WO2013174931A1 - 7-oxo-4,7 -dihydro- pyrazolo [1, 5 -a] pyrimidine derivatives which are useful in the treatment, amelioration or prevention of a viral disease - Google Patents
7-oxo-4,7 -dihydro- pyrazolo [1, 5 -a] pyrimidine derivatives which are useful in the treatment, amelioration or prevention of a viral disease Download PDFInfo
- Publication number
- WO2013174931A1 WO2013174931A1 PCT/EP2013/060634 EP2013060634W WO2013174931A1 WO 2013174931 A1 WO2013174931 A1 WO 2013174931A1 EP 2013060634 W EP2013060634 W EP 2013060634W WO 2013174931 A1 WO2013174931 A1 WO 2013174931A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- optionally substituted
- compound
- optionally
- alkyl
- oxo
- Prior art date
Links
- 230000003612 virological effect Effects 0.000 title claims abstract description 53
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 title claims abstract description 35
- 201000010099 disease Diseases 0.000 title claims abstract description 31
- 238000011282 treatment Methods 0.000 title claims description 11
- 230000002265 prevention Effects 0.000 title claims description 5
- NOSYRIKBTPXMRH-UHFFFAOYSA-N O=C1C=CNC2=CC=NN12 Chemical class O=C1C=CNC2=CC=NN12 NOSYRIKBTPXMRH-UHFFFAOYSA-N 0.000 title 1
- 150000001875 compounds Chemical class 0.000 claims abstract description 351
- 239000000203 mixture Substances 0.000 claims abstract description 60
- 150000003839 salts Chemical class 0.000 claims abstract description 21
- 239000000651 prodrug Substances 0.000 claims abstract description 17
- 229940002612 prodrug Drugs 0.000 claims abstract description 17
- 239000012453 solvate Substances 0.000 claims abstract description 13
- 238000000034 method Methods 0.000 claims description 143
- 239000003112 inhibitor Substances 0.000 claims description 63
- 125000000217 alkyl group Chemical group 0.000 claims description 49
- 229910052760 oxygen Inorganic materials 0.000 claims description 41
- 125000003107 substituted aryl group Chemical group 0.000 claims description 39
- 229910052717 sulfur Inorganic materials 0.000 claims description 37
- 125000005842 heteroatom Chemical group 0.000 claims description 28
- 206010022000 influenza Diseases 0.000 claims description 28
- 125000004432 carbon atom Chemical group C* 0.000 claims description 26
- 239000003814 drug Substances 0.000 claims description 26
- 125000001424 substituent group Chemical group 0.000 claims description 25
- 125000003118 aryl group Chemical group 0.000 claims description 23
- 230000000694 effects Effects 0.000 claims description 21
- 229910052757 nitrogen Inorganic materials 0.000 claims description 20
- 229910052736 halogen Inorganic materials 0.000 claims description 19
- 150000002367 halogens Chemical class 0.000 claims description 15
- 239000008194 pharmaceutical composition Substances 0.000 claims description 15
- 102100031780 Endonuclease Human genes 0.000 claims description 14
- 108010042407 Endonucleases Proteins 0.000 claims description 14
- 125000006272 (C3-C7) cycloalkyl group Chemical group 0.000 claims description 12
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 12
- 239000003443 antiviral agent Substances 0.000 claims description 11
- 238000003556 assay Methods 0.000 claims description 11
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 11
- 230000009467 reduction Effects 0.000 claims description 11
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 10
- 239000003446 ligand Substances 0.000 claims description 9
- 229940123066 Polymerase inhibitor Drugs 0.000 claims description 8
- 125000002950 monocyclic group Chemical group 0.000 claims description 8
- 229910052799 carbon Inorganic materials 0.000 claims description 7
- 125000003367 polycyclic group Chemical group 0.000 claims description 7
- 241000712464 Orthomyxoviridae Species 0.000 claims description 6
- 239000003242 anti bacterial agent Substances 0.000 claims description 6
- 241000711504 Paramyxoviridae Species 0.000 claims description 5
- 229940088710 antibiotic agent Drugs 0.000 claims description 5
- 238000002866 fluorescence resonance energy transfer Methods 0.000 claims description 5
- 229940077274 Alpha glucosidase inhibitor Drugs 0.000 claims description 4
- 241000712892 Arenaviridae Species 0.000 claims description 4
- 241000711950 Filoviridae Species 0.000 claims description 4
- 241000700586 Herpesviridae Species 0.000 claims description 4
- 241000150350 Peribunyaviridae Species 0.000 claims description 4
- 241000709664 Picornaviridae Species 0.000 claims description 4
- 241000712907 Retroviridae Species 0.000 claims description 4
- 241000711931 Rhabdoviridae Species 0.000 claims description 4
- 241000710924 Togaviridae Species 0.000 claims description 4
- 239000003888 alpha glucosidase inhibitor Substances 0.000 claims description 4
- 229940125400 channel inhibitor Drugs 0.000 claims description 4
- 241000711573 Coronaviridae Species 0.000 claims description 3
- 101800004538 Bradykinin Proteins 0.000 claims description 2
- QXZGBUJJYSLZLT-UHFFFAOYSA-N H-Arg-Pro-Pro-Gly-Phe-Ser-Pro-Phe-Arg-OH Natural products NC(N)=NCCCC(N)C(=O)N1CCCC1C(=O)N1C(C(=O)NCC(=O)NC(CC=2C=CC=CC=2)C(=O)NC(CO)C(=O)N2C(CCC2)C(=O)NC(CC=2C=CC=CC=2)C(=O)NC(CCCN=C(N)N)C(O)=O)CCC1 QXZGBUJJYSLZLT-UHFFFAOYSA-N 0.000 claims description 2
- 102100035792 Kininogen-1 Human genes 0.000 claims description 2
- 239000000867 Lipoxygenase Inhibitor Substances 0.000 claims description 2
- OFHCOWSQAMBJIW-AVJTYSNKSA-N alfacalcidol Chemical compound C1(/[C@@H]2CC[C@@H]([C@]2(CCC1)C)[C@H](C)CCCC(C)C)=C\C=C1\C[C@@H](O)C[C@H](O)C1=C OFHCOWSQAMBJIW-AVJTYSNKSA-N 0.000 claims description 2
- 239000002260 anti-inflammatory agent Substances 0.000 claims description 2
- 229940121363 anti-inflammatory agent Drugs 0.000 claims description 2
- QXZGBUJJYSLZLT-FDISYFBBSA-N bradykinin Chemical compound NC(=N)NCCC[C@H](N)C(=O)N1CCC[C@H]1C(=O)N1[C@H](C(=O)NCC(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CO)C(=O)N2[C@@H](CCC2)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)CCC1 QXZGBUJJYSLZLT-FDISYFBBSA-N 0.000 claims description 2
- 229930003827 cannabinoid Natural products 0.000 claims description 2
- 239000003557 cannabinoid Substances 0.000 claims description 2
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 2
- 125000000547 substituted alkyl group Chemical group 0.000 claims description 2
- 125000001183 hydrocarbyl group Chemical group 0.000 claims 5
- 241000710781 Flaviviridae Species 0.000 claims 2
- 238000002648 combination therapy Methods 0.000 abstract description 5
- 239000000843 powder Substances 0.000 description 70
- 239000007787 solid Substances 0.000 description 68
- 238000005160 1H NMR spectroscopy Methods 0.000 description 45
- 239000000243 solution Substances 0.000 description 44
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 42
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical class CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 42
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 39
- 239000002253 acid Substances 0.000 description 36
- 241000700605 Viruses Species 0.000 description 35
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 31
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 29
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 28
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 26
- -1 4-substituted 2,4-dioxobutanoic acid compounds Chemical class 0.000 description 25
- 230000000840 anti-viral effect Effects 0.000 description 23
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 21
- 0 **C(C(C1=C)=CN(*)c2c1nc(*=C)[s]2)=C Chemical compound **C(C(C1=C)=CN(*)c2c1nc(*=C)[s]2)=C 0.000 description 18
- 229940079593 drug Drugs 0.000 description 18
- 239000002244 precipitate Substances 0.000 description 18
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 17
- 102000028391 RNA cap binding Human genes 0.000 description 16
- 108091000106 RNA cap binding Proteins 0.000 description 16
- 238000001816 cooling Methods 0.000 description 16
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 15
- 241000712461 unidentified influenza virus Species 0.000 description 15
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 14
- DFNYGALUNNFWKJ-UHFFFAOYSA-N aminoacetonitrile Chemical compound NCC#N DFNYGALUNNFWKJ-UHFFFAOYSA-N 0.000 description 14
- 125000004494 ethyl ester group Chemical group 0.000 description 14
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 12
- 235000011054 acetic acid Nutrition 0.000 description 12
- 230000008901 benefit Effects 0.000 description 11
- 230000015572 biosynthetic process Effects 0.000 description 11
- 150000002148 esters Chemical class 0.000 description 11
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 11
- 230000002401 inhibitory effect Effects 0.000 description 11
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 10
- 210000004027 cell Anatomy 0.000 description 10
- 230000000120 cytopathologic effect Effects 0.000 description 10
- 239000000126 substance Substances 0.000 description 10
- 238000003786 synthesis reaction Methods 0.000 description 10
- 230000008685 targeting Effects 0.000 description 10
- 230000009385 viral infection Effects 0.000 description 10
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 9
- 230000037396 body weight Effects 0.000 description 9
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 9
- 230000007246 mechanism Effects 0.000 description 9
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 8
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical compound C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 8
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 8
- 229940124639 Selective inhibitor Drugs 0.000 description 8
- 150000002430 hydrocarbons Chemical group 0.000 description 8
- 108020004999 messenger RNA Proteins 0.000 description 8
- 239000011541 reaction mixture Substances 0.000 description 8
- 239000002904 solvent Substances 0.000 description 8
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 7
- 238000005481 NMR spectroscopy Methods 0.000 description 7
- 230000009471 action Effects 0.000 description 7
- 239000000890 drug combination Substances 0.000 description 7
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 7
- 235000019341 magnesium sulphate Nutrition 0.000 description 7
- 230000001850 reproductive effect Effects 0.000 description 7
- 230000004044 response Effects 0.000 description 7
- 239000002911 sialidase inhibitor Substances 0.000 description 7
- 229910052708 sodium Inorganic materials 0.000 description 7
- 239000011734 sodium Substances 0.000 description 7
- 230000005758 transcription activity Effects 0.000 description 7
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 6
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 6
- 150000001412 amines Chemical class 0.000 description 6
- DWRWSNAREGLUHZ-UHFFFAOYSA-N ethyl pyrimidine-4-carboxylate Chemical compound CCOC(=O)C1=CC=NC=N1 DWRWSNAREGLUHZ-UHFFFAOYSA-N 0.000 description 6
- 238000003818 flash chromatography Methods 0.000 description 6
- 230000006870 function Effects 0.000 description 6
- 125000001072 heteroaryl group Chemical group 0.000 description 6
- 125000000623 heterocyclic group Chemical group 0.000 description 6
- 229910052739 hydrogen Inorganic materials 0.000 description 6
- 208000015181 infectious disease Diseases 0.000 description 6
- 239000012044 organic layer Substances 0.000 description 6
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 6
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 6
- 239000000047 product Substances 0.000 description 6
- 230000001932 seasonal effect Effects 0.000 description 6
- 239000000758 substrate Substances 0.000 description 6
- 239000000725 suspension Substances 0.000 description 6
- ORILYTVJVMAKLC-UHFFFAOYSA-N adamantane Chemical class C1C(C2)CC3CC1CC2C3 ORILYTVJVMAKLC-UHFFFAOYSA-N 0.000 description 5
- 239000008346 aqueous phase Substances 0.000 description 5
- WGQKYBSKWIADBV-UHFFFAOYSA-N benzylamine Chemical compound NCC1=CC=CC=C1 WGQKYBSKWIADBV-UHFFFAOYSA-N 0.000 description 5
- 239000012043 crude product Substances 0.000 description 5
- 231100000673 dose–response relationship Toxicity 0.000 description 5
- 230000003834 intracellular effect Effects 0.000 description 5
- 239000012074 organic phase Substances 0.000 description 5
- 229960003752 oseltamivir Drugs 0.000 description 5
- NENPYTRHICXVCS-YNEHKIRRSA-N oseltamivir acid Chemical compound CCC(CC)O[C@@H]1C=C(C(O)=O)C[C@H](N)[C@H]1NC(C)=O NENPYTRHICXVCS-YNEHKIRRSA-N 0.000 description 5
- 108090000623 proteins and genes Proteins 0.000 description 5
- 102000004169 proteins and genes Human genes 0.000 description 5
- 229920006395 saturated elastomer Polymers 0.000 description 5
- 238000006467 substitution reaction Methods 0.000 description 5
- WMPPDTMATNBGJN-UHFFFAOYSA-N 2-phenylethylbromide Chemical compound BrCCC1=CC=CC=C1 WMPPDTMATNBGJN-UHFFFAOYSA-N 0.000 description 4
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 4
- 241000282412 Homo Species 0.000 description 4
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 4
- 241000712431 Influenza A virus Species 0.000 description 4
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 4
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 4
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 4
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 4
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 4
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 4
- UHOVQNZJYSORNB-UHFFFAOYSA-N benzene Substances C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 4
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 4
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 4
- 239000000872 buffer Substances 0.000 description 4
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 4
- 239000003795 chemical substances by application Substances 0.000 description 4
- 238000011161 development Methods 0.000 description 4
- LTMHNWPUDSTBKD-UHFFFAOYSA-N diethyl 2-(ethoxymethylidene)propanedioate Chemical compound CCOC=C(C(=O)OCC)C(=O)OCC LTMHNWPUDSTBKD-UHFFFAOYSA-N 0.000 description 4
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical class C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 4
- 229940042406 direct acting antivirals neuraminidase inhibitors Drugs 0.000 description 4
- 208000035475 disorder Diseases 0.000 description 4
- 239000006185 dispersion Substances 0.000 description 4
- 238000001727 in vivo Methods 0.000 description 4
- 125000005647 linker group Chemical group 0.000 description 4
- 239000002609 medium Substances 0.000 description 4
- 239000003921 oil Substances 0.000 description 4
- 235000019198 oils Nutrition 0.000 description 4
- 238000007911 parenteral administration Methods 0.000 description 4
- 229910000027 potassium carbonate Inorganic materials 0.000 description 4
- 230000000069 prophylactic effect Effects 0.000 description 4
- 125000004943 pyrimidin-6-yl group Chemical group N1=CN=CC=C1* 0.000 description 4
- 239000007921 spray Substances 0.000 description 4
- 230000001225 therapeutic effect Effects 0.000 description 4
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 4
- 230000035899 viability Effects 0.000 description 4
- UBCHPRBFMUDMNC-UHFFFAOYSA-N 1-(1-adamantyl)ethanamine Chemical compound C1C(C2)CC3CC2CC1(C(N)C)C3 UBCHPRBFMUDMNC-UHFFFAOYSA-N 0.000 description 3
- SRXFXCKTIGELTI-UHFFFAOYSA-N 2-(4-chlorophenyl)ethanamine Chemical compound NCCC1=CC=C(Cl)C=C1 SRXFXCKTIGELTI-UHFFFAOYSA-N 0.000 description 3
- OULQGLUDBAFDNN-UHFFFAOYSA-N 2-morpholin-4-yl-7-oxo-4-[(2-phenylphenyl)methyl]-[1,3]thiazolo[5,4-b]pyridine-6-carboxylic acid Chemical compound C1=2SC(N3CCOCC3)=NC=2C(=O)C(C(=O)O)=CN1CC1=CC=CC=C1C1=CC=CC=C1 OULQGLUDBAFDNN-UHFFFAOYSA-N 0.000 description 3
- MSFQEZBRFPAFEX-UHFFFAOYSA-N 4-methoxybenzenesulfonamide Chemical compound COC1=CC=C(S(N)(=O)=O)C=C1 MSFQEZBRFPAFEX-UHFFFAOYSA-N 0.000 description 3
- 125000002373 5 membered heterocyclic group Chemical group 0.000 description 3
- 125000004070 6 membered heterocyclic group Chemical group 0.000 description 3
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 3
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 3
- 241000271566 Aves Species 0.000 description 3
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 description 3
- QGJOPFRUJISHPQ-UHFFFAOYSA-N Carbon disulfide Chemical compound S=C=S QGJOPFRUJISHPQ-UHFFFAOYSA-N 0.000 description 3
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 3
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 3
- 102000005348 Neuraminidase Human genes 0.000 description 3
- 108010006232 Neuraminidase Proteins 0.000 description 3
- 229940123424 Neuraminidase inhibitor Drugs 0.000 description 3
- 229910019142 PO4 Inorganic materials 0.000 description 3
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 3
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 3
- 108020005161 RNA Caps Proteins 0.000 description 3
- IWUCXVSUMQZMFG-AFCXAGJDSA-N Ribavirin Chemical compound N1=C(C(=O)N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 IWUCXVSUMQZMFG-AFCXAGJDSA-N 0.000 description 3
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 3
- DKNWSYNQZKUICI-UHFFFAOYSA-N amantadine Chemical compound C1C(C2)CC3CC2CC1(N)C3 DKNWSYNQZKUICI-UHFFFAOYSA-N 0.000 description 3
- 229960003805 amantadine Drugs 0.000 description 3
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 3
- 229910002092 carbon dioxide Inorganic materials 0.000 description 3
- 238000006243 chemical reaction Methods 0.000 description 3
- 238000003776 cleavage reaction Methods 0.000 description 3
- 125000004122 cyclic group Chemical group 0.000 description 3
- 230000003013 cytotoxicity Effects 0.000 description 3
- 231100000135 cytotoxicity Toxicity 0.000 description 3
- LHHLYOVYBZWIGM-UHFFFAOYSA-N diethyl 2-formylbutanedioate Chemical compound CCOC(=O)CC(C=O)C(=O)OCC LHHLYOVYBZWIGM-UHFFFAOYSA-N 0.000 description 3
- 238000009472 formulation Methods 0.000 description 3
- 239000001257 hydrogen Substances 0.000 description 3
- 125000004435 hydrogen atom Chemical class [H]* 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 238000000338 in vitro Methods 0.000 description 3
- 208000037797 influenza A Diseases 0.000 description 3
