WO2014012964A1 - New antifibrinolytic compounds - Google Patents
New antifibrinolytic compounds Download PDFInfo
- Publication number
- WO2014012964A1 WO2014012964A1 PCT/EP2013/065071 EP2013065071W WO2014012964A1 WO 2014012964 A1 WO2014012964 A1 WO 2014012964A1 EP 2013065071 W EP2013065071 W EP 2013065071W WO 2014012964 A1 WO2014012964 A1 WO 2014012964A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- formula
- optionally substituted
- saturated
- substituents
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/10—Spiro-condensed systems
- C07D491/107—Spiro-condensed systems with only one oxygen atom as ring hetero atom in the oxygen-containing ring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/04—Antihaemorrhagics; Procoagulants; Haemostatic agents; Antifibrinolytic agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C317/00—Sulfones; Sulfoxides
- C07C317/14—Sulfones; Sulfoxides having sulfone or sulfoxide groups bound to carbon atoms of six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C317/00—Sulfones; Sulfoxides
- C07C317/44—Sulfones; Sulfoxides having sulfone or sulfoxide groups and carboxyl groups bound to the same carbon skeleton
- C07C317/48—Sulfones; Sulfoxides having sulfone or sulfoxide groups and carboxyl groups bound to the same carbon skeleton the carbon skeleton being further substituted by singly-bound nitrogen atoms, not being part of nitro or nitroso groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/08—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms
- C07D211/18—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D211/34—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms with hydrocarbon radicals, substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D221/00—Heterocyclic compounds containing six-membered rings having one nitrogen atom as the only ring hetero atom, not provided for by groups C07D211/00 - C07D219/00
- C07D221/02—Heterocyclic compounds containing six-membered rings having one nitrogen atom as the only ring hetero atom, not provided for by groups C07D211/00 - C07D219/00 condensed with carbocyclic rings or ring systems
- C07D221/20—Spiro-condensed ring systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D265/00—Heterocyclic compounds containing six-membered rings having one nitrogen atom and one oxygen atom as the only ring hetero atoms
- C07D265/28—1,4-Oxazines; Hydrogenated 1,4-oxazines
- C07D265/34—1,4-Oxazines; Hydrogenated 1,4-oxazines condensed with carbocyclic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/06—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/10—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains three hetero rings
- C07D471/20—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/12—Systems containing only non-condensed rings with a six-membered ring
- C07C2601/14—The ring being saturated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2603/00—Systems containing at least three condensed rings
- C07C2603/93—Spiro compounds
- C07C2603/94—Spiro compounds containing "free" spiro atoms
Definitions
- the present invention relates to spirocyclic compounds of formula (I), to a process for their preparation, as well as to the intermediates used in this process. It also relates to pharmaceutical or veterinary compositions containing them, and to their use in medicine, in particular as antifibrinolytic and antihemorragic agents.
- the haemostatic system is responsible for maintaining circulatory fluidity and for preventing haemorrhage in response to vascular injury.
- Physiological hemostasis is controlled by mechanisms of coagulation and the formation of fibrin and by those favouring the degradation of fibrin (fibrinolysis).
- Hyperfibrinolytic states caused by congenital or acquired conditions predispose to important haemorrhagic complications, often requiring transfusions and the need for re-exploration having a detrimental effect on patient outcome. Hemorhage is responsible for almost 50% of deaths occurring within 24 hours of traumatic injury and for up to 80% of
- a major goal in surgery as well as in the treatment of major tissue damage is to avoid or minimise bleeding in order to ensure the formation of stable and solid haemostatic plugs that are not easily dissolved by fibrinolytic enzymes. Furthermore, it is of importance to ensure quick and effective formation of such plugs or clots.
- Antifibrinolytic agents are widely used in major surgery to prevent fibrinolysis and reduce blood loss.
- EACA epsilon- aminocaproic acid
- TXA tranexamic acid
- antifibrinolytics commercially available to control bleeding. These agents competitively inhibit activation of plasminogen to plasmin, an enzyme that degrades fibrin clots, fibrinogen and other plasma proteins.
- spirocyclic compounds which comprise spirocyclic ring system containing a carbon atom (spiro atom) attached to a hydroxamic acid and a sulfonyl group, show a significant delay in the lysis time in a
- the spirocyclic compounds of the invention also show an important reduction of the bleeding time in vivo animal models as it will be shown in detail in the examples. These characteristics of the compounds of the invention allow a rapid cessation of hemorrhage; favor an effective formation of plugs or clots; have a sustained action (persistence of the clot and prevention of hemorrhage) and aid in minimizing the adverse effects related to other antifibrinolytic/antihemorrhagic treatments having risk of thrombotic complications.
