WO2015059618A1 - SUBSTITUTED PYRIMIDINE COMPOUNDS AS mPGES-1 INHIBITORS - Google Patents
SUBSTITUTED PYRIMIDINE COMPOUNDS AS mPGES-1 INHIBITORS Download PDFInfo
- Publication number
- WO2015059618A1 WO2015059618A1 PCT/IB2014/065465 IB2014065465W WO2015059618A1 WO 2015059618 A1 WO2015059618 A1 WO 2015059618A1 IB 2014065465 W IB2014065465 W IB 2014065465W WO 2015059618 A1 WO2015059618 A1 WO 2015059618A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- chloro
- trifluoromethyl
- formula
- methyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CC(C)C(*(C)(C)Cc(cc1)cc(C(*(C)C(*2C)=O)=CC2c2ccc(C3CC3)cc2)c1N)=O Chemical compound CC(C)C(*(C)(C)Cc(cc1)cc(C(*(C)C(*2C)=O)=CC2c2ccc(C3CC3)cc2)c1N)=O 0.000 description 3
- CDKPQUCKFSMQEA-PKNBQFBNSA-N CC(C)(C)C(NCc(c(F)c1/C=C/C(c2ccc(C)c(C)c2)=O)ccc1Cl)=O Chemical compound CC(C)(C)C(NCc(c(F)c1/C=C/C(c2ccc(C)c(C)c2)=O)ccc1Cl)=O CDKPQUCKFSMQEA-PKNBQFBNSA-N 0.000 description 1
- CHFIXQQTHCXPKE-UHFFFAOYSA-N CC(C)(C)C(NCc(ccc(Cl)c1C(N2)=CC(c3ccc(C(F)(F)F)nc3)=NC2=O)c1F)=O Chemical compound CC(C)(C)C(NCc(ccc(Cl)c1C(N2)=CC(c3ccc(C(F)(F)F)nc3)=NC2=O)c1F)=O CHFIXQQTHCXPKE-UHFFFAOYSA-N 0.000 description 1
- FFJDUAHDIKIOKH-JXMROGBWSA-N CC(C)(C)C(NCc1ccc(C)c(/C=C/C(c(cc2C(F)(F)F)ccc2Cl)=C)c1F)=O Chemical compound CC(C)(C)C(NCc1ccc(C)c(/C=C/C(c(cc2C(F)(F)F)ccc2Cl)=C)c1F)=O FFJDUAHDIKIOKH-JXMROGBWSA-N 0.000 description 1
- DVAMKGJHLMEIRH-UHFFFAOYSA-N CC(C)C(NCc(cc1C(N2)=CC(c3ccc(C(F)(F)F)cc3)=NC2=O)ccc1Cl)=O Chemical compound CC(C)C(NCc(cc1C(N2)=CC(c3ccc(C(F)(F)F)cc3)=NC2=O)ccc1Cl)=O DVAMKGJHLMEIRH-UHFFFAOYSA-N 0.000 description 1
- MNDQFJBOYXJYRZ-UHFFFAOYSA-N CC(c1c(C)ncc(C#N)c1)=O Chemical compound CC(c1c(C)ncc(C#N)c1)=O MNDQFJBOYXJYRZ-UHFFFAOYSA-N 0.000 description 1
- DBSSIOYDFODMFL-UHFFFAOYSA-N CC1(c2cc(C#N)cnc2C)OCCO1 Chemical compound CC1(c2cc(C#N)cnc2C)OCCO1 DBSSIOYDFODMFL-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/62—Oxygen or sulfur atoms
- C07D213/63—One oxygen atom
- C07D213/64—One oxygen atom attached in position 2 or 6
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/32—One oxygen, sulfur or nitrogen atom
- C07D239/34—One oxygen atom
- C07D239/36—One oxygen atom as doubly bound oxygen atom or as unsubstituted hydroxy radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/46—Two or more oxygen, sulphur or nitrogen atoms
- C07D239/47—One nitrogen atom and one oxygen or sulfur atom, e.g. cytosine
Definitions
- the present application relates to substituted pyrimidine compounds which may be useful as microsomal prostaglandin E synthase-1 (mPGES-1) inhibitors.
- mPGES-1 microsomal prostaglandin E synthase-1
- Inflammatory diseases that affect the population include asthma, inflammatory bowel disease, rheumatoid arthritis, osteoarthritis, rhinitis, conjunctivitis and dermatitis. Inflammation is also a common cause of pain.
- COX cyclooxygenase
- PGE 2 is particularly known to be a strong pro-inflammatory mediator, and is also known to induce fever and pain. Consequently, numerous drugs have been developed with a view to inhibiting the formation of PGE 2 , including "NS AIDs” (non-steroidal anti -inflammatory drugs) and “coxibs” (selective COX-2 inhibitors). These drugs act predominantly by inhibition of COX-1 and/or COX-2, thereby reducing the formation of PGE 2 .
- the inhibition of COXs has the disadvantage of reducing the formation of all metabolites of PGH 2 , thereby decreasing the beneficial properties of some of the metabolites. In view of this, drugs which act by inhibition of COXs are suspected to cause adverse biological effects.
- the non-selective inhibition of COXs by NSAIDs may give rise to gastrointestinal side-effects and affect platelet and renal function.
- Even the selective inhibition of COX-2 by coxibs, whilst reducing such gastrointestinal side-effects, is believed to give rise to cardiovascular problems.
- PGES prostaglandin E synthases
- mPGES-1 and mPGES-2 microsomal prostaglandin E synthases
- cPGES cytosolic prostaglandin E synthase
- mPGES-1 is an inducible PGES after exposure to pro- inflammatory stimuli.
- mPGES-1 is induced in the periphery and CNS by inflammation, and represents therefore a target for acute and chronic inflammatory disorders.
- PGE 2 is a major prostanoid, produced from arachidonic acid liberated by phospholipases (PLAs), which drives the inflammatory processes.
- PHAs phospholipases
- Arachidonic acid is transformed by the action of prostaglandin H synthase (PGH synthase, cycloxygenase) into PGH 2 which is a substrate for mPGES-1, the terminal enzyme transforming PGH 2 to the pro-inflammatory PGE 2 .
- PGH synthase prostaglandin H synthase
- Agents that are capable of inhibiting the action of mPGES-1, and thus reducing the formation of the specific arachidonic acid metabolite PGE 2 are beneficial in the treatment of inflammation. Further, agents that are capable of inhibiting the action of the proteins involved in the synthesis of the leukotrienes are also beneficial in the treatment of asthma and COPD.
- PGE 2 facilitates tumor progression by stimulation of cellular proliferation and angiogenesis and by modulation of immunosupression.
- genetic deletion of mPGES-1 in mice suppresses intestinal tumourogenesis (Nakanishi et. al., Cancer Research 2008, 68(9), 3251-9).
- mPGES-1 is also upregulated in cancers such as colorectal cancer (Schroder, Journal of Lipid Research 2006, 47, 1071-80).
- Myositis is a chronic muscle disorder characterized by muscle weakness and fatigue. Proinflammatory cytokines and prostanoids have been implicated in the development of myositis. In skeletal muscle tissue from patients suffering from myositis an increase in cyclooxygenases and mPGES-1 has been demonstrated, implicating mPGES-1 as a target for treating this condition. (Korotkova, Annals of the Rheumatic Diseases 2008, 67, 1596- 1602).
- Atherosclerosis inflammation of the vasculature leads to atheroma formation that eventually may progress into infarction.
- carotid atherosclerosis an increase in mPGES-1 in plaque regions has been reported (Gomez- Hernandez Atherosclerosis 2006, 187, 139-49).
- mice lacking the mPGES-1 receptor were found to show a retarded atherogenesis and a concomitant reduction in macrophage-derived foam cells together with an increase in vascular smooth muscle cells (Wang, Proceedings of National Academy of Sciences 2006, 103(39), 14507-12).
- the present application is directed to compounds that act as inhibitors of the mPGES-1 enzyme and, therefore, are useful for the treatment of pain and inflammation in a variety of diseases or conditions.
- the present invention relates to compound of formula (I):
- X is O, when dotted line T represents a single bond and dotted line '2' is absent; or X is OR a or R a R b , when dotted line ' 1 ' is absent and dotted line '2' represents a single bond;
- G 2 is N
- G 3 is sleeted from N and CH;
- G 4 is selected from N and CH;
- L is selected from -CO- or -S(0) 2 -;
- W is selected from Ci-salkyl, haloCi-salkyl and hydroxyCi-salkyl;
- R a and R b are each independently selected from hydrogen, Ci -4 alkyl and - C(0)Ci -4 alkyl;
- R 1 is selected from hydrogen, halogen and Ci -4 alkyl
- R 2 is absent when dotted line '2' represents a single bond; or R 2 is hydrogen when dotted line '2' is absent;
- aatt eeaacchh ooccccuurrrreennce R is independently selected from halogen, nitro, cyano, Ci. salkyl and haloCi-salkyl;
- R 4 is independently selected from halogen, Ci-salkyl, haloCi-salkyl and C 3- i 2 cycloalkyl;
- R x and R y are hydrogen
- 'm' is an integer ranging from 0 to 3, both inclusive;
- 'n' is an integer ranging from 1 to 4, both inclusive;
- 'p' is an integer ranging from 1 to 3, both inclusive.
- the compounds of formula (I) may involve one or more embodiments.
- Embodiments of formula (I) include compounds of formula (II), formula (III) and formula (IV) as described hereinafter. It is to be understood that the embodiments below are illustrative of the present invention and are not intended to limit the claims to the specific embodiments exemplified. It is also to be understood that the embodiments defined herein may be used independently or in conjunction with any definition of any other embodiment defined herein. Thus, the invention contemplates all possible combinations and permutations of the various independently described embodiments.
- the invention provides compounds of formula (I) as defined above, wherein R 1 is hydrogen or Ci -4 alkyl (according to an embodiment defined below), L is -CO- (according to another embodiment defined below), and 'p' is 1 (according to yet another embodiment defined below).
- R a is hydrogen
- Ci -4 alkyl e.g. methyl
- -C(0)Ci -4 alkyl e.g. -C(O)methyl
- R b is hydrogen
- R a is -C(0)Ci -4 alkyl (e.g. -C(O)methyl).
- R 1 is hydrogen or Ci -4 alkyl (e.g. methyl).
- R 3 is halogen (e.g. fluorine or chlorine), Ci -8 alkyl (e.g. methyl) or haloCi -8 alkyl (e.g. difluoromethyl).
- R 3 is fluorine, chlorine, methyl or difluoromethyl.
- R 4 is halogen (e.g. chlorine), Ci -8 alkyl (e.g. methyl, isopropyl or ie/ -butyl), haloCi -8 alkyl (e.g. tnfluoromethyl) or C3-i 2 cycloalkyl (e.g. cyclopropyl).
- R 4 is halogen (e.g. chlorine), Ci -8 alkyl (e.g. methyl, isopropyl or ie/ -butyl), haloCi -8 alkyl (e.g. tnfluoromethyl) or C3-i 2 cycloalkyl (e.g. cyclopropyl).
- R 4 is chlorine, methyl, isopropyl, ie/t-butyl, trifluoromethyl or cyclopropyl.
- W is Ci -8 alkyl (e.g. isopropyl or ie/ -butyl).
- X is O, dotted line T represents a single bond and dotted line '2' is absent;
- R 1 is hydrogen or methyl
- R 2 is hydrogen
- R 3 is fluorine, chlorine, methyl or difluoromethyl
- R 4 is isopropyl, tert-butyl, trifluoromethyl or cyclopropyl
- L is -CO-
- W is isopropyl or ie/t-butyl
- R x and R y are hydrogen; m is 1 or 2; n is 1 and p is 1.
- X is OR a or R a R b , dotted line ' 1 ' is absent and dotted line '2' represents a single bond;
- R a is hydrogen, methyl or -C(0)methyl
- R b is hydrogen
- R 1 is hydrogen
- R 2 is absent
- R 3 is fluorine or chlorine
- R 4 is chlorine, methyl or trifluoromethyl.
- L is -CO-
- W is tert-butyl
- R x and R y are hydrogen; m is 1 or 2; n is lor 2 and p is 1.
- compounds of formula (I) that exhibit an IC 50 value of less than 500 nM, preferably, less than 100 nM, more preferably, less than 50 nM with respect to mPGES-1 inhibition.
- the invention specifically provides compounds of formula (II), formula (III) or formula (IV) wherein R 1 is hydrogen or Ci -4 alkyl and consequently there is also provided a compound of formula (I) wherein R 1 is hydrogen or Ci -4 alkyl.
- the invention also provides a compound of formula (II) which is an embodiment of a compound of formula (I).
- G 3 is sleeted from N and CH;
- G 4 is selected from N and CH;
- L is selected from -CO- or -S(0) 2 -;
- W is selected from Ci -8 alkyl, haloCi -8 alkyl and hydroxyCi -8 alkyl;
- R 1 is selected from hydrogen, halogen and
- R 3 is independently selected from halogen, nitro, cyano, Ci. 8 alkyl and haloCi -8 alkyl;
- R 4 is independently selected from halogen, Ci -8 alkyl, haloCi -8 alkyl and C3-i 2 cycloalkyl;
- R x and R y are hydrogen
- 'm' is an integer ranging from 0 to 3, both inclusive;
- 'n' is an integer ranging from 1 to 4, both inclusive;
- 'p' is an integer ranging from 1 to 3, both inclusive.
- the compounds of formula (II) may involve one or more embodiments. It is to be understood that the embodiments below are illustrative of the present invention and are not intended to limit the claims to the specific embodiments exemplified. It is also to be understood that the embodiments defined herein may be used independently or in conjunction with any definition of any other embodiment defined herein. Thus, the invention contemplates all possible combinations and permutations of the various independently described embodiments.
- the invention provides compounds of formula (II) as defined above, wherein R 1 is hydrogen or (according to an embodiment defined below), R 4 is Ci -8 alkyl, haloCi -8 alkyl or C 3- i 2 cycloalkyl (according to another embodiment defined below), and W is Ci -8 alkyl (according to yet another embodiment defined below).
- R 1 is hydrogen or (according to an embodiment defined below)
- R 4 is Ci -8 alkyl, haloCi -8 alkyl or C 3- i 2 cycloalkyl (according to another embodiment defined below)
- W Ci -8 alkyl (according to yet another embodiment defined below).
- G 3 is CH.
- R 1 is hydrogen or Ci -4 alkyl (e.g. methyl).
- R 3 is halogen (e.g. fluorine or chlorine), Ci-salkyl (e.g. methyl) or haloCi-salkyl (e.g. difluoromethyl).
- R 3 is fluorine, chlorine, methyl or difluoromethyl.
- R 4 is Ci-salkyl (e.g. isopropyl or ie/t-butyl), haloCi-salkyl (e.g. trifluoromethyl) or C 3- i 2 Cycloalkyl (e.g. cyclopropyl).
- Ci-salkyl e.g. isopropyl or ie/t-butyl
- haloCi-salkyl e.g. trifluoromethyl
- C 3- i 2 Cycloalkyl e.g. cyclopropyl
- R 4 is isopropyl, tert-butyl, trifluoromethyl or cyclopropyl.
- W is Ci-salkyl (e.g. isopropyl or tert-buty ⁇ ).
- G 3 is N or CH;
- G 4 is N or CH;
- R 1 is hydrogen or methyl
- R 3 is fluorine, chlorine, methyl or difluoromethyl
- R 4 is isopropyl, ie/t-butyl, trifluoromethyl or cyclopropyl
- L is -CO-
- W is isopropyl or ie/t-butyl
- R x and R y are hydrogen; m is 1 or 2; n is 1 and p is 1.
- compounds of formula (II) that exhibit an IC 50 value of less than 500 nM, preferably, less than 100 nM, more preferably, less than 50 nM with respect to mPGES-1 inhibition.
- the invention also provides a compound of formula (III) which is an embodiment of a compound of formula (I).
- W is selected from Ci -8 alkyl, haloCi -8 alkyl and hydroxyCi -8 alkyl;
- R a and R b are each independently selected from hydrogen, Ci -4 alkyl and - C(0)Ci -4 alkyl;
- R 3 is independently selected from halogen, nitro, cyano, Ci. 8 alkyl and haloCi -8 alkyl;
- R 4 is independently selected from halogen, Ci -8 alkyl, haloCi -8 alkyl and C 3- i 2 cycloalkyl;
- 'm' is an integer ranging from 0 to 3, both inclusive; and 'n' is an integer ranging from 1 to 4, both inclusive.
- the compounds of formula (III) may involve one or more embodiments. It is to be understood that the embodiments below are illustrative of the present invention and are not intended to limit the claims to the specific embodiments exemplified. It is also to be understood that the embodiments defined herein may be used independently or in conjunction with any definition of any other embodiment defined herein. Thus, the invention contemplates all possible combinations and permutations of the various independently described embodiments.
- the invention provides compounds of formula (III) as defined above, wherein R b is hydrogen (according to an embodiment defined below), R 4 is halogen, Ci -8 alkyl or haloCi -8 alkyl (according to another embodiment defined below), and W is Ci -8 alkyl (according to yet another embodiment defined below).
- R a is hydrogen, (e.g. methyl) or -C(0)Ci- 4 alkyl (e.g. - C(O)methyl).
- R a is hydrogen, methyl or -C(0)methyl.
- R a is hydrogen, methyl or -C(0)methyl and R b is hydrogen.
- R 3 is halogen (e.g. fluorine or chlorine).
- R 4 is halogen (e.g. chlorine), Ci -8 alkyl (e.g. methyl) or haloCi. 8 alkyl (e.g. trifluoromethyl).
- R 4 is chlorine, methyl or trifluoromethyl.
- W is Ci -8 alkyl (e.g. ie/ -butyl).
- R a is hydrogen, methyl or -C(0)methyl
- R b is hydrogen
- R 3 is fluorine or chlorine; R 4 is chlorine, methyl or trifluorom ethyl.
- W is tert-butyl; m is 1 or 2 and n is lor 2.
- the invention also provides a compound of formula (IV) which is an embodiment of a compound of formula (I).
- W is selected from Ci -8 alkyl, haloCi -8 alkyl and hydroxyCi -8 alkyl;
- R a is -C(0)Ci -4 alkyl
- R 3 is independently selected from halogen, nitro, cyano, Ci. 8 alkyl and haloCi -8 alkyl;
- R 4 is independently selected from halogen, Ci -8 alkyl, haloCi -8 alkyl and C 3- i 2 cycloalkyl;
- 'm' is an integer ranging from 0 to 3, both inclusive;
- 'n' is an integer ranging from 1 to 4, both inclusive.
- the compounds of formula (IV) may involve one or more embodiments. It is to be understood that the embodiments below are illustrative of the present invention and are not intended to limit the claims to the specific embodiments exemplified. It is also to be understood that the embodiments defined herein may be used independently or in conjunction with any definition of any other embodiment defined herein. Thus, the invention contemplates all possible combinations and permutations of the various independently described embodiments.
- the invention provides compounds of formula (III) as defined above, wherein R a is C(0)Ci- 4 alkyl (according to an embodiment defined below), R 4 is haloCi -8 alkyl (according to another embodiment defined below), and W is Ci -8 alkyl (according to yet another embodiment defined below).
- R a is -C(0)C M alkyl (e.g. -C(O)methyl).
- R 3 is halogen (e.g. chlorine).
- R 4 is haloCi -8 alkyl (e.g. trifluoromethyl).
- W is Ci -8 alkyl (e.g. ie/ -butyl).
- R a is -C(0)methyl; R 3 is chlorine; R 4 is trifluoromethyl.
- W is tert-butyl; m is 1 and n is 1.
- compounds of formula (IV) that exhibit an IC 50 value of less than 500 nM, preferably, less than 100 nM, more preferably, less than 50 nM with respect to mPGES-1 inhibition.
- the compounds of formula (II) structurally encompass all tautomeric forms whether such tautomer exists in equilibrium or predominantly in one form.
- Such tautomeric form may be different or the same when the compound is bound to the mPGES-1 enzyme.
- the present application also provides a pharmaceutical composition that includes at least one compound described herein and at least one pharmaceutically acceptable excipient (such as a pharmaceutically acceptable carrier or diluent).
- the pharmaceutical composition comprises a therapeutically effective amount of at least one compound described herein.
- the compounds described herein may be associated with a pharmaceutically acceptable excipient (such as a carrier or a diluent) or be diluted by a carrier, or enclosed within a carrier which can be in the form of a capsule, sachet, paper or other container.
- the compounds and pharmaceutical compositions of the present invention are useful for inhibiting the activity of mPGES-1, which is related to a variety of disease states.
- the present invention further provides a method of inhibiting mPGES-1 in a subject in need thereof by administering to the subject one or more compounds described herein in an amount effective to cause inhibition of such receptor.
- halogen or halo means fluorine (fluoro), chlorine (chloro), bromine (bromo), or iodine (iodo).
- alkyl refers to a hydrocarbon chain radical that includes solely carbon and hydrogen atoms in the backbone, containing no unsaturation, having from one to eight carbon atoms (i.e. Ci -8 alkyl), and which is attached to the rest of the molecule by a single bond, e.g., methyl, ethyl, n-propyl, 1-methylethyl (isopropyl), n- butyl, n-pentyl, and 1,1-dimethylethyl (t-butyl).
- Ci -8 alkyl refers to an alkyl chain having 1 to 6 carbon atoms.
- the term refers to an alkyl chain having 1 to 4 carbon atoms. Unless set forth or recited to the contrary, all alkyl groups described or claimed herein may be straight chain or branched, substituted or unsubstituted.
- alkenyl refers to a hydrocarbon chain containing from 2 to 10 carbon atoms (i.e. C2-ioalkenyl) and including at least one carbon-carbon double bond.
- alkenyl groups include ethenyl, 1-propenyl, 2-propenyl (allyl), z ' so-propenyl, 2-methyl- 1-propenyl, 1-butenyl, and 2-butenyl. Unless set forth or recited to the contrary, all alkenyl groups described or claimed herein may be straight chain or branched, substituted or unsubstituted.
- alkynyl refers to a hydrocarbyl radical having at least one carbon- carbon triple bond, and having 2 to about 12 carbon atoms (with radicals having 2 to about 10 carbon atoms being preferred i.e. C2-ioalkynyl).
- alkynyl groups include ethynyl, propynyl, and butynyl. Unless set forth or recited to the contrary, all alkynyl groups described or claimed herein may be straight chain or branched, substituted or unsubstituted.
- alkoxy denotes an alkyl group attached via an oxygen linkage to the rest of the molecule (i.e. Ci -8 alkoxy). Representative examples of such groups are -OCH3 and -OC 2 H 5 . Unless set forth or recited to the contrary, all alkoxy groups described or claimed herein may be straight chain or branched, substituted or unsubstituted.
- alkoxyalkyl or “alkyloxyalkyl” refers to an alkoxy or alkyloxy group as defined above directly bonded to an alkyl group as defined above (i.e. Ci. 8 alkoxyCi- 8 alkyl or Ci-salkyloxyCi-salkyl).
- alkoxyalkyl moiety includes, but are not limited to, -CH 2 OCH 3 and -CH 2 OC 2 H 5 . Unless set forth or recited to the contrary, all alkoxyalkyl groups described herein may be straight chain or branched, substituted or unsubstituted.
- haloalkyl refers to at least one halo group (selected from F, CI, Br or I), linked to an alkyl group as defined above (i.e. haloCi-salkyl).
- haloalkyl moiety include, but are not limited to, trifluoromethyl, difluoromethyl and fluoromethyl groups. Unless set forth or recited to the contrary, all haloalkyl groups described herein may be straight chain or branched, substituted or unsubstituted.
- haloalkoxy refers to an alkoxy group substituted with one or more halogen atoms (i.e. haloCi-salkoxy).
- haloalkoxy include but are not limited to fluoromethoxy, difluoromethoxy, trifluoromethoxy, 2,2,2-trifluoroethoxy, pentafluoroethoxy, pentachloroethoxy, chloromethoxy, dichlorormethoxy, trichloromethoxy and 1-bromoethoxy.
- all haloalkoxy groups described herein may be straight chain or branched, substituted or unsubstituted.
- haloalkoxyalkyl refers to haloalkoxy group as defined above directly bonded to an alkyl group as defined above (i.e. haloCi-salkoxyCi-salkyl).
- haloCi-salkoxyCi-salkyl examples include but are not limited to (2,2,2- trifluoroethoxy )m ethyl or (2,2-difluoroethoxy)m ethyl. Unless set forth or recited to the contrary, all haloalkoxyalkyl groups described herein may be straight chain or branched, substituted or unsubstituted.
- hydroxyalkyl refers to an alkyl group as defined above wherein one to three hydrogen atoms on different carbon atoms is/are replaced by hydroxyl groups (i.e. hydroxyCi-salkyl).
- hydroxyalkyl moieties include, but are not limited to -CH 2 OH, -C 2 H 4 OH and -CH(OH)C 2 H 4 OH. Unless set forth or recited to the contrary, all hydroxyalkyl groups described herein may be straight chain or branched, substituted or unsubstituted.
- carboxyl means the group -COOH.
- carboxylalkyl refers to an Ci -8 alkyl group as defined above wherein at least one of the hydrogen atoms of the Ci -8 alkyl group is replaced by a carboxyl group (i.e. "carboxylCi -8 alkyl”).
- carboxylalkyl moieties include, but are not limited to carboxylmethyl (-CH 2 -COOH), carboxylethyl (-CH 2 - CH 2 -COOH), carboxylisopropyl (-C(CH 3 ) 2 -COOH) and carboxyltertbutyl (- C(CH 3 ) 2 CH 2 -COOH). Unless set forth or recited to the contrary, all carboxylalkyl groups described herein may be straight chain or branched, substituted or unsubstituted.
- cycloalkyl denotes a non-aromatic mono or multicyclic ring system of 3 to about 12 carbon atoms, (i.e. C 3- i 2 cycloalkyl).
- monocyclic cycloalkyl include but are not limited to cyclopropyl, cyclobutyl, cyclopentyl, and cyclohexyl.
- multicyclic cycloalkyl groups include, but are not limited to, perhydronapthyl, adamantyl and norbornyl groups, bridged cyclic groups or spirobicyclic groups, e.g., spiro(4,4)non-2-yl.
- C 3-6 Cycloalkyl refers to the cyclic ring having 3 to 6 carbon atoms. Unless set forth or recited to the contrary, all cycloalkyl groups described or claimed herein may be substituted or unsubstituted.
- cycloalkylalkyl refers to a non-aromatic cyclic ring-containing radical having 3 to about 8 carbon atoms directly attached to an alkyl group (i.e. C 3- 8 cycloalkylCi -8 alkyl).
- the cycloalkylalkyl group may be attached to the main structure at any carbon atom in the alkyl group that results in the creation of a stable structure.
- Non-limiting examples of such groups include cyclopropylmethyl, cyclobutylethyl, and cyclopentylethyl. Unless set forth or recited to the contrary, all cycloalkylalkyl groups described or claimed herein may be substituted or unsubstituted.
- cycloalkenyl refers to a ccyclic ring-containing radical having 3 to about 8 carbon atoms with at least one carbon-carbon double bond, (i.e. C 3- 8 cycloalkenyl).
- Examples of “cycloalkenyl” include but are not limited to cyclopropenyl, cyclobutenyl, and cyclopentenyl. Unless set forth or recited to the contrary, all cycloalkenyl groups described or claimed herein may be substituted or unsubstituted.
- cycloalkenylalkyl refers to a non-aromatic cyclic ring-containing radical having 3 to about 8 carbon atoms with at least one carbon-carbon double bond, directly attached to an alkyl group, (i.e. C 3-8 cycloalkenylCi -8 alkyl). Unless set forth or recited to the contrary, all cycloalkenylalkyl groups described or claimed herein may be substituted or unsubstituted.
- aryl refers to an aromatic radical having 6 to 14 carbon atoms (i.e. C 6 -i 4 ryl), including monocyclic, bicyclic and tricyclic aromatic systems, such as phenyl, naphthyl, tetrahydronapthyl, indanyl, and biphenyl. Unless set forth or recited to the contrary, all aryl groups described or claimed herein may be substituted or unsubstituted.
- aryloxy refers to an aryl group as defined above attached via an oxygen linkage to the rest of the molecule (i.e. C 6 -i 4 aryloxy).
- aryloxy moieties include, but are not limited to phenoxy and naphthoxy. Unless set forth or recited to the contrary, all aryloxy groups described herein may be substituted or unsubstituted.
- arylalkyl refers to an aryl group as defined above directly bonded to an alkyl group as defined above, i.e. such as -CH 2 C 6 H 5 and - C 2 H 4 C 6 H 5 . Unless set forth or recited to the contrary, all arylalkyl groups described or claimed herein may be substituted or unsubstituted.
- heterocyclic ring or “heterocyclyl” unless otherwise specified refers to substituted or unsubstituted non-aromatic 3 to 15 membered ring radical (i.e. 3 to 15 membered heterocyclyl) which consists of carbon atoms and from one to five hetero atoms selected from nitrogen, phosphorus, oxygen and sulfur.
- the heterocyclic ring radical may be a mono-, bi- or tricyclic ring system, which may include fused, bridged or spiro ring systems, and the nitrogen, phosphorus, carbon, oxygen or sulfur atoms in the heterocyclic ring radical may be optionally oxidized to various oxidation states.
- heterocyclic ring or heterocyclyl may optionally contain one or more olefinic bond(s).
- heterocyclic ring radicals include, but are not limited to azepinyl, azetidinyl, benzodioxolyl, benzodioxanyl, chromanyl, dioxolanyl, dioxaphospholanyl, decahydroisoquinolyl, indanyl, indolinyl, isoindolinyl, isochromanyl, isothiazolidinyl, isoxazolidinyl, morpholinyl, oxazolinyl, oxazolidinyl, oxadiazolyl, 2-oxopiperazinyl, 2- oxopiperidinyl, 2-oxopyrrolidinyl, 2-oxoazepinyl,
- heterocyclylalkyl refers to a heterocyclic ring radical directly bonded to an alkyl group (i.e. 3 to 15 membered heterocyclylCi-salkyl). Unless set forth or recited to the contrary, all heterocyclylalkyl groups described or claimed herein may be substituted or unsubstituted.
- heteroaryl refers to substituted or unsubstituted 5 to 14 membered aromatic heterocyclic ring radical with one or more heteroatom(s) independently selected from N, O or S (i.e. 5 to 14 membered heteroaryl).
- the heteroaryl may be a mono-, bi- or tricyclic ring system.
- heteroaryl ring radicals include, but are not limited to oxazolyl, isoxazolyl, imidazolyl, furyl, indolyl, isoindolyl, pyrrolyl, triazolyl, triazinyl, tetrazoyl, thienyl, thiazolyl, isothiazolyl, pyridyl, pyrimidinyl, pyrazinyl, pyridazinyl, pyrazolyl, benzofuranyl, benzothiazolyl, benzoxazolyl, benzimidazolyl, benzothienyl, benzopyranyl, carbazolyl, quinolinyl, isoquinolinyl, quinazolinyl, cinnolinyl, naphthyridinyl, pteridinyl, purinyl, quinoxalinyl, quinolyl, isoquinolyl, thiadiazol
- heteroarylalkyl refers to a heteroaryl ring radical directly bonded to an alkyl group (i.e. 5 to 14 membered heterarylCi-salkyl). Unless set forth or recited to the contrary, all heteroarylalkyl groups described or claimed herein may be substituted or unsubstituted.
- salts prepared from pharmaceutically acceptable bases or acids including inorganic or organic bases and inorganic or organic acids include, but are not limited to, acetate, benzenesulfonate, benzoate, bicarbonate, bisulfate, bitartrate, borate, bromide, camsylate, carbonate, chloride, clavulanate, citrate, dihydrochloride, edetate, edisylate, estolate, esylate, fumarate, gluceptate, gluconate, glutamate, glycollylarsanilate, hexylresorcinate, hydrabamine, hydrobromide, hydrochloride, hydroxynaphthoate, iodide, isothionate, lactate, lactobionate, laurate, malate, maleate, mandelate, mesylate, methylbromide, methylnitrate, methyl sul
- treating or “treatment” of a state, disorder or condition includes: (a) preventing or delaying the appearance of clinical symptoms of the state, disorder or condition developing in a subject that may be afflicted with or predisposed to the state, disorder or condition but does not yet experience or display clinical or subclinical symptoms of the state, disorder or condition; (b) inhibiting the state, disorder or condition, i.e., arresting or reducing the development of the disease or at least one clinical or subclinical symptom thereof; or (c) relieving the disease, i.e., causing regression of the state, disorder or condition or at least one of its clinical or subclinical symptoms.
- subject includes mammals (especially humans) and other animals, such as domestic animals (e.g., household pets including cats and dogs) and non- domestic animals (such as wildlife).
- domestic animals e.g., household pets including cats and dogs
- non- domestic animals such as wildlife.
- a “therapeutically effective amount” means the amount of a compound that, when administered to a subject for treating a state, disorder or condition, is sufficient to effect such treatment.
- the “therapeutically effective amount” may vary depending on the compound, the disease and its severity and the age, weight, physical condition and responsiveness of the subj ect to be treated.
- Nociceptors are primary sensory afferent (C and ⁇ fibers) neurons that are activated by a wide variety of noxious stimuli including chemical, mechanical, thermal, and proton (pH ⁇ 6) modalities.
- Nociceptors are the nerves which sense and respond to parts of the body which suffer from damage. They signal tissue irritation, impending injury, or actual injury. When activated, they transmit pain signals (via the peripheral nerves as well as the spinal cord) to the brain.
- chronic pain usually refers to pain which persists for 3 months or longer and can lead to significant changes in a patient's personality, lifestyle, functional ability and overall quality of life.
- Chronic pain can be classified as either nociceptive or neuropathic.
- Nociceptive pain includes tissue injury -induced pain and inflammatory pain such as that associated with arthritis.
- Neuropathic pain is caused by damage to the sensory nerves of the peripheral or central nervous system and is maintained by aberrant somatosensory processing. The pain is typically well localized, constant, and often with an aching or throbbing quality.
- Visceral pain is the subtype of nociceptive pain that involves the internal organs. It tends to be episodic and poorly localized.
- Nociceptive pain is usually time limited, meaning when the tissue damage heals, the pain typically resolves (arthritis is a notable exception in that it is not time limited).
- the compounds of the invention are typically administered in the form of a pharmaceutical composition.
- Such compositions can be prepared using procedures known in the pharmaceutical art and comprise at least one compound of the invention.
- the pharmaceutical composition of the present patent application comprises one or more compounds described herein and one or more pharmaceutically acceptable excipients.
- the pharmaceutically acceptable excipients are approved by regulatory authorities or are generally regarded as safe for human or animal use.
- the pharmaceutically acceptable excipients include, but are not limited to, carriers, diluents, glidants and lubricants, preservatives, buffering agents, chelating agents, polymers, gelling agents, viscosifying agents, and solvents.
- suitable carriers include, but are not limited to, water, salt solutions, alcohols, polyethylene glycols, peanut oil, olive oil, gelatin, lactose, terra alba, sucrose, dextrin, magnesium carbonate, sugar, amylose, magnesium stearate, talc, gelatin, agar, pectin, acacia, stearic acid, lower alkyl ethers of cellulose, silicic acid, fatty acids, fatty acid amines, fatty acid monoglycerides and diglycerides, fatty acid esters, and polyoxyethylene.
- the pharmaceutical composition may also include one or more pharmaceutically acceptable auxiliary agents, wetting agents, suspending agents, preserving agents, buffers, sweetening agents, flavoring agents, colorants or any combination of the foregoing.
- compositions may be in conventional forms, for example, capsules, tablets, solutions, suspensions, injectables or products for topical application. Further, the pharmaceutical composition of the present invention may be formulated so as to provide a desired release profile.
- Administration of the compounds of the invention, in pure form or in an appropriate pharmaceutical composition can be carried out using any of the accepted routes of administration of pharmaceutical compositions.
- the route of administration may be any route which effectively transports the active compound of the patent application to the appropriate or desired site of action.
- Suitable routes of administration include, but are not limited to, oral, nasal, buccal, dermal, intradermal, transdermal, parenteral, rectal, subcutaneous, intravenous, intraurethral, intramuscular, or topical.
- Solid oral formulations include, but are not limited to, tablets, capsules (soft or hard gelatin), dragees (containing the active ingredient in powder or pellet form), troches and lozenges.
- Liquid formulations include, but are not limited to, syrups, emulsions, and sterile injectable liquids, such as suspensions or solutions.
- Topical dosage forms of the compounds include ointments, pastes, creams, lotions, powders, solutions, eye or ear drops, and impregnated dressings, and may contain appropriate conventional additives such as preservatives, solvents to assist drug penetration.
- compositions of the present patent application may be prepared by conventional techniques, e.g., as described in Remington: The Science and Practice of Pharmacy, 20 th Ed., 2003 (Lippincott Williams & Wilkins).
- Suitable doses of the compounds for use in treating the diseases and disorders described herein can be determined by those skilled in the relevant art.
- Therapeutic doses are generally identified through a dose ranging study in humans based on preliminary evidence derived from animal studies. Doses are generally sufficient to result in a desired therapeutic benefit without causing unwanted side effects. Mode of administration, dosage forms, and suitable pharmaceutical excipients can also be well used and adjusted by those skilled in the art. All changes and modifications are envisioned within the scope of the present patent application.
- Compounds of the present invention are particularly useful because they may inhibit the activity of prostaglandin E synthases (and particularly microsomal prostaglandin E synthase-1 (mPGES-1)), i.e., they prevent, inhibit, or suppress the action of mPGES-1 or a complex of which the mPGES-1 enzyme forms a part, and/or may elicit mPGES-1 modulating effect.
- mPGES-1 microsomal prostaglandin E synthase-1
- inflammation will be understood by those skilled in the art to include any condition characterized by a localized or a systemic protective response, which may be elicited by physical trauma, infection, chronic diseases, such as those mentioned hereinbefore, and/or chemical and/or physiological reactions to external stimuli (e.g. as part of an allergic response). Any such response, which may serve to destroy, dilute or sequester both the injurious agent and the injured tissue, may be manifest by, for example, heat, swelling, pain, redness, dilation of blood vessels and/or increased blood flow.
- inflammation is also understood to include any inflammatory disease, disorder or condition per se, any condition that has an inflammatory component associated with it, and/or any condition characterized by inflammation as a symptom, including, inter alia, acute, chronic, ulcerative, specific, allergic, infection by pathogens, immune reactions due to hypersensitivity, entering foreign bodies, physical injury, and necrotic inflammation, and other forms of inflammation known to those skilled in the art.
- the term thus also includes, for the purposes of this invention, inflammatory pain, pain generally and/or fever.
- the compounds of the present invention may also be useful in the treatment of asthma, chronic obstructive pulmonary disease, pulmonary fibrosis, inflammatory bowel disease, irritable bowel syndrome, inflammatory pain, chronic pain, acute pain, fever, migraine, headache, low back pain, fibromyalgia, myofascial disorders, viral infections (e.g. influenza, common cold, herpes zoster, hepatitis C and AIDS), bacterial infections, fungal infections, dysmenorrhea, burns, surgical or dental procedures, malignancies (e.g. influenza, common cold, herpes zoster, hepatitis C and AIDS), bacterial infections, fungal infections, dysmenorrhea, burns, surgical or dental procedures, malignancies (e.g.
- hyperprostaglandin E syndrome classic Bartter syndrome, atherosclerosis, gout, arthritis, osteoarthritis, juvenile arthritis, rheumatoid arthritis, juvenile onset rheumatoid arthritis, rheumatic fever, ankylosing spondylitis, Hodgkin's disease, systemic lupus erythematosus, vasculitis, pancreatitis, nephritis, bursitis, conjunctivitis, ulceris, scleritis, uveitis, wound healing, dermatitis, eczema, psoriasis, stroke, diabetes mellitus, neurodegenerative disorders such as Alzheimer's disease and multiple sclerosis, autoimmune diseases, allergic disorders, rhinitis, ulcers, mild to moderately active ulcerative colitis, familial adenomatous polyposis, coronary heart disease, sarcoidosis and any other disease with an inflammatory
- Compounds of the invention may also have effects that are not linked to inflammatory mechanisms, such as in the reduction of bone loss in a subject.
- Conditions that may be mentioned in this regard include osteoporosis, osteoarthritis, Paget's disease and/or periodontal diseases.
- the compounds are useful for the relief of pain, fever and inflammation of a variety of conditions including rheumatic fever, symptoms associated with influenza or other viral infections, common cold, low back and neck pain, dysmenorrhea, headache, migraine (acute and prophylactic treatment), toothache, sprains and strains, myositis, neuralgia, synovitis, arthritis, including rheumatoid arthritis, juvenile rheumatoid arthritis, degenerative joint diseases (osteoarthritis), acute gout and ankylosing spondylitis, acute, subacute and chronic musculoskeletal pain syndromes such as bursitis, burns, injuries, and pain following surgical (post-operative pain) and dental procedures as well as the preemptive treatment of surgical pain.
- rheumatic fever symptoms associated with influenza or other viral infections, common cold, low back and neck pain, dysmenorrhea, headache, migraine (acute and prophylactic treatment), toothache, sprains and strains, myo
- the pain may be mild pain, moderate pain, severe pain, musculoskeletal pain, complex regional pain syndrome, neuropathic pain, back pain such as acute visceral pain, neuropathies, acute trauma, chemotherapy - induced mononeuropathy pain states, polyneuropathy pain states (such as diabetic peripheral neuropathy & chemotherapy induced neuropathy), autonomic neuropathy pain states, pheriphaeral nervous system (PNS) lesion or central nervous system (CNS) lesion or disease related pain states, polyradiculopathies of cervical, lumbar or sciatica type, cauda equina syndrome, piriformis syndrome, paraplegia, quadriplegia, pain states related to various Polyneuritis conditions underlying various infections, chemical injuries, radiation exposure, underlying disease or deficiency conditions (such as beriberi, vitamin deficiencies, hypothyroidism, porphyria, cancer, HIV, autoimmune disease such as multiple sclerosis and spinal-cord injury, fibromyalgia, nerve injury, ischaemia, neuro
- Compounds of the present invention will also inhibit prostanoid-induced smooth muscle contraction by preventing the synthesis of contractile prostanoids and hence may be of use in the treatment of dysmenorrhea, premature labor and asthma.
- cancer includes Acute Lymphoblastic Leukemia, Acute Myeloid Leukemia, Adolescents Cancer, Adrenocortical Carcinoma, Anal Cancer, Appendix Cancer, Astrocytomas, Atypical Teratoid, Basal Cell Carcinoma, Bile Duct Cancer, Extrahepatic, Bladder Cancer, Bone Cancer, Brain Stem Glioma, Brain Tumor, Breast Cancer, Bronchial Tumors, Burkitt Lymphoma, Carcinoid Tumor, Carcinoma of Unknown Primary, Cardiac (Heart) Tumors, Central Nervous System tumors, Cervical Cancer, Childhood Cancers, Chordoma, Chronic Lymphocytic Leukemia, Chronic Myelogenous Leukemia, Chronic
- the compounds of the present invention may be useful in the treatment of disease, disorder, syndrome or condition selected from the group consisting of inflammation, asthma, chronic obstructive pulmonary disease, pulmonary fibrosis, inflammatory bowel disease, irritable bowel syndrome, pain, inflammatory pain, chronic pain, acute pain, fever, migraine, headache, low back pain, fibromyalgia, myofascial disorders, viral infections, influenza, common cold, herpes zoster, hepatitis C, AIDS, bacterial infections, fungal infections, dysmenorrhea, burns, surgical or dental procedures, malignancies hyperprostaglandin E syndrome, classic Bartter syndrome, synovitis, atherosclerosis, gout, arthritis, osteoarthritis, juvenile arthritis, rheumatoid arthritis, juvenile onset rheumatoid arthritis, rheumatic fever, ankylosing spondylitis, Hodgkin's disease, systemic lupus erythematosus, vascu
- the compounds of the present invention may be useful in the treatment of pain, chronic pain, acute pain, rheumatoid arthritic pain or osteoarthritic pain.
- the compounds of the present invention may be useful in the treatment of inflammation, neurodegenerative disorders such as Parkinson's disease, Alzheimer's disease and amyotrophic lateral sclerosis.
- the compounds of the present invention may be useful in the treatment prevention or management of the cancer.
- Compounds of the present invention are indicated both in the therapeutic and/or prophylactic treatment of the above-mentioned conditions.
- the dosage administered will, of course, vary with the compound employed, the mode of administration, the treatment desired and the disorder indicated.
- the daily dosage of the compound of the invention may be in the range from 0.05 mg/kg to 100 mg/kg.
- substituted benzoic ester derivatives of formula (1) undergoes formylation by using n-butyllithium and suitable solvent such as DMF in presence of organic base such as DIPEA or TEA to obtain compound of formula (2).
- the compound of formula (2) can be treated with hydroxyl amine in presence of suitable solvent such as methanol or ethanol to obtain compound of formula (3).
- the compound of formula (3) can be reduced by using reducing reagent such as zinc-dust or Fe-powder in presence of acid such as concentrated hydrochloric acid and suitable solvent such as methanol or ethanol to obtain the compound of formula (4).
- the compound of formula (4) can be reacted with compound of formula (5) in presence of organic base such as DIPEA or TEA in suitable solvent such as THF to obtain compound of formula (6).
- the compound of formula (6) can be converted to compound of formula (7) using reducing reagents such as sodium borohydride or lithium aluminium hydride in presence of suitable solvent such THF.
- the compound of formua (7) can be oxidized to obtain compound of formula (12) by using oxidizing agents such as manganese dioxide or pyridinium chlorochromate in presence of solvent such as DCM.
- the compound of formula (12) can be reacted with substituted acetophenone derivatives of formula (13) and urea in presence of iodine to obtain compound of formula (Il-a).
- the compound of formula (7) can be also prepared from substituted benzoic acid derivative.
- substituted benzoic acid derivatives of formula (8) can be treated with 2,2,2-trifluoro-N-(hydroxymethyl)acetamide to obtain compound of formula (9) under standard conditions.
- the compound of formula (9) can be converted to compound of formula (10) in presence of acid such as sulphuric acid or hydrochloric acid in a suitable solvent such as ethanol or methanol.
- the compound of formula (10) can be reacted with compound of formula (5) in presence of organic base such as DIPEA or TEA in suitable solvent such as THF to obtain compound of formula (1 1).
- the compound of formula (1 1) can be further converted to compound of formula (7) using reducing reagents such as sodium borohydride or lithium aluminium hydride in presence of suitable solvent such THF.
- the substituted benzaldehyde of formula (12) can be reacted with compound of formula (13a) and urea in presence of suitable reagents such as trimethylsilyl chloride and sulphamic acid to obtain compound of formula (14).
- suitable reagents such as trimethylsilyl chloride and sulphamic acid
- the compound of formula (14) can be further converted to the compound of formula (Il-b) by using suitable reagents such as 2,3-dichloro-5,6-dicyano-l,4-benzoquinone (DDQ) in a suitable solvent such as toluene.
- the substituted acetophenone derivatives of formula (15) can be treated with 2,2,2- trifluoro-N-(hydroxymethyl)acetamide, followed by treatment with base such as lithium hydroxide to obtain compound of formula (16).
- the compound of formula (16) can be reacted with compound of formula (5) in presence of organic base such as DIPEA or TEA in presence of suitable solvent such as THF to obtain a compound of formula (17).
- the compound of formula (17) can be reacted with compound of formula (18) and urea in presence of suitable reagents such as trimethylsilyl chloride and sulphamic acid to obtain compound of formula (Il-a).
- substituted acetophenone derivatives of formula (17) can be reacted with compound of formula (18) and urea in presence of suitable reagents such as trimethylsilyl chloride and sulphamic acid to obtain compound of formula (14a).
- suitable reagents such as trimethylsilyl chloride and sulphamic acid
- the compound of formula (14a) can be further converted to the compound of formula
- R y are hydrogen; p is 1 and R 3 , R 4 , W, L, m and n are as defined above with respect to a compound of formula (II)) can be prepared by the procedure as depicted in the
- the substituted nitrile derivatives of formula (19) can be reacted with a compound of formula (18) and urea in presence of suitable reagents such as trimethylsilyl chloride and sulphamic acid to obtain compound of formula (20).
- suitable reagents such as trimethylsilyl chloride and sulphamic acid to obtain compound of formula (20).
- the compound of formula (20) can be further converted to the compound of formula (21) by using suitable reagents such as 2,3-dichloro-5,6-dicyano-l,4-benzoquinone (DDQ) in a suitable solvent such as toluene.
- DDQ 2,3-dichloro-5,6-dicyano-l,4-benzoquinone
- the compound of formula (21) undergoes reduction reaction by using reducing reagent such as Pd/C in presence of solvent such as ethanol or methanol to give the corresponding amine derivative (which is highly unstable).
- the amine derivatives can be protected by using protecting reagents such as di-i3 ⁇ 4rt-buty! di carbonate in presence of base such as DIPEA or TEA in solvent such as DCM to give compound of formula (22).
- the compound of formula (22) can be further deprotected by using methanol: HQ/ ethanol :HC1 to give compound of formula (23).
- the compound of formula (23) can be reacted with compound of formula (5) in presence of organic base such as DIPEA or TEA in a suitable solvent such as THF to obtain compound of formula (II-c).
- the substituted acetophenone derivatives of formula (17a) can be reacted with compound of formula (18) in presence of base such as sodium hydroxide in solvent such as methanol to give compound of formula (24).
- the compound of formula (24) can be reacted with guanidine or substituted guanidine in presence of anion generating reagent such as sodium hydride in a suitable solvent such as dimethyl formamide to give compound of formula (III).
- the compound of formula (III) (wherein R a and R b are hydrogen) can be futher treated with acetic anhydride to obtain the corresponding acetyl derivative.
- substituted aldehyde derivatives of formula (12a) can be reacted with compound of formula (13b) in presence of base such as sodium hydroxide in solvent such as methanol to give compound of formula (25).
- base such as sodium hydroxide in solvent such as methanol
- the compound of formula (25) can be reacted with guanidine or substituted guanidine in presence of anion generating reagent such as sodium hydride in a suitable solvent such as dimethyl formamide to give compound of formula (III).
- compound of formula (26) can be treated with suitable reagent such as acetic anhydride to obtain compound of formyula (IV).
- Step (a) involves reacting the compound of formula (27) with compound of formula (13a) and urea in presence of suitable reagents.
- the suitable reagents may be trimethylsilyl chloride and sulphamic acid.
- the reaction may be carried out at a suitable temperature.
- the suitable temperature may be around 100- 150 °C, preferable 100 °C.
- Step (b) involves conerting the compound of formula (28) to compound of formula (II) using a suitable reagent.
- the suitable reagent may be 2,3-dichloro-5,6-dicyano- 1,4-benzoquinone (DDQ).
- DDQ 2,3-dichloro-5,6-dicyano- 1,4-benzoquinone
- the reaction may be carried out in presence of a suitable solvent.
- the suitable solvent may be toluene.
- work-up includes distribution of the reaction mixture between the organic and aqueous phase indicated within parentheses, separation of layers and drying the organic layer over sodium sulphate (Na 2 S0 4 ), filtration and evaporation of the solvent under reduced pressure.
- Purification includes purification by silica gel chromatographic techniques, in suitable solvents of a suitable polarity as the mobile phase.
- EDCI l-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- DCM or MDC Dichlorom ethane
- LAH Lithium aluminium hydride
- PCC pyridinium chlorochromate
- DEE Diethylether
- DIPEA N,N-Diisopropylethylamine
- DMSO Di-methyl sulfoxide
- THF Tetrahydrofuran
- TEA Triehtylamine
- EDC Ethylene di chloride
- EtOAc or EA Ethyl acetate
- CH 2 C1 2 Dichloromethane
- CHC1 3 Chloroform
- MeOH Methanol
- NaOH Sodium hydroxide
- Na 2 S0 4 Sodium sulfate
- RT or rt
- Step 1 Preparation of 2-chloro- - ⁇ [(trifluoroacetyl)amino]methyl ⁇ benzoic acid
- Step 1 Preparation of methyl 2-chloro-5-(isobutyramidomethyl)benzoate
- Step-3 Preparation of N-(4-chloro-3-formylbenzyl)isobutyramide
- Step-1 Preperation of ethyl 6- chloro-2-fluoro-3-formylbenzoate
- Step-3 Preperation of ethyl 3-(aminomethyl)-6-chloro-2-fluorobenzoate
- Step-5 Preparation of N-(4-chloro-2-fluoro-3(hydroxymethyl)benzyl)pivalamide
- N-(4-chloro-2-fluoro-3-formylbenzyl)pivalamide (Intermediate-3, 0.20 g, 0.73 mmol), 4-trifluoromethyl acetophenone (0.138 g, 0.73 mmol), urea (0.066 g, 1.10 mmol), trimethylsilyl chloride (0.079 g, 0.73 mmol) and sulphamic acid (0.0036 g, 0.0368 mmol) were heated together at 100°C for 6-8 h. After completion of the reaction, the reaction was quenched with ice-cold water and the obtained solid was filtered to afford a crude product.
- Step -1 Preparation of ethyl -chloro-2-fluoro-3-(isobutyramidomethyl)benzoate
- step 3 To a solution of ethyl 3-(aminomethyl)-6-chloro-2-fluorobenzoate (step 3, Intermediate 3) (1.00 g, 4.32 mmol) in THF (30 mL) were added DIPEA (1.67 g, 12.96 mmol) and isobutyryl chloride (690 mg, 6.48 mmol). The reaction mass was stirred at RT for 2 h. After completion of the reaction, the reaction mixture was diluted with EtOAc, washed with water and brine, dried over Na 2 S0 4 and concentrated to afford 1.0 g of the title product.
- Step-2 Preparation of N-(chloro-2-fluoro-3- hydroxymethyl)benzyl) isobutyramide
- Step-3 Preparation of N-(4-chloro-2-fluoro-3-formylbenzyl)isobutyramide
- Step-1 Preparation of N-(3-acetyl-4-chlorobenzyl)-2,2,2-trifluoroacetamide
- step-2 Intermediate- 1 1, 0.600 g, 3.26 mmol
- TEA 2.0 mL
- propane-2-sulfonyl chloride 2.0 mL
- dry THF 10 mL
- Step-2 Preparation of N-(4-chloro-3-(2-oxo-6-(4-(trifluoromethyl)phenyl)-l,2,3,6- tetrahydropyrimidin-4-yl)benzyl)propane-2-sulfonamide
- Step-1 Preparation of 3-(ethoxymethylene)pentane-2,4-dione
- Step-3 Preparation of 6-methyl-5-(2-oxo-6-(4-(trifluoromethyl)phenyl)-l,2,3,6- tetrahydropyrimidin-4-yl)nicotinonitrile
- Step-4 Preparation of 6-methyl-5-(2-oxo-6-(4-(trifluoromethyl)phenyl)-2,3- dihydropyrimidin-4-yl)nicotinonitrile
- Step-5 Preparation of ie/t-butyl ((6-methyl-5-(2-oxo-6-(4-(trifluoromethyl)phenyl)- 2,3-dihydropyrimidin-4-yl)pyridin-3-yl)methyl)carbamate
- Step-6 Preparation of 6-(5-(aminomethyl)-2-methylpyridin-3-yl)-4-(4- (trifluoromethyl)phenyl)pyrimidin-2(lH)-one hydrochloride
- Step-1 Preparation of 6-methyl-5-(2-meth l-l,3-dioxolan-2-yl)nicotinonitrile
- Step-2 Preparation of 6-formyl-5-(2-meth l-l,3-dioxolan-2-yl)nicotinonitrile
- Step 3 Preparation of 6-(difluoromethyl)-5-(2-methyl-l,3-dioxolan-2-yl) nicotinonitrile
- Step-5 Preparation of 6-(difluoromethyl)-5-(2-oxo-6-(4-(trifluoromethyl)phenyl)- l,2,3,6-tetrahydropyrimidin-4-yl)nicotinonitrile
- Step-6 Preparation of 6-(difluoromethyl)-5-(2-oxo-6-(4-(trifluoromethyl)phenyl)-2,3- dihydropyrimidin-4-yl)nicotinonitrile
- Step 7 Preparation of ie/ -butyl ((6-(difluoromethyl)-5-(2-oxo-6-(4- (trifluoromethyl)phenyl)-2,3-dihydropyrimidin-4-yl)pyridin-3-yl)methyl)carbamate
- Step 8 Preparation of 6-(5-(aminomethyl)-2-(difluoromethyl)pyridin-3-yl)-4-(4- (trifluoromethyl)phenyl)pyrimidin-2(lH)-one hydrochloride
- N-(4-chloro-3-formylbenzyl)pivalamide (Intermediate- 1, 0.100 g, 0.37 mmol), 4- trifluoromethyl-acetophenone (0.070g, 0.37 mmol), urea (0.034g, 0.55 mmol) and iodine (0.009 g, 0.037 mmol) were heated together at 100 °C for 6-8 h. After completion of the reaction, the reaction mixture was quenched with ice-cold water and the obtained solid was filtered. The solid was purified by column chromatography on silica, 100-200 mesh eluting with 3-4 % methanol in DCM to afford 0.020 g of the title compound.
- Example-1 A suspension of N-(4-chloro-3-(2-oxo-6-(4-(trifluoromethyl)-2,3-dihydropyrimidin-4- yl)benzyl)pivalamide (Example-1, 0.100 g, 0.215 mmol) in acetic anhydride (4.0 mL) was heated at 100 °C for 16 h. The reaction mass was concentrated. The residue was washed with DEE, filtered and suck dried to afford 0.025 g of title product.
- Example-16 N-(3-(2-amino-6-(4-(trifluoromethyl)phenyl)pyrimidin-4-yl)-4-chloro benzyl)pivalamide (Example-16, 0.100 g) and acetic anhydride (5.0 mL) to afford 0.026 g of the desired compound.
- mPGES-1 microsomal prostaglandin E synthase-1
- PGH 2 prostaglandin H 2
- product PGE 2 prostaglandin E 2
- GSH reduced glutathione
- mPGES-1 inhibitors were screened by assessing their ability to inhibit formation of PGE 2 from PGH 2 in presence of mPGES-1 using an anti-PGE 2 antibody based detection method.
- Recombinant human mPGES-1 was generated in-house by expression in CHO cells (Ouellet M et al. (2002), Protein Expression and Purification 26: 489 - 495).
- the assay was set up using crude microsomal fractions at protein concentration of 40-60 ⁇ g/mL.
- Test compounds were prepared in 100 % dimethyl sulfoxide (DMSO) to obtain 20 mM stock solution and then diluted using assay buffer comprising 0.1 M Potassium phosphate buffer with 2 mM EDTA. The final concentration of DMSO in reaction was 0.5 % (v/v).
- Negative controls were comprised of all assay reagents except the enzyme. Positive controls were comprised of the enzyme reaction in the absence of any inhibitor.
- Test compounds were incubated for 10 minutes in assay buffer containing 2.5 mM GSH and mPGES-1 enzyme followed by addition of PGH 2 at a concentration of 15 ⁇ for 1 minute.
- Inhibition of mPGES-1 enzyme activity was measured using the percent of reaction occurring in the positive control. Concentration response curves were plotted using percent inhibition of maximum enzyme reaction. The IC 50 value was calculated from the concentration response curve by nonlinear regression analysis using GraphPad PRISM software.
- the compounds prepared were tested using the above assay procedure and the results obtained are given in Table 1. Percentage inhibition at concentrations of 1.0 ⁇ and 10.0 ⁇ are given in the table along with IC 50 (nM) details for selected examples.
- the compounds prepared were tested using the above assay procedure and were found to have IC 50 less than 200nM, preferably less than ⁇ , more preferably less than 50nM or most preferably less than 20nM.
- IC 50 (nM) values of the compounds are set forth in Table 1 wherein "A” refers to an IC 50 value of less than 50 nM, “B” refers to IC 50 value in range of 50.01 to 100.0 nM and “C” refers to IC 50 values more than 100.01 nM.
- A549 cell line was monitored as inhibition of IL- ⁇ induced PGE 2 release.
- A549 cells were maintained in DMEM medium with 10% FBS and 1% Penicillin-Streptomycin Solution in 5% C0 2 at 37°C. Cells were seeded 24 h prior to the assay in 96 well plates in DMEM containing 1% Penicillin-Streptomycin and 2% FBS so as to get ⁇ 40,000 cells per well on the day of experiment. The assay was carried out in a total volume of 200 ⁇ L ⁇ . Test compounds were dissolved in dimethyl sulfoxide (DMSO) to prepare 2 mM stock solution and then diluted using plain DMEM.
- DMSO dimethyl sulfoxide
- the final concentration of DMSO in the reaction was 0.55% (v/v).
- Cells were treated with test compounds for 30 minutes followed by addition of IL- ⁇ at a final concentration of 10 ng/mL for 16-20 h. Plates were then centrifuged at 1000 rpm for 10 min at 4°C. Supernatants were collected & analyzed by the addition of PGE 2 -D2 & anti-PGE 2 cryptate conjugate supplied by the CisBio HTRF kit in a 96 well half area Blackwell EIA/RIA plate. The assay plate was incubated overnight at 4-5° C before being read in Artemis (K-101) (Japan) HTRF plate reader and levels of PGE 2 calculated by extrapolation from the standard curve.
- concentration response curves were plotted as % of maximal response obtained in the absence of test antagonist.
- the IC 50 value was calculated from the concentration response curve by nonlinear regression analysis using GraphPad PRISM software.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The present disclosure is directed to substituted pyrimidine compounds of formula (I), and pharmaceutically acceptable salts thereof, as mPGES-1 inhibitors. These compounds are inhibitors of the microsomal prostaglandin E synthase-1 (mPGES-1) enzyme and are therefore useful in the treatment of pain and/or inflammation from a variety of diseases or conditions, such as asthma, osteoarthritis, rheumatoid arthritis, acute or chronic pain and neurodegenerative diseases.
Description
SUBSTITUTED PYRIMIDINE COMPOUNDS AS mPGES-1 INHIBITORS
Related Applications
This application claims the benefit of Indian Provisional Application No. 3304/MUM/2013 filed on October 22, 2013; which is hereby incorporated by reference in its entirety.
Technical Field
The present application relates to substituted pyrimidine compounds which may be useful as microsomal prostaglandin E synthase-1 (mPGES-1) inhibitors. Background of the Invention
There are many diseases or disorders that are inflammatory in their nature. One of the major problems associated with existing treatments of inflammatory conditions is inadequate efficacy and/or the prevalence of side effects. Inflammatory diseases that affect the population include asthma, inflammatory bowel disease, rheumatoid arthritis, osteoarthritis, rhinitis, conjunctivitis and dermatitis. Inflammation is also a common cause of pain.
The enzyme cyclooxygenase (COX) converts arachidonic acid to an unstable intermediate, prostaglandin H2 (PGH2), which is further converted to other prostaglandins, including PGE2, PGF2a, PGD2, prostacyclin and thromboxane A2. These arachidonic acid metabolites are known to have pronounced physiological and pathophysiological activity, including pro-inflammatory effects. The COX enzyme exists in two forms, one that is constitutively expressed in many cells and tissues (COX-1), and another that in most cells and tissues is induced by pro-inflammatory stimuli, such as cytokines, during an inflammatory response (COX-2).
Among all prostaglandin metabolites, PGE2 is particularly known to be a strong pro-inflammatory mediator, and is also known to induce fever and pain. Consequently, numerous drugs have been developed with a view to inhibiting the formation of PGE2, including "NS AIDs" (non-steroidal anti -inflammatory drugs) and "coxibs" (selective COX-2 inhibitors). These drugs act predominantly by inhibition of COX-1 and/or COX-2, thereby reducing the formation of PGE2. However, the inhibition of COXs has the disadvantage of reducing the formation of all metabolites of PGH2, thereby decreasing the beneficial properties of some of the metabolites. In view of this, drugs which act by inhibition of COXs are suspected to cause adverse biological effects. For example, the non-selective inhibition of COXs by NSAIDs
may give rise to gastrointestinal side-effects and affect platelet and renal function. Even the selective inhibition of COX-2 by coxibs, whilst reducing such gastrointestinal side-effects, is believed to give rise to cardiovascular problems.
A combination of pharmacological, genetic and neutralizing antibody approaches demonstrates the importance of PGE2 in inflammation. The conversion of PGH2 to PGE2 by prostaglandin E synthases (PGES) may, therefore, represent a pivotal step in the propagation of inflammatory stimuli. There are two microsomal prostaglandin E synthases (mPGES-1 and mPGES-2), and one cytosolic prostaglandin E synthase (cPGES). mPGES-1 is an inducible PGES after exposure to pro- inflammatory stimuli. mPGES-1 is induced in the periphery and CNS by inflammation, and represents therefore a target for acute and chronic inflammatory disorders. PGE2 is a major prostanoid, produced from arachidonic acid liberated by phospholipases (PLAs), which drives the inflammatory processes. Arachidonic acid is transformed by the action of prostaglandin H synthase (PGH synthase, cycloxygenase) into PGH2 which is a substrate for mPGES-1, the terminal enzyme transforming PGH2 to the pro-inflammatory PGE2.
Agents that are capable of inhibiting the action of mPGES-1, and thus reducing the formation of the specific arachidonic acid metabolite PGE2, are beneficial in the treatment of inflammation. Further, agents that are capable of inhibiting the action of the proteins involved in the synthesis of the leukotrienes are also beneficial in the treatment of asthma and COPD.
Blocking the formation of PGE2 in animal models of inflammatory pain results in reduced inflammation, pain and fever response (Kojima et. al, The Journal of Immunology 2008, 180, 8361-6; Xu et. al., The Journal of Pharmacology and Experimental Therapeutics 2008, 326, 754-63). In abdominal aortic aneurism, inflammation leads to connective tissue degradation and smooth muscle apoptosis ultimately leading to aortic dilation and rupture. In animals lacking mPGES-1 a slower disease progression and disease severity has been demonstrated (Wang et. al., Circulation, 2008, 117, 1302-1309).
Several lines of evidence indicate that PGE2 is involved in malignant growth.
PGE2 facilitates tumor progression by stimulation of cellular proliferation and angiogenesis and by modulation of immunosupression. In support of a role for PGE2 in cancers, genetic deletion of mPGES-1 in mice suppresses intestinal tumourogenesis (Nakanishi et. al., Cancer Research 2008, 68(9), 3251-9). In human beings, mPGES-1
is also upregulated in cancers such as colorectal cancer (Schroder, Journal of Lipid Research 2006, 47, 1071-80).
Myositis is a chronic muscle disorder characterized by muscle weakness and fatigue. Proinflammatory cytokines and prostanoids have been implicated in the development of myositis. In skeletal muscle tissue from patients suffering from myositis an increase in cyclooxygenases and mPGES-1 has been demonstrated, implicating mPGES-1 as a target for treating this condition. (Korotkova, Annals of the Rheumatic Diseases 2008, 67, 1596- 1602).
In atherosclerosis, inflammation of the vasculature leads to atheroma formation that eventually may progress into infarction. In patients with carotid atherosclerosis an increase in mPGES-1 in plaque regions has been reported (Gomez- Hernandez Atherosclerosis 2006, 187, 139-49). In an animal model of atherosclerosis, mice lacking the mPGES-1 receptor were found to show a retarded atherogenesis and a concomitant reduction in macrophage-derived foam cells together with an increase in vascular smooth muscle cells (Wang, Proceedings of National Academy of Sciences 2006, 103(39), 14507-12).
International Publication Nos. WO 2006/063466, WO 2007/059610, WO 2010/034796, WO 2010/100249, WO 2012/055995, WO 2012/110860, WO 2013/038308 and WO 2013/072825 disclose numerous heterocyclic compounds which are stated to be inhibitors of the microsomal prostaglandin E synthase- 1 (mPGES-1) enzyme.
The present application is directed to compounds that act as inhibitors of the mPGES-1 enzyme and, therefore, are useful for the treatment of pain and inflammation in a variety of diseases or conditions.
Summary of the Invention
In one aspect, the present invention relates to compound of formula (I):

