WO2014040639A1 - Silane sulfide modified elastomeric polymers - Google Patents
Silane sulfide modified elastomeric polymers Download PDFInfo
- Publication number
- WO2014040639A1 WO2014040639A1 PCT/EP2012/068120 EP2012068120W WO2014040639A1 WO 2014040639 A1 WO2014040639 A1 WO 2014040639A1 EP 2012068120 W EP2012068120 W EP 2012068120W WO 2014040639 A1 WO2014040639 A1 WO 2014040639A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- polymer
- alkyl
- silane sulfide
- independently selected
- Prior art date
Links
- 229920000642 polymer Polymers 0.000 title claims abstract description 463
- UCKMPCXJQFINFW-UHFFFAOYSA-N Sulphide Chemical compound [S-2] UCKMPCXJQFINFW-UHFFFAOYSA-N 0.000 title claims abstract description 188
- 229910000077 silane Inorganic materials 0.000 title claims abstract description 183
- BLRPTPMANUNPDV-UHFFFAOYSA-N Silane Chemical compound [SiH4] BLRPTPMANUNPDV-UHFFFAOYSA-N 0.000 title claims abstract description 178
- 239000000203 mixture Substances 0.000 claims abstract description 230
- 239000003795 chemical substances by application Substances 0.000 claims abstract description 109
- 239000003607 modifier Substances 0.000 claims abstract description 105
- 150000001875 compounds Chemical class 0.000 claims abstract description 89
- 229920002521 macromolecule Polymers 0.000 claims abstract description 87
- 238000000034 method Methods 0.000 claims abstract description 46
- 238000004073 vulcanization Methods 0.000 claims abstract description 31
- 125000000129 anionic group Chemical group 0.000 claims abstract description 20
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 claims description 112
- 125000000217 alkyl group Chemical group 0.000 claims description 79
- 239000000945 filler Substances 0.000 claims description 72
- 239000007822 coupling agent Substances 0.000 claims description 64
- 239000000377 silicon dioxide Substances 0.000 claims description 63
- PPBRXRYQALVLMV-UHFFFAOYSA-N Styrene Chemical compound C=CC1=CC=CC=C1 PPBRXRYQALVLMV-UHFFFAOYSA-N 0.000 claims description 45
- 238000006116 polymerization reaction Methods 0.000 claims description 44
- 239000002904 solvent Substances 0.000 claims description 44
- KAKZBPTYRLMSJV-UHFFFAOYSA-N Butadiene Chemical compound C=CC=C KAKZBPTYRLMSJV-UHFFFAOYSA-N 0.000 claims description 43
- -1 amine compound Chemical class 0.000 claims description 38
- 239000000178 monomer Substances 0.000 claims description 38
- 239000006229 carbon black Substances 0.000 claims description 37
- RRHGJUQNOFWUDK-UHFFFAOYSA-N Isoprene Chemical compound CC(=C)C=C RRHGJUQNOFWUDK-UHFFFAOYSA-N 0.000 claims description 36
- 229910052710 silicon Inorganic materials 0.000 claims description 31
- 229920001577 copolymer Polymers 0.000 claims description 27
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical group [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 claims description 26
- 239000011541 reaction mixture Substances 0.000 claims description 26
- 229910052718 tin Chemical group 0.000 claims description 25
- 125000003118 aryl group Chemical group 0.000 claims description 24
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical group [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 claims description 23
- 125000002877 alkyl aryl group Chemical group 0.000 claims description 23
- 125000003710 aryl alkyl group Chemical group 0.000 claims description 21
- 239000010703 silicon Substances 0.000 claims description 21
- 238000004519 manufacturing process Methods 0.000 claims description 14
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims description 13
- 239000007795 chemical reaction product Substances 0.000 claims description 13
- 239000005062 Polybutadiene Substances 0.000 claims description 12
- 238000001914 filtration Methods 0.000 claims description 12
- 229920002857 polybutadiene Polymers 0.000 claims description 11
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 claims description 10
- 125000004948 alkyl aryl alkyl group Chemical group 0.000 claims description 9
- 229920001195 polyisoprene Polymers 0.000 claims description 9
- 125000003808 silyl group Chemical group [H][Si]([H])([H])[*] 0.000 claims description 9
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 claims description 8
- 229910052739 hydrogen Inorganic materials 0.000 claims description 7
- 239000001257 hydrogen Substances 0.000 claims description 7
- 239000003505 polymerization initiator Substances 0.000 claims description 7
- 229920001897 terpolymer Polymers 0.000 claims description 7
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 claims description 6
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 6
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 6
- 239000002245 particle Substances 0.000 claims description 6
- LFQCEHFDDXELDD-UHFFFAOYSA-N tetramethyl orthosilicate Chemical compound CO[Si](OC)(OC)OC LFQCEHFDDXELDD-UHFFFAOYSA-N 0.000 claims description 6
- BOTDANWDWHJENH-UHFFFAOYSA-N Tetraethyl orthosilicate Chemical compound CCO[Si](OCC)(OCC)OCC BOTDANWDWHJENH-UHFFFAOYSA-N 0.000 claims description 5
- 229910021627 Tin(IV) chloride Inorganic materials 0.000 claims description 5
- HPGGPRDJHPYFRM-UHFFFAOYSA-J tin(iv) chloride Chemical compound Cl[Sn](Cl)(Cl)Cl HPGGPRDJHPYFRM-UHFFFAOYSA-J 0.000 claims description 5
- 229910000019 calcium carbonate Inorganic materials 0.000 claims description 4
- 239000004927 clay Substances 0.000 claims description 4
- 238000004821 distillation Methods 0.000 claims description 4
- 229920005610 lignin Polymers 0.000 claims description 4
- ZLNQQNXFFQJAID-UHFFFAOYSA-L magnesium carbonate Chemical compound [Mg+2].[O-]C([O-])=O ZLNQQNXFFQJAID-UHFFFAOYSA-L 0.000 claims description 4
- 239000001095 magnesium carbonate Substances 0.000 claims description 4
- 229910000021 magnesium carbonate Inorganic materials 0.000 claims description 4
- 229910003910 SiCl4 Inorganic materials 0.000 claims description 3
- 229910000102 alkali metal hydride Inorganic materials 0.000 claims description 3
- 150000008046 alkali metal hydrides Chemical class 0.000 claims description 3
- XYLMUPLGERFSHI-UHFFFAOYSA-N alpha-Methylstyrene Chemical compound CC(=C)C1=CC=CC=C1 XYLMUPLGERFSHI-UHFFFAOYSA-N 0.000 claims description 3
- 229910052570 clay Inorganic materials 0.000 claims description 3
- 239000011521 glass Substances 0.000 claims description 3
- FDNAPBUWERUEDA-UHFFFAOYSA-N silicon tetrachloride Chemical compound Cl[Si](Cl)(Cl)Cl FDNAPBUWERUEDA-UHFFFAOYSA-N 0.000 claims description 3
- 238000001704 evaporation Methods 0.000 claims description 2
- 230000008020 evaporation Effects 0.000 claims description 2
- PPDADIYYMSXQJK-UHFFFAOYSA-N trichlorosilicon Chemical compound Cl[Si](Cl)Cl PPDADIYYMSXQJK-UHFFFAOYSA-N 0.000 claims description 2
- 125000001302 tertiary amino group Chemical group 0.000 claims 3
- 150000002431 hydrogen Chemical class 0.000 claims 1
- 238000006243 chemical reaction Methods 0.000 abstract description 36
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 127
- GRVDJDISBSALJP-UHFFFAOYSA-N methyloxidanyl Chemical compound [O]C GRVDJDISBSALJP-UHFFFAOYSA-N 0.000 description 118
- 125000002572 propoxy group Chemical group [*]OC([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 95
- 238000012986 modification Methods 0.000 description 72
- 230000004048 modification Effects 0.000 description 66
- 125000000325 methylidene group Chemical group [H]C([H])=* 0.000 description 38
- 239000000243 solution Substances 0.000 description 36
- 230000002829 reductive effect Effects 0.000 description 33
- 239000003921 oil Substances 0.000 description 29
- 235000019198 oils Nutrition 0.000 description 29
- 238000002360 preparation method Methods 0.000 description 28
- 238000005859 coupling reaction Methods 0.000 description 27
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 24
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 24
- 238000002156 mixing Methods 0.000 description 22
- 230000008569 process Effects 0.000 description 20
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 19
- 230000008878 coupling Effects 0.000 description 19
- 238000010168 coupling process Methods 0.000 description 19
- 229920001971 elastomer Polymers 0.000 description 18
- 239000005060 rubber Substances 0.000 description 18
- 239000000047 product Substances 0.000 description 17
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 14
- OFBQJSOFQDEBGM-UHFFFAOYSA-N n-pentane Natural products CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 14
- 241001441571 Hiodontidae Species 0.000 description 13
- 239000002879 Lewis base Substances 0.000 description 13
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 13
- 150000001993 dienes Chemical class 0.000 description 13
- 238000005227 gel permeation chromatography Methods 0.000 description 13
- 150000007527 lewis bases Chemical class 0.000 description 13
- 229920003048 styrene butadiene rubber Polymers 0.000 description 13
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 12
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 12
- 238000005096 rolling process Methods 0.000 description 12
- 229920002554 vinyl polymer Polymers 0.000 description 12
- XLOMVQKBTHCTTD-UHFFFAOYSA-N Zinc monoxide Chemical compound [Zn]=O XLOMVQKBTHCTTD-UHFFFAOYSA-N 0.000 description 11
- 238000009472 formulation Methods 0.000 description 11
- 230000037361 pathway Effects 0.000 description 11
- 239000000523 sample Substances 0.000 description 11
- 229910052717 sulfur Inorganic materials 0.000 description 11
- 239000006057 Non-nutritive feed additive Substances 0.000 description 10
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 10
- 229910052799 carbon Inorganic materials 0.000 description 10
- 239000003999 initiator Substances 0.000 description 10
- 238000005259 measurement Methods 0.000 description 10
- 239000011593 sulfur Substances 0.000 description 10
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 9
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 9
- 238000005481 NMR spectroscopy Methods 0.000 description 9
- 239000006087 Silane Coupling Agent Substances 0.000 description 9
- 230000015572 biosynthetic process Effects 0.000 description 9
- 239000006193 liquid solution Substances 0.000 description 9
- 238000000746 purification Methods 0.000 description 9
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 9
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 8
- UHOVQNZJYSORNB-MZWXYZOWSA-N benzene-d6 Chemical compound [2H]C1=C([2H])C([2H])=C([2H])C([2H])=C1[2H] UHOVQNZJYSORNB-MZWXYZOWSA-N 0.000 description 8
- 238000009826 distribution Methods 0.000 description 8
- 238000005516 engineering process Methods 0.000 description 8
- 229910052744 lithium Inorganic materials 0.000 description 8
- 238000006011 modification reaction Methods 0.000 description 8
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Chemical compound O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 8
- MYRTYDVEIRVNKP-UHFFFAOYSA-N 1,2-Divinylbenzene Chemical compound C=CC1=CC=CC=C1C=C MYRTYDVEIRVNKP-UHFFFAOYSA-N 0.000 description 7
- 101000801643 Homo sapiens Retinal-specific phospholipid-transporting ATPase ABCA4 Proteins 0.000 description 7
- 102100033617 Retinal-specific phospholipid-transporting ATPase ABCA4 Human genes 0.000 description 7
- 238000005299 abrasion Methods 0.000 description 7
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 7
- 150000003512 tertiary amines Chemical group 0.000 description 7
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- 239000004793 Polystyrene Substances 0.000 description 6
- 125000001931 aliphatic group Chemical group 0.000 description 6
- 150000001336 alkenes Chemical class 0.000 description 6
- 239000000470 constituent Substances 0.000 description 6
- 238000001125 extrusion Methods 0.000 description 6
- 229910000103 lithium hydride Inorganic materials 0.000 description 6
- 239000000725 suspension Substances 0.000 description 6
- 239000011787 zinc oxide Substances 0.000 description 6
- 235000014692 zinc oxide Nutrition 0.000 description 6
- UUEWCQRISZBELL-UHFFFAOYSA-N 3-trimethoxysilylpropane-1-thiol Chemical compound CO[Si](OC)(OC)CCCS UUEWCQRISZBELL-UHFFFAOYSA-N 0.000 description 5
- 239000004215 Carbon black (E152) Substances 0.000 description 5
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 5
- 238000013329 compounding Methods 0.000 description 5
- 125000004122 cyclic group Chemical group 0.000 description 5
- 230000003247 decreasing effect Effects 0.000 description 5
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 5
- 229930195733 hydrocarbon Natural products 0.000 description 5
- 150000002430 hydrocarbons Chemical class 0.000 description 5
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 5
- 229920002223 polystyrene Polymers 0.000 description 5
- 239000002244 precipitate Substances 0.000 description 5
- 125000003396 thiol group Chemical class [H]S* 0.000 description 5
- ZRALSGWEFCBTJO-UHFFFAOYSA-N Guanidine Chemical compound NC(N)=N ZRALSGWEFCBTJO-UHFFFAOYSA-N 0.000 description 4
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- 239000000654 additive Substances 0.000 description 4
- MTAZNLWOLGHBHU-UHFFFAOYSA-N butadiene-styrene rubber Chemical compound C=CC=C.C=CC1=CC=CC=C1 MTAZNLWOLGHBHU-UHFFFAOYSA-N 0.000 description 4
- 239000002041 carbon nanotube Substances 0.000 description 4
- 229910021393 carbon nanotube Inorganic materials 0.000 description 4
- 150000001805 chlorine compounds Chemical group 0.000 description 4
- ZSWFCLXCOIISFI-UHFFFAOYSA-N cyclopentadiene Chemical compound C1C=CC=C1 ZSWFCLXCOIISFI-UHFFFAOYSA-N 0.000 description 4
- RJGHQTVXGKYATR-UHFFFAOYSA-L dibutyl(dichloro)stannane Chemical compound CCCC[Sn](Cl)(Cl)CCCC RJGHQTVXGKYATR-UHFFFAOYSA-L 0.000 description 4
- LIKFHECYJZWXFJ-UHFFFAOYSA-N dimethyldichlorosilane Chemical compound C[Si](C)(Cl)Cl LIKFHECYJZWXFJ-UHFFFAOYSA-N 0.000 description 4
- 239000011133 lead Substances 0.000 description 4
- 230000000704 physical effect Effects 0.000 description 4
- 238000012545 processing Methods 0.000 description 4
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 4
- 150000004756 silanes Chemical class 0.000 description 4
- 125000003011 styrenyl group Chemical group [H]\C(*)=C(/[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 4
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 4
- 238000012360 testing method Methods 0.000 description 4
- 125000000101 thioether group Chemical group 0.000 description 4
- 229940086542 triethylamine Drugs 0.000 description 4
- DURPTKYDGMDSBL-UHFFFAOYSA-N 1-butoxybutane Chemical compound CCCCOCCCC DURPTKYDGMDSBL-UHFFFAOYSA-N 0.000 description 3
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 3
- JLBJTVDPSNHSKJ-UHFFFAOYSA-N 4-Methylstyrene Chemical compound CC1=CC=C(C=C)C=C1 JLBJTVDPSNHSKJ-UHFFFAOYSA-N 0.000 description 3
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 3
- 239000004606 Fillers/Extenders Substances 0.000 description 3
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 3
- KWYHDKDOAIKMQN-UHFFFAOYSA-N N,N,N',N'-tetramethylethylenediamine Chemical compound CN(C)CCN(C)C KWYHDKDOAIKMQN-UHFFFAOYSA-N 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 239000001089 [(2R)-oxolan-2-yl]methanol Substances 0.000 description 3
- 150000001298 alcohols Chemical class 0.000 description 3
- 229910052783 alkali metal Inorganic materials 0.000 description 3
- 150000001340 alkali metals Chemical class 0.000 description 3
- 125000003545 alkoxy group Chemical group 0.000 description 3
- 125000002947 alkylene group Chemical group 0.000 description 3
- 125000004106 butoxy group Chemical group [*]OC([H])([H])C([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 3
- 238000007334 copolymerization reaction Methods 0.000 description 3
- 229960004132 diethyl ether Drugs 0.000 description 3
- 230000009977 dual effect Effects 0.000 description 3
- 238000010528 free radical solution polymerization reaction Methods 0.000 description 3
- 239000000446 fuel Substances 0.000 description 3
- 230000006870 function Effects 0.000 description 3
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 3
- 229910052757 nitrogen Inorganic materials 0.000 description 3
- 229940038597 peroxide anti-acne preparations for topical use Drugs 0.000 description 3
- 229920002589 poly(vinylethylene) polymer Polymers 0.000 description 3
- 125000006239 protecting group Chemical group 0.000 description 3
- 230000003014 reinforcing effect Effects 0.000 description 3
- 229910010271 silicon carbide Inorganic materials 0.000 description 3
- 229910052708 sodium Inorganic materials 0.000 description 3
- 239000011734 sodium Substances 0.000 description 3
- 230000003595 spectral effect Effects 0.000 description 3
- 239000003381 stabilizer Substances 0.000 description 3
- TXDNPSYEJHXKMK-UHFFFAOYSA-N sulfanylsilane Chemical class S[SiH3] TXDNPSYEJHXKMK-UHFFFAOYSA-N 0.000 description 3
- BSYVTEYKTMYBMK-UHFFFAOYSA-N tetrahydrofurfuryl alcohol Chemical compound OCC1CCCO1 BSYVTEYKTMYBMK-UHFFFAOYSA-N 0.000 description 3
- 229960002447 thiram Drugs 0.000 description 3
- APPOKADJQUIAHP-GGWOSOGESA-N (2e,4e)-hexa-2,4-diene Chemical compound C\C=C\C=C\C APPOKADJQUIAHP-GGWOSOGESA-N 0.000 description 2
- XWJBRBSPAODJER-UHFFFAOYSA-N 1,7-octadiene Chemical compound C=CCCCCC=C XWJBRBSPAODJER-UHFFFAOYSA-N 0.000 description 2
- BHKKSKOHRFHHIN-MRVPVSSYSA-N 1-[[2-[(1R)-1-aminoethyl]-4-chlorophenyl]methyl]-2-sulfanylidene-5H-pyrrolo[3,2-d]pyrimidin-4-one Chemical compound N[C@H](C)C1=C(CN2C(NC(C3=C2C=CN3)=O)=S)C=CC(=C1)Cl BHKKSKOHRFHHIN-MRVPVSSYSA-N 0.000 description 2
- HLBZWYXLQJQBKU-UHFFFAOYSA-N 4-(morpholin-4-yldisulfanyl)morpholine Chemical compound C1COCCN1SSN1CCOCC1 HLBZWYXLQJQBKU-UHFFFAOYSA-N 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- 102100024133 Coiled-coil domain-containing protein 50 Human genes 0.000 description 2
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 2
- LCGLNKUTAGEVQW-UHFFFAOYSA-N Dimethyl ether Chemical compound COC LCGLNKUTAGEVQW-UHFFFAOYSA-N 0.000 description 2
- 229920002943 EPDM rubber Polymers 0.000 description 2
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 2
- 238000005033 Fourier transform infrared spectroscopy Methods 0.000 description 2
- 101000910772 Homo sapiens Coiled-coil domain-containing protein 50 Proteins 0.000 description 2
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 2
- CHJJGSNFBQVOTG-UHFFFAOYSA-N N-methyl-guanidine Natural products CNC(N)=N CHJJGSNFBQVOTG-UHFFFAOYSA-N 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical group [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- ATUOYWHBWRKTHZ-UHFFFAOYSA-N Propane Chemical compound CCC ATUOYWHBWRKTHZ-UHFFFAOYSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- 229910018540 Si C Inorganic materials 0.000 description 2
- 229910004028 SiCU Inorganic materials 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- 229910000831 Steel Inorganic materials 0.000 description 2
- 239000002174 Styrene-butadiene Substances 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- 238000000862 absorption spectrum Methods 0.000 description 2
