WO2014108407A1 - Pyridone derivatives and their use in the treatment, amelioration or prevention of a viral disease - Google Patents
Pyridone derivatives and their use in the treatment, amelioration or prevention of a viral disease Download PDFInfo
- Publication number
- WO2014108407A1 WO2014108407A1 PCT/EP2014/050166 EP2014050166W WO2014108407A1 WO 2014108407 A1 WO2014108407 A1 WO 2014108407A1 EP 2014050166 W EP2014050166 W EP 2014050166W WO 2014108407 A1 WO2014108407 A1 WO 2014108407A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- methyl
- oxo
- benzyloxy
- pyran
- carboxylic acid
- Prior art date
Links
- 0 *c(cccc1)c1S(Cl)(=O)=O Chemical compound *c(cccc1)c1S(Cl)(=O)=O 0.000 description 4
- KSHZTBSWXIAFCE-UHFFFAOYSA-N CC(C)NC(C(N(C)C(CNS(c1ccc(C)cc1)(=O)=O)=CC1=O)=C1O)=O Chemical compound CC(C)NC(C(N(C)C(CNS(c1ccc(C)cc1)(=O)=O)=CC1=O)=C1O)=O KSHZTBSWXIAFCE-UHFFFAOYSA-N 0.000 description 1
- JKEUAEQMMZPLJV-UHFFFAOYSA-N CN(C(CNS(c1ccccc1)(=O)=O)=CC1=O)C(C(O)=O)=C1O Chemical compound CN(C(CNS(c1ccccc1)(=O)=O)=CC1=O)C(C(O)=O)=C1O JKEUAEQMMZPLJV-UHFFFAOYSA-N 0.000 description 1
- WCDYYZYJUCSUFC-UHFFFAOYSA-N CNC(C(N(C)C(CNS(c1cc(Cl)ccc1)(=O)=O)=CC1=O)=C1O)=O Chemical compound CNC(C(N(C)C(CNS(c1cc(Cl)ccc1)(=O)=O)=CC1=O)=C1O)=O WCDYYZYJUCSUFC-UHFFFAOYSA-N 0.000 description 1
- AQVRQXCCLOCMKK-UHFFFAOYSA-N Cc(cc1)ccc1S(NCC(N(C)C(C(O)=O)=C1O)=CC1=O)(=O)=O Chemical compound Cc(cc1)ccc1S(NCC(N(C)C(C(O)=O)=C1O)=CC1=O)(=O)=O AQVRQXCCLOCMKK-UHFFFAOYSA-N 0.000 description 1
- RTRZUYFXCIQSGP-UHFFFAOYSA-N Cc(cc1)ccc1S(NCC(OC(CO)=C1O)=CC1=O)(=O)=O Chemical compound Cc(cc1)ccc1S(NCC(OC(CO)=C1O)=CC1=O)(=O)=O RTRZUYFXCIQSGP-UHFFFAOYSA-N 0.000 description 1
- LWWNNIQFLLAKKY-UHFFFAOYSA-N Cc(cccc1)c1S(NCC(OC(C(O)=O)=C1OCc2ccccc2)=CC1=O)(=O)=O Chemical compound Cc(cccc1)c1S(NCC(OC(C(O)=O)=C1OCc2ccccc2)=CC1=O)(=O)=O LWWNNIQFLLAKKY-UHFFFAOYSA-N 0.000 description 1
- HWJTXLRWPZUAMU-UHFFFAOYSA-N O=C1C(OCc2ccccc2)=COC(CNS(c(cc2)ccc2Cl)(=O)=O)=C1 Chemical compound O=C1C(OCc2ccccc2)=COC(CNS(c(cc2)ccc2Cl)(=O)=O)=C1 HWJTXLRWPZUAMU-UHFFFAOYSA-N 0.000 description 1
- YQFAIDGZKRWCQC-UHFFFAOYSA-N O=CC(OC(CNS(c1cc(Cl)ccc1)(=O)=O)=CC1=O)=C1OCc1ccccc1 Chemical compound O=CC(OC(CNS(c1cc(Cl)ccc1)(=O)=O)=CC1=O)=C1OCc1ccccc1 YQFAIDGZKRWCQC-UHFFFAOYSA-N 0.000 description 1
- CSKNSYBAZOQPLR-UHFFFAOYSA-N O=S(c1ccccc1)(Cl)=O Chemical compound O=S(c1ccccc1)(Cl)=O CSKNSYBAZOQPLR-UHFFFAOYSA-N 0.000 description 1
- NMLHMQOXIGACAD-UHFFFAOYSA-N OC(C(OC(CNS(c(cc1)ccc1Cl)(=O)=O)=CC1=O)=C1OCc1ccccc1)=O Chemical compound OC(C(OC(CNS(c(cc1)ccc1Cl)(=O)=O)=CC1=O)=C1OCc1ccccc1)=O NMLHMQOXIGACAD-UHFFFAOYSA-N 0.000 description 1
- WJIWILHCZWAPQV-UHFFFAOYSA-N OC1=COC(CNS(c2ccccc2)(=O)=O)=CC1=O Chemical compound OC1=COC(CNS(c2ccccc2)(=O)=O)=CC1=O WJIWILHCZWAPQV-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/81—Amides; Imides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4412—Non condensed pyridines; Hydrogenated derivatives thereof having oxo groups directly attached to the heterocyclic ring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/79—Acids; Esters
Definitions
- the present invention relates to a compound having the general formula (II), optionally in the form of a pharmaceutically acceptable salt, solvate, polymorph, codrug, cocrystal, prodrug, tautomer, racemate, enantiomer, or diastereomer or mixture thereof,
- H5N 1 and related highly pathogenic avian influenza viruses could acquire mutations rendering them more easily transmissible between humans or the new A/H 1 N1 could become more virulent and only a single point mutation would be enough to confer resistance to oseltamivir (Neumann et al., Nature, 2009 (18; 459(7249) 931 -939)); as many seasonal H1 N1 strains have recently done (Dharan et al., The Journal of the American Medical Association, 2009 Mar 1 1 ; 301 (10), 1034-1041 ; Moscona et al., The New England Journal of Medicine, 2009 (Mar 5;360(10) pp 953-956)).
- the delay in generating and deploying a vaccine ( ⁇ 6 months in the relatively favourable case of A/H1 N1 and still not a solved problem for H5N1 ) could have been catastrophically costly in human lives and societal disruption.
- amantadine and rimantadine target the viral M2 ion channel protein, which is located in the viral membrane interfering with the uncoating of the virus particle inside the cell.
- they have not been extensively used due to their side effects and the rapid development of resistant virus mutants (Magden, J. et al., (2005), Appl. Microbiol. Biotechnol., 66, pp. 612-621 ).
- more unspecific viral drugs, such as ribavirin have been shown to work for treatment of influenza and other virus infections (Eriksson, B. et al., (1977), Antimicrob. Agents Chemother., 1 1 , pp. 946-951 ).
- Influenza virus as well as Thogotovirus and isavirus belong to the family of Orthomyxoviridae which, as well as the family of the Bunyaviridae, including the Hantavirus, Nairovirus, Orthobunyavirus, and Phlebovirus, amongst others, are negative stranded RNA viruses.
- RNA dependent RNA polymerase which carries out (i) the initial copying of the single-stranded negative-sense viral RNA (vRNA) into viral mRNAs (i.e. transcription) and (ii) the vRNA replication.
- This enzyme a trimeric complex composed of subunits PA, PB1 and PB2, is central to the life cycle of the virus since it is responsible for the replication and transcription of viral RNA.
- a 5' cap is a modified guanine nucleotide that has been added to the 5' end of a messenger RNA.
- the 5' cap (also termed an RNA cap or RNA m7G cap) consists of a terminal 7-methylguanosine residue which is linked through a 5'-5'-triphosphate bond to the first transcribed nucleotide.
- the viral polymerase binds to the 5' RNA cap of cellular mRNA molecules and cleaves the RNA cap together with a stretch of 10 to 15 nucleotides.
- the capped RNA fragments then serve as primers for the synthesis of viral mRNA (Plotch, S. J. et al., (1981 ), Cell, 23, pp.
- the polymerase complex seems to be an appropriate antiviral drug target since it is essential for synthesis of viral mRNA and viral replication and contains several functional active sites likely to be significantly different from those found in host cell proteins (Magden, J. et al., (2005), Appl. Microbiol. Biotechnol., 66, pp. 612-621 ). Thus, for example, there have been attempts to interfere with the assembly of polymerase subunits by a 25-amino-acid peptide resembling the PA-binding domain within PB1 (Ghanem, A. et al., (2007), J. Virol., 81 , pp. 7801 -7804).
- nucleoside analogs such as 2'-deoxy-2'-fluoroguanosine (Tisdale, M. et al., (1995), Antimicrob. Agents Chemother., 39, pp. 2454-2458). It is an object of the present invention to identify further compounds which are effective against viral diseases and which have improved pharmacological properties.
- the present invention provides a compound having the general formula (II).
- a compound having the general formula (II) encompasses pharmaceutically acceptable salts, solvates, polymorphs, prodrugs, codrugs, cocrystals, tautomers, racemates, enantiomers, or diastereomers or mixtures thereof unless mentioned otherwise.
- a further embodiment of the present invention relates to a pharmaceutical composition
- a pharmaceutical composition comprising a compound having the general formula (II) and optionally one or more pharmaceutically acceptable excipient(s) and/or carrier(s).
- the compounds having the general formula (II) are useful for treating, ameliorating or preventing viral diseases.
- alkyl refers to a saturated straight or branched carbon chain.
- cycloalkyl represents a cyclic version of “alkyl”.
- cycloalkyl is also meant to include bicyclic, tricyclic and polycyclic versions thereof. Unless specified otherwise, the cycloalkyl group can have 3 to 12 carbon atoms.
- “Hal” or “halogen” represents F, CI, Br and I.
- "3- to 7-membered carbo- or heterocyclic ring” refers to a three-, four-, five-, six- or seven- membered ring wherein none, one or more of the carbon atoms in the ring have been replaced by 1 or 2 (for the three-membered ring), 1 , 2 or 3 (for the four-membered ring) 1 , 2,
- heteroatoms are selected from O, N and S.