(I)
or a pharmaceutically acceptable salt thereof,
wherein,
X is O, when dotted line T represents a single bond and dotted line '2' is absent; or X is ORa or RaRb, when dotted line ' 1 ' is absent and dotted line '2' represents a single bond;
G2 is N;
G3 is sleeted from N and CH;
G4 is selected from N and CH;
L is selected from -CO- or -S(0)2-;
W is selected from Ci-salkyl, haloCi-salkyl and hydroxyCi-salkyl;
Ra and Rb are each independently selected from hydrogen, Ci-4alkyl and - C(0)Ci-4alkyl;
R1 is selected from hydrogen, halogen and Ci-4alkyl;
R2 is absent when dotted line '2' represents a single bond; or R2 is hydrogen when dotted line '2' is absent;
aatt eeaacchh ooccccuurrrreennce R is independently selected from halogen, nitro, cyano, Ci. salkyl and haloCi-salkyl;
at each occurrence R4 is independently selected from halogen, Ci-salkyl, haloCi-salkyl and C3-i2cycloalkyl;
at each occurrence of Rx and Ry are hydrogen;
'm' is an integer ranging from 0 to 3, both inclusive;
'n' is an integer ranging from 1 to 4, both inclusive; and
'p' is an integer ranging from 1 to 3, both inclusive.
The compounds of formula (I) may involve one or more embodiments. Embodiments of formula (I) include compounds of formula (II), formula (III) and formula (IV) as described hereinafter. It is to be understood that the embodiments below are illustrative of the present invention and are not intended to limit the claims to the specific embodiments exemplified. It is also to be understood that the embodiments defined herein may be used independently or in conjunction with any definition of any other embodiment defined herein. Thus, the invention contemplates all possible combinations and permutations of the various independently described embodiments. For example, the invention provides compounds of formula (I) as defined above, wherein R1 is hydrogen or Ci-4alkyl (according to an embodiment
defined below), L is -CO- (according to another embodiment defined below), and 'p' is 1 (according to yet another embodiment defined below).
According to an embodiment, specifically provided are compounds of formula (I), in which X is O, dotted line T represents a single bond and dotted line '2' is absent.
According to another embodiment, specifically provided are compounds of formula (I), in which X is RaRb, dotted line ' 1 ' is absent and dotted line '2' represents a single bond. In this embodiment, Ra is hydrogen, Ci-4alkyl (e.g. methyl) or -C(0)Ci-4alkyl (e.g. -C(O)methyl) and Rb is hydrogen.
According to another embodiment, specifically provided are compounds of formula (I), in which X is H2, NHCH3 or NHCOCH3; dotted line ' 1 ' is absent and dotted line '2' represents a single bond.
According to yet another embodiment, specifically provided are compounds of formula (I), in which X is ORa, dotted line T is absent and dotted line '2' represents a single bond. In this embodiment, Ra is -C(0)Ci-4alkyl (e.g. -C(O)methyl).
According to yet another embodiment, specifically provided are compounds of formula (I), in which X is OCOCH3, dotted line T is absent and dotted line '2' represents a single bond.
According to yet another embodiment, specifically provided are compounds of formula (I), in which G3 is CH.
According to yet another embodiment, specifically provided are compounds of formula (I), in which G3 is N.
According to yet another embodiment, specifically provided are compounds of formula (I), in which G4 is CH.
According to yet another embodiment, specifically provided are compounds of formula (I), in which G4 is N.
According to yet another embodiment, specifically provided are compounds of formula (I), in which R1 is hydrogen or Ci-4alkyl (e.g. methyl).
According to yet another embodiment, specifically provided are compounds of formula (I), in which R1 is hydrogen or methyl.
According to yet another embodiment, specifically provided are compounds of formula (I), in which R2 is hydrogen and dotted line '2' is absent.
According to yet another embodiment, specifically provided are compounds of formula (I), in which R2 is absent and dotted line '2' represents a single bond.
According to yet another embodiment, specifically provided are compounds of formula (I), in which R3 is halogen (e.g. fluorine or chlorine), Ci-8alkyl (e.g. methyl) or haloCi-8alkyl (e.g. difluoromethyl).
According to yet another embodiment, specifically provided are compounds of formula (I), in which R3 is fluorine, chlorine, methyl or difluoromethyl.
According to yet another embodiment, specifically provided are compounds of formula (I), in which R4 is halogen (e.g. chlorine), Ci-8alkyl (e.g. methyl, isopropyl or ie/ -butyl), haloCi-8alkyl (e.g. tnfluoromethyl) or C3-i2cycloalkyl (e.g. cyclopropyl).
According to yet another embodiment, specifically provided are compounds of formula (I), in which R4 is chlorine, methyl, isopropyl, ie/t-butyl, trifluoromethyl or cyclopropyl.
According to yet another embodiment, specifically provided are compounds of formula (I), in which L is -CO-.
According to yet another embodiment, specifically provided are compounds of formula (I), in which L is -S(0)2-
According to yet another embodiment, specifically provided are compounds of formula (I), in which W is Ci-8alkyl (e.g. isopropyl or ie/ -butyl).
According to yet another embodiment, specifically provided are compounds of formula (I), in which W is isopropyl or ie/t-butyl.
According to yet another embodiment specifically provided are compounds of formula (I), in which 'm' is 1 or 2.
According to yet another embodiment specifically provided are compounds of formula (I), in which 'n' is 1 or 2.
According to yet another embodiment specifically provided are compounds of formula (I), in which 'p' is 1.
According to yet another embodiment specifically provided are compounds of formula (I), in which
X is O, dotted line T represents a single bond and dotted line '2' is absent;
Gi is CR1; G2 is N; G3 is N or CH; G4 is N or CH;
R1 is hydrogen or methyl;
R2 is hydrogen;
R3 is fluorine, chlorine, methyl or difluoromethyl;
R4 is isopropyl, tert-butyl, trifluoromethyl or cyclopropyl;
L is -CO-;
W is isopropyl or ie/t-butyl;
Rx and Ry are hydrogen; m is 1 or 2; n is 1 and p is 1.
According to yet another embodiment specifically provided are compounds of formula (I), in which
X is ORa or RaRb, dotted line ' 1 ' is absent and dotted line '2' represents a single bond;
Gi is CR1; G2 is N; G3 is CH; G4 is CH;
Ra is hydrogen, methyl or -C(0)methyl; Rb is hydrogen;
R1 is hydrogen;
R2 is absent;
R3 is fluorine or chlorine;
R4 is chlorine, methyl or trifluoromethyl.
L is -CO-;
W is tert-butyl;
Rx and Ry are hydrogen; m is 1 or 2; n is lor 2 and p is 1.
According to an embodiment, specifically provided are compounds of formula (I) that exhibit an IC50 value of less than 500 nM, preferably, less than 100 nM, more preferably, less than 50 nM with respect to mPGES-1 inhibition.
Further embodiments relating to groups G3, G4, R1, R3, R4, R5, Ra, Rb, Rx, Ry, L, W, m, n and p (and groups defined therein) are described hereinafter in relation to the compounds of formula (II), formula (III) and formula (IV). It is to be understood that these embodiments are not limited to use in conjunction with formula (II), formula (III) or formula (IV), but apply independently and individually to the compounds of formula (I). For example, in an embodiment described hereinafter, the invention specifically provides compounds of formula (II), formula (III) or formula (IV) wherein R1 is hydrogen or Ci-4alkyl and consequently there is also provided a compound of formula (I) wherein R1 is hydrogen or Ci-4alkyl.
The invention also provides a compound of formula (II) which is an embodiment of a compound of formula (I).
Accordingly the invention provides a compound of formula (II)

