound that, when administered, is sufficient to prevent development of, or alleviate to some extent, one or more of the symptoms of the disorder being treated.
- therapeutically effective amount also refers to the amount of a compound that is sufficient to elicit the biological or medical response of a cell, tissue, system, animal, or human that is being sought by a researcher, veterinarian, medical doctor, or clinician.
- subject refers to an animal, including, but not limited to, a primate (e.g., human, monkey, chimpanzee, gorilla, and the like), rodents (e.g., rats, mice, gerbils, hamsters, ferrets, and the like), lagomorphs, swine (e.g., pig, miniature pig), equine, canine, feline, and the like.
- a primate e.g., human, monkey, chimpanzee, gorilla, and the like
- rodents e.g., rats, mice, gerbils, hamsters, ferrets, and the like
- lagomorphs e.g., pig, miniature pig
- swine e.g., pig, miniature pig
- equine canine
- feline feline
- combination therapy means the administration of two or more therapeutic agents to treat a therapeutic disorder described in the present disclosure. Such administration encompasses co-administration of these therapeutic agents in a simultaneous manner, such as in a single capsule having a fixed ratio of active ingredients or in multiple, separate capsules for each active ingredient. In addition, such administration also encompasses use of each type of therapeutic agent in a sequential manner. In either case, the treatment regimen will provide beneficial
- VMAT2 refers to vesicular monoamine transporter 2, an integral membrane protein that acts to transport monoamines—particularly neurotransmitters such as dopamine, norepinephrine, serotonin, and histamine— from cellular cytosol into synaptic vesicles.
- VMAT2-mediated disorder refers to a disorder that is characterized by abnormal VMAT2 activity.
- a VMAT2-mediated disorder may be completely or partially mediated by modulating VMAT2.
- a VMAT2-mediated disorder is one in which inhibition of VMAT2 results in some effect on the underlying disorder e.g., administration of a VMAT2 inhibitor results in some improvement in at least some of the patients being treated.
- VMAT2 inhibitor refers to the ability of a compound disclosed herein to alter the function of VMAT2.
- a VMAT2 inhibitor may block or reduce the activity of VMAT2 by forming a reversible or irreversible covalent bond between the inhibitor and VMAT2 or through formation of a noncovalently bound complex. Such inhibition may be manifest only in particular cell types or may be contingent on a particular biological event.
- VMAT2 inhibitor also refers to altering the function of VMAT2 by decreasing the probability that a complex forms between a VMAT2 and a natural substrate
- Tetrabenazine (Nitoman, Xenazine, Ro 1-9569), 1,3,4,6,7,1 lb-Hexahydro- 9,10- dimethoxy-3-(2-methylpropyl)-2H-benzo[a]quinoline, is a vesicular monoamine transporter 2 (VMAT2) inhibitor. Tetrabenazine is commonly prescribed for the treatment of Huntington's disease (Savani et al., Neurology 2007, 68(10), 797; and Kenney et al., Expert Review of
- Tetrabenazine [029] In vivo, tetrabenazine is rapidly and extensively metabolized to its reduced form, dihydrotetrabenazine (CAS # 3466-75-9), 1,3,4,6,7,1 lb-hexahydro-9,10-dimethoxy-3-(2- methylpropyl)-2H-benzo[a]quinolizin-2-ol. Dihydrotetrabenazine is a VMAT2 inhibitor and an active metabolite of tetrabenazine.
- Dihydrotetrabenazine is currently under investigation for the treatment of Huntington's disease, hemiballismus, senile chorea, tic disorders, tardive dyskinesia, dystonia, Tourette's syndrome, depression, cancer, rheumatoid arthritis, psychosis, multiple sclerosis, and asthma.
- NBI-98854 (CAS # 1025504-59-9), (S)-(2R,3R,l lbR)-3-isobutyl-9,10-dimethoxy- 2,3,4,6,7,1 lb-hexahydro-lH-pyrido[2,l-a]isoquinolin-2-yl 2-amino-3-methylbutanoate, is a VMAT2 inhibitor.
- NBI-98854 is currently under investigation for the treatment of movement disorders including tardive dyskinesia. WO 2008058261; WO 2011153157; and US 8,039,627.
- NBI-98854 a valine ester of (+)-a-dihydrotetrabenazine, in humans is slowly hydrolyzed to (+)- a-dihydrotetrabenazine which is an active metabolite of tetrabenazine.
- d6-tetrabenazine and/or d6-dihydrotetrabenazine are metabolites of d6-tetrabenazine and/or d6-dihydrotetrabenazine.
- d6-Tetrabenazine and d 6 - dihydrotetrabenazine, as well as the Ml and M4 metabolites, are VMAT2 inhibitors.
- d 6 - Tetrabenazine and d6-dihydrotetrabenazine are currently under investigation for the treatment of Huntington's disease and other VMAT2-mediated disorders. US 8,524,733, US 20100130480, and US 20120003330.
- Tetrabenazine, dihydrotetrabenazine, and NBI-98854 are subject to extensive oxidative metabolism, including O-demethylation of the methoxy groups, as well as
- tetrabenazine, dihydrotetrabenazine, and/or NBI-98854 Adverse effects associated with the administration of tetrabenazine, dihydrotetrabenazine, and/or NBI-98854 include neuroleptic malignant syndrome, drowsiness, fatigue, nervousness, anxiety, insomnia, agitation, confusion, orthostatic hypotension, nausea, dizziness, depression, and Parkinsonism.
- Tetrabenazine, dyhydrotetrabenazine, and NBI-98854 are VMAT2 inhibitors.
- the carbon-hydrogen bonds of tetrabenazine, dyhydrotetrabenazine, and NBI-98854 contain a naturally occurring distribution of hydrogen isotopes, namely l H or protium (about 99.9844%), 2 H or deuterium (about 0.0156%), and 3 H or tritium (in the range between about 0.5 and 67 tritium atoms per 10 18 protium atoms).
- Increased levels of deuterium incorporation may produce a detectable Deuterium Kinetic Isotope Effect (DKIE) that could affect the pharmacokinetic, pharmacologic and/or toxicologic profiles of tetrabenazine, dyhydrotetrabenazine, and/or NBI- 98854 in comparison with tetrabenazine, dyhydrotetrabenazine, and/or NBI-98854 having naturally occurring levels of deuterium.
- DKIE Deuterium Kinetic Isotope Effect
- tetrabenazine, dyhydrotetrabenazine, and/or NBI-98854 are metabolized in humans at the isobutyl and methoxy groups.
- the current approach has the potential to prevent metabolism at these sites.
- Other sites on the molecule may also undergo transformations leading to metabolites with as-yet-unknown pharmacology/toxicology. Limiting the production of these metabolites has the potential to decrease the danger of the administration of such drugs and may even allow increased dosage and/or increased efficacy. All of these transformations can occur through polymorphically-expressed enzymes, exacerbating interpatient variability.
- Various deuteration patterns can be used to (a) reduce or eliminate unwanted metabolites, (b) increase the half-life of the parent drug, (c) decrease the number of doses needed to achieve a desired effect, (d) decrease the amount of a dose needed to achieve a desired effect, (e) increase the formation of active metabolites, if any are formed, (f) decrease the production of deleterious metabolites in specific tissues, and/or (g) create a more effective drug and/or a safer drug for polypharmacy, whether the polypharmacy be intentional or not.
- the deuteration approach has the strong potential to slow the metabolism of tetrabenazine, dyhydrotetrabenazine, and/or NBI-98854 and attenuate interpatient variability.
- the animal body expresses various enzymes, such as the cytochrome P450 enzymes (CYPs), esterases, proteases, reductases, dehydrogenases, and monoamine oxidases, to react with and convert these foreign substances to more polar intermediates or metabolites for renal excretion.
- CYPs cytochrome P450 enzymes
- esterases proteases
- reductases reductases
- dehydrogenases dehydrogenases
- monoamine oxidases monoamine oxidases
- Such metabolic reactions frequently involve the oxidation of a carbon-hydrogen (C-H) bond to either a carbon- oxygen (C-O) or a carbon-carbon (C-C) -bond.
- C-H carbon-hydrogen
- C-O carbon- oxygen
- C-C carbon-carbon
- the resultant metabolites may be stable or unstable under physiological conditions, and can have substantially different pharmacokinetic, pharmacodynamic, and acute and long-term toxicity profiles relative to
- the transition state in a reaction is a short lived state along the reaction pathway during which the original bonds have stretched to their limit.
- the activation energy Eact for a reaction is the energy required to reach the transition state of that reaction. Once the transition state is reached, the molecules can either revert to the original reactants, or form new bonds giving rise to reaction products.
- a catalyst facilitates a reaction process by lowering the activation energy leading to a transition state. Enzymes are examples of biological catalysts.
- Carbon-hydrogen bond strength is directly proportional to the absolute value of the ground-state vibrational energy of the bond. This vibrational energy depends on the mass of the atoms that form the bond, and increases as the mass of one or both of the atoms making the bond increases. Since deuterium (D) has twice the mass of protium ( l H), a C-D bond is stronger than the corresponding C- 1 ! bond. If a C- 1 ! bond is broken during a rate-determining step in a chemical reaction (i.e. the step with the highest transition state energy), then substituting a deuterium for that protium will cause a decrease in the reaction rate. This phenomenon is known as the Deuterium Kinetic Isotope Effect (DKIE).
- DKIE Deuterium Kinetic Isotope Effect
- the magnitude of the DKIE can be expressed as the ratio between the rates of a given reaction in which a C- 1 ! bond is broken, and the same reaction where deuterium is substituted for protium.
- the DKIE can range from about 1 (no isotope effect) to very large numbers, such as 50 or more. Substitution of tritium for hydrogen results in yet a stronger bond than deuterium and gives numerically larger isotope effects
- Deuterium ( 2 H or D) is a stable and non-radioactive isotope of hydrogen which has approximately twice the mass of protium ( ⁇ ⁇ ), the most common isotope of hydrogen.
- Deuterium oxide looks and tastes like H 2 0, but has different physical properties.
- PD pharmacodynamics
- toxicity profiles has been demonstrated previously with some classes of drugs.
- the DKIE was used to decrease the hepatotoxicity of halothane, presumably by limiting the production of reactive species such as trifluoroacetyl chloride.
- Metabolic switching occurs when xenogens, sequestered by Phase I enzymes, bind transiently and re-bind in a variety of conformations prior to the chemical reaction (e.g., oxidation). Metabolic switching is enabled by the relatively vast size of binding pockets in many Phase I enzymes and the promiscuous nature of many metabolic reactions. Metabolic switching can lead to different proportions of known metabolites as well as altogether new metabolites. This new metabolic profile may impart more or less toxicity. Such pitfalls are non-obvious and are not predictable a priori for any drug class.
- Tetrabenazine, dyhydrotetrabenazine, and NBI-98854 are VMAT2 inhibitors.
- the carbon-hydrogen bonds of tetrabenazine, dyhydrotetrabenazine, and NBI-98854 contain a naturally occurring distribution of hydrogen isotopes, namely X H or protium (about 99.9844%), 2 H or deuterium (about 0.0156%), and 3 H or tritium (in the range between about 0.5 and 67 tritium atoms per 10 18 protium atoms).
- Increased levels of deuterium incorporation may produce a detectable Deuterium Kinetic Isotope Effect (DKIE) that could affect the pharmacokinetic, pharmacologic and/or toxicologic profiles of tetrabenazine, dyhydrotetrabenazine, and/or NBI- 98854 in comparison with tetrabenazine, dyhydrotetrabenazine, and/or NBI-98854 having naturally occurring levels of deuterium.
- DKIE Deuterium Kinetic Isotope Effect
- tetrabenazine, dyhydrotetrabenazine, and/or NBI-98854 are metabolized in humans at the isobutyl and methoxy groups.
- the current approach has the potential to prevent metabolism at these sites.
- Other sites on the molecule may also undergo transformations leading to metabolites with as-yet-unknown pharmacology/toxicology. Limiting the production of these metabolites has the potential to decrease the danger of the administration of such drugs and may even allow increased dosage and/or increased efficacy. All of these transformations can occur through polymorphically-expressed enzymes, exacerbating interpatient variability.
- Various deuteration patterns can be used to (a) reduce or eliminate unwanted metabolites, (b) increase the half-life of the parent drug, (c) decrease the number of doses needed to achieve a desired effect, (d) decrease the amount of a dose needed to achieve a desired effect, (e) increase the formation of active metabolites, if any are formed, (f) decrease the production of deleterious metabolites in specific tissues, and/or (g) create a more effective drug and/or a safer drug for polypharmacy, whether the polypharmacy be intentional or not.
- the deuteration approach has the strong potential to slow the metabolism of tetrabenazine, dyhydrotetrabenazine, and/or NBI-98854 and attenuate interpatient variability.
- R1-R27 are independently selected from the group consisting of hydrogen and deuterium
- At least one of R1-R27 is deuterium.
