WO2013074386A2 - Hcv ns3 protease inhibitors - Google Patents
Hcv ns3 protease inhibitors Download PDFInfo
- Publication number
- WO2013074386A2 WO2013074386A2 PCT/US2012/064270 US2012064270W WO2013074386A2 WO 2013074386 A2 WO2013074386 A2 WO 2013074386A2 US 2012064270 W US2012064270 W US 2012064270W WO 2013074386 A2 WO2013074386 A2 WO 2013074386A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- optionally substituted
- methyl
- cyclopropyl
- carboxamide
- Prior art date
Links
- 229940124771 HCV-NS3 protease inhibitor Drugs 0.000 title description 5
- 208000015181 infectious disease Diseases 0.000 claims abstract description 34
- 101800001838 Serine protease/helicase NS3 Proteins 0.000 claims abstract description 22
- 239000003112 inhibitor Substances 0.000 claims abstract description 14
- 150000001875 compounds Chemical class 0.000 claims description 275
- -1 -(CH^-het! Chemical group 0.000 claims description 226
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 claims description 87
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 69
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 claims description 62
- 150000003839 salts Chemical class 0.000 claims description 55
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 46
- 229910052757 nitrogen Inorganic materials 0.000 claims description 32
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 31
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 26
- 239000003814 drug Substances 0.000 claims description 26
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 claims description 23
- 125000002572 propoxy group Chemical group [*]OC([H])([H])C(C([H])([H])[H])([H])[H] 0.000 claims description 23
- 125000001424 substituent group Chemical group 0.000 claims description 22
- 239000008194 pharmaceutical composition Substances 0.000 claims description 21
- 125000003118 aryl group Chemical group 0.000 claims description 20
- 125000000623 heterocyclic group Chemical group 0.000 claims description 20
- 125000000217 alkyl group Chemical group 0.000 claims description 18
- 229940124597 therapeutic agent Drugs 0.000 claims description 18
- 125000005842 heteroatom Chemical group 0.000 claims description 17
- 229910003827 NRaRb Inorganic materials 0.000 claims description 15
- 229920006395 saturated elastomer Polymers 0.000 claims description 15
- IWUCXVSUMQZMFG-AFCXAGJDSA-N Ribavirin Chemical compound N1=C(C(=O)N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 IWUCXVSUMQZMFG-AFCXAGJDSA-N 0.000 claims description 14
- 239000003443 antiviral agent Substances 0.000 claims description 14
- 229960000329 ribavirin Drugs 0.000 claims description 14
- 125000002950 monocyclic group Chemical group 0.000 claims description 13
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 13
- HZCAHMRRMINHDJ-DBRKOABJSA-N ribavirin Natural products O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1N=CN=C1 HZCAHMRRMINHDJ-DBRKOABJSA-N 0.000 claims description 13
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims description 12
- 125000006432 1-methyl cyclopropyl group Chemical group [H]C([H])([H])C1(*)C([H])([H])C1([H])[H] 0.000 claims description 12
- 229910052799 carbon Inorganic materials 0.000 claims description 12
- 230000002401 inhibitory effect Effects 0.000 claims description 12
- 229910052760 oxygen Inorganic materials 0.000 claims description 12
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 claims description 11
- 125000001153 fluoro group Chemical group F* 0.000 claims description 11
- 125000002619 bicyclic group Chemical group 0.000 claims description 10
- 125000001072 heteroaryl group Chemical group 0.000 claims description 10
- 229910052739 hydrogen Inorganic materials 0.000 claims description 10
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 claims description 10
- 229910052717 sulfur Inorganic materials 0.000 claims description 9
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 9
- 108010050904 Interferons Proteins 0.000 claims description 8
- 102000014150 Interferons Human genes 0.000 claims description 8
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 claims description 8
- 229940079322 interferon Drugs 0.000 claims description 8
- 125000006413 ring segment Chemical group 0.000 claims description 8
- 125000001412 tetrahydropyranyl group Chemical group 0.000 claims description 8
- 229940124772 HCV-NS5B polymerase inhibitor Drugs 0.000 claims description 7
- 229960005475 antiinfective agent Drugs 0.000 claims description 7
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 claims description 7
- 125000005843 halogen group Chemical group 0.000 claims description 7
- 239000002955 immunomodulating agent Substances 0.000 claims description 7
- 239000004599 antimicrobial Substances 0.000 claims description 6
- 230000000694 effects Effects 0.000 claims description 6
- 229940121354 immunomodulator Drugs 0.000 claims description 6
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 claims description 6
- 125000000335 thiazolyl group Chemical group 0.000 claims description 6
- 229940042399 direct acting antivirals protease inhibitors Drugs 0.000 claims description 5
- 125000002757 morpholinyl group Chemical group 0.000 claims description 5
- 125000001624 naphthyl group Chemical group 0.000 claims description 5
- 239000000137 peptide hydrolase inhibitor Substances 0.000 claims description 5
- 125000004076 pyridyl group Chemical group 0.000 claims description 5
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 claims description 4
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical group C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 claims description 4
- 239000005977 Ethylene Chemical group 0.000 claims description 4
- 229910019142 PO4 Inorganic materials 0.000 claims description 4
- 125000002393 azetidinyl group Chemical group 0.000 claims description 4
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 claims description 4
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 4
- 125000004210 cyclohexylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 claims description 4
- 125000004639 dihydroindenyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 claims description 4
- 239000003937 drug carrier Substances 0.000 claims description 4
- ZGEGCLOFRBLKSE-UHFFFAOYSA-N methylene hexane Natural products CCCCCC=C ZGEGCLOFRBLKSE-UHFFFAOYSA-N 0.000 claims description 4
- 239000010452 phosphate Substances 0.000 claims description 4
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 claims description 4
- 125000003386 piperidinyl group Chemical group 0.000 claims description 4
- 125000003367 polycyclic group Chemical group 0.000 claims description 4
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 4
- 229910052705 radium Inorganic materials 0.000 claims description 4
- 229910052701 rubidium Inorganic materials 0.000 claims description 4
- 125000006552 (C3-C8) cycloalkyl group Chemical group 0.000 claims description 3
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 claims description 3
- 125000001246 bromo group Chemical group Br* 0.000 claims description 3
- 125000000592 heterocycloalkyl group Chemical group 0.000 claims description 3
- 125000004193 piperazinyl group Chemical group 0.000 claims description 3
- 125000003226 pyrazolyl group Chemical group 0.000 claims description 3
- 125000000719 pyrrolidinyl group Chemical group 0.000 claims description 3
- 125000000168 pyrrolyl group Chemical group 0.000 claims description 3
- 125000001425 triazolyl group Chemical group 0.000 claims description 3
- HYZJCKYKOHLVJF-UHFFFAOYSA-N 1H-benzimidazole Chemical compound C1=CC=C2NC=NC2=C1 HYZJCKYKOHLVJF-UHFFFAOYSA-N 0.000 claims description 2
- UBEOVNYFKACFOT-UHFFFAOYSA-N 2-cyclononyloxazonane Chemical group C1CCCCCCCC1N1OCCCCCCC1 UBEOVNYFKACFOT-UHFFFAOYSA-N 0.000 claims description 2
- 101800001019 Non-structural protein 4B Proteins 0.000 claims description 2
- 101800001014 Non-structural protein 5A Proteins 0.000 claims description 2
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 claims description 2
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 claims description 2
- 125000005879 dioxolanyl group Chemical group 0.000 claims description 2
- 125000000555 isopropenyl group Chemical group [H]\C([H])=C(\*)C([H])([H])[H] 0.000 claims description 2
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 claims description 2
- 125000006574 non-aromatic ring group Chemical group 0.000 claims description 2
- 125000005961 oxazepanyl group Chemical group 0.000 claims description 2
- 241000711549 Hepacivirus C Species 0.000 abstract description 76
- 230000015572 biosynthetic process Effects 0.000 abstract description 65
- 238000003786 synthesis reaction Methods 0.000 abstract description 62
- 150000002678 macrocyclic compounds Chemical class 0.000 abstract description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 439
- 238000000034 method Methods 0.000 description 187
- 239000000047 product Substances 0.000 description 187
- 235000019439 ethyl acetate Nutrition 0.000 description 153
- 239000000243 solution Substances 0.000 description 152
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 150
- 239000000203 mixture Substances 0.000 description 149
- 239000000543 intermediate Substances 0.000 description 132
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 120
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 117
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 113
- 239000011541 reaction mixture Substances 0.000 description 108
- 238000006243 chemical reaction Methods 0.000 description 105
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 99
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 86
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 79
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 79
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 65
- 239000007787 solid Substances 0.000 description 58
- ZMANZCXQSJIPKH-UHFFFAOYSA-N N,N-Diethylethanamine Substances CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 54
- 238000000746 purification Methods 0.000 description 53
- HPQRQAOVNXWEEQ-UHFFFAOYSA-N quinoline-8-carboxamide Chemical compound C1=CN=C2C(C(=O)N)=CC=CC2=C1 HPQRQAOVNXWEEQ-UHFFFAOYSA-N 0.000 description 53
- 239000012267 brine Substances 0.000 description 51
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 51
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 49
- 235000019341 magnesium sulphate Nutrition 0.000 description 49
- 238000003818 flash chromatography Methods 0.000 description 48
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 44
- WYURNTSHIVDZCO-UHFFFAOYSA-N tetrahydrofuran Substances C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 41
- 239000002904 solvent Substances 0.000 description 38
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 35
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 33
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 30
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 29
- 239000011734 sodium Substances 0.000 description 29
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 27
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 27
- 229960000583 acetic acid Drugs 0.000 description 27
- 235000011054 acetic acid Nutrition 0.000 description 27
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 27
- 239000012044 organic layer Substances 0.000 description 26
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 26
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 24
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 23
- QRDZFPUVLYEQTA-UHFFFAOYSA-M quinoline-8-carboxylate Chemical compound C1=CN=C2C(C(=O)[O-])=CC=CC2=C1 QRDZFPUVLYEQTA-UHFFFAOYSA-M 0.000 description 22
- 238000011282 treatment Methods 0.000 description 22
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 22
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 21
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 21
- 229910052938 sodium sulfate Inorganic materials 0.000 description 21
- 235000011152 sodium sulphate Nutrition 0.000 description 21
- HEMHJVSKTPXQMS-UHFFFAOYSA-M sodium hydroxide Inorganic materials [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 20
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 19
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 19
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 19
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 19
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 18
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 18
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 18
- 238000001816 cooling Methods 0.000 description 18
- 239000010410 layer Substances 0.000 description 18
- 239000003921 oil Substances 0.000 description 18
- 239000000843 powder Substances 0.000 description 18
- 239000000741 silica gel Substances 0.000 description 18
- 229910002027 silica gel Inorganic materials 0.000 description 18
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 17
- 239000012043 crude product Substances 0.000 description 17
- 229910052700 potassium Inorganic materials 0.000 description 17
- 239000011591 potassium Substances 0.000 description 17
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 16
- 239000003153 chemical reaction reagent Substances 0.000 description 16
- 230000002829 reductive effect Effects 0.000 description 16
- 229940086542 triethylamine Drugs 0.000 description 16
- 239000002253 acid Substances 0.000 description 15
- NLKNQRATVPKPDG-UHFFFAOYSA-M potassium iodide Chemical compound [K+].[I-] NLKNQRATVPKPDG-UHFFFAOYSA-M 0.000 description 15
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 14
- 239000007821 HATU Substances 0.000 description 14
- 101710111275 Non-structural 3 Proteins 0.000 description 14
- 125000004432 carbon atom Chemical group C* 0.000 description 14
- 239000006260 foam Substances 0.000 description 13
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 12
- 239000012071 phase Substances 0.000 description 12
- 229910000343 potassium bisulfate Inorganic materials 0.000 description 12
- 238000004007 reversed phase HPLC Methods 0.000 description 12
- 238000003756 stirring Methods 0.000 description 12
- 208000024891 symptom Diseases 0.000 description 12
- 229960000549 4-dimethylaminophenol Drugs 0.000 description 11
- 229910000024 caesium carbonate Inorganic materials 0.000 description 11
- 230000002441 reversible effect Effects 0.000 description 11
- 239000012047 saturated solution Substances 0.000 description 11
- GQHTUMJGOHRCHB-UHFFFAOYSA-N 2,3,4,6,7,8,9,10-octahydropyrimido[1,2-a]azepine Chemical compound C1CCCCN2CCCN=C21 GQHTUMJGOHRCHB-UHFFFAOYSA-N 0.000 description 10
- 102000004190 Enzymes Human genes 0.000 description 10
- 108090000790 Enzymes Proteins 0.000 description 10
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 10
- 239000000758 substrate Substances 0.000 description 10
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 9
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 9
- 238000003556 assay Methods 0.000 description 9
- 125000004122 cyclic group Chemical group 0.000 description 9
- 239000007858 starting material Substances 0.000 description 9
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 8
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 8
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 8
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 8
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 8
- 150000003857 carboxamides Chemical class 0.000 description 8
- 239000000706 filtrate Substances 0.000 description 8
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 8
- 229910052737 gold Inorganic materials 0.000 description 8
- 239000010931 gold Substances 0.000 description 8
- 239000001257 hydrogen Substances 0.000 description 8
- 125000004482 piperidin-4-yl group Chemical group N1CCC(CC1)* 0.000 description 8
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 8
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 8
- 235000017557 sodium bicarbonate Nutrition 0.000 description 8
- 230000000840 anti-viral effect Effects 0.000 description 7
- 239000012298 atmosphere Substances 0.000 description 7
- 239000003795 chemical substances by application Substances 0.000 description 7
- 238000001914 filtration Methods 0.000 description 7
- 239000012458 free base Substances 0.000 description 7
- 239000004615 ingredient Substances 0.000 description 7
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 7
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 description 7
- 229910000029 sodium carbonate Inorganic materials 0.000 description 7
- KZNICNPSHKQLFF-UHFFFAOYSA-N succinimide Chemical compound O=C1CCC(=O)N1 KZNICNPSHKQLFF-UHFFFAOYSA-N 0.000 description 7
- BAUWRHPMUVYFOD-UHFFFAOYSA-N 1-methylpiperidin-4-ol Chemical compound CN1CCC(O)CC1 BAUWRHPMUVYFOD-UHFFFAOYSA-N 0.000 description 6
- YGYGASJNJTYNOL-CQSZACIVSA-N 3-[(4r)-2,2-dimethyl-1,1-dioxothian-4-yl]-5-(4-fluorophenyl)-1h-indole-7-carboxamide Chemical compound C1CS(=O)(=O)C(C)(C)C[C@@H]1C1=CNC2=C(C(N)=O)C=C(C=3C=CC(F)=CC=3)C=C12 YGYGASJNJTYNOL-CQSZACIVSA-N 0.000 description 6
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 6
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 6
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 6
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 6
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 6
- 108091005804 Peptidases Proteins 0.000 description 6
- 239000004365 Protease Substances 0.000 description 6
- 102100037486 Reverse transcriptase/ribonuclease H Human genes 0.000 description 6
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 6
- 125000002947 alkylene group Chemical group 0.000 description 6
- 150000001412 amines Chemical class 0.000 description 6
- 125000003865 brosyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1Br)S(*)(=O)=O 0.000 description 6
- 239000000872 buffer Substances 0.000 description 6
- WMSPXQIQBQAWLL-UHFFFAOYSA-N cyclopropanesulfonamide Chemical compound NS(=O)(=O)C1CC1 WMSPXQIQBQAWLL-UHFFFAOYSA-N 0.000 description 6
- 230000008034 disappearance Effects 0.000 description 6
- 230000005764 inhibitory process Effects 0.000 description 6
- 239000002777 nucleoside Substances 0.000 description 6
- 108090000765 processed proteins & peptides Proteins 0.000 description 6
- 238000010898 silica gel chromatography Methods 0.000 description 6
- 238000010626 work up procedure Methods 0.000 description 6
- IWUCXVSUMQZMFG-RGDLXGNYSA-N 1-[(2s,3s,4r,5s)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]-1,2,4-triazole-3-carboxamide Chemical compound N1=C(C(=O)N)N=CN1[C@@H]1[C@@H](O)[C@@H](O)[C@H](CO)O1 IWUCXVSUMQZMFG-RGDLXGNYSA-N 0.000 description 5
- CVMXEDZZSWLXPB-UHFFFAOYSA-N 4-(2-bromoethyl)morpholine Chemical compound BrCCN1CCOCC1 CVMXEDZZSWLXPB-UHFFFAOYSA-N 0.000 description 5
- HDHQZCHIXUUSMK-UHFFFAOYSA-N 4-hydroxy-2-quinolone Chemical compound C1=CC=C2C(O)=CC(=O)NC2=C1 HDHQZCHIXUUSMK-UHFFFAOYSA-N 0.000 description 5
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 5
- 125000004429 atom Chemical group 0.000 description 5
- 150000002148 esters Chemical class 0.000 description 5
- 238000004128 high performance liquid chromatography Methods 0.000 description 5
- 230000007062 hydrolysis Effects 0.000 description 5
- 238000006460 hydrolysis reaction Methods 0.000 description 5
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 5
- 239000012074 organic phase Substances 0.000 description 5
- 238000002360 preparation method Methods 0.000 description 5
- 238000010791 quenching Methods 0.000 description 5
- LISFMEBWQUVKPJ-UHFFFAOYSA-N quinolin-2-ol Chemical compound C1=CC=C2NC(=O)C=CC2=C1 LISFMEBWQUVKPJ-UHFFFAOYSA-N 0.000 description 5
- 239000002002 slurry Substances 0.000 description 5
- 239000000725 suspension Substances 0.000 description 5
- NQRYJNQNLNOLGT-UHFFFAOYSA-N tetrahydropyridine hydrochloride Natural products C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 5
- 238000004809 thin layer chromatography Methods 0.000 description 5
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 4
- XXMFJKNOJSDQBM-UHFFFAOYSA-N 2,2,2-trifluoroacetic acid;hydrate Chemical compound [OH3+].[O-]C(=O)C(F)(F)F XXMFJKNOJSDQBM-UHFFFAOYSA-N 0.000 description 4
- RKATWUBDSJHPEV-UHFFFAOYSA-N 3,3-difluorocyclobutan-1-amine Chemical compound NC1CC(F)(F)C1 RKATWUBDSJHPEV-UHFFFAOYSA-N 0.000 description 4
- KCBWAFJCKVKYHO-UHFFFAOYSA-N 6-(4-cyclopropyl-6-methoxypyrimidin-5-yl)-1-[[4-[1-propan-2-yl-4-(trifluoromethyl)imidazol-2-yl]phenyl]methyl]pyrazolo[3,4-d]pyrimidine Chemical compound C1(CC1)C1=NC=NC(=C1C1=NC=C2C(=N1)N(N=C2)CC1=CC=C(C=C1)C=1N(C=C(N=1)C(F)(F)F)C(C)C)OC KCBWAFJCKVKYHO-UHFFFAOYSA-N 0.000 description 4
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 4
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 4
- BDAGIHXWWSANSR-UHFFFAOYSA-N Formic acid Chemical compound OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 4
- 238000006751 Mitsunobu reaction Methods 0.000 description 4
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 4
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 4
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 4
- 230000009471 action Effects 0.000 description 4
- 239000004480 active ingredient Substances 0.000 description 4
- 230000029936 alkylation Effects 0.000 description 4
- 238000005804 alkylation reaction Methods 0.000 description 4
- 235000019270 ammonium chloride Nutrition 0.000 description 4
- 229910052786 argon Inorganic materials 0.000 description 4
- HYGWNUKOUCZBND-UHFFFAOYSA-N azanide Chemical compound [NH2-] HYGWNUKOUCZBND-UHFFFAOYSA-N 0.000 description 4
- 230000037396 body weight Effects 0.000 description 4
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 4
- 239000003054 catalyst Substances 0.000 description 4
- DGLFSNZWRYADFC-UHFFFAOYSA-N chembl2334586 Chemical compound C1CCC2=CN=C(N)N=C2C2=C1NC1=CC=C(C#CC(C)(O)C)C=C12 DGLFSNZWRYADFC-UHFFFAOYSA-N 0.000 description 4
- 239000000460 chlorine Substances 0.000 description 4
- 125000000753 cycloalkyl group Chemical group 0.000 description 4
- 201000010099 disease Diseases 0.000 description 4
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 4
- 229940079593 drug Drugs 0.000 description 4
- PQVSTLUFSYVLTO-UHFFFAOYSA-N ethyl n-ethoxycarbonylcarbamate Chemical compound CCOC(=O)NC(=O)OCC PQVSTLUFSYVLTO-UHFFFAOYSA-N 0.000 description 4
- 239000000284 extract Substances 0.000 description 4
- 235000019253 formic acid Nutrition 0.000 description 4
- 229940040692 lithium hydroxide monohydrate Drugs 0.000 description 4
- GLXDVVHUTZTUQK-UHFFFAOYSA-M lithium hydroxide monohydrate Substances [Li+].O.[OH-] GLXDVVHUTZTUQK-UHFFFAOYSA-M 0.000 description 4
- 150000003833 nucleoside derivatives Chemical class 0.000 description 4
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 4
- 125000004159 quinolin-2-yl group Chemical group [H]C1=C([H])C([H])=C2C([H])=C([H])C(*)=NC2=C1[H] 0.000 description 4
- QRDZFPUVLYEQTA-UHFFFAOYSA-N quinoline-8-carboxylic acid Chemical compound C1=CN=C2C(C(=O)O)=CC=CC2=C1 QRDZFPUVLYEQTA-UHFFFAOYSA-N 0.000 description 4
- 238000000926 separation method Methods 0.000 description 4
- 239000011780 sodium chloride Substances 0.000 description 4
- JQWHASGSAFIOCM-UHFFFAOYSA-M sodium periodate Chemical compound [Na+].[O-]I(=O)(=O)=O JQWHASGSAFIOCM-UHFFFAOYSA-M 0.000 description 4
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 4
- YWWDBCBWQNCYNR-UHFFFAOYSA-N trimethylphosphine Chemical compound CP(C)C YWWDBCBWQNCYNR-UHFFFAOYSA-N 0.000 description 4
- DOMQFIFVDIAOOT-ROUUACIJSA-N (2S,3R)-N-[4-(2,6-dimethoxyphenyl)-5-(5-methylpyridin-3-yl)-1,2,4-triazol-3-yl]-3-(5-methylpyrimidin-2-yl)butane-2-sulfonamide Chemical compound COC1=C(C(=CC=C1)OC)N1C(=NN=C1C=1C=NC=C(C=1)C)NS(=O)(=O)[C@@H](C)[C@H](C)C1=NC=C(C=N1)C DOMQFIFVDIAOOT-ROUUACIJSA-N 0.000 description 3
- RTMMSCJWQYWMNK-UHFFFAOYSA-N 2,2,2-trifluoroethyl trifluoromethanesulfonate Chemical compound FC(F)(F)COS(=O)(=O)C(F)(F)F RTMMSCJWQYWMNK-UHFFFAOYSA-N 0.000 description 3
- OISVCGZHLKNMSJ-UHFFFAOYSA-N 2,6-dimethylpyridine Chemical compound CC1=CC=CC(C)=N1 OISVCGZHLKNMSJ-UHFFFAOYSA-N 0.000 description 3
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 3
- XQJMXPAEFMWDOZ-UHFFFAOYSA-N 3exo-benzoyloxy-tropane Natural products CN1C(C2)CCC1CC2OC(=O)C1=CC=CC=C1 XQJMXPAEFMWDOZ-UHFFFAOYSA-N 0.000 description 3
- XYWIPYBIIRTJMM-IBGZPJMESA-N 4-[[(2S)-2-[4-[5-chloro-2-[4-(trifluoromethyl)triazol-1-yl]phenyl]-5-methoxy-2-oxopyridin-1-yl]butanoyl]amino]-2-fluorobenzamide Chemical compound CC[C@H](N1C=C(OC)C(=CC1=O)C1=C(C=CC(Cl)=C1)N1C=C(N=N1)C(F)(F)F)C(=O)NC1=CC(F)=C(C=C1)C(N)=O XYWIPYBIIRTJMM-IBGZPJMESA-N 0.000 description 3
- WPHKPXFQHKEVBK-UHFFFAOYSA-N 6-bromo-4-phenylmethoxy-3-prop-2-enyl-1h-quinolin-2-one Chemical compound C12=CC(Br)=CC=C2NC(=O)C(CC=C)=C1OCC1=CC=CC=C1 WPHKPXFQHKEVBK-UHFFFAOYSA-N 0.000 description 3
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 3
- YVHAIVPPUIZFBA-UHFFFAOYSA-N Cyclopentylacetic acid Chemical compound OC(=O)CC1CCCC1 YVHAIVPPUIZFBA-UHFFFAOYSA-N 0.000 description 3
- QMMFVYPAHWMCMS-UHFFFAOYSA-N Dimethyl sulfide Chemical compound CSC QMMFVYPAHWMCMS-UHFFFAOYSA-N 0.000 description 3
- 239000007995 HEPES buffer Substances 0.000 description 3
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 3
- 108010047761 Interferon-alpha Proteins 0.000 description 3
- 102000006992 Interferon-alpha Human genes 0.000 description 3
- QQXLDOJGLXJCSE-UHFFFAOYSA-N N-methylnortropinone Natural products C1C(=O)CC2CCC1N2C QQXLDOJGLXJCSE-UHFFFAOYSA-N 0.000 description 3
- 101800001020 Non-structural protein 4A Proteins 0.000 description 3
- 229920002594 Polyethylene Glycol 8000 Polymers 0.000 description 3
- QIZDQFOVGFDBKW-DHBOJHSNSA-N Pseudotropine Natural products OC1C[C@@H]2[N+](C)[C@H](C1)CC2 QIZDQFOVGFDBKW-DHBOJHSNSA-N 0.000 description 3
- 102000012479 Serine Proteases Human genes 0.000 description 3
- 108010022999 Serine Proteases Proteins 0.000 description 3
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 3
- RBYGDVHOECIAFC-UHFFFAOYSA-L acetonitrile;palladium(2+);dichloride Chemical compound [Cl-].[Cl-].[Pd+2].CC#N.CC#N RBYGDVHOECIAFC-UHFFFAOYSA-L 0.000 description 3
- 150000001298 alcohols Chemical class 0.000 description 3
- 239000007864 aqueous solution Substances 0.000 description 3
- 239000012131 assay buffer Substances 0.000 description 3
- 125000000051 benzyloxy group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])O* 0.000 description 3
- 125000002837 carbocyclic group Chemical group 0.000 description 3
- 125000000131 cyclopropyloxy group Chemical group C1(CC1)O* 0.000 description 3
- 230000005284 excitation Effects 0.000 description 3
- 101150045736 fliK gene Proteins 0.000 description 3
- 101150005992 fliP gene Proteins 0.000 description 3
- 229910052736 halogen Inorganic materials 0.000 description 3
- 150000002367 halogens Chemical class 0.000 description 3
- 238000010438 heat treatment Methods 0.000 description 3
- 150000004677 hydrates Chemical class 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 230000000155 isotopic effect Effects 0.000 description 3
- 230000001404 mediated effect Effects 0.000 description 3
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 3
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 3
- 229910000027 potassium carbonate Inorganic materials 0.000 description 3
- 239000000651 prodrug Substances 0.000 description 3
- 229940002612 prodrug Drugs 0.000 description 3
- 238000010926 purge Methods 0.000 description 3
- 230000010076 replication Effects 0.000 description 3
- MHOVAHRLVXNVSD-UHFFFAOYSA-N rhodium atom Chemical compound [Rh] MHOVAHRLVXNVSD-UHFFFAOYSA-N 0.000 description 3
- 238000006798 ring closing metathesis reaction Methods 0.000 description 3
- 229910052708 sodium Inorganic materials 0.000 description 3
- 238000006467 substitution reaction Methods 0.000 description 3
- NHKZSTHOYNWEEZ-AFCXAGJDSA-N taribavirin Chemical compound N1=C(C(=N)N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 NHKZSTHOYNWEEZ-AFCXAGJDSA-N 0.000 description 3
- 229950006081 taribavirin Drugs 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- 230000001225 therapeutic effect Effects 0.000 description 3
- CYHOMWAPJJPNMW-JIGDXULJSA-N tropine Chemical compound C1[C@@H](O)C[C@H]2CC[C@@H]1N2C CYHOMWAPJJPNMW-JIGDXULJSA-N 0.000 description 3
- DYLIWHYUXAJDOJ-OWOJBTEDSA-N (e)-4-(6-aminopurin-9-yl)but-2-en-1-ol Chemical compound NC1=NC=NC2=C1N=CN2C\C=C\CO DYLIWHYUXAJDOJ-OWOJBTEDSA-N 0.000 description 2
- 0 **(*)=Cc1ccccc1 Chemical compound **(*)=Cc1ccccc1 0.000 description 2
- VEFLKXRACNJHOV-UHFFFAOYSA-N 1,3-dibromopropane Chemical compound BrCCCBr VEFLKXRACNJHOV-UHFFFAOYSA-N 0.000 description 2
- AZQWKYJCGOJGHM-UHFFFAOYSA-N 1,4-benzoquinone Chemical compound O=C1C=CC(=O)C=C1 AZQWKYJCGOJGHM-UHFFFAOYSA-N 0.000 description 2
- PVOAHINGSUIXLS-UHFFFAOYSA-N 1-Methylpiperazine Chemical compound CN1CCNCC1 PVOAHINGSUIXLS-UHFFFAOYSA-N 0.000 description 2
- YZUPZGFPHUVJKC-UHFFFAOYSA-N 1-bromo-2-methoxyethane Chemical compound COCCBr YZUPZGFPHUVJKC-UHFFFAOYSA-N 0.000 description 2
- ATJVVVCODTXRAE-UHFFFAOYSA-N 1-methylcyclopropane-1-sulfonamide Chemical compound NS(=O)(=O)C1(C)CC1 ATJVVVCODTXRAE-UHFFFAOYSA-N 0.000 description 2
- LTMRRSWNXVJMBA-UHFFFAOYSA-L 2,2-diethylpropanedioate Chemical compound CCC(CC)(C([O-])=O)C([O-])=O LTMRRSWNXVJMBA-UHFFFAOYSA-L 0.000 description 2
- IPOVOSHRRIJKBR-UHFFFAOYSA-N 2-ethylpropanedioyl dichloride Chemical compound CCC(C(Cl)=O)C(Cl)=O IPOVOSHRRIJKBR-UHFFFAOYSA-N 0.000 description 2
- HTLZEVDBZFQNCB-UHFFFAOYSA-N 3-bromo-8-fluoro-4-phenylmethoxy-1h-quinolin-2-one Chemical compound BrC=1C(=O)NC=2C(F)=CC=CC=2C=1OCC1=CC=CC=C1 HTLZEVDBZFQNCB-UHFFFAOYSA-N 0.000 description 2
- 125000004575 3-pyrrolidinyl group Chemical group [H]N1C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- MSHFRERJPWKJFX-UHFFFAOYSA-N 4-Methoxybenzyl alcohol Chemical compound COC1=CC=C(CO)C=C1 MSHFRERJPWKJFX-UHFFFAOYSA-N 0.000 description 2
- PNGPMNXTYUTTTK-UHFFFAOYSA-N 4-[[4-[3-benzoyl-8-(trifluoromethyl)quinolin-4-yl]phenoxy]methyl]benzoic acid Chemical compound C1=CC(C(=O)O)=CC=C1COC1=CC=C(C=2C3=CC=CC(=C3N=CC=2C(=O)C=2C=CC=CC=2)C(F)(F)F)C=C1 PNGPMNXTYUTTTK-UHFFFAOYSA-N 0.000 description 2
- 125000004800 4-bromophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1Br 0.000 description 2
- RBJWVYAOEWLLGM-UHFFFAOYSA-N 4-hydroxy-3-prop-2-enyl-1h-quinolin-2-one Chemical compound C1=CC=C2C(=O)C(CC=C)=C(O)NC2=C1 RBJWVYAOEWLLGM-UHFFFAOYSA-N 0.000 description 2
- 125000004217 4-methoxybenzyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1OC([H])([H])[H])C([H])([H])* 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 2
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 2
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 2
- HTJDQJBWANPRPF-UHFFFAOYSA-N Cyclopropylamine Chemical compound NC1CC1 HTJDQJBWANPRPF-UHFFFAOYSA-N 0.000 description 2
- YZCKVEUIGOORGS-OUBTZVSYSA-N Deuterium Chemical compound [2H] YZCKVEUIGOORGS-OUBTZVSYSA-N 0.000 description 2
- YXHKONLOYHBTNS-UHFFFAOYSA-N Diazomethane Chemical compound C=[N+]=[N-] YXHKONLOYHBTNS-UHFFFAOYSA-N 0.000 description 2
- BWLUMTFWVZZZND-UHFFFAOYSA-N Dibenzylamine Chemical compound C=1C=CC=CC=1CNCC1=CC=CC=C1 BWLUMTFWVZZZND-UHFFFAOYSA-N 0.000 description 2
- 206010016654 Fibrosis Diseases 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- 241000725303 Human immunodeficiency virus Species 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 2
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- 241001465754 Metazoa Species 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 2
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 2
- FEYNFHSRETUBEM-UHFFFAOYSA-N N-[3-(1,1-difluoroethyl)phenyl]-1-(4-methoxyphenyl)-3-methyl-5-oxo-4H-pyrazole-4-carboxamide Chemical compound COc1ccc(cc1)N1N=C(C)C(C(=O)Nc2cccc(c2)C(C)(F)F)C1=O FEYNFHSRETUBEM-UHFFFAOYSA-N 0.000 description 2
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 2
- YZCKVEUIGOORGS-IGMARMGPSA-N Protium Chemical compound [1H] YZCKVEUIGOORGS-IGMARMGPSA-N 0.000 description 2
- 241000720974 Protium Species 0.000 description 2
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 2
- KJTLSVCANCCWHF-UHFFFAOYSA-N Ruthenium Chemical compound [Ru] KJTLSVCANCCWHF-UHFFFAOYSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- TVRCRTJYMVTEFS-ICGCPXGVSA-N [(2r,3r,4r,5r)-5-(4-amino-2-oxopyrimidin-1-yl)-4-hydroxy-2-(hydroxymethyl)-4-methyloxolan-3-yl] (2s)-2-amino-3-methylbutanoate Chemical compound C[C@@]1(O)[C@H](OC(=O)[C@@H](N)C(C)C)[C@@H](CO)O[C@H]1N1C(=O)N=C(N)C=C1 TVRCRTJYMVTEFS-ICGCPXGVSA-N 0.000 description 2
- 239000013543 active substance Substances 0.000 description 2
- 150000001336 alkenes Chemical class 0.000 description 2
- 150000001345 alkine derivatives Chemical class 0.000 description 2
- 125000003545 alkoxy group Chemical group 0.000 description 2
- DKNWSYNQZKUICI-UHFFFAOYSA-N amantadine Chemical compound C1C(C2)CC3CC2CC1(N)C3 DKNWSYNQZKUICI-UHFFFAOYSA-N 0.000 description 2
- 229940024606 amino acid Drugs 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 229940077388 benzenesulfonate Drugs 0.000 description 2
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 235000019445 benzyl alcohol Nutrition 0.000 description 2
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 2
- JHXKRIRFYBPWGE-UHFFFAOYSA-K bismuth chloride Chemical compound Cl[Bi](Cl)Cl JHXKRIRFYBPWGE-UHFFFAOYSA-K 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- UORVGPXVDQYIDP-UHFFFAOYSA-N borane Chemical compound B UORVGPXVDQYIDP-UHFFFAOYSA-N 0.000 description 2
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- 229910052794 bromium Inorganic materials 0.000 description 2
- IYYIVELXUANFED-UHFFFAOYSA-N bromo(trimethyl)silane Chemical compound C[Si](C)(C)Br IYYIVELXUANFED-UHFFFAOYSA-N 0.000 description 2
- AEILLAXRDHDKDY-UHFFFAOYSA-N bromomethylcyclopropane Chemical compound BrCC1CC1 AEILLAXRDHDKDY-UHFFFAOYSA-N 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 230000003197 catalytic effect Effects 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- 125000001309 chloro group Chemical group Cl* 0.000 description 2
- 238000004587 chromatography analysis Methods 0.000 description 2
- 230000007882 cirrhosis Effects 0.000 description 2
- 208000019425 cirrhosis of liver Diseases 0.000 description 2
- 238000004440 column chromatography Methods 0.000 description 2
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 2
- 238000005859 coupling reaction Methods 0.000 description 2
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- HGCIXCUEYOPUTN-UHFFFAOYSA-N cyclohexene Chemical compound C1CCC=CC1 HGCIXCUEYOPUTN-UHFFFAOYSA-N 0.000 description 2
- WXQFLZZSZCDWCV-UHFFFAOYSA-N cyclopropanol Chemical compound OC1[CH]C1 WXQFLZZSZCDWCV-UHFFFAOYSA-N 0.000 description 2
- 230000002939 deleterious effect Effects 0.000 description 2
- 229910052805 deuterium Inorganic materials 0.000 description 2
- JMVOCSLPMGHXPG-UHFFFAOYSA-N dipotassium;dioxido(dioxo)osmium Chemical compound [K+].[K+].[O-][Os]([O-])(=O)=O JMVOCSLPMGHXPG-UHFFFAOYSA-N 0.000 description 2
- FJKIXWOMBXYWOQ-UHFFFAOYSA-N ethenoxyethane Chemical compound CCOC=C FJKIXWOMBXYWOQ-UHFFFAOYSA-N 0.000 description 2
- MPHRJJDVELSFQN-UHFFFAOYSA-N ethyl 3-(4-bromoanilino)-3-oxopropanoate Chemical compound CCOC(=O)CC(=O)NC1=CC=C(Br)C=C1 MPHRJJDVELSFQN-UHFFFAOYSA-N 0.000 description 2
- OAYLNYINCPYISS-UHFFFAOYSA-N ethyl acetate;hexane Chemical compound CCCCCC.CCOC(C)=O OAYLNYINCPYISS-UHFFFAOYSA-N 0.000 description 2
- 239000012065 filter cake Substances 0.000 description 2
- BDHPJXPOQGPUNI-UHFFFAOYSA-N formic acid;morpholine Chemical compound [O-]C=O.C1COCC[NH2+]1 BDHPJXPOQGPUNI-UHFFFAOYSA-N 0.000 description 2
- 230000004927 fusion Effects 0.000 description 2
- 238000007429 general method Methods 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- UYTPUPDQBNUYGX-UHFFFAOYSA-N guanine Chemical class O=C1NC(N)=NC2=C1N=CN2 UYTPUPDQBNUYGX-UHFFFAOYSA-N 0.000 description 2
- 230000036541 health Effects 0.000 description 2
- 150000002431 hydrogen Chemical class 0.000 description 2
- 230000003834 intracellular effect Effects 0.000 description 2
- NNPPMTNAJDCUHE-UHFFFAOYSA-N isobutane Chemical compound CC(C)C NNPPMTNAJDCUHE-UHFFFAOYSA-N 0.000 description 2
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- FMASTMURQSHELY-UHFFFAOYSA-N n-(4-fluoro-2-methylphenyl)-3-methyl-n-[(2-methyl-1h-indol-4-yl)methyl]pyridine-4-carboxamide Chemical compound C1=CC=C2NC(C)=CC2=C1CN(C=1C(=CC(F)=CC=1)C)C(=O)C1=CC=NC=C1C FMASTMURQSHELY-UHFFFAOYSA-N 0.000 description 2
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- NNKPHNTWNILINE-UHFFFAOYSA-N n-cyclopropyl-3-fluoro-4-methyl-5-[3-[[1-[2-[2-(methylamino)ethoxy]phenyl]cyclopropyl]amino]-2-oxopyrazin-1-yl]benzamide Chemical compound CNCCOC1=CC=CC=C1C1(NC=2C(N(C=3C(=C(F)C=C(C=3)C(=O)NC3CC3)C)C=CN=2)=O)CC1 NNKPHNTWNILINE-UHFFFAOYSA-N 0.000 description 2
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 2
- 229920000137 polyphosphoric acid Polymers 0.000 description 2
- IUBQJLUDMLPAGT-UHFFFAOYSA-N potassium bis(trimethylsilyl)amide Chemical compound C[Si](C)(C)N([K])[Si](C)(C)C IUBQJLUDMLPAGT-UHFFFAOYSA-N 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- VVWRJUBEIPHGQF-MDZDMXLPSA-N propan-2-yl (ne)-n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)\N=N\C(=O)OC(C)C VVWRJUBEIPHGQF-MDZDMXLPSA-N 0.000 description 2
- QLULGIRFKAWHOJ-UHFFFAOYSA-N pyridin-4-ylboronic acid Chemical compound OB(O)C1=CC=NC=C1 QLULGIRFKAWHOJ-UHFFFAOYSA-N 0.000 description 2
- 230000000171 quenching effect Effects 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- 230000004044 response Effects 0.000 description 2
- 239000002342 ribonucleoside Substances 0.000 description 2
- 229910052707 ruthenium Inorganic materials 0.000 description 2
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 239000000126 substance Substances 0.000 description 2
- 239000003826 tablet Substances 0.000 description 2
- GFLBASJQJLQUMN-ULKQDVFKSA-N tert-butyl (1s,5r)-9-hydroxy-3-oxa-7-azabicyclo[3.3.1]nonane-7-carboxylate Chemical compound C1OC[C@]2([H])CN(C(=O)OC(C)(C)C)C[C@@]1([H])C2O GFLBASJQJLQUMN-ULKQDVFKSA-N 0.000 description 2
- XRNLYXKYODGLMI-UHFFFAOYSA-N tert-butyl 3-fluoro-4-hydroxypiperidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCC(O)C(F)C1 XRNLYXKYODGLMI-UHFFFAOYSA-N 0.000 description 2
- SEGZJJSZYOEABC-UHFFFAOYSA-N tert-butyl 3-hydroxy-8-azabicyclo[3.2.1]octane-8-carboxylate Chemical compound C1C(O)CC2CCC1N2C(=O)OC(C)(C)C SEGZJJSZYOEABC-UHFFFAOYSA-N 0.000 description 2
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 2
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 2
- DLYUQMMRRRQYAE-UHFFFAOYSA-N tetraphosphorus decaoxide Chemical compound O1P(O2)(=O)OP3(=O)OP1(=O)OP2(=O)O3 DLYUQMMRRRQYAE-UHFFFAOYSA-N 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- 125000001544 thienyl group Chemical group 0.000 description 2
- GYUURHMITDQTRU-UHFFFAOYSA-N tributyl(pyridin-2-yl)stannane Chemical compound CCCC[Sn](CCCC)(CCCC)C1=CC=CC=N1 GYUURHMITDQTRU-UHFFFAOYSA-N 0.000 description 2
- WJKHJLXJJJATHN-UHFFFAOYSA-N triflic anhydride Chemical compound FC(F)(F)S(=O)(=O)OS(=O)(=O)C(F)(F)F WJKHJLXJJJATHN-UHFFFAOYSA-N 0.000 description 2
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 2
- GPRLSGONYQIRFK-MNYXATJNSA-N triton Chemical compound [3H+] GPRLSGONYQIRFK-MNYXATJNSA-N 0.000 description 2
- 229960005486 vaccine Drugs 0.000 description 2
- 230000003612 virological effect Effects 0.000 description 2
- 239000011991 zhan catalyst Substances 0.000 description 2
- NLHROWOWTHQMEZ-HTQZYQBOSA-N (1r,2r)-2-pent-4-ynylcyclopropan-1-ol Chemical compound O[C@@H]1C[C@H]1CCCC#C NLHROWOWTHQMEZ-HTQZYQBOSA-N 0.000 description 1
- UJCIPQBSAOEKMR-MCIONIFRSA-N (2S)-2-(oxan-4-yl)-2-[[(1R,2R)-2-pent-4-ynylcyclopropyl]oxycarbonylamino]acetic acid Chemical compound N([C@H](C(=O)O)C1CCOCC1)C(=O)O[C@@H]1C[C@H]1CCCC#C UJCIPQBSAOEKMR-MCIONIFRSA-N 0.000 description 1
- HSWLXFMSIVDBBD-MPGHIAIKSA-N (2S)-2-[[(1S,2R)-2-but-1-enyl-1-methylcyclopentyl]oxycarbonylamino]-2-cyclopentylacetic acid Chemical compound C(=CCC)[C@@H]1[C@@](CCC1)(C)OC(=O)N[C@H](C(=O)O)C1CCCC1 HSWLXFMSIVDBBD-MPGHIAIKSA-N 0.000 description 1
- PLAQGKKUVFMRFR-SNVBAGLBSA-N (2s)-2-(1-methylcyclohexyl)-2-[(2-methylpropan-2-yl)oxycarbonylamino]acetic acid Chemical compound CC(C)(C)OC(=O)N[C@H](C(O)=O)C1(C)CCCCC1 PLAQGKKUVFMRFR-SNVBAGLBSA-N 0.000 description 1
- ZVCMWNFQYIQWSY-NSHDSACASA-N (2s)-2-[(2-methylpropan-2-yl)oxycarbonylamino]non-8-enoic acid Chemical compound CC(C)(C)OC(=O)N[C@H](C(O)=O)CCCCCC=C ZVCMWNFQYIQWSY-NSHDSACASA-N 0.000 description 1
- TZXKCMDOTUMCCR-DVOMOZLQSA-N (2s)-2-[[(1r,2r)-2-but-3-enyl-1-methylcyclopropyl]oxycarbonylamino]-2-cyclopentylacetic acid Chemical compound N([C@@H](C1CCCC1)C(O)=O)C(=O)O[C@]1(C)C[C@H]1CCC=C TZXKCMDOTUMCCR-DVOMOZLQSA-N 0.000 description 1
- WZSYGMIWHIONBI-PPHPATTJSA-N (2s)-2-amino-2-(2,3-dihydro-1h-inden-2-yl)acetic acid;hydrochloride Chemical compound Cl.C1=CC=C2CC([C@H](N)C(O)=O)CC2=C1 WZSYGMIWHIONBI-PPHPATTJSA-N 0.000 description 1
- NPDBDJFLKKQMCM-SCSAIBSYSA-N (2s)-2-amino-3,3-dimethylbutanoic acid Chemical compound CC(C)(C)[C@H](N)C(O)=O NPDBDJFLKKQMCM-SCSAIBSYSA-N 0.000 description 1
- IVWWFWFVSWOTLP-YVZVNANGSA-N (3'as,4r,7'as)-2,2,2',2'-tetramethylspiro[1,3-dioxolane-4,6'-4,7a-dihydro-3ah-[1,3]dioxolo[4,5-c]pyran]-7'-one Chemical compound C([C@@H]1OC(O[C@@H]1C1=O)(C)C)O[C@]21COC(C)(C)O2 IVWWFWFVSWOTLP-YVZVNANGSA-N 0.000 description 1
- KNTZCGBYFGEMFR-UHFFFAOYSA-N (propan-2-ylazaniumyl)formate Chemical compound CC(C)NC(O)=O KNTZCGBYFGEMFR-UHFFFAOYSA-N 0.000 description 1
- 125000004277 1,3-dioxalan-2-yl group Chemical group [H]C1([H])OC([H])(*)OC1([H])[H] 0.000 description 1
- DQQXBXSKTVBANA-UHFFFAOYSA-N 1,3-dioxepane-2,4,7-trione Chemical compound O=C1CCC(=O)OC(=O)O1 DQQXBXSKTVBANA-UHFFFAOYSA-N 0.000 description 1
- 229940005561 1,4-benzoquinone Drugs 0.000 description 1
- PRBHEGAFLDMLAL-UHFFFAOYSA-N 1,5-Hexadiene Natural products CC=CCC=C PRBHEGAFLDMLAL-UHFFFAOYSA-N 0.000 description 1
- BIAAQBNMRITRDV-UHFFFAOYSA-N 1-(chloromethoxy)-2-methoxyethane Chemical compound COCCOCCl BIAAQBNMRITRDV-UHFFFAOYSA-N 0.000 description 1
- NBKORJKMMVZAOZ-VPCXQMTMSA-N 1-[(2r,3r,4r,5r)-3,4-dihydroxy-5-(hydroxymethyl)-3-methyloxolan-2-yl]pyrimidine-2,4-dione Chemical compound C[C@@]1(O)[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)NC(=O)C=C1 NBKORJKMMVZAOZ-VPCXQMTMSA-N 0.000 description 1
- MDMZMLPPFIHZMD-UHFFFAOYSA-N 1-ethenylcyclopropane-1-carboxamide;hydrochloride Chemical compound Cl.NC(=O)C1(C=C)CC1 MDMZMLPPFIHZMD-UHFFFAOYSA-N 0.000 description 1
- 125000004484 1-methylpiperidin-4-yl group Chemical group CN1CCC(CC1)* 0.000 description 1
- WUIJTQZXUURFQU-UHFFFAOYSA-N 1-methylsulfonylethene Chemical compound CS(=O)(=O)C=C WUIJTQZXUURFQU-UHFFFAOYSA-N 0.000 description 1
- BUMDBYZTJUBAIE-UHFFFAOYSA-N 19-cyclohexyl-9-[2-(dimethylamino)ethyl]-5-methoxy-10-oxo-9,12-diazatetracyclo[10.7.0.02,7.013,18]nonadeca-1(19),2(7),3,5,13(18),14,16-heptaene-15-carboxylic acid Chemical compound C12=CC=C(C(O)=O)C=C2N2CC(=O)N(CCN(C)C)CC3=CC(OC)=CC=C3C2=C1C1CCCCC1 BUMDBYZTJUBAIE-UHFFFAOYSA-N 0.000 description 1
- SKDWJRKCMYHPMY-UHFFFAOYSA-N 19-cyclohexyl-9-[3-(dimethylamino)propyl]-10-oxo-9,12-diazatetracyclo[10.7.0.02,7.013,18]nonadeca-1(19),2,4,6,13(18),14,16-heptaene-15-carboxylic acid Chemical compound C12=CC=C(C(O)=O)C=C2N2CC(=O)N(CCCN(C)C)CC3=CC=CC=C3C2=C1C1CCCCC1 SKDWJRKCMYHPMY-UHFFFAOYSA-N 0.000 description 1
- DNCYBUMDUBHIJZ-UHFFFAOYSA-N 1h-pyrimidin-6-one Chemical compound O=C1C=CN=CN1 DNCYBUMDUBHIJZ-UHFFFAOYSA-N 0.000 description 1
- SXGZJKUKBWWHRA-UHFFFAOYSA-N 2-(N-morpholiniumyl)ethanesulfonate Chemical compound [O-]S(=O)(=O)CC[NH+]1CCOCC1 SXGZJKUKBWWHRA-UHFFFAOYSA-N 0.000 description 1
- KZTWONRVIPPDKH-UHFFFAOYSA-N 2-(piperidin-1-yl)ethanol Chemical compound OCCN1CCCCC1 KZTWONRVIPPDKH-UHFFFAOYSA-N 0.000 description 1
- TXHAHOVNFDVCCC-UHFFFAOYSA-N 2-(tert-butylazaniumyl)acetate Chemical compound CC(C)(C)NCC(O)=O TXHAHOVNFDVCCC-UHFFFAOYSA-N 0.000 description 1
- XNNJMXBWUMKLRR-UHFFFAOYSA-N 2-(trifluoromethoxy)ethyl trifluoromethanesulfonate Chemical compound FC(F)(F)OCCOS(=O)(=O)C(F)(F)F XNNJMXBWUMKLRR-UHFFFAOYSA-N 0.000 description 1
- NQTLZJODEOHALT-UHFFFAOYSA-N 2-amino-4-(trifluoromethyl)benzoic acid Chemical compound NC1=CC(C(F)(F)F)=CC=C1C(O)=O NQTLZJODEOHALT-UHFFFAOYSA-N 0.000 description 1
- LGPVTNAJFDUWLF-UHFFFAOYSA-N 2-amino-4-fluorobenzoic acid Chemical compound NC1=CC(F)=CC=C1C(O)=O LGPVTNAJFDUWLF-UHFFFAOYSA-N 0.000 description 1
- RPGKFFKUTVJVPY-UHFFFAOYSA-N 2-amino-4-methylbenzoic acid Chemical compound CC1=CC=C(C(O)=O)C(N)=C1 RPGKFFKUTVJVPY-UHFFFAOYSA-N 0.000 description 1
- NVKAMPJSWMHVDK-GITKWUPZSA-N 2-amino-9-[(2r,3r,4r,5r)-3,4-dihydroxy-5-(hydroxymethyl)-3-methyloxolan-2-yl]-3h-purin-6-one Chemical compound C[C@@]1(O)[C@H](O)[C@@H](CO)O[C@H]1N1C2=NC(N)=NC(O)=C2N=C1 NVKAMPJSWMHVDK-GITKWUPZSA-N 0.000 description 1
- JBKINHFZTVLNEM-UHFFFAOYSA-N 2-bromoethoxy-tert-butyl-dimethylsilane Chemical compound CC(C)(C)[Si](C)(C)OCCBr JBKINHFZTVLNEM-UHFFFAOYSA-N 0.000 description 1
- FTZQXOJYPFINKJ-UHFFFAOYSA-N 2-fluoroaniline Chemical compound NC1=CC=CC=C1F FTZQXOJYPFINKJ-UHFFFAOYSA-N 0.000 description 1
- XDOFKUHHLNYRPM-UHFFFAOYSA-N 2-hydroxypiperidine-1-carboxylic acid Chemical compound OC1CCCCN1C(O)=O XDOFKUHHLNYRPM-UHFFFAOYSA-N 0.000 description 1
- GSLTVFIVJMCNBH-UHFFFAOYSA-N 2-isocyanatopropane Chemical compound CC(C)N=C=O GSLTVFIVJMCNBH-UHFFFAOYSA-N 0.000 description 1
- 125000004200 2-methoxyethyl group Chemical group [H]C([H])([H])OC([H])([H])C([H])([H])* 0.000 description 1
- XWKFPIODWVPXLX-UHFFFAOYSA-N 2-methyl-5-methylpyridine Natural products CC1=CC=C(C)N=C1 XWKFPIODWVPXLX-UHFFFAOYSA-N 0.000 description 1
- JTNCEQNHURODLX-UHFFFAOYSA-N 2-phenylethanimidamide Chemical compound NC(=N)CC1=CC=CC=C1 JTNCEQNHURODLX-UHFFFAOYSA-N 0.000 description 1
- 125000004105 2-pyridyl group Chemical group N1=C([*])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- MBQIAXJKVVWCEP-UHFFFAOYSA-N 2h-1,8-naphthyridine-1-carboxylic acid Chemical compound C1=CN=C2N(C(=O)O)CC=CC2=C1 MBQIAXJKVVWCEP-UHFFFAOYSA-N 0.000 description 1
- CDBAEFXTCRKJPZ-UHFFFAOYSA-N 3,3-difluoroazetidine;hydron;chloride Chemical compound Cl.FC1(F)CNC1 CDBAEFXTCRKJPZ-UHFFFAOYSA-N 0.000 description 1
- LEHHIPIDKQVNEV-UHFFFAOYSA-N 3,3-difluoropiperidine;hydrochloride Chemical compound Cl.FC1(F)CCCNC1 LEHHIPIDKQVNEV-UHFFFAOYSA-N 0.000 description 1
- GNZBXECGDWQFIZ-UHFFFAOYSA-N 3-(3,3-difluoropiperidin-1-yl)propan-1-ol Chemical compound OCCCN1CCCC(F)(F)C1 GNZBXECGDWQFIZ-UHFFFAOYSA-N 0.000 description 1
- FUEPSWDWSHCZHJ-UHFFFAOYSA-N 3-(azetidin-1-yl)propan-1-ol Chemical compound OCCCN1CCC1 FUEPSWDWSHCZHJ-UHFFFAOYSA-N 0.000 description 1
- PYSGFFTXMUWEOT-UHFFFAOYSA-N 3-(dimethylamino)propan-1-ol Chemical compound CN(C)CCCO PYSGFFTXMUWEOT-UHFFFAOYSA-N 0.000 description 1
- YYFLDZZDOUDZQM-UHFFFAOYSA-N 3-[1-[[4-(3-phenylquinolin-2-yl)phenyl]methyl]piperidin-4-yl]-1h-benzimidazol-2-one Chemical compound O=C1NC2=CC=CC=C2N1C(CC1)CCN1CC(C=C1)=CC=C1C1=NC2=CC=CC=C2C=C1C1=CC=CC=C1 YYFLDZZDOUDZQM-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- JJVRAMNFIQYYMP-UHFFFAOYSA-N 3-bromo-4-hydroxy-1h-quinolin-2-one Chemical compound C1=CC=C2C(=O)C(Br)=C(O)NC2=C1 JJVRAMNFIQYYMP-UHFFFAOYSA-N 0.000 description 1
- UPXOJNKIBHPPBZ-UHFFFAOYSA-N 3-bromo-4-hydroxy-7-(trifluoromethyl)-1h-quinolin-2-one Chemical compound FC(F)(F)C1=CC=C2C(O)=C(Br)C(=O)NC2=C1 UPXOJNKIBHPPBZ-UHFFFAOYSA-N 0.000 description 1
- BOZPFJAKJQPFGS-UHFFFAOYSA-N 3-bromo-4-phenylmethoxy-1h-quinolin-2-one Chemical compound C12=CC=CC=C2NC(=O)C(Br)=C1OCC1=CC=CC=C1 BOZPFJAKJQPFGS-UHFFFAOYSA-N 0.000 description 1
- OFMBZMPBPPCUKD-UHFFFAOYSA-N 3-bromo-4-phenylmethoxy-7-(trifluoromethyl)-1h-quinolin-2-one Chemical compound BrC=1C(=O)NC2=CC(C(F)(F)F)=CC=C2C=1OCC1=CC=CC=C1 OFMBZMPBPPCUKD-UHFFFAOYSA-N 0.000 description 1
- YZYHLIYVVZFYPI-UHFFFAOYSA-N 3-bromo-8-fluoro-4-hydroxy-1h-quinolin-2-one Chemical compound C1=CC=C2C(O)=C(Br)C(=O)NC2=C1F YZYHLIYVVZFYPI-UHFFFAOYSA-N 0.000 description 1
- QGMROEZDWJTIDW-UHFFFAOYSA-N 3-bromopropoxy-tert-butyl-dimethylsilane Chemical compound CC(C)(C)[Si](C)(C)OCCCBr QGMROEZDWJTIDW-UHFFFAOYSA-N 0.000 description 1
- PNPCRKVUWYDDST-UHFFFAOYSA-N 3-chloroaniline Chemical compound NC1=CC=CC(Cl)=C1 PNPCRKVUWYDDST-UHFFFAOYSA-N 0.000 description 1
- PXFUWRWCKSLCLS-UHFFFAOYSA-N 3-fluoroazetidine;hydron;chloride Chemical compound Cl.FC1CNC1 PXFUWRWCKSLCLS-UHFFFAOYSA-N 0.000 description 1
- 125000001137 3-hydroxypropoxy group Chemical group [H]OC([H])([H])C([H])([H])C([H])([H])O* 0.000 description 1
- FREZLSIGWNCSOQ-UHFFFAOYSA-N 3-methylbutanoyl 3-methylbutanoate Chemical compound CC(C)CC(=O)OC(=O)CC(C)C FREZLSIGWNCSOQ-UHFFFAOYSA-N 0.000 description 1
- XCCNRBCNYGWTQX-UHFFFAOYSA-N 3-propan-2-ylaniline Chemical compound CC(C)C1=CC=CC(N)=C1 XCCNRBCNYGWTQX-UHFFFAOYSA-N 0.000 description 1
- LZPWAYBEOJRFAX-UHFFFAOYSA-N 4,4,5,5-tetramethyl-1,3,2$l^{2}-dioxaborolane Chemical compound CC1(C)O[B]OC1(C)C LZPWAYBEOJRFAX-UHFFFAOYSA-N 0.000 description 1
- ROLMZTIHUMKEAI-UHFFFAOYSA-N 4,5-difluoro-2-hydroxybenzonitrile Chemical compound OC1=CC(F)=C(F)C=C1C#N ROLMZTIHUMKEAI-UHFFFAOYSA-N 0.000 description 1
- VJPPLCNBDLZIFG-ZDUSSCGKSA-N 4-[(3S)-3-(but-2-ynoylamino)piperidin-1-yl]-5-fluoro-2,3-dimethyl-1H-indole-7-carboxamide Chemical compound C(C#CC)(=O)N[C@@H]1CN(CCC1)C1=C2C(=C(NC2=C(C=C1F)C(=O)N)C)C VJPPLCNBDLZIFG-ZDUSSCGKSA-N 0.000 description 1
- PPUDLEUZKVJXSZ-VPCXQMTMSA-N 4-amino-1-[(2r,3r,4r,5r)-3,4-dihydroxy-5-(hydroxymethyl)-3-methyloxolan-2-yl]pyrimidin-2-one Chemical compound C[C@@]1(O)[C@H](O)[C@@H](CO)O[C@H]1N1C(=O)N=C(N)C=C1 PPUDLEUZKVJXSZ-VPCXQMTMSA-N 0.000 description 1
- ODLGMSQBFONGNG-JVZYCSMKSA-N 4-amino-1-[(2r,3r,4s,5r)-5-azido-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]pyrimidin-2-one Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@H](O)[C@H](O)[C@](CO)(N=[N+]=[N-])O1 ODLGMSQBFONGNG-JVZYCSMKSA-N 0.000 description 1
- WDFQBORIUYODSI-UHFFFAOYSA-N 4-bromoaniline Chemical compound NC1=CC=C(Br)C=C1 WDFQBORIUYODSI-UHFFFAOYSA-N 0.000 description 1
- PXACTUVBBMDKRW-UHFFFAOYSA-M 4-bromobenzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=C(Br)C=C1 PXACTUVBBMDKRW-UHFFFAOYSA-M 0.000 description 1
- FISQRWQWLJBJJA-UHFFFAOYSA-N 4-hydroxy-7-(trifluoromethyl)-1h-quinolin-2-one Chemical compound FC(F)(F)C1=CC=C2C(O)=CC(=O)NC2=C1 FISQRWQWLJBJJA-UHFFFAOYSA-N 0.000 description 1
- GBHMCXPZLBHGNB-UHFFFAOYSA-N 5-chloro-4-hydroxy-1h-quinolin-2-one Chemical compound ClC1=CC=CC2=NC(O)=CC(O)=C21 GBHMCXPZLBHGNB-UHFFFAOYSA-N 0.000 description 1
- WTDWVLJJJOTABN-UHFFFAOYSA-N 5-cyclopropyl-2-(4-fluorophenyl)-6-[(2-hydroxyethyl)(methylsulfonyl)amino]-n-methyl-1-benzofuran-3-carboxamide Chemical compound C1=C2C(C(=O)NC)=C(C=3C=CC(F)=CC=3)OC2=CC(N(CCO)S(C)(=O)=O)=C1C1CC1 WTDWVLJJJOTABN-UHFFFAOYSA-N 0.000 description 1
- DQFPMEMMMGZBKU-UHFFFAOYSA-N 6-bromo-4-hydroxy-1h-quinolin-2-one Chemical compound C1=C(Br)C=CC2=NC(O)=CC(O)=C21 DQFPMEMMMGZBKU-UHFFFAOYSA-N 0.000 description 1
- OISCAKBSPQHUJG-UHFFFAOYSA-N 6-bromo-4-hydroxy-3-prop-2-enyl-1h-quinolin-2-one Chemical compound C1=C(Br)C=C2C(O)=C(CC=C)C(=O)NC2=C1 OISCAKBSPQHUJG-UHFFFAOYSA-N 0.000 description 1
- GWOWMINWCTWMII-UHFFFAOYSA-N 7-chloro-4-hydroxy-3-prop-2-enyl-1h-quinolin-2-one Chemical compound ClC1=CC=C2C(O)=C(CC=C)C(O)=NC2=C1 GWOWMINWCTWMII-UHFFFAOYSA-N 0.000 description 1
- SCAYTRRWZZPYRH-UHFFFAOYSA-N 7-chloro-4-methoxy-3-prop-2-enyl-1h-quinolin-2-one Chemical compound ClC1=CC=C2C(OC)=C(CC=C)C(O)=NC2=C1 SCAYTRRWZZPYRH-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- CFCWRLAINATSJN-UHFFFAOYSA-N 8-fluoro-4-hydroxy-1h-quinolin-2-one Chemical compound C1=CC=C2C(O)=CC(=O)NC2=C1F CFCWRLAINATSJN-UHFFFAOYSA-N 0.000 description 1
- 208000032484 Accidental exposure to product Diseases 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- 241001120493 Arene Species 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- FERIUCNNQQJTOY-UHFFFAOYSA-M Butyrate Chemical compound CCCC([O-])=O FERIUCNNQQJTOY-UHFFFAOYSA-M 0.000 description 1
- SAZQUIHSXDHCTF-KFWWJZLASA-N C#CCCC[C@H](C1)[C@@H]1OC(N[C@@H](C1CCCCC1)C(O)=O)=O Chemical compound C#CCCC[C@H](C1)[C@@H]1OC(N[C@@H](C1CCCCC1)C(O)=O)=O SAZQUIHSXDHCTF-KFWWJZLASA-N 0.000 description 1
- HGOKOMDQELXENI-NXEZZACHSA-N C#CCCC[C@H](C1)[C@@H]1OC(ON(C(CC1)=O)C1=O)=O Chemical compound C#CCCC[C@H](C1)[C@@H]1OC(ON(C(CC1)=O)C1=O)=O HGOKOMDQELXENI-NXEZZACHSA-N 0.000 description 1
- DIOAMZYNRGSCTE-UHFFFAOYSA-N C=CCc(c(O)nc1c2cccc1)c2OCc1ccccc1 Chemical compound C=CCc(c(O)nc1c2cccc1)c2OCc1ccccc1 DIOAMZYNRGSCTE-UHFFFAOYSA-N 0.000 description 1
- ZPMIEURRUNXTEK-ACIUFKGASA-N C=C[C@H](C1)[C@]1(C(NS(C1CC1)(=O)=O)=O)NC([C@H](C[C@H](C1)Oc2nc(cccc3)c3c(OCCCOP(O)(O)=O)c2C/C=C/CC[C@H](CCC2)[C@@H]2OC(N[C@H]2C3CCCC3)=O)N1C2=O)=O Chemical compound C=C[C@H](C1)[C@]1(C(NS(C1CC1)(=O)=O)=O)NC([C@H](C[C@H](C1)Oc2nc(cccc3)c3c(OCCCOP(O)(O)=O)c2C/C=C/CC[C@H](CCC2)[C@@H]2OC(N[C@H]2C3CCCC3)=O)N1C2=O)=O ZPMIEURRUNXTEK-ACIUFKGASA-N 0.000 description 1
- PMVDGZGMJSLCBC-NLFVJICLSA-N CC(C)(C)CC(N(C[C@@H](C1)Oc2nc3ccccc3c(OC3CCN(CC4CC4)CC3)c2CCCCC[C@H](C2)[C@@H]2OC(N)=O)[C@@H]1C(NC(C1)([C@@H]1C=C)C(NS(C1(C)CC1)(=O)=O)=O)=O)=O Chemical compound CC(C)(C)CC(N(C[C@@H](C1)Oc2nc3ccccc3c(OC3CCN(CC4CC4)CC3)c2CCCCC[C@H](C2)[C@@H]2OC(N)=O)[C@@H]1C(NC(C1)([C@@H]1C=C)C(NS(C1(C)CC1)(=O)=O)=O)=O)=O PMVDGZGMJSLCBC-NLFVJICLSA-N 0.000 description 1
- VLJMOXZQGJFGNW-FMWMJCRMSA-N CC(C)(C)[C@@H](C(N(CCC1)[C@@H]1C(NC(C1)(C1C=C)C(NS(C1(C)CC1)(=O)=O)=O)=O)=O)NC(O)=O Chemical compound CC(C)(C)[C@@H](C(N(CCC1)[C@@H]1C(NC(C1)(C1C=C)C(NS(C1(C)CC1)(=O)=O)=O)=O)=O)NC(O)=O VLJMOXZQGJFGNW-FMWMJCRMSA-N 0.000 description 1
- YJUZHOYAWRCAIR-DEJNBMECSA-N CC(C)(C)[C@@H](C(N(CCC1)[C@@H]1C(NC([C@H](C=C)I)C(NS(C1(C)CC1)(=O)=O)=O)=O)=O)NC(OC1[C@H](CCCCCc(c(O)nc2c3cccc2)c3OCCN2CCOCC2)C1)=O Chemical compound CC(C)(C)[C@@H](C(N(CCC1)[C@@H]1C(NC([C@H](C=C)I)C(NS(C1(C)CC1)(=O)=O)=O)=O)=O)NC(OC1[C@H](CCCCCc(c(O)nc2c3cccc2)c3OCCN2CCOCC2)C1)=O YJUZHOYAWRCAIR-DEJNBMECSA-N 0.000 description 1
- MOPHDEJQIJHKDN-HJFHDIQHSA-N CC(C)(C)[C@@H](C(N(CCC1)[C@@H]1C(O)=O)=O)NC(OC1[C@H](CCCCCc(c(O)nc2c3cccc2OC)c3OCCCN2CCCC2)C1)=O Chemical compound CC(C)(C)[C@@H](C(N(CCC1)[C@@H]1C(O)=O)=O)NC(OC1[C@H](CCCCCc(c(O)nc2c3cccc2OC)c3OCCCN2CCCC2)C1)=O MOPHDEJQIJHKDN-HJFHDIQHSA-N 0.000 description 1
- KSNBBKWXXQZRLU-GCZQVXSOSA-N CC(C)(C)[C@@H](C(N(CCC1)[C@@H]1C(OC)=O)=O)NC(OC1[C@H](CCCCCc(c(O)nc2c3cccc2)c3O)CCC1)=O Chemical compound CC(C)(C)[C@@H](C(N(CCC1)[C@@H]1C(OC)=O)=O)NC(OC1[C@H](CCCCCc(c(O)nc2c3cccc2)c3O)CCC1)=O KSNBBKWXXQZRLU-GCZQVXSOSA-N 0.000 description 1
- UMWMNMKIDZHAPK-IGTSPJGSSA-N CC(C)(C)[C@@H](C(N(C[C@@H](C1)Oc2nc(cccc3)c3c(OC3CCN(CC(F)(F)F)CC3)c2CCCCC[C@H](C2)[C@@H]2O2)[C@@H]1C(OC)=O)=O)NC2=O Chemical compound CC(C)(C)[C@@H](C(N(C[C@@H](C1)Oc2nc(cccc3)c3c(OC3CCN(CC(F)(F)F)CC3)c2CCCCC[C@H](C2)[C@@H]2O2)[C@@H]1C(OC)=O)=O)NC2=O UMWMNMKIDZHAPK-IGTSPJGSSA-N 0.000 description 1
- YQLSCXYGTZKHEB-QQNOCINOSA-N CC(C)(C)[C@@H](C(N(C[C@@H](C1)Oc2nc(cccc3)c3c(OC3CCN(CCOC)CC3)c2CCCCC[C@H](C2)[C@@H]2O2)[C@@H]1C(NC(C1)([C@@H]1C=C)C(NS(C1(C)CC1)(=O)=O)=O)=O)=O)NC2=O Chemical compound CC(C)(C)[C@@H](C(N(C[C@@H](C1)Oc2nc(cccc3)c3c(OC3CCN(CCOC)CC3)c2CCCCC[C@H](C2)[C@@H]2O2)[C@@H]1C(NC(C1)([C@@H]1C=C)C(NS(C1(C)CC1)(=O)=O)=O)=O)=O)NC2=O YQLSCXYGTZKHEB-QQNOCINOSA-N 0.000 description 1
- BUXDUKBMKMEOHQ-VYXMSSRQSA-N CC(C)(C)[C@@H](C(N(C[C@@H](C1)Oc2nc3ccccc3c(OCCCO)c2C/C=C/CC[C@H](CCC2)[C@@H]2O2)[C@@H]1C(N[C@](C1)([C@@H]1C=C)C(NS(C1CC1)(=O)=O)=O)=O)=O)NC2=O Chemical compound CC(C)(C)[C@@H](C(N(C[C@@H](C1)Oc2nc3ccccc3c(OCCCO)c2C/C=C/CC[C@H](CCC2)[C@@H]2O2)[C@@H]1C(N[C@](C1)([C@@H]1C=C)C(NS(C1CC1)(=O)=O)=O)=O)=O)NC2=O BUXDUKBMKMEOHQ-VYXMSSRQSA-N 0.000 description 1
- ZUENMVHFAKPVHZ-JNBOGDBDSA-N CC(C)C[C@@H](C1)[C@@]1(C)OC(N[C@@H](C1CCCC1)C(N(C[C@@H](C1)OC(C)=N)[C@@H]1C(NC([C@H](C=C)C#C)C(NS(C1CC1)(=O)=O)=O)=O)=O)=O Chemical compound CC(C)C[C@@H](C1)[C@@]1(C)OC(N[C@@H](C1CCCC1)C(N(C[C@@H](C1)OC(C)=N)[C@@H]1C(NC([C@H](C=C)C#C)C(NS(C1CC1)(=O)=O)=O)=O)=O)=O ZUENMVHFAKPVHZ-JNBOGDBDSA-N 0.000 description 1
- YEGGOOOZRKVFDN-UHFFFAOYSA-N CC(C)c(cc1NC2=O)ccc1C(OCc1ccccc1)=C2Br Chemical compound CC(C)c(cc1NC2=O)ccc1C(OCc1ccccc1)=C2Br YEGGOOOZRKVFDN-UHFFFAOYSA-N 0.000 description 1
- IJKIBLDLZGIAGJ-UHFFFAOYSA-N CC(NC(c1ccncc11)=O)=C1O Chemical compound CC(NC(c1ccncc11)=O)=C1O IJKIBLDLZGIAGJ-UHFFFAOYSA-N 0.000 description 1
- BPFADORYCUPTPX-RBXDHEIQSA-N CC1(CC1)S(NC(C(C1)([C@@H]1C=C)NC([C@H](C[C@H](C1)Oc2nc(cccc3)c3c(OCCNCCO)c2CCCCC[C@H](C2)[C@@H]2OC(N)=O)N1C(CC1CCCCC1)=O)=O)=O)(=O)=O Chemical compound CC1(CC1)S(NC(C(C1)([C@@H]1C=C)NC([C@H](C[C@H](C1)Oc2nc(cccc3)c3c(OCCNCCO)c2CCCCC[C@H](C2)[C@@H]2OC(N)=O)N1C(CC1CCCCC1)=O)=O)=O)(=O)=O BPFADORYCUPTPX-RBXDHEIQSA-N 0.000 description 1
- PSKJXIRGPCRLTE-NIJPHNKFSA-N CC1(CC1)S(NC(C([C@H](/C=C\CCCCC[C@@H](C(N(C1)[C@H]2C[C@H]1Oc1nc3ccccc3c(OCCCN3CCCCC3)c1/C=C/CCCC1CC1)=O)NC(O)=O)I)NC2=O)=O)(=O)=O Chemical compound CC1(CC1)S(NC(C([C@H](/C=C\CCCCC[C@@H](C(N(C1)[C@H]2C[C@H]1Oc1nc3ccccc3c(OCCCN3CCCCC3)c1/C=C/CCCC1CC1)=O)NC(O)=O)I)NC2=O)=O)(=O)=O PSKJXIRGPCRLTE-NIJPHNKFSA-N 0.000 description 1
- HMYXLPVVDQYNIC-OXLDAUETSA-N CC1(CC1)S(NC(C1(NC([C@H](C[C@H](C2)O)N2C([C@H](C2CCCCC2)NC(O[C@H]2[C@H](CN(C)c(cccc3)c3C(O[C@@H](CCNC3)[C@@H]3F)=C)C2)=O)=O)=O)[IH]C1C=C)=O)(=O)=O Chemical compound CC1(CC1)S(NC(C1(NC([C@H](C[C@H](C2)O)N2C([C@H](C2CCCCC2)NC(O[C@H]2[C@H](CN(C)c(cccc3)c3C(O[C@@H](CCNC3)[C@@H]3F)=C)C2)=O)=O)=O)[IH]C1C=C)=O)(=O)=O HMYXLPVVDQYNIC-OXLDAUETSA-N 0.000 description 1
- ORRMSVXAIPBKCG-MUAKATCZSA-N CC1(CC1)S(NC(C1(NC([C@H](C[C@H](C2)Oc3nc(cccc4)c4c(O[C@@H](CCN(C)C4)[C@@H]4F)c3CCCCC[C@H](C3)[C@@H]3OC(N[C@H]3C4CCCCC4)=O)N2C3=O)=O)[IH]C1C=C)=O)(=O)=O Chemical compound CC1(CC1)S(NC(C1(NC([C@H](C[C@H](C2)Oc3nc(cccc4)c4c(O[C@@H](CCN(C)C4)[C@@H]4F)c3CCCCC[C@H](C3)[C@@H]3OC(N[C@H]3C4CCCCC4)=O)N2C3=O)=O)[IH]C1C=C)=O)(=O)=O ORRMSVXAIPBKCG-MUAKATCZSA-N 0.000 description 1
- DLMNRMMMRHQZQO-RIMPRFFBSA-N CC1(CC1)S(NC([C@@](C1)(C1C=C)NC([C@H](C[C@H](C1)Oc2nc3ccccc3c(OC3CC(CC4)NC4C3)c2CCCCC[C@H](C2)[C@@H]2OC(N[C@H]2C3CCCCC3)=O)N1C2=O)=O)=O)(=O)=O Chemical compound CC1(CC1)S(NC([C@@](C1)(C1C=C)NC([C@H](C[C@H](C1)Oc2nc3ccccc3c(OC3CC(CC4)NC4C3)c2CCCCC[C@H](C2)[C@@H]2OC(N[C@H]2C3CCCCC3)=O)N1C2=O)=O)=O)(=O)=O DLMNRMMMRHQZQO-RIMPRFFBSA-N 0.000 description 1
- ZRYUHBLNLDKLSC-SOPQNWEZSA-N CC1(CC1)S(NC([C@@](C1)([C@@H]1/C=C\CCCCC[C@@H](C(N(C1)[C@H]2C[C@H]1Oc1nc3ccccc3c(OC)c1CCCCCC1CC1)=O)NC(O)=O)NC2=O)=O)(=O)=O Chemical compound CC1(CC1)S(NC([C@@](C1)([C@@H]1/C=C\CCCCC[C@@H](C(N(C1)[C@H]2C[C@H]1Oc1nc3ccccc3c(OC)c1CCCCCC1CC1)=O)NC(O)=O)NC2=O)=O)(=O)=O ZRYUHBLNLDKLSC-SOPQNWEZSA-N 0.000 description 1
- CPOWGZKMDWSYAR-FGBMWSQPSA-N CC1(CC1)S(NC([C@]1(C#C[C@H]1/C=C\CCCCC[C@@H](C(N1[C@H]2CCC1)=O)NC(O)=O)NC2=O)=O)(=O)=O Chemical compound CC1(CC1)S(NC([C@]1(C#C[C@H]1/C=C\CCCCC[C@@H](C(N1[C@H]2CCC1)=O)NC(O)=O)NC2=O)=O)(=O)=O CPOWGZKMDWSYAR-FGBMWSQPSA-N 0.000 description 1
- AHYMSCXHMTYMSR-WZENJKSDSA-N CCOC(C(C(C=C)C=C)NC([C@H](C[C@@H](C1)OS(c(cc2)ccc2Br)(=O)=O)N1C(OC(C)(C)C)=O)=O)=O Chemical compound CCOC(C(C(C=C)C=C)NC([C@H](C[C@@H](C1)OS(c(cc2)ccc2Br)(=O)=O)N1C(OC(C)(C)C)=O)=O)=O AHYMSCXHMTYMSR-WZENJKSDSA-N 0.000 description 1
- VPFALUKSCUROAR-ZMDQLVOHSA-N CCOC([C@@](C1)(C1/C=C\CCCCC[C@@H](C(N(C1)[C@H]2C[C@H]1Oc1nc3ccccc3c(OCCN3CCCCC3)c1/C=C/CCCC1CC1)=O)NC(O)=O)NC2=O)=O Chemical compound CCOC([C@@](C1)(C1/C=C\CCCCC[C@@H](C(N(C1)[C@H]2C[C@H]1Oc1nc3ccccc3c(OCCN3CCCCC3)c1/C=C/CCCC1CC1)=O)NC(O)=O)NC2=O)=O VPFALUKSCUROAR-ZMDQLVOHSA-N 0.000 description 1
- ZLDKLJAOLWOGAE-UZLBHIALSA-N CCOc1c(CC=C)c(O[C@H](C[C@H]2C(OC)=O)CN2C(OC(C)(C)C)=O)nc2ccccc12 Chemical compound CCOc1c(CC=C)c(O[C@H](C[C@H]2C(OC)=O)CN2C(OC(C)(C)C)=O)nc2ccccc12 ZLDKLJAOLWOGAE-UZLBHIALSA-N 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 229910021591 Copper(I) chloride Inorganic materials 0.000 description 1
- HMFHBZSHGGEWLO-SOOFDHNKSA-N D-ribofuranose Chemical compound OC[C@H]1OC(O)[C@H](O)[C@@H]1O HMFHBZSHGGEWLO-SOOFDHNKSA-N 0.000 description 1
- 101710088194 Dehydrogenase Proteins 0.000 description 1
- PEIKTSJIUKYDPC-UHFFFAOYSA-N Diethyl 3-Bromopropylphosphonate Chemical compound CCOP(=O)(OCC)CCCBr PEIKTSJIUKYDPC-UHFFFAOYSA-N 0.000 description 1
- 244000166102 Eucalyptus leucoxylon Species 0.000 description 1
- 235000004694 Eucalyptus leucoxylon Nutrition 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- 229940124683 HCV polymerase inhibitor Drugs 0.000 description 1
- 108091006905 Human Serum Albumin Proteins 0.000 description 1
- 102000008100 Human Serum Albumin Human genes 0.000 description 1
- OAKJQQAXSVQMHS-UHFFFAOYSA-N Hydrazine Chemical compound NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 description 1
- QIGBRXMKCJKVMJ-UHFFFAOYSA-N Hydroquinone Chemical compound OC1=CC=C(O)C=C1 QIGBRXMKCJKVMJ-UHFFFAOYSA-N 0.000 description 1
- GRSZFWQUAKGDAV-KQYNXXCUSA-N IMP Chemical compound O[C@@H]1[C@H](O)[C@@H](COP(O)(O)=O)O[C@H]1N1C(NC=NC2=O)=C2N=C1 GRSZFWQUAKGDAV-KQYNXXCUSA-N 0.000 description 1
- 206010069803 Injury associated with device Diseases 0.000 description 1
- 102000003996 Interferon-beta Human genes 0.000 description 1
- 108090000467 Interferon-beta Proteins 0.000 description 1
- 102000005741 Metalloproteases Human genes 0.000 description 1
- 108010006035 Metalloproteases Proteins 0.000 description 1
- 108060004795 Methyltransferase Proteins 0.000 description 1
- ZKGNPQKYVKXMGJ-UHFFFAOYSA-N N,N-dimethylacetamide Chemical compound CN(C)C(C)=O.CN(C)C(C)=O ZKGNPQKYVKXMGJ-UHFFFAOYSA-N 0.000 description 1
- 150000001204 N-oxides Chemical class 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- RREDYABFXCKTIR-UHFFFAOYSA-N O=C(c1c2cncc1)NC(Br)=C2OCc1ccccc1 Chemical compound O=C(c1c2cncc1)NC(Br)=C2OCc1ccccc1 RREDYABFXCKTIR-UHFFFAOYSA-N 0.000 description 1
- XSUXDJHVNPNNFJ-UHFFFAOYSA-N OBOC=C Chemical compound OBOC=C XSUXDJHVNPNNFJ-UHFFFAOYSA-N 0.000 description 1
- AROURNOOGLAWKR-UHFFFAOYSA-N Oc1nc(cccc2)c2c(OC2C3COCC2CN(CC2CC2)C3)c1CCCCCC1CC1 Chemical compound Oc1nc(cccc2)c2c(OC2C3COCC2CN(CC2CC2)C3)c1CCCCCC1CC1 AROURNOOGLAWKR-UHFFFAOYSA-N 0.000 description 1
- KDUOTEZKYYOPFG-LHHJGKSTSA-N Oc1nc(cccc2)c2c(OCCN2CCCCC2)c1/C=C/CCC1CCCC1 Chemical compound Oc1nc(cccc2)c2c(OCCN2CCCCC2)c1/C=C/CCC1CCCC1 KDUOTEZKYYOPFG-LHHJGKSTSA-N 0.000 description 1
- 229940123066 Polymerase inhibitor Drugs 0.000 description 1
- 244000078856 Prunus padus Species 0.000 description 1
- 101800001554 RNA-directed RNA polymerase Proteins 0.000 description 1
- PYMYPHUHKUWMLA-LMVFSUKVSA-N Ribose Natural products OC[C@@H](O)[C@@H](O)[C@@H](O)C=O PYMYPHUHKUWMLA-LMVFSUKVSA-N 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 1
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 1
- 108010078233 Thymalfasin Proteins 0.000 description 1
- 102400000800 Thymosin alpha-1 Human genes 0.000 description 1
- 241001664469 Tibicina haematodes Species 0.000 description 1
- 229920004890 Triton X-100 Polymers 0.000 description 1
- 239000013504 Triton X-100 Substances 0.000 description 1
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- LXRZVMYMQHNYJB-UNXOBOICSA-N [(1R,2S,4R)-4-[[5-[4-[(1R)-7-chloro-1,2,3,4-tetrahydroisoquinolin-1-yl]-5-methylthiophene-2-carbonyl]pyrimidin-4-yl]amino]-2-hydroxycyclopentyl]methyl sulfamate Chemical compound CC1=C(C=C(S1)C(=O)C1=C(N[C@H]2C[C@H](O)[C@@H](COS(N)(=O)=O)C2)N=CN=C1)[C@@H]1NCCC2=C1C=C(Cl)C=C2 LXRZVMYMQHNYJB-UNXOBOICSA-N 0.000 description 1
- OXLURKCRXVAJQS-UHFFFAOYSA-L [1,3-bis(2,4,6-trimethylphenyl)imidazolidin-2-ylidene]-dichloro-[[5-(dimethylsulfamoyl)-2-propan-2-yloxyphenyl]methylidene]ruthenium Chemical compound CC(C)OC1=CC=C(S(=O)(=O)N(C)C)C=C1C=[Ru](Cl)(Cl)=C1N(C=2C(=CC(C)=CC=2C)C)CCN1C1=C(C)C=C(C)C=C1C OXLURKCRXVAJQS-UHFFFAOYSA-L 0.000 description 1
- WMJMABVHDMRMJA-UHFFFAOYSA-M [Cl-].[Mg+]C1CCCCC1 Chemical compound [Cl-].[Mg+]C1CCCCC1 WMJMABVHDMRMJA-UHFFFAOYSA-M 0.000 description 1
- FYJKEHKQUPSJDH-UHFFFAOYSA-N [dimethyl-(trimethylsilylamino)silyl]methane;potassium Chemical compound [K].C[Si](C)(C)N[Si](C)(C)C FYJKEHKQUPSJDH-UHFFFAOYSA-N 0.000 description 1
- WXIONIWNXBAHRU-UHFFFAOYSA-N [dimethylamino(triazolo[4,5-b]pyridin-3-yloxy)methylidene]-dimethylazanium Chemical compound C1=CN=C2N(OC(N(C)C)=[N+](C)C)N=NC2=C1 WXIONIWNXBAHRU-UHFFFAOYSA-N 0.000 description 1
- WLLIXJBWWFGEHT-UHFFFAOYSA-N [tert-butyl(dimethyl)silyl] trifluoromethanesulfonate Chemical compound CC(C)(C)[Si](C)(C)OS(=O)(=O)C(F)(F)F WLLIXJBWWFGEHT-UHFFFAOYSA-N 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 108010080374 albuferon Proteins 0.000 description 1
- 125000003158 alcohol group Chemical group 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 150000001335 aliphatic alkanes Chemical class 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 238000005865 alkene metathesis reaction Methods 0.000 description 1
- 125000003342 alkenyl group Chemical group 0.000 description 1
- 239000002168 alkylating agent Substances 0.000 description 1
- 229940100198 alkylating agent Drugs 0.000 description 1
- 125000000304 alkynyl group Chemical group 0.000 description 1
- HMFHBZSHGGEWLO-UHFFFAOYSA-N alpha-D-Furanose-Ribose Natural products OCC1OC(O)C(O)C1O HMFHBZSHGGEWLO-UHFFFAOYSA-N 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- 229960003805 amantadine Drugs 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 230000002924 anti-infective effect Effects 0.000 description 1
- 229940124425 anti-infective immunomodulator Drugs 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 229940121357 antivirals Drugs 0.000 description 1
- 239000006286 aqueous extract Substances 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 1
- 125000003725 azepanyl group Chemical group 0.000 description 1
- 125000004567 azetidin-3-yl group Chemical group N1CC(C1)* 0.000 description 1
- KVVMXWRFYAGASO-UHFFFAOYSA-N azetidine-1-carboxylic acid Chemical compound OC(=O)N1CCC1 KVVMXWRFYAGASO-UHFFFAOYSA-N 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 125000003354 benzotriazolyl group Chemical group N1N=NC2=C1C=CC=C2* 0.000 description 1
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 1
- HSDAJNMJOMSNEV-UHFFFAOYSA-N benzyl chloroformate Chemical compound ClC(=O)OCC1=CC=CC=C1 HSDAJNMJOMSNEV-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 125000002618 bicyclic heterocycle group Chemical group 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 239000012472 biological sample Substances 0.000 description 1
- 230000001851 biosynthetic effect Effects 0.000 description 1
- 125000002529 biphenylenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3C12)* 0.000 description 1
- PFYXSUNOLOJMDX-UHFFFAOYSA-N bis(2,5-dioxopyrrolidin-1-yl) carbonate Chemical compound O=C1CCC(=O)N1OC(=O)ON1C(=O)CCC1=O PFYXSUNOLOJMDX-UHFFFAOYSA-N 0.000 description 1
- 239000010836 blood and blood product Substances 0.000 description 1
- 210000001124 body fluid Anatomy 0.000 description 1
- 239000010839 body fluid Substances 0.000 description 1
- 229910000085 borane Inorganic materials 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 125000004452 carbocyclyl group Chemical group 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- IFMWVBVPVXRZHE-UHFFFAOYSA-M chlorotitanium(3+);propan-2-olate Chemical compound [Cl-].[Ti+4].CC(C)[O-].CC(C)[O-].CC(C)[O-] IFMWVBVPVXRZHE-UHFFFAOYSA-M 0.000 description 1
- 125000003016 chromanyl group Chemical group O1C(CCC2=CC=CC=C12)* 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 description 1
- PMMYEEVYMWASQN-IMJSIDKUSA-N cis-4-Hydroxy-L-proline Chemical compound O[C@@H]1CN[C@H](C(O)=O)C1 PMMYEEVYMWASQN-IMJSIDKUSA-N 0.000 description 1
- 238000003776 cleavage reaction Methods 0.000 description 1
- 239000012230 colorless oil Substances 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 230000006957 competitive inhibition Effects 0.000 description 1
- 239000000470 constituent Substances 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- OXBLHERUFWYNTN-UHFFFAOYSA-M copper(I) chloride Chemical compound [Cu]Cl OXBLHERUFWYNTN-UHFFFAOYSA-M 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 125000000000 cycloalkoxy group Chemical group 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- OASCQHPNDMJKTK-UHFFFAOYSA-N cyclopentanol Chemical compound OC1CCCC1.OC1CCCC1 OASCQHPNDMJKTK-UHFFFAOYSA-N 0.000 description 1
- YFPCLQKFNXUAAK-UHFFFAOYSA-N cyclopentyl acetate Chemical compound CC(=O)OC1CCCC1 YFPCLQKFNXUAAK-UHFFFAOYSA-N 0.000 description 1
- AIMMVWOEOZMVMS-UHFFFAOYSA-N cyclopropanecarboxamide Chemical compound NC(=O)C1CC1 AIMMVWOEOZMVMS-UHFFFAOYSA-N 0.000 description 1
- DDDLJYBLWWBBSI-UHFFFAOYSA-N cyclopropanecarboxamide;hydrochloride Chemical compound Cl.NC(=O)C1CC1 DDDLJYBLWWBBSI-UHFFFAOYSA-N 0.000 description 1
- JXCJOPFAIBLEJB-UHFFFAOYSA-N cyclopropylsulfonylcarbamic acid Chemical compound OC(=O)NS(=O)(=O)C1CC1 JXCJOPFAIBLEJB-UHFFFAOYSA-N 0.000 description 1
- DEZRYPDIMOWBDS-UHFFFAOYSA-N dcm dichloromethane Chemical compound ClCCl.ClCCl DEZRYPDIMOWBDS-UHFFFAOYSA-N 0.000 description 1
- 238000007872 degassing Methods 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 235000005911 diet Nutrition 0.000 description 1
- 230000037213 diet Effects 0.000 description 1
- LGTLXDJOAJDFLR-UHFFFAOYSA-N diethyl chlorophosphate Chemical compound CCOP(Cl)(=O)OCC LGTLXDJOAJDFLR-UHFFFAOYSA-N 0.000 description 1
- 125000004177 diethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- UCQFCFPECQILOL-UHFFFAOYSA-N diethyl hydrogen phosphate Chemical compound CCOP(O)(=O)OCC UCQFCFPECQILOL-UHFFFAOYSA-N 0.000 description 1