(Π)
or a pharmaceutically acceptable salt thereof,
wherein,
G3 is sleeted from N and CH;
G4 is selected from N and CH;
L is selected from -CO- or -S(0)2-;
W is selected from Ci-8alkyl, haloCi-8alkyl and hydroxyCi-8alkyl;
R1 is selected from hydrogen, halogen and

at each occurrence R3 is independently selected from halogen, nitro, cyano, Ci. 8alkyl and haloCi-8alkyl;
at each occurrence R4 is independently selected from halogen, Ci-8alkyl, haloCi-8alkyl and C3-i2cycloalkyl;
at each occurrence of Rx and Ry are hydrogen;
'm' is an integer ranging from 0 to 3, both inclusive;
'n' is an integer ranging from 1 to 4, both inclusive; and
'p' is an integer ranging from 1 to 3, both inclusive.
The compounds of formula (II) may involve one or more embodiments. It is to be understood that the embodiments below are illustrative of the present invention and are not intended to limit the claims to the specific embodiments exemplified. It is also to be understood that the embodiments defined herein may be used independently or in conjunction with any definition of any other embodiment defined herein. Thus, the invention contemplates all possible combinations and permutations of the various independently described embodiments. For example, the invention provides compounds of formula (II) as defined above, wherein R1 is hydrogen or
 (according to an embodiment defined below), R4 is Ci-8alkyl, haloCi-8alkyl or C3- i2cycloalkyl (according to another embodiment defined below), and W is Ci-8alkyl (according to yet another embodiment defined below).
According to an embodiment, specifically provided are compounds of formula (II), in which G3 is CH.
(according to an embodiment defined below), R4 is Ci-8alkyl, haloCi-8alkyl or C3- i2cycloalkyl (according to another embodiment defined below), and W is Ci-8alkyl (according to yet another embodiment defined below).
According to an embodiment, specifically provided are compounds of formula (II), in which G3 is CH.
According to another embodiment, specifically provided are compounds of formula (II), in which G3 is N.
According to yet another embodiment specifically provided are compounds of formula (II), in which G4 is CH.
According to yet another embodiment, specifically provided are compounds of formula (II), in which G4 is N.
According to yet another embodiment specifically provided are compounds of formula (II), in which R1 is hydrogen or Ci-4alkyl (e.g. methyl).
According to yet another embodiment specifically provided are compounds of formula (II), in which R1 is hydrogen or methyl.
According to yet another embodiment specifically provided are compounds of formula (II), in which R3 is halogen (e.g. fluorine or chlorine), Ci-salkyl (e.g. methyl) or haloCi-salkyl (e.g. difluoromethyl).
According to yet another embodiment specifically provided are compounds of formula (II), in which R3 is fluorine, chlorine, methyl or difluoromethyl.
According to yet another embodiment specifically provided are compounds of formula (II), in which R4 is Ci-salkyl (e.g. isopropyl or ie/t-butyl), haloCi-salkyl (e.g. trifluoromethyl) or C3-i2Cycloalkyl (e.g. cyclopropyl).
According to yet another embodiment specifically provided are compounds of formula (II), in which R4 is isopropyl, tert-butyl, trifluoromethyl or cyclopropyl.
According to yet another embodiment specifically provided are compounds of formula (II), in which L is -CO-.
According to yet another embodiment, specifically provided are compounds of formula (II), in which L is -S(0)2-
According to yet another embodiment, specifically provided are compounds of formula (II), in which W is Ci-salkyl (e.g. isopropyl or tert-buty\).
According to yet another embodiment, specifically provided are compounds of formula (II), in which W is isopropyl or tert-butyl.
According to yet another embodiment specifically provided are compounds of formula (II), in which 'm' is 1 or 2.
According to yet another embodiment specifically provided are compounds of formula (II), in which 'n' is 1.
According to yet another embodiment specifically provided are compounds of formula (II), in which 'p' is 1.
According to yet another embodiment specifically provided are compounds of formula (II), in which
G3 is N or CH; G4 is N or CH;
R1 is hydrogen or methyl;
R3 is fluorine, chlorine, methyl or difluoromethyl;
R4 is isopropyl, ie/t-butyl, trifluoromethyl or cyclopropyl;
L is -CO-;
W is isopropyl or ie/t-butyl;
Rx and Ry are hydrogen; m is 1 or 2; n is 1 and p is 1.
According to one embodiment, specifically provided are compounds of formula (II) that exhibit an IC50 value of less than 500 nM, preferably, less than 100 nM, more preferably, less than 50 nM with respect to mPGES-1 inhibition.
The invention also provides a compound of formula (III) which is an embodiment of a compound of formula (I).
Accordingly the invention provides a compound of formula (III)

or a pharmaceutically acceptable salt thereof,
wherein,
W is selected from Ci-8alkyl, haloCi-8alkyl and hydroxyCi-8alkyl;
Ra and Rb are each independently selected from hydrogen, Ci-4alkyl and - C(0)Ci-4alkyl;
at each occurrence R3 is independently selected from halogen, nitro, cyano, Ci. 8alkyl and haloCi-8alkyl;
at each occurrence R4 is independently selected from halogen, Ci-8alkyl, haloCi-8alkyl and C3-i2cycloalkyl;
'm' is an integer ranging from 0 to 3, both inclusive; and
'n' is an integer ranging from 1 to 4, both inclusive.
The compounds of formula (III) may involve one or more embodiments. It is to be understood that the embodiments below are illustrative of the present invention and are not intended to limit the claims to the specific embodiments exemplified. It is also to be understood that the embodiments defined herein may be used independently or in conjunction with any definition of any other embodiment defined herein. Thus, the invention contemplates all possible combinations and permutations of the various independently described embodiments. For example, the invention provides compounds of formula (III) as defined above, wherein Rb is hydrogen (according to an embodiment defined below), R4 is halogen, Ci-8alkyl or haloCi-8alkyl (according to another embodiment defined below), and W is Ci-8alkyl (according to yet another embodiment defined below).
According to an embodiment, specifically provided are compounds of formula (III), in which Ra is hydrogen,
 (e.g. methyl) or -C(0)Ci-4alkyl (e.g. - C(O)methyl).
(e.g. methyl) or -C(0)Ci-4alkyl (e.g. - C(O)methyl).
According to yet another embodiment, specifically provided are compounds of formula (III), in which Ra is hydrogen, methyl or -C(0)methyl.
According to yet another embodiment specifically provided are compounds of formula (III), in which Rb is hydrogen.
According to yet another embodiment, specifically provided are compounds of formula (III), in which Ra is hydrogen, methyl or -C(0)methyl and Rb is hydrogen.
According to yet another embodiment, specifically provided are compounds of formula (III), in which R3 is halogen (e.g. fluorine or chlorine).
According to yet another embodiment specifically provided are compounds of formula (III), in which R3 is fluorine or chlorine.
According to yet another embodiment specifically provided are compounds of formula (III), in which R4 is halogen (e.g. chlorine), Ci-8alkyl (e.g. methyl) or haloCi. 8alkyl (e.g. trifluoromethyl).
According to yet another embodiment specifically provided are compounds of formula (III), in which R4 is chlorine, methyl or trifluoromethyl.
According to yet another embodiment, specifically provided are compounds of formula (III), in which W is Ci-8alkyl (e.g. ie/ -butyl).
According to yet another embodiment, specifically provided are compounds of formula (III), in which W is tert-butyl.
According to yet another embodiment specifically provided are compounds of formula (III), in which 'm' is 1 or 2.
According to yet another embodiment specifically provided are compounds of formula (III), in which 'n' is 1 or 2.
According to yet another embodiment specifically provided are compounds of formula (III), in which
Ra is hydrogen, methyl or -C(0)methyl; Rb is hydrogen;
R3 is fluorine or chlorine; R4 is chlorine, methyl or trifluorom ethyl.
W is tert-butyl; m is 1 or 2 and n is lor 2.
According to an embodiment, specifically provided are compounds of formula
(III) that exhibit an IC50 value of less than 500 nM, preferably, less than 100 nM, more preferably, less than 50 nM with respect to mPGES-1 inhibition.
The invention also provides a compound of formula (IV) which is an embodiment of a compound of formula (I).
Accordingly the invention provides a compound of formula (IV)

or a pharmaceutically acceptable salt thereof,
wherein,
W is selected from Ci-8alkyl, haloCi-8alkyl and hydroxyCi-8alkyl;
Ra is -C(0)Ci-4alkyl;
at each occurrence R3 is independently selected from halogen, nitro, cyano, Ci. 8alkyl and haloCi-8alkyl;
at each occurrence R4 is independently selected from halogen, Ci-8alkyl, haloCi-8alkyl and C3-i2cycloalkyl;
'm' is an integer ranging from 0 to 3, both inclusive; and
'n' is an integer ranging from 1 to 4, both inclusive.
The compounds of formula (IV) may involve one or more embodiments. It is to be understood that the embodiments below are illustrative of the present invention and are not intended to limit the claims to the specific embodiments exemplified. It is
also to be understood that the embodiments defined herein may be used independently or in conjunction with any definition of any other embodiment defined herein. Thus, the invention contemplates all possible combinations and permutations of the various independently described embodiments. For example, the invention provides compounds of formula (III) as defined above, wherein Ra is C(0)Ci-4alkyl (according to an embodiment defined below), R4 is haloCi-8alkyl (according to another embodiment defined below), and W is Ci-8alkyl (according to yet another embodiment defined below).
According to an embodiment, specifically provided are compounds of formula (IV), in which Ra is -C(0)CMalkyl (e.g. -C(O)methyl).
According to yet another embodiment, specifically provided are compounds of formula (IV), in which Ra is -C(0)methyl.
According to yet another embodiment specifically provided are compounds of formula (IV), in which R3 is halogen (e.g. chlorine).
According to yet another embodiment specifically provided are compounds of formula (IV), in which R3 is chlorine.
According to yet another embodiment specifically provided are compounds of formula (IV), in which R4 is haloCi-8alkyl (e.g. trifluoromethyl).
According to yet another embodiment specifically provided are compounds of formula (IV), in which R4 is trifluoromethyl.
According to yet another embodiment, specifically provided are compounds of formula (IV), in which W is Ci-8alkyl (e.g. ie/ -butyl).
According to yet another embodiment, specifically provided are compounds of formula (IV), in which W is tert-butyl.
According to yet another embodiment specifically provided are compounds of formula (IV), in which 'm' is 1.
According to yet another embodiment specifically provided are compounds of formula (IV), in which 'n' is 1.
According to yet another embodiment specifically provided are compounds of formula (IV), in which
Ra is -C(0)methyl; R3 is chlorine; R4 is trifluoromethyl.
W is tert-butyl; m is 1 and n is 1.
According to an embodiment, specifically provided are compounds of formula (IV) that exhibit an IC50 value of less than 500 nM, preferably, less than 100 nM, more preferably, less than 50 nM with respect to mPGES-1 inhibition.
Compounds of the present invention include the specific compounds in Examples 1- 23. However, it is to be understood that the specific compounds of the present invention are not intended to limit the claims to the specific compounds exemplified.
It should be understood that the formulas (I) structurally encompass all geometrical isomers, stereoisomers, enantiomers and diastereomers, N-oxides, and pharmaceutically acceptable salts that may be contemplated from the chemical structure of the genera described herein.
According to an embodiment, the compounds of formula (II) structurally encompass all tautomeric forms whether such tautomer exists in equilibrium or predominantly in one form. Such tautomeric form may be different or the same when the compound is bound to the mPGES-1 enzyme.