- 238000010539 anionic addition polymerization reaction Methods 0.000 description 2
- 238000013459 approach Methods 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- 239000002585 base Substances 0.000 description 2
- 230000009286 beneficial effect Effects 0.000 description 2
- 239000001273 butane Substances 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- 150000001735 carboxylic acids Chemical class 0.000 description 2
- 230000000052 comparative effect Effects 0.000 description 2
- 229910052805 deuterium Inorganic materials 0.000 description 2
- 235000014113 dietary fatty acids Nutrition 0.000 description 2
- SWSQBOPZIKWTGO-UHFFFAOYSA-N dimethylaminoamidine Natural products CN(C)C(N)=N SWSQBOPZIKWTGO-UHFFFAOYSA-N 0.000 description 2
- KPUWHANPEXNPJT-UHFFFAOYSA-N disiloxane Chemical compound [SiH3]O[SiH3] KPUWHANPEXNPJT-UHFFFAOYSA-N 0.000 description 2
- 239000002270 dispersing agent Substances 0.000 description 2
- AUZONCFQVSMFAP-UHFFFAOYSA-N disulfiram Chemical compound CCN(CC)C(=S)SSC(=S)N(CC)CC AUZONCFQVSMFAP-UHFFFAOYSA-N 0.000 description 2
- 125000000219 ethylidene group Chemical group [H]C(=[*])C([H])([H])[H] 0.000 description 2
- 239000000194 fatty acid Substances 0.000 description 2
- 229930195729 fatty acid Natural products 0.000 description 2
- 229920001519 homopolymer Polymers 0.000 description 2
- 239000012442 inert solvent Substances 0.000 description 2
- 230000001788 irregular Effects 0.000 description 2
- 125000003253 isopropoxy group Chemical group [H]C([H])([H])C([H])(O*)C([H])([H])[H] 0.000 description 2
- 238000004898 kneading Methods 0.000 description 2
- WGOPGODQLGJZGL-UHFFFAOYSA-N lithium;butane Chemical compound [Li+].CC[CH-]C WGOPGODQLGJZGL-UHFFFAOYSA-N 0.000 description 2
- 230000001404 mediated effect Effects 0.000 description 2
- 238000003801 milling Methods 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 125000001979 organolithium group Chemical group 0.000 description 2
- 150000002978 peroxides Chemical class 0.000 description 2
- OSFBJERFMQCEQY-UHFFFAOYSA-N propylidene Chemical group [CH]CC OSFBJERFMQCEQY-UHFFFAOYSA-N 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 150000004760 silicates Chemical class 0.000 description 2
- 150000003377 silicon compounds Chemical class 0.000 description 2
- 238000001179 sorption measurement Methods 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 239000010959 steel Substances 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 230000008961 swelling Effects 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 150000003573 thiols Chemical class 0.000 description 2
- KUAZQDVKQLNFPE-UHFFFAOYSA-N thiram Chemical compound CN(C)C(=S)SSC(=S)N(C)C KUAZQDVKQLNFPE-UHFFFAOYSA-N 0.000 description 2
- QPBYLOWPSRZOFX-UHFFFAOYSA-J tin(iv) iodide Chemical compound I[Sn](I)(I)I QPBYLOWPSRZOFX-UHFFFAOYSA-J 0.000 description 2
- ILWRPSCZWQJDMK-UHFFFAOYSA-N triethylazanium;chloride Chemical compound Cl.CCN(CC)CC ILWRPSCZWQJDMK-UHFFFAOYSA-N 0.000 description 2
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 2
- 239000003039 volatile agent Substances 0.000 description 2
- 238000010626 work up procedure Methods 0.000 description 2
- RRKODOZNUZCUBN-CCAGOZQPSA-N (1z,3z)-cycloocta-1,3-diene Chemical compound C1CC\C=C/C=C\C1 RRKODOZNUZCUBN-CCAGOZQPSA-N 0.000 description 1
- BOOBDAVNHSOIDB-UHFFFAOYSA-N (2,3-dichlorobenzoyl) 2,3-dichlorobenzenecarboperoxoate Chemical compound ClC1=CC=CC(C(=O)OOC(=O)C=2C(=C(Cl)C=CC=2)Cl)=C1Cl BOOBDAVNHSOIDB-UHFFFAOYSA-N 0.000 description 1
- OGQVROWWFUXRST-FNORWQNLSA-N (3e)-hepta-1,3-diene Chemical compound CCC\C=C\C=C OGQVROWWFUXRST-FNORWQNLSA-N 0.000 description 1
- GGQQNYXPYWCUHG-RMTFUQJTSA-N (3e,6e)-deca-3,6-diene Chemical compound CCC\C=C\C\C=C\CC GGQQNYXPYWCUHG-RMTFUQJTSA-N 0.000 description 1
- PRBHEGAFLDMLAL-GQCTYLIASA-N (4e)-hexa-1,4-diene Chemical compound C\C=C\CC=C PRBHEGAFLDMLAL-GQCTYLIASA-N 0.000 description 1
- OJOWICOBYCXEKR-KRXBUXKQSA-N (5e)-5-ethylidenebicyclo[2.2.1]hept-2-ene Chemical compound C1C2C(=C/C)/CC1C=C2 OJOWICOBYCXEKR-KRXBUXKQSA-N 0.000 description 1
- 125000000008 (C1-C10) alkyl group Chemical group 0.000 description 1
- 125000006732 (C1-C15) alkyl group Chemical group 0.000 description 1
- PMJHHCWVYXUKFD-SNAWJCMRSA-N (E)-1,3-pentadiene Chemical compound C\C=C\C=C PMJHHCWVYXUKFD-SNAWJCMRSA-N 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- MFHNAXHSHOCFEC-UHFFFAOYSA-N 1,1-diphenylguanidine Chemical compound C=1C=CC=CC=1N(C(=N)N)C1=CC=CC=C1 MFHNAXHSHOCFEC-UHFFFAOYSA-N 0.000 description 1
- LZDKZFUFMNSQCJ-UHFFFAOYSA-N 1,2-diethoxyethane Chemical compound CCOCCOCC LZDKZFUFMNSQCJ-UHFFFAOYSA-N 0.000 description 1
- VPBZZPOGZPKYKX-UHFFFAOYSA-N 1,2-diethoxypropane Chemical compound CCOCC(C)OCC VPBZZPOGZPKYKX-UHFFFAOYSA-N 0.000 description 1
- LEEANUDEDHYDTG-UHFFFAOYSA-N 1,2-dimethoxypropane Chemical compound COCC(C)OC LEEANUDEDHYDTG-UHFFFAOYSA-N 0.000 description 1
- CORMBJOFDGICKF-UHFFFAOYSA-N 1,3,5-trimethoxy 2-vinyl benzene Natural products COC1=CC(OC)=C(C=C)C(OC)=C1 CORMBJOFDGICKF-UHFFFAOYSA-N 0.000 description 1
- QTYUSOHYEPOHLV-FNORWQNLSA-N 1,3-Octadiene Chemical compound CCCC\C=C\C=C QTYUSOHYEPOHLV-FNORWQNLSA-N 0.000 description 1
- PRJNEUBECVAVAG-UHFFFAOYSA-N 1,3-bis(ethenyl)benzene Chemical compound C=CC1=CC=CC(C=C)=C1 PRJNEUBECVAVAG-UHFFFAOYSA-N 0.000 description 1
- KPZGRMZPZLOPBS-UHFFFAOYSA-N 1,3-dichloro-2,2-bis(chloromethyl)propane Chemical compound ClCC(CCl)(CCl)CCl KPZGRMZPZLOPBS-UHFFFAOYSA-N 0.000 description 1
- OWRCNXZUPFZXOS-UHFFFAOYSA-N 1,3-diphenylguanidine Chemical compound C=1C=CC=CC=1NC(=N)NC1=CC=CC=C1 OWRCNXZUPFZXOS-UHFFFAOYSA-N 0.000 description 1
- HNSDLXPSAYFUHK-UHFFFAOYSA-N 1,4-bis(2-ethylhexyl) sulfosuccinate Chemical compound CCCCC(CC)COC(=O)CC(S(O)(=O)=O)C(=O)OCC(CC)CCCC HNSDLXPSAYFUHK-UHFFFAOYSA-N 0.000 description 1
- WEERVPDNCOGWJF-UHFFFAOYSA-N 1,4-bis(ethenyl)benzene Chemical compound C=CC1=CC=C(C=C)C=C1 WEERVPDNCOGWJF-UHFFFAOYSA-N 0.000 description 1
- PRBHEGAFLDMLAL-UHFFFAOYSA-N 1,5-Hexadiene Natural products CC=CCC=C PRBHEGAFLDMLAL-UHFFFAOYSA-N 0.000 description 1
- GDXHBFHOEYVPED-UHFFFAOYSA-N 1-(2-butoxyethoxy)butane Chemical compound CCCCOCCOCCCC GDXHBFHOEYVPED-UHFFFAOYSA-N 0.000 description 1
- QMGJMGFZLXYHCR-UHFFFAOYSA-N 1-(2-butoxypropoxy)butane Chemical compound CCCCOCC(C)OCCCC QMGJMGFZLXYHCR-UHFFFAOYSA-N 0.000 description 1
- DHHDFVWSMNNDBJ-UHFFFAOYSA-N 1-dimethylsilyl-2-[(2-dimethylsilyl-2-hydroxyethyl)disulfanyl]ethanol Chemical compound OC(CSSCC(O)[SiH](C)C)[SiH](C)C DHHDFVWSMNNDBJ-UHFFFAOYSA-N 0.000 description 1
- KKRNTLXBOLWRRD-UHFFFAOYSA-N 1-dimethylsilyl-2-[(2-dimethylsilyl-2-hydroxyethyl)tetrasulfanyl]ethanol Chemical compound OC(CSSSSCC(O)[SiH](C)C)[SiH](C)C KKRNTLXBOLWRRD-UHFFFAOYSA-N 0.000 description 1
- OEVVKKAVYQFQNV-UHFFFAOYSA-N 1-ethenyl-2,4-dimethylbenzene Chemical compound CC1=CC=C(C=C)C(C)=C1 OEVVKKAVYQFQNV-UHFFFAOYSA-N 0.000 description 1
- NVZWEEGUWXZOKI-UHFFFAOYSA-N 1-ethenyl-2-methylbenzene Chemical compound CC1=CC=CC=C1C=C NVZWEEGUWXZOKI-UHFFFAOYSA-N 0.000 description 1
- JZHGRUMIRATHIU-UHFFFAOYSA-N 1-ethenyl-3-methylbenzene Chemical compound CC1=CC=CC(C=C)=C1 JZHGRUMIRATHIU-UHFFFAOYSA-N 0.000 description 1
- 238000005160 1H NMR spectroscopy Methods 0.000 description 1
- FXRQXYSJYZPGJZ-UHFFFAOYSA-N 2-[(2-methylpropan-2-yl)oxy]ethenylbenzene Chemical compound CC(C)(C)OC=CC1=CC=CC=C1 FXRQXYSJYZPGJZ-UHFFFAOYSA-N 0.000 description 1
- GTEXIOINCJRBIO-UHFFFAOYSA-N 2-[2-(dimethylamino)ethoxy]-n,n-dimethylethanamine Chemical compound CN(C)CCOCCN(C)C GTEXIOINCJRBIO-UHFFFAOYSA-N 0.000 description 1
- MATDIXOGHXOZDW-UHFFFAOYSA-N 2-butoxyoxolane Chemical compound CCCCOC1CCCO1 MATDIXOGHXOZDW-UHFFFAOYSA-N 0.000 description 1
- PDELBHCVXBSVPJ-UHFFFAOYSA-N 2-ethenyl-1,3,5-trimethylbenzene Chemical compound CC1=CC(C)=C(C=C)C(C)=C1 PDELBHCVXBSVPJ-UHFFFAOYSA-N 0.000 description 1
- JQYYUWHWGCJWTN-UHFFFAOYSA-N 2-ethoxyoxolane Chemical compound CCOC1CCCO1 JQYYUWHWGCJWTN-UHFFFAOYSA-N 0.000 description 1
- GDQKLNXQGUDPKS-UHFFFAOYSA-N 2-hexoxyoxolane Chemical compound CCCCCCOC1CCCO1 GDQKLNXQGUDPKS-UHFFFAOYSA-N 0.000 description 1
- GAODDBNJCKQQDY-UHFFFAOYSA-N 2-methyl-4,6-bis(octylsulfanylmethyl)phenol Chemical compound CCCCCCCCSCC1=CC(C)=C(O)C(CSCCCCCCCC)=C1 GAODDBNJCKQQDY-UHFFFAOYSA-N 0.000 description 1
- OHNPPRNQKABHPI-UHFFFAOYSA-N 2-propoxyoxolane Chemical compound CCCOC1CCCO1 OHNPPRNQKABHPI-UHFFFAOYSA-N 0.000 description 1
- KGIGUEBEKRSTEW-UHFFFAOYSA-N 2-vinylpyridine Chemical compound C=CC1=CC=CC=N1 KGIGUEBEKRSTEW-UHFFFAOYSA-N 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- FCTIELMLTXMNOW-UHFFFAOYSA-N 3-[[dimethylamino-[[dimethylamino-[dimethylsilyl(3-hydroxypropyl)-lambda4-sulfanylidene]methyl]tetrasulfanyl]methylidene]-dimethylsilyl-lambda4-sulfanyl]propan-1-ol Chemical compound OCCCS(=C(N(C)C)SSSSC(N(C)C)=S([SiH](C)C)CCCO)[SiH](C)C FCTIELMLTXMNOW-UHFFFAOYSA-N 0.000 description 1
- VXEGSRKPIUDPQT-UHFFFAOYSA-N 4-[4-(4-methoxyphenyl)piperazin-1-yl]aniline Chemical compound C1=CC(OC)=CC=C1N1CCN(C=2C=CC(N)=CC=2)CC1 VXEGSRKPIUDPQT-UHFFFAOYSA-N 0.000 description 1
- CJSBUWDGPXGFGA-UHFFFAOYSA-N 4-methylpenta-1,3-diene Chemical compound CC(C)=CC=C CJSBUWDGPXGFGA-UHFFFAOYSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- NLHHRLWOUZZQLW-UHFFFAOYSA-N Acrylonitrile Chemical compound C=CC#N NLHHRLWOUZZQLW-UHFFFAOYSA-N 0.000 description 1
- 238000004438 BET method Methods 0.000 description 1
- KJQMOGOKAYDMOR-UHFFFAOYSA-N CC(=C)C=C.CC(=C)C=C Chemical compound CC(=C)C=C.CC(=C)C=C KJQMOGOKAYDMOR-UHFFFAOYSA-N 0.000 description 1
- 229910014455 Ca-Cb Inorganic materials 0.000 description 1
- UGFAIRIUMAVXCW-UHFFFAOYSA-N Carbon monoxide Chemical compound [O+]#[C-] UGFAIRIUMAVXCW-UHFFFAOYSA-N 0.000 description 1
- 241000861718 Chloris <Aves> Species 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 244000043261 Hevea brasiliensis Species 0.000 description 1
- 239000006237 Intermediate SAF Substances 0.000 description 1
- VQTUBCCKSQIDNK-UHFFFAOYSA-N Isobutene Chemical group CC(C)=C VQTUBCCKSQIDNK-UHFFFAOYSA-N 0.000 description 1
- VVQNEPGJFQJSBK-UHFFFAOYSA-N Methyl methacrylate Chemical compound COC(=O)C(C)=C VVQNEPGJFQJSBK-UHFFFAOYSA-N 0.000 description 1
- 241000357292 Monodactylus Species 0.000 description 1
- ZNJLLWWCBTZFSB-UHFFFAOYSA-N OC(CCSSSSCCC(O)[SiH](C)C)[SiH](C)C Chemical compound OC(CCSSSSCCC(O)[SiH](C)C)[SiH](C)C ZNJLLWWCBTZFSB-UHFFFAOYSA-N 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- MITFXPHMIHQXPI-UHFFFAOYSA-N Oraflex Chemical compound N=1C2=CC(C(C(O)=O)C)=CC=C2OC=1C1=CC=C(Cl)C=C1 MITFXPHMIHQXPI-UHFFFAOYSA-N 0.000 description 1
- CBENFWSGALASAD-UHFFFAOYSA-N Ozone Chemical compound [O-][O+]=O CBENFWSGALASAD-UHFFFAOYSA-N 0.000 description 1
- 101150050048 SNCB gene Proteins 0.000 description 1
- 229920000026 Si 363 Polymers 0.000 description 1
- 229910008048 Si-S Inorganic materials 0.000 description 1
- 229910003930 SiCb Inorganic materials 0.000 description 1
- 229910006336 Si—S Inorganic materials 0.000 description 1
- 229910020929 Sn-Sn Inorganic materials 0.000 description 1
- 229910008827 Sn—Sn Inorganic materials 0.000 description 1
- 102100029329 Somatostatin receptor type 1 Human genes 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- PJANXHGTPQOBST-VAWYXSNFSA-N Stilbene Natural products C=1C=CC=CC=1/C=C/C1=CC=CC=C1 PJANXHGTPQOBST-VAWYXSNFSA-N 0.000 description 1
- RYYWUUFWQRZTIU-UHFFFAOYSA-N Thiophosphoric acid Chemical class OP(O)(S)=O RYYWUUFWQRZTIU-UHFFFAOYSA-N 0.000 description 1
- 229910021626 Tin(II) chloride Inorganic materials 0.000 description 1
- 229910021623 Tin(IV) bromide Inorganic materials 0.000 description 1
- GCTFWCDSFPMHHS-UHFFFAOYSA-M Tributyltin chloride Chemical compound CCCC[Sn](Cl)(CCCC)CCCC GCTFWCDSFPMHHS-UHFFFAOYSA-M 0.000 description 1
- 229920000800 acrylic rubber Polymers 0.000 description 1
- 239000008186 active pharmaceutical agent Substances 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 238000005054 agglomeration Methods 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 230000032683 aging Effects 0.000 description 1
- 125000005370 alkoxysilyl group Chemical group 0.000 description 1
- 150000001350 alkyl halides Chemical class 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 229920006318 anionic polymer Polymers 0.000 description 1
- 210000000436 anus Anatomy 0.000 description 1
- 239000010692 aromatic oil Substances 0.000 description 1
- 239000003849 aromatic solvent Substances 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- 208000027697 autoimmune lymphoproliferative syndrome due to CTLA4 haploinsuffiency Diseases 0.000 description 1
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 1
- 230000008033 biological extinction Effects 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- 150000004074 biphenyls Chemical class 0.000 description 1
- 229920001400 block copolymer Polymers 0.000 description 1
- MPMBRWOOISTHJV-UHFFFAOYSA-N but-1-enylbenzene Chemical compound CCC=CC1=CC=CC=C1 MPMBRWOOISTHJV-UHFFFAOYSA-N 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- ZPFKRQXYKULZKP-UHFFFAOYSA-N butylidene Chemical group [CH2+]CC[CH-] ZPFKRQXYKULZKP-UHFFFAOYSA-N 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 238000011088 calibration curve Methods 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 229910002092 carbon dioxide Inorganic materials 0.000 description 1
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 1
- 229920003193 cis-1,4-polybutadiene polymer Polymers 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 238000002485 combustion reaction Methods 0.000 description 1
- 238000007906 compression Methods 0.000 description 1
- 230000006835 compression Effects 0.000 description 1
- 230000001010 compromised effect Effects 0.000 description 1
- 239000007859 condensation product Substances 0.000 description 1
- 238000001816 cooling Methods 0.000 description 1
- 238000012937 correction Methods 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 description 1
- IJKVHSBPTUYDLN-UHFFFAOYSA-N dihydroxy(oxo)silane Chemical compound O[Si](O)=O IJKVHSBPTUYDLN-UHFFFAOYSA-N 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 239000012990 dithiocarbamate Substances 0.000 description 1
- 150000004659 dithiocarbamates Chemical class 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 230000000694 effects Effects 0.000 description 1
- 230000009881 electrostatic interaction Effects 0.000 description 1
- 238000010828 elution Methods 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 238000005265 energy consumption Methods 0.000 description 1
- HQQADJVZYDDRJT-UHFFFAOYSA-N ethene;prop-1-ene Chemical group C=C.CC=C HQQADJVZYDDRJT-UHFFFAOYSA-N 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- BLHLJVCOVBYQQS-UHFFFAOYSA-N ethyllithium Chemical compound [Li]CC BLHLJVCOVBYQQS-UHFFFAOYSA-N 0.000 description 1
- 230000007717 exclusion Effects 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 239000003546 flue gas Substances 0.000 description 1
- 238000007306 functionalization reaction Methods 0.000 description 1
- 230000009477 glass transition Effects 0.000 description 1
- 125000005842 heteroatom Chemical group 0.000 description 1
- PYGSKMBEVAICCR-UHFFFAOYSA-N hexa-1,5-diene Chemical compound C=CCCC=C PYGSKMBEVAICCR-UHFFFAOYSA-N 0.000 description 1
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine hydrate Chemical compound O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 1
- BHEPBYXIRTUNPN-UHFFFAOYSA-N hydridophosphorus(.) (triplet) Chemical compound [PH] BHEPBYXIRTUNPN-UHFFFAOYSA-N 0.000 description 1
- 125000001183 hydrocarbyl group Chemical group 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 230000003116 impacting effect Effects 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 230000010354 integration Effects 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 238000004255 ion exchange chromatography Methods 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 125000002510 isobutoxy group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])O* 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- RLAWWYSOJDYHDC-BZSNNMDCSA-N lisinopril Chemical compound C([C@H](N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(O)=O)C(O)=O)CC1=CC=CC=C1 RLAWWYSOJDYHDC-BZSNNMDCSA-N 0.000 description 1
- 150000002641 lithium Chemical group 0.000 description 1
- UBJFKNSINUCEAL-UHFFFAOYSA-N lithium;2-methylpropane Chemical compound [Li+].C[C-](C)C UBJFKNSINUCEAL-UHFFFAOYSA-N 0.000 description 1
- 238000010551 living anionic polymerization reaction Methods 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 150000002734 metacrylic acid derivatives Chemical class 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- IUJLOAKJZQBENM-UHFFFAOYSA-N n-(1,3-benzothiazol-2-ylsulfanyl)-2-methylpropan-2-amine Chemical compound C1=CC=C2SC(SNC(C)(C)C)=NC2=C1 IUJLOAKJZQBENM-UHFFFAOYSA-N 0.000 description 1
- DZRKBPWATCKLKY-UHFFFAOYSA-N n-benzyl-n-methylprop-2-en-1-amine Chemical compound C=CCN(C)CC1=CC=CC=C1 DZRKBPWATCKLKY-UHFFFAOYSA-N 0.000 description 1
- IJDNQMDRQITEOD-UHFFFAOYSA-N n-butane Chemical compound CCCC IJDNQMDRQITEOD-UHFFFAOYSA-N 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 229920003052 natural elastomer Polymers 0.000 description 1
- 229920001194 natural rubber Polymers 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen(.) Chemical compound [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- SJYNFBVQFBRSIB-UHFFFAOYSA-N norbornadiene Chemical compound C1=CC2C=CC1C2 SJYNFBVQFBRSIB-UHFFFAOYSA-N 0.000 description 1
- 238000010606 normalization Methods 0.000 description 1
- 238000001208 nuclear magnetic resonance pulse sequence Methods 0.000 description 1
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 230000000269 nucleophilic effect Effects 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 125000004115 pentoxy group Chemical group [*]OC([H])([H])C([H])([H])C([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- NHKJPPKXDNZFBJ-UHFFFAOYSA-N phenyllithium Chemical compound [Li]C1=CC=CC=C1 NHKJPPKXDNZFBJ-UHFFFAOYSA-N 0.000 description 1
- PMJHHCWVYXUKFD-UHFFFAOYSA-N piperylene Natural products CC=CC=C PMJHHCWVYXUKFD-UHFFFAOYSA-N 0.000 description 1
- 229920002959 polymer blend Polymers 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 230000003449 preventive effect Effects 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 239000011164 primary particle Substances 0.000 description 1
- 239000001294 propane Substances 0.000 description 1
- 230000001681 protective effect Effects 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 239000012429 reaction media Substances 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 230000002787 reinforcement Effects 0.000 description 1
- 102200090666 rs1556026984 Human genes 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 125000005372 silanol group Chemical group 0.000 description 1
- AIFMYMZGQVTROK-UHFFFAOYSA-N silicon tetrabromide Chemical compound Br[Si](Br)(Br)Br AIFMYMZGQVTROK-UHFFFAOYSA-N 0.000 description 1
- 239000005049 silicon tetrachloride Substances 0.000 description 1
