WO2013175397A1 - New process for the preparation of 2-cyclopentyl-6-methoxy-isonicotinic acid - Google Patents
New process for the preparation of 2-cyclopentyl-6-methoxy-isonicotinic acid Download PDFInfo
- Publication number
- WO2013175397A1 WO2013175397A1 PCT/IB2013/054170 IB2013054170W WO2013175397A1 WO 2013175397 A1 WO2013175397 A1 WO 2013175397A1 IB 2013054170 W IB2013054170 W IB 2013054170W WO 2013175397 A1 WO2013175397 A1 WO 2013175397A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- cyclopentyl
- methoxy
- ethyl
- pyridin
- oxadiazol
- Prior art date
Links
- 0 CC*CCS1(C[*+2]CCC1)C([C@](C)*)=C Chemical compound CC*CCS1(C[*+2]CCC1)C([C@](C)*)=C 0.000 description 4
- NCJNQCZEFXNLLE-UHFFFAOYSA-N COC(c1cc(Cl)nc(C2CCCC2)c1)=O Chemical compound COC(c1cc(Cl)nc(C2CCCC2)c1)=O NCJNQCZEFXNLLE-UHFFFAOYSA-N 0.000 description 1
- KFYXHXWIDPBLSD-UHFFFAOYSA-N COC(c1cc(O)nc(C2CCCC2)c1)=O Chemical compound COC(c1cc(O)nc(C2CCCC2)c1)=O KFYXHXWIDPBLSD-UHFFFAOYSA-N 0.000 description 1
- HNBBOUJRDHVLFJ-UHFFFAOYSA-N COc1nc(C2CCCC2)cc(C(O)=O)c1 Chemical compound COc1nc(C2CCCC2)cc(C(O)=O)c1 HNBBOUJRDHVLFJ-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C45/00—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds
- C07C45/61—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups
- C07C45/67—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton
- C07C45/673—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton by change of size of the carbon skeleton
- C07C45/676—Preparation of compounds having >C = O groups bound only to carbon or hydrogen atoms; Preparation of chelates of such compounds by reactions not involving the formation of >C = O groups by isomerisation; by change of size of the carbon skeleton by change of size of the carbon skeleton by elimination of carboxyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/30—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group
- C07C67/317—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group by splitting-off hydrogen or functional groups; by hydrogenolysis of functional groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C67/00—Preparation of carboxylic acid esters
- C07C67/30—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group
- C07C67/333—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group by isomerisation; by change of size of the carbon skeleton
- C07C67/343—Preparation of carboxylic acid esters by modifying the acid moiety of the ester, such modification not being an introduction of an ester group by isomerisation; by change of size of the carbon skeleton by increase in the number of carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/79—Acids; Esters
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/79—Acids; Esters
- C07D213/803—Processes of preparation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/84—Nitriles
- C07D213/85—Nitriles in position 3
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/06—Systems containing only non-condensed rings with a five-membered ring
- C07C2601/08—Systems containing only non-condensed rings with a five-membered ring the ring being saturated
Definitions
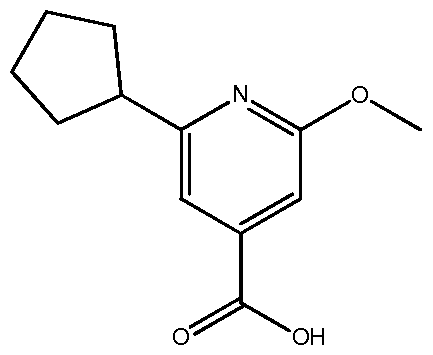
- the present invention relates to new processes for the preparation of 2-cyclopentyl-6-methoxy-isonicotinic acid, which is a useful intermediate for the synthesis of pyridine-4-yl derivatives of formula (PD) disclosed in WO201 1007324 as immunomodulating agent. Moreover, the present invention also relates to new intermediates used in those processes.
- R a represents 3-pentyl, 3-methyl-but-1-yl, cyclopentyl, or cyclohexyl;
- R b represents methoxy
- R c represents 2,3-dihydroxypropoxy, -OCH 2 -CH(OH)-CH 2 -NHCO-CH 2 OH,
- R d represents ethyl or chloro.
- those pyridine-4-yl derivatives are useful for prevention and / or treatment of diseases or disorders associated with an activated immune system, including rejection of transplanted organs such as kidney, liver, heart, lung, pancreas, cornea, and skin; graft-versus-host diseases brought about by stem cell transplantation; autoimmune syndromes including rheumatoid arthritis, multiple sclerosis, inflammatory bowel diseases such as Crohn's disease and ulcerative colitis, psoriasis, psoriatic arthritis, thyroiditis such as Hashimoto's thyroiditis, uveo-retinitis; atopic diseases such as rhinitis, conjunctivitis, dermatitis; asthma; type I diabetes; post-infectious autoimmune diseases including rheumatic fever and post-infectious glomerulonephritis; solid cancers and tumor metastasis.
- 2-Cyclopentyl-6-methoxy-isonicotinic acid which is also disclosed in WO201 1007324, is a useful intermediate for the synthesis of the pyridine-4-yl derivatives of formula (PD), wherein R a is a cyclopentyl group.
- the processes described in the above-listed publications are not efficient for scale-up since they require cryogenic temperatures, expensive starting materials, toxic reagents or many steps.
- the lack of an efficient process to manufacture 1 -cyclopentylethanone is further also mirrored by the difficulty in sourcing this compound on kilogram scale for a reasonable price and delivery time. Therefore, the purpose of the present invention is to provide a new, efficient and cost effective process for the preparation of 2-cyclopentyl-6-methoxy-isonicotinic acid, which is suitable for industrial scale synthesis.
