WO2014085225A1 - Pyrimidine compounds for the treatment of cancer - Google Patents
Pyrimidine compounds for the treatment of cancer Download PDFInfo
- Publication number
- WO2014085225A1 WO2014085225A1 PCT/US2013/071409 US2013071409W WO2014085225A1 WO 2014085225 A1 WO2014085225 A1 WO 2014085225A1 US 2013071409 W US2013071409 W US 2013071409W WO 2014085225 A1 WO2014085225 A1 WO 2014085225A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- mmol
- group
- cancer
- added
- Prior art date
Links
- 0 CCCC*CN=C Chemical compound CCCC*CN=C 0.000 description 5
- OMUMOUPOIWPOTG-LTDTZLOSSA-N CCC(CNC(/C(/C=N\CN(C)CC)=C(\C)/NC)O)/C=C\C([FH+])=C Chemical compound CCC(CNC(/C(/C=N\CN(C)CC)=C(\C)/NC)O)/C=C\C([FH+])=C OMUMOUPOIWPOTG-LTDTZLOSSA-N 0.000 description 1
- LHNWZCQLUGOOCN-DAXSKMNVSA-N CCC/C=N\CCl Chemical compound CCC/C=N\CCl LHNWZCQLUGOOCN-DAXSKMNVSA-N 0.000 description 1
- JSVARSIOVPNQPH-QMMMGPOBSA-N C[O]([C@@H](CC1)[IH]CC1=C)=C Chemical compound C[O]([C@@H](CC1)[IH]CC1=C)=C JSVARSIOVPNQPH-QMMMGPOBSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/46—Two or more oxygen, sulphur or nitrogen atoms
- C07D239/48—Two nitrogen atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/06—Peri-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/08—Bridged systems
Definitions
- the present invention concerns compounds, compositions and methods for the treatment of cancer.
- Acute Lymphoblastic Leukemia is the most common malignancy in children and common varieties are cured by chemotherapy in 75% ⁇ 85% of the cases.
- Collectively the less common T ceil and rare B ceil subsets represent less than 2000 cases yearly and thus can be classified as a rare disease; these subsets have a poorer prognosis.
- resistance to and relapse from therapy is a major cause of pediatric cancer death.
- ALL chemotherapies can cause late complications that are increasingly recognized in pediatric survivor populations.
- a first aspect of the invention is active compounds of Formula 1 or ii:
- Ring A is a 5- or 6-raembered heteroaryl group such as pyridyl, pyrimidyl. thiazol, furanyl, pyridazinyl, pyrazinyl, imidazol, etc..
- the dashed line is an optional double bond, X is N or O. Y is C. S or N and ca move on the ring.
- R is ⁇ R J R°, where R J is a covending bond or CI to C3 aikyl or a linker group (for example, sulfonamide, amide, etc.) and R 6 is cycloalkyl. heterocycloaikyl, aryl, heteroaryl aikylcycloalkyl, alkylbeterocycloalkyl, alkylaryl, aikylheteroaryi, or alkyl, and wherein R 6 is optionally substituted one, two or three times with independently selected polar groups:
- R 4 is -R 7 R S , where R 7 is a covending bond or CI to C3 alkyl and R 8 is cycloalkyl, heterocycloaikyl, aryl, heteroaryl or alkyl, and wherein R 8 is optionally substituted one, two or three times with independently selected polar groups;
- R 3 is selected from the group consisting of H, alkyl, arylalkyl; cycioalkyialkyi, heterocyeloaikylalkyl, heteroaryalkyl, and aikoxyalkyl, each of which is optionally substituted one, two or three times with independently selected polar groups;
- R 4 is H, loweralkyl, halo, or loweraikox'v;
- a further aspect of the invention is an active compound as described herein in a pharmaceutically acceptable carrier.
- a further aspect of the invention is a method of treating cancer in a subject, in need thereof, comprising administering said subject an active compound as described herein in an amount effective to treat the cancer.
- a fuller aspect of the invention is an active compound as described herein for use in treating cancer, and/or for the preparation of a medicament for the treatment of cancer.
- Alkyl refers to a straight or branched chain hydrocarbon containing from 1 to 10 carbon atoms. Representative examples of alky! include, but. are not limited, to, methyl, ethyl, n-propyl, iso-propy!, n-butyl, sec-butyl, iso-b tyl, tert-butyl, n-pentyl, isopentyl, neopentyL n-hexyl, 3-methylhexyl, 2,2- dimethyl entyl, 2,3-dimethylpentyl, n-heptyl, n-octyl, n-nonyl, n-decyl, and the like, "Lower alkyl.” as used herein, is a subset of alkyl, in some embodiments preferred, and refers to a straight or branched chain hydrocarbon grou containing from 1
- lower alkyl include, but are not limited to. methyl, ethyl, n- propyi, iso-propyl, n-butyl, iso-butyl, tert-butyl, and the like.
- alkyl or “loweralkyi” is intended to include both substituted and unsubstituied alkyl or loweraikyl unless otherwise indicated and these groups may be substituted with groups selected from halo (e.g., haloalkyl), alkyl, haloalkyl, alkenyl, a!kynyl, cyeloalkyl, cycloalkylalkyl, aryl, arylalkyl, heterocyclo, heterocycloaikyf hydroxy], aikoxy (thereby creating a polyalkoxy such as polyethylene glycol), aikenyloxy, alkynyloxy, haloaikoxy, cyc!oalkoxy, cycloalkylalkyloxy, aryloxy, aryialkyioxy, heierocyclooxy, heterocycloiaikvloxy, mercapto, alkyl-S(0) m , halo alkyl, al
- heterocyclo-S(0) m heterocycloalkyl- S(0)m, amino, carboxy, aikyiamino, alkenylammo, alkynylarntno, haloalkylamino, eycioalkyiamino, cycloalkyialkylamino, arylamino, aryl aikyiamino, heterocycioamino, heieroeycioalkylamino, disubstituted-axnino, acyl amino, acyloxy, ester, amide, sulfonamide, urea, aikoxyaeylaniino, aminoacyloxy, nitro or cyano where m- Q, 1 , 2 or 3.
- alkenyl refers to a straight or branched chain hydrocarbon containing from 1 to 10 carbon atoms for in Ioweralkeny 1 1 to 4 carbon atoms) which include 1 to 4 double bonds in the normal chain.
- alkenyl include, but are not limited to, vinyl, 2-propenyl, 3-butenyl, 2-butenyl, 4- pentenyl, 3-pentenyl, 2-hexenyl, 3-bexenyl, 2,4-heptadiene, and the like.
- alkenyl or “loweralkenyl” is intended to include both substituted and unsubstituied alkenyl or loweralkenyl unless otherwise indicated and these groups may be substituted with groups as described in connection with alkyl and loweralkyi above.
- Alkynyl refers to a straight or branched chain hydrocarbon containing from 1 to 10 carbon atoms (or in loweralkynyl 1 to 4 carbon atoms) which include 1 triple bond in the normal chain.
- Representative examples of alkynyl include, but are not limited to, 2-propynyl, 3-butynyi, 2- buiynyl, 4-pentynyl. 3- pentynyi, and the like.
- alkynyl or "ioweralkynyl” is intended to include both substituted and unsubstitvtted alkynyl or Ioweralkynyl unless otherwise indicated and these groups may be substituted with the same groups as set forth in connection with alkyl and ioweraikyl above.
- Cycloalkyl refers to a saturated or partially unsaturated cyclic hydrocarbon group containing from 3, 4 or 5 to 6. 7 or 8 carbons (which carbons may be replaced in heterocyclic group as discussed below).
- Representative examples of cycloalkyl include, cyclopropyl, cyclobutyl, cyclopenty!, cyelohexyl, eyeloheptyl. and cyclooctyl. These rings may be optionally substituted with additional substituents as described herein such as halo or Ioweralkyl
- the term "cycloalkyl" is generic and intended to include heterocyclic groups as discussed below unless specified otherwise.
- Heterocyclic group' 1 or “heterocycio” as used herein alone or as part of another group, refers to an aliphatic (e.g., fully or partially saturated heterocyclo) or aromatic (e.g., heteroaryt) monocyclic- or a bicyclic-ring system
- Monocyclic ring systems are exemplified by any 5 or 6 membered ring containing 1 , 2, 3, or 4 heteroatoms independently selected from oxygen, nitrogen and sulfur.
- the 5 membered ring has from 0-2 double bonds and the 6 membered ring has from 0-3 double bonds.
- Representative examples of monocyclic ring systems include, but are not limited to, azetidine.
- azepine aziridine, diazepine, 1 ,3-dioxolane, dioxane, dithiane, furan, imidazole, imidazoline, imidazolidine, isothiazole, isothiazoline, isothiazo!idine, isoxazole, isoxazoline, isoxazolidine, morphoiine, oxadiazole, oxadiazoline, oxadiazoiidme, oxazole, oxazoline, oxazolidine, piperazine, piperidine, pyran, pyrazine, pyrazole, pyrazolone, pyrazolidine, pyridine, pyrimidine.
- Bicyclic ring systems are exemplified by any of the above monocyclic ring systems fused to an aryl group as defined herein, a cycloalkyl group as defined herein, or another monocyclic ring system as defined herein.
- bicyclic ring systems include but are not limited to, for example, benzimidazole, benzoihiazole, benzothiadiazoie, benzothiophene, benzoxadiazole, benzoxazole, benzofuran, benzopyran, benzothiopyran. benzodioxine, 1,3- benzodioxo!e, cinnoline.
- These rings include quaternized derivatives thereof and may be optionally substituted with groups selected from halo, alkyl, haloaikyi, alkenyl, alkynyl, cycloaikyl, cycloalkyialkyl, aryl, arylalkyl, heterocycio, heterocycioalkyl, hydroxy!, alkoxy, alkenyloxy, aikynyioxy, haloalkoxy, eycloalkoxy, cyeioalkylalkyloxy, aryloxy, arylalkyloxy, heterocyclooxy, heterocyclolalkyloxy, mercapto, alkyl ⁇ S(0) m , haioalkyl ⁇ S(0) m , alkenyl- S(0) m , alkynyl -S(0) m , cycloalkyi ⁇ S(Q) mj cycloalkylalkyl-S(0) m
- arylalkyl - S(0) m heterocyclo-S(0) ms heterocycloalkyl-S(0) m , amino, alkylamino, alkenylamino, alkynyl amino, haloalkylamino, cycloalkylamino, cycioaikylalkylamino, arylamino, arylalkylamino, heterocycloamino.
- heterocycloalkyiamino disubstituted-amino, acylaraino, acyloxy, ester, amide, sulfonamide, urea, alkoxyacylammo, ammoacyloxy, nitro or cyano where m - 0, 1 , 2 or 3.
- Aryl refers to a monocyclic carbocyclic ring system or a bicyclic carbocyclic fused ring system having one or more aromatic rings.
- Representative examples of aryl include, azuienyl, indanyl, indenyl, naphthyl, phenyl, tetrahydronaphthyl, and the like.
- aryl is intended to include both substituted and unsubstituted aryl unless otherwise indicated and these groups may be substituted with the same groups as set forth in connection with alkyl and loweralkyl above.
- Arylalkyl refers to an aryl group, as defined herein, appended to the parent molecular moiety through an alkyl group, as defined herein.
- Representative examples of arylalkyl include, but are not limited to, benzyl, 2- phenylethyl, 3-phenylpropyl, 2-naphth-2-ylet;hyl, and the like.
- Heteroaryi as used herein is as described in connection with heterocycio above,
- Alkoxy refers to an alkyl or loweralkyl group, as defined herein (and thus including substituted versions such as polyalkoxy), appended to the parent molecular moiety through an oxy group, -0-.
- alkoxy include, but are not limited, to, methoxy, ethoxy, pro poxy, 2-propoxy, butoxy, iert-butoxy, pentyioxy, hexyloxy and the like.
- Halo refers to any suitable halogen, including -F, -CI, -Br, and -I.
- Cyano as used herein refers to a -CN group.
- Formi refers to a -C(0)H group.
- Carboxylic acid refers to a -C(0)GH group.
- Hydroxyloxy 1 refers to an -OH group.
- Acyl as used herein alone or as part of another group refers to a -C(0)R radical, where R is any suitable substituent such as aryl. aikyl, aikenyi, alkynyi, cycloaikyl or other suitable substituent as described herein.
- Alkylthio refers to an alkyl group, as defined herein, appended to the parent molecular moiety through a thio moiety, as defined herein.
- Representative examples of alkylthio include, but are not limited, methylthio, ethylthio. tert-butylihio, hexylihio, and the like.
- Amino as used herein means the radical ⁇ -N3 ⁇ 4.
- Alkylaniino as used herein alone or as part of another group means the radical - NHR, where R is an alkyl group.
- Arylalkylamino as used herein alone or as part of another group means the radical ⁇ NHR, where R is an aryl alkyl group.
- Disubstituted-amino as used herein alone or as part of another group means the radical -NR s R b , where R a and R lt are independently selected from the groups alkyl, haloalkyl, aikenyi, alkynyi. cycloaikyl, cycloalkyialkyl, aryl, arylalkyl, heterocyclo, heterocycloaikyl.
- Acylamino as used herein alone or as part of another group means the radical - ⁇ NR a Rb, where R a is an acyl group as defined herein and R is selected from the groups hydrogen, alkyl, haloalkyl, aikenyi, alkynyi, cycloaikyl, cycloalkyialkyl, aryl, arylalkyl, heterocyclo , hetero c c 1 o alkyl .
- Alkoxy as used herein alone or as pari of another group means the radical -OR, where R is an acyl group as defined herein.
- Ester as used herein alone or as part of another group refers to a -C(0)OR radical, where R is any suitable substituent such as alkyl, cycloaikyl, aikenyi, alkynyi or aryl.
- Amide as used herein alone or as part of another group refers to a -C(0)NR a R b radical, where R a and R b are any suitable substituent such as alkyl, cycloaikyl, aikenyi, alkynyi or aryl.
- Sulfoxyl refers to a compound of the fomiula -S(0)R. where R is any suitable substituent such as alkyl, cycloaikyl, aikenyi, alkynyi or aryl.
- “Sulfonyl” as used herein refers to a compound of the fomiula -S(0)(0)R, where R is any suitable substituent such as amino, alkyl, cycloaikyl, aikenyi, alkynyi or aryl.
- “Sulfonate” as used herein refers to a compound of the formula -S(0)(0)OR, where R is any suitable substituent such as alkyl, cycloaikyl, alkenyl, aikynyl or aryi,
- Sulfonic acid refers to a compound of the formula -S(0)(0)OH.
- Sulfonamide as used herein alone or as pail of another group refers to a - S(0)2NRaRb radical, where R a and Rb are any suitable substituent such as H, alkyl, cycloaikyl, alkenyl, aikynyl or aryL
- Rea as used herein alone or as part of another group refers to an - (R c )C(0) R a R b radical where R a , R b and e are any suitable substituent such as H, alkyl, cycloaikyl, alkenyl, aikynyl or aryL
- Alkoxyacylamino as used herein alone or as part of another group refers to an - N(Ra)C(0)ORb radical, where R a , R b are any suitable substituent such as H, alkyl, cycloaikyl, alkenyl, aikynyl or aryL
- “Ammoacyioxy” as used herein alone or as part of another group refers to an - OC(0)NR a Rb radical, where R a and Rb are any suitable substituent such as H, alkyl, cycloaikyl, alkenyl, aikynyl or aryL
- Poly group refers to a group wherein the nuclei of the atoms co vaiently bound to each other td form the group do not share the electrons of the eovalent bond (s) joining them equally; that is the electron cloud is denser about one atom than another. This results in one end of the eovalent bond(s) being relatively negative and the other end relatively positive; i.e., there is a negative pole and a positive pole.
- polar groups include, without limitations, halo, hydroxy, alkoxy, carboxy, nitro, cya.no, amino (primary, secondary and tertiary), amido, ureido, sulfonamido, sulfinyl, suifhydryl silyl, S- sulfonamido.
- the polar group can be an ionic group.
- Ionic group as used herein includes anionic and cationic groups, and includes groups (sometimes referred to as "ionogenic” groups) that are uncharged in one form but can be easily converted to ionic groups (for example, by protonation or deprotonation in aqueous solution). Examples include but are not limited to carhoxylate, sulfonate, phosphate, amine, N-oxide, and ammonium (including quaternized heterocyclic amines such as imidazolium and pyridiniurrs) groups. See, e.g., U.S. Pat. Nos. 6,478,863; 6,800,276; and 6,896,246.
- Additional examples include tironic acids, carboxylic acid, sulfonic acid, amine, and moieties such as guanidinium, phosphoric acid, phosphonic acid, phosphatidyl choline, phosphonium, borate, sulfate, etc,
- Deuterium as used herein alone or as part of another group, refers to a safe, nonradioactive relative of hydrogen. Any hydrogen in a group or substitueni described above may be replaced with deuterium to provide a "deuterated” compound, in some embodiments to modify and/or improve metabolic stability, resulting in better safety, toierability and/or efficacy.
- linking group or "linker group” as used herein are generally bivalent aromatic, aliphatic, or mixed aromatic and aliphatic groups.
- linking groups include linear or branched, substituted or imsubstituted aryl, aikyl, alkylaryl, or alkylaryl alkyl linking groups, where the alkyl groups are saturated or unsaturated, and where the alkyl and aryl groups optionally containing independentl selected heteroatoms such as 1 , 2, 3 or 4 heteroatoms selected from the group consisting of N, 0, and S.
- linking groups containing from 2 to 20 carbon atoms are preferred.
- suitable linking groups are known, including but not limited to those described in, US Patents Nos. 8,247,572; 8,097,609: 6,624,317; 6,613,345; 6,596,935; and 6,420,377, the disclosures of which are incorporated by reference herein in their entirety.
- Treat refers to any type of treatment that imparts a benefit to a patient afflicted with a disease, including improvement in the condition of the patient ⁇ e.g., in one or more symptoms), delay in the progression of the disease, delay in onset of the disease, etc.
- “Pharmaceutically acceptable” as used herein means that the compound or composition is suitable for administration to a subject to achieve the treatments described herein, without unduly deleterious side effects in light of the severity of the disease and necessity of the treatment.
- Active compounds of the present invention may optionally be administered in conjunction with other compounds useful in the treatment of cancer.
- the other compounds may optionally be administered concurrently.
- concurrently means sufficiently close in time to produce a combined effect (that is. concurrently may be simultaneously, or it may be two or more events occurring within a short time period before or after each other).
- the present invention is primarily concerned with the treatment of human subjects, but the invention may also be carried out on animal subjects, particularly mammalian subjects such as mice, rats, dogs, cats, livestock and horses for veterinary purposes, and for drug screening and drug development purposes.
- Subjects may be of any age, including infant, juvenile, adolescent, adult, and geriatric subjects.
- R l is -R ⁇ R 6 , where R s is a covending bond, CI to C3 alky! or a linker group (for example, sulfonamide, amide,, etc.) and R 6 is eycloalkyl, heterocycloalkyl, aryi, heteroaryl, alkylcycloalkyi, alkylheterocycloalkyl, al.kyl.aryl, alkyiheteroaryl or alkyl, and wherein R 6 is optionally substituted one, two or three times with independently selected polar groups;
- R A is ⁇ R 7 R S , where R' is a covending bond or CI to C3 alkyl and R 8 is eycloalkyl, heterocycloalkyl, aryl, heteroaryl or alkyl. and wherein R is optionally substituted one, two or three times with independently selected polar groups;
- R is selected from the group consisting of H, alkyl, aryla!kyk cycloalkylaikyl, heterocycioalkylaikyl, heieroaryalkyl, and alkoxyalkyl, each of which is optionally substituted one, two or three times with independently selected polar groups;
- R 4 is H, !oweralkyk halo, or loweralkoxy
- 3 is a covending bond; in other embodiments, R " is CI to C3 alkylene such as -03 ⁇ 4 ⁇ or R is a linker group (for example, sulfonamide, amide, etc.), In some embodiments of the foregoing, ' is a covalent bond; in other embodiments,
- R' is CI to C3 alkyiene such as -(. ' 3 ⁇ 4- ⁇ .
- R is phenyl, piperidyl, or C1-C8 alkyl, or C3 to C8 cycloaikyl, which phenyl, pipyridyl, alkyl, or cycloaikyl alkyl is unsubstituted or substituted from 1 to 3 times with sulfono, halo, amino, nitro, alkyl, alkoxy!, haloaikyl, cycloaikyl. heterocyc!oaikyl, aryl, or heteroaryl.
- R 8 is Cl-CS alkyl or cyclohexyl, which alkyl or cyclohexy! is unsubstituted or substituted from 1 to 3 times with hydroxy! or amino.
- R 3 is CI-C8 alkyl, C3-C8 cycloaikyl, C4-C12 cycloalkylaikyl,
- C3-C8 heteroeycioalkyh C4-C12 heterocycloaikylalkyl, C4-C12 arylalkyl, C4-C12 heteraarylaikyl, each of which is imsubstituted or substituted from, one to three times with hydroxy!, halo, or aikoxy.
- R 4 is H.
- linking groups include, but are not limited to:
- each n and m is independently 0, 1 , 2, 4, 5, or 6; and where each of the linking grou structures illustrated above may optionally be substituted, e.g., substituted one, two, or three times with independently selected polar groups.
- Patic lar examples of compounds of the present invention include but are not limited to those set forth in Tables 1-7 below.
- the active compounds disclosed herein can, as noted above, be provided in the form of their pharmaceutically acceptable salts.
- Pharmaceutically acceptable salts are salts that retain the desired biological activity of the parent compound and do not impart undesired toxieologieai. effects.
- Examples of such salts are (a) acid addition salts formed with inorganic acids, for example hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid, nitric acid and the like; and salts formed with organic acids such as, for example, acetic acid, oxalic acid, tartaric acid, succinic acid, raaleic acid, fumaric acid, gluconic acid, citric acid, malic acid, ascorbic acid, benzoic acid, tannic acid, palmitic acid, alginic acid, polyglutamic acid, naphihalenesulfonic acid, methanesulfonic acid, p-toiuenesulfonic acid, naphthalenedisul
- Active compounds as described herein can be prepared in accordance with known procedures, or variations thereof that will be apparent to those skilled in the art.
- the active compounds described above may be formulated for administration in a pharmaceutical carrier in accordance with known techniques. See, e.g., Remington, The Science And Practice of Pharmacy (9 th Ed. 1995).
- the active compound (including the physiologically acceptable salts thereof) is typically admixed with, inter alia, an acceptable carrier.
- the carrier must, of course, be acceptable in the sense of being compatible with any other ingredients in the formulation and must not be deleterious to the patient.
- the carrier may be a solid or a liquid, or both, and is preferably formulated with the compound as a unit-dose formulation, for example., a tablet, which may contain from 0.01 or 0.5% to 95% or 99% by weight of the active compound.
- One or more active compounds may be incorporated in the formulaiions of the invention, which may be prepared by any of the well. known techniques of pharmacy comprising admixing the components, optionally including one or more accessory ingredients.
- the formulations of the invention include those suitable for oral, rectal, topical, buccal (e.g., sub-lingual), vaginal, parenteral (e.g., subcutaneous, intramuscular, intradermal, or intravenous), topical (i.e., both skin and mucosal surfaces, including airway surfaces), transdermal administration, and intraventricular injection (injection into a ventricle of the brain, e.g., by an implanted catheter or ormnan reservoir, such as in the case of morbid obesity) and although the most suitable route in any given case will depend on the nature and severity of the condition being treated and on the nature of the particular active compound which is being used.
- Formulations suitable for oral administration may be presented in discrete units, such as capsules, cachets, lozenges, or tablets, each containing a predetermined amount of the active compound; as a powder or granules; as a solution or a suspension in an aqueous or non-aqueous liquid; or as an oil-in-water or water-in-oil emulsion.
- Such formulations may be prepared by any suitable method of pharmacy which includes the step of bringing into association the active compound and a suitable carrier (which may contain one or more accessory ingredients as noted above).
- the formulations of the invention are prepared by uniformly and intimately admixing the active compound with a liquid or finely divided solid carrier, or both, and then, if necessary, shaping the resulting mixture.
- a tablet may be prepared by compressing or molding a powder or granules containing the active compound, optionally with one or more accessory ingredients.
- Compressed tablets may be prepared by compressing, in a suitable machine, the compound in a free-flowing form, such .as a powder or granules optionally mixed with a binder, lubricant, inert diluent, and/or surface active/dispersing agent(s).
- Molded tablets may be made by molding, in a suitable machine, the powdered compound moistened with an inert liquid binder,
- Formulations suitable for buccal (sub-lingual) administration include lozenges comprising the active compound in a flavoured base, usually sucrose and acacia or tragacanth; and pastilles comprising the compound in an inert base such as gelatin and glycerin or sucrose and acacia,
- Formulations of the present invention suitable for parenteral administration comprise sterile aqueous and non-aqueous injection solutions of the active compound, which preparations are preferably isotonic with the blood of the intended recipient. These preparations may contain anti-oxidants, buffers, bacteriostais and solutes which render the formulation isotonic with the blood of the intended recipient.
- Aqueous and non-aqueous sterile suspensions may include suspending agents and thickening agents.
- the formulations may be presented in unit ⁇ dose or multi-dose containers, for example sealed ampoules and vials, and may be stored in a freeze-dried (iyophilized) condition requiring only the addition of the sterile liquid carrier, for example, saline or water-for-injection immediately prior to use.
- Extemporaneous injection solutions and suspensions may be prepared from sterile powders, granules and tablets of the kind previously described.
- an injectable, stable, sterile composition comprising a compound of Formula ( ⁇ ) > or a salt thereof, in a unit dosage form in a sealed container.
- the compound o salt is provided in the form of a iyophilizate which is capable of being reconstituted with a suitable pharmaceutically acceptable carrier to form a liquid composition suitable for injection thereof into a subject.
- the unit dosage form typically comprises from about 10 rag to about 1.0 grams of the compound or salt.
- emulsifying agent which is physiologically acceptable may be employed in sufficient quantity to emulsify the compound or salt in an aqueous carrier.
- emulsifying agent is phosphatidyl choline.
- Formulations suitable for rectal administration are preferably presented as unit dose suppositories. These may be prepared by admixing the active compound with one or more conventional solid carriers, for example, cocoa butter, and then shaping the resulting mixture.
- Formulations suitable for topical application to the skin preferably take the form, of an ointment, cream, lotion, paste, gel, spray, aerosol, or oil.
- Carriers which may be used include petroleum jelly, lanoUne, polyethylene glycols, alcohols, transdermal enhancers, and combinations of two or more thereof.
- Formulations suitable for transdermal administration may be presented as discrete patches adapted to remain in intimate contact with the epidermis of the recipient for a prolonged period of time. Formulations suitable for transdermal administration may also be delivered by iontophoresis (see, for example, Pharmaceutical Research 3 (6):318 (1986)) and typically take the form of an optionally buffered aqueous solution of the active compound. Suitable formulations comprise citrate or bis ⁇ tris buffer (pH 6) or ethanol/water and contain from O. i to 0.2M active ingredient.
- the present invention provides liposomal formulations of the compounds disclosed herein and salts thereof.
- the technology for forming liposomal suspensions is well known in the art.
- the compound or salt thereof is an aqueous-soluble salt, using conventional liposome technology, the same may be incorporated into lipid vesicles. In such an instance, due to the water solubility of the compound or salt, the compound or salt will be substantially entrained within the hydrophilic center or core of the liposomes.
- the lipid layer employed may be of any conventional composition and may either contain cholesterol or may be cholesterol-free.
- the salt may be substantially entrained within the hydrophobic lipid bilayer which forms the structure of the liposome.
- the liposomes which are produced may be reduced in size, as through the use of standard soni cation and homogenization techniques.
- liposomal formulations containing the compounds disclosed herein or salts thereof may be Iyophilized to produce a lyophilizate which may be reconstituted with a pharmaceutically acceptable carrier, such as water, to regenerate a liposomal suspension.
- a pharmaceutically acceptable carrier such as water
- compositions may be prepared from the water-insoluble compounds disclosed herein, or salts thereof, such as aqueous base emulsions.
- the composition will contain a sufficient amount of pharmaceutically acceptable emulsifying agent to emulsify the desired amount of the compound or salt thereof.
- Particularly useful emulsifying agents include phosphatidyl cholines, and lecithin.
- the pharmaceutical compositions may contain other additives, such as pB -adjusting additives.
- useful pH-adjusting agents include acids, such as hydrochloric acid, bases or buffers, such as sodium lactate, sodium acetate, sodium phosphate, sodium citrate, sodium borate, or sodium gluconate.
- the compositions may contain microbial preservatives.
- Useful microbial preservatives include methylparaben, propylparaben, and benzyl alcohol. Th microbial preservative is typically employed when the formulation is placed in a vial designed for multidose use.
- the pharmaceutical compositions of the present invention may be Iyophilized using techniques well known in the art,
- the present invention provides pharmaceutical formulations comprising the active compounds (including the pharmaceutically acceptable salts thereof), in pharmaceutically acceptable carriers for oral, rectal, topical, buccal, parenteral, intramuscular, intradermal, or intravenous, and. transdermal administration.
- the therapeutically effective dosage of any specific compound will vary somewhat from compound to compound, and patient to patient, and will depend upon the condition of the patient and the route of delivery.
- a dosage from about 0.1 to about 50 mg kg will have therapeutic efficacy, with all weights being calculated based upon the weight of the active compound, including the cases where a salt is employed.
- Toxicity concerns at the higher level may restrict intravenous dosages to a lower level such as up to about 10 mg/kg, with all weights being calculated based upon, the weight of the active base, including the cases where a salt is employed.
- a dosage from about 10 mg/kg to about 50 mg/kg may be employed for oral administration.
- a dosage from about 0,5 mg/kg to 5 mg/kg may be employed for intramuscular injection, In some embodiments, dosages are 1 pmoi/kg to 50 imo /kg, and more preferabl 22 ⁇ ⁇ kg and 33 ⁇ /kg of the compound for intravenous or oral administration, The duration of the treatment can be once per day for a period of two to three weeks or until the condition is essentially controlled.
- Active compounds may be administered as pharmaceutically acceptable prodrugs, which are those prodrugs of the active compounds of the present invention which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of humans and lower animals without undue toxicity, irritation, allergic response and the like, commensurate with a reasonable risk benefit ratio, and effective for their intended use, as well as the zwitterionie forms, where possible, of the compounds of the invention.
- prodrug refers to compounds that are rapidly transformed in vivo to yield the parent compound of the above formutae, for example, by hydrolysis in blood, A thorough discussion is provided in T. Higuchi and V. Stella, Prodrugs as Novel delivery Systems, Vol. 14 of the A.C.S. Symposium Series and in Edward B.
- Examples include a prodrug that is metabolized in vivo by a subject to an active drug having an activity of active compounds as described herein, wherein the prodrug is an ester of an alcohol or carboxylic acid group, if such a group is present in the compound; an acetal or keial of an alcohol group, if such a group is present in the compound; an N-Mannich base or an inline of an amine group, if such a group is present in the compound; or a Schiff base, oxime, acetal, enol ester, oxazolidine, or thiazolidine of a carbonyl group, if such a group is present in the compound, such as described in US Patent No. 6,680,324 and US Patent No. 6,680,322.
- Example cancers that may be treated by the compounds and methods of the invention inciude, but are not limited to, myeioid leukemia, lymphoblastic leukemia, melanoma, breast, lung, colon, liver, gastric, kidney, ovarian, uterine, and brain cancer.
- i-Qrniihine 232 mg, 1 ,0 mmol
- N.N-diisopropyleihylarnine (194 nig, 1.5 mmol.) were added into a solution of 2,4-dichloro-N-(4-fluorobenzyl)pyrimidine-5-carboxamide (300 mg, 1.0 mmol) i a mixture of isopropyl alcohol and DMF (10 mL, 3:2, v/v) at room temperature.
- the resulting mixture was stirred for overnight.
- 1/3 of the reaction mixture was added to a solution of 1,3-diaminopropane (304 mg, 4.0 mmol) in DMF (1.0 mL) at room temperature.
- Table 1 describes compounds prepared following procedures described in Example 4 (General Procedure D), using appropriate reagents.
- Mer 1C50 ++++ ⁇ means ⁇ 10 nM; ⁇ - means between 10-lOOnM, ++ means between 100 tiM-i uM; + means between 1-30 ⁇ , ⁇ ; - means inactive.
- Table ⁇ describes compounds prepared following procedures described in Example 5 (General Procedure E), using appropriate reagents.
- the reaction was heated at 70 °C for 6 h and the volatiles were removed under a reduced pressure.
- the residue was dissolved in CH2CI2 (4.0 mL) and washed with 3 ⁇ 40 (2.0 mL).
- the 3 ⁇ 40 layer was extracted with CH 2 C1 2 (2 ⁇ 20 mL); the organic layers were combined, dried (NaiSO ⁇ ), and the solvent was removed under a reduced pressure.
- the residue was purified by ISCO silica gel column to provid the title compound (39 mg, 25%) as a white solid. ⁇ N.MR.
- Tlie voiatiles were removed and the residue was purified by IS CO silica gel column to provide the RCM product as a crude brown solid.
- the residue was dissolved in MeOH (1.0 mL) and Pd/C (10 mg) was added. The mixture was stirred at room temperature under a hydrogen atmosphere for 24 h. The mixture was filtered through C elite and washed with MeOH then purified by HPLC to afford the title compound as a white solid (5.3 mg, 16%).
- Table ⁇ describes compounds can be prepared following procedures described in Example 6 (General Procedure F), using appropriate reagents.
- the organic layer was washed with brine (10 mL) and dried to provide the crude mesylate that was used for the next step without further purification.
- the mesylate was dissolved in THF (7,3 mL) and TBAF (1M i THF, 585 L, 0.585 mmol) was added. The reaction mixture was stirred at room temperature overnight. The solvents were removed under a reduced pressure and the residue was purified by ISCO silica gel column to provide the title compound (32 mg, 34%) as a white solid.
- Table j ⁇ describes compounds prepared following procedures described in Example 10 (General Procedure .1), using appropriate reagents.
- reaction mixture was stirred at room temperature for 3.5 h, then was added dropwise to a solution of 6-azidohexan- 1 -amine (0.801 g, 5.64 mmol) in ethanol (4.0 mL) at 50 °C After the reaction was complete (monitored by LCMS), the mixture was diluted with water (10 mL) and concentrated under a reduced pressure and filtered. The yellow solid was washed with water and dried under vacuum to be used in the next step without further purification (0.723 g, 39% over 3 steps).
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Compounds of Formula I or II: are described, along with pharmaceutical compositions containing the same and methods of using such compounds for the treatment of cancer.
Description
PYRJMIDI E COMPOUNDS
FOR THE TREATMENT OF CANCER
Xiaodong Wang, Weihe Zhang, Stephen V. Frye, Dmitri Kireev,
Andrew L. Mclver. and J'ing Liu
Related A p plica lions
This application is related to PCT Applicationd Nod. PCT/US 20.11/036215 filed May 12, 2011 (Attorney Docke No. 5470-549WO); PCT/US2012/0582:98 filed Oct 01, 2012 (Attorney Docket No. S470-610WO); and PCT/US2013/042033, tiled May 21 , 2013 (Attorney Docket No. 5470-627 WO).
Field of the Invention
The present invention, concerns compounds, compositions and methods for the treatment of cancer.
Background of the. iBveatioi.
Acute Lymphoblastic Leukemia (ALL) is the most common malignancy in children and common varieties are cured by chemotherapy in 75%~85% of the cases. Collectively the less common T ceil and rare B ceil subsets represent less than 2000 cases yearly and thus can be classified as a rare disease; these subsets have a poorer prognosis. Unfortunately with either subset, resistance to and relapse from therapy is a major cause of pediatric cancer death. In addition. ALL chemotherapies can cause late complications that are increasingly recognized in pediatric survivor populations. In fact, in pediatric cancer survivors, the incidence of severe !ate effects (neurocognitive sequelae, auditory complications, cardiovascular dysfunction, gastrointestinal/hepatic dysfunction, growth delay, secondary malignancies, and infertility) directly related to therapy is approximately 25%. A better understanding of therapeutic resistance and its reversal could not only help those who relapse but may help lower the dose of chemotherapy needed in ALL patients thus reducing long-term toxicity for future survivors.
Summary of thejDyention
A first aspect of the invention is active compounds of Formula 1 or ii:

Ring A is a 5- or 6-raembered heteroaryl group such as pyridyl, pyrimidyl. thiazol, furanyl, pyridazinyl, pyrazinyl, imidazol, etc.. The dashed line is an optional double bond, X is N or O. Y is C. S or N and ca move on the ring.
R is ~RJR°, where RJ is a covaient bond or CI to C3 aikyl or a linker group (for example, sulfonamide, amide, etc.) and R6 is cycloalkyl. heterocycloaikyl, aryl, heteroaryl aikylcycloalkyl, alkylbeterocycloalkyl, alkylaryl, aikylheteroaryi, or alkyl, and wherein R6 is optionally substituted one, two or three times with independently selected polar groups:
R4 is -R7RS, where R7 is a covaient bond or CI to C3 alkyl and R8 is cycloalkyl, heterocycloaikyl, aryl, heteroaryl or alkyl, and wherein R8 is optionally substituted one, two or three times with independently selected polar groups;
R3 is selected from the group consisting of H, alkyl, arylalkyl; cycioalkyialkyi, heterocyeloaikylalkyl, heteroaryalkyl, and aikoxyalkyl, each of which is optionally substituted one, two or three times with independently selected polar groups;
or " and R" together form a linking group;
R4 is H, loweralkyl, halo, or loweraikox'v;
or a pharmaceutically acceptable salt thereof.
A further aspect of the invention is an active compound as described herein in a pharmaceutically acceptable carrier.
A further aspect of the invention is a method of treating cancer in a subject, in need thereof, comprising administering said subject an active compound as described herein in an amount effective to treat the cancer.
A fuller aspect of the invention is an active compound as described herein for use in treating cancer, and/or for the preparation of a medicament for the treatment of cancer.
Detailed Description of Prefer ed Emboidiments
"Alkyl" as used herein alone or as pariof another group, refers to a straight or branched chain hydrocarbon containing from 1 to 10 carbon atoms. Representative examples of alky! include, but. are not limited, to, methyl, ethyl, n-propyl, iso-propy!, n-butyl, sec-butyl, iso-b tyl, tert-butyl, n-pentyl, isopentyl, neopentyL n-hexyl, 3-methylhexyl, 2,2- dimethyl entyl, 2,3-dimethylpentyl, n-heptyl, n-octyl, n-nonyl, n-decyl, and the like, "Lower alkyl." as used herein, is a subset of alkyl, in some embodiments preferred, and refers to a straight or branched chain hydrocarbon grou containing from 1 to 4 carbon atoms. Representative examples of lower alkyl include, but are not limited to. methyl, ethyl, n- propyi, iso-propyl, n-butyl, iso-butyl, tert-butyl, and the like. The term "alkyl" or "loweralkyi" is intended to include both substituted and unsubstituied alkyl or loweraikyl unless otherwise indicated and these groups may be substituted with groups selected from halo (e.g., haloalkyl), alkyl, haloalkyl, alkenyl, a!kynyl, cyeloalkyl, cycloalkylalkyl, aryl, arylalkyl, heterocyclo, heterocycloaikyf hydroxy], aikoxy (thereby creating a polyalkoxy such as polyethylene glycol), aikenyloxy, alkynyloxy, haloaikoxy, cyc!oalkoxy, cycloalkylalkyloxy, aryloxy, aryialkyioxy, heierocyclooxy, heterocycloiaikvloxy, mercapto, alkyl-S(0)m, halo alkyl- S(0)mj alkenyl-S(0)m> alkynyl-S(0)m, eyeloalkyi-5(0)m, cycloalkyIaikyl-S(0)m, aryi-S(Q)m, arylalkyl-S(0)ms. heterocyclo-S(0)m, heterocycloalkyl- S(0)m, amino, carboxy, aikyiamino, alkenylammo, alkynylarntno, haloalkylamino, eycioalkyiamino, cycloalkyialkylamino, arylamino, aryl aikyiamino, heterocycioamino, heieroeycioalkylamino, disubstituted-axnino, acyl amino, acyloxy, ester, amide, sulfonamide, urea, aikoxyaeylaniino, aminoacyloxy, nitro or cyano where m- Q, 1 , 2 or 3.
"Alkenyl" as used herein alone or as part of another group, refers to a straight or branched chain hydrocarbon containing from 1 to 10 carbon atoms for in Ioweralkeny 1 1 to 4 carbon atoms) which include 1 to 4 double bonds in the normal chain. Representative examples of alkenyl include, but are not limited to, vinyl, 2-propenyl, 3-butenyl, 2-butenyl, 4- pentenyl, 3-pentenyl, 2-hexenyl, 3-bexenyl, 2,4-heptadiene, and the like. The term "alkenyl" or "loweralkenyl" is intended to include both substituted and unsubstituied alkenyl or loweralkenyl unless otherwise indicated and these groups may be substituted with groups as described in connection with alkyl and loweralkyi above.
"Alkynyl" as used herein alone or as part o another group, refers to a straight or branched chain hydrocarbon containing from 1 to 10 carbon atoms (or in loweralkynyl 1 to 4 carbon atoms) which include 1 triple bond in the normal chain. Representative examples of
alkynyl include, but are not limited to, 2-propynyl, 3-butynyi, 2- buiynyl, 4-pentynyl. 3- pentynyi, and the like. The term "alkynyl" or "ioweralkynyl" is intended to include both substituted and unsubstitvtted alkynyl or Ioweralkynyl unless otherwise indicated and these groups may be substituted with the same groups as set forth in connection with alkyl and ioweraikyl above.
"Cycloalkyl" as used herein alone or as part of another group, refers to a saturated or partially unsaturated cyclic hydrocarbon group containing from 3, 4 or 5 to 6. 7 or 8 carbons (which carbons may be replaced in heterocyclic group as discussed below). Representative examples of cycloalkyl include, cyclopropyl, cyclobutyl, cyclopenty!, cyelohexyl, eyeloheptyl. and cyclooctyl. These rings may be optionally substituted with additional substituents as described herein such as halo or Ioweralkyl The term "cycloalkyl" is generic and intended to include heterocyclic groups as discussed below unless specified otherwise.
"Heterocyclic group'1 or "heterocycio" as used herein alone or as part of another group,, refers to an aliphatic (e.g., fully or partially saturated heterocyclo) or aromatic (e.g., heteroaryt) monocyclic- or a bicyclic-ring system, Monocyclic ring systems are exemplified by any 5 or 6 membered ring containing 1 , 2, 3, or 4 heteroatoms independently selected from oxygen, nitrogen and sulfur. The 5 membered ring has from 0-2 double bonds and the 6 membered ring has from 0-3 double bonds. Representative examples of monocyclic ring systems include, but are not limited to, azetidine. azepine, aziridine, diazepine, 1 ,3-dioxolane, dioxane, dithiane, furan, imidazole, imidazoline, imidazolidine, isothiazole, isothiazoline, isothiazo!idine, isoxazole, isoxazoline, isoxazolidine, morphoiine, oxadiazole, oxadiazoline, oxadiazoiidme, oxazole, oxazoline, oxazolidine, piperazine, piperidine, pyran, pyrazine, pyrazole, pyrazolone, pyrazolidine, pyridine, pyrimidine. pyridazine, pyrrole, pyrroline, pyrrolidine, tetrahydro furan, tetrahydrothiophene, tetrazine, tetrazole, thiadiazole, thiadiazoline, thiadiazolidine, thiazole, thiazoline, thiazolidine, ihiophene, thiomorpholine, thiomorpholine sulfone, thiopyran, triazine, triazole, tritbiane, and the like, Bicyclic ring systems are exemplified by any of the above monocyclic ring systems fused to an aryl group as defined herein, a cycloalkyl group as defined herein, or another monocyclic ring system as defined herein. Representative examples of bicyclic ring systems include but are not limited to, for example, benzimidazole, benzoihiazole, benzothiadiazoie, benzothiophene, benzoxadiazole, benzoxazole, benzofuran, benzopyran, benzothiopyran. benzodioxine, 1,3- benzodioxo!e, cinnoline. indazole, indole, indoline, indolizine, naphthyridine, isobenzofuran, isobenzothiophene, isoindole, isoindoline, isoquinoline, phthalazine, purine, pyranopyridine,
quinoline, qinnolizine, quinoxaiine, quinazoline, tetrahydroisoquinoline, tetrahydroquinoline, thiopyranopyridine, and the like. These rings include quaternized derivatives thereof and may be optionally substituted with groups selected from halo, alkyl, haloaikyi, alkenyl, alkynyl, cycloaikyl, cycloalkyialkyl, aryl, arylalkyl, heterocycio, heterocycioalkyl, hydroxy!, alkoxy, alkenyloxy, aikynyioxy, haloalkoxy, eycloalkoxy, cyeioalkylalkyloxy, aryloxy, arylalkyloxy, heterocyclooxy, heterocyclolalkyloxy, mercapto, alkyl~S(0)m, haioalkyl~S(0)m, alkenyl- S(0)m, alkynyl -S(0)m, cycloalkyi~S(Q)mj cycloalkylalkyl-S(0)m, aryl-S(0)1Tl! arylalkyl - S(0)m, heterocyclo-S(0)ms heterocycloalkyl-S(0)m, amino, alkylamino, alkenylamino, alkynyl amino, haloalkylamino, cycloalkylamino, cycioaikylalkylamino, arylamino, arylalkylamino, heterocycloamino. heterocycloalkyiamino, disubstituted-amino, acylaraino, acyloxy, ester, amide, sulfonamide, urea, alkoxyacylammo, ammoacyloxy, nitro or cyano where m - 0, 1 , 2 or 3.
"Aryl" as used herein alone or as part of another group, refers to a monocyclic carbocyclic ring system or a bicyclic carbocyclic fused ring system having one or more aromatic rings. Representative examples of aryl include, azuienyl, indanyl, indenyl, naphthyl, phenyl, tetrahydronaphthyl, and the like. The term "aryl" is intended to include both substituted and unsubstituted aryl unless otherwise indicated and these groups may be substituted with the same groups as set forth in connection with alkyl and loweralkyl above.
"Arylalkyl" as used herein alone or as part of another group, refers to an aryl group, as defined herein, appended to the parent molecular moiety through an alkyl group, as defined herein. Representative examples of arylalkyl include, but are not limited to, benzyl, 2- phenylethyl, 3-phenylpropyl, 2-naphth-2-ylet;hyl, and the like.
"Heteroaryi" as used herein is as described in connection with heterocycio above,
"Alkoxy" as used herein alone or as part of another group, refers to an alkyl or loweralkyl group, as defined herein (and thus including substituted versions such as polyalkoxy), appended to the parent molecular moiety through an oxy group, -0-. Representative examples of alkoxy include, but are not limited, to, methoxy, ethoxy, pro poxy, 2-propoxy, butoxy, iert-butoxy, pentyioxy, hexyloxy and the like.
"Halo" as used herein refers to any suitable halogen, including -F, -CI, -Br, and -I.
"Mercapto" as used herein refers to an. -SB group,
"Azido" as used herein refers to an -N3 group.
"Cyano" as used herein refers to a -CN group.
"Formyi" as used herein refers to a -C(0)H group.
"Carboxylic acid" as used herein refers to a -C(0)GH group.
"Hydroxy 1" as used herein refers to an -OH group.
" itro" as used herein refers to an -NO?, group.
"Acyl" as used herein alone or as part of another group refers to a -C(0)R radical, where R is any suitable substituent such as aryl. aikyl, aikenyi, alkynyi, cycloaikyl or other suitable substituent as described herein.
"Alkylthio" as used herein alone or as part of another group, refers to an alkyl group, as defined herein, appended to the parent molecular moiety through a thio moiety, as defined herein. Representative examples of alkylthio include, but are not limited, methylthio, ethylthio. tert-butylihio, hexylihio, and the like.
"Amino" as used herein means the radical ~-N¾.
"Alkylaniino" as used herein alone or as part of another group means the radical - NHR, where R is an alkyl group.
"Arylalkylamino" as used herein alone or as part of another group means the radical ~ NHR, where R is an aryl alkyl group.
"Disubstituted-amino" as used herein alone or as part of another group means the radical -NRsRb, where Ra and Rlt are independently selected from the groups alkyl, haloalkyl, aikenyi, alkynyi. cycloaikyl, cycloalkyialkyl, aryl, arylalkyl, heterocyclo, heterocycloaikyl.
"Acylamino" as used herein alone or as part of another group means the radical -· NRaRb, where Ra is an acyl group as defined herein and R is selected from the groups hydrogen, alkyl, haloalkyl, aikenyi, alkynyi, cycloaikyl, cycloalkyialkyl, aryl, arylalkyl, heterocyclo , hetero c c 1 o alkyl .
"Aeyioxy" as used herein alone or as pari of another group means the radical -OR, where R is an acyl group as defined herein.
"Ester" as used herein alone or as part of another group refers to a -C(0)OR radical, where R is any suitable substituent such as alkyl, cycloaikyl, aikenyi, alkynyi or aryl.
"Amide" as used herein alone or as part of another group refers to a -C(0)NRaRb radical, where Ra and Rb are any suitable substituent such as alkyl, cycloaikyl, aikenyi, alkynyi or aryl.
"Sulfoxyl" as used herein refers to a compound of the fomiula -S(0)R. where R is any suitable substituent such as alkyl, cycloaikyl, aikenyi, alkynyi or aryl.
"Sulfonyl" as used herein refers to a compound of the fomiula -S(0)(0)R, where R is any suitable substituent such as amino, alkyl, cycloaikyl, aikenyi, alkynyi or aryl.
"Sulfonate" as used herein refers to a compound of the formula -S(0)(0)OR, where R is any suitable substituent such as alkyl, cycloaikyl, alkenyl, aikynyl or aryi,
" Sulfonic acid" as used herein refers to a compound of the formula -S(0)(0)OH.
"Sulfonamide" as used herein alone or as pail of another group refers to a - S(0)2NRaRb radical, where Ra and Rb are any suitable substituent such as H, alkyl, cycloaikyl, alkenyl, aikynyl or aryL
"Urea" as used herein alone or as part of another group refers to an - (Rc)C(0) RaRb radical where Ra, Rb and e are any suitable substituent such as H, alkyl, cycloaikyl, alkenyl, aikynyl or aryL
"Alkoxyacylamino" as used herein alone or as part of another group refers to an - N(Ra)C(0)ORb radical, where Ra, Rb are any suitable substituent such as H, alkyl, cycloaikyl, alkenyl, aikynyl or aryL
"Ammoacyioxy" as used herein alone or as part of another group refers to an - OC(0)NRaRb radical, where Ra and Rb are any suitable substituent such as H, alkyl, cycloaikyl, alkenyl, aikynyl or aryL
"Polar group" as used herein refers to a group wherein the nuclei of the atoms co vaiently bound to each other td form the group do not share the electrons of the eovalent bond (s) joining them equally; that is the electron cloud is denser about one atom than another. This results in one end of the eovalent bond(s) being relatively negative and the other end relatively positive; i.e., there is a negative pole and a positive pole. Examples of polar groups include, without limitations, halo, hydroxy, alkoxy, carboxy, nitro, cya.no, amino (primary, secondary and tertiary), amido, ureido, sulfonamido, sulfinyl, suifhydryl silyl, S- sulfonamido. N-sulfonamido, C-carboxy, O-carboxy, C-amido. N-amido, sulfonyl, N-ferr- butoxycarbonyl (or "r-BOC") groups, phosphono, morpholino, piperazinyL tetrazolo, and the like. See, e.g., U.S. Pat. No. 6,878,733, as well as alcohol, thiol, polyethylene glycol, polyol (including sugar, aminosugar, uronic acid), sulfonamide, carboxamide, hydrazide, N- hydroxycarboxamide, urea, metal chelates (including macrocyclic ligand or crown ether metal chelates). The polar group can be an ionic group.
"Ionic group" as used herein includes anionic and cationic groups, and includes groups (sometimes referred to as "ionogenic" groups) that are uncharged in one form but can be easily converted to ionic groups (for example, by protonation or deprotonation in aqueous solution). Examples include but are not limited to carhoxylate, sulfonate, phosphate, amine, N-oxide, and ammonium (including quaternized heterocyclic amines such as imidazolium
and pyridiniurrs) groups. See, e.g., U.S. Pat. Nos. 6,478,863; 6,800,276; and 6,896,246. Additional examples include tironic acids, carboxylic acid, sulfonic acid, amine, and moieties such as guanidinium, phosphoric acid, phosphonic acid, phosphatidyl choline, phosphonium, borate, sulfate, etc,
"Deuterium" as used herein alone or as part of another group, refers to a safe, nonradioactive relative of hydrogen. Any hydrogen in a group or substitueni described above may be replaced with deuterium to provide a "deuterated" compound, in some embodiments to modify and/or improve metabolic stability, resulting in better safety, toierability and/or efficacy.
"Linking group" or "linker group" as used herein are generally bivalent aromatic, aliphatic, or mixed aromatic and aliphatic groups. Thus linking groups include linear or branched, substituted or imsubstituted aryl, aikyl, alkylaryl, or alkylaryl alkyl linking groups, where the alkyl groups are saturated or unsaturated, and where the alkyl and aryl groups optionally containing independentl selected heteroatoms such as 1 , 2, 3 or 4 heteroatoms selected from the group consisting of N, 0, and S. In some embodiments, linking groups containing from 2 to 20 carbon atoms are preferred. Numerous examples of suitable linking groups are known, including but not limited to those described in, US Patents Nos. 8,247,572; 8,097,609: 6,624,317; 6,613,345; 6,596,935; and 6,420,377, the disclosures of which are incorporated by reference herein in their entirety.
"Treat" as used herein refers to any type of treatment that imparts a benefit to a patient afflicted with a disease, including improvement in the condition of the patient {e.g., in one or more symptoms), delay in the progression of the disease, delay in onset of the disease, etc.
"Pharmaceutically acceptable" as used herein means that the compound or composition is suitable for administration to a subject to achieve the treatments described herein, without unduly deleterious side effects in light of the severity of the disease and necessity of the treatment.
Active compounds of the present invention may optionally be administered in conjunction with other compounds useful in the treatment of cancer. The other compounds may optionally be administered concurrently. As used herein, the word "concurrently" means sufficiently close in time to produce a combined effect (that is. concurrently may be simultaneously, or it may be two or more events occurring within a short time period before or after each other).
The present invention is primarily concerned with the treatment of human subjects, but the invention may also be carried out on animal subjects, particularly mammalian subjects such as mice, rats, dogs, cats, livestock and horses for veterinary purposes, and for drug screening and drug development purposes. Subjects may be of any age, including infant, juvenile, adolescent, adult, and geriatric subjects.