The present application also provides a pharmaceutical composition that includes at least one compound described herein and at least one pharmaceutically acceptable excipient (such as a pharmaceutically acceptable carrier or diluent). Preferably, the pharmaceutical composition comprises a therapeutically effective amount of at least one compound described herein. The compounds described herein may be associated with a pharmaceutically acceptable excipient (such as a carrier or a diluent) or be diluted by a carrier, or enclosed within a carrier which can be in the form of a capsule, sachet, paper or other container.
The compounds and pharmaceutical compositions of the present invention are useful for inhibiting the activity of mPGES-1, which is related to a variety of disease states.
The present invention further provides a method of inhibiting mPGES-1 in a subject in need thereof by administering to the subject one or more compounds described herein in an amount effective to cause inhibition of such receptor. Detailed Description of the Invention
Definitions
The symbol dotted line " " refers to a single bond.
The terms "halogen" or "halo" means fluorine (fluoro), chlorine (chloro), bromine (bromo), or iodine (iodo).
The term "alkyl" refers to a hydrocarbon chain radical that includes solely carbon and hydrogen atoms in the backbone, containing no unsaturation, having from one to eight carbon atoms (i.e. Ci-8alkyl), and which is attached to the rest of the molecule by a single bond, e.g., methyl, ethyl, n-propyl, 1-methylethyl (isopropyl), n- butyl, n-pentyl, and 1,1-dimethylethyl (t-butyl). The term "Ci-6 alkyl" refers to an alkyl chain having 1 to 6 carbon atoms. The term
 refers to an alkyl chain having 1 to 4 carbon atoms. Unless set forth or recited to the contrary, all alkyl groups described or claimed herein may be straight chain or branched, substituted or unsubstituted.
refers to an alkyl chain having 1 to 4 carbon atoms. Unless set forth or recited to the contrary, all alkyl groups described or claimed herein may be straight chain or branched, substituted or unsubstituted.
The term "alkenyl" refers to a hydrocarbon chain containing from 2 to 10 carbon atoms (i.e. C2-ioalkenyl) and including at least one carbon-carbon double bond. Non-limiting examples of alkenyl groups include ethenyl, 1-propenyl, 2-propenyl (allyl), z'so-propenyl, 2-methyl- 1-propenyl, 1-butenyl, and 2-butenyl. Unless set forth or recited to the contrary, all alkenyl groups described or claimed herein may be straight chain or branched, substituted or unsubstituted.
The term "alkynyl" refers to a hydrocarbyl radical having at least one carbon- carbon triple bond, and having 2 to about 12 carbon atoms (with radicals having 2 to about 10 carbon atoms being preferred i.e. C2-ioalkynyl). Non-limiting examples of alkynyl groups include ethynyl, propynyl, and butynyl. Unless set forth or recited to the contrary, all alkynyl groups described or claimed herein may be straight chain or branched, substituted or unsubstituted.
The term "alkoxy" denotes an alkyl group attached via an oxygen linkage to the rest of the molecule (i.e. Ci-8 alkoxy). Representative examples of such groups are -OCH3 and -OC2H5. Unless set forth or recited to the contrary, all alkoxy groups
described or claimed herein may be straight chain or branched, substituted or unsubstituted.
The term "alkoxyalkyl" or "alkyloxyalkyl" refers to an alkoxy or alkyloxy group as defined above directly bonded to an alkyl group as defined above (i.e. Ci. 8alkoxyCi-8alkyl or Ci-salkyloxyCi-salkyl). Example of such alkoxyalkyl moiety includes, but are not limited to, -CH2OCH3 and -CH2OC2H5. Unless set forth or recited to the contrary, all alkoxyalkyl groups described herein may be straight chain or branched, substituted or unsubstituted.
The term "haloalkyl" refers to at least one halo group (selected from F, CI, Br or I), linked to an alkyl group as defined above (i.e. haloCi-salkyl). Examples of such haloalkyl moiety include, but are not limited to, trifluoromethyl, difluoromethyl and fluoromethyl groups. Unless set forth or recited to the contrary, all haloalkyl groups described herein may be straight chain or branched, substituted or unsubstituted.
The term "haloalkoxy" refers to an alkoxy group substituted with one or more halogen atoms (i.e. haloCi-salkoxy). Examples of "haloalkoxy" include but are not limited to fluoromethoxy, difluoromethoxy, trifluoromethoxy, 2,2,2-trifluoroethoxy, pentafluoroethoxy, pentachloroethoxy, chloromethoxy, dichlorormethoxy, trichloromethoxy and 1-bromoethoxy. Unless set forth or recited to the contrary, all haloalkoxy groups described herein may be straight chain or branched, substituted or unsubstituted.
The term "haloalkoxyalkyl" refers to haloalkoxy group as defined above directly bonded to an alkyl group as defined above (i.e. haloCi-salkoxyCi-salkyl). Examples of "haloCi-salkoxyCi-salkyl" include but are not limited to (2,2,2- trifluoroethoxy )m ethyl or (2,2-difluoroethoxy)m ethyl. Unless set forth or recited to the contrary, all haloalkoxyalkyl groups described herein may be straight chain or branched, substituted or unsubstituted.
The term "hydroxyalkyl" refers to an alkyl group as defined above wherein one to three hydrogen atoms on different carbon atoms is/are replaced by hydroxyl groups (i.e. hydroxyCi-salkyl). Examples of hydroxyalkyl moieties include, but are not limited to -CH2OH, -C2H4OH and -CH(OH)C2H4OH. Unless set forth or recited to the contrary, all hydroxyalkyl groups described herein may be straight chain or branched, substituted or unsubstituted.
The term "carboxyl" means the group -COOH.
The term "carboxylalkyl" refers to an Ci-8alkyl group as defined above wherein at least one of the hydrogen atoms of the Ci-8alkyl group is replaced by a carboxyl group (i.e. "carboxylCi-8alkyl"). Examples of carboxylalkyl moieties include, but are not limited to carboxylmethyl (-CH2-COOH), carboxylethyl (-CH2- CH2-COOH), carboxylisopropyl (-C(CH3)2-COOH) and carboxyltertbutyl (- C(CH3)2CH2-COOH). Unless set forth or recited to the contrary, all carboxylalkyl groups described herein may be straight chain or branched, substituted or unsubstituted.
The term "cycloalkyl" denotes a non-aromatic mono or multicyclic ring system of 3 to about 12 carbon atoms, (i.e. C3-i2cycloalkyl). Examples of monocyclic cycloalkyl include but are not limited to cyclopropyl, cyclobutyl, cyclopentyl, and cyclohexyl. Examples of multicyclic cycloalkyl groups include, but are not limited to, perhydronapthyl, adamantyl and norbornyl groups, bridged cyclic groups or spirobicyclic groups, e.g., spiro(4,4)non-2-yl. The term "C3-6Cycloalkyl" refers to the cyclic ring having 3 to 6 carbon atoms. Unless set forth or recited to the contrary, all cycloalkyl groups described or claimed herein may be substituted or unsubstituted.
The term "cycloalkylalkyl" refers to a non-aromatic cyclic ring-containing radical having 3 to about 8 carbon atoms directly attached to an alkyl group (i.e. C3- 8cycloalkylCi-8alkyl). The cycloalkylalkyl group may be attached to the main structure at any carbon atom in the alkyl group that results in the creation of a stable structure. Non-limiting examples of such groups include cyclopropylmethyl, cyclobutylethyl, and cyclopentylethyl. Unless set forth or recited to the contrary, all cycloalkylalkyl groups described or claimed herein may be substituted or unsubstituted.
The term "cycloalkenyl" refers to a ccyclic ring-containing radical having 3 to about 8 carbon atoms with at least one carbon-carbon double bond, (i.e. C3- 8cycloalkenyl). Examples of "cycloalkenyl" include but are not limited to cyclopropenyl, cyclobutenyl, and cyclopentenyl. Unless set forth or recited to the contrary, all cycloalkenyl groups described or claimed herein may be substituted or unsubstituted.
The term "cycloalkenylalkyl" refers to a non-aromatic cyclic ring-containing radical having 3 to about 8 carbon atoms with at least one carbon-carbon double bond, directly attached to an alkyl group, (i.e. C3-8cycloalkenylCi-8alkyl). Unless set forth or
recited to the contrary, all cycloalkenylalkyl groups described or claimed herein may be substituted or unsubstituted.
The term "aryl" refers to an aromatic radical having 6 to 14 carbon atoms (i.e. C6-i4 ryl), including monocyclic, bicyclic and tricyclic aromatic systems, such as phenyl, naphthyl, tetrahydronapthyl, indanyl, and biphenyl. Unless set forth or recited to the contrary, all aryl groups described or claimed herein may be substituted or unsubstituted.
The term "aryloxy" refers to an aryl group as defined above attached via an oxygen linkage to the rest of the molecule (i.e. C6-i4aryloxy). Examples of aryloxy moieties include, but are not limited to phenoxy and naphthoxy. Unless set forth or recited to the contrary, all aryloxy groups described herein may be substituted or unsubstituted.
The term "arylalkyl" refers to an aryl group as defined above directly bonded to an alkyl group as defined above, i.e.
 such as -CH2C6H5 and - C2H4C6H5. Unless set forth or recited to the contrary, all arylalkyl groups described or claimed herein may be substituted or unsubstituted.
such as -CH2C6H5 and - C2H4C6H5. Unless set forth or recited to the contrary, all arylalkyl groups described or claimed herein may be substituted or unsubstituted.
The term "heterocyclic ring" or "heterocyclyl" unless otherwise specified refers to substituted or unsubstituted non-aromatic 3 to 15 membered ring radical (i.e. 3 to 15 membered heterocyclyl) which consists of carbon atoms and from one to five hetero atoms selected from nitrogen, phosphorus, oxygen and sulfur. The heterocyclic ring radical may be a mono-, bi- or tricyclic ring system, which may include fused, bridged or spiro ring systems, and the nitrogen, phosphorus, carbon, oxygen or sulfur atoms in the heterocyclic ring radical may be optionally oxidized to various oxidation states. In addition, the nitrogen atom may be optionally quaternized; also, unless otherwise constrained by the definition the heterocyclic ring or heterocyclyl may optionally contain one or more olefinic bond(s). Examples of such heterocyclic ring radicals include, but are not limited to azepinyl, azetidinyl, benzodioxolyl, benzodioxanyl, chromanyl, dioxolanyl, dioxaphospholanyl, decahydroisoquinolyl, indanyl, indolinyl, isoindolinyl, isochromanyl, isothiazolidinyl, isoxazolidinyl, morpholinyl, oxazolinyl, oxazolidinyl, oxadiazolyl, 2-oxopiperazinyl, 2- oxopiperidinyl, 2-oxopyrrolidinyl, 2-oxoazepinyl, octahydroindolyl, octahydroisoindolyl, perhydroazepinyl, piperazinyl, 4-piperidonyl, pyrrolidinyl, piperidinyl, phenothiazinyl, phenoxazinyl, quinuclidinyl, tetrahydroisquinolyl, tetrahydrofuryl or tetrahydrofuranyl, tetrahydropyranyl, thiazolinyl, thiazolidinyl,
thiamorpholinyl, thiamorpholinyl sulfoxide and thiamorpholinyl sulfone. Unless set forth or recited to the contrary, all heterocyclyl groups described or claimed herein may be substituted or unsubstituted.
The term "heterocyclylalkyl" refers to a heterocyclic ring radical directly bonded to an alkyl group (i.e. 3 to 15 membered heterocyclylCi-salkyl). Unless set forth or recited to the contrary, all heterocyclylalkyl groups described or claimed herein may be substituted or unsubstituted.
The term "heteroaryl" unless otherwise specified refers to substituted or unsubstituted 5 to 14 membered aromatic heterocyclic ring radical with one or more heteroatom(s) independently selected from N, O or S (i.e. 5 to 14 membered heteroaryl). The heteroaryl may be a mono-, bi- or tricyclic ring system. Examples of such heteroaryl ring radicals include, but are not limited to oxazolyl, isoxazolyl, imidazolyl, furyl, indolyl, isoindolyl, pyrrolyl, triazolyl, triazinyl, tetrazoyl, thienyl, thiazolyl, isothiazolyl, pyridyl, pyrimidinyl, pyrazinyl, pyridazinyl, pyrazolyl, benzofuranyl, benzothiazolyl, benzoxazolyl, benzimidazolyl, benzothienyl, benzopyranyl, carbazolyl, quinolinyl, isoquinolinyl, quinazolinyl, cinnolinyl, naphthyridinyl, pteridinyl, purinyl, quinoxalinyl, quinolyl, isoquinolyl, thiadiazolyl, indolizinyl, acridinyl, phenazinyl and phthalazinyl. Unless set forth or recited to the contrary, all heteroaryl groups described or claimed herein may be substituted or unsubstituted.
The term "heteroarylalkyl" refers to a heteroaryl ring radical directly bonded to an alkyl group (i.e. 5 to 14 membered heterarylCi-salkyl). Unless set forth or recited to the contrary, all heteroarylalkyl groups described or claimed herein may be substituted or unsubstituted.
Unless otherwise specified, the term "substituted" as used herein refers to substitution with any one or any combination of the following substituents: hydroxy, halogen, carboxyl, cyano, nitro, oxo (=0), thio (=S), substituted or unsubstituted alkyl, substituted or unsubstituted haloalkyl, substituted or unsubstituted hydroxyl alkyl, substituted or unsubstituted alkoxy, substituted or unsubstituted haloalkoxy, substituted or unsubstituted alkenyl, substituted or unsubstituted alkynyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted cycloalkylalkyl, substituted or unsubstituted cycloalkenyl, substituted or unsubstituted amino, substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, substituted or unsubstituted heteroaryl, substituted or unsubstituted heteroarylalkyl, substituted or unsubstituted
heterocyclylalkyl, substituted or unsubstituted heterocyclic ring, substituted or unsubstiuted guanidine, -COORx', -C(0)Rx', -C(S)RX', -C(0) RxRy', -C(0)O RxRy', - RxCO RyRz, -N(Rx')SORy', -N(Rx')S02Ry', -(=N-N(Rx)Ry), - RxC(0)ORy', - RxRy , -NRxC(0)Ry', - RxC(S)Ry', - RxC(S) Ry,Rz', -SO RxRy, -S02 RxRy', - ORx', -OC(0) RyRz', -OC(0)ORy', -OC(0)Rx', -OC(0) RxRy', -SRX', -SORx', - S02Rx, and -ON02, wherein each occurrence of Rx, Ry and Rz are independently selected from hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted alkoxy, substituted or unsubstituted alkenyl, substituted or unsubstituted alkynyl, substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted cycloalkylalkyl, substituted or unsubstituted cycloalkenyl, substituted or unsubstituted amino, substituted or unsubstituted aryl, substituted or unsubstituted heteroaryl, substituted heterocyclylalkyl, substituted or unsubstituted heteroarylalkyl, and substituted or unsubstituted heterocyclic ring. The substituents in the aforementioned "substituted" groups cannot be further substituted. For example, when the substituent on "substituted alkyl" is "substituted aryl", the substituent on "substituted aryl" can be unsubstituted alkenyl but cannot be "substituted alkenyl".
The term "pharmaceutically acceptable salt" includes salts prepared from pharmaceutically acceptable bases or acids including inorganic or organic bases and inorganic or organic acids. Examples of such salts include, but are not limited to, acetate, benzenesulfonate, benzoate, bicarbonate, bisulfate, bitartrate, borate, bromide, camsylate, carbonate, chloride, clavulanate, citrate, dihydrochloride, edetate, edisylate, estolate, esylate, fumarate, gluceptate, gluconate, glutamate, glycollylarsanilate, hexylresorcinate, hydrabamine, hydrobromide, hydrochloride, hydroxynaphthoate, iodide, isothionate, lactate, lactobionate, laurate, malate, maleate, mandelate, mesylate, methylbromide, methylnitrate, methyl sulfate, mucate, napsylate, nitrate, N-methylglucamine ammonium salt, oleate, oxalate, pamoate (embonate), palmitate, pantothenate, phosphate, diphosphate, polygalacturonate, salicylate, stearate, sulfate, subacetate, succinate, tannate, tartrate, teoclate, tosylate, triethiodide and valerate. Examples of salts derived from inorganic bases include, but are not limited to, aluminum, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, manganic, mangamous, potassium, sodium, and zinc.
The term "treating" or "treatment" of a state, disorder or condition includes: (a) preventing or delaying the appearance of clinical symptoms of the state, disorder
or condition developing in a subject that may be afflicted with or predisposed to the state, disorder or condition but does not yet experience or display clinical or subclinical symptoms of the state, disorder or condition; (b) inhibiting the state, disorder or condition, i.e., arresting or reducing the development of the disease or at least one clinical or subclinical symptom thereof; or (c) relieving the disease, i.e., causing regression of the state, disorder or condition or at least one of its clinical or subclinical symptoms.
The term "subject" includes mammals (especially humans) and other animals, such as domestic animals (e.g., household pets including cats and dogs) and non- domestic animals (such as wildlife).
A "therapeutically effective amount" means the amount of a compound that, when administered to a subject for treating a state, disorder or condition, is sufficient to effect such treatment. The "therapeutically effective amount" may vary depending on the compound, the disease and its severity and the age, weight, physical condition and responsiveness of the subj ect to be treated.
The sensation of pain can be triggered by any number of physical or chemical stimuli and the sensory neurons which mediate the response to this harmful stimulus are termed as "nociceptors". Nociceptors are primary sensory afferent (C and Αδ fibers) neurons that are activated by a wide variety of noxious stimuli including chemical, mechanical, thermal, and proton (pH<6) modalities. Nociceptors are the nerves which sense and respond to parts of the body which suffer from damage. They signal tissue irritation, impending injury, or actual injury. When activated, they transmit pain signals (via the peripheral nerves as well as the spinal cord) to the brain.
The term "chronic pain" usually refers to pain which persists for 3 months or longer and can lead to significant changes in a patient's personality, lifestyle, functional ability and overall quality of life. Chronic pain can be classified as either nociceptive or neuropathic. Nociceptive pain includes tissue injury -induced pain and inflammatory pain such as that associated with arthritis. Neuropathic pain is caused by damage to the sensory nerves of the peripheral or central nervous system and is maintained by aberrant somatosensory processing. The pain is typically well localized, constant, and often with an aching or throbbing quality. Visceral pain is the subtype of nociceptive pain that involves the internal organs. It tends to be episodic and poorly localized. Nociceptive pain is usually time limited, meaning when the
tissue damage heals, the pain typically resolves (arthritis is a notable exception in that it is not time limited).
Pharmaceutical Compositions
The compounds of the invention are typically administered in the form of a pharmaceutical composition. Such compositions can be prepared using procedures known in the pharmaceutical art and comprise at least one compound of the invention. The pharmaceutical composition of the present patent application comprises one or more compounds described herein and one or more pharmaceutically acceptable excipients. Typically, the pharmaceutically acceptable excipients are approved by regulatory authorities or are generally regarded as safe for human or animal use. The pharmaceutically acceptable excipients include, but are not limited to, carriers, diluents, glidants and lubricants, preservatives, buffering agents, chelating agents, polymers, gelling agents, viscosifying agents, and solvents.
Examples of suitable carriers include, but are not limited to, water, salt solutions, alcohols, polyethylene glycols, peanut oil, olive oil, gelatin, lactose, terra alba, sucrose, dextrin, magnesium carbonate, sugar, amylose, magnesium stearate, talc, gelatin, agar, pectin, acacia, stearic acid, lower alkyl ethers of cellulose, silicic acid, fatty acids, fatty acid amines, fatty acid monoglycerides and diglycerides, fatty acid esters, and polyoxyethylene.
The pharmaceutical composition may also include one or more pharmaceutically acceptable auxiliary agents, wetting agents, suspending agents, preserving agents, buffers, sweetening agents, flavoring agents, colorants or any combination of the foregoing.
The pharmaceutical compositions may be in conventional forms, for example, capsules, tablets, solutions, suspensions, injectables or products for topical application. Further, the pharmaceutical composition of the present invention may be formulated so as to provide a desired release profile.
Administration of the compounds of the invention, in pure form or in an appropriate pharmaceutical composition, can be carried out using any of the accepted routes of administration of pharmaceutical compositions. The route of administration may be any route which effectively transports the active compound of the patent application to the appropriate or desired site of action. Suitable routes of administration include, but are not limited to, oral, nasal, buccal, dermal, intradermal,
transdermal, parenteral, rectal, subcutaneous, intravenous, intraurethral, intramuscular, or topical.
Solid oral formulations include, but are not limited to, tablets, capsules (soft or hard gelatin), dragees (containing the active ingredient in powder or pellet form), troches and lozenges.
Liquid formulations include, but are not limited to, syrups, emulsions, and sterile injectable liquids, such as suspensions or solutions.
Topical dosage forms of the compounds include ointments, pastes, creams, lotions, powders, solutions, eye or ear drops, and impregnated dressings, and may contain appropriate conventional additives such as preservatives, solvents to assist drug penetration.
The pharmaceutical compositions of the present patent application may be prepared by conventional techniques, e.g., as described in Remington: The Science and Practice of Pharmacy, 20th Ed., 2003 (Lippincott Williams & Wilkins).
Suitable doses of the compounds for use in treating the diseases and disorders described herein can be determined by those skilled in the relevant art. Therapeutic doses are generally identified through a dose ranging study in humans based on preliminary evidence derived from animal studies. Doses are generally sufficient to result in a desired therapeutic benefit without causing unwanted side effects. Mode of administration, dosage forms, and suitable pharmaceutical excipients can also be well used and adjusted by those skilled in the art. All changes and modifications are envisioned within the scope of the present patent application.
Methods of Treatment
Compounds of the present invention are particularly useful because they may inhibit the activity of prostaglandin E synthases (and particularly microsomal prostaglandin E synthase-1 (mPGES-1)), i.e., they prevent, inhibit, or suppress the action of mPGES-1 or a complex of which the mPGES-1 enzyme forms a part, and/or may elicit mPGES-1 modulating effect. Compounds of the invention are thus useful in the treatment of those conditions treatable by inhibition of a PGES, and particularly mPGES-1.
Compounds of the invention are thus expected to be useful in the treatment of inflammation. The term "inflammation" will be understood by those skilled in the art to include any condition characterized by a localized or a systemic protective response, which may be elicited by physical trauma, infection, chronic diseases, such
as those mentioned hereinbefore, and/or chemical and/or physiological reactions to external stimuli (e.g. as part of an allergic response). Any such response, which may serve to destroy, dilute or sequester both the injurious agent and the injured tissue, may be manifest by, for example, heat, swelling, pain, redness, dilation of blood vessels and/or increased blood flow.
The term "inflammation" is also understood to include any inflammatory disease, disorder or condition per se, any condition that has an inflammatory component associated with it, and/or any condition characterized by inflammation as a symptom, including, inter alia, acute, chronic, ulcerative, specific, allergic, infection by pathogens, immune reactions due to hypersensitivity, entering foreign bodies, physical injury, and necrotic inflammation, and other forms of inflammation known to those skilled in the art. The term thus also includes, for the purposes of this invention, inflammatory pain, pain generally and/or fever.
The compounds of the present invention may also be useful in the treatment of asthma, chronic obstructive pulmonary disease, pulmonary fibrosis, inflammatory bowel disease, irritable bowel syndrome, inflammatory pain, chronic pain, acute pain, fever, migraine, headache, low back pain, fibromyalgia, myofascial disorders, viral infections (e.g. influenza, common cold, herpes zoster, hepatitis C and AIDS), bacterial infections, fungal infections, dysmenorrhea, burns, surgical or dental procedures, malignancies (e.g. breast cancer, colon cancer, and prostate cancer), hyperprostaglandin E syndrome, classic Bartter syndrome, atherosclerosis, gout, arthritis, osteoarthritis, juvenile arthritis, rheumatoid arthritis, juvenile onset rheumatoid arthritis, rheumatic fever, ankylosing spondylitis, Hodgkin's disease, systemic lupus erythematosus, vasculitis, pancreatitis, nephritis, bursitis, conjunctivitis, iritis, scleritis, uveitis, wound healing, dermatitis, eczema, psoriasis, stroke, diabetes mellitus, neurodegenerative disorders such as Alzheimer's disease and multiple sclerosis, autoimmune diseases, allergic disorders, rhinitis, ulcers, mild to moderately active ulcerative colitis, familial adenomatous polyposis, coronary heart disease, sarcoidosis and any other disease with an inflammatory component.
Compounds of the invention may also have effects that are not linked to inflammatory mechanisms, such as in the reduction of bone loss in a subject. Conditions that may be mentioned in this regard include osteoporosis, osteoarthritis, Paget's disease and/or periodontal diseases.
By virtue of the mPGES-1 inhibitory activity of compounds of the present invention, the compounds are useful for the relief of pain, fever and inflammation of a variety of conditions including rheumatic fever, symptoms associated with influenza or other viral infections, common cold, low back and neck pain, dysmenorrhea, headache, migraine (acute and prophylactic treatment), toothache, sprains and strains, myositis, neuralgia, synovitis, arthritis, including rheumatoid arthritis, juvenile rheumatoid arthritis, degenerative joint diseases (osteoarthritis), acute gout and ankylosing spondylitis, acute, subacute and chronic musculoskeletal pain syndromes such as bursitis, burns, injuries, and pain following surgical (post-operative pain) and dental procedures as well as the preemptive treatment of surgical pain. The pain may be mild pain, moderate pain, severe pain, musculoskeletal pain, complex regional pain syndrome, neuropathic pain, back pain such as acute visceral pain, neuropathies, acute trauma, chemotherapy - induced mononeuropathy pain states, polyneuropathy pain states (such as diabetic peripheral neuropathy & chemotherapy induced neuropathy), autonomic neuropathy pain states, pheriphaeral nervous system (PNS) lesion or central nervous system (CNS) lesion or disease related pain states, polyradiculopathies of cervical, lumbar or sciatica type, cauda equina syndrome, piriformis syndrome, paraplegia, quadriplegia, pain states related to various Polyneuritis conditions underlying various infections, chemical injuries, radiation exposure, underlying disease or deficiency conditions (such as beriberi, vitamin deficiencies, hypothyroidism, porphyria, cancer, HIV, autoimmune disease such as multiple sclerosis and spinal-cord injury, fibromyalgia, nerve injury, ischaemia, neurodegeneration, stroke, post stroke pain, inflammatory disorders, oesophagitis, gastroeosophagal reflux disorder (GERD), irritable bowel syndrome, inflammatory bowel disease, pelvic hypersensitivity, urinary incontinence, cystitis, stomach duodenal ulcer, muscle pain, pain due to colicky and referred pain. Compounds of the present invention may also be useful for the treatment or prevention of endometriosis, hemophilic arthropathy and Parkinson's disease.
Compounds of the present invention will also inhibit prostanoid-induced smooth muscle contraction by preventing the synthesis of contractile prostanoids and hence may be of use in the treatment of dysmenorrhea, premature labor and asthma.
In addition, the compounds of the present invention may inhibit cellular neoplastic transformations and metastic tumor growth and hence can be used in the treatment of cancer, and pain associated with cancer. Furthermore, the present
invention provides preferred embodiments of the methods and uses as described herein, in which cancer includes Acute Lymphoblastic Leukemia, Acute Myeloid Leukemia, Adolescents Cancer, Adrenocortical Carcinoma, Anal Cancer, Appendix Cancer, Astrocytomas, Atypical Teratoid, Basal Cell Carcinoma, Bile Duct Cancer, Extrahepatic, Bladder Cancer, Bone Cancer, Brain Stem Glioma, Brain Tumor, Breast Cancer, Bronchial Tumors, Burkitt Lymphoma, Carcinoid Tumor, Carcinoma of Unknown Primary, Cardiac (Heart) Tumors, Central Nervous System tumors, Cervical Cancer, Childhood Cancers, Chordoma, Chronic Lymphocytic Leukemia, Chronic Myelogenous Leukemia, Chronic Myeloproliferative Disorders, Colon Cancer, Colorectal Cancer, Craniopharyngioma, Cutaneous T-Cell Lymphoma, Duct Bile Extrahepatic cancer, Ductal Carcinoma In Situ, Embryonal Tumors, Central Nervous System cancer, Endometrial Cancer, Ependymoma, Esophageal Cancer, Esthesioneuroblastoma, Ewing Sarcoma, Extracranial Germ Cell Tumor, Extragonadal Germ Cell Tumor, Extrahepatic Bile Duct Cancer, Eye Cancer, Fibrous Histiocytoma of Bone, Malignant, and Osteosarcoma, Gall bladder Cancer, Gastric (Stomach) Cancer, Gastrointestinal Carcinoid Tumor, Gastrointestinal Stromal Tumors, Germ Cell Tumor, Gestational Trophoblastic Tumor, Glioma, Hairy Cell Leukemia, Head and Neck Cancer, Heart Cancer, Hepatocellular (Liver) Cancer, Histiocytosis, Langerhans Cell, Hodgkin Lymphoma, Hypopharyngeal Cancer, Intraocular Melanoma, Islet Cell Tumors, Pancreatic Neuroendocrine Tumors, Kaposi Sarcoma, Kidney cancer, Langerhans Cell Histiocytosis, Laryngeal Cancer, Acute Lymphoblastic Leukemia, Acute Myeloid Leukemia, Chronic Lymphocytic Leukemia, Chronic Myelogenous Leukemia, Hairy Cell Leukemia, Lip and Oral Cavity Cancer, Liver Cancer, Lobular Carcinoma In Situ, Lung Cancer, AIDS- Related Lymphoma, Cutaneous T-Cell Lymphoma, Hodgkin Lymphoma, Non- Hodgkin Lymphoma, Primary Central Nervous System (CNS) Lymphoma, Macroglobulinemia, Waldenstrom, Male Breast Cancer, Malignant Fibrous Histiocytoma of Bone and Osteosarcoma, Melanoma, Merkel Cell Carcinoma, Mesothelioma, Malignant, Metastatic Squamous Neck Cancer with Occult Primary, Midline Tract Carcinoma Involving NUT Gene, Mouth Cancer, Multiple Endocrine Neoplasia Syndromes, Multiple Myeloma/Plasma Cell Neoplasm, Mycosis Fungoides, Myelodysplastic Syndromes, Myelodysplastic/Myeloproliferative Neoplasms, Myelogenous Leukemia, Chronic, Myeloid Leukemia Acute, Multiple Myeloma, Chronic Myeloproliferative Disorders, Nasal Cavity and Paranasal Sinus
Cancer, Nasopharyngeal Cancer, Neuroblastoma, Non-Hodgkin Lymphoma, Non- Small Cell Lung Cancer, Oral Cancer, Oral Cavity Cancer, Lip and, Oropharyngeal Cancer, Osteosarcoma and Malignant Fibrous Histiocytoma of Bone, Ovarian Cancer, Pancreatic Cancer, Papillomatosis, Paraganglioma, Paranasal Sinus and Nasal Cavity Cancer, Parathyroid Cancer, Penile Cancer, Pharyngeal Cancer, Pheochromocytoma, Pituitary Tumor, Plasma Cell Neoplasm/Multiple Myeloma, Pleuropulmonary Blastoma, Pregnancy and Breast Cancer, Primary Central Nervous System (CNS) Lymphoma, Prostate Cancer, Rectal Cancer, Renal Cell (Kidney) Cancer, Renal Pelvis and Ureter, Transitional Cell Cancer, Retinoblastoma, Rhabdomyosarcoma, Salivary Gland Cancer, Ewing Sarcoma, Kaposi Sarcoma, Osteosarcoma, Rhadomyosarcoma, Soft Tissue Sarcoma, Uterine Sarcoma, Sezary Syndrome, Skin Cancer, Small Cell Lung Cancer, Small Intestine Cancer, Soft Tissue Sarcoma, Squamous Cell Carcinoma, Squamous Neck Cancer with Occult Primary, Metastatic, Stomach (Gastric) Cancer, T-Cell Lymphoma, Cutaneous, Testicular Cancer, Throat Cancer, Thymoma and Thymic Carcinoma, Thyroid Cancer, Transitional Cell Cancer of the Renal Pelvis and Ureter, Trophoblastic Tumor, Gestational, Unknown Primary, Carcinoma of, Ureter and Renal Pelvis, Transitional Cell Cancer, Urethral Cancer, Uterine Cancer, Endometrial, Uterine Sarcoma, Vaginal Cancer, Vulvar Cancer, Waldenstrom, Macroglobulinemia, Wilms Tumor and Women's Cancers.
The compounds of the present invention may be useful in the treatment of disease, disorder, syndrome or condition selected from the group consisting of inflammation, asthma, chronic obstructive pulmonary disease, pulmonary fibrosis, inflammatory bowel disease, irritable bowel syndrome, pain, inflammatory pain, chronic pain, acute pain, fever, migraine, headache, low back pain, fibromyalgia, myofascial disorders, viral infections, influenza, common cold, herpes zoster, hepatitis C, AIDS, bacterial infections, fungal infections, dysmenorrhea, burns, surgical or dental procedures, malignancies hyperprostaglandin E syndrome, classic Bartter syndrome, synovitis, atherosclerosis, gout, arthritis, osteoarthritis, juvenile arthritis, rheumatoid arthritis, juvenile onset rheumatoid arthritis, rheumatic fever, ankylosing spondylitis, Hodgkin's disease, systemic lupus erythematosus, vasculitis, pancreatitis, nephritis, bursitis, conjunctivitis, iritis, scleritis, uveitis, wound healing, dermatitis, eczema, psoriasis, stroke, diabetes mellitus, cancer, neurodegenerative disorders such as Alzheimer's disease, Parkinson's disease, Amyotrophic lateral sclerosis and multiple sclerosis, autoimmune diseases, allergic disorders, rhinitis,
ulcers, mild to moderately active ulcerative colitis, familial adenomatous polyposis, coronary heart disease, and sarcoidosis.
The compounds of the present invention may be useful in the treatment of pain, chronic pain, acute pain, rheumatoid arthritic pain or osteoarthritic pain.
The compounds of the present invention may be useful in the treatment of inflammation, neurodegenerative disorders such as Parkinson's disease, Alzheimer's disease and amyotrophic lateral sclerosis.
The compounds of the present invention may be useful in the treatment prevention or management of the cancer.
Compounds of the present invention are indicated both in the therapeutic and/or prophylactic treatment of the above-mentioned conditions. For the above- mentioned therapeutic uses the dosage administered will, of course, vary with the compound employed, the mode of administration, the treatment desired and the disorder indicated. The daily dosage of the compound of the invention may be in the range from 0.05 mg/kg to 100 mg/kg.
General Methods of Preparation
The compounds described herein, including compounds of formula (I), (II), (III), (IV) and specific examples are prepared using techniques known to one skilled in the art through the reaction sequences depicted in schemes provided below, as well as by other methods. Furthermore, in the following schemes, where specific acids, bases, reagents, coupling agents, solvents, etc. are mentioned, it is understood that other suitable acids, bases, reagents, coupling agents etc. may be used and are included within the scope of the present invention. Modifications to reaction conditions, for example, temperature, duration of the reaction or combinations thereof, are envisioned as part of the present invention. The compounds obtained by using the general reaction sequences may be of insufficient purity. These compounds can be purified by using any of the methods for purification of organic compounds known to person skilled in the art, for example, crystallization or silica gel or alumina column chromatography using different solvents in suitable ratios. All possible geometrical isomers and stereoisomers are envisioned within the scope of this invention.
The starting materials for the below reaction schemes are commercially available or can be prepared according to methods known to one skilled in the art or by methods disclosed herein. In general, intermediates and compounds of the present
application may be prepared using the reaction scheme as follows, wherein all symbols are as defined above.
A general approach for the preparation of a compound of formula (II) [(II-a) and (II-b)], (wherein G3 is CH, G4 is CH or N, Rx and Ry are hydrogen; p is 1 and, R1, R3, R4, W, L, m and n are as defined above with respect to a compound of formula (II)), is depicted in the Synthetic Schemes-1, 2, 3 and 4.
Synthetic scheme 1

Thus, substituted benzoic ester derivatives of formula (1) undergoes formylation by using n-butyllithium and suitable solvent such as DMF in presence of organic base such as DIPEA or TEA to obtain compound of formula (2). The compound of formula (2) can be treated with hydroxyl amine in presence of suitable solvent such as methanol or ethanol to obtain compound of formula (3). The compound of formula (3) can be reduced by using reducing reagent such as zinc-dust or Fe-powder in presence of acid such as concentrated hydrochloric acid and suitable solvent such as methanol or ethanol to obtain the compound of formula (4). The compound of formula (4) can be reacted with compound of formula (5) in presence of organic base such as DIPEA or TEA in suitable solvent such as THF to obtain compound of formula (6). The compound of formula (6) can be converted to compound of formula (7) using reducing reagents such as sodium borohydride or lithium aluminium hydride in presence of suitable solvent such THF. The compound of formua (7) can be oxidized to obtain compound of formula (12) by using oxidizing agents such as manganese dioxide or pyridinium chlorochromate in presence of solvent such as DCM. The
compound of formula (12) can be reacted with substituted acetophenone derivatives of formula (13) and urea in presence of iodine to obtain compound of formula (Il-a).
The compound of formula (7) can be also prepared from substituted benzoic acid derivative. Thus, substituted benzoic acid derivatives of formula (8) can be treated with 2,2,2-trifluoro-N-(hydroxymethyl)acetamide to obtain compound of formula (9) under standard conditions. The compound of formula (9) can be converted to compound of formula (10) in presence of acid such as sulphuric acid or hydrochloric acid in a suitable solvent such as ethanol or methanol. The compound of formula (10) can be reacted with compound of formula (5) in presence of organic base such as DIPEA or TEA in suitable solvent such as THF to obtain compound of formula (1 1). The compound of formula (1 1) can be further converted to compound of formula (7) using reducing reagents such as sodium borohydride or lithium aluminium hydride in presence of suitable solvent such THF.
Synthetic scheme 2