- a first aspect of the invention relates to a compound of formula (I) (hereinafter also referred as compounds of the invention), or a
- a and B form a spirocyclic ring system wherein the spiro atom connecting A and B is a carbon atom and wherein
- A is a known 3- to 8-membered carbocyclic or heterocyclic monocyclic ring, saturated or partially unsaturated; or alternatively
- A is a known 6- to 18-membered carbocyclic or heterocyclic polycyclic ring system, saturated, partially unsaturated, or partially aromatic; and B is a known 4- to 7-membered carbocyclic or heterocyclic monocyclic ring, saturated or partially unsaturated;
- C is phenyl or a known 5- to 6-membered heteroaromatic ring
- R1-R3 are independently selected from H, halogen, -NO 2 , -CN, R a , -OR a , -OC(Y)R a' , -OC(Y)OR a' , -OC(Y)NR R a , -OSO 2 OR a' , -NR R a , -NR C(Y)R a , -NR C(Y)OR a , -NR C(Y)NR R a , -NR S(O) 2 R a' , -NR SO 2 NR R a , -SR a' , -S(O)R a' , -S(O)OR a' , -SO 2 R a' ,-SO 2 (OR a ), -SO 2 NR R a , -SC(Y)NR R a , -C(Y)R a' ,
- R 4 -R 7 are independently selected from halogen, -NO 2 , -CN, R c , -OR c , -NR d R c , -NR d C(Y)R c , -NR d C(Y)OR c , -NR d C(Y)NR d R c , -NR d S(O) 2 R c , -NR d SO 2 NR d R c , -SR C , -S(O)R c , -S(O)OR c , -SO 2 R c , -SO 2 R(OR c ), -SO 2 NR d R c , -SC(Y)NR d R c , -C(Y)R C , -C(Y)OR c , -C(Y)NR d R c , -C(Y)NR d OR c , and -C(O)NR
- R a is a saturated or unsaturated (Ci-Ci 2 )alkyl optionally substituted with one or more substituents R e and/or one Cy 1 ; or alternatively R a is Cy 2 ;
- Cy 1 and Cy 2 are independently optionally substituted with: one Cy 3 and/or one or more substituents R e , and/or one or more saturated or unsaturated (CrC 6 )alkyl groups optionally substituted with one or more substituents R e and/or one Cy 3 ; and
- any Cy 3 is optionally substituted with one or more substituents independently selected from R e and saturated or unsaturated (CrC 6 )alkyl optionally substituted with one or more substituents R e ; each R a and R are independently H or R a ;
- R c and each R d are independently selected from H, Cy 4 , and saturated or unsaturated (CrC 6 )alkyl optionally substituted with one or more substituents R h and/or one Cy 5 ;
- R h independently selected from R h and saturated or unsaturated (CrC 6 )alkyl optionally substituted with one or more substituents R h ;
- each R e is independently selected from halogen, -NO 2 , -CN, -OR f , -OC(Y)R f , -OC(Y)OR f , -OC(Y)NR 9 R f , -NR 9 R f , -NR 9 C(Y)R f , -NR 9 C(Y)OR f , -NR 9 C(Y)NR 9 R f , -NR 9 S(O) 2 R f , -NR 9 SO 2 NR 9 R f , -SR f , -S(O)R f , -S(O)OR f , -SO 2 R f , -SO 2 (OR f ), -SO 2 NR 9 R f , -SC(Y)NR 9 R f , -
- Cy 6 is optionally substituted with: one Cy 7 , and/or one or more substituents R h , and/or one or more saturated or unsaturated (CrC 6 )alkyl groups optionally substituted with one or more substituents R h and/or one Cy 7 ; and
- any Cy 7 is optionally substituted with one or more substituents independently selected from R h and (Ci-C 4 )alkyl optionally substituted with one or more substituents R h ; each R h is independently selected from halogen, -NO 2 , -CN, -OR', - ⁇ ( ⁇ ) ⁇ , -00(0)0 ⁇ , -OCiOJNR'R 1 , -NR'R', -NR'CiOJR 1 , -NR ⁇ OJOR 1 , -NR'C(O)NR'R', -NR ⁇ O ⁇ R 1 , -NR'SCfeNR'R 1 , -SR 1 , -S ⁇ 0)R -SOzR 1 , -SOziOR 1 ), -SCfeNR'R 1 , -C ⁇ 0)R -0(0)0 ⁇ , -CiOJNR'R 1 , and -C(O)NR'OR' each R 1 is independently H or
- Cy 1 , Cy 2 , Cy 4 and Cy 6 are independently a C or N-attached known ring system selected from 3- to 8-membered carbocyclic or heterocyclic monocyclic ring, saturated or partially unsaturated; phenyl; 5- or 6-membered heteroaromatic ring; and 6- to 18-membered carbocyclic or heterocyclic polycyclic ring system, saturated, partially unsaturated, aromatic or partially aromatic;
- Cy 3 , Cy 5 and Cy 7 are independently a C or N-attached known ring system selected from 3- to 8-membered carbocyclic or heterocyclic monocyclic ring, saturated or partially unsaturated; phenyl; and 5- or 6-membered
- heteroaromatic ring wherein in the carbocyclic rings all ring members are carbon atoms; and in the heterocyclic and heteroaromatic rings one or more ring members are selected from N, O, and S; and wherein in all saturated or partially
- Another aspect of the invention relates to a pharmaceutical or veterinary composition which comprises an effective amount of a compound of formula (I) as defined above, or a pharmaceutically or veterinary acceptable salt thereof, or any stereoisomer either of the compound of formula (I) or of its pharmaceutically or veterinary acceptable salt, together with one or more pharmaceutically or veterinary acceptable excipients or carriers.
- the compounds of the invention are useful as antifibrinolytic and antihemorrhagic agents. Therefore, another aspect of the invention relates to a compound of formula (I) as defined above, or a pharmaceutically or veterinary acceptable salt thereof, or any stereoisomer either of the compound of formula (I) or of its pharmaceutically or veterinary acceptable salt, for use as a medicament.
- Another aspect of the invention relates to a compound of formula (I) as defined above, or a pharmaceutically or veterinary acceptable salt thereof, or any stereoisomer either of the compound of formula (I) or of its
- this aspect relates to the use of a compound of formula (I) as defined above, for the manufacture of a medicament for use as antifibrinolytic and antihemorrhagic agent; and may also be formulated as a method for the treatment and/or prevention of hyperfibrinolysis and/or hemorrhages comprising administering an effective amount of the previously defined compound of formula (I), or a pharmaceutically or veterinary acceptable salt thereof, or any stereoisomer either of the compound of formula (I) or of its pharmaceutically or veterinary acceptable salt, and one or more pharmaceutically or veterinary acceptable excipients or carriers, in a subject in need thereof, including a human.
- Another aspect of the invention relates to a compound of formula (II)
- R is an hydroxamic acid protective group, more particularly an hydroxamic acid protective group selected from the group consisting of tetrahydro-2H-pyran-2-yloxy (THP), benzyl, 1 -naphthylmethyl and dimethyloxybenzyl (DMB).
- THP tetrahydro-2H-pyran-2-yloxy
- DMB dimethyloxybenzyl
- Carbocyclic ring system refers to a known ring system wherein all the ring members are carbon atoms.
- heterocyclic ring system refers to a known ring system wherein one or more of the ring members, preferably 1 , 2, 3, or 4 ring members, are selected from N, O, and S, where chemically possible. Unless otherwise specified, the "heterocyclic” ring system may be attached to the rest of the molecule through a C or a N atom. Both the carbocyclic and heterocyclic rings can be saturated or partially unsaturated, and may be unsubstituted or substituted as described herein, being the susbstituents placed on any available position.
- polycyclic ring refers to a ring system which is formed by two, three or four rings which can be fused, bridged-fused, spiro-fused or can contain different types of fusion.
