WO2013177534A2 - New salicylic acid derivatives, pharmaceutically acceptable salt thereof, composition thereof and method of use thereof - Google Patents
New salicylic acid derivatives, pharmaceutically acceptable salt thereof, composition thereof and method of use thereof Download PDFInfo
- Publication number
- WO2013177534A2 WO2013177534A2 PCT/US2013/042689 US2013042689W WO2013177534A2 WO 2013177534 A2 WO2013177534 A2 WO 2013177534A2 US 2013042689 W US2013042689 W US 2013042689W WO 2013177534 A2 WO2013177534 A2 WO 2013177534A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alk
- compound
- cancer
- alkoxy
- stat3
- Prior art date
Links
- 238000000034 method Methods 0.000 title claims abstract description 138
- 150000003839 salts Chemical class 0.000 title claims abstract description 37
- 239000000203 mixture Substances 0.000 title abstract description 49
- 229940058287 salicylic acid derivative anticestodals Drugs 0.000 title description 2
- 150000003872 salicylic acid derivatives Chemical class 0.000 title description 2
- 150000001875 compounds Chemical class 0.000 claims abstract description 189
- 108010017324 STAT3 Transcription Factor Proteins 0.000 claims abstract description 173
- 206010028980 Neoplasm Diseases 0.000 claims abstract description 89
- 230000000694 effects Effects 0.000 claims abstract description 76
- 108010029477 STAT5 Transcription Factor Proteins 0.000 claims abstract description 60
- 102000001712 STAT5 Transcription Factor Human genes 0.000 claims abstract description 59
- 201000011510 cancer Diseases 0.000 claims abstract description 50
- 239000012453 solvate Substances 0.000 claims abstract description 40
- 239000000651 prodrug Substances 0.000 claims abstract description 31
- 229940002612 prodrug Drugs 0.000 claims abstract description 31
- 102000004495 STAT3 Transcription Factor Human genes 0.000 claims abstract description 21
- 230000002401 inhibitory effect Effects 0.000 claims abstract description 21
- -1 Ci-6 polyhaloalkoxy Chemical group 0.000 claims description 108
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 102
- 125000003118 aryl group Chemical group 0.000 claims description 62
- 125000000217 alkyl group Chemical group 0.000 claims description 59
- 239000007787 solid Substances 0.000 claims description 57
- KQZLRWGGWXJPOS-NLFPWZOASA-N 1-[(1R)-1-(2,4-dichlorophenyl)ethyl]-6-[(4S,5R)-4-[(2S)-2-(hydroxymethyl)pyrrolidin-1-yl]-5-methylcyclohexen-1-yl]pyrazolo[3,4-b]pyrazine-3-carbonitrile Chemical compound ClC1=C(C=CC(=C1)Cl)[C@@H](C)N1N=C(C=2C1=NC(=CN=2)C1=CC[C@@H]([C@@H](C1)C)N1[C@@H](CCC1)CO)C#N KQZLRWGGWXJPOS-NLFPWZOASA-N 0.000 claims description 47
- 125000003545 alkoxy group Chemical group 0.000 claims description 43
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 36
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 36
- 125000006684 polyhaloalkyl group Polymers 0.000 claims description 36
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 33
- 208000005017 glioblastoma Diseases 0.000 claims description 33
- 125000001188 haloalkyl group Chemical group 0.000 claims description 32
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 32
- 125000004663 dialkyl amino group Chemical group 0.000 claims description 29
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 27
- 125000003282 alkyl amino group Chemical group 0.000 claims description 23
- 125000004438 haloalkoxy group Chemical group 0.000 claims description 23
- 229910052739 hydrogen Inorganic materials 0.000 claims description 21
- 208000003174 Brain Neoplasms Diseases 0.000 claims description 20
- 201000005787 hematologic cancer Diseases 0.000 claims description 19
- 125000003342 alkenyl group Chemical group 0.000 claims description 18
- 125000004414 alkyl thio group Chemical group 0.000 claims description 16
- 208000024200 hematopoietic and lymphoid system neoplasm Diseases 0.000 claims description 16
- 125000000592 heterocycloalkyl group Chemical group 0.000 claims description 16
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims description 15
- 206010006187 Breast cancer Diseases 0.000 claims description 15
- 208000026310 Breast neoplasm Diseases 0.000 claims description 15
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 15
- 208000014829 head and neck neoplasm Diseases 0.000 claims description 15
- 239000001257 hydrogen Substances 0.000 claims description 15
- 239000008194 pharmaceutical composition Substances 0.000 claims description 14
- 125000004435 hydrogen atom Chemical class [H]* 0.000 claims description 13
- 208000008443 pancreatic carcinoma Diseases 0.000 claims description 13
- 206010061902 Pancreatic neoplasm Diseases 0.000 claims description 11
- 208000000236 Prostatic Neoplasms Diseases 0.000 claims description 11
- 208000015486 malignant pancreatic neoplasm Diseases 0.000 claims description 11
- 201000002528 pancreatic cancer Diseases 0.000 claims description 11
- 125000006700 (C1-C6) alkylthio group Chemical group 0.000 claims description 10
- 206010060862 Prostate cancer Diseases 0.000 claims description 10
- 230000001154 acute effect Effects 0.000 claims description 10
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 9
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 claims description 8
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 claims description 8
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 claims description 8
- 208000026037 malignant tumor of neck Diseases 0.000 claims description 8
- 125000004890 (C1-C6) alkylamino group Chemical group 0.000 claims description 7
- 208000000453 Skin Neoplasms Diseases 0.000 claims description 7
- 201000007270 liver cancer Diseases 0.000 claims description 7
- 208000014018 liver neoplasm Diseases 0.000 claims description 7
- 201000000849 skin cancer Diseases 0.000 claims description 7
- 125000004737 (C1-C6) haloalkoxy group Chemical group 0.000 claims description 6
- 101100240518 Caenorhabditis elegans nhr-12 gene Proteins 0.000 claims description 6
- 208000034578 Multiple myelomas Diseases 0.000 claims description 6
- 206010035226 Plasma cell myeloma Diseases 0.000 claims description 6
- 208000032839 leukemia Diseases 0.000 claims description 6
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 5
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 claims description 4
- 125000000171 (C1-C6) haloalkyl group Chemical group 0.000 claims description 4
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 4
- SXPRVMIZFRCAGC-UHFFFAOYSA-N 1,2,3,4,5-pentafluoro-6-methylbenzene Chemical group CC1=C(F)C(F)=C(F)C(F)=C1F SXPRVMIZFRCAGC-UHFFFAOYSA-N 0.000 claims description 3
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 claims description 3
- 101100240516 Caenorhabditis elegans nhr-10 gene Proteins 0.000 claims description 3
- 101100516563 Caenorhabditis elegans nhr-6 gene Proteins 0.000 claims description 3
- 101100516568 Caenorhabditis elegans nhr-7 gene Proteins 0.000 claims description 3
- 101100516572 Caenorhabditis elegans nhr-8 gene Proteins 0.000 claims description 3
- 101100079984 Caenorhabditis elegans nhr-9 gene Proteins 0.000 claims description 3
- 229940126208 compound 22 Drugs 0.000 claims description 3
- 125000000262 haloalkenyl group Chemical group 0.000 claims description 3
- 125000000232 haloalkynyl group Chemical group 0.000 claims description 3
- 101150009274 nhr-1 gene Proteins 0.000 claims description 3
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 3
- WWTBZEKOSBFBEM-SPWPXUSOSA-N (2s)-2-[[2-benzyl-3-[hydroxy-[(1r)-2-phenyl-1-(phenylmethoxycarbonylamino)ethyl]phosphoryl]propanoyl]amino]-3-(1h-indol-3-yl)propanoic acid Chemical group N([C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)O)C(=O)C(CP(O)(=O)[C@H](CC=1C=CC=CC=1)NC(=O)OCC=1C=CC=CC=1)CC1=CC=CC=C1 WWTBZEKOSBFBEM-SPWPXUSOSA-N 0.000 claims description 2
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 2
- PNUZDKCDAWUEGK-CYZMBNFOSA-N Sitafloxacin Chemical compound C([C@H]1N)N(C=2C(=C3C(C(C(C(O)=O)=CN3[C@H]3[C@H](C3)F)=O)=CC=2F)Cl)CC11CC1 PNUZDKCDAWUEGK-CYZMBNFOSA-N 0.000 claims description 2
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 claims description 2
- 125000001475 halogen functional group Chemical group 0.000 claims 10
- NPRYCHLHHVWLQZ-TURQNECASA-N 2-amino-9-[(2R,3S,4S,5R)-4-fluoro-3-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-7-prop-2-ynylpurin-8-one Chemical compound NC1=NC=C2N(C(N(C2=N1)[C@@H]1O[C@@H]([C@H]([C@H]1O)F)CO)=O)CC#C NPRYCHLHHVWLQZ-TURQNECASA-N 0.000 claims 1
- 238000011282 treatment Methods 0.000 abstract description 40
- 230000001419 dependent effect Effects 0.000 abstract description 5
- 102100024040 Signal transducer and activator of transcription 3 Human genes 0.000 description 155
- 239000000126 substance Substances 0.000 description 94
- 239000012467 final product Substances 0.000 description 75
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 64
- 210000004027 cell Anatomy 0.000 description 64
- 239000003112 inhibitor Substances 0.000 description 53
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 48
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 47
- 229940125877 compound 31 Drugs 0.000 description 47
- 238000006243 chemical reaction Methods 0.000 description 44
- 238000004128 high performance liquid chromatography Methods 0.000 description 43
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 41
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 39
- 125000004432 carbon atom Chemical group C* 0.000 description 37
- 108090000623 proteins and genes Proteins 0.000 description 37
- 239000000243 solution Substances 0.000 description 36
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 34
- 125000001424 substituent group Chemical group 0.000 description 33
- 239000003814 drug Substances 0.000 description 32
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 30
- 108091000080 Phosphotransferase Proteins 0.000 description 27
- 229940079593 drug Drugs 0.000 description 27
- 102000020233 phosphotransferase Human genes 0.000 description 27
- 239000011734 sodium Substances 0.000 description 27
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 26
- 230000005764 inhibitory process Effects 0.000 description 26
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 25
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 23
- 125000005843 halogen group Chemical group 0.000 description 23
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 23
- 230000027455 binding Effects 0.000 description 22
- 125000000392 cycloalkenyl group Chemical group 0.000 description 22
- 208000035475 disorder Diseases 0.000 description 22
- 239000003921 oil Substances 0.000 description 22
- 235000019198 oils Nutrition 0.000 description 22
- 125000000738 acetamido group Chemical group [H]C([H])([H])C(=O)N([H])[*] 0.000 description 21
- 238000009739 binding Methods 0.000 description 21
- 125000005842 heteroatom Chemical group 0.000 description 21
- 229910052757 nitrogen Inorganic materials 0.000 description 21
- 102000004169 proteins and genes Human genes 0.000 description 21
- 150000003254 radicals Chemical class 0.000 description 21
- 229910052799 carbon Inorganic materials 0.000 description 20
- 230000003389 potentiating effect Effects 0.000 description 20
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 19
- 210000004556 brain Anatomy 0.000 description 19
- 201000010099 disease Diseases 0.000 description 19
- 235000019439 ethyl acetate Nutrition 0.000 description 19
- 125000001072 heteroaryl group Chemical group 0.000 description 18
- 239000002904 solvent Substances 0.000 description 18
- DLFVBJFMPXGRIB-UHFFFAOYSA-N Acetamide Chemical compound CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 description 17
- 239000002253 acid Substances 0.000 description 17
- 125000000304 alkynyl group Chemical group 0.000 description 17
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 17
- 229910052760 oxygen Inorganic materials 0.000 description 17
- 239000001301 oxygen Substances 0.000 description 17
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 17
- 241000699670 Mus sp. Species 0.000 description 16
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 16
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 16
- 125000004429 atom Chemical group 0.000 description 16
- 229910052717 sulfur Inorganic materials 0.000 description 16
- 239000011593 sulfur Substances 0.000 description 16
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 15
- 239000000047 product Substances 0.000 description 15
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 14
- 239000003981 vehicle Substances 0.000 description 14
- FJKROLUGYXJWQN-UHFFFAOYSA-N 4-hydroxybenzoic acid Chemical compound OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 13
- 238000003556 assay Methods 0.000 description 13
- 229910052736 halogen Inorganic materials 0.000 description 13
- 150000002367 halogens Chemical class 0.000 description 13
- 230000002829 reductive effect Effects 0.000 description 13
- 229920006395 saturated elastomer Polymers 0.000 description 13
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 12
- 150000004820 halides Chemical class 0.000 description 12
- 238000002198 surface plasmon resonance spectroscopy Methods 0.000 description 12
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 11
- 210000000481 breast Anatomy 0.000 description 11
- 229960004889 salicylic acid Drugs 0.000 description 11
- 238000001262 western blot Methods 0.000 description 11
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 10
- WPYMKLBDIGXBTP-UHFFFAOYSA-M benzoate Chemical compound [O-]C(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-M 0.000 description 10
- 230000004064 dysfunction Effects 0.000 description 10
- 238000002474 experimental method Methods 0.000 description 10
- 239000000463 material Substances 0.000 description 10
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 10
- 239000000843 powder Substances 0.000 description 10
- 238000004809 thin layer chromatography Methods 0.000 description 10
- 210000004881 tumor cell Anatomy 0.000 description 10
- 102000003688 G-Protein-Coupled Receptors Human genes 0.000 description 9
- 108090000045 G-Protein-Coupled Receptors Proteins 0.000 description 9
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 9
- 125000001931 aliphatic group Chemical group 0.000 description 9
- 230000005754 cellular signaling Effects 0.000 description 9
- 230000002559 cytogenic effect Effects 0.000 description 9
- 238000005516 engineering process Methods 0.000 description 9
- 238000001727 in vivo Methods 0.000 description 9
- 230000001965 increasing effect Effects 0.000 description 9
- 230000004048 modification Effects 0.000 description 9
- 238000012986 modification Methods 0.000 description 9
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 9
- 150000003384 small molecules Chemical class 0.000 description 9
- 150000003573 thiols Chemical class 0.000 description 9
- WZUVPPKBWHMQCE-UHFFFAOYSA-N Haematoxylin Chemical compound C12=CC(O)=C(O)C=C2CC2(O)C1C1=CC=C(O)C(O)=C1OC2 WZUVPPKBWHMQCE-UHFFFAOYSA-N 0.000 description 8
- 241000124008 Mammalia Species 0.000 description 8
- 241000699666 Mus <mouse, genus> Species 0.000 description 8
- 101150058731 STAT5A gene Proteins 0.000 description 8
- 150000001299 aldehydes Chemical class 0.000 description 8
- 239000012267 brine Substances 0.000 description 8
- 238000004440 column chromatography Methods 0.000 description 8
- 231100000135 cytotoxicity Toxicity 0.000 description 8
- 238000001514 detection method Methods 0.000 description 8
- 150000002148 esters Chemical class 0.000 description 8
- 238000002360 preparation method Methods 0.000 description 8
- 230000004044 response Effects 0.000 description 8
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 8
- 230000004083 survival effect Effects 0.000 description 8
- 238000012360 testing method Methods 0.000 description 8
- DTQVDTLACAAQTR-UHFFFAOYSA-N trifluoroacetic acid Substances OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 8
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 7
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 7
- 102100026596 Bcl-2-like protein 1 Human genes 0.000 description 7
- 102000016736 Cyclin Human genes 0.000 description 7
- 108050006400 Cyclin Proteins 0.000 description 7
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 7
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 7
- 239000008280 blood Substances 0.000 description 7
- 210000004369 blood Anatomy 0.000 description 7
- 230000003833 cell viability Effects 0.000 description 7
- 229940125904 compound 1 Drugs 0.000 description 7
- 230000003013 cytotoxicity Effects 0.000 description 7
- 230000003993 interaction Effects 0.000 description 7
- 150000002894 organic compounds Chemical class 0.000 description 7
- 229910052698 phosphorus Inorganic materials 0.000 description 7
- 239000011574 phosphorus Substances 0.000 description 7
- 230000026731 phosphorylation Effects 0.000 description 7
- 238000006366 phosphorylation reaction Methods 0.000 description 7
- 210000002307 prostate Anatomy 0.000 description 7
- 239000000741 silica gel Substances 0.000 description 7
- 229910002027 silica gel Inorganic materials 0.000 description 7
- 238000003756 stirring Methods 0.000 description 7
- 230000001225 therapeutic effect Effects 0.000 description 7
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 6
- 241000283707 Capra Species 0.000 description 6
- 108020004414 DNA Proteins 0.000 description 6
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 6
- 108010044012 STAT1 Transcription Factor Proteins 0.000 description 6
- 102000006381 STAT1 Transcription Factor Human genes 0.000 description 6
- 102100024481 Signal transducer and activator of transcription 5A Human genes 0.000 description 6
- 102000040945 Transcription factor Human genes 0.000 description 6
- 108091023040 Transcription factor Proteins 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- 239000004480 active ingredient Substances 0.000 description 6
- 238000004458 analytical method Methods 0.000 description 6
- 150000001540 azides Chemical class 0.000 description 6
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 6
- 230000004071 biological effect Effects 0.000 description 6
- 230000015572 biosynthetic process Effects 0.000 description 6
- 239000000460 chlorine Substances 0.000 description 6
- 239000012230 colorless oil Substances 0.000 description 6
- 239000013078 crystal Substances 0.000 description 6
- 238000002347 injection Methods 0.000 description 6
- 239000007924 injection Substances 0.000 description 6
- 239000003446 ligand Substances 0.000 description 6
- 238000004519 manufacturing process Methods 0.000 description 6
- YYROPELSRYBVMQ-UHFFFAOYSA-N p-toluenesulfonyl chloride Substances CC1=CC=C(S(Cl)(=O)=O)C=C1 YYROPELSRYBVMQ-UHFFFAOYSA-N 0.000 description 6
- 230000001575 pathological effect Effects 0.000 description 6
- 230000035699 permeability Effects 0.000 description 6
- 230000036515 potency Effects 0.000 description 6
- 238000000746 purification Methods 0.000 description 6
- 238000003753 real-time PCR Methods 0.000 description 6
- 230000011664 signaling Effects 0.000 description 6
- 239000011780 sodium chloride Substances 0.000 description 6
- 238000006467 substitution reaction Methods 0.000 description 6
- 238000003786 synthesis reaction Methods 0.000 description 6
- BQXUPNKLZNSUMC-YUQWMIPFSA-N CCN(CCCCCOCC(=O)N[C@H](C(=O)N1C[C@H](O)C[C@H]1C(=O)N[C@@H](C)c1ccc(cc1)-c1scnc1C)C(C)(C)C)CCOc1ccc(cc1)C(=O)c1c(sc2cc(O)ccc12)-c1ccc(O)cc1 Chemical compound CCN(CCCCCOCC(=O)N[C@H](C(=O)N1C[C@H](O)C[C@H]1C(=O)N[C@@H](C)c1ccc(cc1)-c1scnc1C)C(C)(C)C)CCOc1ccc(cc1)C(=O)c1c(sc2cc(O)ccc12)-c1ccc(O)cc1 BQXUPNKLZNSUMC-YUQWMIPFSA-N 0.000 description 5
- 0 CN(CC(N(Cc1ccc(C2CCCCC2)cc1)c(cc1)ccc1C(O)=O)=O)S(*)(=O)=O Chemical compound CN(CC(N(Cc1ccc(C2CCCCC2)cc1)c(cc1)ccc1C(O)=O)=O)S(*)(=O)=O 0.000 description 5
- KXDHJXZQYSOELW-UHFFFAOYSA-N Carbamic acid Chemical compound NC(O)=O KXDHJXZQYSOELW-UHFFFAOYSA-N 0.000 description 5
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 5
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 5
- 229920001213 Polysorbate 20 Polymers 0.000 description 5
- 101150063267 STAT5B gene Proteins 0.000 description 5
- 230000001594 aberrant effect Effects 0.000 description 5
- 150000001408 amides Chemical class 0.000 description 5
- 125000003277 amino group Chemical group 0.000 description 5
- 239000011324 bead Substances 0.000 description 5
- 230000008901 benefit Effects 0.000 description 5
- 229910052796 boron Inorganic materials 0.000 description 5
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 5
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical compound OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 5
- 230000004663 cell proliferation Effects 0.000 description 5
- 239000003795 chemical substances by application Substances 0.000 description 5
- 238000011161 development Methods 0.000 description 5
- 230000018109 developmental process Effects 0.000 description 5
- 239000011737 fluorine Substances 0.000 description 5
- 229910052731 fluorine Inorganic materials 0.000 description 5
- 230000014509 gene expression Effects 0.000 description 5
- 238000000338 in vitro Methods 0.000 description 5
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 5
- 210000004072 lung Anatomy 0.000 description 5
- 230000009437 off-target effect Effects 0.000 description 5
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 5
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 5
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 5
- 230000008569 process Effects 0.000 description 5
- 108090000765 processed proteins & peptides Proteins 0.000 description 5
- 230000035755 proliferation Effects 0.000 description 5
- 125000000547 substituted alkyl group Chemical group 0.000 description 5
- 239000000758 substrate Substances 0.000 description 5
- 239000000725 suspension Substances 0.000 description 5
- 238000011144 upstream manufacturing Methods 0.000 description 5
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 4
- ALYNCZNDIQEVRV-UHFFFAOYSA-N 4-aminobenzoic acid Chemical compound NC1=CC=C(C(O)=O)C=C1 ALYNCZNDIQEVRV-UHFFFAOYSA-N 0.000 description 4
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 4
- LNSXRXFBSDRILE-UHFFFAOYSA-N Cucurbitacin Natural products CC(=O)OC(C)(C)C=CC(=O)C(C)(O)C1C(O)CC2(C)C3CC=C4C(C)(C)C(O)C(O)CC4(C)C3(C)C(=O)CC12C LNSXRXFBSDRILE-UHFFFAOYSA-N 0.000 description 4
- CVKKIVYBGGDJCR-SXDZHWHFSA-N Cucurbitacin B Natural products CC(=O)OC(C)(C)C=CC(=O)[C@@](C)(O)[C@@H]1[C@@H](O)C[C@]2(C)C3=CC[C@@H]4C(C)(C)C(=O)[C@H](O)C[C@@]4(C)[C@@H]3CC(=O)[C@@]12C CVKKIVYBGGDJCR-SXDZHWHFSA-N 0.000 description 4
- 241000283074 Equus asinus Species 0.000 description 4
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical compound C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 4
- 101000617830 Homo sapiens Sterol O-acyltransferase 1 Proteins 0.000 description 4
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 4
- 238000005481 NMR spectroscopy Methods 0.000 description 4
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 4
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium on carbon Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 4
- 102100024474 Signal transducer and activator of transcription 5B Human genes 0.000 description 4
- 102100021993 Sterol O-acyltransferase 1 Human genes 0.000 description 4
- 101000697584 Streptomyces lavendulae Streptothricin acetyltransferase Proteins 0.000 description 4
- 102000000887 Transcription factor STAT Human genes 0.000 description 4
- 108050007918 Transcription factor STAT Proteins 0.000 description 4
- 230000004913 activation Effects 0.000 description 4
- 150000001412 amines Chemical class 0.000 description 4
- 230000006907 apoptotic process Effects 0.000 description 4
- 230000008499 blood brain barrier function Effects 0.000 description 4
- 239000000872 buffer Substances 0.000 description 4
- 150000001721 carbon Chemical group 0.000 description 4
- 230000010261 cell growth Effects 0.000 description 4
- 239000003153 chemical reaction reagent Substances 0.000 description 4
- 229910052801 chlorine Inorganic materials 0.000 description 4
- 210000001072 colon Anatomy 0.000 description 4
- 229940125851 compound 27 Drugs 0.000 description 4
- 229940126540 compound 41 Drugs 0.000 description 4
- 150000001904 cucurbitacins Chemical class 0.000 description 4
- 230000007423 decrease Effects 0.000 description 4
- 238000003745 diagnosis Methods 0.000 description 4
- PIGAXYFCLPQWOD-UHFFFAOYSA-N dihydrocucurbitacin I Natural products CC12C(=O)CC3(C)C(C(C)(O)C(=O)CCC(C)(O)C)C(O)CC3(C)C1CC=C1C2C=C(O)C(=O)C1(C)C PIGAXYFCLPQWOD-UHFFFAOYSA-N 0.000 description 4
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 4
- 239000000839 emulsion Substances 0.000 description 4
- 210000002950 fibroblast Anatomy 0.000 description 4
- 238000002875 fluorescence polarization Methods 0.000 description 4
- 238000009472 formulation Methods 0.000 description 4
- 201000000459 head and neck squamous cell carcinoma Diseases 0.000 description 4
- 125000000623 heterocyclic group Chemical group 0.000 description 4
- 230000006872 improvement Effects 0.000 description 4
- 239000012044 organic layer Substances 0.000 description 4
- 230000036470 plasma concentration Effects 0.000 description 4
- 238000012216 screening Methods 0.000 description 4
- 239000000377 silicon dioxide Substances 0.000 description 4
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 4
- 210000000130 stem cell Anatomy 0.000 description 4
- 230000001629 suppression Effects 0.000 description 4
- VFUAJMPDXIRPKO-LQELWAHVSA-N (e)-3-(6-bromopyridin-2-yl)-2-cyano-n-[(1s)-1-phenylethyl]prop-2-enamide Chemical compound N([C@@H](C)C=1C=CC=CC=1)C(=O)C(\C#N)=C\C1=CC=CC(Br)=N1 VFUAJMPDXIRPKO-LQELWAHVSA-N 0.000 description 3
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 3
- TVTJUIAKQFIXCE-HUKYDQBMSA-N 2-amino-9-[(2R,3S,4S,5R)-4-fluoro-3-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-7-prop-2-ynyl-1H-purine-6,8-dione Chemical compound NC=1NC(C=2N(C(N(C=2N=1)[C@@H]1O[C@@H]([C@H]([C@H]1O)F)CO)=O)CC#C)=O TVTJUIAKQFIXCE-HUKYDQBMSA-N 0.000 description 3
- WNVSFFVDMUSXSX-UHFFFAOYSA-N 4-[(4-cyclohexylphenyl)methyl-[2-[methyl-(2,3,4,5,6-pentafluorophenyl)sulfonylamino]acetyl]amino]-2-hydroxybenzoic acid Chemical compound FC=1C(F)=C(F)C(F)=C(F)C=1S(=O)(=O)N(C)CC(=O)N(C=1C=C(O)C(C(O)=O)=CC=1)CC(C=C1)=CC=C1C1CCCCC1 WNVSFFVDMUSXSX-UHFFFAOYSA-N 0.000 description 3
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 3
- 102000007469 Actins Human genes 0.000 description 3