- aryl preferably refers to an aromatic monocyclic ring containing 6 carbon atoms, an aromatic bicyclic ring system containing 10 carbon atoms or an aromatic tricyclic ring system containing 14 carbon atoms. Examples are phenyl, naphthyl or anthracenyl, preferably phenyl.
- heteroaryl preferably refers to a five-or six-membered aromatic ring wherein one or more of the carbon atoms in the ring have been replaced by 1 , 2, 3, or 4 (for the five- membered ring) or 1 , 2, 3, 4, or 5 (for the six-membered ring) of the same or different heteroatoms, whereby the heteroatoms are selected from O, N and S.
- heteroaryl group examples include pyrrole, pyrrolidine, oxolane, furan, imidazolidine, imidazole, pyrazole, oxazolidine, oxazole, thiazole, piperidine, pyridine, morpholine, piperazine, and dioxolane.
- hydrocarbon group which contains from 5 to 20 carbon atoms and optionally 1 to 4 heteroatoms selected from O, N and S and which contains at least one ring refers to any group having 5 to 20 carbon atoms and optionally 1 to 4 heteroatoms selected from O, N and 2 as long as the group contains at least one ring.
- the term is also meant to include bicyclic, tricyclic and polycyclic versions thereof. If more than one ring is present, they can be separate from each other or be annelated.
- the ring(s) can be either carbocyclic or heterocyclic and can be saturated, unsaturated or aromatic.
- these groups include -(optionally substituted C 3 _7 cycloalkyl), -(optionally substituted aryl) wherein the aryl group can be, for example, phenyl, -(optionally substituted biphenyl), adamantyl, -(C 3-7 cycloalkyl)-aryl as well as the corresponding compounds with a linker.
- a compound or moiety is referred to as being "optionally substituted", it can in each instance include 1 or more of the indicated substituents, whereby the substituents can be the same or different.
- pharmaceutically acceptable salt refers to a salt of a compound of the present invention.
- suitable pharmaceutically acceptable salts include acid addition salts which may, for example, be formed by mixing a solution of compounds of the present invention with a solution of a pharmaceutically acceptable acid such as hydrochloric acid, sulfuric acid, fumaric acid, maleic acid, succinic acid, acetic acid, benzoic acid, citric acid, tartaric acid, carbonic acid or phosphoric acid.
- suitable pharmaceutically acceptable salts thereof may include alkali metal salts (e.g., sodium or potassium salts); alkaline earth metal salts (e.g., calcium or magnesium salts); and salts formed with suitable organic ligands (e.g., ammonium, quaternary ammonium and amine cations formed using counteranions such as halide, hydroxide, carboxylate, sulfate, phosphate, nitrate, alkyl sulfonate and aryl sulfonate).
- alkali metal salts e.g., sodium or potassium salts
- alkaline earth metal salts e.g., calcium or magnesium salts
- suitable organic ligands e.g., ammonium, quaternary ammonium and amine cations formed using counteranions such as halide, hydroxide, carboxylate, sulfate, phosphate, nitrate, alkyl sulfonate and aryl sul
- compositions include, but are not limited to, acetate, adipate, alginate, ascorbate, aspartate, benzenesulfonate, benzoate, bicarbonate, bisulfate, bitartrate, borate, bromide, butyrate, calcium edetate, camphorate, camphorsulfonate, camsylate, carbonate, chloride, citrate, clavulanate, cyclopentanepropionate, digluconate, dihydrochloride, dodecylsulfate, edetate, edisylate, estolate, esylate, ethanesulfonate, formate, fumarate, gluceptate, glucoheptonate, gluconate, glutamate, glycerophosphate, glycolylarsanilate, hemisulfate, heptanoate, hexanoate, hexylresorcinate
- the structure can contain solvent molecules.
- the solvents are typically pharmaceutically acceptable solvents and include, among others, water (hydrates) or organic solvents. Examples of possible solvates include ethanolates and iso-propanolates.
- codrug refers to two or more therapeutic compounds bonded via a covalent chemical bond.
- a detailed definition can be found, e.g., in N. Das et al., European Journal of Pharmaceutical Sciences, 41 , 2010, 571-588.
- cocrystal refers to a multiple component crystal in which all components are solid under ambient conditions when in their pure form. These components co-exist as a stoichiometric or non-stoichometric ratio of a target molecule or ion (i.e., compound of the present invention) and one or more neutral molecular cocrystal formers.
- the compounds of the present invention can also be provided in the form of a prodrug, namely a compound which is metabolized in vivo to the active metabolite.
- Suitable prodrugs are, for instance, esters. Specific examples of suitable groups are given, among others, in US 2007/0072831 in paragraphs [0082] to [01 18] under the headings prodrugs and protecting groups.
- Preferred examples of the prodrug include compounds in which R 20 is replaced by:
- the group R is H or Ci_ 6 alkyl.
- the present invention provides a compound having the general formula (II).
- the present invention provides a compound having the general formula (II) in which the following definitions apply. r20
- R is -H , a -Ci_6 alkyl group or a -C(0)-Ci_ 6 alkyl group.
- R 20 is -H, or -(optionally substituted Ci_ 6 alkyl); more preferably -H.
- R 21 is -H, a -Ci_6 alkyl group, or a -Ci_ 6 alkyl group which is substituted by one or more halogen atoms; preferably R 21 is -H.
- R 22 is -H, a -Ci_6 alkyl group, or a -Ci_ 6 alkyl group which is substituted by one or more halogen atoms; preferably R 22 is -H.
- R 21 and R 22 can be joined together to form a 3- to 7-membered carbo- or heterocyclic ring.
- R ⁇ is -R , or -X -R . In one embodiment R ⁇ is -R . In an alternative embodiment, R ⁇ is
- _ X 20_ R 26 R is H, or a Ci_ 6 alkyl group.
- R is -H, -(optionally substituted Ci_ 6 alkyl), -(optionally substituted C3-7 cycloalkyl), - (optionally substituted aryl), -C1- alkyl— (optionally substituted C3-7 cycloalkyl), or -C1- alkyl— (optionally substituted aryl).
- R 25 is -H or -(optionally substituted Ci_ 6 alkyl).
- R 26 is -(optionally substituted hydrocarbon group which contains from 5 to 20 carbon atoms and optionally 1 to 4 heteroatoms selected from O, N and S and which contains at least one ring).
- the at least one ring is aromatic such as an aryl or heteroaryl ring.
- R 26 is a hydrocarbon group which contains from 5 to 20 carbon atoms and optionally 1 to 4 heteroatoms and which contains at least two rings, wherein the hydrocarbon group can be optionally substituted.
- at least one of the at least two rings is aromatic such as an aryl or heteroaryl ring.
- Preferred examples of R 26 can be selected from the group consisting of
- X is absent, CH 2 , NH, C(0)NH, S or O.
- Y is CH 2 .
- X and Y can be joined together to form an annulated, carbo- or heterocylic 3- to 8-membered ring which can be saturated or unsaturated.
- Specific examples of X-Y include -CH 2 -, -CH 2 -CH 2 -, -0-, and -NH-.
- Z is O or S.
- R is independently selected from -H, -Ci_ 6 alkyl, -CF 3 , -halogen, -CN, -OH, and -0-Ci_6 alkyl.
- R 27 is -H, -Ci_6 alkyl, or -(CH 2 CH 2 0) r H; preferably R 27 is -H, or -Ci_ 6 alkyl.
- R 28 is -H, or -Ci_6 alkyl.
- R is independently selected from -Ci_ 6 alkyl, -C(0)-Ci_ 6 alkyl, -Hal, -CF 3 , -CN, -COOR 27 , -OR 27 , -(CH 2 ) q NR 27 R 28 , -C(0)-NR 27 R 28 , and -NR 27 -C(0)-Ci_6 alkyl.
- R is -Hal, -CF 3 , or -CN, more preferably -Hal, or -CF 3 .. q is 0 to 4.
- r is 1 to 3.
- the optional substituent of the alkyl group, aryl group, hydrocarbon group and/or cycloalkyi group is selected from the group consisting of one or more substituents R, which includes -Ci_6 alkyl, -C(0)-Ci_ 6 alkyl, -Hal, -CF 3 , -CN, -COOR 27 , -OR 27 , -(CH 2 ) q NR 27 R 28 , -C(O)- NR 27 R 28 , and -NR 27 -C(0)-Ci_ 6 alkyl.
- the optional substituent of the aryl group, hydrocarbon group and/or cycloalkyi group is -halogen (preferably F), -OCH 3 or -CN.
- the optional substituent of the alkyl group is selected from the group consisting of halogen, -CN, -NR 28 R 28 (wherein each R 28 is chosen independently of each other), -OH, and -0-Ci_6 alkyl.
- the substituent of the alkyl group is -halogen, more preferably F.
- the present inventors have surprisingly found that the compounds of the present invention which have a bulky moiety R 23 have improved pharmacological properties compared to corresponding compounds which have a smaller moiety R 23 .
- the viral polymerase protein has a pocket for binding and that the bulky moiety R 23 of the compounds of the present invention fills this pocket to a larger extent.
- the larger moiety R 23 is able to provide more hydrophobic interaction with the pocket than smaller moieties such as methyl.
- the compounds of the present invention can be administered to a patient in the form of a pharmaceutical composition which can optionally comprise one or more pharmaceutically acceptable excipient(s) and/or carrier(s).
- the compounds of the present invention can be administered by various well known routes, including oral, rectal, intragastrical, intracranial and parenteral administration, e.g. intravenous, intramuscular, intranasal, intradermal, subcutaneous, and similar administration routes. Oral, intranasal and parenteral administration are particularly preferred. Depending on the route of administration different pharmaceutical formulations are required and some of those may require that protective coatings are applied to the drug formulation to prevent degradation of a compound of the invention in, for example, the digestive tract.
- a compound of the invention is formulated as a syrup, an infusion or injection solution, a spray, a tablet, a capsule, a capslet, lozenge, a liposome, a suppository, a plaster, a band-aid, a retard capsule, a powder, or a slow release formulation.
- the diluent is water, a buffer, a buffered salt solution or a salt solution and the carrier preferably is selected from the group consisting of cocoa butter and vitebesole.
