WO2014027199A1 - Fak and flt3 inhibitors - Google Patents
Fak and flt3 inhibitors Download PDFInfo
- Publication number
- WO2014027199A1 WO2014027199A1 PCT/GB2013/052163 GB2013052163W WO2014027199A1 WO 2014027199 A1 WO2014027199 A1 WO 2014027199A1 GB 2013052163 W GB2013052163 W GB 2013052163W WO 2014027199 A1 WO2014027199 A1 WO 2014027199A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- mmol
- give
- phenyl
- etoac
- amino
- Prior art date
Links
- 0 *CN(CC1)CCN1I Chemical compound *CN(CC1)CCN1I 0.000 description 12
- TXIOGJHPPVXTOY-UHFFFAOYSA-N CCN1CCN(C)CC1 Chemical compound CCN1CCN(C)CC1 TXIOGJHPPVXTOY-UHFFFAOYSA-N 0.000 description 2
- OLOIFCYZWOTWRO-UHFFFAOYSA-N CC(C)(C)OC(N(CCc1c2)Cc1ccc2N)=O Chemical compound CC(C)(C)OC(N(CCc1c2)Cc1ccc2N)=O OLOIFCYZWOTWRO-UHFFFAOYSA-N 0.000 description 1
- LCJRDURRRLSHJO-UHFFFAOYSA-N CC(c(cc1)ccc1[N+]([O-])=O)=C Chemical compound CC(c(cc1)ccc1[N+]([O-])=O)=C LCJRDURRRLSHJO-UHFFFAOYSA-N 0.000 description 1
- OQFMXDAKDMRDTK-UHFFFAOYSA-N CC1OCCN(CC(CCC2)CN2N)C1 Chemical compound CC1OCCN(CC(CCC2)CN2N)C1 OQFMXDAKDMRDTK-UHFFFAOYSA-N 0.000 description 1
- QCJILPSFGUEKAE-UHFFFAOYSA-N CN(CC1)CCN1c1cccc(Nc2nc(CCc3c(CC(O)=O)cccc3)c(C(F)(F)F)cn2)c1 Chemical compound CN(CC1)CCN1c1cccc(Nc2nc(CCc3c(CC(O)=O)cccc3)c(C(F)(F)F)cn2)c1 QCJILPSFGUEKAE-UHFFFAOYSA-N 0.000 description 1
- CGIQVJDRLWRQEU-UHFFFAOYSA-N CN(CCC1)CC1c(cc1)ccc1N Chemical compound CN(CCC1)CC1c(cc1)ccc1N CGIQVJDRLWRQEU-UHFFFAOYSA-N 0.000 description 1
- ZDZDFCAZHMNHHM-UHFFFAOYSA-N CNC(Cc1c(CCc2c(C(F)(F)F)cnc(Nc3cc(N4CCN(C)CC4)ccc3)n2)cccc1)=O Chemical compound CNC(Cc1c(CCc2c(C(F)(F)F)cnc(Nc3cc(N4CCN(C)CC4)ccc3)n2)cccc1)=O ZDZDFCAZHMNHHM-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
Definitions
- This invention relates to a method of using 2,4,5-substituted pyrimidines that inhibit Focal Adhesion Kinase (FAK), also known as protein tyrosine kinase 2 (PTK2), and F S-like tyrosine kinase (Flt3), also known as Fetal liver kinase 2 (FLK2), Stem ceil kinase 1 (STK1 ) and CD135, for the prevention and/or treatment of Acute Myeloid Leukemia (AML) and other diseases ameliorated by the inhibition of F!t3, or FLt3 and FAK.
- FK Focal Adhesion Kinase
- FLK2 protein tyrosine kinase 2
- Flt3 F S-like tyrosine kinase
- FLK2 Fetal liver kinase 2
- STK1 Stem ceil kinase 1
- CD13 for the prevention and/or treatment of Acute Myeloid Le
- Directional cell migration is important in many physiological and pathological processes including embryonic development, wound healing, angiogenesis, tumour invasion and metastasis.
- Transduction of extracellular signals, that stimulate cells to move directionally may be induced by a number of processes including trans- membrane integrins binding to extra cellular matrix proteins and the action of growth factors (for example EGF, IGF and VEGF) on the extracellular domains of their cognate receptors.
- FAK is a non receptor tyrosine kinase that mediates signals from both trans- membrane integrins and growth factor receptors. FAK has been reported to play a central role in coordinating these diverse extra cellular signals, integrating them in a fashion that results in directional movement of cells through their external environment (Tomar and Schlaepfer. Current Opinion in Ceil Biology: 2009, 21 , 876- 683).
- Integrin clustering or the activation of a growth factor receptor promotes FAK autophosphorylation at Y397.
- a growth factor receptor for example EGFR, IGF-1 R, Her2 and VEGFR
- FAK-Src signaling through cellular apoptosis susceptibility protein leads to the expression of matrix metalioproteases ( MPs) including MP2 and M P9.
- MPs matrix metalioproteases
- FAK-Src activation also promotes cell surface expression of MMP14 via phosphorylation of endophilin A2.
- MP14 then activates MMP2 by cleavage of pro-M P2 to its active form (Siesser and Hanks. Clinical Cancer Research: 2006, 12(11), 3233-3237).
- FAK has been shown to play an important role in ceil survival. Activation of FAK has been shown to result in suppression of anoikis (apopotosis in response to an inappropriate extra cellular matrix environment) (Frisch et al Journal of Cell Biology. 1996, 134(3), 793-799 and Xu et al Cell Growth and Differentiation, 1996, 7(4), 413- 418). Studies have demonstrated that FAK activates multiple downstream pathways to suppress anoikis in both fibroblasts and epithelial ceils (Zouq et al. Journal of Cell Science: 2008, 122, 357-367).
- TAE226 has the structure:
- TAE226 inhibited the phosphorylation of FAK at both Y397 and Y861 sites, inhibited cell growth in a time- and dose-dependent manner, and enhanced docetaxel-mediated growth inhibition by 0- and 20-fold in the taxane-sensitive and taxane-resistant cell lines, respectively.
- FAK inhibition by TAE226 significantly reduced tumour burden in the HeyA8, SKOV3ip1 , and HeyA8- DR models (46-64%) compared with vehicle-treated controls.
- FAK mRNA and/or protein has been reported in numerous human cancers including colorectal cancer (de Heer. European Journal of Surgical Oncology: 2008, 34(11), 1253-1281), prostate cancer (Tremblay, L, W. Hauck, et al. International Journal of Cancer: 1996, 68(2), 164-171), breast cancer (Watermann et al. British Journal of Cancer 2005, 93(6), 694-698) and melanomas (Hess et al. Cancer Research: 2005, 65(21), 9851- 60). Furthermore FAK over expression is frequently correlated with more aggressive phenotypes of these cancers.
- a FAK inhibitor would have application for the reduction of cell adhesion, cell migration, ceil invasion, ceil proliferation and chemo-resistance. Furthermore, a FAK inhibitor would have applicability to induce apoptosis for cells in inappropriate extra cellular matrix environments and reduce angiogenesis.
- FMS-like tyrosine kinase also known as Fetal liver kinase 2 (FLK2), Stem cell kinase 1 (STK1) and CD135, is known as a proto-oncogene.
- the internal tandem duplication mutation of Flt-3 (ITD-Flt3) is a recognized molecular lesion in some forms of acute myeloid leukemia (A ML) and predicts for poor patient prognosis (Chen W, et al., Mol Cancer: (2010) 9, 292; Gu T, et aL PLos One, 201 1 4:6, e19169; Smith C, et al., 2012 Nature 485, 280-263).
- FAK focal adhesion kinase
- splice variants of FAK have also been implicated in poor prognosis AML (Despeaux M, et a!., Stem Ceils (2012) 30, 1597-1610; Recher C, et al., (2004) Cancer Research 64: 3191- 3197; Casanova I, et al., (2008) Int. J. Cancer, 123, 217-226)
- Some patients with AML are predicted to have both ITD-Flt3 mutations and FAK/FAK splice variant expression.
- Flt3 may also be useful in ameliorating autoimmune diseases, such as multiple sclerosis (Wharternby, K, et al., PNAS (2005), 102(46): 16741-16746).
- the inhibition of F!t3 may also be useful in treating mye!odysp!astic syndrome (MDS) and chronic autoimmune diseases (MDS) and chronic autoimmune diseases (MDS).
- CMPDs myeloproliferative diseases
- VEGFR3 the vascular endothelial growth factor receptor VEGFR3 (Flt4) is over expressed in melanoma patients with metastases in regional lymph nodes (Mouavvad et al. European Journal of Cancer: 2009, 45, 1407-1414). Abnormal expression levels of endogenous receptor tyrosine kinase ligands are also observed in many human cancers. For example, the expression levels of vascular endothelial growth factors C and D (VEGF-C and VEGF-D), ligands of VEGFR3, are significantly correlated with lymphatic metastasis and lymphatic vessel invasion in early-stage invasive cervical carcinoma (Journal of Experimental & Clinical Cancer Research 2009, 28). Accordingly, compounds that inhibit Flt3, or Flt3 and FAK, and/or VEGFR3, would be useful for the treatment of proliferative diseases, such as cancer.
- VEGF-C and VEGF-D vascular endothelial growth factors C and D
- PF-562,271 is described in WO2004/056786, WO2004/056807, WO2005/023780, WO2007/063384 and Roberts et al. Cancer Res 2008, 68(6), 1935-1944.
- PF-573,228 is described in Slack-Davis et al. J. Biol. Chem. 2007, 282(20), 14845- 14852.
- further classes of FAK inhibitors are disclosed in WO2008/129380, WO2008/1 5369, WO2009/105498, US2010/1 13475, WO2009/143389, WO2009/071535, WO2010/055 17,
- the present inventors have discovered a particular class of compounds which are effective as FAK inhibitors, and also inhibit FLt3 (including mutants thereof). These compounds may also inhibit VEGFR3. These compounds may exhibit selectivity for FAK over kinases such as VEGFR1 , IGF-1 (insulin-like growth factor 1 receptor), IR (insulin receptor) and CDKs (cyciin-dependent kinases). Additionally, the compounds of the invention may have enhanced selectivity for the inhibition of cytochrome p450 enzymes, specifically the 2C9 and 3A4 isoforms. Furthermore, the compounds of the invention may be less prone to the formation of adducts with glutathione. In a first aspect, the present invention provides the use of compounds of the following formula (i):
- R 1 is selected from: H and
- R N4 is selected from H and CH 3 ;
- R N7 and R N8 are independently selected from H and CH 3 ;
- R 1 and R 2 are H;
- R 4 is selected from CF 3 , halo, CF 2 H and CN; and R b is selected from g
- R 6 is selected from H, (CHR c1 ) n1 C(0)N(R N13 )Z 1 and (CH 2 ) n2 C(0)OZ 2 ; wherein:
- n1 1 ;
- R C1 is H or e
- R N13 is H or CH 3 ;
- Z is H, CH 3 or OCH 3 ;
- n2 is 1 ;
- Z 2 is CH 3 ;
- R 7 if present, is selected from H, and (CH 2 )miC(0)N(R M1 )Y 1 , wherein:
- ml is 0 or 1 ;
- R M is H
- Y 1 is H, Me or OCH 3 ;
- a second aspect of the present invention provides compounds as described in the first aspect for use in a method of treatment of Acute Myeloid Leukemia or a disease ameliorated by the inhibition of Fit3, or Flt3 and FAK.
- a third aspect of the present invention provides a method of treatment of the human or animal body suffering from Acute Myeloid Leukemia or a disease ameliorated by the inhibition of Flt3, or Fit3 and FAK, comprising administering compounds as described in the first aspect, preferably in the form of a pharmaceutical composition.
- Another aspect of the invention provides a method of inhibiting Fit3, or FAK and Fit3, in vitro or in vivo, comprising contacting a cell with an effective amount of an active compound as described herein.
- Compounds of the present invention may also inhibit VEGFR3.
- Each of the groups R 1 to R 8 will be discussed in more detail below.
- R N/ is either H or methyl.
- R N7 and R N8 are independently selected from H and CH 3 .
- R 2 may have one of the following structures:
- R 1 (discussed above) is not H.
- R 1 and R 2 together form the group ⁇ CH2 ⁇ N(R N12 )-C 2 H4 ⁇ ,
- R 4 is selected from CF 3 , halo (i.e. F, CI, Br, I), CF 2 H and CN.
- the halo group is either CI or Br.
- R 6 is selected from H, (CHR c1 ) n1 C(0)N(R N13 )Z 1 and (CH 2 ) n2 C(G)OZ 2 ; wherein: n1 is 1 ;
- R C1 is H or Me
- R N13 is H or CH 3 ;
- Z 1 is H, CH 3 or OCH 3 ;
- n2 is 1 ;
- Z 2 is CH 3 ;
- R N13 and Z 1 may be CH 3 .
- R 6 is H
- R 7 (discussed below) is not H
- R e is (CHR c ) ril C(0)N(R N6 )Z 1 , it may be selected from: CH 2 C(0)NH 2 , CH 2 C(0)NHCH 3j CH 2 C(0)NHOCH 3> CH 2 C(0)NCH 3 OCH 3j CHCH 3 C(0)NH 2 , CHCH 3 C(0)NHCH 3 , CHCH 3 C(0)NHOCH 3 , and CHCH 3 C(0)NCH 3 OCH 3 ,
- R 6 is (CH 2 ) n2 C(0)OZ 2 , if is CH 2 C(0)OCH 3 .
- R 7 is (CH 2 ) n2 C(0)OZ 2 , if is CH 2 C(0)OCH 3 .
- R 7 is selected from H, and (CH 2 ) m1 C(0)N(R 1 )Y 1 , wherein:
- ml is 0 or 1 ;
- R M1 is H; and Y 1 is H, Me or OCH 3 ;
- R 7 is H
- R B is not H
- R 6 is not H.
- R 7 is (CH 2 ) m1 C(0)N(R M1 )Y 1 , it may be selected from C(0)NH 2 , C(0)NHCH 3 , C(0)NHOCH 3) CH 2 C(0)NH 2 , CH 2 C(0)NHCH 3 and CH 2 C(0)NHOCH 3 .
- a reference to carboxyiic acid also includes the anionic (carboxyiate) form (-COO " ), a salt or solvate thereof, as well as conventional protected forms.
- a reference to an amino group includes the protonated form (-N + HR 1 R 2 ), a salt or solvate of the amino group, for example, a hydrochloride salt, as well as conventional protected forms of an amino group.
- a reference to a hydroxyl group also includes the anionic form (-0 " ), a salt or solvate thereof, as well as conventional protected forms of a hydroxyl group.
- Certain compounds may exist in one or more particular geometric, optical, enantiomeric, diasteriomeric, epimeric, stereoisomeric, tautomeric, conformational, or anomeric forms, including but not limited to, cis- and trans-forms; E- and Z-forms; c-, t-, and r- forms; endo- and exo-forms; R-, S-, and meso-forms; D- and L-forms; d- and l-forms; (+) and (-) forms; keto-, enok and enolate-forms; syn- and anti-forms; synclinal- and anticlinal-forms; a- and ⁇ -forms; axial and equatorial forms; boat-, chair-, twist-, envelope-, and haifchair-forms; and combinations thereof, hereinafter collectively referred to as "isomers” (or "isomeric forms").
- isomers are structural (or constitutional) isomers (i.e. isomers which differ in the connections between atoms rather than merely by the position of atoms in space).
- a reference to a methoxy group, -OCH 3 is not to be construed as a reference to its structural isomer, a hydroxymethyi group, -CH 2 OH,
- a reference to ortho-chiorophenyi is not to be construed as a reference to its structural isomer, meta-chlorophenyl.
- a reference to a class of structures may well include structurally isomeric forms falling within that class
- Ci-7 alky! includes n-propyl and iso-propyi; butyl includes n-, iso-, sec-, and tert- butyl; methoxyphenyl includes ortho-, meta-, and para-methoxyphenyl).
- keto/enol illustrated below
- imine/enamine amide/imino alcohol
- amidine/amidine a compound selected from the group consisting of the following tautomeric forms: keto/enol (illustrated below), imine/enamine, amide/imino alcohol, amidine/amidine,
- H may be in any isotopic form, including 1 H, 2 H (D), and 3 H (T); C may be in any isotopic form, including 2 C, 3 C and 4 C; O may be in any isotopic form, including 16 0 and 18 0; and the like.
- a reference to a particular compound includes ail such isomeric forms, including (wholly or partially) racemic and other mixtures thereof.
- Methods for the preparation (e.g. asymmetric synthesis) and separation (e.g., fractional crystallisation and chromatographic means) of such isomeric forms are either known in the art or are readily obtained by adapting the methods taught herein, or known methods, in a known manner.
- a reference to a particular compound also includes ionic, salt, solvate, and protected forms of thereof, for example, as discussed below.
- a salt may be formed with a suitable cation.
- suitable inorganic cations include, but are not limited to, alkali metal ions such as Na + and K + , alkaline earth cations such as Ca2 + and g 2+ , and other cations such as Al 3' ⁇
- suitable organic cations include, but are not limited to, ammonium ion (i.e., NH 4+ ) and substituted ammonium ions (e.g., NH 3 R + , NH 2 R 2+ , NHR 3+ , NR 4+ ).
- substituted ammonium ions examples include those derived from: ethyiamine, diethyiamine, dicyclohexyiamine, triethylamine, butyiamine, ethylenediamine, ethanoiamine, diethanoiamine, piperazine,
- benzylamine phenyibenzylamine, choline, meglumine, and tromethamine, as well as amino acids, such as lysine and arginine.
- An example of a common quaternary ammonium ion is N(CH 3 ) 4+ . If the compound is cationic, or has a functional group which may be cationic (e.g., -NH 2 may be -NH 3+ ), then a salt may be formed with a suitable anion.
- suitable inorganic anions include, but are not limited to, those derived from the following inorganic acids: hydrochloric, hydrobromic, hydroiodic, sulphuric, sulphurous, nitric, nitrous, phosphoric, and phosphorous.
- Suitable organic anions include, but are not limited to, those derived from the following organic acids: acetic, propionic, succinic, glycolic, stearic, palmitic, lactic, malic, pamoic, tartaric, citric, gluconic, ascorbic, ma!eic, hydroxymaieic, phenylacetic, glutamic, aspartic, benzoic, cinnamic, pyruvic, saiicyciic, sulfanilic, 2- acetyoxybenzoic, fumaric, phenylsulfonic, toiuenesuifonic, methanesulfonic, ethanesulfonic, ethane disulfonic, oxalic, pantothenic, isethionic, valeric, lactobionic, and gluconic.
- Suitable polymeric anions include, but are not limited to, those derived from the following polymeric acids: tannic acid, carboxymethyi cellulose. It may be convenient or desirable to prepare, purify, and/or handle a corresponding solvate of the active compound.
- solvate is used herein in the conventional sense to refer to a complex of solute (e.g. active compound, salt of active compound) and solvent. If the solvent is water, the solvate may be conveniently referred to as a hydrate, for example, a mono-hydrate, a di-hydrate, a tri-hydrate, etc. it may be convenient or desirable to prepare, purify, and/or handle the active compound in a chemically protected form.
- chemically protected form pertains to a compound in which one or more reactive functional groups are protected from undesirable chemical reactions, that is, are in the form of a protected or protecting group (also known as a masked or masking group or a blocked or blocking group).
- a protected or protecting group also known as a masked or masking group or a blocked or blocking group.
- the aldehyde or ketone group is readily regenerated by hydrolysis using a large excess of water in the presence of acid.
- an amine group may be protected, for example, as an amide or a urethane, for example, as: a methyl amide ( ⁇ NHCO ⁇ CH 3 ); a benzyloxy amide (-
- a carboxyiic acid group may be protected as an ester for example, as: an Ci-7 alkyl ester (e.g. a methyl ester; a t-butyl ester); a Ci. 7 haioalkyl ester (e.g., a Ci-7 trihaloaikyi ester); a triC alkylsilyl-d.7 alkyl ester; or a C 5 . 2 o aryi-Ci- 7 alkyl ester (e.g. a benzyl ester; a nitrobenzyl ester); or as an amide, for example, as a methyl amide.
- an Ci-7 alkyl ester e.g. a methyl ester; a t-butyl ester
- a Ci. 7 haioalkyl ester e.g., a Ci-7 trihaloaikyi ester
- a prodrug pertains to a compound which, when metabolised (e.g. in vivo), yields the desired active compound.
- the prodrug is inactive, or less active than the active compound, but may provide advantageous handling, administration, or metabolic properties.
- some prodrugs are esters of the active compound (e.g.
- metaboiically labile esters include those wherein R is C1-7 alkyl (e.g. -Me, -Et); Ci_ aminoaikyi (e.g. aminoethyl; 2-(N,N-diethylamino)ethyl; 2- (4- morpho!ino)ethyl); and acyloxy-Ci_ 7 alkyl (e.g. acyloxymethyi; acyioxyethyi; e.g.
- pivaloyloxymethyl acetoxym ethyl; 1-acetoxyethyl; 1-(1-methoxy-1-methyl)ethyl- carbonxyioxyethyl; 1-(benzoyloxy)ethyl; isopropoxy-carbonyloxymethyl; 1- isopropoxy-carbonyloxyethyl; cyclohexy!-carbonyloxymethy!; 1-cyclohexyl- carbonyloxyethyi; cyclohexyloxy-carbonyloxymethyl; 1-cyciohexyioxy- carbonyloxyethyl; (4-tetrahydropyranyioxy) carbonyioxymethyl; 1 ⁇ (4 ⁇
- prodrugs are activated enzymaticaily to yield the active compound, or a compound which, upon further chemical reaction, yields the active compound.
- the prodrug may be a sugar derivative or other glycoside conjugate, or may be an amino acid ester derivative.
- the selectivity of the compounds for inhibiting FAK and Flt3 over other kinases, such as IGF- R, IR and CDKs can be demonstrated by biochemical assay results (see, for example, the FAK kinase assay and FltS assays described below).
- the compounds of the invention may also be selective over VEGFR1 and/or VEGFR2.
- the compounds of the present invention may also inhibit VEGRF3.
- the selectivity of the compounds for FAK over the inhibition of cytochrome p450 enzymes, specifically the 2C9 and 3A4 isoforms may be determined using standard inhibition assays.
- R is H and R is:
- R is H and R is:
- R N2 is selected from H and Cl . 3 alkyl (i.e. methyl, ethyl, prop-1-yl and prop-2-yl).
- R m is selected from H and methyl.
- R N2 is ethyl.
- R 2 is H and R 1 is: , wherein Ni> is selected from H and C1.3 alkyl (i.e. methyl, ethyl, prop-1-y! and prop-2-yl).
- R N3 is selected from H and methyl.
- R N3 is ethyl.
- R 2 is H and R 1 is:
- R N4 is selected from H and methyl, in these embodiments, it may be preferred that R m is H. nts, R 2 is H and R 1 is:
- R and R are both H or both methyl. In some of these embodiments, it may be preferred that R and R are both H I bodiments, R 2 is H and R 1 is:
- R is selected from H and d. 3 alkyl (i.e. methyl, ethyl, prop-1-yi and prop-2-yl).
- R N9 is H.
- R 2 is H and R 1 is: wherein R N1 ° is selected from H and C1.3 alkyl (i.e. methyl, ethyl prop-1-yl and prop-2-yl).
- R mo is selected from H and methyl. in other embodiments, R 2 is H and R 1 is:
- R N11 is selected from H and Ci_ 3 aikyl (i.e. methyl, ethyl, prop-
- R N i is H.
- R 1 is H and R 2 is:
- R is selected from H and d. 3 aikyl (i.e. methyl, ethyl, prop- 1-yl and prop-2-yi). In these embodiments, it may be preferred that R NS is selected from H and methyl.
- R 1 is H and R 2 is:
- R is selected from H and C1.3 alkyl (i.e. methyl, ethyl, prop- 1-yl and prop-2-yi). in these embodiments, it may be preferred that R NS is selected from H and methyl. r preferred that R 1 is H and R 2 is:
- R N 2 is selected from H and C
- R 4 is selected from CF 3 , CI, Br, CF 2 H , and CN.
- R 4 is selected from CF 3 , CI and CF 2 H.
- R 4 is selected from CF 3 and CI. It may be preferred that R 4 is CF 3 . in some embodiments, it may be preferred that R 5 is a group of the following formulae:
- R 5 may be preferably selected from R 5e and R 5c , and maymore preferably be R 5e .
- R 7 is H and R e is (CHR c 1 ) ri C(0)N(R N6 )Z 1 .
- R 7 is H and R 6 is selected from CH 2 C(0)NH 2 , CH 2 C(0)NHCH 3 , CHCH 3 C(0)NH 2 and CHCH 3 C(0)NHCH 3 .
- R 7 is H and R 6 is selected from CH 2 C(0)NH 2) CHCH 3 C(0)NH 2 and CH 2 C(0)NHCH 3 , and more preferably from CH 2 C(0)NH : CHCH 3 C(0)NH 2 .
- R 6 is H and R 7 is (CH 2 ) m1 C(0)N(R M1 )Y 1 .
- R 6 is H and R 7 is selected from C(0)NH 2 , C(0)NHCH CH 2 C(0)NH 2 and CH 2 C(0)NHCH 3 .
- R 6 is H and R 7 is C(0)NH 2 .
- R 8 is H and R 7 is C(0)NH 2 , R 8 is methyl. in some embodiments, it may be preferred that R 5 is a group of the following formula:
- R ' a is selected from:
- N4 s selected from H and CH.
- R 1 b is selected from:
- R N4 is selected from H and CH 3 ;
- R N ' and R N8 are independently selected from H and CH 3 ;
- R N1 , R N2 , R N3 , R N4 , R NJ , R m , R Ng , R M0 and R 1 apply here as well.
- R N2d is selected from H , methyl and ethyl
- R N3B is selected from H and methyl
- R N4B is H ;
- R N9B is H ;
- R N1 ° is selected from H and methyl.
- Embodiments of the inventions are compounds of the examples, including
- Embodiments of particular interest include compounds 4, 5, 8, 11 and 16. Further embodiments of particular interest include compounds 21 , 22, 25, 31 and 36.
- the compounds of the invention can be prepared employing the following general methods and using procedures described in detail in the experimental section.
- the reaction conditions referred to are illustrative and non-limiting.
- 2,4-dichioro-5-(trifluoromethyl)pyrimidine (G1 ) can be selectively reacted with sodium thiomethoxide in the presence of zinc(l l) chloride to give 2-thiomethyl-4-chioro-5-(trifluoromethyl)pyrimidine (G2), 2-Thiomethyl-4-chloro- 5-(trifluoromethyl)pyrimidine (G2) can be further reacted, for example by conversion to 2-thiomethyl-4-iodo-5-(trifluoromethyl)pyrimidine (G3) under Finkelstein conditions and/or by oxidation with mCPBA to give the corresponding suifone if further differentiation of the 2 and 4-position is required or if additional activation is desirable.
- a catalyst for example palladium on charcoal
- the corresponding 4-piperidine analogues of G8 can be prepared by a sequence of reactions starting with the conversion of commercially available fert-butyl 4- oxopiperidine-1-carboxylate (G10) to vinyl triflate G11. Coupling of G11 in a Suzuki type reaction with (4-nitrophenyl)boronic acid (G12) gives tetrahydropyridine (G13). Subsequent reduction via hydrogenation in the presence of a catalyst, for example palladium on charcoal, gives gives ferf-butyl 4-(4-aminophenyi)piperidine-1- carboxylate (G 4).
- a catalyst for example palladium on charcoal
- G18 G17 The corresponding 4- ⁇ 3-aminophenyl)piperidine analogue of G9 can be prepared by a sequence of reactions starting with the conversion of commercially available tert- butyl 4-oxopiperidine-1-carboxyiate (G10) to vinyl trifiate G11. Coupling of G11 in a Suzuki type reaction with (3-nitrophenyl)boronic acid (G15) gives tetrahydropyridine (G16). Subsequent reduction via hydrogenation in the presence of a catalyst, for example palladium on charcoal, gives iert-butyl 4-(3-aminophenyl)piperidine-1- carboxylate (G 7).
- a catalyst for example palladium on charcoal
- G23 pyridin- 2-ylboronic acid
- G23 2-(4-nitrophenyl)pyridine
- Reduction of G23 with hydrogen in the presence of a catalyst, for example platinum oxide gives 4-(piperidin-2-yl)aniline (G24) which may be protected using Boc anhydride to give fe/t-butyl 2-(4-aminophenyl)piperidine-1-carboxylate (G25).
- ferf-Butyl (1-(4-aminophenyl)piperidin-4-yl)carbamate can be prepared by nucieophilic aromatic substitution of commercially available ferf-butyi piperidin-4- ylcarbamate (G26) and 1-fluoro-4-nitrobenzene (G27) under thermal conditions to give ferf-butyl (1-(4-nitrophenyl)piperidin-4-yl)carbamate (G28). Reduction of G28 with hydrogen in the presence of a catalyst, for example 0% palladium on charcoal gives terf-butyl (1-(4-aminophenyl)piperidin-4-yi)carbamate (G29).
- G36 G37 G38 Commercially available terf-butyl 3-oxopyrroiidine-1-carboxylate (G33) can be converted to a mixture of vinyl triflates (G34) and (G35) in the presence of a triflamide and a suitable base, for example NaH DS. Coupling of the mixture with (4- nitrophenyi)boronic acid (G12) under Suzuki conditions gives dihydropyrroies (G38) and (G37), Reduction of this mixture using hydrogen in the presence of a catalyst, for example 10% palladium on charcoal, gives terf-butyl 3-(4-aminophenyi)pyrrolidine-1- carboxylate (G38). cheme L
- R 9 may then be removed to generate compounds of the formula F10.
- R 9 T S or TES potassium carbonate or tetra-n-butyl ammonium fluoride may be employed to induce this transformation.
- Pyrimidines of the formula F3 may be reacted with terminal acetylenes of the formula F10 to give acetylenes of the formula F11 in a Sonagashira type coupling.
- the acetylene in compounds of the formula F11 may be reduced to an alkane of the formula F26 using hydrogen gas in the presence of a transition metal catalyst.
- Carboxyiic acids of the formula F13 can be converted to amides of the formula F14 using a suitable amine or ammonia salt in the presence of a peptide coupling agent, for example HATU.
- a suitable amine or ammonia salt in the presence of a peptide coupling agent, for example HATU.
- esters of the formula F12 may be directly converted to amides of the formula F14 by reaction with an amine at elevated temperatures.
- Compounds of the formula F 8 may then be further modified by derivitisation of the amine functionality.
- heteroaryl analogues of F10 may be prepared as outlined in Schemes T, U and V. These heteroaryl acetylenes can be coupled to compounds of the formula F3, and then further elaborated in an analogous manner to that described above in schemes N, O, P, R and S.
- 2,3-di-ch!oropyrazine (G48) can be reacted with ethyl acetate in the presence of LiHMDS to give ester G47.
- Coupling of G47 with TMS acetylene under Sonagashira conditions gives acetylene G48.
- Removal of the trimethyisiiy! group using TBAF gives ethyl 2 ⁇ (3 ⁇ ethynylpyrazin-2-yl)acetate (G49).
- diethyl succinate (G50) and ethyi formate (G51) can be condensed to give aldehyde G52 in the presence of sodium metal.
- Cyclisation using thiourea gives 4-oxo-2-thioxo ⁇ 1 ,2,3,4-tetrahydropyrimidine (G53).
- Desulfurisation using Raney-nickel gives pyrimidone G54, which can be converted to 4-chloro pyrimidine G55 using phosphorous oxychloride.
- 2-(pyrid!n-3-yi)acetonitriie (G58) can be oxidised to /V-oxide G59.
- Chlorination with phosphorous oxychloride gives 2 ⁇ chloropyridine G80 which can be hydrolysed with sodium hydroxide to acetic acid G61.
- Ester formation using methanol gives 2-chloropyridine ester G62.
- Coupling of TES-acety!ene under Sonagashira conditions, followed by removal of the triethyisiiyl group using TBAF gives methyl 2-(2-ethynylpyridin-3-yl)acetate (G84).
- heteroaryl acetylenes anaiagous to F10 can be hydroborylated to give vinyl boranes as in scheme W.
- These can be coupled using Suzuki chemistry to compounds of the formula F3, then further elaborated in an analogous manner to the described above in schemes N, O, P, R and S.
- Methyl 2-bromoisonicotinate (G65) can be coupled using Sonagashira conditions give acetylene G66. Removal of the trimethylsilyi group with TBAF gives terminal acetylene G67 which can be hydroborylated to give (E)-methyl 2-(2-(4,4,5,5- tetramethyl-1 ,3,2-dioxaborolan-2-yl)vinyl)isonicotinate (G88).
- the present invention provides active compounds, specifically, active 2,4,5- substituted pyrimidines .
- active refers to compounds which are capable of inhibiting FAK activity as well as the activity of Flt3 (and possibly VEGFR3), and specifically includes both compounds with intrinsic activity (drugs) as well as prodrugs of such compounds, which prodrugs may themselves exhibit little or no intrinsic activity.
- Assays which may be used in order to assess the FAK, Flt3 and VEGFR3 inhibition offered by a particular compound are described in the examples below,
- the present invention further provides a method of inhibiting FAK activity, as well as the activity of Flt3, in a cell, comprising contacting said cell with an effective amount of an active compound, preferably in the form of a pharmaceutically acceptable composition. Such a method may be practised in vitro or in vivo. The method may also include inhibiting VEGFR3 in a ceil.
- the present invention further provides active compounds which inhibit FAK activity, as well as the activity of Fit3, as well as methods of methods of inhibiting FAK activity, as well as the activity of Fit3, comprising contacting a cell with an effective amount of an active compound, whether in vitro or in vivo.
- the active compounds may also include inhibit the activity of VEGFR3 in a cell.
- Active compounds may also be used as part of an in vitro assay, for example, in order to determine whether a candidate host is likely to benefit from treatment with the compound in question.
- the invention further provides active compounds for use in a method of treatment of the human or animal body.
- a method may comprise administering to such a subject a therapeutically-effective amount of an active compound, preferably in the form of a pharmaceutical composition.
- treatment pertains generally to treatment and therapy, whether of a human or an animal (e.g. in veterinary applications), in which some desired therapeutic effect is achieved, for example, the inhibition of the progress of the condition, and includes a reduction in the rate of progress, a halt in the rate of progress, amelioration of the condition, and cure of the condition.
- Treatment as a prophylactic measure i.e. prophylaxis is also included.
- terapéuticaally-effective amount refers to that amount of an active compound, or a material, composition or dosage from comprising an active compound, which is effective for producing some desired therapeutic effect, commensurate with a reasonable benefit/risk ratio.
- the present invention provides active compounds which inhibit Focal Adhesion Kinase (FAK), and FIVlS-like tyrosine kinase (Flt3), including mutants thereof.
- FAK Focal Adhesion Kinase
- Flt3 FIVlS-like tyrosine kinase
- the inhibition of these is thought to be useful in the treatment of Acute Myleloid Leukemia (A ML). Therefore, the present invention relates to the treatment of AML with the compounds as described above.
- the present invention relates to the treatement of:
- One of ordinary skill in the art is readily able to determine whether or not a candidate compound treats a cancerous condition for any particular cell type, either alone or in combination.
- the treatment defined hereinbefore may be applied as a sole therapy or may involve, in addition to the compound of the invention, conventional surgery or radiotherapy or chemotherapy.
- Such chemotherapy may include one or more of the following categories of anti-tumour agents:-
- antiproliferative/antineoplastic drugs and combinations thereof as used in medical oncology, such as alkylating agents (for example cisplatin, oxalipiatin, carboplatin, cyclophosphamide, nitrogen mustard, meiphalan, chlorambucil, busulphan, temozolamide and nitrosoureas); antimetabolites (for example
- gemcitabine and antifolates such as fluoropyrimidines like 5 fiuorouracil and tegafur, raltitrexed, methotrexate, cytosine arabinoside, and hydroxyurea); antitumour antibiotics (for example anthracyciines like adriamycin, bleomycin, doxorubicin, daunomycin, epirubicin, idarubicin, mitomycin-C, dactinomycin and mithramycin); antimitotic agents (for example vinca alkaloids like vincristine, vinblastine, vindesine and vinoreibine and taxoids like taxol and docetaxei (Taxotere) and polokinase inhibitors); and topoisomerase inhibitors (for example epipodophyilotoxins like etoposide and teniposide, amsacrine, topotecan and camptothecin);
- antitumour antibiotics for example
- cytostatic agents such as antioestrogens (for example tamoxifen, fulvestrant, toremifene, raloxifene, dro!oxifene and iodoxyfene), antiandrogens (for example bicalutamide, flutamide, nilutamide and cyproterone acetate), LHRH antagonists or LHRH agonists (for example goserelin, ieuprorelin and buserelin), progestogens (for example megestrol acetate), aromatase inhibitors (for example as anastrozole, letrozole, vorazole and exemestane) and inhibitors of 5*-reductase such as finasteride;
- antioestrogens for example tamoxifen, fulvestrant, toremifene, raloxifene, dro!oxifene and iodoxyfene
- anti-invasion agents for example c-Src kinase family inhibitors like 4-(6- chloro-2,3-mefhylenedioxyaniiino)-7-[2-(4-mefhylpiperazin-1-yi)ethoxy]-5- tetrahydropyran-4-yloxyquinazoiine (AZD0530; International Patent Application WO 01/94341), N-(2-chloro-6-methylphenyl)-2- ⁇ 8-[4-(2-hydroxyethyl)piperazin-1-yl]-2- methylpyrirnidin-4-ylamino ⁇ thiazole-5-carboxamide (dasatinib, BMS-354825; J.
- anti-invasion agents for example c-Src kinase family inhibitors like 4-(6- chloro-2,3-mefhylenedioxyaniiino)-7-[2-(4-mefhylpiperazin-1-yi)eth
- inhibitors of growth factor function include growth factor antibodies and growth factor receptor antibodies (for example the anti erbB2 antibody trasfuzumab [HerceptinT], the anti-EGFR antibody panitumumab, the anti erbB1 antibody cetuximab [Erbitux, C225] and any growth factor or growth factor receptor antibodies disclosed by Stern et al. Critical reviews in
- inhibitors also include tyrosine kinase inhibitors, for example inhibitors of the epidermal growth factor family (for example EGFR family tyrosine kinase inhibitors such as N-(3-chloro-4-fluorophenyl)- 7-methoxy ⁇ 8-(3-morpholinopropoxy)quinazolin-4-amine (gefitinib, ZD 839), N-(3- ethynylphenyl)-6,7-bis(2-methoxyethoxy)quinazolin-4-amine (eriotinib, OSI 774) and 6-acrylamido-N-(3-chloro-4-fluorophenyi)-7-(3-morpholinopropoxy)-quinazolin-4- amine (CI 1033), erbB2 tyrosine kinase inhibitors such as iapatin
- IGF receptor insulin-like growth factor
- aurora kinase inhibitors for example AZD1 52, PH739358, VX-680, MLN8054, R763, MP235, P529, VX-528 AND AX39459
- cyclin dependent kinase inhibitors such as CDK2 and/or CDK4 inhibitors
- antiangiogenic agents such as those which inhibit the effects of vascular endothelial growth factor, [for example the anti vascular endothelial cell growth factor antibody bevacizumab (AvastinT) and VEGF receptor tyrosine kinase inhibitors such as 4-(4-bromo-2-fluoroanilino)-6-methoxy-7-(1-methylpiperidin-4- ylmethoxy)quinazoiine (ZD6474; Example 2 within WO 01/32651), 4-(4-fluoro-2- methylindol-5-yloxy)-6-methoxy-7-(3-pyrrolidin-1-ylpropoxy)quinazoline (AZD2171 ; Example 240 within WO 00/47212), vataianib (PTK787; WO 98/35985) and SU11248 (sunitinib; W 01/60814), compounds such as those disclosed in International Patent Applications W097/22598,
- vascular damaging agents such as Combretastatin A4 and compounds disclosed in International Patent Applications WO 99/02166, WO 00/40529, WO 00/41669, WO 01/92224, WO 02/04434 and WO 02/08213;
- antisense therapies for example those which are directed to the targets listed above, such as ISIS 2503, an anti-ras antisense;
- gene therapy approaches including for example approaches to replace aberrant genes such as aberrant p53 or aberrant BRCA1 or BRCA2, GDEPT (gene directed enzyme pro drug therapy) approaches such as those using cytosine deaminase, thymidine kinase or a bacterial nitroreductase enzyme and approaches to increase patient tolerance to chemotherapy or radiotherapy such as multi drug resistance gene therapy; and
- immunotherapy approaches including for example ex vivo and in vivo approaches to increase the immunogenicity of patient tumour cells, such as transfection with cytokines such as interieukin 2, interieukin 4 or granulocyte macrophage colony stimulating factor, approaches to decrease T ceil anergy, approaches using transfected immune cells such as cytokine transfected dendritic cells, approaches using cytokine transfected tumour cell lines and approaches using anti idiotypic antibodies.
- cytokines such as interieukin 2, interieukin 4 or granulocyte macrophage colony stimulating factor
- approaches to decrease T ceil anergy approaches using transfected immune cells such as cytokine transfected dendritic cells
- approaches using cytokine transfected tumour cell lines and approaches using anti idiotypic antibodies approaches ameliorated by inhibition of Fit3, or FLt3 and FAK
- the inhibition of Fit3 may be useful in the treatment of autoimmune diseases, such as multiple sclerosis (Whartenby, K, et ai., PNAS (2005), 102(48): 16741-16746).
- the inhibition of Flt3 (including mutations such as ITD and D835 point mutations) may also be useful in treating myelodyspiastic syndrome (IV1DS) and chronic myeloproliferative diseases (C PDs) (Line, P., et a!., Am J Clin Pathol
- thalidomide and thalidomide analogues such as !enaiidomide and pomalidomide.
- Such combinations may be appropriate in the treatment of myelodyspiastic syndrome ( DS).
- DS myelodyspiastic syndrome
- the active compound or pharmaceutical composition comprising the active compound may be administered to a subject by any convenient route of
- administration whether systemicaily/ peripherally or at the site of desired action, including but not limited to, oral (e.g. by ingestion); topical (including e.g.
- transdermal, intranasal, ocular, buccal, and sublingual pulmonary (e.g. by inhalation or insufflation therapy using, e.g. an aerosol, e.g. through mouth or nose); rectal; vaginal; parenteral, for example, by injection, including subcutaneous, intradermal, intramuscular, intravenous, intraarterial, intracardiac, intrathecal, intraspinal, intracapsular, subcapsular, intraorbital, intraperitoneal, intratracheal, subcuticular, intraarticular, subarachnoid, and intrasternai; by implant of a depot, for example, subcufaneously or intramuscularly.
- parenteral for example, by injection, including subcutaneous, intradermal, intramuscular, intravenous, intraarterial, intracardiac, intrathecal, intraspinal, intracapsular, subcapsular, intraorbital, intraperitoneal, intratracheal, subcuticular, intraarticular, sub
- the subject may be a eukaryote, an animal, a vertebrate animal, a mammal, a rodent (e.g. a guinea pig, a hamster, a rat, a mouse), murine (e.g. a mouse), canine (e.g. a dog), feline (e.g. a cat), equine (e.g. a horse), a primate, simian (e.g. a monkey or ape), a monkey (e.g. marmoset, baboon), an ape (e.g. gorilla, chimpanzee, orang-utan, gibbon), or a human.
- a rodent e.g. a guinea pig, a hamster, a rat, a mouse
- murine e.g. a mouse
- canine e.g. a dog
- feline e.g. a cat
- the active compound While it is possible for the active compound to be administered alone, it is preferable to present it as a pharmaceutical composition (e.g. formulation) comprising at least one active compound, as defined above, together with one or more pharmaceutically acceptable carriers, adjuvants, excipients, diluents, fillers, buffers, stabilisers, preservatives, lubricants, or other materials well known to those skilled in the art and optionally other therapeutic or prophylactic agents.
- a pharmaceutical composition e.g. formulation
- the present invention further provides pharmaceutical compositions, as defined above, and methods of making a pharmaceutical composition comprising admixing at least one active compound, as defined above, together with one or more
- pharmaceutically acceptable refers to compounds, materials, compositions, and/or dosage forms which are, within the scope of sound medical judgement, suitable for use in contact with the tissues of a subject (e.g. human) without excessive toxicity, irritation, allergic response, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
- a subject e.g. human
- Each carrier, excipient, etc. must also be “acceptable” in the sense of being compatible with the other ingredients of the formulation.
- Suitable carriers, excipients, etc. can be found in standard pharmaceutical texts, for example, Remington's Pharmaceutical Sciences, 18th edition, Mack Publishing Company, Easton, Pa,, 1990.
- the formulations may conveniently be presented in unit dosage form and may be prepared by any methods well known in the art of pharmacy. Such methods include the step of bringing into association the active compound with the carrier which constitutes one or more accessory ingredients. In general, the formulations are prepared by uniformly and intimately bringing into association the active compound with liquid carriers or finely divided solid carriers or both, and then if necessary shaping the product.
- Formulations may be in the form of liquids, solutions, suspensions, emulsions, elixirs, syrups, tablets, losenges, granules, powders, capsules, cachets, pills, ampoules, suppositories, pessaries, ointments, gels, pastes, creams, sprays, mists, foams, lotions, oils, boluses, electuaries, or aerosols.
- Formulations suitable for oral administration e.g.
- the active compound may be presented as discrete units such as capsules, cachets or tablets, each containing a predetermined amount of the active compound; as a powder or granules; as a solution or suspension in an aqueous or non-aqueous liquid; or as an oil-in-water liquid emulsion or a water-in-oil liquid emulsion; as a bolus; as an electuary; or as a paste.
- a tablet may be made by conventional means, e.g., compression or moulding, optionally with one or more accessory ingredients.
- Compressed tablets may be prepared by compressing in a suitable machine the active compound in a free- flowing form such as a powder or granules, optionally mixed with one or more binders (e.g. povidone, gelatin, acacia, sorbitol, fragacanth, hydroxypropyi methyl cellulose); fillers or diluents (e.g. lactose, microcrystaiiine cellulose, calcium hydrogen phosphate); lubricants (e.g. magnesium stearate, talc, silica); disinfegrants (e.g.
- Moulded tablets may be made by moulding in a suitable machine a mixture of the powdered compound moistened with an inert liquid diluent. The tablets may optionally be coated or scored and may be formulated so as to provide slow or controlled release of the active compound therein using, for example,
- Formulations suitable for topical administration may be formulated as an ointment, cream, suspension, lotion, powder, solution, past, gel, spray, aerosol, or oil.
- a formulation may comprise a patch or a dressing such as a bandage or adhesive plaster impregnated with active compounds and optionally one or more excipients or diluents.
- Formulations suitable for topical administration in the mouth include losenges comprising the active compound in a flavoured basis, usually sucrose and acacia or tragacanfh; pastilles comprising the active compound in an inert basis such as gelatin and glycerin, or sucrose and acacia; and mouthwashes comprising the active compound in a suitable liquid carrier.
- Formulations suitable for topical administration to the eye also include eye drops wherein the active compound is dissolved or suspended in a suitable carrier, especially an aqueous solvent for the active compound.
- Formulations suitable for nasal administration wherein the carrier is a solid, include a coarse powder having a particle size, for example, in the range of about 20 to about 500 microns which is administered in the manner in which snuff is taken, i.e. by rapid inhalation through the nasal passage from a container of the powder held close up to the nose.
- Suitable formulations wherein the carrier is a liquid for administration as, for example, nasal spray, nasal drops, or by aerosol administration by nebuliser include aqueous or oily solutions of the active compound.
- Formulations suitable for administration by inhalation include those presented as an aerosol spray from a pressurised pack, with the use of a suitable propeilant, such as dichlorodifluoromethane, trichlorofluoromethane, dichoro-tetraf!uoroethane, carbon dioxide, or other suitable gases.
- a suitable propeilant such as dichlorodifluoromethane, trichlorofluoromethane, dichoro-tetraf!uoroethane, carbon dioxide, or other suitable gases.
- Formulations suitable for topical administration via the skin include ointments, creams, and emulsions.
- the active compound When formulated in an ointment, the active compound may optionally be employed with either a paraffinic or a water-miscible ointment base.
- the active compounds may be formulated in a cream with an oil-in- water cream base, if desired, the aqueous phase of the cream base may include, for example, at least about 30% w/w of a poiyhydric alcohol, i.e., an alcohol having two or more hydroxy!
- topical formulations may desirably include a compound which enhances absorption or penetration of the active compound through the skin or other affected areas.
- dermal penetration enhancers include dimethyisulfoxide and related analogues.
- the oily phase may optionally comprise merely an emu!sifier (otherwise known as an emulgent), or it may comprises a mixture of at least one emulsifier with a fat or an oil or with both a fat and an oil.
- an emu!sifier otherwise known as an emulgent
- a hydrophilic emulslfier is included together with a lipophilic emulsifier which acts as a stabiliser, it is also preferred to include both an oil and a fat.
- the emuisifier(s) with or without stabiiiser(s) make up the so-called emulsifying wax
- the wax together with the oil and/or fat make up the so-called emulsifying ointment base which forms the oily dispersed phase of the cream formulations.
- Suitable emulgents and emulsion stabilisers include Tween 60, Span 80, cetosteary! alcohol, myristy! alcohol, glyceryl monostearate and sodium lauryl sulphate.
- Tween 60, Span 80, cetosteary! alcohol, myristy! alcohol, glyceryl monostearate and sodium lauryl sulphate The choice of suitable oiis or fats for the formulation is based on achieving the desired cosmetic properties, since the solubility of the active compound in most oils likely to be used in pharmaceutical emulsion formulations may be very low.
- the cream should preferably be a non-greasy, non-staining and washable product with suitable consistency to avoid leakage from tubes or other containers.
- Straight or branched chain, mono- or dibasic alkyl esters such as di-isoadipate, isocetyi stearate, propylene glycol diester of coconut fatty acids, isopropyl myristate, decyi oieate, isopropyl paimitate, butyl stearate, 2-ethylhexyi palmitate or a blend of branched chain esters known as Crodamoi CAP may be used, the last three being preferred esters. These may be used alone or in combination depending on the properties required.
- mono-isoadipate such as di-isoadipate, isocetyi stearate, propylene glycol diester of coconut fatty acids, isopropyl myristate, decyi oieate, isopropyl paimitate, butyl stearate, 2-ethylhexyi palmitate or a blend of branched chain esters known as Crodamoi CAP may
- high melting point lipids such as white soft paraffin and/or liquid paraffin or other mineral oils can be used.
- Formulations suitable for rectal administration may be presented as a suppository with a suitable base comprising, for example, cocoa butter or a salicylate.
- Formulations suitable for vaginal administration may be presented as pessaries, tampons, creams, gels, pastes, foams or spray formulations containing in addition to the active compound, such carriers as are known in the art to be appropriate.
- Formulations suitable for parenteral administration include aqueous and non-aqueous isotonic, pyrogen-free, sterile injection solutions which may contain anti-oxidants, buffers, preservatives, stabilisers, bacteriostats, and solutes which render the formulation isotonic with the blood of the intended recipient; and aqueous and non-aqueous sterile suspensions which may include suspending agents and thickening agents, and liposomes or other micro particulate systems which are designed to target the compound to blood components or one or more organs.
- suitable isotonic vehicles for use in such formulations include
- the concentration of the active compound in the solution is from about 1 ng/m! to about 10 Mg/mi, for example from about 10 ng/m! to about 1 pg/ml.
- the formulations may be presented in unit-dose or multi-dose sealed containers, for example, ampoules and vials, and may be stored in a freeze-dried (lyophilised) condition requiring only the addition of the sterile liquid carrier, for example water for injections, immediately prior to use.
- Extemporaneous injection solutions and suspensions may be prepared from sterile powders, granules, and tablets.
- Formulations may be in the form of liposomes or other micro particulate systems which are designed to target the active compound to blood components or one or more organs.
- appropriate dosages of the active compounds, and compositions comprising the active compounds can vary from patient to patient. Determining the optimal dosage will generally involve the balancing of the level of therapeutic benefit against any risk or deleterious side effects of the treatments of the present invention.
- the selected dosage level will depend on a variety of factors including, but not limited to, the activity of the particular compound, the route of administration, the time of administration, the rate of excretion of the compound, the duration of the treatment, other drugs, compounds, and/or materials used in combination, and the age, sex, weight, condition, general health, and prior medical history of the patient.
- the amount of compound and route of administration will ultimately be at the discretion of the physician, although generally the dosage will be to achieve local concentrations at the site of action which achieve the desired effect without causing substantial harmful or deleterious side-effects.
- Administration in vivo can be effected in one dose, continuously or intermittently (e.g. in divided doses at appropriate intervals) throughout the course of treatment.
- Methods of determining the most effective means and dosage of administration are well known to those of skill in the art and will vary with the formulation used for therapy, the purpose of the therapy, the target cell being treated, and the subject being treated. Single or multiple administrations can be carried out with the dose level and pattern being selected by the treating physician.
- a suitable dose of the active compound is in the range of about 100 to about 250 mg per kilogram body weight of the subject per day.
- the active compound is a salt, an ester, prodrug, or the like
- the amount administered is calculated on the basis of the parent compound and so the actual weight to be used is increased proportionately.
- Figure 1 shows the tumour volume and % body weight change of mice when treated with a compound of the invention in an ectopic leukemia model.
- Figure 2 shows the CD31 +ve blood vessel density in the tumours treated in the same model as in figure 1.
- Figure 3 shows the inhibition of pTyr591-ITD-FLT3 in the primary tumours treated in the same model as in figure 1.
- Figure 4 show the tumour progression in mice when treated with a compound of the invention in an orthotopic leukemia model.
- Figure 5 shows the whole body bioluminescence imaging of the test subjects treated in the same model as in figure 4.
- EDCI carbodiimide
- DMAP 4-dimethylaminopyridine
- TBAF tetra-n-butylammonium fluoride
- DIPEA /V-Diisopropylethyiamine
- HOBt 1-hydroxybenzotriazoie
- DCE 1,2-dichloroethene
- the multiplicity of a signal is designated by the following
- LC/MS data was generated using either a Finnigan LCQ Advantage Max (LCMS-A), a Waters ZQ 3100 system (LCMS-B) or an Agilent 6100 Series Single Quad LC/MS (LCMS-C).
- LCMS Method A LCMS-A
- Solvent A Water 0.1 % Formic Acid
- Solvent B Acetonitrile 0.1 % Formic Acid Gradient: 10-100% B over 10min
- Ion Source Ion trap
- Solvent A Water 0.1 % Formic Acid
- Solvent B Acetonitrile 0.1 % Formic Acid Gradient: 10-100% B over 0min
- Ion Source Sing!e-quadrupole
- Solvent A Water 0.1 % Formic Acid
- Solvent B Acetonitrile 0.1 % Formic Acid
- Vaporizer temperature 200°C
- Step size 0.1 sec
- Analytical thin-layer chromatography was performed on Merck silica gel 80F254 aluminium-backed plates which were visualised using fluorescence quenching under UV light or acidic anisaidehyde or a basic potassium permanganate dip. Flash chromatography was performed using either a Teiedyne isco CombiF!ash Rf purifi- cation system using standard RediSep® cartridges or a Biotage I solera purification system using either Grace or Biotage silica cartridges.
- anhydrous solvents were prepared using a Braun purification system or purchased from Sigma-Aidrich.
- tert-Butyl 4-(4-nitrophenyl)piperazine-1-carboxyiate (11) (3.24 g, 10.5 mmol) was dissolved in EtOAc (90 mL) under an atmosphere of nitrogen and a slurry of 10% Pd/C (0.500 g) in EtOAc (10 mL) was added. The resulting suspension was then stirred vigorously under an atmosphere of hydrogen at room temperature for 42 hours.
- 2,4-Dichloro-5-(trifluoromethyl)pyrimidine (2.39 g, 11.0 mmol) was stirred in a 1 :1 t- BuOH:1 ,2-dichloroethane mixture (80 mL) at 0 °C and a 1.0 M ZnCI 2 solution in diethyl ether (12.6 mL, 12.6 mmol) was added cautiously over 20 minutes and the reaction was left stirring at 0 °C for 30 minutes.
- 2-(2-iodophenyi)acetic acid (5.00 g, 19.1 mrnoi) was placed into a reaction flask and dissolved in MeOH (150 mL). Sulfuric acid (250 L) was added and reaction mixture was stirred and heated at 80 °C under nitrogen for 16 hours. The resulting mixture was cooled to room temperature and the volatiles removed by evaporation under reduced pressure.
- the reaction was heated under microwave irradiation at 150 °C for 30 minutes then concentrated in vacuo and purified by silica gel chromatography (Biotage Isolera, 12 g Si Cartridge, 0-100% EtOAc in petroleum benzine 40-60 °C, then 0-100% MeOH in EtOAc) to give a pale yellow solid.
- the solid was dissolved in EtOAc (20 mL) and sat. aq. NaHC0 3 (10 mL) and the layers were separated.
- 2,4-Dichloro-5-(trifluoromethyl)pyrimidine (0.355 g, 1.64 mmoi) was stirred in a 1 :1 t- BuOH: 1 ,2-dichloroethane mixture (30 mL) at 0 °C and a 1.0 M ZnCI 2 solution in diethyl ether (1.87 mL, 1.87 mmol) was added cautiously over 20 minutes and the reaction was left stirring at 0 °C for 30 minutes.
- triphenylphosphine (0.026 g, 0.098 mmol), irans-dichlorobis(triphenylphosphine) pailadium(il) (0.046 g, 0.066 mmol) and Cul (0.019 g, 0.098 mmol).
- terf-Butyl 3-ethynylbenzoate (117) (1.50 g, 9.37 mmoi) was dissolved in dry DCM (70 mL) and TFA (35.9 mL, 488 mmoi) was added carefully. The reaction was stirred at room temperature for 3 hours, concentrated in vacuo and toluene was added and then removed in vacuo to give a pale yellow solid. This material was dissolved in methanol (50 mL) and cone. H 2 S0 4 (-1 mL) was added and the resulting solution was stirred at 85 °C for 20 hours.
- the reaction was sealed with a balloon and stirred at room temperature for 18 hours after which the reaction was flushed with nitrogen gas and Pearlman's catalyst (0.150 g) in EtOAc (5 mL) was added. The atmosphere was again changed to hydrogen gas (balloon) and the reaction was sealed with balloon and stirred for 20 hours at room temperature. The catalyst was removed by filtration through Celite, which was washed with EtOAc (5 x 10 mL).
- Ammonium carbonate (0.067 g, 0.69 mmol) was added in one portion, and the reaction was stirred room temperature for 60 hours. The volatiles were removed in vacuo and the residual solution was diluted with EtOAc (70 mL) and sat. aq. NaHC0 3 (70 mL). The layers were separated and the aqueous layer was extracted with EtOAc (70 mL), the combined organic layers were washed with water (70 mL), brine (70 mL), dried (MgSQ 4 ), filtered and concentrated in vacuo to give an off-white solid.
- 2,4-Dichloro-5-(trifluoromethyl)pyrimidine (0.101 g, 0.464 mmol) was stirred in a 1 : 1 t- BuOH: 1 ,2-dichloroethane mixture (10 mL) at 0 °C and a 1.0 M ZnCI 2 solution in diethyl ether (0.530 mL, 0.530 mmol) was added cautiously over 20 minutes.
- 2-iodophenyiacetic acid (2.00 g, 7.83 mrnoi) was dissolved in dry THF (70 mL) and dry D F (10 mL) under an atmosphere of nitrogen.
- To the solution were added 1- hydroxybenzotriazole (1 , 134 g, 8.396 mmol) and EDCI (1.609 g, 8.396 mmol) and W,A -diisopropylethylamine (5.318 mL, 30.53 mmol) and the reaction mixture was stirred at room temperature for 10 minutes.
- Ammonium carbonate (2.933 g, 30.53 mmol) was added in one portion, and the reaction was stirred room temperature for 17 hours.
- reaction mixture was then stirred at room temperature for 8 hours, concentrated in vacuo and purified by silica gel chromatography (Biotage isoiera, 40 g Si cartridge, 0-100% EtOAc in petroleum benzine 40-60 °C) to give a beige solid.
- This material was dissolved in dry THF (25 mL) under an atmosphere of nitrogen and TBAF (1 ,0 M in THF, 2.805 mL, 2.805 mmol) was added dropwise at 0 °C. The solution was stirred at this temperature for 1 hour and 15 minutes after which water (5 mL) was added.
- the reaction mixture was concentrated in vacuo and diluted with EtOAc (100 mL) and sat. aq.
- Lithium diisopropylamide (2 M in heptane/THF/ethyibenzene; 15.1 mL, 30.1 mmol) was added dropwise to a solution of ferf-butyl 4-oxopiperidine-1-carboxylate (3.00 g, 15.1 mmol) in THF (50 mL) at -78 °C and the mixture left to stir for 30 minutes.
- a solution of A-phenyl-bis(trifiuoromethanesuifonimide) (6.46 g, 18.1 mmol) in THF (60 mL) was then added dropwise over 30 minutes to the reaction and mixture left to stir for 30 minutes-78 °C.
- Zinc chloride (1.0 M in Et 2 0) (1.97 mL, 1.97 mmol) was added to a solution of 2,4- dichloro-5-(trifluoromethyl)pyrimidine (0.384 g, 1.77 mmol) in 1 :1 DCE/i-BuOH (10 mL) at 0 °C under a stream of N 2 gas. The mixture was stirred for 1 hour at 0 °C and then feri-butyl 4-(4-aminophenyl)piperidine-1 -carboxylate (144) (0.453 g, 1.64 mmol) in 1 :1 DCE/fBuOH (7 mL) was added.
- the reaction mixture was heated under microwave irradiation at 120 °C for 15 minutes.
- the reaction was cooled and the mixture diluted with EtOAc and passed through a plug of celite and washed through with ethyl acetate (50 mL). Water (50 mL) was added and the layers separated. The aqueous layer was extracted with EtOAc (2 x 50 mL). The combined organic extracts were washed with water (50 mL) and brine (50 mL) and dried over Na 2 S0 4 . After filtration the solvent was removed under reduced pressure to give a dark brown residue. The residue was purified by column chromatography on silica gel (0-20% EtOAc in cyciohexane) to yield the title compound ( 46) (0. 57 g, 80%) as a brown viscous oil.
- Trifluoroacetic acid (0.595 mL, 7.78 mmoi) was added to a solution of feri-butyl 4-(4- ((4-(2-(2-amino-2-oxoethyl)phenethyl)-5-(trifluoromethyi)pyrimidin-2- yi)amino)phenyl)piperidine-1-carboxylate (149) (90.8 mg, 0.156 mmol) in dry DCM (5 mL) under an atmosphere of nitrogen. The reaction was stirred at room temperature for 23 hours. The volatiles were removed in vacuo and the residue partitioned between EtOAc (30 mL) and 2M NaOH (30 mL).
- Trifluoroacetic acid (0.417 mL, 5.46 mmol) was added to a solution of ferf-buiyi 4-(4- ((4-(2-(1-amino-1-oxopropan-2-yl)phenethyl)-5-(trifluoromethyl)pyrimidin-2- yl)amino)phenyl)piperidine-1 -carboxyiate (155) (65.2 mg, 0.109 mmol) in dry DCM (8 mL) under an atmosphere of nitrogen. The reaction was stirred at room temperature for 23 hours. The volatiles were removed in vacuo and the residue partitioned between EtOAc (20 mL) and 2 M NaOH (20 mL).
- Trifluoroacetic acid (0.222 mL, 2.906 mmol) was added to the solution and the reaction was stirred at room temperature for 6 hours. Volatiles were removed in vacuo, EtOAc (70 mL) and 2 M aq. NaOH (70 mL) were added to the residue and the layers were separated. The aqueous layer was extracted with EtOAc (2 x 70 mL), the combined organics were washed with water (50 mL), brine (50 mL), dried (MgS0 4 ), filtered and concentrated in vacuo to give a solid which was taken up in DCM (- 0 mL) and methanol ( ⁇ 1 mL) and concentrated in vacuo.
- Methyl 2-iodo ⁇ 6-methy!benzoate (2.00 g, 7.245 mmoi) and NBS (1.418 g, 7.969 mmol) were stirred in chiorobenzene (50 mL) and benzoyl peroxide (75% w/w, 0.234 g, 0.724 mmol) was added. The reaction was stirred at 90 °C for 18 hours, cooled to room temperature, filtered and the precipitate was washed with cyclohexane (4x10 mL). The combined filtrates were evaporated, and the resulting brown oil was diluted with THF (50 mL).
- Aqueous ammonia solution (20 mL) was added, and the mixture was stirred vigorously for 17 hours.
- the mixture was diluted with water (20 mL) and the THF was removed in vacuo.
- DCM 150 mL was added, the layers were separated and the aqueous layer was extracted with DCM (2x100 mL), the combined organics were washed with brine (100 mL), dried (MgS0 4 ) and filtered.
- Silica gel was added and the voiatiles were removed in vacuo to give the crude material absorbed onto silica gel.
- Formaldehyde (37 % in H 2 0; 15.6 L, 0.210 mmol) was added to a suspension of 2- (2-(2-(2-((4-(piperidin-4-yl)phenyl)amino)-5-(trifluoromethyl)pyrimidin-4- yl)ethyl)phenyl)acetamide (11) (25 mg, 0.053 mmol) in anhydrous methanol (5 mL) under an atmosphere of nitrogen. Sodium triacetoxyborohydride (0.111 g, 0.525 mmol) was then added in one portion to the reaction mixture. The reaction was stirred at room temperature for 1.5 hours.
- Methyl 2-(2-((2-(methyisuifonyi)-5-(trifiuoromethyl)pyrimidin-4- yi)ethynyi)phenyl)acetate (165) (1.50 g, 3.76 mmol) was taken up in DMF (30 mL) then 10% Pd/C (750 mg) was added. The resulting suspension was stirred under H 2 (1 atm) for 16 hours at room temperature. The crude reaction mixture was filtered through Celite, washing with MeOH. The filtrate was evaporated under reduced pressure to give a yellow liquid which was adsorbing onto silica.
- PdCI 2 (PPh 3 ) 2 (0.049 g, 0.070 mmoi), triphenylphosphine (0.054 g, 0.21 mmoi) and copper iodide (0.036 g, 0, 19 mmoi) in DMF (3 mL) was added triethyiamine (0.570 mL, 4.09mmol) and trimethylsiiy!acetyiene (0,210 mL, 1 ,49 mmoi) and the resulting mixture heated under microwave irradiation at 120 °C for 25 minutes.
- 2,4-Dichloro-5-(trifluoromethyi)pyrimidine (0.551 g, 2.54 mmol) was stirred in a 1 :1 t- BuOH: 1 ,2-dichloroethane mixture (30 mL) at 0 °C.
- a 1.0 M ZnCI 2 solution in diethyl ether (2.903 mL, 2.903 mmol) was added cautiously over 0 minutes, after addition the reaction was left stirring at 0 °C for 30 minutes.
- Lithium hydroxide mono hydrate (43.0 mg, 1.03 mmo!) was added to a suspension of methyl 2-(2-(2-((4-(4-((ier L butoxycarbonyl)amino)piperidin-1-yl)phenyl)amino)-5- (trifluoromethyi)pyrimidin-4-yl)ethyl)pheny!acetate (178) (211 mg, 0.344 mmol) in THF (10 mL), MeOH (1.0 mL) and water (1.5 mL) and the resulting mixture was stirred at room temperature for 6 hours. The organics were removed in vacuo then 2 M aqueous NaOH solution (100 mL) was added.
- the cooled mixture was concentrated, co-evaporated with toluene (3x 20 mL) and loaded onto a 10 g SCX cartridge in methanol.
- the cartridge was eluted with methanol (200 mL), then with 1 % methanolic methyiamine (200 mL).
- the methanolic methylamine eluent was concentrated to give a brown oil (0.850 g).
- the oil was dissolved in DCM (5 mL), and Boc anhydride (549 mg, 2.52 mmoi) was added.
- the resulting mixture was stirred under an oil bubbler for 18 hours, then diluted with DCM (50 mL) and washed with water (50 mL).
- the aqueous layer was extracted with DCM (2x 50 mL), and the combined DCM phases dried (phase separation filter) and evaporated.
- trifluoromethanesulfonate (1.21 g, 2.72 mmol) was added. The mixture was stirred for 17 hours, allowing the cooling bath to come to room temperature over this time. Ethyl acetate (200 mL), saturated ammonium chloride (80 mL) and water (20 mL) were added and the layers separated. The aqueous phase was extracted with ethyl acetate (2x100 mL) and the combined ethyl acetate phases were washed with brine, dried (sodium sulfate) and evaporated.
- the mixture was concentrated, evaporated from toluene and loaded onto a 5 g SCX cartridge in methanol (1 mL). The cartridge was washed with methanol (50 mL), and then eluted with 1 % methylamine/methanol (50 mL). The basic eluent was concentrated, and taken up in dichloromethane (5 mL). Boc anhydride (0.062 mL, 0.27 mmol) was added, and the mixture stirred at room temperature for 18 hours.
- Triethyiamine (34.6 ⁇ , 0.248 mmol) and acetic anhydride (23.4 ⁇ , 0.248 mmol) were added to a solution of the 2-(2-(2-(2-((4-(piperidin-4-yl)phenyl)amino)-5- (trifluoromethyl)pyrimidin-4-yl)ethyl)phenyl)acetamide ( 11) (30 mg, 0.062 mmoi) in DMF (10 mL).
- the reaction mixture was stirred at room temperature for 20 hours. The volatiles were removed in vacuo and the residue was diluted with EtOAc (20 mL) and sat. aq. NaHC0 3 (20 mL).
- yi)ethyl)pyrazin-2-yl)acetamide (26) (42 mg, 0.087 mmol) was dissolved in methanol (4 mL) and 37% formaldehyde (26 ⁇ _, 0.35 mmol) was added. After five minutes sodium tri(acetoxy)borohydride (92 mg, 0.44 mmol) was added and the mixture stirred for three hours. The solution was concentrated, and the residue suspended in 10% sodium hydroxide (1 mL). After five minutes brine (2 mL) was added, and the mixture extracted with ethyl acetate (5x3 mL).
- Example 28 2 ⁇ (2 ⁇ (2 ⁇ (2 ⁇ ((2 ⁇ IVlethy ⁇ 1 ,2,3,4-tetrahydroisoquinolin-6-yl)amino)-5- (trif!uorometh l)pyrimsdsn ⁇ 4 ⁇ yl)ethy!phenyl)acetamsde (28)
- the aqueous layer was then acidified to pH 5 using 11 N HCI and extracted with diethyl ether (3x100 mL), the ethereal extracts of the acidified layer were combined, dried (Na 2 S0 4 ) then evaporated to dryness under reduced pressure to give the title compound (1 /05) (18.5 g) as a yellow mobile liquid.
- the crude product was not purified further and was used directly in the following step.
Abstract
The use of a compound of the formula (I): (Formula (I)) in the preparation of a medicament for treating Acute Myeloid Leukemia or a disease ameliorated by the inhibition of Flt3, or Flt3 and FAK.
Description
This invention relates to a method of using 2,4,5-substituted pyrimidines that inhibit Focal Adhesion Kinase (FAK), also known as protein tyrosine kinase 2 (PTK2), and F S-like tyrosine kinase (Flt3), also known as Fetal liver kinase 2 (FLK2), Stem ceil kinase 1 (STK1 ) and CD135, for the prevention and/or treatment of Acute Myeloid Leukemia (AML) and other diseases ameliorated by the inhibition of F!t3, or FLt3 and FAK. Background
Directional cell migration is important in many physiological and pathological processes including embryonic development, wound healing, angiogenesis, tumour invasion and metastasis. Transduction of extracellular signals, that stimulate cells to move directionally, may be induced by a number of processes including trans- membrane integrins binding to extra cellular matrix proteins and the action of growth factors (for example EGF, IGF and VEGF) on the extracellular domains of their cognate receptors.
FAK is a non receptor tyrosine kinase that mediates signals from both trans- membrane integrins and growth factor receptors. FAK has been reported to play a central role in coordinating these diverse extra cellular signals, integrating them in a fashion that results in directional movement of cells through their external environment (Tomar and Schlaepfer. Current Opinion in Ceil Biology: 2009, 21 , 876- 683).
Integrin clustering or the activation of a growth factor receptor (for example EGFR, IGF-1 R, Her2 and VEGFR) promotes FAK autophosphorylation at Y397.
Phosphorylated Y397 FAK then binds to c-Src (referred to as Src herein) and Src mediated phosphorylation of FAK at Y576 and Y577 occurs to give rise to an active FAK-Src complex. Active FAK-Src then facilitates signaling via a number of biochemical pathways which influence processes such as ceil adhesion, migration, invasion, ceil survival, proliferation, acquisition of chemotherapy resistance and metastasis (Brunton and Frame. Current Opinion in Pharmacology: 2008, 8, 437-432 and Chatzizacharias et al. Expert Opinion in Therapeutic Targets: 2007, 11 ( 0), 1315-1328).
Cell adhesion
Functional studies addressing the role of FAK in cell adhesion suggest that it contributes to both focal adhesion assembly (Richardson and Parsons. Nature: 1998, 380, 538-540) and focal adhesion turnover (Fincham et al. Oncogene: 1995, 10(11), 2247-2252). Inhibition of FAK by RNAi in both human and mouse ceil lines, resulting in decreased FAK protein levels, has been shown to reduce cell adhesion to a fibronectin/laminin-coated plate in vitro (Tsutsumi et al. International Journal of Oncology: 2008, 33(1), 215-224).
Ceil migration
There is strong evidence that FAK is a key regulator of ceil migration (Angelucci and Bologna. Current Pharmaceutical Design: 2007, 13, 2129-2145 and itra et al.
Nature Reviews Molecular Ceil Biology: 2005, 8, 56-68). Cells derived from FAK -/- mouse embryos exhibit reduced migration as a result of impaired adhesion turnover (Hie et al. Nature: 1995, 377, 539-544). Moreover, displacement of FAK from focal adhesions reduces cell migration (Gilmore and Romer. Molecular Biology of the Cell: 1996, 7(8), 1209-1224), whilst over-expression in CHO ceils stimulates migration (Gary et al. Journal of Cell Science: 1996, 7, 1787-1794). In addition, inhibition of FAK by RNAi in both human and mouse ceil lines, resulting in decreased FAK protein levels, has been shown to reduce cell migration in an in vitro haptotactic migration assay (Tsutsumi et al. International Journal of Oncology: 2008, 33(1), 215-224).
Cell invasion
FAK activation has been shown to enhance matrix degrading invasive behaviour.
FAK-Src signaling through cellular apoptosis susceptibility protein (CAS) (Liao et al. Journal of Experimental and Clinical Cancer Research: 2008, 27:15) leads to the expression of matrix metalioproteases ( MPs) including MP2 and M P9. FAK-Src activation also promotes cell surface expression of MMP14 via phosphorylation of endophilin A2. MP14 then activates MMP2 by cleavage of pro-M P2 to its active form (Siesser and Hanks. Clinical Cancer Research: 2006, 12(11), 3233-3237).
Highly invasive cancer cells form specialized actin-rich extra cellular matrix degrading membrane protrusions known as invadopodia which are rich in matrix-degrading proteases such as MMPs. Both FAK and Src have been shown to be instrumental in
the formation of invaclopodia (Chan ef al. Journal of Chemical Biology: 2009, 85(2), 357-370).
Cell survival
FAK has been shown to play an important role in ceil survival. Activation of FAK has been shown to result in suppression of anoikis (apopotosis in response to an inappropriate extra cellular matrix environment) (Frisch et al Journal of Cell Biology. 1996, 134(3), 793-799 and Xu et al Cell Growth and Differentiation, 1996, 7(4), 413- 418). Studies have demonstrated that FAK activates multiple downstream pathways to suppress anoikis in both fibroblasts and epithelial ceils (Zouq et al. Journal of Cell Science: 2008, 122, 357-367). In human intestinal crypt cells signalling via the association of FAK with β1 integrin and subsequent binding with Src up regulates expression of the anti-apoptotic proteins Bcl-XL and Mcl-1 via PI3-K/Akt-1 signalling. PI3-K/Akt-1 signalling also down regulates expression of the pro-apoptotic activators Bax and Bak, causes phosphorylation of the pro-apoptotic sensitizer Bad and antagonizes ρ38β activation. Dissociation of FAK/Src results in a sustained/enhanced activation of ρ38β which is an apoptosis/anoikis driver (Bouchard et al. Apoptosis: 2008, 13, 531-542). Ceil proliferation
Reduction in the expression of either FAK or β1 integrin and hence disruption of the βΙ-FAK signalling axis results in decreased initial proliferation of micro-metastatic cells distributed in the lung. Using 3D cultured D2 cells a strong correlation was observed between FAK Y397 and Y861 phosphorylation and proliferative ability (Shibue and Weinberg. PNAS 2009, 106(25), 10290-10295). HL-60 Ceils, transfected to over express FAK, have been shown to double at a rate 1.5 times faster than control HL-60 cells. Studies revealed a marked induction of cyclin D3 expression and CDK activity in the ceils over expressing FAK. Activation of PI3- K Akt-1 signalling, a process associated with FAK activation in a number of studies, was identified as a probable cause of the cyclin expression/activation (Yamamoto et al. Cellular Signaling: 2003, 15. 575-583).
Acquisition of chemotherapy resistance
Exposure of the cisplatin sensitive ovarian cancer cell line OAW42 to repeated cycles of cisplatin treatment and subsequent recovery resulted in the formation of chemo-
resistant QAW42-R cells. Studies aimed at identifying the cause of this chemo- resistance revealed that FAK was constituently active in both the sensitive and chemo-resistant cells. However, inhibition of phosphorylation of Y397 FAK was induced by treatment with cisplatin in OAW42 cells but not in OAW42-R ceils (Poulain and co-workers. Gynaecoiogic oncology: 2006, 101 , 507-519). The effects of FAK inhibition on chemo-resistance has also been studied in vitro and in vivo using the FAK inhibitor TAE226, alone and in combination with docetaxel, in taxane- sensitive (SKOV3ip1 and HeyA8) and taxane-resistant (HeyA8~MDR) ovarian cancer cell lines. TAE226 has the structure:

and is described in WO 2004/080980 and WO 2005/016894. In vitro, TAE226 inhibited the phosphorylation of FAK at both Y397 and Y861 sites, inhibited cell growth in a time- and dose-dependent manner, and enhanced docetaxel-mediated growth inhibition by 0- and 20-fold in the taxane-sensitive and taxane-resistant cell lines, respectively. In vivo, FAK inhibition by TAE226 significantly reduced tumour burden in the HeyA8, SKOV3ip1 , and HeyA8- DR models (46-64%) compared with vehicle-treated controls. However, the greatest efficacy was observed with concomitant administration of TAE226 and docetaxel in all three models (85-97% reduction), in addition, TAE226 in combination with docetaxel significantly prolonged survival in tumour-bearing mice (Haider et al. Cancer Res: 2007, 67(22), 10976- 10983).
Metastatic potential
Several studies have examined the role of FAK protein levels and it's relation to tumor progression in animal models. In a mouse skin carcinogenesis model using FAK +/- mice, reduced FAK protein expression correlated with decreased papilloma formation (46%), compared with FAK +/+ wild-type control mice (McLean et al.
Cancer Research: 2001 , 61 , 8385-8389). Using human breast carcinoma cells, researchers showed that FAK siRNA treated cells were inhibited from metastasizing to the lung after orthotopic implantation in nude mice (Benlimame et al. Journal of Ceil Biology: 2005, 171 , 505-516). Similar experiments using short hairpin RNA
(shRNA) against FAK in 4T1 mouse breast carcinoma cells resulted in an inhibition of metastasis to the lungs after orthotopic implantation in mammary pads (Mitra et al. Oncogene: 2008, 25, 4429-4440). Inhibition of FAK by dominant negative expression in 4T1 mouse breast carcinoma ceils reduced tumour growth and angiogenesis in mice (Mitra et al. Oncogene: 2008, 25, 5969-5984). Use of a C re/I ox P recombination system to disrupt FAK function in the mammary epithelium of a transgenic model of breast cancer has demonstrated that FAK expression is required for the transition of premalignant hyperplasias to carcinomas and their subsequent metastases. The observed decrease in tumor progression was further correlated with impaired mammary epithelial proliferation suggesting that FAK plays a critical role in mammary tumor progression (Lahlou et al. PNAS USA: 2007, 104(51), 20302-20307).
In accordance with the above observations over expression of FAK mRNA and/or protein has been reported in numerous human cancers including colorectal cancer (de Heer. European Journal of Surgical Oncology: 2008, 34(11), 1253-1281), prostate cancer (Tremblay, L, W. Hauck, et al. International Journal of Cancer: 1996, 68(2), 164-171), breast cancer (Watermann et al. British Journal of Cancer 2005, 93(6), 694-698) and melanomas (Hess et al. Cancer Research: 2005, 65(21), 9851- 60). Furthermore FAK over expression is frequently correlated with more aggressive phenotypes of these cancers.
Thus, there is strong evidence to suggest that a FAK inhibitor would have application for the reduction of cell adhesion, cell migration, ceil invasion, ceil proliferation and chemo-resistance. Furthermore, a FAK inhibitor would have applicability to induce apoptosis for cells in inappropriate extra cellular matrix environments and reduce angiogenesis.
It will be appreciated that activity at other tyrosine kinases and serine/threonine kinase in combination with FAK activity may be beneficial for the treatment of proliferative diseases, such as cancer.
For example, FMS-like tyrosine kinase (Fit3), also known as Fetal liver kinase 2 (FLK2), Stem cell kinase 1 (STK1) and CD135, is known as a proto-oncogene.
Mutations of the Fit3 receptor can lead to the development of leukemia, a cancer of bone marrow hematopoietic progenitors. The internal tandem duplication mutation of
Flt-3 (ITD-Flt3) is a recognized molecular lesion in some forms of acute myeloid leukemia (A ML) and predicts for poor patient prognosis (Chen W, et al., Mol Cancer: (2010) 9, 292; Gu T, et aL PLos One, 201 1 4:6, e19169; Smith C, et al., 2012 Nature 485, 280-263).
Furthermore, focal adhesion kinase (FAK) and more particularly, splice variants of FAK, have also been implicated in poor prognosis AML (Despeaux M, et a!., Stem Ceils (2012) 30, 1597-1610; Recher C, et al., (2004) Cancer Research 64: 3191- 3197; Casanova I, et al., (2008) Int. J. Cancer, 123, 217-226) Some patients with AML are predicted to have both ITD-Flt3 mutations and FAK/FAK splice variant expression.
Inhibition of Flt3 may also be useful in ameliorating autoimmune diseases, such as multiple sclerosis (Wharternby, K, et al., PNAS (2005), 102(46): 16741-16746). The inhibition of F!t3 (including mutations such as ITD and D835 point mutations) may also be useful in treating mye!odysp!astic syndrome (MDS) and chronic
myeloproliferative diseases (CMPDs) (Line, P., et aL, Am J Clin Pathol
2006:126:530-533). For example, the vascular endothelial growth factor receptor VEGFR3 (Flt4) is over expressed in melanoma patients with metastases in regional lymph nodes (Mouavvad et al. European Journal of Cancer: 2009, 45, 1407-1414). Abnormal expression levels of endogenous receptor tyrosine kinase ligands are also observed in many human cancers. For example, the expression levels of vascular endothelial growth factors C and D (VEGF-C and VEGF-D), ligands of VEGFR3, are significantly correlated with lymphatic metastasis and lymphatic vessel invasion in early-stage invasive cervical carcinoma (Journal of Experimental & Clinical Cancer Research 2009, 28). Accordingly, compounds that inhibit Flt3, or Flt3 and FAK, and/or VEGFR3, would be useful for the treatment of proliferative diseases, such as cancer.
In particular, compounds that inhibit Flt3, or F!t3 and FAK, including mutants thereof, would be useful in the treatment of acute myeloid leukemia (AML).

PF-582,271 PF-573,228
PF-562,271 is described in WO2004/056786, WO2004/056807, WO2005/023780, WO2007/063384 and Roberts et al. Cancer Res 2008, 68(6), 1935-1944.
PF-573,228 is described in Slack-Davis et al. J. Biol. Chem. 2007, 282(20), 14845- 14852. In addition to these specifically described compounds, further classes of FAK inhibitors are disclosed in WO2008/129380, WO2008/1 5369, WO2009/105498, US2010/1 13475, WO2009/143389, WO2009/071535, WO2010/055 17,
WO2010/058030, WO2010/058032, WO2007/ 40222, and WO2009/024332. Summary of the invention
The present inventors have discovered a particular class of compounds which are effective as FAK inhibitors, and also inhibit FLt3 (including mutants thereof). These compounds may also inhibit VEGFR3. These compounds may exhibit selectivity for FAK over kinases such as VEGFR1 , IGF-1 (insulin-like growth factor 1 receptor), IR (insulin receptor) and CDKs (cyciin-dependent kinases). Additionally, the compounds of the invention may have enhanced selectivity for the inhibition of cytochrome p450 enzymes, specifically the 2C9 and 3A4 isoforms. Furthermore, the compounds of the invention may be less prone to the formation of adducts with glutathione. In a first aspect, the present invention provides the use of compounds of the following formula (i):

in the preparation of a medicament for treating Acute Myeloid Leukemia or a diseas ameliorated by the inhibition of Flt3, or Fit3 and FAK, wherein:
R1 is selected from: H and

RN1 is selected from H, Ci-3 alkyl and C(=0) e;
RN2 is selected from H, Ci-3 a!ky! and C(==0)Me;
RN3 is selected from H, Ci.3 alkyl and C(=0)Me;
RN4 is selected from H and CH3;
RN7 and RN8 are independently selected from H and CH3;
RN9 is selected from H, C1-3 alkyl and C(=0)Me;
RN1° is selected from H, C,..3 alkyl and C(=0)Me;
RN11 is selected from H, d.3 alkyl and C(=0)Me;
2 is selected from H and
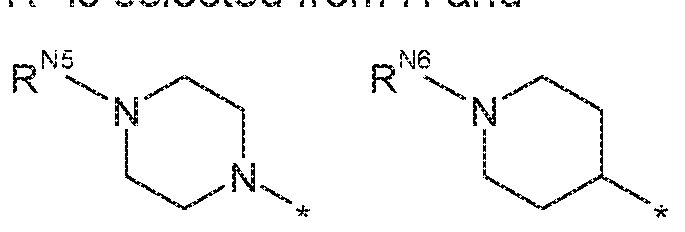 wherein:
wherein:
RNS is selected from H, Ci_3 alkyl and C(=0)Me;
RN6 is selected from H, Ci-3 alkyl and C(=0) e;
and vv'herein only one of R1 and R2 is H;
or R1 and R2 together form the group ~CH2-N(RN12)-C2H -, where RN12 is selected from H, Ci.3 alkyl and C(=0)Me;
R4 is selected from CF3, halo, CF2H and CN; and
Rb is selected from g

R6 is selected from H, (CHRc1)n1C(0)N(RN13)Z1 and (CH2)n2C(0)OZ2; wherein:
n1 is 1 ;
RC1 is H or e;
RN13 is H or CH3;
Z is H, CH3 or OCH3;
n2 is 1 ; and
Z2 is CH3;
and where only one of R^13 and Z1 can be CH3i
R7, if present, is selected from H, and (CH2)miC(0)N(RM1)Y1, wherein:
ml is 0 or 1 ;
RM is H; and
Y1 is H, Me or OCH3;
wherein when both R6 and R7 are present, one is H and the other is not H, and wherein only R6 is present, it is not H; and
R8, if present, is H or, when R7 is C(=0)NH2, R8, if present, is selected from H and C aikyi.
Compounds of formula I are described in co-pending application,
PCT/GB2012/000175, which is incorporated herein by reference.
A second aspect of the present invention provides compounds as described in the first aspect for use in a method of treatment of Acute Myeloid Leukemia or a disease ameliorated by the inhibition of Fit3, or Flt3 and FAK.
A third aspect of the present invention provides a method of treatment of the human or animal body suffering from Acute Myeloid Leukemia or a disease ameliorated by the inhibition of Flt3, or Fit3 and FAK, comprising administering compounds as described in the first aspect, preferably in the form of a pharmaceutical composition.
Another aspect of the invention provides a method of inhibiting Fit3, or FAK and Fit3, in vitro or in vivo, comprising contacting a cell with an effective amount of an active compound as described herein.
Compounds of the present invention may also inhibit VEGFR3. Each of the groups R1 to R8 will be discussed in more detail below. R1
R

(R1!)
When R1 is H, R2 (discussed below) is not H.
Each of RN1 , RN2 and RN3 is independently selected from H, Ci.3 alkyl (i.e. methyl, ethyl, prop-1-yi and prop-2-yi) and C(=0)Me and RN4 is selected from either H or methyl. RN/ is either H or methyl. RN7 and RN8 are independently selected from H and CH3. Each of RN9, RN1° and RN1 i are also independently selected from H, C1.3 alkyl (i.e. methyl, ethyl, prop-1-yl and prop-2-yl) and C(=0)Me.
R2
R2 may have one of the following structures:

When R2 is H, R1 (discussed above) is not H.
RN5 and Rm are independently selected from H, d-3 alkyl (i.e. methyl, ethyl, prop-1-yl and prop-2-yl) and C(=0)Me.
Ft1 and F?
1 and R2 together form the group ~CH2~N(RN12)-C2H4~,

RN 2 is selected from H, d.3 alkyl (i.e. methyl, ethyl, prop-1-yi and prop-2-yl) and C(=0)Me.
Ft
R4 is selected from CF3, halo (i.e. F, CI, Br, I), CF2H and CN. In some embodiments, the halo group is either CI or Br.


R6 is selected from H, (CHRc1)n1C(0)N(RN13)Z1 and (CH2)n2C(G)OZ2; wherein: n1 is 1 ;
RC1 is H or Me;
RN13 is H or CH3;
Z1 is H, CH3 or OCH3;
n2 is 1 ; and
Z2 is CH3;
wherein only one of RN13 and Z1 may be CH3.
When R6 is H, R7 (discussed below) is not H,
If Re is (CHRc )rilC(0)N(RN6)Z1 , it may be selected from: CH2C(0)NH2, CH2C(0)NHCH3j CH2C(0)NHOCH3> CH2C(0)NCH3OCH3j CHCH3C(0)NH2, CHCH3C(0)NHCH3, CHCH3C(0)NHOCH3, and CHCH3C(0)NCH3OCH3,
If R6 is (CH2)n2C(0)OZ2, if is CH2C(0)OCH3. R7
R7 is selected from H, and (CH2)m1C(0)N(R 1)Y1 , wherein:
ml is 0 or 1 ;
RM1 is H; and
Y1 is H, Me or OCH3;
When R7 is H, RB (discussed above) is not H. In addition, when R' is not present, R6 (discussed above) is not H.
When R7 is (CH2)m1C(0)N(RM1)Y1, it may be selected from C(0)NH2, C(0)NHCH3, C(0)NHOCH3) CH2C(0)NH2, CH2C(0)NHCH3and CH2C(0)NHOCH3.
R8
R8 is H, except for when R' is C(=0)NH2, it may alternatively be d.2 aikyl, i.e. methyl or ethyl.
Includes Other Forms
Included in the above are the well known ionic, salt, solvate, and protected forms of these substituents. For example, a reference to carboxyiic acid (-COOH) also includes the anionic (carboxyiate) form (-COO"), a salt or solvate thereof, as well as conventional protected forms. Similarly, a reference to an amino group includes the protonated form (-N+HR1 R2), a salt or solvate of the amino group, for example, a hydrochloride salt, as well as conventional protected forms of an amino group.
Similarly, a reference to a hydroxyl group also includes the anionic form (-0"), a salt or solvate thereof, as well as conventional protected forms of a hydroxyl group.
Isomers, Salts, Solvates, Protected Forms, and Prodrugs
Certain compounds may exist in one or more particular geometric, optical, enantiomeric, diasteriomeric, epimeric, stereoisomeric, tautomeric, conformational, or anomeric forms, including but not limited to, cis- and trans-forms; E- and Z-forms; c-, t-, and r- forms; endo- and exo-forms; R-, S-, and meso-forms; D- and L-forms; d- and l-forms; (+) and (-) forms; keto-, enok and enolate-forms; syn- and anti-forms; synclinal- and anticlinal-forms; a- and β-forms; axial and equatorial forms; boat-, chair-, twist-, envelope-, and haifchair-forms; and combinations thereof, hereinafter collectively referred to as "isomers" (or "isomeric forms").
Note that, except as discussed below for tautomeric forms, specifically excluded from the term "isomers", as used herein, are structural (or constitutional) isomers (i.e. isomers which differ in the connections between atoms rather than merely by the
position of atoms in space). For example, a reference to a methoxy group, -OCH3, is not to be construed as a reference to its structural isomer, a hydroxymethyi group, -CH2OH, Similarly, a reference to ortho-chiorophenyi is not to be construed as a reference to its structural isomer, meta-chlorophenyl. However, a reference to a class of structures may well include structurally isomeric forms falling within that class
(e.g., Ci-7 alky! includes n-propyl and iso-propyi; butyl includes n-, iso-, sec-, and tert- butyl; methoxyphenyl includes ortho-, meta-, and para-methoxyphenyl).
The above exclusion does not pertain to tautomeric forms, for example, keto-, e and enolate-forms, as in, for example, the following tautomeric pairs: keto/enol (illustrated below), imine/enamine, amide/imino alcohol, amidine/amidine,
nitroso/oxime, thioketone/enethiol, N-nitroso/hyroxyazo, and nitro/aci-nitro.

keto eno! enolaie
Note that specifically included in the term "isomer" are compounds with one or more isotopic substitutions. For example, H may be in any isotopic form, including 1H, 2H (D), and 3H (T); C may be in any isotopic form, including 2C, 3C and 4C; O may be in any isotopic form, including 160 and 180; and the like.
Unless otherwise specified, a reference to a particular compound includes ail such isomeric forms, including (wholly or partially) racemic and other mixtures thereof. Methods for the preparation (e.g. asymmetric synthesis) and separation (e.g., fractional crystallisation and chromatographic means) of such isomeric forms are either known in the art or are readily obtained by adapting the methods taught herein, or known methods, in a known manner.
Unless otherwise specified, a reference to a particular compound also includes ionic, salt, solvate, and protected forms of thereof, for example, as discussed below.
It may be convenient or desirable to prepare, purify, and/or handle a corresponding salt of the active compound, for example, a pharmaceutically-acceptable salt Examples of pharmaceutically acceptable salts are discussed in Berge et al. J.
Pharm. Sci., 66, 1-19 (1977).
For example, if the compound is anionic, or has a functional group which may be anionic (e.g., -COOH may be -COO"), then a salt may be formed with a suitable cation. Examples of suitable inorganic cations include, but are not limited to, alkali metal ions such as Na+ and K+, alkaline earth cations such as Ca2+ and g2+, and other cations such as Al3'\ Examples of suitable organic cations include, but are not limited to, ammonium ion (i.e., NH4+) and substituted ammonium ions (e.g., NH3R+, NH2R2+, NHR3+, NR4+). Examples of some suitable substituted ammonium ions are those derived from: ethyiamine, diethyiamine, dicyclohexyiamine, triethylamine, butyiamine, ethylenediamine, ethanoiamine, diethanoiamine, piperazine,
benzylamine, phenyibenzylamine, choline, meglumine, and tromethamine, as well as amino acids, such as lysine and arginine. An example of a common quaternary ammonium ion is N(CH3)4+. If the compound is cationic, or has a functional group which may be cationic (e.g., -NH2 may be -NH3+), then a salt may be formed with a suitable anion. Examples of suitable inorganic anions include, but are not limited to, those derived from the following inorganic acids: hydrochloric, hydrobromic, hydroiodic, sulphuric, sulphurous, nitric, nitrous, phosphoric, and phosphorous. Examples of suitable organic anions include, but are not limited to, those derived from the following organic acids: acetic, propionic, succinic, glycolic, stearic, palmitic, lactic, malic, pamoic, tartaric, citric, gluconic, ascorbic, ma!eic, hydroxymaieic, phenylacetic, glutamic, aspartic, benzoic, cinnamic, pyruvic, saiicyciic, sulfanilic, 2- acetyoxybenzoic, fumaric, phenylsulfonic, toiuenesuifonic, methanesulfonic, ethanesulfonic, ethane disulfonic, oxalic, pantothenic, isethionic, valeric, lactobionic, and gluconic. Examples of suitable polymeric anions include, but are not limited to, those derived from the following polymeric acids: tannic acid, carboxymethyi cellulose. It may be convenient or desirable to prepare, purify, and/or handle a corresponding solvate of the active compound. The term "solvate" is used herein in the conventional sense to refer to a complex of solute (e.g. active compound, salt of active compound) and solvent. If the solvent is water, the solvate may be conveniently referred to as a hydrate, for example, a mono-hydrate, a di-hydrate, a tri-hydrate, etc.
it may be convenient or desirable to prepare, purify, and/or handle the active compound in a chemically protected form. The term "chemically protected form", as used herein, pertains to a compound in which one or more reactive functional groups are protected from undesirable chemical reactions, that is, are in the form of a protected or protecting group (also known as a masked or masking group or a blocked or blocking group). By protecting a reactive functional group, reactions involving other unprotected reactive functional groups can be performed, without affecting the protected group; the protecting group may be removed, usually in a subsequent step, without substantially affecting the remainder of the molecule. See, for example, Protective Groups in Organic Synthesis (T. Green and P. Wuts, Wiley, 1999).
For example, a hydroxy group may be protected as an ether (-OR) or an ester (-OC(=0)R), for example, as: a t-butyi ether; a benzyl, benzhydryl fdiphenylmethyl), or trityl (triphenylmethyl) ether; a trsmethy!sily! or t-butyldimethylsilyl ether; or an acetyl ester (-OC(=0)CH3, -OAc).
For example, an aldehyde or ketone group may be protected as an acetal or ketal, respectively, in which the carbonyl group (>C=0) is converted to a diether (>C(OR)2), by reaction with, for example, a primary alcohol. The aldehyde or ketone group is readily regenerated by hydrolysis using a large excess of water in the presence of acid.
For example, an amine group may be protected, for example, as an amide or a urethane, for example, as: a methyl amide (~NHCO~CH3); a benzyloxy amide (-
NHCO-OCH2C6H5, -NH-Cbz); as a t-butoxy amide (- HCQ-0C(CH3)3, -NH-Boc); a 2- biphenyl-2-propoxy amide (-NHCO-OC CH3 2CeH406H5, -NH-Bpoc), as a 9- fluorenylmethoxy amide (-NH-Fmoc), as a 6-nitroveratryloxy amide (-NH-Nvoc), as a 2-trimethyisiiylethyloxy amide (-NH-Teoc), as a 2,2,2-trichloroethyloxy amide (-NH- Troc), as an ailyioxy amide (-NH-Alloc), as a 2(-phenylsulphonyi)ethyioxy amide (- NH-Psec); or, in suitable cases, as an N-oxide (>ΝΟ·)-
For example, a carboxyiic acid group may be protected as an ester for example, as: an Ci-7 alkyl ester (e.g. a methyl ester; a t-butyl ester); a Ci.7 haioalkyl ester (e.g., a Ci-7 trihaloaikyi ester); a triC alkylsilyl-d.7 alkyl ester; or a C5.2o aryi-Ci-7 alkyl ester
(e.g. a benzyl ester; a nitrobenzyl ester); or as an amide, for example, as a methyl amide.
For example, a thiol group may be protected as a thioether (-SR), for example, as: a benzyl thioether; an acetamidomethyl ether (-S-CH2NHC(=0)CH3). it may be convenient or desirable to prepare, purify, and/or handle the active compound in the form of a prodrug. The term "prodrug", as used herein, pertains to a compound which, when metabolised (e.g. in vivo), yields the desired active compound. Typically, the prodrug is inactive, or less active than the active compound, but may provide advantageous handling, administration, or metabolic properties. For example, some prodrugs are esters of the active compound (e.g. a physiologically acceptable metaboiically labile ester). During metabolism, the ester group (-C(=0)OR) is cleaved to yield the active drug. Such esters may be formed by esterification, for example, of any of the carboxylic acid groups (-C(=0)OH) in the parent compound, with, where appropriate, prior protection of any other reactive groups present in the parent compound, followed by deprotection if required.
Examples of such metaboiically labile esters include those wherein R is C1-7 alkyl (e.g. -Me, -Et); Ci_ aminoaikyi (e.g. aminoethyl; 2-(N,N-diethylamino)ethyl; 2- (4- morpho!ino)ethyl); and acyloxy-Ci_7 alkyl (e.g. acyloxymethyi; acyioxyethyi; e.g. pivaloyloxymethyl; acetoxym ethyl; 1-acetoxyethyl; 1-(1-methoxy-1-methyl)ethyl- carbonxyioxyethyl; 1-(benzoyloxy)ethyl; isopropoxy-carbonyloxymethyl; 1- isopropoxy-carbonyloxyethyl; cyclohexy!-carbonyloxymethy!; 1-cyclohexyl- carbonyloxyethyi; cyclohexyloxy-carbonyloxymethyl; 1-cyciohexyioxy- carbonyloxyethyl; (4-tetrahydropyranyioxy) carbonyioxymethyl; 1~(4~
tetrahydropyranyioxy)carbonyioxyethyi;
(4-tetrahydropyranyl)carbonyloxymethyl; and 1-(4- tetrahydropyranyl)carbonyloxyethyl).
Also, some prodrugs are activated enzymaticaily to yield the active compound, or a compound which, upon further chemical reaction, yields the active compound. For example, the prodrug may be a sugar derivative or other glycoside conjugate, or may be an amino acid ester derivative.
Selectivity
The selectivity of the compounds for inhibiting FAK and Flt3 over other kinases, such as IGF- R, IR and CDKs can be demonstrated by biochemical assay results (see, for example, the FAK kinase assay and FltS assays described below). The compounds of the invention may also be selective over VEGFR1 and/or VEGFR2. The compounds of the present invention may also inhibit VEGRF3.
The selectivity of the compounds for FAK over the inhibition of cytochrome p450 enzymes, specifically the 2C9 and 3A4 isoforms may be determined using standard inhibition assays.
How prone the compounds of the invention may be to the formation of adducts with glutathione may be determined by the protocol described in Walker, et al. Biorg. Med. Chem. Letts. 2008, 18, 6071-6077.
Further Embodiments
The following embodiments and preferences may be combined with one another as appropriate. n some embodiments, R is H and R is:

, wherein RN1 is selected from H, d-3 alkyl (i.e. methyl, ethyl, prop- - yl and prop~2-yl) and C(=0)Me. In some of these embodiments, it may be preferred that RN1 is C(=0)Me. In others of these embodiments, it may be preferred that Rm is H, methyl or ethyl.
In other embodiments, R is H and R is:

, wherein RN2 is selected from H and Cl.3 alkyl (i.e. methyl, ethyl, prop-1-yl and prop-2-yl). In these embodiments, it may be preferred that Rm is selected from H and methyl. In other of these embodiments, it may be preferred that RN2 is ethyl.
in other embodiments, R2 is H and R1 is:
 , wherein Ni> is selected from H and C1.3 alkyl (i.e. methyl, ethyl, prop-1-y! and prop-2-yl). In these embodiments, it may be preferred that RN3 is selected from H and methyl. In other of these embodiments, it may be preferred that RN3 is ethyl.
, wherein Ni> is selected from H and C1.3 alkyl (i.e. methyl, ethyl, prop-1-y! and prop-2-yl). In these embodiments, it may be preferred that RN3 is selected from H and methyl. In other of these embodiments, it may be preferred that RN3 is ethyl.
I odiments, R2 is H and R1 is:

wherein RN4 is selected from H and methyl, in these embodiments, it may be preferred that Rm is H. nts, R2 is H and R1 is:

, wherein R and R are both H or both methyl. In some of these embodiments, it may be preferred that R and R are both H I bodiments, R2 is H and R1 is:

wherein R is selected from H and d.3 alkyl (i.e. methyl, ethyl, prop-1-yi and prop-2-yl). In these embodiments, it may be preferred that RN9 is H. ts, R2 is H and R1 is:
 wherein RN1° is selected from H and C1.3 alkyl (i.e. methyl, ethyl prop-1-yl and prop-2-yl). In these embodiments, it may be preferred that Rmo is selected from H and methyl.
in other embodiments, R2 is H and R1 is:
wherein RN1° is selected from H and C1.3 alkyl (i.e. methyl, ethyl prop-1-yl and prop-2-yl). In these embodiments, it may be preferred that Rmo is selected from H and methyl.
in other embodiments, R2 is H and R1 is:
N1

, wherein RN11 is selected from H and Ci_3 aikyl (i.e. methyl, ethyl, prop-
1-yl and prop-2-yi). In these embodiments, it may be preferred that RN i is H. I ments, R1 is H and R2 is:

, where R is selected from H and d.3 aikyl (i.e. methyl, ethyl, prop- 1-yl and prop-2-yi). In these embodiments, it may be preferred that RNS is selected from H and methyl. iments, R1 is H and R2 is:

, where R is selected from H and C1.3 alkyl (i.e. methyl, ethyl, prop- 1-yl and prop-2-yi). in these embodiments, it may be preferred that RNS is selected from H and methyl. r preferred that R1 is H and R2 is:

In some embodiments, when R1 and R2 together form the group -CH2-N(R 12)-C2H4-, RN 2 is selected from H and C |.3 alkyl (i.e. methyl, ethyl, prop-1-yl and prop~2-yl). in these embodiments, it may be preferred that Rm2 is selected from H and methyl, and it may be more preferred that RN12 is methyl.
In some embodiments, R4 is selected from CF3, CI, Br, CF2H , and CN.
In further embodiments, R4 is selected from CF3, CI and CF2H. In further
embodiments, R4 is selected from CF3 and CI. It may be preferred that R4 is CF3.
in some embodiments, it may be preferred that R5 is a group of the following formulae:

(R5a)
In some embodimen 5 is a group selected from:

(R5c) (R d) (R5s)
In these embodiments, R5 may be preferably selected from R5e and R5c, and maymore preferably be R5e.
In some embodiments, R7 is H and Re is (CHRc 1)ri C(0)N(RN6)Z1.
In further embodiments, R7 is H and R6 is selected from CH2C(0)NH2, CH2C(0)NHCH3, CHCH3C(0)NH2 and CHCH3C(0)NHCH3.
It may be preferred that R7 is H and R6 is selected from CH2C(0)NH2) CHCH3C(0)NH2 and CH2C(0)NHCH3, and more preferably from CH2C(0)NH: CHCH3C(0)NH2.
In some embodiments, R6 is H and R7 is (CH2)m1C(0)N(RM1)Y1.
In further embodiments, R6 is H and R7 is selected from C(0)NH2, C(0)NHCH CH2C(0)NH2 and CH2C(0)NHCH3.
It may be preferred that R6 is H and R7 is C(0)NH2.
In some embodiments where R8 is H and R7 is C(0)NH2, R8 is methyl.
in some embodiments, it may be preferred that R5 is a group of the following formula:

(R 5b\
In selected e

wherein R 'a is selected from:
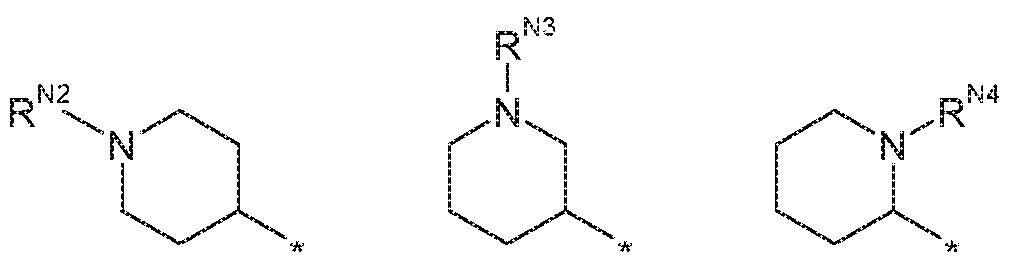
wherein:
•s selected from H, Ci.3 alkyl and C(=G) e;
R s selected from H, C1..3 alkyl and C(=0)Me; and
N4 s selected from H and CH.
The preferences expressed above for R , R and RN4 apply here as we!
In selected e

wherein R1 b is selected from:

RN1 is selected from H, C1..3 alkyl and C(=0)Me;
RN2 is selected from H, C1.3 alkyl and C(=0)Me;
RN3 is selected from H, C1.3 alkyl and C(=0)Me;
RN4 is selected from H and CH3;
RN' and RN8 are independently selected from H and CH3;
RN9 is selected from H, C1.3 alkyl and C(=0) e;
RN1° is selected from H, C1.3 alkyl and C(=0) e; and
RN11 is selected from H, Ο μ3 alkyl and C(=0)Me.
The preferences expressed above for RN1 , RN2, RN3, RN4, RNJ, Rm, RNg, RM0 and R1 apply here as well. In particular, compounds of formula lb where R is selected from:
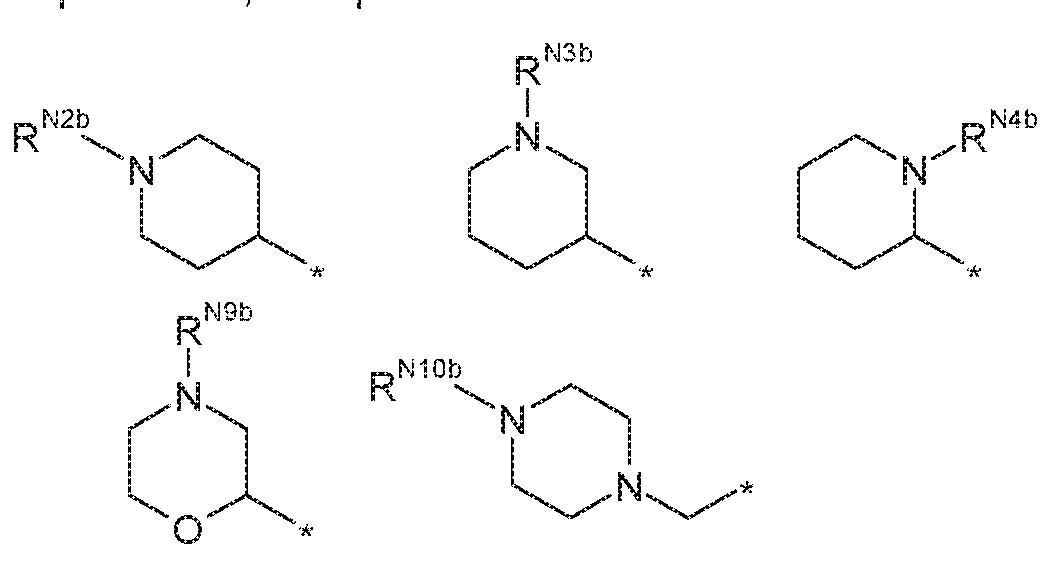
and wherein:
RN2d is selected from H , methyl and ethyl;
RN3B is selected from H and methyl;
RN4B is H ;
RN9B is H ; and
RN1° is selected from H and methyl.
Embodiments of the inventions are compounds of the examples, including
compounds 1 to 40. Embodiments of particular interest include compounds 4, 5, 8, 11 and 16. Further embodiments of particular interest include compounds 21 , 22, 25, 31 and 36.
General synthesis methods
The compounds of the invention can be prepared employing the following general methods and using procedures described in detail in the experimental section. The reaction conditions referred to are illustrative and non-limiting.
Compounds of formula I, as described above, can be prepared by synthetic strategies outlined below, wherein the definitions above apply:
Scheme A

F2 F3
Compounds of formula F1 may be reacted with substituted commercial or synthetic anilines of formula F2 (as prepared in scheme C, D , E, F, G, H, I, J, K and L) to form intermediates of formula F3 where L1 and L2 may be the same or different and include CI, Br, I, SMe, S02Me and R4 = CF3, halogen, CF2H or CN.
An example of a commercial aniline is:

which is useful for preparing compounds where R and R together form the group - CH2-N(RN12)-C2H4-.
Compounds of the formula F1 may be prepared where L1 and L2 are different (see scheme B) to allow regioselective substitution or when L1=L2 suitable reaction conditions can be employed (choice of solvent, reaction temperature, addition of a Lewis acid, for example ZnCi2 in diethyl ether) to allow L1 to be selectively displacec
over L2. Where regiochemical mixtures and di-substitution are obtained the regioisomers may be separated by chromatography.
Compounds of the formula F1 where L*'~L2 are either commercially available, for example 2,4-dichloro-5-(trifluoromethyi)pyrimidine, 2,4-dichloro-5-fluoropyrimidine, 2,4,5-trichloropyrimidine, 2,4-dichloro-5-bromopyrimidine, 2,4-dichloro-5- iodopyrimidine, 2,4-dichloro-5-cyanopyrimidine or may be prepared readily from commercial starting materials. Where R4 = CF3 and differentiation of L1 and L2 is desirable, the method outlined in scheme B may be employed.
Scheme B

Commercially available 2,4-dichioro-5-(trifluoromethyl)pyrimidine (G1 ) can be selectively reacted with sodium thiomethoxide in the presence of zinc(l l) chloride to give 2-thiomethyl-4-chioro-5-(trifluoromethyl)pyrimidine (G2), 2-Thiomethyl-4-chloro- 5-(trifluoromethyl)pyrimidine (G2) can be further reacted, for example by conversion to 2-thiomethyl-4-iodo-5-(trifluoromethyl)pyrimidine (G3) under Finkelstein conditions and/or by oxidation with mCPBA to give the corresponding suifone if further differentiation of the 2 and 4-position is required or if additional activation is desirable.
Scheme C

G4 G5 G6
Commercially available 1-(4-nitrophenyi)piperazine (G4), or a salt thereof, can be reacted with Boc anhydride to give ferf-butyl 4-(4-nitrophenyi)piperazine-1 - carboxylate (G5). Subsequent reduction via hydrogenation in the presence of a
catalyst, for example palladium on charcoal, gives the corresponding aniline, tert- butyi 4-(4-aminophenyi)piperazine-1-carboxylate (G6).
Scheme D

G9
ferf- Butyl 4-(3-aminophenyl)piperazine-1-carboxyiate (G9) can be prepared by coupling of commercially available fe/f-butyl piperazine-1-carboxyiate (G7) and compounds of the formula F4, where L3=l or Br, in a Buchwald type reaction to give terf-butyl 4-(3-nitrophenyl)piperazine-1-carboxyiate (G8). Reduction with hydrogen in the presence of a catalyst, for example palladium on charcoal, gives ieri-bufyl 4~(3~ aminophenyl)piperazine-1-carboxylate (G9).
Scheme E

The corresponding 4-piperidine analogues of G8 can be prepared by a sequence of reactions starting with the conversion of commercially available fert-butyl 4- oxopiperidine-1-carboxylate (G10) to vinyl triflate G11. Coupling of G11 in a Suzuki type reaction with (4-nitrophenyl)boronic acid (G12) gives tetrahydropyridine (G13). Subsequent reduction via hydrogenation in the presence of a catalyst, for example palladium on charcoal, gives gives ferf-butyl 4-(4-aminophenyi)piperidine-1- carboxylate (G 4).
Scheme F

G18 G17
The corresponding 4-{3-aminophenyl)piperidine analogue of G9 can be prepared by a sequence of reactions starting with the conversion of commercially available tert- butyl 4-oxopiperidine-1-carboxyiate (G10) to vinyl trifiate G11. Coupling of G11 in a Suzuki type reaction with (3-nitrophenyl)boronic acid (G15) gives tetrahydropyridine (G16). Subsequent reduction via hydrogenation in the presence of a catalyst, for example palladium on charcoal, gives iert-butyl 4-(3-aminophenyl)piperidine-1- carboxylate (G 7).
Scheme G

The 3-(4-aminophenyi)piperidine regioisomers of G14 can be prepared by reaction of commercially available compounds of the formula F5, where L3=l or Br, with pyridin- 3-ylboronic acid (G18) in a Suzuki type reaction to form 3-(4-nitrophenyl)pyridine (G19). Reduction of G19 with hydrogen in the presence of a catalyst, for example platinum oxide, gives 4-(piperidin-3-yl)aniiine (G20) which may be protected using Boc anhydride to give ferf-butyl 3-(4-aminophenyi)piperidine-1-carboxyiate (G21),


G24 G2S
The 2-(4-aminopheny!)piperidine regioisomer of G14 can be prepared by reaction of commercially available compounds of the formula F5, where L3=l or Br, with pyridin- 2-ylboronic acid (G22) in a Suzuki type reaction to form 2-(4-nitrophenyl)pyridine (G23). Reduction of G23 with hydrogen in the presence of a catalyst, for example platinum oxide, gives 4-(piperidin-2-yl)aniline (G24) which may be protected using Boc anhydride to give fe/t-butyl 2-(4-aminophenyl)piperidine-1-carboxylate (G25).
Scheme I
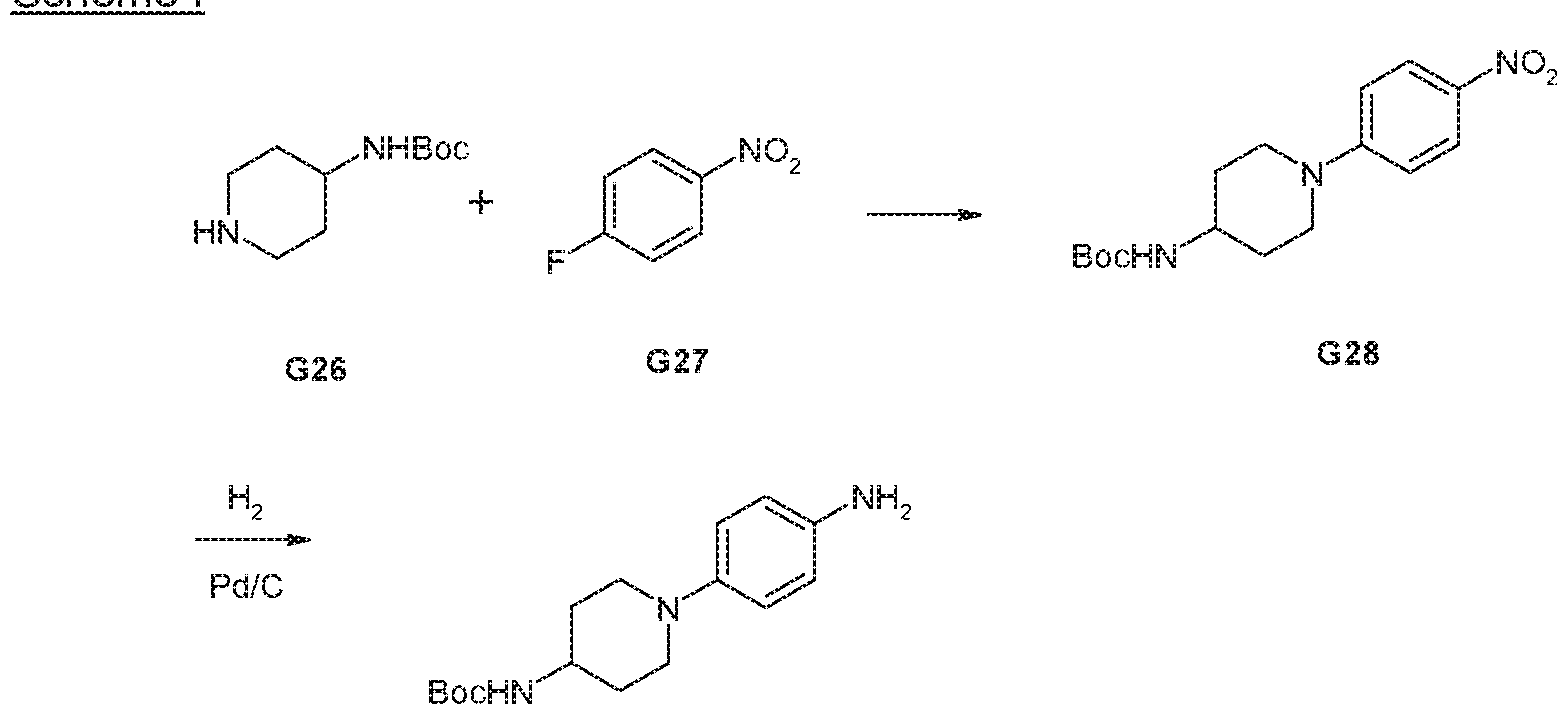
G29
ferf-Butyl (1-(4-aminophenyl)piperidin-4-yl)carbamate (G29) can be prepared by nucieophilic aromatic substitution of commercially available ferf-butyi piperidin-4- ylcarbamate (G26) and 1-fluoro-4-nitrobenzene (G27) under thermal conditions to give ferf-butyl (1-(4-nitrophenyl)piperidin-4-yl)carbamate (G28). Reduction of G28
with hydrogen in the presence of a catalyst, for example 0% palladium on charcoal gives terf-butyl (1-(4-aminophenyl)piperidin-4-yi)carbamate (G29).
Scheme J
 ferf- Butyl 4-(4-aminobenzyi)piperazine-1-carboxylate (G32) can be prepared by the nucleophilic displacement of commercially available 1-(bromomethyl)-4-nitrobenzene (G30) with tert-butyl piperazine-1-carboxylate (G7) to give terf-butyl 4-(4- nitrobenzyi)piperazine-1-carboxylate (G31 ). Subsequent reduction with hydrogen in the presence of a catalyst, for example 10% % palladium on charcoal, gives terf-butyl 4-(4-aminobenzyl)piperazine-1-carboxylate (G32).
ferf- Butyl 4-(4-aminobenzyi)piperazine-1-carboxylate (G32) can be prepared by the nucleophilic displacement of commercially available 1-(bromomethyl)-4-nitrobenzene (G30) with tert-butyl piperazine-1-carboxylate (G7) to give terf-butyl 4-(4- nitrobenzyi)piperazine-1-carboxylate (G31 ). Subsequent reduction with hydrogen in the presence of a catalyst, for example 10% % palladium on charcoal, gives terf-butyl 4-(4-aminobenzyl)piperazine-1-carboxylate (G32).
Scheme K

G36 G37 G38 Commercially available terf-butyl 3-oxopyrroiidine-1-carboxylate (G33) can be converted to a mixture of vinyl triflates (G34) and (G35) in the presence of a triflamide and a suitable base, for example NaH DS. Coupling of the mixture with (4- nitrophenyi)boronic acid (G12) under Suzuki conditions gives dihydropyrroies (G38) and (G37), Reduction of this mixture using hydrogen in the presence of a catalyst, for example 10% palladium on charcoal, gives terf-butyl 3-(4-aminophenyi)pyrrolidine-1- carboxylate (G38).
cheme L

Commercially available 2-bromo-1-(4-nitrophenyl)ethanone (G39) can be reduced and cyclised to give epoxide (G40), Opening of the epoxide with tosylamide followed by cyclisation with (2-bromoefhy!)diphenylsulfonium trifluoromethanesulfonate gives morphoiine (G42). Cleavage of the sulphonamide and subsequent re-protection with Boc anhydride gives carbamate (G44), Reducution using hydrazine in the presence of iron(ill) chloride gives terf-butyi 2-(4-aminophenyl)morpholine-4-carboxylate (G45). Scheme M

Compounds of the formula F6 may be reacted to form esters of the formula F7 where X=Br or I, R8=H or Me and Y is selected from a single bond, -CH2- and -CHCH3~, When R01= f-Bu, Boc anhydride may be employed or where R°1=Me methanol in the presence of an acid, for example sulfuric acid, may be used to form the desired ester. Esters of the formula F7 can be reacted with terminal acetylenes of the formula F8 in a Sonagashira type coupling to give acetylenes of the formula F9 where R9=TMS,
TES or {CH3)2COH. R9 may then be removed to generate compounds of the formula F10. When R9=T S or TES potassium carbonate or tetra-n-butyl ammonium fluoride may be employed to induce this transformation. When R9=(CH3)2C*OH, sodium hydride in refluxed toluene may be used.
Scheme N

F13 Pi2
Pyrimidines of the formula F3 may be reacted with terminal acetylenes of the formula F10 to give acetylenes of the formula F11 in a Sonagashira type coupling. The acetylene in compounds of the formula F11 may be reduced to an alkane of the formula F26 using hydrogen gas in the presence of a transition metal catalyst. The exact choice of catalyst and conditions employed is dependant on the nature of R4. For example, where R =CF3, 10% Pd/C may be used, where R4=CI, platinum oxide is employed. Esters of the formula F12 may then be deprotected to give carboxyiic acids of the formula F13. Where R°1= e, lithium hydroxide solutions may be employed. Where R° =f-Bu, acidic solutions, for example trifiuoroacetic acid in dichioromethane may be used. It will be appreciated that under acidic conditions Boc protecting groups in R1 and R2 will also be cleaved.
cheme O

Carboxyiic acids of the formula F13 can be converted to amides of the formula F14 using a suitable amine or ammonia salt in the presence of a peptide coupling agent, for example HATU.
Scheme P

Alternatively, when R -Me, esters of the formula F12 may be directly converted to amides of the formula F14 by reaction with an amine at elevated temperatures.
Scheme Q

F15 F16
Where molecules with lactams fused to the right hand side aromatic ring are required compounds of the formula F15 can be reacted with terminal acetylenes of the formula F8 in a Sonagashira type coupling to give acetylenes of the formula F16 where R9=TMS, TES or (CH3)2C*OH. R9 may then be removed to generate compounds of the formula F17. When R9=T S or TES, potassium carbonate or tetra- >butyi ammonium fluoride may be employed to induce this transformation. When R9=(CH3)2C*OH, sodium hydride in refluxed toluene may be used.
Compounds of the formula F1 / can then be coupled to compounds of the form (as in Scheme N) and further elaborated as described above.
Scheme R

Compounds of the formula F14, or analogues containing lactams, with Boc protecting groups present in R1 or R2 (in the place of RN1 to RN12) may then be deprotected under acidic conditions, for example using frifluoroacetic acid in dichioromethane solutions, to give the corresponding parent compounds of the formula F18.