- 108010085238 Actins Proteins 0.000 description 3
- KXDAEFPNCMNJSK-UHFFFAOYSA-N Benzamide Chemical compound NC(=O)C1=CC=CC=C1 KXDAEFPNCMNJSK-UHFFFAOYSA-N 0.000 description 3
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 3
- 102000004127 Cytokines Human genes 0.000 description 3
- 241000282326 Felis catus Species 0.000 description 3
- 108010010803 Gelatin Proteins 0.000 description 3
- 108010001336 Horseradish Peroxidase Proteins 0.000 description 3
- 108090000144 Human Proteins Proteins 0.000 description 3
- 102000003839 Human Proteins Human genes 0.000 description 3
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 3
- 229940121730 Janus kinase 2 inhibitor Drugs 0.000 description 3
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 3
- 239000000020 Nitrocellulose Substances 0.000 description 3
- 108700020796 Oncogene Proteins 0.000 description 3
- 241000283973 Oryctolagus cuniculus Species 0.000 description 3
- 229910019142 PO4 Inorganic materials 0.000 description 3
- 229930040373 Paraformaldehyde Natural products 0.000 description 3
- 108010001441 Phosphopeptides Proteins 0.000 description 3
- ABLZXFCXXLZCGV-UHFFFAOYSA-N Phosphorous acid Chemical class OP(O)=O ABLZXFCXXLZCGV-UHFFFAOYSA-N 0.000 description 3
- 229920000776 Poly(Adenosine diphosphate-ribose) polymerase Polymers 0.000 description 3
- 239000004721 Polyphenylene oxide Substances 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- 102000001708 Protein Isoforms Human genes 0.000 description 3
- 108010029485 Protein Isoforms Proteins 0.000 description 3
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 3
- 108060006706 SRC Proteins 0.000 description 3
- 102000001332 SRC Human genes 0.000 description 3
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 description 3
- 239000012190 activator Substances 0.000 description 3
- 125000002947 alkylene group Chemical group 0.000 description 3
- 150000001413 amino acids Chemical class 0.000 description 3
- 239000007864 aqueous solution Substances 0.000 description 3
- 210000001130 astrocyte Anatomy 0.000 description 3
- 239000012298 atmosphere Substances 0.000 description 3
- 239000002585 base Substances 0.000 description 3
- 239000004305 biphenyl Substances 0.000 description 3
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 3
- 229910052794 bromium Inorganic materials 0.000 description 3
- 239000002775 capsule Substances 0.000 description 3
- 238000001516 cell proliferation assay Methods 0.000 description 3
- 239000013626 chemical specie Substances 0.000 description 3
- 239000012043 crude product Substances 0.000 description 3
- 230000001086 cytosolic effect Effects 0.000 description 3
- 230000003247 decreasing effect Effects 0.000 description 3
- 206010012601 diabetes mellitus Diseases 0.000 description 3
- 238000010790 dilution Methods 0.000 description 3
- 239000012895 dilution Substances 0.000 description 3
- 239000006185 dispersion Substances 0.000 description 3
- 238000010494 dissociation reaction Methods 0.000 description 3
- 230000005593 dissociations Effects 0.000 description 3
- 231100000673 dose–response relationship Toxicity 0.000 description 3
- 239000003937 drug carrier Substances 0.000 description 3
- 230000001605 fetal effect Effects 0.000 description 3
- 125000000524 functional group Chemical group 0.000 description 3
- 239000007789 gas Substances 0.000 description 3
- 239000000499 gel Substances 0.000 description 3
- 239000008273 gelatin Substances 0.000 description 3
- 229920000159 gelatin Polymers 0.000 description 3
- 235000019322 gelatine Nutrition 0.000 description 3
- 235000011852 gelatine desserts Nutrition 0.000 description 3
- 125000004366 heterocycloalkenyl group Chemical group 0.000 description 3
- 150000004677 hydrates Chemical class 0.000 description 3
- 150000002430 hydrocarbons Chemical group 0.000 description 3
- 208000013403 hyperactivity Diseases 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 238000003364 immunohistochemistry Methods 0.000 description 3
- 229910052740 iodine Inorganic materials 0.000 description 3
- 230000000670 limiting effect Effects 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 230000001404 mediated effect Effects 0.000 description 3
- 239000012528 membrane Substances 0.000 description 3
- 229910052751 metal Inorganic materials 0.000 description 3
- 238000010172 mouse model Methods 0.000 description 3
- 229920001220 nitrocellulos Polymers 0.000 description 3
- 230000002611 ovarian Effects 0.000 description 3
- 230000002018 overexpression Effects 0.000 description 3
- 229920002866 paraformaldehyde Polymers 0.000 description 3
- 239000002245 particle Substances 0.000 description 3
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 3
- 239000010452 phosphate Substances 0.000 description 3
- 229920000570 polyether Polymers 0.000 description 3
- 229920000642 polymer Polymers 0.000 description 3
- 239000003755 preservative agent Substances 0.000 description 3
- 230000002265 prevention Effects 0.000 description 3
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 3
- 239000011541 reaction mixture Substances 0.000 description 3
- 238000004007 reversed phase HPLC Methods 0.000 description 3
- 239000000523 sample Substances 0.000 description 3
- 238000013207 serial dilution Methods 0.000 description 3
- 229910052708 sodium Inorganic materials 0.000 description 3
- 238000010186 staining Methods 0.000 description 3
- 125000005415 substituted alkoxy group Chemical group 0.000 description 3
- 125000005346 substituted cycloalkyl group Chemical group 0.000 description 3
- 239000003826 tablet Substances 0.000 description 3
- 230000008685 targeting Effects 0.000 description 3
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 3
- 238000002560 therapeutic procedure Methods 0.000 description 3
- FWPIDFUJEMBDLS-UHFFFAOYSA-L tin(II) chloride dihydrate Chemical compound O.O.Cl[Sn]Cl FWPIDFUJEMBDLS-UHFFFAOYSA-L 0.000 description 3
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 3
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Substances C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 3
- 238000013042 tunel staining Methods 0.000 description 3
- QWUWMCYKGHVNAV-UHFFFAOYSA-N 1,2-dihydrostilbene Chemical group C=1C=CC=CC=1CCC1=CC=CC=C1 QWUWMCYKGHVNAV-UHFFFAOYSA-N 0.000 description 2
- KJUGUADJHNHALS-UHFFFAOYSA-N 1H-tetrazole Chemical compound C=1N=NNN=1 KJUGUADJHNHALS-UHFFFAOYSA-N 0.000 description 2
- QRBRVZCEKCBURQ-UHFFFAOYSA-N 2-(1-methyltetrazol-5-yl)pyridine Chemical compound CN1N=NN=C1C1=CC=CC=N1 QRBRVZCEKCBURQ-UHFFFAOYSA-N 0.000 description 2
- KTPONJJKCBOJCQ-UHFFFAOYSA-N 4-(2h-tetrazol-5-yl)aniline Chemical compound C1=CC(N)=CC=C1C1=NNN=N1 KTPONJJKCBOJCQ-UHFFFAOYSA-N 0.000 description 2
- KUHNCPCUPOPMDY-UHFFFAOYSA-N 4-cyclohexylbenzaldehyde Chemical compound C1=CC(C=O)=CC=C1C1CCCCC1 KUHNCPCUPOPMDY-UHFFFAOYSA-N 0.000 description 2
- PRJHEJGMSOBHTO-UHFFFAOYSA-N 7-[(4-chlorophenyl)methyl]-1-(3-hydroxypropyl)-3-methyl-8-[3-(trifluoromethoxy)phenoxy]purine-2,6-dione Chemical compound C=1C=C(Cl)C=CC=1CN1C=2C(=O)N(CCCO)C(=O)N(C)C=2N=C1OC1=CC=CC(OC(F)(F)F)=C1 PRJHEJGMSOBHTO-UHFFFAOYSA-N 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- 241000283690 Bos taurus Species 0.000 description 2
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 description 2
- 241000282472 Canis lupus familiaris Species 0.000 description 2
- 208000005623 Carcinogenesis Diseases 0.000 description 2
- 201000009030 Carcinoma Diseases 0.000 description 2
- 241000700199 Cavia porcellus Species 0.000 description 2
- 206010009944 Colon cancer Diseases 0.000 description 2
- 229940126657 Compound 17 Drugs 0.000 description 2
- 229940126639 Compound 33 Drugs 0.000 description 2
- 229940127007 Compound 39 Drugs 0.000 description 2
- 229940126650 Compound 3f Drugs 0.000 description 2
- 229940126559 Compound 4e Drugs 0.000 description 2
- 108090000695 Cytokines Proteins 0.000 description 2
- 101100239628 Danio rerio myca gene Proteins 0.000 description 2
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 2
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- 241000588724 Escherichia coli Species 0.000 description 2
- 108010009202 Growth Factor Receptors Proteins 0.000 description 2
- 102000009465 Growth Factor Receptors Human genes 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- 206010061218 Inflammation Diseases 0.000 description 2
- 208000022559 Inflammatory bowel disease Diseases 0.000 description 2
- 108090001005 Interleukin-6 Proteins 0.000 description 2
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 2
- 240000007472 Leucaena leucocephala Species 0.000 description 2
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 2
- 238000000134 MTT assay Methods 0.000 description 2
- 231100000002 MTT assay Toxicity 0.000 description 2
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- 238000011789 NOD SCID mouse Methods 0.000 description 2
- 102000007399 Nuclear hormone receptor Human genes 0.000 description 2
- 108020005497 Nuclear hormone receptor Proteins 0.000 description 2
- 108010058765 Oncogene Protein pp60(v-src) Proteins 0.000 description 2
- 206010033128 Ovarian cancer Diseases 0.000 description 2
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 description 2
- 241001494479 Pecora Species 0.000 description 2
- 108091005804 Peptidases Proteins 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- 102000004160 Phosphoric Monoester Hydrolases Human genes 0.000 description 2
- 108090000608 Phosphoric Monoester Hydrolases Proteins 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- 229920002556 Polyethylene Glycol 300 Polymers 0.000 description 2
- 239000004365 Protease Substances 0.000 description 2
- 102000001253 Protein Kinase Human genes 0.000 description 2
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Chemical compound C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 description 2
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 2
- 102100033810 RAC-alpha serine/threonine-protein kinase Human genes 0.000 description 2
- 239000012083 RIPA buffer Substances 0.000 description 2
- 102100037486 Reverse transcriptase/ribonuclease H Human genes 0.000 description 2
- 102000004389 Ribonucleoproteins Human genes 0.000 description 2
- 108010081734 Ribonucleoproteins Proteins 0.000 description 2
- PXIPVTKHYLBLMZ-UHFFFAOYSA-N Sodium azide Chemical compound [Na+].[N-]=[N+]=[N-] PXIPVTKHYLBLMZ-UHFFFAOYSA-N 0.000 description 2
- 101710172711 Structural protein Proteins 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 2
- 238000012288 TUNEL assay Methods 0.000 description 2
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 2
- 102000004243 Tubulin Human genes 0.000 description 2
- 108090000704 Tubulin Proteins 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 125000002015 acyclic group Chemical group 0.000 description 2
- 230000008484 agonism Effects 0.000 description 2
- 125000005431 alkyl carboxamide group Chemical group 0.000 description 2
- 125000005119 alkyl cycloalkyl group Chemical group 0.000 description 2
- 125000005233 alkylalcohol group Chemical group 0.000 description 2
- 229960004050 aminobenzoic acid Drugs 0.000 description 2
- 125000000129 anionic group Chemical group 0.000 description 2
- 230000000259 anti-tumor effect Effects 0.000 description 2
- 238000013459 approach Methods 0.000 description 2
- 206010003246 arthritis Diseases 0.000 description 2
- BLFLLBZGZJTVJG-UHFFFAOYSA-N benzocaine Chemical compound CCOC(=O)C1=CC=C(N)C=C1 BLFLLBZGZJTVJG-UHFFFAOYSA-N 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid group Chemical group C(C1=CC=CC=C1)(=O)O WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- WALNLEVAFXXSSS-UHFFFAOYSA-N benzyl 4-[(4-cyclohexylphenyl)methylamino]-2-phenylmethoxybenzoate Chemical compound C=1C=C(NCC=2C=CC(=CC=2)C2CCCCC2)C=C(OCC=2C=CC=CC=2)C=1C(=O)OCC1=CC=CC=C1 WALNLEVAFXXSSS-UHFFFAOYSA-N 0.000 description 2
- WCGLJFMXFQXBKC-UHFFFAOYSA-N benzyl 4-amino-2-phenylmethoxybenzoate Chemical compound C=1C=CC=CC=1COC1=CC(N)=CC=C1C(=O)OCC1=CC=CC=C1 WCGLJFMXFQXBKC-UHFFFAOYSA-N 0.000 description 2
- 238000005574 benzylation reaction Methods 0.000 description 2
- 208000036815 beta tubulin Diseases 0.000 description 2
- 150000005347 biaryls Chemical group 0.000 description 2
- 125000002619 bicyclic group Chemical group 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 230000031018 biological processes and functions Effects 0.000 description 2
- 229960002685 biotin Drugs 0.000 description 2
- 235000020958 biotin Nutrition 0.000 description 2
- 239000011616 biotin Substances 0.000 description 2
- 235000010290 biphenyl Nutrition 0.000 description 2
- 210000001218 blood-brain barrier Anatomy 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- 210000005103 brain tumor initiating cell Anatomy 0.000 description 2
- IYYIVELXUANFED-UHFFFAOYSA-N bromo(trimethyl)silane Chemical compound C[Si](C)(C)Br IYYIVELXUANFED-UHFFFAOYSA-N 0.000 description 2
- 238000011088 calibration curve Methods 0.000 description 2
- 230000036952 cancer formation Effects 0.000 description 2
- CREMABGTGYGIQB-UHFFFAOYSA-N carbon carbon Chemical group C.C CREMABGTGYGIQB-UHFFFAOYSA-N 0.000 description 2
- 150000007942 carboxylates Chemical group 0.000 description 2
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 2
- 231100000504 carcinogenesis Toxicity 0.000 description 2
- 230000030833 cell death Effects 0.000 description 2
- 230000012292 cell migration Effects 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- OSASVXMJTNOKOY-UHFFFAOYSA-N chlorobutanol Chemical compound CC(C)(O)C(Cl)(Cl)Cl OSASVXMJTNOKOY-UHFFFAOYSA-N 0.000 description 2
- 239000011248 coating agent Substances 0.000 description 2
- 238000000576 coating method Methods 0.000 description 2
- 230000009850 completed effect Effects 0.000 description 2
- 238000010668 complexation reaction Methods 0.000 description 2
- 229940125773 compound 10 Drugs 0.000 description 2
- 229940125797 compound 12 Drugs 0.000 description 2
- 229940126543 compound 14 Drugs 0.000 description 2
- 229940125758 compound 15 Drugs 0.000 description 2
- 229940126142 compound 16 Drugs 0.000 description 2
- 229940125810 compound 20 Drugs 0.000 description 2
- 229940126086 compound 21 Drugs 0.000 description 2
- 229940125833 compound 23 Drugs 0.000 description 2
- 229940125961 compound 24 Drugs 0.000 description 2
- 229940127204 compound 29 Drugs 0.000 description 2
- 229940125878 compound 36 Drugs 0.000 description 2
- 229940125807 compound 37 Drugs 0.000 description 2
- 229940127573 compound 38 Drugs 0.000 description 2
- 229940125796 compound 3d Drugs 0.000 description 2
- 229940125844 compound 46 Drugs 0.000 description 2
- 229940126115 compound 4f Drugs 0.000 description 2
- 229940125880 compound 4j Drugs 0.000 description 2
- 229940125801 compound 7f Drugs 0.000 description 2
- 230000008878 coupling Effects 0.000 description 2
- 238000010168 coupling process Methods 0.000 description 2
- 238000005859 coupling reaction Methods 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- 230000002498 deadly effect Effects 0.000 description 2
- 230000002939 deleterious effect Effects 0.000 description 2
- UQLDLKMNUJERMK-UHFFFAOYSA-L di(octadecanoyloxy)lead Chemical compound [Pb+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O UQLDLKMNUJERMK-UHFFFAOYSA-L 0.000 description 2
- 125000005432 dialkylcarboxamide group Chemical group 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- 239000012897 dilution medium Substances 0.000 description 2
- 238000006471 dimerization reaction Methods 0.000 description 2
- 239000002270 dispersing agent Substances 0.000 description 2
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 2
- 238000007876 drug discovery Methods 0.000 description 2
- 239000003480 eluent Substances 0.000 description 2
- 239000003995 emulsifying agent Substances 0.000 description 2
- HKSZLNNOFSGOKW-UHFFFAOYSA-N ent-staurosporine Natural products C12=C3N4C5=CC=CC=C5C3=C3CNC(=O)C3=C2C2=CC=CC=C2N1C1CC(NC)C(OC)C4(C)O1 HKSZLNNOFSGOKW-UHFFFAOYSA-N 0.000 description 2
- YQGOJNYOYNNSMM-UHFFFAOYSA-N eosin Chemical compound [Na+].OC(=O)C1=CC=CC=C1C1=C2C=C(Br)C(=O)C(Br)=C2OC2=C(Br)C(O)=C(Br)C=C21 YQGOJNYOYNNSMM-UHFFFAOYSA-N 0.000 description 2
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 description 2
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 description 2
- 125000004494 ethyl ester group Chemical group 0.000 description 2
- 208000021045 exocrine pancreatic carcinoma Diseases 0.000 description 2
- 239000012091 fetal bovine serum Substances 0.000 description 2
- ZHNUHDYFZUAESO-UHFFFAOYSA-N formamide Substances NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 2
- 235000011187 glycerol Nutrition 0.000 description 2
- 230000012010 growth Effects 0.000 description 2
- 210000003128 head Anatomy 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- 238000003929 heteronuclear multiple quantum coherence Methods 0.000 description 2
- 230000001976 improved effect Effects 0.000 description 2
- 230000004054 inflammatory process Effects 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- 239000007972 injectable composition Substances 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- 239000011630 iodine Substances 0.000 description 2
- 208000002551 irritable bowel syndrome Diseases 0.000 description 2
- 230000000155 isotopic effect Effects 0.000 description 2
- 150000002576 ketones Chemical class 0.000 description 2
- 239000008101 lactose Substances 0.000 description 2
- 239000010410 layer Substances 0.000 description 2
- GLXDVVHUTZTUQK-UHFFFAOYSA-M lithium;hydroxide;hydrate Chemical compound [Li+].O.[OH-] GLXDVVHUTZTUQK-UHFFFAOYSA-M 0.000 description 2
- 210000004185 liver Anatomy 0.000 description 2
- RENRQMCACQEWFC-UGKGYDQZSA-N lnp023 Chemical compound C1([C@H]2N(CC=3C=4C=CNC=4C(C)=CC=3OC)CC[C@@H](C2)OCC)=CC=C(C(O)=O)C=C1 RENRQMCACQEWFC-UGKGYDQZSA-N 0.000 description 2
- 208000020816 lung neoplasm Diseases 0.000 description 2
- 239000006166 lysate Substances 0.000 description 2
- 238000012423 maintenance Methods 0.000 description 2
- 230000014759 maintenance of location Effects 0.000 description 2
- 108010082117 matrigel Proteins 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 201000001441 melanoma Diseases 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- 125000000250 methylamino group Chemical group [H]N(*)C([H])([H])[H] 0.000 description 2
- 239000000178 monomer Substances 0.000 description 2
- RLKHFSNWQCZBDC-UHFFFAOYSA-N n-(benzenesulfonyl)-n-fluorobenzenesulfonamide Chemical compound C=1C=CC=CC=1S(=O)(=O)N(F)S(=O)(=O)C1=CC=CC=C1 RLKHFSNWQCZBDC-UHFFFAOYSA-N 0.000 description 2
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 description 2
- 239000013642 negative control Substances 0.000 description 2
- 230000035407 negative regulation of cell proliferation Effects 0.000 description 2
- 210000004940 nucleus Anatomy 0.000 description 2
- SBOJXQVPLKSXOG-UHFFFAOYSA-N o-amino-hydroxylamine Chemical compound NON SBOJXQVPLKSXOG-UHFFFAOYSA-N 0.000 description 2
- 230000002246 oncogenic effect Effects 0.000 description 2
- 230000003287 optical effect Effects 0.000 description 2
- 210000003463 organelle Anatomy 0.000 description 2
- 210000000496 pancreas Anatomy 0.000 description 2
- 238000007911 parenteral administration Methods 0.000 description 2
- 230000037361 pathway Effects 0.000 description 2
- 239000000825 pharmaceutical preparation Substances 0.000 description 2
- 238000007747 plating Methods 0.000 description 2
- 229920001281 polyalkylene Polymers 0.000 description 2
- 229920000728 polyester Polymers 0.000 description 2
- 239000013641 positive control Substances 0.000 description 2
- 125000006239 protecting group Chemical class 0.000 description 2
- 108060006633 protein kinase Proteins 0.000 description 2
- 238000010791 quenching Methods 0.000 description 2
- 230000000171 quenching effect Effects 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 238000006722 reduction reaction Methods 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 238000012552 review Methods 0.000 description 2
- 210000003491 skin Anatomy 0.000 description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 2
- HKSZLNNOFSGOKW-FYTWVXJKSA-N staurosporine Chemical compound C12=C3N4C5=CC=CC=C5C3=C3CNC(=O)C3=C2C2=CC=CC=C2N1[C@H]1C[C@@H](NC)[C@@H](OC)[C@]4(C)O1 HKSZLNNOFSGOKW-FYTWVXJKSA-N 0.000 description 2
- CGPUWJWCVCFERF-UHFFFAOYSA-N staurosporine Natural products C12=C3N4C5=CC=CC=C5C3=C3CNC(=O)C3=C2C2=CC=CC=C2N1C1CC(NC)C(OC)C4(OC)O1 CGPUWJWCVCFERF-UHFFFAOYSA-N 0.000 description 2
- 239000008223 sterile water Substances 0.000 description 2
- 230000000638 stimulation Effects 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 235000000346 sugar Nutrition 0.000 description 2
- 150000008163 sugars Chemical class 0.000 description 2
- 230000006103 sulfonylation Effects 0.000 description 2
- 238000005694 sulfonylation reaction Methods 0.000 description 2
- 239000000375 suspending agent Substances 0.000 description 2
- 208000024891 symptom Diseases 0.000 description 2
- 150000003536 tetrazoles Chemical class 0.000 description 2
- 238000011287 therapeutic dose Methods 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- 125000003944 tolyl group Chemical group 0.000 description 2
- 238000011200 topical administration Methods 0.000 description 2
- 238000013518 transcription Methods 0.000 description 2
- 230000035897 transcription Effects 0.000 description 2
- CSRZQMIRAZTJOY-UHFFFAOYSA-N trimethylsilyl iodide Chemical compound C[Si](C)(C)I CSRZQMIRAZTJOY-UHFFFAOYSA-N 0.000 description 2
- 230000004614 tumor growth Effects 0.000 description 2
- 239000000080 wetting agent Substances 0.000 description 2
- ASGMFNBUXDJWJJ-JLCFBVMHSA-N (1R,3R)-3-[[3-bromo-1-[4-(5-methyl-1,3,4-thiadiazol-2-yl)phenyl]pyrazolo[3,4-d]pyrimidin-6-yl]amino]-N,1-dimethylcyclopentane-1-carboxamide Chemical compound BrC1=NN(C2=NC(=NC=C21)N[C@H]1C[C@@](CC1)(C(=O)NC)C)C1=CC=C(C=C1)C=1SC(=NN=1)C ASGMFNBUXDJWJJ-JLCFBVMHSA-N 0.000 description 1
- UAOUIVVJBYDFKD-XKCDOFEDSA-N (1R,9R,10S,11R,12R,15S,18S,21R)-10,11,21-trihydroxy-8,8-dimethyl-14-methylidene-4-(prop-2-enylamino)-20-oxa-5-thia-3-azahexacyclo[9.7.2.112,15.01,9.02,6.012,18]henicosa-2(6),3-dien-13-one Chemical compound C([C@@H]1[C@@H](O)[C@@]23C(C1=C)=O)C[C@H]2[C@]12C(N=C(NCC=C)S4)=C4CC(C)(C)[C@H]1[C@H](O)[C@]3(O)OC2 UAOUIVVJBYDFKD-XKCDOFEDSA-N 0.000 description 1
- AOSZTAHDEDLTLQ-AZKQZHLXSA-N (1S,2S,4R,8S,9S,11S,12R,13S,19S)-6-[(3-chlorophenyl)methyl]-12,19-difluoro-11-hydroxy-8-(2-hydroxyacetyl)-9,13-dimethyl-6-azapentacyclo[10.8.0.02,9.04,8.013,18]icosa-14,17-dien-16-one Chemical compound C([C@@H]1C[C@H]2[C@H]3[C@]([C@]4(C=CC(=O)C=C4[C@@H](F)C3)C)(F)[C@@H](O)C[C@@]2([C@@]1(C1)C(=O)CO)C)N1CC1=CC=CC(Cl)=C1 AOSZTAHDEDLTLQ-AZKQZHLXSA-N 0.000 description 1
- ABJSOROVZZKJGI-OCYUSGCXSA-N (1r,2r,4r)-2-(4-bromophenyl)-n-[(4-chlorophenyl)-(2-fluoropyridin-4-yl)methyl]-4-morpholin-4-ylcyclohexane-1-carboxamide Chemical compound C1=NC(F)=CC(C(NC(=O)[C@H]2[C@@H](C[C@@H](CC2)N2CCOCC2)C=2C=CC(Br)=CC=2)C=2C=CC(Cl)=CC=2)=C1 ABJSOROVZZKJGI-OCYUSGCXSA-N 0.000 description 1
- FANCTJAFZSYTIS-IQUVVAJASA-N (1r,3s,5z)-5-[(2e)-2-[(1r,3as,7ar)-7a-methyl-1-[(2r)-4-(phenylsulfonimidoyl)butan-2-yl]-2,3,3a,5,6,7-hexahydro-1h-inden-4-ylidene]ethylidene]-4-methylidenecyclohexane-1,3-diol Chemical compound C([C@@H](C)[C@@H]1[C@]2(CCCC(/[C@@H]2CC1)=C\C=C\1C([C@@H](O)C[C@H](O)C/1)=C)C)CS(=N)(=O)C1=CC=CC=C1 FANCTJAFZSYTIS-IQUVVAJASA-N 0.000 description 1
- GLGNXYJARSMNGJ-VKTIVEEGSA-N (1s,2s,3r,4r)-3-[[5-chloro-2-[(1-ethyl-6-methoxy-2-oxo-4,5-dihydro-3h-1-benzazepin-7-yl)amino]pyrimidin-4-yl]amino]bicyclo[2.2.1]hept-5-ene-2-carboxamide Chemical compound CCN1C(=O)CCCC2=C(OC)C(NC=3N=C(C(=CN=3)Cl)N[C@H]3[C@H]([C@@]4([H])C[C@@]3(C=C4)[H])C(N)=O)=CC=C21 GLGNXYJARSMNGJ-VKTIVEEGSA-N 0.000 description 1
- LAJAFFLJAJMYLK-CVOKMOJFSA-N (1s,2s,3r,4r)-3-[[5-chloro-2-[[(7s)-4-methoxy-7-morpholin-4-yl-6,7,8,9-tetrahydro-5h-benzo[7]annulen-3-yl]amino]pyrimidin-4-yl]amino]bicyclo[2.2.1]hept-5-ene-2-carboxamide Chemical compound N1([C@H]2CCC3=CC=C(C(=C3CC2)OC)NC=2N=C(C(=CN=2)Cl)N[C@H]2[C@H]([C@@]3([H])C[C@@]2(C=C3)[H])C(N)=O)CCOCC1 LAJAFFLJAJMYLK-CVOKMOJFSA-N 0.000 description 1
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 1
- SHAHPWSYJFYMRX-GDLCADMTSA-N (2S)-2-(4-{[(1R,2S)-2-hydroxycyclopentyl]methyl}phenyl)propanoic acid Chemical compound C1=CC([C@@H](C(O)=O)C)=CC=C1C[C@@H]1[C@@H](O)CCC1 SHAHPWSYJFYMRX-GDLCADMTSA-N 0.000 description 1
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 1
- IUSARDYWEPUTPN-OZBXUNDUSA-N (2r)-n-[(2s,3r)-4-[[(4s)-6-(2,2-dimethylpropyl)spiro[3,4-dihydropyrano[2,3-b]pyridine-2,1'-cyclobutane]-4-yl]amino]-3-hydroxy-1-[3-(1,3-thiazol-2-yl)phenyl]butan-2-yl]-2-methoxypropanamide Chemical compound C([C@H](NC(=O)[C@@H](C)OC)[C@H](O)CN[C@@H]1C2=CC(CC(C)(C)C)=CN=C2OC2(CCC2)C1)C(C=1)=CC=CC=1C1=NC=CS1 IUSARDYWEPUTPN-OZBXUNDUSA-N 0.000 description 1
- YJLIKUSWRSEPSM-WGQQHEPDSA-N (2r,3r,4s,5r)-2-[6-amino-8-[(4-phenylphenyl)methylamino]purin-9-yl]-5-(hydroxymethyl)oxolane-3,4-diol Chemical compound C=1C=C(C=2C=CC=CC=2)C=CC=1CNC1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O YJLIKUSWRSEPSM-WGQQHEPDSA-N 0.000 description 1
- KAFZOLYKKCWUBI-HPMAGDRPSA-N (2s)-2-[[(2s)-2-[[(2s)-1-[(2s)-3-amino-2-[[(2s)-2-[[(2s)-2-(3-cyclohexylpropanoylamino)-4-methylpentanoyl]amino]-5-methylhexanoyl]amino]propanoyl]pyrrolidine-2-carbonyl]amino]-5-(diaminomethylideneamino)pentanoyl]amino]butanediamide Chemical compound N([C@@H](CC(C)C)C(=O)N[C@@H](CCC(C)C)C(=O)N[C@@H](CN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCN=C(N)N)C(=O)N[C@@H](CC(N)=O)C(N)=O)C(=O)CCC1CCCCC1 KAFZOLYKKCWUBI-HPMAGDRPSA-N 0.000 description 1
- STBLNCCBQMHSRC-BATDWUPUSA-N (2s)-n-[(3s,4s)-5-acetyl-7-cyano-4-methyl-1-[(2-methylnaphthalen-1-yl)methyl]-2-oxo-3,4-dihydro-1,5-benzodiazepin-3-yl]-2-(methylamino)propanamide Chemical compound O=C1[C@@H](NC(=O)[C@H](C)NC)[C@H](C)N(C(C)=O)C2=CC(C#N)=CC=C2N1CC1=C(C)C=CC2=CC=CC=C12 STBLNCCBQMHSRC-BATDWUPUSA-N 0.000 description 1
- KRIWIRSMQRQYJG-DLBZAZTESA-N (2s,3s)-3-[[7-(benzylamino)-3-propan-2-ylpyrazolo[1,5-a]pyrimidin-5-yl]amino]butane-1,2,4-triol Chemical compound C=1C(N[C@@H](CO)[C@H](O)CO)=NC2=C(C(C)C)C=NN2C=1NCC1=CC=CC=C1 KRIWIRSMQRQYJG-DLBZAZTESA-N 0.000 description 1
- AEVBPXDFDKBGLT-YOUFYPILSA-N (2s,3s,4r,5r)-n-[2-[4-(diethoxyphosphorylmethyl)anilino]-2-oxoethyl]-5-(2,4-dioxopyrimidin-1-yl)-3,4-dihydroxyoxolane-2-carboxamide Chemical compound C1=CC(CP(=O)(OCC)OCC)=CC=C1NC(=O)CNC(=O)[C@@H]1[C@@H](O)[C@@H](O)[C@H](N2C(NC(=O)C=C2)=O)O1 AEVBPXDFDKBGLT-YOUFYPILSA-N 0.000 description 1
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 description 1