- 238000001802 infusion Methods 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- QWXYZCJEXYQNEI-OSZHWHEXSA-N intermediate I Chemical compound COC(=O)[C@@]1(C=O)[C@H]2CC=[N+](C\C2=C\C)CCc2c1[nH]c1ccccc21 QWXYZCJEXYQNEI-OSZHWHEXSA-N 0.000 description 3
- 239000010410 layer Substances 0.000 description 3
- 239000002502 liposome Substances 0.000 description 3
- 230000001404 mediated effect Effects 0.000 description 3
- 239000002777 nucleoside Substances 0.000 description 3
- 150000003833 nucleoside derivatives Chemical class 0.000 description 3
- 230000000144 pharmacologic effect Effects 0.000 description 3
- 235000021317 phosphate Nutrition 0.000 description 3
- 229960000888 rimantadine Drugs 0.000 description 3
- 239000012266 salt solution Substances 0.000 description 3
- 239000000523 sample Substances 0.000 description 3
- 230000007017 scission Effects 0.000 description 3
- 239000007858 starting material Substances 0.000 description 3
- 230000035897 transcription Effects 0.000 description 3
- 238000013518 transcription Methods 0.000 description 3
- AKGJLIXNRPNPCH-UHFFFAOYSA-N (2,5-dichlorophenyl)methanamine Chemical compound NCC1=CC(Cl)=CC=C1Cl AKGJLIXNRPNPCH-UHFFFAOYSA-N 0.000 description 2
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 2
- IDPURXSQCKYKIJ-UHFFFAOYSA-N 1-(4-methoxyphenyl)methanamine Chemical compound COC1=CC=C(CN)C=C1 IDPURXSQCKYKIJ-UHFFFAOYSA-N 0.000 description 2
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 2
- RQEUFEKYXDPUSK-UHFFFAOYSA-N 1-phenylethylamine Chemical compound CC(N)C1=CC=CC=C1 RQEUFEKYXDPUSK-UHFFFAOYSA-N 0.000 description 2
- JVVRJMXHNUAPHW-UHFFFAOYSA-N 1h-pyrazol-5-amine Chemical compound NC=1C=CNN=1 JVVRJMXHNUAPHW-UHFFFAOYSA-N 0.000 description 2
- WKGDAQXKZUKLAP-UHFFFAOYSA-N 2-(4-benzylpiperazin-1-yl)-7-oxo-4-[(2-phenylphenyl)methyl]-[1,3]thiazolo[5,4-b]pyridine-6-carboxylic acid Chemical compound C1=2SC(N3CCN(CC=4C=CC=CC=4)CC3)=NC=2C(=O)C(C(=O)O)=CN1CC1=CC=CC=C1C1=CC=CC=C1 WKGDAQXKZUKLAP-UHFFFAOYSA-N 0.000 description 2
- YEDUAINPPJYDJZ-UHFFFAOYSA-N 2-hydroxybenzothiazole Chemical compound C1=CC=C2SC(O)=NC2=C1 YEDUAINPPJYDJZ-UHFFFAOYSA-N 0.000 description 2
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 2
- VGUWZCUCNQXGBU-UHFFFAOYSA-N 3-[(4-methylpiperazin-1-yl)methyl]-5-nitro-1h-indole Chemical compound C1CN(C)CCN1CC1=CNC2=CC=C([N+]([O-])=O)C=C12 VGUWZCUCNQXGBU-UHFFFAOYSA-N 0.000 description 2
- FOCJXECLIBAZSA-UHFFFAOYSA-N 3-cyclopentyl-3-oxopropanenitrile Chemical compound N#CCC(=O)C1CCCC1 FOCJXECLIBAZSA-UHFFFAOYSA-N 0.000 description 2
- MLNFMFAMNBGAQT-UHFFFAOYSA-N 4-propan-2-yloxyaniline Chemical compound CC(C)OC1=CC=C(N)C=C1 MLNFMFAMNBGAQT-UHFFFAOYSA-N 0.000 description 2
- OGHAROSJZRTIOK-KQYNXXCUSA-O 7-methylguanosine Chemical compound C1=2N=C(N)NC(=O)C=2[N+](C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O OGHAROSJZRTIOK-KQYNXXCUSA-O 0.000 description 2
- KLSJWNVTNUYHDU-UHFFFAOYSA-N Amitrole Chemical compound NC1=NC=NN1 KLSJWNVTNUYHDU-UHFFFAOYSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 2
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 2
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 2
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 2
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 2
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 2
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 2
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 2
- 229940123734 Endonuclease inhibitor Drugs 0.000 description 2
- SIXHCCPAJIVTOY-UHFFFAOYSA-N Flutimide Natural products CC(C)CC1=NC(=CC(C)C)C(=O)N(O)C1=O SIXHCCPAJIVTOY-UHFFFAOYSA-N 0.000 description 2
- 241000252870 H3N2 subtype Species 0.000 description 2
- 108090000862 Ion Channels Proteins 0.000 description 2
- 102000004310 Ion Channels Human genes 0.000 description 2
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- 229910002651 NO3 Inorganic materials 0.000 description 2
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 2
- FRWASHLBVRTOKM-UHFFFAOYSA-N O=C1C(CC(=O)O)=CNC2=CC(C(F)(F)F)=NN21 Chemical compound O=C1C(CC(=O)O)=CNC2=CC(C(F)(F)F)=NN21 FRWASHLBVRTOKM-UHFFFAOYSA-N 0.000 description 2
- 235000019502 Orange oil Nutrition 0.000 description 2
- 206010034133 Pathogen resistance Diseases 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 102000029797 Prion Human genes 0.000 description 2
- 108091000054 Prion Proteins 0.000 description 2
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 2
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 2
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 240000001068 Thogoto virus Species 0.000 description 2
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 2
- 102000004142 Trypsin Human genes 0.000 description 2
- 108090000631 Trypsin Proteins 0.000 description 2
- 208000036142 Viral infection Diseases 0.000 description 2
- 125000002877 alkyl aryl group Chemical group 0.000 description 2
- 229940024606 amino acid Drugs 0.000 description 2
- 239000004599 antimicrobial Substances 0.000 description 2
- 229940121357 antivirals Drugs 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 239000012131 assay buffer Substances 0.000 description 2
- XMIIGOLPHOKFCH-UHFFFAOYSA-N beta-phenylpropanoic acid Natural products OC(=O)CCC1=CC=CC=C1 XMIIGOLPHOKFCH-UHFFFAOYSA-N 0.000 description 2
- 125000002619 bicyclic group Chemical group 0.000 description 2
- 239000004305 biphenyl Substances 0.000 description 2
- 235000010290 biphenyl Nutrition 0.000 description 2
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 150000001721 carbon Chemical group 0.000 description 2
- 239000001768 carboxy methyl cellulose Substances 0.000 description 2
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 2
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 230000003833 cell viability Effects 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- OSASVXMJTNOKOY-UHFFFAOYSA-N chlorobutanol Chemical compound CC(C)(O)C(Cl)(Cl)Cl OSASVXMJTNOKOY-UHFFFAOYSA-N 0.000 description 2
- 229940110456 cocoa butter Drugs 0.000 description 2
- 235000019868 cocoa butter Nutrition 0.000 description 2
- 229940111134 coxibs Drugs 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- 239000003255 cyclooxygenase 2 inhibitor Substances 0.000 description 2
- 230000001086 cytosolic effect Effects 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- 238000010790 dilution Methods 0.000 description 2
- 239000012895 dilution Substances 0.000 description 2
- 125000005982 diphenylmethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])(*)C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 2
- 230000009977 dual effect Effects 0.000 description 2
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 2
- LMDGTEVHZCLPMN-UHFFFAOYSA-N ethyl 4-[2-(4-chlorophenyl)ethyl]-2-cyclopropyl-7-oxopyrazolo[1,5-a]pyrimidine-6-carboxylate Chemical compound C12=CC(C3CC3)=NN2C(=O)C(C(=O)OCC)=CN1CCC1=CC=C(Cl)C=C1 LMDGTEVHZCLPMN-UHFFFAOYSA-N 0.000 description 2
- WBJINCZRORDGAQ-UHFFFAOYSA-N ethyl formate Chemical compound CCOC=O WBJINCZRORDGAQ-UHFFFAOYSA-N 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- SIXHCCPAJIVTOY-UITAMQMPSA-N flutimide Chemical compound CC(C)CC1=N\C(=C/C(C)C)C(=O)N(O)C1=O SIXHCCPAJIVTOY-UITAMQMPSA-N 0.000 description 2
- 239000012909 foetal bovine serum Substances 0.000 description 2
- 239000012634 fragment Substances 0.000 description 2
- 210000001035 gastrointestinal tract Anatomy 0.000 description 2
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 2
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 2
- 239000004615 ingredient Substances 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 2
- 229940126181 ion channel inhibitor Drugs 0.000 description 2
- GLXDVVHUTZTUQK-UHFFFAOYSA-M lithium;hydroxide;hydrate Chemical compound [Li+].O.[OH-] GLXDVVHUTZTUQK-UHFFFAOYSA-M 0.000 description 2
- 235000015250 liver sausages Nutrition 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 239000006199 nebulizer Substances 0.000 description 2
- 125000004433 nitrogen atom Chemical group N* 0.000 description 2
- 239000002773 nucleotide Substances 0.000 description 2
- 125000003729 nucleotide group Chemical group 0.000 description 2
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 2
- 239000010502 orange oil Substances 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 2
- 239000010452 phosphate Substances 0.000 description 2
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 2
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 2
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 2
- 239000002243 precursor Substances 0.000 description 2
- 230000010076 replication Effects 0.000 description 2
- 229960000329 ribavirin Drugs 0.000 description 2
- HZCAHMRRMINHDJ-DBRKOABJSA-N ribavirin Natural products O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1N=CN=C1 HZCAHMRRMINHDJ-DBRKOABJSA-N 0.000 description 2
- 239000012047 saturated solution Substances 0.000 description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 2
- 239000012312 sodium hydride Substances 0.000 description 2
- 229910000104 sodium hydride Inorganic materials 0.000 description 2
- 159000000000 sodium salts Chemical class 0.000 description 2
- 239000000600 sorbitol Substances 0.000 description 2
- 230000001954 sterilising effect Effects 0.000 description 2
- 238000004659 sterilization and disinfection Methods 0.000 description 2
- 235000000346 sugar Nutrition 0.000 description 2
- 150000008163 sugars Chemical class 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 201000010740 swine influenza Diseases 0.000 description 2
- 208000024891 symptom Diseases 0.000 description 2
- 238000010189 synthetic method Methods 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- 235000012222 talc Nutrition 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- 239000004408 titanium dioxide Substances 0.000 description 2
- 239000012588 trypsin Substances 0.000 description 2
- 229960005486 vaccine Drugs 0.000 description 2
- 229960001028 zanamivir Drugs 0.000 description 2
- ARAIBEBZBOPLMB-UFGQHTETSA-N zanamivir Chemical compound CC(=O)N[C@@H]1[C@@H](N=C(N)N)C=C(C(O)=O)O[C@H]1[C@H](O)[C@H](O)CO ARAIBEBZBOPLMB-UFGQHTETSA-N 0.000 description 2
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 2
- LSPHULWDVZXLIL-UHFFFAOYSA-N (+/-)-Camphoric acid Chemical compound CC1(C)C(C(O)=O)CCC1(C)C(O)=O LSPHULWDVZXLIL-UHFFFAOYSA-N 0.000 description 1
- VCGRFBXVSFAGGA-UHFFFAOYSA-N (1,1-dioxo-1,4-thiazinan-4-yl)-[6-[[3-(4-fluorophenyl)-5-methyl-1,2-oxazol-4-yl]methoxy]pyridin-3-yl]methanone Chemical compound CC=1ON=C(C=2C=CC(F)=CC=2)C=1COC(N=C1)=CC=C1C(=O)N1CCS(=O)(=O)CC1 VCGRFBXVSFAGGA-UHFFFAOYSA-N 0.000 description 1
- AOSZTAHDEDLTLQ-AZKQZHLXSA-N (1S,2S,4R,8S,9S,11S,12R,13S,19S)-6-[(3-chlorophenyl)methyl]-12,19-difluoro-11-hydroxy-8-(2-hydroxyacetyl)-9,13-dimethyl-6-azapentacyclo[10.8.0.02,9.04,8.013,18]icosa-14,17-dien-16-one Chemical compound C([C@@H]1C[C@H]2[C@H]3[C@]([C@]4(C=CC(=O)C=C4[C@@H](F)C3)C)(F)[C@@H](O)C[C@@]2([C@@]1(C1)C(=O)CO)C)N1CC1=CC=CC(Cl)=C1 AOSZTAHDEDLTLQ-AZKQZHLXSA-N 0.000 description 1
- GLGNXYJARSMNGJ-VKTIVEEGSA-N (1s,2s,3r,4r)-3-[[5-chloro-2-[(1-ethyl-6-methoxy-2-oxo-4,5-dihydro-3h-1-benzazepin-7-yl)amino]pyrimidin-4-yl]amino]bicyclo[2.2.1]hept-5-ene-2-carboxamide Chemical compound CCN1C(=O)CCCC2=C(OC)C(NC=3N=C(C(=CN=3)Cl)N[C@H]3[C@H]([C@@]4([H])C[C@@]3(C=C4)[H])C(N)=O)=CC=C21 GLGNXYJARSMNGJ-VKTIVEEGSA-N 0.000 description 1
- SZUVGFMDDVSKSI-WIFOCOSTSA-N (1s,2s,3s,5r)-1-(carboxymethyl)-3,5-bis[(4-phenoxyphenyl)methyl-propylcarbamoyl]cyclopentane-1,2-dicarboxylic acid Chemical compound O=C([C@@H]1[C@@H]([C@](CC(O)=O)([C@H](C(=O)N(CCC)CC=2C=CC(OC=3C=CC=CC=3)=CC=2)C1)C(O)=O)C(O)=O)N(CCC)CC(C=C1)=CC=C1OC1=CC=CC=C1 SZUVGFMDDVSKSI-WIFOCOSTSA-N 0.000 description 1
- FTLYMKDSHNWQKD-UHFFFAOYSA-N (2,4,5-trichlorophenyl)boronic acid Chemical compound OB(O)C1=CC(Cl)=C(Cl)C=C1Cl FTLYMKDSHNWQKD-UHFFFAOYSA-N 0.000 description 1
- KDDNKZCVYQDGKE-UHFFFAOYSA-N (2-chlorophenyl)methanamine Chemical compound NCC1=CC=CC=C1Cl KDDNKZCVYQDGKE-UHFFFAOYSA-N 0.000 description 1
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 1
- NWZSZGALRFJKBT-KNIFDHDWSA-N (2s)-2,6-diaminohexanoic acid;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.NCCCC[C@H](N)C(O)=O NWZSZGALRFJKBT-KNIFDHDWSA-N 0.000 description 1
- BJFPYGGTDAYECS-UHFFFAOYSA-N (3-chlorophenyl)methanamine Chemical compound NCC1=CC=CC(Cl)=C1 BJFPYGGTDAYECS-UHFFFAOYSA-N 0.000 description 1
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 description 1
- OOKAZRDERJMRCJ-KOUAFAAESA-N (3r)-7-[(1s,2s,4ar,6s,8s)-2,6-dimethyl-8-[(2s)-2-methylbutanoyl]oxy-1,2,4a,5,6,7,8,8a-octahydronaphthalen-1-yl]-3-hydroxy-5-oxoheptanoic acid Chemical compound C1=C[C@H](C)[C@H](CCC(=O)C[C@@H](O)CC(O)=O)C2[C@@H](OC(=O)[C@@H](C)CC)C[C@@H](C)C[C@@H]21 OOKAZRDERJMRCJ-KOUAFAAESA-N 0.000 description 1
- YMVFJGSXZNNUDW-UHFFFAOYSA-N (4-chlorophenyl)methanamine Chemical compound NCC1=CC=C(Cl)C=C1 YMVFJGSXZNNUDW-UHFFFAOYSA-N 0.000 description 1
- GHOKWGTUZJEAQD-ZETCQYMHSA-N (D)-(+)-Pantothenic acid Chemical compound OCC(C)(C)[C@@H](O)C(=O)NCCC(O)=O GHOKWGTUZJEAQD-ZETCQYMHSA-N 0.000 description 1
- DEVSOMFAQLZNKR-RJRFIUFISA-N (z)-3-[3-[3,5-bis(trifluoromethyl)phenyl]-1,2,4-triazol-1-yl]-n'-pyrazin-2-ylprop-2-enehydrazide Chemical compound FC(F)(F)C1=CC(C(F)(F)F)=CC(C2=NN(\C=C/C(=O)NNC=3N=CC=NC=3)C=N2)=C1 DEVSOMFAQLZNKR-RJRFIUFISA-N 0.000 description 1
- ZILSBZLQGRBMOR-UHFFFAOYSA-N 1,3-benzodioxol-5-ylmethanamine Chemical compound NCC1=CC=C2OCOC2=C1 ZILSBZLQGRBMOR-UHFFFAOYSA-N 0.000 description 1
- WNXJIVFYUVYPPR-UHFFFAOYSA-N 1,3-dioxolane Chemical compound C1COCO1 WNXJIVFYUVYPPR-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- LKPWGXCMVLJRIK-UHFFFAOYSA-N 1-(2-bromoethyl)-3-chlorobenzene Chemical compound ClC1=CC=CC(CCBr)=C1 LKPWGXCMVLJRIK-UHFFFAOYSA-N 0.000 description 1
- GLVSPVSJMYQIPJ-UHFFFAOYSA-N 1-(2-bromoethyl)-3-fluorobenzene Chemical compound FC1=CC=CC(CCBr)=C1 GLVSPVSJMYQIPJ-UHFFFAOYSA-N 0.000 description 1
- OXHPTABOQVHKLN-UHFFFAOYSA-N 1-(2-bromoethyl)-4-methoxybenzene Chemical compound COC1=CC=C(CCBr)C=C1 OXHPTABOQVHKLN-UHFFFAOYSA-N 0.000 description 1
- YIWGJFPJRAEKMK-UHFFFAOYSA-N 1-(2H-benzotriazol-5-yl)-3-methyl-8-[2-[[3-(trifluoromethoxy)phenyl]methylamino]pyrimidine-5-carbonyl]-1,3,8-triazaspiro[4.5]decane-2,4-dione Chemical compound CN1C(=O)N(c2ccc3n[nH]nc3c2)C2(CCN(CC2)C(=O)c2cnc(NCc3cccc(OC(F)(F)F)c3)nc2)C1=O YIWGJFPJRAEKMK-UHFFFAOYSA-N 0.000 description 1
- SEXZHJJUKFXNDY-UHFFFAOYSA-N 1-(bromomethyl)-2-phenylbenzene Chemical group BrCC1=CC=CC=C1C1=CC=CC=C1 SEXZHJJUKFXNDY-UHFFFAOYSA-N 0.000 description 1
- PVOAHINGSUIXLS-UHFFFAOYSA-N 1-Methylpiperazine Chemical compound CN1CCNCC1 PVOAHINGSUIXLS-UHFFFAOYSA-N 0.000 description 1
- VQHPRVYDKRESCL-UHFFFAOYSA-N 1-bromoadamantane Chemical compound C1C(C2)CC3CC2CC1(Br)C3 VQHPRVYDKRESCL-UHFFFAOYSA-N 0.000 description 1
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 1
- MYOUHONFMXUKQJ-UHFFFAOYSA-N 2,4-dichlorobenzenesulfonamide Chemical compound NS(=O)(=O)C1=CC=C(Cl)C=C1Cl MYOUHONFMXUKQJ-UHFFFAOYSA-N 0.000 description 1
- 125000004201 2,4-dichlorophenyl group Chemical group [H]C1=C([H])C(*)=C(Cl)C([H])=C1Cl 0.000 description 1
- JSOCYZMLYCBVRB-UHFFFAOYSA-N 2-[(2,6-dichlorophenyl)methylamino]-7-oxo-4-[(2-phenylphenyl)methyl]-[1,3]thiazolo[5,4-b]pyridine-6-carboxylic acid Chemical compound C1=2SC(NCC=3C(=CC=CC=3Cl)Cl)=NC=2C(=O)C(C(=O)O)=CN1CC1=CC=CC=C1C1=CC=CC=C1 JSOCYZMLYCBVRB-UHFFFAOYSA-N 0.000 description 1
- RVHXHSAYKUNCDL-UHFFFAOYSA-N 2-[(4-chloro-2-fluorophenyl)sulfonylamino]-7-oxo-4-[(2-phenylphenyl)methyl]-[1,3]thiazolo[5,4-b]pyridine-6-carboxylic acid Chemical compound C1=2SC(NS(=O)(=O)C=3C(=CC(Cl)=CC=3)F)=NC=2C(=O)C(C(=O)O)=CN1CC1=CC=CC=C1C1=CC=CC=C1 RVHXHSAYKUNCDL-UHFFFAOYSA-N 0.000 description 1
- NAMYKGVDVNBCFQ-UHFFFAOYSA-N 2-bromopropane Chemical compound CC(C)Br NAMYKGVDVNBCFQ-UHFFFAOYSA-N 0.000 description 1
- CDTVPABKWVRVSM-UHFFFAOYSA-N 2-cyclopropyl-4-[2-(4-methoxyphenyl)ethyl]-7-oxopyrazolo[1,5-a]pyrimidine-6-carboxylic acid Chemical compound C1=CC(OC)=CC=C1CCN1C2=CC(C3CC3)=NN2C(=O)C(C(O)=O)=C1 CDTVPABKWVRVSM-UHFFFAOYSA-N 0.000 description 1
- ASUDFOJKTJLAIK-UHFFFAOYSA-N 2-methoxyethanamine Chemical compound COCCN ASUDFOJKTJLAIK-UHFFFAOYSA-N 0.000 description 1
- ZVDUZQPVDOPQDQ-UHFFFAOYSA-N 2-methylsulfanyl-7-oxo-4h-[1,3]thiazolo[5,4-b]pyridine-6-carboxylic acid Chemical compound OC(=O)C1=CN=C2SC(SC)=NC2=C1O ZVDUZQPVDOPQDQ-UHFFFAOYSA-N 0.000 description 1
- 229940080296 2-naphthalenesulfonate Drugs 0.000 description 1
- HCDMJFOHIXMBOV-UHFFFAOYSA-N 3-(2,6-difluoro-3,5-dimethoxyphenyl)-1-ethyl-8-(morpholin-4-ylmethyl)-4,7-dihydropyrrolo[4,5]pyrido[1,2-d]pyrimidin-2-one Chemical compound C=1C2=C3N(CC)C(=O)N(C=4C(=C(OC)C=C(OC)C=4F)F)CC3=CN=C2NC=1CN1CCOCC1 HCDMJFOHIXMBOV-UHFFFAOYSA-N 0.000 description 1
- HAEQAUJYNHQVHV-UHFFFAOYSA-N 3-[4-(aminomethyl)-6-(trifluoromethyl)pyridin-2-yl]oxy-N-phenylbenzamide Chemical compound NCC1=CC(=NC(=C1)C(F)(F)F)OC=1C=C(C(=O)NC2=CC=CC=C2)C=CC=1 HAEQAUJYNHQVHV-UHFFFAOYSA-N 0.000 description 1
- ICASMSGEUGPHGI-UHFFFAOYSA-N 3-amino-1h-pyrazole-5-carboxylic acid Chemical compound NC=1C=C(C(O)=O)NN=1 ICASMSGEUGPHGI-UHFFFAOYSA-N 0.000 description 1
- HRZZAOXOXSWVOI-UHFFFAOYSA-N 3-amino-5-[3-(methylamino)propyl]-4h-pyrazole-4-carbonitrile Chemical compound CNCCCC1=NNC(=N)C1C#N HRZZAOXOXSWVOI-UHFFFAOYSA-N 0.000 description 1
- XMZQWZJMTBCUFT-UHFFFAOYSA-N 3-bromopropylbenzene Chemical compound BrCCCC1=CC=CC=C1 XMZQWZJMTBCUFT-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-M 3-carboxy-2,3-dihydroxypropanoate Chemical compound OC(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-M 0.000 description 1
- ALKYHXVLJMQRLQ-UHFFFAOYSA-M 3-carboxynaphthalen-2-olate Chemical compound C1=CC=C2C=C(C([O-])=O)C(O)=CC2=C1 ALKYHXVLJMQRLQ-UHFFFAOYSA-M 0.000 description 1
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 1
- ZRPLANDPDWYOMZ-UHFFFAOYSA-N 3-cyclopentylpropionic acid Chemical compound OC(=O)CCC1CCCC1 ZRPLANDPDWYOMZ-UHFFFAOYSA-N 0.000 description 1
- 125000004180 3-fluorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C(F)=C1[H] 0.000 description 1
- XMIIGOLPHOKFCH-UHFFFAOYSA-M 3-phenylpropionate Chemical compound [O-]C(=O)CCC1=CC=CC=C1 XMIIGOLPHOKFCH-UHFFFAOYSA-M 0.000 description 1