- ABTOQLMXBSRXSM-UHFFFAOYSA-N silicon tetrafluoride Chemical compound F[Si](F)(F)F ABTOQLMXBSRXSM-UHFFFAOYSA-N 0.000 description 1
- JHGCXUUFRJCMON-UHFFFAOYSA-J silicon(4+);tetraiodide Chemical compound [Si+4].[I-].[I-].[I-].[I-] JHGCXUUFRJCMON-UHFFFAOYSA-J 0.000 description 1
- FGEJJBGRIFKJTB-UHFFFAOYSA-N silylsulfanylsilane Chemical group [SiH3]S[SiH3] FGEJJBGRIFKJTB-UHFFFAOYSA-N 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 108010082379 somatostatin receptor type 1 Proteins 0.000 description 1
- 238000004611 spectroscopical analysis Methods 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- KXCAEQNNTZANTK-UHFFFAOYSA-N stannane Chemical compound [SnH4] KXCAEQNNTZANTK-UHFFFAOYSA-N 0.000 description 1
- 229910000080 stannane Inorganic materials 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- PJANXHGTPQOBST-UHFFFAOYSA-N stilbene Chemical compound C=1C=CC=CC=1C=CC1=CC=CC=C1 PJANXHGTPQOBST-UHFFFAOYSA-N 0.000 description 1
- 235000021286 stilbenes Nutrition 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 239000011115 styrene butadiene Substances 0.000 description 1
- 150000003440 styrenes Chemical class 0.000 description 1
- LZOZLBFZGFLFBV-UHFFFAOYSA-N sulfene Chemical compound C=S(=O)=O LZOZLBFZGFLFBV-UHFFFAOYSA-N 0.000 description 1
- 125000004434 sulfur atom Chemical group 0.000 description 1
- 229920003051 synthetic elastomer Polymers 0.000 description 1
- 229920001169 thermoplastic Polymers 0.000 description 1
- 239000004416 thermosoftening plastic Substances 0.000 description 1
- 150000003557 thiazoles Chemical class 0.000 description 1
- 150000003568 thioethers Chemical class 0.000 description 1
- 150000003606 tin compounds Chemical class 0.000 description 1
- LTSUHJWLSNQKIP-UHFFFAOYSA-J tin(iv) bromide Chemical compound Br[Sn](Br)(Br)Br LTSUHJWLSNQKIP-UHFFFAOYSA-J 0.000 description 1
- YUOWTJMRMWQJDA-UHFFFAOYSA-J tin(iv) fluoride Chemical compound [F-].[F-].[F-].[F-].[Sn+4] YUOWTJMRMWQJDA-UHFFFAOYSA-J 0.000 description 1
- 238000004448 titration Methods 0.000 description 1
- 229920003194 trans-1,4-polybutadiene polymer Polymers 0.000 description 1
- 125000005106 triarylsilyl group Chemical group 0.000 description 1
- 150000005671 trienes Chemical class 0.000 description 1
- 125000003258 trimethylene group Chemical group [H]C([H])([*:2])C([H])([H])C([H])([H])[*:1] 0.000 description 1
- 229940070710 valerate Drugs 0.000 description 1
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical compound CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- ABDKAPXRBAPSQN-UHFFFAOYSA-N veratrole Chemical compound COC1=CC=CC=C1OC ABDKAPXRBAPSQN-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/18—Compounds having one or more C—Si linkages as well as one or more C—O—Si linkages
- C07F7/1804—Compounds having Si-O-C linkages
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F236/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, at least one having two or more carbon-to-carbon double bonds
- C08F236/02—Copolymers of compounds having one or more unsaturated aliphatic radicals, at least one having two or more carbon-to-carbon double bonds the radical having only two carbon-to-carbon double bonds
- C08F236/04—Copolymers of compounds having one or more unsaturated aliphatic radicals, at least one having two or more carbon-to-carbon double bonds the radical having only two carbon-to-carbon double bonds conjugated
- C08F236/10—Copolymers of compounds having one or more unsaturated aliphatic radicals, at least one having two or more carbon-to-carbon double bonds the radical having only two carbon-to-carbon double bonds conjugated with vinyl-aromatic monomers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/24—Crosslinking, e.g. vulcanising, of macromolecules
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/54—Silicon-containing compounds
- C08K5/548—Silicon-containing compounds containing sulfur
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/22—Tin compounds
- C07F7/226—Compounds with one or more Sn-S linkages
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2347/00—Characterised by the use of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, at least one having two or more carbon-to-carbon double bonds; Derivatives of such polymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/02—Elements
- C08K3/04—Carbon
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/34—Silicon-containing compounds
- C08K3/36—Silica
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/0008—Organic ingredients according to more than one of the "one dot" groups of C08K5/01 - C08K5/59
- C08K5/0025—Crosslinking or vulcanising agents; including accelerators
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02T—CLIMATE CHANGE MITIGATION TECHNOLOGIES RELATED TO TRANSPORTATION
- Y02T10/00—Road transport of goods or passengers
- Y02T10/80—Technologies aiming to reduce greenhouse gasses emissions common to all road transportation technologies
- Y02T10/86—Optimisation of rolling resistance, e.g. weight reduction
Definitions
- This invention relates to silane sulfide modifier compounds and methods of making them.
- the invention also relates to a silane sulfide modified macromolecular compound obtainable by reacting a living anionic elastomeric polymer and a silane sulfide modifier.
- the silane sulfide modified macromolecular compound may be provided in the form of a polymer composition, and the polymer composition may be vulcanized (crossHnked) by making use of and reaction with at least one vulcanization agent, resulting in a vulcanized polymer composition.
- the invention also provides an article comprising at least one component formed from (constituted by) the vulcanized polymer composition.
- the vulcanized polymer composition has relatively low hysteresis loss and is useful in many articles, including tire treads having low- heat build up, low rolling resistance, good wet grip and ice grip, in combination with a good balance of other desirable physical and chemical properties, for example, abrasion resistance and tensile strength, and excellent processability.
- hysteresis loss of a cross-linked elastomeric polymer composition is related to its Tan ⁇ at 60°C value (see ISO 4664-1 :2005; Rubber, Vulcanized or thermoplastic; Determination of dynamic properties - part 1 : General guidance), in general, vulcanized elastomeric polymer compositions having relatively small Tan ⁇ values at 60°C are preferred as having lower hysteresis loss, in the final tire product, this ' translates into a lower rolling resistance and better fuel economy.
- the tire wet and ice grip of a cross-linked elastonieric polymer composition is related to its Tan ⁇ at 0°C and Tan d at - i 0 ( ' values. It is generally accepted that a lower rolling resistance tire can be made on the expense of deteriorated wet gri properties and vice versa. For example, if, in a random solution styrene- butadiene rubber (random SSBR).
- random SSBR random solution styrene- butadiene rubber
- the polystyrene unit concentration is reduced with respect to the total polvbutadiene unit concentration and the 1,2-polydiene unit concentration is kept constant, both tan delta at 60°C and tan delta at 0°C are reduced, generally corresponding to improved rolling resistance and deteriorated wet grip performance of a tire.
- both tan delta at 60°C and tan delta at 0°C are reduced, generally corresponding to improved rolling resistance and deteriorated wet grip perfoniian.ee of a tire. Accordingly, when assessing the rubber vulcanizate performance correctly, both the rolling resistance, or tan delta at 60°C, and the wet grip, or tan delta at 0°C, should be monitored.
- Coupled polymers as reactants to living polymers more often than, not leads to the formation of polymer blends comprising one fraction, of linear or uncoupled polymers, and one or more fractions comprising more than two polymer arms at the coupling point.
- the reference "'Synthesis of end-functionalized polymer by means of living anionic polymerization.' " Journal of Macromolecular Chemistry and Physics.
- SH functions in the termination reactions, because the corresponding silyl ethers and thioethers are found to be both stable and compatible with anionic living polymers.
- WO2007/047943 describes the use of a silane-sulfide omega chain end modifier.
- a silane sulfide compound is reacted with anionically-initiated, living polymers to produce "chain end modified" polymers, which are subsequently blended with fillers, vulcanizing agents, accelerators or oil extenders, to produce a ' vulcanized elastomeric polymer composition having low hysteresis loss.
- a coupling agent or linking agent
- the modifier is added before, after or during the addition of a coupling agent, and the modification reaction is preferably completed after the addition of the coupling agent. In some embodiments, more than a third of the polymer chain ends are reacted with a coupling agent prior to addition of the modifier.
- WO 2009/148932 describes an elastomeric polymer composition as the reaction product of a living anionic elastomeric polymer with two silane modifier compounds (A) and (B).
- the silane modifier compound (A) is reported to react with at least two polymer chains, forming branched modified polymer macromolecules, while silane modifier compound (B) is reported to react with only one polymer chain, forming chain-end modified polymer macromolecules.
- the resulting cured composition comprising branched -modified and chain-end modified polymer macrom lecules is stated to result in lower "Tan ⁇ at 60°C values, without negatively impacting other physical properties, particularly "Tan ⁇ at 0°C.”
- WO2007/047943 and WO 2009/148932 do not provide Theological information, on filler-containing polymer compositions. Yet, it is reasonable to expect higher viscosities as a result of enhanced polymer-filler associations.
- Two fillers, silica and carbon black, are typically used in the tire production. Standard formulations very often comprise both fillers in varying ratios. Therefore, it would be desirable to have a modified polymer (comprising one or both of branched modified polymer macromolecules and chain end modified polymer macromolecules) which exhibits reduced viscosity in (non-cured) polymer compositions, especially lower Mooney viscosity, and/or improved rolling resistance/grip balance characteristics of the cured compositions.
- a modified polymer comprising one or both of branched modified polymer macromolecules and chain end modified polymer macromolecules
- the present invention provides a silane sulfide modifier represented by the following Fo:
- M is silicon or tin
- x is an. integer selected from 1 , 2 and 3 ;
- s is an integer selected from 2, 3 and 4;
- t is an integer selected from 0, 1 and. 2;
- R 1 is independently selected from. (C Q,) alkyl
- R 2 is independently selected from (Ci-Cje) alkyl, (C 7 -Ci 6 ) alkylaryi and (C7-C16) arvlalkyi;
- R ⁇ is at least divalent and is independently selected from (Cj-Cje) alkyl, (Cg-Cie) alkylarylalkyl, (C7-C16) aiylalkyl and (C 7 -Ci 6 ) alkylaiyl, and each, group may be substituted with one or more of the following groups: tertiary amine group, silyl group, (C 7 -Ci8) aralkyl group and (C 6 -Cig) aryl group;
- R 4 is independently selected from (CrQ 6) alkyl and (C7- 6) alkylaryi;
- X is independently selected from chloride, bromide and -OR. 5 ; wherein R 5 is selected from.
- the invention furthermore provides a method of making the silane sulfide modifier of Formula 1 as defined above, comprising the steps of
- R ⁇ R ⁇ x and y are as defined above;
- R ⁇ R 6 R 7 5 R 8 5 R 9 , R 10 and R 1 1 are each, independently selected from hydrogen, (Q-Qe) alkyl, (C7-C16) alkylaryi; (C 7 -C 16 ) arvlalkyi and (C6-Q 6 ) aryl; and v is an integer selected from 1 to 10;
- step (ii) reacting the mixture resulting from step (i) with, a compound of the following Formul 4
- M is silicon or tin
- u is an integer selected from 2, 3 and 4; R 4 .
- step (iii) optionally isolating the silane sulfide modifier of Formula 1 obtained in step (ii).
- the invention provides a further method of making the silane sulfide modifier of Formula 1 as defined above, comprising the steps of
- step (ii) reacting the reaction product resulting from step (i) with a compound of Formula 4 as defined above in a solvent:
- step (iii) optionally isolating the silane sulfide modifier of Formula 1 obtained in step (ii).
- the invention also provides a silane sulfide modified maeromoleeular compound (also referred to as a silane sulfide modified elastomeiic maeromoleeular compound) obtainable by reacting
- the invention further provides a first polymer composition
- a first polymer composition comprising at least one of said silane sulfide modified maeromoleeular compounds as defined above and one or more further components selected from non-modified elastomerie polymers and elastomerie polymers modified with non-inventive modifiers or coupling agents as described herein.
- the first polymer composition may comprise additives such as stabilizing agents or softeners, including oils, as described herein.
- the first polymer composition is the result of the polymerization process (reaction) employed to provide the silane sulfide modified maeromoleeular compound of the present invention and, thus, comprises the modified maeromoleeular compound and one or more further components selected from components which (i) are added to or formed as a result of the polymerization process and which (ii) remain after solvent removal.
- the invention furthermore provides a second polymer composition comprising at least the following: (i) the first polymer composition as defined above; and
- the invention also provides a vulcanized polymer composition comprising the reaction product of at least the following:
- the vulcanized polymer composition can be produced by reacting the at least one vulcanization agent and the first or second polymer composition as described herein.
- the invention also provides an article comprising at least one component formed from the vulcanized polymer composition as defined above.
- the invention provides a silane sulfide modifier of Formula 1 as defined above.
- M is a silicon atom.
- R is divalent and is (Cj-Cie) alkyl.
- X is -OR 5 ; wherein R is selected from (Ci-Cie) alkyl.
- X is chloride or bromide.
- R and R 4 are independently selected from (C t -C 16 ) alkyl.
- R 2 R 4 and R 3 are independently selected from (C 1 -C4) alkyl
- s and t are each 2 and u is 0.
- s is 3, t is 1 and u is 0.
- x is 2 and y is 1.
- x is 1 and y is 2.
- the invention furthermore provides a method of making the silane sulfide modifier represented by Formula 1 as defined above, comprising the steps of (i) combining (ia) a compound of Formula 2 as defined above and (ib) an amine compound selected from, formula
- step (ii) reacting the mixture resulting from step (i) with a compound of Formula. 4 as defined above in a solvent; and (iii) optionally isolating the silane sulfide modifier of Formula 1 obtained in step (ii).
- R. 3 is divalent and. is (Cj-Cie) alkyl
- X is -OR 5 ; wherein R 5 is selected from (Ci-Cie) alkyl. In another embodiment, X is chloride or bromide.
- R 2 and R 4 are independently selected from (Ci-Cu) alkyl.
- R 1 , R 2 , R 4 and R 5 are independently selected from (C 1 -C4) alkyl.
- s and t are each 2 and u is 0 in Formula 1, and it and t are each 2 in Formula 4.
- s is 3, t is 1 and u is 0.
- x is 2 and y is 1.
- x is 1 and y is 2,
- v is selected from an integer of 2
- R ' , R 6 , R', R s , R 9 , R 10 and R 1 1 are independently selected from (C1 -C4) alkyl.
- the invention provides a iiirther method of making the silane sulfide modifier represented by the Formula 1 as defined above, comprising the steps of (i) reacting a compound of Formula 2 as defined above and an alkali metal hydride in a solvent; (ii) reacting the reaction product resulting from step (i) with a compound of Formula 4 as defined above in a solvent; and optionally (iii) isolating the silane sulfide modifier of Formula 1 obtained in step (ii).
- R 3 is divalent and is (Ci-Cje) alkyl.
- X is -OR 5 ; wherein R 5 is selected from (C C !6 ) alkyl,
- X is chloride or bromide.
- R 2 and R 4 are independently selected from (Ci ⁇ Cj 6 ) alkyl.
- R 1 , R 2 , R 4 and R 5 are independently selected from (C1-C4) alkyl.
- s and t are each 2 and u is 0 in Formula 1
- u and t are each 2 in
- s is 3, t is 1 and u is 0.
- x is 2 and y is 1.
- x is 1 and y is 2.
- v is selected from, an integer of 2,
- R " ⁇ R 6 , R 7 , R 8 , R 9 , R !0 and R 1 1 are independently selected from (C 1 -C4) alkyl.
- the latter compound may be isolated in a respective step (iii) in a conventional manner, for example by separation from the reaction mixture through filtration, evaporation of the solvent or distillation.
- the invention also provides a silane sulfide modified macromoleciilar compound obtainable by reacting:
- one or more living polymer chains are modified at their polymer chain end(s) by one silane sulfide modifier.
- the resulting modified macromoleciilar compound is not necessarily chain end modified.
- the reaction of two polymer chains with one silane sulfide modifier may result in a structure of polymer - silane sulfide - polymer.
- the invention further provides a first polymer composition comprising at least one of said silane sulfide modified macromolecular compounds as defined above and one or more further components selected from non-modified elastomeric polymers and elastomeric polymers modified with non-inventive modifiers or coupling agents as described herein.
- the invention furthermore provides a second polymer composition comprising at least the following:
- the at least one filler is silica.
- the at least one filler is carbon black.
- the second polymer composition further comprises an oil.
- the second polymer composition comprises a vulcanization agent.
- the second polymer composition is the result of a mechanical mixing process involving the first polymer composition and at least one filler.
- the second polymer composition typically includes components which are added to the (solvent-free) first polymer composition and which remain in the composition after completion of the mechanical mixing process. Therefore, the specified components contained in the second polymer composition include at least one filler and. may, but do not have to include, and are not limited to alternative (solvent-free) modified or non-modified polymers, stabilizers and softeners.
- the invention also provides a vulcanized polymer compositioe comprising the reactio product o f at 1 east the followin :
- component 2 is the second polymer composition as described herein,
- the invention also provides a method for making a vulcanized polymer composition comprising reacting at least the following components:
- component 2 is the second polymer composition as described herein.