- UDQTXCHQKHIQMH-KYGLGHNPSA-N (3ar,5s,6s,7r,7ar)-5-(difluoromethyl)-2-(ethylamino)-5,6,7,7a-tetrahydro-3ah-pyrano[3,2-d][1,3]thiazole-6,7-diol Chemical compound S1C(NCC)=N[C@H]2[C@@H]1O[C@H](C(F)F)[C@@H](O)[C@@H]2O UDQTXCHQKHIQMH-KYGLGHNPSA-N 0.000 description 1
- HUWSZNZAROKDRZ-RRLWZMAJSA-N (3r,4r)-3-azaniumyl-5-[[(2s,3r)-1-[(2s)-2,3-dicarboxypyrrolidin-1-yl]-3-methyl-1-oxopentan-2-yl]amino]-5-oxo-4-sulfanylpentane-1-sulfonate Chemical compound OS(=O)(=O)CC[C@@H](N)[C@@H](S)C(=O)N[C@@H]([C@H](C)CC)C(=O)N1CCC(C(O)=O)[C@H]1C(O)=O HUWSZNZAROKDRZ-RRLWZMAJSA-N 0.000 description 1
- YQOLEILXOBUDMU-KRWDZBQOSA-N (4R)-5-[(6-bromo-3-methyl-2-pyrrolidin-1-ylquinoline-4-carbonyl)amino]-4-(2-chlorophenyl)pentanoic acid Chemical compound CC1=C(C2=C(C=CC(=C2)Br)N=C1N3CCCC3)C(=O)NC[C@H](CCC(=O)O)C4=CC=CC=C4Cl YQOLEILXOBUDMU-KRWDZBQOSA-N 0.000 description 1
- VUDZSIYXZUYWSC-DBRKOABJSA-N (4r)-1-[(2r,4r,5r)-3,3-difluoro-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-4-hydroxy-1,3-diazinan-2-one Chemical compound FC1(F)[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)N[C@H](O)CC1 VUDZSIYXZUYWSC-DBRKOABJSA-N 0.000 description 1
- PNHBRYIAJCYNDA-VQCQRNETSA-N (4r)-6-[2-[2-ethyl-4-(4-fluorophenyl)-6-phenylpyridin-3-yl]ethyl]-4-hydroxyoxan-2-one Chemical compound C([C@H](O)C1)C(=O)OC1CCC=1C(CC)=NC(C=2C=CC=CC=2)=CC=1C1=CC=C(F)C=C1 PNHBRYIAJCYNDA-VQCQRNETSA-N 0.000 description 1
- QVBVQHTXLPNXEY-ZMFCMNQTSA-N (4r)-6-[2-[4-(4-fluorophenyl)-6-phenyl-2-propan-2-ylpyridin-3-yl]ethyl]-4-hydroxyoxan-2-one Chemical compound C([C@H](O)C1)C(=O)OC1CCC=1C(C(C)C)=NC(C=2C=CC=CC=2)=CC=1C1=CC=C(F)C=C1 QVBVQHTXLPNXEY-ZMFCMNQTSA-N 0.000 description 1
- VIMMECPCYZXUCI-MIMFYIINSA-N (4s,6r)-6-[(1e)-4,4-bis(4-fluorophenyl)-3-(1-methyltetrazol-5-yl)buta-1,3-dienyl]-4-hydroxyoxan-2-one Chemical compound CN1N=NN=C1C(\C=C\[C@@H]1OC(=O)C[C@@H](O)C1)=C(C=1C=CC(F)=CC=1)C1=CC=C(F)C=C1 VIMMECPCYZXUCI-MIMFYIINSA-N 0.000 description 1
- MNIPVWXWSPXERA-IDNZQHFXSA-N (6r,7r)-1-[(4s,5r)-4-acetyloxy-5-methyl-3-methylidene-6-phenylhexyl]-4,7-dihydroxy-6-(11-phenoxyundecanoyloxy)-2,8-dioxabicyclo[3.2.1]octane-3,4,5-tricarboxylic acid Chemical compound C([C@@H](C)[C@H](OC(C)=O)C(=C)CCC12[C@@H]([C@@H](OC(=O)CCCCCCCCCCOC=3C=CC=CC=3)C(O1)(C(O)=O)C(O)(C(O2)C(O)=O)C(O)=O)O)C1=CC=CC=C1 MNIPVWXWSPXERA-IDNZQHFXSA-N 0.000 description 1
- QOLHWXNSCZGWHK-BWBORTOCSA-N (6r,7r)-1-[(4s,5r)-4-acetyloxy-5-methyl-3-methylidene-6-phenylhexyl]-4,7-dihydroxy-6-(11-phenoxyundecylcarbamoyloxy)-2,8-dioxabicyclo[3.2.1]octane-3,4,5-tricarboxylic acid Chemical compound C([C@@H](C)[C@H](OC(C)=O)C(=C)CCC12[C@@H]([C@@H](OC(=O)NCCCCCCCCCCCOC=3C=CC=CC=3)C(O1)(C(O)=O)C(O)(C(O2)C(O)=O)C(O)=O)O)C1=CC=CC=C1 QOLHWXNSCZGWHK-BWBORTOCSA-N 0.000 description 1
- IGVKWAAPMVVTFX-BUHFOSPRSA-N (e)-octadec-5-en-7,9-diynoic acid Chemical compound CCCCCCCCC#CC#C\C=C\CCCC(O)=O IGVKWAAPMVVTFX-BUHFOSPRSA-N 0.000 description 1
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 1
- WACNXHCZHTVBJM-UHFFFAOYSA-N 1,2,3,4,5-pentafluorobenzene Chemical compound FC1=CC(F)=C(F)C(F)=C1F WACNXHCZHTVBJM-UHFFFAOYSA-N 0.000 description 1
- UGUHFDPGDQDVGX-UHFFFAOYSA-N 1,2,3-thiadiazole Chemical compound C1=CSN=N1 UGUHFDPGDQDVGX-UHFFFAOYSA-N 0.000 description 1
- JYEUMXHLPRZUAT-UHFFFAOYSA-N 1,2,3-triazine Chemical compound C1=CN=NN=C1 JYEUMXHLPRZUAT-UHFFFAOYSA-N 0.000 description 1
- HTJMXYRLEDBSLT-UHFFFAOYSA-N 1,2,4,5-tetrazine Chemical compound C1=NN=CN=N1 HTJMXYRLEDBSLT-UHFFFAOYSA-N 0.000 description 1
- FYADHXFMURLYQI-UHFFFAOYSA-N 1,2,4-triazine Chemical compound C1=CN=NC=N1 FYADHXFMURLYQI-UHFFFAOYSA-N 0.000 description 1
- UDGKZGLPXCRRAM-UHFFFAOYSA-N 1,2,5-thiadiazole Chemical compound C=1C=NSN=1 UDGKZGLPXCRRAM-UHFFFAOYSA-N 0.000 description 1
- FKASFBLJDCHBNZ-UHFFFAOYSA-N 1,3,4-oxadiazole Chemical compound C1=NN=CO1 FKASFBLJDCHBNZ-UHFFFAOYSA-N 0.000 description 1
- MBIZXFATKUQOOA-UHFFFAOYSA-N 1,3,4-thiadiazole Chemical compound C1=NN=CS1 MBIZXFATKUQOOA-UHFFFAOYSA-N 0.000 description 1
- JIHQDMXYYFUGFV-UHFFFAOYSA-N 1,3,5-triazine Chemical compound C1=NC=NC=N1 JIHQDMXYYFUGFV-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- WZZBNLYBHUDSHF-DHLKQENFSA-N 1-[(3s,4s)-4-[8-(2-chloro-4-pyrimidin-2-yloxyphenyl)-7-fluoro-2-methylimidazo[4,5-c]quinolin-1-yl]-3-fluoropiperidin-1-yl]-2-hydroxyethanone Chemical compound CC1=NC2=CN=C3C=C(F)C(C=4C(=CC(OC=5N=CC=CN=5)=CC=4)Cl)=CC3=C2N1[C@H]1CCN(C(=O)CO)C[C@@H]1F WZZBNLYBHUDSHF-DHLKQENFSA-N 0.000 description 1
- ONBQEOIKXPHGMB-VBSBHUPXSA-N 1-[2-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)propan-1-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 ONBQEOIKXPHGMB-VBSBHUPXSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- QWENRTYMTSOGBR-UHFFFAOYSA-N 1H-1,2,3-Triazole Chemical compound C=1C=NNN=1 QWENRTYMTSOGBR-UHFFFAOYSA-N 0.000 description 1
- NRKYWOKHZRQRJR-UHFFFAOYSA-N 2,2,2-trifluoroacetamide Chemical compound NC(=O)C(F)(F)F NRKYWOKHZRQRJR-UHFFFAOYSA-N 0.000 description 1
- UOJCTEGNHXRPKO-UHFFFAOYSA-N 2,3,4,5,6-pentafluorobenzenesulfonyl chloride Chemical compound FC1=C(F)C(F)=C(S(Cl)(=O)=O)C(F)=C1F UOJCTEGNHXRPKO-UHFFFAOYSA-N 0.000 description 1
- FQMZXMVHHKXGTM-UHFFFAOYSA-N 2-(1-adamantyl)-n-[2-[2-(2-hydroxyethylamino)ethylamino]quinolin-5-yl]acetamide Chemical compound C1C(C2)CC(C3)CC2CC13CC(=O)NC1=CC=CC2=NC(NCCNCCO)=CC=C21 FQMZXMVHHKXGTM-UHFFFAOYSA-N 0.000 description 1
- KKKJYDOHVKIIQP-UHFFFAOYSA-N 2-[4-(3-chlorobenzoyl)phenoxy]-N-pyridin-3-ylacetamide Chemical compound ClC=1C=C(C(=O)C2=CC=C(OCC(=O)NC=3C=NC=CC=3)C=C2)C=CC=1 KKKJYDOHVKIIQP-UHFFFAOYSA-N 0.000 description 1
- PYRKKGOKRMZEIT-UHFFFAOYSA-N 2-[6-(2-cyclopropylethoxy)-9-(2-hydroxy-2-methylpropyl)-1h-phenanthro[9,10-d]imidazol-2-yl]-5-fluorobenzene-1,3-dicarbonitrile Chemical compound C1=C2C3=CC(CC(C)(O)C)=CC=C3C=3NC(C=4C(=CC(F)=CC=4C#N)C#N)=NC=3C2=CC=C1OCCC1CC1 PYRKKGOKRMZEIT-UHFFFAOYSA-N 0.000 description 1
- LHASZEBEQGPCFM-CJFMBICVSA-N 2-amino-4-[(1r)-1-[[(6r)-6-[(5-chloro-2-methoxyphenyl)methyl]-7-oxo-3-(phenoxyamino)-5,6-dihydro-2h-1,4-diazepine-1-carbonyl]amino]propyl]benzoic acid Chemical compound C([C@@H]1CNC(CN(C1=O)C(=O)N[C@H](CC)C=1C=C(N)C(C(O)=O)=CC=1)=NOC=1C=CC=CC=1)C1=CC(Cl)=CC=C1OC LHASZEBEQGPCFM-CJFMBICVSA-N 0.000 description 1
- YSUIQYOGTINQIN-UZFYAQMZSA-N 2-amino-9-[(1S,6R,8R,9S,10R,15R,17R,18R)-8-(6-aminopurin-9-yl)-9,18-difluoro-3,12-dihydroxy-3,12-bis(sulfanylidene)-2,4,7,11,13,16-hexaoxa-3lambda5,12lambda5-diphosphatricyclo[13.2.1.06,10]octadecan-17-yl]-1H-purin-6-one Chemical compound NC1=NC2=C(N=CN2[C@@H]2O[C@@H]3COP(S)(=O)O[C@@H]4[C@@H](COP(S)(=O)O[C@@H]2[C@@H]3F)O[C@H]([C@H]4F)N2C=NC3=C2N=CN=C3N)C(=O)N1 YSUIQYOGTINQIN-UZFYAQMZSA-N 0.000 description 1
- QLVGHFBUSGYCCG-UHFFFAOYSA-N 2-amino-n-(1-cyano-2-phenylethyl)acetamide Chemical compound NCC(=O)NC(C#N)CC1=CC=CC=C1 QLVGHFBUSGYCCG-UHFFFAOYSA-N 0.000 description 1
- YLLRPQWLASQXSI-UHFFFAOYSA-N 3-(4b,8a,9,9a-tetrahydro-4aH-pyrido[2,3-b]indol-4-ylamino)phenol Chemical compound Oc1cccc(NC2=CC=NC3NC4C=CC=CC4C23)c1 YLLRPQWLASQXSI-UHFFFAOYSA-N 0.000 description 1
- MWDVCHRYCKXEBY-LBPRGKRZSA-N 3-chloro-n-[2-oxo-2-[[(1s)-1-phenylethyl]amino]ethyl]benzamide Chemical compound N([C@@H](C)C=1C=CC=CC=1)C(=O)CNC(=O)C1=CC=CC(Cl)=C1 MWDVCHRYCKXEBY-LBPRGKRZSA-N 0.000 description 1
- IGPFOKFDBICQMC-UHFFFAOYSA-N 3-phenylmethoxyaniline Chemical compound NC1=CC=CC(OCC=2C=CC=CC=2)=C1 IGPFOKFDBICQMC-UHFFFAOYSA-N 0.000 description 1
- RIYGJSDIJWHXJS-UHFFFAOYSA-N 4-[4-(9-hydroxynonoxy)phenyl]benzonitrile Chemical compound C1=CC(OCCCCCCCCCO)=CC=C1C1=CC=C(C#N)C=C1 RIYGJSDIJWHXJS-UHFFFAOYSA-N 0.000 description 1
- ZCQTYNBTSOIGRA-UHFFFAOYSA-N 4-[[2-(benzenesulfonamido)acetyl]amino]-2-hydroxybenzoic acid Chemical class C1=C(O)C(C(=O)O)=CC=C1NC(=O)CNS(=O)(=O)C1=CC=CC=C1 ZCQTYNBTSOIGRA-UHFFFAOYSA-N 0.000 description 1
- YBAZINRZQSAIAY-UHFFFAOYSA-N 4-aminobenzonitrile Chemical compound NC1=CC=C(C#N)C=C1 YBAZINRZQSAIAY-UHFFFAOYSA-N 0.000 description 1
- WUBBRNOQWQTFEX-UHFFFAOYSA-N 4-aminosalicylic acid Chemical compound NC1=CC=C(C(O)=O)C(O)=C1 WUBBRNOQWQTFEX-UHFFFAOYSA-N 0.000 description 1
- PXACTUVBBMDKRW-UHFFFAOYSA-M 4-bromobenzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=C(Br)C=C1 PXACTUVBBMDKRW-UHFFFAOYSA-M 0.000 description 1
- FIIDVVUUWRJXLF-UHFFFAOYSA-N 4-phenylmethoxyaniline Chemical compound C1=CC(N)=CC=C1OCC1=CC=CC=C1 FIIDVVUUWRJXLF-UHFFFAOYSA-N 0.000 description 1
- NSPMIYGKQJPBQR-UHFFFAOYSA-N 4H-1,2,4-triazole Chemical compound C=1N=CNN=1 NSPMIYGKQJPBQR-UHFFFAOYSA-N 0.000 description 1
- NJYVEMPWNAYQQN-UHFFFAOYSA-N 5-carboxyfluorescein Chemical compound C12=CC=C(O)C=C2OC2=CC(O)=CC=C2C21OC(=O)C1=CC(C(=O)O)=CC=C21 NJYVEMPWNAYQQN-UHFFFAOYSA-N 0.000 description 1
- LDIOUQIXNSSOGU-UHFFFAOYSA-N 8-(3-pentylamino)-2-methyl-3-(2-chloro-4-methoxyphenyl)-6,7-dihydro-5h-cyclopenta[d]pyrazolo[1,5-a]pyrimidine Chemical compound CC1=NN2C(NC(CC)CC)=C3CCCC3=NC2=C1C1=CC=C(OC)C=C1Cl LDIOUQIXNSSOGU-UHFFFAOYSA-N 0.000 description 1
- 241000251468 Actinopterygii Species 0.000 description 1
- 206010048998 Acute phase reaction Diseases 0.000 description 1
- 108010063104 Apoptosis Regulatory Proteins Proteins 0.000 description 1
- 102000010565 Apoptosis Regulatory Proteins Human genes 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 206010003571 Astrocytoma Diseases 0.000 description 1
- 208000023275 Autoimmune disease Diseases 0.000 description 1
- 102100021663 Baculoviral IAP repeat-containing protein 5 Human genes 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- KCBAMQOKOLXLOX-BSZYMOERSA-N CC1=C(SC=N1)C2=CC=C(C=C2)[C@H](C)NC(=O)[C@@H]3C[C@H](CN3C(=O)[C@H](C(C)(C)C)NC(=O)CCCCCCCCCCNCCCONC(=O)C4=C(C(=C(C=C4)F)F)NC5=C(C=C(C=C5)I)F)O Chemical compound CC1=C(SC=N1)C2=CC=C(C=C2)[C@H](C)NC(=O)[C@@H]3C[C@H](CN3C(=O)[C@H](C(C)(C)C)NC(=O)CCCCCCCCCCNCCCONC(=O)C4=C(C(=C(C=C4)F)F)NC5=C(C=C(C=C5)I)F)O KCBAMQOKOLXLOX-BSZYMOERSA-N 0.000 description 1
- AFTFCQCZYDYQDG-UHFFFAOYSA-N CCOP(Cc(cc1)ccc1N(Cc1ccc(C2CCCCC2)cc1)C(CN(C)S(c(c(F)c(c(F)c1F)F)c1F)(=O)=O)=O)(O)=O Chemical compound CCOP(Cc(cc1)ccc1N(Cc1ccc(C2CCCCC2)cc1)C(CN(C)S(c(c(F)c(c(F)c1F)F)c1F)(=O)=O)=O)(O)=O AFTFCQCZYDYQDG-UHFFFAOYSA-N 0.000 description 1
- KPFXQMIKXBBGCB-UHFFFAOYSA-N CN(CC(N(Cc1ccc(C2CCCCC2)cc1)c(cc1)ccc1C(OC)=O)=O)S(c(c(F)c(c(F)c1F)F)c1F)(=O)=O Chemical compound CN(CC(N(Cc1ccc(C2CCCCC2)cc1)c(cc1)ccc1C(OC)=O)=O)S(c(c(F)c(c(F)c1F)F)c1F)(=O)=O KPFXQMIKXBBGCB-UHFFFAOYSA-N 0.000 description 1
- BSWRNVQCOPIFEH-UHFFFAOYSA-N CN(CC(N(Cc1ccc(C2CCCCC2)cc1)c1ccc(CP(O)(O)=O)cc1)=O)S(c(c(F)c(c(F)c1F)F)c1F)(=O)=O Chemical compound CN(CC(N(Cc1ccc(C2CCCCC2)cc1)c1ccc(CP(O)(O)=O)cc1)=O)S(c(c(F)c(c(F)c1F)F)c1F)(=O)=O BSWRNVQCOPIFEH-UHFFFAOYSA-N 0.000 description 1
- SEUPUBCBAVAKEE-UHFFFAOYSA-N CN(CC(O)=O)S(c(c(F)c(c(F)c1F)F)c1F)(=O)=O Chemical compound CN(CC(O)=O)S(c(c(F)c(c(F)c1F)F)c1F)(=O)=O SEUPUBCBAVAKEE-UHFFFAOYSA-N 0.000 description 1
- OKTJSMMVPCPJKN-NJFSPNSNSA-N Carbon-14 Chemical compound [14C] OKTJSMMVPCPJKN-NJFSPNSNSA-N 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- JWPVZOYUZPKCCV-UHFFFAOYSA-N Cc(cc1)ccc1S(N(C)CC(OC)=O)(=O)=O Chemical compound Cc(cc1)ccc1S(N(C)CC(OC)=O)(=O)=O JWPVZOYUZPKCCV-UHFFFAOYSA-N 0.000 description 1
- 238000003591 CyQUANT Cell Proliferation Assay Kit Methods 0.000 description 1
- 230000004568 DNA-binding Effects 0.000 description 1
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 1
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 1
- SNRUBQQJIBEYMU-UHFFFAOYSA-N Dodecane Natural products CCCCCCCCCCCC SNRUBQQJIBEYMU-UHFFFAOYSA-N 0.000 description 1
- 206010059866 Drug resistance Diseases 0.000 description 1
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- 241000283073 Equus caballus Species 0.000 description 1
- RRSNDVCODIMOFX-MPKOGUQCSA-N Fc1c(Cl)cccc1[C@H]1[C@@H](NC2(CCCCC2)[C@@]11C(=O)Nc2cc(Cl)ccc12)C(=O)Nc1ccc(cc1)C(=O)NCCCCCc1cccc2C(=O)N(Cc12)C1CCC(=O)NC1=O Chemical compound Fc1c(Cl)cccc1[C@H]1[C@@H](NC2(CCCCC2)[C@@]11C(=O)Nc2cc(Cl)ccc12)C(=O)Nc1ccc(cc1)C(=O)NCCCCCc1cccc2C(=O)N(Cc12)C1CCC(=O)NC1=O RRSNDVCODIMOFX-MPKOGUQCSA-N 0.000 description 1
- 102000003974 Fibroblast growth factor 2 Human genes 0.000 description 1
- 108090000379 Fibroblast growth factor 2 Proteins 0.000 description 1
- GYHNNYVSQQEPJS-UHFFFAOYSA-N Gallium Chemical compound [Ga] GYHNNYVSQQEPJS-UHFFFAOYSA-N 0.000 description 1
- 206010018338 Glioma Diseases 0.000 description 1
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Polymers OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 1
- 229920002971 Heparan sulfate Polymers 0.000 description 1
- 101001056180 Homo sapiens Induced myeloid leukemia cell differentiation protein Mcl-1 Proteins 0.000 description 1
- 101000932478 Homo sapiens Receptor-type tyrosine-protein kinase FLT3 Proteins 0.000 description 1
- 101000826373 Homo sapiens Signal transducer and activator of transcription 3 Proteins 0.000 description 1
- 101000997832 Homo sapiens Tyrosine-protein kinase JAK2 Proteins 0.000 description 1
- 108010058683 Immobilized Proteins Proteins 0.000 description 1
- 102100026539 Induced myeloid leukemia cell differentiation protein Mcl-1 Human genes 0.000 description 1
- 102000042838 JAK family Human genes 0.000 description 1
- 108091082332 JAK family Proteins 0.000 description 1
- 108010024121 Janus Kinases Proteins 0.000 description 1
- 102000015617 Janus Kinases Human genes 0.000 description 1
- 238000003749 KINOMEscan Methods 0.000 description 1
- 241000270322 Lepidosauria Species 0.000 description 1
- 206010058467 Lung neoplasm malignant Diseases 0.000 description 1
- 206010025323 Lymphomas Diseases 0.000 description 1
- 102000019149 MAP kinase activity proteins Human genes 0.000 description 1
- 108040008097 MAP kinase activity proteins Proteins 0.000 description 1
- 102000043136 MAP kinase family Human genes 0.000 description 1
- 108091054455 MAP kinase family Proteins 0.000 description 1
- 238000000719 MTS assay Methods 0.000 description 1
- 231100000070 MTS assay Toxicity 0.000 description 1
- 101150039798 MYC gene Proteins 0.000 description 1
- 206010064912 Malignant transformation Diseases 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 238000006751 Mitsunobu reaction Methods 0.000 description 1
- 241000713333 Mouse mammary tumor virus Species 0.000 description 1
- 241001529936 Murinae Species 0.000 description 1
- TZYWCYJVHRLUCT-VABKMULXSA-N N-benzyloxycarbonyl-L-leucyl-L-leucyl-L-leucinal Chemical compound CC(C)C[C@@H](C=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(C)C)NC(=O)OCC1=CC=CC=C1 TZYWCYJVHRLUCT-VABKMULXSA-N 0.000 description 1
- 150000001204 N-oxides Chemical class 0.000 description 1
- OPFJDXRVMFKJJO-ZHHKINOHSA-N N-{[3-(2-benzamido-4-methyl-1,3-thiazol-5-yl)-pyrazol-5-yl]carbonyl}-G-dR-G-dD-dD-dD-NH2 Chemical compound S1C(C=2NN=C(C=2)C(=O)NCC(=O)N[C@H](CCCN=C(N)N)C(=O)NCC(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(=O)N[C@H](CC(O)=O)C(N)=O)=C(C)N=C1NC(=O)C1=CC=CC=C1 OPFJDXRVMFKJJO-ZHHKINOHSA-N 0.000 description 1
- 229910004749 OS(O)2 Inorganic materials 0.000 description 1
- 108091034117 Oligonucleotide Proteins 0.000 description 1
- 235000019502 Orange oil Nutrition 0.000 description 1
- 206010061535 Ovarian neoplasm Diseases 0.000 description 1
- 102000003992 Peroxidases Human genes 0.000 description 1
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 description 1
- 102000045595 Phosphoprotein Phosphatases Human genes 0.000 description 1
- 108700019535 Phosphoprotein Phosphatases Proteins 0.000 description 1
- 101100365603 Phytophthora infestans (strain T30-4) SFI2 gene Proteins 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 229920002732 Polyanhydride Polymers 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 229920000954 Polyglycolide Polymers 0.000 description 1
- 229920001710 Polyorthoester Polymers 0.000 description 1
- 239000004743 Polypropylene Substances 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- 108091008611 Protein Kinase B Proteins 0.000 description 1
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 102100020718 Receptor-type tyrosine-protein kinase FLT3 Human genes 0.000 description 1
- 208000007660 Residual Neoplasm Diseases 0.000 description 1
- 108091027981 Response element Proteins 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 102000014400 SH2 domains Human genes 0.000 description 1
- 108050003452 SH2 domains Proteins 0.000 description 1
- 102000000395 SH3 domains Human genes 0.000 description 1
- 108050008861 SH3 domains Proteins 0.000 description 1
- 229940125907 SJ995973 Drugs 0.000 description 1
- 229910006124 SOCl2 Inorganic materials 0.000 description 1
- 108010081691 STAT2 Transcription Factor Proteins 0.000 description 1
- 102000004265 STAT2 Transcription Factor Human genes 0.000 description 1
- 101150099493 STAT3 gene Proteins 0.000 description 1
- 108010019992 STAT4 Transcription Factor Proteins 0.000 description 1
- 102000005886 STAT4 Transcription Factor Human genes 0.000 description 1
- 108010011005 STAT6 Transcription Factor Proteins 0.000 description 1
- SKZKKFZAGNVIMN-UHFFFAOYSA-N Salicilamide Chemical compound NC(=O)C1=CC=CC=C1O SKZKKFZAGNVIMN-UHFFFAOYSA-N 0.000 description 1
- BUGBHKTXTAQXES-UHFFFAOYSA-N Selenium Chemical compound [Se] BUGBHKTXTAQXES-UHFFFAOYSA-N 0.000 description 1
- 102100023980 Signal transducer and activator of transcription 6 Human genes 0.000 description 1
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 108010090804 Streptavidin Proteins 0.000 description 1
- 241000282887 Suidae Species 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 1
- 108010002687 Survivin Proteins 0.000 description 1
- 241000282898 Sus scrofa Species 0.000 description 1
- 241000255588 Tephritidae Species 0.000 description 1
- DHXVGJBLRPWPCS-UHFFFAOYSA-N Tetrahydropyran Chemical compound C1CCOCC1 DHXVGJBLRPWPCS-UHFFFAOYSA-N 0.000 description 1
- DPOPAJRDYZGTIR-UHFFFAOYSA-N Tetrazine Chemical compound C1=CN=NN=N1 DPOPAJRDYZGTIR-UHFFFAOYSA-N 0.000 description 1
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- 102100033444 Tyrosine-protein kinase JAK2 Human genes 0.000 description 1
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 1
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 description 1
- 241000251539 Vertebrata <Metazoa> Species 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- 101100459258 Xenopus laevis myc-a gene Proteins 0.000 description 1
- LJOOWESTVASNOG-UFJKPHDISA-N [(1s,3r,4ar,7s,8s,8as)-3-hydroxy-8-[2-[(4r)-4-hydroxy-6-oxooxan-2-yl]ethyl]-7-methyl-1,2,3,4,4a,7,8,8a-octahydronaphthalen-1-yl] (2s)-2-methylbutanoate Chemical compound C([C@H]1[C@@H](C)C=C[C@H]2C[C@@H](O)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)CC1C[C@@H](O)CC(=O)O1 LJOOWESTVASNOG-UFJKPHDISA-N 0.000 description 1
- LNUFLCYMSVYYNW-ZPJMAFJPSA-N [(2r,3r,4s,5r,6r)-2-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[(2r,3r,4s,5r,6r)-6-[[(3s,5s,8r,9s,10s,13r,14s,17r)-10,13-dimethyl-17-[(2r)-6-methylheptan-2-yl]-2,3,4,5,6,7,8,9,11,12,14,15,16,17-tetradecahydro-1h-cyclopenta[a]phenanthren-3-yl]oxy]-4,5-disulfo Chemical compound O([C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1[C@@H](COS(O)(=O)=O)O[C@H]([C@@H]([C@H]1OS(O)(=O)=O)OS(O)(=O)=O)O[C@@H]1C[C@@H]2CC[C@H]3[C@@H]4CC[C@@H]([C@]4(CC[C@@H]3[C@@]2(C)CC1)C)[C@H](C)CCCC(C)C)[C@H]1O[C@H](COS(O)(=O)=O)[C@@H](OS(O)(=O)=O)[C@H](OS(O)(=O)=O)[C@H]1OS(O)(=O)=O LNUFLCYMSVYYNW-ZPJMAFJPSA-N 0.000 description 1
- PSLUFJFHTBIXMW-WYEYVKMPSA-N [(3r,4ar,5s,6s,6as,10s,10ar,10bs)-3-ethenyl-10,10b-dihydroxy-3,4a,7,7,10a-pentamethyl-1-oxo-6-(2-pyridin-2-ylethylcarbamoyloxy)-5,6,6a,8,9,10-hexahydro-2h-benzo[f]chromen-5-yl] acetate Chemical compound O([C@@H]1[C@@H]([C@]2(O[C@](C)(CC(=O)[C@]2(O)[C@@]2(C)[C@@H](O)CCC(C)(C)[C@@H]21)C=C)C)OC(=O)C)C(=O)NCCC1=CC=CC=N1 PSLUFJFHTBIXMW-WYEYVKMPSA-N 0.000 description 1
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 1
- SMNRFWMNPDABKZ-WVALLCKVSA-N [[(2R,3S,4R,5S)-5-(2,6-dioxo-3H-pyridin-3-yl)-3,4-dihydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl] [[[(2R,3S,4S,5R,6R)-4-fluoro-3,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-hydroxyphosphoryl]oxy-hydroxyphosphoryl] hydrogen phosphate Chemical compound OC[C@H]1O[C@H](OP(O)(=O)OP(O)(=O)OP(O)(=O)OP(O)(=O)OC[C@H]2O[C@H]([C@H](O)[C@@H]2O)C2C=CC(=O)NC2=O)[C@H](O)[C@@H](F)[C@@H]1O SMNRFWMNPDABKZ-WVALLCKVSA-N 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- 230000021736 acetylation Effects 0.000 description 1
- 238000006640 acetylation reaction Methods 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 229910052768 actinide Inorganic materials 0.000 description 1
- 230000004658 acute-phase response Effects 0.000 description 1
- 125000004423 acyloxy group Chemical group 0.000 description 1
- 230000006978 adaptation Effects 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 230000032683 aging Effects 0.000 description 1
- 239000000556 agonist Substances 0.000 description 1
- 238000003349 alamar blue assay Methods 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 150000001340 alkali metals Chemical class 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- 125000004183 alkoxy alkyl group Chemical group 0.000 description 1
- 125000004644 alkyl sulfinyl group Chemical group 0.000 description 1
- 125000004390 alkyl sulfonyl group Chemical group 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 239000012491 analyte Substances 0.000 description 1
- 238000013103 analytical ultracentrifugation Methods 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- 230000001093 anti-cancer Effects 0.000 description 1
- 229940124650 anti-cancer therapies Drugs 0.000 description 1
- 230000003466 anti-cipated effect Effects 0.000 description 1
- 238000011319 anticancer therapy Methods 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 239000002246 antineoplastic agent Substances 0.000 description 1
- 230000001640 apoptogenic effect Effects 0.000 description 1
- 239000008365 aqueous carrier Substances 0.000 description 1
- 239000008135 aqueous vehicle Substances 0.000 description 1
- 125000001204 arachidyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229910052785 arsenic Inorganic materials 0.000 description 1
- RQNWIZPPADIBDY-UHFFFAOYSA-N arsenic atom Chemical compound [As] RQNWIZPPADIBDY-UHFFFAOYSA-N 0.000 description 1
- XRWSZZJLZRKHHD-WVWIJVSJSA-N asunaprevir Chemical compound O=C([C@@H]1C[C@H](CN1C(=O)[C@@H](NC(=O)OC(C)(C)C)C(C)(C)C)OC1=NC=C(C2=CC=C(Cl)C=C21)OC)N[C@]1(C(=O)NS(=O)(=O)C2CC2)C[C@H]1C=C XRWSZZJLZRKHHD-WVWIJVSJSA-N 0.000 description 1
- METKIMKYRPQLGS-UHFFFAOYSA-N atenolol Chemical compound CC(C)NCC(O)COC1=CC=C(CC(N)=O)C=C1 METKIMKYRPQLGS-UHFFFAOYSA-N 0.000 description 1
- HONIICLYMWZJFZ-UHFFFAOYSA-N azetidine Chemical compound C1CNC1 HONIICLYMWZJFZ-UHFFFAOYSA-N 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 230000006399 behavior Effects 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- WXBLLCUINBKULX-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1.OC(=O)C1=CC=CC=C1 WXBLLCUINBKULX-UHFFFAOYSA-N 0.000 description 1
- MFFQVSHNJWZZAK-UHFFFAOYSA-N benzyl 4-[(4-cyclohexylphenyl)methylamino]-2-fluorobenzoate Chemical compound C=1C=C(C(=O)OCC=2C=CC=CC=2)C(F)=CC=1NCC(C=C1)=CC=C1C1CCCCC1 MFFQVSHNJWZZAK-UHFFFAOYSA-N 0.000 description 1
- JYIULPVXCRYPIM-UHFFFAOYSA-N benzyl 4-[(4-cyclohexylphenyl)methylamino]benzoate Chemical compound C=1C=C(NCC=2C=CC(=CC=2)C2CCCCC2)C=CC=1C(=O)OCC1=CC=CC=C1 JYIULPVXCRYPIM-UHFFFAOYSA-N 0.000 description 1
- NWIOEAMCVOFABQ-UHFFFAOYSA-N benzyl 4-amino-2-fluorobenzoate Chemical compound FC1=CC(N)=CC=C1C(=O)OCC1=CC=CC=C1 NWIOEAMCVOFABQ-UHFFFAOYSA-N 0.000 description 1
- DAOPOOMCXJPWPK-UHFFFAOYSA-N benzyl 4-aminobenzoate Chemical compound C1=CC(N)=CC=C1C(=O)OCC1=CC=CC=C1 DAOPOOMCXJPWPK-UHFFFAOYSA-N 0.000 description 1
- DBNUWCZNSWBDST-UHFFFAOYSA-N benzyl 5-[(4-cyclohexylphenyl)methylamino]-2-phenylmethoxybenzoate Chemical compound C=1C(NCC=2C=CC(=CC=2)C2CCCCC2)=CC=C(OCC=2C=CC=CC=2)C=1C(=O)OCC1=CC=CC=C1 DBNUWCZNSWBDST-UHFFFAOYSA-N 0.000 description 1
- XICZFKDDJLPGJR-UHFFFAOYSA-N benzyl 5-amino-2-phenylmethoxybenzoate Chemical compound C=1C=CC=CC=1COC(=O)C1=CC(N)=CC=C1OCC1=CC=CC=C1 XICZFKDDJLPGJR-UHFFFAOYSA-N 0.000 description 1
- 235000019445 benzyl alcohol Nutrition 0.000 description 1
- OGBVRMYSNSKIEF-UHFFFAOYSA-L benzyl-dioxido-oxo-$l^{5}-phosphane Chemical compound [O-]P([O-])(=O)CC1=CC=CC=C1 OGBVRMYSNSKIEF-UHFFFAOYSA-L 0.000 description 1
- 125000000649 benzylidene group Chemical group [H]C(=[*])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 102000000072 beta-Arrestins Human genes 0.000 description 1
- 108010080367 beta-Arrestins Proteins 0.000 description 1
- 239000012148 binding buffer Substances 0.000 description 1
- 229920002988 biodegradable polymer Polymers 0.000 description 1
- 239000004621 biodegradable polymer Substances 0.000 description 1
- 230000008512 biological response Effects 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 125000001246 bromo group Chemical group Br* 0.000 description 1
- NHYXMAKLBXBVEO-UHFFFAOYSA-N bromomethyl acetate Chemical compound CC(=O)OCBr NHYXMAKLBXBVEO-UHFFFAOYSA-N 0.000 description 1
- 239000007853 buffer solution Substances 0.000 description 1
- 239000008366 buffered solution Substances 0.000 description 1
- OSVHLUXLWQLPIY-KBAYOESNSA-N butyl 2-[(6aR,9R,10aR)-1-hydroxy-9-(hydroxymethyl)-6,6-dimethyl-6a,7,8,9,10,10a-hexahydrobenzo[c]chromen-3-yl]-2-methylpropanoate Chemical compound C(CCC)OC(C(C)(C)C1=CC(=C2[C@H]3[C@H](C(OC2=C1)(C)C)CC[C@H](C3)CO)O)=O OSVHLUXLWQLPIY-KBAYOESNSA-N 0.000 description 1
- 125000006309 butyl amino group Chemical group 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 230000004611 cancer cell death Effects 0.000 description 1
- 125000002837 carbocyclic group Chemical group 0.000 description 1
- 238000001460 carbon-13 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 125000002843 carboxylic acid group Chemical group 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 230000001364 causal effect Effects 0.000 description 1
- 238000000423 cell based assay Methods 0.000 description 1
- 230000004709 cell invasion Effects 0.000 description 1
- 239000008004 cell lysis buffer Substances 0.000 description 1
- 230000009087 cell motility Effects 0.000 description 1
- 239000006285 cell suspension Substances 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- 239000012829 chemotherapy agent Substances 0.000 description 1
- 238000004296 chiral HPLC Methods 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 229960004926 chlorobutanol Drugs 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 230000003021 clonogenic effect Effects 0.000 description 1
- 208000029742 colonic neoplasm Diseases 0.000 description 1
- 238000004737 colorimetric analysis Methods 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 230000002860 competitive effect Effects 0.000 description 1
