WO2013132007A1 - Human binding molecules capable of binding to and neutralizing influenza b viruses and uses thereof - Google Patents
Human binding molecules capable of binding to and neutralizing influenza b viruses and uses thereof Download PDFInfo
- Publication number
- WO2013132007A1 WO2013132007A1 PCT/EP2013/054606 EP2013054606W WO2013132007A1 WO 2013132007 A1 WO2013132007 A1 WO 2013132007A1 EP 2013054606 W EP2013054606 W EP 2013054606W WO 2013132007 A1 WO2013132007 A1 WO 2013132007A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- influenza
- binding molecule
- amino acid
- seq
- binding
- Prior art date
Links
- 230000027455 binding Effects 0.000 title claims abstract description 433
- 241000713196 Influenza B virus Species 0.000 title claims abstract description 98
- 241000282414 Homo sapiens Species 0.000 title claims abstract description 83
- 230000003472 neutralizing effect Effects 0.000 title claims abstract description 49
- 101710154606 Hemagglutinin Proteins 0.000 claims abstract description 91
- 101710093908 Outer capsid protein VP4 Proteins 0.000 claims abstract description 91
- 101710135467 Outer capsid protein sigma-1 Proteins 0.000 claims abstract description 91
- 101710176177 Protein A56 Proteins 0.000 claims abstract description 91
- 239000000185 hemagglutinin Substances 0.000 claims abstract description 64
- 150000007523 nucleic acids Chemical class 0.000 claims abstract description 49
- 108020004707 nucleic acids Proteins 0.000 claims abstract description 42
- 102000039446 nucleic acids Human genes 0.000 claims abstract description 42
- 238000011282 treatment Methods 0.000 claims abstract description 23
- 241000712431 Influenza A virus Species 0.000 claims abstract description 18
- 238000003745 diagnosis Methods 0.000 claims abstract description 13
- 238000011321 prophylaxis Methods 0.000 claims abstract description 11
- 125000003275 alpha amino acid group Chemical group 0.000 claims description 152
- 150000001413 amino acids Chemical class 0.000 claims description 46
- 239000012634 fragment Substances 0.000 claims description 44
- 229940127121 immunoconjugate Drugs 0.000 claims description 42
- 238000000034 method Methods 0.000 claims description 42
- 239000000427 antigen Substances 0.000 claims description 34
- 108091007433 antigens Proteins 0.000 claims description 33
- 102000036639 antigens Human genes 0.000 claims description 33
- 206010022000 influenza Diseases 0.000 claims description 28
- 239000008194 pharmaceutical composition Substances 0.000 claims description 25
- 239000003814 drug Substances 0.000 claims description 16
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 12
- 229940117432 influenza b virus antigen Drugs 0.000 claims description 8
- 239000012472 biological sample Substances 0.000 claims description 6
- 239000003937 drug carrier Substances 0.000 claims 1
- 241000712461 unidentified influenza virus Species 0.000 abstract description 69
- 239000000203 mixture Substances 0.000 abstract description 38
- 210000004027 cell Anatomy 0.000 description 106
- 235000001014 amino acid Nutrition 0.000 description 43
- 229940024606 amino acid Drugs 0.000 description 40
- 208000037798 influenza B Diseases 0.000 description 37
- 241000700605 Viruses Species 0.000 description 35
- 108090000623 proteins and genes Proteins 0.000 description 34
- 125000000539 amino acid group Chemical group 0.000 description 29
- 102000004169 proteins and genes Human genes 0.000 description 29
- 125000003729 nucleotide group Chemical group 0.000 description 27
- 235000018102 proteins Nutrition 0.000 description 27
- 239000013598 vector Substances 0.000 description 27
- 208000015181 infectious disease Diseases 0.000 description 24
- 108090000765 processed proteins & peptides Proteins 0.000 description 24
- 230000035772 mutation Effects 0.000 description 23
- 239000002773 nucleotide Chemical group 0.000 description 23
- 108060003951 Immunoglobulin Proteins 0.000 description 19
- 239000003795 chemical substances by application Substances 0.000 description 19
- 102000018358 immunoglobulin Human genes 0.000 description 19
- 102000004196 processed proteins & peptides Human genes 0.000 description 19
- 108091028043 Nucleic acid sequence Proteins 0.000 description 16
- 230000000069 prophylactic effect Effects 0.000 description 16
- 241000699666 Mus <mouse, genus> Species 0.000 description 15
- 238000003556 assay Methods 0.000 description 15
- 239000002609 medium Substances 0.000 description 15
- 241000894006 Bacteria Species 0.000 description 13
- 238000001727 in vivo Methods 0.000 description 13
- 239000000523 sample Substances 0.000 description 13
- 230000001225 therapeutic effect Effects 0.000 description 13
- 238000002965 ELISA Methods 0.000 description 12
- 230000000694 effects Effects 0.000 description 12
- 238000000338 in vitro Methods 0.000 description 12
- 230000004048 modification Effects 0.000 description 12
- 238000012986 modification Methods 0.000 description 12
- 230000009385 viral infection Effects 0.000 description 12
- 241000699670 Mus sp. Species 0.000 description 11
- 239000000872 buffer Substances 0.000 description 11
- 239000000126 substance Substances 0.000 description 11
- 239000006228 supernatant Substances 0.000 description 11
- 108010047041 Complementarity Determining Regions Proteins 0.000 description 10
- 229920001184 polypeptide Polymers 0.000 description 10
- 238000002360 preparation method Methods 0.000 description 10
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 9
- 241001465754 Metazoa Species 0.000 description 9
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 9
- 230000000890 antigenic effect Effects 0.000 description 9
- 238000001514 detection method Methods 0.000 description 9
- 238000009472 formulation Methods 0.000 description 9
- 239000000463 material Substances 0.000 description 9
- 108020003175 receptors Proteins 0.000 description 9
- 102000005962 receptors Human genes 0.000 description 9
- 230000009261 transgenic effect Effects 0.000 description 9
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 8
- 241000831652 Salinivibrio sharmensis Species 0.000 description 8
- 239000000306 component Substances 0.000 description 8
- 238000002474 experimental method Methods 0.000 description 8
- 239000000047 product Substances 0.000 description 8
- 239000000243 solution Substances 0.000 description 8
- 210000003719 b-lymphocyte Anatomy 0.000 description 7
- 230000001413 cellular effect Effects 0.000 description 7
- 230000000295 complement effect Effects 0.000 description 7
- 238000011534 incubation Methods 0.000 description 7
- 230000001404 mediated effect Effects 0.000 description 7
- 238000006467 substitution reaction Methods 0.000 description 7
- 239000000725 suspension Substances 0.000 description 7
- 229940124597 therapeutic agent Drugs 0.000 description 7
- 108020004414 DNA Proteins 0.000 description 6
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 6
- 241000282412 Homo Species 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- 230000003321 amplification Effects 0.000 description 6
- 239000003443 antiviral agent Substances 0.000 description 6
- 230000015572 biosynthetic process Effects 0.000 description 6
- 239000013604 expression vector Substances 0.000 description 6
- 210000004602 germ cell Anatomy 0.000 description 6
- 230000005764 inhibitory process Effects 0.000 description 6
- 238000004519 manufacturing process Methods 0.000 description 6
- 239000012528 membrane Substances 0.000 description 6
- 238000003199 nucleic acid amplification method Methods 0.000 description 6
- 239000002245 particle Substances 0.000 description 6
- 108091033319 polynucleotide Proteins 0.000 description 6
- 102000040430 polynucleotide Human genes 0.000 description 6
- 239000002157 polynucleotide Substances 0.000 description 6
- 238000003127 radioimmunoassay Methods 0.000 description 6
- 230000001932 seasonal effect Effects 0.000 description 6
- 230000009466 transformation Effects 0.000 description 6
- FWMNVWWHGCHHJJ-SKKKGAJSSA-N 4-amino-1-[(2r)-6-amino-2-[[(2r)-2-[[(2r)-2-[[(2r)-2-amino-3-phenylpropanoyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]hexanoyl]piperidine-4-carboxylic acid Chemical compound C([C@H](C(=O)N[C@H](CC(C)C)C(=O)N[C@H](CCCCN)C(=O)N1CCC(N)(CC1)C(O)=O)NC(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 FWMNVWWHGCHHJJ-SKKKGAJSSA-N 0.000 description 5
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 5
- 102000004190 Enzymes Human genes 0.000 description 5
- 108090000790 Enzymes Proteins 0.000 description 5
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 5
- 238000004458 analytical method Methods 0.000 description 5
- 230000001580 bacterial effect Effects 0.000 description 5
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 5
- 229960002685 biotin Drugs 0.000 description 5
- 239000011616 biotin Substances 0.000 description 5
- 238000012217 deletion Methods 0.000 description 5
- 230000037430 deletion Effects 0.000 description 5
- 230000001419 dependent effect Effects 0.000 description 5
- 239000012636 effector Substances 0.000 description 5
- 229960001031 glucose Drugs 0.000 description 5
- 239000008103 glucose Substances 0.000 description 5
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 5
- 239000001963 growth medium Substances 0.000 description 5
- 230000002401 inhibitory effect Effects 0.000 description 5
- 230000003993 interaction Effects 0.000 description 5
- 231100000518 lethal Toxicity 0.000 description 5
- 230000001665 lethal effect Effects 0.000 description 5
- 238000002823 phage display Methods 0.000 description 5
- -1 phosphotriesters Chemical class 0.000 description 5
- 230000002265 prevention Effects 0.000 description 5
- 150000003839 salts Chemical class 0.000 description 5
- 210000001519 tissue Anatomy 0.000 description 5
- 238000001890 transfection Methods 0.000 description 5
- 230000003612 virological effect Effects 0.000 description 5
- 238000001262 western blot Methods 0.000 description 5
- 108091026890 Coding region Proteins 0.000 description 4
- 229920001213 Polysorbate 20 Polymers 0.000 description 4
- 238000007792 addition Methods 0.000 description 4
- AVKUERGKIZMTKX-NJBDSQKTSA-N ampicillin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)=CC=CC=C1 AVKUERGKIZMTKX-NJBDSQKTSA-N 0.000 description 4
- 229960000723 ampicillin Drugs 0.000 description 4
- 235000020958 biotin Nutrition 0.000 description 4
- 230000000903 blocking effect Effects 0.000 description 4
- 210000004369 blood Anatomy 0.000 description 4
- 239000008280 blood Substances 0.000 description 4
- 238000010367 cloning Methods 0.000 description 4
- 150000001875 compounds Chemical class 0.000 description 4
- 238000011161 development Methods 0.000 description 4
- 238000010790 dilution Methods 0.000 description 4
- 239000012895 dilution Substances 0.000 description 4
- 238000009826 distribution Methods 0.000 description 4
- 210000003128 head Anatomy 0.000 description 4
- 230000001900 immune effect Effects 0.000 description 4
- 230000003053 immunization Effects 0.000 description 4
- 238000010166 immunofluorescence Methods 0.000 description 4
- 239000000543 intermediate Substances 0.000 description 4
- 229960003752 oseltamivir Drugs 0.000 description 4
- VSZGPKBBMSAYNT-RRFJBIMHSA-N oseltamivir Chemical compound CCOC(=O)C1=C[C@@H](OC(CC)CC)[C@H](NC(C)=O)[C@@H](N)C1 VSZGPKBBMSAYNT-RRFJBIMHSA-N 0.000 description 4
- 239000013612 plasmid Substances 0.000 description 4
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 4
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 4
- 239000000843 powder Substances 0.000 description 4
- 238000000746 purification Methods 0.000 description 4
- 230000010076 replication Effects 0.000 description 4
- 238000002741 site-directed mutagenesis Methods 0.000 description 4
- 239000011780 sodium chloride Substances 0.000 description 4
- 239000007787 solid Substances 0.000 description 4
- 230000004083 survival effect Effects 0.000 description 4
- 238000012546 transfer Methods 0.000 description 4
- 239000003981 vehicle Substances 0.000 description 4
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 3
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 3
- 229920001817 Agar Polymers 0.000 description 3
- 241000588724 Escherichia coli Species 0.000 description 3
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 3
- SXRSQZLOMIGNAQ-UHFFFAOYSA-N Glutaraldehyde Chemical compound O=CCCCC=O SXRSQZLOMIGNAQ-UHFFFAOYSA-N 0.000 description 3
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 3
- 239000004472 Lysine Substances 0.000 description 3
- 241000283973 Oryctolagus cuniculus Species 0.000 description 3
- 239000002202 Polyethylene glycol Substances 0.000 description 3
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 3
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 3
- 235000014680 Saccharomyces cerevisiae Nutrition 0.000 description 3
- 239000004098 Tetracycline Substances 0.000 description 3
- 239000002671 adjuvant Substances 0.000 description 3
- 239000008272 agar Substances 0.000 description 3
- 238000013459 approach Methods 0.000 description 3
- 230000037396 body weight Effects 0.000 description 3
- 229940098773 bovine serum albumin Drugs 0.000 description 3
- 239000000969 carrier Substances 0.000 description 3
- 239000006143 cell culture medium Substances 0.000 description 3
- 230000008859 change Effects 0.000 description 3
- 239000012707 chemical precursor Substances 0.000 description 3
- 239000002299 complementary DNA Substances 0.000 description 3
- 238000010276 construction Methods 0.000 description 3
- 230000000120 cytopathologic effect Effects 0.000 description 3
- 239000000032 diagnostic agent Substances 0.000 description 3
- 229940039227 diagnostic agent Drugs 0.000 description 3
- 201000010099 disease Diseases 0.000 description 3
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 3
- 229940079593 drug Drugs 0.000 description 3
- 238000004520 electroporation Methods 0.000 description 3
- 238000000605 extraction Methods 0.000 description 3
- 238000001943 fluorescence-activated cell sorting Methods 0.000 description 3
- 230000004927 fusion Effects 0.000 description 3
- 239000011521 glass Substances 0.000 description 3
- 235000013922 glutamic acid Nutrition 0.000 description 3
- 239000004220 glutamic acid Substances 0.000 description 3
- 230000035931 haemagglutination Effects 0.000 description 3
- 230000036541 health Effects 0.000 description 3
- 210000004408 hybridoma Anatomy 0.000 description 3
- 238000002649 immunization Methods 0.000 description 3
- 238000003018 immunoassay Methods 0.000 description 3
- 229940072221 immunoglobulins Drugs 0.000 description 3
- 238000003780 insertion Methods 0.000 description 3
- 230000037431 insertion Effects 0.000 description 3
- 230000034217 membrane fusion Effects 0.000 description 3
- 229910052751 metal Inorganic materials 0.000 description 3
- 239000002184 metal Substances 0.000 description 3
- 238000006386 neutralization reaction Methods 0.000 description 3
- 231100000252 nontoxic Toxicity 0.000 description 3
- 230000003000 nontoxic effect Effects 0.000 description 3
- 238000007911 parenteral administration Methods 0.000 description 3
- 230000037361 pathway Effects 0.000 description 3
- 239000008188 pellet Substances 0.000 description 3
- 229920001223 polyethylene glycol Polymers 0.000 description 3
- 230000008569 process Effects 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- 229960002180 tetracycline Drugs 0.000 description 3
- 229930101283 tetracycline Natural products 0.000 description 3
- 235000019364 tetracycline Nutrition 0.000 description 3
- 150000003522 tetracyclines Chemical class 0.000 description 3
- 238000013519 translation Methods 0.000 description 3
- 229960005486 vaccine Drugs 0.000 description 3
- 230000007502 viral entry Effects 0.000 description 3
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 3
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 2
- OZFAFGSSMRRTDW-UHFFFAOYSA-N (2,4-dichlorophenyl) benzenesulfonate Chemical compound ClC1=CC(Cl)=CC=C1OS(=O)(=O)C1=CC=CC=C1 OZFAFGSSMRRTDW-UHFFFAOYSA-N 0.000 description 2
- UBCHPRBFMUDMNC-UHFFFAOYSA-N 1-(1-adamantyl)ethanamine Chemical compound C1C(C2)CC3CC2CC1(C(N)C)C3 UBCHPRBFMUDMNC-UHFFFAOYSA-N 0.000 description 2
- FWBHETKCLVMNFS-UHFFFAOYSA-N 4',6-Diamino-2-phenylindol Chemical compound C1=CC(C(=N)N)=CC=C1C1=CC2=CC=C(C(N)=N)C=C2N1 FWBHETKCLVMNFS-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- 239000012591 Dulbecco’s Phosphate Buffered Saline Substances 0.000 description 2
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 2
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 2
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 2
- 241000287828 Gallus gallus Species 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- 241000238631 Hexapoda Species 0.000 description 2
- 108700005091 Immunoglobulin Genes Proteins 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 description 2
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 2
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 2
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 2
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 2
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- 206010057249 Phagocytosis Diseases 0.000 description 2
- 239000004698 Polyethylene Substances 0.000 description 2
- 238000012300 Sequence Analysis Methods 0.000 description 2
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 2
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 2
- 239000004473 Threonine Substances 0.000 description 2
- 108700019146 Transgenes Proteins 0.000 description 2
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 2
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 2
- 230000010530 Virus Neutralization Effects 0.000 description 2
- 239000002253 acid Substances 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- DZBUGLKDJFMEHC-UHFFFAOYSA-N acridine Chemical compound C1=CC=CC2=CC3=CC=CC=C3N=C21 DZBUGLKDJFMEHC-UHFFFAOYSA-N 0.000 description 2
- 239000011543 agarose gel Substances 0.000 description 2
- 230000004075 alteration Effects 0.000 description 2
- DKNWSYNQZKUICI-UHFFFAOYSA-N amantadine Chemical compound C1C(C2)CC3CC2CC1(N)C3 DKNWSYNQZKUICI-UHFFFAOYSA-N 0.000 description 2
- 229960003805 amantadine Drugs 0.000 description 2
- 238000010171 animal model Methods 0.000 description 2
- 230000000692 anti-sense effect Effects 0.000 description 2
- 230000000840 anti-viral effect Effects 0.000 description 2
- 239000003963 antioxidant agent Substances 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- 210000004436 artificial bacterial chromosome Anatomy 0.000 description 2
- 210000001106 artificial yeast chromosome Anatomy 0.000 description 2
- VEZXCJBBBCKRPI-UHFFFAOYSA-N beta-propiolactone Chemical compound O=C1CCO1 VEZXCJBBBCKRPI-UHFFFAOYSA-N 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- 229940041514 candida albicans extract Drugs 0.000 description 2
- 239000002738 chelating agent Substances 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 238000003776 cleavage reaction Methods 0.000 description 2
- 239000011248 coating agent Substances 0.000 description 2
- 238000000576 coating method Methods 0.000 description 2
- 230000001268 conjugating effect Effects 0.000 description 2
- 238000012258 culturing Methods 0.000 description 2
- 230000001472 cytotoxic effect Effects 0.000 description 2
- 238000004925 denaturation Methods 0.000 description 2
- 230000036425 denaturation Effects 0.000 description 2
- 239000012502 diagnostic product Substances 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- 229940042406 direct acting antivirals neuraminidase inhibitors Drugs 0.000 description 2
- 235000013601 eggs Nutrition 0.000 description 2
- 230000029117 egress of virus within host cell Effects 0.000 description 2
- 210000001163 endosome Anatomy 0.000 description 2
- 239000000284 extract Substances 0.000 description 2
- 239000012091 fetal bovine serum Substances 0.000 description 2
- 230000002538 fungal effect Effects 0.000 description 2
- 108020001507 fusion proteins Proteins 0.000 description 2
- 102000037865 fusion proteins Human genes 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 210000005260 human cell Anatomy 0.000 description 2
- 238000003384 imaging method Methods 0.000 description 2
- 230000001524 infective effect Effects 0.000 description 2
- 208000037797 influenza A Diseases 0.000 description 2
- 229960003971 influenza vaccine Drugs 0.000 description 2
- 239000004615 ingredient Substances 0.000 description 2
- 239000003112 inhibitor Substances 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 238000011081 inoculation Methods 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 description 2
- 229960000310 isoleucine Drugs 0.000 description 2
- 229960000318 kanamycin Drugs 0.000 description 2
- 229930027917 kanamycin Natural products 0.000 description 2
- SBUJHOSQTJFQJX-NOAMYHISSA-N kanamycin Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CN)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O2)O)[C@H](N)C[C@@H]1N SBUJHOSQTJFQJX-NOAMYHISSA-N 0.000 description 2
- 229930182823 kanamycin A Natural products 0.000 description 2
- 238000002372 labelling Methods 0.000 description 2
- 150000002632 lipids Chemical class 0.000 description 2
- 239000002502 liposome Substances 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 210000004962 mammalian cell Anatomy 0.000 description 2
- 230000035800 maturation Effects 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 230000011987 methylation Effects 0.000 description 2
- 238000007069 methylation reaction Methods 0.000 description 2
- 235000013336 milk Nutrition 0.000 description 2
- 239000008267 milk Substances 0.000 description 2
- 210000004080 milk Anatomy 0.000 description 2
- 239000013642 negative control Substances 0.000 description 2
- 230000008782 phagocytosis Effects 0.000 description 2
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 2
- 239000004033 plastic Substances 0.000 description 2
- 229920003023 plastic Polymers 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 238000012545 processing Methods 0.000 description 2
- 230000000644 propagated effect Effects 0.000 description 2
- ZCCUUQDIBDJBTK-UHFFFAOYSA-N psoralen Chemical compound C1=C2OC(=O)C=CC2=CC2=C1OC=C2 ZCCUUQDIBDJBTK-UHFFFAOYSA-N 0.000 description 2
- RXWNCPJZOCPEPQ-NVWDDTSBSA-N puromycin Chemical compound C1=CC(OC)=CC=C1C[C@H](N)C(=O)N[C@H]1[C@@H](O)[C@H](N2C3=NC=NC(=C3N=C2)N(C)C)O[C@@H]1CO RXWNCPJZOCPEPQ-NVWDDTSBSA-N 0.000 description 2
- 238000005057 refrigeration Methods 0.000 description 2
- 108091008146 restriction endonucleases Proteins 0.000 description 2
- 229960000888 rimantadine Drugs 0.000 description 2
- 230000007017 scission Effects 0.000 description 2
- 238000012216 screening Methods 0.000 description 2
- 238000012163 sequencing technique Methods 0.000 description 2
- 210000002966 serum Anatomy 0.000 description 2
- 239000002911 sialidase inhibitor Substances 0.000 description 2
- 238000011125 single therapy Methods 0.000 description 2
- 238000005063 solubilization Methods 0.000 description 2
- 230000007928 solubilization Effects 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 230000009870 specific binding Effects 0.000 description 2
- 239000012798 spherical particle Substances 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- 208000024891 symptom Diseases 0.000 description 2
- 230000002195 synergetic effect Effects 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 2
- 239000004474 valine Substances 0.000 description 2
- 210000003462 vein Anatomy 0.000 description 2
- 210000002845 virion Anatomy 0.000 description 2
- 239000012138 yeast extract Substances 0.000 description 2
- 229960001028 zanamivir Drugs 0.000 description 2
- ARAIBEBZBOPLMB-UFGQHTETSA-N zanamivir Chemical compound CC(=O)N[C@@H]1[C@@H](N=C(N)N)C=C(C(O)=O)O[C@H]1[C@H](O)[C@H](O)CO ARAIBEBZBOPLMB-UFGQHTETSA-N 0.000 description 2
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- 102100039358 3-hydroxyacyl-CoA dehydrogenase type-2 Human genes 0.000 description 1
- VXGRJERITKFWPL-UHFFFAOYSA-N 4',5'-Dihydropsoralen Natural products C1=C2OC(=O)C=CC2=CC2=C1OCC2 VXGRJERITKFWPL-UHFFFAOYSA-N 0.000 description 1
- 206010069754 Acquired gene mutation Diseases 0.000 description 1
- 229920000936 Agarose Polymers 0.000 description 1
- 241000589158 Agrobacterium Species 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- 102000009027 Albumins Human genes 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 description 1
- 102100026189 Beta-galactosidase Human genes 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- QCMYYKRYFNMIEC-UHFFFAOYSA-N COP(O)=O Chemical class COP(O)=O QCMYYKRYFNMIEC-UHFFFAOYSA-N 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- 102000008186 Collagen Human genes 0.000 description 1
- 108010035532 Collagen Proteins 0.000 description 1
- 241000699802 Cricetulus griseus Species 0.000 description 1
- 101150074155 DHFR gene Proteins 0.000 description 1
- 102000012410 DNA Ligases Human genes 0.000 description 1
- 108010061982 DNA Ligases Proteins 0.000 description 1
- 102000004163 DNA-directed RNA polymerases Human genes 0.000 description 1
- 108090000626 DNA-directed RNA polymerases Proteins 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- 241000255581 Drosophila <fruit fly, genus> Species 0.000 description 1
- 239000004150 EU approved colour Substances 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- YQYJSBFKSSDGFO-UHFFFAOYSA-N Epihygromycin Natural products OC1C(O)C(C(=O)C)OC1OC(C(=C1)O)=CC=C1C=C(C)C(=O)NC1C(O)C(O)C2OCOC2C1O YQYJSBFKSSDGFO-UHFFFAOYSA-N 0.000 description 1
- 241000588722 Escherichia Species 0.000 description 1
- 102000009109 Fc receptors Human genes 0.000 description 1
- 108010087819 Fc receptors Proteins 0.000 description 1
- 241000192125 Firmicutes Species 0.000 description 1
- 238000012413 Fluorescence activated cell sorting analysis Methods 0.000 description 1
- 108700028146 Genetic Enhancer Elements Proteins 0.000 description 1