- the present invention relates to a process for the preparation of 1 -cyclopentylethanone
- One embodiment of the present invention relates to a process according to embodiment (i), comprising the reaction of terf.-butyl acetoacetate (1 ) with 1 ,4-dibromobutane, to obtain terf.-butyl 1 -acetylcyclopentanecarboxylate (2):
- One embodiment of the present invention relates to a process according to embodiment (i) or (ii), wherein 1 -cyclopentylethanone (3) is reacted with an alkyl oxalic acid ester ROC(0)C(0)OR to generate compound (4) which is then reacted with cyanacetamide to enerate compound (5),
- R is ethyl, methyl, butyl, or tert-butyl.
- R is preferably ethyl.
- the process according to embodiment (iii) or (iv) further comprises the reaction of compound (5) with an aqueous acid to give 2-cyclopentyl-6-hydroxyisonicotinic acid 6)
- the process according to embodiment (v) further comprises the reaction of compound (6) with HC(OMe) 3 under acid catalysis to give methyl 2-cyclopentyl-6-hydroxyisonicotinate (7)
- the process according to embodiment (vi) further comprises the reaction of compound (7) with a chlorination reagent to give methyl 2-chloro-6-cyclopentylisonicotinate (8)
- the process according to embodiment (v) further comprises the reaction of compound (6) with phosphorous oxychloride (POCI 3 ), followed by treatment with methanol, to give methyl 2-chloro-6-cyclopentylisonicotinate (8).
- POCI 3 phosphorous oxychloride
- the process according to embodiment (vii) or (viii) further comprises the reaction of compound (8) with NaOMe/MeOH, followed by hydrolysis of the ester, to give 2-cyclo entyl-6-methoxy-isonicotinic acid (I):
- Terf.-butyl acetoacetate (1 ) is converted to terf.-butyl 1 -acetylcyclopentanecarboxylate (2), by reacting compound (1 ) with 1 ,4-dibromobutane in aqueous base such as 20-60%, 25-55%, 25-50% or preferably 32-50% NaOH, most preferably 32% NaOH, in the presence of a phase transfer catalyst such as tetrabutylammonium bromide or iodide, preferably tetrabutylammonium bromide (TBABr).
- a phase transfer catalyst such as tetrabutylammonium bromide or iodide, preferably tetrabutylammonium bromide (TBABr).
- TBABr tetrabutylammonium bromide
- the base potassium carbonate or sodium carbonate in DMSO and in presence of a phase catalyst can be used as well (see Tetrahedron
- the temperature of the mixture is kept at a temperature between 45-65°C, 45-60°C, 45-50°C, or preferably 50°C.
- a temperature of 20 to 30°C, preferably around 25°C is sufficient.
- 1 ,4-Dibromobutane is added to the mixture in 1 equivalent, the phase transfer catalyst in a catalytic amount from 0.03 to 0.1 equivalents, preferably around 0.05 equivalents, and the alkyl acetoacetate is added from 0.8 to 1.2 equivalents, preferably 1 equivalent.
- the aqueous base is added in excess.
- the reaction time is from 1 h to 10 h, from 2 h to 8 h, from 3 h to 7 h, from 4 h to 6 h, or preferably the reaction time is 5 hours.
- the system K 2 C0 3 /DMSO affords a longer reaction time, i.e. 15-25 h, preferably around 20 h.
- the organic layer is separated.
- the organic layer is washed with aqueous acid, for example with 1 N HCI.
- aqueous acid for example with 1 N HCI.
- other acids can be used.
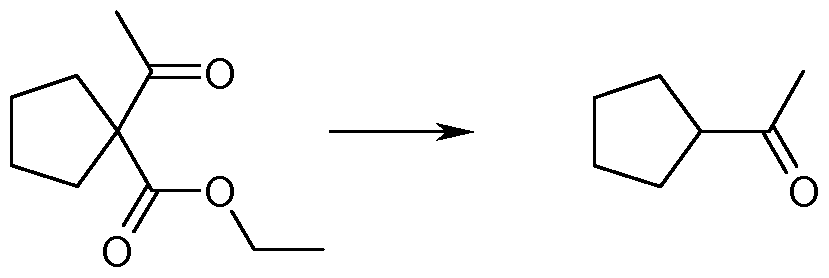
- ieri.-butyl 1 -acetylcyclopentanecarboxylate (2) is converted to 1 -cyclopentylethanone (3) by means of acidic hydrolysis.
- acid such as HCI, aqueous sulphuric acid, or trifluoroacetic acid (TFA).
- HCI hydrochloric acid
- concentrations of 25 to 32% HCI may be used, preferred is concentrated aq. HCI, i.e. 32% HCI.
- non-aqueous HCI solutions may be used as well, for instance 5M HCI in isopropanol.
- 32% HCI is used.
- aqueous concentrations of 40 - 60%, 45-55% and preferably 50% may be used.
- the reaction temperature can range from 50 °C to reflux.
- the reaction temperature is kept at 60°C to 80°C in case of HCI and TFA, and around 120°C for sulphuric acid.
- the work up is done in usual way.
- the mixture is washed with aqueous sodium chloride solution. It may be neutralized with a base before. After drying the organic layer, and filtration, the solvent is evaporated to give crude 1 -cyclopentylethanone (3).