- 229940126214 compound 3 Drugs 0.000 description 1
- 229940125936 compound 42 Drugs 0.000 description 1
- 229940125872 compound 4d Drugs 0.000 description 1
- 238000009833 condensation Methods 0.000 description 1
- 230000005494 condensation Effects 0.000 description 1
- 238000012790 confirmation Methods 0.000 description 1
- 238000012937 correction Methods 0.000 description 1
- 230000002596 correlated effect Effects 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 125000001047 cyclobutenyl group Chemical group C1(=CCC1)* 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000003678 cyclohexadienyl group Chemical group C1(=CC=CCC1)* 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- 125000000058 cyclopentadienyl group Chemical group C1(=CC=CC1)* 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000298 cyclopropenyl group Chemical group [H]C1=C([H])C1([H])* 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 108010057085 cytokine receptors Proteins 0.000 description 1
- 230000009089 cytolysis Effects 0.000 description 1
- 229940127089 cytotoxic agent Drugs 0.000 description 1
- DEZRYPDIMOWBDS-UHFFFAOYSA-N dcm dichloromethane Chemical compound ClCCl.ClCCl DEZRYPDIMOWBDS-UHFFFAOYSA-N 0.000 description 1
- 125000002704 decyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 238000012217 deletion Methods 0.000 description 1
- 230000037430 deletion Effects 0.000 description 1
- 229940009976 deoxycholate Drugs 0.000 description 1
- KXGVEGMKQFWNSR-LLQZFEROSA-N deoxycholic acid Chemical compound C([C@H]1CC2)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(O)=O)C)[C@@]2(C)[C@@H](O)C1 KXGVEGMKQFWNSR-LLQZFEROSA-N 0.000 description 1
- 238000001212 derivatisation Methods 0.000 description 1
- 229910052805 deuterium Inorganic materials 0.000 description 1
- 238000000502 dialysis Methods 0.000 description 1
- MHDVGSVTJDSBDK-UHFFFAOYSA-N dibenzyl ether Chemical compound C=1C=CC=CC=1COCC1=CC=CC=C1 MHDVGSVTJDSBDK-UHFFFAOYSA-N 0.000 description 1
- 125000004915 dibutylamino group Chemical group C(CCC)N(CCCC)* 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- 125000004177 diethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 125000001664 diethylamino group Chemical group [H]C([H])([H])C([H])([H])N(*)C([H])([H])C([H])([H])[H] 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- 150000004683 dihydrates Chemical class 0.000 description 1
- 239000000539 dimer Substances 0.000 description 1
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 1
- UXGNZZKBCMGWAZ-UHFFFAOYSA-N dimethylformamide dmf Chemical compound CN(C)C=O.CN(C)C=O UXGNZZKBCMGWAZ-UHFFFAOYSA-N 0.000 description 1
- 229910001873 dinitrogen Inorganic materials 0.000 description 1
- USIUVYZYUHIAEV-UHFFFAOYSA-N diphenyl ether Chemical compound C=1C=CC=CC=1OC1=CC=CC=C1 USIUVYZYUHIAEV-UHFFFAOYSA-N 0.000 description 1
- 125000004914 dipropylamino group Chemical group C(CC)N(CCC)* 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 238000006073 displacement reaction Methods 0.000 description 1
- UZZWBUYVTBPQIV-UHFFFAOYSA-N dme dimethoxyethane Chemical compound COCCOC.COCCOC UZZWBUYVTBPQIV-UHFFFAOYSA-N 0.000 description 1
- CETRZFQIITUQQL-UHFFFAOYSA-N dmso dimethylsulfoxide Chemical compound CS(C)=O.CS(C)=O CETRZFQIITUQQL-UHFFFAOYSA-N 0.000 description 1
- 125000003438 dodecyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 239000003596 drug target Substances 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 239000008157 edible vegetable oil Substances 0.000 description 1
- 238000002337 electrophoretic mobility shift assay Methods 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 239000012149 elution buffer Substances 0.000 description 1
- ZSWFCLXCOIISFI-UHFFFAOYSA-N endo-cyclopentadiene Natural products C1C=CC=C1 ZSWFCLXCOIISFI-UHFFFAOYSA-N 0.000 description 1
- 150000002085 enols Chemical group 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 238000006345 epimerization reaction Methods 0.000 description 1
- 238000010931 ester hydrolysis Methods 0.000 description 1
- LHWWETDBWVTKJO-UHFFFAOYSA-N et3n triethylamine Chemical compound CCN(CC)CC.CCN(CC)CC LHWWETDBWVTKJO-UHFFFAOYSA-N 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- YWKVMGDEOUPQGN-UHFFFAOYSA-N ethyl 2-[4-[2-(3-hydroxy-1-azabicyclo[2.2.2]octan-3-yl)ethynyl]phenyl]acetate Chemical compound C1=CC(CC(=O)OCC)=CC=C1C#CC1(O)C(CC2)CCN2C1 YWKVMGDEOUPQGN-UHFFFAOYSA-N 0.000 description 1
- OWZFULPEVHKEKS-UHFFFAOYSA-N ethyl 2-chloro-2-oxoacetate Chemical compound CCOC(=O)C(Cl)=O OWZFULPEVHKEKS-UHFFFAOYSA-N 0.000 description 1
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- 125000000031 ethylamino group Chemical group [H]C([H])([H])C([H])([H])N([H])[*] 0.000 description 1
- DEFVIWRASFVYLL-UHFFFAOYSA-N ethylene glycol bis(2-aminoethyl)tetraacetic acid Chemical compound OC(=O)CN(CC(O)=O)CCOCCOCCN(CC(O)=O)CC(O)=O DEFVIWRASFVYLL-UHFFFAOYSA-N 0.000 description 1
- SFNALCNOMXIBKG-UHFFFAOYSA-N ethylene glycol monododecyl ether Chemical compound CCCCCCCCCCCCOCCO SFNALCNOMXIBKG-UHFFFAOYSA-N 0.000 description 1
- OJCSPXHYDFONPU-UHFFFAOYSA-N etoac etoac Chemical compound CCOC(C)=O.CCOC(C)=O OJCSPXHYDFONPU-UHFFFAOYSA-N 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 230000001747 exhibiting effect Effects 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 238000003818 flash chromatography Methods 0.000 description 1
- GNBHRKFJIUUOQI-UHFFFAOYSA-N fluorescein Chemical compound O1C(=O)C2=CC=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 GNBHRKFJIUUOQI-UHFFFAOYSA-N 0.000 description 1
- 150000002222 fluorine compounds Chemical group 0.000 description 1
- 239000012634 fragment Substances 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- JKFAIQOWCVVSKC-UHFFFAOYSA-N furazan Chemical compound C=1C=NON=1 JKFAIQOWCVVSKC-UHFFFAOYSA-N 0.000 description 1
- 229910052733 gallium Inorganic materials 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 229910052732 germanium Inorganic materials 0.000 description 1
- GNPVGFCGXDBREM-UHFFFAOYSA-N germanium atom Chemical compound [Ge] GNPVGFCGXDBREM-UHFFFAOYSA-N 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 230000036433 growing body Effects 0.000 description 1
- 239000003102 growth factor Substances 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- JAXFJECJQZDFJS-XHEPKHHKSA-N gtpl8555 Chemical compound OC(=O)C[C@H](N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@@H]1C(=O)N[C@H](B1O[C@@]2(C)[C@H]3C[C@H](C3(C)C)C[C@H]2O1)CCC1=CC=C(F)C=C1 JAXFJECJQZDFJS-XHEPKHHKSA-N 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 230000002489 hematologic effect Effects 0.000 description 1
- 238000007490 hematoxylin and eosin (H&E) staining Methods 0.000 description 1
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001245 hexylamino group Chemical group [H]N([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 1
- 210000005260 human cell Anatomy 0.000 description 1
- BHEPBYXIRTUNPN-UHFFFAOYSA-N hydridophosphorus(.) (triplet) Chemical group [PH] BHEPBYXIRTUNPN-UHFFFAOYSA-N 0.000 description 1
- 238000007327 hydrogenolysis reaction Methods 0.000 description 1
- 230000007062 hydrolysis Effects 0.000 description 1
- 238000006460 hydrolysis reaction Methods 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 206010051040 hyper-IgE syndrome Diseases 0.000 description 1
- 238000011532 immunohistochemical staining Methods 0.000 description 1
- 239000007943 implant Substances 0.000 description 1
- 238000000099 in vitro assay Methods 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 208000015181 infectious disease Diseases 0.000 description 1
- 238000001361 intraarterial administration Methods 0.000 description 1
- 230000031146 intracellular signal transduction Effects 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- 125000002346 iodo group Chemical group I* 0.000 description 1
- PELJISAVHGXLAL-UHFFFAOYSA-N iodomethyl 2,2-dimethylpropanoate Chemical compound CC(C)(C)C(=O)OCI PELJISAVHGXLAL-UHFFFAOYSA-N 0.000 description 1
- 238000005342 ion exchange Methods 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 125000006316 iso-butyl amino group Chemical group [H]N(*)C([H])([H])C([H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000001972 isopentyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])C([H])([H])* 0.000 description 1
- QQVIHTHCMHWDBS-UHFFFAOYSA-L isophthalate(2-) Chemical compound [O-]C(=O)C1=CC=CC(C([O-])=O)=C1 QQVIHTHCMHWDBS-UHFFFAOYSA-L 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- ZLTPDFXIESTBQG-UHFFFAOYSA-N isothiazole Chemical compound C=1C=NSC=1 ZLTPDFXIESTBQG-UHFFFAOYSA-N 0.000 description 1
- 239000007951 isotonicity adjuster Substances 0.000 description 1
- CTAPFRYPJLPFDF-UHFFFAOYSA-N isoxazole Chemical compound C=1C=NOC=1 CTAPFRYPJLPFDF-UHFFFAOYSA-N 0.000 description 1
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 1
- 125000000468 ketone group Chemical group 0.000 description 1
- 208000017169 kidney disease Diseases 0.000 description 1
- 238000000021 kinase assay Methods 0.000 description 1
- 229940043355 kinase inhibitor Drugs 0.000 description 1
- 229910052747 lanthanoid Inorganic materials 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 231100000518 lethal Toxicity 0.000 description 1
- 231100000636 lethal dose Toxicity 0.000 description 1
- 230000001665 lethal effect Effects 0.000 description 1
- 125000002463 lignoceryl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 244000144972 livestock Species 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 201000005202 lung cancer Diseases 0.000 description 1
- 239000012139 lysis buffer Substances 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- 230000036212 malign transformation Effects 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- 210000001161 mammalian embryo Anatomy 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- 102000006240 membrane receptors Human genes 0.000 description 1
- 108020004084 membrane receptors Proteins 0.000 description 1
- COTNUBDHGSIOTA-UHFFFAOYSA-N meoh methanol Chemical compound OC.OC COTNUBDHGSIOTA-UHFFFAOYSA-N 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 229910021645 metal ion Inorganic materials 0.000 description 1
- 229910052752 metalloid Inorganic materials 0.000 description 1
- 150000002738 metalloids Chemical class 0.000 description 1
- CMERFRIDJKKBEQ-UHFFFAOYSA-N methyl 4-[(4-cyclohexylphenyl)methylamino]benzoate Chemical compound C1=CC(C(=O)OC)=CC=C1NCC1=CC=C(C2CCCCC2)C=C1 CMERFRIDJKKBEQ-UHFFFAOYSA-N 0.000 description 1
- LZXXNPOYQCLXRS-UHFFFAOYSA-N methyl 4-aminobenzoate Chemical compound COC(=O)C1=CC=C(N)C=C1 LZXXNPOYQCLXRS-UHFFFAOYSA-N 0.000 description 1
- 239000004530 micro-emulsion Substances 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 238000000386 microscopy Methods 0.000 description 1
- 238000013508 migration Methods 0.000 description 1
- 230000003278 mimic effect Effects 0.000 description 1
- 231100000324 minimal toxicity Toxicity 0.000 description 1
- 238000000329 molecular dynamics simulation Methods 0.000 description 1
- 239000002808 molecular sieve Substances 0.000 description 1
- 150000004682 monohydrates Chemical class 0.000 description 1
- 229910000402 monopotassium phosphate Inorganic materials 0.000 description 1
- 239000002324 mouth wash Substances 0.000 description 1
- 125000001421 myristyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- PEECTLLHENGOKU-UHFFFAOYSA-N n,n-dimethylpyridin-4-amine Chemical compound CN(C)C1=CC=NC=C1.CN(C)C1=CC=NC=C1 PEECTLLHENGOKU-UHFFFAOYSA-N 0.000 description 1
- PSBGROLQRNSAMY-UHFFFAOYSA-N n-(3-chlorophenyl)-4-methyl-3-(2-pyrazolo[1,5-a]pyrimidin-6-ylethynyl)benzamide Chemical compound C1=C(C#CC2=CN3N=CC=C3N=C2)C(C)=CC=C1C(=O)NC1=CC=CC(Cl)=C1 PSBGROLQRNSAMY-UHFFFAOYSA-N 0.000 description 1
- NPIUPPBNIDMZIM-UHFFFAOYSA-N n-[(4-cyclohexylphenyl)methyl]-4-(diethoxyphosphorylmethyl)aniline Chemical compound C1=CC(CP(=O)(OCC)OCC)=CC=C1NCC1=CC=C(C2CCCCC2)C=C1 NPIUPPBNIDMZIM-UHFFFAOYSA-N 0.000 description 1
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 1
- XZMHJYWMCRQSSI-UHFFFAOYSA-N n-[5-[2-(3-acetylanilino)-1,3-thiazol-4-yl]-4-methyl-1,3-thiazol-2-yl]benzamide Chemical compound CC(=O)C1=CC=CC(NC=2SC=C(N=2)C2=C(N=C(NC(=O)C=3C=CC=CC=3)S2)C)=C1 XZMHJYWMCRQSSI-UHFFFAOYSA-N 0.000 description 1
- LVCDXCQFSONNDO-UHFFFAOYSA-N n-benzylhydroxylamine Chemical class ONCC1=CC=CC=C1 LVCDXCQFSONNDO-UHFFFAOYSA-N 0.000 description 1
- HBEDNENASUYMPO-LJQANCHMSA-N n-hydroxy-4-[[(2r)-3-oxo-2-(thiophen-2-ylmethyl)-2,4-dihydroquinoxalin-1-yl]methyl]benzamide Chemical compound C1=CC(C(=O)NO)=CC=C1CN1C2=CC=CC=C2NC(=O)[C@H]1CC1=CC=CS1 HBEDNENASUYMPO-LJQANCHMSA-N 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- IOMMMLWIABWRKL-WUTDNEBXSA-N nazartinib Chemical compound C1N(C(=O)/C=C/CN(C)C)CCCC[C@H]1N1C2=C(Cl)C=CC=C2N=C1NC(=O)C1=CC=NC(C)=C1 IOMMMLWIABWRKL-WUTDNEBXSA-N 0.000 description 1
- 210000003739 neck Anatomy 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 210000001577 neostriatum Anatomy 0.000 description 1
- 210000001178 neural stem cell Anatomy 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 229910052759 nickel Inorganic materials 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- 229910052756 noble gas Inorganic materials 0.000 description 1
- 125000006574 non-aromatic ring group Chemical group 0.000 description 1
- 239000012457 nonaqueous media Substances 0.000 description 1
- 239000002687 nonaqueous vehicle Substances 0.000 description 1
- 230000009871 nonspecific binding Effects 0.000 description 1
- 231100000956 nontoxicity Toxicity 0.000 description 1
- 125000001400 nonyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000003518 norbornenyl group Chemical group C12(C=CC(CC1)C2)* 0.000 description 1
- 125000002868 norbornyl group Chemical group C12(CCC(CC1)C2)* 0.000 description 1
- XYEOALKITRFCJJ-UHFFFAOYSA-N o-benzylhydroxylamine Chemical compound NOCC1=CC=CC=C1 XYEOALKITRFCJJ-UHFFFAOYSA-N 0.000 description 1
- OIPZNTLJVJGRCI-UHFFFAOYSA-M octadecanoyloxyaluminum;dihydrate Chemical compound O.O.CCCCCCCCCCCCCCCCCC(=O)O[Al] OIPZNTLJVJGRCI-UHFFFAOYSA-M 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- PIDFDZJZLOTZTM-KHVQSSSXSA-N ombitasvir Chemical compound COC(=O)N[C@@H](C(C)C)C(=O)N1CCC[C@H]1C(=O)NC1=CC=C([C@H]2N([C@@H](CC2)C=2C=CC(NC(=O)[C@H]3N(CCC3)C(=O)[C@@H](NC(=O)OC)C(C)C)=CC=2)C=2C=CC(=CC=2)C(C)(C)C)C=C1 PIDFDZJZLOTZTM-KHVQSSSXSA-N 0.000 description 1
- 231100000590 oncogenic Toxicity 0.000 description 1
- 230000000771 oncological effect Effects 0.000 description 1
- 238000005457 optimization Methods 0.000 description 1
- 239000010502 orange oil Substances 0.000 description 1
- 210000000056 organ Anatomy 0.000 description 1
- 150000002895 organic esters Chemical class 0.000 description 1
- WCPAKWJPBJAGKN-UHFFFAOYSA-N oxadiazole Chemical compound C1=CON=N1 WCPAKWJPBJAGKN-UHFFFAOYSA-N 0.000 description 1
- 238000002638 palliative care Methods 0.000 description 1
- 125000000913 palmityl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 235000010603 pastilles Nutrition 0.000 description 1
- 125000004894 pentylamino group Chemical group C(CCCC)N* 0.000 description 1
- 230000010412 perfusion Effects 0.000 description 1
- 230000000737 periodic effect Effects 0.000 description 1
- 108040007629 peroxidase activity proteins Proteins 0.000 description 1
- 230000002085 persistent effect Effects 0.000 description 1
- 229940127557 pharmaceutical product Drugs 0.000 description 1
- 229960003742 phenol Drugs 0.000 description 1
- UEZVMMHDMIWARA-UHFFFAOYSA-M phosphonate Chemical compound [O-]P(=O)=O UEZVMMHDMIWARA-UHFFFAOYSA-M 0.000 description 1
- LFGREXWGYUGZLY-UHFFFAOYSA-N phosphoryl Chemical group [P]=O LFGREXWGYUGZLY-UHFFFAOYSA-N 0.000 description 1
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 230000001766 physiological effect Effects 0.000 description 1
- 230000010287 polarization Effects 0.000 description 1
- 229920001748 polybutylene Polymers 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229940068886 polyethylene glycol 300 Drugs 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 229920001155 polypropylene Polymers 0.000 description 1
- 229920001451 polypropylene glycol Polymers 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 238000002953 preparative HPLC Methods 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 230000002062 proliferating effect Effects 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- 125000006308 propyl amino group Chemical group 0.000 description 1
- 230000004952 protein activity Effects 0.000 description 1
- 238000002731 protein assay Methods 0.000 description 1
- 238000000751 protein extraction Methods 0.000 description 1
- 239000012460 protein solution Substances 0.000 description 1
- 230000004063 proteosomal degradation Effects 0.000 description 1
- 125000002577 pseudohalo group Chemical group 0.000 description 1
- PBMFSQRYOILNGV-UHFFFAOYSA-N pyridazine Chemical compound C1=CC=NN=C1 PBMFSQRYOILNGV-UHFFFAOYSA-N 0.000 description 1
- 230000006340 racemization Effects 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- 238000001959 radiotherapy Methods 0.000 description 1
- 239000011535 reaction buffer Substances 0.000 description 1
- 230000008707 rearrangement Effects 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 238000011084 recovery Methods 0.000 description 1
- 238000006268 reductive amination reaction Methods 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- RWWYLEGWBNMMLJ-YSOARWBDSA-N remdesivir Chemical compound NC1=NC=NN2C1=CC=C2[C@]1([C@@H]([C@@H]([C@H](O1)CO[P@](=O)(OC1=CC=CC=C1)N[C@H](C(=O)OCC(CC)CC)C)O)O)C#N RWWYLEGWBNMMLJ-YSOARWBDSA-N 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 239000012146 running buffer Substances 0.000 description 1
- 229930195734 saturated hydrocarbon Natural products 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 150000003335 secondary amines Chemical class 0.000 description 1
- 229910052711 selenium Inorganic materials 0.000 description 1
- 239000011669 selenium Substances 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 208000002491 severe combined immunodeficiency Diseases 0.000 description 1
- 238000007493 shaping process Methods 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 description 1
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 description 1
- 239000008247 solid mixture Substances 0.000 description 1
- 238000007614 solvation Methods 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 210000001324 spliceosome Anatomy 0.000 description 1
- 108010087686 src-Family Kinases Proteins 0.000 description 1
- 102000009076 src-Family Kinases Human genes 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 239000003206 sterilizing agent Substances 0.000 description 1
- 210000002536 stromal cell Anatomy 0.000 description 1
- 238000005556 structure-activity relationship Methods 0.000 description 1
- 125000005017 substituted alkenyl group Chemical group 0.000 description 1
- 125000003107 substituted aryl group Chemical group 0.000 description 1
- 229940124530 sulfonamide Drugs 0.000 description 1
- 150000003456 sulfonamides Chemical class 0.000 description 1
- 125000005420 sulfonamido group Chemical group S(=O)(=O)(N*)* 0.000 description 1
- 150000003457 sulfones Chemical class 0.000 description 1
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 1
- 150000003462 sulfoxides Chemical class 0.000 description 1
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical compound ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 1
- 239000001117 sulphuric acid Substances 0.000 description 1
- 235000011149 sulphuric acid Nutrition 0.000 description 1
- 239000013589 supplement Substances 0.000 description 1
- 230000008093 supporting effect Effects 0.000 description 1
- 230000003319 supportive effect Effects 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 238000010189 synthetic method Methods 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 238000012911 target assessment Methods 0.000 description 1
- 229910052714 tellurium Inorganic materials 0.000 description 1
- PORWMNRCUJJQNO-UHFFFAOYSA-N tellurium atom Chemical compound [Te] PORWMNRCUJJQNO-UHFFFAOYSA-N 0.000 description 1
- 150000004685 tetrahydrates Chemical class 0.000 description 1
- WROMPOXWARCANT-UHFFFAOYSA-N tfa trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.OC(=O)C(F)(F)F WROMPOXWARCANT-UHFFFAOYSA-N 0.000 description 1
- VLLMWSRANPNYQX-UHFFFAOYSA-N thiadiazole Chemical compound C1=CSN=N1.C1=CSN=N1 VLLMWSRANPNYQX-UHFFFAOYSA-N 0.000 description 1
- 125000004001 thioalkyl group Chemical group 0.000 description 1
- 229930192474 thiophene Natural products 0.000 description 1
- 210000001541 thymus gland Anatomy 0.000 description 1
- 229910052718 tin Inorganic materials 0.000 description 1
- GZNAASVAJNXPPW-UHFFFAOYSA-M tin(4+) chloride dihydrate Chemical compound O.O.[Cl-].[Sn+4] GZNAASVAJNXPPW-UHFFFAOYSA-M 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 238000003354 tissue distribution assay Methods 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 229910052723 transition metal Inorganic materials 0.000 description 1
- 150000003624 transition metals Chemical class 0.000 description 1
- 150000003852 triazoles Chemical class 0.000 description 1
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 1
- 150000004684 trihydrates Chemical class 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 1
- 238000000825 ultraviolet detection Methods 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 239000011534 wash buffer Substances 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/15—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of six-membered aromatic rings
- C07C311/16—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of six-membered aromatic rings having the nitrogen atom of at least one of the sulfonamide groups bound to hydrogen atoms or to an acyclic carbon atom
- C07C311/19—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of six-membered aromatic rings having the nitrogen atom of at least one of the sulfonamide groups bound to hydrogen atoms or to an acyclic carbon atom to an acyclic carbon atom of a hydrocarbon radical substituted by carboxyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/15—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of six-membered aromatic rings
- C07C311/21—Sulfonamides having sulfur atoms of sulfonamide groups bound to carbon atoms of six-membered aromatic rings having the nitrogen atom of at least one of the sulfonamide groups bound to a carbon atom of a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/36—Sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/66—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D233/84—Sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/28—Phosphorus compounds with one or more P—C bonds
- C07F9/38—Phosphonic acids [RP(=O)(OH)2]; Thiophosphonic acids ; [RP(=X1)(X2H)2(X1, X2 are each independently O, S or Se)]
- C07F9/3804—Phosphonic acids [RP(=O)(OH)2]; Thiophosphonic acids ; [RP(=X1)(X2H)2(X1, X2 are each independently O, S or Se)] not used, see subgroups
- C07F9/3882—Arylalkanephosphonic acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/18—Sulfonamides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/63—Compounds containing para-N-benzenesulfonyl-N-groups, e.g. sulfanilamide, p-nitrobenzenesulfonyl hydrazide
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/12—Systems containing only non-condensed rings with a six-membered ring
- C07C2601/14—The ring being saturated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D257/00—Heterocyclic compounds containing rings having four nitrogen atoms as the only ring hetero atoms
- C07D257/02—Heterocyclic compounds containing rings having four nitrogen atoms as the only ring hetero atoms not condensed with other rings
- C07D257/04—Five-membered rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/28—Phosphorus compounds with one or more P—C bonds
- C07F9/38—Phosphonic acids [RP(=O)(OH)2]; Thiophosphonic acids ; [RP(=X1)(X2H)2(X1, X2 are each independently O, S or Se)]
- C07F9/40—Esters thereof
Definitions
- the present invention relates to novel salicylic acid derivative compound, compositions containing same and methods inhibiting STAT3 activity or for treating cancer where STAT3/5 are involved, such as in brain, breast, colon, hematologic, lung, ovarian and prostate cancers using said compounds.
- STAT3 is persistently activated in over a dozen types of human cancers, including all the major carcinomas, including breast, brain, colon, pancreas, ovarian, and squamous cell carcinomas of head and neck (SCCHN) cancers, and melanomas as well as some hematologic tumors (Bowman T, et al (2000) Oncogene 19, 2474-88, and Darnell, J. E. (2005) Nat. Med. 1 1, 595-596).
- SCHN head and neck
- GBM Glioblastoma
- BTSCs brain tumour stem cells
- STAT proteins were originally discovered as latent cytoplasmic transcription factors that mediate cytokine and growth factor responses (Darnell, J. E., Jr. (1996) Recent Prog. Norm. Res. 51, 391-403; Darnell, J. E. (2005) Nat. Med. 1 1, 595-596). Seven members of the family, STAT1, STAT2, STAT3, STAT4, STAT5a and STAT5b, and STAT6, mediate several physiological effects including growth and differentiation, survival, development and inflammation. STATs are SH2 domain-containing proteins.
- STATs Upon ligand binding to cytokine or growth factor receptors, STATs become phosphorylated on critical Tyr residue (Tyr705 for STAT3) by growth factor receptors, cytoplasmic Janus kinases (Jaks) or Src family kinases. Two phosphorylated and activated STAT monomers dimerize through reciprocal pTyr-SH2 domain interactions, translocate to the nucleus, and bind to specific DNA-response elements of target genes, thereby inducing gene transcription (Darnell, J. E., Jr. (1996) Recent Prog. Norm. Res. 51 , 391-403; Darnell, J. E. (2005) Nat. Med. 1 1, 595-596).
- STAT3 protein is one of seven family members of the STAT family of transcription factor proteins.
- STAT3 is activated through phosphorylation of a tyrosine 705 (Y705) that initiates complexation of two phosphorylated STAT3 monomers (pSTAT3).
- pSTAT3 homo- dimers are mediated through reciprocal STAT3 Src Homology 2 (SH2) domain-pY705 STAT3 interactions.
- pSTAT3:pSTAT3 homodimers translocate to the nucleus and bind DNA, promoting STAT3 target gene transcription.
- Targeting STAT3 has been previously achieved with dominant negative constructs, oligonucleotides or, most commonly, phosphopeptidic agents that mimic the native pY705 containing binding sequence.