- fused rings the fusion occurs through one bond which is common to two adjoining rings; in “bridged-fused” rings the fusion occurs through a sequence of atoms (bridgehead) which is common to two rings; and in “spiro-fused” rings, the fusion occurs through only one atom
- spiro atom preferably a carbon atom, which is common to two adjoining rings (including bridged rings)
- the polycyclic ring system can be saturated, partially unsaturated, aromatic (except in the case of ring system A) or partially aromatic; and may be unsubstituted or substituted as described herein, being the susbstituents placed on any available position.
- the term "heteroaromatic" ring refers to a known aromatic ring system, wherein one or more of the ring members, preferably 1 , 2, 3, or 4 ring members, are selected from N, O, and S where chemically possible.
- the heteroaromatic ring and phenyl may be unsubstituted or substituted as described herein, being the susbstituents placed on any available position.
- one or two members of the rings are optionally C(O) and/or C(NH) and/or C[N(C C 4 )alkyl].
- saturated or unsaturated (CrC n )alkyl refers to a saturated branched or linear hydrocarbon chain which contains from 1 to n carbon atoms. When the (C-i-Cn)alkyl is saturated it contains only single bonds. When the
- (d-Cn)alkyl is unsaturated it contains one or two double bonds and/or one or two triple bonds.
- the saturated or unsaturated (CrC n )alkyl may be
- a halogen substituent means fluoro, chloro, bromo or iodo.
- Protective group refers to a grouping of atoms that when attached to a reactive group in a molecule masks, reduces or prevents that reactivity.
- substituted with one or more means that a group can be substituted with one or more, preferably with 1 , 2, 3 or 4 substituents, provided that this group has enough positions susceptible of being
- a first aspect of the invention relates to compounds of formula (I) or pharmaceutically or veterinary acceptable salts thereof, or any stereoisomer either of the compound of formula (I) or of its pharmaceutically or veterinary acceptable salt.
- compositions of formula (I) can be carried out by methods known in the art. For instance, they can be prepared from the parent compound, which contains a basic or acidic moiety, by conventional chemical methods. Generally, such salts are, for example, prepared by reacting the free acid or base forms of these compounds with a stoichiometric amount of the appropriate
- the compounds of the invention may be in crystalline form either as free solvation compounds or as solvates (e.g. hydrates) and it is intended that both forms are within the scope of the present invention.
- Methods of solvation are generally known within the art.
- the solvated forms with pharmaceutically or veterinary acceptable solvents such as water, ethanol and the like are equivalent to the unsolvated form for the purposes of the invention.
- Some compounds of formula (I) can have chiral centres that can give rise to various stereoisomers.
- the present invention relates to each of these stereoisomers and also mixtures thereof.
- some of the compounds of the present invention can show cis/trans isomers.
- the present invention relates to each of the geometric isomers and mixtures thereof.
- Diastereoisomers can be separated by conventional techniques such as chromatography or fractional crystallization.
- Optical isomers can be resolved by conventional techniques of optical resolution to give optically pure isomers. This resolution can be carried out on any chiral synthetic
- the invention refers to a compound of formula (I) as defined above, wherein A is a known 3- to 8-membered carbocyclic or heterocyclic monocyclic ring or a known 6- to 10-membered carbocyclic or heterocyclic bicyclic ring system.
- the invention refers to a compound of formula (I), wherein A is a monocyclic ring selected from a 3- to 6-membered carbocyclic ring, and a 5- to 6-membered heterocyclic ring.
- the invention refers to a compound of formula (I), wherein A is a carbocyclic monocyclic ring; or a polycyclic ring system, preferably a bicyclic ring system, wherein the ring containing the spiro atom attached to the hydroxamic acid and the sulfonyl group is a carbocyclic ring.
- the invention refers to a compound of formula (I), wherein A is selected from cyclopropane, cyclobutane, cyclopentane, cyclohexane, tetrahydrofuran, pyrrolidine, bicyclo[2.2.1 ]heptane, 2,3-dihydro- 1 H-indene, hexahydropyrrolizin-3-one, and 4-azaspiro[4.4]nonane.
- A is selected from cyclopropane, cyclobutane, cyclopentane, cyclohexane, tetrahydrofuran, pyrrolidine, bicyclo[2.2.1 ]heptane, 2,3-dihydro- 1 H-indene, hexahydropyrrolizin-3-one, and 4-azaspiro[4.4]nonane.
- the invention refers to a compound of formula (I), wherein A is unsubstituted, i.e. R 7 is H.
- the invention refers to a compound of formula (I), wherein B is a 6- to 7-membered carbocyclic or heterocyclic monocyclic ring.
- the invention refers to a compound of formula (I), wherein B is a saturated monocyclic ring, carbocyclic or heterocyclic, wherein at least one of the ring members of the heterocyclic ring is NR 4 .
- the invention refers to a compound of formula (I), wherein B is selected from cyclohexane, piperidine, morpholine, azepane, piperazine, pyrrolidine, and azetidine.
- B is piperidine, morpholine, azepane, pyrrolidine, and azetidine, wherein R 4 is placed on the N atom of these rings and R 5 -R 6 are H.
- B is piperazine, wherein R 4 and R 5 are placed on the N atoms and R 6 is H.
- the invention refers to a compound of formula (I), wherein A and B form a spirocyclic ring system selected from the group consisting of:
- the invention refers to a compound of formula (I), wherein C is phenyl.
- C is phenyl substituted with R 1 at the orto, meta or para position, and R 2 and R 3 are independently selected from H, halogen, R a , -OR a' ,and -NR R a ; wherein R a , R a' and R are
- C is independently selected from H and -(Ci-C 4 )alkyl optionally substituted with one or more halogen atoms.
- C is phenyl substituted with R-i at the para position and R 2 and R 3 are H.
- C is phenyl substituted with R 1 at the orto position and R 2 and R 3 are H.
- C is phenyl substituted with R 1 at the meta position and R 2 and R 3 are H.
- C is phenyl substituted with R 1 at the meta position, and R 2 at the para position, R 3 being H; or alternatively, C is phenyl substituted with R 1 at the para position, and R 2 at the meta position, R 3 being H; wherein R 2 is selected from H, halogen, R a , -OR a' ,and -NR R a ; and R a , R a and R are independently selected from H and -(Ci-C 4 )alkyl optionally substituted with one or more halogen atoms.