- 102000005720 Glutathione transferase Human genes 0.000 description 1
- 108010070675 Glutathione transferase Proteins 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 108090000288 Glycoproteins Proteins 0.000 description 1
- 102000003886 Glycoproteins Human genes 0.000 description 1
- 108010043121 Green Fluorescent Proteins Proteins 0.000 description 1
- 102000004144 Green Fluorescent Proteins Human genes 0.000 description 1
- HVLSXIKZNLPZJJ-TXZCQADKSA-N HA peptide Chemical compound C([C@@H](C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](C)C(O)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](N)CC=1C=CC(O)=CC=1)C1=CC=C(O)C=C1 HVLSXIKZNLPZJJ-TXZCQADKSA-N 0.000 description 1
- 239000012981 Hank's balanced salt solution Substances 0.000 description 1
- 108010093488 His-His-His-His-His-His Proteins 0.000 description 1
- 101001035740 Homo sapiens 3-hydroxyacyl-CoA dehydrogenase type-2 Proteins 0.000 description 1
- 108010067060 Immunoglobulin Variable Region Proteins 0.000 description 1
- 102000017727 Immunoglobulin Variable Region Human genes 0.000 description 1
- 241000235058 Komagataella pastoris Species 0.000 description 1
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 1
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 1
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 1
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 1
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 1
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 1
- 108060001084 Luciferase Proteins 0.000 description 1
- 239000005089 Luciferase Substances 0.000 description 1
- 101710175625 Maltose/maltodextrin-binding periplasmic protein Proteins 0.000 description 1
- 108010052285 Membrane Proteins Proteins 0.000 description 1
- 102000018697 Membrane Proteins Human genes 0.000 description 1
- 241001529936 Murinae Species 0.000 description 1
- 241000699660 Mus musculus Species 0.000 description 1
- 241000282341 Mustela putorius furo Species 0.000 description 1
- 108091061960 Naked DNA Proteins 0.000 description 1
- 229930193140 Neomycin Natural products 0.000 description 1
- 239000000020 Nitrocellulose Substances 0.000 description 1
- 239000004677 Nylon Substances 0.000 description 1
- 241000320412 Ogataea angusta Species 0.000 description 1
- 238000012408 PCR amplification Methods 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 108010067902 Peptide Library Proteins 0.000 description 1
- 102000003992 Peroxidases Human genes 0.000 description 1
- 229920002732 Polyanhydride Polymers 0.000 description 1
- 229920002584 Polyethylene Glycol 6000 Polymers 0.000 description 1
- 229920000954 Polyglycolide Polymers 0.000 description 1
- 108010039918 Polylysine Proteins 0.000 description 1
- 229920001710 Polyorthoester Polymers 0.000 description 1
- 239000004793 Polystyrene Substances 0.000 description 1
- 206010036790 Productive cough Diseases 0.000 description 1
- 241000589516 Pseudomonas Species 0.000 description 1
- 108020004511 Recombinant DNA Proteins 0.000 description 1
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 description 1
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 description 1
- 108091028664 Ribonucleotide Proteins 0.000 description 1
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 1
- 241000700584 Simplexvirus Species 0.000 description 1
- 108010003723 Single-Domain Antibodies Proteins 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- 108010090804 Streptavidin Proteins 0.000 description 1
- 210000001744 T-lymphocyte Anatomy 0.000 description 1
- 239000004809 Teflon Substances 0.000 description 1
- 229920006362 Teflon® Polymers 0.000 description 1
- RYYWUUFWQRZTIU-UHFFFAOYSA-N Thiophosphoric acid Chemical class OP(O)(S)=O RYYWUUFWQRZTIU-UHFFFAOYSA-N 0.000 description 1
- 108010022394 Threonine synthase Proteins 0.000 description 1
- 108020004440 Thymidine kinase Proteins 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- 206010058874 Viraemia Diseases 0.000 description 1
- 108010003533 Viral Envelope Proteins Proteins 0.000 description 1
- 108010084455 Zeocin Proteins 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- 230000021736 acetylation Effects 0.000 description 1
- 238000006640 acetylation reaction Methods 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 239000012190 activator Substances 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- 230000006978 adaptation Effects 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 230000009824 affinity maturation Effects 0.000 description 1
- 235000004279 alanine Nutrition 0.000 description 1
- RGCKGOZRHPZPFP-UHFFFAOYSA-N alizarin Chemical compound C1=CC=C2C(=O)C3=C(O)C(O)=CC=C3C(=O)C2=C1 RGCKGOZRHPZPFP-UHFFFAOYSA-N 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 1
- 238000007112 amidation reaction Methods 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 230000010100 anticoagulation Effects 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 235000009582 asparagine Nutrition 0.000 description 1
- 229960001230 asparagine Drugs 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 238000007845 assembly PCR Methods 0.000 description 1
- 230000002238 attenuated effect Effects 0.000 description 1
- 239000011324 bead Substances 0.000 description 1
- 108010005774 beta-Galactosidase Proteins 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 238000012575 bio-layer interferometry Methods 0.000 description 1
- 238000004166 bioassay Methods 0.000 description 1
- 229920000249 biocompatible polymer Polymers 0.000 description 1
- 239000012620 biological material Substances 0.000 description 1
- 238000001574 biopsy Methods 0.000 description 1
- 238000006664 bond formation reaction Methods 0.000 description 1
- DQXBYHZEEUGOBF-UHFFFAOYSA-N but-3-enoic acid;ethene Chemical compound C=C.OC(=O)CC=C DQXBYHZEEUGOBF-UHFFFAOYSA-N 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 150000004657 carbamic acid derivatives Chemical class 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 230000034303 cell budding Effects 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 239000013592 cell lysate Substances 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 235000013339 cereals Nutrition 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- ZYWFEOZQIUMEGL-UHFFFAOYSA-N chloroform;3-methylbutan-1-ol;phenol Chemical compound ClC(Cl)Cl.CC(C)CCO.OC1=CC=CC=C1 ZYWFEOZQIUMEGL-UHFFFAOYSA-N 0.000 description 1
- 210000000349 chromosome Anatomy 0.000 description 1
- 239000013599 cloning vector Substances 0.000 description 1
- 229920001436 collagen Polymers 0.000 description 1
- 238000004040 coloring Methods 0.000 description 1
- 230000002301 combined effect Effects 0.000 description 1
- 238000004590 computer program Methods 0.000 description 1
- 230000021615 conjugation Effects 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 244000038559 crop plants Species 0.000 description 1
- 238000004132 cross linking Methods 0.000 description 1
- 239000003431 cross linking reagent Substances 0.000 description 1
- 230000001186 cumulative effect Effects 0.000 description 1
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 1
- 235000018417 cysteine Nutrition 0.000 description 1
- 230000001086 cytosolic effect Effects 0.000 description 1
- 231100000433 cytotoxic Toxicity 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 231100000517 death Toxicity 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000005860 defense response to virus Effects 0.000 description 1
- 230000018044 dehydration Effects 0.000 description 1
- 238000006297 dehydration reaction Methods 0.000 description 1
- 239000003599 detergent Substances 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 230000029087 digestion Effects 0.000 description 1
- NAGJZTKCGNOGPW-UHFFFAOYSA-N dithiophosphoric acid Chemical class OP(O)(S)=S NAGJZTKCGNOGPW-UHFFFAOYSA-N 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 239000008298 dragée Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 210000003743 erythrocyte Anatomy 0.000 description 1
- 239000005038 ethylene vinyl acetate Substances 0.000 description 1
- 235000013861 fat-free Nutrition 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 238000000684 flow cytometry Methods 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- 238000007499 fusion processing Methods 0.000 description 1
- 238000001502 gel electrophoresis Methods 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 238000001415 gene therapy Methods 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 150000004676 glycans Chemical class 0.000 description 1
- 230000013595 glycosylation Effects 0.000 description 1
- 238000006206 glycosylation reaction Methods 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 239000005090 green fluorescent protein Substances 0.000 description 1
- 239000007902 hard capsule Substances 0.000 description 1
- 238000003306 harvesting Methods 0.000 description 1
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 1
- 238000009396 hybridization Methods 0.000 description 1
- 229910052739 hydrogen Inorganic materials 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- 230000033444 hydroxylation Effects 0.000 description 1
- 238000005805 hydroxylation reaction Methods 0.000 description 1
- 210000002829 igm memory b cell Anatomy 0.000 description 1
- 230000028993 immune response Effects 0.000 description 1
- 210000000987 immune system Anatomy 0.000 description 1
- 238000003125 immunofluorescent labeling Methods 0.000 description 1
- 230000016784 immunoglobulin production Effects 0.000 description 1
- 238000011532 immunohistochemical staining Methods 0.000 description 1
- 238000003364 immunohistochemistry Methods 0.000 description 1
- 239000007943 implant Substances 0.000 description 1
- 230000000415 inactivating effect Effects 0.000 description 1
- 230000002779 inactivation Effects 0.000 description 1
- 239000003701 inert diluent Substances 0.000 description 1
- 230000002458 infectious effect Effects 0.000 description 1
- 239000003978 infusion fluid Substances 0.000 description 1
- 239000002054 inoculum Substances 0.000 description 1
- 150000002484 inorganic compounds Chemical group 0.000 description 1
- 229910010272 inorganic material Inorganic materials 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 239000010410 layer Substances 0.000 description 1
- 231100000636 lethal dose Toxicity 0.000 description 1
- 229960005015 local anesthetics Drugs 0.000 description 1
- 239000007937 lozenge Substances 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 210000004698 lymphocyte Anatomy 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- 230000010534 mechanism of action Effects 0.000 description 1
- 201000001441 melanoma Diseases 0.000 description 1
- 210000004779 membrane envelope Anatomy 0.000 description 1
- 210000001806 memory b lymphocyte Anatomy 0.000 description 1
- 229910021645 metal ion Inorganic materials 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- 229930182817 methionine Natural products 0.000 description 1
- 238000000520 microinjection Methods 0.000 description 1
- 230000003278 mimic effect Effects 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 239000000178 monomer Substances 0.000 description 1
- 238000002703 mutagenesis Methods 0.000 description 1
- 231100000350 mutagenesis Toxicity 0.000 description 1
- 229960004927 neomycin Drugs 0.000 description 1
- 229920001220 nitrocellulos Polymers 0.000 description 1
- 210000004940 nucleus Anatomy 0.000 description 1
- 229920001778 nylon Polymers 0.000 description 1
- 229920002113 octoxynol Polymers 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 239000006186 oral dosage form Substances 0.000 description 1
- 150000002894 organic compounds Chemical group 0.000 description 1
- 230000008520 organization Effects 0.000 description 1
- PGZUMBJQJWIWGJ-ONAKXNSWSA-N oseltamivir phosphate Chemical compound OP(O)(O)=O.CCOC(=O)C1=C[C@@H](OC(CC)CC)[C@H](NC(C)=O)[C@@H](N)C1 PGZUMBJQJWIWGJ-ONAKXNSWSA-N 0.000 description 1
- 229910052762 osmium Inorganic materials 0.000 description 1
- SYQBFIAQOQZEGI-UHFFFAOYSA-N osmium atom Chemical compound [Os] SYQBFIAQOQZEGI-UHFFFAOYSA-N 0.000 description 1
- 210000001672 ovary Anatomy 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 239000003973 paint Substances 0.000 description 1
- 238000004091 panning Methods 0.000 description 1
- 230000005298 paramagnetic effect Effects 0.000 description 1
- 244000052769 pathogen Species 0.000 description 1
- 230000001717 pathogenic effect Effects 0.000 description 1
- 230000007170 pathology Effects 0.000 description 1
- 230000006320 pegylation Effects 0.000 description 1
- 210000005259 peripheral blood Anatomy 0.000 description 1
- 239000011886 peripheral blood Substances 0.000 description 1
- 108040007629 peroxidase activity proteins Proteins 0.000 description 1
- CWCMIVBLVUHDHK-ZSNHEYEWSA-N phleomycin D1 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC[C@@H](N=1)C=1SC=C(N=1)C(=O)NCCCCNC(N)=N)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C CWCMIVBLVUHDHK-ZSNHEYEWSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- 150000008298 phosphoramidates Chemical class 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 230000036470 plasma concentration Effects 0.000 description 1
- 210000004180 plasmocyte Anatomy 0.000 description 1
- 229920001200 poly(ethylene-vinyl acetate) Polymers 0.000 description 1
- 229920000747 poly(lactic acid) Polymers 0.000 description 1
- 230000008488 polyadenylation Effects 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 239000004633 polyglycolic acid Substances 0.000 description 1
- 229920002704 polyhistidine Polymers 0.000 description 1
- 239000004626 polylactic acid Substances 0.000 description 1
- 229920000656 polylysine Polymers 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 229920002223 polystyrene Polymers 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- 230000004481 post-translational protein modification Effects 0.000 description 1
- 230000001323 posttranslational effect Effects 0.000 description 1
- 239000002244 precipitate Substances 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 229960000380 propiolactone Drugs 0.000 description 1
- 230000002797 proteolythic effect Effects 0.000 description 1
- 230000006337 proteolytic cleavage Effects 0.000 description 1
- 210000001938 protoplast Anatomy 0.000 description 1
- 229950010131 puromycin Drugs 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- 239000012857 radioactive material Substances 0.000 description 1
- 239000000941 radioactive substance Substances 0.000 description 1
- 238000002708 random mutagenesis Methods 0.000 description 1
- 239000002994 raw material Substances 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 238000010188 recombinant method Methods 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000002829 reductive effect Effects 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 230000003362 replicative effect Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 230000000241 respiratory effect Effects 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 210000001525 retina Anatomy 0.000 description 1
- 239000002336 ribonucleotide Substances 0.000 description 1
- 125000002652 ribonucleotide group Chemical group 0.000 description 1
- 210000003705 ribosome Anatomy 0.000 description 1
- 238000004626 scanning electron microscopy Methods 0.000 description 1
- 238000007790 scraping Methods 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 238000013207 serial dilution Methods 0.000 description 1
- 125000005629 sialic acid group Chemical group 0.000 description 1
- 239000002356 single layer Substances 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 150000003384 small molecules Chemical group 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 235000017557 sodium bicarbonate Nutrition 0.000 description 1
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 1
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 1
- 230000037439 somatic mutation Effects 0.000 description 1
- 238000000527 sonication Methods 0.000 description 1
- 238000011895 specific detection Methods 0.000 description 1
- 210000003802 sputum Anatomy 0.000 description 1
- 208000024794 sputum Diseases 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 238000010186 staining Methods 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 238000000352 supercritical drying Methods 0.000 description 1
- 238000002198 surface plasmon resonance spectroscopy Methods 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 229940061367 tamiflu Drugs 0.000 description 1
- 238000012956 testing procedure Methods 0.000 description 1
- 230000004797 therapeutic response Effects 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- RTKIYNMVFMVABJ-UHFFFAOYSA-L thimerosal Chemical compound [Na+].CC[Hg]SC1=CC=CC=C1C([O-])=O RTKIYNMVFMVABJ-UHFFFAOYSA-L 0.000 description 1
- 231100000167 toxic agent Toxicity 0.000 description 1
- 239000003440 toxic substance Substances 0.000 description 1
- 230000002110 toxicologic effect Effects 0.000 description 1
- 231100000759 toxicological effect Toxicity 0.000 description 1
- 239000003053 toxin Substances 0.000 description 1
- 231100000765 toxin Toxicity 0.000 description 1
- 108700012359 toxins Proteins 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 230000005026 transcription initiation Effects 0.000 description 1
- 238000011830 transgenic mouse model Methods 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
- 238000011269 treatment regimen Methods 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- 239000013638 trimer Substances 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 239000012137 tryptone Substances 0.000 description 1
- 229910021642 ultra pure water Inorganic materials 0.000 description 1
- 239000012498 ultrapure water Substances 0.000 description 1
- 241000701161 unidentified adenovirus Species 0.000 description 1
- 241000701447 unidentified baculovirus Species 0.000 description 1
- 241001515965 unidentified phage Species 0.000 description 1
- 210000002700 urine Anatomy 0.000 description 1
- 238000002255 vaccination Methods 0.000 description 1
- 238000001291 vacuum drying Methods 0.000 description 1
- 238000009777 vacuum freeze-drying Methods 0.000 description 1
- 238000010200 validation analysis Methods 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 235000013311 vegetables Nutrition 0.000 description 1
- 230000007501 viral attachment Effects 0.000 description 1
- 239000011534 wash buffer Substances 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 210000005253 yeast cell Anatomy 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/08—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from viruses
- C07K16/10—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from viruses from RNA viruses
- C07K16/1018—Orthomyxoviridae, e.g. influenza virus
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/28—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from animals or humans against receptors, cell surface antigens or cell surface determinants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39533—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals
- A61K39/39558—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals against tumor tissues, cells, antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/42—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum viral
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/68—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment
- A61K47/6835—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site
- A61K47/6839—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting material from viruses
- A61K47/6841—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an antibody, an immunoglobulin or a fragment thereof, e.g. an Fc-fragment the modifying agent being an antibody or an immunoglobulin bearing at least one antigen-binding site the antibody targeting material from viruses the antibody targeting a RNA virus
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/16—Antivirals for RNA viruses for influenza or rhinoviruses
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
- C07K16/08—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from viruses
- C07K16/10—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies against material from viruses from RNA viruses
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/569—Immunoassay; Biospecific binding assay; Materials therefor for microorganisms, e.g. protozoa, bacteria, viruses
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/53—Immunoassay; Biospecific binding assay; Materials therefor
- G01N33/569—Immunoassay; Biospecific binding assay; Materials therefor for microorganisms, e.g. protozoa, bacteria, viruses
- G01N33/56983—Viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/21—Immunoglobulins specific features characterized by taxonomic origin from primates, e.g. man
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/30—Immunoglobulins specific features characterized by aspects of specificity or valency
- C07K2317/33—Crossreactivity, e.g. for species or epitope, or lack of said crossreactivity
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/51—Complete heavy chain or Fd fragment, i.e. VH + CH1
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/515—Complete light chain, i.e. VL + CL
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/56—Immunoglobulins specific features characterized by immunoglobulin fragments variable (Fv) region, i.e. VH and/or VL
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/60—Immunoglobulins specific features characterized by non-natural combinations of immunoglobulin fragments
- C07K2317/62—Immunoglobulins specific features characterized by non-natural combinations of immunoglobulin fragments comprising only variable region components
- C07K2317/622—Single chain antibody (scFv)
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/70—Immunoglobulins specific features characterized by effect upon binding to a cell or to an antigen
- C07K2317/76—Antagonist effect on antigen, e.g. neutralization or inhibition of binding
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N2333/00—Assays involving biological materials from specific organisms or of a specific nature
- G01N2333/005—Assays involving biological materials from specific organisms or of a specific nature from viruses
- G01N2333/08—RNA viruses
- G01N2333/11—Orthomyxoviridae, e.g. influenza virus
Definitions
- Human binding molecules capable of binding to and neutralizing influenza B viruses and uses thereof
- the invention relates to medicine.
- the invention in particular relates to human binding molecules, e.g. monoclonal antibodies or antigen-binding fragments thereof, capable of binding to and neutralizing influenza B viruses, in particular neutralizing binding molecules binding to and neutralizing influenza B viruses from both the
- the invention relates to the diagnosis, prophylaxis and/or treatment of infections caused by an influenza B virus, in particular of infections caused by influenza B viruses from the B/Y amagata and B/Victoria lineages.
- Influenza infection also referred to as “influenza” or “the flu”
- the flu is one of the most common diseases known to man causing between three and five million cases of severe illness and between 250,000 and 500,000 deaths every year around the world. Influenza rapidly spreads in seasonal epidemics affecting 5-15% of the population and the burden on health care costs and lost productivity are extensive (World Healthcare Organization (WHO)).
- WHO World Healthcare Organization
- the type A and type B viruses are the agents responsible for the influenza epidemics and pandemics observed in humans.
- antiviral drugs such as oseltamivir (Tamiflu®) can be effective for prevention and treatment of influenza infection.
- the number of influenza virus strains showing resistance against antiviral drugs such as oseltamivir is, however, increasing.
- An alternative approach is the development of antibody-based prophylactic or therapeutic means to neutralize various seasonal influenza viruses.
- cross-neutralizing antibodies recognizing epitopes in the conserved stem- region of HA of influenza A viruses of phylogenetic group 1 have recently been disclosed (e.g. CR6261, see WO2008/028946), as well as cross-neutralizing antibodies recognizing a highly conserved epitope in the stem-region of HA of influenza A viruses of phylogenetic group 2, such as influenza viruses comprising HA of the H3 and/or H7 subtypes (e.g. CR8020, CR8043; see WO 2010/130636).
- antibodies capable of binding to and neutralizing influenza A viruses of both phylogenetic group 1 and group 2 were discovered (e.g. CR9114, described in application no.
- influenza B viruses have been paid to. This may be due to the fact that - primarily being restricted to humans as host - influenza B viruses lack the large animal reservoirs that are key to the emergence of pandemic influenza A strains.
- the cumulative impact of annual epidemics during interpandemic periods exceeds that of pandemics and although the morbidity and mortality rates attributable to influenza B are lower than those of e.g. H3N2 viruses, they are higher than those of H1N1 viruses (Thompson (2003), Thompson (2004).
- influenza B viruses The evolution of influenza B viruses is characterized by co-circulation of antigenically and genetically distinct lineages for extended periods of time. Two lineages, represented by the prototype viruses B/Victoria/2/87 (Victoria lineage) and
- B/Y amagata/16/88 (Yamagata lineage), are currently distinguished (Kanegae (1990), Rota (1990)). B/Y amagata was the major lineage circulating until the 1980s, when B/Victoria lineage viruses appeared. Since then, drift variants of both influenza B lineages have been co-circulating globally, with both lineages concurrently circulating in recent influenza seasons.