- STAT3 inhibitors were designed for treatment of cancers harboring hyperactivated STAT3 protein.
- Acid-based inhibitors have been identified in WO2012/018868 that potently and selectively block STAT3 dimerization and DNA-binding activity, namely, compound 450, also referred to as BP-1-102 (sometimes referred to as compound 1 herein).
- Compound 450 in WO2012018868 potently suppresses multiple oncogenic properties in diverse cultured cancer cells (breast, lung, pancreatic, prostate, lung), including: cell proliferation, anchorage-independent cell growth, migration, invasion and motility.
- STAT3 It is selective for STAT3, with over 10-fold less binding to 93% homologous STAT protein, STAT1. It showed little or no effect on phosphorylation of She, Src, Jak-1/2, Erkl/2 or Akt and had no effect on non-transformed cells (NIH 3T3 cells, STATS null mouse embryo fibroblasts, or mouse thymus stromal cells, nor does it affect transformed cells that do not harbor activated STAT3). Moreover, BP-1-102 exhibited striking anti-tumor effects in vivo in murine xenograft models of lung or breast cancer resulting in dramatic regression in tumor volumes. Western blots of residual tumors from treated mice showed repression in pSTAT3, cMyc, Cyclin Dl, Bcl-xL, Survivin, and VEGF in a dose-dependent manner.
- the STAT3 pathway is a key oncogenic driver in over a dozen types of human cancers, including all the major carcinomas, including breast, brain, colon, pancreas, ovarian, and squamous cell carcinomas of head and neck (SCCHN) cancers, and melanomas as well as some hematologic tumors (Bowman T, et al (2000) Oncogene 19, 2474-88, and Darnell, J. E. (2005) Nat. Med. 1 1 , 595-596) the direct inhibition of STAT3 would provide a molecularly targeted route for effectively managing these cancers and especially aggressive forms such as GBM.
- SCCHN head and neck
- STAT5 signaling like STAT3 signaling, is transiently activated in normal cells and is deactivated by a number of different cytosolic and nuclear regulators, including phosphatases, SOCS, PIAS, and proteasomal degradation. Like STAT3, STAT5 has gained notoriety for its aberrant role in human cancers and tumorigenesis, having been found to be constitutively activated in many cancers, including those of the breast, liver, prostate, blood, skin, head and neck. (Muller, J., et al. ChemBioChem 2008, 9, 723-727). In cancer cells, STAT5 is routinely constitutively phosphorylated which leads to the aberrant expression of STAT5 target genes resulting in malignant transformation.
- STAT5 has been identified as a key regulator in the development and progression of acute myelogenic (AML) and acute lymphoblastic leukemias (ALL; Gouilleux-Gruart, V., et al. Leukemia and Lymphoma 1997, 28, 83-88; Gouilleux-Gruart, V., et al. Blood 1996, 87, 1692-1697; Weber-Nordt, R. M., et al.
- AML acute myelogenic
- ALL acute lymphoblastic leukemias
- inhibitors of upstream STAT5 activators have been shown to exhibit promising anti-cancer properties (Pardanani, A., et al. Leukemia 201 1 , 25, 218-225; Quintas-Cardama, A., et al. Nature Reviews Drug Discovery 201 1 , 10, 127- 140).
- the invention in one aspect, relates to compounds useful as inhibitors of STAT3.
- the disclosed compounds and products of disclosed methods of making, or a pharmaceutically acceptable salt, hydrate, solvate, or polymorph thereof are modulators of STAT3 and/or STAT5 activity, methods of making same, pharmaceutical compositions comprising same, and methods of treating disorders associated with a STAT3 activity dysfunction using same.
- the present invention relates to compounds that bind to STAT3 protein and negatively modulate STAT3 activity.
- the present invention relates to compounds that bind to STAT5 protein and negatively modulate STAT5 activity.
- compositions comprising a therapeutically effective amount of a disclosed compound and a pharmaceutically acceptable earner.
- methods for the treatment of a disorder associated with STAT3/STAT5 activity dysfunction, preferably hyperactivity or over-expression, in a mammal comprising the step of administering to the mammal a therapeutically effective amount of a disclosed compound, or a pharmaceutically acceptable salt, hydrate, solvate, or polymorph thereof.
- Also disclosed are methods for inhibition of STAT3 and/or STAT5 activity in a mammal comprising the step of administering to the mammal a therapeutically effective amount of least one disclosed compound, or a pharmaceutically acceptable salt, hydrate, solvate, or polymorph thereof.
- Also disclosed are methods for inhibiting STAT3 and/or STAT5 activity in at least one cell comprising the step of contacting the at least one cell with an effective amount of least one disclosed compound, or a pharmaceutically acceptable salt, hydrate, solvate, or polymorph thereof.
- a 5 is selected from C 3-6 cycloalkyl, C 3-6 heterocycloalkyl, and aryl, and substituted with 0-3 groups selected from halo, hydroxyl, amino, nitro, cyano, C 1 -6 haloalkyl, Cj -6 polyhaloalkyl, C
- a 6 is selected from C 3- 6 cycloalkyl, C 3-6 heterocycloalkyl, and aryl, and substituted with 0-3 groups selected from halo, hydroxyl, amino, nitro, cyano, Ci collective haloalkyl, C).
- Ci -6 alkoxy Ci -6 alkoxy, C 1 -6 haloalkoxy, Ci -6 polyhaloalkoxy, Cj -6 alkylthio, C i -6 haloalkythio, Ci -6 polyhaloalkylthio, Ci -6 alkylamino, Ci -6 dialkylamino, (Ci- 6 )-alk-(Ci.
- a 7 is selected from C 3-6 cycloalkyl, C3-6 heterocycloalkyl, and aryl, and substituted with 0-3 groups selected from halo, hydroxyl, amino, nitro, cyano, Ci -6 haloalkyl, Ci -6 polyhaloalkyl, Ci-6 alkoxy, Ci -6 haloalkoxy, Ci- polyhaloalkoxy, Ci_ 6 alkylthio, Ci -6 haloalkythio, Ci -6 polyhaloalkylthio, Ci -6 alkylamino, Ci -6 dialkylamino, (Ci -6 )-alk-(C i -6 )-alkoxy, (C].
- R 2 is aryl, and substituted with 0-5 groups independently selected from halo, hydroxyl, amino, nitro, cyano, Cj.
- R 3 is aryl substituted with 0-5 groups independently selected from halo, hydroxyl, amino, nitro, cyano, C 1-6 alkyl, C].
- R 3 is selected from the structure represented by formula:
- R is selected from the group consisting of -OH, -COR , -CN, -CH 2 PO(OH) 2 , -CH 2 P(0) 3 (CH 2 CH 3 ) 2 , -N0 2 , -NHR 17 , and lH-tetrazole;
- R 16 is selected from the group consisting of: -OH, -0-C(i -2 )alkyl, -OCH 2 OC(0)CH 3 , -OCH 2 OC(0)t-Butyl, and -NHOH,
- R 17 is selected from the group consisting of: -H, -C(0)C(0)CH 2 CH 3 , -C(0)C(0)OH, and -C(0)CH 2 -lH-tetrazole
- R 14 is -H or when R 13 is -COR 16 and R 16 is OH, R 14 is -F, -OC(0)CH 3 ;
- R 15 is H or when R 13 is - COR 16 , R 16 is OH, and R 14 is H, R 15 is -F, -OC(0)CH 3
- R 13 is -H when R 14 is -OH or both R 14 and R 15 are -COOH.
- the invention relates to pharmaceutical compositions comprising a pharmaceutically acceptable carrier and an effective amount of a disclosed compound, or a pharmaceutically acceptable salt, hydrate, solvate, or polymorph thereof.
- a pharmaceutical composition comprising a compound as defined herein or a pharmaceutically acceptable salt, solvate or prodrug thereof, and an acceptable excipient.
- a method for inhibiting STAT3 and/or STAT5 activity comprising administering a therapeutically effective amount of a compound as defined herein or a pharmaceutically acceptable salt, solvate or prodrug thereof, to a patient.
- a method for treating or preventing cancer associated with STAT3/STAT5 activity dysfunction comprising administering a therapeutically effective amount of a compound as defined herein, or a pharmaceutically acceptable salt, solvate or prodrug thereof, to a patient.
- the cancer is from solid or hematological tumors.
- the cancer is one harbouring activated STAT3 and/or STAT5.
- Such cancer can be for example breast, liver, prostate, blood, skin, head, neck cancer, glioblastoma or acute myelogenic (AML) and acute lymphoblastic leukemias.
- a compound as defined herein or a pharmaceutically acceptable salt, solvate or prodrug thereof in the manufacture of a medicament for treating or preventing cancer harbouring activated STAT3 and/or STAT5, such as cancer from solid or hematological tumors, breast cancer, liver cancer, prostate cancer, blood cancer, skin cancer, head cancer, neck cancer, glioblastoma or acute myelogenic (AML) and acute lymphoblastic leukemias.
- a compound as defined herein or a pharmaceutically acceptable salt, solvate or prodrug thereof for inhibiting STAT3 and/or STAT5 activity.
- a compound as defined herein or a pharmaceutically acceptable salt, solvate or prodrug thereof for treating or preventing cancer harbouring activated STAT3 and/or STAT5, such as the cancer is from solid or hematological tumors, breast cancer, liver cancer, prostate cancer, blood cancer, skin cancer, head cancer, neck cancer, glioblastoma or acute myelogenic (AML) and acute lymphoblastic leukemiasassociated with STAT3/STAT5 activity dysfunction, such as breast, prostate or brain cancer.
- AML acute myelogenic
- a pharmaceutical composition as defined herein for use in inhibiting STAT3 and/or STAT5 activity in another aspect of the disclosure.
- methods for manufacturing a medicament comprising combining at least one disclosed compound or at least one disclosed product with a pharmaceutically acceptable carrier or diluent.
- the invention relates to the use of a disclosed compound in the manufacture of a medicament for the treatment of a disorder associated with STAT3/STAT5 activity dysfunction (such as hyperactivity or over-expression).
- the invention relates to the use of the disclosed compound in the manufacture of a medicament for the treatment of a cancer harbouring activated STAT3 and/or STAT5, such as the cancer is from solid or hematological tumors, breast cancer, liver cancer, prostate cancer, blood cancer, skin cancer, head cancer, neck cancer, glioblastoma or acute myelogenic (AML) and acute lymphoblastic leukemias.
- FIG. 1A is a graph illustrating a comparison of compound 1 against compounds 22, 31 , 32, and 33 from library BTSC30M;
- Fig. I B is a table illustrating the IC 50 values from the compounds tested in Fig. 1A against BTSCs, 25EF, 67EF, 73E, 84EF, and 127EF;
- FIG. 2A illustrates the insensitivity of a number of compounds against a variety of kinases and structural proteins
- Fig. 2B illustrates SPR curves displaying the binding affinity of the best hits to STAT3/5 proteins
- FIG. 3 illustrates a treespot dendrogram displaying that compound 27 does not inhibit any of the 101 kinases tested against, a hit being defined as >35% inhibition.
- FIG. 4 illustrates a Western blot analysis of the top three compounds against BTSC 147M and their comparison against a well-established Jak2 inhibitor WP1066 at ⁇ ⁇ against BTSCM 73M;
- FIG. 5 illustrates the IC50 determination of control (Staurosporine) against the selected kinases
- FIG. 6 is a table illustrating the measurements of kinase concentration in eluates by qPCR
- FIG. 7 is a calibration curve performed using ⁇ Fam-pYLPQTV and dilutions of STAT3 and STAT1 protein (5.0 ⁇ to 2.4 nM) at a final DMSO concentration of 10%;
- Fig. 8 illustrates the screening of compounds 22, 31 , 32, and 33 against a number of BTSCs, where each compound was screened twice at various concentrations '
- Fig. 9 illustrates the survival of pancreatic 10.05 patient cells treated with STAT3 inhibitors
- Fig. 10 illustrates the effect of Pa03C low passage patient cells pretreated with STAT3 inhibitor 1 hr and then stimulated with 1L6 cytokine for 15 min;
- Fig. 1 1 illustrates the effects of STAT3 inhibitor 31 on low passage patient-derived pancreatic cancer line (Pa03C) plated in a 96-well plate coated with 1% Noble Agar in media containing 3% Matrigel, with and without cancer-associated fibroblasts;
- Fig. 12 illustrates compound 31 potently reducing cell viability in 12 of 15 different multiple myeloma tumor cell lines as assessed by MTT assay with calculated EDso's ranging from between 2-6 ⁇ ;
- Fig. 13 illustrates the hematoxylin/eosin staining - hypercellular tumour dense areas staining (white) with hematoxylin in BT73 cells (A) without and (B) with inhibitor 31 ;
- Fig. 14 illustrates the effect of compound 31 for decreasing pSTAT expression in mice orthotopically xenografted with BT73 brain cancer cells;
- Fig. 15 illustrates the effect of compound 31 for reducing proliferation in BT73 brain tumours
- Fig. 16 illustrates an increased apoptosis (TUNEL staining) in mice treated with compound 3 ⁇
- Fig. 17 illustrates that concentration of compound 31 was found to be present in the brain with different dosing (l Omg/kg and 25 mg/kg) as assessed by LCMS;
- Fig. 18 illustrates the concentration of compound 31 found in the brain as assessed by LCMS
- Figure 19 illustrates the concentration of compound 32 found in the brain as assessed by LCMS
- Fig. 20 illustrates the fluorescence of 2071 NIC Cell Line treated with compound 31 (SH-04- 54) or with BP 1-102;
- Fig. 21 illustrates the fluorescence of various NIC Cell Line treated with compound 31 (SH- 04-54) or with BP1 -102.
- Ranges can be expressed herein as from “about” one particular value, and/or to "about” another particular value. When such a range is expressed, a further aspect includes from the one particular value and/or to the other particular value. Similarly, when values are expressed as approximations, by use of the antecedent "about,” it will be understood that the particular value forms a further aspect. It will be further understood that the endpoints of each of the ranges are significant both in relation to the other endpoint, and independently of the other endpoint. It is also understood that there are a number of values disclosed herein, and that each value is also herein disclosed as "about” that particular value in addition to the value itself. For example, if the value " 10" is disclosed, then “about 10" is also disclosed. It is also understood that each unit between two particular units are also disclosed. For example, if 10 and 15 are disclosed, then 1 1 , 12, 13, and 14 are also disclosed.
- references in the specification and concluding claims to parts by weight of a particular element or component in a composition denotes the weight relationship between the element or component and any other elements or components in the composition or article for which a part by weight is expressed.
- X and Y are present at a weight ratio of 2:5, and are present in such ratio regardless of whether additional components are contained in the compound.
- a weight percent (wt. %) of a component is based on the total weight of the formulation or composition in which the component is included.
- the terms “optional” or “optionally” means that the subsequently described event or circumstance can or cannot occur, and that the description includes instances where said event or circumstance occurs and instances where it does not.
- STAT3 signal transducer and activator of transcription 3 (acute- phase response),” and “signal transducer and activator of transcription 3” can be used interchangeably and refer to a a transcription factor encoded by a gene designated in human as the STAT3 gene, which has a human gene map locus of 17q21 and described by Entrez Gene cytogenetic band: 17q21.31 ; Ensembl cytogenetic band: 17q21.2; and, HGNC cytogenetic band: 17q21.
- STAT3 refers to a human protein that has 770 amino acids and has a molecular weight of about 88,068 Da.
- the term is inclusive of splice isoforms or variants, and also inclusive of that protein referred to by such alternative designations as: APRF, MGC 16063, Acute-phase response factor, DNA-binding protein APRF, HIES as used by those skilled in the art to that protein encoded by human gene STAT3.
- APRF splice isoforms or variants
- MGC 16063 Acute-phase response factor
- APRF DNA-binding protein
- HIES DNA-binding protein encoded by human gene STAT3.
- the term is also inclusive of the non-human ortholog or homolog thereof.
- STAT5 refers to STAT5A and/or STAT5B. If specific reference to either STAT5A or STAT5B is required, the specific term will be used herein.
- STAT5A and “signal transducer and activator of transcription 5A” can be used interchangeably and refer to a a transcription factor encoded by a gene designated in human as the STAT5A gene, which has a human gene map locus described by Entrez Gene cytogenetic band: 17ql 1.2; Ensembl cytogenetic band: 17q21.2; and, HGNC cytogenetic band: 17q 1 1.2.
- STAT5A refers to a human protein that has 794 amino acids and has a molecular weight of about 90,647 Da.
- STAT5B and “signal transducer and activator of transcription 5B” can be used interchangeably and refer to a a transcription factor encoded by a gene designated in human as the STAT5B gene, which has a human gene map locus described by Entrez Gene cytogenetic band: 17ql 1.2; Ensembl cytogenetic band: 17q21.2; and, HGNC cytogenetic band: 17ql 1.2.
- STAT5A refers to a human protein that has 787 amino acids and has a molecular weight of about 89,866 Da.
- the term is inclusive of splice isoforms or variants, and also inclusive of that protein referred to by such alternative designations as transcription factor STAT5B as used by those skilled in the art to that protein encoded by human gene STAT5A.
- the term is also inclusive of the non-human ortholog or homolog thereof.
- the term "subject” can be a vertebrate, such as a mammal, a fish, a bird, a reptile, or an amphibian.
- the subject of the herein disclosed methods can be a human, non-human primate, horse, pig, rabbit, dog, sheep, goat, cow, cat, guinea pig or rodent. The term does not denote a particular age or sex.
- the subject is a mammal.
- a patient refers herein to a subject afflicted with cancer, preferably glioblastoma.
- patient includes human and veterinary subjects.
- treatment refers to the medical management of a patient with the intent to cure, ameliorate, stabilize, or prevent a disease, pathological condition, or disorder.
- This term includes active treatment, that is, treatment directed specifically toward the improvement of a disease, pathological condition, or disorder, and also includes causal treatment, that is, treatment directed toward removal of the cause of the associated disease, pathological condition, or disorder.
- this term includes palliative treatment, that is, treatment designed for the relief of symptoms rather than the curing of the disease, pathological condition, or disorder; preventative treatment, that is, treatment directed to minimizing or partially or completely inhibiting the development of the associated disease, pathological condition, or disorder; and supportive treatment, that is, treatment employed to supplement another specific therapy directed toward the improvement of the associated disease, pathological condition, or disorder.
- the term covers any treatment of a subject, including a mammal (e.g., a human), and includes: (i) preventing the disease from occurring in a subject that can be predisposed to the disease but has not yet been diagnosed as having it; (ii) inhibiting the disease, i.e., arresting its development; or (iii) relieving the disease, i.e., causing regression of the disease.
- the subject is a mammal such as a primate, and, in a further aspect, the subject is a human.
- subject also includes domesticated animals (e.g., cats, dogs, etc.), livestock (e.g., cattle, horses, pigs, sheep, goats, etc.), and laboratory animals (e.g., mouse, rabbit, rat, guinea pig, fruit fly, etc.).
- livestock e.g., cattle, horses, pigs, sheep, goats, etc.
- laboratory animals e.g., mouse, rabbit, rat, guinea pig, fruit fly, etc.
- diagnosisd means having been subjected to a physical examination by a person of skill, for example, a physician, and found to have a condition that can be diagnosed or treated by the compounds, compositions, or methods disclosed herein.
- diagnosis with a disorder treatable by STAT3 inhibition means having been subjected to a physical examination by a person of skill, for example, a physician, and found to have a condition that can be diagnosed or treated by a compound or composition that can inhibit or negatively modulate STAT3.
- diagnosis with a need for inhibition of STAT3 refers to having been subjected to a physical examination by a person of skill, for example, a physician, and found to have a condition characterized by a dysfunction in STAT3 activity.
- a diagnosis can be in reference to a disorder, such as an oncological disorder or disease, cancer and/or disorder of uncontrolled cellular proliferation and the like, as discussed herein.
- the term "diagnosed with a need for inhibition of STAT3 activity” refers to having been subjected to a physical examination by a person of skill, for example, a physician, and found to have a condition that can be diagnosed or treated by inhibition of STAT3 activity.
- diagnosisd with a need for modulation of STAT3 activity means having been subjected to a physical examination by a person of skill, for example, a physician, and found to have a condition that can be diagnosed or treated by modulation of STAT3 activity, e.g. negative modulation.
- diagnosisd with a need for treatment of one or more disorder of uncontrolled cellular proliferation associated with STAT3 dysfunction means having been subjected to a physical examination by a person of skill, for example, a physician, and found to have one or disorders of uncontrolled cellular proliferation, e.g. a cancer, associated with STAT3 dysfunction.
- STAT3 or STAT5 -dependent cancer refers to a cancer harboring constitutively activated STAT3 or STAT5.
- the phrase "identified to be in need of treatment for a disorder," or the like, refers to selection of a subject based upon need for treatment of the disorder.
- a subject can be identified as having a need for treatment of a disorder (e.g., a disorder related to STAT3 activity) based upon an earlier diagnosis by a person of skill and thereafter subjected to treatment for the disorder.
- the identification can, in one aspect, be performed by a person different from the person making the diagnosis.
- the administration can be performed by one who subsequently performed the administration.
- administering refers to any method of providing a pharmaceutical preparation to a subject. Such methods are well known to those skilled in the art and include, but are not limited to, oral administration, transdermal administration, administration by inhalation, nasal administration, topical administration, intravaginal administration, ophthalmic administration, intraaural administration, intracerebral administration, rectal administration, sublingual administration, buccal administration, and parenteral administration, including injectable such as intravenous administration, intra-arterial administration, intramuscular administration, and subcutaneous administration. Administration can be continuous or intermittent.
- a preparation can be administered therapeutically; that is, administered to treat an existing disease or condition.
- a preparation can be administered prophylactically; that is, administered for prevention of a disease or condition.
- contacting refers to bringing a disclosed compound and a cell, target STAT3 protein, or other biological entity together in such a manner that the compound can affect the activity of the target (e.g., spliceosome, cell, etc.), either directly; i.e., by interacting with the target itself, or indirectly; i.e., by interacting with another molecule, co- factor, factor, or protein on which the activity of the target is dependent.
- the target e.g., spliceosome, cell, etc.
- the terms “effective amount” and “amount effective” refer to an amount that is sufficient to achieve the desired result or to have an effect on an undesired condition.
- a “therapeutically effective amount” refers to an amount that is sufficient to achieve the desired therapeutic result or to have an effect on undesired symptoms, but is generally insufficient to cause adverse side effects.
- the specific therapeutically effective dose level for any particular patient will depend upon a variety of factors including the disorder being treated and the severity of the disorder; the specific composition employed; the age, body weight, general health, sex and diet of the patient; the time of administration; the route of administration; the rate of excretion of the specific compound employed; the duration of the treatment; drugs used in combination or coincidental with the specific compound employed and like factors well known in the medical arts. For example, it is well within the skill of the art to start doses of a compound at levels lower than those required to achieve the desired therapeutic effect and to gradually increase the dosage until the desired effect is achieved. If desired, the effective daily dose can be divided into multiple doses for purposes of administration.
- compositions can contain such amounts or submultiples thereof to make up the daily dose.
- the dosage can be adjusted by the individual physician in the event of any contraindications. Dosage can vary, and can be administered in one or more dose administrations daily, for one or several days. Guidance can be found in the literature for appropriate dosages for given classes of pharmaceutical products.
- a preparation can be administered in a "prophylactically effective amount"; that is, an amount effective for prevention of a disease or condition.
- EC 5 0, is intended to refer to the concentration of a substance (e.g., a compound or a drug) that is required for 50% agonism or activation of a biological process, or component of a process, including a protein, subunit, organelle, ribonucleoprotein, etc.
- a substance e.g., a compound or a drug
- an EC5 0 can refer to the concentration of a substance that is required for 50% agonism or activation in vivo, as further defined elsewhere herein.
- EC 50 refers to the concentration of agonist or activator that provokes a response halfway between the baseline and maximum response.
- IC 50 is intended to refer to the concentration of a substance (e.g., a compound or a drug) that is required for 50%> inhibition of a biological process, or component of a process, including a protein, subunit, organelle, ribonucleoprotein, etc.
- a substance e.g., a compound or a drug
- an IC50 can refer to the plasma concentration of a substance that is required for 50% inhibition in vivo, as further defined elsewhere herein. More commonly, IC 5 0 refers to the half maximal (50%) inhibitory concentration (IC) of a substance required to inhibit a process or activity in vitro.
- STAT3 IC 5 0 refers to the concentration of a substance (e.g., a compound or a drug) that is required for 50% inhibition of a STAT3 activity.
- an IC 50 can refer to the plasma concentration of a substance that is required for 50% inhibition of an in vivo activity or process as further defined elsewhere herein, e.g. tumor growth in an animal or human.
- STAT3 IC50 refers the half maximal (50%) inhibitory concentration (IC) of a substance or compound required to inhibit a process or activity an in vitro context, e.g. a cell-free or cell-based assay.
- the STAT3 IC 0 can be in the context of the half-maximal concentration required to inhibit cell growth.
- the response is measured in a cell-line with aberrant STAT3 activity.
- the response is measured in a cell-line with persistently active STAT3.
- the response can be determined using a cell-line derived from a human breast cancer, human pancreatic cancer, and human prostate cancer.
- the response can be measured in a cell-line selected from MDA-MB-231 , Panc-1 , and DU-145.
- Cell-lines transfected with specific genes can also be used.
- the response can be measured in a cell-line transfected with v-Src.
- the cell-line transfected with v-Src is a permanent cell- line.
- the STAT3 IC50 is the half-maximal concentration required to inhibit STAT3 activity in a cell-free assay, e.g. an electrophoretic mobility shift assay ("EMSA").
- the STAT3 IC50 is the half-maximal concentration required to inhibit cell- growth, cell viability or cell migration activity.
- STAT3 KD refers to the binding affinity of a compound or substance for the STAT3 determined in an in vitro assay.
- the KD of a substance for a protein can be determined by a variety of methods known to one skilled in the art, e.g. equilibrium dialysis, analytical ultracentrifugation and surface plasmon resonance ("SPR") analysis.
- SPR surface plasmon resonance
- STAT3 KD is defined as the ratio of association and dissociation rate constants determined using SPR analysis using purified STAT3 protein.
- STAT3 Kj refers to the inhibition constant for the displacement of a STAT3 SH2 probe from STAT3 protein.
- the STAT3 SH2 can be fluorescence-labelled GpYLPQTV.
- the fluorescence label is 5- carboxyfluorescein, although other suitable fluorescence probes can be used as determined to be useful and convenient by one skilled in the art.
- pharmaceutically acceptable describes a material that is not biologically or otherwise undesirable, i.e., without causing an unacceptable level of undesirable biological effects or interacting in a deleterious manner.
- derivative refers to a compound having a structure derived from the structure of a parent compound (e.g., a compound disclosed herein) and whose structure is sufficiently similar to those disclosed herein and based upon that similarity, would be expected by one skilled in the art to exhibit the same or similar activities and utilities as the claimed compounds, or to induce, as a precursor, the same or similar activities and utilities as the claimed compounds.
- exemplary derivatives include salts, esters, amides, salts of esters or amides, and N-oxides of a parent compound.
- aqueous and non-aqueous carriers include water, ethanol, polyols (such as glycerol, propylene glycol, polyethylene glycol and the like), carboxymethylcellulose and suitable mixtures thereof, vegetable oils (such as olive oil) and injectable organic esters such as ethyl oleate.
- Proper fluidity can be maintained, for example, by the use of coating materials such as lecithin, by the maintenance of the required particle size in the case of dispersions and by the use of surfactants.
- These compositions can also contain adjuvants such as preservatives, wetting agents, emulsifying agents and dispersing agents.
- Prevention of the action of microorganisms can be ensured by the inclusion of various antibacterial and antifungal agents such as paraben, chlorobutanol, phenol, sorbic acid and the like. It can also be desirable to include isotonic agents such as sugars, sodium chloride and the like.
- Prolonged absorption of the injectable pharmaceutical form can be brought about by the inclusion of agents, such as aluminum monostearate and gelatin, which delay absorption.
- Injectable depot forms are made by forming microencapsule matrices of the drug in biodegradable polymers such as polylactide-polyglycolide, poly(orthoesters) and poly(anhydrides). Depending upon the ratio of drug to polymer and the nature of the particular polymer employed, the rate of drug release can be controlled. Depot injectable formulations are also prepared by entrapping the drug in liposomes or microemulsions which are compatible with body tissues.
- the injectable formulations can be sterilized, for example, by filtration through a bacterial-retaining filter or by incorporating sterilizing agents in the form of sterile solid compositions which can be dissolved or dispersed in sterile water or other sterile injectable media just prior to use.
- Suitable inert carriers can include sugars such as lactose. Desirably, at least 95% by weight of the particles of the active ingredient have an effective particle size in the range of 0.01 to 10 micrometers.
- a residue of a chemical species refers to the moiety that is the resulting product of the chemical species in a particular reaction scheme or subsequent formulation or chemical product, regardless of whether the moiety is actually obtained from the chemical species.
- the term "substituted" is contemplated to include all permissible substituents of organic compounds.
- the permissible substituents include acyclic and cyclic, branched and unbranched, carbocyclic and heterocyclic, and aromatic and nonaromatic substituents of organic compounds.
- Illustrative substituents include, for example, those described below.
- the permissible substituents can be one or more and the same or different for appropriate organic compounds.
- the heteroatoms such as nitrogen, can have hydrogen substituents and/or any permissible substituents of organic compounds described herein which satisfy the valences of the heteroatoms.
- substitution or “substituted with” include the implicit proviso that such substitution is in accordance with permitted valence of the substituted atom and the substituent, and that the substitution results in a stable compound, e.g., a compound that does not spontaneously undergo transformation such as by rearrangement, cyclization, elimination, etc. It is also contemplated that, in certain aspects, unless expressly indicated to the contrary, individual substituents can be further optionally substituted (i.e., further substituted or unsubstituted).
- alkyl as used herein is a branched or unbranched saturated hydrocarbon group of 1 to 24 carbon atoms, such as methyl, ethyl, n-propyl, isopropyl, n- butyl, isobutyl, s-butyl, i- butyl, n-pentyl, isopentyl, i-pentyl, neopentyl, hexyl, heptyl, octyl, nonyl, decyl, dode cyl, tetradecyl, hexadecyl, eicosyl, tetracosyl, and the like.
- the alkyl group can be cyclic or acyclic.
- the alkyl group can be branched or unbranched.
- the alkyl group can also be substituted or unsubstituted.
- the alkyl group can be substituted with one or more groups including, but not limited to, alkyl, cycloalkyl, alkoxy, amino, ether, halide, hydroxy, nitro, silyl, sulfo-oxo, or thiol, as described herein.
- a "lower alkyl” group is an alkyl group containing from one to six (e.g., from one to four) carbon atoms.
- alkyl is generally used to refer to both unsubstituted alkyl groups and substituted alkyl groups; however, substituted alkyl groups are also specifically referred to herein by identifying the specific substituent(s) on the alkyl group.
- halogenated alkyl or “haloalkyl” specifically refers to an alkyl group that is substituted with one or more halide, e.g., fluorine, chlorine, bromine, or iodine.
- alkoxyalkyl specifically refers to an alkyl group that is substituted with one or more alkoxy groups, as described below.
- alkylamino specifically refers to an alkyl group that is substituted with one or more amino groups, as described below, and the like.