In another approach, the substituted benzaldehyde of formula (12) can be reacted with compound of formula (13a) and urea in presence of suitable reagents such as trimethylsilyl chloride and sulphamic acid to obtain compound of formula (14). The compound of formula (14) can be further converted to the compound of formula (Il-b) by using suitable reagents such as 2,3-dichloro-5,6-dicyano-l,4-benzoquinone (DDQ) in a suitable solvent such as toluene.
Synthetic scheme 3

The substituted acetophenone derivatives of formula (15) can be treated with 2,2,2- trifluoro-N-(hydroxymethyl)acetamide, followed by treatment with base such as lithium hydroxide to obtain compound of formula (16). The compound of formula (16) can be reacted with compound of formula (5) in presence of organic base such as
DIPEA or TEA in presence of suitable solvent such as THF to obtain a compound of formula (17). The compound of formula (17) can be reacted with compound of formula (18) and urea in presence of suitable reagents such as trimethylsilyl chloride and sulphamic acid to obtain compound of formula (Il-a).
Synthetic scheme 4

In another approach, the substituted acetophenone derivatives of formula (17) can be reacted with compound of formula (18) and urea in presence of suitable reagents such as trimethylsilyl chloride and sulphamic acid to obtain compound of formula (14a). The compound of formula (14a) can be further converted to the compound of formula
(Il-a) using suitable reagents such as 2,3-dichloro-5,6-dicyano-l,4-benzoquinone
(DDQ) in a suitable solvent such as toluene.
The compound of formula (II)[(II-c)] (wherein G3 is N, G4 is CH, R1, Rx and
Ry are hydrogen; p is 1 and R3, R4, W, L, m and n are as defined above with respect to a compound of formula (II)) can be prepared by the procedure as depicted in the
Synthetic Scheme- 5.
Synthetic scheme 5

Thus, the substituted nitrile derivatives of formula (19) can be reacted with a compound of formula (18) and urea in presence of suitable reagents such as trimethylsilyl chloride and sulphamic acid to obtain compound of formula (20). The compound of formula (20) can be further converted to the compound of formula (21) by using suitable reagents such as 2,3-dichloro-5,6-dicyano-l,4-benzoquinone (DDQ) in a suitable solvent such as toluene. The compound of formula (21) undergoes reduction reaction by using reducing reagent such as Pd/C in presence of solvent such
as ethanol or methanol to give the corresponding amine derivative (which is highly unstable). The amine derivatives can be protected by using protecting reagents such as di-i¾rt-buty! di carbonate in presence of base such as DIPEA or TEA in solvent such as DCM to give compound of formula (22). The compound of formula (22) can be further deprotected by using methanol: HQ/ ethanol :HC1 to give compound of formula (23). The compound of formula (23) can be reacted with compound of formula (5) in presence of organic base such as DIPEA or TEA in a suitable solvent such as THF to obtain compound of formula (II-c).
The compound of formula (III) (wherein Ra, Rb, R3, R4, W, m and n are as defined above with respect to a compound of formula (III)) can be prepared by the procedure as depicted in the Synthetic Schemes-6 and 7.
Synthetic scheme 6

Thus, the substituted acetophenone derivatives of formula (17a) can be reacted with compound of formula (18) in presence of base such as sodium hydroxide in solvent such as methanol to give compound of formula (24). The compound of formula (24) can be reacted with guanidine or substituted guanidine in presence of anion generating reagent such as sodium hydride in a suitable solvent such as dimethyl formamide to give compound of formula (III). The compound of formula (III) (wherein Ra and Rb are hydrogen) can be futher treated with acetic anhydride to obtain the corresponding acetyl derivative.
Synthetic scheme 7

In another approach, the substituted aldehyde derivatives of formula (12a) can be reacted with compound of formula (13b) in presence of base such as sodium hydroxide in solvent such as methanol to give compound of formula (25). The compound of formula (25) can be reacted with guanidine or substituted guanidine in
presence of anion generating reagent such as sodium hydride in a suitable solvent such as dimethyl formamide to give compound of formula (III).
The compound of formula (IV) (wherein Ra, R3, R4, W, m and n are as defined above with respect to a compound of formula (IV)) can be prepared by the procedure as depicted in the Synthetic Scheme-8.
Synthetic scheme 8

Thus, compound of formula (26) can be treated with suitable reagent such as acetic anhydride to obtain compound of formyula (IV).
A general approach for the preparation of a compound of formula (II) (wherein G3, G4, Rx, Ry, R1, R3, R4, W, L, m and n are as defined above with respect to a compound of formula (II) and p is 1), is depicted in the Synthetic Scheme-9.
Synthetic scheme 9

The process for the preparation of compound of formula (II) or a pharmaceutically acceptable salt thereof comprises:
(a) reacting compound of formula (27) with compound of formula (13a) and urea to obtain compound of formula (28), and
(b) converting the compound of formula (28) to compound of formula (II).
Step (a) involves reacting the compound of formula (27) with compound of formula (13a) and urea in presence of suitable reagents. The suitable reagents may be trimethylsilyl chloride and sulphamic acid. The reaction may be carried out at a suitable temperature. The suitable temperature may be around 100- 150 °C, preferable 100 °C.
Step (b) involves conerting the compound of formula (28) to compound of formula (II) using a suitable reagent. The suitable reagent may be 2,3-dichloro-5,6-dicyano- 1,4-benzoquinone (DDQ). The reaction may be carried out in presence of a suitable solvent. The suitable solvent may be toluene.
Experimental
Unless otherwise stated, work-up includes distribution of the reaction mixture between the organic and aqueous phase indicated within parentheses, separation of layers and drying the organic layer over sodium sulphate (Na2S04), filtration and evaporation of the solvent under reduced pressure. Purification, unless otherwise mentioned, includes purification by silica gel chromatographic techniques, in suitable solvents of a suitable polarity as the mobile phase.
The abbreviations, symbols and terms used in the application have the following meanings: EDCI: l-ethyl-3-(3-dimethylaminopropyl)carbodiimide; DCM or MDC: Dichlorom ethane; LAH: Lithium aluminium hydride; PCC : pyridinium chlorochromate; DEE: Diethylether; DIPEA: N,N-Diisopropylethylamine ; DMSO: Di-methyl sulfoxide; THF: Tetrahydrofuran; TEA: Triehtylamine; EDC: Ethylene di chloride; EtOAc or EA: Ethyl acetate; CH2C12: Dichloromethane; CHC13: Chloroform; MeOH: Methanol; NaOH: Sodium hydroxide; Na2S04: Sodium sulfate; : Coupling constant in units of Hz; RT or rt: room temperature (22-26°C); aq.: aqueous; equiv. or eq.: equivalents; cone. : concentrated; i.e. : that is; e.g. for example; h : hour(s); 1H MR: Proton Nuclear Magnetic Resonance; APCI-MS: Atmospheric Pressure Chemical Ionization Mass Spectrometry.
Preparation of Intermediates
Intermediate - 1
N-(4-chloro-3-formylbenzyl)pivalamide

Step 1 : Preparation of 2-chloro- -{[(trifluoroacetyl)amino]methyl}benzoic acid

To a solution of 2-chlorobenzoic acid (500 mg, 3.49 mmol) in cone. H2S04 was added 2,2,2-trifluoro-N-(hydroxymethyl)acetamide (547 mg, 3.49 mmol). The mixture was stirred at RT for 16 h. The reaction mixture was poured into ice-water and the obtained precipitate was collected by filtration, dried and recrystallized from toluene/butan-2-one (7: 1) to afford 800 mg of the desired product. 1H NMR (300
MHz, DMSO-i¾): δ 13.47 (br s, 1H), 10.06 (br s, 1H), 7.71 (s, 1H), 7.54 (d, J Hz, 1H), 7.43 (d, = 9.9 Hz, 1H), 4.42 (d, = 6.0 Hz, 2H); MS [M+H]+: 282.34. Step 2: Preparation of methyl 5-(aminometh l)-2-chlorobenzoate

To a solution of 2-chloro-5-{ [(trifluoroacetyl)amino]methyl}benzoic acid (800 mg, 2.84 mmol) in MeOH (10 mL) was added cone. H2S04 (0.5 mL) and heated at reflux overnight. The reaction mass was then concentrated, diluted with water and the solid obtained was filtered and suck dried to afford 800 mg of the desired product. 1H MR (300 MHz, DMSO-i¾): δ 7.77(s), 7.65-7.45 (m,2H), 3.85 (s,3H), 3.72 (s,2H); MS [M+H]+: 200.10.
Step 3 : Preparation of methyl 2-chloro-5-(pivalamidomethyl)benzoate

To a solution of methyl 5-(aminomethyl)-2-chlorobenzoate (800 mg, 4.02 mmol) in THF (20 mL) was added TEA (4.0 mL) followed by drop-wise addition of pivaloyl chloride (763 mg, 6.03 mmol) at 0°C. The reaction mixture was stirred at RT for 3 h. The reaction mass was quenched with water and extracted with EtOAc. The organic layer was separated, dried over Na2S04 and concentrated to afford 560 mg of the desired product. 1H NMR (300 MHz, DMSO-i¾): δ 8.16 (m, 1H), 7.65 (s, 1H), 7.53 (d, = 8.4 Hz, 1H), 7.41 (d, = 8.4 Hz, 1H), 4.25 (d, = 5.7 Hz, 2H), 3.85 (s, 3H), 1.15 (s, 9H); MS [M+H]+: 284.32.
Step 4: Preparation of N-(4-chloro-3-(hydroxymethyl)benzyl)pivalamide

To a solution of methyl 2-chloro-5-(pivalamidomethyl)benzoate (500 mg, 1.76 mmol) in THF (5.0 mL) was added LiAlH4 (134 mg, 3.53 mmol) at 0°C and the reaction mass was stirred at RT for 4 h. The reaction mixture was quenched with saturated
Na2S04 solution at 0°C and filtered through celite pad. The organic layer was dried over Na2S04 and concentrated to afford 500 mg of the desired product. 1H NMR (300 MHz, OMSO-d6): δ 8.09 (br t, 1H), 7.42 (s, 1H), 7.33-7.31 (d, J = 7.2 Hz, 1H), 7.12- 7.09 (d, J = 8.1 Hz, 1H), 5.41-5.37 (t, J = 5.4 Hz, 1H), 4.53-4.52 (d, J = 5.4 Hz, 2H), 4.24-4.22 (d, = 6.0 Hz, 2H), 1.12 (s , 9H); MS [M+H]+: 256.10.
Step 5 : Preparation of N-(4-chloro-3-formylbenzyl)pivalamide
To a solution of N-(4-chloro-3-(hydroxymethyl)benzyl)pivalamide (500 mg, 1.96 mmoL) in CH2C12 (10.0 mL) was added Mn02 (1.68 g, 19.60 mmol) under inert atmosphere and the reaction mixture was stirred at RT overnight. The reaction mass was filtered through celite bed and the filtrate was concentrated to afford 320 mg of the title compound. 1H NMR (300 MHz, DMSO-i¾): δ 10.32 (s, 1H), 8.19 (br t, 1H), 7.71 (s, 1H), 7.59-7.51 (m, 2H), 4.29-4.23 (d, J = 6.3Hz, 2H), 1.1 1 (s, , 9H); MS [M+H]+: 258.42.
Intermediate - 2
Preparation of N-(4-chloro-3-formylbenzyl)isobutyramide

Step 1 : Preparation of methyl 2-chloro-5-(isobutyramidomethyl)benzoate

To a solution of methyl 5-(aminomethyl)-2-chlorobenzoate hydrochloride (2.50 g, 10.59 mmol) in THF (20 mL) were added DIPEA (4.78 g, 37.07 mmol) and isobutyryl chloride (1.70 g, 15.89 mmol). The reaction mass was stirred at RT for 2 h.
After completion of the reaction, the reaction mixture was diluted with EtOAc, washed with water and brine, dried over Na2S04 and concentrated to afford 1.2 g of the desired product. 1H NMR (300 MHz, DMSO-i¾): δ 8.35 (t, 1H), 7.66 (s, 1H), 7.55-7.52 (d, J = 8.1 Hz, 1H), 7.43-7.40 (d, J = 8.1 Hz, 1H), 4.26 (d, J = 5.7 Hz, 2H),
3.85 (s, 3H), 2.44 (m, 1H), 1.02 (d, J = 6.9 Hz, 6H); MS [M+H]+: 270.12.
Step-2: Preparation of N-(4-chloro-3-(hydroxymethyl)benzyl)isobutyramide

The title compound was prepared following the procedure described in step-4 of Intermediate- 1 by using methyl 2-chloro-5-(isobutyramidomethyl)benzoate (500 mg, 1.76 mmol), THF (5.0 mL) and LiAlH4 (134 mg, 3.53 mmol) at 0 °C to afford 300 mg of the desired product. 1H NMR (300 MHz, DMSO-i¾): δ 8.29 (br s, 1H), 7.42 (s, 1H), 7.33-7.31 (d, J = 7.2 Hz, 1H), 7.12-7.09 (d, J = 8.1 Hz, 1H), 5.41-5.37 (t, J = 5.4 Hz, 1H), 4.53-4.52 (d, J = 5.4 Hz, 2H), 4.24-4.22 (d, J = 6.0 Hz, 2H), 2.43-2.38 (m, 1H), 1.02 (d, J = 6.9 Hz, 6H); MS [M+H]+: 242.21.
Step-3 : Preparation of N-(4-chloro-3-formylbenzyl)isobutyramide
The title compound was prepared following the procedure described in step-5 of Intermediate- 1 by using N-(4-chloro-3-(hydroxymethyl)benzyl)isobutyramide (500 mg, 2.07 mmoL), DCM (10.0 mL) and PCC (892 mg, 4.14 mmol) at RT to afford 320 mg of the title product. 1H NMR (300 MHz, OMSO-d6): δ 10.32 (s, 1H), 8.39 (br t, 1H), 7.73 (s, 1H), 7.60-7.52 (m, 2H), 4.30-4.28 (d, J = 6.3Hz, 2H), 2.45-2.38 (m, 1H), 1.02 (d, J = 6.9 Hz, 6H); MS [M+H]+: 244.17.
Intermediate 3
Prearation of N-(4-chloro-2-fluoro-3-formylbenzyl)pivalamide

Step-1 : Preperation of ethyl 6- chloro-2-fluoro-3-formylbenzoate

To a solution of DIPEA (6.6 mL, 46.0 mmol) in THF (15 mL) was addeed n-BuLi (27 mL, 43.0 mmol, 1.6 M in hexane) at -78 °C and the reaction mixture was warmed to 0°C over a period of 1 h. The reaction mixture was cooled to -78 °C and a solution of ethyl 2-chloro-6-fluorobenzoate (3.50 g, 19.0 mmol) in THF (56 mL) was added to the reaction mixture dropwise over 30 mins. The resulting mixture was stirred at -78
°C for 2 h. DMF (14 mL, 186 mmol) was added to the reaction mixture. The resulting mixture was further stirred at -78 °C for 1 h and slowly warmed to 0 °C. The reaction mass was quenched with 10% aq. AcOH and extracted with EtOAc. The organic layer was washed with water and brine, dried over Na2S04, concentrated and purified by flash chromatography using 2-8% methanol in DCM to provide ethyl 6-chloro-2- fluoro-3-formylbenzoate. 1H NMR (300 MHz, DMSO-i¾): δ 10.27 (s, 1H), 8.05-8.00 (t, 7 = 7.8 Hz, 1H), 7.72-7.69 (d, 7 = 8.4 Hz, 1H), 4.53-4.46 (q, 7 = 6.9, 14.4 Hz, 2H), 1.41-1.37 (t, 7 = 6.6 Hz, 3H).
Step-2 Preperation of ethyl 6-chloro-2-fluoro-3-((hydroxyimino)methyl)benzoate

A mixture of ethyl 6-chloro-2-fluoro-3-formylbenzoate (4.04 g, 17.52 mmol) and hydroxylamine (50% aqueous solution, 4.29 mL, 70 mmol) in MeOH (60 mL) was stirred at 55 °C for 1.5 h. The reaction mixture was concentrated and the residue was diluted with EtOAc, washed with water and brine. The organic layer was separated, dried over Na2S04 and concentrated to provide ethyl 6-chloro-2-fluoro-3-
((hydroxyimino)methyl)benzoate. 1H NMR (300 MHz, DMSO- 6): δ 11.89 (s, 1H), 8.20 (s, 1H), 7.88-7.83 (t, 7 = 8.1 Hz, 1H), 7.49-7.46 (d, 7 = 8.4 Hz, 1H), 4.44-4.37 (q, 7 = 7.5, 14.1 Hz, 2H), 1.34-1.29 (t, 7 = 7.2 Hz, 3H).
Step-3 : Preperation of ethyl 3-(aminomethyl)-6-chloro-2-fluorobenzoate

A mixture of ethyl 6-chloro-2-fluoro-3-((hydroxyimino)methyl)benzoate (4.21 g, 17.14 mmol), zinc dust (4.48 g, 68.56 mmol), and 10N HC1 (in EtOH) (51.42 mL, 514.2 mmol) in MeOH (200 mL) was heated at reflux for 3 h. Additional zinc dust (2.24 g, 34.25 mmol) was added to the reaction mixture and heated at reflux for 2 h and further stirred at RT for 16 h. The reaction mixture was concentrated and the residue was diluted with EtOAc and filtered through celite bed. The organic layer was washed with a saturated solution of NaHC03 and brine, dried over Na2S04 and concentrated to provide 3.5 g of the desired product. 1H NMR (300 MHz, CDC13): δ 7.54 (t, 7 = 7.8 Hz, 1H), 7.28 (d, 7 = 7.8 Hz, 1H), 4.45 (q, 7 = 7.5, 14.1 Hz, 2H), 4.11 (d, 7 = 3.6 Hz, 2H), 3.37 (br s, 2H), 1.40 (t, 7 = 7.2 Hz, 3H).
Step-4: Preparation of ethyl 6-chloro-2-fluoro-3-(pivalamidomethyl)benzoate

To a solution of ethyl 3-(aminomethyl)-6-chloro-2-fluorobenzoate (1.00 g, 4.32 mmol) in THF (30 mL) was added DIPEA (1.67 g, 12.96 mmol) and pivaloyl chloride (690 mg, 6.48 mmol) at 0°C and the reaction mixture was stirred at RT for 2 h. After completion of the reaction, the reaction mixture was diluted with EtOAc, washed with water and brine, dried over Na2S04, concentrated and purified by flash chromatography using 5-15% EtOAc in hexane to afford 1.0 g of the desired product. 1H NMR (300 MHz, DMSO- 6): δ 8.35 (t, 1H), 7.41 (m, 2H), 4.42-4.35 (q, = 7.2 Hz, 2H), 4.28-4.26 (d, J = 5.7 Hz, 2H), 1.10 (s, 9H); MS [M+H]+: 293.09.
Step-5 : Preparation of N-(4-chloro-2-fluoro-3(hydroxymethyl)benzyl)pivalamide
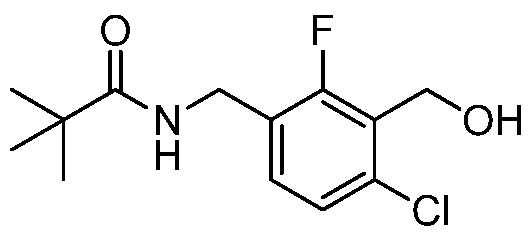
Step-6: Preparation of N-(4-chloro-2-fluoro-3-formylbenzyl)pivalamide
The title compound was prepared following the procedure described in step-5 of Intermediate- 1 using N-(4-chloro-2-fluoro-3-(hydroxymethyl)benzyl)pivalamide (350 mg, 1.35 mmol), pyridinium chlorochromate (581 mg, 2.70 mmol) and CH2C12 (3.0 mL) at RT to afford 250 mg of the desired product. 1H NMR (300 MHz, DMSO d6): δά6): δ 10.32 (s, 1H), 8.39 (m, 1H), 7.73 (s, 1H), 7.60-7.52 (m, 2H), 4.30-4.28 (d, J = 6.3Hz, 2H), 1.12 (s, 9H).
Intermediate-4
Preparation of N-(4-chloro-2-fluoro-3-(2-oxo-6-(4-(trifluoromethyl)phenyl)-l,2,3,6- tetrahydro pyrimidin-4-yl)benzyl)pivalamide

N-(4-chloro-2-fluoro-3-formylbenzyl)pivalamide (Intermediate-3, 0.20 g, 0.73 mmol), 4-trifluoromethyl acetophenone (0.138 g, 0.73 mmol), urea (0.066 g, 1.10 mmol), trimethylsilyl chloride (0.079 g, 0.73 mmol) and sulphamic acid (0.0036 g, 0.0368 mmol) were heated together at 100°C for 6-8 h. After completion of the reaction, the reaction was quenched with ice-cold water and the obtained solid was filtered to afford a crude product. The product was purified by column chromatography on silica gel, 100-200 mesh eluting with 3-6 % methanol in DCM to afford 0.12 g of the title compound. 1HNMR (300MHz, DMSO-i¾): δ 8.82 (s, 1H), 8.1 1 (t, 1H), 7.76 (m, 4H), 7.30 (d, J = 8.1 Hz, 2H), 7.14 (t, 1H), 5.80 (s, 1H), 5.13 (s, 1H), 4.25 (d, J = 5.7 Hz, 2H), 1.1 1 (s, 9H); MS [M+H]+: 484.10.
Intermediate-5
Preparation of N-(4-chloro-2-fluoro-3-(5-methyl-2-oxo-6-(4-(trifluoromethyl) phenyl)-!, 2,3, 6-tetrahydropyrimidin-4-yl)benzyl)pivalamide

The title compound was prepared following the procedure described in Intermediate-4 by using N-(4-chloro-2-fluoro-3-formylbenzyl)pivalamide (Intermediate-3, 0.20 g, 0.73 mmol), 4-trifluoromethyl propiophenone (0.149 g, 0.73 mmol), urea (0.066 g, 1.10 mmol), trimethylsilyl chloride (0.079 g, 0.73 mmol) and sulphamic acid (0.0036 g, 0.0368 mmol) to afford 0.10 g of the desired product. 1HNMR (300MHz, DMSO- d6): δ 8.46 (s, 1H), 8.1 1 (t, 1H), 7.77 (d, J = 7.8 Hz, 2H), 7.51 (d, J = 7.8 Hz, 2H), 7.31 (d, J = 8.4 Hz, 1H), 7.16 (m, 2H), 5.63 (s, 1H), 4.28 (d, J = 5.7 Hz, 2H), 1.30 (s, 3H), 1.13 (s, 9H) ); MS [M+H]+: 498.22.
Intermediate-6
Preparation of N-(4-chloro-2-fluoro-3-formylbenzyl)isobutyramide

Step -1 : Preparation of ethyl -chloro-2-fluoro-3-(isobutyramidomethyl)benzoate

To a solution of ethyl 3-(aminomethyl)-6-chloro-2-fluorobenzoate (step 3, Intermediate 3) (1.00 g, 4.32 mmol) in THF (30 mL) were added DIPEA (1.67 g, 12.96 mmol) and isobutyryl chloride (690 mg, 6.48 mmol). The reaction mass was stirred at RT for 2 h. After completion of the reaction, the reaction mixture was diluted with EtOAc, washed with water and brine, dried over Na2S04 and concentrated to afford 1.0 g of the title product. 1H NMR (300 MHz, DMSO- 6): δ 8.35 (t, 1H), 7.41 (m, 2H), 4.42-4.35 (q, = 7.2 Hz, 2H), 4.28-4.26 (d, = 5.7 Hz, 2H), 2.40 (m, 1H), 1.00 (d, = 6.9 Hz, 6H).
Step-2: Preparation of N-(chloro-2-fluoro-3- hydroxymethyl)benzyl) isobutyramide

The title compound was prepared following the procedure of step-4 of Intermediate- 1 by using ethyl 6-chloro-2-fluoro-3-(isobutyramidomethyl)benzoate (500 mg, 1.66 mmol) and UAIH4 (126 mg, 3.32 mmol) in THF (10.0 mL) to afford 250 mg of the desired product. 1H NMR (300 MHz, DMSO d6): δ 7.26-7.23 (m, 1H), 7.18-7.15 (d, = 7.8 Hz, 1H), 5.85 (br s, 1H), 4.84 (s, 1H), 4.45-4.43 (d, = 5.7 Hz, 2H), 3.48 (s, 2H), 2.39-2.35 (m, 1H), 1.17-1.15 (d, = 6.6 Hz, 6H); MS [M+H]+: 260.
Step-3 : Preparation of N-(4-chloro-2-fluoro-3-formylbenzyl)isobutyramide
The title compound was prepared following the procedure of step-5 of Intermediate- 1 by using N-(4-chloro-2-fluoro-3-(hydroxymethyl)benzyl) isobutyramide (350 mg, 1.35 mmol), pyridinium chlorochromate (581 mg, 2.70 mmol) and CH2C12 (3.0 mL) to afford 250 mg of the desired product. 1H NMR (300 MHz, DMSO d6): δ 10.31 (s, 1H), 8.34 (br s, 1H), 7.53-7.50 (d, = 7.8 Hz, 1H), 7.46-7.43 (d, = 9.0 Hz, 1H), 4.29-4.27 (d, = 5.4 Hz, 2H), 2.44-2.39 (m, 1H), 1.02-1.00 (d, = 6.9 Hz, 6H).
Intermediate-7
Preparation of N-(4-chloro-2-fluoro-3-(5-methyl-2-oxo-6-(4-(trifluoromethyl) phenyl)-!, 2,3, 6 amide

The title compound was prepared following the procedure described in Intermediate-4 by using N-(4-chloro-2-fluoro-3-formylbenzyl)isobutyramide (Intermediate-6, 0.200 g, 0.73 mmol), 4-trifluoromethyl propiophenone (0.153 g, 0.73 mmol), urea (0.069 g, 1.10 mmol), trimethylsilyl chloride (0.084 g, 0.73 mmol) and sulphamic acid (0.0037 g, 0.0368 mmol) to afford 0.100 g of desired product. 1H NMR (300 MHz, DMSO d6): δ 8.44 (s, 1H), 8.31 (m, 1H), 7.75-7.72 (d, = 7.8 Hz, 2H), 7.49-7.47 (d, = 7.8 Hz, 2H), 7.30-7.28 (d, = 8.1 Hz, 1H), 7.22-7.19 (d, = 7.8 Hz, 1H), 7.13 (s, 1H), 5.62 (s, 1H), 4.26 (m, 2H), 2.39 (m, 1H), 1.28 (s, 3H); MS [M+H]+: 484.
Intermediate-8
Preparation of l-(4-cyclopropylphenyl)ethanone

To a solution of l-(4-bromophenyl)ethanone (3.0g, 0.0150 mmol) in mixture of DMSO: water (3 : 1, 30 mL) was added tripotassium phosphate (9.5 g, 0.045 mmol), l, l'-bis(diphenylphosphino)ferrocene-palladium(II)di chloride dichloromethane (0.200 g,0.245 mmol) and cyclopropylboronic acid (1.9 g, 0.022 mmol). The reaction mixture was heated at 100 °C for 48 h. The reaction mass was quenched with water and extracted with EtOAc. The organic layer were washed with water and brine, dried over Na2S04 and concentrated. The obtained solid was purified by column chromatography on silica gel to afford 1.0 g of the title product. 1H NMR (300 MHz, DMSO d6): δ 7.86-7.83 (d, = 7.8 Hz, 2H), 7.13.7.10 (d, = 7.8 Hz, 2H), 2.57 (s, 3H), 1.94 (m, 1H), 1.07-1.05 (q, J = 7.2 Hz, 2H),0.79-0.77 (d, J = 4.8 Hz, 2H).
Intermediate-9
Preparation of N-(4-chloro-3-(6-(4-cyclopropylphenyl)-2-oxo-l,2,3,6-tetrahydro pyrimidin-4-yl)benz l)isobutyramide

The title compound was prepared following the procedure described in Intermediate-4 by using N-(4-chloro-3-formylbenzyl)isobutyramide (Intermediate-2, 0.200 g, 0.835 mmol), l-(4-cyclopropylphenyl)ethanone (Intermediate-8, 0.133 g, 0.835 mmol), urea (0.082 g, 1.25 mmol), trimethylsilyl chloride (0.108 g, 0.835 mmol) and sulphamic acid (0.004 g, 0.041 mmol) to afford 0.090 g of desired product. 1HNMR (300MHz, DMSO- e): δ 8.57 (s, 1H), 8.32 (s, 1H), 7.39-7.34 (m, 4H), 7.18-7.13 (m, 2H), 7.04-7.01 (d, = 7.5 Hz, 2H), 5.47 (s, 1H), 5.01 (s, 1H), 4.23-4.21 (d, = 8.7 Hz, 2H), 2.39-2.37 (m, 1H), 1.89 (s, 1H), 1.06-0.89 (m, 8H), 0.64 (m, 2H); MS [M+H]+: 424.
Intermediate- 10
Preparation of 4-cyclopropylbenzaldehyde
To a solution of 4-bromobenzaldehyde (3.4 g, 0.0180 mmol) in mixture of toluene: water (40 mL: 3.0 mL) was added tripotassium phosphate (9.5 g, 0.045 mmol), triphenylphosphine (0.719 g, 0.002 mmol) and cyclopropylboronic acid (2.3 g, 0.027 mmol). The reaction mixture was heated at 100°C for 18 h. The reaction mass was quenched with water and extracted with EtOAc. The organic layer were washed with water and brine, dried over Na2S04 and concentrated. The obtained solid was purified by column chromatography to afford 1.70 g of the title product. 1H MR (300 MHz, DMSO d6): δ 9.94 (s, 1H), 7.77-7.75 (d, = 8.4 Hz, 2H), 7.20-7.17 (d, = 7.8 Hz, 2H), 1.97 (m, 1H), 1.1 1-1.08 (m, 2H), 0.82-0.80 (m, 2H).
Intermediate- 1 1
Preparation of N-(3-acetyl-4-chlorobenzyl)pivalamide

Step-1 : Preparation of N-(3-acetyl-4-chlorobenzyl)-2,2,2-trifluoroacetamide

A solution of l-(2-chlorophenyl)ethanone (5.0 g, 32.36 mmol) and 2,2,2-trifluoro-N- (hydroxymethyl)acetamide (4.6 g, 32.6 mmol) in conc.H2S04 (50 mL) was stirred at RT for 48 h. The reaction mixture was quenched with water and extracted with EtOAc. The organic layer were washed with water and brine, dried over Na2S04 and concentrated. The obtained solid was purified by column chromatography to afford 1.40 g of the desired product.1H MR (300 MHz, DMSO d6): δ 10.04 (br s, 1H), 7.60 (s, 1H), 7.55-7.52 (d, J = 8.1 Hz, 1H), 7.43-7.40 (d, 7 = 8.4 Hz, 1H), 4.43-4.41 (d, J = 5.7 Hz, 2H), 2.58 (s, 3H).
Step-2 Preparation of l-(5-(aminomethyl)-2-chlorophenyl)ethanone