- DYYVTFCYVZEQDG-UHFFFAOYSA-N 4-(2-bromoethyl)phenol Chemical compound OC1=CC=C(CCBr)C=C1 DYYVTFCYVZEQDG-UHFFFAOYSA-N 0.000 description 1
- KVCQTKNUUQOELD-UHFFFAOYSA-N 4-amino-n-[1-(3-chloro-2-fluoroanilino)-6-methylisoquinolin-5-yl]thieno[3,2-d]pyrimidine-7-carboxamide Chemical compound N=1C=CC2=C(NC(=O)C=3C4=NC=NC(N)=C4SC=3)C(C)=CC=C2C=1NC1=CC=CC(Cl)=C1F KVCQTKNUUQOELD-UHFFFAOYSA-N 0.000 description 1
- CWLVDUBCXAEMMZ-UHFFFAOYSA-N 4-benzhydryl-2-[(4-methoxyphenyl)methylamino]-7-oxo-[1,3]thiazolo[5,4-b]pyridine-6-carboxylic acid Chemical compound C1=CC(OC)=CC=C1CNC1=NC(C(C(C(O)=O)=CN2C(C=3C=CC=CC=3)C=3C=CC=CC=3)=O)=C2S1 CWLVDUBCXAEMMZ-UHFFFAOYSA-N 0.000 description 1
- SVUGMGZTBFEQSC-UHFFFAOYSA-N 4-benzyl-2-cyclopentyl-7-oxopyrazolo[1,5-a]pyrimidine-6-carboxylic acid Chemical compound C12=CC(C3CCCC3)=NN2C(=O)C(C(=O)O)=CN1CC1=CC=CC=C1 SVUGMGZTBFEQSC-UHFFFAOYSA-N 0.000 description 1
- CIMAHKIITQIPHH-UHFFFAOYSA-N 4-benzyl-7-oxo-2-propan-2-ylpyrazolo[1,5-a]pyrimidine-6-carboxylic acid Chemical compound C1=C(C(O)=O)C(=O)N2N=C(C(C)C)C=C2N1CC1=CC=CC=C1 CIMAHKIITQIPHH-UHFFFAOYSA-N 0.000 description 1
- HHHDJHHNEURCNV-UHFFFAOYSA-N 4-chlorobenzenesulfonamide Chemical compound NS(=O)(=O)C1=CC=C(Cl)C=C1 HHHDJHHNEURCNV-UHFFFAOYSA-N 0.000 description 1
- DQAZPZIYEOGZAF-UHFFFAOYSA-N 4-ethyl-n-[4-(3-ethynylanilino)-7-methoxyquinazolin-6-yl]piperazine-1-carboxamide Chemical compound C1CN(CC)CCN1C(=O)NC(C(=CC1=NC=N2)OC)=CC1=C2NC1=CC=CC(C#C)=C1 DQAZPZIYEOGZAF-UHFFFAOYSA-N 0.000 description 1
- 125000004203 4-hydroxyphenyl group Chemical group [H]OC1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- RYFCSYHXNOKHCK-UHFFFAOYSA-N 5-amino-3h-1,3-thiazole-2-thione Chemical compound NC1=CNC(=S)S1 RYFCSYHXNOKHCK-UHFFFAOYSA-N 0.000 description 1
- PXEDPQCHOFQXCW-UHFFFAOYSA-N 5-cyclopentyl-1h-pyrazol-3-amine Chemical compound N1N=C(N)C=C1C1CCCC1 PXEDPQCHOFQXCW-UHFFFAOYSA-N 0.000 description 1
- PWSZRRFDVPMZGM-UHFFFAOYSA-N 5-phenyl-1h-pyrazol-3-amine Chemical compound N1N=C(N)C=C1C1=CC=CC=C1 PWSZRRFDVPMZGM-UHFFFAOYSA-N 0.000 description 1
- INSBBZDRQQVATI-UHFFFAOYSA-N 5-propan-2-yl-1h-pyrazol-3-amine Chemical compound CC(C)C1=CC(N)=NN1 INSBBZDRQQVATI-UHFFFAOYSA-N 0.000 description 1
- RSIWALKZYXPAGW-NSHDSACASA-N 6-(3-fluorophenyl)-3-methyl-7-[(1s)-1-(7h-purin-6-ylamino)ethyl]-[1,3]thiazolo[3,2-a]pyrimidin-5-one Chemical compound C=1([C@@H](NC=2C=3N=CNC=3N=CN=2)C)N=C2SC=C(C)N2C(=O)C=1C1=CC=CC(F)=C1 RSIWALKZYXPAGW-NSHDSACASA-N 0.000 description 1
- FHVDTGUDJYJELY-UHFFFAOYSA-N 6-{[2-carboxy-4,5-dihydroxy-6-(phosphanyloxy)oxan-3-yl]oxy}-4,5-dihydroxy-3-phosphanyloxane-2-carboxylic acid Chemical compound O1C(C(O)=O)C(P)C(O)C(O)C1OC1C(C(O)=O)OC(OP)C(O)C1O FHVDTGUDJYJELY-UHFFFAOYSA-N 0.000 description 1
- QOFYDBAVRABJPA-UHFFFAOYSA-N 7-oxo-2-(1-phenylethylamino)-4-[(2-phenylphenyl)methyl]-[1,3]thiazolo[5,4-b]pyridine-6-carboxylic acid Chemical compound C=1C=CC=CC=1C(C)NC(S1)=NC(C(C(C(O)=O)=C2)=O)=C1N2CC1=CC=CC=C1C1=CC=CC=C1 QOFYDBAVRABJPA-UHFFFAOYSA-N 0.000 description 1
- CYJRNFFLTBEQSQ-UHFFFAOYSA-N 8-(3-methyl-1-benzothiophen-5-yl)-N-(4-methylsulfonylpyridin-3-yl)quinoxalin-6-amine Chemical compound CS(=O)(=O)C1=C(C=NC=C1)NC=1C=C2N=CC=NC2=C(C=1)C=1C=CC2=C(C(=CS2)C)C=1 CYJRNFFLTBEQSQ-UHFFFAOYSA-N 0.000 description 1
- IRBAWVGZNJIROV-SFHVURJKSA-N 9-(2-cyclopropylethynyl)-2-[[(2s)-1,4-dioxan-2-yl]methoxy]-6,7-dihydropyrimido[6,1-a]isoquinolin-4-one Chemical compound C1=C2C3=CC=C(C#CC4CC4)C=C3CCN2C(=O)N=C1OC[C@@H]1COCCO1 IRBAWVGZNJIROV-SFHVURJKSA-N 0.000 description 1
- 241000702423 Adeno-associated virus - 2 Species 0.000 description 1
- 241000202702 Adeno-associated virus - 3 Species 0.000 description 1
- 241000701242 Adenoviridae Species 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 241001533362 Astroviridae Species 0.000 description 1
- 241000972773 Aulopiformes Species 0.000 description 1
- IYHHRZBKXXKDDY-UHFFFAOYSA-N BI-605906 Chemical compound N=1C=2SC(C(N)=O)=C(N)C=2C(C(F)(F)CC)=CC=1N1CCC(S(C)(=O)=O)CC1 IYHHRZBKXXKDDY-UHFFFAOYSA-N 0.000 description 1
- KHBQMWCZKVMBLN-UHFFFAOYSA-N Benzenesulfonamide Chemical compound NS(=O)(=O)C1=CC=CC=C1 KHBQMWCZKVMBLN-UHFFFAOYSA-N 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 1
- 241000776207 Bornaviridae Species 0.000 description 1
- 241001115070 Bornavirus Species 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-M Butyrate Chemical compound CCCC([O-])=O FERIUCNNQQJTOY-UHFFFAOYSA-M 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Natural products CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 1
- KXRDQVZULWYJCO-TUIDNXLVSA-N C/C=C(/CCN(C=C1C(O)=O)c2cc(C3CC3)n[n]2C1=O)\C=C(/C#C)\Cl Chemical compound C/C=C(/CCN(C=C1C(O)=O)c2cc(C3CC3)n[n]2C1=O)\C=C(/C#C)\Cl KXRDQVZULWYJCO-TUIDNXLVSA-N 0.000 description 1
- LBCMLYNDHNPXLC-UHFFFAOYSA-N CC(C(C1NC=C2C=O)Br)=NN1C2=O Chemical compound CC(C(C1NC=C2C=O)Br)=NN1C2=O LBCMLYNDHNPXLC-UHFFFAOYSA-N 0.000 description 1
- UHNRLQRZRNKOKU-UHFFFAOYSA-N CCN(CC1=NC2=C(N1)C1=CC=C(C=C1N=C2N)C1=NNC=C1)C(C)=O Chemical compound CCN(CC1=NC2=C(N1)C1=CC=C(C=C1N=C2N)C1=NNC=C1)C(C)=O UHNRLQRZRNKOKU-UHFFFAOYSA-N 0.000 description 1
- SXDYRYDBDRMWGJ-UHFFFAOYSA-N CCOC(C(C([n]1nc(NCc2ccccc2)nc1NC)=O)=C)=O Chemical compound CCOC(C(C([n]1nc(NCc2ccccc2)nc1NC)=O)=C)=O SXDYRYDBDRMWGJ-UHFFFAOYSA-N 0.000 description 1
- PQFHFQUQQUPCDD-UHFFFAOYSA-N CCOC(C1=CN(CCc2cc(F)ccc2)c2cc(C3CC3)n[n]2C1=O)=O Chemical compound CCOC(C1=CN(CCc2cc(F)ccc2)c2cc(C3CC3)n[n]2C1=O)=O PQFHFQUQQUPCDD-UHFFFAOYSA-N 0.000 description 1
- UTYLIVGNNZMCHT-UHFFFAOYSA-O CC[O](C)C(C1=CN(Cc2ccccc2)C2=CC(C(C)C)=N[NH+]2C1=O)=O Chemical compound CC[O](C)C(C1=CN(Cc2ccccc2)C2=CC(C(C)C)=N[NH+]2C1=O)=O UTYLIVGNNZMCHT-UHFFFAOYSA-O 0.000 description 1
- DLMYHUARHITGIJ-UHFFFAOYSA-N CCc(cccc1)c1-c1ccccc1 Chemical compound CCc(cccc1)c1-c1ccccc1 DLMYHUARHITGIJ-UHFFFAOYSA-N 0.000 description 1
- TYIHAZWTGZKYHB-UHFFFAOYSA-N CN(C)c1n[nH]c(N)c1 Chemical compound CN(C)c1n[nH]c(N)c1 TYIHAZWTGZKYHB-UHFFFAOYSA-N 0.000 description 1
- 229940124638 COX inhibitor Drugs 0.000 description 1
- GAWIXWVDTYZWAW-UHFFFAOYSA-N C[CH]O Chemical group C[CH]O GAWIXWVDTYZWAW-UHFFFAOYSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 241000714198 Caliciviridae Species 0.000 description 1
- 241001493160 California encephalitis virus Species 0.000 description 1
- 241000282465 Canis Species 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 238000003734 CellTiter-Glo Luminescent Cell Viability Assay Methods 0.000 description 1
- 108091006146 Channels Proteins 0.000 description 1
- 241001502567 Chikungunya virus Species 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- HZZVJAQRINQKSD-UHFFFAOYSA-N Clavulanic acid Natural products OC(=O)C1C(=CCO)OC2CC(=O)N21 HZZVJAQRINQKSD-UHFFFAOYSA-N 0.000 description 1
- 241000204955 Colorado tick fever virus Species 0.000 description 1
- 229940126657 Compound 17 Drugs 0.000 description 1
- 241000150230 Crimean-Congo hemorrhagic fever orthonairovirus Species 0.000 description 1
- 241000701022 Cytomegalovirus Species 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- 241000104338 Delitschia confertaspora Species 0.000 description 1
- 241001533413 Deltavirus Species 0.000 description 1
- 241000725619 Dengue virus Species 0.000 description 1
- DKMROQRQHGEIOW-UHFFFAOYSA-N Diethyl succinate Chemical compound CCOC(=O)CCC(=O)OCC DKMROQRQHGEIOW-UHFFFAOYSA-N 0.000 description 1
- 241001520695 Duvenhage lyssavirus Species 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 241001115402 Ebolavirus Species 0.000 description 1
- 241001466953 Echovirus Species 0.000 description 1
- 241000709661 Enterovirus Species 0.000 description 1
- 241000988559 Enterovirus A Species 0.000 description 1
- 241000991587 Enterovirus C Species 0.000 description 1
- 102000004190 Enzymes Human genes 0.000 description 1
- 108090000790 Enzymes Proteins 0.000 description 1
- RRSNDVCODIMOFX-MPKOGUQCSA-N Fc1c(Cl)cccc1[C@H]1[C@@H](NC2(CCCCC2)[C@@]11C(=O)Nc2cc(Cl)ccc12)C(=O)Nc1ccc(cc1)C(=O)NCCCCCc1cccc2C(=O)N(Cc12)C1CCC(=O)NC1=O Chemical compound Fc1c(Cl)cccc1[C@H]1[C@@H](NC2(CCCCC2)[C@@]11C(=O)Nc2cc(Cl)ccc12)C(=O)Nc1ccc(cc1)C(=O)NCCCCCc1cccc2C(=O)N(Cc12)C1CCC(=O)NC1=O RRSNDVCODIMOFX-MPKOGUQCSA-N 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- 241000531123 GB virus C Species 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 241000190708 Guanarito mammarenavirus Species 0.000 description 1
- 241000150562 Hantaan orthohantavirus Species 0.000 description 1
- 241000893570 Hendra henipavirus Species 0.000 description 1
- 241000711549 Hepacivirus C Species 0.000 description 1
- 241000700739 Hepadnaviridae Species 0.000 description 1
- 241000700721 Hepatitis B virus Species 0.000 description 1
- 241000724675 Hepatitis E virus Species 0.000 description 1
- 206010019799 Hepatitis viral Diseases 0.000 description 1
- 241000709721 Hepatovirus A Species 0.000 description 1
- 241001122120 Hepeviridae Species 0.000 description 1
- 244000309467 Human Coronavirus Species 0.000 description 1
- 241000598171 Human adenovirus sp. Species 0.000 description 1
- 241000701085 Human alphaherpesvirus 3 Species 0.000 description 1
- 241000724642 Human astrovirus 1 Species 0.000 description 1
- 241001207270 Human enterovirus Species 0.000 description 1
- 241000713673 Human foamy virus Species 0.000 description 1
- 241001502974 Human gammaherpesvirus 8 Species 0.000 description 1
- 241000725303 Human immunodeficiency virus Species 0.000 description 1
- 241000702617 Human parvovirus B19 Species 0.000 description 1
- 241000829111 Human polyomavirus 1 Species 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- WRYCSMQKUKOKBP-UHFFFAOYSA-N Imidazolidine Chemical compound C1CNCN1 WRYCSMQKUKOKBP-UHFFFAOYSA-N 0.000 description 1
- 241000134304 Influenza A virus H3N2 Species 0.000 description 1
- 102000014150 Interferons Human genes 0.000 description 1
- 108010050904 Interferons Proteins 0.000 description 1
- 241001661732 Isavirus Species 0.000 description 1
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical class CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 1
- 241000710842 Japanese encephalitis virus Species 0.000 description 1
- 241000712890 Junin mammarenavirus Species 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 241000712902 Lassa mammarenavirus Species 0.000 description 1
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 1
- 241000712899 Lymphocytic choriomeningitis mammarenavirus Species 0.000 description 1
- 102000043136 MAP kinase family Human genes 0.000 description 1
- 108091054455 MAP kinase family Proteins 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- 102100024295 Maltase-glucoamylase Human genes 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 241001480504 Mammalian orthoreovirus 1 Species 0.000 description 1
- 241001480506 Mammalian orthoreovirus 2 Species 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 241001115401 Marburgvirus Species 0.000 description 1
- 241000712079 Measles morbillivirus Species 0.000 description 1
- 108060004795 Methyltransferase Proteins 0.000 description 1
- 102100024193 Mitogen-activated protein kinase 1 Human genes 0.000 description 1
- 229920000881 Modified starch Polymers 0.000 description 1
- 241000711386 Mumps virus Species 0.000 description 1
- YWPQKGKQWHBCDH-UHFFFAOYSA-N N=1N2C(=O)C(C(=O)O)=CNC2=CC=1C1CC1 Chemical compound N=1N2C(=O)C(C(=O)O)=CNC2=CC=1C1CC1 YWPQKGKQWHBCDH-UHFFFAOYSA-N 0.000 description 1
- QXWLARXCXIBIDC-UHFFFAOYSA-N N=1N2C(=O)C(CC(=O)O)=CNC2=CC=1C1=CC=CC=C1 Chemical compound N=1N2C(=O)C(CC(=O)O)=CNC2=CC=1C1=CC=CC=C1 QXWLARXCXIBIDC-UHFFFAOYSA-N 0.000 description 1
- NURWYRRGLDKRSX-UHFFFAOYSA-N N=1N2C(=O)C(CC(=O)O)=CNC2=CC=1C1CC1 Chemical compound N=1N2C(=O)C(CC(=O)O)=CNC2=CC=1C1CC1 NURWYRRGLDKRSX-UHFFFAOYSA-N 0.000 description 1
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 description 1
- 241000710944 O'nyong-nyong virus Species 0.000 description 1
- KVQFJDILLCBCIX-UHFFFAOYSA-N O=C1C(C(=O)OCC)=CNC2=CC(C(O)=O)=NN21 Chemical compound O=C1C(C(=O)OCC)=CNC2=CC(C(O)=O)=NN21 KVQFJDILLCBCIX-UHFFFAOYSA-N 0.000 description 1
- KCRIHOOVSNHMLN-UHFFFAOYSA-N O=C1C(C(=O)OCC)=CNC2=CC=NN21 Chemical compound O=C1C(C(=O)OCC)=CNC2=CC=NN21 KCRIHOOVSNHMLN-UHFFFAOYSA-N 0.000 description 1
- JJJDUQYIHZKCFP-UHFFFAOYSA-N O=C1C(CC(=O)OCC)=CNC2=C(Br)C(C)=NN21 Chemical compound O=C1C(CC(=O)OCC)=CNC2=C(Br)C(C)=NN21 JJJDUQYIHZKCFP-UHFFFAOYSA-N 0.000 description 1
- JDQORWARSHCXPQ-UHFFFAOYSA-N O=C1C(CC(=O)OCC)=CNC2=CC(C(F)(F)F)=NN21 Chemical compound O=C1C(CC(=O)OCC)=CNC2=CC(C(F)(F)F)=NN21 JDQORWARSHCXPQ-UHFFFAOYSA-N 0.000 description 1
- PHCMTRQADOUDSH-UHFFFAOYSA-N OC(C1=CN(CCc2ccccc2)c2cc(C3CC3)n[n]2C1=O)=O Chemical compound OC(C1=CN(CCc2ccccc2)c2cc(C3CC3)n[n]2C1=O)=O PHCMTRQADOUDSH-UHFFFAOYSA-N 0.000 description 1
- 108091034117 Oligonucleotide Proteins 0.000 description 1
- 241000713112 Orthobunyavirus Species 0.000 description 1
- 241000150452 Orthohantavirus Species 0.000 description 1
- 241000150218 Orthonairovirus Species 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 description 1
- 101150020891 PRKCA gene Proteins 0.000 description 1
- 208000002606 Paramyxoviridae Infections Diseases 0.000 description 1
- 241000991583 Parechovirus Species 0.000 description 1
- 241000701945 Parvoviridae Species 0.000 description 1
- 241000713137 Phlebovirus Species 0.000 description 1
- 108091000080 Phosphotransferase Proteins 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 241001631648 Polyomaviridae Species 0.000 description 1
- 229920003081 Povidone K 30 Polymers 0.000 description 1
- 241000700625 Poxviridae Species 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 102100038277 Prostaglandin G/H synthase 1 Human genes 0.000 description 1
- 108050003243 Prostaglandin G/H synthase 1 Proteins 0.000 description 1
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 1
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 1
- 241000150264 Puumala orthohantavirus Species 0.000 description 1
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 1
- 241000711798 Rabies lyssavirus Species 0.000 description 1
- 241000702247 Reoviridae Species 0.000 description 1
- 241000725643 Respiratory syncytial virus Species 0.000 description 1
- 102000004389 Ribonucleoproteins Human genes 0.000 description 1
- 108010081734 Ribonucleoproteins Proteins 0.000 description 1
- 241000710942 Ross River virus Species 0.000 description 1
- 241001137860 Rotavirus A Species 0.000 description 1
- 241000710799 Rubella virus Species 0.000 description 1
- 241000192617 Sabia mammarenavirus Species 0.000 description 1
- 241000149820 Sakhalin orthonairovirus Species 0.000 description 1
- 241001135555 Sandfly fever Sicilian virus Species 0.000 description 1
- 241000369753 Sapporo virus Species 0.000 description 1
- 241000150278 Seoul orthohantavirus Species 0.000 description 1
- 241000700584 Simplexvirus Species 0.000 description 1
- 241000150288 Sin Nombre orthohantavirus Species 0.000 description 1
- 108020004459 Small interfering RNA Proteins 0.000 description 1
- 101001039853 Sonchus yellow net virus Matrix protein Proteins 0.000 description 1
- 239000004147 Sorbitan trioleate Substances 0.000 description 1
- PRXRUNOAOLTIEF-ADSICKODSA-N Sorbitan trioleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@@H](OC(=O)CCCCCCC\C=C/CCCCCCCC)[C@H]1OC[C@H](O)[C@H]1OC(=O)CCCCCCC\C=C/CCCCCCCC PRXRUNOAOLTIEF-ADSICKODSA-N 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- 208000000389 T-cell leukemia Diseases 0.000 description 1
- 208000028530 T-cell lymphoblastic leukemia/lymphoma Diseases 0.000 description 1
- 229920002253 Tannate Polymers 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 1
- 241000710771 Tick-borne encephalitis virus Species 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- 241000713152 Uukuniemi virus Species 0.000 description 1
- 241000700647 Variola virus Species 0.000 description 1
- 241000251539 Vertebrata <Metazoa> Species 0.000 description 1
- 241000711975 Vesicular stomatitis virus Species 0.000 description 1
- 108010067390 Viral Proteins Proteins 0.000 description 1
- 108020000999 Viral RNA Proteins 0.000 description 1
- 241000710886 West Nile virus Species 0.000 description 1
- 241000710772 Yellow fever virus Species 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- YMHQNVGWFTZOAO-UHFFFAOYSA-N [1,3]thiazolo[5,4-b]pyridine-6-carboxylic acid Chemical compound OC(=O)C1=CN=C2SC=NC2=C1 YMHQNVGWFTZOAO-UHFFFAOYSA-N 0.000 description 1
- SAHIZENKTPRYSN-UHFFFAOYSA-N [2-[3-(phenoxymethyl)phenoxy]-6-(trifluoromethyl)pyridin-4-yl]methanamine Chemical compound O(C1=CC=CC=C1)CC=1C=C(OC2=NC(=CC(=C2)CN)C(F)(F)F)C=CC=1 SAHIZENKTPRYSN-UHFFFAOYSA-N 0.000 description 1
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 1
- 230000005856 abnormality Effects 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 1
- 108700010877 adenoviridae proteins Proteins 0.000 description 1
- WNLRTRBMVRJNCN-UHFFFAOYSA-L adipate(2-) Chemical compound [O-]C(=O)CCCCC([O-])=O WNLRTRBMVRJNCN-UHFFFAOYSA-L 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 239000000556 agonist Substances 0.000 description 1
- QBYJBZPUGVGKQQ-SJJAEHHWSA-N aldrin Chemical compound C1[C@H]2C=C[C@@H]1[C@H]1[C@@](C3(Cl)Cl)(Cl)C(Cl)=C(Cl)[C@@]3(Cl)[C@H]12 QBYJBZPUGVGKQQ-SJJAEHHWSA-N 0.000 description 1
- 229940072056 alginate Drugs 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 125000003342 alkenyl group Chemical group 0.000 description 1
- 125000003545 alkoxy group Chemical group 0.000 description 1
- 125000000304 alkynyl group Chemical group 0.000 description 1
- 108010028144 alpha-Glucosidases Proteins 0.000 description 1
- AWUCVROLDVIAJX-UHFFFAOYSA-N alpha-glycerophosphate Natural products OCC(O)COP(O)(O)=O AWUCVROLDVIAJX-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 235000019270 ammonium chloride Nutrition 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 239000000427 antigen Substances 0.000 description 1
- 102000036639 antigens Human genes 0.000 description 1
- 108091007433 antigens Proteins 0.000 description 1
- 229940124977 antiviral medication Drugs 0.000 description 1
- 125000003710 aryl alkyl group Chemical group 0.000 description 1