- alkyl is used in one instance and a specific term such as “alkylalcohol” is used in another, it is not meant to imply that the term “alkyl” does not also refer to specific terms such as “alkylalcohol” and the like.
- cycloalkyl refers to both unsubstituted and substituted cycloalkyl moieties
- the substituted moieties can, in addition, be specifically identified herein; for example, a particular substituted cycloalkyl can be referred to as, e.g., an "alkylcycloalkyl.”
- a substituted alkoxy can be specifically referred to as, e.g., a "halogenated alkoxy”
- a particular substituted alkenyl can be, e.g., an "alkenylalcohol,” and the like.
- cycloalkyl is a non-aromatic carbon-based ring composed of at least three carbon atoms.
- examples of cycloalkyl groups include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, norbornyl, and the like.
- heterocycloalkyl is a type of cycloalkyl group as defined above, and is included within the meaning of the term “cycloalkyl,” where at least one of the carbon atoms of the ring is replaced with a heteroatom such as, but not limited to, nitrogen, oxygen, sulfur, or phosphorus.
- the cycloalkyl group and heterocycloalkyl group can be substituted or unsubstituted.
- the cycloalkyl group and heterocycloalkyl group can be substituted with one or more groups including, but not limited to, alkyl, cycloalkyl, alkoxy, amino, ether, halide, hydroxy, nitro, silyl, sulfo-oxo, or thiol as described herein.
- the term "polyalkylene group” as used herein is a group having two or more CH 2 groups linked to one another.
- the polyalkylene group can be represented by the formula- (CH2) A - , where "a" is an integer of from 2 to 500.
- alkoxy and alkoxyl as used herein to refer to an alkyl or cycloalkyl group bonded through an ether linkage; that is, an "alkoxy” group can be defined as- OA 1 where A 1 is alkyl or cycloalkyl as defined above.
- Alkoxy also includes polymers of alkoxy groups as just described; that is, an alkoxy can be a polyether such as- OA 1 - OA 2 or- OA 1 - (OA 2 ) a - 1 2 3
- OA alkyl and/or cycloalkyl groups.
- alkenyl as used herein is a hydrocarbon group of from 2 to 24 carbon atoms with a structural formula containing at least one carbon-carbon double bond.
- the alkenyl group can be substituted with one or more groups including, but not limited to, alkyl, cycloalkyl, alkoxy, alkenyl, cycloalkenyl, alkynyl, cycloalkynyl, aryl, heteroaryl, aldehyde, amino, carboxylic acid, ester, ether, halide, hydroxy, ketone, azide, nitro, silyl, sulfo-oxo, or thiol, as described herein.
- cycloalkenyl as used herein is a non-aromatic carbon-based ring composed of at least three carbon atoms and containing at least one carbon-carbon double bound, i.e.
- cycloalkenyl groups include, but are not limited to, cyclopropenyl, cyclobutenyl, cyclopentenyl, cyclopentadienyl, cyclohexenyl, cyclohexadienyl, norbornenyl, and the like.
- heterocycloalkenyl is a type of cycloalkenyl group as defined above, and is included within the meaning of the term “cycloalkenyl,” where at least one of the carbon atoms of the ring is replaced with a heteroatom such as, but not limited to, nitrogen, oxygen, sulfur, or phosphorus.
- the cycloalkenyl group and heterocycloalkenyl group can be substituted or unsubstituted.
- the cycloalkenyl group and heterocycloalkenyl group can be substituted with one or more groups including, but not limited to, alkyl, cycloalkyl, alkoxy, alkenyl, cycloalkenyl, alkynyl, cycloalkynyl, aryl, heteroaryl, aldehyde, amino, carboxylic acid, ester, ether, halide, hydroxy, ketone, azide, nitro, silyl, sulfo-oxo, or thiol as described herein.
- alkynyl as used herein is a hydrocarbon group of 2 to 24 carbon atoms with a structural formula containing at least one carbon-carbon triple bond.
- the alkynyl group can be unsubstituted or substituted with one or more groups including, but not limited to, alkyl, cycloalkyl, alkoxy, alkenyl, cycloalkenyl, alkynyl, cycloalkynyl, aryl, heteroaryl, aldehyde, amino, carboxylic acid, ester, ether, halide, hydroxy, ketone, azide, nitro, silyl, sulfo-oxo, or thiol, as described herein.
- cycloalkynyl as used herein is a non-aromatic carbon-based ring composed of at least seven carbon atoms and containing at least one carbon-carbon triple bound.
- cycloalkynyl groups include, but are not limited to, cycloheptynyl, cyclooctynyl, cyclononynyl, and the like.
- heterocycloalkynyl is a type of cycloalkenyl group as defined above, and is included within the meaning of the term “cycloalkynyl,” where at least one of the carbon atoms of the ring is replaced with a heteroatom such as, but not limited to, nitrogen, oxygen, sulfur, or phosphorus.
- the cycloalkynyl group and heterocycloalkynyl group can be substituted or unsubstituted.
- the cycloalkynyl group and heterocycloalkynyl group can be substituted with one or more groups including, but not limited to, alkyl, cycloalkyl, alkoxy, alkenyl, cycloalkenyl, alkynyl, cycloalkynyl, aryl, heteroaryl, aldehyde, amino, carboxylic acid, ester, ether, halide, hydroxy, ketone, azide, nitro, silyl, sulfo-oxo, or thiol as described herein.
- aryl as used herein is a group that contains any carbon-based aromatic group including, but not limited to, benzene, naphthalene, phenyl, biphenyl, phenoxybenzene, and the like.
- aryl also includes "heteroaryl,” which is defined as a group that contains an aromatic group that has at least one heteroatom incorporated within the ring of the aromatic group. Examples of heteroatoms include, but are not limited to, nitrogen, oxygen, sulfur, and phosphorus.
- non-heteroaryl which is also included in the term “aryl,” defines a group that contains an aromatic group that does not contain a heteroatom. The aryl group can be substituted or unsubstituted.
- the aryl group can be substituted with one or more groups including, but not limited to, alkyl, cycloalkyl, alkoxy, alkenyl, cycloalkenyl, alkynyl, cycloalkynyl, aryl, heteroaryl, aldehyde, amino, carboxylic acid, ester, ether, halide, hydroxy, ketone, azide, nitro, silyl, sulfo-oxo, or thiol as described herein.
- groups including, but not limited to, alkyl, cycloalkyl, alkoxy, alkenyl, cycloalkenyl, alkynyl, cycloalkynyl, aryl, heteroaryl, aldehyde, amino, carboxylic acid, ester, ether, halide, hydroxy, ketone, azide, nitro, silyl, sulfo-oxo, or thiol as described herein.
- biasing is a specific type of aryl group and is included in the definition of "aryl.”
- Biaryl refers to two aryl groups that are bound together via a fused ring structure, as in naphthalene, or are attached via one or more carbon-carbon bonds, as in biphenyl.
- amine or “amino” as used herein are represented by the formula- NA'A 2 , where A 1 and A 2 can be, independently, hydrogen or alkyl, cycloalkyl, alkenyl, cycloalkenyl, alkynyl, cycloalkynyl, aryl, or heteroaryl group as described herein.
- alkylamino as used herein is represented by the formula- NH(-alkyl) where alkyl is a described herein. Representative examples include, but are not limited to, methylamino group, ethylamino group, propylamino group, isopropylamino group, butylamino group, isobutylamino group, (sec-butyl)amino group, (tert-butyl)amino group, pentylamino group, isopentylamino group, (tert-pentyl)amino group, hexylamino group, and the like.
- dialkylamino as used herein is represented by the formula- N(-alkyl)2 where alkyl is a described herein.
- Representative examples include, but are not limited to, dimethylamino group, diethylamino group, dipropylamino group, diisopropylamino group, dibutylamino group, diisobutylamino group, di(sec-butyl)amino group, di(tert-butyl)amino group, dipentylamino group, diisopentylamino group, di(tert-pentyl)amino group, dihexylamino group, N-ethyl-N-niethylamino group, N-methyl-N-propylamino group, N- ethyl-N- propylamino group and the like.
- carboxylic acid as used herein is represented by the formula -C(0)OH.
- esters as used herein is represented by the formula- OC(0)A' or- C(0)OA', where A 1 can be alkyl, cycloalkyl, alkenyl, cycloalkenyl, alkynyl, cycloalkynyl, aryl, or heteroaryl group as described herein.
- polystyrene resin as used herein is represented by the formula -(A'0-(0)C-A 2 -C(0)0) a - or- (A 1 0(0)C-A 2 -OC(0)) a - , where A 1 and A 2 can be, independently, an alkyl, cycloalkyl, alkenyl, cycloalkenyl, alkynyl, cycloalkynyl, aryl, or heteroaryl group described herein and "a” is an integer from 1 to 500. "Polyester” is as the term used to describe a group that is produced by the reaction between a compound having at least two carboxylic acid groups with a compound having at least two hydroxyl groups.
- ether as used herein is represented by the formula A OA , where A and A can be, independently, an alkyl, cycloalkyl, alkenyl, cycloalkenyl, alkynyl, cycloalkynyl, aryl, or heteroaryl group described herein.
- polyether as used herein is represented by the formula- (A'0-A 2 O) a - , where A 1 and A 2 can be, independently, an alkyl, cycloalkyl, alkenyl, cycloalkenyl, alkynyl, cycloalkynyl, aryl, or heteroaryl group described herein and "a" is an integer of from 1 to 500.
- polyether groups include polyethylene oxide, polypropylene oxide, and polybutylene oxide.
- halide refers to the halogens fluorine, chlorine, bromine, and iodine.
- heterocycle refers to single and multi-cyclic aromatic or non- aromatic ring systems in which at least one of the ring members is other than carbon.
- Heterocycle includes azetidine, dioxane, furan, imidazole, isothiazole, isoxazole, morpholine, oxazole, oxazole, including, 1 ,2,3-oxadiazole, 1 ,2,5-oxadiazole and 1 ,3,4-oxadiazole, piperazine, piperidine, pyrazine, pyrazole, pyridazine, pyridine, pyrimidine, pyrrole, pyrrolidine, tetrahydrofuran, tetrahydropyran, tetrazine, including 1 ,2,4,5-tetrazine, tetrazole, including 1 ,2,3,4-tetrazole and 1 ,2,4,5-tetrazole, thi
- ketone as used herein is represented by the formula A C(0)A , where A and A can be, independently, an alkyl, cycloaikyl, alkenyl, cycloalkenyl, alkynyl, cycloalkynyl, aryl, or heteroaryl group as described herein.
- nitrile as used herein is represented by the formula- CN.
- sulfo-oxo is represented by the formulas- S(0)A' ,- S(0) 2 A' ,-OS(0) 2 A ] , or -OSCCThOA 1 , where A 1 can be hydrogen or an alkyl, cycloaikyl, alkenyl, cycloalkenyl, alkynyl, cycloalkynyl, aryl, or heteroaryl group as described herein.
- sulfonyl is used herein to refer to the sulfo-oxo group represented by the -S(0)2A !
- a 1 can be hydrogen or an alkyl, cycloaikyl, alkenyl, cycloalkenyl, alkynyl, cycloalkynyl, aryl, or heteroaryl group as described herein.
- the term "sulfone" as used herein is represented by the formula A'S(0) 2 A 2 , where A 1 and A 2 can be, independently, an alkyl, cycloaikyl, alkenyl, cycloalkenyl, alkynyl, cycloalkynyl, aryl, or heteroaryl group as described herein.
- sulfoxide as used herein is represented by the formula A 1 S(0)A , where A 1 and A2 can be, independently, an alkyl, cycloaikyl, alkenyl, cycloalkenyl, alkynyl, cycloalkynyl, aryl, or heteroaryl group as described herein.
- thiol as used herein is represented by the formula- SH.
- R 1 ", “R 2 ", “R 3 ", “R n " where n is an integer, as used herein can, independently, possess one or more of the groups listed above.
- R 1 is a straight chain alkyl group
- one of the hydrogen atoms of the alkyl group can optionally be substituted with a hydroxyl group, an alkoxy group, an alkyl group, a halide, and the like.
- a first group can be incorporated within second group or, alternatively, the first group can be pendant (i.e., attached) to the second group.
- an alkyl group comprising an amino group the amino group can be incorporated within the backbone of the alkyl group. Alternatively, the amino group can be attached to the backbone of the alkyl group. The nature of the group(s) that is (are) selected will determine if the first group is embedded or attached to the second group. As described herein, compounds of the invention may contain "optionally substituted” moieties. In general, the term “substituted,” whether preceded by the term “optionally” or not, means that one or more hydrogens of the designated moiety are replaced with a suitable substituent.
- an "optionally substituted" group may have a suitable substituent at each substitutable position of the group, and when more than one position in any given structure may be substituted with more than one substituent selected from a specified group, the substituent may be either the same or different at every position.
- Combinations of substituents envisioned by this invention are preferably those that result in the formation of stable or chemically feasible compounds.
- individual substituents can be further optionally substituted (i.e., further substituted or unsubstituted).
- stable refers to compounds that are not substantially altered when subjected to conditions to allow for their production, detection, and, in certain aspects, their recovery, purification, and use for one or more of the purposes disclosed herein.
- Suitable monovalent substituents on R are independently halogen, - -(haloR * ), -(CH 2 ) 0 . 2 OH, -(CH 2 ) 0 - 2 OR * , -(CH 2 ) 0 - 2 CH(OR * ) 2 ; -0(haloR * ), -CN, -N 3 , -(CH 2 ) 0 - 2 C(O)R * , -(CH 2 )o -2 C(0)OH, -(CH 2 ) 0-2 C(O)OR * , -(CH 2 ) 0 - 2 SR * , -(CH 2 ) 0 .
- each R is unsubstituted or where preceded by "halo" is substituted only with one or more halogens, and is independently selected from Ci -4 aliphatic, -CH 2 Ph, -0(CH 2 )o-iPh, or a 5-6-membered saturated, partially unsaturated, or aryl ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur.
- Suitable divalent substituents that are bound to vicinal substitutable carbons of an "optionally substituted” group include: -0(CR 2 ) 2-3 0-, wherein each independent occurrence of R is selected from hydrogen, Cj. 6 aliphatic which may be substituted as defined below, or an unsubstituted 5-6- membered saturated, partially unsaturated, or aryl ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur.
- Suitable substituents on the aliphatic group of R include halogen, -R , -(haloR ), -OH, -OR , -OQialoR * ), -CN, -C(0)OH, -C(0)OR * , -NH 2 , -NHR * , -NR * 2 , or -N0 2 , wherein each R * is unsubstituted or where preceded by "halo" is substituted only with one or more halogens, and is independently Ci -4 aliphatic, -CH 2 Ph, -0(CH 2 )o-iPh, or a 5-6-membered saturated, partially unsaturated, or aryl ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur.
- Suitable substituents on a substitutable nitrogen of an "optionally substituted" group include - R + , -NR + 2, -C(0)R + , -C(0)OR + , -C(0)C(0)R + , -C(0)CH 2 C(0)R + , - S(0) 2 R + , -S(0) 2 NR + 2 , -C(S)NR + 2 , -C(NH)NR + 2, or -N(R + )S(0) 2 R + ; wherein each R + is independently hydrogen, d_ 6 aliphatic which may be substituted as defined below, unsubstituted -OPh, or an unsubstituted 5-6-membered saturated, partially unsaturated, or aryl ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur, or, notwithstanding the definition above, two independent occurrences of R , taken together with their intervening atom(s) form an unsubstituted 3-12-membered saturated, partially uns
- Suitable substituents on the aliphatic group of R * are independently halogen, -R “ , -(haloR ' ), - OH, -OR-, -O(haloR ' ), -CN, -C(0)OH, -C(0)OR ⁇ -NH 2 , -NHR " , -NR ' 2 , or -N0 2 , wherein each R " is unsubstituted or where preceded by "halo” is substituted only with one or more halogens, and is independently Ci -4 aliphatic, -CH 2 Ph, -O(CH 2 ) 0- iPh, or a 5-6-membered saturated, partially unsaturated, or aryl ring having 0-4 heteroatoms independently selected from nitrogen, oxygen, or sulfur.
- leaving group refers to an atom (or a group of atoms) with electron withdrawing ability that can be displaced as a stable species, taking with it the bonding electrons.
- suitable leaving groups include halides - including chloro, bromo, and iodo - and pseudohalides (sulfonate esters) - including triflate, mesylate, tosylate, and brosylate. It is also contemplated that a hydroxyl moiety can be converted into a leaving group via Mitsunobu reaction.
- hydrolysable group and “hydrolysable moiety” refer to a functional group capable of undergoing hydrolysis, e.g., under basic or acidic conditions.
- hydrolysable residues include, without limitation, acid halides, activated carboxylic acids, and various protecting groups known in the art (see, for example, "Protective Groups in Organic Synthesis,” T. W. Greene, P. G. M. Wuts, Wiley-Interscience, 1999).
- organic residue defines a carbon containing residue, i.e., a residue comprising at least one carbon atom, and includes but is not limited to the carbon-containing groups, residues, or radicals defined hereinabove.
- Organic residues can contain various heteroatoms, or be bonded to another molecule through a heteroatom, including oxygen, nitrogen, sulfur, phosphorus, or the like. Examples of organic residues include but are not limited alkyl or substituted alkyls, alkoxy or substituted alkoxy, mono or di-substituted amino, amide groups, etc.
- Organic residues can preferably comprise 1 to 18 carbon atoms, 1 to 15, carbon atoms, 1 to 12 carbon atoms, 1 to 8 carbon atoms, 1 to 6 carbon atoms, or 1 to 4 carbon atoms.
- an organic residue can comprise 2 to 18 carbon atoms, 2 to 15, carbon atoms, 2 to 12 carbon atoms, 2 to 8 carbon atoms, 2 to 4 carbon atoms, or 2 to 4 carbon atoms.
- a very close synonym of the term “residue” is the term "radical,” which as used in the specification and concluding claims, refers to a fragment, group, or substructure of a molecule described herein, regardless of how the molecule is prepared.
- the radical for example an alkyl
- the radical can be further modified (i.e., substituted alkyl) by having bonded thereto one or more "substituent radicals.”
- substituted alkyl i.e., substituted alkyl
- the number of atoms in a given radical is not critical to the present invention unless it is indicated to the contrary elsewhere herein.
- Organic radicals contain one or more carbon atoms.
- An organic radical can have, for example, 1-26 carbon atoms, 1 - 18 carbon atoms, 1 -12 carbon atoms, 1-8 carbon atoms, 1-6 carbon atoms, or 1 -4 carbon atoms.
- an organic radical can have 2-26 carbon atoms, 2-18 carbon atoms, 2- 12 carbon atoms, 2-8 carbon atoms, 2-6 carbon atoms, or 2-4 carbon atoms.
- Organic radicals often have hydrogen bound to at least some of the carbon atoms of the organic radical.
- an organic radical can contain 1 - 10 inorganic heteroatoms bound thereto or therein, including halogens, oxygen, sulfur, nitrogen, phosphorus, and the like.
- organic radicals include but are not limited to an alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, mono-substituted amino, di-substituted amino, acyloxy, cyano, carboxy, carboalkoxy, alkylcarboxamide, substituted alkylcarboxamide, dialkylcarboxamide, substituted dialkylcarboxamide, alkylsulfonyl, alkylsulfinyl, thioalkyl, thiohaloalkyl, alkoxy, substituted alkoxy, haloalkyl, haloalkoxy, aryl, substituted aryl, heteroaryl, heterocyclic, or substituted heterocyclic radicals, wherein the terms are defined elsewhere herein.
- Inorganic radicals contain no carbon atoms and therefore comprise only atoms other than carbon.
- Inorganic radicals comprise bonded combinations of atoms selected from hydrogen, nitrogen, oxygen, silicon, phosphorus, sulfur, selenium, and halogens such as fluorine, chlorine, bromine, and iodine, which can be present individually or bonded together in their chemically stable combinations.
- Inorganic radicals have 10 or fewer, or preferably one to six or one to four inorganic atoms as listed above bonded together. Examples of inorganic radicals include, but not limited to, amino, hydroxy, halogens, nitro, thiol, sulfate, phosphate, and like commonly known inorganic radicals.
- the inorganic radicals do not have bonded therein the metallic elements of the periodic table (such as the alkali metals, alkaline earth metals, transition metals, lanthanide metals, or actinide metals), although such metal ions can sometimes serve as a pharmaceutically acceptable cation for anionic inorganic radicals such as a sulfate, phosphate, or like anionic inorganic radical.
- Inorganic radicals do not comprise metalloids elements such as boron, aluminum, gallium, germanium, arsenic, tin, lead, or tellurium, or the noble gas elements, unless otherwise specifically indicated elsewhere herein.
- a formula with chemical bonds shown only as solid lines and not as wedges or dashed lines contemplates each possible isomer, e.g., each enantiomer and diastereomer, and a mixture of isomers, such as a racemic or scalemic mixture.
- Compounds described herein can contain one or more asymmetric centers and, thus, potentially give rise to diastereomers and optical isomers.
- the present invention includes all such possible diastereomers as well as their racemic mixtures, their substantially pure resolved enantiomers, all possible geometric isomers, and pharmaceutically acceptable salts thereof. Mixtures of stereoisomers, as well as isolated specific stereoisomers, are also included.
- stereoisomers For a given chemical structure, these compounds, called stereoisomers, are identical except that they are non- superimposable mirror images of one another.
- a specific stereoisomer can also be referred to as an enantiomer, and a mixture of such isomers is often called an enantiomeric mixture.
- a 50:50 mixture of enantiomers is referred to as a racemic mixture.
- Many of the compounds described herein can have one or more chiral centers and therefore can exist in different enantiomeric forms. If desired, a chiral carbon can be designated with an asterisk (*).
- bonds to the chiral carbon are depicted as straight lines in the disclosed formulas, it is understood that both the (R) and (S) configurations of the chiral carbon, and hence both enantiomers and mixtures thereof, are embraced within the formula.
- bonds to the chiral carbon when it is desired to specify the absolute configuration about a chiral carbon, one of the bonds to the chiral carbon can be depicted as a wedge (bonds to atoms above the plane) and the other can be depicted as a series or wedge of short parallel lines is (bonds to atoms below the plane).
- the Cahn-Inglod-Prelog system can be used to assign the (R) or (S) configuration to a chiral carbon.
- Compounds described herein comprise atoms in both their natural isotopic abundance and in non-natural abundance.
- the disclosed compounds can be isotopically- labelled or isotopically-substituted compounds identical to those described, but for the fact that one or more atoms are replaced by an atom having an atomic mass or mass number different from the atomic mass or mass number typically found in nature.
- isotopes that can be incorporated into compounds of the invention include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorous, fluorine and chlorine, such as 2 H, 3 H, 13 C, 14 C, 15 N, 16 0, 17 0, 35 S, 18 F and 36 C1, respectively.
- Compounds further comprise prodrugs thereof and pharmaceutically acceptable salts of said compounds or of said prodrugs which contain the aforementioned isotopes and/or other isotopes of other atoms are within the scope of this invention.
- Certain isotopically-labelled compounds of the present invention for example those into which radioactive isotopes such as H and C are incorporated, are useful in drug and/or substrate tissue distribution assays. Tritiated, i.e., 3 H, and carbon-14, i.e., 14 C, isotopes are particularly preferred for their ease of preparation and detectability.
- Isotopically labelled compounds of the present invention and prodrugs thereof can generally be prepared by carrying out the procedures below, by substituting a readily available isotopically labelled reagent for a non- isotopically labelled reagent.
- the compounds described in the invention can be present as a solvate.
- the solvent used to prepare the solvate is an aqueous solution, and the solvate is then often referred to as a hydrate.
- the compounds can be present as a hydrate, which can be obtained, for example, by crystallization from a solvent or from aqueous solution.
- one, two, three or any arbitrary number of solvate or water molecules can combine with the compounds according to the invention to form solvates and hydrates.
- the invention includes all such possible solvates.
- co-crystal means a physical association of two or more molecules which owe their stability through non-covalent interaction.
- One or more components of this molecular complex provide a stable framework in the crystalline lattice.
- the guest molecules are incorporated in the crystalline lattice as anhydrates or solvates, see e.g. "Crystal Engineering of the Composition of Pharmaceutical Phases. Do Pharmaceutical Co- crystals Represent a New Path to Improved Medicines?" Almarasson, O., et. al., The Royal Society of Chemistry, 1889- 1896, 2004. Examples of co-crystals include p- toluenesulfonic acid and benzenesulfonic acid.
- ketones with an a-hydrogen can exist in an equilibrium of the keto form and the enol form.
- amides with an N-hydrogen can exist in equilibrium of the amide form and the imidic acid form.
- the invention includes all such possible tautomers.
- polymorphic forms It is known that chemical substances form solids which are present in different states of order which are termed polymorphic forms or modifications.
- the different modifications of a polymorphic substance can differ greatly in their physical properties.
- the compounds according to the invention can be present in different polymorphic forms, with it being possible for particular modifications to be metastable. Unless stated to the contrary, the invention includes all such possible polymorphic forms.
- a structure of a compound can be represented by a formula:
- n is typically an integer. That is, R n is understood to represent five independent substituents, R n(a) , R n(b) , R n(c) , R n(d) , and R n(e) .
- independent substituents it is meant that each R substituent can be independently defined. For example, if in one instance R n(a) is halogen, then R n(b) is not necessarily halogen in that instance.
- compositions of the invention Disclosed are the components to be used to prepare the compositions of the invention as well as the compositions themselves to be used within the methods disclosed herein.
- these and other materials are disclosed herein, and it is understood that when combinations, subsets, interactions, groups, etc. of these materials are disclosed that while specific reference of each various individual and collective combinations and permutation of these compounds cannot be explicitly disclosed, each is specifically contemplated and described herein. For example, if a particular compound is disclosed and discussed and a number of modifications that can be made to a number of molecules including the compounds are discussed, specifically contemplated is each and every combination and permutation of the compound and the modifications that are possible unless specifically indicated to the contrary.
- the invention relates to compounds useful as inhibitors of STAT3/STAT5.
- the disclosed compounds and products of disclosed methods of making are modulators of STAT3/STAT5 activity.
- the present invention relates to compounds that bind to a STAT3 protein and negatively modulate STAT3 activity.
- the present invention relates to compounds that bind to a STAT5 protein and negatively modulate STAT5 activity.
- the disclosed compounds exhibit inhibition of STAT3/5 activity.
- the compounds of the invention are useful in the treatment of cancer associated with STAT3/STAT5 activity dysfunction, such as breast, prostate or brain cancer and glioblastoma, and other diseases in which a STAT3/5 protein is involved, as further described herein.
- each disclosed derivative can be optionally further substituted. It is also contemplated that any one or more derivative can be optionally omitted from the invention. It is understood that a disclosed compound can be provided by the disclosed methods. It is also understood that the disclosed compounds can be employed in the disclosed methods of using.
- R 1 is selected from A 1 , A 2 , -(A' )-(A 2 ), -(A 2 )-(A 3 ), -(A 3 )-(A 2 ), -(A 3 )-(A 4 ), -(A 5 )-(A')- (A 7 ), -(A 5 )-(A 2 )- (A 8 ), -(A 5 )-(A 3 )-(A 7 ), and -(A 5 )-(A 6 )-L-(A 7 ); wherein A 1 is C 3 .
- a 2 is C 3 - 6 cycloalkyl or heterocycloalkyl, substituted with 0-3 groups selected from halo, hydroxyl, amino, nitro, cyano, C] -6 haloalkyl, C] _6 polyhaloalkyl, Ci -6 alkoxy, Ci- haloalkoxy, Ci -6 polyhaloalkoxy, C).
- a 3 is aryl, and substituted with 0-3 groups selected from halo, hydroxyl, amino, nitro, cyano, Ci -6 haloalkyl, Ci -6 polyhaloalkyl, C]- 6 alkoxy, C]- haloalkoxy, C] -6 polyhaloalkoxy, Ci -6 alkylthio, C] -6 haloalkythio, Ci -6 polyhaloalkylthio, Ci -6 alkylamino, C 1-6 dialkylamino, (Ci -6 )-alk-(Ci-6)-al
- a 7 is selected from C 3-6 cycloalkyl, C 3-6 heterocycloalkyl, and aryl, and substituted with 0-3 groups selected from halo, hydroxyl, amino, nitro, cyano, Ci_ 6 haloalkyl, Cj -6 polyhaloalkyl, Ci-6 alkoxy, Ci_ 6 haloalkoxy, Ci -6 polyhaloalkoxy, Ci -6 alkylthio, C ] - haloalkythio, Cj.
- R 3 is aryl substituted with 0-5 groups independently selected from halo, hydroxyl, amino, nitro, cyano, Ci-6 alkyl, C ] -6 haloalkyl, C] -6 polyhaloalkyl, Ci -6 alkoxy, Ci_ 6 haloalkoxy, Ci -6 polyhaloalkoxy, Ci -6 alkylamino, Ci -6 dialkylamino, (Ci-6)-alk-(Ci_ 6 )-alkoxy, (Ci -6 )-alk-(C !
- R5, R6, R7, R8, R9, RI O, Rl l , and R n is independently selected from hydrogen, C 1 -6 alkyl, C 1-6 haloalkyl, and Ci-6 polyhaloalkyl; or a pharmaceutically acceptable salt, hydrate, solvate, or polymorph thereof, wherein R 2 is selected from the group consisting of:
- R 3 is selected from the structure represented by formula:
- R 13 is selected from the group consisting of -OH, -COR 16 , -CN, -CH 2 PO(OH) 2 , -CH 2 P(0) 3 (CH 2 CH 3 ) 2 , -NO 2 , -NHR 17 , and lH-tetrazole;
- R 16 is selected from the group consisting of: -OH, -0-C ()-2) alkyl, -OCH 2 OC(0)CH 3 , -OCH 2 OC(0)t-Butyl, and -NIIOH,
- R 17 is selected from the group consisting of: -H, -C(0)C(0)CH 2 CH 3 , -C(0)C(0)OH, and -C(0)CH 2 -lH-tetrazole
- R 14 is -H or when R 13 is -COOH, R 14 is -F, -OC(0)CH 3 ;
- R 13 is -H when R 14 is -OH or both R 14 and R 15 are -COOH.
- alkyl 39-40
- acetoxymethyl AOM
- pivaloyloxymethyl POM
- acetylated prodrug 22, 23
- a phosphotyrosine-like phosphonate derivative of 1 was prepared, 21 and the prodrug analog, 20.
- the relative ring positions of the hydroxy- and carboxylate groups were inversed (38), the hydroxyl substituent replaced with a fluoride (33, 43) or removed entirely and replaced with a hydrogen atom (31, 42).
- the electronegative fluorinated analogs, 33, 43 were prepared to lower inhibitor polarity by reducing the charge on carboxylate and thus, for improving cell permeability. As an additional benefit, the hydroxyl group deletion or replacement with fluorine would preclude phase II glucoronidation.
- an N-hydroxyl amino (32, 44), sulfonamide (12- 15), sulfonamine (16), tetrazole (35, 41) and N-hydoxy-oxamic acid (28, 46) derivatives were prepared.
- Selected prodrug analogs of the bioisosteres were also prepared (17-18).
- Table 1 A focused library of inhibitors consisting of prodrugs and bioisosteres of the salicylic acid functionality
- the library was screened for biological activity against a set of three representative GBM BTSCs derived from GBM patients, including the very aggressive GBM BTSC lines, BTSC73M, BTSC30M and BTSC68EF.