As noted above, the present invention provides active compounds of Formula I or II:

Rl is -R^R6, where Rs is a covaient bond, CI to C3 alky! or a linker group (for example, sulfonamide, amide,, etc.) and R6 is eycloalkyl, heterocycloalkyl, aryi, heteroaryl, alkylcycloalkyi, alkylheterocycloalkyl, al.kyl.aryl, alkyiheteroaryl or alkyl, and wherein R6 is optionally substituted one, two or three times with independently selected polar groups;
RA is ~R7RS, where R' is a covaient bond or CI to C3 alkyl and R8 is eycloalkyl, heterocycloalkyl, aryl, heteroaryl or alkyl. and wherein R is optionally substituted one, two or three times with independently selected polar groups;
R is selected from the group consisting of H, alkyl, aryla!kyk cycloalkylaikyl, heterocycioalkylaikyl, heieroaryalkyl, and alkoxyalkyl, each of which is optionally substituted one, two or three times with independently selected polar groups;
or R" and R" together form a Unking group;
R4 is H, !oweralkyk halo, or loweralkoxy;
or a pharmaceutically acceptable salt thereof.
in some embodiments of the foregoing, 3 is a covaient bond; in other embodiments, R" is CI to C3 alkylene such as -0¾~ or R is a linker group (for example, sulfonamide, amide, etc.),
In some embodiments of the foregoing, ' is a covalent bond; in other embodiments,
R' is CI to C3 alkyiene such as -(.'¾-·.
In some embodiments, R is phenyl, piperidyl, or C1-C8 alkyl, or C3 to C8 cycloaikyl, which phenyl, pipyridyl, alkyl, or cycloaikyl alkyl is unsubstituted or substituted from 1 to 3 times with sulfono, halo, amino, nitro, alkyl, alkoxy!, haloaikyl, cycloaikyl. heterocyc!oaikyl, aryl, or heteroaryl.
In some embodiments, wherein R8 is Cl-CS alkyl or cyclohexyl, which alkyl or cyclohexy! is unsubstituted or substituted from 1 to 3 times with hydroxy! or amino.
in some embodiments, R3 is CI-C8 alkyl, C3-C8 cycloaikyl, C4-C12 cycloalkylaikyl,
C3-C8 heteroeycioalkyh C4-C12 heterocycloaikylalkyl, C4-C12 arylalkyl, C4-C12 heteraarylaikyl, each of which is imsubstituted or substituted from, one to three times with hydroxy!, halo, or aikoxy.
In some embodiments, R4 is H.
Specific examples of linking groups include, but are not limited to:

Patic lar examples of compounds of the present invention include but are not limited to those set forth in Tables 1-7 below.
The active compounds disclosed herein can, as noted above, be provided in the form of their pharmaceutically acceptable salts. Pharmaceutically acceptable salts are salts that retain the desired biological activity of the parent compound and do not impart undesired toxieologieai. effects. Examples of such salts are (a) acid addition salts formed with inorganic acids, for example hydrochloric acid, hydrobromic acid, sulfuric acid, phosphoric acid, nitric acid and the like; and salts formed with organic acids such as, for example, acetic acid, oxalic acid, tartaric acid, succinic acid, raaleic acid, fumaric acid, gluconic acid, citric acid, malic acid, ascorbic acid, benzoic acid, tannic acid, palmitic acid, alginic acid, polyglutamic acid, naphihalenesulfonic acid, methanesulfonic acid, p-toiuenesulfonic acid, naphthalenedisulfonic acid, polygalacturonic acid, and the like; (b) salts formed from elemental anions such as chlorine, bromine, and iodine, and (c) salts derived from bases, such as ammonium salts, alkali metal, salts such as those of sodium and potassium, alkaline earth metal salts such as those of calcium and magnesium, and salts with organic bases such as dicyclohexylamine and N-metliyl-D-glucamine.
Active compounds as described herein can be prepared in accordance with known procedures, or variations thereof that will be apparent to those skilled in the art.
2. Pharmaceutical . formulations.
The active compounds described above may be formulated for administration in a pharmaceutical carrier in accordance with known techniques. See, e.g., Remington, The Science And Practice of Pharmacy (9th Ed. 1995). In the manufacture of a pharmaceutical formulation according to the invention, the active compound (including the physiologically acceptable salts thereof) is typically admixed with, inter alia, an acceptable carrier. The carrier must, of course, be acceptable in the sense of being compatible with any other ingredients in the formulation and must not be deleterious to the patient. The carrier may be a solid or a liquid, or both, and is preferably formulated with the compound as a unit-dose formulation, for example., a tablet, which may contain from 0.01 or 0.5% to 95% or 99% by weight of the active compound. One or more active compounds may be incorporated in the
formulaiions of the invention, which may be prepared by any of the weil. known techniques of pharmacy comprising admixing the components, optionally including one or more accessory ingredients.
The formulations of the invention include those suitable for oral, rectal, topical, buccal (e.g., sub-lingual), vaginal, parenteral (e.g., subcutaneous, intramuscular, intradermal, or intravenous), topical (i.e., both skin and mucosal surfaces, including airway surfaces), transdermal administration, and intraventricular injection (injection into a ventricle of the brain, e.g., by an implanted catheter or ormnan reservoir, such as in the case of morbid obesity) and although the most suitable route in any given case will depend on the nature and severity of the condition being treated and on the nature of the particular active compound which is being used.
Formulations suitable for oral administration may be presented in discrete units, such as capsules, cachets, lozenges, or tablets, each containing a predetermined amount of the active compound; as a powder or granules; as a solution or a suspension in an aqueous or non-aqueous liquid; or as an oil-in-water or water-in-oil emulsion. Such formulations may be prepared by any suitable method of pharmacy which includes the step of bringing into association the active compound and a suitable carrier (which may contain one or more accessory ingredients as noted above). In general, the formulations of the invention are prepared by uniformly and intimately admixing the active compound with a liquid or finely divided solid carrier, or both, and then, if necessary, shaping the resulting mixture. For example, a tablet may be prepared by compressing or molding a powder or granules containing the active compound, optionally with one or more accessory ingredients. Compressed tablets may be prepared by compressing, in a suitable machine, the compound in a free-flowing form, such .as a powder or granules optionally mixed with a binder, lubricant, inert diluent, and/or surface active/dispersing agent(s). Molded tablets may be made by molding, in a suitable machine, the powdered compound moistened with an inert liquid binder,
Formulations suitable for buccal (sub-lingual) administration include lozenges comprising the active compound in a flavoured base, usually sucrose and acacia or tragacanth; and pastilles comprising the compound in an inert base such as gelatin and glycerin or sucrose and acacia,
Formulations of the present invention suitable for parenteral administration comprise sterile aqueous and non-aqueous injection solutions of the active compound, which
preparations are preferably isotonic with the blood of the intended recipient. These preparations may contain anti-oxidants, buffers, bacteriostais and solutes which render the formulation isotonic with the blood of the intended recipient. Aqueous and non-aqueous sterile suspensions may include suspending agents and thickening agents. The formulations may be presented in unit\dose or multi-dose containers, for example sealed ampoules and vials, and may be stored in a freeze-dried (iyophilized) condition requiring only the addition of the sterile liquid carrier, for example, saline or water-for-injection immediately prior to use. Extemporaneous injection solutions and suspensions may be prepared from sterile powders, granules and tablets of the kind previously described. For example, in one aspect of the present invention, there is provided an injectable, stable, sterile composition comprising a compound of Formula (ϊ)> or a salt thereof, in a unit dosage form in a sealed container. The compound o salt is provided in the form of a iyophilizate which is capable of being reconstituted with a suitable pharmaceutically acceptable carrier to form a liquid composition suitable for injection thereof into a subject. The unit dosage form typically comprises from about 10 rag to about 1.0 grams of the compound or salt. When the compound or salt is substantially water-insoluble, a sufficient amount of emulsifying agent which is physiologically acceptable may be employed in sufficient quantity to emulsify the compound or salt in an aqueous carrier. One such useful emulsifying agent is phosphatidyl choline.
Formulations suitable for rectal administration are preferably presented as unit dose suppositories. These may be prepared by admixing the active compound with one or more conventional solid carriers, for example, cocoa butter, and then shaping the resulting mixture.
Formulations suitable for topical application to the skin preferably take the form, of an ointment, cream, lotion, paste, gel, spray, aerosol, or oil. Carriers which may be used include petroleum jelly, lanoUne, polyethylene glycols, alcohols, transdermal enhancers, and combinations of two or more thereof.
Formulations suitable for transdermal administration may be presented as discrete patches adapted to remain in intimate contact with the epidermis of the recipient for a prolonged period of time. Formulations suitable for transdermal administration may also be delivered by iontophoresis (see, for example, Pharmaceutical Research 3 (6):318 (1986)) and typically take the form of an optionally buffered aqueous solution of the active compound. Suitable formulations comprise citrate or bis\tris buffer (pH 6) or ethanol/water and contain from O. i to 0.2M active ingredient.
Further, the present invention provides liposomal formulations of the compounds
disclosed herein and salts thereof. The technology for forming liposomal suspensions is well known in the art. When the compound or salt thereof is an aqueous-soluble salt, using conventional liposome technology, the same may be incorporated into lipid vesicles. In such an instance, due to the water solubility of the compound or salt, the compound or salt will be substantially entrained within the hydrophilic center or core of the liposomes. The lipid layer employed may be of any conventional composition and may either contain cholesterol or may be cholesterol-free. When the compound or salt of interest is water-insoluble, again employing conventional liposome formation technology, the salt may be substantially entrained within the hydrophobic lipid bilayer which forms the structure of the liposome. In either instance, the liposomes which are produced may be reduced in size, as through the use of standard soni cation and homogenization techniques.
Of course, the liposomal formulations containing the compounds disclosed herein or salts thereof, may be Iyophilized to produce a lyophilizate which may be reconstituted with a pharmaceutically acceptable carrier, such as water, to regenerate a liposomal suspension.
Other pharmaceutical compositions may be prepared from the water-insoluble compounds disclosed herein, or salts thereof, such as aqueous base emulsions. In such an instance, the composition will contain a sufficient amount of pharmaceutically acceptable emulsifying agent to emulsify the desired amount of the compound or salt thereof. Particularly useful emulsifying agents include phosphatidyl cholines, and lecithin.
In addition to compounds of formula (I) or their salts, the pharmaceutical compositions may contain other additives, such as pB -adjusting additives. In particular, useful pH-adjusting agents include acids, such as hydrochloric acid, bases or buffers, such as sodium lactate, sodium acetate, sodium phosphate, sodium citrate, sodium borate, or sodium gluconate. Futher, the compositions may contain microbial preservatives. Useful microbial preservatives include methylparaben, propylparaben, and benzyl alcohol. Th microbial preservative is typically employed when the formulation is placed in a vial designed for multidose use. Of course, as indicated, the pharmaceutical compositions of the present invention may be Iyophilized using techniques well known in the art,
3. Dosage aad routes o.LadmmjstrattoB.
As noted above, the present invention provides pharmaceutical formulations comprising the active compounds (including the pharmaceutically acceptable salts thereof),
in pharmaceutically acceptable carriers for oral, rectal, topical, buccal, parenteral, intramuscular, intradermal, or intravenous, and. transdermal administration.
The therapeutically effective dosage of any specific compound, the use of which is in the scope of present invention, will vary somewhat from compound to compound, and patient to patient, and will depend upon the condition of the patient and the route of delivery. As a general proposition, a dosage from about 0.1 to about 50 mg kg will have therapeutic efficacy, with all weights being calculated based upon the weight of the active compound, including the cases where a salt is employed. Toxicity concerns at the higher level may restrict intravenous dosages to a lower level such as up to about 10 mg/kg, with all weights being calculated based upon, the weight of the active base, including the cases where a salt is employed. A dosage from about 10 mg/kg to about 50 mg/kg may be employed for oral administration. In some embodiments, a dosage from about 0,5 mg/kg to 5 mg/kg may be employed for intramuscular injection, In some embodiments, dosages are 1 pmoi/kg to 50 imo /kg, and more preferabl 22 μπ οΐ kg and 33 μτηοΐ/kg of the compound for intravenous or oral administration, The duration of the treatment can be once per day for a period of two to three weeks or until the condition is essentially controlled.
Active compounds may be administered as pharmaceutically acceptable prodrugs, which are those prodrugs of the active compounds of the present invention which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of humans and lower animals without undue toxicity, irritation, allergic response and the like, commensurate with a reasonable risk benefit ratio, and effective for their intended use, as well as the zwitterionie forms, where possible, of the compounds of the invention. The term "prodrug" refers to compounds that are rapidly transformed in vivo to yield the parent compound of the above formutae, for example, by hydrolysis in blood, A thorough discussion is provided in T. Higuchi and V. Stella, Prodrugs as Novel delivery Systems, Vol. 14 of the A.C.S. Symposium Series and in Edward B. Roche, ed., Bioreversible Carriers in Drug Design, American Pharmaceutical Association and Pergamon Press, 1987, both of which are incorporated b reference herein. See also US Patent No, 6,680,299 Examples include a prodrug that is metabolized in vivo by a subject to an active drug having an activity of active compounds as described herein, wherein the prodrug is an ester of an alcohol or carboxylic acid group, if such a group is present in the compound; an acetal or keial of an alcohol group, if such a group is present in the compound; an N-Mannich base or an inline of an amine
group, if such a group is present in the compound; or a Schiff base, oxime, acetal, enol ester, oxazolidine, or thiazolidine of a carbonyl group, if such a group is present in the compound, such as described in US Patent No. 6,680,324 and US Patent No. 6,680,322.
As noted above, the active compounds described herein are usefui for the treatment of cancer. Example cancers that may be treated by the compounds and methods of the invention inciude, but are not limited to, myeioid leukemia, lymphoblastic leukemia, melanoma, breast, lung, colon, liver, gastric, kidney, ovarian, uterine, and brain cancer.
The present invention is explained in greater detail In the following non-limiting Examples,
Examples 1.-11
General Structure I:


2,4 )ichIoro~A^4-(morpho
 A solution of 2s4-dichiorop rin idine-5~carbonyl chloride (422mg, 2,0 mmoi) in dichloromethane (10 mL) was added 4-(morpholinosulfonyl)aniline (508 nig. 2.1 mmoi) and DT.EA (387 mg, 3.0 mmoi) at 0 °C. The resulting mixture was stirred at 0 °C for 1 h, Then, water was added. The resulting mixture was extracted with Et'OAc (3x). The combined organic layers were dried (NaaSQ*), filtered and concentrated. The residue was purified on iSCO to give the title compound as a white solid (701.2 mg, 84%). !H NMR (400 MHz, DMSO-r ) δ 1 1.98 - 1 1.90 (m5 I B), 8.29 (d, J = 6.4 Hz, IH), 7.89 (d, J = 8.8 Hz, 2H), 7.69 (d, J= 8.8 Hz, 2H), 3.65 - 3.56 (m, 4H), 2.87 - 2.78 (m, 4H); MS m/z 418,30 [M+H .
A solution of 2s4-dichiorop rin idine-5~carbonyl chloride (422mg, 2,0 mmoi) in dichloromethane (10 mL) was added 4-(morpholinosulfonyl)aniline (508 nig. 2.1 mmoi) and DT.EA (387 mg, 3.0 mmoi) at 0 °C. The resulting mixture was stirred at 0 °C for 1 h, Then, water was added. The resulting mixture was extracted with Et'OAc (3x). The combined organic layers were dried (NaaSQ*), filtered and concentrated. The residue was purified on iSCO to give the title compound as a white solid (701.2 mg, 84%). !H NMR (400 MHz, DMSO-r ) δ 1 1.98 - 1 1.90 (m5 I B), 8.29 (d, J = 6.4 Hz, IH), 7.89 (d, J = 8.8 Hz, 2H), 7.69 (d, J= 8.8 Hz, 2H), 3.65 - 3.56 (m, 4H), 2.87 - 2.78 (m, 4H); MS m/z 418,30 [M+H .

A solution of 2J4-dichloro-N-(4-(morpholinosulfonyl)p enyl)pyrimidine-5- carboxamide (700rng, 1.68 mmoi) in IPA (15 mL) was added tra«i'-4-aminoeycloliexanol (231.4 mg, 2.2 mmoi) and DIEA (387 mgf 3,0 mmoi) at 0 °C, The resulting mixture was stirred at 0 °C for 50 min and warmed to room temperature and stirred for another 50 min. Then the solvent was removed, the residue was dissolved in a mixture of CH2CL2 and methanol (20 mL, 3:2, v/v), the suspension was filtered though a filter paper to give the title compound as a white solid (683.4 mg, 82%). Ή NMR (400 MHz, DMSO-d6) S 10.71 (s, IH), 8.69 (s, IH), 7.96 7.89 (m, 2H), 7.76 - 7.70 (m, 2H), 4.57 (s, IH), 3.93 - 3.81 (m, 11 1), 3.64 - 3.57 (m, 4H), 3.49 - 3.40 (m, IH), 2.88 - 2.78 (m, 4H), 1.95 - 1.86 (m, 2H), 1.85 - 1.76 (m, 2H), 1.38 ~ 1.20 (m, 4H); MS m/z 496.20 [M+Hf .

A solution of 2-chloro-4-(((lr,4r)-4-hydroxycyc.Sohexy.i)amino)-A;r-(4- (morphoiinosulfonyl)phenyi) pyrimidine-5-carboxamide (86mg, 0.17 mmoi) in IPA (lOmL) was added butyiamine (59,6 mg, 0.81 mmoi) and DIEA (124.7 nig, 0.96 mmoi) at room temperature. The resulting mixture was stirred for 3b. at room temperature. Water was then
added. The resulting mixture was extracted with EtOAc (3X). The combined organic layers were dried (NazSC^ , filtered and concentrated, The residue was purified on ISCO to give the title compound as a white solid {59.3 mg, 64%). !H NMR (400 MHz, CD3OD+CDCI3) S 8.21 (s, 1H), 7.71 - 7.64 (m, 2H), 7,59 - 7.53 (in, 2H), 3.93 - 3.77 (ra, 1H), 3.74 - 3,64 (m, 4H), 3.63 - 3.58 (m, 4H), 3.56 - 3.46 (ra, 1H), 3,26 (t, J- 7.1 Hz, 2H), 2.91 - 2.81 (m, 4H), 2.05■··■ 1.95 (m, '2H), 1 .93 - 1.82 (m, 2H), 1.50 - 1.41 (m, 2H), 1.33 - 1.17 (m, 6H), 0.83 (t, J - 7.3 Hz, 3H); °C NMR (101 MHz, CD3OD+CDCI3) S 166.8, 156,4, 143.5, 128.8, 128.6, 120.3, 120.2, 6.9.2, 66.0, 45.9, 41.0, 33.4, 31.6, 30.2, 20.0, 13.7; MS m/z 533.30 [M+Hf.
Example 2
2~(¾utylamino)-N-(4-fluorobenzyl pvrimidine carboxamide
General Procedure B:

To a solution of 2,4-dichloropyrimidine~5~carbonyl chloride (500 mg, 2.38 mmol) in dichloromethane (30 mL) was added methanol (87.6 mg, 2.73 mmol) and diisopropy!eihylamine (369 mg, 2,86 mmol) at 0 °C. The resulting mixture was stirred for 1 h at 0 °C. Then the solvent was removed. The residue (461rng, 94%) was dissolved in IPA (20 ml,) and followed by the addition of fr ¾y-4-ammocyclohexanol (301.6 mg, 2.62 mmol) then DIEA (461.4 mg, 3.57 mmol) dropwiseiy. The resulting mixture was stirred at 0 °C for 90 min. After which buiyiamine (208,8 mg, 2.86 mmol) was added, followed by DIEA (461.4
mg., 3.57 mmol). The resulting mixture was stirred at room temperature for 3 h. Water was then added. The resulting mixture was extracted with EtOAc (3X). The combined organic layers were dried (NaaSQTj, filtered and concentrated. The residue was purified on ISCO to give methyl 2-(butyiamino)-4-(((?ra»i^^
(682.6 mg, 89% over 3 steps). Ή NMR (400 MHz, CDC13) δ 9.21 (s, 1H), S.77 (s, 1H), 6.29 (s, 1H), 4.81 - 4.64 (m, 1H), 4.51 (s, 3H), 4.46-4.38 (m, 1H), 4.13-4.1 ί (m, 2H), 2.89-2.81 (m, 2H), 2.74 (d, J - 9.7 Hz, 2H), 2.35 - 2.25 (m, 2H), 2.23 - 2.00 (m, 6H), 1 ,67 (t, J- 7.2 Hz, 3H); 33C NMR (101 MHz, CDC13) δ 167.9, 162,5, 161.3, 160.3, 95.5, 69.7, 51.2, 48.3, 41. L 33.8, 31 ,7, 30.3, 20.1, 13,8; MS m/z 323.20 [M+Hf .
2-(Butylamino)-N-f4-fluoro^

A mixture of methyl 2-(butylamino)-4-((( r ra-4-hydiOxycyclohexyl)amino) pyriraidine-5-carboxylate (682.6 mg, 2.12 mmol) and lithium hydroxide monohydrate (888.4 mg, 21.2 mmol) in a mixture of methanol and water (25 mL, 3:2, v/v) was heated at reflux for 2h. Then the reaction mixture was coo!ed to room temperature and acidified by a 4. ON solution of EC) (4N) to PH 3. The resulting mixture was extracted with EtOAc (4x). The combined organic layers were dried (NajSC ) and concentrated to provide 2~(butylaraino)-4- (((lr,4r)~4-hydroxycyeIohexyl)amino)pyrimidine-5-carboxylic acid(647, 1 mg, 99%), which was used in the next step without further purifications. MS m/z 309.30 [M H]+.
A solution of the acid (61 mg, 0.20 mmol), 0-(benzotriazol- 1 -yl)-N,N,N' iV- tetramethyluroniurn tetrafluoroborate (TBTU, 81.4 mg. 0.25 mmol, 1.3 eq) and Ν,Ν· diisopropylethylarmne (DIEA, 77.5 mg, 0.60 mmol, 3,0 eq) in anhydrous DMF (3.0 mL) and was added 4-il orobenzylamine (37.5 mg, 0.30 mmol, l .Seq) in DMF (1.0 mL) dropwisely at room temperature. The resulting mixture was stirred for overnight, then diluted with EtOAc (15 mL) and washed with water (3x). The organic layer was dried (NaiSCi) and concentrated. The residue was purified on HPLC to give the title compound (61.5 mg, 74%) as a yellow solid. 1H NMR (400 MHz, CDCi3) δ 9.57 (d, J = 7.4 Hz, 1H), 8.77 (s, 1H), 8.1 (s, 1H), 7.40 - 7.31 (m, 1H), 7.29 - 7.22 (m, 3H), 7.00 (t, J « 8.6 Hz, 2¾ 4.44 (d, J = 5.2
Hz, 2H), 4.00 (s, IK), 3.77 - 3.62 (m, 1H), 3.40 (dd, J - 13.0, 6.9 Hz, 2B), 2.15 - 2.09 (m, 2H), 2,04 (d, J= 7.6 Hz, 2H), 1.66 - 1.54 (m, 2H), 1.49 - 1.33 (m, 6H), 0.94 (t, J - 7.3 Hz, 3I-I); MS m/z 416.30 [ΜϊΉ]+.

Example 3
( l r,4r)-4-((2-{ButylaiTiiBO)-5-(5-(morphoUnosui.fo
yl)aB ino)cyclohexanol
General Procedure C;


4-((6-ChloropYridln-3-yl)sulibn l)mo^hoiine

A solution of 2-chloropyridine-S-suifonyl chloride (212 mg5 1 .0 mmol) in CH2C12 (10 mL) was added morpholine (87.1 mg, 1.0 mmol) at 0 °C. The resulting mixture was stirred at 0 °C for 10 min and then DIEA (194 mg, 1.5 mmol) was added dropwisely. The mixture was stirred at 0 °C for 2h. Then the mixture was diluted with EtOAc (15 mL) and washed with water (2x). The organic layer was dried (NaaSO^, filtered and concentrated. The residue was purified on ISCO to give the title compound as a white solid (246.1 mg, 94%). !H NMR (400 MHz, CDC13) δ 8.73 - 8.65 (m, 1H), 7.94 (dd, J~ 8.3, 2.5 Hz, IH), 7.49 (dd, J - 8.3, 0.6 Hz, 1H), 3.74 - 3.63 (m, 4H), 2.99 (dd, J = 5.6, 3.9 Hz, 4H); !3C NMR (101 MHz, cdcB) δ 155.9, 148.7, 137.9, 131.1 , 124.9, 65.9, 45.8; MS w/z 263.30 [M+H .