- DENRZWYUOJLTMF-UHFFFAOYSA-N diethyl sulfate Chemical compound CCOS(=O)(=O)OCC DENRZWYUOJLTMF-UHFFFAOYSA-N 0.000 description 1
- 229940008406 diethyl sulfate Drugs 0.000 description 1
- GLUUGHFHXGJENI-UHFFFAOYSA-N diethylenediamine Natural products C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-M dihydrogenphosphate Chemical compound OP(O)([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-M 0.000 description 1
- VAYGXNSJCAHWJZ-UHFFFAOYSA-N dimethyl sulfate Chemical compound COS(=O)(=O)OC VAYGXNSJCAHWJZ-UHFFFAOYSA-N 0.000 description 1
- UXGNZZKBCMGWAZ-UHFFFAOYSA-N dimethylformamide dmf Chemical compound CN(C)C=O.CN(C)C=O UXGNZZKBCMGWAZ-UHFFFAOYSA-N 0.000 description 1
- 150000002012 dioxanes Chemical class 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- PMMYEEVYMWASQN-UHFFFAOYSA-N dl-hydroxyproline Natural products OC1C[NH2+]C(C([O-])=O)C1 PMMYEEVYMWASQN-UHFFFAOYSA-N 0.000 description 1
- CETRZFQIITUQQL-UHFFFAOYSA-N dmso dimethylsulfoxide Chemical compound CS(C)=O.CS(C)=O CETRZFQIITUQQL-UHFFFAOYSA-N 0.000 description 1
- 239000000890 drug combination Substances 0.000 description 1
- 239000003596 drug target Substances 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- ZELRXSQSERBNAV-UHFFFAOYSA-N ethyl 3-(2-fluoroanilino)-3-oxopropanoate Chemical compound CCOC(=O)CC(=O)NC1=CC=CC=C1F ZELRXSQSERBNAV-UHFFFAOYSA-N 0.000 description 1
- VRZVPALEJCLXPR-UHFFFAOYSA-N ethyl 4-methylbenzenesulfonate Chemical compound CCOS(=O)(=O)C1=CC=C(C)C=C1 VRZVPALEJCLXPR-UHFFFAOYSA-N 0.000 description 1
- 125000006437 ethyl cyclopropyl group Chemical group 0.000 description 1
- 125000004494 ethyl ester group Chemical group 0.000 description 1
- OJCSPXHYDFONPU-UHFFFAOYSA-N etoac etoac Chemical compound CCOC(C)=O.CCOC(C)=O OJCSPXHYDFONPU-UHFFFAOYSA-N 0.000 description 1
- 230000007717 exclusion Effects 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- KNPWRSVXYPZCOS-UHFFFAOYSA-N formic acid;piperidine Chemical compound [O-]C=O.C1CC[NH2+]CC1 KNPWRSVXYPZCOS-UHFFFAOYSA-N 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 235000013905 glycine and its sodium salt Nutrition 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 125000001188 haloalkyl group Chemical group 0.000 description 1
- 230000005802 health problem Effects 0.000 description 1
- 108700008776 hepatitis C virus NS-5 Proteins 0.000 description 1
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 1
- 231100000844 hepatocellular carcinoma Toxicity 0.000 description 1
- PYGSKMBEVAICCR-UHFFFAOYSA-N hexa-1,5-diene Chemical compound C=CCCC=C PYGSKMBEVAICCR-UHFFFAOYSA-N 0.000 description 1
- 238000004896 high resolution mass spectrometry Methods 0.000 description 1
- 238000000589 high-performance liquid chromatography-mass spectrometry Methods 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- UVNXNSUKKOLFBM-UHFFFAOYSA-N imidazo[2,1-b][1,3,4]thiadiazole Chemical compound N1=CSC2=NC=CN21 UVNXNSUKKOLFBM-UHFFFAOYSA-N 0.000 description 1
- 125000002632 imidazolidinyl group Chemical group 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 230000002519 immonomodulatory effect Effects 0.000 description 1
- 230000002584 immunomodulator Effects 0.000 description 1
- 238000009169 immunotherapy Methods 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 description 1
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 1
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 229940090438 infergen Drugs 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 235000013902 inosinic acid Nutrition 0.000 description 1
- 108010010648 interferon alfacon-1 Proteins 0.000 description 1
- 229960001388 interferon-beta Drugs 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 229940065638 intron a Drugs 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 229910052740 iodine Inorganic materials 0.000 description 1
- 125000002346 iodo group Chemical group I* 0.000 description 1
- HVTICUPFWKNHNG-UHFFFAOYSA-N iodoethane Chemical compound CCI HVTICUPFWKNHNG-UHFFFAOYSA-N 0.000 description 1
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 1
- LSACYLWPPQLVSM-UHFFFAOYSA-N isobutyric acid anhydride Chemical compound CC(C)C(=O)OC(=O)C(C)C LSACYLWPPQLVSM-UHFFFAOYSA-N 0.000 description 1
- 125000003384 isochromanyl group Chemical group C1(OCCC2=CC=CC=C12)* 0.000 description 1
- 125000004594 isoindolinyl group Chemical group C1(NCC2=CC=CC=C12)* 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 125000004628 isothiazolidinyl group Chemical group S1N(CCC1)* 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- GWYFCOCPABKNJV-UHFFFAOYSA-N isovaleric acid Chemical compound CC(C)CC(O)=O GWYFCOCPABKNJV-UHFFFAOYSA-N 0.000 description 1
- 125000003965 isoxazolidinyl group Chemical group 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 206010023497 kuru Diseases 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 208000019423 liver disease Diseases 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L magnesium chloride Substances [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 238000004949 mass spectrometry Methods 0.000 description 1
- COTNUBDHGSIOTA-UHFFFAOYSA-N meoh methanol Chemical compound OC.OC COTNUBDHGSIOTA-UHFFFAOYSA-N 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 238000005649 metathesis reaction Methods 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- GRWIABMEEKERFV-UHFFFAOYSA-N methanol;oxolane Chemical compound OC.C1CCOC1 GRWIABMEEKERFV-UHFFFAOYSA-N 0.000 description 1
- 125000001160 methoxycarbonyl group Chemical group [H]C([H])([H])OC(*)=O 0.000 description 1
- SUKHIRJFHOIEDY-JKIFEVAISA-N methyl (2s)-2-[[(1r,2r)-2-but-3-enyl-1-methylcyclopropyl]oxycarbonylamino]-2-cyclopentylacetate Chemical compound N([C@H](C(=O)OC)C1CCCC1)C(=O)O[C@]1(C)C[C@H]1CCC=C SUKHIRJFHOIEDY-JKIFEVAISA-N 0.000 description 1
- CUHKBKYWFZPMBG-JTDSTZFVSA-N methyl (2s)-2-[[(1r,2s)-2-but-3-enyl-1-methylcyclopentyl]oxycarbonylamino]-2-cyclopentylacetate Chemical compound N([C@H](C(=O)OC)C1CCCC1)C(=O)O[C@]1(C)CCC[C@H]1CCC=C CUHKBKYWFZPMBG-JTDSTZFVSA-N 0.000 description 1
- GLXVKVOENRRZDF-ZETCQYMHSA-N methyl (2s)-2-amino-2-(oxan-4-yl)acetate Chemical compound COC(=O)[C@@H](N)C1CCOCC1 GLXVKVOENRRZDF-ZETCQYMHSA-N 0.000 description 1
- HSFRPAJFWVHJBW-QMMMGPOBSA-N methyl (2s)-2-cyclopentyl-2-isocyanatoacetate Chemical compound COC(=O)[C@@H](N=C=O)C1CCCC1 HSFRPAJFWVHJBW-QMMMGPOBSA-N 0.000 description 1
- RUZLIIJDZBWWSA-INIZCTEOSA-N methyl 2-[[(1s)-1-(7-methyl-2-morpholin-4-yl-4-oxopyrido[1,2-a]pyrimidin-9-yl)ethyl]amino]benzoate Chemical group COC(=O)C1=CC=CC=C1N[C@@H](C)C1=CC(C)=CN2C(=O)C=C(N3CCOCC3)N=C12 RUZLIIJDZBWWSA-INIZCTEOSA-N 0.000 description 1
- TXBMTCMROZBKLB-UHFFFAOYSA-N methyl 2-acetamido-4-(trifluoromethyl)benzoate Chemical compound COC(=O)C1=CC=C(C(F)(F)F)C=C1NC(C)=O TXBMTCMROZBKLB-UHFFFAOYSA-N 0.000 description 1
- DZICUHOFOOPVFM-UHFFFAOYSA-N methyl 2-amino-4-(trifluoromethyl)benzoate Chemical compound COC(=O)C1=CC=C(C(F)(F)F)C=C1N DZICUHOFOOPVFM-UHFFFAOYSA-N 0.000 description 1
- YDCHPLOFQATIDS-UHFFFAOYSA-N methyl 2-bromoacetate Chemical compound COC(=O)CBr YDCHPLOFQATIDS-UHFFFAOYSA-N 0.000 description 1
- VNXBKJFUJUWOCW-UHFFFAOYSA-N methylcyclopropane Chemical compound CC1CC1 VNXBKJFUJUWOCW-UHFFFAOYSA-N 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 150000004712 monophosphates Chemical class 0.000 description 1
- STUHQDIOZQUPGP-UHFFFAOYSA-N morpholin-4-ium-4-carboxylate Chemical compound OC(=O)N1CCOCC1 STUHQDIOZQUPGP-UHFFFAOYSA-N 0.000 description 1
- 125000004573 morpholin-4-yl group Chemical group N1(CCOCC1)* 0.000 description 1
- 239000004570 mortar (masonry) Substances 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- RTGDFNSFWBGLEC-SYZQJQIISA-N mycophenolate mofetil Chemical compound COC1=C(C)C=2COC(=O)C=2C(O)=C1C\C=C(/C)CCC(=O)OCCN1CCOCC1 RTGDFNSFWBGLEC-SYZQJQIISA-N 0.000 description 1
- 229960004866 mycophenolate mofetil Drugs 0.000 description 1
- VMWJCFLUSKZZDX-UHFFFAOYSA-N n,n-dimethylmethanamine Chemical compound [CH2]N(C)C VMWJCFLUSKZZDX-UHFFFAOYSA-N 0.000 description 1
- WOOWBQQQJXZGIE-UHFFFAOYSA-N n-ethyl-n-propan-2-ylpropan-2-amine Chemical compound CCN(C(C)C)C(C)C.CCN(C(C)C)C(C)C WOOWBQQQJXZGIE-UHFFFAOYSA-N 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- PSACHCMMPFMFAJ-UHFFFAOYSA-N nmm n-methylmorpholine Chemical compound CN1CCOCC1.CN1CCOCC1 PSACHCMMPFMFAJ-UHFFFAOYSA-N 0.000 description 1
- FBUKVWPVBMHYJY-UHFFFAOYSA-N nonanoic acid Chemical compound CCCCCCCCC(O)=O FBUKVWPVBMHYJY-UHFFFAOYSA-N 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- BKIMMITUMNQMOS-UHFFFAOYSA-N normal nonane Natural products CCCCCCCCC BKIMMITUMNQMOS-UHFFFAOYSA-N 0.000 description 1
- 125000003835 nucleoside group Chemical group 0.000 description 1
- VMPITZXILSNTON-UHFFFAOYSA-N o-anisidine Chemical compound COC1=CC=CC=C1N VMPITZXILSNTON-UHFFFAOYSA-N 0.000 description 1
- TVMXDCGIABBOFY-UHFFFAOYSA-N octane Chemical compound CCCCCCCC TVMXDCGIABBOFY-UHFFFAOYSA-N 0.000 description 1
- DJLYFOLCFWQWQG-UHFFFAOYSA-N octane;2,2,2-trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.CCCCCCCC DJLYFOLCFWQWQG-UHFFFAOYSA-N 0.000 description 1
- 239000013110 organic ligand Substances 0.000 description 1
- 230000008520 organization Effects 0.000 description 1
- 229940042443 other antivirals in atc Drugs 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- LMYJGUNNJIDROI-UHFFFAOYSA-N oxan-4-ol Chemical compound OC1CCOCC1 LMYJGUNNJIDROI-UHFFFAOYSA-N 0.000 description 1
- 125000000160 oxazolidinyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- NXJCBFBQEVOTOW-UHFFFAOYSA-L palladium(2+);dihydroxide Chemical compound O[Pd]O NXJCBFBQEVOTOW-UHFFFAOYSA-L 0.000 description 1
- 235000011837 pasties Nutrition 0.000 description 1
- HVAMZGADVCBITI-UHFFFAOYSA-M pent-4-enoate Chemical compound [O-]C(=O)CCC=C HVAMZGADVCBITI-UHFFFAOYSA-M 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 239000008177 pharmaceutical agent Substances 0.000 description 1
- UEZVMMHDMIWARA-UHFFFAOYSA-M phosphonate Chemical compound [O-]P(=O)=O UEZVMMHDMIWARA-UHFFFAOYSA-M 0.000 description 1
- 239000006187 pill Substances 0.000 description 1
- 125000004194 piperazin-1-yl group Chemical group [H]N1C([H])([H])C([H])([H])N(*)C([H])([H])C1([H])[H] 0.000 description 1
- CHKVPAROMQMJNQ-UHFFFAOYSA-M potassium bisulfate Chemical compound [K+].OS([O-])(=O)=O CHKVPAROMQMJNQ-UHFFFAOYSA-M 0.000 description 1
- 159000000001 potassium salts Chemical class 0.000 description 1
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 238000012545 processing Methods 0.000 description 1
- 230000000069 prophylactic effect Effects 0.000 description 1
- JTTWNTXHFYNETH-UHFFFAOYSA-N propyl 4-methylbenzenesulfonate Chemical compound CCCOS(=O)(=O)C1=CC=C(C)C=C1 JTTWNTXHFYNETH-UHFFFAOYSA-N 0.000 description 1
- 239000012264 purified product Substances 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003072 pyrazolidinyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- ABMYEXAYWZJVOV-UHFFFAOYSA-N pyridin-3-ylboronic acid Chemical compound OB(O)C1=CC=CN=C1 ABMYEXAYWZJVOV-UHFFFAOYSA-N 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 150000003242 quaternary ammonium salts Chemical class 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- LEWDKQKVAFOMPI-UHFFFAOYSA-N quinoline-4-carboxamide Chemical compound C1=CC=C2C(C(=O)N)=CC=NC2=C1 LEWDKQKVAFOMPI-UHFFFAOYSA-N 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 230000007017 scission Effects 0.000 description 1
- 238000012216 screening Methods 0.000 description 1
- 238000007423 screening assay Methods 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- WGRULTCAYDOGQK-UHFFFAOYSA-M sodium;sodium;hydroxide Chemical compound [OH-].[Na].[Na+] WGRULTCAYDOGQK-UHFFFAOYSA-M 0.000 description 1
- 239000012439 solid excipient Substances 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 239000012258 stirred mixture Substances 0.000 description 1
- 239000007929 subcutaneous injection Substances 0.000 description 1
- 238000010254 subcutaneous injection Methods 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- AQZLKYPTEAHKNF-IORBWHJUSA-N tert-butyl (2S)-3,3-dimethyl-2-[[(1R,2R)-2-(4-tributylstannylpent-4-enyl)cyclopropyl]oxycarbonylamino]butanoate Chemical compound CCCC[Sn](CCCC)(CCCC)C(=C)CCC[C@@H]1C[C@H]1OC(=O)N[C@H](C(=O)OC(C)(C)C)C(C)(C)C AQZLKYPTEAHKNF-IORBWHJUSA-N 0.000 description 1
- DGDBOFASCDPPMQ-URVQXPFFSA-N tert-butyl (2S,4S)-4-(4-bromophenyl)sulfonyloxy-2-[[(1R,2S)-2-ethenyl-1-ethoxycarbonylcyclopropyl]carbamoyl]pyrrolidine-1-carboxylate Chemical compound O=C([C@H]1N(C[C@H](C1)OS(=O)(=O)C=1C=CC(Br)=CC=1)C(=O)OC(C)(C)C)N[C@]1(C(=O)OCC)C[C@H]1C=C DGDBOFASCDPPMQ-URVQXPFFSA-N 0.000 description 1
- FFYVUQQGPRVGIA-RBSFLKMASA-N tert-butyl (2s)-3,3-dimethyl-2-[[(1r,2r)-2-pent-4-ynylcyclopropyl]oxycarbonylamino]butanoate Chemical compound CC(C)(C)OC(=O)[C@H](C(C)(C)C)NC(=O)O[C@@H]1C[C@H]1CCCC#C FFYVUQQGPRVGIA-RBSFLKMASA-N 0.000 description 1
- HKIGXXRMJFUUKV-QMMMGPOBSA-N tert-butyl (3s)-3-(hydroxymethyl)pyrrolidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CC[C@H](CO)C1 HKIGXXRMJFUUKV-QMMMGPOBSA-N 0.000 description 1
- PWQLFIKTGRINFF-UHFFFAOYSA-N tert-butyl 4-hydroxypiperidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCC(O)CC1 PWQLFIKTGRINFF-UHFFFAOYSA-N 0.000 description 1
- WKBWNRUVSJJEAM-UHFFFAOYSA-N tert-butyl 7-hydroxy-3-oxa-9-azabicyclo[3.3.1]nonane-9-carboxylate Chemical compound C1OCC2CC(O)CC1N2C(=O)OC(C)(C)C WKBWNRUVSJJEAM-UHFFFAOYSA-N 0.000 description 1
- GUDBJYOHDUEFPZ-MEBBXXQBSA-N tert-butyl n-[(1r,2s)-2-ethenyl-1-[(1-methylcyclopropyl)sulfonylcarbamoyl]cyclopropyl]carbamate Chemical compound C1CC1(C)S(=O)(=O)NC(=O)[C@@]1(NC(=O)OC(C)(C)C)C[C@H]1C=C GUDBJYOHDUEFPZ-MEBBXXQBSA-N 0.000 description 1
- IRFHMTUHTBSEBK-QGZVFWFLSA-N tert-butyl n-[(2s)-2-(2,5-difluorophenyl)-3-quinolin-3-ylpropyl]carbamate Chemical compound C1([C@H](CC=2C=C3C=CC=CC3=NC=2)CNC(=O)OC(C)(C)C)=CC(F)=CC=C1F IRFHMTUHTBSEBK-QGZVFWFLSA-N 0.000 description 1
- 125000001981 tert-butyldimethylsilyl group Chemical group [H]C([H])([H])[Si]([H])(C([H])([H])[H])[*]C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- WHRNULOCNSKMGB-UHFFFAOYSA-N tetrahydrofuran thf Chemical compound C1CCOC1.C1CCOC1 WHRNULOCNSKMGB-UHFFFAOYSA-N 0.000 description 1
- 125000003039 tetrahydroisoquinolinyl group Chemical group C1(NCCC2=CC=CC=C12)* 0.000 description 1
- 125000000147 tetrahydroquinolinyl group Chemical group N1(CCCC2=CC=CC=C12)* 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- WROMPOXWARCANT-UHFFFAOYSA-N tfa trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F.OC(=O)C(F)(F)F WROMPOXWARCANT-UHFFFAOYSA-N 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000001984 thiazolidinyl group Chemical group 0.000 description 1
- 125000003396 thiol group Chemical group [H]S* 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- NZVYCXVTEHPMHE-ZSUJOUNUSA-N thymalfasin Chemical compound CC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(N)=O)C(O)=O NZVYCXVTEHPMHE-ZSUJOUNUSA-N 0.000 description 1
- 229960004231 thymalfasin Drugs 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- WUOFQGMXQCSPPV-UHFFFAOYSA-N tributyl(1,3-thiazol-2-yl)stannane Chemical compound CCCC[Sn](CCCC)(CCCC)C1=NC=CS1 WUOFQGMXQCSPPV-UHFFFAOYSA-N 0.000 description 1
- QIWRFOJWQSSRJZ-UHFFFAOYSA-N tributyl(ethenyl)stannane Chemical compound CCCC[Sn](CCCC)(CCCC)C=C QIWRFOJWQSSRJZ-UHFFFAOYSA-N 0.000 description 1
- DBGVGMSCBYYSLD-UHFFFAOYSA-N tributylstannane Chemical compound CCCC[SnH](CCCC)CCCC DBGVGMSCBYYSLD-UHFFFAOYSA-N 0.000 description 1
- NTJBWZHVSJNKAD-UHFFFAOYSA-N triethylazanium;fluoride Chemical compound [F-].CC[NH+](CC)CC NTJBWZHVSJNKAD-UHFFFAOYSA-N 0.000 description 1
- MHNHYTDAOYJUEZ-UHFFFAOYSA-N triphenylphosphane Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 MHNHYTDAOYJUEZ-UHFFFAOYSA-N 0.000 description 1
- UCPYLLCMEDAXFR-UHFFFAOYSA-N triphosgene Chemical compound ClC(Cl)(Cl)OC(=O)OC(Cl)(Cl)Cl UCPYLLCMEDAXFR-UHFFFAOYSA-N 0.000 description 1
- 239000001226 triphosphate Substances 0.000 description 1
- 229910000404 tripotassium phosphate Inorganic materials 0.000 description 1
- 235000019798 tripotassium phosphate Nutrition 0.000 description 1
- 229960004295 valine Drugs 0.000 description 1
- 239000004474 valine Substances 0.000 description 1
- 229950002810 valopicitabine Drugs 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 239000003039 volatile agent Substances 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 239000002023 wood Substances 0.000 description 1
- 239000011992 zhan catalyst-1B Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/08—Tripeptides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4738—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4745—Quinolines; Isoquinolines ortho- or peri-condensed with heterocyclic ring systems condensed with ring systems having nitrogen as a ring hetero atom, e.g. phenantrolines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/675—Phosphorus compounds having nitrogen as a ring hetero atom, e.g. pyridoxal phosphate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/7056—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing five-membered rings with nitrogen as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/005—Enzyme inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/06—Tripeptides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/19—Cytokines; Lymphokines; Interferons
- A61K38/21—Interferons [IFN]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/08—Tripeptides
- C07K5/0802—Tripeptides with the first amino acid being neutral
- C07K5/0804—Tripeptides with the first amino acid being neutral and aliphatic
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/08—Tripeptides
- C07K5/0802—Tripeptides with the first amino acid being neutral
- C07K5/0804—Tripeptides with the first amino acid being neutral and aliphatic
- C07K5/0808—Tripeptides with the first amino acid being neutral and aliphatic the side chain containing 2 to 4 carbon atoms, e.g. Val, Ile, Leu
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/08—Tripeptides
- C07K5/0802—Tripeptides with the first amino acid being neutral
- C07K5/0812—Tripeptides with the first amino acid being neutral and aromatic or cycloaliphatic
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/08—Tripeptides
- C07K5/0821—Tripeptides with the first amino acid being heterocyclic, e.g. His, Pro, Trp
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/12—Cyclic peptides, e.g. bacitracins; Polymyxins; Gramicidins S, C; Tyrocidins A, B or C
Definitions
- the present invention relates to macrocyclic compounds that are useful as inhibitors of the hepatitis C virus (HCV) NS3 protease, the synthesis of such compounds, and the use of such compounds for treating HCV infection and/or reducing the likelihood or severity of symptoms of HCV infection.
- HCV hepatitis C virus
- HCV infection is a major health problem that leads to chronic liver disease, such as cirrhosis and hepatocellular carcinoma, in a substantial number of infected individuals, estimated to be 2-15% of the world's population.
- HCV human immunodeficiency virus
- WHO World Health Organization
- NS3 protease is located in the N-terminal domain of the NS3 protein.
- the NS3 protease is considered a prime drug target.
- Previous research has identified classes of peptides, such as hexapeptides as well as tripeptides discussed in U.S. Patent Application Publications Nos. US2005/0020503, US2004/0229818, and US2004/00229776, showing degrees of activity in inhibiting the NS3 protease. Additional HSV NS3 protease inhibitors have been described in International Patent Applicaton Publication Nos. WO2008/057208 and
- the aim of the present invention is to provide further compounds which exhibit activity against the HCV NS3 protease.
- the present invention relates to novel macrocyclic compounds of formula (I) and/or pharmaceutically acceptable salts or hydrates thereof. These compounds are useful in the inhibition of HCV (hepatitis C virus) NS3 (non-structural 3) protease, the prevention or treatment of one or more of the symptoms of HCV infection, either as compounds or their pharmaceutically acceptable salts or hydrates (when appropriate), or as pharmaceutical composition ingredients.
- HCV hepatitis C virus
- NS3 non-structural 3
- pharmaceutical composition ingredients these compounds, salts and hydrates may be the primary active therapeutic agent, and, when appropriate, may be combined with other therapeutic agents including but not limited to other HCV antivirals, anti- infectives, immunomodulators, antibiotics or vaccines. More particularly, the present invention relates to a compound of formula (I) and/or a pharmaceutically acceptable salt thereof:
- Y is CH or N
- R 1 is:
- alkyl is optionally substituted with 1 or 2 fluoro substituents
- said phosphate group is optionally substituted with 1, 2 or 3 C 1-6 alkyl
- aryl selected from phenyl or napthyl optionally substituted with 1 or 2 substituents selected from -OH, Cr 6 alkyl, or halo;
- heteroaryl selected from 5- and 6-membered aromatic rings having 1, 2 or 3 heteroatoms independently selected from N, O and S, wherein said heteroaryl is attached through a ring atom selected from C or N and optionally substituted with 1 or 2 substituents independently selected from C h alky! and -OH; or
- R a and R b are independently selected from H; d- 6 alkyl; t-Boc; aryl; C 3 - 6 cycloalkyl optionally substituted with 1 or 2 fluoro substituents; C ealko y-Ci-ealkyl; tetrahydropyranyl; C ⁇ ealkyl-OH; C 1 - 6 alkyl-aryl; Q- 6 alkyl-C(OH)-aryl; Q-ealkyl-imidazolyl optionally substituted with methyl, C ealkyl-benzimidazolyl optionally substituted with methyl; Crealkyl-pyrazolyl; Crealkyl-dihydrotriazole optionally substituted with oxo; or d-ealkyl-pyrrolidinyl optionally substituted with oxo;
- m is 0 or 1 to 4;
- aryl is phenyl, naphthalenyl, tetrahydronapthalenyl, or 7-10 membered fused bicyclic ring structure wherein at least one of the rings is aromatic and is optionally substituted with 2 -OH;
- said tetrahydropyranyl is optionally substituted with 1 oxo;
- R 2 is Ci-ealkyl, C 2 - 6 alkenyl, C 3 -C 6 cycloalkyl or NR c R d ;
- the C 3-6 cycloalkyl is optionally substituted with C 1-6 alkyl optionally substituted with -OH, morpholinyl, C 1-6 alkoxy, C 1-6 alkoxy-C 1-6 alkoxy, C 1-6 alkoxy-phenyl, or C 1-6 alkenyl;
- R c and R d are independently H or C 1- alkyl, or may be taken together, with the N to which they are attached, to form a 4-7-membered monocyclic ring;
- R is C ⁇ alkyl, C 2 - alkenyl, C3-C 6 cycloalkyl, CF 2 or CF 3 ;
- R 4 is Ci-8 alkyl, C 3 -g cycloalkyl, Ci-8 alkyl-C 3 - 8 cycloalkyl, adamantyl, dihydroindenyl, or a 4-8 membered heterocycloalkyl having 1 or 2 heteroatoms selected from N, O, or S, wherein R 4 is optionally substituted with one or two substituents independently selected from (Ci- C 6 )alkyl, halo, and -0(C 1 -C 6 )alkyl; or
- Z is C or N
- R 5 is H or d- 6 alkyl; or R 5 is absent when Z is N;
- W is a bond, O or NR
- R is H or d- 6 alkyl
- X is absent or is halo, CF 3 , -OCHF 2 , -OCH 2 F, -OCD 2 F, -OCDF 2 , C C 6 alkyl, Crealkoxy, aryl, heteroaryl, or -0(CH 2 ) 1-6 NR a R b ;
- A is absent, O or N;
- B is (CH 2 ) m ;
- n 1-4.
- the present invention also includes pharmaceutical compositions containing a compound of the present invention and methods of preparing such pharmaceutical compositions.
- the present invention further includes methods of treating or reducing the likelihood or severity of one or more symptoms of HCV infection.
- Other embodiments, aspects and features of the present invention are either further described in or will be apparent from the ensuing description, examples and appended claims.
- the present invention relates to compounds of Formula (I) and pharmaceutically acceptable salts thereof, as defined above and a first embodiment of the invention. Different embodiments further describing Formula (I) variables are described below.
- the present invention relates to c mpounds, or a pharmaceutically acceptable salt thereof, having a formula of
- the present invention relates to compounds of Formula (I), (la), (lb), (Ic), (Id) or (le) and pharmaceutically acceptable salts thereof, wherein Z is C and the other variables are as provided for in the first or second embodiments.
- the present invention relates to compounds of Formula
- the present invention relates to compounds of Formula (I), (la), (lb), (Ic), (Id), (Ie), (If), (Ig) and (Ih) and pharmaceutically acceptable salts thereof, wherein R is ethyl, ethylene, or cyclopropyl and the other variables are as provided for in the first or sixth embodiments.
- R 3 is ethylene.
- the present invention relates to compounds of Formula (I), (la), (lb), (Ic), (Id), (Ie), (If), (Ig) and (Ih) and pharmaceutically acceptable salts thereof, wherein R 4 is propyl, t-butyl, cyclopentyl, cyclohexyl optionally substituted with 1 or 2 F, cyclohexylmethyl, methylcyclohexyl, methylcyclopentyl,
- R 4 is t-butyl, cyclopentyl, or cyclohexyl, 1 -methylcyclohexyl, propan-2-yl, 2,3 -dihydroindenyl, tetrahydro- 2H-pyranyl, or cyclohexylmethyl.
- R 4 is t-butyl, cyclopentyl, or cyclohexyl, 1 -methylcyclohexyl, 2,3 -dihydroindenyl, or tetrahydro-2H-pyranyl.
- the present invention relates to compounds of Formula (I), (la), (lb), (Ic), (Id), (Ie), (If), (Ig) and (Ih) and pharmaceutically acceptable salts thereof, wherein n is 1 or 3, and the other variables are as provided for in any of the first or sixth to eighth embodiments.
- R 2 is cyclopropyl, N(CH 3 ) 2 , (methyl)cyclopropyl,
- R 2 is cyclopropyl, N(C3 ⁇ 4) 2 , (methyl)cyclopropyl, or l-(methoxymethyl)cyclopropyl.
- R a and R b are independently
- heti is phenyl; oxazepanyl; oxooxazolidinyl; pyridinyl; pyrazolyl; pyrrolyl;
- tetrahydropyranyl triazolyl optionally substituted with C 1-6 alkyl; dioxolanyl; oxoimidazolidinyl; morpholinyl optionally substituted with dimethyl or ethyl; pyrrolidinyl optionally substituted with 1 or 2 substituents independently selected from oxo, Boc, C 1-6 alkyl, OH, C(0)NH 2 , dimethylamino, and methylsulfomyl; piperidinyl optionally substituted with 1 or 2 substituents independently selected from C 1-6 alkyl, C 1-6 alkoxy, Q-ealkoxy-C ⁇ alkyl optionally substituted with CF 3 , cyclopropyl-C 1-6 alkyl, cyclopropyl, -(CH 2 )mF, OH, -C 1-6 alkyl-S0 2 C 1-6 alkyl,
- oxaazaspiroheptyl optionally substituted with methoxyethyl; azetidinyl optionally substituted with 1 or 2 substituents independently selected from C 1-6 alkyl, Q-ealkoxy, cyano, fluoro, OH, phenyl and Boc; dioxidothiomorpholinyl; piperazinyl optionally substituted with 1 or 2 substituents independently selected from C 1-6 alkyl, C 1-6 alkyl-cyclopropyl, CF 3 , methylsulfonyl, Boc, and oxo; azabicyclooctyl substituted with C 1-6 alkyl, Q-ealkoxy-C ⁇ alkyl, -COOC 1-6 alkyl, or -(CH 2 ) m CF 3 ; oxaazabicyclononyl optionally substituted with Boc, C 1-6 alkyl, -COOC 1- alkyl, C 1-6 alkoxy-C 1-6 al
- the present invention relates to compounds of Formula (I), (la), (lb), (Ic), (Id), (Ie), (If), (Ig) and (Ih) and pharmaceutically acceptable salts thereof, wherein X is absent or selected from -Br, -CI, -F, methoxy, methyl, propanyl and CF 3 , and the other variables are as provided for in any of the first or sixth through eleventh embodiments.
- compositions comprising an effective amount of a compound of formula (I), in any of the described embodiments, and a pharmaceutically acceptable carrier.
- the second therapeutic agent is ribavirin.
- HCV antiviral agent is an antiviral selected from the group consisting of HCV protease inhibitors and HCV NS5B polymerase inhibitors.
- HCV antiviral agent is an antiviral selected from the group consisting of HCV protease inhibitors and HCV NS5B polymerase inhibitors.
- HCV antiviral agent is an antiviral selected from the group consisting of HCV protease inhibitors and HCV NS5B polymerase inhibitors.
- a method of inhibiting HCV NS3 protease in a subject in need thereof which comprises administering to the subject the pharmaceutical composition of (a), (b), or (c) or the combination of (d) or (e).
- the present invention also includes a compound of the present invention for use (i) in, (ii) as a medicament for, or (iii) in the preparation of a medicament for: (a) inhibiting HCV NS3 protease, or (b) treating HCV infection and/or reducing the likelihood or severity of symptoms of HCV infection.
- the compounds of the present invention can optionally be employed in combination with one or more second therapeutic agents selected from HCV antiviral agents, anti-infective agents, and immunomodulators.
- the second therapeutic agent is ribavirin.
- Additional embodiments of the invention include the pharmaceutical compositions, combinations and methods set forth in (a)-(k) above and the uses set forth in the preceding paragraph, wherein the compound of the present invention employed therein is a compound of one of the embodiments, aspects, classes, sub-classes, or features of the
- the compound may optionally be used in the form of a pharmaceutically acceptable salt or hydrate as appropriate.
- the dotted line is an optional bond.
- the bond in its entirety
- the bond in its entirety
- the bond in its entirety
- the bond in its entirety
- the bond is a single bond.
- Each such bond is independently a single bond or a double bond.
- two such bonds when two such bonds are adjacent to each other, it can represent two single bonds, two double bonds, a single bond adjacent to a double bond, or a double bond adjacent to a single bond.
- alkyl refers to any linear or branched chain alkyl group having a number of carbon atoms in the specified range.
- C 1-6 alkyl (or “Ci-Ce alkyl”) refers to all of the hexyl alkyl and pentyl alkyl isomers as well as n-, iso-, sec- and t-butyl, n- and isopropyl, ethyl and methyl.
- CM alkyl refers to n-, iso-, sec- and t-butyl, n- and isopropyl, ethyl and methyl. Alkyl groups may be substituted as indicated.
- alkoxy refers to an "alkyl-O-" group. Alkoxy groups may be substituted as indicated.
- alkylene refers to any linear or branched chain alkylene group (or alternatively “alkanediyl”) having a number of carbon atoms in the specified range.
- -C ⁇ alkylene- refers to any of the C ⁇ to C 6 linear or branched alkylenes.
- a class of alkylenes of particular interest with respect to the invention is -(CH 2 ) 1-6 -, and sub-classes of particular interest include -( ⁇ 1 ⁇ 4) -, -( ⁇ 3 ⁇ 4) 1-3 -, -(CH 2 ) 1-2 -, and -CH 2 -.
- alkylene -CH(CH 3 )- is also of interest.
- Alkylene groups may be substituted as indicated.
- cycloalkyl refers to any monocyclic or bicyclic ring structure of an alkane or alkene having a number of carbon atoms in the specified range.
- C 3-8 cycloalkyl (or “C 3 -C 8 cycloalkyl”) includes cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, and cyclooctyl.
- cycloalkoxy refers to a "cycloalkyl-O-" group. Cycloalkyl groups may be substituted as indicated.
- Carbocyclyl as used herein, unless otherwise indicated, refers to (i) a C 3 to C 8 monocyclic, saturated or unsaturated ring or (ii) a C 7 to C 12 bicyclic saturated or unsaturated ring system.
- Each ring in (ii) is either independent of, or fused to, the other ring, and each ring is saturated or unsaturated.
- Carbocycle groups may be substituted as indicated, for example with C 1-6 alkyl, C 1-6 alkenyl, C 1-6 alkynyl, aryl, halogen, -NH 2 or -OH.
- the carbocycle may be attached to the rest of the molecule at any carbon atom which results in a stable compound.
- a subset of the fused bicyclic unsaturated carbocycles are those bicyclic carbocycles in which one ring is a benzene ring and the other ring is saturated or unsaturated, with attachment via any carbon atom that results in a stable compound.
- Depicted ring systems include, where appropriate, able to
- Variable R 5 is shown as a floating variable which can be attached to any ring atom, provided that such attachment results in formation of a stable ring.
- each ring in (ii) and (iii) is independent of, or fused to, the other ring or rings and each ring is saturated or unsaturated, and the monocyclic ring, bicyclic ring system or tricyclic ring system contains one or more heteroatoms (e.g., from 1 to 6 heteroatoms, or from 1 to 4 heteroatoms) independently selected from N, O and S and a balance of carbon atoms (the monocyclic ring typically contains at least one carbon atom and the bicyclic and tricyclic ring systems typically contain at least two carbon atoms); and wherein any one or more of the nitrogen and sulfur heteroatoms is optionally oxidized, and any one or more of the nitrogen heteroatoms is optionally quaternized.
- heteroatoms e.g., from 1 to 6 heteroatoms, or from 1 to 4 heteroatoms
- the monocyclic ring typically contains at least one carbon atom and the bicyclic and tricyclic ring systems typically contain at least two carbon atoms
- heterocyclic ring may be attached at any heteroatom or carbon atom, provided that attachment results in the creation of a stable structure.
- Heterocycle groups may be substituted as indicated, and unless otherwise specified, the substituents may be attached to any atom in the ring, whether a heteroatom or a carbon atom, provided that a stable chemical structure results.
- Representative examples include piperidinyl, piperazinyl, azepanyl, pyrrolidinyl, pyrazolidinyl, imidazolidinyl, oxazolidinyl, isoxazolidinyl, morpholinyl, thiomorpholinyl, thiazolidinyl, isothiazolidinyl, and tetrahydrofuryl (or tetrahydrofuranyl).
- heterocyclics include 4-8 membered heterocycloalkyls having 1 to 2 heteroatoms selected from N, O, and S.
- heteroaromatic rings include pyridyl, pyrrolyl, pyrazinyl, pyrimidinyl, pyridazinyl, thienyl (or thiophenyl), thiazolyl, furanyl, imidazolyl, pyrazolyl, triazolyl, tetrazolyl, oxazolyl, isooxazolyl, oxadiazolyl, thiazolyl, isothiazolyl, and thiadiazolyl.
- bicyclic heterocycles include benzotriazolyl, indolyl, isoindolyl, indazolyl, indolinyl, isoindolinyl, quinoxalinyl, quinazolinyl, cinnolinyl, chromanyl, isochromanyl, tetrahydroquinolinyl, quinolinyl, tetrahydroisoquinolinyl, isoquinolinyl,
- 2,3-dihydrobenzofuranyl, 2,3-dihydrobenzo-l,4-dioxinyl i.e., CO ⁇
- imidazo(2,l-6)(l,3)thiazole i.e., )
- aanndd bbeennzzoo--ll,,33--ddiiooxxoollyyll ((ii..ee..,, ).
- phenyl having as a substituent methylenedioxy attached to two adjacent carbon atoms.
- a reference to a compound of formula (I) is a reference to the compound per se, or to any one of its tautomers per se, or to mixtures of two or more tautomers.
- Isotopically-enriched compounds within generic Formula I can be prepared without undue experimentation by conventional techniques well known to those skilled in the art or by processes analogous to those described in the Schemes and Examples herein using appropriate isotopically-enriched reagents and/or intermediates.
- the compounds of the present inventions are useful in the inhibition of HCV protease (e.g., HCV NS3 protease) and the treatment of HCV infection and or reduction of the likelihood or severity of symptoms of HCV infection.
- HCV protease e.g., HCV NS3 protease
- the compounds of this invention are useful in treating infection by HCV after suspected past exposure to HCV by such means as blood transfusion, exchange of body fluids, bites, accidental needle stick, or exposure to patient blood during surgery.
- Suitable salts include acid addition salts which may, for example, be formed by mixing a solution of the compound of the present invention with a solution of a pharmaceutically acceptable acid such as hydrochloric acid, sulfuric acid, acetic acid, trifluoroacetic acid, or benzoic acid.
- a pharmaceutically acceptable acid such as hydrochloric acid, sulfuric acid, acetic acid, trifluoroacetic acid, or benzoic acid.
- suitable pharmaceutically acceptable salts thereof can include alkali metal salts (e.g., sodium or potassium salts), alkaline earth metal salts (e.g., calcium or magnesium salts), and salts formed with suitable organic ligands such as quaternary ammonium salts.
- suitable organic ligands such as quaternary ammonium salts.
- pharmaceutically acceptable esters can be employed to modify the solubility or hydrolysis characteristics of the compound.
- Solid preparations suitable for oral administration can be prepared according to techniques known in the art and can employ such solid excipients as starches, sugars, kaolin, lubricants, binders, disintegrating agents and the like.
- Parenteral compositions can be prepared according to techniques known in the art and typically employ sterile water as a carrier and optionally other ingredients, such as solubility aids.
- injectable solutions can be prepared according to methods known in the art wherein the carrier comprises a saline solution, a glucose solution or a solution containing a mixture of saline and glucose. Further description of methods suitable for use in preparing pharmaceutical compositions of the present invention and of ingredients suitable for use in said compositions is provided in Remington's Pharmaceutical Sciences, 18 edition (ed. A. R. Gennaro, Mack Publishing Co., 1990).
- the present invention also relates to a method of inhibiting HCV NS3 protease, inhibiting HCV replication, treating HCV infection and/or reducing the likelihood or severity of symptoms of HCV infection with a compound of the present invention in combination with one or more therapeutic agents and a pharmaceutical composition comprising a compound of the present invention and one or more therapeutic agents selected from the group consisting of a HCV antiviral agent, an immunomodulator, and an anti-infective agent.
- Amgen's recombinant consensus interferon has the brand name Infergen®.
- Levovirin is the L-enantiomer of ribavirin which has shown immunomodulatory activity similar to ribavirin.
- Viramidine represents an analog of ribavirin disclosed in International Patent Application Publication No. WO 01/60379.
- the individual components of the combination can be administered separately at different times during the course of therapy or concurrently in divided or single combination forms.
- the compounds of the present invention may also be administered in combination with an agent that is an inhibitor of HCV NS3 serine protease.
- HCV NS3 serine protease is an essential viral enzyme and has been described to be an excellent target for inhibition of HCV replication. Both substrate and non-substrate based inhibitors of HCV NS3 protease inhibitors are disclosed in International Patent Application Publication Nos.
- WO 98/22496 WO 98/46630, WO 99/07733, WO 99/07734, WO 99/38888, WO 99/50230, WO 99/64442, WO 00/09543, WO 00/59929, WO 02/48116 and WO 02/48172, British Patent No. GB 2 337 262, and U.S. Patent No. 6,323,180.
- Ribavirin, levovirin, and viramidine may exert their anti-HCV effects by modulating intracellular pools of guanine nucleotides via inhibition of the intracellular enzyme inosine monophosphate dehydrogenase (IMPDH).
- IMPDH inosine monophosphate dehydrogenase
- Ribavirin is readily
- the compounds of the present invention may also be administered in combination with an inhibitor of IMPDH, such as VX-497, which is disclosed in International Patent Application Publication Nos. WO 97/41211 and WO 01/00622; another IMPDH inhibitor, such as that disclosed in International Patent Application Publication No. WO 00/25780; or mycophenolate mofetil. See Allison et i, 1993, Agents Action 44 (Suppl.):165.
- the compounds of the present invention may also be administered in combination with the antiviral agent amantadine (1-aminoadamantane).
- amantadine 1-aminoadamantane
- the compounds of the present invention may also be combined for the treatment of HCV infection with antiviral 2'-C-branched ribonucleosides disclosed in Harry-O'Kuru et ah, 1997, J. Org. Chem. 62:1754-59; Wolfe et al, 1995, Tet. Lett. 36:7611-14; U.S. Patent No. 3,480,613; and International Patent Application Publication Nos. WO 01/90121, WO 01/92282, WO 02/32920, WO 04/002999, WO 04/003000 and WO 04/002422.
- Such 2'-C-branched ribonucleosides include, but are not limited to, 2'-C-methyl-cytidine, 2'-C-methyl-uridine, -C- methyl-adenosine, 2'-C-methyl-guanosine, and 9-(2-C-methyl-P-D-ribofuranosyl)-2,6- diaminopurine, and the corresponding amino acid ester of the ribose C-2', C-3', and C-5' hydroxyls and the corresponding optionally substituted cyclic 1,3 -propanediol esters of the 5'- phosphate derivatives.
- the compounds of the present invention may also be combined for the treatment of HCV infection with other nucleosides having anti-HCV properties, such as those disclosed in International Patent Application Publication Nos. WO 02/51425, WO 01/79246, WO 02/32920, WO 02/48165, WO05/003147 (including R1656, (2'i?)-2'-deoxy-2'-fluoro-2'-C-methylcytidine), WO 01/68663, WO 99/43691, WO 02/18404, WO06/021341, WO 02/100415, WO 03/026589, WO 03/026675, WO 03/093290, WO 04/011478, WO 04/013300 and WO 04/028481, and U.S.
- Patent Application Publication Nos. US2005/0038240 including 4'-azido nucleosides such as R1626, 4'-azidocytidine), US2002/0019363, US2003/0236216, US2004/0006007 and
- the compounds of the present invention may also be administered in combination with an agent that is an inhibitor of HCV NS5B polymerase.
- HCV NS5B polymerase inhibitors that may be used as combination therapy include, but are not limited to, those disclosed in International Patent Application Publication Nos.
- HCV polymerase inhibitors include, but are not limited to, valopicitabine (NM-283; Idenix) and 2'-F-2'-beta-methylcytidine (see also International Patent Application Publication No. WO 05/003147).
- nucleoside HCV NS5B polymerase inhibitors that are used in combination with the present HCV NS3 protease inhibitors are selected from the following compounds: 4-amino-7-(2-C-methyl-P-D-arabinofuranosyl)-7H-pyrrolo[2,3-if]pyrimidine; 4- amino-7-(2-C-methyl-P-D-ribofiiranosyl)-7H-pyrrolo[2,3-i ]pyrimidine; 4-methylamino-7-(2-C- methyl-P-D-ribofuranosyl)-7H-pyrrolo[2,3-£ ]pyrimidine; 4-dimethylamino-7-(2-C-methyl-P-D- ribofuranosyl)-7H-pyrrolo[2,3-cf
- the compounds of the present invention may also be combined for the treatment of HCV infection with non-nucleoside inhibitors of HCV polymerase such as HCV-796 (Viropharma Inc.) and those disclosed in International Patent Application Publication Nos. WO 01/77091; WO 01/47883; WO 02/04425; WO 02/06246; WO 02/20497; and WO 05/016927 (in particular JTK003).
- HCV-796 Viropharma Inc.
- non-nucleoside HCV NS5B polymerase inhibitors that are used in combination with the present HCV NS3 protease inhibitors are selected from the following compounds: 14-cyclohexyl-6-[2-(dimethylamino)ethyl]-7-oxo-5,6,7,8- tetrahydroindolo[2,l-a][2,5]benzodiazocine-l 1-carboxylic acid; 14-cyclohexyl-6-(2-morpholin- 4-ylethyl)-5,6,7,8-tetrahydroindolo[2,l-o][2,5]benzodiazocine-l 1-carboxylic acid; 14- cyclohexyl-6-[2-(dimethylamino)ethyl]-3-methoxy-5,6,7,8-tetrahydroindolo[2,l- ]
- [2,5]benzodiazocine-l 1-carboxylic acid 6-allyl-14-cyclohexyl-3-methoxy-5,6,7,8- tetrahydroindolo[2,l-a][2,5]benzodiazocine-l 1-carboxylic acid; 14-cyclopentyl-6-[2- (dimethylamino)ethyl]-5,6,7,8-tetrahydroindolo[2,l- ][2,5]benzodiazocine-l 1-carboxylic acid; 14-cyclohexyl-6-[2-(dimethylamino)ethyl]-5,6,7,8-tetrahydroindolo[2,l-a][2,5]benzodiazoc 11-carboxylic acid; 13-cyclohexyl-5-methyl-4,5,6,7-tetrahydrofuro[3',2':6,7][l,4]diazocino[l,8-
- the HCV NS3 protease inhibitory activity of the present compounds may be tested using assays known in the art.
- One such assay is HCV NS3 protease time-resolved fluorescence (TRF) assay as described below and in International Patent Application Publication No. WO2006/102087.
- TRF time-resolved fluorescence
- Other examples of such assays are described in e.g., International Patent Application Publication No. WO2005/046712.
- the assay is performed in a final volume of 100 ⁇ in assay buffer containing 50 mM HEPES, pH 7.5, 150 mM NaCl, 15 % glycerol, 0.15 % Triton X-100, 10 mM DTT, and 0.1 % PEG 8000.
- NS3 protease is pre-incubated with various concentrations of inhibitors in DMSO for 30 minutes. The reaction is initiated by adding the TRF peptide substrate (final concentration 100 nM). NS3 mediated hydrolysis of the substrate is quenched after 1 hour at room temperature with 100 ⁇ of 500 mM MES, pH 5.5. Product fluorescence is detected using either a VICTOR V2 or FUSION fluorophotometer (Perkin Elmer Life and Analytical Sciences) with excitation at 340 nm and emission at 615 nm with a 400 ⁇ 8 delay. Testing concentrations of different enzyme forms are selected to result in a signal to background ratio (S/B) of 10-30. IC 50 values are derived using a standard four-parameter fit to the data. Kj values are derived from IC 50 values using the following formula,
- the present invention also includes processes for making compounds of formula (I).
- the compounds of the present invention can be readily prepared according to the following reaction schemes and examples, or modifications thereof, using readily available starting materials, reagents and conventional synthesis procedures. In these reactions, it is also possible to make use of variants which are themselves known to those of ordinary skill in this art, but are not mentioned in greater detail. Furthermore, other methods for preparing compounds of the invention will be readily apparent to the person of ordinary skill in the art in light of the following reaction schemes and examples. Unless otherwise indicated, all variables are as defined above.
- Olefin metathesis catalysts include the following Ruthenium based species:
- Step 1 tert-butyl [(lR,2S)-2-ethenyl-l-( ⁇ [l- carbamoyl)cyclopropyl]carbamate
- Step 2 (lR,2S)-l-amino-2-ethenyl-N- ⁇ [l-(prop-l-en-2-yl)cyclopropyl]sulfonyl ⁇
- Step 1 tert-butyl ⁇ (1 R,2S)-1 -[( ⁇ 1 -[(benzyloxy)methylJcyclopropyl ⁇ sulfonyl)carbamoyl]-2- ethenylcyclopropyljcarbamate
- Step 2 (lR,2S)-l-amino-N-( ⁇ l-[(benzyloxy)methyl]cyclopropyl ⁇ sulfonyl)-2- ethenylcyclopropanecarboxamide hydrochloride
- Step 1 tert-butyl ( ⁇ l-[(2-methoxyethoxy)methyl]cyclopropyl ⁇ sulfonyl)carbamate BuLi 2.5 M in hexanes (7.53 ml) was added dropwise to the solution of tert-butyl
- Step 2 l-[(2-methoxyethoxy)methyl]cyclopropanesulfonamide
- Step 4 (lR,2S)-l-amino-2-ethenyl-N-( ⁇ l-[(2- sulfonyl)cyclopropanecarboxamide hydrochloride
- Step 1 tert-butyl ⁇ (lR,2S)-2-ethenyl-l-[( ⁇ l-[2-(morpholin-4- yl)ethyl]cyclopropyl ⁇ sulfonyl)carbamoyl]cyclopropyl ⁇ carbamate
- Step 2 benzyl ⁇ [l-(2-hydroxypropan-2-yl)cyclopropyl]sulfonyl ⁇ carbamate
- Step 2 methyl (2S)-[( ⁇ [(lR,2S)-2-(but-3-en-l-yl)-l- methylcyclopentyl]oxy ⁇ carbonyl)amino](cyclopentyl)ethanoate and methyl (2S)-[( ⁇ [(lS,2R)-2- (but-3-en-l-yl)-l-methylcyclopentyl]oxy ⁇ carbonyl)amino] (cyclopentyl)ethanoate
- DMAP (0.59 g) was added to a stirred mixture of the product of Step 1 (0.75 g) and methyl (2S)-cyclopentyl(isocyanato)ethanoate (1.34 g) (See International Patent Application Publication No.: WO08/057209) in toluene (24 ml).
- DIPEA (3.40 mL) was then added and the mixture was stirred at 100°C for 22 hours. Since the reaction wasn't complete, methyl (25)- cyclopentyl(isocyanato)ethanoate (0.6 g), DIPEA (1.5 mL) and DMAP (0.3 g) were added and the mixture was stirred at 100°C for 8 hours.
- Step 3 (2S)-[( ⁇ [(lR,2R)-2-(but-3-en-l-yl)-l- methylcyclopropyl]oxy ⁇ carbonyl)amino](cyclopentyl)ethanoic acid and (2S)-[( ⁇ [(lS,2S)-2-(but- -en-l-yl)-l-me ethanoic acid
- Step 2 (2S)-cyclopentyl[( ⁇ [(lR,2R)-2-(pent-4-yn-l-yl)cyclopropyl]oxy ⁇ carbonyl)
- Steps 1-2 methyl (2S)-cyclopentyl[( ⁇ [(lR,2R)-2-(pent-4-yn-l-yl)cyclopropyl]
- Step 1 (4S)-4- ⁇ [(4-bromophenyl)sulfonyl]oxy ⁇ -l-(tert-butoxycarbonyl)-L-proline
- Step 2 tert-butyl (2S,4S)-4- ⁇ [(4-bromophenyl)sulfonyl]oxy ⁇ -2- ⁇ [(lR,2S)-2-ethenyl-l- (ethoxycarbonyl)cyclopropyl]carbamoyl ⁇ pyrrolidine-l-carboxylate
- HATU 49.6 g was added to the solution of the acid from Step 1 (53.4 g), (lR,2S)-cyclopropanecarboxylic acid, l-amino-2-ethenyl-, ethyl ester, hydrochloride
- Step 3 ethyl (lR,2S)-l- ⁇ [(4S)-4- ⁇ [(4-bromophenyl)sulfonyl]oxy ⁇ -L-prolyl]amino ⁇ -2- ethenylcyclopropanecarboxylate
- Step 4 ethyl (lR,2S)-l- ⁇ [(4S)-4- ⁇ [(4-bromophen l)sidfo ⁇
- Step 5 ethyl (lR,2S)-l- ⁇ [(4S)-4- ⁇ [(4-bromophenyl)sulfonyl]oxy ⁇ -l- ⁇ (2S)-2-[(tert- butoxycarbonyl)amino]non-8-enoyl ⁇ -L-prolyl](tert-butoxycarbonyl)amino ⁇ -2- ethenylcyclopropanecarboxylate
- Di-tert-butyl dicarbonate (17.54 ml) was added at 0°C to the solution of the product of Step 4 (37.3 g) and DMAP (1.85 g) in ethyl acetate (504 ml). The solution was stirred at room temperature for 4 hours. At this point, another 2.5 g of di-tert-butyl dicarbonate was added and the solution was stirred at room temperature for 2 hours. The reaction was quenched with water and the mixture was extracted with ethyl acetate (3x). The combined organic fractions were dried over magnesium sulfate, filtered and concentrated.
- Step 6 15-tert-butyl 14a-ethyl (2S,6S, 12Z, 13aS, 14aR, 16aS)-2- ⁇ [(4-bromophenyl)sulfonyl]oxy ⁇ - 6-[ (tert-butoxycarbonyl)amino J-5, 16-dioxo-2, 3, 6, 7, 8, 9, 10,11,13a, 14, 16,16a-dodecahydro c clopropafe Jpyrrolof 1, 2-a][ 1, 4 ]diazacyclopentadecine-14a, 15(1H, 5H)-dicarboxylate
- Step 1 ethyl 3-[(4-bromophenyl)amino]-3-oxopropanoate
- Step 1 The product of Step 1 (16.63 g) was dissolved in THF (150 mL) and 2N NaOH
- Step 2 The product of Step 2 (14.7 g) was added to polyphosphoric acid (55.3 mL) and mixture was heated to 140°C for 3 hrs. The reaction mixture was cooled slightly and poured into 3N HC1 (168 mL). The pH was adjusted to 4 with 3N NaOH and resulting solid was filtered after cooling the reaction mixture to 10°C. The cake was washed with water and then slurried in 400 mL of 50% isopropanol/water for 18 hours. The solids were filtered, air dried to give a pasty solid which was dried in vacuo at 90°C for 4 hours, ground in mortar/pestle and re-dried at 90°C for another 18 hours to give 15 g of the desired product. Step 4: 6-bromo-4-hydroxy-3-(prop-2-en-l-yl)quinolin-2(lH)-one
- Step 5 4-(benzyloxy)-6-bromo-3-(prop-2-en-l-yl)quinolin-2(lH)-one
- Step 1 To the product of Step 1 (3.75 g) in a flask was added a solution of methanesulfonic acid (8 ml) containing phosphorus pentoxide (0.8 g) and the mixture was warmed to 170°C for 1 hour. The reaction was cooled and poured into 50 g of ice. The mixture was allowed to stir then it was diluted with water and solids were filtered. The solid was dissolved with 0.5N sodium hydroxide and washed with toluene (2X). The pH was adjusted to 3 with concentrated HCl to give solids which were filtered and washed with water to give 2.1 g of an orange solid. The material was clean but it was a 1 :1 mixture of regioisomers. LRMS (ES+) m/z (M+H) + 196.1.
- Step 3 7-chloro-3-(prop-2-en-l-yl)quinoline-2,4-diol and 5-chloro-3-(prop-2-en-l-yl)quinoline- 2 4-diol
- Step 4 7-chloro-4-methoxy-3-(prop-2-en-l-yl)quinolin-2-ol (A) and 5-chloro-4-methoxy-3- rop-2-en-l-yl)quinolin-2-ol (B)
- N-bromosuccinimide (2.21 g) was added to the solution of 2,4- dihydroxyquinoline (2 g) in DCM (50 ml). The mixture was stirred at room temperature for 3 days. The mixture was filtrated and the solid was triturated with isopropanol. After filtration, toluene was added to the solid and the solvent was evaporated under reduced pressure to give the desired product (1.92 g) as a beige solid. LRMS (ES+) m/z 240.1 (M+H) + .
- Step 1 ethyl 3-[(2-fluorophenyl)amino]-3-oxopropanoate
- Step 5 4-(benzyloxy)-3-bromo- 7-(trifluoromethyl)quinolin-2(lH)-one
- Step 2 tert-butyl ((lR,2S)-l-(((l-methylcyclopropyl)sulfonyl)carbamoyl)-2- vinylcyclopropyl)carbamate
- the mixture was heated (oil bath at 75°C) overnight.
- the reaction mixture was treated with aq 1M HC1 (20 mL) and water (50 mL).
- the product was extracted into ethyl acetate (400 mL).
- the organic layer was washed with aq 1M HCl/water (1 :2, 80 mL), and brine (80 mL), dried over magnesium sulfate, filtered and concentrated in rotavap.
- Step 3 (lR,2S)-l-amino-N-((l-methylcyclopropyl)sulfonyl)-2-vinylcyclopropanecarboxam hydrochloride
- Boc-L-indanylglycine (Chem-Impex International Inc., Wood Dale, IL) (4.2 g, 14.42 mmol) was treated with 4M hydrochloric acid (80 ml, 320 mmol) in dioxane at room temperature. The resulting slurry was stirred for 2 hours. TLC showed complete reaction and the mixture was concentrated to dryness in rotavap. The residue was dried under vacuum to give the product D5 (3.3 g, 101 %) as a white powder.
- the HCV NS3 protease inhibitory activity was measured using the protease time- resolved fluorescence (TRF) assay as described below and in International Patent Application Publication No. WO 2006/102087.
- TRF protease time- resolved fluorescence
- the assay was performed with HCV genotype lb (BK) NS3 modified enzyme with a R155K mutation and genotype 3a (3A-1).
- the assay was performed in a final volume of 50 ⁇ in assay buffer containing 50 mM HEPES, pH 7.5, 150 mM NaCl, 15% glycerol, 0.15% TRITON X-100, 10 mM DTT, and 0.1 % PEG 8000.
- NS3 and NS4A protease is pre-incubated with various concentrations of inhibitors in DMSO for 10 minutes.
- the reaction was initiated by adding the TRF peptide substrate (final concentration 25 nM) and NS3 mediated hydrolysis of the substrate proceeds for 6 hours at room temperature.
- Product fluorescence is detected using an Envision plate reader (Perkin Elmer) with excitation at 340 nm and emission at 615 nm with a 400 delay. Testing concentrations of the enzymes were selected to result in a signal to background ratio (S/B) of 5- 20.
- S/B signal to background ratio
- the Ki values can be obtained using the following protocol:
- the assay is performed in a final volume of 100 ⁇ in assay buffer containing 50 mM HEPES, pH 7.5, 150 mM NaCl, 15% glycerol, 0.15% TRITON X-100, 10 mM DTT, and 0.1 % PEG 8000.
- NS3 and NS4A protease is pre-incubated with various concentrations of inhibitors in DMSO for 30 minutes. The reaction is initiated by adding the TRF peptide substrate (final concentration 100 nM).
- NS3 mediated hydrolysis of the substrate is quenched after 1 hour at room temperature with 100 ⁇ of 500 mM MES, pH 5.5.
- Example 1 (3a ?JS,10S.12i?.21E.24aS)-7-cvclopentyl-N-story?,2S)-l- (cvclopropylsulfonyl carbamoyll-2-ethenylcvclopropy -19-ethoxy-5,8-dioxo- 1.2.3.3a.5.6.7.8.11.12,20,23,24,24a-tetradecahvdro-10H-9.12-methanocvclo
- Step 1 1-tert-butyl 2 -methyl (2S,4R)-4- ⁇ [4-ethoxy-3-(prop-2-en-l-yl)quinolin-2- yl]oxy ⁇ pyrrolidine-l,2-dicarboxylate
- Step 3 methyl (4R)-l- ⁇ (2S)-2-[( ⁇ [(lR,2S)-2-(but-3-en-l-yl)cyclopentyl]oxy ⁇ carbonyl) cyclopentylacetyl ⁇ -4- ⁇ [4-ethoxy-3-(prop-2-en-l-yl)quinolin-2-yl]oxy ⁇ -L-prolinate
- Step 4 methyl (3aR, 7S,10S,12R,21Z,24aS)-7-cyclopentyl-19-ethoxy-5,8-dioxo- 1, 2, 3, 3a, 5, 6, 7, 8, 11,12, 20, 23, 24, 24a-tetradecahydro-l OH-9, 12-methanocyclopenta
- Step 5 (3aR, 7S, 1 OS, 12R,21E, 24aS)- 7-cyclopentyl-19-ethoxy-5, 8-dioxo-l, 2, 3, 3a, 5, 6, 7, 8, 11, 12,20, 23, 24, 24a-tetradecahydro-l OH-9, 12-methanocyclopenta[18,19][l,10, 3, 6]
- Example 8 (19 mg) was treated with TFA (1 ml) in a sealed tube and warmed to 55°C. After 30 minutes, the reaction was concentrated and the residue was dissolved in ACN and purified by reverse phase HPLC to provide the desired product as a white foam (13 mg). LRMS (ES+) m/z (M+H) + 790.2.
- Example 18 (3ai?.7S.10S.12i?.21E,25aj? -7-cvclopentyl-N-(aj?,25)-l-r(cvclopropyl sulfonvncarbamoyll ⁇ -ethenylcvclopro yU-ig-hvdroxy-S.S-dioxo ⁇ .S a.S.ej.S.l l. .
- Example 19 riai?.5 ⁇ 8 .10i?J9E,22ai?V5-cvclopentyl-N- ⁇ (lj?.2S)-l-r(cvclopropyl sulfonyl)carbamoyl]-2-ethenylcyclopropyl ⁇ - 17-hydroxy- 1 a-methyl-3 ,6-dioxo-
- Example 21 (3ai?.7 l S l ,10S.12i?.21E.24a ⁇ -7-cvclopentyl-N-r(lS.2igV2- (cvclopropyl sulfonyDcarbamoyl -l.r-bi cyclopropyD ⁇ -yll- ⁇ -hvdroxy-S ⁇ -dioxo- 1.2.3 ,3a.5.6.7.8.11.12.20.23.24.24a-tetradecahvdro- 1 OH-9.12-methanocyclopentar 18, 191
- Example 22 (3ai?JSq0Sa2i?,21E,24aS)-17-bromo-7-cvclopentyl-N-i( ' li?,2S)-l- (cyclopropylsulfonvncarbamovn-2-ethenylcyclopropyl ⁇ -19-hydroxy-5,8-dioxo- l,2,3,3a,5,6J.8Jia2.20,23,24.24a-tetradecahvdro-10H-9,12-methanocvclopentari8J91 ⁇ ⁇ 03 ,6]dioxadiazacvclononadecino 11 , 12-b] quinoline- 10-carboxamide
- Example 23 (3ai?JSJ0SJ2i?.21E.24ay)-7-cvclopentyl-N-r(li?,2 ⁇ -2-ethenyl-l- ⁇ r(l- methylcvclopropyl)sulfonyl1carbamoyl>cvclopropyl]-19-hvdroxy-5,8-dioxo-L2,3, 3a.5.6.7.8.11.12,20,23, 24.24a-tetradecahvdro- 10H-9.12-methanocvclopentar 18.191
- Example 24 r3a j ?.7 ⁇ .10S.12i?,24ai?)-7-cvclopentyl-N-(ni?,2 ⁇ -l-r(cvclopropyl sulfonvDcarbarnoyll-2-ethenylcyclopropyU - 19-ethoxy-5,8-dioxo- 1 ,2,3 ,3a,5,6,7,8, 1 1,12, 20,21 ,22.23 ,24.24a-hexadecahvdro- 1 OH-9.12-methanocvclopentar 18, 191 ⁇ 1 , 10.3.61dioxa diazacyclononadecino ⁇ 11 , 12-61quinoline- 10-carboxamide
- Step 1 methyl (3aR, 7S,10S,12R,24aR)-7-cyclopentyl-19-ethoxy-5,8-dioxo-
- Step 2 (3aR, 7S,10S,12R,24aR)-7-cyclopentyl-19-ethoxy-5,8-dioxo-l,2,3,3a,5,6, 7,8, 11,
- Step 3 (3aR, 7S, 10S,12R,24aR)-7-cyclopentyl-N- ⁇ (lR,2S)-l-[(cyclopropylsulfonyl) carbamoyl] - 2-ethenylcyclopropyl ⁇ -19-ethoxy-5, 8-dioxo-l, 2, 3, 3a, 5, 6, 7, 8, 11,12, 20, 21, 22, 23, 24, 24a-hexadeca hydro-10H-9, 12-methanocyclopenta[18, 19] [1, 10,3, 6] dioxadiazacyclononadecinofl 1, 12-b] quinoline-10-carboxamide
- Example 29 (3aig.7S.10S.12i?.24ai?V7-cvclopentyl-N-(rii?.2S)-l-rrcvclopropylsulfonvn carbamoyl]-2-ethenylcvclopropyl ⁇ -19-hvdroxy-3a-methyl-5,8-dioxo-l,2,3,3a,5,6,7,8J l, 12,20,21 ,22,23 ,24,24a-hexadecahydro- lQH-9,12-methano cvclopentai 18,191 ⁇ 1.10.3 ,6] dioxadiazacyclononadecino [11,12-b] quinoline- 10-carboxamide
- Step 1 (3aR, 7S, 10S, 12R,24aR)-7-cyclopentyl-19-hydroxy-3a-methyl-5,8-dioxo-
- Step 2 (3aR, 7S, 10S, 12R,24aR)-7-cydopentyl ⁇ N- ⁇ (lR,2S)-l-[(cyclopropylsulfonyl) carbamoyl]- 2-ethenylcyclopropyl ⁇ -19-hydroxy-3a-methyl-5, 8-dioxo-l, 2, 3, 3a, 5, 6, 7, 8, 11,
- Example 29 By following the procedures outlined in Example 29 and using the appropriate A, B and C intermediates (depicted below the structure as Int.), the following compounds were prepared.
- Example 38 (-3ai?.7S.10SJ2 ⁇ ,21E.24aS)-7-cvclopentyl-10-ridj?.2S)-l- [(cvclopropylsulfonyl carbamoyl]-2-ethenylcvclopropyl
- Example 39 r3ai?.7S.10S.12i?.21E,24aS)-7-cvclopentyl-10-( ' i(1 ⁇ .2S -l-r( ' cvclopropyl sulfonyl)carbamoyl1-2-ethenylcvclopropyl
- Example 40 r3a ? J ⁇ 10 l S.12i?.21E.24a ⁇ -7-cvclopentyl-10-r ⁇ nig.2 ( ⁇ -l-r(cvclopropyl sulfonyf)carbamoyll-2-ethenylcyclopropyl ⁇ carbamoyl ' )-5,8-dioxo-L2,3,3a,5,6,7,8,l l,
- Example 41 r3ai?.7S.10S.12j?.21E.24aS -7-cvclopentyl-10-(((lj?.2S)-l-r( ' cvclopropyl sulfonyl)carbamoyll-2-ethenylcvclopropyl ⁇ carbamoylV5.8-dioxo-1.2.3.3a.5.6.7.8.11.
- Example 42 r3a ?.7S.10S.12i;.21E,24aS)-7-cvclopentyl-N- ⁇ rii?.2S)-l-rrcvclopropyl sulfonyl)carbamoyll-2-ethenylcvclopropyl ⁇ -5,8-dioxo-19- 2-(2-oxo-l,3-oxazolidin-3- vnethoxyl-1.2 .3a.5.6.7.8.11.12,20,23,24,24a-tetradecahvdro-10H-9,12-methanocvclo
- Example 42 By following the procedures outlined in Example 42 and using the appropriate A, B and C intermediates and reagent (depicted below the structure as Int. and Rg., respectively), the following compounds were prepared.
- Example 61 (3ai?JSJ0S.12i?.24ai? -7-tert-butyl-N-r( ' li?.2S)-2-ethenyl-l-( ' ⁇ ri-(methoxy methvDcvclopropyll sulfonyl ⁇ carbamo vDcyclopropyl] - 19- [2-(morpholin-4-yl')ethoxyl -5 ,8-dioxo- 1.2.3.3 a.5.6.7,8, 11 , 12,20.21.22.23 ,24.24a-hexadecahvdro- 1 OH-9.12-methano
- Example 61 By following the procedures outlined in Example 61 and using the appropriate A, B and C intermediates and reagent (depicted below the structure as Int. and Rg., respectively), the following compounds were prepared.
- Example 66 (r(3aj?.7S.10S.12i?.21E.24aS -7-cvclopentyl-10-( ⁇ (lj?.2S)-l-r(cvclor>ropyl sulfonyl carbamoyl]-2-ethenylcvclopropyl
- Step 1 methyl ⁇ [(3aR,7S,10S,12R,21E,24aS)-7-cyclopentyl-10-( ⁇ (lR,2S)-l- [(cyclopropylsulfonyl)carbamoyl]-2-ethenylcyclopropyl ⁇ carbamoyl)-5,8-dioxo- 1 ,2,3 ,3a,5,6,7,8, 11 , 12,20,23 ,24,24a-tetradecahydro- 1 OH-9, 12-methanocyclopenta
- Step 2 ⁇ [(3aR, 7S, 10S, 12R,21E,24aS)-7-cyclopentyl-10-( ⁇ (lR,2S)-l- [( cyclopropylsulfonyl)carbamoyl ]-2-ethenylcyclopropyl ⁇ carbamoyl)-5, 8-dioxo- 1, 2, 3, 3a, 5, 6, 7, 8, 11,12, 20, 23, 24, 24a-tetradecahydro-l OH-9, 12-methanocyclopenta
- Step 1 The product of Step 1 (43 mg) was dissolved in THF (1 ml) and methanol (0.2 ml). Water (0.5 ml) and LiOH (11.95 mg) were added and the reaction was stirred until complete conversion. The reaction was quenched with IN HC1 (0.4 mL) and 5% KHS0 4 was added until the pH was 3. The mixture was extracted with ether then ethyl acetate. The combined organics were washed with water (5x) then brine, dried over magnesium sulfate, filtered and concentrated to yield 42 mg of pure product. HRMS (ES+) m/z 848.3522 (M+H) + .
- Example 67 (3ai?,7S,10SJ2i?,21E,24aS)-7-cvclopentyl-N- ⁇ (l ?,2S)-l-r(cvclopropyl sulfonyl)carbamoyll-2-ethenylcvclopropyU-5,8-dioxo-19- 2-oxo-2-(pyrrolidin-l-yl)ethoxyl- 1 ,2.3.3a.5.6,7,8, 11.12.20,23 ,24.24a-tetradecahvdro- 1 OH-9.12-methanocvclopenta
- Step 1 (SaR, 7S,10S,12R,21E,24aS)-19-(4- ⁇ [tert-butyl(dimethyl)silyl]oxy ⁇ butoxy)-7-cyclopentyl- N- ⁇ (lR,2S)-l-[(cyclopropylsulfonyl)carbamoyl]-2-ethenylcyclopropyl ⁇ -5,8-dioxo- 1, 2, 3, 3a, 5, 6, 7, 8, 11,12, 20, 23, 24, 24a-tetradecahydro-l OH-9, 12-methanocyclo
- Step 2 (3aR, 7S,10S,12R,21E,24aS)-7-cyclopentyl-N- ⁇ (lR,2S)-l-[(cyclopropyl
- Example 69 r3ai?.7 ⁇ 10 ,12i?.21E.24aS , )-7-cvclopentyl-N-i(Ti?,2 -l-rfcvclopropyl sulfonyl)carbamoyll-2-ethenylcvclopropyl
- Example 70 (Z ⁇ R SSQSA2R ⁇ E2 ⁇ aS)-l-tert ⁇ uty ⁇ -N-U ⁇ R2S) - ( c ⁇ o )roO ⁇
- the title compound was prepared using the same method as Example 70, using 3- bromopropanol-TBS ether and (3ai?,7S,105,12 ⁇ ,21E,24aS)-7-tert-butyl-N- ⁇ (lJ?,2S)-l- [(cyclopropylsulfonyl)carbamoyl]-2-ethenylcyclopropyl ⁇ -19-hydroxy-5,8-dioxo- l,2,3,3a,5,6,7,8,l l ,12,20,23,24,24a-tetradecahydro-10H-9,12-methanocyclo
- Example 71 (3aR SA0SA2R21E24aS)-7-tert-butyl-N- ⁇ (lR2S)-2-et enylA-m ⁇ methylcvclopropyDsulfonyllcarbamoyllcvclopropy ⁇ -ig-O-hvdroxypropoxy ⁇ -S ⁇ -dioxo-
- Example 72 r3aig.7SJ0S,12i?.21E,24aS)-7-cvclopentyl-N-(rii?,2 ⁇ )-l-rrcvclopropyl sulfonvDcarbamoyll -2-ethenylcyclopropyl ⁇ - 19-(3-hydroxypropoxy)-5 , 8-dioxo- L2,3 a,5.6.7,8,lia2.20.23.24.24a-tetradecahvdro-10H-9J2-methanocyclo
- Example 74 3-t ⁇ (3aR SA0SA2R2 ⁇ E24aS)-7-CYc ⁇ opentyl-l0-(UlR2S)-l- lYcyclopropyl sulfonvDcarbamoyll -2-ethenylcvclopropyl ⁇ carbamoyl)-5 , 8-dioxo- 1.2.3.3 a.5.6.7.8.11.12,20.23.24,24a-tetradecahydro- 1 OH-9.12-methanocvclo
- Example 72 To a solution of Example 72 (31 mg) in dichloromethane (0.5 mL) was added ⁇ , ⁇ -dimethylglycine (11.3 mg) then triethylamine (0.015 ml), A N'-dicyclo hexylcarbodiimide (18.9 mg) and DMAP (1.1 mg). The solution was stirred at room temperature for 3 days. The mixture was diluted with ether and the solids that were formed were filtered off. The filtrate was concentrated in vacuo. Purification by reverse phase HPLC (30-100% ACN/water w/ 0.15% TFA) yielded 21 mg of the desired product after workup with NaHC0 3 and ethyl acetate.
- Example 75 By following the procedures outlined in Example 75 and using the appropriate A, B and C intermediates and reagent (depicted below the structure as Int. and Rg., respectively), the following compounds were prepared.
- Example 93 ( , 3ai?.7S,10Sa2i?,21E,24aS)-7-cvclopentyl-10- rii?.2S -l-r( ' cvclo propylsulfonvDcarbamoyl] -2-ethenylcyclopropyl ⁇ carbamovD-5 ,8-dioxo- 1 ,2 ,3 ,3a, 5,6,7,8,1 l,12,20,23.24.24a-tetradecahvdro-10H-9,12-methanocvclopentari8.191 ⁇ 1 , 10,3 ,6]dioxadiazacyclononadecino [11,12-6]quinolin- 19-yl propan-2-ylcarbamate
- Example 17 enta[18,19][l ,10,3,6]dioxadiazacyclononadecino[l l,12- )]quinoline-10-carboxamide (Example 17) (30 mg) in dichloroethane (0.6 mL) was added isopropylisocyanate (0.037 mL) then DMAP (4.6 mg). The reaction mixture was heated to 50°C for 1 hour. After cooling back to room temperature, the mixture was diluted with ethyl acetate and water was added. The mixture was extracted with ethyl acetate (3x). The combined organics were dried over magnesium sulfate, filtered and concentrated.
- Example 94 (3ai?JS,10 ⁇ ,12ig.21E,24ay)-19-(3-aminopropoxyV7-cvclopentyl-N- ⁇ (li?,25 f )-l- [(cvclopropylsulfonyl)carbamoyll -2-ethenylcyclopropyl > -5 , 8 -dioxo-
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Organic Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Epidemiology (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- Biophysics (AREA)
- Genetics & Genomics (AREA)
- Immunology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Gastroenterology & Hepatology (AREA)
- Virology (AREA)
- Zoology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Peptides Or Proteins (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
The present invention relates to macrocyclic compounds of formula (I) that are useful as inhibitors of the hepatitis C virus (HCV) NS3 protease, their synthesis, and their use for treating or preventing HCV infections.
Description
TITLE OF THE APPLICATION
HCV NS3 PROTEASE INHIBITORS
FIELD OF THE INVENTION
The present invention relates to macrocyclic compounds that are useful as inhibitors of the hepatitis C virus (HCV) NS3 protease, the synthesis of such compounds, and the use of such compounds for treating HCV infection and/or reducing the likelihood or severity of symptoms of HCV infection. BACKGROUND OF THE INVENTION
Hepatitis C virus (HCV) infection is a major health problem that leads to chronic liver disease, such as cirrhosis and hepatocellular carcinoma, in a substantial number of infected individuals, estimated to be 2-15% of the world's population. There are an estimated 3.9 million infected people in the United States alone, according to the U.S. Center for Disease Control, roughly five times the number of people infected with the human immunodeficiency virus (HIV). According to the World Health Organization, there are more than 170 million infected individuals worldwide, with at least 3 to 4 million people being infected each year. Once infected, about 20% of people clear the virus, but about 80% of those infected harbor HCV the rest of their lives. Ten to 20% of chronically infected individuals eventually develop liver- destroying cirrhosis or cancer. The viral disease is transmitted parenterally by contaminated blood and blood products, contaminated needles, or sexually and vertically from infected mothers or carrier mothers to their off-spring.
Current treatments for HCV infection, which are restricted to immunotherapy with recombinant interferon-a alone or in combination with the nucleoside analog ribavirin, are of limited clinical benefit. Moreover, there is no established vaccine for HCV. Consequently, there is an urgent need for improved therapeutic agents that effectively combat chronic HCV infection. The current state of the art in the treatment of HCV infection has been discussed in the following references: Dymock et al, 2000, Antiviral Chem. & Chemotherapy 11 :79-96; Rosen et al, 1999, Molec. Med. Today 5:393-399; Moradpour et al, 1999, Euro. J. Gastroenterol.
Hepatol. 11 :1189-1202; Bartenschlager, 1997, Intervirology 40(5-6):378-393; Lauer et al, 2001, N. Engl. J. Med. 345:41-52; Dymock, 2001, Emerging Drugs 6:13-42; and Crabb, 2001, Science 294:506-507.
Several virally-encoded enzymes are putative targets for therapeutic intervention, including a metalloprotease (NS2-3), a serine protease (NS3), a helicase (NS3), and an RNA- dependent R A polymerase (NS5B). The NS3 protease is located in the N-terminal domain of the NS3 protein. Because it is responsible for an intramolecular cleavage at the NS3/4A site and for downstream intermolecular processing at the NS4A 4B, NS4B/5A and NS5A/5B junctions, the NS3 protease is considered a prime drug target. Previous research has identified classes of peptides, such as hexapeptides as well as tripeptides discussed in U.S. Patent Application Publications Nos. US2005/0020503, US2004/0229818, and US2004/00229776, showing degrees of activity in inhibiting the NS3 protease. Additional HSV NS3 protease inhibitors have been described in International Patent Applicaton Publication Nos. WO2008/057208 and
WO2008/057209. The aim of the present invention is to provide further compounds which exhibit activity against the HCV NS3 protease.
SUMMARY OF THE INVENTION
The present invention relates to novel macrocyclic compounds of formula (I) and/or pharmaceutically acceptable salts or hydrates thereof. These compounds are useful in the inhibition of HCV (hepatitis C virus) NS3 (non-structural 3) protease, the prevention or treatment of one or more of the symptoms of HCV infection, either as compounds or their pharmaceutically acceptable salts or hydrates (when appropriate), or as pharmaceutical composition ingredients. As pharmaceutical composition ingredients, these compounds, salts and hydrates may be the primary active therapeutic agent, and, when appropriate, may be combined with other therapeutic agents including but not limited to other HCV antivirals, anti- infectives, immunomodulators, antibiotics or vaccines. More particularly, the present invention relates to a compound of formula (I) and/or a pharmaceutically acceptable salt thereof:

(I) wherein:
Y is CH or N;
R1 is:
-OH,
-OCi-6alkyl,
-OC1-6alkyl-het1,
-Od-6alkyl-OH,
-OC 6alkyl-NRaRb,
-O-het!,
-OC1-6alkylC02H,
-OC1-6alkylC(=0)-het1,
-0(CH2)1-6OC(=0)CH2NRaRb,
-OC i -6alkyl-C i -6alkoxy,
-OC \ -6alkyl-C \ -6alkoxy-C \ -6alkoxy,
-OC(0)Cr6alkyl,
-OC(0)NRaRb,
-Od-ealkyl-S-het!,
-OC i - alkyl-phosphate, a phosphate group,
-(CH2)1-6-het!,
pyridinyl, or
thiazolyl;
said alkyl is optionally substituted with 1 or 2 fluoro substituents, said phosphate group is optionally substituted with 1, 2 or 3 C1-6alkyl;

a) aryl selected from phenyl or napthyl optionally substituted with 1 or 2 substituents selected from -OH, Cr6alkyl, or halo;
b) heteroaryl selected from 5- and 6-membered aromatic rings having 1, 2 or 3 heteroatoms independently selected from N, O and S, wherein said heteroaryl is attached through a ring atom selected from C or N and optionally substituted with 1 or 2 substituents independently selected from Chalky! and -OH; or
c) heterocycle selected from 4-7 membered monocyclic or 6-10
membered polycyclic bridged, linearly fused or spirocyclic saturated or unsaturated non-aromatic rings having 1, 2, 3 or 4 heteroatoms independently selected from N, O and S, wherein said heterocycle is attached through a ring atom selected from C or N and optionally substituted with 1 or 2 substituents independently selected from C1-6alkyl, oxo, -(CH2)mF, Boc, -(CH2)mCF3,
-(C¾)mOCF3, -OH, -NRaRb, -C1-6alkoxy, -(CH2)mS02CH3, aryl, -Q-ealkoxy-Ci-ealkyl, -C1-6alkyl-C1-6alkoxy optionally substituted with CF3, cyano, C(=0)NH2, C3-6cycloalkyl,
-C1-6alkyl-C3-6cycloalkyl, -COOC1-6alkyl,
-C1-6alkyl-S02C1-6alkyl, and benzimidazolyl wherein the benzimidazolyl is optionally substituted with F;
Ra and Rb are independently selected from H; d-6 alkyl; t-Boc; aryl; C3-6cycloalkyl optionally substituted with 1 or 2 fluoro substituents; C ealko y-Ci-ealkyl; tetrahydropyranyl; C^ealkyl-OH; C1-6alkyl-aryl; Q-6 alkyl-C(OH)-aryl; Q-ealkyl-imidazolyl optionally substituted with methyl, C ealkyl-benzimidazolyl optionally substituted with methyl; Crealkyl-pyrazolyl; Crealkyl-dihydrotriazole optionally substituted with oxo; or d-ealkyl-pyrrolidinyl optionally substituted with oxo;
wherein
m is 0 or 1 to 4;
said aryl is phenyl, naphthalenyl, tetrahydronapthalenyl, or 7-10 membered fused bicyclic ring structure wherein at least one of the rings is aromatic and is optionally substituted with 2 -OH;
said tetrahydropyranyl is optionally substituted with 1 oxo;
R2 is Ci-ealkyl, C2-6alkenyl, C3-C6cycloalkyl or NRcRd;
wherein
the C3-6cycloalkyl is optionally substituted with C1-6alkyl optionally substituted with -OH, morpholinyl, C1-6alkoxy, C1-6alkoxy-C1-6alkoxy, C1-6alkoxy-phenyl, or C1-6alkenyl;
Rc and Rd are independently H or C1- alkyl, or may be taken together, with the N to which they are attached, to form a 4-7-membered monocyclic ring;
R is C^alkyl, C2- alkenyl, C3-C6cycloalkyl, CF2 or CF3;
R4 is Ci-8 alkyl, C3-g cycloalkyl, Ci-8 alkyl-C3-8 cycloalkyl, adamantyl, dihydroindenyl, or a 4-8 membered heterocycloalkyl having 1 or 2 heteroatoms selected from N, O, or S, wherein R4 is optionally substituted with one or two substituents independently selected from (Ci- C6)alkyl, halo, and -0(C1-C6)alkyl; or
R3 and R4 together form heptene;
Z is C or N;
R5 is H or d-6alkyl; or R5 is absent when Z is N;
W is a bond, O or NR;
R is H or d-6alkyl;
X is absent or is halo, CF3, -OCHF2, -OCH2F, -OCD2F, -OCDF2, C C6alkyl, Crealkoxy, aryl, heteroaryl, or -0(CH2)1-6NRaRb;
A is absent, O or N;
B is (CH2)m; and
n is 1-4.
The present invention also includes pharmaceutical compositions containing a compound of the present invention and methods of preparing such pharmaceutical compositions. The present invention further includes methods of treating or reducing the likelihood or severity of one or more symptoms of HCV infection.
Other embodiments, aspects and features of the present invention are either further described in or will be apparent from the ensuing description, examples and appended claims. DETAILED DESCRIPTION OF THE INVENTION
The present invention relates to compounds of Formula (I) and pharmaceutically acceptable salts thereof, as defined above and a first embodiment of the invention. Different embodiments further describing Formula (I) variables are described below.
In a second embodiment of the invention, the present invention relates to c mpounds, or a pharmaceutically acceptable salt thereof, having a formula of


and pharmaceutically acceptable salts thereof, where all variables as provided for in the first embodiment.
In a third embodiment of the invention, the present invention relates to compounds of Formula (I), (la), (lb), (Ic), (Id) or (le) and pharmaceutically acceptable salts thereof, wherein Z is C and the other variables are as provided for in the first or second embodiments.
In a fourth embodiment of the invention, the present invention relates to compounds of Formula

and pharmaceutically acceptable salts thereof, wherein the variables are as provided for in the first embodiment.
In a fifth embodiment of the invention, the present invention relates to a compound having the formula of
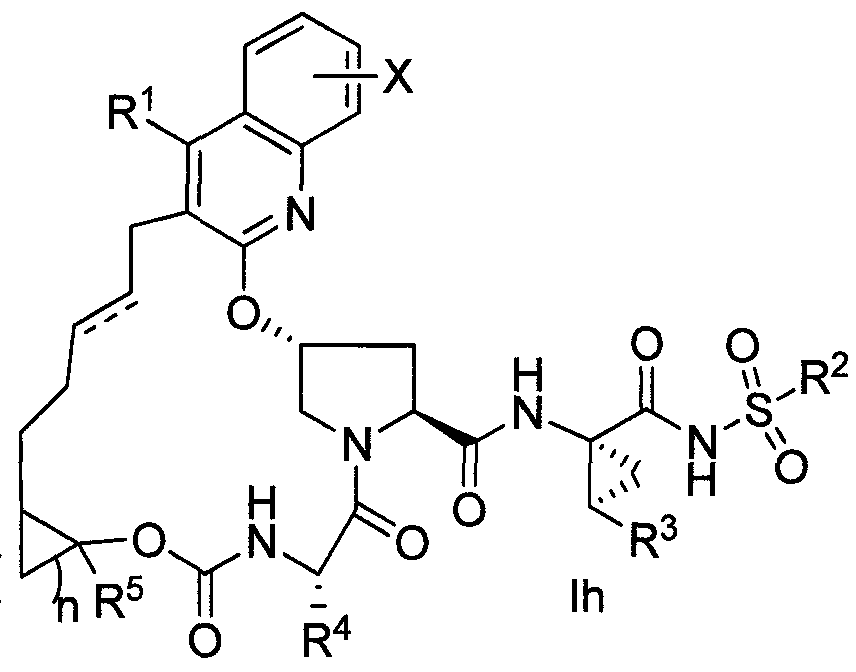
and pharmaceutically acceptable salts thereof, wherein the variables are as provided for in the first embodiment.
In a sixth embodiment of the invention, the present invention relates to compounds of Formula (I), (la), (lb), (Ic), (Id), (Ie), (If), (Ig) and (Ih) and pharmaceutically acceptable salts thereof, wherein R5 is H or CH3 wherein the other variables are as provided for in the first embodiment
In a seventh embodiment of the invention, the present invention relates to compounds of Formula (I), (la), (lb), (Ic), (Id), (Ie), (If), (Ig) and (Ih) and pharmaceutically acceptable salts thereof, wherein R is ethyl, ethylene, or cyclopropyl and the other variables are as provided for in the first or sixth embodiments. In one aspect of this embodiment, R3 is ethylene.
In a eighth embodiment of the invention, the present invention relates to compounds of Formula (I), (la), (lb), (Ic), (Id), (Ie), (If), (Ig) and (Ih) and pharmaceutically acceptable salts thereof, wherein R4 is propyl, t-butyl, cyclopentyl, cyclohexyl optionally substituted with 1 or 2 F, cyclohexylmethyl, methylcyclohexyl, methylcyclopentyl,
dihydroindenyl, or tetrahydro-2H-pyranyl, and the other variables are as provided for in any of the first, sixth or seventh embodiments. In one aspect of this embodiment, R4 is t-butyl, cyclopentyl, or cyclohexyl, 1 -methylcyclohexyl, propan-2-yl, 2,3 -dihydroindenyl, tetrahydro- 2H-pyranyl, or cyclohexylmethyl. In another aspect of this embodiment, R4 is t-butyl, cyclopentyl, or cyclohexyl, 1 -methylcyclohexyl, 2,3 -dihydroindenyl, or tetrahydro-2H-pyranyl.
In a ninth embodiment of the invention, the present invention relates to compounds of Formula (I), (la), (lb), (Ic), (Id), (Ie), (If), (Ig) and (Ih) and pharmaceutically acceptable salts thereof, wherein n is 1 or 3, and the other variables are as provided for in any of the first or sixth to eighth embodiments.
In a tenth embodiment of the invention, the present invention relates to compounds of Formula (I), (la), (lb), (Ic), (Id), (le), (If), (Ig) and (Ih) and pharmaceutically acceptable salts thereof, wherein R is cyclopropyl, N(CH3)2, or azetidinyl, wherein the cyclopropyl is optionally substituted with methyl, CH(CH3)2, C(CH3)=CH2; C(CH3)2OH, CH2CH2-morpholinyl, CH2OCH3, CH2OCH2CH2OCH3, or CH2OCH2-phenyl, and the other variables are as provided for in any of the first or sixth through ninth embodiments. In one aspect of this embodiment, R2 is cyclopropyl, N(CH3)2, (methyl)cyclopropyl,
(methoxymethyl)cyclopropyl, [(benzyloxy)methyl] cyclopropyl, l-(prop-l -en-2-yl)cyclopropyl, or l-[2-(morpholin-4-yl)ethyl]cyclopropyl. In another aspect of this embodiment, R2 is cyclopropyl, N(C¾)2, (methyl)cyclopropyl, or l-(methoxymethyl)cyclopropyl.
In an eleventh embodiment of the invention, the present invention relates to compounds of Formula (I), (la), (lb), (Ic), (Id), (le), (If), (Ig) and (Ih) and pharmaceutically acceptable salts thereof, wherein R1 is is -OH; -0-C1-6alkyl; -OC(0)Ci-6alkyl; -OC!-6alkyl- heti; -OCrealkyl-d-ealkoxy; -OCrealkyl-Crealkoxy-C ealkoxy; -OCH2C(=0)-het1;
-0(CH2)1-6 OC(=0)CH2NRaRb; -OC(0)NRaRb; -OCrealkyl-phosphate; -OCrealkyl-S-heti ; -O-heti; -O-d-ealkyl-OH optionally substituted with 1 or 2 fluoro substituents; or
-Od-ealkyl-NR'R1'
wherein Ra and Rb are independently
H,
Ci-6 alkyl, t-Boc,
C3-6cycloalkyl optionally substituted with 1 or 2 fluoro substituents,
Ci-6 alkyl-Ci-6alkoxy,
C ealkyl-OH, phenyl,
Q-ealkyl-phenyl, tetrahydropyranyl, Ci-6alkyl-C(OH>-phenyl,
naphthalenyl,
C i -6alkyl-naphthalenyl,
C i -6alkyl-dihydrooxopyrrolidinyl,
Crealkyl-benzimidazolyl optionally substituted with methyl, C i -6alkyl-pyrazolyl, d-ealkyl-triazole optionally substituted with oxo, or d-ealkyl-imidazolyl optionally substituted with methyl, and the other variables are as provided for in any of the first or sixth through tenth embodiments. In one aspect of this embodiment, heti is phenyl; oxazepanyl; oxooxazolidinyl; pyridinyl; pyrazolyl; pyrrolyl;
tetrahydropyranyl, triazolyl optionally substituted with C1-6alkyl; dioxolanyl; oxoimidazolidinyl; morpholinyl optionally substituted with dimethyl or ethyl; pyrrolidinyl optionally substituted with 1 or 2 substituents independently selected from oxo, Boc, C1-6alkyl, OH, C(0)NH2, dimethylamino, and methylsulfomyl; piperidinyl optionally substituted with 1 or 2 substituents independently selected from C1-6alkyl, C1-6alkoxy, Q-ealkoxy-C^alkyl optionally substituted with CF3, cyclopropyl-C1-6alkyl, cyclopropyl, -(CH2)mF, OH, -C1-6alkyl-S02C1-6alkyl,
-(CH2)mCF3, -COOC1-6alkyl, Boc, and benzimidazol; imidizolyl; thiazolyl optionally substituted with methyl; azabicycloheptyl; azaspiroheptyl; azaspirononyl; oxaazabiocycloheptyl;
oxaazaspiroheptyl optionally substituted with methoxyethyl; azetidinyl optionally substituted with 1 or 2 substituents independently selected from C1-6alkyl, Q-ealkoxy, cyano, fluoro, OH, phenyl and Boc; dioxidothiomorpholinyl; piperazinyl optionally substituted with 1 or 2 substituents independently selected from C1-6alkyl, C1-6alkyl-cyclopropyl, CF3, methylsulfonyl, Boc, and oxo; azabicyclooctyl substituted with C1-6alkyl, Q-ealkoxy-C^alkyl, -COOC1-6alkyl, or -(CH2)mCF3; oxaazabicyclononyl optionally substituted with Boc, C1-6alkyl, -COOC1- alkyl, C1-6alkoxy-C1-6alkyl or cyclopropylC1-6alkyl; or azabicyclooctanyl optionally substituted with C1-6alkyl. In one aspect of the invention, R1 is -OH; -OC(0)CH3; methoxy; ethoxy;
2-(tert-butylamino)ethoxy; 3-aminopropoxy; 2-methoxyethoxy; 3-methoxypropoxy;
3-(2-methoxyethoxy)propoxy; 3-(2-azabicyclo[2.2.1 ]hept-2-yl)propoxy;
2- (2-azaspiro[3.3]hept-2-yl)ethoxy; 3-(2-azaspiro[3.3]hept-2-yl)propoxy;
3- (2-azaspiro[4.4]non-2-yl)propoxy; azetidin-3-yloxy; 2-(azetidin-l-yl)ethoxy;
3 -(azetidin- 1 -yl)propoxy ; 3 - { [2-( 1 H-benzimidazol-2-yl)ethyl] (ethyl)amino }propoxy;
3-[benzyl(2-hydroxyethyl)amino]propoxy; 3-[bis(2-methoxyethyl)amino]propoxy;
3-[(2i?)-2-carbamoylpyrrolidin-l-yl]propoxy; 3-(3-cyanoazetidin-l-yl)propoxy;
3 -(cyclobutylamino)propoxy; 3 -(cyclopentylamino)propoxy; 2-(cyclopropylamino)ethoxy ;
[ 1 -(cyclopropylmethyl)piperidin-4-yl]oxy; ( 1 -cyclopropylpiperidin-4-yl)oxy;
3-(cyclopropylamino)propoxy; [l-(cyclopropylmethyl)azetidin-3-yl]oxy;
3 -(3 ,3 -difluoroazetidin- 1 -yl)propoxy ; 2,2,-difluoro-3 -hydroxypropoxy ;
3-{[2-(3,4-dihydroxyphenyl)-2-hydroxyethyl](propan-2-yl)amino}propoxy;
2-(dimethylamino)ethoxy; 3-(dimethylamino)propoxy; 2-(2,2-dimethylmorpholin-4-yl)ethoxy; ( 1 ,4-dimethylpiperazin-2-yl)methoxy; 3 -(dimethylamino)pyrrolidin- 1 -yl;
2- ( 1 ,3 -dioxalan-2-yl)ethoxy; 2-( 1 , 1 -dioxidothiomorpholin-4-yl)ethoxy ;
3- (l , 1 -dioxidothiomorpholin-4-yl)propoxy; 2-(2,5-dioxopyrrolidin- 1 -yl)ethoxy;
(4-ethylmorpholin-2-yl)methoxy; 1 -ethylpiperidin-3-yl)methoxy;
(1 -ethylpiperidin-4-yl)methoxy; ( 1 -ethylpiperidin-4-yl)oxy; 3 -(3 -fluoroazetidin- 1 -yl)propoxy; 3- [2-(5-fluoro- 1 H-benzimidazol-2-yl)piperidin- 1 -yl]propoxy; 3 -(3 -fluoropiperidin- 1 -yl)propoxy ;
2- (3-hydroxyazetidin-l-yl)ethoxy; 4-hydroxybutoxy; 2-hydroxyethoxy;
3 - hydroxy-3 -methylbutoxy; 2-(4-hydroxy-4-methylpiperidin-l-yl)ethoxy;
3-(3 -hydroxy-3 -methylpyrrolidin- 1 -yl)propoxy ; 3-(3 -hydroxy-3 -phenylazetidin- 1 -yl)propoxy ; 3 - [( 1 -hydroxy- 1 -phenylpropan-2-yl)(methyl)amino]propoxy; 3 -hydroxypropoxy;
3 -[(3if)-3 -hydroxy pyrrolidin-1 -yljpropoxy; 2-(lH-imidazol-l-yl)ethoxy;
3-[(lH-imidazol-2-ylmethyl)(methyl)amino]propoxy; 3-(lH-imidazol-l-yl)propoxy;
3 -(3 -methoxy azetidin- 1 -yl)propoxy ;
[(li?,4i?,5i?)-2-(2-methoxyethyl)-2-azabicyclo[2.2.1]hept-5-yl]oxy;
1 -(2-methoxyethyl)piperidin-4-yl]methoxy; [1 -(2-methoxyethyl)piperidin-4-yl]oxy;
3 -(4-methoxypiperidin- 1 -yl)propoxy; 3 - [( 1 -methoxypropan-2-yl)amino]propoxy;
3-(methylamino)propoxy; (l-methylazetidin-3-yl)methoxy;
3 - {methyl [(5 -methyl- 1 H-benzimidazol-2-yl)methyl] amino } propoxy ;
3 - { methyl [(5 -methyl 1 H-imidazol-2-yl)methyl] amino } propoxy;
3-{methyl[(5-oxo-4,5-dihydro-lH-l,2,4-triazol-3-yl)methyl]amino}propoxy;
3-(4-methyl-3-oxopiperazin-l-yl)propoxy;
3-{methyl[(5-oxopyrrolidin-2-yl)methyl]amino}propoxy; 3-(4-methylpiperazin-l-yl)propoxy; ( 1 -methylpiperidin-2-yl)oxy; ( 1 -methylpiperidin-4-yl)oxy ;
3-[methyl(lH-pyrazol-5-ylmethyl)amino]propoxy; (l-methylpyrrolidin-3-yl)methoxy;
3-[4-(methylsulfonyl)piperazin-l-yl]propoxy; 3-[3-(methyl sulfonyl)pyrrolidin-l -yljpropoxy; 3-[(4-methyl-4H-l,2,4-triazol-3-yl)sulfanyl]propoxy; (2i?)-morpholin-2-ylmethoxy;
2-(morpholin-4-yl)ethoxy ; 3 -(morpholin-4-yl)propoxy; 4-(morpholin-4-yl)butoxy;
3 - { [2-(naphthalen- 1 -yl)ethyl] amino } propoxy ;
3-[(lS,4S)-2-oxa-5-azabicyclo[2.2.1]hept-5-yl]propoxy;
3-(2-oxa-6-azaspiro[3.3]hept-6-yl)propoxy; 3-(l ,4-oxazepan-4-yl)propoxy;
3 -(2-oxoimidazolidin- 1 -yl-propoxy ; (2-oxo- 1 ,3 -oxazolidin-3-yl)ethoxy;
2-oxo-2-(pyrrolidin-l-yl)ethoxy; 2-(2-oxopyrrolidin-l-yl)ethoxy;
3- { [2-(2-oxopyrrolidin- 1 -yl)ethyl]amino}propoxy; 3-(2-oxopyrrolidin- 1 -yl)propoxy;
3-[(4-phenylbutyl)amino]propoxy; 2-(piperidin-l -yl)ethoxy; 3-(piperidin-l -yl)propoxy;
3 -(propan-2-ylamino)propoxy; 2-( 1 H-pyrazol- 1 -yl)ethoxy; 2-(pyridin-2-yl)ethoxy;
2- (pyridin-3-yl)ethoxy; 2-(pyridine-4-yl)ethoxy; 2-(lH-pyrrol-l-yl)ethoxy;
3-(lH-pyrrol-l-yl)propoxy; 2-(pyrrolidin-l-yl)ethoxy; 3-(pyrrolidin-l-yl)propoxy;
3- (l,2,3, 4-tetrahydronaphthalen-l-ylamino)propoxy;
3-(tetrahydro-2H-pyran-4-ylamino)propoxy; 3-(4H-l,2,4-triazol-4-yl)propoxy;
3-[3-(trifluoromethyl)piperazin-l-yl]propoxy; -0(CH2)3OC(=0)CH2N(CH3)2; -0(CH2)3NHBoc;
-OC(=0)NHCH(CH3)2; -0(CH2)3OP(-0)(OH)2;
[l-(tert-butoxycarbonyl)pyrrolidin-4-yl]methoxy, [l-(2,2,2-trifluoroethyl)piperidin-4-yl]oxy;
[ 1 -(tert-butoxycarbonyl)piperidin-4-yl]oxy ; 3 - [(3 ,3 -difluorocyclobutyl)amino]propoxy;
pyridin-2-yl; pyridine-3-yl; pyridine-4-yl; l,3-thiazol-2-yl; (morpholin-4-yl)methoxy;
tetrahydro-2H-pyran-4-yloxy; [7-(tert-butoxycarbonyl)-3-oxa-7-azabicyclo[3.3.1]non-9-yl]oxy;
[7-(cyclopropylmethyl)-3 -oxa-7-azabicyclo[3.3.1 ]non-9-yl]oxy;
[7-(2-methoxyethyl)-3-oxa-7-azabicyclo[3.3.1 ]non-9-yl]oxy;
3-oxa-7-azabicyclo[3.3.1 ]non-9-yl} oxy; (7-ethyl-3-oxa-7-azabicyclo[3.3.1 ]non-9-yl)oxy;
{l-[2-(trifluoromethoxy)ethyl]piperidin-4-yl}oxy; (8-methyl-8-azabicyclo[3.2.1]oct-3-yl)oxy;
[ 1 -(tert-butoxycarbonyl)piperazin-4-yl]propoxy; (4-methylpiperazin- 1 -yl)methoxy;
piperidin-4-yloxy; { 1 -[2-(methylsulfonyl)ethyl]piperidin-4-yl}oxy;
[(2-fluoroethyl)piperidin-4-yl]oxy; [8-(tert-butoxycarbonyl)-8-azabicyclo[3.2.1 ]oct-3-yl]oxy;
(8-azabicyclo[3.2.1]oct-3-yl)oxy; [8-(2,2,2-trifluoroethyl)-8-azabicyclo[3.2.1]oct-3-yl]oxy;
[8-(2-methoxyethyl)-8-azabicyclo[3.2.1]oct-3-yl]oxy; (8-ethyl-8-azabicyclo[3.2.1]oct-3-yl)oxy;
[9-(tert-butoxycarbonyl)-3-oxa-9-azabicyclo[3.3.1 ]non-7-yl]oxy;
3 -oxa-9-azabicyclo [3.3.1 ]non-7-yloxy ;
[9-(2-methoxyethyl)-3-oxa-9-azabicyclo[3.3.1]non-7-yl]oxy;
[9-ethyl)-3-oxa-9-azabicyclo[3.3.1]non-7-yl]oxy;
[l-(tert-butoxycarbonyl)-3-fluoro-piperidin-4-yl]oxy; (3-fluoropiperidin-4-yl)oxy; and (3 -fluoro- 1 -methylpiperidin-4-yl)oxy .
In a twelveth embodiment of the invention, the present invention relates to compounds of Formula (I), (la), (lb), (Ic), (Id), (Ie), (If), (Ig) and (Ih) and pharmaceutically acceptable salts thereof, wherein X is absent or selected from -Br, -CI, -F, methoxy, methyl, propanyl and CF3, and the other variables are as provided for in any of the first or sixth through eleventh embodiments.
In a thirteenth embodiment of the invention, the compound of the invention is selected from the exemplary species depicted in Examples 1 through 306 shown below (or a pharmaceutically acceptable salt thereof).
In a fourteenth embodiment of the invention, the compound of the invention is





In a fifteenth embodiment of the invention, the compound of the invention is one of the following compounds:
( 1 aR,5S, 1 1 Z, 12aS, 13aR, 16S, 19R,27E,31 aR)-N-[(l -methylcyclopropyl)sulfonyl]- 3,15,33-trioxo-26-[2-^iperidin-l-yl)ethoxy]-l ,la,3,4,5,6,7,8,9,10,12a,13,15,16,l ^
31 a-icosahydro-5,17: 16,19-dimethanodicyclopropa[12,13:28,29][l,20,3,14,17]
dioxatriazacyclononacosino [21 ,22-b]quinoline- 13a(l 4H)-carboxamide;
( 1 ai?,5S, 11 Z, 12aS, 13 ai?, 16S, 19i?,27E,31 ^
26-[2-(morpholin-4-yl)ethoxy]-3,15,33-trioxo-l ,la,3,4,5,6,7,8,9,10,12a,13,15,16,18,19,29,30,3 31a- icosahydro-5,17: 16,19-dimethanodicyclopropa[12,13:28,29][l,20,3,14,17]
dioxatriazacyclononacosino [21 ,22-6] quinoline- 13 a( 14H)-carboxamide;
(1 &R,5S, 1 lZ,12aS, 13aR,l 6S, 19i?,27E,31 a ?)-N-[(l -methylcyclopropyl)sulfonyl]- 26-[3-(morpholin-4-yl)propoxy]-3,15,33-trioxo-l,la,3,4,5,6,7,8,9,10,12a,13,15,16,18,19,2 31,31a-icosahydro-5,17: 16,19-dimethanodicyclopropa[12,13:28,29][l,20,3,14,17]
dioxatriazacyclononacosino[21,22-0]quinoline-13a(14H)-carboxamide; (la^,55,l lZ,12a5,13ai?,16S,19i?,27E,31ai?)-N-[(l-methylcyclopropyl)sulfonyl] 3,15,33-trioxo-26-[2-(pyrrolidin-l-yl)ethoxy]-l,la,3,4,5,6,7,8,9,10,12a,13,15,16,18,^
31a-icosahydro-5,17:16,19-dimethanodicyclopropa[12,13:28,29][l,20,3, 14,17]
dioxatriazacyclononacosino[21 ,22-Z>] quinoline- 13a( 14H)-carboxamide;
(loR,5S, 11Z, 12aS,l 3aR, 16S, 19i?,27E,31 a£)-26-methoxy-N-[(l - methylcyclopropyl)sulfonyl]-3,15,33-trioxo-l,la,3,4,5,6,7,8,9,10,12a,13,15,16,18,19,2
31 a-icosahydro-5 , 17:16,19-dimethanodicyclopropa[ 12, 13 :28,29] [ 1 ,20,3 , 14, 17]
dioxatriazacyclononacosino [21 ,22-£] quinoline- 13a( 14H)-carboxamide;
(1 a ,5S, 11Z,12aS, 13aR, 16S, 19R,3 laR)-N-[(l -methylcyclopropyl)sulfonyl]-26- [2-(morpholin-4-yl)ethoxy^
31,31a-docosahydro-5,17:16,19-dimethanodicyclopropa[12,13:28,29][l,20,3, 14,17]
dioxatriazacyclononacosino[21 ,22-b]quinoline- 13a(l 4H)-carboxamide;
( 1 aR,5S, 11 Z, 12aS, 13aJ?, 16S, 19i?,27E,31 aR)-N-[( 1 -methylcyclopropyl)sulfonyl]- 3,15,33-trioxo-26-[3-(piperidin-l^
31 a-icosahydro-5 , 17:16,19-dimethanodicyclopropa[ 12, 13 :28,29] [ 1 ,20,3 , 14, 17]
dioxatriazacyclononacosino [21 ,22-b] quinoline- 13a( 14H)-carboxamide;
(1 aR,5S, 11Z, 12aS, 13ai?,l 6S, 19i?,27E,31 ai?)-N-[(l -methylcyclopropyl)sulfonyl]- 3,15,33-trioxo-26-[3-(pyrrolidin-l-yl)propoxy]-l,la,3,4,5,6,7,8,9,10,12a,13, 15,16,18,19,29, 30,31 ,31 a-icosahydro-5, 17: 16, 19-dimethanodicyclopropa[l 2, 13 :28,29] [1 ,20,3, 14, 17]dioxatriaza cyclononacosino[21 ,22-Z>] quinoline- 13a(l 4H)-carboxamide; or
(la#,5S,l \Z,\2aS,l3aR,l6S,l9R,27E,3 lai?)-N-[(l-methylcyclopropyl)sulfonyl]-
26-[3-(4-methylpiperazin-l-yl)propoxy]-3,15,33-trioxo-l,la,3,4,5,6,7,8,9,10,12a,13,15,l^ 19,29,30,31,31a-icosahydro-5,17:16,19-dimethanodicyclopropa[12,13:28,29][l,20,3,14,17] dioxatriazacyclononacosino [21 ,22-b] quinoline- 13a( 14H)-carboxamide, or
a pharmaceutically acceptable salt of any of the above.
Other embodiments of the present invention include the following:
(a) A pharmaceutical composition comprising an effective amount of a compound of formula (I), in any of the described embodiments, and a pharmaceutically acceptable carrier.
(b) The pharmaceutical composition of (a), further comprising a second therapeutic agent selected from the group consisting of HCV antiviral agents,
immunomodulators, and anti-infective agents. In one aspect of the invention, the second therapeutic agent is ribavirin.
(c) The pharmaceutical composition of (b), wherein the HCV antiviral agent is an antiviral selected from the group consisting of HCV protease inhibitors and HCV NS5B polymerase inhibitors.
(d) A pharmaceutical combination which is (i) a compound of formula (I) and (ii) a second therapeutic agent selected from the group consisting of HCV antiviral agents, immunomodulators, and anti-infective agents; wherein the compound of formula (I) and the second therapeutic agent are each employed in an amount that renders the combination effective for inhibiting HCV NS3 protease, or for treating HCV infection and/or reducing the likelihood or severity of symptoms of HCV infection.
(e) The combination of (d), wherein the HCV antiviral agent is an antiviral selected from the group consisting of HCV protease inhibitors and HCV NS5B polymerase inhibitors.
(f) A method of inhibiting HCV NS3 protease in a subject in need thereof which comprises administering to the subject an effective amount of a compound of formula (I).
(g) A method of treating HCV infection and/or reducing the likelihood or severity of symptoms of HCV infection in a subject in need thereof which comprises
administering to the subject an effective amount of a compound of formula (I).
(h) The method of (g), wherein the compound of formula (I) is administered in combination with an effective amount of at least one second therapeutic agent selected from the group consisting of HCV antiviral agents, immunomodulators, and anti- infective agents.
(i) The method of (h), wherein the HCV antiviral agent is an antiviral selected from the group consisting of HCV protease inhibitors and HCV NS5B polymerase inhibitors.
(j) A method of inhibiting HCV NS3 protease in a subject in need thereof which comprises administering to the subject the pharmaceutical composition of (a), (b), or (c) or the combination of (d) or (e).
(k) A method of treating HCV infection and/or reducing the likelihood or severity of symptoms of HCV infection in a subject in need thereof which comprises
administering to the subject the pharmaceutical composition of (a), (b), or (c) or the combination of (d) or (e).
In the embodiments of the compounds provided above, it is to be understood that each embodiment may be combined with one or more other embodiments, to the extent that such a combination provides a stable compound and is consistent with the description of the embodiments. It is further to be understood that the embodiments of compositions and methods provided as (a) through (k) above are understood to include all embodiments of the compounds, including such embodiments as result from combinations of embodiments.
The present invention also includes a compound of the present invention for use (i) in, (ii) as a medicament for, or (iii) in the preparation of a medicament for: (a) inhibiting HCV NS3 protease, or (b) treating HCV infection and/or reducing the likelihood or severity of symptoms of HCV infection. In these uses, the compounds of the present invention can optionally be employed in combination with one or more second therapeutic agents selected from HCV antiviral agents, anti-infective agents, and immunomodulators. In one aspect of the invention, the second therapeutic agent is ribavirin.
Additional embodiments of the invention include the pharmaceutical compositions, combinations and methods set forth in (a)-(k) above and the uses set forth in the preceding paragraph, wherein the compound of the present invention employed therein is a compound of one of the embodiments, aspects, classes, sub-classes, or features of the
compounds described above. In all of these embodiments, the compound may optionally be used in the form of a pharmaceutically acceptable salt or hydrate as appropriate.
As used herein, all ranges are inclusive, and all sub-ranges are included within such ranges, although not necessarily explicitly set forth. In addition, the term "or," as used herein, denotes alternatives that may, where appropriate, be combined; that is, the term "or" includes each listed alternative separately as well as their combination.
As used herein, represents a bond where the dotted line is an optional bond. When the optional bond is present, the bond (in its entirety) is a double bond. When the
optional bond is absent, the bond (it its entirety) is a single bond. Each such bond is independently a single bond or a double bond. Thus, when two such bonds are adjacent to each other, it can represent two single bonds, two double bonds, a single bond adjacent to a double bond, or a double bond adjacent to a single bond.
As used herein, the term "alkyl" refers to any linear or branched chain alkyl group having a number of carbon atoms in the specified range. Thus, for example, "C1-6 alkyl" (or "Ci-Ce alkyl") refers to all of the hexyl alkyl and pentyl alkyl isomers as well as n-, iso-, sec- and t-butyl, n- and isopropyl, ethyl and methyl. As another example, "CM alkyl" refers to n-, iso-, sec- and t-butyl, n- and isopropyl, ethyl and methyl. Alkyl groups may be substituted as indicated.
The term "halogenated" refers to a group or molecule in which a hydrogen atom has been replaced by a halogen. Similarly, the term "haloalkyl" refers to a halogenated alkyl group. The term "halogen" (or "halo") refers to atoms of fluorine, chlorine, bromine and iodine
(alternatively referred to as fluoro, chloro, bromo, and iodo).
The term "alkoxy" refers to an "alkyl-O-" group. Alkoxy groups may be substituted as indicated.
The term "alkylene" refers to any linear or branched chain alkylene group (or alternatively "alkanediyl") having a number of carbon atoms in the specified range. Thus, for example, "-C^ alkylene-" refers to any of the C\ to C6 linear or branched alkylenes. A class of alkylenes of particular interest with respect to the invention is -(CH2)1-6-, and sub-classes of particular interest include -(Ώ¼) -, -(ϋ¾)1-3-, -(CH2)1-2-, and -CH2-. Also of interest is the alkylene -CH(CH3)-. Alkylene groups may be substituted as indicated.
The term "cycloalkyl" refers to any monocyclic or bicyclic ring structure of an alkane or alkene having a number of carbon atoms in the specified range. Thus, for example, "C3-8 cycloalkyl" (or "C3-C8 cycloalkyl") includes cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, and cyclooctyl. The term "cycloalkoxy" refers to a "cycloalkyl-O-" group. Cycloalkyl groups may be substituted as indicated.
The term "carbocycle" (and variations thereof such as "carbocyclic" or
"carbocyclyl") as used herein, unless otherwise indicated, refers to (i) a C3 to C8 monocyclic, saturated or unsaturated ring or (ii) a C7 to C12 bicyclic saturated or unsaturated ring system.
Each ring in (ii) is either independent of, or fused to, the other ring, and each ring is saturated or unsaturated. Carbocycle groups may be substituted as indicated, for example with C1-6 alkyl,
C1-6 alkenyl, C1-6 alkynyl, aryl, halogen, -NH2 or -OH. The carbocycle may be attached to the rest of the molecule at any carbon atom which results in a stable compound. The fused bicyclic carbocycles are a subset of the carbocycles; i.e., the term "fused bicyclic carbocycle" generally refers to a C7 to C10 bicyclic ring system in which each ring is saturated or unsaturated and two adjacent carbon atoms are shared by each of the rings in the ring system. A fused bicyclic carbocycle in which both rings are saturated is a saturated bicyclic ring system. Saturated carbocyclic rings are also referred to as cycloalkyl rings, e.g., cyclopropyl, cyclobutyl, etc. A fused bicyclic carbocycle in which one or both rings are unsaturated is an unsaturated bicyclic ring system. A subset of the fused bicyclic unsaturated carbocycles are those bicyclic carbocycles in which one ring is a benzene ring and the other ring is saturated or unsaturated, with attachment via any carbon atom that results in a stable compound. Representative examples

Depicted ring systems include, where appropriate, able to
which a particular ring atom is attached. For example, the indole
 shows ring atom 2 is directly attached to variable X and ring atom 4 is directly attached to variable Z.
shows ring atom 2 is directly attached to variable X and ring atom 4 is directly attached to variable Z.
Variable R5 is shown as a floating variable which can be attached to any ring atom, provided that such attachment results in formation of a stable ring.
The term "aryl" refers to aromatic mono- and poly-carbocyclic ring systems, also referred to as "arenes," wherein the individual carbocyclic rings in the polyring systems are fused or attached to each other via a single bond. Suitable aryl groups include phenyl, naphthyl, and biphenylenyl. Aryl groups may be substituted as indicated.
Unless indicated otherwise, the term "heterocycle" (and variations thereof such as
"heterocyclic" or "heterocyclyl") broadly refers to (i) a stable 4- to 8-membered, saturated or unsaturated monocyclic ring, (ii) a stable 7- to 12-membered bicyclic ring system, or (iii) a stable
11- to 15-membered tricyclic ring sty stem, wherein each ring in (ii) and (iii) is independent of, or fused to, the other ring or rings and each ring is saturated or unsaturated, and the monocyclic
ring, bicyclic ring system or tricyclic ring system contains one or more heteroatoms (e.g., from 1 to 6 heteroatoms, or from 1 to 4 heteroatoms) independently selected from N, O and S and a balance of carbon atoms (the monocyclic ring typically contains at least one carbon atom and the bicyclic and tricyclic ring systems typically contain at least two carbon atoms); and wherein any one or more of the nitrogen and sulfur heteroatoms is optionally oxidized, and any one or more of the nitrogen heteroatoms is optionally quaternized. Unless otherwise specified, the heterocyclic ring may be attached at any heteroatom or carbon atom, provided that attachment results in the creation of a stable structure. Heterocycle groups may be substituted as indicated, and unless otherwise specified, the substituents may be attached to any atom in the ring, whether a heteroatom or a carbon atom, provided that a stable chemical structure results.
Saturated heterocyclics form a subset of the heterocycles. Unless expressly stated to the contrary, the term "saturated heterocyclic" generally refers to a heterocycle as defined above in which the entire ring system (whether mono- or poly-cyclic) is saturated. The term "saturated heterocyclic ring" refers to a 4- to 8-membered saturated monocyclic ring, a stable 7- to 12-membered bicyclic ring system, or a stable 11- to 15-membered tricyclic ring system, which consists of carbon atoms and one or more heteroatoms independently selected from N, O and S. Representative examples include piperidinyl, piperazinyl, azepanyl, pyrrolidinyl, pyrazolidinyl, imidazolidinyl, oxazolidinyl, isoxazolidinyl, morpholinyl, thiomorpholinyl, thiazolidinyl, isothiazolidinyl, and tetrahydrofuryl (or tetrahydrofuranyl). Saturated
heterocyclics include 4-8 membered heterocycloalkyls having 1 to 2 heteroatoms selected from N, O, and S.
Unsaturated heterocyclics form another subset of the heterocycles. Unless expressly stated to the contrary, the term "unsaturated heterocyclic" generally refers to a heterocycle as defined above in which the entire ring system (whether mono- or poly-cyclic) is not saturated, i.e., such rings are either unsaturated or partially unsaturated. Unless expressly stated to the contrary, the term "heteroaromatic ring" refers a stable 5- or 6-membered monocyclic aromatic ring, a stable 7- to 12-membered bicyclic ring system, or a stable 11- to 15-membered tricyclic ring system, which consists of carbon atoms and one or more heteroatoms selected from N, O and S. In the case of substituted heteraromatic rings containing at least one nitrogen atom (e.g., pyridine), such substitutions can be those resulting in N-oxide formation. Representative examples of heteroaromatic rings include pyridyl, pyrrolyl, pyrazinyl,
pyrimidinyl, pyridazinyl, thienyl (or thiophenyl), thiazolyl, furanyl, imidazolyl, pyrazolyl, triazolyl, tetrazolyl, oxazolyl, isooxazolyl, oxadiazolyl, thiazolyl, isothiazolyl, and thiadiazolyl.
Representative examples of bicyclic heterocycles include benzotriazolyl, indolyl, isoindolyl, indazolyl, indolinyl, isoindolinyl, quinoxalinyl, quinazolinyl, cinnolinyl, chromanyl, isochromanyl, tetrahydroquinolinyl, quinolinyl, tetrahydroisoquinolinyl, isoquinolinyl,
2,3-dihydrobenzofuranyl, 2,3-dihydrobenzo-l,4-dioxinyl (i.e., CO υ ), imidazo(2,l-6)(l,3)thiazole, (i.e.,
 )),, aanndd bbeennzzoo--ll,,33--ddiiooxxoollyyll ((ii..ee..,, ). In
)),, aanndd bbeennzzoo--ll,,33--ddiiooxxoollyyll ((ii..ee..,, ). In
 is alternatively referred to as phenyl having as a substituent methylenedioxy attached to two adjacent carbon atoms.
is alternatively referred to as phenyl having as a substituent methylenedioxy attached to two adjacent carbon atoms.
Unless expressly stated to the contrary, all ranges cited herein are inclusive. For example, a heteroaryl ring described as containing from "1 to 3 heteroatoms" means the ring can contain 1, 2, or 3 heteroatoms. It is also to be understood that any range cited herein includes within its scope all of the sub-ranges within that range. The oxidized forms of the heteroatoms N and S are also included within the scope of the present invention.
When any variable (e.g., R7 and R10) occurs more than one time in any constituent or in formula (I) or in any other formula depicting and describing compounds of the invention, its definition on each occurrence is independent of its definition at every other occurrence. Also, combinations of substituents and/or variables are permissible only if such combinations result in stable compounds.
Unless expressly stated to the contrary, substitution by a named substituent is permitted on any atom in a ring (e.g., aryl, a heteroaromatic ring, or a saturated heterocyclic ring) provided such ring substitution is chemically allowed and results in a stable compound. A "stable" compound is a compound which can be prepared and isolated and whose structure and properties remain or can be caused to remain essentially unchanged for a period of time sufficient to allow use of the compound for the purposes described herein (e.g., therapeutic or prophylactic administration to a subject).
As a result of the selection of substituents and substituent patterns, certain of the compounds of the present invention can have asymmetric centers and can occur as mixtures of
stereoisomers, or as individual diastereomers, or enantiomers. All isomeric forms of these compounds, whether isolated or in mixtures, are within the scope of the present invention.
As would be recognized by one of ordinary skill in the art, certain of the compounds of the present invention can exist as tautomers. For the purposes of the present invention a reference to a compound of formula (I) is a reference to the compound per se, or to any one of its tautomers per se, or to mixtures of two or more tautomers.
In the compounds of generic Formula I, the atoms may exhibit their natural isotopic abundances, or one or more of the atoms may be artificially enriched in a particular isotope having the same atomic number, but an atomic mass or mass number different from the atomic mass or mass number predominantly found in nature. The present invention is meant to include all suitable isotopic variations of the compounds of generic Formula I. For example, different isotopic forms of hydrogen (H) include protium ( H) and deuterium ( H). Protium is the predominant hydrogen isotope found in nature. Enriching for deuterium may afford certain therapeutic advantages, such as increasing in vivo half-life or reducing dosage requirements, or may provide a compound useful as a standard for characterization of biological samples.
Isotopically-enriched compounds within generic Formula I can be prepared without undue experimentation by conventional techniques well known to those skilled in the art or by processes analogous to those described in the Schemes and Examples herein using appropriate isotopically-enriched reagents and/or intermediates.
The compounds of the present inventions are useful in the inhibition of HCV protease (e.g., HCV NS3 protease) and the treatment of HCV infection and or reduction of the likelihood or severity of symptoms of HCV infection. For example, the compounds of this invention are useful in treating infection by HCV after suspected past exposure to HCV by such means as blood transfusion, exchange of body fluids, bites, accidental needle stick, or exposure to patient blood during surgery.
The compounds of this invention are useful in the preparation and execution of screening assays for antiviral compounds. For example, the compounds of this invention are useful for isolating enzyme mutants, which are excellent screening tools for more powerful antiviral compounds. Furthermore, the compounds of this invention are useful in establishing or determining the binding site of other antivirals to HCV protease, e.g., by competitive inhibition. Thus, the compounds of this invention may be commercial products to be sold for these purposes.
The compounds of the present invention may be administered in the form of pharmaceutically acceptable salts. The term "pharmaceutically acceptable salt" refers to a salt which possesses the effectiveness of the parent compound and which is not biologically or otherwise undesirable (e.g., is neither toxic nor otherwise deleterious to the recipient thereof). Suitable salts include acid addition salts which may, for example, be formed by mixing a solution of the compound of the present invention with a solution of a pharmaceutically acceptable acid such as hydrochloric acid, sulfuric acid, acetic acid, trifluoroacetic acid, or benzoic acid. Many of the compounds of the invention carry an acidic moiety, in which case suitable pharmaceutically acceptable salts thereof can include alkali metal salts (e.g., sodium or potassium salts), alkaline earth metal salts (e.g., calcium or magnesium salts), and salts formed with suitable organic ligands such as quaternary ammonium salts. Also, in the case of an acid (-COOH) or alcohol group being present, pharmaceutically acceptable esters can be employed to modify the solubility or hydrolysis characteristics of the compound.
The term "administration" and variants thereof (e.g., "administering" a compound) in reference to a compound of the invention mean providing the compound or a prodrug of the compound to the individual in need of treatment. When a compound of the invention or a prodrug thereof is provided in combination with one or more other active agents (e.g., antiviral agents useful for treating HCV infection), "administration" and its variants are each understood to include concurrent and sequential provision of the compound or salt (or hydrate) and other agents.
As used herein, the term "prodrug" is intended to encompass an inactive drug form or compound that is converted into an active drug form or compound by the action of enzymes, chemicals or metabolic processes in the body of an individual to whom it is administered.
As used herein, the term "composition" is intended to encompass a product comprising the specified ingredients, as well as any product which results, directly or indirectly, from combining the specified ingredients.
By "pharmaceutically acceptable" is meant that the ingredients of the
pharmaceutical composition must be compatible with each other and not deleterious to the recipient thereof.
The term "subject" (alternatively referred to herein as "patient") as used herein refers to an animal, preferably a mammal, most preferably a human, who has been the object of treatment, observation or experiment.
The term "effective amount" as used herein means that amount of active compound or pharmaceutical agent that elicits the biological or medicinal response in a tissue, system, animal or human that is being sought by a researcher, veterinarian, medical doctor or other clinician. In one embodiment, the effective amount is a "therapeutically effective amount" for the alleviation of one or more symptoms of the disease or condition being treated. In another embodiment, the effective amount is a "prophylactically effective amount" for reduction of the severity or likelihood of one or more symptoms of the disease or condition. The term also includes herein the amount of active compound sufficient to inhibit HCV NS3 protease and thereby elicit the response being sought (i.e., an "inhibition effective amount"). When the active compound (i.e., active ingredient) is administered as the salt, references to the amount of active ingredient are to the free acid or free base form of the compound.
For the purpose of inhibiting HCV NS3 protease and treating HCV infection and/or reducing the likelihood or severity of symptoms of HCV infection, the compounds of the present invention, optionally in the form of a salt or a hydrate, can be administered by any means that produces contact of the active agent with the agent's site of action. They can be
administered by any conventional means available for use in conjunction with pharmaceuticals, either as individual therapeutic agents or in a combination of therapeutic agents. They can be administered alone, but typically are administered with a pharmaceutical carrier selected on the basis of the chosen route of administration and standard pharmaceutical practice. The compounds of the invention can, for example, be administered orally, parenterally (including subcutaneous injections, intravenous, intramuscular, intrasternal injection or infusion
techniques), by inhalation (such as in a spray form), or rectally, in the form of a unit dosage of a pharmaceutical composition containing an effective amount of the compound and conventional non-toxic pharmaceutically-acceptable carriers, adjuvants and vehicles. Liquid preparations suitable for oral administration (e.g., suspensions, syrups, elixirs and the like) can be prepared according to techniques known in the art and can employ any of the usual media such as water, glycols, oils, alcohols and the like. Solid preparations suitable for oral administration (e.g., powders, pills, capsules and tablets) can be prepared according to techniques known in the art and can employ such solid excipients as starches, sugars, kaolin, lubricants, binders,
disintegrating agents and the like. Parenteral compositions can be prepared according to techniques known in the art and typically employ sterile water as a carrier and optionally other ingredients, such as solubility aids. Injectable solutions can be prepared according to methods known in the art wherein the carrier comprises a saline solution, a glucose solution or a solution containing a mixture of saline and glucose. Further description of methods suitable for use in preparing pharmaceutical compositions of the present invention and of ingredients suitable for use in said compositions is provided in Remington's Pharmaceutical Sciences, 18 edition (ed. A. R. Gennaro, Mack Publishing Co., 1990).
The compounds of this invention can be administered orally in a dosage range of 0.001 to 1000 mg/kg of mammal (e.g., human) body weight per day in a single dose or in divided doses. One dosage range is 0.01 to 500 mg/kg body weight per day orally in a single dose or in divided doses. Another dosage range is 0.1 to 100 mg/kg body weight per day orally in single or divided doses. For oral administration, the compositions can be provided in the form of tablets or capsules containing 1.0 to 500 mg of the active ingredient, particularly 1, 5, 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 400, and 500 mg of the active ingredient for the symptomatic adjustment of the dosage to the patient to be treated. The specific dose level and frequency of dosage for any particular patient may be varied and will depend upon a variety of factors including the activity of the specific compound employed, the metabolic stability and length of action of that compound, the age, body weight, general health, sex, diet, mode and time of administration, rate of excretion, drug combination, the severity of the particular condition, and the host undergoing therapy.
As noted above, the present invention also relates to a method of inhibiting HCV NS3 protease, inhibiting HCV replication, treating HCV infection and/or reducing the likelihood or severity of symptoms of HCV infection with a compound of the present invention in combination with one or more therapeutic agents and a pharmaceutical composition comprising a compound of the present invention and one or more therapeutic agents selected from the group consisting of a HCV antiviral agent, an immunomodulator, and an anti-infective agent. Such therapeutic agents active against HCV include, but are not limited to, ribavirin, levovirin, viramidine, thymosin alpha- 1, R7025 (an enhanced interferon (Roche)), interferon-β, interferon- a, pegylated interferon-a (peginterferon-a), a combination of interferon- and ribavirin, a combination of peginterferon-a and ribavirin, a combination of interferon-α and levovirin, and a combination of peginterferon-α and levovirin. Interferon-α includes, but is not limited to,
recombinant interferon-a2a (such as Roferon interferon available from Hoffmann-LaRoche, Nutley, NJ), pegylated interferon-a2a (Pegasys™), interferon-a2b (such as Intron-A interferon available from Schering Corp., Kenilworth, NJ), pegylated interferon-a2b (Peglntron™), a recombinant consensus interferon (such as interferon alphacon-1), albuferon (interferon-a bound to human serum albumin (Human Genome Sciences), and a purified interferon-α product.
Amgen's recombinant consensus interferon has the brand name Infergen®. Levovirin is the L-enantiomer of ribavirin which has shown immunomodulatory activity similar to ribavirin. Viramidine represents an analog of ribavirin disclosed in International Patent Application Publication No. WO 01/60379. In accordance with the method of the present invention, the individual components of the combination can be administered separately at different times during the course of therapy or concurrently in divided or single combination forms.
For the treatment of HCV infection, the compounds of the present invention may also be administered in combination with an agent that is an inhibitor of HCV NS3 serine protease. HCV NS3 serine protease is an essential viral enzyme and has been described to be an excellent target for inhibition of HCV replication. Both substrate and non-substrate based inhibitors of HCV NS3 protease inhibitors are disclosed in International Patent Application Publication Nos. WO 98/22496, WO 98/46630, WO 99/07733, WO 99/07734, WO 99/38888, WO 99/50230, WO 99/64442, WO 00/09543, WO 00/59929, WO 02/48116 and WO 02/48172, British Patent No. GB 2 337 262, and U.S. Patent No. 6,323,180.
Ribavirin, levovirin, and viramidine may exert their anti-HCV effects by modulating intracellular pools of guanine nucleotides via inhibition of the intracellular enzyme inosine monophosphate dehydrogenase (IMPDH). IMPDH is the rate-limiting enzyme on the biosynthetic route in de novo guanine nucleotide biosynthesis. Ribavirin is readily
phosphorylated intracellularly and the monophosphate derivative is an inhibitor of IMPDH. Thus, inhibition of IMPDH represents another useful target for the discovery of inhibitors of HCV replication. Therefore, the compounds of the present invention may also be administered in combination with an inhibitor of IMPDH, such as VX-497, which is disclosed in International Patent Application Publication Nos. WO 97/41211 and WO 01/00622; another IMPDH inhibitor, such as that disclosed in International Patent Application Publication No. WO 00/25780; or mycophenolate mofetil. See Allison et i, 1993, Agents Action 44 (Suppl.):165.
For the treatment of HCV infection, the compounds of the present invention may also be administered in combination with the antiviral agent amantadine (1-aminoadamantane).
For a comprehensive description of this agent, see Kirschbaum, 1983, Anal. Profiles Drug Subs. 12:1-36.
For the treatment of HCV infection, the compounds of the present invention may also be administered in combination with the antiviral agent polymerase inhibitor R7128
(Roche).
The compounds of the present invention may also be combined for the treatment of HCV infection with antiviral 2'-C-branched ribonucleosides disclosed in Harry-O'Kuru et ah, 1997, J. Org. Chem. 62:1754-59; Wolfe et al, 1995, Tet. Lett. 36:7611-14; U.S. Patent No. 3,480,613; and International Patent Application Publication Nos. WO 01/90121, WO 01/92282, WO 02/32920, WO 04/002999, WO 04/003000 and WO 04/002422. Such 2'-C-branched ribonucleosides include, but are not limited to, 2'-C-methyl-cytidine, 2'-C-methyl-uridine, -C- methyl-adenosine, 2'-C-methyl-guanosine, and 9-(2-C-methyl-P-D-ribofuranosyl)-2,6- diaminopurine, and the corresponding amino acid ester of the ribose C-2', C-3', and C-5' hydroxyls and the corresponding optionally substituted cyclic 1,3 -propanediol esters of the 5'- phosphate derivatives.
The compounds of the present invention may also be combined for the treatment of HCV infection with other nucleosides having anti-HCV properties, such as those disclosed in International Patent Application Publication Nos. WO 02/51425, WO 01/79246, WO 02/32920, WO 02/48165, WO05/003147 (including R1656, (2'i?)-2'-deoxy-2'-fluoro-2'-C-methylcytidine), WO 01/68663, WO 99/43691, WO 02/18404, WO06/021341, WO 02/100415, WO 03/026589, WO 03/026675, WO 03/093290, WO 04/011478, WO 04/013300 and WO 04/028481, and U.S. Patent Application Publication Nos. US2005/0038240 (including 4'-azido nucleosides such as R1626, 4'-azidocytidine), US2002/0019363, US2003/0236216, US2004/0006007 and
US2004/0063658.
For the treatment of HCV infection, the compounds of the present invention may also be administered in combination with an agent that is an inhibitor of HCV NS5B polymerase. Such HCV NS5B polymerase inhibitors that may be used as combination therapy include, but are not limited to, those disclosed in International Patent Application Publication Nos.
WO 02/057287, WO 02/057425, WO 03/068244, WO 04/000858, WO 04/003138 and
WO 04/007512; U.S. Patent No. 6,777,392 and U.S. Patent Application Publication No.
US2004/0067901. Other such HCV polymerase inhibitors include, but are not limited to,
valopicitabine (NM-283; Idenix) and 2'-F-2'-beta-methylcytidine (see also International Patent Application Publication No. WO 05/003147).
In one embodiment, nucleoside HCV NS5B polymerase inhibitors that are used in combination with the present HCV NS3 protease inhibitors are selected from the following compounds: 4-amino-7-(2-C-methyl-P-D-arabinofuranosyl)-7H-pyrrolo[2,3-if]pyrimidine; 4- amino-7-(2-C-methyl-P-D-ribofiiranosyl)-7H-pyrrolo[2,3-i ]pyrimidine; 4-methylamino-7-(2-C- methyl-P-D-ribofuranosyl)-7H-pyrrolo[2,3-£ ]pyrimidine; 4-dimethylamino-7-(2-C-methyl-P-D- ribofuranosyl)-7H-pyrrolo[2,3-cf|pyrimidine; 4-cyclopropylamino-7-(2-C-methyl-P-D- ribofuranosyl)-7H-pyrrolo[2,3-fii]pyrimidine; 4-amino-7-(2-C-vinyl-P-D-ribofuranosyl)-7H- pyrrolo[2,3-fif]pyrimidine; 4-amino-7-(2-C-hydroxymethyl-P-D-ribofuranosyl)-7H- pyrrolo[2,3-fii]pyrimidine; 4-amino-7-(2-C-fluoromethyl-P-D-ribofuranosyl)-7H- pyrrolo[2,3-i ]pyrimidine; 4-amino-5-methyl-7-(2-C-methyl-P-D-ribofuranosyl)-7H-
 4-amino-7-(2-C-methyl-P-D-ribofuranosyl)-7//- pyrrolo[2,3-i¾pyrimidine-5-carboxylic acid; 4-amino-5-bromo-7-(2-C-methyl-P-D- ribofuranosyl)-7H-pyrrolo[2,3-i ]pyrimidine; 4-amino-5-chloro-7-(2-C-methyl-P-D- ribofuranosyl)-7H-pyrrolo[2,3-<i]pyrimidine; 4-amino-5-fluoro-7-(2-C-methyl-P-D- ribofuranosyl)-7H-pyrrolo[2,3-<flpyrimidine; 2,4-diamino-7-(2-C-methyl-P-D-ribofuranosyl)- 7H-pyrrolo[2,3-cf]pyrimidine; 2-amino-7-(2-C-methyl-P-D-ribofuranosyl)-7H- pyrrolo[2,3-(i]pyrimidine; 2-amino-4-cyclopropylamino-7-(2-C-methyl-P-D-ribofuranosyl)-7H- pyrrolo[2,3-i ]pyrimidine; 2-amino-7-(2-C-methyl-P-D-ribofuranosyl)-7H- pyrroloP^-^pyrimidin^pHj-one; 4-amino-7-(2-C-ethyl-P-D-ribofuranosyl)-7H- pyrrolo[2,3-i ]pyrimidine; 4-amino-7-(2-C,2-0-dimethyl-P-D-ribofuranosyl)-7H- pyrrolo[2,3-<i]pyrimidine; 7-(2-C-methyl-P-D-ribofuranosyl)-7H-pyn-olo[2,3-^pyrimidin-4(3H)- one; 2-amino-5-methyl-7-(2-C, 2-0-dimethyl-P-D-ribof iranosyl)-7H-pyrrolo[2,3-i ]pyrimidin- 4(3H)-one; 4-amino-7-(3-deoxy-2-C-methyl-P-D-ribofuranosyl)-7H-pyrrolo[2,3-^pyrimidine; 4-amino-7-(3-deoxy-2-C-methyl-P-D-arabinofuranosyl)-7H-pyrrolo[2,3-i ]pyrimidine; 4-amino- 2-fluoro-7-(2-C-methyl-p-D-ribofuranosyl)-7H-pyrrolo[2,3-i/]pyrimidine; 4-amino-7-(3-C- methyl-P-D-ribofuranosyl)-7H-pyrrolo[2,3-i ]pyrimidine; 4-amino-7-(3-C-methyl-P-D- xylofuranosyl)-7H-pyrrolo[2,3-^pyrimidine; 4-amino-7-(2,4-di-C-methyl-P-D-ribofuranosyl)- 7H-pyrrolo[2,3- ]pyrimidine; 4-amino-7-(3-deoxy-3-fluoro-2-C-methyl-P-D-ribofuranosyl)-7H- pyrrolo[2,3-c¾pyrimidine; and the corresponding 5'-triphosphates; or a pharmaceutically acceptable salt thereof.
The compounds of the present invention may also be combined for the treatment of HCV infection with non-nucleoside inhibitors of HCV polymerase such as HCV-796 (Viropharma Inc.) and those disclosed in International Patent Application Publication Nos. WO 01/77091; WO 01/47883; WO 02/04425; WO 02/06246; WO 02/20497; and WO 05/016927 (in particular JTK003).
4-amino-7-(2-C-methyl-P-D-ribofuranosyl)-7//- pyrrolo[2,3-i¾pyrimidine-5-carboxylic acid; 4-amino-5-bromo-7-(2-C-methyl-P-D- ribofuranosyl)-7H-pyrrolo[2,3-i ]pyrimidine; 4-amino-5-chloro-7-(2-C-methyl-P-D- ribofuranosyl)-7H-pyrrolo[2,3-<i]pyrimidine; 4-amino-5-fluoro-7-(2-C-methyl-P-D- ribofuranosyl)-7H-pyrrolo[2,3-<flpyrimidine; 2,4-diamino-7-(2-C-methyl-P-D-ribofuranosyl)- 7H-pyrrolo[2,3-cf]pyrimidine; 2-amino-7-(2-C-methyl-P-D-ribofuranosyl)-7H- pyrrolo[2,3-(i]pyrimidine; 2-amino-4-cyclopropylamino-7-(2-C-methyl-P-D-ribofuranosyl)-7H- pyrrolo[2,3-i ]pyrimidine; 2-amino-7-(2-C-methyl-P-D-ribofuranosyl)-7H- pyrroloP^-^pyrimidin^pHj-one; 4-amino-7-(2-C-ethyl-P-D-ribofuranosyl)-7H- pyrrolo[2,3-i ]pyrimidine; 4-amino-7-(2-C,2-0-dimethyl-P-D-ribofuranosyl)-7H- pyrrolo[2,3-<i]pyrimidine; 7-(2-C-methyl-P-D-ribofuranosyl)-7H-pyn-olo[2,3-^pyrimidin-4(3H)- one; 2-amino-5-methyl-7-(2-C, 2-0-dimethyl-P-D-ribof iranosyl)-7H-pyrrolo[2,3-i ]pyrimidin- 4(3H)-one; 4-amino-7-(3-deoxy-2-C-methyl-P-D-ribofuranosyl)-7H-pyrrolo[2,3-^pyrimidine; 4-amino-7-(3-deoxy-2-C-methyl-P-D-arabinofuranosyl)-7H-pyrrolo[2,3-i ]pyrimidine; 4-amino- 2-fluoro-7-(2-C-methyl-p-D-ribofuranosyl)-7H-pyrrolo[2,3-i/]pyrimidine; 4-amino-7-(3-C- methyl-P-D-ribofuranosyl)-7H-pyrrolo[2,3-i ]pyrimidine; 4-amino-7-(3-C-methyl-P-D- xylofuranosyl)-7H-pyrrolo[2,3-^pyrimidine; 4-amino-7-(2,4-di-C-methyl-P-D-ribofuranosyl)- 7H-pyrrolo[2,3- ]pyrimidine; 4-amino-7-(3-deoxy-3-fluoro-2-C-methyl-P-D-ribofuranosyl)-7H- pyrrolo[2,3-c¾pyrimidine; and the corresponding 5'-triphosphates; or a pharmaceutically acceptable salt thereof.
The compounds of the present invention may also be combined for the treatment of HCV infection with non-nucleoside inhibitors of HCV polymerase such as HCV-796 (Viropharma Inc.) and those disclosed in International Patent Application Publication Nos. WO 01/77091; WO 01/47883; WO 02/04425; WO 02/06246; WO 02/20497; and WO 05/016927 (in particular JTK003).
In one embodiment, non-nucleoside HCV NS5B polymerase inhibitors that are used in combination with the present HCV NS3 protease inhibitors are selected from the following compounds: 14-cyclohexyl-6-[2-(dimethylamino)ethyl]-7-oxo-5,6,7,8- tetrahydroindolo[2,l-a][2,5]benzodiazocine-l 1-carboxylic acid; 14-cyclohexyl-6-(2-morpholin- 4-ylethyl)-5,6,7,8-tetrahydroindolo[2,l-o][2,5]benzodiazocine-l 1-carboxylic acid; 14- cyclohexyl-6-[2-(dimethylamino)ethyl]-3-methoxy-5,6,7,8-tetrahydroindolo[2,l- ]
[2,5]benzodiazocine-l 1-carboxylic acid; 14-cyclohexyl-3-methoxy-6-methyl-5,6,7,8- tetrahydroindolo[2,l-a][2,5]benzodiazocine-l 1-carboxylic acid; methyl ({[(14-cyclohexyl-3- methoxy-6-methyl-5,6,7,8-tetrahydroindolo[2,l-a][2,5]benzodiazocin-l l- yl)carbonyl] amino } sulfonyl)acetate; ( { [( 14-cyclohexyl-3 -memoxy-6-methyl-5 ,6,7,8- tetrahydroindolo[2,l-a][2,5]benzodiazocin-l l-yl)carbonyl] amino }sulfonyl)acetic acid; 14- cyclohexyl-N-[(dimethylamino)sulfonyl]-3-methoxy-6-methyl-5,6,7,8-tetrahydroindolo[2,l-a] [2,5]benzodiazocine-l l-carboxamide; 3-chloro-14-cyclohexyl-6-[2-(dimethylamino)ethyl]-7- oxo-5,6,7,8-tetrahydroindolo[2,l-a][2,5]benzodiazocine 11-carboxylic acid; iV-(l l-carboxy-14- cyclohexyl-7,8-dihydro-6H-indolo[l,2-e][l,5]benzoxazocin-7-yl)-N,iV-dimethylethane-l,2- diaminium bis(trifluoroacetate); 14-cyclohexyl-7,8-dihydro-6H-indolo[ 1 ,2-e] [1,5]
benzoxazocine-11-carboxylic acid; 14-cyclohexyl-6-methyl-7-oxo-5,6,7,8-tetrahydroindolo [2, 1 -a] [2,5]benzodiazocine-l 1-carboxylic acid; 14-cyclohexyl-3-methoxy-6-methyl-7-oxo- 5,6,7,8-tetrahydroindolo[2, 1 -a] [2,5]benzodiazocine-l 1-carboxylic acid; 14-cyclohexyl-6-[2- (dimethylamino)ethyl]-3-methoxy-7-oxo-5,6,7,8-tetrahydroindolo[2, 1 -a] [2,5]benzodiazocine- 11-carboxylic acid; 14-cyclohexyl-6-[3-(dimethylamino)propyl]-7-oxo-5,6,7,8-tetrahydroindolo [2,1 -a] [2,5]benzodiazocine- 11 -carboxylic acid; 14-cyclohexyl-7-oxo-6-(2-piperidin- 1 -ylethyl)- 5,6,7,8-tetrahydroindolo[2,l- ][2,5]benzodiazocine-l 1-carboxylic acid; 14-cyclohexyl-6-(2- morpholin-4-ylethyl)-7-oxo-5, 6,7,8-tetrahydroindolo [2,1 -a] [2,5]benzodiazocine-l 1-carboxylic acid; 14-cyclohexyl-6-[2-(diethylamino)ethyl]-7-oxo-5,6,7,8-tetrahydroindolo[2,l-a]
[2,5]benzodiazocine-l 1-carboxylic acid; 14-cyclohexyl-6-(l-methylpiperidin-4-yl)-7-oxo- 5,6,7,8-tetrahydroindolo[2,l-a][2,5]benzodiazocine-l 1-carboxylic acid; 14-cyclohexyl-N-
[(dimethylamino)sulfonyl]-7-oxo-6-(2-piperidin-l-ylethyl)-5,6,7,8-tetrahydroind
[2,5]benzodiazocine-l l-carboxamide; 14-cyclohexyl-6-[2-(dimethylamino)ethyl]-N- [(dimethylamino)sulfonyl]-7-oxo-5,6,7,8-tetrahydroindolo[2,l-a][2,5]benzodiazocm^ carboxamide; 14-cyclopentyl-6-[2-(dimethylamino)ethyl]-7-oxo-5,6,7,8-tetrahydroindolo[2,l-a] [2,5]benzodiazocine-l 1 -carboxylic acid; 14-cyclohexyl-5,6,7,8-tetrahydroindolo[2,l -a]
[2,5]benzodiazocine-l 1-carboxylic acid; 6-allyl-14-cyclohexyl-3-methoxy-5,6,7,8- tetrahydroindolo[2,l-a][2,5]benzodiazocine-l 1-carboxylic acid; 14-cyclopentyl-6-[2- (dimethylamino)ethyl]-5,6,7,8-tetrahydroindolo[2,l- ][2,5]benzodiazocine-l 1-carboxylic acid; 14-cyclohexyl-6-[2-(dimethylamino)ethyl]-5,6,7,8-tetrahydroindolo[2,l-a][2,5]benzodiazoc 11-carboxylic acid; 13-cyclohexyl-5-methyl-4,5,6,7-tetrahydrofuro[3',2':6,7][l,4]diazocino[l,8- a] indole- 10-carboxy lie acid; 15-cyclohexyl-6-[2-(dimethylamino)ethyl]-7-oxo-6,7,8,9- tetrahydro-5H-indolo[2,l-«][2,6]benzodiazonine-12-carboxylic acid; 15-cyclohexyl-8-oxo- 6,7,8,9-tetrahydro-5H-indolo[2,l- ][2,5]benzodiazonine-12-carboxylic acid; 13-cyclohexyl-6- oxo-6,7-dihydro-5H-indolo[l,2-if][l,4]benzodiazepine-10-carboxylic acid; and pharmaceutically acceptable salts thereof.
The HCV NS3 protease inhibitory activity of the present compounds may be tested using assays known in the art. One such assay is HCV NS3 protease time-resolved fluorescence (TRF) assay as described below and in International Patent Application Publication No. WO2006/102087. Other examples of such assays are described in e.g., International Patent Application Publication No. WO2005/046712. The assay is performed in a final volume of 100 μΐ in assay buffer containing 50 mM HEPES, pH 7.5, 150 mM NaCl, 15 % glycerol, 0.15 % Triton X-100, 10 mM DTT, and 0.1 % PEG 8000. NS3 protease is pre-incubated with various concentrations of inhibitors in DMSO for 30 minutes. The reaction is initiated by adding the TRF peptide substrate (final concentration 100 nM). NS3 mediated hydrolysis of the substrate is quenched after 1 hour at room temperature with 100 μΐ of 500 mM MES, pH 5.5. Product fluorescence is detected using either a VICTOR V2 or FUSION fluorophotometer (Perkin Elmer Life and Analytical Sciences) with excitation at 340 nm and emission at 615 nm with a 400 μ8 delay. Testing concentrations of different enzyme forms are selected to result in a signal to background ratio (S/B) of 10-30. IC50 values are derived using a standard four-parameter fit to the data. Kj values are derived from IC50 values using the following formula,
IC5o = Ki (l + [S] / KM), Eqn (l),
where [S] is the concentration of substrate peptide in the reaction and KM is the Michaelis
constant. See Gallinari et al, 1999, Biochem. 38:5620-32; Gallinari et al., 1998, J Virol.
72:6758-69; Taliani et al, 1996, Anal. Biochem. 240:60-67.
The present invention also includes processes for making compounds of formula (I). The compounds of the present invention can be readily prepared according to the following reaction schemes and examples, or modifications thereof, using readily available starting materials, reagents and conventional synthesis procedures. In these reactions, it is also possible to make use of variants which are themselves known to those of ordinary skill in this art, but are not mentioned in greater detail. Furthermore, other methods for preparing compounds of the invention will be readily apparent to the person of ordinary skill in the art in light of the following reaction schemes and examples. Unless otherwise indicated, all variables are as defined above.
Olefin metathesis catalysts include the following Ruthenium based species:
Miller et al, 1996, J Am. Chem. Soc. 118:9606; Kingsbury et al, 1999, J Am. Chem. Soc.
121 :791; Scholl et al, 1999, Org. Lett. 1 :953; U.S. Patent Application Publication
US2002/0107138; Furstner et al, 1999, J Org. Chem. 64:8275. The utility of these catalysts in ring closing metathesis is well known in the literature (e.g. Trnka et al, 2001, Acc. Chem. Res.