- the invention refers to a compound of formula (I), wherein in R (relating to Ri-R 3 ), Cy 1 and Cy 2 are independently optionally substituted with one or more substituents selected from R e and saturated or unsaturated (CrC 6 )alkyl optionally substituted with one or more substituents
- R h independently selected from R h and saturated or unsaturated (CrC 6 )alkyl optionally substituted as previously defined, more particularly with one or more substituents R h .
- the invention refers to a compound of formula (I), wherein in R (relating to Ri-R 3 ), R is H and saturated or unsaturated (Ci-Ci 2 )alkyl optionally substituted with one or more substituents R e , more particularly wherein in R e , R f and each R 9 are independently selected from H and saturated or unsaturated (CrC 6 )alkyl optionally substituted with one or more fluorine atoms.
- the invention refers to a compound of formula (I), wherein in R R 3 , R e is selected from halogen, -NO 2 , -CN, -OR f , -OC(O)R f , -OC(O)OR f , -OC(O)NR 9 R f , -NR 9 R f , -NR 9 C(O)R f , -NR 9 C(O)OR f ,
- the invention refers to a compound of formula (I), wherein in Ri-R 3 , R f and each R 9 are independently selected from H and saturated or unsaturated (CrC 6 )alkyl optionally substituted with one or more fluorine atoms.
- the invention refers to a compound of formula (I), wherein in Ri-R 3 , Cy 1 and Cy 2 are independently optionally substituted with one or more substituents selected from R e and saturated or unsaturated (CrC 6 )alkyl optionally substituted as previously defined; and Cy 6 is optionally substituted with one or more substituents independently selected from R h and saturated or unsaturated (CrC 6 )alkyl optionally substituted as previously defined.
- the invention refers to a compound of formula (I), wherein R R 3 are independently selected from H, halogen, -NO 2 , -CN, R a , -OR 3' , -OC(O)R a' , -OC(O)OR a' , -OC(O)NR R a' , -NR R a , -NR C(O)R a ,
- the invention refers to a compound of formula (I), wherein in R 4 -R 7 , R h is selected from halogen, -NO 2 , -CN, -OR', - ⁇ ( ⁇ ) ⁇ , -00(0)0 ⁇ , -OCiOJNR'R 1 , -N R 1 , -NR'CiO) ⁇ , -NRtyOJOR 1 , -NR'C(O)NR'R', -NR ⁇ O), ⁇ , -S , -S(O)R, -SO 2 R, -SO 2 NRR, -C(O)R, -C(O)OR, and
- the invention refers to a compound of formula (I), wherein R 4 -R 7 are independently selected from halogen, -NO 2 , -CN, R c , -OR c , -NR d R c , -NR d C(O)R c , -NR d C(O)OR c , -NR d C(O)NR d R c , -NR d S(O) 2 R c , -SR C , -S(O)R c , -SO 2 R c , -SO 2 NR d R c , -C(O)R c , -C(O)OR c , and -C(O)NR d R c .
- the invention refers to a compound of formula (I), wherein R 2 and R 3 are independently selected from H, halogen, R a , -OR a ,and -NR R a ; and R 5 -R 7 are independently selected from H, halogen, R c , -OR c , and -NR d R c , wherein R a , R a' , R , R c and R d are independently selected from H and -(Ci-C 4 )alkyl optionally substituted with one or more fluorine atoms.
- the invention refers to a compound of formula (I), wherein is selected from H, halogen, -NO 2 , -CN, R a , -OR 3' , -OR 3' ,
- R 4 is selected from halogen, -NO 2 , -CN, R c , -OR c , -NR d R c , -NR d C(O)R c , -NR d C(O)OR c , -NR d C(O)OR 3' ;
- the present invention also relates to the combination of any of the specific embodiments defined above for any of the variables A, B, C, and R 1 -R 7 .
- the compound of formula (I) is selected from the group consisting of:
- R is an hydroxamic acid protective group, more particularly an hydroxamic acid protective group selected from the group consisting of tetrahydro-2H-pyran-2-yloxy (THP), benzyl,
- the first conversion can be carried out in the presence of an activating agent such as 1 -ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride
- EDC.HCI Hydroxybenzotriazole
- HOBt Hydroxybenzotriazole
- a base such as N-methylmorpholine (NMM)
- NMM N-methylmorpholine
- suitable solvent such as dichloromethane, chloroform or dimethylformamide
- hydroxy protective groups include those where the hydroxy group is either acylated or alkylated such as benzyl, and trityl ethers as well as alkyl ethers, tetrahydropyranyl ethers, trialkylsilyl ethers and allyl ethers.
- the hydroxamic acid protective group is tetrahydro-2H-pyran-2-yloxy (THP), benzyl,
- DMB dimethyloxybenzyl
- (CrC 6 )alkyl benzyl, p-methoxyphenyl, trimethylsilyl, or [2-(Trimethylsilyl)- ethoxy]methyl (SEM).
- the carboxy protective group is (CrC 6 )alkyl
- the deprotection is carried out in basic medium, for example with LiOH in a suitable solvent such as tetrahydrofuran-methanol.