- Particular preferred pharmaceutical forms for the administration of a compound of the invention are forms suitable for injectionable use and include sterile aqueous solutions or dispersions and sterile powders for the extemporaneous preparation of sterile injectable solutions or dispersions. In all cases the final solution or dispersion form must be sterile and fluid. Typically, such a solution or dispersion will include a solvent or dispersion medium, containing, for example, water-buffered aqueous solutions, e.g. biocompatible buffers, ethanol, polyol, such as glycerol, propylene glycol, polyethylene glycol, suitable mixtures thereof, surfactants or vegetable oils.
- a compound of the invention can also be formulated into liposomes, in particular for parenteral administration. Liposomes provide the advantage of increased half life in the circulation, if compared to the free drug and a prolonged more even release of the enclosed drug.
- Sterilization of infusion or injection solutions can be accomplished by any number of art recognized techniques including but not limited to addition of preservatives like anti-bacterial or anti-fungal agents, e.g. parabene, chlorobutanol, phenol, sorbic acid or thimersal. Further, isotonic agents, such as sugars or salts, in particular sodium chloride, may be incorporated in infusion or injection solutions.
- Production of sterile injectable solutions containing one or several of the compounds of the invention is accomplished by incorporating the respective compound in the required amount in the appropriate solvent with various ingredients enumerated above as required followed by sterilization. To obtain a sterile powder the above solutions are vacuum-dried or freeze-dried as necessary.
- Preferred diluents of the present invention are water, physiological acceptable buffers, physiological acceptable buffer salt solutions or salt solutions.
- Preferred carriers are cocoa butter and vitebesole.
- Excipients which can be used with the various pharmaceutical forms of a compound of the invention can be chosen from the following non-limiting list: a) binders such as lactose, mannitol, crystalline sorbitol, dibasic phosphates, calcium phosphates, sugars, microcrystalline cellulose, carboxymethyl cellulose, hydroxyethyl cellulose, polyvinyl pyrrolidone and the like;
- lubricants such as magnesium stearate, talc, calcium stearate, zinc stearate, stearic acid, hydrogenated vegetable oil, leucine, glycerids and sodium stearyl fumarates
- disintegrants such as starches, croscarmellose, sodium methyl cellulose, agar, bentonite, alginic acid, carboxymethyl cellulose, polyvinyl pyrrolidone and the like.
- the formulation is for oral administration and the formulation comprises one or more or all of the following ingredients: pregelatinized starch, talc, povidone K 30, croscarmellose sodium, sodium stearyl fumarate, gelatin, titanium dioxide, sorbitol, monosodium citrate, xanthan gum, titanium dioxide, flavoring, sodium benzoate and saccharin sodium.
- a compound of the invention may be administered in the form of a dry powder inhaler or an aerosol spray from a pressurized container, pump, spray or nebulizer with the use of a suitable propellant, e.g., dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane, a hydrofluoro- alkane such as 1 ,1 ,1 ,2-tetrafluoroethane (HFA 134ATM) or 1 ,1 ,1 ,2,3,3,3-heptafluoropropane (HFA 227EATM), carbon dioxide, or another suitable gas.
- a suitable propellant e.g., dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane, a hydrofluoro- alkane such as 1 ,1 ,1 ,2-tetrafluoroethane (HFA
- the pressurized container, pump, spray or nebulizer may contain a solution or suspension of the compound of the invention, e.g., using a mixture of ethanol and the propellant as the solvent, which may additionally contain a lubricant, e.g., sorbitan trioleate.
- a lubricant e.g., sorbitan trioleate.
- Other suitable excipients can be found in the Handbook of Pharmaceutical Excipients, published by the American Pharmaceutical Association, which is herein incorporated by reference. It is to be understood that depending on the severity of the disorder and the particular type which is treatable with one of the compounds of the invention, as well as on the respective patient to be treated, e.g. the general health status of the patient, etc., different doses of the respective compound are required to elicit a therapeutic or prophylactic effect.
- the dosage of a compound of the invention in the therapeutic or prophylactic use of the invention should be in the range of about 0.1 mg to about 1 g of the active ingredient (i.e. compound of the invention) per kg body weight.
- a compound of the invention is administered to a subject in need thereof in an amount ranging from 1 .0 to 500 mg/kg body weight, preferably ranging from 1 to 200 mg/kg body weight.
- the duration of therapy with a compound of the invention will vary, depending on the severity of the disease being treated and the condition and idiosyncratic response of each individual patient.
- from 10 mg to 200 mg of the compound are orally administered to an adult per day, depending on the severity of the disease and/or the degree of exposure to disease carriers.
- the pharmaceutically effective amount of a given composition will also depend on the administration route. In general, the required amount will be higher if the administration is through the gastrointestinal tract, e.g., by suppository, rectal, or by an intragastric probe, and lower if the route of administration is parenteral, e.g., intravenous.
- a compound of the invention will be administered in ranges of 50 mg to 1 g/kg body weight, preferably 10 mg to 500 mg/kg body weight, if rectal or intragastric administration is used and in ranges of 1 to 100 mg/kg body weight if parenteral administration is used. For intranasal administration, 1 to 100 mg/kg body weight are envisaged.
- a person is known to be at risk of developing a disease treatable with a compound of the invention, prophylactic administration of the biologically active blood serum or the pharmaceutical composition according to the invention may be possible.
- the respective compound of the invention is preferably administered in above outlined preferred and particular preferred doses on a daily basis. Preferably, from 0.1 mg to 1 g/kg body weight once a day, preferably 10 to 200 mg/kg body weight. This administration can be continued until the risk of developing the respective viral disorder has lessened. In most instances, however, a compound of the invention will be administered once a disease/disorder has been diagnosed. In these cases it is preferred that a first dose of a compound of the invention is administered one, two, three or four times daily.
- the compounds of the present invention are particularly useful for treating, ameliorating, or preventing viral diseases.
- the type of viral disease is not particularly limited.
- examples of possible viral diseases include, but are not limited to, viral diseases which are caused by Poxviridae, Herpesviridae, Adenoviridae, Papillomaviridae, Polyomaviridae, Parvoviridae, Hepadnaviridae, Retroviridae, Reoviridae, Filoviridae, Paramyxoviridae, Rhabdoviridae, Orthomyxoviridae, Bunyaviridae, Arenaviridae, Coronaviridae, Picornaviridae, Hepeviridae, Caliciviridae, Astroviridae, Togaviridae, Flaviviridae, Deltavirus, Bornaviridae, and prions.
- viral diseases which are caused by Herpesviridae, Retroviridae, Filoviridae, Paramyxoviridae, Rhabdoviridae, Orthomyxoviridae, Bunyaviridae, Arenaviridae, Coronaviridae, Picornaviridae, Togaviridae, Flaviviridae, more preferably viral diseases which are caused by orthomyxoviridae.
- Herpesviridae Herpes simplex virus
- Hepadnaviridae Hepatitis B virus Family Virus (preferred examples)
- Picornaviridae Human enterovirus types A-D (Poliovirus, Echovirus,
- Hepeviridae Hepatitis E virus Family Virus (preferred examples)
- the compounds of the present invention are capable of inhibiting endonuclease activity, particularly that of influenza virus. More specifically it is assumed that they directly interfere with the N-terminal part of the influenza virus PA protein, which harbors endonuclease activity and is essential for influenza virus replication. Influenza virus replication takes place inside the cell within the nucleus.
- compounds designed to inhibit PA endonuclease activity need to cross both the cellular and the nuclear membrane, a property which strongly depends on designed-in physico-chemical properties of the compounds.
- the present invention shows that the claimed compounds have in vitro endonuclease inhibitory activity and have antiviral activity in vitro in cell-based assays.
- a possible measure of the in vitro endonuclease inhibitory activity of the compounds having the formula (II) is the FRET (fluorescence-resonance energy transfer)-based endonuclease activity assay disclosed herein.
- the compounds exhibit a % reduction of at least about 50 % at 25 ⁇ in the FRET assay.
- the % reduction is the % reduction of the initial reaction velocity (vO) measured as fluorescence increase of a dual-labelled RNA substrate cleaved by the influenza virus endonuclease subunit (PA-Nter) upon compound treatment compared to untreated samples.
- the compounds exhibit an IC 50 of less than about 40 ⁇ , more preferably less than about 20 ⁇ , in this assay.
- the half maximal inhibitory concentration (IC 50 ) is a measure of the effectiveness of a compound in inhibiting biological or biochemical function and was calculated from the initial reaction velocities (vO) in a given concentration series ranging from maximum 100 ⁇ to at least 2 nM.
- the compounds having the general formula (II) can be used in combination with one or more other medicaments.
- the type of the other medicaments is not particularly limited and will depend on the disorder to be treated.
- the other medicament will be a further medicament which is useful in treating, ameliorating or preventing a viral disease, more preferably a further medicament which is useful in treating, ameliorating or preventing influenza that has been caused by influenza virus infection and conditions associated with this viral infection such as viral pneumonia or secondary bacterial pneumonia and medicaments to treat symptoms such as chills, fever, sore throat, muscle pains, severe headache, coughing, weakness and fatigue.
- the compounds having the general formula (I) can be used in combination with anti-inflammatories.
- the combination with endonuclease and cap-binding inhibitors are not particularly limited and can be any endonuclease inhibitor, particularly any viral endonuclease inhibitor.
- Preferred endonuclease inhibitors are those as defined in the US applications with the serial numbers 61/550,045 (filed on October 21 , 201 1 ), 61/650,713 (filed on May 23, 2012), 61/650,725 (filed on May 23, 2012) and 61/679,968 (filed on August 6, 2012).
- the complete disclosure of these applications is incorporated herein by reference.
- all descriptions with respect to the general formula of the compounds according to these US applications, the preferred embodiments of the various substituents as well as the medical utility and advantages of the compounds are incorporated herein by reference.
- the cap-binding inhibitors are not particularly limited either and can be any cap-binding inhibitor, particularly any viral cap-binding inhibitor.
- Preferred cap-binding inhibitors are those having the general formula (II) as defined in US application 61/550,057 (filed on October 21 , 201 1 ) and/or the compounds disclosed in WO201 1/000566, the complete disclosure of which is incorporated by reference.
- all descriptions with respect to the general formula of the compounds according to US 61/550,057 or WO201 1/000566, the preferred embodiments of the various substituents as well as the medical utility and advantages of the compounds are incorporated herein by reference.