- the vulcanized polymer composition is the result of a reactive polymer-polymer crosslink forming process which is performed on the first or second polymer composition comprising at least one vulcanization agent. Therefore, the reactive process converts an essentially uncrosslinked elastomeric polymer composition into a crosslinked elastomeric polymer composition, i.e. the vulcanized polymer composition.
- the invention also provides an article comprising at least one component formed, from the vulcanized polymer composition as defined above.
- the article is a tire or tire tread.
- the polymer portion of the silane sulfide modified macromolecular compound of the present invention is selected from the group consisting of modified styrene- butadiene copolymers, modified polybutadiene, modified biitadiene-isoprene copolymers, modified polyisoprene and modified butadiene-styrene-isoprene terpolymers.
- the first or second polymer composition in accordance with the present invention further comprises at least one polymer selected from the group consisting of styrene- butadiene copolymers, including but not limited to solution styrene-butadiene rubber (SSBR) and emulsion styrene-butadiene rubber (ESBR); polybutadiene, including polybutadiene ' with a 1 ,4-cis-polybutadiene concentration ranging from 90 to 99 percent, from 30 to 70 percent, or from 2 to 25 percent, based on weight; butadiene-isoprene copolymers; polyisoprene; b u tad i en e- st yren e - i opren e terpolymers; and combinations thereof.
- SSBR solution styrene-butadiene rubber
- ESBR emulsion styrene-butadiene rubber
- polybutadiene
- the invention encompasses, within its scope, any combinations of two or more specific or preferred features as defined herein, unless such combination is technically or logically excluded.
- the living anionic elastomeric polymer used in the present invention is obtained by polymerization of one or more monomers, as is conventionally known in the art.
- General information about applicable polymerization technologies including polymerization initiator compounds; randomizer agents (also called polar coordinator compounds) and accelerators, each to increase the reactivity of the initiator, to randomly arrange aromatic vinyl compounds, to randomly arrange 1 ,2 -polybutadiene or 1 ,2-polyisoprene or 3,4-polyisoprene units introduced in the polymer; the amounts of each compound; monomer(s); and suitable process conditions are described in WO 2009/148932 fully incorporated herein by reference, Solution polymerizations normally take place at lower pressures, preferably below 10 MPa, preferably in a temperature range of from 0 to 120°C.
- the polymerization is generally conducted under batch, continuous or semi-continuous polymerization conditions.
- the polymerization process is preferably conducted as a solution polynieri.zat.ion, wherein the polymer formed is substantially soluble in the reaction, mixture, or as a suspension/slurry polymerization, wherein the polymer formed is substantially insoluble in the reaction medium.
- preferred randomizer agents also called polar coordinator compounds
- accelerators are listed in WO 2009/148932
- ionic initiators such as lithium initiators
- anionic solution polymerization Such polymerizations proceed according to an anionic polymerization mechanism, wherein the reaction of the monomers is by nucleophilic initiation to form and propagate a polymeric structure.
- the active center is typically a carbon ion with a partial or total negative charge.
- the polymer structure is ionic or "'living".
- the polymer structure has at least one reactive or "living" end. This is the context of the term, "living,” as used herein, to describe those uncross! inked eiastomeric polymers prepared by an anionic solution polymerization technology.
- a living anionic eiastomeric polymer is prepared by an anionic polymerization, as discussed herein.
- Polymerization of the monomers, as described herein, is, in case of anionic living type polymerization reactions, typically initiated with an anionic initiator, such as, but not limited to. an organo metal compound having at least one lithium, sodium, or potassium atom, and where the organo metal compounds contain from 1 to about 20 carbon atoms.
- an anionic initiator such as, but not limited to. an organo metal compound having at least one lithium, sodium, or potassium atom, and where the organo metal compounds contain from 1 to about 20 carbon atoms.
- the organo metal compound has at least one lithium atom, such as ethyl lithium, propyl, lithium, n -butyl lithium, sec -butyl lithium, tert-butyl lithium, phenyl lithium, he yl lithium, 1,4-diIithio-n- butane, l ,3-di(2-lithio-2-hexyl)benzene, and preferably n-butyl lithium and sec-butyl lithium.
- These organo lithium initiators may be used alone or in combination as a mixture of two or more different kinds.
- the amount of organo lithium initiator used varies, based upon the monomers being polymerized .arid on the target molecular weight of the produced polymer; however, the amount is typically from 0.05 to 5 mmol, preferably from 0.2 to 3 mmol per 100 grams of monomer.
- Lewis bases may optionally be added, to the polymerization mixture to adjust the microstructure (the content of vinyl bonds) of the conjugated diolefin portion, of diolefin-type homo-, co- or terpolymer, or to adjust the composition distribution of the aromatic vinyl compound in the conjugated dienc monomer-containing co- or terpolymer, and thus for example to serve as a randomizer component.
- Lewis bases are, for example, but not limited to, ether compounds, such as diethyl ether, di-n-butyl ether, ethylene glycol diethyl ether, ethylene glycol dibutyl ether, diethylene glycol dimethyl ether, propylene glycol dimethyl ether, propylene glycol diethyl ether, propylene glycol dibutylether, alkyltetrahydroforylethers, such as, m ethyl tetrah.ydrofu.ryl ether, ethyltetrahydrofuryl ether, propyltetrahydrofurylether, butyltetrahydrofurylether, hexyltetrahydrofurylether, octyltetrahydrofurylether, tetrahydrofuran, 2,2-(bistetrahydrofurfuryl)propane, bistetrahydrofurfuryl formal, methyl ether of tetrahydr
- Coupling agents include tin tetrachloride, tin tetrabromide, tin tetrafluoride, tin tetraiodide, silicon tetrachloride, silicon tetrabromide, silicon tetrafluoride, silicon tetraiodide, alkyl tin and alkyl silicon trihalides or dialkyl tin and dialkyl silicon dilialides.
- Polymers coupled with tin or silicon tetrahalides have a maximum of four arms
- polymers coupled with alkyl tin and alkyl silicon trihalides have a maximum of three arms
- polymers coupled with dialkyl tin and dialkyl silicon dihalides have a maximum of two arms.
- Hexahalo disilanes or hexahalo disiloxanes can, also be used as coupling agents resulting in polymers with a maximum of six arms.
- Useful tin and silicon haiides coupling agents include: 8n €l 4 , (Rj) 2 SnCl 2 , R 1 S11CI3, SiCU, (Ri) 2 SiCl 2 , R, SiCl 3 , Cl 3 Si-SiCl 3 , Cl 3 Si ⁇ 0-$iC3 ⁇ 4, Ci 3 Sn-Sn €3 ⁇ 4 and Cl 3 Sn-0-SnCl 3 wherein R
- Examples of tin and silicon alkoxides coupling agents further include: Sn(OMe)4, Si(OMe) 4 , Sn(OEt) 4 and Si(OEt) 4 .
- the most preferred coupling agents are: SnC , SiCU, 5n(OMe) 4 and Si(OMe)4.
- the coupling agents may be added intermittently (or at regular or irregular intervals) or continuously during the polymerization, but are preferably added at a conversion rate of the polymerization of more than 80 percent, and ore preferably at a conversion rate of more than 90 percent.
- a coupling agent can be continuously added during the polymerization, in cases where asymmetrical coupling is desired. This continuous addition is normally done in a reaction zone separate from the zone where the bulk of the polymerization is occurring.
- the coupling agent can be added in a hydrocarbon solution, for example, in cyclohexane, to the polymerization admixture, with suitable mixing for distribution and reaction.
- the coupling agent will typically be added only after a high degree of conversion has already been attained. For instance, the coupling agent will normally be added only after a monomer conversion of greater than about 80 percent has been realized. It will typically be preferred for the monomer conversion to reach at least about 90 percent before the coupling agent is added.
- Polymers coupled with coupling agents have a minimum of two polymer chain arms.
- a substantial, amount of the living polymer chai ends is not terminated with termination agents (i.e. water, alcohols, inorganic or organic acids, such as hydrochloric acid, sulfuric acid .and carboxylic acids, preferably alcohols or water) or is not reacted with the silane sulfide modifier of the present invention prior to the reaction with the coupling agent. That is, living polymer chain, ends are present and. capable of reacting with the coupling agent in a polymer chain coupling reaction.
- the coupling agent mediated coupling reaction occurs before, after or during the addition of the silane sulfide modifier.
- the coupling agent mediated coupling reaction is completed prior to the addition, of the silane sulfide modifier.
- 80 percent or less of the living polymer chains, as determined " by GPC, are reacted with the coupling agent.
- 65 percent or less of the polymer chains are reacted with the coupling agent, and more preferably 50 percent or less of the polymer chains are reacted with the coupling agent.
- between 10 and. 30 percent of the living polymer chain ends, as determined by GPC, are reacted with coupling agent(s), prior to the addition of the silane sulfide modification agent. In other embodiments, between 20 and 35 percent of the living polymer chain ends are reacted with coupling agent(s), prior to the addition of the silane sulfide modifier. In yet other embodiment, between 35 and 50 percent of the living polymer chain, ends are reacted with coupling agent(s), prior to the addition of the silane sulfide modifier.
- the coupling agent may be directly added into the polymer solution without dilution; however, it may be beneficial to provide addition of the coupling agent in solution, such as in an inert solvent (for example, cyclohexane).
- an inert solvent for example, cyclohexane
- the coupling agent is utilized for every 4,0 moles of living and thus anionic polymer chain ends.
- a combination of different coupling agents such as Bu 2 SnCl 2 and Sn.Cl 4 ; Me 2 SiCl 2 and. Si(OMe) ; Me 2 SiCl 2 and SiCl 4 ; SnCl 4 and. Si(OMe) 4 ; SnC and. SiC can also be used to couple polymer chains. It is particularly desirable to utilize a combination of tin and silicon coupling, agents in tire tread compounds that, contain both silica and carbon black. In such cases, the molar ratio of the tin.
- the silicon compound employed, for coupling the el.astom.eric polymer will normally be withi the range of from. 20:80 to 95:5; more typically from 40:60 to 90:10, and preferably from 60:40 to 85:15. Most typically, a range of from about 0.001 to 4.5 mmol of coupling agent (tin and silicon compound, silicon coupling agents) is employed, per 100 grams of the elastomeiic polymer. It is normally preferred to utilize from about 0.05 to about 0.5 mmoi of the coupling agent per 100 grams of polymer to obtain the desired Mooney viscosity and to enable subsequent chain-end functionalization of the remaining living polymer fraction. Larger quantities tend to produce polymers containing terminally reactive groups or insufficient coupling and only enable an insufficient chain end-modification.
- the coupling agent in one embodiment, from 0.01 to less than 5.0 mol, preferably from 0.05 to 2.5 mol, and more preferably from 0.1 to 1.5 mol, of the coupling agent is utilized for every 10.0 moles of living lithium polymer chain ends.
- the coupling agent can be added in a hydrocarbon solution (e.g. in cyclohexane) to the polymerization admixture in the reactor, with suitable mixing for distribution and reaction.
- the polymer coupling reaction may be carried out in a temperature range of from 0°C to 150°C, preferably from 15°C to 120°C. and even more preferably from 40°C to 100°C. There is no limitation for the duration of the coupling reaction. However, with respect to an economical polymerization process, for example, in the case of a batch polymerization process, the coupling reaction is usually stopped at about 5 to 60 minutes after the addition of the coupling agent.
- the coupling agent can be added in a hydrocarbon solution, for example in cyclohexane, to the polymerization admixture in the reactor with suitable mixing for distribution and reaction.
- si lane sulfide modification agents For control of polymer properties, si lane sulfide modification agents (also referred to as silane sulfide modifiers or modifier agents) are employed in accordance with the present invention.
- silane sulfide modification agent includes the silane sulfide modifiers of Formula 1 of the present invention, including compounds of Formula 5 and Formula 6 as further specified below.
- silane sulfide modification agent of the present invention encompasses compounds of the following Formula 5:
- M is a silicon atom or a tin atom
- IV is at least divalent and is (C 8 -C ) 6 ) alkyl aryl alkyl, (C7-C16) arylalkyl, (C 7 ⁇ C U -,) alkylaryl, or
- (Ci-Ci(,) alkyl and each group may be substituted with one or more of the following groups: tertiary amine group, silyl group, (C7-C18) aralkyl group and (Q-Cig) aryl group;
- R and R are each independently selected from (C r C 4 ) alkyl
- R 2 and R 1 ' ' are each independently selected from (Ci-Ct 6 ) alkyl, (C7-C16) alkylaryl and (C7-C16) arylalkyl:
- R 4 and R 14 are each independently selected from (CrCj f ,) alkyl and (C7-C16) alkylaryl:
- R is a (CrCi 6 ) divalent alkyl group or (Cg-Cte) divalent alkyl arylalkyl group.
- R 3 is alkyleiie.
- the alkylene is selected from -CH 2 - (methylene). -(CH 2 ) 2 - (ethylidene), -(CH 2 )3- (propylidene) and -(CH 2 )4- ( utyl i dene).
- R " is a divalent aralkylene group.
- the aralkylene group is selected from -CH2-C6H -CH2- (xylidene) and -CeH4-C(CH3) 2 -CftH -.
- R 2 , R 4 . R i3 and R 14 are each independently a (Q-Cie) alkyl.
- the alkyl is selected from C3 ⁇ 4- (methyl), CH3-CH 2 - (ethyl), CH 3 -(CH 2 )2- (propyl), CH 3 -(CH 2 )3 (n-butyl) and CH 3 -C(CH 3 )2 (tert-butyl).
- R is selected from the group consisting of linear Q- C 10 alkyl (divalent), cyclic C 6 -C 12 alkyl (divalent), C Cjs aryl (divalent) and C7-Q2 alkylaryl (divalent).
- M is a silicon atom; a and c are each an integer selected from 2 and 3; and b and d axe each an integer selected from 0 and 1 .
- silane sulfide modifiers of the present invention also encompass their corresponding Lewis base adducts (for example, with solvent molecules tetrahydrofuran, diethyl ether, dimethoxyethane coordinated with silicon atoms).
- silane sulfide modification agent of the present invention include the following compounds and their corresponding Lewis base adducts:
- si lane sulfide modification agent of the present invention furthermore includes compounds of the following Formula 6 :
- M is a silicon atom or a tin atom
- R “ ' is at least divalent an.d is (CS-CM) alkyiaryl alkyl, (C7-C16) ar lalk l, (C7-C16) alkyiaryl, or (Ci-Cie) alkyl, and each group may be substituted with one or more of the following groups: tertiary amine group, silyl group, (C 7 -Cig) aralkyl group and (C « > -Ctg) aryl group;
- R ⁇ R I2 and R 16 are each independently selected from (C1-C4) alkyl
- R , R and R. ,J are each independently selected from. (Ci-Cje) alkyl. (C7-C16) alkyiaryl and (C 7 - C
- R 3 is a (Cj-Cie) divalent alkyl group or (Cg-Cu) divalent alylaral alkyl. group.
- R is alkylene.
- the alkylene is selected from -CH,2- (methylene), -(CH 2 ) 2 - (ethylidene), -(CH 2 ) 3 - (propylidene) and -(CH 2 )4- (butylidene).
- R is a divalent aralkylene group.
- the aralkylene group is selected from (xylidene) and
- R 2 , R 4 , R 13 and R 1 5 are each independently a (Ci-Cie) alkyl
- the alkyl is selected from CH3- (methyl), CH3-CH2- (ethyl), CH 3 -(CH 2 ) 2 - (propyi), CH 3 -(CH 2 ) 3 (n-butyl) and CH 3 -C(CH 3 ) 2 (tert-buty!).
- R " is selected from the group consisting of linear C
- b, d and f are each independently selected from an integer of 0 and 1 ; a, c and e axe each independently selected from an integer of 2 and 3.
- M is a silicon atom; a, c and e are each an integer selected from 2 and 3; and b, d and f are each an integer selected from 0 and 1 .
- silane sulfide modifiers of the present invention may also include their corresponding Lewis base add cts
- silane sulfide modifier of the present invention include the following compounds and their corresponding Lewis base adducts:
- the silane sulfide modification agents may be added intermittently (or at regular or irregular intervals) or continuously during the polymerization, but are preferably added at a conversion rate of the polymerization of more than 80 percent, and more preferably at a conversion rate of more than 90 percent.
- a substantial amount of the polymer chain ends is not terminated prior to the reaction with the silane sulfide modification agent; that is,
- the living polymer chain ends are present and capable of reacting with the silane sulfide end modification agent.
- the silane sulfide modification reaction may occur before, after or during the addition of the couplin agent (if used) or any other further modification agent (if used).
- the silane sulfide modification reaction is completed after the addition of the coupling agent. See, for example, WO
- the reaction with the silane sulfide modifier of the invention is preferably completed prior to the addition of the further modification agent.
- the preferred order of addition causes the silane sulfide modification agents of the invention to react with more than one of the living polymer chains forming modified coupled macromolecular compounds (provided the ratio of the number of living polymer chains to the number of silane sulfide modification agents is significantly larger than 1, preferably larger than 2), while the further modification, agents form essentially linear modified macromolecular compounds (provided the ratio of the number of living polymer chains still available after completion of the reaction with the silane sulfide modification agent of the invention to the number of further modification agents is relatively close to 1 , such as lower than 1.4 or more preferably lower than 1.0),
- more than 20 percent, preferably more than 35 percent, and even more preferably more than 50 percent of the living polymer chains, as determined by GPC, formed in the course of the polymerization process, are linked with a silane sulfide modification agent in the process of polymer silane sulfide modification.
- more than 20 percent of the living polymer chain ends, as determined by GPC, are reacted with coupling agent(s), prior to the addition of the silane sulfide modification agent(s). In yet other embodiments, more than 35 percent of the living polymer chain ends are reacted with coupling agent(s), prior to the addition of the silane sulfide modification agent(s).
- between 20 and 35 percent of the living polymer chain ends, as determined by GPC, are reacted with coupling agent(s), prior to the addition of silane sulfide modification agent(s). In other embodiments, between 35 and 50 percent of the living polymer chain ends, as determined by GPC, are reacted with coupling agent(s), prior to the addition of silane sulfide modification agent(s). In yet other embodiment, between 50 and 80 percent of the living polymer chain ends are reacted with coupling agent(s), prior to the addition of silane sulfide modification agent(s).
- Silane sulfide modified polymer macromolecules comprise a functionality derived from the silane sulfide modification, agent.
- silane sulfide modifier of Formula I of the present invention may be prepared by reacting a sulfur containing compound of Formula 2:
- M is silicon or tin
- u is an. integer selected from 2, 3 and 4
- R. 4 , X and t are as defined above
- t + u 4, optionally in the presence of a strong Lewis base, such as for example but not limited to a compound of Formula 3 a and Formula 3b,
- the Lewis base is selected, from compound of Formula. 3a. In one preferred embodiment the Lewis base is selected from a compound of Formula 3a and
- R and R are each independently selected from methyl (Me), ethyl (Et). propyl (Pr) and butyl (Bu).
- u in Formula 4 is 2.
- silane sulfide modifier of Formula 1 of the present invention may be also prepared by reacting a sulfur containing compound of Formula 1 1 :
- M * is lithium, sodium or potassium and the other symbols have the same meaning as defined with respect to Formula 1 , with a compound of Formula 4:
- M is silicon or tin
- u in Formula 4 is 2.
- the silane sulfide end-modification agent may be directly added to a solution of the living anionic elastomeric polymer; however, it may be beneficial to add the agent in dissolved form, such as in an inert solvent (e.g. cyclohexane).
- the amount of silane sulfide modification agent added to the living anionic elastomeric polymer may vary depending on the kind of monomer species, kind and amount of optional coupling agent, kind and amount of further modification agent, reaction conditions, and desired end properties, but is generally from 0.05 to 5 mol-equivalent, preferably from 0.1 to 2.0 mol-equivalent, and most preferably from 0.2 to 1.5 mol-equivalent, per mol equivalent of alkali metal in the initiator compound.
- the silane sulfide modification agent is used to form, a branched modified macromolecular compound and is employed in an amount from 0.1 to 0.5 mol-equivalent per mol equivalent of alkali metal in the initiator compound.
- the silane sulfide modification agent is used to form a linear modified macromolecular compound and is employed in an amount from 0.6 to 1.5 mol-equivalent per mol equivalent of alkali metal in the initiator compound.
- the polymer silane sulfide modification reaction may be carried out in a temperature range of from 0°C to 150°C, preferably of from 15°C to 120°C, and even more preferably of from 40°C to 100°C.
- the duration of the silane sulfide modification reaction is no limitation for the duration of the silane sulfide modification reaction.