A solution of N-(3-acetyl-4-chlorobenzyl)-2,2,2-trifluoroacetamide (1.4 g, 5.01 mmol) in mixture of THF: water (5 : 1, 10 mL) was added lithium hydroxide (0.420 g, 10.0 mmol). The reaction mixture was stirred at RT for 3-4 h. The reaction mixture was quenched with water and extracted with EtOAc. The organic layer were washed with water and brine, dried over Na2S04 and concentrated to afford 0.800 g of the desired product. 1H NMR (300 MHz, DMSO d6): δ 7.44-7.22 (m, 3H), 4.62 (br s, 1H), 4.15 (br s, 2H), 3.76 (br s, 1H), 2.23 (br s, 3H).
Step-3 Preparation of N-(3-acetyl-4-chlorobenzyl)pivalamide
The title compound was prepared following the procedure described in step-3 of Intermediate- 1 using l-(5-(aminomethyl)-2-chlorophenyl)ethanone (0.800 g, 4.37
mmol), TEA (1.5 mL), pivaloyl chloride (2.0 mL) and dry THF (10 mL) to afford 0.800 g of the desired product. 1H NMR (300 MHz, DMSO d6): δ 8.14 (br s, 1H), 7.48-7.45 (m, 2H), 7.33-7.30 (d, = 8.1 Hz, 1H), 4.25-4.23 (d, = 6.0 Hz, 2H), 2.54 (s, 3H), 1.10 (s, 9H).
Intermediate- 12
Preparation of l-(6-(trifluorometh l)pyridin-3-yl)ethanone

To a solution of 6-(trifluoromethyl)nicotinonitrile (2.0 g, 0.01 1 mmol) in diethyl ether (30 mL) was added methyl magnesium bromide solution (3M in THF) (5.6 ml) at 0 °C. The reaction mixture was stirred at RT for 3 h. Concentrated HCl was added to the reaction mixture and was heated at 60 °C for 2 h. The reaction mixture was concentrated and neutrallised with sat NaHC03 and extracted with EtOAc. The organic layers were concentrated to afford 0.500 g of the title product.
Intermediate- 13
Preparation of N-(4-chloro-2-fluoro-3-(2-oxo-6-(6-(trifluoromethyl)pyridin-3-yl)- 1,2,3, 6-tetrahydropyrimidin-4- l)benzyl)pivalamide

The title compound was prepared following the procedure described in Intermediate-4 by using N-(4-chloro-2-fluoro-3-formylbenzyl)pivalamide (Intermediate-3, 0.200 g, 0.730 mmol), l-(6-(trifluoromethyl)pyridin-3-yl)ethanone (Intermediate- 12, 0.139 g, 0.730 mmol), urea (0.072 g, 1.5 mmol), trimethylsilyl chloride (0.078 g, 0.730 mmol) and sulphamic acid (0.0035 g, 0.036 mmol) to afford 0.092 g of desired product. 1HNMR (300MHz, DMSO- 6): δ 9.15 (m, 1H), 8.97-8.92 (m, 1H), 8.13 (m, 2H), 7.91-7.84 (m, 2H), 7.33-7.15 (m, 2H), 5.83-5.78 (m, 1H), 5.33 (br s, 1H), 4.26 (br s, 2H), 1.1 1 (s, 9H); MS [M+H]+:485.
Intermediate- 14
Preparation of N-(4-chloro-3-(2-oxo-6-(4-(trifluoromethyl)phenyl)-l,2,3,6- tetrahydropyrimidin -4-yl)benzyl)propane-2-sulfonamide

The title compound was prepared following the procedure described in step-3 of Intermediate- 1 by using l-(5-(aminomethyl)-2-chlorophenyl)ethanone (step-2, Intermediate- 1 1, 0.600 g, 3.26 mmol), TEA (2.0 mL), propane-2-sulfonyl chloride (2.0 mL) and dry THF (10 mL) to afford a crude product. The obtained product was purified by column chromatography to afford 0.400 g of the desired compound. 1H MR (300 MHz, CDC13): δ 7.51 (s, 1H), 7.42 (s, 2H), 4.60 (br s, 1H), 4.33-4.31 (d, J = 6.0 Hz, 2H), 3.15-3.1 1 (m, 1H), 2.65 (s, 3H), 1.39-1.37 (d, J = 6.9 Hz, 6H).
Step-2: Preparation of N-(4-chloro-3-(2-oxo-6-(4-(trifluoromethyl)phenyl)-l,2,3,6- tetrahydropyrimidin-4-yl)benzyl)propane-2-sulfonamide
The title compound was prepared following the procedure described in Intermediate-4 by using N-(3-acetyl-4-chlorobenzyl)propane-2-sulfonamide (0.400 g, 1.38 mmol), 4- (trifluoromethyl)benzaldehyde (0.240 mg, 1.38 mmol), urea (0.136 g, 2.07 mmol), trimethylsilyl chloride (0.149 g, 1.38 mmol) and sulphamic acid (0.007 g, 0.069 mmol) to afford 0.200 g of the desired product. 1HNMR (300MHz, DMSO- 6): δ 8.36 (d, 1H), 7.93 (d, 2H), 7.62-7.54 (m, 6H), 6.02 (s, 1H), 4.24 (br s, 2H), 3.16 (m, 1H), 1.23-1.22 (d, 7 = 6.9 Hz, 6H).
Intermediate- 15
Preparation of N-(3-(6-(4-(ieri-butyl)phenyl)-2-oxo-l,2,3,6-tetrahydropyrimidin-4- yl)-4-chloro-2-fluorobenzyl)pival amide

The title compound was prepared following the procedure described in Intermediate-4 by using N-(4-chloro-2-fluoro-3-formylbenzyl)pivalamide (Intermediate-3, 0.200 g,
0.73 mmol), l-(4-(ie/ -butyl)phenyl)ethanone (0.130 g, 0.730 mmol), urea (0.066 g, 1.10 mmol), trimethylsilyl chloride (0.079 g, 0.73 mmol) and sulphamic acid (0.0035 g, 0.036 mmol) to afford 0.070 g of the desired product. 1HNMR (300MHz, DMSO- d6): δ 8.58-8.52 (d, 1H), 8.1 1 (br s, 1H), 7.43-7.18 (m, 7H), 5.77 (br s, 1H), 4.89 (br s, 1H), 4.25 (m, 2H), 1.26 (s, 9H), 1.1 1 (s, 9H); MS [M+H]+:472.
Intermediate- 16
Preparation of N-(4-chloro-2-fluoro-3-(6-(4-isopropylphenyl)-2-oxo-l,2,3,6- tetrahydropyrimidin-4-yl)benzyl)pivalamide

The title compound was prepared following the procedure described in Intermediate-4 by using N-(4-chloro-2-fluoro-3-formylbenzyl)pivalamide (Intermediate-3, 0.200 g, 0.73 mmol), l-(4-isopropylphenyl)ethanone (0.1 19 g, 0.730 mmol), urea (0.066 g, 1.10 mmol), trimethylsilyl chloride (0.079 g, 0.73 mmol) and sulphamic acid (0.0035 g, 0.036 mmol) to afford 0.080 g of the desired product. MS [M+H]+: 458.
Intermediate- 17
Preparation of 6-(5-(aminomethyl)-2-methylpyridin-3-yl)-4-(4-(trifluoromethyl) phenyl)pyrimidin-2(lH -one hy rochloride

Step-1 : Preparation of 3-(ethoxymethylene)pentane-2,4-dione

A mixture of pentane-2,4-dione (15 g, 150 mmol) and triethyl orthoformate (44.8 g, 302 mmol) in acetic anhydride (100 mL) was heated at 70-80 °C for 16 h. The reaction mass was concentrated to afford 20 g of title product. 1HNMR (300MHz,
DMSO- e): δ 7.82 (s, 1H), 4.32-4.25 (d, J = 7.2 Hz, 2H), 2.24 (s, 3H), 2.19 (s, 3H), 1.30-1.26 (t, / = 6.9 Hz, 3H).
Step-2: Preparation of 5-acetyl-6-methylnicotinonitrile

A solution of 3-(ethoxymethylene)pentane-2,4-dione (20 g, 108 mmol) and (E)-4- (dimethylamino)but-2-enenitrile (10.43 g, 108 mmol) in DMF (100 mL) was heated at 100 °C for 24 h. Ammonium acetate (12.47 g, 161 mmol) was added to it and the reaction was further stirred at the same tempreture for 6 h. The reaction was quenched with ice-cold water and extracted with DCM. The organic layers were dried over Na2S04 and concentrated to afford 4.5 g of the desired product. 1HNMR (300MHz, CDC13): δ 8.84 (s, 1H), 8.19 (s, 1H), 2.80 (s, 3H), 2.62 (s, 3H).
Step-3 : Preparation of 6-methyl-5-(2-oxo-6-(4-(trifluoromethyl)phenyl)-l,2,3,6- tetrahydropyrimidin-4-yl)nicotinonitrile

The title compound was prepared following the procedure described in Intermediate-4 by using 5-acetyl-6-methylnicotinonitrile (2.0 g, 12.5 mmol), 4- (trifluoromethyl)benzaldehyde (2.1 g, 12.5 mmol), urea (1.2 g, 13.50 mmol), trimethylsilyl chloride (1.3 mL, 12.5 mmol) and sulphamic acid (0.060 g, 0.61 mmol) to afford 1.00 g of the desired product. 1HNMR (300MHz, CDC13): δ 8.73 (s, 1H), 8.04 (d, 1H), 7.87 (s, 1H), 7.77-7.74 (d, = 7.5 Hz, 1H), 7.63-7.60 (d, = 6.9 Hz, 2H), 7.47-7.44 (d, J = 7.5 Hz, 2H), 5.32 (s, 1H), 4.77 (s, 1H), 2.65 (s, 3H).
Step-4: Preparation of 6-methyl-5-(2-oxo-6-(4-(trifluoromethyl)phenyl)-2,3- dihydropyrimidin-4-yl)nicotinonitrile

Step-5 : Preparation of ie/t-butyl ((6-methyl-5-(2-oxo-6-(4-(trifluoromethyl)phenyl)- 2,3-dihydropyrimidin-4-yl)pyridin-3-yl)methyl)carbamate

To a solution of 6-methyl-5-(2-oxo-6-(4-(trifluoromethyl)phenyl)-2,3- dihydropyrimidin-4-yl)nicotinonitrile (0.500 g, 1.40 mmol) in EtOH (10 mL) was added TEA (2.0 mL), di-feri-butyl dicarbonate (2.0 mL) and 10% Pd/C (0.050 g). The reaction mixture was hydrogenated in Parr appatarus under 40 psi pressure for 16 h. The reaction mass was filtered and the filtrate was concentrated to afford 0.300 g of the desired product.
Step-6: Preparation of 6-(5-(aminomethyl)-2-methylpyridin-3-yl)-4-(4- (trifluoromethyl)phenyl)pyrimidin-2(lH)-one hydrochloride
A solution of ie/t-butyl ((6-methyl-5-(2-oxo-6-(4-(trifluoromethyl)phenyl)-2,3- dihydropyrimidin-4-yl)pyridin-3-yl)methyl)carbamate (0.300 g) in EtOH: HC1 (10 mL) was stirred at RT for 16 h. The reaction mass was filtered and concentrated to afford 0.100 g of the desired product. 1HNMR (300MHz, DMSO d6): δ 8.95 (s, 1H), 8.81 (br s, 3H), 8.42-8.40 (d, = 7.8 Hz, 2H), 7.96-7.93 (d, = 7.8 Hz, 2H), 7.82 (s, 1H), 6.40 (br s, 2H), 4.24 (s, 2H), 2.86 (s, 3H); MS [M+H]+: 361.
Intermediate- 18
Preparation of 6-(5-(aminomethyl)-2-(difluoromethyl)pyridin-3-yl)-4-(4-
(trifluoromethyl)phenyl)pyrimidin-2(lH)-one hydrochloride

Step-1 : Preparation of 6-methyl-5-(2-meth l-l,3-dioxolan-2-yl)nicotinonitrile

To a solution of 5-acetyl-6-methylnicotinonitrile (10.0 g, 62.0 mmol) in dry toluene (100 mL) was added /?-toluenesulphonic acid (12.9 g, 68.0 mmol) and ethylene glycol (7.7 g, 124 mmol). The reaction mixture was heated to reflux for 16 h. The reaction mass was concentrated, diluted with water and extracted with EtOAc. The organic layer was washed with saturated NaHC03 solution and concentrated to afford 9.0 g of the desired compound. 1HNMR (300MHz, DMSO d6): δ 8.85 (s, 1H), 8.14 (s, 1H), 4.02 (s, 2H), 3.69 (s, 2H), 2.71 (s, 3H), 1.64 (s, 3H); MS [M+H]+: 219.
Step-2: Preparation of 6-formyl-5-(2-meth l-l,3-dioxolan-2-yl)nicotinonitrile

To a solution of 6-methyl-5-(2-methyl-l,3-dioxolan-2-yl)nicotinonitrile (9.0 g, 98.0 mmol) in 1,4-dioxane (100 mL) was added selenium dioxide (21.5 g, 196 mmol). The reaction mixture was heated to reflux for 16 h. The reaction mass was concentrated, diluted with water and extracted with EtOAc. The organic layer was washed with water and concentrated to afford 7.0 g of the desired compound. 1HNMR (300MHz, CDC13): δ 10.55 (s, 1H), 8.97 (s, 1H), 8.34 (s, 1H), 4.15-4.13 (br s, 2H), 3.76-3.74 (br s, 2H), 1.85 (s, 3H).
Step 3 : Preparation of 6-(difluoromethyl)-5-(2-methyl-l,3-dioxolan-2-yl) nicotinonitrile

Step 4: Preparation of 5-acetyl-6-(difluorometh l)nicotinonitrile

A solution of 6-(difluoromethyl)-5-(2-methyl-l,3-dioxolan-2-yl)nicotinonitrile (4.0 g, 16.52 mmol) in methanolic HC1 (40 mL) was heated at 50-60 °C for 16 h. The reaction mass was concentrated, diluted with water and extracted with EtOAc. The organic layer was washed with saturated NaHC03 solution and concentrated to afford 2.5 g of desired compound. 1HNMR (300MHz, CDC13): δ 9.04 (s, 1H), 8.27 (s, 1H), 7.09-6.91 (m, 1H), 2.69 (s, 3H).
Step-5 : Preparation of 6-(difluoromethyl)-5-(2-oxo-6-(4-(trifluoromethyl)phenyl)- l,2,3,6-tetrahydropyrimidin-4-yl)nicotinonitrile

The title compound was prepared following the procedure described in Intermediate-4 by using 5-acetyl-6-(difluoromethyl)nicotinonitrile (2.5 g, 12.0 mmol), 4- (trifluoromethyl)benzaldehyde (2.20 g, 12.0 mmol), urea (1.0 g, 16.00 mmol), trimethylsilyl chloride (1.2 g, 11.0 mmol) and sulphamic acid (0.058 g, 0.59 mmol) to afford 1.00 g of desired product. 1HNMR (300MHz, DMSO- 6): δ 9.15 (m, 1H), 8.82 (s, 1H), 8.55 (s, 1H), 7.78-7.75 (d, J = 8.1 Hz, 2H), 7.64-7.61 (d, 7 = 8.4 Hz, 2H), 7.48 (s, 1H), 7.25-76.90 (m, 1H), 5.28 (s, 1H), 4.89 (s, 1H).
Step-6: Preparation of 6-(difluoromethyl)-5-(2-oxo-6-(4-(trifluoromethyl)phenyl)-2,3- dihydropyrimidin-4-yl)nicotinonitrile

The title compound was prepared following the procedure described in step-4 of Intermediate- 17 using 6-(difluoromethyl)-5-(2-oxo-6-(4-(trifluoromethyl)phenyl)- 1,2,3, 6-tetrahydropyrimidin-4-yl)nicotinonitrile (4.0 g, 1.01 mmol), 2,3-dichloro-5,6- dicyano-l,4-benzoquinone (DDQ) (0.230 g, 1.01 mmol) and dry toluene (5.0 mL) to afford 0.250 g of desired product. 1HNMR (300MHz, DMSO- 6): δ 12.49 (br s, 1H), 9.32 (s, 1H), 8.95 (s, 1H), 8.37 (m, 2H), 7.98-7.95 (d, = 7.8 Hz, 2H), 7.70-7.35 (m, 2H); MS [M+H]+: 393.
Step 7: Preparation of ie/ -butyl ((6-(difluoromethyl)-5-(2-oxo-6-(4- (trifluoromethyl)phenyl)-2,3-dihydropyrimidin-4-yl)pyridin-3-yl)methyl)carbamate

The title compound was prepared following the procedure described in step-5 of Intermediate- 17 using 6-(difluoromethyl)-5-(2-oxo-6-(4-(trifluoromethyl)phenyl)-2,3- dihydropyrimidin-4-yl)nicotinonitrile (0.300 g, 0.76 mmol), TEA (2.0 mL), di-tert- butyl dicarbonate (2.0 mL) and 10 Pd/C (50 mg) in EtOH (10.0 mL) to afford 0.200 g of the desired product. 1HNMR (300MHz, CDC13): δ 8.75 (s, 1H), 8.61 (s, 1H), 8.19- 8.17 (m, 2H), 8.0 (m, 1H), 7.81 (br s, 1H), 7.74-7.71 (d, = 8.7 Hz, 1H), 7.63 (br s, 1H), 7.55 (m, 1H), 4.40 (m, 2H), 1.25 (s, 9H); MS [M+H]+: 497.
Step 8: Preparation of 6-(5-(aminomethyl)-2-(difluoromethyl)pyridin-3-yl)-4-(4- (trifluoromethyl)phenyl)pyrimidin-2(lH)-one hydrochloride
The title compound was prepared following the procedure described in step-6 of Intermediate- 17 using ie/t-butyl ((6-(difluoromethyl)-5-(2-oxo-6-(4- (trifluoromethyl)phenyl)-2,3-dihydropyrimidin-4-yl)pyridin-3-yl)methyl)carbamate (0.300 g), MeOH:HCl (10 mL) to afford 0.100 g of the desired compound.
Intermediate- 19
Preparation of l-(2-amino-2-oxoethyl)pyridin-l-ium chloride

A solution of pyridine (8.0 g) and 2-chloroacetamide (10.0 g) in acetonitrile (20 mL) was heated at reflux for 10 h. The reaction mass was cooled to 22 °C and filtered. The obtained solid was washed with hexane and recrystallized from EtOH. 1HNMR (300MHz, DMSO- e): δ 9.07-9.06 (d, 7 = 5.4 Hz, 2H), 8.67-8.62 (t, 7 = 7.2 Hz, 1H), 8.38 (br s, 1H), 8.20-8.15 (t, 7 = 7.2 Hz, 2H), 7.68 (s, 1H), 5.53 (s, 2H).
Intermediate-20
Preparation of (E)-N-(4-chloro-3-(3-(4-(trifluoromethyl)phenyl)acryloyl)benzyl) pi val amide

To a solution of N-(3-acetyl-4-chlorobenzyl)pivalamide
(Intermediate-1 1, 0.200 g, 0.74 mmol) in MeOH (10 mL) was added 4- (trifluoromethyl)benzaldehyde (0.130g, 0.74 mmol) and sodium hydroxide (0.059 g, 1.45 mmol) at 0°C. The reaction mass was stirred at same tempreture for 2 h. The reaction mass was concentrated at low tempreture and the obtained solid was filtered to afford 0.150 g of the title compound. 1HNMR (300MHz, DMSO- 6): δ 8.15 (t, 1H), 8.01-7.98 (d, 7 = 8.4 Hz, 2H), 7.81-7.78 (d, 7 = 8.4 Hz, 2H), 7.56-7.54 (d, 7 = 4.5 Hz, 1H), 7.49 (s, 1H), 7.44-7.38 (m, 3H), 4.30-4.28 (d, 7 = 6.0 Hz, 2H), 1.18 (s, 9H); MS [M-H]": 421.
Intermediate-21
Preparation of (E)-N-(4-chloro-3-(3-(4-(trifluoromethyl)phenyl)acryloyl)benzyl) pi val amide

Intermediate-22
Preparation of (E)-N-(4-chloro-3-(3-(3,4-dichlorophenyl)-3-oxoprop-l-en-l-yl)-2- fluorob enzy l)pi val ami de

The title compound was prepared following the procedure described in Intermediate- 20 using N-(4-chloro-2-fluoro-3-formylbenzyl)pivalamide (Intermediate-3, 0.500 g, 1.84 mmol), l-(3,4-dichlorophenyl)ethanone (0.348 g, 1.84 mmol), sodium hydroxide (0.073 g, 1.84 mmol) and MeOH (15 mL) to afford 0.500 g of the desired compound. 1HNMR (300MHz, DMSO- 6): δ 8.14 (br s, 1H), 7.91-7.78 (m, 2H), 7.60 (m, 1H), 7.46-6.75 (m, 4H), 4.25 (m, 2H), 1.09 (s, 9H).
Intermediate-23
Preparation of ((E)-N-(4-chloro-3-(3-(4-chloro-3-methylphenyl)-3-oxoprop-l-en-l- yl)-2-fluorobenzyl)pival ami

The title compound was prepared following the procedure described in Intermediate- 20 using N-(4-chloro-2-fluoro-3-formylbenzyl)pivalamide (Intermediate-3, 0.500 g,
1.84 mmol), l-(4-chloro-3-methylphenyl)ethanone (0.309 g, 1.84 mmol), sodium hydroxide (0.073 g, 1.84 mmol) and MeOH (15 mL) to afford 0.500 g of the desired compound. 1HNMR (300MHz, DMSO- 6): δ 8.14 (t, 1H), 8.03 (s, 1H), 7.92-7.86 (m, 2H), 7.81-7.79 (d, = 5.4 Hz, 1H), 7.62-7.65 (m, 1H), 7.45-7.42 (d, = 8.7 Hz, 1H), 7.31-7.28 (m, 1H), 4.32-4.30 (d, = 6.0 Hz, 2H), 2.41 (s, 3H), 1.1 1 (s, 9H); MS [M+H]+: 422.
Intermediate-24
Preparation of (E)-N-(4-chloro-3-(3-(4-chloro-3-(trifluoromethyl)phenyl)-3-oxoprop- 1 -en- 1 -yl)-2-fluorobenzyl)pival amide

The title compound was prepared following the procedure described in Intermediate- 20 using N-(4-chloro-2-fluoro-3-formylbenzyl)pivalamide (Intermediate-3, 0.500 g, 1.84 mmol), l-(4-chloro-3-(trifluoromethyl)phenyl)ethanone (0.408 g, 1.84 mmol), sodium hydroxide (0.073 g, 1.84 mmol) and MeOH (15 mL) to afford 0.500 g of the desired compound. 1H MR (300MHz, DMSO- 6): δ 8.26-8.21 (m, 1H), 8.13 (br s, 1H), 7.99-7.89 (m, 2H), 7.67-7.03 (m, 4H), 4.25 (br s, 2H), 1.06 (s, 9H).
Intermediate-25
Preparation of (E)-N-(4-chloro-3 -(3 -(3 ,4-dimethylphenyl)-3 -oxoprop- 1 -en- 1 -yl)-2- fluorob enzy l)pi val ami de

The title compound was prepared following the procedure described in Intermediate- 20 using N-(4-chloro-2-fluoro-3-formylbenzyl)pivalamide (Intermediate-3, 0.500 g, 1.84 mmol), l-(3,4-dimethylphenyl)ethanone (0.272 g, 1.84 mmol), sodium hydroxide (0.073 g, 1.84 mmol) and MeOH (15 mL) to afford 0.500 g of the desired
compound. 1HNMR (300MHz, DMSO- 6): δ 8.14 (t, 2H), 7.83-7.66 (m, 3H), 7.46- 7.26 (m, 3H), 4.33-4.32 (d, J = 4.8 Hz, 2H), 2.32(s, 3H), 2.28 (s, 3H), 1.13 (s, 9H); MS [M+H]+: 402.
Examples
Example- 1
N-(4-chloro-3-(2-oxo-6-(4-(trifluoromethyl)-2,3-dihydropyrimidin-4-yl)benzyl) pi val amide

Example-2
N-(4-chloro-(2-oxo-6-(4-(trifluoromethyl)phenyl)-2,3-dihydropyrimidin-4-yl)- benzyl)-isobutyramide

The title compound was prepared following the procedure described in Example- 1 by using N-(4-chloro-3-formylbenzyl)isobutyramide (Intermediate-2, 0.100 g, 0.39 mmol), 4-trifluoromethyl acetophenone (0.073g, 0.39 mmol), urea (0.034g, 0.58 mmol) and iodine (0.009g, 0.039 mmol) at 100 °C for 8 h to afford 0.20g of the desired product. 1HNMR (300MHz, DMSO- 6): 58.34 (d, J = 8.0 Hz, 2H), 8.30 (t, 1H), 7.90 (d, J = 7.8 Hz, 2H), 7.59 (s, J = 8.4 Hz, 1H), 7.51 (s, 1H), 7.37 (d, J = 8.4
Hz, 2H), 4.31 (d, = 5.7 Hz, 2H), 2.4 (m, 1H), 1.03 (d, = 6.9 Hz, 6H); MS [M+H]+: 450.18.
Example-3
N-(4-chloro-2-fluoro-3-(2-oxo-6-(4-(trifluoromethyl)phenyl)-2,3-dihydro pyrimidin- 4-yl)benzyl)-pivalamid

2,3-Dichloro-5,6-dicyano-l,4-benzoquinone (DDQ) (0.105 g, 0.464 mmol) was added to N-(4-chloro-2-fluoro-3-(2-oxo-6-(4-(trifluoromethyl)phenyl)-l,2,3,6-tetrahydro pyrimidin-4-yl)- benzyl)pival amide (Intermediate-4, 0.150 g, 0.309 mmol) in dry- toluene at room temperature and stirred for 5 h at same temperature. After completion of the reaction, the reaction was quenched with ice-cold water. The reaction mass was extracted with ethyl acetate. The organic layer was washed with 1% NaOH solution, water and brine, dried over Na2S04 and concentrated. The crude product was purified by column chromatography on silica, 100-200 mesh, eluting with 3-4 % methanol in DCM to afford 0.065 g of the title compound. 1HNMR (300MHz, DMSO- 6): δ 8.35 (d, J = 7.2 Hz, 2H), 8.19 (t, 1H), 7.92 (d, J = 7.2 Hz, 2H), 7.48 (m, 2H), 7.38 (t, 1H), 4.30 (d, J = 5.7 Hz, 2H), 1.14 (s, 9H); MS [M+H]+: 482.33.
Example-4
N-(4-chloro-2-fluoro-3-(5-methyl-2-oxo-6-(4-(trifluoromethyl)phenyl)-2,3- dihydropyrimidin-4-yl)benzyl)pival amide

The title compound was prepared following the procedure described in Example-3 by using N-(4-chloro-2-fluoro-3-(5-methyl-2-oxo-6-(4-(trifluoromethyl)phenyl)- 1,2,3,6- tetrahydropyrimidin -4-yl)benzyl)pivalamide (Intermediate-5, 0.10 g, 0.20 mmol), 2,3-dichloro-5-6,dicyano-l,4-benzoquinone (DDQ) (0.068 g, 0.30 mmol) and dry toluene (5 mL) to afford 0.045 g of desired product. 1HNMR (300MHz, DMSO-i¾): δ
8.16 (t, 1H), 7.85 (m, 4H), 7.51 (d, 1H), 7.40 (t, 1H), 4.33 (d, J = 5.7 Hz, 2H), 1.81 (s, 3H), 1.13 (s, 9H); MS [M+H]+: 496.28.
Example-5
N-(4-chloro-2-fluoro-3-(5-methyl-2-oxo-6-(4-(trifluoromethyl)phenyl)-2,3- dihydropyrimidin-4-yl)benzyl)isobutyramide

The title compound was prepared following the procedure described in Example-3 by using N-(4-chloro-2-fluoro-3-(5-methyl-2-oxo-6-(4-(trifluoromethyl)phenyl)-l,2,3,6- tetrahydropyrimidin-4-yl)benzyl)isobutyramide (Intermediate-7, 0.080 g, 0.160 mmol), 2,3-dichloro-5-6,dicyano-l,4-benzoquinone (DDQ) (0.080 g, 0.30 mmol) and dry toluene (5 mL) to obtain a crude product. The product was purified by column chromatography on silica, 100-200 mesh, eluting with 4 % methanol in DCM to afford 0.035 g of the desired product. 1H MR (300 MHz, DMSO d6): δ 12.32 (s, 1H), 8.36 (s, 1H), 7.91-7.85 (m, 4H), 7.51-7.43 (m, 2H), 4.33-4.31 (d, = 5.1 Hz, 2H), 2.41 (m, 1H), 1.81 (s, 3H), 1.04-1.02 (s, 6H); MS [M+H]+: 482.
Example-6
N-(4-chloro-3-(6-(4-cyclopropylphenyl)-2-oxo-2,3-dihydropyrimidin-4-yl)benzyl) isobutyramide

The title compound was prepared following the procedure described in Example-3 by using N-(4-chloro-3-(6-(4-cyclopropylphenyl)-2-oxo-l,2,3,6-tetrahydropyrimidin-4- yl)benzyl)isobutyramide (Intermediate-9, 0.080 g, 0.188 mmol), 2,3-dichloro-5- 6,dicyano-l,4-benzoquinone (DDQ) (0.064 g, 0.283 mmol) and dry toluene (10 mL) to afford 0.040 g of the desired product. 1HNMR (300MHz, DMSO- ): δ 12.15 (br s, 1H), 8.36 (t, 1H), 7.99 (br s, 2H), 7.54-7.56 (d, J = 8.4 Hz, 1H), 7.84 (s, 1H), 7.39- 7.37 (d, J = 8.1 Hz, 1H), 7.23-7.20 (d, J = 7.8 Hz, 3H), 4.30-4.28 (d, J = 6.0 Hz, 2H),
2.45-2.40 (m, 1H), 2.00 (m, 1H), 1.03-1.01 (d, = 6.9 Hz, 8H), 0.78 (d, 2H). MS [M+H]+: 422.
Example-7
N-(4-chloro-3-(6-(4-cyclopropylphenyl)-2-oxo-2,3-dihydropyrimidin-4- y l)b enzy l)pi val ami dei de

The title compound was prepared following the procedure described in Intermediate-4 by using N-(3-acetyl-4-chlorobenzyl)pivalamide (Intermediate-1 1, 0.200 g, 0.747 mmol), 4-cyclopropylbenzaldehyde (Intermediate- 10, 0.109 g, 0.747 mmol), urea (0.074 g, 1.12 mmol), trimethylsilyl chloride (0.080 g, 0.747 mmol) and sulphamic acid (0.0036 g, 0.037 mmol) to afford 0.080 g of the desired product. 1HNMR (300MHz, DMSO- e): δ 12.13 (br s, 1H), 8.16 (t, 1H), 8.00-7.98 (d, = 6.9 Hz, 2H), 7.58-7.55 (d, J = 8.4 Hz, 1H), 7.47 (s, 1H), 7.37-7.34 (d, 7 = 8.1 Hz, 1H), 7.23-7.20 (d, J = 8.4 Hz, 2H), 7.12 (m, 1H), 4.30-4.28 (d, J = 6.0 Hz, 2H), 2.0 (m, 1H), 1.12 (s, 9H), 1.01-0.97 (m, 2H), 0.77-0.76 (m, 2H). MS [M+H]+: 436.
Example-8
N-(4-chloro-2-fluoro-3-(2-oxo-6-(6-(trifluoromethyl)pyridin-3-yl)-2,3-dihydro pyrimidin-4-yl)benzyl) ival amide