G H j
(Zhan catalyst 1 A, Zannan Pharma Ltd.)

K Zhan ruthenium metathesis catalyst RC-303
(Zhan catalyst IB, RC-303,
Zannan Pharma Ltd.)
The following reaction schemes and examples serve only to illustrate the invention and its practice. The examples are not to be construed as limitations on the scope or spirit of the invention.
List of Abbreviations
ACN Acetonitrile
Aq. Aqueous
ACN Acetonitrile
Bn Benzyl
BOC (also Boc) t-Butyloxycarbonyl
BuLi Butyl lithium
CDC13 Deuterio-trichloromethane
DBU l,8-Diazabicyclo[5.4.0]undec-7-ene
DCM Dichloromethane
DIPEA Diisopropylethylamine
DMA Dimethylacetamide
DMAP 4-Dimethylamine pyridine
DMF Dimethylformamide
DMSO Dimethyl sulfoxide
ES Electronspray ionization
Et20 Diethyl ether
EtOAc Ethyl acetate
HC1 Hydrochloric acid
HATU O-(7-Azabenzotriazol- 1 -yl)-N,N,N',N'-tetramethyluronium
hexafluorophosphate
HF-TEA Hydrogen fluoride triethylamine
HPLC High performance liquid chromatography
HRMS High resolution mass spectrometry
Int. Intermediate
KHMDS Potassium hexamethyldisilazane
KHSO4 Potassium bisulfate
LiOH Lithium hydroxide
LCMS High performance liquid chromatography - mass spectrometry
LRMS Low resolution mass spectrometry
MeOH Methanol
MES 2-(N-morpholino)ethanesulfonic acid
NaOH Sodium hydroxide
NaHC03 Sodium hydrogen carbonate (sodium bicarbonate)
Na2C03 Sodium carbonate
NMM N-methylmorpholine
Pd/C Palladium on carbon
Pd(Ph3P)4 Tetrakis(triphenylphosphine)palladium(0)
PPh3 Triphenylphosphine
Rh/C Rhodium on carbon
TBS tert-Butyldimethylsilyl
TEA Triethylamine
TFA Trifluoroacetic acid
THF Tetrahydrofuran
TLC Thin Layer Chromatography
Tosyl p-Toluenesulfonyl
General methods
For Intermediates A, various salt forms have been used for the coupling reaction.
These include but are not limited to tosylate and TFA salts.
For the final products, they have been isolated either in their free form or a salt derivative (TFA", MeS(0)20", K+, Na+, etc.).
General methods





-39-
Synthesis of Intermediates
Intermediates A

Intermediate A6 : ai?,2S)-l-amino-2-ethenyl-iV-(ri- (methoxymethyl cvclopropyl] sulfonyl } cyclopropanecarboxamide trifluoroacetate


A solution of (li?,2S)-l-[(tert-butoxycarbonyl)amino]-2- ethenylcyclopropanecarboxylic acid (132 mg) in THF (3 mL) was added 1,1'- carbonyldiimidazole (283 mg). The mixture was stirred 4 hours at reflux. The mixture was cooled to room temperature and a solution of l-(methoxymethyl)cyclopropanesulfonamide (125 mg; Li et al, 2006, Synlett 5:725) and DBU (0.438 mL) in THF (3 mL) was added via cannula. The mixture was stirred for 40 hours at room temperature. The reaction was quenched with IN HC1 and the mixture was extracted twice with ethyl acetate. The combined organic layers were washed with brine, dried over anhydrous magnesium sulfate, filtered and concentrated under reduced pressure. The resulting residue was purified by flash chromatography (ISCO) to give the desired product (120 mg). Ή NMR (400 MHz, CDC13): 5.71-5.62 (m, 1H), 5.29 (d, 1H), 5.16 (d, 1H), 3.36 (s, 2H), 2.15 (dt, 1H), 1.89 (dd, 1H), 1.75-1.68 (m, 2H), 1.49 (s, 9H), 1.34- 1.30 (m, 1H), 1.08-1.03 (m, 2H).
Step 2 : (lR,2S)-l-amino-2-ethenyl-N-{[l-
(methoxymethyl)cyclopropyl]sulfonyl}cyclopropanecarboxamide trifluoroacetate

To a 0°C solution of the product of Step 1 (120 mg) in DCM (1 mL) at 0°C was added TFA (ImL). The mixture was slowly warmed to room temperature and stirred for 3 hours. The solvent was removed in vacuo and the crude product was used in the next step. Ή NMR (400 MHz, CDC13): δ (ppm) 5.82 (ddd, 1 H), 5.44 (d, 1H), 5.43 (d, 1 H), 3.67 (d, 1H), 3.63 (d, 1H), 3.35 (s, 3H), 2.64 (dt, 1H), 1.92 (dd, 1H), 1.83 (dd, 1H), 1.76-1.70 (m, 1H), 1.67-1.60 (m, 1H), 1.14-1.02 (m, 2H).
Intermediate A7 : ai?,2y)-l-amino-2-ethenyl-N-(il-(prop-l-en-2-vncvclopropyl1
sulfonyl cyclopropanecarboxamide hydrochloride

Step 1 : tert-butyl [(lR,2S)-2-ethenyl-l-({[l-(prop-l-en-2-yl)cyclopropyl]sulfonyl}
carbamo l) cyclopropyl] carbamate

The title compound was prepared using the same method as described for
Intermediate A6, Step 1 using l-(prop-l-en-2-yl)cyclopropanesulfonamide. Ή NMR (500 MHz, CDC13): δ (ppm) 5.66-5.59 (m, 1 H), 5.30 (d, IH), 5.24 (s, 1 H), 5.22 (s, IH), 5.16 (d, IH), 5.16 (s, IH), 2.14 (dt, IH), 1.96 (s, 3H), 1.90-1.80 (m, 3H), 1.48 (s, 9H), 1.33-1.30 (m, 1 H), 1.12- 1.08 (s, 2 H).
Step 2 : (lR,2S)-l-amino-2-ethenyl-N-{[l-(prop-l-en-2-yl)cyclopropyl]sulfonyl}
cyclopropanecarboxamide hydrochloride
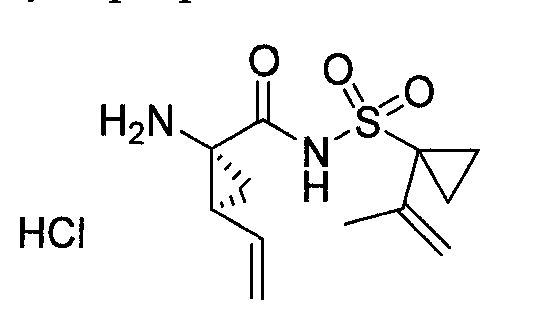
The product of step 1 (136 mg) was dissolved in a solution of HC1 in dioxane (1.5 mL). The mixture was stirred for 3 hours at room temperature. The solvent was removed in vacuo and the crude product was used in the next step. Ή NMR (400 MHz, CDCI3): δ (ppm) 5.69-5.62 (m, IH), 5.41 (d, IH), 5.26 (d, IH), 5.23 (s, IH), 5.21 (s, IH), 2.77-2.72 (m, IH), 2.02- 1.98 (m, IH), 1.95 (s, 3H), 1.93-1.84 (m, 2H), 1.74-1.69 (m, IH), 1.17-1.06 (m, 2H).

ethenylcyclopropanecarboxamide hydrochloride
Step 1 : tert-butyl {(1 R,2S)-1 -[({1 -[(benzyloxy)methylJcyclopropyl}sulfonyl)carbamoyl]-2- ethenylcyclopropyljcarbamate

The title compound was prepared using the same method as described for Intermediate A6, Step 1 using l-[(benzyloxy)methyl]cyclopropanesulfonamide. See
International Patent Publication No. WO 09/061699. Ή NMR (500 MHz, CDC13): δ (ppm) 7.40-7.33 (m, 5Η), 5.67-5.57 (m, 1 Η), 5.25 (d, IH), 5.12 (d, IH), 4.55 (d, IH), 4.50 (d, IH), 3.85 (d, IH), 3.77 (d, IH), 1.96 (br s, 1 H), 1.80 (dd, IH), 1.74-1.72 (m, 2 H), 1.46 (s, 9H), 1.12 (dd, IH), 1.08-1.01 (m, 2 H).
Step 2 : (lR,2S)-l-amino-N-({l-[(benzyloxy)methyl]cyclopropyl}sulfonyl)-2- ethenylcyclopropanecarboxamide hydrochloride

The title compound was prepared using the same method as described for Intermediate A7, Step 2. The crude product was used without further purification. Intermediate A9: (lig.2S -l-amino-2-ethenyl-N-((l-r(2-
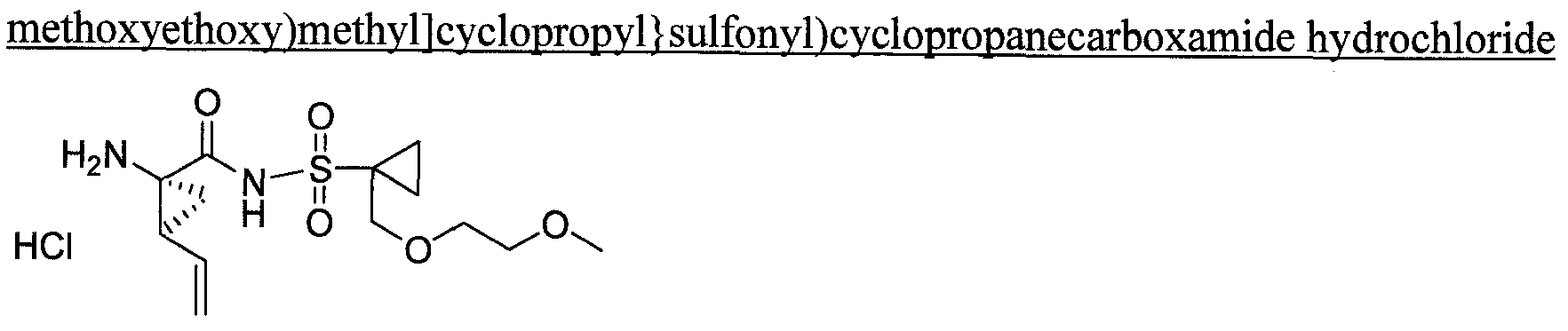
Step 1 : tert-butyl ({l-[(2-methoxyethoxy)methyl]cyclopropyl}sulfonyl)carbamate
 BuLi 2.5 M in hexanes (7.53 ml) was added dropwise to the solution of tert-butyl
BuLi 2.5 M in hexanes (7.53 ml) was added dropwise to the solution of tert-butyl
(cyclopropylsulfonyl)carbamate (1.81 g) in THF (40 ml) at -78°C. This solution was stirred at -78°C for 1 hour then 2-methoxyethoxymethyl chloride (1.85 ml) was added dropwise. The reaction was allowed to warm to room temperature and was stirred for 3 days. The solvent was evaporated under reduced pressure and the residue was diluted with ethyl acetate. At this point, HC1 (1 M) was added and the mixture was extracted with ethyl acetate (3x). The combined organic layers were washed with brine, dried over magnesium sulfate, filtered and the solvent was evaporated under reduced pressure. The residue was purified by flash chromatography (ISCO, 0 to 100% ethyl acetate in hexanes) to give the desired product (0.630 g) as a clear oil. LRMS (ES+) m/z 332.2 (M+Na)+.
Step 2: l-[(2-methoxyethoxy)methyl]cyclopropanesulfonamide

TFA (5 ml) was added to the solution of the product of Step 1 (0.63 g) in dichloromethane (5 ml) at 0°C. The reaction was allowed to warm to room temperature and was stirred for 1.5 hour. The solvent was removed under reduced pressure to give the desired product (0.426 g) as a light brown oil. Ή NMR (400 MHz, CDC13): δ (ppm) 6.80-6.21 (m, 2H), 3.75 (s, 2H), 3.66-3.64 (m, 2H), 3.56-3.53 (m, 2H), 3.36 (s, 3H), 1.42-1.39 (m, 2H), 0.91-0.89 (m, 2H) Step 3: tert-butyl {(lR,2S)-2-ethenyl-l-[({l-[(2- methox ethoxy)methyl]cyclopropyl}sulfonyl)carbamoyl]cyclopropyl}carbamate

The title compound was prepared using the same method as described for
Intermediate A6, Step 1. LRMS (ES+) m/z 441.2 (M+Na)+.
Step 4: (lR,2S)-l-amino-2-ethenyl-N-({l-[(2- sulfonyl)cyclopropanecarboxamide hydrochloride

Intermediate A7, Step 2. LRMS (ES+) m/z 341.2 (M+Na)+.
Intermediate A10: (lR.2S l-amino-2-ethenyl-N-((l-r2-(mo holin-4- yDethyl] cyclopropyl } sulfonvOcyclopropanecarboxamide dihydrochloride
Step 1 : tert-butyl {(lR,2S)-2-ethenyl-l-[({l-[2-(morpholin-4- yl)ethyl]cyclopropyl}sulfonyl)carbamoyl]cyclopropyl}carbamate
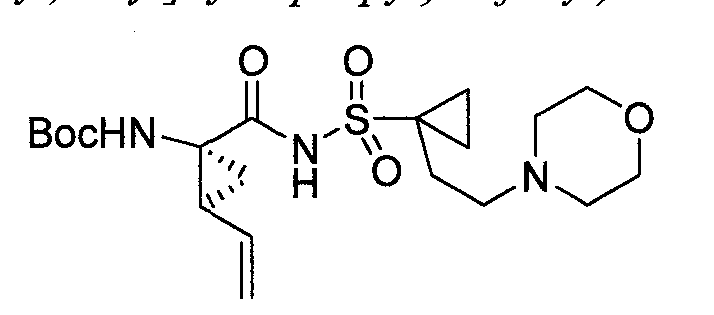
The title compound was prepared using the same method as described for
th 4-(2-bromoethyl)morpholine. LRMS (ES+) m/z 444.4 (M+H)+.

H-CI
The title compound was prepared using the same method as described for Intermediate A7, Step 2. The crude product was used directly in the next step. Intermediate Al l : 3- 4H-l,2,4-triazol-4-yl)propyl 4-methylbenzenesulfonate

To a solution of 3-(4H-l,2,4-triazol-4-yl)propan-l-ol (100 mg) in pyridine (2.62 mL) under nitrogen at 0 °C was added Tosyl-Cl (165 mg). The reaction was stirred for 18 hours. Pyridine was removed in vacuo and the remaining oil and solids were dissolved in ethyl acetate and washed several times with an aqueous solution of KHS04. The combined aqueous layers were back extracted with ethyl acetate. The combined organics were dried over magnesium sulfate, filtered and concentrated to provide a clear oil. Since the product was still in the aqueous layer, the pH of the aqueous phases was adjusted to 10 using 4.0 N NaOH. The mixture was extracted (3x) with ethyl acetate. The combined organics were dried over magnesium sulfate,
filtered and concentrated. Purification of the reaction mixture by flash chromatography (ISCO, 0 to 50% ethyl acetate in hexanes) provided the desired product (50 mg). LRMS (ES+) m/z 282.2 (M+H)+.
Intermediate A12 : 3-(3,3-difluoropiperidin-l-yl)propan-l-ol
-(3-{[tert-butyl(dimethyl)silyl]oxy}propyl)-3!3-difluoropiperidine

To a solution of 3,3-difluoropiperidine hydrochloride (518 mg) in DMF (15 mL) was added cesium carbonate (3.21 g) and (3-bromopropoxy)-tert-butyldimethylsilane (0.838 mL). The resulting reaction mixture was heated to 50°C for 1.5 hours before cooling back to room temperature. Water and ethyl acetate were added to the reaction mixture. The mixture was extracted with ethyl acetate (2x). The combined organics were washed with water (2x), brine, dried over magnesium sulfate, filtered and concentrated. Purification by flash chromatography (ISCO, 0 to 40% ethyl acetate in hexanes with 1% triethylamine) gave the desired product (612 mg). Ή NMR (400 MHz, CDC13): δ (ppm) 4.20 (t, 1H), 3.71-3.64 (m, 2H), 3.46 (t, 1H), 2.62 (t, 1H), 2.50-2.47 (m, 1H), 2.44-2.42 (m, 1H), 2.05-1.98 (m, 1H), 1.91-1.82 (m, 2H), 1.77-1.67 (m, 4H), 0.89 (d, 9H), 0.05 (s, 3H), 0.04 (s, 3H).
-(3,3-difluoropiperidin-l-yl)propan-l-0l

To a solution of the product of Step 1 (612 mg) in THF (20 mL) was added HF-
TEA (13.58 mL) at room temperature. The solution was heated to 50°C for 45 minutes. The reaction mixture was concentrated to remove THF and then diluted with ethyl acetate (100 mL) and water was added (100 mL). To that mixture was added Na2C03 (14.4 g) portion wise at 0°C When the quench was complete, the layers were separated. The organic layer was washed with 10% Na2C03, water and brine. The aqueous layer was re-extracted with ethyl acetate (2x). The combined organics were dried over magnesium sulfate, filtered and concentrated. The crude product (350 mg) was used directly in the next step. HRMS (ES+) m/z 180.1202 (M+H)+.
Intermediate A13 : l-(2-hvdroxypropan-2-yl cvclopropanesulfonamide
ulfonyl) carbamate

To a solution of cyclopropanesulfonamide (6.51 g, triethylamine (40 mL) and
DMAP (0.656 g) in DCM (150 mL) was slowly added benzyl chloroformate (12 mL). The mixture was stirred at room temperature for 18 hours. The mixture was washed with 1 N HC1. The aqueous layer (pH 1) was extracted with EtOAc (x2). The combined organics were washed with brine, dried over anhydrous MgS04 and concentrated. The resulting residue was purified by flash chromatography (ISCO, 0-10% methanol in dichloromethane) to afford the desired product (7.8 g). Ή NMR (400 MHz, CDC13): δ (ppm) 7.56-7.44 (m, 1H), 7.38 (s, 5H), 5.22 (s, 2H), 2.92-2.84 (m, 1H), 1.40-1.35 (m, 2H), 1.12-1.06 (m, 2H).
Step 2 : benzyl {[l-(2-hydroxypropan-2-yl)cyclopropyl]sulfonyl}carbamate

The title compound was prepared using the same method as described for
Intermediate A9, Step 1 using acetone. Ή NMR (400 MHz, CDC13): δ (ppm) 8.03-7.83 (m, 1H), 7.36 (s, 5H), 5.18 (s, 2H), 1.72-1.68 (m, 2H), 1.36 (s, 6H), 1.10-1.06 (m, 2H).
-(2-hydroxypropan-2-yl)cyclopropanesulfonamide

To a solution of the product of Step 2 (995 mg) in methanol (30 mL) under nitrogen was added Pd/C (169 mg). The flask was purged with hydrogen and stirred for 3 hours. Celite was added to the reaction mixture and it was filtered through a pad of Celite. The solvent was removed in vacuo. The resulting residue was purified by flash chromatography (ISCO, 0 to 10 % methanol in dichloromethane) to afford the desired product (391 mg). Ή NMR (400 MHz, CDC13): 5 (ppm) 4.88 (br s, 2H), 2.58 (s, 1H), 1.44-1.41 (m, 2H), 1.41 (s, 6H), 1.06-1.03 (m,
2H).
Intermediate A14 : l-(r({(lig.2j?V2-rr4E)-5-(4,4,5,5-tetramethyl-1.3.2-dioxaborolan-2-vnpent-4- - 1 -yl] cyclopropyl } oxy)carbonyll oxy) pyrrolidine-2 ,5 -dione

-[({[(lR,2R)-2-(pent-4-yn-l-yl)cyclopropyl]oxy}carbonyl)oxy]pyrrolidine-2,5-dione

To a solution of (li?,2i?)-2-(pent-4-en-l-yl)cyclopropanol (see International Patent Application Publication No. WO 08/057209) (17.1 g) in acetonitrile (193 mL) was added Ν,Ν'- disuccinimidyl carbonate (49.3 g) then triethylamine (53.7 mL). The mixture was heated to 40°C for 18 hours. The reaction mixture was cooled to room temperature and the solids were removed by filtration. The solvent was removed in vacuo. The residue was purified by flash chromatography (ISCO, 10 to 70% ethyl acetate in hexanes) to give the desired product (18 g). Ή NMR (500 MHz, CDC13): δ (ppm) 4.06-4.03 (m, IH), 2.87 (s, 4H), 2.28-2.25 (m, 2H), 1.98- 1.97 (m, IH), 1.72-1.63 (m, 2H), 1.44-1.39 (m, 2H), 1.30-1.25 (m, IH), 1.12-1.08 (m, IH), 0.72- 0.68 (m, IH).
Step 2 : l-{[({(lR,2R)-2-[(4E)-5-(4 ,5,5-tetramethyl-l,3,2-dioxaboro
yl ]cyclopropyl}oxy)carbonyl ]oxy}pyrrolidine-2, 5-dione

Cyclohexene (1.09 mL) was added to a 10 M dimethyl sulfide solution of borane (0.58 mL). Upon addition, a white solid had formed and 3 mL of degassed heptane was added to suspend it. The mixture was stirred for 5 minutes at room temperature. At this point, alkyne from Step 1 (15.5 g) in THF (55 mL) was added dropwise to the reaction mixture. After complete addition, the mixture was warmed to 40 °C for 20 minutes. Pinacolborane (8.48 mL) was added slowly to reaction mixture and the heating was continued for 2 hours at 40°C. The reaction mixture was cooled to room temperature and quenched with brine (100 mL). The mixture was extracted with ethyl acetate (3x). The combined organics were dried over magnesium sulfate, filtered and concentrated to provide the desired compound. Ή NMR (500 MHz, CDC13): δ (ppm) 6.66-6.58 (m, 1H), 5.45 (d, 1H), 4.02-3.99 (m, 1H), 2.87 (s, 4H), 2.22- 2.17 (m, 2H), 1.61-1.54 (m, 3H), 1.32-1.23 (m, 2H), 1.29 (s, 12H), 1.09-1.05 (m, 1H), 0.68-0.63 (m, 1H). Intermediate Al 5: l-(r({(Tj?,2i?V2-r5-(4,4,5,5-tetramethyl-L3,2-dioxaborolan-2- vPpentyl] cyclopropyl > oxy)carbonyll oxy } p yrrolidine-2, 5 -dione

To a degassed suspension of Pd/C (812 mg) in ethyl acetate (51 mL) was added vinyl boronate (intermediate A14, 10 g). After degassing, the mixture was purged and re-filled with hydrogen (balloon, 1 atm). The reaction mixture was stirred at room temperature for 2
hours. The atmosphere was carefully exchanged for nitrogen and the mixture was filtered over celite (rinsing with ethyl acetate). The solvent was removed in vacuo. The residue was purified by flash chromatography (ISCO, 0 to 50% ethyl acetate in hexanes) to give the desired product (7 g). 'HNMR (500 MHz, CDC13): δ (ppm) 4.03-4.00 (m, 1H), 2.87 (s, 4H), 1.46-1.39 (m, 4H), 1.35-1.24 (m, 5H), 1.26 (s, 12H), 1.07-1.03 (m, 1H), 0.81-0.77 (m, 2H), 0.67-0.63 (m, 1H).
Synthesis of Intermediates B


To a stirred solution of (li?,2S)-2-(but-3-en-l-yi)cyclopentanol (see International Patent Application Publication No. WO2009/134624) (2.1 g) and DIPEA (5.3 mL) in anhydrous 1,4-dioxane (51 ml), at 10°C and under nitrogen, was added a solution of triphosgene (1.5 g) in dioxane (51 ml). This reaction was stirred at 22°C for 1.5 hour and 3 -methyl-L- valine (2 g) and NaOH (1M, 30.5 mL) were added, then stirred at 70°C for 15 hours. At 22 °C, the reaction solution was acidified to pH 3 with 1 N HC1 and extracted with (3 x 100 ml) ether. The combined organic layer were washed with water (100 ml), brine (70 ml), dried over Na2S04, filtered and concentrated. The residue was purified by flash chromatography (silica gel, eluting with 10 to 100% EtOAc in hexane) to give the desired product (3.26 g). LRMS (ES+) M/Z (M+Na)+ 320.1.
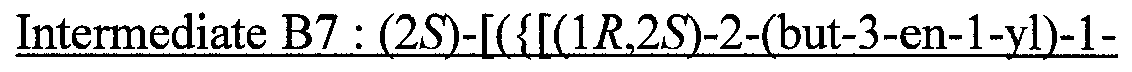
methylcvclopentylloxylcarbonvDaminoi cvclopentvnethanoic acid and (2S)- ({\(lS2R)-2-(but- -en- 1 -vD- 1 -methylcyclopentylloxy} carbonypaminol (cyclopentyl ethanoic acid
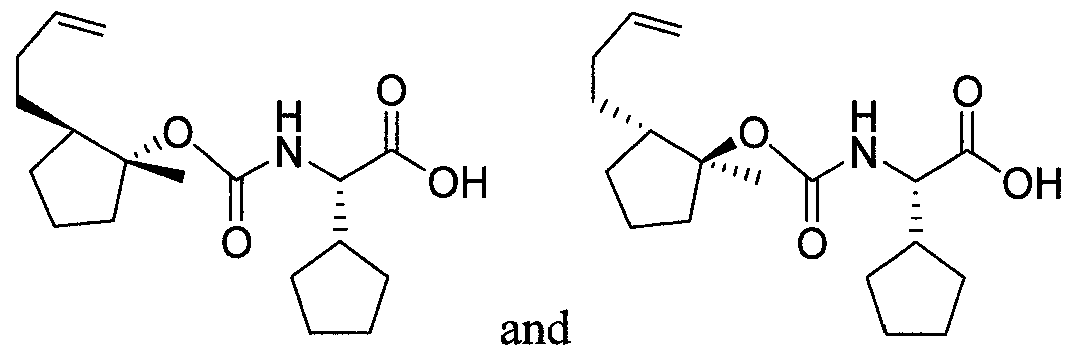
±)-(lR,2S)-2-(but-3-en-l-yl)-l-methylcyclopentanol
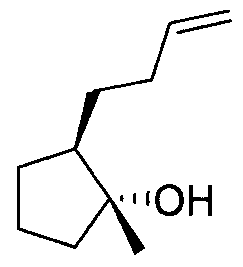
(+/-)
To a 0°C solution of butenyl magnesium chloride (45.6 mL, 0.5 M) was added copper(I) chloride (0.113 g) followed by methylcyclopropane oxide (1.12 g) in diethyl ether (5 ml). The reaction mixture was warmed to room temperature and stirred for 20 hours. The
mixture was cooled back to 0°C before pouring into a saturated solution of ammonium chloride (at 0°C). The mixture was extracted with ethyl acetate (3x). The combined organic fractions were dried over magnesium sulfate, filtered and concentrated carefully. The residue was purified by flash chromatography (ISCO, 0 to 30% ethyl acetate in hexanes) to give the desired product (1.3 g). Ή NMR (400 MHz, CDC13): δ (ppm) 5.86-5.79 (m, 1H), 5.05-4.99 (m, 1H), 4.97-4.93 (m, 1H), 2.20-2.09 (m, 1H), 2.02-1.92 (m, 2H), 1.73-1.50 (m, 7H), 1.31-1.10 (m, 5H).
Step 2 : methyl (2S)-[({[(lR,2S)-2-(but-3-en-l-yl)-l- methylcyclopentyl]oxy}carbonyl)amino](cyclopentyl)ethanoate and methyl (2S)-[({[(lS,2R)-2- (but-3-en-l-yl)-l-methylcyclopentyl]oxy}carbonyl)amino] (cyclopentyl)ethanoate

DMAP (0.59 g) was added to a stirred mixture of the product of Step 1 (0.75 g) and methyl (2S)-cyclopentyl(isocyanato)ethanoate (1.34 g) (See International Patent Application Publication No.: WO08/057209) in toluene (24 ml). DIPEA (3.40 mL) was then added and the mixture was stirred at 100°C for 22 hours. Since the reaction wasn't complete, methyl (25)- cyclopentyl(isocyanato)ethanoate (0.6 g), DIPEA (1.5 mL) and DMAP (0.3 g) were added and the mixture was stirred at 100°C for 8 hours. The reaction mixture was worked up with ethyl acetate and 5% KHS04. The organic layer was washed with a saturated solution of sodium bicarbonate, then brine, dried over magnesium sulfate and the solvent was removed in vacuo to give crude product. The residue was purified by flash chromatography (ISCO, 0 to 20% ethyl acetate in hexanes) to give the desired product (1.17 g) as a 1 :1 mixture of diastereomers. LRMS (ES+) M/Z (M+H)+ 360.4.
Step 3 : (2S)-[({[(lR2S)-2-(but-3-en-l-yl)-l- methylcyclopentyl]oxy}carbonyl)amino](cyclopentyl)ethanoic acid and (2S)-[({[(lS,2R)-2-(but- -en-l-yl)-l-methylcyclopentyl]oxy}carbonyl)amino](cyclopentyl)ethanoic acid
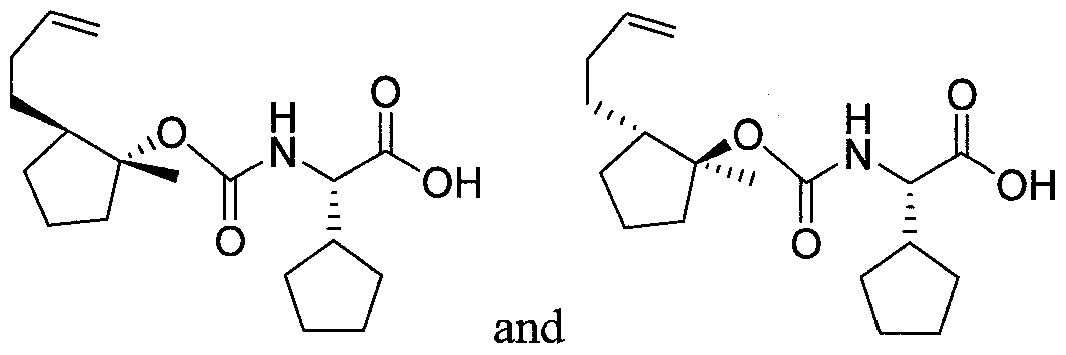
Intermediate B8 : (2^-Γ((Γ(ϋ?.2^ -2-^ΐ-3-6η-1-ν1)-1- methylcvclopropyl oxy)carbonyl aminol(cvclopentyl)ethanoic acid and (25f)-[({[(lS,2S)-2-(but- -en- 1 -yl)- 1 -methylcyclopropylloxy} carbonyl aminol(cyclopentyl ethanoic acid

±)-(lR,2R)-2-(but-3-en-l-yl)-l-methylcyclopropanol

(+/-)
In a dry sure seal, ethyl acetate (2 mL) was added to THF (204 mL) followed by 1,5-hexadiene (7.28 ml) and chlorotitanium triisopropoxide (20.43 ml). At this point, cyclohexylmagnesium chloride (46.0 mL) was added via a syringe pump over 1 hour. The mixture was stirred 1 hour.
It was then slowly poured into ice water and ether. The mixture was extracted with ether (3x). The combined organic layers were washed with brine, dried over magnesium sulfate and the solvent was removed in vacuo. The residue was purified by flash
chromatography on silica gel (0 to 40% diethyl ether in hexanes) to give the desired product (1.4 g). Ή NMR (400 MHz, CDC13): δ (ppm) 5.88-5.79 (m, 1H), 5.05-5.00 (m, 1H), 4.98-4.94 (m,
1H), 2.17-2.12 (m, 2H), 1.79 (s. 1H), 1.46-1.39 (m, 2H), 1.32-1.15 (m, 3H), 1.04-0.96 (m, 1H),
0.90-0.84 (m, 1H), 0.10-0.07 (m, 1H).
Step 2 : methyl (2S)-[({[(lR,2R)-2-(but-3-en-l-yl)-l- methylcyclopropyl]oxy}carbonyl)amino](cyclopentyl)ethanoate and methyl (2S)-[({[(lS,2S)-2- but-3-en-l -yl)-l -methylcyclopropyl]oxy}carbonyl)amino] ' (cyclopentyl) ethanoate

The title compound was prepared using the same method as described for
Intermediate B6, Step 2. LRMS (ES+) M/Z (M+H)+ 310.4.
Step 3 : (2S)-[({[(lR,2R)-2-(but-3-en-l-yl)-l- methylcyclopropyl]oxy}carbonyl)amino](cyclopentyl)ethanoic acid and (2S)-[({[(lS,2S)-2-(but- -en-l-yl)-l-me ethanoic acid

The title compound was prepared using the same method as described for Intermediate B7, Step 3. LRMS (ES+) M/Z (M+K)+ 334.1417.
Intermediate B9 : iV-(([(l ?,2i? -2-(but-3-en-l-ylVl-methylcvclopropylloxy|carbonyl)-3-methyl- L-valine and N-( ( [( 1 S2S)-2-(but-3 -en- 1 -ylV 1 -methylcyclopropylloxy ) carbonyl)-3 -methyl-L- valine

The title compound was prepared using the same method as described for Intermediate B8, Steps 1 to 3 using methyl 3 -methyl -N-(oxomethylidene)-L-valinate (See International Patent Application Publication No. WO 10/11566). LRMS of ester (ES+) M/Z (M+K)+ 336.1573.
Intermediate BIO : 3-methyl-iV-({[(li?,2i?')-2-(pent-4-vn-l-yl')cvclopropyl]oxylcarbonyl)-L- valine - N-benzyl-l-phenylmethanamine (1 :1)

The title compound was synthesized in a manner similar to the synthesis of the corresponding terminal alkene-bearing intermediate described in WO2008/057209.
Intermediate Bl l : (2S)-cyclohexyl[({[(li?,2i?)-2-(pent-4-vn-l-yl)cvclopropylloxy} carbonyl) amino ethanoic acid

-[({[(lR R)-2-^ent-4-yn-l-yl)cyclopropyl]oxy}carbonyl)oxy]pyrrolidine-2,5-dione

To a solution of (li?,2R)-2-(pent-4-en-l-yl)cyclopropanol (See International Patent Application Publication No. WO08/057209) (2.13 g; 70 % wt) in acetonitrile (20 ml) was added pyridine (1.5 ml), followed by Ν,Ν'-disuccinimidyl carbonate (3.74 g) and a crystal of DMAP. The mixture was stirred at 40°C overnight. After cooling to room temperature, the reaction mixture was diluted with ether and washed with HC1 IN, water and brine and dried over
sodium sulfate. The organic layer was filtered and concentrated to provide the desired product (2.88 g) as an oil. Ή NMR (500 MHz, CDC13): δ (ppm) 4.06-4.03 (m, 1H), 2.87 (s, 4H), 2.28- 2.25 (m, 2H), 1.98-1.97 (m, 1H), 1.72-1.65 (m, 2H), 1.46-1.40 (m, 2H), 1.31-1.24 (m, 1H), 1.12- 1.08 (m, 1H), 0.72-0.68 (m, 1H).
Step 2 : (2S)-cyclopentyl[({[(lR,2R)-2-(pent-4-yn-l-yl)cyclopropyl]oxy}carbonyl)
amino] ethanoic acid

To a solution of the product of Step 1 (457 mg) in acetonitrile (6 ml) was added (25)-amino(cyclohexyl)ethanoic acid (352 mg) followed by triethylamine (0.720 ml) and water (6.00 ml). The mixture was stirred at room temperature for 18 hours. The mixture was diluted with ethyl acetate and washed with HCl IN, water and brine. The organic layer was dried over sodium sulfate, filtered and concentrated. This provided the desired product (510 mg) as an oil. LRMS (ES+) M/Z (M+H)+ 308.2. Intermediate B12: (2S -cvclopentvirr(r(lig,2i?)-2-(pent-4-vn-l-vncvclopropylloxy}
carbonvDamino] ethanoic acid

Steps 1-2: methyl (2S)-cyclopentyl[({[(lR,2R)-2-(pent-4-yn-l-yl)cyclopropyl]
oxyjcarbonyl) amino Jethanoate

The title compound was prepared using the same method as described for Intermediate BIO, Steps 1 to 2 using methyl (25)-amino(cyclopentyl)ethanoate. Ή NMR (500 MHz, CDC13): δ (ppm) 5.14-5.09 (m, IH), 4.33-4.29 (m, IH), 3.74 (s, 3H), 2.25-2.20 (m, 2H), 1.94-1.93 (m, IH), 1.74-1.49 (m, 9H), 1.44-1.27 (m, 4H), 1.23-1.20 (m, IH), 1.02-0.97 (m, IH), 0.85-0.78 (m, IH), 0.56-0.49 (m, IH).
Step 3 : (28)-€γαΙορεηίγΙ[({[(1 ,2 )-2- βηί-4-γη-1-γ1)€γαΙορΓοργ1]οχγ}οα^οηγΙ)αηιϊηο]

The title compound was prepared using the same method as described for
Intermediate B7, Step 3. LRMS (ES+) M/Z (M+H)+ 294.1.
Intermediates B13-B15
Using the same method as described for either Intermediate Bl 1 (amino acid) or Intermediate B12 (amino ester), the following intermediates were synthesized:


*B14 NMR characterization : Ή NMR (400 MHz, CDC13): δ (ppm) 5.16-5.11 (m, IH), 4.40- 4.33 (m, IH), 3.82-3.79 (m, IH), 2.29-2.23 (m, 3Η), 1.97-1.95 (m, IH), 1.72-1.61 (m, 2H), 1.46- 1.31 (m, 2H), 1.07-0.93 (m, 8H), 0.89-0.83 (m, IH), 0.60-0.52 (m, IH).
Intermediate B16: 15-tert-butyl 14a-ethyl (2S.6Sa2Z.13aSa4aRJ6aSV2-ir(4- bromophenyl sulfonyl1oxy}-6-[(tert-butoxycarbonyl')aminol-5J6-dioxo- 2,3 ,6,7,8,9, 10 , 11 , 13a, 14, 16 , 16a-dodecahydrocyclopropa[elpyrrolo[ 1 ,2- a [1 ,4]diazacyclopentadecine- 14a, 15(1 H,5H -dicarboxylate

Step 1 : (4S)-4-{[(4-bromophenyl)sulfonyl]oxy}-l-(tert-butoxycarbonyl)-L-proline

LiOH (1M in water, 250 ml) was added to the solution of 1-tert-butyl 2-methyl (25,45)-4-{[(4-bromophenyl)sulfonyl]oxy}pyrrolidine-l,2-dicarboxylate (56.37 g) in THF (250 ml). The mixture was stirred at room temperature for 5 hours. The solution was acidified to pH = 1 with HC1 IN and extracted with ethyl acetate (3x). The combined organic fractions were washed with brine, dried over magnesium sulfate, filtered and concentrated. The desired acid was obtained as a white solid (53.4 g). LRMS (ES+) m/z 471.9 (M+Na)+.
Step 2 : tert-butyl (2S,4S)-4-{[(4-bromophenyl)sulfonyl]oxy}-2-{[(lR,2S)-2-ethenyl-l- (ethoxycarbonyl)cyclopropyl]carbamoyl}pyrrolidine-l-carboxylate

HATU (49.6 g) was added to the solution of the acid from Step 1 (53.4 g), (lR,2S)-cyclopropanecarboxylic acid, l-amino-2-ethenyl-, ethyl ester, hydrochloride
(Intermediate A4, 34.1 g) and DIPEA (62.2 ml) in DMF (475 ml). The solution was stirred at room temperature until disappearance of the starting material. HC1 1 N and water were added and the mixture was extracted with ether (3x). The combined organic fractions were washed with brine, dried over magnesium sulfate, filtered and concentrated. The residue was purified on a pad of silica gel (hexanes: ethyl acetate 100:0 to 40:60). The residue was further purified by flash chromatography (ISCO, 0 to 100% ethyl acetate in hexanes) to give the desired product (40 g) as a white solid. LRMS (ES+) m/z 609.0 (M+Na)+.
Step 3: ethyl (lR,2S)-l-{[(4S)-4-{[(4-bromophenyl)sulfonyl]oxy}-L-prolyl]amino}-2- ethenylcyclopropanecarboxylate

HC1 (4M in dioxane, 300 ml) was added to the product of Step 2 (40 g) and the solution was stirred at room temperature for 30 min. The solvent was evaporated under reduced pressure. The residue was dissolved in ethyl acetate and a saturated sodium bicarbonate solution was added. The phases were separated. The precipitated solid was filtrated from the organic layer to give the desired product (33.8 g) as a white solid. LRMS (ES+) m/z 487.0 (M+H)+. Step 4 : ethyl (lR,2S)-l-{[(4S)-4-{[(4-bromophen l)sidfo^
butox carbonyl)amino]non-8-enoyl}-L-prolyl]amino}

HATU (27.3 g) was added to the solution of (2S)-2-[(tert- butoxycarbonyl)amino]non-8-enoic acid (19.5 g) in DMF (100 ml) and the solution was stirred 15 minutes. The reaction mixture was cooled to 0°C. Then, the amine from Step 3 (31.8 g) in DMF (200 ml) (cooled to 0°C) was added followed by DIPEA (11.40 ml). The solution was stirred at 0°C for 1 hour. Hydrochloric acid (1M) was added and the mixture was extracted with diethyl ether (3x). The combined organic fractions were washed with brine, dried over magnesium sulfate, filtered and concentrated. The residue was purified by flash chromatography (ISCO, 0 to 100% ethyl acetate in hexanes) to give the desired product (40.2 g) as a white foam. LRMS (ES+) m/z 762.2 (M+Na)+.
Step 5: ethyl (lR,2S)-l-{[(4S)-4-{[(4-bromophenyl)sulfonyl]oxy}-l-{(2S)-2-[(tert- butoxycarbonyl)amino]non-8-enoyl}-L-prolyl](tert-butoxycarbonyl)amino}-2- ethenylcyclopropanecarboxylate

Di-tert-butyl dicarbonate (17.54 ml) was added at 0°C to the solution of the product of Step 4 (37.3 g) and DMAP (1.85 g) in ethyl acetate (504 ml). The solution was stirred at room temperature for 4 hours. At this point, another 2.5 g of di-tert-butyl dicarbonate was added and the solution was stirred at room temperature for 2 hours. The reaction was quenched with water and the mixture was extracted with ethyl acetate (3x). The combined organic fractions were dried over magnesium sulfate, filtered and concentrated. The residue was purified by flash chromatograph (ISCO, 0 to 40% ethyl acetate in hexanes) to give the desired product (35.8 g) as a white foam. LRMS (ES+) m/z 862.2 (M+Na)+.
Step 6 : 15-tert-butyl 14a-ethyl (2S,6S, 12Z, 13aS, 14aR, 16aS)-2-{[(4-bromophenyl)sulfonyl]oxy}- 6-[ (tert-butoxycarbonyl)amino J-5, 16-dioxo-2, 3, 6, 7, 8, 9, 10,11,13a, 14, 16,16a-dodecahydro c clopropafe Jpyrrolof 1, 2-a][ 1, 4 ]diazacyclopentadecine-14a, 15(1H, 5H)-dicarboxylate

The stirred solution of di-ene from Step 5 (15 g) in dichloroethane (1784 ml) was bubbled with nitrogen for 1 hour. 1 ,4 benzoquinone (0.19 g) and Zhan catalyst 1 B ( 1.31 g) were added and the solution was purged with nitrogen. The solution was then stirred at 75°C for 2 hours under a nitrogen flow. The reaction mixture was cooled to room temperature and ethyl vinyl ether (1.71 ml) was added to quench the catalyst. At this point, the solvent was evaporated under reduced pressure. The residue was purified by flash chromatography (ISCO, 0 to 40% ethyl acetate in hexanes) to give the desired product (12.3 g) as a white solid. LRMS (ES+) m/z 834.2 (M+Na)+.
Synthesis of intermediates C
Intermediate CI : 4-methoxy-3-(prop-2-en-l-vDquinolin-2-ol

-(prop-2-en-l-yl)quinoline-2,4-diol

To a degassed solution of 2,4-dihydroxyquinoline (10.0 g) in DMF (100 ml) was added triethylamine (9.51 ml), Pd(Ph3P)4 (2.151 g), followed by allyl acetate (7.43 ml). The mixture was allowed to stir at 60°C under nitrogen for 18 hours. The reaction mixture was cooled to room temperature and quenched into water (600 mL). The pH was adjusted to 12 using a saturated sodium carbonate solution. Dichloromethane was added and the layers were cut. The aqueous layer was extracted with dichloromethane once more. Then, the aqueous layer was cooled with ice and the pH was slowly adjusted to 2.5 with 12N HC1 to give a pink solid. The solids were filtered and washed with water to give 11.5 g (92% yield). LRMS (ES+) M/Z (M+H)+ 202.0.
-methoxy-3-(prop-2-en-l-yl)quinolin-2-ol

Dimethyl sulfate (0.237 ml) was added to a mixture of 3-(prop-2-en-l- yl)quinoline-2,4-diol (1.0 g) and potassium carbonate (1.37 g) in acetone (100 ml) and the mixture was stirred at room temperature for 3 hours then warmed to 40°C for 30 minutes. The reaction mixture was cooled and the solids were removed by filtration. The filtrate was concentrated in vacuo. Purification by flash chromatography (ISCO, 4-10% acetone/DCM) gave the desired product (0.70 g). LRMS (ES+) M/Z (M+H)+ 216.0.
Intermediate C2 : 4-ethoxy-3-(prop-2-en-l-yl quinolin-2-ol

The title compound was prepared using the same method as described for
Intermediate CI using diethyl sulfate. LRMS (ES+) m/z 230.1 (M+H)+. Intermediate C3 : 4-(benzyloxy -3-(prop-2-en-l-vf)quinolin-2-ol

To a mixture of 3-(prop-2-en-l-yl)quinoline-2,4-diol (4.0 g),
triphenylphosphine(6.78 g) and benzyl alcohol (2.27 ml) in THF (240 ml) at 0°C was added dropwise a THF (12 mL) solution of diisopropylazodicarboxylate (5.02 ml). Upon completion of addition, the mixture was allowed to stir at room temperature for 60 minutes. The reaction mixture was concentrated in vacuo to give a thick oil. Oil dissolved in DCM (20 mL) and solids started to precipitate. The solids were filtered and washed with DCM (10 mL). This provided 2.0 g of the desired product. Purification of the mother liquors using flash chromatography (ISCO, 1-8% acetone/DCM) gave an oil that was a mixture of products. This oil was dissolved in 15 mL of diethyl ether and seeded with the desired product. The mixture was stirred overnight and crystallization occurred. The solids were filtered and washed with ether to provide the desired product (1.06 g) as a white solid for a total of 3.06 g of product. LRMS (ES+) M/Z (M+H)+ 292.1. Intermediate C4 : 4-(benzyloxy)-6-bromo-3-(prop-2-en-l-yl)quinolin-2(lH)-one
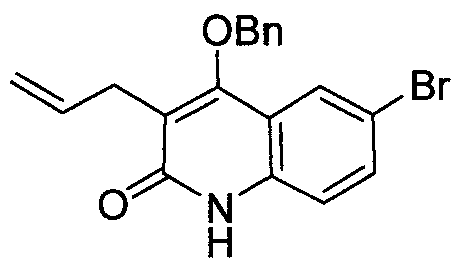
Step 1: ethyl 3-[(4-bromophenyl)amino]-3-oxopropanoate

4-Bromoaniline (10.0 g) was dissolved in benzene (70 mL) and cooled to 0°C. A solution of ethyl malonyl chloride in benzene (70 mL) was added over 15 minutes. The reaction was allowed to warm to room temperature and was stirred for 1 hour. The reaction mixture was diluted with an aqueous saturated sodium carbonate solution (50 mL) and stirred for 1 hour. The reaction was complete by LCMS. The reaction mixture was diluted with water and the layers were separated. The aqueous layer was re-extracted with diethyl ether. The combined organics were washed with brine, dried over sodium sulfate and concentrated to give 18 g of a cake that was used directly in the next step.
-[(4-bromophenyl)amino]-3-oxopropanoic acid

The product of Step 1 (16.63 g) was dissolved in THF (150 mL) and 2N NaOH
(145 mL) was added over 15 minutes. The reaction was stirred for 24 hours. The reaction mixture was concentrated to remove the THF. Diethyl ether (20 mL) was added and the layers were separated. The aqueous layer was acidified to pH = 2.2 with IN HC1 which resulted in the precipitation of solids. Ethyl acetate was added until the solids were dissolved and the layers were separated. The organic extract was dried with sodium sulfate, filtered and concentrated to give 14.7 g of a cake.
Step 3: 6-bromo-4-hydroxyquinolin-2(lH)-one

The product of Step 2 (14.7 g) was added to polyphosphoric acid (55.3 mL) and mixture was heated to 140°C for 3 hrs. The reaction mixture was cooled slightly and poured into 3N HC1 (168 mL). The pH was adjusted to 4 with 3N NaOH and resulting solid was filtered after cooling the reaction mixture to 10°C. The cake was washed with water and then slurried in 400 mL of 50% isopropanol/water for 18 hours. The solids were filtered, air dried to give a pasty solid which was dried in vacuo at 90°C for 4 hours, ground in mortar/pestle and re-dried at 90°C for another 18 hours to give 15 g of the desired product.
Step 4: 6-bromo-4-hydroxy-3-(prop-2-en-l-yl)quinolin-2(lH)-one

The title compound was prepared using the same method as described for Intermediate CI, Step 1 using the product of step 3. LRMS (ES+) m/z 280.0 (M+H)+.
Step 5: 4-(benzyloxy)-6-bromo-3-(prop-2-en-l-yl)quinolin-2(lH)-one

The title compound was prepared using the same method as Intermediate C3, g the product of step 4. LRMS (ES+) m/z 371.9 (M+H)+. Intermediate C5: 7-chloro-4-methoxy-3-(prop-2-en-l-vf)quinolin-2-ol (A and 5-chloro-4- methox -3 -(prop-2-en- 1 -yl)quinolin-2-ol (B
3-chloroaniline (5.01 g) and diethyl malonate (2.51 ml) were combined in a flask and heated to 220°C utilizing a short path condenser with nitrogen bleed over the reaction to remove reaction ethanol. The reaction was stirred for 18 hours and then cooled to room temperature. The solid that had formed in the flask was broken up in diethyl ether 30 mL to give free flowing orange solid upon filtration and washing with ether (3.75 g). LRMS (ES+) m/z (M+H)+ 322.9.
-chloroquinoline-2,4-diol and 5-chloroquinoline-2,4-diol
To the product of Step 1 (3.75 g) in a flask was added a solution of methanesulfonic acid (8 ml) containing phosphorus pentoxide (0.8 g) and the mixture was warmed to 170°C for 1 hour. The reaction was cooled and poured into 50 g of ice. The mixture was allowed to stir then it was diluted with water and solids were filtered. The solid was dissolved with 0.5N sodium hydroxide and washed with toluene (2X). The pH was adjusted to 3 with concentrated HCl to give solids which were filtered and washed with water to give 2.1 g of an orange solid. The material was clean but it was a 1 :1 mixture of regioisomers. LRMS (ES+) m/z (M+H)+ 196.1.
Step 3: 7-chloro-3-(prop-2-en-l-yl)quinoline-2,4-diol and 5-chloro-3-(prop-2-en-l-yl)quinoline- 2 4-diol

The title compound was prepared using the same method as described for Intermediate CI, Step 1 using the product of step 2. LRMS (ES+) m/z 236.1 (M+H)+.
Step 4 : 7-chloro-4-methoxy-3-(prop-2-en-l-yl)quinolin-2-ol (A) and 5-chloro-4-methoxy-3- rop-2-en-l-yl)quinolin-2-ol (B)

The title compound was prepared using the same method as described for Intermediate CI, Step 2 using the product of step 3. The separation of isomers was done by flash chromatography (15-60% ethyl acetate/hexanes) to provide product A (570 mg) and B (350 mg). (A) LRMS (ES+) m/z 250.0 (M+H)+. (B) LRMS (ES+) m/z 250.0 (M+H)+.
Intermediate C6 : 4-(benzyloxyV3-bromoquinolin-2(lH)-one

Step 1: 3-bromo-4-hydroxyquinolin-2(lH)-one

N-bromosuccinimide (2.21 g) was added to the solution of 2,4- dihydroxyquinoline (2 g) in DCM (50 ml). The mixture was stirred at room temperature for 3 days. The mixture was filtrated and the solid was triturated with isopropanol. After filtration, toluene was added to the solid and the solvent was evaporated under reduced pressure to give the desired product (1.92 g) as a beige solid. LRMS (ES+) m/z 240.1 (M+H)+.
Step 2: 4-(benzyloxy)-3-bromoq inolin-2(lH)-one

To a 0°C solution of PPh3 (1.64 g) in THF (42 mL) was added
diisopropylazodicarboxylate (1.21 mL) dropwise. The mixture was stirred at 0°C for 15 minutes before the addition of benzyl alcohol (0.52 mL) followed by quinoline alcohol from Step 1 (1.0 g). The mixture was stirred 15 minutes at 0°C then 5 hours at room temperature. A suspension had formed at that point. The solid was filtered and washed with cold isopropanol to afford the desired product (0.78 g). LRMS (ES+) m/z 352.1 (M+Na)+.
Intermediate C7: 4-(benzyloxyV3-bromo-8-fluoroquinolin-2(lHVone

Step 1: ethyl 3-[(2-fluorophenyl)amino]-3-oxopropanoate

To a solution of 2-fluoroaniline (8.67 ml) in ethyl acetate (266 ml) was added water (200 ml) and sodium bicarbonate (15.12 g). Ethyl malonyl chloride (13.82 ml) was added and the solution was stirred at room temperature for 1 hour. The two layers were separated and the organic phase was washed with a saturated aqueous solution of sodium bicarbonate, water and brine, dried over sodium sulfate and evaporated to provide a brownish oil. The crude reaction mixture was used directly in the next step. LRMS (ES+) m/z 226.1 (M+H)+.
Step 2 : 8-fluoro-4-hydroxyquinolin-2(lH)-one

A solution of the product from step 1 (20.27 g) in DCM (100 ml) was added to polyphosphoric acid (43.7 ml). Dichloromefhane was distilled from the reaction mixture by increasing the temperature slowly and then the brownish gummy solution was stirred at 120°C for 3 hours and then left at room temperature for 16 hours. The reaction mixture was quenched by adding ice cooled water and the product was filtered. The filter cake was stirred with 500 mL of water and filtered to give the desired product (9.38 g) as a colorless solid. LRMS (ES+) m/z 180.1 (M+H)+.
Step 3: 3-bromo-8-fluoro-4-hydroxyquinolin-2(lH)-one

The title compound was prepared using the same method as described for Intermediate C6, Step 1 using the product of step 2. LRMS (ES+) m/z 257.95 (M+H)+.
Step 4: 4-(benzyloxy)-3-bromo-8-fluoroquinolin-2(lH)-one

Intermediate C8 : 4-(benzyloxy -3-bromo-8-methoxyquinolin-2(lH)-one

The title compound was prepared using the same method as described for Intermediate C7 starting with o-anisidine. LRMS (ES+) m/z 382.1 (M+Na)+.
Intermediate C9 : 4-(benzyloxy)-3-bromo-7-(propan-2-yl quinolin-2(lH)-one

The title compound was prepared using the same method as described for Intermediate C7 starting with 3-(propan-2-yl)aniline. LRMS (ES+) m z 372.1 (M+Na)+.
Intermediate CIO : 4-(benzyloxy -3-bromo-7- trifluoromethyl)quinolin-2(lH)-one

methyl 2-amino-4-(trifluoromethyl)benzoate

To a solution of 2-amino-4-(trifluoromethyl)benzoic acid (5 g) in THF (85 ml) was added diazomethane (48.7 ml) in ether until completion of the reaction. Nitrogen was bubbled into the reaction mixture for 15 minutes to remove the excess of diazomethane and the solvent was removed under reduced pressure to provide a light brown solid (5.34 g). The compound was used in the next without purification. LRMS (ES+) m/z 220.1 (M+H)+.
Step 2: methyl 2-(acetylamino)-4-(trifluoromethyl)benzoate

To a solution of the product from Step 1 (5.34 g) in dioxane (25 ml) was added acetic anhydride (6 ml) and pyridine (4 ml). After 60 hours of stirring at 80°C, the solution was concentrated under reduced pressure and the residue was dissolved into ethyl acetate. The organic layer was washed with 2M sodium carbonate, 10% aqueous HCl, water and brine, dried over sodium sulfate and evaporated. The product was purified by flash chromatography (ISCO, 5%-20% ethyl acetate in hexanes) to provide a beige solid (5.27 g). LRMS (ES+) m/z 262.0 (M+H)+.
Step 3: 4-hydroxy-7-(trifluoromethyl)quinolin-2(lH)-one

To a solution of the product of step 2 (3.0 g) in THF (60 ml) was added 0.5 M
KHMDS in toluene (108 ml) dropwise at -78°C. After the addition was completed, the mixture was kept at -78°C for 40 minutes, then it was allowed to warm up slowly at room temperature. The mixture was quenched with H20 and the product was extracted twice with water. The combined aqueous layers were washed with ethyl acetate twice and then acidified with 6N HCl. The solid was filtered and the filter cake was washed with water and dried by vacuum aspiration to get a beige solid which was triturated into ethyl acetate to get a white solid (1.7 g). LRMS (ES+) m/z 230.05 (M+H)+.
Step 4: 3-bromo-4-hydroxy-7-(trifluoromethyl)quinolin-2(lH)-one

The title compound was prepared using the same method as described for Intermediate C6, Step 1 using the product of step 3. LRMS (ES+) m/z 308.2 (M+H)+.
Step 5: 4-(benzyloxy)-3-bromo- 7-(trifluoromethyl)quinolin-2(lH)-one

The title compound was prepared using the same method as described for Intermediate C6, Step 2 using the product of step 4. LRMS (ES+) m/z 420.2 (M+Na)+. Intermediate Cl l : 4-(benzyloxyV3-bromo-7-methylquinolin-2(lH)-one

The title compound was prepared using the same method as described for Intermediate CIO starting from 2-amino-4-methylbenzoic acid. LRMS (ES+) m/z 344.05 (M+H)+.
Intermediate C12 : 4-(benzyloxyV3-bromo-7-fluoroquinolin-2(lH -one

The title compound was prepared using the same method as described for Intermediate CIO starting from 2-amino-4-fluorobenzoic acid. LRMS (ES+) m/z 348.0 (Μ+Η)4
Intermediate C13 : 3-bromo-4-[(4-methoxybenzyl oxylquinolin-2(lH -one
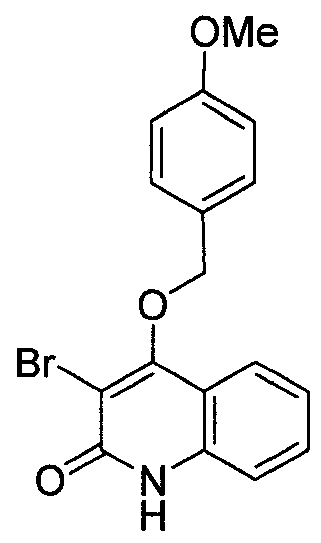
The title compound was prepared using the same method as described for Intermediate C6 using (4-methoxyphenyl)methanol. LCMS (ES+) m/z 382.0 (M+Na)+.

A round-bottom flask was charged with (S)-2-(tert-butoxycarbonylamino)-2-(l- methylcyclohexyl)acetic acid (synthesized according to procedures described in Tetrahedron Lett. 2007, 48(36):6343-6347) (10 g, 36.9 mmol) and 4M hydrochloric acid (40 ml, 160 mmol) in dioxane. The mixture was stirred for 1 hour and then concentrated to dryness in rotavap to give intermediate Dl (7.6 g, 36.6 mmol, 99 % yield) as a white powder. No further purification was carried out.
Intermediate D2: (2S,4R)-l-tert-butyl 2-methyl 4-((4-(benzyloxy)-3-bromoquinolin-2- yl)oxy pyrrolidine- 1 ,2-dicarboxylate
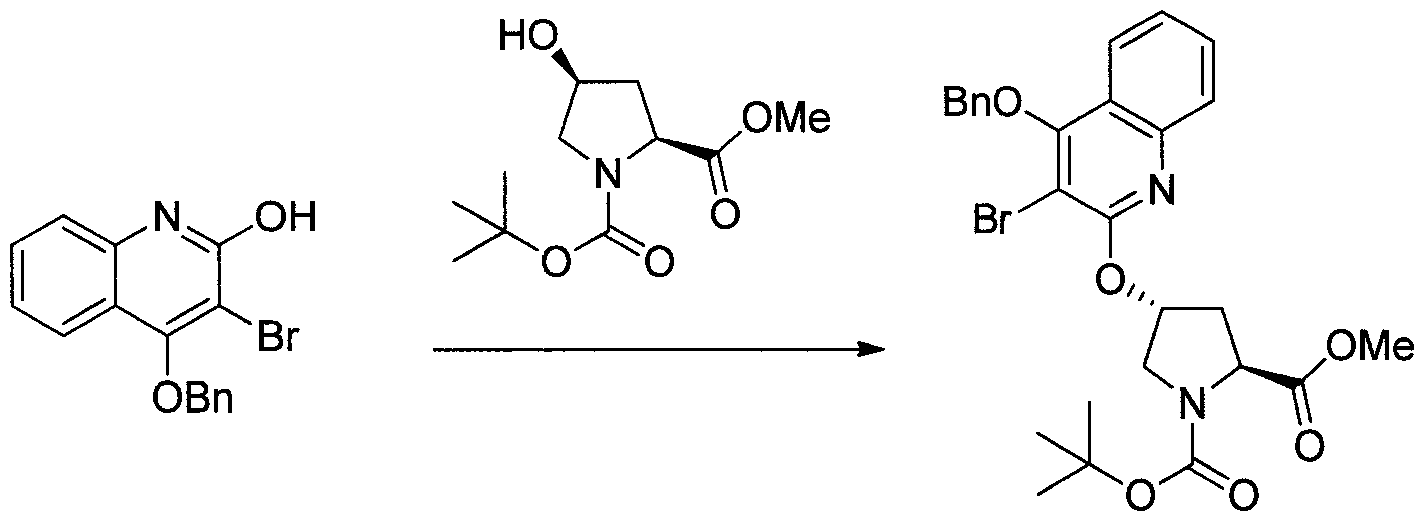
A round-bottom flask was charged with triphenylphosphine (3.61 g, 13.77 mmol), L-cis-Boc-4-hydroxyproline methyl ester (BaChem) (3.25 g, 13.26 mmol) and 4-(benzyloxy)-3- bromoquinolin-2-ol (3.37 g, 10.2 mmol). Dry THF (68.0 ml) was added under anhydrous conditions and the resulting slurry was stirred at 0°C. Diisopropyl azodicarboxylate (2.67 ml, 13.77 mmol) was added dropwise and the slurry was stirred for 10 min. The cooling bath was removed and the mixture was stirred for 2 hour (reaction mixture became homogeneous after approximately 30 min). The mixture was diluted with ethyl acetate (500 mL) and washed with aq. 1M HCl (100 mL), aq saturated sodium bicarbonate (2 x 100 mL) and brine (100 mL), dried over magnesium sulfate, filtered and concentrated in rotavap. The residue was purified on RediSep® (330 g; Teledyne Isco, Inc., Lincoln, NE) silica gel column (gradient: 0 to 50 % ethyl acetate in hexanes) to give intermediate D2 (5.42 g, 9.72 mmol, 95 % yield) as a colorless foam.
Intermediate D3: 2,5-dioxopyrrolidin-l-yl ((l -(pent-4-vn-l-yl)cyclopropyl carbonate:

A solution of (lR,2R)-2-(pent-4-ynyl)cyclopropanol (8.5 g, 68.4 mmol) in dry acetonitrile (68.4 ml) was treated with pyridine (6.64 ml, 82 mmol) and N,N'-disuccinimidyl carbonate (17.53 g, 68.4 mmol). The slurry was stirred for 10 min at room temp and then heated at 40°C overnight. The mixture was diluted with ethyl acetate (1.2 L) and washed with water (200 mL), aq 1M HC1 (200 mL), aq. saturated sodium bicarbonate (200 mL), and brine (200 mL), dried over magnesium sulfate, filtered and concentrated in rotavap to give intermediate D3 (14.56 g, 54.9 mmol, 80 % yield) as a slightly yellow gum. No further purification was carried out.
Intermediate D4: ( 1 R,2S)- 1 -amino-N-((l -methylcvclopropyl)sulfonyl)-2- vinylcyclopropanecarboxamide hydrochloride
Ste 1: (lR,2S)-l-((tert-butoxycarbonyl)amino)-2-vin lcyclopropanecarboxylic

A round-bottom flask was charged with (lR,2S)-ethyl l-((tert- butoxycarbonyl)amino)-2-vinylcyclopropanecarboxylate (4 g, 15.67 mmol) and lithium hydroxide monohydrate (2.63 g, 62.7 mmol). Methanol (52.2 ml), THF (52.2 ml) and water (52.2 ml) were added. The mixture was heated (oil bath at 45°C) overnight. The reaction mixture was concentrated to half-its volume in rotavap and the pH of the mixture was adjusted to pH = 2-3 with aq 1M HC1. The mixture was extracted with dichloromethane (3 x 150 mL). The combined organic extracts were washed with brine (50 mL), dried over magnesium sulfate, filtered and concentrated in rotavap to give the title compound (3.5 g, 15.40 mmol, 98 % yield) as a white powder. No further purification was carried out.
Step 2: tert-butyl ((lR,2S)-l-(((l-methylcyclopropyl)sulfonyl)carbamoyl)-2- vinylcyclopropyl)carbamate

A round-bottom flask was charged with the carboxylic acid product of step 1 (2 g, 8.80 mmol) and Ι,Γ-carbonyldiimidazole (2.141 g, 13.20 mmol). Dry THF (44.0 ml) was added under anhydrous conditions and the mixture was heated (oil bath at 85°C) for 2 hours with exclusion of moisture. The mixture was cooled to room temp and a solution of 1- methylcyclopropane-1 -sulfonamide (2.379 g, 17.60 mmol) in dry THF (10 mL) was added followed by l,8-diazabicyclo[5.4.0]undec-7-ene (2.63 ml, 17.60 mmol). The mixture was heated (oil bath at 75°C) overnight. The reaction mixture was treated with aq 1M HC1 (20 mL) and water (50 mL). The product was extracted into ethyl acetate (400 mL). Upon separation, the organic layer was washed with aq 1M HCl/water (1 :2, 80 mL), and brine (80 mL), dried over magnesium sulfate, filtered and concentrated in rotavap. The residue was purified on a gold cap RediSep® (120 g) silica gel column (gradient: 0 to 25% ethyl acetate in dichloromethane) to give the the title compound (2.25 g, 6.53 mmol, 74.2 % yield) as a white powder.
Step 3: (lR,2S)-l-amino-N-((l-methylcyclopropyl)sulfonyl)-2-vinylcyclopropanecarboxam hydrochloride

The N-Boc protected amine product of step 2 (2.25 g, 6.53 mmol) was dissolved in 4M hydrochloric acid (20 ml, 80 mmol) in dioxane and stirred for 30 minutes. TLC showed complete reaction. The reaction mixture was concentrated to dryness to afford Intermediate D4 (1.85 g, 6.59 mmol, 101 % yield) as a white powder. No further purification was carried out.
Intermediate D5: (S)-2-amino-2-(2,3-dihydro-lH-inden-2-yl)acetic acid hydrochloride

NS3 protease enzymatic activity
The HCV NS3 protease inhibitory activity was measured using the protease time- resolved fluorescence (TRF) assay as described below and in International Patent Application Publication No. WO 2006/102087. The assay was performed with HCV genotype lb (BK) NS3 modified enzyme with a R155K mutation and genotype 3a (3A-1).
The assay was performed in a final volume of 50 μΐ in assay buffer containing 50 mM HEPES, pH 7.5, 150 mM NaCl, 15% glycerol, 0.15% TRITON X-100, 10 mM DTT, and 0.1 % PEG 8000. NS3 and NS4A protease is pre-incubated with various concentrations of inhibitors in DMSO for 10 minutes. The reaction was initiated by adding the TRF peptide substrate (final concentration 25 nM) and NS3 mediated hydrolysis of the substrate proceeds for 6 hours at room temperature. Product fluorescence is detected using an Envision plate reader (Perkin Elmer) with excitation at 340 nm and emission at 615 nm with a 400 delay. Testing concentrations of the enzymes were selected to result in a signal to background ratio (S/B) of 5- 20. IC5o values are derived using a standard four-parameter fit to the data.
Alternatively, the Ki values can be obtained using the following protocol:
The assay is performed in a final volume of 100 μΐ in assay buffer containing 50 mM HEPES, pH 7.5, 150 mM NaCl, 15% glycerol, 0.15% TRITON X-100, 10 mM DTT, and 0.1 % PEG 8000. NS3 and NS4A protease is pre-incubated with various concentrations of inhibitors in DMSO for 30 minutes. The reaction is initiated by adding the TRF peptide substrate (final concentration 100 nM). NS3 mediated hydrolysis of the substrate is quenched after 1 hour at room temperature with 100 μΐ of 500 mM MES, pH 5.5. Product fluorescence is detected using either a VICTOR V2 or FUSION fluorophotometer (Perkin Elmer Life and Analytical Sciences) with excitation at 340 nm and emission at 615 nm with a 400 μ8 delay. Testing concentrations of the enzymes are selected to result in a signal to background ratio (S/B) of 8-30. IC50 values are derived using a standard four-parameter fit to the data. Ki values are derived from IC50 values using the following formula,
IC50=Ki (l+[S]/KM), Eqn (l), where [S] is the concentration of substrate peptide in the reaction and KM is the Michaelis constant. See Gallinari et al., 1999, Biochem. 38:5620-32; Gallinari et al, 1998, J Virol. 72:6758-69; and Taliani et al, 1996, Anal. Biochem. 240:60-67. ND means not determined.

