WO2014015088A1 - Amide, urea or sulfone amide linked benzothiazole inhibitors of endothelial lipase - Google Patents
Amide, urea or sulfone amide linked benzothiazole inhibitors of endothelial lipase Download PDFInfo
- Publication number
- WO2014015088A1 WO2014015088A1 PCT/US2013/050972 US2013050972W WO2014015088A1 WO 2014015088 A1 WO2014015088 A1 WO 2014015088A1 US 2013050972 W US2013050972 W US 2013050972W WO 2014015088 A1 WO2014015088 A1 WO 2014015088A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- independently
- pyrid
- substituted
- haloalkyl
- Prior art date
Links
- -1 sulfone amide Chemical class 0.000 title claims description 94
- 102100031375 Endothelial lipase Human genes 0.000 title description 51
- 101710087274 Endothelial lipase Proteins 0.000 title description 50
- 239000003112 inhibitor Substances 0.000 title description 16
- IOJUPLGTWVMSFF-UHFFFAOYSA-N benzothiazole Chemical compound C1=CC=C2SC=NC2=C1 IOJUPLGTWVMSFF-UHFFFAOYSA-N 0.000 title description 8
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 title description 7
- 150000001408 amides Chemical class 0.000 title description 7
- 239000004202 carbamide Substances 0.000 title description 4
- 150000001875 compounds Chemical class 0.000 claims abstract description 329
- 125000000217 alkyl group Chemical group 0.000 claims description 242
- 125000001188 haloalkyl group Chemical group 0.000 claims description 62
- 150000003839 salts Chemical class 0.000 claims description 55
- 125000003545 alkoxy group Chemical group 0.000 claims description 50
- 229910052736 halogen Inorganic materials 0.000 claims description 41
- 150000002367 halogens Chemical class 0.000 claims description 41
- 125000000623 heterocyclic group Chemical group 0.000 claims description 34
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 30
- 229910052739 hydrogen Inorganic materials 0.000 claims description 28
- 125000004432 carbon atom Chemical group C* 0.000 claims description 27
- 125000004438 haloalkoxy group Chemical group 0.000 claims description 26
- 229910052799 carbon Inorganic materials 0.000 claims description 22
- 125000003342 alkenyl group Chemical group 0.000 claims description 20
- 125000006297 carbonyl amino group Chemical group [H]N([*:2])C([*:1])=O 0.000 claims description 20
- 239000008194 pharmaceutical composition Substances 0.000 claims description 19
- 125000005842 heteroatom Chemical group 0.000 claims description 18
- 229910052717 sulfur Inorganic materials 0.000 claims description 18
- 229910052760 oxygen Inorganic materials 0.000 claims description 16
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 claims description 14
- 125000001072 heteroaryl group Chemical group 0.000 claims description 14
- 125000000714 pyrimidinyl group Chemical group 0.000 claims description 14
- 125000003226 pyrazolyl group Chemical group 0.000 claims description 11
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 10
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 9
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 claims description 8
- 125000000842 isoxazolyl group Chemical group 0.000 claims description 8
- 125000004076 pyridyl group Chemical group 0.000 claims description 8
- 125000004193 piperazinyl group Chemical group 0.000 claims description 7
- 125000005843 halogen group Chemical group 0.000 claims description 6
- 125000000565 sulfonamide group Chemical group 0.000 claims description 6
- 125000006570 (C5-C6) heteroaryl group Chemical group 0.000 claims description 5
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 5
- 125000004528 pyrimidin-5-yl group Chemical group N1=CN=CC(=C1)* 0.000 claims description 5
- MDFFNEOEWAXZRQ-UHFFFAOYSA-N aminyl Chemical compound [NH2] MDFFNEOEWAXZRQ-UHFFFAOYSA-N 0.000 claims description 4
- 125000001255 4-fluorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1F 0.000 claims description 3
- 125000006367 bivalent amino carbonyl group Chemical group [H]N([*:1])C([*:2])=O 0.000 claims description 3
- 125000004186 cyclopropylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C1([H])[H] 0.000 claims description 3
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 claims description 2
- 229910002091 carbon monoxide Inorganic materials 0.000 claims description 2
- 125000004312 morpholin-2-yl group Chemical group [H]N1C([H])([H])C([H])([H])OC([H])(*)C1([H])[H] 0.000 claims description 2
- 229910052702 rhenium Inorganic materials 0.000 claims description 2
- 239000000203 mixture Substances 0.000 abstract description 39
- 239000003814 drug Substances 0.000 abstract description 35
- 229940124770 endothelial lipase inhibitor Drugs 0.000 abstract description 3
- 238000000034 method Methods 0.000 description 189
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical class OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 148
- 239000002904 solvent Substances 0.000 description 101
- 229910001868 water Inorganic materials 0.000 description 100
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 97
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 88
- 238000004128 high performance liquid chromatography Methods 0.000 description 73
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 70
- 238000005160 1H NMR spectroscopy Methods 0.000 description 68
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 64
- ZMXDDKWLCZADIW-UHFFFAOYSA-N dimethylformamide Substances CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 59
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 53
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 51
- 239000011541 reaction mixture Substances 0.000 description 50
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 47
- 230000002829 reductive effect Effects 0.000 description 42
- 229910002092 carbon dioxide Inorganic materials 0.000 description 39
- 239000003795 chemical substances by application Substances 0.000 description 38
- 229910052796 boron Inorganic materials 0.000 description 37
- 239000012453 solvate Substances 0.000 description 36
- 238000011282 treatment Methods 0.000 description 32
- DLFVBJFMPXGRIB-UHFFFAOYSA-N Acetamide Chemical class CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 28
- 102000019267 Hepatic lipases Human genes 0.000 description 28
- 108050006747 Hepatic lipases Proteins 0.000 description 28
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 27
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 26
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 23
- 239000007787 solid Substances 0.000 description 23
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 22
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 22
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 22
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 21
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 21
- 102000015779 HDL Lipoproteins Human genes 0.000 description 20
- 108010010234 HDL Lipoproteins Proteins 0.000 description 20
- HEMHJVSKTPXQMS-UHFFFAOYSA-M sodium hydroxide Inorganic materials [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 20
- 239000000243 solution Substances 0.000 description 20
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 19
- 235000019439 ethyl acetate Nutrition 0.000 description 19
- 238000006243 chemical reaction Methods 0.000 description 18
- 230000000694 effects Effects 0.000 description 18
- 229940124597 therapeutic agent Drugs 0.000 description 18
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 16
- 208000032928 Dyslipidaemia Diseases 0.000 description 16
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 16
- 229910052757 nitrogen Inorganic materials 0.000 description 16
- 239000012071 phase Substances 0.000 description 16
- 208000029078 coronary artery disease Diseases 0.000 description 15
- 238000011321 prophylaxis Methods 0.000 description 15
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N silicon dioxide Inorganic materials O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 15
- 239000003643 water by type Substances 0.000 description 15
- 201000001320 Atherosclerosis Diseases 0.000 description 14
- 238000002360 preparation method Methods 0.000 description 14
- 239000000047 product Substances 0.000 description 14
- 125000001424 substituent group Chemical group 0.000 description 14
- 239000004480 active ingredient Substances 0.000 description 13
- 229940107161 cholesterol Drugs 0.000 description 13
- 201000010099 disease Diseases 0.000 description 13
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 13
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 12
- 125000003118 aryl group Chemical group 0.000 description 12
- 235000012000 cholesterol Nutrition 0.000 description 12
- 239000003937 drug carrier Substances 0.000 description 12
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 12
- 239000011734 sodium Substances 0.000 description 12
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 11
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 11
- 239000000651 prodrug Substances 0.000 description 11
- 229940002612 prodrug Drugs 0.000 description 11
- 229920006395 saturated elastomer Polymers 0.000 description 11
- 0 *c1ccc2nc(C(C(N*)=O)S(*)(=O)=O)[s]c2c1 Chemical compound *c1ccc2nc(C(C(N*)=O)S(*)(=O)=O)[s]c2c1 0.000 description 10
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 10
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 10
- 238000004519 manufacturing process Methods 0.000 description 10
- 238000004007 reversed phase HPLC Methods 0.000 description 10
- 239000000741 silica gel Substances 0.000 description 10
- 229910002027 silica gel Inorganic materials 0.000 description 10
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 10
- 241000282414 Homo sapiens Species 0.000 description 9
- 125000004429 atom Chemical group 0.000 description 9
- 238000004587 chromatography analysis Methods 0.000 description 9
- 125000004433 nitrogen atom Chemical group N* 0.000 description 9
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 8
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 8
- 229940121710 HMGCoA reductase inhibitor Drugs 0.000 description 8
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 8
- 239000002253 acid Substances 0.000 description 8
- 150000001412 amines Chemical class 0.000 description 8
- 229910052786 argon Inorganic materials 0.000 description 8
- 238000003556 assay Methods 0.000 description 8
- 239000002471 hydroxymethylglutaryl coenzyme A reductase inhibitor Substances 0.000 description 8
- 150000002632 lipids Chemical class 0.000 description 8
- 239000000463 material Substances 0.000 description 8
- 125000002950 monocyclic group Chemical group 0.000 description 8
- 239000002245 particle Substances 0.000 description 8
- 238000002953 preparative HPLC Methods 0.000 description 8
- 238000010898 silica gel chromatography Methods 0.000 description 8
- 238000002560 therapeutic procedure Methods 0.000 description 8
- 108090001060 Lipase Proteins 0.000 description 7
- 239000012267 brine Substances 0.000 description 7
- 150000002148 esters Chemical class 0.000 description 7
- 230000008569 process Effects 0.000 description 7
- WRIKHQLVHPKCJU-UHFFFAOYSA-N sodium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([Na])[Si](C)(C)C WRIKHQLVHPKCJU-UHFFFAOYSA-N 0.000 description 7
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 7
- 239000000126 substance Substances 0.000 description 7
- 239000005695 Ammonium acetate Substances 0.000 description 6
- 241000937413 Axia Species 0.000 description 6
- 208000024172 Cardiovascular disease Diseases 0.000 description 6
- 239000007821 HATU Substances 0.000 description 6
- 102000004882 Lipase Human genes 0.000 description 6
- 208000017170 Lipid metabolism disease Diseases 0.000 description 6
- ZMXDDKWLCZADIW-YYWVXINBSA-N N,N-dimethylformamide-d7 Chemical compound [2H]C(=O)N(C([2H])([2H])[2H])C([2H])([2H])[2H] ZMXDDKWLCZADIW-YYWVXINBSA-N 0.000 description 6
- 238000005481 NMR spectroscopy Methods 0.000 description 6
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 description 6
- 235000019257 ammonium acetate Nutrition 0.000 description 6
- 229940043376 ammonium acetate Drugs 0.000 description 6
- 239000003472 antidiabetic agent Substances 0.000 description 6
- 239000003524 antilipemic agent Substances 0.000 description 6
- 125000005605 benzo group Chemical group 0.000 description 6
- 230000015572 biosynthetic process Effects 0.000 description 6
- 239000003153 chemical reaction reagent Substances 0.000 description 6
- 208000035475 disorder Diseases 0.000 description 6
- 239000001257 hydrogen Substances 0.000 description 6
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 6
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 6
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 6
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 5
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 5
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 5
- 241000124008 Mammalia Species 0.000 description 5
- 208000008589 Obesity Diseases 0.000 description 5
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 5
- 239000002585 base Substances 0.000 description 5
- 125000002618 bicyclic heterocycle group Chemical group 0.000 description 5
- 206010012601 diabetes mellitus Diseases 0.000 description 5
- 229940079593 drug Drugs 0.000 description 5
- 125000001041 indolyl group Chemical group 0.000 description 5
- 235000020824 obesity Nutrition 0.000 description 5
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 5
- 229940096701 plain lipid modifying drug hmg coa reductase inhibitors Drugs 0.000 description 5
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 5
- 230000002441 reversible effect Effects 0.000 description 5
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 5
- 239000000725 suspension Substances 0.000 description 5
- 238000003786 synthesis reaction Methods 0.000 description 5
- 125000001544 thienyl group Chemical group 0.000 description 5
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 5
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 4
- PAQZWJGSJMLPMG-UHFFFAOYSA-N 2,4,6-tripropyl-1,3,5,2$l^{5},4$l^{5},6$l^{5}-trioxatriphosphinane 2,4,6-trioxide Chemical compound CCCP1(=O)OP(=O)(CCC)OP(=O)(CCC)O1 PAQZWJGSJMLPMG-UHFFFAOYSA-N 0.000 description 4
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 4
- 208000000563 Hyperlipoproteinemia Type II Diseases 0.000 description 4
- 206010020772 Hypertension Diseases 0.000 description 4
- 239000004367 Lipase Substances 0.000 description 4
- 102000004895 Lipoproteins Human genes 0.000 description 4
- 108090001030 Lipoproteins Proteins 0.000 description 4
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 4
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 4
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 4
- 239000013543 active substance Substances 0.000 description 4
- 230000000879 anti-atherosclerotic effect Effects 0.000 description 4
- 239000003529 anticholesteremic agent Substances 0.000 description 4
- 229940125708 antidiabetic agent Drugs 0.000 description 4
- 239000002830 appetite depressant Substances 0.000 description 4
- 239000012298 atmosphere Substances 0.000 description 4
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 4
- 125000002527 bicyclic carbocyclic group Chemical group 0.000 description 4
- 125000002619 bicyclic group Chemical group 0.000 description 4
- ILAHWRKJUDSMFH-UHFFFAOYSA-N boron tribromide Chemical compound BrB(Br)Br ILAHWRKJUDSMFH-UHFFFAOYSA-N 0.000 description 4
- 235000013877 carbamide Nutrition 0.000 description 4
- 150000001721 carbon Chemical group 0.000 description 4
- 210000004027 cell Anatomy 0.000 description 4
- 208000026106 cerebrovascular disease Diseases 0.000 description 4
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 4
- 230000034994 death Effects 0.000 description 4
- 231100000517 death Toxicity 0.000 description 4
- 230000018109 developmental process Effects 0.000 description 4
- 239000002552 dosage form Substances 0.000 description 4
- 201000001386 familial hypercholesterolemia Diseases 0.000 description 4
- 239000000706 filtrate Substances 0.000 description 4
- 238000009472 formulation Methods 0.000 description 4
- 125000002541 furyl group Chemical group 0.000 description 4
- 125000002883 imidazolyl group Chemical group 0.000 description 4
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 description 4
- 230000002401 inhibitory effect Effects 0.000 description 4
- 239000000543 intermediate Substances 0.000 description 4
- 235000019421 lipase Nutrition 0.000 description 4
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 4
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 4
- 125000001624 naphthyl group Chemical group 0.000 description 4
- 125000002971 oxazolyl group Chemical group 0.000 description 4
- 125000004430 oxygen atom Chemical group O* 0.000 description 4
- HXITXNWTGFUOAU-UHFFFAOYSA-N phenylboronic acid Chemical compound OB(O)C1=CC=CC=C1 HXITXNWTGFUOAU-UHFFFAOYSA-N 0.000 description 4
- 235000011007 phosphoric acid Nutrition 0.000 description 4
- 239000004033 plastic Substances 0.000 description 4
- 229910000027 potassium carbonate Inorganic materials 0.000 description 4
- 229910000160 potassium phosphate Inorganic materials 0.000 description 4
- 235000011009 potassium phosphates Nutrition 0.000 description 4
- 102000004169 proteins and genes Human genes 0.000 description 4
- 108090000623 proteins and genes Proteins 0.000 description 4
- 125000003373 pyrazinyl group Chemical group 0.000 description 4
- 108020003175 receptors Proteins 0.000 description 4
- 102000005962 receptors Human genes 0.000 description 4
- 229910000029 sodium carbonate Inorganic materials 0.000 description 4
- 239000011780 sodium chloride Substances 0.000 description 4
- 229910000104 sodium hydride Inorganic materials 0.000 description 4
- LPXPTNMVRIOKMN-UHFFFAOYSA-M sodium nitrite Chemical compound [Na+].[O-]N=O LPXPTNMVRIOKMN-UHFFFAOYSA-M 0.000 description 4
- 125000004434 sulfur atom Chemical group 0.000 description 4
- 125000003831 tetrazolyl group Chemical group 0.000 description 4
- 125000000335 thiazolyl group Chemical group 0.000 description 4
- 210000001519 tissue Anatomy 0.000 description 4
- 125000004306 triazinyl group Chemical group 0.000 description 4
- UFTFJSFQGQCHQW-UHFFFAOYSA-N triformin Chemical compound O=COCC(OC=O)COC=O UFTFJSFQGQCHQW-UHFFFAOYSA-N 0.000 description 4
- CXNIUSPIQKWYAI-UHFFFAOYSA-N xantphos Chemical compound C=12OC3=C(P(C=4C=CC=CC=4)C=4C=CC=CC=4)C=CC=C3C(C)(C)C2=CC=CC=1P(C=1C=CC=CC=1)C1=CC=CC=C1 CXNIUSPIQKWYAI-UHFFFAOYSA-N 0.000 description 4
- KZPYGQFFRCFCPP-UHFFFAOYSA-N 1,1'-bis(diphenylphosphino)ferrocene Chemical compound [Fe+2].C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=C[C-]1P(C=1C=CC=CC=1)C1=CC=CC=C1 KZPYGQFFRCFCPP-UHFFFAOYSA-N 0.000 description 3
- ZIIUUSVHCHPIQD-UHFFFAOYSA-N 2,4,6-trimethyl-N-[3-(trifluoromethyl)phenyl]benzenesulfonamide Chemical compound CC1=CC(C)=CC(C)=C1S(=O)(=O)NC1=CC=CC(C(F)(F)F)=C1 ZIIUUSVHCHPIQD-UHFFFAOYSA-N 0.000 description 3
- OISVCGZHLKNMSJ-UHFFFAOYSA-N 2,6-dimethylpyridine Chemical compound CC1=CC=CC(C)=N1 OISVCGZHLKNMSJ-UHFFFAOYSA-N 0.000 description 3
- DGUVEYAZTOUCEJ-UHFFFAOYSA-N 2-aminoethanesulfonamide;hydron;chloride Chemical compound Cl.NCCS(N)(=O)=O DGUVEYAZTOUCEJ-UHFFFAOYSA-N 0.000 description 3
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 3
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 3
- 108010023302 HDL Cholesterol Proteins 0.000 description 3
- 101000941275 Homo sapiens Endothelial lipase Proteins 0.000 description 3
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- 108010028554 LDL Cholesterol Proteins 0.000 description 3
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 3
- 108010064785 Phospholipases Proteins 0.000 description 3
- 102000015439 Phospholipases Human genes 0.000 description 3
- 239000004793 Polystyrene Substances 0.000 description 3
- 208000006011 Stroke Diseases 0.000 description 3
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 3
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 3
- 239000000556 agonist Substances 0.000 description 3
- 125000001931 aliphatic group Chemical group 0.000 description 3
- 125000002947 alkylene group Chemical group 0.000 description 3
- 125000000304 alkynyl group Chemical group 0.000 description 3
- 239000000883 anti-obesity agent Substances 0.000 description 3
- 229940125710 antiobesity agent Drugs 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- KXDAEFPNCMNJSK-UHFFFAOYSA-N benzene carboxamide Natural products NC(=O)C1=CC=CC=C1 KXDAEFPNCMNJSK-UHFFFAOYSA-N 0.000 description 3
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 3
- 210000004369 blood Anatomy 0.000 description 3
- 239000008280 blood Substances 0.000 description 3
- 229910000024 caesium carbonate Inorganic materials 0.000 description 3
- 239000002775 capsule Substances 0.000 description 3
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 3
- 238000007796 conventional method Methods 0.000 description 3
- OPQARKPSCNTWTJ-UHFFFAOYSA-L copper(ii) acetate Chemical compound [Cu+2].CC([O-])=O.CC([O-])=O OPQARKPSCNTWTJ-UHFFFAOYSA-L 0.000 description 3
- 230000002596 correlated effect Effects 0.000 description 3
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 3
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 3
- NLUNLVTVUDIHFE-UHFFFAOYSA-N cyclooctylcyclooctane Chemical compound C1CCCCCCC1C1CCCCCCC1 NLUNLVTVUDIHFE-UHFFFAOYSA-N 0.000 description 3
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 3
- 238000011161 development Methods 0.000 description 3
- UAOMVDZJSHZZME-UHFFFAOYSA-N diisopropylamine Chemical compound CC(C)NC(C)C UAOMVDZJSHZZME-UHFFFAOYSA-N 0.000 description 3
- 210000002889 endothelial cell Anatomy 0.000 description 3
- 238000003818 flash chromatography Methods 0.000 description 3
- 235000019253 formic acid Nutrition 0.000 description 3
- 230000036541 health Effects 0.000 description 3
- 230000007062 hydrolysis Effects 0.000 description 3
- 238000006460 hydrolysis reaction Methods 0.000 description 3
- 230000003301 hydrolyzing effect Effects 0.000 description 3
- 125000002632 imidazolidinyl group Chemical group 0.000 description 3
- JYGFTBXVXVMTGB-UHFFFAOYSA-N indolin-2-one Chemical compound C1=CC=C2NC(=O)CC2=C1 JYGFTBXVXVMTGB-UHFFFAOYSA-N 0.000 description 3
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 3
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 3
- 230000000670 limiting effect Effects 0.000 description 3
- 238000004949 mass spectrometry Methods 0.000 description 3
- 230000004060 metabolic process Effects 0.000 description 3
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Natural products C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 3
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 3
- 150000007522 mineralic acids Chemical class 0.000 description 3
- 125000002757 morpholinyl group Chemical group 0.000 description 3
- 208000010125 myocardial infarction Diseases 0.000 description 3
- 231100000252 nontoxic Toxicity 0.000 description 3
- 230000003000 nontoxic effect Effects 0.000 description 3
- 125000001715 oxadiazolyl group Chemical group 0.000 description 3
- 125000000160 oxazolidinyl group Chemical group 0.000 description 3
- 239000000123 paper Substances 0.000 description 3
- 239000000546 pharmaceutical excipient Substances 0.000 description 3
- 150000003904 phospholipids Chemical class 0.000 description 3
- 125000003386 piperidinyl group Chemical group 0.000 description 3
- 125000003367 polycyclic group Polymers 0.000 description 3
- 229920000642 polymer Polymers 0.000 description 3
- 230000009862 primary prevention Effects 0.000 description 3
- 125000006239 protecting group Chemical group 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- 125000000168 pyrrolyl group Chemical group 0.000 description 3
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 3
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 3
- 230000009863 secondary prevention Effects 0.000 description 3
- 238000010189 synthetic method Methods 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 3
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 3
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 3
- CXWXQJXEFPUFDZ-UHFFFAOYSA-N tetralin Chemical compound C1=CC=C2CCCCC2=C1 CXWXQJXEFPUFDZ-UHFFFAOYSA-N 0.000 description 3
- 230000001225 therapeutic effect Effects 0.000 description 3
- 230000009466 transformation Effects 0.000 description 3
- 238000000844 transformation Methods 0.000 description 3
- 150000003626 triacylglycerols Chemical class 0.000 description 3
- 125000001425 triazolyl group Chemical group 0.000 description 3
- DCXXMTOCNZCJGO-UHFFFAOYSA-N tristearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(OC(=O)CCCCCCCCCCCCCCCCC)COC(=O)CCCCCCCCCCCCCCCCC DCXXMTOCNZCJGO-UHFFFAOYSA-N 0.000 description 3
- 208000019553 vascular disease Diseases 0.000 description 3
- MKXNIPKWBZUUAJ-UHFFFAOYSA-N (3-propan-2-yl-1,2-oxazol-5-yl)methanamine Chemical compound CC(C)C=1C=C(CN)ON=1 MKXNIPKWBZUUAJ-UHFFFAOYSA-N 0.000 description 2
- ZGGHKIMDNBDHJB-NRFPMOEYSA-M (3R,5S)-fluvastatin sodium Chemical compound [Na+].C12=CC=CC=C2N(C(C)C)C(\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O)=C1C1=CC=C(F)C=C1 ZGGHKIMDNBDHJB-NRFPMOEYSA-M 0.000 description 2
- 125000006656 (C2-C4) alkenyl group Chemical group 0.000 description 2
- 125000004514 1,2,4-thiadiazolyl group Chemical group 0.000 description 2
- RBIZQDIIVYJNRS-UHFFFAOYSA-N 1,3-benzothiazole-5-carboxylic acid Chemical compound OC(=O)C1=CC=C2SC=NC2=C1 RBIZQDIIVYJNRS-UHFFFAOYSA-N 0.000 description 2
- BDNKZNFMNDZQMI-UHFFFAOYSA-N 1,3-diisopropylcarbodiimide Chemical compound CC(C)N=C=NC(C)C BDNKZNFMNDZQMI-UHFFFAOYSA-N 0.000 description 2
- NZMICYAXDXTDJV-UHFFFAOYSA-N 1-(2-methoxyethyl)-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrazole Chemical compound C1=NN(CCOC)C=C1B1OC(C)(C)C(C)(C)O1 NZMICYAXDXTDJV-UHFFFAOYSA-N 0.000 description 2
- DFPYXQYWILNVAU-UHFFFAOYSA-N 1-hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1.C1=CC=C2N(O)N=NC2=C1 DFPYXQYWILNVAU-UHFFFAOYSA-N 0.000 description 2
- GELKGHVAFRCJNA-UHFFFAOYSA-N 2,2-Dimethyloxirane Chemical compound CC1(C)CO1 GELKGHVAFRCJNA-UHFFFAOYSA-N 0.000 description 2
- FIZJAXWVKMBXTN-UHFFFAOYSA-N 2-[1-methylsulfonyl-2-oxo-2-(2-sulfamoylethylamino)ethyl]-1,3-benzothiazole-5-carboxylic acid Chemical compound OC(=O)C1=CC=C2SC(C(C(=O)NCCS(N)(=O)=O)S(=O)(=O)C)=NC2=C1 FIZJAXWVKMBXTN-UHFFFAOYSA-N 0.000 description 2
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 2
- SSLMAEFYOURYEK-UHFFFAOYSA-N 2-[6-[4-(morpholine-4-carbonyl)phenyl]-1,3-benzothiazol-2-yl]-2-(2-morpholin-4-ylethylsulfonyl)-n-(2-sulfamoylethyl)acetamide Chemical compound C1COCCN1CCS(=O)(=O)C(C(=O)NCCS(=O)(=O)N)C(SC1=C2)=NC1=CC=C2C(C=C1)=CC=C1C(=O)N1CCOCC1 SSLMAEFYOURYEK-UHFFFAOYSA-N 0.000 description 2
- ILPUOPPYSQEBNJ-UHFFFAOYSA-N 2-methyl-2-phenoxypropanoic acid Chemical class OC(=O)C(C)(C)OC1=CC=CC=C1 ILPUOPPYSQEBNJ-UHFFFAOYSA-N 0.000 description 2
- 239000005541 ACE inhibitor Substances 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- 102000057234 Acyl transferases Human genes 0.000 description 2
- 108700016155 Acyl transferases Proteins 0.000 description 2
- 208000024827 Alzheimer disease Diseases 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 2
- 206010002383 Angina Pectoris Diseases 0.000 description 2
- 229940123413 Angiotensin II antagonist Drugs 0.000 description 2
- 108010059886 Apolipoprotein A-I Proteins 0.000 description 2
- 102000005666 Apolipoprotein A-I Human genes 0.000 description 2
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 2
- XUKUURHRXDUEBC-KAYWLYCHSA-N Atorvastatin Chemical compound C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CC[C@@H](O)C[C@@H](O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-KAYWLYCHSA-N 0.000 description 2
- XUKUURHRXDUEBC-UHFFFAOYSA-N Atorvastatin Natural products C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CCC(O)CC(O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-UHFFFAOYSA-N 0.000 description 2
- KZMGYPLQYOPHEL-UHFFFAOYSA-N Boron trifluoride etherate Chemical compound FB(F)F.CCOCC KZMGYPLQYOPHEL-UHFFFAOYSA-N 0.000 description 2
- JBPPSLURCSFQDH-UHFFFAOYSA-N CC(N1Cc2ccccc2CC1)=O Chemical compound CC(N1Cc2ccccc2CC1)=O JBPPSLURCSFQDH-UHFFFAOYSA-N 0.000 description 2
- YJRQBOOMJGYUPI-UHFFFAOYSA-N CN1c(cccc2)c2OCC1 Chemical compound CN1c(cccc2)c2OCC1 YJRQBOOMJGYUPI-UHFFFAOYSA-N 0.000 description 2
- IFCGSCOEBXOAKM-UHFFFAOYSA-N CS(C(C(NCCS(N)(=O)=O)=O)c([s]c1c2)nc1ccc2Br)(=O)=O Chemical compound CS(C(C(NCCS(N)(=O)=O)=O)c([s]c1c2)nc1ccc2Br)(=O)=O IFCGSCOEBXOAKM-UHFFFAOYSA-N 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- 102000012336 Cholesterol Ester Transfer Proteins Human genes 0.000 description 2
- 108010061846 Cholesterol Ester Transfer Proteins Proteins 0.000 description 2
- 229920001268 Cholestyramine Polymers 0.000 description 2
- 229920002911 Colestipol Polymers 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- 239000004131 EU approved raising agent Substances 0.000 description 2
- 208000037487 Endotoxemia Diseases 0.000 description 2
- 239000007995 HEPES buffer Substances 0.000 description 2
- 206010019280 Heart failures Diseases 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 2
- 102000004286 Hydroxymethylglutaryl CoA Reductases Human genes 0.000 description 2
- 108090000895 Hydroxymethylglutaryl CoA Reductases Proteins 0.000 description 2
- 208000035150 Hypercholesterolemia Diseases 0.000 description 2
- 206010021024 Hypolipidaemia Diseases 0.000 description 2
- 102000036770 Islet Amyloid Polypeptide Human genes 0.000 description 2
- 108010041872 Islet Amyloid Polypeptide Proteins 0.000 description 2
- 238000008214 LDL Cholesterol Methods 0.000 description 2
- 102000000853 LDL receptors Human genes 0.000 description 2
- 108010001831 LDL receptors Proteins 0.000 description 2
- 108010013563 Lipoprotein Lipase Proteins 0.000 description 2
- 102000043296 Lipoprotein lipases Human genes 0.000 description 2
- 102100024640 Low-density lipoprotein receptor Human genes 0.000 description 2
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- PCZOHLXUXFIOCF-UHFFFAOYSA-N Monacolin X Natural products C12C(OC(=O)C(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 PCZOHLXUXFIOCF-UHFFFAOYSA-N 0.000 description 2
- HSHXDCVZWHOWCS-UHFFFAOYSA-N N'-hexadecylthiophene-2-carbohydrazide Chemical compound CCCCCCCCCCCCCCCCNNC(=O)c1cccs1 HSHXDCVZWHOWCS-UHFFFAOYSA-N 0.000 description 2
- 150000001204 N-oxides Chemical class 0.000 description 2
- DFPAKSUCGFBDDF-UHFFFAOYSA-N Nicotinamide Chemical compound NC(=O)C1=CC=CN=C1 DFPAKSUCGFBDDF-UHFFFAOYSA-N 0.000 description 2
- TZOYXRMEFDYWDQ-UHFFFAOYSA-N O=C(CC1)Nc2c1cccc2 Chemical compound O=C(CC1)Nc2c1cccc2 TZOYXRMEFDYWDQ-UHFFFAOYSA-N 0.000 description 2
- 229910002666 PdCl2 Inorganic materials 0.000 description 2
- 208000005764 Peripheral Arterial Disease Diseases 0.000 description 2
- 208000030831 Peripheral arterial occlusive disease Diseases 0.000 description 2
- 208000018262 Peripheral vascular disease Diseases 0.000 description 2
- TUZYXOIXSAXUGO-UHFFFAOYSA-N Pravastatin Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(O)C=C21 TUZYXOIXSAXUGO-UHFFFAOYSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- 206010063837 Reperfusion injury Diseases 0.000 description 2
- YASAKCUCGLMORW-UHFFFAOYSA-N Rosiglitazone Chemical compound C=1C=CC=NC=1N(C)CCOC(C=C1)=CC=C1CC1SC(=O)NC1=O YASAKCUCGLMORW-UHFFFAOYSA-N 0.000 description 2
- RYMZZMVNJRMUDD-UHFFFAOYSA-N SJ000286063 Natural products C12C(OC(=O)C(C)(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 RYMZZMVNJRMUDD-UHFFFAOYSA-N 0.000 description 2
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- 239000005864 Sulphur Substances 0.000 description 2
- 229910021626 Tin(II) chloride Inorganic materials 0.000 description 2
- 206010045261 Type IIa hyperlipidaemia Diseases 0.000 description 2
- 108010062497 VLDL Lipoproteins Proteins 0.000 description 2
- 206010047249 Venous thrombosis Diseases 0.000 description 2
- RAFKCLFWELPONH-UHFFFAOYSA-N acetonitrile;dichloromethane Chemical compound CC#N.ClCCl RAFKCLFWELPONH-UHFFFAOYSA-N 0.000 description 2
- 229960001138 acetylsalicylic acid Drugs 0.000 description 2
- 230000002378 acidificating effect Effects 0.000 description 2
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 2
- 125000004450 alkenylene group Chemical group 0.000 description 2
- 125000004419 alkynylene group Chemical group 0.000 description 2
- VSCWAEJMTAWNJL-UHFFFAOYSA-K aluminium trichloride Chemical compound Cl[Al](Cl)Cl VSCWAEJMTAWNJL-UHFFFAOYSA-K 0.000 description 2
- 235000011114 ammonium hydroxide Nutrition 0.000 description 2
- 239000002333 angiotensin II receptor antagonist Substances 0.000 description 2
- 229940044094 angiotensin-converting-enzyme inhibitor Drugs 0.000 description 2
- 239000003957 anion exchange resin Substances 0.000 description 2
- 229940125709 anorectic agent Drugs 0.000 description 2
- 230000001708 anti-dyslipidemic effect Effects 0.000 description 2
- 230000001346 anti-hypertriglyceridemic effect Effects 0.000 description 2
- 229940121363 anti-inflammatory agent Drugs 0.000 description 2
- 239000002260 anti-inflammatory agent Substances 0.000 description 2
- 230000002253 anti-ischaemic effect Effects 0.000 description 2
- 230000003063 anti-neuropathic effect Effects 0.000 description 2
- 230000002946 anti-pancreatic effect Effects 0.000 description 2
- 230000002769 anti-restenotic effect Effects 0.000 description 2
- 239000003146 anticoagulant agent Substances 0.000 description 2
- 229940125682 antidementia agent Drugs 0.000 description 2
- 239000002220 antihypertensive agent Substances 0.000 description 2
- 229940030600 antihypertensive agent Drugs 0.000 description 2
- 239000003963 antioxidant agent Substances 0.000 description 2
- 230000003078 antioxidant effect Effects 0.000 description 2
- 235000006708 antioxidants Nutrition 0.000 description 2
- 229940127218 antiplatelet drug Drugs 0.000 description 2
- 229960004676 antithrombotic agent Drugs 0.000 description 2
- 229960005370 atorvastatin Drugs 0.000 description 2
- 125000004604 benzisothiazolyl group Chemical group S1N=C(C2=C1C=CC=C2)* 0.000 description 2
- 125000004603 benzisoxazolyl group Chemical group O1N=C(C2=C1C=CC=C2)* 0.000 description 2
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 2
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 2
- 125000004935 benzoxazolinyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 2
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 2
- 125000005512 benztetrazolyl group Chemical group 0.000 description 2
- 239000002876 beta blocker Substances 0.000 description 2
- 229940097320 beta blocking agent Drugs 0.000 description 2
- 229960000516 bezafibrate Drugs 0.000 description 2
- IIBYAHWJQTYFKB-UHFFFAOYSA-N bezafibrate Chemical compound C1=CC(OC(C)(C)C(O)=O)=CC=C1CCNC(=O)C1=CC=C(Cl)C=C1 IIBYAHWJQTYFKB-UHFFFAOYSA-N 0.000 description 2
- 229920000080 bile acid sequestrant Polymers 0.000 description 2
- 229940096699 bile acid sequestrants Drugs 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- 210000004204 blood vessel Anatomy 0.000 description 2
- 125000001246 bromo group Chemical group Br* 0.000 description 2
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 2
- 201000011510 cancer Diseases 0.000 description 2
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 2
- 125000002837 carbocyclic group Chemical group 0.000 description 2
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 2
- 238000001460 carbon-13 nuclear magnetic resonance spectrum Methods 0.000 description 2
- 239000011111 cardboard Substances 0.000 description 2
- 238000001023 centrifugal evaporation Methods 0.000 description 2
- GPUADMRJQVPIAS-QCVDVZFFSA-M cerivastatin sodium Chemical compound [Na+].COCC1=C(C(C)C)N=C(C(C)C)C(\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O)=C1C1=CC=C(F)C=C1 GPUADMRJQVPIAS-QCVDVZFFSA-M 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- 239000007795 chemical reaction product Substances 0.000 description 2
- 230000001906 cholesterol absorption Effects 0.000 description 2
- 125000003016 chromanyl group Chemical group O1C(CCC2=CC=CC=C12)* 0.000 description 2
- 229960001214 clofibrate Drugs 0.000 description 2
- KNHUKKLJHYUCFP-UHFFFAOYSA-N clofibrate Chemical compound CCOC(=O)C(C)(C)OC1=CC=C(Cl)C=C1 KNHUKKLJHYUCFP-UHFFFAOYSA-N 0.000 description 2
- AGVAZMGAQJOSFJ-WZHZPDAFSA-M cobalt(2+);[(2r,3s,4r,5s)-5-(5,6-dimethylbenzimidazol-1-yl)-4-hydroxy-2-(hydroxymethyl)oxolan-3-yl] [(2r)-1-[3-[(1r,2r,3r,4z,7s,9z,12s,13s,14z,17s,18s,19r)-2,13,18-tris(2-amino-2-oxoethyl)-7,12,17-tris(3-amino-3-oxopropyl)-3,5,8,8,13,15,18,19-octamethyl-2 Chemical compound [Co+2].N#[C-].[N-]([C@@H]1[C@H](CC(N)=O)[C@@]2(C)CCC(=O)NC[C@@H](C)OP(O)(=O)O[C@H]3[C@H]([C@H](O[C@@H]3CO)N3C4=CC(C)=C(C)C=C4N=C3)O)\C2=C(C)/C([C@H](C\2(C)C)CCC(N)=O)=N/C/2=C\C([C@H]([C@@]/2(CC(N)=O)C)CCC(N)=O)=N\C\2=C(C)/C2=N[C@]1(C)[C@@](C)(CC(N)=O)[C@@H]2CCC(N)=O AGVAZMGAQJOSFJ-WZHZPDAFSA-M 0.000 description 2
- 239000005515 coenzyme Substances 0.000 description 2
- 230000019771 cognition Effects 0.000 description 2
- 229960002604 colestipol Drugs 0.000 description 2
- GMRWGQCZJGVHKL-UHFFFAOYSA-N colestipol Chemical compound ClCC1CO1.NCCNCCNCCNCCN GMRWGQCZJGVHKL-UHFFFAOYSA-N 0.000 description 2
- 239000003636 conditioned culture medium Substances 0.000 description 2
- 239000006071 cream Substances 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- 125000001162 cycloheptenyl group Chemical group C1(=CCCCCC1)* 0.000 description 2
- NXQGGXCHGDYOHB-UHFFFAOYSA-L cyclopenta-1,4-dien-1-yl(diphenyl)phosphane;dichloropalladium;iron(2+) Chemical compound [Fe+2].Cl[Pd]Cl.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1.[CH-]1C=CC(P(C=2C=CC=CC=2)C=2C=CC=CC=2)=C1 NXQGGXCHGDYOHB-UHFFFAOYSA-L 0.000 description 2
- DEZRYPDIMOWBDS-UHFFFAOYSA-N dcm dichloromethane Chemical compound ClCCl.ClCCl DEZRYPDIMOWBDS-UHFFFAOYSA-N 0.000 description 2
- NNBZCPXTIHJBJL-UHFFFAOYSA-N decalin Chemical compound C1CCCC2CCCCC21 NNBZCPXTIHJBJL-UHFFFAOYSA-N 0.000 description 2
- FMGSKLZLMKYGDP-USOAJAOKSA-N dehydroepiandrosterone Chemical compound C1[C@@H](O)CC[C@]2(C)[C@H]3CC[C@](C)(C(CC4)=O)[C@@H]4[C@@H]3CC=C21 FMGSKLZLMKYGDP-USOAJAOKSA-N 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 2
- BPHQZTVXXXJVHI-UHFFFAOYSA-N dimyristoyl phosphatidylglycerol Chemical compound CCCCCCCCCCCCCC(=O)OCC(COP(O)(=O)OCC(O)CO)OC(=O)CCCCCCCCCCCCC BPHQZTVXXXJVHI-UHFFFAOYSA-N 0.000 description 2
- 150000002170 ethers Chemical class 0.000 description 2
- NRVKXPLEMJKYKS-UHFFFAOYSA-N ethyl 2-(6-bromo-1,3-benzothiazol-2-yl)acetate Chemical compound C1=C(Br)C=C2SC(CC(=O)OCC)=NC2=C1 NRVKXPLEMJKYKS-UHFFFAOYSA-N 0.000 description 2
- 239000000284 extract Substances 0.000 description 2
- 229960002297 fenofibrate Drugs 0.000 description 2
- YMTINGFKWWXKFG-UHFFFAOYSA-N fenofibrate Chemical compound C1=CC(OC(C)(C)C(=O)OC(C)C)=CC=C1C(=O)C1=CC=C(Cl)C=C1 YMTINGFKWWXKFG-UHFFFAOYSA-N 0.000 description 2
- 229940125753 fibrate Drugs 0.000 description 2
- 239000002319 fibrinogen receptor antagonist Substances 0.000 description 2
- 238000001914 filtration Methods 0.000 description 2
- 125000001153 fluoro group Chemical group F* 0.000 description 2
- 229960003765 fluvastatin Drugs 0.000 description 2
- BTCSSZJGUNDROE-UHFFFAOYSA-N gamma-aminobutyric acid Chemical compound NCCCC(O)=O BTCSSZJGUNDROE-UHFFFAOYSA-N 0.000 description 2
- 239000007789 gas Substances 0.000 description 2
- 210000001035 gastrointestinal tract Anatomy 0.000 description 2
- 239000003292 glue Substances 0.000 description 2
- ZNNLBTZKUZBEKO-UHFFFAOYSA-N glyburide Chemical compound COC1=CC=C(Cl)C=C1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)NC2CCCCC2)C=C1 ZNNLBTZKUZBEKO-UHFFFAOYSA-N 0.000 description 2
- 150000002430 hydrocarbons Chemical group 0.000 description 2
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 2
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 2
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 2
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 2
- 208000006575 hypertriglyceridemia Diseases 0.000 description 2
- 208000029498 hypoalphalipoproteinemia Diseases 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 239000000411 inducer Substances 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- 239000004615 ingredient Substances 0.000 description 2
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- 210000000936 intestine Anatomy 0.000 description 2
- 125000004936 isatinoyl group Chemical group N1(C(=O)C(=O)C2=CC=CC=C12)C(=O)* 0.000 description 2
- 208000028867 ischemia Diseases 0.000 description 2
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 2
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 2
- 238000002955 isolation Methods 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 125000001786 isothiazolyl group Chemical group 0.000 description 2
- 125000000468 ketone group Chemical group 0.000 description 2
- 150000002596 lactones Chemical group 0.000 description 2
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 2
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 2
- 210000004185 liver Anatomy 0.000 description 2
- 229960004844 lovastatin Drugs 0.000 description 2
- PCZOHLXUXFIOCF-BXMDZJJMSA-N lovastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 PCZOHLXUXFIOCF-BXMDZJJMSA-N 0.000 description 2
- QLJODMDSTUBWDW-UHFFFAOYSA-N lovastatin hydroxy acid Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(C)C=C21 QLJODMDSTUBWDW-UHFFFAOYSA-N 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 230000006883 memory enhancing effect Effects 0.000 description 2
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 2
- 208000031225 myocardial ischemia Diseases 0.000 description 2
- 235000005152 nicotinamide Nutrition 0.000 description 2
- 239000011570 nicotinamide Substances 0.000 description 2
- 229960003966 nicotinamide Drugs 0.000 description 2
- 229960003512 nicotinic acid Drugs 0.000 description 2
- 235000001968 nicotinic acid Nutrition 0.000 description 2
- 239000011664 nicotinic acid Substances 0.000 description 2
- 239000012457 nonaqueous media Substances 0.000 description 2
- 239000002664 nootropic agent Substances 0.000 description 2
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 2
- 125000004930 octahydroisoquinolinyl group Chemical group C1(NCCC2CCCC=C12)* 0.000 description 2
- 150000007524 organic acids Chemical class 0.000 description 2
- 235000005985 organic acids Nutrition 0.000 description 2
- 239000012074 organic phase Substances 0.000 description 2
- 125000004095 oxindolyl group Chemical group N1(C(CC2=CC=CC=C12)=O)* 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 2
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 2
- DHHVAGZRUROJKS-UHFFFAOYSA-N phentermine Chemical compound CC(C)(N)CC1=CC=CC=C1 DHHVAGZRUROJKS-UHFFFAOYSA-N 0.000 description 2
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 2
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 2
- HYAFETHFCAUJAY-UHFFFAOYSA-N pioglitazone Chemical compound N1=CC(CC)=CC=C1CCOC(C=C1)=CC=C1CC1C(=O)NC(=O)S1 HYAFETHFCAUJAY-UHFFFAOYSA-N 0.000 description 2
- 239000000106 platelet aggregation inhibitor Substances 0.000 description 2
- 239000003880 polar aprotic solvent Substances 0.000 description 2
- 125000005575 polycyclic aromatic hydrocarbon group Chemical group 0.000 description 2
- 229920002223 polystyrene Polymers 0.000 description 2
- 235000011056 potassium acetate Nutrition 0.000 description 2
- 229960002965 pravastatin Drugs 0.000 description 2
- TUZYXOIXSAXUGO-PZAWKZKUSA-N pravastatin Chemical compound C1=C[C@H](C)[C@H](CC[C@@H](O)C[C@@H](O)CC(O)=O)[C@H]2[C@@H](OC(=O)[C@@H](C)CC)C[C@H](O)C=C21 TUZYXOIXSAXUGO-PZAWKZKUSA-N 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- 230000003449 preventive effect Effects 0.000 description 2
- FYPMFJGVHOHGLL-UHFFFAOYSA-N probucol Chemical compound C=1C(C(C)(C)C)=C(O)C(C(C)(C)C)=CC=1SC(C)(C)SC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 FYPMFJGVHOHGLL-UHFFFAOYSA-N 0.000 description 2
- 229960003912 probucol Drugs 0.000 description 2
- 230000001737 promoting effect Effects 0.000 description 2
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 2
- 125000002098 pyridazinyl group Chemical group 0.000 description 2
- JUJWROOIHBZHMG-RALIUCGRSA-N pyridine-d5 Chemical compound [2H]C1=NC([2H])=C([2H])C([2H])=C1[2H] JUJWROOIHBZHMG-RALIUCGRSA-N 0.000 description 2
- ZUFQODAHGAHPFQ-UHFFFAOYSA-N pyridoxine hydrochloride Chemical compound Cl.CC1=NC=C(CO)C(CO)=C1O ZUFQODAHGAHPFQ-UHFFFAOYSA-N 0.000 description 2
- GZUITABIAKMVPG-UHFFFAOYSA-N raloxifene Chemical compound C1=CC(O)=CC=C1C1=C(C(=O)C=2C=CC(OCCN3CCCCC3)=CC=2)C2=CC=C(O)C=C2S1 GZUITABIAKMVPG-UHFFFAOYSA-N 0.000 description 2
- 229960004622 raloxifene Drugs 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 208000037803 restenosis Diseases 0.000 description 2
- 229960002855 simvastatin Drugs 0.000 description 2
- RYMZZMVNJRMUDD-HGQWONQESA-N simvastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)C(C)(C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 RYMZZMVNJRMUDD-HGQWONQESA-N 0.000 description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 2
- 239000012312 sodium hydride Substances 0.000 description 2
- 235000010288 sodium nitrite Nutrition 0.000 description 2
- 229910052938 sodium sulfate Inorganic materials 0.000 description 2
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 2
- 235000011152 sodium sulphate Nutrition 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 150000003413 spiro compounds Chemical class 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- 238000013268 sustained release Methods 0.000 description 2
- 239000012730 sustained-release form Substances 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 238000003419 tautomerization reaction Methods 0.000 description 2
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 2
- DSBSHPTYSMATNZ-UHFFFAOYSA-N tert-butyl 2-(2-ethoxy-2-oxoethyl)-1,3-benzothiazole-6-carboxylate Chemical compound C1=C(C(=O)OC(C)(C)C)C=C2SC(CC(=O)OCC)=NC2=C1 DSBSHPTYSMATNZ-UHFFFAOYSA-N 0.000 description 2
- DPKBAXPHAYBPRL-UHFFFAOYSA-M tetrabutylazanium;iodide Chemical compound [I-].CCCC[N+](CCCC)(CCCC)CCCC DPKBAXPHAYBPRL-UHFFFAOYSA-M 0.000 description 2
- 125000003039 tetrahydroisoquinolinyl group Chemical group C1(NCCC2=CC=CC=C12)* 0.000 description 2
- 125000001712 tetrahydronaphthyl group Chemical group C1(CCCC2=CC=CC=C12)* 0.000 description 2
- 125000000147 tetrahydroquinolinyl group Chemical group N1(CCCC2=CC=CC=C12)* 0.000 description 2
- CZDYPVPMEAXLPK-UHFFFAOYSA-N tetramethylsilane Chemical compound C[Si](C)(C)C CZDYPVPMEAXLPK-UHFFFAOYSA-N 0.000 description 2
- 125000004525 thiadiazinyl group Chemical group S1NN=C(C=C1)* 0.000 description 2
- 125000001113 thiadiazolyl group Chemical group 0.000 description 2
- 238000004809 thin layer chromatography Methods 0.000 description 2
- AXZWODMDQAVCJE-UHFFFAOYSA-L tin(II) chloride (anhydrous) Chemical compound [Cl-].[Cl-].[Sn+2] AXZWODMDQAVCJE-UHFFFAOYSA-L 0.000 description 2
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 description 2
- 125000000725 trifluoropropyl group Chemical group [H]C([H])(*)C([H])([H])C(F)(F)F 0.000 description 2
- ONDSBJMLAHVLMI-UHFFFAOYSA-N trimethylsilyldiazomethane Chemical compound C[Si](C)(C)[CH-][N+]#N ONDSBJMLAHVLMI-UHFFFAOYSA-N 0.000 description 2
- 229910000404 tripotassium phosphate Inorganic materials 0.000 description 2
- 238000000825 ultraviolet detection Methods 0.000 description 2
- 230000002792 vascular Effects 0.000 description 2
- 239000011782 vitamin Substances 0.000 description 2
- 235000013343 vitamin Nutrition 0.000 description 2
- 229940088594 vitamin Drugs 0.000 description 2
- 229930003231 vitamin Natural products 0.000 description 2
- 239000011715 vitamin B12 Substances 0.000 description 2
- 239000011726 vitamin B6 Substances 0.000 description 2
- UGOMMVLRQDMAQQ-UHFFFAOYSA-N xphos Chemical compound CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C1=CC=CC=C1P(C1CCCCC1)C1CCCCC1 UGOMMVLRQDMAQQ-UHFFFAOYSA-N 0.000 description 2
- DBGIVFWFUFKIQN-VIFPVBQESA-N (+)-Fenfluramine Chemical compound CCN[C@@H](C)CC1=CC=CC(C(F)(F)F)=C1 DBGIVFWFUFKIQN-VIFPVBQESA-N 0.000 description 1
- DBGIVFWFUFKIQN-UHFFFAOYSA-N (+-)-Fenfluramine Chemical compound CCNC(C)CC1=CC=CC(C(F)(F)F)=C1 DBGIVFWFUFKIQN-UHFFFAOYSA-N 0.000 description 1
- OGNSCSPNOLGXSM-UHFFFAOYSA-N (+/-)-DABA Natural products NCCC(N)C(O)=O OGNSCSPNOLGXSM-UHFFFAOYSA-N 0.000 description 1
- XFQNWPYGEGCIMF-HCUGAJCMSA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].[Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 XFQNWPYGEGCIMF-HCUGAJCMSA-N 0.000 description 1
- WXGBZJJAGLSBPR-UHFFFAOYSA-N (2-fluoropyridin-4-yl)boronic acid Chemical compound OB(O)C1=CC=NC(F)=C1 WXGBZJJAGLSBPR-UHFFFAOYSA-N 0.000 description 1
- FTRWOTNVAQWBED-HYARGMPZSA-N (2E)-2-[6-(6-fluoropyridin-3-yl)-1,3-benzothiazol-2-yl]-2-hydroxyimino-N-(2-sulfamoylethyl)acetamide Chemical compound C1=C2SC(/C(=N/O)C(=O)NCCS(=O)(=O)N)=NC2=CC=C1C1=CC=C(F)N=C1 FTRWOTNVAQWBED-HYARGMPZSA-N 0.000 description 1
- XUFXOAAUWZOOIT-SXARVLRPSA-N (2R,3R,4R,5S,6R)-5-[[(2R,3R,4R,5S,6R)-5-[[(2R,3R,4S,5S,6R)-3,4-dihydroxy-6-methyl-5-[[(1S,4R,5S,6S)-4,5,6-trihydroxy-3-(hydroxymethyl)-1-cyclohex-2-enyl]amino]-2-oxanyl]oxy]-3,4-dihydroxy-6-(hydroxymethyl)-2-oxanyl]oxy]-6-(hydroxymethyl)oxane-2,3,4-triol Chemical compound O([C@H]1O[C@H](CO)[C@H]([C@@H]([C@H]1O)O)O[C@H]1O[C@@H]([C@H]([C@H](O)[C@H]1O)N[C@@H]1[C@@H]([C@@H](O)[C@H](O)C(CO)=C1)O)C)[C@@H]1[C@@H](CO)O[C@@H](O)[C@H](O)[C@H]1O XUFXOAAUWZOOIT-SXARVLRPSA-N 0.000 description 1
- QJHHTOGVYWVZFA-UHFFFAOYSA-N (3,3-difluoroazetidin-1-yl)-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]methanone Chemical compound O1C(C)(C)C(C)(C)OB1C1=CC=C(C(=O)N2CC(F)(F)C2)C=C1 QJHHTOGVYWVZFA-UHFFFAOYSA-N 0.000 description 1
- QXUKUCKOYMDDRY-UHFFFAOYSA-N (5-methyl-1,3,4-oxadiazol-2-yl)methanamine;hydrochloride Chemical compound Cl.CC1=NN=C(CN)O1 QXUKUCKOYMDDRY-UHFFFAOYSA-N 0.000 description 1
- 125000006272 (C3-C7) cycloalkyl group Chemical group 0.000 description 1
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 1
- 125000004605 1,2,3,4-tetrahydroisoquinolinyl group Chemical group C1(NCCC2=CC=CC=C12)* 0.000 description 1
- 125000004607 1,2,3,4-tetrahydroquinolinyl group Chemical group N1(CCCC2=CC=CC=C12)* 0.000 description 1
- FNQJDLTXOVEEFB-UHFFFAOYSA-N 1,2,3-benzothiadiazole Chemical class C1=CC=C2SN=NC2=C1 FNQJDLTXOVEEFB-UHFFFAOYSA-N 0.000 description 1
- 125000004502 1,2,3-oxadiazolyl group Chemical group 0.000 description 1
- 125000004511 1,2,3-thiadiazolyl group Chemical group 0.000 description 1
- 125000001399 1,2,3-triazolyl group Chemical group N1N=NC(=C1)* 0.000 description 1
- 125000004504 1,2,4-oxadiazolyl group Chemical group 0.000 description 1
- 125000001376 1,2,4-triazolyl group Chemical group N1N=C(N=C1)* 0.000 description 1
- 125000004506 1,2,5-oxadiazolyl group Chemical group 0.000 description 1
- 125000004517 1,2,5-thiadiazolyl group Chemical group 0.000 description 1
- 125000001781 1,3,4-oxadiazolyl group Chemical group 0.000 description 1
- 125000004520 1,3,4-thiadiazolyl group Chemical group 0.000 description 1
- LVEYOSJUKRVCCF-UHFFFAOYSA-N 1,3-Bis(diphenylphosphino)propane Substances C=1C=CC=CC=1P(C=1C=CC=CC=1)CCCP(C=1C=CC=CC=1)C1=CC=CC=C1 LVEYOSJUKRVCCF-UHFFFAOYSA-N 0.000 description 1
- BOVGTQGAOIONJV-BETUJISGSA-N 1-[(3ar,6as)-3,3a,4,5,6,6a-hexahydro-1h-cyclopenta[c]pyrrol-2-yl]-3-(4-methylphenyl)sulfonylurea Chemical compound C1=CC(C)=CC=C1S(=O)(=O)NC(=O)NN1C[C@H]2CCC[C@H]2C1 BOVGTQGAOIONJV-BETUJISGSA-N 0.000 description 1
- FOZVXADQAHVUSV-UHFFFAOYSA-N 1-bromo-2-(2-bromoethoxy)ethane Chemical compound BrCCOCCBr FOZVXADQAHVUSV-UHFFFAOYSA-N 0.000 description 1
- MICMHFIQSAMEJG-UHFFFAOYSA-N 1-bromopyrrolidine-2,5-dione Chemical compound BrN1C(=O)CCC1=O.BrN1C(=O)CCC1=O MICMHFIQSAMEJG-UHFFFAOYSA-N 0.000 description 1
- LLJFMFZYVVLQKT-UHFFFAOYSA-N 1-cyclohexyl-3-[4-[2-(7-methoxy-4,4-dimethyl-1,3-dioxo-2-isoquinolinyl)ethyl]phenyl]sulfonylurea Chemical compound C=1C(OC)=CC=C(C(C2=O)(C)C)C=1C(=O)N2CCC(C=C1)=CC=C1S(=O)(=O)NC(=O)NC1CCCCC1 LLJFMFZYVVLQKT-UHFFFAOYSA-N 0.000 description 1
- 125000006432 1-methyl cyclopropyl group Chemical group [H]C([H])([H])C1(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000006017 1-propenyl group Chemical group 0.000 description 1
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 1
- KIPSRYDSZQRPEA-UHFFFAOYSA-N 2,2,2-trifluoroethanamine Chemical compound NCC(F)(F)F KIPSRYDSZQRPEA-UHFFFAOYSA-N 0.000 description 1
- 125000004206 2,2,2-trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 1
- QZMHSVLOZTUEJK-UHFFFAOYSA-N 2-(2-aminoethylsulfonyl)-2-(6-bromo-1,3-benzothiazol-2-yl)-n-(2-sulfamoylethyl)acetamide;hydrobromide Chemical compound Br.C1=C(Br)C=C2SC(C(C(=O)NCCS(N)(=O)=O)S(=O)(=O)CCN)=NC2=C1 QZMHSVLOZTUEJK-UHFFFAOYSA-N 0.000 description 1
- JNYSVFXJKTWZIO-UHFFFAOYSA-N 2-(4-fluoro-6-phenyl-1,3-benzothiazol-2-yl)-n-(2-sulfamoylethyl)acetamide Chemical compound C1=C2SC(CC(=O)NCCS(=O)(=O)N)=NC2=C(F)C=C1C1=CC=CC=C1 JNYSVFXJKTWZIO-UHFFFAOYSA-N 0.000 description 1
- GZGROMZYMLYCHA-UHFFFAOYSA-N 2-(6-bromo-1,3-benzothiazol-2-yl)-2-(2-morpholin-4-ylethylsulfonyl)-n-(2-sulfamoylethyl)acetamide Chemical compound N=1C2=CC=C(Br)C=C2SC=1C(C(=O)NCCS(=O)(=O)N)S(=O)(=O)CCN1CCOCC1 GZGROMZYMLYCHA-UHFFFAOYSA-N 0.000 description 1
- ZYWDWAHHOFHILG-UHFFFAOYSA-N 2-(6-bromo-1,3-benzothiazol-2-yl)-n-(2-sulfamoylethyl)acetamide Chemical compound C1=C(Br)C=C2SC(CC(=O)NCCS(=O)(=O)N)=NC2=C1 ZYWDWAHHOFHILG-UHFFFAOYSA-N 0.000 description 1
- LJVDUOZUBQPMNG-UHFFFAOYSA-N 2-(6-phenyl-1,3-benzothiazol-2-yl)-n-(2-sulfamoylethyl)acetamide Chemical compound C1=C2SC(CC(=O)NCCS(=O)(=O)N)=NC2=CC=C1C1=CC=CC=C1 LJVDUOZUBQPMNG-UHFFFAOYSA-N 0.000 description 1
- MPIYNHSFBXWYDD-UHFFFAOYSA-N 2-[6-(2-fluoropyridin-4-yl)-1,3-benzothiazol-2-yl]-n-(2-methylsulfonylethyl)acetamide Chemical compound C1=C2SC(CC(=O)NCCS(=O)(=O)C)=NC2=CC=C1C1=CC=NC(F)=C1 MPIYNHSFBXWYDD-UHFFFAOYSA-N 0.000 description 1
- UBVKYNGNNCHTPI-UHFFFAOYSA-N 2-[6-(2-fluoropyridin-4-yl)-1,3-benzothiazol-2-yl]-n-[2-(2,2,2-trifluoroethylsulfamoyl)ethyl]acetamide Chemical compound C1=NC(F)=CC(C=2C=C3SC(CC(=O)NCCS(=O)(=O)NCC(F)(F)F)=NC3=CC=2)=C1 UBVKYNGNNCHTPI-UHFFFAOYSA-N 0.000 description 1
- PBRZLXGFNJRJSM-UHFFFAOYSA-N 2-[6-(3,3-difluoropyrrolidine-1-carbonyl)-1,3-benzothiazol-2-yl]-n-(2-sulfamoylethyl)-2-(3,3,3-trifluoropropylsulfonyl)acetamide Chemical compound C1=C2SC(C(C(=O)NCCS(=O)(=O)N)S(=O)(=O)CCC(F)(F)F)=NC2=CC=C1C(=O)N1CCC(F)(F)C1 PBRZLXGFNJRJSM-UHFFFAOYSA-N 0.000 description 1
- UYBHZZFFVSUVGX-UHFFFAOYSA-N 2-[6-(4-fluorophenyl)-1,3-benzothiazol-2-yl]-n-[(5-methyl-1,3,4-oxadiazol-2-yl)methyl]acetamide Chemical compound O1C(C)=NN=C1CNC(=O)CC1=NC2=CC=C(C=3C=CC(F)=CC=3)C=C2S1 UYBHZZFFVSUVGX-UHFFFAOYSA-N 0.000 description 1
- YSVSVEOTXKCPHT-UHFFFAOYSA-N 2-[6-(6-fluoropyridin-3-yl)-1,3-benzothiazol-2-yl]-n-(2-sulfamoylethyl)acetamide Chemical compound C1=C2SC(CC(=O)NCCS(=O)(=O)N)=NC2=CC=C1C1=CC=C(F)N=C1 YSVSVEOTXKCPHT-UHFFFAOYSA-N 0.000 description 1
- GSVXOWTXTTWPFD-UHFFFAOYSA-N 2-[6-[4-(3,3-difluoroazetidine-1-carbonyl)phenyl]-1,3-benzothiazol-2-yl]-2-methylsulfonyl-n-(2-sulfamoylethyl)acetamide Chemical compound C1=C2SC(C(C(=O)NCCS(N)(=O)=O)S(=O)(=O)C)=NC2=CC=C1C(C=C1)=CC=C1C(=O)N1CC(F)(F)C1 GSVXOWTXTTWPFD-UHFFFAOYSA-N 0.000 description 1
- WEWQIQYOLOFSDO-UHFFFAOYSA-N 2-[6-[4-(3,3-difluoroazetidine-1-carbonyl)phenyl]-1,3-benzothiazol-2-yl]-n-(2-sulfamoylethyl)acetamide Chemical compound C1=C2SC(CC(=O)NCCS(=O)(=O)N)=NC2=CC=C1C(C=C1)=CC=C1C(=O)N1CC(F)(F)C1 WEWQIQYOLOFSDO-UHFFFAOYSA-N 0.000 description 1
- KAFFNZFGJICJKK-UHFFFAOYSA-N 2-[6-[4-(3-hydroxy-3-methylazetidine-1-carbonyl)phenyl]-1,3-benzothiazol-2-yl]-2-methylsulfonyl-n-(2-sulfamoylethyl)acetamide Chemical compound C1C(C)(O)CN1C(=O)C1=CC=C(C=2C=C3SC(=NC3=CC=2)C(C(=O)NCCS(N)(=O)=O)S(C)(=O)=O)C=C1 KAFFNZFGJICJKK-UHFFFAOYSA-N 0.000 description 1
- YQVHZJPEZQYJLX-UHFFFAOYSA-N 2-[[2-[6-(6-fluoropyridin-3-yl)-1,3-benzothiazol-2-yl]-2-(2-methoxyethylsulfonyl)acetyl]amino]acetic acid Chemical compound C1=C2SC(C(C(=O)NCC(O)=O)S(=O)(=O)CCOC)=NC2=CC=C1C1=CC=C(F)N=C1 YQVHZJPEZQYJLX-UHFFFAOYSA-N 0.000 description 1
- 125000005273 2-acetoxybenzoic acid group Chemical group 0.000 description 1
- UCZNOTCZJGYDDD-UHFFFAOYSA-N 2-amino-n-(2,2,2-trifluoroethyl)ethanesulfonamide Chemical compound NCCS(=O)(=O)NCC(F)(F)F UCZNOTCZJGYDDD-UHFFFAOYSA-N 0.000 description 1
- MVQXBXLDXSQURK-UHFFFAOYSA-N 2-aminoethanesulfonamide Chemical compound NCCS(N)(=O)=O MVQXBXLDXSQURK-UHFFFAOYSA-N 0.000 description 1
- 125000004974 2-butenyl group Chemical group C(C=CC)* 0.000 description 1
- QLAKWSXVXQRGDB-UHFFFAOYSA-N 2-chloro-1,3-benzothiazole-6-carboxylic acid Chemical compound OC(=O)C1=CC=C2N=C(Cl)SC2=C1 QLAKWSXVXQRGDB-UHFFFAOYSA-N 0.000 description 1
- 125000006040 2-hexenyl group Chemical group 0.000 description 1
- YEDUAINPPJYDJZ-UHFFFAOYSA-N 2-hydroxybenzothiazole Chemical compound C1=CC=C2SC(O)=NC2=C1 YEDUAINPPJYDJZ-UHFFFAOYSA-N 0.000 description 1
- KOHBEDRJXKOYHL-UHFFFAOYSA-N 2-methoxy-n-methylethanamine Chemical compound CNCCOC KOHBEDRJXKOYHL-UHFFFAOYSA-N 0.000 description 1
- DUNNNVSEUCJEOY-UHFFFAOYSA-N 2-methoxyethanesulfonyl chloride Chemical compound COCCS(Cl)(=O)=O DUNNNVSEUCJEOY-UHFFFAOYSA-N 0.000 description 1
- UVHGBTYSDWIAPE-UHFFFAOYSA-N 2-methyl-2-(6-phenyl-1,3-benzothiazol-2-yl)-n-[(3-propan-2-yl-1,2-oxazol-5-yl)methyl]propanamide Chemical compound O1N=C(C(C)C)C=C1CNC(=O)C(C)(C)C1=NC2=CC=C(C=3C=CC=CC=3)C=C2S1 UVHGBTYSDWIAPE-UHFFFAOYSA-N 0.000 description 1
- 125000006022 2-methyl-2-propenyl group Chemical group 0.000 description 1
- XWKFPIODWVPXLX-UHFFFAOYSA-N 2-methyl-5-methylpyridine Natural products CC1=CC=C(C)N=C1 XWKFPIODWVPXLX-UHFFFAOYSA-N 0.000 description 1
- KJGIDIVEURYMDI-UHFFFAOYSA-N 2-methylsulfonyl-2-(6-phenyl-1,3-benzothiazol-2-yl)-n-(2-sulfamoylethyl)acetamide Chemical compound C1=C2SC(C(C(=O)NCCS(N)(=O)=O)S(=O)(=O)C)=NC2=CC=C1C1=CC=CC=C1 KJGIDIVEURYMDI-UHFFFAOYSA-N 0.000 description 1
- VFWVPVQLGGZEDL-UHFFFAOYSA-N 2-methylsulfonyl-2-[6-(1-piperidin-4-ylpyrazol-4-yl)-1,3-benzothiazol-2-yl]-n-(2-sulfamoylethyl)acetamide Chemical compound C1=C2SC(C(C(=O)NCCS(N)(=O)=O)S(=O)(=O)C)=NC2=CC=C1C(=C1)C=NN1C1CCNCC1 VFWVPVQLGGZEDL-UHFFFAOYSA-N 0.000 description 1
- 125000006024 2-pentenyl group Chemical group 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- CDBAEFXTCRKJPZ-UHFFFAOYSA-N 3,3-difluoroazetidine;hydron;chloride Chemical compound Cl.FC1(F)CNC1 CDBAEFXTCRKJPZ-UHFFFAOYSA-N 0.000 description 1
- SRVXSISGYBMIHR-UHFFFAOYSA-N 3-[3-[3-(2-amino-2-oxoethyl)phenyl]-5-chlorophenyl]-3-(5-methyl-1,3-thiazol-2-yl)propanoic acid Chemical compound S1C(C)=CN=C1C(CC(O)=O)C1=CC(Cl)=CC(C=2C=C(CC(N)=O)C=CC=2)=C1 SRVXSISGYBMIHR-UHFFFAOYSA-N 0.000 description 1
- 125000004975 3-butenyl group Chemical group C(CC=C)* 0.000 description 1
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 1
- 125000006041 3-hexenyl group Chemical group 0.000 description 1
- WEAZZOCWAJWPRL-UHFFFAOYSA-N 3-methylazetidin-3-ol;hydrochloride Chemical compound Cl.CC1(O)CNC1 WEAZZOCWAJWPRL-UHFFFAOYSA-N 0.000 description 1
- KOZLFGJOHMAYIA-UHFFFAOYSA-N 3-oxo-1,2-benzothiazole-2-carboxamide Chemical class C1=CC=C2C(=O)N(C(=O)N)SC2=C1 KOZLFGJOHMAYIA-UHFFFAOYSA-N 0.000 description 1
- GWRSATNRNFYMDI-UHFFFAOYSA-N 4-[(9-cyclopentyl-7,7-difluoro-5-methyl-6-oxo-8h-pyrimido[4,5-b][1,4]diazepin-2-yl)amino]-2-fluoro-5-methoxy-n-(1-methylpiperidin-4-yl)benzamide Chemical compound FC=1C=C(NC=2N=C3N(C4CCCC4)CC(F)(F)C(=O)N(C)C3=CN=2)C(OC)=CC=1C(=O)NC1CCN(C)CC1 GWRSATNRNFYMDI-UHFFFAOYSA-N 0.000 description 1
- ZAQIZLQBMSQOBE-UHFFFAOYSA-N 4-[2-[1-methylsulfonyl-2-oxo-2-(2-sulfamoylethylamino)ethyl]-1,3-benzothiazol-6-yl]benzoic acid Chemical compound C1=C2SC(C(C(=O)NCCS(N)(=O)=O)S(=O)(=O)C)=NC2=CC=C1C1=CC=C(C(O)=O)C=C1 ZAQIZLQBMSQOBE-UHFFFAOYSA-N 0.000 description 1
- BFQSQUAVMNHOEF-UHFFFAOYSA-N 4-bromo-2,6-difluoroaniline Chemical compound NC1=C(F)C=C(Br)C=C1F BFQSQUAVMNHOEF-UHFFFAOYSA-N 0.000 description 1
- UIVXXFYJRYVRKJ-UHFFFAOYSA-N 4-fluorobenzohydrazide Chemical compound NNC(=O)C1=CC=C(F)C=C1 UIVXXFYJRYVRKJ-UHFFFAOYSA-N 0.000 description 1
- 125000006042 4-hexenyl group Chemical group 0.000 description 1
- 125000004217 4-methoxybenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1OC([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000003119 4-methyl-3-pentenyl group Chemical group [H]\C(=C(/C([H])([H])[H])C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000005986 4-piperidonyl group Chemical group 0.000 description 1
- 125000002471 4H-quinolizinyl group Chemical group C=1(C=CCN2C=CC=CC12)* 0.000 description 1
- 125000004608 5,6,7,8-tetrahydroquinolinyl group Chemical group N1=C(C=CC=2CCCCC12)* 0.000 description 1
- IDMZCFVZYIPOJF-UHFFFAOYSA-N 5-bromo-1-(2-hydroxy-2-methylpropyl)pyridin-2-one Chemical compound CC(C)(O)CN1C=C(Br)C=CC1=O IDMZCFVZYIPOJF-UHFFFAOYSA-N 0.000 description 1
- NDMZZQRNZFWMEZ-UHFFFAOYSA-N 5-bromo-1h-pyridin-2-one Chemical compound OC1=CC=C(Br)C=N1 NDMZZQRNZFWMEZ-UHFFFAOYSA-N 0.000 description 1
- 125000006043 5-hexenyl group Chemical group 0.000 description 1
- 125000006163 5-membered heteroaryl group Chemical group 0.000 description 1
- FFUYMSMYJBBDJW-UHFFFAOYSA-N 6-bromo-2-chloro-4-ethyl-1,3-benzothiazole Chemical compound CCC1=CC(Br)=CC2=C1N=C(Cl)S2 FFUYMSMYJBBDJW-UHFFFAOYSA-N 0.000 description 1
- ITZMJCSORYKOSI-AJNGGQMLSA-N APGPR Enterostatin Chemical compound C[C@H](N)C(=O)N1CCC[C@H]1C(=O)NCC(=O)N1[C@H](C(=O)N[C@@H](CCCN=C(N)N)C(O)=O)CCC1 ITZMJCSORYKOSI-AJNGGQMLSA-N 0.000 description 1
- 239000000275 Adrenocorticotropic Hormone Substances 0.000 description 1
- 229940124225 Adrenoreceptor agonist Drugs 0.000 description 1
- 101150102415 Apob gene Proteins 0.000 description 1
- 108010071619 Apolipoproteins Proteins 0.000 description 1
- 102000007592 Apolipoproteins Human genes 0.000 description 1
- 229910015845 BBr3 Inorganic materials 0.000 description 1
- 229940123208 Biguanide Drugs 0.000 description 1
- 102000013585 Bombesin Human genes 0.000 description 1
- 108010051479 Bombesin Proteins 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- KFGYAWVTAZVUAD-UHFFFAOYSA-N C1=C(C(O)=O)C=C2SC(CC(=O)NCCS(=O)(=O)N)=NC2=C1 Chemical compound C1=C(C(O)=O)C=C2SC(CC(=O)NCCS(=O)(=O)N)=NC2=C1 KFGYAWVTAZVUAD-UHFFFAOYSA-N 0.000 description 1
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 description 1
- BNLKVTSQZBYJFX-UHFFFAOYSA-N CC(C)(C)OC(c1ccc2[s]c(C(C(NCCS(N)(=O)=O)=O)S(C)(=O)=O)nc2c1)=O Chemical compound CC(C)(C)OC(c1ccc2[s]c(C(C(NCCS(N)(=O)=O)=O)S(C)(=O)=O)nc2c1)=O BNLKVTSQZBYJFX-UHFFFAOYSA-N 0.000 description 1
- RNNRWQQHAFWPLH-UHFFFAOYSA-N CC(NC1COC1)=O Chemical compound CC(NC1COC1)=O RNNRWQQHAFWPLH-UHFFFAOYSA-N 0.000 description 1
- TVOJIBGZFYMWDT-UHFFFAOYSA-N CC1(C)OB(c2c[nH]nc2)OC1(C)C Chemical compound CC1(C)OB(c2c[nH]nc2)OC1(C)C TVOJIBGZFYMWDT-UHFFFAOYSA-N 0.000 description 1
- WYZDCUGWXKHESN-UHFFFAOYSA-N CN(Cc1ccccc1)Cc1ccccc1 Chemical compound CN(Cc1ccccc1)Cc1ccccc1 WYZDCUGWXKHESN-UHFFFAOYSA-N 0.000 description 1
- QZMOEVMIFJSOCO-UHFFFAOYSA-N COC(C1)CN1C(c(cc1)ccc1-c(cc1)cc2c1nc(C(C(NCCS(N)(=O)=O)=O)S(Cc1ccc(C(F)(F)F)cc1)(=O)=O)[s]2)=O Chemical compound COC(C1)CN1C(c(cc1)ccc1-c(cc1)cc2c1nc(C(C(NCCS(N)(=O)=O)=O)S(Cc1ccc(C(F)(F)F)cc1)(=O)=O)[s]2)=O QZMOEVMIFJSOCO-UHFFFAOYSA-N 0.000 description 1
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 1
- 229930186147 Cephalosporin Natural products 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- RKWGIWYCVPQPMF-UHFFFAOYSA-N Chloropropamide Chemical compound CCCNC(=O)NS(=O)(=O)C1=CC=C(Cl)C=C1 RKWGIWYCVPQPMF-UHFFFAOYSA-N 0.000 description 1
- 108010004103 Chylomicrons Proteins 0.000 description 1
- 108020004635 Complementary DNA Proteins 0.000 description 1
- 101800000414 Corticotropin Proteins 0.000 description 1
- FMGSKLZLMKYGDP-UHFFFAOYSA-N Dehydroepiandrosterone Natural products C1C(O)CCC2(C)C3CCC(C)(C(CC4)=O)C4C3CC=C21 FMGSKLZLMKYGDP-UHFFFAOYSA-N 0.000 description 1
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 1