- 125000005228 aryl sulfonate group Chemical group 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- 206010064097 avian influenza Diseases 0.000 description 1
- OISFUZRUIGGTSD-LJTMIZJLSA-N azane;(2r,3r,4r,5s)-6-(methylamino)hexane-1,2,3,4,5-pentol Chemical compound N.CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO OISFUZRUIGGTSD-LJTMIZJLSA-N 0.000 description 1
- 239000002585 base Substances 0.000 description 1
- 239000000440 bentonite Substances 0.000 description 1
- 229910000278 bentonite Inorganic materials 0.000 description 1
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 229940050390 benzoate Drugs 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000005540 biological transmission Effects 0.000 description 1
- IULFXBLVJIPESI-UHFFFAOYSA-N bis(methylsulfanyl)methylidenecyanamide Chemical compound CSC(SC)=NC#N IULFXBLVJIPESI-UHFFFAOYSA-N 0.000 description 1
- 239000012267 brine Substances 0.000 description 1
- OQROAIRCEOBYJA-UHFFFAOYSA-N bromodiphenylmethane Chemical compound C=1C=CC=CC=1C(Br)C1=CC=CC=C1 OQROAIRCEOBYJA-UHFFFAOYSA-N 0.000 description 1
- UUWSLBWDFJMSFP-UHFFFAOYSA-N bromomethylcyclohexane Chemical compound BrCC1CCCCC1 UUWSLBWDFJMSFP-UHFFFAOYSA-N 0.000 description 1
- 239000000337 buffer salt Substances 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- FATUQANACHZLRT-KMRXSBRUSA-L calcium glucoheptonate Chemical compound [Ca+2].OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)C([O-])=O.OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)C([O-])=O FATUQANACHZLRT-KMRXSBRUSA-L 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 125000002837 carbocyclic group Chemical group 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical compound OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 1
- 150000007942 carboxylates Chemical class 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 238000003570 cell viability assay Methods 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- 229960004926 chlorobutanol Drugs 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 229940090805 clavulanate Drugs 0.000 description 1
- HZZVJAQRINQKSD-PBFISZAISA-N clavulanic acid Chemical compound OC(=O)[C@H]1C(=C/CO)/O[C@@H]2CC(=O)N21 HZZVJAQRINQKSD-PBFISZAISA-N 0.000 description 1
- 239000012230 colorless oil Substances 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 229940125797 compound 12 Drugs 0.000 description 1
- 229940126543 compound 14 Drugs 0.000 description 1
- 229940125758 compound 15 Drugs 0.000 description 1
- 238000005564 crystal structure determination Methods 0.000 description 1
- 239000003260 cyclooxygenase 1 inhibitor Substances 0.000 description 1
- NISGSNTVMOOSJQ-UHFFFAOYSA-N cyclopentanamine Chemical compound NC1CCCC1 NISGSNTVMOOSJQ-UHFFFAOYSA-N 0.000 description 1
- 150000001940 cyclopentanes Chemical class 0.000 description 1
- 231100000433 cytotoxic Toxicity 0.000 description 1
- 230000001472 cytotoxic effect Effects 0.000 description 1
- 238000006731 degradation reaction Methods 0.000 description 1
- ACYGYJFTZSAZKR-UHFFFAOYSA-J dicalcium;2-[2-[bis(carboxylatomethyl)amino]ethyl-(carboxylatomethyl)amino]acetate Chemical compound [Ca+2].[Ca+2].[O-]C(=O)CN(CC([O-])=O)CCN(CC([O-])=O)CC([O-])=O ACYGYJFTZSAZKR-UHFFFAOYSA-J 0.000 description 1
- 125000004177 diethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- SPCNPOWOBZQWJK-UHFFFAOYSA-N dimethoxy-(2-propan-2-ylsulfanylethylsulfanyl)-sulfanylidene-$l^{5}-phosphane Chemical compound COP(=S)(OC)SCCSC(C)C SPCNPOWOBZQWJK-UHFFFAOYSA-N 0.000 description 1
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 239000001177 diphosphate Substances 0.000 description 1
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical compound [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 1
- 235000011180 diphosphates Nutrition 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 239000002612 dispersion medium Substances 0.000 description 1
- 229940000406 drug candidate Drugs 0.000 description 1
- 238000007876 drug discovery Methods 0.000 description 1
- 239000013583 drug formulation Substances 0.000 description 1
- 239000003596 drug target Substances 0.000 description 1
- 229940112141 dry powder inhaler Drugs 0.000 description 1
- 241001493065 dsRNA viruses Species 0.000 description 1
- 229940009662 edetate Drugs 0.000 description 1
- 229950005627 embonate Drugs 0.000 description 1
- 206010014599 encephalitis Diseases 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- OAIUHDMUDJPZAZ-UHFFFAOYSA-N ethyl 2-(2-anilino-7-oxo-1h-[1,2,4]triazolo[1,5-a]pyrimidin-6-yl)acetate Chemical compound N=1N2C(=O)C(CC(=O)OCC)=CNC2=NC=1NC1=CC=CC=C1 OAIUHDMUDJPZAZ-UHFFFAOYSA-N 0.000 description 1
- GEKFSDQHRFPVNL-UHFFFAOYSA-N ethyl 2-(2-cyclopropyl-4-methyl-7-oxopyrazolo[1,5-a]pyrimidin-6-yl)acetate Chemical compound N=1N2C(=O)C(CC(=O)OCC)=CN(C)C2=CC=1C1CC1 GEKFSDQHRFPVNL-UHFFFAOYSA-N 0.000 description 1
- HPPLUDBPKKDPSR-UHFFFAOYSA-N ethyl 2-[(4-ethoxycarbonylphenyl)sulfonylamino]-7-oxo-4-[(2-phenylphenyl)methyl]-[1,3]thiazolo[5,4-b]pyridine-6-carboxylate Chemical compound C1=CC(C(=O)OCC)=CC=C1S(=O)(=O)NC1=NC(C(C(C(=O)OCC)=CN2CC=3C(=CC=CC=3)C=3C=CC=CC=3)=O)=C2S1 HPPLUDBPKKDPSR-UHFFFAOYSA-N 0.000 description 1
- XHFSMKZTDHZLDA-UHFFFAOYSA-N ethyl 2-[(4-methoxyphenyl)methylamino]-7-oxo-4-[(2-phenylphenyl)methyl]-[1,3]thiazolo[5,4-b]pyridine-6-carboxylate Chemical compound C1=2SC(NCC=3C=CC(OC)=CC=3)=NC=2C(=O)C(C(=O)OCC)=CN1CC1=CC=CC=C1C1=CC=CC=C1 XHFSMKZTDHZLDA-UHFFFAOYSA-N 0.000 description 1
- GGTKTOGNUZJMQZ-UHFFFAOYSA-N ethyl 2-[(4-methoxyphenyl)sulfonylamino]-7-oxo-4-[(2-phenylphenyl)methyl]-[1,3]thiazolo[5,4-b]pyridine-6-carboxylate Chemical compound C1=2SC(NS(=O)(=O)C=3C=CC(OC)=CC=3)=NC=2C(=O)C(C(=O)OCC)=CN1CC1=CC=CC=C1C1=CC=CC=C1 GGTKTOGNUZJMQZ-UHFFFAOYSA-N 0.000 description 1
- MVPICKVDHDWCJQ-UHFFFAOYSA-N ethyl 3-pyrrolidin-1-ylpropanoate Chemical compound CCOC(=O)CCN1CCCC1 MVPICKVDHDWCJQ-UHFFFAOYSA-N 0.000 description 1
- WCTQEHGXBKFLKG-UHFFFAOYSA-N ethyl 4-(aminomethyl)benzoate Chemical compound CCOC(=O)C1=CC=C(CN)C=C1 WCTQEHGXBKFLKG-UHFFFAOYSA-N 0.000 description 1
- CTLARIZXXZCEEM-UHFFFAOYSA-N ethyl 4-[2-(3-chlorophenyl)ethyl]-2-cyclopropyl-7-oxopyrazolo[1,5-a]pyrimidine-6-carboxylate Chemical compound C12=CC(C3CC3)=NN2C(=O)C(C(=O)OCC)=CN1CCC1=CC=CC(Cl)=C1 CTLARIZXXZCEEM-UHFFFAOYSA-N 0.000 description 1
- BBUYDEGLTXGIEL-UHFFFAOYSA-N ethyl 4-benzyl-2-cyclopentyl-7-oxopyrazolo[1,5-a]pyrimidine-6-carboxylate Chemical compound C12=CC(C3CCCC3)=NN2C(=O)C(C(=O)OCC)=CN1CC1=CC=CC=C1 BBUYDEGLTXGIEL-UHFFFAOYSA-N 0.000 description 1
- QJULLHHLCIBXHI-UHFFFAOYSA-N ethyl 4-benzyl-2-cyclopropyl-7-oxopyrazolo[1,5-a]pyrimidine-6-carboxylate Chemical compound C12=CC(C3CC3)=NN2C(=O)C(C(=O)OCC)=CN1CC1=CC=CC=C1 QJULLHHLCIBXHI-UHFFFAOYSA-N 0.000 description 1
- DFTXGSKTFDBVQM-UHFFFAOYSA-N ethyl 4-sulfamoylbenzoate Chemical compound CCOC(=O)C1=CC=C(S(N)(=O)=O)C=C1 DFTXGSKTFDBVQM-UHFFFAOYSA-N 0.000 description 1
- KUDYZMHPDZSHRO-UHFFFAOYSA-N ethyl 7-oxo-4-(2-phenylethyl)-2-propan-2-ylpyrazolo[1,5-a]pyrimidine-6-carboxylate Chemical compound C12=CC(C(C)C)=NN2C(=O)C(C(=O)OCC)=CN1CCC1=CC=CC=C1 KUDYZMHPDZSHRO-UHFFFAOYSA-N 0.000 description 1
- 125000000816 ethylene group Chemical group [H]C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 230000007717 exclusion Effects 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- ZCGNOVWYSGBHAU-UHFFFAOYSA-N favipiravir Chemical compound NC(=O)C1=NC(F)=CNC1=O ZCGNOVWYSGBHAU-UHFFFAOYSA-N 0.000 description 1
- 229950008454 favipiravir Drugs 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-L fumarate(2-) Chemical class [O-]C(=O)\C=C\C([O-])=O VZCYOOQTPOCHFL-OWOJBTEDSA-L 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 230000002538 fungal effect Effects 0.000 description 1
- 108020001507 fusion proteins Proteins 0.000 description 1
- 102000037865 fusion proteins Human genes 0.000 description 1
- LNTHITQWFMADLM-UHFFFAOYSA-N gallic acid Chemical compound OC(=O)C1=CC(O)=C(O)C(O)=C1 LNTHITQWFMADLM-UHFFFAOYSA-N 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 229960001731 gluceptate Drugs 0.000 description 1
- KWMLJOLKUYYJFJ-VFUOTHLCSA-N glucoheptonic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)[C@@H](O)C(O)=O KWMLJOLKUYYJFJ-VFUOTHLCSA-N 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- PKWIYNIDEDLDCJ-UHFFFAOYSA-N guanazole Chemical compound NC1=NNC(N)=N1 PKWIYNIDEDLDCJ-UHFFFAOYSA-N 0.000 description 1
- UYTPUPDQBNUYGX-UHFFFAOYSA-N guanine Chemical class O=C1NC(N)=NC2=C1N=CN2 UYTPUPDQBNUYGX-UHFFFAOYSA-N 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 230000003862 health status Effects 0.000 description 1
- 208000006454 hepatitis Diseases 0.000 description 1
- 231100000283 hepatitis Toxicity 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 1
- XGIHQYAWBCFNPY-AZOCGYLKSA-N hydrabamine Chemical compound C([C@@H]12)CC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC[C@@]1(C)CNCCNC[C@@]1(C)[C@@H]2CCC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC1 XGIHQYAWBCFNPY-AZOCGYLKSA-N 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine monohydrate Substances O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 1
- 239000008172 hydrogenated vegetable oil Substances 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 1
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 239000000411 inducer Substances 0.000 description 1
- 230000002458 infectious effect Effects 0.000 description 1
- 230000001524 infective effect Effects 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 230000002452 interceptive effect Effects 0.000 description 1
- 229940079322 interferon Drugs 0.000 description 1
- 238000007917 intracranial administration Methods 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 150000002500 ions Chemical class 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 239000007951 isotonicity adjuster Substances 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 229940099584 lactobionate Drugs 0.000 description 1
- JYTUSYBCFIZPBE-AMTLMPIISA-N lactobionic acid Chemical compound OC(=O)[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O JYTUSYBCFIZPBE-AMTLMPIISA-N 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 238000004020 luminiscence type Methods 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 208000030194 mouth disease Diseases 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- OREFDBCAINNFMR-UHFFFAOYSA-N n-[2-(4-chlorophenyl)ethyl]-3-nitro-1h-pyrazole-5-carboxamide Chemical compound N1C([N+](=O)[O-])=CC(C(=O)NCCC=2C=CC(Cl)=CC=2)=N1 OREFDBCAINNFMR-UHFFFAOYSA-N 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-M naphthalene-2-sulfonate Chemical compound C1=CC=CC2=CC(S(=O)(=O)[O-])=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-M 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 235000001968 nicotinic acid Nutrition 0.000 description 1
- 239000011664 nicotinic acid Substances 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 108700013356 oplunofusp Proteins 0.000 description 1
- 239000013110 organic ligand Substances 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- PGZUMBJQJWIWGJ-ONAKXNSWSA-N oseltamivir phosphate Chemical compound OP(O)(O)=O.CCOC(=O)C1=C[C@@H](OC(CC)CC)[C@H](NC(C)=O)[C@@H](N)C1 PGZUMBJQJWIWGJ-ONAKXNSWSA-N 0.000 description 1
- RZXMPPFPUUCRFN-UHFFFAOYSA-N p-toluidine Chemical compound CC1=CC=C(N)C=C1 RZXMPPFPUUCRFN-UHFFFAOYSA-N 0.000 description 1
- 229940014662 pantothenate Drugs 0.000 description 1
- 235000019161 pantothenic acid Nutrition 0.000 description 1
- 239000011713 pantothenic acid Substances 0.000 description 1
- 239000002245 particle Substances 0.000 description 1
- 230000001717 pathogenic effect Effects 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- XRQDFNLINLXZLB-CKIKVBCHSA-N peramivir Chemical compound CCC(CC)[C@H](NC(C)=O)[C@@H]1[C@H](O)[C@@H](C(O)=O)C[C@H]1NC(N)=N XRQDFNLINLXZLB-CKIKVBCHSA-N 0.000 description 1
- 229960001084 peramivir Drugs 0.000 description 1
- 230000008447 perception Effects 0.000 description 1
- JRKICGRDRMAZLK-UHFFFAOYSA-L peroxydisulfate Chemical compound [O-]S(=O)(=O)OOS([O-])(=O)=O JRKICGRDRMAZLK-UHFFFAOYSA-L 0.000 description 1
- 229940124531 pharmaceutical excipient Drugs 0.000 description 1
- 229960003742 phenol Drugs 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 102000020233 phosphotransferase Human genes 0.000 description 1
- 229940075930 picrate Drugs 0.000 description 1
- OXNIZHLAWKMVMX-UHFFFAOYSA-M picrate anion Chemical compound [O-]C1=C([N+]([O-])=O)C=C([N+]([O-])=O)C=C1[N+]([O-])=O OXNIZHLAWKMVMX-UHFFFAOYSA-M 0.000 description 1
- CYJAWBVQRMVFEO-UHFFFAOYSA-N piperazine-2,6-dione Chemical class O=C1CNCC(=O)N1 CYJAWBVQRMVFEO-UHFFFAOYSA-N 0.000 description 1
- HDOWRFHMPULYOA-UHFFFAOYSA-N piperidin-4-ol Chemical compound OC1CCNCC1 HDOWRFHMPULYOA-UHFFFAOYSA-N 0.000 description 1
- 239000011505 plaster Substances 0.000 description 1
- 229920001223 polyethylene glycol Polymers 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 159000000001 potassium salts Chemical class 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- 239000011253 protective coating Substances 0.000 description 1
- 230000005180 public health Effects 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 125000001453 quaternary ammonium group Chemical group 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 238000009877 rendering Methods 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 229940085605 saccharin sodium Drugs 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- 235000019515 salmon Nutrition 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- XGVXKJKTISMIOW-ZDUSSCGKSA-N simurosertib Chemical compound N1N=CC(C=2SC=3C(=O)NC(=NC=3C=2)[C@H]2N3CCC(CC3)C2)=C1C XGVXKJKTISMIOW-ZDUSSCGKSA-N 0.000 description 1
- 239000002356 single layer Substances 0.000 description 1
- AWUCVROLDVIAJX-GSVOUGTGSA-N sn-glycerol 3-phosphate Chemical compound OC[C@@H](O)COP(O)(O)=O AWUCVROLDVIAJX-GSVOUGTGSA-N 0.000 description 1
- WXMKPNITSTVMEF-UHFFFAOYSA-M sodium benzoate Chemical compound [Na+].[O-]C(=O)C1=CC=CC=C1 WXMKPNITSTVMEF-UHFFFAOYSA-M 0.000 description 1
- 239000004299 sodium benzoate Substances 0.000 description 1
- 235000010234 sodium benzoate Nutrition 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 229940045902 sodium stearyl fumarate Drugs 0.000 description 1
- YQZWHIDCXMDDKM-UHFFFAOYSA-M sodium;2-(1,3-benzodioxol-5-ylmethylamino)-7-oxo-1h-[1,2,4]triazolo[1,5-a]pyrimidine-6-carboxylate Chemical compound [Na+].C1=C2OCOC2=CC(CNC2=NC3=NC=C(C(N3N2)=O)C(=O)[O-])=C1 YQZWHIDCXMDDKM-UHFFFAOYSA-M 0.000 description 1
- HSFQBFMEWSTNOW-UHFFFAOYSA-N sodium;carbanide Chemical group [CH3-].[Na+] HSFQBFMEWSTNOW-UHFFFAOYSA-N 0.000 description 1
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 235000019337 sorbitan trioleate Nutrition 0.000 description 1
- 229960000391 sorbitan trioleate Drugs 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 125000005346 substituted cycloalkyl group Chemical group 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 1
- 150000003462 sulfoxides Chemical class 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- HKYHBMLIEAMWRO-UHFFFAOYSA-N sy002454 Chemical compound OC(=O)C1=CC([N+]([O-])=O)=NN1 HKYHBMLIEAMWRO-UHFFFAOYSA-N 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 229940061367 tamiflu Drugs 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 229950002757 teoclate Drugs 0.000 description 1
- RYYWUUFWQRZTIU-UHFFFAOYSA-K thiophosphate Chemical compound [O-]P([O-])([O-])=S RYYWUUFWQRZTIU-UHFFFAOYSA-K 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 238000011277 treatment modality Methods 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical class [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- CYRMSUTZVYGINF-UHFFFAOYSA-N trichlorofluoromethane Chemical compound FC(Cl)(Cl)Cl CYRMSUTZVYGINF-UHFFFAOYSA-N 0.000 description 1
- 229940029284 trichlorofluoromethane Drugs 0.000 description 1
- 239000001226 triphosphate Substances 0.000 description 1
- 235000011178 triphosphate Nutrition 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- ZDPHROOEEOARMN-UHFFFAOYSA-N undecanoic acid Chemical compound CCCCCCCCCCC(O)=O ZDPHROOEEOARMN-UHFFFAOYSA-N 0.000 description 1
- 229940070710 valerate Drugs 0.000 description 1
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical compound CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 230000029812 viral genome replication Effects 0.000 description 1
- 230000006490 viral transcription Effects 0.000 description 1
- 210000002845 virion Anatomy 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000000230 xanthan gum Substances 0.000 description 1
- 229920001285 xanthan gum Polymers 0.000 description 1
- 235000010493 xanthan gum Nutrition 0.000 description 1
- 229940082509 xanthan gum Drugs 0.000 description 1
- 229940051021 yellow-fever virus Drugs 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- XOOUIPVCVHRTMJ-UHFFFAOYSA-L zinc stearate Chemical compound [Zn+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O XOOUIPVCVHRTMJ-UHFFFAOYSA-L 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/496—Non-condensed piperazines containing further heterocyclic rings, e.g. rifampin, thiothixene or sparfloxacin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4985—Pyrazines or piperazines ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/16—Antivirals for RNA viruses for influenza or rhinoviruses
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/04—Ortho-condensed systems
Definitions
- the present invention relates to a compound having the general formula (C), optionally in the form of a pharmaceuticaiiy acceptable salt, solvate, polymorph, codrug, cocrystal, prodrug, tautomer, racemate, enantiomer, or diastereomer or mixture thereof,
- H5N1 could have been more easily transmissible between humans or the new A/H1 N1 could have been more virulent and could have carried the single point mutation that confers Tamiflu resistance (Neumann et al., Nature, 2009 (18; 459(7249) 931-939)), as many seasonal H1 N1 strains have recently done (Dharan et al., The Journal of the American Medical Association, 2009 Mar 11 ; 301 (10), 1034-1041; Moscona et al., The New England Journal of Medicine, 2009 (Mar 5;360( 0) pp 953-956)).