Compounds of the formula F 8 may then be further modified by derivitisation of the amine functionality. For example, compounds of the formula F19 where RN1, RN2, RN3 R 4, R 5 , R 6, R 7, RN8, RN9, RN °, RN11 or RN12 = Me may be prepared by reductive alkylation with formaldehyde in the presence of sodium triacetoxyborohydride.
Derivatives were RN1, RN2, RN3, RN5, Rm, RN7, RtiB, RN9, RN1°, RN11 or RN12 = Et may be prepared by reductive alkylation with acetaldehyde in the presence of sodium triacetoxyborohydride. Compounds of the formula F19 where RN1, RNZ, RN3, RNs, R 6,
RN'', RN8, RN9, RN °, RN 1 or RN12 = acetyl may be prepared by reaction of compounds of the formula F 8 with a suitable acyiating agent, for example acetic anhydride.
Alternatively, when compounds in which R5=heteroary! are desired heteroaryl analogues of F10 may be prepared as outlined in Schemes T, U and V. These heteroaryl acetylenes can be coupled to compounds of the formula F3, and then further elaborated in an analogous manner to that described above in schemes N, O, P, R and S.
Scheme T

G49
For pyrazine containing analogues, 2,3-di-ch!oropyrazine (G48) can be reacted with ethyl acetate in the presence of LiHMDS to give ester G47. Coupling of G47 with TMS acetylene under Sonagashira conditions gives acetylene G48. Removal of the trimethyisiiy! group using TBAF gives ethyl 2~(3~ethynylpyrazin-2-yl)acetate (G49).
cheme U

G57
For pyrimidine analogues, diethyl succinate (G50) and ethyi formate (G51) can be condensed to give aldehyde G52 in the presence of sodium metal. Cyclisation using thiourea gives 4-oxo-2-thioxo~1 ,2,3,4-tetrahydropyrimidine (G53). Desulfurisation using Raney-nickel gives pyrimidone G54, which can be converted to 4-chloro pyrimidine G55 using phosphorous oxychloride. Coupling of TES-acety!ene under Sonagashira conditions, followed by removal of the triethyisiiyl group using TBAF gives ethyl 2-(4-ethynylpyrimidin-5-yl)acetate (G57).
Scheme V

G82 G63 G64
For 3-pyridyl acetates, 2-(pyrid!n-3-yi)acetonitriie (G58) can be oxidised to /V-oxide G59. Chlorination with phosphorous oxychloride gives 2~chloropyridine G80 which can be hydrolysed with sodium hydroxide to acetic acid G61. Ester formation using
methanol gives 2-chloropyridine ester G62. Coupling of TES-acety!ene under Sonagashira conditions, followed by removal of the triethyisiiyl group using TBAF gives methyl 2-(2-ethynylpyridin-3-yl)acetate (G84).
Alternatively, heteroaryl acetylenes anaiagous to F10 can be hydroborylated to give vinyl boranes as in scheme W. These can be coupled using Suzuki chemistry to compounds of the formula F3, then further elaborated in an analogous manner to the described above in schemes N, O, P, R and S.
Scheme W

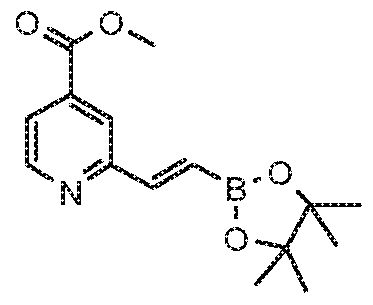
G68
Methyl 2-bromoisonicotinate (G65) can be coupled using Sonagashira conditions give acetylene G66. Removal of the trimethylsilyi group with TBAF gives terminal acetylene G67 which can be hydroborylated to give (E)-methyl 2-(2-(4,4,5,5- tetramethyl-1 ,3,2-dioxaborolan-2-yl)vinyl)isonicotinate (G88).
An alternate strategy for the formation of compounds of the formula F12, where R4=CF3 and R8=H, is to prepare compounds of the formula F22, as outlined in scheme X.
Scheme X

Coupling of esters of the formula F10, where R™H, with 4-iodo-2-(methylthio)-5- (trifiuoromethyl)pyrimidine (G3) under Sonagashira conditions gives acetylenes of the formula F20, Oxidation, using CPBA, gives su!fones of the formula F21 , Reduction of the acetylene using hydrogen, in the presence of a catalyst, for example 10% palladium on charcoal, gives compounds of the formula F22. Compounds of the formla F22 can be reacted with anilines of the formula F2 under acidic conditions, for example in the presence of trifluoro acetic acid to give compounds of the formula F12 which can then be further elaborated as described above. Use of Compounds of the Invention
The present invention provides active compounds, specifically, active 2,4,5- substituted pyrimidines .
The term "active", as used herein, pertains to compounds which are capable of inhibiting FAK activity as well as the activity of Flt3 (and possibly VEGFR3), and specifically includes both compounds with intrinsic activity (drugs) as well as prodrugs of such compounds, which prodrugs may themselves exhibit little or no intrinsic activity.
Assays which may be used in order to assess the FAK, Flt3 and VEGFR3 inhibition offered by a particular compound are described in the examples below, The present invention further provides a method of inhibiting FAK activity, as well as the activity of Flt3, in a cell, comprising contacting said cell with an effective amount of an active compound, preferably in the form of a pharmaceutically acceptable composition. Such a method may be practised in vitro or in vivo. The method may also include inhibiting VEGFR3 in a ceil.
The present invention further provides active compounds which inhibit FAK activity, as well as the activity of Fit3, as well as methods of methods of inhibiting FAK activity, as well as the activity of Fit3, comprising contacting a cell with an effective amount of an active compound, whether in vitro or in vivo. The active compounds may also include inhibit the activity of VEGFR3 in a cell.
Active compounds may also be used as part of an in vitro assay, for example, in order to determine whether a candidate host is likely to benefit from treatment with the compound in question.
The invention further provides active compounds for use in a method of treatment of the human or animal body. Such a method may comprise administering to such a subject a therapeutically-effective amount of an active compound, preferably in the form of a pharmaceutical composition.
The term "treatment", as used herein in the context of treating a condition, pertains generally to treatment and therapy, whether of a human or an animal (e.g. in veterinary applications), in which some desired therapeutic effect is achieved, for example, the inhibition of the progress of the condition, and includes a reduction in the rate of progress, a halt in the rate of progress, amelioration of the condition, and cure of the condition. Treatment as a prophylactic measure (i.e. prophylaxis) is also included.
The term "therapeutically-effective amount" as used herein, pertains to that amount of an active compound, or a material, composition or dosage from comprising an
active compound, which is effective for producing some desired therapeutic effect, commensurate with a reasonable benefit/risk ratio.
Acute M elo Leukemia
The present invention provides active compounds which inhibit Focal Adhesion Kinase (FAK), and FIVlS-like tyrosine kinase (Flt3), including mutants thereof. The inhibition of these is thought to be useful in the treatment of Acute Myleloid Leukemia (A ML). Therefore, the present invention relates to the treatment of AML with the compounds as described above.
In particular, the present invention relates to the treatement of:
(i) Acute Myeloid leukemia in patients who express activated forms of F!t3;
(ii) Acute Myeloid leukemia in patients who express ITD-Flt3;
(iii) Acute Myeloid leukemia in patients who express ITD-Flt3 and FAK; and/or
(iv) Acute Myeloid leukemia in patients who express ITD~Flt3 and constitutively active splice variants of FAK,
One of ordinary skill in the art is readily able to determine whether or not a candidate compound treats a cancerous condition for any particular cell type, either alone or in combination.
The treatment defined hereinbefore may be applied as a sole therapy or may involve, in addition to the compound of the invention, conventional surgery or radiotherapy or chemotherapy. Such chemotherapy may include one or more of the following categories of anti-tumour agents:-
(i) other antiproliferative/antineoplastic drugs and combinations thereof, as used in medical oncology, such as alkylating agents (for example cisplatin, oxalipiatin, carboplatin, cyclophosphamide, nitrogen mustard, meiphalan, chlorambucil, busulphan, temozolamide and nitrosoureas); antimetabolites (for example
gemcitabine and antifolates such as fluoropyrimidines like 5 fiuorouracil and tegafur, raltitrexed, methotrexate, cytosine arabinoside, and hydroxyurea); antitumour antibiotics (for example anthracyciines like adriamycin, bleomycin, doxorubicin, daunomycin, epirubicin, idarubicin, mitomycin-C, dactinomycin and mithramycin); antimitotic agents (for example vinca alkaloids like vincristine, vinblastine, vindesine and vinoreibine and taxoids like taxol and docetaxei (Taxotere) and polokinase
inhibitors); and topoisomerase inhibitors (for example epipodophyilotoxins like etoposide and teniposide, amsacrine, topotecan and camptothecin);
(ii) cytostatic agents such as antioestrogens (for example tamoxifen, fulvestrant, toremifene, raloxifene, dro!oxifene and iodoxyfene), antiandrogens (for example bicalutamide, flutamide, nilutamide and cyproterone acetate), LHRH antagonists or LHRH agonists (for example goserelin, ieuprorelin and buserelin), progestogens (for example megestrol acetate), aromatase inhibitors (for example as anastrozole, letrozole, vorazole and exemestane) and inhibitors of 5*-reductase such as finasteride;
(iii) anti-invasion agents (for example c-Src kinase family inhibitors like 4-(6- chloro-2,3-mefhylenedioxyaniiino)-7-[2-(4-mefhylpiperazin-1-yi)ethoxy]-5- tetrahydropyran-4-yloxyquinazoiine (AZD0530; International Patent Application WO 01/94341), N-(2-chloro-6-methylphenyl)-2-{8-[4-(2-hydroxyethyl)piperazin-1-yl]-2- methylpyrirnidin-4-ylamino}thiazole-5-carboxamide (dasatinib, BMS-354825; J. Med. Chem,, 2004, 47, 6858-6661 and and 4-((2,4-dichloro-5-methoxyphenyl)amino)-6- methoxy-7-(3-(4-methylpiperazin-1-yl)propoxy)quinoline-3-carbonitrile (bosutinib, SKi-606; Cancer research (2003), 63(2), 375-81), and metailoproteinase inhibitors like marimastat, inhibitors of urokinase plasminogen activator receptor function or antibodies to Heparanase);
(iv) inhibitors of growth factor function: for example such inhibitors include growth factor antibodies and growth factor receptor antibodies (for example the anti erbB2 antibody trasfuzumab [HerceptinT], the anti-EGFR antibody panitumumab, the anti erbB1 antibody cetuximab [Erbitux, C225] and any growth factor or growth factor receptor antibodies disclosed by Stern et al. Critical reviews in
oncology/haemato!ogy, 2005, Vol. 54, pp11-29); such inhibitors also include tyrosine kinase inhibitors, for example inhibitors of the epidermal growth factor family (for example EGFR family tyrosine kinase inhibitors such as N-(3-chloro-4-fluorophenyl)- 7-methoxy~8-(3-morpholinopropoxy)quinazolin-4-amine (gefitinib, ZD 839), N-(3- ethynylphenyl)-6,7-bis(2-methoxyethoxy)quinazolin-4-amine (eriotinib, OSI 774) and 6-acrylamido-N-(3-chloro-4-fluorophenyi)-7-(3-morpholinopropoxy)-quinazolin-4- amine (CI 1033), erbB2 tyrosine kinase inhibitors such as iapatinib, inhibitors of the hepatocyte growth factor family, inhibitors of the platelet-derived growth factor family such as imatinib, inhibitors of serine/threonine kinases (for example Ras/Raf signalling inhibitors such as farnesyl transferase inhibitors, for example sorafenib (BAY 43-9006)), inhibitors of cell signalling through MEK and/or AKT kinases,
inhibitors of the hepatocyte growth factor family, c-kit inhibitors, ab! kinase inhibitors, IGF receptor (insulin-like growth factor) kinase inhibitors; aurora kinase inhibitors (for example AZD1 52, PH739358, VX-680, MLN8054, R763, MP235, P529, VX-528 AND AX39459) and cyclin dependent kinase inhibitors such as CDK2 and/or CDK4 inhibitors;
(v) antiangiogenic agents such as those which inhibit the effects of vascular endothelial growth factor, [for example the anti vascular endothelial cell growth factor antibody bevacizumab (AvastinT) and VEGF receptor tyrosine kinase inhibitors such as 4-(4-bromo-2-fluoroanilino)-6-methoxy-7-(1-methylpiperidin-4- ylmethoxy)quinazoiine (ZD6474; Example 2 within WO 01/32651), 4-(4-fluoro-2- methylindol-5-yloxy)-6-methoxy-7-(3-pyrrolidin-1-ylpropoxy)quinazoline (AZD2171 ; Example 240 within WO 00/47212), vataianib (PTK787; WO 98/35985) and SU11248 (sunitinib; W 01/60814), compounds such as those disclosed in International Patent Applications W097/22598, WO 97/30035, WO 97/32858 and WO 98/13354 and compounds that work by other mechanisms (for example iinomide, inhibitors of integrin avb3 function and angiostatin)];
(vi) vascular damaging agents such as Combretastatin A4 and compounds disclosed in International Patent Applications WO 99/02166, WO 00/40529, WO 00/41669, WO 01/92224, WO 02/04434 and WO 02/08213;
(vii) antisense therapies, for example those which are directed to the targets listed above, such as ISIS 2503, an anti-ras antisense;
(viii) gene therapy approaches, including for example approaches to replace aberrant genes such as aberrant p53 or aberrant BRCA1 or BRCA2, GDEPT (gene directed enzyme pro drug therapy) approaches such as those using cytosine deaminase, thymidine kinase or a bacterial nitroreductase enzyme and approaches to increase patient tolerance to chemotherapy or radiotherapy such as multi drug resistance gene therapy; and
(ix) immunotherapy approaches, including for example ex vivo and in vivo approaches to increase the immunogenicity of patient tumour cells, such as transfection with cytokines such as interieukin 2, interieukin 4 or granulocyte macrophage colony stimulating factor, approaches to decrease T ceil anergy, approaches using transfected immune cells such as cytokine transfected dendritic cells, approaches using cytokine transfected tumour cell lines and approaches using anti idiotypic antibodies.
Diseases ameliorated by inhibition of Fit3, or FLt3 and FAK
The inhibition of Fit3 may be useful in the treatment of autoimmune diseases, such as multiple sclerosis (Whartenby, K, et ai., PNAS (2005), 102(48): 16741-16746). The inhibition of Flt3 (including mutations such as ITD and D835 point mutations) may also be useful in treating myelodyspiastic syndrome (IV1DS) and chronic myeloproliferative diseases (C PDs) (Line, P., et a!., Am J Clin Pathol
2006:126:530-533), These diseases may also be treated with combinations as describe above.
Particularly relevant may be the combinations with thalidomide and thalidomide analogues such as !enaiidomide and pomalidomide. Such combinations may be appropriate in the treatment of myelodyspiastic syndrome ( DS). Administration
The active compound or pharmaceutical composition comprising the active compound may be administered to a subject by any convenient route of
administration, whether systemicaily/ peripherally or at the site of desired action, including but not limited to, oral (e.g. by ingestion); topical (including e.g.
transdermal, intranasal, ocular, buccal, and sublingual); pulmonary (e.g. by inhalation or insufflation therapy using, e.g. an aerosol, e.g. through mouth or nose); rectal; vaginal; parenteral, for example, by injection, including subcutaneous, intradermal, intramuscular, intravenous, intraarterial, intracardiac, intrathecal, intraspinal, intracapsular, subcapsular, intraorbital, intraperitoneal, intratracheal, subcuticular, intraarticular, subarachnoid, and intrasternai; by implant of a depot, for example, subcufaneously or intramuscularly. The subject may be a eukaryote, an animal, a vertebrate animal, a mammal, a rodent (e.g. a guinea pig, a hamster, a rat, a mouse), murine (e.g. a mouse), canine (e.g. a dog), feline (e.g. a cat), equine (e.g. a horse), a primate, simian (e.g. a monkey or ape), a monkey (e.g. marmoset, baboon), an ape (e.g. gorilla, chimpanzee, orang-utan, gibbon), or a human.
Formulations
While it is possible for the active compound to be administered alone, it is preferable to present it as a pharmaceutical composition (e.g. formulation) comprising at least one active compound, as defined above, together with one or more pharmaceutically
acceptable carriers, adjuvants, excipients, diluents, fillers, buffers, stabilisers, preservatives, lubricants, or other materials well known to those skilled in the art and optionally other therapeutic or prophylactic agents. Thus, the present invention further provides pharmaceutical compositions, as defined above, and methods of making a pharmaceutical composition comprising admixing at least one active compound, as defined above, together with one or more
pharmaceutically acceptable carriers, excipients, buffers, adjuvants, stabilisers, or other materials, as described herein.
The term "pharmaceutically acceptable" as used herein pertains to compounds, materials, compositions, and/or dosage forms which are, within the scope of sound medical judgement, suitable for use in contact with the tissues of a subject (e.g. human) without excessive toxicity, irritation, allergic response, or other problem or complication, commensurate with a reasonable benefit/risk ratio. Each carrier, excipient, etc. must also be "acceptable" in the sense of being compatible with the other ingredients of the formulation.
Suitable carriers, excipients, etc. can be found in standard pharmaceutical texts, for example, Remington's Pharmaceutical Sciences, 18th edition, Mack Publishing Company, Easton, Pa,, 1990.
The formulations may conveniently be presented in unit dosage form and may be prepared by any methods well known in the art of pharmacy. Such methods include the step of bringing into association the active compound with the carrier which constitutes one or more accessory ingredients. In general, the formulations are prepared by uniformly and intimately bringing into association the active compound with liquid carriers or finely divided solid carriers or both, and then if necessary shaping the product.
Formulations may be in the form of liquids, solutions, suspensions, emulsions, elixirs, syrups, tablets, losenges, granules, powders, capsules, cachets, pills, ampoules, suppositories, pessaries, ointments, gels, pastes, creams, sprays, mists, foams, lotions, oils, boluses, electuaries, or aerosols.
Formulations suitable for oral administration (e.g. by ingestion) may be presented as discrete units such as capsules, cachets or tablets, each containing a predetermined amount of the active compound; as a powder or granules; as a solution or suspension in an aqueous or non-aqueous liquid; or as an oil-in-water liquid emulsion or a water-in-oil liquid emulsion; as a bolus; as an electuary; or as a paste.
A tablet may be made by conventional means, e.g., compression or moulding, optionally with one or more accessory ingredients. Compressed tablets may be prepared by compressing in a suitable machine the active compound in a free- flowing form such as a powder or granules, optionally mixed with one or more binders (e.g. povidone, gelatin, acacia, sorbitol, fragacanth, hydroxypropyi methyl cellulose); fillers or diluents (e.g. lactose, microcrystaiiine cellulose, calcium hydrogen phosphate); lubricants (e.g. magnesium stearate, talc, silica); disinfegrants (e.g. sodium starch glycolate, cross-linked povidone, cross-linked sodium carboxymethyi cellulose); surface-active or dispersing or wetting agents (e.g. sodium iauryi sulfate); and preservatives (e.g. methyl p-hydroxybenzoate, propyl p-hydroxybenzoate, sorbic acid). Moulded tablets may be made by moulding in a suitable machine a mixture of the powdered compound moistened with an inert liquid diluent. The tablets may optionally be coated or scored and may be formulated so as to provide slow or controlled release of the active compound therein using, for example,
hydroxypropy!methyi cellulose in varying proportions to provide the desired release profile. Tablets may optionally be provided with an enteric coating, to provide release in parts of the gut other than the stomach. Formulations suitable for topical administration (e.g. transdermal, intranasal, ocular, buccal, and sublingual) may be formulated as an ointment, cream, suspension, lotion, powder, solution, past, gel, spray, aerosol, or oil. Alternatively, a formulation may comprise a patch or a dressing such as a bandage or adhesive plaster impregnated with active compounds and optionally one or more excipients or diluents.
Formulations suitable for topical administration in the mouth include losenges comprising the active compound in a flavoured basis, usually sucrose and acacia or tragacanfh; pastilles comprising the active compound in an inert basis such as gelatin
and glycerin, or sucrose and acacia; and mouthwashes comprising the active compound in a suitable liquid carrier.
Formulations suitable for topical administration to the eye also include eye drops wherein the active compound is dissolved or suspended in a suitable carrier, especially an aqueous solvent for the active compound.
Formulations suitable for nasal administration, wherein the carrier is a solid, include a coarse powder having a particle size, for example, in the range of about 20 to about 500 microns which is administered in the manner in which snuff is taken, i.e. by rapid inhalation through the nasal passage from a container of the powder held close up to the nose. Suitable formulations wherein the carrier is a liquid for administration as, for example, nasal spray, nasal drops, or by aerosol administration by nebuliser, include aqueous or oily solutions of the active compound.
Formulations suitable for administration by inhalation include those presented as an aerosol spray from a pressurised pack, with the use of a suitable propeilant, such as dichlorodifluoromethane, trichlorofluoromethane, dichoro-tetraf!uoroethane, carbon dioxide, or other suitable gases.
Formulations suitable for topical administration via the skin include ointments, creams, and emulsions. When formulated in an ointment, the active compound may optionally be employed with either a paraffinic or a water-miscible ointment base. Alternatively, the active compounds may be formulated in a cream with an oil-in- water cream base, if desired, the aqueous phase of the cream base may include, for example, at least about 30% w/w of a poiyhydric alcohol, i.e., an alcohol having two or more hydroxy! groups such as propylene glycol, butane-1 ,3-diol, mannitol, sorbitol, glycerol and polyethylene glycol and mixtures thereof. The topical formulations may desirably include a compound which enhances absorption or penetration of the active compound through the skin or other affected areas. Examples of such dermal penetration enhancers include dimethyisulfoxide and related analogues.
When formulated as a topical emulsion, the oily phase may optionally comprise merely an emu!sifier (otherwise known as an emulgent), or it may comprises a mixture of at least one emulsifier with a fat or an oil or with both a fat and an oil.
Preferably, a hydrophilic emulslfier is included together with a lipophilic emulsifier which acts as a stabiliser, it is also preferred to include both an oil and a fat.
Together, the emuisifier(s) with or without stabiiiser(s) make up the so-called emulsifying wax, and the wax together with the oil and/or fat make up the so-called emulsifying ointment base which forms the oily dispersed phase of the cream formulations.
Suitable emulgents and emulsion stabilisers include Tween 60, Span 80, cetosteary! alcohol, myristy! alcohol, glyceryl monostearate and sodium lauryl sulphate. The choice of suitable oiis or fats for the formulation is based on achieving the desired cosmetic properties, since the solubility of the active compound in most oils likely to be used in pharmaceutical emulsion formulations may be very low. Thus the cream should preferably be a non-greasy, non-staining and washable product with suitable consistency to avoid leakage from tubes or other containers. Straight or branched chain, mono- or dibasic alkyl esters such as di-isoadipate, isocetyi stearate, propylene glycol diester of coconut fatty acids, isopropyl myristate, decyi oieate, isopropyl paimitate, butyl stearate, 2-ethylhexyi palmitate or a blend of branched chain esters known as Crodamoi CAP may be used, the last three being preferred esters. These may be used alone or in combination depending on the properties required.
Alternatively, high melting point lipids such as white soft paraffin and/or liquid paraffin or other mineral oils can be used. Formulations suitable for rectal administration may be presented as a suppository with a suitable base comprising, for example, cocoa butter or a salicylate.
Formulations suitable for vaginal administration may be presented as pessaries, tampons, creams, gels, pastes, foams or spray formulations containing in addition to the active compound, such carriers as are known in the art to be appropriate.
Formulations suitable for parenteral administration (e.g. by injection, including cutaneous, subcutaneous, intramuscular, intravenous and intradermal), include aqueous and non-aqueous isotonic, pyrogen-free, sterile injection solutions which may contain anti-oxidants, buffers, preservatives, stabilisers, bacteriostats, and
solutes which render the formulation isotonic with the blood of the intended recipient; and aqueous and non-aqueous sterile suspensions which may include suspending agents and thickening agents, and liposomes or other micro particulate systems which are designed to target the compound to blood components or one or more organs. Examples of suitable isotonic vehicles for use in such formulations include
Sodium Chloride Injection, Ringers Solution, or Lactated Ringer's injection. Typically, the concentration of the active compound in the solution is from about 1 ng/m! to about 10 Mg/mi, for example from about 10 ng/m! to about 1 pg/ml. The formulations may be presented in unit-dose or multi-dose sealed containers, for example, ampoules and vials, and may be stored in a freeze-dried (lyophilised) condition requiring only the addition of the sterile liquid carrier, for example water for injections, immediately prior to use. Extemporaneous injection solutions and suspensions may be prepared from sterile powders, granules, and tablets. Formulations may be in the form of liposomes or other micro particulate systems which are designed to target the active compound to blood components or one or more organs.
Dosage
It will be appreciated that appropriate dosages of the active compounds, and compositions comprising the active compounds, can vary from patient to patient. Determining the optimal dosage will generally involve the balancing of the level of therapeutic benefit against any risk or deleterious side effects of the treatments of the present invention. The selected dosage level will depend on a variety of factors including, but not limited to, the activity of the particular compound, the route of administration, the time of administration, the rate of excretion of the compound, the duration of the treatment, other drugs, compounds, and/or materials used in combination, and the age, sex, weight, condition, general health, and prior medical history of the patient. The amount of compound and route of administration will ultimately be at the discretion of the physician, although generally the dosage will be to achieve local concentrations at the site of action which achieve the desired effect without causing substantial harmful or deleterious side-effects.
Administration in vivo can be effected in one dose, continuously or intermittently (e.g. in divided doses at appropriate intervals) throughout the course of treatment.
Methods of determining the most effective means and dosage of administration are well known to those of skill in the art and will vary with the formulation used for therapy, the purpose of the therapy, the target cell being treated, and the subject
being treated. Single or multiple administrations can be carried out with the dose level and pattern being selected by the treating physician.
In general, a suitable dose of the active compound is in the range of about 100
 to about 250 mg per kilogram body weight of the subject per day. Where the active compound is a salt, an ester, prodrug, or the like, the amount administered is calculated on the basis of the parent compound and so the actual weight to be used is increased proportionately. FIGURES
to about 250 mg per kilogram body weight of the subject per day. Where the active compound is a salt, an ester, prodrug, or the like, the amount administered is calculated on the basis of the parent compound and so the actual weight to be used is increased proportionately. FIGURES
Figure 1 shows the tumour volume and % body weight change of mice when treated with a compound of the invention in an ectopic leukemia model.
Figure 2 shows the CD31 +ve blood vessel density in the tumours treated in the same model as in figure 1.
Figure 3 shows the inhibition of pTyr591-ITD-FLT3 in the primary tumours treated in the same model as in figure 1. Figure 4 show the tumour progression in mice when treated with a compound of the invention in an orthotopic leukemia model.
Figure 5 shows the whole body bioluminescence imaging of the test subjects treated in the same model as in figure 4.
EXAMPLES
The following are examples are provided solely to illustrate the present invention and are not intended to limit the scope of the invention, as described herein. Acronyms
For convenience, many chemical moieties are represented using well known abbreviations, including but not limited to, methyl ( e), ethyl (Et), n-propyl (nPr), iso- propyl (iPr), n-butyl (nBu), tert-butyl (tBu), n-hexyi (nHex), cyclohexyl (cHex), phenyl (Ph), biphenyl (biPh), benzyl (Bn), naphthyi (naph), methoxy ( eO), ethoxy (EtO), benzoyl (Bz), and acetyl (Ac).
For convenience, many chemical compounds are represented using well known abbreviations, including but not limited to, methanol (MeOH), ethanoi (EtOH), iso- propanoi (i-PrOH), methyl ethyl ketone (MEK), ether or diethyl ether (Et20), acetic acid (AcOH), dichioromethane (methylene chloride, DCM), trifluoroacetic acid (TFA), dimethylformamide (DMF), tetrahydrofuran (THF), dimethylsuifoxide (D SO), meta- ch I oroperox benzoic acid (mCPBA), tert-butyloxycarbonyl (Boc), trimethylsilyi (TMS), triethylsilyl (TES), 2-(1 H-7-azabenzotriazol-1-yl)-1 , 1 ,3,3-tetramethyluronium hexafluorophosphate (HATU), diphenyiphosphoryi azide (DPPA), 1 ,8- diazabicycio[5.4.0]undec-7-ene (DBU), 1-ethyl-3-(3-dimethylaminopropyl)
carbodiimide (EDCI), 4-dimethylaminopyridine (DMAP), tetra-n-butylammonium fluoride (TBAF), /V-Diisopropylethyiamine (DIPEA), 1-hydroxybenzotriazoie (HOBt), and 1 ,2-dichloroethene (DCE). General Experimental Details
Unless otherwise stated the following generalisations apply.
1NMR spectra were recorded on either a Bruker Avance DRX300 (300 MHz), a Bruker Ultrasheild plus (400 MHz) or a Varian Unity inova 600 (600 MHz)
spectrometer. The multiplicity of a signal is designated by the following
abbreviations: s, singlet; d, doublet; t, triplet; q, quartet; br, broad; m, multiplet. All observed coupling constants, J, are reported in Hertz. 1iC NMR were recorded on a Bruker Avance DRX300 (75 MHz), a Bruker Ultrasheild plus (100 MHz) or a Varian Unity inova 600 ( 50 MHz) spectrometer in a broad band decoupled mode.
LC/MS data was generated using either a Finnigan LCQ Advantage Max (LCMS-A), a Waters ZQ 3100 system (LCMS-B) or an Agilent 6100 Series Single Quad LC/MS (LCMS-C). LCMS Method A (LCMS-A )
Instrument: Finnigan LCQ Advantage Max
Pump: Finnigan Surveyor LC Pump
Finnigan Surveyor Autosampler
Finnigan Surveyor PDA Detector
LC conditions:
Reverse Phase HPLC analysis
Column: Gemini 3μ C18 20x4.Gmm 110A Injection Volume 10μί
Solvent A: Water 0.1 % Formic Acid
Solvent B: Acetonitrile 0.1 % Formic Acid Gradient: 10-100% B over 10min
Detection: 100-600nm S conditions:
Ion Source: Ion trap
Ion Mode: ES positive
Temp: 300°C
Capillary V- 25
Detection: Ion counting
Scan Range: 80-1000A mu
Scan Time: 0.2 sec
Acquisition time: 1Qmin LCMS Method B (LCMS-B)
Instrument: Waters ZQ 3100 -Mass Detector Waters 2545-Pump
Waters SFO System Fluidics Organizer Waters 2996 Diode Array Detector
Waters 2767 Sample Manager
LC conditions:
Reverse Phase HPLC analysis
Column: XBridge TM C18 5μηΊ 4.6x100mm Injection Volume 10μί
Solvent A: Water 0.1 % Formic Acid
Solvent B: Acetonitrile 0.1 % Formic Acid Gradient: 10-100% B over 0min
Flow rate: 1.5 mL/min
Detection: 100-600nm
MS conditions:
Ion Source: Sing!e-quadrupole
Ion Mode: ES positive
Source Temp: 150°C
Desolvation Temp: 350°C
Detection: Ion counting
Cpil!ary (KV)-3.00
Cone (V): 30
Extractor (V): 3
RF Lens (V): 0.1
Scan Range: 100-1000 Amu
Scan Time: 0.5 sec
Acquisition time: 10min
Gas Flow
Desolvation L/hr-650
LCMS Method C (LCMS-C)
Instrument: Agilent 6100 Series Single Quad LC/MS
Agilent 1200 Series HPLC
Pump: 1200 Series G1311A Quaternary pump
Autosampler: 1200 Series G1329A Thermostatted Autosampler
Detector: 1200 Series G1314B Variable Wavelength Detector LC conditions:
Reverse Phase HPLC analysis
Column: Luna C8(2) 5μ 50X 4.6mm 100A
Column temperature: 30°C
Injection Volume: 5μί
Solvent A: Water 0.1 % Formic Acid
Solvent B: Acetonitrile 0.1 % Formic Acid
Gradient: 5-100% B over 10min
Detection: 254 nm or 214 nm MS conditions:
ion Source: Quadrupole
Ion Mode: ultirnode-ES
Drying gas temp: 30Q°C
Vaporizer temperature: 200°C
Capillary voltage (V): 2000 (positive)
Capillary voltage (V): 4000 (negative)
Scan Range: 100-1000
Step size: 0.1 sec
Acquisition time: 1Qmin
Analytical thin-layer chromatography was performed on Merck silica gel 80F254 aluminium-backed plates which were visualised using fluorescence quenching under UV light or acidic anisaidehyde or a basic potassium permanganate dip. Flash chromatography was performed using either a Teiedyne isco CombiF!ash Rf purifi- cation system using standard RediSep® cartridges or a Biotage I solera purification system using either Grace or Biotage silica cartridges.
Where necessary, anhydrous solvents were prepared using a Braun purification system or purchased from Sigma-Aidrich.
Example 1 : 2-{2-{2-{2-{(4-(Piperazsn-1 -y!)phenyf)amino)-5- (triff uorometh l)pyrimidsn-4-yl)ethyf)pheny!)acetamide (1 )

iS 1
(a) tert- Butyl 4-(4-nitrophenyl)piperazine- 1 -carboxylate (11)
4-(4'-Nitrophenyl)piperazine hydrochloride (5.00 g, 20.5 mmo!) was dissolved in DCM (100 mL) and treated with triethy!amine (7.15 mL, 51.3 mmol) followed by Boc anhydride (4.93 g, 22.6 mmol) and the reaction was stirred at room temperature for 20 hours. To the mixture was added water (100 mL) and DCM (70 mL) and the layers were separated. The aqueous layer was extracted with DCM (100 mL), the organics were combined and washed with brine (100 mL), dried (Na2S04), filtered and concentrated in vacuo to give a ye!low-orange solid. The product was purified by silica gel chromatography (Biotage Isolera, 120 g Si cartridge, 0-100% EtOAc in petroleum benzine 40-60 °C) to give the title compound (//) (4.895 g, 78% yield) as a yellow solid; 1H NMR (400 MHz, o¾-DMSO) δ 8.10 - 8.04 (m, 2H), 7.04 - 6.97 (m,
2H), 3.48 (m, 8H), 1.42 (s, 9H). LC S Method C: rt 8.13 min; m/z 208.2 [M- Boc+2H]4.
(b) tert-Butyl 4-(4-aminophenyl)piperazine- 1-carboxylate (12)
tert-Butyl 4-(4-nitrophenyl)piperazine-1-carboxyiate (11) (3.24 g, 10.5 mmol) was dissolved in EtOAc (90 mL) under an atmosphere of nitrogen and a slurry of 10% Pd/C (0.500 g) in EtOAc (10 mL) was added. The resulting suspension was then stirred vigorously under an atmosphere of hydrogen at room temperature for 42 hours. The catalyst was removed by filtration through Celite, which was washed with EtOAc (7 x 10 mL) and the solvent was removed in vacuo to give the title compound (12) (2.92 g, 99 % yield) as a pale pink solid; 1H N R (400 MHz, afe-DMSO) δ 6.72 - 6.66 (m, 2H), 6.52 - 6.45 (m, 2H), 4.60 (s, 2H), 3.44 - 3.39 (m, 4H), 2.87 - 2.79 (m, 4H), 1.41 (s, 9H). LCMS Method C: rt 4.40 min; m/z 278.2 [M+H]+. (c) tert-Buiy! 4-(4-((4-chloro-5-(trifluoromethyl)pyrimidin-2- yl)amino)phenyl)piperazine- 1 -carboxy!ate (13)
2,4-Dichloro-5-(trifluoromethyl)pyrimidine (2.39 g, 11.0 mmol) was stirred in a 1 :1 t- BuOH:1 ,2-dichloroethane mixture (80 mL) at 0 °C and a 1.0 M ZnCI2 solution in diethyl ether (12.6 mL, 12.6 mmol) was added cautiously over 20 minutes and the reaction was left stirring at 0 °C for 30 minutes. A solution of tert-butyl 4- (4- aminophenyl)piperazine-1-carboxylate (12) (2.92 g, 10.5 mmol) in 1 : 1 f-BuOH:1 ,2- dichloroethane (40 mL) was added drop-wise over 15 minutes at 0 °C followed by a solution of triethyiamine (1.76 mL, 12.6 mmol) in 1 : 1 ?-BuOH: 1 ,2-dichloroethane (40 mL) and the reaction was allowed to warm to room temperature and was stirred for 18 hours. The organic solvents were evaporated in vacuo and the crude yellow oily solid was suspended in water (400 mL), the suspension was sonicated for 30 minutes and the product was collected by filtration, the solid was washed with water (10 x 20 mL) and dried under a high vacuum to give the title compound (13) (4.75 g, 98% yield) as a beige solid; 1H NMR (400 MHz, cfe-DMSO) δ 10.45 (s, 1 H), 8.72 (s, 1 H), 7.50 (d, J = 8.5 Hz, 2H), 6.96 (d, J = 9.0 Hz, 2H), 3.50 - 3.42 (m, 4H), 3.09 - 3.02 (m, 4H), 1.42 (s, 9H). LCMS Method C: rt 6.56 min; m/z 456.2, 458.1 [M-H]'.
(d) tert-Butyl 4-(4-((4-((2-(2-methoxy-2-oxoethyl)phenyl)ethynyl)-5- (trifluoromethyl)pyrimidin-2-yl)amino)phenyl)piperazine- 1 -carboxylate (15)
To a nitrogen de-gassed solution of methyl 2-(2-ethynylphenyl)acetate (14: prepared according to the procedure of Peng, C, et at, Adv. Synth. Catai 2008, 350, 2359 - 2364 or as detailed below) (0.114 g, 0.653 mmol) in dry D F (6 mL) were added triethylamine (0.280 mL, 2.01 mmoi) followed by terf-butyl 4-(4-((4-chloro-5- (trifluoromethyl)pyrimidin-2-yl)amino)phenyl)piperazine-1-carboxylate (13) (0.230 g, 0.502 mmol), triphenylphosphine (0.020 g, 0.075 mmoi), trans- dichlorobis(friphenylphosphine) paliadium(i!) (0.035 g, 0.050 mmol) and Cul (0.014 g, 0.075 mmoi). The reaction mixture was heated under microwave irradiation at 120 °C for 20 minutes and then concentrated to dryness in vacuo and purified by silica gel chromatography (Biotage isolera, 40 g Si cartridge, 0-80% EtOAc in cyclohexane) to give the title compound (15) (0.267 g, 89% yield) as an orange glassy solid; 1H N R (400 MHz, dff-DMSO) δ 10.27 (br s, 1 H), 8.76 (s, 1 H), 7.65 - 7.49 (m, 4H), 7.49 - 7.38 (m, 2H), 6.95 (d, J = 9.1 Hz, 2H), 3.94 (s, 2H), 3.61 (s, 3H), 3.51 - 3.42 (m, 4H), 3.09 - 3.00 (m, 4H), 1.42 (s, 9H). LCMS Method C: rt 6.67 min; m/z 596.3 [M+H]+.
(e) tert-Butyl 4-(4-((4-(2-(2-methoxy-2-oxoethyl)phenethyl)-5- (trifluoromethyl)pyrimidin-2-yl)amino)phenyl)piperazine- 1 -carboxylate (16)
terf-Butyl 4-(4-((4-((2-(2-methoxy-2-oxoethyl)phenyl)ethynyl)-5-
(trifluoromethyl)pyrimidin-2-yl)amino)phenyl)piperazine-1 -carboxylate (15) (0.250 g, 0.420 mmol) was dissolved in EtOAc (8 mL) and absolute ethanol (10 mL) under an atmosphere of nitrogen. 10% Pd/C (0.200 g) in EtOAc (4 mL) was added to the solution and the atmosphere was changed to hydrogen gas (balloon). The reaction was sealed with a balloon and stirred at room temperature for 18 hours. The catalyst was removed by filtration through Celite, which was washed with EtOAc (7 x 10 mL). The solvent was removed in vacuo to give the title compound (16) (0.211 g, 84% yield) as a yellow solid. LCMS Method C: rt 6.78 min; m/z 600.3 [M+H]+. if) Lithium 2-(2-(2-(2-((4 - (4 - (tert-butoxycarbonyl)piperazin- 1 -yl)phenyl)amino) -5-
( trifiuoromethyl)pyrimidin -4 ~yl)ethyl)phenyi)acetate ( 17)
LiOH.H20 (0.044 g, 1.06 mmol) was added to ier-buty! 4-(4-((4-(2-(2-methoxy-2- oxoethyl)phenethyi)-5-(trifluoromethyl)pyrimidin-2-yl)am!no)phenyi)piperazine-1- carboxylate (16) (0.211 g, 0.352 mmol) in THF (10 mL), water (2.5 mL) and methanol (1 mL). The resulting mixture was allowed to stir for 3 hours at 40 °C. The volatiies
were removed in vacuo and the residue was diluted with EtOAc (100 mL) and 2 M aq. NaOH (100 mL). The layers were separated and the aqueous layer was extracted with EtOAc (70 mL), the organic layers were combined, washed with brine (100 mL), dried (MgS04), filtered and concentrated under reduced pressure to give the title compound (17) (0.195 g, 96% yield) as a yellow solid; 1H NMR (400 MHz, cfg-DMSO) δ 10.02 (s, 1 H), 8.61 (s, 1 H), 7.64 - 7.55 (m, 2H), 7.26 - 7.14 (m, 4H), 6.97 - 6.91 (m, 2H), 3.65 (s, 2H), 3.50 - 3.41 (m, 4H), 3.10 - 2.93 (m, 8H), 1.42 (s, 9H). LCMS Method C: rt 6,32 min; m/z 586.3 [M+Hf . (g) teri-Butyi 4-(4-((4-(2-(2-amino-2-oxoethyl)phenethyl)-5-(trifluorom
2-yl)amino)phenyl)piperazine- 1 -carboxylate (18)
Lithium 2-(2-(2-(2-((4-(4-(ieri-butoxycarbonyl)piperazin-1-yl)phenyl)amino)-5- (trifluoromethyl) pyrimidin-4-yl)ethyl)phenyl)acetate (17) (0.195 g, 0.333 mmol) was dissolved in dry THF (10 mL) and dry D F (2 mL) under an atmosphere of nitrogen. To the solution were added 1-hydroxybenzofriazole (0.049 g, 0.37 mmol) and EDCI (0.070 g, 0.37 mmol) and /V-diisopropyiethylamine (0.232 mL, 1.33 mmol) and the reaction mixture was stirred at room temperature for 10 minutes. Ammonium carbonate (0.128 g, 1.332 mmol) was added in one portion, and the reaction was stirred room temperature for 20 hours. The volatiles were removed in vacuo and the residual solution was diluted with EtOAc (70 mL) and sat. aq. NaHC03 (70 mL). The layers were separated and the organic layer was washed with water (70 mL), brine (70 mL), dried (MgS04), filtered and concentrated in vacuo to give the crude product which was purified by silica gel chromatography (Biotage Isolera, 12 g Si Cartridge, 0-100% EtOAc in petroleum benzine 40-60 °C, then 0-10% methanol in EtOAc) to give the title compound (18) (0.133 g, 68% yield) as a pale yellow solid; 1H NMR (400 MHz, cf6~DMSO) δ 10.01 (s, 1 H), 8.61 (s, 1 H), 7.65 - 7.56 (m, 2H), 7.44 (s, 1 H), 7.26 - 7.13 (m, 4H), 6.98 - 6.89 (m, 3H), 3.52 - 3.41 (m, 6H), 3.13 - 2.95 (m, 8H), 1.42 (s, 9H). LCMS Method C: rt 6.18 min; m/z 585.3 [M+H]+. (h) 2-(2-(2-(2-((4-(Piperazin- 1 -yl)phenyl)amino)-5-(trifluoromethyl)pyrimidin-4- yl)ethyl)phenyl) acetamide (1)
ierf-Butyl 4-(4-((4-(2-(2-amino-2-oxoethyl)phenethyl)-5-(trifluoromethyl)pyrimidin-2- yl)amino) pheny!)piperazine~1~earboxyiate (18) (0.131 g, 0.224 mmol) was dissolved in DCM (7 mL) under an atmosphere of nitrogen. Trifluoroacetic acid (0.857 mL, 11.2 mmol) was added to the solution and the reaction was stirred at room temperature for
24 hours. Volatiles were removed in vacuo, EtOAc (70 mL) and 2 M aq. NaOH (70 mL) were added to the oil and the layers were separated. The aqueous layer was extracted with EtOAc (50 mL), the combined organics were washed with brine (50 mL), dried (Na2S04), filtered and concentrated in vacuo. The resulting solid was taken up in DCM (~ 5 mL) and methanol (~ 1 mL) and concentrated in vacuo. The process was repeated with only DCM twice to give the title compound ( 1) (0.104 g, 96% yield) as a pale yellow solid; 1H N MR (400 MHz, efe-DMSO) δ 9.96 (s, 1 H), 8.60 (s, 1 H), 7.61 - 7.53 (m, 2H), 7.43 (br s, 1 H), 7.25 - 7.13 (m, 4H), 6.98 - 6.86 (m, 3H), 3.50 (s, 2H), 3.12 - 3.05 (m, 2H), 3.04 - 2.94 (m, 6H), 2.86 - 2.78 (m, 4H). LC S Method C: rt 4.774 min; m/z 485.2 [M+H]+.
S

I9 110 14
(a) Methyl 2-(2-iodophenyl)acetate (19)
2-(2-iodophenyi)acetic acid (5.00 g, 19.1 mrnoi) was placed into a reaction flask and dissolved in MeOH (150 mL). Sulfuric acid (250 L) was added and reaction mixture was stirred and heated at 80 °C under nitrogen for 16 hours. The resulting mixture was cooled to room temperature and the volatiles removed by evaporation under reduced pressure. The residue was taken up in ethyl acetate (100 mL), washed with 10% NaHC03 (100 mL), dried (MgS04) and evaporated under reduced pressure to give the title compound (19) (5,20g, 99%) as a dear liquid; 1 H NMR (400 MHz, CDCI3) δ 7.85 (dd, J = 7.9, 1.0 Hz, 1 H), 7.35 - 7.27 (m, 2H), 6.97 (ddd, J = 7.9, 7.0, 2.1 Hz, 1 H), 3.81 (s, 2H), 3.72 (s, 3H). (b) Methyl 2-(2-((trimethylsilyl)ethynyl)phenyl)acetate (110)
Methyl 2-(2-iodophenyl)acetate {19) (4.65 g, 16.8 mmol), PdCI2(PPh3)2 (295 mg, 421 pmoi) and Cu(l)l (80.0 mg, 421 pmoi) were placed into an oven dried reaction flask under nitrogen. (Trimethylsilyl)acetylene (2.80 mL, 20.2 mmol), dry degassed THF (20 mL) and triethyiamine (20 mL) were added and the reaction mixture was stirred at room temperature for 16 hours. The volatiles were removed under reduced pressure to give a black residue which was adsorbed onto silica then
chromatographed on silica gel (0-5% ethyl acetate/petroleum benzine 40-60 °C) to
give the title compound (HO) (4.83 g, 99%) as a light brown liquid; 1H NMR (400 Hz, CDCI3) δ 7.48 (dd, J = 7.5, 0.8 Hz, 1 H), 7.32 - 7.14 (m, 3H), 3.84 (s, 2H), 3.71 (s, 3H), 0.26 (s, 9H). LC S Method C: rt 6.64 min.
(c) Methyl 2-(2-ethynylphenyl)acetate (14)
Methyl 2-(2-((trimethyisilyi)ethynyi)phenyl)acetate (110) (4.63 g, 19.0 mmol) was dissolved in DC (200 mL) and TBAF (1.0 M in THF) (28.5 mL, 28.5 mmol, 1.5 eq) was added at 0 °C. The resulting solution was stirred at room temperature for 1 hour before washing with 10% NaHC03 ( 00 mL). The organic layer was dried (MgS04) then evaporated under reduced pressure to give a dark brown/black residue. The residue was adsorbed onto silica and then cbromatographed on silica gel (0-10% ethyl acetate/petroleum benzine 40-60 °C) to give the title compound (14) (2.76 g, 83%) as a red liquid; 1 H NMR (400 MHz, CDCI3) δ 7.52 (dd, J = 7.6, 1.1 Hz, 1 H), 7.43 - 7.16 (m, 3H), 3.88 (d, J = 9.6 Hz, 2H), 3.77 - 3.52 (m, 3H), 3.28 (s, 1 H).
Example 2: 3-{2-{2-{(4-(4-IVIethy!piperazsn-1-yS)phenyl)am!no)-5-{tnf!uoromethyl) pyri idin-4-yi}ethyl)ben_;amsdle (2)

121 2
(a) 1-Methy!-4-(4-nitropheny!)piperazine (111)
To 4-(4'-nitrophenyl)piperazine hydrochloride (1.00 g, 4.10 mmol) was added formic acid (1.55 mL, 41.0 mmol) and 37% aq. formaldehyde (3.06 mL, 41.0 mmol) in a microwave vessel and the reaction was heated at 120 °C for 3 minutes. To the cooled reaction mixture was added EtOAc (100 mL) and 2 M aq. NaOH (70 mL). The layers were separated and the organic layer was washed with brine (50 mL), the layers were separated and the aqueous brine layer was extracted with EtOAc (50 mL), the organic layers were combined and dried (Na2S04), filtered and concentrated in vacuo
to give the crude product which was purified by silica gei chromatography (Biotage Isoiera, 40 g Si cartridge, 0-80% methanol (containing 1 % ammonia solution) in EtOAc) to give the title compound (111) (0,636 g, 70% yield) as a yellow solid; 1H NMR (400 MHz, ofe-DMSO) δ 8.07 - 8.01 (m, 2H), 7.05 - 6.99 (m, 2H), 3.48 - 3.40 (m, 4H), 2.46 - 2.39 (m, 4H), 2.21 (s, 3H). LCMS Method C: rt 1.45 min; m/z 222.2 [M+H]+.
(b) 1-4-(4-Methylpiperazin-1-yl)aniline (112)
1-Methyi-4-(4-nitrophenyi)piperazine (111) (0.632 g, 2.86 mmol) was dissolved in EtOAc (45 mL) under an atmosphere of nitrogen and a slurry of 10% Pd/C (0.200 g) in EtOAc (5 mL) was added. The resulting suspension was then stirred vigorously under an atmosphere of hydrogen at room temperature for 18 hours. The catalyst was removed by filtration through Ceiite, which was washed with EtOAc (7x10 mL) and the solvent was removed in vacuo to give the title compound (112) (0.537 g, 98% yield) as a pink solid; 1H NMR (400 MHz, oV-DMSO) δ 6.70 - 6.64 (m, 2H), 6.51 - 6.45 (m, 2H), 4.54 (s, 2H), 2,94 - 2.84 (m, 4H), 2.46 - 2.36 (m, 4H), 2.19 (s, 3H). LCMS Method C: rt 0.98 min; m/z 192.3 [M+H]+.
(c) 4-Chloro-2-(methylthio)-5-(trifluoromethyl)pyrimidine (113)
To a solution of the 2,4-dichloro-5-(trifiuoromethyl)pyrimidine (2.50 g, 11.5 mmol) in
THF (50 mL) in an ice bath under nitrogen was added zinc(li) chloride (1.0 M in ether, 13.8 mL, 13.8 mmol) dropwise. The mixture was stirred in the ice bath for two hours, then sodium methanethioiate (0.888 g, 12.7 mmol) was added. The mixture was stirred overnight, allowing the reaction to slowly come to room temperature. After 18 hours the reaction was quenched with 2 M HCI ( 5 mL), and the organics removed by evaporation under reduced pressure. The aqueous residue was diluted with brine (15 mL), and extracted with DCM (3x30 mL). The combined organic phases were dried (phase separator) and carefully evaporated to give a pale yellow oil.
Chromatography (Biotage isoiera, 2x40g silica cartridge, 0-20% DCM/n-hexane) followed by carefully evaporation of solvent (40°C@400 mmHg then room
temperature@200 mmHg) gave the title compound (113) (2.149 g, 82% yield) as a colourless oil; 1 H NMR (600 MHz, CDCI3) δ 8.66 (s, 1 H), 2.61 (s, 3H). LCMS Method C: rt: 7.95 min; m z 229.1 [M+H]+. Note: 113 \s volatile.
(d) 4-lodo-2-(methylthio)-5-(trifluoromethyl)pyrimidine (114)
4-Chloro-2-(methylthio)-5-(trifluoromethyl)pyrimidine (113) (5.00 g, 21.9 mmol) was placed into a reaction flask then sodium iodide (9.80 g, 65.6 mmol) and hydroiodic acid (58%; 70 mL) were added. The reaction mixture was stirred for 48 hours in darkness then diluted with water (200 mL) where upon a colourless solid precipitated. The precipitate was collected by filtration and was washed with 10% NaHC03 solution until neutral. The resulting solid was washed with water (100 mL) then suction dried for 2 hours to give the title compound (114) (3.956 g, 57%) as a pale yellow solid; 1H NMR (400 MHz, CDCI3) δ 8.42 (s, 1 H), 2.58 (s, 3H). LC S Method C: rt 6.30 min, m/z 321.0 [ +H]+.
(e) tert-Buty! 3-iodobenzoate (115)
To a solution of 3-iodobenzoic acid (5.06 g, 20.4 mmol) in DCM (25 mL) was added a solution of Boc20 (4.90 g, 22.5 mmol) in dichloromethane (10 mL) and 4-DMAP (0.624 g, 5.1 1 mmol) in dichloromethane (5 mL). The resulting solution was stirred at room temperature under a nitrogen atmosphere for 64 hours. The resulting mixture was partitioned between water (100 mL) and dichloromethane (50 mL) and the layers separated. The organic layer was washed with water (2x100 mL) before being concentrated under reduced pressure. The resulting residue was purifed using silica gel column chromatography (0-50% dichloromethane/petroleum benzene 40-60 °C) to give the title compound (115) (65% 4.02 g) as a colourless oil; 1H NMR (400 MHz, CDCI3) δ 8.30 (dd, J = 1.6, 1.6 Hz, 1 H), 7.95 (ddd, J = 7.8, 1.5, 1.1 Hz, 1 H), 7.85 (ddd, J = 7.9, 1.8, 1.1 Hz, 1 H), 7.16 (ddd, J = 7.8, 7.8, 0.2 Hz, 1 H), 1.59 (s, 9H). LCMS Method C: rt 6.88 min.
(f) tert-Butyl 3-((trimethylsilyl)ethynyl)benzoate (116)
A mixture of ferf-butyi 3-iodobenzoate (115) (4.02 g, 13.2 mmol), Pd(PPh3)4 (0.38 g, 0.33 mmol) and copper(l) iodide (0.13 g, 0.68 mmol) was dissolved in anhydrous THF (50 mL) under a nitrogen atmosphere and the resulting solution degassed by bubbling nitrogen through it. Triethylamine (9,2 mL, 66 mmol) was added and the mixture was then stirred for 10 minutes before addition of TMS-acetylene (3.8 mL, 27 mmol). The resulting mixture was then stirred for 18 hours . The mixture was concentrated under reduced pressure and purified using silica gel column
chromatography (0-10% ethyl acetate/petroleum benzine 40-60 °C) to give the title compound (116) (3.57 g, 99%) as a cream crystalline solid; 1 H NMR (400 MHz,
CDCI3) δ 8.06 (ddd, J = 1.7, 1.7, 0.5 Hz, 1 H), 7.92 (ddd, J = 7.9, 1.3, 1.3 Hz, 1 H), 7.60 (ddd, J = 7.7, 1.4, 1.4 Hz, 1 H), 7.35 (ddd, J = 7.8, 7.8, 0.5 Hz, 1 H), 1.59 (s, 9H), 0.26 (s, 9H). LC S Method C: rt 7.46 min. (g) tert-Buty 13-ethynylbenzoate (117)
To a solution of ferf-butyl 3-((trimethylsilyl)ethynyl)benzoate (116) (4.40 g, 16.0 mmol) in THF (100 mL) at 0 °C under nitrogen was added 1.0 M TBAF in THF (20.0 mL, 20.0 mmo!). The mixture was then stirred at 0 °C for 1 hour then 16 hours at room temperature. The mixture was then concentrated under reduced pressure, diluted with ethyl acetate (100 mL) and washed with water (3x100 mL). The organic extract was then purified using silica gel column chromatogaphy (0-5% EtOAc/petroieum benzine 40-60 °C) to give the title compound (117) (2.72 g, 84%) as a pale yellow oil; 1H N (400 MHz, CDCI3) δ 8.09 (dd, J = 1.5, 1.5 Hz, 1 H), 7.97 (ddd, J = 7.9, 1.5, 1.5 Hz, H), 7.63 (ddd, J = 7.7, 1.4, 1.4 Hz, 1 H), 7.38 (ddd, J = 7.8, 7.8, 0.5 Hz, 1 H), 3.11 (s, 1 H), 1.59 (s, 9H).
(h) tert-Buty 13- ((2- (methylthio) -5-(trifluoromethyl)pyrimidin -4 -yl) ethynyl)benzoate (118)
i-Butyl 3-ethynyibenzoate (117) (1.603 g, 7.93 mmol), 4-iodo-2-(methylthio)-5- (trifiuoromethyl)pyrimidine (114) (1.647 g, 5.15 mmol), PdCI2(PPh3)2 (0.316 g, 0.45 mmoi), PPh3 (0.355 g, 1.35 mmol), Cu(i)i (0.232 g, 1.22 mmol) and trethyiamine (4.00 mL, 28.7 mmoi) were combined in D F (20 mL) and the resulting mixture heated at 120 °C under microwave irradiation for 25 minutes. The mixture was then concentrated under reduced pressure and purified twice using silica gel column chromatography (10-20% EtOAc/petroleum benzine 40-60 °C then 50-100%
DCM/petroleum benzine 40-60 °C) to give the title compound (118) (0.624 g, 31 %) as a yellow solid; 1H NMR (400 MHz, CDCI3) δ 8.73 (d, J = 0,8 Hz, H), 8.25 (m, H), 8.08 (m, 1 H), 7.78 (m, 1 H), 7.48 (m, 1 H), 2.63 (s, 3H), 1.61 (s, 9H). (i) tert-Butyl 3-(2-(2-(methylthio)-5-(trifluoromethyl)pyrimidifr (119) fe/f-Butyl 3-((2-(methylthio)-5-(trifluoromethyi)pyrimid!n-4-yi)ethynyi)benzoate (118) (0.624 g, 1.58 mmol) and 10% Pd/C (0.206 g) was taken up in THF (20 mL) and H2 bubbled through the mixture for 5 minutes before stirring at room temperature for 20 hours under a hydrogen atmosphere. The mixture was filtered through celite and concentrated under reduced pressure. This procedure was repeated twice with 0%
Pd/C (0.212 g) and 20% Pearlman's caia!ysi (0.316 g) respectively to give the title compound (119) as a yellow oil that was reacted without further purification. LC S Method C: rt 7.14 min. (j) tert-Butyl 3-(2-(2-(methylsulfonyl) -5-(trifluoromethyl)pyrimidin-4-yl)ethyl)benzoate
(120)
A mixture of iert-butyl 3-(2-(2-(methylthio)-5-(trifluoromethyl)pyrimidin-4- yl)ethyl)benzoate (119) (0.630 g, 1.58 mmol) and MCPBA (0.975 g, 3.96 mmol) were dissolved in DCM (20 mL) at 0 °C. The resulting solution was allowed to warm to room temperature, at which stirring was continued for 16 hours. The voiatiles were evaporated under reduced pressure and the residue triturated with DCM . The resulting suspension was filtered and the filtrate was evaporated to dryness. The residue was triturated a second time with DCM and the precipitate removed via filtration. The filtrate was evaporated to dryness and the residue purified using silica gel column chromatography (20-100% DCM/petroleum benzine 40-60 °C, 0-50%
EtOAc/petroleum benzine 40-60 °C) to give the title compound (120) (0.360 g, 53%) as a yellow semi solid in 70% purity by NM ; 1 H NMR (400 MHz, CDCi3) δ 9.06 (s, 1 H), 7.85 (m, 2H), 7.38 (m, 2H), 3.42 (m, 2H), 3.36 (s, 3H), 3.24 (m, 2H), 1.60 (s, 10H). LCMS Method C: rt 6.40 min; m/z 357.1 [M- ?-BuO]+, 453.1 [M+Naf .
(k) tert-Butyl 3-(2-(2-((4-(4-methylpiperazin- 1-yl)phenyl)amino)-5- (trifluoromethyl)pyrimidin-4-yl)ethyl)benzoate (121 )
1-4-(4-Methylpiperazin-1 -yl)aniline (112) (0.071 g, 0.372 mmol), iert-butyl 3-(2-(2- (methylsulfonyl)-5-(trifluoromethyl)pyrirriidin-4-yl)ethyl)benzoate (120) (0.100 g, 0.232 mmol) and tosic acid monohydrate (0,088 g, 0.465 mmol) were combined in a microwave vessel and dry dioxane (3 mL) was added. The reaction was heated under microwave irradiation at 150 °C for 30 minutes then concentrated in vacuo and purified by silica gel chromatography (Biotage Isolera, 12 g Si Cartridge, 0-100% EtOAc in petroleum benzine 40-60 °C, then 0-100% MeOH in EtOAc) to give a pale yellow solid. The solid was dissolved in EtOAc (20 mL) and sat. aq. NaHC03 (10 mL) and the layers were separated. The aqueous layer was extracted with EtOAc (20 mL), the organics were combined and washed with wafer (20 mL), brine (20 mL), dried (MgS04), filtered and concentrated in vacuo to give the title compound (121) (0.025 g, 20% yield) as a pale yellow solid; 1 H NMR (400 MHz, afe-DMSO) δ 9.98 (s, 1 H), 8.59 (s, 1 H), 7.76 - 7.69 (m, 2H), 7.57 - 7.37 (m, 4H), 6.90 (d, J = 9.1 Hz, 2H),
3.19 - 3.00 (m, 8H), 2.48 - 2.40 (m, 4H), 2.21 (s, 3H), 1.52 (s, 9H). LC S Method C: rt 5.41 min; m/z 542.3 [ +H .
(!) 3-(2-(2-((4-(4-Methylpiperazin-1-yl)phenyl)amino)-5-(trifluorom pyrimidin-4- yi)eihyi)benzamide (2)
ferf- Butyl 3-(2-(2-((4-(4-methylpiperazin-1-yl)phenyl)amino)-5-
(trifluoromethy!)pyrimidin-4-yl)ethyl)benzoate (121) (0.025 g, 0.046 mmol) was dissolved in dry DCM (3 mL) under an atmosphere of nitrogen. Trifluoroacetic acid (0.177 mL, 2.31 mmol) was added to the solution and the reaction was stirred at 35 °C for 2 hours. The mixture was concentrated to dryness, toluene (~2 mL) was added to the residue and the solvent was removed in vacuo to give a yellow solid. This material was dissolved in dry THF (3 mL) and dry DMF (0.2 mL) under an
atmosphere of nitrogen. To the solution were added 1-hydroxybenzotriazole (0.009 g, 0.064 mmol) and EDCI (0.012 g, 0.064 mmol) and A ,/V-diisopropylethylamine (0.048 mL, 0,276 mmol) and the reaction mixture was stirred at room temperature for 10 minutes. Ammonium carbonate (0.018 g, 0.18 mmol) was added in one portion, and the reaction was stirred room temperature for 20 hours. The volatiies were removed in vacuo and the residual solution was diluted with EtOAc (20 mL) and saturated aq. NaHC03 (10 mL). The layers were separated and the organic layer was washed with water (10 mL), brine (10 mL), dried ( gS04), filtered and concentrated in vacuo to give a pale yellow solid. The product was dissolved in DCM (~ 4 mL) and MeOH (-1 mL) and the solvents were removed in vacuo. The process was repeated 3 times with DCM only and to give the title compound (2) (0.017 g, 76% yield over 2 steps) as a pale yellow solid; 1H NMR (400 MHz, cfe-DMSO) δ 9.97 (s, 1 H), 8.60 (s, 1 H), 7.95 (br s, 1 H), 7.79 (s, 1 H), 7.75 - 7.66 (m, 1 H), 7.57 - 7.47 (m, 2H), 7.40 - 7.30 (m, 3H), 6.94 - 6.86 (m, 2H), 3.14 - 3.01 (m, 8H), 2.47 - 2.42 (m, 4H), 2.21 (s, 3H). LCMS Method C: rt 4.75 min; m/z 485.3 [M+H]÷.
Examp 3: 2-{2-{2-{2-{(4-(4-IVIethy!piperazsn-1-yS)phenyf)amino)-5- (tri -4-yl)ethy!)phenyl)acetamide (3)

To a suspension of 2-(2-(2-(2-((4-(piperazin-1-yl)phenyl)amino)-5- (trifluoromethyl)pyrimidin-4-yl)ethyl)phenyl)acetamide ( 1) (0.015 g, 0.031 mmo!) in anhydrous methanol (1.5 mL) were added a 37% aq. solution of formaldehyde (0,005 mL, 0.062 mmol) followed by sodium triacetoxyborohydride (0.033 g, 0.155 mmol) under an atmosphere of nitrogen. The reaction was stirred at room temperature for 1.5 hours, the volatiles were removed in vacuo and the residue was diluted with EtOAc (15 mL) and sat. aq, NaHC03 (10 mL). The layers were separated and the aqueous layer was extracted with EtOAc (2 x 10 mL), the combined organic layers were washed with water (10 mL), brine (10 mL), dried ( gS04), filtered and concentrated in vacuo to give the title compound (3) (14.5 mg, 97% yield) as a pale yellow solid; 1H N (400 MHz, cfe-DMSO) δ 9.97 (s, 1 H), 8.60 (s, 1 H), 7.57 (d, J = 9.0 Hz, 2H), 7.43 (s, 1 H), 7.26 - 7.13 (m, 4H), 6.96 - 6.87 (m, 3H), 3.50 (s, 2H), 3.12 - 3.03 (m, 6H), 3.03 - 2.94 (m, 2H), 2.47 - 2.42 (m, 4H), 2.22 (s, 3H). LCIV1S Method C: rf 4.74 min; m/z 499.3 [M+H]+.
Example 4: 2-{2-{2-{2-{(4-(Pipend!n-3-y!)phenyl)amino)-5-