- Formula I can include a single enantiomer, a mixture of the (+)-enantiomer and the (-)-enantiomer, a mixture of about 90% or more by weight of the (-)- enantiomer and about 10% or less by weight of the (+) -enantiomer, a mixture of about 90% or more by weight of the (-i-)-enantiomer and about 10% or less by weight of the (-)-enantiomer, an individual diastereomer, or a mixture of diastereomers thereof.
- R28-R46 and R48-R56 are independently selected from the group consisting of hydrogen and deuterium;
- R47 is selected from the group consisting of hydrogen, deuterium, -C(0)0-alkyl and - C(0)-Ci-6alkyl, or a group cleavable under physiological conditions, wherein said alkyl or Ci-6alkyl is optionally substituted with one or more substituents selected from the group consisting of -NH-C(NH)NH2, -CO2H, -C0 2 alkyl, -SH, -C(0)NH 2 , -NH 2 , phenyl, -OH, 4-hydroxyphenyl, imidazolyl, and indolyl, and any R46 substituent is further optionally substituted with deuterium; and
- At least one of R28-R56 is deuterium or contains deuterium.
- the compounds of Formula II have alpha stereochemistry.
- the compounds of Formula II have beta stereochemistry.
- the compounds of Formula II are a mixture of alpha and beta stereoisomers.
- the ratio of alpha/beta stereoisomers is at least 100: 1, at least 50: 1, at least 20: 1, at least 10: 1, at least 5: 1, at least 4: 1, at least 3: 1, or at least 2: 1.
- the ratio of beta/alpha stereoisomers is at least 100: 1, at least 50: 1, at least 20: 1, at least 10: 1, at least 5: 1, at least 4: 1, at least 3: 1, or at least 2: 1.
- R50-R56 are deuterium, at least one of R1-R49 is deuterium.
- compounds have structural Formula
- R57-R83 are independently selected from the group consisting of hydrogen and deuterium
- At least one of R57-R83 is deuterium.
- R84-R110 are independently selected from the group consisting of hydrogen and deuterium
- At least one of R84-R110 is deuterium.
- Certain compounds disclosed herein may possess useful VMAT2 inhibiting activity, and may be used in the treatment or prophylaxis of a disorder in which VMAT2 plays an active role.
- certain embodiments also provide pharmaceutical compositions comprising one or more compounds disclosed herein together with a pharmaceutically acceptable carrier, as well as methods of making and using the compounds and compositions.
- Certain embodiments provide methods for inhibiting VMAT2.
- Other embodiments provide methods for treating a VMAT2- mediated disorder in a patient in need of such treatment, comprising administering to said patient a therapeutically effective amount of a compound or composition according to the present invention.
- Also provided is the use of certain compounds disclosed herein for use in the manufacture of a medicament for the prevention or treatment of a disorder ameliorated by the inhibition of VMAT2.
- the compounds as disclosed herein may also contain less prevalent isotopes for other elements, including, but not limited to, 13 C or 14 C for carbon, 33 S, 34 S, or 36 S for sulfur, 15 N for nitrogen, and 17 0 or 18 0 for oxygen.
- the compound disclosed herein may expose a patient to a maximum of about 0.000005% D 2 0 or about 0.00001 DHO, assuming that all of the C-D bonds in the compound as disclosed herein are metabolized and released as D 2 0 or DHO.
- the levels of D 2 0 shown to cause toxicity in animals is much greater than even the maximum limit of exposure caused by administration of the deuterium enriched compound as disclosed herein.
- the deuterium-enriched compound disclosed herein should not cause any additional toxicity due to the formation of D 2 0 or DHO upon drug metabolism.
- the deuterated compounds disclosed herein maintain the beneficial aspects of the corresponding non-isotopically enriched molecules while substantially increasing the maximum tolerated dose, decreasing toxicity, increasing the half-life (Ti/ 2 ), lowering the maximum plasma concentration (C ma x) of the minimum efficacious dose (MED), lowering the efficacious dose and thus decreasing the non-mechanism-related toxicity, and/or lowering the probability of drug-drug interactions.
- deuterium enrichment refers to the percentage of incorporation of deuterium at a given position in a molecule in the place of hydrogen. For example, deuterium enrichment of 1% at a given position means that 1% of molecules in a given sample contain deuterium at the specified position. Because the naturally occurring distribution of deuterium is about 0.0156%, deuterium enrichment at any position in a compound synthesized using non- enriched starting materials is about 0.0156%. The deuterium enrichment can be determined using conventional analytical methods known to one of ordinary skill in the art, including mass spectrometry and nuclear magnetic resonance spectroscopy.
- deuterium when used to describe a given position in a molecule such as Ri-Rno or the symbol "D", when used to represent a given position in a drawing of a molecular structure, means that the specified position is enriched with deuterium above the naturally occurring distribution of deuterium.
- deuterium enrichment is no less than about 1%, in another no less than about 5%, in another no less than about 10%, in another no less than about 20%, in another no less than about 50%, in another no less than about 70%, in another no less than about 80%, in another no less than about 90%, or in another no less than about 98% of deuterium at the specified position.
- isotopic enrichment refers to the percentage of incorporation of a less prevalent isotope of an element at a given position in a molecule in the place of the more prevalent isotope of the element.
- non-isotopically enriched refers to a molecule in which the percentages of the various isotopes are substantially the same as the naturally occurring percentages.
- alpha-dihydrotetrabenazine refers to either of the dihydrotetrabenazine stereoisomers having the structural formulas shown below, or a mixture thereof:
- alpha or alpha stereoisomer or the symbol “a” as applied to a compound of Formula II refers to either of the stereoisomers of compounds of Formula II shown below, or a mixture thereof
- beta-dihydrotetrabenazine refers to either of the dihydrotetrabenazine stereoisomers having the structural formulas shown below, or a mixture thereof:
- (+)-beta-dihydrotetrabenazine (-)-beta-dihydrotetrabenazine.
- beta or “beta stereoisomer” or the symbol “ ⁇ ” as applied to a compound of Formula II refers to either of the stereoisomers of compounds of Formula II shown below, or a mixture thereof:
- 3S,1 IbS enantiomer or the term “3R,1 IbR enantiomer” refers to either of the d6-tetrabenazine M4 metabolite stereoisomers having the structural formulas shown below:
- a chemical structure may be drawn as either the 3S,1 IbS enantiomer or the 3R,1 IbR enantiomer, but the text of the specification may indicate that the 3S,1 IbS enantiomer, the 3R,1 IbR enantiomer, a racemic mixture thereof, or all of the foregoing may be intended to be described.
- mixture of diastereomers refers to either of the d6-tetrabenazine Ml metabolite stereoisomers having the structural formulas shown below:
- a chemical structure may be drawn as one of the diastereomers shown above, but the text of the specification may indicate that each individual diastereomer or a mixture thereof, or all of the foregoing may be intended to be described.
- mixture of diastereomers refers to a mixture of the stereoisomers of com ounds of Formula IV shown below:
- the present invention provides a method of treating abnormal muscular activity in a subject in need thereof comprising the steps of:
- At least one accelerometer is used to detect muscular activity.
- Accelerometers are well known in the art.
- An accelerometer may sense changes in velocity directly through interrogation or receipt of signal from an inertial transducer.
- An accelerometer may also calculate changes in velocity from data received from position sensing transducers.
- Accelerometers are often electromechanical devices and can measure the static gravitational force or dynamic forces caused by changes in speed and/or direction (changes in velocity).
- Accelerometers can utilize the piezoelectric effect and can detect acceleration in three orthogonal axis, as well as rotation about the axis. Accelerometers have been utilized in medical devices as well - see for example issued US patent serial numbers 5,233,984 and US 5,593,431.
- Multiple accelerometers may detect activity or motion at separate locations on a subject. For example, as a subject moves, an accelerometer located on the torso of a subject detects the motion of the torso, and an accelerometer located on the head detects the motion of the head of subject. In the case in which accelerometers comprise multi-axis accelerometers, the accelerometers detect the motion of the head and torso in terms of magnitude and direction. The accelerometers may generate signals as a function of the detected motion, and a processor may compare the motion of the head relative to the torso. The accelerometers may be located elsewhere on a subject, such as a limb.
- the frame of reference is another accelerometer.
- This new frame of reference from the perspective of another accelerometer allows the processor to ignore motions that are experienced by all portions of the subject, thus making it easier to detect, for example, motions that represent conditions of abnormal muscular activity.
- the processor may ignore motion that is experienced by both the accelerometers. For the example, if a subject experiences a bumpy plane ride, both of the activity sensors experience the motion due to the turbulence.
- these detected motions When compared to one another (e.g., subtracted) these detected motions may be substantially eliminated, leaving only the motions of the accelerometers that are different, such as the motions caused by a tremor or a seizure. In this manner, the new frame of reference provided by analyzing relative motion allows for more accurate detection of movement disorders.
- a processor may process the measured muscular activity data to distinguish between normal muscular activity and abnormal muscular activity in the subject.
- the processor may compare the magnitudes of the signals generated by the two or more accelerometers, the directionality of the signals generated by the two or more accelerometers, the frequency of signals generated by accelerometers or a combination thereof to calculate the relative motion.
- the processor may then analyze the relative motion to detect a condition of a movement disorder. For example, a processor may analyze a plurality of relative motion measurements computed over a window of time, e.g., over 15-20 relative motion measurements.
- the processor may detect abnormal muscular activity, such as a tremor or seizure, when the magnitude, frequency and/or the directionality of the relative motion measurements exceed a threshold for a consecutive number of measurements.
- the processor may detect a condition of the movement disorder when relative motion is detected between the two sensors for a threshold number of times over a period.
- the processor may compare the relative motion over a window of time to one or more pre-defined patterns, and detect a condition of abnormal muscular activity when the relative motion measurements over the window of time substantially match one of the predefined patterns.
- the processor may determine the pre-defined patterns based on relative motion measurements computed during previous episodes, e.g., previous tremors or seizures, of a subject. Pre-defined patterns may also be determined based on sensor signals obtained from a population of subjects and/or clinical subjects during symptomatic movement episodes, e.g., tremors or seizures.
- the processor may employ or include a neural network for identifying symptomatic movement. The neural network may be trained based on prior patient episodes and/or episodes gathered from other patients/subjects.
- the processor may compute the pre-defined patterns based on a basic body model.
- the body model may, for example, represent information regarding a subject (e.g., height, weight and age), the position of accelerometers within the subject, or the like.
- the processor may, for example, receive the body model information from a physician or subject via one of programmers e.g., during initial configuration of the device.
- programmers may compute look-up tables based on the body model.
- the processor may use the body model information or other information generated from the body model to compute relative motions between two or more accelerometers or to analyze the relative motion to identify abnormal muscular activity.
- the processor may use a variety of algorithms based on kinesiology and the biomechanics of the human body to predict relative motion measurements that are indicative of a condition of a muscular disorder for the particular subject. Such computations may account for other variables such as age, weight and height of patient.
- a remote access device may receive the signal generated by accelerometers and compare the signals to compute the relative motion between the
- the remote access device may analyze the relative motion using the techniques described above to determine whether the relative motion is indicative of a symptom of the abnormal muscular activity.
- the magnitude, duration, and frequency of the abnormal muscular activity may be determined using the methods described above.
- the present method also anticipates methods that determine whether abnormal muscular activity occurs at all or occurs above a threshold (e.g., a control threshold).
- a threshold e.g., a control threshold.
- the method may provide a "yes or no” result without necessarily providing quantification of abnormal muscular activity is within the scope of the present disclosure.
- the method may involve quantitative or qualitative assessment of abnormal muscular activity.
- a subject is determined to have a high level of abnormal muscular activity, the subject may be treated to reduce the level of abnormal muscular activity. Treating the subject may include administering a therapeutically effective amount of a therapeutic agent to the subject.
- the dosage amount and frequency of the therapeutic agent may be adjusted based upon the magnitude, duration, and frequency of the determined level of abnormal muscular activity.
- the amounts and frequencies of dosage may be adjusted to minimize any side effects from the therapeutic agent.
- this method may be used to monitor abnormal muscular activity associated with a therapy's unwanted side effect, and treatment adjusted based upon the level of the unwanted side effect.
- the abnormal muscular activity is associated with at least one of bradykinesia, dyskinesia, and hyperkinesia.
- the abnormal muscular activity is associated with Huntington's disease.
- the abnormal muscular activity is associated with at least one of bradykinesia, dyskinesia, and hyperkinesia.
- the abnormal muscular activity is associated with Huntington's disease.
- treating the subject may include administering a therapeutically effective amount of tetrabenazine or its metabolites.
- treating the subject may include administering a therapeutically effective amount of a deuterium enriched tetrabenazine analogue as described herein.
- release controlling excipient refers to an excipient whose primary function is to modify the duration or place of release of the active substance from a dosage form as compared with a conventional immediate release dosage form.
- nonrelease controlling excipient refers to an excipient whose primary function do not include modifying the duration or place of release of the active substance from a dosage form as compared with a conventional immediate release dosage form.