- the silane sulfide modification reaction is usually stopped at about 5 to 60 minutes after the addition of the modifier.
- the invention also provides a method for making the silane sulfide modified elastomeric macromolecular compound comprising the following steps A through D.
- Step A reacting the polymerization initiator, as described herein, with one or more monomer types, and preferably monomers selected from butadiene, styrene, isoprene, alpha methyl-styrene and combinations thereof, in a polymerization solvent to form a reaction mixture A.
- Suitable polymerization solvents include non-polar aliphatic and non-polar aromatic solvents, preferably hexane, heptane, butane, pentane, isopar, cyclohexane, toluene and benzene.
- Step B optionally reacting reaction mixture A with at least one coupling agent, preferably selected from the group consisting of SnCl 4 , (R ⁇ SnCI, (R' SnCl,, R'SnCb, SiCl 4 , (R') 3 SiCL (R ⁇ SiCb,, R ⁇ iCU, ClsSi-SiCfj, CbjSi-O-SiCb, ChjSn-SnC , CI 3 Sn-0-SnCl 3 , wherein R ! is as defined above, Sn(OMe) 4 , Si(OMe) 4 , Sn(OEt) 4 and Si(OEt) 4 to form a reaction mixture B.
- at least one coupling agent preferably selected from the group consisting of SnCl 4 , (R ⁇ SnCI, (R' SnCl,, R'SnCb, SiCl 4 , (R') 3 SiCL (R ⁇ SiCb,, R ⁇ iCU,
- Step C reacting the reaction mixture A or B with at least one silane sulfide modification agent of Formula 1 as defined above, including Formula 5 and Formula 6, to produce a silane sulfide modified macromolecular compound of the invention, usually in the form of a polymer composition C
- Step D optionally reacting the silane sulfide modified macromolecular compound or polymer' composition C obtained in Step C with a further modification agent, preferably with a compound of Formula 7.
- the polymerization initiator compound is reacted first with monomers to form a living polymer (step A). Some of these polymer molecules are optionally reacted with coupling agent to form branched polymer molecules (optional step B). In step C, some of the living polymer molecules are reacted with the silane sulfide chain-end modification agent to form linear silane sulfide modified macromolecular compound(s). Linear silane sulfide modified macromolecular ' compounds are formed when one equivalent of living polymer chains are modified (or reacted) with one equivalent of silane sulfide modification agent.
- the polymerization initiator compound is reacted first with monomers to form a living polymer (step A). Some of the polymer molecules are reacted with silane sulfide modification agent to form branched modified polymer molecules (step C). Branched silane sulfide modified macromolecular compounds are formed when two or more equivalents of living polymer chains are modified (or reacted) with one equivalent of silane sulfide modification agent. For example, if three (equivalents of) living polymer chains are modi.fi. ed with one (equivalent of) silane sulfide modification agent, a silane sulfide modified macromolecuiar compound is formed, which comprises three polymer chain anus. In optional. step D, some of the living polymer molecules are reacted with the further modification agent to form linear modified macromolecuiar compound(s).
- the optional coupling agent is selected from the following: SnCLt, Bu 3 SnCl, Bu 2 8nCl 2 , BuSnCl 3 , Me 3 SiCl, Me 2 SlC1 ⁇ 2, C%Si-SiCl 3 , Cl 3 Si-0-SjCl 3 ,
- the silane sulfide modification agent is the compound of Fomiula 5
- the silane sulfide modification agent is the compound of Fomiula 6.
- Monomers useful for preparing living anionic elastonieric polymers and, thus, the silane sulfide modified macromolecuiar compounds of the present invention and the polymer compositions comprising said macromolecuiar compound(s) include conjugated olefins and olefins selected from a-olefins, internal olefins, cyclic olefins, polar olefins and nonconjugated diolefms.
- Suitable conjugated unsaturated monomers are preferably conjugated dienes, such as
- Preferred olefins are C2-20 a-olefins, including, but not limited to, long chain macromolecuiar a-olefins, more especially an aromatic vinyl compound.
- Preferred aromatic vinyl compounds are styrene, including CM alky! substituted styrene, such as 2-methylstyrene, 3-methylstyrene, 4-methylstyrene, 2,4-dimethylstyrene, 2,4,6-trimethylstyrene, a-m ethyl styrene and stilbene,
- Suitable polar olefins included acrylonitrile, methacrylates, methylmethacrylate.
- Suitable nonconjugated olefins include C -20 diolefins, especially norbornadiene, ethylidenenorbornene, 1,4-hexadiene, 1,5-hexadiene, 1 ,7-octadiene, 4- vinylcyelohexene, divinylbenzene including 1 ,2-divinylbenzene, 1,3-divinylbenzene and 1 ,4- divinylbenzene and mixtures thereof.
- Preferred conjugated dienes include butadiene, isoprene and cyclopentadiene
- preferred aromatic a-olefins include: styrene and 4-methylstyrene,
- Silane sulfide modified macromolecular compound is intended to mean the reaction product of one or more living polymer chairi(s) with the silane sulfide modifier agents of the present invention.
- Silane sulfide modified macromolecular compounds may be represented by the following Formula PI
- P is a polymer chain comprising monomer units derived from at least one of the following monomer .groups; butadiene, isoprene, styrene and alpha-methylstyrene, the number of monomer units per macromolecule ranging from 10 to 50,000, preferably from 20 to 40.000; M is a silicon, atom or a tin atom;
- R 1 is independently selected from a hydrogen atom and (C r C 6 ) alkyl
- R is independently selected from (C 1 -C 1 0) alkyl, (C 7 -Ci 6 ) alkylaryl and (C 7 -Ci 6 ) arylalkyl;
- R 3 is at least divalent and is independently selected from ( €
- R 4 is independently selected from (C 1 -C 15) alkyl and (C 7 -C i6) alkylaryl;
- X is independently selected from chloride, bromide and -OR 5 ; wherein R 5 is selected from
- R is divalent and is (Q-C ⁇ ) alkyl.
- X is -OR 5 ; wherein R 5 is selected from. (Ci-C l 6 ) alkyl.
- X is chloride or bromide.
- R 4 are independently selected from (Ci-Cie) alkyl.
- R , R ⁇ R and R are independently selected from. (C 1 -C4) alkyl.
- s and. t are each. 2 and u is 0.
- s is 3, t is 1 and u is 0.
- r is 1
- x is 1
- y is 1
- Silane sulfide modified macromolecular compounds may be exemplified by the following Formulas P2 to P6:
- P is a polymer chain comprising monomer units derived from, at least one of the following monomer groups: butadiene, isoprene, styrene and alpha-rnethylstyrene, the number of monomer units per polymer macromolecule ranging from 10 to 50,000, preferably from 20 to 40.000;
- R 3 is at least divalent and is (Cg-Cie) alkylarylalkyi (C 7 -C 16 ) aryiaikyl, (C7-C 1 6) alkylaryl, or (Ci-Ci 6 ) alkyl, and each group may be substituted with one or more of the following groups: tertiary amine group, silyl group, (C 7 -Cig) aralkyl group and (C 6 - Cis) aiyl group;
- M is a silicon atom or a tin atom
- R ! , R° and R 16 are each independently selected from a hydrogen atom and (C
- R 4 is selected from (Ci-C ! ( ,) alkyl and (C 7 ⁇ C 16 ) alkylaryl;
- R 2 , R 13 and R 1 S are each independently selected from. (Cj-Cig) alkyl, (C7-C16) alkylaryl and (C 7 - Ci 6 ) aryiaikyl;
- p, o and i are each independently selected from an integer of 1 , 2 and 3; g, Ii and j are each independently selected from an integer of 0, 1 and. 2; b, d and. f are each independently selected from an integer of 0, 1 and 2; and
- R 3 is divalent and is (Ci-C ie) alkyl; and R ⁇ R. 4 , R 13 and R 14 are each independently selected from (Ci- € ) 6 ) alkyl; and R 1 and R 12 are each independently selected from (C 1 -C4) alkyl; and.
- p and o are each independently selected from, an integer of 1 and 2; g and h. are each independently selected from an. integer of 1 and 2; b and d are each independently selected from an integer of 0 and 1 ,
- R is divalent and is (Cj-Cie) alkyl; and R , R , R 13 and R 15 are each independently selected from (Ci-Ci & ) alkyl; and R 1 , R 12 and R 16 are each independently selected, from (C1-C4) alkyl; and p, o and i are each independently selected from an integer of 1 and 2; g, h and ] are each independently selected from an integer of 1 and 2; b, d and f are each independently selected from an integer of 0 and 1 ,
- modified macromolecular compounds include the following polymers (and their corresponding Lewis base adducts):
- dihydrocarbylsilendiyl including dialkylsilendiyi, diaralkylsilendiyl and diarylsilettdiyl
- dihydrocarbylstannendiyl including dialkylstannendiyl, diaralkylstannendiyl and diarylstannendiyl groups
- hydrocarbylsilentriyl including alkylsilentriyl, aralkylsilentriyl and.
- arylsilentriyl hydrocarbylstannentriyl, including alkylstannentriyl, aralkylstannentriyl and arylstannentriyl groups of Formulas PI , P2, P3, P4, P5 and P6 are each believed to function as a protective group, which prevents iiointeoded subsequent reaction.
- steam stripping of a polymer solution containing the modified macromolecuiar compounds of Fonnula PI , P2, P3, P4, P5 and P6 will remove a certain percentage of the protecting trihydrocarbyl groups, including trialkyl, triaralkyl, or triarylsilyl groups, resulting in the unprotected thiol (-SH) group and forming a. certain percentage of compounds of Formula P7 (corresponding to Formula I ), Formula P8. Formula P9 or Formula P10.
- P, x, y, r, R'; . and R are as defined in Formula PI , and
- P is a polymer chain comprising monomer units derived from at least one of the following monomer . groups: butadiene, isoprene, styrene and alplia-m.ethylstyrene, the number of monomer units per macromolecule ranging from 10 to 50,000 g mol, preferably from 20 to 40,000 g/mol; O is an oxygen atom; Si is a.
- R is at least divalent and is (Ct-Cjj) alkyl, whic may be substituted with one or more of the following groups: tertiary amine group, silyl group, (C7-C18) aralkyl group and (C( ⁇ -C[g) aryl group:
- R l , R l2 and. R 16 are independently selected from a hydrogen atom and (C 1 -C4) alkyl;
- R ⁇ R 13 and R. 15 are each independently selected from (Ci-C] g ) alkyl
- g, h and j are each, independently selected, from, an integer of 0, 1 and 2; b, d and f are each independently selected, from an integer of 0, 1 and 2; and
- R. 3 is divalent and is (Cj-Cig) alkyl;
- R 1 , R 6 or R !0 are independently selected ixom hydrogen and (Cj-Cig) alkyl;
- p. o or i are selected from an integer of 1 and 2:
- g, h or j are selected from. an. integer of 1 and 2;
- b, d or f are selected from an integer of 0 and 1 ,
- silane sulfide modified macromolecular compounds of the present invention include their corresponding Lewis base adducts.
- a water- free work up procedure can be used for the preparation of the silane sulfi.de modified macromolecular compounds of Formula 1 , Formula P2, Formula P3, Formula P4. Formula P5 and Formula P6.
- hydrocarbyloxysilyl (-SiOR) group of the modified macromolecular compound is reactive with fillers, such as silica and/or carbon black, preferably with silica, present.
- fillers such as silica and/or carbon black, preferably with silica, present. This interaction, is believed to result in the formation of bonds with, fillers, or in the case of some fillers, in electrostatic interactions, which result in more homogeneous distributions of filler within the polymer compositions.
- modified macromolecular compounds based on Formula PI to Formula P6 include the following polymers (and their corresponding Lewis base adducts):
- the reaction product i.e. the polymer composition comprising si lane sulfide modified macroniolecular compound(s) of the present invention, typically comprises one or more alkoxysilyl or silanol group-containing compounds as represented by Formula P7, Formula P8, Formula P9 and Formula PIO, typically in a total amount from 0.0001 to 1.50 mmol/gram of polymer, preferably from 0.0005 to 0.9 mmol/gram, and more preferably from 0.0010 to 0.5 mmol/gram, and even more preferably from 0.0020 to 0.1 mmol/gram of polymer.
- the polymer composition comprising si Jane sulfide modified macroniolecular compound(s) of the invention preferably comprises sulfide group-containing compounds, and the sulfide groups, in the form of hydrocarbylsilyl or hydrocarbylstannyl protective groups and/or thiol groups (sulfide and thiol groups), are typically present in a total amount of from 0.0001 to 0.50 mmol/gram of polymer, preferably from 0.0005 to 0.30 mmol/gram, and more preferably from 0.0010 to 0.20 mmol/gram, and even more preferably from 0.0020 to 0.10 mmol/gram of polymer.
- the sulfide groups are present in an amount ranging from 0.0001 to 0.50 mmol/gram of polymer, preferably ranging from 0.0005 to 0.30 mmol/gram, and more preferably ranging from. 0.0010 to 0.20 mmol/gram, and even more preferably from ranging from 0.0020 to 0.10 mmol/gram of polymer.
- the thiol groups are present in an amount ranging from 0.0001 to 0.50 mmol/gram of polymer, preferably ranging from 0.0005 to 0.30 mmol/gram, and more preferably ranging from 0,0010 to 0.20 mmol/gram, and even more preferably from ranging from 0.0020 to 0.10 mmol/gram of polymer.
- the si lane sulfide modified macromolecular compound is preferably a homopolymer derived from a conjugated diolefin, a copolymer derived from a conjugated diolefin monomer with an aromatic vinyl monomer, and/or a terpolymer of one or two types of conjugated diolefins with one or two types of aromatic vinyl compounds.
- vinyl bonds 1,2-bonds and/or 3,4- bonds (hereinafter called "vinyl bonds”) of the conjugated diolefin portion in the polymer composition comprising the silane sulfide modified macromolecular compound(s) of the invention
- the vinyl bond content is preferably from. 10 to 90 % by weight and. particularly preferably from. 15 to 80 % by weight (based on the total weight of the polymer). If the vinyl bond content in a polymer composition, is less than 10 % by weight, the resulting product may have inferior wet skid resistance.
- the product may exhibit compromised, tensile strength and abrasion resistance and a relatively large hysteresis loss.
- the aromatic vinyl monomers constitute from 5 to 60 % by weight of the total monomer content, and more preferably from 10 to 50 % by weight (based on the total weight of the polymer). Values of less than 5 % by weight may lead to reduced wet skid properties, abrasion resistance, and tensile strength; whereas values of more than 60 % by weight may lead to increased hysteresis loss.
- the silane sulfide modified macromolecular compound may be a block or random, copolymer, and preferably 40 % by weight or more of the aromatic vinyl compound units are linked singly, and 10 % by weight or less are "blocks" in. which eight or more aromatic vinyl compounds are linked successively. Copolymers falling outside this range often, exhibit increased hysteresis.
- the length, of successively linked aromatic vinyl units can be measured by an ozonolysis-gel permeation chromatography method developed by Tanaka et al. (Polymer, Vol. 22, Pages 1721-1723 (1 81)).
- the first polymer composition comprising at least one silane sulfide modified macromolecular compound of the present invention, such as the polymer product obtained in the method of making said silane sulfide modified macromolecular compound, preferably has a Mooney viscosity (ML 1 +4, 100°C, as measured in accordance with A.STM D 1.646 (.2004), in. the range of from 0 to 150, preferably from 0 to 100, and more preferably in the range of from 20 to 100, as determined using a Monsanto MV2000 instrument.
- ML 1 +4, 100°C as measured in accordance with A.STM D 1.646 (.2004)
- Mooney viscosity (ML 1+4, 100°C) of the polymer is more than 150 MU, the processability (filler incorporation and heat build-up in the internal mixer, banding on the roll mill, extrusion rate, extrudate die swell, smoothness, etc.) is likely to be negatively affected because the compounding machinery used by the tire manufacturers are not designed, to handle such high Mooney rubber grades, and the cost of processing increases.
- a Mooney viscosity (ML 1 +4, ! 00°C) of less than 20 may not be preferred, due to increased tack and. cold flow of the uncrosslinked. elastomeric polymer, resulting in difficult handling, poor green strength and poor dimensional stability during storage.
- the first polymer compositions comprising at least one silane sul.fi.de modified macromolecular compound are used, as a softener, eompatibilizer or processing aid in polymer formulations, a Mooney viscosity (ML 1+4, 100°C) of less than 20 may be preferred.
- the preferred, molecular weight distribution of the total polymer contained in the first polymer composition comprising at least one silane sulfide modified macromolecular compound, represented by the ratio of the weight average molecular weight to the number average molecular weight (M w /M n ), ranges from 1.0 to 10.0, preferably from. 1.1 to 8.0 and more preferably from 1.2 to 4.5.
- the first polymer composition comprising at least one silane sulfide modified macromolecular compound
- fiUer(s) selected from, silica, carbon-silica dual phase filler, carbon black, carbon nano-tube filler, lignin, glass filler, layered silicates, such as magadiite, in some preferred embodiments comprising silica as main filler component, and vulcanization agent and, optionally, additional constituents, including, but not limited to, processing aids, oils, vulcanization agents, silane coupling agents and unmodified uncrosslinked elastomeric polymers, thus forming a second polymer composition comprising filler.
- the first polymer composition comprises at least one silane sulfide modified macromolecular compound and optionally one or both of (i) oil (often referred to as oil extended polymer) and (ii) polymer which is not identical with the modified macromolecular compounds according to the invention.
- Polymers which are not identical with the modified macromolecular compounds of the invention may be by-produced in the process of preparation of the modified macromolecular compound (see above) and may result from blending modified macromolecular compounds) (for example in the form as obtained after polymerization) in solution with another polymer solution, followed by solvent removal.
- the first polymer composition preferably contains at least 25 % by weight of the silane sulfide modified macromolecular compound, based on the total polymer contained in the composition, more preferably at least 35 % by weight and even more preferably at least 45% by weight.
- the remaining portion of the polymer contained in the polymer composition is unmodified elastomeric polymer or polymer modified in a manner other than according to the invention.
- Examples of preferred unmodified elastomeric. polymers are listed in WO 2009/148932 and preferably include styrene-butadiene copolymer, natural rubbers, polyisopreoe and polybutadiene. It is desirable that the unmodified polymers have a Mooney viscosity (ML 1 +4, 100 °C as measured in accordance with ASTM D 1646 (2004)) in the range of from. 20 to 200, preferably from 25 to 150.
- Oils may be used in combination with the uncrosslinked elastomeric polymers to reduce viscosity or Mooney values, or to improve processabiiity of the first and the second polymer compositions and various performance properties of (vulcanized) second polymer compositions,
- Oil(s) can be added to the modified macromolecular compound prior to the end of the preparation process, or as a separate component of the first or second polymer composition preparation process.
- Oil(s) see WO 2009/148932 and U.S. 2005/0159513, each of which is incorporated herein by reference in its entirety.
- Representative oils include but are not limited to MES (Mild Extraction Solvate), TDAE (Treated Distillate Aromatic Extract), RAE (Residual Aromatic Extract) including but not limited to T-RAE and S-RAE, DAE including T-DAE and NAP (light and heavy naphthenic oils), including but not limited to Nytex 4700, Nytex 8450, Nytex 5450, Nytex 832. Tufflo 2000, and Tufflo 1200.
- native oils including but not limited to vegetable oils, can he used as extender oils.
- Representative oils also include functionalized variations of the aforementioned oils, particularly epoxidized or hydroxylated oils.
- oils comprise varying concentrations of polycyclic aromatic compounds, paraffinics, naphthenics and aromatics and have different glass transition temperatures.
- the above mentioned types of oils have been characterized (Kautsch k Kunststoff Kunsmoffe, vol. 52, pages 799-805).
- the MES, RAE and TDAE are extender oils for rubber.
- Processing aids can optionally be added to the first and to the second polymer compositions, but preferably to the second polymer composition of the present invention. Processing aids are usually added to reduce the first and/or second polymer composition viscosity. As a result, the mixing period is decreased and/or the number of mixing steps is reduced and, consequently, less energy is consumed and/or a higher throughput in the course of the rubber compound extrusion process is achieved.
- processing aids which can optionally be used as a component in the first polymer compositions of the present invention are described in Rubber Handbook, SGF, The Swedish Institution of Rubber Technology 2000 and in Werner KJeemann, Kurt Weber, Elast kauKenn nuances und Be sacredsmetJioden, Deutscher Veiiag fur Grandstoffindustrie (Leipzig, 1990), each of which is incorporated herein by reference in its entirety.