(a) 3-(4-Nitrophenyi)pyridine (122)
To a solution of 1-iodo-4-nitrobenzene (1.00 g, 4.02 mmol) in nitrogen degassed dry DMF (20 mL) was added 3-pyridineboronic acid (0,592 g, 4.82 mmol), Cs2C03 (5.23 g, 18.1 mmol), iriphenylphosphine (0.158 g, 0.602 mmol) and Pd(OAc)2 (0.090 g, 0.40 mmol). The reaction mixture was heated at 80 °c for 18 hours, cooled to room temperature and concentrated to dryness in vacuo. The crude material was purified by silica gel chromatography (Biotage Isolera, 40 g Si cartridge, 20-100% EtOAc in petroleum benzine 40-60 °C) to give the title compound {122) (0.590 g, 73% yield) as a yellow solid; 1 H N R (400 MHz, CDCi3) δ 8.90 (d, J = 1.8 Hz, 1 H), 8.70 (dd, J = 4.8, 1.5 Hz, 1 H), 8.39 - 8.32 (m, 2H), 7.93 (ddd, J = 7.9, 2.4, 1.7 Hz, H), 7.78 - 7.72 (m, 2H), 7.44 (ddd, J = 7,9, 4.8, 0.8 Hz, 1 H), LCMS Method C: rt 4,62 min; m/z 201.1 [M+Hf.
(b) tert- Butyl 3-(4-aminophenyl)piperidine-1 -carboxylate (123)
To a solution of 3-(4-nitrophenyi)pyridine {122) (0.590 g, 2.947 mmoi) in 1 HCl (3 mL) and methanol (30 mL) was added Pt02 (0.059 g) under an atmosphere of nitrogen. The reaction was then subjected to a 40 psi hydrogen atmosphere in a Parr hydrogenator for 24 hours, the catalyst was removed by filtration, and the solvents were removed in vacuo. The resulting yellow solid was again dissolved in 1 HCl (3 mL) and methanol (30 mL) and Pt02 (0.059 g) was added under an atmosphere of nitrogen. The reaction was subjected to a 40 psi hydrogen atmosphere in a Parr hydrogenator for 24 hours, the reaction mixture was filtered through celite which was washed with EtOAc (3 x 10 mL) and water (3 x 10 mL) and the filtrate was concentrated in vacuo to give the crude material (0.720 g) as a pale brown glassy solid. This material was dissolved in DC (25 mL), DMF (5 mL) and methanol (20 mL) and treated with triethylamine (1.438 mL, 10.315 mmol) followed by Boc anhydride (0.675 g, 3.094 mmol). The reaction was stirred at room temperature for 20 hours, then concentrated in vacuo and EtOAc (100 mL) and sat. aq. NaHC03 (50 mL) were added and the layers were separated. The aqueous layer was extracted with EtOAc (70 mL), the organics were combined and washed with water (100 mL), brine (100 mL), water (100 mL), brine (100 mL), dried (Na2S04), filtered and concentrated in vacuo to give a pink foam. The crude product was purified by silica gel chromatography (Biotage I solera, 40 g Si cartridge, 0-55% EtOAc in petroleum benzine 40-60 °C) to give the title compound ( 23) (0.435 g, 53% yield over 2 steps) as a pink solid; N R (400 MHz, cfe-DMSO) δ 6.91 - 6.86 (m, 2H), 6.52 - 6.47 (m, 2H), 4.88 (s, 2H), 3.99 - 3.80 (m, 2H), 2.80 - 2.54 (m, 2H), 2.37 (tt, J = 11.6, 3.7 Hz, 1 H), 1.85 - 1.75 (m, 1 H), 1.70 - 1.62 (m, 1 H), 1.53 (ddd, J = 24.4, 12.4, 3.3 Hz, 1 H), 1.46 - 1.33 (m, 10H). LC S Method C: rt 4.86 min; m/z 177.2 [M-Boc+2H]+.
(c) tert-Butyl 3-(4-((4-chloro-5-(trifluoromethyl)pyrimidin-2-yl)am
1 -carboxyiate (124)
2,4-Dichloro-5-(trifluoromethyl)pyrimidine (0.355 g, 1.64 mmoi) was stirred in a 1 :1 t- BuOH: 1 ,2-dichloroethane mixture (30 mL) at 0 °C and a 1.0 M ZnCI2 solution in diethyl ether (1.87 mL, 1.87 mmol) was added cautiously over 20 minutes and the reaction was left stirring at 0 °C for 30 minutes. A solution of ferf-buty! 3~(4~ aminophenyl)piperidine-1-carboxylate {123) (0.431 g, 1.56 mmol) in 1 : 1 f-BuOH:1 ,2- dichloroefhane (10 mL) was added drop-wise over 15 minutes at 0 °C followed by a solution of triethylamine (0.261 μί, 1.871 mmoi) in 1 : 1 f-BuOH: 1 ,2-dichloroethane
(10 mL). The reaction was allowed to warm to room temperature and was stirred for 60 hours. Voiatiles were evaporated in vacuo and the resulting oily residue was suspended in water (200 mL), the suspension was sonicated for 30 minutes and the product was collected by filtration, the solid was washed with water (5 x 10 mL) and dried under a high vacuum to give the title compound (124) (0.638 g, 90% yield) as a pale pink solid; 1 H NMR (400 MHz, DMSQ) δ 10.62 (s, 1 H), 8.78 (s, 1 H), 7.61 (d, J = 8.5 Hz, 2H), 7.26 (d, J = 8.6 Hz, 2H), 4.05 - 3.87 (m, 2H), 2.94 - 2.63 (m, 2H), 2.57 (ddd, J = 11.2, 7.7, 3,7 Hz, 1 H), 1.87 (d, J = 12.8 Hz, 1 H), 1.75 - 1.54 (m, 2H), 1.51 - 1.35 (m, 10H). LCMS Method C: rt 6.89 min; m/z 455.3 M-H]\
(d) tert-Butyl 3~(4~((4~((2~(2~methoxy-2-oxoeihyl)phenyl)ethynyl}-5- (trifluommethyi)pyrimidin-2-yi)amino)phenyi)piperidine- 1-carboxylate (125)
To a nitrogen de-gassed solution of methyl 2-(2-ethynylphenyl)acetate (14) (0.137 g, 0.788 mmol) in dry DMF (7 mL) were added triethyiamine (0.366 mL, 2.63 mmol) followed by ieri-bufyi 3-(4-((4-chloro-5-(trifluoromethyl)pyrimidin-2- yl)amino)phenyl)piperidine-1-carboxylate (124) (0.300 g, 0.657 mmol),
triphenylphosphine (0.026 g, 0.098 mmol), irans-dichlorobis(triphenylphosphine) pailadium(il) (0.046 g, 0.066 mmol) and Cul (0.019 g, 0.098 mmol). The reaction mixture was heated under microwave irradiation at 120 °C for 20 minutes and then concentrated to dryness in vacuo and purified by silica gel chromatography (Biotage I solera, 40 g Si cartridge, 0-70% EtOAc in petroleum benzine 40-60 °C) to give the title compound (125) (0.310 g, 79% yield) as a yellow sticky oil; flH NMR (400 MHz, drDMSO) δ 10.44 (s, 1 H), 8.81 (s, 1 H), 7.73 - 7.60 (m, 3H), 7.54 (td, J = 7.6, 1.3 Hz, 1 H), 7.50 - 7.34 (m, 2H), 7.24 (d, J = 8.6 Hz, 2H), 4.02 - 3.87 (m, 4H), 3.61 (s, 3H), 2.90 - 2.51 (m, 4H), 1.88 (d, J = 12,0 Hz, 1 H), 1.76 - 1.55 (m, 2H), 1.51 - 1.34 (m, 10H). LCMS Method C: rt 6.98 min; m/z 539.2 [M-C4H9 (f-Bu)+H]+.
(e) tert-Butyl 3-(4-((4-(2-(2-methoxy-2-oxoethyl)phenethyl)-5-
(trifluoromethyi)pyrimidin-2-yl)amino)phenyl)piperidine- 1-carb (126)
terf-Butyl 3-(4-((4-((2-(2-methoxy-2-oxoethyl)phenyi)ethynyi)-5-
(trifluoromethyl)pyrimidin-2-yl)amino)phenyl)piperidine-1-carboxylate (125) (0.302 g, 0.508 mmol) was dissolved in EtOAc ( 2 mL) and absolute ethanol (8 mL) under an atmosphere of nitrogen. 10% Pd/C (0.250 g) in EtOAc (4 mL) was added to the solution and the atmosphere was changed to hydrogen gas (balloon). The reaction was sealed with a balloon and stirred at room temperature for 18 hours after which
the catalyst was removed by filtration through Celite and the solvent was removed in vacuo. The resulting solid was again dissolved in EtOAc (12 mL) and absolute ethanol (8 mL) under an atmosphere of nitrogen and 10% Pd/C (0.250 g) in EtOAc (4 mL) was added to the solution and the atmosphere was changed to hydrogen gas (balloon). The reaction was sealed with a balloon and stirred at room temperature for 24 hours. The catalyst was removed by filtration through Celite, which was washed with EtOAc (7 x 10 mL) and the solvent was removed in vacuo to give a pale yellow viscous oil. The crude product was purified by silica gel chromatography (Biotage Isolera, 40 g Si Cartridge, 0-50% EtOAc in petroleum benzine 40-60 °C) to give the title compound (126) (0.249 g, 82% yield) as a pale yellow viscous oil; H N R (400 MHz, de-DMSO) δ 10.18 (s, 1 H), 8.67 (s, 1 H), 7.72 - 7.64 (m, 2H), 7.26 - 7.16 (m, 6H), 4.02 - 3.89 (m, 2H), 3.76 (s, 2H), 3.57 (s, 3H), 3.10 - 2,95 (m, 4H), 2.91 - 2.66 (m, 2H), 2.61 - 2.51 (m, 1 H), 1.88 (d, J = 11.8 Hz, 1 H), 1.75 - 1.54 (m, 2H), 1.43 (m, 10H). LC S Method C: rt 7.11 min; m/z 599.3 [M+H]+.
(f) Lithium 2-(2-(2-(2-((4-(1-(tert-butoxycarbonyl)piperidin-3-yl)phenyl)am
(trifluoromethyl) pyrimidin-4-yl)ethyl)phenyl)acetate (127)
LiOH.H20 (0.052 g, 1.25 mmol) was added to iert-butyl 3-(4-((4-(2-(2-methoxy-2- oxoethyl)phenethyi)-5-(trifiuoromethyl)pyrimidin-2-yl)amino)phenyi)piperidine-1- carboxylate (126) (0.249 g, 0.416 mmol) in THF (10 mL), water (2.5 mL) and methanol (1 mL). The resulting mixture was allowed to stir for 2 hours at 40 °C and then 20 hours at room temperature. The volatiles were removed in vacuo and the residue was diluted with EtOAc (70 mL) and 2 M aq. NaOH (50 mL). The layers were separated and the aqueous layer was extracted with EtOAc (70 mL), the organic layers were combined, washed with brine (70 mL), dried (MgS04), filtered and concentrated under reduced pressure to give the title compound (127) (0.250 g, 100% yield) as an off-white oily solid; 1 H NMR (400 MHz, d DMSO) δ 10.23 (s, 1 H), 8.64 (s, 1 H), 7.72 - 7.60 (m, 2H), 7.25 - 7.00 (m, 6H), 4.01 - 3.88 (m, 2H), 3.48 (s, 2H), 3.13 - 2.92 (m, 4H), 2.86 - 2.63 (m, 2H), 1.91 - 1.81 (m, 1 H), 1 ,72 - 1.54 (m, 2H), 1.48 - 1.34 (m, 10H). LCMS Method C: rt 6.71 min; m/z 585.3 [M+H]+.
(g) tert-Butyl 3-(4-((4-(2-(2-amino-2-oxoethyi)phenethyi)-5-(trifiuoromethy
2-yl)amino)phenyl)piperidine- 1 -carboxylate (128)
Lithium 2-(2-(2-(2-((4-(1-(ierf-butoxycarbonyl)piperidin-3-yl)phenyl)amino)-5- (trifluoromethyl) pyrimidin-4-yl)ethyl)phenyl)acetate (127) (0.246 g, 0.416 mmol) was
dissolved in dry THF (10 mL) and dry DMF (2 mL) under an atmosphere of nitrogen. To the solution were added 1-hydroxybenzotriazole (0.067 g, 0.50 mmoi) and EDCI (0.096 g, 0.50 mmol) and A/,A -diisopropylethylamine (0.290 mL, 1.66 mmol) and the reaction mixture was stirred at room temperature for 0 minutes. Ammonium carbonate (0.160 g, 1.66 mmol) was added in one portion, and the reaction was stirred at room temperature for 45 hours. The volatiles were removed in vacuo and the residual solution was diluted with EtOAc (70 mL) and sat. aq. NaHC03 (70 mL). The layers were separated and the aqueous layer was extracted with EtOAc (70 mL), the combined organic layers were washed with water (70 mL), brine (70 mL), dried ( gS04), filtered and concentrated in vacuo to give an off-white solid. The crude product was purified by silica gel chromatography (Biotage isolera, 40 g Si Cartridge, 0-100% EtOAc in petroleum benzine 40-60 °C, then 0-10% methanol in EtOAc) to give the title compound {128) (0.186 g, 77% yield) as a white solid; 1H N R (400 MHz, cfe-DMSO) δ 10.17 (s, 1 H), 8.67 (s, 1 H), 7.69 (d, J = 8.6 Hz, 2H), 7.44 (br s, 1 H), 7.28 - 7.11 (m, 6H), 6.93 (br s, 1 H), 4.02 - 3.89 (m, 2H), 3.50 (s, 2H), 3.15 - 3.07 (m, 2H), 3.06 - 2.98 (m, 2H), 2.89 - 2,67 (m, 2H), 2,61 - 2.51 (m, 1 H), 1.88 (d, J = 11.1 Hz, 1 H), 1.74 - 1.55 (m, 2H), 1.50 - 1.35 (m, 10H). LCMS Method C: rt 6.57 min; m/z 584.3 [M+H]+. (h) 2-(2-(2-(2-((4-(Piperidin-3-yl)phenyl)amino)-5-(trifluo
yi)ethyi)phenyi) acetamide (4)
ferf-Butyl 3-(4-((4-(2-(2-amino-2-oxoethyl)phenethyl)-5-(trifluoromethyl)pyrirnidin-2- yi)amino)phenyl)piperid!ne-1-carboxylate {128) (0.184 g, 0.315 mmoi) was dissolved in DCM (10 mL) under an atmosphere of nitrogen. Trifiuoroacetic acid (1.21 mL, 15.8 mmoi) was added to the solution and the reaction was stirred at room temperature for 1 hour. Volatiles were removed in vacuo, EtOAc (100 mL) and 2 M aq. NaOH (70 mL) were added to the residue and the layers were separated. The aqueous layer was extracted with EtOAc (70 mL), the combined organics were washed with water (50 mL), brine (50 mL), dried (MgS04), filtered and concentrated in vacuo to give a white solid. The solid was taken up in DCM (~ 7 mL) and methanol (~ 1 mL) and concentrated in vacuo. The process was repeated with only DCM twice to give the title compound (4) (0.110 g, 72% yield) as a white solid; 1H NMR (400 MHz, cfe- DMSO) δ 10.13 (s, 1 H), 8.65 (s, 1 H), 7.69 - 7.62 (m, 2H), 7.44 (s, 1 H), 7.26 - 7,12 (m, 6H), 6.93 (s, 1 H), 3.50 (s, 2H), 3.15 - 3.06 (m, 2H), 3.06 - 2.90 (m, 4H), 2.60 - 2.43 (m, 3H), 1.85 (d, J = 11.1 Hz, 1 H), 1.69 - 1.61 (m, 1 H), 1.61 - 1.41 (m, 2H). 3
Aliphatic protons obscured by residual D SO. LC S Method C: rt 4.81 min; m/z 484.3 [M+H]+.
Example 5: 2-(2-(2-(2-((4-(1 -IVIethylpsperidin-3-yl}phenyl)amsno}-5- ft

To a suspension of 2-(2-(2-(2-((4-(piperidin-3-yl)phenyl)amino)-5- (trifluoromethyl)pyrimidin-4-yl)ethyl)phenyl) acetamide (4) (0.021 g, 0.043 mmol) in anhydrous methanol (2 mL) were added a 37% aq. solution of formaldehyde (0.006 g, 0.195 mmol) followed by sodium triacetoxyborohydride (0.046 g, 0,217 mmol) under an atmosphere of nitrogen. The reaction was stirred at room temperature for 1 hour, the volatiles were removed in vacuo and the residue was diluted with EtOAc (15 mL) and sat. aq. NaHC03 (10 mL). The layers were separated and the aqueous layer was extracted with EtOAc (15 mL), the combined organic layers were washed with water (10 mL), brine (10 mL), dried (MgS04), filtered and concentrated in vacuo to give the title compound (5) (20 mg, 93% yield) as a an off-white solid; 1H NMR (400 Hz, drMeOD) δ 8.54 (s, 1 H), 7.62 (d, J = 8.5 Hz, 2H), 7.30 - 7.15 (m, 6H), 3.67 (s, 2H), 3.19 - 3.12 (m, 2H), 3.12 - 3.02 (m, 2H), 2.96 (d, J = 11.1 Hz, 2H), 2.85 - 2.74 (m, 1 H), 2.34 (s, 3H), 2.13 - 2.03 (m, 2H), 1.96 - 1.67 (m, 3H), 1.49 (ddd, J = 24.8, 12.5, 3.9 Hz, 1 H). LCMS Method C: rt 4.86 min; m/z 498.3 [M+Hf.
Example 6: 3~(2~(2~((3-(4- ethylpsperazm~1 ~y!)phenyl)amino)

!30
(a) tert-Butyl 3-(2-(2-((3-(4-methylpiperazin- 1-yl)phenyl)amino)-5- (trifluoromethyl)pyrimidin-4-yl)ethyl)benzoate (129)
A mixture of feri-butyi 3-(2-(2-(methylsulfonyl)-5-(trifluoromethyl)pyrimidin-4- yl)ethyl)benzoate (120) (0.072 g, 0.17 mmol), Tosic acid monohydrate (0.044 g, 0.231 mmol), and 3-(4-metbyipiperazinyi-1-y!)aniiine (0.055 g, 0.29 mmol) in dioxane (3.0 mL) was heated to 140-150X under microwave irradiation for 30 minutes. The mixture was then concentrated under reduced pressure and purified using silica gel column chromatography (0-10% MeOH/EtOAc with 1 % NH3 aq.) to give the title compound (129) (0.048 g, 52%); 1H NMR (400 MHz, CDCI3) δ 8.54 (s, 1 H), 7.88 (s, 1 H), 7.84 (ddd, J = 7.4, 1.6, 1.6 Hz, 1 H), 7.37 (m, 4H), 7.23 (m, 1 H), 7.04 (dd, J = 7.9, 1.4 Hz, 1 H), 6.69 (dd, J = 8.2, 1.8 Hz, 1 H), 3.27 (m, 4H), 3.14 (m, 4H), 2.58 (m, 4H), 2.35 (s, 3H), 1 ,59 (s, 9H). LC S Method C: rt 5.45 min; m/z 542,3 [M+H]+.
(b) 3~(2~(2~( (3-(4-Methylpiperazin- 1 -yl)phenyl)amino)-5-(trifluoromethyl)pyrimidin-4- yl)ethyl)benzoic acid (130)
To a solution of ferf-butyl 3-(2-(2-((3-(4-methylpiperazin-1-yl)phenyl)amino)-5-
(trifluoromethyl)pyrimidin-4-yl)ethyl)benzoate (129) (0.048 g, 0.088 mmol) in DCM (3 mL) was added TFA (0.5 mL); the reaction was then stirred for 35 minutes at room temperature before concentrating under reduced pressure. The residue was taken up in toluene (2x10 mL) and concentrated under reduced pressure. The resulting residue was taken up in DCM (3 mL) and TFA (0.5 mL) was added. The resulting solution was stirred at room temperature for 1 hour then the volatiies removed by evaporation under reduced pressure. The residue was taken up in toluene (2x20 mL) and concentrated under reduced pressure to give the title compound (130) in qualitative yield; 1 H NMR (400 MHz, cfe-Acetone) δ 9.14 (s, 1 H), 8.62 (s, 1 H), 7.96 (s, 1 H), 7.89 (d, J = 7.7 Hz, 1 H), 7.74 (bs, 1 H), 7.54 (d, J = 7.7 Hz, 1 H), 7.44 (dd, J = 7.6, 7.6 Hz, 1 H), 7.34 (bd, J = 8.0 Hz, 1 H), 7.25 (dd, J = 8.1 , 8.1 Hz, 1 H), 6.77 (dd, J = 8.1 , 1.8 Hz, 1 H), 3.60 (m, 4H), 3.47 (m, 4H), 3.20 (m, 4H), 2.94 (s, 3H). LCMS Method C: rt 4.93 min; m/z 486.2 [M+H]+.
(c) 3-(2-(2-((3-(4-Methylpiperazin-1-yl)phenyl)amino)-5-(triH^
yl)ethyl)benzamide (6)
To a solution of 3-(2-(2-((3-(4-methylpiperazin-1-yl)phenyi)amino)-5- (trifiuoromethyl)pyrimidin-4-yl)ethyl)benzoic acid (130) (0.048 g, 0.10 mmol) and HATU (0.051 g, 0.13 mmol) in DMF (2 mL) was added DIPEA (0.068 mL, 0.39
mmol), the resulting solution was then stirred for 10 minutes before addition of NH4OH (0.2 mL). The resulting mixture was then stirred overnight (18 hours) at room temperature. The mixture was diluted with water and extracted with EfOAc (2x20 mL). The combined organic extracts were dried using a phase separation cartridge before concentrating under reduced pressure. The organic residues were then purified using silica gel column chromatography (0-30% MeOH/EtOAc with 1 % NH4OH in the eOH) to give the title compound (6)(0.028 mg, 58%) as a white solid; 1H NMR (400 Hz, d6-DMSO) δ 10.07 (s, 1 H), 8.67 (s, 1 H), 7.95 (s, 1 H), 7.78 (s, 1 H), 7.72 (m, 1 H), 7.54 (s, 1 H), 7.38 (m, 2H), 7.34 (m, 1 H), 7.14 (d, J = 5.2 Hz, 2H), 6.64 (m, 1 H), 3.10 (m, 8H), 2.42 (m, 4H), 2.19 (s, 3H). LCMS Method C: rt 4.80 min; m/z 485.3 [M+H]4.
Example 7: 2-(2-(2-(2-((4-(1 -IVIethylpsperidin-3-yl}phenyl)amsno}-5- ftnf!uoromethy!) pyrimidsn-4-yS)ethyl)pheny!)acetamide (7)

terf-Butyl 3-ethynylbenzoate (117) (1.50 g, 9.37 mmoi) was dissolved in dry DCM (70 mL) and TFA (35.9 mL, 488 mmoi) was added carefully. The reaction was stirred at room temperature for 3 hours, concentrated in vacuo and toluene was added and then removed in vacuo to give a pale yellow solid. This material was dissolved in methanol (50 mL) and cone. H2S04 (-1 mL) was added and the resulting solution was stirred at 85 °C for 20 hours. Upon cooling to room temperature, the volatiies were removed in vacuo and the residue was diluted with EtOAc (200 mL) and sat. aq. NaHCO3 (100 mL) was added slowly. The layers were separated and the aqueous layer was extracted with EtOAc (200 mL), the organic layers were combined and washed with water (100 mL), brine (100 mL), dried ( gS04), filtered and concentrated in vacuo to give the title compound (131) (1.136 g, 96% yield over 2 steps) as a pale yellow solid; 1H NMR (400 MHz, CDCI3) δ 8.17 (t, J = 1.5 Hz, 1 H), 8.03 - 8.00 (m, 1 H), 7.66 (dt, J = 7.7, 1.4 Hz, 1 H), 7.41 (td, J = 7.8, 0.4 Hz, 1 H), 3.93 (s, 3H), 3.12 (s, 1 H). LCMS Method C: rt 5.84 min.
(b) tert-Butyl 4-(4-( (4-((3-(methoxycarbonyl)phenyl)ethynyl)-5- (trifluoromethyl)pyrimidin-2-yl)amino)phenyl)piperazine- 1 -carboxylate (132)
To a nitrogen de-gassed solution of methyl 3-ethynylbenzoate (131) (0.105 g, 0.655 mmoi) in dry DMF (6 mL) were added triethylamine (0.308 mL, 2.18 mmoi) followed by ferf-butyl 4-(4-((4-chloro-5-(trifluoromethyl)pyrimidin-2-yl)amino)phenyl)piperazine- 1-carboxylate (13) (0.250 g, 0.546 mmoi), triphenyiphosphine (0.021 g, 0.082 mmoi), frans-dichlorobis(triphenyiphosphine) palladium(ll) (0.038 g, 0.055 mmoi) and Cu(l)l (0.016 g, 0.082 mmoi). The reaction mixture was heated under microwave irradiation at 120 °C for 20 minutes, concentrated to dryness in vacuo and purified by silica gel chromatography (Biotage Isoiera, 40 g Si cartridge, 0-50% EtOAc in petroleum benzine 40-60 °C) to give the title compound (132) (0.182 g, 57% yield) as an orange solid; 1H NMR (400 MHz, d^DMSO) δ 10.28 (br s, 1 H), 8.78 (s, 1 H), 8.16 - 8.03 (m, 2H), 7.90 (d, J = 7.8 Hz, 1 H), 7.69 (t, J = 7.9 Hz, 1 H), 7.55 (d, J = 9.0 Hz, 2H), 6.96 (d, J = 9.0 Hz, 2H), 3.90 (s, 3H), 3.50 - 3.41 (m, 4H), 3.11 - 2.99 (m, 4H), 1.42 (s, 9H). LCMS Method C: rt 6.82 min; m/z 582.2 [M+H]+.
(c) tert- Butyl 4--(4--((4-(3-(methoxycarbonyl)phenethyl)-5-(trif!uom
yl)amino)phenyl)piperazine- 1 -carboxylate (133)
fe/f-Butyl 4-(4-((4-((3-(methoxycarbonyl)phenyl)ethynyl)-5-(trifluoromethyl)pyrimidin- 2-yl)amino)phenyl)piperazine-1 -carboxylate (132) (0.180 g, 0.309 mmol) was dissolved in dry D F (10 mL) under an atmosphere of nitrogen. 10% Pd/C (0.100 g) in EtOAc (10 mL) was added to the solution and the atmosphere was changed to hydrogen gas (balloon). The reaction was sealed with a balloon and stirred at room temperature for 18 hours after which the reaction was flushed with nitrogen gas and Pearlman's catalyst (0.150 g) in EtOAc (5 mL) was added. The atmosphere was again changed to hydrogen gas (balloon) and the reaction was sealed with balloon and stirred for 20 hours at room temperature. The catalyst was removed by filtration through Celite, which was washed with EtOAc (5 x 10 mL). The solvent was removed in vacuo to give a yellow oil which was purified by silica gel chromatography (Biotage Isolera, 40 g Si Cartridge, 0-40% EtOAc in petroleum benzine 40-80 °C) to give the title compound (133) (0.120 g, 66% yield) as a yellow foam; 1H NMR (400 MHz, o¾- DMSO) δ 10.01 is, 1 H), 8.60 (s, 1 H), 7.88 - 7.77 (m, 2H), 7.61 - 7.39 (m, 4H), 6.93 (d, J = 9.1 Hz, 2H), 3.85 (s, 3H), 3.52 - 3.42 (m, 4H), 3.21 - 2.99 (m, 8H), 1.42 (s, 9H). LCMS Method C: rt 6.86 min; m/z 586.3 [M+H]+. (d) Lithium 3-(2-(2-((4-(4-(tert-butoxycarbonyl)piperazin-1-yl)phenyl)amino)-5- ( trifluoromethyl)pyrimidin -4 -yl)ethyl)benzoate (134)
LiOH.H20 (0.025 g, 0.60 mmol) was added to iert-butyl 4-(4-((4-(3- (methoxycarbonyi)phenethyl)-5-(trifluoromethyl)pyrim!din-2- yl)amino)phenyl)piperazine-1-carboxylate (133) (0.117 g, 0.200 mmol) in THF (7 mL), water (1.5 mL) and methanol (1 mL). The resulting mixture was allowed to stir at room temperature for 17 hours, the voiatiies were removed in vacuo and the residue was diluted with EtOAc (100 mL) and sat. aq. NaHC03 (80 mL). The layers were separated and the aqueous layer was extracted with EtOAc (2 x 80 mL), the organic layers were combined, washed with brine (70 mL), dried (MgS04), filtered and concentrated under reduced pressure to give the title compound (134) (0.105 g, 91 % yield) as a pale yellow solid; 1H NMR (400 MHz, cfe-DMSO) δ 10.00 (s, 1 H), 8.60 (s, 1 H), 7.85 - 7.74 (m, 2H), 7.56 - 7.48 (m, 2H), 7.47 - 7.35 (m, 2H), 6.93 (d, J = 9.1 Hz, 2H), 3.50 - 3,41 (m, 4H), 3, 17 - 2.98 (m, 8H), 1.42 is, 9H). LCMS Method C: rt 6.30 min; m/z 572.3 [M+H .
(e) tert-Butyl 4-(4-((4-(3-carbamoylphenethyl)-5-(trifluorom
yl)amino)phenyl)piperazine-1-carboxylate (135)
Lithium 3-(2-(2-((4-(4-(iert-butoxycarbonyl)piperazin-1-yl)phenyl)amino)-5- (trifluoromethyl) pyrimidin-4-yl)ethyl)benzoate (134) (0,100 g, 0.173 mmol) was dissolved in dry THF (7 mL) and dry DMF (1 mL) under an atmosphere of nitrogen. To the solution were added 1-hydroxybenzotriazole (0.028 g, 0.21 mmol) and EDCI (0.040 g, 0.21 mmol) and A/-diisopropyiethylamine (0.121 mL, 0.893mmo!) and the reaction mixture was stirred at room temperature for 10 minutes. Ammonium carbonate (0.067 g, 0.69 mmol) was added in one portion, and the reaction was stirred room temperature for 60 hours. The volatiles were removed in vacuo and the residual solution was diluted with EtOAc (70 mL) and sat. aq. NaHC03 (70 mL). The layers were separated and the aqueous layer was extracted with EtOAc (70 mL), the combined organic layers were washed with water (70 mL), brine (70 mL), dried (MgSQ4), filtered and concentrated in vacuo to give an off-white solid. The crude product was purified by silica gel chromatography (Biotage Isolera, 12 g Si Cartridge, 0-100% EtOAc in petroleum benzine 40-60 °C) to give the title compound (135) (0.073 g, 74% yield) as an off-white solid; 1 H NMR (400 MHz, cfe-D SO) δ 10.00 (s, 1 H), 8.61 (s, 1 H), 7.95 (s, 1 H), 7.79 (s, 1 H), 7.75 - 7.68 (m, 1 H), 7.54 (d, J = 8.6 Hz, 2H), 7.40 - 7.29 (m, 3H), 6.93 (d, J = 9.1 Hz, 2H), 3.50 - 3.41 (m, 4H), 3.14 - 3.00 (m, 8H), 1.42 (s, 9H).
(f) 2-(2-(2-(2-((4-(1-Methylpiperidin-3-yl)phenyl)amino)-5-(triflu^ pyrimidin-4- y!)ethyi)phenyi)aceta ide (7)
ferf- Butyl 4-(4-((4-(3-carbamoylphenethyl)-5-(trifluoromethyl)pyrimidin-2- yl)amino)phenyl) piperazine-1-carboxylate (135) (0.070 g, 0.12 mmol) was dissolved in DCM (5 mL) under an atmosphere of nitrogen. Trifiuoroacetic acid (0.282 mL, 3.68 mmol) was added to the solution and the reaction was stirred at room temperature for 18 hours. Volatiles were removed in vacuo, EtOAc (50 mL) and 2 M aq. NaOH (50 mL) were added to the residue and the layers were separated. The aqueous layer was extracted with EtOAc (50 mL), the combined organics were washed with water (50 mL), brine (50 mL), dried (MgSOA), filtered and concentrated in vacuo to give a pale yellow solid. The solid was taken up in DCM (- 10 mL) and methanol (~ 1 mL) and concentrated in vacuo. The process was repeated with only DCM twice to give the title compound ( 7) (0.043 g, 75% yield) as a pale yellow solid; ΊΗ NMR (400 MHz, de-DMSO) δ 9.96 (s, 1 H), 8.60 (s, 1 H), 7.95 (s, 1 H), 7.79 (s, 1 H), 7.74 - 7.66 (m, H),
7.56 - 7.47 (m, 2H), 7.40 - 7.29 (m, 3H), 6.94 - 6.82 (m, 2H), 3.15 - 2.96 (m, 8H), 2.86 - 2.79 (m, 4H). LCMS Method C: rt 4.71 min; m/z 471.2 [ +Hf .
Example 8: 2-(2-(2-(2-((4-(Pspendin-2-yf}phenyl)amsno}-5- ftnf!uorometh S)pyrimidin-4-yl)ethy!)pheny!)acetamide (8)

(a) tert-Butyl 2-(4-aminophenyl)piperidine-1-carboxylate (136)
To a solution of 2-chloropyridine (0.816 g, 7.19 mmol) in nitrogen degassed dry DMF (20 mL) was added 4-nitrophenyiboronic acid (1.00 g, 5.99 mmol), Cs2C03 (7.19 g, 24.0 mmol), triphenylphosphine (0.236 g, 0.899 mmol) and Pd(OAc)2 (0.134 g, 0.599 mmol). The reaction mixture was heated at 80 °C for 24 hours. The reaction was cooled to room temperature and concentrated to dryness in vacuo. The crude material absorbed onto silica gel and purified by silica gel chromatography (Biotage Isolera, 40 g Si cartridge, 0-100% EtOAc in petroleum benzine 40-60 °C) to give a pale yellow solid. This material (0.365 g) was dissolved in 1 M HCI (1.5 mL) and methanol (15 mL) and Pt02 (0.036 g) was added under an atmosphere of nitrogen. The reaction was then subjected to a 40 psi hydrogen atmosphere in a Parr
hydrogenator for 24 hours, and then filtered through ceiife which was washed with EtOAc (3 x 10 mL) and water (3 x 10 mL). The combined filtrate was concentrated in vacuo to give the crude material (0,460 g) as a pale brown-pink oily solid. This material was dissolved in anhydrous methanol (20 mL) and treated with triethyiamine (0.889 mL, 8.38 mmol) followed by Boc anhydride (0.418 g, 1.91 mmoi). The reaction was stirred at room temperature for 20 hours, concentrated in vacuo and EtOAc (100 mL) and sat. aq. NaHC03 (50 mL) were added and the layers were separated. The aqueous layer was extracted with EtOAc (70 mL), the organics were combined and washed with water (50 mL), brine (50 mL), dried ( gS04), filtered and concentrated in vacuo to give a yellow solid. The crude product was purified by silica gel chromatography (Biotage isolera, 12 g Si cartridge, 0-100% EtOAc in petroleum benzine 40-60 °C), to give the title compound (136) (0, 22 g, 8% yield over 3 steps) as a white solid; 1 H NMR (400 MHz, c 6-DMSO) 5 6.85 - 6.78 (m, 2H), 6.57 - 6.51 (m, 2H), 5.14 (d, J = 3.7 Hz, 1 H), 4.98 - 4.89 (m, 2H), 3.85 (d, J = 13.0 Hz, 1 H), 2.70 - 2.57 (m, 1 H), 2.19 (d, J = 13.9 Hz, 1 H), 1.73 - 1.61 (m, 1 H), 1 ,57 - 1.44 (m, 2H), 1.44 - 1.27 (m, 1 H). LCMS Method C: rt 4.94 min; m/z 177.3 [M~Boc+2H]+
(b) tert-Butyl 2-(4-((4-chloro-5-(trifluoro ethyl)pyri idin-2-yl)a
1 -carboxylate (137)
2,4-Dichloro-5-(trifluoromethyl)pyrimidine (0.101 g, 0.464 mmol) was stirred in a 1 : 1 t- BuOH: 1 ,2-dichloroethane mixture (10 mL) at 0 °C and a 1.0 M ZnCI2 solution in diethyl ether (0.530 mL, 0.530 mmol) was added cautiously over 20 minutes. After addition, the reaction was left stirring at 0 °C for 30 minutes and a solution of tert- butyl 2-(4-aminophenyl)piperidine-1-carboxylate (136) (0.122 g, 0.441 mmol) in 1 : 1 /- BuOH:1 ,2-dichloroethane (4 mL) was added drop-wise over 5 minutes at 0 °C followed by a solution of triethyiamine (0.074 μί, 0.530 mmoi) in 1 :1 f-BuOH: 1 ,2- dichloroethane (4 mL) and the reaction was allowed to warm to room temperature and was stirred for 60 hours. Voiafiles were evaporated in vacuo and the resulting residue was suspended in water (40 mL), the suspension was sonicated for 40 minutes and the product was collected by filtration, the solid was washed with water (5 x 10 mL) and dried under a high vacuum to give the title compound (137) (0.135 g, 67% yield) as a pale pink solid. LCMS Method C: rt 6.96 min; m/z 455,2, 457.2 M-H]".
(c) 2-(2-lodophenyl)acetamide (138)
2-iodophenyiacetic acid (2.00 g, 7.83 mrnoi) was dissolved in dry THF (70 mL) and dry D F (10 mL) under an atmosphere of nitrogen. To the solution were added 1- hydroxybenzotriazole (1 , 134 g, 8.396 mmol) and EDCI (1.609 g, 8.396 mmol) and W,A -diisopropylethylamine (5.318 mL, 30.53 mmol) and the reaction mixture was stirred at room temperature for 10 minutes. Ammonium carbonate (2.933 g, 30.53 mmol) was added in one portion, and the reaction was stirred room temperature for 17 hours. The volatiles were removed in vacuo and the residual solution was diluted with EtOAc (150 mL) and sat. aq. NaHC03 (100 mL). The layers were separated and the organic layer were washed with water (100 mL), brine (100 mL), dried (MgS04), filtered and concentrated in vacuo to give the title compound (138) (1.755 g, 88% yield) as a beige solid; H N R (400 MHz, e/g-DMSG) δ 7,82 (dd, J = 7.9, 0.9 Hz, 1 H), 7.42 (s, 1 H), 7.36 - 7.28 (m, 2H), 7.02 - 6.94 (m, 2H), 3.55 (s, 2H). LC S Method C: rt 4.77 min; m/z 262.0 [M+H]+.
(d) 2-(2-Ethynylphenyl)acetamide (139)
To a nitrogen de-gassed solution of 2-(2-iodophenyl)acetamide (138) (1.75 g, 6.70 mmol) in dry THF (50 mL) and dry DMF (10 mL) was added Pd(PPh3)4 (0.194 g, 0.168 mmol) and Cu(i)i (0.064 g, 0.34 mmol), triethylamine (3.27 mL, 23.5 mmol). The mixture was stirred for 10 minutes and T S-acetylene (1.52 mL, 10.7 mmol) was added. The reaction mixture was then stirred at room temperature for 8 hours, concentrated in vacuo and purified by silica gel chromatography (Biotage isoiera, 40 g Si cartridge, 0-100% EtOAc in petroleum benzine 40-60 °C) to give a beige solid. This material was dissolved in dry THF (25 mL) under an atmosphere of nitrogen and TBAF (1 ,0 M in THF, 2.805 mL, 2.805 mmol) was added dropwise at 0 °C. The solution was stirred at this temperature for 1 hour and 15 minutes after which water (5 mL) was added. The reaction mixture was concentrated in vacuo and diluted with EtOAc (100 mL) and sat. aq. NaHC03 ( 00 mL). The layers were separated and aqueous layer was extracted with EtOAc ( 00 mL), the organic layers were combined and washed with water (100 mL), brine (100 mL), dried (MgS04), filtered and concentrated in vacuo to give the crude product. The material was purified by silica gel chromatography (Biotage Isoiera, 40 g Si cartridge, 0-100% EtOAc in petroleum benzine 40-60 °C, then 0-20% methanol in EtOAc) to give the title compound (139) (0.239 g, 22% yield over 2 steps) as a beige solid; H NMR (400 MHz, 6-DMSO) δ
7.21 (dd, J = 7.6, 1.1 Hz, 1 H), 7.18 - 7.05 (m, 3H), 7.04 - 6.98 (m, 1 H), 6.70 (s, 1 H), 4.08 (s, 1 H), 3.36 (s, 2H). LC S Method C: rt 4.71 min; m/z 160.2 [M+H]+.
(e) tert-Butyl 2-(4-((4-( (2-(2-amino-2-oxoethyl)phenyl)ethynyl)-5- (trifiuoromethyl)pyrimidin~2-yi)amino)phenyl)piperidine- 1 -carboxylate (140)
To a nitrogen de-gassed solution of 2-(2-ethynylphenyl)acetarnide (139) (0.054 g, 0.788 mmol) and ie -butyl 2-(4-((4-chloro-5-(trifiuoromethyl)pyrimidin-2- yl)amino)phenyl)piperidine-1-carboxylate {137) in dry DMF (4 mL) were added triethylamine (0.159 mL, 1.138 mmol), triphenylphosphine (0.011 g, 0.043 mmol), frans-dichlorobis(triphenylphosphine) paliadium(ii) (0.020 g, 0.028 mmoi) and Cul (0.008 g, 0.04 mmol). The reaction mixture was heated under microwave irradiation at 120 °C for 20 minutes. The reaction was concentrated to dryness in vacuo and purified by silica gel chromatography (Biotage Isolera, 40 g Si cartridge, 0-100% EtOAc in petroleum benzine 40-60 °C, then 0-5% methanol in EtOAc) to give the title compound {140) (0.122 g, 74% yield) as a yellow glassy solid; 1H NMR (400 MHz, dg- DMSO) δ 10.48 (s, 1 H), 8.82 (s, 1 H), 7.73 (d, J = 8.7 Hz, 2H), 7.65 - 7.55 (m, 1 H), 7.55 - 7.47 (m, 1 H), 7.47 - 7,33 (m, 3H), 7,16 (d, J = 8.5 Hz, 2H), 7.01 (s, 1 H), 5.29 - 5.22 (m, 1 H), 3.92 (d, J = 13.1 Hz, 1 H), 3.70 (s, 2H), 2.71 (t, J = 13.3 Hz, 1 H), 2.34 - 2.24 (m, 1 H), 1.82 - 1.68 (m, 1 H), 1.54 (d, J = 11.3 Hz, 2H), 1.45 - 1.22 (m, 11 H). LCMS Method C: rt 6.52 min; m/z 578.3 [M-H]\
(f) tert-Butyl 2-(4-((4-(2-(2-amino-2-oxoethyl)phenethyl)-5-(trifluoro
yl)amino)phenyl)piperidine- 1 -carboxylate (141)
ferf- Butyl 2-(4-((4-((2-(2-amino-2-oxoethyl)phenyi)ethynyi)-5- (trifluoromethyl)pyrimidin-2-yl)am!no)phenyi)piper!dine-1 -carboxylate (140) (0,120 g, 0.207 mmol) was dissolved in dry D F (5 mL) under an atmosphere of nitrogen. 20% Pearlman's catalyst (0.060 g) in EtOAc (5 mL) was added to the solution and the atmosphere was changed to hydrogen gas (balloon). The reaction was sealed with a balloon and stirred at room temperature for 20 hours at room temperature. The catalyst was removed by filtration through Celite, which was washed with EtOAc (5 x 10 mL). The solvent was removed in vacuo to give a pale yellow gum which was purified by silica gel chromatography (Biotage, Isolera 12 g Si Cartridge, 0-100% EtOAc in petroleum benzine 40-60 °C) to give the title compound (141) (0.090 g, 74% yield) as a pale yellow solid; 1H NMR (400 MHz, cf6-DMSO) δ 10.20 (s, 1 H), 8.67 (s, 1 H), 7.73 (d, J = 8.7 Hz, 2H), 7.43 (s, 1 H), 7.26 - 7.21 (m, 1 H), 7.20 - 7.09 (m, 5H),
6.89 (s, 1H), 5.31 -5.19 (m, 1H), 3.92 (d, J= 13.2 Hz, 1H), 3.49 (s, 2H), 3.16-2.98 (m, 4H), 2.71 (t, J= 11.8 Hz, 1H), 2.29 (d, J= 13.2 Hz, 1H), 1.75 (t, J= 11.0 Hz, 1H), 1.58-1.50 (m, 2H), 1.46-1.23 (m, 11 H). LC S Method C: rt6.59 min; m/z 584.3 [M+H]+.
(g) 2-(2-(2-(2-((4-(Piperidin-2-yl)pheny!)amino)-5-(trif!uorome
yl)ethyl)phenyl) acetamide (8)
iert-Butyl 2-(4-((4-(2-(2-amino-2-oxoethyl)phenethyl)-5-(trifluoromethyl)pyrimidin-2- y!)amino) phenyl)piperidine-1-carboxylate (141) (0.087 g, 0.15 mmol) was dissolved in dry DCM (5 mL) under an atmosphere of nitrogen. Trifluoroacetic acid (0.351 mL, 4.58 mmol) was added to the solution and the reaction was stirred at room temperature for 22 hours. Volati!es were removed in vacuo, EtOAc (20 mL) and sat, aq. NaHC03 (15 mL) were added to the residue and the layers were separated. The aqueous layer was extracted with EtOAc (15 mL), the combined organics were washed with water (50 mL), brine (50 mL), dried (MgS04), filtered and concentrated in vacuo to give a pale yellow solid which was taken up in DCM (-10 mL) and methanol (~ 1 mL) and concentrated in vacuo. The process was repeated with only DCM twice to give the title compound (8) (0.064 g, 89% yield) as a pale yellow solid; 1H NMR (400 MHz, cfe-DMSO) δ 10.15 (s, 1H), 8.66 (s, 1H), 7.70-7.62 (m, 2H), 7.44 (s, 1H), 7.30 (d, J= 8.6 Hz, 2H), 7.26-7.12 (m, 4H), 6.92 (s, 1H), 3.54-3.47 (m, 3H), 3.15-2.97 (m, 5H), 2.71 -2.59 (m, 1H), 1.84-1.76 (m, 1H), 1.67 (d, J= 12,4 Hz, 1 H), 1.61 - 1.50 (m, 1 H), 1.46 - 1.27 (m, 3H). LCMS Method C: rt 4.84 min; m/z 484.3 [M+H]+.
Example 9: 2-(2-(2-(2-((4-(1-methy!pipend!n-2-y!)pheny}amino}
(

To a suspension of 2-(2-(2-(2-((4-piperidin-2-yl)phenyl)amino)-5- (trifluoromethyl)pyrimidin-4-yl)ethyl)phenyl) acetamide (8) (0.023 g, 0.048 mmol) in anhydrous methanol (1.5 mL) were added a 37% aq. solution of formaldehyde (0.014
mL, 0.19 mmol) and sodium triaceioxyborohydride (0.050 g, 0.24 mmol) under an atmosphere of nitrogen. The reaction was stirred at room temperature for 2.5 hours, the volatiles were removed in vacuo and the residue was diluted with EtOAc (15 mL) and sat, aq. NaHC03 (10 mL). The layers were separated and the aqueous layer was extracted with EtOAc (10 mL), the combined organic layers were washed with water (10 mL), brine (10 mL), dried ( gSC ), filtered and concentrated in vacuo to give a solid which was taken up in DCM (- 10 mL) and methanol (~ 1 mL) and concentrated in vacuo. The process was repeated with only DCM twice after which the sample was further dried on high-vacuum to give the title compound (9) (0.022 g, 91 % yield) as an off-white solid; 1H N R (400 MHz, cfe-DMSO) δ 10.17 (s, 1 H), 8.66 (s, 1 H), 7.68 (d, J = 8.6 Hz, 2H), 7.43 (s, 1 H), 7.27 - 7.13 (m, 6H), 6.92 (s, 1 H), 3.49 (s, 2H), 3.15 - 2.98 (m, 4H), 2.97 - 2.90 (m, 1 H), 2.75 - 2.69 (m, 1 H), 2.07 - 1.97 (m, 1 H), 1.90 (s, 3H), 1.73 (d, J = 12.8 Hz, 1 H), 1.66 - 1.54 (m, 3H), 1.50 - 1.38 (m, 1 H), 1.36 - 1.27 (m, 1 H). LCMS Method C: rt 4.88 min; m/z 498.3 [M+H .
Example 10: 2~(2~(2~(2~((4~(4~Ethylpsperazm~1~y!)phenyl)amino)-5~

To a suspension of 2-(2-(2-(2-((4-(piperazin-1-yi)phenyl)amino)-5- (trifluoromethyl)pyrimidin-4-yl)ethyl)phenyl)acetamide ( 1) (0.020 g, 0.041 mmol) in anhydrous methanol (1.5 mL) were added acetaldehyde (0.0090 mL, 0.17 mmol) and sodium triacetoxyborohydride (0.044 g, 0.21 mmol) under an atmosphere of nitrogen The reaction was stirred at room temperature for 18 hours, the volatiles were removed in vacuo and the residue was diluted with EtOAc (15 mL) and sat. aq. NaHC03 (10 mL). The layers were separated and the aqueous layer was extracted with EtOAc (10 mL), the combined organic layers were washed with water (10 mL), brine (10 mL), dried (MgS04), filtered and concentrated in vacuo to give a solid which was taken up in DCM (- 10 mL) and methanol (~ 1 mL) and concentrated in vacuo. The process was repeated with only DCM twice after which the sample was further dried on high-vacuum to give the title compound ( 10) (0.016 g, 76% yield) as an off-
white solid; 1H NMR (400 MHz, cfe-DMSO) δ 9.97 (s, 1 H), 8.60 (s, 1 H), 7.57 (d, J = 8.9 Hz, 2H), 7.43 (s, 1 H), 7.26 - 7.13 (m, 4H), 6.97 - 6.87 (m, 3H), 3.50 (s, 2H), 3.13 - 2.94 (m, 8H), 2.36 (q, J = 7.2 Hz, 2H), 1.03 (t, J = 7.2 Hz, 3H). 4 Aliphatic protons obscured by the residual DMSO. LCMS Method C: rt 4.82 min; m/z 513.3 [M+H]+.
Example 11 : 2-(2-(2-(2-((4-(piperid!n-4-yl)phenyl)amino)-5- (trifluororneth l)pyrimidm~4~y!)ethyl)phenyl)acetarnide (11 )

!49 11
(a) tert-Butyl 4-(((trifluoromethyl)sulfonyl)oxy)-5,6-dihydrop
(142)
Lithium diisopropylamide (2 M in heptane/THF/ethyibenzene; 15.1 mL, 30.1 mmol) was added dropwise to a solution of ferf-butyl 4-oxopiperidine-1-carboxylate (3.00 g, 15.1 mmol) in THF (50 mL) at -78 °C and the mixture left to stir for 30 minutes. A solution of A-phenyl-bis(trifiuoromethanesuifonimide) (6.46 g, 18.1 mmol) in THF (60 mL) was then added dropwise over 30 minutes to the reaction and mixture left to stir for 30 minutes-78 °C. The resulting mixture was then allowed to warm to room temperature and was stirred for 24 hours. The solvent was partially removed (ca 80 mL) and the reaction mixture quenched with saturated NaHC03 solution (50 mL).
DCM (50 mL) was added to the solution and the layers separated. The aqueous layer was then extracted with DCM (2 x 50 mL). The organic layers were combined and washed with 0.2 M citric acid solution (50 mL), 1 M NaOH (50 mL), brine (50 mL) and dried over Na2S04, The solvent was removed under reduced pressure to give a brown oil which was purified by column chromatography on silica gel (0-10% diethyl ether in petroleum benzine 40-80 °C) to afford the title compound (142) (2.48 g, 50%) as an orange oil which crystallized on cooling to - 8 °C; 1 H NMR (400 MHz, CDCi3) δ 5.76 (s, 1 H), 4,05 - 4.04 (m, 2H), 3.83 (t, J = 5.6 Hz, 2H), 2.46 - 2.43 (m, 2H), .47 (s, 9H).
(b) tert-Butyl 4-(4-nitrophenyl)-5,6-dihydropyridine- 1(2H)-carboxylate (143)
A solution of 2 M Na2C03 (5.66 mL, 1.3 mmol) was added to a mixture of 4- nitrophenyiboronic acid (0.831 g, 4.98 mmol), fe/f -butyl 4- (((trifluoromethyi)sulfonyl)oxy)-5,6-dihydropyridine-1 (2H)-carboxyiate (142) (1.50 g, 4.53 mmol), LiCi (0.384 g, 9.06 mmol) and Pd(PPh3) (1.308 g, 1.132 mmol) in 1 ,4- dioxane (20 mL). The reaction mixture was stirred at 85 - 90 °C for 4 hours. The resulting mixture was dissolved in EtOAc (100 mL) and the organic layer was washed with H20 (50 mL), brine (50 mL) and dried over Na2S04 to yield a dark red oil. The oil was purified by column chromatography on silica gel (0-20% EtOAc in petroleum benzine 40-60 °C) to yield the title compound {143) (0.683 g, 50%) as a pale brown solid; 1H NMR (400 MHz, CDCI3) δ 8.24 - 8.16 (m, 2H), 7.55 - 7.47 (m, 2H), 6.23 (s, 1 H), 4.14 - 4.12 (m, 2H), 3.66 (t, J = 5.7 Hz, 2H), 2.55 (bs, 2H), 1.50 (s, 9H). LCMS Method C: rt 6.39 min; m/z 249 [M-Boc+2H]+, 205 [M-iButyi+2H]+. (c) tert-Butyl 4-(4-aminophenyl)piperidine- 1-carboxylate (144)
A solution of ferf -butyl 4-(4-nitrophenyl)-5,6-dihydropyridine-1 (2H)-carboxylate (143) (0.570 g, 1.87 mmol) in EtOH (5 mL) and DMF (5 mL) was added to a solution of 10% Pd/C (200 mg) in DMF (10 mL). The reaction was stirred at room temperature for 24 hours under an atmosphere of hydrogen. The reaction was filtered through a pad of celite and washed through with EtOAc (130 mL). The solvent was removed in vacuo to yield a brown oil which was purified by column chromatography on silica gel (0-50% EtOAc in petroleum benzine 40-60 °C) to afford the title compound {144) (0.46 g, 89%) as a crystalline solid; 1 H NMR (400 MHz, CDCI3) δ 7.02 - 8.98 (m, 2H), 6.67 - 6.61 (m, 2H), 4.21 (bs, 2H), 3.57 (s, 2H), 2.77 (t, J = 12.2 Hz, 2H), 2.53 (tt, J = 12.1 , 3.5 Hz, 1 H), 1.77 (d, J = 13.3 Hz, 2H), 1.64 - 1.50 (m, peak obscured by
solvent), 1.48 (s, 9H). LC S Method C: rt 4.77 min; m/z 221 [M-fButyl+2H]+, 177 [M- Boc+2H]4.
(d) tert-Butyl 4-(4-((4-chloro-5-(trifluoromethyl)pyrimidin-2-yl)am
/ -carboxylate (145)
Zinc chloride (1.0 M in Et20) (1.97 mL, 1.97 mmol) was added to a solution of 2,4- dichloro-5-(trifluoromethyl)pyrimidine (0.384 g, 1.77 mmol) in 1 :1 DCE/i-BuOH (10 mL) at 0 °C under a stream of N2 gas. The mixture was stirred for 1 hour at 0 °C and then feri-butyl 4-(4-aminophenyl)piperidine-1 -carboxylate (144) (0.453 g, 1.64 mmol) in 1 :1 DCE/fBuOH (7 mL) was added. A solution of NEt3 (0.251 mL, 1.80 mmol) in 1 : 1 DCE/f-BuOH (8 mL) was next added dropwise at 0 °C. The reaction mixture was vigorously stirred for a further 30 minutes at 0 °C after the final addition and then at room temperature for 24 hours. The solvent was removed in vacuo to afford a brown oily residue which was purified by column chromatography on silica gel (0-20% EtOAc in petroleum benzine 40-60 °C) to yield a pale yellow solid. The solid was suspended in MeOH (10 mL) and water (10 mL). The resulting precipitate was filtered to afford the title compound (145) (0.658 g, 88%) as a white solid; 1H N (400 MHz, afe-DMSO) δ 10.60 (s, 1 H), 8.77 (d, J = 0.5 Hz, 1 H), 7.59 (d, J = 8.5 Hz, 2H), 7.23 (d, J = 8.6 Hz, 2H), 4.13 - 3.98 (m, 2H), 2.80 (bs, 2H), 2.69 - 2.61 (m, 1 H), 1.74 (d, J = 12.4 Hz, 2H), 1.53 - 1.39 (m, 11 H). LCMS Method C: rt 8.81 min; m/z 401 [M-iButyl+2H]÷, 357 [M-Boc+2H]+.
(e) tert-Butyl 4-(4-((4-((2-(2-methoxy-2-oxoethyl)phenyl)ethynyl)-5- (trifluoromethyl)pyrimidin-2-yl)amino)phenyl)piperidine- 1 -carboxylate (146)
A solution of methyl 2-(2-ethynylphenyl)acetate (14) (0.069 g, 0.394 mmol) in dimethylformamide (2 mL) and friethyiamine (0.183 mL, 1.31 mmol) was added to a mixture of ferf-butyi 4-(4-((4-chloro-5-(trifiuoromethyi)pyrimidin-2- yi)amino)phenyl)piperidine-1 -carboxylate (145) (0.150 g, 0.328 mmol), Pd(PPh3)2CI2 (0.023 g, 0.033 mmol), Cu(l)l (0.0090 g, 0.049 mmol) and triphenylphosphine (0,013 g, 0.049 mmol) in dimethylformamide (2 mL). The reaction mixture was heated under microwave irradiation at 120 °C for 15 minutes. The reaction was cooled and the mixture diluted with EtOAc and passed through a plug of celite and washed through with ethyl acetate (50 mL). Water (50 mL) was added and the layers separated. The aqueous layer was extracted with EtOAc (2 x 50 mL). The combined organic extracts were washed with water (50 mL) and brine (50 mL) and dried over Na2S04. After
filtration the solvent was removed under reduced pressure to give a dark brown residue. The residue was purified by column chromatography on silica gel (0-20% EtOAc in cyciohexane) to yield the title compound ( 46) (0. 57 g, 80%) as a brown viscous oil.
(f) tert-Butyl 4-(4-((4-(2-(2-methoxy-2-oxoethyl)phenethyl)-5- (thfluoromethyl)pyrimidin-2-yl)amino)phenyl)piperidine- 1 -carboxyiate (147)
A solution of ferf-butyl 4-(4-((4-((2-(2-methoxy-2-oxoethyl)phenyl)ethynyl)-5- (trifluoromethyl)pyrimidin-2-yl)amino)phenyl)piperidine-1-carboxylate (146) (0.157 g, 0.264 mmol) in D F (15 mL) was added to a solution of 10% Pd/C (95 mg) in DMF (5 mL). The reaction was stirred at room temperature for 24 hours under an atmosphere of hydrogen. The reaction was filtered through a pad of ceiite and washed through with EtOAc (100 mL). The solvent was removed in vacuo to afford a pale yellow oil which was purified by column chromatography on silica gel (0-20% EtOAc in petroleum benzine 40-60 °C) to yield the title compound {147) (0.128 g,
81 %) as a pale yellow viscous oil; 1H NMR (400 MHz, CDCI3) 5 8.53 (d, J = 0.4 Hz, 1 H), 7.59 - 7.54 (m, 2H), 7.39 (s, 1 H), 7,28 -- 7.17 (m, peaks obscured by CDCI3), 4.25 (bs, 2H), 3.75 (s, 2H), 3.68 (s, 3H), 3.17 - 3.04 (m, 4H), 2.81 (t, J= 12.1 Hz, 2H), 2.64 (it, J = 11.8, 3.4 Hz, 1 H), 1.83 (d, J = 13.0 Hz, 2H), 1.67-1.59 (m, 2H), 1.49 (s, 9H). LCMS Method C: rt 7.02 min; m/z 621 [M+IMa]+, 599 [M+H]+, 543 [M-Butyl+2H]+, 499 [M-Boc+2H]+.
(g) Lithium 2~(2~(2~(2~((4~( 1 -(tert-butoxycarbonyl)piperidin-4-yl)phenyl)amino)-5- (trifluoromethyl)pyrimidin-4-yl)ethyl)phenyl)acetate (148)
LiOH.H20 (0.027 g, 0,647 mmol) was added to a solution of ferf-butyl 4-(4-((4-(2-(2- methoxy-2-oxoethyl)phenethyl)-5-(trifluoromethyl)pyrimidin-2- yi)amino)phenyl)piperidine-1-carboxylate (147) (0.128 g, 0.214 mmol) in THF (7 mL), water (1.5 mL) and methanol (1 mL). The resulting mixture was allowed to stir at room temperature for 20 hours. The voiatiles were removed in vacuo and the residue was diluted with EtOAc (50 mL) and sat. aq. NaHC03 (50 mL). The layers were separated and the aqueous layer was extracted with EtOAc (2 x 50 mL), the organic layers were combined, washed with brine (50 mL), dried over Na2S04, filtered and concentrated under reduced pressure to give the title compound (148) (0.130 g) as a pale yellow viscous oil.
(h) teri-Buty! 4-(4-((4-(2-(2-amino-2-oxoethyl)phenethyl)-5-(trifluorom 2-yl)amino)phenyl)piperidine- 1 -carboxylaie (149)
1-Hydroxybenzotriazole (32.8 mg, 0.243 mmol), EDCI (46.6 mg, 0.243 mmoi) and N,A -diisopropylethylamine (84.6 μί, 0.486 mmol) were added to a solution of lithium 2-(2-(2-(2-((4-(1-(ieAi-butoxycarbonyl)piperidin-4-yl)phenyl)amino)-5-
(trifluoromethyl)pyrimidin-4-yl)ethyl)phenyl)acetate (148) (0.130 g, 0.221 mmoi) in dry THF (6 mL) and dry D F (1 mL) under an atmosphere of nitrogen. Ammonium carbonate (84,8 mg, 0.883 mmol) was added in one portion to the stirred reaction mixture after 10 minutes. The reaction was left stirred at room temperature for 18 hours. The volatiles were removed in vacuo and the residual solution was diluted with EtOAc (50 mL), transferred to a separating funnel and washed with saturated NaHC03 (50 mL). The aqueous layer was extracted with EtOAc (2x50 mL). The combined organic layers were washed with water (50 mL) and brine (2x50 mL) and dried over Na2S04. After filtration the solvent was removed in vacuo to afford a pale yellow solid. The crude material was purified by column chromatography on silica gel (0-80% EtOAc in petroleum benzine 40-60 °C) to afford the title compound (149) (90,8 mg, 70%) as a white foamy soiid; ! H NMR (400 MHz, CDCi3) 6 8,53 (s, 1 H), 7.53 (m, 3H), 7.31 - 7.23 (m, peaks obscured by CDCI3), 7.20 (d, J = 8.5 Hz, 2H), 5.37 (s, 1 H), 5.29 (s, 1 H), 4.25 (b s, 2H), 3.72 (s, 2H), 3.15 - 3.03 (m, 4H), 2.80 (t, J = 12.4 Hz, 2H), 2.69 - 2.59 (m, 1 H), 1.83 (d, J = 12.6 Hz, 2H), 1.68 - 1.55 (m, peaks obscured by water peak), 1.49 (s, 9H), LCMS Method C: rt 6,48 min; m/z 606
[M÷Na]+, 584 [M÷H]+, 528 [M-iButyl+2H]+, 484 [M-Boc+2H]+.
(i) 2-(2-(2-(2-((4-(Piperidin-4-yl)phenyl)amino)-5-(trifluorom
yi)ethyi)pheny!)acetamide (11)
Trifluoroacetic acid (0.595 mL, 7.78 mmoi) was added to a solution of feri-butyl 4-(4- ((4-(2-(2-amino-2-oxoethyl)phenethyl)-5-(trifluoromethyi)pyrimidin-2- yi)amino)phenyl)piperidine-1-carboxylate (149) (90.8 mg, 0.156 mmol) in dry DCM (5 mL) under an atmosphere of nitrogen. The reaction was stirred at room temperature for 23 hours. The volatiles were removed in vacuo and the residue partitioned between EtOAc (30 mL) and 2M NaOH (30 mL). The two layers were separated and the aqueous layer was extracted with EtOAc (2x30 mL). The combined organic layers were washed with water (30 mL), brine (30 mL), dried over Na2S04. After filtration the solvent was removed under reduced pressure to give a white solid which was suspended in DCM (2 mL) and cyciohexane (10 mL). The resulting precipitate
was filtered to afford title compound ( 11) (63 mg, 84%) as an off-white solid; 1 H NMR (400 MHz, cfe-D SO) δ 10.13 (s, 1 H), 8.65 (s, 1 H), 7.67-7.64 (m, 2H), 7.44 (s, 1 H), 7.27 - 7.12 (m, 6H), 6.93 (s, 1 H), 3.50 (s, 2H), 3.14 - 3.06 (m, 2H), 3.02 - 2.99 (m, 4H), 2.62 - 2.46 (m, peaks obscured by DMSO), 1.67 (d, J = 11.4 Hz, 2H), 1.49 (qd, J = 12.5, 3.9 Hz, 2H). LC S Method C: rt 4.84 min; m/z 484 [M+Hf.
Example 12: 2-(2-(2-(2-((4-(piperidin-4-yl)phenyl)amino)-5-
(trifluoromethyl)pyrimidm~4~y!)ethyl)phenyl)propanamide (12)