- influenza B viruses are the major cause of seasonal influenza epidemics every 2 - 4 years, and in view of the severity of the respiratory illness caused by certain influenza B viruses, as well has the high economic impact of the seasonal epidemics, there is an ongoing need for alternative and effective means for the prevention and treatment influenza B subtypes. There is thus a need for binding molecules, preferably broadly neutralizing human binding molecules, capable of cross-neutralizing influenza B viruses.
- the invention provides binding molecules, in particular human binding molecules, capable of specifically binding to and neutralizing influenza B virus strains from both the B/Y amagata and B/Victoria lineages.
- the binding molecules do not bind to influenza A virus subtypes.
- the invention also pertains to immunoconjugates and/or pharmaceutical compositions comprising the binding molecules, as well as to nucleic acid molecules encoding at least the binding region of the human binding molecules.
- binding molecules, immunoconjugates and/or nucleic acid molecules of the invention are suitable for use as a universal prophylactic, diagnostic and/or treatment agent for influenza B viruses, irrespective of the causative influenza B virus subtype.
- FIG. 1 is a schematic epitope map based on competition experiments. Anti-influenza B antibodies identified in the invention cluster into 4 groups based on binding/competition to influenza B HA.
- FIG. 2 shows the results of an immunofluorescence entry assay designed to analyze the ability of the binding molecules to block receptor binding and internalization of the influenza virus.
- FIG. 3 shows inhibition of viral egress by the binding molecule of the invention.
- FIG. 4 shows the results of scanning EM of influenza B infected cells.
- FIG. 5 shows in vivo protection by CR8033 against lethal influenza B infection
- mice (B/Florida/04/2006 and B/Malaysia/2506/2004) in mice.
- binding molecule refers to an intact immunoglobulin including monoclonal antibodies, such as chimeric, humanized or human monoclonal antibodies, or to an antigen-binding and/or variable domain comprising fragment of an immunoglobulin that competes with the intact immunoglobulin for specific binding to the binding partner of the immunoglobulin, e.g. HA. Regardless of structure, the antigen- binding fragment binds with the same antigen that is recognized by the intact
- An antigen-binding fragment can comprise a peptide or polypeptide comprising an amino acid sequence of at least 2, 5, 10, 15, 20, 25, 30, 35, 40, 50, 60, 70, 80, 90, 100, 125, 150, 175, 200, or 250 contiguous amino acid residues of the amino acid sequence of the binding molecule.
- binding molecule includes all immunoglobulin classes and subclasses known in the art. Depending on the amino acid sequence of the constant domain of their heavy chains, binding molecules can be divided into the five major classes of intact antibodies: IgA, IgD, IgE, IgG, and IgM, and several of these may be further divided into subclasses (isotypes), e.g., IgAl, IgA2, IgGl, IgG2, IgG3 and IgG4.
- Antigen-binding fragments include, inter alia, Fab, F(ab'), F(ab')2, Fv, dAb, Fd, complementarity determining region (CDR) fragments, single-chain antibodies (scFv), bivalent single-chain antibodies, single-chain phage antibodies, diabodies, triabodies, tetrabodies, (poly)peptides that contain at least a fragment of an immunoglobulin that is sufficient to confer specific antigen binding to the (polypeptide, etc.
- the above fragments may be produced synthetically or by enzymatic or chemical cleavage of intact immunoglobulins or they may be genetically engineered by recombinant DNA techniques.
- a binding molecule or antigen-binding fragment thereof may have one or more binding sites. If there is more than one binding site, the binding sites may be identical to one another or they may be different.
- the binding molecule can be a naked or unconjugated binding molecule but can also be part of an immunoconjugate.
- a naked or unconjugated binding molecule is intended to refer to a binding molecule that is not conjugated, operatively linked or otherwise physically or functionally associated with an effector moiety or tag, such as inter alia a toxic substance, a radioactive substance, a liposome, an enzyme. It will be understood that naked or unconjugated binding molecules do not exclude binding molecules that have been stabilized, multimerized, humanized or in any other way manipulated, other than by the attachment of an effector moiety or tag.
- naked and unconjugated binding molecules are included herewith, including where the modifications are made in the natural binding molecule- producing cell environment, by a recombinant binding molecule -producing cell, and are introduced by the hand of man after initial binding molecule preparation.
- naked or unconjugated binding molecule does not exclude the ability of the binding molecule to form functional associations with effector cells and/or molecules after administration to the body, as some of such interactions are necessary in order to exert a biological effect.
- the lack of associated effector group or tag is therefore applied in definition to the naked or unconjugated binding molecule in vitro, not in vivo.
- biological sample encompasses a variety of sample types, including blood and other liquid samples of biological origin, solid tissue samples such as a biopsy specimen or tissue cultures, or cells derived there from and the progeny thereof.
- the term also includes samples that have been manipulated in any way after their procurement, such as by treatment with reagents, solubilization, or enrichment for certain components, such as proteins or polynucleotides.
- the term encompasses various kinds of clinical samples obtained from any species, and also includes cells in culture, cell supernatants and cell lysates.
- complementarity determining regions as used herein means sequences within the variable regions of binding molecules, such as immunoglobulins, that usually contribute to a large extent to the antigen binding site which is
- the CDR regions can be specific for linear epitopes, discontinuous epitopes, or conformational epitopes of proteins or protein fragments, either as present on the protein in its native conformation or, in some cases, as present on the proteins as denatured, e.g., by solubilization in SDS.
- Epitopes may also consist of posttranslational modifications of proteins.
- deletion denotes a change in either amino acid or nucleotide sequence in which one or more amino acid or nucleotide residues,
- expression-regulating nucleic acid sequence refers to polynucleotide sequences necessary for and/or affecting the expression of an operably linked coding sequence in a particular host organism.
- the expression-regulating nucleic acid sequences such as inter alia appropriate transcription initiation, termination, promoter, enhancer sequences; repressor or activator sequences; efficient RNA processing signals such as splicing and polyadenylation signals; sequences that stabilize cytoplasmic mR A; sequences that enhance translation efficiency (e.g., ribosome binding sites); sequences that enhance protein stability; and when desired, sequences that enhance protein secretion, can be any nucleic acid sequence showing activity in the host organism of choice and can be derived from genes encoding proteins, which are either homologous or heterologous to the host organism.
- the identification and employment of expression-regulating sequences is routine to the person skilled in the art.
- the term "functional variant”, as used herein, refers to a nucleic acid molecule or binding molecule that comprises a nucleotide and/or amino acid sequence that is altered by one or more nucleotides and/or amino acids compared to the nucleotide and/or amino acid sequences of the reference nucleic acid molecule or binding molecule.
- a functional variant of a binding molecule according to the invention is capable of competing for binding to the binding partner, i.e. the influenza virus, with the reference binding molecule.
- the modifications in the amino acid and/or nucleotide sequence of the reference binding molecule do not significantly affect or alter the binding characteristics of the binding molecule encoded by the nucleotide sequence or containing the amino acid sequence, i.e.
- the binding molecule is still able to recognize and bind its target.
- the functional variant may have conservative sequence modifications including nucleotide and amino acid substitutions, additions and deletions. These modifications can be introduced by standard techniques known in the art, such as site-directed mutagenesis and random PCR-mediated mutagenesis, and may comprise natural as well as non-natural nucleotides and amino acids.
- Conservative amino acid substitutions include the ones in which the amino acid residue is replaced with an amino acid residue having similar structural or chemical properties. Families of amino acid residues having similar side chains have been defined in the art. These families include amino acids with basic side chains (e.g., lysine, arginine, histidine), acidic side chains (e.g., aspartic acid, glutamic acid), uncharged polar side chains (e.g., asparagine, glutamine, serine, threonine, tyrosine, cysteine, tryptophan), non-polar side chains (e.g., glycine, alanine, valine, leucine, isoleucine, proline, phenylalanine, methionine), beta-branched side chains (e.g., threonine, valine, isoleucine) and aromatic side chains (e.g., tyrosine, phenylalanine, tryptophan).
- basic side chains e.g.
- a variant may have non-conservative amino acid substitutions, e.g., replacement of an amino acid with an amino acid residue having different structural or chemical properties. Similar minor variations may also include amino acid deletions or insertions, or both. Guidance in determining which amino acid residues may be substituted, inserted, or deleted without abolishing immunological activity may be found using computer programs well known in the art.
- a mutation in a nucleotide sequence can be a single alteration made at a locus (a point mutation), such as transition or transversion mutations, or alternatively, multiple nucleotides may be inserted, deleted or changed at a single locus. In addition, one or more alterations may be made at any number of loci within a nucleotide sequence.
- the mutations may be performed by any suitable method known in the art.
- the term "influenza virus subtype" in relation to influenza A viruses refers to influenza A virus variants that are characterized by various combinations of the hemagglutinin (H) and neuramidase (N) viral surface proteins.
- Influenza A virus subtypes may be referred to by their H number, such as for example “influenza virus comprising HA of the HI or H3 subtype", or “HI influenza virus” "H3 influenza virus”, or by a combination of an H number and an N number, such as for example “influenza virus subtype H3N2" or “H3N2".
- influenza virus "subtype” specifically includes all individual influenza virus “strains” within each subtype, which usually result from mutations and show different pathogenic profiles. Such strains may also be referred to as various "isolates” of a viral subtype. Accordingly, as used herein, the terms “strains” and “isolates” may be used interchangeably.
- neutralizing refers to a binding molecule that inhibits an influenza virus from replication, in vitro and/or within a subject, regardless of the mechanism by which neutralization is achieved.
- neutralization can e.g. be achieved by inhibiting the attachment or adhesion of the virus to the cell surface, or by inhibition of the fusion of viral and cellular membranes following attachment of the virus to the target cell, or by inhibiting viral egress from infected cells, and the like.
- cross-neutralizing or “cross-neutralization” as used herein in relation to the binding molecules of the invention refers to the ability of the binding molecules of the invention to neutralize influenza B viruses from both the B/Yamagata and the B/Victoria lineage, and/or different influenza B virus strains within these lineages.
- the term "host”, as used herein, is intended to refer to an organism or a cell into which a vector such as a cloning vector or an expression vector has been introduced.
- the organism or cell can be prokaryotic or eukaryotic.
- the hosts isolated host cells e.g. host cells in culture.
- the term "host cells” merely signifies that the cells are modified for the (over)-expression of the binding molecule of the invention and include B-cells that originally express these binding molecule and which cells have been modified to over-express the binding molecule by immortalization, amplification, enhancement of expression etc.
- the term host is intended to refer not only to the particular subject organism or cell but to the progeny of such an organism or cell as well. Because certain modifications may occur in succeeding generations due to either mutation or environmental influences, such progeny may not, in fact, be identical to the parent organism or cell, but are still included within the scope of the term "host” as used herein.
- human when applied to binding molecules as defined herein, refers to molecules that are either directly derived from a human or based upon a human germ line sequence. When a binding molecule is derived from or based on a human sequence and subsequently modified, it is still to be considered human as used throughout the specification. In other words, the term human, when applied to binding molecules is intended to include binding molecules having variable and constant regions derived from human germline immunoglobulin sequences or based on variable or constant regions occurring in a human or human lymphocyte and modified in some form.
- the human binding molecules may include amino acid residues not encoded by human germline immunoglobulin sequences, comprise substitutions and/or deletions (e.g., mutations introduced by for instance random or site-specific mutagenesis in vitro or by somatic mutation in vivo). "Based on” as used herein refers to the situation that a nucleic acid sequence may be exactly copied from a template, or with minor mutations, such as by error-prone PCR methods, or synthetically made matching the template exactly or with minor modifications.
- insertion also known as the term “addition” denotes a change in an amino acid or nucleotide sequence resulting in the addition of one or more amino acid or nucleotide residues, respectively, as compared to the parent sequence.
- binding molecules when applied to binding molecules as defined herein, refers to binding molecules that are substantially free of other proteins or polypeptides, particularly free of other binding molecules having different antigenic specificities, and are also substantially free of other cellular material and/or chemicals.
- the binding molecules when they are recombinantly produced, they are preferably substantially free of culture medium components, and when the binding molecules are produced by chemical synthesis, they are preferably substantially free of chemical precursors or other chemicals, i.e., they are separated from chemical precursors or other chemicals which are involved in the synthesis of the protein.
- isolated when applied to nucleic acid molecules encoding binding molecules as defined herein, is intended to refer to nucleic acid molecules in which the nucleotide sequences encoding the binding molecules are free of other nucleotide sequences, particularly nucleotide sequences encoding binding molecules that bind other binding partners.
- isolated refers to nucleic acid molecules that are substantially separated from other cellular components that naturally accompany the native nucleic acid molecule in its natural host, e.g., ribosomes, polymerases, or genomic sequences with which it is naturally associated.
- isolated nucleic acid molecules, such as cDNA molecules can be substantially free of other cellular material, or culture medium when produced by recombinant techniques, or substantially free of chemical precursors or other chemicals when chemically synthesized.
- monoclonal antibody refers to a preparation of antibody molecules of single specificity.
- a monoclonal antibody displays a single binding specificity and affinity for a particular epitope.
- human monoclonal antibody refers to an antibody displaying a single binding specificity which has variable and constant regions derived from or based on human germline
- the method of preparing the monoclonal antibody is not relevant for the binding specificity.
- naturally occurring refers to the fact that an object or compound can be found in nature.
- a polypeptide or polynucleotide sequence that is present in an organism that can be isolated from a source in nature and which has not been intentionally modified by man in the laboratory is naturally occurring.
- nucleic acid molecule refers to a polymeric form of nucleotides and includes both sense and anti-sense strands of RNA, cDNA, genomic DNA, and synthetic forms and mixed polymers of the above.
- a nucleotide refers to a ribonucleotide, deoxynucleotide or a modified form of either type of nucleotide. The term also includes single- and double-stranded forms of DNA.
- a polynucleotide may include either or both naturally occurring and modified nucleotides linked together by naturally occurring and/or non- naturally occurring nucleotide linkages.
- the nucleic acid molecules may be modified chemically or biochemically or may contain non-natural or derivatized nucleotide bases, as will be readily appreciated by those of skill in the art. Such modifications include, for example, labels, methylation, substitution of one or more of the naturally occurring nucleotides with an analogue, internucleotide modifications such as uncharged linkages (e.g., methyl phosphonates, phosphotriesters, phosphoramidates, carbamates, etc.), charged linkages (e.g., phosphorothioates, phosphorodithioates, etc.), pendent moieties (e.g., polypeptides), intercalate rs (e.g., acridine, psoralen, etc.), chelators, alkylators, and modified linkages (e.g., alpha anomeric nucleic acids, etc.).
- uncharged linkages e.g., methyl phosphonates, phosphotriesters,
- the above term is also intended to include any topological conformation, including single-stranded, double-stranded, partially duplexed, triplex, hairpinned, circular and padlocked conformations.
- synthetic molecules that mimic polynucleotides in their ability to bind to a designated sequence via hydrogen bonding and other chemical interactions. Such molecules are known in the art and include, for example, those in which peptide linkages substitute for phosphate linkages in the backbone of the molecule.
- a reference to a nucleic acid sequence encompasses its complement unless otherwise specified.
- a reference to a nucleic acid molecule having a particular sequence should be understood to encompass its complementary strand, with its complementary sequence.
- the complementary strand is also useful, e.g., for anti-sense therapy, hybridisation probes and PCR primers.
- operably linked refers to two or more nucleic acid sequence elements that are usually physically linked and are in a functional relationship with each other.
- a promoter is operably linked to a coding sequence, if the promoter is able to initiate or regulate the transcription or expression of a coding sequence, in which case the coding sequence should be understood as being "under the control of the promoter.
- pharmaceutically acceptable excipient any inert substance that is combined with an active molecule such as a drug, agent, or binding molecule for preparing an agreeable or convenient dosage form.
- the "pharmaceutically acceptable excipient” is an excipient that is non-toxic to recipients at the used dosages and concentrations, and is compatible with other ingredients of the formulation comprising the drug, agent or binding molecule.
- Pharmaceutically acceptable excipients are widely applied and known in the art.
- the term “specifically binding”, as used herein, in reference to the interaction of a binding molecule, e.g. an antibody, and its binding partner, e.g. an antigen, means that the interaction is dependent upon the presence of a particular structure, e.g.
- the antibody preferentially binds or recognizes the binding partner even when the binding partner is present in a mixture of other molecules or organisms.
- the binding may be mediated by covalent or non-covalent interactions or a combination of both.
- binding means immunospecifically binding to an antigenic determinant or epitope and not immunospecifically binding to other antigenic determinants or epitopes.
- a binding molecule that immunospecifically binds to an antigen may bind to other peptides or polypeptides with lower affinity as determined by, e.g., radioimmunoassays (RIA), enzyme-linked immunosorbent assays (ELISA), BIACORE, or other assays known in the art. Binding molecules or fragments thereof that immunospecifically bind to an antigen may be cross-reactive with related antigens, carrying the same epitope.
- binding molecules or fragments thereof that immunospecifically bind to an antigen do not cross-react with other antigens.
- substitution denotes the replacement of one or more amino acids or nucleotides by different amino acids or nucleotides, respectively.
- terapéuticaally effective amount refers to an amount of the binding molecule as defined herein that is effective for preventing, ameliorating and/or treating a condition resulting from infection with an influenza B virus. Ameloriation as used in herein may refer to the reduction of visible or perceptible disease symptoms, viremia, or any other measurable manifestation of influenza infection.
- treatment refers to therapeutic treatment as well as prophylactic or preventative measures to cure or halt or at least retard disease progress.
- Those in need of treatment include those already inflicted with a condition resulting from infection with influenza virus as well as those in which infection with influenza virus is to be prevented.
- Subjects partially or totally recovered from infection with influenza virus might also be in need of treatment.
- Prevention encompasses inhibiting or reducing the spread of influenza virus or inhibiting or reducing the onset, development or progression of one or more of the symptoms associated with infection with influenza virus.
- vector denotes a nucleic acid molecule into which a second nucleic acid molecule can be inserted for introduction into a host where it will be replicated, and in some cases expressed.
- a vector is capable of transporting a nucleic acid molecule to which it has been linked.
- Vectors include, but are not limited to, plasmids, cosmids, bacterial artificial chromosomes (BAC) and yeast artificial chromosomes (YAC) and vectors derived from bacteriophages or plant or animal (including human) viruses.
- Vectors comprise an origin of replication recognized by the proposed host and in case of expression vectors, promoter and other regulatory regions recognized by the host.
- a vector containing a second nucleic acid molecule is introduced into a cell by transformation, transfection, or by making use of viral entry mechanisms.
- vectors are capable of autonomous replication in a host into which they are introduced (e.g., vectors having a bacterial origin of replication can replicate in bacteria). Other vectors can be integrated into the genome of a host upon introduction into the host, and thereby are replicated along with the host genome.
- the present invention provides binding molecules capable of specifically binding to hemagglutinin (HA) of influenza B virus strains of the
- the binding molecules do not bind to HA of influenza A viruses.
- the binding molecules are capable of neutralizing influenza B viruses both in vitro and in vivo.
- the binding molecules are human binding molecules.
- the binding molecules are human antibodies, or antigen-binding fragments thereof.
- the binding molecules bind to a different epitope as compared to the epitope of CR9114 (as described in the co-pending application
- EP11 173953.8 comprising a heavy chain variable region comprising the amino acid sequence of SEQ ID NO: 116, and a light chain variable region comprising the amino acid sequence of SEQ ID NO: 117.
- CR91 14 has been shown to be capable of binding to and in vivo neutralizing influenza A viruses of both phylogenetic group 1 and 2, as well as influenza B viruses.
- the binding molecules bind to the head region of the HA protein of influenza B viruses, in particular to the head region of HAlof influenza B viruses.
- the binding molecules block the cellular receptor binding of influenza B viruses of the B/Yamagata lineage and/or the B/Victoria lineage.
- the binding molecules do not block the cellular receptor binding of influenza viruses of the B/Y amagata lineage and/or the B/Victoria lineage.
- the binding molecules block egress of influenza B viruses, in particular of influenza virus strains of both the B/Victoria and the B/Yamagata lineage, from infected cells.
- the isolated binding molecules are capable of specifically binding to the hemagglutinin protein (HA) of an influenza B virus and capable of neutralizing influenza B virus strains of both the B/Victoria/2/87 lineage and the B/Y amagata/16/88 lineage, wherein the binding molecules do not bind to the HA protein of influenza A virus subtypes, and comprise a heavy chain variable region comprising the amino acid sequence set forth in SEQ ID NO: 71 or a sequence of amino acids having at least or at least about 80 %, 90 %, 91 %, 92 %, 93 %, 94 %, 95 %, 96 %, 97 %, 98 %, 99 % or more sequence identity thereto.
- HA hemagglutinin protein
- the binding molecules comprise a light chain variable region comprising the amino acid sequence set forth in SEQ ID NO: 73, or a sequence of amino acids having at least or at least about 80 %, 90 %, 91 %, 92 %, 93 %, 94 %, 95 %, 96 %, 97 %, 98 %, 99 % or more sequence identity thereto.
- the binding molecule comprises a heavy chain variable region comprising the amino acid sequence of SEQ ID NO: 71 and a light chain variable region comprising the amino acid sequence of SEQ ID NO: 73.
- the invention also provides binding molecules capable of specifically binding to the hemagglutinin protein (HA) of an influenza B virus and capable of neutralizing influenza B virus strains of both the B/Victoria/2/87 lineage and the B/Y amagata/16/88 lineage, wherein the binding molecules do not bind to the HA protein of influenza A virus subtypes, and wherein the binding molecules comprise a heavy chain CDR1, comprising the sequence of amino acid residues set forth in SEQ ID NO: 1; a heavy chain CDR2, comprising the sequence of amino acid residues set forth in SEQ ID NO:2, and a heavy chain CDR3, comprising the sequence of amino acid residues set forth in SEQ ID NO: 3.
- CDR regions are according to Kabat et al. (1991) as described in Sequences of Proteins of Immunological Interest.
- the binding molecules comprise a light chain CDR1 , comprising the sequence of amino acid residues set forth in SEQ ID NO: 4, a light chain CDR2, comprising the sequence of amino acid residues set forth in SEQ ID NO: 5, and a light chain CDR3, comprising the sequence of amino acid residues set forth in SEQ ID NO: 6.
- the binding molecule comprises a heavy chain CDR1 comprising the amino acid sequence of SEQ ID NO: 1 , a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 2, and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 3, and a light chain CDR1 comprising the amino acid sequence of SEQ ID NO: 4, a light chain CDR2 comprising the amino acid sequence of SEQ ID NO: 5, and a light chain CDR3 comprising the amino acid sequence of SEQ ID NO: 6.
- the invention further provides binding molecules that immunospecifically bind to the same epitope on an influenza B virus HA protein as a binding molecule, comprising a heavy chain variable sequence comprising the amino acid sequence of SEQ ID NO: 71 and a light chain variable region comprising the amino acid sequence of SEQ ID NO: 73.
- the binding molecule comprises a heavy chain CDR1 comprising the amino acid sequence of SEQ ID NO: 14, a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 15, and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 16, and a light chain CDR1 comprising the amino acid sequence of SEQ ID NO: 17, a light chain CDR2 comprising the amino acid sequence of SEQ ID NO: 18, and a light chain CDR3 comprising the amino acid sequence of SEQ ID NO: 19.
- the binding molecule comprises a heavy chain CDR1 comprising the amino acid sequence of SEQ ID NO: 26, a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 27, and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 28, and a light chain CDR1 comprising the amino acid sequence of SEQ ID NO: 29, a light chain CDR2 comprising the amino acid sequence of SEQ ID NO: 24, and a light chain CDR3 comprising the amino acid sequence of SEQ ID NO:30.