0.12 1.4 184 0.057 1.1
1.3 4.9 185 0.032 0.36
4.1 23 186 0.078 1.9
0.54 9.2 187 0.24 6.1
0.32 22 188 0.033 0.85
0.10 1.8 189 0.081 3.4
0.15 2.0 190 0.074 1.5
0.15 3.2 191 0.16 1.5
0.16 6.0 192 0.10 0.71
1.2 94 193 0.16 1.5
0.13 3.5 194 0.032 0.38
1.3 27 195 0.049 0.66
6.6 73 196 0.10 0.85
0.073 1.5 197 0.027 0.31
0.28 9.9 198 0.050 0.77
0.25 7.9 199 0.057 0.74
0.10 0.50 200 0.074 0.96
0.15 4.0 201 0.079 1.3
0.12 2.9 202 0.050 0.95
0.12 0.76 203 0.13 4.9
0.16 1.5 204 0.15 2.4
0.23 6.6 205 0.19 1.9
0.21 1.9 206 0.20 2.6
1.1 10 207 0.11 1.3
0.11 0.55 208 0.11 1.0
0.02 0.45 209 0.056 0.55
0.22 3.9 210 0.046 0.29
0.09 0.95 211 0.033 0.30
0.11 2.3 212 0.22 1.9
0.10 0.57 213 0.36 3.8
0.11 0.45 214 0.021 1.2
0.61 12 215 1.7 55
0.70 20 216 0.24 6.4
1.6 85 217 0.27 7.1
0.35 7.2 218 0.035 2.6
0.43 5.5 219 0.12 1.9
0.56 16 220 0.29 3.9
0.077 25 221 0.050 1.4
0.081 6.6 222 0.045 0.44
0.16 1.6 223 0.055 1.2
<0.016 1.8 224 0.061 0.67
0.066 0.59 225 0.079 0.60
<0.016 0.42 226 0.11 1.6
<0.016 1.1 227 0.095 0.36
0.14 2.5 228 0.11 0.47
0.10 1.3 229 0.034 0.25
0.28 2.3 230 0.065 0.38
0.09 0.71 231 0.13 1.8
0.09 0.72 232 0.020, 0.040 0.275
0.08 0.54 233 0.015, 0.080 4.2
0.06 1.8 234 0.08, 1.38 29.12
0.20 2.4 235 0.53, 4.36 203.15
0.29 4.6 236 0.33, 5.44 109.95
0.18 2.5 237 0.013, 0.098 0.80
1.1 23 238 0.031, 0.047 0.90
0.24 7.2 239 0.016, 0.064 0.278
0.11 0.56 240 0.019, 0.28 2.904
0.23 4.4 241 0.021, 0.162 2.787
0.24 3.2 242 0.013, 0.072 0.935
0.34 4.4 243 0.033, 0.275 3.305
0.07 0.32 244 1.589, 1.653 296.200
0.15 1.6 245 0.546, 1.414 128.100
0.16 1.0 246 1.016, 1.136 211.500
1.6 19 247 ND ND
0.26 2.8 248 0.100, 0.348 40.220
0.093 0.59 249 0.026, 0.500 7.550
0.18 1.0 250 0.024, 0.160 4.450
0.15 0.78 251 0.015, 0.051 1.150
0.08 073 252 0.009, 0.022 0.088
0.18 2.9 253 0.026, 0.265 11.000
0.28 2.8 254 0.116, 2.304 47.531
100 0.24 2.0 255 0.120, 2.206 59.112
101 0.090 0.96 256 0.021, 0.122 5.450
102 0.21 3.2 257 0.032, 0.230 3.900
103 0.77 8.5 258 0.012, 0.079 2.750
104 1.3 26 259 0.030, 0.350 7.400
105 0.43 14 260 0.019, 0.093 2.400
106 0.78 17 261 0.026, 0.155 3.450
107 0.74 23 262 0.018, 0.120 1.200
108 0.93 23 263 0.006, 0.011 0.041
109 4.1 122 264 0.037, 1.400 7.700
110 14 488 265 0.016, 0.110 1.150
111 11 272 266 0.020, 0.044 7.500
112 24 620 267 0.114, 4.600 56.000
113 0.090 1.6 268 0.012, 0.034 0.200
114 0.051 0.25
115 0.057 0.25 270 0.021, 0.180 1.400
116 0.17 1.6 271 0.023, 0.215 5.200
117 0.080 0.93 272 0.015, 0.128 2.120
118 0.066 0.89 273 0.100, 1.850 68.000
119 0.12 3.2 274 0.015, 0.015 0.094
120 0.062 1.1 275 0.110, 1.150 16.000
121 0.38 12 276 0.013, 0.019 0.140
122 0.33 8.8 277 0.008, 0.013 0.078
123 0.49 6.1 278 0.860, 20.000 740.000
124 0.79 10 279 0.012, 0.145 2.600
125 4.1 114 280 0.024, 0.280 4.650
126 0.40 7.7 281 0.016, 0.125 1.245
127 0.11 2.1 282 0.140, 5.300 140.000
128 0.41 13 283 0.014, 0.022 0.108
129 0.51 8.7 284 0.023, 0.053 0.280
130 2.2 18 285 0.042, 0.104 0.410
131 0.11 1.3 286 0.022, 0.094 0.515
132 0.39 6.8 287 0.092, 2.550 58.000
133 0.11 1.0 288 0.018, 0.050 0.224
134 0.56 6.1 289 0.030, 0.197 3.275
135 0.20 1.8 290 0.012, 0.125 1.144
136 0.58 5.1 291 0.010, 0.085 0.774
137 0.14 1.2 292 0.120, 0.733 49.400
138 0.63 7.0 293 0.029, 0.138 1.300
139 0.10 0.38 294 0.015, 0.046 2.270
140 0.26 3.0 295 0.082 1.070
141 0.066 0.33 296 0.27 6.0
142 0.70 15 297 0.27 4.3
143 0.059 0.64 298 0.49 4.0
144 0.61 6.2 299 0.19 3.8
145 0.079 1.2 300 0.23 7.1
146 0.054 0.68 301 0.35 5.9
147 0.26 2.2 302 0.16 2.0
148 0.065 0.74 303 0.28 3.6
149 0.041 0.74 304 0.078 1.2
150 0.12 1.3 305 0.066 2.0
151 0.074 0.62 306 0.083 0.72
152 0.25 1.5
153 0.059 0.61
154 0.11 1.5
155 0.11 1.4
Example 1 : (3a ?JS,10S.12i?.21E.24aS)-7-cvclopentyl-N-irii?,2S)-l- (cvclopropylsulfonyl carbamoyll-2-ethenylcvclopropy -19-ethoxy-5,8-dioxo- 1.2.3.3a.5.6.7.8.11.12,20,23,24,24a-tetradecahvdro-10H-9.12-methanocvclo
penta l 8 Λ 91 Γ 1 0,3,61dioxadiazacyclononadecino l 1 , 12-61quinoline- 10-carboxamide


To a mixture of 1 -tert-butyl 2-methyl (2S,4S)-4-{[(4- bromophenyl)sulfonyl]oxy}pyrrolidine-l,2-dicarboxylate (1337 mg) and Intermediate C2 (600 mg) in N-methyl-2-pyrrolidinone (12 ml) was added cesium carbonate (2558 mg) and the mixture was stirred at 60°C for 1 hour. The reaction was not complete and additional brosylate (250 mg) and cesium carbonate (450 mg) were added. The mixture was stirred at 60°C for 1 more hour. The reaction mixture was then cooled before quenching into water (150 mL) and aqueous KHS04 (pH = 3.5). The product was extracted into ethyl acetate (150 mL). The organic layer was washed with aq. NaHC03 and brine, dried over sodium sulfate, filtered and concentrated. The material was dissolved in ACN and purified by reverse phase HPLC. After extractive workup (aq. NaHC03) with ethyl acetate, the desired product was obtained (1.0 g). LRMS (ES+) M/Z (M+H)+ 457.0.
-ethoxy-3-(prop-2-en-l-yl)quinolin-2-yl]oxy}-L-prolinate

To a 0°C solution of the product of Step 1 (500 mg) in dichoromefhane (10 ml) was added TFA (10 ml) and the mixture was stirred at room temperature for 1 hour. The reaction mixture was concentrated in vacuo. The residue was dissolved in dichloromethane and the organic was washed with aq. NaHC03 then brine, dried over sodium sulfate, filtered and concentrated to give the desired product (0.39 g). LRMS (ES+) M/Z(M+H)+ 357.0.
Step 3: methyl (4R)-l-{(2S)-2-[({[(lR,2S)-2-(but-3-en-l-yl)cyclopentyl]oxy} carbonyl) cyclopentylacetyl}-4-{[4-ethoxy-3-(prop-2-en-l-yl)quinolin-2-yl]oxy}-L-prolinate

The product of Step 2 (390 mg), DMAP (66.8 mg), DIPEA (0.573 ml) and Intermediate B2 (406 mg) were combined in DMF (10 mL) and stirred 5 minutes before adding HATU (541 mg). The reaction was stirred for 1 hour before quenching with aq. KHS04 and water (60 mL). The product was extracted into ethyl acetate (70 mL). The organic layer was washed with 10% aq. NaHC03 then brine. The aqueous extracts were re-extracted with ethyl acetate. The combined organics were dried over sodium sulfate, filtered and concentrated. Purification by flash chromatography (ISCO, 10-50% ethyl acetate/hexanes) gave the desired product as a foam (0.662 g). LRMS (ES+) M/Z( +H)+ 648.2.
Step 4 : methyl (3aR, 7S,10S,12R,21Z,24aS)-7-cyclopentyl-19-ethoxy-5,8-dioxo- 1, 2, 3, 3a, 5, 6, 7, 8, 11,12, 20, 23, 24, 24a-tetradecahydro-l OH-9, 12-methanocyclopenta
[18, 19] [1 , 10,3,6]dioxadiazacyclononadecino[l 1 ,12-bJquinoline-l 0-carboxylate (A) and methyl (3aR, 7S, 1 OS, 12R, 2 IE, 24aS)-7-cyclopentyl-19-ethoxy-5, 8-dioxo-l, 2, 3, 3a, 5, 6, 7, 8, 11, 12,
20,23, 24, 24a-tetradecahydro-10H-9, 12-methanocyclopenta[ 18,19] [1, 10,3,6]
dioxadiazacyclononadecinof 11, 12-b ]quinoline-l 0-carboxylate (B)

To degassed solution of the product of Step 3 (660 mg) in dichloroethane (150 ml) was added -benzoquinone (44.1 mg) followed by Zhan IB (150 mg). The mixture was stirred at room temperature for 4 hours. The reaction mixture was concentrated in vacuo. Purification by flash chromatography (ISCO, 1-8 % acetone/DCM) provided 60 mg of a hi-RF product (cis, A) and 440 mg of a low-RF product (trans,B). LRMS (ES+) M/Z (M+H)+ 620.1. Step 5 : (3aR, 7S, 1 OS, 12R,21E, 24aS)- 7-cyclopentyl-19-ethoxy-5, 8-dioxo-l, 2, 3, 3a, 5, 6, 7, 8, 11, 12,20, 23, 24, 24a-tetradecahydro-l OH-9, 12-methanocyclopenta[18,19][l,10, 3, 6]
dioxadiazacyclononadecinof 11, 12-b Jquinol ine-10-carboxylic acid

To a solution of the ester from Step 4 (100 mg) in THF (4 ml) was added 1 M NaOH (1.291 ml) and the mixture was stirred 18 hours at room temperature. The reaction mixture was diluted with IN HC1 (1.35 mL) and 10% aq. KHS04. The product was extracted into ethyl acetate. The combined organics were dried over sodium sulfate, filtered and concentrated to give the desired acid as a foam (98 mg). LRMS (ES+) M/Z (M+H)+ 606.1. Step 6 : (3aR, 7S,10S,12R,21E,24aS)-7-cyclopentyl-N-{(lR,2S)-l-[(cyclopropyl
sulfonyl)carbamoyl]-2-ethenylcyclopropyl}-19-ethoxy-5, 8-dioxo-l, 2, 3, 3 a, 5, 6, 7, 8, 11,
12,20, 23, 24, 24a-tetradecahydro-l OH-9, 12-methanocyclopenta[18, 19] [1,10, 3, 6]
ne-l O-carboxamide

The acid from Step 5 (0.040 g), DMAP (4.03 mg), DIPEA (0.035 ml) and Intermediate Al (0.040 g) were combined in DMF (2 mL) and stirred 5 minutes before adding HATU (33 mg). The reaction was stirred for 2 hours. The reaction mixture was purified by reverse phase HPLC to provide the desired product as a foam (40 mg). LRMS (ES+) M/Z (M+H)+ 818.2. Examples 2-16
By following the procedures outlined in Example 1 and using the appropriate A, B and C intermediates and indicated reaction schemes (depicted below the structure as Int. and Rx., respectively), the following compounds were prepared.
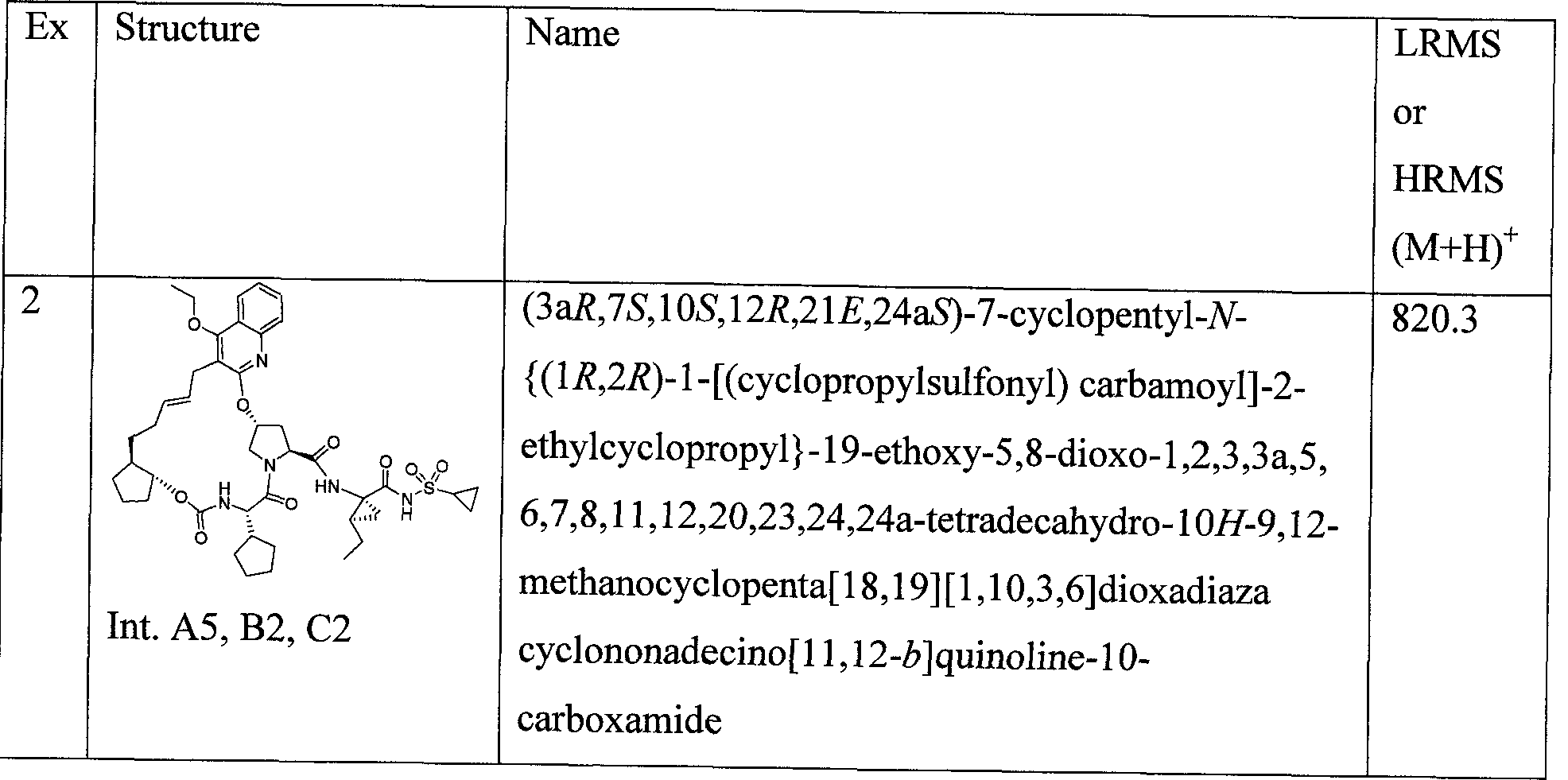

l,2,3,3a,5,6,7,8,l 1,12, 20,23, 24,24a-tetradecahydro-
Int. A1, B6, CI 1 OH-9, 12-methanocyclopenta[l 8, 19][1 , 10,3,6]dioxa
diazacyclononadecino [11,12-6] quinoline- 10- carboxamide
(3ai?,7S, 1 OS, 127?,2 lE,24aS 19-(benzyloxy)-7-cyclo 880.3 pentyl-N- { ( 1 R,2S)- 1 - [(cyclopropylsulfonyl)
carbamoyl] -2-ethenylcyclopropyl } -5 ,8-dioxo- 1 ,2,3 , 3a,5,6,7,8,l 1,12,20,23,24, 24a-tetradecahydro-10H-
° 6 } 9, 12-methanocyclopenta[ 18, 19] [ 1 , 10,3 ,6]dioxa
Int. A1,B2, C3 diazacyclononadecino [11 ,12-6]quinoline-10- carboxamide
(3ai?,7S, 1 OS, 12i?,2 lE,24aS)- 16-chloro-7- 838.3
XX cyclopentyl-N- {( li?,2S)- 1 - [(cyclopropylsulfonyl)
carbamoyl] -2-ethenylcyclopropyl } - 19-methoxy- 5,8-dioxo-l,2,3,3a,5,6,7,8,l l, 12,20,23,24,24a-
° 6 /} tetradecahydro- 1 OH-9, 12-methanocyclopenta
Int. Al, B2, C5 [ 18, 19] [ 1 , 10,3 ,6]dioxadiazacyclononadecino
Rx. A [11,12-b] quinoline- 10-carboxamide
(3 ai?,7S, 1 OS, 12i?,21 E,24aS)- 18-chloro-7- 838.3 cyclopentyl-N- {(1 R,2S)- 1 -[(cyclopropylsulfonyl) carbamoyl] -2-ethenylcyclopropyl } - 19-methoxy- 5,8-dioxo-l,2,3,3a,5,6,7,8,l 1, 12,20,23,24,24a-
° 6 /} tetradecahydro- 1 OH-9, 12-methanocyclopenta
Int. Al, B2, C5 [ 18, 19] [ 1 , 10,3 ,6]dioxadiazacyclononadecino
Rx. B [11,12-b] quinoline- 10-carboxamide
(1 ai?,5S,8S, 1 OR, 19E,22ai?)-5-cyclo pentyl-N- 776.8 { ( 1 R,2S)- 1 - [(cyclopropylsulfonyl) carbamoyl]-2- ethenylcyclo propyl } - 17-methoxy-3 ,6-dioxo- 1,1a,
3,4,5,6,9,10,18, 21,22,22a-dodeca hydro-8H-7,10- methanocyclopropa[l 8,19] [1 , 10,3,6]dioxadiaza
Int. Al, Bl, CI cyclononadecino[l 1,12-6] quinoline-8-carboxamide

quinoline- 10-carboxamide

Example 17: (3&R SA0SA2R2lE24aS)-7-CYc\opentyl-N-UlR2S)-l-\(cYcloOropyl
sulfonvncarbamoyl -2-ethenylcyclopropyl|-19-hvdroxy-5,8-dioxo-L2,3,3a,5,6,7,8, 11,12, 20,23 ,24.24a-tetradecahvdro- 1 OH-9, 12-methanocvclopentar 18, 191 Γ 1.10.3.61dioxadiaza

Example 8 (19 mg) was treated with TFA (1 ml) in a sealed tube and warmed to 55°C. After 30 minutes, the reaction was concentrated and the residue was dissolved in ACN and purified by reverse phase HPLC to provide the desired product as a white foam (13 mg). LRMS (ES+) m/z (M+H)+ 790.2.
Example 18: (3ai?.7S.10S.12i?.21E,25aj? -7-cvclopentyl-N-(aj?,25)-l-r(cvclopropyl sulfonvncarbamoyll^-ethenylcvclopro yU-ig-hvdroxy-S.S-dioxo^.S a.S.ej.S.l l. .
20.23.24.25 ,25a-tetradecahvdro- 1 H.1 OH-9.12-methanocvclopentar 19,201 Γ 1 ,10,3 ,61dioxa diazacvcloicosino [11,12- >lquinoline- 10-carboxamide

QaR S, 1 OS, 12R,2 \E,25&R)- 19-(benzyloxy)-7-cyclopentyl-N- { (\R,2S)- 1 - [(cyclopropylsulfonyl)carbamoyl]-2-ethenylcyclopropyl}-5,8-dioxo-2,3,3a,5,6,7,8, 11,12, 20,23 ,24,25 ,25a-tetradecahydro- 1H, 1 OH-9, 12-methanocyclopenta[ 19,20] [ 1 , 10,3 ,6]dioxa diazacycloicosino[ 11 , 12-6]quinoline- 10-carboxamide prepared by the method described for Example 1 using intermediates Al, B4, and C3 was deprotected using the method described for Example 17. HRMS (ES+) m/z 804.3639 (M+H)+.
Example 19 : riai?.5^8 .10i?J9E,22ai?V5-cvclopentyl-N-{(lj?.2S)-l-r(cvclopropyl sulfonyl)carbamoyl]-2-ethenylcyclopropyl } - 17-hydroxy- 1 a-methyl-3 ,6-dioxo-
1.1 a.3.4.5.6.9.10.18,21.22.22a-dodecahydro-8H-7, 10-methanocyclopropar 18.191

(1 aR,5S,8S, 1 OR, 19E,22ai?)- 17-(benzyloxy)-5-cyclopentyl-N- {(IR,2S)- 1 - [(cyclopropylsulfonyl)carbamoyl]-2-ethenylcyclopropyl} -1 a-methyl-3, 6-dioxo- 1 , 1 a,3 ,4,5 ,6,9, 10, 18 ,21 ,22,22a-dodecahydro-8H-7, 10-methanocyclopropa[ 18,19]
[l,10,3,6]dioxadiazacyclononadecino[l l,12-6]quinoline-8-carboxamide prepared by the method
described for Example 1 using intermediates Al, B7, and C3 (with separation of isomers after ring closing metathesis) was deprotected using the method described for Example 17. HRMS (ES+) m/z 776.3309 (M+H)+. Example 20 : (la ,5^8^.10j?.19E.22a )-5-cvclopentyl-N-{f l^S l-ITcyclopropyl
sulfonyl')carbamoyl]-2-ethenylcvclopropyl } - 17-hydroxy- 1 a-methyl-3.6-dioxo- 1.1 a.3.4.5.6.9.10,18.21.22.22a-dodecahydro-8H-7, 10-methanocvclopropar 18, 191
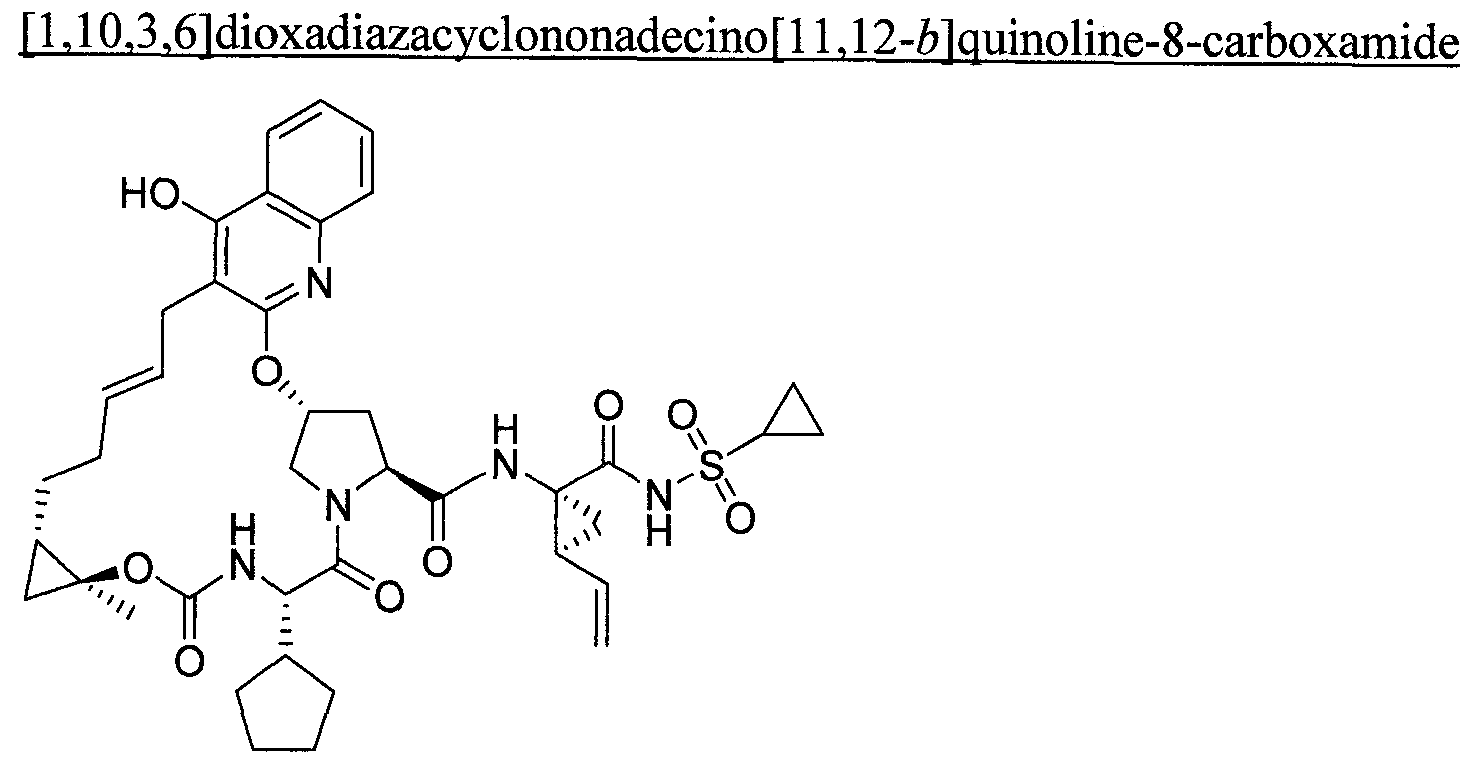
( 1 aS,5S,8S, 1 OR, 19£,22aS)- 17-(benzyloxy)-5-cyclopentyl-N- { (1 R,2S)- 1 -
[(cyclopropylsulfonyl)carbamoyl]-2-ethenylcyclopropyl}-la-methyl-3,6-dioxo-
I , 1 a,3 ,4,5,6,9, 10, 18,21 ,22,22a-dodecahydro-8H-7, 10-methanocyclopropa[ 18,19]
[l,10,3,6]dioxadiazacyclononadecino[l l,12-6]quinoline-8-carboxamide prepared by the method described for Example 1 using intermediates A1,B7,C3 (with separation of isomers after ring closing metathesis) was deprotected using the method described for Example 17. LRMS (ES+) m/z 776.7 (M+H)+. )+.
Example 21 : (3ai?.7lSl,10S.12i?.21E.24a^-7-cvclopentyl-N-r(lS.2igV2- (cvclopropyl sulfonyDcarbamoyl -l.r-bi cyclopropyD^-yll-^-hvdroxy-S^-dioxo- 1.2.3 ,3a.5.6.7.8.11.12.20.23.24.24a-tetradecahvdro- 1 OH-9.12-methanocyclopentar 18, 191
II.10.3.61dioxadiazacyclononadecinor 11.12-fcl quinoline- 10-carboxamide

(3a^,75,105,12i?,21E,24aS)-19-(benzyloxy)-7-cyclopentyl-N-[(15,2i?)-2- [(cyclopropylsulfonyl)carbamoyl]-l,r-bi(cyclopropyl)-2-yl]-5,8-dioxo-l,2,3,3a,5,6,7,8,
11,12,20,23 ,24,24a-tetradecahydro- 1 OH-9, 12-methanocyclopenta[ 18, 19] [ 1 , 10,3 ,6]
dioxadiazacyclononadecino [ 11 , 12-b] quinoline- 10-carboxamide prepared by the method described for Example 1 using intermediates A3, B2, and C3 was deprotected using the method described for Example 17. LRMS (ES+) m/z 804.8 (M+H)+.
Example 22 : (3ai?JSq0Sa2i?,21E,24aS)-17-bromo-7-cvclopentyl-N-i('li?,2S)-l- (cyclopropylsulfonvncarbamovn-2-ethenylcyclopropyl}-19-hydroxy-5,8-dioxo- l,2,3,3a,5,6J.8Jia2.20,23,24.24a-tetradecahvdro-10H-9,12-methanocvclopentari8J91 Γ ΙΛ 03 ,6]dioxadiazacvclononadecino 11 , 12-b] quinoline- 10-carboxamide

(3ai?,7S, 1 OS, 12i?,2 lE,24aS> 19-(benzyloxy)- 17-bromo-7-cyclopentyl-N- {( IR,2S)- 1 - [(cyclopropylsulfonyl)carbamoyl]-2-ethenylcyclopropyl } -5 ,8-dioxo-
1 ,2,3 ,3a,5 ,6,7, 8, 11 , 12,20,23 ,24,24a-tetradecahydro- 1 OH-9, 12-methanocyclopenta[ 18,19]
[l,10,3,6]dioxadiazacyclononadecino[l l,12-Z>]quinoline-l 0-carboxamide prepared by the method described for Example 1 using intermediates Al, B2, and C4 was deprotected using method described for Example 17. LRMS (ES+) m/z 868.2602 (M+H)+.
Example 23 : (3ai?JSJ0SJ2i?.21E.24ay)-7-cvclopentyl-N-r(li?,2^-2-ethenyl-l-{r(l- methylcvclopropyl)sulfonyl1carbamoyl>cvclopropyl]-19-hvdroxy-5,8-dioxo-L2,3, 3a.5.6.7.8.11.12,20,23, 24.24a-tetradecahvdro- 10H-9.12-methanocvclopentar 18.191
[1,10,3 ,61dioxadiazacyclononadecino [ 11 , 12-61 quinoline- 10-carboxamide
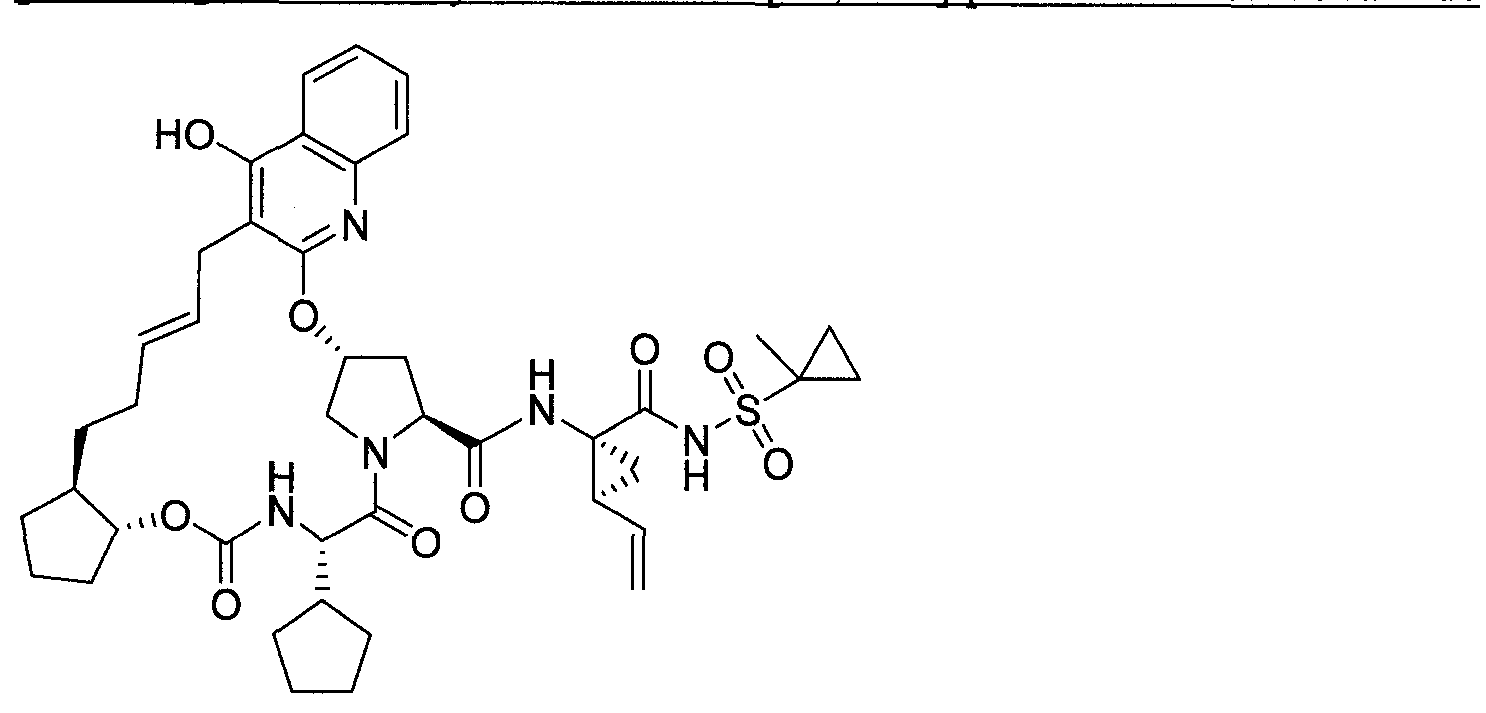
The title compound was prepared using the same method as described for
Example 17 using Example 15. HRMS (ES+) m/z 804.3642 (M+H)+.
Example 24 : r3aj?.7^.10S.12i?,24ai?)-7-cvclopentyl-N-(ni?,2^-l-r(cvclopropyl sulfonvDcarbarnoyll-2-ethenylcyclopropyU - 19-ethoxy-5,8-dioxo- 1 ,2,3 ,3a,5,6,7,8, 1 1,12, 20,21 ,22.23 ,24.24a-hexadecahvdro- 1 OH-9.12-methanocvclopentar 18, 191 Γ 1 , 10.3.61dioxa diazacyclononadecino Γ 11 , 12-61quinoline- 10-carboxamide

1, 2, 3, 3a, 5, 6, 7, 8, 11,12, 20,21, 22, 23, 24, 24a-hexadecahydro-10H-9, 12-methanocyclo
diazacyclononadecino[ 11, 12-b Jquinoline-10-carboxylate

To a solution of methyl (3ai?,7S,10S,12i?,21E,24aS)-7-cyclopentyl-19-ethoxy-5,8- dioxo-l,2,3,3a,5,6,7,8,l l,12,20,23,24,24a-tetradecahydro-10H-9,12- methanocyclopenta[ 18, 19] [1,10,3 ,6]dioxadiazacyclononadecino[ 11 , 12-Z>]quinoline- 10- carboxylate (Example 1, Step 4) (100 mg) in ethyl acetate (20 ml) and methanol (20 ml) was added 5% Rh/C (20 mg) and the mixture was stirred for 18 hours under hydrogen atmosphere. After exchanging the atmosphere for nitrogen, the reaction mixture was filtered and concentrated to give the desired product as a foam (100 mg). LRMS (ES+) M/Z (M+H)+ 622.1.
Step 2 : (3aR, 7S,10S,12R,24aR)-7-cyclopentyl-19-ethoxy-5,8-dioxo-l,2,3,3a,5,6, 7,8, 11,
12, 20, 21, 22, 23, 24, 24a-hexadecahydro-l OH-9, 12-methanocyclopenta[18, 19] [1, 10, 3, 6] dioxadiazacyclononadecinofl 1, 12-b Jquinoline-10-carboxylic acid

Using the product from Step 1, the title compound was prepared according to the procedure in Example 1, Step 5. LRMS (ES+) m/z 608.2 (M+H)+.
Step 3 : (3aR, 7S, 10S,12R,24aR)-7-cyclopentyl-N-{(lR,2S)-l-[(cyclopropylsulfonyl) carbamoyl] - 2-ethenylcyclopropyl}-19-ethoxy-5, 8-dioxo-l, 2, 3, 3a, 5, 6, 7, 8, 11,12, 20, 21, 22, 23, 24, 24a-hexadeca hydro-10H-9, 12-methanocyclopenta[18, 19] [1, 10,3, 6] dioxadiazacyclononadecinofl 1, 12-b] quinoline-10-carboxamide

Using the product from Step 2, the title compound was prepared according to the procedure in Example 1, Step 6. LRMS (ES+) m/z 820.3 (M+H)+.
Examples 25-28
By following the procedures outlined in Example 24 and using the appropriate A,
B and C intermediates (depicted below the structure as Int.), the following compounds were prepared.


Example 29 : (3aig.7S.10S.12i?.24ai?V7-cvclopentyl-N-(rii?.2S)-l-rrcvclopropylsulfonvn carbamoyl]-2-ethenylcvclopropyl}-19-hvdroxy-3a-methyl-5,8-dioxo-l,2,3,3a,5,6,7,8J l, 12,20,21 ,22,23 ,24,24a-hexadecahydro- lQH-9,12-methano cvclopentai 18,191 Γ 1.10.3 ,6] dioxadiazacyclononadecino [11,12-b] quinoline- 10-carboxamide

Step 1 : (3aR, 7S, 10S, 12R,24aR)-7-cyclopentyl-19-hydroxy-3a-methyl-5,8-dioxo-
1, 2, 3, 3a, 5, 6, 7, 8, 11, 12, 20, 21, 22, 23, 24, 24a-hexadecahydro-10H-9, 12-methanocyclopenta
cyclononadecino[l 1 , 12-b]quinoline-l O-carboxylic acid

To a solution of (3ai?,75',105',12i?,21E,24aS)-19-(benzyloxy)-7-cyclopentyl-3a- methyl-5,8-dioxo-l,2,3,3a,5,6,7,8,l l ,12,20,23,24,24a-tetradecahydro-10H-9,12-methano cyclopenta[l 8, 19] [ 1 , 10,3,6]dioxadiazacyclononadecino[ 1 1 , 12-b]quinoline- 1 O-carboxylic acid (synthesized as described in Example 1 with intermediates B7 and C3) (1 13 mg) in ethyl acetate (5 ml) was added 10% Pd/C (20 mg) and the mixture was stirred for 18 hours under hydrogen atmosphere. After exchanging the atmosphere for nitrogen, the reaction mixture was filtered and concentrated to give the desired product (93 mg). LRMS (ES+) M/Z (M+H)+ 594.7.
Step 2 : (3aR, 7S, 10S, 12R,24aR)-7-cydopentyl~N-{(lR,2S)-l-[(cyclopropylsulfonyl) carbamoyl]- 2-ethenylcyclopropyl}-19-hydroxy-3a-methyl-5, 8-dioxo-l, 2, 3, 3a, 5, 6, 7, 8, 11,
12,20,21, 22, 23, 24, 24a-hexadecahydro-l OH-9, 12-methanocyclopenta [ 18, 19] [1, 10,3, 6] dioxadiazacyclononadecino[l 1, 12-b Jquinoline-10-carboxamide
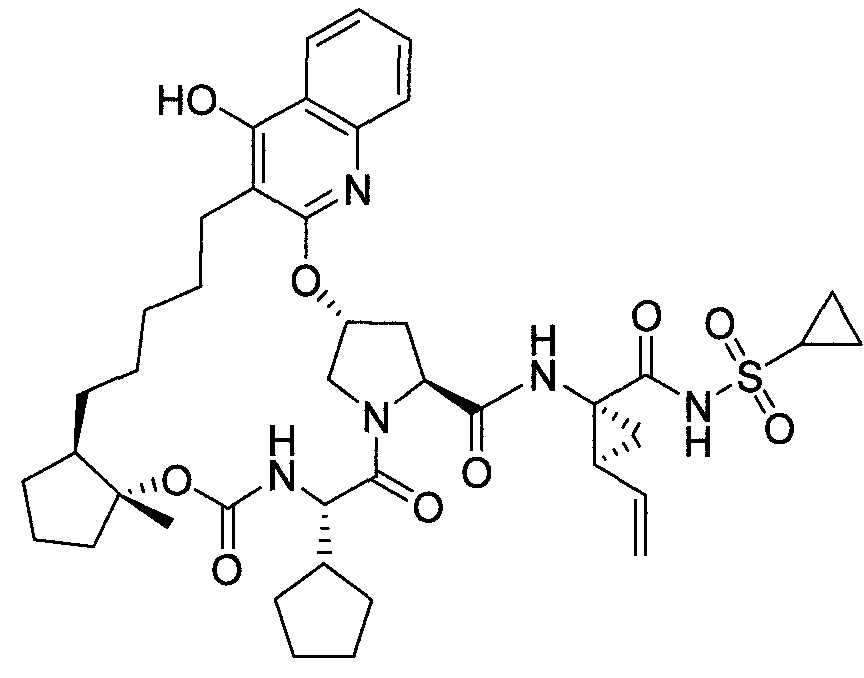
Using the product from Step 1, the title compound was prepared according to the procedure in Example 1 Step 6 using Intermediate Al . The two diastereoisomers were separated by reverse phase HPLC (40%-100% ACN/water/0.15%TFA) to provide the desired product (38 mg) HRMS (ES+) m/z 806.3802 (M+H)+.
Examples 30-34
By following the procedures outlined in Example 29 and using the appropriate A,
B and C intermediates (depicted below the structure as Int.), the following compounds were prepared.

32 QaR S, 105, 12i?,24ai?)-7-cyclo pentyl-N- 808.4
{(li?,2i?)-l-[(cyclopropyl sulfonyl) carbamoyl]-2-
X H YW ethylcyclopropyl } - 19-hy droxy-3 a-methyl-5 , 8 - dioxo- 1 ,2,3 ,3a,5,6, 7,8, 11 , 12,20,21 ,22,23,24,24a-
° 0 hexadeca hydro- 1 OH-9, 12-methanocyclopenta
Int. A5, B7, C3
[ 18, 19] [ 1 , 10,3 ,6]dioxadiazacyclononadecino
[11,12-6] quinoline- 10-carboxamide
33 (2R,4S,7S, 135)-7-cyclopentyl-N- { ( 1 R,2S)- 1 - 835.8
rV [(cyclopropylsulfonyl) carbamoyl] -2-ethenyl
cyclopropyl}-21-ethoxy-6,9-dioxo-3,4,6,7, 8,9,12,
13,15,16,17,18,19,20-tetradecahydro-2H, 11 H- Q 2,5: 10,13 -dimethano [ 1 , 14,5 ,7, 10]dioxatriazacyclo
Int. Al, B3, C2 docosino [ 15 , 16-b] quinoline-4-carboxamide
34 (3aR,7S, 1 OS, 12 ,24aK)-7-cyclo pentyl-N- 792.3
{(li?,2S)-l -[(cyclopropyl sulfonyl) carbamoyl]-2-
/— °- ethenylcyclopropyl}-19-hydroxy-5,8-dioxo- 1 ,2,3,3a,5,6,7,8,l 1,12, 20,21 ,22,23,24,24a- ° 6 /} hexadecahydro- 1 OH-9, 12-methanocyclopenta
Int. Al, B2, C3
[18, 19] [1,10,3,6] dioxadiazacyclononadecino
[11,12-6]quinoline- 10-carboxamide
Examples 35-37
By following the procedures outlined in Example 29 and using the appropriate A, B and C intermediates (depicted below the structure as Int.), the following compounds were prepared.

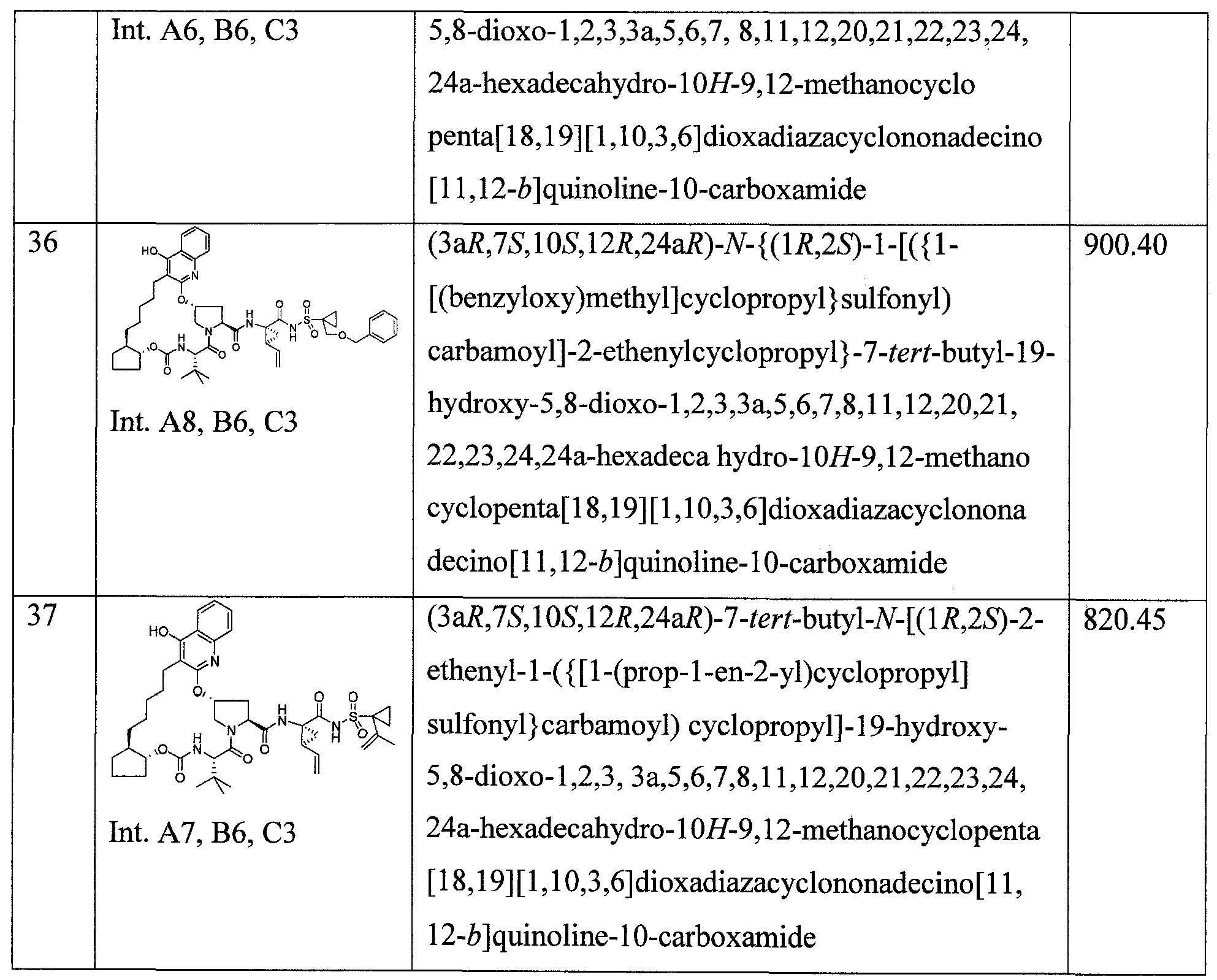
Example 38 : (-3ai?.7S.10SJ2^,21E.24aS)-7-cvclopentyl-10-ridj?.2S)-l- [(cvclopropylsulfonyl carbamoyl]-2-ethenylcvclopropyl|carbamoyl)-5,8-dioxo- 1.2.3.3a.5.6.7.8.11.12,20,23,24,24a-tetradecahvdro-10H-9,12-methanocvclopentari8J91 [l,10,3,6]dioxadiazacyclononadecino[l 1 2-6]quinorin-19-yl diethyl phosphate
To a solution of (3ai?,7S',10S,12i?,21E,24aS)-7-cyclopentyl-N-{(li?,2S)-l-
[(cyclopropylsulfonyl)carbamoyl]-2-ethenylcyclopropyl}-19-hydroxy-5,8-dioxo- l,2,3,3a,5,6,7,8,l l,12,20,23,24,24a-tetradecahydro-10H-9,12-methanocyclopenta
[18,19][l,10,3,6]dioxadiazacyclononadecino[l l,12-i>]quinoline-10-carboxamide (Example 17) (35 mg) in dichloromethane (0.44 mL) was added triethylamine (123 μΐ) followed by diethyl chlorophosphate (64 μΐ). The reaction mixture was stirred at room temperature until disappearance of the starting material. The reaction was quenched with water and extracted with ethyl acetate (3x). The combined organic fractions were dried over magnesium sulfate, filtered and concentrated. Purification by reverse phase HPLC (40-100% ACN/0.015% TFA-water) afforded the desired product (20 mg). LRMS (ES+) m/z 926.4 (M+H)+.
Example 39 : r3ai?.7S.10S.12i?.21E,24aS)-7-cvclopentyl-10-('i(1^.2S -l-r('cvclopropyl sulfonyl)carbamoyl1-2-ethenylcvclopropyl|carbamoyl)-5,8-dioxo-l,2,3,3a,5,6,7,8J l,12,
20.23.24.24a-tetradecahvdro- 1 OH-9.12-methanocvclopentar 18.191 IT .10.3.61
dioxadiazacyclononadecinofl l,12-61quinolin-19-yl acetate

To a solution of (3ai?,7S,10S,12i?,21E,24aS)-7-cyclopentyl-N-{(li?,2S)-l-
[(cyclopropylsulfonyl)carbamoyl]-2-ethenylcyclopropyl}-19-hydroxy-5,8-dioxo- l,2,3,3a,5,6,7,8,l l,12,20,23,24,24a-tetradecahydro-10H-9,12-methanocyclopenta[18,19]
[l,10,3,6]dioxadiazacyclononadecino[l l,12- >]quinoline-10-carboxamide (Example 17) (35 mg) in pyridine (0.44 mL) was added acetic anhydride (42 μΐ). The reaction mixture was stirred at room temperature until disappearance of the starting material. The reaction was quenched with water and extracted with ethyl acetate (3x). The combined organic fractions were dried over magnesium sulfate, filtered and concentrated. Purification by reverse phase HPLC (40%
ACN/0.05%TFA water to 100%) afforded the desired product (26 mg) after workup with NaHC03 and ethyl acetate. LRMS (ES+) m/z 832.8 (M+H)+.
Example 40 : r3a ? J^10lS.12i?.21E.24a^-7-cvclopentyl-10-r{nig.2(^-l-r(cvclopropyl sulfonyf)carbamoyll-2-ethenylcyclopropyl}carbamoyl')-5,8-dioxo-L2,3,3a,5,6,7,8,l l,
12.20.23.24.24a-tetradecahvdro- 1 OH-9.12-methanocvclopentar 18.191 \ 1.10.3.61

The title compound was prepared using the same method as described in Example 39, using isobutyric anhydride. HRMS (ES+) m/z 860.3863 (M+H)+.
Example 41 : r3ai?.7S.10S.12j?.21E.24aS -7-cvclopentyl-10-(((lj?.2S)-l-r('cvclopropyl sulfonyl)carbamoyll-2-ethenylcvclopropyl}carbamoylV5.8-dioxo-1.2.3.3a.5.6.7.8.11.
12.20.23.24.24a-tetradecahvdro- 1 OH-9.12-methanocvclopentar 18.191Π.10.3.61
dioxadiazacvclononadecino [ 11.12-6]quinolin- 19-yl 3-methylbutanoate

The title compound was prepared using the same method as described for Example 39, using isovaleric anhydride. HRMS (ES+) m/z 874.4027 (M+H)+.
Example 42 : r3a ?.7S.10S.12i;.21E,24aS)-7-cvclopentyl-N-{rii?.2S)-l-rrcvclopropyl sulfonyl)carbamoyll-2-ethenylcvclopropyl}-5,8-dioxo-19- 2-(2-oxo-l,3-oxazolidin-3- vnethoxyl-1.2 .3a.5.6.7.8.11.12,20,23,24,24a-tetradecahvdro-10H-9,12-methanocvclo

To a solution of (3aR,7S,10S,\2R,2\E,24aS)-7-cyclopentyl-N-{(lR,2S)-\-
[(cyclopropylsulfonyl)carbamoyl]-2-ethenylcyclopropyl}-19-hydroxy-5,8-dioxo- l,2,3,3a,5,6,7,8,l l,12,20,23,24,24a-tetradecahydro-10H-9,12-methanocyclopenta[18,19]
[l,10,3,6]dioxadiazacyclononadecino[l l,12-0]quinoline-l 0-carboxamide (Example 17) (40 mg) in DMF (0.75 mL) under nitrogen was added cesium carbonate (495 mg) followed by 2-(2-oxo- l,3-oxazolidin-3-yl)ethyl 4-methylbenzenesulfonate (144 mg). The reaction mixture was stirred at room temperature for 30 minutes and at 50°C for 3 hours. After cooling back to room temperature, the reaction was quenched with water and extracted with ethyl acetate (3x). The combined organic fractions were dried over magnesium sulfate, filtered and concentrated.
Purification by reverse phase HPLC (40%-100% ACN/0.15% TFA-water) afforded the desired product (39 mg) after workup with NaHC03 and ethyl acetate. HRMS (ES+) m/z 903.3933 (M+H)+.
Examples 43-60
By following the procedures outlined in Example 42 and using the appropriate A, B and C intermediates and reagent (depicted below the structure as Int. and Rg., respectively), the following compounds were prepared.

carboxamide

benzenesulfonate 1 nonadecino [11,12-6] quinoline- 10-carboxamide

benzene sulfonate (All) 10-carboxamide

carboxamide

Example 61 : (3ai?JSJ0S.12i?.24ai? -7-tert-butyl-N-r('li?.2S)-2-ethenyl-l-('{ ri-(methoxy methvDcvclopropyll sulfonyl } carbamo vDcyclopropyl] - 19- [2-(morpholin-4-yl')ethoxyl -5 ,8-dioxo- 1.2.3.3 a.5.6.7,8, 11 , 12,20.21.22.23 ,24.24a-hexadecahvdro- 1 OH-9.12-methano
cyclopenta[ 18, 191 [ 1 , 10,3.61dioxadiazacyclononadecino [11.12- >lquinoline- 10-carboxamide

The title compound was prepared using the same method as described for Example 42 using 4-(2-bromoethyl)morpholine and (3ai?,7S,10S,12i?,24ai?)-7-tert-butyl-N- [( 1 i?,25)-2-ethenyl- 1 -( { [ 1 -(methoxymethyl)cyclopropyl] sulfonyl } carbamoyl) cyclopropyl] -19- hydroxy-5,8-dioxo-l,2,3,3a,5,6,7,8,l l,12,20,21,22,23,24,24a-hexadecahydro-10H-9,12- methanocyclopenta[l 8,19][l,10,3,6]dioxadiazacyclo nonadecino[l l,12-£]quinoline-10- carboxamide (Example 35). Purification by flash chromatography (ISCO) afforded the desired product (21.5 mg). LRMS (ES+) m/z 937.3 (M+H)+. Examples 62-65
By following the procedures outlined in Example 61 and using the appropriate A, B and C intermediates and reagent (depicted below the structure as Int. and Rg., respectively), the following compounds were prepared.


Example 66 : (r(3aj?.7S.10S.12i?.21E.24aS -7-cvclopentyl-10-({(lj?.2S)-l-r(cvclor>ropyl sulfonyl carbamoyl]-2-ethenylcvclopropyl|carbamoylV5.8-dioxo-l,2.3,3a.5.6.7.8.11,12. 20,23 ,24,24a-tetradecahvdro- 1 OH-9.12-methanocyclopentar 18.191Π.10.3.61
dioxadiazacyclononadecinor 11 , 12-6]quinolin- 19-ylloxy) acetic acid

Step 1 : methyl {[(3aR,7S,10S,12R,21E,24aS)-7-cyclopentyl-10-({(lR,2S)-l- [(cyclopropylsulfonyl)carbamoyl]-2-ethenylcyclopropyl}carbamoyl)-5,8-dioxo- 1 ,2,3 ,3a,5,6,7,8, 11 , 12,20,23 ,24,24a-tetradecahydro- 1 OH-9, 12-methanocyclopenta
[ 18, 19] [ 1 , 10,3 ,6]dioxadiazacyclononadecino [11, 12-b]quinolin- 19-yl]oxy } acetate

The title compound was prepared using the same method as described in Example 42, using methyl bromoacetate. LRMS (ES+) m/z 862.6 (M+H)+.
Step 2 : {[(3aR, 7S, 10S, 12R,21E,24aS)-7-cyclopentyl-10-({(lR,2S)-l- [( cyclopropylsulfonyl)carbamoyl ]-2-ethenylcyclopropyl}carbamoyl)-5, 8-dioxo- 1, 2, 3, 3a, 5, 6, 7, 8, 11,12, 20, 23, 24, 24a-tetradecahydro-l OH-9, 12-methanocyclopenta
[18,19][1, 10, 3, 6]dioxadiazacyclononadecino[ll, 12-b]quinolin-19-yl]oxy}acetic acid

The product of Step 1 (43 mg) was dissolved in THF (1 ml) and methanol (0.2 ml). Water (0.5 ml) and LiOH (11.95 mg) were added and the reaction was stirred until complete conversion. The reaction was quenched with IN HC1 (0.4 mL) and 5% KHS04 was added until the pH was 3. The mixture was extracted with ether then ethyl acetate. The combined organics were washed with water (5x) then brine, dried over magnesium sulfate, filtered and concentrated to yield 42 mg of pure product. HRMS (ES+) m/z 848.3522 (M+H)+.
Example 67 : (3ai?,7S,10SJ2i?,21E,24aS)-7-cvclopentyl-N-{(l ?,2S)-l-r(cvclopropyl sulfonyl)carbamoyll-2-ethenylcvclopropyU-5,8-dioxo-19- 2-oxo-2-(pyrrolidin-l-yl)ethoxyl- 1 ,2.3.3a.5.6,7,8, 11.12.20,23 ,24.24a-tetradecahvdro- 1 OH-9.12-methanocvclopenta
Γ 18, 19] [ 1.10,3 ,6]dioxadiazacyclononadecino Γ 11.12-61 quinoline- 10-carboxamide

To a solution of {[(3aR,7S,10S,12R,21E,24aS)-7-cyclopentyl-10-({(lR,2S)-l- [(cyclopropy lsulfonyl)carbamoyl] -2-ethenylcyclopropyl } carbamoyl)- 5 , 8-dioxo- l,2,3,3a,5,6,7,8,l l,12,20,23,24,24a-tetradecahydro-10H-9,12-methano
cyclopenta[ 18, 19] [ 1 , 10,3 ,6]dioxadiazacyclononadecino [11,12-b]quinolin- 19-yl]oxy } acetic acid (Example 66) (27 mg) in DMF (0.5 mL) was added pyrrolidine (5.27 μΐ), DIPEA (0.028 mL)
- Ill -
and HATU (14.53 mg). The reaction mixture was stirred at room temperature for 15 minutes. The reaction was quenched with water and 5% KHS04 until pH = 3. More water was added and the mixture was stirred 5 minutes. The white solid was filtered (washed with water). This solid was dissolved in ethyl acetate and the mixture was dried over magnesium sulfate, filtered and concentrated to afford the desired product (26 mg). HRMS (ES+) m/z 901.4109 (M+H)+.
Example 68 : (,3ai?JS,10^12i?,21E,24aS -7-cyclopentyl-N-{(li?,2.S)-l-r('cvclopropyl
sulfonyl)carbamoyl1-2-ethenylcvclopropyl}-19-(4-hydroxybutoxy)-5,8-dioxo- l,2,3,3a,5,6,7,8,l l,12,20,23,24,24a-tetradecahvdro-10H-9J2-methanocyclopenta

Step 1 : (SaR, 7S,10S,12R,21E,24aS)-19-(4-{[tert-butyl(dimethyl)silyl]oxy}butoxy)-7-cyclopentyl- N-{(lR,2S)-l-[(cyclopropylsulfonyl)carbamoyl]-2-ethenylcyclopropyl}-5,8-dioxo- 1, 2, 3, 3a, 5, 6, 7, 8, 11,12, 20, 23, 24, 24a-tetradecahydro-l OH-9, 12-methanocyclo
12-b]quinoline-l 0-carboxamide

42, using 3-chlorobutanol-TBS ether and heating to 100°C for 8 hours. LRMS (ES+) m/z 976.9 (M+H)+.
Step 2 : (3aR, 7S,10S,12R,21E,24aS)-7-cyclopentyl-N-{(lR,2S)-l-[(cyclopropyl
sulfonyl)carbamoyl]-2-ethenylcyclopropyl}-19-(4-hydroxybutoxy)-5,8-dioxo-
1, 2, 3, 3a, 5, 6, 7, 8, 11,12, 20, 23, 24, 24a-tetradecahydro-10H-9, 12-methanocyclopenta
[18, 19] [1, 10, 3, 6]dioxadiazacyclononadecino[l 1, 12-bJquinoline-l 0-carboxamide

To a solution of the product of Step 1 (50 mg) in THF (0.5 mL) was added HF- TEA (0.334 mL) at room temperature. The solution was heated to 50°C for 30 minutes. The reaction mixture was concentrated to remove THF and then diluted with EtOAc (10 mL) and water was added (10 mL). To that mixture was added Na2C03 (353 mg) portion wise at 0°C. When the quench was complete, the layers were separated. The organic layer was washed with 10% Na2C03, water and brine. The aqueous layer was re-extracted with ethyl acetate (2x). The combined organics were dried over magnesium sulfate, filtered and concentrated. Purification by reverse phase HPLC (40-100% ACN/water with 0.15% TFA) afforded the desired product (7.2 mg) after workup with NaHC03 and ethyl acetate. HRMS (ES+) m/z 862.4073 (M+H)+.
Example 69 : r3ai?.7^10 ,12i?.21E.24aS,)-7-cvclopentyl-N-i(Ti?,2 -l-rfcvclopropyl sulfonyl)carbamoyll-2-ethenylcvclopropyl|-19-(2-hydroxyethoxy -5,8-dioxo-
1 ,2,3 ,3a,5 ,6,7,8, 11 , 12,20,23 ,24,24a-tetradecahvdro- 1 OH-9, 12-methanocyclopenta
Γ 18, 19] 1 , 10,3 ,61dioxadiazacyclononadecino l 1 , 12- ?lquinoline- 10-carboxamide

The title compound was prepared using the same method as described in Example 68, using the (2-bromoethoxy)-tert-butyldimethylsilane (See Zink et al., 2006, J Org. Chem. 71 :202). LRMS (ES+) m/z 834.6 (M+H)+.
Example 70 : (Z^R SSQSA2R \E2^aS)-l-tert^uty\-N-U\R2S) - ( c\o )roO \
sulfonyl carbamoyl]-2-ethenylcvclopropyl>-19-(3-hydroxypropoxy')-5.8-dioxo- l,2,3,3a.5.6,7,8,l 1,12,20,23 ,24,24a-tetradecahydro- 1 OH-9, 12-methanocyclopenta
[11, 12-&lquinoline- 10-carboxamide
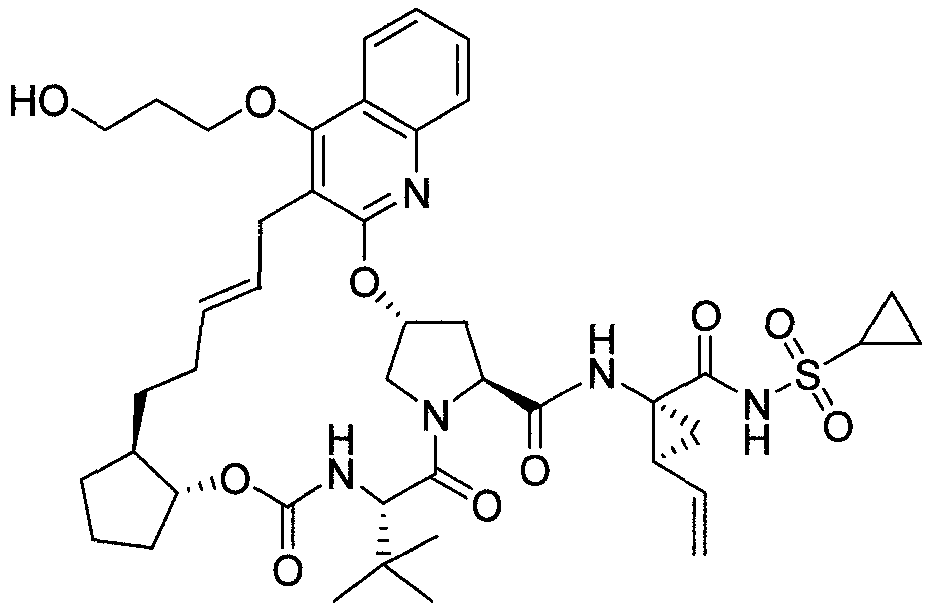
The title compound was prepared using the same method as Example 70, using 3- bromopropanol-TBS ether and (3ai?,7S,105,12^,21E,24aS)-7-tert-butyl-N-{(lJ?,2S)-l- [(cyclopropylsulfonyl)carbamoyl]-2-ethenylcyclopropyl}-19-hydroxy-5,8-dioxo- l,2,3,3a,5,6,7,8,l l ,12,20,23,24,24a-tetradecahydro-10H-9,12-methanocyclo
penta[18,19][l,10,3,6]dioxadiazacyclononadecino[l l,12-£]quinoline-l 0-carboxamide that was synthesized by the method described in Example 17 with intermediates Al, B6, and C3. HRMS (ES+) m/z 836.3922 (M+H)+.
Example 71 : (3aR SA0SA2R21E24aS)-7-tert-butyl-N-\(lR2S)-2-et enylA-m^ methylcvclopropyDsulfonyllcarbamoyllcvclopropy^-ig-O-hvdroxypropoxy^-S^-dioxo-
1.2.3.3a.5.6.7.8.11.12.20.23.24,24a-tetradecahvdro-10H-9.12-methanocvclo
~ldioxadiazacyclononadecino[ 11 , 12- >lquinoline- 10-carboxamide

The title compound was prepared using the same method as Example 68, using 3- bromopropanol-TBS ether and (3a ?,7S,10S,12i?,21J£:,24aS)-7-tert-butyl-N-[(li?,2S)-2-ethenyl-l- { [( 1 -methylcyclopropyl)sulfonyl] carbamoyl } cyclopropyl] - 19-hydroxy-5 ,8-dioxo- 1 ,2,3 ,3a,5 ,6,7,8, 11 , 12,20,23 ,24,24a-tetradecahydro- 1 OH-9, 12- methanocyclopenta[ 18, 19] [ 1 , 10,3 ,6]dioxadiazacyclononadecino[ 11 , 12-6]quinoline- 10- carboxamide that was synthesized by the method described in Example 17 with intermediates A2, B6, and C3. HRMS (ES+) m/z 850.5 (M+H)+.
Example 72 : r3aig.7SJ0S,12i?.21E,24aS)-7-cvclopentyl-N-(rii?,2^)-l-rrcvclopropyl sulfonvDcarbamoyll -2-ethenylcyclopropyl } - 19-(3-hydroxypropoxy)-5 , 8-dioxo- L2,3 a,5.6.7,8,lia2.20.23.24.24a-tetradecahvdro-10H-9J2-methanocyclo
pentaf 18, 19] [1 , 10,3,61dioxadiazacyclononadecino[ 11 , 12-¾]quinoline- 10-carboxamide

68 using 3-bromopropanol-TBS ether. HRMS (ES+) m/z 848.3923 (M+H) .
Example 73 : (3sLR SA0SA2R2lE,24aS)-7-cYc\omn yl-N-UlR2S)-l-\(cYcloOrom\
sulfonyl')carbamoyl]-2-ethenylcvclopropyl>-19-(2,2-difluoro-3-hvdroxypropoxyV5,8-dioxo- L2,33a,5,6,7,8J lJ2,20,23,24,24a-tetradecahvdro-10H-9.12-methanocvclo
penta[l 8, 191 [1 , 10,3 ,6]dioxadiazacyclononadecino[ 11 , 12- >]quinoline- 10-carboxamide

The title compound was prepared using the same method as described in Example 68 using 3-{[tert-butyl(dimethyl)silyl]oxy}-2,2-difluoropropyltrifluoro methanesulfonate (See International Patent Application Publication No. WO 2009/101917). HRMS (ES+) m/z
884.3722 (M+H)+.
Example 74 : 3-t\(3aR SA0SA2R2\E24aS)-7-CYc\opentyl-l0-(UlR2S)-l- lYcyclopropyl sulfonvDcarbamoyll -2-ethenylcvclopropyl } carbamoyl)-5 , 8-dioxo- 1.2.3.3 a.5.6.7.8.11.12,20.23.24,24a-tetradecahydro- 1 OH-9.12-methanocvclo
pentaf 18.19] [1,10,3 ,6]dioxadiazacvclononadecino [11,12-^1quinolin- 19-yl"|oxy } propyl N,N- dimethylglycinate

To a solution of Example 72 (31 mg) in dichloromethane (0.5 mL) was added Ν,Ν-dimethylglycine (11.3 mg) then triethylamine (0.015 ml), A N'-dicyclo hexylcarbodiimide (18.9 mg) and DMAP (1.1 mg). The solution was stirred at room temperature for 3 days. The mixture was diluted with ether and the solids that were formed were filtered off. The filtrate was concentrated in vacuo. Purification by reverse phase HPLC (30-100% ACN/water w/ 0.15% TFA) yielded 21 mg of the desired product after workup with NaHC03 and ethyl acetate.
HRMS (ES+) m/z 933.4454 (M+H)+. Example 75 : (3aR SA0SA2R2lE24s )-7-tert-but\\-N-UlR2S)-U(c\cloOrop\l
sulfonyl carbamoyll-2-ethenylcvclopropyl>-5,8-dioxo-19-[2-(piperidin-l-yl)ethoxy]- 12 a,5 ,6 J,8 Λ 1 Λ 2,20,23 ,24,24a-tetradecahydro- 1 OH-9, 12-methanocvclo
vclononadecino [11,12-b] quinoline- 10-carboxamide

To a solution of (3aR,7S, 105,12i?,21E,24a5)-7-tert-butyl-N-{(li?,2S)-l-
[(cyclopropylsulfonyl)carbamoyl] -2-ethenylcyclopropyl } - 19-hydroxy-5 , 8 -dioxo- 1 ,2,3 ,3 a,5 ,6,7,8, 11 , 12,20,23 ,24,24a-tetradecahydro- 10H-9, 12-niethanocyclopenta
[18,19][l,10,3,6]dioxadiazacyclononadecino[l l,12-Z)]quinoline-10-carboxamide (synthesized by the method described in Example 17 with intermediates Al, B6, C3) (40 mg) in THF (0.8 mL)
under nitrogen was added 2-(piperidin-l-yl)ethanol (0.137 niL), trimethylphosphine (1.028 mL) and diisopropylazodicarboxylate (0.200 mL). After stirring for 18 hours, the mixture was diluted with ethyl acetate and water was added. The mixture was extracted with ethyl acetate (3x). The combined organics were dried over magnesium sulfate, filtered and concentrated. Purification by reverse phase HPLC (30%-100% ACN/0.015% TFA-water) yielded 13.2 mg of the desired product. HRMS (ES+) m/z 889.4523 (M+H)+.
Examples 76-92
By following the procedures outlined in Example 75 and using the appropriate A, B and C intermediates and reagent (depicted below the structure as Int. and Rg., respectively), the following compounds were prepared.


carboxamide

10-carboxamide

carboxamide

2 See US Pat. No. 2,794,806
Example 93 : (,3ai?.7S,10Sa2i?,21E,24aS)-7-cvclopentyl-10- rii?.2S -l-r('cvclo propylsulfonvDcarbamoyl] -2-ethenylcyclopropyl } carbamovD-5 ,8-dioxo- 1 ,2 ,3 ,3a, 5,6,7,8,1 l,12,20,23.24.24a-tetradecahvdro-10H-9,12-methanocvclopentari8.191 Γ 1 , 10,3 ,6]dioxadiazacyclononadecino [11,12-6]quinolin- 19-yl propan-2-ylcarbamate

To a solution of (3a ?,7S,105,12i?,21E,24a5)-7-cyclopentyl-N-{(li?,2,S)-l- [(cyclopropylsulfonyl)carbamoyl]-2-ethenylcyclopropyl}-19-hydroxy-5,8-dioxo- 1 ,2,3 ,3a,5,6,7,8, 11 , 12,20,23,24,24a-tetradecahydro- 1 OH-9, 12-methanocyclo
enta[18,19][l ,10,3,6]dioxadiazacyclononadecino[l l,12- )]quinoline-10-carboxamide (Example 17) (30 mg) in dichloroethane (0.6 mL) was added isopropylisocyanate (0.037 mL) then DMAP (4.6 mg). The reaction mixture was heated to 50°C for 1 hour. After cooling back to room temperature, the mixture was diluted with ethyl acetate and water was added. The mixture was extracted with ethyl acetate (3x). The combined organics were dried over magnesium sulfate, filtered and concentrated. Purification by reverse phase HPLC (40% ACN- 100% ACN/0.05% TF A/water) yielded 27 mg of the desired product after workup with NaHC03 and ethyl acetate. HRMS (ES+) m/z 875.3981 (M+H)+.
Example 94 : (3ai?JS,10^,12ig.21E,24ay)-19-(3-aminopropoxyV7-cvclopentyl-N-{(li?,25f)-l- [(cvclopropylsulfonyl)carbamoyll -2-ethenylcyclopropyl > -5 , 8 -dioxo-
1 ,2,3 ,3a,5,6,7,8, 11,12,20,23 ,24,24a-tetradecahydro- 1 OH-9, 12-methanocyclopenta
1 , 12-6]quinoline- 10-carboxamide

Example 95 : 3-i\(3aR SA0SA2RaiE24aS)-7-cyclopentYl-l0-(UlR2S)-l-\(c\cloproOyl sulfonvDcarbamoyll^-ethenylcvclopropyllcarbamovD-S^-dioxo-l^J a^^ ^, 11,12,20, 23 ,24,24a-tetradecahydro- 1 OH-9, 12-methanocyclopental 8, 19] [1 , 10,3 ,6]dioxadiazacvclo nonadecino|T l,12-blquinolin-19-ylloxy}propyl dihydrogen phosphate

Step 1 : diethyl (3-{[(3aR,7S,10S,12R,21E,24aS)-7-cyclopentyl-10-({(lR,2S)-l- [(cyclopropylsulfonyl)carbamoyl]-2-ethenylcyclopropyl}carbamoyl)-5,8-dioxo- 1, 2,3,3a,5, 6,7,8, 11, 12,20,23,24 ,24a-tetradecahydro-10H-9,12-methanocyclopenta
[ 18, 19] [ 1 , 10,3 ,6]dioxadiazacyclononadecino [11, 12-b]quinolin- 19-yl]oxy} propyl) phosphonate

The title compound was prepared using the same method as Example 42, using diethyl (3-bromopropyl)phosphonate and stirring at room temperature. HRMS (ES+) m/z 968.4214 (M+H)+.
Step 2 : 3-{[(3aR, 7S,10S,12R,21E,24aS)-7-cyclopentyl-10-({(lR,2S)-l-[(cyclopropyl
sulfonyl)carbamoyl]-2-ethenylcyclopropyl}carbamoyl)-5, 8-dioxo-l, 2, 3, 3 a, 5, 6, 7, 8, 11,
12,20,23,24,24a-tetradecahydro-10H-9, 12-methanocyclopenta[18, 19] [1,10,3, 6]
opyl dihydrogen phosphate

To a solution of the product of Step 1 (43 mg) in dichloromethane (1 mL) was added trimethylsilyl bromide (0.115 mL). After 1 hour at room temperature, another 0.115 mL of trimethylsilyl bromide was added and the mixture was stirred for 2 hours. The reaction was quenched with 0.1 mL water and 1.5 mL ethanol. The solvents were removed in vacuo. The crude product was dissolved in dichloromethane and ether was added to precipitate a white solid. The solid was filtered and washed with ether to yield 36 mg of the desired product. HRMS (ES+) m/z 912.3603 (M+H)+.
Example 96 : (3siR SA0SA2R24aR)-7-tert-butYl-N- (lR2S)-2-et enyl- {\l- CmethoxymethyDcvclopropyll sulfonyl ) carbamoyl cyclopropyll -5 ,8-dioxo- 19-Γ3 -(piperidin- 1 - ynpropoxyl-1.2,3,3a,5,6J,8.11.12.20,21,22,23,24.24a-hexadecahydro-10H-9,12- methanocyclopentaf 18 , 19] [ 1J 0,3 ,61dioxadiazacyclononadecino|T 1 , 12-Z>lquinoline- 10- carboxamide

Step 1 : methyl (3aR, 7S,10S,12R,24aR)-7-tert-butyl-19-hydroxy-5,8-dioxo-l,2,3,3a,5,
6, 7, 8, 11,12, 20, 21, 22, 23, 24, 24a-hexadecahydro-l OH-9, 12-methanocyclopenta[18,19]
[1, 10,3,6 Jdioxadiazacyclononadecinof 11, 12-b Jq inoline-10-carboxylate
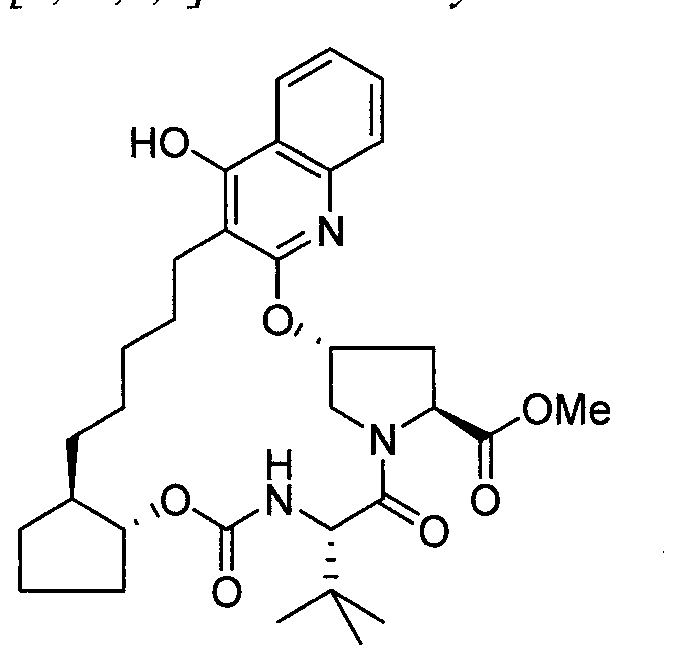
To a solution of methyl (3 aR S, 1 OS, 12^,21 E,24aS 19-(benzyloxy)-7-tert-butyl- 5,8-dioxo-l,2,3,3a,5,6,7,8,l l,12,20,23,24,24a-tetradecahydro-10H-9,12- methanocyclopenta[ 18, 19] [ 1 , 10,3 ,6]dioxadiazacyclononadecino[ 11 , 12-6]quinoline- 10- carboxylate (synthesized as in Example 1 with intermediates B6 and C3) (1.16 g) in THF (8.7 ml) and methanol (8.7 mL) was added 10% Pd/C (92 mg) and the mixture was stirred for 18 hours under hydrogen atmosphere. The atmosphere was changed to nitrogen and the reaction was carefully filtered through celite to give 0.88 g of the desired product. LRMS (ES+) m/z 582.40 (M+H)+.
Step 2 : methyl (3aR, 7S 0S, 12R,24aR)-7-tert-butyl-5,8-dioxo-19-[3-(piperidin-l-yl)propoxy]- 1, 2, 3, 3a, 5, 6, 7, 8, 11,12, 20, 21, 22, 23, 24, 24a-hexadecahydro-l OH-9, 12- methanocyclopentafl 8,19][1,10,3,6 Jdioxadiazacyclononadecinof 11,12-b Jq inoline-10- carboxylate

The title compound was prepared using the same method as described in Example 42 using l-(3-chloropropyl)piperidine hydrochloride as the alkylating agent with the product of Step 1. LRMS (ES+) m/z 707.5 (M+H)+.
Step 3 : (3aR, 7S,10S,12R,24aR)-7-tert-butyl-5,8-dioxo-i9-[3-(piperidin-l-yl)propoxy]- 1,2, 3, 3a, 5, 6, 7, 8, 11,12, 20, 21, 22, 23, 24, 24a-hexadecahydro-l OH-9, 12-methanocyclo
lononadecino[l 1, 12-b ]quinoline-l 0-carboxylic acid

To a solution of the ester from Step 2 (55.8 mg) in THF (0.8 mL) and ethanol (0.2 mL) was added 2M LiOH (0.4 mL). After 1 hour at room temperature, the reaction was done. The mixture was diluted with ethyl acetate and water. Then, acetic acid was added until pH = 5. The mixture was extracted with ethyl acetate (3x). The combined organics were dried over sodium sulfate, filtered and concentrated. LRMS (ES-) m/z 691.3 (M-H)".
Step 4 : (3aR, 7S,10S,12R,24aR)-7-tert-butyl-N-[(lR,2S)-2-ethenyl-l-({[l- (methoxymethyl)cyclopropyl ]sulfonyl}carbamoyl)cyclopropyl]-5, 8-dioxo-19-[3-(piperidin-l-
yl)propoxy]-l, 2, 3, 3a, 5, 6, 7, 8, 11,12, 20, 21, 22, 23, 24, 24a-hexadecahydro-l 0H-9, 12- methanocyclopentafl 8, 19] [1, 10, 3, 6]dioxadiazacyclononadecino[ 11, 12-b ]quinoline-l 0- carboxamide

1, Step 6 with intermediate A6. Purification by flash chromatography (ISCO, 0 to 10% methanol in DCM) afforded the desired product (44.6 mg). LRMS (ES+) m/z 949.5 (M+H)+.
Examples 97-105
By following the procedures outlined in Example 96 and using the appropriate A,
B and C intermediates and reagent (depicted below the structure as Int. and Rg., respectively), the following compounds were prepared.


carboxamide

[1,10,3 ,6]dioxadiazacvclononadecino[ 11.12-&lquinoline- 10-carboxamide

Step 1: ethyl (lR,2S)-l-[({(3aR S 0S 2R,24aR)-7-tert-butyl-19-[2-(morphoto
5, 8-dioxo-l, 2, 3, 3a, 5, 6, 7, 8, 11,12, 20, 21, 22, 23, 24, 24a-hexadecahydro-l OH-9, 12- methanocyclopentafl 8,19 ][1, 10,3,6 ]dioxadiazacyclononadecino[ 11,12-b Jquinolin-10- yl}carbonyl)amino]-2-ethenylcyclopropanecarboxylate
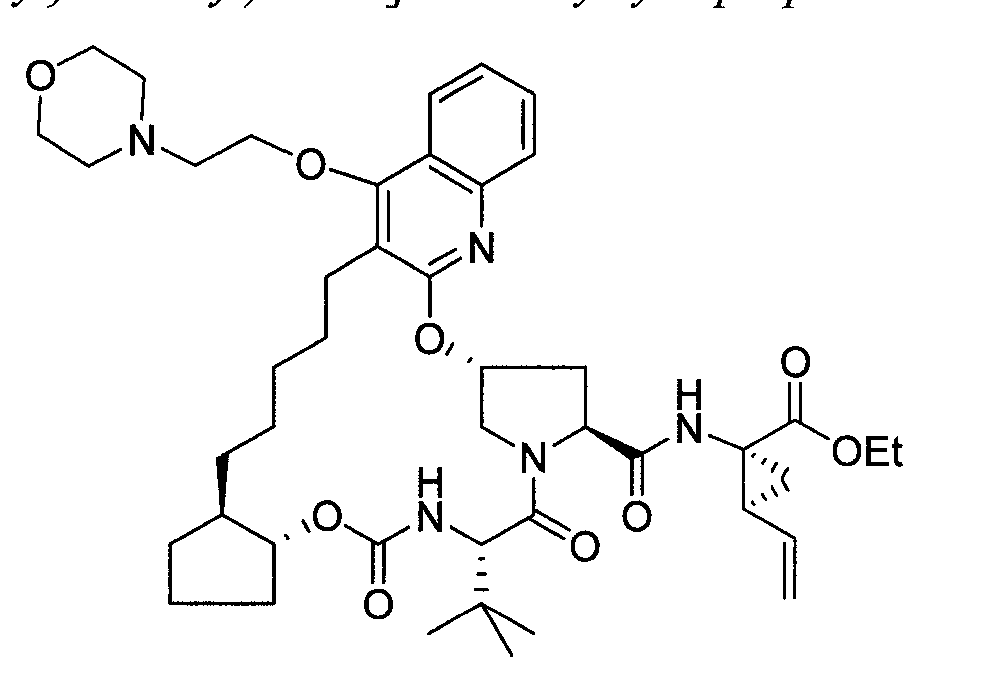
The title compound was prepared using the same method as described in Example
1, Step 6 with intermediate A4 and (3a7^,7S,10S,12 ^,24a ^)-7-tgrt-butyl-19-[2-(moφholin-4- yl)ethoxy]-5,8-dioxo-l,2,3,3a,5,6,7,8,l l,12,20,21,22,23,24,24a-hexadecahydro-10H-9,12- methanocyclopenta[ 18, 19] [ 1 , 10,3 ,6] dioxadiazacyclononadecino [11,12- >]quinoline- 10- carboxylic acid (Synthesized by the method described for Example 96, Steps 1-3 with 4-(2- bromoethyl)morpholine)). LRMS (ES+) m/z 818.5 (M+H)+.
Step 2 : (lR,2S)-l-[({(3aR, 7S,10S,12R,24aR)-7-tert-butyl-19-[2-(morpholin-4-yl)ethoxy]-5,8- dioxo-1, 2, 3, 3 a, 5, 6, 7, 8,11,12, 20,21, 22, 23, 24, 24a-hexadecahydro-l OH-9, 12-
methanocyclopenta[18, 19] [1,10,3, 6]dioxadiazacyclononadecino[ll, 12-b]quinol
boxylic acid

The title compound was prepared using the same method as Example 96, Step 3 but the reaction was stirred for 18 hours. LRMS (ES+) m/z 790.60 (M+H)+.
Step 3: (3aR, 7S,10S,12R,24aR)-7-tert-butyl-N-{(lR,2S)-l-[(dimethylsulfamoyl) carbamoyl]-2- ethenylcyclopropyl}-19-[2-(morpholin-4-yl)ethoxy]-5,8-dioxo-
1, 2, 3, 3a, 5, 6, 7, 8, 11,12, 20, 21, 22, 23, 24, 24a-hexadecahydro-10H-9, 12-methanocyclo
penta[18, 19] [1,10,3, 6]dioxadiazacyclononadecino[l 1 , 12-b]quinoline-l 0-carboxamide
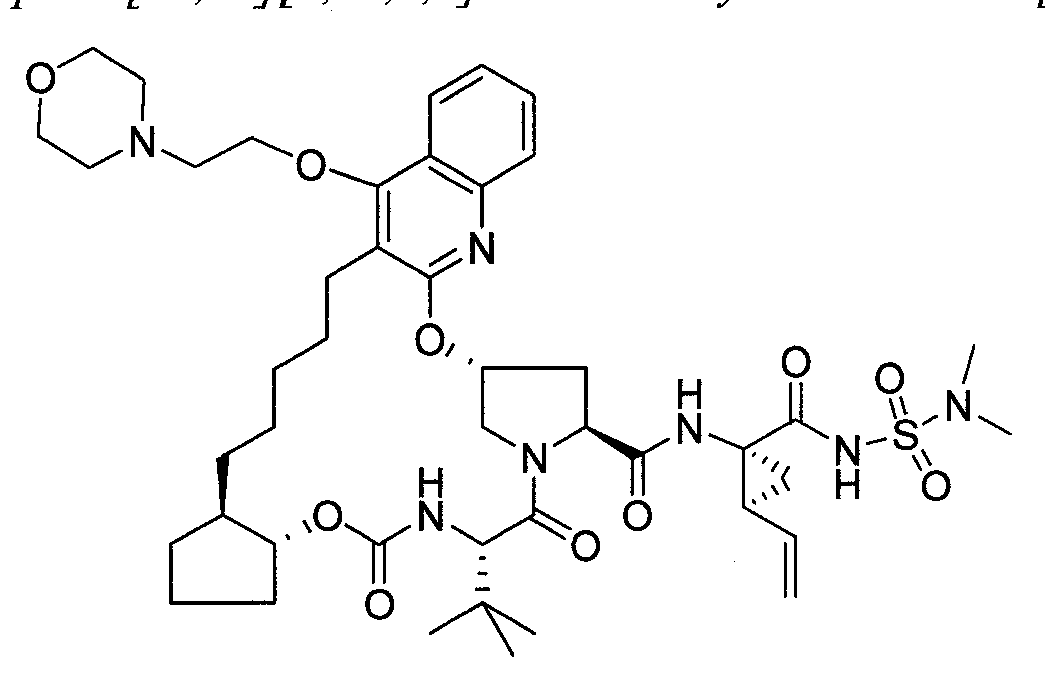
To a solution of the acid from Step 2 (46.6 mg) and Ν,Ν-dimethylsulfuric diamide (29.3 mg) in DMF (lmL) was added DIPEA (52 μΐ), DMAP (28.8 mg) and DBU (40 μΐ,). The reaction mixture was stirred 5 minutes before adding HATU (26.9 mg). The mixture was stirred at room temperature for 18 hours. The reaction mixture was diluted with Et20 and ethyl acetate and quenched with water and acetic acid (pH = 4). The mixture was extracted with diethyl ether (3x). The combined organic phases were washed with water (3x), dried over magnesium sulfate, filtered and concentrated. Purification by flash chromatography (ISCO, 0 to 10 % methanol in dichloromethane) afforded the desired product (34.2 mg). LRMS (ES+) m/z 896.50 (M+H)+.
By following the procedures outlined in Example 106 and using the appropriate reagent (depicted below the structure as Rg.), the following compounds were prepared.