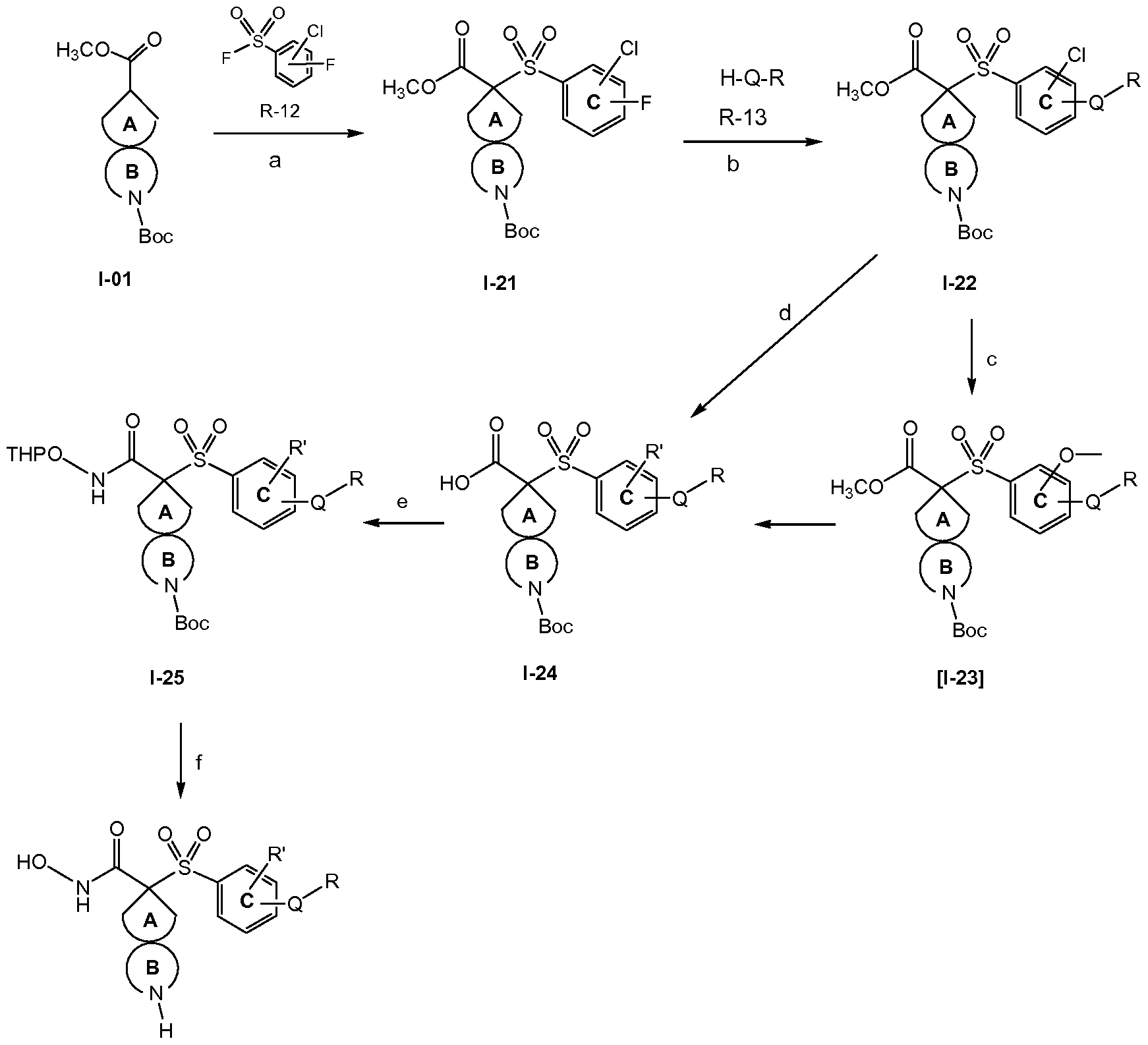
- a compound of formula (lla) can be reacted with a compound of formula H-Q-alk (VIII) in the presence of a base, such as NaH, optionally in a suitable solvent or without solvent, and preferably heating. After removal of the protective group in the resulting compound of formula (lie), compound of fomula (lb) is obtained.
- a base such as NaH
- a compound of formula (lla) can be reacted with a compound of formula H-NCy 2 R a (X), optionally in a suitable solvent or without solvent, and preferably heating. After removal of the protective group in the resulting compound of formula (lie), compound of formula (Id) is obtained.
- R 1 is -NR C(Y)R a , -NR C(Y)OR a ,
- -NR C(Y)NR R a , -NR S(O) 2 R a' , -NR SO 2 NR R a' may be obtained from a compound of formula (llg), which can be prepared as shown in the following scheme:
- compound of formula (Ila) is reacted with benzylamine, preferably heating to give a compound of formula (llf) which can be
- a compound of formula (llg) hydrogenated in the presence of Pd/C to give a compound of formula (llg).
- This compound can be reacted for example with an acyl halide in the presence of a base, such as triethylamine, in a suitable solvent, such as tetrahydrofuran, at a temperature comprised from room temperature to the reflux temperature of the solvent, to give an amide; or with an isocyanate to give a urea.
- a base such as triethylamine
- a suitable solvent such as tetrahydrofuran
- compound of formula (Ila) is reacted with benzyl alcohol in the presence of a base, such as NaH, in a suitable solvent, such as
- This compound can be reacted for example with an acyl halide in the presence of a base, such as triethylamine, in a suitable solvent, preferably at room temperature to give an ester.
- a base such as triethylamine
- the reactions described above can be carried out in a different order.
- the reactions carried out on compounds of formula (II) i.e. on the protected hydroxamic acid
- compounds of formula (III) i.e. the carboxylic ester
- the present invention also relates to a pharmaceutical or veterinary composition
- a pharmaceutical or veterinary composition comprising an effective amount of a compound of formula (I) as defined above, or a pharmaceutically or veterinary acceptable salt thereof, or any stereoisomer either of the compound of formula (I) or of its
- an effective amount refers to the amount of a compound that, when administered, is sufficient to prevent development of, or alleviate to some extent, one or more of the symptoms of the disease which is addressed.
- the specific dose of the compound of the invention to obtain a therapeutic benefit may vary depending on the particular circumstances of the individual patient including, among others, the size, weight, age and sex of the patient, the nature and stage of the disease, the aggressiveness of the disease, and the route of administration. For example, a dose of from about 0.01 to about 300 mg/kg may be used.
- pharmaceutically or veterinary acceptable excipients or carriers refers to pharmaceutically or veterinary acceptable materials, compositions or vehicles. Each component must be pharmaceutically or veterinary acceptable in the sense of being compatible with the other ingredients of the pharmaceutical or veterinary composition. It must also be suitable for use in contact with the tissue or organ of humans and animals without excessive toxicity, irritation, allergic response, immunogenicity or other problems or complications commensurate with a reasonable benefit/risk ratio.
- the election of the pharmaceutical or veterinary formulation will depend upon the nature of the active compound and its route of administration. Any route of administration may be used, for example oral, parenteral and topical administration.
- the pharmaceutical or veterinary composition may be formulated for oral administration and may contain one or more physiologically
- compatible carriers or excipients in solid or liquid form.
- These preparations may contain conventional ingredients such as binding agents, fillers, lubricants, and acceptable wetting agents.
- the pharmaceutical or veterinary composition may be formulated for parenteral administration in combination with conventional injectable liquid carriers, such as water or suitable alcohols.
- conventional pharmaceutical or veterinary excipients for injection such as stabilizing agents, solubilizing agents, and buffers, may be included in such compositions.