- prodrug refers to a compound functional derivative of the compound as disclosed herein and is readily convertible into the parent compound in vivo. Prodrugs are often useful because, in some situations, they may be easier to administer than the parent compound. They may, for instance, be bioavailable by oral administration whereas the parent compound is not. The prodrug may also have enhanced solubility in pharmaceutical compositions over the parent compound. A prodrug may be converted into the parent drug by various mechanisms, including enzymatic processes and metabolic hydrolysis. See Harper, Progress in Drug Research 1962, 4, 221-294; Morozowich et al. in "Design of Biopharmaceutical Properties through Prodrugs and Analogs," Roche Ed., APHA Acad. Pharm. Sci.
- the compounds disclosed herein can exist as therapeutically acceptable salts.
- the term "therapeutically acceptable salt,” as used herein, represents salts or zwitterionic forms of the compounds disclosed herein which are therapeutically acceptable as defined herein.
- the salts can be prepared during the final isolation and purification of the compounds or separately by reacting the appropriate compound with a suitable acid or base.
- Therapeutically acceptable salts include acid and basic addition salts.
- Suitable acids for use in the preparation of pharmaceutically acceptable salts include, but are not limited to, acetic acid, 2,2-dichloroacetic acid, acylated amino acids, adipic acid, alginic acid, ascorbic acid, L-aspartic acid, benzenesulfonic acid, benzoic acid, 4- acetamidobenzoic acid, boric acid, (+)-camphoric acid, camphorsulfonic acid, (+)-(lS)-camphor- 10-sulfonic acid, capric acid, caproic acid, caprylic acid, cinnamic acid, citric acid, cyclamic acid, cyclohexanesulfamic acid, dodecylsulfuric acid, ethane- 1,2-disulfonic acid, ethanesulfonic acid, 2-hydroxy-ethanesulfonic acid, formic acid, fumaric acid, galactaric acid, gentisic acid, glucohe
- Suitable bases for use in the preparation of pharmaceutically acceptable salts including, but not limited to, inorganic bases, such as magnesium hydroxide, calcium hydroxide, potassium hydroxide, zinc hydroxide, or sodium hydroxide; and organic bases, such as primary, secondary, tertiary, and quaternary, aliphatic and aromatic amines, including L-arginine, benethamine, benzathine, choline, deanol, diethanolamine, diethylamine, dimethylamine, dipropylamine, diisopropylamine, 2-(diethylamino)-ethanol, ethanolamine, ethylamine, ethylenediamine, isopropylamine, N-methyl-glucamine, hydrabamine, lH-imidazole, L-lysine, morpholine, 4-(2-hydroxyethyl)-morpholine, methylamine, piperidine, piperazine, propylamine, pyrrolidine, l-
- compositions which comprise one or more of certain compounds disclosed herein, or one or more pharmaceutically acceptable salts, prodrugs, or solvates thereof, together with one or more pharmaceutically acceptable carriers thereof and optionally one or more other therapeutic ingredients.
- pharmaceutical compositions which comprise one or more of certain compounds disclosed herein, or one or more pharmaceutically acceptable salts, prodrugs, or solvates thereof, together with one or more pharmaceutically acceptable carriers thereof and optionally one or more other therapeutic ingredients.
- Proper formulation is dependent upon the route of administration chosen. Any of the well-known techniques, carriers, and excipients may be used as suitable and as understood in the art; e.g., in Remington's
- compositions disclosed herein may be any pharmaceutical compositions disclosed herein.
- compositions may also be formulated as a modified release dosage form, including delayed-, extended-, prolonged-, sustained-, pulsatile-, controlled-, accelerated- and fast-, targeted-, programmed-release, and gastric retention dosage forms.
- dosage forms can be prepared according to conventional methods and techniques known to those skilled in the art (see, Remington: The Science and Practice of Pharmacy, supra; Modified- Release Drug Delivery Technology, Rathbone et al., Eds., Drugs and the Pharmaceutical Science, Marcel Dekker, Inc., New York, NY, 2002; Vol. 126).
- compositions include those suitable for oral, parenteral (including subcutaneous, intradermal, intramuscular, intravenous, intraarticular, and intramedullary), intraperitoneal, transmucosal, transdermal, rectal and topical (including dermal, buccal, sublingual and intraocular) administration although the most suitable route may depend upon for example the condition and disorder of the recipient.
- parenteral including subcutaneous, intradermal, intramuscular, intravenous, intraarticular, and intramedullary
- intraperitoneal including transmucosal, transdermal, rectal and topical (including dermal, buccal, sublingual and intraocular) administration although the most suitable route may depend upon for example the condition and disorder of the recipient.
- the compositions may conveniently be presented in unit dosage form and may be prepared by any of the methods well known in the art of pharmacy. Typically, these methods include the step of bringing into association a compound of the subject invention or a pharmaceutically salt, prodrug, or solvate thereof ("active ingredient”) with the
- compositions are prepared by uniformly and intimately bringing into association the active ingredient with liquid carriers or finely divided solid carriers or both and then, if necessary, shaping the product into the desired formulation.
- formulations of the compounds disclosed herein suitable for oral administration may be presented as discrete units such as capsules, cachets or tablets each containing a
- the active ingredient may also be presented as a bolus, electuary or paste.
- compositions which can be used orally include tablets, push fit capsules made of gelatin, as well as soft, sealed capsules made of gelatin and a plasticizer, such as glycerol or sorbitol. Tablets may be made by compression or molding, optionally with one or more accessory ingredients. Compressed tablets may be prepared by compressing in a suitable machine the active ingredient in a free-flowing form such as a powder or granules, optionally mixed with binders, inert diluents, or lubricating, surface active or dispersing agents. Molded tablets may be made by molding in a suitable machine a mixture of the powdered compound moistened with an inert liquid diluent.
- the tablets may optionally be coated or scored and may be formulated to provide slow or controlled release of the active ingredient therein. All formulations for oral administration should be in dosages suitable for such administration.
- the push fit capsules can contain the active ingredients in admixture with filler such as lactose, binders such as starches, and/or lubricants such as talc or magnesium stearate and, optionally, stabilizers.
- the active compounds may be dissolved or suspended in suitable liquids, such as fatty oils, liquid paraffin, or liquid polyethylene glycols.
- suitable liquids such as fatty oils, liquid paraffin, or liquid polyethylene glycols.
- stabilizers may be added. Dragee cores are provided with suitable coatings.
- concentrated sugar solutions may be used, which may optionally contain gum arabic, talc, polyvinyl pyrrolidone, carbopol gel, polyethylene glycol, and/or titanium dioxide, lacquer solutions, and suitable organic solvents or solvent mixtures.
- Dyestuffs or pigments may be added to the tablets or dragee coatings for identification or to characterize different combinations of active compound doses.
- the compounds may be formulated for parenteral administration by injection, e.g., by bolus injection or continuous infusion.
- Formulations for injection may be presented in unit dosage form, e.g., in ampoules or in multi dose containers, with an added preservative.
- the compositions may take such forms as suspensions, solutions or emulsions in oily or aqueous vehicles, and may contain formulatory agents such as suspending, stabilizing and/or dispersing agents.
- the formulations may be presented in unit-dose or multi-dose containers, for example sealed ampoules and vials, and may be stored in powder form or in a freeze-dried (lyophilized) condition requiring only the addition of the sterile liquid carrier, for example, saline or sterile pyrogen-free water, immediately prior to use.
- sterile liquid carrier for example, saline or sterile pyrogen-free water
- Extemporaneous injection solutions and suspensions may be prepared from sterile powders, granules and tablets of the kind previously described.
- Formulations for parenteral administration include aqueous and non-aqueous (oily) sterile injection solutions of the active compounds which may contain antioxidants, buffers, bacteriostats and solutes which render the formulation isotonic with the blood of the intended recipient; and aqueous and non-aqueous sterile suspensions which may include suspending agents and thickening agents.
- Suitable lipophilic solvents or vehicles include fatty oils such as sesame oil, or synthetic fatty acid esters, such as ethyl oleate or triglycerides, or liposomes.
- Aqueous injection suspensions may contain substances which increase the viscosity of the suspension, such as sodium carboxymethyl cellulose, sorbitol, or dextran.
- the suspension may also contain suitable stabilizers or agents which increase the solubility of the compounds to allow for the preparation of highly concentrated solutions.
- the compounds may also be formulated as a depot preparation. Such long acting formulations may be administered by implantation (for example subcutaneously or intramuscularly) or by intramuscular injection.
- the compounds may be formulated with suitable polymeric or hydrophobic materials (for example, as an emulsion in an acceptable oil) or ion exchange resins, or as sparingly soluble derivatives, for example, as a sparingly soluble salt.
- compositions may take the form of tablets, lozenges, pastilles, or gels formulated in conventional manner.
- Such compositions may comprise the active ingredient in a flavored basis such as sucrose and acacia or tragacanth.
- the compounds may also be formulated in rectal compositions such as suppositories or retention enemas, e.g., containing conventional suppository bases such as cocoa butter, polyethylene glycol, or other glycerides.
- Certain compounds disclosed herein may be administered topically, that is by non- systemic administration. This includes the application of a compound disclosed herein externally to the epidermis or the buccal cavity and the instillation of such a compound into the ear, eye and nose, such that the compound does not significantly enter the blood stream.
- systemic administration refers to oral, intravenous, intraperitoneal and intramuscular administration.
- Formulations suitable for topical administration include liquid or semi-liquid preparations suitable for penetration through the skin to the site of inflammation such as gels, liniments, lotions, creams, ointments or pastes, and drops suitable for administration to the eye, ear or nose.
- compounds may be delivered from an insufflator, nebulizer pressurized packs or other convenient means of delivering an aerosol spray.
- Pressurized packs may comprise a suitable propellant such as dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane, carbon dioxide or other suitable gas.
- a suitable propellant such as dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane, carbon dioxide or other suitable gas.
- the dosage unit may be determined by providing a valve to deliver a metered amount.
- the compounds according to the invention may take the form of a dry powder composition, for example a powder mix of the compound and a suitable powder base such as lactose or starch.
- the powder composition may be presented in unit dosage form, in for example, capsules, cartridges, gelatin or blister packs from which the powder may be administered with the aid of an inhalator or insufflator.
- an extended-release pharmaceutical formulation comprising, in a solid dosage form for oral delivery of between about 100 mg and about 1 g total weight:
- the diluent or diluents are chosen from mannitol, lactose, and microcrystalline cellulose; the binder is a polyvinylpyrrolidone; and the surfactant is a polysorbate.
- the extended-release pharmaceutical formulation comprises between about 2.5% and about 11% of a compound as disclosed herein.
- the extended-release pharmaceutical formulation comprises: between about 60% and about 70% mannitol or lactose; between about 15% and about 25% microcrystalline cellulose
- the extended-release pharmaceutical formulation comprises: between about 4% and about 9% of a compound as disclosed herein;
- an extended-release pharmaceutical formulation comprising, in a solid dosage form for oral delivery of between about 100 mg and about 1 g total weight:
- sustained-release polymer between about 5% and about 20% of sustained-release polymer
- the extended-release pharmaceutical formulation comprises: between about 5% and about 15% of one or more spray-dried mannitol or spray-dried lactose;
- sustained-release polymer between about 5% and about 20% of sustained-release polymer; and between about 0.5 and about 2% of a magnesium stearate.
- the sustained-release polymer is chosen from a polyvinyl acetate-polyvinylpyrrolidone mixture and a poly(ethylene oxide) polymer.
- the sustained-release polymer is chosen from Kollidon® SR, POLYOX® N60K, and Carbopol®.
- the sustained-release polymer is Kollidon® SR.
- the sustained-release polymer is POLYOX® N60K.
- the sustained-release polymer is Carbopol®.
- the extended-release pharmaceutical formulation comprises from about 5 mg to about 100 mg of a compound as disclosed herein.
- the compounds disclosed herein can be formulated as extended-release pharmaceutical formulations as described in U.S. Patent Application No.
- Preferred unit dosage formulations are those containing an effective dose, as herein below recited, or an appropriate fraction thereof, of the active ingredient.
- Compounds may be administered orally or via injection at a dose of from 0.1 to 500 mg/kg per day.
- the dose range for adult humans is generally from 5 mg to 2 g/day.
- Tablets or other forms of presentation provided in discrete units may conveniently contain an amount of one or more compounds which is effective at such dosage or as a multiple of the same, for instance, units containing 5 mg to 500 mg, usually around 10 mg to 200 mg.
- the amount of active ingredient that may be combined with the carrier materials to produce a single dosage form will vary depending upon the host treated and the particular mode of administration.
- the compounds can be administered in various modes, e.g. orally, topically, or by injection.
- the precise amount of compound administered to a patient will be the responsibility of the attendant physician.
- the specific dose level for any particular patient will depend upon a variety of factors including the activity of the specific compound employed, the age, body weight, general health, sex, diets, time of administration, route of administration, rate of excretion, drug combination, the precise disorder being treated, and the severity of the disorder being treated. Also, the route of administration may vary depending on the disorder and its severity.
- the administration of the compounds may be administered chronically, that is, for an extended period, including throughout the duration of the patient's life in order to ameliorate or otherwise control or limit the symptoms of the patient' s disorder.
- the administration of the compounds may be given continuously or suspended for a certain length of time (i.e., a "drug holiday").
- a maintenance dose is administered if necessary.