International Patent Application Publication No.: WO2003/029226)
Example 113 : aai?,5S,8SJ0i?,22aj?V5-fert-butyl-N-ra^.2S)-2-ethenyl-l-{rri-methyl cyclopropyPsulfonyl] carbamoyl } cyclopropyl] - 17- [2-(morpholin-4-yl)ethoxy| -3 ,6-dioxo- 1.1 a.3.4.5.6.9, 10,18.19.20,21.22.22a-tetradecahydro-8H-7.10-methanocyclopropar 18.19] [1,10.3.6]dioxadiazacyclononadecino[l 1 , 12-Z?]quinoline-8-carboxamide

Step 1: 1-tert-butyl 2-methyl (2S,4R)-4-{[4- enzyloxy)-3-bromoquinolin-2-yl]oxy}pyrrolidine- 1, 2-dicarboxylate

To a 0°C solution of triphenylphosphine (2.54 g) in THF (75 ml) was added dropwise diisopyl azodicarboxylate (1.820 ml). The resulting mixture was stirred at 0 °C for 10 minutes then 1-tert-butyl 2-methyl (2S,4S)-4-hydroxypyrrolidine-l, 2-dicarboxylate (2.01g) followed by intermediate C6 (2.06 g) were added to the mixture. The mixture was stirred another 15 minutes at 0°C then allowed to reach room temperature and stirred for 2 hours. Silica gel was added and the adsorbed product was purified by flash chromatography (ISCO) to provide the desired product (2.88 g). LRMS (ES+) m/z 557.0 (M+H)+.
Step 2 : N-({[(lR )-2-{5-[4-(benzyloxy)-2-{[(3R,5S)-l-(tert-butoxycarbonyl)-5- (methoxycarbonyl)pyrrolidin-3-yl]oxy}quinolin-3-ylJpent-4-yn-l-yl}cyclopropylJ oxyjcarbonyl)- 3-methyl-L-valine
C02Me

PdCl2(MeCN)2 (51 mg), tri-t-butylphosphonium tetrafluoroborate (170
K2C03 (1.35 g) and intermediate BIO (2.43 g) were added to a reaction flask. The reaction mixture was degassed and refilled with nitrogen (3x). In another flask, bromide from step 1 (2.18 g) was dissolved in 33 mL acetonitrile. This solution was degassed and refilled with nitrogen (3x). Then, this solution was added to the reaction mixture. The complete reaction mixture was degassed and refilled with nitrogen (3x) and stirred at 75°C for 18 hours. After the reaction mixture was cooled back to room temperature, the acetonitrile was removed in vacuo. The crude reaction mixture was dissolved in ethyl acetate and IN HC1 was slowly added. The reaction mixture was extracted 3x with ethyl acetate. The combined organics were washed with brine, dried over sodium sulfate, filtered and concentrated. The crude mixture was used directly in the next step. LRMS (ES+) m/z 758.3 (M+H)+.
Step 3 : N-({[(lR,2R)-2-{5-[4-(benzyloxy)-2-{[(3R,5S)-5-(methoxycarbonyl) pyrrolidinium-3- cyclopropyl]oxy}carbonyl)-3-methyl-L-valine chloride

Step 4 : methyl (laR,5S,8S,10R,22aR)-17-(benzyloxy)-5-tert-butyl-3,6-dioxo-18,19-didehydro- 1,1a, 3, 4, 5, 6, 9, 10, 20, 21, 22, 22a-dodecahydro-8H- 7, 10-methanocyclopropa
lononadecino[ll, 12-b]quinoline-8-carboxylate

To a solution of the product of step 3 in DMF (52 mL) at 0°C was added DIPEA
(3.1 mL) and HATU (1.64 g). The mixture was stirred at room temperature for 2 hours until disappearance of the starting material. The reaction mixture was diluted with Et20 and quenched with water and HC1 (IN). The mixture was extracted (3x) with ether. The combined organic phases were washed with water then brine, dried over sodium sulfate, filtered and concentrated.
Purification by flash chromatography (ISCO, 0 to 100% ethyl acetate in hexanes) afforded the desired product (1.3 g). LRMS (ES+) m/z 640.45 (M+H)+.
Step 5 : methyl (laR, 5S, 8S, 1 OR, 22aR)-5-tert-butyl-l 7-hydroxy-3, 6-dioxo-
1, la, 3, 4, 5, 6, 9, 10,18,19, 20, 21, 22, 22a-tetradecahydro-8H- 7, 10-methanocyclopropa
cyclononadecino[ 11,12-b ]quinoline-8-carboxylate
The title compound was prepared using the same method as Example 96, Step 1.
The reaction was stirred for 4 days at room temperature and additional Pd/C (1 mol %) was added after the third day. LRMS (ES+) m/z 554.35 (M+H)+.
Step 6 : methyl (laR,5S,8S,10R,22aR)-5-tert-butyl-17-[2-(morpholin-4-yl)ethoxy]-3,6-dioxo- 1,1a, 3, 4, 5, 6, 9, 10,18,19, 20, 21, 22, 22a-tetradecahydro-8H-7, 10-methanocyclopropa
[18, 19] [1,10, 3, 6]dioxadiazacyclononadecino[ll, 12-b]quinoline-8-carboxylate

The title compound was prepared using the same method as described in Example
42. LRMS (ES+) m/z 667.45 (M+H)+.
Step 7 : (laR, 5S, 8S, 1 OR, 22aR)-5-tert-butyl-l 7-[2-(morpholin-4-yl)ethoxy]-3, 6-dioxo- 1,1 a, 3, 4, 5, 6,9, 10, 18, 19,20,21, 22, 22a-tetradecahydro-8H- 7, 10-methanocyclopropa
[18,19] [1,10,3,6 ]dioxadiazacyclononadecino[ 11,12-b [quinol ine-8-carboxylic acid

The title compound was prepared using the same method as described in Example 96, Step 3. LRMS (ES+) m/z 653.50 (M+H)+.
Step 8 : (laR, 5S, 8S, 1 OR, 22aR)-5-tert-butyl-N-[(lR, 2S)-2-ethenyl-l-{[(l- methylcyclopropyl)sulfonyl ]carbamoyl}cyclopropyl]-l 7-[2-(morpholin-4-yl)ethoxy]-3, 6-dioxo- 1,1 a, 3, 4, 5,6,9, 10, 18, 19,20,21, 22, 22a-tetradecahydro-8H- 7, 10-methanocyclopropa[l 8,19]
[1,10,3,6 Jdioxadiazacyclononadecinofl 1,12-b ]quinoline-8-carboxamide

The title compound was prepared using the same method as Example 1 , Step 6 with intermediate A2. Purification by flash chromatography (ISCO, 0 to 10% methanol in DCM) afforded the desired product. LRMS (ES+) m/z 879.40 (M+H)+.
Examples 114-130
By following the procedures outlined in Example 113 and using the appropriate A, B and C intermediates and reagent (depicted below the structure as Int. and Rg., respectively), the following compounds were prepared.


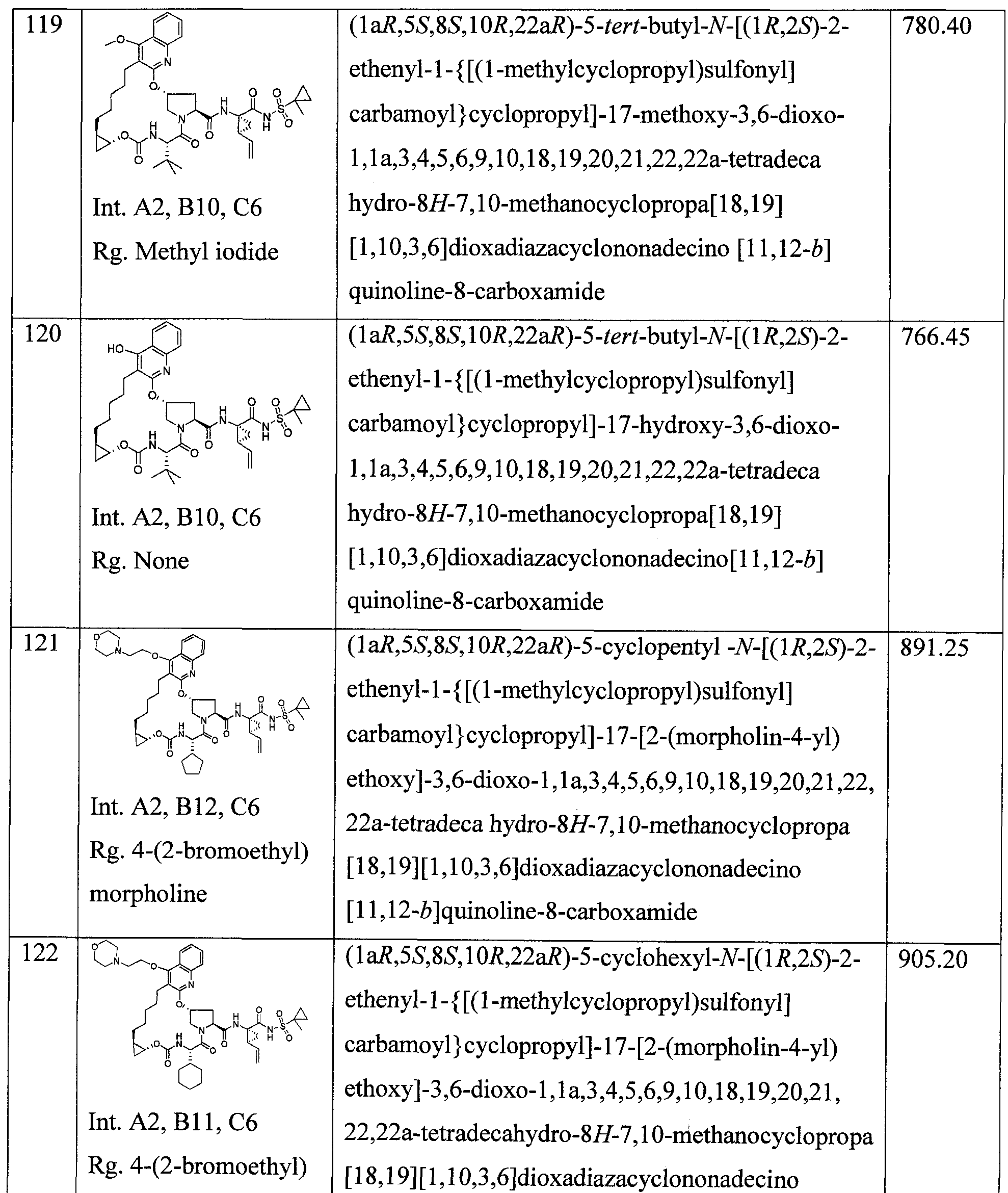
morpholine [11,12-Z>]quinoline-8-carboxamide

morpholine cyclononadecino[l 1 ,12-&]quinoline-8-carboxamide

nonadecino [ 11 , 12-b] quinoline-8-carboxamide
Example 131 : (lai?.5S.8S.10i?,22ai?V5-tgrt-butyl-N-r(li?,2S)-2-ethenyl-l-ira-methyl cvclopropyl sulfonyl]carbamoyUcyclopropyl1-17- 3-(4-methylpiperazin-l-yl)propoxy]-3,6-
dioxo- 1.1 a,3.4.5.6.9.10.18.19.20.21.22,22a-tetradecahvdro-8H-7, 10-methano
cyclopropa|T 8, 191 Γ 1.10.3,6]dioxadiazacvclononadecinof 11 , 12-&1quinoline-8-carboxamide

The title compound was prepared using the same method as described in Example 42, using the l-(3-bromopropyl)-4-methylpiperazine and (la/?,5S,8S,10/?, 22ai?)-5-tert-butyl-N- [( 1 J?,25)-2-ethenyl- 1 - { [( 1 -methylcyclopropyl)sulfonyl] carbamoyl } cyclopropyl] - 17-hydroxy- 3 ,6-dioxo-l , 1 a,3,4,5,6,9, 10,18,19,20,21 ,22,22a-tetradecahydro-8H-7, 10- methanocyclopropa[l 8, 19] [1 , 10,3,6]dioxadiazacyclononadecino[l 1 , 12-&]quinoline-8- carboxamide (Example 120). LRMS (ES+) m/z 906.40 (M+H)+.
Example 132 : naj?.5S.8S.10i?.22ai?V5-tert-butyl-N-r(lj?.25' -2-ethenyl-l-{r(l-methyl cvclopropyl)sulfonyl]carbamovUcvclopropyl]-17- 2-(morpholin-4-yl ethoxy]-3,6-dioxo-14- rpropan-2-ylVl.la.3.4.5.6.9.10.18.19.20.21.22,22a-tetradecahvdro-8H-7.10-methano

The title compound was prepared using the same method as described in Example 42, using the 4-(2-bromoethyl)morpholine and (UR,5S,SS,\0R,22aR)-5-tert-bnty\-N-[(lR,2S)-2- ethenyl- 1 - { [( 1 -methylcyclopropyl)sulfonyl]carbamoyl} cyclopropyl] - 17-hydroxy-3 ,6-dioxo- 14- (propan-2-yl)- 1 , 1 a,3 ,4,5 ,6,9, 10, 18, 19,20,21 ,22,22a-tetradecahydro-8H-7, 10-
methanocyclopropa[ 18, 19] [ 1 , 10,3,6]dioxadiazacyclononadecino [11,12-6]quinoline-8- carboxamide (synthesized in the same way as Example 120 but using intermediates A2, BIO and C9). LRMS (ES+) m/z 921.55 (M+H)+.

ethenyl- !-{[(! -methylcyclopropyDsulfonyl] carbamoyl} cyclopropyl]-3,6-dioxo-
1 , 1 a,3 ,4.5 ,6.9, 10, 18, 19,20.21 ,22.22a-tetradecahydro-8H-7, 10-methano

The title compound was prepared using the same method as described in Example
75, using the 3-(azetidin-l-yl)propan-l-ol and (la/?,55,81S,10i?,22ai?)-5-tert-butyl-iV-[(li?,2S)-2- ethenyl-l-{[(l-methylcyclopropyl)sulfonyl]carbamoyl}cyclopropyl]-17-hydroxy-3,6-dioxo- 1 , 1 a,3 ,4,5 ,6,9, 10, 18, 19,20,21 ,22,22a-tetradecahydro-8H-7, 10-methanocyclopropa[ 18,19]
[l,10,3,6]dioxadiazacyclononadecino[l l,12-b]quinoline-8-carboxamide (Example 120). LRMS (ES+) m/z 863.50 (M+H)+.
Examples 134-136
By following the procedures outlined in Example 133 and using the appropriate reagent (depicted below the structure as Rg.), the following compounds were prepared.


Example 137 : tert-butyl (3S -3-(,{[riai?,5^8^10i?,22a^ -5-tert-butyl-8-{[('lj;,2S -2-ethenyl-l- {[(l-methylcyclopropyl)sulfonyl]carbamoyl}cvclopropyl]carbamoyll-3,6-dioxo- 1.1 a.3.4.5.6.9.10.18.19.20.21.22.22a-tetradecahydro-8H-7.10-methanocvclo
propa[ 18, 19] [ 1 , 10.3 ,6]dioxadiazacyclononadecino [11.12-6]quinolin- 17-ylloxy}
methyDpyrrolidine- 1 -carboxylate

The title compound was prepared using the same method as described in Example 75, using the tert-butyl (3S)-3-(hydroxymethyl)pyrrolidine-l -carboxylate and
(1 aR,5S,8S, 10i?,22ai?)-5-tert-butyl-N-[(li?,25)-2-ethenyl-l -{ [(1 -methylcyclopropyl)
sulfonyl] carbamoyl } cyclopropyl] - 17-hydroxy-3 ,6-dioxo- 1 , 1 a,3 ,4,5 ,6,9, 10, 18, 19,20,21 , 22,22a- tetradecahydro-8H-7, 10-methanocyclopropa[ 18, 19] [ 1 , 10,3 ,6]dioxadiazacyclononadecino [11,12- b]quinoline-8-carboxamide (Example 120). The method was slightly modified as
triphenylphosphine was used instead of trimethylphosphine and the reaction was done at a 0.3 M concentration LRMS (ES+) m/z 949.5 (M+H)+.
Examples 138-142
By following the procedures outlined in Example 137 and using the appropriate reagent (depicted below the structure as Rg.), the following compounds were prepared.


azetidine- 1 -carboxylate
Example 143 : riaJ?.5 .8^J0jg,22ai?V5-fert-butyl-N- ri^.2,$l)-2-ethenyl-l-ira-methyl cyclopropyPsulfonyllcarbamoyl } cyclopropyll- 13 -methoxy-3 ,6-dioxo- 17- [3 -(pyrrolidin- 1 - vnpropoxyl - 1.1 a.3.4.5.6,9.10.18.19.20,21.22.22a-tetradecahvdro-8H-7.10-methano
cvclopropaf 18.19] [ 1.10.3.61dioxadiazacyclononadecino [ 11.12-b] quinoline-8-carboxamide

Step 1 : methyl (laR,5S,8S,10R,22aR)-17-(3-bromopropoxy)-5-tert-butyl-13-methoxy-3,6-dioxo-
1, la, 3, 4, 5, 6, 9, 10,18,19, 20, 21, 22, 22a-tetradecahydro-8H- 7, 10-methano
cyclopropafl 8, 19] [1,10,3, 6]dioxadiazacyclononadecino[ll, 12-b]quinoline-8-carboxylate
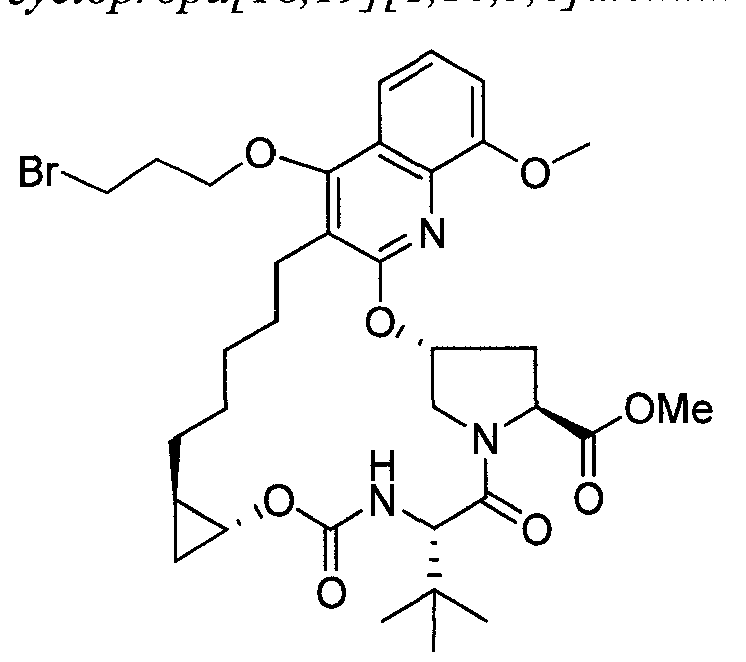
To a solution of methyl (lai?,55,85',10i?,22a ?)-5-tert-butyl-17-hydroxy-13- methoxy-3, 6-dioxo-l , 1 a,3,4,5,6,9, 10, 18, 19,20,21 ,22,22a-tetradecahydro-8H-7, 10-methano cyclopropafl 8,19][l,10,3,6]dioxadiazacyclononadecino[l l,12-Z>]quinoline-8-carboxylate (253 mg) (prepared by the same method as Example 113, Steps 1-5 with intermediates B10 and C8) and 1,3-dibromopropane (500 μΐ) in DMF (5 ml) was added cesium carbonate (700 mg). After 90 minutes of stirring at room temperature, the reaction mixture was partitioned between water/brine 1 : 1 and ethyl acetate. The product was extracted twice with ethyl acetate and the combined organic phases were washed with water and brine, dried over sodium sulfate and evaporated. The crude residue was purified by flash chromatography (ISCO 10%-50% ethyl acetate in hexanes) to afford a white solid (231 mg). LRMS (ES+) m/z 704.30 (M+H)+.
Step 2 : methyl (laR,5S,8S,10R,22aR)-5-tert-butyl-13-methoxy-3,6-dioxo-17-[3-(pyrrolidin-l- yl)propoxy]-l, la, 3, 4, 5, 6, 9, 10,18,19, 20, 21, 22, 22a-tetradecahydro-8H- 7, 10-methano
nonadecino[l 1 , 12-b]quinoline-8-carboxylate

To a solution of the product resulting from step 1 (72 mg) in DMSO (1.5 ml) were added pyrrolidine (85 μΐ) and DIPEA (25 μΐ). After 2 hours of stirring at 50°C, the reaction mixture was poured into water and the product was extracted twice with ethyl acetate. The combined organic phases were washed with water and brine, dried over sodium sulfate and evaporated. The crude product was used as such for the next step. LRMS (ES+) m/z 695.50 (M+H)+.
Step 3 : (laR,5S,8S,10R,22aR)-5-tert-butyl-13-methoxy-3,6-dioxo-17-[3-(pyrrolidin-l-yl) propoxyj-l, la, 3, 4, 5, 6, 9,10,18,19, 20,21, 22, 22a-tetradecahydro-8H- 7, 10-methano
cyclopropaf 18, 19] [1, 10,3,6 ]dioxadiazacyclononadecino[l 1, 12-b ]quinoline-8-carboxylic acid

The title compound was prepared using the same method as described in Example
113, Step 7. LRMS (ES+) m/z 681.40 (M+H)+.
Step 4 : (laR,5S,8S,10R,22aR)-5-tert-butyl-N-[(lR,2S)-2-ethenyl-l-{[(l-methylcycl^
propyl)sulfonyl]carbamoyl}cyclopropyl]-13-methoxy-3,6-dioxo-17-[3-(pyrrolidin-l-yl)
propoxy J-l, 1 a, 3, 4, 5, 6, 9,10,18, 19,20,21, 22, 22a-tetradecahydro-8H- 7, 10-methanocyclo
[ 11, 12-b ]quinoline-8-carboxamide
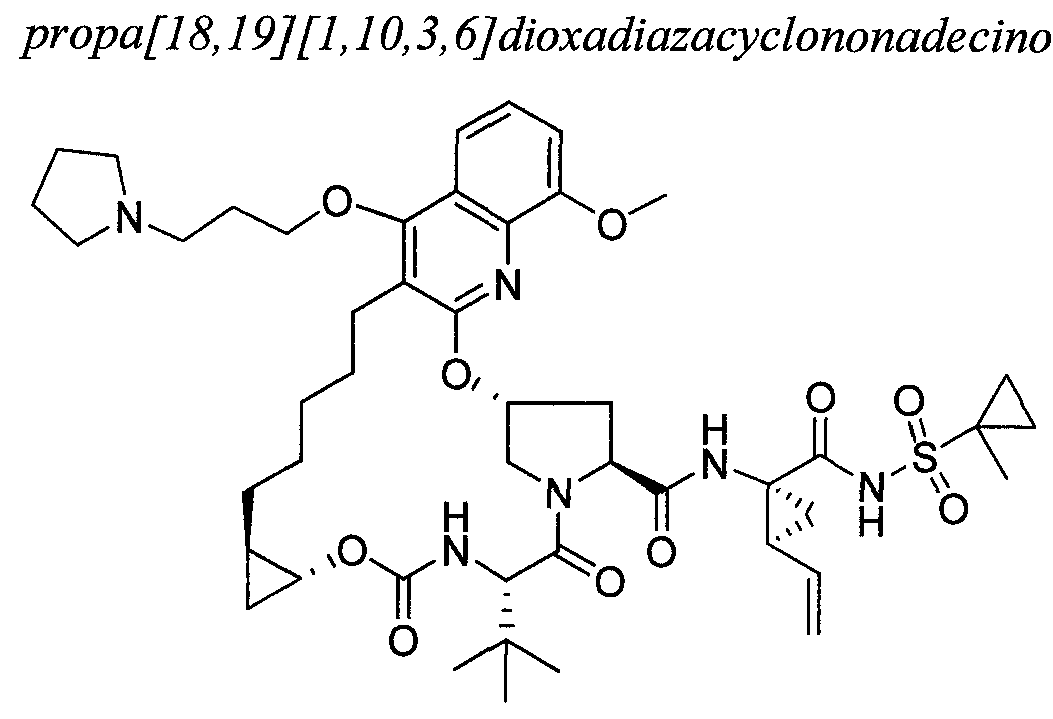
The title compound was prepared using the same method as described in Example 113, Step 8. Purification by flash chromatography (ISCO reverse phase, 15%-70% ACN in water (0.1% TFA buffer)) afforded the desired product (40.5 mg). LRMS (ES+) m/z 907.30 (M+H)+.
Example 144 : (laig.5S.8S.10i?.22a^ -5-tgrt-butyl-N-rn ?.2S -2-ethenyl-l-( ri-methyl cycloprop vDsulfonyll carbamoyl } cycloprop yl] - 17- [3 -d H-imidazol- 1 - vDpropoxy] - 13 -methoxy- 3 ,6-dioxo- 1.1 a.3 A5.6.9.10.18.19.20.21.22.22a-tetradecahvdro-8H-7.10-methano
cyclopropafl 8.19][1.10,3,61dioxadiazacyclononadecino[l 1.12-6]quinoline-8-carboxamide

The title compound was prepared using the same method as described in Example 143 using imidazole. LRMS (ES+) m/z 904.45 (M+H)+.
Example 145 : (lai?.5S.8^.10i?.22ai?)-5-tgrt-butyl-17-r3-(cvclopropylamino propoxyl-N-
[Y li?,2S)-2-ethenyl- 1 - { [Y 1 -methylcyclopropyDsulfonyllcarbamoyl } cyclopropyl]- 14-methyl-3.6-
dioxo- 1.1 a.3.4.5.6.9.10,18.19.20.21.22.22a-tetradecahvdro-8H-7, 10-methano

Step 1 : (laR,5S,8S,10R,22aR) 7-(3-bromopropoxy)-5-tert-butyl-N-[(lR,2S)-2-ethenyl-l-{[(l- methylcyclopropyl)sulfonyl]carbamoyl}cyclopropyl]-14-methyl-3,6-dioxo-
1, la, 3, 4, 5, 6, 9, 10,18,19, 20,21, 22, 22a-tetradecahydro-8H- 7, 10-methanocyclopropa
[18,19 ][ 1, 10, 3, 6]dioxadiazacyclononadecino[ 11, 12-b Jquinoline-8-carboxamide

(lai?,5S,85,10i?,22ai?)-5-rert-butyl-iV-[(li?,25)-2-ethenyl-l-{[(l- methylcyclopropyl)sulfonyl]carbamoyl}cyclopropyl]-17-hydroxy-14-methyl-3,6-dioxo- 1 , 1 a,3 ,4,5,6,9, 10,18,19,20,21 ,22,22a-tetradecahydro-8H-7, 10-methanocyclopropa[ 18,19]
[l,10,3,6]dioxadiazacyclononadecino[l l,12-6]qmnoline-8-carboxamide (synthesized by the method described for Example 120 using intermediates A2,B10,C11) was alkylated using the method for example 143, step 1. LRMS (ES+) m/z 900.30 (M+H)+.
Step 2 : (laR,5S,8S,10R,22aR)-5-tert-butyl-17-[3-(cyclopropylamino)propoxy]-N-[(lR,2S)^ ethenyl-l-{[(l-methylcyclopropyl)s lfonyl]carbamoyl}cyclopropylJ-14-methyl-3,6-dioxo- 1,1 a, 3, 4,5, 6,9,10,18,19,20,21, 22, 22a-tetradecahydro-8H- 7, 10-methano
cyclopropa[18, 19] [1,10,3, 6]dioxadiazacyclononadecino[ll,12-b]quinoline-8-carboxamide

The title compound was prepared using the same method as described in Example 143, Step 2 using cyclopropyl amine. Purification by flash chromatography (ISCO reverse phase, 20%-80% ACN in water (0.1% TFA buffer)) afforded the desired product (40.5 mg). LRMS (ES+) m/z 877.30 (M+H)+.
Examples 146-209
By following the procedures outlined in Example 145 and using the appropriate
A, B and C intermediates and reagent (depicted below the structure as Int. and Rg., respectively), the following compounds were prepared.


[11,12-&]quinoline-8-carboxamide

[11,12-b]quinoline-8-carboxamide

hydrochloride decino [ 11 , 12-b] quinoline- 8 -carboxamide

quinoline- 8-carboxamide

sulfonyl) pyrrolidine cyclononadecino[ 11,12-b]quinoline-8-carboxamide

hydrochloride

cyclononadecino[l 1 ,12-Z>]quinoline-8-carboxamide

[11,12-£]quinoline-8-carboxamide

[ 11 , 12-b] quinoline-8-carboxamide

[11 , 12-0]quinoline-8-carboxamide

quinoline-8-carboxamide

cyclononadecino[ 11,12-b]quinoline-8-carboxamide
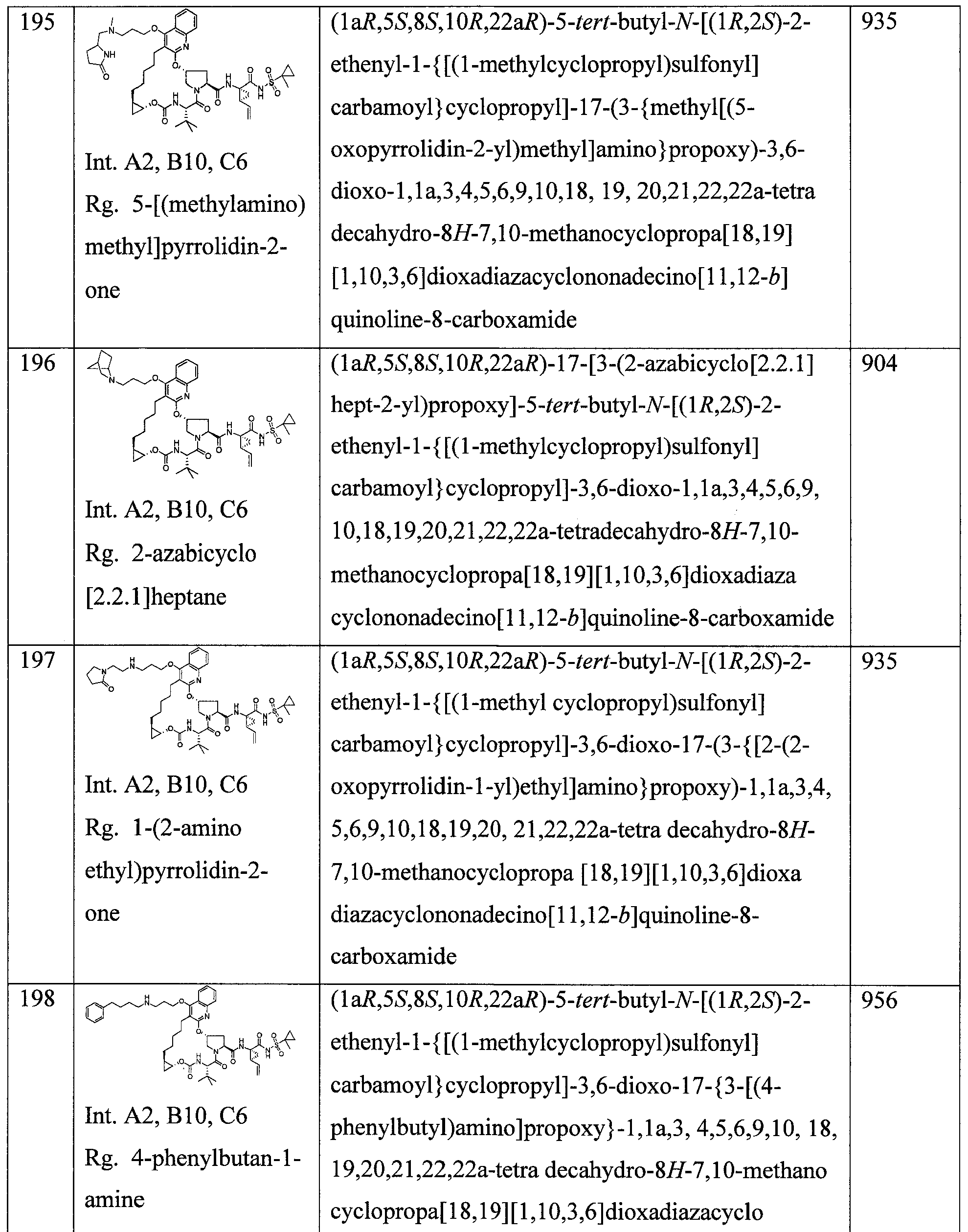
nonadecino[l 1 ,12-Z>]quinoline-8-carboxamide

quinoline- 8 -carboxamide

carboxamide

carboxamide

cyclopropyDsulfonyl] carbamoyl } cyclopropyl] - 17- [Yl -methylpiperidin-4- vDoxy] -3.6-dioxo-
1.1 a.3.4.5.6.9.10, 18, 19,20.21.22.22a-tetradecahydro-8H-7, 10-methanocvclopropa

Step 1 : methyl (laR,5S,8S,10R,22aR)-5-tert-butyl-17-[(l-methylpiperidin-4-yl)oxy]-3,6-dioxo- l,la,3,4,5,6,9,10,18,19,20,21, 22, 22a-tetradecahydro-8H- 7, 10-methanocyclopropa
[18, 19] [1,10, 3, 6]dioxadiazacyclononadecino[ll, 12-b]quinoline-8-carboxylate

Methyl (la/?,55,85,10i?,22ai?)-5-tert-butyl-17-hydroxy-3,6-dioxo- l,la,3,4,5,6,9,10,18,19,20,21 ,22,22a-tetradecahydro-8H-7, 10-methanocyclopropa
[18,19][l,10,3,6]dioxadiazacyclononadecino[l l,12-£]quinoline-8-carboxylate synthesized in example 113, step 5 was reacted with l-methylpiperidin-4-ol using the procedure described for Example 137 (heating was required: 40°C for 18 hours). LRMS (ES+) m/z 651.55 (M+H)+. Step 2 : (1 aR, 5S,8S, 1 OR, 22aR)-5-tert-butyl-l 7-[(l -methylpiperidin-4-yl)oxy] -3,6-dioxo- 1, la, 3, 4, 5, 6, 9, 10,18,19, 20, 21, 22, 22a-tetradecahydro-8H-7, 10-methanocyclopropa
[18,19] [1,10, 3, 6]dioxadiazacyclononadecino[l l,12-b]quinoline-8-carboxylic acid

The title compound was prepared using the same method as described for
Example 96, Step 3. LRMS (ES+) m/z 637.45 (M+H)+.
Step 3 : (laR,5S,8S,10R,22 R)-5-tert-butyl-N-[(lR,2S)-2-ethenyl-l-{[(l-methyl
cyclopropyl)sulfonyl]carbamoyl}cyclopropyl]-l 7-[(l-methylpiperidin-4-yl)oxy]-3, 6-dioxo- 1, la, 3, 4, 5, 6, 9, 10,18,19, 20, 21, 22, 22a-tetradecahydro-8H- 7, 10-methanocyclo
ino[ll, 12-b]quinoline-8-carboxamide

The title compound was prepared using the same method as described in Example 1, Step 6 with intermediate A2. Purification by flash chromatography (ISCO, 0 to 10% methanol in DCM, loading with 0.5% acetic acid in DCM) afforded the desired product. LRMS (ES+) m/z 863.50 (M+H)+.
Examples 211-220
By following the procedures outlined in Example 210 and using the appropriate
A, B and C intermediates and reagent (depicted below the structure as Int. and Rg., respectively), the following compounds were prepared.

cyclononadecino[ 11,12-Z>]quinoline-8-carboxamide

[11,12-b]quinoline-8-carboxamide

Example 221 : (lai?.51y,85,a0i?,22ai?V5-tgrt-butyl-N-[rii?,2S)-2-ethenyl-l-{[(,l-methyl cvclopropyl)sulfonyl]carbamoyl|cvclopropyl1-17-[(2i?')-morpholin-2-ylmethoxy1-3,6-dioxo- 1,1 a,3 ,4,5,6,9, 10,18,19,20,21 ,22,22a-tetradecahydro-8H-7, 10-methanocvclopropa
11 , 12-b]quinoline-8-carboxamide

Step 1 : tert-butyl (2R)-2-({[(laR,5S,8S, 10R,22aR)-5-tert-butyl-8-{[(lR,2S)-2-ethenyl-l-{[(l- methylcyclopropyl)sulfonyl]carbamoyl}cyclopropyl]carbamoyl}-3, 6-dioxo-l, la, 3, 4, 5, 6, 9, 10, 18,19, 20, 21, 22, 22a-tetradecahydro-8H- 7, 10-methanocyclopropa [18,19] [1, 10,3, 6]dioxadiaza cyclononadecino [11, 12-b]quinolin-l 7 -yl] oxy [methyl) morpholine-4-carboxylate

The title compound was prepared using the same method as described in Example 210, Step 1-3 with tert-butyl (2i?)-2-(hydroxymethyl)morpholine-4-carboxylate as the reagent for the Mitsunobu reaction. LRMS (ES+) m/z 965.50 (M+H)+.
Step 2 : (laR,5S,8S,10R,22aR)-5-tert-butyl-N-[(lR,2S)-2-ethenyl-l-{[(l-methylcyclo propyl sulfonyl] carbamoyl}cyclopropyl] -17 -[(2R)-morpholin-2-ylmethoxy] -3 ,6-dioxo- 1, la, 3, 4, 5, 6, 9, 10,18,19, 20, 21, 22, 22a-tetradecahydro-8H- 7, 1 O-methanocyclopropa
[18,19][1,10, 3, 6]dioxadiazacyclononadecino[ll, 12-b]quinoline-8-carboxamide

To a solution of the product from step 1 (92 mg) in dichloromethane (0.5 mL) was added trifluoroacetic acid (0.5 mL). The resulting solution was stirred for 3 hours at room temperature until disappearance of the starting material. The solvent was removed in vacuo. The reaction mixture was dissolved in ethyl acetate and a saturated solution of sodium
bicarbonate was added slowly. After extracting 3x with ethyl acetate, the combined organics were dried with sodium sulfate, filtered and concentrated to afford the desired product (77.9 mg). LRMS (ES+) m/z 865.45 (M+H)+.
Example 222 : (laig.5^8^J0ig,22ai? -17-(azetidin-3-yloxyV5-fert-butyl-N-r(li?.2S)-2-ethenyl-l- { Γ( 1 -methylcvclopropyl sulfonyl]carbamoyl } cvclopropyl] -3 ,6-dioxo-
1.1 a.3.4.5.6.9, 10.18,19,20,21 ,22,22a-tetradecahydro-8H-7.1 O-methanocvclopropa
"| [ 1 , 10.3 ,6]dioxadiazacvclononadecino [11, 12-&lquinoline-8-carboxamide

The title compound was prepared using the same method as described in Example 221 using ter -butyl 3 -hydroxyazetidine-l-carboxylate for the Mitsunobu reaction. Purification by flash chromatography (ISCO reverse phase, 25 to 70% ACN in water (0.1% TFA buffer)) afforded the desired product LRMS (ES+) m/z 821.50 (M+H)+.
Example 223 : nai?,5S,8S,10i?,22ai? -5-tgrt-butyl-17-(ri-rcvclopropylmethyl azetidin-3- ylloxy}-N-[(lj?,2S)-2-ethenyl-l-{[(l-methylcvclopropyl)sulfonyl]carbamoyl> cyclopropyl]-3,6- dioxo- 1 , 1 a,3 ,4,5 ,6,9, 10, 18, 19,20,21 ,22.22a-tetradecahvdro-8H-7.10- methanocyclopropa[ 18, 19] [ 1 , 10,3 ,61dioxadiazacvclononadecino[l 1 , 12-6]quinoline-8- carboxamide

To a solution of Example 222 (100 mg) in DMF (0.5 ml) were added cyclopropyl methyl bromide (28.9 mg) and DIPEA (100 μΐ). After 18 hours of stirring at 50°C, the reaction mixture was diluted in DMSO (1.5 mL) and it was loaded over a CI 8 column and purified by flash chromatography (ISCO, 10%-70% ACN in water (0.1% TFA buffer)) to afford a white solid (44 mg of TFA salt). LRMS (ES+) m/z 875.35 (M+H)+.
Example 224 : (laR.5S.8S,10R,22aR)-5-tert-butyl-N-rriR.2SV2-ethenyl-l-ira- methylcyclopropyDsulfonyl]carbamoyl) cyclopropyl] - 17- [( 1 -methylazetidin-3 -yl) methoxy] -3,6- dioxo- 1.1 a.3.4.5.6.9, 10, 18, 19,20,21 ,22,22a-tetradecahydro-8H-7, 10- methanocyclopropaf 18, 19] Γ 1 , 10,3 ,6]dioxadiazacyclononadecinof 11 , 12-b]quinoline-8- carboxamide

Step 1 : (laR,5S,8S 0R,22aR)-17-(azetidin-3-ylmethoxy)-5-tert-butyl-N-^
{[(l-methylcyclopropyl)sulfonyl]carbamoyl}cyclopropyl]-3,6-dioxo- 1, 1 a, 3, 4, 5, 6,9, 10, 18, 19,20,21, 22, 22a-tetradecahydro-8H- 7, 10-methanocyclopropa
1, 12-b]quinoline-8-carboxamide

The title compound was prepared using the same method as described in Example
221, Step 2 with Example 142. LRMS (ES+) m/z 835.45 (M+H)+.
Step 2 .■ (laR,5S,8S 0R,22aR)-5-tert-butyl-N-[(lR,2S)-2-ethenyl-l-{[(l-methyl
cydopropyl)sulfonyl]carbamoyl}cyclopropyl]-17-[(l-methylazetidin-3-yl)meth^
1, la, 3, 4, 5, 6, 9, 10, 18,19, 20, 21, 22, 22a-tetradecahydro-8H-7, 10-methanocyclopropa
[18, 19] [1, 10, 3,6]dioxadiazacyclononadecino[ll, 12-b]quinoline-8-carboxamide

To a suspension of the amine from Step 1 (20.6 mg) in methanol (1 mL) was added formaldehyde (10 μΐ). Sodium borohydride (5.6 mg) was added slowly. The clear reaction mixture was stirred overnight at room temperature. The solvent was removed in vacuo and the product was dissolved in ethyl acetate and water (with acetic acid to a pH=5). The mixture was extracted (3x) with ethyl acetate. The combined organics were dried with sodium sulfate, filtered and concentrated. Purification by flash chromatography (ISCO reverse phase, 0 to 95 % water in acetonitrile followed by ISCO : 0 to 20 % methanol in DCM) gave the desired product (1.4 mg). LRMS (ES+) m/z 849.55 (M+H)+.
Example 225 : (lai?,5S,8SJ0i?,22a^ -5-tgrt-butyl-N-rni?,2^-2-ethenyl-l-{r(l-methyl cvclopropyDsulfonyllcarbamoyl } cyclopropyll - 17- { |Ί -(2-methoxyethyl piperidin-4- yl]oxy } -3,6- dioxo- 1 , 1 a,3 A5 ,6,9, 10.18 , 19.20.21.22.22a-tetradecahvdro-8H-7, 10-methano
cyclopropaf 18, 19] [ 1.10.3,6]dioxadiazacvclononadecino[ 11 , 12-Z>1quinoline-8-carboxamide

Step 1 : methyl (laR,5S,8S,10R,22aR)-17-{[l-(tert-butoxycarbonyl)piperidin-4-yl]oxy}-5-tert- butyl-3, 6-dioxo-l, la, 3, 4, 5, 6, 9,10,18,19, 20,21, 22, 22a-tetradecahydro-8H- 7, 10-
methanocyclopropa[18, 19] [1, 10, 3, 6]dioxadiazacyclononadecino[ 11, 12-b Jquinoline-8- carboxylate

The title compound was prepared using the same method as described in Example 210, Step 1 with methyl ( 1 ai?,5S,8S, 10i?,22ai?)-5-tert-butyl- 17-hydroxy-3 ,6-dioxo- 1 , 1 a,3 , 4,5,6,9,10,18, 19,20,21 ,22,22a-tetradecahydro-8H-7, 10-methanocyclopropa
[18,19][l,10,3,6]dioxadiazacyclononadecino[l l,12-b]quinoline-8-carboxylate (Example 113, Step 5) and l-Boc-4-hydroxypiperidine. LRMS (ES+) m/z 737.5 (M+H)+.
Step 2 : methyl (laR,5S,8S,10R,22aR)-5-tert-butyl-3,6-dioxo-17-(piperidin-4-yloxy)- 1, 1 a, 3, 4, 5, 6, 9, 10, 18, 19, 20, 21, 22, 22a-tetradecahydro-8H- 7, 10-methanocyclopropa
[18,19 ][1, 10,3,6 Jdioxadiazacyclononadecinofl 1,12-b Jquinoline-8-carboxylate trifluoroacetate

To a solution of the product from step 1 in dichloromethane (2 mL) was added trifluoroacetic acid (2 mL). The resulting solution was stirred for 1 hour at room temperature until disappearance of the starting material. The solvent was removed in vacuo. Purification of the residue by flash chromatography (ISCO reverse phase, 5 to 95 % acetonitrile in water (0.5% TFA buffer)) gave the desired product (170 mg). LRMS (ES+) m/z 637.5 (M+H)+.
Step 3 : methyl (laR,5S,8S,10R,22aR)-5-tert-butyl-17-{[l-(2-methoxyethyl)piperidin-4-yl]oxy}- 3, 6-dioxo-l,l , 3, 4, 5, 6, 9, 10,18,19, 20, 21, 22, 22a-tetradecahydro-8H- 7, 10-methano
cyclopropafl 8,19 ][1, 10,3,6 ]dioxadiazacyclononadecino[ 11,12-b Jquinol ine-8-carboxylate

The amine from Step 2 (70.9 mg) was dissolved in DMF (0.3 mL) and triethylamine (53 μΐ), potassium iodide (1.6 mg) and 2-bromoethyl methyl ether (20 μΐ) were added sequentially. The reaction mixture was stirred at 60°C for 24 hours. The reaction mixture was then cooled to room temperature and quenched with a saturated solution of sodium bicarbonate. The mixture was extracted (3x) with ethyl acetate and the combined organics were dried with sodium sulfate, filtered and concentrated. Purification of the residue by flash chromatography (ISCO, 0 to 10% methanol in DCM) gave the desired product (50.6 mg).
LRMS (ES+) m/z 695.5 (M+H)+.
Step 4 : (laR,5S,8S,10R,22aR)-5-tert-butyl-17-{[l-(2-methoxyethyl)piperidin-4-yl]oxy dioxo-1, la, 3, 4, 5, 6, 9,10,18,19, 20, 21, 22, 22a-tetradecahydro-8H- 7, 10-methanocyclo
propa[l 8, 19] [1 , 10,3,6]dioxadiazacyclononadecino[l 1 , 12-b]quinoline-8-carboxylic acid

The title compound was prepared using the same method as Example 96, Step 3. LRMS (ES+) m/z 681.5 (M+H)+.
Step 5 : (laR,5S,8S 0R R)-5-tert-butyl-N-[(lR,2S)-2-ethenyl-l-{[(l-methylcyclo propyl)sulfonyl]c rbamoyl}cyclopropyl]-17-{[l-(2-methoxyethyl)piperidin-4-ylJox
1, la,3,4,5,6,9, 10, 18, 19,20,21, 22,22a-tetradecahydro-8H-7, 10-methanocyclo
2-b]quinoline-8-carboxamide

The title compound was prepared using the same method as described in Example 1, Step 6 with intermediate A2. Purification of the residue by flash chromatography (ISCO reverse phase, 5 to 95% acetonitrile in water (0.5% TFA buffer)) gave the desired product. LRMS (ES+) m/z 907.45 (M+H)+.
Examples 226
By following the procedures outlined in Example 225 and using the appropriate reagent (depicted below the structure as Rg.), the following compounds were prepared.


ethyliodide

Example 232 : (1 aR,5S,8S, 10i?,22ai? -N-[(lig,2S)-2-ethenyl- !-{[(! -methylcyclopropyn sulfonyl] carbamoyl } cyclopropyl] -5-( 1 -methylcyclohexyD- 17- [(1 -methylpiperidin-4-yDoxy] - 3 ,6-dioxo- 1.1 a,3 ,4.5.6,9,10,18,19,20,21 ,22,22a-tetradecahydro-8H-7, 10-methanocyclopropa [18, 19][1, 10,3,6] dioxadiazacyclononadecino [ 11 , 12-b] quinoline-8-carboxamide

aminojacetic acid

A solution of intermediate D3 (4.3 g, 16.21 mmol) in acetohitrile (81 ml) was treated with intermediate Dl (3.37 g, 16.21 mmol) and triethylamine (9.04 ml, 64.8 mmol). Water (81 ml) was added to facilitate dissolution. The mixture was stirred at room temperature for 4 hours. The mixture was concentrated to half its volume and then diluted with ethyl acetate (500 mL), washed with aq 1M HCI (2 x 100 mL) and brine (100 mL). The organic layer was dried over magnesium sulfate, filtered and concentrated in the rotavap to give the title compound as a crude product (5.3 g, 16.49 mmol, 102 % yield) as a slightly yellow oil.
Step 2: (S)-2-((((lR, 2R)-2-(5-(4-(benzyloxy)-2-(((3R, 5S)-l-(tert-butoxycarbonyl)-5- (methoxycarbonyl)pyrrolidin-3-yl)oxy)quinolin-3-yl)pent-4-yn-l-yl)cycloprop
carbonyl)amino)-2-(l-methylcyclohexyl)acetic acid

A reaction vessel was charged with the product of step 1, (643 mg, 2 mmol), cesium carbonate (1629 mg, 5.00 mmol), bis(acetonitrile)dichloropalladium(II) (51.9 mg, 0.200 mmol) and tri-tert-butylphosphonium tetrafluoroborate (174 mg, 0.600 mmol). Acetonitrile (5000 μΐ) was added followed by dibenzylamine (387 μΐ, 2.000 mmol). The reaction mixture was purged with argon followed by addition of a solution of intermediate D2 (1115 mg, 2.000
mmol) in acetonitrile (5000 μΐ). The reaction mixture was purged with argon and the tube was sealed. The reaction was heated in an oil bath (80°C) overnight. Ethyl acetate was added (150 mL) and the mixture was washed with aq 1M HC1 (2 x 50 mL) and brine (50 mL), dried over magnesium sulfate, filtered and concentrated in rotavap to give the crude product. The crude product was purified on a gold cap Redisep (220 g) silica gel column (gradient: 0 to 30% solvent B in dichloromethane (solvent B: 20% methanol in ethyl acetate)) to give the title compound (840 mg, 1.053 mmol, 52.6 % yield) as a slighlty yellow foam.
Step 3: (S)-2-((((lR,2R)-2-(5-(4-(benzyloxy)-2-(((3R,5S)-5-(methoxycarbonyl) pyrrolidin-3-yl)

The N-Boc protected product from Step 2 (1.8 g, 2.256 mmol) was dissolved in dichloromethane (15.04 ml) and treated with TFA (7.52 ml). The mixture was stirred at room temp and monitored by LCMS. Reaction was completed after 45 min. The mixture was concentrated to dryness in rotavap. The residual TFA was azeotropically removed with toluene and the title compound (1.83 g, 2.254 mmol, 100 % yield) was dried under vacuum. No further purification was carried out.
Step 4: Methyl (laR,5S,8S,10R,22aR)-17-(benzyloxy)-5-(l-methylcyclohexyl)-3,6-dioxo-18,19- didehydro-l, la, 3, 4, 5, 6, 9, 10, 20, 21, 22, 22a-dodecahydro-8H- 7, 10-methanocyclopropa
[18, 19] [1, 10, 3,6]dioxadiazacyclononadecino[ll, 12-b]quinoline-8-carboxylate

A round-bottom flask was charged with HATU (1711 mg, 4.50 mmol) and dry DMF (2.25E+04 μΐ) at 0 °C. N,N-diisopropylethylamine (1531 μΐ, 9.00 mmol) was added followed by addition of a solution of the product of step 3 (1827 mg, 2.25 mmol) in dry DMF (2.25E+04 μΐ) via syringe pump over 1 hour. After addition was complete the reaction was stirred at room temperature for 2 hours. LCMS and MS analyses showed a complete reaction. The reaction mixture was concentrated to almost dryness in rotavap (high vacuum) and the residue was diluted with ethyl acetate (200 mL). The mixture was washed with water (50 mL), aq. 1M HC1 (50 mL), half-saturated sodium bicarbonate (50 mL) and brine (50 mL). The organic layer was dried over magnesium sulfate, filtered and concentrated in rotavap. The residue was purified on a Redisep (120 g) silica gel column (gradient: 0 to 40 % ethyl acetate in hexanes) to give the title compound (900 mg, 1.324 mmol, 58.8 % yield) as a white powder. Step 5: Methyl (laR,5S,8S,10R,22aR)-5-(l-methylcyclohexyl)-17-hydroxy-3,6-dioxo- 1, la, 3, 4, 5, 6, 9, 10,18,19, 20, 21, 22, 22a-tetradecahydro-8H- 7, 10-methanocyclopropa
[18, 19] [1, 10, 3, 6]dioxadiazacyclononadecino[ 11, 12-b ]quinoline-8-carboxylate

Step 6: Methyl (laR, 5S, 8S, 1 OR, 22aR)-5-(l-methylcyclohexyl)-l 7-[ (l-methylpiperidin-4-yl)oxy]- 3, 6-dioxo-l,la,3,4,5, 6,9, 10, 18, 19,20,21, 22, 22a-tetradecahydro-8H-7, 10-methano
cyclopropaf 18, 19] [1, 10, 3, 6]dioxadiazacyclononadecino[ 11, 12-b ]quinoline-8-carboxylate

A reaction tube was charged with the product of step 5 (400 mg, 0.674 mmol) and triphenylphosphine (1414 mg, 5.39 mmol). The tube was sealed and THF (6737 μΐ) was added via syringe at 0°C. 4-hydroxy-l-methylpiperidine (633 μΐ, 5.39 mmol) was added followed by slow addition of diisopropyl azodicarboxylate (1044 μΐ, 5.39 mmol). After 5 min, the cooling bath was removed and the mixture was allowed to reach room temperature and stirred for 10 minutes. The reaction tube was heated at 40°C. LCMS showed complete reaction after 2 hours. The reaction mixture was concentrated to dryness in rotavap and the residue was purified on a gold cap silica gel (120 g) column (gradient: 0 to 50% solvent B in dichloromethane (solvent B: 20%) methanol in ethyl acetate)) to give the title compound (450 mg, 0.651 mmol, 97 %> yield) as a white powder.
Step 7: (laR, 5S, 8S, 1 OR, 22aR)-5-(l-methylcyclohexyl)-l 7-[(l-methylpiperidin-4-yl)oxy]-3, 6- dioxo-1, la, 3, 4, 5, 6, 9,10,18,19, 20,21, 22, 22a-tetradecahydro-8H- 7, 10-methanocyclo
propaf 18,19 ][1, 10,3,6 Jdioxadiazacyclononadecinof 11, 12-b Jquinoline-8-carboxylic acid

A round-bottom flask was charged with the product of step 6 (450 mg, 0.651 mmol) and lithium hydroxide monohydrate (137 mg, 3.26 mmol). MeOH (2171 μΐ), THF (2171 μΐ) and water (2171 μΐ) were successively added and the mixture was stirred at room temperature LCMS showed complete reaction after 2 hours. The reaction was quenched by addition of acetic acid (373 μΐ, 6.51 mmol). Water was added (20 mL) and the mixture was extracted with dichloromethane (2 x 20 mL) and ethyl acetate (20 mL). The combined organic extracts were dried over magnesium sulfate, filtered and concentrated in rotavap. Residual acetic acid was azeotropically removed with toluene to give the title compound as a crude product (450 mg, 0.665 mmol, 102 % yield) as a white powder. No further purification was carried out.
Step 8: (laR,5S,8S,10R,22aR)-N-[(lR,2S)-2-ethenyl-l-{[(l-methylcyclopropyl)su^
carbamoyl) cyclopropyl] -5 -(1 -methylcyclohexyl)-! 7-[(l -methylpiperidin-4-yl)oxy]-3,6-dioxo- 1,1 a, 3, 4, 5,6,9,10,18, 19,20,21, 22, 22a-tetradecahydro-8H- 7, 10-methanocyclopropa[ 18,19]
[1,10, 3, 6]dioxadiazacyclononadecino[l 1, 12-b ]quinoline-8-carboxamide

A round-bottom flask was charged with the product of step 7 (1.1 g, 1.625 mmol), intermediate D4 (0.684 g, 2.438 mmol) and HATU (0.927 g, 2.438 mmol). Dry DMF (16.25 ml) was added by syringe at 0°C followed by N,N-diisopropylethylamine (1.106 ml, 6.50 mmol). The cooling bath was removed after 10 minutes and the reaction mixture was stirred overnight.
The mixture was concentrated to one third of its volume in rotavap and the residue was diluted with ethyl acetate (200 mL). The organic layer was washed with water (2 x 25 mL) and brine (25 mL), dried over magnesium sulfate, filtered and concentrated in rotavap. The residue was purified on a gold cap RediSep® (220 g) silica gel column (gradient: 0 to 50 % solvent B in dichloromethane (solvent B: 20% MeOH in ethyl acetate)) to give the title compound (810 mg, 0.897 mmol, 55.2 % yield) as a white powder. LRMS (ESI) Calcd for C48H67N609S [M + H]+ 903.46; found 902.8
Example 233 : (lai?,5S,8S.10i?.22ai? -5-(23-dihvdro-lH-inden-2-yl)-17-r3-(dimethylamino propoxy|-N-|Y 1 R.2S)-2-ethenyl- 1 - { \( 1 -methylcyclopropyl) sulfonyl] carbamoyl } cvclopropyl] - 3 ,6-dioxo- 1.1 a.3.4.5.6.9.10.18.19.20.21.22.22a-tetradecahvdro-8H-7.10-methano

Step l: (S)-2-(2,3-dihydro-lH-inden-2-yl)-2-((((lR,2R)-2-(pent-4- l)cyclopropoxy)carbonyl)amino)acetic acid


A reaction vessel was charged with the product of step 1 (1990 mg, 5.83 mmol), potassium carbonate (1831 mg, 13.25 mmol), bis(acetonitrile)dichloro palladium(II) (68.7 mg, 0.265 mmol), and tri-tert-butylphosphonium tetrafluoroborate (231 mg, 0.795 mmol).
Acetonitrile (1.77E+04 μΐ) was added followed by dibenzylamine (1017 μΐ, 5.30 mmol). The reaction mixture was purged with argon followed by addition of a solution of intermediate D2 (2954 mg, 5.3 mmol) in acetonitrile (8833 μΐ). The reaction mixture was purged with argon and the tube was sealed. The reaction was heated in an oil bath (80°C) overnight. Ethyl acetate was added (300 mL) and the mixture was washed with aq. 1M HCl (2 x 50 mL) and brine (50 mL), dried over magnesium sulfate, filtered and concentrated in rotavap. The crude product was purified on a gold cap ediSep® (220 g) silica gel column (gradient: 0 to 30% solvent B in dichloromethane (solvent B: 20% methanol in ethyl acetate)). The fractions containing the
product were combined and fractions containing impure product were combined, evaporated and purified again under the same conditions (120 g column) to give a second batch of product. The purified products were combined to give the title compound (950 mg, 22%) as a colorless foam. Step 3: (S)-2-((((lR, 2R)-2-(5-(4-(benzyloxy)-2-(((3R, 5S)-5-(methoxycarbonyl)pyrrolidin-3- yl)oxy)quinolin-3-yl)pent-4-yn-l-yl)cyclopropoxy)carbonyl)amino)-2
yl)acetic acid

A solution of the product of step 2 (750 mg, 0.917 mmol) in CH2C12 (4.58 ml) was treated with trifiuoroacetic acid (4.5 ml, 58.8 mmol) and stirred at room temperature.
LCMS showed complete reaction after 30 minutes. The mixture was concentrated to dryness in rotavap and residual TFA was azeotropically removed with toluene. The title compound as crude product (760 mg, 100%) was dried under vacuum and used without further purification. Step 4: Methyl (laR,5S,8S,10R,22aR)-17^enzyloxy)-5-(2,3-dihydro-lH^nden-2-yl)-3,6-dioxo- 18,19-didehydro-l, 1 a, 3, 4, 5,6,9,10,20,21, 22, 22a-dodecahydro-8H- 7, 1 -methano
cyclopropafl 8, 19] [ 1, 10, 3, 6]dioxadiazacyclononadecino[ 11, 12-b Jquinoline-8-carboxylate

A round-bottom flask was charged with HATU (695 mg, 1.828 mmol) and dry DMF (9140 μΐ) at 0°C. N,N-diisopropylethylamine (637 μΐ, 3.66 mmol) was added followed by addition of a solution of the product of step 3 (760 mg, 0.914 mmol) in dry DMF (9140 μΐ) via syringe pump over 1 hour. After addition was complete the reaction was stirred at room temperature for 1 hour. LCMS and MS analyses showed a complete reaction. The reaction mixture was concentrated to almost dryness in rotavap (high vacuum) and the residue was diluted with ethyl acetate (100 mL). The mixture was washed with water (40 mL), aq. 1M HCl (40 mL), half-saturated sodium bicarbonate (30 mL) and brine (30 mL). The organic layer was dried over magnesium sulfate, filtered and concentrated in rotavap. The residue was purified on a RediSep® (80 g) silica gel column (gradient: 0 to 50 % ethyl acetate in hexanes) to give the title compound (510 mg, 0.729 mmol, 80 % yield) as a white powder.
Step 5: Methyl (laR,5S,8S, 10R,22aR)-5-(2, 3-dihydro-lH-inden-2-yl)-17-hydroxy-3, 6-dioxo- 1, 1 a, 3, 4,5, 6,9, 10, 18, 19,20,21, 22, 22a-tetradecahydro-8H- 7, 1 -methanocyclopropa[l 8, 19]
[1, 10, 3, 6]dioxadiazacyclononadecino[l 1, 12-b Jquinoline-8-carboxylate

A solution of the product of step 4 (480 mg, 0.686 mmol) in MeOH (9146 μΐ) and THF (4573 μΐ) was treated with a catalytic amount of 10% palladium on carbon (36 mg). The mixture was hydrogenated at 20 psi. After 2 hours LCMS showed >70% conversion.
Hydrogenation was continued for another 2 h. LCMS showed >90% conversion. The mixture was diluted with ethyl acetate (50 mL) and the solids were removed by filtration. The filtrate was concentrated in rotavap and the residue was purified on a RediSep® (80 g) silica gel column (gradient: 0 to 50 % ethyl acetate in hexanes) to give the title compound (420 mg, 0.684 mmol, 100 % yield) as a white powder.
Step 6: Methyl (laR, 5S, 8S, 1 OR, 22aR)-5-(2, 3-dihydro-l H-inden-2-yl)-l 7-[ 3-(dimethylamino) propoxy]-3, 6-dioxo-l, la, 3, 4, 5, 6, 9, 10,18,19, 20, 21, 22, 22a-tetradecahydro-8H- 7, 10-methano cyclopropafl 8,19 ][1, 10,3,6] dioxadiazacyclononadecinof 11,12-b ]quinoline-8-carboxylate

A reaction tube was charged with the product of step 5 (200 mg, 0.326 mmol) and triphenylphosphine (855 mg, 3.26 mmol). The tube was sealed and THF (3259 μΐ) was added
via syringe at 0°C. 3-dimethylamino-l-propanol (381 μΐ, 3.26 mmol) was added followed by slow addition of diisopropyl azodicarboxylate (631 μΐ, 3.26 mmol). After 5 minutes the cooling bath was removed and the mixture was allowed to reach room temperature and stirred for 10 minutes. The reaction tube was heated at 40°C for 3 hours. The reaction mixture was concentrated to dryness in rotavap and the residue was purified on a gold cap silica gel (80 g) column (gradient: 0 to 60% solvent B in dichloromethane (solvent B: 20% methanol in ethyl acetate)) to give the title compound (210 mg, 0.300 mmol, 92 % yield) as a white powder. Step 7: (laR, 5S, 8S, 1 OR, 22aR)-5-(2, 3-dihydro-l H-inden-2-yl)-l 7-[3-(dimethylamino)propoxy]- 3, 6-dioxo-l, la, 3, 4, 5, 6, 9, 10,18,19, 20, 21, 22, 22a-tetradecahydro-8H- 7, 10-methano
boxylic acid

A solution of the product of step 6 (210 mg, 0.300 mmol) in MeOH (3005 μΐ), THF (1502 μΐ) and eater (1502 μΐ) was treated with lithium hydroxide monohydrate (63.0 mg, 1.502 mmol). The reaction mixture was stirred at room temperature. After 5 hour the reaction was quenched by addition of acetic acid (172 μΐ, 3.00 mmol) and water (10 mL). The mixture was extracted into dichloromethane (2 x 10 mL) and ethyl acetate (10 mL). The combined organic extracts were dried over magnesium sulfate filtered and concentrated in rotavap to give the title compound (215 mg, 0.314 mmol, 104 % yield) as a white powder. No further purification was carried out for the product.
Step 8: (laR, 5S, 8S, 1 OR, 22aR)-5-(2, 3-dihydro-l H-inden-2-yl)-l 7-[3-(dimethylamino)propoxy]- N-[ (lR,2S)-2-ethenyl-l-{[ (l-methylcyclopropyl)sulfonyl] carbamoyl} cyclopropyl] -3, 6-dioxo- 1, la, 3, 4, 5, 6, 9, 10,18,19, 20, 21, 22, 22a-tetradecahydro-8H-7, 10-methanocyclopropa
[18,19] [1, 10,3, 6]dioxadiazacyclononadecino[ll,12-b]quinoline-8-carboxamide

A round-bottom flask was charged with the product of step 7 (105 mg, 0.153 mmol), intermediate D4 (53.8 mg, 0.192 mmol) and HATU (72.9 mg, 0.192 mmol). Dry DMF (3066 μΐ) was added by syringe followed by 4-methylmorpholine (67.4 μΐ, 0.613 mmol). The reaction mixture was stirred for 22 hours. Ethyl acetate was added (20 mL) and the mixture was washed with water (2 x mL), brine (5 mL), dried over magnesium sulfate, filtered and concentrated in rotavap. The residue was purified on a gold cap RediSep® (12 g) silica gel column (gradient: 0 to 60 % solvent B in dichloromethane (solvent B: 20% MeOH in ethyl acetate)) to give the title compound (52 mg, 0.057 mmol, 37.2 % yield) as a white powder. LRMS (ESI) Calculated for C49H63N609S [M + H]+ 911.4; found 911.2
Example 234 : ( 1 aR,5S,8S.10R.22aR)-5-tert-butyl-N-r( 1 R.2S)-2-ethenyl- 1 - ( Γ( 1 -methyl cyclopropyl sulfonyl]carbamov cyclopropyll-3 ,6-dioxo- 17- ( [ 1 -(2.2.2-trifluoroethyl)piperidin- 4-ylloxy } - 1 , 1 a ,3 Λ5.6,9.10.18.19.20.21.22.22a-tetradecahvdro-8H-7.10-methano
cyclopropa|T 8.19] 1.10.3.61dioxadiazacyclononadecinor 11 , 12-b]quinoline-8-carboxamide

Step 1: methyl (laR,5S,8S,10R,22aR)-5-tert-butyl-17-{[l-(2,2,2-trifluoroethyl)piperidin-4- yl Joxy}-3, 6-dioxo-l, la, 3, 4, 5, 6, 9,10,18,19, 20,21, 22, 22a-tetradecahydro-8H- 7, 10-methano cyclopropa [18, 19] [1,10,3, 6]dioxadiazacyclononadecino[ll, 12-b]quinoline-8-carboxylate