- Examples of representative possessing aids which can optionally be used as component in. the first polymer compositions of the present invention can be classified as follows: (A) fatty acids including but not limited to oleic acid, priolene, pristerene and stearic acid;
- (B) fatty acid salts including but not limited to Aktiplast GT, PP, ST, T, T-60, 8, F; Deoflow S: ettlitz Dispergator FL, FL Plus; Dispergum 18, C, E, K, L, N, T, R; Polyplastol 6, 15, 19, 21, 23; Struktol A50P, A60, EF44, EF66, EM 16, EM50, WA48, WB16, WB42, WS180, WS280 and ZEHDL;
- (C) dispersing agents and processing aids including but not limited to Afhix 12, 16, 42, 54, 25; Deoflow A, D; Deogum 80; Deosol H; Kettlitz Dispergator DS, KB, OX; Kettlitz- Mediaplast 40, 50, Pertac/GR; Kettlitz-Dispergator SI; Struktol FL and WB 212; and
- silane coupling agents Bifunctionaiized silanes and monofunctional silanes (herein also called “silane coupling agents”) are also occasionally referred to as processing aids but are separately described below, SILANE COUPLING AGENTS
- a silane coupling agent (used for compatibilization of pol mer and fillers) is added to the polymer composition which contains at least one silane sulfide modified macroiiiolecular compound as described herein and silica, layered silicate (such as but not limited to magadiite) or carbon-silica dual -phase filler, which may be used as filler component.
- the typical amount of a silane coupling agent added is from about 1 to about 20 parts by weight and, in some embodiments, from about 5 to about 15 parts by weight for 100 parts by weight of the total amount of silica and/or carbon-silica dual-phase filler.
- Silane coupling agents can be classified according to Fritz Rotheme er, Franz Sommer: Kautsckuk Technologic, (Carl Hanser Verlag 2006):
- (B) monofunctional silanes including but not limited to Si 203 (EtO) 3 -Si-C 3 H7, and Si 208 (EtO) 3 -Si-C 8 Hi7.
- silane coupling agents include but are not limited to bis-(3-hydroxy-dimethylsilyl-propyl)tetrasulfide 5 bis-(3-hydroxy-dimethylsilyl- propyl)-disuIfide, bis-(2-hydroxy-dimethylsilyl-ethyl)tetrasulfide, bis-(2-hydroxy- dimethylsilyl-ethyl)disulfide, 3-hydroxy-dimethylsilyl-propyl-N,N-dimethyl- thiocarbamoyltetrasulfide and 3 -hydroxy-dimethyl silyl-propylbenzothiazole tetrasulfide.
- Vulcanization agents as described herein, are added to the first or second polymer composition, as described herein.
- the addition of the vulcanization agents to the first or second polymer composition represents the key criteria for the of the formation of the vulcanized polymer composition.
- Sulfur, sulfur-containing compounds acting as sulfur-donors, sulfur- accelerator systems and peroxides are the most common vulcanizing agents.
- sulfur-containing compounds acting as sulfur-donors include but are not limited to dithiodimorpholine (DTDM), tetramethylthiuramdisulphide (TMTD), tetraethylthiuramdisulphide (TETD), and dipentarnethylenthturamtetrasulphide (DPTT).
- sulfur accelerators include but are not limited to amine derivates, guanidine derivates, aldehydeamine condensation products, thiazoles, thiuram sulphides, dithiocarbamates and thiophosphates.
- peroxides used as vulcanizing agents include but are not limited to di-ierf.-butyl-peroxides, di-(ier/ * .-butyl- peroxy-trimethyl-cyclohexane), di-(ferf. -butyl -peroxy-isopropyl-)benzene, dichloro- benzoylperoxide, dicumylperoxides, iert.-butyl-cumyl-peroxide, dimethyl-di(3 ⁇ 4rf. -butyl - peroxylhexane. d i m eth y 1 -d i ( ⁇ c/ .
- vulcanizing agents can be found in Kirk-Othrner, Encyclopedia of Chemical technology 3 rd , Ed., (Wiley Interscience, N.Y. 1 82), volume 20, pp. 365-468, (specifically "Vulcanizing Agents and Auxiliary Materials” pp. 390-402).
- a vulcanizing accelerator of the sulfene amide-type, guanidine -type, or thiuram-type can. be used together with a vulcanizing agent as required. Other additives such as zinc white, vulcanization auxiliaries, aging preventives, processing adjuvants and the like may optionally be added.
- a vulcanizing agent is typically added, to the polymer composition in an amount of from 0.5 to 10 parts by weight and, in some embodiments, from 1 to 6 parts by weight per 100 parts by weight of the total elastomeric polymer. Examples of vulcanizing accelerators and the amount of accelerator added with respect to the total polymer are given in WO 2009/148932.
- the expression ''total polymer refers to the sum of all individual amounts of different polymer types, including the silane sulfide modified (elastomeric) macromolecular compound.
- Sulfur-accelerator systems may or may not comprise zinc oxide.
- zinc oxide is used as a component of the sulfur-accelerator system.
- Second polymer compositions are added to first polymer compositions for forming second polymer compositions.
- Second polymer compositions once cured, form filler-containing vulcanized polymer compositions.
- second polymer compositions and products made therefiOm as well as vulcanized polymer compositions made from, second polymer compositions and products containing such vulcanized polymer compositions include filler which serves as a reinforcement agent.
- Carbon black, silica, carbon-silica dual-phase filler, clay (layered silicates), calcium carbonate, magnesium carbonate, lignin, carbon-nano-tubes, amorphous fillers, such as glass particle based fillers, starch based fillers and the like and combinations thereof are examples of suitable fillers.
- Carbon black is manufactured by a furnace method, and in some embodiments a nitrogen adsorption specific surface area of 50-200 m 2 /g and DBF oil absorption of 80-200 ml/100 grams, for example, FEF; HAF, ISAF, or SAF class carbon black, is used, in some embodiments, high agglomeration type carbon black is used.
- Carbon black is t pically added in an amount of from 2 to 100 parts by weight, in some embodiments from 5 to 100 parts by weight, in some embodiments from 10 to 100 parts by weight, and in some embodiments from 10 to 95 parts by weight per 100 parts by weight of the total elastomeric polymer.
- silica fillers include but are not limited to wet process silica, dry process silica, synthetic silicate-type silica and combinations thereof.
- Silica with a small particle diameter and high surface area exhibits a high reinforcing effect.
- Small diameter, high agglomeration-type silica i.e., having a large surface area and high oil absorptivity
- An average particle diameter of silica, in terms of a primary particle diameter is in some embodiments from 5 to 60 nm, and in some embodiments from 10 to 35 nm.
- the specific surface area of the silica particles is in some embodiments from 35 to 300 ⁇ /g.
- Silica is added in an amount of from 10 to 100 parts by weight in some embodiments from 30 to 100 parts by weight, and in some embodiments from 30 to 95 parts by weight for 100 parts by weight of the total elastomeric polymer.
- Silica fillers can be used in combinations with other fillers including but not limited to carbon black, carbon-silica dual-phase-filler, clay, calcium carbonate, carbon - nano-tubes, magnesium carbonate and. combinations thereof.
- Carbon black and silica may be added together; in which case the total amount of carbon black and silica added is from 30 to 100 parts by weight and, in some embodiments, from 30 to 95 parts by weight per 100 parts by weight of the total elastomeric polymer.
- fillers are homogeneously dispersed in the elastomeric composition, increasing quantities (within the above ranges) result in compositions having excellent rolling and extruding processability and vulcanized products (products comprising vulcanized polymer compositions) exhibiting favorable hysteresis loss properties, rolling resistance, improved wet skid resistance, abrasion resistance and tensile strength.
- Carbon-silica dual-phase-filler may be used either independently or in combination with carbon, black and or silica in accordance with the present teachings. Carbon-silica dual-pha.se- fi!ier can exhibit the same effects as those obtained by the combined use of carbon black and silica, even in the case where it is added alone. Carbon-silica dual-phase-filler is so called silica-coated-carbon black made by coating silica over the surface of carbon black, and is commercially available under the trademark CRX2000, CRX2002 or CRX2006 (products of Cabot Co.). Carbon-silica dual-phase-filler is added in the same amounts as described above with respect to silica.
- Carbon-silica dual-phase-filler can be used in combinations with other fillers including but not limited to carbon black, silica, clay, calcium carbonate, carbon-nano- tubes, magnesium carbonate and combinations thereof, in some embodiments, carbon black and silica, either individually or in combination, are used.
- Silica, carbon, black or carbon black-silica dual-phase-fillers or combinations thereof can be used in combination with natural fillers including but not limited to starch or lignin.
- the silica incorporated in second polymer compositions, in products and vulcanized polymer compositions made from second polymer compositions, and in products comprising such, vulcanized polymer compositions has a surface area determined, by nitrogen adsorption (hereinafter referred, to as "N2A") of from 150 to 300 m 2 /g.
- N2A nitrogen adsorption
- A. silica having an N2A of less than 150 m 2 /g may lead to an unfavorably low reinforcing effect.
- A. silic having an N2A of more than 300 m 2 /g may provide a. rubber compound with. an. increased viscosity and a. deteriorated, processability.
- an N2A from 60 to 150 m /g is suitable.
- Carbon black having an. N2A. of less than 60 m7g leads to a low reinforcing effect.
- Carbon, black having an. N2A of more than 150 ⁇ /g provides products and vulcanized polymer compositions made from second polymer compositions and products comprising such vulcanized poiymer compositions with an increased hysteresis loss and a deteriorated processability.
- the second polymer composition comprising filler in accordance with the present invention can be prepared by kneading the above-described first polymer composition (containing at least one modified macromolecular compound according to the invention as defined above and including oil-containing first polymer composition varieties), unmodified polymers or polymers modified in a manner other than according to the invention (including oil extended varieties) and filler(s) (carbon black, silica, carbon-silica dual-phase filler, etc..) and optionally processing aids, oils, silane coupling agents and other additives, in a kneader at 140 C to 180 °C, to form a "first stage" second composition containing filler.
- first polymer composition containing at least one modified macromolecular compound according to the invention as defined above and including oil-containing first polymer composition varieties
- unmodified polymers or polymers modified in a manner other than according to the invention including oil extended varieties
- filler(s) carbon black, silica, carbon-silica dual-phase filler, etc
- second polymer compositions in accordance with the present invention can be prepared by kneading a polymer composition already containing at least one of the fillers (for example, carbon black, silica, carbon-silica dual-phase filler, etc.). formed as result of the modified macromolecular compound manufacturing process, and optionally processing aids, oils, silane coupling agents, fillers (for example, carbon black, silica, carbon-silica dual- phase filler, etc.) and other additives in a kneader at 140 °C to 1 0 °C to form a "first stage" second polymer composition containing filler.
- the formation of the "first stage" second polymer composition may comprise one or more mixing steps, preferably 2 to 7 mixing steps.
- the silane sulfide modified macromolecular compound contained in the second polymer composition allows mixing at the given process conditions at a relatively low Mooney viscosity compared with a corresponding polymer composition not containing the silane sulfide modified macromolecular compound, the mixing throughput can be increased and/or the energy consumption per time unit can be decreased.
- vulcanizing agents such as sulfur, vulcanizing accelerators, optionally zinc oxide and the like are added to the "first stage" second composition, and the resulting mixture, also referred to as “second stage” second composition, is blended using a Brabender mixer, Banbury mixer or open roll mill to form the desired shape.
- the "second stage” second composition is then vulcanized at 140 °C to 180 °C to obtain a vulcanized article, also referred to as "vulcanized polymer composition” or "vulcanized elastomeric polymer composition”.
- vulcanizing agents such as sulfur, vulcanizing accelerators, optionally zinc oxide and the like can be added to the aforementioned first polymer composition, and the resulting mixture is blended using a Brabender mixer, Banbury mixer or open roll mill to form the desired shape, and the mixture is vulcanized at 140 °C to 180 °C, to obtain a vulcanized article, also referred to as "vulcanized polymer composition” or as “vulcanized elastomeric polymer composition",
- the "vulcanized elastomeric polymer compositions" of the present invention exhibit low rolling resistance, low dynamic heat build-up and superior wet skid performance. They are well suited for use in preparing tires, tire treads, side walls, and tire carcasses as well as other industrial products such as belts, hoses, vibration dampers and footwear components.
- the living anionic elastomeric polymer is produced ("living polymer").
- a portion, or all, of the living polymer is modified with the silane sulfide modifier of the present invention to produce the modified macromolecular compound of the invention.
- Non- modified polymer may be produced as well in the reaction.
- the resulting polymer composition comprises both the modified macromolecular compound(s) of the invention and further modified or n on -modified polymers ("further polymer").
- Examples of the living anionic elastomeric polymer to be modified in accordance with the present invention, and of the further polymers include homopolymers of conjugated dienes, especially butadiene or isoprene, and random or block co- and terpolymers of at least one conjugated diene, especially butadiene or isoprene, with at least one conjugated diene or with at least one aromatic a-olefin, and especially styrene and 4-methylstyrene, aromatic diolefin, especially divinylbenzene.
- the random copolymerization optionally terpolymerization, of at least one conjugated diene with at least one aromatic a-olefin, and optionally at least one aromatic diolefin or aliphatic a-olefin, and especially butadiene or isoprene with styrene, 4-raethylstyrene and/or divinylbenzene.
- the random copolymerization of butadiene with isoprene especially preferred is the random copolymerization of butadiene with isoprene.
- Examples of the living anionic elastomeric polymer to be modified and of the further polymers include the following: BR - polybutadiene; butadiene/Cl-C4-alkyl acrylate copolymers; IR - polyisoprene; SBR - styrene/butadiene copolymers with styren contents of 1 to 60.
- EPDM ethylene/propylene/diene copolymer
- EPDM represents an ethylene/propylene/diene copolymer.
- the living polymer or further polymer is a polybutadiene.
- the living polymer or further polymer is a butadiene/styrene copolymer (SSBR) prepared in solution.
- SSBR butadiene/styrene copolymer
- the living polymer or further polymer is a isoprene/styrene copolymer (SSIR) prepared in solution.
- SSIR isoprene/styrene copolymer
- the living polymer or further polymer is a butadiene/isoprene copolymer (BRIR) prepared in solution.
- BRIR butadiene/isoprene copolymer
- the living polymer or further polymer is a polyisoprene, including synthetic polyisoprene.
- the living polymer or further polymer is a styrene/butadiene copolymer with a styrene unit content from 1 to 60 weight percent, preferably from 10 to 50 weight percent, based on the total weight of the copolymer.
- the living polymer or further polymer is a styrene/butadiene copolymer with a 1.2-polybutadiene unit content from 5 to 70 weight percent, preferably from 50 to 70, or 5 to 25 weight percent, based on the total weight of polybutadiene unit fraction of the copolymer.
- the living polymer or further polymer is a styrene/isoprene copolymer with a styrene unit content from 1 to 60 weight percent, preferably from 10 to 50 weight percent, based on the total weight of the copolymer.
- the living polymer or further polymer is a styrene/isoprene copolymer with a 1 ,2-polyisoprene unit content from 5 to 70 weight percent, preferably from 50 t 70, or 5 to 25 weight percent, based on the total weight of pol ybutadiene unit fraction of the copolymer.
- the living polymer or further polymer is a butadiene/isoprene copolymer with an isoprene unit content from 0.1 to 70 weight percent, preferably from 5 to 50 weight percent, based on the total weight of the copolymer.
- the living polymer or further polymer is a isobutylene/isoprene copolymer. In another embodiment, the living polymer or further polymer is a partially Jiydrogenated butadiene.
- the living polymer or further polymer is a partially hydrogenated styrene-butadiene copolymer.
- alkyl refers to an aliphatic group.
- the alkyl group may be linear, branched, cyclic or contain a combination of linear, branched and/or cyclic parts, and may be saturated or unsaturated.
- straight chain aliphatic hydrocarbon groups include methyl (Me), ethyl (Et), n-propyl (Pr), n-butyl (Bu), n-pentyl and n-hexyl
- examples of branched aliphatic hydrocarbon groups include isopropyl and tert-butyl.
- aryl refers to an aromatic group which may contain two or more aromatic rings. Examples include phenyls, biphenyls and other benzenoid compounds, each optionally substituted with alkyl, alkoxy or other heteroatoms, such as oxygen-, nitrogen-, sulfur- and/or phosphorous-containing moieties.
- alkoxy is understood to include methoxy (MeO), ethoxy (EtO), propoxy (PrO), butoxy (BuO), isopropoxy (iPrO), isobutoxy (i ' BuO), pentoxy, and the like.
- aralkyl refers to a group containing at least one aromatic ring to an alkyl group.
- (C a -C b ), for example (Ci-Cu), as used herein is intended to mean a range of the number of carbon atoms of from a to b and includes all individual values and subranges from a to b.
- hydrocarbon groups is understood to include any group, including saturated, unsaturated, linear, branched, cyclic and aromatic groups, which only consists of the elements hydrogen and carbon.
- the following Examples are provided in order to further illustrate the invention, and are not to be construed as limiting.
- the examples describe the preparation of silane sulfide modifiers; the preparation and testing of modified (elastomeric) polymers (i.e. polymer compositions containing a silane sulfide modified macromolecular compound of the present invention); and the preparation and testing of mncrosslinked polymer compositions, including the first polymer composition and second, polymer composition, as well as of crosslinked or cured polymer compositions, also referred to as vulcanized polymer compositions. Unless stated otherwise, all parts and percentages are expressed on a weight basis.
- the term “overnight” refers to a time of approximately 16-18 hours, and "room temperature” refers to a temperature of about 20 ⁇ 25°C, The polymerizations were performed under exclusion of moisture and oxygen, in a nitrogen atmosphere.
- the vinyl content in the conjugated diolefin part was additionally determined by 1R absorption spectrum (Morello method, IFS 66 FT-IR spectrometer of Bmker Analytic GmbH).
- the IR samples were prepared using CS 2 as swelling agent.
- Bonded styrene content A calibration curve was prepared by IR absorption spectrum (IR (IFS 66 FT-IR spectrometer of Broker Analytik GmbH). The IR samples were prepared using CS; as swelling agent.). For the IR determination of the bound styrene in styrene- butadiene copolymers, four bands are assessed: a) band for trans- 1,4-polybutadiene units at 966 cm "1 , b) band for cis- 1,4-polybutadiene units at 730 cm "1 , c) band for 1 ,2-polybutadiene units at 910 cm "1 and band for bound styrene (styrene aromatic bond) at 700 cm "1 .
- the band heights are normalized according to the appropriate extinction coefficients and summarized to a total of 100%.
- the normalization is done via ⁇ - and " C-NMR.
- the ID NMR spectra were collected on a BRUKER Avance 200 NMR spectrometer
- Table 1 1 D-NMK acquisition parameters using BRUKER standard pulse sequences 1H-NMR 13C-NMR 29Si- MR
- GPC-Method SEC calibratcc with narrow distri 5uted polystyrene standard.
- Injection volume 100.00 urn (GPC-method B 50.00 ⁇ )
- each GPC-device 3 columns were used in an connected mode.
- the length of each of the columns 300 mm;
- Mw/Mn Polydispersity
- Mw and Mn weight average molecular weight (Mw) and number average molecular weight (Mn)
- Mp3 correspond to the (maximum peak) molecular weight measured at the first, second or third peaks of the GPC curve [the first peak Mpl (lowest molecular weight) is located on the right side of the curve, and the last peak (highest molecular weight) is located on the left side of the curve], respectively.
- Maximum peak molecular weight means the molecular weight of the peak at the position of maximum peak intensity.
- the Mp2 and Mp3 are two or three polymer chains coupled to one macromolecule.
- Mpl is one polymer chain (base molecular weight - no coupling of two or more polymer chains to one macromolecule).
- the total coupling rate represents the sum of the weight fractions of coupled polymers relative to the total polymer weight, including the sum of the weight fractions of all coupled polymer and the uncoupled polymer.
- the individual coupling rate (e.g. two polymer arms coupled corresponding to the peak with peak maximum Mp2) is calculated as depicted below:
- CR(2arms) (Area fraction of peak with peak maximum Mp2)/ ( ⁇ Area fraction of all peaks [Peak with peak maximum Mpl to peak with highest indexed peak maximum]).
- (-Si-OMe) signal at 3.3-3.5.
- ppm and
- (- SiMe 3 ) signal at 0.1 -0.2 ppm.
- Mn number average molecular weight
- Modification efficiency with sulfanylsilan.es was also determined via sulfur content as sulfate.
- the procedure required combustion of the sample in an automatic oven (Combustor 02 of the company GAM.AB, Germany, Bad Durrenberg) followed by absorption of the flue gas in water with 0.1% hydrazinium hydroxide and subsequent determination of the sulfate concentration with ion chromatography (Metrohm, column: Dionex IonPac AS 12 A.