- the dosage or the frequency of administration, or both can be reduced, as a function of the symptoms, to a level at which the improved disorder is retained. Patients can, however, require intermittent treatment on a long-term basis upon any recurrence of symptoms.
- the compounds disclosed herein may also be combined or used in combination with other agents useful in the treatment of VMAT2-mediated disorders.
- the therapeutic effectiveness of one of the compounds described herein may be enhanced by administration of an adjuvant (i.e., by itself the adjuvant may only have minimal therapeutic benefit, but in combination with another therapeutic agent, the overall therapeutic benefit to the patient is enhanced).
- Such other agents, adjuvants, or drugs may be administered, by a route and in an amount commonly used therefor, simultaneously or sequentially with a compound as disclosed herein.
- a pharmaceutical composition containing such other drugs in addition to the compound disclosed herein may be utilized, but is not required.
- the compounds disclosed herein can be combined with one or more dopamine precursors, DOPA decarboxylase inhibitors, catechol-O-methyl transferase (COMT) inhibitors, dopamine receptor agonists, neuroprotective agents, NMDA antagonists, and anti-psychotics.
- DOPA decarboxylase inhibitors DOPA decarboxylase inhibitors
- catechol-O-methyl transferase (COMT) inhibitors DOPA decarboxylase inhibitors
- dopamine receptor agonists e.g., dopamine receptor agonists, neuroprotective agents, NMDA antagonists, and anti-psychotics.
- the compounds disclosed herein can be combined with one or more dopamine precursors selected from the group consisting of levodopa and deuterated L- DOPA.
- said deuterated L-DOPA has the structural formula:
- said deuterated L-DOPA has the structural formula:
- deuterated L-DOPA comprises a composition of compounds of structural formula V
- R70-R72 are independently selected from the group consisting of hydrogen and deuterium;
- composition has deuterium enrichment of at least 10% at each of the positions R70-R72 in the compounds of Formula I;
- the deuterium enrichment at the positions R71 and R72 is different from each other by at least 5%.
- R70 has deuterium enrichment of no less than 90%.
- R70 has deuterium enrichment of no less than 98%.
- R72 has deuterium enrichment of no less than 90%.
- R72 has deuterium enrichment of no less than 98%.
- R71 has deuterium enrichment of between about 78% and about 95%
- R71 has deuterium enrichment of between about 78% and about 82%
- R71 has deuterium enrichment of between about 88% and about 92% [0142] In certain embodiments, said DOPA decarboxylase inhibitor is carbidopa.
- said catechol-O-methyl transferase (COMT) inhibitor is selected from the group consisting of entacapone and tolcapone.
- said dopamine receptor agonist is selected from the group consisting of apomorphine, bromocriptine, ropinirole, and pramipexole.
- said neuroprotective agent is selected from the group consisting of selegeline and riluzole.
- said NMDA antagonist is amantidine.
- said anti-psychotic is clozapine.
- the compounds disclosed herein can be combined with one or more anti-psychotics, including, but not limited to, chlorpromazine, levomepromazine, promazine, acepromazine, triflupromazine, cyamemazine, chlorproethazine, dixyrazine, fluphenazine, perphenazine, prochlorperazine, thiopropazate, trifluoperazine, acetophenazine, thioproperazine, butaperazine, perazine, periciazine, thioridazine, mesoridazine, pipotiazine, haloperidol, trifluperidol, melperone, moperone, pipamperone, bromperidol, benperidol, droperidol, fluanisone, oxypertine, molindone, sertindole, ziprasidone, flupentixol, clopen
- mosapramine zotepine
- pripiprazole pripiprazole
- paliperidone
- the compounds disclosed herein can be combined with one or more benzodiazepines ("minor tranquilizers"), including, but not limited to alprazolam, adinazolam, bromazepam, camazepam, clobazam, clonazepam, clotiazepam, cloxazolam, diazepam, ethyl loflazepate, estizolam, fludiazepam, flunitrazepam, halazepam, ketazolam, lorazepam, medazepam, dazolam, nitrazepam, nordazepam, oxazepam, potassium clorazepate, pinazepam, prazepam, tofisopam, triazolam, temazepam, and chlordiazepoxide.
- minor tranquilizers including, but not limited to alprazolam, adinazolam, bromaze
- the compounds disclosed herein can be combined with olanzapine or pimozide.
- the compounds disclosed herein can also be administered in combination with other classes of compounds, including, but not limited to, anti-retroviral agents; CYP3A inhibitors; CYP3A inducers; protease inhibitors; adrenergic agonists; anti-cholinergics; mast cell stabilizers; xanthines; leukotriene antagonists; glucocorticoids treatments; local or general anesthetics; nonsteroidal anti-inflammatory agents (NSAIDs), such as naproxen; antibacterial agents, such as amoxicillin; cholesteryl ester transfer protein (CETP) inhibitors, such as anacetrapib; anti-fungal agents, such as isoconazole; sepsis treatments, such as drotrecogin- steroidals, such as hydrocortisone; local or general anesthetics, such as ketamine; norepinephrine reuptake inhibitors (NRIs) such as atomoxetine; dopamine
- squalene synthetase inhibitors fibrates; bile acid sequestrants, such as questran; niacin; anti- atherosclerotic agents, such as ACAT inhibitors; MTP Inhibitors; calcium channel blockers, such as amlodipine besylate; potassium channel activators; alpha-muscarinic agents; beta-muscarinic agents, such as carvedilol and metoprolol; antiarrhythmic agents; diuretics, such as
- chlorothiazide hydrochlorothiazide, flumethiazide, hydroflumethiazide, bendroflumethiazide, methylchlorothiazide, trichioromethiazide, polythiazide, benzothlazide, ethacrynic acid, tricrynafen, chlorthalidone, furosenilde, musolimine, bumetanide, triamterene, amiloride, and spironolactone; thrombolytic agents, such as tissue plasminogen activator (tPA), recombinant tPA, streptokinase, urokinase, prourokinase, and anisoylated plasminogen streptokinase activator complex (APSAC); anti-diabetic agents, such as biguanides (e.g.
- metformin glucosidase inhibitors
- glucosidase inhibitors e.g., acarbose
- insulins meglitinides (e.g., repaglinide), sulfonylureas (e.g., glimepiride, glyburide, and glipizide), thiozolidinediones (e.g. troglitazone, rosiglitazone and pioglitazone), and PPAR-gamma agonists; mineralocorticoid receptor antagonists, such as spironolactone and eplerenone; growth hormone secretagogues; aP2 inhibitors;
- glucosidase inhibitors e.g., acarbose
- insulins e.g., meglitinides (e.g., repaglinide), sulfonylureas (e.g., glimepiride,
- phosphodiesterase inhibitors such as PDE III inhibitors (e.g., cilostazol) and PDE V inhibitors (e.g., sildenafil, tadalafil, vardenafil); protein tyrosine kinase inhibitors; antiinflammatories; antiproliferatives, such as methotrexate, FK506 (tacrolimus, Prograf), mycophenolate mofetil; chemotherapeutic agents; immunosuppressants; anticancer agents and cytotoxic agents (e.g., alkylating agents, such as nitrogen mustards, alkyl sulfonates, nitrosoureas, ethylenimines, and triazenes); antimetabolites, such as folate antagonists, purine analogues, and pyrridine analogues; antibiotics, such as anthracyclines, bleomycins, mitomycin, dactinomycin, and plicamycin;
- enzymes such as L-asparaginase; farnesyl-protein transferase inhibitors; hormonal agents, such as glucocorticoids (e.g., cortisone), estrogens/antiestrogens, androgens/antiandrogens, progestins, and luteinizing hormone-releasing hormone anatagonists, and octreotide acetate; microtubule- disruptor agents, such as ecteinascidins; microtubule- stablizing agents, such as pacitaxel, docetaxel, and epothilones A-F; plant-derived products, such as vinca alkaloids,
- cytotoxic drugs such as azathiprine and cyclophosphamide
- TNF-alpha inhibitors such as tenidap
- anti-TNF antibodies or soluble TNF receptor such as etanercept, rapamycin, and leflunimide
- COX-2 cyclooxygenase-2
- celecoxib and rofecoxib miscellaneous agents such as, hydroxyurea, procarbazine, mitotane, hexamethylmelamine, gold compounds, platinum coordination complexes, such as cisplatin, satraplatin, and carboplatin.
- certain embodiments provide methods for treating VMAT2- mediated disorders in a subject in need of such treatment comprising administering to said subject an amount of a compound disclosed herein effective to reduce or prevent said disorder in the subject, in combination with at least one additional agent for the treatment of said disorder.
- certain embodiments provide therapeutic compositions comprising at least one compound disclosed herein in combination with one or more additional agents for the treatment of VMAT2-mediated disorders.
- Isotopic hydrogen can be introduced into a compound as disclosed herein by synthetic techniques that employ deuterated reagents, whereby incorporation rates are pre-determined; and/or by exchange techniques, wherein incorporation rates are determined by equilibrium conditions, and may be highly variable depending on the reaction conditions.
- Synthetic techniques where tritium or deuterium is directly and specifically inserted by tritiated or deuterated reagents of known isotopic content, may yield high tritium or deuterium abundance, but can be limited by the chemistry required.
- Exchange techniques on the other hand, may yield lower tritium or deuterium incorporation, often with the isotope being distributed over many sites on the molecule.
- the compounds as disclosed herein can be prepared by methods known to one of skill in the art and routine modifications thereof, and/or following procedures similar to those described in the Example section herein and routine modifications thereof, and/or procedures found in WO 2005077946; WO 2008/058261; EP 1716145; Lee et al., J. Med. Chem., 1996, (39), 191-196; Kilbourn et al., Chirality, 1997, (9), 59-62; Boldt et al., Synth. Commun., 2009, (39), 3574-3585; Rishel et al., J. Org. Chem., 2009, (74), 4001-4004; DaSilva et al., Appl.
- Compound 1 is reacted with compound 2 in an appropriate solvent, such as nitromethane, in the presence of an appropriate acid, such as ammonium acetate, at an elevated temperature to give compound 3.
- Compound 3 is reacted with compound 4 in the presence of an appropriate base, such as potassium carbonate, in an appropriate solvent, such as N,N- dimethylformamide, at an elevated temperature to afford compound 5.
- Compound 5 is reacted with an appropriate reducing reagent, such as lithium aluminum hydride, in an appropriate solvent, such as tetrahyrdofuran, at an elevated temperature to give compound 6.
- Compound 6 is reacted with compound 7 in the presence of an appropriate acid, such as trifluoroacetic acid, in an appropriate solvent, such as acetic acid, at an elevated temperature to give compound 8.
- Compound 9 is reacted with compound 10 and compound 11, in an appropriate solvent, such as methanol, at an elevated temperature to afford compound 12.
- Compound 12 is reacted with an appropriate methylating agent, such as methyl iodide, in an appropriate solvent, such as ethyl acetate, to give compound 13.
- Compound 8 is reacted with compound 13 in an appropriate solvent, such as ethanol, at an elevated temperature to give compound 14.
- Compound 14 is reacted with an appropriate reducing agent, such as sodium borohydride, in an appropriate solvent, such as methanol, to give compound 15 of Formula I.
- Deuterium can be incorporated to different positions synthetically, according to the synthetic procedures as shown in Scheme I, by using appropriate deuterated intermediates.
- compound 4 with the corresponding deuterium substitutions can be used.
- compound 1 with the corresponding deuterium substitutions can be used.
- compound 2 with the corresponding deuterium substitutions can be used.
- compound 10 with the corresponding deuterium substitutions can be used.
- compound 7 with the corresponding deuterium substitution can be used.
- compound 9 with the corresponding deuterium substitutions can be used.
- sodium borodeuteride can be used.
- Deuterium can be incorporated to various positions having an exchangeable proton, such as the hydroxyl O-H, via proton-deuterium equilibrium exchange.
- this proton may be replaced with deuterium selectively or nonselective ⁇ through a proton-deuterium exchange method known in the art.
- Compound 14 is reacted with an appropriate reducing agent, such as lithium tri-sec- butyl borohydride, in an appropriate solvent, such as ethanol, to give a mixture of compounds 16 and 17 of Formula II.
- an appropriate dehydrating reagent such as phosphorous pentachloride, in an appropriate solvent, such as dichloromethane to afford a mixture of compounds 18 and 19.
- Mixtures of compounds 16 and 17 or 20 and 21 can be separated by chiral preparative chromatography of through the preparation of Mosher' s esters (wherein the mixture is treated with R-(+)-3,3,3-trifluoro-2-methoxy-2-phenylpropanoic acid, an appropriate chlorinating agent, such as oxalyl chloride, and an appropriate base, such as 4- dimethylaminopyridine, in an appropriate solvent, such as dichloromethane, to give an epimeric mixture of R-(+)-3,3,3-trifluoro-2-methoxy-2-phenylpropanoate esters), which can be isolated via chromatography and then converted to the desired alcohol via hydrolysis (the Mosher' s esters are treated with an appropriate base, such as sodium hydroxide, in an appropriate solvent, such as methanol, to give the desired compounds of Formula II).