- the delay in generating and deploying a vaccine -6 months in the relatively favourable case of A/H1 N1 and still not a solved problem for H5N1 ) could have been catastrophically costly in human lives and societal disruption.
- ribavirin is only approved in a few countries, probably due to severe side effects (Furuta et al., ANTIMICROBIAL AGENTS AND CHEMOTHERAPY, 2005, p. 981-986).
- new antiviral compounds are needed, preferably directed against different targets. Influenza virus as well as Thogotovirus belong to the family of Orthomyxoviridae which, as well as the family of the Bunyaviridae, including the Hantavirus, Nairovirus, Orthobunyavirus, and Phlebovirus, are negative stranded RNA viruses.
- RNA dependent RNA polymerase which carries out (i) the initial copying of the single-stranded virion RNA (vRNA) into viral mRNAs and (ii) the vRNA replication.
- This enzyme a trimeric complex composed of subunits PA, PB1 and PB2, is central to the life cycle of the virus since it is responsible for the replication and transcription of viral RNA.
- a 5' cap (also termed an RNA cap, RNA 7-methylguanosine cap or an RNA m7G cap) is a modified guanine nucleotide that has been added to the 5' end of a messenger RNA.
- the 5' cap consists of a terminal 7-methylguanosine residue which is linked through a 5'-5'- triphosphate bond to the first transcribed nucleotide.
- the viral polymerase binds to the 5' RNA cap of cellular mRNA molecules and cleaves the RNA cap together with a stretch of 10 to 15 nucleotides. The capped RNA fragments then serve as primers for the synthesis of viral mRNA.
- the polymerase complex seems to be an appropriate antiviral drug target since it is essential for synthesis of viral mRNA and viral replication and contains several functional active sites likely to be significantly different from those found in host cell proteins (Magden, J. et al., (2005), Appl. Microbiol. BiotechnoL, 66, pp. 612-621 ), Thus, for example, there have been attempts to interfere with the assembly of polymerase subunits by a 25-amino-acid peptide resembling the PA-binding domain within PB1 (Ghanem, A. et al., (2007), J. Virol., 81 , pp. 7801 -7804).
- nucleoside analogs such as 2'-deoxy-2 ! -fiuoroguanosine (Tisdaie, M. et al., (1995), Antimicrob. Agents Chemother., 39, pp. 2454-2458).
- V. L. Rusinov et al. described the synthesis and antiviral activity of nucleoside analogs based on 1 ,2,4-triazoio[3,2-c][1 ,2,4]triazsn-7-ones in the Russian Chemical Bulletin, international Edition, 59(1), 2010, 136-143.
- the present invention relates a compound having the general formula (C) wherein the compound is for use in the treatment, amelioration or prevention of a virai disease.
- a compound having the genera! formula (C) encompasses pharmaceutically acceptable salts, solvates, polymorphs, prodrugs, tautomers, racemates, enantiomers, or diastereomers or mixtures thereof unless mentioned otherwise.
- alky refers to a saturated straight or branched carbon chain.
- cycioalkyl represents a cyclic version of “alkyi”.
- cycloalkyl is aiso meant to include bicyc!ic, tricyclic and poiycyclic versions thereof. Uniess specified otherwise, the cycioalkyl group can have 3 to 12 carbon atoms.
- Hal or "halogen” represents F, CI, Br and I.
- aryl prefer ⁇ teJy refers to an aromatic monocyclic ring containing 6 carbon atoms, an aromatic bicyclic ring system containing 10 carbon atoms or an aromatic tricyclic ring system containing 14 carbon atoms. Examples are phenyl, naphthyl or anthraceny!, preferably phenyl.
- heteroaryi preferably refers to a five or six-membered aromatic ring wherein one or more of the carbon atoms in the ring have been replaced by 1 , 2, 3, or 4 (for the five membered ring) or 1 , 2, 3, 4, or 5 (for the six membered ring) of the same or different heteroatoms, whereby the heteroatoms are selected from O, N and S.
- heteroaryi group include pyrrole, pyrrolidine, oxolane, furan, imidazolidine, imidazole, pyrazole, oxazotidine, oxazole, thiazole, piperidine, pyridine, morphoiine, piperazine, and dioxolane.
- hydrocarbon group which contains from 5 to 20 carbon atoms and optionally 1 to 4 heteroatoms selected from O, N and S and which contains at least one ring refers to any group having 5 to 20 carbon atoms and optionally 1 to 4 heteroatoms selected from O, N and 2 as long as the group contains at least one ring.
- the term is also meant to include bicyclic, tricyclic and poiycyciic versions thereof. If more than one ring is present, they can be separate from each other or be annelated.
- the ring(s) can be either carbocycfic or heterocyclic and can be saturated, unsaturated or aromatic.
- these groups include -(optionally substituted C 3 _ 7 cycloalkyl), -(optionally substituted aryl) wherein the aryl group can be, for example, phenyl, -(optionally substituted biphenyl), adamantyl, - ⁇ C 3-7 cycloafkyl)-aryl as well as the corresponding compounds with a linker.
- the term "(optionally substituted mono- or poiycyciic group containing 3 to 20 carbon atoms and optionally 1 to 4 heteroatoms selected from O, N and S)" refers to any mono- or poiycyciic group containing 3 to 20 carbon atoms and optionally 1 to 4 heteroatoms selected from O, N and S. This term includes monocyclic, bicyclic, tricyclic and poiycyciic versions thereof. If more than one ring is present, they can be separate from each other or be annelated. The ring(s) can be either carbocyclic or heterocyclic and can be saturated, unsaturated or aromatic.
- these groups include -(optionally substituted cycloalkyl), and -(optionally substituted aryi) wherein the aryl group can be, for example, phenyl or anthracenyi as well as the corresponding compounds with a linker.
- a compound or moiety is referred to as being "optionally substituted", it can in each instance include 1 or more of the indicated substituents, whereby the substituents can be the same or different.
- pharmaceutically acceptable salt refers to a salt of a compound of the present invention.
- suitable pharmaceutically acceptable salts include acid addition salts which may, for example, be formed by mixing a solution of compounds of the present invention with a solution of a pharmaceutically acceptable acid such as hydrochloric acid, sulfuric acid, fumaric acid, maleic acid, succinic acid, acetic acid, benzoic acid, citric acid, tartaric acid, carbonic acid or phosphoric acid.
- suitable pharmaceutically acceptable salts thereof may include alkali metal salts (e.g., sodium or potassium salts); alkaline earth metal salts (e.g., calcium or magnesium salts); and salts formed with suitable organic ligands (e.g., ammonium, quaternary ammonium and amine cations formed using counteranions such as halide, hydroxide, carboxylate, sulfate, phosphate, nitrate, alky!
- alkali metal salts e.g., sodium or potassium salts
- alkaline earth metal salts e.g., calcium or magnesium salts
- suitable organic ligands e.g., ammonium, quaternary ammonium and amine cations formed using counteranions such as halide, hydroxide, carboxylate, sulfate, phosphate, nitrate, alky!
- illustrative examples of pharmaceutically acceptable salts include, but are not limited to, acetate, adipate, alginate, ascorbaie, aspartate, benzenesulfonate, benzoate, bicarbonate, bisulfate, bitartrate, borate, bromide, butyrate, calcium edetate, camphorate, camphorsulfonate, camsylate, carbonate, chloride, citrate, clavulanate, cyclopentanepropionate, digluconate, dihydrochloride, dodecylsuifate, edetate, edisylate, estoiate, esylate, ethanesulfonate, formate, fumarate, gluceptate, glucoheptonate, gluconate, giutamate, glycerophosphate, glycolylarsanilate, hemisulfate, hepian
- the structure can contain solvent moiecuies.
- the solvents are typically pharmaceutically acceptable solvents and include, among others, water (hydrates) or organic solvents. Examples of possible solvates include ethanolates and iso-propanolates.
- codrug refers to two or more therapeutic compounds bonded via a covIER chemical bond
- cocrystal refers to a multiple component crystal in which ail components are soiid under ambient conditions when in their pure form. These components co-exist as a stoichiometric or non-stoichiometric ratio of a target molecule or ion (i.e., compound of the present invention) and one or more neutral molecular cocrystal formers.
- the compounds of the present invention can also be provided in the form of a prodrug, namely a compound which is metabolized in vivo to the active metabolite.
- Suitable prodrugs are, for instance, esters. Specific examples of suitable groups are given, among others, in US 2007/0072831 In paragraphs [0082] to [01 18] under the headings prodrugs and protecting groups. If X 1 is O or S, preferred examples of the prodrug include compounds in which R 2 is replaced by one of the following groups;
- R 6 can be the same or different.
- R 9 is a cyclic group such as an aryl group or a C 3- _7 cycloalkyl group, p is 2 to 8.
- X 1 is NR 8
- preferred examples of the prodrug include compounds in which R 2 and R 8 are not both H.
- a compound having the general formula "(C)” encompasses pharmaceutically acceptable salts, solvates, polymorphs, prodrugs, tautomers, racemates, enantiomers, or diastereomers or mixtures thereof uniess mentioned otherwise.
- V is N, or CR 6
- X 1 is O, S, or NR 8 , preferably X 1 is O.
- X 2 is NR 5 , N(R 5 )C(0), C(0)NR 5 , O, C(O), C(0)0, OC(O); S, SO, S0 2 , S0 2 N(R 5 ) or
- N(R 5 )S0 2 is N(R 5 )S0 2 .
- X 2 is NR 5 or N(R 5 )S0 2 .
- R * is -H, -Hal, -(optionally substituted d-e alkyl), -(optionally substituted mono- or polycyclic group containing 3 to 20 carbon atoms and optionally 1 to 4 heteroatoms 5 selected from O, N and S), -d-4 aikyl-(optionaliy substituted mono- or po!ycyclic group containing 3 to 20 carbon atoms and optionally 1 to 4 heteroatoms selected from O, N and S), or -X 2 -R 1 .
- R * is H, -(optionally substituted d-e alkyl),
- R 1 is -C 1-4 alkyl-(optionaliy substituted mono- or polycyclic group containing 3 to 20 carbon atoms and optionally 1 to 4 heteroatoms selected
- R 2 is -H, -(optionally substituted Ci_e alkyl), -(optionally substituted C3..7 cycioalkyl), -(optionally substituted ary!), -d-4 alkyl— (optionally substituted C 3 _ 7 cycioalkyl), or -C 1-J ⁇ alkyi-(optionaily substituted aryl) or if X 1 is NR' then R 2 can also be -OH.
- R 2 is -H or -d-e alkyl.
- R 3 is -H, -(optionally substituted d-e alkyl), -R 7 , or -X 2 -R 7 .
- R 3 is -H, -C ⁇ alkyl— (optionally substituted aryl) or -S0 2 -R 5 .
- R 3 is -H.
- R 4 is -H, -(optionally substituted d-e alkyl), -(optionally substituted C ⁇ 7 cycioalkyl), - (optionally substituted aryl), -d-* alkyl ⁇ -(optionally substituted C 3 _ 7 cycioalkyl), or -d-4 alkyl— (optionally substituted aryl).
- R 4 is -H, or -(optionally substituted
- R 5 is -H, -(optionaiiy substituted Ci_6 alkyl), - ⁇ optionally substituted C 3 _ 7 cycloaikyl), -(optionally substituted aryl), -Ci_4 alkyl-(optionatiy substituted cycloaikyl), or -Ci_4 alkyl— (optionally substituted aryl).
- R 5 is -C -4 alkyl— (optionally substituted aryl) or -(optionally substituted cycloaikyl).
- R 6 H, -Ci_6 alkyl, -aryl, halogen or CN.
- R 6 is H or -aryl.
- R 7 is -(optionally substituted hydrocarbon group which contains from 5 to 20 carbon atoms and optionally 1 to 4 heteroatoms selected from O, N and S and which contains at least one ring).
- R 7 is -C 1-4 alkyl— (optionally substituted aryl).
- R 8 is -H, -Ci_ 6 alkyl or -C ⁇ alkyl-(optionally substituted aryl).
- R 8 is -Ci_e alkyl or -C-i_4 alkyi-(optionaiiy substituted aryl).
- n is 0 to 4, preferably 0 or 1.
- the optional substituent of the alkyl group can be selected from the group consisting of halogen, -CN, -NR 5 R 5 , -OH, and -0-C ⁇ 6 alkyl.
- the optional substituent of the cycloaikyl group, the aryl group, the mono- or poiycyciic group or the hydrocarbon group can be selected from the group consisting of -Ci_e alkyl, halogen, -CF 3 , -CN, -X 2 -C_6 alkyl and -d_ 6 alkyl— aryl.
- the present inventors have surprisingly found that the compounds of the present invention which have a carbon atom im position 5 have improved pharmacological properties compared to corresponding compounds which have a nitrogen atom in this position. Without wishing to be bound by theory it is assumed that the viral polymerase protein has a pocket for binding and that carbon atom of the compounds of the present invention has improved binding compared to a nitrogen atom. This could not have been predicted or expected based on the art.
- the compounds of the present invention can be administered to a patient in the form of a pharmaceutical composition which can optionally comprise one or more pharmaceutically acceptable excipient(s) and/or carrier(s).
- the compounds of the present invention can be administered by various well known routes, including oral, rectal, intragastrical, intracranial and parenteral administration, e.g. intravenous, intramuscular, intranasal, intradermal, subcutaneous, and similar administration routes. Oral, intranasal and parenteral administration are particularly preferred. Depending on the route of administration different pharmaceutical formulations are required and some of those may require that protective coatings are applied to the drug formulation to prevent degradation of a compound of the invention in, for example, the digestive tract.
- a compound of the invention is formulated as a syrup, an infusion or injection solution, a spray, a tablet, a capsule, a capslet, lozenge, a liposome, a suppository, a plaster, a band-aid, a retard capsule, a powder, or a stow release formulation.
- the diluent is water, a buffer, a buffered salt solution or a salt solution and the carrier preferably is selected from the group consisting of cocoa butter and vitebesole.
- Particular preferred pharmaceutical forms for the administration of a compound of the invention are forms suitable for injectionable use and include sterile aqueous solutions or dispersions and sterile powders for the extemporaneous preparation of sterile injectable solutions or dispersions. In all cases the final solution or dispersion form must be sterile and fluid. Typically, such a solution or dispersion will include a solvent or dispersion medium, containing, for example, water-buffered aqueous solutions, e.g. biocompatible buffers, ethanoi, polyol, such as glycerol, propylene glycol, polyethylene glycol, suitable mixtures thereof, surfactants or vegetable oils.
- a compound of the invention can also be formulated into liposomes, in particular for parenteral administration. Liposomes provide the advantage of increased half life in the circulation, if compared to the free drug and a prolonged more even release of the enclosed drug.
- Sterilization of infusion or injection solutions can be accomplished by any number of art recognized techniques including but not limited to addition of preservatives like anti-bacterial or anti-fungal agents, e.g. parabene, chlorobutanol, phenol, sorbic acid or thimersaf. Further, isotonic agents, such as sugars or salts, in particular sodium chloride, may be incorporated in infusion or injection solutions.
- preservatives like anti-bacterial or anti-fungal agents, e.g. parabene, chlorobutanol, phenol, sorbic acid or thimersaf.
- isotonic agents such as sugars or salts, in particular sodium chloride, may be incorporated in infusion or injection solutions.
- steriie injectable solutions containing one or several of the compounds of the invention is accomplished by incorporating the respective compound in the required amount in the appropriate solvent with various ingredients enumerated above as required followed by sterilization. To obtain a sterile powder the above solutions are vacuum-dried or freeze-dried as necessary.
- Preferred diluents of the present invention are water, physiological acceptable buffers, physiological acceptable buffer salt solutions or salt solutions.
- Preferred carriers are cocoa butter and vitebesole.
- Excipients which can be used with the various pharmaceutical forms of a compound of the invention can be chosen from the following non-limiting list: a) binders such as lactose, mannitol, crystalline sorbitol, dibasic phosphates, calcium phosphates, sugars, microcrystaSline cellulose, carboxymethyl cellulose, hydroxyethyl celiulose, polyvinyl pyrrolidone and the like;
- binders such as lactose, mannitol, crystalline sorbitol, dibasic phosphates, calcium phosphates, sugars, microcrystaSline cellulose, carboxymethyl cellulose, hydroxyethyl celiulose, polyvinyl pyrrolidone and the like;
- lubricants such as magnesium stearate, talc, calcium stearate, zinc stearate, stearic acid, hydrogenated vegetable oil, leucine, glycerids and sodium siearyl fumarates
- disintegrants such as starches, croscarrnellose, sodium methyl cellulose, agar, bentonite, aiginic acid, carboxymethyl cellulose, polyvinyl pyrrolidone and the like.
- the formulation is for oral administration and the formulation comprises one or more or all of the following ingredients: pregelatinized starch, talc, povidone K 30, croscarrnellose sodium, sodium stearyl fumarate, gelatin, titanium dioxide, sorbitol, monosodtum citrate, xanthan gum, titanium dioxide, flavoring, sodium benzoate and saccharin sodium.
- a compound of the invention may be administered in the form of a dry powder inhaler or an aerosol spray from a pressurized container, pump, spray or nebulizer with the use of a suitable prope!lant, e.g., dichiorodifluoromethane, trichlorofluoromethane, dichloroteirafluoroethane, a hydrofluoro- a!kane such as 1 , 1 ,1 ,2 etrafluoroethane (HFA 134ATM) or 1 , 1 ,1 ,2,3,3, 3-heptafiuoropropane (HFA 227EATM), carbon dioxide, or another suitable gas.
- a suitable prope!lant e.g., dichiorodifluoromethane, trichlorofluoromethane, dichloroteirafluoroethane, a hydrofluoro- a!kane such as 1 , 1 ,1 ,2 etrafluor
- the pressurized container, pump, spray or nebulizer may contain a solution or suspension of the compound of the invention, e.g., using a mixture of ethanol and the propeliant as the solvent, which may additionally contain a lubricant, e.g., sorbitan trioleate.
- a lubricant e.g., sorbitan trioleate.
- the dosage of a compound of the invention in the therapeutic or prophylactic use of the invention should be in the range of about 0.1 mg to about 1 g of the active ingredient (i.e. compound of the invention) per kg body weight.
- a compound of the invention is administered to a subject in need thereof in an amount ranging from 1.0 to 500 mg/kg body weight, preferably ranging from 1 to 200 mg/kg body weight.
- the duration of therapy with a compound of the invention will vary, depending on the severity of the disease being treated and the condition and idiosyncratic response of each individual patient.
- from 10 mg to 200 mg of the compound are orally administered to an adult per day, depending on the severity of the disease and/or the degree of exposure to disease carriers.
- the pharmaceutically effective amount of a given composition will also depend on the administration route. In general the required amount will be higher, if the administration is through the gastrointestinal tract, e.g., by suppository, rectal, or by an intragastric probe, and lower if the route of administration is parenteral, e.g., intravenous.
- a compound of the invention will be administered in ranges of 50 mg to 1 g/kg body weight, preferably 10 mg to 500 mg/kg body weight, if rectal or intragastric administration is used and in ranges of 1 to 100 mg/kg body weight if parenteral administration, is used. For intranasal administration, 1 to 100 mg/kg body weight are envisaged.
- a person is known to be at risk of developing a disease treatable with a compound of the invention, prophylactic administration of the biologically active blood serum or the pharmaceutical composition according to the invention may be possible.
- the respective compound of the invention is preferably administered in above outlined preferred and particular preferred doses on a daily basis. Preferably, from 0.1 mg to 1 g/kg body weight once a day, preferably 10 to 200 mg/kg body weight. This administration can be continued until the risk of developing the respective viral disorder has lessened. In most instances, however, a compound of the invention will be administered once a disease/disorder has been diagnosed. In these cases it is preferred that a first dose of a compound of the invention is administered one, two, three or four times daily.
- the compounds of the present invention are particularly useful for treating, ameliorating, or preventing viral diseases.
- the type of viral disease is not particularly limited.
- examples of possible virai diseases include, but are noi limited to, viral diseases which are caused by Poxviridae, Herpesviridae, Adenoviridae, Papii!omaviridae, Polyomaviridae, Parvoviridae, Hepadnaviridae, Retroviridae, Reoviridae, Filoviridae, Paramyxoviridae, Rhabdoviridae, Orthomyxoviridae, Bunyavi idae, Arenaviridae, Coronavindae, Picornaviridae, Hepeviridae, Caliciviridae, Astroviridae, Togaviridae, Fiaviviridae, Deltavirus, Bornaviridae, and prions.
- viral diseases which are caused by Herpesviridae, Retroviridae, Filoviridae, Paramyxoviridae, Rhabdoviridae, Orthomyxoviridae, Bunyaviridae, Arenaviridae, Coronaviridae, Picornaviridae, Togaviridae, Fiaviviridae, more preferably viral diseases which are caused by orthomyxoviridae.
- Herpesviridae Herpes simplex virus
- Picornaviridae Human enterovirus types A-D (Poliovirus, Echovirus,
- Hepatitis G virus Hepatitis GB virus
- influenza includes influenza A, B, C, isavirus and thogotovirus and also covers bird flu and swine flu.
- the subject to be treated is not particularly restricted and can be any vertebrate, such as birds and mammals (including humans).
- the compounds of the present invention are capable of inhibiting endonuclease activity, particularly of the influenza virus. More specifically it is assumed that they directiy interfere with the N-terminal part of the influenza PA protein, which harbours endonuclease activity.
- delivery of a compound into a cell may represent a problem depending on, e.g., the solubility of the compound or its capabilities to cross the cell membrane.