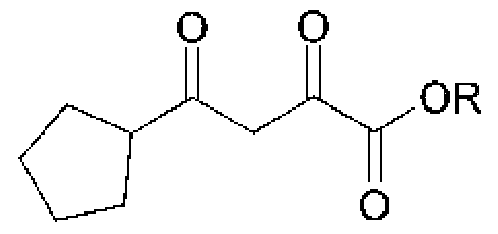
- 1 -Cyclopentylethanone (3) is converted to the alkyl 4-cyclopentyl-2,4-dioxobutanoate (4) by reacting it with a dialkyl oxalate (dialkyl oxalic acid ester), in a solvent such as THF or Methyl THF in the presence of a base, such as KOtBu, NaOEt or NaOMe. Preferred are THF and KOtBu.
- the base is added in an amount of 1 to 1.3 equivalents, preferred is 1.1 equivalent.
- Cyclopentylethanone (3) is added in an amount of 1 equivalent, and the dialkyl oxalate is added in an amout of 0.8 to 1 .2 equivalents, preferably 1 equivalent.
- the person skilled in the art is well aware of the reaction conditions such as temperatures.
- the initial temperature range in the above reaction is from -23°C to less than -18°C, and then kept at -23°C to -5°C, from -20 to -10°C, or from -18°C to -10°C.
- the initial temperature range is kept for a time depending on the scale of synthesis. For instance, it could be around 10 minutes to 1 hour. Afterwards, the reaction mixture is allowed to warm up to around 10 to 20 °C, preferably around 15°C.
- the reaction time may be from around 16 to around 25 hours, for instance around 20 hours.
- the reaction is performed at ambient temperature, for instance at 20 to 25 °C, preferably at around 22°C.
- reaction mixture To the reaction mixture is then added water, and the reaction mixture is concentrated by removing most of the solvent and water.
- Oxalic acid esters ROC(0)C(0)OR may be selected from diethyl oxalate, dimethyl oxalate, dibutyl oxalate, or di-tert-butyl oxalate, preferred is diethyl oxalate.
- the following intermediates (4) are obtained:
- R is ethyl, i.e. compound (4) is ethyl 4-cyclopentyl-2,4-dioxobutanoate, and compound (5) is ethyl 2-hydroxy-3-cyano-6-cyclopentyl-isonicotinate.
- the obtained alkyl 2-hydroxy-3-cyano-6-cyclopentyl-isonicotinate (5) is further converted to 2-cyclopentyl-6-hydroxyisonicotinic acid (6) with and aqueous acid, for example HCI at a concentration of 30% to 34%, preferably 32%.
- the reaction is performed at a temperature ranging from 90 to 100°C, preferably at 100°C.
- the reaction time may be from around 20 to around 25 hours, for example around 22 hours.
- the work up is known by a person skilled in the art.
- around half of the solvent is removed and the obtained suspension is diluted with water and cooled to around 5°C to 15°C, preferably 10°C, before being filtered to obtain 2-cyclopentyl-6-hydroxyisonicotinic acid (6).
- the conversion of Compound (3) to Compound (6) via Compound (4) and (5) can be performed sequentially without further purifying Compounds (4) and (5).
- 2-Cyclopentyl-6-hydroxyisonicotinic acid (6) is converted to methyl 2-cyclopentyl-6-hydroxyisonicotinate (7), by reacting compound (6) with trimethylorthoformiate in methanol in the presence of a acid, such as sulphuric acid or with MeOH and sulphuric acid without trimethylorthoformiate.
- a acid such as sulphuric acid or with MeOH and sulphuric acid without trimethylorthoformiate.
- 2-Cyclopentyl-6-hydroxyisonicotinic acid (6) is added in an amount of 1 equivalent, trimethylorthoformiate is added in an amount of 1.9 to 2.2 equivalents, preferably 2 equivalents, and the acid is added in an amount of 1 to 1.4 equivalents.
- the solvent is added in excess.
- the skilled person is aware of the temperature ranges and in particular of the reaction times.
- the reaction is preferably kept at reflux temperature, or a temperature in the range of 60 to 65 °C.
- the work up is known to the skilled in the art.
- the solvent is removed (under reduced pressure) and water is added to obtain a suspension containing the product, which can be isolated by filtration, preferably at a temperature below 15°C, preferably around
- the obtained methyl 2-cyclopentyl-6-hydroxyisonicotinate (7) is converted to methyl 2-chloro-6-cyclopentylisonicotinate (8), by reacting Compound (7) with a chlorination reagent, for example with phenylphosphonic dichloride, phosphoryl chloride or thionyl chloride, and preferably with phenylphosphonic dichloride.
- Methyl 2-cyclopentyl-6-hydroxyisonicotinate (7) is added in an amount of 1 equivalent, the chlorination reagent is added in an amount of 1.5 to 2.5 equivalents, depending also on the nature of the chlorination reagent. For example, the chlorination reagent is added in an amount of 2 equivalents.
- the reaction temperature is kept at a range from 120 to 140°C, preferably at around 130°C.
- the work up is known to the person skilled in the art.
- the reaction mixture is added to a mixture of an aqueous buffer and an organic solvent.
- an aqueous buffer for example, potassium phosphates in water and isopropyl acetate can be used.
- the organic fraction is collected and purified, for example by distillation.
- 2-Cyclopentyl-6-hydroxyisonicotinic acid (6) is converted to methyl 2-chloro-6-cyclopentylisonicotinate (8), by reacting Compound 6 with phosphorous oxychloride (POCI 3 ), followed by treatment with methanol, to give compound (8).
- POCI 3 phosphorous oxychloride
- 2-Cyclopentyl-6-hydroxyisonicotinic acid (6) is added in an amount of 1 equivalent
- phosphorous oxychloride (POCI 3 ) is added in an amount of 1.5 to 12 equivalents or 8 to 12 equivalents, preferably in an amount of around 10 equivalents.