The title compound was prepared following the procedure described in Example-3 by using N-(4-chloro-2-fluoro-3-(2-oxo-6-(6-(trifluoromethyl)pyridin-3-yl)- 1,2,3,6- tetrahydropyrimidin-4-yl)benzyl)pivalamide (Intermediate- 13, 0.200 g, 0.413 mmol), 2,3-dichloro-5-6,dicyano-l,4-benzoquinone (DDQ) (0.141 g, 0.620 mmol) and dry toluene (10 mL) to afford 0.018 g of desired product. 1H MR (300MHz, DMSO-<¾): δ 12.67 (br s, 1H), 9.46 (s, 1H), 8.78 (d, 1H), 8.20 (m, 2H), 8.1 1-8.09 (d, J = 8.4 Hz, 1H), 7.50 (m, 1H), 7.40 (m, 2H), 4.30 (d, 2H), 1.14 (s, 9H); MS [M+H]+: 485.
Example-9
N-(4-chloro-3-(2-oxo-6-(4-(trifluoromethyl)phenyl)-2,3-dihydropyrimidin-4- yl)benzyl)propane-2-sulfonamide

The title compound was prepared following the procedure described in Example-3 by using N-(4-chloro-3-(2-oxo-6-(4-(trifluoromethyl)phenyl)-l,2,3,6-tetrahydro pyrimidin-4-yl)benzyl)propane-2-sulfonamide (Intermediate- 14, 0.200 g, 0.413 mmol), 2,3-dichloro-5-6,dicyano-l,4-benzoquinone (DDQ) (0.141 g, 0.620 mmol) and dry toluene (10 mL) to afford 0.030 g of the desired product. 1HNMR (300MHz, DMSO- e): δ 12.44 (br s, 1H), 8.35 (m, 2H), 7.92-7.90 (d, J = 7.8 Hz, 2H), 7.62 (m, 2H), 7.55 (m, 3H), 4.24 (d, 2H), 3.15 (m, 1H), 1.24-1.21 (d, = 6.6 Hz, 6H); MS [M+H]+: 486.
Example- 10
4-(2-chloro-5-(pivalamidomethyl)phenyl)-6-(4-(trifluoromethyl)phenyl)pyrimidin-2- yl acetate

A suspension of N-(4-chloro-3-(2-oxo-6-(4-(trifluoromethyl)-2,3-dihydropyrimidin-4- yl)benzyl)pivalamide (Example-1, 0.100 g, 0.215 mmol) in acetic anhydride (4.0 mL) was heated at 100 °C for 16 h. The reaction mass was concentrated. The residue was washed with DEE, filtered and suck dried to afford 0.025 g of title product. 1HNMR (300MHz, CDC13): δ 8.25-8.23 (d, J = 6.6 Hz, 2H), 8.14 (s, 1H), 7.81-7.78 (d, J = 7.8 Hz, 2H), 7.66 (s, 1H), 7.52-7.49 (d, = 8.1 Hz, 1H), 7.39 (d, 1H), 6.04 (m, 1H), 4.86 (d, 2H), 2.44 (s, 3H), 1.24 (s, 9H); MS [M+H]+: 506.
Example-1 1
N-(3-(6-(4-(tert-butyl)phenyl)-2-oxo-2,3-dihydropyrimidin-4-yl)-4-chloro-2- fluorob enzy l)pi val ami de

The title compound was prepared following the procedure described in Example-3 by using N-(3-(6-(4-(ieri-butyl)phenyl)-2-oxo-l,2,3,6-tetrahydropyrimidin-4-yl)-4- chloro-2-fluorobenzyl)pivalamide (Intermediate-15, 0.060 g, 0.139 mmol), 2,3- dichloro-5-6,dicyano-l,4-benzoquinone (DDQ) (0.060 g, 0.260 mmol) and dry toluene (4 mL) to afford 0.020 g of the desired product. 1HNMR (300MHz, DMSO- d6): δ 12.29 (br s, 1H), 8.19 (s, 1H), 8.09 (br s, 2H), 7.57-7.54 (d, J = 8.4 Hz, 2H), 7.47 (m, 1H), 7.36 (m, 2H), 4.31-4.30 (d, J = 5.4 Hz, 2H), 1.32 (s, 9H), 1.14 (s, 9H); MS [M+H]+: 470.
Example- 12
N-(4-chloro-2-fluoro-3-(6-(4-isopropylphenyl)-2-oxo-2,3-dihydropyrimidin-4- yl)benzyl)pivalamide

The title compound was prepared following the procedure described in Example-3 by using N-(4-chloro-2-fluoro-3-(6-(4-isopropylphenyl)-2-oxo-l,2,3,6-tetrahydro pyrimidin-4-yl)benzyl)pivalamide (Intermediate- 16, 0.050 g, 0.10 mmol), 2,3- dichloro-5-6,dicyano-l,4-benzoquinone (DDQ) (0.050 g, 0.220 mmol) and dry toluene (4 mL) to afford 0.020 g of desired product. 1HNMR (300MHz, DMSO- 6): δ 12.28 (br s, 1H), 8.19-8.09 (m, 2H), 7.42-7.25 (m, 6H), 4.31 (s, 2H), 2.97 (m, 1H), 1.24 (s, 6H), 1.14 (s, 9H); MS [M+H]+: 456.
Example- 13
N-((6-methyl-5-(2-oxo-6-(4-(trifluoromethyl)phenyl)-2,3-dihydropyrimidin-4- yl)pyridin-3-yl)methyl)isobutyramide

The title compound was prepared following the procedure described in step-3 of Intermediate- 1 by using 6-(5-(aminomethyl)-2-methylpyridin-3-yl)-4-(4- (trifluoromethyl)phenyl)pyrimidin-2(lH)-one hydrochloride (Intermediate- 17, 0.050 g, 0.12 mmol), TEA (1.0 mL), isobutyryl chloride (0.050 g, 0.47 mmol) and dry THF (10 mL) to afford 0.025 g of the desired product. 1HNMR (300MHz, CDC13): δ 8.74 (s, 2H), 8.12 (br s, 2H), 7.78 (br s, 2H), 7.28 (s, 1H), 6.74 (s, 1H) 4.49 (br s, 2H), 3.08-2.92 (s, 3H), 2.45 (m, 1H), 1.19 (m, 6H); MS [M+H]+: 431.
Example- 14
N-((6-(difluoromethyl)-5-(2-oxo-6-(4-(trifluoromethyl)phenyl)-2,3-dihydropyrimidin- 4-yl)pyridin-3-yl)methyl)isobutyramide

The title compound was prepared following the procedure described in step-3 of Intermediate- 1 by using 6-(5-(aminomethyl)-2-(difluoromethyl)pyridin-3-yl)-4-(4- (trifluoromethyl)phenyl)pyrimidin-2(lH)-one hydrochloride (Intermediate- 18, 0.100 g, 0.23 mmol), TEA (1.0 mL), isobutyryl chloride (0.5 mL) and dry THF (10 mL) to afford 0.030 g of the desired product. 1HNMR (300MHz, CDC13): δ 8.77 (br s, 1H), 8.17 (br s, 3H), 7.83 (br s, 2H), 6.90 (m, 4H), 4.62 (m, 2H), 3.0 (m, 1H), 1.24 (s, 3H), 1.15 (s, 3H).
Example- 15
N-(4-chloro-3-(6-hydroxy-4-(4-(trifluoromethyl)phenyl)pyridin-2-yl)benzyl) pi val amide

Example- 16
N-(3-(2-amino-6-(4-(trifluoromethyl)phenyl)pyrimidin-4-yl)-4-chlorobenzyl) pivalamide

To a solution of (E)-N-(4-chloro-3-(3-(4-(trifluoromethyl)phenyl)acryloyl) benzyl)pivalamide (Intermediate-20, 0.400 g, 0.944 mmol) in dry DMF (5.0 mL) was added guanidine hydrochloride (0.180 g, 1.88 mmol) and sodium hydride (0.056 g, 1.41 mmol, 60% mineral oil). The reaction mixture was heated at reflux for 3 h. The reaction mass was concentred and diluted with water. The obtained precipitate was filtered to afford 0.050 g of the title compound. 1HNMR (300MHz, DMSO- 6): δ 8.32-8.30 (d, J = 8.1 Hz, 2H), 8.15 (t, 1H), 7.89-7.87 (d, J = 8.4 Hz, 2H), 7.55-7.52 (d, J = 7.8 Hz, 1H), 7.46 (s, 1H), 7.40 (s, 1H), 7.33-7.30 (d, J = 8.1 Hz, 1H), 6.99 (s, 2H), 4.29-4.28 (d, J = 5.7 Hz, 2H), 1.12 (s, 9H); MS [M+H]+: 463.
Example- 17
N-(3-(2-acetamido-6-(4-(trifluoromethyl)phenyl)pyrimidin-4-yl)-4-chlorobenzyl) pivalamide

Example- 18
N-(3-(2-amino-6-(4-(trifluoromethyl)phenyl)pyrimidin-4-yl)-4-chloro-2-fluoro benzyl)pivalamide

The title compound was prepared following the procedure described in Example-16 using (E)-N-(4-chloro-3-(3-(4-(trifluoromethyl)phenyl)acryloyl)benzyl)pivalamide (Intermediate-21, 0.500 g, 1.13 mmol), guanidine hydrochloride (0.216 g, 2.26 mmol), sodium hydride (0.068 g, 1.69 mmol, 60% mineral oil) in DMF (10 mL) to afford 0.020 g of the desired compound. 1HNMR (300MHz, DMSO- 6): δ 8.35-8.33 (d, = 8.4 Hz, 2H), 8.16 (t, 1H), 7.89-7.86 (d, = 8.1 Hz, 2H), 7.45-7.42 (d, = 8.4 Hz, 2H), 7.37-7.32 (m, 1H), 7.06 (s, 2H), 4.31-4.29 (d, = 6.0 Hz, 2H), 1.14 (s, 9H); MS [M+H]+: 481.
Example- 19
N-(4-chloro-2-fluoro-3-(2-(methylamino)-6-(4-(trifluoromethyl)phenyl)pyrimidin-4- yl)benzyl)pivalamide

The title compound was prepared following the procedure described in Example-16 using (E)-N-(4-chloro-3-(3-(4-(trifluoromethyl)phenyl)acryloyl)benzyl)pivalamide (Intermediate-21, 0.500 g, 1.13 mmol), 1-methylguanidine hydrochloride (0.248 g, 2.26 mmol), sodium hydride (0.068 g, 1.69 mmol, 60% mineral oil) in DMF (10 mL)
to afford 0.040 g of the desired compound. 1HNMR (300MHz, DMSO- 6): δ 8.38 (br s, 2H), 8.17 (t, 1H), 7.89-7.86 (d, J = 8.4 Hz, 2H), 7.50-7.33 (m, 4H), 4.31-4.29 (d, = 5.4 Hz, 2H), 2.95 (m, 3H), 1.14 (s, 9H); MS [M+H]+: 495.
Example-20
N-(3-(2-amino-6-(3,4-dichlorophenyl)pyrimidin-4-yl)-4-chloro-2-fluorobenzyl) pi val amide

The title compound was prepared following the procedure described in Example- 16 using (E)- V-(4-chloro-3 -(3 -(3 ,4-dichlorophenyl)-3 -oxoprop- 1 -en- 1 -yl)-2-fluoro benzyl)pivalamide (Intermediate-22, 0.500 g, 1.13 mmol), guanidine hydrochloride (0.216 g, 2.26 mmol), sodium hydride (0.068 g, 1.69 mmol, 60% mineral oil) in DMF (10 mL) to afford 0.030 g of the desired compound. 1HNMR (300MHz, DMSO- 6): δ 8.41 (s, 1H), 8.15-8.12 (m, 2H), 7.79-7.76 (d, = 8.7 Hz, 1H), 7.44-7.31 (m, 3H), 7.04 (br s, 2H), 4.30-4.28 (d, J = 5.1 Hz, 2H), 1.14 (s, 9H); MS [M+H]+: 481.
Example-21
N-(3-(2-amino-6-(4-chloro-3-methylphenyl)pyrimidin-4-yl)-4-chloro-2-fluoro b enzy l)pi val ami de

The title compound was prepared following the procedure described in Example- 16 using ((E)-N-(4-chloro-3 -(3 -(4-chloro-3 -methylphenyl)-3 -oxoprop- 1 -en- 1 -yl)-2- fluorobenzyl)pivalamide (Intermediate-23, 0.500 g, 1.18 mmol), guanidine hydrochloride (0.225 g, 2.36 mmol), sodium hydride (0.071 g, 1.69 mmol, 60% mineral oil) in DMF (10 mL) to afford 0.026 g of the desired compound. 1HNMR (300MHz, DMSO- e): δ 8.14 (m, 2H), 8.00-7.97 (d, J = 7.2 Hz, 1H), 7.55-7.52 (d, J = 8.7 Hz, 1H), 7.44-7.41 (d, = 8.7 Hz, 1H), 7.33-7.27 (m, 2H), 6.95 (br s, 2H), 4.30- 4.28 (d, J = 5.4 Hz, 2H), 2.49 (s, 3H), 1.14 (s, 9H); MS [M+H]+: 461.
Example-22
N-(3-(2-amino-6-(4-chloro-3-(trifluoromethyl)phenyl)pyrimidin-4-yl)-4-chloro-2- fluorob enzy l)pi val ami de

The title compound was prepared following the procedure described in Example- 16 using (E)-N-(4-chloro-3 -(3 -(4-chloro-3 -(trifluoromethyl)phenyl)-3 -oxoprop- 1 -en- 1 - yl)-2-fluorobenzyl)pivalamide (Intermediate-24, 0.500 g, 1.05 mmol), guanidine hydrochloride (0.201 g, 2.10 mmol), sodium hydride (0.062 g, 1.56 mmol, 60% mineral oil) in DMF (10 mL) to afford 0.035 g of the desired compound. 1HNMR (300MHz, DMSO- e): δ 8.59 (s, 1H), 8.45-8.42 (d, J = 8.4 Hz, 1H), 8.16-8.14 (d, J = 6.0 Hz, 1H), 7.89-7.86 (d, J = 8.4 Hz, 1H), 7.48 (s, 1H), 7.45-7.42 (d, J = 8.4 Hz, 1H), 7.35-7.29 (t, 7 = 7.8 Hz, 1H), 7.10 (s, 2H), 4.30-4.28 (d, J = 6.0 Hz, 2H), 1.14 (s, 9H).
Example-23
N-(3-(2-amino-6-(3,4-dimethylphenyl)pyrimidin-4-yl)-4-chloro-2-fluorobenzyl) pi val amide

The title compound was prepared following the procedure described in Example- 16 using (E)- V-(4-chloro-3 -(3 -(3 ,4-dimethylphenyl)-3 -oxoprop- 1 -en- 1 -yl)-2- fluorobenzyl)pivalamide (Intermediate-25, 0.500 g, 1.05 mmol), guanidine hydrochloride (0.201 g, 2.10 mmol), sodium hydride (0.062 g, 1.56 mmol, 60% mineral oil) in DMF (10 mL) to afford 0.035 g of the desired compound. 1HNMR (300MHz, DMSO- e): δ 8.15 (t, 1H), 7.87-7.84 (d, J = 7.8 Hz, 1H), 7.43-7.40 (d, J = 8.7 Hz, 1H), 7.33-7.20 (m, 3H), 6.86 (s, 2H), 4.30-4.28 (d, J = 5.4 Hz, 2H), 2.28 (s, 6H), 1.14 (s, 9H).
Pharmacological activity
In-vitro Protocol for screening of mPGES-1 inhibitors:
mPGES-1 (microsomal prostaglandin E synthase-1) is a microsomal enzyme that converts endoperoxide substrate PGH2 (prostaglandin H2) to product PGE2
(prostaglandin E2) by isomerization in the presence of reduced glutathione (GSH). mPGES-1 inhibitors were screened by assessing their ability to inhibit formation of PGE2 from PGH2 in presence of mPGES-1 using an anti-PGE2 antibody based detection method. Recombinant human mPGES-1 was generated in-house by expression in CHO cells (Ouellet M et al. (2002), Protein Expression and Purification 26: 489 - 495). The assay was set up using crude microsomal fractions at protein concentration of 40-60 μg/mL. Test compounds were prepared in 100 % dimethyl sulfoxide (DMSO) to obtain 20 mM stock solution and then diluted using assay buffer comprising 0.1 M Potassium phosphate buffer with 2 mM EDTA. The final concentration of DMSO in reaction was 0.5 % (v/v). Negative controls were comprised of all assay reagents except the enzyme. Positive controls were comprised of the enzyme reaction in the absence of any inhibitor. Test compounds were incubated for 10 minutes in assay buffer containing 2.5 mM GSH and mPGES-1 enzyme followed by addition of PGH2 at a concentration of 15 μΜ for 1 minute. The reaction was stopped by addition of Stannous chloride (l lmg/ml) and PGE2 levels were measured (Masse F et al. (2005), Journal of Biomolecular Screening 10(6) 599 - 605., Goedken RE et al. (2008), Journal of Biomolecular Screening 13(7): 619 - 625) by HTRF kit (CisBio)).
Inhibition of mPGES-1 enzyme activity was measured using the percent of reaction occurring in the positive control. Concentration response curves were plotted using percent inhibition of maximum enzyme reaction. The IC50 value was calculated from the concentration response curve by nonlinear regression analysis using GraphPad PRISM software.
The compounds prepared were tested using the above assay procedure and the results obtained are given in Table 1. Percentage inhibition at concentrations of 1.0 μΜ and 10.0 μΜ are given in the table along with IC50 (nM) details for selected examples. The compounds prepared were tested using the above assay procedure and were found to have IC50 less than 200nM, preferably less than ΙΟΟηΜ, more preferably less than 50nM or most preferably less than 20nM.
The IC50 (nM) values of the compounds are set forth in Table 1 wherein "A" refers to an IC50 value of less than 50 nM, "B" refers to IC50 value in range of 50.01 to 100.0 nM and "C" refers to IC50 values more than 100.01 nM.
Table 1 :
Sr. Example No. Percentage inhibition at IC50 value
No. Ι μΜ 10 μΜ
1 Example- 1 100.00 100.00 A
2 Example-2 99.09 100.00 A
3 Example-3 94.81 97.99 A
4 Example-4 12.12 87.00 -
5 Example- 5 49.91 98.47 -
6 Example-6 98.64 100.00 A
7 Example-7 95.47 100.00 A
8 Example- 8 99.56 95.67 A
9 Example-9 98.93 98.85 A
10 Example- 10 99.47 98.23 A
11 Example- 11 99.58 96.56 A
12 Example- 12 98.17 97.67 A
13 Example- 13 88.83 100.00 B
14 Example- 14 99.45 98.58 A
15 Example- 15 96.93 94.48 A
16 Example- 16 94.83 96.76 A
17 Example- 17 69.67 91.98 C
18 Example- 18 96.76 98.40 A
19 Example- 19 100.00 93.37 A
20 Example-20 95.45 99.36 A
21 Example-21 91.37 96.04 A
22 Example-22 91.51 97.75 A
23 Example-23 91.15 97.13 A
(-): Not determined
Screening for mPGES-1 inhibitors using the A549 cell based assay
The inhibition of mPGES-1 enzyme in A549 cell line was monitored as inhibition of IL-Ιβ induced PGE2 release. A549 cells were maintained in DMEM medium with 10% FBS and 1% Penicillin-Streptomycin Solution in 5% C02 at 37°C. Cells were seeded 24 h prior to the assay in 96 well plates in DMEM containing 1% Penicillin-Streptomycin and 2% FBS so as to get ~ 40,000 cells per well on the day of
experiment. The assay was carried out in a total volume of 200 μL·. Test compounds were dissolved in dimethyl sulfoxide (DMSO) to prepare 2 mM stock solution and then diluted using plain DMEM. The final concentration of DMSO in the reaction was 0.55% (v/v). Cells were treated with test compounds for 30 minutes followed by addition of IL-Ιβ at a final concentration of 10 ng/mL for 16-20 h. Plates were then centrifuged at 1000 rpm for 10 min at 4°C. Supernatants were collected & analyzed by the addition of PGE2-D2 & anti-PGE2 cryptate conjugate supplied by the CisBio HTRF kit in a 96 well half area Blackwell EIA/RIA plate. The assay plate was incubated overnight at 4-5° C before being read in Artemis (K-101) (Japan) HTRF plate reader and levels of PGE2 calculated by extrapolation from the standard curve.
The concentration response curves were plotted as % of maximal response obtained in the absence of test antagonist. The IC50 value was calculated from the concentration response curve by nonlinear regression analysis using GraphPad PRISM software.
Claims
1. A compound of formula (I)

(I)
or a pharmaceutically acceptable salt thereof,
wherein,
X is O, when dotted line T represents a single bond and dotted line '2' is absent; or X is ORa or RaRb, when dotted line ' 1 ' is absent and dotted line '2' represents a single bond;
G2 is N;
G3 is sleeted from N and CH;
G4 is selected from N and CH;
L is selected from -CO- or -S(0)2-;
W is selected from Ci-salkyl, haloCi-salkyl and hydroxyCi-salkyl;
Ra and Rb are each independently selected from hydrogen, Ci-4alkyl and - C(0)Ci-4alkyl;
R1 is selected from hydrogen, halogen and Ci-4alkyl;
R2 is absent when dotted line '2' represents a single bond; or R2 is hydrogen when dotted line '2' is absent;
at each occurrence R3 is independently selected from halogen, nitro, cyano, Ci. salkyl and haloCi-salkyl;
at each occurrence R4 is independently selected from halogen, Ci-salkyl, haloCi-salkyl and C3-i2cycloalkyl;
at each occurrence of Rx and Ry are hydrogen;
'm' is an integer ranging from 0 to 3, both inclusive;
'n' is an integer ranging from 1 to 4, both inclusive; and
'p' is an integer ranging from 1 to 3, both inclusive.
2. The compound according to claim 1, wherein X is O, dotted line T represents a single bond and dotted line '2' is absent.
3. The compound according to claim 1, wherein X is H2, HCH3 or HCOCH3; dotted line T is absent and dotted line '2' represents a single bond.
4. The compound according to claim 1, wherein X is OCOCH3, dotted line T is absent and dotted line '2' represents a single bond.
5. The compound according to any one of claims 1 to 4, wherein R1 is hydrogen or methyl.
6. The compound according to any one of claims 1 to 5, wherein R3 is fluorine, chlorine, methyl or difluorom ethyl.
7. The compound according to any one of claims 1 to 6, wherein R4 is chlorine, methyl, isopropyl, ie/t-butyl, trifluoromethyl or cyclopropyl.
8. The compound according to to any one of claims 1 to 7, wherein W is isopropyl or tert-butyl.
9. The compound according to any one of claims 1 to 8, wherein L is -CO-.
10. The compound according to claim 1, wherein
X is O, dotted line T represents a single bond and dotted line '2' is absent; Gi is CR1; G2 is N; G3 is N or CH; G4 is N or CH;
R1 is hydrogen or methyl;
R2 is hydrogen;
R3 is fluorine, chlorine, methyl or difluoromethyl;
R4 is isopropyl, ie/t-butyl, trifluoromethyl or cyclopropyl;
L is -CO-;
W is isopropyl or ie/t-butyl;
Rx and Ry are hydrogen; m is 1 or 2; n is 1 and p is 1.
11. The compound according to claim 1, wherein
X is ORa or RaRb, dotted line ' 1 ' is absent and dotted line '2' represents a single bond;
Gi is CR1; G2 is N; G3 is CH; G4 is CH;
Ra is hydrogen, methyl or -C(0)methyl; Rb is hydrogen;
R1 is hydrogen;
R2 is absent;
R3 is fluorine or chlorine;
R is chlorine, methyl or trifluoromethyl.
L is -CO-;
W is tert-butyl;
Rx and Ry are hydrogen; m is 1 or 2; n is lor 2 and p is 1.
12. A compound of formula (II)

(Π)
or a pharmaceutically acceptable salt thereof,
wherein,
G3 is sleeted from N and CH;
G4 is selected from N and CH;
L is selected from -CO- or -S(0)2-;
W is selected from Ci-8alkyl, haloCi-8alkyl and hydroxyCi-8alkyl;
R1 is selected from hydrogen, halogen and Ci-4alkyl;
at each occurrence R3 is independently selected from halogen, nitro, cyano, Ci. 8alkyl and haloCi-8alkyl;
at each occurrence R4 is independently selected from halogen, Ci-8alkyl, haloCi-8alkyl and C3-i2cycloalkyl;
at each occurrence of Rx and Ry are hydrogen;
'm' is an integer ranging from 0 to 3, both inclusive;
'n' is an integer ranging from 1 to 4, both inclusive; and
'p' is an integer ranging from 1 to 3, both inclusive.
13. The compound according to claim 12, wherein R3 is fluorine, chlorine, methyl or difluoromethyl.
14. The compound according to claim 12 or 13, wherein R4 is isopropyl, tert- butyl, trifluoromethyl or cyclopropyl.
15. The compound according to any one of claims 12 to 14, wherein W is isopropyl or tert-butyl.
16. The compound according to claim 12, wherein
G3 is N or CH; G4 is N or CH;
R1 is hydrogen or methyl;
R3 is fluorine, chlorine, methyl or difluoromethyl;
R4 is isopropyl, ie/t-butyl, trifluoromethyl or cyclopropyl;
L is -CO-;
W is isopropyl or ie/t-butyl;
Rx and Ry are hydrogen; m is 1 or 2; n is 1 and p is 1.
17. A compound of formula (III)

or a pharmaceutically acceptable salt thereof,
wherein,
W is selected from Ci-8alkyl, haloCi-8alkyl and hydroxyCi-8alkyl;
Ra and Rb are each independently selected from hydrogen, Ci-4alkyl and - C(0)Ci-4alkyl;
at each occurrence R3 is independently selected from halogen, nitro, cyano, Ci. 8alkyl and haloCi-8alkyl;
at each occurrence R4 is independently selected from halogen, Ci-8alkyl, haloCi-8alkyl and C3-i2cycloalkyl;
'm' is an integer ranging from 0 to 3, both inclusive; and
'n' is an integer ranging from 1 to 4, both inclusive.
18. The compound according to claim 17, wherein Ra is hydrogen, methyl or - C(0)methyl and Rb is hydrogen.
19. The compound according to claim 17 or 18, wherein R3 is fluorine or chlorine.
20. The compound according to any one of claims 17 to 19, wherein R4 is chlorine, methyl or trifluoromethyl.
21. The compound according to any one of claims 17 to 20, wherein W is tert- butyl.
22. The compound according to claim 17, wherein
Ra is hydrogen, methyl or -C(0)methyl; Rb is hydrogen;
R3 is fluorine or chlorine; R4 is chlorine, methyl or trifluoromethyl.
W is tert-butyl; m is 1 or 2 and n is lor 2.
23. A compound selected from
N-(4-chloro-3-(2-oxo-6-(4-(trifluoromethyl)-2,3-dihydropyrimidin-4-yl)benzyl) pival amide;
N-(4-chloro-(2-oxo-6-(4-(trifluoromethyl)phenyl)-2,3-dihydropyrimidin-4-yl)- benzyl)-isobutyramide;
N-(4-chloro-2-fluoro-3-(2-oxo-6-(4-(trifluoromethyl)phenyl)-2,3-dihydro pyrimidin-4-yl)benzyl)-pivalamide;
N-(4-chloro-2-fluoro-3-(5-methyl-2-oxo-6-(4-(trifluoromethyl)phenyl)-2,3- dihydropyrimidin-4-yl)benzyl)pival amide;
N-(4-chloro-2-fluoro-3-(5-methyl-2-oxo-6-(4-(trifluoromethyl)phenyl)-2,3- dihydropyrimidin-4-yl)benzyl)isobutyramide;
N-(4-chloro-3-(6-(4-cyclopropylphenyl)-2-oxo-2,3-dihydropyrimidin-4- yl)benzyl) isobutyramide;
N-(4-chloro-3-(6-(4-cyclopropylphenyl)-2-oxo-2,3-dihydropyrimidin-4- yl)benzyl)pivalamideide;
N-(4-chloro-2-fluoro-3-(2-oxo-6-(6-(trifluoromethyl)pyridin-3-yl)-2,3-dihydro pyrimidin-4-yl)benzyl)pival amide;
N-(4-chloro-3-(2-oxo-6-(4-(trifluoromethyl)phenyl)-2,3-dihydropyrimidin-4- yl)benzyl)propane-2-sulfonamide;
4-(2-chloro-5-(pivalamidomethyl)phenyl)-6-(4-(trifluoromethyl)phenyl) pyrimidin-2-yl acetate;
N-(3-(6-(4-(tert-butyl)phenyl)-2-oxo-2,3-dihydropyrimidin-4-yl)-4-chloro-2- fluorob enzy l)pi val ami de;
N-(4-chloro-2-fluoro-3-(6-(4-isopropylphenyl)-2-oxo-2,3-dihydropyrimidin-4- yl)benzyl)pivalamide;
N-((6-methyl-5-(2-oxo-6-(4-(trifluoromethyl)phenyl)-2,3-dihydropyrimidin-4- yl)pyridin-3-yl)methyl)isobutyramide;
N-((6-(difluoromethyl)-5-(2-oxo-6-(4-(trifluoromethyl)phenyl)-2,3-dihydro pyrimidin-4-yl)pyridin-3-yl)methyl)isobutyramide;
N-(4-chloro-3-(6-hydroxy-4-(4-(trifluoromethyl)phenyl)pyridin-2-yl)benzyl) pival amide;
N-(3-(2-amino-6-(4-(trifluoromethyl)phenyl)pyrimidin-4-yl)-4-chlorobenzyl) pival amide;
N-(3-(2-acetamido-6-(4-(trifluoromethyl)phenyl)pyrimidin-4-yl)-4- chlorobenzyl) pivalamide;
N-(3-(2-amino-6-(4-(trifluoromethyl)phenyl)pyrimidin-4-yl)-4-chloro-2-fluoro benzyl)pival amide;
N-(4-chloro-2-fluoro-3-(2-(methylamino)-6-(4-(trifluoromethyl)
phenyl)pyrimidin-4-yl)benzyl)pival amide;
N-(3-(2-amino-6-(3,4-dichlorophenyl)pyrimidin-4-yl)-4-chloro-2- fluorobenzyl) pivalamide;
N-(3-(2-amino-6-(4-chloro-3-methylphenyl)pyrimidin-4-yl)-4-chloro-2-fluoro benzyl)pival amide;
N-(3-(2-amino-6-(4-chloro-3-(trifluoromethyl)phenyl)pyrimidin-4-yl)-4- chl oro-2-fluorob enzy l)pi val ami de and
N-(3-(2-amino-6-(3,4-dimethylphenyl)pyrimidin-4-yl)-4-chloro-2- fluorobenzyl) pivalamide;
or pharmaceutically acceptable salts thereof.
24. A compound of formula