- 102000004980 Dopamine D2 Receptors Human genes 0.000 description 1
- 108090001111 Dopamine D2 Receptors Proteins 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- OTMSDBZUPAUEDD-UHFFFAOYSA-N Ethane Chemical compound CC OTMSDBZUPAUEDD-UHFFFAOYSA-N 0.000 description 1
- 102000019432 Galanin Human genes 0.000 description 1
- 101800002068 Galanin Proteins 0.000 description 1
- FAEKWTJYAYMJKF-QHCPKHFHSA-N GlucoNorm Chemical compound C1=C(C(O)=O)C(OCC)=CC(CC(=O)N[C@@H](CC(C)C)C=2C(=CC=CC=2)N2CCCCC2)=C1 FAEKWTJYAYMJKF-QHCPKHFHSA-N 0.000 description 1
- 108010080123 HDL-triglyceride Proteins 0.000 description 1
- 102000004384 Histamine H3 receptors Human genes 0.000 description 1
- 108090000981 Histamine H3 receptors Proteins 0.000 description 1
- 208000031226 Hyperlipidaemia Diseases 0.000 description 1
- 102000004877 Insulin Human genes 0.000 description 1
- 108090001061 Insulin Proteins 0.000 description 1
- 229940122199 Insulin secretagogue Drugs 0.000 description 1
- 229940122355 Insulin sensitizer Drugs 0.000 description 1
- 229930194542 Keto Natural products 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 229940127470 Lipase Inhibitors Drugs 0.000 description 1
- ZPXSCAKFGYXMGA-UHFFFAOYSA-N Mazindol Chemical compound N12CCN=C2C2=CC=CC=C2C1(O)C1=CC=C(Cl)C=C1 ZPXSCAKFGYXMGA-UHFFFAOYSA-N 0.000 description 1
- 239000000637 Melanocyte-Stimulating Hormone Substances 0.000 description 1
- 108010007013 Melanocyte-Stimulating Hormones Proteins 0.000 description 1
- 101710151321 Melanostatin Proteins 0.000 description 1
- IBAQFPQHRJAVAV-ULAWRXDQSA-N Miglitol Chemical compound OCCN1C[C@H](O)[C@@H](O)[C@H](O)[C@H]1CO IBAQFPQHRJAVAV-ULAWRXDQSA-N 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- 102400000064 Neuropeptide Y Human genes 0.000 description 1
- PXZQEOJJUGGUIB-UHFFFAOYSA-N O=C1NCc2c1cccc2 Chemical compound O=C1NCc2c1cccc2 PXZQEOJJUGGUIB-UHFFFAOYSA-N 0.000 description 1
- 101150014691 PPARA gene Proteins 0.000 description 1
- 229930182555 Penicillin Natural products 0.000 description 1
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 1
- 101000941277 Rattus norvegicus Endothelial lipase Proteins 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 229940100389 Sulfonylurea Drugs 0.000 description 1
- 108700012920 TNF Proteins 0.000 description 1
- 229940123464 Thiazolidinedione Drugs 0.000 description 1
- 208000007536 Thrombosis Diseases 0.000 description 1
- JLRGJRBPOGGCBT-UHFFFAOYSA-N Tolbutamide Chemical compound CCCCNC(=O)NS(=O)(=O)C1=CC=C(C)C=C1 JLRGJRBPOGGCBT-UHFFFAOYSA-N 0.000 description 1
- YZCKVEUIGOORGS-NJFSPNSNSA-N Tritium Chemical compound [3H] YZCKVEUIGOORGS-NJFSPNSNSA-N 0.000 description 1
- FZNCGRZWXLXZSZ-CIQUZCHMSA-N Voglibose Chemical compound OCC(CO)N[C@H]1C[C@](O)(CO)[C@@H](O)[C@H](O)[C@H]1O FZNCGRZWXLXZSZ-CIQUZCHMSA-N 0.000 description 1
- 239000012391 XPhos Pd G2 Substances 0.000 description 1
- WGOBPPNNYVSJTE-HSZRJFAPSA-N [(2r)-1-diphenylphosphanylpropan-2-yl]-diphenylphosphane Chemical compound C([C@@H](C)P(C=1C=CC=CC=1)C=1C=CC=CC=1)P(C=1C=CC=CC=1)C1=CC=CC=C1 WGOBPPNNYVSJTE-HSZRJFAPSA-N 0.000 description 1
- BDHMWOQIBZOHFK-UHFFFAOYSA-N [1-[1-[(2-methylpropan-2-yl)oxycarbonyl]piperidin-4-yl]pyrazol-4-yl]boronic acid Chemical compound C1CN(C(=O)OC(C)(C)C)CCC1N1N=CC(B(O)O)=C1 BDHMWOQIBZOHFK-UHFFFAOYSA-N 0.000 description 1
- QRGVHOKYRIQQTN-UHFFFAOYSA-N [4-(2-methoxyethylcarbamoyl)phenyl]boronic acid Chemical compound COCCNC(=O)C1=CC=C(B(O)O)C=C1 QRGVHOKYRIQQTN-UHFFFAOYSA-N 0.000 description 1
- FNAWJOBKLWLHTA-UHFFFAOYSA-N [4-(trifluoromethyl)benzoyl] 4-(trifluoromethyl)benzoate Chemical compound C1=CC(C(F)(F)F)=CC=C1C(=O)OC(=O)C1=CC=C(C(F)(F)F)C=C1 FNAWJOBKLWLHTA-UHFFFAOYSA-N 0.000 description 1
- CKUAXEQHGKSLHN-UHFFFAOYSA-N [C].[N] Chemical group [C].[N] CKUAXEQHGKSLHN-UHFFFAOYSA-N 0.000 description 1
- 229960002632 acarbose Drugs 0.000 description 1
- XUFXOAAUWZOOIT-UHFFFAOYSA-N acarviostatin I01 Natural products OC1C(O)C(NC2C(C(O)C(O)C(CO)=C2)O)C(C)OC1OC(C(C1O)O)C(CO)OC1OC1C(CO)OC(O)C(O)C1O XUFXOAAUWZOOIT-UHFFFAOYSA-N 0.000 description 1
- 229960001466 acetohexamide Drugs 0.000 description 1
- VGZSUPCWNCWDAN-UHFFFAOYSA-N acetohexamide Chemical compound C1=CC(C(=O)C)=CC=C1S(=O)(=O)NC(=O)NC1CCCCC1 VGZSUPCWNCWDAN-UHFFFAOYSA-N 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 125000000641 acridinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3C=C12)* 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- 239000011149 active material Substances 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 210000000577 adipose tissue Anatomy 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 125000006177 alkyl benzyl group Chemical group 0.000 description 1
- 125000004414 alkyl thio group Chemical group 0.000 description 1
- 208000026935 allergic disease Diseases 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- XKMRRTOUMJRJIA-UHFFFAOYSA-N ammonia nh3 Chemical compound N.N XKMRRTOUMJRJIA-UHFFFAOYSA-N 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- 150000008064 anhydrides Chemical class 0.000 description 1
- 239000005557 antagonist Substances 0.000 description 1
- 125000002178 anthracenyl group Chemical group C1(=CC=CC2=CC3=CC=CC=C3C=C12)* 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 1
- 239000012131 assay buffer Substances 0.000 description 1
- 230000003143 atherosclerotic effect Effects 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 125000002393 azetidinyl group Chemical group 0.000 description 1
- 125000004931 azocinyl group Chemical group N1=C(C=CC=CC=C1)* 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 125000004618 benzofuryl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid group Chemical group C(C1=CC=CC=C1)(=O)O WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 125000004190 benzothiazol-2-yl group Chemical group [H]C1=C([H])C([H])=C2N=C(*)SC2=C1[H] 0.000 description 1
- WGNZRLMOMHJUSP-UHFFFAOYSA-N benzotriazol-1-yloxy(tripyrrolidin-1-yl)phosphanium Chemical compound C1CCCN1[P+](N1CCCC1)(N1CCCC1)ON1C2=CC=CC=C2N=N1 WGNZRLMOMHJUSP-UHFFFAOYSA-N 0.000 description 1
- 125000003354 benzotriazolyl group Chemical group N1N=NC2=C1C=CC=C2* 0.000 description 1
- CRVCWUXOKRGDGD-UHFFFAOYSA-N benzyl n-(2-chlorosulfonylethyl)carbamate Chemical compound ClS(=O)(=O)CCNC(=O)OCC1=CC=CC=C1 CRVCWUXOKRGDGD-UHFFFAOYSA-N 0.000 description 1
- BYJKUXDUDCOJSP-UHFFFAOYSA-N benzyl n-[2-(2,2,2-trifluoroethylsulfamoyl)ethyl]carbamate Chemical compound FC(F)(F)CNS(=O)(=O)CCNC(=O)OCC1=CC=CC=C1 BYJKUXDUDCOJSP-UHFFFAOYSA-N 0.000 description 1
- UOWDVAJAXBWPAM-UHFFFAOYSA-N benzyl n-[2-[1-(6-bromo-1,3-benzothiazol-2-yl)-2-oxo-2-(2-sulfamoylethylamino)ethyl]sulfonylethyl]carbamate Chemical compound N=1C2=CC=C(Br)C=C2SC=1C(C(=O)NCCS(=O)(=O)N)S(=O)(=O)CCNC(=O)OCC1=CC=CC=C1 UOWDVAJAXBWPAM-UHFFFAOYSA-N 0.000 description 1
- 150000004283 biguanides Chemical class 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 239000012867 bioactive agent Substances 0.000 description 1
- 230000031018 biological processes and functions Effects 0.000 description 1
- IPWKHHSGDUIRAH-UHFFFAOYSA-N bis(pinacolato)diboron Chemical compound O1C(C)(C)C(C)(C)OB1B1OC(C)(C)C(C)(C)O1 IPWKHHSGDUIRAH-UHFFFAOYSA-N 0.000 description 1
- 230000015624 blood vessel development Effects 0.000 description 1
- DNDCVAGJPBKION-DOPDSADYSA-N bombesin Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(N)=O)NC(=O)CNC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CC=1NC2=CC=CC=C2C=1)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)CNC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H]1NC(=O)CC1)C(C)C)C1=CN=CN1 DNDCVAGJPBKION-DOPDSADYSA-N 0.000 description 1
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 239000000872 buffer Substances 0.000 description 1
- 244000309464 bull Species 0.000 description 1
- 125000000480 butynyl group Chemical group [*]C#CC([H])([H])C([H])([H])[H] 0.000 description 1
- 239000001110 calcium chloride Substances 0.000 description 1
- 235000011148 calcium chloride Nutrition 0.000 description 1
- 229910001628 calcium chloride Inorganic materials 0.000 description 1
- 125000004452 carbocyclyl group Chemical group 0.000 description 1
- 125000004623 carbolinyl group Chemical group 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 150000001735 carboxylic acids Chemical group 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 229940124587 cephalosporin Drugs 0.000 description 1
- 150000001780 cephalosporins Chemical class 0.000 description 1
- BJDCWCLMFKKGEE-CMDXXVQNSA-N chembl252518 Chemical compound C([C@@](OO1)(C)O2)C[C@H]3[C@H](C)CC[C@@H]4[C@@]31[C@@H]2O[C@H](O)[C@@H]4C BJDCWCLMFKKGEE-CMDXXVQNSA-N 0.000 description 1
- 125000003636 chemical group Chemical group 0.000 description 1
- 239000007810 chemical reaction solvent Substances 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- RSLSVURFMXHEEU-UHFFFAOYSA-M chloropalladium(1+);dicyclohexyl-[3-[2,4,6-tri(propan-2-yl)phenyl]phenyl]phosphane;2-phenylaniline Chemical compound [Pd+]Cl.NC1=CC=CC=C1C1=CC=CC=[C-]1.CC(C)C1=CC(C(C)C)=CC(C(C)C)=C1C1=CC=CC(P(C2CCCCC2)C2CCCCC2)=C1 RSLSVURFMXHEEU-UHFFFAOYSA-M 0.000 description 1
- 229960001761 chlorpropamide Drugs 0.000 description 1
- 150000001840 cholesterol esters Chemical class 0.000 description 1
- 125000004230 chromenyl group Chemical group O1C(C=CC2=CC=CC=C12)* 0.000 description 1
- YZFWTZACSRHJQD-UHFFFAOYSA-N ciglitazone Chemical compound C=1C=C(CC2C(NC(=O)S2)=O)C=CC=1OCC1(C)CCCCC1 YZFWTZACSRHJQD-UHFFFAOYSA-N 0.000 description 1
- 229950009226 ciglitazone Drugs 0.000 description 1
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 239000013066 combination product Substances 0.000 description 1
- 229940127555 combination product Drugs 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 239000002299 complementary DNA Substances 0.000 description 1
- 235000008504 concentrate Nutrition 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- IDLFZVILOHSSID-OVLDLUHVSA-N corticotropin Chemical compound C([C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(N)=O)C(=O)NCC(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(O)=O)NC(=O)[C@@H](N)CO)C1=CC=C(O)C=C1 IDLFZVILOHSSID-OVLDLUHVSA-N 0.000 description 1
- 229960000258 corticotropin Drugs 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 125000001047 cyclobutenyl group Chemical group C1(=CCC1)* 0.000 description 1
- XSYZCZPCBXYQTE-UHFFFAOYSA-N cyclodecylcyclodecane Chemical compound C1CCCCCCCCC1C1CCCCCCCCC1 XSYZCZPCBXYQTE-UHFFFAOYSA-N 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000522 cyclooctenyl group Chemical group C1(=CCCCCCC1)* 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 125000004856 decahydroquinolinyl group Chemical group N1(CCCC2CCCCC12)* 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- OKKJLVBELUTLKV-MICDWDOJSA-N deuteriomethanol Chemical compound [2H]CO OKKJLVBELUTLKV-MICDWDOJSA-N 0.000 description 1
- 229910052805 deuterium Inorganic materials 0.000 description 1
- 229960004597 dexfenfluramine Drugs 0.000 description 1
- ASWXNYNXAOQCCD-UHFFFAOYSA-N dichloro(triphenyl)-$l^{5}-phosphane Chemical compound C=1C=CC=CC=1P(Cl)(C=1C=CC=CC=1)(Cl)C1=CC=CC=C1 ASWXNYNXAOQCCD-UHFFFAOYSA-N 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 235000001434 dietary modification Nutrition 0.000 description 1
- 229960004890 diethylpropion Drugs 0.000 description 1
- XXEPPPIWZFICOJ-UHFFFAOYSA-N diethylpropion Chemical compound CCN(CC)C(C)C(=O)C1=CC=CC=C1 XXEPPPIWZFICOJ-UHFFFAOYSA-N 0.000 description 1
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 description 1
- 102000038379 digestive enzymes Human genes 0.000 description 1
- 108091007734 digestive enzymes Proteins 0.000 description 1
- 229940043279 diisopropylamine Drugs 0.000 description 1
- BGRWYRAHAFMIBJ-UHFFFAOYSA-N diisopropylcarbodiimide Natural products CC(C)NC(=O)NC(C)C BGRWYRAHAFMIBJ-UHFFFAOYSA-N 0.000 description 1
- JMRYOSQOYJBDOI-UHFFFAOYSA-N dilithium;di(propan-2-yl)azanide Chemical compound [Li+].CC(C)[N-]C(C)C.CC(C)N([Li])C(C)C JMRYOSQOYJBDOI-UHFFFAOYSA-N 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 1
- UXGNZZKBCMGWAZ-UHFFFAOYSA-N dimethylformamide dmf Chemical compound CN(C)C=O.CN(C)C=O UXGNZZKBCMGWAZ-UHFFFAOYSA-N 0.000 description 1
- DLNKOYKMWOXYQA-UHFFFAOYSA-N dl-pseudophenylpropanolamine Natural products CC(N)C(O)C1=CC=CC=C1 DLNKOYKMWOXYQA-UHFFFAOYSA-N 0.000 description 1
- UZZWBUYVTBPQIV-UHFFFAOYSA-N dme dimethoxyethane Chemical compound COCCOC.COCCOC UZZWBUYVTBPQIV-UHFFFAOYSA-N 0.000 description 1
- CETRZFQIITUQQL-UHFFFAOYSA-N dmso dimethylsulfoxide Chemical compound CS(C)=O.CS(C)=O CETRZFQIITUQQL-UHFFFAOYSA-N 0.000 description 1
- 238000009510 drug design Methods 0.000 description 1
- 238000009509 drug development Methods 0.000 description 1
- 238000002651 drug therapy Methods 0.000 description 1
- 230000008406 drug-drug interaction Effects 0.000 description 1
- 230000004064 dysfunction Effects 0.000 description 1
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 150000002081 enamines Chemical class 0.000 description 1
- 210000003038 endothelium Anatomy 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 125000002587 enol group Chemical group 0.000 description 1
- 239000002702 enteric coating Substances 0.000 description 1
- 238000009505 enteric coating Methods 0.000 description 1
- 238000007824 enzymatic assay Methods 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 125000004185 ester group Chemical group 0.000 description 1
- OCLXJTCGWSSVOE-UHFFFAOYSA-N ethanol etoh Chemical compound CCO.CCO OCLXJTCGWSSVOE-UHFFFAOYSA-N 0.000 description 1
- KBWBBXURHZCVSO-UHFFFAOYSA-N ethyl 2-(4-fluoro-6-phenyl-1,3-benzothiazol-2-yl)acetate Chemical compound C1=C2SC(CC(=O)OCC)=NC2=C(F)C=C1C1=CC=CC=C1 KBWBBXURHZCVSO-UHFFFAOYSA-N 0.000 description 1
- JJGILQIDFZDNBM-UHFFFAOYSA-N ethyl 2-(6-bromo-1,3-benzothiazol-2-yl)-2-methylsulfonylacetate Chemical compound C1=C(Br)C=C2SC(C(C(=O)OCC)S(C)(=O)=O)=NC2=C1 JJGILQIDFZDNBM-UHFFFAOYSA-N 0.000 description 1
- OPHDTSWKDLXXQP-UHFFFAOYSA-N ethyl 2-(6-bromo-1,3-benzothiazol-2-yl)pent-4-enoate Chemical compound C1=C(Br)C=C2SC(C(CC=C)C(=O)OCC)=NC2=C1 OPHDTSWKDLXXQP-UHFFFAOYSA-N 0.000 description 1
- MJKNPFKZYHSUCP-UHFFFAOYSA-N ethyl 2-(6-bromo-4-fluoro-1,3-benzothiazol-2-yl)acetate Chemical compound C1=C(Br)C=C2SC(CC(=O)OCC)=NC2=C1F MJKNPFKZYHSUCP-UHFFFAOYSA-N 0.000 description 1
- XUUZAIXBNLIZFF-UHFFFAOYSA-N ethyl 2-(6-phenyl-1,3-benzothiazol-2-yl)acetate Chemical compound C1=C2SC(CC(=O)OCC)=NC2=CC=C1C1=CC=CC=C1 XUUZAIXBNLIZFF-UHFFFAOYSA-N 0.000 description 1
- WGGJVTLPAUASLG-UHFFFAOYSA-N ethyl 2-(6-phenyl-1,3-benzothiazol-2-yl)pent-4-enoate Chemical compound C1=C2SC(C(CC=C)C(=O)OCC)=NC2=CC=C1C1=CC=CC=C1 WGGJVTLPAUASLG-UHFFFAOYSA-N 0.000 description 1
- BHTSHRPBMDTHHX-UHFFFAOYSA-N ethyl 2-[6-(2-fluoropyridin-4-yl)-1,3-benzothiazol-2-yl]acetate Chemical compound C1=C2SC(CC(=O)OCC)=NC2=CC=C1C1=CC=NC(F)=C1 BHTSHRPBMDTHHX-UHFFFAOYSA-N 0.000 description 1
- QHOKAVKYKSJRAD-UHFFFAOYSA-N ethyl 2-[6-(4-fluorophenyl)-1,3-benzothiazol-2-yl]acetate Chemical compound C1=C2SC(CC(=O)OCC)=NC2=CC=C1C1=CC=C(F)C=C1 QHOKAVKYKSJRAD-UHFFFAOYSA-N 0.000 description 1
- UDMVPVBAUTVSTA-UHFFFAOYSA-N ethyl 2-methylsulfonyl-2-(6-phenyl-1,3-benzothiazol-2-yl)acetate Chemical compound C1=C2SC(C(C(=O)OCC)S(C)(=O)=O)=NC2=CC=C1C1=CC=CC=C1 UDMVPVBAUTVSTA-UHFFFAOYSA-N 0.000 description 1
- KWFADUNOPOSMIJ-UHFFFAOYSA-N ethyl 3-chloro-3-oxopropanoate Chemical compound CCOC(=O)CC(Cl)=O KWFADUNOPOSMIJ-UHFFFAOYSA-N 0.000 description 1
- OAYLNYINCPYISS-UHFFFAOYSA-N ethyl acetate;hexane Chemical compound CCCCCC.CCOC(C)=O OAYLNYINCPYISS-UHFFFAOYSA-N 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- OJCSPXHYDFONPU-UHFFFAOYSA-N etoac etoac Chemical compound CCOC(C)=O.CCOC(C)=O OJCSPXHYDFONPU-UHFFFAOYSA-N 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 229960001582 fenfluramine Drugs 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 125000003983 fluorenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 description 1
- 238000002866 fluorescence resonance energy transfer Methods 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 125000003709 fluoroalkyl group Chemical group 0.000 description 1
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 description 1
- PTCGDEVVHUXTMP-UHFFFAOYSA-N flutolanil Chemical compound CC(C)OC1=CC=CC(NC(=O)C=2C(=CC=CC=2)C(F)(F)F)=C1 PTCGDEVVHUXTMP-UHFFFAOYSA-N 0.000 description 1
- 239000011888 foil Substances 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 238000001640 fractional crystallisation Methods 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 125000003838 furazanyl group Chemical group 0.000 description 1
- 229960003692 gamma aminobutyric acid Drugs 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 230000014509 gene expression Effects 0.000 description 1
- 238000007429 general method Methods 0.000 description 1
- 230000007614 genetic variation Effects 0.000 description 1
- 229960004580 glibenclamide Drugs 0.000 description 1
- 229960000346 gliclazide Drugs 0.000 description 1
- 229960004346 glimepiride Drugs 0.000 description 1
- WIGIZIANZCJQQY-RUCARUNLSA-N glimepiride Chemical compound O=C1C(CC)=C(C)CN1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)N[C@@H]2CC[C@@H](C)CC2)C=C1 WIGIZIANZCJQQY-RUCARUNLSA-N 0.000 description 1
- 229960001381 glipizide Drugs 0.000 description 1
- ZJJXGWJIGJFDTL-UHFFFAOYSA-N glipizide Chemical compound C1=NC(C)=CN=C1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)NC2CCCCC2)C=C1 ZJJXGWJIGJFDTL-UHFFFAOYSA-N 0.000 description 1
- 229960003468 gliquidone Drugs 0.000 description 1
- 239000003862 glucocorticoid Substances 0.000 description 1
- 229940120105 glynase Drugs 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 125000004995 haloalkylthio group Chemical group 0.000 description 1
- 125000001475 halogen functional group Chemical group 0.000 description 1
- 125000006343 heptafluoro propyl group Chemical group 0.000 description 1
- 125000005980 hexynyl group Chemical group 0.000 description 1
- 238000004896 high resolution mass spectrometry Methods 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 150000004680 hydrogen peroxides Chemical class 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 1
- UWYVPFMHMJIBHE-OWOJBTEDSA-N hydroxymaleic acid group Chemical group O/C(/C(=O)O)=C/C(=O)O UWYVPFMHMJIBHE-OWOJBTEDSA-N 0.000 description 1
- 238000003384 imaging method Methods 0.000 description 1
- JXXWJMUPNZDILL-UHFFFAOYSA-N imidazo[4,5-b]pyridin-2-one Chemical class C1=CC=NC2=NC(=O)N=C21 JXXWJMUPNZDILL-UHFFFAOYSA-N 0.000 description 1
- 125000002636 imidazolinyl group Chemical group 0.000 description 1
- 150000002466 imines Chemical class 0.000 description 1
- 230000028993 immune response Effects 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000004926 indolenyl group Chemical group 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 229910052500 inorganic mineral Inorganic materials 0.000 description 1
- 229940125396 insulin Drugs 0.000 description 1
- 230000031891 intestinal absorption Effects 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 229940125425 inverse agonist Drugs 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 125000002346 iodo group Chemical group I* 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- SYJRVVFAAIUVDH-UHFFFAOYSA-N ipa isopropanol Chemical compound CC(C)O.CC(C)O SYJRVVFAAIUVDH-UHFFFAOYSA-N 0.000 description 1
- 230000007794 irritation Effects 0.000 description 1
- 125000001977 isobenzofuranyl group Chemical group C=1(OC=C2C=CC=CC12)* 0.000 description 1
- 125000003384 isochromanyl group Chemical group C1(OCCC2=CC=CC=C12)* 0.000 description 1
- 239000012948 isocyanate Substances 0.000 description 1
- 150000002513 isocyanates Chemical class 0.000 description 1
- YDNLNVZZTACNJX-UHFFFAOYSA-N isocyanatomethylbenzene Chemical compound O=C=NCC1=CC=CC=C1 YDNLNVZZTACNJX-UHFFFAOYSA-N 0.000 description 1
- 125000005438 isoindazolyl group Chemical group 0.000 description 1
- 125000004594 isoindolinyl group Chemical group C1(NCC2=CC=CC=C12)* 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003253 isopropoxy group Chemical group [H]C([H])([H])C([H])(O*)C([H])([H])[H] 0.000 description 1
- 125000005956 isoquinolyl group Chemical group 0.000 description 1
- 150000004797 ketoamides Chemical class 0.000 description 1
- 230000003907 kidney function Effects 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- CFHGBZLNZZVTAY-UHFFFAOYSA-N lawesson's reagent Chemical compound C1=CC(OC)=CC=C1P1(=S)SP(=S)(C=2C=CC(OC)=CC=2)S1 CFHGBZLNZZVTAY-UHFFFAOYSA-N 0.000 description 1
- 210000000265 leukocyte Anatomy 0.000 description 1
- 235000019626 lipase activity Nutrition 0.000 description 1
- 230000002366 lipolytic effect Effects 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 239000012280 lithium aluminium hydride Substances 0.000 description 1
- 230000003908 liver function Effects 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 108010030696 low density lipoprotein triglyceride Proteins 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 235000019341 magnesium sulphate Nutrition 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- 229960000299 mazindol Drugs 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- COTNUBDHGSIOTA-UHFFFAOYSA-N meoh methanol Chemical compound OC.OC COTNUBDHGSIOTA-UHFFFAOYSA-N 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 229910052987 metal hydride Inorganic materials 0.000 description 1
- 150000004681 metal hydrides Chemical class 0.000 description 1
- XZWYZXLIPXDOLR-UHFFFAOYSA-N metformin Chemical compound CN(C)C(=N)NC(N)=N XZWYZXLIPXDOLR-UHFFFAOYSA-N 0.000 description 1
- 229960003105 metformin Drugs 0.000 description 1
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 1
- NQMRYBIKMRVZLB-UHFFFAOYSA-N methylamine hydrochloride Chemical compound [Cl-].[NH3+]C NQMRYBIKMRVZLB-UHFFFAOYSA-N 0.000 description 1
- 125000002816 methylsulfanyl group Chemical group [H]C([H])([H])S[*] 0.000 description 1
- 229960001110 miglitol Drugs 0.000 description 1
- 235000010755 mineral Nutrition 0.000 description 1
- 239000011707 mineral Substances 0.000 description 1
- SLZIZIJTGAYEKK-CIJSCKBQSA-N molport-023-220-247 Chemical compound C([C@@H](C(=O)N[C@@H](C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=1N=CNC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC=1N=CNC=1)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C)C(N)=O)NC(=O)[C@H]1N(CCC1)C(=O)CNC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)CN)[C@@H](C)O)C1=CNC=N1 SLZIZIJTGAYEKK-CIJSCKBQSA-N 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- PSHKMPUSSFXUIA-UHFFFAOYSA-N n,n-dimethylpyridin-2-amine Chemical compound CN(C)C1=CC=CC=N1 PSHKMPUSSFXUIA-UHFFFAOYSA-N 0.000 description 1
- PEECTLLHENGOKU-UHFFFAOYSA-N n,n-dimethylpyridin-4-amine Chemical compound CN(C)C1=CC=NC=C1.CN(C)C1=CC=NC=C1 PEECTLLHENGOKU-UHFFFAOYSA-N 0.000 description 1
- PEYGYLANDJMCGX-UHFFFAOYSA-N n-(2-methoxyethyl)-4-[2-[2-oxo-2-(2-sulfamoylethylamino)ethyl]-1,3-benzothiazol-6-yl]benzamide Chemical compound C1=CC(C(=O)NCCOC)=CC=C1C1=CC=C(N=C(CC(=O)NCCS(N)(=O)=O)S2)C2=C1 PEYGYLANDJMCGX-UHFFFAOYSA-N 0.000 description 1
- AZLZCIUGEZSLOM-UHFFFAOYSA-N n-(2-methoxyethyl)-n-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzamide Chemical compound C1=CC(C(=O)N(C)CCOC)=CC=C1B1OC(C)(C)C(C)(C)O1 AZLZCIUGEZSLOM-UHFFFAOYSA-N 0.000 description 1
- KYPXZZNACVMSGT-UHFFFAOYSA-N n-(2-methoxyethyl)-n-methyl-4-[2-[1-methylsulfonyl-2-oxo-2-(2-sulfamoylethylamino)ethyl]-1,3-benzothiazol-6-yl]benzamide Chemical compound C1=CC(C(=O)N(C)CCOC)=CC=C1C1=CC=C(N=C(S2)C(C(=O)NCCS(N)(=O)=O)S(C)(=O)=O)C2=C1 KYPXZZNACVMSGT-UHFFFAOYSA-N 0.000 description 1
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003506 n-propoxy group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])O* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- KHSSPZJNBPLHRP-UHFFFAOYSA-N n-tert-butyl-2,2,2-trichloroacetamide Chemical compound CC(C)(C)NC(=O)C(Cl)(Cl)Cl KHSSPZJNBPLHRP-UHFFFAOYSA-N 0.000 description 1
- 239000006070 nanosuspension Substances 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- OELFLUMRDSZNSF-BRWVUGGUSA-N nateglinide Chemical compound C1C[C@@H](C(C)C)CC[C@@H]1C(=O)N[C@@H](C(O)=O)CC1=CC=CC=C1 OELFLUMRDSZNSF-BRWVUGGUSA-N 0.000 description 1
- 229960000698 nateglinide Drugs 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 125000002868 norbornyl group Chemical group C12(CCC(CC1)C2)* 0.000 description 1
- 238000005016 nuclear Overhauser enhanced spectroscopy Methods 0.000 description 1
- URPYMXQQVHTUDU-OFGSCBOVSA-N nucleopeptide y Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(N)=O)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](N)CC=1C=CC(O)=CC=1)C1=CC=C(O)C=C1 URPYMXQQVHTUDU-OFGSCBOVSA-N 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 239000007935 oral tablet Substances 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 239000012044 organic layer Substances 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- AHLBNYSZXLDEJQ-FWEHEUNISA-N orlistat Chemical compound CCCCCCCCCCC[C@H](OC(=O)[C@H](CC(C)C)NC=O)C[C@@H]1OC(=O)[C@H]1CCCCCC AHLBNYSZXLDEJQ-FWEHEUNISA-N 0.000 description 1
- 229960001243 orlistat Drugs 0.000 description 1
- 230000002018 overexpression Effects 0.000 description 1
- 150000004866 oxadiazoles Chemical class 0.000 description 1
- 239000007800 oxidant agent Substances 0.000 description 1
- LXNAVEXFUKBNMK-UHFFFAOYSA-N palladium(II) acetate Substances [Pd].CC(O)=O.CC(O)=O LXNAVEXFUKBNMK-UHFFFAOYSA-N 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 239000004031 partial agonist Substances 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 229940049954 penicillin Drugs 0.000 description 1
- 125000006340 pentafluoro ethyl group Chemical group FC(F)(F)C(F)(F)* 0.000 description 1
- 125000002255 pentenyl group Chemical group C(=CCCC)* 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 125000005981 pentynyl group Chemical group 0.000 description 1
- 239000002304 perfume Substances 0.000 description 1
- 239000008024 pharmaceutical diluent Substances 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 229940127557 pharmaceutical product Drugs 0.000 description 1
- 230000003285 pharmacodynamic effect Effects 0.000 description 1
- 125000004934 phenanthridinyl group Chemical group C1(=CC=CC2=NC=C3C=CC=CC3=C12)* 0.000 description 1
- 125000004625 phenanthrolinyl group Chemical group N1=C(C=CC2=CC=C3C=CC=NC3=C12)* 0.000 description 1
- 125000001791 phenazinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3N=C12)* 0.000 description 1
- 125000001484 phenothiazinyl group Chemical group C1(=CC=CC=2SC3=CC=CC=C3NC12)* 0.000 description 1
- 125000005954 phenoxathiinyl group Chemical group 0.000 description 1
- 125000001644 phenoxazinyl group Chemical group C1(=CC=CC=2OC3=CC=CC=C3NC12)* 0.000 description 1
- 229960003562 phentermine Drugs 0.000 description 1
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical compound OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 1
- OAHKWDDSKCRNFE-UHFFFAOYSA-N phenylmethanesulfonyl chloride Chemical compound ClS(=O)(=O)CC1=CC=CC=C1 OAHKWDDSKCRNFE-UHFFFAOYSA-N 0.000 description 1
- 229960000395 phenylpropanolamine Drugs 0.000 description 1
- DLNKOYKMWOXYQA-APPZFPTMSA-N phenylpropanolamine Chemical compound C[C@@H](N)[C@H](O)C1=CC=CC=C1 DLNKOYKMWOXYQA-APPZFPTMSA-N 0.000 description 1
- 125000001557 phthalyl group Chemical group C(=O)(O)C1=C(C(=O)*)C=CC=C1 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 229960005095 pioglitazone Drugs 0.000 description 1
- 125000004928 piperidonyl group Chemical group 0.000 description 1
- 229960005235 piperonyl butoxide Drugs 0.000 description 1
- 125000004591 piperonyl group Chemical group C(C1=CC=2OCOC2C=C1)* 0.000 description 1
- LYKMMUBOEFYJQG-UHFFFAOYSA-N piperoxan Chemical compound C1OC2=CC=CC=C2OC1CN1CCCCC1 LYKMMUBOEFYJQG-UHFFFAOYSA-N 0.000 description 1
- 230000036470 plasma concentration Effects 0.000 description 1
- 230000000291 postprandial effect Effects 0.000 description 1
- 235000015320 potassium carbonate Nutrition 0.000 description 1
- 235000011181 potassium carbonates Nutrition 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 229960003611 pramlintide Drugs 0.000 description 1
- 108010029667 pramlintide Proteins 0.000 description 1
- NRKVKVQDUCJPIZ-MKAGXXMWSA-N pramlintide acetate Chemical compound C([C@@H](C(=O)NCC(=O)N1CCC[C@H]1C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](C(C)C)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CS)NC(=O)[C@@H](N)CCCCN)[C@@H](C)O)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 NRKVKVQDUCJPIZ-MKAGXXMWSA-N 0.000 description 1
- 229960002847 prasterone Drugs 0.000 description 1
- 239000012041 precatalyst Substances 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 108010070701 procolipase Proteins 0.000 description 1
- AAHFRWRQQDVWPX-UHFFFAOYSA-N prop-2-ene-1-sulfonyl chloride Chemical compound ClS(=O)(=O)CC=C AAHFRWRQQDVWPX-UHFFFAOYSA-N 0.000 description 1
- QLNJFJADRCOGBJ-UHFFFAOYSA-N propionamide Chemical compound CCC(N)=O QLNJFJADRCOGBJ-UHFFFAOYSA-N 0.000 description 1
- 125000002568 propynyl group Chemical group [*]C#CC([H])([H])[H] 0.000 description 1
- 230000004224 protection Effects 0.000 description 1
- 239000003586 protic polar solvent Substances 0.000 description 1
- 125000001042 pteridinyl group Chemical group N1=C(N=CC2=NC=CN=C12)* 0.000 description 1
- 125000004309 pyranyl group Chemical group O1C(C=CC=C1)* 0.000 description 1
- 125000003072 pyrazolidinyl group Chemical group 0.000 description 1
- 125000002755 pyrazolinyl group Chemical group 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000001422 pyrrolinyl group Chemical group 0.000 description 1
- FCACETKNGAHGMV-UHFFFAOYSA-N pyrrolo[3,2-b]pyridin-3-one Chemical class C1=CN=C2C(=O)C=NC2=C1 FCACETKNGAHGMV-UHFFFAOYSA-N 0.000 description 1
- 150000003242 quaternary ammonium salts Chemical class 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 125000004621 quinuclidinyl group Chemical group N12C(CC(CC1)CC2)* 0.000 description 1
- 230000033300 receptor internalization Effects 0.000 description 1
- 239000003488 releasing hormone Substances 0.000 description 1
- 229960002354 repaglinide Drugs 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 238000004366 reverse phase liquid chromatography Methods 0.000 description 1
- 238000012552 review Methods 0.000 description 1
- 125000006413 ring segment Chemical group 0.000 description 1
- 229960004586 rosiglitazone Drugs 0.000 description 1
- 238000007789 sealing Methods 0.000 description 1
- 239000008299 semisolid dosage form Substances 0.000 description 1
- 229960004425 sibutramine Drugs 0.000 description 1
- UNAANXDKBXWMLN-UHFFFAOYSA-N sibutramine Chemical compound C=1C=C(Cl)C=CC=1C1(C(N(C)C)CC(C)C)CCC1 UNAANXDKBXWMLN-UHFFFAOYSA-N 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 108091008012 small dense LDL Proteins 0.000 description 1
- 230000000391 smoking effect Effects 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 235000010265 sodium sulphite Nutrition 0.000 description 1
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 1
- WGRULTCAYDOGQK-UHFFFAOYSA-M sodium;sodium;hydroxide Chemical compound [OH-].[Na].[Na+] WGRULTCAYDOGQK-UHFFFAOYSA-M 0.000 description 1
- 239000007909 solid dosage form Substances 0.000 description 1
- 238000007614 solvation Methods 0.000 description 1
- 239000011877 solvent mixture Substances 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 238000010922 spray-dried dispersion Methods 0.000 description 1
- 230000006641 stabilisation Effects 0.000 description 1
- 238000011105 stabilization Methods 0.000 description 1
- 235000011150 stannous chloride Nutrition 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 230000000365 steroidogenetic effect Effects 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- 229910021653 sulphate ion Inorganic materials 0.000 description 1
- 235000011149 sulphuric acid Nutrition 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 229940037128 systemic glucocorticoids Drugs 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 238000002626 targeted therapy Methods 0.000 description 1
- ROEYCXPPEWFKRS-UHFFFAOYSA-N tert-butyl 2-(2-ethoxy-2-oxoethyl)-1,3-benzothiazole-5-carboxylate Chemical compound CC(C)(C)OC(=O)C1=CC=C2SC(CC(=O)OCC)=NC2=C1 ROEYCXPPEWFKRS-UHFFFAOYSA-N 0.000 description 1
- BUKGEFFXGBQUIX-UHFFFAOYSA-N tert-butyl 2-[[2-[6-(6-fluoropyridin-3-yl)-1,3-benzothiazol-2-yl]-2-(2-methoxyethylsulfonyl)acetyl]amino]acetate Chemical compound C1=C2SC(C(C(=O)NCC(=O)OC(C)(C)C)S(=O)(=O)CCOC)=NC2=CC=C1C1=CC=C(F)N=C1 BUKGEFFXGBQUIX-UHFFFAOYSA-N 0.000 description 1
- SVDLMOXBYRIHHL-UHFFFAOYSA-N tert-butyl 2-[[2-[6-(6-fluoropyridin-3-yl)-1,3-benzothiazol-2-yl]acetyl]amino]acetate Chemical compound C1=C2SC(CC(=O)NCC(=O)OC(C)(C)C)=NC2=CC=C1C1=CC=C(F)N=C1 SVDLMOXBYRIHHL-UHFFFAOYSA-N 0.000 description 1
- SJMDMGHPMLKLHQ-UHFFFAOYSA-N tert-butyl 2-aminoacetate Chemical compound CC(C)(C)OC(=O)CN SJMDMGHPMLKLHQ-UHFFFAOYSA-N 0.000 description 1
- BDTGRLLMTJQUOQ-UHFFFAOYSA-N tert-butyl 2-chloro-1,3-benzothiazole-6-carboxylate Chemical compound CC(C)(C)OC(=O)C1=CC=C2N=C(Cl)SC2=C1 BDTGRLLMTJQUOQ-UHFFFAOYSA-N 0.000 description 1
- WHRNULOCNSKMGB-UHFFFAOYSA-N tetrahydrofuran thf Chemical compound C1CCOC1.C1CCOC1 WHRNULOCNSKMGB-UHFFFAOYSA-N 0.000 description 1
- WROMPOXWARCANT-UHFFFAOYSA-N tfa trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.OC(=O)C(F)(F)F WROMPOXWARCANT-UHFFFAOYSA-N 0.000 description 1
- 231100001274 therapeutic index Toxicity 0.000 description 1
- 238000011285 therapeutic regimen Methods 0.000 description 1
- 125000004627 thianthrenyl group Chemical group C1(=CC=CC=2SC3=CC=CC=C3SC12)* 0.000 description 1
- 150000001467 thiazolidinediones Chemical class 0.000 description 1
- 125000005309 thioalkoxy group Chemical group 0.000 description 1
- 125000005403 thiohaloalkoxy group Chemical group 0.000 description 1
- 230000002537 thrombolytic effect Effects 0.000 description 1
- 229960002277 tolazamide Drugs 0.000 description 1
- OUDSBRTVNLOZBN-UHFFFAOYSA-N tolazamide Chemical compound C1=CC(C)=CC=C1S(=O)(=O)NC(=O)NN1CCCCCC1 OUDSBRTVNLOZBN-UHFFFAOYSA-N 0.000 description 1
- 229960005371 tolbutamide Drugs 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 230000032258 transport Effects 0.000 description 1
- FAQYAMRNWDIXMY-UHFFFAOYSA-N trichloroborane Chemical compound ClB(Cl)Cl FAQYAMRNWDIXMY-UHFFFAOYSA-N 0.000 description 1
- 125000003866 trichloromethyl group Chemical group ClC(Cl)(Cl)* 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 1
- 230000001960 triggered effect Effects 0.000 description 1
- 235000019798 tripotassium phosphate Nutrition 0.000 description 1
- 229910052722 tritium Inorganic materials 0.000 description 1
- GXPHKUHSUJUWKP-UHFFFAOYSA-N troglitazone Chemical compound C1CC=2C(C)=C(O)C(C)=C(C)C=2OC1(C)COC(C=C1)=CC=C1CC1SC(=O)NC1=O GXPHKUHSUJUWKP-UHFFFAOYSA-N 0.000 description 1
- 229960001641 troglitazone Drugs 0.000 description 1
- GXPHKUHSUJUWKP-NTKDMRAZSA-N troglitazone Natural products C([C@@]1(OC=2C(C)=C(C(=C(C)C=2CC1)O)C)C)OC(C=C1)=CC=C1C[C@H]1SC(=O)NC1=O GXPHKUHSUJUWKP-NTKDMRAZSA-N 0.000 description 1
- 150000003672 ureas Chemical class 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- PXXNTAGJWPJAGM-UHFFFAOYSA-N vertaline Natural products C1C2C=3C=C(OC)C(OC)=CC=3OC(C=C3)=CC=C3CCC(=O)OC1CC1N2CCCC1 PXXNTAGJWPJAGM-UHFFFAOYSA-N 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 238000012800 visualization Methods 0.000 description 1
- 229960001729 voglibose Drugs 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 238000010626 work up procedure Methods 0.000 description 1
- 125000001834 xanthenyl group Chemical group C1=CC=CC=2OC3=CC=CC=C3C(C12)* 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/60—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings condensed with carbocyclic rings or ring systems
- C07D277/62—Benzothiazoles
- C07D277/64—Benzothiazoles with only hydrocarbon or substituted hydrocarbon radicals attached in position 2
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/10—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Definitions
- the present invention provides novel amide, urea or sulfone amide linked benzothiazole compounds and analogues, which are endothelial lipase (EL) inhibitors, compositions containing them, and methods of using them, for example, for the treatment and/or prophylaxis of dyslipidemias and the sequelae thereof.
- EL endothelial lipase
- Cardiovascular disease is a major health risk throughout the industrialized world.
- Atherosclerosis the most prevalent of cardiovascular diseases, is the principal cause of heart attack, and stroke, and thereby the principal cause of death in the United States.
- Atherosclerosis is a complex disease involving many cell types and molecular factors (for a detailed review, see Ross, R., Nature, 362(6423):801-809 (1993)).
- HDL high density lipoprotein
- TG lipase and triacylglycerol (TG) lipase family of proteins which hydrolyze triglycerides, phospholipids (PL), and cholesteryl esters (CE), generating fatty acids to facilitate intestinal absorption, energy production, or storage.
- TA lipases lipoprotein lipase (LPL) influences the metabolism of HDL cholesterol by hydro lyzing triglycerides in triglyceride-rich lipoproteins, resulting in the transfer of lipids and apolipoproteins to HDL and is responsible for hydrolyzing chylomicron and very low density lipoprotein (VLDL) in muscle and adipose tissues.
- VLDL very low density lipoprotein
- Hepatic lipase hydrolyzes HDL triglyceride and phospholipids, generating smaller, lipid-depleted HDL particles, and plays a role in the uptake of HDL cholesterol (Jin, W. et al., Trends Endocrinol. Metab., 13(4): 174-178 (2002); Wong, H. et al, J. Lipid Res., 43:993-999 (2002)).
- Endothelial lipase also known as EDL, EL, LIPG, endothelial-derived lipase, and endothelial cell-derived lipase
- EDL EDL
- EL endothelial-derived lipase
- endothelial cell-derived lipase endothelial cell-derived lipase
- Recombinant endothelial lipase protein has substantial phospholipase activity but has been reported to have less hydrolytic activity toward triglyceride lipids (Hirata, K. et al, J. Biol. Chem., 274(20): 14170-14175 (1999); Jaye, M. et al, Nat. Genet., 21 :424- 428 (1999)).
- endothelial lipase does exhibit triglyceride lipase activity ex vivo in addition to its HDL phospholipase activity, and endothelial lipase was found to hydrolyze HDL more efficiently than other lipoproteins (McCoy, M.G.
- endothelial lipase is a relatively new member in the lipase gene family
- a full understanding of the potential of endothelial lipase inhibitors to human health, as well as the inhibitors of other lipases in general, requires more studies.
- the present disclosure provides amide, urea or sulfone amide linked benzothiazole compounds and their analogues, including stereoisomers, tautomers, pharmaceutically acceptable salts, or solvates thereof, which are useful as EL inhibitors.
- the present invention also provides processes and intermediates for making the compounds of the present invention.
- the present invention also provides pharmaceutical compositions comprising a pharmaceutically acceptable carrier and at least one of the compounds of the present invention or stereoisomers, tautomers, pharmaceutically acceptable salts, or solvates thereof.
- the compounds of the invention may be used in the treatment and/or prophylaxis of dyslipidemias and the sequelae thereof.
- the compounds of the invention may be used in therapy.
- the compounds of the invention may be used for the manufacture of a medicament for the treatment and/or prophylaxis of dyslipidemias and the sequelae thereof.
- the compounds of the invention can be used alone, in combination with other compounds of the present invention, or in combination with one or more, preferably one to two other agent(s).
- the present invention provides, inter alia, a compound of Formula (I):
- R 1 is inde endently selected from: halogen, CN, C0 2 (C 1 . 4 alkyl), -CO-RJ,
- R 2 is, independently at each occurrence, selected from: halogen, OH, C ⁇ alkyl, C j .4 alkoxy, C ⁇ haloalkyl, C ⁇ haloalkoxy, CN, NH 2 , N0 2 , ⁇ ( ⁇ _ 4 alkyl),
- R 3 is independently selected from: -S0 2 R 5 and -NHCOR 6 ;
- R 4 independently selected from:
- R 5 is independently selected from: Ci _g alkyl substituted with 0-1 R 9 ,
- R 6 is independently selected from: C ⁇ g alkyl, C 2 . 6 alkenyl, C ⁇ g alkoxy,
- R 7 is independently selected from: CONH 2 , CONH(C!_ 4 alkyl),
- alkoxy ? ? phenyl and a 5 _ to 6-membered heteroaryl comprising: carbon atoms and 1-4 heteroatoms selected from N, NR C , O, and S(0) p ; wherein said phenyl and heteroaryl are is substituted with 0-2 R 1 1 ;
- R 9 is independently selected from: halogen, C1 .4 alkoxy, C 1 .4 haloalkyl, C haloalkoxy, NH 2 , C0 2 H, CO ⁇ C ⁇ alkyl), S0 3 H, CONHR d , NHCONHR d ,
- NHC0 2 R d s and 5- to 6-membered heterocycle comprising: carbon atoms and 1-4 heteroatoms selected from N, NR8, O, and S(0) p ;
- R 10 is independently selected from: OH, Ci _g alkyl, C 2 _g alkenyl, NH 2 ,
- R 1 1 is, independently at each occurrence, selected from: halogen, C1 .4 alkyl, Ci .4 haloalkyl, NH 2 , and phenyl;
- R a is, independently at each occurrence, selected from: halogen, Ci _g alkyl substituted with 0-1 R f , Ci .4 alkoxy substituted with 0-1 R f , Ci .g haloalkyl,
- R b is, independently at each occurrence, selected from: halogen, C 1 .4 alkyl, C alkoxy, C haloalkyl, C haloalkoxy, OH, CN, NH 2 , N0 2 , NH(C!_ 4 alkyl), (C!. 4 alkyl) 2 , C0 2 H, CO ⁇ C ⁇ alkyl), S0 2 (C M alkyl), CONH 2 , and
- R c is, independently at each occurrence, selected from: H, Ci _g alkyl substituted with 0-1 R e , CO(C alkyl), C0 2 (C 1 . 4 alkyl), COBn, C0 2 Bn, -(CH 2 ) t -piperidinyl, -(CH 2 ) t -morpholinyl, -(CH 2 ) t -piperazinyl, pyrimidinyl,
- R d is, independently at each occurrence, selected from: Ci _g alkyl and
- R e is, independently at each occurrence, selected from: halogen, Ci .4 alkyl, C1 .4 alkoxy, C 1 .4 haloalkyl, and C1 .4 haloalkoxy;
- R f is, independently at each occurrence, selected from: OH, halogen, C1 .4 alkyl, C1 .4 alkoxy, C 1 .4 haloalkyl, and C1 .4 haloalkoxy;
- R8 is, independently at each occurrence, selected from: H and C ⁇ alkyl
- R h is, independently at each occurrence, selected from: H, Ci _g haloalkyl and Ci _6 alkyl substituted with 0-1 R f ;
- R 1 is, independently at each occurrence, selected from: Ci .4 haloalkyl and
- RJ is, independently at each occurrence: C3.6 carbocycle or a 4- to 6-membered heterocycle comprising: carbon atoms and 1-4 heteroatoms selected from N, NR ⁇ , O, and S(0) p ; wherein said carbocycle and heterocycle are substituted with 0-2 R f ;
- n and t are, independently at each occurrence, selected from 0, 1 , 2, and 3;
- n is, independently at each occurrence, selected from 0 and 1 ;
- p is, independently at each occurrence, selected from 0, 1 , and 2;
- s is, independently at each occurrence, selected from 1 , 2, and 3.
- the present invention includes a compound of Formula (la) or (lb):
- n and t are, independently at each occurrence, selected from 0, 1 , and 2; and s is, independently at each occurrence, selected from 1 and 2.
- the present invention includes a compound of Formula (Ila) or (lib):
- R 2 is independently selected from: halogen and C ⁇ alkyl.
- the present invention includes a compound of Formula (la) (Ila) or (lib), or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof, within the scope of the first, second and third aspects, wherein:
- R4 is ⁇ so2NH2 ;
- R 6 is independently selected from: C X alkyl, -CHF 2 , NHBn, 4-CF 3 -Ph, and pyrid-3-yl.
- the present invention includes a compound of Formula (Ilia) or (nib):
- R 2 is independently selected from: halogen, C ⁇ _4 alkyl and C ⁇ _4 haloalkyl.
- the present invention includes a compound of Formula (I), (lb), (Ilia) or (Illb), or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof, within the scope of any of the first, second and fifth aspects, wherein:
- R 1 is inde endently selected from: H)
- R 4 is independently selected from: ⁇ " ⁇ SO ⁇ C ⁇ alkyl) ? ⁇ S0 2 NH 2 ?
- R 5 is independently selected from: Ci .4 alkyl substituted with 0-1 R 9 ,
- R 9 is independently selected from: C 1 .4 alkoxy, C 1 .4 haloalkyl, NH 2 , and NHC0 2 Bn;
- R a is, independently at each occurrence, selected from: OH, halogen, CN, C .4 alkyl substituted with 0-1 OH, C ⁇ alkoxy, -0(CH 2 ) 1 . 2 0(C 1 . 4 alkyl),
- halogen and Ci .4 haloalkyl
- R c is, independently at each occurrence, selected from: H, Ci .4 alkyl,
- R f is, independently at each occurrence, selected from: OH, halogen, C ⁇ alkyl, C ⁇ _4 haloalkyl, C ⁇ _4 alkoxy, and C ⁇ _4 haloalkoxy.
- the present invention includes a compound of Formula (I), (lb), (Ilia) or (Illb), or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof, within the scope of any of the first, second, fifth and sixth aspects, wherein:
- R 1 is independently selected from: Ph, 3-halo-Ph, 4-halo-Ph, 3- ⁇ _ 4 alkoxy-Ph, 4-CH 2 OH-Ph, 3-CN-Ph, 4-CN-Ph, 4-C0 2 H-Ph, 4-C0 2 (C 1 . 4 alkyl)-Ph,
- R 4 is independently selected from: ⁇ S0 2 NH 2 ? ⁇ S0 2 NH(C 2 . 6 alkenyl) ;
- R 5 is independently selected from: C ⁇ alkyl substituted with 0-1 R 9 , and
- R 9 is independently selected from: C 1.4 alkoxy, C1.4 haloalkyl, NH 2 , and
- the present invention includes a compound of Formula (I), (lb), (Ilia) or (Illb), or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof, within the scope of any of the first, second, fifth, sixth and seventh aspects, wherein:
- R 1 is independently selected from: Ph, 3-halo-Ph, 4-halo-Ph, 3- ⁇ _ 4 alkoxy-Ph, 4-CH 2 OH-Ph, 3-CN-Ph, 4-CN-Ph, 4-C0 2 H-Ph, 4-C0 2 (C 1 . 4 alkyl)-Ph, 3-NHCO C alkyl)-Ph, 4-NHCO(C 1 _ alkyl)-Ph, 4-(pyrazol-l-yl)-Ph, pyrimidinyl and
- R 4 is ii ⁇ S02NH2 ⁇
- R 5 is independently selected from: C ⁇ alkyl substituted with 0-1 R 9 , and
- R 9 is independently selected from: C 1 .4 alkoxy, C1 .4 haloalkyl, NH 2 , and NHC0 2 Bn.
- the present invention includes a compound of Formula (I), (lb), (Ilia) or (Illb), or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or solvate thereof, within the scope of any of the first, second, fifth, sixth, seventh and eighth aspects, wherein:
- R 1 is independently selected from: Ph, 3-F-Ph, 4-F-Ph, 3-OMe-Ph, 4-CH 2 OH-Ph, 3-CN-Ph, 4-CN-Ph, 4-C0 2 H-Ph, 4-C0 2 Me-Ph 3-NHCOMe-Ph, 4-NHCOMe-Ph,
- R 5 is independently selected from: Me, Et, Pr, z ' -Bu, ⁇ 5 ⁇ , Bn, 4-F-Bn,
- the present invention includes a compound of Formula (I), (lb), (Ilia) or (Illb), or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or solvate thereof, within the scope of any of the first, second, fifth, sixth and seventh aspects, wherein:
- R 1 is Ph; t idependently selected from: ⁇ S0 2 NH(C 2 . 6 alkenyl) ? an( j
- R 5 is independently selected from: C 1 .4 alkyl, C 2 _4 alkenyl, -(CH 2 ) 2 0(Ci and -(CH 2 ) 2 NHC0 2 Bn.
- the present invention includes a compound of Formula (I), (lb), (Ilia) or (Illb), or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof, within the scope of any of the first, second, fifth and sixth aspects, wherein: independently selected from: H)
- R 4 is independently selected from: "Ji ⁇ S0 2 (C 1 _ 4 alkyl) ? " J ⁇ SOsNHs ?
- R 5 is independently selected from: Ci .4 alkyl substituted with 0-1 R 9
- R 9 is independently selected from: C 1 .4 alkoxy, C1 .4 haloalkyl, NH 2 , and NHC0 2 Bn;
- R a is, independently at each occurrence, selected from: OH, halogen, C1.4 alkyl substituted with 0- 1 OH, C _ 4 alkoxy, -0(CH 2 ) 2 0(C _ 4 alkyl), C _ 4 haloalkyl,
- R b is independently selected from: CN, halogen and C ⁇ haloalkyl
- R c is, independently at each occurrence, selected from: H, Ci .4 alkyl substituted with 0-1 Ci .4 alkoxy, -(CH 2 )o -2 - henyl substituted with 0-1 halo), -(CH 2 )o -2 -piperidinyl,
- R f is, independently at each occurrence, selected from: OH, CN, halogen,
- the present invention includes a compound of Formula (I), (lb), (Ilia) or (Illb), or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof, within the scope of any of the first, second, fifth, sixth and twelfth aspects, wherein:
- R 1 is independently selected from: 3-(CH 2 ) 2 OH-Ph, 4-(CH 2 ) 2 OH-Ph,
- the present invention includes a compound of Formula (I), (lb), (Ilia) or (Illb), or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof, within the scope of any of the first, second, fifth, sixth, twelfth and thirteenth aspects, wherein:
- R 1 is independently selected from: 3-(CH 2 ) 2 OH-Ph, 4-(CH 2 ) 2 OH-Ph, 4-0(CH 2 ) 2 OMe-Ph, 4-OCF 3 -Ph, 4-NH 2 -Ph, 4-CONH 2 -Ph, 4-CONH(t-Bu)-Ph,
- R 4 is independently selected from: ⁇ S0 2 Me ? ! ⁇ S0 2 NH 2 ?
- R 5 is independently selected from: Me, Pr, z ' -Pr, z ' -Bu, -(CH 2 ) 2 OMe, -(CH 2 )2CF 3 , -(CH 2 ) 3 CF 3 , -(CH 2 ) 2 NH 2 , cyclopropylmethyl, Bn, 4-F-Bn, 4-CF 3 -Bn,
- the present invention provides a compound of Formula (I), or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof, within the scope of the first aspect, wherein:
- R 1 is independently selected from: phenyl and a 5- to 6-membered heteroaryl comprising: carbon atoms and 1 -4 heteroatoms selected from N, NR C , O, and S(0) p ; wherein each phenyl and heteroaryl are substituted with 0-3 R a ;
- R 2 is, independently at each occurrence, selected from: halogen, OH, Ci _ 4 alkyl, C alkoxy, C haloalkyl, C haloalkoxy, CN, NH 2 , N0 2 , NH(C alkyl),
- R 3 is independently selected from: -S0 2 R 5 and -NHCOR 6 ;
- R 4 independently selected from: s and 4 ;
- R 5 is independently selected from: Ci .g alkyl, C 2 -6 alkenyl, -(CH 2 ) m CHF 2 , -(CH 2 ) m CF 3 , -(CH 2 ) m -(0) n -(C 3 _ 6 carbocycle substituted with 0-3 R b ),
- R 6 is independently selected from: Ci _g alkyl, C 2.6 alkenyl, Ci_ 6 alkoxy,
- R 7 is independently selected from: CONH 2 , CO H(C!_ 4 alkyl),
- R a is, independently at each occurrence, selected from: halogen, Ci _g alkyl substituted with 0-1 OH, Ci .4 alkoxy, Ci .g haloalkyl, Ci.g haloalkoxy, OH, CN, N0 2 , NH 2 , NH(C alkyl), N(C alkyl) 2 , C0 2 H, C0 2 (C 1 . 4 alkyl), CONH 2 ,
- R b is, independently at each occurrence, selected from: halogen, C 1 .4 alkyl, C alkoxy, C haloalkyl, C haloalkoxy, OH, CN, NH 2 , N0 2 , NH(C alkyl), N(Ci_ 4 alkyl) 2 , C0 2 H, C0 2 (C alkyl), S0 2 (C M alkyl), CONH 2 , and
- R c is, independently at each occurrence, selected from: H, Ci _g alkyl,
- R d is, independently at each occurrence, selected from: Ci.g alkyl and
- R e is, independently at each occurrence, selected from: halogen, Ci .4 alkyl,
- s is, independently at each occurrence, selected from 1 , 2, and 3.
- the present invention includes a compound of Formula (I), (la) or (lb), or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof, within the scope of the first or second aspect, wherein: 4 is independently selected from: ⁇ SC ⁇ C ⁇ alkyl) ? ⁇ S0 2 NH 2 ?
- the present invention includes a compound of Formula (I), (lb) or (Ilia), or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof, wherein:
- R 1 is independently selected from: Ph, 4 ⁇ 0 2 ( ⁇ _ 4 alkyl)-Ph, 4-NH 2 -Ph, 4-CONH 2 -Ph, 4-S02 (C alkyl) 2 -Ph, 4- ⁇ 0 2 ( ⁇ _ 4 alkyl)-Ph, 4-(pyrazol-l-yl)-Ph, 3-C j _4 alkyl-pyrid-4-yl, 6-C _4 alkoxy-pyrid-3-yl, 6-halo-pyrid-3-yl, 3-halo-pyrid-4-yl,
- 6-CF 3 -pyrid-3-yl, and c i- alkyl 4 is independently selected from: ⁇ SC ⁇ C ⁇ alkyl) ? ⁇ S0 2 NH 2 ?
- R 5 is independently selected from: C 1 .4 alkyl, C 2 _4 alkenyl, Bn, 4-CF 3 -Bn, and -(CH 2 ) 1 . 3 CF 3 .
- the present invention includes a compound of Formula (I), (lb) or (Ilia), or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof, wherein:
- R 1 is independently selected from: Ph, 4-C0 2 (C 1.4 alkyl)-Ph and
- R4 is ⁇ so 2 NH 2 ;
- the present invention includes a compound of Formula (I), (lb) or (Ilia), or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof, wherein:
- R 1 is independently selected from: Ph, 4-C02Me-Ph and 4-(pyrazol-l-yl)-Ph; and i independently selected from: Me and
- the present invention includes a compound of Formula (I), (lb) or (Ilia), or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof, within the scope of any of the first, second, third, sixth and seventh aspects, wherein:
- 6-C1 alkoxy-pyrid-3-yl, 6-halo-pyrid-3-yl, 3-halo-pyrid-4-yl, 6-CF 3 -pyrid-3-yl,
- the present invention includes a compound of Formula (I), (lb) or (Ilia), or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof, within the scope of any of the first, second, third, sixth and tenth aspects, wherein:
- R 1 is independently selected from: 4-NH 2 -Ph, 4-CONH 2 -Ph, 4-S0 2 N(Me) 2 -Ph, 4-NHC0 2 Me-Ph, 3-Me-pyrid-4-yl, 6-OMe-pyrid-3-yl, 6-F-pyrid-3-yl, 3-F-pyrid-4-yl,
- 3-Cl-pyrid-4-yl, 6-CF 3 -pyrid-3-yl, 4 is independently selected from: ⁇ S0 2 Me ? ! ⁇ S0 2 NH 2 ?
- R 5 is independently selected from: Me, Pr, z ' -Pr, Bn, 4-CF 3 -Bn, and -(CH 2 ) 2 CF 3 [0037]
- the present invention includes a compound of Formula (I), (lb), (Ilia) or (Illb), or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof, wherein:
- R 1 is independently selected from: 4-C0 2 H-Ph, 4-NHCO(C 1 . 4 alkyl)-Ph,
- R 4 is independently selected from: so 2 NH 2 and
- R 5 is independently selected from: C ⁇ alkyl substituted with 0-1 R 9 ,
- R 9 is independently selected from: C 1 .4 alkoxy, and C1 .4 haloalkyl
- R b is independently selected from: CN, halogen C ⁇ haloalkyl and
- the present invention includes a compound of Formula (I), (lb), (Ilia) or (Illb), or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof, wherein:
- R 1 is independently selected from: 4-C0 2 H-Ph, 4-NHCOMe-Ph, 4-CONH 2 -Ph, 4-CONH(CH 2 ) 2 _ 3 OH-Ph, 4-CONH(CH 2 ) 2 OMe-Ph, 4-CONH(CH 2 )C(Me) 2 OH-Ph, 4-CONH(cyclopropyl)-Ph, 3-F-4-CON(Me) 2 -Ph, 6-F-pyrid-3-yl, 6-OH-pyrid-2-yl, -OH-pyrid-3-yl, 2-OH-pyrid-4-yl, 2-OH-3-F-pyrid-4-yl, 2-OH-5-F-pyrid-4-yl,
- R 4 is independently selected from: ; and R 5 is independently selected from: Me, z ' -Pr, -(C ⁇ OMe, -(CH 2 )2CF 3 , cyclopropylmethyl, Bn, 3-CN-Bn, 3-F-Bn, 4-F-Bn, 4-CF 3 -Bn, and 4-OCF 3 -Bn.
- the present invention provides a compound selected from the exemplified examples or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof.
- the present invention provides a compound selected from any subset list of compounds within the scope of the twelfth aspect.
- the compounds of the present invention have EL IC50 values ⁇ 500 11M.
- the compounds of the present invention have EL IC50 values ⁇ 100 11M.
- the compounds of the present invention have EL IC50 values ⁇ 50 11M.
- the compounds of the present invention have EL IC50 values ⁇ 25 11M.
- the compounds of the present invention have EL IC50 values ⁇ 10 11M.
- the present invention provides a composition comprising at least one of the compounds of the present invention or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a pharmaceutically acceptable carrier and at least one of the compounds of the present invention or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof.
- the present invention provides a pharmaceutical composition, comprising: a pharmaceutically acceptable carrier and a therapeutically effective amount of at least one of the compounds of the present invention or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof.
- the present invention provides a process for making a compound of the present invention.
- the present invention provides an intermediate for making a compound of the present invention.
- the present invention provides a pharmaceutical composition as defined above further comprising additional therapeutic agent(s).
- the present invention provides a method for the treatment and/or prophylaxis of dyslipidemias and the sequelae thereof comprising administering to a patient in need of such treatment and/or prophylaxis a therapeutically effective amount of at least one of the compounds of the present invention, alone, or, optionally, in combination with another compound of the present invention and/or at least one other type of therapeutic agent.
- diseases or disorders associated with the activity of endothelial lipase that can be prevented, modulated, or treated according to the present invention include, but are not limited to, atherosclerosis, coronary heart disease, coronary artery disease, coronary vascular disease, cerebrovascular disorders, Alzheimer's disease, venous thrombosis, peripheral vascular disease, dyslipidemia, hyperbetalipoproteinemia, hypoalphalipoproteinemia, hypercholesterolemia, hypertriglyceridemia, familial- hypercholesterolemia, cardiovascular disorders, angina, ischemia, cardiac ischemia, stroke, myocardial infarction, reperfusion injury, angioplastic restenosis, hypertension, vascular complications of diabetes, obesity or endotoxemia.
- the present invention provides a method for the treatment and/or prophylaxis of atherosclerosis, coronary heart disease, cerebrovascular disorders and dyslipidemia, comprising administering to a patient in need of such treatment and/or prophylaxis a therapeutically effective amount of at least one of the compounds of the present invention, alone, or, optionally, in combination with another compound of the present invention and/or at least one other type of therapeutic agent.
- the present invention provides a compound of the present invention for use in therapy.
- the present invention provides a compound of the present invention for use in therapy for the treatment and/or prophylaxis of dyslipidemias and the sequelae thereof.
- the present invention also provides the use of a compound of the present invention for the manufacture of a medicament for the treatment and/or prophylaxis of dyslipidemias and the sequelae thereof.
- the present invention provides a method for the treatment and/or prophylaxis of dyslipidemias and the sequelae thereof, comprising: administering to a patient in need thereof a therapeutically effective amount of a first and second therapeutic agent, wherein the first therapeutic agent is a compound of the present invention.
- the present invention provides a combined
- the present invention provides a combined
- the compounds of the present invention may be employed in combination with additional therapeutic agent(s) selected from one or more, preferably one to three, of the following therapeutic agents: anti-atherosclerotic agents, anti-dyslipidemic agents, anti-diabetic agents, anti-hyperglycemic agents, anti-hyperinsulinemic agents, anti-thrombotic agents, anti-retinopathic agents, anti-neuropathic agents,
- additional therapeutic agent(s) selected from one or more, preferably one to three, of the following therapeutic agents: anti-atherosclerotic agents, anti-dyslipidemic agents, anti-diabetic agents, anti-hyperglycemic agents, anti-hyperinsulinemic agents, anti-thrombotic agents, anti-retinopathic agents, anti-neuropathic agents,
- anti-nephropathic agents anti-ischemic agents, anti-hypertensive agents, anti-obesity agents, anti-hyperlipidemic agents, anti-hypertriglyceridemic agents, anti-hypercholesterolemic agents, anti-restenotic agents, anti-pancreatic agents, lipid lowering agents, anorectic agents, memory enhancing agents, anti-dementia agents, cognition promoting agents, appetite suppressants, treatments for heart failure, treatments for peripheral arterial disease, treatment for malignant tumors, and anti-inflammatory agents.
- additional therapeutic agent(s) used in combined pharmaceutical compositions or combined methods or combined uses are selected from one or more, preferably one to three, of the following therapeutic agents in treating atherosclerosis: anti-hyperlipidemic agents, plasma HDL-raising agents,
- anti-hypercholesterolemic agents cholesterol biosynthesis inhibitors (such as HMG CoA reductase inhibitors), acyl-coenzyme Axholesterol acyltransferase (ACAT) inhibitors, LXR agonist, probucol, raloxifene, nicotinic acid, niacinamide, cholesterol absorption inhibitors, bile acid sequestrants (such as anion exchange resins, or quaternary amines (e.g., cholestyramine or colestipol)), low density lipoprotein receptor inducers, clofibrate, fenofibrate, benzofibrate, cipofibrate, gemfibrizol, vitamin B 6 , vitamin B 12 , anti-oxidant vitamins, ⁇ -blockers, anti-diabetes agents, angiotensin II antagonists, angiotensin converting enzyme inhibitors, platelet aggregation inhibitors, fibrinogen receptor antagonists, aspirin
- additional therapeutic agent(s) used in combined pharmaceutical compositions or combined methods or combined uses are selected from one or more, preferably one to three, of the following therapeutic agents in treating cholesterol biosynthesis inhibitor, particularly an HMG-CoA reductase inhibitor.
- HMG-CoA reductase inhibitors include, but are not limited to, lovastatin, simvastatin, pravastatin, fluvastatin, atorvastatin, and rivastatin.
- Optically active forms may be prepared by resolution of racemic forms or by synthesis from optically active starting materials. All processes used to prepare compounds of the present invention and intermediates made therein are considered to be part of the present invention. When enantiomeric or diastereomeric products are prepared, they may be separated by conventional methods, for example, by
- the end products of the present invention are obtained either in free (neutral) or salt form. Both the free form and the salts of these end products are within the scope of the invention. If so desired, one form of a compound may be converted into another form. A free base or acid may be converted into a salt; a salt may be converted into the free compound or another salt; a mixture of isomeric compounds of the present invention may be separated into the individual isomers.
- Compounds of the present invention, free form and salts thereof may exist in multiple tautomeric forms, in which hydrogen atoms are transposed to other parts of the molecules and the chemical bonds between the atoms of the molecules are consequently rearranged. It should be understood that all tautomeric forms, insofar as they may exist, are included within the invention.
- alkyl or “alkylene” is intended to include both branched and straight-chain saturated aliphatic hydrocarbon groups having the specified number of carbon atoms.
- C ⁇ to C ⁇ Q alkyl or “C ⁇ Q alkyl” (or alkylene) is intended to include C , C2, C3, C4, C5, Cg, C 7 , Cg, C9, and C Q alkyl groups.
- C ⁇ to Cg alkyl or "Ci _g alkyl” denotes alkyl having 1 to 6 carbon atoms.
- Alkyl group can be unsubstituted or substituted with at least one hydrogen being replaced by another chemical group.
- Example alkyl groups include, but are not limited to, methyl (Me), ethyl (Et), propyl (e.g., n-propyl and isopropyl), butyl (e.g., n-butyl, isobutyl, t-butyl), and pentyl (e.g., n-pentyl, isopentyl, neopentyl).
- Cg alkyl or "C 0 alkylene” is used, it is intended to denote a direct bond.
- alkenyl or “alkenylene” is intended to include hydrocarbon chains of either straight or branched configuration having the specified number of carbon atoms and one or more, preferably one to two, carbon-carbon double bonds that may occur in any stable point along the chain.
- C2 to Cg alkenyl or “C 2 _g alkenyl” (or alkenylene) is intended to include C 2 , C 3 , C 4 , C 5 , and C 6 alkenyl groups.
- alkenyl examples include, but are not limited to, ethenyl, 1-propenyl, 2-propenyl, 2-butenyl, 3-butenyl, 2-pentenyl, 3, pentenyl, 4-pentenyl, 2-hexenyl, 3-hexenyl, 4-hexenyl, 5-hexenyl, 2-methyl-2-propenyl, and 4-methyl-3-pentenyl.
- Alkynyl or “alkynylene” is intended to include hydrocarbon chains of either straight or branched configuration having one or more, preferably one to three, carbon-carbon triple bonds that may occur in any stable point along the chain.
- C 2 to C 6 alkynyl or “C 2- 6 alkynyl” (or alkynylene) is intended to include C 2 , C 3 , C 4 , C 5 , and C 6 alkynyl groups; such as ethynyl, propynyl, butynyl, pentynyl, and hexynyl.
- alkoxy refers to an -O-alkyl group.
- C to Cg alkoxy or “Ci _g alkoxy” (or alkyloxy) is intended to include C , C 2 , C3, C4, C5, and Cg alkoxy groups.