- the reaction temperature is kept at a range from 110 to 120°C, preferably around 1 15°C.
- the reaction time is from 3 to 5 h, preferably 4 h.
- the reaction mixture is concentrated by distilling off the excess phosphorous oxychloride (POCI 3 ).
- An organic solvent can be added for diluting the concentrate and methanol is added in order to produce the methyl ester.
- Methyl 2-chloro-6-cyclopentylisonicotinate (8) is reacted with NaOMe/MeOH, followed by hydrolysis of the ester, to give 2-cyclopentyl-6-methoxy-isonicotinic acid (I).
- Methyl 2-chloro-6-cyclopentylisonicotinate (8) is added in an amount of 1 equivalent, and sodium methanolate in methanol is added in excess, e.g. in a range of 8 equivalent to 15 equivalents, preferably around 10 equivalents.
- the reaction temperature may be kept at reflux temperature. Reaction time may range from 10 to 48 hours.
- the residue is acidified, for example to a pH of around 1 to 1 .5, preferably to about 1.
- Aqueous hydrochloric acid may be used.
- the present invention further relates to a preferred intermediate of the process for the preparation of 2-cyclopentyl-6-methoxy-isonicotinic acid, which is ethyl
- the present invention further relates to a preferred intermediate of the process for the preparation of 2-cyclopentyl-6-methoxy-isonicotinic acid, which is
- the present invention further relates to a preferred intermediate of the process for the preparation of 2-cyclopentyl-6-methoxy-isonicotinic acid, which is methyl
- the present invention further relates to a preferred intermediate of the process for the preparation of 2-cyclopentyl-6-methoxy-isonicotinic acid, which is methyl
- the present invention further relates to a process for the preparation of the pyridine-4-yl derivatives of formula (PD), wherein R a is a cyclopentyl group, comprising the process according to any one of embodiments i) to ix).
- the preparation of pyridine-4-yl derivatives of formula (PD), wherein R a is cyclopentyl, from 2-cyclopentyl-6-methoxy-isonicotinic acid is described in detail in WO 201 1/007324.
- 5-pyridin-4-yl-[1 ,2,4]oxadiazole derivatives of formula (PD), wherein R a is a cyclopentyl group may be prepared by reacting a compound of Structure 9 in a solvent such as toluene, pyridine, DMF, THF, dioxane, DME, etc. at rt or elevated temperatures in the presence or absence of auxiliaries such as acids (e.g. TFA, acetic acid, HCI, etc.), bases (e.g.
- Compounds of Structure 9 may be prepared by reacting 2-cyclopentyl-6-methoxy-isonicotinic acid with a compound of Structure 10 in a solvent such as DMF, THF, DCM, etc. in the presence of one or more coupling agents such as TBTU, DCC, EDC, HBTU, CDI, etc. and in the presence or absence of a base such as NEt 3 , DIPEA, NaH, K 2 C0 3 , etc. (Lit.: e.g. A. Hamze, J.-F. Hernandez, P. Fulcrand, J. Martinez, J. Org. Chem. 68 (2003) 7316-7321 ).
- Pyridine-4-yl derivatives of formula (PD), which are readily prepared by using 2-cyclopentyl-6-methoxy-isonicotinic acid, include:
- Preferred pyridine-4-yl derivatives of formula (PD), which are readily prepared by using 2-cyclopentyl-6-methoxy-isonicotinic acid, include:
- 2-Cyclopentyl-6-methoxy-isonicotinic acid is especially suitable for the preparation of 5-pyridin-4-yl-[1 ,2,4]oxadiazole derivatives of formula (PD), wherein R a is a cyclopentyl group.
- PD 5-pyridin-4-yl-[1 ,2,4]oxadiazole derivatives of formula (PD), wherein R a is a cyclopentyl group.
- Any reference hereinbefore or hereinafter to a compound is to be understood as referring also to salts, especially pharmaceutically acceptable salts, of such compound, as appropriate and expedient.
- pharmaceutically acceptable salts refers to non-toxic, inorganic or organic acid and/or base addition salts. Reference can be made to "Salt selection for basic drugs", Int. J. Pharm. (1986), 33, 201 -217.
- the purpose of the present invention is to provide a new, efficient and cost effective process for the preparation of 2-cyclopentyl-6-methoxy-isonicotinic acid, which is suitable for industrial scale synthesis. Even though the process to prepare 1-cyclopentylethanone (3) is a key step for this purpose, also as described in particular in embodiment (i) above, it is still to be said that
- the present invention relates to a process for the preparation of 2-cyclopentyl-6-methoxy-isonicotinic acid
- One embodiment of the present invention relates to a process according to embodiment (a), comprising a reaction sequence b) of converting 1 -cyclopentylethanone (3) to 2-cyclopentyl-6-hydroxyisonicotinic acid (6):
- One embodiment of the present invention relates to a process according to embodiment (b), wherein in reaction sequence b), 1 -cyclopentylethanone (3) is reacted with an alkyl oxalic acid ester ROC(0)C(0)OR to generate compound (4)
- R is ethyl, methyl, butyl, or tert-butyl
- R is preferably ethyl.
- Another embodiment of present invention relates to a process according to embodiment (b), (c), or (d) wherein 2-cyclopentyl-6-hydroxyisonicotinic acid (6) is converted to methyl 2-chloro-6-cyclopentylisonicotinate 8):
- One embodiment of present invention relates to a process according to embodiment (e), wherein 2-cyclopentyl-6-hydroxyisonicotinic acid (6) is reacted with HC(OMe) 3 under acid catalysis to give methyl 2-cyclopentyl-6-hydroxyisonicotinate (7)
- One embodiment of present invention relates to a process according to embodiment (e), wherein 2-cyclopentyl-6-hydroxyisonicotinic acid (6) is reacted with phosphorous oxychloride (POCI 3 ), followed by treatment with methanol, to give compound (8).