Ϊ55 12
(a) Methyl 2-(2-((trimethylsilyl)ethynyl)phenyl)propanoate (150)
2 M LDA solution (1.24 mL, 2.48 mmol) was added to solution of methyl 2-(2- ((trimethylsilyi)ethynyi)phenyl)acetate (110) (0.306 g, 1.24 mmol) in THF (10 mL) at - 78 °C and the mixture stirred for 30 minutes. Methyl iodide was then added (0.155 mL, 2,48 mmol) and the reaction mixture slowly warmed to room temperature over 1.5 hours. The reaction mixture was then left to stir at room temperature for 18 hours before quenching with a saturated solution of NH4CI (20 mL). EtOAc (20 mL) was then added and the layers separated. The aqueous layer was further extracted with
EtOAc (2 x 20 mL). The solvent was removed in vacuo to give a brown oil which was purified by column chromatography on silica gel (0-5% EtOAc in petroleum benzine 40-60 °C) to afford the title compound (150) (0.297 g, 92%) as a yellow oil; 1H NMR (400 MHz, CDC!3) <5 7.49 - 7.44 (m, 1 H), 7.33 - 7.24 (m, peaks obscured by CDCi3), 7.21-7.17 (m, 1 H), 4.25 (q, J = 7.2 Hz, 1 H), 3.67 (s, 3H), 1.51 (d, J = 7.2 Hz, 3H), 0.26 (s, 9H).
(b) Methyl 2-(2-ethynylphenyl)propanoate (151)
A solution of TBAF (1 M solution in THF; 2.28 mL, 2.28 mmol) was added to a solution of methyl 2-(2-((trimethylsilyl)ethynyl)phenyl)propanoate (150) (0.297 g, 1.14 mmoi) in THF (10 mL) at 0 °C. The reaction was stirred for 50 minutes at 0 °C then concentrated under reduced pressure and the residue taken up in EtOAc (20 mL). The organic solution was washed with saturated NaHC03 (20 mL), water (20 mL) and dried over Na2SC¾. The solvent was removed in vacuo to yield a brown oily residue. The oil was purified using column chromatography on silica gel (0-5% EtOAc in cyclohexane) to afford the title compound (151) (0.192 g, 89%) as a pale yellow oil; 1H NMR (400 MHz, CDC!3) <5 7.53 - 7.48 (m, 1 H), 7.36 - 7.28 (m, 2H), 7.24-7.20 (m, 1 H), 4.31 (q, J = 7.2 Hz, 1 H), 3.67 (s, 3H), 3.28 (s, 1 H), 1.50 (d, J = 7.2 Hz, 3H). LC S Method C: rt 5.92 min; m/z 189 [M+H]+.
(c) tert-Butyl 4-(4-((4-((2-(1 '-methoxy- 1 -oxopropan-2-yl)phenyl)ethynyl)-5- (trifluoromethyl)pyrimidin-2-yl)amino)phenyl)piperidine-1-c^ (152)
A solution of methyl 2-(2-ethynylphenyl)propanoate (151) (0.074 g, 0.39 mmoi) in dimethylformamide (2 mL) and triethyiamine (0.183 mL, 1.31 mmol) was added to a mixture of terf-butyi 4-(4-((4-chloro-5-(trifluoromethyl)pyrimidin-2- yl)amino)phenyl)piperidine-1-carboxylate (145) (0.150 g, 0.328 mmol), Pd(PPh3)2CI2 (0.023 g, 0.033 mmoi), Cul (0.0090 g, 0.049 mmoi) and triphenylphosphine (0.013 g, 0.049 mmol) in dimethylformamide (2 mL). The reaction mixture was heated under microwave irradiation at 120 °C for 15 minutes. The reaction was cooled and the mixture diluted with EtOAc and passed through a plug of Celite washing with ethyl acetate (50 mL). Water (50 mL) was added and the layers separated. The aqueous layer was extracted with EtOAc (2x50 mL). The combined organic extracts were washed with water (50 mL) and brine (50 mL) and dried over Na2S04. After filtration the solvent was removed under reduced pressure to give a dark brown residue. The
residue was purified by column chromatography on silica gel (0-20% EtOAc in cyciohexane) to yield the title compound (152) (0.108 g, 54%) as a brown viscous oi
(d) tert-Butyl 4-(4-((4-(2-( 1 -methoxy- 1-oxopropan-2-yl)phenethyl)-5- (trifiuoromethy!)pyrimidin-2-^i)amino)pheny!)piperidine~ 1 -carboxylate (153)
A solution of ier?-butyi 4-(4-((4-((2-(1-methoxy-1-oxopropan-2-yl)phenyi)ethynyi)-5- (trifluoromethyl)pyrimidin-2-yl)amino)phenyl)piperidine-1 -carboxylate (152) (0.108 g, 0.177 mmol) in EtOAc (5 mL) and D F (10 mL) was added to a suspension of 10% Pd/C (80 mg) in EtOAc (5 mL). The reaction was stirred at room temperature for 24 hours under an atmosphere of hydrogen. The reaction was filtered and the solvent removed in vacuo to yield a brown residue. The residue was redissolved in DMF (15 mL) and added to a suspension of 10% Pd/C (55 mg) in DMF (5 mL). The reaction was stirred at room temperature for 24 hours under an atmosphere of hydrogen. The reaction was filtered through a pad of ceiite washing with EtOAc (100 mL). Removal of the solvent under reduced pressure yielded a brown viscous oil which was purified by column chromatography on silica gel (0-15% EtOAc in petroleum benzine 40-60 °C) to afford the title compound (153) (84.3 mg, 77%) as a pale yellow viscous oil; 1H NMR (400 MHz, CDCI3) δ 8.54 (s, 1 H), 7.59 - 7.54 (m, 2H), 7.38 (s, 1 H), 7.33 - 7.30 (m, 1 H), 7.25 - 7.17 (m, 5H), 4.25 (s, 2H), 4.11 (q, J = 7.1 Hz, 1 H), 3.64 (s, 3H), 3.23 - 3.02 (m, 4H), 2.81 (t, J = 12.0 Hz, 2H), 2.64 (tt, J = 12.0, 3.4 Hz, 1 H), 1.83 (d, J = 12.9 Hz, 2H), 1.67 - 1.59 (m, 2H), 1.50 - 1.48 (d, J = 7.0 Hz, 3H; s, 9H). LCMS Method C: rt 7.15 min; m/z 635 [M+Na]÷, 613 [M+H]+, 557 [M-¾utyl+2H]+.
(e) Lithium 2-(2-(2-(2-((4-( 1 -(tert-butoxycarbonyl)piperidin-4-yl)phenyl)amino)-5- (trifluoromethyl)pyrimidin-4-yl)ethyl)phenyl)propanoate (154)
LiOH.H20 (17.3 mg, 0.413 mmol) was added to a solution of fe/f-butyl 4-(4-((4-(2-(1- methoxy-1-oxopropan-2-yl)phenethyl)-5-(trifluoromethyi)pyrimidin-2- yi)amino)phenyl)piperidine-1-carboxylate (153) (0.084 g, 0.137 mmol) in THF (7 mL), water (1.5 mL) and methanol (1 mL). The resulting mixture was allowed to stir at room temperature for 20 hours. The voiatiles were removed in vacuo and the residue diluted with EtOAc (50 mL) and sat. aq. NaHC03 (50 mL). The layers were separated and the aqueous layer was extracted with EtOAc (1 x 50 mL). The organic layers were combined, washed with brine (50 mL), dried with Na2S04, filtered and concentrated under reduced pressure to give the title compound (154) (86 mg) as a pale yellow viscous oil.
(f) tert-Butyl 4-(4-( (4-(2-( 1 -amino- 1 -oxopropan-2-yi)phenethyl)-5- (trifiuoromethy!)pyrimidin-2-y!)amino)phenyi)piperidine- 1 -carboxyiate (155)
1- Hydroxybenzotriazole (21.1 mg, 0.158 mmol), EDCI (30.0 mg, 0, 158 mmol) and W,A -diisopropylethylamine (54.5 pL, 0.313 mmo!) were added to a soluiion of lithium
2- (2-(2-(2-((4-(1-(fer?-butoxycarbonyl)piperidin-4-yl)phenyl)amino)-5- (trifluoromethyl)pyrimidin-4-yl)ethyl)phenyl)propanoate (154) (0.086 g, 0.142 mmol) in dry THF (6 mL) and dry DMF (1 mL) under an atmosphere of nitrogen. Ammonium carbonate (54.6 mg, 0.569 mmol) was added in one portion to the stirred reaction mixture after 10 minutes. The reaction was left stirred at room temperature for 18 hours. The volatiles were removed in vacuo and the residual solution was diluted with EtOAc (50 mL) then washed with saturated NaHC03 (50 mL). The aqueous layer was extracted with EtOAc (2x50 mL). The combined organic layers were washed with water (50 mL) and brine (50 mL) and dried over Na2S04. After filtration the solvent was removed in vacuo to afford a pale yellow solid. The crude material was purified by column chromatography on silica gel (0-85% EtOAc in petroleum benzine 40-60 °C) to afford the title compound (155) (65.2 mg, 77%) as a white foamy solid; 1H N R (400 MHz, CDCI3) 6 8.54 (s, 1 H), 7.51 (d, J = 8.5 Hz, 2H), 7.47 (s, 1 H), 7.40 - 7.36 (m, 1 H), 7.30 - 7.17 (m, peaks obscured by CDCi3), 5.37 (s, 1 H), 5.20 (s, 1 H), 4.25 (b s, 2H), 4.01 (q, J = 7.1 Hz, 1 H), 3.17 - 3.02 (m, 4H), 2.80 (t, J = 12.3 Hz, 2H), 2.64 (it, J = 12,3, 3.6 Hz, 1 H), 1.83 (d, J = 12.7 Hz, 2H), 1 ,67 - 1.59 (m, peaks obscured by water peak), 1.56 (d, J = 7.2 Hz, 3H), 1.49 (s, 9H). LCMS Method C: rt 6.60 min; m/z 620 [M+Na]+, 598 [M+H]+, 542 [M-iButyi+2Hf, 498 [M-Boc+2Hf. (g) 2-(2-(2-(2-((4-(Piperidin-4-y!)phenyi)aminQ)-5-(trifiuommethy!)pyrimidi
yi)ethyi)pheny!)propanamide ( 12)
Trifluoroacetic acid (0.417 mL, 5.46 mmol) was added to a solution of ferf-buiyi 4-(4- ((4-(2-(1-amino-1-oxopropan-2-yl)phenethyl)-5-(trifluoromethyl)pyrimidin-2- yl)amino)phenyl)piperidine-1 -carboxyiate (155) (65.2 mg, 0.109 mmol) in dry DCM (8 mL) under an atmosphere of nitrogen. The reaction was stirred at room temperature for 23 hours. The volatiles were removed in vacuo and the residue partitioned between EtOAc (20 mL) and 2 M NaOH (20 mL). The layers were separated and the aqueous layer was extracted with EtOAc (2x20 mL). The combined organic layers were washed with water (20 mL), brine (20 mL), dried over Na2S04. After filtration the solvent was removed under reduced pressure to give a pale yellow solid which was
dissolved in EtOAc (2 mL) to which was added cyciohexane (10 mL). The resulting precipitate was collected by filtration to afford the title compound ( 12) (25 mg, 47%) as an off-white solid; H N R (400 MHz, 6~DMSO) δ 10.14 (s, 1 H), 8.67 (s, 1 H), 7.68 - 7.65 (m, 2H), 7.42 - 7.34 (m, 1 H), 7.26 - 7, 12 (m, 6H), 6,87 (s, 1 H), 3.86 (q, J = 7.0 Hz, H), 3.44 - 3.18 (m, peaks obscured by water peak), 3.14 - 2.95 (m, 5H), 2.64 - 2.46 (m, peaks obscured by D SO), 1.68 (d, J = 13.0 Hz, 2H), 1.49 (qd, J = 12.1 , 2.4 Hz, 2H), 1.31 (d, J = 7.0 Hz, 3H). LCMS Method C: rt 4.91 min; m/z 498 [M+H]÷. Example 13: 2-(2-(2-(2-({4-{4-Acetylpiperaz!n-1-yl)pheny!)am!no)-5- (triffuoromethyl) pyrimidin-4-yl)ethy!)phenyl)a

13
2-(2-(2-(2-((4-(Piperazin-1-yl)phenyl)amino)-5-(trifluoromethyl)pyrimidin-4- y!)ethy!)phenyl) acetamide ( 1) (0.020 g, 0. 041 mmoi) was dissolved in dry DCM (2 mL), dry THF (1 mL) and dry DMF (1 mL) then triethylamine (0.012 mL, 0.083 mmoi) followed by acetic anhydride (0.008 mL, 0.083 mmoi) were added. The reaction was then stirred at room temperature for 5 hours, the voiafiles were removed in vacuo and the residue was diluted with EtOAc (15 mL) and sat. aq. NaHC03 (10 mL). The layers were separated and the aqueous layer was extracted with EtOAc (10 mL), the combined organic layers were washed with water (10 mL), brine (10 mL), water (10 mL), brine (10 mL), dried (MgS04), filtered and concentrated in vacuo to give a solid which was taken up in DCM (- 10 mL) and methanol (~ 1 mL) and concentrated in vacuo. The process was repeated with only DCM twice after which the sample was further dried on high-vacuum to give the title compound ( 13) (0.019 g, 87% yield) as an off-white solid; 1H NMR (400 MHz, d^-DMSO) δ 10.01 (s, 1 H), 8.61 (s, 1 H), 7.63 - 7.56 (m, 2H), 7.44 (s, 1 H), 7.26 - 7, 13 (m, 4H), 6,98 - 6.90 (m, 3H), 3.61 - 3.54 (m, 4H), 3.50 (s, 2H), 3.13 - 2.95 (m, 8H), 2.04 (s, 3H). LCMS Method C: rt 5.62 min; m/z 527.2 [M+H]+.
Example 14: IV-IVlethyf-2-(2-(2-(2-((4-(piperazsn-1 -yS)phenyf)amino)-5- (triffuoromethyl) pyrimidin-4-yl)ethy!)phenyl)acetamide (14)


(a) tert-Butyl 4-(4-((4-(2-(2-(methylamino)-2-oxoethyl)phenethyl)-5- (trifluoromethyl)pyrimidin-2-yl)amino)phenyl)pipe 1 -carboxylate (156)
Lithium 2-(2-(2-(2-((4-(4-(tert-butoxycarbonyi)piperazin-1-yl)phenyi)amino}-5- (trifluoromethyl) pyrimidin~4-yl)ethyl)pheny!)acetate (17) (0. 30 g, 0.220 mmol) was dissolved in dry THF (7 mL) and dry DMF (1 mL) under an atmosphere of nitrogen. To the solution were added 1-hydroxybenzotriazole (0.036 g, 0.26 mmol) and EDCI (0.051 g, 0.26 mmol) and A/,A -diisopropylethylamine (0.153 mL, 0.879 mmol) and the reaction mixture was stirred at room temperature for 10 minutes. Methylamine hydrochloride (0.059 g, 0.88 mmol) was added in one portion, and the reaction was stirred at room temperature for 60 hours. The volatiles were removed in vacuo and the residual solution was diluted with EtOAc (100 mL) and sat. aq. NaHC03 (80 mL). The layers were separated and the aqueous layer was extracted with EtOAc (70 mL), the organic layers were combined and washed with water (100 mL), brine (100 mL), dried (MgS04), filtered and concentrated in vacuo to give a pale yellow solid. The crude product was purified by silica gel chromatography (Biotage isoiera, 40 g Si Cartridge, 0-100% EtOAc in petroleum benzine 40-60 °C, then 0-60% methanol in EtOAc) to give the title compound (156) (0.091 g, 69% yield) as a pale yellow solid; 1H N (400 MHz, cfe-DMSO) δ 10.00 (s, 1 H), 8.61 (s, 1 H), 7.93 - 7.86 (m, 1 H), 7.63 - 7.56 (m, 2H), 7.24 - 7.12 (m, 4H), 6.96 - 6.89 (m, 2H), 3.54 - 3.42 (m, 6H), 3.14 - 2.93 (m, 8H), 2.56 (d, J = 4.6 Hz, 3H), 1.42 (s, 9H). LC S Method C: rt 6.27 min; m/z 599.3 [M+H]+.
(b) N-Methyl-2-(2-(2-(2-((4-(piperazin- 1 -yl)phenyl)amino)-5-(trifluoromethyl) pyrimidin- 4-yl)ethyl)phenyl)acetamide (14)
terf-Butyl 4-(4-((4-(2-(2-(methylamino)-2-oxoethyl)phenethyl)-5- (trifluoromethyl)pyrimidin-2-yl)amino)phenyl)piperazine-1-carboxylate (156) (0,087 g, 0.15 mmol) was dissolved in DCM (5 mL) under an atmosphere of nitrogen.
Trifluoroacetic acid (0.222 mL, 2.906 mmol) was added to the solution and the reaction was stirred at room temperature for 6 hours. Volatiles were removed in vacuo, EtOAc (70 mL) and 2 M aq. NaOH (70 mL) were added to the residue and the layers were separated. The aqueous layer was extracted with EtOAc (2 x 70 mL), the combined organics were washed with water (50 mL), brine (50 mL), dried (MgS04), filtered and concentrated in vacuo to give a solid which was taken up in DCM (- 0 mL) and methanol (~ 1 mL) and concentrated in vacuo. The process was repeated with only DCM twice after which the sample was further dried on high-vacuum to give the title compound ( 14) (0.088 g, 94% yield) as a pale yellow solid; 1 H N R (400 MHz, cfe-DMSO) δ 9.97 (s, H), 8.60 (s, H), 7.93 - 7.85 (m, 1 H), 7.60 - 7.53 (m, 2H), 7.23 " 7.12 (m, 4H), 6.91 - 6.86 (m, 2H), 3.50 (s, 2H), 3.14 - 3,06 (m, 2H), 3,02 - 2.93 (m, 6H), 2.87 - 2.80 (m, 4H), 2.56 (d, J = 4.6 Hz, 3H). LCMS Method A: rt 4.37 min; m/z 499.6 [M+Hf.
Example 15: IV-IVlethyf-2-(2-(2-(2-((4-(piperazsn-1 -yS)phenyf)amino)-5-

15
(a) 2-iodo-6-methyibenzoic acid (157)
To a solution of o-toluic acid (2.00 g, 14.7 mmoi) in dry D F (60 mL) under an atmosphere of nitrogen was added A/-iodosuccinimide (3.64 g, 16.2 mmol) followed by Pd(OAc)2 (0.330 g, 1.47 mmoi). The resulting reaction mixture was heated to 100 °C and stirred for 17 hours. Upon cooling to room temperature the reaction was diluted with water (100 mL) and EtOAc (150 mL), the layers were separated and the aqueous layer was extracted with EtOAc (2 x 100 mL), the combined organics were washed with water (100 mL), brine (100 mL), water (100 mL), brine (100 mL), dried (MgS04), filtered and concentrated in vacuo to give the title compound {157) (3.56 g, 92% yield) as a brown oily solid; 1H N R (400 MHz, CDCi3) δ 7.69 (d, J = 7.9 Hz, 1 H), 7.21 (d, J = 7.6 Hz, 1 H), 7.02 (t, J = 7.8 Hz, 1 H), 2.44 (s, 3H). LC S Method C: rt 5.36 min.
(b) Methyl 2-iodo-6-methylbenzoate (158)
A solution of 2-iodo-6-methylbenzoic acid (157) (2.50 g, 9.54 mmoi) in DCM (30 mL) and methanol (8 mL) under an atmosphere of nitrogen was cooled to 0 °C and trimethylsilyldiazomethane (2,0 M in diethyl ether, 9.54 mL, 19.1 mmol) was added dropwise. The reaction was stirred at 0 °C for 45 minutes and then quenched with 2 M aq. HCI (50 mL). DCM (150 mL) was added to the quenched reaction and the layers were separated. The aqueous layer was extracted with DCM (100 mL), the organics were combined and washed with sat. aq. NaHC03 (100 mL), water (100 mL), brine (100 mL), dried (MgS04), filtered and concentrated in vacuo to give a yellow oil. The crude product was purified by silica gel chromatography (Biotage
I solera, 40 g Si Cartridge, 0-20% EtOAc in petroleum benzine 40-60 °C) to give the title compound (158) (2.00 g, 76% yield) as a colourless oil; H NMR (400 MHz, CDCI3) δ 7.65 (ddd, J = 7.9, 1.0, 0.5 Hz, 1 H), 7.19 - 7.15 (m, 1 H), 6.99 (t, J = 7.8 Hz, 1 H), 3.95 (s, 3H), 2.33 (s, 3H). LCMS Method C: rt 6.08 min; m/z 277.0 [M+H]+.
(c) 7-lodoisoindoiin-1 -one (159)
Methyl 2-iodo~6-methy!benzoate (158) (2.00 g, 7.245 mmoi) and NBS (1.418 g, 7.969 mmol) were stirred in chiorobenzene (50 mL) and benzoyl peroxide (75% w/w, 0.234 g, 0.724 mmol) was added. The reaction was stirred at 90 °C for 18 hours, cooled to room temperature, filtered and the precipitate was washed with cyclohexane (4x10 mL). The combined filtrates were evaporated, and the resulting brown oil was diluted with THF (50 mL). Aqueous ammonia solution (20 mL) was added, and the mixture was stirred vigorously for 17 hours. The mixture was diluted with water (20 mL) and the THF was removed in vacuo. DCM (150 mL) was added, the layers were separated and the aqueous layer was extracted with DCM (2x100 mL), the combined organics were washed with brine (100 mL), dried (MgS04) and filtered. Silica gel was added and the voiatiles were removed in vacuo to give the crude material absorbed onto silica gel. The material was purified by silica gel chromatography (Biotage Isolera, 40 g Si cartridge, 0- 00% EtOAc in petroleum benzine 40-60 °C, then 0-20% methanol in EtOAc) to give the title compound (159) (0.757 g, 40% yield) as a beige solid; 1H NMR (400 MHz, CDCI3) δ 7.93 (dd, J = 7.8, 0.7 Hz, 1 H), 7.46 (dd, J = 7.5, 0.8 Hz, 1 H), 7.26 - 7.21 (m, 1 H), 7.10 (br s, 1 H), 4.37 (d, J = 0.6 Hz, 2H). LCMS Method C: rt 5,06 min; m/z 260.0 [M+Hf .
(d) 7-((Trimethylsilyl)ethynyl)isoindolin-1-one (160)
To a nitrogen de-gassed solution of 7-iodoisoindoiin-1-one (159) (0.233 g, 0.899 mmol) in dry DMF (6 mL) were added triethylamine (0.501 mL, 3.598 mmol) followed by triphenylphosphine (0.035 g, 0.14 mmol), irans-dichlorobis(triphenylphosphine) palladium(ll) (0.063 g, 0.090 mmol), Cul (0.026 g, 0.14 mmol) and finally
(trimethyisilyi)acetylene (0.292 mL, 1.63 mmol). The reaction mixture was then heated under microwave irradiation at 100 °C for 30 then 10 minutes. The reaction mixture was concentrated in vacuo, then absorbed onto silica gel and purified by silica gel chromatography (Biotage Isolera, 40 g Si cartridge, 0-46% EtOAc in dichloromethane) to give the title compound (160) (0.120 g, 49% yield) as an off-white solid; 1H IMMR (400 MHz, d^DMSO) δ 8.47 (s, 1 H), 7.60 - 7.51 (m, 2H), 7.49 (dd, J = 6.7, 2.0 Hz, 1 H), 4.32 (s, 2H), 1.03 (t, J = 7.9 Hz, 9H), 0.66 (q, J = 7.9 Hz, 6H). LCMS Method C: rt 6.44 min; m/z 272.2 [M+H]+. (e) 7-Eihynyiisoindoiin- 1-one (161)
To a solution of 7-((trimethyisi!y!)ethyny!)isoindolin-1-one (160) (0.172 g, 0.634 mmol) in dry THF (8 mL) under an atmosphere of nitrogen was added TBAF (1.0 M in THF, 0.697 mL, 0.697 mmol) dropwise at 0 °C. The solution was stirred at this temperature for 1.5 hours and then quenched by the addition of water (2 mL). The reaction mixture was concentrated in vacuo and diluted with DCM (100 mL) and sat. aq.
NaHC03 (70 mL). The layers were separated and the aqueous layer was extracted with DCM (70 mL), the combined organic layers were washed with water (100 mL), brine (100 mL), dried (MgS04), filtered and concentrated in vacuo to give a beige solid. The crude product was purified by silica gel chromatography (Biotage Isolera, 12 g Si Cartridge, 0-80% EtOAc in DCM) to give the title compound (161) (0.076 g,
76% yield) as an off-white solid; NMR (400 MHz, cfe-DMSO) δ 8.59 (s, 1 H), 7.61 - 7.50 (m, 3H), 4.39 (s, 1 H), 4.33 (s, 2H). LCMS Method C: rt 4.56 min, m/z 158.1 [M+H (f) tert-Butyl 4-(4-((4-((3-oxoisoindolin-4-yl)ethynyl)-5-(trifluorom
yi)amino)pheny! )piperazine- 1 -carboxylate (162)
To a nitrogen de-gassed solution of 7-ethynylisoindolin-1-one (161) (0.074 g, 0.47 mmol) and terf-butyl 4-(4-((4-chloro-5-(trifluoromethyl)pyrimidin-2- yl)amino)phenyl)piperazine-1-carboxylate (13) (0.180 g, 0.393 mmol), in dry DMF (7 mL) were added triethylamine (0.219 mL, 1.57 mmol) followed by triphenylphosphine
(0.015 g, 0.059 mmol), irans-dichlorobis(triphenylphosphine) palladium(ll) (0.028 g, 0.039 mmol) and Cul (0.011 g, 0.059 mmol). The reaction mixture was heated under microwave irradiation at 120 °C for 20 minutes and then concentrated to dryness in vacuo and purified by silica gel chromatography (Biotage Isolera, 40 g Si cartridge, 0- 100% EtOAc in petroleum benzine 40-60 °C, then 0-5% methanol in EtOAc) to give the title compound (162) (0.151 g, 86% yield) as an orange gum; 1H NMR (400 MHz, dff-D SO) δ 10.32 (s, 1 H), 8.77 (s, 1 H), 8.72 (s, 1 H), 7.75 - 7.67 (m, 2H), 7.67 - 7.64 (m, 1 H), 7.60 - 7.56 (m, 2H),), 6,95 (d, J = 9.1 Hz, 2H), 4.40 (s, 2H), 3.51 - 3.42 (m, 4H), 3.11 - 3.00 (m, 4H), 1.42 (s, 9H). LCMS Method C: rt 6.23 min, m/z 579.2 [M+H]+.
(g) tert-Butyl 4-(4-((4-(2-(3-oxoisoindolin-4-yl)ethyl)-5-(trifiuorom
yl)amino)phenyl)piperazine- 1 -carboxylate (163)
fert- Butyl 4-(4-((4-((3-oxoisoindolin-4-yl)ethynyl)-5-(trifluoromethyl)pyrimidin-2- yljamino) phenyl) piperazine-1 -carboxylate (162) (0.149 g, 0.258 mmol) was dissolved in dry DMF (6 mL) under an atmosphere of nitrogen, 20% Pearlman's catalyst (0.090 g) in EtOAc (6 mL) was added to the solution and the atmosphere was changed to hydrogen gas (balloon). The reaction was sealed with a balloon and stirred at room temperature for 18 hours at room temperature. The catalyst was removed by filtration through Celite, which was washed with EtOAc (5 x 10 mL). The solvent was removed in vacuo to give a yellow solid which was purified by silica gel chromatography (Biotage isolera, 40 g Si Cartridge, 0-100% EtOAc in petroleum benzine 40-60 °C, then 0-5% methanol in EtOAc) to give the title compound (163) (0.084 g, 56% yield) as an off-white solid; 1H NMR (400 MHz, cfg-DMSO) δ 9.98 (s, 1 H), 8.57 (s, 1 H), 8.48 (s, 1 H), 7.62 - 7.55 (m, 2H), 7.44 (t, J = 7.4 Hz, 1 H), 7,38 (d, J = 7.3 Hz, 1 H), 7.17 (br d, J = 6.0 Hz, 1 H), 6.92 (d, J = 9.0 Hz, 2H), 4.31 (s, 2H), 3.56 (t, J = 7.6 Hz, 2H), 3.51 - 3.41 (m, 4H), 3.12 (t, J = 7.5 Hz, 2H), 3.08 - 2.98 (m, 4H), 1.42 (s, 9H). LCMS Method A: rt 5.86 min; m/z 583.5 [M+H]*. (h) 7-(2-(2-((4-(Piperazin- 1 -yl)phenyl)amino)-5-(trifluoromethyl)pyrimidin-4- yi)eihyi)isoindo!in- 1 -one ( 15}
ferf- Butyl 4-(4-((4-(2-(3-oxoisoindolin-4-yl)ethyl)-5-(trifluoromethyl)pyrimidin-2- yl)amino) phenyl) piperazine- -carboxylate (163) (0,080 g, 0.137 mmol) was dissolved in DCM (5 mL) under an atmosphere of nitrogen. Trifiuoroacetic acid (0.210 mL, 2.75 mmol) was added to the solution and the reaction was stirred at room
temperature for 1 hour and then at 35 °C for another 30 minutes. More trifiuoroacetic acid (0.100 mL) was added and the reaction was further stirred at 35 °C for another 30 minutes. Volatiles were removed in vacuo then EtOAc (70 mL) and 2 M aq. NaOH (70 mL) were added to the residue and the layers were separated. The aqueous layer was extracted with EtOAc (2x70 mL), the combined organics were washed with water (50 mL), brine (50 mL), dried (MgS04), filtered and concentrated in vacuo to give an oily solid which was taken up in DCM (-10 mL) and concentrated in vacuo. The process was repeated twice after which the sample was further dried on high- vacuum to give the title compound ( 15) (0.050 g, 75% yield) as an off-white solid; 1 H NMR (400 MHz, frD SO δ 9.94 (s, 1 H), 8.56 (s, 1 H), 8.48 (s, 1 H), 7.59 - 7.52 (m, 2H), 7.47 - 7.36 (m, 2H), 7.23 - 7.11 (m, 1 H), 6.87 (d, J = 9.0 Hz, 2H), 4.31 (s, 2H), 3.56 (t, J = 7.6 Hz, 2H), 3.11 (t, J = 7.4 Hz, 2H), 3.01 - 2.95 (m, 4H), 2.86 - 2.79 (m, 4H). LCMS Method A: rt 4.4Q min; m/z 483.8 [M÷H]+. Example 16: 2-(2-(2-f2-f{4-{1 -methylpiperidsn-4-yl)phenyt)amino)-5- (trifluoromethyl)pyrimidm~4~y!)ethyl)phenyl)acetamide (16)

11 16
Formaldehyde (37 % in H20; 15.6 L, 0.210 mmol) was added to a suspension of 2- (2-(2-(2-((4-(piperidin-4-yl)phenyl)amino)-5-(trifluoromethyl)pyrimidin-4- yl)ethyl)phenyl)acetamide (11) (25 mg, 0.053 mmol) in anhydrous methanol (5 mL) under an atmosphere of nitrogen. Sodium triacetoxyborohydride (0.111 g, 0.525 mmol) was then added in one portion to the reaction mixture. The reaction was stirred at room temperature for 1.5 hours. The volatiles were removed in vacuo and the residue was diluted with EtOAc (25 mL) and saturated aq. NaHC03 (25 mL). The layers were separated and the aqueous layer was extracted with EtOAc (2 x 25 mL), the combined organic layers were washed with water (25 mL), brine (25 mL) and dried over Na2S04. The solvent was removed under reduced pressure yield a white solid. The solid was suspended in DCM (2 mL) and cyclohexane (10 mL) then filtered to afford the title compound ( 16) (19 mg, 73%) as a white solid; 1H NMR (400 MHz, de-DMSO) δ 10.14 (s, 1 H), 8.65 (s, 1 H), 7.70 - 7.61 (m, 2H), 7.44 (s, 1 H), 7,27 - 7.12 (m, 6H), 6.93 (s, 1 H), 3.50 (s, 2H), 3.14 - 2.97 (m, 4H), 2.88 (d, J = 10.6 Hz,
2H), 2.46 - 2.36 (m, 1 H), 2.22 (s, 3H), 2.07 - 1.93 (m, 2H), 1.78 - 1.58 (m, 4H). LC S Method C: rt 4.86 min; m/z 498 [M+H]+.
Example 17: 2-{2-{2-{2-{{3-{4-rnethy!piperazsn~1 ~yl)pheny!)amino)-5- ftnf!uoromethyS)pyrimidin-4-yl)ethy!)pheny!)acetamide (17)

17
(a) Methyl 2-(2-((2-(methylthio)-5-(trifluoromethyl)pyrimidin-4- yl)ethynyl)phenyl)acetate (164)
4-lodo-2-(methylthio)-5-(trifluoroiT!ethyl)pyriiT!idine (114) (2.00 g, 6.24 mmol), PdCI2(PPh3)2 (438 mg, 625 μπιοΙ), Cul (119 mg, 625 μπιοΙ) and triphenylphosphine (164 mg, 625 mol) were placed into an oven dried microwave reaction vial under nitrogen. Methyl 2-(2-ethynylphenyi)acetate (14) (1.31 g, 7.49 mmol), THF (20 mL) and TEA (10 mL) were added and the resulting mixture was stirred at 100 °C under microwave irradiation for 10 minutes. The volatiles were evaporated under reduced pressure then the residue was adsorbed onto silica from DCM, The pre-adsorbed material was chromatographed on silica gel (0-25% ethyl acetate/ petroleum benzine 40-60 °C) to give the title compound (164) (1.571 g, 69%) as an orange solid; 1H
NMR (400 MHz, CDC!3) δ 8.71 (d, J = 0.8 Hz, 1 H), 7.68 (dd, J = 7.7, 1.1 Hz, 1 H), 7.50 - 7.29 (m, 3H), 3.93 (s, 2H), 3.71 (d, J = 3.4 Hz, 3H), 2.62 (d, J = 3.4 Hz, 3H).
(b) Methyl 2-(2-((2-(methylsulfonyl ) -5- ( trifluoromethyl)pyrimidin - - yl)ethynyl)phenyl)acetate (I65)
Methyl 2-(2-((2-(methylthio)-5-(trifluoromethyl)pyrimidin-4-yl)ethynyl)phenyl)acetate {164) (3.14 g, 8.57 mmo!) was dissolved in DCM (150 mL) and the resulting solution cooled to 0 °C. mCPBA (70%; 4.65 g, 18.9 mmo!) vv'as added then the reaction mixture was allowed to warm to room temperature, at which, stirring was continued overnight. The crude mixture was washed with 10% NaHC03 (200 mL) and the layers were separated. The organics were dried (MgS04) then evaporated under reduced pressure to give a light yellow solid. The solid was adsorbed onto silica then chromatographed on silica gel (0-50% ethyl acetate/ petroleum benzine 40-60 °C) to give the title compound (165) (2.876 g, 84%) as a yellow solid; 1H NMR (400 MHz, CDCI3) δ 9.13 (d, J = 0.7 Hz, 1 H), 7.73 (dd, J = 7.6, 0.9 Hz, 1 H), 7.54 - 7.46 (m, 1 H), 7.44 - 7.32 (m, 2H), 3.94 (s, 2H), 3.77 - 3,67 (m, 3H), 3,43 (s, 3H). LCMS Method C: rt 5,90 min; m/z 421.0 (M+Na), 399.1 (M+1), 367,0 (M-OMe), 339, 1 (M-COOMe).
(c) Methyl 2-(2-(2-(2-(methylsulfonyl)-5-(trifluoromethyl)pyrimidin-4^
yl)ethyl)phenyl)acetate (166)
Methyl 2-(2-((2-(methyisuifonyi)-5-(trifiuoromethyl)pyrimidin-4- yi)ethynyi)phenyl)acetate (165) (1.50 g, 3.76 mmol) was taken up in DMF (30 mL) then 10% Pd/C (750 mg) was added. The resulting suspension was stirred under H2 (1 atm) for 16 hours at room temperature. The crude reaction mixture was filtered through Celite, washing with MeOH. The filtrate was evaporated under reduced pressure to give a yellow liquid which was adsorbing onto silica. The silica adsorbed material was chromatographed on silica gel (0-100% ethyl acetate/ petroleum benzine 40-60 °C) to give the title compound (166) (1.38 g, 91 %) as a yellow solid; Ή NMR (400 MHz, CDCI3) δ 9,07 (d, J = 0.7 Hz, 1 H), 7.30 - 7.12 (m, 4H), 3.72 (s, 2H), 3.68 (s, 3H), 3.41 - 3.35 (m, 2H), 3.35 (s, 3H), 3.20 (dd, J = 9.6, 6.3 Hz, 2H). LCMS Method C: rt 5.92 min; m/z 425.1 (M+Na), 403.1 (M+1), 401.1 (M-1), 371.1 (M-OMe), 343.1 (M-COOMe).
(d) Methyl 2-(2-(2-(2-((3-(4-methylpiperazin- 1 -yl)phenyl)amino)-5- (trifluoromethyl)pyrimidin-4-yl)ethyl)phenyl)acetate (167)
3-(4-Methylpiperizin-1-nyi)aniline (36,0 mg, 186 μπιοΙ) was dissolved in
trifluoroethanol (1 mL), then methyl 2-(2-(2-(2-(methylsulfonyl)-5- (trifluoromethyi)pyrimidin-4-yl)ethyl)phenyl)acetate (166) (50 mg, 124 μηΊθΙ) was added followed by trifluoroacetic acid (48 μί). The resulting mixture was stirred at 100 °C under microwave irradiation for 10 minutes. The resulting mixture was adsorbed onto silica then chromatographed on silica gel (0-10% MeOH/DC ) to give the title compound (167) as a yellow liquid (69 mg). LC S Method C: rt 5.10 min; m/z 5 4.3 ( +1). This procedure was repeated and the reaction products combined for progression into the following synthetic step.
(e) 2-(2-(2-(2-((3-(4-Methylpiperazin-1-yl)phenyl)amino)-5-(tr^
4-yl)ethyl)phenyl)acetic acid (168)
Methyl 2-(2-(2-(2-((3-(4-methylpiperazin-1-yl)phenyl)amino)-5-
(trifluoromethyl)pyrimidin-4-yl)ethyl)phenyl)acetate (167) (150 mg, 292 μρηο!) was dissolved in a THF (10 mL), then LiOH.H2G (36,7 mg, 876 μΓηοΙ) was added. Water (2 mL) then MeOH (1 mL) was added. The resulting mixture was stirred at room temperature for 16 hours then the voiatiies were removed by evaporation under reduced pressure. The residue was dissolved in MeOH (3 mL) then acidified with concentrated HCi to pH 2-3. The voiatiies were evaporated under reduced pressure to give an orange residue. The residue was chromatographed on silica gel (0-20% MeOH/DCM) to give the title compound (168) (120 mg, 82%) as a yellow crystalline solid; 1H NMR (400 MHz, d6-DMSO) δ 11.04 (bs, 1 H), 10,12 (s, 1 H), 8.68 (s, 1 H), 7.44 (m, 1 H), 7.35 (m, 1 H), 7.24-7,18 (m, 5H), 6,72 (m, 1 H), 3,77-3,74 (m, partially obscured by residual water signal), 3.44 (m, 3H), 3.17-3.03 (m, 8H), 2.78 (s, 3H). LCMS Method C: rt 4.92 min; m/z 500.3 (M+1 ), 498.2 (M-1), 454.3 (M-COOH).
(f) 2-(2-(2-(2-((3-(4-Methylpiperazin-1-yl)phenyl)amino)-5-(tri^
yl)ethyl)phenyl)acetamide (17)
2-(2-(2-(2-((3-(4-Methylpiperazin-1-yi)phenyl)amino)-5-(trifluoromethyi)pyrimidin-4- yl)ethyl)phenyl)acetic acid (168) (75.0 mg, 169 μιηοΙ) was dissolved in DMF (1 mL) then HATU (129 mg, 339 μπηοΙ), DIPEA (57 uL, 339 μιηοΙ) and ammonium chloride (181 mg, 3.39 mmol) were added. The resulting mixture was stirred at room temperature overnight then the voiatiies were evaporated under reduced pressure.
The residue was diluted with ethyl acetate and washed with 10% NaHC03, the layers were separated and the organic layer was dried (MgSCv) then evaporated under reduced pressure to give a cream solid. The cream solid was adsorbed onto silica and chromatographed on silica gel (0-10% eOH/DCM) to give the title compound ( 17) (23.3 mg, 28%) as a cream solid; 1 H NMR (400 M Hz, CD3CN) δ 8.57 (s, 1 H),
8.34 (s, 1 H), 7.58 (s, 1 H), 7.30 - 7.16 (m, 5H), 7.09 (d, J = 8.0 Hz, 1 H), 6.71 (dd, J = 8.2, 1 .9 Hz, 1 H), 3.61 (s, 2H), 3.27 (t, J = 4.9 Hz, 4H), 3.19 - 3.03 (m, 4H), 2.92 (bs, 4H), 2.91 (s, 3H). LCMS Method C: rt 4.43 min; m/z 499.7. Example 18: W-IVlethyf-2-(2-(2-(2-((3-(4-methy!p^eraz!n-1 -y!}phenyS)arnsno)-5- (triff u

S68 18
2-(2-(2-(2-((3-(4- ethylp!perazin-1-yi)phenyl)amino)-5-(trifluoromethyi)pyrimidin-4- yi)ethyi)phenyl)acetic acid (168) (75.0 mg, 169 μηιοΙ) was dissolved in DMF (1 mL) then HATU (129 mg, 339 μηιοΙ), DI PEA (57 μ[_, 339 μηιοΙ) and methylamine (8.0 M in ethanol; 200 μΙ_) were added. The resulting mixture was stirred at room temperature overnight then the volatiles were evaporated under reduced pressure. The residue was diluted with ethyl acetate and washed with 10% NaHCQ3, the layers were separated and the organic layer was dried (MgS04) then evaporated under reduced pressure to give a cream solid. The cream solid was adsorbed onto silica and chromatographed on silica gel (0-10% MeOH/DCM) to give the title compound ( 18) (9.0 mg, 10%) as a solid; 1 H NMR (400 MHz, CD3CIM) δ 8.56 (d, J = 0.6 Hz, 1 H), 8.36 - 8.28 (m, 1 H), 7.39 (s, 1 H), 7.20 (ddd, J = 5.6, 4.4, 1 .9 Hz, 6H), 6.71 - 6,65 (m, 1 H), 6.33 - 6.23 (m, 1 H), 3.56 (s, 2H), 3.22 - 3.15 (m, 4H), 3.10 (s, 2H), 3.07 (s, 2H), 2.60 (d, J = 4.7 Hz, 3H), 2.49 - 2.43 (m, 4H). LCMS Method C: rt 4.86 min; m/z 513.3.
Example 19: 2-!Vlethyf-5-(2-(2-((4-(p!peraz!n-1-y!)phenyl)amino)-5- (triff uorometh l}pyrimidsn-4-y!)ethyl)benzamide (19}

Ϊ73 19
(a) 5-Bromo-2-methylbenzamide (169)
To a mixture of 5-bromo-2-methylbenzoic acid (0,538 g, 2,50 mmo!) and HATU
(1.289 g, 3.390 mmoi) in DMF (8 mL) was added D!PEA (0.800 mL, 4.59 mmoi). The mixture was stirred for 10 minutes before addition of NH4OH (0.50 mL) and then left stirring for 16 hours at room temperature. The mixture was poured in to water and cooled at 0 °C for 20 minutes before collecting the resulting precipitate via vacuum filtration to give the title compound (169) (0.292 g, 55%); H N R (400 MHz, CDCI3) δ 7.58 (d, J = 2.1 Hz, 1 H), 7.45 (dd, J = 8.2, 2.1 Hz, 1 H), 7.12 (d, J = 8.2 Hz, 1 H), 5.68 (bs, 2H), 2.44 (s, 3H). LCMS Method C: rt 4.89 min; m/z 214, 216 [M+H]+.
(b) 2-Methyl-5-( ( trimethylsilyl)ethynyl)benzamide (170)
To a mixture of 5-bromo-2-methylbenzamide (169) (0.292 g, 1.36 mmoi),
PdCI2(PPh3)2 (0.049 g, 0.070 mmoi), triphenylphosphine (0.054 g, 0.21 mmoi) and copper iodide (0.036 g, 0, 19 mmoi) in DMF (3 mL) was added triethyiamine (0.570 mL, 4.09mmol) and trimethylsiiy!acetyiene (0,210 mL, 1 ,49 mmoi) and the resulting mixture heated under microwave irradiation at 120 °C for 25 minutes. The resulting mixture was concentrated under reduced pressure and purified using silica gel column chromatography (0-20% EtOAc/ petroleum benzine 40-60 °C) to give the title compound (170) (0, 190 g, 60%); 1H NMR (400 MHz, d6-DMSO) δ 7.80 (s, 1 H), 7,40 (m, 3H), 7.24 (d, J = 7.9 Hz, 1 H), 2.36 (s, 3H), 0.22 (s, 9H).
(c) 5-Ethynyl-2-methylbenzamide (171)
To a solution of 2-methyl-5-((trimethylsilyl)ethynyl)benzamide (170) (0.190 g, 0.819 mmol) in THF (4 mL) at 0 °C was added 1.0 M solution TBAF in THF (0.5 mL). The mixture was then stirred under N2 at 0 °C for 10 minutes and then at room
temperature for 3 hours. The resulting mixture was concentrated under reduced pressure then diluted with water. The resulting precipitate was collected by vacuum filtration to give the title compound (171) (0.087 g, 67%); 1H NMR (400 MHz, 6~ DMSO) δ 7.80 (s, 1 H), 7.41 (m, 3H), 7.25 (d, J = 7.7 Hz, 1 H), 4.17 (s, 1 H), 2.36 (s, 3H).
(d) tert-Butyl 4-(4-((4-((3-carbamoyl-4-methylphenyl)ethynyl)-5-
(trifluoromethyl)pyrimidin-2-yl)amino)phenyl)piperazine- 1 -carboxylate (172)
To a mixture of 5-ethynyl-2-methylbenzamide (171) (0.041 g, 0.25 mmol), ferf-butyl 4-
(4-((4-chioro-5-(trifluoromethyl)pyrimidin-2-yl)amino)phenyi)piperazine-1-carboxyiate (13) (0.105 g, 0.230 mmol), PdCI2(PPh3)2 (0.011 g, 0.016 mmol), triphenyiphosphine (0.010 g, 0.038 mmo!) and copper(l) iodide (0.010 g, 0.53 mmo!) in DMF (2 mL) was added triethylamine (0.091 mL, 0.65 mmol). The mixture was then heated under microwave irradiation at 120 °C for 30 minutes. The resulting mixture was concentrated under reduced pressure then purified using silica gel column chromatography (0-100% EtOAc/petro!eum benzine 40-60 °C) to give title compound (172) (0.092 g, 69%); !H NMR (400 MHz, afe-DMSO) δ 10.24 (s, 1 H), 8.75 (s, 1 H), 8.31 (s, 1 H), 7.90 (s, 1 H), 7.58 (m, 4H), 7.39 (m, 1 H), 6.96 (d, J = 9.0 Hz, 2H), 3.46 (dd, J = 6.3, 3.3 Hz, 4H), 3.06 (m, 4H), 2.42 (s, 3H), 1.42 (s, 9H). LCMS Method C: rt 6.22 min; m/z 581.2 [M+H]+.
(e) tert-Butyl 4-(4-((4-(3-carbamoyl-4-methylphenethyl)-5-(trifluo
yl)amino)phenyl)piperazine- 1 -carboxylate ( 173)
A mixture of feri-buty! 4-(4-((4-((3-carbamoyl-4-methylphenyl)ethynyl)-5- (trifluoromethyl)pyrimidin-2-yl)amino)phenyl)piperazine-1 -carboxylate (172) (0,092 g, 0.16 mmol) and 10% Pd/C (0.116 g) in DMF (5 mL) was stirred over night under a H2 atmosphere. The mixture was filtered through Celite then concentrated under reduced pressure. The residue was then purified using silica gel column
chromatography (0-100% EtOAc/ petroleum benzine 40-60 °C) and the product triturated with methanol. The resulting precipitate was collected by vacuum filtration to give the title compound (173) (29.5 mg, 32%); 1H NMR (400 MHz, cfe-D SO) δ
10.00 (s, 1 H), 8.61 (s, 1 H), 7.66 (s, 1 H), 7.55 (d, J = 9.0 Hz, 2H), 7.33 (s, 1 H), 7.25 (s, 1 H), 7.16 (m, 2H), 6.94 (d, J = 9.1 Hz, 2H), 3.46 (m, 4H), 3.04 (m, 8H), 2.32 (s, 3H), 1.42 (s, 9H). LC S Method C: rt 8.18 min; m/z 585.3 [ +Hf. if) 2-Methyl-5-(2-(2-((4-(piperazin- 1 -yl)phenyl)amino)-5-(trifluoromethyl)pyrimidin-4- yi)eihyi)benzamide (19)
To ferf-butyl 4-(4-((4-(3-carbamoyl-4-methylphenethyl)-5-(trifluoromethyl)pyrimidin-2- yl)amino)phenyl)piperazine-1-carboxylate (173) (0.029 g, 0.054 mmol) in DCM (2 mL) was added TFA (0.100 mL, 1.36 mmo!) and the mixture stirred overnight (16 hours) at room temperature. The resulting mixture was then concentrated under reduced pressure and the residue taken up in EtOAc (10 mL). The resulting organic suspension was washed with 10% aqueous NaOH (10 mL) and the aqueous layer extracted with EtOAc (2x10 mL). The combined organics were washed with water (15 mL) then concentrated under reduced pressure. The residue was taken up in DCM (4 mL) and TFA (0.200 mL) and stirred overnight (16 hours). The resulting mixture was concentrated under reduced pressure then diluted with EtOAc (15 mL). The resulting solution was washed with 10% aqueous NaOH (15 mL) then the aqueous layer was extracted with EtOAc (15 mL). The combined organic extracts were washed with water (20 mL) then dried using a phase separation cartridge before concentrating under reduced pressure to give the title compound ( 19) (5.1 mg, 21 %); 1 H NMR (400 MHz, de-DMSO) δ 9.96 (s, H), 8,60 (s, 1 H), 7.67 (s, 1 H), 7.53 (d, J = 9.0 Hz, 2H), 7.33 (s, 1 H), 7.25 (s, 1 H), 7.16 (m, 2H), 6.90 (d, J = 9.1 Hz, 2H), 3.01 (m, 8H), 2.83 (m, 4H), 2.32 (s, 3H). LCMS Method C: rt 4.76 min; m/z 485.1 [M+H]+.
Example 20: 2-(2-(2-{2-{{4-{4-Am!nop!peridin-1-yS)phenyl)am!no)-5- (triffuoromethyl)pyrimidsn-4-yl)ethyf)pheny!}acetamid
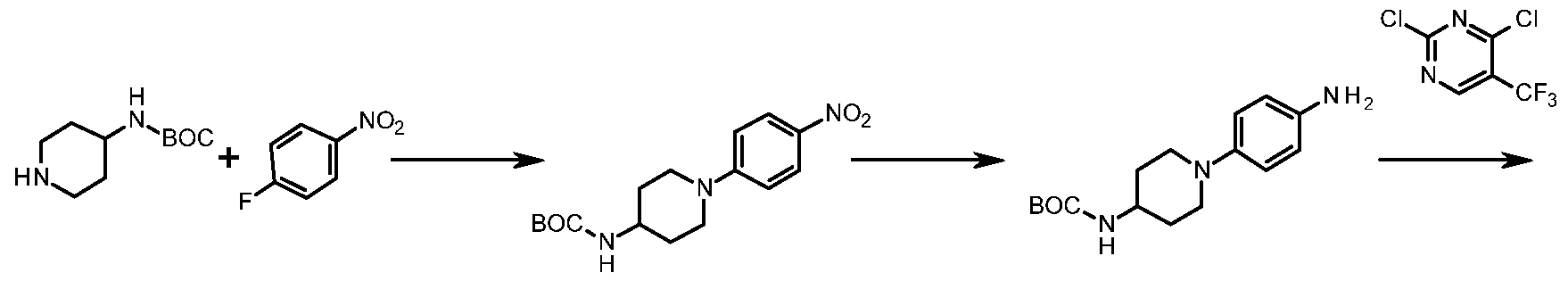
!74 I75

(a) tert-Butyl (1-(4-nitrophenyl)piperidin-4-yl)carbamate (174)
terf-Butyl piperidin-4-ylcarbamate (1.200 g, 5.992 mmol) and 4-fluoronitrobenzene
(0.705 g, 4.99 mmol) were placed in a 30 mL microwave vial then aceionitriie (20 mL) followed by diisopropy!ethy!amine (1.778 mL, 9.986 mmol) were added. The reaction was heated under microwave irradiation at 150 °C for 15 minutes. The reaction mixture was diluted with EtOAc (200 mL) and 2 M aq. HCI (150 mL), the layers were separated and the aqueous layer was extracted with EtOAc (2 * 100 mL), the organics were combined and washed with brine (150 mL), dried (MgS04), filtered and concentrated in vacuo to give the title compound (174) (1.040 g, 65%) as a yellow solid; 1H N R (400 MHz, cfe-DMSO) δ 8.06 - 7.99 (m, 2H), 7.03 - 6.97 (m, 2H), 6.88 (d, J = 7.5 Hz, 1 H), 4.01 - 3.93 (m, 2H), 3.62 - 3.47 (m, 1 H), 3.13 - 3.02 (m, 2H), 1.85 - 1.74 (m, 2H), 1.44 - 1.30 (m, 11 H).
(b) tert- Butyl (1~(4~aminophenyi)piperidin~4~yi)carbamate (175)
?e/i-Butyl (1-(4-nitrophenyi)piperidin-4-yi)carbamate (174) (1.038 g, 3.230 mmol) was dissolved in dry D F (15 mL), EiOAc (15 mL) and absolute EtOH (15 mL) under an atmosphere of nitrogen. 10% Pd/C (0.200 g) in EtOAc (5 mL) was added to the solution and the atmosphere was changed to hydrogen gas (balloon). The reaction was sealed with balloon and stirred at room temperature for 18 hours. The catalyst was removed by filtration through celite, which was washed with EtOAc (5 x10 mL). The solvent was removed in vacuo to give a pink solid which was purified by silica gel chromatography using a gradient of 0-80% ethyl acetate in petroleum benzine 40- 60 °C to give the title compound (175) (0.730 g, 78%) as a purple-brown solid;
1H IMMR (400 MHz, CDCI3) δ 6.81 (d, J = 8.6 Hz, 2H), 6.64 (d, J = 8.7 Hz, 2H), 4,55 - 4.41 (m, 1 H), 3.64 - 3.32 (m, 5H), 2.72 (t, J = 11.0 Hz, 2H), 2.08 - 1.98 (m, 2H), 1.56 (ddd, J = 23.7, 11.3, 3.9 Hz, 2H), 1.45 (s, 9H). ). LC S Method C: rt 0.38 min; m/z 292.0 [l 3÷H]+
(c) tert-Butyi ( 1 -(4-((4-chloro-5-(trifluoromethyl)pyrimidin-2-yl)amino)pheny
4-yl)carbamate (176)
2,4-Dichloro-5-(trifluoromethyi)pyrimidine (0.551 g, 2.54 mmol) was stirred in a 1 :1 t- BuOH: 1 ,2-dichloroethane mixture (30 mL) at 0 °C. A 1.0 M ZnCI2 solution in diethyl ether (2.903 mL, 2.903 mmol) was added cautiously over 0 minutes, after addition the reaction was left stirring at 0 °C for 30 minutes. A solution of fe/f-butyi (1~(4~ aminophenyl)piperidin-4-yl)carbamate (175) (0.705 g, 2.42 mmol) in 1 : 1 f-BuOH:1 ,2- dichloroethane (15 mL) was added drop-wise over 15 minutes at 0 °C, a solution of NEt3 (0.405 mL, 2.903 mmol) in 1 : 1 i-BuOH: 1 ,2-dichloroethane (15 mL) was added drop-wise over 15 minutes and the reaction was allowed to warm to room
temperature and was stirred for 8 hours. The organic solvents were evaporated in vacuo and the crude oily solid was suspended in water (200 mL), the suspension was sonicated for 30 minutes and the product was separated by filtration, the solid was washed with water (10x20 mL) and dried under a high vacuum. The material was further purified by silica gel chromatography using a gradient of 0-50% ethyl acetate in petroleum benzine 40-60 °C to give the title compound (176) (0.730 g, 64%) as a yellow solid; 1H NMR (400 MHz, dff-DMSO) δ 10.41 (s, 1 H), 8.71 (s, 1 H), 7.46 (d, J = 7.7 Hz, 2H), 6.92 (d, J = 8.9 Hz, 2H), 6,84 (d, J = 7.1 Hz, 1 H), 3.60 (d, J = 12.4 Hz, 2H), 2.69 (t, J = 10.9 Hz, 2H), 1.78 (d, J = 10.9 Hz, 2H), 1.54 - 1.34 (m, 11 H). LCMS Method C: rt 6.36 min; m/z 474.1 [M+H]+.
(d) Methyl 2-(2-((2-((4-(4-((tert-butoxycarbonyl)amino)piperidin- (trifluoromethyl)pyrimidin-4-yl)ethynyl)phenyl)acetate (177)
A suspension of fe/f-butyl (1-(4-((4-chloro-5-(trifluoromethyl)pyrimidin-2- yl)amino)phenyl)piperidin-4-yl)carbamate (176) (550 mg, 1.17 mmol), Cul (15 mg,
0.12 mmol), triphenyiphosphine (31 mg, 0.117 mmol) and triethyiamine (487 μΐ_, 3.50 mmol) in DMF (6 mL) was sonicated for 5 minutes in a 10 mL microwave vial, to this PdCi2(PPh3)2 (54.0 mg, 0.077 mmol) and methyl 2-(2-ethynylphenyl)acetate (14) (305 mg, 1.75 mmol) were added and the reaction heated to 120°C for 20 minutes under microwave irradiation. Upon cooling the reaction mixture was chromatographed on silica gel using gradient eiution (0-100% ethyl acetate in petroleum benzine 40-60 °C) to yield a mixture of the title compound (177) and homo-coupled acetylene which was used without further purification. (e) Methyl 2-(2-(2-(2-((4-(4-((tert-butoxycarbonyl)amino)piperidin- 1 -yl)phenyl)amino)- 5- ( trifluoromethyl)pyrimidin -4 -yl)ethyl)phenyl)acetate (178)
To a solution of crude methyl 2-(2-((2-((4-(4-((ie t-butoxycarbonyl)amino)piperidin-1- yl)phenyl)amino)-5-(frifluoromefhyl)pyrimjdin-4-yi)ethynyl)phenyl)acetate (177) in DMF (10 mL) and triethyiamine (1 mL) was added 20% Pd(OH)2 (0.92 g) and the resulting suspension was stirred at room temperature overnight under an atmosphere of hydrogen. The reaction mixture was filtered through celite and the filter cake washed with EtOAc (3*75 mL). The combined filtrates were evaporated to dryness to give a brown solid, which was suspended in eOH (25 mL) and then sonicated. The resulting suspension was filtered to give the title compound (178) (2 1 mg, 29%) as a tan solid; 1H N R (400 MHz, cfe-DMSO) δ 9.97 (s, 1 H), 8.60 (s, 1 H), 7.55 (d, J = 9,0 Hz, 2H), 7.29 - 7.15 (m, 4H), 6.96 - 6.78 (m, 3H), 3.76 (s, 2H), 3.64 - 3.49 (m, 5H), 3.38 (s, 2H), 3.11 - 3.00 (m, 2H), 3.00 - 2.91 (m, 2H), 2.68 (t, J = 12.0 Hz, 2H), 1.80 (d, J = 10.7 Hz, 2H), 1.48 (ddd, J = 15.0, 12.2, 3.5 Hz, 2H), 1.39 (s, 9H). LCMS Method C: rt 5.87 min; m/z 614 [M+H]+
( f) tert-Butyl ( 1 - (4- ((4 - (2- (2-amino-2-oxoethyl)phenethyl) -5-(trifluoromethyl)pyrimidin - 2-yl)amino)phenyl)piperidin-4-yl)carbamate (179)
Lithium hydroxide mono hydrate (43.0 mg, 1.03 mmo!) was added to a suspension of methyl 2-(2-(2-(2-((4-(4-((ierLbutoxycarbonyl)amino)piperidin-1-yl)phenyl)amino)-5- (trifluoromethyi)pyrimidin-4-yl)ethyl)pheny!)acetate (178) (211 mg, 0.344 mmol) in THF (10 mL), MeOH (1.0 mL) and water (1.5 mL) and the resulting mixture was stirred at room temperature for 6 hours. The organics were removed in vacuo then 2 M aqueous NaOH solution (100 mL) was added. The resulting solution was extracted with EtOAc (2x 100 mL), then the combined organic extracts were dried ( gS04) and the solvent removed in vacuo to yield a white solid. The solid was dissolved in dry THF (10 mL) and dry DMF (2 mL) under an atmosphere of nitrogen. To this solution were added 1-hydroxybenzotriazole (72 mg, 0.53 mmol), EDCi (101 g, 0.529 mmol) and /V,A-diisopropylethylamine (246 L, 1.41 mmol) and the resulting mixture was stirred at room temperature for 10 minutes. Ammonium carbonate (203 mg, 2.12 mmol) was added in one portion and the reaction was stirred at room temperature for 3 days. The volatiies were removed in vacuo and the residue was taken up in EtOAc (100 mL) and saturated aqueous NaHC03 (100 mL), The layers were separated and the aqueous layer was extracted with EtOAc (70 mL). The combined organic layers were washed with water (2x 00 mL), brine (50 mL), dried (MgS04) and concentrated in vacuo to give a yellow solid which was dissolved in a small amount of acetone and precipitated with petroleum benzine 40-60 °C to yield the title compound (179) ( 38 mg, 65%) as a yellow solid; 1H N R (400 MHz, cf6-DMSO) δ 9.97 (s, H), 8.60 (s, 1 H), 7.56 (d, J = 8.9 Hz, 2H), 7.44 (s, 1 H), 7.27 - 7.12 (m, 4H), 6.96 - 6.81 (m, 4H), 3.58 (d, J = 12.7 Hz, 2H), 3.50 (s, 2H), 3.15 - 2.93 (m, J = 15.2, 6.3 Hz, 4H), 2.67 (t, J = 12.0 Hz, 2H), 1.79 (d, J = 11.4 Hz, 2H), 1 ,55 - 1.42 (m, 2H), 1.39 (s, 9H). LCMS Method C: rt 5.30 min; m/z 599 [M+H]+.
(g 2-(2-(2-(2-((4-(4-Aminopiperidin-1-yi)phenyi)amino)-5-(trif ^
yl)ethyl)phenyl)acetamide (20)
terf-Butyl (1-(4-((4-(2-(2-amino-2-oxoethyl)phenethyl)-5-(trifluoromethyl)pyrimidin-2- yi)amino)phenyl)piperidin-4-yl)carbamate (179) (138 mg, 0.231 mmol) was dissolved in dry dichioromethane (10 mL) under an atmosphere of nitrogen. Trifluoroacetic acid (1.0 mL, 13 mmol) was added to the solution and the reaction was stirred at room temperature overnight. The volatiies were removed in vacuo, EtOAc (50 mL) and sat. aq. NaHC03 (50 mL) were added to the residue and the layers were separated.
The aqueous layer was extracted with EtOAc (50 mL), the combined organics were washed with water (50 mL), brine (50 mL), dried (MgSCv) and concentrated in vacuo to give a yellow solid which was taken up in acetone (~ 2 mL) and precipitated with petroleum benzine 40-60 °C to yield the title compound (20) (47 mg, 41 %) as a white solid; 1H N R (400 MHz,cfe-DMSO) δ 9.96 (d, J = 6.3 Hz, 1 H), 8.60 (d, J = 1.4 Hz, 1 H), 7.56 (t, j = 7.8 Hz, 2H), 7.44 (s, 1 H) 7.32 - 7.10 (m, 4H), 7.03 - 6.84 (m, 3H), 3.70 - 3.53 (m, 2H), 3.50 (s, 2H), 3.18 - 2.91 (m, 4H), 2.85 - 2.60 (m, J = 34.4, 8.4 Hz, 3H), 1.84 - 1 ,70 (m, 2H), 1 ,69 - 1.54 (m, J = 15.3, 6.6 Hz, 2H). LC S Method C: rt 4.64 min; m/z 499 [M+H]+.
Example 21 : 2-(2-(2-(2-({1 , 2,3,4- Tetrahydro!Soqu!not!n-6-yf)amino)-S- {tnf uorometh !)pyrimidsn-4-yl)ethy )phenyl)acetamide (21)

21
(a ) tert-Butyl 6-((4-(2-(2-methoxy-2-oxoethyl)phenethyl)-5-(trifluoromethyl)p^
2-yl)amino)-3,4-dihydroisoquinoline-2(1H)-carboxylate (180)
fe/f-Butyl 6-amino-3,4-dihydroisoquinoline-2(1 H)-carboxylate (0.500 g, 2.01 mmol), methyl 2-(2-(2-(2-(methylsulfonyl)-5-(trifiuoromethyl)pyrimidin-4- yi)ethyi)phenyl)acetate (166) (0.675 g, 1.68 mmol), trifiuoroethanol (3 mL), and TFA (0.3 mL) were loaded into a microwave tube, sonicated for two minutes, then heated under microwave irradiation at 100 °C for 20 minutes. The cooled mixture was concentrated, co-evaporated with toluene (3x 20 mL) and loaded onto a 10 g SCX cartridge in methanol. The cartridge was eluted with methanol (200 mL), then with 1 % methanolic methyiamine (200 mL). The methanolic methylamine eluent was
concentrated to give a brown oil (0.850 g). The oil was dissolved in DCM (5 mL), and Boc anhydride (549 mg, 2.52 mmoi) was added. The resulting mixture was stirred under an oil bubbler for 18 hours, then diluted with DCM (50 mL) and washed with water (50 mL). The aqueous layer was extracted with DCM (2x 50 mL), and the combined DCM phases dried (phase separation filter) and evaporated.
Chromatography (isoiera, 40 g silica cartridge, 0-50% ethyl acetate/petroleum benzine 40-80 °C) gave the title compound (/SO) (520 mg, 54%) as a yellow syrup; 1H NMR (400 MHz, CDCI3) δ 8.54 (s, 1 H), 7.45 (s, 2H), 7.38 (s, 1 H), 7.28 - 7.18 (m, overlaps with CDCI3), 7.10 (d, J = 8.5 Hz, 1 H), 4.56 (s, 2H), 3.75 (s, 2H), 3.70 - 3.62 (m, 5H), 3.17 - 3.03 (m, 4H), 2.85 (t, J = 5.6 Hz, 2H), 1.50 (s, 9H). LCMS Method C: rt 6.93 min; m/z 571.1 [M+H] +, m/z 515.0 [M+tBu+2H] +.
(b) Lithium 2- (2- (2- (2-( (2-( tert-butoxycarbonyl) -1,2, 3,4-tetrahydroisoquino!in -6- yi) amino ) -5-(trifluoromethyl)pyrimidin - 4 -yl)ethyl)phenyl)acetate ( 181)
ferf-Butyl 6-((4-(2-(2-methoxy-2-oxoethyl)phenethyl)-5-(trifluoromethyl)pyrimidin-2- yl)amino)-3,4-dihydroisoquinoline-2(1 H)-carboxylate (180) (520 mg, 0.91 mmoi) was dissolved in THF (20 mL) and a solution of lithium hydroxide hydrate (76 mg. 1.8 mmoi) in water (5 mL) was added. After 18 hours the THF was removed under reduced pressure, the mixture was diluted with water ( 0 mL) and extracted with ethyl acetate (3x50 mL). The combined ethyl acetate phases were washed with brine, dried (sodium sulfate) and evaporated to give the title compound (181) (414 mg, 81 % yield) as a yellow solid; H NMR (400 MHz, d6~DMSO) δ 10.36 (s, 1 H), 8.65 (s, 1 H), 7.64 (s, H), 7.59 (d, J = 8.4 Hz, 1 H), 7.20 - 7.02 (m, 5H), 4.45 (s, 2H), 3.54 (t, J = 5.8 Hz, 2H), 3.17 (s, 2H), 3.14 - 2.98 (m, 4H), 2.76 (t, J = 5.7 Hz, 2H), 1.43 (s, 9H). LCMS Method C: rt 8.51 min; m/z 557.1 [M-Li+2H] +, 501.1 [M-tBu-Li+3H] +, 457.1 [M- Li-Boc+3H] +; m/z 555.1 [M-Li] .
(c) tert-Butyl 6-((4-(2-(2-amino-2-oxoethyl)phenethyl)-5-(triflu^
yl)amino)-3,4-dihydroisoquinoline-2( 1 H)-carboxylate (182)
Lithium 2-(2-(2-(2-((2-(tert-butoxycarbonyl)-1 ,2,3,4-tetrahydroisoquinoiin-6-yl)amino)- 5-(trifluoromethyl)pyrimidin-4-yl)ethyl)phenyl)acetate (181) (205 mg, 0.37 mmoi) was dissolved in DMF (3 mL) and HATU (154 mg, 0.41 mmoi) was added. After stirring for 3 minutes, ammonium carbonate (212 mg, 2.20 mmoi) and DIPEA (0.26 mL, 1.5 mmoi) were added and the mixture was stirred at room temperature for 18 hours. The resulting mixture was added to water (50 mL) and saturated sodium bicarbonate
(10 mL) then extracted with ethyl acetate (3x 50 mL). The organic extracts were washed with brine (2x 50 mL), dried (sodium sulphate) and evaporated to dryness. The residue was chromatographed (12 g silica cartridge, 0-100% ethyl
acetate/petroleum benzine 40-60 °C) and the product triturated with diethyl ether to give the title compound {182) (138 mg, 87%) as an off-white foam; 1H N R (400 MHz, CDCI3) δ 8.53 (s, H), 7.57 (s, H), 7.43 (d, J = 8.9 Hz, 2H), 7.29 - 7.26 (m, overlaps with CHCI3), 7.10 (d, J = 8.4 Hz, 1 H), 5.36 (d, J = 14.4 Hz, 2H), 4.55 (s, 2H), 3.73 (s, 2H), 3,65 (s, 2H), 3.15 - 3.04 (m, 4H), 2.85 (t, J = 5.8 Hz, 2H), 1.50 is, 9H). LCMS Method C: rt 6.36 min; /z 556.1 [ ÷H] ÷, 500.0 [M-tBu+2H] +, 456.1 [M- Boc÷2H] +; m/z 554.2 [M-H]\
(d) 2-(2-(2-(2-((1,2,3 -Tetrahydroisoquinolin-6-yl)amino)-5-(tri^
4-yl)ethyl)phenyl)acetamide (21)
iert-Butyl 6-((4-(2-(2-amino-2-oxoethyl)phenethyl)-5-(trifluoromethyl)pyrimidin-2- yl)amino)-3,4-dihydroisoquinoline-2(1 H)-carboxylate (182) (136 mg, 0.245 mmol),
DCM (10 mL) and TFA (1 mL) were stirred together at room temperature. After three hours the solution was concentrated and the residue treated with 1 M sodium hydroxide (25 mL). The resulting suspension was extracted with ethyl acetate (3x50 mL) and the combined organic extracts washed with brine (100 mL), dried and evaporated to give the title compound (21) (81.6 mg, 74% yield) as a white solid; 1H NMR (400 MHz, ofe-DMSO) δ 10.06 (s, 1 H), 8,65 (s, H), 7,51 (s, 1 H), 7.48 (dd, J = 8.3, 2.1 Hz, 1 H), 7.42 (s, 1 H), 7.26 - 7.21 (m, 1 H), 7.19 - 7.13 (m, 3H), 6.96 (d, J = 8.3 Hz, 1 H), 6.90 (s, 1 H), 3.81 (s, 2H), 3.49 (s, 2H), 3.17 (d, J = 3.1 Hz, 1 H), 3.14 - 3.06 (m, 2H), 3.05 - 2.97 (m, 2H), 2.94 (t, J = 5.9 Hz, 2H), 2.67 (t, J = 5.6 Hz, 2H). LCMS Method C: rt: 4.76 min; m/z 456.1 [M+H]+; m/z 454.1 [M-H]".
Example 22: 2-(2-(2-(2-({4-{ orpho!in-2-yl)phenyS)amino)-S-