- Example alkoxy groups include, but are not limited to, methoxy, ethoxy, propoxy (e.g., n-propoxy and isopropoxy), and t-butoxy.
- alkylthio or
- thioalkoxy represents an alkyl group as defined above with the indicated number of carbon atoms attached through a sulphur bridge; for example methyl-S- and ethyl-S-.
- Halo or halogen includes fluoro, chloro, bromo, and iodo.
- Haloalkyl is intended to include both branched and straight-chain saturated aliphatic hydrocarbon groups having the specified number of carbon atoms, substituted with 1 or more halogens.
- haloalkyl include, but are not limited to, fluoromethyl, difluoromethyl, trifluoromethyl, trichloromethyl, pentafluoroethyl, pentachloroethyl, 2,2,2-trifluoroethyl, heptafluoropropyl, and heptachloropropyl.
- haloalkyl also include "fluoroalkyl” that is intended to include both branched and straight-chain saturated aliphatic hydrocarbon groups having the specified number of carbon atoms, substituted with 1 or more fluorine atoms.
- Haloalkoxy or "haloalkyloxy” represents a haloalkyl group as defined above with the indicated number of carbon atoms attached through an oxygen bridge.
- C ⁇ to C 6 haloalkoxy or "C ⁇ .g haloalkoxy” is intended to include C 1 ? C 2 , C 3 ,
- haloalkoxy examples include, but are not limited to, trifluoromethoxy, 2,2,2-trifluoroethoxy, and pentafluorothoxy.
- haloalkylthio or “thiohaloalkoxy” represents a haloalkyl group as defined above with the indicated number of carbon atoms attached through a sulphur bridge; for example trifluoromethyl-S-, and pentafluoroethyl-S-.
- cycloalkyl refers to cyclized alkyl groups, including mono-, bi- or poly-cyclic ring systems.
- C 3 to C 7 cycloalkyl or “C 3 _ 7 cycloalkyl” is intended to include C 3 , C 4 , C 5 , C 6 , and C 7 cycloalkyl groups.
- Example cycloalkyl groups include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, and norbornyl.
- Branched cycloalkyl groups such as 1-methylcyclopropyl and 2-methylcyclopropyl are included in the definition of "cycloalkyl”.
- carbocycle As used herein, “carbocycle,” “carbocyclyl,” or “carbocyclic residue” is intended to mean any stable 3-, 4-, 5-, 6-, 7-, or 8-membered monocyclic or bicyclic or 7-, 8-, 9-, 10-, 1 1-, 12-, or 13-membered bicyclic or tricyclic ring, any of which may be saturated, partially unsaturated, unsaturated or aromatic.
- carbocycles include, but are not limited to, cyclopropyl, cyclobutyl, cyclobutenyl, cyclopentyl, cyclopentenyl, cyclohexyl, cycloheptenyl, cycloheptyl, cycloheptenyl, adamantyl, cyclooctyl, cyclooctenyl, cyclooctadienyl, [3.3.0]bicyclooctane, [4.3.0]bicyclononane, [4.4.0]bicyclodecane (decalin), [2.2.2]bicyclooctane, fluorenyl, phenyl, naphthyl, indanyl, adamantyl, anthracenyl, and tetrahydronaphthyl (tetralin).
- bridged rings are also included in the definition of carbocycle (e.g., [2.2.2]bicyclooctane).
- carbocycles e.g., [2.2.2]bicyclooctane
- Preferred carbocycles are cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, phenyl, indanyl, and tetrahydronaphthyl.
- carbocycle When the term “carbocycle” is used, it is intended to include “aryl.”
- a bridged ring occurs when one or more, preferably one to three, carbon atoms link two non-adjacent carbon atoms.
- Preferred bridges are one or two carbon atoms. It is noted that a bridge always converts a monocyclic ring into a tricyclic ring. When a ring is bridged, the substituents recited for the ring may also be present on the bridge.
- bicyclic carbocycle or "bicyclic carbocyclic group” is intended to mean a stable 9- or 10-membered carbocyclic ring system that contains two fused rings and consists of carbon atoms. Of the two fused rings, one ring is a benzo ring fused to a second ring; and the second ring is a 5- or 6-membered carbon ring which is saturated, partially unsaturated, or unsaturated.
- the bicyclic carbocyclic group may be attached to its pendant group at any carbon atom which results in a stable structure.
- the bicyclic carbocyclic group described herein may be substituted on any carbon if the resulting compound is stable.
- bicyclic carbocyclic group examples include naphthyl, 1 ,2-dihydronaphthyl, 1 ,2,3,4-tetrahydronaphthyl, and indanyl.
- Aryl groups refer to monocyclic or polycyclic aromatic hydrocarbons, including, for example, phenyl, naphthyl, and phenanthranyl. Aryl moieties are well known and described, for example, in Lewis, R.J., ed., Hawley's Condensed Chemical Dictionary, 13th Edition, John Wiley & Sons, Inc., New York (1997). "Cg or C Q aryl” or “Cg.i o aryl” refers to phenyl and naphthyl.
- aryl may be unsubstituted or substituted with 1 to 5 groups, preferably 1 to 3 groups, selected from -OH, -OCH 3 , -CI, -F, -Br, -I, -CN, -N0 2 , -NH 2 , -N(CH 3 )H, -N(CH 3 ) 2 , -CF 3 , -OCF 3 , -C(0)CH 3 , -SCH 3 , -S(0)CH 3 , -S(0) 2 CH 3 , -CH 3 , -CH 2 CH 3 , -C0 2 H, and -C0 2 CH 3 .
- heterocycle As used herein, the term “heterocycle,” “heterocyclyl,” or “heterocyclic group” is intended to mean a stable 3-, 4-, 5-, 6-, or 7-membered monocyclic or bicyclic or 7-, 8-, 9-, 10-, 1 1-, 12-, 13-, or 14-membered polycyclic heterocyclic ring that is saturated, partially unsaturated, or fully unsaturated, and that contains carbon atoms and 1, 2, 3 or 4 heteroatoms independently selected from the group consisting of N, O and S; and including any polycyclic group in which any of the above-defined heterocyclic rings is fused to a benzene ring.
- the nitrogen and sulfur heteroatoms may optionally be oxidized (i.e., N ⁇ 0 and S(0) p , wherein p is 0, 1 or 2).
- the nitrogen atom may be substituted or unsubstituted (i.e., N or NR wherein R is H or another substituent, if defined).
- the heterocyclic ring may be attached to its pendant group at any heteroatom or carbon atom that results in a stable structure.
- the heterocyclic rings described herein may be substituted on carbon or on a nitrogen atom if the resulting compound is stable.
- a nitrogen in the heterocycle may optionally be quaternized. It is preferred that when the total number of S and O atoms in the heterocycle exceeds 1 , then these heteroatoms are not adjacent to one another. It is preferred that the total number of S and O atoms in the heterocycle is not more than 1.
- heterocycle it is intended to include heteroaryl.
- heterocycles include, but are not limited to, acridinyl, azetidinyl, azocinyl, benzimidazolyl, benzofuranyl, benzothiofuranyl, benzothiophenyl,
- benzisoxazolyl benzisothiazolyl, benzimidazolinyl, carbazolyl, 4aH-carbazolyl, carbolinyl, chromanyl, chromenyl, cinnolinyl, decahydroquinolinyl,
- Examples of 5- to 10-membered heterocycles include, but are not limited to, pyridinyl, furanyl, thienyl, pyrrolyl, pyrazolyl, pyrazinyl, piperazinyl, piperidinyl, imidazolyl, imidazolidinyl, indolyl, tetrazolyl, isoxazolyl, morpholinyl, oxazolyl, oxadiazolyl, oxazolidinyl, tetrahydrofuranyl, thiadiazinyl, thiadiazolyl, thiazolyl, triazinyl, triazolyl, benzimidazolyl, lH-indazolyl, benzofuranyl, benzothiofuranyl, benztetrazolyl, benzotriazolyl, benzisoxazolyl, benzoxazolyl, oxindolyl, benzoxazoliny
- Examples of 5- to 6-membered heterocycles include, but are not limited to, pyridinyl, furanyl, thienyl, pyrrolyl, pyrazolyl, pyrazinyl, piperazinyl, piperidinyl, imidazolyl, imidazolidinyl, indolyl, tetrazolyl, isoxazolyl, morpholinyl, oxazolyl, oxadiazolyl, oxazolidinyl, tetrahydrofuranyl, thiadiazinyl, thiadiazolyl, thiazolyl, triazinyl, and triazolyl. Also included are fused ring and spiro compounds containing, for example, the above heterocycles.
- bicyclic heterocycle or "bicyclic heterocyclic group” is intended to mean a stable 9- or 10-membered heterocyclic ring system which contains two fused rings and consists of carbon atoms and 1, 2, 3, or 4 heteroatoms independently selected from the group consisting of N, O and S. Of the two fused rings, one ring is a 5- or 6-membered monocyclic aromatic ring comprising a 5-membered heteroaryl ring, a 6-membered heteroaryl ring or a benzo ring, each fused to a second ring.
- the second ring is a 5- or 6-membered monocyclic ring which is saturated, partially unsaturated, or unsaturated, and comprises a 5-membered heterocycle, a 6-membered heterocycle or a carbocycle (provided the first ring is not benzo when the second ring is a carbocycle).
- the bicyclic heterocyclic group may be attached to its pendant group at any heteroatom or carbon atom which results in a stable structure.
- the bicyclic heterocyclic group described herein may be substituted on carbon or on a nitrogen atom if the resulting compound is stable. It is preferred that when the total number of S and O atoms in the heterocycle exceeds 1, then these heteroatoms are not adjacent to one another. It is preferred that the total number of S and O atoms in the heterocycle is not more than 1.
- Examples of a bicyclic heterocyclic group are, but not limited to, quinolinyl, isoquinolinyl, phthalazinyl, quinazolinyl, indolyl, isoindolyl, indolinyl, lH-indazolyl, benzimidazolyl, 1 ,2,3,4-tetrahydroquinolinyl, 1 ,2,3,4-tetrahydroisoquinolinyl,
- aromatic heterocyclic group or "heteroaryl” is intended to mean stable monocyclic and polycyclic aromatic hydrocarbons that include at least one heteroatom ring member such as sulfur, oxygen, or nitrogen.
- Heteroaryl groups include, without limitation, pyridyl, pyrimidinyl, pyrazinyl, pyridazinyl, triazinyl, furyl, quinolyl, isoquinolyl, thienyl, imidazolyl, thiazolyl, indolyl, pyrroyl, oxazolyl, benzofuryl, benzothienyl, benzthiazolyl, isoxazolyl, pyrazolyl, triazolyl, tetrazolyl, indazolyl, 1,2,4-thiadiazolyl, isothiazolyl, purinyl, carbazolyl, benzimidazolyl, indolinyl, benzo
- Heteroaryl groups are substituted or unsubstituted.
- the nitrogen atom is substituted or unsubstituted (i.e., N or NR wherein R is H or another substituent, if defined).
- the nitrogen and sulfur heteroatoms may optionally be oxidized (i.e., N ⁇ 0 and S(0) p , wherein p is 0, 1 or 2).
- Bridged rings are also included in the definition of heterocycle.
- a bridged ring occurs when one or more, preferably one to three, atoms (i.e., C, O, N, or S) link two non-adjacent carbon or nitrogen atoms.
- Examples of bridged rings include, but are not limited to, one carbon atom, two carbon atoms, one nitrogen atom, two nitrogen atoms, and a carbon-nitrogen group. It is noted that a bridge always converts a monocyclic ring into a tricyclic ring. When a ring is bridged, the substituents recited for the ring may also be present on the bridge.
- counter ion is used to represent a negatively charged species such as chloride, bromide, hydroxide, acetate, and sulfate.
- a dotted ring is used within a ring structure, this indicates that the ring structure may be saturated, partially saturated or unsaturated.
- substituted means that at least one hydrogen atom is replaced with a non-hydrogen group, provided that normal valencies are maintained and that the substitution results in a stable compound.
- 2 hydrogens on the atom are replaced.
- Keto substituents are not present on aromatic moieties.
- a ring system e.g., carbocyclic or heterocyclic
- nitrogen atoms e.g. , amines
- these may be converted to N-oxides by treatment with an oxidizing agent (e.g., mCPBA and/or hydrogen peroxides) to afford other compounds of this invention.
- an oxidizing agent e.g., mCPBA and/or hydrogen peroxides
- shown and claimed nitrogen atoms are considered to cover both the shown nitrogen and its N-oxide (N— >0) derivative.
- any variable occurs more than one time in any constituent or formula for a compound, its definition at each occurrence is independent of its definition at every other occurrence.
- a group is shown to be substituted with 0-3 R groups, then said group may optionally be substituted with up to three R groups, and at each occurrence R is selected independently from the definition of R.
- phrases "pharmaceutically acceptable” is employed herein to refer to those compounds, materials, compositions, and/or dosage forms that are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, and/or other problem or complication, commensurate with a reasonable benefit/risk ratio.
- pharmaceutically acceptable salts refer to derivatives of the disclosed compounds wherein the parent compound is modified by making acid or base salts thereof.
- examples of pharmaceutically acceptable salts include, but are not limited to, mineral or organic acid salts of basic groups such as amines; and alkali or organic salts of acidic groups such as carboxylic acids.
- the pharmaceutically acceptable salts include the conventional non-toxic salts or the quaternary ammonium salts of the parent compound formed, for example, from non-toxic inorganic or organic acids.
- such conventional non-toxic salts include those derived from inorganic acids such as hydrochloric, hydrobromic, sulfuric, sulfamic, phosphoric, and nitric; and the salts prepared from organic acids such as acetic, propionic, succinic, glycolic, stearic, lactic, malic, tartaric, citric, ascorbic, pamoic, maleic, hydroxymaleic, phenylacetic, glutamic, benzoic, salicylic, sulfanilic, 2-acetoxybenzoic, fumaric, toluenesulfonic,
- inorganic acids such as hydrochloric, hydrobromic, sulfuric, sulfamic, phosphoric, and nitric
- organic acids such as acetic, propionic, succinic, glycolic, stearic, lactic, malic, tartaric, citric, ascorbic, pamoic, maleic, hydroxymaleic, phenylacetic, glutamic,
- the pharmaceutically acceptable salts of the present invention can be synthesized from the parent compound that contains a basic or acidic moiety by conventional chemical methods. Generally, such salts can be prepared by reacting the free acid or base forms of these compounds with a stoichiometric amount of the appropriate base or acid in water or in an organic solvent, or in a mixture of the two; generally, nonaqueous media like ether, ethyl acetate, ethanol, isopropanol, or acetonitrile are preferred. Lists of suitable salts are found in Remington: The Science and Practice of Pharmacy, 22 nd Edition, Allen, L. V. Jr., Ed.; Pharmaceutical Press, London, UK (2012), the disclosure of which is hereby incorporated by reference.
- compounds of Formula (I), Formula (II), or Formula (III) may have prodrug forms. Any compound that will be converted in vivo to provide the bioactive agent (i.e., a compound of Formula (I), Formula (II) or Formula (III)) is a prodrug within the scope and spirit of the invention.
- a prodrug within the scope and spirit of the invention.
- Various forms of prodrugs are well known in the art. For examples of such prodrug derivatives, see:
- hydrolyzable esters that serve as prodrugs by being hydrolyzed in the body to yield Formula (I), (Ila), (lib), (Ilia) or (Illb) compounds per se.
- prodrugs are preferably administered orally since hydrolysis in many instances occurs principally under the influence of the digestive enzymes. Parenteral administration may be used where the ester per se is active, or in those instances where hydrolysis occurs in the blood.
- physiologically hydrolyzable esters of compounds of the present invention include C ⁇ to Cg alkyl, C ⁇ to Cg alkylbenzyl, 4-methoxybenzyl, indanyl, phthalyl, methoxymethyl, Ci .g alkanoyloxy-C ⁇ .g alkyl (e.g., acetoxymethyl, pivaloyloxymethyl or propionyloxymethyl), C ⁇ to Cg alkoxycarbonyloxy-Cj to Cg alkyl (e.g., methoxycarbonyl-oxymethyl or ethoxycarbonyloxymethyl, glycyloxymethyl,
- esters used, for example, in the penicillin and cephalosporin arts.
- esters may be prepared by conventional techniques known in the art.
- prodrugs are well known in the art and described in, for example, King, F.D., ed., Medicinal Chemistry: Principles and Practice, The Royal Society of Chemistry, Cambridge, UK (2 nd edition, reproduced, 2006); Testa, B. et al, Hydrolysis in Drug and Prodrug Metabolism. Chemistry, Biochemistry and Enzymology, VCHA and Wiley- VCH, Zurich, Switzerland (2003); Wermuth, C.G., ed., The Practice of Medicinal Chemistry, 3 rd edition, Academic Press, San Diego, CA (2008).
- the present invention is intended to include all isotopes of atoms occurring in the present compounds.
- Isotopes include those atoms having the same atomic number but different mass numbers.
- isotopes of hydrogen include deuterium and tritium.
- isotopes of carbon include 13 C and 14 C.
- “Stable compound” and “stable structure” are meant to indicate a compound that is sufficiently robust to survive isolation to a useful degree of purity from a reaction mixture, and formulation into an efficacious therapeutic agent. It is preferred that compounds of the present invention do not contain a N-halo, S(0) 2 H, or S(0)H group.
- solvate means a physical association of a compound of this invention with one or more solvent molecules, whether organic or inorganic. This physical association includes hydrogen bonding. In certain instances the solvate will be capable of isolation, for example when one or more, preferably one to three, solvent molecules are incorporated in the crystal lattice of the crystalline solid.
- the solvent molecules in the solvate may be present in a regular arrangement and/or a non-ordered arrangement.
- the solvate may comprise either a stoichiometric or nonstoichiometric amount of the solvent molecules.
- “Solvate” encompasses both solution-phase and isolable solvates. Exemplary solvates include, but are not limited to, hydrates, ethanolates, methanolates, and isopropanolates. Methods of solvation are generally known in the art.
- HATU (2-(7-aza- 1 H-benzotriazole- 1 -yl)- 1 , 1 ,3 ,3 -tetramethyluronium hexafluorophosphate)
- the compounds of the present invention can be prepared in a number of ways known to one skilled in the art of organic synthesis.
- the compounds of the present invention can be synthesized using the methods described below, together with synthetic methods known in the art of synthetic organic chemistry, or by variations thereon as appreciated by those skilled in the art. Preferred methods include, but are not limited to, those described below.
- the reactions are performed in a solvent or solvent mixture appropriate to the reagents and materials employed and suitable for the transformations being affected. It will be understood by those skilled in the art of organic synthesis that the functionality present on the molecule should be consistent with the transformations proposed. This will sometimes require a judgment to modify the order of the synthetic steps or to select one particular process scheme over another in order to obtain a desired compound of the invention.
- novel compounds of this invention may be prepared using the reactions and techniques described in this section. Also, in the description of the synthetic methods described below, it is to be understood that all proposed reaction conditions, including choice of solvent, reaction atmosphere, reaction temperature, duration of the experiment and workup procedures, are chosen to be the conditions standard for that reaction, which should be readily recognized by one skilled in the art. Restrictions to the substituents that are compatible with the reaction conditions will be readily apparent to one skilled in the art and alternate methods must then be used.
- Step 1 describes the preparation of compounds of Formula (Gib) by reacting amides of Formula (Gla) with a sodium nitrite.
- a preferred solvent is water, and a preferred acid is acetic acid.
- Step 2 describes the preparation of amides and ureas of Formula (I) by reacting compounds of the Formula (Gib) with an anhydride or isocyanate and Pd/C in a hydrogen atmosphere.
- Preferred solvents are polar protic solvents, such as MeOH and EtOH.
- Step 1 describes the preparation of compounds of Formula (I) by reacting a compound of Formula (G2a) with a sulfonylating reagent R 5 -S0 2 C1.
- Preferred solvents are polar aprotic solvents (such as ⁇ , ⁇ -dimethylformamide ) and ethers (such as tetrahydrofuran, dioxane and the like).
- Preferred bases include metal hydrides (such as sodium hydride and the like), metal amides (such as sodium bis(trimethylsilyl)amide and lithium diisopropylamide and the like) and organic amines (such as DBU, triethylamine and the like).
- Step 1 describes the preparation of a compound of Formula (G3b) from a compound of Formula (G3a) and is analogous to Step 1 in Scheme 1.
- Step 2 describes the preparation of a compound of Formula (G3b) from a compound of Formula (G3a) and is analogous to Step 1 in Scheme 1.
- Step 2 describes the preparation of a compound of Formula (I) from a compound of Formula (G3b) by direct exchange of an ester group with an amine of formula H 2 NR 4 .
- Preferred reaction solvents are polar aprotic solvents such as ethers (such as tetrahydrofuran, dioxane and the like), DMF and NMP.
- Bases such as an organic amine (such as triethylamine, diisopropylamine, DBU, 2,6-lutidine and the like) can be used.
- PHENOMENEX® Luna CI 8 column (4.6 x 50 mm) eluted at 4 mL/min with a 4 min gradient from 100% A to 100% B (A: 10% acetonitrile, 89.9% water, 0.1% TFA; B: 10% water, 89.9% acetonitrile, 0.1% TFA, UV 220 nm) or Method C: PHENOMENEX® Luna C 18 column (4.6 x 50 mm or 4.6 x 75 mm) eluted at 4 mL/min with a 2, 4 or 8 min gradient from 100% A to 100% B (A: 10% methanol, 89.9% water, 0.1% H3PO4; B:
- PHENOMENEX® Luna CI 8 column (4.6 x 50 mm or 4.6 x 75 mm) eluted at 4 mL/min with a 2, 4 or 8 min gradient from 100% A to 100% B (A: 10% methanol, 89.9% water, 0.1% NH 4 OAc; B: 10% water, 89.9% methanol, 0.1% NH 4 OAc, UV 220 nm).
- Method A YMC Sunfire 5 ⁇ C18 30 x 100 mm column with a 10 min gradient at 40 mL/min from 100% A to 100% B (A: 10% methanol, 89.9% water, 0.1% TFA; B: 10% water, 89.9% methanol, 0.1% TFA, UV 220 nm), Method B: PHENOMENEX® Axia Luna 5 ⁇ C18 30 x 100 mm column with a 10 min gradient at 40 mL/min from 100% A to 100% B (A: 10% acetonitrile, 89.9% water, 0.1% TFA; B: 10% water, 89.9% acetonitrile, 0.1% TFA, UV 220 nm), Method C : PHENOMENEX® Luna 5 ⁇ C 18 30 x 100 mm column with a 10 min gradient at 40 mL/min from 100% A to 100% B (A: 10% acetonitrile, 89.9% water, 0.1% TFA; B: 10% water, 89.9% acetonitrile
- Method D PHENOMENEX® Luna 5 ⁇ C18 30 x 100 mm column with a 10 min gradient at 40 mL/min from 100% A to 100% B (A: 10% methanol, 89.9% water, 0.1% TFA; B: 10% water, 89.9% methanol, 0.1% TFA, UV 220 nm).
- LCMS chromatograms were obtained on a Shimadzu HPLC system running Discovery VP software, coupled with a Waters ZQ mass spectrometer running MassLynx version 3.5 software and using the following respective methods. Unless specified otherwise, for each method, the LC column was maintained at room temperature and UV detection was set to 220 nm.
- Method A A linear gradient using solvent A (10% methanol, 90% water, 0.1% of TFA) and solvent B (90% methanol, 10% water, 0.1% of TFA); 0-100% of solvent B over 4 min and then 100% of solvent B over 1 min.
- Method B A linear gradient using solvent A (10% methanol, 90% water, 0.1% of TFA) and solvent B (90% methanol, 10% water, 0.1% of TFA); 0-100% of solvent B over 2 min and then 100% of solvent B over 1 min.
- PHENOMENEX® Luna 5 nm C18 (2.0 x 30 mm). Flow rate was 1 mL/min.
- Method C A linear gradient using solvent A (10% acetonitrile, 90% water, 10 mM NH 4 OAc) and solvent B (90% acetonitrile, 10% water, 10 mM NH 4 OAc); 0- 100% of solvent B over 4 min and then 100% of solvent B over 1 min.
- Method D A linear gradient using solvent A (10% acetonitrile, 90% water, 0.05% of TFA) and solvent B (90% acetonitrile, 10% water, 0.05% of TFA); 0-100% of solvent B over 2 min and then 100% of solvent B over 1 min.
- Method E A linear gradient using solvent A (10% MeOH, 90% water, 10 mM NH 4 OAc) and solvent B (90% MeOH, 10% water, 10 mM NH 4 OAc); 0-100% of solvent B over 4 min and then 100% of solvent B over 1 min.
- Method F A linear gradient using solvent A (10 mM NH4OAC, 95% water,
- Method G A linear gradient using solvent A (10%> acetonitrile, 90%> water, 0.1% TFA) and solvent B (90% acetonitrile, 10% water, 0.1% TFA); 0-100% of solvent
- Method H A linear gradient using solvent A (10% methanol, 90% water, 0.1%) of formic acid) and solvent B (90%> methanol, 10%> water, 0.1% of formic acid); 0- 100% of solvent B over 2 min and then 100% of solvent B over 1 min.
- Method J A linear gradient using solvent A (10% methanol, 90% water, 0.1% of formic acid) and solvent B (90% methanol, 10% water, 0.1% of TFA); 0-100% of solvent B over 4 min and then 100% of solvent B over 1 min.
- Method L A linear gradient using solvent A (5 % methanol, 95 % water, 0.05% of TFA) and solvent B (95% methanol, 5% water, 0.05% of TFA); 0-100% of solvent B over 4 min and then 100% of solvent B over 1 min.
- Method M A linear gradient using of Solvent A (0.05% TFA, 100% water) and Solvent B (0.05% TFA, 100% ACN); 2 to 98% B over 1 min, with 0.5 min hold time at 98% B.
- Method N A linear gradient using solvent A (5% ACN, 95% water, 10 mM NH 4 OAc) and solvent B (95% ACN, 5% water, 10 mM NH 4 OAc); 0-100% of solvent B over 3 min and then 100% of solvent B over 1 min.
- Method O A linear gradient using solvent A (5% ACN, 95% water, 0.05% of TFA) and solvent B (95% ACN, 5% water, 0.05% of TFA); 0-100% of solvent B over 3 min and then 100% of solvent B over 1 min.
- Method P A linear gradient using solvent A (5% ACN, 95% water, 10 mM NH 4 OAc) and solvent B (95% ACN, 5% water, 10 mM NH 4 OAc); 0-100% of solvent B over 4 min and then 100% of solvent B over 1 min.
- Method Q A linear gradient using solvent A (10% MeOH, 90% water, 0.1% TFA) and solvent B (90% MeOH, 10% water, 0.1% TFA); 0-100% of solvent B over 2 min and then 100% of solvent B over 1 min.
- Method R Column: Waters BEH CI 8, 2.0 x 50 mm, 1.7- ⁇ particles; Mobile Phase A: 5:95 acetonitrile: water with 10 mM ammonium acetate; Mobile Phase B: 95 :5 acetonitrile: water with 10 mM ammonium acetate; Temperature: 40 °C; Gradient: 0.5 min hold at 0%B, 0-100% B over 4 minutes, then a 0.5-minute hold at 100% B; Flow: 1 mL/min.
- Method A Linear gradient of 0 to 100% B over 10 min, with 5 min hold time at 100%) B; Shimadzu LC-8A binary pumps
- Solvent A 0.1% TFA, 10% ACN, 90% water
- Solvent B 0.1% TFA, 90% ACN, 10% water NMR Employed in Characterization of Examples
- 3 ⁇ 4 NMR spectra were obtained with Bruker or JEOL Fourier transform spectrometers operating at frequencies as follows: 3 ⁇ 4 NMR: 400 MHz (Bruker or JEOL) or 500MHz (JEOL). 13 C NMR: 100 MHz (Bruker or JEOL). Spectra data are reported in the format: chemical shift (multiplicity, coupling constants, number of hydrogens).
- the endothelium occupies a pivotal position at the interface between the circulating humoral and cellular elements of the blood, and the solid tissues which constitute the various organs.
- endothelial cells regulate a large number of critical processes, including leukocyte adherence and transit through the blood vessel wall, local control of blood vessel tone, modulation of the immune response, the balance between thrombosis and thrombolysis, and new blood vessel development.
- endothelial cell dysfunction has been postulated as a central feature of vascular diseases such as hypertension and atherosclerosis.
- Atherosclerosis and its associated coronary artery disease is the leading cause of mortality in the industrialized world.
- CAD coronary artery disease
- CHD coronary heart disease
- cardiovascular disease accounted for 33% of all deaths in the U.S., and ⁇ 1 of every 6 deaths were specifically caused by atherosclerotic coronary heart disease ⁇ Circulation 125:e2-e220 (2012)).
- LDL-C low density lipoprotein- cholesterol
- HDL-C high density lipoprotein-cholesterol
- dyslipidemia is not a unitary risk profile for CHD but may be comprised of one or more, preferably one to three, lipid aberrations.
- HDL cholesterol levels At least 50% of the variation in HDL cholesterol levels is genetically determined.
- the phenotype of elevated HDL cholesterol is often dominantly inherited, but homozygous deficiency of HL or of the cholesteryl ester transfer protein (CETP), which result in elevated HDL cholesterol, are recessive conditions.
- CETP cholesteryl ester transfer protein
- endothelial lipase-mediated binding and uptake of HDL particles and the selective uptake of HDL-derived cholesterol esters have been reported to be independent of its enzymatic lipolytic activity (Strauss, J.G. et al, Biochem. J, 368:69-79 (2002)).
- an agent which inhibits EL activity in humans are expected to be useful for the treatment, prevention, the arrestment and/or regression of atherosclerosis, coronary heart disease, cerebrovascular disorders etc., especially those (but not restricted thereto) which are characterized by one or more of the following factors: (a) high plasma triglyceride concentrations, high postprandial plasma triglyceride concentrations; (b) low HDL cholesterol concentration; (c) low apoAl lipoprotein concentrations; (d) high LDL cholesterol concentrations; (e) high levels of small dense LDL cholesterol particles; and (f) high apoB lipoprotein concentrations.
- modulator refers to a chemical compound with capacity to either enhance (e.g., "agonist” activity) or partially enhance (e.g., “partial agonist” activity) or inhibit (e.g., "antagonist” activity or "inverse agonist” activity) a functional property of biological activity or process (e.g., enzyme activity or receptor binding); such
- enhancement or inhibition may be contingent on the occurrence of a specific event, such as activation of a signal transduction pathway, receptor internalization, and/or may be manifest only in particular cell types.
- the term “patient” encompasses all mammalian species.
- the term “subject” refers to any human or non-human organism that could potentially benefit from treatment with an anti-atherosclerosis agent, e.g., an endothelial lipase inhibitor.
- exemplary subjects include human beings of any age with risk factors for atherosclerosis and its associated coronary artery disease. Common risk factors include, but are not limited to, age, sex, weight, and family history.
- treating cover the treatment of a disease-state in a mammal, particularly in a human, and include: (a) inhibiting the disease-state, i.e., arresting it development; and/or (b) relieving the disease-state, i.e., causing regression of the disease state.
- prophylaxis covers the preventive treatment of a subclinical disease-state in a mammal, particularly in a human, aimed at reducing the probability of the occurrence of a clinical disease-state. Patients are selected for preventative therapy based on factors that are known to increase risk of suffering a clinical disease state compared to the general population. "Prophylaxis” therapies can be divided into (a) primary prevention and (b) secondary prevention. Primary prevention is defined as treatment in a subject that has not yet presented with a clinical disease state, whereas secondary prevention is defined as preventing a second occurrence of the same or similar clinical disease state.
- risk reduction covers therapies that lower the incidence of development of a clinical disease state.
- primary and secondary prevention therapies are examples of risk reduction.
- “Therapeutically effective amount” is intended to include an amount of a compound of the present invention that is effective when administered alone or in combination to inhibit endothelial lipase and/or to prevent or treat the disorders listed herein. When applied to a combination, the term refers to combined amounts of the active ingredients that result in the preventive or therapeutic effect, whether administered in combination, serially, or simultaneously.
- Endothelial lipase (EL) and hepatic lipase (HL) activities were measured using a fluorescent substrate, A10070, (Invitrogen, CA) doped into an artificial vesicle containing DMPG (Avanti Polar Lipids) as the excipient.
- Vesicles were prepared by combining 571 ⁇ . of 29 mM DMPG in a 1 :1 mixture of MeOH and CHC1 3 with 2000 ⁇ . of 1 mM A 10070 in a 1 : 1 mixture of MeOH and CHCI3.
- the mixture was dried under nitrogen in multiple vials then resuspended in 20 mL total volume of 50 mM HEPES pH 8.0 buffer containing 50 mM NaCl and 0.2 mM EDTA.
- the sample was allowed to sit at room temperature for 15 min and then was sonicated 3 x 4 mins on ice with a Branson Sonicator using duty cycle 1. This preparation provides vesicles with a mole fraction of 0.11 for the FRET substrate.
- the enzymatic assay was measured using 384-well white Optiplates. Each well contained 20 ⁇ of assay buffer (50 mM HEPES pH 8.0, 50 mM NaCl and 1 mM CaCl2) and 0.25 ⁇ ⁇ of a DMSO solution containing a compound of interest.
- assay buffer 50 mM HEPES pH 8.0, 50 mM NaCl and 1 mM CaCl2
- 0.25 ⁇ ⁇ of a DMSO solution containing a compound of interest EL or HL
- the source of EL was conditioned media obtained from HT-1080 cells that were transformed using RAGE technology (Athersys) to overexpress endogenous EL, and HL was partially purified from conditioned media obtained from COS cells overexpressing HL.
- the reaction was started by the addition of 10 ⁇ of a 1 : 10 dilution of vesicles. The final total reaction volume was 20.25 ⁇ ⁇ .
- the reaction rates were measured on a Gemini plate reader with an excitation wavelength of 490 nm and an emission wavelength of 530 nm. Readings were taken over a period of 60 minutes, and the slope between 300 and 900 sees of the readout was used to calculate the rate of the reaction.
- Example Al to Example A9 and Example Bl to Example 205 and Example 207 to Example B397, disclosed in the present invention were tested in the EL assay described above.
- Examples A1-A9 B1-B205, and B207- B397 were found having a range of EL IC 50 values of ⁇ 0.3 ⁇ (300 nM), as shown below.
- the compounds of the present invention can be administered to mammals, preferably humans, for the treatment of a variety of conditions and disorders, including, but not limited to, atherosclerosis, coronary heart disease, coronary artery disease, coronary vascular disease, cerebrovascular disorders, Alzheimer's disease, venous thrombosis, peripheral vascular disease, dyslipidemia, hyperbetalipoproteinemia, hypoalphalipoproteinemia, hypercholesterolemia, hypertriglyceridemia, familial- hypercholesterolemia, cardiovascular disorders, angina, ischemia, cardiac ischemia, stroke, myocardial infarction, reperfusion injury, angioplastic restenosis, hypertension, vascular complications of diabetes, obesity or endotoxemia.
- the compounds of this invention can be administered for any of the uses described herein by any suitable means, for example, orally, such as tablets, capsules,
- each of which includes sustained release or timed release formulations pills, powders, granules, elixirs, tinctures, suspensions, (including nanosuspensions, microsuspensions, spray-dried dispersions), syrups, and emulsions; sublingually; bucally; parenterally, such as by subcutaneous, intravenous, intramuscular, or intrasternal injection, or infusion techniques (e.g., as sterile injectable aqueous or non-aqueous solutions or suspensions); nasally, including administration to the nasal membranes, such as by inhalation spray; topically, such as in the form of a cream or ointment; or rectally such as in the form of suppositories. They can be administered alone, but generally will be administered with a pharmaceutical carrier selected on the basis of the chosen route of administration and standard pharmaceutical practice.
- composition means a composition comprising a compound of the invention in combination with at least one additional pharmaceutically acceptable carrier.
- a “pharmaceutically acceptable carrier” refers to media generally accepted in the art for the delivery of biologically active agents to animals, in particular, mammals, including, i.e., adjuvant, excipient or vehicle, such as diluents, preserving agents, fillers, flow regulating agents, disintegrating agents, wetting agents, emulsifying agents, suspending agents, sweetening agents, flavoring agents, perfuming agents, antibacterial agents, anti-fungal agents, lubricating agents and dispensing agents, depending on the nature of the mode of administration and dosage forms.
- Pharmaceutically acceptable carriers are formulated according to a number of factors well within the purview of those of ordinary skill in the art. These include, without limitation: the type and nature of the active agent being formulated; the subject to which the agent-containing composition is to be administered; the intended route of administration of the
- compositions include both aqueous and non-aqueous liquid media, as well as a variety of solid and semi-solid dosage forms.
- Such carriers can include a number of different ingredients and additives in addition to the active agent, such additional ingredients being included in the formulation for a variety of reasons, e.g., stabilization of the active agent, binders, etc., well known to those of ordinary skill in the art. Descriptions of suitable
- the dosage regimen for the compounds of the present invention will, of course, vary depending upon known factors, such as the pharmacodynamic characteristics of the particular agent and its mode and route of administration; the species, age, sex, health, medical condition, and weight of the recipient; the nature and extent of the symptoms; the kind of concurrent treatment; the frequency of treatment; the route of administration, the renal and hepatic function of the patient, and the effect desired.
- the daily oral dosage of each active ingredient when used for the indicated effects, will range between about 0.01 to about 5000 mg per day, preferably between about 0.1 to about 1000 mg per day, and most preferably between about 0.1 to about 250 mg per day.
- the most preferred doses will range from about 0.01 to about 10 mg/kg/minute during a constant rate infusion.
- Compounds of this invention may be administered in a single daily dose, or the total daily dosage may be administered in divided doses of two, three, or four times daily.
- the compounds are typically administered in admixture with suitable pharmaceutical diluents, excipients, or carriers (collectively referred to herein as pharmaceutical carriers) suitably selected with respect to the intended form of
- oral administration e.g., oral tablets, capsules, elixirs, and syrups, and consistent with conventional pharmaceutical practices.
- Dosage forms suitable for administration may contain from about 1 milligram to about 2000 milligrams of active ingredient per dosage unit.
- the active ingredient will ordinarily be present in an amount of about 0.1-95% by weight based on the total weight of the composition.
- a typical capsule for oral administration contains at least one of the compounds of the present invention (250 mg), lactose (75 mg), and magnesium stearate (15 mg). The mixture is passed through a 60 mesh sieve and packed into a No. 1 gelatin capsule.
- a typical injectable preparation is produced by aseptically placing at least one of the compounds of the present invention (250 mg) into a vial, aseptically freeze-drying and sealing. For use, the contents of the vial are mixed with 2 mL of physiological saline, to produce an injectable preparation.
- the present invention includes within its scope pharmaceutical compositions comprising, as an active ingredient, a therapeutically effective amount of at least one of the compounds of the present invention, alone or in combination with a pharmaceutical carrier.
- compounds of the present invention can be used alone, in
- the compounds of the present invention may be employed in combination with other EL inhibitors or one or more, preferably one to three, other suitable therapeutic agents useful in the treatment of the aforementioned disorders including:
- anti-atherosclerotic agents anti-dyslipidemic agents, anti-diabetic agents,
- anti-hyperglycemic agents anti-hyperinsulinemic agents, anti-thrombotic agents, anti-retinopathic agents, anti-neuropathic agents, anti-nephropathic agents, anti-ischemic agents, anti-hypertensive agents, anti-obesity agents, anti-hyperlipidemic agents, anti-hypertriglyceridemic agents, anti-hypercholesterolemic agents, anti-restenotic agents, anti-pancreatic agents, lipid lowering agents, anorectic agents, memory enhancing agents, anti-dementia agents, cognition promoting agents, appetite suppressants, treatments for heart failure, treatments for peripheral arterial disease, treatment for malignant tumors, and anti-inflammatory agents.
- the compounds of the present invention may be employed in combination with additional therapeutic agent(s) selected from one or more, preferably one to three, of the following therapeutic agents in treating atherosclerosis: anti-hyperlipidemic agents, plasma HDL-raising agents, anti-hypercholesterolemic agents, cholesterol biosynthesis inhibitors (such as HMG CoA reductase inhibitors), acyl-coenzyme Axholesterol acyltransferase (ACAT) inhibitors, LXR agonist, probucol, raloxifene, nicotinic acid, niacinamide, cholesterol absorption inhibitors, bile acid sequestrants (such as anion exchange resins, or quaternary amines ⁇ e.g., cholestyramine or colestipol)), low density lipoprotein receptor inducers, clofibrate, fenofibrate, benzofibrate, cipofibrate, gemfibrizol, vitamin B 6 , vitamin B
- the compounds of the present invention may be employed in combination with additional therapeutic agent(s) selected from one or more, preferably one to three, of the following therapeutic agents in treating cholesterol biosynthesis inhibitor, particularly an HMG-CoA reductase inhibitor.
- additional therapeutic agent(s) selected from one or more, preferably one to three, of the following therapeutic agents in treating cholesterol biosynthesis inhibitor, particularly an HMG-CoA reductase inhibitor.
- suitable HMG-CoA reductase inhibitors include, but are not limited to, lovastatin, simvastatin, pravastatin, fluvastatin, atorvastatin, and rivastatin.
- HMG-CoA reductase inhibitor is intended to include all
- the compounds of the invention may be used in combination with one or more, preferably one to three, of the following anti-diabetic agents depending on the desired target therapy. Studies indicate that diabetes and hyperlipidemia modulation can be further improved by the addition of a second agent to the therapeutic regimen.
- anti-diabetic agents include, but are not limited to, sulfonylureas (such as chlorpropamide, tolbutamide, acetohexamide, tolazamide, glyburide, gliclazide, glynase, glimepiride, and glipizide), biguanides (such as metformin), thiazolidinediones (such as ciglitazone, pioglitazone, troglitazone, and rosiglitazone), and related insulin sensitizers, such as selective and non-selective activators of PPARa, ⁇ and PPARy;
- sulfonylureas such as chlorpropamide, tolbutamide, acetohexamide, tolazamide, glyburide, gliclazide, glynase, glimepiride, and glipizide
- biguanides such as metformin
- dehydroepiandrosterone also referred to as DHEA or its conjugated sulphate ester, DHEA-SO4
- DHEA dehydroepiandrosterone
- anti-glucocorticoids include TNFa inhibitors; a-glucosidase inhibitors (such as acarbose, miglitol, and voglibose), pramlintide (a synthetic analog of the human hormone amylin), other insulin secretagogues (such as repaglinide, gliquidone, and nateglinide), insulin, as well as the therapeutic agents discussed above for treating atherosclerosis.
- TNFa inhibitors such as acarbose, miglitol, and voglibose
- pramlintide a synthetic analog of the human hormone amylin
- other insulin secretagogues such as repaglinide, gliquidone, and nateglinide
- the compounds of the invention may be used in combination with one or more, preferably one to three, of the following anti-obesity agents selected from phenylpropanolamine, phentermine, diethylpropion, mazindol, fenfluramine,
- dexfenfluramine phentiramine, P3-adrenoreceptor agonist agents
- sibutramine gastrointestinal lipase inhibitors (such as orlistat), and leptins.
- Other agents used in treating obesity or obesity-related disorders include neuropeptide Y, enterostatin, cholecytokinin, bombesin, amylin, histamine H3 receptors, dopamine D2 receptor modulators, melanocyte stimulating hormone, corticotrophin releasing factor, galanin and gamma amino butyric acid (GAB A).
- the compound of the present invention and a second therapeutic agent are combined in a single dosage unit they are formulated such that although the active ingredients are combined in a single dosage unit, the physical contact between the active ingredients is minimized (that is, reduced).
- one active ingredient may be enteric coated. By enteric coating one of the active ingredients, it is possible not only to minimize the contact between the combined active ingredients, but also, it is possible to control the release of one of these components in the gastrointestinal tract such that one of these components is not released in the stomach but rather is released in the intestines.
- One of the active ingredients may also be coated with a material that affects a sustained-release throughout the gastrointestinal tract and also serves to minimize physical contact between the combined active ingredients.
- the sustained-released component can be additionally enteric coated such that the release of this component occurs only in the intestine.
- Still another approach would involve the formulation of a combination product in which the one component is coated with a sustained and/or enteric release polymer, and the other component is also coated with a polymer such as a low viscosity grade of hydroxypropyl methylcellulose (HPMC) or other appropriate materials as known in the art, in order to further separate the active components.
- HPMC hydroxypropyl methylcellulose
- the polymer coating serves to form an additional barrier to interaction with the other component.
- the compounds of the present invention can be administered alone or in combination with one or more, preferably one to three, additional therapeutic agents.
- administered in combination or “combination therapy” it is meant that the compound of the present invention and one or more, preferably one to three, additional therapeutic agents are administered concurrently to the mammal being treated.
- each component may be administered at the same time or sequentially in any order at different points in time. Thus, each component may be administered separately but sufficiently closely in time so as to provide the desired therapeutic effect.
- the compounds of the present invention are also useful as standard or reference compounds, for example as a quality standard or control, in tests or assays involving the endothelial lipase.
- Such compounds may be provided in a commercial kit, for example, for use in pharmaceutical research involving endothelial lipase or HDL activity.
- a compound of the present invention could be used as a reference in an assay to compare its known activity to a compound with an unknown activity. This would ensure the experimenter that the assay was being performed properly and provide a basis for comparison, especially if the test compound was a derivative of the reference compound.
- compounds according to the present invention could be used to test their effectiveness.
- the compounds of the present invention may also be used in diagnostic assays involving endothelial lipase.
- the present invention also encompasses an article of manufacture.
- article of manufacture is intended to include, but not be limited to, kits and packages.
- the article of manufacture of the present invention comprises: (a) a first container; (b) a pharmaceutical composition located within the first container, wherein the composition, comprises: a first therapeutic agent, comprising: a compound of the present invention or a pharmaceutically acceptable salt form thereof; and, (c) a package insert stating that the pharmaceutical composition can be used for the treatment and/or prophylaxis of dyslipidemias and the sequelae thereof.
- the package insert states that the pharmaceutical composition can be used in combination (as defined previously) with a second therapeutic agent for the treatment and/or prophylaxis of dyslipidemias and the sequelae thereof.
- the article of manufacture can further comprise: (d) a second container, wherein components (a) and (b) are located within the second container and component (c) is located within or outside of the second container. Located within the first and second containers means that the respective container holds the item within its boundaries.
- the first container is a receptacle used to hold a pharmaceutical composition.
- This container can be for manufacturing, storing, shipping, and/or individual/bulk selling.
- First container is intended to cover a bottle, jar, vial, flask, syringe, tube (e.g., for a cream preparation), or any other container used to manufacture, hold, store, or distribute a pharmaceutical product.
- the second container is one used to hold the first container and, optionally, the package insert.
- the second container include, but are not limited to, boxes (e.g., cardboard or plastic), crates, cartons, bags (e.g., paper or plastic bags), pouches, and sacks.
- the package insert can be physically attached to the outside of the first container via tape, glue, staple, or another method of attachment, or it can rest inside the second container without any physical means of attachment to the first container.
- the package insert is located on the outside of the second container. When located on the outside of the second container, it is preferable that the package insert is physically attached via tape, glue, staple, or another method of attachment. Alternatively, it can be adjacent to or touching the outside of the second container without being physically attached.
- the package insert is a label, tag, marker, etc. that recites information relating to the pharmaceutical composition located within the first container.
- the information recited will usually be determined by the regulatory agency governing the area in which the article of manufacture is to be sold (e.g., the United States Food and Drug
- the package insert specifically recites the indications for which the pharmaceutical composition has been approved.
- the package insert may be made of any material on which a person can read information contained therein or thereon.
- the package insert is a printable material (e.g., paper, plastic, cardboard, foil, adhesive-backed paper or plastic, etc.) on which the desired information has been formed (e.g., printed or applied).
- PdCl 2 (dppf)-CH 2 Cl 2 (21.6 mg, 0.026 mmol) was added to a solution of Compound Ala (100 mg, 0.26 mmol), potassium phosphate (84 mg, 0.40 mmol) and 2- fluoro-5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)pyridine (88 mg, 0.40 mmol) in dioxane (4 mL).
- the reaction mixture was purged with argon, and the reaction vessel was sealed and heated at 110°C for 18 hrs.
- Example A2 to Example A8 were prepared by the general procedures described for Example Al .
- WO2011/074560 500 mg, 1.59 mmol) in THF (4 mL) was treated with sodium hydroxide (IN NaOH, 1.9 mL, 1.9 mmol). After 2 hours, the reaction mixture was concentrated to dryness under reduced pressure. The residue was co-evaporated with toluene (5 mL), re-dissolved in DMF (4 mL), and treated with (5 -methyl- 1,3, 4-oxadiazol- 2-yl)methanamine hydrochloride (284 mg, 1.90 mmol), TEA (2.2 mL, 16 mmol) and PyBOP (990 mg, 1.9 mmol). After 16 hours, the reaction mixture was diluted with water (20 mL), extracted with DCM (3 X 10 mL), dried over Na 2 S0 4 and concentrated under reduced pressure. The residue was purified by silica gel (eluting with 0-20%
- Example B1 Compound Bla. Ethyl 2-(methylsulfonyl)-2-(6-phenylbenzo[d]thiazol-2-yl)acetate
- Example B2 Compound B2a. 2-(6-Phenylbenzo[d]thiazol-2-yl)-N-(2-sulfamoylethyl)acetamide
- Example B3 to Example B22 were prepared as described in the general procedure given for Example B2.
- Example B27 to Example B29 were prepared as described in the general procedure given for Example B26.
- Compound B30c (10 mg, 11%>) was prepared from Compound B30b and
- Example B32 to Example B97 were prepared as described in the general procedure given for Example B31.
- Example B99 (10 mg, 31% yield) was prepared from Compound B99a and benzyl sulfonylchloride as described in the general procedure given for Compound B31a.
- HPLC: RT 0.83 min (LCMS Method M).
- MS (ES): m/z 630.8 [M+H] + .
- Example B 100 to Example B156 were prepared as described in the general procedure given for Example B99.
- Example B 157 (18 mg, 49% yield) was prepared from Compound B 157c and 4-(morpholine-4-carbonyl)phenyl)boronic acid as described in the general procedure given for Compound B2a.
- Example B 158 to Example B161 were prepared as described in the general procedure given for Example B157.
- Example B162 (9 mg, 50% yield) was prepared from Compound B 162a and Compound B31a as described in the general procedure given for Example B2a.
- HPLC: RT 0.7 min (LCMS Method M).
- MS (ES): m/z 501.8 [M+H] + .
- Example B 163 to Example B165 were prepared as described in the general procedure given for Example B162.
- Example B 167 to Example B171 were prepared as described in the general procedure given for Example B166.
- Example B172 (2.2 mg, 7% yield) was prepared from Compound B172a as described in the general procedure given for Compound B2a.
- HPLC: RT 1.22 min (LCMS Method N).
- MS (ES): m/z 569.1 [M+H] + .
- Example B 157 (101 mg, 0.18 mmol) in DCM (2.0 mL) was added TFA (1.0 mL, 13 mmol). The mixture was stirred at room temperature for 3 hours. The mixture was concentrated under reduced pressure. The residue was dissolved in
- Example B174 (15 mg, 55%) was prepared from Example B173 and 3- methylazetidin-3-ol hydrochloride as described in the general procedure given for Example B166.
- HPLC: RT 1.0 min (LCMS Method O).
- MS (ES): m/z 567.0 [M+H] + .
- Example B 175 to Example B179 were prepared as described in the general procedure given for Example B174.
- Example B180 (3.8 mg, 14%) was prepared from Compound B31a and 5- (4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)indolin-2-one as described in the general procedure given for Example 166.
- Example B 181 to Example B 186 were prepared as described in the general procedure given for Example B180.
- Example B 188 to Example B195 were prepared as described in the general procedure given for Example B187.
- Example B197 (6.2 mg, 21% yield) was prepared from Example B 196 as described in the general procedure given for Example B174.
- HPLC: RT 2.16 (LCMS Method O).
- MS (ES): m/z 593.05 [M+H] + .
- 1H NMR (500MHz, DMSO-d 6 ) ⁇ 8.15 (d, J 8.5 Hz, 1H), 6.99 (m, 2H), 4.04 - 3.53 (m, 8H), 3.27 - 3.13 (m, 2H), 2.94 - 2.80 (m, 2H), 2.50 - 2.35 (m, 2H).
- Example B 198 to Example B201 were prepared as described in the general procedure given for Example B197.
- Example B203 (4.1 mg, 14.3 % yield) was prepared from Compound B203a as described in the general procedure given for Example B99.
- HPLC: RT 0.70 (LCMS Method M).
- MS (ES): m/z 472.8 [M+H] + .
- Example B204 to Example B205 were prepared as described in the general procedure given for Example B2.
- Example B207 to Example B281 were prepared as described in the general procedure given for Example B31.
- Example B282 to Example B312 were prepared as described in the general procedure given for Example B99.
- Example B313 to Example B316 were prepared as described in the general procedure given for Example B157.
- Example B317 was prepared as described in the general procedure given for Example B162.
- Example B318 to Example B324 were prepared as described in the general procedure given for Example B166.
- Example B325 to Example B327 were prepared as described in the general procedure given for Example B173.
- Example B328 to Example B351 were prepared as described in the general procedure given for Example B174.
- Example B352 to Example B374 were prepared as described in the general procedure given for Example B 180.
- Example B375 to Example B385 were prepared as described in the general procedure given for Example B187.
- Example B386 was prepared as described in the general procedure given for Example B31.
- Example B387 (2.2 mg, 9.0% yield) was prepared from Compound B387c (prepared as described in the general procedure given for Example B31) and Compound B387b as described in the general procedure given for Example B180.
- EL IC 50 13.6 nM.
- HPLC: RT 1.4 min (LCMS Method Q).
- MS (ES): m/z 687.1 [M+H] + .
- 1H NMR 500MHz, DMSO-d 6 ) ⁇ 14.04 and 11.46 and 6.15 (s, 1H), 9.12 - 9.02 and 8.61 (1H), 8.19 (br.
- Example B388 to Example B390 were prepared as described in the g procedure given for Example B388.
- Example B391 10 mg, 28% yield.
- EL IC 50 25 nM.
- HPLC: RT 1.72 min (LCMS Method B).
- MS (ES): m/z 478
- Example B392 (5.9 mg, 43 % yield) was prepared from Compound B392a and (3-isopropylisoxazol-5-yl)methanamine as described in the general procedure given for Example B197.
- HPLC: RT 1.24 min.
- MS (ES): m/z 478 [M+H] + .
- 1H NMR 500MHz, DMSO-d 6 ) ⁇ 9.35 (br. s., 1H), 8.99 (br.
- Example B393 to Example B394 were prepared as described in the general procedure given for Example B392.
- Example B397 was prepared from Compound B396d as described in the general procedure given for Example B396 (8 mg, 49%).
- RT 1.63 min (LCMS Method B).
- MS (ES): m/z 495.1 [M+H] + .
Abstract
The present invention provides compounds of Formula (I): as defined in the specification and compositions comprising any of such novel compounds. These compounds are endothelial lipase inhibitors which may be used as medicaments.
Description
AMIDE, UREA OR SULFONE AMIDE LINKED BENZOTHIAZOLE INHIBITORS
OF ENDOTHELIAL LIPASE
FIELD OF THE INVENTION
[0001] The present invention provides novel amide, urea or sulfone amide linked benzothiazole compounds and analogues, which are endothelial lipase (EL) inhibitors, compositions containing them, and methods of using them, for example, for the treatment and/or prophylaxis of dyslipidemias and the sequelae thereof. BACKGROUND OF THE INVENTION
[0002] Cardiovascular disease is a major health risk throughout the industrialized world. Atherosclerosis, the most prevalent of cardiovascular diseases, is the principal cause of heart attack, and stroke, and thereby the principal cause of death in the United States.
[0003] Atherosclerosis is a complex disease involving many cell types and molecular factors (for a detailed review, see Ross, R., Nature, 362(6423):801-809 (1993)). Results from epidemiologic studies have clearly established an inverse relationship between levels of high density lipoprotein (HDL), which transports endogenous cholesterol from tissues to the liver as well as mediating selective cholesteryl ester delivery to
steroidogenic tissues, and the risk for atherosclerosis (Gordon, D.J. et al, N. Engl. J. Med., 321(19): 1311-1316 (1989)).
[0004] The metabolism of HDL is influenced by several members of the
phospholipase and triacylglycerol (TG) lipase family of proteins, which hydrolyze triglycerides, phospholipids (PL), and cholesteryl esters (CE), generating fatty acids to facilitate intestinal absorption, energy production, or storage. Of the TG lipases, lipoprotein lipase (LPL) influences the metabolism of HDL cholesterol by hydro lyzing triglycerides in triglyceride-rich lipoproteins, resulting in the transfer of lipids and apolipoproteins to HDL and is responsible for hydrolyzing chylomicron and very low density lipoprotein (VLDL) in muscle and adipose tissues. Hepatic lipase (HL) hydrolyzes HDL triglyceride and phospholipids, generating smaller, lipid-depleted HDL particles, and plays a role in the uptake of HDL cholesterol (Jin, W. et al., Trends Endocrinol. Metab., 13(4): 174-178 (2002); Wong, H. et al, J. Lipid Res., 43:993-999
(2002)). Endothelial lipase (also known as EDL, EL, LIPG, endothelial-derived lipase, and endothelial cell-derived lipase) is synthesized in endothelial cells, a characteristic that distinguishes it from the other members of the family.
[0005] Recombinant endothelial lipase protein has substantial phospholipase activity but has been reported to have less hydrolytic activity toward triglyceride lipids (Hirata, K. et al, J. Biol. Chem., 274(20): 14170-14175 (1999); Jaye, M. et al, Nat. Genet., 21 :424- 428 (1999)). However, endothelial lipase does exhibit triglyceride lipase activity ex vivo in addition to its HDL phospholipase activity, and endothelial lipase was found to hydrolyze HDL more efficiently than other lipoproteins (McCoy, M.G. et al., J. Lipid Res., 43:921-929 (2002)). Overexpression of the human endothelial lipase gene in the livers of mice markedly reduces plasma concentrations of HDL cholesterol and its major protein, apolipoprotein A-I (apoA-I) (Jaye, M. et al, Nat. Genet., 21 :424-428 (1999)).
[0006] Various types of compounds have been reported to modulate the expression of endothelial lipase, for example, 3-oxo-l,3-dihydro-indazole-2-carboxamides (WO
2004/093872, US 2006/0211755A1), 3-oxo-3-H-benzo[d]isoxazole-2-carboxamides (WO 2004/094393, U.S. Patent No. 7,217,727), and benzisothiazol-3-one-2-carboxamides (WO 2004/094394, U.S. Patent No. 7,595,403) by Eli Lilly & Co.; diacylindazole derivatives (WO 2007/042178, US 2008/0287448A1) and imidazopyridin-2-one derivatives (WO 2007/110215, US 2009/0076068A1), and azolopyridin-3-one derivatives (WO 2007/110216, US 2009/0054478A1) by Sanofi-Aventis; heterocyclic derivatives (WO 2009/123164), keto-amide derivatives (WO 2009/133834), acetic acid amide derivatives (WO20/ 10/44441, US 2011/0251386A1), oxadiazole derivatives (WO
2011074560, US2012253040 Al), benzothiazole and azabenzothiazole derivatives (WO 2012081563) and amino derivatives (WO2012173099) by Shionogi & Co., Ltd.
However, because endothelial lipase is a relatively new member in the lipase gene family, a full understanding of the potential of endothelial lipase inhibitors to human health, as well as the inhibitors of other lipases in general, requires more studies.
[0007] Thus, there is a clear need for new types of compounds capable of inhibiting the activity of lipases, particularly endothelial lipase, that would constitute effective treatments to the diseases or disorders associated with the activity of such lipases.
SUMMARY OF THE INVENTION
[0008] The present disclosure provides amide, urea or sulfone amide linked benzothiazole compounds and their analogues, including stereoisomers, tautomers, pharmaceutically acceptable salts, or solvates thereof, which are useful as EL inhibitors.
[0009] The present invention also provides processes and intermediates for making the compounds of the present invention.
[0010] The present invention also provides pharmaceutical compositions comprising a pharmaceutically acceptable carrier and at least one of the compounds of the present invention or stereoisomers, tautomers, pharmaceutically acceptable salts, or solvates thereof.
[0011] The compounds of the invention may be used in the treatment and/or prophylaxis of dyslipidemias and the sequelae thereof.
[0012] The compounds of the invention may be used in therapy.
[0013] The compounds of the invention may be used for the manufacture of a medicament for the treatment and/or prophylaxis of dyslipidemias and the sequelae thereof.
[0014] The compounds of the invention can be used alone, in combination with other compounds of the present invention, or in combination with one or more, preferably one to two other agent(s).
[0015] Other features and advantages of the invention will be apparent from the following detailed description and claims.
DETAILED DESCRIPTION OF THE INVENTION I. COMPOUNDS OF THE INVENTION
[0016] In a first aspect, the present invention provides, inter alia, a compound of Formula (I):