- phosphorous oxychloride POCI 3
- One embodiment of present invention relates to a process according to embodiment (e), (f) or (g), wherein methyl 2-chloro-6-cyclopentylisonicotinate (8) is reacted with NaOMe/MeOH, followed by hydrolysis of the ester, to give 2-cyclopentyl-6-methoxy-isonicotinic acid I):
- One preferred embodiment of present invention relates to a process for the preparation of 1 -cyclopentylethanone (3):
- Carrier gas Helium
- a 10 L reactor was charged with potassium terf.-butylate (220 g, 1.1 eq.) and THF (3 L). The solution was cooled to -20 °C.
- a mixture of diethyloxalate (260 g, 1 eq.) and 1 -cyclopentylethanone (200 g, 1 .78 mol, 1 eq.) was added at a temperature below -18 °C.
- the reaction mixture was stirred at -10 °C for 30 min and then warmed to 15 °C.
- To the mixture was added cyano acetamide (180 g, 1.2 eq.). The mixture was stirred for 20 h at 22 °C.
- Methyl 2-cyclopentyl-6-hydroxyisonicotinate 50 g, 0.226 mol, 1 eq.
- phenylphosphonic dichloride 70 mL, 2 eq.
- the reaction mixture was added to a solution of potassium phosphate (300 g) in water (600 mL) and isopropyl acetate (600 mL) at 0 °C.
- the mixture was filtered over kieselguhr (i.e. diatomite, CeliteTM) (50 g).
- the aq. layer was separated and discarded.
- the org. layer was washed with water (500 mL).
- the org. layer was concentrated to dryness at 65 °C and reduced pressure to obtain a black oil; yield: 50.4 g (93%); purity (LC-MS): 94% a/a.
- Methyl 2-chloro-6-cyclopentylisonicotinate 40 g, 0.168 mol, 1 eq.
- MeOH 320 mL, 10 eq.
- Water 250 mL was added carefully at 80 °C external temperature.
- Methanol was distilled off at 60 °C and reduced pressure (300 mbar).
- the residue was acidified with 32% HCI (150 mL) and the pH was adjusted to 1 .
- the mixture was extracted with isopropyl acetate (300 mL). The aq. layer was discarded.
- the org. layer was washed with water (200 mL). The org.
- Triethylamine (1 12 mL, 1 eq.) and cyanoacetamide (67.9 g, 1 eq.) was heated in ethanol to 65 °C.
- Ethyl 4-cyclopentyl-2,4-dioxobutanoate (171 g, 0.807 mol, 1 eq.) was added to the mixture at 65 °C.
- the mixture was stirred for 3 h at 65 °C.
- the mixture was cooled to 20 °C and filtered.
- the product was washed with TBME (2 x 200 mL).
- ketonic ester ethyl 1-acetylcyclopentanecarboxylate (19.5 g) was refluxed for 24 h with a considerable excess of potash (19 g) in alcohol (150 cc), two-thirds of the alcohol then distilled off, the residue refluxed for 3 h, the bulk of the alcohol finally removed, saturated brine added, and the ketone extracted with ether.
- the oil obtained from the extract distilled at 150-160 760 mm and yielded nearly 4 g of a colourless oil, b.p. 153-155 760 mm, on redistillation.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pyridine Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
Description





































Claims


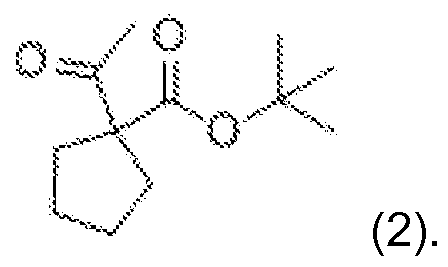






Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015513330A JP5753333B1 (en) | 2012-05-22 | 2013-05-21 | Novel process for the preparation of 2-cyclopentyl-6-methoxy-isonicotinic acid |
| EP13734849.6A EP2852575A1 (en) | 2012-05-22 | 2013-05-21 | New process for the preparation of 2-cyclopentyl-6-methoxy-isonicotinic acid |
| CN201380025969.8A CN104321311A (en) | 2012-05-22 | 2013-05-21 | New process for the preparation of 2-cyclopentyl-6-methoxy-isonicotinic acid |
| US14/402,159 US20150133669A1 (en) | 2012-05-22 | 2013-05-21 | New process for the preparation of 2-cyclopentyl-6-methoxy-isonicotinic acid |
| KR1020147035745A KR20150021056A (en) | 2012-05-22 | 2013-05-21 | New process for the preparation of 2-cyclopentyl-6-methoxy-isonicotinic acid |
| MX2014014138A MX2014014138A (en) | 2012-05-22 | 2013-05-21 | New process for the preparation of 2-cyclopentyl-6-methoxy-isonic otinic acid. |
| CA2873439A CA2873439A1 (en) | 2012-05-22 | 2013-05-21 | New process for the preparation of 2-cyclopentyl-6-methoxy-isonicotinic acid |
| IL235802A IL235802A0 (en) | 2012-05-22 | 2014-11-20 | New process for the preparation of 2-cyclopentyl-6-methoxy-isonicotinic acid |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP12168823 | 2012-05-22 | ||
| EP12168823.8 | 2012-05-22 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013175397A1 true WO2013175397A1 (en) | 2013-11-28 |
Family
ID=48748310