- M2 ion channel inhibitors adamantanes
- neuraminidase inhibitors e.g. oseltamivir
- H3N2 strains are resistant to adamantanes (rimantadine and amantadine), and for oseltamivir, the most widely prescribed neuraminidase inhibitor (NAI), the WHO reported on significant emergence of influenza A/H1 N1 resistance starting in the influenza season 2007/2008; and for the second and third quarters of 2008 in the southern hemisphere. Even more serious numbers were published for the fourth quarter of 2008 (northern hemisphere) where 95% of all tested isolates revealed no oseltamivir- susceptibility. Considering the fact that now most national governments have been stockpiling NAIs as part of their influenza pandemic preparedness plan, it is obvious that the demand for new, effective drugs is growing significantly.
- NAI neuraminidase inhibitor
- Influenza virus polymerase inhibitors are novel drugs targeting the transcription activity of the polymerase. Selective inhibitors against the cap-binding and endonuclease active sites of the viral polymerase severely attenuate virus infection by stopping the viral reproductive cycle. These two targets are located within distinct subunits of the polymerase complex and thus represent unique drug targets. Due to the fact that both functions are required for the so-called "cap-snatching" mechanism which is essential for viral transcription, concurrent inhibition of both functions is expected to act highly synergistically. This highly efficient drug combination would result in lower substance concentrations and hence improved dose-response-relationships and better side effect profiles.
- the combination of an endonuclease inhibitor and a cap-binding inhibitor or a dual specific polymerase inhibitor targeting both the endonuclease active site and the cap- binding domain would be effective against virus strains resistant against adamantanes and neuraminidase inhibitors and moreover combine the advantage of low susceptibility to resistance generation with activity against a broad range of virus strains.
- the combination of inhibitors of different antiviral targets (particularly targeting influenza virus) focusing on the combination with (preferably influenza virus) polymerase inhibitors as dual or multiple combination therapy.
- Influenza virus polymerase inhibitors are novel drugs targeting the transcription and replication activity of the polymerase. Selective inhibitors against the viral polymerase severely attenuate virus infection by stopping the viral reproductive cycle.
- the combination of a polymerase inhibitor specifically addressing a viral intracellular target with an inhibitor of a different antiviral target is expected to act highly synergistically. This is based on the fact that these different types of antiviral drugs exhibit completely different mechanisms of action requiring different pharmacokinetics properties which act advantageously and synergistically on the antiviral efficacy of the combination.
- At least one compound selected from the first group of polymerase inhibitors e.g., cap-binding and endonuclease inhibitors
- at least one compound selected from the second group of polymerase inhibitors is combined with at least one compound selected from the second group of polymerase inhibitors.
- the first group of polymerase inhibitors which can be used in this type of combination therapy includes, but is not limited to, the compounds having the formula (II).
- the second group of polymerase inhibitors which can be used in this type of combination therapy includes, but is not limited to, the compounds having the general formula (I) as defined in the US application with the serial number 61/550,045 filed on October 21 , 201 1 , the compounds having the general formula (II) as defined in US application 61/550,057 filed on October 21 , 201 1 , the compounds disclosed in WO 201 1/000566, WO 2010/1 10231 , WO 2010/1 10409, WO 2006/030807 or US 5,475,109 as well as flutimide and analogues, favipiravir and analogues, epigallocatechin gallate and analogues, as well as nucleoside analogs such as ribavirine.
- the combination of polymerase inhibitors with neuraminidase inhibitors include, but is not limited to, the compounds having the general formula (I) as defined in the US application with the serial number 61/550,045 filed on October 21 ,
- Influenza virus polymerase inhibitors are novel drugs targeting the transcription and replication activity of the polymerase.
- the combination of a polymerase inhibitor specifically addressing a viral intracellular target with an inhibitor of a different extracellular antiviral target, especially the (e.g., viral) neuraminidase is expected to act highly synergistically. This is based on the fact that these different types of antiviral drugs exhibit completely different mechanisms of action requiring different pharmacokinetic properties which act advantageously and synergistically on the antiviral efficacy of the combination.
- At least one compound selected from the above mentioned first group of polymerase inhibitors is combined with at least one neuraminidase inhibitor.
- the neuraminidase inhibitor (particularly influenza neuramidase inhibitor) is not specifically limited. Examples include zanamivir, oseltamivir, peramivir, KDN DANA, FANA, and cyclopentane derivatives.
- Influenza virus polymerase inhibitors are novel drugs targeting the transcription and replication activity of the polymerase.
- the combination of a polymerase inhibitor specifically addressing a viral intracellular target with an inhibitor of a different extracellular and cytoplasmic antiviral target, especially the viral M2 ion channel, is expected to act highly synergistically. This is based on the fact that these different types of antiviral drugs exhibit completely different mechanisms of action requiring different pharmacokinetic properties which act advantageously and synergistically on the antiviral efficacy of the combination.
- This highly efficient drug combination would result in lower substance concentrations and hence improved dose-response-relationships and better side effect profiles.
- advantages described above for polymerase inhibitors would prevail for combinations of inhibitors of different antiviral targets with polymerase inhibitors.
- At least one compound selected from the above mentioned first group of polymerase inhibitors is combined with at least one M2 channel inhibitor.
- the M2 channel inhibitor (particularly influenza M2 channel inhibitor) is not specifically limited. Examples include amantadine and rimantadine.
- Influenza virus polymerase inhibitors are novel drugs targeting the transcription and replication activity of the polymerase.
- the combination of a polymerase inhibitor specifically addressing a viral intracellular target, with an inhibitor of a different host-cell target, especially alpha glucosidase, is expected to act highly synergistically. This is based on the fact that these different types of antiviral drugs exhibit completely different mechanisms of action requiring different pharmacokinetic properties which act advantageously and synergistically on the antiviral efficacy of the combination.
- At least one compound selected from the above-mentioned first group of polymerase inhibitors is combined with at least one alpha glucosidase inhibitor.
- the alpha glucosidase inhibitor is not specifically limited. Examples include the compounds described in Chang et al., Antiviral Research 201 1 , 89, 26-34. ⁇ The combination of polymerase inhibitors with ligands of other influenza targets
- Influenza virus polymerase inhibitors are novel drugs targeting the transcription and replication activity of the polymerase.
- the combination of a polymerase inhibitor specifically addressing a viral intracellular target with an inhibitor of different extracellular, cytoplasmic or nucleic antiviral targets is expected to act highly synergistically. This is based on the fact that these different types of antiviral drugs exhibit completely different mechanisms of action requiring different pharmacokinetic properties which act advantageously and synergistically on the antiviral efficacy of the combination.
- At least one compound selected from the above mentioned first group of polymerase inhibitors is combined with at least one ligand of another influenza target.
- the ligand of another influenza target is not specifically limited.
- examples include compounds acting on the sialidase fusion protein (e.g., Fludase (DAS181 ), siRNAs and phosphorothioate oligonucleotides), signal transduction inhibitors (e.g., ErbB tyrosine kinase, Abl kinase family, MAP kinases, PKCa-mediated activation of ERK signalling) as well as interferon (inducers).
- sialidase fusion protein e.g., Fludase (DAS181 ), siRNAs and phosphorothioate oligonucleotides
- signal transduction inhibitors e.g., ErbB tyrosine kinase, Abl kinase family, MAP kinases, PKCa-mediated activation of ERK signalling
- interferon inducers
- influenza polymerase inhibitors preferably influenza polymerase inhibitors with a compound used as an adjuvant to minimize the symptoms of the disease
- antibiotics anti-inflammatory agents like COX inhibitors (e.g., COX-1/COX-2 inhibitors, selective COX-2 inhibitors), lipoxygenase inhibitors, EP ligands (particularly EP4 ligands), bradykinin ligands, and/or cannabinoid ligands (e.g., CB2 agonists)
- Influenza virus polymerase inhibitors are novel drugs targeting the transcription and replication activity of the polymerase..
- the combination of a polymerase inhibitor specifically addressing a viral intracellular target with a compound used as an adjuvance to minimize the symptoms of the disease address the causative and symptomatic pathological consequences of viral infection.
- This combination is expected to act synergistically because these different types of drugs exhibit completely different mechanisms of action requiring different pharmacokinetic properties which act advantageously and synergistically on the antiviral efficacy of the combination.
- influenza A virus IAV PA-Nter fragment (amino acids 1 - 209) harboring the influenza endonuclease activity was generated and purified as described in Dias et al., Nature 2009; Apr 16; 458(7240), 914-918.
- the protein was dissolved in buffer containing 20mM Tris pH 8.0, 100mM NaCI and 10mM ⁇ -mercaptoethanol and aliquots were stored at -20 °C.
- RNA oligo with 5 ' -FAM fluorophore and 3 ' -BHQ1 quencher was used as a substrate to be cleaved by the endonuclease activity of the PA-Nter. Cleavage of the RNA substrate frees the fluorophore from the quencher resulting in an increase of the fluorescent signal. All assay components were diluted in assay buffer containing 20mM Tris-HCI pH 8.0, 100mM NaCI, 1 mM MnCI 2 , 10mM MgCI 2 and 10mM ⁇ -mercaptoethanol. The final concentration of PA- Nter was 0.5 ⁇ and 1 .6 ⁇ RNA substrate.
- test compounds were dissolved in DMSO and generally tested at two concentrations or a concentration series resulting in a final plate well DMSO concentration of 0.5 %. In those cases where the compounds were not soluble at that concentration, they were tested at the highest soluble concentration.