- Dissociated BTSC spheres were seeded at 1500 cells/ 96-well and treated with drug compound or vehicle (DMSO) one day after plating. Cell viability following drug treatment was assessed after 72 hrs using the alamarBlueTM assay (Invitrogen) according to the manufacturer's instructions. All culture experiments were performed in triplicate with a minimum of three wells per condition.
- IC50 values obtained from the results of these experiments were compared to BP-1-102 compound (also referred in these experiments as compound 1), and Cucurbitacin, a JAK2 inhibitor and the most potent inhibitor of BTSCs to date, as references (Table 2).
- the top four ranked compounds, 22, 31, 32, and 33, which displayed IC50 values ranging from 100 nM to 3.8 ⁇ were further investigated against BTSCs, 25M, 67EF, 73EF, 84EF and 127EF which represent the molecular heterogeneity of human GBMs (Figs. 1A and IB).
- BTSCs 25M, 67EF, 73EF, 84EF and 127EF which represent the molecular heterogeneity of human GBMs
- BTSCs Across, all 8 BTSCs evaluated, compounds 22 and 31-33 showed remarkable activity, with EC50S ranging from 66-1 145 nM.
- compound 31, equipped with a benzoic acid substituent exhibited significantly higher potency against a larger number of BTSCs.
- nM cytoxicity Given the nM cytoxicity, these compounds were assayed for biological effects in healthy cells.
- a series of normal human fetal astrocytes were subjected to top three inhibitors at concentrations up to 5 ⁇ (10-20 fold higher than reported IC 5 os against BTSCs).
- compounds 22, 31 and 32 showed minimal toxicity at concentrations of up to 5 ⁇ in normal human cells, identifying a clear therapeutic window for these agents in GBM.
- compound 31 represents the most potent, non-phosphorylated STAT3 inhibitor reported to-date, with low nM K D values against STAT3 protein.
- Studies reported on Fig. 2B were done at multiple concentrations, i.e. 5.000, 1.667, 0.556, and 0.185 ⁇ .
- BTSC spheres were dissociated to single cells and 1 x 10 6 cells were treated with drug or vehicle (DMSO) at different concentrations and time points. 15 g of protein were loaded on 7.5% or 10% SDS- PAGE gels and trans-blotted to nitrocellulose membranes.
- BTSCs were treated to varying concentrations of lead inhibitors (0, 0.1 , 0.5, 1.0 and 5.0 ⁇ ) and lysates subjected to Western Blot analysis.
- lead inhibitors (0, 0.1 , 0.5, 1.0 and 5.0 ⁇ ) and lysates subjected to Western Blot analysis.
- dose-dependent decreases in pSTAT3 levelSAvere observed, as well as potent inhibition of downstream targets involved in cell growth and survival, Cyclin Dl and Bcl-xL
- compound 31 the most potent compound against BTSCs, exhibited concentration-dependant decreases in pSTAT3 levels that correlated well with observed cytotoxicity and downstream target suppression.
- treatment with compound 31 at 500 nM completely silenced pSTAT3 signalling.
- Compounds 22 and 32 whilst slightly less potent, still exhibited nM inhibition of pSTAT3.
- kinome screen 101 diverse kinases, DiscoveRxTM KINOMEscan
- protein and receptor screen 21 biologically important G protein-coupled receptors (GPCRs)
- kinome screening kinases labelled with DNA were treated with compound 31 (500 nM) and incubated with an immobilized ligand designed to capture the target kinase.
- Ultra-sensitive quantitative PCR qPCR was used to measure levels of immobilized kinases after treatment with compound 31. The immobilized kinase levels were compared to control samples and hits were designated when the quantity of the seized kinase fell below a 35 % threshold.
- the GPCR screening employed the widely applied PathHunter ⁇ -arrestin GPCR assay platform (DiscoveRx) to evaluate the activity of compound 31.
- the GPCRs are widely regarded as a most important family of membrane receptors, due to their master regulatory role in intracellular signal transduction. As a result, GPCRs are among the most heavily investigated drug targets in the pharmaceutical industry. Encouragingly, compound 32, showed no off- target activity against any of the 21 GPCRs tested (500 nM). Moreover, compound 31 at same concentration (500 nM), showed negligible effects against the representative family of kinases including a large number of both SH2 (JaKl and JaK2) and SH3 (Fes, Fer, Fyn) domain-containing kinases.
- compound 31 exhibited unprecedented cytotoxicity across a range of human BTSCs, exhibited no toxicity in human fetal astrocytes at concentrations 15-20-fold higher than IC50S, potently suppressed pSTAT3 at nM concentrations correlating well with in vitro nM K-Q values (SPR), inhibited STAT3's downstream targets and showed no discernible off-target effects at therapeutic doses as assessed by kinome and GPCR screens.
- STAT3 SH2 domain inhibitors have been synthesized and tested herein.
- their application in GBM BTSCs recently reported to harbor high levels of hyperactivated STAT3, which critically, has been shown to be the major driver in brain cancer tumourogenesis and BTSC drug resistance, has been validated.
- the inhibitors presented herein effectively suppress STAT3 phosphorylation and its downstream protein targets (Cyclin Dl and Bcl-xL).
- the potential of STAT3 inhibitors have now been clearly demonstrated for the first time for the treatment of BTSCs and its clinical efficacy for GBM clinical application.
- the amount of a compound of the invention required for use in treatment will vary not only with the particular compound selected but also with the route of administration, the nature of the condition for which treatment is required and the age and condition of the patient and will be ultimately at the discretion of the attendant physician.
- the amount administered will be empirically determined, typically in the range of about 10 ⁇ g to 100 mg/kg body weight of the recipient.
- compositions include, without limitation, those suitable for oral, (including buccal and sub-lingual), transdermal, or parenteral (including intramuscular, sub-cutaneous and intravenous) administration or in a form suitable for administration by inhalation.
- compositions suitable for oral administration may conveniently be presented as discrete units such as capsules, cachets or tablets each containing a predetermined amount of the active ingredient; as a powder or granules; as a solution, a suspension or as an emulsion. Tablets and capsules for oral administration may contain conventional excipients such as binding agents, fillers, lubricants, disintegrants, or wetting agents.
- Oral liquid preparations may be in the form of, for example, aqueous or oily suspensions, solutions, emulsions, syrups or elixirs, or may be presented as a dry product for constitution with water or other suitable vehicle before use.
- Such liquid preparations may contain conventional additives such as suspending agents, emulsifying agents, non-aqueous vehicles (which may include edible oils), or preservatives.
- the compounds and combinations as defined herein may also be formulated for parenteral administration (e.g.
- compositions may take such forms as suspensions, solutions, or emulsions in oily or aqueous vehicles, and may contain formulatory agents such as suspending, stabilizing and/or dispersing agents.
- the active ingredient may be in powder form, obtained by aseptic isolation of sterile solid or by lyophilisation from solution, for constitution with a suitable vehicle, e.g. sterile water or saline, before use.
- compositions suitable for topical administration in the mouth include lozenges comprising the active ingredient in a flavoured base, usually sucrose and acacia or tragacanth; pastilles comprising the active ingredient in an inert base such as gelatin and glycerin or sucrose and acacia; and mouthwashes comprising the active ingredient in a suitable liquid carrier.
- the compounds and combinations as defined herein may take the form of a dry powder composition, for example a powder mix of the compound and a suitable powder base such as lactose or starch.
- the powder composition may be presented in unit dosage form in, for example, capsules or cartridges or e.g. gelatin or blister packs from which the powder may be administered with the aid of an inhalator or insufflator.
- the compounds as defined herein may include a chiral center which gives rise to enantiomers.
- the compounds may thus exist in the form of two different optical isomers, that is (+) or (-) enantiomers. All such enantiomers and mixtures thereof, including racemic or other ratio mixtures of individual enantiomers, are included within the scope of the invention.
- the single enantiomer can be obtained by methods well known to those of ordinary skill in the art, such as chiral HPLC, enzymatic resolution and chiral auxiliary derivatization.
- the compounds in accordance with the present disclosure can contain more than one chiral centre.
- the compounds of the present invention may thus exist in the form of different diastereomers. All such diastereomers and mixtures thereof are included within the scope of the invention.
- the single diastereomer can be obtained by methods well known in the art, such as HPLC, crystalisation and chromatography.
- Solvate means that a compound as defined herein incorporates one or more pharmaceutically acceptable solvents including water to give rise to hydrates.
- the solvate may contain one or more molecules of solvent per molecule of compound or may contain one or more molecules of compound per molecule of solvent.
- Illustrative non-limiting examples of hydrates include monohydrate, dihydrate, trihydrate and tetrahydrate or semi-hydrate.
- the solvent may be held in the crystal in various ways and thus, the solvent molecule may occupy lattice positions in the crystal, or they may form bonds with salts of the compounds as described herein.
- the solvate(s) must be "acceptable” in the sense of not being deleterious to the recipient thereof. The solvation may be assessed by methods known in the art such as Loss on Drying techniques (LOD).
- LOD Loss on Drying techniques
- HMQC Heteronuclear multiple quantum coherence mCPBA meto-chloroperbenzoic acid
- the compounds of the present disclosure can be prepared according to the procedures denoted in the following reaction Schemes 1 and 2 and Examples or modifications thereof using readily available starting materials, reagents, and conventional procedures or variations thereof well-known to a practitioner of ordinary skill in the art of synthetic organic chemistry. Specific definitions of variables in the Schemes are given for illustrative purposes only and are not intended to limit the procedures described.
- TsCl para- toluenesulfonyl chloride
- DIPEA N,N-diisopropylethylamine
- DMF N,N- dimethylformamide
- TFA Trifluoroacetic acid
- (COCl) 2 oxalyl chloride
- THF tetrahydrofuran, LiOH H20, lithium hydroxide hydrate.
- Ligand purity was confirmed using linear gradients from 50 % A and 50 % B to 100 % B after an initial 2 minute period of 100 % A, and a second linear gradient of 100% A to 100% B.
- the linear gradient consisted of a changing solvent composition of either (I) 5.2 % per minute and UV detection at 254nm or (II) 1.8 % per minute and detection at 254nm, each ending with 5 minutes of 100% B.
- retention times for each condition are written followed by their purities in their respective order.
- Biologically evaluated compounds are > 95 % chemical purity as measured by HPLC. Traces of the HPLC results are provided in supporting information.
- Example 76 Kinase and GPCR Screenings Test compound was evaluated at a single dose of 5 ⁇ in duplicate mode. Control compound was tested in a 10-dose IC50 with 3-fold serial dilution starting at 20 ⁇ . Reactions were carried out at 10 ⁇ ATP.
- Preliminary screen against AKT, c-Src, ERKl , JaKl and JaK2 was performed at Reaction Biology Corporation (www.reactionbiology.com, Malvern, PA) using the "HotSpot” assay platform.
- Specific kinase/substrate pairs along with their required cofactors were prepared in reaction buffer; 20 raM Hepes pH 7.5, 10 niM MgCl 2 , 1 mM EGTA, 0.02% Brij35, 0.02 mg/ml BSA, 0.1 raM Na 3 V0 4 , 2 mM DTT, 1 % DMSO.
- the screen was completed in duplicate at a single concentration of 5 ⁇ .
- reaction well with the subject compound was treated with a mixture of ATP (Sigma, St. Louis MO) and P ATP (Perkin Elmer, Waltham MA) to a final concentration of ⁇ ⁇ .
- Control compound Staurosporine
- P81 ion exchange filter paper Whatman Inc., Piscataway, NJ. Any unbound phosphate was removed through extensive washing of filters using 0.75% phosphoric acid.
- kinase activity data was expressed as the percent remaining kinase activity in test samples compared to vehicle (dimethyl sulfoxide) reactions. All IC 5 o values (table 3) and curve fitting were produced using Prism (GraphPad Software) (Fig. 5).
- Second screen against 101 selected kinases was performed. The test compound was evaluated at a single dose of 500 nM.
- qPCR quantitative PCR
- Magnetic beads coated with Streptavidin were treated with biotinylated small molecule ligands for 30 minutes at room temperature to generate the affinity resins for needed kinase assays.
- the liganded beads were blocked with excess biotin and washed with blocking buffer (SeaBlock (Pierce), 1 % BSA, 0.05 % Tween 20, 1 mM DTT) in order to remove unbound ligand and to also reduce non-specific phage binding.
- Binding reactions were assembled by combining kinases, liganded affinity beads, and 31 in lx binding buffer (20 % SeaBlock, 0.17x PBS, 0.05 % Tween 20, 6 mM DTT).
- Compound 31 was prepared as 40x stocks in 100% DMSO and was directly diluted into the assay. All reactions were performed in polypropylene 384-well plates in a final volume of 40 ⁇ . The assay plates were shaked at room temperature for 1 hour and the affinity beads were washed with wash buffer (lx PBS, 0.05 % Tween 20). Following the wash, beads were re-suspended in elution buffer (lx PBS, 0.05 % Tween 20, 0.5 ⁇ non-biotinylated affinity ligand) and incubated at room temperature with shaking for 30 minutes. Kinase concentration in eluates was measured by qPCR (Fig. 6).
- BTSC spheres were dissociated to single cells with the enzyme Accumax (Innovative Technologies), seeded at 1500 cells/ 96-well and treated with drug or vehicle (DMSO) one day after plating. Cytotoxicity studies were repeated independently at Sickkids Hospital using BTSC lines 25M, 67EF, 73EF, 84EF and 127EF. BTSC spheres were dissociated to single cells as above and plated in 96 well plates in triplicate at 3000 cells/ 96-well. In both sets of experiments drugs were used as serial dilutions within the range of 5uM to 100 nM in the first set and 25 uM to 10 nM.
- Table 4 screen of the library against a number of BTSCs using alamarBlueTM assay
- BTSC spheres were lysed in modified RIPA buffer supplemented with Complete Mini protease (Roche) and Halt phosphatase (Thermo Scientific) inhibitor cocktails.
- BTSC spheres were dissociated to single cells and 1 x 10 6 cells were treated with drug or vehicle (DMSO) for 2, 24 or 72 hours. 15 ⁇ g of protein were loaded on 7.5% or 10% SDS-PAGE gels and transblotted to nitrocellulose membranes.
- Blots were stained with the following antibodies: phospho-STAT3 Y705 (1 : 1000, Cell Signalling Technology), phospho-STAT3 S727 (1 : 1000, Cell Signalling Technology), STAT3 (1 : 1000, Santa Cruz Biotechnology), Bcl-xL (1 : 1000, Cell Signalling Technology), cyclin Dl (1 : 1000, Cell Signalling Technology) PARP (1 : 1000, Cell Signalling Technology) and Actin (1 :2500, Santa Cruz Biotechnology).
- HRP conjugated secondary antibodies, goat anti-rabbit (Cell Signalling Technology) were used at 1 :2000 and donkey anti-mouse, donkey anti-goat, and goat anti-rabbit, (Calbiochem) were used at 1 :6000. Bands were visualized with the ECL Plus Western Blotting Detection System and Hyperfilm (Amersham).
- GBM BTSCs were cultured from a series of tumor specimens obtained following informed consent from adult GBM patients during their operative procedure as previously described (Kelly et al, Stem Cells 2009) and approved by the University of Calgary Ethics Review Board. Briefly, BTSC cultures were initiated in serum-free defined culture medium (SFM) and gave rise to nonadherent spheres after 7 to 21 days in culture. Primary tumor spheres were expanded for several passages and then cryopreserved in ⁇ 0% DMSO (Sigma Aldrich) in serum-free defined medium until use in experiments. Human fetal neural stem cells were also cultured as previously described and induced to differentiate into astrocytes by addition of 10% fetal bovine serum and removal of EGF, FGF-2 and heparan sulfate.
- SFM serum-free defined culture medium
- Human fetal neural stem cells were also cultured as previously described and induced to differentiate into astrocytes by addition of 10% fetal bovine serum and removal of EGF, FGF-2
- Fluorescence Polarization Assay STAT3, STAT5 and STAT1 The fluorescence polarization studies were performed on an Infinite M l 000 (Tecan, Crailsheim, Germany) machine using black 384-round bottom well plates (Corning).
- the buffer used contained 50 mM NaCl, 10 mM Hepes, pH 7.5, ImM EDTA and 2 mM dithiothreitol and a final concentration of 5% DMSO.
- Test compounds were dissolved in DMSO and diluted using a dilution medium consisting of 50mM NaCl, l OmM Hepes, pH 7.5, ImM EDTA, and 2mM dithiothreitol.
- Fluorescently- labelled pYLPQTV peptide (FAM-pYLPQTV) was kept at a final concentration of ⁇ ⁇ in ⁇ the buffered solution.
- Non-labelled pYLPQTV peptide was dissolved in DMSO and diluted using the dilution medium for use as a positive control.
- a calibration curve was performed using ⁇ ⁇ Fam-pYLPQTV and dilutions of STAT3 and STAT1 protein (5.0 ⁇ , to 2.4 nM) at a final DMSO concentration of 10% (Fig. 7).
- the midpoint of the saturation curve was used to determine the STAT3 concentration required for the competitive fluorescent polarization assay.
- 7.5 ⁇ ⁇ of FAM-pYLPQTV peptides was added to the already plated 15 ⁇ , of a 750 nM STAT3 protein solution in 384- flat well plates.
- 7.5 ⁇ , of inhibitory molecules (and positive control) solution were individually added to the plate.
- the final inhibitory concentrations were 100 ⁇ , 50 ⁇ , 25 ⁇ ,, 12.5 ⁇ ,, 6.3 ⁇ ,, or 3.1 uL.
- the 10% DMSO buffer solution was incubated for 15-30 minutes before signal measurements by the Infinite M1000. Results for compounds 22, 3 1 , 32, and 33 are illustrated in Fig. 8.
- SPR Surface Plasmon Resonance
- the IC 50 for compounds 22, 31 , 32, 23, et 33 (also referred to in the table as SH-04-08, SH-04-54, SH-05-07, SH-05-19, AND SH-05-23, respectively) was determined against other STAT3 dependent cancers such as Breast cancer cells (MDA-MB-468), Prostate (PPCl or DU145) and leukemia cancer cells (AML-2) in parallel with BTSC 30M. The results are reported in Table 6 below.
- Fig. 9 illustrates the potent biological effects in a patient-derived pancreatic cancer cell line of the STAT3 inhibitors tested.
- ED 50 (IC 50 ) values calculated for compounds 31, 41 , 42, and 38 are respectively of 6.2, 16.4, 16.7, and 4.8 ⁇ .
- Low passage patient-derived pancreatic cancer lines (Pa03C and Panel 0.05) were plated in 6- well plates (2xl0 5 and 8xl0 4 , respectively) and allowed to attach overnight.
- STAT3 inhibitors were added for 1 hr prior to IL-6 stimulation (50ng/mL, 15 min). STAT3 inhibitors were present during IL-6 stimulation.
- Cells were harvested and lysed for Western blotting.
- phospho-Specific STAT3 antibody (Y705) was used to look at STAT3 phosphorylation and total STAT3 antibody was used to normalize activation of STAT3.
- Antibodies were from Cell Signaling and used at a 1 : 1000 dilution.
- Fig. 1 1 illustrates the effects of STAT3 inhibitor 31 on low passage patient-derived pancreatic cancer line (Pa03C) plated in a 96-well plate coated with 1% Noble Agar in media containing 3% Matrigel, with and without cancer-associated fibroblasts.
- the ratio of the tumor to CAFs is 1 :4.
- Tumor spheres begin to form around Day 3, and STAT3 inhibitor 31 added at Day 4 and 8. Cytotoxicity is assessed by fluorescence using Alamar Blue assay on Day 1 1.
- HMCL cells were seeded in 96-well plates in triplicate at a density of 20,000 cells/well and incubated with increasing concentrations of 16i and 21h or DMSO control and cell viability assessed by MTT (3-(4,5-dimethylthiazol)-2,5-diphenyl tetrazolium) colorimetric analysis. Results are reported at Fig. 12.
- Fig. 12 illustrates compound 31 potently reducing cell viability in 12 of 15 different multiple myeloma tumor cell lines as assessed by MTT assay with calculated EDso's ranging from between 2-6 ⁇ .
- BTSC spheres were dissociated with Accumax (eBioscience) and plated at 10 6 cells/1.5 mL of media. Cells were treated with vehicle (DMSO) or drug and pelleted at select time points.
- DMSO vehicle
- BTICs were lysed in modified RIPA buffer (50 mM Tris, 150 mM NaCl, 0.1% SDS, 0.5% Na Deoxycholate, 1% NP-40) supplemented with Complete Mini protease (Roche) and Halt phosphatase (Thermo Scientific) inhibitor cocktails.
- Protein concentrations were quantified using the BioRad protein assay; 1 1 -15 ⁇ g of protein were loaded on 7.5% or 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS PAGE) gels and transblotted to nitrocellulose membranes. Blots were stained with primary antibodies followed by horseradish peroxidase (HRP)-conjugated secondary antibodies.
- HRP horseradish peroxidase
- mice Seven days following implants, mice were randomized into vehicle and drug cohorts and injected i.p., four days on, three days off, for a total of 10 doses.
- Drug-treated mice received a 10 mg/kg dose of compound 31 re-suspended in 100 ⁇ . 50% polyethylene glycol 300 (PEG 300, Sigma-Aldrich) in water. Vehicle-treated mice received an equal volume of 50% PEG 300 in water. Mice were euthanized with a lethal dose of sodium pentobarbitol, followed by transcardiac perfusion of 4% paraformaldehyde (PFA) two hours following injection of the last drug dose.
- PEG 300 polyethylene glycol 300
- PFA paraformaldehyde
- H&E Hematoxylin and eosin staining was performed according to standard protocols. Immunohistochemistry was performed by incubating with primary antibodies overnight at 4 °C followed by the Vectastain Elite mouse IgG or rabbit IgG kits (Vector Laboratories) and detection with DAB substrate and hematoxylin counterstaining (Sigma-Aldrich) according to the manufacturer's instructions. Primary antibodies included - STAT3 S727 (Cell Signaling Technology) and Ki67 (Novocastra).
- Fig. 13 illustrating sections of NOD-SCID mouse brains showing tumours from BT73 cells in control mice and in mice treated with inhibitor 31. Treatment with compound 31 resulted in significant decrease in tumour cells.
- Fig. 14 illustrates the effect of compound 31 for decreasing pSTAT expression in mice orthotopically xenografted with BT73 brain cancer cells. As can be seen from Fig. 14, compound 31 displays on target effectiveness at reducing activated phospho-STAT as demonstrated by reduced immunohistochemical staining in treated tumours. (A) vehicle treated and (B) inhibitor 31 treated brains. Bottom panels are higher magnification of tumour regions in top panels.
- Fig. 15 illustrates the effect of compound 31 for reducing proliferation in BT73 brain tumours.
- Decreased expression of proliferation marker Ki67 is observed in mice treated with inhibitor 31 (A) vehicle treated and (B) inhibitor 31 treated brains. Bottom panels are higher magnification of tumour regions in top panels.
- Fig. 16 illustrates an increased apoptosis (TUNEL staining) in mice treated with compound 31.
- Increased TUNEL staining demonstrating apoptic cell death is observed in the tumours of mice treated with compound 31.
- Panel (A) is vehicle treated and panel (B) is compound 31 treated brains. Bottom panels are higher magnification of tumour regions in top panels.
- compound 31 is effective at reducing activated phospho- STAT3 in tumours generated by orthotopically xenografted human brain tumour stem cells in the brains of NOD-SCID mice, accompanied by a decrease in proliferation and increased cell death.
- the other compounds of the present invention are anticipated to have the same properties. In fact, other tests as reported hereinbefore have shown that compound 31 behave like other compounds tested.
- compound 31 was used to demonstrate the general behavior of the compounds of the present invention.
- compound 31 was found to be present in the brain with different dosing (lOmg/kg and 25 mg/kg) as assessed by LCMS.
- Compound 31 accumulates up to 0.7 ⁇ concentrations in the brain in mice models.
- compound 31 accumulates up to 300 nM concentrations in the brain in mice models at 10 mg/kg dosing.
- compound 32 accumulates up to -1.2 ⁇ concentrations in the brain in mice models at 10 mg/kg dosing.
- Plasma concentration of compound 31 increased dose dependently from 0.24 ⁇ to 0.58 ⁇ for 10 and 25 mg/kg, respectively. However, no compound was detected after 5 hr at both doses indicating a fast clearance. Plasma concentration did not change significantly between 30 min. and 300 min for both compounds 31 and 32.
- about 0.3 ⁇ compound was seen in the brain of 10 mg/kg dosed mouse.
- the brain concentration of compound 32 was at -1.2 ⁇ for 10 mg/kg dosed mouse. The above is a clear indication that the compounds of the present invention can cross the blood brai barrier.
- NIC ErbB2 mammary breast tumour cell lines
- NIC Mouse Mammary Tumour Virus ErbB2 mammary tumour cell lines
- NIC Three individual Mouse Mammary Tumour Virus ErbB2 mammary tumour cell lines (NIC) were resuspended in 10% FBS plus single quots DMEM media to a final concentration of 40,000 cells per lmL.
- Cells were plated at a concentration of 4,000 cells in ⁇ ⁇ of media per well on 96 well NUNC plates and incubated for 24h hours at 37°C plus 5% C0 2 . After 24 hours the media was aspirated and replaced with either normal media, media + DMSO (1 : 1000) or media containing the appropriate concentration of drug (BPl -102 5 ⁇ , 10 ⁇ or 20 ⁇ or compound 31 , also referred to herein as SH-04-54 5 ⁇ , 10 ⁇ or 20 ⁇ ). Each condition was repeated in quadruplicate. At predetermined time points; at treatment, 24h, 48h and 72h post treatment, the media was removed from the cells and the plates were stored at -
- the Cell lysis buffer was diluted 1 :20 with water and then the CyQuant GR solution 1 :400 in the lysis buffer. 200 ⁇ 1, of the Cyquant solution was added to each well and the plate protected from light. The plates were then incubated for 2-5 minutes protected from light, then read on a fluorescence microplate reader with filters for 480nm excitation and 520nm emission maxima. Quadruplicates were averaged and the error bars represent the S.E.M. In some cases the absorbance was normalized to the DMSO control. In Fig.
- Fig. 21 cells were plated at 4,000 cells per well in a 96 well plate, incubated for 24 hours then treated with BPl-102 or compound 31 at 5 ⁇ for 24, 48 and 72 hours. Plates were stored at -80°C. Plates were thawed and treated with Invitrogen's Cyquant Cell Proliferation Assay which measures DNA content. Fluorescence was measured using a fluorescence microplate reader with filters at 480nm and 520nm. Each data point was done in quadruplicate and error bars represent S.E.M.
- compound 31 is potent at 5 ⁇ in MMTV ErbB2 mammary tumour cell lines (NIC), completely inhibiting cell proliferation in 2071 NIC cells.