- compositions may be injected intramuscularly, intraperitoneal ⁇ , or intravenously.
- the pharmaceutical or veterinary composition may be formulated for topical administration.
- Formulations include creams, lotions, gels, powders, solutions and patches wherein the compound is dispersed or dissolved in suitable excipients.
- the topical compositions of the invention may be administered by means of a carrier material, which can be a solid support.
- a topical composition comprising a carrier material, which can be a solid support.
- solid supports include intelligent textiles, dressings, coatings, sponges, band-aids, sanitary pads, compresses, plasters, etc.
- the manufacture of such compositions can be obtained by conventional methods, for example, by mixing the
- compositions may be in any form, including, among others, tablets, pellets, capsules, aqueous or oily solutions,
- suspensions, emulsions, or dry powdered forms suitable for reconstitution with water or other suitable liquid medium before use, for immediate or retarded release can readily be determined by those skilled in the art according to the type of formulation being prepared.
- the compounds of the present invention are useful as antihemorrhagic and antifibrinolytic agents and can be used in a broad range of therapeutic applications.
- antifibrinolytic agents in addition to reducing postoperative hemorrhage, can be an alternative to blood transfusion and other hemoderivatives for example in heart, liver and orthopedic surgery, and also in the setting of oncologic surgery in organs rich in fibrinolysis activators (prostate, uterus).
- antifibrinolytic agents can reduce all- cause mortality and death due to bleeding.
- the antifibrinolytic agents of the invention can also be used to control bleeding in trombolytic therapy, e.g.
- the antifibrinolytic agents of the invention are useful in the treatment of local hemorrhages, e.g. after teeth extraction, in particular in patients with congenital coagulopathies, such as hemophilia, or patients with diabetes; in the treatment of menorrhage in women associated with congenital or acquired coagulopathies, as well as in post-partum haemorrhage, and in the treatment of hemorrhages of
- the HPLC measurement was performed using Gilson 281 from 233 pump (binary), an autosampler, and a UV detector. The fractions was detected by
- the MS detector was configured with an electrospray ionization source.
- the source temperature was maintained at 300-350 oC.
- Reverse phase HPLC was carried out on Luna C18 (100x30 mm; 4um).
- Solvent A water with 0.075% trifluoroacetic acid
- Solvent B acetonitrile with 0.075% trifluoroacetic acid.
- Gradient At room temperature, 20% of B to 40% of B within 6 min at 25 mL/min; then 40% B at 25 mL/min over 2 min, UV detector.
- PE Petroleum ether
- Q is O or S
- Q' is Q or SO 2
- Cy is phenyl or a 5- to 6- membered heteroaryl and can be optionally subtituted.
- R is a hydrocarbon chain which can be optionally subtituted.
- R is a hydrocarbon chain optionally substituted, a carbocyclic or heteroaliphatic ring optionally substituted, a phenyl or 5- to 6- membered heteroaryl optionally substituted.
- B is boronic acids, boronate esters or trifluoroborate salts and R is phenyl or a 5- to 6- membered heteroaryl or a 3- to 7- heterocyclic or carbocyclic aliphatic ring or a hydrocarbon chain which can be optionally substituted.
- R-10a 4-bromobenzenesulfonyl fluoride
- R and R' are hydrogen, X is a halogen and R" is phenyl or a 5- to 6- membered heteroaryl or a 3- to 7- heterocyclic or carbocyclic aliphatic ring or a hydrocarbon chain which can be optionally substituted.
- Q is O or NH
- R is a 3- to 7- heterocyclic or carbocyclic aliphatic ring or a hydrocarbon chain which can be optionally substituted and R' is halogen or alcoxy.
- Q is O or S and Cy is phenyl or a 5- to 6- membered heteroaryl and can be optionally substituted.
- Q is O or NH
- R is hydrogen
- X is a halogen
- R' is phenyl or a 5- to 6- membered heteroaryl or a 3- to 7- heterocyclic or carbocyclic aliphatic ring or a hydrocarbon chain which can be optionally substituted.
- Synthesized compounds are obtained as racemic mixtures.
- Corresponding isomers are purified by supercritical fluid chromatography (SFC) to obtain two enantiomers from each racemic compound.
- Thromboelastometry is a viscoelastometric method for haemostasis testing in whole blood.
- TEM® measures the interactions of coagulation factors, inhibitors and cellular components during the phases of clotting and
- tPA Tissue plasminogen activator
- CaCI 2 start-tern reagent
- CMs tested compounds
- Table 3 shows the results in human blood as effective concentration to delay lysis time by 50% (EC 50 LT); where, EC 50 LT ⁇ 25 ⁇ (+), 1 0 ⁇ ⁇ EC 50 LT ⁇ 25 ⁇ (++), 1 ⁇ ⁇ ECSO LT ⁇ 1 0 ⁇ (+++), EC 50LT ⁇ 1 ⁇ (++++) at all the assayed concentrations (1 000-0.2 ⁇ ).
- Example EC50LT Example EC50LT
- Table 4 shows the results in mice blood as effective concentration to delay lysis time by 50% (EC 50 LT); where, EC 50 LT ⁇ 10 ⁇ (+),1 ⁇ ⁇ EC 50 LT ⁇ 0 ⁇ (++),1 nM ⁇ EC50LT ⁇ 1 ⁇ (+++) and EC 50LT ⁇ 1 nM (++++) for all the assayed concentrations (1000-0.2 ⁇ ).
- Antifibrinolvtic effect in vivo tail bleeding assay
- Hyperfibrinolytic bleeding model consisted in injection of 0.5 mg/kg tPA into the ocular plexus to prolong bleeding time due to excessive fibrinolysis.
- tPA 0.5 mg/kg tPA
- polyurethane catheter (Microcannula 72-9030, Harvard Apparatus) for agents administration.
- the catheter was connected to a syringe pump (AL-1000, WPI) for the infusion of 200 ⁇ _ (10% bolus, 90% perfusion during 40 minutes) of tested agents.
- tPA 0.5 mg/kg
- saline or the different compounds was infused through the femoral catheter to ensure systemic distribution of all the agents.
- Reference compounds, TXA and Aprotinin were administered at 300 and 10 mg/Kg respectively; however, all compounds of the invention were administered at 1 mg/Kg. Five minutes later, 5 mm of tail tip were removed using a scalpel blade and the tail tip bathed in 1 mL of sterile saline at 37 °C.