- the present invention not only shows that the claimed compounds have in vitro polymerase inhibitory activity but also in vivo antiviral activity.
- a possible measure of the in vitro polymerase inhibitory activity of the compounds having the formula (A) and/or (C) is the FRET endonuclease activity assay disclosed herein.
- the compounds exhibit a % reduction of at least about 50 % at 25 ⁇ in the FRET assay.
- the % reduction is the % reduction of the initial reaction velocity (vO) of substrate cleavage of compound-treated samples compared to untreated samples.
- the compounds exhibit an IC S0 of at least about 40 ⁇ , more preferably at least about 20 ⁇ , in the FRET assay.
- the half maximal inhibitory concentration (IC 50 ) is a measure of the effectiveness of a compound in inhibiting biological or biochemical function and was calculated from the initial reaction velocities (vO) in a given concentration series ranging from maximum 100 ⁇ to at least 2 nM.
- a possible measure of the in vivo antiviral activity of the compounds having the formula (A) and/or (C) is the CPE assay disclosed herein.
- the compounds exhibit a % reduction of at least about 30 % at 50 ⁇ .
- the reduction in the virus-mediated cytopathic effect (CPE) upon treatment with the compounds was calculated as follows: The ceil viability of infected-treated and uninfected-treated cells was determined using an ATP- based cell viability assay (Promega).
- the response in relative luminescent units (RLU) of Infected-untreated samples was subtracted from the response (RLU) of the Infected-treated samples and then normalized to the viability of the corresponding uninfected sample resulting in % CPE reduction.
- the compounds exhibit an IC 50 of at least about 45 ⁇ , more preferably at least about 10 ⁇ , in the CPE assay.
- the half maximal inhibitory concentration (IC5 0 ) is a measure of the effectiveness of a compound in inhibiting biological or biochemical function and was calculated from the RLU response in a given concentration series ranging from maximum 00 ⁇ to at least 100 nM.
- the compounds having the general formula (C) can be used in combination with one or more other medicaments.
- the type of the other medicaments is not particularly iimited and wiii depend on the disorder to be treated.
- the other medicament will be a further medicament which is useful in treating, ameloriating or preventing a viral disease, more preferably a further medicament which is useful in treating, ameloriating or preventing influenza.
- the following combinations of medicaments are envisaged as being particularly suitable:
- endonuclease and cap binding inhibitors particularly targeting influenza.
- the endonuclease inhibitors are not particularly limited and can be any endonuclease inhibitor, particularly any viral endonuclease inhibitor.
- Preferred endonuclease inhibitors are those having the general formula (I) as defined in the US application with the serial number 61/550,045, filed on October 21, 2011 , the complete disclosure of which is incorporated by reference. In particular, all descriptions with respect to the general formula of the compounds according to US 61/550,045, the preferred embodiments of the various substituents as well as the medical utility and advantages of the compounds are incorporated herein by reference.
- the compounds having the general formula (I) of this reference can optionally be in the form of a pharmaceutically acceptable salt, solvate, polymorph, codrug, co crystal, prodrug, tautomer, racemate, enantiomer, or diastereomer or mixture thereof. They are defined as follows (wherein the definitions of the various moieties given in this earlier application apply): wherein
- R 1 is selected from -H, -C,_ 6 alkyi, -(C 3 diligent 7 cycloalkyl) and -C 2 -(C : ⁇ 7 cycloalkyl);
- R 2 is selected from -H, , -d_e alkyl, -Hal, -(C 3 _ 7 cycloalkyl), -CH 2 -(C 3 _ 7 cycloalkyl), - ⁇ CH ⁇ -ioptionally substituted aryl), -(optionally substituted 5- or 6- membered heterocyclic ring which contains at least one heteroatom selected from N, O and S, wherein the substituent is selected from -C ⁇ alkyl, -halogen, -CN, -CHal 3 , -aryl, ⁇ NR 6 R 7 , and -CONR 6 R 7 ;
- R 3 is selected from -H, -C ⁇ e alkyl- ⁇ (CH 2 ⁇ n -NR 6 R 8 ,
- R 4 is -H
- R 5 is selected from the group consisting of -H or -(CH 2 ) n -(optional!y substituted aryl), wherein the substituent is selected from -Hal and -C ⁇ a!kyl; or wherein R 4 and R 5 together form a methylene group -CH 2 -, ethylene group -CH 2 CH 2 - or ethyne group -CHCH-, which can be optionally substituted by -C-i_4 aikyl, -halogen, -CHal 3 , -R 6 R 7 , -OR 6 , -CONR 6 R 7 , -S0 2 R 6 R 7 , aryl or heteroaryS; R 6 is selected from -H and -C-, ⁇ alkyl;
- R 7 is selected from -H and -C 1- alkyl
- R 8 is selected from -H, -C ⁇ e alkyl, -(CH 2 ) n -(optionaily substituted aryt), -S0 2 --(CH 2 ) n - (optionaily substituted aryl), -S0 2 - ⁇ CH 2 )n-(optionally substituted 5- to 10-membered mono- or bicyciic heteroring which contains at least one heteroatom selected from N, O and S), -(CH 2 )n-(opt!onaliy substituted 5- or 6-membered heterocyclic ring which contains at least one heteroatom selected from N, O and S), wherein the substituent is selected from -Hal, -CF 3 , -C-M alkyl, and -(CH 2 ) n -aryl;
- R 9 is selected from -H, -C 1-4 alkyl, and -C ⁇ alkylene-NR 1 11 ;
- R 1G is selected from -H, alkylene-NR 11 R 11 ;
- R 11 is selected from -H, -CF 3 , and -C 1- alkyl; each m is 0 or 1 ; and each n is independently 0, 1 , 2, or 3.
- Further preferred endonuclease inhibitors are those having the general formula (C) as defined in the copending application with attorney's docket number T3450 US which was filed on even date herewith, the complete disclosure of which is incorporated by reference.
- the compounds having the general formula (C) can be optionally in the form of a pharmaceutically acceptable salt, solvate, polymorph, codrug, cocrystal, prodrug, tautomer, racemate, enantiomer, or diastereomer or mixture thereof. They are defined below.
- the cap binding inhibitors are not are not particularly limited either and can be any cap binding inhibitor, particularly any viral cap binding inhibitor.
- Preferred cap binding inhibitors are those having the general formuia (ii) as defined in US application 61/550,057 and/or the compounds disclosed in WO201 1 /000566, the complete disciosure of which is incorporated by reference, in particular, all descriptions with respect to the general formula of the compounds according to US 61/550,057 or WO201 1/000566, the preferred embodiments of the various substituents as well as the medical utility and advantages of the compounds are incorporated herein by reference.
- the compound having the general formuia (II) can be optionally in the form of a pharmaceutically acceptable salt, solvate, polymorph, codrug, cocrystal, prodrug, tautomer, racemate, enantiomer, or diastereomer or mixture thereof. It is defined as follows:
- R 21 is selected from -H, -C ⁇ alkyl, -(CH 2 ) q -aryl, - ⁇ CH 2 ) q -heterocyclyl, -(CH 2 ) q -cycloalkyl, -(CH ⁇ -OR 25 , and -(CH 2 ) P -NR 25 R 26 ;
- R 22 is selected from -H, -d_ 6 alky!, -(CH 2 ) q -cycioalkyl, -Hal, -CF 3 and -CN;
- R 23 is selected from -aryl, -heterocyclyl, -cycloalkyl, -C( ⁇ R 28 )(-R 29 )-aryl, -C(-R 28 )(-R 29 )-heterocyclyi, and -C(-R 28 )(-R 9 )-cyc!oalkyl;
- R 25 is selected from -H, -Ci_e alkyi, and -(CH 2 CH 2 0) r H;
- R 26 is selected from -H, and -Ci_e alkyl
- R 27 is independently selected from -C ⁇ alkyl, - ⁇ C ⁇ 0) ⁇ C Q alkyl, -Hal, -CF 3 , -CN, -COOR 25 , -OR 25 , -(CH 2 ) q NR 25 R 26 , -C(0)-NR 25 R 26 , and -NR 25 -C(0)-C ⁇ alkyl;
- R 10 , R 10' and R 0" are each individuaiiy seiected from the group consisting of hydrogen, d-Ce-alkyl, C 2 -C 6 -alkenyi, C 2 -C 8 -aikynyl, - ⁇ CH 2 ) n C(0)OH,
- R 11 is selected from the group consisting of hydrogen, d-Ce-alkyl, -CF 3 , d-Ce-aikenyl, C 2 -C 8 -alkynyI, -(CH 2 )n-cycloalkyi, -(CH 2 )n-aryl, -(CH 2 ) n -heterocycloalkyl and -(CH ⁇ - heteroaryl; optionally substituted;
- R 12 is selected from the group consisting of Ci-C 6 -alkyl, -CF 3 , C 2 -C 6 -alkenyl, C 2 -C e - alkynyl, -(CH 2 ) n -cycIoalkyl, - ⁇ CH 2 ) n -heterocycloalkyl, - ⁇ CH 2 ) n -aryI, -NR ie R 17 , and -(CH 2 ) n -heteroaryl; optionally substituted;
- R 16 and R 17 are independently selected from the group consisting of d-Ce-alkyl, C 2 -C 6 - alkenyl, C 2 -C 6 -alkynyi, -(CH 2 ) n -cycloalkyl, -(CH 2 ) n -aryl, -CF 3 , -C(0)R 18 and -S ⁇ 0) 2 R 18 ; optionally substituted;
- R is independently selected from the group consisting of d-Ce-alkyl, C 2 -C 6 -alkenyl, Ca-Cg-alkynyl, -(CH 2 ) n -cycloalkyl and -CF 3 ; optionally substituted; and n is in each instance selected from 0, 1 and 2.
- substituents e.g. 1 , 2, 3, 4, 5, 6, 7, 8, 9, or 10 substituents which are in each instance preferably independently selected from the group consisting of halogen, in particular F, CI, Br or I; -N0 2 , -CN, -OR', -NR'R", -(CO)OR', - ⁇ CO)OR" ⁇ -(COJNR'R", -NR'COR”", -NR'COR', -NR"CONR'R",
- R' and R" are each independently selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, -OE, cycloalkyl, heterocycloaikyl, aryl, heteroaryl, and aralkyl or together form a heteroaryl, or heterocycloaikyl; optionally substituted;
- FT and R"" are each independently selected from the group consisting of alkyl, aikenyl, aikyny!, cycioa!kyl, heterocyc!oalkyi, alkoxy, aryl, aralky!, heteroaryl, and -NR'R"; and
- E is selected from the group consisting of aikyl, aikenyl, cycloalkyi, a!koxy, aikoxyalkyl, heterocycloaikyl, an alicydic system, aryl and heteroaryl; optionally substituted.
- Influenza virus polymerase inhibitors are novel drugs targeting the transcription activity of the polymerase. Selective inhibitors against the cap-binding and endonuclease active sites of the viral polymerase severely attenuate virus infection by stopping the viral reproductive cycle. These two targets are located within distinct subunits of the polymerase complex and thus represent unique drug targets. Due to the fact thai both functions are required for the so-called "cap-snatching" mechanism mandatory for viral transcription, concurrent inhibition of both functions is expected to act highly synergistically. This highly efficient drug combination would result in lower substance concentrations and hence improved dose-response-reSationships and better side effect profiles.
- an endonudease inhibitor and a cap-binding inhibitor or a dual specific polymerase inhibitor targeting both the endonudease active site and the cap- binding domain would be effective against virus strains resistant against adamantanes and neuraminidase inhibitors and moreover combine the advantage of Sow susceptibility to resistance generation with activity against a broad range of virus strains.
- Influenza virus polymerase inhibitors are novel drugs targeting the transcription activity of the polymerase. Selective inhibitors against the cap- binding and endonudease active sites of the viral polymerase severely attenuate virus infection by stopping the viral reproductive cycle.
- the combination of a polymerase inhibitor specifically addressing a viral intracellular target with an inhibitor of a different antiviral target is expected to act highly synergistically. This is based on the fact that these different types of antiviral drugs exhibit completely different mechanisms of action and pharmacokinetics properties which act advantageously and synergistically on the antiviral efficacy of the combination.
- At least one compound selected from the first group of polymerase inhibitors is combined with at least one compound selected from the second group of polymerase inhibitors.
- the first group of polymerase inhibitors which can be used in this type of combination therapy includes, but is not limited to, the compounds having the formula (A) or (C).
- the second group of polymerase inhibitors which can be used in this type of combination therapy includes, but is not limited to, the compounds having the general formula (I), the compounds having the general formula (II), the compounds disclosed in WO 2011/000566, WO 2010/110231 , WO 2010/1 10409, WO 2006/030807 or US 5,475,109 as well as flutimide and analogues, favipiravir and analogues, epigaliocatechin gallate and analogues, as well as nucleoside analogs such as ribavirine.
- Influenza virus polymerase inhibitors are novel drugs targeting the transcription activity of the polymerase. Selective inhibitors against the cap-binding and endonuclease active sites of the viral polymerase severely attenuate virus infection by stopping the viral reproductive cycle.
- the combination of a polymerase inhibitor specifically addressing a viral intracellular target with an inhibitor of a different extracellular antiviral target, especially the (e.g., viral) neuraminidase is expected to act highly synergistica!ly. This is based on the fact that these different types of antiviral drugs exhibit completely different mechanisms of action and pharmacokinetic properties which act advantageously and synergistica!ly on the antiviral efficacy of the combination.
- polymerase inhibitors would prevail for combinations of inhibitors of different antiviral targets with polymerase inhibitors.
- at least one compound selected from the above-mentioned first group of polymerase inhibitors is combined with at least one neuramidase inhibitor.
- the neuraminidase inhibitor (particularly influenza neuramidase inhibitor) is not specifically limited. Examples include zanamivir, ose!tamivir, peramivir, KDN, DANA, FANA, and cyclopentane derivatives.
- Influenza virus polymerase inhibitors are novel drugs targeting the transcription activity of the polymerase. Selective inhibitors against the cap-binding and endonuclease active sites of the viral polymerase severely attenuate virus infection by stopping the viral reproductive cycle.
- the combination of a polymerase inhibitor specifically addressing a viral intracellular target with an inhibitor of a different extracellular and cytoplasmic antiviral target, especially the viral 2 ion channel, is expected to act highly synergistically. This is based on the fact that these different types of antiviral drugs exhibit completely different mechanisms of action and pharmacokinetic properties which act advantageously and synergistically on the antiviral efficacy of the combination.
- At least one compound selected from the above-mentioned first group of polymerase inhibitors is combined with at least one M2 channel inhibitor.
- the M2 channel inhibitor (particularly influenza M2 channel inhibitor) is not specifically limited. Examples include amantadine and rimantadine.
- Influenza virus polymerase inhibitors are novel drugs targeting the transcription activity of the polymerase. Selective inhibitors against the cap-binding and endonuclease active sites of the viral polymerase severely attenuate virus infection by stopping the viral reproductive cycle.
- the combination of a polymerase inhibitor specifically addressing a viral intracellular target, with an inhibitor of a different extracellular target, especially alpha glucosidase, is expected to act highly synergistically. This is based on the fact that these different types of antiviral drugs exhibit completely different mechanisms of action and pharmacokinetic properties which act advantageously and synergistically on the antiviral efficacy of the combination.
- At least one compound selected from the above-mentioned first group of polymerase inhibitors is combined with at least one alpha glucosidase inhibitor.
- the alpha glucosidase inhibitor (particularly influenza alpha glucosidase inhibitor) is not specifically limited. Examples include the compounds described in Chang et aL, Antiviral Research 2011, 89, 26-34.
- the combination of polymerase inhibitors with ligands of other influenza targets influenza virus polymerase inhibitors are novel drugs targeting the transcription activity of the polymerase. Selective inhibitors against the cap-binding and endonuclease active sites of the viral polymerase severely attenuate virus infection by stopping the viral reproductive cycle.
- the combination of a polymerase inhibitor specifically addressing a viral intraceilular target with an inhibitor of different extracellular, cytoplasmic or nucleic antiviral targets is expected to act highly synergistically. This is based on the fact that these different types of antiviral drugs exhibit completely different mechanisms of action and pharmacokinetic properties which act advantageously and synergistically on the antiviral efficacy of the combination.
- Typicaliy at least one compound selected from the above mentioned first group of polymerase inhibitors is combined with at least one ligand of another influenza target.
- the ligand of another influenza target is not specifically limited.
- examples include compounds acting on the sialidase fusion protein, e.g. Fludase (DAS 181), siRNAs and phosphorothioate oligonucleotides, signal transduction inhibitors (ErbB tyrosine kinase, Abl kinase family, MAP kinases, PKCa-mediated activation of ERK signaling as well as interferon (inducers).
- influenza polymerase inhibitors preferably influenza polymerase inhibitors with a compound used as an adjuvance to minimize the symptoms of the disease
- antibiotics anti-inflammatory agents like COX inhibitors (e.g., COX-1/COX-2 inhibitors, selective COX-2 inhibitors), lipoxygenase inhibitors, EP ligands (particularly EP4 !igands), bradykinin ligands, and/or cannabinoid ligands (e.g., CB2 agonists).
- Influenza virus polymerase inhibitors are novel drugs targeting the transcription activity of the po!ymerase. Selective inhibitors against the cap-binding and endonuclease active sites of the viral polymerase severely attenuate virus infection by stopping the viral reproductive cycle.
- the combination of a polymerase inhibitor specifically addressing a viral intracellular target with an compound used as an adjuvance to minimize the symptoms of the disease address the causative and symptomatic pathological consequences of viral infection.
- This combination is expected to act synergistically because these different types of drugs exhibit completely different mechanisms of action and pharmacokinetic properties which act advantageously and synergistically on the antiviral efficacy of the combination.
- the present invention discloses a compound having the general formula (A).
- a compound having the general formula (A) encompasses pharmaceutically acceptable salts, solvates, polymorphs, prodrugs, iautomers, racemates, enantiomers, or diastereomers or mixtures thereof unless mentioned otherwise.
- R * is -H, -Hal, -(optionally substituted d_6 a!kyl), -(optionally substituted C 3 _ cycloalkyl), -(optionally substituted aryl), -C M alkyi— (optionally substituted C 3 _ 7 cycloalkyl), -C 1-4 aikyl— (optionally substituted aryl) or -X 1 -R 1 .
- R * is -Hal, -(optionally substituted Ci rating 6 alkyl) (wherein the optional substituent of the alkyl group is preferably Hal, more preferably F); -C -4 alkyl— (optionally substituted aryl) (wherein the optional substituent of the aryl group is preferably halogen) or -X 1 -R 1 .
- R* is X -R 1 .
- X 1 is O, C(O), C(0)0, OC(O); S, SO, S0 2 , NR 4 , N(R 5 )C(0), C(0 ⁇ NR 5 , preferably X 1 is O, or NR 4 , more preferably X 1 is NR 4 , In one preferred embodiment, X 1 is NR 4 and R and R 4 are joined together to form a 5- to 7-membered ring, which can optionally contain O, S or further N. In another preferred embodiment, X 1 is NR 4 and R 1 is -SO2-R 4 . X 2 is O, S, NR 4 , preferably X 2 is O. is O or S, preferably X 3 is O. is O or S, preferably X 4 is 0.
- R 1 is -H, -(optionally substituted alkyl), -(optionally substituted C 3 ⁇ 7 cycloaikyl), - (optionaily substituted aryl), -Ci_4 alkyi-(optionaliy substituted C 3 _ 7 cycioa!kyl), -C 1 ⁇ a!kyl-(optional!y substituted aryl).
- R 1 is -H, -(optionally substituted Ci_6 alkyl), -(optionally substituted benzyl), more preferably, R 1 is -H or -(optionally substituted benzyl).
- R ? is a hydrocarbon group which contains from 5 to 20 carbon atoms and optionaily 1 to 4 heteroatoms and which contains at least two rings, wherein the hydrocarbon group can be optionally substituted.
- at least one of the at least two rings is aromatic such as an aryl or heteroaryl ring.
- R 2 can be selected from the group consisting of
- X is absent, CH 2 , NH, C(0)NH, S or O.
- Y is CH 2 .
- X and Y can be joined together to form an annulated, carbo- or heterocyiic 3 to 8 membered ring which can be saturated or unsaturated.
- Specific examples of X-Y include -CH 2 ⁇ , -CH 2 -CH 2 -, -0-, and -NH-.
- R is independently selected from H, -Ci_e aikyl, halogen, -CN, -OH, and -0-C -J3 alkyl.
- R 3 is -H, -(optiona!!y substituted Ci-e aikyf), -(optionally substituted C 3 _ 7 cycloaikyi), - (optionally substituted aryl), or -C-,_4 alkyl-(optionally substituted aryl) if X 2 is NR 4 then R 3 can also be -OH, preferably R 3 is -H, -C ⁇ alky] or Bz.
- R 4 is -H, -(optionally substituted d_ e alkyl), -(optionally substituted cycloaikyi), - (optionally substituted aryl), -C ⁇ aikyl-(optionally substituted C 3 _ 7 cycloaikyi), or -C ⁇ alkyl ⁇ -(optionally substituted aryl) or if X 1 is NR 4 then R 4 and R 1 can be joined together to form a 5- to 7-membered ring, which can optionally contain O, S or further N or if X 2 is NR 4 then R 4 and R 3 can be joined together to form a 5- to 7-membered ring, which can optionally contain O, S or further N.
- R 4 is -H, -(optionally substituted aryl), or
- R 4 is -H or -(optionally substituted benzyl).
- R 5 is -H, -(optionally substituted Ct_s alkyl), -(optionally substituted C3-7 cycloaikyi), - (optionally substituted aryl), -C ⁇ alkyl-(optiona!ly substituted C3_ 7 cycloaikyi), or -C 1-Jt aikyl-(optionatiy substituted aryl).
- R 5 is -H.
- R 6 is -H, or -C ⁇ alkyl.
- the optional substituent of the alkyl group is selected from the group consisting of halogen, - CN, -NR 6 R S , -OH, and -0-Ci_ 6 alkyl.
- the substituent is -halogen, more preferably F.