191 22
(a) 2-(4-Nitrophenyl)oxirane (183)
2-Bromo-1-(4-nitrophenyl)ethanone (1.9 g, 7.9 mmo!) was stirred in methanol (30 mL) and the suspension cooled in an ice bath. Sodium borohydride (0.33 g, 8.7 mmol) was added in one portion and after five minutes the ice bath was removed and the mixture stirred at room temperature. After three hours potassium carbonate (1.1 g, 7.9 mmol) was added, and the mixture stirred at room temperature for a further 18 hours. The methanol was evaporated, water (50 mL) was added, and the mixture extracted with DC (3x100 mL). The combined DC phases were washed with brine, dried (sodium sulfate) and evaporated to give the title compound (183) (1.296 g, 99% yield) as a yellow solid; 1H N R (400 MHz, CDCI3) δ 8.25 - 8.18 (m, 2H), 7.50 - 7.41 (m, 2H), 3.96 (dd, J = 4.1 , 2.5 Hz, 1 H), 3.23 (dd, J = 5.5, 4.1 Hz, 1 H), 2.78 (dd, J = 5.5, 2.5 Hz, 1 H). LC S Method C: rt 5.42 min.
(b) N-(2-Hydroxy-2-(4-nitrophenyl)ethyl)-4-methylbenzenesulfonamide (184)
Tosyiamide (0.69 g, 4.0 mmol), 2-(4-nitrophenyl)oxirane (183) (0.33 g, 2.0 mmol), benzyltriethylammonium chloride (46 mg, 0.20 mmol) and potassium carbonate (28 mg, 0.20 mmol) were suspended in dioxane (1.0 mL) and the resulting mixture was stirred at 90°C. After four hours the mixture was cooled to room temperature and poured into DCM (15 mL). The resulting mixture was filtered and evaporated. The residue was chromatographed (Isoiera, 40 g silica cartridge, 0-100% ethyl acetate/ petroleum benzine 40-60 °C) to give the title compound (184) as an orange solid (363 mg, 38% yield); 1H NMR (400 MHz, ef6~DMSO) δ 8.19 - 8.13 (m, 2H), 7.64 - 7.59 (m, 2H), 7.57 - 7.52 (m, 2H), 7.34 (d, J = 7.9 Hz, 2H), 7.27 (s, H), 5.82 (d, J = 4.5 Hz, 1 H), 4.71 (dd, J = 10.7, 6.0 Hz, 1 H), 2.92 (td, J = 6.2, 1.7 Hz, 2H), 2.36 (s, 3H).
LCMS Method C: rt 5,55 min, m/z 335.0 [M-H]\
(c) 2-(4-Nitrophenyl)-4-tosylmorpholine (185)
/V-(2-Hydroxy-2-(4-nitrophenyl)ethyl)-4-methylbenzenesulfonamide (184) (0.610 g,
1.34 mmol) was sonicated in DCM (20 mL) for five minutes and cooled to 0 °C under nitrogen. A 60% dispersion of NaH (0.220 g, 5.44 mmol) was added and the mixture stirred for five minutes before (2-bromoethyl)diphenylsulfonium
trifluoromethanesulfonate (1.21 g, 2.72 mmol) was added. The mixture was stirred for 17 hours, allowing the cooling bath to come to room temperature over this time. Ethyl acetate (200 mL), saturated ammonium chloride (80 mL) and water (20 mL) were added and the layers separated. The aqueous phase was extracted with ethyl acetate (2x100 mL) and the combined ethyl acetate phases were washed with brine, dried (sodium sulfate) and evaporated. The residue was chromatographed (Isoiera, 40 g silica cartridge, 0-40% ethyl acetate/petroleum benzine 40-60 °C) to give the title compound (185) (433 mg, 88%) as a white solid; 1H NMR (400 MHz, CDCI3) δ 8.22 - 8.18 (m, 2H), 7.63 - 7.59 (m, 2H), 7.52 - 7.48 (m, 2H), 7.33 (dd, J = 8.5, 0.6 Hz, 2H), 4.71 (dd, J = 10.2, 2.6 Hz, 1 H), 4.10 (ddd, J = 11.6, 3.4, 1.4 Hz, 1 H), 3.87 (td, J = 11.6, 2.7 Hz, 1 H), 3.82 - 3.77 (m, 1 H), 3.65 (ddt, J = 11.6, 2.8, 1.6 Hz, 1 H), 2.51 (td, J = 11.6, 3.4 Hz, 1 H), 2.43 (s, 3H), 2.18 (dd, J = 11.5, 10.3 Hz, 1 H). LCMS Method C: rt 6.20 min; m/z 363.0 [M+H]+.
(d) 2-(4-Nitrophenyl)morpholine (186)
A mixture of 2-(4-Nitrophenyl)-4-tosyimorpholine (185) (430 mg, 1.19 mmol), phenol (670 mg, 7.12 mmol) and 33% HBr/AcOH (2.2 mL) was heated in a sealed tube
(microwave iube, conventional heating) at 75 °C for twenty hours. The cooled mixture was concentrated and the residue loaded onto a 10 g SCX cartridge in methanol. The cartridge was washed with methanol (200 mL), then eiuted with 1 % methanoiic methylamine (100 mL). The methylamine eluent was evaporated to give the title compound (186) (204 mg, 83%) as a yellow oil; 1 H N R (400 MHz, CDCI3) δ 8.27 - 8.18 (m, 2H), 7.57 - 7.51 (m, 2H), 4.63 (dd, J = 10.3, 2.5 Hz, 1 H), 4.13 - 4.04 (m, 1 H), 3.82 (td, J = 11 .3, 3.1 Hz, 1 H), 3.13 (dd, J = 12.3, 2.5 Hz, 1 H), 3.06 - 2.92 (m, 2H), 2.74 (dd, J = 12.3, 10.3 Hz, 1 H). LCMS Method C: rt 1.49, 1 .58 min; m/z 209.1 [M+Hf.
(e) tert-Butyl 2-(4-nitrophenyl)morpholine-4-carboxylate (187)
2-(4-Nitrophenyl)morpholine (186) (200 mg, 0.961 mmol) was dissolved in DCM (5 mL) then DMAP (12 mg, 10 mol%) and Boc anhydride (0.265 mL, 1 .15 mmol) were added. After one hour the mixture was diluted with DCM (20 mL), and washed with water (20 mL). The aqueous phase was extracted with DCM (2x20 mL), and the combined DCM extracts were dried (hydrophobic frit) and evaporated. The residue was chromatographed (12g silica cartridge, 0-40% ethyl acetate/petroleum benzine 40-60 °C) to give the title compound (187) (0.230 g, 78%) as a white solid; 1 H NMR (400 MHz, CDCI3) δ 8.26 - 8.19 (m, 2H), 7.60 - 7.53 (m, 2H), 4.53 (dd, J = 10.5, 2.6 Hz, 1 H), 4.17 (br s, 1 H), 4.05 (dd, J = 11.5, 2.4 Hz, 1 H), 3.97 (br s, 1 H), 3.70 (td, J = 11.7, 2.8 Hz, 1 H), 3.06 (t, J = 11.2 Hz, 1 H), 2.84 - 2.67 (m, 1 H), 1 .49 (s, 9H). LCMS Method C: rt 6.23 min; m/z 209.1 [M-Boc+2H]÷.
(f) tert-Buty! 2-(4-aminophenyl)morpholine-4-carboxylate (188)
ferf-Butyl 2-(4-nitrophenyl)morpholine-4-carboxylate (187) (100 mg, 0.324 mmol), activated charcoal (20 mg), iron(l i i) chloride hexahydrate (9 mg, 10 moi%), methanol (1 mL) and hydrazine hydrate (162 mg, 1 .62 mmol @ 50%) were refiuxed together for five hours. The mixture was filtered through cotton, and the cotton washed with DCM (5 mL). The filtrate was evaporated, and redissolved in 95% ethanol (3 mL) and ethyl acetate (2 mL). A solution of ammonium chloride ( 73 mg. 3.24 mmol) in water (1 mL) was added, followed by indium powder (153 mg, 1 .30 mmol). The mixture was refiuxed for four hours then filtered. The collected solids were washed with DCM (20 mL) and the combined filtrates then diluted with water (10 mL) and saturated sodium bicarbonate (10 mL). The aqueous phase was washed with DCM (2x 25 mL), the combined DCM extracts dried (phase separation filter) and evaporated. The residue
was dissolved in 95% ethanol (3 mL), and treated at reflux with further indium powder (153 mg, 1.30 mmol) and ammonium chloride (173 mg. 3.24 mmol) in water (1 mL). After three hours the mixture was diluted with water (10 mL) and filtered. The collected solids were washed sequentially with ethyl acetate (25 mL) and saturated sodium bicarbonate (10 mL). The filtrate aqueous phase was separated, and washed with ethyl acetate (2x25 mL). The combined organic extracts were washed with brine (50 mL), dried and evaporated. The residue was chromafographed (12 g silica cartridge, 0-100% ethyl acetate/ petroleum benzine 40-60 °C) to give the title compound (188) (51.4 mg, 57% yield) as a yellow oil; ! H N R (400 MHz, CDCl3) δ 7.19 - 7.12 (m, 2H), 8.71 - 6.63 (m, 2H), 4.33 - 4.25 (m, 1 H), 4.03 - 3.85 (m, 3H), 3.73 - 3.60 (m, 3H), 3.02 (s, 1 H), 2.84 (s, 1 H). LCMS Method C: rt: 4.72 min; m/z 179.1 [M-Boc+2H]+.
(g) tert-Butyl 2-(4-((4-(2-(2-methoxy-2-oxoethyl)phenethyl)-5- (trifluoromethyl)pyrimidin-2-yl)amino)phenyl)m (189) ferf-Butyl 2-(4-aminophenyl)morpholine-4-carboxylate (188) (50 mg, 0.18 mmol) and methyl 2-(2-(2-(2-(methylsulfonyl)-5-(trifluoromethyl)pyrimidin-4- yl)efhyl)phenyl)acetate (167) (72 mg, 0.18 mmol) were heated in trifiuoroefhanol (1.2 mL) and TFA (0.12 mL) under microwave irradiation (100 °C/20 minutes). The mixture was concentrated, evaporated from toluene and loaded onto a 5 g SCX cartridge in methanol (1 mL). The cartridge was washed with methanol (50 mL), and then eluted with 1 % methylamine/methanol (50 mL). The basic eluent was concentrated, and taken up in dichloromethane (5 mL). Boc anhydride (0.062 mL, 0.27 mmol) was added, and the mixture stirred at room temperature for 18 hours. The mixture was evaporated onto silica gel, and chromatographed (12 g silica cartridge, 0-60% ethyl acetate/ petroleum benzine 40-60 °C) to give the title compound (189) (46 mg, 42% yield) as a pale yellow oil; 1H NMR (400 MHz, CDCI3) δ 8.55 (s, 1 H), 7.64 (d, J = 8.6 Hz, 2H), 7.47 (s, 1 H), 7.37 (d, J = 8.5 Hz, 2H), 7.29 - 7.18 (m, overlaps with CHCI3), 4.41 (d, J = 8.4 Hz, 1 H), 4.08 (m, 3H), 3.76 (s, 2H), 3.73 - 3.64 (m, 4H), 3.16 - 2.98 (m, 5H), 2.85 (s, 1 H), 1.51 - 1.47 (m, 9H). LCMS Method C: rt 6.88 min; m/z 601.1 [M+H]+, 545.1 [M-tBu+2H]+.
(h) 2-(2-(2-(2-((4-(4-(tert-Butoxycarbonyl)morpholin-2-yl)phenyl)am
(trifluoromethy!)pyrimidin-4-yl}ethyl}phenyl)acetic acid (190)
feii-Buiyl 2-(4-((4-(2-(2-methoxy-2-oxoethyl)phenethyl)-5-(trifluoromethyl)pyrimidin-2^ yl)amino)phenyl)morpholine-4-carboxylate (189) (46 mg, 0.077 mmo!) was dissolved in THF (2 mL), and lithium hydroxide hydrate (6.0 mg, 0.15 mmoi) in water (0.5 mL) was added. After 18 hours the mixture was concentrated, diluted with water (5 mL) and the pH adjusted to 3 with 6 M HCI. The mixture was extracted with ethyl acetate (3x10 mL), and the combined organic extracts washed with brine (20 mL), dried over sodium sulfate and evaporated to give the title compound (190) (39 mg, 85% yield) as a pale yellow syrup; ΊΗ NMR (400 MHz, CDCI3) δ 8.52 (s, 1 H), 7.54 (d, J = 8.4 Hz, 2H), 7.35 - 7.27 (m, 4H), 4.38 (d, J = 10.1 Hz, 1 H), 4.07 - 3.80 (m, 5H), 3.67 (td, J = 11.7, 2.7 Hz, 1 H), 3.06 (s, 4H), 2.84 (s, 2H), 1.47 (s, 9H). LCMS Method C: rt 6.49 min; m/z 587.1 [M+Hf, 531.0 [M-tBu+2H] \ 487.1 [M-Boc+2H]+; m/z 585.2 [M-H]\ (i) tert-Butyl 2-(4-((4-(2-(2-amino-2-oxoethyl)phenethyl)-5-(trifluom
yl)amino)phenyl)morpholine-4-carboxylate (191}
2-(2-(2-(2-((4-(4-(tert-Butoxycarbonyl)morpholin-2-yl)phenyl)amino)-5-
(trifiuoromethyl)pyrimidin-4-yl)ethyl)phenyi)acetic acid (190) (39 mg, 0.066 mmoi) was dissolved in DMF (3 mL) and ammonium carbonate (38 mg, 0.40 mmoi), HATU (28 mg, 0.073 mmoi) and DIPEA (0.046 mL, 0.27 mmoi) were added. The yellow mixture was stirred at room temperature for 18 hours then added to water (30 mL) and brine (10 mL). The mixture was extracted with ethyl acetate (3x30 mL), and the combined ethyl acetate phases washed with brine (50 mL), dried over sodium sulfate and evaporated. The residue was chromotographed (4 g deactivated* silica cartridge, 0- 100% 1 % isopropylamine in ethyl acetate/petroleum benzine 40-60 °C) to give the title compound (191) (23.3 mg, 60% yieid)as a colourless glass; 1 H NMR (400 MHz, CDCI3) δ 8.53 (s, 1 H), 7.78 (s, 1 H), 7.61 (d, J = 8.4 Hz, 2H), 7.37 (d, J = 8.5 Hz, 2H), 7.31 - 7.19 (m, 5H, overlaps with CHCI3), 5.58 (s, 1 H), 5.42 (s, 1 H), 4.40 (d, J = 8.5 Hz, 1 H), 4,08 - 3.84 (m, 3H), 3.74 - 3,63 (m, 3H), 3, 16 - 3.00 (m, 5H), 2.85 (s, 1 H), 1.48 (s, 9H). LCMS Method C: rt 6.29 min; m/z 586.1 [M+H]+, 530.1 [M Bu+2H]+, 608.1 [M+Na]+; m/z 584.1 [M-H]\
* cartridge deactivated by treating with 3 volumes of 1 % isopropylamine in ethyl acetate followed by rinsing with a 3 volume gradient of 100-0% of 1 % isopropylamine in ethyl acetate/ petroleum benzine 40-60 °C.
(j) 2-(2-(2-(2-((4-(morpholin-2-yl)phenyl)amino)-5-(trifluorome
yl)ethyl)phenyl)acetamide (22)
fe/f-Butyi 2-(4-((4-(2-(2-amino-2-oxoethyl)phenethyl)-5-(trifluoromethyl)pyrimidin-2- yl)amino)phenyl)morpholine-4-carboxyiate (191) (23 mg, 0.039 mmol) was stirred with DCM (3 mL) and TFA (0.3 mL). After 18 hours the mixture was concentrated and the residue suspended in 10% aqueous NaOH (1 mL) and brine (1 mL). The mixture was extracted with ethyl acetate (5x3 mL), and the combined ethyl acetate phases washed with brine (20 mL), dried over sodium sulfate and evaporated to give the title compound (22) (17 mg, 89%) as a white solid; 1 H HMR (400 MHz, g-DMSO) δ 10.19 (s, 1 H), 8.67 (s, 1 H), 7.74 - 7.67 (m, 2H), 7.42 (s, 1 H), 7.27 (d, J = 8.6 Hz, 2H), 7.25 - 7.21 (m, 1 H), 7.19 - 7.14 (m, 3H), 6.91 (s, 1 H), 4.32 (dd, J = 10.1 , 2.1 Hz, 1 H), 3.86 (d, J = 10.9 Hz, 1 H), 3.58 (dt, J = 11.2, 7.2 Hz, 1 H), 3.50 (s, 2H), 3.15 - 2.98 (m, 4H), 2.89 (dd, J = 12.3, 2.2 Hz, 1 H), 2.72 (d, J = 5.2 Hz, 2H). LCMS Method C: rt 4.75 min; m/z 486.1 [M+H]+, 508.0 [M+Na]*; m/z 484.1 [M-H]".
Example 23: 2~(2~(2~(2~((4~(1 ~Acety!piperid!n-4-y!)phenyf)am!no)~5~
(tnfluorometh l}pynmidsn-4-y!)ethyl)pheny!}acetamide

Triethyiamine (34.6 μί, 0.248 mmol) and acetic anhydride (23.4 μί, 0.248 mmol) were added to a solution of the 2-(2-(2-(2-((4-(piperidin-4-yl)phenyl)amino)-5- (trifluoromethyl)pyrimidin-4-yl)ethyl)phenyl)acetamide ( 11) (30 mg, 0.062 mmoi) in DMF (10 mL). The reaction mixture was stirred at room temperature for 20 hours. The volatiles were removed in vacuo and the residue was diluted with EtOAc (20 mL) and sat. aq. NaHC03 (20 mL). The layers were separated and the aqueous layer was extracted with EtOAc (2x20 mL). The combined organic layers were washed with water (20 mL), brine (20 mL) and dried over Na2S04. The solution was filtered and concentrated in vacuo to give a white solid which was suspended in DCM (ca 2 mL) and cyciohexane (ca 10 mL). The suspension was filtered to give the title compound (23) (24 mg, 72%) as an off-white solid; 1 H NMR (400 MHz, ce-DMSO) δ 10.14 (s,
1 H), 8.66 (s, 1 H), 7.71 - 7.63 (m, 2H), 7.42 (s, 1 H), 7.27 - 7.12 (m, 6H), 6.91 (s, 1 H), 4.57 - 4.48 (m, 1 H), 3.93 - 3.90 (m, 1 H), 3.50 (s, 2H), 3.16 - 3.06 (m, 3H), 3.06 - 2.97 (m, 2H), 2.77 - 2.65 (m, H), 2.62 - 2.51 (m, peak obscured by solvent), 2.03 (s, 3H), 1.81 - 1.74 (m, 2H), 1.57 (qd, J = 12.6, 4.2 Hz, 1 H), 1.49 - 1.35 (m, 1 H); LCMS Method C: rt 5.89 min; m/z 526 [M+H]+.
Example 24: 2~(2~(2~(2~((4~(4~(Dsmethylamino)piperidsn~1~y^)phenyl)amino)-5-

20 24 To a suspension of 2-(2-(2-(2-((4-(4-aminopiperidin-1-yl)phenyl)amino)-5-
(trifluoromethyl)pyrimidin-4-yl)ethyl)phenyl)acetamide (20) (30m g, 0.060 mmol) in anhydrous methanol (2 mL) was added a 37% aq. solution of formaldehyde (7 μΙ_, 0.06 mmol) and sodium triacetoxyborohydride (0.038 g, 0.18 mmol) under an atmosphere of nitrogen. The resulting suspension was stirred at room temperature for 2.5 hours. The voiatiles were removed in vacuo and the residue partitioned between in EtOAc (50 mL) and water (50 mL), The organic layer was separated, dried (MgS04) and the solvent removed in vacuo to yield a crude yellow oil. The crude oil was chromatographed on silica gel (Biotage isolera: 0-100% MeOH in EtOAc) to yield the title compound (24) (11 mg, 35%) as a yellow solid; 1H NMR (400 MHz.df- eOD) δ 8.51 (s, 1 H), 7.57 - 7.51 (m, 2H), 7.31 - 7.18 (m, 4H), 7.04 - 6.99 (m, 2H), 3.76 - 3.67 (m, 4H), 3.20 - 3.13 (m, 2H), 3.09 - 3.03 (m, J = 9.9, 5.5 Hz, 2H), 2.76 - 2.64 (m, 2H), 2.34 (s, 7H), 2.01 (d, J = 12.7 Hz, 2H), 1.66 (qd, J = 12.4, 3.9 Hz, 2H). LCMS Method C: rt 4.77 min; m/z = 527 [M+1]+.
Example 25: 2-(2-(2-(2-({4-{Piperaz!n-1-ylmethy!)phenyS)am!no)-5-
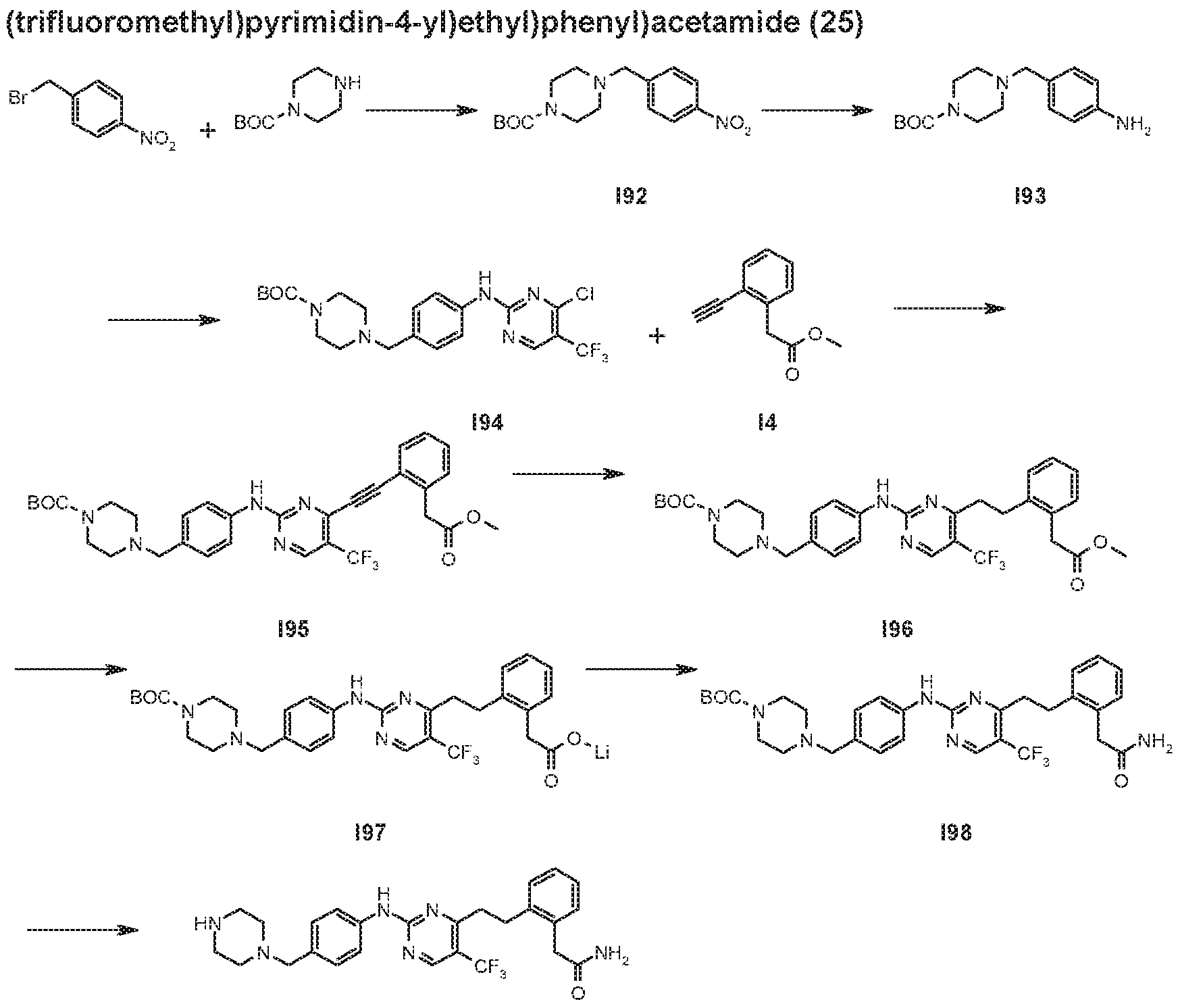
25 (a) tert-Buty! 4-(4-nitrobenzyl)piperazine- 1-carboxylate (192)
1-(Bromomethyl)-4-nitrobenzene (1.08 g, 5.00 mmol) was added to a vigorously stirred mixture of ferf-butyi piperazine-1-carboxylate (1.02 g, 5.50 mmol) and sodium carbonate (0.583 g, 5.50 mmol) in DMF (5 mL) at room temperature and the resulting mixture stirred for two hours. Water (25 mL) was added, and the resulting suspension was allowed to stand for five minutes then filtered. The collected solid was washed with water (25 mL) and air-dried to give the title compound (192) (1.523 g, 95% yield); 1H N R (400 MHz, CDCI3) δ 8.32 - 8.10 (m, 2H), 7.51 (d, J = 8.8 Hz, 2H), 3.59 (s, 2H), 3.52 - 3.39 (m, 4H), 2.43 - 2.34 (m, 4H), 1.45 (s, 9H). LCMS Method C: rt 4.58 min, m/z 268, 1 [ -tBu+2H]+, 222.1 [M-Boc+2H]÷.
(b) tert-Butyl 4-(4-aminobenzyl)piperazine-1 -carboxylate (193)
terf-Butyl 4-(4-nitrobenzyl)piperazine-1-carboxylate (192) (0.500 g, 1.58 mmol), ethyl acetate (100 mL) and 10% Pd/C (150 mg) were agitated under a hydrogen atmosphere at 50 psi. After two hours the mixture was filtered through celite and concentrated. The residue was chromatographed (12 g silica cartridge, 0-60% ethyl acetate/petroleum benzine 40-60 °C) to give the title compound {193) (327 mg, 72% yield) as a white solid; 1H N R (400 Hz, CDCI3) δ 7.11 - 7.05 (m, 2H), 6.67 - 6.61 (m, 2H), 3.62 (s, 2H), 3.43 - 3.37 (m, 6H), 2.40 - 2.30 (m, 4H), 1.45 (s, 9H). LCMS Method C: rt 1.80 min; m/z 292.1 [M+H]+.
( c) tert-Butyl 4-(4-(( 4 -chloro -5- ( trifluoromethyl)pyrimidin-2- yl)amino)benzyl)piperazine- 1 -carboxylate (194)
2,4-Dichloro-5-(trifluoromethyl)pyrimidine (0.546 g, 2.52 mmol) in 1 : 1 dichioroethane: fe/f-butanol was cooled to 0 °C under nitrogen. A 1.0 M solution of zinc(ll) chloride in diethyl ether (3.43 mL, 3.34 mmol) was added, and the mixture stirred for one hour at 0 °C. iert-Butyl 4-(4-aminobenzyi)piperazine-1-carboxylate (193) (0.667 g, 2.29 mmol) in 1 : 1 dichioroethane: ferf-butanol (20 mL) vv'as added dropwise over thirty minutes, followed by triethyiamine (0.351 mL, 2.52 mmol) in 1 :1 dichioroethane: ferf-butanol (10 mL). The mixture was stirred overnight, allowing the ice bath to come to room temperature over this time. The mixture was concentrated onto silica gel and chromatographed (40 g silica cartridge, 0-100% ethyl acetate/petroleum benzine 40- 60 °C) to give a residue which was triturated with petroleum benzine 40-60 °C to give the title compound {194) (0.976 g, 90%) as an off white solid; 1H NMR (400 MHz, dr MeOD) δ 8.68 (d, J = 0.6 Hz, 1 H), 7.85 (d, J = 8.6 Hz, 2H), 7.51 (d, J = 8.6 Hz, 2H), 4.30 (s, 2H), 3.27 - 3,00 (br, overlaps with solvent), 1.47 (s, 9H). LCMS Method C: 5.08 min; m/z 472.1 [M+H]+; m/z 470.1 [M-H]\
(d) tert-Butyl 4-(4-((4-((2-(2-methoxy-2-oxoethyl)phenyl)ethynyl)-5- (triiluoromethyl)pyrimidin-2-yl)amino)benzyl)piperazine- 1 -carboxylate (195) ferf-Butyl 4-(4-((4-chloro-5-(trifluoromethyl)pyrimidin-2-yl)amino)benzyl)piperazine-1- carboxylate (194) (0.500 g, 1.06 mmol) and methyl 2-(2-ethynyiphenyl)acetate (14) (0.203 g, 1.17 mmol) were dissolved in DMF (10 mL) and
bis(triphenylphosphine)palladium(ll) chloride (37 mg, 5 mol%) was added. The mixture was degassed with nitrogen for ten minutes, then copper(l) iodide (10 mg, 5 mol%) and triethyiamine (0.738 mL, 5.30 mmol) were added. The mixture was
heated under microwave irradiation (100 °C/ 20 minutes) then concentrated.
Chromatography of the residue (12 g silica cartridge, 0-100% ethyl acetate/petroleum benzine 40-60 °C) gave the title compound (195) (192 mg, 30% yield) as a brown oil; 1H N (400 Hz, CDCI3) δ 8.63 (s, 1 H), 7.68 (dd, J = 7.6, 1.0 Hz, 1 H), 7.59 (d, J = 8.4 Hz, 2H), 7.50 (s, 1 H), 7.42 (dd, J = 7.5, 1.4 Hz, 1 H), 7.38 - 7.29 (m, 4H), 3.95 (s, 2H), 3.70 (s, 3H), 3.50 (s, 2H), 3.45 - 3.40 (m, 4H), 2.43 - 2.35 (m, 4H), .45 (s, 9H). LCMS Method C: rt 5.30 min; /n z 610.1 [M+H]*, m/z 608.2 [M-H]".
(e) tert-Butyl 4-(4-((4-(2-(2-methoxy-2-oxoethyl)phenethyl)-5- (trifluoromethyl)pyrimidin-2-yl)amino)benzyl)piperazm^ (196) terf-Butyl 4-(4-((4-((2-(2-methoxy-2-oxoethyl)phenyl)ethynyl)-5- (trifluoromethyl)pyrimidin-2-yl)amino)benzyl)piperazine-1-carboxylate (195) (190 mg, 0.312 mmol) was stirred vigorously with 10% Pd/C (100 mg) in DMF (10 mL) under an atmosphere of hydrogen. After two the reaction was transferred to a Parr tube with the aid of ethyl acetate (10 mL) and the mixture was hydrogenated at 45 psi. After 18 hours Pearlman's catalyst (100 mg) and triethylamine (0.2 mL) were added, and the mixture agitated under hydrogen at 40 psi. After three hours the mixture was diluted with ethyl acetate (50 mL) and filtered through celite, washing the ceiife with ethyl acetate (2x25 mL). The filtrate was diluted with ethyl acetate (100 mL) and washed with 5% lithium chloride solution (3x100 mL). The organic phase was dried over sodium sulfate and evaporated to give the title compound (196) (127 mg, 66% yield) as a yellow syrup; H NMR (400 MHz, CDCI3) δ 8.54 (s, 1 H), 7.63 - 7.56 (m, 2H), 7.46 (s, 1 H), 7.31 (d, J = 8.5 Hz, 2H), 7.26 - 7.17 (m, 4H), 3.75 (s, 2H), 3.68 (s, 3H), 3.50 (s, 2H), 3.46 - 3.40 (m, 4H), 3.17 - 3.05 (m, 4H), 2.45 - 2.34 (m, 4H), 1.45 (s, 9H), LCMS Method C: rt 5,63 min; m/z 614.2 [M+Hf; m/z 612.1 [M-H]".
(f) Lithium 2-(2-(2-(2-((4-((4-(tert-butoxycarbony!)piperazin-1- yl)methyl)phenyl)amino)-5-(trifluoromethyl)pyrimitf^ (197) fe/f-Butyl 4-(4-((4-(2-(2-methoxy-2-oxoethyl)phenethyl)-5-(trifluoromethyl)pyrimidin-2- yl)amino)benzyl)piperazine-1-carboxylate (196) (120 mg, 0.196 mmol) was dissolved in THF (4 mL), and a solution of lithium hydroxide hydrate (25 mg, 0.59 mmol) in water (1 mL) was added. The mixture was stirred at room temperature for 18 hours then concentrated and the residue evaporated twice from toluene to give the title compound (197) as a tan solid which was used without purification. LCMS Method C: rt 5.17 min; m/z 600.2 [M-Li+2H]+; 544.1 [M-tBu-Li+3H]*; m/z 598.2 [M-Li]".
(g) tert-Butyi 4-(4-((4-(2-(2-amino-2-oxoethyl)phenethyl)-5-(trifluorom 2-yl)amino)benzyl)piperazine- 1 -carboxy!ate (198)
The crude lithium 2-(2-(2-(2-((4-((4-(tert-butoxycarbonyl)piperazin-1- yl)methyi)pheny!)amino)-5-(trjfluoromethyi)pyrimidin-4-yi)ethyi)pheny!)acetate (197) was dissolved in D F (2 mL) and ammonium chloride (210 mg, 3.92 mmol), HATU (149 mg, 0.392 mmol) and DIPEA (68.0 μΙ_, 0.392 mmol) were added. The resulting mixture was stirred at room temperature for 18 hours, then concentrated. The residue was partitioned between saturated sodium bicarbonate (50 mL) and ethyl acetate (50 mL); the aqueous phase was extracted with further ethyl acetate (2x50 mL) and the combined ethyl acetate phases washed with brine (3x50 mL), dried over sodium sulfate and evaporated. The residue was ehromatographed (4 g silica cartridge, 20- 100% gradient of 1 % isopropylamine in ethyi acetate /petroleum benzine 40-60 °C, then 0-5% gradient methanol/ 1 % isopropylamine in ethyl acetate) to give a residue which was ehromatographed (12 g silica cartridge, 80-100% gradient of 1 % isopropylamine in ethyl acetate/petroleum benzine 40-60 °C) to give the title compound (198) (34 mg, 29% yield over two steps from ester) as a white foam; 1H NMR (400 MHz, CDCI3) δ 8.54 (s, 1 H), 7.62 (s, 1 H), 7.57 (d, J = 8.5 Hz, 2H), 7.31 (d, J = 8.5 Hz, 2H), 7.29 - 7.20 (m, 8H), 5.38 (s, 2H), 3.73 (s, 2H), 3.49 (s, 2H), 3.46 - 3.39 (m, 4H), 3.16 - 3.04 (m, 4H), 2.44 - 2.34 (m, 4H), 1.45 (s, 9H). LC S Method C: rt 5.08 min; m/z 599.1 [M+H]+, 499.1 [M-Boc+2H]+; m/z 597.2 [M-H]".
(h) 2-(2-(2-(2-((4-(Piperazin-1-ylmethyl)phenyl)amino)-5-(trifluorom
yl)ethyl)phenyl)acetamide (25)
ferf-Butyl 4-(4-((4-(2-(2-amino-2-oxoethyl)phenethyl)-5-(trifluoromethyl)pyrimidin-2- yl)amino)benzyi)piperazine-1-carboxyiate (198) (34 mg, 0.057 mmol) was dissolved in DC (4 mL) and TFA (0.4 mL) was added. The resulting mixture was stirred for 16 hours at room temperature then concentrated under reduced pressure. The residue was suspended in 10% aqueous NaOH (2 mL) and brine (3 mL) then extracted with ethyi acetate (4x5 mL). Tthe combined oragnic phases were washed with brine, dried (sodium sulphate) and evaporated to dryness. The residue was triturated with diethyl ether to give the title compound (25) (27.5 mg, 98%) as a yellow solid; 1H NMR (400 MHz, £¾-DMSO) δ 10.17 (s, 1 H), 8.66 (s, 1 H), 7.72 - 7.66 (m, 2H), 7.26 - 7.20 (m, 3H), 7.20 - 7.13 (m, 3H), 6.90 (s, 1 H), 3.49 (s, 2H), 3.37 (s, 2H), 3.14 - 2.98 (m, 4H),
2.67 (t, J = 4.7 Hz, 4H), 2.26 (s, 4H). LC S Method C: rt 4.52 min; m/z 499.1 [M÷H]+; m/z 497.1 [M-H]".
Example 26: 2-{3-{2-{2-{{4-{P!peridin-4-y )pheny!)amino)-5- ftnf!uorometh !)pyrimidin-4-yl)ethy!)pyrazin-2-yf)acetamide (26)

1104 26
(a) Ethyl 2-(3-chloropyrazin-2-yl)acetate (199)
To a 1.0 M solution of LiHMDS in toluene (14.8 mL, 14.8 mmol) under nitrogen at 0 °C was added 2,3-dichloropyrazine (0.699 mL, 6.71 mmol) and ethyl acetate (0.725 mL, 7.38 mmol). The mixture was stirred overnight for 18 hours, allowing the ice bath to warm to room temperature. The mixture was poured into saturated ammonium chloride (100 mL), and extracted with diethyl ether (3x100 mL). The combined ether extracts were washed with brine, dried (sodium sulphate) and evaporated. The residue was chromatographed (40 g silica cartridge, 0-25% ethyl acetate/petroleum benzine 40-60 °C) to give the title compound (199) (0.414 g, 31 % yield) as a pale yellow oil; 1 H NMR (400 MHz, CDCI3) δ 8.46 (d, J = 2.5 Hz, 1 H), 8.31 (d, J = 2.5 Hz,
1 H), 4.22 (q, J = 7.1 Hz, 2H), 4.03 (s, 2H), 1.27 (t, J = 7.1 Hz, 3H). LCMS Method C: rt 5.16 min.
(b) Ethyl 2-(3-((trimethylsilyl)ethynyl)pyrazin-2-yl)acetate (1100)
A mixture of the ethyl 2-(3-chloropyrazin-2-yl)acetate (199) (0.410 g, 2.04 mmol), DMF (6 mL), trlethylamine (2 mL), bis(trlphenyiphosphine)palladium(il) chloride (72 mg, 5 mo!%) and copper(l) iodide ( 9 mg, 5 mol%) in a Schlenk tube was degassed with three vacuum/nitrogen cycles, then trimethylsily!acety!ene (0.866 mL, 6.13 mmol) was added under nitrogen. The tube was flushed with nitrogen, sealed and heated to 90 °C. After 18 hours the mixture was cooled and poured into water (50 mL). Saturated ammonium chloride (50 mL) was added, and the mixture was extracted with diethyl ether (3x100 mL). The combined ether phases were washed with brine, dried (sodium sulphate) and evaporated. The residue was
chromatographed (40 g silica cartridge, 0-30% ethyl acetate/petroleum benzine 40- 60 °C) to give the title compound (1100) (0.386 g, 72% yield) as a yellow oil; 1H NMR (400 MHz, CDCi3) δ 8.47 - 8.41 (m, 2H), 4.20 (q, J = 7.1 Hz, 2H), 4,05 (s, 2H), 1.26 (t, J = 7.1 Hz), 0.28 (s, 9H). LCMS Method C: rt 6.20 min; m/z 263.1 [M+H]+.
(c) Ethyl 2-(3-ethynylpyrazin-2-yl)acetate (1101)
Ethyl 2-(3-((trimethylsilyl)ethynyl)pyrazin-2-yl)acetate (1100) (0.386 g, 1.47 mmol) in THF (15 mL) was cooled to 0 °C and a 1.0 M solution of TBAF in THF (1.84 mL, 1 ,84 mmol) was added. The mixture was stirred for two minutes then poured into water (150 mL). The resulting mixture was extracted with diethyl ether (2x150 mL) and the combined ether phases washed with brine, dried (sodium sulphate) and evaporated to give the title compound (1101) (0.209 g, 75% yield) as a yellow-brown oil; 1 H NMR (400 MHz, CDCi3) δ 8.50 - 8.47 (m, 2H), 4.21 (q, J = 7.1 Hz, 2H), 4.08 (s, 2H), 3.49 (s, 1 H), 1.26 (t, J = 7.1 Hz, 3H). LCMS Method C: rt 4.88 min; m/z 191.1 [M+HJ\
(d) tert-Butyl 4-(4-((4-((3-(2-ethoxy-2-oxoethyl)pyrazin-2-yl)ethynyl)-5- (trifluoromethyl)pyrimidin-2-yl)amino)phenyl)pipe 1 -carboxylate (! 102) iert-Butyl 4-(4-((4-chloro-5-(trifluoromethyl)pyrimidin-2-yl)amino)phenyl)piperidine-1- carboxylate (143) (140 mg, 0.306 mmol), ethyl 2-(3-ethynylpyrazin-2-yl)acetate (1101)
(64 mg, 0.34 mmol), b!s(triphenyiphosphine)palladium(il) chloride (11 mg, 5 mo!%) and DMF (2 mL) were loaded into a microwave tube and degassed with a nitrogen for ten minutes. Copper(i) iodide (3 mg, 5 mol%) and trlethylamine (0.192 mL, 1.38
mmoi) were added under nitrogen and the resulting mixture heated under microwave irradiation (120 °C/15 minutes). The cooled mixture was concentrated, and the residue evaporated onto silica gel. Chromatography (12 g silica cartridge, 0-100% ethyl acetate/petroleum benzine 40-60 °C) gave the title compound (1102) (95.5 mg, 52% yield) as a yellow solid; 1 H N R (400 MHz, /frDMSO) δ 10.47 (s, 1 H), 8.87 (d, J = 0.5 Hz, H), 8.76 - 8.71 (m, 2H), 7.63 (d, J = 8.6 Hz, 2H), 7.23 (d, J = 8.6 Hz, 2H), 4.15 - 3.99 (m, 6H), 2.80 (s, 2H), 2.70 - 2.63 (m, 1 H), 1.80 - 1.70 (m, 2H), 1.55 - 1.44 (m, 2H), 1.42 (s, 9H), 1.14 (t, J = 7.1 Hz, 3H). LCMS Method C: rt 6.73 min; m/z 633.1 [M+Naf , 555.0 [M-tBu+2H]+, 511.1 [M-Boc+2H]+; m/z 609.1 [M-H]\
(e) tert-Butyl 4~(4~( (4-(2-(3-(2-ethoxy-2-oxoethyl)pyrazin-2-yl)ethyl)-5-
(trifiuoromethy!}pyrimidin-2-yi)amino)phenyi)piperidine- 1-carbox (1103) terf-Butyl 4-(4-((4-((3-(2-ethoxy-2-oxoethyi)pyrazin-2-yi)ethynyi)-5- (trifluoromethyl)pyrimidin-2-yl)amino)phenyl)piperidine-1-carboxylate (1102) (187 mg, 0.306 mmoi) was dissolved in DMF (20 mL), triethylamine (0.1 mL) and a slurry of 10% Pd/C (0.100 g) in DMF (2 mL) was added. The mixture was purged with 3xvacuum/hydrogen cycles, and then stirred vigorously under a hydrogen
atmosphere. After 17 hours the mixture was filtered through celite, and the ceiite washed with ethyl acetate (200 mL). The combined filtrates were washed with 1 : 1 wafer: saturated brine (4x100 mL), dried (sodium sulphate) and evaporated to give the title compound (1103) (142 mg, 76%) as a yellow oil; 1 H NMR (400 MHz,d4- MeOD) δ 8.51 (d, J = 0.6 Hz, 1 H), 8.42 (d, J = 2.6 Hz, 1 H), 8.34 (d, J = 2.6 Hz, 1 H), 7.48 (d, J = 8.3 Hz, 2H), 7.20 - 7.13 (m, 2H), 4.21 (dd, J = 11.4, 1.8 Hz, 2H), 4.15 (q, J = 7.1 Hz, 2H), 3.97 (s, 2H), 3.37 (s, 4H), 2.87 (br s), 2.70 (tt, J = 11.9, 3.3 Hz, 1 H), 1.83 (d, J = 12.4 Hz, 2H), 1.59 (ddd, J = 25.6, 12.9, 4.4 Hz, 2H), 1.48 (s, 9H), 1.21 (t, J = 7.1 Hz, 3H). LCMS Method C: rt 6.76 min; m/z 615.1 [M+Hf, 559.1 [M-tBu+2H]+, 515.1 [M-Boc+2H]+.
(f) tert-Butyl 4-(4-((4-(2-(3-(2-amino-2-oxoethyl)pyrazin-2-yl)ethyl)-5- (trifluoromethyl)pyrimidin-2-yl)amino)phenyl)pipe 1 -carboxylaie (1104) iert-Butyl 4-(4-((4-(2-(3-(2-ethoxy-2-oxoethyi)pyrazin-2-yi)ethyi)-5- (trifluoromethyl)pyrimidin-2-yl)amino)phenyl)piperidine-1-carboxylate (1103) (142 mg, 0.231 mmoi) was dissolved in THF (10 mL) and methanol (5 mL) then a solution of lithium hydroxide monohydrate (48.0 mg, 1.16 mmoi) in water (2.5 mL) was added. The mixture was stirred at room temperature for 17 hours, and then concentrated.
The residue was evaporated twice from toluene then dissolved in DMF (20 mL) and ammonium chloride (82 mg, 1.2 mmol), HOBt (47 mg, 0.35 mmo!), PyBQP (181 mg, 0.347 mmol) and DIPEA (0.161 mL, 0.924 mmol) were added. After two hours the mixture was quenched with water (1 mL), concentrated and the residue partitioned between 1 :3 saturated brine:water (30 mL) and ethyl acetate (20 mL). The aqueous phase was washed with ethyl acetate (3x20 mL) then the combined ethyl acetate phases were washed with brine (50 mL), dried (sodium sulfate) and evaporated. The residue was chromatographed (12 g silica cartridge, 20-100% ethyl
acetate/petroleum benzine 40-60 °C then 100% ethyl acetate for 10 column volumes) to give the title compound {1104) (68.2 mg, 49% yield) as a colourless syrup; 1H N R (400 MHz, drMeOD) δ 8.40 (d, J = 0.6 Hz, H), 8.29 (d, J = 2.6 Hz, 1 H), 8.24 (d, J = 2.5 Hz, 1 H), 7.40 (d, J = 8,3 Hz, 2H), 7.09 - 7.01 (m, 2H), 4.10 (d, J = 13.3 Hz, 2H), 3.81 - 3.75 (m, 2H), 3.34 - 3.23 (m, 4H), 2.76 (br s), 2.58 (tt, J = 12.0, 3.4 Hz, 1 H), 1.71 (d, J = 12.2 Hz, 2H), 1.47 (ddd, J = 25.5, 12.8, 4.3 Hz, 2H), 1.38 (s, 9H). LCMS Method C: 6.17 min; m/z 586.1 [M-s-H]4, 530.1 [M-tBu+2H]+, 486.1 [M~Boc+2H]4; m/z 584.2 [M-H]\
(g) 2-(3-(2-(2-((4-(Piperidin-4-yl)phenyl)amino)-5-(trifluorom
yl)ethyl)pyrazin-2-yl)acetamide (26)
terf-Butyl 4-(4-((4-(2-(3-(2-amino-2-oxoethyl)pyrazin-2-yl)ethyl)-5-
(trifluoromethyl)pyrimidin-2-yl)amino)phenyl)piperidine-1-carboxylate (1104) (66 mg,
0.11 mmol), DCM (20 mL) and TFA (2 mL) were stirred at room temperature for 17 hours, then concentrated. The residue was suspended in 10% NaOH (10 mL) and brine (10 mL), and the mixture extracted with ethyl acetate (4x 20 mL). The combined ethyl acetate phases were washed with brine, dried (sodium sulphate) and evaporated to give the title compound (26) (47 mg, 85% yield) as a yellow solid; 1H NMR (400 MHz, d DMSO) δ 10.05 (s, 1 H), 8.64 (s, 1 H), 8.42 (d, J = 2.5 Hz, 1 H), 8.39 (d, J = 2.5 Hz, 1 H), 7.61 (s, 1 H), 7.54 (d, J = 6.7 Hz, 2H), 7.14 (d, J = 8.4 Hz, 2H), 7.08 (s, 1 H), 3,78 (s, 2H), 3,07 - 2.97 (m, 2H), 2.63 - 2.53 (m, overlaps with solvent), 1.67 (d, J = 12.1 Hz, 2H), 1.55 - 1.43 (m, 2H), 1.23 (s, 1 H). LCMS Method C: rt 4.36 min; m/z 486.1 [M+HJ4; m/z 484.1 [ -HJ".
Example 27: 2-(3-(2-(2-({4-{1 -IVlethyfp!pend!n-4-yl)pheny!)am!no)-5- {triffuorometh l}pyrimidsn-4-yl)ethyf)pyrazin-2-y!)acetam!de (27)

26 27
2-(3-(2-(2-((4-(Piperidin-4-yl)pb^
yi)ethyl)pyrazin-2-yl)acetamide (26) (42 mg, 0.087 mmol) was dissolved in methanol (4 mL) and 37% formaldehyde (26 μΙ_, 0.35 mmol) was added. After five minutes sodium tri(acetoxy)borohydride (92 mg, 0.44 mmol) was added and the mixture stirred for three hours. The solution was concentrated, and the residue suspended in 10% sodium hydroxide (1 mL). After five minutes brine (2 mL) was added, and the mixture extracted with ethyl acetate (5x3 mL). The combined ethyl acetate phases were washed with brine, dried (sodium sulphate) and evaporated to give the title compound (27) (34 mg, 77% yield) as an off-white solid; 1H N R (400 MHz, d6- DMSO) δ 10.07 (s, 1 H), 8.64 (s, 1 H), 8.42 (d, J = 2.6 Hz, 1 H), 8.38 (d, J = 2.6 Hz, 1 H), 7.61 (s, 1 H), 7.55 (d, J = 7.3 Hz, 2H), 7.15 (d, J = 8.5 Hz, 2H), 7.08 (s, 1 H), 4.09 (d, J = 4.7 Hz, 1 H), 3.77 (s, 2H), 3.17 (d, J = 4.3 Hz, 2H), 3.05 - 2.92 (m, 2H), 2.33 (s, 3H), 2.20 (s, 2H), 1.82 - 1.61 (m, 4H), LCMS Method C: rt 4,53 min; m/z 500.1 [M÷H]+; m/z 498.2 [M-H]".
Example 28: 2~(2~(2~(2~((2~IVlethy ~1 ,2,3,4-tetrahydroisoquinolin-6-yl)amino)-5- (trif!uorometh l)pyrimsdsn~4~yl)ethy!)phenyl)acetamsde (28)

2-(2-(2-(2-((1 ,2,3,4-tetrahydroisoquinoiin-6-yl)amino)-5-(trif!uoromethyi)pyrimidin-4- yi)ethyi)phenyl)acetamide (21) (25 mg, 0.055 mmol) was suspended in methanol (2 mL) and 37% aqueous formaldehyde (0.016 mL, 0.22 mmol) was added. The mixture was stirred for five minutes then sodium tri(acetoxy)borohydride (58 mg, 0.27 mmol) was added. After stirring for 2 hours at room temperature the mixture was
concentrated and the residue treated with 10% aqueous NaOH (1 mL) for five minutes. Brine (2 mL) was added, and the mixture was extracted with ethyl acetate (5x5 mL). The combined ethyl acetate extracts were washed with brine, dried (sodium sulphate) and evaporated to give the title compound (28) (22.8 mg, 88% yield) as an off-white solid; 1H N R (400 MHz, drMeOD) δ 8.54 (d, J = 0.5 Hz, 1 H), 7.52 (d, J = 1.9 Hz, 1 H), 7.47 (dd, J = 8.3, 2.2 Hz, 1 H), 7.28 - 7.16 (m, 4H), 7.04 (d, J = 8.3 Hz, 1 H), 3.86 (s, 2H), 3.60 (s, 2H), 3.21 - 3.11 (m, 2H), 3.11 - 3.02 (m, 2H), 2.96 (t, J = 6.0 Hz, 2H), 2.76 (t, J = 6.1 Hz, 2H), 2.46 (s, 3H). LC S Method C: rt 4.78 min; m/z 470.1 [M+H]+; m/z 468.1 [M-H]".
Example 29: 2-(4-(2-{2-{{4-{P^erid!n-4-yl)pheny!)amino)-5-(trifluoromethy!) pynmidin-4-y!}ethyl)pyrimidin-5-yt)aceta!Ti!de (29)