- the time of bleeding was defined as the interval between initial transections and the visual cessation of bleeding, that was measured up to 30 minutes. A value of 30 min was assigned to those animals bleeding longer than the observation period.
- Table 6 shows the results reporting bleeding time (BT); where BT > 20 minutes (+),10 minutes ⁇ BT ⁇ 20 minutes (++), 5 minutes ⁇
- tested compounds of the invention show a very significant reduction of the bleeding time when compared to the control or TXA. In all the cases the dose of tested compounds were lower than TXA or Aprotinin doses.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Life Sciences & Earth Sciences (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Plural Heterocyclic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Other In-Based Heterocyclic Compounds (AREA)
Abstract
Description





































Claims





Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14/415,474 US9440989B2 (en) | 2012-07-18 | 2013-07-17 | Antifibrinolytic compounds |
| JP2015522082A JP6203257B2 (en) | 2012-07-18 | 2013-07-17 | New antifibrinolytic compounds |
| EP13737275.1A EP2877446B9 (en) | 2012-07-18 | 2013-07-17 | New antifibrinolytic compounds |
| AU2013292076A AU2013292076B2 (en) | 2012-07-18 | 2013-07-17 | New antifibrinolytic compounds |
| ES13737275.1T ES2603740T3 (en) | 2012-07-18 | 2013-07-17 | New antifibrinolytic compounds |
| CA2917604A CA2917604C (en) | 2012-07-18 | 2013-07-17 | New antifibrinolytic compounds |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP12382285 | 2012-07-18 | ||
| EP12382285.0 | 2012-07-18 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014012964A1 true WO2014012964A1 (en) | 2014-01-23 |
Family
ID=48793275
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2013/065071 WO2014012964A1 (en) | 2012-07-18 | 2013-07-17 | New antifibrinolytic compounds |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US9440989B2 (en) |
| EP (1) | EP2877446B9 (en) |
| JP (1) | JP6203257B2 (en) |
| AU (1) | AU2013292076B2 (en) |
| CA (1) | CA2917604C (en) |
| ES (1) | ES2603740T3 (en) |
| WO (1) | WO2014012964A1 (en) |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016071216A1 (en) * | 2014-11-03 | 2016-05-12 | Bayer Pharma Aktiengesellschaft | Piperidinylpyrazolopyrimidinones and their use |
| US10202331B2 (en) | 2014-11-03 | 2019-02-12 | Thrombolytics, Llc | Antifibrinolytic compounds |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB201216127D0 (en) * | 2012-09-11 | 2012-10-24 | Jaguar Cars | A method for determining the change in a vehicle battery |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3868411A (en) * | 1969-07-22 | 1975-02-25 | En Nom Collectif Science Union | Isobutyl cyclohexenyl compounds alpha (p-isobutyl-cyclohexenyl) alkanoic acids |
| US4483867A (en) * | 1981-11-17 | 1984-11-20 | Kabivitrum Ab | Antifibrinolytically active derivatives of tranexamic acid |
| US20020099035A1 (en) * | 2001-01-24 | 2002-07-25 | Sandanayaka Vincent P. | Method for preparing alpha-sulfonyl hydroxamic acid derivatives |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR20000075809A (en) * | 1997-02-27 | 2000-12-26 | 윌리암 에이취 캘넌, 에곤 이 버그 | N-Hydroxy-2-(alkyl, aryl or heteroaryl sulfanyl, sulfinyl or sulfonyl)-3-substituted alkyl, aryl or heteroarylamides as matrix metalloproteinase inhibitors |
| US6197791B1 (en) * | 1997-02-27 | 2001-03-06 | American Cyanamid Company | N-hdroxy-2-(alkyl, aryl, or heteroaryl, sulfanyl, sulfinyl or sulfonyl)-3-substituted alkyl, aryl or heteroarylamides as matrix metalloproteinase inhibitors |
| US6172057B1 (en) * | 1997-02-27 | 2001-01-09 | American Cyanamid Company | N-Hydroxy-2-(alkyl, aryl, or heteroaryl sulfanyl, sulfinyl or sulfonyl)-3-substituted alkyl, aryl or heteroarylamides as matrix metalloproteinase inhibitors |
| IL137566A0 (en) * | 1998-02-19 | 2001-07-24 | American Cyanamid Co | N-hydroxy-2-(alkyl, aryl or heteroaryl sulfanyl, sulfinyl or sulfonyl)-3-substituted-alkyl, aryl or heteroarylamides as matrix metalloproteinase inhibitors |
| US6225311B1 (en) * | 1999-01-27 | 2001-05-01 | American Cyanamid Company | Acetylenic α-amino acid-based sulfonamide hydroxamic acid tace inhibitors |
| US20040024024A1 (en) * | 2001-05-11 | 2004-02-05 | Freskos John N. | Aromatic sulfone hydroxamates and their use as protease inhibitors |
-
2013
- 2013-07-17 ES ES13737275.1T patent/ES2603740T3/en active Active
- 2013-07-17 JP JP2015522082A patent/JP6203257B2/en active Active
- 2013-07-17 CA CA2917604A patent/CA2917604C/en active Active
- 2013-07-17 WO PCT/EP2013/065071 patent/WO2014012964A1/en active Application Filing
- 2013-07-17 EP EP13737275.1A patent/EP2877446B9/en active Active
- 2013-07-17 US US14/415,474 patent/US9440989B2/en active Active
- 2013-07-17 AU AU2013292076A patent/AU2013292076B2/en active Active
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3868411A (en) * | 1969-07-22 | 1975-02-25 | En Nom Collectif Science Union | Isobutyl cyclohexenyl compounds alpha (p-isobutyl-cyclohexenyl) alkanoic acids |
| US4483867A (en) * | 1981-11-17 | 1984-11-20 | Kabivitrum Ab | Antifibrinolytically active derivatives of tranexamic acid |
| US20020099035A1 (en) * | 2001-01-24 | 2002-07-25 | Sandanayaka Vincent P. | Method for preparing alpha-sulfonyl hydroxamic acid derivatives |