- IC 50 half maximal inhibitory concentration
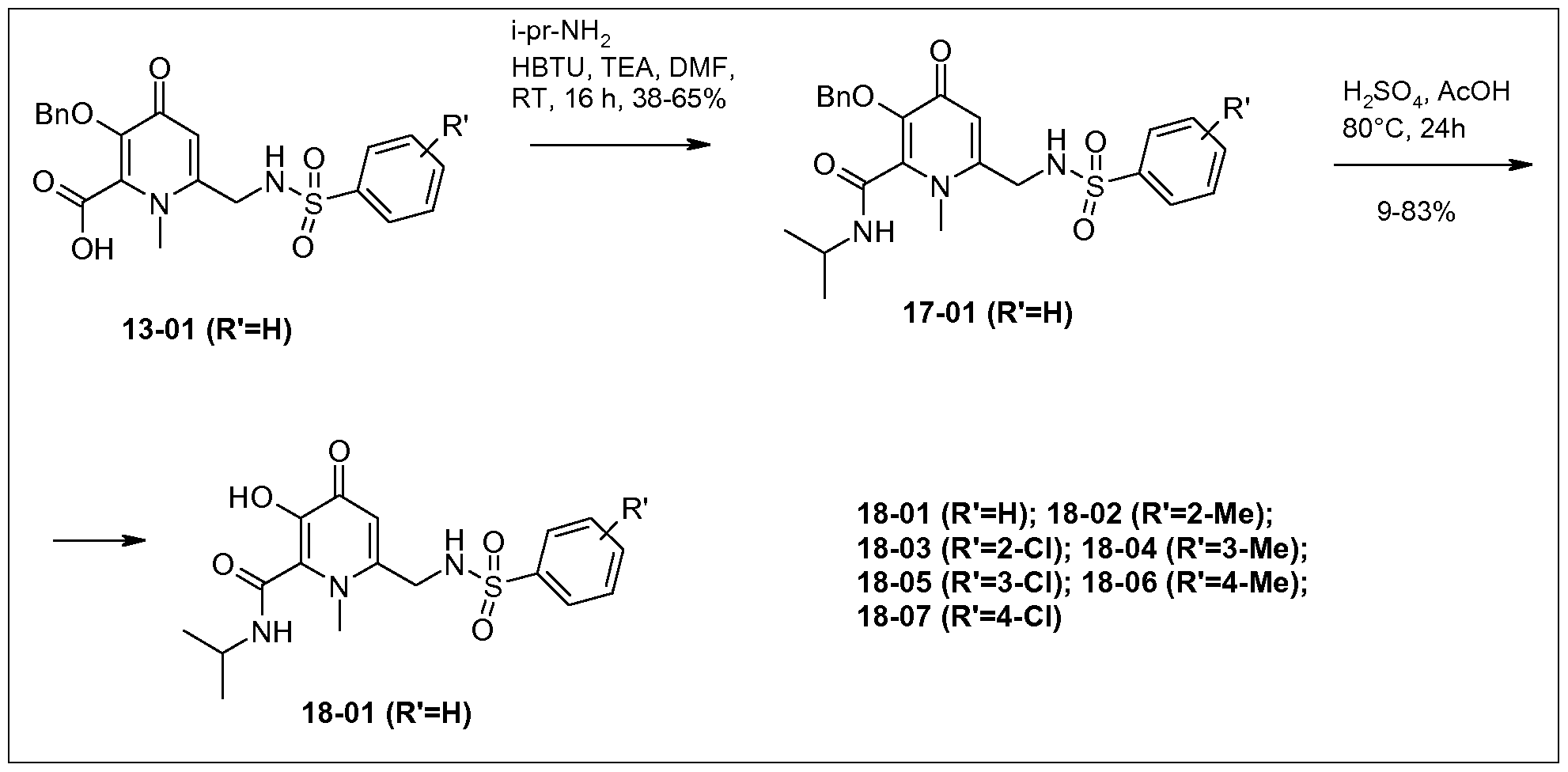
- N-(5-Hydroxy-4-oxo-4H-pyran-2-ylmethyl)-benzenesulfonamide (2.0 g, 5.39 mmol) was dissolved in acetic acid (25.0 mL) and sulfuric acid (0.058 mL) was added, then the reaction mixture was heated up to 80 °C for 24 h. After completion of the reaction, the mixture was cooled to room temperature and concentrated under vacuum. It was then diluted with water and extracted with ethyl acetate.
- 6-(benzene sulfonyl amino-methyl)-3-benzyloxy-4-oxo-4H-pyran-2- carboxylic acid (12-01 ) 640.0 mg, 1 .54 mmol
- MeOH 5.0 mL
- methylamine (2 M in methanol, 2.0 mL
- N-(5-Benzyloxy-4-oxo-4H-pyran-2-ylmethyl)-2-methyl-benzenesulfonamide N-(5-Benzyloxy-4-oxo-4H-pyran-2-ylmethyl)-2-methyl-benzenesulfonamide (7-02) (22.0 g, 64.32 %) was synthesized as a light brown solid from 2-aminomethyl-5-benzyloxy-pyran-4-one (5) (20.5 g, 88.74 mmol) and 2-methyl-benzenesulfonyl chloride (6-02) (20.23 g, 106.49 mmol) following the procedure described for N-(5-benzyloxy-4-oxo-4H-pyran-2-ylmethyl)- benzenesulfonamide (7-01 ).
- N-(5-Hydroxy-4-oxo-4H-pyran-2-ylmethyl)-2-methyl-benzenesulfonamide N-(5-Hydroxy-4-oxo-4H-pyran-2-ylmethyl)-2-methyl-benzenesulfonamide (8-02) (14.0 g, crude) was synthesized as a light brown solid from N-(5-benzyloxy-4-oxo-4H-pyran-2- ylmethyl)-2-methyl-benzenesulfonamide (7-02) (22.0 g, 57.14 mmol) following the procedure described for N-(5-hydroxy-4-oxo-4H-pyran-2-ylmethyl)-benzenesulfonamide (8-01 ). LC-MS: 296.2 (M+H).
- N-(5-Hydroxy-6-hydroxymethyl-4-oxo-4H-pyran-2-ylmethyl)-2-methyl-benzenesulfonamide N-(5-Hydroxy-6-hydroxymethyl-4-oxo-4 (9- 02) (9.0 g, 62.78 %) was synthesized as a white solid from N-(5-hydroxy-4-oxo-4H-pyran-2- ylmethyl)-2-methyl-benzenesulfonamide (8-02) (13.0 g, 44.06 mmol) following the procedure described for N-(5-hydroxy-6-hydroxymethyl-4-oxo-4H-pyran-2-ylmethyl)-benzenesulfonamide (9-01 ).
- N-(5-Benzyloxy-6-hydroxymethyl-4-oxo-4H-pyran-2-ylmethyl)-2-methyl-benzenesulfonamide N-(5-Benzyloxy-6-hydroxymethyl-4-oxo-4H-pyran-2-ylmethyl)-2-methyl-benzenesulfonamide (10-02) (4.7 g, 40.85 %) was synthesized as a white solid from N-(5-hydroxy-6-hydroxymethyl- 4-oxo-4H-pyran-2-ylmethyl)-2-methyl-benzenesulfonamide (9-02) (9.0 g, 27.69 mmol) following the procedure described for N-(5-benzyloxy-6-hydroxymethyl-4-oxo-4H-pyran-2- ylmethyl)-benzenesulfonamide (10-01 ).
- N-(5-Benzyloxy-6-formyl-4-oxo-4H-pyran-2-ylmethyl)-2-methyl-benzenesulfonamide N-(5-Benzyloxy-6-formyl-4-oxo-4H-pyran-2-ylmethyl)-2-methyl-benzenesulfonami (11 -02) (2.8 g, 66.92 %) was synthesized as a white solid from N-(5-benzyloxy-6-hydroxymethyl-4- oxo-4H-pyran-2-ylmethyl)-2-methyl-benzenesulfonamide (10-02) (4.2 g, 10.12 mmol) following the procedure described for N-(5-benzyloxy-6-formyl-4-oxo-4H-pyran-2-ylmethyl)- benzenesulfonamide (11 -01 ).
- N-(5-Benzyloxy-4-oxo-4H-pyran-2-ylmethyl)-2-chloro-benzenesulfonamide (7-03) (26.0 g, 59.19 %) was synthesized as a light brown solid from 2-aminomethyl-5-benzyloxy-pyran-4-one (5) (25.0 g, 108.2 mmol) and 2-chloro-benzenesulfonyl chloride (6-03) (27.2 g,129.87 mmol) following the procedure described for N-(5-benzyloxy-4-oxo-4H-pyran-2-ylmethyl)- benzenesulfonamide (7-01 ).
- LC-MS 406.0 (M+H).
- N-(5-Benzyloxy-6-hydroxymethyl-4-oxo-4H-pyran-2-ylmethyl)-2-chloro-benzenesulfonamide (10-03) (3.5 g, 35.0 %) was synthesized as a white solid from 2-chloro-N-(5-hydroxy-6- hydroxymethyl-4-oxo-4H-pyran-2-ylmethyl)-benzenesulfonamide (9-03) (8.0 g, 23.18 mmol) following the procedure described for N-(5-benzyloxy-6-hydroxymethyl-4-oxo-4H-pyran-2- ylmethyl)-benzenesulfonamide (10-01 ).
- N-(5-Benzyloxy-6-formyl-4-oxo-4H-pyran-2-ylmethyl)-2-chloro-benzenesulfonamide (11 -03) (2.8 g, 85.07 %) was synthesized as a brown sticky solid from N-(5-benzyloxy-6- hydroxymethyl-4-oxo-4H-pyran-2-ylmethyl)-2-chloro-benzenesulfonamide (10-03) (3.3 g, 7.58 mmol) following the procedure described for N-(5-benzyloxy-6-formyl-4-oxo-4H-pyran-2- ylmethyl)-benzenesulfonamide (11 -01 ).
- N-(5-Benzyloxy-4-oxo-4H-pyran-2-ylmethyl)-3-methyl-benzenesulfonamide (7-04) (24.0 g, 57.60 %) was synthesized as a light brown solid from 2-aminomethyl-5-benzyloxy-pyran-4-one (5) and 3-methyl-benzenesulfonyl chloride (6-04) (24.67 g, 129.87 mmol) following the procedure described for N-(5-benzyloxy-4-oxo-4H-pyran-2-ylmethyl)-benzenesulfonamide (7- 01 ).
- N-(5-Hydroxy-6-hydroxymethyl-4-oxo-4H-pyran-2-ylmethyl)-3-methyl-benzenesulfonamide (9- 04) (7.5 g, 45.34 %) was synthesized as a white solid from N-(5-hydroxy-4-oxo-4H-pyran-2- ylmethyl)-3-methyl-benzenesulfonamide (8-04) (15.0 g, 50.84 mmol) following the procedure described for N-(5-hydroxy-6-hydroxymethyl-4-oxo-4H-pyran-2-ylmethyl)-benzenesulfonamide (9-01 ).