- an appropriate base such as sodium hydroxide
- an appropriate solvent such as methanol
- Deuterium can be incorporated to different positions synthetically, according to the synthetic procedures as shown in Scheme II, by using appropriate deuterated intermediates.
- deuterium at one or more positions of Ri-Rn and R21-R29 compound 14 with the corresponding deuterium substitutions can be used.
- deuterium at Ri8 lithium tri-sec -butyl borodeuteride can be used.
- trideuteroborane can be used.
- Deuterium can be incorporated to various positions having an exchangeable proton, such as the hydroxyl O-H, via proton-deuterium equilibrium exchange.
- this proton may be replaced with deuterium selectively or nonselective ⁇ through a proton-deuterium exchange method known in the art.
- Mixtures of compounds 24 and 25 can be separated by chiral preparative chromatography of through the preparation of Mosher's esters (wherein the mixture is treated with R-(+)-3,3,3-trifluoro-2-methoxy-2-phenylpropanoic acid, an appropriate chlorinating agent, such as oxalyl chloride, and an appropriate base, such as 4-dimethylaminopyridine, in an appropriate solvent, such as dichloromethane, to give an epimeric mixture of R-(+)-3,3,3- trifluoro-2-methoxy-2-phenylpropanoate esters), which can be isolated via chromatography and then converted to the desired alcohol via hydrolysis (the Mosher's esters are treated with an appropriate base, such as sodium hydroxide, in an appropriate solvent, such as methanol, to give the desired compounds of Formula II).
- an appropriate base such as sodium hydroxide
- an appropriate solvent such as methanol
- Deuterium can be incorporated to different positions synthetically, according to the synthetic procedures as shown in Scheme III, by using appropriate deuterated intermediates.
- deuterium at one or more positions of Ri-Ri8 and R21-R29 compounds 18 and 19 with the corresponding deuterium substitutions can be used.
- trideuteroborane can be used.
- Deuterium can be incorporated to various positions having an exchangeable proton, such as the hydroxyl O-H, via proton-deuterium equilibrium exchange.
- this proton may be replaced with deuterium selectively or nonselective ⁇ through a proton-deuterium exchange method known in the art.
- Compound 26 is reacted with an appropriate alcohol, such as compound 27, in the presence of an appropriate base, such as 4-dimethylaminopyridine, to give compound 28 of Formula II (where R22 is -C(0))-alkyl).
- an appropriate base such as 4-dimethylaminopyridine
- Deuterium can be incorporated to different positions synthetically, according to the synthetic procedures as shown in Scheme IV, by using appropriate deuterated intermediates.
- deuterium at one or more positions of R1-R19 and R21-R29 compound 16 with the corresponding deuterium substitutions can be used.
- compound 27 with the corresponding deuterium substitutions can be used.
- Compound 29 is reacted with an appropriate protecting agent, such as di-tert-butyl dicarbonate, in an appropriate solvent, such as a mixture of tetrathydrofuran and water, in the presence of an appropriate base, such as sodium carbonate, to give compound 30.
- Compound 30 is reacted with compound 4 in the presence of an appropriate base, such as potassium carbonate, in the presence of an appropriate catalyst, such as 18-crown-6, in an appropriate solvent, such as acetone, to afford compound 31.
- Compound 31 is reacted with an appropriate deprotecting agent, such as hydrogen chloride, in an appropriate solvent, such as ethyl acetate, to give compound 6.
- Compound 6 is reacted with compound 32 at an elevated temperature to give compound 33.
- Compound 33 is reacted with an appropriate dehydrating agent, such as phosphorous oxychloride, at an elevated temperature to afford compound 8.
- Compound 8 is reacted with compound 13 in an appropriate solvent, such as methanol, at an elevated
- Deuterium can be incorporated to different positions synthetically, according to the synthetic procedures as shown in Scheme V, by using appropriate deuterated intermediates.
- compound 4 with the corresponding deuterium substitutions can be used.
- compound 29 with the corresponding deuterium substitutions can be used.
- compound 32 with the corresponding deuterium substitution can be used.
- compound 13 with the corresponding deuterium substitutions can be used.
- Compound 9 is reacted with compound 11 and compound 34 (paraformaldehyde and/or formaldehyde) in an appropriate solvent, such as ethanol, in the presence of an appropriate acid, such as hydrochloric acid, at an elevated temperature to give compound 12.
- Compound 12 is reacted with an appropriate methylating agent, such as methyl iodide, in an appropriate solvent, such as ethyl acetate, to give compound 13.
- compound 8 is reacted with compound 13 in an appropriate solvent, such as dichloromethane, to give compound 13.
- Deuterium can be incorporated to different positions synthetically, according to the synthetic procedures as shown in Scheme VI, by using appropriate deuterated intermediates.
- compound 10 with the corresponding deuterium substitutions can be used.
- compound 9 with the corresponding deuterium
- Compound 35 is reacted with compound 36 in an appropriate solvent, such as tetrahydrofuran, in the presence of an appropriate catalyst, such as cuprous iodide, and an appropriate co-solvent, such as hexamethylphosphorous triamide, then reacted with an appropriate protecting agent, such as trimethylsilyl chloride, and an appropriate base, such as triethylamine, to give compound 37.
- an appropriate mannich base such as N-methyl-N-methylenemethanaminium iodide
- an appropriate solvent such as acetonitrile
- Compound 12 is reacted with an appropriate methylating agent, such as methyl iodide, in an appropriate solvent, such as diethyl ether, to give compound 13.
- Deuterium can be incorporated to different positions synthetically, according to the synthetic procedures as shown in Scheme VII, by using appropriate deuterated intermediates.
- compound 35 with the corresponding deuterium substitutions can be used.
- compound 36 with the corresponding deuterium substitutions can be used.
- Compound 38 is reacted with an appropriate reducing agent, such as sodium borohydride, in an appropriate solvent, such as ethanol, to give compound 39 of Formula II having predominantly (-4: 1) alpha stereochemistry.
- an appropriate reducing agent such as sodium borohydride
- an appropriate solvent such as ethanol
- the alpha stereoisomer can be further enriched by recrystalization from an appropriate solvent, such as ethanol.
- Deuterium can be incorporated to different positions synthetically, according to the synthetic procedures as shown in Scheme I, by using appropriate deuterated intermediates.
- deuterium can be introduced at one or more positions of Ri-Rn, R99, and R21-R29.
- compound 38 with the corresponding deuterium substitutions can be used.
- sodium borodeuteride can be used.
- Deuterium can be incorporated to various positions having an exchangeable proton, such as the hydroxyl O-H, via proton-deuterium equilibrium exchange.
- this proton may be replaced with deuterium selectively or nonselective ⁇ through a proton-deuterium exchange method known in the art.
- Compound 38 is reacted with an appropriate reducing agent, such as potassium tri- sec-butyl borohydride (K-selectride), in an appropriate solvent, such as tetrahydrofuran, to give compound 40 of Formula I having beta stereochemistry.
- an appropriate reducing agent such as potassium tri- sec-butyl borohydride (K-selectride)
- K-selectride potassium tri- sec-butyl borohydride
- solvent such as tetrahydrofuran
- Deuterium can be incorporated to different positions synthetically, according to the synthetic procedures as shown in Scheme I, by using appropriate deuterated intermediates.
- deuterium at one or more positions of Ri-Rn, R99, and R21-R29 compound 38 with the corresponding deuterium substitutions can be used.
- potassium tri-sec -butyl borodeuteride can be used.
- Deuterium can be incorporated to various positions having an exchangeable proton, such as the hydroxyl O-H, via proton-deuterium equilibrium exchange.
- this proton may be replaced with deuterium selectively or nonselective ⁇ through a proton-deuterium exchange method known in the art.
- Compound 40 is reacted with compound 41 (wherein P.G. is an appropriate protecting group, such as carboxybenzoyl) in the presence of an appropriate coupling agent, such as dicyclohexylcarbodiimide (DCC), an appropiate catalyst, such as 4-dimethylaminopyridine (DMAP), in an appropriate solvent, such as dichloromethane, to give compound 42.
- an appropriate deprotecting agent such as a combination of hydrogen and an appropriate catalyst, such as palladium on carbon, in an appropriate solvent, such as methanol, to give compound 43 of Formula II.
- Deuterium can be incorporated to different positions synthetically, according to the synthetic procedures as shown in Scheme I, by using appropriate deuterated intermediates. For example, to introduce deuterium at one or more positions of R1-R19 and R21-R29, compound 40 with the corresponding deuterium substitutions can be used. To indroduce deuterium at one or more positions of R30-R37, compound 41 with the corresponding deuterium substitutions can be used.
- Deuterium can be incorporated to various positions having an exchangeable proton, such as the hydroxyl O-H or amine N-Hs, via proton-deuterium equilibrium exchange.
- an exchangeable proton such as the hydroxyl O-H or amine N-Hs
- these protons may be replaced with deuterium selectively or non- selectively through a proton-deuterium exchange method known in the art.
- Compound 44 is reacted with compound 45 in the presence of an appropriate base, such as potassium carbonate, in the presence of an appropriate phase transfer catalyst, such as a combination of potassium iodide and tetrabutylammonium bromide, in an appropriate solvent, such as N,N-dimethylformamide, at an elevated temperature to afford compound 46.
- an appropriate base such as potassium carbonate
- an appropriate phase transfer catalyst such as a combination of potassium iodide and tetrabutylammonium bromide
- an appropriate solvent such as N,N-dimethylformamide
- compound 49 in the presence of an appropriate acid, such as hydrochloric acid, and an appropriate phase transfer catalyst, such as tetrabutylammonium bromide, in an appropriate solvent, such as water, to afford compound 49.
- an appropriate methylating agent such as methyl iodide
- an appropriate solvent such as methyl tert-butyl ether
- compound 50 Compound 8 is reacted with compound 50 in an appropriate solvent, such as a mixture of methanol and water, at an elevated temperature to give compound 51.
- compound 51 is reacted with an appropriate acid, such as sulfuric acid, in an appropriate solvent, such as water, to give compound 52 of Formula III.
- Deuterium can be incorporated to different positions synthetically, according to the synthetic procedures as shown in Scheme I, by using appropriate deuterated intermediates.
- compound 8 with the corresponding deuterium substitutions can be used.
- compound 47 with the corresponding deuterium substitutions can be used.
- compound 44 with the corresponding deuterium substitutions can be used.
- compound 45 with the corresponding deuterium substitutions can be used.
- D2SO4 and/or D2O can be used.
- Deuterium can be incorporated to various positions having an exchangeable proton, such as the hydroxyl O-H, via proton-deuterium equilibrium exchange.
- this proton may be replaced with deuterium selectively or nonselective ⁇ through a proton-deuterium exchange method known in the art.
- Compound 53 is reacted with an appropriate reducing agent, such as lithium tri-sec- butyl borohydride, in an appropriate solvent, such as tetrahydrofuran, to give compound 54.
- Compound 54 is reacted with an appropriate protecting agent, such as benzyl bromide, in the presence of an appropriate base, such as sodium hydride, in an appropriate solvent, such as tetrahydrofuran to give compound 55.
- Compound 55 is reacted with an appropriate reducing agent, such as lithium tri-sec- butyl borohydride, in an appropriate solvent, such as tetrahydrofuran, to give compound 54.
- an appropriate protecting agent such as benzyl bromide
- an appropriate base such as sodium hydride
- an appropriate solvent such as tetrahydrofuran
- hydroborating reagent such as borane-dimethylsulfide complex
- an appropriate solvent such as tetrahyrdofuran
- an appropriate base such as aqueous sodium hydroxide
- compound 56 is reacted with an appropriate oxidizing agent, such as Jones reagent (an aqueous solution of chromium trioxide and sulfuric acid), in an appropriate solvent, such as acetone, to give compound 57.
- compound 57 is reacted with an appropriate deprotecting agent, such as a mixute of palladium on carbon and hydrogen gas, in an appropriate solvent, such as methanol, to give compound 58 of Formula IV.
- Deuterium can be incorporated to different positions synthetically, according to the synthetic procedures as shown in Scheme II, by using appropriate deuterated intermediates.
- compound 53 with the corresponding deuterium substitutions can be used.
- lithium tri-sec -butyl borodeuteride can be used.
- trideuteroborane can be used.
- Deuterium can be incorporated to various positions having an exchangeable proton, such as the hydroxyl O-H or carboxyl O-H, via proton-deuterium equilibrium exchange.
- these protons may be replaced with deuterium selectively or non-selectively through a proton-deuterium exchange method known in the art.
- Compound 60 is reacted with compound 61 in the presence of an appropriate base, such as potassium carbonate, in an appropriate solvent, such as dichloromethane, at an elevated temperature to afford compound 62.
- Compound 62 is reacted with an appropriate base, such as sodium hydroxide, in an appropriate solvent, such as a mixture of ethanol and water, to afford compound 63.
- Compound 63 is heated to an elevated temperature in an appropriate solvent, such as a mixture of dimethylsulf oxide and water, to give compound 64.