To a solution of the product from Example 225 Step 2 (90 mg, 0.141 mmol) in 2 mL acetonitrile were added cesium carbonate (92 mg, 0.283 mmol) followed by 2,2,2- trifluoroethyl trifluoromethanesulfonate (65.6 mg, 0.283 mmol). The reaction was stirred at room temperature for 2 hours at which stage it was judged to be complete by LCMS. The reaction mixture was filtered through celite, washed with EtOAc, concentrated and purified by PTLC (40% EtOAc/Hexane) to give the title compound (75 mg, 0.104 mmol, 73.8 % yield). Step 2: (laR, 5S, 8S, 1 OR, 22aR)-5-tert-butyl-N-[ (1R, 2S)-2-ethenyl-l-{[ (1 -methyl
cyclopropyl)sulfonyl]carbamoyl}cyclopropyl]-3, 6-dioxo-l 7-{[l-(2, 2, 2-trifluoroethyl)piperidin-4- yl]oxy}-l, 1 a, 3, 4, 5, 6, 9,10,18, 19,20,21, 22, 22a-tetradecahydro-8H- 7, 10-methanocyclopropa
[18, 19] [1,10, 3, 6]dioxadiazacyclononadecino[l 1, 12-b]quinoline-8-carboxamide
The product from Step 1 was converted to Example 234 using the previously described procedures for the synthesis of Example 225. The title compound was purified by PTLC (3% MeOH in CH2C12). LRMS m/z 931.4 (M+H)+
Example 235: tert-butyl 4-{rriaR,5S,8S,10R.22aRV5-cvclohexyl-8-{r(TR.2S)-2-ethenyl-l-irri- methylcvclopropyl)sulfonyl1carbamoyl}cvclopropyllcarbamoyll-3,6-dioxo-l,la,3,4,5,6,9,10, 18.19,20.21 ,22.22a-tetradecahvdro-8H-7, 10-methanocvclopropar 18,191 [ 1 , 10 ,3 ,61
dioxadiazacyclononadecino[ 11 , 12-blquinolin-l 7-yl] oxylpiperidine-1 -carboxylate

Example 235 was prepared by utilizing the procedures described for the synthesis of Example 211. Tert-butyl 4-hydroxypiperidine-l -carboxylate was used instead of 1 - methylpiperidine-4-ol for the Mitsunobu reaction. LRMS m/z 975.4 (M+H)+
Example 236: 4-(rflaR.5S,8Sa0R.22aRV5-cvclohexyl-8-(r(lR.2SV2-ethenyl-l-{r(l- methylcvclopropyl)sulfonyll carbamoyl 1 cyclopropyll carbamoyl} -3,6-dioxo- 1 , 1 a,3,4,5,6,9, 10, 18,19,20,21 ,22,22a-tetradecahydro-8H-7, 10-methanocyclopropa[ 18, 19"] [ 1 , 10,3 ,61dioxadiaza cyclononadecino [11,12-b]quinolin- 17-yl] oxyj - 1 -(2,2,2-trifluoroethvDpiperidinium formate

Step 1 : 4-{[ (laR, 5S, 8S, 1 OR, 22aR)-5-cyclohexyl-8-{[(lR, 2S)-2-ethenyl-l-{[(l -methylcyclopropyl) sulfonyl ]carbamoyl}cyclopropyl]carbamoyl}-3, 6-dioxo-l, la, 3, 4, 5, 6, 9, 10,18,19, 20, 21, 22, 22a- tetradecahydroSH- 7, 10-methanocyclopropaf 18,19][1,10,3, 6Jdioxadiazacyclononadecino
[11, 12-bJquinolin-l 7-yl]oxy}-piperidinium trifluoroacetate

To a solution of Example 235 (100 mg, 0.103 mmol) in dichloromethane (5 ml) was added trifluoroacetic acid (1.025 ml, 0.103 mmol). The reaction was stirred at room temperature for 2 hours at which stage LCMS indidcated complete hydrolysis of the NBoc group. The volatiles were evaporated under reduced pressure and the residue was diluted with DCM and azeotroped twice with toluene. The resulting residue (-101 mg) was dried under vacuum and used for the next step without purification.
Step 2: 4-{[(laR, 5S, 8S, 1 OR, 22aR)-5-cyclohexyl-8-{[ (1R, 2S)-2-ethenyl-l-{[ (1-methylcyclopropyl) sulfonyl]carbamoyl}cyclopropyl]carbamoyl}-3, 6-dioxo-l, la, 3, 4, 5, 6, 9,10,18, 19, 20,21, 22, 22a- tetradecahydro-8H- 7, 10-methanocyclopropaf 18,19] [1,10,3,6 ] dioxadiazacyclononadecino
[11, 12-bJquinolin-l 7-yl]oxy}-l-(2,2,2-trifluoroethyl)piperidinium formate
To a solution of the product from Step 1 (101 mg, 0.102 mmol) in acetonitrile (5 mL) was added Hunig's base (0.089 mL, 0.511 mmol) followed by 2,2,2 -trifluoroethyl trifluoromethanesulfonate (71.1 mg, 0.306 mmol). The reaction was stirred at 55°C for 3 hours after which it was quenched with water and extracted with EtOAc. Purification by PTLC (3%
MeOH in CH2C12) followed by reverse phase HPLC (0 to 90% acetonitrile in water; with 0.1%
HCOOH) provided the title compound.
Example 237: 4- { ΓΓ1 aR,5S,8S, 10R,22aR -5-cyclohexyl-8-{ r(lR,2SV2-ethenyl-l -{ ΓΠ -methyl cyclopropyl)sulfonyllcarbamoyl}cyclopropyllcarbamoyl}-3,6-dioxo-lJa,3,4,5,6,9J0,18, 19,20,21 , 22,22a-tetradecahydro-8H-7, 10-methanocvclopropa[ 18 Λ 9} \ 1 0,3,6]dioxadiaza cyclononadecino[ 11 , 12-blquinolin- 17-yl]oxy } - 1 -(2-methoxyethyl piperidinium formate

To a solution of the product from Step 1, Example 236 (100 mg, 0.101 mmol) in DMF (3 mL) was added triethylamine (0.070 mL, 0.506 mmol), potassium iodide (3.36 mg, 0.020 mmol) and 2-bromoethylmethylether (42.2 mg, 0.303 mmol). The reaction was heated to 55°C and stirred for 4 hours at which stage LCMS indicated no starting material. After cooling to room temperature, water was added and the reaction was extracted with ethyl acetate. The combined organic fractions were washed with brine, dried (Na2S04) arid concentrated to give an oily residue, which was purified by HPLC (0 to 90% acetonitrile in water; with 0.1 % HCOOH) to furnish the title compound.
Example 238: 4-i r(laR.5S.8S.10R.22aRV5-cvclohexyl-8-( r(lR.2SV2-ethenyl-l-{ r(l-methyl cyclopropyDsulfonyl"]carbamovU cyclopropyll carbamoyl} -3,6-dioxo- 1 , 1 a,3 ,4,5,6,9, 10, 18, 19, 20,21 ,22,22a-tetradecahydro-8H-7, 10-methanocyclopropa[ 18,191 IT, 10,3,61dioxadiaza

Example 239: 4-U(laR.5S.8S.10R.22aRV5-cvclohexyl-8-U(lR.2SV2-ethenyl-l-(r(l- methylcyclopropyDsulfonyllcarbamoyl } cyclopropyllcarbamoyl } -3.6-dioxo- 1,1 a.3 ,4,5 ,6.9, 10. 18,19,20,21 ,22.22a-tetradecahvdro-8H-7.1 O-methanocvclopropar 18.191 Γ 1.10,3,61
dioxadiazacyclononadecino[l 1 , 12-blquinolin- 17-ylloxy} - 1 -ethylpiperidinium formate

Example 23 was prepared by utilizing procedures described for the synthesis of Example 237. lodoethane was used instead of l-bromo-2-methoxyethane and potassium iodide was not utilized in the last step to furnish the title compound.
Example 240: 4-(3-{raaR,5S,8S.10R.22aRV5-cyclohexyl-8-{rriR.2SV2-ethenyl-l-(rri- methylcvclopropyDsulfonyll carbamoyl } cyclopropyll carbamoyl) -3 ,6-dioxo- 1 , 1 a,3 ,4,5.6,9. 10,18.19.20.21.22.22a-tetradecahvdro-8H-7.10-methanocvclopropar 18.191Π ,10.3.61dioxadiaza cyclononadecino [11.12-bl quinolin- 17-yll oxy} propyl)morpholin-4-ium formate

19,20,21, 22, 22a-tetradecahydro-8H- 7, 10-methanocyclopropa[ 18,19] [1,10,3, 6 Jdioxadiaza cyclononadecinof 11, 12-b ]quinoline-8-carboxamide

The title compound in step 1 was prepared by utilizing procedures similar to that described for the synthesis of the intermediate obtained in Example 145, Step 1.
Step 2: 4-(3-{[(laR,5S,8S,10R,22aR)-5-cyclohexyl-8-{[(lR,2S)-2-ethenyl-l-{[(l-methyl cyclopropyl)sulfonyl]carbamoyl}cyclopropyl]carbamoyl}-3, 6-dioxo-l, la, 3, 4, 5, 6, 9, 10, 18, 19, 20,21, 22, 22a-tetradecahydro-8H- 7, 10-methanocyclopropa[ 18,19] [1,10, 3, 6]dioxadiaza cyclononadecino[l 1 , 12-b]quinolin-l 7-yl]oxy}propyl)morpholin-4-ium formate
A solution of the intermediate from Step 1 (60 mg, 0.066 mmol), morpholine (28.6 mg, 0.329 mmol), triethylamine (0.046 mL, 0.329 mmol) and potassium iodide (21.82 mg, 0.131 mmol) in DMF (2 mL) was stirred at 55 °C for 4 hours at which stage LCMS indicated complete conversion to the desired product. After cooling the reaction to room temperature, water was added the reaction was extracted with ethyl acetate. The combined organic fractions were dried over Na2S04 and concentrated under reduced pressure. Purification by HPLC (0 to 90% acetonitrile in water; with 0.1% HCOOH) provided the title compound. LRMS m/z 919.4 (M+H)+.
Example 241 : N-(3-{ r(laR,5S,8S,10R,22aRV5-cvclohexyl-8-{ r(lR.2SV2-ethenyl-l-i Γ(1- memylcyclopropyl)sulfonyllcarbamoyl>cvclopropyl1carbarnoyl}-3,6-dioxo-
1.1 a.3.4.5.6.9.10.18.19.20.21 ,22,22a-tetradecahydro-8H-7.10-
methanocyclopropa[ 18, 19] [ 1 , 10.3 ,61dioxadiazacvclononadecino [11,12-blquinolin- 17- "|oxy|propyDcvclopropanaminium formate

A solution of the intermediate from Example 240, Step 1 (85 mg, 0.093 mmol) and cyclopropyl amine (80 mg, 1.401 mmol) in DMF (2 mL) was stirred at 50°C for 3 hours; LCMS indicated complete conversion. After cooling the reaction to room temperature, water was added the reaction was extracted with ethyl acetate. The combined organic fractions were dried over Na2S04 and concentrated under reduced pressure. Purification by HPLC (0 to 90% acetonitrile in water; with 0.1% HCOOH) furnished the title compound. LRMS m/z 889.4 (M+H)+.
Example 242: N-(3-ir(laR.5S,8SJ0R,22aRV5-cvclohexyl-8-(r(lR.2SV2-ethenyl-l-{r(l- methylcvclopropyl)sulfonyl]carbamoyl| cvclopropyl]carbamoyl) -3 ,6-dioxo- 1.1 a.3.4.5,6,9, 10.18.19.20.21 ,22.22a-tetradecahvdro-8H-7, 10-methano
cyclopropaf 18,191 [1,10,3,6] dioxadiazacyclononadecino [ 11 , 12-b] quinolin- 17-

Example 243: riaR.5S.8S.10R.22aRV5-cvclohexyl-17-{3-r(,3.3-difluorocvclobutvnaminol propoxy}-N- (lR,2S)-2-ethenyl-l-{ (l-methylcyclopropyl sulfonyllcarbamoyl|cyclopropyll- 3.6-dioxo- 1.1 a.3.4.5.6.9.10.18.19.20.21.22.22a-tetradecahydro-8H-7.10-methanocvclopropa
, 12-b] quinoline-8-carboxamide

A solution of the intermediate from Example 240, Step 1 (50 mg, 0.055 mmol), 3,3-difluorocyclobutanamine (46.9 mg, 0.438 mmol), triethylamine (0.076 mL, 0.548 mmol) and potassium iodide (91 mg, 0.548 mmol) was stirred at 55 °C for 4h at which stage LCMS indicated complete conversion to the desired product. After cooling the reaction to room temperature, water was added the reaction was extracted with ethyl acetate. The combined organic fractions were dried over Na2S04 and concentrated under reduced pressure. Purification by PTLC (5% MeOH in CH2C12) furnished the title compound. LRMS m/z 939.4 (M+H)+.
Example 244 : (laR.5S.8S.10R.22aR>5-tert-butyl-N-ri 1 R.2S >2-ethenyl- 1 - { Γ(1 - methylcvclopropyl)sulfonyl1carbamoyl}cvclopropyl1-3,6-dioxo-17-(pyridin-4-yl)- 1.1 a.3.4.5.6.9.10.18.19.20.21.22.22a-tetradecahydro-8H-7.10-methano
cyclopropaf 18.19] [ 1.10.3.61dioxadiazacvclononadecino [11.12-b]quinoline-8-carboxamide

Step 1: methyl (laR,5S,8S,10R,22aR)-5-tert-butyl-17-{[(trifluoromethyl)sulfonyl]oxy}-3,6-dioxo- 1, la, 3, 4, 5, 6, 9, 10,18,19, 20, 21, 22, 22a-tetradecahydro-8H-7, 10-methanocyclopropa
cyclononadecino[l 1, 12-b ]quinoline-8-carboxylate

To a solution of the intermediate from Example 113, step 5 (200 mg, 0.361 mmol) in pyridine (2 ml) was added triflic anhydride (0.305 ml, 1.806 mmol) at 0°C. The reaction was stirred at 0°C for 15 minutes and then at room temperature for 15 minutes after which water was added and an extraction was performed with EtOAc. The combined organics were washed with saturated aq. NH4C1 and then with 10% aq. KHS04 solution followed by brine. Purification by PTLC (30% EtOAc/hexanes) provided the title compound.
Step 2: methyl (laR,5S,8S,10R,22aR)-5-tert-butyl-17-(pyridin-4-yl)-3,6-dioxo- 1, la, 3, 4, 5, 6, 9, 10,18,19, 20, 21, 22, 22a-tetradecahydro-8H- 7, 10-methanocyclopropa
[18, 19] [1,10, 3, 6]dioxadiazacyclononadecino[l 1 , 12-b]quinoline-8-carboxylate

To a solution of the product from step 1 (50 mg, 0.073 mmol) in dioxane (5 ml) was added pyridine-4-boronic acid, (35.9 mg, 0.292 mmol), potassium phosphate tribasic (38.7 mg, 0.182 mmol) and tetrakis (16.85 mg, 0.015 mmol). After purging with N2 for 5 min, the reaction was stirred at 80°C for 16 hours. After cooling to room temperature, water was added and the reaction was extracted with ethyl acetate, washed with brine, dried (Na2S04) and concentrated. Purification by PTLC (40% EtOAc hexane) provided the desired compound. Step 3: (laR, 5S, 8S, 1 OR, 22aR)-5-tert-butyl-N-[ (1R, 2S)-2-ethenyl-l-{[ (1 -methyl
cyclopropyl)sulfonyl]carbamoyl}cyclopropyl]-3, 6-dioxo-l 7 -(pyridin-4-yl)-l , la, 3, 4, 5, 6, 9, 10,18,19, 20, 21, 22, 22a-tetradecahydro-8H- 7, 10-methanocyclopropa[18, 19 ][1, 10,3,6]
noline-8-carboxamide

The product from step 2 was converted to Example 244 using the procedures described for the synthesis of Example 113, steps 7 and 8. LRMS m/z 827.2 (M+H)+.
Example 245 : ( 1 aR,5 S,8S, 10R.22aRV5-tert-butyl-N-r(l R.2SV2-ethenyl- 1 - { Γ(Τ - methylcyclopropyDsulfonyl] carbamoyl} cyclopropyl] -3 ,6-dioxo- 17-(pyridin-2-yl - 1,1 a,3 , 4,5,6,9,10,18, 19,20,21 ,22,22a-tetradecahydro-8H-7, 10-methanocyclopropa
[ 18, 19] [ 1 , 10,3 ,6] dioxadiazacvclononadecino Γ 11 , 12-blquinoline-8-carboxamide

To a solution of the product from Example 244 step 1 (50 mg, 0.073 mmol) in dioxane (3 ml) was added 2-(tributylstannyl)pyridine (53.7 mg, 0.146 mmol), copper(I)iodide (2.78 mg, 0.015 mmol) and tetrakis (16.85 mg, 0.015 mmol). After purging with N2 for 5 minutes, the reaction was stirred at 80°C for 16 hours. After cooling to room temperature, water was added and the reaction was extracted with ethyl acetate, washed with brine, dried (Na2S04) and concentrated. Purification by PTLC (40% EtOAc/hexane) provided the desired compound, which was converted to Example 245 using the procedures described for the synthesis of Example 244. LRMS m/z 827.2 (M+H)+.
Example 246: (1 aR,5S.8S, 10R.22aRV 5-tert-butyl-N-rq R.2S -2-ethenyl- 1 - { Γ( 1 - methylcvclopropyl)sulfonyl]carbamoyl|cvclopropyl]-3,6-dioxo-17-(pyridin-3-yl)- 1.1 a.3.4.5.6.9.10.18.19.20.21.22.22a-tetradecahvdro-8H-7.10-methanocvclo
propa|" 18.19] [ 1.10.3 ,6 dioxadiazacyclononadecino[ 11.12-b]quinoline-8-carboxamide

Example 246 was prepared using the procedures described for the synthesis of Example 244. Pyridin-3-ylboronic acid was used instead of pyridin-4-ylboronic acid in the coupling step. LRMS m/z 827.2 (M+H)+.
Example 247 : Π aR.5S.8S.10R.22aRV5-tert-butyl-N-r( 1 R.2S V2-ethenyl- 1 - { \(\ - methylcyclopropyDsulfonyl] carbamoyl } cyclopropyl] -3.6-dioxo- 17-( 1 ,3 -thiazol-2-ylV
1.1 a.3.4.5.6.9.10.18.19.20.21.22.22a-tetradecahydro-8H-7.1 O-methanocvclopropa
[18, 19] [ 1.10.3.61dioxadiazacyclononadecino[ 11.12-blquinoline-8-carboxamide

Example 247 was prepared using the procedures described for the synthesis of Example 245. 2-(Tributylstannyl)thiazole was used instead of 2-(tributylstannyl)pyridine. LRMS m/z 833.2 (M+H)+ Example 248: potassium (r(lR.2S -l-riraaR.5S.8S.10R.22aRV5-tert-butyl-17-(morpholin-4- ylmethvn-3.6-dioxo- 1.1 a.3.4.5.6.9.10.18.19.20.21.22.22a-tetradecahydro-8H-7.10- methanocyclopropafl 8, 19] [ 1 , 10,3,6]dioxadiazacyclononadecino 11 , 12-b]quinolin-8- yl] carbonyl } amino -2-ethenylcyclopropyl1 carbonyl } [( 1 -methylcyclopropyDsulfonyl] azanide

Step 1 : methyl (laR, 5S, 8S, 1 OR, 22aR)-5-tert-butyl-l 7-vinyl-3, 6-dioxo-
1, la, 3, 4, 5,6,9,10,18,19,20,21, 22, 22a-tetradecahydro-8H- 7, 10-methanocyclopropa
[18,19] [1, 10, 3, 6]dioxadiazacyclononadecino[ll,12-b]quinoline-8-carboxylate

To a solution of the product from Example 244 step 1 (100 mg, 0.146 mmol) in dioxane (3 ml) was added tributyl(vinyl)stannane (92 mg, 0.292 mmol), copper (I) iodide (5.55 mg, 0.029 mmol) and tetrakis (33.7 mg, 0.029 mmol). After purging with N for 5 min, the reaction was stirred at 80°C for 16 hours. After cooling to room temperature, water was added and the reaction was extracted with ethyl acetate, washed with brine, dried (Na2S04) and concentrated. Purification by PTLC (30% EtOAc/hexane) provided the desired compound. Step 2: methyl (laR,5S,8S,10R,22aR)-5-tert-butyl-17-formyl-3,6-dioxo- 1, la, 3, 4, 5, 6, 9, 10,18,19, 20, 21, 22, 22a-tetradecahydro-8H- 7, 10-methanocyclopropa
cyclononadecino[l 1, 12-b]quinoline-8-carboxylate

To a solution of the product from Step 1 (30 mg, 0.053 mmol) in acetone (1 ml) and water (ImL) was added potassium osmate dehydrate (19.61 mg, 0.053 mmol). The reaction was stirred at room temperature for 10 minutes after which sodium periodate (114 mg, 0.532 mmol) was added. After stirring for 2 hours, an additional ~15 mg of potassium osmate and 115 mg of sodium periodate were added. After stirring 16 hours, the reaction was filtered and the solid was washed with acetone. The filtrate was concentrated, diluted with EtOAc and washed with Na2S203 and NaHC03 aq. soln. Purification by PTLC (30% EtOAc-hexane) provided the desired aldehyde.
Step 3: methyl (laR,5S,8S,10R,22aR)-5-tert-butyl-17-(morpholinomethyl)-3,6-dioxo- 1,1 a, 3, 4, 5, 6, 9,10,18,19, 20, 21, 22, 22a-tetradecahydro-8H- 7, 10-methanocyclopropa
clononadecino[ll, 12-b]quinoline-8-carboxylate

To a solution of the product from Step 2 (34 mg, 0.060 mmol) in CH2C12 (3 ml) was added morpholine (0.016 ml, 0.180 mmol) and acetic acid (10.32 μΐ, 0.180 mmol) followed by sodium triacetoxyborohydride (38.2 mg, 0.180 mmol). The reaction was complete in 30 minutes at which stage it was quenched with water and extracted with dichloromethane.
Purification by PTLC (40% EtOAc/hexane) provided the desired compound.
Step 4: potassium {[ (1R, 2S)-l-({[ (laR, 5S, 8S, 1 OR, 22aR)-5-tert-butyl-l 7-(morpholin-4-ylmethyl)- 3, 6-dioxo-l,la, 3, 4, 5, 6, 9, 10,18,19, 20, 21, 22, 22a-tetradecahydro-8H- 7, 10- methanocyclopropa[18, 19] [1, 10, 3, 6]dioxadiazacyclononadecino[l 1, 12-b ]quinolin-8- onyl}[(l-methylcyclopropyl)sulfony

Steps 7 and 8 described for the synthesis of Example 113 were performed on the product from step 3 and the resulting compound was converted to Example 248 after treatment with 1 equivalent of 0.1N aqueous KOH solution. LRMS m/z 849.2 (M+H)+.
Example 249: daR.5S,8S.10R.22aRV5-cvclohexyl-17-r3-(,3,3-difluoroazetidin-l-vnpropoxyl- N- [d R.2 S)-2 -ethenyl- 1 - { [Y 1 -methylcyclopropyDsulfonyl] carbamoyl I cvclopropyl] -3.6-dioxo- 1, 1 a.3 ,4,5,6.9.10.18.19.20.21.22.22a-tetradecahvdro-8H-7.10-methanocvclopropa
18.191 1.10.3.61dioxadiazacyclononadecino l l,12-b1quinoline-8-carboxamide

Example 249 was prepared using the procedures described for the synthesis of Example 243. 3,3-Difluoroazetidine hydrochloride was used instead of 3,3- difluorocyclobutanamine in the final alkylation step to provide the title compound. LRMS m/z 925.6 (M+H)+
Example 250: aaR.5S.8S.10R.22aRV5-cvclohexyl-N-r(lR.2SV2-ethenyl-l-(ra -methyl cyclopropyDsulfonyl] carbamoyl } cvclopropyl] - 17- [3 -(3 -fluoroazetidin- 1 - vDpropoxy] -3.6-dioxo- 1.1 a.3.4.5.6.9.10.18.19.20.21 ,22,22a-tetradecahvdro-8H-7.10-methanocvclopropa
18, 19] [ 1 , 10,3 ,6]dioxadiazacyclononadecino Γ 11 , 12-b] quinoline-8-carboxamide

Example 251 : (laR,5S,8S J0R,22aRV5-cvclohexyl-N-r(lR,2SV2-ethenyl-l-{rn -methyl cyclopropyDsulfonyl] carbamoyl } cyclopropyl] - 17- [3 -(4-methylpiperazin- 1 - yDpropoxy] -3,6- dioxo- 1,1 a,3 ,4,5,6,9, 10, 18, 19,20,21 ,22,22a-tetradecahydro-8H-7, 10-methanocyclo
propafl 8, 19] [1 , 10,3,6]dioxadiazacyclononadecino[l 1 , 12-blquinoline-8-carboxamide

Example 251 was prepared using the procedures described for the synthesis of Example 243. N-methylpiperazine was used instead of 3,3-difluorocyclobutanamine in the final alkylation step to provide the title compound. LRMS m/z 932.6 (M+H)+
Example 252: potassium {[qR.2SV2-ethenyl-l-((r(laR,5S.8S.10R.22aRV17-r(l-methyl piperidin-4-vnoxyl -3.6-dioxo-5-(tetrahydro-2H-pyran-4-vn- 1.1 a.3 ,4.5.6.9.10.18.19.20,21.22. 22a-tetradecahydro-8H-7, 10-methanocyclopropan 8.19] [1.10.3.61dioxadiazacyclo
nonadecino Γ 11.12-b] quinolin- 8 - yll carbonyl } amino'jcvclopropyl] carbonyl } [( 1 - methylcvclopropyPsulfonyllazanide

Step 1: (S)-methyl 2-((((lR,2R)-2^ent-4-yn-l-yl)cyclopropoxy)carbonyl)amino)-2-(tetrahydro- -pyran-4-yl)acetate

The title compound was prepared using the procedures described for Intermediate
Bl 1, Step 2. (S)-methyl 2-amino-2-(tetrahydro-2H-pyran-4-yl)acetate was reacted with the product obtained after Step 1 in the preparation of Intermediate Bl 1.
Step 2: (S)-2-(( ((1R, 2R)-2-(pent-4-yn-l-yl)cyclopropoxy)carbonyl)amino)-2-(tetrahydro-2H- pyran-4-yl)acetic acid

To a solution of the product from Step 1 (1.88 g, 5.81 mmol) in THF (20 ml) and
MeOH (10 mL) was added an aq. 2M solution of lithium hydroxide monohydrate (14.53 ml, 29.1 mmol). The reaction was stirred at 50°C for 20 hours at which stage TLC indicated no more starting material. The reaction was treated with 10% aq. KHS04 and extracted with EtOAc. The organic fractions were washed with brine, dried over sodium sulfate, evaporated and dried to provide the desired compound, which was used for the next step without purification.
Step 3: potassium {[(1R, 2S)-2-ethenyl-l-({[ (laR, 5S, 8S, 1 OR, 22aR)-l 7-[ (l-methylpiperidin-4- yl)oxy]-3, 6-dioxo-5-(tetrahydro-2H-pyran-4-yl)-l, la, 3, 4, 5, 6, 9, 10,18,19, 20, 21, 22, 22a- tetradecahydroSH- 7, 10-methanocyclopropa[ 18, 19] [1,10,3, 6Jdioxadiazacyclo
nonadecino[ll,12-b]quinolin-8-yl]carbonyl}amino)cyclopropyl]carbonyl}[(l-

The product from Step 2 was converted to Example 252 using the procedures described for the synthesis of Example 210; the potassium salt was prepared upon treatment of the parent compound with 1 equivalent of 0.1N aqueous KOH solution. LRMS m/z 891.2
(M+H)+
Example 253: (1 aR,5S,8S, 10R,22aRV5-cvclohexyl-N-r(l R.2S V2-ethenyl- 1 - { Γ(1 -methyl cyclopropyDsulfonyllcarbamoyl|cyclopropyll-3,6-dioxo-17-('tetrahvdro-2H-pyran-4-yloxy)- 1 , 1 a,3 ,4,5 ,6,9, 10, 18, 19,20,21 ,22,22a-tetradecahydro-8H-7, 10-methanocyclopropa
[ 18 J 9] [ 1 , 10,3 ,61dioxadiazacyclononadecino [11,12-blquinoline-8-carboxamide

Example 254: tert-butyl 9-(r(laR,5S.8SJ0 ,22aRV5-tert-butyl-8-l|'flR.2SV2-etfaenyl-l-ir(l- memylcyclopropyl)sulfonyl]carbamoyl}cyclopropyl carbamoy -3,6-dioxo-
1 , 1 a,3 ,4,5 ,6.9.10.18.19.20,21.22,22a-tetradecahvdro-8H-7, 10- methanocyclopropa 18, 19] (T , 10,3 ,6]dioxadiazacyclononadecino[ 11 , 12-b]quinolin- 17-yl]oxy} -3- oxa-7-azabicyclo 3.3.1 lnonane-7-carboxylate

Step 1: methyl (1 aR,5S,8S, 10R,22aR)-5-tert-butyl-l 7-{syn-[7-(tert-butoxycarbonyl)-S-oxa-7- azabicyclof 3.3.1 ]non-9-yl]-oxy}-3, 6-dioxo-l, la, 3, 4, 5, 6, 9, 10,18,19, 20, 21, 22, 22a-tetradecahydro- 8H- 7, 10-methanocyclopropa [18, 19] [1, 10, 3, 6]dioxadiazacyclononadecino[l 1, 12-b ]quinoline-8- carboxylate

The title compound was prepared using the procedure described for the synthesis of Example 210, Step 1. (lR,5S)-tert-butyl 9-hydroxy-3-oxa-7-azabicyclo[3.3.1]nonane-7- carboxylate (1:2 mixture of anti:syn alcohols; prepared as described in International Patent Publication No. WO2009055331) was used instead of l-methylpiperidin-4-ol. The title
compound (syn isomer) was separated from the anti isomer by column chromatography (0-40% EtOAc in hexane); the syn isomer eluted first and was collected as the minor isomer.
Step 2: tert-butyl 9-{[(laR,5S,8S,10R,22aR)-5-tert-butyl-8-{[(lR,2S)-2-ethenyl-l-{[(l- methylcyclopropyl)sulfonyl]carbamoyl}cyclopropyl]carbamoyl}-3,6-dioxo- 1,1a, 3, 4, 5, 6, 9, 10,18,19, 20, 21, 22, 22a-tetradecahydro-8H- 7, 10- methanocyclopropa[18, 19] [1,10,3, 6]dioxadiazacyclononadecino[ll, 12-b]quinolin-l 7-yl]oxy}- -oxa- 7-azabicyclo[3.3.1 Jnonane- 7-carboxylate

Example 254 was prepared from the product of Step 1 by utilizing the procedi described for the synthesis of Example 210. LRMS m/z 991.2 (M+H)+
Example 255: tert-butyl 9-{raaR,5S,8S,10R,22aRV5-tert-butyl-8-{rnR,2SV2-ethenyl-l-{r(l- methylcvclopropyl sulfonyl]carbamoyl}cyclopropyl1carbamoyl|-3,6-dioxo-lJa,3,4,5,6,9J0, 18, 19.20.21.22.22a-tetradecahydro-8H-7.10-methanocyclopropaf 18.191 Γ 1.10.3 ,61
dioxadiazac yclononadecino 11 , 12-b] quinolin- 17-yl] oxy } -3 -oxa-7-azabicyclo [3.3.1 ]nonane-7- carboxylate

Step 1: methyl (laR,5S,8S,10R,22aR)-5-tert-butyl-17-{anti-[7-(tert-butoxycarbonyl)-3-oxa-7- azabicyclo[3.3.1 ]non-9-yl]-oxy}-3, 6-dioxo-l, la, 3, 4, 5, 6, 9, 10,18,19, 20, 21, 22, 22a-tetradecahydro- 8H- 7, 10-methanocyclopropa [18, 19] [1, 10,3,6 ]dioxadiazacyclononadecino[ 11, 12-b ] quinoline-8- carboxylate

The title compound was prepared using the procedure described for the synthesis of Example 210, Step 1. (lR,5S)-tert-butyl 9-hydroxy-3-oxa-7-azabicyclo[3.3.1]nonane-7- carboxylate (1:2 mixture of anti:syn alcohols; prepared as described in International Patent Publication No. WO2009055331) was used instead of l-methylpiperidin-4-ol. The title compound (anti isomer) was separated from the syn isomer by column chromatography (0-40% EtOAc in hexane); the anti isomer eluted after the syn isomer and was collected as the major isomer.
Step 2: tert-butyl 9-{[(laR,5S,8S 0R,22aR)-5-tert-butyl-8-{[(lR,2S)-2-ethenyl-l-{[(l- methylcyclopropyl)sulfonyl]carbamoyl}cyclopropyl ]carbamoyl}-3, 6-dioxo- 1, 1 a, 3, 4, 5, 6, 9,10,18, 19, 20, 21, 22, 22a-tetradecahydro-8H- 7, 10- methanocyclopropafl 8, 19] [1,10,3, 6] dioxadiazacyclononadecino [11 , 12-bJquinolin-l 7-ylJoxy}- -oxa-7-azabicyclo[3.3.1 Jnonane-7-carboxylate

Example 255 was prepared from the product of Step 1 by utilizing the procedures described for the synthesis of Example 210. LRMS m/z 991.2 (M+H)+
Example 256: 9-i r(laR,5S,8SJ0R,22aR)-5-tert-butyl-8-irriR,2S)-2-ethenyl-l-{ r(l- methylcvclopropyl sulfonyllcarbamoyl|cvclopropyl]carbamovU-3,6-dioxo- 1 , 1 a,3 A5 ,6,9 , 10.18.19,20,21 ,22.22a-tetradecahvdro-8H-7.10-
methanocyclopropa[ 18, 19] [ 1,10,3 ,6]dioxadiazacyclononadecino [11,12-b]quinolin- 17-νΠοχν} -7-

Example 256 was prepared from Example 255 by utilizing the procedures described for the synthesis of Example 238. LRMS m/z 945.2 (M+H)+
Example 257: 9-{r(laR.5S.8S.10R.22aRV5-tert-butyl-8-(rflR.2SV2-ethenyl-l-{r(l- methylcyclopropyDsulfonyllcarbamoyl} cyclopropyl] carbamoyl} -3 ,6-dioxo- 1 , 1 a.3.4.5.6.9.10.18.19,20,21 ,22,22a-tetradecahydro-8H-7.10- methanocyclopropa[ 18 , 19] [ 1 , 10,3,6] dioxadiazacyclononadecino [ 11 , 12-b] quinolin- 17-yl] oxy ) -7- 2-methoxyethyl)-3-oxa-7-azoniabicyclo[3.3.nnonane formate

Example 257 was prepared from Example 255 by utilizing the procedures described for the synthesis of Example 237. LRMS m/z 949.2 (M+H)+
Example 258: 9-(rriaR.5S.8S.10R.22aRV5-tert-butyl-8-(rflR.2SV2-ethenyl-l-{r(l- methylcyclopropyl)sulfonyllcarbamovUcvclopropyl]carbarnoyl}-3,6-dioxo-
1.1 a.3.4.5.6.9.10.18.19,20,21 ,22.22a-tetradecahydro-8H-7.10- methanocyclopropa[18,191[l,10,3,61dioxadiazacvclononadecino[l l,12-blquinolin-17-ylloxy}-3- oxa-7-azoniabicyclo [3.3.1 ]nonane trifluoroacetate

Example 258 was prepared from Example 255 by utilizing the procedure described for the synthesis of Example 236, Step 1. LRMS m/z 891.2 (M+H)+. Example 259: 9-{rriaR.5S.8S.10R.22aR)-5-tert-butyl-8-{rriR.2SV2-ethenyl-l-irri-methyl cyclopropyl)sulfonyl1carbamoyl)cvclopropyl carbamovU-3,6-dioxo-l , 1 a,3,4,5,6,9, 10, 18, 19,20.21 ,22,22a-tetradecahydro-8H-7, 10-methanocyclopropa[ 18, 19] [ 1.10,3 ,6]dioxadiaza cyclononadecino [11,12-b] quinolin- 17-yl]oxy) -3-oxa-7-azoniabicyclo [3.3.1 ]nonane trifluoroacetate

Example 259 was prepared from Example 254 by utilizing the procedure described for the synthesis of Example 236, Step 1. LRMS m/z 891.2 (M+H)+
Example 260: (laR.5S.8S, 10R,22aRV5-tert-butyl-N-r(lR.2SV2-ethenyl-l - ( ΓΠ -methyl cvclopropyDsulfonyl]carbamovU cyclopropyl]- 17-[(7-ethyl-3 -oxa-7-azabicyclo[3.3.1 ]non-9-yl oxy|-3 ,6-dioxo- 1 , 1 a,3.4,5.6,9, 10.18.19.20.21 ,22,22a-tetradecahydro-8H-7.10-methano cyclopropa[ 18, 191 [ 1.10,3.6] dioxadiazacyclononadecino [ 11.12-b] quinoline- 8 -carboxamide

Example 260 was prepared from Example 254 by utilizing the procedure described for the synthesis of Example 239. LRMS m/z 919.2 (M+H)+. Example 261 : (laR.5S.8S.10R.22aRV5-tert-butyl-N-rriR.2SV2-ethenyl-l-(r(l-methyl cvclopropyl sulfonyllcarbamoyl>cvclopropyn-17-([7-('2-methoxyethyl -3-oxa-7-azabicvclo Γ3.3.11non-9-yll oxyl -3.6-dioxo- 1.1 a.3.4.5.6.9.10.18.19.20.21.22.22a-tetradecahydro-8H-7, 10- methanocyclopropa[ 18, 19] [ 1 , 10.3,61dioxadiazacyclononadecino[ 11.12-b1quinoline-8- carboxamide

Example 261 was prepared from Example 254 by utilizing the procedures described for the synthesis of Example 237. LRMS m z 949.2 (M+H)+.
Example 262: potassium {rriR.2SVl-({r(laR.5S.8S.10R.22aRV5-cyclohexyl-3.6-dioxo-17-r(l- r2-(trifluoromethoxy ethyllpiperidin-4-yl } oxy )- 1.1 a.3.4.5.6.9.10.18.19.20,21.22.22a- tetradecahydro-8H-7, 10-methanocyclopropa 18, 191 [1 , 10.3 ,61dioxadiazacyclononadecino l 1,12- blquinolin-S-yllcarbonyllamino'l^-ethenylcvclopropyncarbonyll d- methylcyclopropyDsulfonyllazanide

Example 262 was prepared by utilizing the procedures described for the synthesis of Example 236. 2-(Trifluoromethoxy)ethyl trifluoromethanesulfonate was used instead of 2,2,2-trifluoroethyl trifluoromethanesulfonate in the final alkylation step. LRMS m/z 987.2 (M+H)+.
Example 263: (laR.5S.8S.10R.22aRVN-r(,lR.2SV2-ethenyl-l-ira-methylcvclopropyn sulfonyl]carbamoyl}cvclopropyl]-17- (8-methyl-8-azabicvclo[3.2.1]oct-3-yl)oxy]-5-(l-methyl cyclohexylV3.6-dioxo- 1.1 a.3.4.5.6.9.10.18, 19,20,21.22.22a-tetradecahydro-8H-7.10-methano cyclopropaf 18, 19] [ 1 , 10,3,6]dioxadiazacyclononadecino|"l 1.12-b]quinoline-8-carboxamide

Example 263 was prepared by utilizing the procedures described for the synthesis of Example 232; tropine was used instead of l-methylpiperidin-4-ol. LRMS m/z 929.2 (M+H)+. Example 264: tert-butyl 4-(3-{rdaR.5S.8S.10R.22aRV5-cvclohexyl-8-{rriR.2SV2-ethenyl-l- { [(1 -methylcy clopropyf)sulfonyll carbamoyl } cyclopropyl] carbamoyl } -3 ,6-dioxo- 1.1 a.3.4.5.6.9.10.18,19,20.21.22.22a-tetradecahydro-8H-7.10-methanocyclopropa
[18, 19] [1,10,3,6] dioxadiazacyclononadecino [ 11 , 12-b] quinolin- 17- yl] oxy } propy Dpiperazine- 1 - carboxylate

Example 264 was prepared by utilizing the procedures described for the synthesis of Example 251; NBoc-piperazine was used instead of N-methylpiperazine. LRMS m/z 1019.2 (M+H)+.
Example 265: potassium ({(lR,2SVl-r(((laR,5S,8S.10R.22aRV5-cvclohexyl-3.6-dioxo-17-r3- (piperazin- 1 -vflpropoxyl -1,1 a,3,4,5,6,9, 10,18,19.20.21.22.22a-tetradecahvdro-8H-7.10- methanocyclopropa 18, 19] [ 1 , 10,3 ,6] dioxadiazacyclononadecino [11,12-blquinolin-8- yl } carbonvDamino] -2-ethenylcyclopropyl } carbonyl) Γ( 1 -methylcyclopropyDsulfonyl] azanide

Example 265 was prepared from Example 262 by utilizing the procedure described for the synthesis of Example 236, Step 1; the potassium salt was prepared upon treatment of the parent compound with 0.1N aqueous KOH solution. LRMS m/z 918.4 (M+H)+. Example 266: potassium fi(lR,2S -l-rr{naR.5S,8S.10R.22aRV5-cvclohexyl-17-rf4- methylpiperazin- 1 -vDmethyll -3 ,6-dioxo- 1.1 a.3.4.5.6.9.10.18.19.20.21.22.22a-tetradecahydro-
8H-7, 10-methanocyclopropa[l 8, 191 Π , 10,3 ,61dioxadiazacyclononadecinoi 11 , 12-blquinolin-8- yl|carbonvnamino1-2-ethenylcvclopropyl}carbonyl) (l-methylcyclopropynsulfonyl1azanide

Example 266 was prepared by utilizing the procedures described for the synthesis of Example 248. LRMS m/z 888.3 (M+H)+.
Example 267: tert-butyl 4-{r(laR,5S,8SJ0R.22aRV8-{r(lR.2SV2-ethenyl-l-{rri- methylcvclopropyDsulfonyll carbamoyl } cyclopropyl] carbamoyl} -5 -( 1 -methylcvclohexyiy 3 ,6- dioxo- 1.1 a.3 ,4,5,6,9, 10,18.19.20.21.22,22a-tetradecahvdro-8H-7, 10- methanocyclopropal" 18.19] 1 , 10,3 ,61dioxadiazacyclononadecino [11,12-b]quinolin- 17- yl1oxy}piperidine- 1 -carboxylate

Example 267 was prepared by utilizing the procedures described for Example 263; NBoc-piperidine was used instead of tropine. LRMS m/z 989.2 (M+H)+.
Example 268 : ( 1 aR.5S.8S, 10R.22aRVN-r(l R,2SV2-ethenyl- 1 - { \( 1 -methylcyclopropyD sulfonyl1carbamoyl}cvclopropyll-5-(l-methylcyclohexyl)-3,6-dioxo-17-(piperidin-4-yloxyV 1.1 a.3 ,4.5.6.9, 10,18.19,20.21.22,22a-tetradecahydro-8H-7.10-methanocvclopropar 18.191
[l,10.3,6]dioxadiazacvclononadecino l l,12-b1quinoline-8-carboxamide

Example 268 was prepared from Example 267 by utilizing the procedure described for the synthesis of Example 236, Step 1. LRMS m/z 889.2 (M+H)+. Example 270: (1 aR,5 S.8S, 10R.22aRVN-f(l R,2SV2-ethenyl- 1 - i Γ(Ί -methylcyclopropyn sulfonyl] carbamoyl } cyclopropyll -5-d -methylcyclohexyl)- 17- [3 -(1 ,4-oxazepan-4-vDpropoxy1 - 3,6-dioxo- 1 , 1 a,3,4,5,6,9, 10,18,19,20,21 ,22,22a-tetradecahydro-8H-7, 10-methanocyclopropa [ 18, 19] [ 1 , 10,3 ,61dioxadiazacyclononadecino [11, 12-blquinoline-8-carboxamide

Example 270 was prepared by utilizing the procedures described for the synthesis of Example 240. LRMS m/z 947.2 (M+H)+.
Example 271 : riaR.5S,8S,10R,22aRV5-cvclohexyl-N-rriR.2S -2-ethenyl-l-{ rri-methyl cvclopropyDsulfonyl]carbamoyUcvclopropyll-17-({l-[2-(methylsulfonyl)ethyl piperidin-4- yl I oxy)-3 ,6-dioxo- 1 , 1 a.3 ,4,5 ,6,9, 10, 18 J 9,20.21 ,22,22a-tetradecahydro-8H-7.10-methano cyclopropa[ 18,19"||"1,10,3.6 dioxadiazacyclononadecino [ 11.12-b] quinoline- 8 -carboxamide

To a solution of the product from Step 1, Example 236 (50 mg, 0.057 mmol) in CH2C12 (3 ml) was added DIPEA (0.030 ml, 0.171 mmol) followed by methyl vinyl sulfone (12.10 mg, 0.114 mmol). The reaction was stirred in a sealed vial for 16h at which stage LCMS indicated complete conversion to one peak corresponding to a product with desired mass. After removing the solvent the crude mixture was purified by PTLC (5% MeOH in CH2CI2) and then repurified by PTLC using 40% acetone in hexane to furnish the title compound. LRMS m/z 981.6 (M+H)+. Example 272: 4-ir(laR,5S,8SJ0R,22aRV5-cyclohexyl-8-{ (lR.2SV2-ethenyl-l-(r(l- methylcyclopropyl)sulfonyllcarbamoyl}cvclopropyl]carbamoyl}-3,6-dioxo- 1 , 1 a,3 ,4,5 ,6,9.10, 18, 19.20,21 ,22,22a-tetradecahydro-8H-7.10-methanocyclopropa
[18, 19] [1 , 10,3 ,61dioxadiazacyclononadecino|~ 11 , 12-b]quinolin- 17-ν1]οχνΙ - 1 -(2- fluoroethvDpiperidinium formate

The product from Step 1, Example 236 was reacted with l-bromo-2-fluoro ethane as described for the synthesis of Example 237 to give Example 272. LRMS m/z 921.2 (M+H)+.
Example 273: tert-butyl 3-{rriaR.5S.8S.10R.22aR -5-tert-butyl-8-(r(lR.2S -2- ethenyl- 1 - ( |Y 1 -methylcyclopropyPsulfonyl] carbamoyl ) cyclopropyll carbamoyl } -3 ,6-dioxo- 1 , 1 a,3.4,5,6.9,10,18, 19.20.21 ,22,22a-tetradecahydro-8H-7.10-
methanocyclopropa[ 18, 19] [1,103.6] dioxadiazacyclononadecino f 11 , 12-b]quinolin- 17-yl]oxy| -8- azabicyclo [3.2.1] octane-8-carboxylate

Example 273 was prepared using the procedures described for the synthesis of Example 210. The endo isomer of tert-butyl 3-hydroxy-8-azabicyclo[3.2.1]octane-8-carboxylate (as prepared in International Patent Application No. WO2009055331) was used instead of 1- methylpiperidin-4-ol. LRMS m/z 975.4 (M+H)+.
Example 274: 3-( Γ(Ί aR.5S.8S, 10R,22aRV5-tert-butyl-8-{ [(lR.2S -2-ethenyl-l -{ [(1 - methylcyclopropyl sulfonyl]carbamoyl} cyclopropyl] carbamoyl} -3 ,6-dioxo- 1.1 a.3.4.5.6.9, 10.18.19.20.21.22.22a-tetradecahydro-8H-7.10- methanocyclopropa[ 18.19] [1.10,3.6] dioxadiazacyclononadecino [11.12-b]quinolin- 17-yl]oxy} -8- azoniabicyclo [3.2.1] octane trifluoroacetate

Example 274 was prepared from Example 273 by utilizing the procedure described for the synthesis of Example 236, Step 1. LRMS m/z 875.4 (M+H)+
Example 275: potassium iraR^SVl-rirdaR.SS^S.lOR^aR S-tert-butyl-S.e-dio o-n-irS- (2,2,2-trifluoroethvn-8-azabicvclo[3.2.11oct-3-yl1oxy}-l.la,3,4,5,6,9,10,18,19,20,21.22,22a- tetradecahydro-8H-7, 10-methanocyclopropa[ 18 , 19] [ 1.10.3.6]dioxadiazacyclononadecino [11.12- b] quinolin- 8 -yl] carbonyl } amino)-2-ethenylcvclopropyl] carbonyl } [( 1 - methylcvclopropyl)sulfonyl]azanide

Example 274 was converted into Example 275 by utilizing the procedures described for the synthesis of Example 236; the potassium salt was prepared upon treatment of the free base with 0. IN aqueous KOH solution. LRMS m/z 957.4 (M+H)+.
Example 276: potassium ir(lR.2S)-l-rir(laR,5S,8SJ0R.22aRV5-tert-butyl-17-ir8-(2- methoxyethylV8-azabicvclor3.2.11oct-3-yl1oxy>-3.6-dioxo-l.la.3A5.6.9.10.18.19.20.2L22.22a- tetradecahydro-8H-7, 10-methanocyclopropa[ 18, 19] [ 1 , 10,3 ,6]dioxadiazacyclononadecino [11,12- b] quinolin- 8 - yl] carbonyl 1 amino)-2-ethenylcyclopropyl] carbonyl } [( 1 - methylcyclopropyDsulfonyllazanide

Example 274 was converted into Example 276 by utilizing the procedures described for the synthesis of Example 237; the potassium salt was prepared upon treatment of the free base with 0. IN aqueous KOH solution. LRMS m/z 933.4 (M+H)+.
Example 277: potassium ({(lR.2SVl-r((riaR.5S.8S.10R,22aRV5-tert-butyl-17-r(8-ethyl-8- azabicvclor3.2.11oct-3-vnoxyl-3.6-dioxo-lJa,3.4.5.6,9,10.18a9,20.21,22.22a-tetradecahvdro- 8H-7, 10-methanocyclopropa[l 8, 19] [ 1 , 10,3,61dioxadiazacyclononadecino[ 11 , 12-blquinolin-8- yl } carbony amino] -2-ethenylcvclopropyl) carbonyD ( 1 -methylcyclopropynsulfonyllazanide

Example 274 was converted into Example 277 by utilizing the procedures described for the synthesis of Example 238; the potassium salt was prepared upon treatment of the free base with 0. IN aqueous KOH solution. LRMS m/z 903.4 (M+H)+.
Example 278: tert-butyl 3-iff laR.5S.8S 0R.22aRV5-1^-b ityl-8-(r(lR^SV2-ethenyl-l-{rf 1- methylcvclopropyDsulfonyllcarbamoyl) cyclopropyl] carbamoyl > -3 ,6-dioxo- 1 , 1 a.3.4.5.6.9.10, 18, 19.20,21.22.22a-tetradecahvdro-8H-7.10- methanocyclopropa[ 18.191 |T .10,3 ,61dioxadiazacyclononadecino|T 1 , 12-blquinolin- 17-yl]oxy> -8- azabicyclo[3.2.1 ]octane-8-carboxylate

Example 210. The exo isomer of tert-butyl 3-hydroxy-8-azabicyclo[3.2.1]octane-8-carboxylate (as prepared in International Patent Publication No. WO2009055331) was used instead of 1- methylpiperidin-4-ol. LRMS m/z 975.4 (M+H)+.
Example 279: 3-{ (laR.5S.8S.10R.22aRV5-tert-butyl-8-(rriR.2SV2-ethenyl-l-i[n- methylcvclopropyl)sulfonyllcarbamoyl}cvclopropyllcarbamoyl}-3,6-dioxo- 1.1 a.3.4.5.6.9.10.18.19,20,21 ,22,22a-tetradecahydro-8H-7.10- methanocyclopropa 18, 19] [ 1 , 10.3.6]dioxadiazacyclononadecino [11.12-blquinolin- 17-ν1]οχν> -8- azoniabicyclor3.2.1 ]octane trifluoroacetate

Example 279 was prepared from Example 278 by utilizing the procedure described for the synthesis of Example 236, Step 1. LRMS m/z 875.4 (M+H)+. Example 280: potassium i r(lR.2SVl-({rdaR,5S,8S.10R.22aRV5-tert-butyl-17-( r8-r2- methoxyethylV8-azabicvclor3.2.noct-3-yl1oxyl-3.6-dioxo-l,la.3,4,5,6,9,10a8,19,20,21,22,22a- tetradecahydro-8H-7, 10-methanocyclopropari 8, 19] [1 , 10,3 ,61dioxadiazacyclononadecino[ 11,12- b]quinolin-8-yllcarbonyl } aminoV 2-ethenylcyclopropyllcarbonyll |Y 1 - methylcyclopropyDsulfonyl azanide
MeQ

Example 279 was converted into Example 280 by utilizing the procedures described for the synthesis of Example 237; the potassium salt was prepared upon treatment of the free base with 0. IN aqueous KOH solution. LRMS m/z 933.5 (M+H)+. Example 281 : potassium (((lR.2SVl-r((flaR.5S.8S.10R.22aRV5-tert-butyl-17-r(8-etfayl-8- azabicvclon .2.11οοτ-3-νΓ)οχν1-3 ,6-dioxo- 1.1 a,3 ,4.5.6.9, 10.18.19,20.21 ,22.22a-tetradecahvdro- 8H-7, 10-methanocyclopropa[ 18 , 191 [ 1 , 10,3 ,61dioxadiazacyclononadecino [1 1,12-blquinolin-8- yl } carbonvDaminol -2-ethenylcyclopropyl 1 carbonyl') [( 1 -methylcyclopropyDsulfonyllazanide

Example 279 was converted into Example 281 by utilizing the procedures described for the synthesis of Example 238; the potassium salt was prepared upon treatment of the free base with 0. IN aqueous KOH solution. LRMS m/z 903.4 (M+H)+.
Example 282: tert-butyl 3-i r(,laR.5S,8Sa0R,22aRV5-cvclohexyl-8-ir(lR,2SV2-ethenyl-l-(r(l- methylcvclopropyl)sulfonyllcarbamoyl)cvclopropyl1carbamov -3,6-dioxo- l.la.3.4,5,6,9a0.18a9,20,21,22,22a-tetradecahvdro-8H-7.10- methanocyclopropaf 18, 19] [ 1.10,3,61dioxadiazacyclononadecino[ 1 1 , 12-b]quinolin- 17-yl]oxyl -8- azabic clo[3.2.11octane-8-carboxylate

Example 282 was prepared by utilizing the procedure described for the synthesis of Example 273. LRMS m/z 945.3 (M+H-tBu)+. Example 283: potassium ( rfl R.2SVl-f(rflaR.5S.8S.10R.22aRV17-f8-azabicvclor3.2.noct-3- yloxy>5-cvclohexyl-3.6-dioxo- 1.1 a .3.4.5,6,9.10.18.19.20.21 ,22.22a-tetradecahvdro-8H-7.10- methanocyclopropa[ 18.19] [ 1 , 10,3 ,61dioxadiazacyclononadecinor 11 , 12-blquinolin-8- yl]carbonvUaminoV2-ethenylcvclopropyl]carbonyl| [(l-methylcyclopropynsulfonyl1azanide

Example 283 was prepared from Example 282 by utilizing the procedure described for the synthesis of Example 274; the potassium salt was prepared upon treatment of the free base with 0. IN aqueous KOH solution. LRMS m/z 901.2 (M+H)+.
Example 284 : (1 aR.5 S.8S.10R,22aRV5-cvclohexyl-N-i( 1 R.2S V2-ethenyl- 1 - { Γ(1 -methyl cvclopropyl)sulfonyl]carbamoyllcyclopropyl]-17-{[8-(2-methoxyethylV8-azabicyclo[3.2.1]oct- 3 -ylloxyl -3.6-dioxo- 1.1 a,3 ,4.5 ,6,9.10.18.19.20.21.22.22a-tetradecahvdro-8H-7, 10-methano cyclopropa l 8, 19] [ 1 , 10,3,61dioxadiazacyclononadecinof 11 , 12-blquinoline-8-carboxamide

Example 284 was prepared from Example 283 by utilizing the procedure described for the synthesis of Example 276. LRMS m/z 959.4 (M+H)+.
Example 285 : flaR.5S.8S.10R.22aRV5-cyclohexyl-N-r(lR.2SV2-ethenyl-l -{ Γίΐ -methyl cvclopropyf)sulfonyllcarbamoyl}cyclopropyll-17-r(8-methyl-8-azabicyclor3.2.11oct-3-yl)oxyl- 3 ,6-dioxo- 1 , 1 a.3.4,5.6.9, 10, 18, 19,20.21 ,22.22a-tetradecahydro-8H-7.10-methano
cyclopropa 18, 19] 1.10,3,6] dioxadiazacyclononadecino 11 , 12-b] quinoline-8 -carboxamide

Example 285 was prepared by utilizing the procedures described for the synthesis of Example 211; tropine was used instead of 1 -methylpiperidin-4-ol. LRMS m/z 915.2 (M+H)+. Example 286: daR.5S.8S.10R,22aR -5-cvclohexyl-N-[aR.2SV2-ethenyl-l-ira -methyl cyclopropyDsulfonyl] carbamoyl > cycloprop yl] - 13 -fluoro- 17- |Y 1 -methylpiperidin-4-yl)oxy] -3,6- dioxo- 1 , 1 a,3 ,4,5 ,6,9, 10, 18, 19,20,21 ,22,22a-tetradecahydro-8H-7, 10-methanocyclopropa

Example 286 was prepared by utilizing the procedures described for the synthesis of Example 211. LRMS m/z 907.6 (M+H)+.
Example 287: tert-butyl 7-(rnaR.5S,8S,10R,22aRV5-cvclohexyl-8-{r(lR.2SV2-ethenyl-l-{r(l- methylcvclopropyPsulfonyllcarbamoyl } cyclopropyl] carbamoyl } -3 ,6-dioxo- 1 , 1 a.3 ,4.5.6.9, 10,18,19.20,21 ,22,22a-tetradecahydro-8H-7.10- methanocyclopropaf 18,19] [1,10,3,6] dioxadiazacyclononadecino 11 , 12-b] quinolin- 17-yl] oxy> -3 - oxa-9-azabicyclo [3.3.1 lnonane-9-carboxylate

The syn isomer of tert-butyl 7-hydroxy-3-oxa-9-azabicyclo[3.3.1]nonane-9- carboxylate (as prepared in International Patent Publication No. WO2009055331) was converted into Example 287 by utilizing the procedures described for the synthesis of Example 282.
LRMS m/z 961.2 (M+H-tBu)+.
Example 288: (laR,5S.8S.10R.22aRV5-cvclohexyl-N-r(lR.2SV2-ethenyl-l-{rq- methylcyclopropyDsulfonyll carbamoyl } cyclopropyl]- 17-(3-oxa-9-azabicvclo[3.3.1 ]non-7- yloxy V 3.6-dioxo- 1 , 1 a,3 ,4,5.6,9, 10,18,19,20,21 ,22,22a-tetradecahydro-8H-7, 10- methanocyclopropa 18, 19] [" 1 , 10,3 ,61dioxadiazacyclononadecinor 11 , 12-b]quinoline-8- carboxamide

Example 288 was prepared by utilizing the procedure described for the synthesis of Example 274. LRMS m/z 917.2 (M+H)+.
Example 289: (laR.5S.8S.10R.22aR)-5-cyclohexyl-N-r(lR.2SV2-ethenyl-l-i (,l-methyl cyclopropyl)sulfonyllcarbamoyl>cyclopropyn-17-([9-(2-methoxyethylV3-oxa-9-azabicyclo Γ3.3.1 lnon-7-yll oxyl -3 ,6-dioxo- 1.1 a.3 ,4.5 ,6.9, 10, 18, 19,20.21 ,22.22a-tetradecahydro-8H-7, 10- methanocyclopropaf 18, 19] [ 1 , 10,3,61dioxadiazacyclononadecino l 1.12-blquinoline-8- carboxamide

Example 288 was converted into Example 289 by utilizing the procedures described for the synthesis of Example 47. LRMS m/z 975.2 (M+H)+. Example 290: 7-(r(laR.5S.8S.10R.22aRV5-cvclohexyl-8-irriR,2SV2-ethenyl-l-{ra- methylcvclopropyDsulfonyl] carbamoyl ) cyclopropyl] carbamoyl } -3 ,6-dioxo- 1.1 a.3.4.5.6.9.10,18,19,20.21.22.22a-tetradecahydro-8H-7.10- methanocyclopropa[ 18, 19 [1 , 10,3 ,61dioxadiazacyclononadecino[ 11 , 12-blquinolin- 17-yl"|oxy} -9-

Example 288 was converted into Example 290 by utilizing the procedures described for the synthesis of Example 48. LRMS m/z 945.2 (M+H)+.
Example 291 : 3-i [flaR,5S,8S,10R.22aRV5-cyclohexyl-8-{rqR,2SV2-ethenyl-l-{[ri- methylcyclopropyl)sulfonyl]carbamoyl } cyclopropyl] carbamoyl } -3 ,6-dioxo- 1.1 a.3.4.5.6.9.10.18.19.20.21.22.22a-tetradecahvdro-8H-7.10- methanocyclopropa[ 18, 19] [ 1 , 10,3 ,6 dioxadiazacyclononadecino [11,12-blquinolin- 17-yl]oxyl - 1 - methylpiperidinium formate

Example 291 was prepared by utilizing the procedures described for the synthesis of Example 211; l-methylpiperidin-3-ol was used instead of l-methylpiperidin-4-ol. LRMS m/z 889.2 (M+H)+.
Example 292: tert-butyl (3S.4RV4-irriaR.5S.8S.10R.22aRV5-cvclohexyl-8-{r(lR.2S)-2- ethenyl- 1 - { [Y 1 -methylcyclopropyDsulfonyl] carbamoyl } cyclopropyl] carbamoyl } -3 ,6-dioxo- 1 , 1 a ,3 ,4,5,6,9, 10,18,19,20,21.22.22a-tetradecahvdro-8H-7.10- methanocyclopropa 18, 19] [ 1,10,3,61dioxadiazacyclononadecino|T 1 , 12-b]quinolin- 17-yl]oxy} -3- fluoropiperidine- 1 -carboxylate

The trans isomer of tert-butyl 3 -fluoro-4-hydroxypiperidine-l -carboxylate (as prepared in International Patent Publication No. WO 2011036576) was converted into Example 292 by utilizing the procedures described for the synthesis of Example 282. The substituents at the 3 and 4 position of the piperidine group in the title compound are in cis orientation; only relative stereochemistry is shown. Example 292 is a diastereomeric mixture. LRMS m/z 993.2 (M+H-fBu)+.
Example 293: r3S,4RV4-{raaR,5S,8S.10R.22aRV5-cvclohexyl-8-{rdR.2SV2-ethenyl-l-ir(l- methylcvclopropyl sulfonyl1carbamoyl}cvclopropyllcarbamoyl}-3,6-dioxo-
1.1 a.3.4.5.6.9.10, 18, 19,20.21 ,22.22a-tetradecahydro-8H-7.10- methanocyclopropa[ 18, 19] [ 1.10,3 ,61dioxadiazacyclononadecino[ 11 , 12-b]quinolin- 17-yl]oxy} -3- fluoropiperidinium chloride

Example 292 was converted into Example 293 by utilizing the procedure described for the synthesis of Example 236, Step 1. LRMS m/z 893.2 (M+H)+. Example 294: potassium (r(lR.2SVl-((rriaR.5S,8S.10R.22aRV5-cyclohexyl-17-ir(3S.4RV3- fluoro- 1 -methylpiperidin-4- yl"|oxy} -3 ,6-dioxo- 1 , 1 a.3.4.5.6.9.10.18.19.20.21 ,22,22a- tetradecahydro-8H-7, 10-methanocyclopropa 18, 19] 1 , 10,3,6]dioxadiazacyclononadecino[ 11,12- blquinolin-8-yl1carbonyl}aminoV2-ethenylcyclopropyllcarbonyl}[(l-

Step 1: methyl (1 aR,5S,8S 0R,22aR)-5-cyclohexyl-l 7-[(cis-3-fluoropiperidin-4-yl)oxy]-3,6- dioxo-1, la, 3, 4, 5, 6, 9, 10,18,19, 20, 21, 22, 22a-tetradecahydro-8H- 7, 10-methanocyclopropa
[18,19] '[1, 10, 3, 6]dioxadiazacyclononadecino[l 1, 12-b Jq inoline-8-carboxylate

The trans isomer of tert-butyl 3-fluoro-4-hydroxypiperidine-l-carboxylate (as prepared in International Patent Publication No. WO 2011036576) was converted into the title compound by utilizing the procedures described in Example 225, steps 1 and 2.
Step 2: methyl (laR, 5S, 8S, 1 OR, 22aR)-5-cyclohexyl-l 7-[(cis-3-fluoro-l-methylpiperidin-4- yl)oxy]-3, 6-dioxo-l, la, 3, 4, 5, 6, 9, 10,18,19, 20, 21, 22, 22a-tetradecahydro-8H- 7, 10- methanocyclopropa [18,19][1,10,3, 6]dioxadiazacyclononadecino[ll, 12-b]quinoline-8- carboxylate

The product from step 1 (90 mg, 0.132 mmol), formaldehye (0.107 ml, 1.322 mmol), and acetic acid (11.91 mg, 0.198 mmol) were dissolved in dichloromethane (1.322 ml) and allowed to stir for 30 minutes. Sodium triacetoxyborohydride (84 mg, 0.397 mmol) was added and the reaction was stirred overnight at room temperature, after which it was quenched with water and extracted (x3) with ethyl acetate. The combined organics were washed with brine, dried over sodium sulfate, and concentrated under reduced pressure. Purification by PTLC (25% acetone in hexane) provided the title compound.
Step 3: potassium {[(1R, 2S)-l-({[ (laR, 5S, 8S, 1 OR, 22aR)-5-cyclohexyl-l 7-{[(3S, 4R)-3-fluoro-l- methylpiperidin-4-yl ]oxy}-3, 6-dioxo-l, la, 3, 4, 5, 6, 9,10,18,19, 20, 21, 22, 22a-tetradecahydro-8H-
7, 10-methanocyclopropa[ 18, 19] [1, 10,3,6 Jdioxadiazacyclononadecinof 11, 12-b ]quinolin-8- yl ] carbonyl}amino)-2-ethenylcyclopropyl] carbonyl} [ ( I -methylcyclopropyl)sulfonyl Jazanide

The product from Step 2 was converted into Example 294 by utilizing the procedures described for the synthesis of Example 225, steps 4 and 5.
Example 295: riaS.5S.8S.10R,22aSV5-tert-butyl-N-iriR.2SV2-ethenyl-l-r(l- methylcyclopropanesulfonamido carbonyllcyclopropyl 1 - 17- |Y 1 -methylpiperidin-4-v oxy] -3,6- dioxo- Ua.3.4.5.6.9.10.18.19,20,21 ,22.22a-tetradecahvdro-8H-7.10- methanocyclopropa[ 18, 19] [ 1 , 10,3 ,6]dioxadiazacyclononadecino [ 11 , 12-b] [ 1 ,61naphthyridine-8- carboxamide


The sodium salt lc from previous step was dissolved in hydrobromic acid (100 mL) and heated at reflux for 12 hours. The reaction mixture was concentrated in vacuo and the solid Id was used as it is in next step.
Step 3: 3-bromo-4-hydroxy-2,6-naphthyridin-l(2H)-one hydrobromide

A solution of naphtyridinone Id (241 mg, 1.49 mmol) in acetic acid (3.00 mL) was treated with bromine (77 micro liters, 1.49 mmol) and stirred at room temperature for 2 hours. The reaction mixture was concentrated in vacuo, triturated with ether and filtered. The filtered solid le (1.25 g, 84%) was used as it is in the next step.
Step 4: 4-(benzyloxy)-3-bromo-2,6-naphthyridin-l(2H)-one

A suspension of le (2.75 g, 6.83 mmol) in THF (20 mL) was treated with potassium tert-butoxide (2.30 g, 20.48 mmol), benzyl bromide (1.17 g, 6.83 mmol) and stirred at room temperature overnight. The reaction mixture was concentrated in vacuo and treated with
water. The solid separating out was filtered and dried in vacuo. The residue If (1.25, 55.3%) was used as it is in next step.
Step 5: (2S,4R)-l-tert-butyl 2-methyl 4-((4-(benzyloxy)-3-bromo-l,6-naphthyridin-2- yl)oxy)p rrolidine-l,2-dicarboxylate

A solution of naphtyridine If (1.25 g, 3.77 mmol), triphenylphosphine (1.98 g,
7.55 mmol), and cis-4-hydroxyproline lg (1.29 g) in CH2CI2 (40 mL) was cooled to 0°C and treated with a solution of DIAD (1.145 g) in CH2C12 (10 mL). The reaction mixture was stirred at room temperature for overnight concentrated in vacuo and purified by silica gel
chromatography using Acetone/Hexanes to yield product lh (610 mg, 29%) as a yellow solid. Step 6: (S)-tert-butyl 3,3-dimethyl-2-((((lR,2R)-2-(pent-4-yn-l- yl)cyclopropoxy)carbonyl)amino)butanoate

A solution of succinyl carbonate li (5.42 g, 20.43 mmol) in dry acetonitrile (50 mL) was treated with tert-butylglycine ferr-butyl ester (4.57 g, 20.43 mmol) and cooled to 0°C. The reaction mixture was treated with triethyl amine (2.95 mL, 20.43 mmol) and stirred at room temperature for 36 hours. The reaction mixture was concentrated in vacuo and extracted into EtOAc (300 mL). The combined organic layers were dried (MgS04), filtered, concentrated in vacuo and purified by silica gel chromatography to yield product lj (4.2 g, 61%).
Step 7: (S)-tert-butyl 3,3-dimethyl-2-((((lR,2R)-2-((E)-5-(tributylstannyl)pent-4^
yl)cyclopropoxy)carbonyl)amino)butanoate and (S)-tert-butyl 3,3-dimethyl-2-((((lR,2R)-2-(4- (tributylstannyl)pent-4-en-l-yl)cyclopropoxy)carbonyl)amino)butanoate

A solution of alkyne lj (2.00 g, 5.93 mmol) in THF (50 mL) was cooled to 0°C and treated with Pd(PPh3)2Cl2 and tributyltin hydride (1.73 g, 5.93 mmol). The reaction mixture was stirred at room temperature for 0.5 hours and concentrated in vacuo. The residue was taken in hexane and filtered through a plug of celite. The filtrate was concentrated in vacuo and purified by silica gel chromatography to yield stannanes lk and 11 as a inseparable mixture (1.99 g, 53%).
Step 8: (2S,4R)-l-tert-butyl 2-methyl 4-((4-(benzyloxy)-3-((E)-5-((lR,2R)-2-((((S)-l-(tert- butoxy)-3,3-dimethyl-l-oxobutan-2-yl)carbamoyl)oxy)cyclopropyl)pent-l-en-l-yl)-l,6- naphthyridin-2-yl)oxy)pyrrolidine-l,2-dicarboxylate and (2S,4R)-l-tert-butyl 2-methyl 4-((4- (benzyloxy)-3-(5-((lR,2R)-2-((((S)-l-(tert-butoxy)-3,3-dimethyl-l-oxobutan-2-yl)
carbamoyl)oxy)cyclopropyl)pent-l-en-2-yl)-l,6-naphthyridin-2-yl)oxy)pyrrolidine-l,2- dicarboxylate

A solution of stannanes lk & 11 (225 mg, 0.358 mmol), bromide lh (100 mg, 0.179 mmol) and Pd(PPh3)2Cl2 (20.7 mg) in dioxane (3.0 mL) was degassed and heated at 115°C for 14 hours. The reaction mixture was cooled and taken in EtOAc, filtered through a plug of
celite. The filtrate was concentrated in vacuo and purified by silica gel chromatography to yield coupled product lm (60 mg).
Step 9: (2S,4R)-l-tert~butyl 2-methyl 4-((3-(5-((lR,2R)-2-((((S)-l-(tert-butoxy)-3,3-dimethyl-l- oxobutan-2-yl)carbamoyl)oxy)cyclopropyl)pentyl)-4-hydroxy-l,6-naphthyridin-2- yl)oxy)pyrrolidine-l,2-dicarboxylate

A solution of benzylated derivative lm (130 mg, 0.159 mmol) in methanol was treated with palladium hydroxide on carbon (10%, 130 mg) and hydrogenated with hydrogen in a balloon for 12 hours. The reaction mixture was filtered through a plug of celite, concentrated in vacuo and purified by silica gel chromatography (Acetone, Hexanes) to yield reduced product lo.
Step 10: (2S,4R)-l-tert-butyl 2-methyl 4-((3-(5-((lR,2R)-2-((((S)-l-(tert-butoxy)-3,3-dimethyl-l- oxobutan-2-yl)carbamoyl)oxy)cyclopropyl)pentyl)-4-((l-methylpiperidin-4-yl)oxy)-l,6- naphthyridin-2-yl)oxy)pyrrolidine-l,2-dicarboxylate

A solution of lo (60 mg, 0.082 mmol), triphenylphosphine (216 mg, 0.823 mmol), 4-hydroxy-N-methylpiperidine (95 mg, 0.823 mmol) in THF (3.00 mL) in a two necked flask was filled with nitrogen and treated drop wise with DIAD (166 mg, 0.823 mmol). The reaction mixture was stirred at 40°C for 3 hours. The reaction mixture was concentrated in vacuo and purified by silica gel chromatorgraphy to yield lp. After first purification, the product co-eluted with N-methylpiperidinol. It was therefore subjected to second purification using methylene chloride and ammoniacal methanol, to yield product still containing some N- methylpiperidinol (162 mg).
Step 11: Methyl-(laS,5S,8S,10R,22aS)-5-tert-butyl- 17-[(l-methylpiperidin-4-yl)oxy]-3,6-dioxo- 1, 1 a, 3, 4, 5,6,9,10,18,19,20,21, 22, 22a-tetradecahydro-8H- 7, 10-methanocyclopropa
[18, 19] [1, 10,3,6 ]dioxadiazacyclononadecino[l 1, 12-b ][ 1, 6]naphthyridine-8-carboxylate

A solution of lp (85 mg, 0.103 mmol) dissolved in CH2C12 (2.0 mL) and TFA (2.0 mL) and stirred at room temperature for 2 hours. The reaction mixture was concentrated in vacuo and used as it is in next step. The crude mixture was dried in vacuo for 48 hours, dissolved in DMF (2.00 mL) and cooled to 0°C. It was treated with NMM (41.6 mg, 0.412 mmol) and HATU (117 mg, 0.31 mmol) and stirred at 0°C for 0.5 hours and room temperature for 1 hour. The reaction mixture was diluted with 30 mL aqueous sodium bicarbonate solution and extracted into EtOAc (90 mL). The combined organic layers were dried (MgS04), filtered, concentrated in vacuo, and purified by silica gel chromatography using (CH2C12, and 10% methanol in CH2CI2) to yield cyclized product l as colorless solid (30 mg).
Step 12: Methyl-(laS,5S,8S, 10R,22aS)-5-tert-butyl- 17-[(l-methylpiperidin-4-yl)oxy]-3,6-dioxo-
1, 1 a, 3, 4, 5,6,9,10, 18, 19,20,21, 22, 22a-tetradecahydro-8H- 7, 10-methanocyclopropa
[18, 19 ][1, 10,3, 6 Jdioxadiazacyclononadecinof 11,12-b Jfl, 6]naphthyridine-8-carboxylic acid

A solution of lq (30.0 mg, 0.046 mmol) in water, methanol THF (0.6 mL each) was treated with aqueous solution of lithium hydroxide (0.5 M, 0.276 mL) and stirred overnight. The reaction mixture was quenched with acetic acid (30 μΐ) and extracted into CH2C12 (7x20 mL) and EtOAc (2x20 mL). The combined organic layers were dried (MgS04), filtered, concentrated in vacuo and used as it is in next step.
Step 13: (laS,5S,8S,10R,22aS)-5-tert-butyl-N-{(lR,2S)-2-ethenyl-l-[(l-methyl
cyclopropanesulfonamido)carbonyl]cyclopropyl}-l 7-[(l-methylpiperidin-4-yl)oxy]-3, 6-dioxo- l,la,3,4,5,6,9,10,18,19,20,21, 22, 22a-tetradecahydro-8H- 7, 10-methanocyclo
ide

A solution of lr (22 mg, 0.034 mmol), amine Is (12.6 mg, 0.052 mmol) and HATU (26.2 mg, 0.069 mmol) in DMF (0.6 mL) and CH2C12 (0.6 mL) was stirred at room temperature for 10 minutes and treated with 4-methyl morpholine (20 μΐ). The reaction mixture was stirred at room temperature overnight and quenched with 20 μΐ of acetic acid. The reaction mixture was extracted with methylene chloride and the combined organic layers were dried (MgS04), filtered, concentrated in vacuo and purified by silica gel chromatography (CH2C12,
ethanol) to yield 1 (13 mg, 44%) as a colorless solid. LR-MS (ESI) Calculated for C44H62N709S (M+H)+ 864.43; Found 864.45.
Example 296: (laR.5£l lZJ2aS.13aj?a6Sa9i?,27E31aj?VN-r(l-methylcvclopropyl)sulfonvn- 3 J5 3-trioxo-26-r2-(piperidin-l-yl ethoxy1-l.la.3.4.5.6.7.8.9.10,12a.l3.15.16.18.19,29, 30.31.31a-icosahvdro-5.17:16.19-dimethanodicvclopropari2.13:28.29iri.20.3.14.171

Step 1 : 15-tert-butyl 14a-ethyl (2R,6S, 12Z,13aS,14aR,16aS)-2-{[4-(ben∑yloxy)-3- bromoquinolin-2-yl]oxy}-6-[(tert-butoxycarbonyl)amino J -5, 16-dioxo- 2, 3, 6, 7, 8, 9, 10,11,13 a, 14,16, 16a-dodecahydrocyclopropa[e Jpyrrolof 1, 2- a][l,4 Jdiazacyc pentadecine-l 4a, 15(lH, 5H)-dicarboxylate

To a solution of Intermediate C6 (1.48 g) and Intermediate B16 (2.03 g) in DMA (12.5 mL) was added cesium carbonate (1.02 g). The reaction mixture was heated to 60°C for 5 hours. After cooling to room temperature, the reaction mixture was diluted with ethyl acetate and water. The mixture was extracted (3x) with ethyl acetate. The combined organic layers were washed with water, then brine, dried over magnesium sulfate, filtered and concentrated. The residue was suspended in dichloromethane and filtered to remove the insoluble Intermediate C6. The mother liquors were concentrated and the residue was purified by flash chromatography
(ISCO, 0 to 100% ethyl acetate in hexanes) to give the title compound (2.06 g). LRMS (ES+) m/z 905.0 (M+H)+.
Step 2 : ethyl (2R,6S, 12Z,13aS, 14aR,16aS)-6-amino-2-[(3-bromo-4-hydroxyquinolin-2-yl)oxy]- 5, 16-dioxo-l, 2, 3, 6, 7, 8, 9, 10,11,13 a, 14, 15,16, 16a-tetradecahydrocyclopropa[ Jpyrrolof 1, 2- [1 ,4]diazacyclopentadecine-14a(5H)-carboxylate

The product of Step 1 (2.06 g) was dissolved in TFA (22.7 mL) and the reaction was stirred for 5 hours at room temperature. The solvent was removed in vacuo. The residue was dissolved in ethyl acetate and a saturated solution of sodium bicarbonate was added slowly. The layers were separated and the organic layer was washed again with a saturated solution of sodium bicarbonate then with brine, dried over magnesium sulfate, filtered and concentrated.
The product was used without further purification. LRMS (ES+) m/z 615.2 (M+H)+.
Step 3 : ethyl (2R,6S, 12Z, 13aS, 14aR, 16aS)-2-[(3-bromo-4-hydroxyquinolin-2-yl)oxy]-5,16- dioxo-6-{[( {(1R, 2R)-2-[ (4E)-5-(4, 4, 5, 5-tetramethyl-l, 3, 2-dioxaborolan-2-yl)pent-4-en-l- yl]cyclopropyl}oxy)carbonyl Jamino -1, 2, 3, 6, 7, 8, 9, 10, 11,13a, 14, 15,16,16a- tetradecahydrocyclopropaf e Jpyrrolofl, 2-a][l, 4]diazacyclopentadecine-14a(5H)-carboxylate

To a solution of the amine from Step 2 (1.58 g) and Intermediate A14 (1.11 g) in acetonitrile (12.8 mL) was added triethylamine (1.78 mL). The reaction mixture was stirred
overnight at room temperature. The solvent was removed in vacuo. The residue was dissolved in ethyl acetate and water was added. The mixture was extracted (3x) with ethyl acetate. The combined organics were dried over sodium sulfate, filtered and concentrated. The residue was purified by flash chromatography (ISCO, 0 to 10% methanol in dichloromethane) to give the title compound (1.53 g) as a white solid. LRMS (ES+) m/z 893.3 (M+H)+.
Step 4 : ethyl (2R,6S,12Z,13aS,l 4aR,l 6aS)-2-({3-bromo-4-[2-(piperidin-l -yl)ethoxy]quinolin-2- yl}oxy)-5, 16-dioxo-6-{[ ({(1R, 2R)-2-[(4E)-5-(4, 4, 5, 5-tetramethyl-l, 3, 2-dioxaborolan-2-yl)pent-4- en-l-yl ]cyclopropyl}oxy)carbonyl]amino}-l, 2, 3, 6, 7, 8, 9,10,11, 13a, 14, 15,16,16a- tetradecahydrocyclopropaf e Jpyrrolof 1, 2-a][ 1, 4]diazacyclopentadecine-14a(5H)-carboxylate

To a solution of phenol from Step 3 (0.513 g) in DMF (5.7 mL) was added l-(2- bromoethyl)piperidine (0.39 g) and cesium carbonate (1.5 g). The reaction mixture was stirred at room temperature for 18 hours. The reaction was quenched with water and the reaction mixture was diluted with ethyl acetate. The mixture was extracted (3x) with ethyl acetate. The combined organics were washed with water (2x), then brine, dried over magnesium sulfate, filtered and concentrated. The residue was purified by flash chromatography (ISCO, 0 to 10% methanol in dichloromethane) to give the title compound (456 mg) as a colorless oil. LRMS (ES+) m/z 1004.4 (M+H)+.
Step 5 : ethyl (laR,5S,HZ,12aS,13aR,16S,19R,27E,31aR)-3,15,33-trioxo-26-[2-(piperidin-l- yl)ethoxy]-l, la, 3, 4, 5, 6, 7, 8, 9, 10, 12a, 13, 15,16,18,19, 29, 30,31, 31a-icosahydro-5, 17: 16,19- dimethanodicyclopropa[12, 13:28,29J[1,20,3, 14,17]dioxatriazacyclononacosino[21,22- b ]quinoline-13a( 14H)-carboxylate

In a reaction flask, boronate from Step 4 (456 mg), Catacxium A (35.4 mg), cesium carbonate (483 mg) were dissolved in 10.3 mL of dioxane and 2.1 mL of water. The reaction mixture was degassed with nitrogen (3 cycles) before the addition of palladium acetate (11.1 mg). The reaction mixture was once again degassed with nitrogen (3 cycles) and heated to 100°C for 1 hour. After cooling to room temperature, the reaction was quenched with a saturated solution of ammonium chloride. The mixture was extracted (3x) with ethyl acetate. The combined organics were dried over magnesium sulfate, filtered and concentrated. The residue was purified by flash chromatography (ISCO, 0 to 10% methanol in dichloromethane) to give the title compound (232 mg) as a yellow foam. LRMS (ES+) m/z 798.35 (M+H)+.
Step 6 : (laR,5S,llZ,12aS,13aR,16S,19R,27E,31aR)-3,15,33-trioxo-26-[2-(piperidin-l- yl)ethoxy]-l, la, 3, 4, 5, 6, 7, 8, 9, 10, 12a, 13, 15,16,18,19, 29, 30,31, 31a-icosahydro-5, 17: 16,19- dimethanodicyclopropafl 2, 13:28, 29] [1, 20, 3,14,17]dioxatriazacyclononacosino[21, 22-b ] quinoline-13a(14H)-carboxylic acid

Ester from step 5 (231 mg) was dissolved in THF (1.4 mL) and methanol (0.7 mL). LiOH (121 mg) in 0.7 mL of water was then added to the reaction mixture. The reaction was stirred overnight at room temperature. The reaction mixture was acidified to pH = 4 with acetic acid. The mixture was extracted (3x) with ethyl acetate. The combined organics were
dried over magnesium sulfate, filtered and concentrated. The residue was purified by flash chromatography (ISCO reverse phase, 5 to 95% acetonitrile in water) to give the desired product (87.3 mg). LRMS (ES+) m/z 770.60 (M+H)+.
Step 7 : (laR,5S,l lZ,12aS,13aR,16S,19R,27E,31aR)-N-[(l-methylcyclopropyl)sulfonyl]-3, 15,33- trioxo-26-[2-(piperidin-l -yl)ethoxy]-l, la, 3, 4, 5, 6, 7, 8, 9, 10, 12a, 13, 15,16,18,19, 29, 30,31,3 la- icosahydro-5, 17:16, 19-dimethanodicyclopropa[12, 13:28, 29] [1, 20,3, 14, 17]
3a(14H)-carboxamide

To a solution of acid from Step 6 (87.3 mg) in THF (0.6 mL) was added 1,1'- carbonyldiimidazole (28 mg) and the reaction mixture was heated to 40°C for 1 hour. The reaction mixture was cooled to room temperature. At this point, 1- methylcyclopropanesulfonamide (61 mg) and DBU (85 μΐ) were added. The reaction mixture was heated to 40°C for 18 hours. Once the mixture cooled to room temperature, it was diluted with ethyl acetate. Water was added and the mixture was acidified to pH = 4. The mixture was extracted (3x) with ethyl acetate. The combined organics were dried over magnesium sulfate, filtered and concentrated. The residue was purified by flash chromatography (ISCO reverse phase, 5 to 95% acetonitrile in water) to give the desired product (40.8 mg). LRMS (ES+) m/z 887.40 (M+H)+.
Examples 297-299
By following the procedures outlined in Example 296 and using the appropriate reagents (depicted below the structure as Rg.), the following compounds were prepared.