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2013/054170 WO2013175397A1 (en) | 2012-05-22 | 2013-05-21 | New process for the preparation of 2-cyclopentyl-6-methoxy-isonicotinic acid |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US20150133669A1 (en) |
| EP (1) | EP2852575A1 (en) |
| JP (1) | JP5753333B1 (en) |
| KR (1) | KR20150021056A (en) |
| CN (1) | CN104321311A (en) |
| CA (1) | CA2873439A1 (en) |
| IL (1) | IL235802A0 (en) |
| MX (1) | MX2014014138A (en) |
| WO (1) | WO2013175397A1 (en) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2018521001A (en) * | 2015-05-20 | 2018-08-02 | イドーシア ファーマシューティカルズ リミテッドIdorsia Pharmaceuticals Ltd | Compound (S) -3- {4- [5- (2-Cyclopentyl-6-methoxy-pyridin-4-yl)-[1,2,4] oxadiazol-3-yl] -2-ethyl-6 -Methyl-phenoxy} -propane-1,2-diol crystal form |
| WO2021084068A1 (en) | 2019-10-31 | 2021-05-06 | Idorsia Pharmaceuticals Ltd | Combination of a cxcr7 antagonist with an s1p1 receptor modulator |
| WO2021148314A1 (en) | 2020-01-20 | 2021-07-29 | Idorsia Pharmaceuticals Ltd | Accelerated elimination of (s)-3-{4-[5-(2-cyclopentyl-6-methoxy-pyridin-4-yl)-[1,2,4]oxadiazol-3-yl]-2-ethyl-6-methyl-phenoxy}-propane-1,2-diol |
| EP4212156A1 (en) | 2022-01-13 | 2023-07-19 | Abivax | Combination of 8-chloro-n-(4-(trifluoromethoxy)phenyl)quinolin-2-amine and its derivatives with a s1p receptor modulator |
| WO2024056631A1 (en) | 2022-09-14 | 2024-03-21 | Idorsia Pharmaceuticals Ltd | S1p 1 receptor modulators for use in the treatment of type 1 ifn mediated diseases |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN105753682B (en) * | 2016-03-08 | 2018-01-09 | 中国科学院成都有机化学有限公司 | A kind of preparation method of phenylcyclopentyl ketone |
| CN107382965A (en) * | 2017-08-14 | 2017-11-24 | 河南科技大学第附属医院 | The synthetic method of the receptor stimulating agent drug molecules of new S1P 1 with antitumor activity |
| CN107311994A (en) * | 2017-08-14 | 2017-11-03 | 淄博职业学院 | A kind of novel method for synthesizing of the receptor stimulating agent drug molecules of S1P 1 |
| KR101998523B1 (en) | 2018-08-31 | 2019-07-09 | (주)바이오액츠 | A complex of fluorescent dye and amino acid residue and the method for preparing the same |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5001140A (en) | 1989-04-17 | 1991-03-19 | Hoffmann-La Roche Inc. | Cycloalkylthiazoles |
| WO2004074270A2 (en) | 2003-02-21 | 2004-09-02 | Pfizer Inc. | Inhibitors of hepatitis c virus rna-dependent rna polymerase, and compositions and treatments using the same |
| US20040220186A1 (en) | 2003-04-30 | 2004-11-04 | Pfizer Inc. | PDE9 inhibitors for treating type 2 diabetes,metabolic syndrome, and cardiovascular disease |
| US20060199853A1 (en) | 2005-02-18 | 2006-09-07 | Charles Mioskowski | Analogs of 4-hydroxyisoleucine and uses thereof |
| US20060223884A1 (en) | 2005-03-22 | 2006-10-05 | Nicolas Chapal | Compounds and compositions for use in the prevention and treatment of obesity and related syndromes |
| US20080242661A1 (en) | 2006-12-11 | 2008-10-02 | Graeme James Dykes | Novel compounds useful for the treatment of degenerative & inflammatory diseases |
| WO2009071707A1 (en) | 2007-12-07 | 2009-06-11 | Galapagos Nv | Novel compounds useful for the treatment of degenerative & inflammatory diseases |
| WO2011007324A1 (en) | 2009-07-16 | 2011-01-20 | Actelion Pharmaceuticals Ltd | Pyridin-4-yl derivatives |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP3820827B2 (en) * | 1999-12-20 | 2006-09-13 | 宇部興産株式会社 | Production method of ketone |
| AU2008205642B2 (en) * | 2007-01-12 | 2013-06-06 | Msd K.K. | Spirochromanon derivatives |
| CN106316812A (en) * | 2009-07-24 | 2017-01-11 | Dpx精细化学奥地利两合公司 | Indane derivatives for use as intermediates |
| JP5623835B2 (en) * | 2010-09-08 | 2014-11-12 | Jx日鉱日石エネルギー株式会社 | Dicarbonyl compound, intermediate thereof and method for producing the same |
-
2013
- 2013-05-21 WO PCT/IB2013/054170 patent/WO2013175397A1/en active Application Filing
- 2013-05-21 MX MX2014014138A patent/MX2014014138A/en unknown
- 2013-05-21 CA CA2873439A patent/CA2873439A1/en not_active Abandoned
- 2013-05-21 KR KR1020147035745A patent/KR20150021056A/en active Search and Examination
- 2013-05-21 EP EP13734849.6A patent/EP2852575A1/en not_active Withdrawn
- 2013-05-21 CN CN201380025969.8A patent/CN104321311A/en active Pending
- 2013-05-21 US US14/402,159 patent/US20150133669A1/en not_active Abandoned
- 2013-05-21 JP JP2015513330A patent/JP5753333B1/en not_active Expired - Fee Related