or pharmaceutically acceptable salts thereof.
25. A compound of formula

26. A compound of formula

or pharmaceutically acceptable salts thereof.
27. A pharmaceutical composition comprising a compound according to any one of of claims 1 to 26 and a pharmaceutically acceptable excipient.
28. The pharmaceutical composition according to claim 27, wherein the pharmaceutically acceptable excipient is a carrier or diluent.
29. A method of treating a mPGES-1 mediated disease, disorder or syndrome in a subject comprising administering an effective amount of a compound according to any one of claims 1 to 26.
30. A method of treatment of disease, disorder, syndrome or condition selected from the group consisting of inflammation, asthma, chronic obstructive pulmonary disease, pulmonary fibrosis, inflammatory bowel disease, irritable bowel syndrome, pain, inflammatory pain, chronic pain, acute pain, fever, migraine, headache, low back pain, fibromyalgia, myofascial disorders, viral infections, influenza, common cold, herpes zoster, hepatitis C, AIDS, bacterial infections, fungal infections, dysmenorrhea, burns, pain associated with surgical or dental procedures, dental pain, malignancies hyperprostaglandin E syndrome, classic Bartter syndrome, synovitis, atherosclerosis, gout, arthritis, osteoarthritis, juvenile arthritis, rheumatoid arthritis, juvenile onset rheumatoid arthritis, rheumatic fever, ankylosing spondylitis, Hodgkin's disease, systemic lupus erythematosus, vasculitis, pancreatitis, nephritis, bursitis, conjunctivitis, iritis, scleritis, uveitis, wound healing, dermatitis, eczema, psoriasis, stroke, diabetes mellitus, cancer, neurodegenerative disorders, autoimmune diseases, multiple sclerosis, allergic disorders, rhinitis, ulcers, mild to moderately active ulcerative colitis, familial adenomatous polyposis, coronary heart disease, and sarcoidosis comprising administering a compound according to any one of claims 1 to
26.
31. The method according to claim 30, wherein the disease, disorder, syndrome or condition is pain.
32. The method according to claim 30, wherein the disease, disorder, syndrome or condition is chronic or acute pain.
33. The method according to claim 30, wherein the disease, disorder, syndrome or condition is rheumatoid arthritic pain, osteoarthritic pain or dental pain
34. The method according to claim 30, wherein the disease, disorder, syndrome or condition is inflammation, asthma or chronic obstructive pulmonary disease.
35. The method according to claim 30, wherein the disease, disorder, syndrome or condition is a neurodegenerative disorder selected from Parkinson's disease, Alzheimer's disease and amyotrophic lateral sclerosis.
36. A method of treating, preventing or managing cancer comprising administering to a subject in need of such treatment an effective amount of a compound according to any one of claims 1 to 26.
37. A process for preparing a compound of formula (II)

or a pharmaceutically acceptable salt thereof, which comprises
(a) reacting a compound of formula (27) with compound of formula (13a) and urea to form compound of formula (28);
(b) converting compound of formula (28) to compound of formua (II)

wherein,
G3 is sleeted from N and CH;
G4 is selected from N and CH;
L is selected from -CO- or -S(0)2-;
W is selected from Ci-8alkyl, haloCi-8alkyl and hydroxyCi-8alkyl;
R1 is selected from hydrogen, halogen and Ci-4alkyl;
at each occurrence R3 is independently selected from halogen, nitro, cyano, Ci. 8alkyl and haloCi-8alkyl;
at each occurrence R4 is independently selected from halogen, Ci-8alkyl, haloCi-8alkyl and C3.i2cycloalkyl;
at each occurrence of Rx and Ry are hydrogen;
'm' is an integer ranging from 0 to 3, both inclusive;
'n' is an integer ranging from 1 to 4, both inclusive; and
'p' is 1.
38. The process according to claim 37, wherein step (a) is carried out in presence of trimethylsilyl chloride and sulphamic acid.
39. The process according to claim 37, wherein step (b) is carried out using 2,3- dichloro-5,6-dicyano-l,4-benzoquinone (DDQ).
40. The process according to claim 37, wherein step (b) is carried out in presence of toluene.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN3304/MUM/2013 | 2013-10-22 | ||
| IN3304MU2013 | 2013-10-22 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2015059618A1 true WO2015059618A1 (en) | 2015-04-30 |
Family
ID=51947404
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2014/065465 Ceased WO2015059618A1 (en) | 2013-10-22 | 2014-10-20 | SUBSTITUTED PYRIMIDINE COMPOUNDS AS mPGES-1 INHIBITORS |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2015059618A1 (en) |
Cited By (45)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2017039714A (en) * | 2015-08-17 | 2017-02-23 | 日本たばこ産業株式会社 | Hydroxytriazine compound and pharmaceutical use thereof |
| WO2017073709A1 (en) | 2015-10-29 | 2017-05-04 | あすか製薬株式会社 | Pyrimidine derivative |
| WO2019044868A1 (en) * | 2017-08-30 | 2019-03-07 | あすか製薬株式会社 | Pyrimidine derivative |
| WO2020263830A1 (en) | 2019-06-25 | 2020-12-30 | Gilead Sciences, Inc. | Flt3l-fc fusion proteins and methods of use |
| WO2021076908A1 (en) | 2019-10-18 | 2021-04-22 | Forty Seven, Inc. | Combination therapies for treating myelodysplastic syndromes and acute myeloid leukemia |
| WO2021087064A1 (en) | 2019-10-31 | 2021-05-06 | Forty Seven, Inc. | Anti-cd47 and anti-cd20 based treatment of blood cancer |
| WO2021096860A1 (en) | 2019-11-12 | 2021-05-20 | Gilead Sciences, Inc. | Mcl1 inhibitors |
| WO2021130638A1 (en) | 2019-12-24 | 2021-07-01 | Carna Biosciences, Inc. | Diacylglycerol kinase modulating compounds |
| WO2021163064A2 (en) | 2020-02-14 | 2021-08-19 | Jounce Therapeutics, Inc. | Antibodies and fusion proteins that bind to ccr8 and uses thereof |
| WO2021222522A1 (en) | 2020-05-01 | 2021-11-04 | Gilead Sciences, Inc. | Cd73 inhibiting 2,4-dioxopyrimidine compounds |
| RU2772907C2 (en) * | 2015-08-17 | 2022-05-27 | Джапан Тобакко Инк. | Hydroxytriazine compound and its application for medical purposes |
| JPWO2022168808A1 (en) * | 2021-02-02 | 2022-08-11 | ||
| WO2022221304A1 (en) | 2021-04-14 | 2022-10-20 | Gilead Sciences, Inc. | CO-INHIBITION OF CD47/SIRPα BINDING AND NEDD8-ACTIVATING ENZYME E1 REGULATORY SUBUNIT FOR THE TREATMENT OF CANCER |
| WO2022245671A1 (en) | 2021-05-18 | 2022-11-24 | Gilead Sciences, Inc. | Methods of using flt3l-fc fusion proteins |
| WO2022271677A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271650A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271684A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271659A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| US20230011968A1 (en) * | 2014-02-20 | 2023-01-12 | Japan Tobacco Inc. | Triazine compounds and pharmaceutical use thereof |
| WO2023077030A1 (en) | 2021-10-29 | 2023-05-04 | Gilead Sciences, Inc. | Cd73 compounds |
| WO2023076983A1 (en) | 2021-10-28 | 2023-05-04 | Gilead Sciences, Inc. | Pyridizin-3(2h)-one derivatives |
| WO2023122615A1 (en) | 2021-12-22 | 2023-06-29 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| WO2023122581A2 (en) | 2021-12-22 | 2023-06-29 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| WO2023127883A1 (en) | 2021-12-28 | 2023-07-06 | 日本新薬株式会社 | Indazole compound and pharmaceutical |
| WO2023147418A1 (en) | 2022-01-28 | 2023-08-03 | Gilead Sciences, Inc. | Parp7 inhibitors |
| EP4245756A1 (en) | 2022-03-17 | 2023-09-20 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| WO2023183817A1 (en) | 2022-03-24 | 2023-09-28 | Gilead Sciences, Inc. | Combination therapy for treating trop-2 expressing cancers |
| WO2023196784A1 (en) | 2022-04-05 | 2023-10-12 | Gilead Sciences, Inc. | Combinations of antibody therapies for treating colorectal cancer |
| WO2023205719A1 (en) | 2022-04-21 | 2023-10-26 | Gilead Sciences, Inc. | Kras g12d modulating compounds |
| WO2024006929A1 (en) | 2022-07-01 | 2024-01-04 | Gilead Sciences, Inc. | Cd73 compounds |
| WO2024010015A1 (en) | 2022-07-06 | 2024-01-11 | あすか製薬株式会社 | Pyrimidine derivatives |
| WO2024064668A1 (en) | 2022-09-21 | 2024-03-28 | Gilead Sciences, Inc. | FOCAL IONIZING RADIATION AND CD47/SIRPα DISRUPTION ANTICANCER COMBINATION THERAPY |
| WO2024137852A1 (en) | 2022-12-22 | 2024-06-27 | Gilead Sciences, Inc. | Prmt5 inhibitors and uses thereof |
| WO2024215754A1 (en) | 2023-04-11 | 2024-10-17 | Gilead Sciences, Inc. | Kras modulating compounds |
| WO2024220917A1 (en) | 2023-04-21 | 2024-10-24 | Gilead Sciences, Inc. | Prmt5 inhibitors and uses thereof |
| WO2025006720A1 (en) | 2023-06-30 | 2025-01-02 | Gilead Sciences, Inc. | Kras modulating compounds |
| WO2025024663A1 (en) | 2023-07-26 | 2025-01-30 | Gilead Sciences, Inc. | Parp7 inhibitors |
| WO2025024811A1 (en) | 2023-07-26 | 2025-01-30 | Gilead Sciences, Inc. | Parp7 inhibitors |
| WO2025054347A1 (en) | 2023-09-08 | 2025-03-13 | Gilead Sciences, Inc. | Kras g12d modulating compounds |
| WO2025054530A1 (en) | 2023-09-08 | 2025-03-13 | Gilead Sciences, Inc. | Pyrimidine-containing polycyclic derivatives as kras g12d modulating compounds |
| WO2025096589A1 (en) | 2023-11-03 | 2025-05-08 | Gilead Sciences, Inc. | Prmt5 inhibitors and uses thereof |
| US12319655B2 (en) | 2021-12-09 | 2025-06-03 | Deciphera Pharmaceuticals, Llc | RAF kinase inhibitors and methods of use thereof |
| WO2025137640A1 (en) | 2023-12-22 | 2025-06-26 | Gilead Sciences, Inc. | Azaspiro wrn inhibitors |
| WO2025245003A1 (en) | 2024-05-21 | 2025-11-27 | Gilead Sciences, Inc. | Prmt5 inhibitors and uses thereof |
| WO2026039365A1 (en) | 2024-08-12 | 2026-02-19 | Gilead Sciences, Inc. | Kras modulating compounds |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006063466A1 (en) | 2004-12-17 | 2006-06-22 | Merck Frosst Canada Ltd. | 2-(phenyl or heterocyclic)-1h-phenantrho[9,10-d]imidazoles as mpges-1 inhibitors |
| WO2007059610A1 (en) | 2005-11-23 | 2007-05-31 | Merck Frosst Canada Ltd. | 2-(phenyl or heterocyclic)-1h-phenantrho[9,10-d]imidazoles as mpges-1 inhibitors |
| WO2010034796A1 (en) | 2008-09-25 | 2010-04-01 | Boehringer Ingelheim International Gmbh | 1h-benz imidazole-5-carboxamides as anti-inflammatory agents |
| WO2010100249A1 (en) | 2009-03-05 | 2010-09-10 | Boehringer Ingelheim International Gmbh | 3h-imidazo [4, 5 -c] pyridine- 6 -carboxamides as anti- inflammatory agents |
| WO2012055995A1 (en) | 2010-10-29 | 2012-05-03 | Glenmark Pharmaceuticals S.A. | Tricyclic compounds as mpges-1 inhibitors |
| WO2012110860A1 (en) | 2011-02-17 | 2012-08-23 | Glenmark Pharmaceuticals S.A. | TRICYCLIC COMPOUNDS AS mPGES-1 INHIBITORS |
| WO2013038308A1 (en) | 2011-09-15 | 2013-03-21 | Glenmark Pharmaceuticals S.A. | SUBSTITUTED BICYCLIC HETEROARYL COMPOUNDS AS mPGES-1 INHIBITORS |
| WO2013072825A1 (en) | 2011-11-16 | 2013-05-23 | Glenmark Pharmaceuticals S.A. | Phtalazinone derivatives as mpegs -1 inhibitors |
-
2014
- 2014-10-20 WO PCT/IB2014/065465 patent/WO2015059618A1/en not_active Ceased
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006063466A1 (en) | 2004-12-17 | 2006-06-22 | Merck Frosst Canada Ltd. | 2-(phenyl or heterocyclic)-1h-phenantrho[9,10-d]imidazoles as mpges-1 inhibitors |
| WO2007059610A1 (en) | 2005-11-23 | 2007-05-31 | Merck Frosst Canada Ltd. | 2-(phenyl or heterocyclic)-1h-phenantrho[9,10-d]imidazoles as mpges-1 inhibitors |
| WO2010034796A1 (en) | 2008-09-25 | 2010-04-01 | Boehringer Ingelheim International Gmbh | 1h-benz imidazole-5-carboxamides as anti-inflammatory agents |
| WO2010100249A1 (en) | 2009-03-05 | 2010-09-10 | Boehringer Ingelheim International Gmbh | 3h-imidazo [4, 5 -c] pyridine- 6 -carboxamides as anti- inflammatory agents |
| WO2012055995A1 (en) | 2010-10-29 | 2012-05-03 | Glenmark Pharmaceuticals S.A. | Tricyclic compounds as mpges-1 inhibitors |
| WO2012110860A1 (en) | 2011-02-17 | 2012-08-23 | Glenmark Pharmaceuticals S.A. | TRICYCLIC COMPOUNDS AS mPGES-1 INHIBITORS |
| WO2013038308A1 (en) | 2011-09-15 | 2013-03-21 | Glenmark Pharmaceuticals S.A. | SUBSTITUTED BICYCLIC HETEROARYL COMPOUNDS AS mPGES-1 INHIBITORS |
| WO2013072825A1 (en) | 2011-11-16 | 2013-05-23 | Glenmark Pharmaceuticals S.A. | Phtalazinone derivatives as mpegs -1 inhibitors |
Non-Patent Citations (14)
| Title |
|---|
| "Remington: The Science and Practice of Pharmacy", 2003, LIPPINCOTT WILLIAMS & WILKINS |
| FLORIAN RÖRSCH ET AL: "Structure-Activity Relationship of Nonacidic Quinazolinone Inhibitors of Human Microsomal Prostaglandin Synthase 1 (mPGES 1)", JOURNAL OF MEDICINAL CHEMISTRY, vol. 55, no. 8, 26 April 2012 (2012-04-26), pages 3792 - 3803, XP055116286, ISSN: 0022-2623, DOI: 10.1021/jm201687d * |
| GOEDKEN RE ET AL., JOURNAL OF BIOMOLECULAR SCREENING, vol. 13, no. 7, 2008, pages 619 - 625 |
| GOMEZ-HERNANDEZ, ATHEROSCLEROSIS, vol. 187, 2006, pages 139 - 49 |
| KOJIMA, THE JOURNAL OF IMMUNOLOGY, vol. 180, 2008, pages 8361 - 6 |
| KOROTKOVA: "Annals of the Rheumatic Diseases", vol. 67, 2008, pages: 1596 - 1602 |
| MASSE F ET AL., JOURNAL OF BIOMOLECULAR SCREENING, vol. 10, no. 6, 2005, pages 599 - 605 |
| NAKANISHI, CANCER RESEARCH, vol. 68, no. 9, 2008, pages 3251 - 9 |
| OUELLET M ET AL., PROTEIN EXPRESSION AND PURIFICATION, vol. 26, 2002, pages 489 - 495 |
| SCHRODER, JOURNAL OF LIPID RESEARCH, vol. 47, 2006, pages 1071 - 80 |
| WANG J ET AL: "Selective inducible microsomal prostaglandin E2 synthase-1 (mPGES-1) inhibitors derived from an oxicam template", BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, PERGAMON, AMSTERDAM, NL, vol. 20, no. 5, 1 March 2010 (2010-03-01), pages 1604 - 1609, XP026911315, ISSN: 0960-894X, [retrieved on 20100125], DOI: 10.1016/J.BMCL.2010.01.060 * |
| WANG, CIRCULATION, vol. 117, 2008, pages 1302 - 1309 |
| WANG, PROCEEDINGS OF NATIONAL ACADEMY OF SCIENCES, vol. 103, no. 39, 2006, pages 14507 - 12 |
| XU, THE JOURNAL OF PHARMACOLOGY AND EXPERIMENTAL THERAPEUTICS, vol. 326, 2008, pages 754 - 63 |
Cited By (80)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20230011968A1 (en) * | 2014-02-20 | 2023-01-12 | Japan Tobacco Inc. | Triazine compounds and pharmaceutical use thereof |
| US12398111B2 (en) * | 2014-02-20 | 2025-08-26 | Japan Tobacco Inc. | Triazine compounds and pharmaceutical use thereof |
| EP4046991A1 (en) * | 2015-08-17 | 2022-08-24 | Japan Tobacco Inc. | Hydroxytriazine compounds and pharmaceutical use thereof |
| WO2017030115A1 (en) * | 2015-08-17 | 2017-02-23 | 日本たばこ産業株式会社 | Hydroxytriazine compound and medical use thereof |
| RU2772907C2 (en) * | 2015-08-17 | 2022-05-27 | Джапан Тобакко Инк. | Hydroxytriazine compound and its application for medical purposes |
| CN108137515B (en) * | 2015-08-17 | 2021-07-06 | 日本烟草产业株式会社 | Hydroxytriazine compounds and their medicinal uses |
| JP2024019449A (en) * | 2015-08-17 | 2024-02-09 | 日本たばこ産業株式会社 | Hydroxytriazine compounds and their pharmaceutical uses |
| CN108137515A (en) * | 2015-08-17 | 2018-06-08 | 日本烟草产业株式会社 | Hydroxyl triaizine compounds and its medical usage |
| JP7640658B2 (en) | 2015-08-17 | 2025-03-05 | 日本たばこ産業株式会社 | Hydroxytriazine compounds and their medical uses |
| TWI704139B (en) * | 2015-08-17 | 2020-09-11 | 日商日本煙草產業股份有限公司 | Hydroxytriazine compound and pharmaceutical use thereof |
| JP2017039714A (en) * | 2015-08-17 | 2017-02-23 | 日本たばこ産業株式会社 | Hydroxytriazine compound and pharmaceutical use thereof |
| JP2020193235A (en) * | 2015-08-17 | 2020-12-03 | 日本たばこ産業株式会社 | Hydroxytriazine compounds and their pharmaceutical uses |
| US12492182B2 (en) | 2015-08-17 | 2025-12-09 | Japan Tobacco Inc. | Hydroxytriazine compounds and pharmaceutical use thereof |
| AU2016309337B2 (en) * | 2015-08-17 | 2021-03-04 | Japan Tobacco Inc. | Hydroxytriazine compound and medical use thereof |
| WO2017073709A1 (en) | 2015-10-29 | 2017-05-04 | あすか製薬株式会社 | Pyrimidine derivative |
| CN108137513B (en) * | 2015-10-29 | 2022-03-01 | Aska 制药株式会社 | Pyrimidine derivatives |
| US10710967B2 (en) | 2015-10-29 | 2020-07-14 | Aska Pharmaceutical Co., Ltd. | Pyrimidine derivative |
| JPWO2017073709A1 (en) * | 2015-10-29 | 2018-08-23 | あすか製薬株式会社 | Pyrimidine derivative |
| KR20180067678A (en) | 2015-10-29 | 2018-06-20 | 아스카 세이야쿠 가부시키가이샤 | Pyrimidine derivative |
| CN108137513A (en) * | 2015-10-29 | 2018-06-08 | Aska 制药株式会社 | Pyrimidine derivatives |
| KR102649802B1 (en) | 2015-10-29 | 2024-03-20 | 아스카 세이야쿠 가부시키가이샤 | Pyrimidine derivatives |
| WO2019044868A1 (en) * | 2017-08-30 | 2019-03-07 | あすか製薬株式会社 | Pyrimidine derivative |
| JPWO2019044868A1 (en) * | 2017-08-30 | 2020-10-01 | あすか製薬株式会社 | Pyrimidine derivative |
| JP7237002B2 (en) | 2017-08-30 | 2023-03-10 | あすか製薬株式会社 | pyrimidine derivative |
| WO2020263830A1 (en) | 2019-06-25 | 2020-12-30 | Gilead Sciences, Inc. | Flt3l-fc fusion proteins and methods of use |
| WO2021076908A1 (en) | 2019-10-18 | 2021-04-22 | Forty Seven, Inc. | Combination therapies for treating myelodysplastic syndromes and acute myeloid leukemia |
| EP4349413A2 (en) | 2019-10-18 | 2024-04-10 | Forty Seven, Inc. | Combination therapies for treating myelodysplastic syndromes and acute myeloid leukemia |
| WO2021087064A1 (en) | 2019-10-31 | 2021-05-06 | Forty Seven, Inc. | Anti-cd47 and anti-cd20 based treatment of blood cancer |
| WO2021096860A1 (en) | 2019-11-12 | 2021-05-20 | Gilead Sciences, Inc. | Mcl1 inhibitors |
| EP4445902A2 (en) | 2019-12-24 | 2024-10-16 | Carna Biosciences, Inc. | Diacylglycerol kinase modulating compounds |
| WO2021130638A1 (en) | 2019-12-24 | 2021-07-01 | Carna Biosciences, Inc. | Diacylglycerol kinase modulating compounds |
| US11692038B2 (en) | 2020-02-14 | 2023-07-04 | Gilead Sciences, Inc. | Antibodies that bind chemokine (C-C motif) receptor 8 (CCR8) |
| WO2021163064A2 (en) | 2020-02-14 | 2021-08-19 | Jounce Therapeutics, Inc. | Antibodies and fusion proteins that bind to ccr8 and uses thereof |
| US12297282B2 (en) | 2020-02-14 | 2025-05-13 | Gilead Sciences, Inc. | Nucleic acids encoding, and methods of producing, antibodies that bind human chemokine (C—C motif) receptor 8 (CCR8) |
| WO2021222522A1 (en) | 2020-05-01 | 2021-11-04 | Gilead Sciences, Inc. | Cd73 inhibiting 2,4-dioxopyrimidine compounds |
| RU2852458C1 (en) * | 2021-02-02 | 2025-12-08 | Аска Фармасьютикал Ко., Лтд. | Pyrimidine derivative |
| JPWO2022168808A1 (en) * | 2021-02-02 | 2022-08-11 | ||
| WO2022168808A1 (en) | 2021-02-02 | 2022-08-11 | あすか製薬株式会社 | Pyrimidine derivative |
| JP7277676B2 (en) | 2021-02-02 | 2023-05-19 | あすか製薬株式会社 | pyrimidine derivative |
| KR20230142739A (en) | 2021-02-02 | 2023-10-11 | 아스카 세이야쿠 가부시키가이샤 | Pyrimidine derivatives |
| US12552769B2 (en) | 2021-02-02 | 2026-02-17 | Aska Pharmaceutical Co., Ltd. | Pyrimidine derivative |
| CN116867774A (en) * | 2021-02-02 | 2023-10-10 | Aska 制药株式会社 | Pyrimidine derivatives |
| WO2022221304A1 (en) | 2021-04-14 | 2022-10-20 | Gilead Sciences, Inc. | CO-INHIBITION OF CD47/SIRPα BINDING AND NEDD8-ACTIVATING ENZYME E1 REGULATORY SUBUNIT FOR THE TREATMENT OF CANCER |
| WO2022245671A1 (en) | 2021-05-18 | 2022-11-24 | Gilead Sciences, Inc. | Methods of using flt3l-fc fusion proteins |
| WO2022271650A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271677A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271684A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2022271659A1 (en) | 2021-06-23 | 2022-12-29 | Gilead Sciences, Inc. | Diacylglyercol kinase modulating compounds |
| WO2023076983A1 (en) | 2021-10-28 | 2023-05-04 | Gilead Sciences, Inc. | Pyridizin-3(2h)-one derivatives |
| WO2023077030A1 (en) | 2021-10-29 | 2023-05-04 | Gilead Sciences, Inc. | Cd73 compounds |
| US12319655B2 (en) | 2021-12-09 | 2025-06-03 | Deciphera Pharmaceuticals, Llc | RAF kinase inhibitors and methods of use thereof |
| WO2023122615A1 (en) | 2021-12-22 | 2023-06-29 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| EP4667056A1 (en) | 2021-12-22 | 2025-12-24 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| WO2023122581A2 (en) | 2021-12-22 | 2023-06-29 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| WO2023127883A1 (en) | 2021-12-28 | 2023-07-06 | 日本新薬株式会社 | Indazole compound and pharmaceutical |
| KR20240128981A (en) | 2021-12-28 | 2024-08-27 | 니뽄 신야쿠 가부시키가이샤 | Indazole compounds and pharmaceuticals |
| WO2023147418A1 (en) | 2022-01-28 | 2023-08-03 | Gilead Sciences, Inc. | Parp7 inhibitors |
| EP4245756A1 (en) | 2022-03-17 | 2023-09-20 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| EP4464703A2 (en) | 2022-03-17 | 2024-11-20 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| WO2023178181A1 (en) | 2022-03-17 | 2023-09-21 | Gilead Sciences, Inc. | Ikaros zinc finger family degraders and uses thereof |
| WO2023183817A1 (en) | 2022-03-24 | 2023-09-28 | Gilead Sciences, Inc. | Combination therapy for treating trop-2 expressing cancers |
| WO2023196784A1 (en) | 2022-04-05 | 2023-10-12 | Gilead Sciences, Inc. | Combinations of antibody therapies for treating colorectal cancer |
| WO2023205719A1 (en) | 2022-04-21 | 2023-10-26 | Gilead Sciences, Inc. | Kras g12d modulating compounds |
| WO2024006929A1 (en) | 2022-07-01 | 2024-01-04 | Gilead Sciences, Inc. | Cd73 compounds |
| KR20250030486A (en) | 2022-07-06 | 2025-03-05 | 아스카 세이야쿠 가부시키가이샤 | Pyrimidine derivatives |
| WO2024010015A1 (en) | 2022-07-06 | 2024-01-11 | あすか製薬株式会社 | Pyrimidine derivatives |
| WO2024064668A1 (en) | 2022-09-21 | 2024-03-28 | Gilead Sciences, Inc. | FOCAL IONIZING RADIATION AND CD47/SIRPα DISRUPTION ANTICANCER COMBINATION THERAPY |
| WO2024137852A1 (en) | 2022-12-22 | 2024-06-27 | Gilead Sciences, Inc. | Prmt5 inhibitors and uses thereof |
| WO2024215754A1 (en) | 2023-04-11 | 2024-10-17 | Gilead Sciences, Inc. | Kras modulating compounds |
| WO2024220917A1 (en) | 2023-04-21 | 2024-10-24 | Gilead Sciences, Inc. | Prmt5 inhibitors and uses thereof |
| WO2025006720A1 (en) | 2023-06-30 | 2025-01-02 | Gilead Sciences, Inc. | Kras modulating compounds |
| WO2025024811A1 (en) | 2023-07-26 | 2025-01-30 | Gilead Sciences, Inc. | Parp7 inhibitors |
| WO2025024663A1 (en) | 2023-07-26 | 2025-01-30 | Gilead Sciences, Inc. | Parp7 inhibitors |
| WO2025054530A1 (en) | 2023-09-08 | 2025-03-13 | Gilead Sciences, Inc. | Pyrimidine-containing polycyclic derivatives as kras g12d modulating compounds |
| WO2025054347A1 (en) | 2023-09-08 | 2025-03-13 | Gilead Sciences, Inc. | Kras g12d modulating compounds |
| WO2025096589A1 (en) | 2023-11-03 | 2025-05-08 | Gilead Sciences, Inc. | Prmt5 inhibitors and uses thereof |
| WO2025137640A1 (en) | 2023-12-22 | 2025-06-26 | Gilead Sciences, Inc. | Azaspiro wrn inhibitors |
| WO2025245003A1 (en) | 2024-05-21 | 2025-11-27 | Gilead Sciences, Inc. | Prmt5 inhibitors and uses thereof |
| WO2026039365A1 (en) | 2024-08-12 | 2026-02-19 | Gilead Sciences, Inc. | Kras modulating compounds |
| US12612369B1 (en) | 2025-12-15 | 2026-04-28 | King Faisal University | Eco-friendly synthesis of a novel nicotinonitrile derivative (MNPN) with antibacterial and anti-inflammatory activities for preventing pressure ulcers in individuals with disabilities |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2015059618A1 (en) | SUBSTITUTED PYRIMIDINE COMPOUNDS AS mPGES-1 INHIBITORS | |
| WO2014167444A1 (en) | SUBSTITUTED BICYCLIC COMPOUNDS AS mPGES-1 INHIBITORS | |
| US10821100B2 (en) | Triazolone compounds as mPGES-1 inhibitors | |
| US9006257B2 (en) | Bicyclic compounds as mPGES-1 inhibitors | |
| WO2013072825A1 (en) | Phtalazinone derivatives as mpegs -1 inhibitors | |
| JP7789662B2 (en) | MTA-cooperative PRMT5 inhibitor | |
| WO2013153535A1 (en) | TRICYCLIC COMPOUNDS AS mPGES-1 INHIBITORS | |
| AU2013339167B2 (en) | Novel amine derivative or salt thereof | |
| WO2013038308A1 (en) | SUBSTITUTED BICYCLIC HETEROARYL COMPOUNDS AS mPGES-1 INHIBITORS | |
| JP6319747B2 (en) | Substituted 4-pyridones and their use as neutrophil elastase activity inhibitors | |
| DE102008022221A1 (en) | Inhibitors of human aldosterone synthase CYP11B2 | |
| KR101280809B1 (en) | Anthranilamide pyridinureas as vegf receptor kinase inhibitors | |
| BG63778B1 (en) | Aromatic compounds and pharmaceutical compositions containing them | |
| KR20200038473A (en) | New heteroaryl amide derivatives as selective inhibitors of histone deacetylases 1 and 2 (HDAC1-2) | |
| WO2018136437A2 (en) | Compounds useful as inhibitors of indoleamine 2,3-dioxygenase and/or tryptophan dioxygenase | |
| JP2024542145A (en) | 2-Aminoimidazole Derivatives as PRMT5 Inhibitors | |
| US20110098298A1 (en) | New Pyridin-3-Amine Derivatives | |
| KR20070083906A (en) | New Anthranylamide Pyridineurea As Vascular Endothelial Growth Factor (VEF) Receptor Kinase Inhibitor | |
| EP4332101A1 (en) | Methionine adenosyltransferase inhibitor, preparation method therefor and application thereof | |
| US20140228396A1 (en) | Triazolopyridyl compounds as aldosterone synthase inhibitors | |
| JP5069119B2 (en) | Nicotinamide pyridine urea as a vascular endothelial growth factor (VEGF) receptor kinase inhibitor | |
| US8273880B2 (en) | Pyrazole compounds with inhibitory activity against ROS kinase | |
| WO2016128905A1 (en) | Thienopyrrole compounds as s-nitrosoglutathione reductase inhibitors | |
| CA2447675A1 (en) | Novel pyridylmethylaminopyrimidines | |
| CN118344325A (en) | PI3K selective inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14802500 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 14802500 Country of ref document: EP Kind code of ref document: A1 |