- N-(5-Benzyloxy-6-hydroxymethyl-4-oxo-4H-pyran-2-ylmethyl)-3-methyl-benzenesulfonamide N-(5-Benzyloxy-6-hydroxymethyl-4-oxo-4H-pyran-2-ylmethyl)-3-methyl-benzenesulfonamide (10-04) (2.6 g, 27.12 %) was synthesized as a white solid from N-(5-hydroxy-6-hydroxymethyl- 4-oxo-4H-pyran-2-ylmethyl)-3-methyl-benzenesulfonamide (9-04) (7.5 g, 23.07 mmol) following the procedure described for N-(5-benzyloxy-6-hydroxymethyl-4-oxo-4H-pyran-2- ylmethyl)-benzenesulfonamide (10-01 ).
- N-(5-Benzyloxy-6-formyl-4-oxo-4H-pyran-2-ylmethyl)-3-methyl-benzenesulfonamide (11 -04) (1 .8 g, 75.28 %) was synthesized as a white solid from N-(5-benzyloxy-6-hydroxymethyl-4- oxo-4H-pyran-2-ylmethyl)-3-methyl-benzenesulfonamide (10-04) (2.4 g, 5.78 mmol) following the procedure described for N-(5-benzyloxy-6-formyl-4-oxo-4H-pyran-2-ylmethyl)- benzenesulfonamide (11 -01 ).
- N-(5-Benzyloxy-4-oxo-4H-pyran-2-ylmethyl)-3-chloro-benzenesulfonamide (7-05) (25.0 g, 56.92 %) was synthesized as a brown solid from 2-aminomethyl-5-benzyloxy-pyran-4-one (5) (25 g, 108.22 mmol) and 3-chloro-benzenesulfonyl chloride (6-05) (22.98 mL, 162.33 mmol) following the procedure described for N-(5-benzyloxy-4-oxo-4H-pyran-2-ylmethyl)- benzenesulfonamide (7-01 ).
- LC-MS 406.0 (M+H).
- N-(5-Benzyloxy-6-hydroxymethyl-4-oxo-4H-pyran-2-ylmethyl)-3-chloro-benzenesulfonamide N-(5-Benzyloxy-6-hydroxymethyl-4-oxo-4H-pyran-2-ylmethyl)-3-chloro-benzenesulfonamide (10-05) (2.2 g, 24.88 %) was synthesized as a white solid from 3-chloro-N-(5-hydroxy-6- hydroxymethyl-4-oxo-4H-pyran-2-ylmethyl)-benzenesulfonamide (9-05) (7.0 g, 20.29 mmol) following the procedure described for N-(5-benzyloxy-6-hydroxymethyl-4-oxo-4H-pyran-2- ylmethyl)-benzenesulfonamide (10-01 ).
- N-(5-Benzyloxy-6-formyl-4-oxo-4H-pyran-2-ylmethyl)-3-chloro-benzenesulfonamide (11 -05) (2.3 g, 82.36 %) was synthesized as a white solid from N-(5-benzyloxy-6-hydroxymethyl-4- oxo-4H-pyran-2-ylmethyl)-3-chloro-benzenesulfonamide (10-05) (2.8 g, 6.44 mmol) following the procedure described for N-(5-benzyloxy-6-formyl-4-oxo-4H-pyran-2-ylmethyl)- benzenesulfonamide (11 -01 ).
- N-(5-Benzyloxy-4-oxo-4H-pyran-2-ylmethyl)-4-methyl-benzenesulfonamide (7-06) (20.0 g, 57.08 %) was synthesized as a light brown solid from 2-aminomethyl-5-benzyloxy-pyran-4-one (5) (21 .0 g, 90.90 mmol) and 4-methyl-benzenesulfonyl chloride (6-06) (20.7 g, 109.09 mmol) following the procedure described for N-(5-benzyloxy-4-oxo-4H-pyran-2-ylmethyl)- benzenesulfonamide (7-01 ).
- LC-MS 386.0 (M+H).
- N-(5-Hydroxy-4-oxo-4H-pyran-2-ylmethyl)-4-methyl-benzenesulfonamide (8-06) (14.0 g, crude) was synthesized as a light brown solid from N-(5-benzyloxy-4-oxo-4H-pyran-2- ylmethyl)-4-methyl-benzenesulfonamide (7-06) (25.0 g, 64.93 mmol) following the procedure described for N-(5-hydroxy-4-oxo-4H-pyran-2-ylmethyl)-benzenesulfonamide (8-01 ).
- N-(5-Hydroxy-6-hydroxymethyl-4-oxo-4H-pyran-2-ylmethyl)-4-methyl-benzenesulfonamide (9- 06) (7.0 g, 45.34 %) was synthesized as a white solid from N-(5-hydroxy-4-oxo-4H-pyran-2- ylmethyl)-4-methyl-benzenesulfonamide (8-06) (14.0 g, 47.45 mmol) following the procedure described for N-(5-hydroxy-6-hydroxymethyl-4-oxo-4H-pyran-2-ylmethyl)-benzenesulfonamide (9-01 ).
- N-(5-Benzyloxy-6-hydroxymethyl-4-oxo-4H-pyran-2-ylmethyl)-4-methyl-benzenesulfonamide N-(5-Benzyloxy-6-hydroxymethyl-4-oxo-4H-pyran-2-ylmethyl)-3-methyl-benzenesulfonamide (10-06) (3.7 g, 41.35 %) was synthesized as a white solid from N-(5-hydroxy-6-hydroxymethyl- 4-oxo-4H-pyran-2-ylmethyl)-4-methyl-benzenesulfonamide (9-06) (7.0 g, 21.54 mmol) following the procedure described for N-(5-benzyloxy-6-hydroxymethyl-4-oxo-4H-pyran-2- ylmethyl)-benzenesulfonamide (10-01 ).
- N-(5-Benzyloxy-6-formyl-4-oxo-4H-pyran-2-ylmethyl)-4-methyl-benzenesulfonamide (11 -06) (2.2 g, 59.62 %) was synthesized as a yellow sticky solid from N-(5-benzyloxy-6- hydroxymethyl-4-oxo-4H-pyran-2-ylmethyl)-4-methyl-benzenesulfonamide (10-06) (3.7 g, 8.92 mmol) following the procedure described for N-(5-benzyloxy-6-formyl-4-oxo-4H-pyran-2- ylmethyl)-benzenesulfonamide (11 -01 ).
- N-(5-Benzyloxy-4-oxo-4H-pyran-2-ylmethyl)-4-chloro-benzenesulfonamide (7-07) (20.0 g, 56.92 %) was synthesized as a brown solid from 2-aminomethyl-5-benzyloxy-pyran-4-one (5) (20.0 g, 86.58 mmol) and 4-chloro-benzenesulfonyl chloride (6-07) (45.67 g, 216.45 mmol) following the procedure described for N-(5-benzyloxy-4-oxo-4H-pyran-2-ylmethyl)- benzenesulfonamide (7-01 ).
- N-(5-Benzyloxy-6-hydroxymethyl-4-oxo-4H-pyran-2-ylmethyl)-4-chloro-benzenesulfonamide (10-07) (3.5 g, 46.17 %) was synthesized as a white solid from 4-chloro-N-(5-hydroxy-6- hydroxymethyl-4-oxo-4H-pyran-2-ylmethyl)-benzenesulfonamide (9-07) ( 6.0 g, 17.39 mmol) following the procedure described for N-(5-benzyloxy-6-hydroxymethyl-4-oxo-4H-pyran-2- ylmethyl)-benzenesulfonamide (10-01 ).
- LC-MS 436.2 (M+H).
- N-(5-Benzyloxy-6-formyl-4-oxo-4H-pyran-2-ylmethyl)-4-chloro-benzenesulfonamide (11 -07) (3.4 g, 85.22 %) was synthesized as a white solid from N-(5-benzyloxy-6-hydroxymethyl-4- oxo-4H-pyran-2-ylmethyl)-4-chloro-benzenesulfonamide (10-07) (4.0 g, 9.19 mmol) following the procedure described for N-(5-benzyloxy-6-formyl-4-oxo-4H-pyran-2-ylmethyl)- benzenesulfonamide (11 -01 ).
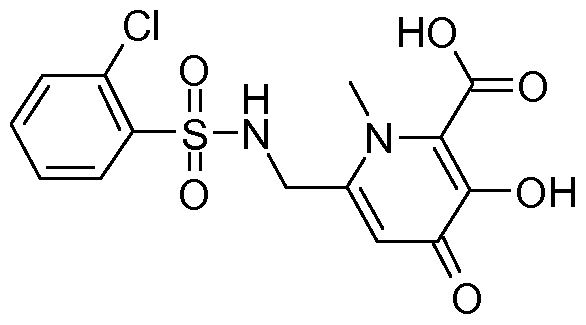
- 6-[(4-Chloro-benzenesulfonylamino)-methyl]-3-hydroxy-1 -methyl-4-oxo-1 ,4-dihydro-pyridine-2- carboxylic acid (14-07) (140.0 mg, 31.55 %) was synthesized as a brown solid from 3- benzyloxy-6-[(4-chloro-benzenesulfonylamino)-methyl]-1 -methyl-4-oxo-1 ,4-dihydro-pyridine-2- carboxylic acid (13-07) (550.0 mg, 1 .19 mmol) following the procedure described for 6- (benzenesulfonylamino-methyl)-3-hydroxy-1 -methyl-4-oxo-1 ,4-dihydro-pyridine-2-carboxylic acid (14-01 ).
- 6-[(2-Chloro-benzenesulfonylamino)-methyl]-3-hydroxy-1 -methyl-4-oxo-1 ,4-dihydro-pyridine-2- carboxylic acid methylamide (16-03) (1 10.0 mg, 52.09 %, purified by Prep-HPLC) was synthesized as a yellow solid from 3-benzyloxy-6-[(2-chloro-benzenesulfonylamino)-methyl]-1 - methyl-4-oxo-1 ,4-dihydro-pyridine-2-carboxylic acid methylamide (15-03) (260.0 mg, 0.547 mmol) following the procedure described for 6-(benzene sulfonyl amino-methyl)-3-hydroxy-1 - methyl-4-oxo-1 ,4-dihydro-pyridine-2-carboxylic acid (14-01 ).