- the invention also provides binding molecules, capable of specifically binding to the hemagglutinin protein (HA) and capable of neutralizing influenza B virus strains of the B/Victoria lineage, in particular the influenza B virus strain B/Malaysia/2506/2004, when the amino acid on position 168 of HA of the influenza B virus, in particular the influenza B virus strain B/Malaysia/2506/2004, is proline (P), and is not capable of neutralizing influenza B virus strains of the B/Victoria lineage, in particular
- HA hemagglutinin protein
- P proline
- B/Malaysia/2506/2004 when the amino acid on position 168 of the HA of the influenza B virus, in particular B/Malaysia/2506/2004, is glutamine (Q).
- the invention provides binding molecules, capable of specifically binding to the hemagglutinin protein (HA) and capable of neutralizing influenza B virus strains of the B/Yamagata lineage, in particular the influenza B virus strain B/Florida/04/2006, when the amino acid on position 38 of HA of the influenza B virus, in particular the influenza B virus strain B/Florida/04/200, is lysine (K), and is also capable of neutralizing influenza B virus strains of the B/Yamagata lineage, in particular B/Florida/04/2006, when the amino acid on position 38 of HA of the influenza B virus, in particular B/Florida/04/2006, is glutamic acid (E).
- HA hemagglutinin protein
- K lysine
- E glutamic acid
- the invention further provides binding molecules that are capable of specifically binding to the hemagglutinin protein (HA) of an influenza B virus and capable of neutralizing influenza B virus strains of both the B/Victoria/2/87 lineage and the B/Y amagata/16/88 lineage, and do not bind to the HA protein of influenza A virus subtypes, and comprise a heavy chain variable region comprising the amino acid sequence set forth in SEQ ID NO: 75, or a sequence of amino acids having at least or at least about 80 %, 90 %, 91 %, 92 %, 93 %, 94 %, 95 %, 96 %, 97 %, 98 %, 99 % or more sequence identity thereto.
- HA hemagglutinin protein
- the binding molecules comprise a light chain variable region comprising the amino acid sequence set forth in SEQ ID NO: 77, or a sequence of amino acids having at least or at least about 80 %, 90 %, 91 %, 92 %, 93 %, 94 %, 95 %, 96 %, 97 %, 98 %, 99 % or more sequence identity thereto.
- the binding molecules comprise heavy chain variable region comprising the amino acid sequence of SEQ ID NO: 75 and a light chain variable region comprising the amino acid sequence of SEQ ID NO: 77.
- the binding molecule comprises a heavy chain variable region consisting of the amino acid sequence of SEQ ID NO: 78 and a light chain variable region consisting of the amino acid sequence of SEQ ID NO: 79.
- the invention provides binding molecules capable of specifically binding to the hemagglutinin protein (HA) of an influenza B virus and capable of neutralizing influenza B virus strains of both the B/Victoria/2/87 lineage and the B/Y amagata/ 16/88 lineage, wherein the binding molecules do not bind to the HA protein of influenza A virus subtypes, and wherein the binding molecules comprise a heavy chain CDR1 , comprising the sequence of amino acid residues set forth in SEQ ID NO: 7; a heavy chain CDR2, comprising the sequence of amino acid residues set forth in SEQ ID NO: 8, and a heavy chain CDR3, comprising the sequence of amino acid residues set forth in SEQ ID NO: 9.
- HA hemagglutinin protein
- the binding molecules comprise a light chain CDR1 , comprising the sequence of amino acid residues set forth in SEQ ID NO: 10, a light chain CDR2, comprising the sequence of amino acid residues set forth in SEQ ID NO: 11 , and a light chain CDR3, comprising the sequence of amino acid residues set forth in SEQ ID NO: 12 or 13.
- the binding molecule comprises a heavy chain CDR1 comprising the amino acid sequence of SEQ ID NO: 7, a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 8, and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 9, and a light chain CDR1 comprising the amino acid sequence of SEQ ID NO: 10, a light chain CDR2 comprising the amino acid sequence of SEQ ID NO: 11 , and a light chain CDR3 comprising the amino acid sequence of SEQ ID NO: 12.
- the binding molecule comprises a heavy chain CDR1 comprising the amino acid sequence of SEQ ID NO: 7, a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 8, and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 9, and a light chain CDRl comprising the amino acid sequence of SEQ ID NO: 10, a light chain CDR2 comprising the amino acid sequence of SEQ ID NO: 11 , and a light chain CDR3 comprising the amino acid sequence of SEQ ID NO: 13.
- the invention further provides binding molecules that immunospecifically bind to the same epitope on an influenza B virus HA protein as a binding molecule, comprising a heavy chain variable sequence comprising the amino acid sequence of SEQ ID NO: 75 and a light chain variable region comprising the amino acid sequence of SEQ ID NO: 77.
- the binding molecule comprises a heavy chain CDRl comprising the amino acid sequence of SEQ ID NO: 20, a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 21 , and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 22, and a light chain CDRl comprising the amino acid sequence of SEQ ID NO: 23, a light chain CDR2 comprising the amino acid sequence of SEQ ID NO: 24, and a light chain CDR3 comprising the amino acid sequence of SEQ ID NO: 25.
- the binding molecule comprises a heavy chain CDRl comprising the amino acid sequence of SEQ ID NO: 31 , a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 32, and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 33, and a light chain CDRl comprising the amino acid sequence of SEQ ID NO: 4, a light chain CDR2 comprising the amino acid sequence of SEQ ID NO: 5, and a light chain CDR3 comprising the amino acid sequence of SEQ ID NO:34.
- the invention provides binding molecules, capable of specifically binding to the hemagglutinin protein (HA) and capable of neutralizing influenza B virus strains of the B/Victoria lineage, in particular the influenza B virus strain B/Malaysia/2506/2004, when the amino acid on position 168 of HA of the influenza B virus, in particular the influenza B virus strain B/Malaysia/2506/2004, is proline (P), and also capable of neutralizing influenza B virus strains of the B/Victoria lineage, in particular B/Malaysia/2506/2004, when the amino acid on position 168 of the HA of the influenza B virus, in particular B/Malaysia/2506/2004, is glutamine (Q).
- HA hemagglutinin protein
- Q glutamine
- the invention provides binding molecules, capable of specifically binding to the hemagglutinin protein (HA) and capable of neutralizing influenza B virus strains of the B/Yamagata lineage, in particular the influenza B virus strain B/Florida/04/2006, when the amino acid on position 38 of HA of the influenza B virus, in particular the influenza B virus strain B/Florida/04/200, is lysine (K), and not capable of neutralizing influenza B virus strains of the B/Yamagata lineage, in particular B/Florida/04/2006, when the amino acid on position 38 of HA of the influenza B virus, in particular B/Florida/04/2006, is glutamic acid (E).
- HA hemagglutinin protein
- K lysine
- E glutamic acid
- the present invention further provides binding molecules, capable of specifically binding to the hemagglutinin protein (HA) of an influenza B virus and capable of neutralizing influenza B virus strains of both the B/Victoria/2/87 lineage and the
- binding molecules do not bind to the HA protein of influenza A virus subtypes, and wherein the binding molecules comprise a heavy chain variable region comprising the amino acid sequence set forth in SEQ ID NO: 113 or a sequence of amino acids having at least or at least about 80 %, 90 %, 91 %, 92 %, 93 %, 94 %, 95 %, 96 %, 97 %, 98 %, 99 % or more sequence identity thereto.
- the binding molecules comprise a light chain variable region comprising the amino acid sequence set forth in SEQ ID NO: 115, or a sequence of amino acids having at least or at least about 80 %, 90 %, 91 %, 92 %, 93 %, 94 %, 95 %, 96 %, 97 %, 98 %, 99 % or more sequence identity thereto.
- the binding molecule comprises a heavy chain variable region comprising the amino acid sequence of SEQ ID NO: 113 and a light chain variable region comprising the amino acid sequence of SEQ ID NO: 115.
- the invention provides binding molecules capable of specifically binding to the hemagglutinin protein (HA) of an influenza B virus and capable of neutralizing influenza B virus strains of both the B/Victoria/2/87 lineage and the B/Y amagata/16/88 lineage, wherein the binding molecules do not bind to the HA protein of influenza A virus subtypes, and wherein the binding molecules comprise a heavy chain CDR1 , comprising the sequence of amino acid residues set forth in SEQ ID NO:54; a heavy chain CDR2, comprising the sequence of amino acid residues set forth in SEQ ID NO:55 and a heavy chain CDR3, comprising the sequence of amino acid residues set forth in SEQ ID NO:56.
- HA hemagglutinin protein
- the binding molecules comprise a light chain CDR1 , comprising the sequence of amino acid residues set forth in SEQ ID NO: 57, a light chain CDR2, comprising the sequence of amino acid residues set forth in SEQ ID NO: 5, and a light chain CDR3, comprising the sequence of amino acid residues set forth in SEQ ID NO: 58.
- the binding molecule comprises a heavy chain CDR1 comprising the amino acid sequence of SEQ ID NO: 54, a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 55, and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 56, and a light chain CDR1 comprising the amino acid sequence of SEQ ID NO: 57 a light chain CDR2 comprising the amino acid sequence of SEQ ID NO: 5, and a light chain CDR3 comprising the amino acid sequence of SEQ ID NO:58.
- the invention further provides binding molecules that immunospecifically bind to the same epitope on an influenza B virus HA protein as a binding molecule, comprising a heavy chain variable sequence comprising the amino acid sequence of SEQ ID NO: 113 and a light chain variable region comprising the amino acid sequence of SEQ ID NO: 115.
- Influenza B viruses like influenza A viruses, infect cells by binding to sialic acid residues on the cell surface of target cells and following transfer into endosomes, by fusing their membranes with the endosomal membranes and releasing the genome - transcriptase complex into the cell. Both receptor binding and membrane fusion process are mediated by the HA glycoprotein.
- the HA of both influenza A and B viruses comprises two structurally distinct regions, i.e. a globular head region, which contains a receptor binding site which is responsible for virus attachment to the target cell, and which is involved in the haemagglutination activity of HA, and a stem region, containing a fusion peptide which is necessary for membrane fusion between the viral envelope and the endosomal membrane of the cell.
- the HA protein is a trimer in which each monomer consists of two disulphide - linked glycopolypeptides, HA1 and HA2, that are produced during infection by proteolytic cleavage of a precursor (HAO). Cleavage is necessary for virus infectivity since it is required to prime the HA for membrane fusion, to allow conformational change. Activation of the primed molecule occurs at low pH in endosomes, between pH5 and pH6, and requires extensive changes in HA structure.
- the binding molecules are capable of specifically binding to the HAl subunit of the HA protein, in particular to the head region of the HAl subunit.
- the binding molecules may be capable of specifically binding to linear or structural and/or conformational epitopes on the HAl subunit of the HA protein.
- the HA molecule may be purified from viruses or recombinantly produced and optionally isolated before use. Alternatively, HA may be expressed on the surface of cells.
- the binding molecules of the invention may be capable of specifically binding to influenza B viruses that are viable, living and/or infective or that are in
- inactivated/attenuated form e.g. influenza viruses
- Methods for inactivating/attenuating virus include, but are not limited to, treatment with formalin, ⁇ -propiolactone (BPL), merthiolate, and/or ultraviolet light.
- BPL ⁇ -propiolactone
- the binding molecules of the invention may also be capable of specifically binding to one or more fragments of the influenza B viruses, such as inter alia a preparation of one or more proteins and/or (poly)peptides, derived from subtypes of influenza B viruses or one or more recombinantly produced proteins and/or polypeptides of influenza B viruses.
- the nucleotide and/or amino acid sequence of proteins of various influenza B strains can be found in the GenBank-database, NCBI Influenza Virus Sequence Database, Influenza Sequence Database (ISD), EMBL-database and/or other databases. It is well within the reach of the skilled person to find such sequences in the respective databases.
- binding molecules of the invention are capable of specifically binding to a fragment of the above-mentioned proteins and/or polypeptides, wherein the fragment at least comprises an epitope recognized by the binding molecules of the invention.
- An "epitope” as used herein is a moiety that is capable of binding to a binding molecule of the invention with sufficiently high affinity to form a detectable antigen-binding molecule complex.
- the binding molecules of the invention can be intact immunoglobulin molecules such as monoclonal antibodies, or the binding molecules can be antigen-binding fragments thereof, including, but not limited to, heavy and light chain variable regions, Fab, F(ab'), F(ab') 2 , Fv, dAb, Fd, complementarity determining region (CDR) fragments, single-chain antibodies (scFv), bivalent single-chain antibodies, single-chain phage antibodies, diabodies, triabodies, tetrabodies, and (poly)peptides that contain at least a fragment of an immunoglobulin that is sufficient to confer specific antigen binding to influenza virus strains or a fragment thereof.
- the binding molecules of the invention are human monoclonal antibodies, and/or antigen-binding fragments thereof.
- the binding molecules may also be nanobodies, alphabodies, affibodies, FN3-domain scaffolds and other scaffolds based on domains in (human) repeat proteins, like Adnectins, Anticalins, Darpins, etc, or other scaffolds comprising epitope binding sequences.
- the binding molecules are intact antibodies comprising complete heavy and light chain variable regions as well as complete heavy and light chain constant regions.
- the binding molecules have complement-dependent cytotoxic activity (CDC) and/or antibody-dependent cell-mediated cytotoxic (ADCC) activity.
- the binding molecules of the invention can be used in non-isolated or isolated form. Furthermore, the binding molecules of the invention can be used alone or in a mixture comprising at least one binding molecule (or variant or fragment thereof) of the invention, and one or more other binding molecules that bind to influenza and have influenza virus inhibiting effect. In other words, the binding molecules can be used in combination, e.g. , as a pharmaceutical composition comprising two or more binding molecules, variants or fragments thereof. For example, binding molecules having different, but complementary activities can be combined in a single therapy to achieve a desired prophylactic, therapeutic or diagnostic effect, but alternatively, binding molecules having identical activities can also be combined in a single therapy to achieve a desired prophylactic, therapeutic or diagnostic effect.
- the mixture may also comprise at least one binding molecule according to the invention and at least one other therapeutic agent.
- the therapeutic agent such as, e.g., M2 inhibitors (e.g., amantidine, rimantadine) and/or neuraminidase inhibitors (e.g., zanamivir, oseltamivir) is useful in the prophylaxis and/or treatment of an influenza virus infection
- binding molecule according to the invention can bind to its binding partners, i.e. an influenza B virus of the B/Yamagata and/or B/Victoria lineage, and/or fragments thereof, with an affinity constant (3 ⁇ 4- value) that is lower than 0.2x10 -4 M, 1.0x10 -5 M, 1.0x10 -6 M, 1.0x10 -7 M, preferably lower than 1.0x10 -8 M, more preferably lower than 1.OxlO "9 M, more preferably lower than 1.OxlO "10 M, even more preferably lower than 1.0x10 - " 11 M, and i ⁇ n particular lower than 1.0x10 - " 12 M.
- the affinity constants can vary for antibody isotypes.
- affinity binding for an IgM isotype refers to a binding affinity of at least about 1.0x10 " M.
- Affinity constants can for instance be measured using surface plasmon resonance, for example using the BIACORE system (Pharmacia Biosensor AB, Uppsala, Sweden).
- the binding molecules of the invention exhibit neutralizing activity.
- Neutralizing activity can for instance be measured as described herein.
- Alternative assays measuring neutralizing activity are described in for instance WHO Manual on Animal Influenza Diagnosis and Surveillance, Geneva: World Health Organization, 2005, version 2002.5.
- the binding molecules according to the invention have a neutralizing activity of 50 ⁇ g/ml or less, preferably 20 ⁇ g/ml or less, more preferably a neutralizing activity of 10 ⁇ g/ml or less, even more preferably 5 ⁇ g/ml or less, as determined in an in vitro virus neutralization assay (VNA) as described in Example 6.
- VNA in vitro virus neutralization assay
- the binding molecules according to the invention may bind to influenza virus or a fragment thereof in soluble form such as for instance in a sample or in suspension or may bind to influenza viruses or fragments thereof bound or attached to a carrier or substrate, e.g., micro titer plates, membranes and beads, etc.
- Carriers or substrates may be made of glass, plastic (e.g., polystyrene), polysaccharides, nylon, nitrocellulose, or Teflon, etc.
- the surface of such supports may be solid or porous and of any convenient shape.
- the binding molecules may bind to influenza virus in purified/isolated or non-purified/non-isolated form.
- the present invention in certain embodiments provides isolated human binding molecules that are able to recognize and bind to an epitope in the influenza haemagglutinin protein (HA) of influenza B viruses, wherein said binding molecules have neutralizing activity against influenza B viruses of both the B/Y amagata and/or B/Victoria lineages, both in vitro and in vivo.
- binding molecules of the present invention cross-neutralize influenza virus subtypes belonging to both phylogenetic lineages.
- the skilled person can determine whether an antibody indeed cross-reacts with HA proteins from different subtypes and can also determine whether they are able to neutralize influenza viruses of different subtypes in vitro and/or in vivo.
- Another aspect of the invention includes functional variants of the binding molecule as defined above.
- Molecules are considered to be functional variants of a binding molecule according to the invention, if the variant binding molecules are capable of competing for immunospecifically binding to an influenza virus or a fragment thereof with the "parental” or “reference” binding molecules.
- molecules are considered to be functional variants of a binding molecule according to the invention when the functional variants are still capable of binding to the same or overlapping epitope of the influenza virus or a fragment thereof.
- parental and “reference” will be used as synonyms meaning that the information of the reference or parental molecule, or the physical molecule itself may form the basis for the variation.
- Functional variants include, but are not limited to, derivatives that are substantially similar in primary structural sequence, including those that have
- modifications in the Fc receptor or other regions involved with effector functions and/or which contain e.g. in vitro or in vivo modifications, chemical and/or biochemical, that are not found in the parental binding molecule.
- modifications include inter alia acetylation, acylation, covalent attachment of a nucleotide or nucleotide derivative, covalent attachment of a lipid or lipid derivative, cross-linking, disulfide bond formation, glycosylation, hydroxylation, methylation, oxidation, pegylation, proteolytic processing, phosphorylation, and the like.
- functional variants can be binding molecules as defined in the present invention comprising an amino acid sequence containing substitutions, insertions, deletions or combinations thereof of one or more amino acids compared to the amino acid sequences of the parental binding molecules.
- functional variants can comprise truncations of the amino acid sequence at either or both the amino or carboxyl termini.
- Functional variants according to the invention may have the same or different, either higher or lower, binding affinities compared to the parental binding molecule but are still capable of binding to the influenza virus or a fragment thereof.
- functional variants according to the invention may have increased or decreased binding affinities for an influenza virus or a fragment thereof compared to the parental binding molecules.
- the amino acid sequences of the variable regions including, but not limited to, framework regions, hypervariable regions, in particular the CDR3 regions, are modified.
- the light chain and the heavy chain variable regions comprise three hypervariable regions, comprising three CDRs, and more conserved regions, the so-called framework regions (FRs).
- the hypervariable regions comprise amino acid residues from CDRs and amino acid residues from hypervariable loops.
- Functional variants intended to fall within the scope of the present invention have at least about 80% to about 99%, preferably at least about 70% to about 99%, more preferably at least about 80% to about 99%, even more preferably at least about 90%) to about 99%, most preferably at least about 95% to about 99%, in particular at least about 97% to about 99% amino acid sequence identity and/or homology with the parental binding molecules as defined herein.
- Computer algorithms such as inter alia Gap or Bestfit known to a person skilled in the art can be used to optimally align amino acid sequences to be compared and to define similar or identical amino acid residues.
- Functional variants can be obtained by altering the parental binding molecules or parts thereof by general molecular biology methods known in the art including, but not limited to, error-prone PCR, oligonucleotide-directed mutagenesis, site-directed mutagenesis and heavy and/or light chain shuffling.
- the functional variants of the invention have neutralizing activity against influenza B viruses.
- the neutralizing activity may either be identical, or be higher or lower compared to the parental binding molecules.
- the term (human) binding molecule when used in this application, this also encompasses functional variants of the (human) binding molecule.
- Assays for verifying if a variant binding molecule has neutralizing activity are well known in the art (see WHO Manual on Animal Influenza Diagnosis and Surveillance, Geneva: World Health Organisation, 2005 version 2002.5).
- the functional variants are binding molecules comprising a heavy chain variable sequence comprising one or more amino acid mutations, such as one, two, three, four, five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen or fifteen amino acid mutations, as compared to SEQ ID NO: 71 and/or a light chain variable region comprising one or more amino acid mutations, such as one, two, three, four, five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen or fifteen amino acid mutations as compared to SEQ ID NO: 73.
- the functional variants are binding molecules comprising a heavy chain variable sequence comprising one or more amino acid mutations, such as one, two, three, four, five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen or fifteen amino acid mutations, as compared to SEQ ID NO: 75 and/or a light chain variable region comprising one or more amino acid mutations, such as one, two, three, four, five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen or fifteen amino acid mutations as compared to SEQ ID NO: 77.
- the functional variants are binding molecules comprising a heavy chain variable sequence comprising one or more amino acid mutations, such as one, two, three, four, five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen or fifteen amino acid mutations, as compared to SEQ ID NO: 113 and/or a light chain variable region comprising one or more amino acid mutations, such as one, two, three, four, five, six, seven, eight, nine, ten, eleven, twelve, thirteen, fourteen or fifteen amino acid mutations as compared to SEQ ID NO: 115.
- a binding molecule according to the invention is selected from the group consisting of binding molecules comprising:
- a heavy chain CDR1 comprising the amino acid sequence of SEQ ID NO: 59, a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 2, and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 3, and a light chain CDR1 comprising the amino acid sequence of SEQ ID NO: 17, a light chain CDR2 comprising the amino acid sequence of SEQ ID NO: 18, and a light chain CDR3 comprising the amino acid sequence of SEQ ID NO: 60;
- a heavy chain CDR1 comprising the amino acid sequence of SEQ ID NO: 61 , a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 2, and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 3, and a light chain CDRl comprising the amino acid sequence of SEQ ID NO: 62, a light chain CDR2 comprising the amino acid sequence of SEQ ID NO: 18, and a light chain CDR3 comprising the amino acid sequence of SEQ ID NO: 63;
- a heavy chain CDRl comprising the amino acid sequence of SEQ ID NO: 59, a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 2, and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 3, and a light chain CDRl comprising the amino acid sequence of SEQ ID NO: 64, a light chain CDR2 comprising the amino acid sequence of SEQ ID NO: 65, and a light chain CDR3 comprising the amino acid sequence of SEQ ID NO: 66;
- a heavy chain CDRl comprising the amino acid sequence of SEQ ID NO: 59, a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 2, and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 3, and a light chain CDRl comprising the amino acid sequence of SEQ ID NO: 67, a light chain CDR2 comprising the amino acid sequence of SEQ ID NO: 68, and a light chain CDR3 comprising the amino acid sequence of SEQ ID NO: 69;
- a heavy chain CDRl comprising the amino acid sequence of SEQ ID NO: 35, a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 36 and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 37, and a light chain CDRl comprising the amino acid sequence of SEQ ID NO: 4, a light chain CDR2 comprising the amino acid sequence of SEQ ID NO: 38, and a light chain CDR3 comprising the amino acid sequence of SEQ ID NO: 39;
- a heavy chain CDRl comprising the amino acid sequence of SEQ ID NO: 40, a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 41, and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 42, and a light chain CDRl comprising the amino acid sequence of SEQ ID NO: 43, a light chain CDR2 comprising the amino acid sequence of SEQ ID NO: 5, and a light chain CDR3 comprising the amino acid sequence of SEQ ID NO: 44;
- CDR3 comprising the amino acid sequence of SEQ ID NO: 47, and a light chain CDRl comprising the amino acid sequence of SEQ ID NO: 48, a light chain CDR2 comprising the amino acid sequence of SEQ ID NO: 38, and a light chain CDR3 comprising the amino acid sequence of SEQ ID NO: 49;
- a heavy chain CDRl comprising the amino acid sequence of SEQ ID NO: 45, a heavy chain CDR2 comprising the amino acid sequence of SEQ ID NO: 50, and a heavy chain CDR3 comprising the amino acid sequence of SEQ ID NO: 47, and a light chain CDRl comprising the amino acid sequence of SEQ ID NO: 51 , a light chain CDR2 comprising the amino acid sequence of SEQ ID NO: 52, and a light chain CDR3 comprising the amino acid sequence of SEQ ID NO: 53.