-
2014
- 2014-11-20 IL IL235802A patent/IL235802A0/en unknown
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5001140A (en) | 1989-04-17 | 1991-03-19 | Hoffmann-La Roche Inc. | Cycloalkylthiazoles |
| WO2004074270A2 (en) | 2003-02-21 | 2004-09-02 | Pfizer Inc. | Inhibitors of hepatitis c virus rna-dependent rna polymerase, and compositions and treatments using the same |
| US20040220186A1 (en) | 2003-04-30 | 2004-11-04 | Pfizer Inc. | PDE9 inhibitors for treating type 2 diabetes,metabolic syndrome, and cardiovascular disease |
| US20060199853A1 (en) | 2005-02-18 | 2006-09-07 | Charles Mioskowski | Analogs of 4-hydroxyisoleucine and uses thereof |
| US20060223884A1 (en) | 2005-03-22 | 2006-10-05 | Nicolas Chapal | Compounds and compositions for use in the prevention and treatment of obesity and related syndromes |
| US20080242661A1 (en) | 2006-12-11 | 2008-10-02 | Graeme James Dykes | Novel compounds useful for the treatment of degenerative & inflammatory diseases |
| WO2009071707A1 (en) | 2007-12-07 | 2009-06-11 | Galapagos Nv | Novel compounds useful for the treatment of degenerative & inflammatory diseases |
| WO2011007324A1 (en) | 2009-07-16 | 2011-01-20 | Actelion Pharmaceuticals Ltd | Pyridin-4-yl derivatives |
Non-Patent Citations (16)
| Title |
|---|
| "Salt selection for basic drugs", INT. J. PHARM., vol. 33, 1986, pages 201 - 217 |
| A. ALBECK ET AL: "Carbon-13 NMR studies of model compounds for bacteriorhodopsin: factors affecting the retinal chromophore chemical shifts and absorption maximum", JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, vol. 114, no. 7, 1 March 1992 (1992-03-01), pages 2400 - 2411, XP055077277, ISSN: 0002-7863, DOI: 10.1021/ja00033a015 * |
| A. HAMZE; J.-F. HERNANDEZ; P. FULCRAND; J. MARTINEZ, J. ORG. CHEM., vol. 68, 2003, pages 7316 - 7321 |
| A. R. GANGLOFF; J. LITVAK; E. J. SHELTON; D. SPERANDIO; V. R. WANG; K. D. RICE, TETRAHEDRON LETT., vol. 42, 2001, pages 1441 - 1443 |
| B. KABOUDIN; K. NAVAEE, HETEROCYCLES, vol. 60, 2003, pages 2287 - 2292 |
| BULL. SOC. CHIM. FR., 1967, pages 3722 - 3729 |
| DATABASE REGISTRY [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; 15 April 2012 (2012-04-15), XP002712107, Database accession no. RN-1368348-22-4 * |
| E. O. JOHN; J. M. SHREEVE, INORGANIC CHEMISTRY, vol. 27, 1988, pages 3100 - 3104 |
| GOLDSWORTHY, J. CHEM. SOC., 1934, pages 377 - 378 |
| GOLDSWORTHY, LEONARD J.: "Reaction of ethyl sodioacetoacetate and tetramethylene dibromide", JOURNAL OF THE CHEMICAL SOCIETY 377 -8 CODEN: JCSOA9; ISSN: 0368-1769, 1934, XP002712108 * |
| J. AM. CHEM. SOC., vol. 105, 1983, pages 4008 - 4017 |
| R. F. POULAIN; A. L. TARTAR; B. P. DEPREZ, TETRAHEDRON LETT., vol. 42, 2001, pages 1495 - 1498 |
| R. M. SRIVASTAVA; F. J. S. OLIVEIRA; D. S. MACHADO; R. M. SOUTO-MAIOR, SYNTHETIC COMMUN., vol. 29, 1999, pages 1437 - 1450 |
| T. SUZUKI; K. IWAOKA; N. IMANISHI; Y. NAGAKURA; K. MIYTA; H. NAKAHARA; M. OHTA; T. MASE, CHEM. PHARM. BULL., vol. 47, 1999, pages 120 - 122 |
| TETRAHEDRON LETTERS, vol. 46, 2005, pages 635 - 638 |
| ZHANG, PANG; LI, LIAN-CHU, SYNTH. COMMUN., vol. 16, 1986, pages 957 - 966 |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2018521001A (en) * | 2015-05-20 | 2018-08-02 | イドーシア ファーマシューティカルズ リミテッドIdorsia Pharmaceuticals Ltd | Compound (S) -3- {4- [5- (2-Cyclopentyl-6-methoxy-pyridin-4-yl)-[1,2,4] oxadiazol-3-yl] -2-ethyl-6 -Methyl-phenoxy} -propane-1,2-diol crystal form |
| US10385043B2 (en) | 2015-05-20 | 2019-08-20 | Idorsia Pharmaceuticals Ltd | Crystalline form of the compound (S)-3-{4-[5-(2-cyclopentyl-6-methoxy-pyridin-4-yl)-[1,2,4]oxadiazol-3-yl]-2-ethyl-6-methyl-phenoxy}-propane-1,2-diol |
| US10836754B2 (en) | 2015-05-20 | 2020-11-17 | Idorsia Pharmaceuticals Ltd | Crystalline form of the compound (S)-3-{4-[5-(2-cyclopentyl-6-methoxy-pyridin-4-yl)-[1,2,4]oxadiazol-3-yl]-2-ethyl-6-methyl-phenoxy}-propane-1,2-diol |
| US11390615B2 (en) | 2015-05-20 | 2022-07-19 | Idorsia Pharmaceuticals Ltd | Crystalline form of the compound (S)-3-{4-[5-(2-cyclopentyl-6-methoxy-pyridin-4-yl)-[1,2,4]oxadiazol-3-yl]-2-ethyl-6-methyl-phenox |
| US11834443B2 (en) | 2015-05-20 | 2023-12-05 | Idorsia Pharmaceuticals Ltd | Crystalline form of the compound (s)-3-{4-[5-(2-cyclopentyl-6-methoxy-pyridin-4-yl)-[1,2,4]oxadiazol-3-yl]-2-ethyl-6-methyl-phenoxy}-propane-1,2-diol |