A 10 mL microwave tube was charged with 2,4-dichIoropyrimidine-5-boronic acid pinacol ester (55 mg, 0.20 mmol), K2C03 (41.5 mg, 0.30 mmol) and THF (2.0 mL). The resulting mixture was then heated to 80 °C for 15 min under Microwave irradiation. Then 4- i(6-chloropyridin-3~yl)sui.fonyl)moipholine (52.5 mg, 0.20 mmol), Pd(PPi¾)4 (23.1. mg, 0.02 mmol), K2CO3 (41 .5 mg, 0.30 mmol), butylamine (1 16.8 mg, 1.60 mmol) and water (0.5 mL)
were added sequentially. The resulting mixture was heated to !50 °C for 30 min under Microwave irradiation. The mixture was diluted with EtOAc (15 mL) and washed with water (2x), The organic layer was dried (NaaSOd), filtered and concentraied. The residue was purified on ISCO and HPLC to give the title compound as a white solid {27.4 mg, 28%) and (lr}4r)-4-((2-(butylamino)pyrimidin-4-yl)amino)cyclohexanol (25.7 mg, 49%). Ή NMR (400 MHz, CDCla) δ 10.65 (d, J = 7.1 Hz, IH), 9,46 (t, J = 5,4 Hz, 1H), 8,87 (d, J - 2.2 Hz, 1H), 8.31 (s, III), 8.10 (dd, J = 8.6, 2.3 Hz, 1H), 7.77 (d, J - 8.7 Hz, 1 H), 4, 19 - 4.09 (m, 1H), 3.84 -- 3.68 (m5 5H), 3.51 - 3,44 (m, 2H), 3, 15 - 3,06 (m, 4H), 2.86 - 2.66 (m, 3H), 2.28 - 2.15 (m, 2H), 2,13 - 2,01 (m, 2H), 1.72 - 1 ,61 (m, 2H), 1.56 - 1.36 (m, 6H), 0.96 (t, J = 7.4 Hz, 3H); "C NMR (101 MHz, CDC¾) δ 159.4, 156.9, 153.1, 146.6, 142,8, 136.8, 130.2, 1 18,9, 103.4, 69.2, 66.0, 49.7, 45.8, 41.3, 33.3, 31.0, 29.5, 20.1, 13.7; MS m/z 491.30 [M+Hf,
Example 4
UNC2427A
General Procedure D:

2,,4-pichloro-,N-l4-fl
 4-Fluorobenzy!amine (1.78g, 14.2 mmol) in dicMoromeihane (5.0 mL) was added into a solution of 2,4-dich1oropyrimidine-5-carbonyl chloride (3.0 g, 14.2 mmol) in dicMoromeihane (30 mL) at 0 °C. Then N,N-diisopropylethylamine (2.75g, 21.3 mmol) and 4-(dime yiamino)pyridme (17.1 mg, 0.14 mmol) were added into the mixture. The resulting mixture was allowed to warm to room temperature arid stirred for 20 min. The solvent was removed and the residue was dissolved in dichloromethane and washed with water (2x). Solvent was removed and the residue was purified on ISCO to provide the title compound (3.96g, 93%). !H NMR (400 MHz, CD3OD) δ 8.73 (s, 1H), 7.42-7.35 (in, 2H)S 7.12-7.02 (m, 2H), 4.53 (s, 2H); MS m/z 301 ,30 [M+Hf. y)-5-f(2-((3-Arn.!nopropyi)amino)-5-((4-fluorobenzyl)
4-Fluorobenzy!amine (1.78g, 14.2 mmol) in dicMoromeihane (5.0 mL) was added into a solution of 2,4-dich1oropyrimidine-5-carbonyl chloride (3.0 g, 14.2 mmol) in dicMoromeihane (30 mL) at 0 °C. Then N,N-diisopropylethylamine (2.75g, 21.3 mmol) and 4-(dime yiamino)pyridme (17.1 mg, 0.14 mmol) were added into the mixture. The resulting mixture was allowed to warm to room temperature arid stirred for 20 min. The solvent was removed and the residue was dissolved in dichloromethane and washed with water (2x). Solvent was removed and the residue was purified on ISCO to provide the title compound (3.96g, 93%). !H NMR (400 MHz, CD3OD) δ 8.73 (s, 1H), 7.42-7.35 (in, 2H)S 7.12-7.02 (m, 2H), 4.53 (s, 2H); MS m/z 301 ,30 [M+Hf. y)-5-f(2-((3-Arn.!nopropyi)amino)-5-((4-fluorobenzyl)
((te rz-butoxvcarbon vl).amino)pentanoi c acid

i-Qrniihine (232 mg, 1 ,0 mmol), N.N-diisopropyleihylarnine (194 nig, 1.5 mmol.) were added into a solution of 2,4-dichloro-N-(4-fluorobenzyl)pyrimidine-5-carboxamide (300 mg, 1.0 mmol) i a mixture of isopropyl alcohol and DMF (10 mL, 3:2, v/v) at room temperature. The resulting mixture was stirred for overnight. Then 1/3 of the reaction mixture was added to a solution of 1,3-diaminopropane (304 mg, 4.0 mmol) in DMF (1.0 mL) at room temperature. The reaction mixture was heated at 45 °C for 2h, The solvent was removed and the residue was purified on HPLC to give the title compound as TFA salt (92 nig, 34%). }H NMR (400 MHz, CD3OD) δ 8.22 (s, IH), 7.38-7.28 (m, 2H), 7.06-6.95 (m, 2H), 4.45 (s, 2H), 4.18-4.09 (m, I H), 3.69-3.53 (m, 4H), 3.08-2.99 (m, 2H), 2.05-1.97 (m, 2H), 1.91 - 1.83 (m, IH), 1.80-1.66 (m, 3H), 1.41 (s, 9H); MS m/z 534.30 [ M H i] ' .
UNC2427A

butoxycarbonyl)amino)peniaiioic acid (70 mg, 0.085 rnmol) in DMF (50 mL). The resulting mixture was diluted with dichloromethane (60 mL) and stirred a room temperature for overnight. Then the solvent was removed, the residue was dissolved in dichloromethane (5.0 mL) followed by the addition of TFA (1.0 mL). The resulting mixture was stirred at room temperature for 2.0 h. Then the solvent was removed and the residue was purified on HPLC to provide the title compound (UNC2427A) as TFA salt (27.4 mg, 46%); lH NMR. (400 MHz, CD3OD) δ 8.19 (s, I H), 7.37-7.26 (m, 2H), 7,06-6.97 (m, 2H), 4,44 (s, 2H), 4,02-3.97 (m, IH), 3.97-3.89 (m, I H), 3.81-3,71 (m, !H), 3.68-3.60 (m, I H), 3.28-3.12 (m, 2H), 2.87- 2.80 (m, 1 H), 2.01-1.92 (m, 2H), 1 ,91-1.82 (m, i H). 1.80-1.64 (m, 3B). MS m z 416.25 [M+Hf,
Table 1 describes compounds prepared following procedures described in Example 4 (General Procedure D), using appropriate reagents. (Note: Mer 1C50: ++++· means < 10 nM; ■ - means between 10-lOOnM, ++ means between 100 tiM-i uM; + means between 1-30 μ,Μ; - means inactive.)

(m, 3H), 1.73- 1.52 (rn, 5H), 1 .48-1.34 (m, 3H); MS m/z 444.30 f +Hf.
Ή NMR (400 MHz, CD3O.D) δ 8.20 (d, J - 0.9 Hz, IH), 7.38-7.29 (ra, 2H), 7.07-6.99 1 (m, 2H), 4.44 (s, 2H), 3.88- 3.66 (m, 4H), 3.51-3.32 (m, 2H), 3.21-3.1 1 (ra, I H), 2.98- 2.93 (m, 4H), 2,89-2.77 (m, I H), 1 ,98" Ϊ .85 (ra, 2H): 1.84- 5.58 (m, 5H), 1.56--1 .48 (m, HI), 1.46- 1.29 (m, 5H); MS m/z 458.30 [ +H] ,
UNC2479A ¾ NMR (400 MHz, CDjOD) o 8.2] (s, IH}, 7.36-7.30 (m, 2H), 7.07-6.99 (m, 2H), 4.45 (s, 2H), 3.94-3,85 (m, IH), 3.80-3,74 (m, IH), 3.74-3.65 (m, ΪΗ), 3.55-3.45 (m, 2H),
 3.38 -3.30 (ra, IH), 3.23-3.11
3.38 -3.30 (ra, IH), 3.23-3.11
(m, IH), 2.96 (s, H), 2.89- 2.80 (m, IH), 1.97- 1,81 (m, 2H), .77-1.55 (m, 5H), 1.54-
1.43 (m. 2H), 1.43-1.21 (m,
7H); MS w¾ 472,30 [M-HKf.
UNC2431A Ή NMR (400 MHz. CD3OD)
<58.22 (s, IH), 7.37-7.28 (m, 2H), 7.07-6.97 (m, 2H), 4.45 (s, 2H), 3.89-3.79 (m, 2H),
3.71-3.58 (m, 2H), 3.58-3.49 (m, IH), 3.44-3.34 (m, IH),
 2.95 (s, IH), 2.90-2.80 (m,
2.95 (s, IH), 2.90-2.80 (m,
IH), 1.98-1.84 (m, 2H), 1.74- 1.66 (rn, 2H), 1,65-1.57 (m, 2H), 1.56-1.51 (m, IH), 1.50- 1.42 (m, IH), 1.39-1.25 (m, 8H); MS m/z 486.30 [M+Hf.
UNC2342A ¾ NMR (400 MHz, CD3OD)
<5 8.44 (d, J - 2.5 Hz, IH), 7.81-7.73 (in, 2H), 7.53-7.46 (ni 2H), 4.33 (s, 2H), 4.07- 3,93 (m, 4H); 3,82-3.64 (m, 4H), 3,41-3.34 (m, 2H), 3.28-
 3,07 (m, 4H), 2.90-2.82 (m,
3,07 (m, 4H), 2.90-2.82 (m,
IH), 2,04-1.95 (ra, 2H), 1,94-


(ddd, (ai, 4.45 (ta, 4.46


Example S
UNC2324A
General Procedure E:

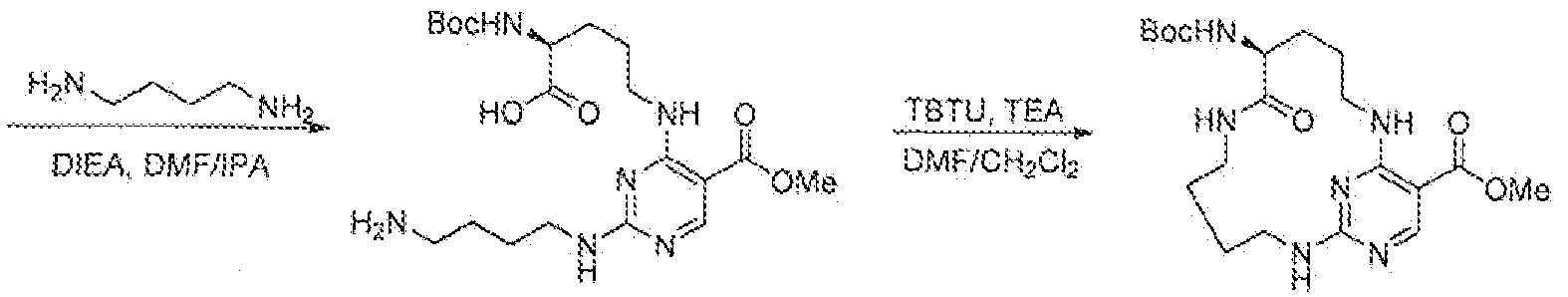
UNC2343A
LiOH
THF,'H20*"

UNC2340A UNC2324A
UNC2343A

To a solution of 254-dich-k>ropyriB idine--5-carbonyI chloride (500 rag, 2.38 mmol) in dichloromethane (30 mL) was added methanol (87.6 mg, 2.73 mmol) and dnsopropylethylamme (369 mg, 2.86 mmol) at 0 °C. The resulting mixture was stirred for at 0 °C 1 h. Then the solvent was removed. The residue (412mg, 2.0 mmol) was dissolved in 1ΡΛ (20 mL) followed by the addition of Z -Ornithine (465 nig, 2,0 mmol) and N,N- diisopropylethylaraine (388 mg, 3,0 mmol). The resulting mixture was stirred at 0 °C for 90 min, then dichloromethane (5.0 mL) was added. The resulting mixture was stirred at 0 °C for 1.0 h and at room temperature overnight. Solvent was removed and the residue (MS m/z 403,30 [M+H]*) was dissolved in DMF (5.0 mL) and was added dropwise into a solution of 1 ,4-diaminobutane (1.68g, 19.1 mmol) in DMF (1.0 mL) at room temperature. The resulting mixture was heated to 45 °C for Ih, The solvent was removed and the residue was dissolved in ethyl acetate (35 mL) and washed with water (3x). The organic layer was dried (Na2S04), filtered and concentrated. The residue was purified on HPLC to provide (5)-5-((2-((4- aminobuty3)amino)-5-(methoxycarb^
amino)pentanoic acid as a semisolid (MS m/z 455.30 [M+H +). The semisolid (595 mg, 1.31 mmol) was dissolved in DMF (150 mL), then TBTU (546.4 mg, 1.70 mmol) and DIEA (508 mg, 3.93 mmol) were added sequentially. The resulting mixture was stirred at room temperature overnight. The solvent was removed. The residue was dissolved in ethyl acetate and washed with water (3x). The organic layer was dried
 filtered and concentrated. The residue was purified on HPLC to provide the totle compound (UNC2343A). l MR (400 MHz, CD3OP) 3 8.42 (s, 1H), 4.08-3.99 (m, IH), 3.90-3,84 (m, 3H), 3.59-3.47 (m, 2H), 3.46-3.31 (m, 2H), 3.28-3.21 (m, IK), 3.09-2.95 (m, 1H), 1.90-1.52 (m, 8H), 1.51- 1.32 (ra, 9H); MS m/z 437.30 [M+H1+.
filtered and concentrated. The residue was purified on HPLC to provide the totle compound (UNC2343A). l MR (400 MHz, CD3OP) 3 8.42 (s, 1H), 4.08-3.99 (m, IH), 3.90-3,84 (m, 3H), 3.59-3.47 (m, 2H), 3.46-3.31 (m, 2H), 3.28-3.21 (m, IK), 3.09-2.95 (m, 1H), 1.90-1.52 (m, 8H), 1.51- 1.32 (ra, 9H); MS m/z 437.30 [M+H1+.
UNC2340A

U C2324A

4-Fluorobenz lamine (65.1 mg, 0.5 mmol), TBTU (128.2 mg, 0.4 mmol) and triethylaniine (68 mg, 0.67 mmol) were added to a solution of U C2340A (40 mg, 0,095 mmol) in anhydrous DMF (20 mL) at room temperature. The resulting mixture was stirred for 2h, then solvent was removed, the residue was dissolved in CFi2(¾ (30 mL) and washed with water (2x). TFA (1.0 mL) was then added and stirred at room temperature for 2,0 h, condensed and purified on HPLC to provide the title compound (UNC2324A) (26 mg, 52%). Hi NMR (400 MHz, CD.,OD) S 8.19 (s, IH), 7.37-7.26 (ms 2H), 7.07-6.96 (m, 2H)5 4.44 (s, 2H)S 3.88-3.82 (m, IH), 3.77-3.68 (m, IH), 3.58-3.47 (m, 2H), 3.43 -3.35 (m, I H), 3.26-3.13 (m, 2H), 2.98-2.93 (m, 2H), 2.92-2.84 (m, I H), 1.92-1.81 (m, 3H), 1.81 -1.53 (m, 5H), 1.44- ί .29 (m, IH); MS m/z 430.20 [M+H]+.
Table § describes compounds prepared following procedures described in Example 5 (General Procedure E), using appropriate reagents.
Structure Compound_ID Mer Physical Data
IC50 MS m z (M+l) or/and Ή
NMR
'UNC2589A ++ lH NMR (400 MHz, CD3OD)
δ 8.21 (s, IH), 7.39-7.35 (m,
HN 4H), 4,50-4.47 (m, 2H), 3.97- 3.9 ! (m, 3H), 3.87-3.83 (m, ! H), 3.40-3.33 (ra, 5H), 3,09- 3.01 (m, 4H), 2.87 (s, 4H), 1. 3-1.82 (m, 4H)5 1.79-1.70 {ra, 2H), 1 .69-1.57 (m, 3H), 1.44-1.30 (in, 2H); MS m/z
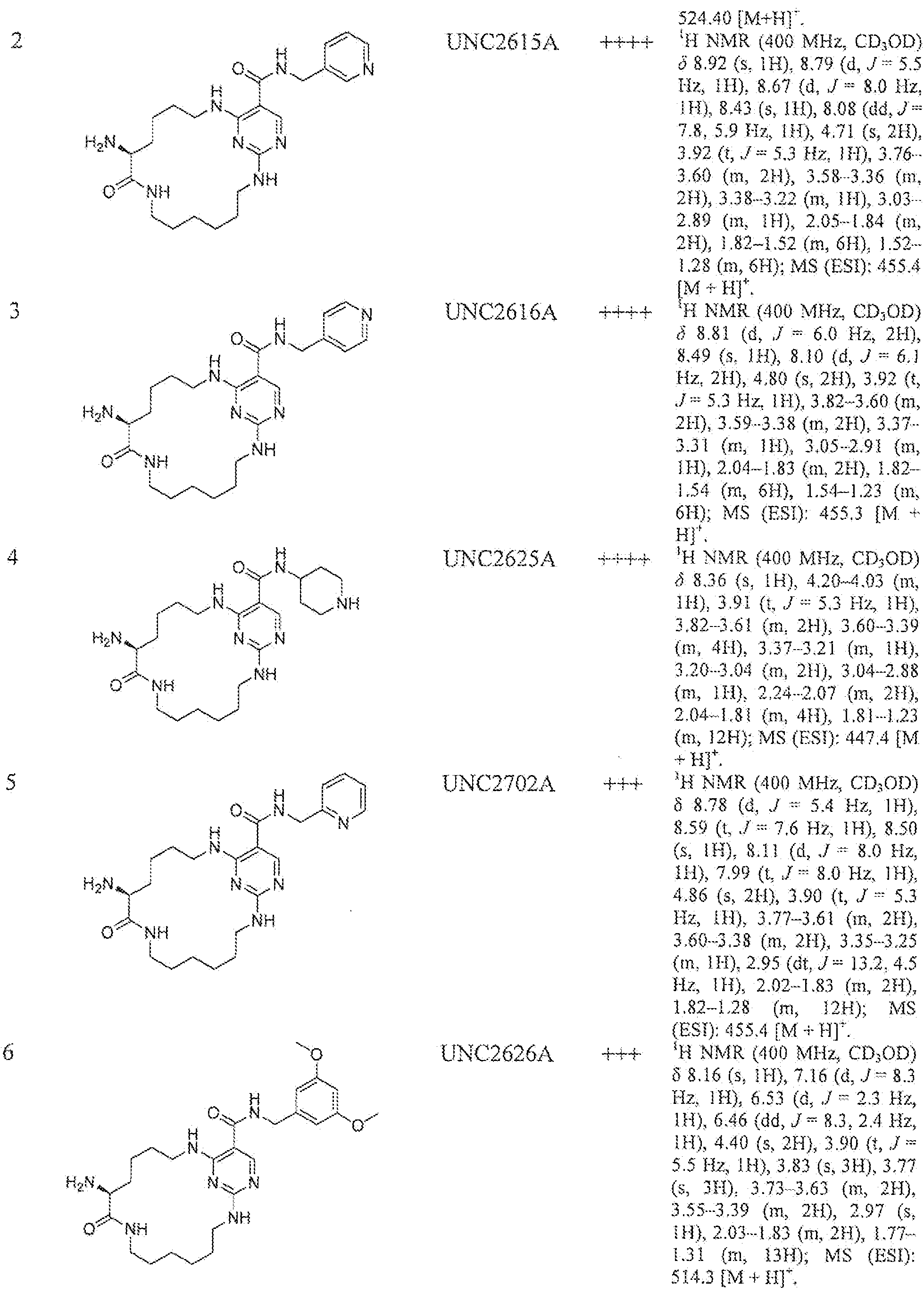
 δ
δ
Hz, H), (dd, J On,
(s, H), 13.3 6H),



Example 6
UNC3263A
General Procedure F:

UNC3263A
2,4-Dich ro-Ar- (4-fluoroph

A solution of 2}4-dichloropyrimidine-5-carbonyl chloride (3.35 g, 15.8 mrno!) in dichioromethane (53 mL) was added (4-iIuorophenyl)methanaxnine (1.32 g, 10.56 mmol) and
DiEA (3.7 mL, 21 mmoi) at 0 °C. The resulting mixture was stirred at 0 °C for 1 h, the solvent was removed, and the residue was purified by ISCO silica gel column, to provide the title compound (1.97 g, 62%) as a yellow solid.