- the binding molecule is selected from the group consisting of:
- a binding molecule comprising a heavy chain variable region of SEQ ID NO: 119 and a light chain variable region of SEQ ID NO: 121;
- a binding molecule comprising a heavy chain variable region of SEQ ID NO: 123 and a light chain variable region of SEQ ID NO: 125;
- a binding molecule comprising a heavy chain variable region of SEQ ID NO: 127 and a light chain variable region of SEQ ID NO: 129;
- a binding molecule comprising a heavy chain variable region of SEQ ID NO: 131 and a light chain variable region of SEQ ID NO: 133;
- a binding molecule comprising a heavy chain variable region of SEQ ID NO: 77 and a light chain variable region of SEQ ID NO: 79;
- a binding molecule comprising a heavy chain variable region of SEQ ID NO: 101 and a light chain variable region of SEQ ID NO: 103;
- a binding molecule comprising a heavy chain variable region of SEQ ID NO: 105 and a light chain variable region of SEQ ID NO: 107;
- a binding molecule comprising a heavy chain variable region of SEQ ID NO: 109 and a light chain variable region of SEQ ID NO: 11 1.
- the binding molecules according to the invention are for a use as a medicament, and preferably for use in the therapeutic and/or prophylactic treatment of influenza infection caused by influenza B viruses.
- the influenza virus that causes the influenza infection and that can be treated using the binding molecules of the present invention may be an influenza B virus of the B/Y amagata and/or B/Victoria lineage.
- the present invention also relates to pharmaceutical compositions comprising at least one binding molecule according to the invention, and at least a pharmaceutically acceptable excipient.
- the invention relates to the use of a binding molecule according to the invention in the preparation of a medicament for the prophylaxis, and/or treatment of an influenza virus infection.
- influenza virus infections that can be prevented and/or treated using the binding molecules of the invention may occur in small populations, but can also spread around the world in seasonal epidemics or, worse, in global pandemics where millions of individuals are at risk.
- the invention provides binding molecules that can neutralize the infection of influenza strains that cause such seasonal epidemics, as well as potential pandemics.
- the invention provides immunoconjugates, i.e. molecules comprising at least one binding molecule as defined herein and further comprising at least one tag, such as inter alia a detectable moiety/agent. Also contemplated in the present invention are mixtures of immunoconjugates according to the invention or mixtures of at least one immunoconjugates according to the invention and another molecule, such as a therapeutic agent or another binding molecule or immunoconjugate. In further embodiments, the immunoconjugates of the invention may comprise more than one tag. These tags can be the same or distinct from each other and can be joined/conjugated non- covalently to the binding molecules.
- the tag(s) can also be joined/conjugated directly to the human binding molecules through covalent bonding. Alternatively, the tag(s) can be joined/conjugated to the binding molecules by means of one or more linking compounds. Techniques for conjugating tags to binding molecules are well known to the skilled artisan.
- the tags of the immunoconjugates of the present invention may be therapeutic agents, but they can also be detectable moieties/agents.
- Tags suitable in therapy and/or prevention may be toxins or functional parts thereof, antibiotics, enzymes, other binding molecules that enhance phagocytosis or immune stimulation.
- Immunoconjugates comprising a detectable agent can be used diagnostically to, for example, assess if a subject has been infected with an influenza virus or to monitor the development or progression of an influenza virus infection as part of a clinical testing procedure to, e.g., determine the efficacy of a given treatment regimen. However, they may also be used for other detection and/or analytical and/or diagnostic purposes.
- Detectable moieties/agents include, but are not limited to, enzymes, prosthetic groups, fluorescent materials, luminescent materials, bioluminescent materials, radioactive materials, positron emitting metals, and non-radioactive paramagnetic metal ions.
- the tags used to label the binding molecules for detection and/or analytical and/or diagnostic purposes depend on the specific detection/analysis/diagnosis techniques and/or methods used such as inter alia immunohistochemical staining of (tissue) samples, flow cytometric detection, scanning laser cytometric detection, fluorescent immunoassays, enzyme-linked immunosorbent assays (ELISAs), radioimmunoassays (RIAs), bioassays ⁇ e.g., phagocytosis assays), Western blotting applications, etc.
- Suitable labels for the detection/analysis/diagnosis techniques and/or methods known in the art are well within the reach of the skilled artisan.
- the human binding molecules or immunoconjugates of the invention can also be attached to solid supports, which are particularly useful for in vitro immunoassays or purification of influenza viruses or fragments thereof.
- solid supports might be porous or nonporous, planar or non-planar.
- the binding molecules of the present invention can be fused to marker sequences, such as a peptide to facilitate purification. Examples include, but are not limited to, the hexa-histidine tag, the hemagglutinin (HA) tag, the myc tag or the flag tag.
- an antibody can be conjugated to a second antibody to form an antibody heteroconjugate.
- the binding molecules of the invention may be conjugated/attached to one or more antigens.
- these antigens are antigens which are recognized by the immune system of a subject to which the binding molecule-antigen conjugate is administered.
- the antigens may be identical, but may also differ from each other.
- Conjugation methods for attaching the antigens and binding molecules are well known in the art and include, but are not limited to, the use of cross-linking agents.
- the binding molecules of the invention will bind to influenza virus HA and the antigens attached to the binding molecules will initiate a powerful T-cell attack on the conjugate, which will eventually lead to the destruction of the influenza virus.
- the immunoconjugates can be produced as fusion proteins comprising the binding molecules of the invention and a suitable tag. Fusion proteins can be produced by methods known in the art such as, e.g., recombinantly by constructing nucleic acid molecules comprising nucleotide sequences encoding the binding molecules in frame with nucleotide sequences encoding the suitable tag(s) and then expressing the nucleic acid molecules.
- nucleic acid molecules encoding at least a binding molecule, functional variant or immunoconjugate according to the invention.
- Such nucleic acid molecules can be used as intermediates for cloning purposes, e.g. in the process of affinity maturation as described above.
- the nucleic acid molecules are isolated or purified.
- nucleic acid molecules are also intended to be a part of the present invention.
- Functional variants are nucleic acid sequences that can be directly translated, using the standard genetic code, to provide an amino acid sequence identical to that translated from the parental nucleic acid molecules.
- the nucleic acid molecules encode binding molecules comprising the CDR regions as described above.
- the nucleic acid molecules encode binding molecules comprising two, three, four, five or even all six CDR regions of the binding molecules of the invention.
- nucleic acid molecules encode binding molecules comprising a heavy chain comprising the variable heavy chain sequences as described above. In another embodiment the nucleic acid molecules encode binding molecules comprising a light chain comprising the variable light chain sequences as described above.
- the nucleotide sequences and the amino acid sequences of the heavy and light chain variable regions of the binding molecules of the invention are given below.
- vectors i.e. nucleic acid constructs, comprising one or more nucleic acid molecules according to the present invention.
- Vectors can be derived from plasmids such as inter alia F, Rl, RP1, Col, pBR322, TOL, Ti, etc; cosmids; phages such as lambda, lambdoid, Ml 3, Mu, PI, P22, ⁇ ) ⁇ , T-even, T- odd, T2, T4, T7, etc; plant viruses.
- Vectors can be used for cloning and/or for expression of the binding molecules of the invention and might even be used for gene therapy purposes.
- Vectors comprising one or more nucleic acid molecules according to the invention operably linked to one or more expression-regulating nucleic acid molecules are also covered by the present invention.
- the choice of the vector is dependent on the recombinant procedures followed and the host used. Introduction of vectors in host cells can be effected by inter alia calcium phosphate transfection, virus infection, DEAE- dextran mediated transfection, lipofectamin transfection or electroporation. Vectors may be autonomously replicating or may replicate together with the chromosome into which they have been integrated.
- the vectors contain one or more selection markers. The choice of the markers may depend on the host cells of choice, although this is not critical to the invention as is well known to persons skilled in the art.
- vectors comprising one or more nucleic acid molecules encoding the human binding molecules as described above operably linked to one or more nucleic acid molecules encoding proteins or peptides that can be used to isolate the human binding molecules are also covered by the invention.
- proteins or peptides include, but are not limited to, glutathione-S-transferase, maltose binding protein, metal-binding polyhistidine, green fluorescent protein, luciferase and beta-galactosidase.
- Hosts containing one or more copies of the vectors mentioned above are an additional aspect of the present invention.
- the hosts are host cells.
- Host cells include, but are not limited to, cells of mammalian, plant, insect, fungal or bacterial origin.
- Bacterial cells include, but are not limited to, cells from Gram-positive bacteria or Gram-negative bacteria such as several species of the genera Escherichia, such as E. coli, and Pseudomonas.
- yeast cells are used in the group of fungal cells. Expression in yeast can be achieved by using yeast strains such as inter alia Pichia pastoris, Saccharomyces cerevisiae and Hansenula polymorpha.
- insect cells such as cells from Drosophila and Sf9 can be used as host cells.
- the host cells can be plant cells such as inter alia cells from crop plants such as forestry plants, or cells from plants providing food and raw materials such as cereal plants, or medicinal plants, or cells from ornamentals, or cells from flower bulb crops.
- Transformed (transgenic) plants or plant cells are produced by known methods, for example, Agrobacterium-mediated gene transfer, transformation of leaf discs, protoplast transformation by polyethylene glycol- induced DNA transfer, electroporation, sonication, microinjection or holistic gene transfer.
- a suitable expression system can be a baculovirus system.
- Mammalian cells such as Chinese Hamster Ovary (CHO) cells, COS cells, BHK cells, NSO cells or Bowes melanoma cells are preferred in the present invention.
- Mammalian cells provide expressed proteins with posttranslational
- the host cells are human cells.
- human cells are inter alia HeLa, 911 , AT1080, A549, 293 and HEK293T cells.
- the human producer cells comprise at least a functional part of a nucleic acid sequence encoding an adenovirus El region in expressible format.
- said host cells are derived from a human retina and immortalized with nucleic acids comprising adenoviral El sequences, such as 911 cells or the cell line deposited at the European Collection of Cell Cultures (ECACC), CAMR, Salisbury, Wiltshire SP4 OJG, Great Britain on 29 February 1996 under number 96022940 and marketed under the trademark PER.C6 ® (PER.C6 is a registered trademark of Crucell Holland B.V.).
- PER.C6 cells refers to cells deposited under number 96022940 or ancestors, passages up-stream or downstream as well as descendants from ancestors of deposited cells, as well as derivatives of any of the foregoing.
- Production of recombinant proteins in host cells can be performed according to methods well known in the art.
- the use of the cells marketed under the trademark PER.C6 ® as a production platform for proteins of interest has been described in WO 00/63403 the disclosure of which is incorporated herein by reference in its entirety.
- a method of producing a binding molecule according to the invention is an additional aspect of the invention.
- the method comprises the steps of a) culturing a host according to the invention under conditions conducive to the expression of the binding molecule, and b) optionally, recovering the expressed binding molecule.
- the expressed binding molecules can be recovered from the cell free extract, but preferably they are recovered from the culture medium.
- the above method of producing can also be used to make functional variants of the binding molecules and/or immunoconjugates of the present invention. Methods to recover proteins, such as binding molecules, from cell free extracts or culture medium are well known to the man skilled in the art. Binding molecules, functional variants and/or immunoconjugates obtainable by the above- described method are also a part of the present invention.
- the binding molecules and immunoconjugates of the invention can be produced synthetically by conventional peptide synthesizers or in cell-free translation systems using R A nucleic acid derived from DNA molecules according to the invention. Binding molecules and immunoconjugates as obtainable by the above described synthetic production methods or cell-free translation systems are also a part of the present invention.
- binding molecules of the present invention can also be produced in transgenic, non-human, mammals such as inter alia rabbits, goats or cows, and secreted into for instance the milk thereof.
- binding molecules according to the present invention may be generated by transgenic non-human mammals, such as for instance transgenic mice or rabbits that express human immunoglobulin genes.
- the transgenic non-human mammals have a genome comprising a human heavy chain transgene and a human light chain transgene encoding all or a portion of the human binding molecules as described above.
- the transgenic non-human mammals can be immunized with a purified or enriched preparation of influenza virus or a fragment thereof. Protocols for immunizing non-human mammals are well established in the art. See Using Antibodies: A Laboratory Manual, Edited by: E. Harlow, D.
- Immunization protocols often include multiple immunizations, either with or without adjuvants such as Freund's complete adjuvant and Freund's incomplete adjuvant, but may also include naked DNA immunizations.
- the human binding molecules are produced by B-cells, plasma and/or memory cells derived from the transgenic animals.
- the human binding molecules are produced by hybridomas, which are prepared by fusion of B-cells obtained from the above-described transgenic non-human mammals to immortalized cells.
- B-cells, plasma cells and hybridomas as obtainable from the above- described transgenic non-human mammals and human binding molecules as obtainable from the above-described transgenic non-human mammals, B-cells, plasma and/or memory cells and hybridomas are also a part of the present invention.
- the invention provides compositions comprising at least a binding molecule, preferably a human monoclonal antibody, according to the invention, at least a functional variant thereof, at least an immunoconjugate according to the invention and/or a combination thereof.
- the compositions may comprise inter alia stabilizing molecules, such as albumin or polyethylene glycol, or salts.
- the salts used are salts that retain the desired biological activity of the binding molecules and do not impart any undesired toxicological effects.
- the human binding molecules of the invention may be coated in or on a material to protect them from the action of acids or other natural or non-natural conditions that may inactivate the binding molecules.
- compositions comprising at least a nucleic acid molecule as defined in the present invention.
- the compositions may comprise aqueous solutions such as aqueous solutions containing salts ⁇ e.g., NaCl or salts as described above), detergents ⁇ e.g., SDS) and/or other suitable components.
- the present invention pertains to pharmaceutical compositions comprising at least a binding molecule, such as a human monoclonal antibody, of the invention (or functional fragment or variant thereof), at least an immunoconjugate according to the invention, at least a composition according to the invention, or combinations thereof.
- the pharmaceutical composition of the invention further comprises at least one pharmaceutically acceptable excipient.
- Pharmaceutically acceptable excipients are well known to the skilled person.
- the pharmaceutical composition according to the invention may further comprise at least one other therapeutic agent. Suitable agents are also well known to the skilled artisan.
- the pharmaceutical composition according to the invention comprises at least one additional binding molecule, i.e. the pharmaceutical composition can be a cocktail or mixture of binding molecules.
- the pharmaceutical composition may comprise at least two binding molecules according to the invention, or at least one binding molecule according to the invention and at least one further influenza virus binding and/or neutralizing molecule, such as another antibody directed against the HA protein or against other antigenic structures present on influenza viruses, such as M2, and/or a binding molecules neutralizing one or more other pathogens.
- the additional binding molecule may be formulated for simultaneous separate or sequential administration.
- the binding molecules exhibit synergistic neutralizing activity, when used in combination.
- synergistic means that the combined effects of the binding molecules when used in combination are greater than their additive effects when used individually.
- the synergistically acting binding molecules may bind to different structures on the same or distinct fragments of influenza virus. A way of calculating synergy is by means of the combination index. The concept of the combination index (CI) has been described by Chou and Talalay (1984).
- the compositions may e.g. comprise one binding molecule having neutralizing activity and one non-neutralizing binding molecule.
- the non-neutralizing and neutralizing binding molecules may also act synergistically in neutralizing influenza virus.
- the pharmaceutical composition may comprise at least one binding molecule according to the invention and at least one further binding molecule, preferably a further influenza virus neutralizing binding molecule.
- the binding molecules in the pharmaceutical composition preferably are capable of reacting with influenza viruses of different subtypes.
- the binding molecules may be of high affinity and have a broad specificity.
- both binding molecules are cross-neutralizing molecules in that they each neutralize influenza viruses of different subtypes.
- the pharmaceutical composition comprises at least one other prophylactic and/or therapeutic agent.
- said further therapeutic and/or prophylactic agents are agents capable of preventing and/or treating an influenza virus infection and/or a condition resulting from such an infection.
- Therapeutic and/or prophylactic agents include, but are not limited to, anti-viral agents.
- agents can be binding molecules, small molecules, organic or inorganic compounds, enzymes, polynucleotide sequences, anti-viral peptides, etc.
- Other agents that are currently used to treat patients infected with influenza viruses are M2 inhibitors (e.g. , amantidine, rimantadine) and/or neuraminidase inhibitors (e.g., zanamivir, oseltamivir). These can be used in combination with the binding molecules of the invention.
- “In combination” herein means simultaneously, as separate formulations, or as one single combined formulation, or according to a sequential administration regimen as separate formulations, in any order.
- Agents capable of preventing and/or treating an infection with influenza virus and/or a condition resulting from such an infection that are in the experimental phase might also be used as other therapeutic and/or prophylactic agents useful in the present invention.
- binding molecules or pharmaceutical compositions of the invention can be tested in suitable animal model systems prior to use in humans.
- animal model systems include, but are not limited to, mouse, ferret and monkey.
- compositions typically must be sterile and stable under the conditions of manufacture and storage.
- the binding molecules, immunoconjugates, or compositions of the present invention can be in powder form for reconstitution in the appropriate pharmaceutically acceptable excipient before or at the time of delivery.
- the preferred methods of preparation are vacuum drying and freeze-drying (lyophilization) that yield a powder of the active ingredient plus any additional desired ingredient from a previously sterile-filtered solution thereof.
- the binding molecules, immunoconjugates, or compositions of the present invention can be in solution and the appropriate pharmaceutically acceptable excipient can be added and/or mixed before or at the time of delivery to provide a unit dosage injectable form.
- the pharmaceutically acceptable excipient used in the present invention is suitable to high drug concentration, can maintain proper fluidity and, if necessary, can delay absorption.
- the choice of the optimal route of administration of the pharmaceutical compositions will be influenced by several factors including the physicochemical properties of the active molecules within the compositions, the urgency of the clinical situation and the relationship of the plasma concentrations of the active molecules to the desired therapeutic effect.
- the binding molecules of the invention can be prepared with carriers that will protect them against rapid release, such as a controlled release formulation, including implants, transdermal patches, and microencapsulated delivery systems.
- Biodegradable, biocompatible polymers can inter alia be used, such as ethylene vinyl acetate, polyanhydrides, polyglycolic acid, collagen, polyorthoesters, and polylactic acid.
- binding molecules may be necessary to coat the binding molecules with, or co-administer the binding molecules with, a material or compound that prevents the inactivation of the human binding molecules.
- the binding molecules may be administered to a subject in an appropriate carrier, for example, liposomes or a diluent.
- the routes of administration can be divided into two main categories, oral and parenteral administration.
- the preferred administration route is intravenous or by inhalation.
- Oral dosage forms can be formulated inter alia as tablets, troches, lozenges, aqueous or oily suspensions, dispersible powders or granules, emulsions, hard capsules, soft gelatin capsules, syrups or elixirs, pills, dragees, liquids, gels, or slurries.
- These formulations can contain pharmaceutically excipients including, but not limited to, inert diluents, granulating and disintegrating agents, binding agents, lubricating agents, preservatives, colouring, flavouring or sweetening agents, vegetable or mineral oils, wetting agents, and thickening agents.
- compositions of the present invention can also be formulated for parenteral administration.
- Formulations for parenteral administration can be inter alia in the form of aqueous or non-aqueous isotonic sterile non-toxic injection or infusion solutions or suspensions.
- the solutions or suspensions may comprise agents that are nontoxic to recipients at the dosages and concentrations employed such as 1,3-butanediol, Ringer's solution, Hank's solution, isotonic sodium chloride solution, oils, fatty acids, local anaesthetic agents, preservatives, buffers, viscosity or solubility increasing agents, water-soluble antioxidants, oil-soluble antioxidants and metal chelating agents.
- the binding molecules such as human monoclonal antibodies (functional fragments and variants thereof), immunoconjugates, compositions, or pharmaceutical compositions of the invention can be used as a medicament or diagnostic agent. So, methods of diagnosis, treatment and/or prevention of an influenza virus infection using the binding molecules, immunoconjugates, compositions, or
- compositions of the invention are another aspect of the present invention.
- the above-mentioned molecules can inter alia be used in the diagnosis, prophylaxis, treatment, or combination thereof, of an influenza virus infection caused by an influenza B virus. They are suitable for treatment of yet untreated patients suffering from an influenza virus infection and patients who have been or are treated for an influenza virus infection.
- binding molecules such as human monoclonal antibodies (or functional variants thereof), immunoconjugates, compositions or pharmaceutical compositions of the invention can be co-administered with a vaccine against influenza virus (if available).
- the vaccine may also be administered before or after administration of the molecules of the invention.
- anti-viral agents can also be employed in conjunction with the binding molecules of the present invention. Suitable anti-viral agents are mentioned above.
- the molecules are typically formulated in the compositions and pharmaceutical compositions of the invention in a therapeutically or diagnostically effective amount. Alternatively, they may be formulated and administered separately. For instance the other molecules such as the anti-viral agents may be applied systemically, while the binding molecules of the invention may be applied intravenously.
- Treatment may be targeted at patient groups that are susceptible to influenza infection.
- patient groups include, but are not limited to e.g., the elderly (e.g. > 50 years old, > 60 years old, and preferably > 65 years old), the young (e.g. ⁇ 5 years old, ⁇ 1 year old), hospitalized patients and already infected patients who have been treated with an antiviral compound but have shown an inadequate antiviral response.
- Dosage regimens can be adjusted to provide the optimum desired response (e.g. , a therapeutic response).
- a suitable dosage range may for instance be 0.01-100 mg/kg body weight, preferably 0.1-50 mg/kg body weight, preferably 0.01-15 mg/kg body weight.
- a single bolus may be administered, several divided doses may be administered over time or the dose may be proportionally reduced or increased as indicated by the exigencies of the therapeutic situation.
- the molecules and compositions according to the present invention are preferably sterile. Methods to render these molecules and compositions sterile are well known in the art.
- the other molecules useful in diagnosis, prophylaxis and/or treatment can be administered in a similar dosage regimen as proposed for the binding molecules of the invention.
- the other molecules may be administered to a patient prior to (e.g. , 2 min, 5 min, 10 min, 15 min, 30 min, 45 min, 60 min, 2 hrs, 4 hrs, 6 hrs, 8 hrs, 10 hrs, 12 hrs, 14 hrs, 16 hrs, 18 hrs, 20 hrs, 22 hrs, 24 hrs, 2 days, 3 days, 4 days, 5 days, 7 days, 2 weeks, 4 weeks or 6 weeks before), concomitantly with, or subsequent to (e.g., 2 min, 5 min, 10 min, 15 min, 30 min, 45 min, 60 min, 2 hrs, 4 hrs, 6 hrs, 8 hrs, 10 hrs, 12 hrs, 14 hrs, 16 hrs, 18 hrs, 20 hrs, 22 hrs, 24 hrs, 2 days, 3 days, 4 days, 5 days, 7 days, 2 weeks, 4 weeks or 6 weeks after) the administration of one or more of the human binding molecules or pharmaceutical compositions of the invention.
- the exact dosing regimen is
- Human binding molecules and pharmaceutical compositions comprising the human binding molecules are particularly useful, and often preferred, when to be administered to human beings as in vivo therapeutic agents, since recipient immune response to the administered antibody will often be substantially less than that occasioned by administration of a monoclonal murine, chimeric or humanized binding molecule.