or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof, wherein:
R1 is inde endently selected from: halogen, CN, C02(C1.4 alkyl), -CO-RJ,


(a 5- to 6-membered heteroaryl comprising: carbon atoms and 1-4 heteroatoms selected from N, NRC, O, and S(0)p; wherein said heteroaryl is substituted with 0-3 Ra);
R2 is, independently at each occurrence, selected from: halogen, OH, C^ alkyl, Cj.4 alkoxy, C^ haloalkyl, C^ haloalkoxy, CN, NH2, N02, ΝΗ(^_4 alkyl),
N(C alkyl)2, C02H, C02(C1.4 alkyl), and CONH2;
R3 is independently selected from: -S02R5 and -NHCOR6;
R4 independently selected from:

R5 is independently selected from: Ci _g alkyl substituted with 0-1 R9,
C2_6 alkenyl, Ci .g haloalkyl, Ci .g alkoxy, -(CH2)m-(0)n-(C3_6 carbocycle substituted with 0-3 Rb), and -(CH2)m-(0)n-(5- to 6-membered heterocycle comprising: carbon atoms and 1-4 heteroatoms selected from N, NRC, O, and S(0)p; wherein said heterocycle is substituted with 0-2 Rb);
R6 is independently selected from: C^g alkyl, C2.6 alkenyl, C^g alkoxy,
Cj.4 haloalkyl, -(CH2)m-(C3.6 carbocycle substituted with 0-3 Rb),
-(CH2)m-(pyridyl substituted with 0-2 Rb), -ΝΗ(^_4 alkyl), -NHCH2C02(C1.4 alkyl),
alkyl
-NH(4-halo-Ph), -NHBn, and
 ;
;
R7 is independently selected from: CONH2, CONH(C!_4 alkyl),
CON(C alkyl)2, S02NHS02(C1.4 alkyl), S02NH(CH2) 2-4C02(C!.4 alkyl),
NHS02NH2, NHS02(C alkyl), N(C!_4 alkyl)S02NH2,
H
ON
N(C02C alkyl)S02(C1.4 alkyl), S02R10, and O ^


R9 is independently selected from: halogen, C1 .4 alkoxy, C 1 .4 haloalkyl, C haloalkoxy, NH2, C02H, CO^C^ alkyl), S03H, CONHRd , NHCONHRd,
NHC02Rd,
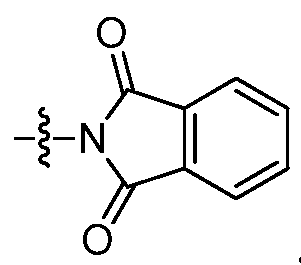 s and 5- to 6-membered heterocycle comprising: carbon atoms and 1-4 heteroatoms selected from N, NR8, O, and S(0)p;
s and 5- to 6-membered heterocycle comprising: carbon atoms and 1-4 heteroatoms selected from N, NR8, O, and S(0)p;
R10 is independently selected from: OH, Ci _g alkyl, C2_g alkenyl, NH2,
H Ci.e alkyl substituted with 0-1 C02(C1.4 alkyl)), NH(C2.6 alkyl),
NH(Ci _4 haloalkyl), NHPh, phenyl substituted with 0-2 halogens, and

R1 1 is, independently at each occurrence, selected from: halogen, C1 .4 alkyl, Ci .4 haloalkyl, NH2, and phenyl;
Ra is, independently at each occurrence, selected from: halogen, Ci _g alkyl substituted with 0-1 Rf, Ci .4 alkoxy substituted with 0-1 Rf, Ci .g haloalkyl,
C 6 haloalkoxy, OH, CN, N02, C02H, CO^C^ alkyl), NR8Rh, CONR8Rh,
CONRgRJ, NHCORi, NHC02Ri, S02NR8Rh, -(0)n-(CH2)t-RJ, and -CO-RJ;
Rb is, independently at each occurrence, selected from: halogen, C 1 .4 alkyl, C alkoxy, C haloalkyl, C haloalkoxy, OH, CN, NH2, N02, NH(C!_4 alkyl), (C!.4 alkyl)2, C02H, CO^C^ alkyl), S02(CM alkyl), CONH2, and
CONH(C alkyl);
Rc is, independently at each occurrence, selected from: H, Ci _g alkyl substituted with 0-1 Re, CO(C alkyl), C02(C1.4 alkyl), COBn, C02Bn, -(CH2)t-piperidinyl, -(CH2)t-morpholinyl, -(CH2)t-piperazinyl, pyrimidinyl,
alkyl— O /
-(CH2)r(C3.6 carbocycle substituted with 0-2 Re), and ° — ;
Rd is, independently at each occurrence, selected from: Ci _g alkyl and
-(CH2)t-(phenyl substituted with 0-2 Re);
Re is, independently at each occurrence, selected from: halogen, Ci .4 alkyl, C1 .4 alkoxy, C 1 .4 haloalkyl, and C1 .4 haloalkoxy;
Rf is, independently at each occurrence, selected from: OH, halogen, C1 .4 alkyl, C1 .4 alkoxy, C 1 .4 haloalkyl, and C1 .4 haloalkoxy;
R8 is, independently at each occurrence, selected from: H and C^ alkyl;
Rh is, independently at each occurrence, selected from: H, Ci _g haloalkyl and Ci _6 alkyl substituted with 0-1 Rf;
R1 is, independently at each occurrence, selected from: Ci .4 haloalkyl and
Ci .4 alkyl substituted with 0-1 Rf;
RJ is, independently at each occurrence: C3.6 carbocycle or a 4- to 6-membered heterocycle comprising: carbon atoms and 1-4 heteroatoms selected from N, NR§, O, and S(0)p; wherein said carbocycle and heterocycle are substituted with 0-2 Rf;
m and t are, independently at each occurrence, selected from 0, 1 , 2, and 3;
n is, independently at each occurrence, selected from 0 and 1 ;
p is, independently at each occurrence, selected from 0, 1 , and 2; and
s is, independently at each occurrence, selected from 1 , 2, and 3.
[0017] In a second aspect, the present invention includes a compound of Formula (la) or (lb):


or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof, within the scope of the first aspect, wherein:
m and t are, independently at each occurrence, selected from 0, 1 , and 2; and s is, independently at each occurrence, selected from 1 and 2.
[0018] In a third aspect, the present invention includes a compound of Formula (Ila) or (lib):

or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof, within the scope of any of the first and second aspects; wherein:
R2 is independently selected from: halogen and C^ alkyl.
[0019] In a fourth aspect, the present invention includes a compound of Formula (la) (Ila) or (lib), or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof, within the scope of the first, second and third aspects, wherein:
1 is independently selected from: 4-halo-Ph, 6-halo-pyrid-3-yl, and
R4 is ^so2NH2 ; and
R6 is independently selected from: CX alkyl, -CHF2, NHBn, 4-CF3-Ph, and pyrid-3-yl.
[0020] In a fifth aspect, the present invention includes a compound of Formula (Ilia) or (nib):


or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof, within the scope of any of the first and second aspects; wherein:
R2 is independently selected from: halogen, C^_4 alkyl and C^_4 haloalkyl.
[0021] In a sixth aspect, the present invention includes a compound of Formula (I), (lb), (Ilia) or (Illb), or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof, within the scope of any of the first, second and fifth aspects, wherein:
R1 is inde endently selected from:
 H)
H)

R4 is independently selected from: ^"^SO^C^ alkyl) ? ^^S02NH2 ?
^/^S02NH(C2-6 alkenyl) "i^^^^^SC^NHCI^CFa ^^S02NH(CH2)3C02(C1^ alkyl)
^/^S02NHS02(C1_4 alkyl) ^^-^SC^NHPh ^^S02(4-halo-Ph)
C-|_4 alkoxy
 , and
, and
N-N
C-|_4 alkyl .
R5 is independently selected from: Ci .4 alkyl substituted with 0-1 R9,
C -4 alkenyl, -(CH2)o-i-C3.6 cycloalkyl, -(CH2)o-r(phenyl substituted with 0-1 Rb) and

R9 is independently selected from: C1 .4 alkoxy, C1 .4 haloalkyl, NH2, and NHC02Bn;
Ra is, independently at each occurrence, selected from: OH, halogen, CN, C .4 alkyl substituted with 0-1 OH, C^ alkoxy, -0(CH2)1.20(C1.4 alkyl),
C haloalkyl, CM haloalkoxy, NH2, C02H, C02(C1.4 alkyl), CONH2,
CONH(C alkyl), CONH(CH2)2.3OH, CONH(CH2)1.30(C1.4 alkyl),
CON(C alkyl)(CH2)1.30(C1.4 alkyl), CONHCC^ haloalkyl), NHCO(C alkyl), HCO(C!.4 haloalkyl), NHC02(C1.4 alkyl), S02N(CM alkyl)2, S0 NH(CH2)2.3OH,
pyrazolyl, -(CH2)0-2-morpholinyl, -CO-morpholinyl, piperazinyl,

is independently selected from: halogen and Ci .4 haloalkyl;
Rc is, independently at each occurrence, selected from: H, Ci .4 alkyl,
-(CH2)2(Ci .4 alkoxy), -(CH2)0-2-(phenyl substituted with 0-1 halo),
-(CH2)o-2-piperidinyl, -(CH2)0-2-morpholinyl, and
 Rf is, independently at each occurrence, selected from: OH, halogen, C^ alkyl, C}_4 haloalkyl, C^_4 alkoxy, and C^_4 haloalkoxy.
Rf is, independently at each occurrence, selected from: OH, halogen, C^ alkyl, C}_4 haloalkyl, C^_4 alkoxy, and C^_4 haloalkoxy.
[0022] In a seventh aspect, the present invention includes a compound of Formula (I), (lb), (Ilia) or (Illb), or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof, within the scope of any of the first, second, fifth and sixth aspects, wherein:
R1 is independently selected from: Ph, 3-halo-Ph, 4-halo-Ph, 3-^_4 alkoxy-Ph, 4-CH2OH-Ph, 3-CN-Ph, 4-CN-Ph, 4-C02H-Ph, 4-C02(C1.4 alkyl)-Ph,
3-ΝΙΚΟ(^_4 alkyl)-Ph, 4-NHCO(C1.4 alkyl)-Ph, 4-(pyrazol-l-yl)-Ph,
2-Ci_4 alkoxy-pyridyl, pyrimidinyl and

R4 is independently selected from: ^^S02NH2 ? ^S02NH(C2.6 alkenyl) ;
? and

R5 is independently selected from: C^ alkyl substituted with 0-1 R9, and
C2-4 alkenyl, Bn, and 4-halo-Bn; and
R9 is independently selected from: C 1.4 alkoxy, C1.4 haloalkyl, NH2, and
NHC02Bn. [0023] In an eighth aspect, the present invention includes a compound of Formula (I), (lb), (Ilia) or (Illb), or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof, within the scope of any of the first, second, fifth, sixth and seventh aspects, wherein:
R1 is independently selected from: Ph, 3-halo-Ph, 4-halo-Ph, 3-^_4 alkoxy-Ph, 4-CH2OH-Ph, 3-CN-Ph, 4-CN-Ph, 4-C02H-Ph, 4-C02(C1.4 alkyl)-Ph,
3-NHCO C alkyl)-Ph, 4-NHCO(C1_ alkyl)-Ph, 4-(pyrazol-l-yl)-Ph, pyrimidinyl and

R4 is ii^ S02NH2 ·
R5 is independently selected from: C^ alkyl substituted with 0-1 R9, and
C2.4 alkenyl, Bn, and 4-halo-Bn; and
R9 is independently selected from: C 1 .4 alkoxy, C1 .4 haloalkyl, NH2, and NHC02Bn.
[0024] In a ninth aspect, the present invention includes a compound of Formula (I), (lb), (Ilia) or (Illb), or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or solvate thereof, within the scope of any of the first, second, fifth, sixth, seventh and eighth aspects, wherein:
R1 is independently selected from: Ph, 3-F-Ph, 4-F-Ph, 3-OMe-Ph, 4-CH2OH-Ph, 3-CN-Ph, 4-CN-Ph, 4-C02H-Ph, 4-C02Me-Ph 3-NHCOMe-Ph, 4-NHCOMe-Ph,
4-(pyrazol-l-yl)-Ph, pyrimidin-5-yl, and
 ; and
; and
R5 is independently selected from: Me, Et, Pr, z'-Bu, ^^^5^, Bn, 4-F-Bn,
-(CH2)2OMe, -(CH2)2CF3, -(CH2)2NH2, and -(CH2)2NHC02Bn.
[0025] In a tenth aspect, the present invention includes a compound of Formula (I), (lb), (Ilia) or (Illb), or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or solvate thereof, within the scope of any of the first, second, fifth, sixth and seventh aspects, wherein:
R1 is Ph;
tidependently selected from: ^^S02NH(C2 . 6 alkenyl) ? an(j

R5 is independently selected from: C 1 .4 alkyl, C2_4 alkenyl, -(CH2)20(Ci and -(CH2)2NHC02Bn.
[0026] In an eleventh aspect, the present invention includes a compound of Formula (lb), (Ilia) or (Illb), or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof, within the scope of any of the first, second, fifth, sixth, seventh and tenth aspects, wherein: 4 is independently selected from: . ^^S02NH(CH2)4CH=CH2

-(CH2)2NHC02Bn.
[0027] In a twelfth aspect, the present invention includes a compound of Formula (I), (lb), (Ilia) or (Illb), or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof, within the scope of any of the first, second, fifth and sixth aspects, wherein:
independently selected from:
 H)
H)

(phenyl substituted with 0-2 Ra), and (a heteroaryl substituted with 0-2 Ra and selected from: isoxazolyl, pyrazolyl, l-Rc-pyrazolyl, pyridyl, and pyrimidinyl);
R4 is independently selected from: "Ji ^S02(C1_4 alkyl) ? "J^^SOsNHs ?
^^S02NH(C2_6 alkenyl) !^^^S02NHCH2CF3 ^^S02NH(CH2)3C02(C1 _4 alkyl)

R5 is independently selected from: Ci .4 alkyl substituted with 0-1 R9
-(CH2)o-i-C3_6 cycloalkyl, benzyl substituted with 0-1 Rb, and

R9 is independently selected from: C 1 .4 alkoxy, C1 .4 haloalkyl, NH2, and NHC02Bn;
Ra is, independently at each occurrence, selected from: OH, halogen, C1.4 alkyl substituted with 0- 1 OH, C _4 alkoxy, -0(CH2)20(C _4 alkyl), C _4 haloalkyl,
C haloalkoxy, NH2, CONH2, CONH(C!_4 alkyl), CONH(CH2)2.3OH,
CONH(CH2)1.30(C1.4 alkyl), CONCC^ alkyl)(CH2)1.30(C1.4 alkyl),
CONH(C haloalkyl), NHC02(C1.4 alkyl), S02N(CM alkyl)2, S02NH(CH2)2.3OH,
- CH2)0_2-morpholinyl, -CO-morpholinyl, piperazinyl,


Rb is independently selected from: CN, halogen and C^ haloalkyl;
Rc is, independently at each occurrence, selected from: H, Ci .4 alkyl substituted with 0-1 Ci .4 alkoxy, -(CH2)o-2- henyl substituted with 0-1 halo), -(CH2)o-2-piperidinyl,

Rf is, independently at each occurrence, selected from: OH, CN, halogen,
C}_4 alkyl, and C^ alkoxy.
[0028] In a thirteenth aspect, the present invention includes a compound of Formula (I), (lb), (Ilia) or (Illb), or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof, within the scope of any of the first, second, fifth, sixth and twelfth aspects, wherein:
R1 is independently selected from: 3-(CH2)2OH-Ph, 4-(CH2)2OH-Ph,
4-0(CH2)20(C1_4 alkyl)-Ph, 4-C haloalkoxy-Ph, 4-NH2-Ph, 4-CONH2-Ph,
4-CONH(C1_4 alkyl)-Ph, 4-CONH(CH2)2OH-Ph, 3-CONH(CH2)20(C1.4 alkyl)-Ph, 4-CONH(CH2)20(C1_4 alkyl)-Ph, 4-CONH(C1.4 haloalkyl)-Ph,
3^0Ν(^_4 alkyl)(CH2)20(C1.4 alkyl)-Ph, 4-NHC02(C1.4 alkyl)-Ph,
4-S02N(C1_ alkyl)2-Ph, 4-S02NH(CH2)2OH-Ph, lH- -C^ haloalkyl-pyrazol-4-yl, I-C1 .4 alkyl-pyrazol-4-yl, l-Bn-pyrazol-4-yl, 3-C 1.4 alkyl-pyrid-4-yl, 6-OH-pyrid-2-yl, 6-OH-pyrid-3-yl, 2-OH-pyrid-4-yl, 6-C 1 .4 alkoxy-pyrid-2-yl, 2-C1 .4 alkoxy-pyrid-3-yl, 6-C1 .4 alkoxy-pyrid-3-yl, 2-C 1 .4 alkoxy-pyrid-4-yl, 6-halo-pyrid-3-yl, 2-halo-pyrid-4-yl, 6-C1 .4 haloalkyl-pyrid-3-yl, 5-Ci .4 alkyl-6-halo-pyrid-3-yl,
3-halo-6-Ci_4 alkoxy-pyrid-4-yl, pyrimidin-5-yl,

- 16-
[0029] In a fourteenth aspect, the present invention includes a compound of Formula (I), (lb), (Ilia) or (Illb), or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof, within the scope of any of the first, second, fifth, sixth, twelfth and thirteenth aspects, wherein:
R1 is independently selected from: 3-(CH2)2OH-Ph, 4-(CH2)2OH-Ph, 4-0(CH2)2OMe-Ph, 4-OCF3-Ph, 4-NH2-Ph, 4-CONH2-Ph, 4-CONH(t-Bu)-Ph,
4-CONH(CH2)2OH-Ph, 3-CONH(CH2)2OMe-Ph, 4-CONH(CH2)2OMe-Ph,
4-CON(Me)(CH2)2OMe-Ph, 4-CONHCH2CF3-Ph, 4-NHC02Me-Ph,
4-NHC02(t-Bu)-Ph, 4-S02N(Me)2-Ph, 4-S02NH(CH2)2OH-Ph, lH-3-CF3-pyrazol-4-yl,
1- Me-pyrazol-4-yl, l-(z'-Bu)-pyrazol-4-yl, l-Bn-pyrazol-4-yl, 3-Me-pyrid-4-yl,
6-OH-pyrid-2-yl, 6-OH-pyrid-3-yl, 2-OH-pyrid-4-yl, 6-OMe-pyrid-2-yl,
2- OMe-pyrid-3-yl, 6-OMe-pyrid-3-yl, 2-OMe-pyrid-4-yl, 6-F-pyrid-3-yl, 2-F-pyrid-4-yl, 2-Cl- rid-4-yl, 6-CF3-pyrid-3-yl, 5-Me-6-F-pyrid-3-yl, 3-F-6-OMe-pyrid-4-yl,


and
R4 is independently selected from: ^^S02Me ? !^S02NH2 ?
ii^^S02NHCH2CF3 i-/^S02NHS02Me p;-/^S02NHPh ^^S02(4-F-Ph)

R5 is independently selected from: Me, Pr, z'-Pr, z'-Bu, -(CH2)2OMe, -(CH2)2CF3, -(CH2)3CF3, -(CH2)2NH2, cyclopropylmethyl, Bn, 4-F-Bn, 4-CF3-Bn,
-(CH2)2NHC02Bn, and ^ '°
[0030] In another aspect, the present invention provides a compound of Formula (I), or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof, within the scope of the first aspect, wherein:
R1 is independently selected from: phenyl and a 5- to 6-membered heteroaryl comprising: carbon atoms and 1 -4 heteroatoms selected from N, NRC, O, and S(0)p; wherein each phenyl and heteroaryl are substituted with 0-3 Ra;
R2 is, independently at each occurrence, selected from: halogen, OH, Ci _4 alkyl, C alkoxy, C haloalkyl, C haloalkoxy, CN, NH2, N02, NH(C alkyl),
N(C alkyl)2, C02H, C02(C1.4 alkyl), and CONH2;
R3 is independently selected from: -S02R5 and -NHCOR6; R4 independently selected from: s and 4 ;
R5 is independently selected from: Ci .g alkyl, C2-6 alkenyl, -(CH2)mCHF2, -(CH2)mCF3, -(CH2)m-(0)n-(C3_6 carbocycle substituted with 0-3 Rb),
-(CH2)m-(0)n-(pyridyl substituted with 0-2 Rb), -(CH2)sNHCONHRd,
-(CH2)sNHC02Rd
 s -ΟΓ1-2 ? _y , and
s -ΟΓ1-2 ? _y , and
R6 is independently selected from: Ci _g alkyl, C2.6 alkenyl, Ci_6 alkoxy,
-(CH2)mCHF2, -(CH2)mCF3, -(CH2)m-(C3.6 carbocycle substituted with 0-3 Rb), -(CH2)m-(pyridyl substituted with 0-2 Rb), -NH(C!_4 alkyl), -NHCH2C02(C1.4 alkyl),
H alkyl
-NH(4-halo-Ph), -NHBn, and °1 "4 alky ;
R7 is independently selected from: CONH2, CO H(C!_4 alkyl),
CON(C!.4 alkyl)2, S02OH, S02(4-halo-Ph), S02NH2, S02NHCH2CF3, S02NHPh, NHS02NH2, NHS02(Ci.4 alkyl), N(C alkyl)S02NH2,
CN
N(C02C1.4 alkyl)S02(C alkyl), and O ^ ;
inde endently selected



Ra is, independently at each occurrence, selected from: halogen, Ci _g alkyl substituted with 0-1 OH, Ci .4 alkoxy, Ci .g haloalkyl, Ci.g haloalkoxy, OH, CN, N02, NH2, NH(C alkyl), N(C alkyl)2, C02H, C02(C1.4 alkyl), CONH2,
CONH(C alk l), S02NH2, S02N(C1.4 alk l)2, CONH(CH2)1.3CF3, pyrazolyl,
ΝΛ-!· ,

Rb is, independently at each occurrence, selected from: halogen, C 1 .4 alkyl, C alkoxy, C haloalkyl, C haloalkoxy, OH, CN, NH2, N02, NH(C alkyl), N(Ci_4 alkyl)2, C02H, C02(C alkyl), S02(CM alkyl), CONH2, and
CONH(Ci_4 alkyl);
Rc is, independently at each occurrence, selected from: H, Ci _g alkyl,
CO(C alkyl), C02(C alkyl), COBn, C02Bn,
 , pyrimidinyl and
, pyrimidinyl and
-(CH2)t-(C3.6 carbocycle substituted with 0-2 Re);
Rd is, independently at each occurrence, selected from: Ci.g alkyl and
-(CH2)t-(phenyl substituted with 0-2 Re);
Re is, independently at each occurrence, selected from: halogen, Ci .4 alkyl,
C1 .4 alkoxy, C 1 .4 haloalkyl, and C1 .4 haloalkoxy;
m and t are, independently at each occurrence, selected from 0, 1, 2, and 3; n is, independently at each occurrence, selected from 0 and 1;
p is, independently at each occurrence, selected from 0, 1 , and 2; and
s is, independently at each occurrence, selected from 1 , 2, and 3.
[0031] In another aspect, the present invention includes a compound of Formula (I), (la) or (lb), or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof, within the scope of the first or second aspect, wherein: 4 is independently selected from: ^^SC^C^ alkyl) ? ^^S02NH2 ?

[0032] In another aspect, the present invention includes a compound of Formula (I), (lb) or (Ilia), or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof, wherein:
R1 is independently selected from: Ph, 4^02(^_4 alkyl)-Ph, 4-NH2-Ph, 4-CONH2-Ph, 4-S02 (C alkyl)2-Ph, 4-ΝΙΚ02(^_4 alkyl)-Ph, 4-(pyrazol-l-yl)-Ph, 3-Cj_4 alkyl-pyrid-4-yl, 6-C _4 alkoxy-pyrid-3-yl, 6-halo-pyrid-3-yl, 3-halo-pyrid-4-yl,
C-|_ alkyl
6-CF3-pyrid-3-yl,
 and ci- alkyl 4 is independently selected from: ^^SC^C^ alkyl) ? ^S02NH2 ?
and ci- alkyl 4 is independently selected from: ^^SC^C^ alkyl) ? ^S02NH2 ?
R5 is independently selected from: C 1 .4 alkyl, C2_4 alkenyl, Bn, 4-CF3-Bn, and -(CH2)1.3CF3.
[0033] In another aspect, the present invention includes a compound of Formula (I), (lb) or (Ilia), or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof, wherein:
R1 is independently selected from: Ph, 4-C02(C1.4 alkyl)-Ph and
4-(pyrazol-l-yl)-Ph;
R4 is ^so2NH2 ; and
independently selected from: C 1 .4 alkyl and C2-4 alkenyl.
[0034] In another aspect, the present invention includes a compound of Formula (I), (lb) or (Ilia), or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof, wherein:
R1 is independently selected from: Ph, 4-C02Me-Ph and 4-(pyrazol-l-yl)-Ph; and i independently selected from: Me and
[0035] In another aspect, the present invention includes a compound of Formula (I), (lb) or (Ilia), or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof, within the scope of any of the first, second, third, sixth and seventh aspects, wherein:
independently selected from: 4-NH2-Ph, 4-CONH2-Ph,
4-S02N(C1_4 alkyl)2-Ph, 4-NHC02(Ci_4 alkyl)-Ph, 3-C 4 alkyl-pyrid-4-yl,
6-C1 . alkoxy-pyrid-3-yl, 6-halo-pyrid-3-yl, 3-halo-pyrid-4-yl, 6-CF3-pyrid-3-yl,

independently selected from: C^ alkyl, Bn, 4-CF3-Bn, and -(CH2)1
[0036] In another aspect, the present invention includes a compound of Formula (I), (lb) or (Ilia), or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof, within the scope of any of the first, second, third, sixth and tenth aspects, wherein:
R1 is independently selected from: 4-NH2-Ph, 4-CONH2-Ph, 4-S02N(Me)2-Ph, 4-NHC02Me-Ph, 3-Me-pyrid-4-yl, 6-OMe-pyrid-3-yl, 6-F-pyrid-3-yl, 3-F-pyrid-4-yl,
3-Cl-pyrid-4-yl, 6-CF3-pyrid-3-yl,
 4 is independently selected from: ^^S02Me ? !^S02NH2 ?
4 is independently selected from: ^^S02Me ? !^S02NH2 ?
R5 is independently selected from: Me, Pr, z'-Pr, Bn, 4-CF3-Bn, and -(CH2)2CF3 [0037] In another aspect, the present invention includes a compound of Formula (I), (lb), (Ilia) or (Illb), or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof, wherein:
R1 is independently selected from: 4-C02H-Ph, 4-NHCO(C1.4 alkyl)-Ph,
4- CONH2-Ph, 4-CONH(CH2)2_3OH-Ph, 4-CONH(CH2)20(C1_4 alkyl)-Ph,
4-CONH(CH2)C(C!.4 alkyl) 2OH-Ph, 4-CONH(C3.6 cycloalkyl)-Ph,
5- halo^-CON^!^ alkyl)2-Ph, 6-halo-pyrid-3-yl, 6-OH-pyrid-2-yl,
6- OH-pyrid-3-y -OH-pyrid-4-yl, 2-OH-3-halo-pyrid-4-yl, 2-OH-5-halo-pyrid-4-yl,
pyrimidin-5-yl,
 R4 is independently selected from: so2NH2 and
R4 is independently selected from: so2NH2 and
R5 is independently selected from: C^ alkyl substituted with 0-1 R9,
-CH2-C3.6 cycloalkyl, and benzyl substituted with 0-1 Rb;
R9 is independently selected from: C 1 .4 alkoxy, and C1 .4 haloalkyl; and
Rb is independently selected from: CN, halogen C^ haloalkyl and
C}_4 haloalkoxy.
[0038] In another aspect, the present invention includes a compound of Formula (I), (lb), (Ilia) or (Illb), or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof, wherein:
R1 is independently selected from: 4-C02H-Ph, 4-NHCOMe-Ph, 4-CONH2-Ph, 4-CONH(CH2)2_3OH-Ph, 4-CONH(CH2)2OMe-Ph, 4-CONH(CH2)C(Me) 2OH-Ph, 4-CONH(cyclopropyl)-Ph, 3-F-4-CON(Me)2-Ph, 6-F-pyrid-3-yl, 6-OH-pyrid-2-yl, -OH-pyrid-3-yl, 2-OH-pyrid-4-yl, 2-OH-3-F-pyrid-4-yl, 2-OH-5-F-pyrid-4-yl,