Non-Patent Citations (8)
| Title |
|---|
| "PROTECTIVE GROUPS IN ORGANIC CHEMISTRY", pages: 369 - 451 |
| BOUYSSI D ET AL: "Rearrangement of oxaspiroheptanes to cyclohexanones mediated by lithium iodide", SYNLETT 2000 DE, no. 5, 2000, pages 749 - 751, XP008157852, ISSN: 0936-5214 * |
| D. BOUYSSI ET AL.: "Rearrangement of oxaspiroheptanes to cyclohexanones mediated by lithium iodide", SYNLETT, vol. 5, 2000, pages 749 - 751 |
| GREEN; P. G. M. WUTS: "Protective Groups in Organic Chemistry", 1999, WILEY, pages: 17 - 200 |
| OSAMU KITAGAWA ET AL.: "Stereoselective Iodine Atom Transfer [3 + 2] Cycloaddition Reaction with Alkenes Using Unsymmetrical Allylated Active Methine Radicals", THE JOURNAL OF ORGANIC CHEMISTRY, vol. 69, 2004, pages 2607 - 2610 |
| OSAMU KITAGAWA ET AL: "Stereoselective Iodine Atom Transfer [3 + 2] Cycloaddition Reaction with Alkenes Using Unsymmetrical Allylated Active Methine Radicals", THE JOURNAL OF ORGANIC CHEMISTRY, vol. 69, no. 7, 1 April 2004 (2004-04-01), pages 2607 - 2610, XP055043559, ISSN: 0022-3263, DOI: 10.1021/jo035771a * |
| T. W. GREEN; P. G. M. WUTS: "Protective Groups in Organic Chemistry", 1999, WILEY, pages: 17 - 200 |
| T. W. GREEN; P. G. M. WUTS: "Protective Groups in Organic Chemistry", 1999, WILEY, pages: 369 - 451 |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016071216A1 (en) * | 2014-11-03 | 2016-05-12 | Bayer Pharma Aktiengesellschaft | Piperidinylpyrazolopyrimidinones and their use |
| CN107108638A (en) * | 2014-11-03 | 2017-08-29 | 拜耳制药股份公司 | Piperidyl PyrazolopyrimidinonecGMP and application thereof |
| JP2017533235A (en) * | 2014-11-03 | 2017-11-09 | バイエル ファーマ アクチエンゲゼルシャフト | Piperidinylpyrazolopyrimidinones and their use |
| US10118930B2 (en) | 2014-11-03 | 2018-11-06 | Bayer Pharma Aktiengesellschaft | Piperidinylpyrazolopyrimidinones and their use |
| US10202331B2 (en) | 2014-11-03 | 2019-02-12 | Thrombolytics, Llc | Antifibrinolytic compounds |
| RU2713937C2 (en) * | 2014-11-03 | 2020-02-11 | Байер Фарма Акциенгезельшафт | Piperidinylpyrazolopyrimidinones and use thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| US20150175618A1 (en) | 2015-06-25 |
| JP2015530362A (en) | 2015-10-15 |
| AU2013292076A1 (en) | 2015-02-19 |
| CA2917604C (en) | 2021-08-10 |
| AU2013292076B2 (en) | 2017-02-23 |
| US9440989B2 (en) | 2016-09-13 |
| EP2877446B1 (en) | 2016-09-14 |
| JP6203257B2 (en) | 2017-09-27 |
| EP2877446B9 (en) | 2017-01-25 |
| ES2603740T3 (en) | 2017-03-01 |
| CA2917604A1 (en) | 2014-01-23 |
| EP2877446A1 (en) | 2015-06-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN103936763B (en) | Oxazolidinone compounds and its production and use | |
| JP4860698B2 (en) | Novel cycloalkanecarboxamides as factor Xa inhibitors | |
| JP4053597B2 (en) | Substituted N-[(aminoiminomethyl or aminomethyl) phenyl] propylamide | |
| US5227490A (en) | Fibrinogen receptor antagonists | |
| AU755805B2 (en) | Benzamide and sulfonamide substituted aminoguanidines and alkoxyguanidines as protease inhibitors | |
| JP2003500376A (en) | Factor Xa inhibitors | |
| MX2007010682A (en) | 2- [ isoquinolin-s - carbonyl ) amino] -propionic acid derivatives as inhibitors of factors xi and ix for the treatment of thrombosis. | |
| US4873253A (en) | Phenylalanine derivative and proteinase inhibitor | |
| JP2001508796A (en) | Thrombin inhibitors | |
| RU2502736C2 (en) | MACROCYCLIC UREA AND SULPHAMIDE DERIVATIVES AS TAFIa INHIBITORS | |
| JP2013543850A (en) | NOVEL COMPOUND, PROCESS FOR PRODUCTION AND USE THEREOF | |
| JPH06509076A (en) | 2-[3-(4-amidino-phenyl)]-propionic acid derivatives, their preparation and use | |
| RU2709810C2 (en) | Pyrazolo[3,4-c]pyridine derivatives | |
| EP2877446B1 (en) | New antifibrinolytic compounds | |
| JP2003528077A (en) | Substituted biphenyl derivatives | |
| FR2758329A1 (en) | New imidazole-4-butane-boronic acid derivatives | |
| US5206428A (en) | Tetrahydronaphthalene derivatives and preparation thereof | |
| JP2005525413A (en) | Substituted 5-membered polycyclic compounds useful for selective inhibition of the coagulation cascade | |
| WO2015104343A1 (en) | New antifibrinolytic compounds | |
| FR2728570A1 (en) | 1-OXO-2- (PHENYLSULFONYLAMINO) PENTYLPIPERIDINE DERIVATIVES, THEIR PREPARATION AND THEIR THERAPEUTIC USE | |
| US5494921A (en) | Fibrinogen receptor antagonists | |
| JP4373223B2 (en) | Novel crystals of 5-hydroxycarbamimidoyl-2-hydroxybenzenesulfonamide derivatives | |
| JP2005519866A (en) | 6-membered heterocyclic compounds useful for selective inhibition of the coagulation cascade | |
| JP2005068152A (en) | Amino acid derivative and drug containing the same as active ingredient | |
| JPH01186855A (en) | Phenoxyacetic acid derivative |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13737275 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14415474 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 2015522082 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2013737275 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013737275 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2013292076 Country of ref document: AU Date of ref document: 20130717 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2917604 Country of ref document: CA |