- the invention concerns the use of the binding molecules such as neutralizing human monoclonal antibodies (functional fragments and variants thereof), immunoconjugates, nucleic acid molecules, compositions or pharmaceutical
- compositions according to the invention in the preparation of a medicament for the diagnosis, prophylaxis, treatment, or combination thereof, of an influenza virus infection, in particular an influenza virus infection caused by influenza B viruses.
- kits comprising at least a binding molecule such as a neutralizing human monoclonal antibody (functional fragments and variants thereof), at least an immunoconjugate, at least a nucleic acid molecule, at least a composition, at least a pharmaceutical composition, at least a vector, at least a host according to the invention or a combination thereof are also an aspect of the present invention.
- the above- described components of the kits of the invention are packed in suitable containers and labelled for diagnosis, prophylaxis and/or treatment of the indicated conditions.
- the above-mentioned components may be stored in unit or multi-dose containers as an aqueous, preferably sterile, solution or as a lyophilised, preferably sterile, formulation for reconstitution.
- the containers may be formed from a variety of materials such as glass or plastic and may have a sterile access port (for example, the container may be an intravenous solution bag or a vial having a stopper pierceable by a hypodermic injection needle).
- the kit may further comprise more containers comprising a pharmaceutically acceptable buffer. It may further include other materials desirable from a commercial and user standpoint, including other buffers, diluents, filters, needles, syringes, culture medium for one or more of the suitable hosts and, possibly, even at least one other therapeutic, prophylactic or diagnostic agent.
- kits can be instructions customarily included in commercial packages of therapeutic, prophylactic or diagnostic products, that contain information about for example the indications, usage, dosage, manufacture, administration, contra-indications and/or warnings concerning the use of such therapeutic, prophylactic or diagnostic products.
- binding molecules according to the present invention can also be any binding molecules.
- the invention thus further pertains to a method of detecting influenza B subtype influenza virus in a sample, wherein the method comprises the steps of
- the biological sample may be a biological sample including, but not limited to blood, serum, stool, sputum, nasopharyngal aspirates, bronchial lavages, urine, tissue or other biological material from (potentially) infected subjects, or a non-biological sample such as water, drink, etc.
- the (potentially) infected subjects may be human subjects, but also animals that are suspected as carriers of influenza virus might be tested for the presence of the virus using the human binding molecules or immunoconjugates of the invention.
- the sample may first be manipulated to make it more suitable for the method of detection.
- Manipulation means inter alia treating the sample suspected to contain and/or containing the virus in such a way that the virus will disintegrate into antigenic components such as proteins, (polypeptides or other antigenic fragments.
- the human binding molecules or immunoconjugates of the invention are contacted with the sample under conditions which allow the formation of an immunological complex between the human binding molecules and the virus or antigenic components thereof that may be present in the sample.
- the formation of an immunological complex, if any, indicating the presence of the virus in the sample is then detected and measured by suitable means.
- suitable means include, inter alia, homogeneous and heterogeneous binding immunoassays, such as radio-immunoassays (RIA), ELISA,
- the binding molecules or immunoconjugates of the invention are conveniently bonded to the inside surface of microtiter wells.
- the binding molecules or immunoconjugates of the invention may be directly bonded to the microtiter well.
- maximum binding of the binding molecules or immunoconjugates of the invention to the wells might be accomplished by pre-treating the wells with polylysine prior to the addition of the binding molecules or immunoconjugates of the invention.
- the binding molecules or immunoconjugates of the invention may be covalently attached by known means to the wells.
- the binding molecules or immunoconjugates are used in a concentration between 0.01 to 100 ⁇ g/ml for coating, although higher as well as lower amounts may also be used. Samples are then added to the wells coated with the binding molecules or immunoconjugates of the invention.
- the invention further provides methods of treating or preventing an influenza B virus infection in a subject, comprising administering to the subject a therapeutically or prophylactically effective amount of the binding molecules, immunoconjugates and/or pharmaceutical compositions of the invention.
- the subject is a mammal, preferably a human.
- binding molecules of the invention can be used to identify specific binding structures of influenza virus.
- the binding structures can be epitopes on proteins and/or polypeptides. They can be linear, but also structural and/or conformational.
- the binding structures can be analysed by means of PEPSCAN analysis (see inter alia WO 84/03564, WO 93/09872, Slootstra et ah, 1996).
- a random peptide library comprising peptides from a protein of influenza virus can be screened for peptides capable of binding to the binding molecules of the invention.
- First round amplification on the respective cDNA yielded seven, six and nine products of about 650 base pairs for VH, Vkappa and Vlambda regions, respectively.
- the OCM constant primer IgM constant heavy chain specific
- the thermal cycling program for first round amplifications was: 2 min 96°C (denaturation step), 35 cycles of 30 sec 96°C/ 30 sec 60°CV 50 sec 72°C, 10 min 72°C final elongation and 6°C refrigeration.
- the products were loaded on and isolated from a 1% agarose gel using gel- extraction columns (Macherey-Nagel, MN) and eluted in 50 ⁇ 5 mM Tris-HCl pH 8.0. Ten percent of first round products (5 ⁇ ) were subjected to second round amplification. These primers were extended with restriction sites enabling the directional cloning of the respective VL and VH regions into phage display vector PDV-C06.
- the PCR program for second round amplifications was as follows: 2 min 96°C (denaturation step), 30 cycles of 30 sec 96°C/ 30 sec 60°C/ 50 sec 72°C, 10 min 72°C final elongation and 6°C refrigeration.
- the second round products (-350 base pairs) were first loaded on gel and extracted from the agarose as above. Then the fragments were pooled according to natural occurrence of J segments found in immunoglobulin gene products, resulting in seven, six and nine pools for respectively the VH, Vkappa and Vlambda variable regions as shown in Table 1 and 2.
- VL pool 5 ⁇ g was digested with Sail and Notl restriction enzymes, loaded on and isolated from a 1.5% agarose gel (-350 base pairs) using MN-extraction columns and ligated in Sall-Notl cut PDV-C06 vector (-5000 base pairs) as follows: 500ng PDV-C06 vector, 70 ng VL insert, 5 ⁇ 10X ligation buffer (NEB), 2.5 T4 DNA Ligase (400 U/ ⁇ ) (NEB), and ultrapure water was added up to a total volume of 50 ⁇ 1 (vector to insert ratio was 1 :2).
- Ligation was performed overnight in a water bath of 16°C. Next, the volume was doubled with water, extracted with an equal volume of phenol-chloroform-isoamylalcohol (75:24: 1) (Invitrogen) followed by chloroform (Merck) extraction and precipitated with 1 ⁇ Pellet Paint (Novogen), 10 ⁇ sodium acetate (3 M pH 5.0) and 100 ⁇ isopropanol for 2 hrs at -20°C. The obtained sample was subsequently centrifuged at 20.000xg for 30 min at 4°C. The obtained precipitate was washed with 70% ethanol and centrifuged for 10 min at 20.000xg at room temperature.
- Ethanol was removed and the pellet was air dried for several min and then dissolved in 50 ⁇ buffer containing 10 mM Tris-HCl, pH 8.0. 2 ⁇ ligation mixture was used for the transformation of 40 ⁇ TG-1 electro-competent cells (Agilent) in a chilled 0.1 cm electroporation cuvette (Biorad) using a Genepulser II apparatus (Biorad) set at 1.7 kV, 200 Ohm, 25 ⁇ (time constant -4,5 msec). Directly after pulse, the bacteria were flushed from the cuvette with 750 ⁇ SOC medium
- the transformed bacteria were plated over large 240 mm square petridishes (NUNC) containing 200 ml 2TY agar (16 g/1 bacto-tryptone, 10 g/1 bacto-yeast extract, 5 g/1 NaCl, 15 g/1 agar, pH 7.0) supplemented with 50 ⁇ g/ml ampicillin and 5% (w/v) glucose (Sigma).
- NUNC small mm square petridishes
- 200 ml 2TY agar (16 g/1 bacto-tryptone, 10 g/1 bacto-yeast extract, 5 g/1 NaCl, 15 g/1 agar, pH 7.0
- a 1 to 1000 and a 1 to 10.000 dilution were plated for counting purposes on 15 cm petridishes containing the same medium. This transformation procedure was repeated sequentially twenty times and the complete library was plated over a total of ten large square petridishes and grown overnight in a 37°C culture stove.
- VL light chain library was harvested from the plates by mildly scraping the bacteria into 12 ml 2TY medium per plate. The cell mass was determined by OD600 measurement and two times 500 OD of bacteria was used for maxi plasmid DNA preparation using two maxiprep columns (MN) according to manufacturer's instructions. Analogous to the VL variable regions, the second round VH-JH products were first mixed together to obtain the normal J segment usage distribution (see Table 2), resulting in 7 VH subpools called PHI to PH7.
- the pools were mixed to acquire a normalized sequence distribution using the percentages depicted in Table 2, obtaining one VH fraction that was digested with Sfil and Xhol restriction enzymes and ligated in Sfil-Xhol cut PDV-VL intermediate library obtained as described above.
- the ligation setup, purification method, subsequent transformation of TGI and harvest of bacteria was exactly as described for the VL intermediate library (see above), with the exception of the number of 240 mm plates used.
- twenty plates were used, resulting in approximately 2x10 cfu.
- the final library was checked for insert frequency with a colony PCR using a primer set flanking the inserted VH-VL regions (100-150 single colonies).
- HA antigens were diluted in PBS (5.0 ⁇ ), added to
- the blocked phage library was added to the immunotubes, incubated for 2 hrs at room temperature, and washed with wash buffer (0.05% (v/v) Tween-20 in PBS) to remove unbound phages.
- Bound phages were eluted from the respective antigen by incubation with 1 ml of 100 mM triethylamine (TEA) for 10 min at room temperature. Subsequently, the eluted phages were mixed with 0.5 ml of 1 M Tris-HCl pH 7.5 to neutralize the pH. This mixture was used to infect 5 ml of an XL 1 -Blue E.coli culture that had been grown at 37°C to an OD 600 nm of approximately 0.3.
- the phages were allowed to infect the XLl-Blue bacteria for 30 min at 37°C. Then, the mixture was centrifuged for 10 min at 3000xg at room temperature and the bacterial pellet was resuspended in 0.5 ml 2-trypton yeast extract (2TY) medium. The obtained bacterial suspension was divided over two 2TY agar plates supplemented with tetracycline, ampicillin and glucose. After incubation overnight of the plates at 37°C, the colonies were scraped from the plates and used to prepare an enriched phage library, essentially as described by De Kruif et al. (1995a) and in WO 02/103012.
- scraped bacteria were used to inoculate 2TY medium containing ampicillin, tetracycline and glucose and grown at a temperature of 37°C to an OD 600 nm of -0.3.
- CT helper phages were added and allowed to infect the bacteria after which the medium was changed to 2TY containing ampicillin, tetracycline and kanamycin. Incubation was continued overnight at 30°C. The next day, the bacteria were removed from the 2TY medium by centrifugation after which the phages in the medium were precipitated using polyethylene glycol (PEG) 6000/NaCl.
- PEG polyethylene glycol
- the phages were dissolved in 2 ml of PBS with 1% bovine serum albumin (BSA), filter-sterilized and used for the next round of selection.
- BSA bovine serum albumin
- Selected supernatants containing single-chain phage antibodies that were obtained in the screenings described above were validated in ELISA for specificity, i.e. binding to different HA antigens.
- baculovirus-expressed recombinant influenza B HA (B/Ohio/01/2005, B/Malaysia/2506/2004, B/Jilin/20/2003, B/Brisbane/60/2008 and B/Florida/04/2006) (Protein Sciences, CT, USA) was coated (O ⁇ g/ml) to MaxisorpTM ELISA plates. After coating, the plates were washed three times with PBS containing 0.1% v/v Tween-20 and blocked in PBS containing 2% ELK for 1 hr at room
- the selected single-chain phage antibodies were incubated for 1 hr in an equal volume of PBS containing 4% ELK to obtain blocked phage antibodies.
- the plates were emptied, washed three times with PBS/0.1% Tween-20 and the blocked single-chain phage antibodies were added to the wells. Incubation was allowed to proceed for one hour; the plates were washed five times with PBS/0.1% Tween-20.
- Bound phage antibodies were detected (using OD 492nm measurement) using an anti-M13 antibody conjugated to peroxidase. As a control, the procedure was performed simultaneously without single-chain phage antibody and with an unrelated negative control single-chain phage antibody.
- scFv single-chain phage antibodies
- the heavy chain variable region (VH) of the scFvs was cloned by restriction digestion (Sfil/Xhol) for expression in the IgG expression vector pIg-C911-HCgammal, which was digested with the same enzymes.
- the light variable region (VL) was also cloned into its IgG designated expression vector pIG-C909-Ckappa , or pIg-C910- Clambda using Sall/Notl for the insert fragment and Xhol/Notl for the target vector, as described previously in WO 2008/028946.
- a single amino acid mutant antibody (CR8071) was generated by assembly PCR. Two overlapping PCR fragments that each contained the desired mutation were generated. These fragments were mixed in equimolar ratios and served as template in a second round PCR to obtain the full length LC sequence. Nucleotide sequences for all constructs were verified using standard sequencing techniques. The resulting expression constructs encoding the human IgGl heavy and light chains were transiently expressed together in HEK293T cells. After one week, the supernatants containing human IgGl antibodies were obtained and processed using standard purification procedures.
- the human IgGl antibodies were titrated in a concentration range of between 10 to 0.003 ⁇ g/ml against influenza B HA antigen (data not shown). An unrelated antibody was included as a control antibody.
- the amino acid sequence of the CDRs of both, heavy and light chain, of the selected immunoglobulin molecules is given in Table 5. The nucleotide sequence and amino acid sequence of the heavy and light chain variable regions are given below.
- the selected anti-influenza B antibodies were used to test breadth of binding by FACS analysis.
- full-length recombinant influenza B expression vectors coding for HA (B/Mississippi/04/2008, B/Houston/B60/1997, B/Nashville/45/1991 , B/Florida/01/2009, B/Mississippi/07/2008 and B/Ohio/01/2005) were transfected into PER.C6 ® cells using lipofectamin (Invitrogen) in a 1 to 5 ratio. 48 hour after transfection, the PER.C6 ⁇ cells expressing the Influenza B HA on the surface were analysed by FACS (CantoII, BD bioscience).
- B/Florida/04/2006 and B/Jillin 20/2003 were labeled with biotin using the EZ-link Sulpho-NHS-LC-LC-biotin kit (Pierce). 1 ⁇ of the 10 mM biotin solution was added to 110 ⁇ g of recombinant HA, which is a six-fold molar excess of biotin, and incubated for 30 to 40 minutes at room temperature. The free unincorporated biotin was removed using an Amicon Ultra centrifugal filter (0.5 ml, 10K Ultracel-IOK membrane; Millipore, cat#: UFC501096).
- Amicon Ultra centrifugal filter 0.5 ml, 10K Ultracel-IOK membrane; Millipore, cat#: UFC501096
- Antibodies CR10023 and CR10049 compete for binding CR8033.
- Antibodies CR10032 and CR10051 compete for binding with CR8059.
- Antibody CR10049 competes for binding with CR10032. None of the tested antibodies compete with stem-binding antibody CR9114. These results indicate the presence of at least three to four different epitopes on the influenza B HA (Fig. 1).
- VNAs virus neutralization assays
- influenza B Yamagata-like (B/Harbin/7/1994 and B/Florida/04/2006) and Victoria-like (B/Malaysia/2506/2004 and B/Brisbane/60/2008) strains used in the assay were all diluted to a titer of 5,7 xlO 3 TCID50/ml (50% tissue culture infective dose per ml), with the titer calculated according to the method of Spearman and Karber.
- the IgG preparations (100 ⁇ g/ml) were serially 2-fold diluted (1 :2 - 1 :512) in complete MEM medium in quadruplicate wells.
- ⁇ 1 of the respective IgG dilution was mixed with 50 ⁇ 1 of virus suspension (100 ⁇ 50/35 ⁇ 1) and incubated for one hr at 37°C.
- the suspension was then transferred in quadruplicate into 96-well plates containing confluent MDCK cultures in ⁇ complete MEM medium.
- MDCK cells Prior to use, MDCK cells were seeded at 2x10 4 cells per well in MDCK cell culture medium, grown until cells had reached confluence, washed with 300-350 ⁇ PBS, pH 7.4 and finally ⁇ complete MEM medium was added to each well.
- the inoculated cells were cultured for 3 - 4 days at 37°C and observed daily for the development of cytopathogenic effect (CPE). CPE was compared to the positive control.
- CPE cytopathogenic effect
- CR8032, CR8033, CR8034, CR8035, CR8059, CR8071 , CR10023, CR10032, CR10049, CR10051 , CR11035, CR1 1036, CR11038 and CR11039 all showed cross- neutralizing activity to representative strains of both, Yamagata and Victoria- like influenza B virus strains. See Table 9.
- haemagglutination inhibition (HI) assays were performed.
- B/Brisbane/60/2008 virus strains were diluted to 8 HA units, as determined in an HAU assay, and combined with an equal volume of serially diluted IgG and incubated for 1 hr at room temperature. An equal volume of 0.5% Turkey red blood cells (TRBC) was added to the wells and incubation continued for 30 min. Button formation was scored as evidence of hemagglutination.
- TRBC Turkey red blood cells
- CR8059, CR8071 , CR10032, CR10051 and CR11036 did not show HI activity to any of the tested influenza B virus strains (>10 ⁇ g/ml for CR11036, > 50 ⁇ g/ml for the other antibodies), indicating that they do not block the receptor binding.
- Antibodies CR8033 and CR10023 show HI activity to representative strains of only the Yamagata-, but not the Victoria-like influenza B virus strains.
- Antibody CR11035 shows HI activity to a representative strain of only the Victoria-, but not the Yamagata-like influenza B virus strains.
- Antibodies CR10049, CR1 1038 and CR1 1039 show HI activity to representative strains of both Yamagata and Victoria-like influenza B virus strains. See Table 10.
- an immunofluorescence entry assay was designed to analyze the ability of a given antibody to block receptor binding and internalization of the virus. Therefore, the virus was pre-incubated with the antibody in serial, two-fold dilution steps before being added to a confluent monolayer of MDCK cells plated in a 96-well dish in infection medium (DMEM+200mM glutamine) for two to three hours. The inoculum was subsequently removed and replaced with antibody at indicated concentrations for 16 - 18 hrs at 37 °C, 5% C02.
- an egress assay was designed to analyze the amount of virus particles released into the supernatant 18 hrs post infection under antibody treatment conditions.
- the detection (or absence) of an anti-HA signal after gel electrophoresis followed by Western blot of such supernatants is taken as indication for the presence (or absence) of released virus particles.
- MDCK cells Four hours prior to the experiment, 40,000 MDCK cells per well were seeded in DMEM/glutamine into 96-well plates. The amount of virus needed to achieve 90-100% infection was titrated in a separate experiment. The required amount of virus was added to the cells and incubated at 37 °C, 5% C02. After three hours, the supernatants were removed and cells were washed thrice with PBS to remove non-internalized virus particles. Cells were replenished with infection medium containing mAbs (serial dilution starting at 20 ⁇ g/ml).
- MDCK cells were seeded on glass coverslips one day prior to the experiment. The next day, cells were infected with different amounts of virus to determine the amount that yielded 90 - 100% infected cells after 18 hrs post infection. Three hours after the initial infection, the supernatants were removed; cells were washed thrice with PBS, before media containing the indicated concentration of antibodies were added. After an additional 15 - 18 hrs, the cell culture medium was removed and cells were fixed in 2.5% glutaraldehyde buffer and stored at 4 °C until further analysis. The samples were subjected to further chemical fixation using glutaraldehyde (GA) and/or osmium tetroxid (Os04).
- G glutaraldehyde
- Os04 osmium tetroxid
- the specimens Prior to SEM imaging, the specimens were subjected to acetone dehydration and critical-point-drying. Finally, the cells were be mounted on alumina stubs and coated with thin layer of carbon and examined in a Zeiss Ultra 55 SEM microscope.
- Fig. 4b The surface of influenza B infected MDCK cells is covered with electron dense spherical particles (Fig. 4b), in contrast to uninfected controls (Fig. 4a).
- Incubation with antibody CR8059 does not prevent the formation of these spherical particles (Fig. 4c) whereas incubation with antibody CR8033 greatly diminishes the formation of particles (Fig. 4d).
- Fig. 4e and f In contrast to CR8059 incubated cells, budding virions can not readily be detected on CR8033 incubated cells (Fig. 4e and f).
- MAbs CR8033 and CR8071 were tested for prophylactic efficacy in a mouse lethal challenge model with mouse adapted influenza B/Florida/04/2006 virus.
- the B/Florida/04/2006 virus was adapted to mice after 5 lung-to-lung passages.
- MAbs CR8033 and CR8071 were dosed at 0.06, 0.2, 0.6, 1.7, and 5 mg/kg intravenously into the tail vein ⁇ vena coccygeus) at day -1 before challenge, assuming an average weight of 18 g per mouse and a fixed dose volume of 0.2 ml.
- a control group was taken along dosed with vehicle control.
- the mice were then challenged at day 0 with 25 LD 50 mouse adapted B/Florida/04/2006 influenza B virus by intranasal inoculation.
- Fig 5 shows the survival rates of the mice, following mAb administration.
- mAbs CR8033 and CR8071 were tested for prophylactic efficacy in a mouse lethal challenge model with mouse adapted influenza B/Malaysia/2506/2004 virus.
- the B/Malaysia/2506/2004 virus was adapted to mice after 4 lung-to-lung passages.
- MAbs CR8033 and CR8071 were dosed at 0.06, 0.2, 0.6, 1.7 and 5 mg/kg intravenously in the tail vein (vena coccygeus) at day -1 before challenge, assuming an average weight of 18 g per mouse and a fixed dose volume of 0.2 ml.
- a control group was taken along dosed with vehicle control.
- the mice were then challenged at day 0 with 25 LD 50 mouse adapted B/Malaysia 2506/2004 influenza B virus by intranasal inoculation.