- Compound 64 is reacted with an appropriate silating agent, such as trimethylsilyl iodide, in the presence of an appropriate base, such as hexamethyldisilazide, to give an intermediate silyl enol ether which is reacted with compound 65 in an appropriate solvent, such as acetonitrile, to afford compound 66.
- Compound 66 is reacted with an appropriate methylating agent, such as methyl iodide, to give compound 67.
- Compound 67 is reacted with compound 68 in an appropriate solvent, such as ethanol, at an elevated temperature to give compound 69.
- Compound 69 is reacted with an appropriate base, such as lithium hydroxide, in an appropriate solvent, such as a mixture of tetrahydrofuran and water, to afford compound 70.
- Compound 70 is reacted with an appropriate reducing agent, such as potassium tri-sec -butyl borohydride (K-selectride), in an appropriate solvent, such as tetrahydrofuran, to give compound 71 as a mixture of diastereomers.
- Compound 71 is recrystalized from water, to give compound 72 of Formula III.
- Deuterium can be incorporated to different positions synthetically, according to the synthetic procedures as shown in Scheme I, by using appropriate deuterated intermediates.
- compound 60 with the corresponding deuterium substitutions can be used.
- compound 61 with the corresponding deuterium substitutions can be used.
- deuterium at R55 D2O can be used.
- compound 65 with the corresponding deuterium substitutions can be used.
- compound 68 with the corresponding deuterium substitutions can be used, introduce deuterium at R55, potassium tri- sec-butyl borodeuteride can be used.
- Deuterium can be incorporated to various positions having an exchangeable proton, such as the hydroxyl O-H or carboxyl O-H, via proton-deuterium equilibrium exchange.
- these protons may be replaced with deuterium selectively or non-selectively through a proton-deuterium exchange method known in the art.
- D6-fert-butyl 3,4-dimethoxyphenethylcarbamate A solution of ieri-butyl 3,4- dihydroxyphenethylcarbamate (127 g, 397 mmol, 1.00 equiv), potassium carbonate (359.3 g, 2.604 mmol, 3.00 equiv) and 18-crown-6 (1,4,7,10,13,16-hexaoxacyclooctadecane ) (68.64 g, 0.26 mmol, 0.03 equiv) in acetone (800 mL) was stirred at 38°C.
- CD 3 I (362 g, 2.604 mmol, 3.00 equiv) was added to the reaction, and the mixture was stirred at 38°C for 12 h. Then an additional CD3I (120 g, 0.868 mmol, 1.00 equiv) was added to the solution and the solution was stirred for 5 h. Then the mixture was cooled to room temperature and the solid was filtered. The filtrate was concentrated under vacuum. The resultant solid was dissolved in H2O (300 mL) and extracted with EA (3x300 mL), the organic layers was combined and concentrated under vacuum to give 114 g (79%) of de-tert-butyl 3,4-dimethoxyphenethylcarbamate as white solid.
- D6-2-(3,4-dimethoxyphenyl)ethanamine A solution of de-tert-butyl 3,4- dimethoxyphenethylcarbamate (128 g, 455.26 mmol, 1.00 equiv) in ethyl acetate (1.5 L) was stirred at room temperature. Then HC1 gas was introduced into the reaction mixture for 2h. The precipitated solid was isolated by filtration. The solid was dissolved in 300 mL of water. The pH value of the solution was adjusted to 12 with sodium hydroxide (solid). The resulting solution was stirred for 1 h at 5-10°C.
- D6-N-r2-(3,4-dimethoxy-phenyl)ethyllformamide A solution of d 6 -2-(3,4- dimethoxyphenyl)ethanamine (69 g, 368 mmol, 1.00 equiv) in ethyl formate(250 mL) was heated under reflux overnight. The solution was concentrated under vacuum to give 71 g (91%) of d6-N-[2-(3,4-dimethoxy-phenyl)ethyl]formamide as yellow solid. The crude solid was used in next step without purification.
- D6-6,7-dimethoxy-3,4-dihvdroisoguinoline A solution of d6-N-[2-(3,4-dimethoxy- phenyl)ethyl]formamide (71 g, 329 mmol, 1.00 equiv) in phosphorus oxychloride (100 mL) was stirred at 105°C for 1 h. Then the solution was concentrated under vacuum to remove
- Trimethyl(5-methylhex-2-en-2-yloxy)silane To a cold (-78°C), stirred solution of j-PrMgBr (500 mL of 2 M solution in tetrahydrofuran, 1 mol, 1.00 equiv) in anhydrous tetrahydrofuran (1 L) was added Cul (19.02 g, 0.1 mol, 0.10 equiv) and the resultant mixture was stirred for 15 min at -78°C.
- ⁇ - ( ⁇ ) -tetrabenazine A solution of d6-6,7-dimethoxy-3,4-dihydroisoquinoline (33.4 g, 169 mmol, 1.10 equiv) and 2-acetyl-N,N,N,4-tetramethylpentan-l-aminium iodide (48 g, 153 mmol, 1.00 equiv) in 300ml of methanol was heated under reflux for 48 h. Then 150 mL water was added. The solution was cooled to room temperature. The precipitated solid was isolated by filtration and dried under vacuum to give 38 g of crude d6-tetrabenazine as yellow solid.
- the product A was diluted with 3 L ethyl acetate, and 50 g toluenesulfonic acid was added, then the solution was stirred overnight at rt. The precipitated solid was removed. The filtrate was washed with water (2x400 mL) and 5% aqueous NaOH (200 mL).
- the product B was diluted with 3.5 L ethyl acetate, and 200 g toluenesulfonic acid was added, then the solution was stirred overnight at rt. The precipitated solid was removed and the filtrate was washed with water (2x400 mL) and 5% aqueous NaOH (200 mL).
- dichloromethane (10 L) was dropwised a solution of methyl iodide (2 kg, 14.12 mol, 1.1 equiv) in dichloromethane (2 L) at 5-10°C. Then the solution was stirred overnight at rt. The reaction was monitored by LCMS until completion of reaction (3-[(dimethylamino)methyl]-5- methylhexan-2-one ⁇ 5.0%). The precipitated solid was isolated by filtration and dried under vacuum to give 3.5 kg (87%) of 2-Acetyl-N,N,N,4-tetramethylpentan-l-aminium iodide as white solid.
- Ethyl 2-acetyl-4-methylpent-4-enoate To a solution of ethyl acetoacetate (500 g, 3.84 mol, 1.00 eq), potassium iodide (63.8 g, 0.384 mol, 0.10 eq), tetrabutylammonium bromide (136.2 g, 0.422 mol, 0.11 eq), and K 2 C0 3 (631.9 g, 4.57 mol, 1.19 eq) in dimethylformamide (1.5 L) was heated to 40-50 °C. At this temperature, 3-chloro-2-methyl-l-propene (382.6 g, 4.22 mol, 1.10 eq) was added.
- reaction mixture was heated to 65-75°C and stirred for 6 hrs. Then the reaction mixture was cool to 25-35 °C and quenched with water (5.00 L). The product was extracted with toluene (2x2.00 L), and the combined toluene layers were washed with water (2x1.5 L) and concentrated under vacuum at 50-55 °C to give 707 g of ethyl 2-acetyl-4- methylpent-4-enoate (quantitative yield) as a brown liquid.
- reaction mixture was heated to 40-45 °C for 30 hrs. Then the reaction mixture was cooled to room temperature (25-35 °C) and water was added (105 mL). The reaction mixture was stirred for 30 minutes. The precipitated solid was filtered, washed with water (105 mL), and dried to give 42 g of crude d 6 - (3S,l lbS)-9,10-dimethoxy-3-(2-methylallyl)-3,4,6,7-tetrahydro-lH-pyrido[2,l-a]isoquinolin- 2(1 lbH)-one as a yellow solid.
- the reaction mixture was cooled to 0-5 °C and adjusted to pH to 9-10 by using 5% NaOH solution.
- the product was extracted with ethyl acetate (2x75 mL).
- the ethyl acetate layer was washed with water (2x25 mL).
- reaction mixture was slowly heated to 25-35 °C and stirred for 1 hr.
- Benzyl bromide (8.14 mL, 0.06811, 1.00 eq) was added to the reaction mass at 0-5 °C over 20 minutes and stirred for 30 minutes.
- the reaction mixture was quenched with cold water (440 mL) at 0-5 °C and the compound was extracted with ethyl acetate (2x 220 mL and 1x110 mL).
- reaction mixture was stirred overnight at 25-30 °C.
- the reaction mixture was quenched with 3M NaOH solution (22 mL) at 0-5 °C.
- the reaction mixture was concentrated under vacuum at 40 °C until complete removal of tetrahydrofuran and co-distilled twice with diethyl ether (2x110 mL).
- 3 M aqueous NaOH solution 55 mL was added to the remaining residue and heated to 80-90 °C for 2 hrs.
- the reaction mixture was cooled to 25-30 °C and the product was extracted with ethyl acetate (3x110 mL).
- the reaction mixture was stirred at 20 °C for 30 minutes.
- the liquid layer was decanted and to the remaining green color gummy mass, acetone (64 mL) was added, stirred for 30 minutes, and decanted.
- the pH of the combined acetone layers were adjusted to 7 using saturated sodium bicarbonate solution (20 mL).
- the solids were filtered and washed with acetone (60 mL).
- the filtrate was distilled under vacuum at 35 °C until complete removal of acetone.
- the remaining aqueous layer was saturated with sodium chloride and extracted with ethyl acetate (5x60 mL).
- 1,3-Diethyl 2-methyl-2-(3-oxobutyl)propanedioate To a solution of 1,3-diethyl 2- methylpropanedioate (500 g, 2.87 mol, 1.00 equiv) in dichloromethane (5000 mL) were added but-3-en-2-one (302 g, 4.31 mol, 1.50 equiv) and potassium carbonate (793 g, 5.74 mol, 2.00 equiv). The resulting solution was stirred for 48 h at 25 °C. The reaction mixture was then quenched by the addition of water (5 L). The dichloromethane layer was separated.
- Ethyl 2-methyl-5-oxohexanoate 2-(ethoxycarbonyl)-2-methyl-5-oxohexanoic acid (350 g, 1.62 mol, 1.00 equiv) was dissolved in dimethylsulfoxide (2000 mL) and water (20 mL). The resulting solution was stirred for 2 h at 160 °C. The reaction mixture was then quenched by the addition of water/ice (3000 mL).
- Ethyl (4Z)-2-methyl-5 (trimethylsilyl)oxylhex-4-enoate To a solution of ethyl 2- methyl-5-oxohexanoate (180 g, 1.05 mol, 1.00 equiv), in dichloromethane (2000 mL) was added hexamethyldisilazide (505 g, 3.13 mol, 3.00 equiv) under an atmosphere of nitrogen followed by the addition of trimethylsilyl iodide (209 g, 1.04 mol, 1.00 equiv) dropwise with stirring at -30 to -20 °C in 30 min. The reaction temperature was allowed to rise to 25 °C and stirred for 5 h at 25 °C. The reaction mixture was then quenched by the addition of cooled sat. NaHC0 3 (2 L).
- Ethyl 4 (dimethylamino)methyl1-2-methyl-5-oxohexanoate To a solution of (4Z)- 2-methyl-5-[(trimethylsilyl)oxy]hex-4-enoate (230 g, 941.07 mmol, 1.00 equiv) in acetonitrile (1500 mL) was added dimethyl(methylidene)azanium iodide (174.4 g, 942.67 mmol, 1.00 equiv) in several batches at 0 °C in 20 min. The resulting solution was stirred for 20 h at 25 °C under an atmosphere of nitrogen. The resulting mixture was concentrated under vacuum.
- the resulting suspension was stirred for 2 h at - 10-0 °C and turned into a solution.
- the reaction progress was monitored by LCMS.
- the reaction mixture was then quenched by the addition of of water/ice (300 mL).
- the reaction mixture was concentrated under vacuum to remove THF.
- the resulting aqueous solution was extracted with dichloromethane (3 x 100 mL) and the pH of the aqueous layers were adjusted to 6 with hydrogen chloride (2N).
- the solid was precipitated from water, then the pH value of the suspension was adjusted to 4 with hydrochloric acid (0.5 N). The solid was dissolved. A sodium hydroxide solution (0.5 N) was used to adjust the pH od the solution to 7 immediately. The solid precipitated and were collected by filtration. LCMS showed the purity of the product was 87%. This process was repeated two times and the purity of the product was 96% in LCMS. Then the product was suspended in ethanol (200 mL) and stirred for 20 min at 70 °C.
- Non-limiting examples include the following compounds and pharmaceutically acceptable salts thereof:
- PAMSysTM is a precise platform for long-term objective evaluation of physical activity during everyday life (1).
- PAMSysTM allows for the collection of posture (sitting, standing, walking, or lying down), postural transitions (duration, time of occurrence), gait (duration, number of steps, cadence and step time variability), and fall (number of falls, time of occurrence) information.
- the PAMSysTM technology is based on over 10 years of research supported in part by the National Institutes of Health (NIH) and uses advanced signal processing algorithms and novel biomechanical models of human motion to identify a complete physical activity map for the user from data measured by a single, lightweight, wearable motion sensor.
- PAMSys-XTM allows for synchronized monitoring of multiple body segment movements.