R4 is independently selected from:
 ; and R5 is independently selected from: Me, z'-Pr, -(C ^OMe, -(CH2)2CF3, cyclopropylmethyl, Bn, 3-CN-Bn, 3-F-Bn, 4-F-Bn, 4-CF3-Bn, and 4-OCF3-Bn.
; and R5 is independently selected from: Me, z'-Pr, -(C ^OMe, -(CH2)2CF3, cyclopropylmethyl, Bn, 3-CN-Bn, 3-F-Bn, 4-F-Bn, 4-CF3-Bn, and 4-OCF3-Bn.
[0039] In a fifteenth aspect, the present invention provides a compound selected from the exemplified examples or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof.
[0040] In another aspect, the present invention provides a compound selected from any subset list of compounds within the scope of the twelfth aspect.
[0041] In another embodiment, the compounds of the present invention have EL IC50 values < 500 11M.
[0042] In another embodiment, the compounds of the present invention have EL IC50 values < 100 11M.
[0043] In another embodiment, the compounds of the present invention have EL IC50 values < 50 11M.
[0044] In another embodiment, the compounds of the present invention have EL IC50 values < 25 11M.
[0045] In another embodiment, the compounds of the present invention have EL IC50 values < 10 11M.
II. OTHER EMBODIMENTS OF THE INVENTION
[0046] In another embodiment, the present invention provides a composition comprising at least one of the compounds of the present invention or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof.
[0047] In another embodiment, the present invention provides a pharmaceutical composition comprising a pharmaceutically acceptable carrier and at least one of the compounds of the present invention or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof.
[0048] In another embodiment, the present invention provides a pharmaceutical composition, comprising: a pharmaceutically acceptable carrier and a therapeutically effective amount of at least one of the compounds of the present invention or a stereoisomer, a tautomer, a pharmaceutically acceptable salt, or a solvate thereof.
[0049] In another embodiment, the present invention provides a process for making a compound of the present invention.
[0050] In another embodiment, the present invention provides an intermediate for making a compound of the present invention.
[0051] In another embodiment, the present invention provides a pharmaceutical composition as defined above further comprising additional therapeutic agent(s).
[0052] In another embodiment, the present invention provides a method for the treatment and/or prophylaxis of dyslipidemias and the sequelae thereof comprising administering to a patient in need of such treatment and/or prophylaxis a therapeutically effective amount of at least one of the compounds of the present invention, alone, or, optionally, in combination with another compound of the present invention and/or at least one other type of therapeutic agent.
[0053] Examples of diseases or disorders associated with the activity of endothelial lipase that can be prevented, modulated, or treated according to the present invention include, but are not limited to, atherosclerosis, coronary heart disease, coronary artery disease, coronary vascular disease, cerebrovascular disorders, Alzheimer's disease, venous thrombosis, peripheral vascular disease, dyslipidemia, hyperbetalipoproteinemia, hypoalphalipoproteinemia, hypercholesterolemia, hypertriglyceridemia, familial- hypercholesterolemia, cardiovascular disorders, angina, ischemia, cardiac ischemia, stroke, myocardial infarction, reperfusion injury, angioplastic restenosis, hypertension, vascular complications of diabetes, obesity or endotoxemia.
[0054] In one embodiment, the present invention provides a method for the treatment and/or prophylaxis of atherosclerosis, coronary heart disease, cerebrovascular disorders and dyslipidemia, comprising administering to a patient in need of such treatment and/or prophylaxis a therapeutically effective amount of at least one of the compounds of the present invention, alone, or, optionally, in combination with another compound of the present invention and/or at least one other type of therapeutic agent.
[0055] In another embodiment, the present invention provides a compound of the present invention for use in therapy.
[0056] In another embodiment, the present invention provides a compound of the present invention for use in therapy for the treatment and/or prophylaxis of dyslipidemias and the sequelae thereof.
[0057] In another embodiment, the present invention also provides the use of a compound of the present invention for the manufacture of a medicament for the treatment and/or prophylaxis of dyslipidemias and the sequelae thereof.
[0058] In another embodiment, the present invention provides a method for the treatment and/or prophylaxis of dyslipidemias and the sequelae thereof, comprising: administering to a patient in need thereof a therapeutically effective amount of a first and second therapeutic agent, wherein the first therapeutic agent is a compound of the present invention.
[0059] In another embodiment, the present invention provides a combined
preparation of a compound of the present invention and additional therapeutic agent(s) for simultaneous, separate or sequential use in therapy.
[0060] In another embodiment, the present invention provides a combined
preparation of a compound of the present invention and additional therapeutic agent(s) for simultaneous, separate or sequential use in the treatment and/or prophylaxis of dyslipidemias and the sequelae thereof.
[0061] The compounds of the present invention may be employed in combination with additional therapeutic agent(s) selected from one or more, preferably one to three, of the following therapeutic agents: anti-atherosclerotic agents, anti-dyslipidemic agents, anti-diabetic agents, anti-hyperglycemic agents, anti-hyperinsulinemic agents, anti-thrombotic agents, anti-retinopathic agents, anti-neuropathic agents,
anti-nephropathic agents, anti-ischemic agents, anti-hypertensive agents, anti-obesity
agents, anti-hyperlipidemic agents, anti-hypertriglyceridemic agents, anti-hypercholesterolemic agents, anti-restenotic agents, anti-pancreatic agents, lipid lowering agents, anorectic agents, memory enhancing agents, anti-dementia agents, cognition promoting agents, appetite suppressants, treatments for heart failure, treatments for peripheral arterial disease, treatment for malignant tumors, and anti-inflammatory agents.
[0062] In another embodiment, additional therapeutic agent(s) used in combined pharmaceutical compositions or combined methods or combined uses, are selected from one or more, preferably one to three, of the following therapeutic agents in treating atherosclerosis: anti-hyperlipidemic agents, plasma HDL-raising agents,
anti-hypercholesterolemic agents, cholesterol biosynthesis inhibitors (such as HMG CoA reductase inhibitors), acyl-coenzyme Axholesterol acyltransferase (ACAT) inhibitors, LXR agonist, probucol, raloxifene, nicotinic acid, niacinamide, cholesterol absorption inhibitors, bile acid sequestrants (such as anion exchange resins, or quaternary amines (e.g., cholestyramine or colestipol)), low density lipoprotein receptor inducers, clofibrate, fenofibrate, benzofibrate, cipofibrate, gemfibrizol, vitamin B6, vitamin B12, anti-oxidant vitamins, β-blockers, anti-diabetes agents, angiotensin II antagonists, angiotensin converting enzyme inhibitors, platelet aggregation inhibitors, fibrinogen receptor antagonists, aspirin or fibric acid derivatives.
[0063] In another embodiment, additional therapeutic agent(s) used in combined pharmaceutical compositions or combined methods or combined uses, are selected from one or more, preferably one to three, of the following therapeutic agents in treating cholesterol biosynthesis inhibitor, particularly an HMG-CoA reductase inhibitor.
Examples of suitable HMG-CoA reductase inhibitors include, but are not limited to, lovastatin, simvastatin, pravastatin, fluvastatin, atorvastatin, and rivastatin.
[0064] The present invention may be embodied in other specific forms without departing from the spirit or essential attributes thereof. This invention encompasses all combinations of preferred aspects of the invention noted herein. It is understood that any and all embodiments of the present invention may be taken in conjunction with any other embodiment or embodiments to describe additional embodiments. It is also understood that each individual element of the embodiments is its own independent embodiment.
Furthermore, any element of an embodiment is meant to be combined with any and all other elements from any embodiment to describe an additional embodiment.
III. CHEMISTRY
[0065] Throughout the specification and the appended claims, a given chemical formula or name shall encompass all stereo and optical isomers and racemates thereof where such isomers exist. Unless otherwise indicated, all chiral (enantiomeric and diastereomeric) and racemic forms are within the scope of the invention. Many geometric isomers of C=C double bonds, C=N double bonds, ring systems, and the like can also be present in the compounds, and all such stable isomers are contemplated in the present invention. Cis- and trans- (or E- and Z-) geometric isomers of the compounds of the present invention are described and may be isolated as a mixture of isomers or as separated isomeric forms. The present compounds can be isolated in optically active or racemic forms. Optically active forms may be prepared by resolution of racemic forms or by synthesis from optically active starting materials. All processes used to prepare compounds of the present invention and intermediates made therein are considered to be part of the present invention. When enantiomeric or diastereomeric products are prepared, they may be separated by conventional methods, for example, by
chromatography or fractional crystallization. Depending on the process conditions the end products of the present invention are obtained either in free (neutral) or salt form. Both the free form and the salts of these end products are within the scope of the invention. If so desired, one form of a compound may be converted into another form. A free base or acid may be converted into a salt; a salt may be converted into the free compound or another salt; a mixture of isomeric compounds of the present invention may be separated into the individual isomers. Compounds of the present invention, free form and salts thereof, may exist in multiple tautomeric forms, in which hydrogen atoms are transposed to other parts of the molecules and the chemical bonds between the atoms of the molecules are consequently rearranged. It should be understood that all tautomeric forms, insofar as they may exist, are included within the invention.
[0066] As used herein, the term "alkyl" or "alkylene" is intended to include both branched and straight-chain saturated aliphatic hydrocarbon groups having the specified number of carbon atoms. For example, "C^ to C^Q alkyl" or "C ^Q alkyl" (or alkylene), is
intended to include C , C2, C3, C4, C5, Cg, C7, Cg, C9, and C Q alkyl groups.
Additionally, for example, "C^ to Cg alkyl" or "Ci _g alkyl" denotes alkyl having 1 to 6 carbon atoms. Alkyl group can be unsubstituted or substituted with at least one hydrogen being replaced by another chemical group. Example alkyl groups include, but are not limited to, methyl (Me), ethyl (Et), propyl (e.g., n-propyl and isopropyl), butyl (e.g., n-butyl, isobutyl, t-butyl), and pentyl (e.g., n-pentyl, isopentyl, neopentyl). When "Cg alkyl" or "C0 alkylene" is used, it is intended to denote a direct bond.
[0067] "Alkenyl" or "alkenylene" is intended to include hydrocarbon chains of either straight or branched configuration having the specified number of carbon atoms and one or more, preferably one to two, carbon-carbon double bonds that may occur in any stable point along the chain. For example, "C2 to Cg alkenyl" or "C2_g alkenyl" (or alkenylene), is intended to include C2, C3, C4, C5, and C6 alkenyl groups. Examples of alkenyl include, but are not limited to, ethenyl, 1-propenyl, 2-propenyl, 2-butenyl, 3-butenyl, 2-pentenyl, 3, pentenyl, 4-pentenyl, 2-hexenyl, 3-hexenyl, 4-hexenyl, 5-hexenyl, 2-methyl-2-propenyl, and 4-methyl-3-pentenyl.
[0068] "Alkynyl" or "alkynylene" is intended to include hydrocarbon chains of either straight or branched configuration having one or more, preferably one to three, carbon-carbon triple bonds that may occur in any stable point along the chain. For example, "C2 to C6 alkynyl" or "C2-6 alkynyl" (or alkynylene), is intended to include C2, C3, C4, C5, and C6 alkynyl groups; such as ethynyl, propynyl, butynyl, pentynyl, and hexynyl.
[0069] The term "alkoxy" or "alkyloxy" refers to an -O-alkyl group. "C to Cg alkoxy" or "Ci _g alkoxy" (or alkyloxy), is intended to include C , C2, C3, C4, C5, and Cg alkoxy groups. Example alkoxy groups include, but are not limited to, methoxy, ethoxy, propoxy (e.g., n-propoxy and isopropoxy), and t-butoxy. Similarly, "alkylthio" or
"thioalkoxy" represents an alkyl group as defined above with the indicated number of carbon atoms attached through a sulphur bridge; for example methyl-S- and ethyl-S-.
[0070] "Halo" or "halogen" includes fluoro, chloro, bromo, and iodo. "Haloalkyl" is intended to include both branched and straight-chain saturated aliphatic hydrocarbon groups having the specified number of carbon atoms, substituted with 1 or more halogens. Examples of haloalkyl include, but are not limited to, fluoromethyl,
difluoromethyl, trifluoromethyl, trichloromethyl, pentafluoroethyl, pentachloroethyl, 2,2,2-trifluoroethyl, heptafluoropropyl, and heptachloropropyl. Examples of haloalkyl also include "fluoroalkyl" that is intended to include both branched and straight-chain saturated aliphatic hydrocarbon groups having the specified number of carbon atoms, substituted with 1 or more fluorine atoms.
[0071] "Haloalkoxy" or "haloalkyloxy" represents a haloalkyl group as defined above with the indicated number of carbon atoms attached through an oxygen bridge. For example, "C^ to C6 haloalkoxy" or "C^.g haloalkoxy", is intended to include C1 ? C2, C3,
C4, C5, and Cg haloalkoxy groups. Examples of haloalkoxy include, but are not limited to, trifluoromethoxy, 2,2,2-trifluoroethoxy, and pentafluorothoxy. Similarly,
"haloalkylthio" or "thiohaloalkoxy" represents a haloalkyl group as defined above with the indicated number of carbon atoms attached through a sulphur bridge; for example trifluoromethyl-S-, and pentafluoroethyl-S-.
[0072] The term "cycloalkyl" refers to cyclized alkyl groups, including mono-, bi- or poly-cyclic ring systems. "C3 to C7 cycloalkyl" or "C3_7 cycloalkyl" is intended to include C3, C4, C5, C6, and C7 cycloalkyl groups. Example cycloalkyl groups include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, and norbornyl. Branched cycloalkyl groups such as 1-methylcyclopropyl and 2-methylcyclopropyl are included in the definition of "cycloalkyl".
[0073] As used herein, "carbocycle," "carbocyclyl," or "carbocyclic residue" is intended to mean any stable 3-, 4-, 5-, 6-, 7-, or 8-membered monocyclic or bicyclic or 7-, 8-, 9-, 10-, 1 1-, 12-, or 13-membered bicyclic or tricyclic ring, any of which may be saturated, partially unsaturated, unsaturated or aromatic. Examples of such carbocycles include, but are not limited to, cyclopropyl, cyclobutyl, cyclobutenyl, cyclopentyl, cyclopentenyl, cyclohexyl, cycloheptenyl, cycloheptyl, cycloheptenyl, adamantyl, cyclooctyl, cyclooctenyl, cyclooctadienyl, [3.3.0]bicyclooctane, [4.3.0]bicyclononane, [4.4.0]bicyclodecane (decalin), [2.2.2]bicyclooctane, fluorenyl, phenyl, naphthyl, indanyl, adamantyl, anthracenyl, and tetrahydronaphthyl (tetralin). As shown above, bridged rings are also included in the definition of carbocycle (e.g., [2.2.2]bicyclooctane). Preferred carbocycles, unless otherwise specified, are cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, phenyl, indanyl, and tetrahydronaphthyl. When the term "carbocycle" is used, it is intended to include "aryl." A bridged ring occurs when one or more, preferably
one to three, carbon atoms link two non-adjacent carbon atoms. Preferred bridges are one or two carbon atoms. It is noted that a bridge always converts a monocyclic ring into a tricyclic ring. When a ring is bridged, the substituents recited for the ring may also be present on the bridge.
[0074] As used herein, the term "bicyclic carbocycle" or "bicyclic carbocyclic group" is intended to mean a stable 9- or 10-membered carbocyclic ring system that contains two fused rings and consists of carbon atoms. Of the two fused rings, one ring is a benzo ring fused to a second ring; and the second ring is a 5- or 6-membered carbon ring which is saturated, partially unsaturated, or unsaturated. The bicyclic carbocyclic group may be attached to its pendant group at any carbon atom which results in a stable structure. The bicyclic carbocyclic group described herein may be substituted on any carbon if the resulting compound is stable. Examples of a bicyclic carbocyclic group are, but not limited to, naphthyl, 1 ,2-dihydronaphthyl, 1 ,2,3,4-tetrahydronaphthyl, and indanyl.
[0075]
[0076] "Aryl" groups refer to monocyclic or polycyclic aromatic hydrocarbons, including, for example, phenyl, naphthyl, and phenanthranyl. Aryl moieties are well known and described, for example, in Lewis, R.J., ed., Hawley's Condensed Chemical Dictionary, 13th Edition, John Wiley & Sons, Inc., New York (1997). "Cg or C Q aryl" or "Cg.i o aryl" refers to phenyl and naphthyl. Unless otherwise specified, "aryl", "Cg or Ci o aryl," "Cg.i Q aryl," or "aromatic residue" may be unsubstituted or substituted with 1 to 5 groups, preferably 1 to 3 groups, selected from -OH, -OCH3, -CI, -F, -Br, -I, -CN, -N02, -NH2, -N(CH3)H, -N(CH3)2, -CF3, -OCF3, -C(0)CH3, -SCH3, -S(0)CH3, -S(0)2CH3, -CH3, -CH2CH3, -C02H, and -C02CH3.
[0077] The term "benzyl," as used herein, refers to a methyl group on which one of the hydrogen atoms is replaced by a phenyl group, wherein said phenyl group may optionally be substituted with 1 to 5 groups, preferably 1 to 3 groups, OH, OCH3, CI, F, Br, I, CN, N02, NH2, N(CH3)H, N(CH3)2, CF3, OCF3, C(=0)CH3, SCH3, S(=0)CH3, S(=0)2CH3, CH3, CH2CH3, C02H, and C02CH3.
[0078] As used herein, the term "heterocycle," "heterocyclyl," or "heterocyclic group" is intended to mean a stable 3-, 4-, 5-, 6-, or 7-membered monocyclic or bicyclic or 7-, 8-, 9-, 10-, 1 1-, 12-, 13-, or 14-membered polycyclic heterocyclic ring that is saturated,
partially unsaturated, or fully unsaturated, and that contains carbon atoms and 1, 2, 3 or 4 heteroatoms independently selected from the group consisting of N, O and S; and including any polycyclic group in which any of the above-defined heterocyclic rings is fused to a benzene ring. The nitrogen and sulfur heteroatoms may optionally be oxidized (i.e., N→0 and S(0)p, wherein p is 0, 1 or 2). The nitrogen atom may be substituted or unsubstituted (i.e., N or NR wherein R is H or another substituent, if defined). The heterocyclic ring may be attached to its pendant group at any heteroatom or carbon atom that results in a stable structure. The heterocyclic rings described herein may be substituted on carbon or on a nitrogen atom if the resulting compound is stable. A nitrogen in the heterocycle may optionally be quaternized. It is preferred that when the total number of S and O atoms in the heterocycle exceeds 1 , then these heteroatoms are not adjacent to one another. It is preferred that the total number of S and O atoms in the heterocycle is not more than 1. When the term "heterocycle" is used, it is intended to include heteroaryl.
[0079] Examples of heterocycles include, but are not limited to, acridinyl, azetidinyl, azocinyl, benzimidazolyl, benzofuranyl, benzothiofuranyl, benzothiophenyl,
benzoxazolyl, benzoxazolinyl, benzthiazolyl, benztriazolyl, benztetrazolyl,
benzisoxazolyl, benzisothiazolyl, benzimidazolinyl, carbazolyl, 4aH-carbazolyl, carbolinyl, chromanyl, chromenyl, cinnolinyl, decahydroquinolinyl,
2H,6H-l,5,2-dithiazinyl, dihydrofuro[2,3-£]tetrahydrofuran, furanyl, furazanyl, imidazolidinyl, imidazolinyl, imidazolyl, lH-indazolyl, imidazolopyridinyl, indolenyl, indolinyl, indolizinyl, indolyl, 3H-indolyl, isatinoyl, isobenzofuranyl, isochromanyl, isoindazolyl, isoindolinyl, isoindolyl, isoquinolinyl, isothiazolyl, isothiazolopyridinyl, isoxazolyl, isoxazolopyridinyl, methylenedioxyphenyl, morpholinyl, naphthyridinyl, octahydroisoquinolinyl, oxadiazolyl, 1,2,3-oxadiazolyl, 1,2,4-oxadiazolyl,
1,2,5-oxadiazolyl, 1,3,4-oxadiazolyl, oxazolidinyl, oxazolyl, oxazolopyridinyl, oxazolidinylperimidinyl, oxindolyl, pyrimidinyl, phenanthridinyl, phenanthrolinyl, phenazinyl, phenothiazinyl, phenoxathiinyl, phenoxazinyl, phthalazinyl, piperazinyl, piperidinyl, piperidonyl, 4-piperidonyl, piperonyl, pteridinyl, purinyl, pyranyl, pyrazinyl, pyrazolidinyl, pyrazolinyl, pyrazolopyridinyl, pyrazolyl, pyridazinyl, pyridooxazolyl, pyridoimidazolyl, pyridothiazolyl, pyridinyl, pyrimidinyl, pyrrolidinyl, pyrrolinyl, 2-pyrrolidonyl, 2H-pyrrolyl, pyrrolyl, quinazolinyl, quinolinyl, 4H-quinolizinyl,
quinoxalinyl, quinuclidinyl, tetrazolyl, tetrahydrofuranyl, tetrahydroisoquinolinyl, tetrahydroquinolinyl, 6H-l,2,5-thiadiazinyl, 1,2,3-thiadiazolyl, 1,2,4-thiadiazolyl, 1,2,5-thiadiazolyl, 1,3,4-thiadiazolyl, thianthrenyl, thiazolyl, thienyl, thiazolopyridinyl, thienothiazolyl, thienooxazolyl, thienoimidazolyl, thiophenyl, triazinyl, 1,2,3-triazolyl, 1,2,4-triazolyl, 1,2,5-triazolyl, 1,3,4-triazolyl, and xanthenyl. Also included are fused ring and spiro compounds containing, for example, the above heterocycles.
[0080] Examples of 5- to 10-membered heterocycles include, but are not limited to, pyridinyl, furanyl, thienyl, pyrrolyl, pyrazolyl, pyrazinyl, piperazinyl, piperidinyl, imidazolyl, imidazolidinyl, indolyl, tetrazolyl, isoxazolyl, morpholinyl, oxazolyl, oxadiazolyl, oxazolidinyl, tetrahydrofuranyl, thiadiazinyl, thiadiazolyl, thiazolyl, triazinyl, triazolyl, benzimidazolyl, lH-indazolyl, benzofuranyl, benzothiofuranyl, benztetrazolyl, benzotriazolyl, benzisoxazolyl, benzoxazolyl, oxindolyl, benzoxazolinyl, benzthiazolyl, benzisothiazolyl, isatinoyl, isoquinolinyl, octahydroisoquinolinyl, tetrahydroisoquinolinyl, tetrahydroquinolinyl, isoxazolopyridinyl, quinazolinyl, quinolinyl, isothiazolopyridinyl, thiazolopyridinyl, oxazolopyridinyl, imidazolopyridinyl, and pyrazolopyridinyl.
[0081] Examples of 5- to 6-membered heterocycles include, but are not limited to, pyridinyl, furanyl, thienyl, pyrrolyl, pyrazolyl, pyrazinyl, piperazinyl, piperidinyl, imidazolyl, imidazolidinyl, indolyl, tetrazolyl, isoxazolyl, morpholinyl, oxazolyl, oxadiazolyl, oxazolidinyl, tetrahydrofuranyl, thiadiazinyl, thiadiazolyl, thiazolyl, triazinyl, and triazolyl. Also included are fused ring and spiro compounds containing, for example, the above heterocycles.
[0082] As used herein, the term "bicyclic heterocycle" or "bicyclic heterocyclic group" is intended to mean a stable 9- or 10-membered heterocyclic ring system which contains two fused rings and consists of carbon atoms and 1, 2, 3, or 4 heteroatoms independently selected from the group consisting of N, O and S. Of the two fused rings, one ring is a 5- or 6-membered monocyclic aromatic ring comprising a 5-membered heteroaryl ring, a 6-membered heteroaryl ring or a benzo ring, each fused to a second ring. The second ring is a 5- or 6-membered monocyclic ring which is saturated, partially unsaturated, or unsaturated, and comprises a 5-membered heterocycle, a 6-membered heterocycle or a carbocycle (provided the first ring is not benzo when the second ring is a carbocycle).
[0083] The bicyclic heterocyclic group may be attached to its pendant group at any heteroatom or carbon atom which results in a stable structure. The bicyclic heterocyclic group described herein may be substituted on carbon or on a nitrogen atom if the resulting compound is stable. It is preferred that when the total number of S and O atoms in the heterocycle exceeds 1, then these heteroatoms are not adjacent to one another. It is preferred that the total number of S and O atoms in the heterocycle is not more than 1.
[0084] Examples of a bicyclic heterocyclic group are, but not limited to, quinolinyl, isoquinolinyl, phthalazinyl, quinazolinyl, indolyl, isoindolyl, indolinyl, lH-indazolyl, benzimidazolyl, 1 ,2,3,4-tetrahydroquinolinyl, 1 ,2,3,4-tetrahydroisoquinolinyl,
5,6,7,8-tetrahydro-quinolinyl, 2,3-dihydro-benzofuranyl, chromanyl,
1 ,2,3,4-tetrahydro-quinoxalinyl, and 1 ,2,3,4-tetrahydro-quinazolinyl.
[0085] As used herein, the term "aromatic heterocyclic group" or "heteroaryl" is intended to mean stable monocyclic and polycyclic aromatic hydrocarbons that include at least one heteroatom ring member such as sulfur, oxygen, or nitrogen. Heteroaryl groups include, without limitation, pyridyl, pyrimidinyl, pyrazinyl, pyridazinyl, triazinyl, furyl, quinolyl, isoquinolyl, thienyl, imidazolyl, thiazolyl, indolyl, pyrroyl, oxazolyl, benzofuryl, benzothienyl, benzthiazolyl, isoxazolyl, pyrazolyl, triazolyl, tetrazolyl, indazolyl, 1,2,4-thiadiazolyl, isothiazolyl, purinyl, carbazolyl, benzimidazolyl, indolinyl, benzodioxolanyl, and benzodioxane. Heteroaryl groups are substituted or unsubstituted. The nitrogen atom is substituted or unsubstituted (i.e., N or NR wherein R is H or another substituent, if defined). The nitrogen and sulfur heteroatoms may optionally be oxidized (i.e., N→0 and S(0)p, wherein p is 0, 1 or 2).
[0086] Bridged rings are also included in the definition of heterocycle. A bridged ring occurs when one or more, preferably one to three, atoms (i.e., C, O, N, or S) link two non-adjacent carbon or nitrogen atoms. Examples of bridged rings include, but are not limited to, one carbon atom, two carbon atoms, one nitrogen atom, two nitrogen atoms, and a carbon-nitrogen group. It is noted that a bridge always converts a monocyclic ring into a tricyclic ring. When a ring is bridged, the substituents recited for the ring may also be present on the bridge.
[0087] The term "counter ion" is used to represent a negatively charged species such as chloride, bromide, hydroxide, acetate, and sulfate.
[0088] When a dotted ring is used within a ring structure, this indicates that the ring structure may be saturated, partially saturated or unsaturated.
[0089] As referred to herein, the term "substituted" means that at least one hydrogen atom is replaced with a non-hydrogen group, provided that normal valencies are maintained and that the substitution results in a stable compound. When a substituent is keto (i.e., =0), then 2 hydrogens on the atom are replaced. Keto substituents are not present on aromatic moieties. When a ring system (e.g., carbocyclic or heterocyclic) is said to be substituted with a carbonyl group or a double bond, it is intended that the carbonyl group or double bond be part (i.e., within) of the ring. Ring double bonds, as used herein, are double bonds that are formed between two adjacent ring atoms (e.g., C=C, C=N, or N=N).
[0090] In cases wherein there are nitrogen atoms (e.g. , amines) on compounds of the present invention, these may be converted to N-oxides by treatment with an oxidizing agent (e.g., mCPBA and/or hydrogen peroxides) to afford other compounds of this invention. Thus, shown and claimed nitrogen atoms are considered to cover both the shown nitrogen and its N-oxide (N— >0) derivative.
[0091] When any variable occurs more than one time in any constituent or formula for a compound, its definition at each occurrence is independent of its definition at every other occurrence. Thus, for example, if a group is shown to be substituted with 0-3 R groups, then said group may optionally be substituted with up to three R groups, and at each occurrence R is selected independently from the definition of R.
[0092] Also, combinations of substituents and/or variables are permissible only if such combinations result in stable compounds.
[0093] When a bond to a substituent is shown to cross a bond connecting two atoms in a ring, then such substituent may be bonded to any atom on the ring. When a substituent is listed without indicating the atom in which such substituent is bonded to the rest of the compound of a given formula, then such substituent may be bonded via any atom in such substituent. Combinations of substituents and/or variables are permissible only if such combinations result in stable compounds.
[0094] As a person of ordinary skill in the art would be able to understand that imine and carbonyl groups in a molecule may tautomerize to their enamine and enol forms, and
the double bond can exist as geometrical (E and Z) isomers as shown in the following e

tautomerization
tautomerization

[0095] Thus, this disclosure is intended to cover all possible tautomers even when a structure depicts only one of them.
[0096] The phrase "pharmaceutically acceptable" is employed herein to refer to those compounds, materials, compositions, and/or dosage forms that are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, allergic response, and/or other problem or complication, commensurate with a reasonable benefit/risk ratio.
[0097] As used herein, "pharmaceutically acceptable salts" refer to derivatives of the disclosed compounds wherein the parent compound is modified by making acid or base salts thereof. Examples of pharmaceutically acceptable salts include, but are not limited to, mineral or organic acid salts of basic groups such as amines; and alkali or organic salts of acidic groups such as carboxylic acids. The pharmaceutically acceptable salts include the conventional non-toxic salts or the quaternary ammonium salts of the parent compound formed, for example, from non-toxic inorganic or organic acids. For example, such conventional non-toxic salts include those derived from inorganic acids such as hydrochloric, hydrobromic, sulfuric, sulfamic, phosphoric, and nitric; and the salts prepared from organic acids such as acetic, propionic, succinic, glycolic, stearic, lactic, malic, tartaric, citric, ascorbic, pamoic, maleic, hydroxymaleic, phenylacetic, glutamic, benzoic, salicylic, sulfanilic, 2-acetoxybenzoic, fumaric, toluenesulfonic,
methanesulfonic, ethane disulfonic, oxalic, and isethionic.
[0098] The pharmaceutically acceptable salts of the present invention can be synthesized from the parent compound that contains a basic or acidic moiety by conventional chemical methods. Generally, such salts can be prepared by reacting the
free acid or base forms of these compounds with a stoichiometric amount of the appropriate base or acid in water or in an organic solvent, or in a mixture of the two; generally, nonaqueous media like ether, ethyl acetate, ethanol, isopropanol, or acetonitrile are preferred. Lists of suitable salts are found in Remington: The Science and Practice of Pharmacy, 22nd Edition, Allen, L. V. Jr., Ed.; Pharmaceutical Press, London, UK (2012), the disclosure of which is hereby incorporated by reference.
[0099] In addition, compounds of Formula (I), Formula (II), or Formula (III) may have prodrug forms. Any compound that will be converted in vivo to provide the bioactive agent (i.e., a compound of Formula (I), Formula (II) or Formula (III)) is a prodrug within the scope and spirit of the invention. Various forms of prodrugs are well known in the art. For examples of such prodrug derivatives, see:
a) Bundgaard, FL, ed., Design of Prodrugs, Elsevier (1985), and Widder, K. et al, eds., Methods in Enzymology, 112:309-396, Academic Press (1985);
b) Bundgaard, FL, Chapter 5, "Design and Application of Prodrugs,"
Krosgaard-Larsen, P. et al, eds., A Textbook of Drug Design and Development, pp. 113- 191, Harwood Academic Publishers (1991);
c) Bundgaard, FL, Adv. Drug Deliv. Rev., 8: 1-38 (1992);
d) Bundgaard, H. et al, J. Pharm. Sci., 77:285 (1988);
e) Kakeya, N. et al, Chem. Pharm. Bull, 32:692 (1984); and
f) Rautio, J (Editor). Prodrugs and Targeted Delivery (Methods and
Principles in Medicinal Chemistry) , Vol 47, Wiley -VCH, 2011.
[00100] Compounds containing a carboxy group can form physiologically
hydrolyzable esters that serve as prodrugs by being hydrolyzed in the body to yield Formula (I), (Ila), (lib), (Ilia) or (Illb) compounds per se. Such prodrugs are preferably administered orally since hydrolysis in many instances occurs principally under the influence of the digestive enzymes. Parenteral administration may be used where the ester per se is active, or in those instances where hydrolysis occurs in the blood.
Examples of physiologically hydrolyzable esters of compounds of the present invention include C\ to Cg alkyl, C\ to Cg alkylbenzyl, 4-methoxybenzyl, indanyl, phthalyl, methoxymethyl, Ci .g alkanoyloxy-C^.g alkyl (e.g., acetoxymethyl, pivaloyloxymethyl or propionyloxymethyl), C\ to Cg alkoxycarbonyloxy-Cj to Cg alkyl (e.g.,
methoxycarbonyl-oxymethyl or ethoxycarbonyloxymethyl, glycyloxymethyl,
phenylglycyloxymethyl, (5-methyl-2-oxo-l,3-dioxolen-4-yl)-methyl), and other well known physiologically hydrolyzable esters used, for example, in the penicillin and cephalosporin arts. Such esters may be prepared by conventional techniques known in the art.
[00101] Preparation of prodrugs is well known in the art and described in, for example, King, F.D., ed., Medicinal Chemistry: Principles and Practice, The Royal Society of Chemistry, Cambridge, UK (2nd edition, reproduced, 2006); Testa, B. et al, Hydrolysis in Drug and Prodrug Metabolism. Chemistry, Biochemistry and Enzymology, VCHA and Wiley- VCH, Zurich, Switzerland (2003); Wermuth, C.G., ed., The Practice of Medicinal Chemistry, 3rd edition, Academic Press, San Diego, CA (2008).
[00102] The present invention is intended to include all isotopes of atoms occurring in the present compounds. Isotopes include those atoms having the same atomic number but different mass numbers. By way of general example and without limitation, isotopes of hydrogen include deuterium and tritium. Isotopes of carbon include 13C and 14C.
Isotopically-labeled compounds of the invention can generally be prepared by
conventional techniques known to those skilled in the art or by processes analogous to those described herein, using an appropriate isotopically-labeled reagent in place of the non-labeled reagent otherwise employed. Such compounds have a variety of potential uses, e.g., as standards and reagents in determining the ability of a potential
pharmaceutical compound to bind to target proteins or receptors, or for imaging compounds of this invention bound to biological receptors in vivo or in vitro.
[00103] "Stable compound" and "stable structure" are meant to indicate a compound that is sufficiently robust to survive isolation to a useful degree of purity from a reaction mixture, and formulation into an efficacious therapeutic agent. It is preferred that compounds of the present invention do not contain a N-halo, S(0)2H, or S(0)H group.
[00104] The term "solvate" means a physical association of a compound of this invention with one or more solvent molecules, whether organic or inorganic. This physical association includes hydrogen bonding. In certain instances the solvate will be capable of isolation, for example when one or more, preferably one to three, solvent molecules are incorporated in the crystal lattice of the crystalline solid. The solvent molecules in the solvate may be present in a regular arrangement and/or a non-ordered
arrangement. The solvate may comprise either a stoichiometric or nonstoichiometric amount of the solvent molecules. "Solvate" encompasses both solution-phase and isolable solvates. Exemplary solvates include, but are not limited to, hydrates, ethanolates, methanolates, and isopropanolates. Methods of solvation are generally known in the art.
[00105] Abbreviations as used herein, are defined as follows: "1 x" for once, "2 x" for twice, "3 x" for thrice, "°C" for degrees Celsius, "eq" for equivalent or equivalents, "g" for gram or grams, "mg" for milligram or milligrams, "L" for liter or liters, "mL" for milliliter or milliliters, "μί" for microliter or microliters, "N" for normal, "M" for molar, "nM" for nanomolar, "mol" for mole or moles, "mmol" for millimole or millimoles,
"min" for minute or minutes, "h" for hour or hours, "rt" for room temperature, "RT" for retention time, "arm" for atmosphere, "psi" for pounds per square inch, "cone." for concentrate, "sat" or "sat'd " for saturated, "MW" for molecular weight, "mp" for melting point, "MS" or "Mass Spec" for mass spectrometry, "ESI" for electrospray ionization mass spectroscopy, "HR" for high resolution, "HRMS" for high resolution mass spectrometry, "LCMS" for liquid chromatography mass spectrometry, "HPLC" for high pressure liquid chromatography, "RP HPLC" for reverse phase HPLC, "TLC" or "tic" for thin layer chromatography, "NMR" for nuclear magnetic resonance spectroscopy, "nOe" for nuclear Overhauser effect spectroscopy, M lH" for proton, "δ" for delta, "s" for singlet, "d" for doublet, "t" for triplet, "q" for quartet, "m" for multiplet, "br" for broad, "Hz" for hertz, and "α", "β", "R", "S", "E", and "Z" are stereochemical designations familiar to one skilled in the art.
AcOH or HO Ac acetic acid
AICI3 aluminum chloride
Alk alkyl
BBr3 boron tribromide
BCI3 boron trichloride
Bn benzyl
Boc fert-butyloxycarbonyl
BOP reagent benzotriazol-l-yloxytris(dimethylamino)phosphonium hexafluorophosphate
Bu butyl
z'-Bu isobutyl
t-Bu tert-butyl
t-BuOH tert-butanol
Cbz carbobenzyloxy
CDCI3 deutero-chloroform
CD3OD deutero-methanol
CH2Cl2 dichloromethane
CH3CN or ACN acetonitrile
CHCI3 chloroform
C02 carbon dioxide
mCPBA or m-CPBA meto-chloroperbenzoic acid
Cs2C03 cesium carbonate
Cu(OAc)2 copper (II) acetate
DBU 1 ,8-diazabicyclo [5.4.0]undec-7-ene
DCE 1 ,2-dichloroethane
DCM dichloromethane
DEA diethylamine
DIC or DIPCDI diisopropylcarbodiimide
DIEA, DIPEA or diisopropylethylamine
Hunig's base
DMAP 4-dimethylaminopyridine
DME 1 ,2-dimethoxyethane
DMF dimethyl formamide
DMSO dimethyl sulfoxide
cDNA complimentary DNA
Dppp (R)-(+)- 1 ,2-bis(diphenylphosphino)propane
EDC N-(3-dimthylaminopropyl)-N'-ethylcarbodiimide
EDTA ethylenediammetetraacetic acid
Et ethyl
Et3N or TEA triethylamine
Et20 diethyl ether
EtOAc ethyl acetate
EtOH ethanol
HATU (2-(7-aza- 1 H-benzotriazole- 1 -yl)- 1 , 1 ,3 ,3 -tetramethyluronium hexafluorophosphate)
HC1 hydrochloric acid
HOBt or HOBT 1-hydroxybenzotriazole
HPLC high-performance liquid chromatography
H3PO4 phosphoric acid
H2SO4 sulfuric acid
K2CO3 potassium carbonate
KOAc potassium acetate
K3PO4 potassium phosphate
LAH lithium aluminum hydride
LDA lithium diisopropylamide
LG leaving group
LiOH lithium hydroxide
Me methyl
MeOH methanol
MgS04 magnesium sulfate
MsOH or MSA methylsulfonic acid
NaCl sodium chloride
Na2C03 sodium carbonate
NaH sodium hydride
NaHB(OAc)3 sodium triacetoxyborohydride
NaHCC"3 sodium bicarbonate
NaHMDS sodium hexamethyldisilazane
NaOH sodium hydroxide
NaOMe sodium methoxide
Na2S03 sodium sulfite
Na2S04 sodium sulfate
NBS N-bromosuccinimide
NH3 ammonia
NH4CI ammonium chloride
NH4OAc ammonium acetate
NH4OH ammonium hydroxide
OTf trifiate or trifluoromethanesulfonate
Pd2(dba)3 tris(dibenzylideneacetone)dipalladium(0)
Pd(OAc)2 palladium(II) acetate
Pd/C palladium on carbon
Pd(dppf)Cl2 [1,1 '-bis(diphenylphosphino)- ferrocene]dichloropalladium(II)
Pli3PCl2 triphenylphosphine dichloride
PG protecting group
Ph phenyl
PMB p-methoxybenzyl
POCI3 phosphorus oxychloride
pr propyl
/_pr isopropyl
z'-PrOH or IPA isopropanol
PS polystyrene
PS-Pd(Ph3)4 tetrakis(triphenylphosphine)palladium (0) on polystyrene support
PyBOP (benzotriazol- 1 -yloxy)tripyrrolidinophosphonium
hexafiuorophosphate
Si02 silica oxide
SnCl2 tin(II) chloride
TBAF tetra-n-butylammonium fluoride
TBAI tetra-n-butylammonium iodide
TEA triethylamine
TFA trifluoroacetic acid
THF tetrahydrofuran
TMSCHN2 trimethylsilyldiazomethane
T3P 1-propanephosphonic acid cyclic anhydride
Xantphos or X-Phos 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene
SYNTHESIS
[00106] The compounds of the present invention can be prepared in a number of ways known to one skilled in the art of organic synthesis. The compounds of the present invention can be synthesized using the methods described below, together with synthetic methods known in the art of synthetic organic chemistry, or by variations thereon as appreciated by those skilled in the art. Preferred methods include, but are not limited to, those described below. The reactions are performed in a solvent or solvent mixture appropriate to the reagents and materials employed and suitable for the transformations being affected. It will be understood by those skilled in the art of organic synthesis that the functionality present on the molecule should be consistent with the transformations proposed. This will sometimes require a judgment to modify the order of the synthetic steps or to select one particular process scheme over another in order to obtain a desired compound of the invention.
[00107] A particularly useful compendium of synthetic methods which may be applicable to the preparation of compounds of the present invention may be found in Larock, R.C., Comprehensive Organic Transformations, VCH Publishers, Inc., New York (1999). Preferred methods include, but are not limited to, those described below. All references cited herein are hereby incorporated in their entirety herein by reference.
[00108] The novel compounds of this invention may be prepared using the reactions and techniques described in this section. Also, in the description of the synthetic methods described below, it is to be understood that all proposed reaction conditions, including choice of solvent, reaction atmosphere, reaction temperature, duration of the experiment and workup procedures, are chosen to be the conditions standard for that reaction, which should be readily recognized by one skilled in the art. Restrictions to the substituents that
are compatible with the reaction conditions will be readily apparent to one skilled in the art and alternate methods must then be used.
[00109] It will also be recognized that another major consideration in the planning of any synthetic route in this field is the judicious choice of the protecting group used for protection of the reactive functional groups present in the compounds described in this invention. An authoritative account describing the many alternatives to the trained practitioner is Wuts, P. G. M. and Greene, T. W {Protective Groups in Organic Synthesis, 4th Edition, Wiley (2007)). GENERIC SCHEMES
Scheme 1

[00110] Step 1 describes the preparation of compounds of Formula (Gib) by reacting amides of Formula (Gla) with a sodium nitrite. A preferred solvent is water, and a preferred acid is acetic acid. Step 2
[00111] Step 2 describes the preparation of amides and ureas of Formula (I) by reacting compounds of the Formula (Gib) with an anhydride or isocyanate and Pd/C in a hydrogen atmosphere. Preferred solvents are polar protic solvents, such as MeOH and EtOH.

Step 1
[00112] Step 1 describes the preparation of compounds of Formula (I) by reacting a compound of Formula (G2a) with a sulfonylating reagent R5-S02C1. Preferred solvents are polar aprotic solvents (such as Ν,Ν-dimethylformamide ) and ethers (such as tetrahydrofuran, dioxane and the like). Preferred bases include metal hydrides (such as sodium hydride and the like), metal amides (such as sodium bis(trimethylsilyl)amide and lithium diisopropylamide and the like) and organic amines (such as DBU, triethylamine and the like).
Scheme 3
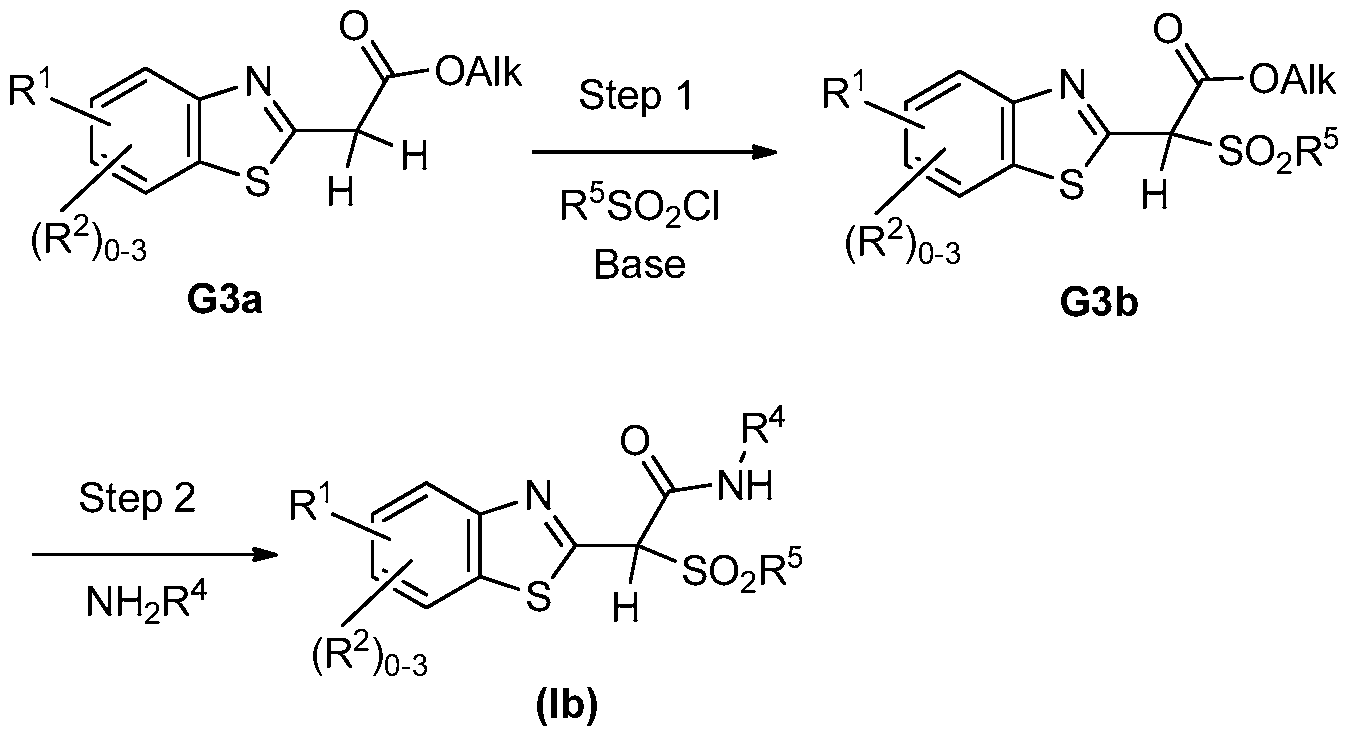
Step 1
[00113] Step 1 describes the preparation of a compound of Formula (G3b) from a compound of Formula (G3a) and is analogous to Step 1 in Scheme 1.
Step 2
[00114] Step 2 describes the preparation of a compound of Formula (I) from a compound of Formula (G3b) by direct exchange of an ester group with an amine of formula H2NR4. Preferred reaction solvents are polar aprotic solvents such as ethers (such as tetrahydrofuran, dioxane and the like), DMF and NMP. Bases such as an organic amine (such as triethylamine, diisopropylamine, DBU, 2,6-lutidine and the like) can be used.
GENERAL METHODS
[00115] The following methods were used in the exemplified Examples, except where noted otherwise.
[00116] Products were analyzed by reverse phase analytical HPLC carried out on a Shimadzu Analytical HPLC system running Discovery VP software using Method A:
PHENOMENEX® Luna CI 8 column (4.6 x 50 mm or 4.6 x 75 mm) eluted at 4 mL/min with a 2, 4 or 8 min gradient from 100% A to 100% B (A: 10% methanol, 89.9% water, 0.1% TFA; B: 10% water, 89.9% methanol, 0.1% TFA, UV 220 nm), or Method B:
PHENOMENEX® Luna CI 8 column (4.6 x 50 mm) eluted at 4 mL/min with a 4 min gradient from 100% A to 100% B (A: 10% acetonitrile, 89.9% water, 0.1% TFA; B: 10% water, 89.9% acetonitrile, 0.1% TFA, UV 220 nm) or Method C: PHENOMENEX® Luna C 18 column (4.6 x 50 mm or 4.6 x 75 mm) eluted at 4 mL/min with a 2, 4 or 8 min gradient from 100% A to 100% B (A: 10% methanol, 89.9% water, 0.1% H3PO4; B:
10% water, 89.9% methanol, 0.1% H3PO4, UV 220 nm) or Method D:
PHENOMENEX® Luna CI 8 column (4.6 x 50 mm or 4.6 x 75 mm) eluted at 4 mL/min with a 2, 4 or 8 min gradient from 100% A to 100% B (A: 10% methanol, 89.9% water, 0.1% NH4OAc; B: 10% water, 89.9% methanol, 0.1% NH4OAc, UV 220 nm).
[00117] Purification of intermediates and final products was carried out via either normal or reverse phase chromatography. Normal phase chromatography was carried out using prepacked S1O2 cartridges eluted with gradients of hexanes and ethyl acetate or methylene chloride and methanol. Reverse phase preparative HPLC was carried out using a Shimadzu Preparative HPLC system running Discovery VP software using
Method A: YMC Sunfire 5 μιη C18 30 x 100 mm column with a 10 min gradient at 40
mL/min from 100% A to 100% B (A: 10% methanol, 89.9% water, 0.1% TFA; B: 10% water, 89.9% methanol, 0.1% TFA, UV 220 nm), Method B: PHENOMENEX® Axia Luna 5 μιη C18 30 x 100 mm column with a 10 min gradient at 40 mL/min from 100% A to 100% B (A: 10% acetonitrile, 89.9% water, 0.1% TFA; B: 10% water, 89.9% acetonitrile, 0.1% TFA, UV 220 nm), Method C : PHENOMENEX® Luna 5 μιη C 18 30 x 100 mm column with a 10 min gradient at 40 mL/min from 100% A to 100% B (A: 10% acetonitrile, 89.9% water, 0.1% TFA; B: 10% water, 89.9% acetonitrile, 0.1% TFA, UV
220 nm), or Method D: PHENOMENEX® Luna 5 μιη C18 30 x 100 mm column with a 10 min gradient at 40 mL/min from 100% A to 100% B (A: 10% methanol, 89.9% water, 0.1% TFA; B: 10% water, 89.9% methanol, 0.1% TFA, UV 220 nm).
[00118] Alternatively, reverse phase preparative HPLC was carried out using a Varian ProStar Preparative HPLC System running Star 6.2 Chromatography Workstation software using Method E: Dynamax 10 μιη CI 8 41.4 x 250 mm column with a 30 min gradient at 30 mL/min from 10%B to 100% B (A 98% water, 2% acetonitrile, 0.05% TFA; B: 98% acetonitrile, 2% water, 0.05% TFA, UV 254 nm).
[00119] LCMS chromatograms were obtained on a Shimadzu HPLC system running Discovery VP software, coupled with a Waters ZQ mass spectrometer running MassLynx version 3.5 software and using the following respective methods. Unless specified otherwise, for each method, the LC column was maintained at room temperature and UV detection was set to 220 nm.
[00120] Method A: A linear gradient using solvent A (10% methanol, 90% water, 0.1% of TFA) and solvent B (90% methanol, 10% water, 0.1% of TFA); 0-100% of solvent B over 4 min and then 100% of solvent B over 1 min. Column:
PHENOMENEX® Luna 5 μιη CI 8 (4.5 x 50 mm). Flow rate was 4 mL/min.
[00121] Method B: A linear gradient using solvent A (10% methanol, 90% water, 0.1% of TFA) and solvent B (90% methanol, 10% water, 0.1% of TFA); 0-100% of solvent B over 2 min and then 100% of solvent B over 1 min. Column:
PHENOMENEX® Luna 5 nm C18 (2.0 x 30 mm). Flow rate was 1 mL/min.
[00122] Method C: A linear gradient using solvent A (10% acetonitrile, 90% water, 10 mM NH4OAc) and solvent B (90% acetonitrile, 10% water, 10 mM NH4OAc); 0-
100% of solvent B over 4 min and then 100% of solvent B over 1 min. Column:
PHENOMENEX® Luna 5 μιη CI 8 (4.5 x 50 mm). Flow rate was 4 mL/min.
[00123] Method D: A linear gradient using solvent A (10% acetonitrile, 90% water, 0.05% of TFA) and solvent B (90% acetonitrile, 10% water, 0.05% of TFA); 0-100% of solvent B over 2 min and then 100% of solvent B over 1 min. Column:
PHENOMENEX®Luna 5 μι Ο8 (4.5 χ 30 mm). Flow rate was 1 mL/min.
[00124] Method E: A linear gradient using solvent A (10% MeOH, 90% water, 10 mM NH4OAc) and solvent B (90% MeOH, 10% water, 10 mM NH4OAc); 0-100% of solvent B over 4 min and then 100% of solvent B over 1 min. Column:
PHENOMENEX® Luna 5 μιη C 18 (4.5 x 50 mm). Flow rate was 4 mL/min.
[00125] Method F: A linear gradient using solvent A (10 mM NH4OAC, 95% water,
5% ACN) and solvent B (10 mM NH4OAc, 95% ACN, 5% water); 0-100% of solvent B over 4 min and then 100% of solvent B over 1 min. Column: Mac-Mod Halo (CI 8, 4.6 x 50 mm). Flow rate was 4 mL/min.
[00126] Method G: A linear gradient using solvent A (10%> acetonitrile, 90%> water, 0.1% TFA) and solvent B (90% acetonitrile, 10% water, 0.1% TFA); 0-100% of solvent
B over 4 min and then 100% of solvent B over 1 min. Column: PHENOMENEX® Luna 3μιη C18 (2.0 x 50 mm). Flow rate was 4 mL/min.
[00127] Method H: A linear gradient using solvent A (10% methanol, 90% water, 0.1%) of formic acid) and solvent B (90%> methanol, 10%> water, 0.1% of formic acid); 0- 100% of solvent B over 2 min and then 100% of solvent B over 1 min. Column:
PHENOMENEX® Luna 3μι Ο8 (2.0 χ 30 mm). Flow rate was 1 mL/min.
[00128] Method I: A linear gradient using solvent A (10% MeOH, 90% water, 10 mM
NH4OAc) and solvent B (90% MeOH, 10% water, 10 mM NH4OAc); 0-100% of solvent B over 2 min and then 100% of solvent B over 1 min. Column: PHENOMENEX® Luna 3μιη C18 (2.0 x 30 mm). Flow rate was 1 mL/min.
[00129] Method J: A linear gradient using solvent A (10% methanol, 90% water, 0.1% of formic acid) and solvent B (90% methanol, 10% water, 0.1% of TFA); 0-100% of solvent B over 4 min and then 100% of solvent B over 1 min. Column:
PHENOMENEX® Luna 5μιη C 18 (4.5 x 50 mm). Flow rate was 4 mL/min.
[00130] Method K: A linear gradient using solvent A (10 mM NH4OAC, 95% water, 5% ACN) and solvent B (10 mM NH4OAc, 95% ACN, 5% water); 0-100% of solvent B over 5.5 min and then 100% of solvent B over 1.5 min. Column: SUPELCO® Ascentis 4.6x50mm 2.7μιη CI 8. Flow rate was 4 mL/min.
[00131] Method L: A linear gradient using solvent A (5 % methanol, 95 % water, 0.05% of TFA) and solvent B (95% methanol, 5% water, 0.05% of TFA); 0-100% of solvent B over 4 min and then 100% of solvent B over 1 min. Column: WATERS® XBridge C18 (4.6 x 50 mm, 5μιη). Flow rate was 4 mL/min. The LC column was maintained at 35 °C.
[00132] Method M: A linear gradient using of Solvent A (0.05% TFA, 100% water) and Solvent B (0.05% TFA, 100% ACN); 2 to 98% B over 1 min, with 0.5 min hold time at 98% B. Column: WATERS® BEH CI 8 (2.1 x 50 mm). Flow rate: 0.8 mL/min.
[00133] Method N: A linear gradient using solvent A (5% ACN, 95% water, 10 mM NH4OAc) and solvent B (95% ACN, 5% water, 10 mM NH4OAc); 0-100% of solvent B over 3 min and then 100% of solvent B over 1 min. Column: WATERS® BEH C 18 (2.1 x 50 mm). Flow rate: 1.1 mL/min.
[00134] Method O: A linear gradient using solvent A (5% ACN, 95% water, 0.05% of TFA) and solvent B (95% ACN, 5% water, 0.05% of TFA); 0-100% of solvent B over 3 min and then 100% of solvent B over 1 min. Column: WATERS® BEH CI 8 (2.1 x 50 mm). Flow rate: 1.1 mL/min.
[00135] Method P: A linear gradient using solvent A (5% ACN, 95% water, 10 mM NH4OAc) and solvent B (95% ACN, 5% water, 10 mM NH4OAc); 0-100% of solvent B over 4 min and then 100% of solvent B over 1 min. Column: WATERS® XBridge C18 (4.6 x 50 mm, 5μιη). Flow rate was 4 mL/min.
[00136] Method Q: A linear gradient using solvent A (10% MeOH, 90% water, 0.1% TFA) and solvent B (90% MeOH, 10% water, 0.1% TFA); 0-100% of solvent B over 2 min and then 100% of solvent B over 1 min. Column: PHENOMENEX® Luna 3μιη CI 8 (2.0 x 50 mm). Flow rate was 1 mL/min.
[00137] Method R: Column: Waters BEH CI 8, 2.0 x 50 mm, 1.7-μιη particles; Mobile Phase A: 5:95 acetonitrile: water with 10 mM ammonium acetate; Mobile Phase B: 95 :5 acetonitrile: water with 10 mM ammonium acetate; Temperature: 40 °C; Gradient: 0.5
min hold at 0%B, 0-100% B over 4 minutes, then a 0.5-minute hold at 100% B; Flow: 1 mL/min.
[00138] Preparative HPLC methods employed in the purification of products:
[00139] Method A: Linear gradient of 0 to 100% B over 10 min, with 5 min hold time at 100%) B; Shimadzu LC-8A binary pumps
Waters ZQ mass spectrometer using Waters Masslynx 4.0 SP4 MS software
UV visualization at 220 nm
Column: WATERS® XBridge 19 x 150 mm 5μιη C18
Flow rate: 20 mL/min
Peak collection triggered by mass spectrometry
Solvent A: 0.1% TFA, 10% ACN, 90% water
Solvent B: 0.1% TFA, 90% ACN, 10% water NMR Employed in Characterization of Examples
[00140] ¾ NMR spectra were obtained with Bruker or JEOL Fourier transform spectrometers operating at frequencies as follows: ¾ NMR: 400 MHz (Bruker or JEOL) or 500MHz (JEOL). 13C NMR: 100 MHz (Bruker or JEOL). Spectra data are reported in the format: chemical shift (multiplicity, coupling constants, number of hydrogens).
Chemical shifts are specified in ppm downfield of a tetramethylsilane internal standard (δ units, tetramethylsilane = 0 ppm) and/or referenced to solvent peaks, which in ¾ NMR spectra appear at 2.49 ppm for CD2HSOCD3, 3.30 ppm for CD2HOD, and 7.24 ppm for
CHC13, and which in 13C NMR spectra appear at 39.7 ppm for CD3SOCD3, 49.0 ppm for CD3OD, and 77.0 ppm for CDC13. All 13C NMR spectra were proton decoupled.
IV. BIOLOGY
[00141] The endothelium occupies a pivotal position at the interface between the circulating humoral and cellular elements of the blood, and the solid tissues which constitute the various organs. In this unique position, endothelial cells regulate a large number of critical processes, including leukocyte adherence and transit through the blood vessel wall, local control of blood vessel tone, modulation of the immune response, the balance between thrombosis and thrombolysis, and new blood vessel development. Thus,
endothelial cell dysfunction has been postulated as a central feature of vascular diseases such as hypertension and atherosclerosis. (WO 1999/032611 and references cited therein, e.g., Folkman, J. et al, Science, 235:442-447 (1987); Yanagisawa, M. et al, Nature, 332(6163):411-415 (1988); Folkman, J. et al, J. Biol. Chem., 267(16): 10931-10934 (1992); Janssens, S.P. et al, J. Biol. Chem., 267(21): 14519-14522 (1992); Lamas, S. et al, Proc. Natl. Acad. Sci. U.S.A., 89(14):6348-6352 (1992); Luscher, T.F. et al,
Hypertension, 19(2): 117-130 (1992); Williams et al, Am. Rev. Respir. Dis., 146:S45-S50 (1992); and Bevilacqua, M.P. et al, J. Clin. Invest., 91(2):379-387 (1993)).
[00142] Atherosclerosis and its associated coronary artery disease (CAD) is the leading cause of mortality in the industrialized world. Despite attempts to modify secondary risk factors (smoking, obesity, lack of exercise) and treatment of dyslipidemia with dietary modification and drug therapy, coronary heart disease (CHD) remains the most common cause of death in the U.S. In 2008, cardiovascular disease accounted for 33% of all deaths in the U.S., and ~1 of every 6 deaths were specifically caused by atherosclerotic coronary heart disease {Circulation 125:e2-e220 (2012)).
[00143] Risk for development of atherosclerosis has been shown to be strongly correlated with certain plasma lipid levels. While elevated low density lipoprotein- cholesterol (LDL-C) may be the most recognized form of dyslipidemia, it is by no means the only significant lipid associated contributor to CHD. A low level of high density lipoprotein-cholesterol (HDL-C) is also a known risk factor for CHD (Gordon, D.J. et al, Circulation, 79(1):8-15 (1989)).
[00144] High LDL-C and triglyceride levels are positively correlated, while high levels of HDL-C are negatively correlated with the risk for developing cardiovascular diseases. Thus, dyslipidemia is not a unitary risk profile for CHD but may be comprised of one or more, preferably one to three, lipid aberrations.
[00145] At least 50% of the variation in HDL cholesterol levels is genetically determined. The phenotype of elevated HDL cholesterol is often dominantly inherited, but homozygous deficiency of HL or of the cholesteryl ester transfer protein (CETP), which result in elevated HDL cholesterol, are recessive conditions. Recently, several genetic variations in the human endothelial lipase gene have been identified, six of which potentially produce functional variants of the protein, and the frequencies of these variants were found to be associated with elevated levels of HDL cholesterol in human
subjects (deLemos, A.S. et al, Circulation, 106(11): 1321-1326 (2002)). Notably, the endothelial lipase-mediated binding and uptake of HDL particles and the selective uptake of HDL-derived cholesterol esters have been reported to be independent of its enzymatic lipolytic activity (Strauss, J.G. et al, Biochem. J, 368:69-79 (2002)).
[00146] Because of the beneficial effects widely associated with elevated HDL levels, an agent which inhibits EL activity in humans, by virtue of its HDL increasing ability, are expected to be useful for the treatment, prevention, the arrestment and/or regression of atherosclerosis, coronary heart disease, cerebrovascular disorders etc., especially those (but not restricted thereto) which are characterized by one or more of the following factors: (a) high plasma triglyceride concentrations, high postprandial plasma triglyceride concentrations; (b) low HDL cholesterol concentration; (c) low apoAl lipoprotein concentrations; (d) high LDL cholesterol concentrations; (e) high levels of small dense LDL cholesterol particles; and (f) high apoB lipoprotein concentrations.
[00147] The term "modulator" refers to a chemical compound with capacity to either enhance (e.g., "agonist" activity) or partially enhance (e.g., "partial agonist" activity) or inhibit (e.g., "antagonist" activity or "inverse agonist" activity) a functional property of biological activity or process (e.g., enzyme activity or receptor binding); such
enhancement or inhibition may be contingent on the occurrence of a specific event, such as activation of a signal transduction pathway, receptor internalization, and/or may be manifest only in particular cell types.
[00148] It is also desirable and preferable to find compounds with advantageous and improved characteristics compared with known anti-atherosclerosis agents, in one or more of the following categories that are given as examples, and are not intended to be limiting: (a) pharmacokinetic properties, including oral bioavailability, half life, and clearance; (b) pharmaceutical properties; (c) dosage requirements; (d) factors that decrease blood drug concentration peak-to-trough characteristics; (e) factors that increase the concentration of active drug at the receptor; (f) factors that decrease the liability for clinical drug-drug interactions; (g) factors that decrease the potential for adverse side- effects, including selectivity versus other biological targets; and (h) improved therapeutic index.
[00149] As used herein, the term "patient" encompasses all mammalian species.
[00150] As used herein, the term "subject" refers to any human or non-human organism that could potentially benefit from treatment with an anti-atherosclerosis agent, e.g., an endothelial lipase inhibitor. Exemplary subjects include human beings of any age with risk factors for atherosclerosis and its associated coronary artery disease. Common risk factors include, but are not limited to, age, sex, weight, and family history.
[00151] As used herein, "treating" or "treatment" cover the treatment of a disease-state in a mammal, particularly in a human, and include: (a) inhibiting the disease-state, i.e., arresting it development; and/or (b) relieving the disease-state, i.e., causing regression of the disease state.
[00152] As used herein, "prophylaxis" or "prevention" covers the preventive treatment of a subclinical disease-state in a mammal, particularly in a human, aimed at reducing the probability of the occurrence of a clinical disease-state. Patients are selected for preventative therapy based on factors that are known to increase risk of suffering a clinical disease state compared to the general population. "Prophylaxis" therapies can be divided into (a) primary prevention and (b) secondary prevention. Primary prevention is defined as treatment in a subject that has not yet presented with a clinical disease state, whereas secondary prevention is defined as preventing a second occurrence of the same or similar clinical disease state.
[00153] As used herein, "risk reduction" covers therapies that lower the incidence of development of a clinical disease state. As such, primary and secondary prevention therapies are examples of risk reduction.
[00154] "Therapeutically effective amount" is intended to include an amount of a compound of the present invention that is effective when administered alone or in combination to inhibit endothelial lipase and/or to prevent or treat the disorders listed herein. When applied to a combination, the term refers to combined amounts of the active ingredients that result in the preventive or therapeutic effect, whether administered in combination, serially, or simultaneously.
BIOLOGICAL ACTIVITY
[00155] Endothelial lipase (EL) and hepatic lipase (HL) activities were measured using a fluorescent substrate, A10070, (Invitrogen, CA) doped into an artificial vesicle containing DMPG (Avanti Polar Lipids) as the excipient. Vesicles were prepared by
combining 571 μΐ. of 29 mM DMPG in a 1 :1 mixture of MeOH and CHC13 with 2000 μΐ. of 1 mM A 10070 in a 1 : 1 mixture of MeOH and CHCI3. The mixture was dried under nitrogen in multiple vials then resuspended in 20 mL total volume of 50 mM HEPES pH 8.0 buffer containing 50 mM NaCl and 0.2 mM EDTA. The sample was allowed to sit at room temperature for 15 min and then was sonicated 3 x 4 mins on ice with a Branson Sonicator using duty cycle 1. This preparation provides vesicles with a mole fraction of 0.11 for the FRET substrate.
[00156] The enzymatic assay was measured using 384-well white Optiplates. Each well contained 20 μΕ of assay buffer (50 mM HEPES pH 8.0, 50 mM NaCl and 1 mM CaCl2) and 0.25 μΐ^ of a DMSO solution containing a compound of interest. EL or HL
(10 μί) was added and allowed to incubate with the compound for 30 min at 37 °C. The source of EL was conditioned media obtained from HT-1080 cells that were transformed using RAGE technology (Athersys) to overexpress endogenous EL, and HL was partially purified from conditioned media obtained from COS cells overexpressing HL. The reaction was started by the addition of 10 μί of a 1 : 10 dilution of vesicles. The final total reaction volume was 20.25 μΐ^. The reaction rates were measured on a Gemini plate reader with an excitation wavelength of 490 nm and an emission wavelength of 530 nm. Readings were taken over a period of 60 minutes, and the slope between 300 and 900 sees of the readout was used to calculate the rate of the reaction.
REFERENCE COMPOUNDS
[00157] The following reference compounds and their preparations are described below. The EL IC50 values were measured using the EL assay described above.