- carboxylic acid methylamide (16-07) (375.0 mg, 65.95 %, purified by Prep-HPLC) was synthesized as a light pink solid from 3-benzyloxy-6-[(4-chloro-benzenesulfonylamino)- methyl]-1 -methyl-4-oxo-1 ,4-dihydro-pyridine-2-carboxylic acid methylamide (15-07) (700.0 mg, 1 .47 mmol) following the procedure described for 6-(benzene sulfonyl amino-methyl)-3- hydroxy-1 -methyl-4-oxo-1 ,4-dihydro-pyridine-2-carboxylic acid (14-01 ).
- carboxylic acid isopropylamide 3-Benzyloxy-1 -methyl-4-oxo-6-[(toluene-2-sulfonylamino)-methyl]-1 ,4-dihydro-pyridine-2- carboxylic acid isopropylamide (17-02) (250.0 mg, 45.70 %) was synthesized as a yellow solid from 3-benzyloxy-1 -methyl-4-oxo-6-[(toluene-2-sulfonylamino)-methyl]-1 ,4-dihydro-pyridine-2- carboxylic acid (13-02) (500.0 mg, 1 .13 mmol) following the procedure described for 6- (benzenesulfonylamino-methyl)-3-benzyloxy-1 -methyl-4-oxo-1 ,4-dihydro-pyridine-2-carboxylic acid isopropylamide (17-01 ).
Abstract
Description


































































































Claims



Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| RU2015125570A RU2015125570A (en) | 2013-01-08 | 2014-01-07 | PYRIDONE DERIVATIVES AND THEIR APPLICATION IN TREATMENT, REDUCING THE INTENSITY OF SYMPTOMS OR PREVENTING A VIRAL DISEASE |
| EP14703276.7A EP2943469A1 (en) | 2013-01-08 | 2014-01-07 | Pyridone derivatives and their use in the treatment, amelioration or prevention of a viral disease |
| KR1020157017812A KR20150103034A (en) | 2013-01-08 | 2014-01-07 | Pyridone derivatives and their use in the treatment, amelioration or prevention of a viral disease |
| CA2894452A CA2894452A1 (en) | 2013-01-08 | 2014-01-07 | Pyridone derivatives and their use in the treatment, amelioration or prevention of a viral disease |
| BR112015016184A BR112015016184A2 (en) | 2013-01-08 | 2014-01-07 | pyridone derivatives and their use in the treatment, amelioration or prevention of a viral disease |
| JP2015551200A JP2016508145A (en) | 2013-01-08 | 2014-01-07 | Pyridone derivatives and their use in the treatment, amelioration or prevention of viral diseases |
| MX2015008288A MX2015008288A (en) | 2013-01-08 | 2014-01-07 | Pyridone derivatives and their use in the treatment, amelioration or prevention of a viral disease. |
| CN201480004181.3A CN104903294A (en) | 2013-01-08 | 2014-01-07 | Pyridone derivatives and their use in treatment, amelioration or prevention of viral disease |
| HK15112096.1A HK1211287A1 (en) | 2013-01-08 | 2015-12-08 | Pyridone derivatives and their use in the treatment, amelioration or prevention of a viral disease |
| HK16100551.3A HK1212688A1 (en) | 2013-01-08 | 2016-01-19 | Pyridone derivatives and their use in the treatment, amelioration or prevention of a viral disease |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201361750023P | 2013-01-08 | 2013-01-08 | |
| US61/750,023 | 2013-01-08 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014108407A1 true WO2014108407A1 (en) | 2014-07-17 |
Family
ID=50070508
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2014/050166 WO2014108407A1 (en) | 2013-01-08 | 2014-01-07 | Pyridone derivatives and their use in the treatment, amelioration or prevention of a viral disease |
Country Status (11)
| Country | Link |
|---|---|
| US (1) | US8952039B2 (en) |
| EP (1) | EP2943469A1 (en) |
| JP (1) | JP2016508145A (en) |
| KR (1) | KR20150103034A (en) |
| CN (1) | CN104903294A (en) |
| BR (1) | BR112015016184A2 (en) |
| CA (1) | CA2894452A1 (en) |
| HK (2) | HK1211287A1 (en) |
| MX (1) | MX2015008288A (en) |
| RU (1) | RU2015125570A (en) |
| WO (1) | WO2014108407A1 (en) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20170131651A (en) | 2015-04-28 | 2017-11-29 | 시오노기세야쿠 가부시키가이샤 | Substituted polycyclic pyridone derivatives and prodrugs thereof |
| WO2018030463A1 (en) | 2016-08-10 | 2018-02-15 | 塩野義製薬株式会社 | Substituted polycyclic pyridone derivative and pharmaceutical composition containing prodrug thereof |
| KR20190049916A (en) | 2015-04-28 | 2019-05-09 | 시오노기세야쿠 가부시키가이샤 | Substituted polycyclic pyridone derivative and prodrug thereof |
| US11040048B2 (en) | 2015-12-15 | 2021-06-22 | Shionogi & Co., Ltd. | Medicament for treating influenza characterized by combining a Cap-dependent endonuclease inhibitor and an anti-influenza drug |
| US11827627B2 (en) | 2021-06-04 | 2023-11-28 | Vertex Pharmaceuticals Incorporated | N-(hydroxyalkyl (hetero)aryl) tetrahydrofuran carboxamides as modulators of sodium channels |
| US11834441B2 (en) | 2019-12-06 | 2023-12-05 | Vertex Pharmaceuticals Incorporated | Substituted tetrahydrofurans as modulators of sodium channels |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10676428B2 (en) | 2015-12-17 | 2020-06-09 | Ramot At Tel-Aviv University Ltd. | Antiviral agents for amantadine-resistant influenza A |
| US10889556B2 (en) * | 2016-03-08 | 2021-01-12 | The Regents Of The University Of California | Compositions and methods for inhibiting influenza RNA polymerase PA endonuclease |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5475109A (en) | 1994-10-17 | 1995-12-12 | Merck & Co., Inc. | Dioxobutanoic acid derivatives as inhibitors of influenza endonuclease |
| WO2005074513A2 (en) * | 2004-01-30 | 2005-08-18 | Merck & Co., Inc. | N-benzyl-3,4-dihyroxypyridine-2-carboxamide and n-benzyl-2,3-dihydroxypyridine-4-carboxamide compounds useful as hiv integrase inhibitors |
| WO2006030807A1 (en) | 2004-09-15 | 2006-03-23 | Shionogi & Co., Ltd. | Carbamoylpyridone derivative having hiv integrase inhibitory activity |
| US20070072831A1 (en) | 2005-05-16 | 2007-03-29 | Gilead Sciences, Inc. | Integrase inhibitor compounds |
| WO2010110231A1 (en) | 2009-03-26 | 2010-09-30 | 塩野義製薬株式会社 | Substituted 3-hydroxy-4-pyridone derivative |
| WO2010110409A1 (en) | 2009-03-26 | 2010-09-30 | 塩野義製薬株式会社 | Process for producing pyrone and pyridone derivatives |
| WO2011000566A2 (en) | 2009-06-30 | 2011-01-06 | Savira Pharmaceuticals Gmbh | Compounds and pharmaceutical compositions for the treatment of negative-sense ssrna virus infections |
-
2014
- 2014-01-07 KR KR1020157017812A patent/KR20150103034A/en not_active Application Discontinuation
- 2014-01-07 BR BR112015016184A patent/BR112015016184A2/en not_active IP Right Cessation
- 2014-01-07 JP JP2015551200A patent/JP2016508145A/en active Pending
- 2014-01-07 RU RU2015125570A patent/RU2015125570A/en not_active Application Discontinuation
- 2014-01-07 CA CA2894452A patent/CA2894452A1/en not_active Abandoned
- 2014-01-07 CN CN201480004181.3A patent/CN104903294A/en active Pending
- 2014-01-07 EP EP14703276.7A patent/EP2943469A1/en not_active Withdrawn
- 2014-01-07 MX MX2015008288A patent/MX2015008288A/en unknown
- 2014-01-07 US US14/149,218 patent/US8952039B2/en not_active Expired - Fee Related
- 2014-01-07 WO PCT/EP2014/050166 patent/WO2014108407A1/en active Application Filing
-
2015
- 2015-12-08 HK HK15112096.1A patent/HK1211287A1/en unknown
-
2016
- 2016-01-19 HK HK16100551.3A patent/HK1212688A1/en unknown
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5475109A (en) | 1994-10-17 | 1995-12-12 | Merck & Co., Inc. | Dioxobutanoic acid derivatives as inhibitors of influenza endonuclease |
| WO2005074513A2 (en) * | 2004-01-30 | 2005-08-18 | Merck & Co., Inc. | N-benzyl-3,4-dihyroxypyridine-2-carboxamide and n-benzyl-2,3-dihydroxypyridine-4-carboxamide compounds useful as hiv integrase inhibitors |
| WO2006030807A1 (en) | 2004-09-15 | 2006-03-23 | Shionogi & Co., Ltd. | Carbamoylpyridone derivative having hiv integrase inhibitory activity |
| US20070072831A1 (en) | 2005-05-16 | 2007-03-29 | Gilead Sciences, Inc. | Integrase inhibitor compounds |
| WO2010110231A1 (en) | 2009-03-26 | 2010-09-30 | 塩野義製薬株式会社 | Substituted 3-hydroxy-4-pyridone derivative |
| WO2010110409A1 (en) | 2009-03-26 | 2010-09-30 | 塩野義製薬株式会社 | Process for producing pyrone and pyridone derivatives |
| EP2412708A1 (en) * | 2009-03-26 | 2012-02-01 | Shionogi&Co., Ltd. | Substituted 3-hydroxy-4-pyridone derivative |
| WO2011000566A2 (en) | 2009-06-30 | 2011-01-06 | Savira Pharmaceuticals Gmbh | Compounds and pharmaceutical compositions for the treatment of negative-sense ssrna virus infections |
Non-Patent Citations (26)
| Title |
|---|