- Rubber compounds were prepared by combining the constituents listed below in Tables 4, 8 and 12 in. a "380 cc Banbury mixer (Labstation 35 OS from Brabender GmbH&Co KG)," following a two-stage mixing process. Stage 1 - mixed all. components together, except the components of the vulcanization package, to form a stage 1 formulation. Stage 2 - components of vulcanization package were mixed into stage 1 formulation to form a stage 2 formulation. Mooney viscosity was measured according to ASTM D 1646 (2004), with a preheating time of one minute and a rotor operation time of 4 minutes, at a temperature of 100°C [ML1 +4(100 °C)], on a MV 2000E from Alpha. Technologies UK.
- the rubber Mooney viscosity measurement is performed on dry (solvent free) raw polymer (uwvulcanized rubber).
- the Mooney values of the raw polymers are listed in Table 3.
- the Compound Moony viscosity is measured on an tmcured (iinvulcanized) second state polymer compound sample prepai'ed according to Tables 4, 8 and 12.
- the Compound Mooney values are listed, in Tables 7, 1 1 and 15.
- Measurement of unvuleanizcd rheological properties was performed according to ASTM D 5289-95 (reapproved .2001 ), using a rotor-less shear rheometer (MDR 2000 E from Alpha Technologies UK) to measure Scorch Time (TS) and Time to Cure (TC).
- the rheometer measurement was performed at a constant temperature of 160°C on a non-vulcanized second stage polymer formulation, according to Tables 5, 9 and 13.
- the amount of polymer sample is about 4.5 g.
- Sample shape and shape preparation are standardized and defined by the measurement device (MDR 2000 E from Alpha Technologies UK).
- the "TC 50 " and "TC 90" values are the respective times required to achieve 50% and 90% conversion of the vulcanization reaction.
- the torque is measured as a function of time of reaction.
- the vulcanization conversion is automatically calculated, from, the generated torque versus time curve.
- the "TS 1" and '"TS 2" values are the respective times required to increase the torque by 1 dNni and 2 dNm above the respective torque minimum (ML) during vulcanization.
- TS 1 is > 1.5 minute
- TS 2 is > 2.5 minute
- TC 50 is from 3 to 8 minutes
- TC 90 is from 8 to 1 minutes.
- the abrasion, measurement was performed on a vulcanized, second stage polymer formulation according to Tables 5, 9 and 13.
- TS 1 is > 1.5 minute
- TS 2 is > 2.5 minute
- TC 50 is from 3 to 8 minutes
- TC 90 is from 8 to 19 minutes.
- Modifier Preparation Six Modifiers and one Coupling Agent were used in the Examples. The structural formula and method of preparation (or source for obtaining) are provided below. Modifiers 3, 4, 5 and 6 are representative of those of the present invention, whereas modifiers 1 and 2 are for comparative purposes.
- Coupling Agent CI is represented by Formula CI .
- Tie tetrachloride (CI ) was purchased from Aldrich.
- Modifier 1 is represented by Formula Ml below and was prepared as follows: (Formula Ml)
- Modifier 2 is represented by Formula M2 below and was prepared as follows:
- Modifier 3 is represented by Formula M3 below and was prepared as follows:
- Modifier 4 is represented by Formula M4 below and was prepared as follows:
- Modifier 5 is represented by Formula MS below and was prepared as follows:
- Modifier 6 is represented by Formula M below and was prepared as follows:
- Gamma-mercaptopropyl trirnethoxy silane represented, by Formula PR2 below, and was purchased from Cromton GmbH.
- the co-polymerizations were performed, in. a double wall 20 liter steel reactor which was first purged with, nitrogen before the addition, of organic solvent, monomers, polar coordinator compound, initiator compound or other components.
- the polymerization reactor was tempered to 40°C unless stated otherwise.
- the following components were then added in the following order: cyclohexane solvent (9000 grams); butadiene mon.oi.iier, styrene monomer, tetramethylethylene diamine (TMEDA), and the mixture was stirred, for one hour followed by titration with n -butyl lithium to remove traces of moisture or other impurities. Additional n-butyl lithium was added as to start the polymerization, reaction.
- the polymerization was performed for 80 minutes not allowing the polymerization temperature to exceed 60°C. Afterwards, 0.5% of the total butadiene mononter amount was added followed by the addition of either the coupling agent or the modifier (1 , 2, 3, 4, 5 or 6) unless stated otherwise.
- the polymer solution was transferred after 45 minutes into a separate double wall steel reactor containing 100 mL ethanol and 1.4 g of concentrated HCl (concentration 36 %) .and 5 g Irganox 1520 as stabilizer for the polymer. This mixture was stirred for 15 minutes. The resulting polymer solution was than stripped with steam for 1 hour to remove solvent and other volatiles and dried in an oven at 70°C for 30 minutes and another one to three days at room temperature.
- the polymerization components are summarized in Table 2, the resulting polymer characteristics in Table 3, the components of the polymer composition are summarized in Tables 4, 8 and 12 below. Unless otherwise stated, quantities in Table 2 are expressed in mrnols. Quantities in Tables 4, 8 and 12 are expressed in phr (parts per hundred parts of the total rubber component of the composition). Examples of polymers applied to the preparation of polymer compositions according to the recipes shown in Tables 4, 8 and 1 2 under identical process conditions (in the same Banbury mixer, under identical process temperatures and timelines, on the same day by the same operator) are designated with identical letters adjacent to the Example number (e.g. 3 ⁇ , 4 ⁇ ).
- Table 4 Polymer Composition using polymers 9, 10, 1 1 , 12, 13, 14, 15, 16, 19, 20 and 21.
- Table 8 Polymer Composition using polymers 1 , 2, 3, 4, 5, 6, 7 and 8.
- Table 12 Polymer Composition using 17 and 1 8.
- silane sulfide modified macromolecular compounds of Formula PI, P2, P3, P4, P5 or P6, in combination, with silane sulfide modification agents of Formula 1, including Formula 5 and Formula 6, optionally coupling agents and optionally further modification agent(s), preferably selected from those of Formula 7 » form polymers which can be used for the preparation of elastomeric polymer compositions and vulcanized elastomeric polymer compositions.
- the present invention provides (second) polymer compositions comprising silica and/or carbon black as well as silane sulfide modified macromolecular compound(s) which have a lower Mooney viscosity increase during mixing of the individual components as compared with corresponding polymer compositions not containing silane sulfide modified macromolecular compound(s).
- the reduced Mooney viscosity enables an increased mixing throughput or a. reduction of the number of individual mixing steps and also enables an increased extrusion speed of the finalized polymer composition.
- silane sulfide chain end-modification agents according to Formula 1 , Formula 5 and Formula 6 the structures of silane sulfide chain end-modification agents M3, M4, M5 and M6 were confirmed by H- and C-NMR spectroscopy (as described above) and the method of preparation of M3, M4, M5 and M6 is described above. Subsequently, the silane sulfide chain end-modification agents formed were used for the preparation of polymers.
- silane sulfide modification agents led to higher polymer chain coupling degrees than polymers modified with alternative (non-inventive) modification agents in equivalent concentrations.
- inventive modified polymer 3 in Table 3 made by using subject siiane sulfide chain end-modification agent M3, has a coupling degree of 55,9% of the total amount of living polymer chains respectively, while reference polymers 1 .and 2 not being made by using subject modification agents resulted in coupling degrees of 24.9 and 1 .6%, It is generally accepted that an increased polymer coupling degree leads to relatively reduced polymer solution viscosities if the weight average molecular weight (Mw) of the polymer or the corresponding solvent free polymer Mooney viscosity is kept constant.
- Mw weight average molecular weight
- an increased polymer coupling degree enables the manufacturing of polymers of increased weight average molecular weight (Mw) or of increased solvent free polymer Mooney viscosity but identical polymer solution viscosity compared with polymers having a relatively lower coupling degree.
- Polymers of increased weight average molecular weight (Mw) or of high solvent free Mooney viscosity are usually more difficult to process in mechanical mixers, roll mills or during extrusion of filler- containing polymer compositions.
- siiane sulfide modified macromolecular compounds when mixed with silica or carbon black.
- the conditions for example the presence of water at elevated temperature, during mechanical mixing of siiane sulfide modified macromolecular compounds with fillers, such as for example carbon black and silica, lead to the cleavage of Si-S and Sn-S bonds present in the silane-sulfide modified macromolecular compounds of the present invention.
- siiane sulfide modified macromolecular compounds having a relatively high coupling degree are transformed during mechanical mixing with fillers into siiane sulfide modified macromolecular compounds having a relatively lower coupling degree and/or linear modified macromolecular compounds of relatively lower weight average molecular weight (Mw) or relatively lower Mooney viscosity value. Therefore, the processing of siiane sulfide modified macromolecular compounds during formation of polymer-filler compositions in mechanical mixers, as well as roll milling or extrusion of polymer-filler compositions is significantly simplified, and the mixing, roll milling or extrusion throughput can be increased.
- Mw weight average molecular weight
- the macromolecular compounds of the invention are comprised in a first composition.
- the first compositions may be converted into "second polymer compositions" (first stage mixing and second stage mixing according to Tables 4, 8 and 12 by addition of carbon black or silica filler to the modified macromolecular compound of the invention), then further converted into a vulcanized polymer composition, which is formed, for example, if the second stage mixing result according to Tables 4, 8 and 12 is cured at 160°C for 20 min.
- the second polymer compositions (as listed in Tables 5, 9 and 13) and the vulcanized polymer compositions (as listed in Tables 6, 10 and 14), which are prepared under identical conditions at the same day by the identical operator, are identified with a capital letter, e.g.
- vulcanized polymer composition is reflected by the polymer number, e.g. 1 , 2, etc..
- polymer number e.g. 1 , 2, etc.
- vulcanized polymer composition series such asl A, 2A, 3A and 4A which can be directly compared with each other
- the second polymer compositions based on macromolecular compounds made by using silane sulfide modifiers of the invention have relatively lower Mooney viscosity increase, calculated as the difference of the Mooney viscosity of the second polymer composition comprising silane sulfide modified macromolecular compound and carbon black (also referred to as Compound Mooney in Tables 7 and 11) and of the Mooney viscosity of the silane sulfide modified macromolecular compound (also referred to as Polymer Mooney), as compared to corresponding polymer compositions comprising silica or carbon black and with reference polymers not containing silane sulfide modified macromolecular compounds.
- the second polymer compositions according to examples 16G and HE in Table 7 contain carbon black filler as shown in Table 4. More particular, the Mooney viscosity increase resulting from the addition of carbon black to a polymer was reduced by 25.6 or 37.5 Mooney units (MU), respectively, when reference polymers 14G or 15G were replaced by silane sulfide modified composition 16G. Furthermore, the Mooney viscosity increase which results when carbon black is added to a polymer was reduced by 24.7 or 27.3 MU, respectively, when reference polymers 9E or 10E were replaced by silane sulfide modified composition HE.
- the second polymer compositions according to example 3A and 8C in Table 1 1 contain silica filler as shown in Table 8.
- the Mooney viscosity increase which results when silica is added to a polymer was reduced by 17.1 or 16.1 MU, respectively, when reference polymers 1A or 2A were replaced by silane sulfide modified polymer 3A. Furthermore, the Mooney viscosity increase which results when silica is added to a polymer was reduced by 6.1 MU, when reference polymer 7C was replaced by silane sulfide modified polymer 8C.
- silane sulfide modified macromolecular compounds compared with reference polymers not containing the silane sulfide modification, enables the production of vulcanized polymer compositions according to Option 2 having at least one of the properties lower "Tan ⁇ at 60°C” values, higher "Tan ⁇ at 0°C” values and higher "Tan ⁇ at -10°C” values improved, while the other properties are at a comparable level.
- the processes of the present invention combining A) polymerization initiator compounds with B) a silane sulfide modifier, optionally C) coupling agents and optionally D) other modification agent, provides an increased degree of polymer modification and an improved performance.
- vulcanized carbon black-containing polymer compositions based on polymers made with the sslane sulfide modifiers of the invention have relatively lower (or reduced) values for tan ⁇ at 60°C; relatively higher (or increased) values for tan ⁇ at 0°C; relatively higher (or increased) values for tan ⁇ at -10°C and relatively decreased tire heat built up, as compared with am elastomeric vulcanized polymer composition, based on other polymers (see example 12F in Tables 5 and 6),
- Exemplary vulcanized composition 13F, based on silane sulfide modified polymer 13, obtained with silane sulfide modifier M4 of the invention and with further modifier Ml has a heat built up value of 85.5°C, a tan ⁇ value at 60°C of 0.079, a tan ⁇ value at 0°C of 0.730 and a tan 5 value at -10°C of
- the vulcanized silica-containing polymer compositions based on polymers made with the silane sulfide modifier of the invention have relatively higher (or increased) values for tan ⁇ at -10°C .and relatively decreased tire heat built up, when compared with an elastomeric vulcanized polymer compositions based on other polymers (see example 1A and 2A in Tables 9 and 10).
- Exemplary vulcanized composition 3 A based on silane sulfide modified polymer 3, obtained with silane sulfide modifier M3 of the invention, has a heat built up value of 88.4°C, and a tan ⁇ value at -10°C of 0.571, while vulcanized compositions 1A and 2 A, both based on non-modified polymers 1 and 2, modified with other modifier Ml or modifiers Ml and M2, have a relatively higher heat built up value of 90.6°C and 95.5°C and a relatively lower tan ⁇ value at -10°C of 0.564 and 0.548, respectively.
- modified elastomeric polymers lies in the preparation of elastomeric polymer compositions, which again, are specifically used for tire treads and which can have one or more of the following key characteristics; reduced viscosity increase during manufacture; reduced rolling resistance; relatively decreased tire heat built up; increased wet grip; increased ice skid.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Addition Polymer Or Copolymer, Post-Treatments, Or Chemical Modifications (AREA)
Abstract
Description









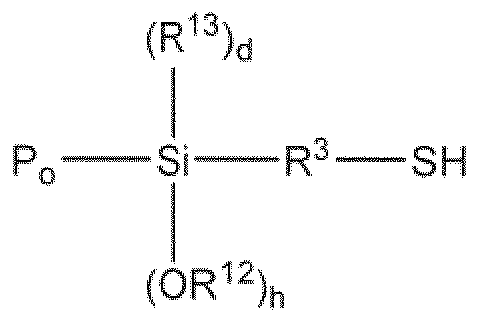
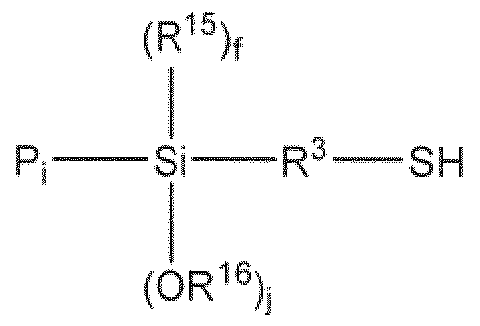

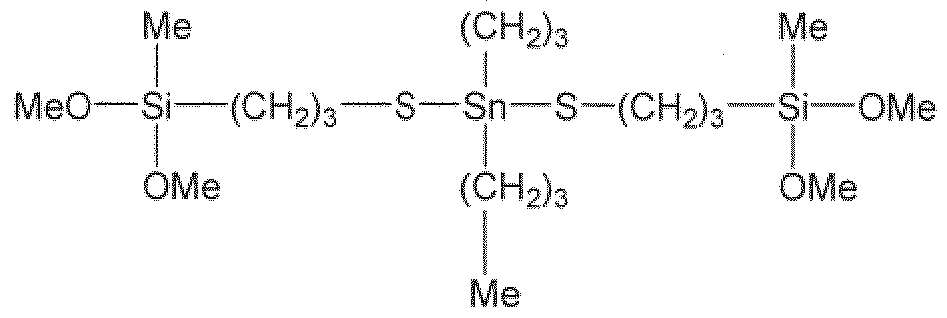




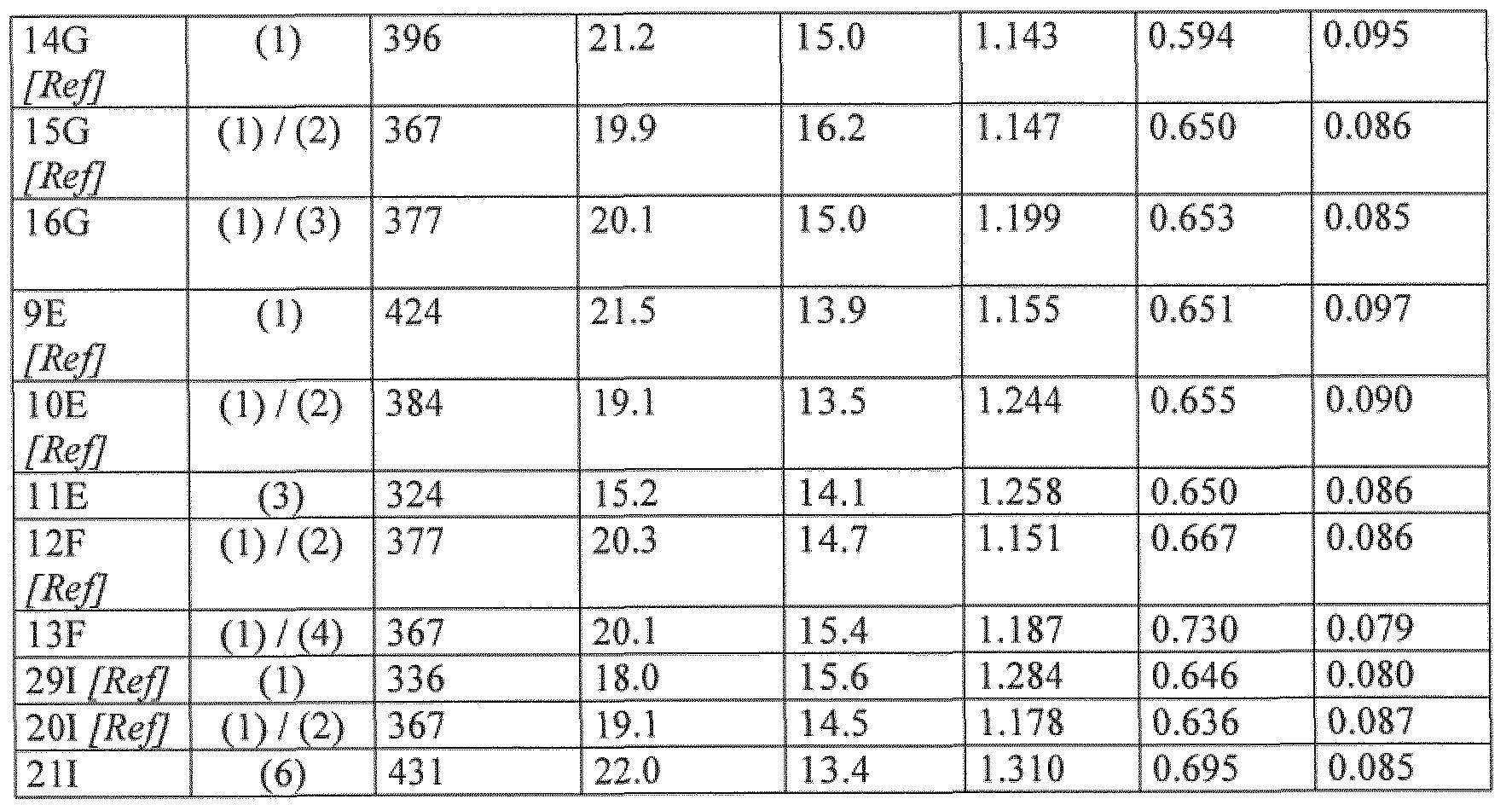





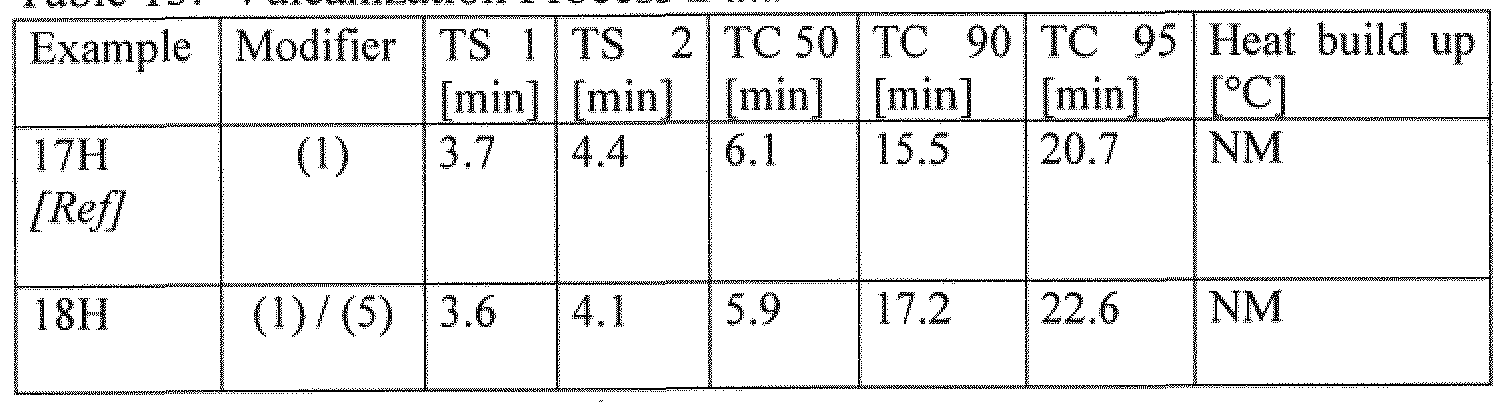

Claims


Priority Applications (15)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/EP2012/068120 WO2014040639A1 (en) | 2012-09-14 | 2012-09-14 | Silane sulfide modified elastomeric polymers |
| SG11201501777XA SG11201501777XA (en) | 2012-09-14 | 2012-09-14 | Silane sulfide modified elastomeric polymers |
| MX2015003424A MX356323B (en) | 2012-09-14 | 2012-09-14 | Silane sulfide modified elastomeric polymers. |