A solution of 2,4-dichIoro-N-[(4-fI«orophenyl)methyl]pyrimidme-5-carboxaraide (97 mg, 0.325 mmol) in -PrOH (5 mL) was added 7-aminohept-l-en-4-ol (42 mg, 0.325 mmoi) and DIEA (85 ,uL, 0.488 mmoi) at 0 °C. The mixture was allowed to warm to room temperature and stirred overnight. To the reaction was added oct-7-en-l -amine (207 mg, 0.975 mmol) and DIEA (85 LIL, 0.488 mmol). The reaction was heated at 70 °C for 6 h and the volatiles were removed under a reduced pressure. The residue was dissolved in CH2CI2 (4.0 mL) and washed with ¾0 (2.0 mL). The ¾0 layer was extracted with CH2C12 (2 χ 20 mL); the organic layers were combined, dried (NaiSO^), and the solvent was removed under a reduced pressure. The residue was purified by ISCO silica gel column to provid the title compound (39 mg, 25%) as a white solid. Ή N.MR. (400 MHz, CDCI.3) δ 8.70 (ss 1H), 8.09 (ss 1H), 7.29 (dd, J = 8.7, 5.3 Hz, 2H), 7.07-6.98 (m, 2H), 6.08 (s, 1H), 5.90-5.71 (m, 2H), 5.18-5.04 (m, 1H), 5.03-4.89 (m, 2H), 4.51 (d, J ~ 5.7 Hz, 2H), 3.70 (s, 1 H), 349 (s, 2H), 3.38 (dt, J = 9.7, 4.8 Hz, 211), 2.35-2.12 (m, 2I¾ 1.80 .1.68 (m, 2H), 1 ,59 (s, 9H), 1.44-1.31 (m, 5H).
Ar"[(4-Fluorophenyl)methyij-6-h ydroxy-2, 16, ί 8,21 -t traazabicyclof 15.3.1 Ihenieosa- 1(20), 17 3263A)

A solution of N-[(4-iluorophenyl)methyl]-4-[(4-hydroxyhept-6~en-l-y!)amino]-2- [(oet-7-en-l-yl)amino]pyrimidine-5-carboxamide (34 mg. 0.07 mmol) in toluene (35 mL) was added benzoquinone (1.5 mg, 0.014 mmoi) and Grubb's 2nd generation catalyst (6.0 mg, 0.007 mraol), The mixture was refluxed under Ar ovemight. After which more catalyst was
added (6.0 mg), and heated at reflux for 8 h. Tlie voiatiles were removed and the residue was purified by IS CO silica gel column to provide the RCM product as a crude brown solid. The residue was dissolved in MeOH (1.0 mL) and Pd/C (10 mg) was added. The mixture was stirred at room temperature under a hydrogen atmosphere for 24 h. The mixture was filtered through C elite and washed with MeOH then purified by HPLC to afford the title compound as a white solid (5.3 mg, 16%). lH NMR (400 MHz, CD3OD) δ 8.21 (s, IH), 7.37 (dd, J = 8.8, 5,4 Hz, 211), 7.05 (t, J - 8.8 Hz, 2H), 4.47 (s, 2H), 3.73-3.60 (m, 2H), 3.58-3.41 (ra, 3H), 1 ,84-1.72 (m, 2H), 1.72-4.62 (m, 2H), 1.61-1.45 (m, 5H), 1.43-1.26 (ra, 11H). MS m/z 458.3 [M+Hf .
Table | describes compounds prepared following procedures described in Example 6

Table § describes compounds can be prepared following procedures described in Example 6 (General Procedure F), using appropriate reagents.
Structure Compound ID Mer Physical Data
ICso_ -MS /z (M-i-1) or/and ¾

Example 7
UNC3017A, U C3018A & U C3019A
General Procedure G:
UNC301 A

UNC30f9A

A solution of (S -6ramino-2-((i«rf-b toxycarbonyl)ainmo)hexanoic acid (0.042 g, 0,10 nmiol) in DMSO was added hexane~l ,6~diamme (0.046 g. 0,40 mmol). The resulting mixture: was heated at 100 °C for 2 h. After coo!ed to room temperature, the solvent was removed under reduced pressure. The resulting residue was purified by reverse phase ISCO column to provide title compound (0.041 g, 79%). lH NMR (400 MHz, C δ 7,91 (s, 1H), 4.13-4.02 (111, I H), 3.57 (t, J- 12, 2H), 3.51-3.38 (bs, 2H), 2.94 (t, J- 12, 2H), 1.92- 1.80 (m, IH), 1.75-1.62 (m, 7H), 1.57-1.36 (m, 15H); MS m/s 517.3 [M+Hf.

A sol ution of {S)-6-((2-((6-aminohexyl)amino)-5-bromopyrimidin-4-yI)amino)-2-((tert- butoxycarbonyl)amino)hexanoic acid (2.1 1 g, 4.08 mmol) arid DiEA (1 .78 mL, 10.19 mmol) in 20 mL DMF and a solution of HATU (2,01. g, 5.28 mmol) in 20 mL DMF were added to 200 mL of DMF in 16 h. After addition, the resulting mixture was stirred for I h. The solvent was removed under reduced pressure. The residue was purified by ISCO silica gel column to provide the desired bromide (0,87 g, 43%) containmated with small amount of impurities. MS m/z 499.3 [M+Hf .

A solution of the bromide (0.66 g, ί .32 mmol) in DMF (6.6 mL) was added PdCl2(dpp 'CH2Cl2 (0.1 1 g, 0.13 mmol), CuBr (0.036 g, 0.26 mmol), 2C03 (0.56 g, 9.96 mmol), (5~( 1 ,3-dioxolan-2-yl)pyridin-2-yl)boronic acid lithium hydroxide (0.86 g, 9.96 mmol) and ¾0 (1 ,7 mL) at. room temperature. The resulting mixture was heated at 120 °C for 30 min under. After cooled to room temperature, the mixture was filtered over Ceiite. The solvents were removed under reduced pressure. The crude residue was purified by ISCO silica gel column to provide the desired acetal (0.76 g, 63%) containraated with small amount of impurities. MS m/z 570,4 [M HT .

A solution of the the acetai (0.47 g, 0.83 mmol) in acetone (10 mL) was added p~TsOH'¾0 (0.24 g, 1.25 mrnol) and H20 (2.0 mL). After stirring at room temperature for 16 h, the reaction mixture was quenched with a sat, aq solution of ΝαΗ€03 and extracted with EtOAc (3X). The combined organic layers were dried (NajSO ) and concentrated. The resulting residue was purified by ISCQ silica gel column to provide the desried aldehyde (0.42 g, 96%) contaminated with small amount of impurities. MS m/z 526.4 [M+H]+.

A solution of the aldehyde (0.53 g, 0.1 mmol) in CH2CI2 (2.0 mL) was added morpholfne (0.013 mL, 0.15 mmol) and acetic acid (0.04 mL). The resulting solution was stirred at. room temperature for 2 h, then NaB(OAc)jH (0.043 g, 0, 15 mmol) was added. After stirred at room temperature for 16 h, the reaction was quenched with a sat. aq. solution of NaHCOs and extracted with EtOAc (3X), The combined organic layers were dried (Na2S0 ) and concentrated. The residue was dissolved in a mixture of CH2CI2 (2.0 mL) and TFA (1.0 mL). After stirred at loom temperature for 2.0 h. the reaction was concentrated and the residue was purified by prep-HPLC to provide the desired product UNC3017A (0.046 g,
93%). 1H NMR (400 MHz, CD3OD) δ 8.88 (s, 1H), 8.41 (s, 1 H), 8.22 (d, J - 8,4 Hz, ΪΗ), 8.01 (d, J = 8.4 Hz, 1H), 4.52 (s, 2H), 4.06 (d, J = 10.7 Hz, 2H), 3.96 3.79 (m, 4H), 3.77- 3.67 (m, I H). 3.57 (td, J= 12.2, 5.2 Hz, 2H), 3.45 (d, J = 12.4 Hz, 2H), 3.39-3.23 (m, 3H)3
3.02-2.93 (m, 1H), 1.96 (dd, J = 10.2, 4.6 Hz, 2H), 1.86-1.51 (m, 7H), 1.42 (d, J = 4.7 Hz, 5H); MS m/z 497.4 [M+Hj\
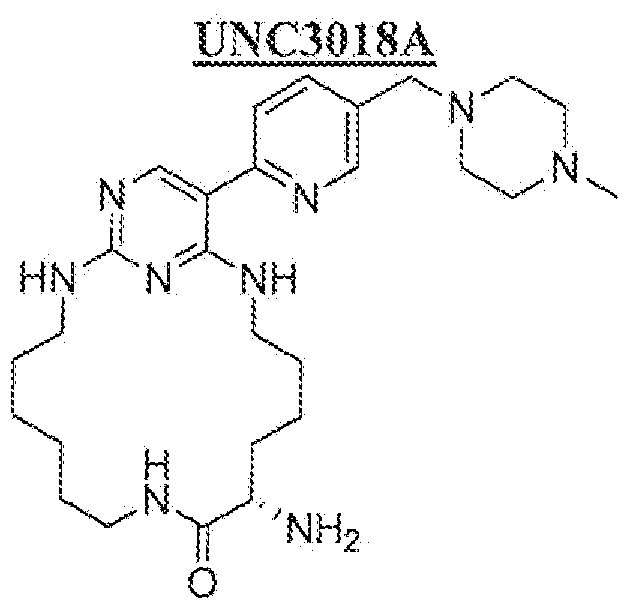
A solution of the above aldehyde (0.53 g> 0.1 mmol) in CH2CI2 (2.0 mL) was added N-methylpiperazine (0.017 rnL, 0.15 mmol) and acetic acid (0.04 mL). The resulting solution was stirred at room temperature for 2 h, then NaB{OAc>3H (0,043 g, 0.15 mmoi) was added. After stirred at room temperature for 16 h, the reaction was quenched with, a sat. aq. solution of NaHC(¾ and extracted with EtOAc (3X). The combined organic layers were dried (NaiSO/j and concentrated. The residue was dissolved in a mixture of CH2CI2 (2.0 mL) and TFA (1,0 mL). After stirred at loom temperature for 2.0 h. the reaction was concentrated and the residue was purified by prep-HPLC to give the desired product U €3018A(0.Q49 g, 98%). !H NMR (400 MHz, CD3OD) δ 8.91 (d, J - 1.7 Hz, IH), 8.40 (s, 1H), 8.27 (dd, J - 8.5, 2,0 Hz, Hi), 8.01 (d, J - 8.5 Hz, I H), 4.61 (s, 2H), 3.91 (t, J = 5.4 Hz, I H), 3.88-3.61 (m, 10H), 3.61-3.51 (m, 2H), 3.39-3.34 (m, IB), 3.02 (s, 3H), 3.00-2.92 (m, IH), 2.03-1.89 (m, 2H), 1.87-1.50 (m, 7H), 1.48-1.35 (in, 5H); MS m/z 510,4 M+H .

A mixture of aldehyde (0.053 g, 0.1 mmol) in 10 mL /BuOH was added NaCiOi (0,034 g, 80%, 0.3 mmol), Na¾P04 (0.041 g, 0.3 mmol), 2-Me~2-Butene (0, 1 1 mL, 1 mmol) and 2 mL H2O. After stirring 16 h at room temperature, the reaction was quenched with sat.
aq. Na2S203, acidified with diluted aq, HCl to pH 4 and extracted with EtOAc (3X). The combined organic layers were dried (Na2S04) and concentrated, The residue was dissolved in 3 ml, DMF. To this solution was added HATU (0.042 g, 0.11 mraol), followed by Λ\Ι~ dimethylpiperidin-4-amine (0,014 g, 0.1 1 mmol). After stirring for 16 at room temperature, the reaction mixture was condensed under vacuum and purified by Prep-HPLC, The Boc protected intermediate was dissolved in 2 mL C¾C12 and 1 n L TFA and stirred for 2 h. After evaporating the solvents, the residue was purified by Prep-HPLC to provide the desired product U C3019A (0.026 g, 47%). Ή NMR (400 MHz, CD3OD) δ 8.70 (s, 1H), 8.41 (s, 1 H), 8.02-7.93 (m, 2H), 3.92-3.50 (m, 8H), 3.39-3.33 (m, 1H), 3.23 (bs, i l l), 3.10-2.75 (m, 8H)5 2.24 (dd, J - 24.3, 1 1 .4 Hz, 2H), 2.15-2.02 (m, 2H), 2.02-1.88 (m, 2H), 1.87-1.48 (m, 8H), 1.45-1.37 (m, 4H); MS m/z 552,4 [M+H ,
Example 8
UNC3588A
General Procedure H


5-Bromo-A¾.-.(but-3-en-.l-y.l)-iV¾-.(5-((te
2.4-diamine

A solution of
 ert-butyldimethy!siiyl)oxy)dec-9- en-l-yl)pyrimidine-2,4-diamine (0.26 g, 0.50 mmol) and benzoquinone (0.009 g, 0.08 mmol) in toluene (20 ml,) was added Greta's catalyst (0.027 g, 0.04 mmol) in toluene (5.0 mL) over 1. h at 110 l'C. The resulting dark brown solution was heated at 1 10 °C for another hour. After cooled to 60 °C, 2-mercaptonicotinic acid (0.39 g, 2.5 mmol) was added and stirred at 60 °C for 2 h. After cooled to room temperature, the reaction mixture was filtered over Ceiite and washed with CH2CI2, The filtrate was concentrated and the residue was purified by ISCO silica gel column to provide the macrocyclic intermediate as a mixture of E and Z olefin isomers (0.19 g, 80%). MS m/z 483.3 [M+Hf .
ert-butyldimethy!siiyl)oxy)dec-9- en-l-yl)pyrimidine-2,4-diamine (0.26 g, 0.50 mmol) and benzoquinone (0.009 g, 0.08 mmol) in toluene (20 ml,) was added Greta's catalyst (0.027 g, 0.04 mmol) in toluene (5.0 mL) over 1. h at 110 l'C. The resulting dark brown solution was heated at 1 10 °C for another hour. After cooled to 60 °C, 2-mercaptonicotinic acid (0.39 g, 2.5 mmol) was added and stirred at 60 °C for 2 h. After cooled to room temperature, the reaction mixture was filtered over Ceiite and washed with CH2CI2, The filtrate was concentrated and the residue was purified by ISCO silica gel column to provide the macrocyclic intermediate as a mixture of E and Z olefin isomers (0.19 g, 80%). MS m/z 483.3 [M+Hf .
A schlenk flask containing LiCi (0.028 g, 0.66 mmol) and stir bar was flame dried. After cooled to room temperature, CuCl (0.054 g, 0,55 mmol) and Pd(PPl¾)4 (0. 13 g, 0.01 1 mmol) was added. The reaction mixture was vacuumed and refilled with argon (4X). The intermediate from previous step (0.054 g, 0.11 mmol) in DMSO (4,0 mL), and Bi¾SnPy (0,049 g, 0,13 mmol) were added under argon. The resulting reaction mixture was freeze- thawed under argon (4X). The mixture was heated at 60 °C for 2 d, then diluted with EiGAc.
quenched with brine and a saturated aq, solution of NaHCOj. The organic layer was washed wit a sat. aq solution o£ NaHC(¾ (3X). The combined aqueous layers were extracted with EtOAc (3X). The combined organic layers were dried (NaiSC^) and concentrated. The residue was purified by ISCO silica gel column to provide Stille coupling products (0.040 g, 75%). MS m/z 482.4 [M+H ,
A solution of the Stille coupling product (0.040 g. 0.083 mmoi) in MeOH (5.0 mL) was added a cone. HC1 solution (0.50 .mL). The resulting mixture was stirred for I h and concentrated under reduced pressure. After zoetrope with MeOH (3X), the residue was dissolved in MeOH (5.0 mL) and AcOH (0.50 mL) and was added Pd/C (0.009 g, 1.0 wt%). The reaction mixture was stirred at room temperature under hydrogen for 1 d, then filtered over Celite and washed with MeOH. The filtrate was concentrated and the residue was purified by reverse-phase HPLC to provide the desired product UNC3588A (0.025 g, 82% over 2 steps). lH NMR (400 MHz, CD3OD) δ 8.62 (ddds J - 5.0, 1.6, 0.9 Hz, 1H), 8.28 (s, 1 H), 7.96-7.82 (m, 2H), 7.39 (ddd, J - 7.3, 5.0, 1.1 Hz, 1H), 3.91-3.79 (m, IE), 3.71-3.53 (m, 3H), 3.49-3.38 (ms 1H), 1.92-1.78 (m, 1H), 1.76-1.65 (m, 3H), 1.62-1.34 (m, 14H). MS m/z 370.3 M+H]+.