- NIC ErbB2 mammary tumour cell lines
- compound 31 was shown to be more potent over three different breast cancer NIC cell lines, 2071, 8518 and 9822.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Public Health (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Oncology (AREA)
- Hematology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Quinoline Compounds (AREA)
Abstract
Description













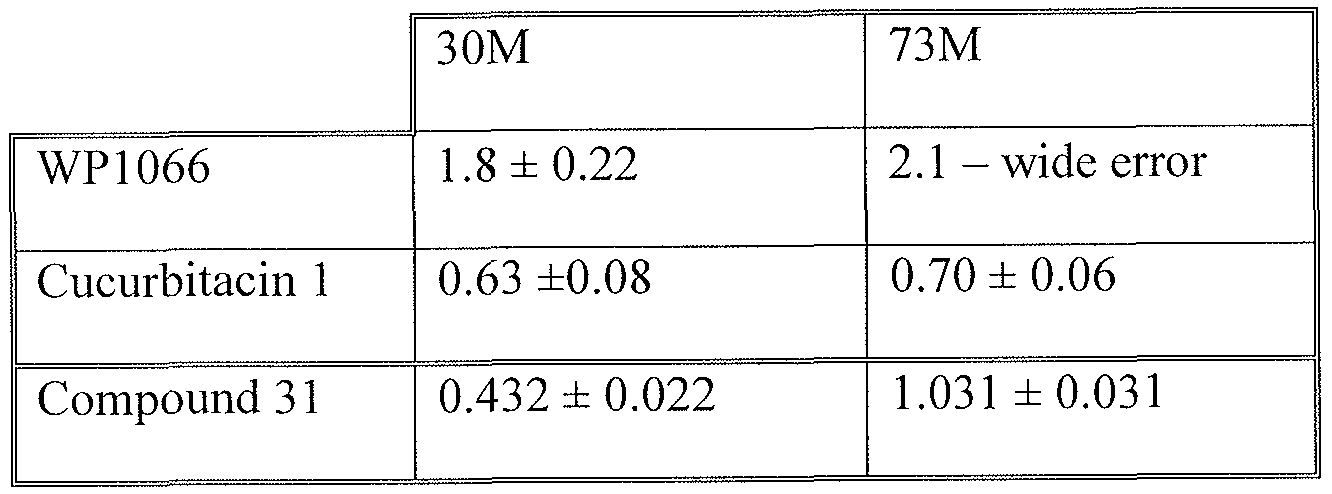
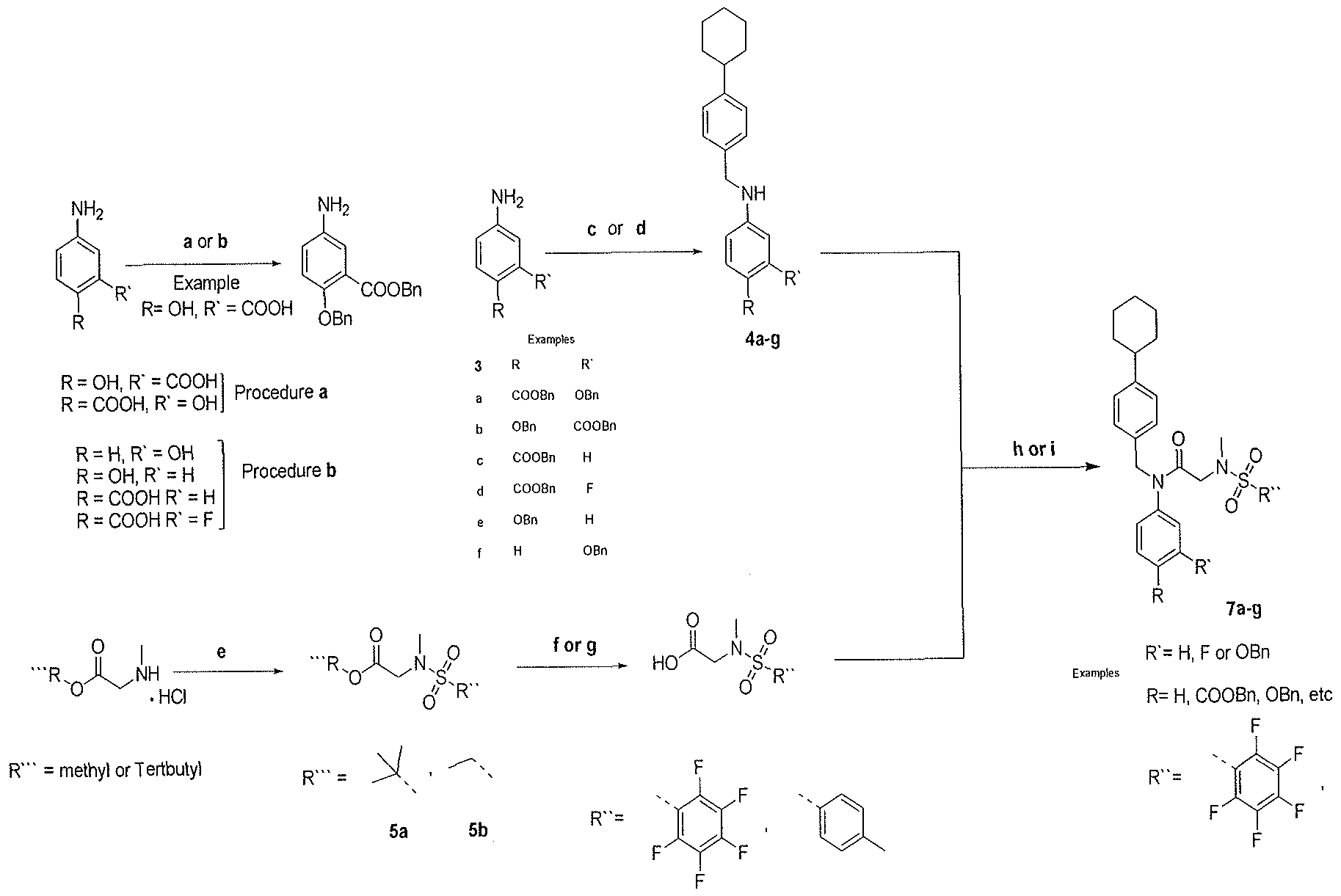






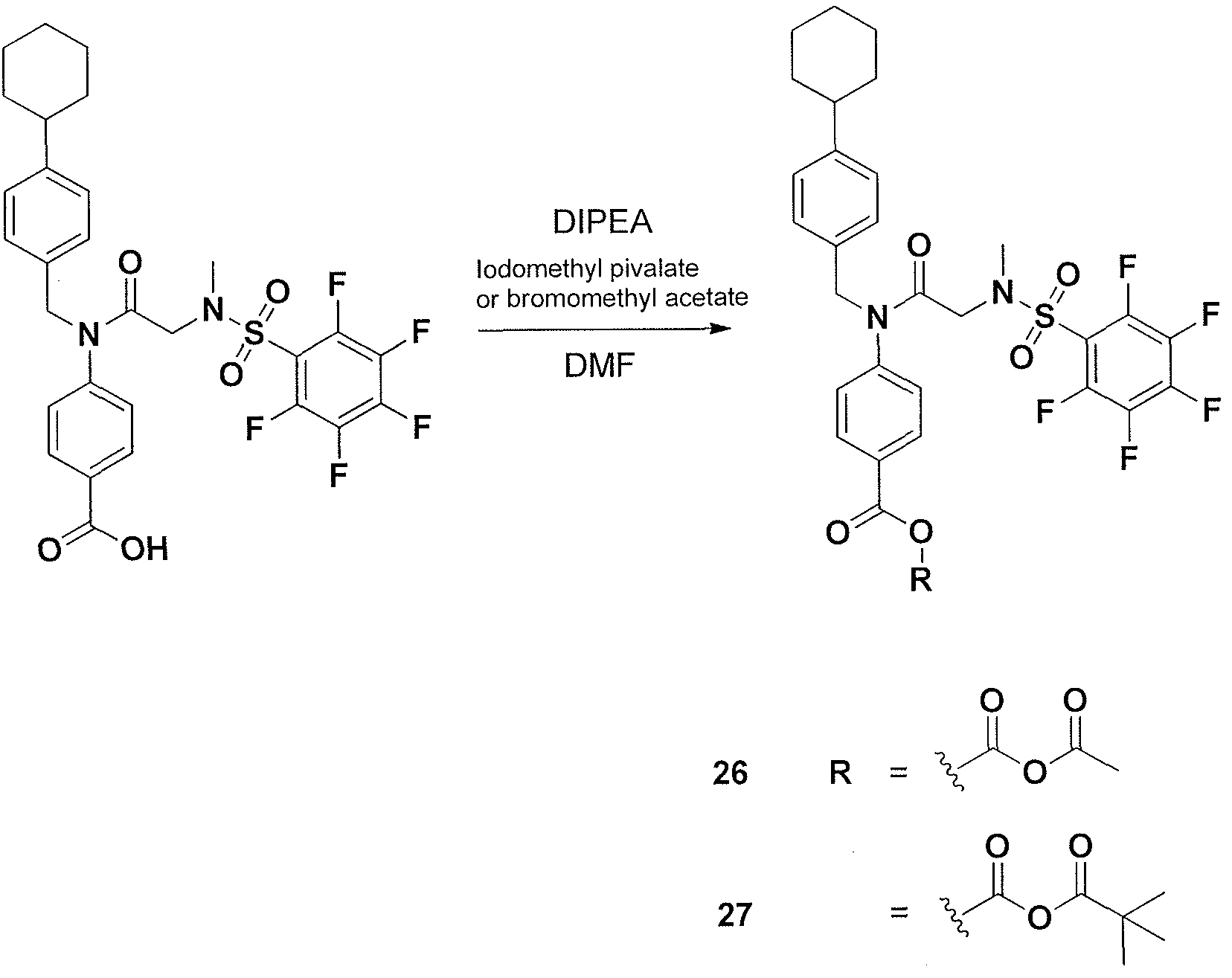

















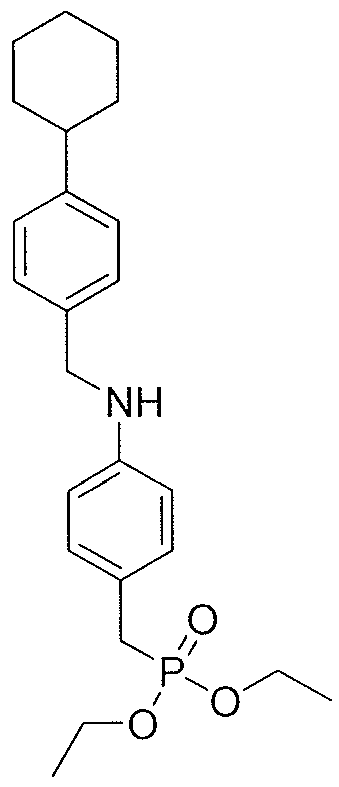







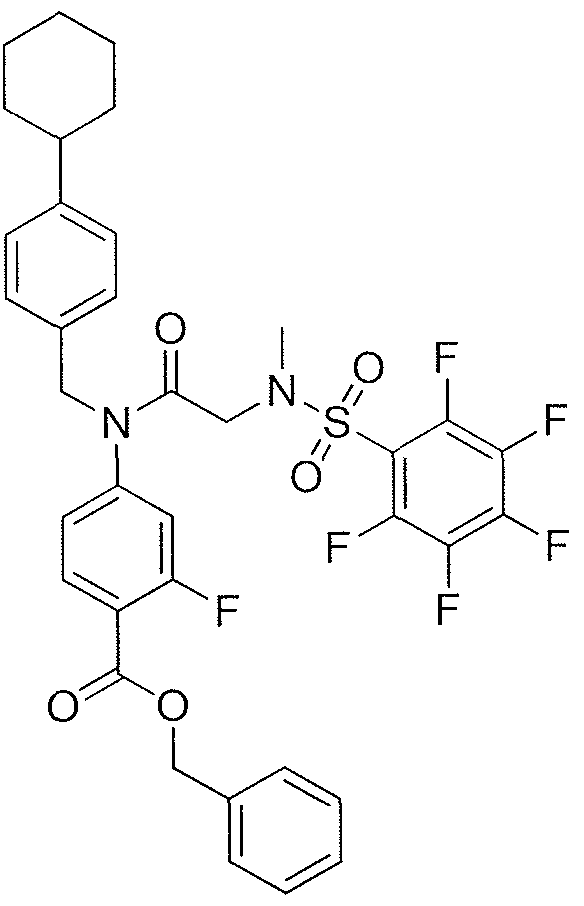











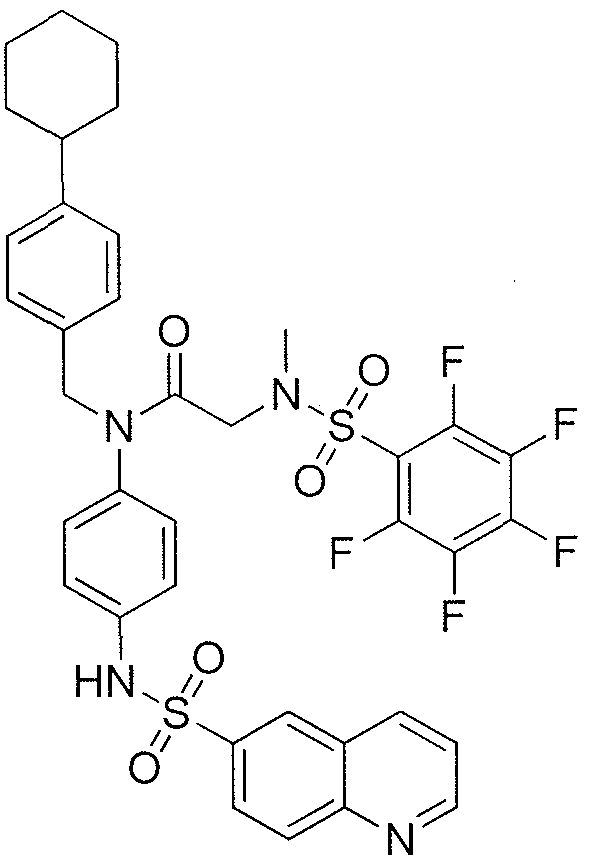



















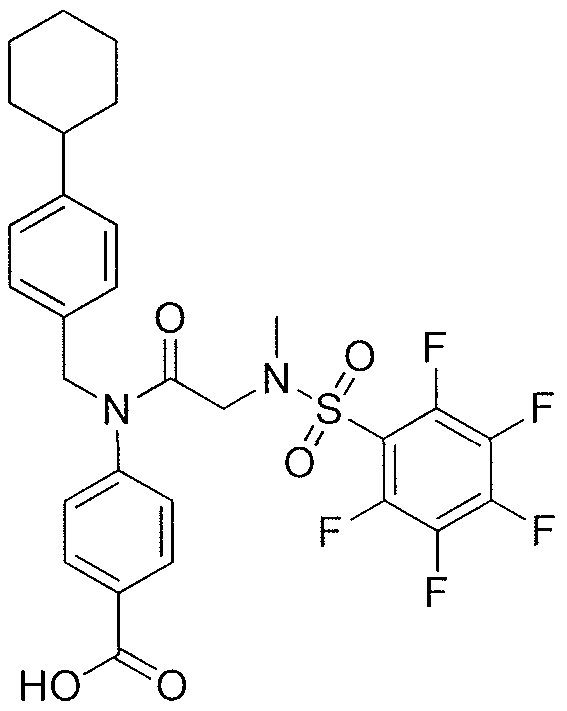





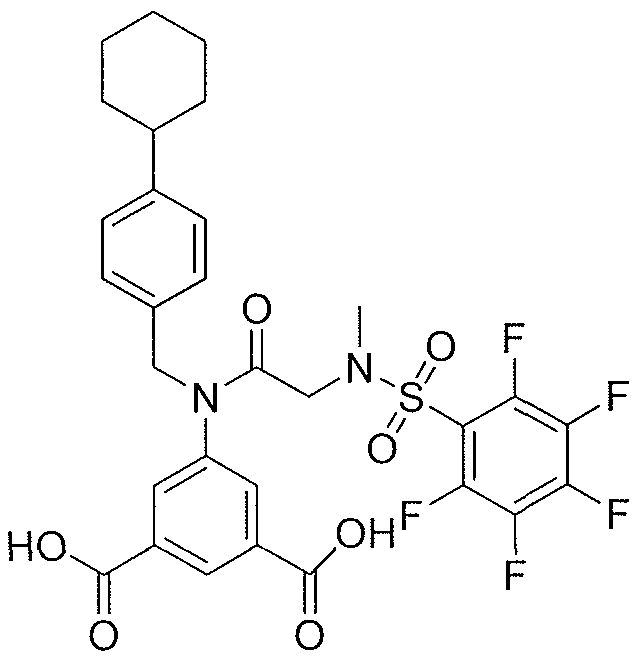















Claims




Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| RU2014152697A RU2641903C2 (en) | 2012-05-25 | 2013-05-24 | New salicylic acid derivatives, their pharmaceutically acceptable salt, compositions and method for application |
| EP13794722.2A EP2854819B1 (en) | 2012-05-25 | 2013-05-24 | Salicylic acid derivatives, pharmaceutically acceptable salt thereof, composition thereof and medical use thereof |
| JP2015514226A JP6290871B2 (en) | 2012-05-25 | 2013-05-24 | New salicylic acid derivatives, pharmaceutically acceptable salts thereof, compositions thereof and methods of use thereof |
| CN201380038486.1A CN105120854B (en) | 2012-05-25 | 2013-05-24 | New salicyclic acid derivatives, its pharmaceutically acceptable salt, its composition and its application method |
| CA2874057A CA2874057A1 (en) | 2012-05-25 | 2013-05-24 | New salicylic acid derivatives, pharmaceutically acceptable salt thereof, composition thereof and method of use thereof |
| US14/550,293 US9650399B2 (en) | 2012-05-25 | 2013-05-24 | Salicylic acid derivatives, pharmaceutically acceptable salt thereof, composition thereof and method of use thereof |
| BR112014029439A BR112014029439A2 (en) | 2012-05-25 | 2013-05-24 | salicylic acid derivatives, pharmaceutically acceptable salt thereof, composition thereof and method of use thereof |
| HK16106053.3A HK1218709A1 (en) | 2012-05-25 | 2016-05-27 | New salicylic acid derivatives, pharmaceutically acceptable salt thereof, composition thereof and method of use thereof |
| US15/475,108 US10377780B2 (en) | 2012-05-25 | 2017-03-30 | Salicylic acid derivatives, pharmaceutically acceptable salt thereof, composition thereof and method of use thereof |
| US16/451,883 US20190382422A1 (en) | 2012-05-25 | 2019-06-25 | Salicylic acid derivatives, pharmaceutically acceptable salt thereof, composition thereof and method of use thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261651757P | 2012-05-25 | 2012-05-25 | |
| US61/651,757 | 2012-05-25 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US14/550,293 A-371-Of-International US9650399B2 (en) | 2012-05-25 | 2013-05-24 | Salicylic acid derivatives, pharmaceutically acceptable salt thereof, composition thereof and method of use thereof |
| US15/475,108 Continuation US10377780B2 (en) | 2012-05-25 | 2017-03-30 | Salicylic acid derivatives, pharmaceutically acceptable salt thereof, composition thereof and method of use thereof |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2013177534A2 true WO2013177534A2 (en) | 2013-11-28 |
| WO2013177534A3 WO2013177534A3 (en) | 2015-05-28 |
Family
ID=49624538
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2013/042689 WO2013177534A2 (en) | 2012-05-25 | 2013-05-24 | New salicylic acid derivatives, pharmaceutically acceptable salt thereof, composition thereof and method of use thereof |
Country Status (9)
| Country | Link |
|---|---|
| US (3) | US9650399B2 (en) |
| EP (1) | EP2854819B1 (en) |
| JP (2) | JP6290871B2 (en) |
| CN (2) | CN105120854B (en) |
| BR (1) | BR112014029439A2 (en) |
| CA (1) | CA2874057A1 (en) |
| HK (1) | HK1218709A1 (en) |
| RU (1) | RU2641903C2 (en) |
| WO (1) | WO2013177534A2 (en) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014153495A2 (en) | 2013-03-22 | 2014-09-25 | University Of Hawaii | Novel stat3 inhibitors |
| JP2017517521A (en) * | 2014-05-30 | 2017-06-29 | ザ ガバニング カウンシル オブ ザ ユニバーシティ オブ トロント | Sulfonamide compounds and their use as STAT5 inhibitors |
| CN111356456A (en) * | 2017-01-23 | 2020-06-30 | 夏威夷大学 | 2-arylsulfonamido-iV-arylacetamide derived Stat3 inhibitor |
| US11485750B1 (en) | 2019-04-05 | 2022-11-01 | Kymera Therapeutics, Inc. | STAT degraders and uses thereof |
| CN115551496A (en) * | 2020-03-05 | 2022-12-30 | 詹皮克斯有限公司 | STAT inhibitory compounds and compositions |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP3898582A4 (en) * | 2018-12-21 | 2022-08-24 | The Governing Council of the University of Toronto | New salicylic acid derivatives, pharmaceutically acceptable salt thereof, composition thereof and method of use thereof |
| US20230137956A1 (en) * | 2020-03-05 | 2023-05-04 | Janpix Limited | Alpha substituted stat inhibitors and compositions thereof |
| CN112941036B (en) * | 2021-02-08 | 2023-06-09 | 吉林惠康生物药业有限公司 | Method for improving replication level of rabies virus in human diploid cells |
| CN114751927B (en) * | 2022-03-08 | 2023-10-31 | 中山大学 | Boric acid compound, preparation method and application |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010117438A2 (en) | 2009-04-06 | 2010-10-14 | University Of Central Florida Research Foundation, Inc. | Compounds that suppress cancer cells and exhibit antitumor activity |
| WO2012018868A1 (en) | 2010-08-02 | 2012-02-09 | University Of Central Florida Research Foundation, Inc. | Substituted 2-hydroxy-4-(2-(phenylsulfonamido)acetamido)benzoic acid analogs as inhibitors of stat proteins |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2004298511B2 (en) * | 2003-12-11 | 2011-08-11 | Board Of Regents, The University Of Texas System | Compounds for treatment of cell proliferative diseases |
| US8779151B2 (en) * | 2006-03-31 | 2014-07-15 | The Board Of Regents Of The University Of Texas System | Orally bioavailable caffeic acid related anticancer drugs |
| WO2007136858A2 (en) | 2006-05-19 | 2007-11-29 | H. Lee Moffitt Cancer Center & Research Institute | Small molecule inhibitors of stat3 with anti-tumor activity |
| UA95978C2 (en) * | 2006-10-02 | 2011-09-26 | Оцука Фармас'Ютікел Ко., Лтд. | Stat3/5 activation inhibitor |
| PL2325181T3 (en) * | 2008-07-10 | 2017-09-29 | General Incorporated Association Pharma Valley Project Supporting Organization | Stat3 inhibitor containing quinolinecarboxamide derivative as active ingredient |
| WO2014153495A2 (en) * | 2013-03-22 | 2014-09-25 | University Of Hawaii | Novel stat3 inhibitors |
-
2013
- 2013-05-24 US US14/550,293 patent/US9650399B2/en active Active
- 2013-05-24 JP JP2015514226A patent/JP6290871B2/en not_active Expired - Fee Related
- 2013-05-24 CN CN201380038486.1A patent/CN105120854B/en not_active Expired - Fee Related
- 2013-05-24 BR BR112014029439A patent/BR112014029439A2/en not_active Application Discontinuation
- 2013-05-24 CA CA2874057A patent/CA2874057A1/en not_active Abandoned
- 2013-05-24 RU RU2014152697A patent/RU2641903C2/en not_active IP Right Cessation
- 2013-05-24 EP EP13794722.2A patent/EP2854819B1/en active Active
- 2013-05-24 CN CN201910222566.1A patent/CN110003104A/en active Pending
- 2013-05-24 WO PCT/US2013/042689 patent/WO2013177534A2/en active Application Filing
-
2016
- 2016-05-27 HK HK16106053.3A patent/HK1218709A1/en unknown
-
2017
- 2017-03-30 US US15/475,108 patent/US10377780B2/en active Active
-
2018
- 2018-02-08 JP JP2018021248A patent/JP6543366B2/en not_active Expired - Fee Related
-
2019
- 2019-06-25 US US16/451,883 patent/US20190382422A1/en not_active Abandoned
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010117438A2 (en) | 2009-04-06 | 2010-10-14 | University Of Central Florida Research Foundation, Inc. | Compounds that suppress cancer cells and exhibit antitumor activity |
| WO2012018868A1 (en) | 2010-08-02 | 2012-02-09 | University Of Central Florida Research Foundation, Inc. | Substituted 2-hydroxy-4-(2-(phenylsulfonamido)acetamido)benzoic acid analogs as inhibitors of stat proteins |
Non-Patent Citations (34)
| Title |
|---|
| ALMARASSON, O.: "Crystal Engineering of the Composition of Pharmaceutical Phases. Do Pharmaceutical Co-crystals Represent a New Path to Improved Medicines?", THE ROYAL SOCIETY OF CHEMISTRY, 2004, pages 1889 - 1896, XP002415977, DOI: doi:10.1039/b402150a |
| BOWMAN T ET AL., ONCOGENE, vol. 19, 2000, pages 2474 - 88 |
| BOWMAN, T. ET AL., ONCOGENE, vol. 19, no. 21, 2000, pages 2474 - 2488 |
| BUETTNER ET AL., CLIN. CANCER RES., vol. 8, no. 4, 2002, pages 945 - 954 |
| CARRO ET AL., NATURE, vol. 463, no. 7279, 2010, pages 318 - 325 |
| DARNELL, J. E., J, RECENT PROG. NORM. RES., vol. 51, 1996, pages 391 - 403 |
| DARNELL, J. E., JR., RECENT PROG. NORM. RES., vol. 51, 1996, pages 391 - 403 |
| DARNELL, J. E., NAT. MED., vol. 1 1, 2005, pages 595 - 596 |
| DARNELL, J. E., NAT. MED., vol. 1, no. 1, 2005, pages 595 - 596 |
| DARNELL, J. E., NAT. MED., vol. 11, no. 6, 2005, pages 595 - 596 |
| DEBONERA, F. ET AL., J. SURG. RES., vol. 96, no. 2, 2001, pages 289 - 295 |
| GOUILLEUX-GRUART, V. ET AL., BLOOD, vol. 87, 1996, pages 1692 - 1697 |
| GOUILLEUX-GRUART, V. ET AL., LEUKEMIA AND LYMPHOMA, vol. 28, 1997, pages 83 - 88 |
| HARRIS, T. J. ET AL., IMMUNOL., vol. 179, no. 7, 2007, pages 4313 - 4317 |
| HAURA, E. B. ET AL., NAT. CLIN. PRACT. ONCOL., vol. 2, no. 6, 2005, pages 315 - 324 |
| MASHILI, F. ET AL., DIABETES, vol. 62, no. 2, 2013, pages 457 - 465 |
| MIYAMOTO. T ET AL., ARTHRITIS RESEARCH & THERAPY, vol. 14, no. 1, 2012, pages 43 |
| MULLER, J. ET AL., CHEMBIOCHEM, vol. 9, 2008, pages 723 - 727 |
| PARDANANI, A. ET AL., LEUKEMIA, vol. 25, 2011, pages 218 - 225 |
| QUINTAS-CARDAMA, A. ET AL., NATURE REVIEWS DRUG DISCOVERY, vol. 10, 2011, pages 127 - 140 |
| SCHUST, J. ET AL., ANAL. BIOCHEM., vol. 330, 2004, pages 114 - 118 |
| See also references of EP2854819A4 |
| SIDDIQUEE, K. ET AL., PROC. NATL. ACAD. SCI. U.S.A., vol. 104, 2007, pages 7391 - 7396 |
| T. W. GREENE; P. G. M. WUTS: "Protective Groups in Organic Synthesis", 1999, WILEY-INTERSCIENCE |
| TURKSON, J. ET AL., J. BIOL. CHEM., vol. 276, no. 48, 2001, pages 45443 - 45455 |
| TURKSON, J. ET AL., J. BIOL. CHEM., vol. 280, no. 38, 2005, pages 32979 - 32988 |
| TURKSON, J. ET AL., MOL. CANCER THER., vol. 3, 2004, pages 1533 - 1542 |
| TURKSON, J. ET AL., MOL. CANCER THER., vol. 3, no. 3, 2004, pages 261 - 269 |
| TURKSON, J. EXPERT OPIN. THER. TARGETS, vol. 8, 2004, pages 409 - 422 |
| WEBER-NORDT, R. M. ET AL., BLOOD, vol. 88, 1996, pages 809 - 816 |
| WEIMBS, T., JAK-STAT, vol. 2, no. 2, 2013, pages 0 - 1 |
| WORLD J GASTROENTEROL, vol. 14, no. 33, 2008, pages 5110 - 5114 |
| WU, P. ET AL., ANAL. BIOCHEM., vol. 249, 1997, pages 29 - 36 |
| YU, H.; JOVE. R., NAT. REV. CANCER, vol. 4, no. 2, 2004, pages 97 - 105 |
Cited By (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9802888B2 (en) * | 2013-03-22 | 2017-10-31 | University Of Hawaii | STAT3 inhibitors |
| WO2014153495A3 (en) * | 2013-03-22 | 2015-02-19 | University Of Hawaii | Novel stat3 inhibitors |
| US20160068478A1 (en) * | 2013-03-22 | 2016-03-10 | William K. Richardson | Novel stat3 inhibitors |
| JP2016517843A (en) * | 2013-03-22 | 2016-06-20 | ユニバーシティ・オブ・ハワイUniversity Of Hawaii | Novel Stat3 inhibitor |
| EP2975936A4 (en) * | 2013-03-22 | 2017-03-15 | University of Hawaii | Novel stat3 inhibitors |
| WO2014153495A2 (en) | 2013-03-22 | 2014-09-25 | University Of Hawaii | Novel stat3 inhibitors |
| US10519107B2 (en) | 2014-05-30 | 2019-12-31 | The Governing Council Of The University Of Toronto | Sulfonamide compounds and their use as STAT5 inhibitors |
| RU2707094C2 (en) * | 2014-05-30 | 2019-11-22 | Зе Гавернинг Каунсл Оф Зе Юниверсити Оф Торонто | Sulphonamide compounds and use thereof as stat5 inhibitors |
| JP2017517521A (en) * | 2014-05-30 | 2017-06-29 | ザ ガバニング カウンシル オブ ザ ユニバーシティ オブ トロント | Sulfonamide compounds and their use as STAT5 inhibitors |
| CN111356456A (en) * | 2017-01-23 | 2020-06-30 | 夏威夷大学 | 2-arylsulfonamido-iV-arylacetamide derived Stat3 inhibitor |
| EP3570836A4 (en) * | 2017-01-23 | 2020-08-19 | The University of Hawaii | 2-arylsulfonamido-n-arylacetamide derivatized stat3 inhibitors |
| US11299480B2 (en) | 2017-01-23 | 2022-04-12 | University Of Hawaii | 2-arylsulfonamido-N-arylacetamide derivatized STAT3 inhibitors |
| US11485750B1 (en) | 2019-04-05 | 2022-11-01 | Kymera Therapeutics, Inc. | STAT degraders and uses thereof |
| US11746120B2 (en) | 2019-04-05 | 2023-09-05 | Kymera Therapeutics, Inc. | Stat degraders and uses thereof |
| US12077555B2 (en) | 2019-04-05 | 2024-09-03 | Kymera Therapeutics, Inc. | STAT degraders and uses thereof |
| CN115551496A (en) * | 2020-03-05 | 2022-12-30 | 詹皮克斯有限公司 | STAT inhibitory compounds and compositions |
| EP4114377A4 (en) * | 2020-03-05 | 2024-04-24 | Centessa Pharmaceuticals (UK) Limited | Stat inhibitory compounds and compositions |
Also Published As
| Publication number | Publication date |
|---|---|
| CN110003104A (en) | 2019-07-12 |
| HK1218709A1 (en) | 2017-03-10 |
| JP2015522551A (en) | 2015-08-06 |
| US20190382422A1 (en) | 2019-12-19 |
| JP6290871B2 (en) | 2018-03-07 |
| US9650399B2 (en) | 2017-05-16 |
| BR112014029439A2 (en) | 2017-06-27 |
| CA2874057A1 (en) | 2013-11-28 |
| JP6543366B2 (en) | 2019-07-10 |
| RU2014152697A (en) | 2016-07-20 |
| US20170267704A1 (en) | 2017-09-21 |
| JP2018109017A (en) | 2018-07-12 |
| CN105120854B (en) | 2019-04-09 |
| EP2854819A4 (en) | 2016-06-15 |
| EP2854819A2 (en) | 2015-04-08 |
| EP2854819B1 (en) | 2020-01-15 |
| US20150158894A1 (en) | 2015-06-11 |
| RU2641903C2 (en) | 2018-01-23 |
| WO2013177534A3 (en) | 2015-05-28 |
| US10377780B2 (en) | 2019-08-13 |
| CN105120854A (en) | 2015-12-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6543366B2 (en) | Novel salicylic acid derivative, pharmaceutically acceptable salt thereof, composition thereof and method of use thereof | |
| CN110423212B (en) | Hydroxy indole carboxylic acid-based inhibitors of SHP2 | |
| US10954258B2 (en) | STAT3 dimerization inhibitors | |
| BR112012002511B1 (en) | ANALOG COMPOUNDS OF ARYL SPHINGOSINE 1-BICYCLIC PHOSPHATE, AND ITS PHARMACEUTICAL COMPOSITION | |
| US9428465B2 (en) | Acid ceramidase inhibitors and their use as medicaments | |
| TWI574686B (en) | 1,5-diphenyl-penta-1,4-dien-3-one compounds | |
| EP1786773A1 (en) | Isoindolin-1-one derivatives | |
| EA019882B1 (en) | Modulators of the prostacyclin (pgi2) receptor useful for the treatment of disorders related thereto | |
| WO2007136858A2 (en) | Small molecule inhibitors of stat3 with anti-tumor activity | |
| JP2013537535A (en) | Substituted 2-hydroxy-4- (2- (phenylsulfonamido) acetamido) benzoic acid analogs as inhibitors of STAT protein | |
| WO2009067856A1 (en) | Histone deacetylase inhibitor, composition and use thereof | |
| RU2707094C2 (en) | Sulphonamide compounds and use thereof as stat5 inhibitors | |
| JP2015533778A (en) | Novel phenyl-pyridine / pyrazine amide for the treatment of cancer | |
| US20220089531A1 (en) | New salicylic acid derivatives, pharmaceutically acceptable salt thereof, composition thereof and method of use thereof | |
| ES2348838T3 (en) | CYCLALKYLIDENE COMPOUNDS AS SLECTIVE MODULATORS OF THE STRESS RECEIVER. | |
| WO2013178545A1 (en) | Acid ceramidase inhibitors and their use as medicaments | |
| WO2013116948A1 (en) | Indolizine derivatives | |
| CA3140417A1 (en) | Substituted fluorine-containing imidazole salt compound, preparation method therefor, pharmaceutical composition thereof and use thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13794722 Country of ref document: EP Kind code of ref document: A2 |
|
| ENP | Entry into the national phase |
Ref document number: 2874057 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14550293 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 2015514226 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013794722 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2014152697 Country of ref document: RU Kind code of ref document: A |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112014029439 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112014029439 Country of ref document: BR Kind code of ref document: A2 Effective date: 20141125 |