[00158] The exemplified compounds, Example Al to Example A9 and Example Bl to Example 205 and Example 207 to Example B397, disclosed in the present invention were tested in the EL assay described above. Surprisingly, Examples A1-A9 B1-B205, and B207- B397 were found having a range of EL IC50 values of < 0.3 μΜ (300 nM), as shown below.
[00159] Accordingly, the compounds of the present invention can be administered to mammals, preferably humans, for the treatment of a variety of conditions and disorders, including, but not limited to, atherosclerosis, coronary heart disease, coronary artery disease, coronary vascular disease, cerebrovascular disorders, Alzheimer's disease, venous thrombosis, peripheral vascular disease, dyslipidemia, hyperbetalipoproteinemia, hypoalphalipoproteinemia, hypercholesterolemia, hypertriglyceridemia, familial- hypercholesterolemia, cardiovascular disorders, angina, ischemia, cardiac ischemia, stroke, myocardial infarction, reperfusion injury, angioplastic restenosis, hypertension, vascular complications of diabetes, obesity or endotoxemia.
VI. PHARMACEUTICAL COMPOSITIONS, FORMULATIONS AND
COMBINATIONS
[00160] The compounds of this invention can be administered for any of the uses described herein by any suitable means, for example, orally, such as tablets, capsules
(each of which includes sustained release or timed release formulations), pills, powders,
granules, elixirs, tinctures, suspensions, (including nanosuspensions, microsuspensions, spray-dried dispersions), syrups, and emulsions; sublingually; bucally; parenterally, such as by subcutaneous, intravenous, intramuscular, or intrasternal injection, or infusion techniques (e.g., as sterile injectable aqueous or non-aqueous solutions or suspensions); nasally, including administration to the nasal membranes, such as by inhalation spray; topically, such as in the form of a cream or ointment; or rectally such as in the form of suppositories. They can be administered alone, but generally will be administered with a pharmaceutical carrier selected on the basis of the chosen route of administration and standard pharmaceutical practice.
[00161] The term "pharmaceutical composition" means a composition comprising a compound of the invention in combination with at least one additional pharmaceutically acceptable carrier. A "pharmaceutically acceptable carrier" refers to media generally accepted in the art for the delivery of biologically active agents to animals, in particular, mammals, including, i.e., adjuvant, excipient or vehicle, such as diluents, preserving agents, fillers, flow regulating agents, disintegrating agents, wetting agents, emulsifying agents, suspending agents, sweetening agents, flavoring agents, perfuming agents, antibacterial agents, anti-fungal agents, lubricating agents and dispensing agents, depending on the nature of the mode of administration and dosage forms. Pharmaceutically acceptable carriers are formulated according to a number of factors well within the purview of those of ordinary skill in the art. These include, without limitation: the type and nature of the active agent being formulated; the subject to which the agent-containing composition is to be administered; the intended route of administration of the
composition; and the therapeutic indication being targeted. Pharmaceutically acceptable carriers include both aqueous and non-aqueous liquid media, as well as a variety of solid and semi-solid dosage forms. Such carriers can include a number of different ingredients and additives in addition to the active agent, such additional ingredients being included in the formulation for a variety of reasons, e.g., stabilization of the active agent, binders, etc., well known to those of ordinary skill in the art. Descriptions of suitable
pharmaceutically acceptable carriers, and factors involved in their selection, are found in a variety of readily available sources such as, for example, Allen, L. V. Jr. et al.
Remington: The Science and Practice of Pharmacy (2 Volumes), 22nd Edition (2012), Pharmaceutical Press.
[00162] The dosage regimen for the compounds of the present invention will, of course, vary depending upon known factors, such as the pharmacodynamic characteristics of the particular agent and its mode and route of administration; the species, age, sex, health, medical condition, and weight of the recipient; the nature and extent of the symptoms; the kind of concurrent treatment; the frequency of treatment; the route of administration, the renal and hepatic function of the patient, and the effect desired.
[00163] By way of general guidance, the daily oral dosage of each active ingredient, when used for the indicated effects, will range between about 0.01 to about 5000 mg per day, preferably between about 0.1 to about 1000 mg per day, and most preferably between about 0.1 to about 250 mg per day. Intravenously, the most preferred doses will range from about 0.01 to about 10 mg/kg/minute during a constant rate infusion.
Compounds of this invention may be administered in a single daily dose, or the total daily dosage may be administered in divided doses of two, three, or four times daily.
[00164] The compounds are typically administered in admixture with suitable pharmaceutical diluents, excipients, or carriers (collectively referred to herein as pharmaceutical carriers) suitably selected with respect to the intended form of
administration, e.g., oral tablets, capsules, elixirs, and syrups, and consistent with conventional pharmaceutical practices.
[00165] Dosage forms (pharmaceutical compositions) suitable for administration may contain from about 1 milligram to about 2000 milligrams of active ingredient per dosage unit. In these pharmaceutical compositions the active ingredient will ordinarily be present in an amount of about 0.1-95% by weight based on the total weight of the composition.
[00166] A typical capsule for oral administration contains at least one of the compounds of the present invention (250 mg), lactose (75 mg), and magnesium stearate (15 mg). The mixture is passed through a 60 mesh sieve and packed into a No. 1 gelatin capsule.
[00167] A typical injectable preparation is produced by aseptically placing at least one of the compounds of the present invention (250 mg) into a vial, aseptically freeze-drying and sealing. For use, the contents of the vial are mixed with 2 mL of physiological saline, to produce an injectable preparation.
[00168] The present invention includes within its scope pharmaceutical compositions comprising, as an active ingredient, a therapeutically effective amount of at least one of the compounds of the present invention, alone or in combination with a pharmaceutical carrier. Optionally, compounds of the present invention can be used alone, in
combination with other compounds of the invention, or in combination with one or more, preferably one to three, other therapeutic agent(s), e.g., HMG-CoA reductase inhibitors or other pharmaceutically active material.
[00169] The compounds of the present invention may be employed in combination with other EL inhibitors or one or more, preferably one to three, other suitable therapeutic agents useful in the treatment of the aforementioned disorders including:
anti-atherosclerotic agents, anti-dyslipidemic agents, anti-diabetic agents,
anti-hyperglycemic agents, anti-hyperinsulinemic agents, anti-thrombotic agents, anti-retinopathic agents, anti-neuropathic agents, anti-nephropathic agents, anti-ischemic agents, anti-hypertensive agents, anti-obesity agents, anti-hyperlipidemic agents, anti-hypertriglyceridemic agents, anti-hypercholesterolemic agents, anti-restenotic agents, anti-pancreatic agents, lipid lowering agents, anorectic agents, memory enhancing agents, anti-dementia agents, cognition promoting agents, appetite suppressants, treatments for heart failure, treatments for peripheral arterial disease, treatment for malignant tumors, and anti-inflammatory agents.
[00170] The compounds of the present invention may be employed in combination with additional therapeutic agent(s) selected from one or more, preferably one to three, of the following therapeutic agents in treating atherosclerosis: anti-hyperlipidemic agents, plasma HDL-raising agents, anti-hypercholesterolemic agents, cholesterol biosynthesis inhibitors (such as HMG CoA reductase inhibitors), acyl-coenzyme Axholesterol acyltransferase (ACAT) inhibitors, LXR agonist, probucol, raloxifene, nicotinic acid, niacinamide, cholesterol absorption inhibitors, bile acid sequestrants (such as anion exchange resins, or quaternary amines {e.g., cholestyramine or colestipol)), low density lipoprotein receptor inducers, clofibrate, fenofibrate, benzofibrate, cipofibrate, gemfibrizol, vitamin B6, vitamin B12, anti-oxidant vitamins, β-blockers, anti-diabetes agents, angiotensin II antagonists, angiotensin converting enzyme inhibitors, platelet aggregation inhibitors, fibrinogen receptor antagonists, aspirin or fibric acid derivatives.
[00171] The compounds of the present invention may be employed in combination with additional therapeutic agent(s) selected from one or more, preferably one to three, of the following therapeutic agents in treating cholesterol biosynthesis inhibitor, particularly an HMG-CoA reductase inhibitor. Examples of suitable HMG-CoA reductase inhibitors include, but are not limited to, lovastatin, simvastatin, pravastatin, fluvastatin, atorvastatin, and rivastatin.
[00172] The term HMG-CoA reductase inhibitor is intended to include all
pharmaceutically acceptable salt, ester, free acid and lactone forms of compounds which have HMG-CoA reductase inhibitory activity and, therefore, the use of such salts, esters, free acids and lactone forms is included within the scope of this invention. Compounds which have inhibitory activity for HMG-CoA reductase can be readily identified using assays well-known in the art.
[00173] The compounds of the invention may be used in combination with one or more, preferably one to three, of the following anti-diabetic agents depending on the desired target therapy. Studies indicate that diabetes and hyperlipidemia modulation can be further improved by the addition of a second agent to the therapeutic regimen.
Examples of anti-diabetic agents include, but are not limited to, sulfonylureas (such as chlorpropamide, tolbutamide, acetohexamide, tolazamide, glyburide, gliclazide, glynase, glimepiride, and glipizide), biguanides (such as metformin), thiazolidinediones (such as ciglitazone, pioglitazone, troglitazone, and rosiglitazone), and related insulin sensitizers, such as selective and non-selective activators of PPARa, ΡΡΑΡνβ and PPARy;
dehydroepiandrosterone (also referred to as DHEA or its conjugated sulphate ester, DHEA-SO4); anti-glucocorticoids; TNFa inhibitors; a-glucosidase inhibitors (such as acarbose, miglitol, and voglibose), pramlintide (a synthetic analog of the human hormone amylin), other insulin secretagogues (such as repaglinide, gliquidone, and nateglinide), insulin, as well as the therapeutic agents discussed above for treating atherosclerosis.
[00174] The compounds of the invention may be used in combination with one or more, preferably one to three, of the following anti-obesity agents selected from phenylpropanolamine, phentermine, diethylpropion, mazindol, fenfluramine,
dexfenfluramine, phentiramine, P3-adrenoreceptor agonist agents; sibutramine, gastrointestinal lipase inhibitors (such as orlistat), and leptins. Other agents used in treating obesity or obesity-related disorders include neuropeptide Y, enterostatin,
cholecytokinin, bombesin, amylin, histamine H3 receptors, dopamine D2 receptor modulators, melanocyte stimulating hormone, corticotrophin releasing factor, galanin and gamma amino butyric acid (GAB A).
[00175] The above other therapeutic agents, when employed in combination with the compounds of the present invention may be used, for example, in those amounts indicated in the Physicians ' Desk Reference, as in the patents set out above, or as otherwise determined by one of ordinary skill in the art.
[00176] Particularly when provided as a single dosage unit, the potential exists for a chemical interaction between the combined active ingredients. For this reason, when the compound of the present invention and a second therapeutic agent are combined in a single dosage unit they are formulated such that although the active ingredients are combined in a single dosage unit, the physical contact between the active ingredients is minimized (that is, reduced). For example, one active ingredient may be enteric coated. By enteric coating one of the active ingredients, it is possible not only to minimize the contact between the combined active ingredients, but also, it is possible to control the release of one of these components in the gastrointestinal tract such that one of these components is not released in the stomach but rather is released in the intestines. One of the active ingredients may also be coated with a material that affects a sustained-release throughout the gastrointestinal tract and also serves to minimize physical contact between the combined active ingredients. Furthermore, the sustained-released component can be additionally enteric coated such that the release of this component occurs only in the intestine. Still another approach would involve the formulation of a combination product in which the one component is coated with a sustained and/or enteric release polymer, and the other component is also coated with a polymer such as a low viscosity grade of hydroxypropyl methylcellulose (HPMC) or other appropriate materials as known in the art, in order to further separate the active components. The polymer coating serves to form an additional barrier to interaction with the other component.
[00177] These as well as other ways of minimizing contact between the components of combination products of the present invention, whether administered in a single dosage form or administered in separate forms but at the same time by the same manner, will be readily apparent to those skilled in the art, once armed with the present disclosure.
[00178] The compounds of the present invention can be administered alone or in combination with one or more, preferably one to three, additional therapeutic agents. By "administered in combination" or "combination therapy" it is meant that the compound of the present invention and one or more, preferably one to three, additional therapeutic agents are administered concurrently to the mammal being treated. When administered in combination, each component may be administered at the same time or sequentially in any order at different points in time. Thus, each component may be administered separately but sufficiently closely in time so as to provide the desired therapeutic effect.
[00179] The compounds of the present invention are also useful as standard or reference compounds, for example as a quality standard or control, in tests or assays involving the endothelial lipase. Such compounds may be provided in a commercial kit, for example, for use in pharmaceutical research involving endothelial lipase or HDL activity. For example, a compound of the present invention could be used as a reference in an assay to compare its known activity to a compound with an unknown activity. This would ensure the experimenter that the assay was being performed properly and provide a basis for comparison, especially if the test compound was a derivative of the reference compound. When developing new assays or protocols, compounds according to the present invention could be used to test their effectiveness. The compounds of the present invention may also be used in diagnostic assays involving endothelial lipase.
[00180] The present invention also encompasses an article of manufacture. As used herein, article of manufacture is intended to include, but not be limited to, kits and packages. The article of manufacture of the present invention, comprises: (a) a first container; (b) a pharmaceutical composition located within the first container, wherein the composition, comprises: a first therapeutic agent, comprising: a compound of the present invention or a pharmaceutically acceptable salt form thereof; and, (c) a package insert stating that the pharmaceutical composition can be used for the treatment and/or prophylaxis of dyslipidemias and the sequelae thereof. In another embodiment, the package insert states that the pharmaceutical composition can be used in combination (as defined previously) with a second therapeutic agent for the treatment and/or prophylaxis of dyslipidemias and the sequelae thereof. The article of manufacture can further comprise: (d) a second container, wherein components (a) and (b) are located within the second container and component (c) is located within or outside of the second container.
Located within the first and second containers means that the respective container holds the item within its boundaries.
[00181] The first container is a receptacle used to hold a pharmaceutical composition. This container can be for manufacturing, storing, shipping, and/or individual/bulk selling. First container is intended to cover a bottle, jar, vial, flask, syringe, tube (e.g., for a cream preparation), or any other container used to manufacture, hold, store, or distribute a pharmaceutical product.
[00182] The second container is one used to hold the first container and, optionally, the package insert. Examples of the second container include, but are not limited to, boxes (e.g., cardboard or plastic), crates, cartons, bags (e.g., paper or plastic bags), pouches, and sacks. The package insert can be physically attached to the outside of the first container via tape, glue, staple, or another method of attachment, or it can rest inside the second container without any physical means of attachment to the first container. Alternatively, the package insert is located on the outside of the second container. When located on the outside of the second container, it is preferable that the package insert is physically attached via tape, glue, staple, or another method of attachment. Alternatively, it can be adjacent to or touching the outside of the second container without being physically attached.
[00183] The package insert is a label, tag, marker, etc. that recites information relating to the pharmaceutical composition located within the first container. The information recited will usually be determined by the regulatory agency governing the area in which the article of manufacture is to be sold (e.g., the United States Food and Drug
Administration). Preferably, the package insert specifically recites the indications for which the pharmaceutical composition has been approved. The package insert may be made of any material on which a person can read information contained therein or thereon. Preferably, the package insert is a printable material (e.g., paper, plastic, cardboard, foil, adhesive-backed paper or plastic, etc.) on which the desired information has been formed (e.g., printed or applied).
[00184] Other features of the invention should become apparent in the course of the above descriptions of exemplary embodiments that are given for illustration of the invention and are not intended to be limiting thereof.
EXAMPLES
[00185] The following Examples have been prepared, isolated and characterized using the methods disclosed herein. The following examples demonstrate a partial scope of the invention and are not meant to be limiting of the scope of the invention.
Example Al
N-(l-(6-(6-Fluoropyridin-3-yl)benzo[d]thiazol-2-yl)-2-oxo-2
lfamoylethyl)amino)ethyl)-4-(trifluoromethyl)benzamide
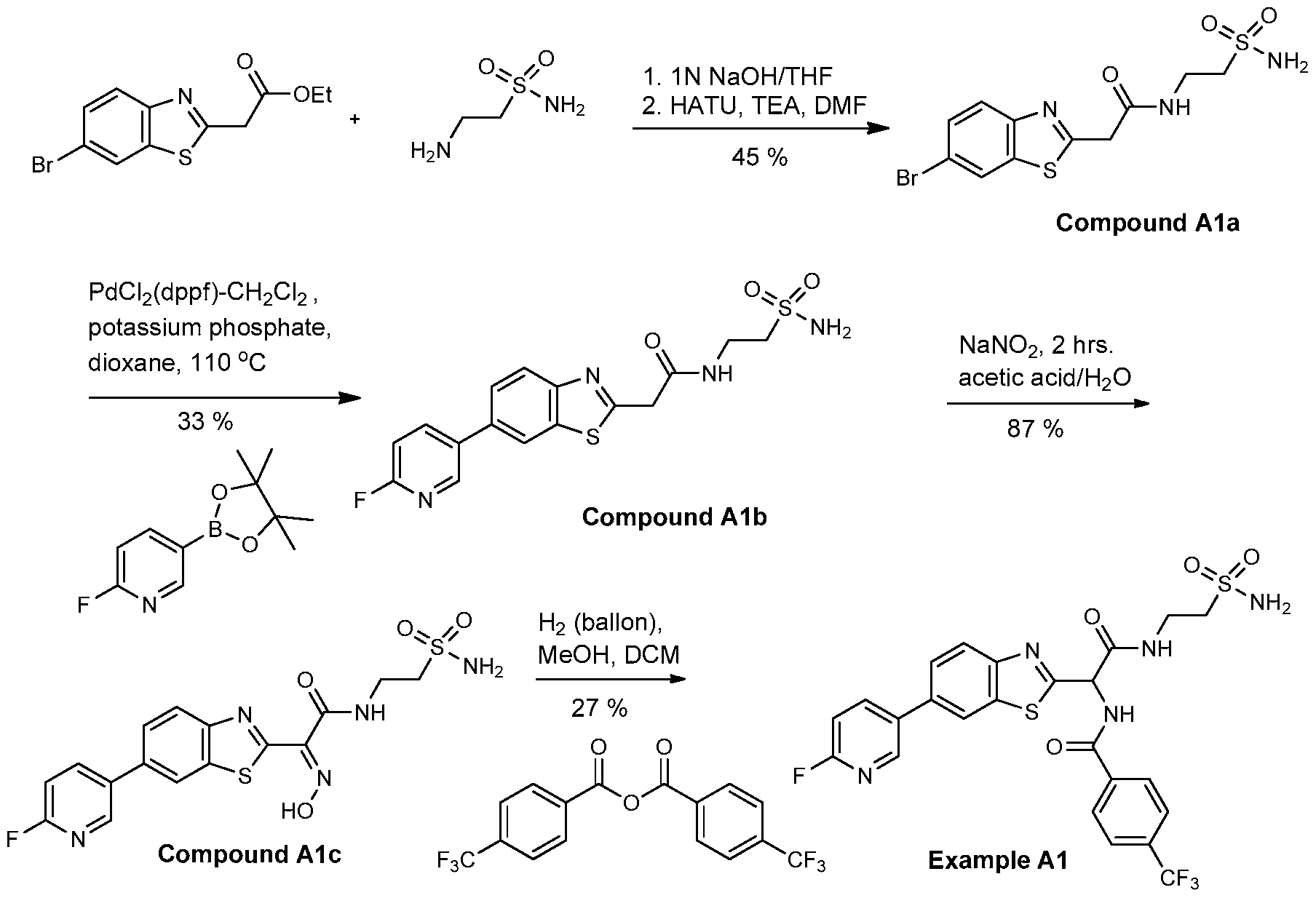
Compound Ala. 2-(6-Bromobenzo[d]thiazol-2-yl)-N-(2-sulfamoylethyl)acetamide
[00186] Ethyl 2-(6-bromobenzo[d]thiazol-2-yl)acetate (described in WO2011/074560, 250 mg, 0.83 mmol) in THF (4 mL) was treated with sodium hydroxide (IN NaOH, 0.92 mL, 0.92 mmol). After 2 hours, the reaction mixture was concentrated to dryness under reduced pressure, and the residue was co-evaporated with toluene (5 mL), re-dissolved in DMF (4 mL), and treated with 2-aminoethanesulfonamide hydrochloride (201 mg, 1.30 mmol), TEA (1.2 mL, 8.3 mmol) and HATU (475 mg, 1.30 mmol). After 16 hours, the reaction mixture was diluted with water (20 mL), extracted with DCM (3 X 10 mL), dried over Na2S04, and concentrated under reduced pressure. The residue was dissolved in
DMF/methanol and purified using reverse phase HPLC (Phenomemenx Luna AXIA 5 micron CI 8, 21.2X100 mm, 30 to 100% B over 15 minutes with 10 minute hold time, solvent A: 90% water/ ACN/ 0.1 %TF A, solvent B:90% ACN/water/0.1 %TFA, Flow rate 20 mL/min; detector at 254). The pooled fractions were lyophilized to Compound Ala, (170 mg, 45 % yield) as a white solid. HPLC: RT = 0.75 (LCMS Method O). MS (ES): m/z = 379.7 [M+H]+. 1H NMR (400MHz, DMF-d7) δ 8.40 (d, J=2.0 Hz, 1H), 7.92 (d, J=8.8 Hz, 1H), 7.69 (dd, J=8.8, 2.0 Hz, 1H), 6.97 (s, 2H), 3.82 - 3.61 (m, 2H), 3.46 (s, 3H), 3.33 (dd, J=7.9, 6.4 Hz, 3H). Compound Alb. 2-(6-(6-Fluoropyridin-3-yl)benzo[d]thiazol-2-yl)-N-(2- sulfamoylethyl)acetamide
[00187] PdCl2(dppf)-CH2Cl2 (21.6 mg, 0.026 mmol) was added to a solution of Compound Ala (100 mg, 0.26 mmol), potassium phosphate (84 mg, 0.40 mmol) and 2- fluoro-5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)pyridine (88 mg, 0.40 mmol) in dioxane (4 mL). The reaction mixture was purged with argon, and the reaction vessel was sealed and heated at 110°C for 18 hrs. The reaction mixture was allowed to cool to room temperature, diluted with water (20 mL), extracted with DCM (3 X 15 mL), dried over Na2S04, and concentrated under reduced pressure. The residue was purified using reverse phase HPLC (Waters Sunfire 5 micron 19x10 mm; 15min. Gradient, Flow rate 20 mL / min. ; wavelength at 220 nm; start % B = 20 & final % B = 100); Solvent A = 10% MeOH - 90%H2O - 0.1% NH40Ac : Solvent B = 90% MeOH - 10%H2O - 0.1% NH4Oac). The fractions containing product were combined and concentrated under reduced pressure to give Compound Alb (34 mg, 33 % yield) as a white solid. HPLC: RT = 0.76 (LCMS Method O). MS (ES): m/z = 395.1 [M+H]+. 1H NMR (400MHz, DMF-d7) δ 8.76 - 8.68 (m, 1H), 8.54 - 8.49 (m, 1H), 8.48 - 8.36 (m, 1H), 8.12 - 8.07 (m, 2H), 7.96 - 7.85 (m, 1H), 7.41 - 7.30 (m, 1H), 7.05 - 6.94 (m, 1H), 5.83 (s, 1H), 4.22 (s, 2H), 3.80 - 3.67 (m, 2H), 3.38 - 3.32 (m, 2H).
Compound Ale. (E)-2-(6-(6-Fluoropyridin-3-yl)benzo[d]thiazol-2-yl)-2- (hydroxyimino)-N-(2-sulfamoylethyl)acetamide
[00188] To a suspension of Compound Alb (100 mg, 0.25 mmol) in acetic acid (4 mL) and water (0.4 mL) was added sodium nitrite (21 mg, 0.30 mmol), and the reaction
stirred at room temperature for 2 hours. A precipitate formed and the reaction mixture was filtered, the solids were washed successively with water, dichloromethane, ether and dried under reduced pressure to give Compound Ale (93 mg, 87 % yield) as a yellow solid. HPLC: RT = 0.77 (LCMS Method O). MS (ES): m/z = 424.1 [M+H]+. 1H NMR (400MHz, DMF-dy) δ 8.71 (d, J=2.5 Hz, 1H), 8.59 - 8.36 (m, 2H), 8.18 (d, J=8.5 Hz, 1H), 7.95 (dd, J=8.4, 1.6 Hz, 1H), 7.34 (dd, J=8.5, 3.0 Hz, 1H), 4.04 (br. s., 1H), 3.93 - 3.76 (m, 1H), 3.64 - 3.34 (m, 2H).
Example Al
[00189] To a suspension of Compound Ale (34 mg, 0.080 mmol) in MeOH (10 mL) was added 4-(trifluoromethyl)benzoic anhydride (116 mg, 0.320 mmol) and 10% Pd/C (3 mg, 0.05mmol). The reaction mixture was degased with argon and stirred under hydrogen atmosphere (ballon) for 2 hours at room temperature. The reaction mixture was diluted with DMF (2 mL), filtered, concentrated under reduced pressure and purified by preparatory HPLC (Sunfire 5μ 19 x 100mm; 25 min. gradient; inj. vol. 2 mL, Flow rate 20mL/min. ; wavelength 220 nm; start % B = 20, final % B = 100, Solvent A = 10 % MeOH - 90 % H20 - 0.1% TFA, Solvent B = 90% MeOH - 10%H2O - 0.1% TFA) to give Example Al (13 mg, 27 % yield). HPLC: RT = 0.9 (LCMS Method O). MS (ES): m/z = 581.9 [M+H]+. 1H NMR (400MHz, DMSO-d6) δ 9.78 (s, 1H), 8.64 (d, J=2.5 Hz, 1H), 8.49 (d, J=1.8 Hz, 1H), 8.37 (d, J=2.8 Hz, 1H), 8.17 (d, J=8.0 Hz, 2H), 8.09 (d, J=8.5 Hz, 1H), 7.95 - 7.80 (m, 3H), 7.32 (dd, J=8.7, 2.9 Hz, 1H), 6.93 (s, 2H), 6.14 (d, J=7.8 Hz, 1H), 3.57 (t, J=6.5 Hz, 2H), 3.26 - 3.09 (m, 2H). EL IC50 35 nM.
[00190] The following compounds, Example A2 to Example A8 were prepared by the general procedures described for Example Al .



Example A9
2-(3-Benzylureido)-2-(6-(4-fluorophenyl)benzo[d]thiazol-2-yl)-N-((5-methyl-l,3,4- oxadiazol-2-yl)methyl)acetamide

Compound A9a. 2-(6-(4-Fluorophenyl)benzo[d]thiazol-2-yl)-N-((5 -methyl- 1,3,4- oxadiazol-2-yl)methyl)acetamide
[00191] Ethyl 2-(6-(4-fluorophenyl)benzo[d]thiazol-2-yl)acetate (described in
WO2011/074560, 500 mg, 1.59 mmol) in THF (4 mL) was treated with sodium hydroxide (IN NaOH, 1.9 mL, 1.9 mmol). After 2 hours, the reaction mixture was concentrated to dryness under reduced pressure. The residue was co-evaporated with toluene (5 mL), re-dissolved in DMF (4 mL), and treated with (5 -methyl- 1,3, 4-oxadiazol- 2-yl)methanamine hydrochloride (284 mg, 1.90 mmol), TEA (2.2 mL, 16 mmol) and PyBOP (990 mg, 1.9 mmol). After 16 hours, the reaction mixture was diluted with water (20 mL), extracted with DCM (3 X 10 mL), dried over Na2S04 and concentrated under reduced pressure. The residue was purified by silica gel (eluting with 0-20%
DCM/MeOH) to give Compound A9a (485 mg, 80.0 % yield) as an oil.
HPLC: RT = 0.88 (LCMS Method O). MS (ES): m/z = 383.2 [M+H]+. 1H NMR
(400MHz, DMF-dy) δ 9.12 (br. s., 1H), 8.41 (d, J=1.8 Hz, 1H), 7.93 - 7.76 (m, 3H), 7.43 - 7.27 (m, 2H), 4.69 (d, J=5.8 Hz, 2H), 4.28 (s, 2H), 2.50 (s, 3H).
Compound A9b. (Z)-2-(6-(4-Fluorophenyl)benzo[d]thiazol-2-yl)-2-(hydroxyimino)-N- ((5-methyl-l,3,4-oxadiazol-2-yl)methyl)acetamide
[00192] Compound A9b) 50 mg, 47% yield) was prepared from Compound A9a as described in the general procedure given for Compound Ale. HPLC: RT = 0.93 (LCMS Method O). MS (ES): m/z = 412.2 [M+H]+. 1H NMR (400MHz, DMF-d7) δ 8.34 - 8.27 (m, 1H), 7.98 - 7.93 (m, 1H), 7.89 - 7.84 (m, 2H), 7.82 - 7.78 (m, 1H), 7.41 - 7.28 (m, 2H), 5.04 - 4.95 (m, 2H), 2.54 (s, 3H).
Example A9
[00193] To a suspension of Compound A9b (50 mg, 0.12 mmol) in EtOH (10 mL) was added (isocyanatomethyl) benzene (16 mg, 0.12 mmol) and 10% Pd/C (3 mg, 0.05 mmol). The mixture was degased with argon and stirred under hydrogen atomsphere
(balloon) at room termperature for 2 hours. The reaction mixture was diluted with DMF (2 mL), filtered, concentrated under reduced pressure and purified by preparatory HPLC (Phenomenex AXIA Luna 5μ 75 x 30mm; 35 min. gradient; inj. vol. 2 mL, Flow rate 40 mL / min. ; wavelength 220nm; start %B = 20, final %B = 100) Solvent A = 10% MeOH - 90% H20 - 0.1 % TFA : Solvent B = 90% MeOH - 10% H20 - 0.1 % TFA) to give
Example A9 (10 mg, 15 % yield). HPLC: RT = 0.95 (LCMS Method O). MS (ES): m/z = 531.2 [M+H]+. 1H NMR (400MHz, DMSO-d6) δ 8.41 (d, J=1.8 Hz, 1H), 8.02 (d, J=8.5 Hz, 1H), 7.86 - 7.74 (m, 3H), 7.40 - 7.23 (m, 9H), 6.98 (s, 1H), 5.87 (d, J=8.0 Hz, 1H), 4.65 - 4.48 (m, 2H), 4.27 (d, J=5.8 Hz, 2H), 2.42 (s, 3H). EL IC50 50 nM.
Example Bl
2-(Methylsulfonyl)-2-(6-phenylbenzo[d]thiazol-2-yl)-N-(2-sulfamoylethyl)acetamide

Compound B1a
D

Example B1
Compound Bla. Ethyl 2-(methylsulfonyl)-2-(6-phenylbenzo[d]thiazol-2-yl)acetate
[00194] Ethyl 2-(6-phenylbenzo[d]thiazol-2-yl)acetate (described in WO2011/074560, 550 mg, 1.9 mmol) was dissolved in DMF (10 mL) and cooled to 0 °C. DBU (0.61 mL, 4.1 mmol) was added. After 10 minutes, methanesulfonyl chloride (0.2 mL, 3 mmol) was added dropwise. The reaction mixture was removed from the ice bath and stirred at room temperature. After 4 hours, the reaction mixture was cooled back to 0 °C, and additional methane sulfonyl chloride (0.11 mL, 1.4 mmol) was added. Following addition, the reaction mixture was removed from ice bath and stirred at room temperature. After 3 days, the reaction mixture was quenched by the addition of water (20 mL), and the resulting solid was collected by filtration. The solids were dissolved in DCM, washed with aqueous 10 % LiCl, IN HCl, brine, dried over MgS04, concentrated under reduced pressure, and purified by silica gel chromatography (eluting with 0-20% DCM/MeOH) to give Compound Bla (250 mg, 36 % yield) as a yellow oil. HPLC: RT = 1.03 min (LCMS Method O). MS (ES): m/z = 376.0 [M+H]+. 1H NMR (400MHz, pyridine-d5) δ 8.07 (br. s., 1H), 7.79 - 7.64 (m, 4H), 7.51 (t, J=7.7 Hz, 2H), 7.40 (s, 1H), 4.39 (d, J=7.0 Hz, 2H), 3.49 (s, 3H), 1.29 (t, J=7.0 Hz, 3H).
Example B 1
[00195] Compound Bla (110 mg, 0.29 mmol) was dissolved in dioxane (1 mL) and TEA (1 mL). 2-Aminoethanesulfonamide hydrochloride (94 mg, 0.59 mmol) was added, and the reaction mixture was sealed in a pressure vessel and heated at 135 °C for 18 hours. Additional 2-aminoethanesulfonamide hydrochloride (94 mg, 0.59 mmol) was added, and the reaction mixture was heated for an additional 3 hours at 165 °C. The reaction mixture was allowed to cool to room temperature, concentrated under reduced pressure, and the residue dissolved in DMF and purified by reverse phase HPLC (Waters Sunfire 5μ 19x100mm; lOmin. Gradient/5 -75mg; inj. vol. 2mL, Flow rate 20mL/min. ; wavelength 220nm; start %B = 80 & final %B = 100) Solvent A = 10% MeOH - 90% H20 - 0.1% NH4OAc : Solvent B = 90% MeOH - 10% H20 - 0.1% NH4Oac) to give Example Bl (20 mg, 15 % yield) as a white solid. HPLC: RT = 0.86 (LCMS Method O). MS (ES): m/z = 453.9 [M+H]+. 1H NMR (400MHz, pyridine-d5) δ 10.14 (s, 2H), 9.63 (br. s., 1H), 9.33 - 9.12 (m, 3H), 9.09 (s, 1H), 8.98 (t, J=7.7 Hz, 2H), 8.87 (d, J=7.3 Hz, 1H), 5.76 (d, J=6.0 Hz, 2H), 5.34 (t, J=6.3 Hz, 2H), 5.01 (br. s., 2H). EL IC50 <10 nM.
Example B2
2- Allylsulfonyl)-2-(6-phenylbenzo[d]thiazol-2-yl)-N-(2-sulfamoylethyl)acetamide

Example B2 Compound B2a. 2-(6-Phenylbenzo[d]thiazol-2-yl)-N-(2-sulfamoylethyl)acetamide
[00196] Pd(PPh3)4 (51 mg, 0.044 mmol) was added to dioxane (2 mL) / 2M Na2C03 (1 mL) solution of Compound Ala (73 mg, 0.15 mmol) and phenylboronic acid (27 mg, 0.22 mmol). The reaction mixture was purged with argon gas and heated at 100 °C. After 90 minutes, the reaction mixture was diluted with water (20 mL), extracted with DCM (3 X 15 mL), and the combined organic portions dried over Na2S04, filtered and concentrated under reduced pressure. The residue was dissolved in DMF/methanol and purified using reverse phase HPLC (Phenomemenx Luna AXIA 5 micron CI 8, 21.2 X 100 mm, 30 to 100% B over 15 minutes with 10 minute hold time, solvent A: 90% water/ ACN/ 0.1 %TF A, solvent B:90% ACN/water/ 0.1 %TFA, Flow rate 20 mL/min; detector at 254). The pooled fractions were diluted with saturated NaHC03 (20 mL), extracted with DCM (3 X 15 mL), dried over Na2S04, and concentrated under reduced pressure to give Compound B2a (23 mg, 41 % yield) as a white solid. HPLC: RT = 0.83 min (LCMS Method M). MS (ES): m/z = 375.9 [M+H]+. 1H NMR (500 MHz, DMSO- d6) δ 8.44 - 8.68 (1 H, m), 8.36 (1 H, d, J=1.93 Hz), 7.88 (1 H, d, J=8.80 Hz), 7.56 - 7.70 (1 H, m), 6.90 (2 H, s), 4.06 (2 H, s), 3.43 - 3.62 (2 H, m), 3.07 - 3.22, (2 H, m).
Example B2
[00197] Compound B2a (22 mg, 0.059 mmol) was dissolved in DMF (1 mL) and treated with NaH (60% in nujol, 11.7 mg, 0.300 mmol) at 0 °C. The reaction mixture was maintained at 0 °C for 2 minutes, allowed to reach room temperature for 3 minutes, and cooled back to 0 °C. After 2 minutes, prop-2-ene-l-sulfonyl chloride (12.4 mg,
0.0900 mmol) was added. After 20 minutes, the reaction mixture was quenched by the addition of 0.1 mL acetic acid, diluted with DMF/methanol and purified using reverse phase HPLC (Phenomemenx Luna AXIA 5 micron CI 8, 21.2 X 100 mm, 30 to 100% B over 15 minutes with 10 minute hold time, solvent A: 90% water/ACN/0.1%TFA, solvent B: 90% ACN/water/ 0.1 %TF A, Flow rate 20 mL/min; detector at 254). The fractions containing product were lyophilized to yield Example B2 (8.1 mg, 29 % yield) as an off- white solid. HPLC: RT = 0.93 min (LCMS Method M). MS (ES): m/z = 479.8 [M+H]+. 1H NMR (500MHz, DMSO-d6) δ 9.16 - 8.93 (m, IH), 8.48 (d, J=1.4 Hz, IH), 8.15 (d, J=8.5 Hz, IH), 7.88 (d, J=1.7 Hz, IH), 7.77 (dd, J=8.3, 1.1 Hz, 2H), 7.51 (d, J=8.0 Hz, 2H), 7.50 - 7.31 (m, IH), 6.95 (s, 2H), 6.06 (s, IH), 5.91 - 5.74 (m, IH), 5.64 - 5.45 (m, 2H), 4.30 - 4.13 (m, 2H), 3.65 - 3.51 (m, 2H), 3.19 (d, J=1.4 Hz, 2H). EL IC50 <10 iiM
[00198] The following compounds, Example B3 to Example B22 were prepared as described in the general procedure given for Example B2.
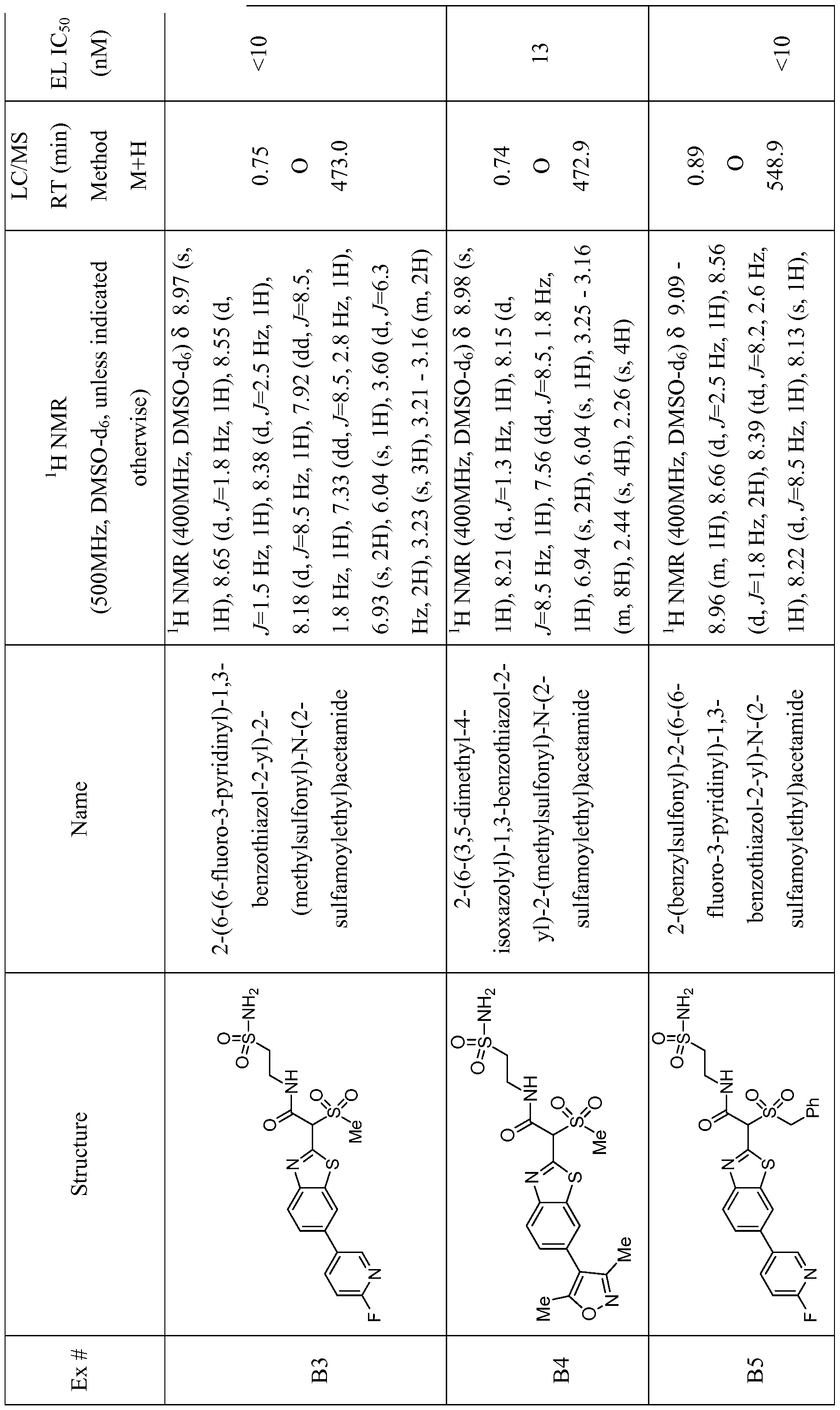
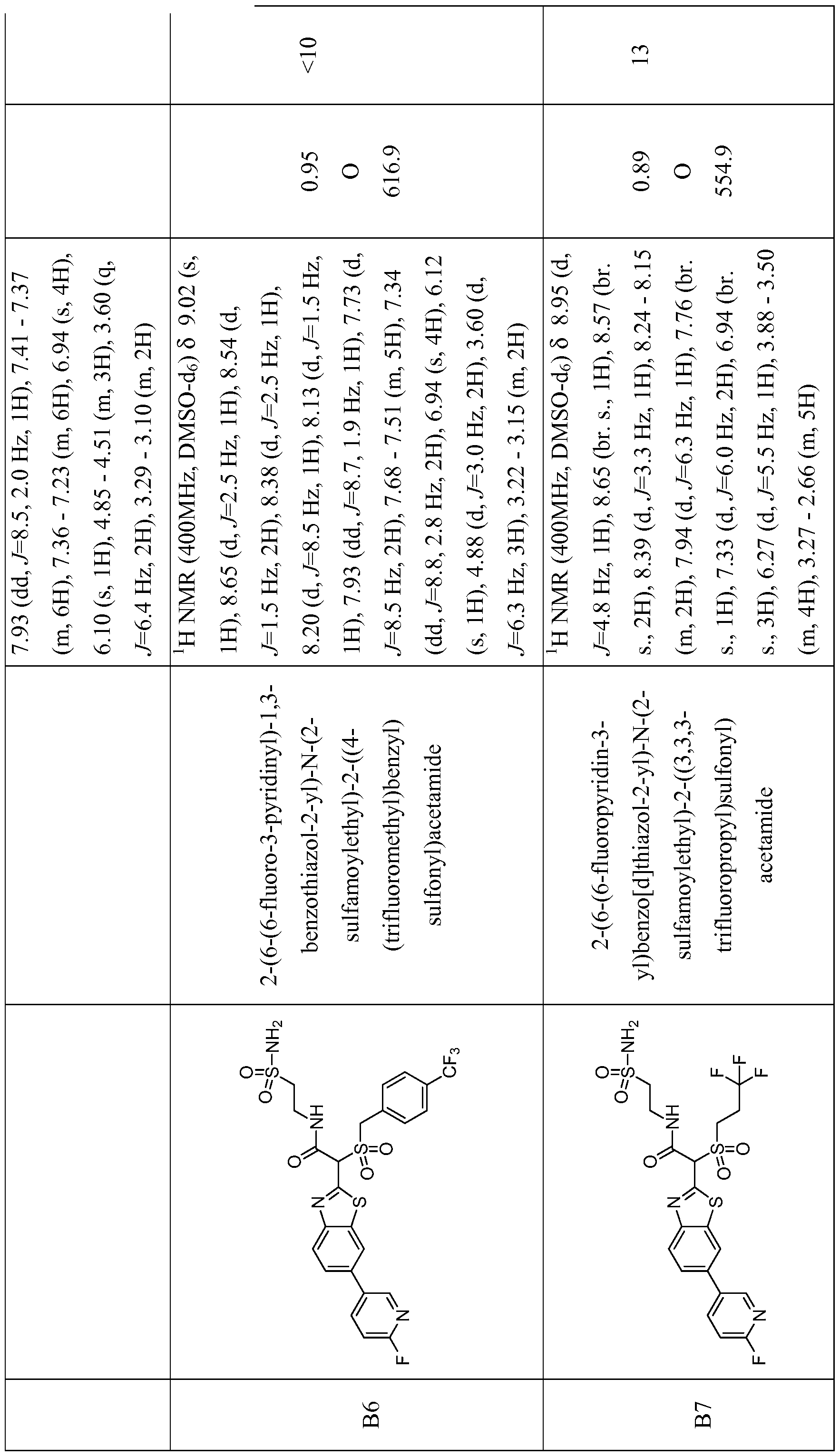





Example B23
2-(6-(2-Fluoro-4-pyridinyl)-l,3-benzothiazol-2-yl)-2-(methylsulfonyl)-N-(2- methylsulfonyl)ethyl)acetamide

Compound B23a. Ethyl 2-(6-(2-fluoropyridin-4-yl)benzo[d]thiazol-2-yl)acetate
[00199] SiliaCat DPP-Pd (0.26 mmol/g loading, from Silicycle, 100 mg, 0.67 mmol), ethyl 2-(6-bromobenzo[d]thiazol-2-yl)acetate (described in WO2011/074560, 200 mg, 0.67 mmol), potassium phosphate (348 mg, 2.00 mmol) and (2-fluoropyridin-4- yl)boronic acid (141 mg, 1.00 mmol) in dioxane (5 mL)/water (0.2 mL) were purged with argon for 5 minutes and then heated at 90 °C. After 4 hours, the reaction mixture was diluted with water (50 mL), extracted with DCM (3 X 50 mL), dried over MgSC^, and concentrated under reduced pressure. The residue was purified by silica gel
chromatography (0 to 100% ethyl acetate/hexanes over 15 minutes) to give Compound B23a (120 mg, 57 % yield) as a brown solid. HPLC: RT = 0.95 min (LCMS Method M). MS (ES): m/z = 316.9 [M+H]+. 1H NMR (500MHz, DMSO-d6) δ 8.68 (d, J=1.7 Hz,
IH), 8.35 (d, J=5.2 Hz, IH), 8.12 (d, J=8.5 Hz, IH), 8.02 (dd, J=8.5, 1.9 Hz, IH), 7.81 (d, J=5.2 Hz, IH), 7.64 (s, IH), 4.38 (s, 2H), 4.19 (q, J=7.1 Hz, 2H), 1.24 (t, J=7.0 Hz, 3H).
Compound B23b. 2-(6-(2-Fluoropyridin-4-yl)benzo[d]thiazol-2-yl)-N-(2- (methylsulfonyl)ethyl)acetamide
[00200] Compound B23b (16 mg, 16 %> yield) was prepared from Compound B23a as described in the general procedure given for Compound Ala. HPLC: RT = 0.73 min (LCMS Method M). MS (ES): m/z = 393.8 [M+H]+. 1H NMR (500MHz, DMSO-d6) δ 8.69 - 8.62 (m, 2H), 8.34 (d, J=5.5 Hz, IH), 8.08 (d, J=8.5 Hz, IH), 8.00 (dd, J=8.5, 1.9 Hz, IH), 7.81 (d, J=5.2 Hz, IH), 7.64 (s, IH), 4.13 (s, 2H), 3.58 - 3.51 (m, 2H), 3.31 (t, J=6.9 Hz, 2H), 3.03 (s, 3H).
Example B23
Example B23 (2.3 mg, 9.6 % yield) was prepared from Compound B23b as described in the general procedure given for Example B2. HPLC: RT = 0.76 min (LCMS Method M) MS (ES): m/z = 471.7 [M+H]+. 1H NMR (500MHz, DMSO-d6) δ 9.08 (s, IH), 8.75 (d, J=1.7 Hz, IH), 8.37 (d, J=5.5 Hz, IH), 8.23 (d, J=8.5 Hz, IH), 8.07 (dd, J=8.7, 1.8 Hz, IH), 7.86 - 7.79 (m, IH), 7.65 (s, IH), 6.09 (s, IH), 3.65 (d, J=5.8 Hz, 2H), 3.37 - 3.32 (m, 2H), 3.26 (s, 3H), 3.04 (s, 3H). EL IC50 210 nM. [00201] The following compounds, Example B24 and Example B25 were prepared as described in the general procedure given for Example B24.

Example B26
2-(4-Fluoro-6-phenylbenzo[d]thiazol-2-yl)-N-(2-sulfamoylethyl)-2-((3,3,3- trifiuoropropyl)sulfonyl)acetamide
Lawesson's

Compound B26a

Compound B26b Compound B26c

Compound B26d Example B26
Compound B26a. Ethyl 3-((4-bromo-2,6-difiuorophenyl)amino)-3-oxopropanoate
[00202] To a solution of 4-bromo-2,6-difluoroaniline (10 g, 48 mmol) in DCM (15 mL) was added ethyl 3-chloro-3-oxopropanoate (6.8 mL, 53 mmol) and DIEA (9.2 mL, 53 mmol) at room temperature. After 1 hour, the reaction mixture was partitioned between H20 and DCM. The organic phase was washed with saturated NH4C1, H20, dried over Na2S04, and filtered. The filtrate was concentrated under reduced pressure to give Compound B26a (10 g, 65 % yield). HPLC: RT = 1.6 (LCMS Method Q). MS (ES): m/z = 321.9 [M+H]+. 1H NMR (500MHz, CHLOROFORM-d) δ 8.89 (br. s., 1H), 7.20 - 7.15 (m, 2H), 4.29 (q, J=7.2 Hz, 2H), 3.54 (s, 2H), 1.36 - 1.31 (m, 3H).
Compound B26b. Ethyl 2-(6-bromo-4-fluorobenzo[d]thiazol-2-yl)acetate
[00203] To a solution of Compound B26a (2.0 g, 6.2 mmol) in toluene (20 mL) was added Lawesson's reagent (1.5 g, 3.7 mmol), and the reaction mixture was heated at reflux. After 3 hours, CS2CO3 (5.1 g, 16 mmol) was added, and the reaction mixture was heated at reflux. After 16 hours, the reaction mixture was filtered, concentrated under reduced pressure and purified using silica gel chromatography (eluting with 0 to 100%
EtOAc in hexanes) to give Compound B26b (1.2 g, 61 % yield). HPLC: RT = 2.02 (LCMS Method Q). MS (ES): m/z = 319.9 [M+H]+. 1H NMR (500 MHz,
CHLOROFORM-d) δ ppm 7.93 (1 H, d), 7.66 (1 H, d, J=1.7 Hz), 4.28 (2 H, q, J=7.2 Hz), 4.22 - 4.24 (2 H, m), 1.33 (3 H, t, J=7.2 Hz).
Compound B26c. Ethyl 2-(4-fluoro-6-phenylbenzo[d]thiazol-2-yl)acetate
[00204] To a solution of Compound B26b (1.0 g, 3.1 mmol) in dioxane (20 mL) was added phenylboronic acid (460 mg, 3.8 mmol), potassium phosphate, (1.7 g, 7.9 mmol), and Pd(PPh3)4 (730 mg, 0.63 mmol). The reaction mixture was purged with argon and heated at 105 °C for 16 hours. The reaction mixture was allowed to cooled to room temperature and filtered. The filtrate was concentrated under reduced pressure and purified by silica gel chromatography (30 minutes gradient from 0 to 100% EtOAc in DCM) to give Compound B26c (810 mg, 82 % yield). HPLC: RT = 2.2 min (LCMS Method Q). MS (ES): m/z = 316 [M+H]+. 1H NMR (500 MHz, CHLOROFORM-d) δ ppm 7.88 (1 H, d, J=1.4 Hz), 7.61 - 7.67 (2 H, m), 7.47 - 7.54 (2 H, m), 7.39 - 7.47 (2 H, m), 4.30 (2 H, q, J=7.2 Hz), 4.26 (2 H, s), 1.33 - 1.38 (3 H, m).
Compound B26d. 2-(4-Fluoro-6-phenylbenzo[d]thiazol-2-yl)-N-(2- sulfamoylethyl)acetamide
[00205] Compound B26d (14.5 mg, 45 % yield) was prepared from Compound B26c as described in the general procedure given for Compound Ala. HPLC: RT = 1.81 min (LCMS Method Q). MS (ES): m/z = 394.9 [M+H]+. 1H NMR (500MHz, METHANOL- d4) δ 8.12 (d, J=1.4 Hz, 1H), 7.81 - 7.70 (m, 2H), 7.59 (dd, J=l 1.8, 1.7 Hz, 1H), 7.52 (t, J=7.7 Hz, 2H), 7.44 (d, J=7.4 Hz, 1H), 4.17 (s, 2H), 3.71 (t, J=6.7 Hz, 2H), 2.64 (dt, J=3.8, 1.8 Hz, 2H).
Example B26
Example B26 (2.6 mg, 18 % yield) was prepared from Compound B26d as described in the general procedure given for Example B2. HPLC: RT = 2.0 (LCMS Method Q). MS (ES): m/z = 554.1 [M+H]+. 1H NMR (500MHz, METHANOL-d4) δ 8.18 - 8.15 (m, 1H), 7.77 - 7.72 (m, 2H), 7.70 - 7.66 (m, 1H), 7.63 (dd, J=l 1.8, 1.4 Hz, 1H), 7.54 - 7.48
(m, 2H), 7.46 - 7.39 (m, 1H), 3.82 (td, J=6.7, 4.4 Hz, 2H), 3.77 (dt, J=9.8, 5.9 Hz, 2H), 3.40 - 3.35 (m, 2H), 2.88 - 2.74 (m, 2H). EL IC50 <10 iiM.
[00206] The following compounds, Example B27 to Example B29 were prepared as described in the general procedure given for Example B26.

Example B30
2-(6-(2-Fluoro-4-pyridinyl)-l,3-benzothiazol-2-yl)-2-(methylsulfonyl)-N-(2-((2,2,2- trifluoroethyl)sulfamoyl)ethyl)acetamide

Example B30
Compound B30a. Benzyl (2-(N-(2,2,2-trifluoroethyl)sulfamoyl)ethyl)carbamate
[00207] To a dioxane (1 mL) solution of 2,2,2-trifluoroethanamine (80 mg, 0.81 mmol) and DIEA (0.28 mL, 1.6 mmol) at 0°C was added benzyl (2- (chlorosulfonyl)ethyl)carbamate (150 mg, 0.54 mmol) in acetonitrile (1 mL). The reaction was allowed to reach room temperature over a period of 20 minutes. The reaction mixture was concentrated under reduced pressure, and the residue purified using reverse phase HPLC (YMC Sunfire 5u (CI 8) 30 x 100 mm, 0-100% ACN (90% in H20, 0.1% TFA) using gradient over 16 min with flow rate 40 mL/min and UV detection at 220 nm) to give Compound B30a (95 mg, 39 % yield). HPLC: RT = 3.84 (LCMS Method M). MS (ES): m/z = 340.9 [M+H]+. 1H NMR (500MHz, CHLOROFORM-d) d 7.43 - 7.34 (m, 5H), 5.28 (br. s., 2H), 5.15 (s, 2H), 3.86 - 3.68 (m, 4H), 3.35 - 3.24 (m, 2H).
Compound B30b. 2-Amino-N-(2,2,2-trifluoroethyl)ethanesulfonamide
[00208] HBr (1 mL, 33% solution in acetic acid) was added to Compound B30a (95 mg, 0.28 mmol) at room temperature. After 5 minutes, the reaction mixture was diluted with diethyl ether (5 mL), and the resulting solid, Compound B30b (42 mg, 41%) was isolated by filtration. HPLC: RT = 0.39 (LCMS Method M). MS (ES): m/z = 207.0 [M+H]+. 1H NMR (500MHz, DMSO-d6) d 8.46 (br. s, 1H), 7.87 (br. s., 2H), 3.87 (td, J=6.3, 3.0 Hz, 2H), 3.40 (t, J=7.4 Hz, 2H), 3.17 (br. s., 2H)
Compound B30c. 2-(6-(2-Fluoropyridin-4-yl)benzo[d]thiazol-2-yl)-N-(2-(N-(2,2,2- trifluoroethyl)sulfamoyl)ethyl)acetamide
[00209] Compound B30c (10 mg, 11%>) was prepared from Compound B30b and
Compound B23a as described in the general procedure given for Compound Ala.
HPLC: RT = 0.83 (LCMS Method M). MS (ES): m/z = 476.8 [M+H]+. 1H NMR
(500MHz, DMSO-de) δ 8.66 (d, J=1.9 Hz, 1H), 8.58 (s, 1H), 8.34 (d, J=5.2 Hz, 1H), 8.17 (t, J=6.7 Hz, 1H), 8.08 (d, J=8.5 Hz, 1H), 8.03 - 7.98 (m, 1H), 7.83 - 7.78 (m, 1H),
7.64 (s, 1H), 4.13 (s, 2H), 3.82 (dd, J=9.6, 6.9 Hz, 2H), 3.54 - 3.47 (m, 2H), 3.30 - 3.26
(m, 2H).
Example B30
[00210] Example B31 (2.0 mg, 18 %> yield) was prepared from Compound B31c as described in the general procedure given for Example B2. HPLC: RT = 0.83 (LCMS Method M). MS (ES): m/z = 476.8 [M+H]+. 1H NMR (500MHz, DMSO-d6) δ 8.66 (d, J=1.9 Hz, 1H), 8.58 (s, 1H), 8.34 (d, J=5.2 Hz, 1H), 8.17 (t, J=6.7 Hz, 1H), 8.08 (d, J=8.5 Hz, 1H), 8.03 - 7.98 (m, 1H), 7.83 - 7.78 (m, 1H), 7.64 (s, 1H), 4.13 (s, 2H), 3.82 (dd, J=9.6, 6.9 Hz, 2H), 3.54 - 3.47 (m, 2H), 3.30 - 3.26 (m, 2H). EL IC50 277 nM.
Example B31
N-tert-Butyl-4-(2-(l-(methylsulfonyl)-2-oxo-2-((2-sulfamoylethyl)amino)ethyl)-l,3- benzothiazol-6-yl)benzamide

Compound B31a

Compound B31a. Ethyl 2-(6-bromobenzo[d]thiazol-2-yl)-2-(methylsulfonyl)acetate
[00211] 1 M NaHMDS in THF (0.40 mL, 0.40 mmol) was added dropwise to a cooled (-20 °C) DMF (3 mL) solution of Compound Ala (100 mg, 0.26 mmol). After 3 minutes, methanesulfonyl chloride (0.03 mL, 0.40 mmol) was added. After 5 minutes, the reaction was quenched with acetic acid (0.2 mL), diluted with methanol (0.5 mL), and purified using reverse HPLC (Phenomemenx Luna AXIA 5 micron CI 8, 21.2X100, UV at 220 nm, 10 to 70% B over 15 minutes with 10 minute hold time, solvent A: 90%
water/ACNl/0.1%TFA, solvent B:90% ACN/water/ 0.1 %TF A, Flow rate 20 mL/min; detector at 254) to isolate Compound B31a (43 mg, 36%> yield) as a white solid.
HPLC: Rt = 0.80 min (LCMS Method M). MS (ES): m/z = 457.6 [M+H]+.
1H NMR (500MHz, DMSO-d6) δ 8.97 (t, J=5.8 Hz, IH), 8.47 (d, J=1.9 Hz, IH), 8.02 (d, J=8.5 Hz, IH), 7.72 (dd, J=8.8, 1.9 Hz, IH), 6.94 (s, 2H), 6.01 (s, IH), 3.78 - 3.48 (m, 2H), 3.21 (s, 3H), 3.20 - 3.13 (m, 2H).
Example B31
[00212] Example B31 (14 mg, 45%> yield) was prepared from Compound B3 la as described in the general procedure given for Compound B2a. EL IC50 < 10 11M. HL IC50 257 11M. HPLC: RT = 0.86 min (LCMS Method M). MS (ES): m/z = 552.8 [M+H]+.
1H NMR (500MHz, DMSO-d6) δ 8.98 (t, J=5.8 Hz, IH), 8.55 (d, J=1.4 Hz, IH), 8.17 (d, J=8.8 Hz, IH), 8.00 - 7.90 (m, 3H), 7.88 - 7.77 (m, 3H), 6.95 (s, 2H), 6.03 (s, IH), 3.61 (q, J=6.8 Hz, 2H), 3.24 (s, 3H), 3.22 - 3.15 (m, 2H), 1.47 - 1.33 (m, 9H).
[00213] Example B32 to Example B97 were prepared as described in the general procedure given for Example B31.
Example B98
2-(Methylsulfonyl)-2-(6-( 1 -(4-piperidinyl)- 1 H-pyrazol-4-yl)- 1 ,3 -benzothiazol-2-yl)-N- (2-sulfamoylethyl)acetamide

55 %
Compound B31a

Example B98
[00214] A mixture of Compound B3 la, (l-(l-(tert-butoxycarbonyl)piperidin-4-yl)-lH- pyrazol-4-yl)boronic acid (15.5 mg, 0.0530 mmol) and Pd(PPh3)4 (12.2 mg, 10.5 μιηοΐ) in dioxane (2 mL) and 2M Na2C03 (1 mL) was purged with argon and heated at 90 °C for 2 hours. The reaction mixture was concentrated under reduced pressure, dissolved in DMF (2 mL) and filtered. The filtrate was concentrated under reduced pressure, dissolved in DCM (1 mL) and treated with TFA (1 mL). After 16 hours, the reaction mixture was concentrated under reduced pressure, disolved in DMF/methanol and purifed by reverse phase HPLC (Column: Waters X Bridge CI 8, 19 x 200 mm, 5-μιη particles; Mobile Phase A: 5:95 acetonitrile: water with 10-mM ammonium acetate; Mobile Phase B: 95:5 acetonitrile: water with 10-mMammonium acetate; Gradient: 0-25% B over 20 minutes, then a 5-minute hold at 100% B; Flow: 20 mL/min) to give Example B98 (12.4 mg, 54.0 % yield). EL IC50 <10 nM. HL IC50 464 nM. HPLC: RT = 1.0 min (LCMS Method N). MS (ES): m/z = 527.2 [M+H]+. 1H NMR (500MHz, DMSO-d6) δ 9.38 - 8.85 (m, 1H), 8.51 - 8.13 (m, 2H), 8.13 - 7.67 (m, 2H), 7.40 (br. s., 1H), 6.88 (br. s., 2H),
4.42 (br. s., 1H), 3.66 - 3.58 (m, 3H), 3.19 (br. s., 5H), 2.97 (br. s., 2H), 2.90 (br. s., 1H), 2.18 (d, J=12.1 Hz, 2H), 2.06 (d, J=10.5 Hz, 2H).
Example B99
4-(2-(l-(Benzylsulfonyl)-2-oxo-2-((2-sulfamoylethyl)amino)ethyl)-l,3-benzothiazol-6- yl)-N- 2-methoxyethyl)benzamide