Example
UNC32 5A
General Procedure I:

UNC329SA
A solution of the bromide intermediate (0.17 g, 0.34 mmol) in DMF (2,0 mL) was added Pd(dppf)Cl2-CH2Cl2 (0,028 g, 0.034 mmol), CuBr (0.098 g, 0,68 mmol), K2C03 (0, 14 g, 1 ,02 mmol), (5-(l53~dioxoian-2-yl)pyridin-2-yl)boiOnic acid lithium hydroxide (0.22 g, 1.02 mmol) and water (0,50 mL). The resulting mixture was heated at 120 °C for 30 min open to air. The mixture was filtered over Celite at room temperature. The solvents were removed under a reduced pressure and the residue was purified by ISCO silica gel column to provide the desired coupling product (0.053 g, 28%) contaminated with small amount impurities; MS m/z [M+Hf.
A solution of the coupling product (0.053 g, 0.10 mmol) in acetone (10 mL) was added >~TsOH,¾0 (0.029 g, 0.15 mmol) and hhO (0,80 mL), The reaction mixture was stirred at room temperature for 16 h, then quenched with a sat, aq solution of NaHCOj and extracted with EtOAc (3X). The combined organic layers were dried (Na2S04) and concentrated. The resulting residue was purified by ISCO silica gel column to provide the desired aldehyde intermediate (0.031 g, 78%) contaminated with small amount impurities; MS m/z 554.4 M+Hf .
A solution of the aldehyde (0.031 g, 0,078 mmol) in C¾C¾ (2,0 mL) was added morphoiine (0.01 mL, 0.12 mmol) and 0,08 mL acetic acid. The resulting solution was stirred at room temperature for 2 h, then NaB(OAc)3H (0.033 g, 0.16 mmol) was added. The reaction mixture was stirred at room temperature for 16 h, quenched by a sat. aq, solution of NaHCOs and extracted with EtOAc (3X). The combined organic layers were dried (NaaSOT) and concentrated. The residue was dissolved in MeOH (5,0 mL) and was added Pd/C (0.015 g„ 5 wt% Pd C). The reaction mixture was stirred under hydrogen atmosphere at room temperature for 16 h, then .filtrated over Celite and washed with MeOH. The filtrate was concentrated and the residue was purified with reverse-phase HPLC to provide the desired product UNC3295A (0.0096 g, 26%). Ή NM (400 MHz, CD3OD) δ 8.73 (d, J - 1 ,8 Hz, IB), 8.40 (s, 1H), 8.06 (dd, J- 8,5, 2.2 Hz. 1H), 7.96 (d J= 8.5 Hz, 1 H), 4.40 (s, 2H), 3.98- 3.80 (ra, 5H), 3.70-3.5 Ϊ (m, 3H), 3.49-3.38 (m, 1H), 3.29 (bs> 4H), 1.90-1.79 (m, 1H), 1.77- 1.65 (m, 3H), 1.62-1.36 (m, 14H); MS m/z 469.4 [M+H]+.
Table 6 describes compounds prepared following procedures described in Example 9 (GeneraJ Procedure I), using appropriate reagents.
Structure Compound_JD Physical Data

Example 10
UNC3096A
General Procedure J:


A solution of methyl 2>4-dichloropyrimidine-5-carboxylate (252 mg, 1.22 mmol) in /"- PrOH (6 mL) was added 2-(2-aminoethoxy)ethanol (122 μΐ,, 1.22 mmol) and IEA (319 pL, 1.83 mmol) at 0 °C. After 1 ,5 h the volatiies were removed imder a reduced pressure and the residue was purified by ISCO silica gel column to provide the title compound (243 mg, 72 %) as a white solid. 1H N R (400 MHz, CDC13) δ 8.67 (s, 1H), 3.90 (s, 3H), 3.82-3.74 (m, 4H), 3.70 (t, J - 5.2 Hz, 2H), 3.67-3.61 (m} 2H), 2.17 (t, J - 6.3 Hz, 1H).
Methyl 2-chloro~4-(8.,8,9 ^etrame^
carboxylate

A solution of methyl 2-chloro~4- { [2-(2-h.ydroxyethoxy)ethyl]am.ino}pyrimidine-5- carboxylate (99 mg, 0.36 mmol) in DMF (720 uL) was added imidazole (49 mg, 0.72 mmol) followed by TBD SC1 (60 mg, 0.396 mmol). The reaction mixture was stirred at room temperature for 1 h, diluted with ¾G (2 raL) and extracted with C¾C (3 x 2 mL), The combined organic layers were washed with brine (2 x 2 mL), dried (Na2S0 ) and concentrated. The residue was purified by ISCO silica gel column to provide the title compound (137 nig. 98 %) as a colorless oil.
Methyl 2-|'(5-hydroxypentyl)amino]-4-(8.8.9,9-tetramethyl-4,7-dioxa- 1 -aza-8-siladecan- 1 - yi)pyrimidine-5-carboxylate


A solution of methyl 2-[(5¾droxypentyl)amino j-4-(8,8,939~tetramethyl-4,7-dioxa-l- a .a-8-siladecaii-l-yl)pyrimidine-5-carboxylate (150 mg, 0.328 mmol) in 0¼(¾ (3.3 mL) was added DIEA (143 μΤ, 0.82 mmol) then methane sulfonylchloride (28 μΐ, 0.361) at 0 °C. The reaction mixture was stirred for 30 mm and diluted with J¾0 (12 mL). The organic layer was washed with brine (10 mL) and dried to provide the crude mesylate that was used for the next step without further purification. The mesylate was dissolved in THF (7,3 mL) and TBAF (1M i THF, 585 L, 0.585 mmol) was added. The reaction mixture was stirred at room temperature overnight. The solvents were removed under a reduced pressure and the residue was purified by ISCO silica gel column to provide the title compound (32 mg, 34%) as a white solid. Ή NMR (400 MHz, CDC13) 6 8.57 (s, 1 H , 3.86-3.79 (ms 7K), 3.79-3.73 (ra, 2H), 3.72-3.66 (m, 5H), 3,64-3.59 (m, 2H), 2.32 (dd, J- 9.3, 3.6 Hz, 1H)5 1 .72-1.64 (m, 2H), 1.59 (dd, J- 10.8, 5.7 Hz, 4H).
5 ,8-Dioxa-2 -14 j 6.19-tetraazabicyclo[l 3.3., 1 jnonadega-l (18), 15(19), 16-triene-l 8-carboxylic acid


A solution of 5,8-dioxa-2s 14,16,19-tetraazabicycio[l 3.3.1 jnonadeca-l (18), 15(19),16- triene-18-carbox lic acid (30 mg, 0.099 mmol) in DMF (1.0 mL) was added [6~(lH-pyrazol- l-yl)pyridm»3-yl]inemanamine (17 mg, 0.099 mmol) and DIEA (34 uL, 0.197 mmol) followed by HATU (41 mg, 0.108 mmol). The reaction mixture was stirred at room temperature overnight, quenched with a 1.0 M NaOH solution (2.0 mL) and extracted with CH2CI2 (2 x 2 mL). The combined organic layers were dried (NaiSO^) and concentrated. The residue was purified by HPLC to afford the title compound (37 mg, 67%) as a light yellow solid. !H NMR (400 MHz, CD3OD) δ 8.62 (dd, J= 2.7, 0.4 Hz, 1H), 8.50 (d, J ^ 1.9 Hz, 1 H), 8.32 (s, 1H), 8.15 (dd, J™ 8.6, 2.3 Hz, 1H), 8.03 (d, J~ 8.5 Hz, 1H), 7,85 (d, J= 1.3 Hz, 1H), 6.61 (dd, J = 2.7, 1.7 Hz, 1H), 4.59 (s, 2H), 3,85-3.63 (m, 10H), 3.63-3.55 (m, 2H), 1.81- 1.65 (m, 6H). MS m/z 467.3 [M+H .
Table j§ describes compounds prepared following procedures described in Example 10 (General Procedure .1), using appropriate reagents.
Structure Compound_iD Mer Physical Data
ICso MS m/z (M+l ) or/and lH
NMR

Example 11
UNC3040A
General Procedure K:

Ethyl 2.8.9.10.17.19.22-heptaazatricvcIoi 16.3.1.1 7,i01tficosa- 1 (21V7(23),8.18f 22), 19- entaene-2. -carboxylate

A solution of 2,4-dichloropyrimidine~5-earbonyi chloride (1.00 g, 4.76 mmol) in ethanol (6.0 mL) was added DIPEA (2.5 raL, 14,4 mmol) slowly at 0 °C under nitrogen. After 30 mm, hex-5-yn-I -amine (0.476 g, 4.91 mmol) was added in one portion. The reaction mixture was stirred at room temperature for 3.5 h, then was added dropwise to a solution of 6-azidohexan- 1 -amine (0.801 g, 5.64 mmol) in ethanol (4.0 mL) at 50 °C After the reaction was complete (monitored by LCMS), the mixture was diluted with water (10 mL) and
concentrated under a reduced pressure and filtered. The yellow solid was washed with water and dried under vacuum to be used in the next step without further purification (0.723 g, 39% over 3 steps).
A solution of the yellow solid (0.130 g, 0.336 mmol) in dry CH2CI2 (25 mL) was added TBTA (1.6 mL, 5 mg/niL in CH2C12, 8.0 mg, 0.016 mmol) and Cu(MeCN)4BF4 (5.2 mg, 0.016 mmol). The reactio mixture was stirred under nitrogen at 45 °C for 24 h, then cooled to room temperature and concentrate. The residue was purified by silica gel column to provide the title compound (0.104 g, 80%). Ή NMR (400 MHz, CD3OD) δ 8.34 (s, 2H), 7.58 (s, 1H), 4,36 (s, 2H). 4.22 (q, J~ 7.1 Hz, 2H), 3.34 (s, 2H), 3.19 (s, 2H), 2.82-2.66 (m, 2H), 1.84 (s, 2H), 1.67 (s, 2H), 1.62 1.44 (m, 4H), 1.40-1.02 (m, 7H).
2.8.9.10 J 7 J 9,22-Heptaazatricyclon 6.3.1.17, l0]tricosa- 1 (2 l\7f23 .8.18(22), 19-pentaene-21 - carboxy lie acid

A solution of ethyl 2,8,9, 10, 17, 19,22-heptaazatricyclofl 6;3.1.17,l0]tricosa- l(21),7(23)i8.18(22),19-pentaene-21-carboxylate (31 mg, 0.081 mmol) in MeOH (2,0 mL) was added an aq. OH solution (10%, 0.50 mL). The reaction was stirred at 60 °C for 12 h. Then the reaction mixture was cooled to room temperature and adjusted to pK-3 by the addition of a HCi solution. The resulting solution was extracted by a mixture of /-PrOH/ C¾C¾ (1 :3, 10 mL x 3). The combined organic layers were washed with water and brine and concentrated under vacuum to afford the desired product (0.029 g, quant.) as a white solid. Ή NMR (400 MHz, CD3OD) 3 8.41-8.22 (m, 1 H)S 5.65 (s9 2H), 4.63-4.46 (m, 1H), 3.55-3.43 (m, 1H), 3.39-3.33 (ms 1 H), 3.02 (t, J ~ 6.9 Hz, 3H), 2.97-2.83 (m, I B), 2.58-2.29 (m, 3H), 2.03-1.89 (m, 1H), 1.85-1.71 (m, lH)t 1.71-1.50 (m, 2H), 1.49-0.97 (m, 3H). jV-f l-benzylpiperidin-4-vl)-2.8,9.10.17.19,22-heptaazatrioyclof 16.3.1.17, ,Qltricosa-
1(21 ),7(23 ,8, 18(22U 9-pentaene-21 -carboxamid (UNC3040A)

A solution of 2I859,10 i7!19s22-HepiaazatTicy lo[16.3.1 , l7,t0]tricosa« l(21),7(23)58,18(22),19-pent¾ene-21 -carboxylic acid (30 mg, 0.084 .mmol) in DMF (2.0 niL) was added 1 -benzylpiperidin-4-amine (19.8 mg, 0.10 mmol) followed by HATU (38 mg, 0.10 mmol}. The reaction mixture was stirred at room temperature overnight, quenched with a 1.0 M NaOH solution (2.0 mL) and extracted with CH2C12 (2 χ 2 ml). The combined organic layers were dried ( a2SG4) and concentrated. The residue was purified by HPLC to afford the title compound UNC3040A (6.0 mg, 13%) as a light yellow solid. H NMR (400 MHz, CD3OD) 8 8.24 (s5 1 H), 8.08-7.93 (in, 2H), 7.64-7.46 (m, 4H), 4.54-4.46 (m, 2H), 4.33 (s, 1H), 3,59-3.40 (m, 4H), 3.22-3.05 (m, 2H), 3.02-2.93 (ra, 3H), 2.91-2.77 (m, 3H), 2.37-2.26 (m, 1H)5 2.25-2.13 (m, 2H), 1.98-1.86 (m, 3H), 1.79-1.70 (m, 2H), 1.66-1.50 (m, 4H), 1.30-1.10 (m, 4H); MS (ESI): 554.5 [M + Naf .
The foregoing is illustrative of the present invention, and is not to be construed as limiting thereof. The invention is defined by the following claims., with equivalents of the claims to be included therein.
Claims
That which is claimed is:
1 , A compound of Formula Ϊ or Formula .11:

wherein:
ring A is a 5~ or 6-membered heteroaryl group, the dashed line is an optional double bond, X is N or 0. and Y is C, S or N in any location in the ring;
R! is -R5R6 5 where R5 is a covalent bond, CI to C3 alkyi or a linker group and Rfi is eycloalkyl, heterocycloalkyl, aryl, heteroaryl, aikylcycloalkyl, aikyiheterocyc!oalkyl, alkylaryk alkyiheteroaryl or alkyi, and wherein R6 is optionally substituted one, two or three times with independently selected polar groups;
R2 and R3 together form a linking group;
R4 is H, loweralkyi, halo, or loweralkoxy;
or a pharmaceutically acceptable salt thereof,
2. The compound of claim 1 , wherein R" is a covalent bond.
3. The compound of claim I, wherein R5 is CI to C3 aikyiene.
4. The compound of claim 1 , wherein R5 is ~C¾-.
5. The compound of claim 1 to 4, wherein R6 is phenyl, piperidyl, C1-C8 alkyi, or C3- C8 eycloalkyl, which phenyl, pipyridyl, alkyi or eycloalkyl is unsubstituted or substituted from 1 to 3 times with sulfono, halo, amino, nitro, alkyi, alkoxyl, haloalkyl, eycloalkyl, heterocycloalkyl, aryl, or heteroaryl
6. The compound of claim 1 to 5, wherein R4 is H,
7. The compound of claim 1, wherein said compound has a structure selected from the group consisting of:
UNC2427A