| WO2021084068A1 (en) | 2019-10-31 | 2021-05-06 | Idorsia Pharmaceuticals Ltd | Combination of a cxcr7 antagonist with an s1p1 receptor modulator |
| WO2021148314A1 (en) | 2020-01-20 | 2021-07-29 | Idorsia Pharmaceuticals Ltd | Accelerated elimination of (s)-3-{4-[5-(2-cyclopentyl-6-methoxy-pyridin-4-yl)-[1,2,4]oxadiazol-3-yl]-2-ethyl-6-methyl-phenoxy}-propane-1,2-diol |
| EP4212156A1 (en) | 2022-01-13 | 2023-07-19 | Abivax | Combination of 8-chloro-n-(4-(trifluoromethoxy)phenyl)quinolin-2-amine and its derivatives with a s1p receptor modulator |
| WO2023135207A1 (en) | 2022-01-13 | 2023-07-20 | Abivax | Combination of 8-chloro-n-(4-(trifluoromethoxy)phenyl)quinolin-2-amine and its derivatives with a s1p receptor modulator |
| WO2024056631A1 (en) | 2022-09-14 | 2024-03-21 | Idorsia Pharmaceuticals Ltd | S1p 1 receptor modulators for use in the treatment of type 1 ifn mediated diseases |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2015522550A (en) | 2015-08-06 |
| IL235802A0 (en) | 2015-02-01 |
| JP5753333B1 (en) | 2015-07-22 |
| EP2852575A1 (en) | 2015-04-01 |
| MX2014014138A (en) | 2015-02-24 |
| CA2873439A1 (en) | 2013-11-28 |
| KR20150021056A (en) | 2015-02-27 |
| US20150133669A1 (en) | 2015-05-14 |
| CN104321311A (en) | 2015-01-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2013175397A1 (en) | New process for the preparation of 2-cyclopentyl-6-methoxy-isonicotinic acid | |
| CN101547901B (en) | Process for the preparation of 2-substituted-5-(1-alkylthio)alkylpyridines | |
| CN101835757A (en) | Diacylglycerol acyltransferase inhibitors | |
| JP2010510191A (en) | 2-Carboxythiophene derivatives as antiviral agents | |
| KR20150011003A (en) | Substituted pyrazole compounds as lpar antagonists | |
| KR20170083535A (en) | Processes and Intermediates for Preparing α,ω-Dicarboxylic Acid-Terminated Dialkane Ethers | |
| JP6505756B2 (en) | Process for isolating 4-chloro-2-fluoro-3-substituted-phenylboronic acid | |
| CN102643194B (en) | Preparation method of posaconazole intermediate | |
| MX2015002624A (en) | Non-annulated thiophenylamides as inhibitors of fatty acid binding proteini(fabp) 4 and/or 5. | |
| TW201245114A (en) | Process for the preparation of pyrazole carboxylic acid amides | |
| JP2021509685A (en) | Process for preparing chrysaborol and its intermediates | |
| MXPA06000369A (en) | Process for the preparation and purification of 2-(alkoxyalkylidene)-3-ketoalkanoic acid esters from 3-ketoalkanoic acid esters. | |
| TW201348190A (en) | New process for the preparation of 2-cyclopentyl-6-methoxy-isonicotinic acid | |
| JP4240978B2 (en) | Aryloxymethyloxadiazole derivatives | |
| US20010049445A1 (en) | Process for the preparation of thiazolidinedione derivatives | |
| JP6256469B2 (en) | Process for the preparation of spiro [2.5] octane-5,7-dione | |
| CN102557941B (en) | Preparation method for intermediate compound of derivative of spiro-propyl formyl | |
| EP2812300B1 (en) | Process for preparing carboxamidine compounds | |
| KR20030021263A (en) | Method for the Production of Imidazo-(1,2-c)(2,3)-Benzodiazepines and Intermediates in the Production thereof | |
| JP4333156B2 (en) | Process for producing 2-substituted-tetrahydropyran-4-ol, its intermediate and process for its production | |
| JP2003026640A (en) | Method for producing aromatic ketone | |
| KR101209572B1 (en) | Potassium organocarbonyltrifluoroborate derivatives and method for producing the same | |
| US20060199965A1 (en) | Process for the preparation of nicotinaldehydes | |
| CN117551043A (en) | Method for preparing uracil compound containing carboxylic ester fragment by using isocyanate as raw material | |
| JP2024509536A (en) | Method for preparing alkyl 4-oxotetrahydrofuran-2-carboxylate |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13734849 Country of ref document: EP Kind code of ref document: A1 |
|
| REEP | Request for entry into the european phase |
Ref document number: 2013734849 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013734849 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2873439 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14402159 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 235802 Country of ref document: IL Ref document number: MX/A/2014/014138 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 2015513330 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20147035745 Country of ref document: KR Kind code of ref document: A |