- the optional substituent of the cycloaikyi group, the aryl group or the hydrocarbon group is selected from the group consisting of -C, ⁇ alkyl, halogen, -CF>, -CN, -X -R 5 and -C1-4 alkyl— aryl.
- the substituent is -halogen (preferably F), -OCH 3 or -CN.
- the present inventors have surprisingly found that the compounds having the formula (A) which have a bulky moiety R 2 have improved pharmacological properties compared to corresponding compounds which have a smaller moiety R 2 .
- the viral polymerase protein has a pocket for binding and that the bulky moiety R 2 of the compounds of the present invention fills this pocket to a larger extent.
- the larger moiety R 2 is able to provide more hydrophobic interaction with the pocket than smaller moieties such as methyl.
- influenza A virus PA-Nter fragment (amino acids 1 - 209) harbouring the influenza endonudease activity was generated a nd purified as described in Dtas et a!., Nature 2009; Apr 16; 458(7240), 914-918.
- the protein was dissolved in buffer containing 20mM Tris pH 8.0, 100m NaCI and 10mM ⁇ -mercaptoethanol and aliquots were stored at -20 °C.
- RNA oligo with 5'-FAM fluorophore and 3'-BHQ1 quencher was used as a substrate to be cleaved by the endonudease activity of the PA-Nter. Cleavage of the RNA substrate frees the fluorophore from the quencher resulting in an increase of the fluorescent signal.
- IC 50 half maximal inhibitory concentration
- influenza A virus was obtained from American Tissue Culture Collection (A Aichi/2/68 (H3N2); VR-547). Virus stocks were prepared by propagation of virus on Mardin- Darby canine kidney ( DCK; ATCC CCL-34) ceils and infectious titres of virus stocks were determined by the 50 % tissue culture infective dose (TCID 50 ) analysis as described in Reed, L J., and H, Muench. 1938, Am. J. Hyg. 27:493-497.
- TCID 50 tissue culture infective dose
- MDCK cells were seeded in 96-weIi plates at 2> ⁇ 10 4 cells/well using DMEM/Ham's F- 2 (1 :1 ) medium containing 10 % foetal bovine serum (FBS), 2 mM L-g!utamine and 1 % antibiotics (all from PAA). Until infection the cells were incubated for 5 hrs at 37 °C, 5.0 % C0 2 to form a -80 % confluent monolayer on the bottom of the well. Each test compound was dissolved in DMSO and generally tested at 25 ⁇ and 250 ⁇ . In those cases where the compounds were not soluble at that concentration they were tested at the highest soluble concentration.
- the compounds were diluted in infection medium ⁇ DMEM/Ham's F-12 (1 :1 ) containing 5 pg/ml trypsin, and 1 % antibiotics) for a final plate well DMSO concentration of 1 %.
- the virus stock was diluted in infection medium (DMEM/Ham's F-12 (1 :1 ) containing 5 pg/ml Trypsin, 1 % DMSO, and 1 % antibiotics) to a theoretical multiplicity of infection (MOi) of 0.05. After removai of the culture medium and one washing step with PBS, virus and compound were added together to the ceils.- In the wells used for cytotoxicity determination (i.e. in the absence of viral infection), no virus suspension was added.
- Relative cell viability values of uninfected-treated versus uninfected-untreated cells were used to evaluate cytotoxicity of the compounds. Substances with a relative viability below 80 % at the tested concentration were regarded as cytotoxic and retested at lower concentrations.
- Reduction in the virus-mediated cytopathic effect (CPE) upon treatment with the compounds was calculated as follows: The response (RLU) of infected-untreated samples was subtracted from the response (RLU) of the infected-treated samples and then normalized to the viability of the corresponding uninfected sample resulting in % CPE reduction.
- the half maximal inhibitory concentration (IC 60 ) is a measure of the effectiveness of a compound in inhibiting biological or biochemical function and was calculated from the RLU response in a given concentration series ranging from maximum 100 ⁇ to at least 00 nM.
- the compound 1-3 (117 g, 0.801 mmol) was dissolved in ethanol (400 mi) and diethyl ethoxymethylenemalonate was added. This reaction mixture was stirred for 3 h at reflux. Then the mixture was cooled to r.t.. The precipitate was filtered to afford the product 1-4 as brown solid 163 g, yield 64%.
- the compound 1-4 (20 g, 63.6 mmoi) was added to diphenyi ether (150 mL). The mixture was heated to 250 °C for 40 min. Then the mixture was cooled to r.t.. and was added to petrolether (PE). The precipitate was filtered to afford the product I -5 as brown solid 16 g, yield 94 %.
- the ethyl ester precursor of 14 was treated with methanamine according to the representative method to obtain compound 117 as a pale white soiid.
- the ethyl ester precursor of IS was treated with methanamine according to the representative method to obtain compound 118 as a pale white solid.
- i-7 (1-7') was treated with methyisulfonamide according to the representative method to obtain compound 122 as a paie white solid.
- Step 1 To a suspension of sodium hydride (350 mg, 8.8 mrnoi, 1.2 eq) in 1 ,4-dioxane (10 mL) was added acetonitrile (450 ⁇ _, 8.8 mmol, 1.2 eq). The mixture was stirred at room temperature for 30 min. Then cyclopentanecarboxy!ic acid ethyl ester (660 ⁇ _, 7.3 mmol, 1 eq) was added. After stirring for 30 min at room temperature, the mixture was heated at 105°C during 16 h. After cooling, the solvent was evaporated to dryness and water was added (30 mL).
- Step 1
- the expected compound was obtained according to general procedure A using benzyiamine.
- the expected compound was isolated as white powder.
- the expected compound was obtained according to general procedure A using 4-bromo- benzyiamine.
- the expected compound was isolated as white powder.
- the expected compound was obtained according to general procedure A using C-(2,3- dihydro-naphthalen-1 -y!-methyiamine.
- the expected compound was isolated as white powder.
- the expected compound was obtained according to general procedure A using 4- isopropoxy-phenylamine.
- the expected compound was isolated as pale ye!low powder.
- the expected compound was obtained according to general procedure A using N-(4- phenyl)-acetamide.
- the expected compound was isolated as off-white powder.
- the expected compound was obtained according to general procedure A using 3-chloro ⁇ 4- methyl-phenylamine.
- the expected compound was isolated as white powder.
- Step 1
- the expected compound was obtained according to general procedure B using 4- isopropoxy-phenylamine.
- the expected compound was isolated as pale yellow powder.
- Step 1
- the expected compound was obtained according to general procedure C step 1 using 4- bromo-5-methyl-2H ⁇ pyrazoi-3-ylamine.
- the expected compound was isolated as pale yeliow powder.
- the expected compound was obtained according to general procedure C step 1 using 5- imino-3-(3-methylamino-propyl ⁇ -4,5-dihydro-1 H-pyrazole-4-carbonitrile.
- the expected compound was isolated as white powder.
- the expected compound was obtained according to genera! procedure C using 5- ⁇ 4-ethoxy- phenyi)-2H ⁇ pyrazol-3-ylamine.
- the expected compound was isoiated as white powder.
- the expected compound was obtained according to general procedure C using 5-isopropyl 2H-pyrazoi-3-ylamine. The expected compound was isolated as white powder.
- the expected compound was obtained according to general procedure C using 5- cyclopentyl-2H-pyrazol-3-ylamine.
- the expected compound was isolated as white powder, MS: 248.1
- the expected compound was obtained according to general procedure C using 5- trifiuoromethyi-2H-pyrazol-3-yiamine.
- the expected compound was isolated as white powder. MS: 248,0
- the expected compound was obtained according to general procedure D using Key Intermediate If and phenethyl bromide.
- the expected compound was isolated as white powder.
- the expected compound was obtained according to genera! procedure D using Key Intermediate II and 1-(2-bromo-ethyf)-4-chioro-benzene.
- the expected compound was isolated as white powder,
- the expected compound was obtained according to general procedure D using Key Intermediate li and 1-(2-bromo-ethyl)-3-chloro-benzene.
- the expected compound was isolated as white powder.
- the expected compound was obtained according to general procedure D using Key Intermediate II and 1-(2-bromo-ethyl)-3-fluoro-benzene.
- the expected compound was isolated as white powder.
- the expected compound was obtained according to general procedure 0 using Key intermediate II and 1-(2-bromo-ethyl)-3 rifiuoromethyl-benzene.
- the expected compound was isolated as white powder.
- the expecied compound was obtained according to general procedure D using Key Intermediate III and phenethyl bromide.
- the expected compound was isolated as white powder.
- the expected compound was obtained according to genera! procedure D using Key Intermediate III and (3-bromo-propyi)-benzene.
- the expected compound was isolated as colorless oil
- the expected compound was obtained according to general procedure D using Key Intermediate !V and phenethyl bromide.
- the expected compound was isolated as white powder.
- the expected compound was obtained according to general procedure E using 5-phenyl-2H- pyrazol-3-ylamine.
- the expected compound was isolated as white powder.
- Step 1
- the expected compound was obtained according to genera! procedure G using 2-(4 ⁇ isopropoxy-phenylamino)-7-oxo-4,7-dihydro-[1 ,2,4]triazolo[1 ,5-a]pyrimidSne-6-carboxylic acid ethyl ester described in example 61.
- the expected compound was isolated as yeliow powder. MS: 330.1
- the expected compound was obtained according to general procedure G using 2- benzyiamino- -oxo ⁇ .y-dihydro-fl ⁇ jtriazolofl ⁇ -ajpyrimidine-e-carboxyiic acid ethyl ester described in example 58.
- the expected compound was isolated as pale yellow powder.
- the expected compound was obtained according to general procedure G using 2- [(naphthaien-l-ylmethylJ-aminoj- -oxo ⁇ -dihydrofl ⁇ . ⁇ triazoioCl ⁇ ajpyrimidine-B-carboxyiic acid ethyl ester described in example 60.
- the expected compound was isolated as pate orange powder.
- the expected compound was obtained according to general procedure G using 2- [(benzo[1 ,3]dioxol-5-ylmethyl)-amino]-7-oxo-4,7-dihydro-[1 ,2,4]triazolo[1.5-a]pyrimidine-6- carboxyiic acid ethyl ester.
- This starting material was obtained according to general procedure A using C-benzo[1 ,3]dioxol-5-yl-methylamine.
- the expected acid was isolated without treatment as sodium salt and as yellow powder.
- the expected compound was obtained according to general procedure G using [2-(4- isopropoxy-phenylamino)-7-oxo-4,7-dihydro-[1 ,2,4]triazoio[1 ,5-a3pyrimidin-6-yl]-acetic acid ethyl ester described in example 65.
- the expected compound was obtained according to general procedure G using 4-benzyl-2- cyciopropyl-7-oxo-4,7-dihydro-pyrazolo[1 ,5-a]pyrimidine-6-carboxylic acid ethyl ester described in example 74.
- the expected compound was isolated as beige powder.
- the expected compound was obtained according to general procedure G using 2- cydopropyl-7-oxo-4-phenethyl-4,7-dihydro-pyrazolo[1 ,5-a]pyrimidine-6-carboxyfic acid ethyi ester described in example 75.
- the expected compound was isolated as beige powder.
- the expected compound was obtained according to general procedure G using 2- cyciopropyl-4-[2- ⁇ 4-hydroxy-phenyl)-ethyi]-7-oxo-4,7-dihydro-pyrazoio[1 ,5-a]pyrimidine-6- carboxylic acid ethyl ester described in example 76.
- the expected compound was isolated as white powder.
- the expected compound was obtained according to genera! procedure G using 4-[2-(4- chloro-phenyl)-ethyl]-2-cyclopropyl-7-oxo-4,7-dihydro-pyrazolo[1 ,5-a]pyrimidine-6-carboxylic acid ethyl ester described in example 77, The expected compound was isolated as white powder.
- the expected compound was obtained according to general procedure G using 2- cyclopropyl-4-[2-(4-methoxy-phenyl)-ethy[ -7-oxo-4,7-dihydro-pyrazolo[1 ,5-a]pyrimidine-6- carboxylic acid ethyl ester described in example 78.
- the expected compound was isolated as white powder.
- the expected compound was obtained according to general procedure G using 2- cyclopropyl-7-oxo-4-[2-(4-trifluoromethyl-phenyl)-ethyl]-4,7-dihydro-pyrazoio[ l 5-a]pyrimidine- 6-carboxylic acid ethyl ester.
- the starting materia! was obtained according to general procedure D using Key intermediate i! and 1 -(2-bromo-ethyl)-4-irifiuoromethy!-benzene.
- the expected compound was isolated as white powder.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Virology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pulmonology (AREA)
- Molecular Biology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description

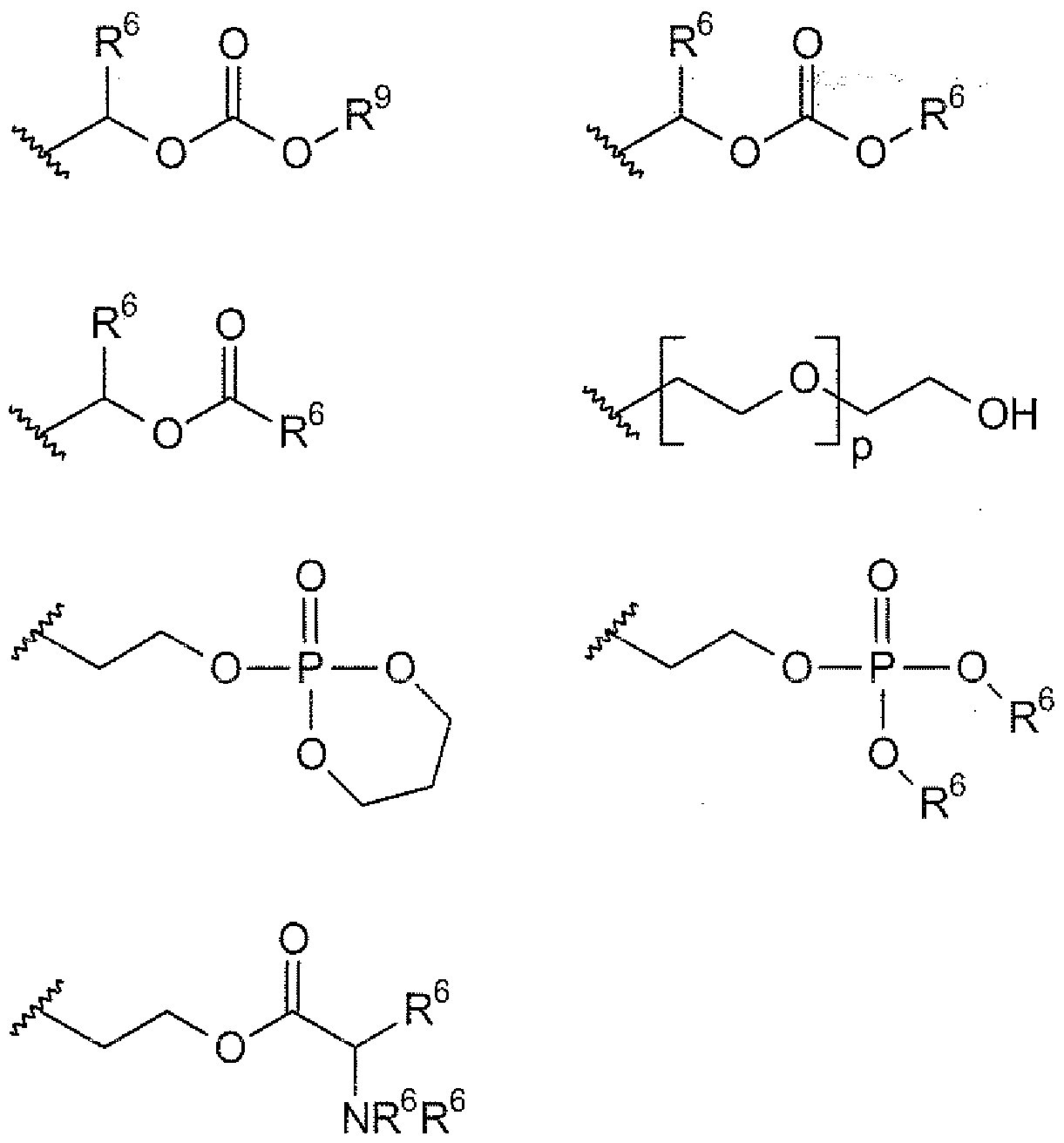























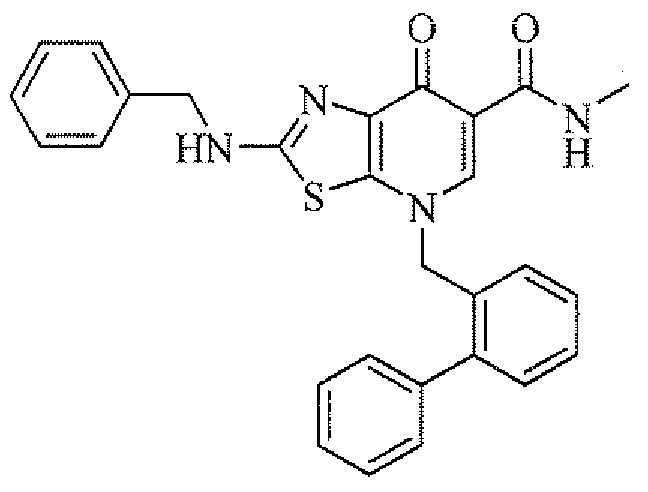









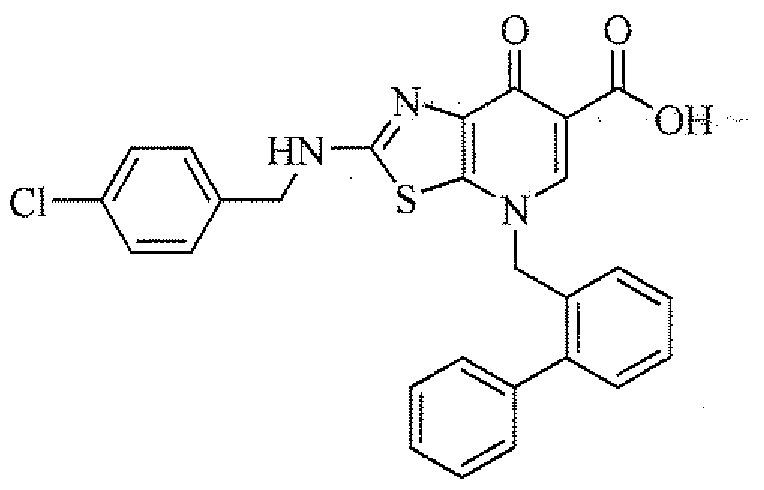











































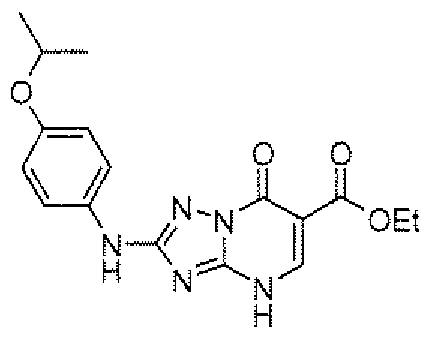

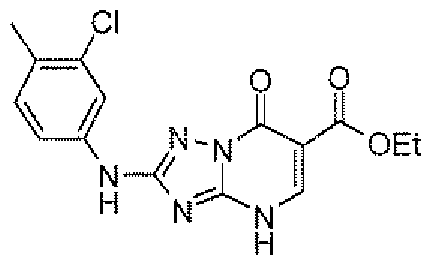









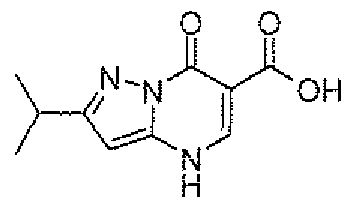


















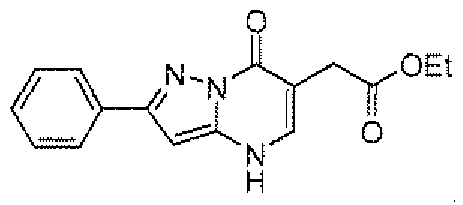










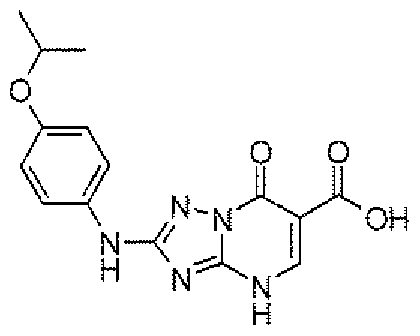



















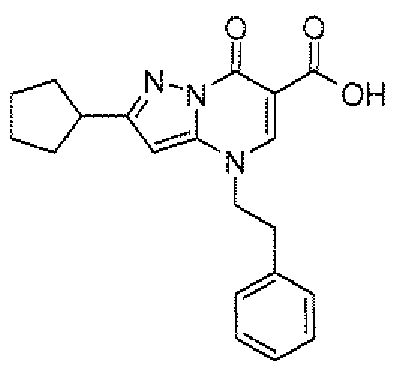

















Claims


Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| RU2014146778A RU2014146778A (en) | 2012-05-23 | 2013-05-23 | 7-OXO-4,7-DIHYDROPYRAZOLO [1,5-A] PYRIMIDINE DERIVATIVES, WHICH ARE SUITABLE FOR TREATMENT, REDUCED INTENSITY, OR PREVENTION OF VIRAL DISEASE |
| KR1020147035547A KR20150014506A (en) | 2012-05-23 | 2013-05-23 | 7-Oxo-4,7-Dihydro-Pyrazolo[1,5-A]Pyrimidine Derivatives Which Are Useful in the Treatment, Amelioration or Prevention of a Viral Disease |
| CA2874253A CA2874253A1 (en) | 2012-05-23 | 2013-05-23 | 7-oxo-4,7-dihydro-pyrazolo [1,5-a] pyrimidine derivatives which are useful in the treatment, amelioration or prevention of a viral disease |
| BR112014029006A BR112014029006A2 (en) | 2012-05-23 | 2013-05-23 | 7-oxo-4,7-dihydro-pyrazolo [1,5-a] pyrimidine derivatives that are useful in treating, ameliorating or preventing a viral disease. |
| JP2015513183A JP2015521189A (en) | 2012-05-23 | 2013-05-23 | 7-oxo-4,7-dihydro-pyrazolo [1,5-a] pyrimidine derivatives useful for the treatment, amelioration or prevention of viral diseases |
| CN201380025121.5A CN104507481B (en) | 2012-05-23 | 2013-05-23 | 7-oxo-4, 7-dihydro-pyrazolo [1,5-a ] pyrimidine derivatives for the treatment, amelioration or prevention of a viral disease |
| EP13730131.3A EP2861232A1 (en) | 2012-05-23 | 2013-05-23 | 7-oxo-4,7 -dihydro- pyrazolo [1, 5 -a]pyrimidine derivatives which are useful in the treatment, amelioration or prevention of a viral disease |
| MX2014014109A MX2014014109A (en) | 2012-05-23 | 2013-05-23 | 7-oxo-4,7 -dihydro- pyrazolo [1, 5 -a] pyrimidine derivatives which are useful in the treatment, amelioration or prevention of a viral disease. |
| HK15105893.0A HK1204987A1 (en) | 2012-05-23 | 2015-06-19 | 7-oxo-4,7 -dihydro- pyrazolo [1, 5 -a]pyrimidine derivatives which are useful in the treatment, amelioration or prevention of a viral disease 7--47--[15-a] |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261650725P | 2012-05-23 | 2012-05-23 | |
| US61/650,725 | 2012-05-23 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013174931A1 true WO2013174931A1 (en) | 2013-11-28 |
Family
ID=48669866
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2013/060634 WO2013174931A1 (en) | 2012-05-23 | 2013-05-23 | 7-oxo-4,7 -dihydro- pyrazolo [1, 5 -a] pyrimidine derivatives which are useful in the treatment, amelioration or prevention of a viral disease |
Country Status (11)
| Country | Link |
|---|---|
| US (2) | US20130317021A1 (en) |
| EP (1) | EP2861232A1 (en) |
| JP (1) | JP2015521189A (en) |
| KR (1) | KR20150014506A (en) |
| CN (1) | CN104507481B (en) |
| BR (1) | BR112014029006A2 (en) |
| CA (1) | CA2874253A1 (en) |
| HK (1) | HK1204987A1 (en) |
| MX (1) | MX2014014109A (en) |
| RU (1) | RU2014146778A (en) |
| WO (1) | WO2013174931A1 (en) |
Cited By (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014139326A1 (en) | 2013-03-13 | 2014-09-18 | Genentech, Inc. | Pyrazolo compounds and uses thereof |