- the baseline assessment will utilize surveys to obtain demographic information, medical and HD history, current medications, and familiarity with technology. These surveys will be completed by participants while on-site and will be stored using the secure, web-based REDCap (Research Electronic Data Capture) survey application (2). Research staff will place 1 PAMSysTM (near the chest) and 4 PAMSys-XTM (on the wrists and ankles) sensors on the subjects. Participants will then complete the Q-motor finger-tap and force transducer assessments (3), the motor portion of the UHDRS (4), and the Montreal Cognitive Assessment (MoCA) (5). Participants will also be video-recorded while performing a standardized motor assessment wearing the five BioSensics mobile sensors. (Table 1)
- the standardized motor assessment for the BioSensics sensors will consist of six tasks that participants will perform while wearing the equipment. When wearing the five BioSensics sensors participants will complete the following tasks: 20 seconds each of static sitting and standing, a Timed Up and Go (TUG) test (6), 30 seconds of tandem walking, 15 seconds of finger tapping, 5 instances of a drinking motion using a cup or glass, and 15 seconds of pronation and supination of the hands. When wearing only the trunk sensor, participants will only complete the first three standard assessments (static sitting/standing, Timed Up and Go test, and tandem walking). The standard assessments will be completed twice per day during the week of study
- the principal outcome measures include remote data collected from the BioSensics devices from two in-person ssessments.
- Clinical outcome measures include clinical characteristics (i.e. UHDRS and Q-motor exams) and comparison of data from individuals with HD to controls.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Epidemiology (AREA)
- Neurology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Chemical & Material Sciences (AREA)
- Physical Education & Sports Medicine (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Psychology (AREA)
- Pain & Pain Management (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Measuring And Recording Apparatus For Diagnosis (AREA)
- Thiazole And Isothizaole Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Indole Compounds (AREA)
Abstract
Description











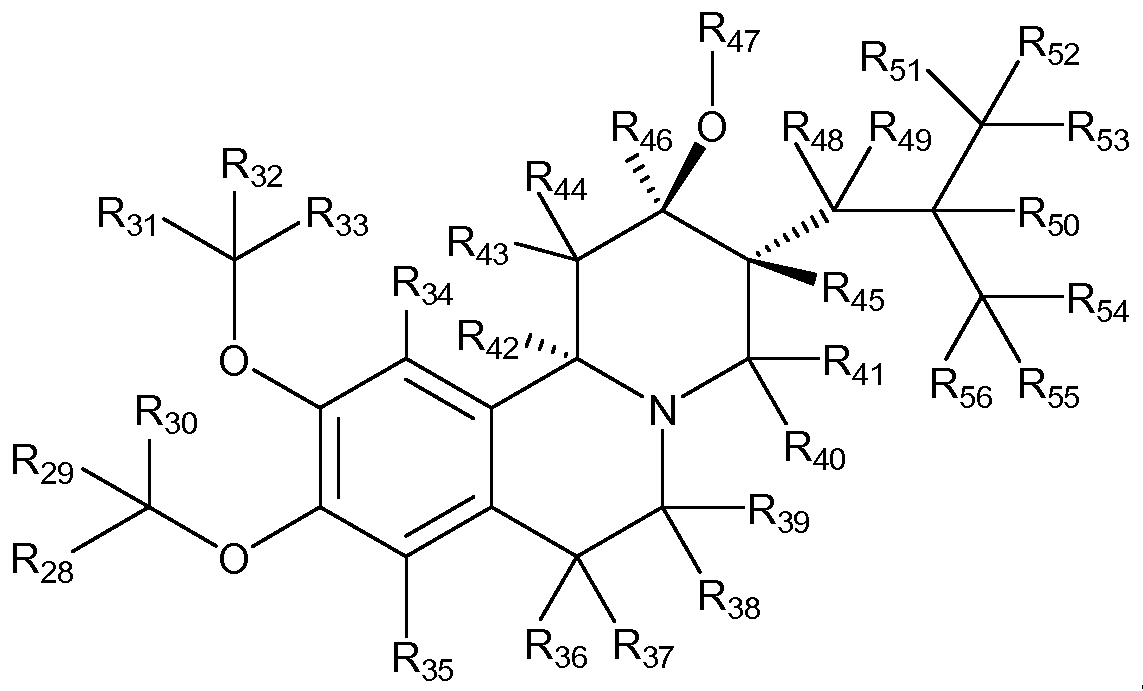















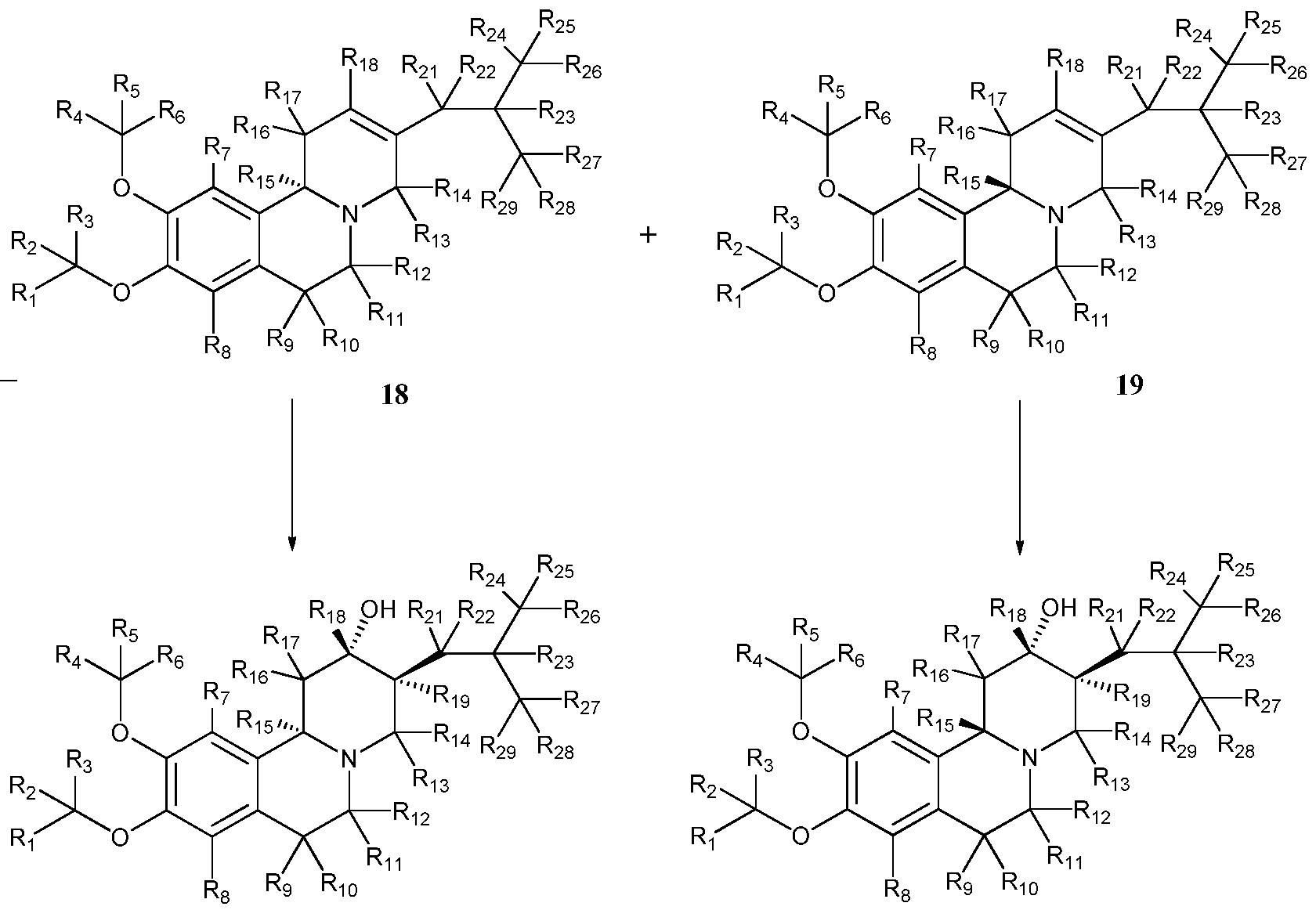






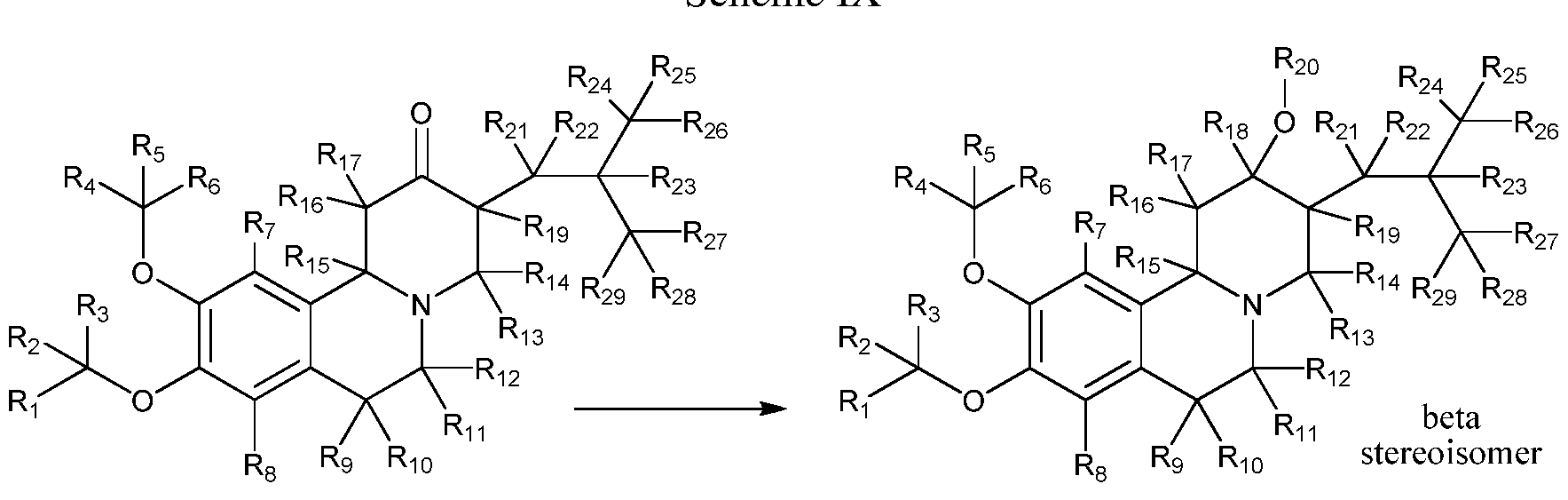



















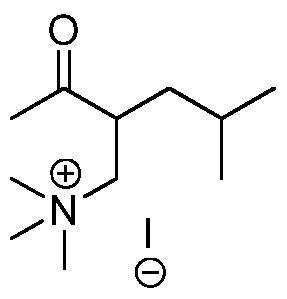

















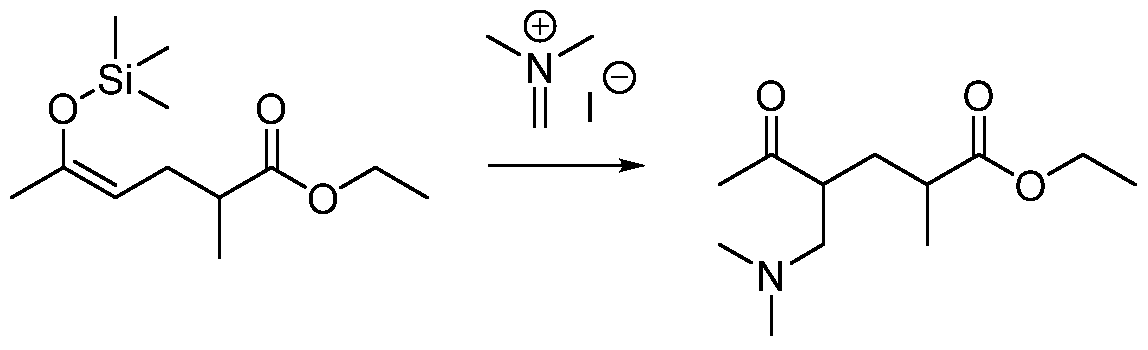







































































Claims




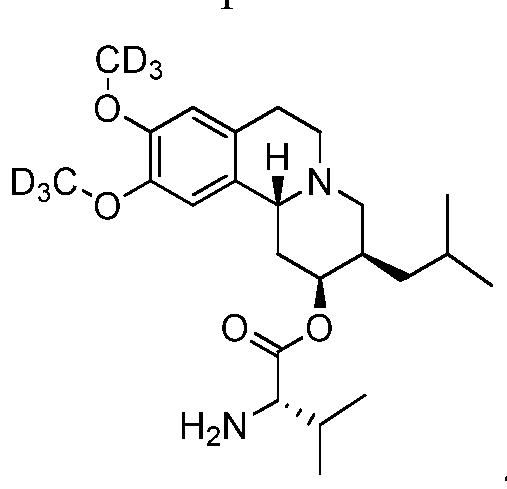






Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA2930167A CA2930167A1 (en) | 2013-11-22 | 2014-11-21 | Methods of treating abnormal muscular activity |
| JP2016533116A JP2017503756A (en) | 2013-11-22 | 2014-11-21 | How to treat abnormal muscle activity |
| US15/037,465 US20160303110A1 (en) | 2013-11-22 | 2014-11-21 | Methods of treating abnormal muscular activity |
| EP14864919.7A EP3071565A4 (en) | 2013-11-22 | 2014-11-21 | Methods of treating abnormal muscular activity |
| MX2016006622A MX2016006622A (en) | 2013-11-22 | 2014-11-21 | Methods of treating abnormal muscular activity. |
| IL245538A IL245538A0 (en) | 2013-11-22 | 2016-05-08 | Methods of treating abnormal muscular activity |
| HK16112663.3A HK1224294A1 (en) | 2013-11-22 | 2016-11-03 | Methods of treating abnormal muscular activity |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201361907675P | 2013-11-22 | 2013-11-22 | |
| US61/907,675 | 2013-11-22 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015077520A1 true WO2015077520A1 (en) | 2015-05-28 |
Family
ID=53180159
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2014/066740 WO2015077520A1 (en) | 2013-11-22 | 2014-11-21 | Methods of treating abnormal muscular activity |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US20160303110A1 (en) |
| EP (1) | EP3071565A4 (en) |
| JP (1) | JP2017503756A (en) |
| CA (1) | CA2930167A1 (en) |
| HK (1) | HK1224294A1 (en) |
| IL (1) | IL245538A0 (en) |
| MX (1) | MX2016006622A (en) |
| WO (1) | WO2015077520A1 (en) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015136003A3 (en) * | 2014-03-11 | 2015-11-26 | Imphar Aktiengesellschaft | Composition comprising a dopamine agonist and an l-dopa derivative for treating parkinson's disease |
| US9233959B2 (en) | 2012-09-18 | 2016-01-12 | Auspex Pharmaceuticals, Inc. | Formulations and pharmacokinetics of deuterated benzoquinoline inhibitors of vesicular monoamine transporter 2 |
| US9550780B2 (en) | 2012-09-18 | 2017-01-24 | Auspex Pharmaceuticals, Inc. | Formulations pharmacokinetics of deuterated benzoquinoline inhibitors of vesicular monoamine transporter 2 |
| US10047077B2 (en) | 2016-04-13 | 2018-08-14 | Skyline Antiinfectives, Inc. | Deuterated O-sulfated beta-lactam hydroxamic acids and deuterated N-sulfated beta-lactams |
| US10513488B2 (en) | 2013-12-03 | 2019-12-24 | Auspex Pharmaceuticals, Inc. | Methods of manufacturing benzoquinoline compounds |
| US10959996B2 (en) | 2015-03-06 | 2021-03-30 | Auspex Pharmaceuticals, Inc. | Methods for the treatment of abnormal involuntary movement disorders |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10786202B2 (en) | 2016-09-28 | 2020-09-29 | International Business Machines Corporation | Quantifying grip strength and characterizing movement idioms |
Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20080050312A1 (en) * | 2006-05-02 | 2008-02-28 | The Trustees Of The University Of Pennsylvania | Radiolabeled dihydrotetrabenazine derivatives and their use as imaging agents |
| US20080212839A1 (en) * | 2003-11-26 | 2008-09-04 | Ge Medical Systems Global Technology Company, Llc | Method and system for determining a period of interest using multiple inputs |
| US20080306267A1 (en) * | 2007-06-08 | 2008-12-11 | General Electric Company | Method for making tetrabenazine compounds |
| US20090018191A1 (en) * | 2006-02-17 | 2009-01-15 | Rudolf-Giesbert Alken | Deuterated Catecholamine Derivatives and Medicaments Comprising Said Compounds |
| US20110182818A1 (en) * | 2008-07-01 | 2011-07-28 | Fallon Joan M | Methods and compositions for the treatment of symptoms of neurological and mental health disorders |
| US20110206782A1 (en) * | 2010-02-24 | 2011-08-25 | Auspex Pharmaceuticals, Inc. | Piperidine modulators of dopamine receptor |
| US20120003330A1 (en) * | 2010-06-01 | 2012-01-05 | Auspex Pharmaceuticals, Inc. | Benzoquinolone inhibitors of vmat2 |
| US20130197067A1 (en) * | 2010-07-08 | 2013-08-01 | The Brigham And Women's Hospital, Inc. | Neuroprotective molecules and methods of treating neurological disorders and inducing stress granules |
| WO2013142816A1 (en) * | 2012-03-23 | 2013-09-26 | Cardero Therapeutics, Inc. | Compounds and compositions for the treatment of muscular disorders |