| "Helvetica Chimica Acta", 1995, article "A multilingual glossary of biotechnological terms: (IUPAC Recommendations" |
| CHANG ET AL., ANTIVIRAL RESEARCH, vol. 89, 2011, pages 26 - 34 |
| CIANCI C ET AL: "IDENTIFICATION OF N-HYDROXAMIC ACID AND N-HYDROXYIMIDE COMPOUNDS THAT INHIBIT THE INFLUENZA VIRUS POLYMERASE", ANTIVIRAL CHEMISTRY & CHEMOTHERAPY, BLACKWELL SCIENTIFIC PUBL., LONDON, GB, vol. 7, no. 6, 1 January 1996 (1996-01-01), pages 353 - 360, XP002925548, ISSN: 0956-3202 * |
| D. J. GOOD ET AL., CRYST. GROWTH DES, vol. 9, no. 5, 2009, pages 2252 - 2264 |
| DHARAN ET AL., THE JOURNAL OF THE AMERICAN MEDICAL ASSOCIATION, vol. 301, no. 10, 11 March 2009 (2009-03-11), pages 1034 - 1041 |
| DIAS ET AL., NATURE, vol. 458, 2009, pages 914 - 918 |
| DIAS ET AL., NATURE, vol. 458, no. 7240, 16 April 2009 (2009-04-16), pages 914 - 918 |
| ERIKSSON, B. ET AL., ANTIMICROB. AGENTS CHEMOTHER., vol. 11, 1977, pages 946 - 951 |
| FURUTA ET AL., ANTIMICROBIAL AGENTS AND CHEMOTHERAPY, 2005, pages 981 - 986 |
| GHANEM, A. ET AL., J. VIROL., vol. 81, 2007, pages 7801 - 7804 |
| GUILLIGAY ET AL., NATURE STRUCTURAL & MOLECULAR BIOLOGY, vol. 15, no. 5, May 2008 (2008-05-01), pages 500 - 506 |
| KUKKONEN, S. K. ET AL., ARCH. VIROL., vol. 150, 2005, pages 533 - 556 |
| LEAHY, M. B. ET AL., J. VIROL., vol. 71, 2005, pages 8347 - 8351 |
| LIU Z D ET AL: "SYNTHESIS OF 2-AMIDO-3-HYDROXYPYRIDIN-4(1H)-ONES: NOVEL IRON CHELATORS WITH ENHANCED PFE3+ VALUES", BIOORGANIC & MEDICINAL CHEMISTRY, PERGAMON, GB, vol. 9, no. 3, 1 January 2001 (2001-01-01), pages 563 - 573, XP001121004, ISSN: 0968-0896, DOI: 10.1016/S0968-0896(00)00273-X * |
| MAGDEN, J. ET AL., APPL. MICROBIOL. BIOTECHNOL., vol. 66, 2005, pages 612 - 621 |
| MOSCONA ET AL., THE NEW ENGLAND JOURNAL OF MEDICINE, vol. 360, no. 10, 5 March 2009 (2009-03-05), pages 953 - 956 |
| N. DAS ET AL., EUROPEAN JOURNAL OF PHARMACEUTICAL SCIENCES, vol. 41, 2010, pages 571 - 588 |
| NEUMANN ET AL., NATURE, vol. 18, no. 7249, 2009, pages 931 - 939 |
| NING SHAN ET AL., DRUG DISCOVERY TODAY, vol. 13, no. 9/10, 2008, pages 440 - 446 |
| NOAH, D. L. ET AL., ADV. VIRUS RES., vol. 65, 2005, pages 121 - 145 |
| PLOTCH, S. J. ET AL., CELL, vol. 23, 1981, pages 847 - 858 |
| S. M. BERGE ET AL.: "Pharmaceutical Salts", J. PHARM. SCI., vol. 66, 1977, pages 1 - 19 |
| TISDALE, M. ET AL., ANTIMICROB. AGENTS CHEMOTHER., vol. 39, 1995, pages 2454 - 2458 |
| TOMASSINI, J. ET AL., ANTIMICROB. AGENTS CHEMOTHER., vol. 38, 1994, pages 2827 - 2837 |
| TOMASSINI, J. ET AL., ANTIMICROB. AGENTS CHEMOTHER., vol. 40, 1996, pages 1189 - 1193 |
| VON ITZSTEIN, M. ET AL., NATURE, vol. 363, 1993, pages 418 - 423 |
Cited By (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10392406B2 (en) | 2015-04-28 | 2019-08-27 | Shionogi & Co., Ltd. | Substituted polycyclic pyridone derivatives and prodrugs thereof |
| EP4219508A1 (en) | 2015-04-28 | 2023-08-02 | Shionogi & Co., Ltd | Substituted polycyclic pyridone derivative and prodrug thereof |
| KR20170131651A (en) | 2015-04-28 | 2017-11-29 | 시오노기세야쿠 가부시키가이샤 | Substituted polycyclic pyridone derivatives and prodrugs thereof |
| KR20190002742A (en) | 2015-04-28 | 2019-01-08 | 시오노기세야쿠 가부시키가이샤 | Substituted polycyclic pyridone derivative and prodrug thereof |
| US10633397B2 (en) | 2015-04-28 | 2020-04-28 | Shionogi & Co., Ltd. | Substituted polycyclic pyridone derivatives and prodrugs thereof |
| KR20190049916A (en) | 2015-04-28 | 2019-05-09 | 시오노기세야쿠 가부시키가이샤 | Substituted polycyclic pyridone derivative and prodrug thereof |
| EP3428170A1 (en) | 2015-04-28 | 2019-01-16 | Shionogi & Co., Ltd | Anti-influenza polycyclic pyridone derivative and prodrug thereof |
| US20180118760A1 (en) | 2015-04-28 | 2018-05-03 | Shionogi & Co., Ltd. | Substituted polycyclic pyridone derivatives and prodrugs thereof |
| US11040048B2 (en) | 2015-12-15 | 2021-06-22 | Shionogi & Co., Ltd. | Medicament for treating influenza characterized by combining a Cap-dependent endonuclease inhibitor and an anti-influenza drug |
| US10759814B2 (en) | 2016-08-10 | 2020-09-01 | Shionogi & Co., Ltd. | Pharmaceutical compositions containing substituted polycyclic pyridone derivatives and prodrug thereof |
| KR20190018469A (en) | 2016-08-10 | 2019-02-22 | 시오노기세야쿠 가부시키가이샤 | Substituted polycyclic pyridone derivative and pharmaceutical composition containing prodrug thereof |
| KR20190007517A (en) | 2016-08-10 | 2019-01-22 | 시오노기세야쿠 가부시키가이샤 | A pharmaceutical composition containing a substituted polycyclic pyridone derivative and a prodrug thereof |
| US11306106B2 (en) | 2016-08-10 | 2022-04-19 | Shionogi & Co., Ltd. | Pharmaceutical compositions containing substituted polycyclic pyridone derivatives and prodrug thereof |
| WO2018030463A1 (en) | 2016-08-10 | 2018-02-15 | 塩野義製薬株式会社 | Substituted polycyclic pyridone derivative and pharmaceutical composition containing prodrug thereof |
| US11834441B2 (en) | 2019-12-06 | 2023-12-05 | Vertex Pharmaceuticals Incorporated | Substituted tetrahydrofurans as modulators of sodium channels |
| US11919887B2 (en) | 2019-12-06 | 2024-03-05 | Vertex Pharmaceuticals Incorporated | Substituted tetrahydrofurans as modulators of sodium channels |
| US11827627B2 (en) | 2021-06-04 | 2023-11-28 | Vertex Pharmaceuticals Incorporated | N-(hydroxyalkyl (hetero)aryl) tetrahydrofuran carboxamides as modulators of sodium channels |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2894452A1 (en) | 2014-07-17 |
| US8952039B2 (en) | 2015-02-10 |
| BR112015016184A2 (en) | 2017-07-11 |
| US20140194476A1 (en) | 2014-07-10 |
| RU2015125570A (en) | 2017-02-10 |
| EP2943469A1 (en) | 2015-11-18 |
| KR20150103034A (en) | 2015-09-09 |
| HK1212688A1 (en) | 2016-06-17 |
| MX2015008288A (en) | 2016-06-02 |
| CN104903294A (en) | 2015-09-09 |
| HK1211287A1 (en) | 2016-05-20 |
| JP2016508145A (en) | 2016-03-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8921388B2 (en) | Dihydroxypyrimidine carbonic acid derivatives and their use in the treatment, amelioration or prevention of a viral disease | |
| EP2864338B1 (en) | 7-oxo-thiazolopyridine carbonic acid derivatives and their use in the treatment, amelioration or prevention of a viral disease | |
| US8952039B2 (en) | Pyridone derivatives and their use in the treatment, ameloriation or prevention of a viral disease | |
| CA2874253A1 (en) | 7-oxo-4,7-dihydro-pyrazolo [1,5-a] pyrimidine derivatives which are useful in the treatment, amelioration or prevention of a viral disease | |
| EP2794616B1 (en) | Pyrimidin-4-one derivatives and their use in the treatment, amelioration or prevention of a viral disease | |
| CA2852750A1 (en) | Heteroaryl hydroxamic acid derivatives and their use in the treatment, amelioration or prevention of a viral disease | |
| US9505758B2 (en) | Substituted 1,5-naphthyridines as endonuclease inhibitors | |
| US20170081331A1 (en) | Pyrazolopyrazines and their use in the treatment, amelioration or prevention of a viral disease | |
| WO2017046318A1 (en) | Triazolones derivatives for use in the treatment, amelioration or prevention of a viral disease | |
| US20160002226A1 (en) | Pyridopyrazine compounds and their use in the treatment, amelioration or prevention of influenza | |
| US20170081324A1 (en) | Triazolones derivatives and their use in the treatment, amelioration or prevention of a viral disease |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 14703276 Country of ref document: EP Kind code of ref document: A1 |
|
| DPE1 | Request for preliminary examination filed after expiration of 19th month from priority date (pct application filed from 20040101) | ||
| ENP | Entry into the national phase |
Ref document number: 2894452 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2015/008288 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 20157017812 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2015551200 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2014703276 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2015125570 Country of ref document: RU Kind code of ref document: A |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112015016184 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112015016184 Country of ref document: BR Kind code of ref document: A2 Effective date: 20150706 |