UNC2343A

UNC2340A

UNC2324A


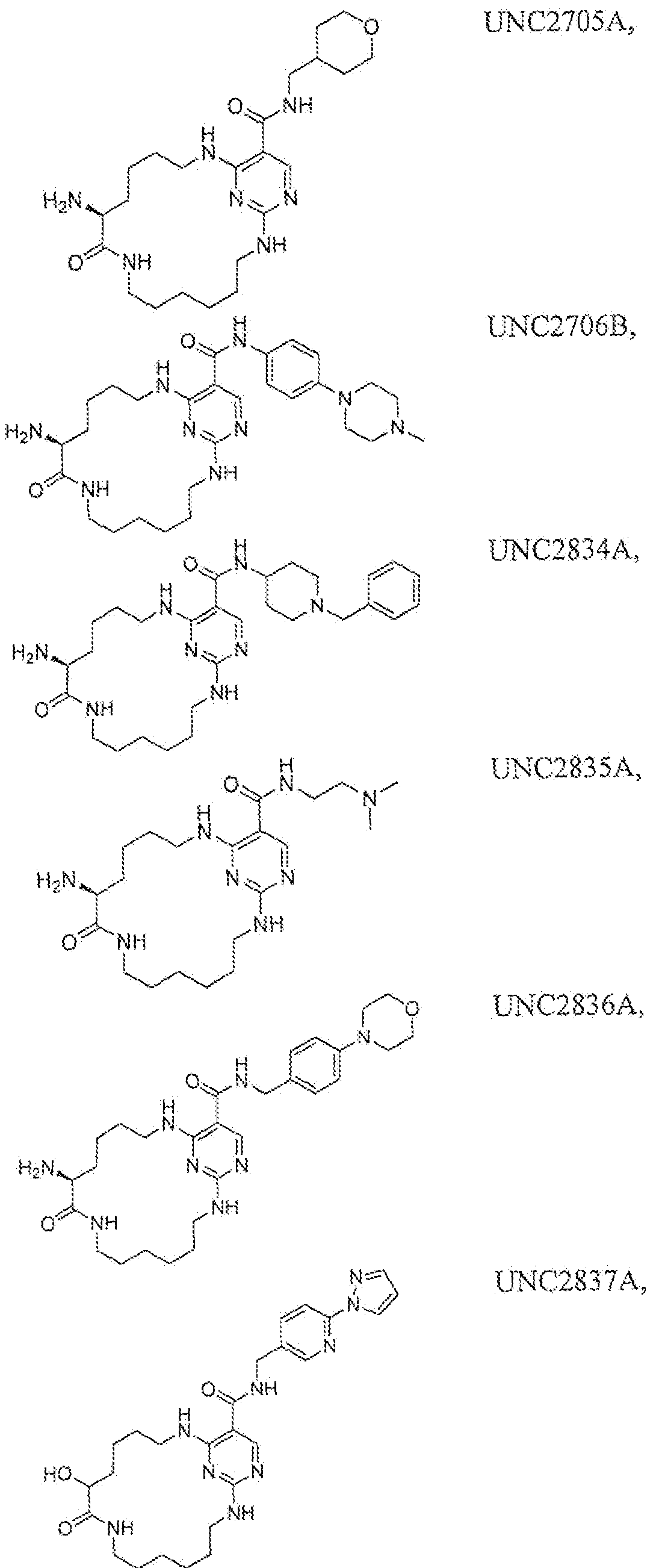




UNC3017A

UNC3588A

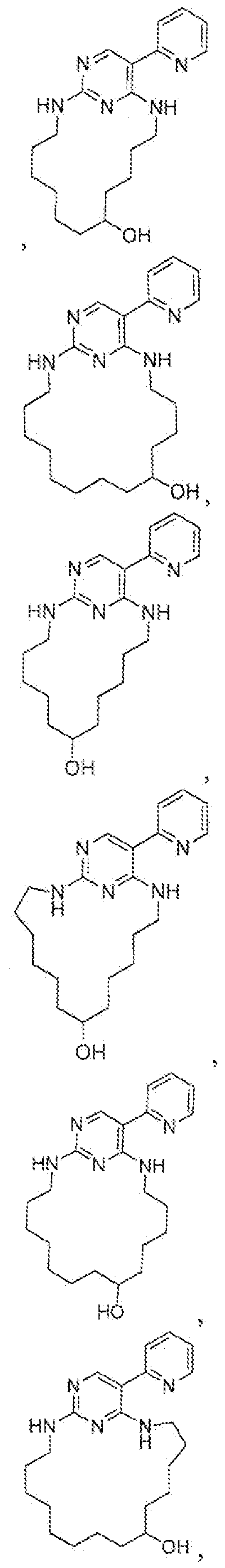



UNC3040A
and pharmaceutically acceptable salts thereof,
8. A composition comprising a compound of claim 1 to 7 in a pharmaceutically acceptable carrier.
9. A method of treating cancer in a subject in need thereof, comprising administering said subject, a compound of claim 1 to 7 in an amoimt effective to treat said cancer,
10. The method of claim 9, wherein said cancer is selected .from the group consisting of myeloid leukemia, lymphoblastic leukemia, melanoma, breast, lung, colon, liver, gastric, kidney, ovarian, uterine, and brain cancer.
1 1. A compound of claim 1 to 7 for use in the treatment of cancer or for the preparation of a. medicament for treating cancer.
12. The use of claim 11, wherein said cancer is selected from the group consisting of myeloid leukemia, lymphoblastic leukemia, melanoma, breast, lung, colon, liver, gastric, kidney, ovarian, uterine, and brain cancer.
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP13858929.6A EP2925752A4 (en) | 2012-11-27 | 2013-11-22 | Pyrimidine compounds for the treatment of cancer |
| US14/647,733 US9771330B2 (en) | 2012-11-27 | 2013-11-22 | Pyrimidine compounds for the treatment of cancer |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261730179P | 2012-11-27 | 2012-11-27 | |
| US61/730,179 | 2012-11-27 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014085225A1 true WO2014085225A1 (en) | 2014-06-05 |
Family
ID=50828366
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2013/071409 WO2014085225A1 (en) | 2012-11-27 | 2013-11-22 | Pyrimidine compounds for the treatment of cancer |
Country Status (3)
| Country | Link |
|---|---|
| US (1) | US9771330B2 (en) |
| EP (1) | EP2925752A4 (en) |
| WO (1) | WO2014085225A1 (en) |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9273056B2 (en) | 2011-10-03 | 2016-03-01 | The University Of North Carolina At Chapel Hill | Pyrrolopyrimidine compounds for the treatment of cancer |
| US9555030B2 (en) | 2014-04-11 | 2017-01-31 | The University Of North Carolina At Chapel Hill | Therapeutic uses of selected pyrazolopyrimidine compounds with anti-Mer tyrosine kinase activity |
| US9562047B2 (en) | 2012-10-17 | 2017-02-07 | The University Of North Carolina At Chapel Hill | Pyrazolopyrimidine compounds for the treatment of cancer |
| US9567326B2 (en) | 2012-05-22 | 2017-02-14 | The University Of North Carolina At Chapel Hill | Pyrimidine compounds for the treatment of cancer |
| US9744172B2 (en) | 2010-05-19 | 2017-08-29 | The University Of North Carolina At Chapel Hill | Pyrazolopyrimidine compounds for the treatment of cancer |
| US9771330B2 (en) | 2012-11-27 | 2017-09-26 | The University Of North Carolina At Chapel Hill | Pyrimidine compounds for the treatment of cancer |
| WO2017197046A1 (en) | 2016-05-10 | 2017-11-16 | C4 Therapeutics, Inc. | C3-carbon linked glutarimide degronimers for target protein degradation |
| WO2017197036A1 (en) | 2016-05-10 | 2017-11-16 | C4 Therapeutics, Inc. | Spirocyclic degronimers for target protein degradation |
| WO2017197055A1 (en) | 2016-05-10 | 2017-11-16 | C4 Therapeutics, Inc. | Heterocyclic degronimers for target protein degradation |
| WO2020132561A1 (en) | 2018-12-20 | 2020-06-25 | C4 Therapeutics, Inc. | Targeted protein degradation |
| US10709708B2 (en) | 2016-03-17 | 2020-07-14 | The University Of North Carolina At Chapel Hill | Method of treating cancer with a combination of MER tyrosine kinase inhibitor and an epidermal growth factor receptor (EGFR) inhibitor |
| CN113278027A (en) * | 2021-06-02 | 2021-08-20 | 浙江合聚生物医药有限公司 | Preparation method of antitumor drug Laolatinib |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000039101A1 (en) * | 1998-12-24 | 2000-07-06 | Astrazeneca Ab | Pyrimidine compounds |
| WO2005037801A1 (en) * | 2003-10-21 | 2005-04-28 | Sankyo Company, Limited | Pyrimidine compound |
| WO2007041379A1 (en) * | 2005-09-30 | 2007-04-12 | Bristol-Myers Squibb Company | Met kinase inhibitors |
| WO2011090760A1 (en) * | 2009-12-29 | 2011-07-28 | Avila Therapeutics, Inc. | Heteroaryl compounds and uses thereof |
Family Cites Families (65)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5958930A (en) | 1991-04-08 | 1999-09-28 | Duquesne University Of The Holy Ghost | Pyrrolo pyrimidine and furo pyrimidine derivatives |
| WO1997049706A1 (en) | 1996-06-25 | 1997-12-31 | Novartis Ag | SUBSTITUTED 7-AMINO-PYRROLO[3,2-d]PYRIMIDINES AND THE USE THEREOF |
| US20080248046A1 (en) | 1997-03-17 | 2008-10-09 | Human Genome Sciences, Inc. | Death domain containing receptor 5 |
| ES2309206T3 (en) | 2001-10-02 | 2008-12-16 | Smithkline Beecham Corporation | CHEMICAL COMPOUNDS. |
| DE10239042A1 (en) * | 2002-08-21 | 2004-03-04 | Schering Ag | New fused macrocyclic pyrimidine derivatives, useful as e.g. cyclin-dependent kinase inhibitors for treating e.g. cancer, autoimmune, cardiovascular or neurodegenerative diseases or viral infections |
| WO2004106340A2 (en) | 2003-05-28 | 2004-12-09 | Università Degli Studi Di Siena | 4-SUBSTITUTED DERIVATIVES OF PYRAZOLO [3,4-d] PYRIMIDINE AND PYRROLO [2,3-d] PYRIMIDINE AND USES THEREOF |
| US7504396B2 (en) | 2003-06-24 | 2009-03-17 | Amgen Inc. | Substituted heterocyclic compounds and methods of use |
| WO2005028434A2 (en) | 2003-09-18 | 2005-03-31 | Conforma Therapeutics Corporation | Novel heterocyclic compounds as hsp90-inhibitors |
| AU2005209231B8 (en) | 2004-01-21 | 2011-07-28 | Emory University | Compositions and methods of use for tyrosine kinase inhibitors to treat pathogenic infection |
| WO2005074603A2 (en) | 2004-02-03 | 2005-08-18 | Abbott Laboratories | Aminobenzoxazoles as therapeutic agents |
| WO2005080393A1 (en) | 2004-02-14 | 2005-09-01 | Irm Llc | Compounds and compositions as protein kinase inhibitors |
| WO2005095382A1 (en) | 2004-03-30 | 2005-10-13 | Kyowa Hakko Kogyo Co., Ltd. | Anti-tumor agent |
| CA2562244A1 (en) | 2004-04-07 | 2005-10-27 | Takeda Pharmaceutical Company Limited | Cyclic compounds |
| GB0420719D0 (en) | 2004-09-17 | 2004-10-20 | Addex Pharmaceuticals Sa | Novel allosteric modulators |
| AU2005288864B2 (en) | 2004-09-30 | 2012-08-23 | Janssen Sciences Ireland Uc | HIV inhibiting 5-heterocyclyl pyrimidines |
| US7906528B2 (en) | 2004-10-05 | 2011-03-15 | Novartis International Pharmaceutical Ltd. | Pyrrolo-pyridine, pyrrolo-pyrimidine and related heterocyclic compounds |
| US7994172B2 (en) | 2004-12-28 | 2011-08-09 | Exelixis, Inc. | [1H-pyrazolo[3, 4-D]pyrimidin-4-yl]-piperidine or -piperazine compounds as serine-theoronine kinase modulators (P70s6k, Atk1 and Atk2) for the treatment of immunological, inflammatory and proliferative diseases |
| EP1710246A1 (en) | 2005-04-08 | 2006-10-11 | Schering Aktiengesellschaft | Sulfoximine-pyrimidine Macrocycles and the salts thereof, a process for making them, and their pharmaceutical use against cancer |
| WO2007032445A1 (en) | 2005-09-16 | 2007-03-22 | Kyowa Hakko Kogyo Co., Ltd. | Protein kinase inhibitors |
| US20070105874A1 (en) | 2005-09-23 | 2007-05-10 | Conforma Therapeutics Corporation | Anti-Tumor Methods Using Multi Drug Resistance Independent Synthetic HSP90 Inhibitors |
| CA2624826A1 (en) | 2005-10-06 | 2007-04-19 | Schering Corporation | Pyrazolopyrimidines as protein kinase inhibitors |
| AU2006321841C1 (en) | 2005-12-08 | 2013-01-24 | E. R. Squibb & Sons, L.L.C. | Human monoclonal antibodies to protein tyrosine kinase 7 ( PTK7 ) and their use |
| EP1979002A2 (en) | 2005-12-19 | 2008-10-15 | OSI Pharmaceuticals, Inc. | Combination of igfr inhibitor and anti-cancer agent |
| EP1803723A1 (en) | 2006-01-03 | 2007-07-04 | Bayer Schering Pharma Aktiengesellschaft | (2,4,9-triaza-1(2,4)-pyrimidina-3(1,3)-benzenacyclononaphan-3^4-yl)-sulfoximide derivatives as selective inhibitors of the aurora kinase for the treatment of cancer |
| JP5078986B2 (en) | 2006-03-30 | 2012-11-21 | テイボテク・フアーマシユーチカルズ | 5-Amido substituted pyrimidines that inhibit HIV |
| WO2007125405A2 (en) | 2006-05-01 | 2007-11-08 | Pfizer Products Inc. | Substituted 2-amino-fused heterocyclic compounds |
| GB0610242D0 (en) | 2006-05-23 | 2006-07-05 | Novartis Ag | Organic compounds |
| TWI398252B (en) | 2006-05-26 | 2013-06-11 | Novartis Ag | Pyrrolopyrimidine compounds and their uses |
| CA2680122A1 (en) | 2007-03-05 | 2008-09-18 | Kyowa Hakko Kirin Co., Ltd. | Pharmaceutical composition |
| AU2008296479A1 (en) | 2007-08-28 | 2009-03-12 | Dana Farber Cancer Institute | Amino substituted pyrimidine, pyrollopyridine and pyrazolopyrimidine derivatives useful as kinase inhibitors and in treating proliferative disorders and diseases associated with angiogenesis |
| WO2009047359A1 (en) | 2007-10-12 | 2009-04-16 | Ingenium Pharmaceuticals Gmbh | Inhibitors of protein kinases |
| EP2902495B1 (en) | 2007-11-09 | 2019-12-25 | The Salk Institute for Biological Studies | Use of tam receptor activators as immunosuppressors |
| US8399206B2 (en) | 2008-07-10 | 2013-03-19 | Nodality, Inc. | Methods for diagnosis, prognosis and methods of treatment |
| WO2010014755A1 (en) | 2008-07-29 | 2010-02-04 | The Regents Of The University Of Colorado | Methods and compounds for enhancing anti-cancer therapy |
| US8569466B2 (en) | 2008-09-10 | 2013-10-29 | Nnochiri Ekwuribe | Aromatic carboxylic acid derivatives for treatment and prophylaxis of gastrointestinal diseases including colon cancers |
| AR073760A1 (en) | 2008-10-03 | 2010-12-01 | Astrazeneca Ab | HETEROCICLIC DERIVATIVES AND METHODS OF USE OF THE SAME |
| GB0819105D0 (en) | 2008-10-17 | 2008-11-26 | Chroma Therapeutics Ltd | Pyrrolo-pyrimidine compounds |
| WO2010068863A2 (en) | 2008-12-12 | 2010-06-17 | Cystic Fibrosis Foundation Therapeutics, Inc. | Pyrimidine compounds and methods of making and using same |
| WO2010085597A1 (en) | 2009-01-23 | 2010-07-29 | Incyte Corporation | Macrocyclic compounds and their use as kinase inhibitors |
| WO2010117425A1 (en) | 2009-03-31 | 2010-10-14 | Biogen Idec Ma Inc. | Certain substituted pyrimidines, pharmaceutical compositions thereof, and methods for their use |
| UA107791C2 (en) | 2009-05-05 | 2015-02-25 | Dow Agrosciences Llc | Pesticidal compositions |
| WO2010129802A1 (en) | 2009-05-06 | 2010-11-11 | Portola Pharmaceuticals, Inc. | Inhibitors of jak |
| WO2011020024A2 (en) | 2009-08-13 | 2011-02-17 | The Johns Hopkins University | Methods of modulating immune function |
| JP2013504543A (en) | 2009-09-10 | 2013-02-07 | ノバルティス アーゲー | Ether derivatives of bicyclic heteroaryls |
| WO2011065800A2 (en) | 2009-11-30 | 2011-06-03 | 주식회사 오스코텍 | Pyrimidine derivative, method for preparing same and pharmaceutical composition containing same |
| US9433621B2 (en) | 2010-02-18 | 2016-09-06 | Merck Sharp & Dohme Corp. | Substituted pyridine and pyrimidine derivatives and their use in treating viral infections |
| GB201004311D0 (en) | 2010-03-15 | 2010-04-28 | Proximagen Ltd | New enzyme inhibitor compounds |
| US9133123B2 (en) | 2010-04-23 | 2015-09-15 | Cytokinetics, Inc. | Certain amino-pyridines and amino-triazines, compositions thereof, and methods for their use |
| US9290499B2 (en) | 2010-05-19 | 2016-03-22 | The University Of North Carolina At Chapel Hill | Pyrazolopyrimidine compounds for the treatment of cancer |
| WO2011153553A2 (en) | 2010-06-04 | 2011-12-08 | The Regents Of The University Of California | Methods and compositions for kinase inhibition |
| JP2014005206A (en) | 2010-10-22 | 2014-01-16 | Astellas Pharma Inc | Arylamino heterocyclic carboxamide compound |
| JP5898687B2 (en) | 2010-11-18 | 2016-04-06 | カシナ ライラ イノバ ファーマシューティカルズ プライベート リミテッド | Substituted 4- (selenophene-2 (or 3) -ylamino) pyrimidine compounds and methods of use thereof |
| US8362023B2 (en) | 2011-01-19 | 2013-01-29 | Hoffmann-La Roche Inc. | Pyrazolo pyrimidines |
| US8889684B2 (en) | 2011-02-02 | 2014-11-18 | Boehringer Ingelheim International Gmbh | Azaindolylphenyl sulfonamides as serine/threonine kinase inhibitors |
| EP2489663A1 (en) | 2011-02-16 | 2012-08-22 | Almirall, S.A. | Compounds as syk kinase inhibitors |
| US9376438B2 (en) | 2011-05-17 | 2016-06-28 | Principia Biopharma, Inc. | Pyrazolopyrimidine derivatives as tyrosine kinase inhibitors |
| RU2014111823A (en) | 2011-08-29 | 2015-10-10 | Инфинити Фармасьютикалз, Инк. | HETEROCYCLIC COMPOUNDS AND THEIR APPLICATIONS |
| AU2012311184A1 (en) | 2011-09-22 | 2014-03-06 | Pfizer Inc. | Pyrrolopyrimidine and purine derivatives |
| EP2763988B1 (en) | 2011-10-03 | 2017-09-20 | The University of North Carolina At Chapel Hill | Pyrrolopyrimidine compounds for the treatment of cancer |
| EP2817328A1 (en) | 2012-02-21 | 2014-12-31 | Institut National de la Santé et de la Recherche Médicale | Tam receptors as virus entry cofactors |
| NO2840080T3 (en) | 2012-04-17 | 2018-05-05 | ||
| WO2013157022A1 (en) | 2012-04-20 | 2013-10-24 | Advinus Therapeutics Limited | Substituted hetero-bicyclic compounds, compositions and medicinal applications thereof |
| US9567326B2 (en) | 2012-05-22 | 2017-02-14 | The University Of North Carolina At Chapel Hill | Pyrimidine compounds for the treatment of cancer |
| US9562047B2 (en) | 2012-10-17 | 2017-02-07 | The University Of North Carolina At Chapel Hill | Pyrazolopyrimidine compounds for the treatment of cancer |
| EP2925752A4 (en) | 2012-11-27 | 2016-06-01 | Univ North Carolina | Pyrimidine compounds for the treatment of cancer |
-
2013
- 2013-11-22 EP EP13858929.6A patent/EP2925752A4/en not_active Withdrawn
- 2013-11-22 US US14/647,733 patent/US9771330B2/en active Active
- 2013-11-22 WO PCT/US2013/071409 patent/WO2014085225A1/en active Application Filing
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000039101A1 (en) * | 1998-12-24 | 2000-07-06 | Astrazeneca Ab | Pyrimidine compounds |
| WO2005037801A1 (en) * | 2003-10-21 | 2005-04-28 | Sankyo Company, Limited | Pyrimidine compound |
| WO2007041379A1 (en) * | 2005-09-30 | 2007-04-12 | Bristol-Myers Squibb Company | Met kinase inhibitors |
| WO2011090760A1 (en) * | 2009-12-29 | 2011-07-28 | Avila Therapeutics, Inc. | Heteroaryl compounds and uses thereof |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP2925752A4 * |
Cited By (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9744172B2 (en) | 2010-05-19 | 2017-08-29 | The University Of North Carolina At Chapel Hill | Pyrazolopyrimidine compounds for the treatment of cancer |
| US10179133B2 (en) | 2011-10-03 | 2019-01-15 | The University Of North Carolina At Chapel Hill | Pyrrolopyrimidine compounds for the treatment of cancer |
| US9795606B2 (en) | 2011-10-03 | 2017-10-24 | The University Of North Carolina At Chapel Hill | Pyrrolopyrimidine compounds for the treatment of cancer |
| US9273056B2 (en) | 2011-10-03 | 2016-03-01 | The University Of North Carolina At Chapel Hill | Pyrrolopyrimidine compounds for the treatment of cancer |
| US9567326B2 (en) | 2012-05-22 | 2017-02-14 | The University Of North Carolina At Chapel Hill | Pyrimidine compounds for the treatment of cancer |
| US9562047B2 (en) | 2012-10-17 | 2017-02-07 | The University Of North Carolina At Chapel Hill | Pyrazolopyrimidine compounds for the treatment of cancer |
| US9771330B2 (en) | 2012-11-27 | 2017-09-26 | The University Of North Carolina At Chapel Hill | Pyrimidine compounds for the treatment of cancer |
| CN106459063A (en) * | 2014-04-11 | 2017-02-22 | 北卡罗来纳-查佩尔山大学 | MerTK-specific pyrimidine compounds |
| US10004755B2 (en) | 2014-04-11 | 2018-06-26 | The University Of North Carolina At Chapel Hill | Therapeutic uses of selected pyrrolopyrimidine compounds with anti-mer tyrosine kinase activity |
| US9603850B2 (en) | 2014-04-11 | 2017-03-28 | The University Of North Carolina At Chapel Hill | MerTK-specific pyrazolopyrimidine compounds |
| US9555031B2 (en) | 2014-04-11 | 2017-01-31 | The University Of North Carolina At Chapel Hill | Therapeutic uses of selected pyrrolopyrimidine compounds with anti-mer tyrosine kinase activity |
| US9649309B2 (en) | 2014-04-11 | 2017-05-16 | The University Of North Carolina At Chapel Hill | Therapeutic uses of selected pyrimidine compounds with anti-Mer tyrosine kinase activity |
| US9555030B2 (en) | 2014-04-11 | 2017-01-31 | The University Of North Carolina At Chapel Hill | Therapeutic uses of selected pyrazolopyrimidine compounds with anti-Mer tyrosine kinase activity |
| EP3129377A4 (en) * | 2014-04-11 | 2017-12-20 | The University of North Carolina at Chapel Hill | Mertk-specific pyrimidine compounds |
| US10709708B2 (en) | 2016-03-17 | 2020-07-14 | The University Of North Carolina At Chapel Hill | Method of treating cancer with a combination of MER tyrosine kinase inhibitor and an epidermal growth factor receptor (EGFR) inhibitor |
| WO2017197046A1 (en) | 2016-05-10 | 2017-11-16 | C4 Therapeutics, Inc. | C3-carbon linked glutarimide degronimers for target protein degradation |
| WO2017197055A1 (en) | 2016-05-10 | 2017-11-16 | C4 Therapeutics, Inc. | Heterocyclic degronimers for target protein degradation |
| WO2017197036A1 (en) | 2016-05-10 | 2017-11-16 | C4 Therapeutics, Inc. | Spirocyclic degronimers for target protein degradation |
| WO2020132561A1 (en) | 2018-12-20 | 2020-06-25 | C4 Therapeutics, Inc. | Targeted protein degradation |
| CN113278027A (en) * | 2021-06-02 | 2021-08-20 | 浙江合聚生物医药有限公司 | Preparation method of antitumor drug Laolatinib |
| CN113278027B (en) * | 2021-06-02 | 2022-05-31 | 浙江合聚生物医药有限公司 | Preparation method of antitumor drug Laolatinib |
Also Published As
| Publication number | Publication date |
|---|---|
| US9771330B2 (en) | 2017-09-26 |
| US20150322019A1 (en) | 2015-11-12 |
| EP2925752A1 (en) | 2015-10-07 |
| EP2925752A4 (en) | 2016-06-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2925752A1 (en) | Pyrimidine compounds for the treatment of cancer | |
| US10179133B2 (en) | Pyrrolopyrimidine compounds for the treatment of cancer | |
| AU2013266438B2 (en) | Pyrimidine compounds for the treatment of cancer | |
| ES2928169T3 (en) | 4-(3H-indol-5-yl)-N-(pyridin-2-yl)pyrimidin-2-amine derivatives as protein kinase inhibitors, and method of preparation and medical use thereof | |
| US9744172B2 (en) | Pyrazolopyrimidine compounds for the treatment of cancer | |
| US9562047B2 (en) | Pyrazolopyrimidine compounds for the treatment of cancer | |
| ES2879441T3 (en) | Protein kinase inhibitors related to ataxia telangiectasia and RAD3 (ATR) | |
| BR112021011147A2 (en) | PYRAZOLYL-AMINO-PYRIMIDINIL DERIVATIVES BENZAMIDES AND COMPOSITIONS AND METHODS THEREOF | |
| CN114409653A (en) | Bridged ring pyrimidine-fused ring compound and application thereof | |
| TW201043630A (en) | Purine derivatives and anticancer agent using the same |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13858929 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14647733 Country of ref document: US |
|
| REEP | Request for entry into the european phase |
Ref document number: 2013858929 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013858929 Country of ref document: EP |