8114
(a) Diethyl 2-formylsuccinate (1105)
A mixture of diethyl succinate (26.1 g, 25.0 mL, 0.150 mol) and ethyl formate (11.1 g, 12.1 mL, 0.150 mol) was added drop wise over 1.5 hours to a stirred suspension of sodium (3.40 g, 0.150 mol) in diethyl ether (120 mL) at 0 °C under nitrogen. On completion of addition, stirring was continued at room temperature for 17 hours. Water (120 mL) was cautiously added to the resulting suspension and stirring continued until all the solids were dissolved. The layers were separated and the aqueous layer was washed with diethyl ether (100 mL). The aqueous layer was then acidified to pH 5 using 11 N HCI and extracted with diethyl ether (3x100 mL), the
ethereal extracts of the acidified layer were combined, dried (Na2S04) then evaporated to dryness under reduced pressure to give the title compound (1 /05) (18.5 g) as a yellow mobile liquid. The crude product was not purified further and was used directly in the following step.
(b) Ethyl 2-(4-oxo-2-thioxo-1,2,3,4-tetrahydropyrimidin-5-yl)acetate (1106)
Sodium ethoxide (6.105 g, 89.71 mmol) was added to absolute ethanoi (150 mL) under an atmosphere of nitrogen followed by diethyl-2-formylsuccinate (1105) (crude, 16.5 g) in absolute ethanoi (30 mL) and thiourea (6.829 g, 89.71 mmol). The reaction mixture was heated at reflux for 1 hour then cooled to room temperature at which stirring was continued for 16 hours. The volatiles were evaporated under reduced pressure to give a brown oily solid. Cold aqueous acetic acid solution (15%; 120 mL) was added and the resulting mixture was sonicated and then stirred at 0 °C until all the residue was in suspension. The resulting precipitate was collected by filtration. The filter cake was washed with water (100 mL) and dried to give the title compound [1106) (6.31 g, 19% yield over 2 steps) as an off-white solid; 1H N R (400 MHz, cfe- DMSO) δ 7.44 (s, 1 H), 4.05 (q, J = 7.1 Hz, 2H), 3,28 (s, 2H), 1.17 (t, J = 7,1 Hz, 3H). LCMS Method C: rt 2.92 min; m/z 213.0 [M-H]\ (c) Ethyl 2-(6-oxo- 1 , 6-dihydropyrimidin-5-yl)acetate (1107)
A Raney nickel suspension in water (Aldrich; 25 mL) was added to a stirred suspension of ethyl 2-(4-oxo-2-thioxo-1 ,2,3,4-tetrahydropyrimidin-5-yl)acetate (1106) (3.0 g, 14 mmol) in water (200 mL) at room temperature. The resulting suspension was heated at reflux for 20 hours and then stirred at room temperature for another 70 hours. The mixture was filtered through a thin pad of celite, the filter cake was washed with hot water (200 mL) and the combined filtrates were evaporated under reduced pressure to give a pale blue solid (-1.8 g). Dichioromethane (250 mL) was added and the resulting suspension was sonicated in an ultrasound bath until a fine suspended solid was obtained. The fine suspension was heated at reflux with vigorous stirring for 1 hour then filtered hot through a pad of celite, washing the filter cake with hot dichioromethane (200 mL). The filtrates were combined and evaporated to dryness under reduced pressure to give the title compound (1107) (1.01 g, 40% yield) as a white fluffy solid; 1H NMR (400 MHz, CDCI3) δ 8.12 (s, 1 H), 8.00 - 7.97 (m, 1 H), 4.19 (q, J = 7.1 Hz, 2H), 3.49 (s, 2H), 1.28 (t, J = 7.1 Hz, 3H). LCMS Method C: rt 2.05 min; m/z 183.1 [M+H]+.
(d) Ethyl 2-(4-chloropyrimidin-5-yl)acetate (1108)
To ethy! 2-(6-oxo-1 ,6-dihydropyrimidin-5-yl)acetate {1107) (0.868 g, 4.77 mmol) was added POCI3 (6 mL) under an atmosphere of nitrogen and the resulting mixture was heated to reflux for 5 minutes and then cooled to room temperature. The reaction was slowly added to water (300 mL), the aqueous solution was extracted with DC (3x100 mL), the combined organics were washed with brine ( 00 mL), dried
(MgS04), filtered and concentrated in vacuo to give the title compound {1108} (0,885 g, 93% yield) as a pale yellow oil; 1H NMR (400 MHz, CDCI3) δ 8.93 (s, 1 H), 8.61 (s, 1 H), 4.21 (q, J = 7.1 Hz, 2H), 3.76 (s, 2H), 1.28 (t, J = 7.1 Hz, 3H). LC S Method C: rt 5.09 min; m/z 201.1 , 203.1 [M+H]+.
(e) Ethyl 2-(4-((triethylsilyl)ethynyl)pyrimidin-5-yl)acetate (1109)
To a nitrogen de-gassed solution of ethyl 2-(4-chloropyrimidin-5-yi)acetate {1108) (0.823 g, 4.10 mmol) in dry DMF (15 mL) were added triethylamine (1.715 mL, 12.31 mmoi) followed by triphenylphosphine (0.124 g, 0.473 mmol), trans- dichlorobis(triphenyl-phosphine)pailadium(ll) (0.144 g, 0.205 mmoi), Cu(l)l (0,078 g, 0.410 mmol) and finally (triethyisilyi)acetylene (1.470 mL, 8.204 mmol). The reaction mixture was then heated under microwave irradiation at 120 °C for 25 minutes, concentrated in vacuo and purified by silica gel chromatography (isoiera Biofage, 40 g Si cartridge, 0-30% EtOAc in petroleum benzine 40-60 °C) to give the title compound (1109) (1.176 g, 94% yield) as a yellow-orange oil; 1H NMR (400 MHz, CDCI3) δ 9.08 (s, 1 H), 8.68 (s, 1 H), 4.18 (q, J = 7.1 Hz, 2H), 3.80 (s, 2H), 1.26 (t, J = 7.1 Hz, 3H), 1.10 - 1.01 (m, 9H), 0.77 - 0.67 (m, 6H) LCMS Method C: rt 6.64 min; m/z 305.1 [M+H]+.
(f) Ethyl 2-(4-ethynylpyrimidin-5-yl)acetate (1110)
To a solution of ethyl 2-(4-((triethylsilyl)ethynyl)pyrimidin-5-yl)acetate (1109) (1.174 g, 3.856 mmol) in dry THF (40 mL) under an atmosphere of nitrogen was added acetic acid (0.243 mL, 4.24 mmol) followed by TBAF (1.0 M in THF, 4.049 mL, 4.049 mmoi) dropwise at 0 °C. The reaction was stirred at this temperature for 5 minutes and was then poured into sat. aq. NaHC03 (100 mL) and DCM (100 mL). The layers were separated and the aqueous layer was extracted with DCM (2x100 mL). The combined organics were washed with water (100 mL), brine (100 mL), dried
( gSC ), filtered and concentrated in vacuo to give a brown oil. The crude material
was purified by silica gel chromatography (isolera Biotage, 40 g Si Cartridge, 0-60% EtOAc in petroleum benzine 40-80 °C) to give the title compound (1110) (0.397 g, 54% yield) as a brown oil; 1H N R (400 MHz, CDCI3) δ 9.11 (s, 1 H), 8.72 (s, 1 H), 4.20 (q, J = 7.1 Hz, 2H), 3,81 (s, 2H), 3.58 (s, 1 H), 1.27 (t, J = 7, 1 Hz, 3H). LCMS Method C: rt 4.78 min; m/z 191.1 [M+H]+.
(g) tert-Butyl 4-(4-((4-((5-(2-ethoxy-2-oxoethyl)pyrimidin-4-y!) ethynyl)-5-
(trifluoromethyl) pyrimidin-2-yl)amino)phenyl)piperidine- 1-carboxylate (1111}
To a nitrogen de-gassed solution of ethyl 2-(4-ethynylpyrimidin-5-yl)acetate (1110) (0.146 g, 0.788 rnmoi) and ferf-butyi 4-(4-((4-chioro-5-(trifluoromethyi)pyrimidin-2- yl)amino) phenyl)piperidine~1~carboxylate (145) (0.250 g, 0.547 mmol) in dry DMF (15 mL) were added triethylamine (0.305 mL, 2.19 mmo!), tri-fert-butylphosphonium tefrafluoroborate (0.016 g, 0.055 mmol), trans- dichlorobis(triphenylphosphine)palladium(ll) (0.019 g, 0.027 mmol) and Cu(i)l (0.010 g, 0.055 mmol). The reaction mixture was heated under microwave irradiation at 120 °C for 20 minutes and then concentrated in vacuo to give a brown gum. The crude material was purified by silica gel chromatography (Isolera Biotage, 40 g Si cartridge, 0-70% EtOAc in petroleum benzine 40-60 °C) to give the title compound (1111) (0.093 g, 27% yield) as a yellow solid; 1H NMR (400 Hz, d6-DMSO) δ 10.50 (s, 1 H), 9.23 (s, 1 H), 8.97 (s, 1 H), 8.88 (s, H), 7.83 (d, J = 8.6 Hz, 2H), 7.23 (d, J = 8.8 Hz, 2H), 4.14™ 4.04 (m, 4H), 3.96 (s, 2H), 2,81 (br s, 2H), 2.70 - 2.61 (m, 1 H), 1.75 (d, J = 11.9 Hz, 2H), 1.53 - 1.37 (m, 11 H), 1.14 (t, J = 7.1 Hz, 3H). LCMS Method C: rt 6.64 min; m/z 609.2 [M-H]\ (h) tert-Butyl 4-(4-((4-(2-(5-(2-ethoxy-2-oxoethyl)pyrimidin-4-yl)ethyl)-5-
(trif!uoromethy!) pyrimidin-2-yl)amino)phenyl)piperidine- 1 -carboxyiate (i 112) tert-Butyl 4-(4-((4-((5-(2-ethoxy-2-oxoethyl)pyrimidin-4-yl) ethynyi)-5-(trifluoromethyl) pyrimidin-2-yl)amino)phenyl)piperidine-1-carboxylate (1111) (0.108 g, 0.177 mmol) was dissolved in dry DMF (10 mL) under an atmosphere of nitrogen. Pd/C (10 wt. %; 0.040 g) in EtOAc (2 mL) was added to the solution and the atmosphere was changed to hydrogen gas (balloon). The reaction was sealed with balloon and stirred at room temperature for 22 hours. The catalyst was removed by filtration through Celite, which was washed with EtOAc (5x20 mL). The solvent was removed in vacuo to give a greyish semi-solid which was purified by silica gel chromatography (Isolera Biotage, 12 g Si Cartridge, 0-70% EtOAc in petroleum benzine 40-60 °C) to give the
title compound {1112) (0.096 g, 88% yield) as a pale yellow foam; H N R (400 MHz, de-DIViSO) δ 10.06 (s, 1 H), 9.00 (s, 1 H), 8.64 (s, 1 H), 8.59 (s, 1 H), 7.56 (d, J = 8.4 Hz, 2H), 7.16 (d, J = 8.6 Hz, 2H), 4.11 - 4.04 (m, 4H), 3.83 (s, 2H), 3.28 - 3.21 (m, 4H), 2.79 (br s, 2H), 2.68 - 2.58 (m, 1 H), 1.74 (d, J = 13.1 Hz, 2H), 1.52 - 1.38 (m, 11 H), 1.14 (t, J = 7.1 Hz, 3H). LCMS Method C: rt 6.65 min; m z 615.1 [M+Hf.
(i) Lithium 2-(4-(2-(2-((4-( 1-(tert-butoxycarbonyi)piperidin-4-yi)phenyi)amino)-5-
(triiiuoromethyi)pyrimidin-4-yi)ethyi)pyrimidin-5-yi)acetat^ (1113)
LiOH.H20 (0.020 g, 0.468 mmoi) was added to a solution of tert-butyl 4-(4-((4-(2-(5- (2-ethoxy-2-oxoethyl)pyrimidiri-4-yl)ethyl)-5-(trifluoromethyl) pyrimidin-2- yi)amino)phenyl) piperidine~1-carboxyiate (1112) (0.096 g, 0.156 mmoi) in THF (7 mL), water (1.5 mL) and methanol (1 mL) and the resulting mixture was allowed to stir at room temperature for 18 hours. The voiatiles were removed in vacuo and the residue was diluted with EtOAc (70 mL) and 2 M aqueous NaOH (80 mL). The layers were separated and the aqueous layer was extracted with EtOAc (2x70 mL), the combined organics were washed with brine (70 mL), dried (MgS04), filtered and concentrated under reduced pressure to give the title compound (1113) (0.096 g, quantitative) as a pale yellow solid. LCMS Method C: rt 6.19 min; rn/z 587.1 [M÷H]+ (j) tert-Butyl 4-(4-((4-(2-(5-(2-amino-2-oxoethyl)pyrimidin^
pyrimidin-2-yl)amino )phenyi)piperidine- 1 -carboxylate (1114)
Lithium 2-(4-(2-(2-((4-(1-(tert-butoxycarbonyl)piperidin-4-yl)phenyl)amino)-5-(trifluoro- methyl)pyrimidin-4-yl)ethyl)pyrimidin-5-yl)acetate {1113) (0.092 g, 0.16 mmoi) was dissolved in dry THF (7 mL) and dry DMF (1 mL) under an atmosphere of nitrogen. To the solution were added 1-hydroxybenzotriazole (0.023 g, 0.17 mmoi) and EDCI (0.033 g, 0.17 mmoi) and W,A -diisopropylethylamine (0.109 mL, 0.624 mmoi) and the reaction mixture was stirred at room temperature for 10 minutes. Ammonium carbonate (0.075 g, 0.78 mmoi) was added in one portion, and the reaction was stirred room temperature for 24 hours. More reagents were added, 1- hydroxybenzotriazole (0.011 g) and EDCI (0.016 g) and A ,A -diisopropylethylamine (0.055 mL) and ammonium carbonate (0.035 g) and the reaction was stirred for another 20 hours at 35 °C. The voiatiles were removed in vacuo and the residual solution was diluted with EtOAc (70 mL) and sat. aq. NaHC03 (70 mL). The layers were separated and the aqueous layer was extracted with EtOAc (2x70 mL), the organic layers were combined and washed with water (100 mL), brine (100 mL),
dried (MgS04), filtered and concentrated in vacuo to give an oily solid. The crude product was purified by silica gel chromatography (Isolera Biotage, 12 g Si Cartridge, 0-100% EtOAc in petroleum benzine 40-60 °C, then 0-10% methanol in EtOAc) to give the title compound (1114) (0.076 g, 83% yield) as a pale yellow foam; Ή NMR (400 MHz, rDMSO) δ 10.03 (s, 1 H), 8.94 (s, 1 H), 8.64 (s, 1 H), 8.53 (s, 1 H), 7.64 (br s, 1 H), 7.55 (d, J = 7.6 Hz, 2H), 7.16 (d, J = 8.6 Hz, 2H), 7.11 (br s, 1 H), 4.12 - 4.04 (m, 2H), 3.57 (s, 2H), 3.30 - 3.21 (m, 4H), 2.77 (br s, 2H), 2.68 - 2.58 (m, 1 H), 1.74 (d, J = 12.7 Hz, 2H), 1.53 - 1.37 (m, 11 H). LCMS Method C: rt 6.12 min; m/z 586.1 [M+Hf.
(k) 2-(4-(2-(2-((4-(Piperidin-4-yl)phenyl)amino)-5-(trifl^
yl)ethyl)pyrimidin-5-yl)acetamide (29)
ferf-Butyl 4-(4-((4-(2-(5-(2-amino-2-oxoethyl)pyrimidin-4-yl)ethyl)-5-(trifluoromethyi) pyrimidin-2-yl)amino)phenyl)piperidine-1-carboxylate (1114) (0.076 g, 0.13 mmol) was dissolved in DCM (5 mL) under an atmosphere of nitrogen. Trifluoroacetic acid (0.298 mL, 3,89 mmol) was added to the solution and the reaction was stirred at room temperature for 23 hours. Vo!atiles were removed in vacuo, EtOAc (70 mL) and 2 M aq. NaOH (70 mL) were added to the residue and the layers were separated. The aqueous layer was extracted with EtOAc (2 x 70 mL), the combined organics were washed with brine (2 x 50 mL), dried ( gS04), filtered and concentrated in vacuo to give a pale yellow gum. Methanol (-5 mL) and cyclohexane (- 15 mL) were added to the product, some of the volatiles were removed in vacuo (~ 50 %) which gave a yellow oil that separated from the solvent solution and was carefully transferred to a new flask with a pipette. The removed solution was concentrated in vacuo and then further dried under high-vacuum to give a pale yellow gum. Diethyl ether (5 mL) and methanol (1 mL) were added and the solution was concentrated in vacuo. The process was repeated twice with diethyl ether to give the title compound (29) (35 mg, 56% yield) as a pale yellow solid; H NMR (400 MHz, o MeOD) δ 8.91 (s, 1 H), 8.53 - 8.50 (m, 2H), 7.52 (d, J = 8.2 Hz, 2H), 7.21 - 7.16 (m, 2H), 3.70 (s, 2H), 3.42 - 3.34 (m, 4H), 3.27 - 3.20 (m, 2H), 2.84 (td, J = 12.5, 2.6 Hz, 2H), 2.70 (tt, J = 12.2, 3.9 Hz, 1 H), 1.89 (d, J = 13.7 Hz, 2H), 1.72 (ddd, J = 16.4, 12.9, 4.0 Hz, 2H). LCMS Method C: rt 4.46 min; m/z 486.1 [M+Hf.
Example 30: 2-(4-(2-{2-{{4-{1-IVlethylp!pend!n-4-y!)phenyl}am
meth !}pynmsdsn-4-yl)ethyf)pyrimidin-5-y!)acetam!de (30)
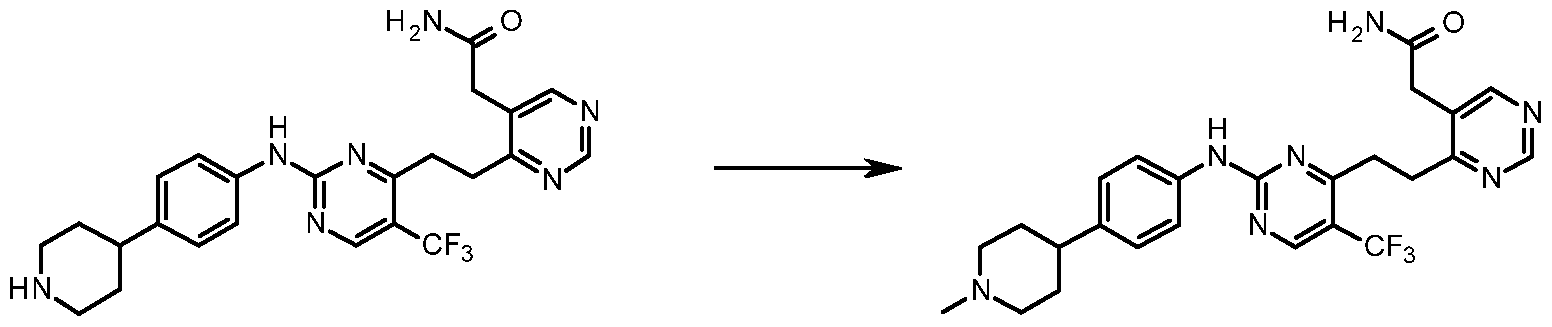
29 30 To a suspension of 2-(4-(2-(2-((4-(piperidin-4-y!)phenyl)arnino)-5-(tr!fluorornethy!) pyrimidin-4-yl)eihyl)pyrimidin-5-yl)aceiamide (29) (0.029 g, 0.060 mmoi) in anhydrous methanol (2 mL) was added a 37% aq. solution of formaldehyde (0.018 mL, 0.24 mmol) under an atmosphere of nitrogen, followed by sodium triacetoxyborohydride (0.063 g, 0.30 mmol). The reaction was stirred at room temperature for 2.5 hours. The volatiies were removed in vacuo and the residue was diluted with EtOAc (50 mL) and sat. aq. NaHC03 (30 mL). The layers were separated and the aqueous layer was extracted with EtOAc (2x30 mL), the combined organic layers were washed with water (40 mL), brine (40 mL), dried (MgS04), filtered and concentrated in vacuo to give a solid which was taken up in DCM (-10 mL) and methanol (-1 mL) and concentrated in vacuo. The process was repeated with only DCM twice. The resulting off-white solid was suspended in DCM (5 mL) and cyciohexane was added (-10 mL). The suspension was sonicated and the product was collected by filtration. The solid was washed with petroleum benzine 40-60 °C (5x10 mL), air-dried and subsequently dried under high-vacuum to give the title compound (30) (0.017 g, 57% yield) as an off-white solid; 1H NMR (400 MHz, d6-DMSO) δ 10.01 (s, 1 H), 8.94 (s, 1 H), 8.63 (s, 1 H), 8.53 (s, 1 H), 7.64 (s, 1 H), 7.54 (d, J = 8.0 Hz, 2H), 7.15 (d, J = 8.5 Hz, 2H), 7.11 (s, 1 H), 3.57 (s, 2H), 3.28 - 3.22 (m, 4H), 2.87 (d, J = 11.2 Hz, 2H), 2.46 - 2.36 (m, 1 H), 2.20 (s, 3H), 1.98 (t, J = 10.6 Hz, 2H), 1.76 - 1.58 (m, 4H). LCMS Method C: rt 4.52 min; m/z 500.1 [M+H]'\
Example 31 : 2-(2-(2-(2-({4-{1 -Ethy!pipend!n-4-y!)phenyl)amino)-5- (triffuoromethyl)pyrimidsn-4-yl)ethyf)pheny!}acetamide (31)
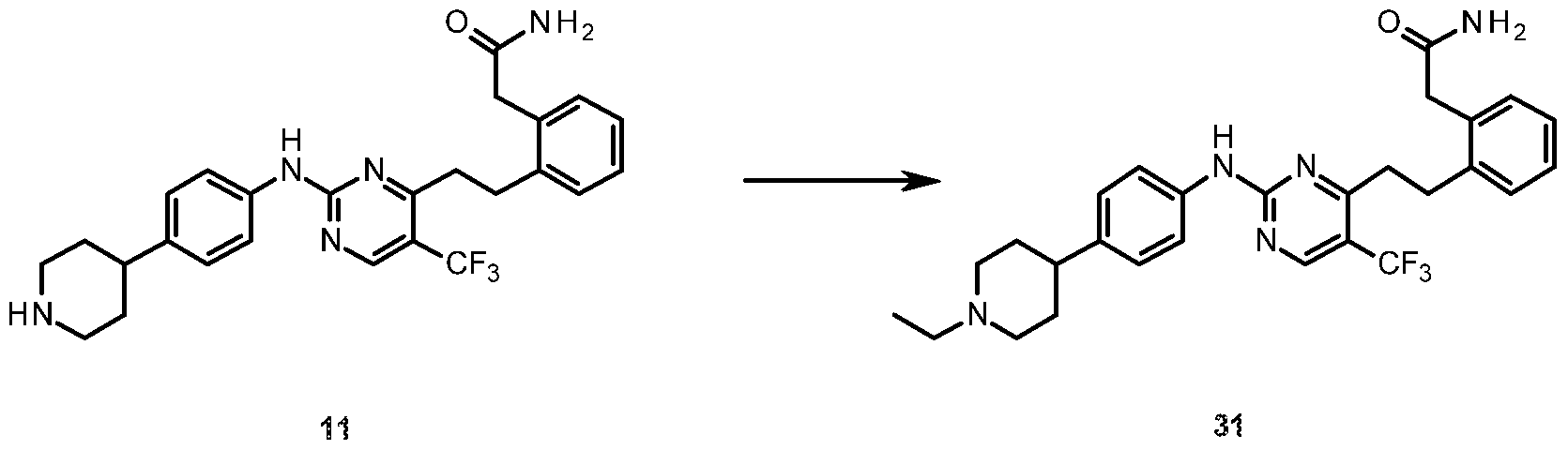
Aceia!dehyde (84.5 μί, 1.51 mmol) was added to a suspension of 2- (2- (2- (2- ((4- (piperidin-4-yl)phenyl)amino)-5-(trifluorome
( 11) (91.0 mg, 0.188 mmol) in anhydrous methanol (10 mL) under an atmosphere of nitrogen. Sodium triacetoxyborohydride (0.838 g, 3.01 mmol) was then added in one portion and the reaction was stirred at room temperature for 24 hours. The volatiles were removed in vacuo and the residue was diluted with EtOAc (35 mL) and sat. aq. NaHC03 (40 mL). The layers were separated and the aqueous layer was extracted with EtOAc (2x35 mL), the combined organic layers were washed with water (30 mL), brine (30 mL) and dried over Na2S04. The solvent was removed under reduced pressure and the residue dissolved in DCM (5 mL) and MeOH (2 mL). Cyclohexane (20 mL) was added and the solvent removed in vacuo to afford a tan solid which was purified by column chromatography on silica gel (0-100% EtOAc in petroleum benzine 40-60 °C; then 0-75% MeOH in EtOAc) to yield a white solid. The solid was suspended in DCM (5 mL) and cyclohexane (20 mL) and filtered to afford the title compound (31) (40.7 mg, 42%) as an off-white solid; 1 H NMR (400 MHz, cfe-DMSO) δ 10.13 (s, 1 H), 8.65 (s, 1 H), 7.68 (d, J = 8.6 Hz, 2H), 7.43 (s, 1 H), 7.26 - 7.12 (m, 6H), 6.92 (s, 1 H), 3.50 (s, 2H), 3.14 - 3.06 (m, 2H), 3.05 - 2.92 (m, 4H), 2.43 (tt, J = 11.7, 3.7 Hz, 1 H), 2.33 (q, J = 7.2 Hz, 2H), 1.93 (td, J = 11.5, 2.1 Hz, 2H), 1.73 (dd, J = 11.8, 2.0 Hz, 2H), 1.62 (ddd, J = 24.8, 12.4, 3.6 Hz, 2H), 1.01 (t, J = 7.2 Hz, 3H); LCMS Method C: rt 4.90 min; m/z 512 [M+H]+.
Example 32: 2-(2-(2-(2-({4-{Piperid!n-4-yl)pheny!)am!no)-5- {t


32
(a) (1-Oxypyridin-3-yl)acetonitrile (1115)
30% Hydrogen peroxide (12 mL) was added to a solution of 3-pyridylacetonitrile (7.50 g, 63.5 mmol) in acetic acid (40 mL) and the mixture heated at 95 °C for 20 hours. The reaction mixture was then cooled and stirred at room temperature for 72 hours. Water (35 mL) was then added and the solution concentrated under reduced pressure. Wafer (2x100 mL) was added to the residue and solution concentrated under reduced pressure. Residual water was removed azeotropically using toluene (2x100 mL) to yield the title compound (1115) (8.3 g, 97%) as a pale yellow solid which was used without further purification.
(b) (2-Chloropyridin-3-yl)acetonitrile (116)
(1-Oxypyridin-3-yl)acetonitrile (1115) (4.00 g, 29.8 mmol) was added slowly to a stirred solution of POCI3 (50 mL). The mixture was heated to 80 °C in 5 - 7 °C increments every 10 -15 minutes. The reaction was then heated at reflux for 3 hours. Excess POCI3 was removed by distillation and the brown residue carefully poured on to cold water (200 mL). A saturated solution NaHC03 (300 mL) was then added carefully. Solid NaHC03 was added in portions to the aqueous mixture until the evolution of gas ceased. The aqueous layer was separated in to two portions (250 mL each) and each portion was extracted with EtOAc (3x100 mL). The combined organic layers were washed with brine (200 mL) and dried over Na2S04. The solvent was removed and the residue purified by column chromatography on silica gel (0- 100% EtOAc in petroleum benzine) to afford a mixture of two isomeric compounds. The mixture was re-purified by column chromatography on silica gel (0-40% diethyl ether in petroleum benzine 40-80 °C) to afford the title compound (1116) (0.932 g, 20%) as a white solid; 1 H NMR (400 MHz, CDCI3) 6 8.41 (dd, J = 4.8, 1.8 Hz, 1 H),
7.90 (ddt, J = 7,6, 1.7, 0.7 Hz, 1 H), 7.34 (dd, J = 7,6, 4.8 Hz, 1 H), 3.87 (s, 2H), LCMS Method C: rt 4,50 min; m/z 153 [M+H]+.
(c) (2-Chioropyridin-3~y!)acetic acid (1117)
A solution of 15% w/w NaOH (15 mL) was added to (2-chioropyridin-3-yl)acetonitrile (1116) (0.932 g, 6.11 mmol). The mixture was heated at reflux for 35 minutes then cooled to room temperature. The mixture was further cooled to 0 °C and then acidified with cone. HCi (ca 5 mL) to pH 1. The suspension was left to stand for 1 hour in an ice bath. The precipitate was filtered and washed with cold propan-2~ol (3x15 mL) to yield the title compound (1117) (1.05 g, 100%) as an off-white solid; 1H NMR (400 MHz, cfe-DMSO) δ 12.63 (s, 1 H), 8.32 (dd, J = 4.8, 1.9 Hz, 1 H), 7.86 (dd, J = 7.5, 1.9 Hz, 1 H), 7.41 (dd, J = 7.5, 4.8 Hz, 1 H), 3.75 (s, 2H). LCMS Method C: rt 4.06 min; m/z 172 [M+H]+. (d) Methyl 2-(2-chloropyridin-3-yl)acetate (1118)
Acetyl chloride (0.651 mL, 9.18 mmol) was added to a suspension of (2- chloropyridin-3-yi)acetic acid (1117) (1.048 g, 6.108 mmol) in MeOH (30 mL). The mixture was heated at reflux for 20 hours. The voiatiles were removed in vacuo and the residue partitioned between DCM (100 mL) and sat. NaHC03 (100 mL). The layers were separated and the aqueous layer extracted with DCM (2x100 mL). The
combined organic layers were washed with brine (100 mL), dried (Na2S04) and the solvent removed under reduced pressure to yield an oil which was purified by column chromatography on silica gel (0-40% EtOAc in petroleum benzine 40-60 °C) to afford the title compound (1118) (0.863 g, 76%) as a pale yellow oil; 1H NMR (400 MHz, d6~ DMSO) δ 8.34 (dd, J = 4.8, 1.9 Hz, 1 H), 7.83 (dd, J = 7.5, 1.9 Hz, 1 H), 7.43 (dd, J = 7.5, 4.8 Hz, 1 H), 3.86 (s, 2H), 3.65 (s, 3H). LC S Method C: rt 5.04 min; m/z 186 [M+H]+.
(e) Methyi 2-(2-((triethylsilyl)ethynyl)pyridin-3-yl)acetate (1119)
A solution of triethylsiiyi acetylene (0.579 mL, 3.23 mmol) in degassed DMF (3 mL) and triethyiamine (0.901 mL, 6.47 mmol) were added to a mixture of methyl 2-(2- chloropyridin-3-y!)acetate (1118) (0.200 g, 1.08 mmol), Pd(PPh3)2Ci2 (75.6 mg, 0.108 mmol), Cu(l)l (30.8 mg, 0.162 mmol) and triphenylphosphine (42.4 mg, 0.162 mmol) in degassed DMF (4 mL) and the resulting mixture was heated at 90 °C for 20 hours. The cooled mixture was diluted with EtOAc and passed through a plug of celite, washing with ethyl acetate (100 mL), Water (75 mL) was added to the filtrate and the layers separated. The aqueous layer was extracted with EtOAc (2x75 mL). The combined organic extracts were washed with brine (100 mL) and dried over Na2S04. After filtration the solvent was removed under reduced pressure to give a dark brown residue. The residue was purified by column chromatography on silica gel (0-50% EtOAc in cyclohexane) to yield the title compound (1119) (0.353 g) as a brown oil which was used without further purification; 1 H NMR (400 MHz, CDCI3) δ 8.50 (dd, J = 4.8, 1.6 Hz, 1 H), 7.62 (dd, J = 7.8, 1.6 Hz, 1 H), 7.22 (dd, J = 7.8, 4.8 Hz, H), 3.86 (s, 2H), 3.70 (s, 3H), 1.05 (t, J = 7.9 Hz, 9H), 0.75 - 0.67 (m, 6H). LCMS Method C: rt 6.55 min; m/z 290 [M+H .
(f) Methyl 2-(2-ethynylpyridin-3-yl)acetate (1120)
A solution of TBAF (1 M solution in THF; 0.207 mL, 0.207 mmol) was added to a solution of methyl 2-(2-((triethylsilyl)ethynyl)pyridin-3-yl)acetate (1119) (50.0 mg, 0.173 mmol) in THF (2 mL) at 0 °C. The reaction was stirred for 2 minutes at 0 °C then diluted with saturated NaHC03 (20 mL). EtOAc (20 mL) was then added and the layers separated. The aqueous layer was extracted with EtOAc (2x20 mL) then the combined organic layers were washed with water 20 mL), brine (20 mL) and dried over Na2S04. The solvent was removed in vacuo to yield a brown oily residue. The oil was purified using column chromatography on silica gel (0-55% EtOAc in
cyclohexane) to afford the title compound (1120) (25.9 mg, 86%) as an orange oil; 1H NMR (400 MHz, CDCI3) δ 8.52 (dd, J = 4.8, 1.6 Hz, 1 H), 7.65 (dd, J = 7.9, .6 Hz, 1 H), 7.29 - 7.24 (m, peak obscured by solvent), 3.87 (s, 2H), 3.72 (s, 3H), 3.35 (s, 1 H). LC S Method C: rt 4.74 min; m/z 176 [M+H]+.
(g) tert-Butyl 4-(4-((4-((3-(2-methoxy-2-oxoethyl)pyridin-2-yl)ethynyl)-5^
(trifluoromethyl)pyrimidin-2-yl)amino)phenyl)piperidine- 1^ (1121)
A solution of methyl 2-(2-ethynylpyridin-3-yl)acetate (1120) (43.6 mg, 0,249 mmol) in dimethylformamide (2 mL) and triethylamine (107 μί, 0.765 mmol) were added to a mixture of ferf-butyi 4-(4-((4-chloro-5-(trifluoromethyl)pyrimidin-2-yl)amino) phenyl)piperidine-1-carboxylate {145) (87.4 mg, 0.191 mmol), Pd(PPh3)2CI2 (13 mg, 0.019 mmol), Cu(l)l (5.5 mg, 0.029 mmol) and triphenylphosphine (7,5 mg, 0.017 mmol) in dimethylformamide (2 mL). The reaction mixture was heated under microwave irradiation at 120 °C for 10 minutes. The cooled mixture was diluted with EtOAc and passed through a plug of celite, washing with ethyl acetate (100 mL). The solvent was removed under reduced pressure and the residue partitioned between EtOAc (70 mL) and water (50 mL). The layers separated and the aqueous layer extracted with EtOAc (2x50 mL). The combined organic extracts were washed with brine (70 mL) and dried over Na2S04. After filtration the solvent was removed under reduced pressure to give a dark brown residue. The residue was purified by column chromatography on silica gel (0-40% EtOAc in petroleum benzine 40-60 °C) to yield the title compound (1121) (81.5 mg, 72%) as a brown viscous oil which was used without further purification. (h) tert-Butyl 4-(4-((4-(2-(3-(2-methoxy-2-oxoethyl)pyridin-2-yl)ethyl)-5-
(trifluoromethyl)pyrimidin -2-yl)amino)phenyl)piperidine- 1 -carboxylate (1122)
A solution of ferf-butyi 4-(4-((4-((3-(2-methoxy-2-oxoethyl)pyridin-2-yl)ethynyl)-5- (trifluoromethyl)pyrimidin-2-yl)amino)phenyl)piperidine-1-carboxylate (1121) (81.5 mg, 0.137 mmol) in DMF (10 mL) was added to a solution of 10% Pd/C (170 mg) in DMF (7 mL). The reaction was stirred at room temperature for 24 hours under an atmosphere of hydrogen. The reaction was filtered through a pad of celite, washimg with EtOAc (100 mL). The solvent was removed in vacuo to afford an oil which was purified on by column chromatography on silica gel (0-40% EtOAc in petroleum benzine 40-60 °C) to yield the title compound (1122) (61.4 mg, 75%) as a viscous oil; 1H NMR (400 MHz, CDCI3) 6 8.51 (s, 1 H), 8.46 (dd, J = 4.8, 1.7 Hz, 1 H), 7.56 - 7.50
(m, 3H), 7.49 (s, 1 H), 7.19 - 7.14 (m, 2H), 7.12 (dd, J = 7.7, 4.8 Hz, 1 H), 4.23 (bs, 2H), 3.72 (s, 2H), 3.70 (s, 3H), 3.40 - 3.33 (m, 2H), 3.32 - 3.23 (m, 2H), 2.80 (t, J = 12.1 Hz, 2H), 2.62 (tt, J = 12.1 , 3.5 Hz, 1 H), 1.81 (d, J = 12.9 Hz, 2H), 1.65 - 1.55 (m, 2H), 1.48 (s, 9H). LC S Method C: rt 6.01 min; m/z 622 [M+Na]+, 600 [M+H]+, 544 [M-iButyi+2H]+.
(i) Lithium 2-(2-(2-(2-((4-( 1-(tert-butoxycarbonyl)piperidin-4-yl)phenyl)amino)-5- (thfluoromethyl)pyrimidin-4-yl)ethyl)pyridin-3-yl)acetate (1123)
LiOH.H20 (19.1 mg, 0.455 mmol) was added to a solution of iert-butyl 4-(4-((4-(2-(3- (2-methoxy-2-oxoethyl)pyridin-2-yl)ethyl)-5-(trifluoromethyl)pyrimidin-2- yi)amino)phenyl)piperid!ne-1-carboxylate (1122) (90.9 mg, 0.152 mmol) in THF (7 mL), water (1 ,5 mL) and methanol (1 mL). The resulting mixture was allowed to stir at room temperature for 20 hours. The volatiies were removed in vacuo and the residue was diluted with EtOAc (50 mL) and sat. aq. NaHC03 (50 mL). The layers were separated and the aqueous layer was extracted with EtOAc (2x50 mL), the organic layers were combined, washed with brine (50 mL), dried over Na2S0 , filtered and concentrated under reduced pressure to give the title compound {1123) (81.3 mg, 91 %) as a pale yellow viscous oil. (j) tert-Butyl 4-(4-((4-(2-(3-(2-amino-2-oxoethyl)pyridin-2-yl)ethyl)-5-
(trifluoromethyl)pyrimidin-2-yl)amino)phenyl)piperidine- 1 -carboxylate (1124)
1- Hydroxybenzofriazoie (20.4 mg, 0.151 mmol), EDCi (29.0 mg, 0.151 mmol) and A/,A -diisopropylethylamine (0.192 mL, 1.10 mmol) were added to a solution of lithium
2- (2-(2-(2-((4-(1-(iert-butoxycarbonyl)piperidin-4-yl)phenyl)amino)-5- (trifluoromethyl)pyrimidin-4-yl)ethyl)pyridin-3-yl)acetate (1123) (81.3 mg, 0.137 mmol) in dry THF (6 mL) and dry DMF (1 mL) under an atmosphere of nitrogen. Ammonium carbonate (106 mg, 1.10 mmol) was added in one portion to the stirred reaction mixture after 10 minutes. The reaction was left to stir at room temperature for 8 hours. The volatiies were removed in vacuo and the residue was diluted with EtOAc (50 mL) and washed with saturated NaHC03 (50 mL). The aqueous layer was extracted with EtOAc (2x50 mL). The combined organic layers were washed with brine (50 mL) and dried over Na2S04. After filtration the solvent was removed in vacuo to afford a pale yellow oil. The crude material was purified by column chromatography on silica gel (0-100% EtOAc in petroleum benzine 40-60 °C) to afford the title compound (1124) (66.3 mg, 83%) as a white semi-solid; 1 H N R (400
MHz, CDCI3) δ 8.50 (d, J = 0.4 Hz, 1 H), 8.49 (dd, J = 4.8, .7 Hz, 1 H), 7.62 (s, 1 H), 7.54 (dd, J = 7.7, 1.7 Hz, 1 H), 7.51 (d, J = 8.7 Hz, 2H), 7.19 - 7.13 (m, 3H), 5.40 - 5.35 (m, 2H), 4.23 (s, 2H), 3.68 (s, 2H), 3.41 - 3.35 (m, 2H), 3.32 - 3.24 (m, 2H), 2.80 (t, J = 12.6 Hz, 2H), 2.63 (tt, J = 12.0, 3.4 Hz, 1 H), 1.81 (d, J = 12,5 Hz, 2H), 1.65 - 1.57 (m, peaks obscured by solvent), 1.48 (s, 9H). LCMS Method C: rt 5.25 min; m/z 007 [M+Naf , 585 [M+H]+, 529 [M-iButyl÷2H]+.
(k) 2-(2-(2-(2-((4-(Piperidin-4-yl)phenyl)amino)-5-(trifluo
yl)ethyl)pyridin-3-y!)acetamide (32)
Tnfiuoroacetic acid (0.337 rnL, 4.40 mmol) was added to a solution feri-butyl 4-(4-((4- (2-(3-(2-amino-2-oxoethyl)pyridin-2-yl)ethyl)-5-(trifluoromethyl)pyrimidin-2- yl)amino)phenyl)piperidine-1-carboxylate (1124) (64.3 mg, 0.110 mmol) in dry DCM (15 mL) under an atmosphere of nitrogen and the reaction was stirred at room temperature for 23 hours. The volatiies were removed in vacuo and the residue partitioned between EtOAc (40 mL) and 2 M NaOH (50 mL). The layers were separated and the aqueous layer was extracted with EtOAc (2x30 mL). The combined organic layers were washed with water (40 mL), brine (40 mL) and dried over Na2S04. After filtration the solvent was removed under reduced pressure to give a yellow solid which was suspended in DCM (5 mL) and cyciohexane (15 mL). The precipitate was filtered to afford the title compound (32) (35.5 mg, 53%) as yellow solid; 1H NM (400 MHz, cfe-DMSO) «5 10.05 (s, 1 H), 8.64 (s, 1 H), 8.36 (dd, J = 4.8, 1.7 Hz, 1 H), 7.62 - 7.55 (m, 3H), 7.53 (s, 1 H), 7.18 (dd, J = 7.6, 4.8 Hz, 1 H), 7.15 (d, J = 8.6 Hz, 2H), 7.00 (s, 1 H), 3.53 (s, 2H), 3.24 (s, 4H), 3.02 (d, J = 11.9 Hz, 2H), 2.61 - 2.52 (m, peaks obscured by solvent), 1.67 (d, J = 11.3 Hz, 2H), 1.49 (qd, J = 12.2, 3.8 Hz, 2H). LCMS Method C: rt 1 ,50, 1.59 min; m/z 485 [M+Hf .
Example 33: 2-(2-(2-(2-({4-{1 -IVlethyfp!pend!n-4-yl)pheny!)am!no)-5-triffuoromethyl}pyrimidsn-4-yl)ethyf)pyndsn-3-y!)acetamide (33)

32 33 Formaldehyde (37 wt % in H20; 16.5 pL, 0.222 mmo!) was added to a suspension of 2-(2-(2-(2-((4-(piperidin-4-yl)phenyl)amino)-5-(trifluoromethyl)pyrimidin-4- yl)ethyl)pyridin-3-yl)acetamide (32) (22 mg, 0.044 mmol) in anhydrous methanol (5 mL) under an atmosphere of nitrogen. Sodium triacetoxyborohydride (94.0 mg, 0.444 mmol) was then added in one portion and the reaction was stirred at room temperature for 2 hours. The voiatiles were removed in vacuo and the residue was diluted with EtOAc (25 mL) and sat. aq. NaHC03 (25 mL). The layers were separated and the aqueous layer was extracted with EtOAc (2x25 mL), the combined organic layers were washed with water (25 mL), brine (25 mL) and dried over Na2S04. The solvent was removed under reduced pressure to yield a white solid. The solid was suspended in DCM (5 mL) and cyciohexane (20 mL) and filtered to afford the title compound {33} (16 mg, 72%) as an off-white solid; 1H NMR (400 MHz, cf6-DMSO) δ 10.06 (s, 1 H), 8.64 (s, 1 H), 8.36 (dd, J = 4.8, 1.7 Hz, 1 H), 7.59 (dd, J = 7.7, 1.2 Hz, 3H), 7.53 (s, 1 H), 7.20 - 7.13 (m, 3H), 7.00 (s, 1 H), 3.53 (s, 2H), 3.24 (s, 4H), 2.91 (d, J = 10.6 Hz, 2H), 2.47 - 2.39 (m, 1 H), 2.24 (s, 3H), 2.12 - 1.99 (m, 2H), 1 ,78 - 1.59 (m, 4H). LCMS Method C: rt 1.51 , 1.579 min; m/z 499 [M+H]+.
Example 34: 2-(2-(2-({4-{1 -IVlethylp!pend!n-4-yl)pheny!}am!no)-5- (triffuorometh l}pyrimidsn-4-y!)ethyl)isonicotinani!de (34)

34
(a) Methyl 2-((trimethylsilyl)ethynyl)isonicotinate (1125)
Methyl 2-bromonicotinate (1.00 g, 4.63 mmol), PdCi2(PPh3)2 (162 mg, 0.231 mmol) triphenyi phosphine (60.7 mg, 0.231 mmol,) and Cu(l)l (44.1 mg, 0.231 mmol) were placed into oven dried reaction flask under nitrogen then TMS-acety!ene (785 μΙ_, 5.55 mmol), dry, degassed THF (5 mL) and triethyiamine (5 mL) were added. The resulting mixture was stirred at room temperature for 16 hours then evaporated under reduced pressure to give a black residue which was adsorbed onto silica gel.
Chromatography (Si02, 0-20% ethyl acetate/petroleum benzine 40-60 °C) gave the title compound (H25) (708.6 mg, 66% yield) as a dark coloured liquid; 1H NMR (400 MHz, CDCI3) δ 8.75 - 8.61 (m, 1 H), 7.98 (dd, J = 1.0, 0.4 Hz, 1 H), 7.74 (dd, J = 5.1 ,
1.6 Hz, 1 H), 3.94 (s, 3H), 0.26 (s, 9H). LC S eihod C: r! 6.38 min; m/z 234.1
(b) Methyl 2-ethynylisonicotinate (1126)
To a solution of methyl 2-((trimethylsilyl)ethynyl)isonicotinate {1125) (8.00 g, 34.2 mmol) in THF (150 mL) was added TBAF (1.0 M in THF) (51.4 mL, 51.4 mmol) at 0 °C. The resulting solution was allowed to warm to room temperature at which stirring was continued for 1 hour. The reaction mixture was diluted with ethyl acetate (50 mL) and washed with 10% NaHC03 (50 mL). The organic layer was dried ( gS04) and evaporated under reduced pressure to give a dark brown/black residue. The residue was adsorbed onto silica gel and purified by chromatography (Si02, 0-20% ethyl acetate/petroleum benzine 40-60 °C) to give the title compound (1126) (4.5 g, 81 %) as a yellow solid; 1H NMR (400 MHz, CDCI3) δ 8.73 (dd, J = 5.1 , 0.9 Hz, 1 H), 8.01 (dd, J = 1.5, 0.9 Hz, 1 H), 7.80 (dd, J = 5.1 , 1.6 Hz, 1 H), 3.95 (s, 3H), 3.22 (s, 1 H).
(c) (E)-Methyl 2-(2-(4A,5,5-tetramethyl-1,3!2-dioxaborolan-2-yl)
(1127)
A solution of methyl 2-ethynyiisonicofinate [1126) (100 mg, 0.621 mmol), Cu(l)CI (1.84 mg, 0.0186 mmol), NaOfBu (3.6 mg, 0.037 mmol), bispinacolatodiboron (189 mg, 3.74 mmol), Xantphos (10.7 mg, 0.0937 mmol) and methanol (40 mg, 1.2 mmol) in THF (5 mL) was stirred at room temperature for 4 hours under nitrogen. The crude reaction mixture was adsorbed onto silica gel and solvents removed by evaporation under reduced pressure. Purification by chromatography (Si02, 0-20% ethyl acetate/petroleum benzine 40-60 °C) gave the title compound (1127) (141.3 mg, 79%) as a yellow liquid; 1 H NMR (400 MHz, CDCI3) δ 8.70 (dd, J = 5.0, 0.6 Hz, 1 H), 7.93 - 7.90 (m, 1 H), 7.67 (dt, J = 4.4, 2.2 Hz, 1 H), 7.46 (d, J = 18.3 Hz, 1 H), 6.67 (d, J = 18.3 Hz, 1 H), 3.91 (s, 3H), 1.27 (s, 12H).
(d) (E) -Methyl 2-(2-(2-((4-(1-(tert-butoxycarbonyl)piperidin-4-yl)phenyl)amm^
(trifluoromethyl)pyrimidin-4-yl)vinyl)isonicotinate (1128)
An aqueous 2.0 M solution of Na2C03 (0.3 mL) was added to a solution of ferf-butyl 4-(4-((4-chloro-5-(trifluoromethyl)pyrimidin-2-yl)amino)phenyl)piperidine-1- carboxylate (145) (100 mg, 0.103 mmol), (£)-methyl 2-(2-(4,4,5,5-tetramethyl-1 ,3,2- dioxaborolan-2-yl)vinyl)isonicotinate (1127) (76 mg, 0.26 mmol), Pd(PPh3)4 (75 mg, 0.66 mmol), LiCi (28 mg, 0.66 mmol) in 1 ,4-dioxane (5 mL) and the resulting mixture
was stirred under nitrogen at 90 °C for 18 hours. The crude mixture was evaporated under reduced pressure and adsorbed onto silica gel. Chromatography (Si02> 0-50% ethyl acetate/petroleum benzine 40-60 °C) gave the title compound (1128) (110 mg, 86%) as a yellow solid; 1H NMR (400 MHz, CDCI3) δ 8.84 (d, J = 4,9 Hz, 1 H), 8.64 (s, 1 H), 8.14 (d, J = 15.1 Hz, 1 H), 8.02 (s, 1 H), 7.88 (dd, J = 15.1 , 1.6 Hz, 1 H), 7.80 (dd, J = 4.9, 1.5 Hz, 1 H), 7.60 (d, J = 8.5 Hz, 2H), 7.36 (s, 1 H), 7.25 (m, 1 H), 4.33 - 4.19 (m, 2H), 4.00 (s, 3H), 2.82 (m, 2H), 2.67 (m, 1 H), 1.85 (d, J = 12.7 Hz, 2H), 1.64 (dd, J = 12.5, 4.0 Hz, 2H), 1.59 (s, 9H). LC S Method C: rt 6.93 min; m/z 584.21 [M+1]+, 582.1 [M-1]-, 528.1 [M-tBu+2]+.
(e) Methyl 2~(2~(2~((4~( 1 -(tert-butoxycarbonyl)piperidin-4-yl)phenyl)amino)-5- (trifluoromethyl)pyrimidin-4-yl)ethyl)isonicotinate (1129)
A suspension of (E)-methyl 2-(2-(2-((4-(1-(feri-butoxycarbonyl)piperidin-4- yl)phenyl)amino)-5-(trifluoromethyl)pyrimidin-4-yl)vinyl)isonicotinate (1128) (110 mg, 0.188 mmol), and 10% Pd/C (20 mg) in MeOH (10 mL) was stirred under an atmosphere of hydrogen at atmospheric pressure for 16 hours. The resulting mixture was filtered and the filtrate evaporated under reduced pressure to give the title compound (1129) (70 mg, 63%) as a yellow liquid; 1 H NMR (400 MHz, CDCI3) δ 8.68 (dd, J = 5.1 , 0.7 Hz, 1 H), 8.51 (s, 1 H), 7.76 (s, 1 H), 7.66 (dd, J = 5.1 , 1.5 Hz, 1 H), 7.54 - 7.47 (m, 3H), 7.18 (t, J = 5.5 Hz, 2H), 4.22 (s, 2H), 3.93 (d, J = 3.9 Hz, 3H), 3.43 - 3.35 (m, 2H), 3.34 - 3.26 (m, 2H), 2.80 (t, J = 12.1 Hz, 2H), 2.63 (d, J = 3,5 Hz, 1 H), 1.82 (d, J = 12.3 Hz, 2H), 1.67 - 1.54 (m, 2H), 1.48 (d, J = 7.1 Hz, 3H). LCMS Method C: rt 6.75 min; m/z 586.1 [M+1f, 584.2 [M-1]". (f) tert-Butyl 4-(4-((4-(2-(4-carbamoylpyridin-2-yl)ethyl)-5-(trifluo
yl)amino)phenyl)piperidine-1-carboxylate (1130)
A solution of methyl 2-(2-(2-((4-(1-(tert-butoxycarbonyl)piperidin-4-yl)phenyl)amino)- 5-(trifluoromethyl)pyrimidin-4-yl)ethyl)isonicotinate (1129) (70.0 mg, 0.120 mmol), and LiOH.H20 (15 mg, 0.36 mmol) in THF (5 mL), water (1 mL) and MeOH (0.5 mL) was stirred at room temperature for 4 hours. The volatiles were evaporated under reduced pressure to give a yellow solid which was dissolved in dry DMF (4 mL). HATU (133 mg, 0.350 mmol), DIPEA (60 μί, 0.034 mmol) and ammonium chloride (187 mg, 3.50 mmol) were added and the resulting mixture was stirred at room temperature overnight. The volatiles were evaporated under reduced pressure and the residue diluted with ethyl acetate. The resulting solution was washed with 10%
aqueous NaHC03, then the organic layer was dried (MgS04) and voiatiies removed by evaporation under reduced pressure. The residue was adsorbed onto silica gel and purified by chromatography (Si02, 0-100% ethyl acetate/petroleum benzine 40- 60 °C) to give the title compound (1130) (50 mg, 50%) as a colourless solid; 1H N (400 MHz, CDC ) δ 8.67 (dd, J = 5.1 , 0.7 Hz, 1 H), 8.51 (s, 1 H), 7.60 (s, 1 H), 7.55 (s, 1 H), 7.53 (d, J = 8.6 Hz, 2H), 7.44 (dd, J = 5.1 , 1.6 Hz, 1 H), 7.18 (d, J = 8.5 Hz, 2H), 6.37 - 6.20 (m, 1 H), 6.18 - 5.96 (m, 1 H), 4.22 (s, 2H), 3.34 (tt, J = 10.4, 5.0 Hz, 4H), 2.63 (s, 1 H), 1.81 (d, J = 12.7 Hz, 2H), 1.73 (s, 2H), 1.60 (dd, J = 12.7, 4.1 Hz, 2H), 1.48 (s, 9H). LCMS Method C: rt 6.04 min; m/z 571.2 [M+1 C, 569.2 [M-1]", 515.2 [M- f-Bu+2]+.
(g) 2-(2-(2-((4-(Piperidin-4-yl)phenyl)amino)-5-(trifluo
yl)ethyl)isonicotinamide (1131)
Trifiuoroacetic acid (100 μί, 0.131 mmol) was added to a solution of fert-butyl 4-(4- ((4-(2-(4-carbamoylpyridin-2-yl)ethyl)-5-(trifluoromethyl)pyrirnidin-2- yl)amino)phenyl)piperidine-1-carboxylate (1130) (50 mg, 88 mol) in DCM (4 mL) and the resulting mixture was stirred for 1 hour at room temperature. The voiatiies were evaporated under reduced pressure and the residue purified by chromatography (Si02, 0-50% MeOH/DCM) to give the title compound (1131) (28 mg, 68%) as a colourless solid; 1H NMR (400 MHz, d -MeOD) δ 8.55 (d, J = 5.2 Hz, 1 H), 8.49 (s,
1 H), 7.74 (s, 1 H), 7,61 (dd, J = 5.2, 1.3 Hz, 1 H), 7.53 (d, J = 8.3 Hz, 2H), 7.17 (d, J = 8.5 Hz, 2H), 3.46 (d, J = 12.7 Hz, 2H), 3.37 (t, J = 7.0 Hz, 2H), 3.27 (dt, J = 3.2, 1.6 Hz, 5H), 3.18 - 3.04 (m, 2H), 2.83 (ddd, J = 12.1 , 8.7, 3.6 Hz, 1 H), 2.03 (d, J = 13.8 Hz, 2H), 1.88 (ddd, J = 16.7, 13.6, 3.8 Hz, 2H). LCMS Method C: rt 4.44 min; m/z 471.1 [M+1]+, 469.1 [M-1]-.
(h) 2-(2-(2-((4-(1-Methylpiperidin-4-yl)phenyl)amino)-5-(trifluorom
yl)ethyl)isonicotinamide (34)
Formaldehyde solution (37% aq; 24 μί, 0.30 mmol) was added to a solution of 2- (2- (2-((4-(Piperidin-4-yl)phenyl)amino)-5-(trifluoromethyl)pyrimidin-4- yi)ethyi)ison!Cotinamide (1131) (28 mg, 60 moi) in dry MeOH (2 mL). Sodium triacetoxyborohydride (63 mg, 0.30 mmol) was added and the resulting mixture stirred at room temperature for 2 hours under nitrogen. Ethyl acetate was added and the resulting solution adsorbed onto silica gel. Chromatography (Si02i 0-50%
MeOH/DCM) gave the title compound (34) (14 mg, 49%) as a coiouiess solid; 1 H
NMR (400 MHz, drMeOD) δ 8.58 (d, J= 4.9 Hz, 1H), 8.51 (s, 1H), 7.73 (s, 1H), 7.61 (dd, J= 5.2, 1.6 Hz, 1H), 7.53 (d, J= 8.5 Hz, 2H), 7.18 (d, J= 8.6 Hz, 2H), 3.51 - 3.31 (m, 4H), 2.99 (d, J = 11.7 Hz, 2H), 2.56-2.44 (m, 1H), 2.32 (s, 3H), 2.15 (d, J = 2.8 Hz, 2H), 1.87-1.70 (m, 4H). LCMS Method C: rt4.50 min; m/z 485,1 [M+1]+, 483.1 [M-1]-.
Example 35; 2~(2~(2~(2~((4~(pyrro din~3-yl)phenyl)amino)~5~

!132 !133 1135

35
(a) tert-Butyl 4-(((trifluoromethyl)sulfonyl)oxy)-2,3-dihydro- 1 H-pyrrole- 1 -carboxylate
(1132 and ί 133)
To a stirred solution of sodium bis(trimethylsilyl) amide (1.01 g, 5.50 mmol) in THF (20 mL) was added dropwise a solution of fe/t-butyl 3-oxopyrrolidine-1 -carboxylate (0.925 g, 5.00 mmol) in THF (7 mL) at -78 °C. After being stirred for 15 minutes, N- phenyl-bis(trifluoromethanesulfonimide) (1.79 g, 5.00 mmol) in THF (12 mL) was added and the reaction mixture was stirred at -78 °C for an additional 3 hours, and then at room temperature for 1 hour. The reaction mixture was quenched with 10 % aqueous NaHC03 and extracted with EtOAc. The organic layer was washed with brine, dried over gS04, and concentrated in vacuo. The resulting residue was purified by column chromatography on silica gel (20-80 % dichioromefhane in petroleum benzine 40-60 °C) to give a mixture of the title compounds {1132 and 1133) (1.44 g, 90 %) as a pale yellow oil; 1H N R (400 MHz, CDCI3) δ 8.65 (s, 1 H), 5.66 (d, J = 17.5 Hz, 2H), 4.21 - 4.09 (m, 8H), 1.41 (s, 18H).
(b) tert-Butyl 4-(4-nitrophenyl)-2,3-dihydro-1H-pyrrole-1 -carboxylate (1134 and 1135) A solution of 2 M aqueous Na2C03 (5.70 mL, 9.09 mmol) was added to a degassed mixture of 4-nitrophenylboronic acid (0.909 g, 1.52 mmol, 1.2 eq), tert-butyl 4- (((trifluoromethyl)sulfonyl)oxy)-2,3-dihydro-1 H-pyrrole-1-carboxylate { 134 and 135} (1.44 g, 4.54 mmol), LiCI (0.385 g, 9.08 mmol) and Pd(PPh3)4 ( .57 g, 1.36 mmol) in 1 ,4-dioxane (10 mL). The reaction mixture was stirred at 80-90 °C for 4 hours. The resulting mixture was dissolved in EtOAc (70 mL) and the organic layer was washed with H20 (50 mL), brine (50 mL) and dried over Na2S04 to yield a dark red oil. The oil was purified by column chromatography on silica gel (0-20% EtOAc in petroleum benzine 40-60 °C) to give a mixture of the title compounds (1134 and 1135) (0.442 g, 34 %) as a light yellow solid; 1 H NMR (400 MHz, CDCi3) δ 8.41 - 8.32 (m, 2H), 8.24 - 8.19 (m, 2H), 7.81 - 7.73 (m, 2H), 7.52 (dd, J = 8.8, 2.4 Hz), 6.43 - 6.33 (m, 1 H), 4.58 - 4.48 (m, 4H), 4.42 - 4.31 (m, 4H), 1.52 (s, 9H), .51 (s, 9H). (c) tert-Butyl 3-(4-aminophenyl)pyrrolidine- 1 -carboxylate (1136)
A solution of ferf-butyl 4-(4-nitrophenyi)-2,3-dihydro-1 H-pyrroie-1-carboxylate (1134 and 1135) (0.442 g, 1.52 mmol) in EtOH (10 mL) and DMF (10 mL) was added to a solution of 10% Pd/C (255 mg) in DMF (10 mL), The reaction was stirred at room temperature for 17 hours under an atmosphere of hydrogen. The reaction was filtered through a pad of celite and washed through with EtOAc (130 mL). The solvent
was removed in vacuo to yield a brown oii which was purified by column
chromatography on silica gel (0-50% EtOAc in petroleum benzine 40-60 °C) to give the title compound (1136) (0.307 g, 77 %) as a brown oii; Ή N R (400 MHz, CDCI3) δ 7.02 (d, J = 8.4 Hz, 2H), 6.64 (d, J = 8.4 Hz, 2H), 3.85 - 3.49 (m, 4H), 3.43 - 3.29 (m, 1 H), 3.29 - 3.15 (m, 2H), 2.18 (d, J = 6.5 Hz, 1 H), 1.97 - 1.85 (m, 1 H), 1.47 (d, J = 4.7 Hz, 9H). LC S Method C: rt 4.88 min; m/z 163.2 [M-Boc +H]+.
(d) tert-Butyl 3-(4-( (4-chloro-5-(trifluoromethyl)pyrimidin-2- yl)amino)phenyl)pyrrolidine- 1 -carboxylate (1137)
Zinc chloride (1.0 M in Et20) (1.40 mL, 1.40 mmol) was added to a solution of 2,4- dichloro-5-(trifluoromethyl)pyrimidine (180 μί, 1.29 mmol) in 1 :1 DCE/i-BuOH (10 mL) at 0 °C under a stream of nitrogen gas. The mixture was stirred for 1 hour at 0 °C and then ferf-buty! 3-(4-aminophenyl)pyrroiidine-1-carboxylate (1136) (0.307 g, 1.17 mmol) in 1 :1 DCE/ BuOH (10 mL) was added. A solution of NEt3 (0.180 mL, 1.29 mmol, 1.1 eq) in 1 : 1 DCE/i-BuOH (5 mL) was next added dropwise at 0 °C. The reaction mixture was vigorously stirred for a further 30 minutes at 0 °C after the final addition and then at room temperature for 16 hours. The solvent was removed in vacuo to afford a brown oily residue which was purified by column chromatography on silica gel (0-50% EtOAc in petroleum benzine 40-60 °C) to yield a pale yellow solid. The solid was suspended in MeOH (15 mL) and water (15 mL). The precipitate was filtered to afford the title compound (1137) (0.449 g, 87%) as a pale yellow solid; 1H NMR (400 MHz, CDCI3) 1H NMR (400 MHz, CDCI3) δ 8.56 (s, 1 H), 7.53 (d, J = 8.5 Hz, 2H), 7.40 (s, 1 H), 7.26 (s, 2H), 3.92 - 3.73 (m, 1 H), 3.69 - 3.51 (m, 1 H), 3.48 - 3.22 (m, 3H), 2.26 (s, H), 2.03 - .90 (m, H), .48 (s, 9H). LCMS Method C: rt 6.68 min; m/z 443.0 [M+H]+, 441.1 [M-H]\
(e) tert-Butyl 3-(4-((4-((2-(2-methoxy-2-oxoethyl)phenyl)ethynyl)-5- (trifluoromethyl)pyrimidin-2-yl)amino)phenyl)pyrrolidine- 1 -carboxylate (1138)
A solution of methyl 2-ethynyibenzoate (14) (0.21 g, 1.2 mmol) in dimethylformamide (3 mL) and triethyiamine (0.57 mL, 4.1 mmol) were added to a mixture of ferf-buty! 3- (4-((4-chioro-5-(trifluoromethyl)pyrimidin-2-yl)amino)phenyi)pyrrolidine-1-carboxyiate (1137) (0.45 g, 1.0 mmol, 1 eq), Pd(PPh3)2C!2 (71 mg, 0.10 mmol), Cu(!)l (30 mg, 0.15 mmol) and triphenyiphosphine (40 mg, 0.15 mmol, 0.15) in dimethylformamide (4 mL). The reaction mixture was heated under microwave irradiation at 120 °C for 15 minutes. The reaction was cooled and the mixture diluted with EtOAc and passed
through a plug of celite and washed through with EtOAc (60 mL). Water (50 mL) was added and the layers separated. The aqueous layer was extracted with EtOAc (2x50 mL). The combined organic extracts were washed with water (50 mL) and brine (50 mL) and dried over Na2S04. After filtration the solvent was removed in vacuo to give a dark brown residue. The residue was purified by column chromatography on silica gel (0-20 then 20-50% EtOAc in in petroleum benzine 40-80 °C) to give the title compound (1138) (0.53 g, 90%) as an orange oil; Ή N R (400 MHz, CDCI3) δ 8.62 (s, 1 H), 7.68 (dd, J = 7.7, 1.0 Hz, 1 H), 7.58 (d, J = 8.4 Hz, 2H), 7.47 - 7.41 (m, 2H), 7.39 - 7.30 (m, 4H), 7.24 (s, 1 H), 3.96 (s, 2H), 3.87 - 3.75 (m, 1 H), 3.70 (s, 3H), 3.68 - 3.52 (m, 1 H), 3.48 - 3.22 (m, 3H), 2.30-2.21 (m, 1 H), 2.02 - 1.94 (m, 1 H), 1.49 (s, 9H). LCMS Method C: rt 6.82 min; m/z 581.1 [M+H]+.
(f) tert-Butyl 3-(4-((4-(2-(2-methoxy-2-oxoethyl)phenethyl)-5- (trifluoromethyl)pyrimidin-2-yl)amino)phenyl)pyrrolidine- 1 -carboxylate (1139}
A solution of iert-butyl 3-(4-((4-((2-(2-methoxy-2-oxoethyl)phenyl)ethynyl)-5-
(trifluoromethyl)pyrimidin-2-yl)amino)phenyl)pyrrolidine-1-carboxylate (1138) (0.527 g, 0.906 mmol) in EtOH (10 mL) was added to a solution of 10% Pd/C (0.500 g) in DMF (6 mL). The reaction was stirred at room temperature for 24 hours under an atmosphere of hydrogen. The reaction was filtered through a pad of celite and washed through with EtOAc (80 mL). The solvent was removed in vacuo to afford a yellow oil. This was taken up in DMF/EtOH (1 : 1 , 10 mL) and a slurry of 10% Pd/C (0.500 g, 1 eq) in DMF was added. The reaction was stirred under an atmosphere of H2 at room temperature for an additional 24 hours. The reaction was filtered through a pad of celite and washed through with EtOAc (80 mL). The solvent removed in vacuo to afford a yellow oil which was purified by column chromatography on silica gel (0-45% EtOAc in petroleum benzine 40-60 °C) to yield the title compound (1139) (0.126 g, 24 %) as a brown oil; 1H NMR (400 MHz, CDCI3) δ 8.54 (s, 1 H), 7.58 (d, J = 8.2 Hz, 2H), 7.39 (s, 1 H), 7.30 - 7.17 (m, 6H), 3.91 - 3.72 (m, 2H), 3.75 (s, 2H), 3.68 (s, 3H), 3.66 - 3.52 (m, 1 H), 3.47 - 3,21 (m, 3H), 3,17 - 3.04 (m, 4H), 2.23 (d, J = 20.4 Hz, 1 H), 1.97 (dd, J = 21.1 , 10.2 Hz, 1 H), 1.48 (d, J = 3.1 Hz, 9H). LCMS Method C: rt 6.91 min; m/z 585 [M+Hf , 607 [M+Na]+.
(g) Lithium 2-(2-(2-(2-((4-( 1 -(tert-butoxycarbonyl)pyrrolidin-3-yl)phenyl)amino)-5- (trifluoromethyl)pyrimidin-4-yl)ethyl)phenyl)acetate (1140)
LiOH.H20 (0,015 g, 0.65 mmo!, 3 eq) was added to a solution of ieri-butyi 3-(4-((4-(2- (2-methoxy-2-oxoethyl)phenethyl)-5-(trifluoromethyl)pyrirnidin-2- yl)amino)phenyl)pyrro!idine-1-carboxylate {1139) (0.13 g, 0.22 mmoi) in THF (7 mL), water (1.5 mL) and methanol (1 mL) and the resulting mixture was allowed to stir at room temperature for 70 hours. The volatiles were removed in vacuo and the residue was diluted with EtOAc (50 mL) and saturated aqueous NaHC03 (50 mL). The layers were separated and the aqueous layer was extracted with EtOAc (2x50 mL). The combined organic layers were washed with brine (50 mL), dried over gSC and the solvent removed in vacuo to give the title compound {1140) (0.12 g, 99 %) as a pale yellow oil; LC S Method C: rt 6.56 min; m/z 571.1 [M-I_i+2H]+, 515, 1 [M-Li-Buty!+2Hf, 471.1 [M-Li-Boc+2H]+, 569.2 [M-Li-H]". (h) tert-Butyl 3-(4-((4-(2-(2-amino-2-oxoethyl)phenethyl)-5-(trifluorome
2-yl)amino)phenyl)pyrrolidine- 1-carboxylate (1141)
1-Hydroxybenzotriazole (38.0 mg, 0.28 mmoi), EDCI (50.0 mg, 0.32 mmoi) and N,N- diisopropylethylamine (187 uL, 1.08 mmoi) were added to a solution of lithium 2-(2- (2-(2-((4-(1-(ie "i-butoxycarbonyl)pyrrolidin-3-yl)phenyl)amino)-5- (trifluoromethyl)pyrimidin-4-yl)ethyl)phenyl)acetate {1140) (124 mg, 0.215 mmoi) in dry THF (6 mL) and dry DMF (1 mL) under an atmosphere of nitrogen. Ammonium carbonate (101 mg, 1.08 mmoi) was added in one portion to the stirred reaction mixture after 10 minutes and the reaction was stirred at room temperature for 23 hours. The volatiles were removed in vacuo and the residual solution was diluted with EtOAc (65 mL) and washed with saturated NaHC03 (65 mL). The aqueous layer was extracted with EtOAc (2x50 mL). The combined organic layers were washed with brine (50 mL) and dried over MgS04. The solvent was removed in vacuo to afford a pale yellow oil. The crude material was purified by column chromatography on silica gel (0-80 % EtOAc in petroleum benzine 40-60 °C) to give the title compound (1141) (76 mg, 62 %) as a white solid; Ή NMR (400 MHz, CDCI3) δ 8.54 (s, 1 H), 8.02 (s, 1 H), 7.56 (d, J = 7.4 Hz, 2H), 7.29 - 7.20 (m, 4H, obscured by solvent), 5.36 (d, J = 21.2 Hz, 2H), 3.89 - 3.76 (m, 1 H), 3.66 - 3.51 (m, 2H), 3.45 - 3.27 (m, 4H), 3.15 - 3.04 (m, 4H), 2.29-2,25 (m, 1 H), 2,01-1 ,94 (m, 1 H), 1 ,49 (s, 9H). LCMS Method C: rt 6.37 min; m/z 570.1 [M+H]+, 568.2 [M-H]\
(i) 2-(2-(2-(2-((4-(Pyrrolidin-3-yl)phenyl)amino)-5-(trifluorom
y!)ethy!)pheny!)aceiamide (35)
To a solution of ie/f-butyl 3-(4-((4~(2~(2~amino-2-oxoethyl)phenethyi)-5- (trifluoromethyl)pyrimidin-2-yl)amino)phenyl)pyrrolidine-1-carboxylate (1141) in DCM (20 mL) was added TFA (4.0 mL) and the reaction mixture was stirred at room temperature for 30 minutes. The solvent was removed in vacuo and the residue taken up in EtOAc (10 mL) and 2 M NaOH (10 mL). The organic layer was extracted with EtOAc (2x 10 mL), and the combined layers washed with water (10 mL), brine (10 mL) and dried over MgS04. The solvent was removed in vacuo to give a yellow solid. The solid was suspended in cydohexane and filtered to give a brown solid (42 mg). The product was purified further by RP-HPLC ( Waters, 0-80 % CH3CN in H20 over 20 minutes at a flow rate of 10 mL/min). Fractions containing product were basified (pH 10) with 2 M NaOH and extracted with EtOAc. The combined organic layers were washed with brine (10 mL), dried ( gS04) and the solvent removed in vacuo to give the title compound (35) (17 mg, 27 %) as a brown oil; 1 H-NMR (400
MHz, £¾~MeGD) δ 8.56 (d, J = 0.8 Hz, 1 H), 7.73 - 7.68 (m, 2H), 7.33 - 7.16 (m, 6H), 3.67 (s, 2H), 3,60 - 3.46 (m, 2H), 3.42 - 3,33 (m, 2H), 3, 14 (m, 5H), 2,45 (dtd, J = 10.1 , 7.1 , 3.4 Hz, 1 H), 2.18 - 2.05 (m, 1 H); LC S Method C: rt 4.84 min; m/z 470.1 [M+Hf.
Example 36: 2-{2-{2-{2-{{4-{(4- ethyf sperazin-1 -yl)methyl)pheny!)amino)-5- ftnf!uorometh S)pyrimidin-4-yl)ethy!)pheny!)acetamide (38)

25 36
2-(2-(2-(2-((4-(Piperazin~1~ylmethyl)phenyi)amino)-5-(trifluoromethyl)pyrimidin-4- yi)ethyi)phenyl)acetamide (25) (11 mg, 0.022 mmol) was dissolved in methanol (1 mL). 37% Formaldehyde solution (7 μί) was added followed by sodium
triacetoxyborohydride (24 mg, 0.11 mmol). The mixture was stirred vigorously at room temperature for two hours then concentrated. The residue was suspended in 10% sodium hydroxide (1 mL) and brine (2 mL) then extracted with ethyl acetate (5x2 mL). The combined ethyl acetate phases were washed with brine, dried over sodium sulfate, evaporated and the residue evaporated from DCM to give the title compound
(36) (10.3 mg, 94% yield) as an off-white solid; H N R (400 MHz, drMeOD) δ 8.46 (d, J = 0.5 Hz, 1 H), 7.60 - 7.54 (m, 2H), 7.20 (d, J = 8.6 Hz, 2H), 7.18 - 7.13 (m, 1 H), 7.13 - 7.07 (m, 3H), 3.57 (s, 2H), 3.46 (s, 2H), 3.06 (qd, J = 6.8, 3.1 Hz, 2H), 3.01 - 2.94 (m, 2H), 2.54 (s, 8H), 2.29 (s, 3H). LCMS Method C: 4.70 min; m/z 513.2
[M+H+]; m/z 511.0 [M-H]\
Example 37: 3~(2~(2~((4~(Piperazin-1 -ylmethy )pheny!)amino)-5- (trifluoromethyl)pyrimidsn~4~y^ethyl)benzamide (37)