Example 300 : aai?.5S.l lZa2aS.13ai?.16S.19i?.27E.31ai?V26-methoxy-N-rri-methyl cvclopropynsulfonyll-3.15.33-trioxo-l.la.3.4.5.6J.8.9.10.12a. 13.15.16.18.19.29.30,31.31a- icosahvdro-5.17:16.19-dimethanodicvclopropa Γ 12.13 :28.29Ί Γ 1.20.3.14.1 1
dioxatriazacyclononacosino \21.22-Z?lquinoline- 13a( 14H)-carboxamide

Step 1 : ethyl (2R,6S, 12Z, 13aS, 14aR, 16aS)-2-[(3-bromo-4-methoxyquinolin-2-yl)oxyJ-5, 16- dioxo-6-{[({ (1R, 2R)-2-[ (4E)-5-(4, 4, 5, 5-tetramethyl-l, 3, 2-dioxaborolan-2-yl)pent-4-en-l- yl ]cyclopropyl}oxy)carbonyl] amino}- 1, 2, 3, 6, 7, 8, 9, 10,11,13a, 14, 15,16, 16a- tetradecahydrocyclopropa[e]pyrrolo[l,2-a] [1 ,4] diazacyclopentadecine-14a(5H)-carboxylate

To a solution of phenol from Example 296, Step 3 (402 mg) in DMF (2.2 mL) was added methyl iodide (84 μΐ) and DIPEA (314 μΐ). The reaction mixture was stirred at room temperature for 18 hours. The reaction was quenched with water and the reaction mixture was diluted with ethyl acetate. The mixture was extracted (3x) with ethyl acetate. The combined organics were washed with brine (2x), dried over magnesium sulfate, filtered and concentrated. The residue was purified by flash chromatography (ISCO, 0 to 10% methanol in
dichloromethane) to give the title compound (403 mg, 99%) as a yellow foam. LRMS (ES+) m/z 907.3 (M+H)+.
Steps 2 to 4 : (laR,5S, llZ, 12aS, 13aR,16S, 19R,27E,31aR)-26-methoxy-N-[(l-methyl cyclopropyl)sulfonyl]-3, 15, 33-trioxo-l, la, 3, 4, 5, 6, 7, 8, 9, 10, 12a, 13, 15, 16, 18, 19, 29, 30,31, 31a- icosahydro-5, 17: 16, 19-dimethanodicyclopropafl 2, 13 : 28,29] [1 ,20,3, 14, 17]dioxatriaza cyclononacosino[21, 22-b ]quinoline-13a(14H)-carboxamide

The title compound was prepared using the same method as Example 296, Steps 5-7 using the product of step 1. LCMS (ES+) m/z 790.25 (M+H)+. Example 301 : riaR.5S.l lZ.12aS.13aR.16S.19R.31aRVN-rri-methylcvclopropynsulfonyll-26- r2-rmon?holin-4-vnethoxy1-3J533-trioxo-lJa3A5.6.7.8.9,10.12a,13.15J6,18,19,27,28, 29.30,31.31 a-docosahvdro-5, 17:16.19-dimethanodicvclopropa[ 12.13:28.29"|[1.20.3.14.17]

Step 1 : 15-tert-butyl 14a-ethyl (2R, 6S, 12Z, 13aS,14aR,16aS)-2-({3-bromo-4-[(4- methoxybenzyl)oxy]quinolin-2-yl}oxy)-6-[(tert-butoxycarbonyl)amino]-5,16-dioxo- 2, 3,6, 7,8,9,10,11,13a, 14, 16,16a-dodecahydrocyclopropa[e ]pyrrolo[ 1,2- a][l,4 Jdiazacyclopentadecine-14a, 15(1H, 5H) -dicarboxylate

The title compound was prepared using the same method as Example 296, Step 1 using Intermediate CI 3. LCMS (ES+) m/z 957.4 (M+Na)+.
Step 2 : 15-tert-butyl 14a-ethyl (2R,6S, 12Z, 13aS, 14aR, 16aS)-6-amino-2-({3-bromo-4-[(4- methoxybenzyl)oxy]quinolin-2-yl } oxy)-5, 16-dioxo-2, 3, 6, 7, 8, 9, 10,11,13a, 14, 16,16a- dodecahydrocyclopropafe ]pyrrolo[ 1, 2-a][ 1, 4]diazacyclopentadecine-14a, 15(1H, 5H)- dicarboxylate

Tert-butyldimethylsilyl trifluoromethanesulfonate (0.655 mL) was added to the solution of 2,6-lutidine (0.31 mL) and the product from Step 1 (1.91 g) in DCM (40 mL). The solution was stirred at room temperature for 1 hour. Tetra-butyl ammonium fluoride (1.0M in THF, 3.06 mL) was added and the solution was stirred at room temperature for 30 minutes. A saturated solution of ammonium chloride was added and the mixture was extracted with dichloromethane (3x). The combined organic fractions were washed with saturated solution of sodium bicarbonate, dried over magnesium sulfate, filtered and concentrated. The crude product was used directly in the next step. LCMS (ES+) m/z 835.3 (M+H)+.
Step 3 : 15-tert-butyl 14a-ethyl (2R,6S, 12Z, 13aS, 14aR, 16aS)-2-({3-bromo-4-[(4-methoxy benzyl)oxy]quinolin-2-yl}oxy)-5, 16-dioxo-6-{[({(lR, 2R)-2-[5-(4, 4, 5, 5-tetramethyl-l, 3, 2- dioxaborolan-2-yl)pentyl ]cyclopropyl}oxy)carbonyl ]amino}-2, 3, 6, 7, 8, 9,10,11,13a, 14,16,16a-
dodecahydrocyclopropa[e]pyrrolo[ 1, 2-a][l, 4]diazacyclopentadecine-14a, 15(1H, 5H)- dicarboxylate

The title compound was prepared using the same method as described in Example 296, Step 3 using intermediate A15.
Step 4 : 14-tert-butyl lSa-ethyl (laR,5S,llZ,12aS,13aR,19R,31aR)-26-[(4-methoxybenzyl)oxy]- 3, 15, 33-trioxo-l, la, 3, 4, 5, 6, 7, 8, 9, 10, 12a, 13,15,16,18,19,27, 28, 29, 30,31, 31a-docosahydro- 5, 17: 16,19-dimethanodicyclopropa[ 12, 13:28,29] [1,20,3,14,17]dioxatriazacyclononacosino [21 ,22-b]quinoline-l 3a, 14-dicarboxylate

The title compound was prepared using the same method as described in Example
296, Step 5 except that cataxium A and palladium acetate were pre-mixed before addition to the reaction mixture. The reaction mixture was heated at 100°C for 9 hours. LCMS (ES+) m/z 909.45 (M+H)+.
Step 5 : ethyl (laR, 5S, 11Z, 12aS, 13aR, 19R, 31aR)-26-hydroxy-3, 15, 33-trioxo-
1, la, 3, 4, 5, 6, 7, 8, 9,10, 12a, 13,15,16, 18,19,27, 28,29, 30, 31, 31a-docosahydro-5, 17:16, 19- dimethanodicyclopropa[12, 13:28,29] [1,20, 3,14, 17]dioxatriazacyclononacosino[21,22- b]quinoline-13a(14H)-carboxylate

TFA (1.51 ml) was added to the solution of bis-macrocycle from Step 4 (0.412 g) in DCM (3.0 ml) at room temperature. The solution was stirred for 3 hours. The solvent was evaporated under reduced pressure and the residue was purified by flash chromatography (ISCO, reverse phase) to afford the desired product (0.122 g) as a white solid. LCMS (ES+) m/z 689.5 (M+H)+.
Step 6 : ethyl (laR,5S,llZ,12aS,13aR,16S,19R,31aR)-26-[2-(morpholin-4-yl)ethoxy]-3, 15,33- trioxo-1, la, 3, 4, 5, 6, 7, 8, 9, 10, 12a, 13,15,16,18, 19,27, 28, 29,30, 31, 31a-docosahydro-5, 17: 16,19- dimethanodicyclopropaf 12,13: 28, 29 ][1, 20,3,14,17]dioxatriazacyclononacosino[21, 22- b]quinoline-13a(14H)-carboxylate

4-(2-Bromoethyl)morpholine (0.069 g) was added to the mixture of phenol from
Step 5 (0.122 g) and cesium carbonate (0.462 g) in DMF (1.77 ml). The mixture was stirred at room temperature for 1 hour 30 minutes. At this point, water was added and the mixture was extracted with ethyl acetate (3x). The combined organic fractions were dried over magnesium sulfate, filtered and concentrated. The residue was purified by flash chromatography (ISCO reverse phase) to give the undesired epimer (19 mg) as a white powder and the desired product (12.9 mg) as a white powder. LCMS (ES+) m/z 802.45 (M+H)+.
Step 7-8 : (laR,5S,llZ,12aS,13aR,16S,19R,31aR)-N-[(l-methylcyclopropyl)sulfonyl]-26-[2- (morpholin-4-yl)ethoxy]-3,15, 33-trioxo-l, la, 3, 4, 5, 6, 7, 8, 9, 10, 12a, 13,15,16,18,19,27,
28,29,30, 31,31a-docosahydro-5, 17 : 16, 19-dimethanodicyclopropa[12, 13:28,29] [1,20,3, 14, 17] dioxatriazacyclononacosino[21,22-b]quinoline-13a(14H)-carboxamide

The title compound was prepared using the same method as Example 296, Steps 6-7. LCMS (ES+) m/z 891.40 (M+H)+.
Example 302: aaj?.5S.l lZ.12aS.13aiU6S.19i?.27£.31^
3,15,33-trioxo-26-[3-(piperidin-l-vnpropoxy1-l,la.3.4.5,6.7,8.9,10,12a,13,15,16.
18.19.29.30.31.31 a-icosahvdro-5.17:16.19-dimethanodicvclopropar 12.13 :28.291 Γ 1.20.3.14.171 dioxatriazacyclononacosino \21.22- >lquinoline- 13aC 14H)-carboxamide

Step 1 : ethyl (2R,6S,12Z,13aS,14aR 6aS)-2-{[3-bromo-4-(3-bromopropoxy)quinolin-2-yl]oxy}- 5, 16-dioxo-6-{[ ({ (1R, 2R)-2-[ (4E)-5-(4, 4, 5, 5-tetramethyl-l, 3, 2-dioxaborolan-2-yl)pent-4-en-l- yl] cyclopropyl}oxy)carbonyl] amino}- 1, 2, 3, 6, 7, 8, 9,10,11,13a, 14, 15,16,16a- tetradecahydrocyclopropa[e]pyrrolo[l ,2-a] [1 , 4]diazacyclopentadecine-14a(5H)-carboxylate

To a 0°C solution of phenol from Example 296, Step 3 (868 mg) in DMF (6 mL) was added sodium hydride (38 mg). The mixture was stirred 10 minutes and 1,3- dibromopropane (1.98 mL) was added. The reaction mixture was stirred at the same temperature for 30 minutes and then it was warmed up to room temperature and stirred for 18 hours. The reaction was quenched with a saturated solution of ammonium chloride and extracted with ethyl acetate (3x). The combined organic fractions were dried over magnesium sulfate, filtered and concentrated. The residue was purified by flash chromatography (ISCO, 0 to 10 % methanol in dichloromethane) to give the title compound (602 mg) as a white foam.
Step 2 : ethyl (2R,6S,12Z,13aS,l 4aR,l 6aS)-2-({3-bromo-4-[3-(piperidin-l -yl)propoxy] quinolin- 2-yl}oxy)-5,16-dioxo-6-{[({(lR,2R)-2-[(4E)-5-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)pent- 4-en-l-yl]cyclopropyl}oxy)carbonyl] amino}- 1, 2, 3, 6, 7,8,9,10,11, 13a, 14,15,16,16a- ntadecine-14a(5H)-carboxylate

Piperidine (317 μΐ) was added to a solution of bromide from Step 1 (326 mg) in DMSO (2 mL) and the resulting mixture was stirred for 2 hours at room temperature. The
reaction was quenched with water and extracted with ethyl acetate (3x). The combined organic fractions were washed with water then brine, dried over magnesium sulfate, filtered and concentrated. The residue was purified by flash chromatography (ISCO reverse phase, 0 to 95 % acetonitrile in water) to give the title compound (262 mg) as a mixture with the boronic acid. Step 3-5 : (laR,5S lZ 2aS,13aR,16S,19R,27E,31aR)-N-[(l-methylcyclopropyl)sulfonyl]- 3, 15, 33-trioxo-26-[3-(piperidin-l-yl)propoxy]-l, la, 3, 4, 5, 6, 7, 8, 9,10, 12a, 13, 15,16,18,
19, 29, 30, 31, 31a-icosahydro-5, 17:16, 19-dimethanodicyclopropa[l 2,13:28, 29] [1, 20, 3, 14, 17]
4H)-carboxamide

The title compound was prepared using the same method as Example 296, Steps 5-7. LCMS (ES+) m/z 901.20 (M+H)+.
Examples 303-304
By following the procedures outlined in Example 7 and using the appropriate reagents (depicted below the structure as Rg.), the following compounds were prepared.

[(1 -methylcyclopropyl) sulfonyl]-26-[3-(4-methyl
piperazin- 1 -yl)propoxy] -3 ,15,33 -trioxo- 1 , 1 a,3 ,4,
Rg. 1 -methyl 5,6,7,8,9,10,12a,13, 15,16,18,19,29,30,31,31a- piperazine icosahydro-5, 17:16,19-dimethanodicyclopropa
[ 12, 13 :28,29] [ 1 ,20,3 , 14, 17]dioxatriazacyclonona
cosino[21 ,22-b quinoline- 13 a( 14H)-carboxamide
Example 305 : (laj?,5SJ lZ,12aSJ3ai?,16S,19i?,31ai?V26-methoxy-iV-ra- methylcvclopropynsulfonyll-3.15.33-trioxo-l,la,3,4.5.6.7.8.9.10J2a,13J5.16.18.19,
27,28,29,30,31 1a-docosahvdro-5.17:16,19-dimethanodicvclopropan2.13:28.29in.20 J4J71 dioxatriazacvclononacosino 21,22- ?1quinoline-13a(14H)-carboxamide

Bismuth trichloride (91 mg) was added to a solution of Example 300 (22.8 mg) in ethanol (2.9 mL) and cooled to 0°C. Then, potassium borohydride (156 mg) was added in small portions. The mixture was stirred at 0°C for 45 minutes then at room temperature for 18 hours. The reaction mixture was diluted with ethyl acetate and acidified with 4N HC1 until pH = 3. The mixture was filtered over celite, rinsing with ethyl acetate. The solvent was removed in vacuo. The residue was purified by flash chromatography (ISCO, 0 to 10% methanol in
dichloromethane) to give the title compound (5.3 mg) as a white solid. LRMS (ES+) m/z 792.25 (M+H)+.
Example 306 : (laj?.5Sa iZ.12aSa3a^J6Sa9ig31a^VN-r(l-methylcvclopropynsulfonyll- 3.15.33-trioxo-26-r2-(pyrrolidin-l-vnethoxyl-l,la.3A5,6,7.8.9.10.12a.l3.15.16.18.
19.27,28.29.30,31 ,31 a-docosahvdro-5.17: 16, 19-dimethanodicvclopropar 12.13 :28.291
Γ 1 ,20.3.14.17]dioxatriazacyclononacosino [21.22-^lquinoline- 13 a( 14H)-carboxamide

The title compound was prepared using the same method as Example 305 with Example 299 except that 200 equivalents of potassium borohydride and 20 equivalents of bismuth trichloride were used. LRMS (ES+) m/z 875.50 (M+H)+.
It will be appreciated that various of the above-discussed and other features and functions, or alternatives thereof, may be desirably combined into many other different systems or applications. Also that various presently unforeseen or unanticipated alternatives, modifications, variations or improvements therein may be subsequently made by those skilled in the art which are also intended to be encompassed by the following claims.
Claims
1. A compound of formula (I) or a pharmaceutically acceptable salt thereof:

Y is CH or N;
R1 is:
-OH,
-OCrealkyl,
-OCrealkyl-heth
-Od-ealkyl-OH,
-Od-6alkyl-NRaRb,
-O-heti,
-OCr6alkylC02H,
-Od-ealkylC^-het
-0(CH2)1-6OC(=0)CH2NRaRb,
-OC i -6alkyl-C i -6alkoxy,
-OC i -6alkyl-C i -6alkoxy-C i - alkoxy,
-OC(0)Ci-6alkyl,
-OC(0)NRaRb,
-Od-ealkyl-S-heti,
-OC i -6alkyl-phosphate,
a phosphate group,
-(CH^-het!, pyridinyl, or
thiazolyl;
wherein
said alkyl is optionally substituted with 1 or 2 fluoro substituents, said phosphate group is optionally substituted with 1, 2 or 3 C1-6alkyl; said hett is:
a) aryl selected from phenyl or napthyl optionally substituted with 1 or 2 substituents selected from -OH, C!-6alkyl, or halo;
b) heteroaryl selected from 5- and 6-membered aromatic rings having 1, 2 or 3 heteroatoms independently selected from N, O and S, wherein said heteroaryl is attached through a ring atom selected from C or N and optionally substituted with 1 or 2 substituents independently selected from d-6alkyl and -OH; or c) heterocycle selected from 4-7 membered monocyclic or 6-10 membered polycyclic bridged, linearly fused or spirocyclic saturated or unsaturated non-aromatic rings having 1, 2, 3 or 4 heteroatoms independently selected from N, O and S, wherein said heterocycle is attached through a ring atom selected from C or N and optionally substituted with 1 or 2 substituents independently selected from C1-6alkyl, oxo, -(CH2)mF, Boc,
-(CH2)mCF3, -(CH2)mOCF3, -OH, -NRaRb, -C1-6alkoxy,
-(CH2)mS02CH3, aryl, -C1-6alkoxy-C1-6alkyl, -C1-6alkyl-d- 6alkoxy optionally substituted with CF3, cyano, C(=0)NH2,
C3-6cycloalkyl, -C1-6alkyl-C3-6cycloalkyl, -COOC1-6alkyl, -Ci- 6alkyl-S02C1-6alkyl, and benzimidazolyl wherein the benzimidazolyl is optionally substituted with F;
Ra and Rb are independently selected from H; d-6 alkyl; t-Boc; aryl; C3-6cycloalkyl optionally substituted with 1 or 2 fluoro; d-6alkoxy- d-6alkyl; tetrahydropyranyl; d-6alkyl-OH; d-6alkyl-arylA; d-6 alkyl- C(OH)-arylA; d-ealkyl-imidazolyl optionally substituted with methyl, d-6alkyl-benzimidazolyl optionally substituted with methyl; d-6alkyl- pyrazolyl; C ealkyl-dihydrotriazole optionally substituted with oxo; or
Q-ealkyl-pyrrolidinyl optionally substituted with oxo;
wherein
m is 0 or 1 to 4;
said arylA is phenyl, naphthalenyl, tetrahydronapthalenyl, or 7-10 membered fused bicyclic ring structure wherein at least one of the rings is aromatic and is optionally substituted with 2 -OH;
said tetrahydropyranyl is optionally substituted with 1 oxo;
R2 is C 6alkyl, C2-6alkenyl, C3-C6cycloalkyl or NRcRd;
wherein
the C3-6cycloalkyl is optionally substituted with C1-6alkyl optionally substituted with -OH, morpholinyl, C1-6alkoxy, Ci-ealkoxy-C^alkoxy, C1-6alkoxy-phenyl, or Ci^alkenyl;
R° and Rd are independently H or C1-6alkyl, or may be taken together, with the N to which they are attached, to form a 4-7-membered monocyclic ring;
R3 is Ci-6alkyl, C2-6alkenyl, C3-C6cycloalkyl, CF2 or CF3;
R4 is C s alkyl, C3-8 cycloalkyl, Ci-g alkyl-C3-8 cycloalkyl, adamantyl, dihydroindenyl, or a 4-8 membered heterocycloalkyl having 1 or 2 heteroatoms selected from N, O, or S, wherein R4 is optionally substituted with one or two substituents independently selected from (Q- C6)alkyl, halo, and -0(C1-C6)alkyl; or
R3 and R4 together form heptene;
Z is C or N;
R5 is H or C!-6alkyl; or R5 is absent when Z is N;
W is a bond, O or NR;
R is H or C ealkyl;
X is absent or is halo, CF3, -OCHF2, -OCH2F, -OCD2F, -OCDF2, d-Qalkyl, Cj-6alkoxy, aryl, heteroaryl, or -0(CH2)1-6NRaRb;
A is absent, O or N;
B is (CH2)m; and
n is 1-4.
2. The compound according to claim 1, or a pharmaceutically acceptable salt thereof having a formula of

3. The compound according to claims 1 or 2, or a pharmaceutically acceptable salt thereof, wherein Z is C.
4. The compound according to claim 1, or a pharmaceutically acceptable salt thereof, having a formula of

5. The compound according to claim 1 , or a pharmaceutically acceptable salt thereof, having a formula of

7. The compound according to any one of claims 1 to 6, or a pharmaceutically acceptable salt thereof, wherein R3 is ethyl, ethylene, or cyclopropyl.
8. The compound according to claims 7, or a pharmaceutically acceptable salt thereof, wherein R3 is ethylene.
9. The compound according to any one of claims 1 to 8, or a pharmaceutically acceptable salt thereof, wherein R4 is propyl, t-butyl, cyclopentyl, cyclohexyl optionally substituted with 1 or 2 F, cyclohexylmethyl, methylcyclohexyl, methylcyclopentyl, dihydroindenyl, or tetrahydro-2H-pyranyl.
10. The compound according to claim 9, or a pharmaceutically acceptable salt thereof, wherein R4 is t-butyl, cyclopentyl, or cyclohexyl, 1-methylcyclohexyl, propan-2-yl, 2,3- dihydroindenyl, tetrahydro-2H-pyranyl, or cyclohexylmethyl.
11. The compound according to claim 9, or a pharmaceutically acceptable salt thereof, wherein R4 is t-butyl, cyclopentyl, or cyclohexyl, 1-methylcyclohexyl, 2,3- dihydroindenyl, or tetrahydro-2H-pyranyl.
12. The compound according to any one of claims 1 to 11 , or a pharmaceutically acceptable salt thereof, wherein n is 1 to 3.
13. The compound according to any one of claims 1 to 12, or a pharmaceutically acceptable salt thereof, wherein R is cyclopropyl, N(CH3)2, or azetidinyl, wherein the cyclopropyl is optionally substituted with methyl, CH(CH3)2, C(CH3)=CH2;
C(CH3)2OH, CH2CH2-morpholinyl, CH2OCH3, CH2OCH2CH2OCH3, or CH2OCH2-phenyl.
14. The compound according to claim 13, of a pharmaceutically acceptable salt thereof, wherein R2 is cyclopropyl, N(CH3)2, (methyl)cyclopropyl,
(methoxymethyl)cyclopropyl, [(benzyloxy)methyl] cyclopropyl, l-(prop-l-en-2-yl)cyclopropyl, or 1 -[2-^ο ηο1ΐη-4^1)6ΐ1^1^ο1ορπ^1.
15. The compound according to claim 13, or a pharmaceutically acceptable salt thereof, wherein R is cyclopropyl, N(CH3)2, (methyl)cyclopropyl, or 1- (methoxymethyl)cyclopropyl.
16. The compound according to any one of claims 1 to 15, or a pharmaceutically acceptable salt thereof, wherein R1 is -OH; -0-C1-6alkyl; -OC(0)C1-6alkyl;
-OC l -ealkyl-hett ; -OC \ -6alkyl-C \ -6alkoxy ; -OC t-ealkyl-C i -6alkoxy-C i -6alkoxy ;
-OCH2C(=0)-het1; -0(CH2)1-6 OC(=0)CH2NRaRb; -OC(0)NRaRb; -Od-ealkyl-phosphate;
-OC ealkyl-S-heti; -O-heti; -0-C1-6alkyl-OH optionally substituted with 1 or 2 fluoro substituents; or -OC1-6alkyl-NRaRb
wherein Ra and Rb are independently
H,
Ci-6 alkyl,
t-Boc, C3-6cycloalkyl optionally substituted with 1 or 2 fluoro substituents,
C 6 alkyl-Ci-6alkoxy,
Ci-6alkyl-OH, phenyl,
C i -6alkyl-pheny 1 ,
tetrahydropyranyl,
C1-6alkyl-C(OH)-phenyl,
naphthalenyl,
C t -6alkyl-naphthalenyl,
C i -6alkyl-dihydrooxopyrrolidinyl,
Crealkyl-benzimidazolyl optionally substituted with methyl, C i -6alkyl-pyrazolyl,
Crealkyl-triazole optionally substituted with oxo, or
Ci-ealkyl-imidazolyl optionally substituted with methyl.
17. The compound according to any one of claims 1 to 15, or a pharmaceutically acceptable salt thereof, wherein the heti is: phenyl; oxazepanyl;
oxooxazolidinyl; pyridinyl; pyrazolyl; pyrrolyl; tetrahydropyranyl, triazolyl optionally substituted with C1-6alkyl; dioxolanyl; oxoimidazolidinyl; morpholinyl optionally substituted with dimethyl or ethyl; pyrrolidinyl optionally substituted with 1 or 2 substituents independently selected from oxo, Boc, Ci-6alkyl, OH, C(0)NH2, dimethylamino, and methylsulfornyl;
piperidinyl optionally substituted with 1 or 2 substituents independently selected from C1-6alkyl, C1-6alkoxy, C1-6alkoxy-C1-6alkyl optionally substituted with CF3, cyclopropyl-C1-6alkyl, cyclopropyl, -(CH2)mF, OH, -Ci-6alkyl-S02C1-6alkyl, -(CH2)mCF3, -COOC1-6alkyl, Boc, and benzimidazol; imidizolyl; thiazolyl optionally substituted with methyl; azabicycloheptyl;
azaspiroheptyl; azaspirononyl; oxaazabiocycloheptyl; oxaazaspiroheptyl optionally substituted with methoxyethyl; azetidinyl optionally substituted with 1 or 2 substituents independently selected from C1-6alkyl, Ci-6alkoxy, cyano, fluoro, OH, phenyl and Boc; dioxidothiomorpholinyl; piperazinyl optionally substituted with 1 or 2 substituents independently selected from C1-6alkyl, C1-6alkyl-cyclopropyl, CF3, methylsulfonyl , Boc, and oxo; azabicyclooctyl substituted with Q. 6alkyl, C^ealkoxy-C^alkyl, -COOC1-6alkyl, or -(CH2)mCF3; oxaazabicyclononyl optionally substituted with Boc, C1-6alkyl, -COOC1-6alkyl, C1- alkoxy-C1-6alkyl or cyclopropylC1-6alkyl; or azabicyclooctanyl optionally substituted with C1-6alkyl.
18. The compound according to any one of claims 1 to 17, or a pharmaceutically acceptable salt thereof, wherein X is absent or selected from -Br, -CI, -F, methoxy, methyl, propanyl and CF3.
A compound according to claim 1 selected from:





20. A compound according to claim 1 selected from:
( 1 aR,5 S, 11 Z, 12aS, 13aR, 16S, 19R,27E,31 aR)-N-[(l -methylcyclopropyl)sulfonyl]- 3,15,33-trioxo-26-[2-(piperidin-l-yl)ethoxy]-l,la,3,4,5,6,7,8,9,10,12a,13,^
31 a-icosahydro-5 , 17:16,19-dimethanodicyclopropa[ 12,13 :28,29] [ 1 ,20,3 , 14, 17]
dioxatriazacyclononacosino [21 ,22-b]quinoline- 13a(l 4H)-carboxamide;
(lai?,5S,l lZ,12aS,13a£,16S,19i?,27E,31aR^
26-[2-(moφholin-4-yl)ethoxy]-3,15,33-trioxo-l,la,3,4,5,6,7,8,9,10,12a,13,15,16,18,19,29^ 31 a- icosahydro-5, 17: 16, 19-dimethanodicyclopropa[12, 13 :28,29] [1 ,20,3, 14, 17]
dioxatriazacyclononacosino [21 ,22-b]quinoline- 13a( 14H)-carboxamide;
( 1 ai?,5S, 11 Z, 12aS, 13afl, 16S, 19i?,27£,31 aR)-N-[( 1 -methylcyclopropyl)sulfonyl] - 26-[3-(mo holin-4-yl)propoxy]-3,15,33-trioxo-l,la,3,4,5,6,7,8,9,10,12a,13, 15,16,18,19,29,30, 31,31 a-icosahydro-5 , 17: 16,19-dimethanodicyclopropa[ 12,13 :28,29] [ 1 ,20,3 , 14, 17]
dioxatriazacyclononacosino [21 ,22-b] quinoline- 13a( 14H)-carboxamide; ( 1 aR,5S, 11 Z, 12aS, 13 aR, 16S, 19i?,27£,31 aR)-N- [( 1 -methylcyclopropyl)sulfonyl]-
3,15,33-trioxo-26-[2-^yrrolidin-l-yl)ethoxy]-l,la,3,4,5,6,7,8,9,10,12a,13,15,16,18,19,29,30,31^ 31 a-icosahydro-5 , 17:16,19-dimethanodicyclopropa[ 12,13 :28,29] [ 1 ,20,3 ,14,17]
dioxatriazacyclononacosino [21 ,22-6]quinoline- 13a( 14H)-carboxamide;
(1 ai?,5S, 11 Z, 12aS 13a^, 16S, 19i?,27E,31 ai?)-26-methoxy-N-[( 1 - methylcyclopropyl)sulfonyl]-3,15,33-trioxo-l,la,3,4,5,6,7,8,9,10,12a,13,15,16,18,19,29,30,31, 31 a-icosahydro-5 , 17:16,19-dimethanodicyclopropa[ 12,13 :28,29] [ 1 ,20,3 , 14, 17]
dioxatriazacyclononacosino[21,22-6]quinoline-13a(14H)-carboxamide;
" (laR,5S,l lZ,12aS,13aR,16S,19R,31aR)-N-[(l-methylcyclopropyl)sulfonyl]-26- [2-(morpholin-4-yl)ethoxy]-3,15,33-trioxo-l,la,3,4,5,6,7,8,9,10,12a,13,15,16,18,19,2
31,31 a-docosahydro-5, 17:16,19-dimethanodicyclopropa[ 12, 13 :28,29] [ 1 ,20,3 , 14, 17] dioxatriazacyclononacosino [21 ,22-b]quinoline- 13a( 14H)-carboxamide;
(laR,5S, 11Z, 12aS,l 3aR, 16S, 19i?,27E,31 aR)-N-[(\ -methylcyclopropyl)sulfonyl]- 3,15,33-trioxo-26-[3-(piperidin-l-yl)propoxy]-l,la,3,4,5,6,7,8,9,10,12a,13,15,16,18,l^ 31 a-icosahydro-5, 17:16,19-dimethanodicyclopropa[l 2, 13 :28,29] [1 ,20,3, 14, 17]
dioxatriazacyclononacosino [21 ,22-&]quinoline- 13a( 14H)-carboxamide;
( 1 aR,5S, 11 Z, 12aS, 13aR, 16S, 19i?,27E,31 aR)-N- [( 1 -methylcyclopropyl)sulfonyl]- 3,15,33 -trioxo-26-[3 -(pyrrolidin- 1 -yl)propoxy] - l,la,3,4,5,6,7,8,9,10,12a,13,15,16,18,19,29,30,31,31a-icosahydro-5,17:16,19- dimethanodicyclopropa[ 12,13 :28,29] [1 ,20,3 , 14, 17]dioxatriazacyclononacosino[21 ,22- 0]quinoline-13a(14H)-carboxamide; or
(1 aR,5S, 11Z,12aS,l 3ai?, 16S, 19i?,27E,31 aR)-N-[(l -methylcyclopropyl)sulfonyl]- 26-[3-(4-methylpiperazin-l-yl)propoxy]-3,15,33-trioxo-l,la,3,4,5,6,7,8,9,10,12a,13,15,16,18, 19,29,30,31,31a-icosahydro-5,17:16,19-dimethanodicyclopropa[12,13:28,29][l,20,3, 14,17] dioxatriazacyclononacosino [21 ,22-Z>]quinoline- 13a( 14H)-carboxamide,
or a pharmaceutically acceptable salt thereof.
21. A pharmaceutical composition comprising an effective amount of the compound according to any one of claims 1 to 20, and a pharmaceutically acceptable carrier.
22. The pharmaceutical composition according to claim 21, further comprising a second therapeutic agent selected from the group consisting of HCV antiviral agents, immunomodulators, and anti-infective agents.
23. The pharmaceutical composition according to claim 22, further comprising a second therapeutic agent selected from the group consisting of HCV protease inhibitors, HCV NS5B polymerase inhibitors, HCV NS5A and NS4B inhibitors, ribavarin and pegylated interferon.
24. The pharmaceutical composition according to claim 22, wherein the second therapeutic agent is ribavirin.
25. Use of the compound according to any one of claims 1 to 20 for inhibiting HCV NS3 protease activity in a subject in need thereof.
26. Use of the compound according to any one of claims 1 to 20 for preventing or treating infection by HCV in a subject in need thereof.
27. The use of claim 25 or 26 wherein the use is with interferon and ribavirin.
28. The pharmaceutical composition of any one of claims 21 to 24 for inhibiting HCV NS3 protease activity in a subject in need thereof.
29. The pharmaceutical composition of any one of claims 21 to 24 for preventing of treating infection by HCV in a subject in need thereof.
30. The pharmaceutical composition of any one of claims 21 to 24 for use as a medicament.
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US14/358,050 US9328138B2 (en) | 2011-11-15 | 2012-11-09 | HCV NS3 protease inhibitors |
| EP12850713.4A EP2780026B1 (en) | 2011-11-15 | 2012-11-09 | Hcv ns3 protease inhibitors |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201161560042P | 2011-11-15 | 2011-11-15 | |
| US61/560,042 | 2011-11-15 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2013074386A2 true WO2013074386A2 (en) | 2013-05-23 |
| WO2013074386A3 WO2013074386A3 (en) | 2015-06-18 |
Family
ID=48430338
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2012/064270 WO2013074386A2 (en) | 2011-11-15 | 2012-11-09 | Hcv ns3 protease inhibitors |
Country Status (3)
| Country | Link |
|---|---|
| US (1) | US9328138B2 (en) |
| EP (1) | EP2780026B1 (en) |
| WO (1) | WO2013074386A2 (en) |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8957203B2 (en) | 2011-05-05 | 2015-02-17 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| WO2015076310A1 (en) | 2013-11-20 | 2015-05-28 | 株式会社 三和化学研究所 | Novel 3-azabicyclo[3.1.0]hexane derivative and use thereof for medical purposes |
| US9296782B2 (en) | 2012-07-03 | 2016-03-29 | Gilead Sciences, Inc. | Inhibitors of hepatitis C virus |
| US9334279B2 (en) | 2012-11-02 | 2016-05-10 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9409943B2 (en) | 2012-11-05 | 2016-08-09 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9499550B2 (en) | 2012-10-19 | 2016-11-22 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9580463B2 (en) | 2013-03-07 | 2017-02-28 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9598433B2 (en) | 2012-11-02 | 2017-03-21 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9617310B2 (en) | 2013-03-15 | 2017-04-11 | Gilead Sciences, Inc. | Inhibitors of hepatitis C virus |
| US9643999B2 (en) | 2012-11-02 | 2017-05-09 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| EP3623364A1 (en) * | 2014-02-13 | 2020-03-18 | Ligand Pharmaceuticals, Inc. | Prodrug compounds and their uses |
| US11970482B2 (en) | 2018-01-09 | 2024-04-30 | Ligand Pharmaceuticals Inc. | Acetal compounds and therapeutic uses thereof |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2956138B1 (en) | 2013-02-15 | 2022-06-22 | Kala Pharmaceuticals, Inc. | Therapeutic compounds and uses thereof |
| ES2831625T3 (en) | 2013-02-20 | 2021-06-09 | Kala Pharmaceuticals Inc | Therapeutic compounds and their uses |
| US9688688B2 (en) | 2013-02-20 | 2017-06-27 | Kala Pharmaceuticals, Inc. | Crystalline forms of 4-((4-((4-fluoro-2-methyl-1H-indol-5-yl)oxy)-6-methoxyquinazolin-7-yl)oxy)-1-(2-oxa-7-azaspiro[3.5]nonan-7-yl)butan-1-one and uses thereof |
| US9890173B2 (en) | 2013-11-01 | 2018-02-13 | Kala Pharmaceuticals, Inc. | Crystalline forms of therapeutic compounds and uses thereof |
| MX355330B (en) | 2013-11-01 | 2018-04-16 | Kala Pharmaceuticals Inc | CRYSTALLINE FORMS OF THERAPEUTIC COMPOUNDS and USES THEREOF. |
| EP3455218A4 (en) | 2016-05-10 | 2019-12-18 | C4 Therapeutics, Inc. | C3-carbon linked glutarimide degronimers for target protein degradation |
| EP3454856B1 (en) | 2016-05-10 | 2024-09-11 | C4 Therapeutics, Inc. | Heterocyclic degronimers for target protein degradation |
| WO2017197036A1 (en) | 2016-05-10 | 2017-11-16 | C4 Therapeutics, Inc. | Spirocyclic degronimers for target protein degradation |
| JP2019533641A (en) | 2016-09-08 | 2019-11-21 | カラ ファーマシューティカルズ インコーポレイテッド | Crystalline forms of therapeutic compounds and uses thereof |
| EP3509423A4 (en) | 2016-09-08 | 2020-05-13 | Kala Pharmaceuticals, Inc. | Crystalline forms of therapeutic compounds and uses thereof |
| WO2018048747A1 (en) | 2016-09-08 | 2018-03-15 | Kala Pharmaceuticals, Inc. | Crystalline forms of therapeutic compounds and uses thereof |
| RU2650610C1 (en) | 2017-02-28 | 2018-04-16 | Васильевич Иващенко Александр | Antiviral composition and method of its application |
Citations (73)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3480613A (en) | 1967-07-03 | 1969-11-25 | Merck & Co Inc | 2-c or 3-c-alkylribofuranosyl - 1-substituted compounds and the nucleosides thereof |
| WO1997041211A1 (en) | 1996-04-30 | 1997-11-06 | Vertex Pharmaceuticals Incorporated | Molecules comprising an impdh-like binding pocket and encoded data storage medium capable of graphically displaying them |
| WO1998022496A2 (en) | 1996-11-18 | 1998-05-28 | F. Hoffmann-La Roche Ag | Antiviral peptide derivatives |
| WO1998046630A1 (en) | 1997-04-16 | 1998-10-22 | Peptide Therapeutics Limited | Hepatitis c ns3 protease inhibitors |
| WO1999007733A2 (en) | 1997-08-11 | 1999-02-18 | Boehringer Ingelheim (Canada) Ltd. | Hepatitis c inhibitor peptides |
| WO1999007734A2 (en) | 1997-08-11 | 1999-02-18 | Boehringer Ingelheim (Canada) Ltd. | Hepatitis c inhibitor peptide analogues |
| WO1999038888A2 (en) | 1998-02-02 | 1999-08-05 | Istituto Di Ricerche Di Biologia Molecolare | Peptide inhibitors of the serine protease activity associated to the ns3 protein of hcv, relevant uses and process of production |
| WO1999043691A1 (en) | 1998-02-25 | 1999-09-02 | Emory University | 2'-fluoronucleosides |
| WO1999050230A1 (en) | 1998-03-31 | 1999-10-07 | Vertex Pharmaceuticals Incorporated | Inhibitors of serine proteases, particularly hepatitis c virus ns3 protease |
| GB2337262A (en) | 1998-03-30 | 1999-11-17 | Hoffmann La Roche | Antiviral peptide derivatives |
| WO1999064442A1 (en) | 1998-06-10 | 1999-12-16 | Istituto Di Ricerche Di Biologia Molecolare P Angeletti S.P.A. | Peptide inhibitors of hepatitis c virus ns3 protease |
| WO2000009543A2 (en) | 1998-08-10 | 2000-02-24 | Boehringer Ingelheim (Canada) Ltd. | Hepatitis c inhibitor tri-peptides |
| WO2000025780A1 (en) | 1998-10-29 | 2000-05-11 | Bristol-Myers Squibb Company | Compounds derived from an amine nucleus that are inhibitors of impdh enzyme |
| WO2000059929A1 (en) | 1999-04-06 | 2000-10-12 | Boehringer Ingelheim (Canada) Ltd. | Macrocyclic peptides active against the hepatitis c virus |
| WO2001000622A1 (en) | 1999-06-25 | 2001-01-04 | Vertex Pharmaceuticals Incorporated | Prodrugs of carbamate inhibitors of impdh |
| WO2001047883A1 (en) | 1999-12-27 | 2001-07-05 | Japan Tobacco Inc. | Fused-ring compounds and use thereof as drugs |
| WO2001060379A1 (en) | 2000-02-15 | 2001-08-23 | Ribapharm Corp. | Nucleoside analogs with carboxamidine modified monocyclic base |
| WO2001068663A1 (en) | 2000-03-15 | 2001-09-20 | Ribapharm Corp. | Nucleoside compounds and uses thereof |
| WO2001077091A2 (en) | 2000-04-05 | 2001-10-18 | Tularik Inc. | Ns5b hcv polymerase inhibitors |
| WO2001079246A2 (en) | 2000-04-13 | 2001-10-25 | Pharmasset, Ltd. | 3'-or 2'-hydroxymethyl substituted nucleoside derivatives for treatment of hepatitis virus infections |
| WO2001090121A2 (en) | 2000-05-23 | 2001-11-29 | Idenix (Cayman) Limited | Methods and compositions for treating hepatitis c virus |
| WO2001092282A2 (en) | 2000-05-26 | 2001-12-06 | Idenix (Cayman) Limited | Methods and compositions for treating flaviviruses and pestiviruses |
| WO2002004425A2 (en) | 2000-07-06 | 2002-01-17 | Boehringer Ingelheim (Canada) Ltd. | Viral polymerase inhibitors |
| WO2002006246A1 (en) | 2000-07-19 | 2002-01-24 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti S.P.A. | Dihydroxypyrimidine carboxylic acids as viral polymerase inhibitors |
| US20020019363A1 (en) | 2000-02-18 | 2002-02-14 | Ismaili Hicham Moulay Alaoui | Method for the treatment or prevention of flavivirus infections using nucleoside analogues |
| WO2002018404A2 (en) | 2000-08-30 | 2002-03-07 | F. Hoffmann-La Roche Ag | Nucleoside derivatives for the treatment of hepatitis c |
| WO2002020497A1 (en) | 2000-09-01 | 2002-03-14 | Shionogi & Co., Ltd. | Compounds having anti-hepatitis c virus effect |
| WO2002032920A2 (en) | 2000-10-18 | 2002-04-25 | Pharmasset Limited | Modified nucleosides for treatment of viral infections and abnormal cellular proliferation |
| WO2002040019A1 (en) | 2000-11-15 | 2002-05-23 | Banyu Pharmaceutical Co.,Ltd. | Benzimidazole derivatives |
| WO2002048165A2 (en) | 2000-12-15 | 2002-06-20 | Pharmasset Ltd. | Antiviral agents for treatment of flaviviridae infections |
| WO2002048172A2 (en) | 2000-12-12 | 2002-06-20 | Schering Corporation | Diaryl peptides as ns3-serine protease inhibitors of hepatits c virus |
| WO2002048116A2 (en) | 2000-12-13 | 2002-06-20 | Bristol-Myers Squibb Pharma Company | Inhibitors of hepatitis c virus ns3 protease |
| WO2002051425A1 (en) | 2000-12-26 | 2002-07-04 | Mitsubishi Pharma Corporation | Remedies for hepatitis c |
| WO2002057287A2 (en) | 2001-01-22 | 2002-07-25 | Merck & Co., Inc. | Nucleoside derivatives as inhibitors of rna-dependent rna viral polymerase |
| US20020107138A1 (en) | 2000-08-10 | 2002-08-08 | Hoveyda Amir H. | Recyclable metathesis catalysts |
| WO2002100415A2 (en) | 2001-06-12 | 2002-12-19 | F. Hoffmann-La Roche Ag | 4'-substituted nucleosides for the treatment of diseases mediated by the hepatitis c virus |
| WO2003026589A2 (en) | 2001-09-28 | 2003-04-03 | Idenix (Cayman) Limited | Methods and compositions for treating hepatitis c virus using 4'-modified nucleosides |
| WO2003026675A1 (en) | 2001-09-28 | 2003-04-03 | Idenix (Cayman) Limited | Methods and compositions for treating flaviviruses and pestiviruses using 4'-modified nucleoside |
| WO2003029226A1 (en) | 2001-09-26 | 2003-04-10 | Basf Aktiengesellschaft | Heterocyclyl substituted phenoxyalkyl-, phenylthioalkyl-, phenylaminoalkyl- and phenylalkyl-sulfamoylcarboxamides |
| WO2003068244A1 (en) | 2002-02-13 | 2003-08-21 | Merck & Co., Inc. | Methods of inhibiting orthopoxvirus replication with nucleoside compounds |
| WO2003093290A2 (en) | 2002-05-06 | 2003-11-13 | Genelabs Technologies, Inc. | Nucleoside derivatives for treating hepatitis c virus infection |
| WO2004000858A2 (en) | 2002-06-21 | 2003-12-31 | Merck & Co., Inc. | Nucleoside derivatives as inhibitors of rna-dependent rna viral polymerase |
| WO2004002422A2 (en) | 2002-06-28 | 2004-01-08 | Idenix (Cayman) Limited | 2’-c-methyl-3’-o-l-valine ester ribofuranosyl cytidine for treatment of flaviviridae infections |
| WO2004003000A2 (en) | 2002-06-28 | 2004-01-08 | Idenix (Cayman) Limited | 1’-, 2'- and 3'- modified nucleoside derivatives for treating flaviviridae infections |
| WO2004002999A2 (en) | 2002-06-28 | 2004-01-08 | Idenix (Cayman) Limited | Modified 2' and 3' -nucleoside produgs for treating flaviridae infections |
| WO2004003138A2 (en) | 2002-06-27 | 2004-01-08 | Merck & Co., Inc. | Nucleoside derivatives as inhibitors of rna-dependent rna viral polymerase |
| WO2004007512A2 (en) | 2002-07-16 | 2004-01-22 | Merck & Co., Inc. | Nucleoside derivatives as inhibitors of rna-dependent rna viral polymerase |
| WO2004011478A2 (en) | 2002-07-25 | 2004-02-05 | Micrologix Biotech Inc. | Anti-viral 7-deaza d-nucleosides and uses thereof |
| WO2004013300A2 (en) | 2002-08-01 | 2004-02-12 | Pharmasset Inc. | Compounds with the bicyclo[4.2.1]nonane system for the treatment of flaviviridae infections |
| WO2004028481A2 (en) | 2002-09-30 | 2004-04-08 | Genelabs Technologies, Inc. | Nucleoside derivatives for treating hepatitis c virus infection |
| US6777392B2 (en) | 2002-03-28 | 2004-08-17 | Council Of Scientific And Industrial Research | 8-(C-β-D-glucopyranosyl)-7, 3', 4'-trihydroxyflavone, process of isolation thereof, pharmaceutical composition and method for the treatment of diabetes |
| US20040229818A1 (en) | 2003-03-05 | 2004-11-18 | Boehringer Ingelheim International Gmbh | Hepatitis C inhibitor compound |
| US20040229776A1 (en) | 2003-04-02 | 2004-11-18 | Boehringer Ingelheim International Gmbh | Pharmaceutical compositions for hepatitis C viral protease inhibitors |
| WO2005003147A2 (en) | 2003-05-30 | 2005-01-13 | Pharmasset, Inc. | Modified fluorinated nucleoside analogues |
| US20050020503A1 (en) | 2003-05-21 | 2005-01-27 | Boehringer Ingelheim International Gmbh | Hepatitis C inhibitor compounds |
| US20050038240A1 (en) | 2003-06-19 | 2005-02-17 | Roche Palo Alto Llc | Processes for preparing 4'-azido-nucleoside derivatives |
| WO2005016927A1 (en) | 2003-08-13 | 2005-02-24 | Japan Tobacco Inc. | Nitrogenous condensed-ring compound and use thereof as hiv integrase inhibitor |
| WO2005046712A1 (en) | 2003-11-12 | 2005-05-26 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2006021341A1 (en) | 2004-08-23 | 2006-03-02 | F. Hoffmann-La Roche Ag | Antiviral 4’-azido-nucleosides |
| WO2006102087A2 (en) | 2005-03-22 | 2006-09-28 | Merck & Co., Inc. | Hcv protease substrates |
| WO2008051514A2 (en) | 2006-10-24 | 2008-05-02 | Merck & Co., Inc. | Hcv ns3 protease inhibitors |
| WO2008057209A1 (en) | 2006-10-27 | 2008-05-15 | Merck & Co., Inc. | Hcv ns3 protease inhibitors |
| WO2008057208A2 (en) | 2006-10-27 | 2008-05-15 | Merck & Co., Inc. | Hcv ns3 protease inhibitors |
| WO2009055331A2 (en) | 2007-10-22 | 2009-04-30 | Schering Corporation | Bicyclic heterocycle derivatives and their use as modulators of the activity of gpr119 |
| WO2009061699A1 (en) | 2007-11-05 | 2009-05-14 | Schering Corporation | Gamma secretase modulators |
| WO2009101917A1 (en) | 2008-02-13 | 2009-08-20 | Eisai R & D Management Co., Ltd. | Bicycloamine derivative |
| WO2009108507A1 (en) | 2008-02-25 | 2009-09-03 | Merck & Co., Inc. | Therapeutic compounds |
| WO2009134624A1 (en) | 2008-04-28 | 2009-11-05 | Merck & Co., Inc. | Hcv ns3 protease inhibitors |
| WO2010011566A1 (en) | 2008-07-22 | 2010-01-28 | Merck & Co., Inc. | Macrocyclic quinoxaline compounds as hcv ns3 protease inhibitors |
| US7879792B2 (en) | 2005-06-02 | 2011-02-01 | The Regents Of The University Of Michigan | Synthetic peptide inhibitors of thrombin and thrombin activation of protease activated receptors 1 and 4 |
| WO2011014487A1 (en) | 2009-07-30 | 2011-02-03 | Merck Sharp & Dohme Corp. | Hepatitis c virus ns3 protease inhibitors |
| WO2011036576A1 (en) | 2009-09-23 | 2011-03-31 | Pfizer Inc. | Gpr 119 modulators |
| WO2012040040A1 (en) | 2010-09-21 | 2012-03-29 | Merck Sharp & Dohme Corp. | Hcv ns3 protease inhibitors |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2794806A (en) | 1954-04-29 | 1957-06-04 | Eastman Kodak Co | Nu-substituted piperidines |
| US5059609A (en) * | 1987-10-19 | 1991-10-22 | Pfizer Inc. | Substituted tetralins, chromans and related compounds in the treatment of asthma, arthritis and related diseases |
| US6982080B2 (en) | 2002-03-15 | 2006-01-03 | Wyeth | Hydroxyethyl starch—containing polypeptide compositions |
| MY140680A (en) | 2002-05-20 | 2010-01-15 | Bristol Myers Squibb Co | Hepatitis c virus inhibitors |
| US7135462B2 (en) | 2003-11-20 | 2006-11-14 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| CA2606195C (en) | 2005-05-02 | 2015-03-31 | Merck And Co., Inc. | Hcv ns3 protease inhibitors |
| TW200831085A (en) | 2006-12-13 | 2008-08-01 | Merck & Co Inc | Non-nucleoside reverse transcriptase inhibitors |
| AU2009303483A1 (en) | 2008-10-15 | 2010-04-22 | Intermune, Inc. | Therapeutic antiviral peptides |
-
2012
- 2012-11-09 WO PCT/US2012/064270 patent/WO2013074386A2/en active Application Filing
- 2012-11-09 EP EP12850713.4A patent/EP2780026B1/en not_active Not-in-force
- 2012-11-09 US US14/358,050 patent/US9328138B2/en active Active
Patent Citations (82)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3480613A (en) | 1967-07-03 | 1969-11-25 | Merck & Co Inc | 2-c or 3-c-alkylribofuranosyl - 1-substituted compounds and the nucleosides thereof |
| WO1997041211A1 (en) | 1996-04-30 | 1997-11-06 | Vertex Pharmaceuticals Incorporated | Molecules comprising an impdh-like binding pocket and encoded data storage medium capable of graphically displaying them |
| WO1998022496A2 (en) | 1996-11-18 | 1998-05-28 | F. Hoffmann-La Roche Ag | Antiviral peptide derivatives |
| WO1998046630A1 (en) | 1997-04-16 | 1998-10-22 | Peptide Therapeutics Limited | Hepatitis c ns3 protease inhibitors |
| WO1999007733A2 (en) | 1997-08-11 | 1999-02-18 | Boehringer Ingelheim (Canada) Ltd. | Hepatitis c inhibitor peptides |
| WO1999007734A2 (en) | 1997-08-11 | 1999-02-18 | Boehringer Ingelheim (Canada) Ltd. | Hepatitis c inhibitor peptide analogues |
| WO1999038888A2 (en) | 1998-02-02 | 1999-08-05 | Istituto Di Ricerche Di Biologia Molecolare | Peptide inhibitors of the serine protease activity associated to the ns3 protein of hcv, relevant uses and process of production |
| WO1999043691A1 (en) | 1998-02-25 | 1999-09-02 | Emory University | 2'-fluoronucleosides |
| GB2337262A (en) | 1998-03-30 | 1999-11-17 | Hoffmann La Roche | Antiviral peptide derivatives |
| WO1999050230A1 (en) | 1998-03-31 | 1999-10-07 | Vertex Pharmaceuticals Incorporated | Inhibitors of serine proteases, particularly hepatitis c virus ns3 protease |
| WO1999064442A1 (en) | 1998-06-10 | 1999-12-16 | Istituto Di Ricerche Di Biologia Molecolare P Angeletti S.P.A. | Peptide inhibitors of hepatitis c virus ns3 protease |
| US6323180B1 (en) | 1998-08-10 | 2001-11-27 | Boehringer Ingelheim (Canada) Ltd | Hepatitis C inhibitor tri-peptides |
| WO2000009543A2 (en) | 1998-08-10 | 2000-02-24 | Boehringer Ingelheim (Canada) Ltd. | Hepatitis c inhibitor tri-peptides |
| WO2000025780A1 (en) | 1998-10-29 | 2000-05-11 | Bristol-Myers Squibb Company | Compounds derived from an amine nucleus that are inhibitors of impdh enzyme |
| WO2000059929A1 (en) | 1999-04-06 | 2000-10-12 | Boehringer Ingelheim (Canada) Ltd. | Macrocyclic peptides active against the hepatitis c virus |
| WO2001000622A1 (en) | 1999-06-25 | 2001-01-04 | Vertex Pharmaceuticals Incorporated | Prodrugs of carbamate inhibitors of impdh |
| WO2001047883A1 (en) | 1999-12-27 | 2001-07-05 | Japan Tobacco Inc. | Fused-ring compounds and use thereof as drugs |
| WO2001060379A1 (en) | 2000-02-15 | 2001-08-23 | Ribapharm Corp. | Nucleoside analogs with carboxamidine modified monocyclic base |
| US20020019363A1 (en) | 2000-02-18 | 2002-02-14 | Ismaili Hicham Moulay Alaoui | Method for the treatment or prevention of flavivirus infections using nucleoside analogues |
| WO2001068663A1 (en) | 2000-03-15 | 2001-09-20 | Ribapharm Corp. | Nucleoside compounds and uses thereof |
| WO2001077091A2 (en) | 2000-04-05 | 2001-10-18 | Tularik Inc. | Ns5b hcv polymerase inhibitors |
| WO2001079246A2 (en) | 2000-04-13 | 2001-10-25 | Pharmasset, Ltd. | 3'-or 2'-hydroxymethyl substituted nucleoside derivatives for treatment of hepatitis virus infections |
| WO2001090121A2 (en) | 2000-05-23 | 2001-11-29 | Idenix (Cayman) Limited | Methods and compositions for treating hepatitis c virus |
| WO2001092282A2 (en) | 2000-05-26 | 2001-12-06 | Idenix (Cayman) Limited | Methods and compositions for treating flaviviruses and pestiviruses |
| WO2002004425A2 (en) | 2000-07-06 | 2002-01-17 | Boehringer Ingelheim (Canada) Ltd. | Viral polymerase inhibitors |
| WO2002006246A1 (en) | 2000-07-19 | 2002-01-24 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti S.P.A. | Dihydroxypyrimidine carboxylic acids as viral polymerase inhibitors |
| US20020107138A1 (en) | 2000-08-10 | 2002-08-08 | Hoveyda Amir H. | Recyclable metathesis catalysts |
| WO2002018404A2 (en) | 2000-08-30 | 2002-03-07 | F. Hoffmann-La Roche Ag | Nucleoside derivatives for the treatment of hepatitis c |
| WO2002020497A1 (en) | 2000-09-01 | 2002-03-14 | Shionogi & Co., Ltd. | Compounds having anti-hepatitis c virus effect |
| WO2002032920A2 (en) | 2000-10-18 | 2002-04-25 | Pharmasset Limited | Modified nucleosides for treatment of viral infections and abnormal cellular proliferation |
| WO2002040019A1 (en) | 2000-11-15 | 2002-05-23 | Banyu Pharmaceutical Co.,Ltd. | Benzimidazole derivatives |
| WO2002048172A2 (en) | 2000-12-12 | 2002-06-20 | Schering Corporation | Diaryl peptides as ns3-serine protease inhibitors of hepatits c virus |
| WO2002048116A2 (en) | 2000-12-13 | 2002-06-20 | Bristol-Myers Squibb Pharma Company | Inhibitors of hepatitis c virus ns3 protease |
| WO2002048165A2 (en) | 2000-12-15 | 2002-06-20 | Pharmasset Ltd. | Antiviral agents for treatment of flaviviridae infections |
| WO2002051425A1 (en) | 2000-12-26 | 2002-07-04 | Mitsubishi Pharma Corporation | Remedies for hepatitis c |
| WO2002057287A2 (en) | 2001-01-22 | 2002-07-25 | Merck & Co., Inc. | Nucleoside derivatives as inhibitors of rna-dependent rna viral polymerase |
| WO2002057425A2 (en) | 2001-01-22 | 2002-07-25 | Merck & Co., Inc. | Nucleoside derivatives as inhibitors of rna-dependent rna viral polymerase |
| US20040067901A1 (en) | 2001-01-22 | 2004-04-08 | Balkrishen Bhat | Nucleoside derivatives as inhibitors of RNA-dependent RNA viral polymerase |
| WO2002100415A2 (en) | 2001-06-12 | 2002-12-19 | F. Hoffmann-La Roche Ag | 4'-substituted nucleosides for the treatment of diseases mediated by the hepatitis c virus |
| US20030236216A1 (en) | 2001-06-12 | 2003-12-25 | Devos Rene Robert | 4'-substituted nucleoside derivatives as inhibitors of HCV RNA replication |
| WO2003029226A1 (en) | 2001-09-26 | 2003-04-10 | Basf Aktiengesellschaft | Heterocyclyl substituted phenoxyalkyl-, phenylthioalkyl-, phenylaminoalkyl- and phenylalkyl-sulfamoylcarboxamides |
| WO2003026675A1 (en) | 2001-09-28 | 2003-04-03 | Idenix (Cayman) Limited | Methods and compositions for treating flaviviruses and pestiviruses using 4'-modified nucleoside |
| US20040006007A1 (en) | 2001-09-28 | 2004-01-08 | Gilles Gosselin | Methods and compositions for treating hepatitis C virus using 4'-modified nucleosides |
| WO2003026589A2 (en) | 2001-09-28 | 2003-04-03 | Idenix (Cayman) Limited | Methods and compositions for treating hepatitis c virus using 4'-modified nucleosides |
| WO2003068244A1 (en) | 2002-02-13 | 2003-08-21 | Merck & Co., Inc. | Methods of inhibiting orthopoxvirus replication with nucleoside compounds |
| US6777392B2 (en) | 2002-03-28 | 2004-08-17 | Council Of Scientific And Industrial Research | 8-(C-β-D-glucopyranosyl)-7, 3', 4'-trihydroxyflavone, process of isolation thereof, pharmaceutical composition and method for the treatment of diabetes |
| WO2003093290A2 (en) | 2002-05-06 | 2003-11-13 | Genelabs Technologies, Inc. | Nucleoside derivatives for treating hepatitis c virus infection |
| US20040063658A1 (en) | 2002-05-06 | 2004-04-01 | Roberts Christopher Don | Nucleoside derivatives for treating hepatitis C virus infection |
| WO2004000858A2 (en) | 2002-06-21 | 2003-12-31 | Merck & Co., Inc. | Nucleoside derivatives as inhibitors of rna-dependent rna viral polymerase |
| WO2004003138A2 (en) | 2002-06-27 | 2004-01-08 | Merck & Co., Inc. | Nucleoside derivatives as inhibitors of rna-dependent rna viral polymerase |
| WO2004002999A2 (en) | 2002-06-28 | 2004-01-08 | Idenix (Cayman) Limited | Modified 2' and 3' -nucleoside produgs for treating flaviridae infections |
| WO2004003000A2 (en) | 2002-06-28 | 2004-01-08 | Idenix (Cayman) Limited | 1’-, 2'- and 3'- modified nucleoside derivatives for treating flaviviridae infections |
| WO2004002422A2 (en) | 2002-06-28 | 2004-01-08 | Idenix (Cayman) Limited | 2’-c-methyl-3’-o-l-valine ester ribofuranosyl cytidine for treatment of flaviviridae infections |
| WO2004007512A2 (en) | 2002-07-16 | 2004-01-22 | Merck & Co., Inc. | Nucleoside derivatives as inhibitors of rna-dependent rna viral polymerase |
| WO2004011478A2 (en) | 2002-07-25 | 2004-02-05 | Micrologix Biotech Inc. | Anti-viral 7-deaza d-nucleosides and uses thereof |
| WO2004013300A2 (en) | 2002-08-01 | 2004-02-12 | Pharmasset Inc. | Compounds with the bicyclo[4.2.1]nonane system for the treatment of flaviviridae infections |
| WO2004028481A2 (en) | 2002-09-30 | 2004-04-08 | Genelabs Technologies, Inc. | Nucleoside derivatives for treating hepatitis c virus infection |
| US20040229818A1 (en) | 2003-03-05 | 2004-11-18 | Boehringer Ingelheim International Gmbh | Hepatitis C inhibitor compound |
| US20040229776A1 (en) | 2003-04-02 | 2004-11-18 | Boehringer Ingelheim International Gmbh | Pharmaceutical compositions for hepatitis C viral protease inhibitors |
| US20050020503A1 (en) | 2003-05-21 | 2005-01-27 | Boehringer Ingelheim International Gmbh | Hepatitis C inhibitor compounds |
| WO2005003147A2 (en) | 2003-05-30 | 2005-01-13 | Pharmasset, Inc. | Modified fluorinated nucleoside analogues |
| US20050038240A1 (en) | 2003-06-19 | 2005-02-17 | Roche Palo Alto Llc | Processes for preparing 4'-azido-nucleoside derivatives |
| WO2005016927A1 (en) | 2003-08-13 | 2005-02-24 | Japan Tobacco Inc. | Nitrogenous condensed-ring compound and use thereof as hiv integrase inhibitor |
| WO2005046712A1 (en) | 2003-11-12 | 2005-05-26 | Bristol-Myers Squibb Company | Hepatitis c virus inhibitors |
| WO2006021341A1 (en) | 2004-08-23 | 2006-03-02 | F. Hoffmann-La Roche Ag | Antiviral 4’-azido-nucleosides |
| WO2006102087A2 (en) | 2005-03-22 | 2006-09-28 | Merck & Co., Inc. | Hcv protease substrates |
| US7879792B2 (en) | 2005-06-02 | 2011-02-01 | The Regents Of The University Of Michigan | Synthetic peptide inhibitors of thrombin and thrombin activation of protease activated receptors 1 and 4 |
| US20100093779A1 (en) | 2006-10-24 | 2010-04-15 | Liverton Nigel J | Hcv ns3 protease inhibitors |
| WO2008051514A2 (en) | 2006-10-24 | 2008-05-02 | Merck & Co., Inc. | Hcv ns3 protease inhibitors |
| WO2008057209A1 (en) | 2006-10-27 | 2008-05-15 | Merck & Co., Inc. | Hcv ns3 protease inhibitors |
| US20100286185A1 (en) | 2006-10-27 | 2010-11-11 | Nigel J Liverton | HCV NS3 Protease Inhibitors |
| WO2008057208A2 (en) | 2006-10-27 | 2008-05-15 | Merck & Co., Inc. | Hcv ns3 protease inhibitors |
| WO2009055331A2 (en) | 2007-10-22 | 2009-04-30 | Schering Corporation | Bicyclic heterocycle derivatives and their use as modulators of the activity of gpr119 |
| WO2009061699A1 (en) | 2007-11-05 | 2009-05-14 | Schering Corporation | Gamma secretase modulators |
| WO2009101917A1 (en) | 2008-02-13 | 2009-08-20 | Eisai R & D Management Co., Ltd. | Bicycloamine derivative |
| WO2009108507A1 (en) | 2008-02-25 | 2009-09-03 | Merck & Co., Inc. | Therapeutic compounds |
| WO2009134624A1 (en) | 2008-04-28 | 2009-11-05 | Merck & Co., Inc. | Hcv ns3 protease inhibitors |
| WO2010011566A1 (en) | 2008-07-22 | 2010-01-28 | Merck & Co., Inc. | Macrocyclic quinoxaline compounds as hcv ns3 protease inhibitors |
| WO2011014487A1 (en) | 2009-07-30 | 2011-02-03 | Merck Sharp & Dohme Corp. | Hepatitis c virus ns3 protease inhibitors |
| US20120121624A1 (en) | 2009-07-30 | 2012-05-17 | Merck Sharp & Dohme Corp | Hepatitis c virus ns3 protease inhibitors |
| WO2011036576A1 (en) | 2009-09-23 | 2011-03-31 | Pfizer Inc. | Gpr 119 modulators |
| WO2012040040A1 (en) | 2010-09-21 | 2012-03-29 | Merck Sharp & Dohme Corp. | Hcv ns3 protease inhibitors |
Non-Patent Citations (23)
| Title |
|---|
| A. R. GENNARO: "Remington's Pharmaceutical Sciences, 18th ed", 1990, MACK PUBLISHING CO. |
| ALLISON ET AL., AGENTS ACTION, vol. 44, 1993, pages 165 |
| BARTENSCHLAGER, INTERVIROLOGY, vol. 40, no. 5-6, 1997, pages 378 - 393 |
| CRABB, SCIENCE, vol. 294, 2001, pages 506 - 507 |
| DYMOCK ET AL., ANTIVIRAL CHEM. & CHEMOTHERAPY, vol. 11, 2000, pages 79 - 96 |
| DYMOCK, EMERGING DRUGS, vol. 6, 2001, pages 13 - 42 |
| FURSTNER ET AL., J. ORG. CHEM., vol. 64, 1999, pages 8275 |
| GALLINARI ET AL., BIOCHEM., vol. 38, 1999, pages 5620 - 32 |
| GALLINARI ET AL., J. VIROL., vol. 72, 1998, pages 6758 - 69 |
| HARRY-O'KURU ET AL., J. ORG. CHEM., vol. 62, 1997, pages 1754 - 59 |
| KINGSBURY ET AL., J. AM. CHEM. SOC., vol. 121, 1999, pages 791 |
| KIRSCHBAUM, ANAL. PROFILES DRUG SUBS., vol. 12, 1983, pages 1 - 36 |
| LAUER ET AL., N. ENGL. J. MED., vol. 345, 2001, pages 41 - 52 |
| LI ET AL., SYNLETT, vol. 5, 2006, pages 725 |
| MILLER ET AL., J. AM. CHEM. SOC., vol. 118, 1996, pages 9606 |
| MORADPOUR ET AL., EURO. J. GASTROENTEROL. HEPATOL., vol. 11, 1999, pages 1189 - 1202 |
| ROSEN ET AL., MOLEC. MED. TODAY, vol. 5, 1999, pages 393 - 399 |
| SCHOLL ET AL., ORG. LETT., vol. 1, 1999, pages 953 |
| TALIANI ET AL., ANAL. BIOCHEM., vol. 240, 1996, pages 60 - 67 |
| TETRAHEDRON LETT., vol. 48, no. 36, 2007, pages 6343 - 6347 |
| TRNKA ET AL., ACC. CHEM. RES., vol. 34, 2001, pages 18 |
| WOLFE ET AL., TET. LETT., vol. 36, 1995, pages 7611 - 14 |
| ZINK ET AL., J. ORG. CHEM., vol. 71, 2006, pages 202 |
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9527885B2 (en) | 2011-05-05 | 2016-12-27 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US8957203B2 (en) | 2011-05-05 | 2015-02-17 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US10603318B2 (en) | 2012-07-03 | 2020-03-31 | Gilead Pharmasset Llc | Inhibitors of hepatitis C virus |
| US9296782B2 (en) | 2012-07-03 | 2016-03-29 | Gilead Sciences, Inc. | Inhibitors of hepatitis C virus |
| US10335409B2 (en) | 2012-07-03 | 2019-07-02 | Gilead Pharmasset Llc | Inhibitors of hepatitis C virus |
| US9499550B2 (en) | 2012-10-19 | 2016-11-22 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9598433B2 (en) | 2012-11-02 | 2017-03-21 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9643999B2 (en) | 2012-11-02 | 2017-05-09 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9334279B2 (en) | 2012-11-02 | 2016-05-10 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9409943B2 (en) | 2012-11-05 | 2016-08-09 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9580463B2 (en) | 2013-03-07 | 2017-02-28 | Bristol-Myers Squibb Company | Hepatitis C virus inhibitors |
| US9617310B2 (en) | 2013-03-15 | 2017-04-11 | Gilead Sciences, Inc. | Inhibitors of hepatitis C virus |
| US9663463B2 (en) | 2013-11-20 | 2017-05-30 | Sanwa Kagaku Kenkyusho Co., Ltd. | 3-azabicyclo[3.1.0]hexane derivative and use thereof for medical purpose |
| WO2015076310A1 (en) | 2013-11-20 | 2015-05-28 | 株式会社 三和化学研究所 | Novel 3-azabicyclo[3.1.0]hexane derivative and use thereof for medical purposes |
| EP3623364A1 (en) * | 2014-02-13 | 2020-03-18 | Ligand Pharmaceuticals, Inc. | Prodrug compounds and their uses |
| JP2020073466A (en) * | 2014-02-13 | 2020-05-14 | リガンド・ファーマシューティカルズ・インコーポレイテッド | Prodrug compound and application thereof |
| US11278559B2 (en) | 2014-02-13 | 2022-03-22 | Ligand Pharmaceuticals Incorporated | Prodrug compounds and their uses |
| US11970482B2 (en) | 2018-01-09 | 2024-04-30 | Ligand Pharmaceuticals Inc. | Acetal compounds and therapeutic uses thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2780026B1 (en) | 2019-10-23 |
| US9328138B2 (en) | 2016-05-03 |
| EP2780026A4 (en) | 2016-06-08 |
| EP2780026A2 (en) | 2014-09-24 |
| WO2013074386A3 (en) | 2015-06-18 |
| US20140296136A1 (en) | 2014-10-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2780026B1 (en) | Hcv ns3 protease inhibitors | |
| EP2083844B1 (en) | Hcv ns3 protease inhibitors | |
| EP2086982B1 (en) | Hcv ns3 protease inhibitors | |
| JP4705984B2 (en) | Macrocyclic peptides as HCV NS3 protease inhibitors | |
| AU2006242475B2 (en) | HCV NS3 protease inhibitors | |
| AU2007309488B2 (en) | HCV NS3 protease inhibitors | |
| US8461107B2 (en) | HCV NS3 protease inhibitors | |
| CA2667146C (en) | Hcv ns3 protease inhibitors | |
| ES2446015T3 (en) | HCV NS3 protease inhibitors | |
| EP2410844B1 (en) | Inhibitors of hepatitis c virus replication | |
| AU2009217551B2 (en) | Therapeutic compounds | |
| EP2618665A1 (en) | Hcv ns3 protease inhibitors | |
| UA74546C2 (en) | Macrocyclic peptides having activity relative to hepatitis c virus, a pharmaceutical composition and use of the pharmaceutical composition | |
| KR20100038417A (en) | Antiviral compounds | |
| TW201307351A (en) | Hepatitis C virus inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12850713 Country of ref document: EP Kind code of ref document: A2 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14358050 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2012850713 Country of ref document: EP |