- Fig. 5 shows the survival rates of the mice, following mAb administration. Mice dosed with dosages as low as 0.2 mg/kg for CR8033 and 0.6 mg/kg for CR8071 showed significantly higher survival rates than the vehicle treated control animals.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Virology (AREA)
- Immunology (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Organic Chemistry (AREA)
- Biochemistry (AREA)
- Biomedical Technology (AREA)
- Genetics & Genomics (AREA)
- Biophysics (AREA)
- Microbiology (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Urology & Nephrology (AREA)
- Hematology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Biotechnology (AREA)
- Epidemiology (AREA)
- Communicable Diseases (AREA)
- Physics & Mathematics (AREA)
- Pulmonology (AREA)
- Food Science & Technology (AREA)
- Cell Biology (AREA)
- Tropical Medicine & Parasitology (AREA)
- Analytical Chemistry (AREA)
- General Physics & Mathematics (AREA)
- Pathology (AREA)
- Mycology (AREA)
- Oncology (AREA)
- General Engineering & Computer Science (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
Abstract
Description

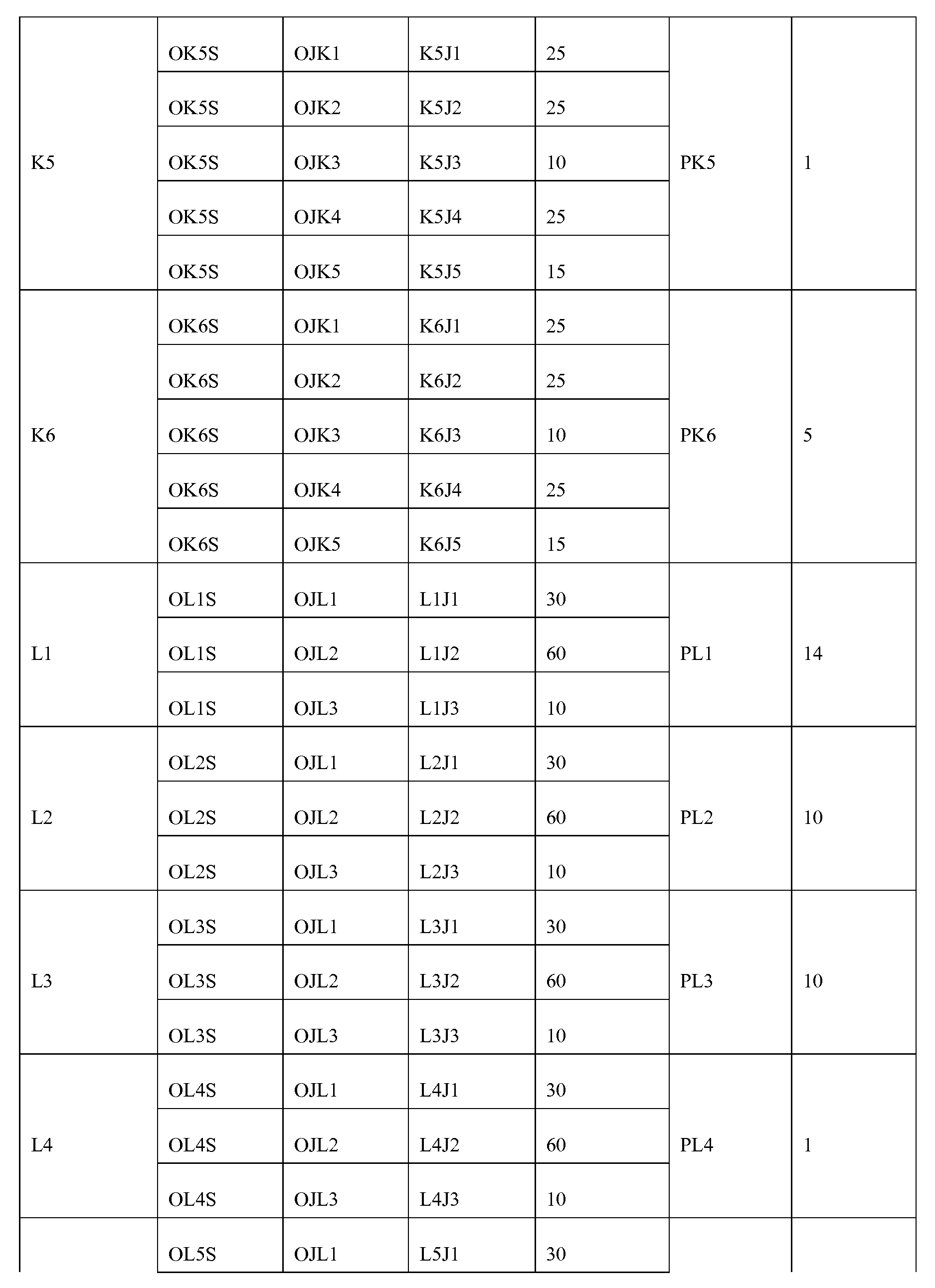
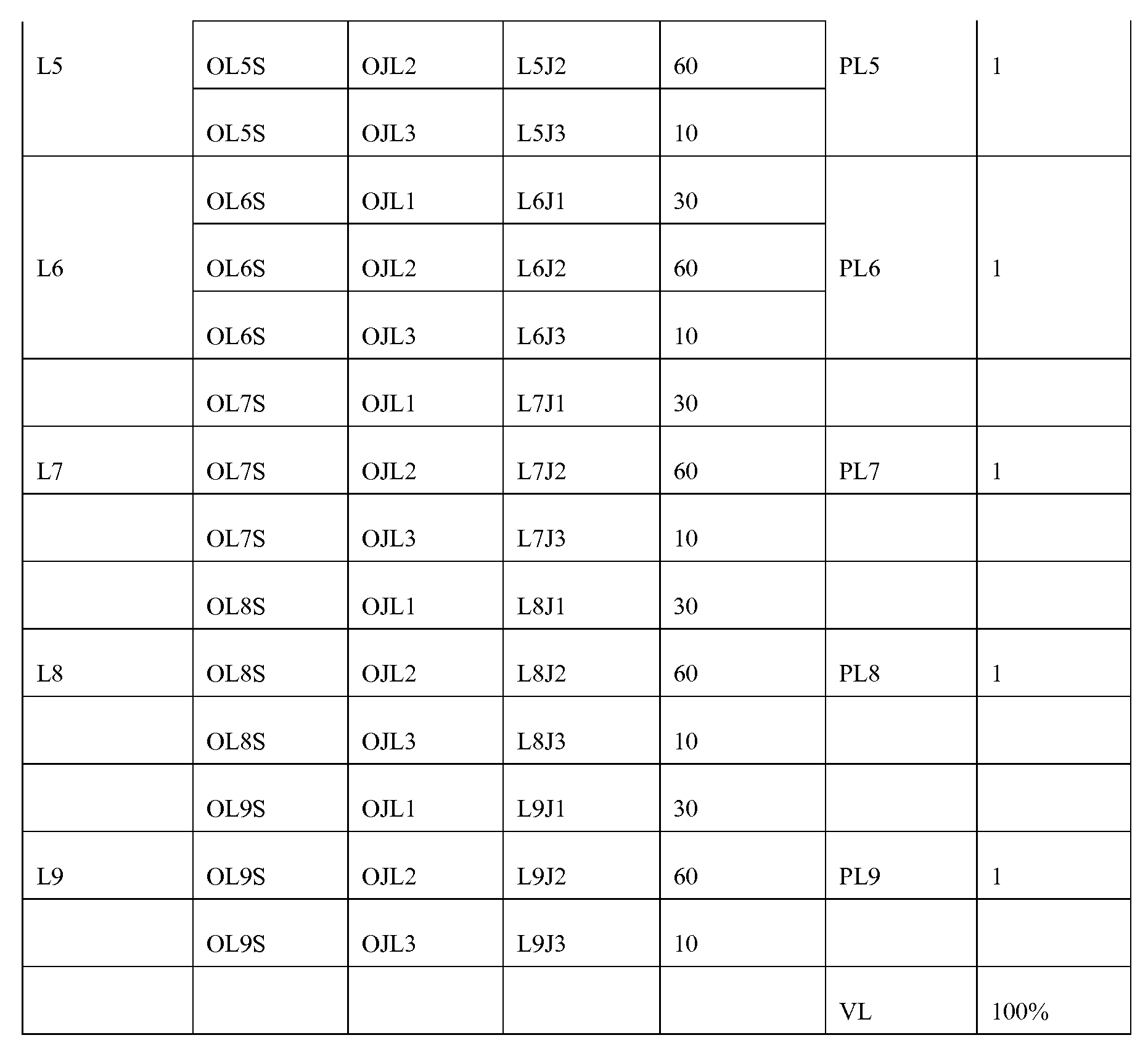
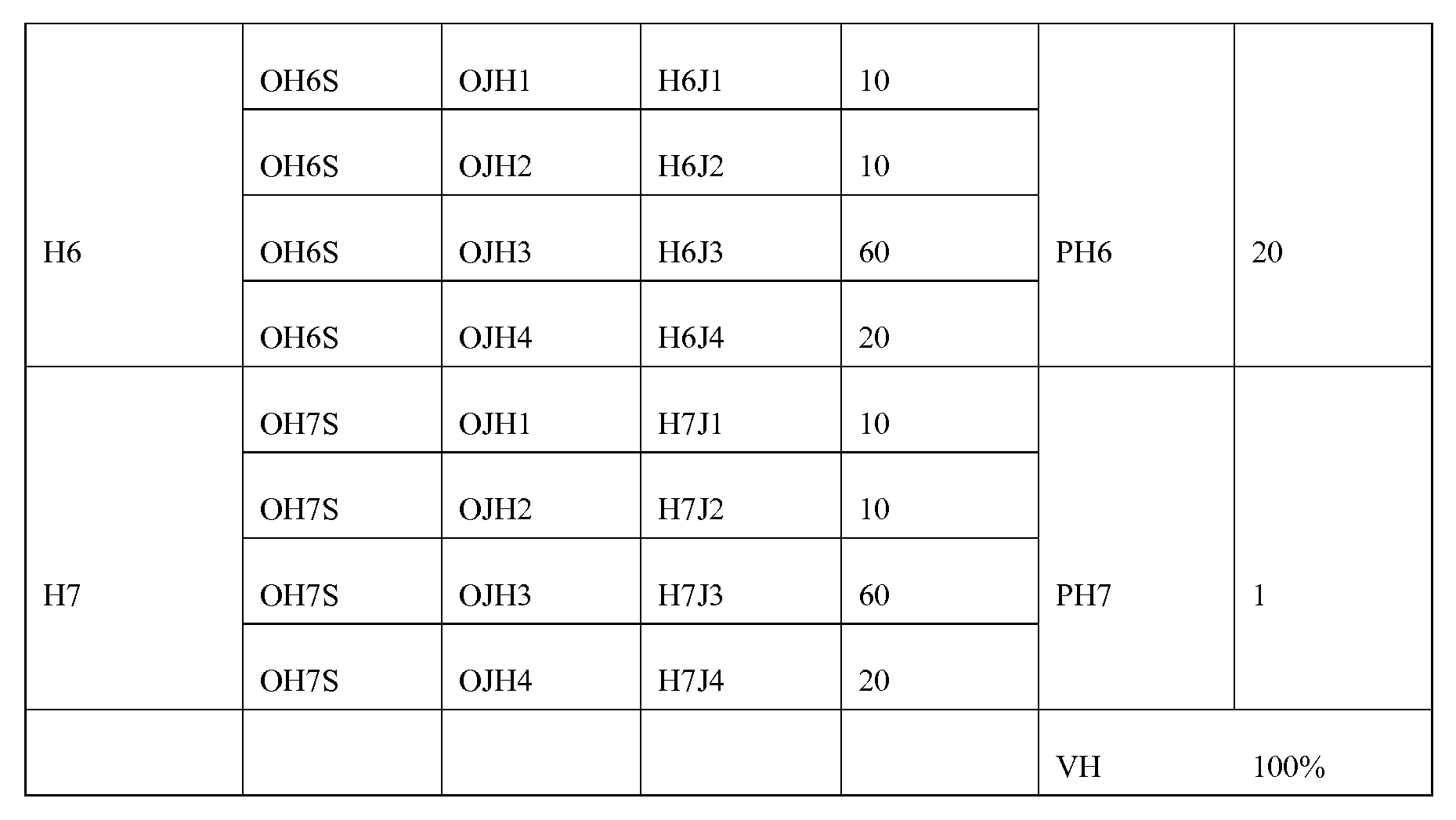














Claims
Priority Applications (18)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP13708411.7A EP2822968B1 (en) | 2012-03-08 | 2013-03-07 | Human binding molecules capable of binding to and neutralizing influenza b viruses and uses thereof |
| CN201380012721.8A CN104169298B (en) | 2012-03-08 | 2013-03-07 | It can combine and neutralize mankind's binding molecule of Type B influenza virus and application thereof |
| SG11201405226TA SG11201405226TA (en) | 2012-03-08 | 2013-03-07 | Human binding molecules capable of binding to and neutralizing influenza b viruses and uses thereof |
| CA2865594A CA2865594C (en) | 2012-03-08 | 2013-03-07 | Human binding molecules capable of binding to and neutralizing influenza b viruses and uses thereof |
| ES13708411.7T ES2664625T3 (en) | 2012-03-08 | 2013-03-07 | Human binding molecules that can bind to influenza b viruses and neutralize them, and their uses |
| DK13708411.7T DK2822968T3 (en) | 2012-03-08 | 2013-03-07 | HUMAN BINDING MOLECULES ABLE TO BIND AND NEUTRALIZE INFLUENZA-B-VIRA, AND APPLICATIONS THEREOF |
| AU2013229488A AU2013229488B2 (en) | 2012-03-08 | 2013-03-07 | Human binding molecules capable of binding to and neutralizing influenza B viruses and uses thereof |
| EA201491656A EA028433B1 (en) | 2012-03-08 | 2013-03-07 | Antibody that binds to influenza b viruses and use thereof |
| JP2014560375A JP6328061B2 (en) | 2012-03-08 | 2013-03-07 | Human binding molecule capable of binding to and neutralizing influenza B virus and use thereof |
| NZ628382A NZ628382A (en) | 2012-03-08 | 2013-03-07 | Human binding molecules capable of binding to and neutralizing influenza b viruses and uses thereof |
| KR1020147025832A KR102050615B1 (en) | 2012-03-08 | 2013-03-07 | Human binding molecules capable of binding to and neutralizing influenza b viruses and uses thereof |
| MX2014010755A MX360056B (en) | 2012-03-08 | 2013-03-07 | Human binding molecules capable of binding to and neutralizing influenza b viruses and uses thereof. |
| PH12014501776A PH12014501776B1 (en) | 2012-03-08 | 2014-08-07 | Human binding molecules capable of binding to and neutralizing influenza b viruses and uses thereof |
| ZA2014/05947A ZA201405947B (en) | 2012-03-08 | 2014-08-13 | Human binding molecules capable of binding to and neutralizing influenza b viruses and uses thereof |
| IL234446A IL234446B (en) | 2012-03-08 | 2014-09-02 | Human binding molecules capable of binding to and neutralizing influenza b viruses and uses thereof |
| HK15100646.1A HK1200176A1 (en) | 2012-03-08 | 2015-01-20 | Human inding molecules capable of inding to and neutralizing influenza viruses and uses thereof |
| PH12015502553A PH12015502553A1 (en) | 2012-03-08 | 2015-11-09 | Human binding molecules capable of binding to and neutralizing influenza b viruses and uses thereof |
| AU2018201647A AU2018201647B2 (en) | 2012-03-08 | 2018-03-07 | Human binding molecules capable of binding to and neutralizing influenza B viruses and uses thereof |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261608414P | 2012-03-08 | 2012-03-08 | |
| EP12158525 | 2012-03-08 | ||
| EP12158525.1 | 2012-03-08 | ||
| US61/608,414 | 2012-03-08 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013132007A1 true WO2013132007A1 (en) | 2013-09-12 |
Family
ID=49115966
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2013/054606 WO2013132007A1 (en) | 2012-03-08 | 2013-03-07 | Human binding molecules capable of binding to and neutralizing influenza b viruses and uses thereof |
Country Status (19)
| Country | Link |
|---|---|
| US (5) | US8834881B2 (en) |
| EP (1) | EP2822968B1 (en) |
| JP (1) | JP6328061B2 (en) |
| KR (1) | KR102050615B1 (en) |
| CN (2) | CN108640989B (en) |
| AR (1) | AR091316A1 (en) |
| AU (2) | AU2013229488B2 (en) |
| CA (1) | CA2865594C (en) |
| DK (1) | DK2822968T3 (en) |
| EA (1) | EA028433B1 (en) |
| ES (1) | ES2664625T3 (en) |
| HK (1) | HK1200176A1 (en) |
| MX (1) | MX360056B (en) |
| MY (1) | MY170407A (en) |
| NZ (1) | NZ628382A (en) |
| PH (2) | PH12014501776B1 (en) |
| SG (1) | SG11201405226TA (en) |
| TW (1) | TWI628190B (en) |
| WO (1) | WO2013132007A1 (en) |
Cited By (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8877200B2 (en) | 2012-05-10 | 2014-11-04 | Visterra, Inc. | HA binding agents |
| WO2015148806A1 (en) * | 2014-03-27 | 2015-10-01 | Genentech, Inc. | Anti-influenza b virus hemagglutinin antibodies and methods of use |
| WO2015120097A3 (en) * | 2014-02-04 | 2015-10-08 | Contrafect Corporation | Antibodies useful in passive influenza immunization, and compositions, combinations and methods for use thereof |
| WO2016011035A2 (en) | 2014-07-15 | 2016-01-21 | Medlmmune, Llc | Neutralizing anti-influenza b antibodies and uses thereof |
| EP3076993A1 (en) * | 2013-12-05 | 2016-10-12 | Janssen Vaccines & Prevention B.V. | Process for preparing influenza vaccines |
| WO2016196470A1 (en) * | 2015-06-01 | 2016-12-08 | Medimmune, Llc | Neutralizing anti-influenza binding molecules and uses thereof |
| US9718875B2 (en) | 2013-03-14 | 2017-08-01 | Contrafect Corporation | Composition and methods based on neutralizing antibodies delivered intranasally for enhanced therapeutic efficacy |
| WO2017148889A1 (en) | 2016-03-01 | 2017-09-08 | Janssen Vaccines & Prevention B.V. | Human neutralizing antibodies binding to influenza b neuraminidase |
| AU2016270147B2 (en) * | 2015-06-03 | 2018-08-23 | Xiamen University | Broad-spectrum monoclonal anti-flu B antibody and uses thereof |
| US10494419B2 (en) | 2013-10-02 | 2019-12-03 | Medimmune, Llc | Neutralizing anti-influenza A antibodies and uses thereof |
| US10513553B2 (en) | 2015-11-13 | 2019-12-24 | Visterra, Inc. | Compositions and methods for treating and preventing influenza |
| US10639370B2 (en) | 2014-02-04 | 2020-05-05 | Contrafect Corporation | Antibodies useful in passive influenza immunization, and compositions, combinations and methods for use thereof |
| US10654915B2 (en) | 2011-12-05 | 2020-05-19 | Trellis Bioscience, Llc | Antibodies useful in passive influenza immunization |
| US10786568B2 (en) | 2017-02-28 | 2020-09-29 | The Trustees Of The University Of Pennsylvania | AAV mediated influenza vaccines |
| US11136379B2 (en) | 2015-02-05 | 2021-10-05 | Janssen Vaccines & Prevention B.V. | Binding molecules directed against influenza hemagglutinin and uses thereof |
| US11230593B2 (en) | 2019-03-25 | 2022-01-25 | Visterra, Inc. | Compositions and methods for treating and preventing influenza |
| RU2784915C2 (en) * | 2014-07-15 | 2022-12-01 | МЕДИММЬЮН, ЭлЭлСи | Neutralizing antibodies to influenza b virus and their application methods |
| US11535665B2 (en) | 2015-05-13 | 2022-12-27 | The Trustees Of The University Of Pennsylvania | AAV-mediated expression of anti-influenza antibodies and methods of use thereof |
| US11547756B2 (en) | 2016-01-13 | 2023-01-10 | Medimmune, Llc | Method of treating influenza A |
| US11578341B2 (en) | 2017-02-28 | 2023-02-14 | The Trustees Of The University Of Pennsylvania | Compositions useful in treatment of spinal muscular atrophy |
| US11827906B2 (en) | 2017-02-28 | 2023-11-28 | The Trustees Of The University Of Pennsylvania | Adeno-associated virus (AAV) clade f vector and uses therefor |
Families Citing this family (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BR112014012681A8 (en) | 2011-11-28 | 2017-06-20 | Crucell Holland Bv | polypeptide, designed to provide a polypeptide, nucleic acid, and immunogenic composition |
| CA2865594C (en) | 2012-03-08 | 2021-07-27 | Crucell Holland B.V. | Human binding molecules capable of binding to and neutralizing influenza b viruses and uses thereof |
| US9624272B2 (en) | 2013-03-14 | 2017-04-18 | University Of Washington Through Its Center For Commercialization | Polypeptides for treating and/or limiting influenza infection |
| TWI702229B (en) | 2014-12-19 | 2020-08-21 | 美商再生元醫藥公司 | Human antibodies to influenza hemagglutinin |
| CA2985402A1 (en) * | 2015-05-11 | 2016-11-17 | Janssen Vaccines & Prevention B.V. | Influenza virus neutralizing peptidomimetic compounds |
| JP2017062300A (en) * | 2015-09-24 | 2017-03-30 | セイコーエプソン株式会社 | Semiconductor device, system, electronic equipment, and voice recognizing method |
| BR112019002039A2 (en) * | 2016-08-05 | 2019-05-07 | Medimmune, Llc | anti-o2 antibodies and their use |
| AU2017346488A1 (en) | 2016-10-19 | 2019-05-30 | Humabs Biomed Sa | Anti-O1 antibodies and uses thereof |
| WO2018189705A1 (en) | 2017-04-13 | 2018-10-18 | Cadila Healthcare Limited | Novel peptide based pcsk9 vaccine |
| KR20200115517A (en) | 2018-01-26 | 2020-10-07 | 리제너론 파마슈티칼스 인코포레이티드 | Human antibodies to influenza hemagglutinin |
| MX2021000281A (en) * | 2018-07-11 | 2021-11-12 | Momenta Pharmaceuticals Inc | Compositions and methods related to engineered fc-antigen binding domain constructs targeted to pd-l1. |
| WO2021195326A1 (en) * | 2020-03-26 | 2021-09-30 | Vanderbilt University | Human monoclonal antibodies to severe acute respiratory syndrome coronavirus 2 (sars-cov-2) |
| WO2021201677A1 (en) | 2020-04-01 | 2021-10-07 | Kiadis Pharma Intellectual Property B.V. | Compositions and methods targeting influenza |
| BR112022021789A2 (en) | 2020-04-27 | 2023-03-07 | Twist Bioscience Corp | CORONAVIRUS VARIANT NUCLEIC ACID LIBRARIES |
Citations (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1984003564A1 (en) | 1983-03-08 | 1984-09-13 | Commw Serum Lab Commission | Method of determining antigenically active amino acid sequences |
| WO1993009872A1 (en) | 1991-11-21 | 1993-05-27 | Seed Capital Investments (Sci) B.V. | Test device comprising a plate containing a multiplicity of wells with an associated metering device, as well as a kit which comprises these devices and use of the devices |
| WO2000063403A2 (en) | 1999-04-15 | 2000-10-26 | Crucell Holland B.V. | Recombinant protein production in a human cell using sequences encoding adenovirus e1 protein |
| EP1117395A1 (en) | 1998-10-01 | 2001-07-25 | Duquesne University Of The Holy Ghost | Compounds for the treatment of estrogen-dependent illnesses and methods for making and using the same |
| WO2002103012A1 (en) | 2001-06-15 | 2002-12-27 | Crucell Holland B.V. | Chimaeric phages |
| WO2003057857A2 (en) * | 2002-01-07 | 2003-07-17 | Abgenix, Inc. | Antibodies directed to pdgfd and uses thereof |
| WO2006124269A2 (en) * | 2005-05-16 | 2006-11-23 | Amgen Fremont Inc. | Human monoclonal antibodies that bind to very late antigen-1 for the treatment of inflammation and other disorders |
| WO2008028946A2 (en) | 2006-09-07 | 2008-03-13 | Crucell Holland B.V. | Human binding molecules capable of neutralizing influenza virus h5n1 and uses thereof |
| WO2009002380A2 (en) * | 2007-06-26 | 2008-12-31 | The Trustees Of The University Of Pennsylvania | Isolation of anti-desmoglein 1 antibodies by phage display of pemphigus foliaceus autoantibodies |
| WO2010054265A2 (en) * | 2008-11-07 | 2010-05-14 | Galaxy Biotech, Llc. | Monoclonal antibodies to fibroblast growth factor receptor 2 |
| WO2010130636A1 (en) | 2009-05-11 | 2010-11-18 | Crucell Holland B.V. | Human binding molecules capable of neutralizing influenza virus h3n2 and uses thereof |
| US20100330103A1 (en) * | 2006-08-03 | 2010-12-30 | Astrazeneca Ab | Antibodies Directed to Alpha V Beta 6 And Uses Thereof |
| WO2011038302A2 (en) * | 2009-09-25 | 2011-03-31 | Xoma Technology Ltd. | Novel modulators |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2600730C (en) * | 2005-03-08 | 2014-11-25 | Medimmune, Inc. | Influenza hemagglutinin and neuraminidase variants |
| FR2921928B1 (en) * | 2007-10-08 | 2011-03-04 | Sanofi Pasteur | MONOCLONAL ANTIBODIES SPECIFIC TO INFLUENZA VIRUS HEMAGGLUTININ |
| MX2011000768A (en) * | 2008-07-25 | 2011-10-05 | Inst Research In Biomedicine | Neutralizing anti-influenza a virus antibodies and uses thereof. |
| EP2179811B1 (en) | 2008-10-21 | 2018-10-03 | Agie Charmilles SA | Method and apparatus for controlling an electric discharge machining process |
| JP5780762B2 (en) * | 2008-12-25 | 2015-09-16 | 国立大学法人大阪大学 | Anti-human influenza virus / human antibody |
| AU2010254136B2 (en) * | 2009-05-26 | 2016-09-29 | Mount Sinai School Of Medicine | Monoclonal antibodies against influenza virus generated by cyclical administration and uses thereof |
| CN104245731B (en) * | 2012-01-31 | 2017-04-05 | 株式会社医学生物学研究所 | The extensive protectiveness human monoclonal antibodies of anti-Influenza B viruss and its using method |
| CA2865594C (en) | 2012-03-08 | 2021-07-27 | Crucell Holland B.V. | Human binding molecules capable of binding to and neutralizing influenza b viruses and uses thereof |
-
2013
- 2013-03-07 CA CA2865594A patent/CA2865594C/en active Active
- 2013-03-07 CN CN201810486503.2A patent/CN108640989B/en active Active
- 2013-03-07 ES ES13708411.7T patent/ES2664625T3/en active Active
- 2013-03-07 MY MYPI2014702487A patent/MY170407A/en unknown
- 2013-03-07 DK DK13708411.7T patent/DK2822968T3/en active
- 2013-03-07 US US13/789,284 patent/US8834881B2/en active Active
- 2013-03-07 NZ NZ628382A patent/NZ628382A/en not_active IP Right Cessation
- 2013-03-07 AU AU2013229488A patent/AU2013229488B2/en not_active Ceased
- 2013-03-07 SG SG11201405226TA patent/SG11201405226TA/en unknown
- 2013-03-07 WO PCT/EP2013/054606 patent/WO2013132007A1/en active Application Filing
- 2013-03-07 CN CN201380012721.8A patent/CN104169298B/en active Active
- 2013-03-07 JP JP2014560375A patent/JP6328061B2/en active Active
- 2013-03-07 EA EA201491656A patent/EA028433B1/en not_active IP Right Cessation
- 2013-03-07 MX MX2014010755A patent/MX360056B/en active IP Right Grant
- 2013-03-07 KR KR1020147025832A patent/KR102050615B1/en active IP Right Grant
- 2013-03-07 EP EP13708411.7A patent/EP2822968B1/en active Active
- 2013-03-08 TW TW102108417A patent/TWI628190B/en not_active IP Right Cessation
- 2013-03-08 AR ARP130100759 patent/AR091316A1/en active IP Right Grant
- 2013-10-10 US US14/051,365 patent/US8852595B2/en active Active
-
2014
- 2014-07-03 US US14/323,908 patent/US9005621B2/en active Active
- 2014-08-07 PH PH12014501776A patent/PH12014501776B1/en unknown
- 2014-10-13 US US14/513,162 patent/US9200064B2/en active Active
-
2015
- 2015-01-20 HK HK15100646.1A patent/HK1200176A1/en not_active IP Right Cessation
- 2015-09-17 US US14/857,587 patent/US20160002318A1/en not_active Abandoned
- 2015-11-09 PH PH12015502553A patent/PH12015502553A1/en unknown
-
2018
- 2018-03-07 AU AU2018201647A patent/AU2018201647B2/en not_active Ceased
Patent Citations (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1984003564A1 (en) | 1983-03-08 | 1984-09-13 | Commw Serum Lab Commission | Method of determining antigenically active amino acid sequences |
| WO1993009872A1 (en) | 1991-11-21 | 1993-05-27 | Seed Capital Investments (Sci) B.V. | Test device comprising a plate containing a multiplicity of wells with an associated metering device, as well as a kit which comprises these devices and use of the devices |
| EP1117395A1 (en) | 1998-10-01 | 2001-07-25 | Duquesne University Of The Holy Ghost | Compounds for the treatment of estrogen-dependent illnesses and methods for making and using the same |
| WO2000063403A2 (en) | 1999-04-15 | 2000-10-26 | Crucell Holland B.V. | Recombinant protein production in a human cell using sequences encoding adenovirus e1 protein |