(a) 3-iodobenzamide (1142)
3-lodobenzoic acid (2.00 g, 8.06 mrnol) was dissolved in THF (20 mL) then oxalyl chloride (1.4 mL, 16 mrnol) and DMF (0.05 mL) were added and the resulting mixture stirred for 2 hours. The volatiies were evaporated under reduced pressure and the residue was dissolved in THF (20 mL) and concentrated aqueous ammonia (10 mL). After 60 minutes water (200 mL) was added and after a further 30 minutes the resulting precipitate was collected by filtration and air dried to give the title compound (1142) (1.97 g, 99% yield) as a white powder; 1H NMR (400 MHz, oVMeOH) δ 8.23 (t,
J = 1.8 Hz, 1 H), 7.90 (ddd, J = 7.9, 1.7, 1.0 Hz, 1 H), 7.86 (ddd, J = 7.8, 1.7, 1.0 Hz, 1 H), 7.24 (t, J = 7.8 Hz, 1 H). LCMS Method C: rt 5.29 min, m/z 248.1 [M+Hf .
(b) 3-((Trimethylsilyl)ethynyl)benzamide (1143)
3-iodobenzamide (J142) (1.00 g, 4.05 mmol), bis(triphenylphosphine)pailadium(l!) chloride (0.142 g, 5 mol%), copper(l) iodide (0.077 g, 10 moi%), DMF (4 mL) and diisopropylamine (12 mL) were loaded into a microwave tube. The mixture was degassed for ten minutes with nitrogen, then trimethylsi!ylacety!ene (0.89 mL, 4.9 mmol) was added and the resulting mixture heated under microwave irradiation at 120 °C for 15 minutes. The volatiles were evaporated under reduced pressure and the residue chromatographed (Biotage Isolera, 40 g silica cartridge, 0-100% ethyl acetate/ petroleum benzine 40-60 °C) to give the title compound (1143) (0.569 g, 65% yield) as a brown solid; 1H NMR (400 MHz, CDCI3) δ 7.88 (td, J = 1.8, 0.5 Hz, 1 H), 7.78 (ddd, J = 7.8, 1.8, 1.2 Hz, 1 H), 7.63 - 7.59 (m, 1 H), 7.40 (td, J = 7.8, 0.5 Hz, 1 H), 6.05 (br s, 1 H), 5.67 (br s, 1 H), 0.26 (s, 9H). LCMS Method C: rt 5.94 min, m/z 218.2 [M+Hf,
(c) 3-Ethynylbenzamide (1144)
3-((Trimethylsilyl)ethynyl)benzamide (1143) (0.565 g, 2.60 mmol) was dissolved in THF (33 mL), and 1 M TBAF in THF (3.25 mL, 3.25 mmol) was added. After two hours the reaction was poured into water (200 mL) and the resulting solution was extracted with diethyl ether (3x200 mL). The combined ether phases were washed with brine (200 mL), dried over sodium sulfate then evaporated to give the title compound (i144) (0.357 g, 95% yield) as a tan solid; 1 H NMR (400 MHz, CDCI3) δ 7.92 (t, J = 1.5 Hz, 1 H), 7.83 - 7.79 (m, 1 H), 7.64 (dt, J = 7.7, 1.4 Hz, 1 H), 7.42 (td, J = 7.8, 0.5 Hz, 1 H), 6.07 (br s, 1 H), 5.77 (br s, 1 H), 3.13 (s, 1 H). LCMS Method C: rt 4.74 min, m/z 146.2 [M+H]'\
(d) tert-Butyl 4-(4-((4-((3-carbamoy!pheny!)ethyny!)-5-(trif!uoromethyl)pyri yljamino )benzyl)piperazine - 1 -carboxylate (1145)
fert- Butyl 4-(4-((4-chloro-5-(trifluoromethyl)pyrimidin-2-yl)amino)benzyl)piperazine-1- carboxylate (194) (0.30 g, 0.64 mmol), 3-ethynylbenzamide (1144) (102 mg, 0.70 mmol), copper(l) iodide (12 mg, 10 mo!%), triphenylphosphine (17 mg, 10 mo!%), bis(triphenylphosphine)palladium(ll) chloride (22 mg, 5 mol%), DMF (3 mL) and triethylamine (0.443 mL, 3.18 mmol) were loaded into a microwave tube and
degassed with nitrogen for five minutes. The resulting mixture was heated under microwave irradiation at 120 °C for 15 minutes then cooled to room temperature. The cooled mixture was added to 5% aqueous potassium carbonate (150 mL) and the resulting mixture extracted with ethyl acetate (3x 150 mL). The combined ethyl acetate phases were washed with wafer (200 mL), brine (200 mL), dried then evaporated. The residue was chromatographed (Biotage isoiera: 12 g silica cartridge, 20-100% ethyl acetate/ petroleum benzine 40-60 °C then 0-10% methanol/ ethyl acetate) to give the title compound (1145) (107 mg, 29% yield) as a yellow solid; 1 H NMR (400 MHz, c/6-DMSO) δ 10.46 (s, H), 8.84 (s, 1 H), 8.17 (s, 1 H), 8.09 (t, J = 1.7 Hz, 1 H), 8.04 (dt, J = 7.9, 1.4 Hz, 1 H), 7.77 (dt, J = 7.8, 1.3 Hz, 1 H), 7.71 - 7.68 (m, 2H), 7.66 - 7.52 (m), 7.27 (d, J = 8.6 Hz, 2H), 3.44 (s, 2H), 2.33 - 2.26 (m, 4H), 1.38 (s, 9H). LCMS Method C: rt 5.06 min; m/z 581.1 [M+H]+, 525.1 [M~tBu+2H]+; m/z 579.2 [M-H]-. (e) tert-Butyl 4-(4-((4-(3-carbamoylphenethyi)-5-(trifluoromethyl)pyrimid^
yl)amino)benzyl)piperazine- 1 -carboxylate (I 146)
A suspension of ferf-buty! 4-(4-((4-((3-carbamoyiphenyi)ethynyi)-5- (trifiuoromethyl)pyrimidin-2-yl)amino)benzyl)piperazine-1 -carboxylate {1145) (105 mg, 0.18 mmol) and Pd/C (75 mg) was stirred in DMF (5 mL) and triethylamine (0.15 mL) at 30 °C under hydrogen for 18 hours. After filtration the voiatiles were evaporated under reduced pressure and the residue chromatographed (Biotage isoiera: 4 g silica cartridge, 0-2% methanol/ ethyl acetate) to give the title compound (1146) (66.6 mg, 63% yield) as a white solid; 1 H NMR (400 MHz, d DMSO) δ 10.20 (s, 1 H), 8.67 (s, 1 H), 7.93 (s, 1 H), 7.79 (s, 1 H), 7.74 - 7.50 (m, 8H), 7.40 - 7.35 (m, 2H), 7.33 (s, 1 H), 7.24 (d, J = 8.6 Hz, 2H), 3.43 (s, 2H), 3.17 - 3,04 (m, 4H), 2.36 - 2.23 (m, 4H), 1.38 (s, 9H). LCMS Method C: rt 5.05 min; m/z 585.2 [M+Hf; m/z 583.2 [M-H]-.
(f) 3-(2-(2-((4-(Piperazin-1-ylmethyl)phenyl)amino)-5-(trifluoro
yi)ethyi)benzamide (37)
ferf-Butyl 4-(4-((4-(3-carbamoylphenefhyl)-5-(trifiuoromethyl)pyrimidin-2- yl)amino)benzyl)piperazine-1 -carboxylate (1146) (65 mg, 0.11 mmol) was dissolved in DCM (10 mL) then TFA (1 mL) was added and the resulting mixture stirred at room temperature for 8 hours. The voiatiles were evaporated under reduced pressure and the residue partitioned between 10% sodium hydroxide (25 mL) and ethyl acetate (25 mL). The aqueous phase was extracted with ethyl acetate (3x25 mL) then the
combined ethyl acetate phases washed with brine, dried over sodium sulfate and evaporated. The residue was washed with toluene (2x2 mL) and DC (2x0.5 mL) to give the title compound (37) (22 mg, 40% yield) as a white solid; 1 H N R (400 MHz, 6-DMSO) δ 10.19 (s, 1 H), 8.86 (s, 1 H), 7.94 (s, 1 H), 7.78 (s, 1 H), 7.74 - 7.64 (m, 3H), 7.40 - 7.35 (m, 2H), 7.35 - 7.29 (m, 1 H), 7.23 (d, J = 8.2 Hz, 2H), 3.38 (s, 2H), 3.17 - 3.02 (m, 4H), 2.75 - 2.61 (m, 4H), 2.29 (s, 4H). LCIV1S Method C: rt 4.52 min; m/z 485.1 [M+H]+; m/z 483.1 [M-H]\
Example 38: 2~(2~(2~(2~((4~(1 ~Ethy!piperidin~4~y!)pheny!)amino)-5- (tnfluoromethyl}pynmidsn-4-y!)ethyl)pyndsn-3-yS)aceia!Ti!de (38)

A mixture of 2-(2-(2-(2-((4-(piperidin-4-yl)phenyl)amino)-5-(trifluoromethyi)pyrimidin- 4-yl)ethyl)pyridin-3-yl)acetamide (32) (74 mg, 0.15 mmol), bromoethane (12 L, 0.16 mmol) and potassium carbonate (63 mg, 0.46 mmol) in DMF (5 mL) was stirred for 48 h at room temperature under an inert atmosphere. The volatiies were removed under reduced pressure and the residue partitioned between ethyl acetate (20 mL) and saturated sodium hydrogen carbonate solution (20 mL). The aqueous layer was extracted with ethyl acetate (2*30 mL) and the combined organics were washed with brine then dried (MgS04). The solvent was removed under reduced pressure and the resulting solid was chromatographed (Biotage isoiera: C-18 reverse phase column, 0-100 % MeCN in H20) to give the title compound (38) (22 mg, 28%) as an off-white solid; 1H NMR (400 MHz, g-DMSG) δ 10,07 (s, H), 8.65 (s, 1 H), 8.37 (dd, J = 4.8,
I .7 Hz, 1 H), 7.65 - 7.57 (m, 3H), 7.55 (s, 1 H), 7.26 - 7.13 (m, 3H), 7.02 (s, 1 H), 3.54 (s, 2H), 3.25 (s, 4H), 2.98 (d, J = 11.3 Hz, 2H), 2.48 - 2.30 (m, 3H), 1.95 (q, J = 11.7,
II .3 Hz, 2H), 1.73 (d, J = 10.5 Hz, 2H), 1.63 (qd, J = 12.4, 3.6 Hz, 2H), 1.03 (t, J = 7.2 Hz, 3H). LCMS Method C: rt 4.18 min; m/z 513 [M+H]+.
Example 39: 2-(2-(2-(2-({4-{Piperid!n-3-yl)pheny!)am!no)-5- {triffuoromethyl}pyrimidsn-4-yl)ethyf)pyndsn-3-y!)acetamide (39)


(a) tert-Butyl 3-(4-((4-((3-(2-methoxy-2-oxoethyl)pyridin-2-yl)ethynyl)-5- (trifluoromethyl)pyrimidin-2-yl)amino)phenyl)piperidm^ (1147)
A solution of methyl 2~(2~ethynylpyridin~3~yl)acetate (1120) (0,100 g, 0.571 mmol) in THF (1 mL) and triethylamine (0.199 mL, 1.43 mmol) was added to a mixture of tert- butyl 3-(4-((4-chloro-5-(trifluoromethyi)pyrimidin-2-yi)amino)phenyl)piperidine-1- carboxylate (124) (0.217 g, 0.476 mmol), Pd(PPh3)2Ci2 (0.033 g, 0.048 mmol), Cu(i)i (0.014 g, 0.071 mmol) and triphenylphosphine (0.019 g, 0,071 mmol) in
dimethyiformamide (3 mL). The resulting mixture was heated under microwave irradiation at 120 °C for 20 minutes then cooled, degassed for 10 minutes and heated under microwave irradiation at 20 °C for a further 20 minutes. The cooled mixture was diluted with ethyl acetate and the resulting solution passed through a plug of
Celiie, washing with ethyl acetate (250 mL). The volatiles were removed to give a brown solid which was chromatographed (Biotage Isolera: 25 g silica cartridge, 0- 40% EtOAc in petroleum benzine 40-80 °C) to give the title compound (1147)
(0.061 g, 22 %); 1H NMR (400 MHz, CDCI3) δ 8.63 (s, 1 H), 8.62 (d, J = 1.2 Hz, 1 H), 7.72 (dd, J = 7.9, 1.3 Hz, 1 H), 7.57 (d, J = 8.4 Hz, 2H), 7.54 (s, 1 H), 7.35 (dd, J = 7.9, 4.7 Hz, 1 H), 7.23 (d, J = 8.5 Hz, 1 H), 4.30-4.06 (m) 3.95 (s, 2H), 3.71 (s, 3H), 2.84 - 2.58 (m, 3H), 2.06-1.98 (m), 1.70- .66 (m, 2H), 1.69 - 1.51 (m, 2H), 1.47 (s, 9H).
(b) tert-Butyl 3-(4-((4-(2-(3-(2-methoxy-2-oxoethyl)pyridin-2-yl)ethyl)-5- (trifluoromethyl)pyrimidin-2-yl)amino)phenyl)piperidi (1148)
To a solution of ie -butyl 3-(4-((4-((3-(2-methoxy-2-oxoethyl)pyridin-2-yl)ethynyl)-5- (trifluoromethyl)pyrimidin-2-yl)amino)phenyl)piperidine-1-carboxylate (1147) (0.061 g, 0.10 mmol) in DMF (9 mL) and triethylamine (1 mL) was added a slurry of Pd/C (0.070 g) in DMF (3 mL). The resulting mixture was stirred under an atmosphere of hydrogen at room temperature for 16 hours. The crude reaction mixture was diluted with ethyl acetate then filtered through a pad of Celite. The Celite was washed with ethyl acetate and the filtrates combined. The solvent was removed in vacuo to give a brown oil which was taken up in DMF (9 mL) and triethylamine (1 mL). A slurry of Pd/C (0.070 g) in DMF (3 mL) was added and the resulting mixture was stirred under an atmosphere of hydrogen at room temperature for a further 19 hours. The crude reaction mixture was diluted with ethyl acetate then filtered through a pad of Celite. The Celite was washed with ethyl acetate and the filtrates combined. The solvent was removed in vacuo to give a brown oil which was chromatographed (Biotage Isolera: 25 g silica cartridge, 0-50 % EtOAc in petroleum benzine 40-80 °C) to give the title compound (1148) (0.030 g, 49 %) as a yellow oil; 1H NMR (400 MHz, CDCI3) 6 8.50 (s, 1 H), 8.46 (dd, J = 4.8, 1.7 Hz, 1 H), 7.61 - 7.46 (m, 4H, NH), 7.19 (d, J = 8.6 Hz, 2H), 7.12 (dd, J = 7.7, 4.8 Hz, 1 H), 4.35 - 3.99 (m), 3.72 (s, 2H), 3.70 (s, 3H), 3.37 ( J = 7.4 Hz, 2H), 3.28 (t, J = 6.9 Hz, 2H), 2.80 - 2.57 (m, 3H), 2.07 - 1.95 (m), 1.91 - 1.69 (m, 2H), 1.69 - 1.50 (m, 3H), 1.46 (s, 9H). LCMS Method C: rt = 6.00 min, m/z 600.2 [M+H]+.
(c) 2-(2-(2-(2-((4-( 1 -(tert-Butoxycarbonyl)piperidin-3-yl)phenyl)amino)-5- (trifluoromethyl)pyrimidin-4-yl)ethyl)pyridin-3-yl)acetic acid (1149)
To a solution of ferf-butyl 3-(4-((4-(2-(3-(2-methoxy-2-oxoethyl)pyridin-2-yl)ethyl)-5- (trifluoromethyl)pyrimidin-2-yl)amino)phenyl)piperidine-1-carboxylate (1148) (0.030 g,
0.050 mmol) in THF (7 mL), water (1.5 mL) and eOH (1 mL) was added lithium hydroxide monohydrate (0.020 g, 0.48 mmol). The reaction mixture was stirred at room temperature for 17 hours then the volatiles were removed in vacuo and the residue was partitioned between ethyl acetate (10 mL) and saturated aqueous sodium hydrogen carbonate (10 mL). The layers were separated and the aqueous layer was extracted with ethyl acetate (2^ 10 mL). The combined organic layers were washed with brine ( 0 mL), dried (MgS04) then the solvent removed in vacuo to give the title compound (1149) (0.029 g, 99 %) as a white solid. LC S Method C: rt 5.50 min, m/z = 586.1 [M+Hf, 584.2 [M-H]\
(d) tert-Butyl 3~(4~( (4-(2-(3-(2-amino-2-oxoethyl)pyridin-2-yl)ethyl)-5- (trifluoromethyl)pyrimidin-2-yl)amino)phenyl)piperidine- 1^ (1150)
1-Hydroxybenzotriazoie (20 mg, 0.15 mmol), EDCi.HCI (32 mg, 0.17 mmol) and N,N- diisopropylethyiamine (45 pL, 0.26 mmol) were added to a solution of 2-(2-(2-(2-((4- (1 -(tert-butoxycarbonyi)piperidin-3-yl)phenyl)amino)-5-(trifluoromethyl)pyrimidin-4- yl)ethyl)pyridin-3-yl)acetic acid (1149) (29 mg, 0.051 mmol) in dry DMF (5 mL) under an atmosphere of nitrogen. Ammonium carbonate (50 mg, 0.52 mmol) was added in one portion after 10 minutes then stirring was continued at room temperature for 17 hours. Further portions of 1-hydroxybenzotriazole (20.0 mg, 0.15 mmol), EDCI.HCI (32 mg, 0.17 mmol) and A ,A -diisopropylethylamine (45 L, 0.26 mmol) were added, then ammonium carbonate (50 mg, 0.52 mmol) was added in one portion after 10 min. The resulting solution was stirred at 25 °C for 24 hours. The volatiles were removed in vacuo and the residue was partitioned between ethyl acetate (10 mL) and saturated sodium hydrogen carbonate (10 mL). The aqueous layer was extracted with ethyl acetate (2x10 mL) then the combined organic layers were washed with brine (10 mL), dried (MgS04) and evaporated to dryness. The residue was chromatographed (Biotage isoiera: 10 g silica cartridge, 0-100% EtOAc/ petroleum benzine 40-60 °C then 10 g silica cartridge, 50-100% EtOAc/ petroleum benzine 40- 60 °C) to give the title compound [1150) (0.023 g, 77 %) as a white solid; 1H NMR (400 MHz, CDC ) δ 8.49 (s, 1 H), 8.46 (dd, J = 4.8, 1.6 Hz, 1 H), 7.77 (br s, 1 H), 7.57 - 7.44 (m, 3H), 7.19 (d, J = 8.5 Hz, 2H), 7.14 (dd, J = 7.6, 4.8 Hz, 1 H), 5.63 (d, J = 22.0 Hz, 2H), 4.35 - 3.96 (m), 3.67 (s, 2H), 3.41 - 3.33 (m, 2H), 3.27 (t, J = 6.9 Hz, 2H), 2.83 - 2.58 (m, 3H), 2.01-1.99 (m, 1 H), 1.80 - 1.71 (m, 1 H), 1.68 (s, 3H), 1.65 - 1.52 (m, 2H), 1.46 (s, 9H).
(e) 2-(2-(2-(2-((4-(Piperidin-3-yl)phenyl)amino)-5-(trifluorom
yl)ethyl)pyridin-3-yl)acetamide (39)
To a solution of tert-butyl 3-(4-((4-(2-(3-(2-amino-2-oxoethyl)pyridin-2-y!)ethy!)-5- (trifluoromethyl)pyrimidin-2-yl)amino)phenyl)piperidine-1-carboxylate {1150) (23 mg, 0.039 mmol) in DCM (7 mL) was added TFA (1 mL). The resulting solution was stirred at room temperature for 17 hours then the voiatiles were removed in vacuo. The residue was partitioned between ethyl acetate (50 mL) and a 2.0 M solution of NaOH (50 mL) then the organic layer was washed with water (50 mL), brine (50 mL) and dried over MgS04 before being evaporated in vacuo to give the title compound (39) (18 mg, 94 %) as a white solid; H N R (400 MHz, cf4-MeOH) δ 8.52 (s, 1 H), 8.37 (dd, J = 4.9, 1.6 Hz, 1 H), 7.67 (dd, J = 7.7, 1.6 Hz, 1 H), 7.61 (d, J = 8.6 Hz, 2H) 7.27 - 7.17 (m, 3H), 3.68 (s, 2H), 3,39 - 3.32 (m, 2H), 3.27 (d, J = 8,7 Hz, 2H), 3.21- 3.14 (m, 2H), 2.82 - 2.70 (m, 3H), 2.07 - 1.94 (m, 1 H), 1.94 - 1.83 (m, 1 H), 1.80 - 1.64 (m, 2H). LCMS Method C: rt 4.12 min, m/z = 485.1 [M+Hf, 483.1 [M-H]\
Example 40: 2~(2~(2~(2~((4~(1 ~Ethylpsperidin~3~yl)phenyl)am sno)~5~
(trif!uoromethyl}pyrimsdsn~4~yl)ethy!)pyridsn-3~y!)acetam!de (40)

39 40 A/,A/-Diisopropylethylamine (18.0 μί, 0.103 mmol) was added to a solution of 2~(2~(2~ (2-((4-(Piperidin-3-yl)phenyl)amino)-5-(trifluoromethyl)pyrimidin-4-yl)ethyl)py yl)acetamide (39) (17 mg, 0.034 mmol) in DMF (5 mL). The mixture was stirred for 5 min then bromoethane (4.0 μΐ., 0.051 mmol) was added and stirring was continued for 24 h at room temperature. A further portion of bromoethane (4.0 μί., 0.051 mmol) was added and the reaction mixture was stirred for an additional 16 hours at room temperature. The voiatiles were evaporated in vacuo and the residue partitioned between ethyl acetate (10 mL) and saturated aqueous sodium hydrogen carbonate (10 mL). The layers were separated and the aqueous layer extracted with ethyl acetate (2x10 mL). The combined organic layers were washed with water (10 mL), brine (10 mL) and dried over Na2S04. Filtration then removal of the solvent under
reduced pressure afforded a beige solid which was purified by mass-directed preparative HPLC to afford the title compound (40) (6.2 rng, 36 %) as a white solid; 1H IMMR (400 MHz, 4-MeOH) δ 8.54 (s, 1 H), 8.38 (dd, J = 4.8, 1.5 Hz, 1 H), 7.69- 7.66 (m, 3H), 7.28 - 7.19 (m, 3H), 3.68 (s, 2H), 3.59 - 3.46 (m, 2H), 3.38 - 3.22 (m, partially obscured by drMeOH), 3.19 - 3.08 (m, 2H), 3.04 - 2.80 (m, 3H), 2.12 -
1.99 (m, 2H), 1.99 - 1.70 (m, 2H), 1.34 (t, J = 7.3 Hz, 3H). LC S Method C: rt = 4.20 min, m/z = 513.2 [M+H]+, 511.2 [M-H]\
Biological Assays
The activity of compounds of the invention can be profiled using biochemical and cellular assays.
Primary potency at FAK can be assessed using an Alpha Screen™ technology biochemical assay.
The kinetics of this binding may be further studied using a surface plasmon resonance (SPR) technology assay using a Biacore™ S51 sensor to establish Ka, kd and consequently KD. When off rates from the protein greatly exceed on rates, as may occur for highly potent compounds, KD gives an accurate measure of protein- ligand binding affinity.
The ability of compounds of the invention to inhibit FAK within ceils can be assessed with an EL!SA -type assay performed using a Meso Scale Discovery SECTOR Imager 6000 instrument, in this assay the ability of compounds of the invention to inhibit phosphorylation of Y397-FAK is determined.
The effect of compounds of the invention on inhibition of cellular proliferation resulting from non-FAK activity may be assessed using a 2D proliferation assay using a suitable cell line. This gives an indication of off-target activities and potential toxicity arising from them. Therefore, comparing inhibition of phosphorylation of Y397-FAK and 2D proliferation gives a measure of FAK specific mediated effects and also of potential toxicity resulting from off-target activity.
Primary potency at VEGFR3 can be assessed using an Alpha Screen "''' technology biochemical assay.
The ability of compounds of the invention to inhibit VEGFR3 within cells can be assessed with an ELISA type assay. FAK biochemical Alpha Screen™ assay
A biotin labeled peptide is used as substrate (amino acid sequence: Biotin-Glu-Gly- Pro-Trp-Leu-Glu-Giu-Glu-Giu-Giu-Aia-Tyr-Gly-Trp-Met-Asp-Phe-NH2). FAK enzyme was expressed in insect ceils as catalytic domain (amino acids 411-686) N-termina!ly tagged with six histidine amino acids and a Tobacco Etch Virus (TeV) cleavage sequence. After lysing the cells by sonication, the kinase was purified by Ni-
Immobilised Metal Affinity Chromatography chromatography, TeV cleavage leaving a N-terminal glycine, and gel filtration. The 15 μΙ assay reactions are run in Greiner brand white 384-weli low volume plates. Ail reactions contained 10 mM HEPES pH 7.4, 25 mM NaCI, 10 mM MgCI2, 0.01 % (v/v) Tween-20, 50 μΜ Na3V04, 0.01 % (w/v) albumin from chicken egg white, 111 nM peptide substrate, 80 μΜ ATP, and
4 ng/reaction FAK enzyme, with the enzyme being omitted from negative control reactions. Compounds were added in a volume of 100 ni from dilution series made up in DMSO, positive and negative control reactions receiving the same volume DMSO without compounds. The plates were sealed with adhesive seals and incubated for 90 minutes at 30°C. The reactions were stopped with the detection reagents added at the same time. Product formation was quantified as amplified luminescence between PerkinE!mer AlphaScreen™ beads, using Streptavidin-coated donor and anti-phosphotyrosine (P-Tyr-100) acceptor beads. To each reaction, 5 μΙ containing 10 mM HEPES pH 7.4, 25 mM NaCI, 100 mM EDTA, 0.01 % (v/v) Tween- 20, and 6.25 μg/ml of each bead type were added. Plates were incubated for 6 hours before being read on a PerkinEimer EnVision™ plate reader in HTS Alphascreen™ mode. ICso values were obtained by calculating percent inhibition (%l) for each reaction relative to controls on the same plate (%I=(I-CN)/(CP-CN) where CN/ CP are the averages of the negative positive reactions, respectively), then fitting the %! data vs. compound concentration [I] to %I=(A+((B-A)/(1 +((C/[I])AD)))) where A is the lower asymptote, B is the upper asymptote, C is the iC50 value, and D is the slope factor.
Results
IC50 (nM)
1 2.1
2 2.5
3 1.9
4 1.5
5 2.4
6 4.2
7 9.6
8 6.1
9 7.0
10 2.9
11 3.0
12 6.3
13 0.60
14 21
15 18
16 2.0
17 3.4
18 13
19 7.7
20 0.81
21 2.4
22 2.8
23 0.39
24 0.77
25 3.0
26 20
27 7.2
28 0.30
29 11
30 11
31 3.6
32 3.5
33 6.2
34 5.7
35 7.7
36 2.3
37 15
38 23
39 14
40 51
FAK Biacore1 M SPR assay
Binding parameters of compounds were determined using a Biacore™ S51 sensor. An anti-GST antibody was immobilized onto a C 5 chip by primary amine-coupiing in accordance with the manufacturer's recommendations. in running buffer (0.01 M HEPES pH 7.4, 0.15 M NaCi, 0.005% Surfactant P20, 10 mM gCI2, and 1 % DMSO) N-terminaily GST-fused purified FAK enzyme was captured on both spot 1 and 2. Spot 1 was subsequently blocked by loading with 30 nM PF-562,271 at the beginning of each cycle. Concentration series' of the test compounds were injected over the spots at 25°C. The specific binding was calculated as difference between spot 2 and 1 signals followed by solvent correction. Fitting to a one site binding model yielded the kinetic rate constants ¾ and ka and the equilibrium binding constant KO ~krjka.
For compounds with an expected KD < 5 nM N-terminally GST-fused purified FAK enzyme was captured on spot 2 of the anti-GST antibody coated chip only. After the injection cycle of a compound the chip surface was regenerated with 0 mM glycine- HCi, pH2.2 before capturing the enzyme again. The binding sensorgrams were analysed as described before.
Results
Compound D (nM)
1 0.49
2 1.5
3 0.91
4 0.73
5 0.57
6 6.3
7 2.3
8 1.0
9 7.0
10 0.96
11 0.61
12 0.92
13 0.47
14 6.1
15 5.0
16 1.3
17 3.5
18 13
19 7.8
20 0.63
21 0.44
22 0.99
23 1.3
24 0.77
25 1.1
26 14
27 16.6
28 0.94
29 12.1
30 5.7
31 1.0
32 2.5
33 5.6
34 5.0
35 1.6
36 0.48
P397Y-FAK Inhibition !VSSD platform ce u!ar biomarker assay
Compounds of ihe invention may be tested for in vitro activity in the following assay: 96-well plates (cat#MA6000, Meso Scale Discovery) are coated with 30μίΛνβΙΙ of mouse monoclonal FAK antibody [63D5] (cat#ab72140, Abeam) pre-diluted in PBS to a concentration of 1 mg/mL. The plates are sealed with adhesive film and incubated for 16 hours at 4°C. The antibody is then flicked out of the plates and 150μί of 3% [w/v] Blocker A (cat#R93AA-1 , Meso Scale Discovery) is added. The plates are reseaied with adhesive film and incubated at room temperature on a shaker set at medium speed for 2 hours. The plates are then washed three times with a solution containing 5Gm Tris-HCI pH 7.5, 0.15M NaCI and 0.02% Tween-20, before cell lysate addition described below. Ceils are split 1 :2 into T150 cell culture flasks 2 days prior to compound treatment. On the day prior to compound treatment, 200 L media containing 20,000 cells is seeded into ail wells of white, clear-bottom, TC treated, clear, 96-well microtitre plates (cat#655098, Greiner Bio-One), and the plates are incubated at 37°C and 5% C02 for 36 hours. 1 pL/weli of compound is then added from dilution series prepared in D SO. Negative control wells receive the same volume of DMSO without compounds, and positive control wells receive 2μΜ of a control compound in the same volume of DMSO. Ceils are treated for 1 hour at 37°C and 5% C02. The media/compounds are then flicked off and 55pL/wel! of ice-cold complete lysis buffer is added. Complete lysis buffer is prepared by adding 1 tablet PhosSTOP complete phosphatase inhibitor (cat#04906837001 , Roche) and 1 tablet Complete, Mini,
EDTA-free, protease inhibitor (cat#04693159001 , Roche) per 1QmL of incomplete lysis buffer (150mM NaCI, 20mM Tris-HCI pH 7.5, 1 mM EDTA, 1 mlv1 EGTA, 1 % Triton-X 100). Plates are incubated on ice for 30 minutes, with 30 seconds high speed plate shaking every 5 minutes. 40μΙ_/ννβΙΙ of cell lysate is transferred to the coated, blocked and washed 96-well microtitre plates described above. The 96-well plates are sealed with adhesive film and incubated for 16 hours at 4°C. The plates are then washed three times with a solution containing 50mM Tris-HCI pH 7.5, 0.15M NaCI and 0.02% Tween-20 and tapped dry. 25 Uweli of detection solution (1 % [w/v] Blocker A (cat#R93AA-1 , Meso Scale Discovery) in 50mM Tris-HCI pH 7.5, 0.15M NaCI and 0.02% Tween-20, with 1 :600 rabbit polyclonal FAK phospho Y397 antibody
(cat#ab39987, Abeam), 1 :1000 anti-rabbit sulfo-tag antibody (cat#R32AB-1 Meso Scale Discovery) and 1 :40 reconstituted Blocker D-M (cat#D609-0100, Rockland Immunochemicals for Research)) is added, and the plates resealed with adhesive film and incubated for I hour at room temperature on a plate shaker set to medium speed. Plates are then washed three times with a solution containing 50mM Tris-HCI pH 7.5, 0.15M NaCI and 0.02% Tween-20 and tapped dry. 150pL/weii of Read Buffer T + Surfactant (cat#R92TC-1 , Meso Scale Discovery) is then added, and pFAK-397 levels quantified using a Meso Scale Discovery SECTOR Imager 6000 instrument. ICso values are determined by first calculating percent inhibition (%l) for each iysate relative to controls on the same plate (%I=(S-CP)/(CN-CP)) where S is the sample result, CN is the average result of DMSO only treated negative controls, and CP is the average result of 2μΜ treated positive controls. %l is plotted against compound concentration [I] and the data fitted using the following equation, %I=(A+((B- A)/(1+((C/[I])AD)))), where A is the lower asymptote, B is the upper asymptote, C is the ICSO value, and D is the slope factor.
Results for MDA -231 -LNA ceils
% response of
Compound ^ so (nM)
control at 2 μΜ
1 59 114
2 58 96
3 27 99
4 140 05
5 70 109
Ό 440 108
7 280 114
8 124 105
9 214 05
10 37 109
11 116 112
12 522 106
13 26 111
14 572 71
16 36 111
17 60 89
18 880 98
20 13 111
21 1 87
22 23 91
23 8 120
24 12 07
25 25 85
27 341 07
28 7 86
30 281 119
31 39 139
33 24 84
2D Cellular proliferation assay
Cells are split 1 :4 into T75 cell culture flasks two days prior to cell seeding. A variety of cancer cell lines can be utilized in this assay.
On the day of cell seeding 100μΙ_ΛνβΙΙ of media containing 1000-5000 cells are added to 96-weli microtitre plates (Cat.#655 180, greiner bio-one) except wells G12 and H12 to which 100μΙ of media is added. In a second plate, a single row of cells is seeded at the same concentration. This second plate is known as the t=0 plate and is used to calculate the relative ceil number prior to addition of test agent. The plates containing cells are incubated for 24 hours at 37°C/ 5%C02. 0.5μΙ_Λ/νβΙΙ of compound is then added from dilution series prepared in D SO. A compound with known potency is included for each set of plates in order to assess assay performance. Negative control wells receive the same volume of DMSO without compounds.
Background signal is determined from wells containing media alone. The t=0 plate is read using addition of a resazurin-based reagent (see below) on the day that other plates have compound added to them. Plates containing cells to which compound has been added are then incubated for 3 days at 37°C and 5% C02.
After 3 days of incubation, cell proliferation is quantified by addition of 20 μΙ/weil of a resazurin-based reagent with a typical composition as follows: Resazurin, Sigma# R7017-1G, 0.015% w/v; methylene blue, Sigma# MB-1 (25g), 0.0025% w/v;
potassium hexacyanoferrate (ill), Sigma# P8131-100G, 0.033 w/v; potassium hexacyanoferrate (II) trihydrate, Sigma# P9387-100G, 0.042% w/v; in PBS buffer. Plates are incubated with resazurin-based reagent for 1-4 hours (37°C, 5% C02) prior to the determination of fluorescence at, or near (579Ex/584Em).
Percentage inhibition of proliferation (%l) for each treated well relative to controls on the same plate is calculated using the equation %I=(S-B)-(T0-B)/(CN-B)-(T0-B) where S is the sample result B is the background fluorescence, T0 is the t=0 value and CN is the average result of DMSO only treated negative controls. For IC50
determination, %\ is plotted against compound concentration [I] and the data fitted using the following equation, %I=(AH-((B-A)/(1 +((C/[I])AD)))), where A is the lower asymptote, B is the upper asymptote, C is the IC50 value, and D is the slope factor.
Results for MDA -231-LNA cells
ί^* ^fS !C50 (μ )
1 1.15
2 3.21
3 1.63
4 1.73
5 1.54
6 3.91
7 1.73
8 1.16
9 3.58
10 1.43
11 2.25
12 1.70
13 >1.0
14 4.49
15 2.37
16 2.00

VEGFR3 Biochemical asay
Compounds of ihe invention may be tested for in vitro activity in the following assay: A biotin labeled peptide is used as substrate (amino acid sequence: Biotin-Glu-Gly- Pro-Trp-Leu-Glu-Glu-Glu-Glu-Glu-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NHz). VEGFR3 cytoplasmic domain (amino acids 798-1298) was purchased as N-terminal GST- fusion protein ("the enzyme"). The 15 μΙ assay reactions are run in Greiner brand white 384-we!l low volume plates. All reactions contained 10 mM HEPES pH 7.4, 10 mM gClz, 0.01 % (v/v) Tween-20, 50 μ Na3V04, 0.01 % (w/v) albumin from chicken egg white, 1 mM Dithiothreitol, 111 n peptide substrate, 500 μΜ ATP, and 3.8 ng/reaction enzyme, with the enzyme being omitted from negative control reactions. Compounds were added in a volume of 100 ni from dilution series
prepared in DMSO, positive and negative control reactions receiving the same volume DMSO without compound. The plates were sealed with adhesive seals and incubated for 90 minutes at 30 degree Celsius. The reactions were stopped with the detection reagents added at the same time as follows: Product formation was quantified as amplified luminescence between PerkinElmer AlphaScreen™ beads, using Streptavidin-coated donor and anti-phosphotyrosine (P-Tyr-100) acceptor beads. To each reaction, 5 μΙ containing 10 mM HEPES pH 7.4, 25 m NaCI, 100 mM EDTA, 0.01 % (v/v) Tween-20, and 6.25 Mg/m! of each bead type were added. Plates were incubated for 6 hours before being read on a PerkinElmer EnVision™ plate reader in HTS Alphascreen™ mode. iC50 values were obtained by calculating percent inhibition (%l) for each reaction relative to controls on the same plate (%I=(I-CN)/(CP-CN) where CN/ CP are the averages of the negative/ positive reactions, respectively), then fitting the %l data vs. compound concentration [I] to %I=(A+((B-A)/(1+((C/[IJ)AD)))) where A is the lower asymptote, B is the upper asymptote, C is the IC50 value, and D is the slope factor.
The above assay was also run in a slightly modified form in some cases (indicated below with *). in these cases, VEGFR3 cytoplasmic domain (amino acids 818-1177, lacking 949-1002 of UniProt accession number P35916) was expressed and purified as N~terminai Hexa-His-fusion protein ("the enzyme"), rather than using the N- terminal GST-fusion protein.
Results
Compound SCso (nM)
1 10
2 120*
3 1.1
4 4.0
5 12
6 52
7 48
8 4.8
9 2.5
10 4.0
11 2.5
12 12
13 70
14 101
15 37
16 11
17 31
18 664
19 37
20 5.0
21 25
22 15
23 12
24 3.9
25 14
26 51
27 30
28 10
29 68
30 7
31 12
32 5.1*
33 16*
34 184*
35 53*
36 16
37 254
38 23*
39 23*
40 46*
Rt4 Phospho EL!ISA assay
Compounds of ihe invention may be tested for in vitro activity in the following assay:
Adult human dermal lymphatic microvascular endothelial cells (HMVEC-dLyAD) (Cat# CC-28 0, Lonza) were seeded into clear-bottom, TC treated 12 well plates (Cat # 685180, Greiner Bio-One) in Endogro MV complete (Cat# SC E004,
Mil!ipore) at 200,000 cells/well (volume 1 mL), and the plates incubated at 37°C and 5% C02 for 6 hours. The media was replaced with Endogro Basal (Cat # SC E-B , iliipore) + 0.1 % BSA (Cat# A8412, Sigma) and cells incubated for a further period (overnight at 37°C and 5% C<¼).
96 well axisorp immuno plates (Cat # 439454, Nunc) were coated with 100μΙ_ of Total VEGFR2 capture antibody (Part # 84 888, Human Total VEGFR3/FK4 EL!SA Kit, Cat # DYC3491 , R&D Systems), or Phospho VEGF R3 Capture antibody (Part # 841885, Human Phospho VEGF R3/FK4 EL!SA Kit, Cat# DYC2724, R&D Systems). The plates were covered and incubated at room temperature overnight.
The coating antibody was flicked out and the plates washed three times with Wash Buffer (Phosphate buffered saline (137mM NaCI, 2.7n KCL, 8.1 nM Na2HP04, 1.5mL KH2P04, pH7.2-7.4), 0.05% Tween 20). SOOpL of Blocking buffer (5% v/v Tween 20, 5% w/v sucrose in PBS) was then added to wells and plate incubated for 2 hours at room temperature. Blocking solution is flicked out and plates washed three times and tapped dry.
Compound dilution series were prepared in Endogro basal (Cat # SCME-BM, Miliipore) + 0.1 % BSA (Cat# A8412, Sigma) with constant 0.1 % DMSO concentration. 439μΙ_ of sample or vehicle control was added to the ceil monolayers. Cells are treated for 1 hour at 37°C and 5% C02. 250ng/mL Recombinant human VEGF-C (Cat # 2179-VC, R & D Systems) added to wells and plates incubated for an additional 10 minutes at 37°C and 5% C02.
The media and compounds were removed and the cell monolayer washed once in Duibecco's Phosphate Buffered Saline (Cat # 21600-044, Invitrogen). 130μΙ_ of Lysis buffer added to wells and cell lysate harvested and transferred to tubes and stored on ice. Complete lysis buffer was prepared by adding 10μΙ_ Protease inhibitor Cocktail (Cat # P8340, Sigma-Aidrich), 10μί_ PMSF (Phenylmethanesulfonyl fluoride, Cat # P7626, Sigma-Aldrich, prepared as 5QQmM DMSO stock) per 1 mL of
PhosphosafeTM Extraction Reagent (Cat # 71296, Merck).
The harvested samples were then diluted 1 :2 in IC Diluent #18 (5% Tween 20/PBS) and 100μΙ_ transferred to the Total and Phospho VEGFR3 coated, blocked and washed 96 well plates and incubated for 2 hours at room temperature. The plates were then washed three times in wash buffer as described above and tapped dry. For detection of Total VEGFR3 100μ!_ of Detection antibody (Total VEGFR3
Detection Antibody Part# 841888 in Total VEGFR3 kit) diluted in IC Diluent #1 (1 % w/v BSA (Cat # A7906, Sigma-Aldrich)/PBS) was added to wells and the plate incubated for 2 hours at room temperature. The plate was then washed three times in wash buffer and tapped dry. 100μ!_ of streptavidin-HPR diluted in IC diluent #1 Streptavidin-HRP, Part # 890803 in Total VEGFR3 kit) was added to wells and incubated at room temperature for 20 minutes followed by washing as described above.100 μΐ_ Substrate solution (3,3',5,5'-Tetramethylbenzidine (TMB) Liquid Substrate System for ELISA, Cat # T0440, Sigma-Aldrich) was added and the plate incubated for 20 minutes in the dark at room temperature followed by the addition of 50μί. stop solution (2N H2S04).
Total VEGFR3 levels were quantified using a Mu!tiskan Ascent plate reader and Ascent software fitted with 450nm filter. For detection of Phospho VEGFR3, 100 L of Detection antibody (Anti-Phospho- Tyrosine-HRP Detection Antibody, Part # 841403 in Phospho VEGFR3 kit) was diluted in IC Diluent #1 (1 % w/v BSA/PBS), added to the wells and the plate incubated for 2 hours at room temperature. The plate was then washed three times in wash buffer as described above and tapped dry. 100 L Substrate solution (3,3',5,5!- Tetramethylbenzidine (TMB) Liquid Substrate System for ELISA, Cat # T0440,
Sigma-Aldrich) was added and the plate incubated for 20 minutes in the dark at room temperature followed by the addition of 50μί stop solution (2N H2S04).
Phospho VEGFR3 levels were quantified using a Multiscan ascent plate reader and ascent software fitted with 450nm filter.
ICso values are determined by first calculating the level of phospho VEGFR3 relative to Total VEGFR3 according to the following formula:
where SRP is the Sample Relative Phospho level, SP is Phospho VEGFR3 reading and ST is Total VEGFR3 reading.
Percent inhibition (%l) for each lysate relative to vehicle control (VEGF-C stimulated) is then calculated according to the following formula:
SMP¥sk&&& -SRP Tes
= - 1 f
SS Vektefa where SRP is the Sample Relative Phospho level as calculated above.
%i is plotted against compound concentration and data fitted using a Sigmoidal dose response curve (GraphPad Prism 4 for Windows) with the following equation (Y=Bottom + (Top-Bottom) / (1 +10Λ (LogECSG-X))} where X is the logarithm of the concentration, Y is the response. Y starts at Bottom and goes to Top with a sigmoid shape.
Results
Compound IC50 (πΜ)
3 80
5 240
8 127
11 67
12 81
16 36
17 44
20 30
21 113
22 30
24 71
25 66
30 658
31 67
33 81
36 169
Flt3 Biochemical Assay
Compounds of the invention may be tested for in vitro activity in the following assay: A biotin labelled peptide is used as substrate (amino acid sequence: Biotin-Glu-Gly- Pro-Trp-Leu-Glu-Glu-Glu-Glu-Glu-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2). Flt3 cytoplasmic domain (amino acids 584-993, NCBI Reference sequence
NP__004 10.1) was purchased from Carna Biosciences as GST-fusion protein ("the enzyme"). The 15 μΙ_ assay reactions are run in Greiner brand white 384-weli low volume plates. All reactions contained 10 mM HEPES pH 7.4, 10 mM MgCI2, 25 mM NaCl, 0.01 % (v/v) Tween-20, 50 μ Na3V04, 0.01 % (w/v) albumin from chicken egg white, 1 mM Dithiothreitol, 111 nM peptide substrate, 94 μΜ ATP, and 45 pg/reaction enzyme, with the enzyme being omitted from negative control reactions. Compounds were added in a volume of 100 nL from dilution series prepared in DMSO, positive and negative control reactions receiving the same volume DMSO without compound. The plates were sealed with adhesive seals and incubated for 90 minutes at 30 degree Celsius. The reactions were stopped with the detection reagents added at the same time as follows: Product formation was quantified as amplified luminescence between PerkinElmer AlphaScreen™ beads, using Streptavidin-coated donor and anti-phosphotyrosine (P-Tyr-100) acceptor beads. To each reaction, 5 μί. containing 10 mM HEPES pH 7.4, 25 mM NaCl, 100 mM EDTA, 0.01 % (v/v) Tween-20, and 6.25 pg/ml of each bead type were added. Plates were incubated for 4-6 hours before being read on a PerkinElmer EnVision™ plate reader in HTS Alphascreen™ mode. IC50 values were obtained by calculating percent inhibition (%l) for each reaction relative to controls on the same plate (%I=(I-CN)/(CP-CN) where CN/ CP are the averages of the negative/ positive reactions, respectively), then fitting the %l data vs. compound concentration [I] to %I=(A+((B-A)/(1 +((C/[I])AD)))) where A is the lower asymptote, B is the upper asymptote, C is the 1C50 value, and D is the slope factor.
Results
Compound iC50 (nM)
1 16
3 3
4 5
5 15
7 57
8 31
11 7
12 9
14 43
16 9
21 16
22 46
24 23
25 68
26 40
27 10
29 69
30 5
31 19
32 6
33 2
36 40
Commercial Flt3 Biochemical Assays
Compound 16 was also tested for inhibition in the following commercially available assays, where n is the number of repeats:
Kinase Assay performed by ICso (nM)
Flt3 CEREP 10 (n=1)
Flt3 Kinomescan 4.6 (n=2)
Flt3 (D835Y) CEREP 16 (n=1)
Fit3 (D835Y) Kinomescan 4.2 (n=2)
Flt3 (D835H) Kinomescan 6.6 (rn=2)
Flt3 (ITD) Kinomescan 3 (n=2)
Flt3 (K663Q) Kinomescan 2.6 (n=2)
Fit3 (N841 I) Kinomescan 1500 (n=2)
Fit3 (R834Q) Kinomescan 60 (n=2)
Flt3-autoi inhibited Kinomescan 310 (n=2)
Activity in leukemia cell Sines
Methods
MOLM-13 cells (ITD-Flt3 heierozygous), MV4-11 cells (ITD-Flt3 homozygous) and K562 cells (BCR-Abl +ve, negative control line) were used in 3 day proliferation assays to determine the anti-proiiferative potency of compound 18.
In brief, on the day of cell seeding 100mL well of media containing 1000-5000 cells are added to 96-well microtitre plates (Cat#855 180, greiner bio-one) except wells G12 and H12 to which 100μΙ of media is added. In a second plate, a single row of cells is seeded at the same concentration. This second plate is known as the t=0 plate and is used to calculate the relative cell number prior to addition of test agent. The plates containing cells are incubated for 1 h at 37°C7 5%C02. 0.5mL well of compound is then added from dilution series prepared in D SO. A compound with known potency is included for each set of plates in order to assess assay
performance. Negative control wells receive the same volume of DMSO without compounds. Background signal is determined from wells containing media alone. The t=0 plate is read using addition of a resazurin-based reagent (see below) on the day that other plates have compound added to them. Plates containing ceils to which compound has been added are then incubated for 3 days at 37°C and 5% C02.
After 3 days of incubation, cell proliferation is quantified by addition of 20 μΙ/well of a resazurin-based reagent with a typical composition as follows: Resazurin, Sigma# R7017-1G, 0.015% w/v; methylene blue, Sigma# MB-1 (25g), 0.0025% w/v;
potassium hexacyanoferrate (111), Sigma# P8131-100G, 0.033 w/v; potassium hexacyanoferrate (II) trihydrate, Sigma# P9387-100G, 0.042% w/v; in PBS buffer. Plates are incubated with resazurin-based reagent for 1-4 hours (37°C, 5% C02) prior to the determination of fluorescence at, or near (579Ex/584Em)-
Percentage inhibition of proliferation (%l) for each treated well relative to controls on the same plate is calculated using the equation %I=(S-B)-(T0-B)/(CN-B)-(T0-B) where S is the sample result B is the background fluorescence, T0 is the t=0 value and CN is the average result of DMSO only treated negative controls. For IC50 determination, %! is plotted against compound concentration [I] and the data fitted using the following equation, %l=(A+((B-A)/(1 ÷((C/[i])AD)))), where A is the lower asymptote, B is the upper asymptote, C is the 1C50 value, and D is the slope factor.
Results
Compound 16 inhibited leukemia cell growth with an IC50 in MV4-11 , MOLM-13 and K562 cells of: 65 ± 8 nfvi, 110 ±12 nfvi, 4,0 ± 0.43 μ (n = 3), respectively,
Activity in ectopic teukemia model
Female Balb/c nu/nu mice (6-8 weeks of age, 9-10 mice per group) were injected subcutaneously with 5 x 106 MV4-11 human acute myeloid leukemia (AML) cells in sterile phosphate buffered saline and mafrige! (1 : 1). When tumours were -250 mm3, mice were randomized to receive vehicle (0.2 ml hydroxypropylmethyi cellulose p.o. BID) or compound 16 equivalent to 25, 50 or 75 mg/kg p.o. BID. Tumour volumes were determined from regular caliper measurements during the experiment and mice were weighed every few days. Following 22 days of treatment, mice were killed by cervical dislocation 12 hours following the final dose of compound or vehicle and tumours harvested for measurement CD31 +ve blood vessel density and p-FLT3 levels by ELISA.
Statistical analysis was performed using a one-way ANOVA with a Dunnett post-hoc test.
Figure 1 (a) shows the tumour volume as follows: Vehicle (closed circles); compound 16 - 25 mg/kg BID (closed squares), 50 mg/kg BID (open circles), 75 mg/kg BID (closed triangles). The symbols represent the mean ± SEM of 9-10 mice. ** p≤ 0.01 , *** p < 0.001 compared to vehicle at day 22.
Figure 1 (b) shows the % body weight change of the mice studied, where the symbols represent the same as in Figure 1 (a).
Figure 2 shows the CD31 +ve blood vessel density in the tumours, as follows:
Vehicle (solid); compound 16 - 25 mg/kg BID (chequered), 50 mg/kg BID (horizontal strips), 75 mg/kg BID (vertical stripes). Bars represent the mean ± SEM of 9-10 mice per group. ** p < 0,01 , *** p < 0.001 compared to vehicle.
Figure 3 shows the inhibition of pTyr591-ITD-FLT3 in the primary tumours treated with compound 16 compared to primary tumours treated with the vehicle only, as
follows: Vehicle (solid); compound 16 - 25 mg/kg BID (chequered), 50 mg/kg BID (horizontal strips), 75 mg/kg BID (vertical stripes). Bars represent the mean ± SE of 9-10 mice per group. *** p < 0.001 compared to vehicle. These results demonstrate that compound 16 demonstrates efficacy in a preclinical ectopic model of ITD-FLT3 positive A L.
Activity in orthotopic leukemia modei
Female NOD SCID mice (6-8 weeks of age, 5 per group) were treated with cyclophosphamide (150 mg/kg i.p.) and 48 hours later, MV4-11 cells stably transduced to express iuciferase and m-cherry (MV4-11-luc, 5 x 106 cells) were injected i.v. in sterile saline. On day 20 after injection of cells, mice were randomized based on whole-body bioluminescence signal, to receive vehicle (0.2 ml
hydroxypropylmethyi cellulose p.o. BID) or compound 16 equivalent to 75 mg/kg p.o. BID. Mice were monitored daily and disease progression was determined twice weekly using bioluminescence imaging. Mice were weighed 2-3 times per week. Individual mice were killed when > 20% body weight was reached or when mice displayed signs of severe illness in accordance with approved ethical endpoinfs. Statistical analysis was performed using a one-way ANOVA with a Dunnett post-hoc test.
Figure 4 shows the tumour progression in the mice treated with either the vehicle alone or with compound 16. Figure 5 shows the whole body bioluminescence imaging of the test subjects, which corresponds to the disease progression, as follows: Vehicle (closed circles) and compound 16 (75 mg/kg BID; closed squares). Symbols represent the mean ± SEM of 4-5 mice per group. These results demonstrate that compound 16 demonstrates efficacy in a preclinical orthotopic model of ITD-FLT3 positive AML.
Claims
1. The use of a compound of the formula (I):

R is selected from: H and

RN1 is selected from H, C .3 aikyi and C(=0) e;
RN2 is selected from H, C 3 alkyl and C(=0) e;
RN3 is selected from H, C1-3 alkyl and C(=0) e;
RN4 is selected from H and CH3;
RN7 and RN8 are independently selected from H and CH3
RN9 is selected from H, d.3 alkyl and C(=0) e;
RN1° is selected from H, C 3 aikyi and C(=0)Me;
RN1 i is selected from H, C1..3 alkyl and C(=0) e; 2 is selected from H and
 wherein:
wherein:
RN5 is selected from H, C .3 aikyi and C(=0) e;
RN6 is selected from H, 01-3 alkyl and C(=0) e;
and wherein only one of R1 and R2 is H;
or R1 and R2 together form the group - H2-N(RN12)-C2H4-, where RN12 is selected from H, Ci-3 alkyi and C(=0)Me;
R4 is selected from CF3, halo, CF2H and CN; and
Rs is selected from

R6 is selected from H, (CHRc )n1C(0)N(RNe)Z1 and (CH2)n2C(0)OZ2; wherein: n1 is 1 ;
RC1 is H or Me;
RN12 is H or CH3;
Z1 is H, CH3 or OCH3;
n2 is 1 ; and
Z2 is CH3;
and where only one of RN12 and Z1 can be CH3,
R7, if present, is selected from H, and (CH2)miC(0)N(RM1)Y1, wherein:
ml is 0 or 1 ;
RM1 is H; and
Y1 is H, Me or OCH3;
and one of R6 and R7 is not H; and
R8, if present, is H or, when R' is C(=0)NH2, R8 is selected from H and C .2 al according to claim 1 , wherein R2 is H and R1 is:

, wherein RN is C(=0)Me.
according to claim 1 , wherein R2 is H and R1 is:

wherein RN is H, methyl or ethyl
4. The use according to claim 1 , wherein R2 is H and R1 is:
RN

, wherein R is selected from H, methyl and ethyl
The use according to claim 1 , wherein R is H and R ! is:

, wherein Rm is selected from H and methyl. use according to claim 1 , wherein R2 is H and R1 is:

, wherein R is selected from H and methyl. cording to claim 1 , wherein R2 is H and R is:

, wherein R and R are both H or both methyl e use according to claim 1 , wherein R2 is H and R1 is:
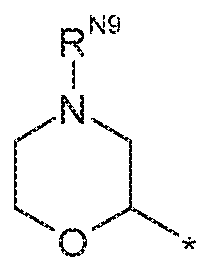
. wherein RN9 is H
9. The use according to claim 1 , wherein R2 is H and R is:
 , wherein R is selected from H and methyl
, wherein R is selected from H and methyl
10. The use according to claim 1 , wherein R is H and R ' is:

, wherein R is H
11. The use according to claim 1 , wherein R1 is H and R2 is:
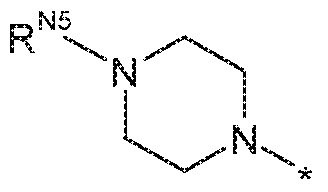
where R is selected from H and methyl.
12. The use according to claim 1 , wherein R is H and is:
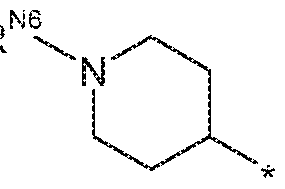
, where R is selected from H and methyl.
13. The use according to any one of claims 1 to 12, wherein R is selected from CF3, CI and CF2H .
14. The use according to claim 13, wherein R4 is CF3.
15. The use according to any one of claims 1 to 14, wherein R5 is a group of the following formula:

16. The use according to any one of claims 1 to 14, wherein R" is a group selected from:

(R5C) (Ra (R5e)
17. The use according to either claim 15 or claim 16, wherein R is H and R° is selected from CH2C(0)NH2, CH2C(0)NHCH3, CHCH3C(0)NH2 and
CHCH3C(0)NHCH3.
18. The use according to claim 17, wherein R7 is H and R6 is selected from CH2C(0)NH2, CHCH3C(0)NH2 and CH2C(0)NHCH3.
19. The use according to claim 18, wherein R7 is H and R6 is selected from CH2C(0)NH2, and CHCH3C(0)NH2. 20. The use according to either claim 15 or claim 16, wherein R6 is H and R7 is selected from C(0)NH2, C(0)NHCH3, CH2C(0)NH2 and CH2C(0)NHCH3.
21. The use according to claim 20, wherein R6 is H and R'* is C(0)NH2. 22. The use according to claim 21 , wherein R is methyl.
23. The use according to any one of claims 1 to 14, wherein R5 is a group of the following formula:

24. A compound as described in any one of claims 1 to 23 for use in ihe method of treatment of Acute Myeioid Leukemia or a disease ameliorated by the inhibition of F!t3, or Fit3 and FAK, 25. A method of treatment of the human or animal body suffering from Acute
Myeloid Leukemia or a disease ameliorated by the inhibition of Fit3, or Flt3 and FAK, including mutants thereof, comprising administering a compound as described in any one of claims 1 to 23. 28. The use according to any one of claims 1 to 23, a compound according to claim 24 or a method of treatment according to claim 25, wherein the Acute Myeloid leukemia is in patients who express activated forms of Flt3.
27. The use according to any one of claims 1 to 23, a compound according to claim 24 or a method of treatment according to claim 25, wherein the Acute Myeioid leukemia is in patients who express iTD-Flt3.
28. The use according to any one of claims 1 to 23, a compound according to claim 24 or a method of treatment according to claim 25, wherein the Acute Myeioid leukemia is in patients who express ITD~Flt3 and FAK.
29. The use according to any one of claims 1 to 23, a compound according to claim 24 or a method of treatment according to claim 25, wherein the Acute Myeloid leukemia is in patients who express ITD-Flt3 and constitutively active splice variants of FAK.
30. A method of inhibiting Flt3, or Flt3 and FAK in vitro or in vivo, comprising contacting a cell with an effective amount of a compound as described in any one of claims 1 to 23.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261682791P | 2012-08-14 | 2012-08-14 | |
| US61/682,791 | 2012-08-14 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014027199A1 true WO2014027199A1 (en) | 2014-02-20 |
Family
ID=49003936
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB2013/052163 WO2014027199A1 (en) | 2012-08-14 | 2013-08-14 | Fak and flt3 inhibitors |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2014027199A1 (en) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20140073620A1 (en) * | 2012-08-17 | 2014-03-13 | Cancer Therapeutics Crc Pty Limited | Vegfr3 inhibitors |
| US20140080798A1 (en) * | 2012-08-17 | 2014-03-20 | Cancer Therapeutics Crc Pty Limited | Vegfr3 inhibitors |
| WO2018124001A1 (en) | 2016-12-27 | 2018-07-05 | 国立研究開発法人理化学研究所 | Bmp-signal-inhibiting compound |
| WO2021237273A1 (en) * | 2020-05-28 | 2021-12-02 | Amplia Therapeutics Pty Ltd | Methods of treating pulmonary fibrosis |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010055117A1 (en) * | 2008-11-14 | 2010-05-20 | Boehringer Ingelheim International Gmbh | 2,4-diaminopyrimidine derivates as ptk2- inhibitors for the treatment of abnormal cell growth |
| WO2012022408A1 (en) * | 2010-08-18 | 2012-02-23 | Merck Patent Gmbh | Pyrimidine derivatives as fak inhibitors |
| WO2012110773A1 (en) * | 2011-02-17 | 2012-08-23 | Cancer Therapeutics Crc Pty Limited | Fak inhibitors |
| WO2012110774A1 (en) * | 2011-02-17 | 2012-08-23 | Cancer Therapeutics Crc Pty Limited | Selective fak inhibitors |
-
2013
- 2013-08-14 WO PCT/GB2013/052163 patent/WO2014027199A1/en active Application Filing
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010055117A1 (en) * | 2008-11-14 | 2010-05-20 | Boehringer Ingelheim International Gmbh | 2,4-diaminopyrimidine derivates as ptk2- inhibitors for the treatment of abnormal cell growth |
| WO2012022408A1 (en) * | 2010-08-18 | 2012-02-23 | Merck Patent Gmbh | Pyrimidine derivatives as fak inhibitors |
| WO2012110773A1 (en) * | 2011-02-17 | 2012-08-23 | Cancer Therapeutics Crc Pty Limited | Fak inhibitors |
| WO2012110774A1 (en) * | 2011-02-17 | 2012-08-23 | Cancer Therapeutics Crc Pty Limited | Selective fak inhibitors |
Non-Patent Citations (3)
| Title |
|---|
| ALEXANDER SCHULTZE & WALTER FIEDLER: "Therapeutic potential and limitations of new FAK inhibitors in the treatment of cancer", EXPERT OPINION ON INVESTIGATIONAL DRUGS, ASHLEY PUBLICATIONS LTD., LONDON, GB, vol. 19, no. 6, 1 June 2010 (2010-06-01), pages 777 - 788, XP008142458, ISSN: 1354-3784, DOI: 10.1517/13543784.2010.489548 * |
| ISOLDA CASANOVA ET AL: "A celecoxib derivative inhibits focal adhesion signaling and induces caspase-8-dependent apoptosis in human acute myeloid leukemia cells", INTERNATIONAL JOURNAL OF CANCER, vol. 123, no. 1, 1 July 2008 (2008-07-01), pages 217 - 226, XP055084347, ISSN: 0020-7136, DOI: 10.1002/ijc.23516 * |
| VITA M GOLUBOVSKAYA: "Focal Adhesion Kinase as a Cancer Therapy Target", ANTI-CANCER AGENTS IN MEDICINAL CHEMISTRY, BENTHAM SCIENCE PUBLISHERS LTD, NL, 1 December 2010 (2010-12-01), pages 1 - 13, XP007922333, ISSN: 1871-5206 * |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20140073620A1 (en) * | 2012-08-17 | 2014-03-13 | Cancer Therapeutics Crc Pty Limited | Vegfr3 inhibitors |
| US20140080798A1 (en) * | 2012-08-17 | 2014-03-20 | Cancer Therapeutics Crc Pty Limited | Vegfr3 inhibitors |
| EP2885292A4 (en) * | 2012-08-17 | 2015-07-01 | Cancer Therapeutics Crc Pty Ltd | Vegfr3 inhibitors |
| US9238644B2 (en) * | 2012-08-17 | 2016-01-19 | Cancer Therapeutics Crc Pty Limited | VEGFR3 inhibitors |
| US9266864B2 (en) * | 2012-08-17 | 2016-02-23 | Cancer Therapeutics Crc Pty Limited | VEGFR3 inhibitors |
| WO2018124001A1 (en) | 2016-12-27 | 2018-07-05 | 国立研究開発法人理化学研究所 | Bmp-signal-inhibiting compound |
| WO2021237273A1 (en) * | 2020-05-28 | 2021-12-02 | Amplia Therapeutics Pty Ltd | Methods of treating pulmonary fibrosis |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2012216893B2 (en) | FAK inhibitors | |
| AU2012216894B2 (en) | Selective FAK inhibitors | |
| EP3596073B1 (en) | Tricyclic compounds for use in treatment of proliferative disorders. | |
| EP3630749B1 (en) | 2-quinolone derived inhibitors of bcl6 | |
| KR20160090786A (en) | Certain chemical entities, compositions, and methods | |
| US9238644B2 (en) | VEGFR3 inhibitors | |
| US9266864B2 (en) | VEGFR3 inhibitors | |
| WO2014027199A1 (en) | Fak and flt3 inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13750922 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 13750922 Country of ref document: EP Kind code of ref document: A1 |