| US20130296360A1 (en) * | 2008-09-18 | 2013-11-07 | Auspex Pharmaceuticals, Inc. | Benzoquinoline inhibitors of vesicular monoamine transporter 2 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2727555C (en) * | 2008-06-12 | 2016-10-04 | Global Kinetics Corporation Pty Ltd | Detection of hypokinetic and/or hyperkinetic states |
-
2014
- 2014-11-21 CA CA2930167A patent/CA2930167A1/en not_active Abandoned
- 2014-11-21 MX MX2016006622A patent/MX2016006622A/en unknown
- 2014-11-21 US US15/037,465 patent/US20160303110A1/en not_active Abandoned
- 2014-11-21 WO PCT/US2014/066740 patent/WO2015077520A1/en active Application Filing
- 2014-11-21 EP EP14864919.7A patent/EP3071565A4/en not_active Withdrawn
- 2014-11-21 JP JP2016533116A patent/JP2017503756A/en active Pending
-
2016
- 2016-05-08 IL IL245538A patent/IL245538A0/en unknown
- 2016-11-03 HK HK16112663.3A patent/HK1224294A1/en unknown
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20080212839A1 (en) * | 2003-11-26 | 2008-09-04 | Ge Medical Systems Global Technology Company, Llc | Method and system for determining a period of interest using multiple inputs |
| US20090018191A1 (en) * | 2006-02-17 | 2009-01-15 | Rudolf-Giesbert Alken | Deuterated Catecholamine Derivatives and Medicaments Comprising Said Compounds |
| US20080050312A1 (en) * | 2006-05-02 | 2008-02-28 | The Trustees Of The University Of Pennsylvania | Radiolabeled dihydrotetrabenazine derivatives and their use as imaging agents |
| US20080306267A1 (en) * | 2007-06-08 | 2008-12-11 | General Electric Company | Method for making tetrabenazine compounds |
| US20110182818A1 (en) * | 2008-07-01 | 2011-07-28 | Fallon Joan M | Methods and compositions for the treatment of symptoms of neurological and mental health disorders |
| US20130296360A1 (en) * | 2008-09-18 | 2013-11-07 | Auspex Pharmaceuticals, Inc. | Benzoquinoline inhibitors of vesicular monoamine transporter 2 |
| US20110206782A1 (en) * | 2010-02-24 | 2011-08-25 | Auspex Pharmaceuticals, Inc. | Piperidine modulators of dopamine receptor |
| US20120003330A1 (en) * | 2010-06-01 | 2012-01-05 | Auspex Pharmaceuticals, Inc. | Benzoquinolone inhibitors of vmat2 |
| US20130197067A1 (en) * | 2010-07-08 | 2013-08-01 | The Brigham And Women's Hospital, Inc. | Neuroprotective molecules and methods of treating neurological disorders and inducing stress granules |
| WO2013142816A1 (en) * | 2012-03-23 | 2013-09-26 | Cardero Therapeutics, Inc. | Compounds and compositions for the treatment of muscular disorders |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3071565A4 * |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11033540B2 (en) | 2012-09-18 | 2021-06-15 | Auspex Pharmaceuticals, Inc. | Formulations and pharmacokinetics of deuterated benzoquinoline inhibitors of vesicular monoamine transporter 2 |
| US9233959B2 (en) | 2012-09-18 | 2016-01-12 | Auspex Pharmaceuticals, Inc. | Formulations and pharmacokinetics of deuterated benzoquinoline inhibitors of vesicular monoamine transporter 2 |
| US9296739B2 (en) | 2012-09-18 | 2016-03-29 | Auspex Pharmaceuticals, Inc. | Formulations and pharmacokinetics of deuterated benzoquinoline inhibitors of vesicular monoamine transporter 2 |
| US9346800B2 (en) | 2012-09-18 | 2016-05-24 | Auspex Pharmaceuticals, Inc. | Formulations pharmacokinetics of deuterated benzoquinoline inhibitors of vesicular monoamine transporter 2 |
| US9550780B2 (en) | 2012-09-18 | 2017-01-24 | Auspex Pharmaceuticals, Inc. | Formulations pharmacokinetics of deuterated benzoquinoline inhibitors of vesicular monoamine transporter 2 |
| US9814708B2 (en) | 2012-09-18 | 2017-11-14 | Auspex Pharmaceuticals, Inc. | Formulations and pharmacokinetics of deuterated benzoquinoline inhibitors of vesicular monoamine transporter 2 |
| US11666566B2 (en) | 2012-09-18 | 2023-06-06 | Auspex Pharmaceuticals, Inc. | Formulations and pharmacokinetics of deuterated benzoquinoline inhibitors of vesicular monoamine transporter 2 |
| US10513488B2 (en) | 2013-12-03 | 2019-12-24 | Auspex Pharmaceuticals, Inc. | Methods of manufacturing benzoquinoline compounds |
| WO2015136003A3 (en) * | 2014-03-11 | 2015-11-26 | Imphar Aktiengesellschaft | Composition comprising a dopamine agonist and an l-dopa derivative for treating parkinson's disease |
| US10959996B2 (en) | 2015-03-06 | 2021-03-30 | Auspex Pharmaceuticals, Inc. | Methods for the treatment of abnormal involuntary movement disorders |
| US11357772B2 (en) | 2015-03-06 | 2022-06-14 | Auspex Pharmaceuticals, Inc. | Methods for the treatment of abnormal involuntary movement disorders |
| US11446291B2 (en) | 2015-03-06 | 2022-09-20 | Auspex Pharmaceuticals, Inc. | Methods for the treatment of abnormal involuntary movement disorders |
| US11564917B2 (en) | 2015-03-06 | 2023-01-31 | Auspex Pharmaceuticals, Inc. | Methods for the treatment of abnormal involuntary movement disorders |
| US11648244B2 (en) | 2015-03-06 | 2023-05-16 | Auspex Pharmaceuticals, Inc. | Methods for the treatment of abnormal involuntary movement disorders |
| US10093666B2 (en) | 2016-04-13 | 2018-10-09 | Arixa Pharmaceuticals, Inc. | Deuterated O-sulfated beta lactam hydroxamic acids and deuterated N-sulfated beta lactams |
| US10047077B2 (en) | 2016-04-13 | 2018-08-14 | Skyline Antiinfectives, Inc. | Deuterated O-sulfated beta-lactam hydroxamic acids and deuterated N-sulfated beta-lactams |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2017503756A (en) | 2017-02-02 |
| HK1224294A1 (en) | 2017-08-18 |
| EP3071565A1 (en) | 2016-09-28 |
| MX2016006622A (en) | 2016-12-09 |
| IL245538A0 (en) | 2016-06-30 |
| US20160303110A1 (en) | 2016-10-20 |
| EP3071565A4 (en) | 2017-08-02 |
| CA2930167A1 (en) | 2015-05-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2017200352B2 (en) | Benzoquinolone inhibitors of VMAT2 | |
| AU2019204534A1 (en) | Benzoquinoline inhibitors of vesicular monoamine transporter 2 | |
| EP3345905B1 (en) | Deuterated benzoquinolizine derivatives as inhibitors of vesicular monoamine transporter 2 | |
| AU2021201629A1 (en) | Benzoquinolone inhibitors of VMAT2 | |
| EP3071565A1 (en) | Methods of treating abnormal muscular activity | |
| US20110206780A1 (en) | Morphinan modulators of nmda receptors, sigma1 receptors, sigma2 receptors, and/or a3b4 nicotinic receptors | |
| WO2015077521A1 (en) | Benzoquinoline inhibitors of vesicular monoamine transporter 2 | |
| US20100105755A1 (en) | Substituted benzamide modulators of dopamine receptor | |
| CA2925562A1 (en) | Benzoquinolone inhibitors of vmat2 | |
| WO2010077730A2 (en) | Indanone inhibitors of acetylcholinesterase |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14864919 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 245538 Country of ref document: IL |
|
| ENP | Entry into the national phase |
Ref document number: 2930167 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 15037465 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 2016533116 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2016/006622 Country of ref document: MX |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2014864919 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014864919 Country of ref document: EP |