| WO2002103012A1 (en) | 2001-06-15 | 2002-12-27 | Crucell Holland B.V. | Chimaeric phages |
| WO2003057857A2 (en) * | 2002-01-07 | 2003-07-17 | Abgenix, Inc. | Antibodies directed to pdgfd and uses thereof |
| WO2006124269A2 (en) * | 2005-05-16 | 2006-11-23 | Amgen Fremont Inc. | Human monoclonal antibodies that bind to very late antigen-1 for the treatment of inflammation and other disorders |
| US20100330103A1 (en) * | 2006-08-03 | 2010-12-30 | Astrazeneca Ab | Antibodies Directed to Alpha V Beta 6 And Uses Thereof |
| WO2008028946A2 (en) | 2006-09-07 | 2008-03-13 | Crucell Holland B.V. | Human binding molecules capable of neutralizing influenza virus h5n1 and uses thereof |
| WO2009002380A2 (en) * | 2007-06-26 | 2008-12-31 | The Trustees Of The University Of Pennsylvania | Isolation of anti-desmoglein 1 antibodies by phage display of pemphigus foliaceus autoantibodies |
| WO2010054265A2 (en) * | 2008-11-07 | 2010-05-14 | Galaxy Biotech, Llc. | Monoclonal antibodies to fibroblast growth factor receptor 2 |
| WO2010130636A1 (en) | 2009-05-11 | 2010-11-18 | Crucell Holland B.V. | Human binding molecules capable of neutralizing influenza virus h3n2 and uses thereof |
| WO2011038302A2 (en) * | 2009-09-25 | 2011-03-31 | Xoma Technology Ltd. | Novel modulators |
| US20110076284A1 (en) * | 2009-09-25 | 2011-03-31 | Xoma Technology Ltd. | Novel Modulators |
Non-Patent Citations (24)
| Title |
|---|
| "WHO Manual on Animal Influenza Diagnosis and Surveillance", 2005, WORLD HEALTH ORGANISATION |
| : E. HARLOW AND D, LANE: "Antibodies: A Laboratory Manual", 1988, COLD SPRING HARBOR LABORATORY |
| : E. HARLOW AND D, LANE: "Using Antibodies: A Laboratory Manual", 1998, COLD SPRING HARBOR LABORATORY |
| BROCHET ET AL., NUCL. ACIDS RES., vol. 36, 2008, pages W503 - 508 |
| DATABASE Geneseq [online] 12 May 2011 (2011-05-12), "Ab LC XPA.015.141 that strengthens/weakens INSR binding SEQ ID:37.", retrieved from EBI accession no. GSP:AZG68048 Database accession no. AZG68048 * |
| DATABASE Geneseq [online] 14 April 2011 (2011-04-14), "Human anti-alpha Vbeta 6 integrin germline antibody VL protein SEQ ID:61.", retrieved from EBI accession no. GSP:AYN41222 Database accession no. AYN41222 * |
| DATABASE Geneseq [online] 22 March 2007 (2007-03-22), "Human anti-VLA-1 antibody heavy chain variable region #102.", retrieved from EBI accession no. GSP:AER52563 Database accession no. AER52563 * |
| DATABASE Geneseq [online] 5 March 2009 (2009-03-05), "Human anti-desmoglein-1 VH antibody protein, SEQ ID:234.", retrieved from EBI accession no. GSP:AUZ94850 Database accession no. AUZ94850 * |
| DATABASE Geneseq [online] 6 May 2004 (2004-05-06), "Anti-human PDGF-D antibody heavy chain protein sequence.", retrieved from EBI accession no. GSP:ADK18618 Database accession no. ADK18618 * |
| DE KRUIF J ET AL., PROC. NATL. ACAD. SCI. USA, vol. 92, 1995, pages 3938 |
| DREYFUS CYRILLE ET AL: "Highly conserved protective epitopes on influenza B viruses.", SCIENCE (NEW YORK, N.Y.) 14 SEP 2012, vol. 337, no. 6100, 14 September 2012 (2012-09-14), pages 1343 - 1348, XP002693350, ISSN: 1095-9203 * |
| EKIERT DAMIAN C ET AL: "A highly conserved neutralizing epitope on group 2 influenza A viruses.", SCIENCE (NEW YORK, N.Y.) 12 AUG 2011, vol. 333, no. 6044, 12 August 2011 (2011-08-12), pages 843 - 850, XP002693349, ISSN: 1095-9203 * |
| EKIERT DAMIAN C ET AL: "Antibody Recognition of a Highly Conserved Influenza Virus Epitope", SCIENCE, AMERICAN ASSOCIATION FOR THE ADVANCEMENT OF SCIENCE, WASHINGTON, DC; US, vol. 324, no. 5924, 1 April 2009 (2009-04-01), pages 246 - 251, XP009144786, ISSN: 0036-8075 * |
| J. C. KRAUSE ET AL: "A Broadly Neutralizing Human Monoclonal Antibody That Recognizes a Conserved, Novel Epitope on the Globular Head of the Influenza H1N1 Virus Hemagglutinin", JOURNAL OF VIROLOGY, vol. 85, no. 20, 15 October 2011 (2011-10-15), pages 10905 - 10908, XP055044176, ISSN: 0022-538X, DOI: 10.1128/JVI.00700-11 * |
| J.E. COLIGAN, A.M. KRUISBEEK, D.H. MARGULIES, E.M. SHEVACH, W. STROBER: "Current Protocols in Immunology", 2001, JOHN WILEY & SONS INC. |
| KABAT ET AL., SEQUENCES OF PROTEINS OF IMMUNOLOGICAL INTEREST, 1991 |
| KANEGAE ET AL., J. VIROL., vol. 64, 1990, pages 2860 - 2865 |
| KUBOTA-KOKETSU ET AL., BIOCHEM. BIOPHYS. RES. COMM., vol. 387, 2009, pages 180 - 185 |
| ROTA ET AL., J. GEN. VIROL., vol. 73, 1992, pages 2737 - 2742 |
| THOMPSON ET AL., JAMA, vol. 289, no. 2, 2003, pages 179 - 186 |
| THOMPSON ET AL., JAMA, vol. 292, no. 11, 2004, pages 1333 - 1340 |
| THROSBY M ET AL: "Heterosubtypic neutralizing monoclonal antibodies cross-protective against H5N1 and H1N1 recovered from human IgM+ memory B cells", PLOS ONE, PUBLIC LIBRARY OF SCIENCE, US, vol. 3, no. 12, 16 December 2008 (2008-12-16), pages 1 - 15, XP002672518, ISSN: 1932-6203, DOI: 10.1371/JOURNAL.PONE.0003942 * |
| WHITTLE JAMES R R ET AL: "Broadly neutralizing human antibody that recognizes the receptor-binding pocket of influenza virus hemagglutinin.", PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES OF THE UNITED STATES OF AMERICA 23 AUG 2011, vol. 108, no. 34, 23 August 2011 (2011-08-23), pages 14216 - 14221, XP002693351, ISSN: 1091-6490 * |
| WRAMMERT ET AL., NATURE, vol. 453, 2008, pages 667 - 672 |
Cited By (66)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10654915B2 (en) | 2011-12-05 | 2020-05-19 | Trellis Bioscience, Llc | Antibodies useful in passive influenza immunization |
| US9096657B2 (en) | 2012-05-10 | 2015-08-04 | Visterra, Inc. | HA binding agents |
| US12024552B2 (en) | 2012-05-10 | 2024-07-02 | Visterra, Inc. | Ha binding agents |
| US10800835B2 (en) | 2012-05-10 | 2020-10-13 | Visterra, Inc. | HA binding agents |
| US9969794B2 (en) | 2012-05-10 | 2018-05-15 | Visterra, Inc. | HA binding agents |
| US8877200B2 (en) | 2012-05-10 | 2014-11-04 | Visterra, Inc. | HA binding agents |
| US9718875B2 (en) | 2013-03-14 | 2017-08-01 | Contrafect Corporation | Composition and methods based on neutralizing antibodies delivered intranasally for enhanced therapeutic efficacy |
| US11827693B2 (en) | 2013-03-14 | 2023-11-28 | Contrafect Corporation | Composition and methods based on neutralizing antibodies delivered intranasally for enhanced therapeutic efficacy |
| US10494419B2 (en) | 2013-10-02 | 2019-12-03 | Medimmune, Llc | Neutralizing anti-influenza A antibodies and uses thereof |
| US11932682B2 (en) | 2013-10-02 | 2024-03-19 | Medimmune, Llc | Neutralizing anti-influenza A antibodies and uses thereof |
| US11186627B2 (en) | 2013-10-02 | 2021-11-30 | Medimmune, Llc | Neutralizing anti-influenza A antibodies and uses thereof |
| EP3076993A1 (en) * | 2013-12-05 | 2016-10-12 | Janssen Vaccines & Prevention B.V. | Process for preparing influenza vaccines |
| US20160304586A1 (en) * | 2013-12-05 | 2016-10-20 | Crucell Holland B.V. | Process for preparing influenza vaccines |
| AU2015214146B2 (en) * | 2014-02-04 | 2020-09-17 | Contrafect Corporation | Antibodies useful in passive influenza immunization, and compositions, combinations and methods for use thereof |
| JP2017511692A (en) * | 2014-02-04 | 2017-04-27 | コントラフェクト コーポレイション | Antibodies useful for passive influenza immunization and compositions, combinations and methods for their use |
| JP7023466B2 (en) | 2014-02-04 | 2022-02-22 | コントラフェクト コーポレイション | Antibodies useful for influenza passive immunity and compositions, combinations and methods for their use |
| US11246928B2 (en) | 2014-02-04 | 2022-02-15 | Contrafect Corporation | Antibodies useful in passive influenza immunization, and compositions, combinations and methods for use thereof |
| CN106795216A (en) * | 2014-02-04 | 2017-05-31 | 康特拉费克特公司 | Can be used for immune antibody of passive influenza and combinations thereof, combination and application method |
| RU2720282C1 (en) * | 2014-02-04 | 2020-04-28 | Контрафект Корпорейшн | Antibodies suitable for passive immunization against influenza, and compositions thereof, combinations and methods of using |
| JP2020096618A (en) * | 2014-02-04 | 2020-06-25 | コントラフェクト コーポレイション | Antibodies useful in passive influenza immunization, and compositions, combinations and methods for use |
| WO2015120097A3 (en) * | 2014-02-04 | 2015-10-08 | Contrafect Corporation | Antibodies useful in passive influenza immunization, and compositions, combinations and methods for use thereof |
| US10639370B2 (en) | 2014-02-04 | 2020-05-05 | Contrafect Corporation | Antibodies useful in passive influenza immunization, and compositions, combinations and methods for use thereof |
| US9745365B2 (en) | 2014-03-27 | 2017-08-29 | Genentech, Inc. | Anti-influenza B virus hemagglutinin antibodies and methods of use |
| CN106132987A (en) * | 2014-03-27 | 2016-11-16 | 豪夫迈·罗氏有限公司 | Anti-influenza B virus hemagglutinin antibodies and methods of use |
| JP2017512471A (en) * | 2014-03-27 | 2017-05-25 | ジェネンテック, インコーポレイテッド | Anti-influenza B virus hemagglutinin antibodies and methods of use |
| WO2015148806A1 (en) * | 2014-03-27 | 2015-10-01 | Genentech, Inc. | Anti-influenza b virus hemagglutinin antibodies and methods of use |
| JP2017523805A (en) * | 2014-07-15 | 2017-08-24 | メディミューン,エルエルシー | Neutralization of anti-type B influenza antibody and use thereof |
| RU2739952C2 (en) * | 2014-07-15 | 2020-12-30 | МЕДИММЬЮН, ЭлЭлСи | Neutralizing antibodies to influenza virus b and ways of their application |
| RU2711932C2 (en) * | 2014-07-15 | 2020-01-23 | МЕДИММЬЮН, ЭлЭлСи | Neutralizing antibodies to influenza virus b and ways for application thereof |
| WO2016011035A2 (en) | 2014-07-15 | 2016-01-21 | Medlmmune, Llc | Neutralizing anti-influenza b antibodies and uses thereof |
| WO2016011035A3 (en) * | 2014-07-15 | 2016-03-10 | Medlmmune, Llc | Neutralizing anti-influenza b antibodies and uses thereof |
| US10294292B2 (en) | 2014-07-15 | 2019-05-21 | Medimmune, Llc | Neutralizing anti-influenza B antibodies and uses thereof |
| JP7453430B2 (en) | 2014-07-15 | 2024-03-19 | メディミューン,エルエルシー | Neutralization of anti-influenza B antibody and its use |
| US11787853B2 (en) | 2014-07-15 | 2023-10-17 | Medimmune, Llc | Neutralizing anti-influenza b antibodies and uses thereof |
| JP7209754B2 (en) | 2014-07-15 | 2023-01-20 | メディミューン,エルエルシー | Neutralizing anti-influenza B antibodies and uses thereof |
| CN113563462A (en) * | 2014-07-15 | 2021-10-29 | 免疫医疗有限责任公司 | Neutralizing anti-influenza b antibodies and uses thereof |
| US10519221B2 (en) | 2014-07-15 | 2019-12-31 | Medimmune, Llc | Neutralizing anti-influenza B antibodies and uses thereof |
| RU2784915C2 (en) * | 2014-07-15 | 2022-12-01 | МЕДИММЬЮН, ЭлЭлСи | Neutralizing antibodies to influenza b virus and their application methods |
| CN106573154A (en) * | 2014-07-15 | 2017-04-19 | 免疫医疗有限责任公司 | Neutralizing anti-influenza b antibodies and uses thereof |
| CN113173990A (en) * | 2014-07-15 | 2021-07-27 | 免疫医疗有限责任公司 | Neutralizing anti-influenza b antibodies and uses thereof |
| US11174304B2 (en) | 2014-07-15 | 2021-11-16 | Medimmune, Llc | Neutralizing anti-influenza B antibodies and uses thereof |
| JP2021074013A (en) * | 2014-07-15 | 2021-05-20 | メディミューン,エルエルシー | Neutralizing anti-influenza b antibodies and uses thereof |
| CN106573154B (en) * | 2014-07-15 | 2021-06-15 | 免疫医疗有限责任公司 | Neutralizing anti-influenza b antibodies and uses thereof |
| US11136379B2 (en) | 2015-02-05 | 2021-10-05 | Janssen Vaccines & Prevention B.V. | Binding molecules directed against influenza hemagglutinin and uses thereof |
| US11535665B2 (en) | 2015-05-13 | 2022-12-27 | The Trustees Of The University Of Pennsylvania | AAV-mediated expression of anti-influenza antibodies and methods of use thereof |
| US11524993B2 (en) | 2015-06-01 | 2022-12-13 | Medimmune, Llc | Neutralizing anti-influenza binding molecules and uses thereof |
| WO2016196470A1 (en) * | 2015-06-01 | 2016-12-08 | Medimmune, Llc | Neutralizing anti-influenza binding molecules and uses thereof |
| US10882897B2 (en) | 2015-06-01 | 2021-01-05 | Medimmune, Llc | Neutralizing anti-influenza binding molecules and uses thereof |
| JP2018518475A (en) * | 2015-06-01 | 2018-07-12 | メディミューン,エルエルシー | Neutralizing anti-influenza binding molecule and use thereof |
| US10442854B2 (en) | 2015-06-01 | 2019-10-15 | Medimmune, Llc | Neutralizing anti-influenza binding molecules and uses thereof |
| RU2721706C2 (en) * | 2015-06-01 | 2020-05-21 | МЕДИММЬЮН, ЭлЭлСи | Neutralizing binding molecules directed against influenza virus, and methods of their application |
| JP2021112193A (en) * | 2015-06-01 | 2021-08-05 | メディミューン,エルエルシー | Neutralizing anti-influenza binding molecules and uses thereof |
| EP3960761A1 (en) * | 2015-06-01 | 2022-03-02 | MedImmune, LLC | Neutralizing anti-influenza binding molecules and uses thereof |
| US11926657B2 (en) | 2015-06-01 | 2024-03-12 | Medimmune, Llc | Neutralizing anti-influenza binding molecules and uses thereof |
| AU2016270147B2 (en) * | 2015-06-03 | 2018-08-23 | Xiamen University | Broad-spectrum monoclonal anti-flu B antibody and uses thereof |
| US10696736B2 (en) | 2015-06-03 | 2020-06-30 | Xiamen University | Broad-spectrum monoclonal anti-flu B antibody and uses thereof |
| EP3305811A4 (en) * | 2015-06-03 | 2019-01-09 | Xiamen University | Broad-spectrum monoclonal anti-flu b antibody and uses thereof |
| JP2018524974A (en) * | 2015-06-03 | 2018-09-06 | シャアメン ユニバーシティ | Broad spectrum monoclonal anti-FluB antibodies and uses thereof |
| US10513553B2 (en) | 2015-11-13 | 2019-12-24 | Visterra, Inc. | Compositions and methods for treating and preventing influenza |
| US11547756B2 (en) | 2016-01-13 | 2023-01-10 | Medimmune, Llc | Method of treating influenza A |
| US10703803B2 (en) | 2016-03-01 | 2020-07-07 | Janssen Vaccines & Prevention B.V. | Human neutralizing antibodies binding to influenza B neuraminidase |
| WO2017148889A1 (en) | 2016-03-01 | 2017-09-08 | Janssen Vaccines & Prevention B.V. | Human neutralizing antibodies binding to influenza b neuraminidase |
| US11578341B2 (en) | 2017-02-28 | 2023-02-14 | The Trustees Of The University Of Pennsylvania | Compositions useful in treatment of spinal muscular atrophy |
| US11827906B2 (en) | 2017-02-28 | 2023-11-28 | The Trustees Of The University Of Pennsylvania | Adeno-associated virus (AAV) clade f vector and uses therefor |
| US10786568B2 (en) | 2017-02-28 | 2020-09-29 | The Trustees Of The University Of Pennsylvania | AAV mediated influenza vaccines |
| US11230593B2 (en) | 2019-03-25 | 2022-01-25 | Visterra, Inc. | Compositions and methods for treating and preventing influenza |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2018201647B2 (en) | Human binding molecules capable of binding to and neutralizing influenza B viruses and uses thereof | |
| JP5683752B2 (en) | Human binding molecule capable of neutralizing influenza A virus of strain group 1 and strain 2 and influenza B virus | |
| US10005831B2 (en) | Human binding molecules capable of neutralizing influenza virus H5N1 and uses thereof | |
| EP2430046B1 (en) | Human binding molecules capable of neutralizing influenza virus h3n2 and uses thereof | |
| EP2455399A2 (en) | Human binding molecules capable of neutralizing influenza virus H5N1 and uses thereof | |
| NZ729856B2 (en) | Human binding molecules capable of binding to and neutralizing influenza b viruses and uses thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13708411 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12014501776 Country of ref document: PH |
|
| ENP | Entry into the national phase |
Ref document number: 2865594 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2014560375 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013708411 Country of ref document: EP Ref document number: MX/A/2014/010755 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 20147025832 Country of ref document: KR Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2013229488 Country of ref document: AU Date of ref document: 20130307 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 201491656 Country of ref document: EA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: IDP00201406086 Country of ref document: ID |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112014021931 Country of ref document: BR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12015502553 Country of ref document: PH |
|
| ENP | Entry into the national phase |
Ref document number: 112014021931 Country of ref document: BR Kind code of ref document: A2 Effective date: 20140904 |