Compound B99a. N-(2-Methoxyethyl)-4-(2-(2-oxo-2-((2- sulfamoylethyl)amino)ethyl)benzo[d]thiazol-6-yl)benzamide
[00215] Compound B99a (26 mg, 52% yield) was prepared from Compound Ala and (4-((2-methoxyethyl)carbamoyl)phenyl)boronic acid as described in the general procedure given for Compound B2a. HPLC: RT = 0.83 min (LCMS Method M). MS (ES): m/z = 375.9 [M+H]+. 1H NMR (500 MHz, DMSO-d6) δ 8.44 - 8.68 (1 H, m), 8.36 (1 H, d, J=1.93 Hz), 7.88 (1 H, d, J=8.80 Hz), 7.56 - 7.70 (1 H, m), 6.90 (2 H, s), 4.06 (2 H, s), 3.43 - 3.62 (2 H, m), 3.07 - 3.22, (2 H, m).
Example B100
[00216] Example B99 (10 mg, 31% yield) was prepared from Compound B99a and benzyl sulfonylchloride as described in the general procedure given for Compound B31a.
EL IC50 < 10 iiM. HL IC50 55 iiM. HPLC: RT = 0.83 min (LCMS Method M). MS (ES): m/z = 630.8 [M+H]+. 1H NMR (500MHz, DMSO-d6) δ 9.02 (t, J=5.8 Hz, 1H), 8.65 - 8.50 (m, 1H), 8.25 - 8.13 (m, 1H), 7.99 (d, J=8.5 Hz, 1H), 7.95 (dt, J=8.5, 1.7 Hz, 1H), 7.88 (d, J=8.5 Hz, 1H), 7.78 (br. s., 1H), 7.40 (s, 2H), 7.37 - 7.14 (m, 2H), 7.00 - 6.90 (m, 2H), 6.10 (s, 1H), 4.74 (q, J=13.5 Hz, 1H), 4.68 - 4.51 (m, 1H), 3.71 - 3.56 (m, 2H), 3.52 - 3.42 (m, 4H), 3.29 - 3.27 (m, 3H), 3.24 - 3.08 (m, 2H).
[00217] Example B 100 to Example B156 were prepared as described in the general procedure given for Example B99.
Example B157
2- {6-[4-(4-Morpholinylcarbonyl)phenyl]- 1 ,3-benzothiazol-2-yl} -2- {[2-(4- morpholinyl)ethyl]sulfonyl}-N-(2-sulfamoylethyl)acetamide

Compound A1a
Compound B157a

Compound B157b

Compound B158c
 Compound B157a. Benzyl (2-((l-(6-bromobenzo[d]thiazol-2-yl)-2-oxo-2-((2- sulfamoylethyl)amino)ethyl)sulfonyl)ethyl)carbamate
Compound B157a. Benzyl (2-((l-(6-bromobenzo[d]thiazol-2-yl)-2-oxo-2-((2- sulfamoylethyl)amino)ethyl)sulfonyl)ethyl)carbamate
[00218] Compound B 157a (260 mg, 46% yield) was prepared from Compound Ala as described in the general procedure given for Compound B31a. HPLC: RT = 1.12 min (LCMS Method M). MS (ES): m/z = 619.1 [M+H]+. 1H NMR (400MHz, DMSO-d6) δ 8.96 (s, 1H), 8.48 (d, J=2.0 Hz, 1H), 8.02 (d, J=8.6 Hz, 1H), 7.73 (dd, J=8.6, 2.0 Hz, 1H), 7.47 (br. s., 1H), 7.40 - 7.22 (m, 5H), 6.90 (s, 2H), 5.03 (s, 2H), 3.74 - 3.34 (m, 6H), 3.25 - 3.02 (m,2H). Compound B157b. 2-((2-Aminoethyl)sulfonyl)-2-(6-bromobenzo[d]thiazol-2-yl)-N-(2- sulfamoylethyl)acetamide hydrobromide
[00219] To Compound B157a (253 mg, 0.41 mmol) was added 33% HBr in AcOH (4 mL). After 1 h, the reaction mixture was diluted with Et20 (~50 mL) generating a white precipitate, which was filtered and dried under reduced pressure to give Compound B157b (230 mg, 100 % yield) as a brown solid. HPLC: RT = 0.84 min (LCMS Method M). MS (ES): m/z = 487.0 [M+H]+. 1H NMR (400MHz, DMF) δ 9.52 (t, J=5.6 Hz, 1H), 8.56 (d, J=2.0 Hz, 1H), 8.06 - 8.03 (m., 1H), 7.80 (dd, J=8.6, 2.0 Hz, 1H), 7.33 (s, 2H), 7.05 (s, 3H), 6.74 (s, 1H), 4.27 - 4.10 (m, 2H), 3.86 - 3.77 (m, 2H), 3.77 - 3.66 (m, 4H), 3.47 - 3.33 (m, 2H).
Compound B 157c. 2-(6-Bromobenzo[d]thiazol-2-yl)-2-((2-morpholinoethyl)sulfonyl)-N- (2-sulfamoylethyl)acetamide
[00220] To a solution of Compound B 157b (105 mg, 0.190 mmol) in acetonitrile (7 mL) was added 2,2'-dibromodiethyl ether (0.027 mL, 0.20 mmol) followed by K2C03 (130 mg, 0.93 mmol), and the reaction mixture heated at 65 °C for 2 days. The reaction mixture was concentrated under reduced pressure and purified by silica gel
chromatography eluting with 0.5 to 10%> MeOH/DCM to give Compound B 157c (31 mg, 30 % yield) as a white solid. HPLC: RT = 1.39 min (LCMS Method B). MS (ES): m/z = 557.0 [M+H]+. 1H NMR (400MHz, DMF) δ 8.80 (br. s., 1H), 8.43 (br. s., 1H), 8.00 (d, J=8.6 Hz, 1H), 7.72 (d, J=7.9 Hz, 1H), 7.03 (br. s., 2H), 3.82 (d, J=5.9 Hz, 2H), 3.66 (d, J=6.4 Hz, 2H), 3.65 - 3.44 (m, 4H), 3.44 - 3.31 (m, 2H), 2.89 - 2.81 (m, 2H), 2.48 (br. s., 4H).
Example B157
[00221] Example B 157 (18 mg, 49% yield) was prepared from Compound B 157c and 4-(morpholine-4-carbonyl)phenyl)boronic acid as described in the general procedure given for Compound B2a. EL IC50 <10 iiM. HL IC50 147 iiM
HPLC: RT = 1.36 min (LCMS Method B). MS (ES): m/z = 666.1 [M+H]+. 1H NMR (400MHz, METHANOL-d4) δ 8.00 (br. s., 1H), 7.76 (s, 6H), 7.55 (d, J=8.1 Hz, 1H), 4.09 - 3.47 (m, 25H). [00222] Example B 158 to Example B161 were prepared as described in the general procedure given for Example B157.
Example B162
2-(6-( 1 -(2-Methoxy ethyl)- 1 H-pyrazol-4-yl)- 1 ,3 -benzothiazol-2-yl)-2-(methylsulfonyl)-N- -sulfamoylethyl)acetamide

Compound B162a

Example B162
Compound B 162a. 1 -(2-Methoxyethyl)-4-(4,4,5 ,5-tetramethyl- 1 ,3 ,2-dioxaborolan-2-yl)- lH-pyrazole
[00223] 4-(4,4,5,5-Tetramethyl-l,3,2-dioxaborolan-2-yl)-lH-pyrazole (100 mg, 0.52 mmol), l-chloro-2-methoxyethane (0.056 mL, 0.62 mmol) and cesium carbonate (252 mg, 0.73 mmol) in DMF (1 mL) were heated in microwave reactor at 160 °C for 30 min. The reaction mixture was concentrated under reduced pressure and purified by silica gel chromatography (ISCO, hexanes/ethyl acetate 0-100% over 15 min) to isolate Compound
B162a (130 mg, 100% yield). HPLC: RT = 1.0 min (LCMS Method M). MS (ES): m z = 253.0 [M+H]+. 1H NMR (500MHz, CHLOROFORM-d) δ 8.04 (s, IH), 7.83 (d, J=18.4 Hz, IH), 4.37 (t, J=5.2 Hz, IH), 3.78 (t, J=5.2 Hz, IH), 3.38 - 3.31 (m, 2H), 2.98 (s, 3H), 2.90 (s, 3H), 1.34 (s, 6H), 1.26 (s, 3H).
Example B162
[00224] Example B162 (9 mg, 50% yield) was prepared from Compound B 162a and Compound B31a as described in the general procedure given for Example B2a. EL IC50 <10 nM. HL IC50 995 nM. HPLC: RT = 0.7 min (LCMS Method M). MS (ES): m/z = 501.8 [M+H]+. 1H NMR (500MHz, DMSO-d6) δ 8.97 (s, IH), 8.36 (d, J=1.4 Hz, IH), 8.27 (s, IH), 8.05 (d, J=8.5 Hz, IH), 8.00 (d, J=0.6 Hz, IH), 7.79 (dd, J=8.5, 1.9 Hz, IH), 6.95 (s, 2H), 5.99 (s, IH), 4.30 (t, j=5.2 Hz, 3H), 3.74 (t, j=5.4 Hz, 3H), 3.65 - 3.56 (m, 6H), 3.28 - 3.24 (m, 4H), 3.21 - 3.15 (m, 3H). [00225] Example B 163 to Example B165 were prepared as described in the general procedure given for Example B162.
Example B166
2-(6-(4-(3,3-Difluoroazetidine-l-carbonyl)phenyl)benzo[d]thiazol-2-yl)-2- (methylsulfonyl)-N-(2-sulfamoylethyl)acetamide

[00226] A mixture of 4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)benzoic acid (490 mg, 2.0 mmol), 3,3-difluoroazetidine hydrochloride (260 mg, 2.00 mmol), DIPEA (0.686 mL, 3.93 mmol) and HATU (900 mg, 2.4 mmol) in DMF (4 mL) was stirred at room temperature for 3 days. The reaction mixture was diluted with EtOAc, the organic portion washed successively with water and brine, dried over sodium sulfate and filtered. The filtrate was concentrated under reduced pressure. The residue was purified by silica gel chromatography (eluting with 0-100% EtOAc/hexane) to give Compound B 166a (360 mg, 57 % yield) as a white solid. HPLC: RT = 1.9 min (LCMS Method Q). MS (ES): m/z = 324.0 [M+H]+. 1H NMR (400MHz, CHLOROFORM-d) δ 7.87 (s, 2H), 7.62 (d, J=8.1 Hz, 2H), 4.53 (t, J=12.0 Hz, 4H), 1.36 (s, 12H).
Compound B 166b. 2-(6-(4-(3,3-difluoroazetidine- 1 -carbonyl)phenyl)benzo[d]thiazol-2- yl)-N-(2-sulfamoylethyl)acetamide
[00227] A mixture of Compound B166a (140 mg, 0.37 mmol), Compound Ala (144 mg, 0.44 mmol), K3PO4 (196 mg, 0.93 mmol) and chloro(2-dicyclohexylphosphino- 2',4',6'-triisopropyl- 1 , 1 '-biphenyl)[2-(2'-amino- 1 , 1 '-biphenyl)]palladium(II) (2nd generation X-Phos precatalyst, 29 mg, 0.037 mmol) in dioxane (3 mL) and water (1 mL) was purged with argon gas and heated at 95 °C for 1 hour. The reaction mixture was concentrated under reduced pressure. The residue was purified by silica gel
chromatography (eluting with 0-10% MeOH/DCM) to give Compound B166b (116 mg, 62.0% yield) as a white solid. HPLC: RT = 1.6 min (LCMS Method Q). MS (ES): m/z = 495 [M+H]+. 1H NMR (400MHz, DMSO-d6) δ 8.55 (t, J=5.4 Hz, 1H), 8.48 (d, J=1.5 Hz, 1H), 8.05 (d, J=8.6 Hz, 1H), 7.90 - 7.85 (m, 3H), 7.83 (s, 2H), 6.92 (s, 2H), 5.05 - 4.33 (m, 4H), 4.15 - 4.09 (m, 2H), 3.60 - 3.44 (m, 2H), 3.18 (d, J=5.3 Hz, 2H).
Example B166
[00228] Example B 166 (16.4 mg, 30%> yield) was prepared from Compound B166b as described in the general procedure given for Compound B3 la. EL IC50 < 10 nM. HL IC50 13 nM. HPLC: RT = 1.4 min (LCMS Method O). MS (ES): m/z = 573.0 [M+H]+.
1H NMR (500MHz, DMSO-d6) δ 8.98 (t, J=5.6 Hz, 1H), 8.57 (s, 1H), 8.18 (d, J=8.5 Hz, 1H), 7.97 - 7.90 (m, 2H), 7.89 - 7.85 (m, 2H), 7.85 - 7.81 (m, 2H), 6.98 - 6.82 (m, 2H), 6.04 (s, 1H), 3.61 (q, J=6.9 Hz, 2H), 3.25 - 3.23 (m, 3H), 3.22 - 3.16 (m, 2H).
[00229] Example B 167 to Example B171 were prepared as described in the general procedure given for Example B166.
Example B172
N-(2-Methoxyethyl)-N-methyl-4-(2-(l-(methylsulfonyl)-2-oxo-2-((2- sulfamoylethyl)amino)ethyl)- 1 ,3 -benzothiazol-6-yl)benzamide

Example B172
Compound B 172a. N-(2-methoxyethyl)-N-methyl-4-(4,4,5 ,5-tetramethyl- 1,3,2- dioxaborolan-2-yl)benzamide
[00230] EDC (310 mg, 1.6 mmol) was added to DMF (4 mL) solution of 4-(4,4,5,5- tetramethyl-l,3,2-dioxaborolan-2-yl)benzoic acid (200 mg, 0.81 mmol), 2-methoxy-N- methylethanamine (144 mg, 1.6 mmol), TEA (0.23 mL, 1.6 mmol) and HOBT hydrate (247 mg, 1.60 mmol). After 3 days at room temperature, the reaction mixture was diluted in water (15 mL), extracted with DCM (3 X 10 mL), the combined organic portions dried over Na2S04, filtered, concentrated under reduced pressure and purified by reverse phase HPLC (Phenomemenx Luna AXIA 5 micron CI 8, 21.2X100, UV at 220 nm, 10 to 70% B over 30 minutes with 10 minute hold time, solvent A: 90% water/ACNl/0.1%TFA, solvent B:90% ACN/water/ 0.1 %>TF A, Flow rate 20 mL/min; detector at 254) to isolate Compound B172a (47 mg, 18 % yield) as a clear oil. HPLC: RT = 0.96 min (LCMS Method M). MS (ES): m/z = 319.7 [M+H]+. 1H NMR (500MHz, CHLOROFORM-d) δ
9.45 - 8.71 (m, 1H), 7.85 (d, J=7.2 Hz, 2H), 7.40 (d, J=8.0 Hz, 2H), 3.89 - 3.64 (m, 2H), 3.42 (d, J=12.1 Hz, 3H), 3.34 - 3.12 (m, 3H), 3.04 (s, 2H), 1.36 (s, 12H).
Example B172
[00231] Example B172 (2.2 mg, 7% yield) was prepared from Compound B172a as described in the general procedure given for Compound B2a. EL IC50 < 10 nM. HL IC50 77 nM. HPLC: RT = 1.22 min (LCMS Method N). MS (ES): m/z = 569.1 [M+H]+. 1H NMR (500MHz, DMSO-d6) δ 9.02 (br. s., 1H), 8.54 (s, 1H), 8.17 (d, J=8.5 Hz, 1H), 7.99 - 7.88 (m, 1H), 7.83 (br. s., 2H), 7.52 (d, J=7.7 Hz, 2H), 6.97 (s, 2H), 6.04 (s, 1H), 3.67 - 3.55 (m, 4H), 3.44 (br. s., 5H), 3.31 (br. s., 2H), 3.22 - 3.14 (m, 3H), 3.00 (br. s., 3H)
Example B173
4-(2-(l-(Methylsulfonyl)-2-oxo-2-(2-sulfamoylethylamino)ethyl)benzo[d]thiazol-6- yl)benzoic acid

[00232] To a solution of Example B 157 (101 mg, 0.18 mmol) in DCM (2.0 mL) was added TFA (1.0 mL, 13 mmol). The mixture was stirred at room temperature for 3 hours. The mixture was concentrated under reduced pressure. The residue was dissolved in
DMF (2 mL), filtered and purified by preparative HPLC (Column: Waters XBridge CI 8, 19 x 200 mm, 5-μιη particles; Mobile Phase A: 5:95 acetonitrile: water with 10-mM ammonium acetate; Mobile Phase B: 95:5 acetonitrile: water with 10-mM ammonium acetate; Gradient: 0-30% B over 20 minutes, then a 5-minute hold at 100% B; Flow: 20 mL/min). Fractions containing the desired product were combined and dried via centrifugal evaporation to give Example B173 (79 mg, 87% yield). EL IC50 < 10 nM. HL IC50 274 nM. HPLC: RT = 1.1 min (LCMS Method O). MS (ES): m/z = 498.0 [M+H]+. 1H NMR (500MHz, DMSO-d6) δ 13.05 (br. s., 1H), 9.03 (br. s., 1H), 8.60 (br. s., 1H),
8.20 (d, J=7.7 Hz, 1H), 8.10 - 8.04 (m, 2H), 7.97 - 7.89 (m, 3H), 6.99 (br. s., 2H), 6.06 (br. s., 1H), 3.62 (br. s., 2H), 3.26 (br. s., 3H), 3.22 - 3.17 (m, 2H).
Example B174
2-(6-(4-(3-Hydroxy-3-methylazetidine-l-carbonyl)phenyl)benzo[d]thiazol-2-yl)-2- (methylsulfonyl)-N-(2-sulfamoylethyl)acetamide

[00233] Example B174 (15 mg, 55%) was prepared from Example B173 and 3- methylazetidin-3-ol hydrochloride as described in the general procedure given for Example B166. EL IC50 < 10 iiM. HL IC50 402 iiM. HPLC: RT = 1.0 min (LCMS Method O). MS (ES): m/z = 567.0 [M+H]+. 1H NMR (500MHz, DMSO-d6) δ 9.00 (br. s., 1H), 8.56 (br. s., 1H), 8.19 (d, J=8.5 Hz, 1H), 7.99 - 7.90 (m, 2H), 7.86 (d, J=7.2 Hz, 2H), 7.77 (d, J=6.6 Hz, 2H), 6.96 (br. s., 2H), 6.06 (br. s., 1H), 5.70 (br. s., 1H), 4.20 (d, J=16.5 Hz, 2H), 4.00 - 3.85 (m, 4H), 3.62 (br. s., 2H), 3.27 - 3.24 (m, 3H), 3.23 - 3.17 (m, 2H).
[00234] Example B 175 to Example B179 were prepared as described in the general procedure given for Example B174.
Example B 180
2-(Methylsulfonyl)-2-(6-(2-oxo-2,3-dihydro-lH-indol-5-yl)-l,3-benzothiazol-2-yl)-N-(2- sulfamoylethyl)acetamide

Example B180
[00235] Example B180 (3.8 mg, 14%) was prepared from Compound B31a and 5- (4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)indolin-2-one as described in the general procedure given for Example 166. EL IC50 < 10 nM. HL IC50 170 nM.
HPLC: RT = 1.1 min (LCMS Method Q). MS (ES): m/z = 509.1 [M+H]+.
1H NMR (500MHz, DMSO-d6) δ 10.54 (br. s., 1H), 9.02 (br. s., 1H), 8.41 (br. s., 1H), 8.12 (d, J=8.5 Hz, 1H), 7.82 (d, J=8.5 Hz, 1H), 7.69 - 7.56 (m, 2H), 7.03 - 6.97 (m, 2H), 6.03 (br. s., 1H), 3.92 (br. s., 1H), 3.64 - 3.54 (m, 4H), 3.30 - 3.10 (m, 5H).
[00236] Example B 181 to Example B 186 were prepared as described in the general procedure given for Example B180.
Example B187
2-(Methylsulfonyl)-2-(6-(6-oxo- 1 ,6-dihydro-3-pyridinyl)- 1 ,3-benzothiazol-2-yl)-N-(2- sulfamo lethyl)acetamide

[00237] A mixture of Example B19 (12 mg, 0.025 mmol), Nal (11 mg, 0.074 mmol) and TMS-C1 (0.016 mL, 0.12 mmol) in acetonitrile (1.5 mL) was stirred at 80 °C for 2 hours. The reaction mixture was quenched with water and purified by reverse phase
preparative HPLC (method A) to give Example B187 (6 mg, 50% yield). EL IC50 16 nM. HL IC50 155 nM. HPLC: RT = 1.2 min (LCMS Method Q). MS (ES): m/z = 471.0 [M+H]+. 1H NMR (400MHz, DMSO-d6) δ 11.97 (br. s., 1H), 9.01 (t, J=5.6 Hz, 1H), 8.39 (s, 1H), 8.18 - 8.06 (m, 1H), 7.98 - 7.92 (m, 1H), 7.88 - 7.82 (m, 1H), 7.79 (dd, J=8.6, 1.8 Hz, 1H), 6.99 (s, 2H), 6.02 (s, 1H), 3.66 - 3.56 (m, 2H), 3.23 (s, 3H), 3.21 - 3.13 (m, 2H).
[00238] Example B 188 to Example B195 were prepared as described in the general procedure given for Example B187. Example B 196
tert-Butyl 2-(2-oxo-2-((2-sulfamoylethyl)amino)- 1 - ((3,3trifluoropropyl)sulfonyl)ethyl)benzo[d]thiazole-6-carboxylate
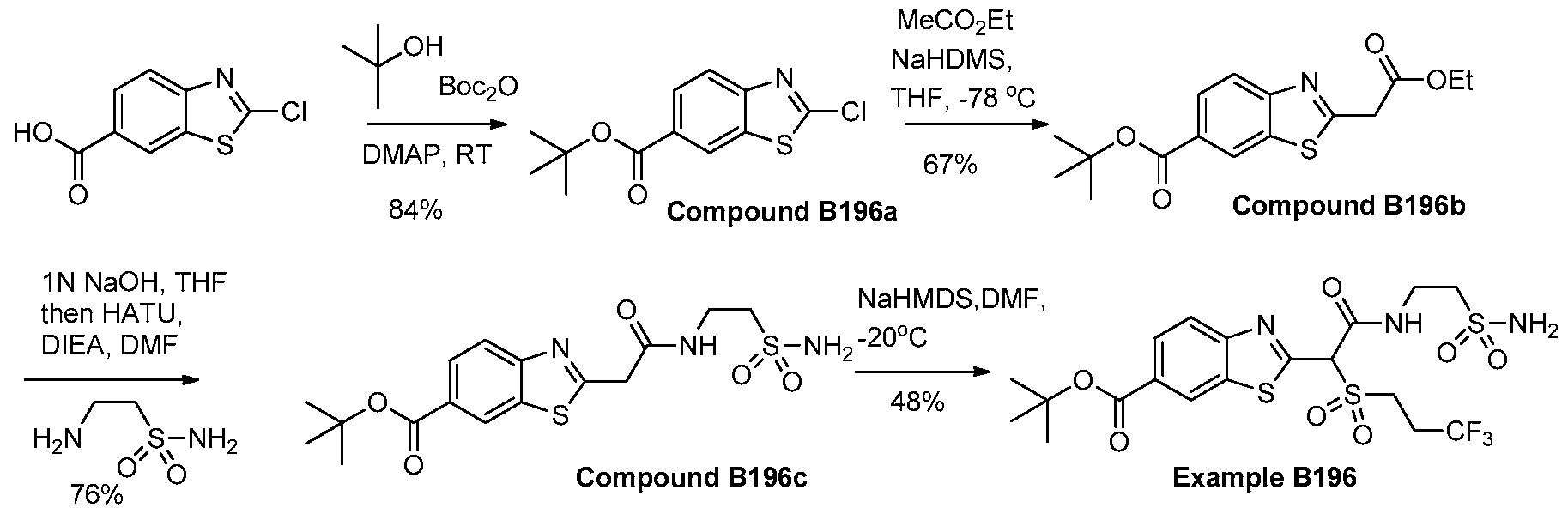
[00239] To a solution of di-tert-butyl dicarbonate (1.2 mL, 5.0 mmol) and 2- chlorobenzo[d]thiazole-6-carboxylic acid (530 mg, 2.5 mmol) in anhydrous t-butanol (10 mL) was added dimethylaminopyridine (91 mg, 0.74 mmol) at room temperature. After 1 hour, the reaction mixture was concentrated under reduced pressure and purified by silica gel chromatography (ISCO: 0-50% EtOAc/Hexane) to afford Compound B196a (560 mg, 84 % yield) as a clear oil. HPLC: RT = 2.22 min (LCMS Method B). MS (ES): m/z = 270.0 [M+H]+. 1H NMR (400MHz, CHLOROFORM-d) δ 8.45 (d, J=1.5 Hz, 1H), 8.12 (dd, J=8.6, 1.8 Hz, 1H), 7.97 (d, J=8.6 Hz, 1H), 1.55 - 1.42 (m, 9H). Compound B196b. tert-Butyl 2-(2-ethoxy-2-oxoethyl)benzo[d]thiazole-6-carboxylate
[00240] Ethyl acetate (0.26 mL, 2.67 mmol) was added to cooled (-78° C) solution of NaHMDS (1 M in THF, 4.9 mL, 4.9 mmol) in toluene (10 mL) over a period of 15 minutes. After one hour, Compound B196a (0.6 g, 2.2 mmol in toluene (5 mL) was added over a period of 15 minutes. After one hour, the reaction mixture was warmed to - 5° C over a period of 90 minutes. The reaction mixture was diluted with IN HC1 (50 mL) and extracted with ethyl acetate (3 X 50 mL). The organic phase was washed with brine (100 mL), dried over Na2S04, filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography (0 to 50% ethyl acetate/hexanes) to isolate Compound B196b (480 mg, 67% yield) as a white solid. HPLC: RT = 2.13 min (LCMS Method B). MS (ES): m/z = 322.0 [M+H]+. 1H NMR (400MHz,
CHLOROFORM-d) δ 8.57 - 8.52 (m, 1H), 8.11 (dd, J=8.6, 1.5 Hz, 1H), 8.04 - 7.98 (m, 1H), 4.32 - 4.25 (m, 2H), 4.20 (s, 2H), 1.63 (s, 9H), 1.32 (t, J=7.2 Hz, 3H).
Compound B196c. tert- utyl 2-(2-oxo-2-((2- sulfamoylethyl)amino)ethyl)benzo [d]thiazole-6-carboxylate
[00241] Compound B 196c (810 mg, 76% yield) was prepared from Compound B 196b as described in the general procedure given for Compound Ala. HPLC: RT = 1.88 min (LCMS Method B). MS (ES): m/z = 399.9 [M+H]+. 1H NMR (400MHz,
CHLOROFORM-d) d 8.51 - 8.47 (m, 1H), 8.07 (dd, J=8.6, 1.5 Hz, 1H), 7.97 (dd, J=8.6, 0.4 Hz, 1H), 4.11 (s, 2H), 3.83 (m, 2H), 3.39 (m, 2H), 1.62 (s, 9H).
Example B196
[00242] Example B 196 (120 mg, 48% yield) was prepared from Compound B 196c as described in the general procedure given for Compound B3 la. EL IC50 65 nM. HL IC50 379 nM. HPLC: RT = 2.16 min (LCMS Method B). MS (ES): m/z = 560.0 [M+H]+. 1H NMR (400MHz, CHLOROFORM-d) δ 7.78 - 7.74 (m, 1H), 7.33 (dd, J=8.6, 1.5 Hz, 1H), 7.23 (dd, J=8.6, 0.4 Hz, 1H), 3.37 (s, 2H), 3.09 (m, 2H), 2.96 (m, 2H), 0.88 (s, 9H).
Example B197
2-(6-(3,3-Difluoropyrrolidine-l-carbonyl)benzo[d]thiazol-2-yl)-N-(2-sulfamoylethyl)-2- ((3,3,3-trifluoropropyl)sulfonyl)acetamide
TFA/DCM then

[00243] Example B197 (6.2 mg, 21% yield) was prepared from Example B 196 as described in the general procedure given for Example B174. EL IC50 < 10 11M. HL IC50 10,100 11M. HPLC: RT = 2.16 (LCMS Method O). MS (ES): m/z = 593.05 [M+H]+. 1H NMR (500MHz, DMSO-d6) δ 8.15 (d, J=8.5 Hz, 1H), 6.99 (m, 2H), 4.04 - 3.53 (m, 8H), 3.27 - 3.13 (m, 2H), 2.94 - 2.80 (m, 2H), 2.50 - 2.35 (m, 2H).
[00244] Example B 198 to Example B201 were prepared as described in the general procedure given for Example B197.
Example B202
N-(2-methoxyethyl)-3 -(2-(2-oxo-2-((2-sulfamoylethyl)amino)- 1
trifluoropropyl)sulfonyl)ethyl)benzo[d]thiazol-6-yl)benzamide

ompound B202a

xamp e
[00245] Example B202 (1.7 mg, 5.0 % yield) was prepared from Compound B202a as described in the general procedure given for Example B99. EL IC50 24 11M. HL IC50 170 11M. HPLC: RT = 1,51 (LCMS Method O). MS (ES): m/z = 637.1 [M+H]+. 1H NMR
(500MHz, DMSO-d6) δ 8.19 - 8.09 (m, 2H), 7.92 - 7.80 (m, 3H), 7.58 - 7.48 (m, 2H), 3.55 (m, 4H), 3.41 (m, 4H), 3.21 (s, 3H), 3.10-3.13 (m, 2H), 2.81 - 2.75 (m, 2H).
Example B203
2-(5-methyl-6-(l -methyl- lH-pyrazol-4-yl)-l,3-benzothiazol-2-yl)-2-(methylsulfonyl)-N- -sulfamoylethyl)acetamide

[00246] Example B203 (4.1 mg, 14.3 % yield) was prepared from Compound B203a as described in the general procedure given for Example B99. EL IC50 10 11M. HL IC50 141 11M. HPLC: RT = 0.70 (LCMS Method M). MS (ES): m/z = 472.8 [M+H]+. 1H NMR (500 MHz, METHANOL-d4) δ 7.98 (s, 1H), 7.95 (s, 1H), 7.90 (s, 1H), 7.76 (d, J=0.55 Hz, 1H), 5.49 (s, 1H), 3.97-4.00 (m, 3H), 3.80 (d, J=2.75 Hz, 1H), 3.59 (d, J=7.98 Hz, 1H), 3.19-3.22 (m, 2H), 3.17 (s, 3H), 2.44-2.55 (m, 3H).
[00247] Example B204 to Example B205 were prepared as described in the general procedure given for Example B2.
[00248] Example B207 to Example B281 were prepared as described in the general procedure given for Example B31.
[00249] Example B282 to Example B312 were prepared as described in the general procedure given for Example B99.
[00250] Example B313 to Example B316 were prepared as described in the general procedure given for Example B157.
[00251] Example B317 was prepared as described in the general procedure given for Example B162.
[00252] Example B318 to Example B324 were prepared as described in the general procedure given for Example B166.
[00253] Example B325 to Example B327 were prepared as described in the general procedure given for Example B173.
[00254] Example B328 to Example B351 were prepared as described in the general procedure given for Example B174.
[00255] Example B352 to Example B374 were prepared as described in the general procedure given for Example B 180.
[00256] Example B375 to Example B385 were prepared as described in the general procedure given for Example B187.
[00257] Example B386 was prepared as described in the general procedure given for Example B31.
Example B387
2- {6-[ 1 -(2-Hydroxy-2-methylpropyl)-2-oxo- 1 ,2-dihydropyridin-4-yl]- 1 ,3-benzothiazol-2- yl} -N-(2-sulfamoylethyl)-2- { [4-(trifluoromethyl)phenyl]methanesulfonyl} acetamide

Compound B387a

Compound B387b Compound B387c CF3
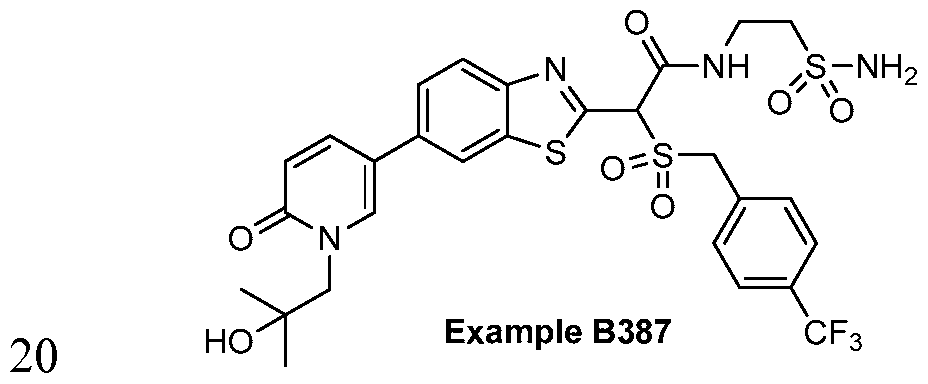 Compound B387a. 5-bromo-l -(2-hydroxy-2-methylpropyl)pyridin-2(lH)-one
Compound B387a. 5-bromo-l -(2-hydroxy-2-methylpropyl)pyridin-2(lH)-one
[00258] A mixture of 5-bromopyridin-2(lH)-one (600 mg, 3.5 mmol), 2,2- dimethyloxirane (750 mg, 10 mmol) and K2CO3 (960 mg, 6.9 mmol) in DMF (3 mL) was heated at 100 °C for 2 hours. The reaction mixture was allowed to cool to room temperature. The reaction mixture was diluted with ethyl acetate (80 mL), washed with water, brine, and dried over MgS04. The organic layer was filtered and concentrated under reduced pressure. The residue was purified by silica gel chromatography (eluting with 10 to 50% EtOAc/hexane) to give Compound B387a (650 mg, 76% yield). 1H NMR (400MHz, CHLOROFORM-d) δ 7.50 (d, J=2.6 Hz, 1H), 7.40 (dd, J=9.6, 2.8 Hz, 1H), 6.54 (d, J=9.7 Hz, 1H), 3.99 (s, 2H), 3.57 - 3.39 (s, 1H), 1.27 (s, 6H).
Compound B387b. l-(2-hydroxy-2-methylpropyl)-5-(4,4,5,5-tetramethyl-l,3,2- dioxaborolan-2-yl)pyridin-2(lH)-one
[00259] To a solution of Compound B387a (500 mg, 2.0 mmol) was added
bis(pinacolato)diboron (1.0 g, 4.1 mmol), PdCl2(dppf)-CH2Cl2 adduct (166 mg, 0.200 mmol) and potassium acetate (995 mg, 10.1 mmol). The reaction mixture was heated at 100 °C for 16 hours. The reaction mixture was allowed to cool to room temperatuer and concentrated under reduced pressure. The residue was purified by silica gel
chromatography (eluting with 0-10% MeOH) to give Compound B387b (360 mg, 1.2 mmol, 60 % yield). 1H NMR (400MHz, CHLOROFORM-d) δ 7.83 - 7.59 (m, 2H), 6.59 (d, J=9.0 Hz, 1H), 4.10 - 3.94 (m, 2H), 1.46 - 1.19 (m, 18H).
Example B387
[00260] Example B387 (2.2 mg, 9.0% yield) was prepared from Compound B387c (prepared as described in the general procedure given for Example B31) and Compound B387b as described in the general procedure given for Example B180. EL IC50 13.6 nM. HL IC50 6544 nM. HPLC: RT = 1.4 min (LCMS Method Q). MS (ES): m/z = 687.1 [M+H]+. 1H NMR (500MHz, DMSO-d6) δ 14.04 and 11.46 and 6.15 (s, 1H), 9.12 - 9.02 and 8.61 (1H), 8.19 (br. s., 1H), 7.97 (s, 1H), 7.82 - 7.55 (m, 6H), 6.97 (br. s., 2H), 6.82 and 6.64 - 6.56 (1H), 6.77 - 6.66 (m, 1H), 6.53 (s, 1H), 6.15 (s, 1H), 4.93 - 4.71 (m, 2H), 4.03 - 3.91 (m, 2H), 3.68 - 3.57 (m, 2H), 3.25 - 3.14 (m, 2H), 1.13 (d, J=6.9 Hz, 6H).
[00261] Example B388 to Example B390 were prepared as described in the g procedure given for Example B388.
Example B391
tert-Butyl 2-(l-(methylsulfonyl)-2-oxo-2-((2-sulfamoylethyl)amino)ethyl)-l ,3- benzothiazole-5-carboxylate

Compound B391c 27 % Example B391 Compound B391b. tert-Butyl 2-(2-ethoxy-2-oxoethyl)benzo[d]thiazole-5-carboxylate
[00262] To a solution of Compound B391a (1.49 mmol, 395 mg, prepared as described in the general procedure given for Example B26) in dichloromethane (20 mL) was added tert-butyl 2,2,2-trichloroacetamidate (1.6 g, 7.4 mmol). Boron trifluoride etherate (38 mL, 0.30 mmol) was added, and the mixture was stirred at room temperature. After 7 hours, the mixture was loaded onto Celite and purified by ISCO flash chromatography (120 g silica gel cartridge, 0-50% ethyl acetate - hexane gradient over 24 min., 85 mL/min.). The fractions containing the product were combined, and the solvent removed under reduced pressure to provide Compound B391b (184 mg, 38.0% yield). HPLC: RT = 2.07 min (LCMS Method B). MS (ES): m/z = 322 [M+H]+. 1H NMR (400MHz, CHLOROFORM-d) δ 8.64 (d, J=l .l Hz, 1H), 8.03 (dd, J=8.4, 1.5 Hz, 1H), 7.90 (d,
J=8.4 Hz, 1H), 4.27 (q, J=7.3 Hz, 2H), 4.19 (s, 2H), 1.63 (s, 9H), 1.31 (t, J=7.2 Hz, 3H).
Compound B391 c. tert-Butyl 2-(2-oxo-2-((2-sulfamoylethyl)amino)ethyl)
benzo[d]thiazole-5-carboxylate
[00263] Compound B391b (184 mg, 0.570 mmol) was dissolved in THF (2 mL) and sodium hydroxide (0.57 mL, 0.57 mmol., 1.0 M ) was added. After 1 hour, the solvent was removed under reduced pressure, and the residue dried in vacuo. The residue was dissolved in DMF (2 mL) and 2-aminoethanesulfonamide HC1 salt was added.
Diisopropylethyl amine (0.20 mL, 1.2 mmol) and HATU (240 mg, 0.63 mmol) were added, and the mixture was stirred at room temperature for 7 hours. DMF was removed in vacuo, and the residue was purified by ISCO flash chromatography (12 g silica gel cartridge, 0-100% acetonitrile - dichloromethane gradient over 11 min., 30 mL/min.). The fractions containing the product were combined, and the solvent removed to provide Compound B391c (131 mg, 53.0% yield). HPLC: RT = 1.71 min (LCMS Method B).
MS (ES): m/z = 400 [M+H]+. 1H NMR (400MHz, CHLOROFORM-d) δ 8.61 (d, J=0.9 Hz, 1H), 8.05 (dd, J=8.4, 1.5 Hz, 1H), 7.90 (dd, J=8.4, 0.4 Hz, 1H), 7.68 (br. s., 1H), 5.05 (br. s., 2H), 4.10 (s, 2H), 3.87 (q, J=6.1 Hz, 2H), 3.40 - 3.31 (m, 2H), 1.64 (s, 9H). Example B391
[00264] Compound B391c (28 mg, 0.070 mmol) was dissolved in DMF (2 mL), and the mixture was cooled to -25 °C. Sodium bis(trimethylsilyl)amide (0.10 mL, 0.10 mmol, 1.0 M in THF) was added dropwise. After 10 min, methanesulfonyl chloride (8\L, 0.1 mmol) was added. After 30 min., the reaction was quenched by the addition of acetic acid, and the solvent was removed in vacuo. The residue was purified by ISCO flash chromatography (4 g silica gel cartridge, 0-100% acetonitrile - dichloromethane gradient over 11 min., 18 mL/min.). The fractions containing the product were combined and the solvent removed to provide Example B391 (10 mg, 28% yield). EL IC50 25 nM. HL IC50 53.4 nM. HPLC: RT = 1.72 min (LCMS Method B). MS (ES): m/z = 478
[M+H]+. 1H NMR (400MHz, DMSO-d6) δ 8.98 (t, J=5.7 Hz, 1H), 8.57 - 8.54 (m, 1H), 8.30 (d, J=8.4 Hz, 1H), 8.02 (dd, J=8.4, 1.5 Hz, 1H), 6.96 (s, 2H), 6.08 (s, 1H), 3.64 - 3.57 (m, 2H), 3.24 (s, 3H), 3.22 - 3.16 (m, 2H), 1.60 (s, 9H).
Example B392
2-(l-(Methylsulfonyl)-2-oxo-2-((2-sulfamoylethyl)amino)ethyl)benzo[d]thiazole-5- carboxylic acid
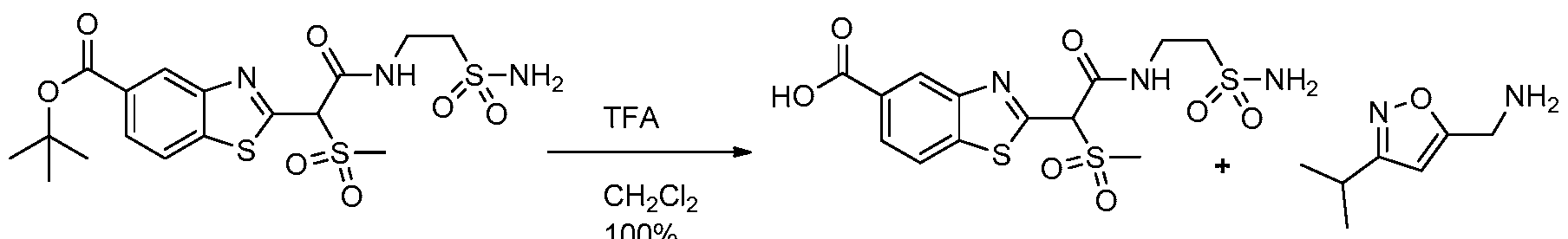

Example B392
Compound B392a. 2-(l -(Methylsulfonyl)-2-oxo-2-((2-sulfamoylethyl)amino)ethyl) benzo[d]thiazole-5-carboxylic acid
[00265] Compound B392a was synthesized by cleaving the tert-butyl ester of Example B391 using the procedure described for Example B197. HPLC: RT = 1.14 min (LCMS Method B). MS (ES): m/z = 421.9 [M+H]+. The product was used without further purification or characterization assuming 100% yield. Example B392
[00266] Example B392 (5.9 mg, 43 % yield) was prepared from Compound B392a and (3-isopropylisoxazol-5-yl)methanamine as described in the general procedure given for Example B197. EL IC50 3.1 nM. HL IC50 92.7 nM. HPLC: RT = 1.24 min. (LCMS Method N). MS (ES): m/z = 478 [M+H]+. 1H NMR (500MHz, DMSO-d6) δ 9.35 (br. s., 1H), 8.99 (br. s., 1H), 8.63 (s, 1H), 8.29 (d, J=8.3 Hz, 1H), 8.03 (d, J=8.3 Hz, 1H), 7.97 (s, 1H), 6.96 (s, 2H), 6.37 (s, 1H), 6.09 (s, 1H), 4.63 (d, J=5.0 Hz, 2H), 3.62 (m, 2H), 3.25 (s, 3H), 3.21 (br. m., 2H), 3.03 - 2.93 (m, 1H), 1.22 (d, J=6.9 Hz, 6H).
[00267] Example B393 to Example B394 were prepared as described in the general procedure given for Example B392.
Example B395
2-(6-(5-(4-Fluorophenyl)-l,3,4-oxadiazol-2-yl)benzo[d]thiazol-2-yl)-2-(methylsulfonyl)- N-(2-sulfamoylethyl)acetamide
- I l l -

Compound B395a

Example B395
[00268] Compound B395a (prepared following the general procedure given for Example B197) and 4-fluorobenzohydrazide (10 mg, 0.065 mmol) were dissolved in dioxane (1.0 mL) and treated with DIEA (0.036 mL, 0.21 mmol) and 1- propanephosphonic acid cyclic anhydride (0.12 mL, 0.21 mmol, 50% in ethyl acetate). The reaction mixture was heated at 70° C for 1 hour, allowed to cool to room temperature and concentrated under reduced pressure. The material was purified via preparative HPLC (Column: Waters XBridge CI 8, 19 x 200 mm, 5-μιη particles; Mobile Phase A: 5:95 acetonitrile: water with 0.1% TFA; Mobile Phase B: 95:5 acetonitrile: water with 0.1% TFA; Gradient: 10-55% B over 20 minutes, then a 5-minute hold at 100% B; Flow: 20 mL/min). Fractions containing the desired product were combined and dried via centrifugal evaporation to afford Example B395 (1 mg, 3% yield) as a clear oil. EL IC50 148 nM. HL IC50 275.5 nM. HPLC: RT = 1.50 min (LCMS Method N). MS (ES): m/z = 540.15 [M+H]+. 1H NMR (500MHz, DMSO-d6) d 9.08 - 8.99 (m, 1H), 8.31 (s, 1H), 8.29 - 8.21 (m, 2H), 7.58 - 7.45 (m, 2H), 6.95 (m, 1H), 3.68 - 3.56 (m, 2H), 3.26 (s, 3H), 3.23 - 3.16 (m, 2H).
Example B396
2-[6-(6-fluoropyridin-3-yl)-l,3-benzothiazol-2-yl]-2-(2-methoxyethanesulfonyl)-N- [(methylcarbamoyl)methyl] acetamide

Compound B396a Compound B396b

(41 %)
Compound B396c

Compound B396b. tert-butyl 2-(2-(6-(6-fluoropyridin-3-yl)benzo[d]thiazol-2- yl)acetamido)acetate
[00269] To a solution of Compound B396a (440 mg, 1.4 mmol, prepared as described in the general procedure given for Compound B23a) in THF (10 mL) at 0 °C was added IN NaOH (1.5 mL, 1.5 mmol). The ice bath was removed and the mixture stirred for 4h. The mixture was evaporated under reduced pressure and the residue evaporated from toluene (2x) under reduced pressure. The residue was dissolved in DMF (7 mL) at 0 °C then DIPEA (0.73 mL, 4.2 mmol) added followed by glycine tert-butyl ester
hydrochloride (280 mg, 1.7 mmol) and HATU (690 mg, 1.8 mmol). The mixture was stirred for 1 h then poured into saturated NH4C1 and extracted with EtOAc (3x). The combined organic extracts were washed with brine, dried (Na2S04) filtered and concentrated under reduced pressure. The residue was purified by silica gel
chromatography eluting with 10 to 75% EtOAc/DCM to give Compound B396b (190 mg, 34 % yield) as a yellow solid. RT = 1.85 min (LCMS Method B). MS (ES): m/z = 403.1 [M+H]+. 1H NMR (400MHz, CDC13) δ 8.48 (d, J=2.0 Hz, 1H), 8.12 (d, J=8.6 Hz, 1H),
8.07 - 7.97 (m, 2H), 7.65 (dd, J=8.5, 1.7 Hz, 1H), 7.45 (br. s., 1H), 7.05 (dd, J=8.5, 3.0 Hz, 1H), 4.14 (s, 2H), 4.01 (d, J=5.1 Hz, 2H), 1.47 (s, 9H).
Compound B396c. tert-butyl 2-(2-(6-(6-fluoropyridin-3-yl)benzo[d]thiazol-2-yl)-2-((2- methoxyethyl)sulfonyl)acetamido)acetate
[00270] To a solution of Compound B396b (190 mg, 0.47 mmol) in DMF (5 mL) at brine/ice bath temperature was added IN NaHMDS in THF (0.71 mL, 0.71 mmol) and the mixture stirred for 2 min. A solution of 2-methoxyethanesulfonyl chloride (113 mg, 0.710 mmol) in DMF (0.5 mL) was added dropwise and the reaction stirred for 5 min. The reaction was quenched by the addition of saturated NH4C1 and extracted with 50% EtOAc/hexanes (3x). The combined organic extracts were washed with brine then dried (Na2S04) filtered and concentrated. The residue was purified by silica gel
chromatography eluting with 5 to 50%> EtOAc/DCM to give Compound B396c (100 mg, 41 % yield) as a brown solid. RT = 1.93 min (LCMS Method B). MS (ES): m/z = 524.1 [M+H]+. 1H NMR (400MHz, CDC13) δ 14.41 (s, 1H), 8.42 (d, J=2.6 Hz, 1H), 8.13 (t, J=5.3 Hz, 1H), 8.00 - 7.90 (m, 1H), 7.78 - 7.71 (m, 1H), 7.57 - 7.49 (m, 1H), 7.44 - 7.36 (m, 1H), 7.03 (dd, J=8.8, 2.9 Hz, 1H), 4.09 - 3.96 (m, 2H), 3.84 (t, J=6.2 Hz, 2H), 3.55 - 3.47 (m, 2H), 3.28 (s, 3H), 1.50 (s, 9H). Compound B396d. 2-(2-(6-(6-fluoropyridin-3-yl)benzo[d]thiazol-2-yl)-2-((2- methoxyethyl)sulfonyl)acetamido)acetic acid
[00271] To a flask charged with Compound B396c (95 mg, 0.18 mmol) was added 50% TFA in DCM (1 mL) and the mixture stirred for lh. The mixture was evaporated under reduced pressure and the residue evaporated from toluene (2x) under reduced pressure to give Compound B396d (84 mg, 99 %> yield) as a brown solid. RT = 1.60 min (LCMS Method B). MS (ES): m/z = 468.0 [M+H]+. 1H NMR (400MHz, DMSO-d6) δ 9.09 (t, J=5.6 Hz, 1H), 8.66 (d, J=2.4 Hz, 1H), 8.56 (d, J=1.8 Hz, 1H), 8.39 (td, J=8.2, 2.5 Hz, 1H), 8.20 (d, J=8.6 Hz, 1H), 7.95 - 7.90 (m, 1H), 7.35 (dd, J=8.4, 2.9 Hz, 1H), 6.19 (s, 1H), 3.98 (dd, J=10.8, 5.7 Hz, 2H), 3.80 - 3.70 (m, 4H), 3.28 (s, 3H).
Example B396
[00272] To a solution of Compound B396d (15 mg, 0.032 mmol) in DMF (0.5 mL) was added methylamine hydrochloride (3.3 mg, 0.048 mmol) followed by DIEA (0.017 mL, 0.10 mmol) and HATU (16 mg, 0.042 mmol) and the mixture stirred for 1.5h. Reaction mixture was diluted with DMF and filtered. The material was purified via prepative HPLC (Column : Phenomex Axia Luna CI 8, 30 x 100mm, 5-μιη particles; Mobile Phase A: 10:90 acetonitrile: water with 0.1% TFA; Mobile Phase B: 90: 10 acetonitrile: water with 0.1% TFA; Gradient: 20-100% B over 20 minutes, then a 5- minute hold at 100% B; Flow: 20 mL/min). The fractions containing product were combined and concentrated under reduced pressure to give Example 396 (10 mg, 64 %> yield) as a yellow solid. RT = 1.56 min (LCMS Method B). MS (ES): m/z = 481.0 [M+H]+. 1H NMR (400MHz, DMSO-d6) δ 9.03 (t, J=5.4 Hz, IH), 8.66 (d, J=2.2 Hz, IH), 8.57 (d, J=1.5 Hz, IH), 8.40 (td, J=8.1, 2.6 Hz, IH), 8.20 (d, J=8.6 Hz, IH), 7.95 - 7.91 (m, 2H), 7.35 (dd, J=8.6, 2.6 Hz, IH), 6.21 (s, IH), 3.87 (dd, J=15.5, 5.6 Hz, 2H), 3.82 - 3.75 (m, 2H), 3.75 - 3.69 (m, 2H), 3.28 (s, 3H), 2.62 (d, J=4.6 Hz, 3H).
Example B397
N-[(ethylcarbamoyl)methyl]-2-[6-(6-fluoropyridin-3-yl)-l,3-benzothiazol-2-yl]-2-(2- methoxyethanesulfonyl)acetamide

[00273] Example B397 was prepared from Compound B396d as described in the general procedure given for Example B396 (8 mg, 49%). EL IC50 109 iiM. HL IC50 460 iiM. RT = 1.63 min (LCMS Method B). MS (ES): m/z = 495.1 [M+H]+. 1H NMR (400MHz, DMSO-de) δ 9.02 (t, J=5.6 Hz, IH), 8.66 (d, J=2.4 Hz, IH), 8.57 (d, J=1.8 Hz, IH), 8.40 (td, J=8.1, 2.6 Hz, IH), 8.20 (d, J=8.6 Hz, IH), 7.95 - 7.91 (m, 2H), 7.35 (dd, J=8.7, 2.8 Hz, IH), 6.22 (s, IH), 3.89 - 3.84 (m, 2H), 3.78 (dt, J=5.0, 2.7 Hz, 2H), 3.75 - 3.69 (m, 2H), 3.28 (s, 3H), 3.16 - 3.05 (m, 2H), 1.03 (t, J=7.3 Hz, 3H).

































1H NMR (400MHz, DMSO-de) δ
9.05 - 8.88 (m, 1H), 8.63 - 8.56
N-(2-methoxyethyl)-4-(2- (m, 2H), 8.20 - 8.13 (m, 1H), 8.03 (2-oxo-2-((2-sulfamoyl
- 7.97 (m, 2H), 7.97 - 7.92 (m, ethyl)amino)- 1 -((3 ,3 ,3 - 1H), 7.90 - 7.85 (m, 2H), 7.84 -
B110 trifluoropropyl)sulfonyl)
7.72 (m, 1H), 7.04 - 6.92 (m, 2H), ethyl)-l ,3-benzothiazol-6-
 6.32 - 6.24 (m, 1H), 3.84 - 3.56 yl)benzamide
6.32 - 6.24 (m, 1H), 3.84 - 3.56 yl)benzamide
(m, 4H), 3.52 - 3.40 (m, 4H), 3.29
- 3.27 (m, 3H), 3.22 - 3.12 (m, 2H), 2.92 - 2.77 (m, 2H)
5 9.14 - 8.92 (m, 1H), 8.52 (br. s., 1H), 8.30 - 8.10 (m, 1H), 8.01 -
2 -((cy clopropy Imethy 1)
7.81 (m, 4H), 7.80 - 7.66 (m, 1H), sulfonyl)-2-(6-(4-(4- 7.55 (d, J=7.7 Hz, 2H), 6.94 (br. morpholinylcarbonyl)
Bi l l s., 2H), 3.59-3.62 (m., 10H), 3.28 phenyl)- 1 ,3-benzothiazol- (br. s., 2H), 3.19 (br. s., 2H), 1.21 2-yl)-N-(2-sulfamoylethyl)

1H), 0.55 - 0.34 (m, 2H), 0.31 - 0.20 (m, 1H)