| BR112015005407A BR112015005407A2 (en) | 2012-09-14 | 2012-09-14 | silane sulfide modifier, method for making a silane sulfide modifier, silane sulfide modified elastomeric macromolecular compound, method for making a silane sulfide modified elastomeric macromolecular compound, polymer composition, vulcanized polymer composition, method for making a vulcanized polymer composition and article |
| JP2015531470A JP2015529735A (en) | 2012-09-14 | 2012-09-14 | Silane sulfide modified elastomeric polymer |
| HUE12761728A HUE039558T2 (en) | 2012-09-14 | 2012-09-14 | Silane sulfide modified elastomeric polymers |
| EP12761728.0A EP2895494B1 (en) | 2012-09-14 | 2012-09-14 | Silane sulfide modified elastomeric polymers |
| CN201280075795.1A CN105008377B (en) | 2012-09-14 | 2012-09-14 | Silane vulcanized object modified elastomer polymer |
| RU2015113563A RU2617403C2 (en) | 2012-09-14 | 2012-09-14 | Silane-sulfide modified elastomeric polymers |
| ES12761728.0T ES2687689T3 (en) | 2012-09-14 | 2012-09-14 | Silane sulfide modified elastomeric polymers |
| PL12761728T PL2895494T3 (en) | 2012-09-14 | 2012-09-14 | Silane sulfide modified elastomeric polymers |
| KR1020157007569A KR101972633B1 (en) | 2012-09-14 | 2012-09-14 | Silane sulfide modified elastomeric polymers |
| US14/427,025 US9586980B2 (en) | 2012-09-14 | 2012-09-14 | Silane sulfide modified elastomeric polymers |
| TW102130575A TWI583691B (en) | 2012-09-14 | 2013-08-27 | Silane sulfide modified elastomeric polymers |
| SA113340846A SA113340846B1 (en) | 2012-09-14 | 2013-09-12 | Silane sulfide modified elastomeric polymers |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/EP2012/068120 WO2014040639A1 (en) | 2012-09-14 | 2012-09-14 | Silane sulfide modified elastomeric polymers |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014040639A1 true WO2014040639A1 (en) | 2014-03-20 |
Family
ID=46881056
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2012/068120 WO2014040639A1 (en) | 2012-09-14 | 2012-09-14 | Silane sulfide modified elastomeric polymers |
Country Status (15)
| Country | Link |
|---|---|
| US (1) | US9586980B2 (en) |
| EP (1) | EP2895494B1 (en) |
| JP (1) | JP2015529735A (en) |
| KR (1) | KR101972633B1 (en) |
| CN (1) | CN105008377B (en) |
| BR (1) | BR112015005407A2 (en) |
| ES (1) | ES2687689T3 (en) |
| HU (1) | HUE039558T2 (en) |
| MX (1) | MX356323B (en) |
| PL (1) | PL2895494T3 (en) |
| RU (1) | RU2617403C2 (en) |
| SA (1) | SA113340846B1 (en) |
| SG (1) | SG11201501777XA (en) |
| TW (1) | TWI583691B (en) |
| WO (1) | WO2014040639A1 (en) |
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN104592315A (en) * | 2014-12-29 | 2015-05-06 | 北京彤程创展科技有限公司 | Siloxy thio-metal compound nanometer material and preparation method thereof |
| EP3059099A1 (en) | 2015-02-18 | 2016-08-24 | Trinseo Europe GmbH | A polymer blend for a tire |
| EP3059240A1 (en) | 2015-02-18 | 2016-08-24 | Trinseo Europe GmbH | Multivinylaminosilanes as branching agents for functionalized elastomeric polymers |
| EP3059256A1 (en) | 2015-02-18 | 2016-08-24 | Trinseo Europe GmbH | A functionalized polymer blend for a tire |
| EP3159346A1 (en) | 2015-10-21 | 2017-04-26 | Trinseo Europe GmbH | Aminosilane-functionalized dienes for use in functionalization of elastomeric polymers |
| WO2017116263A1 (en) * | 2015-12-29 | 2017-07-06 | Public Joint Stock Company "Sibur Holding" | Method for producing rubbers having reduced cold flow |
| EP3241853A1 (en) | 2016-05-04 | 2017-11-08 | Trinseo Europe GmbH | Elastomeric polymers with thioether modified spine |
| WO2017190859A1 (en) | 2016-05-04 | 2017-11-09 | Continental Reifen Deutschland Gmbh | Rubber mixture, vulcanizate of the rubber mixture, and vehicle tires |
| WO2017211823A1 (en) | 2016-06-07 | 2017-12-14 | Pirelli Tyre S.P.A. | Tire for vehicle wheels |
| WO2017211876A1 (en) | 2016-06-07 | 2017-12-14 | Trinseo Europe Gmbh | Rubber composition |
| WO2017216344A1 (en) | 2016-06-17 | 2017-12-21 | Trinseo Europe Gmbh | Silane-mediated enhancement of rubber storage stability |
| JP2018507282A (en) * | 2015-01-14 | 2018-03-15 | トリンゼオ ヨーロッパ ゲゼルシャフト ミット ベシュレンクテル ハフツング | Functionalized elastic polymer composition, method for producing the same, and crosslinked rubber composition thereof |
| US9938305B2 (en) | 2014-07-14 | 2018-04-10 | Trinseo Europe Gmbh | Aminosilyl-substituted diarylethene compounds for anionic polymerisation |
| US10077279B2 (en) | 2013-10-18 | 2018-09-18 | Trinseo Europe Gmbh | Vinylsilanes for use in functionalized elastomeric polymers |
| EP3434699A1 (en) | 2017-07-27 | 2019-01-30 | Trinseo Europe GmbH | Use of specific aminosilyl monomers in the manufacture of rubber |
| US11065914B2 (en) | 2015-04-30 | 2021-07-20 | Bridgestone Americas Tire Operations, Llc | Rubber-covered textile cords, tires containing same, and related methods |
| EP4186911A1 (en) | 2021-11-30 | 2023-05-31 | Trinseo Europe GmbH | Amine-containing vinyldisiloxanes in the manufacture of elastomeric polymers |
| WO2023227763A1 (en) | 2022-05-27 | 2023-11-30 | Arlanxeo Deutschland Gmbh | Rubber compounds of functionalized conjugated diene rubbers and silica fillers |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE102018214229A1 (en) * | 2018-07-30 | 2020-01-30 | Evonik Operations Gmbh | Thioether silanes, process for their preparation and their use |
| US11884760B2 (en) * | 2019-12-12 | 2024-01-30 | Asahi Kasei Kabushiki Kaisha | Production method for branched conjugated diene-based polymer, production method for rubber composition, production method for tire, branched conjugated diene-based polymer, and branched conjugated diene-based polymer composition |
| CN111763300B (en) * | 2020-07-03 | 2021-03-30 | 武汉长盈鑫科技有限公司 | Preparation method of UV resin with surface rapidly cured |
| CN111848666B (en) * | 2020-07-03 | 2021-06-08 | 武汉长盈鑫科技有限公司 | Catalyst for reaction of isocyanate and sulfydryl and preparation method thereof |
| RU2767539C1 (en) * | 2020-10-25 | 2022-03-17 | Публичное акционерное общество «СИБУР Холдинг» | Block copolymer composition and method for its preparation |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998028256A1 (en) * | 1996-12-20 | 1998-07-02 | Ciba Specialty Chemicals Holding Inc. | Transesterification catalysts fixed to solid support materials |
| US6229036B1 (en) * | 1998-09-29 | 2001-05-08 | Degussa-Hüls Aktiengesellschaft | Sulfanylsilanes |
| US20050124740A1 (en) * | 2003-11-21 | 2005-06-09 | Degussa Ag | Rubber mixtures |
| WO2007047943A2 (en) * | 2005-10-19 | 2007-04-26 | Dow Global Technologies Inc. | Silane-sulfide chain end modified elastomeric polymers |
| WO2009148932A1 (en) * | 2008-06-06 | 2009-12-10 | Dow Global Technologies Inc. | Modified elastomeric polymers |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN1241174A (en) * | 1996-12-20 | 2000-01-12 | 西巴特殊化学品控股有限公司 | Transesterification catalysts fixed to solid support materials |
| DE19860439C1 (en) * | 1998-12-28 | 2000-07-06 | Degussa | Process for the preparation of silylalkylthiols |
| DE10122269A1 (en) * | 2001-05-08 | 2002-11-21 | Degussa | Silane-modified biopolymer, bio-oligomeric, oxidic or silicate filler, process for its production and its use |
| DE10327624B3 (en) | 2003-06-20 | 2004-12-30 | Degussa Ag | Organosilicon compounds, process for their preparation, and their use |
| DE102006008670A1 (en) * | 2006-02-24 | 2007-08-30 | Degussa Gmbh | Filled styrene-butadiene mix for molding, tire, tire tread, cable or roller covering, hose, drive or conveyor belt, tire, shoe sole, sealing ring or damping element contains organo-silane polysulfide with long-chain alkyl-polyether groups |
-
2012
- 2012-09-14 SG SG11201501777XA patent/SG11201501777XA/en unknown
- 2012-09-14 RU RU2015113563A patent/RU2617403C2/en not_active IP Right Cessation
- 2012-09-14 JP JP2015531470A patent/JP2015529735A/en active Pending
- 2012-09-14 US US14/427,025 patent/US9586980B2/en active Active
- 2012-09-14 WO PCT/EP2012/068120 patent/WO2014040639A1/en active Application Filing
- 2012-09-14 ES ES12761728.0T patent/ES2687689T3/en active Active
- 2012-09-14 PL PL12761728T patent/PL2895494T3/en unknown
- 2012-09-14 CN CN201280075795.1A patent/CN105008377B/en active Active
- 2012-09-14 BR BR112015005407A patent/BR112015005407A2/en not_active Application Discontinuation
- 2012-09-14 EP EP12761728.0A patent/EP2895494B1/en active Active
- 2012-09-14 MX MX2015003424A patent/MX356323B/en active IP Right Grant
- 2012-09-14 HU HUE12761728A patent/HUE039558T2/en unknown
- 2012-09-14 KR KR1020157007569A patent/KR101972633B1/en active IP Right Grant
-
2013
- 2013-08-27 TW TW102130575A patent/TWI583691B/en not_active IP Right Cessation
- 2013-09-12 SA SA113340846A patent/SA113340846B1/en unknown
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1998028256A1 (en) * | 1996-12-20 | 1998-07-02 | Ciba Specialty Chemicals Holding Inc. | Transesterification catalysts fixed to solid support materials |
| US6229036B1 (en) * | 1998-09-29 | 2001-05-08 | Degussa-Hüls Aktiengesellschaft | Sulfanylsilanes |
| US20050124740A1 (en) * | 2003-11-21 | 2005-06-09 | Degussa Ag | Rubber mixtures |
| WO2007047943A2 (en) * | 2005-10-19 | 2007-04-26 | Dow Global Technologies Inc. | Silane-sulfide chain end modified elastomeric polymers |
| WO2009148932A1 (en) * | 2008-06-06 | 2009-12-10 | Dow Global Technologies Inc. | Modified elastomeric polymers |
Non-Patent Citations (1)
| Title |
|---|
| ROSALES A ET AL: "Synthesis of dibutyl tin di [(3-thiopropyl) trimethoxysilane and its evaluation as thermal stabilizer for PVC", POLYMER DEGRADATION AND STABILITY, BARKING, GB, vol. 63, no. 3, 1 March 1999 (1999-03-01), pages 359 - 363, XP004294608, ISSN: 0141-3910, DOI: 10.1016/S0141-3910(98)00112-8 * |
Cited By (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10077279B2 (en) | 2013-10-18 | 2018-09-18 | Trinseo Europe Gmbh | Vinylsilanes for use in functionalized elastomeric polymers |
| US9938305B2 (en) | 2014-07-14 | 2018-04-10 | Trinseo Europe Gmbh | Aminosilyl-substituted diarylethene compounds for anionic polymerisation |
| CN104592315A (en) * | 2014-12-29 | 2015-05-06 | 北京彤程创展科技有限公司 | Siloxy thio-metal compound nanometer material and preparation method thereof |
| JP2018507282A (en) * | 2015-01-14 | 2018-03-15 | トリンゼオ ヨーロッパ ゲゼルシャフト ミット ベシュレンクテル ハフツング | Functionalized elastic polymer composition, method for producing the same, and crosslinked rubber composition thereof |
| US10344147B2 (en) | 2015-02-18 | 2019-07-09 | Trinseo Europe Gmbh | Functionalized polymer blend for a tire |
| US10858377B2 (en) | 2015-02-18 | 2020-12-08 | Trinseo Europe Gmbh | Multivinylaminosilanes as branching agents for functionalized elastomeric polymers |
| WO2016131913A1 (en) | 2015-02-18 | 2016-08-25 | Trinseo Europe Gmbh | A functionalized polymer blend for a tire |
| EP3059256A1 (en) | 2015-02-18 | 2016-08-24 | Trinseo Europe GmbH | A functionalized polymer blend for a tire |
| EP3059240A1 (en) | 2015-02-18 | 2016-08-24 | Trinseo Europe GmbH | Multivinylaminosilanes as branching agents for functionalized elastomeric polymers |
| EP3059099A1 (en) | 2015-02-18 | 2016-08-24 | Trinseo Europe GmbH | A polymer blend for a tire |
| US11065914B2 (en) | 2015-04-30 | 2021-07-20 | Bridgestone Americas Tire Operations, Llc | Rubber-covered textile cords, tires containing same, and related methods |
| EP3159346A1 (en) | 2015-10-21 | 2017-04-26 | Trinseo Europe GmbH | Aminosilane-functionalized dienes for use in functionalization of elastomeric polymers |
| WO2017067877A1 (en) | 2015-10-21 | 2017-04-27 | Trinseo Europe Gmbh | Aminosilane-functionalized dienes for use in functionalization of elastomeric polymers |
| RU2686097C1 (en) * | 2015-12-29 | 2019-04-24 | Публичное акционерное общество "СИБУР Холдинг" | Method of producing rubber with low cold flow |
| WO2017116263A1 (en) * | 2015-12-29 | 2017-07-06 | Public Joint Stock Company "Sibur Holding" | Method for producing rubbers having reduced cold flow |
| WO2017190859A1 (en) | 2016-05-04 | 2017-11-09 | Continental Reifen Deutschland Gmbh | Rubber mixture, vulcanizate of the rubber mixture, and vehicle tires |
| DE102016207744A1 (en) | 2016-05-04 | 2017-11-09 | Continental Reifen Deutschland Gmbh | Rubber mixture, vulcanizate of the rubber mixture and vehicle tires |
| WO2017191174A1 (en) | 2016-05-04 | 2017-11-09 | Trinseo Europe Gmbh | Elastomeric polymers having a thioether-modified backbone |
| EP3241853A1 (en) | 2016-05-04 | 2017-11-08 | Trinseo Europe GmbH | Elastomeric polymers with thioether modified spine |
| CN109195810A (en) * | 2016-06-07 | 2019-01-11 | 倍耐力轮胎股份公司 | Tire for wheel |
| WO2017211823A1 (en) | 2016-06-07 | 2017-12-14 | Pirelli Tyre S.P.A. | Tire for vehicle wheels |
| CN109195810B (en) * | 2016-06-07 | 2021-02-02 | 倍耐力轮胎股份公司 | Tyre for vehicle wheels |
| WO2017211876A1 (en) | 2016-06-07 | 2017-12-14 | Trinseo Europe Gmbh | Rubber composition |
| US11732115B2 (en) | 2016-06-07 | 2023-08-22 | Pirelli Tyre S.P.A. | Tire for vehicle wheels |
| WO2017216344A1 (en) | 2016-06-17 | 2017-12-21 | Trinseo Europe Gmbh | Silane-mediated enhancement of rubber storage stability |
| WO2019020752A1 (en) | 2017-07-27 | 2019-01-31 | Trinseo Europe Gmbh | Use of specific aminosilyl monomers in the manufacture of rubber |
| EP3434699A1 (en) | 2017-07-27 | 2019-01-30 | Trinseo Europe GmbH | Use of specific aminosilyl monomers in the manufacture of rubber |
| US10954330B2 (en) | 2017-07-27 | 2021-03-23 | Trinseo Europe Gmbh | Use of specific aminosilyl monomers in the manufacture of rubber |
| EP4186911A1 (en) | 2021-11-30 | 2023-05-31 | Trinseo Europe GmbH | Amine-containing vinyldisiloxanes in the manufacture of elastomeric polymers |
| WO2023099462A1 (en) | 2021-11-30 | 2023-06-08 | Trinseo Europe Gmbh | Amine-containing vinyldisiloxanes in the manufacture of elastomeric polymers |
| WO2023227763A1 (en) | 2022-05-27 | 2023-11-30 | Arlanxeo Deutschland Gmbh | Rubber compounds of functionalized conjugated diene rubbers and silica fillers |
Also Published As
| Publication number | Publication date |
|---|---|
| PL2895494T3 (en) | 2018-12-31 |
| TWI583691B (en) | 2017-05-21 |
| KR20150054857A (en) | 2015-05-20 |
| MX356323B (en) | 2018-05-23 |
| EP2895494A1 (en) | 2015-07-22 |
| BR112015005407A2 (en) | 2017-07-04 |
| US20150322098A1 (en) | 2015-11-12 |
| HUE039558T2 (en) | 2019-01-28 |
| CN105008377A (en) | 2015-10-28 |
| CN105008377B (en) | 2018-07-06 |
| JP2015529735A (en) | 2015-10-08 |
| MX2015003424A (en) | 2015-09-23 |
| SA113340846B1 (en) | 2015-12-17 |
| RU2015113563A (en) | 2016-11-10 |
| KR101972633B1 (en) | 2019-04-25 |
| RU2617403C2 (en) | 2017-04-25 |
| ES2687689T3 (en) | 2018-10-26 |
| EP2895494B1 (en) | 2018-07-18 |
| SG11201501777XA (en) | 2015-05-28 |
| TW201418272A (en) | 2014-05-16 |
| US9586980B2 (en) | 2017-03-07 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US9458253B2 (en) | Amino silane-modified polymers | |
| US9586980B2 (en) | Silane sulfide modified elastomeric polymers | |
| JP6276405B2 (en) | Polymerization initiator | |
| JP6291060B2 (en) | Silane modified elastomeric polymer | |
| US8217103B2 (en) | Silane-sulfide chain end modified elastomeric polymers | |
| US8729167B2 (en) | Modified elastomeric polymers | |
| JP6480490B2 (en) | Silane sulfide modified elastomeric polymer | |
| US10294314B2 (en) | Functionalized elastomeric polymer compositions, their preparation methods and crosslinked rubber compositions thereof | |
| WO2018150043A1 (en) | Metallated benzylsilanes and their use as polymerization initiators | |
| US20150087768A1 (en) | Elastomeric Polymer Compositions |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12761728 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2015531470 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2015/003424 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P335/2015 Country of ref document: AE |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20157007569 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012761728 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14427025 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 2015113563 Country of ref document: RU Kind code of ref document: A |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112015005407 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112015005407 Country of ref document: BR Kind code of ref document: A2 Effective date: 20150311 |