| US8957078B2 (en) | 2013-03-15 | 2015-02-17 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US8969360B2 (en) | 2013-03-15 | 2015-03-03 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US9309250B2 (en) | 2011-06-22 | 2016-04-12 | Vertex Pharmaceuticals Incorporated | Substituted pyrrolo[2,3-b]pyrazines as ATR kinase inhibitors |
| US9340546B2 (en) | 2012-12-07 | 2016-05-17 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| WO2017046362A1 (en) * | 2015-09-18 | 2017-03-23 | F. Hoffmann-La Roche Ag | Pyrazolopyrazines and their use in the treatment, amelioration or prevention of a viral disease |
| US9663519B2 (en) | 2013-03-15 | 2017-05-30 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US9670215B2 (en) | 2014-06-05 | 2017-06-06 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US20180118760A1 (en) | 2015-04-28 | 2018-05-03 | Shionogi & Co., Ltd. | Substituted polycyclic pyridone derivatives and prodrugs thereof |
| US10160760B2 (en) | 2013-12-06 | 2018-12-25 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US10738056B2 (en) | 2017-09-15 | 2020-08-11 | Aduro Biotech Inc. | Pyrazolopyrimidinone compounds and uses thereof |
| US10759814B2 (en) | 2016-08-10 | 2020-09-01 | Shionogi & Co., Ltd. | Pharmaceutical compositions containing substituted polycyclic pyridone derivatives and prodrug thereof |
| US11040048B2 (en) | 2015-12-15 | 2021-06-22 | Shionogi & Co., Ltd. | Medicament for treating influenza characterized by combining a Cap-dependent endonuclease inhibitor and an anti-influenza drug |
| US11179394B2 (en) | 2014-06-17 | 2021-11-23 | Vertex Pharmaceuticals Incorporated | Method for treating cancer using a combination of Chk1 and ATR inhibitors |
| US11464774B2 (en) | 2015-09-30 | 2022-10-11 | Vertex Pharmaceuticals Incorporated | Method for treating cancer using a combination of DNA damaging agents and ATR inhibitors |
Families Citing this family (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2874250A1 (en) * | 2012-05-23 | 2013-11-28 | European Molecular Biology Laboratory | 7-oxo-thiazolopyridine carbonic acid derivatives and their use in the treatment, amelioration or prevention of a viral disease |
| EP3164406A4 (en) | 2014-07-01 | 2018-04-04 | Rempex Pharmaceuticals, Inc. | Boronic acid derivatives and therapeutic uses thereof |
| EP3166951A1 (en) | 2014-07-07 | 2017-05-17 | Savira Pharmaceuticals GmbH | Dihydropyridopyrazine-1,8-diones and their use in the treatment, amelioration or prevention of viral diseases |
| WO2018001948A1 (en) | 2016-06-29 | 2018-01-04 | F. Hoffmann-La Roche Ag | Pyridazinone-based broad spectrum anti-influenza inhibitors |
| EP3525785A4 (en) * | 2016-10-12 | 2020-03-25 | Merck Sharp & Dohme Corp. | Kdm5 inhibitors |
| WO2018071282A1 (en) * | 2016-10-12 | 2018-04-19 | Merck Sharp & Dohme Corp. | Kdm5 inhibitors |
| EP3746444B1 (en) * | 2018-01-31 | 2023-09-27 | Janssen Sciences Ireland Unlimited Company | Cycloalkyl substituted pyrazolopyrimidines having activity against rsv |
| CN113620977B (en) * | 2021-08-24 | 2024-02-02 | 江苏弘和药物研发有限公司 | Synthesis method of thiazolopyrimidinone acetic acid |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5475109A (en) | 1994-10-17 | 1995-12-12 | Merck & Co., Inc. | Dioxobutanoic acid derivatives as inhibitors of influenza endonuclease |
| WO2002004444A2 (en) * | 2000-07-12 | 2002-01-17 | Pharmacia & Upjohn Company | Heterocycle carboxamides as antiviral agents |
| WO2006030807A1 (en) | 2004-09-15 | 2006-03-23 | Shionogi & Co., Ltd. | Carbamoylpyridone derivative having hiv integrase inhibitory activity |
| US20070072831A1 (en) | 2005-05-16 | 2007-03-29 | Gilead Sciences, Inc. | Integrase inhibitor compounds |
| WO2007146813A2 (en) * | 2006-06-08 | 2007-12-21 | Cylene Pharmaceuticals, Inc. | Pyridinone analogs as cell proliferation inhibitors |
| WO2010110231A1 (en) | 2009-03-26 | 2010-09-30 | 塩野義製薬株式会社 | Substituted 3-hydroxy-4-pyridone derivative |
| WO2010110409A1 (en) | 2009-03-26 | 2010-09-30 | 塩野義製薬株式会社 | Process for producing pyrone and pyridone derivatives |
| WO2011000566A2 (en) | 2009-06-30 | 2011-01-06 | Savira Pharmaceuticals Gmbh | Compounds and pharmaceutical compositions for the treatment of negative-sense ssrna virus infections |
| WO2011035518A1 (en) * | 2009-09-23 | 2011-03-31 | 辽宁利锋科技开发有限公司 | Pyrimidine derivatives and analogs, preparation method and use thereof |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1511751B1 (en) * | 2002-06-04 | 2008-03-19 | Neogenesis Pharmaceuticals, Inc. | Pyrazolo[1,5-a]pyrimidine compounds as antiviral agents |
| CA2874250A1 (en) * | 2012-05-23 | 2013-11-28 | European Molecular Biology Laboratory | 7-oxo-thiazolopyridine carbonic acid derivatives and their use in the treatment, amelioration or prevention of a viral disease |
-
2013
- 2013-05-23 MX MX2014014109A patent/MX2014014109A/en unknown
- 2013-05-23 JP JP2015513183A patent/JP2015521189A/en active Pending
- 2013-05-23 RU RU2014146778A patent/RU2014146778A/en not_active Application Discontinuation
- 2013-05-23 KR KR1020147035547A patent/KR20150014506A/en not_active Application Discontinuation
- 2013-05-23 CN CN201380025121.5A patent/CN104507481B/en not_active Expired - Fee Related
- 2013-05-23 BR BR112014029006A patent/BR112014029006A2/en not_active IP Right Cessation
- 2013-05-23 CA CA2874253A patent/CA2874253A1/en not_active Abandoned
- 2013-05-23 US US13/900,940 patent/US20130317021A1/en not_active Abandoned
- 2013-05-23 EP EP13730131.3A patent/EP2861232A1/en not_active Withdrawn
- 2013-05-23 WO PCT/EP2013/060634 patent/WO2013174931A1/en active Application Filing
-
2015
- 2015-06-19 HK HK15105893.0A patent/HK1204987A1/en unknown
-
2016
- 2016-06-24 US US15/191,865 patent/US20160367557A1/en not_active Abandoned
Patent Citations (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5475109A (en) | 1994-10-17 | 1995-12-12 | Merck & Co., Inc. | Dioxobutanoic acid derivatives as inhibitors of influenza endonuclease |
| WO2002004444A2 (en) * | 2000-07-12 | 2002-01-17 | Pharmacia & Upjohn Company | Heterocycle carboxamides as antiviral agents |
| WO2006030807A1 (en) | 2004-09-15 | 2006-03-23 | Shionogi & Co., Ltd. | Carbamoylpyridone derivative having hiv integrase inhibitory activity |
| US20070072831A1 (en) | 2005-05-16 | 2007-03-29 | Gilead Sciences, Inc. | Integrase inhibitor compounds |
| WO2007146813A2 (en) * | 2006-06-08 | 2007-12-21 | Cylene Pharmaceuticals, Inc. | Pyridinone analogs as cell proliferation inhibitors |
| WO2010110231A1 (en) | 2009-03-26 | 2010-09-30 | 塩野義製薬株式会社 | Substituted 3-hydroxy-4-pyridone derivative |
| WO2010110409A1 (en) | 2009-03-26 | 2010-09-30 | 塩野義製薬株式会社 | Process for producing pyrone and pyridone derivatives |
| WO2011000566A2 (en) | 2009-06-30 | 2011-01-06 | Savira Pharmaceuticals Gmbh | Compounds and pharmaceutical compositions for the treatment of negative-sense ssrna virus infections |
| WO2011035518A1 (en) * | 2009-09-23 | 2011-03-31 | 辽宁利锋科技开发有限公司 | Pyrimidine derivatives and analogs, preparation method and use thereof |
| US20120178915A1 (en) * | 2009-09-23 | 2012-07-12 | Lifeng Xu | Pyrimidine derivatives and analogs, preparation method and use thereof |
Non-Patent Citations (29)
| Title |
|---|
| "Helvetica Chimica Acta", 1995, article "A multilingual glossary of biotechnological terms: (IUPAC Recommendations" |
| CHANG, ANTIVIRAL RESEARCH, vol. 89, 2011, pages 26 - 34 |
| D. J. GOOD, CRYST. GROWTH DES., vol. 9, no. 5, 2009, pages 2252 - 2264 |
| DHARAN ET AL., THE JOURNAL OF THE AMERICAN MEDICAL ASSOCIATION, vol. 301, no. 10, 11 March 2009 (2009-03-11), pages 1034 - 1041 |
| DIAS, NATURE, vol. 458, 2009, pages 914 - 918 |
| DIAS, NATURE, vol. 458, no. 7240, 16 April 2009 (2009-04-16), pages 914 - 918 |
| ERIKSSON, B. ET AL., ANTIMICROB. AGENTS CHEMOTHER., vol. 11, 1977, pages 946 - 951 |
| FURUTA ET AL., ANTIMICROBIAL AGENTS AND CHEMOTHERAPY, 2005, pages 981 - 986 |
| GHANEM, A. ET AL., J. VIROL., vol. 81, 2007, pages 7801 - 7804 |
| GUILLIGAY, NATURE STRUCTURAL & MOLECULAR BIOLOGY, vol. 15, no. 5, May 2008 (2008-05-01), pages 500 - 506 |
| H. A. AI-KHAMEES ET AL.: "the synthesis of 2-substituted-1,2,4-triazolo[1,5-a]-pyrimidine and 1,2,4-triazolo[4,3-a]pyrimidine derivatives as potential antimicrobial agents", INDIAN JOURNAL OF HETEROCYCLIC CHEMISTRY, vol. 2, 1993, pages 237 - 244 |
| KUKKONEN, S. K. ET AL., ARCH. VIROL., vol. 150, 2005, pages 533 - 556 |
| LEAHY, M. B. ET AL., J. VIROL., vol. 71, 2005, pages 8347 - 8351 |
| MAGDEN, J., APP!. MICROBIOL. BIOTECHNOL., vol. 66, 2005, pages 612 - 621 |
| MAGDEN, J., APPL. MICROBIOL. BIOTECHNOL., vol. 66, 2005, pages 612 - 621 |
| MOSCONA ET AL., THE NEW ENGLAND JOURNAL OF MEDICINE, vol. 360, no. 10, 5 March 2009 (2009-03-05), pages 953 - 956 |
| N. DAS ET AL., EUROPEAN JOURNAL OF PHARMACEUTICAL SCIENCES, vol. 41, 2010, pages 571 - 588 |
| NEUMANN ET AL., NATURE, vol. 459, no. 7249, 18 December 2008 (2008-12-18), pages 931 - 939 |
| NING SHAN ET AL., DRUG DISCOVERY TODAY, vol. 13, no. 9/10, 2008, pages 440 - 446 |
| NOAH, D. L., ADV. VIRUS RES., vol. 65, 2005, pages 121 - 145 |
| PLOTCH, S. J. ET AL., CELL, vol. 23, 1981, pages 847 - 858 |
| REED, L. J.; H. MUENCH, AM. J. HYG., vol. 27, 1938, pages 493 - 497 |
| RUSINOV V L ET AL: "Synthesis and antiviral activity of nucleoside analogs based on 1,2,4-triazolo[3,2-c][1,2,4]triazin-7-ones", RUSSIAN CHEMICAL BULLETIN, KLUWER ACADEMIC PUBLISHERS-PLENUM PUBLISHERS, NE, vol. 59, no. 1, 19 April 2010 (2010-04-19), pages 136 - 143, XP019829845, ISSN: 1573-9171 * |
| S. M. BERGE ET AL.: "Pharmaceutical Salts", J. PHARM. SCI., vol. 66, 1977, pages 1 - 19, XP002675560, DOI: doi:10.1002/jps.2600660104 |
| TISDALE, M. ET AL., ANTIMICROB. AGENTS CHEMOTHER., vol. 39, 1995, pages 2454 - 2458 |
| TOMASSINI, J. ET AL., ANTIMICROB. AGENTS CHEMOTHER., vol. 38, 1994, pages 2827 - 2837 |
| TOMASSINI, J. ET AL., ANTIMICROB. AGENTS CHEMOTHER., vol. 40, 1996, pages 1189 - 1193 |
| V. L. RUSINOV ET AL.: "the synthesis and antiviral activity of nucleoside analogs based on 1,2,4-triazolo[3,2-c][1,2,4]triazin-7-ones in the Russian Chemical Bulletin", INTERNATIONAL EDITION, vol. 59, no. 1, 2010, pages 136 - 143 |
| VON LTZSTEIN, M. ET AL., NATURE, vol. 363, 1993, pages 418 - 423 |
Cited By (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9309250B2 (en) | 2011-06-22 | 2016-04-12 | Vertex Pharmaceuticals Incorporated | Substituted pyrrolo[2,3-b]pyrazines as ATR kinase inhibitors |
| US11370798B2 (en) | 2012-12-07 | 2022-06-28 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US9340546B2 (en) | 2012-12-07 | 2016-05-17 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US9650381B2 (en) | 2012-12-07 | 2017-05-16 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US11117900B2 (en) | 2012-12-07 | 2021-09-14 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US9718827B2 (en) | 2012-12-07 | 2017-08-01 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US10787452B2 (en) | 2012-12-07 | 2020-09-29 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US10392391B2 (en) | 2012-12-07 | 2019-08-27 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| EP2970307A4 (en) * | 2013-03-13 | 2016-10-05 | Genentech Inc | Pyrazolo compounds and uses thereof |
| WO2014139326A1 (en) | 2013-03-13 | 2014-09-18 | Genentech, Inc. | Pyrazolo compounds and uses thereof |
| US10035801B2 (en) | 2013-03-13 | 2018-07-31 | Genentech, Inc. | Pyrazolo compounds and uses thereof |
| US8969360B2 (en) | 2013-03-15 | 2015-03-03 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US8957078B2 (en) | 2013-03-15 | 2015-02-17 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US9663519B2 (en) | 2013-03-15 | 2017-05-30 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US10160760B2 (en) | 2013-12-06 | 2018-12-25 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US10815239B2 (en) | 2013-12-06 | 2020-10-27 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US11485739B2 (en) | 2013-12-06 | 2022-11-01 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US9670215B2 (en) | 2014-06-05 | 2017-06-06 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US10800781B2 (en) | 2014-06-05 | 2020-10-13 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US10093676B2 (en) | 2014-06-05 | 2018-10-09 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US11179394B2 (en) | 2014-06-17 | 2021-11-23 | Vertex Pharmaceuticals Incorporated | Method for treating cancer using a combination of Chk1 and ATR inhibitors |
| US20180118760A1 (en) | 2015-04-28 | 2018-05-03 | Shionogi & Co., Ltd. | Substituted polycyclic pyridone derivatives and prodrugs thereof |
| US10633397B2 (en) | 2015-04-28 | 2020-04-28 | Shionogi & Co., Ltd. | Substituted polycyclic pyridone derivatives and prodrugs thereof |
| US10392406B2 (en) | 2015-04-28 | 2019-08-27 | Shionogi & Co., Ltd. | Substituted polycyclic pyridone derivatives and prodrugs thereof |
| WO2017046362A1 (en) * | 2015-09-18 | 2017-03-23 | F. Hoffmann-La Roche Ag | Pyrazolopyrazines and their use in the treatment, amelioration or prevention of a viral disease |
| US11464774B2 (en) | 2015-09-30 | 2022-10-11 | Vertex Pharmaceuticals Incorporated | Method for treating cancer using a combination of DNA damaging agents and ATR inhibitors |
| US11040048B2 (en) | 2015-12-15 | 2021-06-22 | Shionogi & Co., Ltd. | Medicament for treating influenza characterized by combining a Cap-dependent endonuclease inhibitor and an anti-influenza drug |
| US10759814B2 (en) | 2016-08-10 | 2020-09-01 | Shionogi & Co., Ltd. | Pharmaceutical compositions containing substituted polycyclic pyridone derivatives and prodrug thereof |
| US11306106B2 (en) | 2016-08-10 | 2022-04-19 | Shionogi & Co., Ltd. | Pharmaceutical compositions containing substituted polycyclic pyridone derivatives and prodrug thereof |
| US10738056B2 (en) | 2017-09-15 | 2020-08-11 | Aduro Biotech Inc. | Pyrazolopyrimidinone compounds and uses thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| BR112014029006A2 (en) | 2017-06-27 |
| JP2015521189A (en) | 2015-07-27 |
| CN104507481B (en) | 2017-08-04 |
| HK1204987A1 (en) | 2015-12-11 |
| CA2874253A1 (en) | 2013-11-28 |
| US20160367557A1 (en) | 2016-12-22 |
| MX2014014109A (en) | 2016-03-31 |
| KR20150014506A (en) | 2015-02-06 |
| US20130317021A1 (en) | 2013-11-28 |
| EP2861232A1 (en) | 2015-04-22 |
| CN104507481A (en) | 2015-04-08 |
| RU2014146778A (en) | 2016-07-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2013174931A1 (en) | 7-oxo-4,7 -dihydro- pyrazolo [1, 5 -a] pyrimidine derivatives which are useful in the treatment, amelioration or prevention of a viral disease | |
| EP2864338B1 (en) | 7-oxo-thiazolopyridine carbonic acid derivatives and their use in the treatment, amelioration or prevention of a viral disease | |
| US8921388B2 (en) | Dihydroxypyrimidine carbonic acid derivatives and their use in the treatment, amelioration or prevention of a viral disease | |
| ES2589459T3 (en) | Pyrimidin-4-one derivatives and their use in the treatment, improvement or prevention of a viral disease | |
| WO2016005330A1 (en) | Dihydropyridopyrazine-1,8-diones and their use in the treatment, amelioration or prevention of viral diseases | |
| US8952039B2 (en) | Pyridone derivatives and their use in the treatment, ameloriation or prevention of a viral disease | |
| EP2776396A2 (en) | Heteroaryl hydroxamic acid derivatives and their use in the treatment, amelioration or prevention of a viral disease | |
| US9505758B2 (en) | Substituted 1,5-naphthyridines as endonuclease inhibitors | |
| WO2017046318A1 (en) | Triazolones derivatives for use in the treatment, amelioration or prevention of a viral disease | |
| WO2017060470A1 (en) | Pyrrolopyrazine derivatives for use in the treatment, amelioration or prevention of influenza |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13730131 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2874253 Country of ref document: CA Ref document number: 2015513183 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2014/014109 Country of ref document: MX |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 20147035547 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2014146778 Country of ref document: RU Kind code of ref document: A |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112014029006 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 112014029006 Country of ref document: BR Kind code of ref document: A2 Effective date: 20141121 |