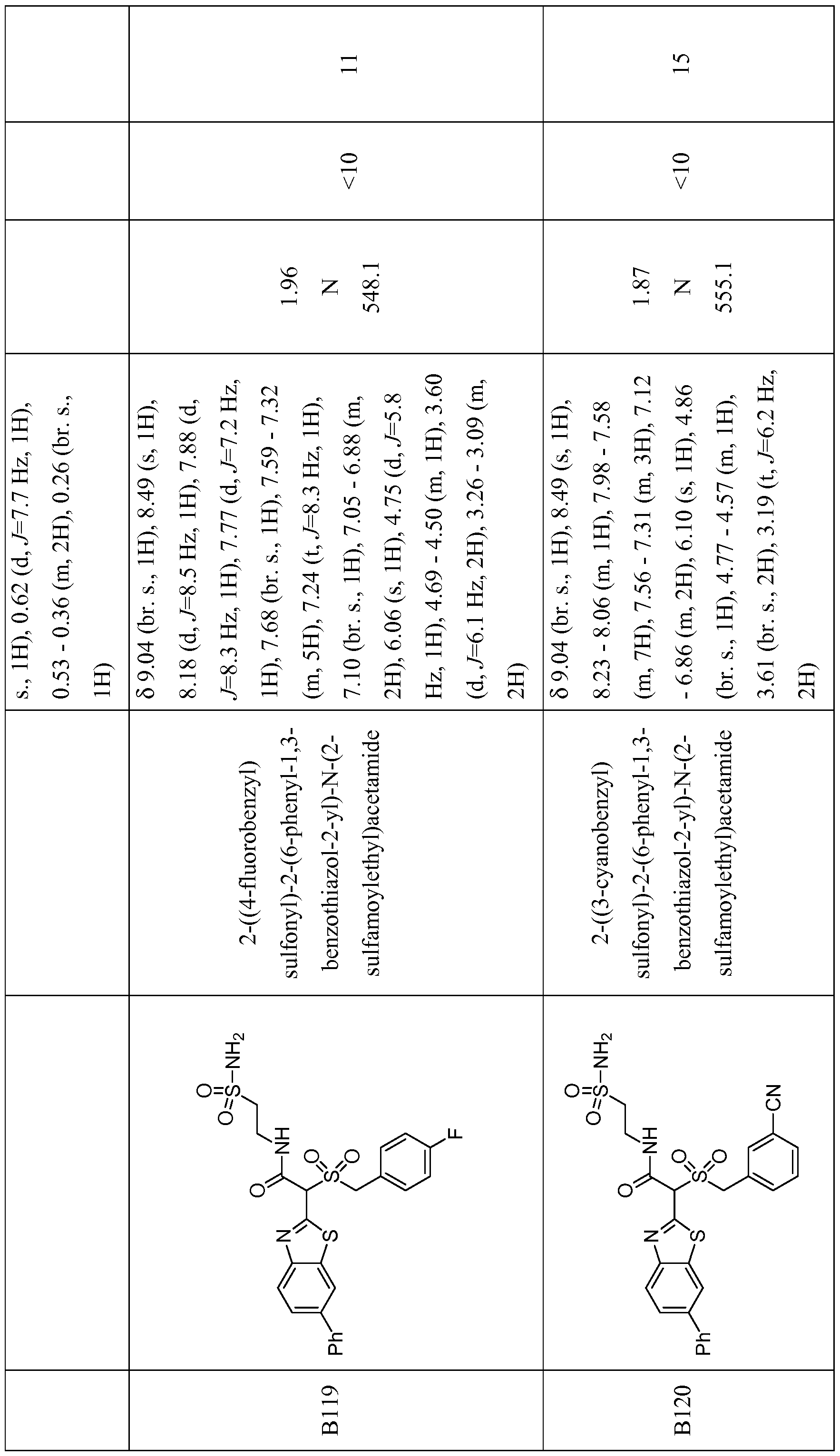
































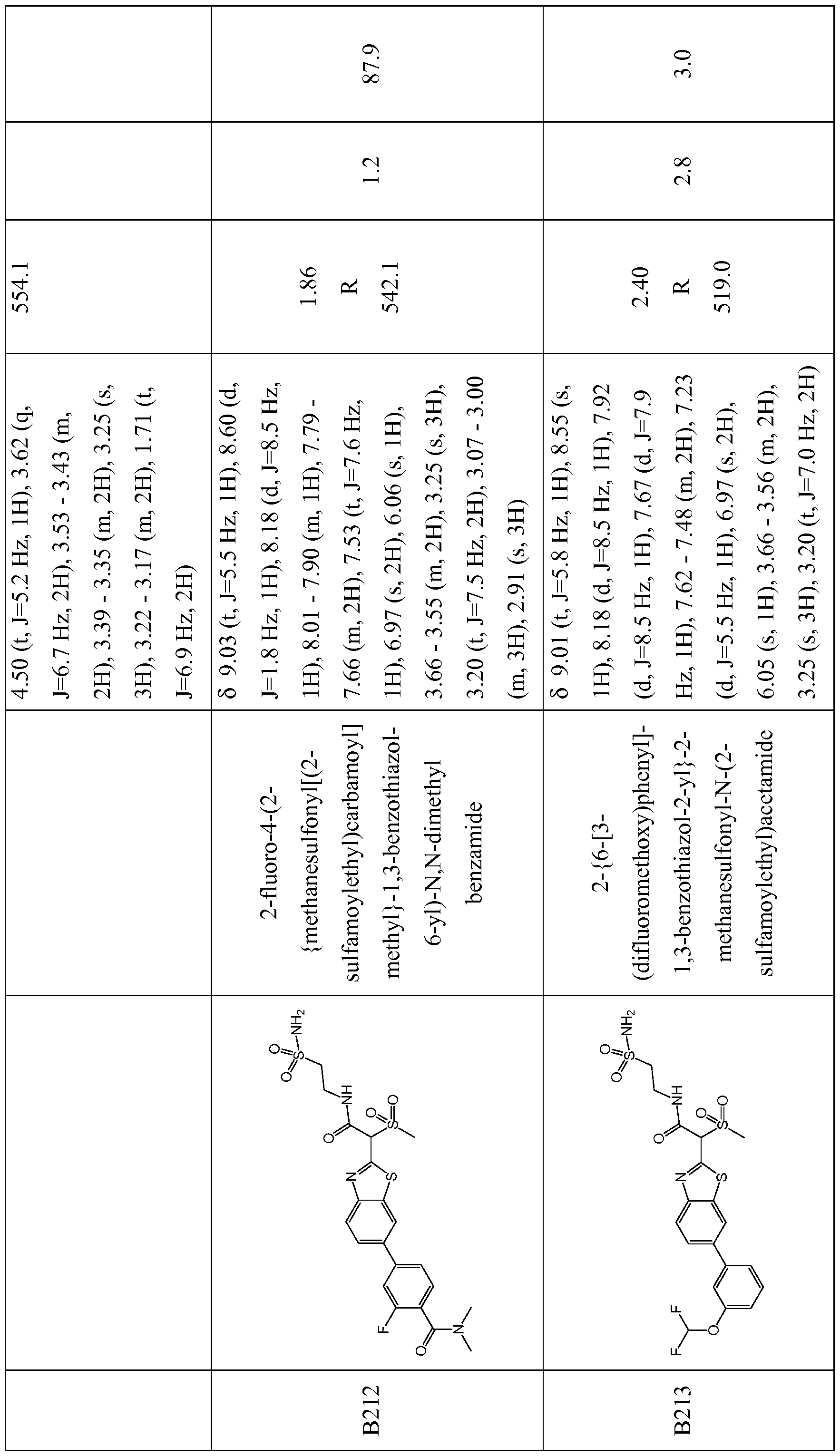

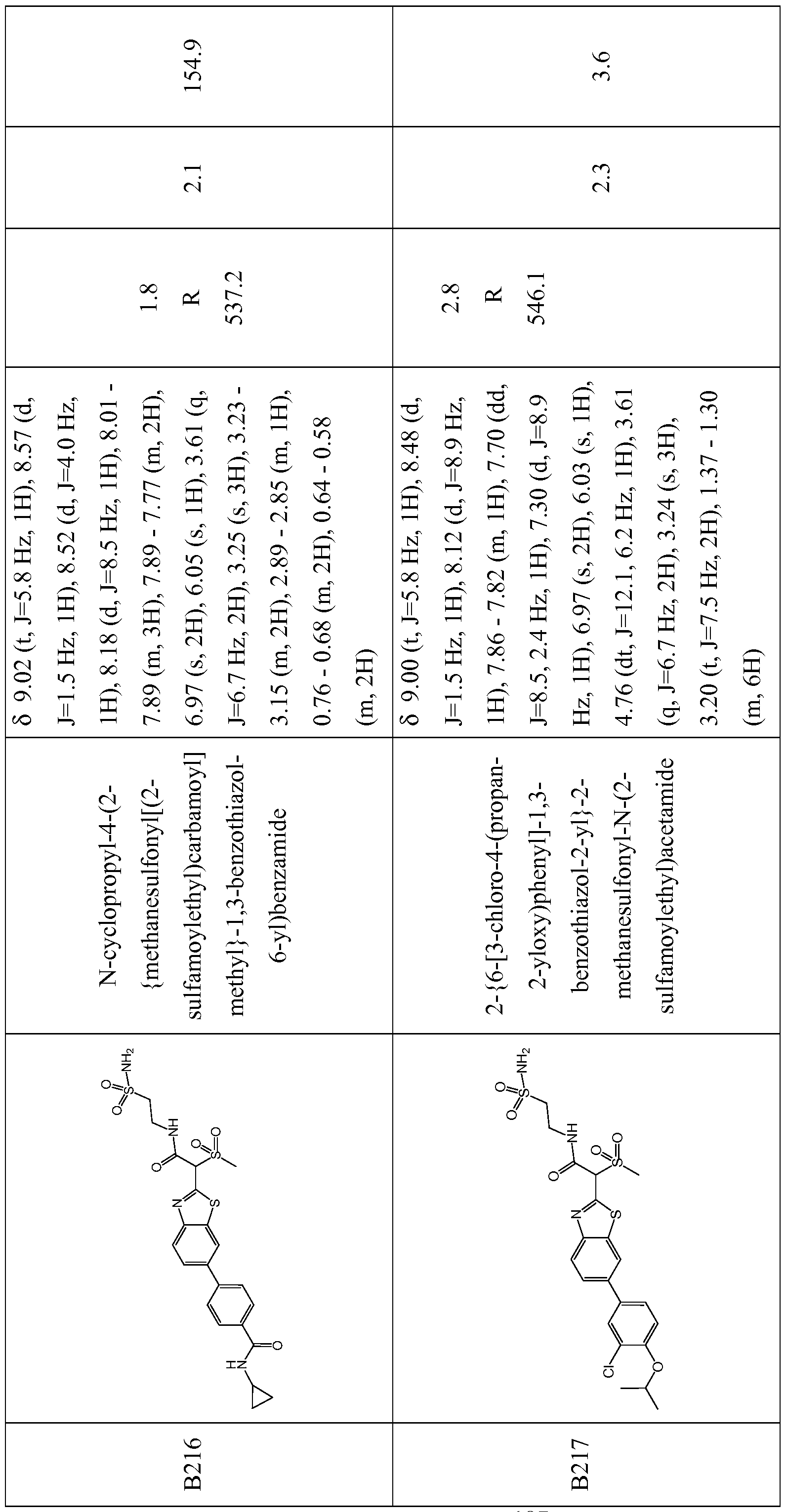





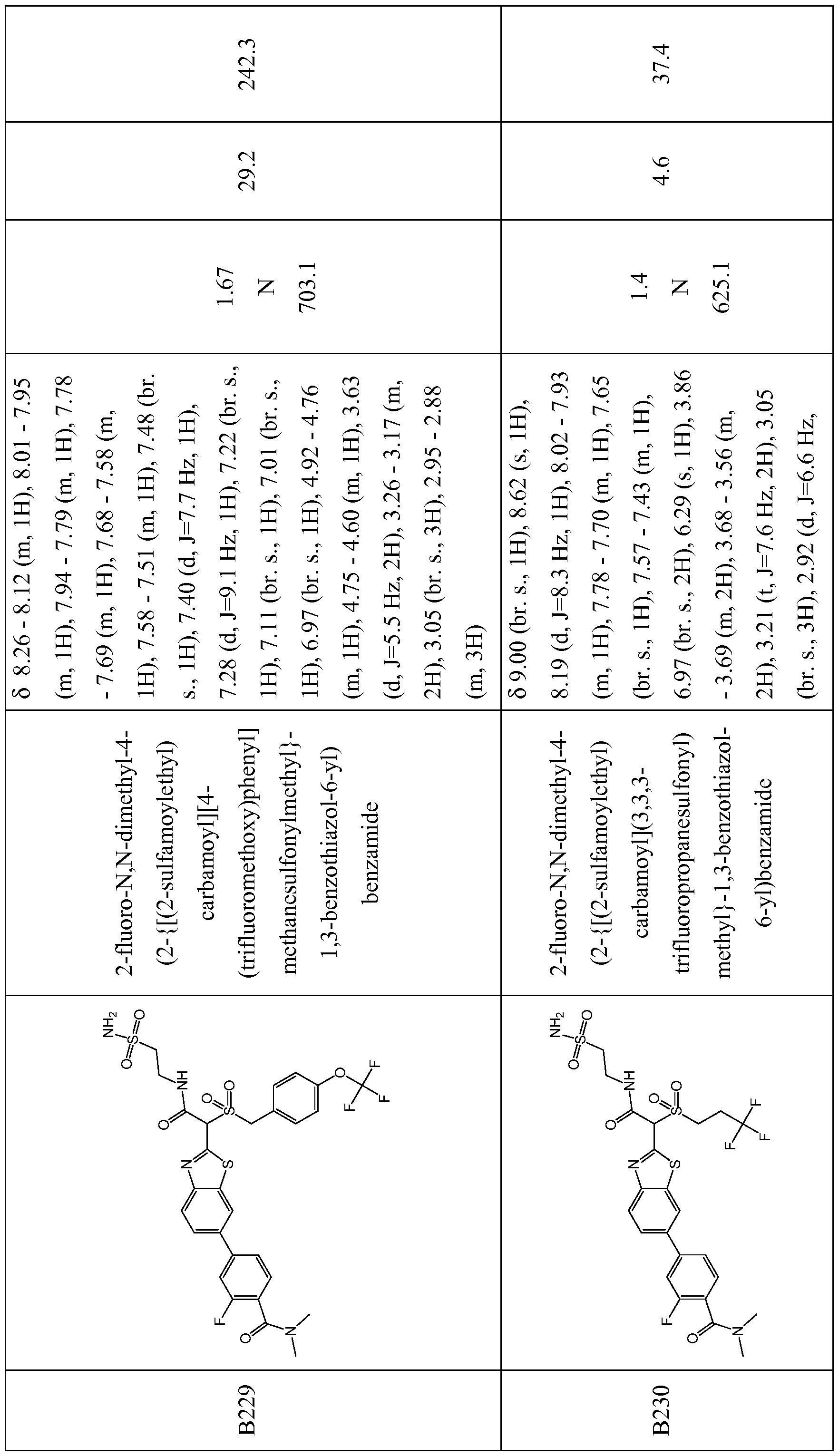





























1H NMR (400MHz, DMSO- d6) δ
2-{6-[4-(3-methoxy 9.17 - 8.53 (m, 1H), 8.27 - 8.10 azetidine- 1 -carbonyl) (m, 1H), 7.85 (s, 3H), 7.76 (d, phenyl]- 1 ,3-benzothiazol- J=9.5 Hz, 4H), 7.67 - 7.58 (m,
B299 2-yl} -N-(2-sulfamoylethyl) 2H), 6.96 (s, 1H), 4.93 - 4.70 (m,
-2- { [4-(trifluoromethyl) 2H), 4.51 (br. s., 1H), 4.33 - 4.11 phenyl] methanesulfony 1 } (m, 3H), 3.88 (br. s., 1H), 3.62 (d,

acetamide J=6.2 Hz, 2H), 3.29 (s, 2H), 3.25
(d, J=1.8 Hz, 3H), 3.21 (s, 1H δ 9.15 - 8.52 (m, 1H), 8.32 - 8.10
2-{6-[4-(3-methoxy
(m, 1H), 7.99 - 7.66 (m, 6H), 7.59 azetidine- 1 -carbonyl)
- 7.37 (m, 3H), 7.28 (dd, J=18.7, phenyl]- 1 ,3-benzothiazol- 6.9 Hz, 1H), 7.03 - 6.90 (m, 2H),
B300 2-yl}-N-(2-sulfamoyl
4.97 - 4.60 (m, 2H), 4.57 - 4.43 ethyl)-2-{[4-(trifluoro
(m, 1H), 4.31 - 4.17 (m, 3H), 3.94 methoxy)pheny 1] methane

2H), 3.28 - 3.16 (m, 5H
















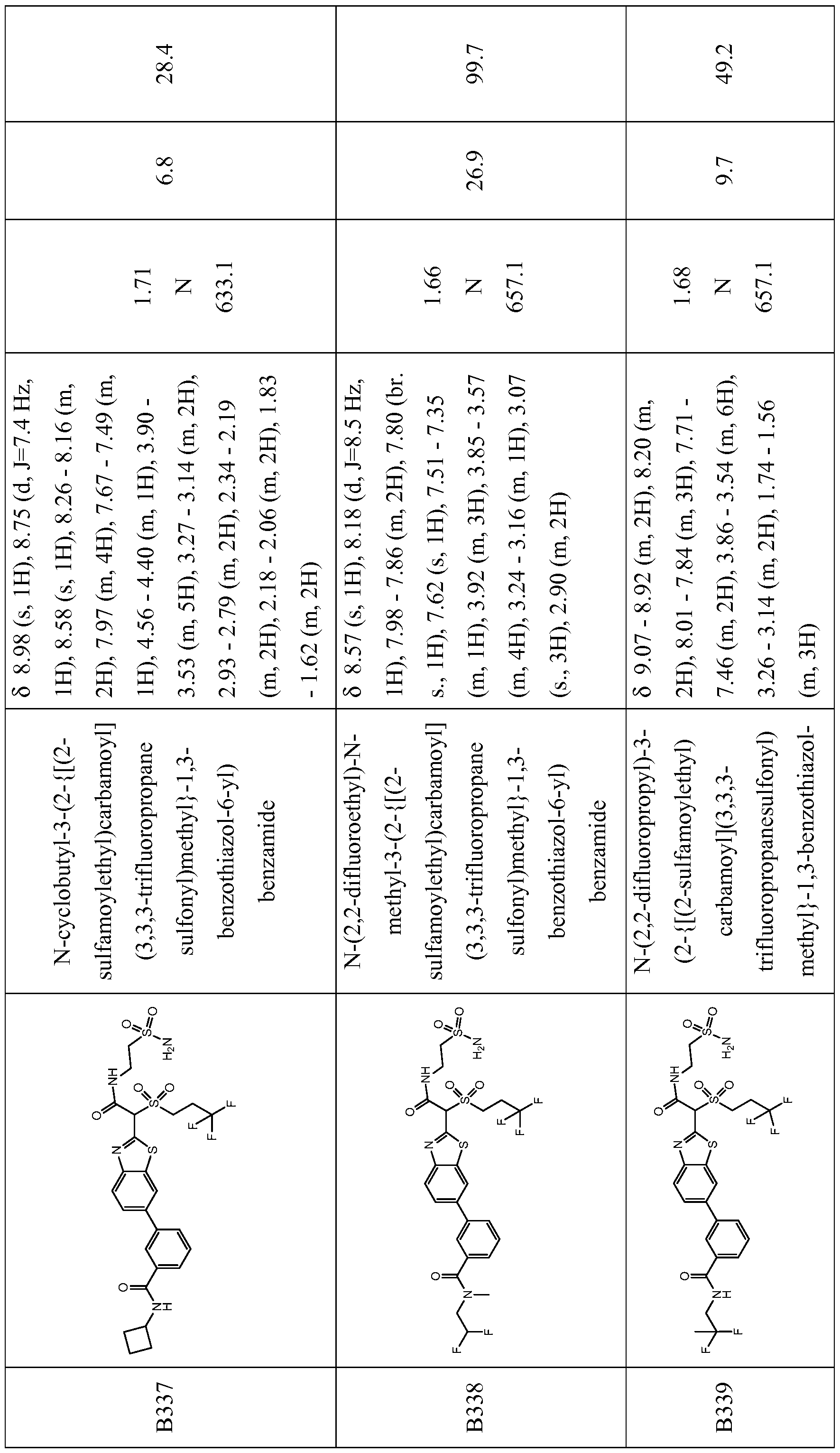
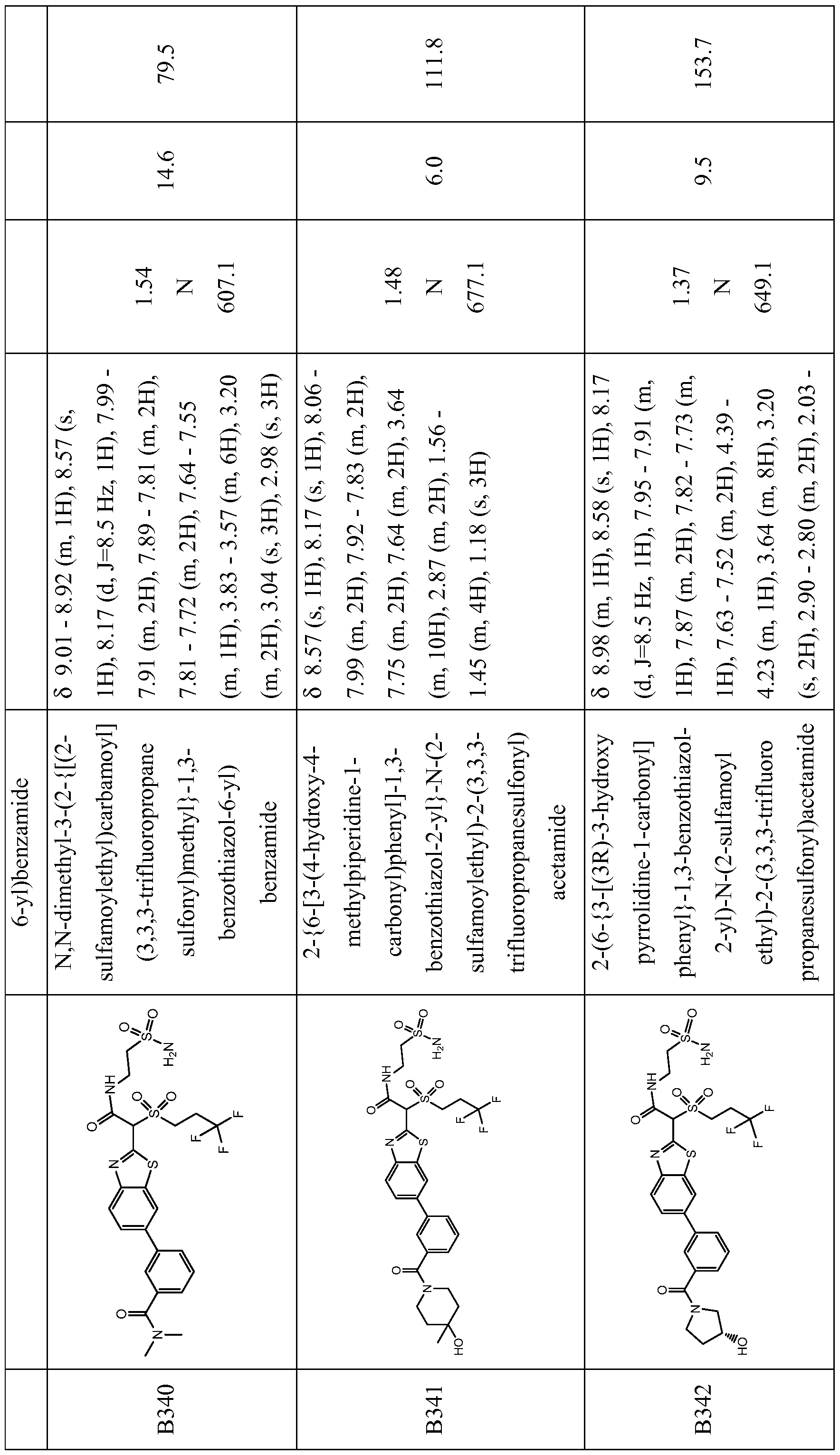











δ 14.31 - 8.88 (m, 1H), 8.47 - 8.26
2-[6-( 1 -methyl-6-oxo- 1 ,6- (m, 1H), 8.19 - 8.07 (m, 1H), 8.00
dihydropyridin-3 -yl)- 1,3- - 7.93 (m, 1H), 7.89 - 7.49 (m,
benzothiazol-2-yl]-N-(2- 6H), 6.97 (br. s., 2H), 6.63 - 6.43 1.50
B366 sulfamoylethyl)-2-{[3- 4.4 219.1
(m, 1H), 6.11 (s, 1H), 5.06 - 4.66 O
(trifluoromethyl)phenyl]
(m, 2H), 3.68 - 3.59 (m, 2H), 3.54 629.1
methanesulfonyl}
(d, J=16.5 Hz, 3H), 3.21 (t, J=6.9

Hz, 2H)
δ 14.40 - 1 1.02 (m, 1H), 9.04 (t,
J=5.6 Hz, 1H), 8.59 (d, J=1.7 Hz,
1H), 8.23 - 8.10 (m, 2H), 7.95 -
2- {6-[ 1 -(2-methoxyethyl)- 7.91 (m, 1H), 7.75 (d, J=7.2 Hz,
2-oxo- 1 ,2-dihydropyridin- 4H), 7.71 - 7.69 (m, 1H), 7.67 - 4-yl]-l,3-benzothiazol-2- 7.63 (m, 2H), 7.59 (s, 3H), 7.04 - 1.57
B367 yl} -N-(2-sulfamoylethyl)- 13.5 2208.
6.94 (m, 5H), 6.82 - 6.73 (m, 1H), O
2- { [4-(trifluoromethyl)
6.72 - 6.63 (m, 2H), 6.63 - 6.53 673.1
phenyl] methanesulfonyl }

acetamide
(m, 2H), 4.81 - 4.68 (m, 2H), 4.18
- 4.06 (m, 4H), 3.92 (s, 1H), 3.70 - 3.56 (m, 9H), 3.28 (s, 3H), 3.28 -





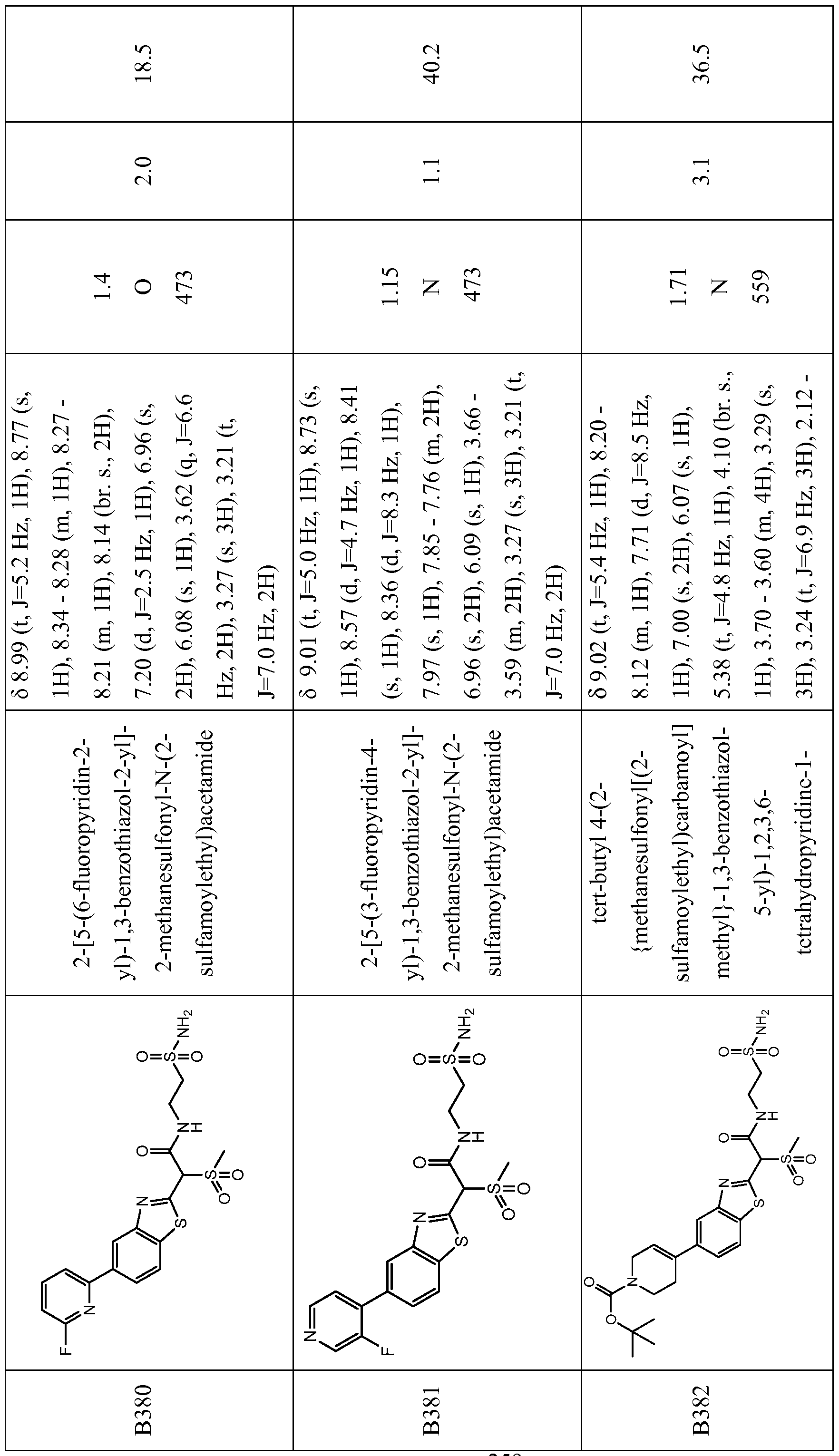




Reference 1
2-(6-Phenyl-l,3-benzothiazol-2-yl)-N-(2-sulfamoylethyl)-4-pentenamide

Compound Rla. Ethyl 2-(6-bromobenzo[d]thiazol-2-yl)pent-4-enoate
[00274] Compound Rla (2.4 g, 35% yield) was prepared from ethyl 6-bromo-2- chlorobenzo[d]thiazole as described in the general procedure given for Compound B196b. HPLC: RT = 1.15 (LCMS Method M). MS (ES): m/z = 341.7 [M+H]+. 1H
NMR (500MHz, CHLOROFORM-d) δ 8.01 (d, J=1.9 Hz, IH), 7.94 - 7.79 (m, IH), 7.58 (dd, J=8.5, 1.9 Hz, IH), 5.80 (ddt, J=17.1, 10.2, 6.9 Hz, IH), 5.15 (dq, J=17.1, 1.5 Hz, IH), 5.08 (dd, J=10.2, 1.4 Hz, IH), 4.34 - 4.18 (m, 3H), 3.03 - 2.90 (m, IH), 2.90 - 2.73 (m, IH), 1.35 - 1.22 (m, 3H).
Compound Rib. Ethyl 2-(6-phenylbenzo[d]thiazol-2-yl)pent-4-enoate
[00275] Compound Rib (120 mg, 29%) was prepared from Compound Rla as described in the general procedure given for Compound Ala. HPLC: RT = 0.86 (LCMS Method M). MS (ES): m/z = 419.7 [M+H]+. 1H NMR (500MHz, DMSO-d6) δ 8.70 - 8.54 (m, IH), 8.36 (d, J=1.9 Hz, IH), 7.89 (d, J=8.8 Hz, IH), 7.68 - 7.59 (m, IH), 6.89 (s,
2H), 5.82 - 5.66 (m, IH), 5.08 (dd, J=17.1, 1.7 Hz, IH), 5.04 - 4.94 (m, lH), 4.18 (dd, J=8.0, 7.2 Hz, IH), 3.55 - 3.37 (m, 2H), 3.16 - 3.04 (m, 2H), 2.76 (d, J=l . l Hz, IH), 2.66 (d, J=7.2 Hz, IH). Reference 1
[00276] Reference 1 (14 mg, 46%) was prepared from Compound Rib as described in the general procedure given for Compound B2a. HPLC: RT = 0.93 (LCMS Method M). MS (ES): m/z = 416.0 [M+H]+. 1H NMR (500MHz, DMSO-d6) δ 8.63 (t, J=5.8 Hz, IH), 8.38 (d, J=l .4 Hz, IH), 8.03 (d, J=8.5 Hz, IH), 7.80 (dd, J=8.4, 1.8 Hz, IH), 7.78 - 7.70 (m, 2H), 7.56 - 7.46 (m, 2H), 7.44 - 7.33 (m, IH), 6.91 (s, 2H), 5.87 - 5.70 (m, IH), 5.12 (dd, J=17.2, 1.8 Hz, IH), 5.07 - 4.97 (m, IH), 4.28 - 4.16 (m, IH), 3.49 (td, J=13.9, 7.4 Hz, 2H), 3.18 (d, J=5.0 Hz, IH), 3.13 (t, J=7.3 Hz, 2H), 2.87 - 2.76 (m, IH), 2.77 - 2.65 (m, IH). Reference 2
N-((3-isopropylisoxazol-5-yl)methyl)-2-methyl-2-(6-phenylbenzo[d]thiazol-2- l)propanamide

[00277] Reference 2 was prepared as described in the general procedure given for Reference 1. HPLC: RT = 2.16 (LCMS Method B). MS (ES): m/z = 420.1 [M+H]+. 1H NMR (400MHz, CHLOROFORM-d) δ 8.11 - 8.05 (m, 2H), 7.90 (m, IH), 7.75 (dd, J=8.5, 1.8 Hz, IH), 7.68 - 7.62 (m, 2H), 7.52 - 7.46 (m, 2H), 7.44 - 7.37 (m, IH), 5.93 (s, IH), 4.55 (dd, J=5.9, 0.6 Hz, 2H), 3.07 - 2.95 (m, IH), 1.84 (s, 6H), 1.24 (s, 3H), 1.22 (s, 3H).
Reference 3
N-((3-isopropylisoxazol-5-yl)methyl)-2-methyl-2-(6-phenylbenzo[d]thiazol-2- yl)propanamide

[00278] Reference 3 was prepared as described in the general procedure given for Reference l . HPLC: RT = 1.97 (LCMS Method B). MS (ES): m/z = 393.1 [M+H]+. NMR (400MHz, CHLOROFORM-d) δ 8.12 - 8.06 (m, 2H), 7.77 (dd, J=8.5, 1.8 Hz, 1H), 7.67 - 7.62 (m, 2H), 7.53 - 7.46 (m, 2H), 7.44 - 7.38 (m, 1H), 4.70 (d, J=6.0 Hz, 2H), 2.55 (s, 3H), 1.86 (s, 6H).
Claims
1. A compound of Formula I):

or a stereoisomer, a tautomer, or a pharmaceutically acceptable salt thereof, wherein:
R1 is inde endently selected from: halogen, CN, C02(Ci_4 alkyl), -CO-RJ,

(a 5- to 6-membered heteroaryl comprising: carbon atoms and 1-4 heteroatoms selected from N, NRC, O, and S(0)p; wherein said heteroaryl is substituted with 0-3 Ra);
R2 is, independently at each occurrence, selected from: halogen, OH, C 1.4 alkyl, C1 - alkoxy, C1 - haloalkyl, C1 - haloalkoxy, CN, NH2, N02, NH(C1 - alkyl),
(C!.4 alkyl)2, C02H, CO^C^ alkyl), and CONH2;
R3 is independently selected from: -S02R5 and -NHCOR6; R4 independently selected from: s and 4 ;
R5 is independently selected from: C^g alkyl substituted with 0-1 R9,
C2_6 alkenyl, Ci .g haloalkyl, Ci .g alkoxy, -(CH2)m-(0)n-(C3_6 carbocycle substituted with 0-3 Rb), and -(CH2)m-(0)n-(5- to 6-membered heterocycle comprising: carbon atoms and 1-4 heteroatoms selected from N, NRC, O, and S(0)p; wherein said heterocycle is substituted with 0-2 Rb);
R6 is independently selected from: Ci .g alkyl, C2_g alkenyl, Ci .g alkoxy,
C _4 haloalkyl, -(CH2)m-(C3_6 carbocycle substituted with 0-3 Rb),
-(CH2)m-(pyridyl substituted with 0-2 R ), -NH(C alkyl), -NHCH2C02(C1.4 alkyl), alkyl
-NH(4-halo-Ph), -NHBn, and
 ;
;
R7 is independently selected from: CONH2, CONHCC^ alkyl),
CONCC^ alkyl)2, SC^NHSO^C^ alkyl), S02NH(CH2) 2.4C02(C1.4 alkyl), NHS02NH2, NHS02(C1.4 alkyl), N(C alkyl)S02NH2,
ON
N(C02Ci.4 alkyl)S02(CM alkyl), S02R10, and O ^ ; 8 is independently selected from *pr-*3 ,
 , s
, s

R9 is independently selected from: halogen, C1.4 alkoxy, C 1.4 haloalkyl, C haloalkoxy, NH2, C02H, C02(C1.4 alkyl), S03H, CONHRd , NHCONHRd,
NHC02Rd,
 , and 5- to 6-membered heterocycle comprising: carbon atoms and 1-4 heteroatoms selected from N, NR8, O, and S(0)p;
, and 5- to 6-membered heterocycle comprising: carbon atoms and 1-4 heteroatoms selected from N, NR8, O, and S(0)p;
R10 is independently selected from: OH, Ci _g alkyl, C2_g alkenyl, NH2,
NH(C!.6 alkyl substituted with 0-1 C02(CM alkyl)), NH(C2-6 alkyl),
NH(Ci .4 haloalkyl), NHPh, phenyl substituted with 0-2 halogens, and

R1 1 is, independently at each occurrence, selected from: halogen, C1 .4 alkyl, Ci .4 haloalkyl, NH2, and phenyl;
Ra is, independently at each occurrence, selected from: halogen, Ci _g alkyl substituted with 0-1 Rf, Ci .4 alkoxy substituted with 0-1 Rf, Ci .g haloalkyl,
C 6 haloalkoxy, OH, CN, N02, C02H, CO^C^ alkyl), NR8Rh, CONR8Rh,
CONRgRJ, NHCORi, NHC02Ri, S02NR8Rh, -(0)n-(CH2)t-RJ, and -CO-RJ;
Rb is, independently at each occurrence, selected from: halogen, C 1 .4 alkyl, Cx.4 alkoxy, C haloalkyl, Cl haloalkoxy, OH, CN, NH2, N02, H(C!.4 alkyl), N(C alkyl)2, C02H, C02(C!.4 alkyl), S02(CM alkyl), CONH2, and
CONH(C alkyl);
Rc is, independently at each occurrence, selected from: H, Ci _g alkyl substituted with 0-1 Re, CO(C!.4 alkyl), C02(C1.4 alkyl), COBn, C02Bn, -(CH2)t-piperidinyl, -(CH2)t-morpholinyl, -(CH2)t-piperazinyl, pyrimidinyl,
(CH2)r(C3_6 carbocycle substituted with 0-2 Re), and

Rd is, independently at each occurrence, selected from: C^
(CH2)t-(phenyl substituted with 0-2 Re);
Re is, independently at each occurrence, selected from: halogen, Ci .4 alkyl, C1.4 alkoxy, C 1.4 haloalkyl, and C1.4 haloalkoxy;
Rf is, independently at each occurrence, selected from: OH, halogen, C1.4 alkyl, C1.4 alkoxy, C 1.4 haloalkyl, and C1.4 haloalkoxy;
R8 is, independently at each occurrence, selected from: H and C 1.4 alkyl;
Rh is, independently at each occurrence, selected from: H, Ci _g haloalkyl and Ci _6 alkyl substituted with 0-1 Rf;
R1 is, independently at each occurrence, selected from: Ci .4 haloalkyl and Ci .4 alkyl substituted with 0-1 Rf;
RJ is, independently at each occurrence: C3.6 carbocycle or a 4- to 6-membered heterocycle comprising: carbon atoms and 1-4 heteroatoms selected from N, NR8, O, and S(0)p; wherein said carbocycle and heterocycle are substituted with 0-2 Rf;
m and t are, independently at each occurrence, selected from 0, 1, 2, and 3;
n is, independently at each occurrence, selected from 0 and 1;
p is, independently at each occurrence, selected from 0, 1, and 2; and
s is, independently at each occurrence, selected from 1,
2, and 3.
A compound according to claim 1, wherein the compound is of Formula (la) or



or a stereoisomer, a tautomer, or a pharmaceutically acceptable salt thereof, wherein: m and t are, independently at each occurrence, selected from 0, 1, and 2; and
s is, independently at each occurrence, selected from 1 and 2.
3. A compound according to claim 1 or claim 2, wherein wherein the compound is of Formula Ila) or (lib):

or a stereoisomer, a tautomer, or a pharmaceutically acceptable salt thereof, wherein:
R2 is independently selected from: halogen and Ci .4 alkyl. 4. A compound according to any one of claims 1 to 3, wherein:
1 is independently selected from: 4-halo-Ph, 6-halo-pyrid-3-yl, and
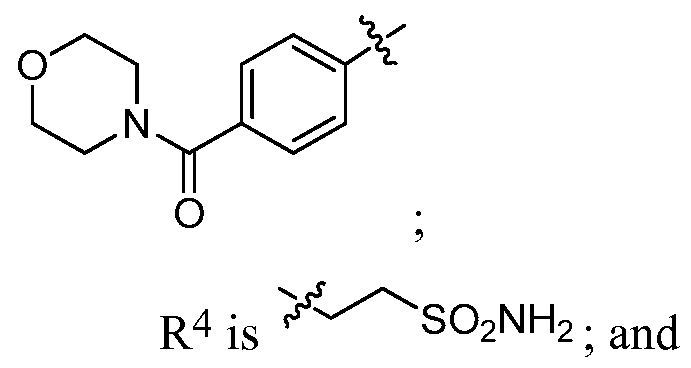
R6 is independently selected from: CX alkyl, -CHF2, NHBn,
4-CF3-Ph, and pyrid-3-yl.
5. A compound according to claim 1 or claim 2, wherein wherein the compound is of Formula (Ilia) or (Illb):


or a stereoisomer, a tautomer, or a pharmaceutically acceptable salt thereof, wherein:
R2 is independently selected from: halogen, C^_4 alkyl and C^_4 haloalkyl.
6. A compound according to any one of claims 1, 2 and 5, wherein:
i inde endently selected from:

OH)

4 is independently selected from: ^^^SO^C^ alkyl) ? ^^302ΝΗ2 ? alkyl)

^^S02NHS02(C1_4 alkyl) ?;-/^S02NHPh ^^S02(4-halo-Ph)

C alkenyl, -(CH2)o-i-C3.6 cycloalkyl, -(CH2)o-r(phenyl substituted with 0-1 Rb) and

R9 is independently selected from: C1 .4 alkoxy, C1 .4 haloalkyl, NH2, and NHC02Bn;
Ra is, independently at each occurrence, selected from: OH, halogen, CN, C alkyl substituted with 0-1 OH, C^ alkoxy, -0(CH2)1.20(C1.4 alkyl),
C haloalkyl, CM haloalkoxy, NH2, C02H, C02(C1.4 alkyl), CONH2,
CONH(C alkyl), CONH(CH2)2.3OH, CONH(CH2)1.30(C1.4 alkyl),
CO (Ci_4 alkyl)(CH2)1.30(C1.4 alkyl), CO H(Ci_4 haloalkyl), NHCO(Ci_4 alkyl), NHCO(C!_4 haloalkyl), NHC02(C alkyl), S02N(CM alkyl)2, S0 NH(CH2)2.3OH,
pyrazolyl, -(CH2)0-2-morpholinyl, -CO-morpholinyl, piperazinyl,
 ,
,

is independently selected from: halogen and Ci .4 haloalkyl;
Rc is, independently at each occurrence, selected from: H, Ci .4 alkyl,
-(CH2)2(Ci .4 alkoxy), -(CH2)0-2-(phenyl substituted with 0-1 halo),
C1 -4 alkyl— O /
-(CH2)0-2-piperidinyl, -(CH2)0-2-morpholinyl, and υ ; and
Rf is, independently at each occurrence, selected from: OH, halogen, C1 .4 alkyl, Ci .4 haloalkyl, Ci .4 alkoxy, and Ci .4 haloalkoxy.
7. A compound according to any one of claims 1 , 2, 5 and 6, wherein:
R1 is independently selected from: Ph, 3-halo-Ph, 4-halo-Ph, 3-Ci _4 alkoxy-Ph, 4-CH2OH-Ph, 3-CN-Ph, 4-CN-Ph, 4-C02H-Ph, 4-C02(Ci.4 alkyl)-Ph,
3-NHCO(C alkyl)-Ph, 4-NHCO(C1_4 alk l)-Ph, 4-(pyrazol-l-yl)-Ph,
2-Ci .4 alkoxy-pyridyl, pyrimidinyl and

R4 is independently selected from: . li^^S02NH2 ^/^S02NH(C2-6 alkenyl)
? and

Pv5 is independently selected from: C i .4 alkyl substituted with 0-1 R9, and
C2_4 alkenyl, Bn, and 4-halo-Bn; and
R9 is independently selected from: C ^ alkoxy, C^ haloalkyl, NH2, and NHC02Bn.
8. A compound according to any one of claims 1, 2, 5, 6 and 7, wherein:
R1 is independently selected from: Ph, 3-halo-Ph, 4-halo-Ph, 3-^_4 alkoxy-Ph,
4- CH2OH-Ph, 3-CN-Ph, 4-CN-Ph, 4-C02H-Ph, 4-C02(C1.4 alkyl)-Ph,
5- NHCOCC alkyl)-Ph, 4-NHCO(C1.4 alkyl)-Ph, 4-(pyrazol-l-yl)-Ph, pyrimidinyl and

R4 is ^^S02NH2 ·
R5 is independently selected from: C i .4 alkyl substituted with 0-1 R9, and
C2_4 alkenyl, Bn, and 4-halo-Bn; and
R9 is independently selected from: C 1 .4 alkoxy, C1 .4 haloalkyl, NH2, and NHC02Bn.
9. A compound according to any one of claims 1, 2, 5, 6, 7 and 8, wherein:
R1 is independently selected from: Ph, 3-F-Ph, 4-F-Ph, 3-OMe-Ph, 4-CH2OH-Ph,
3- CN-Ph, 4-CN-Ph, 4-C02H-Ph, 4-C02Me-Ph 3-NHCOMe-Ph, 4-NHCOMe-Ph,
4- (pyrazol-l-yl)-Ph, pyrimidin-5-yl, and
 and
and
R5 is independently selected from: Me, Et, Pr, z'-Bu, ^^5^, Bn, 4-F-Bn, -(CH2)2OMe, -(CH2)2CF3, -(CH2)2NH2, and -(CH2)2NHC02Bn.
10. A compound according to any one of claims 1, 2, 5, 6 and 7, wherein:
R1 is Ph;
R4 is independently selected from: ^^S02NH(C2_6 alkenyl) ?
? and

R5 is independently selected from: C^ alkyl, C2.4 alkenyl, -(CH2)20(C1.4 alkyl), and -(CH2)2NHC02Bn.
11. A compound according to any one of claims 1, 2, 5, 6, 7 and 10, wherein: R4 is independently selected from: S02NH(CH2)4CH=CH2 ?

R5 is independently selected from: Me, , -(CH2)2OMe, and
-(CH2)2NHC02Bn.
12. A compound according to any one of claims 1, 2, 5 and 6, wherein:
independently selected from:
 H)
H)

(phenyl substituted with 0-2 Ra), and (a heteroaryl substituted with 0-2 Ra and selected from: isoxazolyl, pyrazolyl, l-Rc-pyrazolyl, pyridyl, and pyrimidinyl);
R4 is independently selected from: "Ji ^S02(C1_4 alkyl) ? "J^^SOsNHs ?
^^S02NH(C2_6 alkenyl) !^^^S02NHCH2CF3 ^^S02NH(CH2)3C02(C1 _4 alkyl)

R5 is independently selected from: Ci .4 alkyl substituted with 0-1 R9
-(CH2)o-i-C3_6 cycloalkyl, benzyl substituted with 0-1 Rb, and

R9 is independently selected from: C 1 .4 alkoxy, C1 .4 haloalkyl, NH2, and NHC02Bn;
Ra is, independently at each occurrence, selected from: OH, halogen, C1.4 alkyl substituted with 0- 1 OH, C _4 alkoxy, -0(CH2)20(C _4 alkyl), C _4 haloalkyl,
C haloalkoxy, NH2, CONH2, CONH(C!_4 alkyl), CONH(CH2)2.3OH,
CONH(CH2)1.30(C1.4 alkyl), CONCC^ alkyl)(CH2)1.30(C1.4 alkyl),
CONH(C haloalkyl), NHC02(C1.4 alkyl), S02N(CM alkyl)2, S02NH(CH2)2.3OH,
- CH2)0_2-morpholinyl, -CO-morpholinyl, piperazinyl,


Rb is independently selected from: CN, halogen and C^ haloalkyl;
Rc is, independently at each occurrence, selected from: H, Ci .4 alkyl substituted with 0-1 Ci .4 alkoxy, -(CH2)o-2-( henyl substituted with 0-1 halo), -(CH2)o-2-piperidinyl,

Rf is, independently at each occurrence, selected from: OH, CN, halogen,
Ci .4 alkyl, and Ci .4 alkoxy.
13. A compound according to any one of claims 1, 2, 5, 6 and 12, wherein:
R1 is independently selected from: 3-(CH2)2OH-Ph, 4-(CH2)2OH-Ph,
4-0(CH2)20(C1_4 alkyl)-Ph, 4-C haloalkoxy-Ph, 4-NH2-Ph, 4-CONH2-Ph,
4-CONH(C1_4 alkyl)-Ph, 4-CONH(CH2)2OH-Ph, 3-CONH(CH2)20(C1.4 alkyl)-Ph, 4-CONH(CH2)20(C1_4 alkyl)-Ph, 4-CONH(C1.4 haloalkyl)-Ph,
S-CONCC^ alkyl)(CH2)20(C1.4 alkyl)-Ph, 4-NHC02(C1.4 alkyl)-Ph,
4-S02N(C1_4 alkyl)2-Ph, 4-S02NH(CH2)2OH-Ph, \Η-Ί-€γ_ haloalkyl-pyrazol-4-yl, 1-Ci _4 alkyl-pyrazol-4-yl, l-Bn-pyrazol-4-yl, 3-^_4 alkyl-pyrid-4-yl, 6-OH-pyrid-2-yl, 6-OH-pyrid-3-yl, 2-OH-pyrid-4-yl, 6-Ci_4 alkoxy-pyrid-2-yl, 2-^_4 alkoxy-pyrid-3-yl, 6-CJ.4 alkoxy-pyrid-3-yl, 2-^_4 alkoxy-pyrid-4-yl, 6-halo-pyrid-3-yl, 2-halo-pyrid-4-yl, 6-C1.4 haloalkyl-pyrid-3-yl, 5-Cj_4 alkyl-6-halo-pyrid-3-yl,
3-halo-6-Ci_4 alkoxy-pyrid-4-yl, pyrimidin-5-yl,

-278 -
14. A compound according to any one of claims 1 , 2, 5, 6, 12 and 13, wherein:
R1 is independently selected from: 3-(CH2)2OH-Ph, 4-(CH2)2OH-Ph,
4-0(CH2)2OMe-Ph, 4-OCF3-Ph, 4-NH2-Ph, 4-CONH2-Ph, 4-CONH(t-Bu)-Ph,
4-CONH(CH2)2OH-Ph, 3-CONH(CH2)2OMe-Ph, 4-CONH(CH2)2OMe-Ph,
4-CON(Me)(CH2)2OMe-Ph, 4-CONHCH2CF3-Ph, 4-NHC02Me-Ph,
4-NHC02(t-Bu)-Ph, 4-S02N(Me)2-Ph, 4-S02NH(CH2)2OH-Ph, lH-3-CF3-pyrazol-4-yl,
1- Me-pyrazol-4-yl, l-(z'-Bu)-pyrazol-4-yl, l-Bn-pyrazol-4-yl, 3-Me-pyrid-4-yl,
6-OH-pyrid-2-yl, 6-OH-pyrid-3-yl, 2-OH-pyrid-4-yl, 6-OMe-pyrid-2-yl,
2- OMe-pyrid-3-yl, 6-OMe-pyrid-3-yl, 2-OMe-pyrid-4-yl, 6-F-pyrid-3-yl, 2-F-pyrid-4-yl, 2-Cl- rid-4-yl, 6-CF3-pyrid-3-yl, 5-Me-6-F-pyrid-3-yl, 3-F-6-OMe-pyrid-4-yl,



R5 is independently selected from: Me, Pr, z'-Pr, z'-Bu, -(CH2)2OMe, - CH2)3CF3, -(CH2)2NH cyclopropylmethyl, Bn, 4-F-Bn, 4-CF3-Bn,

-(CH2)2NHC02Bn, and
15. A pharmaceutical composition, comprising: a pharmaceutically acceptable and a compound of any one of claims 1-14, or a stereoisomer, a tautomer, or a pharmaceutically acceptable salt thereof.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP13742364.6A EP2875007B1 (en) | 2012-07-19 | 2013-07-18 | Amide, urea or sulfone amide linked benzothiazole inhibitors of endothelial lipase |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261673488P | 2012-07-19 | 2012-07-19 | |
| US61/673,488 | 2012-07-19 | ||
| US201361770523P | 2013-02-28 | 2013-02-28 | |
| US61/770,523 | 2013-02-28 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014015088A1 true WO2014015088A1 (en) | 2014-01-23 |
Family
ID=48877578
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2013/050972 WO2014015088A1 (en) | 2012-07-19 | 2013-07-18 | Amide, urea or sulfone amide linked benzothiazole inhibitors of endothelial lipase |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US8987314B2 (en) |
| EP (1) | EP2875007B1 (en) |
| AR (1) | AR093281A1 (en) |
| TW (1) | TW201408653A (en) |
| WO (1) | WO2014015088A1 (en) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015105749A1 (en) * | 2014-01-07 | 2015-07-16 | Bristol-Myers Squibb Company | Sulfone amide linked benzothiazole inhibitors of endothelial lipase |
| WO2017108282A1 (en) | 2015-12-22 | 2017-06-29 | Kancera Ab | Bicyclic hydroxamic acids useful as inhibitors of mammalian histone deacetylase activity |
| WO2017117447A1 (en) | 2015-12-31 | 2017-07-06 | Karyopharm Therapeutics Inc. | Multicyclic compounds and uses thereof |
| JP2018511626A (en) * | 2015-04-15 | 2018-04-26 | セルジーン クオンティセル リサーチ,インク. | Bromodomain inhibitor |
| US10899764B2 (en) | 2015-04-21 | 2021-01-26 | Jiangsu Hengrui Medicine Co., Ltd. | Imidazo isoindole derivative, preparation method therefor and medical use thereof |
Citations (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999032611A1 (en) | 1997-12-19 | 1999-07-01 | Progenitor, Inc. | A lipase expressed in endothelial cells and methods for its use |
| WO2004093872A1 (en) | 2003-03-31 | 2004-11-04 | Eli Lilly And Company | 3-oxo-1, 3-dihydro-indazole-2-carboxylic acid amide derivatives as phospholipase inhibitors |
| WO2004094393A1 (en) | 2003-04-01 | 2004-11-04 | Eli Lilly And Company | Phospholipase inhibitors |
| WO2004094394A1 (en) | 2003-04-01 | 2004-11-04 | Eli Lilly And Company | Benzisothiazol-3-one-carboxylic acid amides as phospholipase inhibitors |
| WO2007042178A1 (en) | 2005-10-12 | 2007-04-19 | Sanofi-Aventis | Diacyl indazol derivatives as lipase and phospholipase inhibitors |
| WO2007110215A1 (en) | 2006-03-28 | 2007-10-04 | Sanofi-Aventis | Imidazopyridin-2-one derivatives as inhibitors of endothelial lipase |
| WO2007110216A1 (en) | 2006-03-28 | 2007-10-04 | Sanofi-Aventis | Azolopyridin-3-one derivatives as inhibitors of endothelial lipase |
| WO2009123164A1 (en) | 2008-04-02 | 2009-10-08 | 塩野義製薬株式会社 | Heterocyclic derivative having inhibitory activity on endothelial lipase |
| WO2009133834A1 (en) | 2008-04-28 | 2009-11-05 | 塩野義製薬株式会社 | Keto-amide derivative having inhibitory activity on endothelial lipase |
| WO2010044441A1 (en) | 2008-10-17 | 2010-04-22 | 塩野義製薬株式会社 | Acetic acid amide derivative having inhibitory activity on vascular endothelial lipase |
| WO2011074560A1 (en) | 2009-12-15 | 2011-06-23 | 塩野義製薬株式会社 | Oxadiazole derivative having endothelial lipase inhibitory activity |
| WO2012081563A1 (en) | 2010-12-14 | 2012-06-21 | 塩野義製薬株式会社 | Benzothiazole and azabenzothiazole derivative having endothelial lipase inhibitory activity |
| WO2012173099A1 (en) | 2011-06-14 | 2012-12-20 | 塩野義製薬株式会社 | Endothelial-lipase-inhibiting amino derivative |
-
2013
- 2013-07-18 WO PCT/US2013/050972 patent/WO2014015088A1/en active Application Filing
- 2013-07-18 US US13/944,921 patent/US8987314B2/en active Active
- 2013-07-18 AR ARP130102559A patent/AR093281A1/en unknown
- 2013-07-18 EP EP13742364.6A patent/EP2875007B1/en not_active Not-in-force
- 2013-07-19 TW TW102125982A patent/TW201408653A/en unknown
Patent Citations (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999032611A1 (en) | 1997-12-19 | 1999-07-01 | Progenitor, Inc. | A lipase expressed in endothelial cells and methods for its use |
| WO2004093872A1 (en) | 2003-03-31 | 2004-11-04 | Eli Lilly And Company | 3-oxo-1, 3-dihydro-indazole-2-carboxylic acid amide derivatives as phospholipase inhibitors |
| US20060211755A1 (en) | 2003-03-31 | 2006-09-21 | Eli Lilly And Company Patent Division | 3-oxo-1, 3-dihydro-indazole-2-carboxylic acid amide derivatives as phospholipase inhibitors |
| WO2004094393A1 (en) | 2003-04-01 | 2004-11-04 | Eli Lilly And Company | Phospholipase inhibitors |
| WO2004094394A1 (en) | 2003-04-01 | 2004-11-04 | Eli Lilly And Company | Benzisothiazol-3-one-carboxylic acid amides as phospholipase inhibitors |
| US7217727B2 (en) | 2003-04-01 | 2007-05-15 | Eli Lilly And Company | Phospholipase inhibitors |
| US7595403B2 (en) | 2003-04-01 | 2009-09-29 | Eli Lilly And Company | Benzisothiazol-3-one-carboxylic acid amides as phospholipase inhibitors |
| US20080287448A1 (en) | 2005-10-12 | 2008-11-20 | Sanofi-Aventis | Diacyl indazole derivatives as lipase and phospholipase inhibitors |
| WO2007042178A1 (en) | 2005-10-12 | 2007-04-19 | Sanofi-Aventis | Diacyl indazol derivatives as lipase and phospholipase inhibitors |
| US20090054478A1 (en) | 2006-03-28 | 2009-02-26 | Sanofi-Aventis | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2007110216A1 (en) | 2006-03-28 | 2007-10-04 | Sanofi-Aventis | Azolopyridin-3-one derivatives as inhibitors of endothelial lipase |
| US20090076068A1 (en) | 2006-03-28 | 2009-03-19 | Sanofi-Aventis | Imidazopyridin-2-one derivatives as inhibitors of lipases and phospholipases |
| WO2007110215A1 (en) | 2006-03-28 | 2007-10-04 | Sanofi-Aventis | Imidazopyridin-2-one derivatives as inhibitors of endothelial lipase |
| WO2009123164A1 (en) | 2008-04-02 | 2009-10-08 | 塩野義製薬株式会社 | Heterocyclic derivative having inhibitory activity on endothelial lipase |
| WO2009133834A1 (en) | 2008-04-28 | 2009-11-05 | 塩野義製薬株式会社 | Keto-amide derivative having inhibitory activity on endothelial lipase |
| WO2010044441A1 (en) | 2008-10-17 | 2010-04-22 | 塩野義製薬株式会社 | Acetic acid amide derivative having inhibitory activity on vascular endothelial lipase |
| US20110251386A1 (en) | 2008-10-17 | 2011-10-13 | Shionogi & Co., Ltd. | Acetic acid amide derivative having inhibitory activity on endothelial lipase |
| WO2011074560A1 (en) | 2009-12-15 | 2011-06-23 | 塩野義製薬株式会社 | Oxadiazole derivative having endothelial lipase inhibitory activity |
| CA2784710A1 (en) * | 2009-12-15 | 2011-06-23 | Shionogi & Co., Ltd. | Oxadiazole derivative having endothelial lipase inhibitory activity |
| US20120253040A1 (en) | 2009-12-15 | 2012-10-04 | Shionogi & Co., Ltd. | Oxadiazole derivative having endothelial lipase inhibitory activity |
| WO2012081563A1 (en) | 2010-12-14 | 2012-06-21 | 塩野義製薬株式会社 | Benzothiazole and azabenzothiazole derivative having endothelial lipase inhibitory activity |
| WO2012173099A1 (en) | 2011-06-14 | 2012-12-20 | 塩野義製薬株式会社 | Endothelial-lipase-inhibiting amino derivative |
Non-Patent Citations (34)
| Title |
|---|
| "Design ofProdrugs", 1985, ELSEVIER |
| "Hawley's Condensed Chemical Dictionary", 1997, JOHN WILEY & SONS, INC. |
| "Medicinal Chemistry: Principles and Practice", 2006, THE ROYAL SOCIETY OF CHEMISTRY |
| "Methods in Enzymology", vol. 112, 1985, ACADEMIC PRESS, pages: 309 - 396 |
| "Prodrugs and Targeted Delivery", vol. 47, 2011, WILEY-VCH |
| "Remington: The Science and Practice of Pharmacy", 2012, PHARMACEUTICAL PRESS |
| "The Practice of'Medicinal Chemistry", 2008, ACADEMIC PRESS |
| ALLEN, L. V. JR. ET AL.: "Remington: The Science and Practice ofPhannacy", vol. 2, 2012, PHARMACEUTICAL PRESS |
| BEVILACQUA, M.P. ET AL., J. CLIN. INVEST., vol. 91, no. 2, 1993, pages 379 - 387 |
| BUNDGAARD, H, ADV. DRUG DELIV. REV., vol. 8, 1992, pages 1 - 38 |
| BUNDGAARD, H. ET AL., J. PHARM. SCI., vol. 77, 1988, pages 285 |
| BUNDGAARD, H. ET AL.: "A Textbook of Drug Design and Development", 1991, HARWOOD ACADEMIC PUBLISHERS, article "Design and Application ofProdrugs", pages: 113 - 191 |
| CIRCULATION, vol. 106, no. 11, 2002, pages 1321 - 1326 |
| CIRCULATION, vol. 125, 2012, pages E2 - E220 |
| FOLKMAN, J. ET AL., J. BIOL. CHEM., vol. 267, no. 16, pages 10931 - 10934 |
| FOLKMAN, J. ET AL., SCIENCE, vol. 235, 1987, pages 442 - 447 |
| GORDON, D.J ET AL., N. ENGL. J. MED., vol. 321, no. 19, 1989, pages 1311 - 1316 |
| GORDON, D.J. ET AL., CIRCULATION, vol. 79, no. 1, 1989, pages 8 - 15 |
| HIRATA, K. ET AL., J. BIOL. CHEM., vol. 274, no. 20, 1999, pages 14170 - 14175 |
| JANSSENS, S.P. ET AL., BIOL. CHEM., vol. 267, no. 21, 1992, pages 14519 - 14522 |
| JAYE, M. ET AL., NAT. GENET., vol. 21, 1999, pages 424 - 428 |
| JIN, W. ET AL., TRENDS ENDOCRINOL. METAB., vol. 13, no. 4, 2002, pages 174 - 178 |
| KAKEYA, N. ET AL., CHEM. PHARM. BULL., vol. 32, 1984, pages 692 |
| LAMAS, S. ET AL., PROC. NATL. ACAD. SCI. U.S.A., vol. 89, no. 14, 1992, pages 6348 - 6352 |
| LAROCK, R.C.: "Comprehensive Organic Transfonnat.ions", 1999, VCH PUBLISHERS, INC. |
| LUSCHER, T.F. ET AL., HYPERTENSION, vol. 19, no. 2, 1992, pages 117 - 130 |
| MCCOY, M.G., J. LIPID RES., vol. 43, 2002, pages 921 - 929 |
| ROSS, R., NATURE, vol. 362, no. 6423, 1993, pages 801 - 809 |
| STRAUSS, J.G. ET AL., BIOCHEM. J., vol. 368, 2002, pages 69 - 79 |
| TESTA, B. ET AL.: "Hydrolysis in Drug and Prodrug Metabolism. Chemistry, Biochemistry and Enzymology", 2003, VCHA AND WILEY-VCH, ZURICH |
| WILLIAMS ET AL., AM. REV. RESPIR. DIS., vol. 146, pages S45 - S50 |
| WONG, H. ET AL., J. LIPID RES., vol. 43, 2002, pages 993 - 999 |
| WUTS, P. G. M; GREENE, T. W: "Protective Groups in Organic Synthesis", 2007, WILEY |
| YANAGISAWA, M. ET AL., NATURE, vol. 332, no. 6163, 1988, pages 411 - 415 |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2015105749A1 (en) * | 2014-01-07 | 2015-07-16 | Bristol-Myers Squibb Company | Sulfone amide linked benzothiazole inhibitors of endothelial lipase |
| US10173991B2 (en) | 2014-01-07 | 2019-01-08 | Bristol-Myers Squibb Company | Sulfone amide linked benzothiazole inhibitors of endothelial lipase |
| JP2018511626A (en) * | 2015-04-15 | 2018-04-26 | セルジーン クオンティセル リサーチ,インク. | Bromodomain inhibitor |
| US10494371B2 (en) | 2015-04-15 | 2019-12-03 | Celgene Quanticel Research, Inc. | Bromodomain inhibitors |
| US10807982B2 (en) | 2015-04-15 | 2020-10-20 | Celgene Quanticel Research, Inc. | Bromodomain inhibitors |
| US10899764B2 (en) | 2015-04-21 | 2021-01-26 | Jiangsu Hengrui Medicine Co., Ltd. | Imidazo isoindole derivative, preparation method therefor and medical use thereof |
| WO2017108282A1 (en) | 2015-12-22 | 2017-06-29 | Kancera Ab | Bicyclic hydroxamic acids useful as inhibitors of mammalian histone deacetylase activity |
| US10654814B2 (en) | 2015-12-22 | 2020-05-19 | Kancera Ab | Bicyclic hydroxamic acids useful as inhibitors of mammalian histone deacetylase activity |
| WO2017117447A1 (en) | 2015-12-31 | 2017-07-06 | Karyopharm Therapeutics Inc. | Multicyclic compounds and uses thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| US20140045811A1 (en) | 2014-02-13 |
| TW201408653A (en) | 2014-03-01 |
| US8987314B2 (en) | 2015-03-24 |
| EP2875007A1 (en) | 2015-05-27 |
| AR093281A1 (en) | 2015-05-27 |
| EP2875007B1 (en) | 2016-08-24 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| TWI794297B (en) | Use of a compound in the manufacture of a medicament for use in combination with a second anti-respiratory syncytial virus agent for treating a respiratory syncytial virus infection and pharmaceutical composition | |
| EP3402790B1 (en) | Spiroheptane salicylamides and related compounds as inhibitors of rock | |
| EP3652164B1 (en) | Phenylacetamides as inhibitors of rock | |
| KR20180041142A (en) | Benzodiazepine derivatives as RSV inhibitors | |
| KR20180069788A (en) | Compositions comprising PI3K inhibitors and HDAC inhibitors | |
| EA034395B1 (en) | Phthalazinones and isoquinolinones as rock inhibitors | |
| AU2010303567A1 (en) | Heterocyclic compounds useful as PDK1 inhibitors | |
| EP2875007B1 (en) | Amide, urea or sulfone amide linked benzothiazole inhibitors of endothelial lipase | |
| MX2014015691A (en) | 1-[m-carboxamido(hetero)aryl-methyl]-heterocyclyl-carboxamide derivatives. | |
| WO2013151923A1 (en) | Pyrimidinone carboxamides as inhibitors of endothelial lipase | |
| CN116940354A (en) | Fused heteroaryl compounds and their use as CaMKII inhibitors | |
| WO2016040417A1 (en) | Triazolopyridine and triazolopyrimidine inhibitors of myeloperoxidase | |
| EP2870153B1 (en) | Sulfonyl containing benzothiazole inhibitors of endothelial lipase | |
| US10173991B2 (en) | Sulfone amide linked benzothiazole inhibitors of endothelial lipase | |
| US9169240B2 (en) | Ketone linked benzothiazole inhibitors of endothelial lipase | |
| US10597370B2 (en) | 2-(benzothiazol-2-yl)-2-cyano-acetamide derivatives and their use as endothelial lipase inhibitors | |
| EP2870152B1 (en) | Amide or urea substituted benzothiazole derivatives as inhibitors of endothelial lipase | |
| EP2844643A1 (en) | Pyrimidinedione carboxamide inhibitors of endothelial lipase |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13742364 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2013742364 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013742364 Country of ref document: EP |